CN101501054A - 氨基磷酸酯烷化剂前体药物 - Google Patents
氨基磷酸酯烷化剂前体药物 Download PDFInfo
- Publication number
- CN101501054A CN101501054A CNA2006800300828A CN200680030082A CN101501054A CN 101501054 A CN101501054 A CN 101501054A CN A2006800300828 A CNA2006800300828 A CN A2006800300828A CN 200680030082 A CN200680030082 A CN 200680030082A CN 101501054 A CN101501054 A CN 101501054A
- Authority
- CN
- China
- Prior art keywords
- alkyl
- assorted
- group
- aryl
- alkylsulfonyloxy
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 239000000651 prodrug Substances 0.000 title claims abstract description 381
- 229940002612 prodrug Drugs 0.000 title claims abstract description 381
- PTMHPRAIXMAOOB-UHFFFAOYSA-L phosphoramidate Chemical compound NP([O-])([O-])=O PTMHPRAIXMAOOB-UHFFFAOYSA-L 0.000 title abstract description 404
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 250
- 201000011510 cancer Diseases 0.000 claims abstract description 187
- 150000001875 compounds Chemical class 0.000 claims description 251
- 229910052760 oxygen Inorganic materials 0.000 claims description 174
- 238000011282 treatment Methods 0.000 claims description 164
- 238000000034 method Methods 0.000 claims description 152
- 239000001301 oxygen Substances 0.000 claims description 132
- -1 heterocyclic radical Chemical class 0.000 claims description 131
- 229910052739 hydrogen Inorganic materials 0.000 claims description 119
- 125000000217 alkyl group Chemical group 0.000 claims description 115
- 239000001257 hydrogen Substances 0.000 claims description 107
- 125000003118 aryl group Chemical group 0.000 claims description 95
- 150000002431 hydrogen Chemical class 0.000 claims description 81
- 125000001072 heteroaryl group Chemical group 0.000 claims description 52
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 50
- 229910052717 sulfur Inorganic materials 0.000 claims description 45
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 41
- 125000003368 amide group Chemical group 0.000 claims description 37
- 125000005278 alkyl sulfonyloxy group Chemical group 0.000 claims description 35
- 150000003839 salts Chemical class 0.000 claims description 35
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 34
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 claims description 33
- 238000006243 chemical reaction Methods 0.000 claims description 33
- 229910052736 halogen Inorganic materials 0.000 claims description 31
- 229910052757 nitrogen Inorganic materials 0.000 claims description 30
- 125000003435 aroyl group Chemical group 0.000 claims description 29
- 125000000623 heterocyclic group Chemical group 0.000 claims description 29
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 29
- 125000006823 (C1-C6) acyl group Chemical group 0.000 claims description 28
- 125000002252 acyl group Chemical group 0.000 claims description 28
- 239000000460 chlorine Substances 0.000 claims description 28
- 150000002367 halogens Chemical class 0.000 claims description 25
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 23
- 229910052801 chlorine Inorganic materials 0.000 claims description 23
- 239000000203 mixture Substances 0.000 claims description 23
- 229910052794 bromium Inorganic materials 0.000 claims description 21
- 229910052799 carbon Inorganic materials 0.000 claims description 20
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 20
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 19
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 18
- 125000001118 alkylidene group Chemical group 0.000 claims description 16
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 16
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 13
- ZLRAAUUPULJGTL-UHFFFAOYSA-N diaminophosphinous acid Chemical compound NP(N)O ZLRAAUUPULJGTL-UHFFFAOYSA-N 0.000 claims description 12
- 150000003003 phosphines Chemical class 0.000 claims description 12
- 239000012453 solvate Substances 0.000 claims description 11
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 7
- 125000005604 azodicarboxylate group Chemical group 0.000 claims description 7
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 6
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 6
- JRTIVBKQIMBXIJ-UHFFFAOYSA-N P(OCCCl)(N)N Chemical compound P(OCCCl)(N)N JRTIVBKQIMBXIJ-UHFFFAOYSA-N 0.000 claims description 5
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 5
- 229910052698 phosphorus Inorganic materials 0.000 claims description 5
- 125000004454 (C1-C6) alkoxycarbonyl group Chemical group 0.000 claims description 4
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 4
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 4
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 claims description 4
- DIAYCDKWIYBLKQ-UHFFFAOYSA-N [O].NS(N)(=O)=O Chemical compound [O].NS(N)(=O)=O DIAYCDKWIYBLKQ-UHFFFAOYSA-N 0.000 claims description 4
- 230000015572 biosynthetic process Effects 0.000 claims description 4
- 239000000825 pharmaceutical preparation Substances 0.000 claims description 4
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 3
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 claims description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 2
- 125000005905 mesyloxy group Chemical group 0.000 claims description 2
- 125000004205 trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 claims description 2
- YUCFVHQCAFKDQG-UHFFFAOYSA-N fluoromethane Chemical compound F[CH] YUCFVHQCAFKDQG-UHFFFAOYSA-N 0.000 claims 5
- 125000006619 (C1-C6) dialkylamino group Chemical group 0.000 claims 2
- 125000003968 arylidene group Chemical group [H]C(c)=* 0.000 claims 2
- 239000002246 antineoplastic agent Substances 0.000 abstract description 10
- 229940034982 antineoplastic agent Drugs 0.000 abstract 1
- 206010021143 Hypoxia Diseases 0.000 description 103
- 210000004027 cell Anatomy 0.000 description 99
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 82
- 239000003814 drug Substances 0.000 description 82
- 239000002585 base Substances 0.000 description 73
- 239000002168 alkylating agent Substances 0.000 description 68
- 229940100198 alkylating agent Drugs 0.000 description 65
- 208000018875 hypoxemia Diseases 0.000 description 53
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 46
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 44
- 201000010099 disease Diseases 0.000 description 42
- 230000001146 hypoxic effect Effects 0.000 description 41
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 39
- 239000011541 reaction mixture Substances 0.000 description 37
- 230000000694 effects Effects 0.000 description 34
- 239000000243 solution Substances 0.000 description 31
- 239000003005 anticarcinogenic agent Substances 0.000 description 30
- 239000003795 chemical substances by application Substances 0.000 description 30
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 29
- 229940079593 drug Drugs 0.000 description 29
- 230000001988 toxicity Effects 0.000 description 29
- 231100000419 toxicity Toxicity 0.000 description 29
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical class OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 28
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Substances [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 28
- 125000000753 cycloalkyl group Chemical group 0.000 description 27
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 25
- 230000004913 activation Effects 0.000 description 25
- 229910052697 platinum Inorganic materials 0.000 description 25
- 238000002360 preparation method Methods 0.000 description 25
- 231100000135 cytotoxicity Toxicity 0.000 description 24
- 239000000126 substance Substances 0.000 description 23
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 23
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 21
- 210000001519 tissue Anatomy 0.000 description 21
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 20
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 20
- 229960001101 ifosfamide Drugs 0.000 description 20
- 230000009467 reduction Effects 0.000 description 20
- 230000002829 reductive effect Effects 0.000 description 20
- 230000003327 cancerostatic effect Effects 0.000 description 19
- 230000003013 cytotoxicity Effects 0.000 description 19
- 239000002904 solvent Substances 0.000 description 19
- 235000019508 mustard seed Nutrition 0.000 description 18
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 18
- 239000003513 alkali Substances 0.000 description 17
- 230000002062 proliferating effect Effects 0.000 description 17
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 17
- 125000004429 atom Chemical group 0.000 description 16
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 description 16
- 239000003112 inhibitor Substances 0.000 description 16
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 16
- 206010006187 Breast cancer Diseases 0.000 description 15
- 208000026310 Breast neoplasm Diseases 0.000 description 15
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 15
- 206010061535 Ovarian neoplasm Diseases 0.000 description 15
- 230000007541 cellular toxicity Effects 0.000 description 15
- 238000002512 chemotherapy Methods 0.000 description 15
- PSVUJBVBCOISSP-SPFKKGSWSA-N (2s,3r,4s,5s,6r)-2-bis(2-chloroethylamino)phosphoryloxy-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound OC[C@H]1O[C@@H](OP(=O)(NCCCl)NCCCl)[C@H](O)[C@@H](O)[C@@H]1O PSVUJBVBCOISSP-SPFKKGSWSA-N 0.000 description 14
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 14
- BKCJZNIZRWYHBN-UHFFFAOYSA-N Isophosphamide mustard Chemical compound ClCCNP(=O)(O)NCCCl BKCJZNIZRWYHBN-UHFFFAOYSA-N 0.000 description 14
- 206010033128 Ovarian cancer Diseases 0.000 description 14
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 14
- 238000002648 combination therapy Methods 0.000 description 14
- 229950011595 glufosfamide Drugs 0.000 description 14
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 14
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 13
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 13
- 229960004562 carboplatin Drugs 0.000 description 13
- 239000008103 glucose Substances 0.000 description 13
- 238000002560 therapeutic procedure Methods 0.000 description 13
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 12
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 238000004440 column chromatography Methods 0.000 description 12
- 230000004060 metabolic process Effects 0.000 description 12
- 208000024891 symptom Diseases 0.000 description 12
- FBTUMDXHSRTGRV-ALTNURHMSA-N zorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(\C)=N\NC(=O)C=1C=CC=CC=1)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 FBTUMDXHSRTGRV-ALTNURHMSA-N 0.000 description 12
- VEEGZPWAAPPXRB-BJMVGYQFSA-N (3e)-3-(1h-imidazol-5-ylmethylidene)-1h-indol-2-one Chemical compound O=C1NC2=CC=CC=C2\C1=C/C1=CN=CN1 VEEGZPWAAPPXRB-BJMVGYQFSA-N 0.000 description 11
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical group C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 11
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 11
- 239000002253 acid Substances 0.000 description 11
- 125000003545 alkoxy group Chemical group 0.000 description 11
- 239000004037 angiogenesis inhibitor Substances 0.000 description 11
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 11
- 229960002949 fluorouracil Drugs 0.000 description 11
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 11
- RLOWWWKZYUNIDI-UHFFFAOYSA-N phosphinic chloride Chemical compound ClP=O RLOWWWKZYUNIDI-UHFFFAOYSA-N 0.000 description 11
- 229960001866 silicon dioxide Drugs 0.000 description 11
- 231100000331 toxic Toxicity 0.000 description 11
- 230000002588 toxic effect Effects 0.000 description 11
- 229960000641 zorubicin Drugs 0.000 description 11
- 101710183280 Topoisomerase Proteins 0.000 description 10
- 150000001721 carbon Chemical group 0.000 description 10
- 231100000433 cytotoxic Toxicity 0.000 description 10
- 231100000599 cytotoxic agent Toxicity 0.000 description 10
- 230000003203 everyday effect Effects 0.000 description 10
- 125000005647 linker group Chemical group 0.000 description 10
- 125000003884 phenylalkyl group Chemical group 0.000 description 10
- 239000000741 silica gel Substances 0.000 description 10
- 229910002027 silica gel Inorganic materials 0.000 description 10
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 10
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 10
- 229960004528 vincristine Drugs 0.000 description 10
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 9
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 9
- 206010025323 Lymphomas Diseases 0.000 description 9
- JZTPOMIFAFKKSK-UHFFFAOYSA-N O-phosphonohydroxylamine Chemical compound NOP(O)(O)=O JZTPOMIFAFKKSK-UHFFFAOYSA-N 0.000 description 9
- 230000008859 change Effects 0.000 description 9
- 230000001472 cytotoxic effect Effects 0.000 description 9
- 229910052805 deuterium Inorganic materials 0.000 description 9
- 235000019439 ethyl acetate Nutrition 0.000 description 9
- 229960005420 etoposide Drugs 0.000 description 9
- 230000007954 hypoxia Effects 0.000 description 9
- 210000004185 liver Anatomy 0.000 description 9
- 229960001924 melphalan Drugs 0.000 description 9
- 230000004048 modification Effects 0.000 description 9
- 238000012986 modification Methods 0.000 description 9
- 238000011275 oncology therapy Methods 0.000 description 9
- 239000012044 organic layer Substances 0.000 description 9
- 230000002194 synthesizing effect Effects 0.000 description 9
- 230000009466 transformation Effects 0.000 description 9
- 210000004881 tumor cell Anatomy 0.000 description 9
- 230000003442 weekly effect Effects 0.000 description 9
- 108020004414 DNA Proteins 0.000 description 8
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 8
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 8
- 206010060862 Prostate cancer Diseases 0.000 description 8
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 8
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 8
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 8
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 8
- 230000002354 daily effect Effects 0.000 description 8
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 8
- 229940087004 mustargen Drugs 0.000 description 8
- 230000008569 process Effects 0.000 description 8
- 102000004169 proteins and genes Human genes 0.000 description 8
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 8
- 229960001196 thiotepa Drugs 0.000 description 8
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 7
- 206010009944 Colon cancer Diseases 0.000 description 7
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 7
- 229940122255 Microtubule inhibitor Drugs 0.000 description 7
- 208000034578 Multiple myelomas Diseases 0.000 description 7
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 7
- 229930012538 Paclitaxel Natural products 0.000 description 7
- 206010035226 Plasma cell myeloma Diseases 0.000 description 7
- 206010039491 Sarcoma Diseases 0.000 description 7
- 230000008901 benefit Effects 0.000 description 7
- 230000000973 chemotherapeutic effect Effects 0.000 description 7
- 229960004630 chlorambucil Drugs 0.000 description 7
- 201000010989 colorectal carcinoma Diseases 0.000 description 7
- 229940127089 cytotoxic agent Drugs 0.000 description 7
- 239000002254 cytotoxic agent Substances 0.000 description 7
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 7
- 230000036541 health Effects 0.000 description 7
- 238000001727 in vivo Methods 0.000 description 7
- 238000002347 injection Methods 0.000 description 7
- 239000007924 injection Substances 0.000 description 7
- 208000020816 lung neoplasm Diseases 0.000 description 7
- 231100000782 microtubule inhibitor Toxicity 0.000 description 7
- 239000003921 oil Substances 0.000 description 7
- 229960001592 paclitaxel Drugs 0.000 description 7
- 239000000047 product Substances 0.000 description 7
- 239000003223 protective agent Substances 0.000 description 7
- 108090000623 proteins and genes Proteins 0.000 description 7
- 150000003254 radicals Chemical class 0.000 description 7
- 238000000926 separation method Methods 0.000 description 7
- 230000004083 survival effect Effects 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- 230000004614 tumor growth Effects 0.000 description 7
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 6
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- 206010005003 Bladder cancer Diseases 0.000 description 6
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 6
- 241000699666 Mus <mouse, genus> Species 0.000 description 6
- 238000005481 NMR spectroscopy Methods 0.000 description 6
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 6
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 6
- ITXSBKZSECZAGA-UHFFFAOYSA-N [amino(chlorooxy)phosphoryl] hypochlorite Chemical compound NP(=O)(OCl)OCl ITXSBKZSECZAGA-UHFFFAOYSA-N 0.000 description 6
- 150000001408 amides Chemical class 0.000 description 6
- 229910052786 argon Inorganic materials 0.000 description 6
- 125000003710 aryl alkyl group Chemical group 0.000 description 6
- 230000037396 body weight Effects 0.000 description 6
- 229960002092 busulfan Drugs 0.000 description 6
- 229960005243 carmustine Drugs 0.000 description 6
- 230000001413 cellular effect Effects 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 229940125846 compound 25 Drugs 0.000 description 6
- 230000000857 drug effect Effects 0.000 description 6
- 238000003818 flash chromatography Methods 0.000 description 6
- 239000007789 gas Substances 0.000 description 6
- 230000012010 growth Effects 0.000 description 6
- 125000005842 heteroatom Chemical group 0.000 description 6
- 238000010253 intravenous injection Methods 0.000 description 6
- 208000032839 leukemia Diseases 0.000 description 6
- 201000005202 lung cancer Diseases 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- OSTGTTZJOCZWJG-UHFFFAOYSA-N nitrosourea Chemical compound NC(=O)N=NO OSTGTTZJOCZWJG-UHFFFAOYSA-N 0.000 description 6
- 238000005406 washing Methods 0.000 description 6
- FTBBGQKRYUTLMP-UHFFFAOYSA-N 2-nitro-1h-pyrrole Chemical compound [O-][N+](=O)C1=CC=CN1 FTBBGQKRYUTLMP-UHFFFAOYSA-N 0.000 description 5
- 108010006654 Bleomycin Proteins 0.000 description 5
- 108010092160 Dactinomycin Proteins 0.000 description 5
- VSNHCAURESNICA-UHFFFAOYSA-N Hydroxyurea Chemical compound NC(=O)NO VSNHCAURESNICA-UHFFFAOYSA-N 0.000 description 5
- 208000007766 Kaposi sarcoma Diseases 0.000 description 5
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 5
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 5
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 5
- 150000001336 alkenes Chemical class 0.000 description 5
- 229940041181 antineoplastic drug Drugs 0.000 description 5
- 230000000981 bystander Effects 0.000 description 5
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 5
- 229960003901 dacarbazine Drugs 0.000 description 5
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 5
- 125000006575 electron-withdrawing group Chemical group 0.000 description 5
- 208000021045 exocrine pancreatic carcinoma Diseases 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 5
- 229910052744 lithium Inorganic materials 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 206010061289 metastatic neoplasm Diseases 0.000 description 5
- 238000010172 mouse model Methods 0.000 description 5
- 239000002777 nucleoside Substances 0.000 description 5
- 150000003833 nucleoside derivatives Chemical class 0.000 description 5
- MUMZUERVLWJKNR-UHFFFAOYSA-N oxoplatinum Chemical compound [Pt]=O MUMZUERVLWJKNR-UHFFFAOYSA-N 0.000 description 5
- 239000008194 pharmaceutical composition Substances 0.000 description 5
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 5
- 238000001959 radiotherapy Methods 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 238000010189 synthetic method Methods 0.000 description 5
- 229960004964 temozolomide Drugs 0.000 description 5
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 5
- 238000012546 transfer Methods 0.000 description 5
- 201000005112 urinary bladder cancer Diseases 0.000 description 5
- 230000002792 vascular Effects 0.000 description 5
- 229960003048 vinblastine Drugs 0.000 description 5
- 238000002689 xenotransplantation Methods 0.000 description 5
- JCZPMGDSEAFWDY-SQOUGZDYSA-N (2r,3s,4r,5r)-2,3,4,5,6-pentahydroxyhexanamide Chemical compound NC(=O)[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO JCZPMGDSEAFWDY-SQOUGZDYSA-N 0.000 description 4
- VRYALKFFQXWPIH-PBXRRBTRSA-N (3r,4s,5r)-3,4,5,6-tetrahydroxyhexanal Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)CC=O VRYALKFFQXWPIH-PBXRRBTRSA-N 0.000 description 4
- HNXQXTQTPAJEJL-UHFFFAOYSA-N 2-aminopteridin-4-ol Chemical compound C1=CN=C2NC(N)=NC(=O)C2=N1 HNXQXTQTPAJEJL-UHFFFAOYSA-N 0.000 description 4
- QSKPIOLLBIHNAC-UHFFFAOYSA-N 2-chloro-acetaldehyde Chemical compound ClCC=O QSKPIOLLBIHNAC-UHFFFAOYSA-N 0.000 description 4
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 4
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 4
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 4
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 4
- KCBAMQOKOLXLOX-BSZYMOERSA-N CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O Chemical compound CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O KCBAMQOKOLXLOX-BSZYMOERSA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 4
- 102000010911 Enzyme Precursors Human genes 0.000 description 4
- 108010062466 Enzyme Precursors Proteins 0.000 description 4
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 4
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 4
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 4
- LSDPWZHWYPCBBB-UHFFFAOYSA-N Methanethiol Chemical compound SC LSDPWZHWYPCBBB-UHFFFAOYSA-N 0.000 description 4
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 4
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 4
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 4
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 4
- 206010041067 Small cell lung cancer Diseases 0.000 description 4
- 239000005864 Sulphur Substances 0.000 description 4
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 4
- HGINCPLSRVDWNT-UHFFFAOYSA-N acrylaldehyde Natural products C=CC=O HGINCPLSRVDWNT-UHFFFAOYSA-N 0.000 description 4
- 150000001350 alkyl halides Chemical class 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 150000003863 ammonium salts Chemical class 0.000 description 4
- 150000001454 anthracenes Chemical class 0.000 description 4
- 230000001028 anti-proliverative effect Effects 0.000 description 4
- YTKUWDBFDASYHO-UHFFFAOYSA-N bendamustine Chemical compound ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 YTKUWDBFDASYHO-UHFFFAOYSA-N 0.000 description 4
- 229960002707 bendamustine Drugs 0.000 description 4
- 239000006227 byproduct Substances 0.000 description 4
- 125000004432 carbon atom Chemical group C* 0.000 description 4
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 230000022534 cell killing Effects 0.000 description 4
- 230000004663 cell proliferation Effects 0.000 description 4
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 4
- 229940125833 compound 23 Drugs 0.000 description 4
- 239000012141 concentrate Substances 0.000 description 4
- 238000009792 diffusion process Methods 0.000 description 4
- 239000003937 drug carrier Substances 0.000 description 4
- 229960004756 ethanol Drugs 0.000 description 4
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 4
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 4
- 208000005017 glioblastoma Diseases 0.000 description 4
- 229930182470 glycoside Natural products 0.000 description 4
- 208000014829 head and neck neoplasm Diseases 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 4
- 230000003211 malignant effect Effects 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 4
- 208000011645 metastatic carcinoma Diseases 0.000 description 4
- 229960000485 methotrexate Drugs 0.000 description 4
- 239000003607 modifier Substances 0.000 description 4
- 201000005962 mycosis fungoides Diseases 0.000 description 4
- 125000004433 nitrogen atom Chemical group N* 0.000 description 4
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 4
- 108020004707 nucleic acids Proteins 0.000 description 4
- 150000007523 nucleic acids Chemical class 0.000 description 4
- 102000039446 nucleic acids Human genes 0.000 description 4
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 4
- 230000010412 perfusion Effects 0.000 description 4
- 238000001050 pharmacotherapy Methods 0.000 description 4
- PTMHPRAIXMAOOB-UHFFFAOYSA-N phosphoramidic acid Chemical compound NP(O)(O)=O PTMHPRAIXMAOOB-UHFFFAOYSA-N 0.000 description 4
- 229960000624 procarbazine Drugs 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N pyridine Substances C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 4
- 229960004641 rituximab Drugs 0.000 description 4
- 208000000587 small cell lung carcinoma Diseases 0.000 description 4
- APSBXTVYXVQYAB-UHFFFAOYSA-M sodium docusate Chemical compound [Na+].CCCCC(CC)COC(=O)CC(S([O-])(=O)=O)C(=O)OCC(CC)CCCC APSBXTVYXVQYAB-UHFFFAOYSA-M 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 239000003053 toxin Substances 0.000 description 4
- 231100000765 toxin Toxicity 0.000 description 4
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 4
- STBLNCCBQMHSRC-BATDWUPUSA-N (2s)-n-[(3s,4s)-5-acetyl-7-cyano-4-methyl-1-[(2-methylnaphthalen-1-yl)methyl]-2-oxo-3,4-dihydro-1,5-benzodiazepin-3-yl]-2-(methylamino)propanamide Chemical compound O=C1[C@@H](NC(=O)[C@H](C)NC)[C@H](C)N(C(C)=O)C2=CC(C#N)=CC=C2N1CC1=C(C)C=CC2=CC=CC=C12 STBLNCCBQMHSRC-BATDWUPUSA-N 0.000 description 3
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 3
- 125000006700 (C1-C6) alkylthio group Chemical group 0.000 description 3
- UPGZJCURPNZRSN-UHFFFAOYSA-N (chloroamino)phosphonic acid Chemical compound OP(O)(=O)NCl UPGZJCURPNZRSN-UHFFFAOYSA-N 0.000 description 3
- ZSJLQEPLLKMAKR-YDEIVXIUSA-N 1-methyl-1-nitroso-3-[(3r,4r,5s,6r)-2,4,5-trihydroxy-6-(hydroxymethyl)oxan-3-yl]urea Chemical compound O=NN(C)C(=O)N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-YDEIVXIUSA-N 0.000 description 3
- CQOQDQWUFQDJMK-SSTWWWIQSA-N 2-methoxy-17beta-estradiol Chemical compound C([C@@H]12)C[C@]3(C)[C@@H](O)CC[C@H]3[C@@H]1CCC1=C2C=C(OC)C(O)=C1 CQOQDQWUFQDJMK-SSTWWWIQSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 3
- 208000036762 Acute promyelocytic leukaemia Diseases 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 3
- 229940122815 Aromatase inhibitor Drugs 0.000 description 3
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 208000003174 Brain Neoplasms Diseases 0.000 description 3
- 239000004215 Carbon black (E152) Substances 0.000 description 3
- 201000009030 Carcinoma Diseases 0.000 description 3
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 3
- 101710112752 Cytotoxin Proteins 0.000 description 3
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 3
- 201000010915 Glioblastoma multiforme Diseases 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- LPGWZGMPDKDHEP-HLTPFJCJSA-N Leurosine Chemical compound C([C@]1([C@@H]2O1)CC)N(CCC=1C3=CC=CC=C3NC=11)C[C@H]2C[C@]1(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC LPGWZGMPDKDHEP-HLTPFJCJSA-N 0.000 description 3
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 3
- 206010027336 Menstruation delayed Diseases 0.000 description 3
- 102000029749 Microtubule Human genes 0.000 description 3
- 108091022875 Microtubule Proteins 0.000 description 3
- 206010029260 Neuroblastoma Diseases 0.000 description 3
- 229940121682 Osteoclast inhibitor Drugs 0.000 description 3
- HFVNWDWLWUCIHC-GUPDPFMOSA-N Prednimustine Chemical compound O=C([C@@]1(O)CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)[C@@H](O)C[C@@]21C)COC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 HFVNWDWLWUCIHC-GUPDPFMOSA-N 0.000 description 3
- 208000033826 Promyelocytic Acute Leukemia Diseases 0.000 description 3
- 208000004403 Prostatic Hyperplasia Diseases 0.000 description 3
- 201000004681 Psoriasis Diseases 0.000 description 3
- 208000015634 Rectal Neoplasms Diseases 0.000 description 3
- 208000034189 Sclerosis Diseases 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 208000005718 Stomach Neoplasms Diseases 0.000 description 3
- OUUQCZGPVNCOIJ-UHFFFAOYSA-M Superoxide Chemical compound [O-][O] OUUQCZGPVNCOIJ-UHFFFAOYSA-M 0.000 description 3
- NAVMQTYZDKMPEU-UHFFFAOYSA-N Targretin Chemical compound CC1=CC(C(CCC2(C)C)(C)C)=C2C=C1C(=C)C1=CC=C(C(O)=O)C=C1 NAVMQTYZDKMPEU-UHFFFAOYSA-N 0.000 description 3
- 206010047115 Vasculitis Diseases 0.000 description 3
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 3
- 125000003282 alkyl amino group Chemical group 0.000 description 3
- 125000004656 alkyl sulfonylamino group Chemical group 0.000 description 3
- PMMURAAUARKVCB-UHFFFAOYSA-N alpha-D-ara-dHexp Natural products OCC1OC(O)CC(O)C1O PMMURAAUARKVCB-UHFFFAOYSA-N 0.000 description 3
- CWNKMHIETKEBCA-UHFFFAOYSA-N alpha-Ethylaminohexanophenone Chemical compound CCCCC(NCC)C(=O)C1=CC=CC=C1 CWNKMHIETKEBCA-UHFFFAOYSA-N 0.000 description 3
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 239000005557 antagonist Substances 0.000 description 3
- 238000013459 approach Methods 0.000 description 3
- 239000003886 aromatase inhibitor Substances 0.000 description 3
- 229910052785 arsenic Inorganic materials 0.000 description 3
- RQNWIZPPADIBDY-UHFFFAOYSA-N arsenic atom Chemical compound [As] RQNWIZPPADIBDY-UHFFFAOYSA-N 0.000 description 3
- 229940120638 avastin Drugs 0.000 description 3
- 229960002756 azacitidine Drugs 0.000 description 3
- 230000033228 biological regulation Effects 0.000 description 3
- 206010005084 bladder transitional cell carcinoma Diseases 0.000 description 3
- 201000001528 bladder urothelial carcinoma Diseases 0.000 description 3
- 229960001561 bleomycin Drugs 0.000 description 3
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 3
- 208000003362 bronchogenic carcinoma Diseases 0.000 description 3
- 229960000590 celecoxib Drugs 0.000 description 3
- 230000010261 cell growth Effects 0.000 description 3
- 230000001684 chronic effect Effects 0.000 description 3
- 210000001072 colon Anatomy 0.000 description 3
- 229940125878 compound 36 Drugs 0.000 description 3
- 239000002619 cytotoxin Substances 0.000 description 3
- 229960000975 daunorubicin Drugs 0.000 description 3
- 230000007812 deficiency Effects 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 238000009826 distribution Methods 0.000 description 3
- 229960003668 docetaxel Drugs 0.000 description 3
- 229940088598 enzyme Drugs 0.000 description 3
- 239000000262 estrogen Substances 0.000 description 3
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 3
- 238000011156 evaluation Methods 0.000 description 3
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 3
- 229960000390 fludarabine Drugs 0.000 description 3
- 201000010536 head and neck cancer Diseases 0.000 description 3
- 229930195733 hydrocarbon Natural products 0.000 description 3
- 125000001183 hydrocarbyl group Chemical group 0.000 description 3
- 229960002411 imatinib Drugs 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 3
- 239000000138 intercalating agent Substances 0.000 description 3
- 238000010255 intramuscular injection Methods 0.000 description 3
- 239000007927 intramuscular injection Substances 0.000 description 3
- DKYWVDODHFEZIM-UHFFFAOYSA-N ketoprofen Chemical compound OC(=O)C(C)C1=CC=CC(C(=O)C=2C=CC=CC=2)=C1 DKYWVDODHFEZIM-UHFFFAOYSA-N 0.000 description 3
- WDRYRZXSPDWGEB-UHFFFAOYSA-N lonidamine Chemical compound C12=CC=CC=C2C(C(=O)O)=NN1CC1=CC=C(Cl)C=C1Cl WDRYRZXSPDWGEB-UHFFFAOYSA-N 0.000 description 3
- 231100000682 maximum tolerated dose Toxicity 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 201000001441 melanoma Diseases 0.000 description 3
- 230000002503 metabolic effect Effects 0.000 description 3
- 210000004688 microtubule Anatomy 0.000 description 3
- 244000309715 mini pig Species 0.000 description 3
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 3
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 description 3
- 230000007935 neutral effect Effects 0.000 description 3
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 3
- 230000000269 nucleophilic effect Effects 0.000 description 3
- 210000001672 ovary Anatomy 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 230000036961 partial effect Effects 0.000 description 3
- 229960002340 pentostatin Drugs 0.000 description 3
- 230000035699 permeability Effects 0.000 description 3
- 230000003285 pharmacodynamic effect Effects 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- 150000008298 phosphoramidates Chemical group 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 229960004694 prednimustine Drugs 0.000 description 3
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 3
- 239000002599 prostaglandin synthase inhibitor Substances 0.000 description 3
- 206010038038 rectal cancer Diseases 0.000 description 3
- 201000001275 rectum cancer Diseases 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 206010039073 rheumatoid arthritis Diseases 0.000 description 3
- 239000012266 salt solution Substances 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 238000007614 solvation Methods 0.000 description 3
- 201000000498 stomach carcinoma Diseases 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- 150000003512 tertiary amines Chemical class 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 210000001550 testis Anatomy 0.000 description 3
- 229930192474 thiophene Natural products 0.000 description 3
- 229960003087 tioguanine Drugs 0.000 description 3
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 3
- 229960005267 tositumomab Drugs 0.000 description 3
- AYNNSCRYTDRFCP-UHFFFAOYSA-N triazene Chemical compound NN=N AYNNSCRYTDRFCP-UHFFFAOYSA-N 0.000 description 3
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 3
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 3
- 150000004917 tyrosine kinase inhibitor derivatives Chemical class 0.000 description 3
- ZOCKGBMQLCSHFP-KQRAQHLDSA-N valrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC(OC)=C4C(=O)C=3C(O)=C21)(O)C(=O)COC(=O)CCCC)[C@H]1C[C@H](NC(=O)C(F)(F)F)[C@H](O)[C@H](C)O1 ZOCKGBMQLCSHFP-KQRAQHLDSA-N 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- XRASPMIURGNCCH-UHFFFAOYSA-N zoledronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CN1C=CN=C1 XRASPMIURGNCCH-UHFFFAOYSA-N 0.000 description 3
- AADVCYNFEREWOS-UHFFFAOYSA-N (+)-DDM Natural products C=CC=CC(C)C(OC(N)=O)C(C)C(O)C(C)CC(C)=CC(C)C(O)C(C)C=CC(O)CC1OC(=O)C(C)C(O)C1C AADVCYNFEREWOS-UHFFFAOYSA-N 0.000 description 2
- ABJSOROVZZKJGI-OCYUSGCXSA-N (1r,2r,4r)-2-(4-bromophenyl)-n-[(4-chlorophenyl)-(2-fluoropyridin-4-yl)methyl]-4-morpholin-4-ylcyclohexane-1-carboxamide Chemical compound C1=NC(F)=CC(C(NC(=O)[C@H]2[C@@H](C[C@@H](CC2)N2CCOCC2)C=2C=CC(Br)=CC=2)C=2C=CC(Cl)=CC=2)=C1 ABJSOROVZZKJGI-OCYUSGCXSA-N 0.000 description 2
- NECZZOFFLFZNHL-XVGZVFJZSA-N (2s)-2-amino-5-[[(2r)-3-[2-[bis[bis(2-chloroethyl)amino]-oxidophosphaniumyl]oxyethylsulfonyl]-1-[[(r)-carboxy(phenyl)methyl]amino]-1-oxopropan-2-yl]amino]-5-oxopentanoic acid;hydron;chloride Chemical compound Cl.ClCCN(CCCl)P(=O)(N(CCCl)CCCl)OCCS(=O)(=O)C[C@H](NC(=O)CC[C@H](N)C(O)=O)C(=O)N[C@@H](C(O)=O)C1=CC=CC=C1 NECZZOFFLFZNHL-XVGZVFJZSA-N 0.000 description 2
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 2
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 description 2
- QFWCYNPOPKQOKV-UHFFFAOYSA-N 2-(2-amino-3-methoxyphenyl)chromen-4-one Chemical compound COC1=CC=CC(C=2OC3=CC=CC=C3C(=O)C=2)=C1N QFWCYNPOPKQOKV-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- ONRREFWJTRBDRA-UHFFFAOYSA-N 2-chloroethanamine;hydron;chloride Chemical compound [Cl-].[NH3+]CCCl ONRREFWJTRBDRA-UHFFFAOYSA-N 0.000 description 2
- FGSHJLJPYBUBHO-UHFFFAOYSA-N 2-chloroethyl(methyl)azanium;chloride Chemical compound [Cl-].C[NH2+]CCCl FGSHJLJPYBUBHO-UHFFFAOYSA-N 0.000 description 2
- YZEUHQHUFTYLPH-UHFFFAOYSA-N 2-nitroimidazole Chemical compound [O-][N+](=O)C1=NC=CN1 YZEUHQHUFTYLPH-UHFFFAOYSA-N 0.000 description 2
- JIZRGGUCOQKGQD-UHFFFAOYSA-N 2-nitrothiophene Chemical compound [O-][N+](=O)C1=CC=CS1 JIZRGGUCOQKGQD-UHFFFAOYSA-N 0.000 description 2
- 125000005809 3,4,5-trimethoxyphenyl group Chemical group [H]C1=C(OC([H])([H])[H])C(OC([H])([H])[H])=C(OC([H])([H])[H])C([H])=C1* 0.000 description 2
- WUIABRMSWOKTOF-OYALTWQYSA-N 3-[[2-[2-[2-[[(2s,3r)-2-[[(2s,3s,4r)-4-[[(2s,3r)-2-[[6-amino-2-[(1s)-3-amino-1-[[(2s)-2,3-diamino-3-oxopropyl]amino]-3-oxopropyl]-5-methylpyrimidine-4-carbonyl]amino]-3-[(2r,3s,4s,5s,6s)-3-[(2r,3s,4s,5r,6r)-4-carbamoyloxy-3,5-dihydroxy-6-(hydroxymethyl)ox Chemical compound OS([O-])(=O)=O.N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C WUIABRMSWOKTOF-OYALTWQYSA-N 0.000 description 2
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 2
- ULTTYPMRMMDONC-UHFFFAOYSA-N 5-[(2,5-dihydroxyphenyl)methyl-[(2-hydroxyphenyl)methyl]amino]-2-hydroxybenzoic acid Chemical compound C1=C(O)C(C(=O)O)=CC(N(CC=2C(=CC=CC=2)O)CC=2C(=CC=C(O)C=2)O)=C1 ULTTYPMRMMDONC-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- SHGAZHPCJJPHSC-ZVCIMWCZSA-N 9-cis-retinoic acid Chemical compound OC(=O)/C=C(\C)/C=C/C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-ZVCIMWCZSA-N 0.000 description 2
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 108010079709 Angiostatins Proteins 0.000 description 2
- 102000012936 Angiostatins Human genes 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- KAKZBPTYRLMSJV-UHFFFAOYSA-N Butadiene Chemical compound C=CC=C KAKZBPTYRLMSJV-UHFFFAOYSA-N 0.000 description 2
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 2
- 241000282693 Cercopithecidae Species 0.000 description 2
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 2
- 208000016192 Demyelinating disease Diseases 0.000 description 2
- 206010012305 Demyelination Diseases 0.000 description 2
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 2
- AADVCYNFEREWOS-OBRABYBLSA-N Discodermolide Chemical compound C=C\C=C/[C@H](C)[C@H](OC(N)=O)[C@@H](C)[C@H](O)[C@@H](C)C\C(C)=C/[C@H](C)[C@@H](O)[C@@H](C)\C=C/[C@@H](O)C[C@@H]1OC(=O)[C@H](C)[C@@H](O)[C@H]1C AADVCYNFEREWOS-OBRABYBLSA-N 0.000 description 2
- 239000004278 EU approved seasoning Substances 0.000 description 2
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 2
- 208000006168 Ewing Sarcoma Diseases 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 230000005526 G1 to G0 transition Effects 0.000 description 2
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- 102100032742 Histone-lysine N-methyltransferase SETD2 Human genes 0.000 description 2
- 101001046870 Homo sapiens Hypoxia-inducible factor 1-alpha Proteins 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 2
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 2
- 102000015696 Interleukins Human genes 0.000 description 2
- 108010063738 Interleukins Proteins 0.000 description 2
- 102000004195 Isomerases Human genes 0.000 description 2
- 108090000769 Isomerases Proteins 0.000 description 2
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 2
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 2
- 206010027476 Metastases Diseases 0.000 description 2
- 125000003047 N-acetyl group Chemical group 0.000 description 2
- 102000004960 NAD(P)H dehydrogenase (quinone) Human genes 0.000 description 2
- 108020000284 NAD(P)H dehydrogenase (quinone) Proteins 0.000 description 2
- 108010045510 NADPH-Ferrihemoprotein Reductase Proteins 0.000 description 2
- BLXXJMDCKKHMKV-UHFFFAOYSA-N Nabumetone Chemical compound C1=C(CCC(C)=O)C=CC2=CC(OC)=CC=C21 BLXXJMDCKKHMKV-UHFFFAOYSA-N 0.000 description 2
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- SHGAZHPCJJPHSC-UHFFFAOYSA-N Panrexin Chemical compound OC(=O)C=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-UHFFFAOYSA-N 0.000 description 2
- 101150003085 Pdcl gene Proteins 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- 201000000582 Retinoblastoma Diseases 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 208000021712 Soft tissue sarcoma Diseases 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 108700011582 TER 286 Proteins 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- 208000033781 Thyroid carcinoma Diseases 0.000 description 2
- 208000024770 Thyroid neoplasm Diseases 0.000 description 2
- 102000004243 Tubulin Human genes 0.000 description 2
- 108090000704 Tubulin Proteins 0.000 description 2
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 2
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 2
- 150000001242 acetic acid derivatives Chemical class 0.000 description 2
- DZBUGLKDJFMEHC-UHFFFAOYSA-N acridine Chemical compound C1=CC=CC2=CC3=CC=CC=C3N=C21 DZBUGLKDJFMEHC-UHFFFAOYSA-N 0.000 description 2
- 229930183665 actinomycin Natural products 0.000 description 2
- 125000004442 acylamino group Chemical group 0.000 description 2
- 229940009456 adriamycin Drugs 0.000 description 2
- 229960001445 alitretinoin Drugs 0.000 description 2
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 2
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 2
- 229960000473 altretamine Drugs 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 2
- 230000001093 anti-cancer Effects 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- GOLCXWYRSKYTSP-UHFFFAOYSA-N arsenic trioxide Inorganic materials O1[As]2O[As]1O2 GOLCXWYRSKYTSP-UHFFFAOYSA-N 0.000 description 2
- 125000005161 aryl oxy carbonyl group Chemical group 0.000 description 2
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 2
- FZCSTZYAHCUGEM-UHFFFAOYSA-N aspergillomarasmine B Natural products OC(=O)CNC(C(O)=O)CNC(C(O)=O)CC(O)=O FZCSTZYAHCUGEM-UHFFFAOYSA-N 0.000 description 2
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 2
- 125000004069 aziridinyl group Chemical group 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 2
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 2
- 229960002938 bexarotene Drugs 0.000 description 2
- 208000002352 blister Diseases 0.000 description 2
- 210000004204 blood vessel Anatomy 0.000 description 2
- 210000000988 bone and bone Anatomy 0.000 description 2
- 239000003560 cancer drug Substances 0.000 description 2
- 230000000711 cancerogenic effect Effects 0.000 description 2
- 239000004202 carbamide Substances 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 2
- 231100000315 carcinogenic Toxicity 0.000 description 2
- 229940047495 celebrex Drugs 0.000 description 2
- 230000030833 cell death Effects 0.000 description 2
- 230000032823 cell division Effects 0.000 description 2
- 230000004700 cellular uptake Effects 0.000 description 2
- 229960001701 chloroform Drugs 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 238000009643 clonogenic assay Methods 0.000 description 2
- 231100000096 clonogenic assay Toxicity 0.000 description 2
- 229940125904 compound 1 Drugs 0.000 description 2
- 229940125961 compound 24 Drugs 0.000 description 2
- 229940125898 compound 5 Drugs 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- 229960000640 dactinomycin Drugs 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- DIOQZVSQGTUSAI-UHFFFAOYSA-N decane Chemical compound CCCCCCCCCC DIOQZVSQGTUSAI-UHFFFAOYSA-N 0.000 description 2
- 238000000354 decomposition reaction Methods 0.000 description 2
- 230000007850 degeneration Effects 0.000 description 2
- 229960001259 diclofenac Drugs 0.000 description 2
- DCOPUUMXTXDBNB-UHFFFAOYSA-N diclofenac Chemical compound OC(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl DCOPUUMXTXDBNB-UHFFFAOYSA-N 0.000 description 2
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 239000001177 diphosphate Substances 0.000 description 2
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 2
- 235000011180 diphosphates Nutrition 0.000 description 2
- XEYBHCRIKKKOSS-UHFFFAOYSA-N disodium;azanylidyneoxidanium;iron(2+);pentacyanide Chemical compound [Na+].[Na+].[Fe+2].N#[C-].N#[C-].N#[C-].N#[C-].N#[C-].[O+]#N XEYBHCRIKKKOSS-UHFFFAOYSA-N 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 108010067396 dornase alfa Proteins 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- HKSZLNNOFSGOKW-UHFFFAOYSA-N ent-staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(C)O1 HKSZLNNOFSGOKW-UHFFFAOYSA-N 0.000 description 2
- 239000002532 enzyme inhibitor Substances 0.000 description 2
- 229940125532 enzyme inhibitor Drugs 0.000 description 2
- 229960001433 erlotinib Drugs 0.000 description 2
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 229960001842 estramustine Drugs 0.000 description 2
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 229960000961 floxuridine Drugs 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 229940060037 fluorine Drugs 0.000 description 2
- VVIAGPKUTFNRDU-ABLWVSNPSA-N folinic acid Chemical compound C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-ABLWVSNPSA-N 0.000 description 2
- 235000011194 food seasoning agent Nutrition 0.000 description 2
- 238000004108 freeze drying Methods 0.000 description 2
- 229960002258 fulvestrant Drugs 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- CHPZKNULDCNCBW-UHFFFAOYSA-N gallium nitrate Chemical compound [Ga+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O CHPZKNULDCNCBW-UHFFFAOYSA-N 0.000 description 2
- 201000008361 ganglioneuroma Diseases 0.000 description 2
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 2
- 229960002584 gefitinib Drugs 0.000 description 2
- QTQAWLPCGQOSGP-GBTDJJJQSA-N geldanamycin Chemical group N1C(=O)\C(C)=C/C=C\[C@@H](OC)[C@H](OC(N)=O)\C(C)=C/[C@@H](C)[C@@H](O)[C@H](OC)C[C@@H](C)CC2=C(OC)C(=O)C=C1C2=O QTQAWLPCGQOSGP-GBTDJJJQSA-N 0.000 description 2
- 229960005277 gemcitabine Drugs 0.000 description 2
- 229940045109 genistein Drugs 0.000 description 2
- 235000006539 genistein Nutrition 0.000 description 2
- TZBJGXHYKVUXJN-UHFFFAOYSA-N genistein Natural products C1=CC(O)=CC=C1C1=COC2=CC(O)=CC(O)=C2C1=O TZBJGXHYKVUXJN-UHFFFAOYSA-N 0.000 description 2
- ZCOLJUOHXJRHDI-CMWLGVBASA-N genistein 7-O-beta-D-glucoside Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=C2C(=O)C(C=3C=CC(O)=CC=3)=COC2=C1 ZCOLJUOHXJRHDI-CMWLGVBASA-N 0.000 description 2
- 229960002989 glutamic acid Drugs 0.000 description 2
- 210000003714 granulocyte Anatomy 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 229940022353 herceptin Drugs 0.000 description 2
- 229940088013 hycamtin Drugs 0.000 description 2
- 206010020718 hyperplasia Diseases 0.000 description 2
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 2
- 150000002460 imidazoles Chemical class 0.000 description 2
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 230000002757 inflammatory effect Effects 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 2
- 229960004768 irinotecan Drugs 0.000 description 2
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 2
- 230000002045 lasting effect Effects 0.000 description 2
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 2
- 229960001691 leucovorin Drugs 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 229960002247 lomustine Drugs 0.000 description 2
- 229960003538 lonidamine Drugs 0.000 description 2
- 229920002521 macromolecule Polymers 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 2
- 230000009401 metastasis Effects 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 229960001156 mitoxantrone Drugs 0.000 description 2
- 230000002969 morbid Effects 0.000 description 2
- 208000025189 neoplasm of testis Diseases 0.000 description 2
- IAIWVQXQOWNYOU-FPYGCLRLSA-N nitrofural Chemical compound NC(=O)N\N=C\C1=CC=C([N+]([O-])=O)O1 IAIWVQXQOWNYOU-FPYGCLRLSA-N 0.000 description 2
- 229960001907 nitrofurazone Drugs 0.000 description 2
- 235000016709 nutrition Nutrition 0.000 description 2
- 230000035764 nutrition Effects 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 201000008968 osteosarcoma Diseases 0.000 description 2
- 229940026778 other chemotherapeutics in atc Drugs 0.000 description 2
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 2
- 229960001756 oxaliplatin Drugs 0.000 description 2
- 230000004783 oxidative metabolism Effects 0.000 description 2
- 238000002638 palliative care Methods 0.000 description 2
- WRUUGTRCQOWXEG-UHFFFAOYSA-N pamidronate Chemical compound NCCC(O)(P(O)(O)=O)P(O)(O)=O WRUUGTRCQOWXEG-UHFFFAOYSA-N 0.000 description 2
- 201000002530 pancreatic endocrine carcinoma Diseases 0.000 description 2
- 229940061584 phosphoramidic acid Drugs 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 230000004962 physiological condition Effects 0.000 description 2
- 150000003053 piperidines Chemical class 0.000 description 2
- 210000002381 plasma Anatomy 0.000 description 2
- 230000036470 plasma concentration Effects 0.000 description 2
- 229940063179 platinol Drugs 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical compound CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 2
- 229940117820 purinethol Drugs 0.000 description 2
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 2
- 150000003217 pyrazoles Chemical class 0.000 description 2
- 150000003233 pyrroles Chemical class 0.000 description 2
- AECPBJMOGBFQDN-YMYQVXQQSA-N radicicol Chemical compound C1CCCC(=O)C[C@H]2[C@H](Cl)C(=O)CC(=O)[C@H]2C(=O)O[C@H](C)C[C@H]2O[C@@H]21 AECPBJMOGBFQDN-YMYQVXQQSA-N 0.000 description 2
- 210000000664 rectum Anatomy 0.000 description 2
- 230000000306 recurrent effect Effects 0.000 description 2
- 238000006479 redox reaction Methods 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 208000037803 restenosis Diseases 0.000 description 2
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 2
- OHRURASPPZQGQM-GCCNXGTGSA-N romidepsin Chemical compound O1C(=O)[C@H](C(C)C)NC(=O)C(=C/C)/NC(=O)[C@H]2CSSCC\C=C\[C@@H]1CC(=O)N[C@H](C(C)C)C(=O)N2 OHRURASPPZQGQM-GCCNXGTGSA-N 0.000 description 2
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 238000000526 short-path distillation Methods 0.000 description 2
- 238000011125 single therapy Methods 0.000 description 2
- 210000003491 skin Anatomy 0.000 description 2
- 201000000849 skin cancer Diseases 0.000 description 2
- 201000008261 skin carcinoma Diseases 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229940083618 sodium nitroprusside Drugs 0.000 description 2
- PFNFFQXMRSDOHW-UHFFFAOYSA-N spermine Chemical compound NCCCNCCCCNCCCN PFNFFQXMRSDOHW-UHFFFAOYSA-N 0.000 description 2
- 206010041823 squamous cell carcinoma Diseases 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- HKSZLNNOFSGOKW-FYTWVXJKSA-N staurosporine Chemical compound C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1[C@H]1C[C@@H](NC)[C@@H](OC)[C@]4(C)O1 HKSZLNNOFSGOKW-FYTWVXJKSA-N 0.000 description 2
- TYFQFVWCELRYAO-UHFFFAOYSA-N suberic acid Chemical compound OC(=O)CCCCCCC(O)=O TYFQFVWCELRYAO-UHFFFAOYSA-N 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 150000003457 sulfones Chemical class 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- AYUNIORJHRXIBJ-TXHRRWQRSA-N tanespimycin Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(NCC=C)C(=O)C=C1C2=O AYUNIORJHRXIBJ-TXHRRWQRSA-N 0.000 description 2
- 229960001278 teniposide Drugs 0.000 description 2
- CIHOLLKRGTVIJN-UHFFFAOYSA-N tert‐butyl hydroperoxide Chemical compound CC(C)(C)OO CIHOLLKRGTVIJN-UHFFFAOYSA-N 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 238000007079 thiolysis reaction Methods 0.000 description 2
- 201000002510 thyroid cancer Diseases 0.000 description 2
- 208000013077 thyroid gland carcinoma Diseases 0.000 description 2
- UPSPUYADGBWSHF-UHFFFAOYSA-N tolmetin Chemical compound C1=CC(C)=CC=C1C(=O)C1=CC=C(CC(O)=O)N1C UPSPUYADGBWSHF-UHFFFAOYSA-N 0.000 description 2
- 231100000820 toxicity test Toxicity 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000001131 transforming effect Effects 0.000 description 2
- 238000013519 translation Methods 0.000 description 2
- IUCJMVBFZDHPDX-UHFFFAOYSA-N tretamine Chemical compound C1CN1C1=NC(N2CC2)=NC(N2CC2)=N1 IUCJMVBFZDHPDX-UHFFFAOYSA-N 0.000 description 2
- 229950001353 tretamine Drugs 0.000 description 2
- 150000003852 triazoles Chemical class 0.000 description 2
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical compound CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- YWWDBCBWQNCYNR-UHFFFAOYSA-N trimethylphosphine Chemical compound CP(C)C YWWDBCBWQNCYNR-UHFFFAOYSA-N 0.000 description 2
- 229960001099 trimetrexate Drugs 0.000 description 2
- NOYPYLRCIDNJJB-UHFFFAOYSA-N trimetrexate Chemical compound COC1=C(OC)C(OC)=CC(NCC=2C(=C3C(N)=NC(N)=NC3=CC=2)C)=C1 NOYPYLRCIDNJJB-UHFFFAOYSA-N 0.000 description 2
- 150000004043 trisaccharides Chemical class 0.000 description 2
- 229960000653 valrubicin Drugs 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- GBABOYUKABKIAF-IELIFDKJSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-IELIFDKJSA-N 0.000 description 2
- 229960002066 vinorelbine Drugs 0.000 description 2
- 229960004276 zoledronic acid Drugs 0.000 description 2
- QCPDBEXGCHOIDE-UHFFFAOYSA-N (-)-6xi-Methyl-(2ar,4axi,8at,12bt,12ct)-2a,3,4,4a,5,6,7,8a,12b,12c-decahydro-5xi,12dxi-aethano-furo[4',3',2';4,10]anthra[9,1-bc]oxepin-2,9,12-trion Natural products CC1COC2C(C(C=CC3=O)=O)=C3C3C4C22CCC1C2CCC4C(=O)O3 QCPDBEXGCHOIDE-UHFFFAOYSA-N 0.000 description 1
- NNJPGOLRFBJNIW-HNNXBMFYSA-N (-)-demecolcine Chemical compound C1=C(OC)C(=O)C=C2[C@@H](NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-HNNXBMFYSA-N 0.000 description 1
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- WDQLRUYAYXDIFW-RWKIJVEZSA-N (2r,3r,4s,5r,6r)-4-[(2s,3r,4s,5r,6r)-3,5-dihydroxy-4-[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-6-[[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl]oxy-6-(hydroxymethyl)oxane-2,3,5-triol Chemical compound O[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)[C@H](O)[C@@H](CO[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)O1 WDQLRUYAYXDIFW-RWKIJVEZSA-N 0.000 description 1
- RDJGLLICXDHJDY-NSHDSACASA-N (2s)-2-(3-phenoxyphenyl)propanoic acid Chemical compound OC(=O)[C@@H](C)C1=CC=CC(OC=2C=CC=CC=2)=C1 RDJGLLICXDHJDY-NSHDSACASA-N 0.000 description 1
- FLWWDYNPWOSLEO-HQVZTVAUSA-N (2s)-2-[[4-[1-(2-amino-4-oxo-1h-pteridin-6-yl)ethyl-methylamino]benzoyl]amino]pentanedioic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1C(C)N(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FLWWDYNPWOSLEO-HQVZTVAUSA-N 0.000 description 1
- CGMTUJFWROPELF-YPAAEMCBSA-N (3E,5S)-5-[(2S)-butan-2-yl]-3-(1-hydroxyethylidene)pyrrolidine-2,4-dione Chemical compound CC[C@H](C)[C@@H]1NC(=O)\C(=C(/C)O)C1=O CGMTUJFWROPELF-YPAAEMCBSA-N 0.000 description 1
- FEOHYDSNGHIXOM-WLDMJGECSA-N (3R,4R,5S,6R)-3-amino-6-(hydroxymethyl)-2-methyloxane-2,4,5-triol Chemical compound CC1(O)[C@H](N)[C@@H](O)[C@H](O)[C@H](O1)CO FEOHYDSNGHIXOM-WLDMJGECSA-N 0.000 description 1
- VRYALKFFQXWPIH-RANCGNPWSA-N (3r,4s,5r)-3,4,5,6-tetrahydroxy-2-tritiohexanal Chemical compound O=CC([3H])[C@@H](O)[C@H](O)[C@H](O)CO VRYALKFFQXWPIH-RANCGNPWSA-N 0.000 description 1
- NDSTUUPEXNCUNW-UHFFFAOYSA-N (5-nitrofuran-2-yl)methanol Chemical compound OCC1=CC=C([N+]([O-])=O)O1 NDSTUUPEXNCUNW-UHFFFAOYSA-N 0.000 description 1
- XRBSKUSTLXISAB-XVVDYKMHSA-N (5r,6r,7r,8r)-8-hydroxy-7-(hydroxymethyl)-5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydrobenzo[f][1,3]benzodioxole-6-carboxylic acid Chemical compound COC1=C(OC)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@H](O)[C@@H](CO)[C@@H]2C(O)=O)=C1 XRBSKUSTLXISAB-XVVDYKMHSA-N 0.000 description 1
- XRBSKUSTLXISAB-UHFFFAOYSA-N (7R,7'R,8R,8'R)-form-Podophyllic acid Natural products COC1=C(OC)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C(CO)C2C(O)=O)=C1 XRBSKUSTLXISAB-UHFFFAOYSA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- MUMXDRRTIYLYMY-YJKCNMNRSA-N (Z)-[dodecyl-[6-(dodecylazaniumyl)hexyl]amino]-oxido-oxidoiminoazanium Chemical compound CCCCCCCCCCCC[NH2+]CCCCCCN(CCCCCCCCCCCC)[N+](\[O-])=N\[O-] MUMXDRRTIYLYMY-YJKCNMNRSA-N 0.000 description 1
- AGNGYMCLFWQVGX-AGFFZDDWSA-N (e)-1-[(2s)-2-amino-2-carboxyethoxy]-2-diazonioethenolate Chemical compound OC(=O)[C@@H](N)CO\C([O-])=C\[N+]#N AGNGYMCLFWQVGX-AGFFZDDWSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- CSNIZNHTOVFARY-UHFFFAOYSA-N 1,2-benzothiazole Chemical compound C1=CC=C2C=NSC2=C1 CSNIZNHTOVFARY-UHFFFAOYSA-N 0.000 description 1
- KTZQTRPPVKQPFO-UHFFFAOYSA-N 1,2-benzoxazole Chemical compound C1=CC=C2C=NOC2=C1 KTZQTRPPVKQPFO-UHFFFAOYSA-N 0.000 description 1
- MPCAJMNYNOGXPB-SLPGGIOYSA-N 1,5-anhydro-D-glucitol Chemical compound OC[C@H]1OC[C@H](O)[C@@H](O)[C@@H]1O MPCAJMNYNOGXPB-SLPGGIOYSA-N 0.000 description 1
- OJYIBEYSBXIQOP-UHFFFAOYSA-N 1-methoxy-4-[2-(4-methoxyphenyl)propan-2-yl]benzene Chemical compound C1=CC(OC)=CC=C1C(C)(C)C1=CC=C(OC)C=C1 OJYIBEYSBXIQOP-UHFFFAOYSA-N 0.000 description 1
- HBMINVNVQUDERA-UHFFFAOYSA-N 1-methyl-2-nitroimidazole Chemical compound CN1C=CN=C1[N+]([O-])=O HBMINVNVQUDERA-UHFFFAOYSA-N 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 1
- KAESVJOAVNADME-UHFFFAOYSA-N 1H-pyrrole Natural products C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 1
- AMFYRKOUWBAGHV-UHFFFAOYSA-N 1h-pyrazolo[4,3-b]pyridine Chemical compound C1=CN=C2C=NNC2=C1 AMFYRKOUWBAGHV-UHFFFAOYSA-N 0.000 description 1
- ADVGKWPZRIDURE-UHFFFAOYSA-N 2'-Hydroxyacetanilide Chemical compound CC(=O)NC1=CC=CC=C1O ADVGKWPZRIDURE-UHFFFAOYSA-N 0.000 description 1
- BOMZMNZEXMAQQW-UHFFFAOYSA-N 2,5,11-trimethyl-6h-pyrido[4,3-b]carbazol-2-ium-9-ol;acetate Chemical compound CC([O-])=O.C[N+]1=CC=C2C(C)=C(NC=3C4=CC(O)=CC=3)C4=C(C)C2=C1 BOMZMNZEXMAQQW-UHFFFAOYSA-N 0.000 description 1
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 1
- VZMIDQINDQOVMF-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;methylaminomethanetriol Chemical compound CNC(O)(O)O.OCC(N)(CO)CO VZMIDQINDQOVMF-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 1
- VNBAOSVONFJBKP-UHFFFAOYSA-N 2-chloro-n,n-bis(2-chloroethyl)propan-1-amine;hydrochloride Chemical compound Cl.CC(Cl)CN(CCCl)CCCl VNBAOSVONFJBKP-UHFFFAOYSA-N 0.000 description 1
- CVOFKRWYWCSDMA-UHFFFAOYSA-N 2-chloro-n-(2,6-diethylphenyl)-n-(methoxymethyl)acetamide;2,6-dinitro-n,n-dipropyl-4-(trifluoromethyl)aniline Chemical compound CCC1=CC=CC(CC)=C1N(COC)C(=O)CCl.CCCN(CCC)C1=C([N+]([O-])=O)C=C(C(F)(F)F)C=C1[N+]([O-])=O CVOFKRWYWCSDMA-UHFFFAOYSA-N 0.000 description 1
- IFMNWWRDKCBFTO-UHFFFAOYSA-N 2-chloro-n-dichlorophosphorylethanamine Chemical compound ClCCNP(Cl)(Cl)=O IFMNWWRDKCBFTO-UHFFFAOYSA-N 0.000 description 1
- 125000001340 2-chloroethyl group Chemical group [H]C([H])(Cl)C([H])([H])* 0.000 description 1
- LPWYIMUYYPTXFO-UHFFFAOYSA-N 2-chloroethyl(cyclopropyl)azanium;chloride Chemical compound Cl.ClCCNC1CC1 LPWYIMUYYPTXFO-UHFFFAOYSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 1
- KYVQCVBMLZMRKL-UHFFFAOYSA-N 2-ethoxy-5-(pyrrolidine-1-carbonylamino)benzenesulfonyl chloride Chemical compound C1=C(S(Cl)(=O)=O)C(OCC)=CC=C1NC(=O)N1CCCC1 KYVQCVBMLZMRKL-UHFFFAOYSA-N 0.000 description 1
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- CTRPRMNBTVRDFH-UHFFFAOYSA-N 2-n-methyl-1,3,5-triazine-2,4,6-triamine Chemical compound CNC1=NC(N)=NC(N)=N1 CTRPRMNBTVRDFH-UHFFFAOYSA-N 0.000 description 1
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 1
- ALNUOSXDMYFQNQ-UHFFFAOYSA-N 2-nitro-1,3-thiazole Chemical group [O-][N+](=O)C1=NC=CS1 ALNUOSXDMYFQNQ-UHFFFAOYSA-N 0.000 description 1
- FUBFWTUFPGFHOJ-UHFFFAOYSA-N 2-nitrofuran Chemical group [O-][N+](=O)C1=CC=CO1 FUBFWTUFPGFHOJ-UHFFFAOYSA-N 0.000 description 1
- RVBUGGBMJDPOST-UHFFFAOYSA-N 2-thiobarbituric acid Chemical class O=C1CC(=O)NC(=S)N1 RVBUGGBMJDPOST-UHFFFAOYSA-N 0.000 description 1
- DWHVZCLBMTZRQM-UHFFFAOYSA-N 2H-pyrazolo[4,3-b]quinoxalin-3-amine Chemical compound C1=CC=CC2=NC3=C(N)NN=C3N=C21 DWHVZCLBMTZRQM-UHFFFAOYSA-N 0.000 description 1
- ZZVDXRCAGGQFAK-UHFFFAOYSA-N 2h-oxazaphosphinine Chemical compound N1OC=CC=P1 ZZVDXRCAGGQFAK-UHFFFAOYSA-N 0.000 description 1
- PWMYMKOUNYTVQN-UHFFFAOYSA-N 3-(8,8-diethyl-2-aza-8-germaspiro[4.5]decan-2-yl)-n,n-dimethylpropan-1-amine Chemical compound C1C[Ge](CC)(CC)CCC11CN(CCCN(C)C)CC1 PWMYMKOUNYTVQN-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- GIGCDIVNDFQKRA-LTCKWSDVSA-N 4-[(2s)-2-amino-2-carboxyethyl]-n,n-bis(2-chloroethyl)benzeneamine oxide;dihydrochloride Chemical compound Cl.Cl.OC(=O)[C@@H](N)CC1=CC=C([N+]([O-])(CCCl)CCCl)C=C1 GIGCDIVNDFQKRA-LTCKWSDVSA-N 0.000 description 1
- GQGVBSHMRYHBTF-UOWFLXDJSA-N 4-amino-1-[(2r,4r,5r)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,3,5-triazin-2-one Chemical compound O=C1N=C(N)N=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 GQGVBSHMRYHBTF-UOWFLXDJSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- IDPUKCWIGUEADI-UHFFFAOYSA-N 5-[bis(2-chloroethyl)amino]uracil Chemical compound ClCCN(CCCl)C1=CNC(=O)NC1=O IDPUKCWIGUEADI-UHFFFAOYSA-N 0.000 description 1
- VYDWQPKRHOGLPA-UHFFFAOYSA-N 5-nitroimidazole Chemical compound [O-][N+](=O)C1=CN=CN1 VYDWQPKRHOGLPA-UHFFFAOYSA-N 0.000 description 1
- RXQZLSRIOOYKLF-UHFFFAOYSA-N 5H-pyrazolo[4,3-d]triazine Chemical compound N1=NN=C2C=NNC2=C1 RXQZLSRIOOYKLF-UHFFFAOYSA-N 0.000 description 1
- KDOPAZIWBAHVJB-UHFFFAOYSA-N 5h-pyrrolo[3,2-d]pyrimidine Chemical compound C1=NC=C2NC=CC2=N1 KDOPAZIWBAHVJB-UHFFFAOYSA-N 0.000 description 1
- SUPXSFXAMJPEPH-UHFFFAOYSA-N 5h-pyrrolo[3,2-d]triazine Chemical compound N1=NC=C2NC=CC2=N1 SUPXSFXAMJPEPH-UHFFFAOYSA-N 0.000 description 1
- WYXSYVWAUAUWLD-SHUUEZRQSA-N 6-azauridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=N1 WYXSYVWAUAUWLD-SHUUEZRQSA-N 0.000 description 1
- VVIAGPKUTFNRDU-UHFFFAOYSA-N 6S-folinic acid Natural products C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-UHFFFAOYSA-N 0.000 description 1
- HDZZVAMISRMYHH-UHFFFAOYSA-N 9beta-Ribofuranosyl-7-deazaadenin Natural products C1=CC=2C(N)=NC=NC=2N1C1OC(CO)C(O)C1O HDZZVAMISRMYHH-UHFFFAOYSA-N 0.000 description 1
- 206010000050 Abdominal adhesions Diseases 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 1
- 206010000830 Acute leukaemia Diseases 0.000 description 1
- 208000003200 Adenoma Diseases 0.000 description 1
- 206010001233 Adenoma benign Diseases 0.000 description 1
- CEIZFXOZIQNICU-UHFFFAOYSA-N Alternaria alternata Crofton-weed toxin Natural products CCC(C)C1NC(=O)C(C(C)=O)=C1O CEIZFXOZIQNICU-UHFFFAOYSA-N 0.000 description 1
- 241000208327 Apocynaceae Species 0.000 description 1
- 102000003916 Arrestin Human genes 0.000 description 1
- 108090000328 Arrestin Proteins 0.000 description 1
- 208000033116 Asbestos intoxication Diseases 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 206010003571 Astrocytoma Diseases 0.000 description 1
- 208000004300 Atrophic Gastritis Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 235000016068 Berberis vulgaris Nutrition 0.000 description 1
- VGGGPCQERPFHOB-MCIONIFRSA-N Bestatin Chemical compound CC(C)C[C@H](C(O)=O)NC(=O)[C@@H](O)[C@H](N)CC1=CC=CC=C1 VGGGPCQERPFHOB-MCIONIFRSA-N 0.000 description 1
- 241000335053 Beta vulgaris Species 0.000 description 1
- 229940122361 Bisphosphonate Drugs 0.000 description 1
- 208000010392 Bone Fractures Diseases 0.000 description 1
- 241000219198 Brassica Species 0.000 description 1
- 235000003351 Brassica cretica Nutrition 0.000 description 1
- 235000003343 Brassica rupestris Nutrition 0.000 description 1
- 206010055113 Breast cancer metastatic Diseases 0.000 description 1
- 206010006458 Bronchitis chronic Diseases 0.000 description 1
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- SHHKQEUPHAENFK-UHFFFAOYSA-N Carboquone Chemical compound O=C1C(C)=C(N2CC2)C(=O)C(C(COC(N)=O)OC)=C1N1CC1 SHHKQEUPHAENFK-UHFFFAOYSA-N 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- AOCCBINRVIKJHY-UHFFFAOYSA-N Carmofur Chemical compound CCCCCCNC(=O)N1C=C(F)C(=O)NC1=O AOCCBINRVIKJHY-UHFFFAOYSA-N 0.000 description 1
- 240000001829 Catharanthus roseus Species 0.000 description 1
- XCDXSSFOJZZGQC-UHFFFAOYSA-N Chlornaphazine Chemical compound C1=CC=CC2=CC(N(CCCl)CCCl)=CC=C21 XCDXSSFOJZZGQC-UHFFFAOYSA-N 0.000 description 1
- MKQWTWSXVILIKJ-LXGUWJNJSA-N Chlorozotocin Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](C=O)NC(=O)N(N=O)CCCl MKQWTWSXVILIKJ-LXGUWJNJSA-N 0.000 description 1
- 206010008609 Cholangitis sclerosing Diseases 0.000 description 1
- 208000006332 Choriocarcinoma Diseases 0.000 description 1
- 206010009137 Chronic sinusitis Diseases 0.000 description 1
- 208000006344 Churg-Strauss Syndrome Diseases 0.000 description 1
- QCDFBFJGMNKBDO-UHFFFAOYSA-N Clioquinol Chemical compound C1=CN=C2C(O)=C(I)C=C(Cl)C2=C1 QCDFBFJGMNKBDO-UHFFFAOYSA-N 0.000 description 1
- 208000015943 Coeliac disease Diseases 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 102100031162 Collagen alpha-1(XVIII) chain Human genes 0.000 description 1
- 208000032170 Congenital Abnormalities Diseases 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 102100021906 Cyclin-O Human genes 0.000 description 1
- LVZWSLJZHVFIQJ-UHFFFAOYSA-N Cyclopropane Chemical compound C1CC1 LVZWSLJZHVFIQJ-UHFFFAOYSA-N 0.000 description 1
- HTJDQJBWANPRPF-UHFFFAOYSA-N Cyclopropylamine Chemical compound NC1CC1 HTJDQJBWANPRPF-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- WEAHRLBPCANXCN-UHFFFAOYSA-N Daunomycin Natural products CCC1(O)CC(OC2CC(N)C(O)C(C)O2)c3cc4C(=O)c5c(OC)cccc5C(=O)c4c(O)c3C1 WEAHRLBPCANXCN-UHFFFAOYSA-N 0.000 description 1
- NNJPGOLRFBJNIW-UHFFFAOYSA-N Demecolcine Natural products C1=C(OC)C(=O)C=C2C(NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-UHFFFAOYSA-N 0.000 description 1
- 108010002156 Depsipeptides Proteins 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- 206010012438 Dermatitis atopic Diseases 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 101001031598 Dictyostelium discoideum Probable serine/threonine-protein kinase fhkC Proteins 0.000 description 1
- 208000002699 Digestive System Neoplasms Diseases 0.000 description 1
- RWSOTUBLDIXVET-UHFFFAOYSA-N Dihydrogen sulfide Chemical compound S RWSOTUBLDIXVET-UHFFFAOYSA-N 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- 102100031480 Dual specificity mitogen-activated protein kinase kinase 1 Human genes 0.000 description 1
- 101710146526 Dual specificity mitogen-activated protein kinase kinase 1 Proteins 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 206010014759 Endometrial neoplasm Diseases 0.000 description 1
- 108010079505 Endostatins Proteins 0.000 description 1
- SAMRUMKYXPVKPA-VFKOLLTISA-N Enocitabine Chemical compound O=C1N=C(NC(=O)CCCCCCCCCCCCCCCCCCCCC)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 SAMRUMKYXPVKPA-VFKOLLTISA-N 0.000 description 1
- 208000018428 Eosinophilic granulomatosis with polyangiitis Diseases 0.000 description 1
- XOZIUKBZLSUILX-SDMHVBBESA-N Epothilone D Natural products O=C1[C@H](C)[C@@H](O)[C@@H](C)CCC/C(/C)=C/C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C XOZIUKBZLSUILX-SDMHVBBESA-N 0.000 description 1
- 229930189413 Esperamicin Natural products 0.000 description 1
- KGWDUNBJIMUFAP-KVVVOXFISA-N Ethanolamine Oleate Chemical compound NCCO.CCCCCCCC\C=C/CCCCCCCC(O)=O KGWDUNBJIMUFAP-KVVVOXFISA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- 102000018711 Facilitative Glucose Transport Proteins Human genes 0.000 description 1
- 201000006107 Familial adenomatous polyposis Diseases 0.000 description 1
- CWYNVVGOOAEACU-UHFFFAOYSA-N Fe2+ Chemical compound [Fe+2] CWYNVVGOOAEACU-UHFFFAOYSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- MPJKWIXIYCLVCU-UHFFFAOYSA-N Folinic acid Natural products NC1=NC2=C(N(C=O)C(CNc3ccc(cc3)C(=O)NC(CCC(=O)O)CC(=O)O)CN2)C(=O)N1 MPJKWIXIYCLVCU-UHFFFAOYSA-N 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- 208000036495 Gastritis atrophic Diseases 0.000 description 1
- 206010017993 Gastrointestinal neoplasms Diseases 0.000 description 1
- 206010051066 Gastrointestinal stromal tumour Diseases 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- JRZJKWGQFNTSRN-UHFFFAOYSA-N Geldanamycin Natural products C1C(C)CC(OC)C(O)C(C)C=C(C)C(OC(N)=O)C(OC)CCC=C(C)C(=O)NC2=CC(=O)C(OC)=C1C2=O JRZJKWGQFNTSRN-UHFFFAOYSA-N 0.000 description 1
- 208000007569 Giant Cell Tumors Diseases 0.000 description 1
- 208000007465 Giant cell arteritis Diseases 0.000 description 1
- 206010018364 Glomerulonephritis Diseases 0.000 description 1
- 108091052347 Glucose transporter family Proteins 0.000 description 1
- 102000009465 Growth Factor Receptors Human genes 0.000 description 1
- 108010009202 Growth Factor Receptors Proteins 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 101710113864 Heat shock protein 90 Proteins 0.000 description 1
- 102100034051 Heat shock protein HSP 90-alpha Human genes 0.000 description 1
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 1
- 208000032843 Hemorrhage Diseases 0.000 description 1
- 201000004331 Henoch-Schoenlein purpura Diseases 0.000 description 1
- 206010019617 Henoch-Schonlein purpura Diseases 0.000 description 1
- 239000004705 High-molecular-weight polyethylene Substances 0.000 description 1
- 101000897441 Homo sapiens Cyclin-O Proteins 0.000 description 1
- 101001016865 Homo sapiens Heat shock protein HSP 90-alpha Proteins 0.000 description 1
- 101000582950 Homo sapiens Platelet factor 4 Proteins 0.000 description 1
- 208000037147 Hypercalcaemia Diseases 0.000 description 1
- 206010058490 Hyperoxia Diseases 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- 208000031814 IgA Vasculitis Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical compound C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 102000006992 Interferon-alpha Human genes 0.000 description 1
- 108010047761 Interferon-alpha Proteins 0.000 description 1
- 102000003996 Interferon-beta Human genes 0.000 description 1
- 108090000467 Interferon-beta Proteins 0.000 description 1
- 102000008070 Interferon-gamma Human genes 0.000 description 1
- 108010074328 Interferon-gamma Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 102000000588 Interleukin-2 Human genes 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- 208000005125 Invasive Hydatidiform Mole Diseases 0.000 description 1
- 208000011200 Kawasaki disease Diseases 0.000 description 1
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- CZQHHVNHHHRRDU-UHFFFAOYSA-N LY294002 Chemical compound C1=CC=C2C(=O)C=C(N3CCOCC3)OC2=C1C1=CC=CC=C1 CZQHHVNHHHRRDU-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 229920001491 Lentinan Polymers 0.000 description 1
- 208000034624 Leukocytoclastic Cutaneous Vasculitis Diseases 0.000 description 1
- 208000032514 Leukocytoclastic vasculitis Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 241000282553 Macaca Species 0.000 description 1
- 241000282560 Macaca mulatta Species 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000883509 Mammillaria Species 0.000 description 1
- 208000007054 Medullary Carcinoma Diseases 0.000 description 1
- XOGTZOOQQBDUSI-UHFFFAOYSA-M Mesna Chemical compound [Na+].[O-]S(=O)(=O)CCS XOGTZOOQQBDUSI-UHFFFAOYSA-M 0.000 description 1
- VFKZTMPDYBFSTM-KVTDHHQDSA-N Mitobronitol Chemical compound BrC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-KVTDHHQDSA-N 0.000 description 1
- HRHKSTOGXBBQCB-UHFFFAOYSA-N Mitomycin E Natural products O=C1C(N)=C(C)C(=O)C2=C1C(COC(N)=O)C1(OC)C3N(C)C3CN12 HRHKSTOGXBBQCB-UHFFFAOYSA-N 0.000 description 1
- 241000282341 Mustela putorius furo Species 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 208000009525 Myocarditis Diseases 0.000 description 1
- 201000002481 Myositis Diseases 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 1
- 208000000592 Nasal Polyps Diseases 0.000 description 1
- 208000008846 Neurocytoma Diseases 0.000 description 1
- 208000005890 Neuroma Diseases 0.000 description 1
- RDXLYGJSWZYTFJ-UHFFFAOYSA-N Niridazole Chemical compound S1C([N+](=O)[O-])=CN=C1N1C(=O)NCC1 RDXLYGJSWZYTFJ-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- IOVCWXUNBOPUCH-UHFFFAOYSA-M Nitrite anion Chemical compound [O-]N=O IOVCWXUNBOPUCH-UHFFFAOYSA-M 0.000 description 1
- SYNHCENRCUAUNM-UHFFFAOYSA-N Nitrogen mustard N-oxide hydrochloride Chemical compound Cl.ClCC[N+]([O-])(C)CCCl SYNHCENRCUAUNM-UHFFFAOYSA-N 0.000 description 1
- 102000004459 Nitroreductase Human genes 0.000 description 1
- KGTDRFCXGRULNK-UHFFFAOYSA-N Nogalamycin Natural products COC1C(OC)(C)C(OC)C(C)OC1OC1C2=C(O)C(C(=O)C3=C(O)C=C4C5(C)OC(C(C(C5O)N(C)C)O)OC4=C3C3=O)=C3C=C2C(C(=O)OC)C(C)(O)C1 KGTDRFCXGRULNK-UHFFFAOYSA-N 0.000 description 1
- 108091005461 Nucleic proteins Proteins 0.000 description 1
- MSHZHSPISPJWHW-UHFFFAOYSA-N O-(chloroacetylcarbamoyl)fumagillol Chemical compound O1C(CC=C(C)C)C1(C)C1C(OC)C(OC(=O)NC(=O)CCl)CCC21CO2 MSHZHSPISPJWHW-UHFFFAOYSA-N 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 102000004316 Oxidoreductases Human genes 0.000 description 1
- 108090000854 Oxidoreductases Proteins 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 206010033645 Pancreatitis Diseases 0.000 description 1
- 241001504519 Papio ursinus Species 0.000 description 1
- 208000000821 Parathyroid Neoplasms Diseases 0.000 description 1
- 208000031481 Pathologic Constriction Diseases 0.000 description 1
- 206010034277 Pemphigoid Diseases 0.000 description 1
- 108010057150 Peplomycin Proteins 0.000 description 1
- 241000009328 Perro Species 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 1
- KMSKQZKKOZQFFG-HSUXVGOQSA-N Pirarubicin Chemical compound O([C@H]1[C@@H](N)C[C@@H](O[C@H]1C)O[C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1CCCCO1 KMSKQZKKOZQFFG-HSUXVGOQSA-N 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 208000006994 Precancerous Conditions Diseases 0.000 description 1
- 206010036774 Proctitis Diseases 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-IGMARMGPSA-N Protium Chemical compound [1H] YZCKVEUIGOORGS-IGMARMGPSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 208000006265 Renal cell carcinoma Diseases 0.000 description 1
- 208000018569 Respiratory Tract disease Diseases 0.000 description 1
- NCYCYZXNIZJOKI-OVSJKPMPSA-N Retinaldehyde Chemical compound O=C\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C NCYCYZXNIZJOKI-OVSJKPMPSA-N 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 241000282695 Saimiri Species 0.000 description 1
- 206010039705 Scleritis Diseases 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- 206010070834 Sensitisation Diseases 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- 201000010001 Silicosis Diseases 0.000 description 1
- 229920000519 Sizofiran Polymers 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 239000004141 Sodium laurylsulphate Substances 0.000 description 1
- 108010017622 Somatomedin Receptors Proteins 0.000 description 1
- 102000013275 Somatomedins Human genes 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 241000529895 Stercorarius Species 0.000 description 1
- 241001655322 Streptomycetales Species 0.000 description 1
- 208000010513 Stupor Diseases 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 101150057615 Syn gene Proteins 0.000 description 1
- 206010042971 T-cell lymphoma Diseases 0.000 description 1
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- CGMTUJFWROPELF-UHFFFAOYSA-N Tenuazonic acid Natural products CCC(C)C1NC(=O)C(=C(C)/O)C1=O CGMTUJFWROPELF-UHFFFAOYSA-N 0.000 description 1
- DPOPAJRDYZGTIR-UHFFFAOYSA-N Tetrazine Chemical compound C1=CN=NN=N1 DPOPAJRDYZGTIR-UHFFFAOYSA-N 0.000 description 1
- 244000299461 Theobroma cacao Species 0.000 description 1
- 235000009470 Theobroma cacao Nutrition 0.000 description 1
- 102000002938 Thrombospondin Human genes 0.000 description 1
- 108060008245 Thrombospondin Proteins 0.000 description 1
- 208000031737 Tissue Adhesions Diseases 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- 206010066901 Treatment failure Diseases 0.000 description 1
- UMILHIMHKXVDGH-UHFFFAOYSA-N Triethylene glycol diglycidyl ether Chemical compound C1OC1COCCOCCOCCOCC1CO1 UMILHIMHKXVDGH-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 241000863480 Vinca Species 0.000 description 1
- 241000863486 Vinca minor Species 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 1
- 208000008383 Wilms tumor Diseases 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- IFJUINDAXYAPTO-UUBSBJJBSA-N [(8r,9s,13s,14s,17s)-17-[2-[4-[4-[bis(2-chloroethyl)amino]phenyl]butanoyloxy]acetyl]oxy-13-methyl-6,7,8,9,11,12,14,15,16,17-decahydrocyclopenta[a]phenanthren-3-yl] benzoate Chemical compound C([C@@H]1[C@@H](C2=CC=3)CC[C@]4([C@H]1CC[C@@H]4OC(=O)COC(=O)CCCC=1C=CC(=CC=1)N(CCCl)CCCl)C)CC2=CC=3OC(=O)C1=CC=CC=C1 IFJUINDAXYAPTO-UUBSBJJBSA-N 0.000 description 1
- USDJGQLNFPZEON-UHFFFAOYSA-N [[4,6-bis(hydroxymethylamino)-1,3,5-triazin-2-yl]amino]methanol Chemical compound OCNC1=NC(NCO)=NC(NCO)=N1 USDJGQLNFPZEON-UHFFFAOYSA-N 0.000 description 1
- IKWTVSLWAPBBKU-UHFFFAOYSA-N a1010_sial Chemical compound O=[As]O[As]=O IKWTVSLWAPBBKU-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- ZOZKYEHVNDEUCO-XUTVFYLZSA-N aceglatone Chemical compound O1C(=O)[C@H](OC(C)=O)[C@@H]2OC(=O)[C@@H](OC(=O)C)[C@@H]21 ZOZKYEHVNDEUCO-XUTVFYLZSA-N 0.000 description 1
- 229950002684 aceglatone Drugs 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 229930188522 aclacinomycin Natural products 0.000 description 1
- USZYSDMBJDPRIF-SVEJIMAYSA-N aclacinomycin A Chemical compound O([C@H]1[C@@H](O)C[C@@H](O[C@H]1C)O[C@H]1[C@H](C[C@@H](O[C@H]1C)O[C@H]1C[C@]([C@@H](C2=CC=3C(=O)C4=CC=CC(O)=C4C(=O)C=3C(O)=C21)C(=O)OC)(O)CC)N(C)C)[C@H]1CCC(=O)[C@H](C)O1 USZYSDMBJDPRIF-SVEJIMAYSA-N 0.000 description 1
- 229960004176 aclarubicin Drugs 0.000 description 1
- 208000017733 acquired polycythemia vera Diseases 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000009056 active transport Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 208000009956 adenocarcinoma Diseases 0.000 description 1
- 201000005179 adrenal carcinoma Diseases 0.000 description 1
- 201000005188 adrenal gland cancer Diseases 0.000 description 1
- 229940064305 adrucil Drugs 0.000 description 1
- 229940013181 advil Drugs 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- IAJILQKETJEXLJ-RSJOWCBRSA-N aldehydo-D-galacturonic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-RSJOWCBRSA-N 0.000 description 1
- IAJILQKETJEXLJ-QTBDOELSSA-N aldehydo-D-glucuronic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-QTBDOELSSA-N 0.000 description 1
- 229940060515 aleve Drugs 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 229930013930 alkaloid Natural products 0.000 description 1
- 150000003797 alkaloid derivatives Chemical class 0.000 description 1
- 229940098174 alkeran Drugs 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 239000004411 aluminium Substances 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 229960001220 amsacrine Drugs 0.000 description 1
- XCPGHVQEEXUHNC-UHFFFAOYSA-N amsacrine Chemical compound COC1=CC(NS(C)(=O)=O)=CC=C1NC1=C(C=CC=C2)C2=NC2=CC=CC=C12 XCPGHVQEEXUHNC-UHFFFAOYSA-N 0.000 description 1
- 229960002932 anastrozole Drugs 0.000 description 1
- BBDAGFIXKZCXAH-CCXZUQQUSA-N ancitabine Chemical compound N=C1C=CN2[C@@H]3O[C@H](CO)[C@@H](O)[C@@H]3OC2=N1 BBDAGFIXKZCXAH-CCXZUQQUSA-N 0.000 description 1
- 229950000242 ancitabine Drugs 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- 229940045799 anthracyclines and related substance Drugs 0.000 description 1
- VGQOVCHZGQWAOI-YQRHFANHSA-N anthramycin Chemical compound N1[C@H](O)[C@@H]2CC(\C=C\C(N)=O)=CN2C(=O)C2=CC=C(C)C(O)=C12 VGQOVCHZGQWAOI-YQRHFANHSA-N 0.000 description 1
- PYKYMHQGRFAEBM-UHFFFAOYSA-N anthraquinone Natural products CCC(=O)c1c(O)c2C(=O)C3C(C=CC=C3O)C(=O)c2cc1CC(=O)OC PYKYMHQGRFAEBM-UHFFFAOYSA-N 0.000 description 1
- 150000004056 anthraquinones Chemical class 0.000 description 1
- 230000001772 anti-angiogenic effect Effects 0.000 description 1
- 229940124650 anti-cancer therapies Drugs 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 238000011319 anticancer therapy Methods 0.000 description 1
- 238000010913 antigen-directed enzyme pro-drug therapy Methods 0.000 description 1
- 229950006345 antramycin Drugs 0.000 description 1
- 239000000010 aprotic solvent Substances 0.000 description 1
- 229940078010 arimidex Drugs 0.000 description 1
- 229960002594 arsenic trioxide Drugs 0.000 description 1
- 150000004646 arylidenes Chemical group 0.000 description 1
- 206010003441 asbestosis Diseases 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 201000008937 atopic dermatitis Diseases 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 229950011321 azaserine Drugs 0.000 description 1
- 150000001541 aziridines Chemical class 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 229940125717 barbiturate Drugs 0.000 description 1
- HNYOPLTXPVRDBG-UHFFFAOYSA-N barbituric acid Chemical compound O=C1CC(=O)NC(=O)N1 HNYOPLTXPVRDBG-UHFFFAOYSA-N 0.000 description 1
- 238000003287 bathing Methods 0.000 description 1
- XFILPEOLDIKJHX-QYZOEREBSA-N batimastat Chemical compound C([C@@H](C(=O)NC)NC(=O)[C@H](CC(C)C)[C@H](CSC=1SC=CC=1)C(=O)NO)C1=CC=CC=C1 XFILPEOLDIKJHX-QYZOEREBSA-N 0.000 description 1
- 229950001858 batimastat Drugs 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 229950005567 benzodepa Drugs 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- VFIUCBTYGKMLCM-UHFFFAOYSA-N benzyl n-[bis(aziridin-1-yl)phosphoryl]carbamate Chemical compound C=1C=CC=CC=1COC(=O)NP(=O)(N1CC1)N1CC1 VFIUCBTYGKMLCM-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 229940108502 bicnu Drugs 0.000 description 1
- 210000000013 bile duct Anatomy 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 238000010170 biological method Methods 0.000 description 1
- 238000001815 biotherapy Methods 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 230000007698 birth defect Effects 0.000 description 1
- QKSKPIVNLNLAAV-UHFFFAOYSA-N bis(2-chloroethyl) sulfide Chemical compound ClCCSCCCl QKSKPIVNLNLAAV-UHFFFAOYSA-N 0.000 description 1
- 229950008548 bisantrene Drugs 0.000 description 1
- 150000004663 bisphosphonates Chemical class 0.000 description 1
- 201000001531 bladder carcinoma Diseases 0.000 description 1
- 201000000053 blastoma Diseases 0.000 description 1
- 230000036770 blood supply Effects 0.000 description 1
- 238000010322 bone marrow transplantation Methods 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- VQFAIAKCILWQPZ-UHFFFAOYSA-N bromoacetone Chemical compound CC(=O)CBr VQFAIAKCILWQPZ-UHFFFAOYSA-N 0.000 description 1
- 206010006451 bronchitis Diseases 0.000 description 1
- 208000000594 bullous pemphigoid Diseases 0.000 description 1
- 229940043232 butyl acetate Drugs 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 1
- 230000004611 cancer cell death Effects 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 230000005907 cancer growth Effects 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 229960002115 carboquone Drugs 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 208000002458 carcinoid tumor Diseases 0.000 description 1
- XREUEWVEMYWFFA-CSKJXFQVSA-N carminomycin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=C(O)C=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XREUEWVEMYWFFA-CSKJXFQVSA-N 0.000 description 1
- XREUEWVEMYWFFA-UHFFFAOYSA-N carminomycin I Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=C(O)C=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XREUEWVEMYWFFA-UHFFFAOYSA-N 0.000 description 1
- 229960003261 carmofur Drugs 0.000 description 1
- 210000000845 cartilage Anatomy 0.000 description 1
- 229950001725 carubicin Drugs 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 208000010353 central nervous system vasculitis Diseases 0.000 description 1
- 201000003565 cervix uteri carcinoma in situ Diseases 0.000 description 1
- 230000000739 chaotic effect Effects 0.000 description 1
- OGEBRHQLRGFBNV-RZDIXWSQSA-N chembl2036808 Chemical compound C12=NC(NCCCC)=NC=C2C(C=2C=CC(F)=CC=2)=NN1C[C@H]1CC[C@H](N)CC1 OGEBRHQLRGFBNV-RZDIXWSQSA-N 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 229940124444 chemoprotective agent Drugs 0.000 description 1
- 238000009104 chemotherapy regimen Methods 0.000 description 1
- 208000012191 childhood neoplasm Diseases 0.000 description 1
- 229950008249 chlornaphazine Drugs 0.000 description 1
- 229960001480 chlorozotocin Drugs 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- ZYVSOIYQKUDENJ-WKSBCEQHSA-N chromomycin A3 Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@@H]1OC(C)=O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@@H](O)[C@H](O[C@@H]3O[C@@H](C)[C@H](OC(C)=O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@@H]1C[C@@H](O)[C@@H](OC)[C@@H](C)O1 ZYVSOIYQKUDENJ-WKSBCEQHSA-N 0.000 description 1
- 208000016644 chronic atrophic gastritis Diseases 0.000 description 1
- 208000007451 chronic bronchitis Diseases 0.000 description 1
- 208000027157 chronic rhinosinusitis Diseases 0.000 description 1
- 229960002436 cladribine Drugs 0.000 description 1
- 208000029664 classic familial adenomatous polyposis Diseases 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 230000004154 complement system Effects 0.000 description 1
- 229940125758 compound 15 Drugs 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 229940088547 cosmegen Drugs 0.000 description 1
- ZYGHJZDHTFUPRJ-UHFFFAOYSA-N coumarin Chemical compound C1=CC=C2OC(=O)C=CC2=C1 ZYGHJZDHTFUPRJ-UHFFFAOYSA-N 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 229940111134 coxibs Drugs 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 1
- 239000000824 cytostatic agent Substances 0.000 description 1
- 230000001085 cytostatic effect Effects 0.000 description 1
- 229940070230 daypro Drugs 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 229960005052 demecolcine Drugs 0.000 description 1
- 229940070968 depocyt Drugs 0.000 description 1
- 201000001981 dermatomyositis Diseases 0.000 description 1
- XOZIUKBZLSUILX-UHFFFAOYSA-N desoxyepothilone B Natural products O1C(=O)CC(O)C(C)(C)C(=O)C(C)C(O)C(C)CCCC(C)=CCC1C(C)=CC1=CSC(C)=N1 XOZIUKBZLSUILX-UHFFFAOYSA-N 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- WTWPQOWYFZRSFA-UHFFFAOYSA-N diamino hydrogen phosphate Chemical compound NOP(O)(=O)ON WTWPQOWYFZRSFA-UHFFFAOYSA-N 0.000 description 1
- WVYXNIXAMZOZFK-UHFFFAOYSA-N diaziquone Chemical compound O=C1C(NC(=O)OCC)=C(N2CC2)C(=O)C(NC(=O)OCC)=C1N1CC1 WVYXNIXAMZOZFK-UHFFFAOYSA-N 0.000 description 1
- 229950002389 diaziquone Drugs 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 230000001079 digestive effect Effects 0.000 description 1
- 125000006371 dihalo methyl group Chemical group 0.000 description 1
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical group C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 229960000533 dornase alfa Drugs 0.000 description 1
- ZWAOHEXOSAUJHY-ZIYNGMLESA-N doxifluridine Chemical compound O[C@@H]1[C@H](O)[C@@H](C)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ZWAOHEXOSAUJHY-ZIYNGMLESA-N 0.000 description 1
- 229950005454 doxifluridine Drugs 0.000 description 1
- 229940115080 doxil Drugs 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 238000002651 drug therapy Methods 0.000 description 1
- 210000001198 duodenum Anatomy 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- VLCYCQAOQCDTCN-UHFFFAOYSA-N eflornithine Chemical compound NCCCC(N)(C(F)F)C(O)=O VLCYCQAOQCDTCN-UHFFFAOYSA-N 0.000 description 1
- 229960002759 eflornithine Drugs 0.000 description 1
- 229940121647 egfr inhibitor Drugs 0.000 description 1
- 238000002848 electrochemical method Methods 0.000 description 1
- 239000012039 electrophile Substances 0.000 description 1
- 229940087477 ellence Drugs 0.000 description 1
- 229950000549 elliptinium acetate Drugs 0.000 description 1
- 201000008184 embryoma Diseases 0.000 description 1
- 229940000733 emcyt Drugs 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 201000003914 endometrial carcinoma Diseases 0.000 description 1
- 210000001163 endosome Anatomy 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 229950011487 enocitabine Drugs 0.000 description 1
- 230000006353 environmental stress Effects 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 150000003885 epothilone B derivatives Chemical class 0.000 description 1
- XOZIUKBZLSUILX-GIQCAXHBSA-N epothilone D Chemical compound O1C(=O)C[C@H](O)C(C)(C)C(=O)[C@H](C)[C@@H](O)[C@@H](C)CCC\C(C)=C/C[C@H]1C(\C)=C\C1=CSC(C)=N1 XOZIUKBZLSUILX-GIQCAXHBSA-N 0.000 description 1
- LJQQFQHBKUKHIS-WJHRIEJJSA-N esperamicin Chemical compound O1CC(NC(C)C)C(OC)CC1OC1C(O)C(NOC2OC(C)C(SC)C(O)C2)C(C)OC1OC1C(\C2=C/CSSSC)=C(NC(=O)OC)C(=O)C(OC3OC(C)C(O)C(OC(=O)C=4C(=CC(OC)=C(OC)C=4)NC(=O)C(=C)OC)C3)C2(O)C#C\C=C/C#C1 LJQQFQHBKUKHIS-WJHRIEJJSA-N 0.000 description 1
- IIUMCNJTGSMNRO-VVSKJQCTSA-L estramustine sodium phosphate Chemical compound [Na+].[Na+].ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)OP([O-])([O-])=O)[C@@H]4[C@@H]3CCC2=C1 IIUMCNJTGSMNRO-VVSKJQCTSA-L 0.000 description 1
- MJVRPOLCSCYIRG-UHFFFAOYSA-N ethanol;furan Chemical compound CCO.C=1C=COC=1 MJVRPOLCSCYIRG-UHFFFAOYSA-N 0.000 description 1
- 229940031098 ethanolamine Drugs 0.000 description 1
- QSRLNKCNOLVZIR-KRWDZBQOSA-N ethyl (2s)-2-[[2-[4-[bis(2-chloroethyl)amino]phenyl]acetyl]amino]-4-methylsulfanylbutanoate Chemical compound CCOC(=O)[C@H](CCSC)NC(=O)CC1=CC=C(N(CCCl)CCCl)C=C1 QSRLNKCNOLVZIR-KRWDZBQOSA-N 0.000 description 1
- 229940093499 ethyl acetate Drugs 0.000 description 1
- 238000003810 ethyl acetate extraction Methods 0.000 description 1
- 125000000219 ethylidene group Chemical group [H]C(=[*])C([H])([H])[H] 0.000 description 1
- 229960005237 etoglucid Drugs 0.000 description 1
- LIQODXNTTZAGID-OCBXBXKTSA-N etoposide phosphate Chemical compound COC1=C(OP(O)(O)=O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 LIQODXNTTZAGID-OCBXBXKTSA-N 0.000 description 1
- 229960000752 etoposide phosphate Drugs 0.000 description 1
- 208000021126 eustachian tube disease Diseases 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 210000001723 extracellular space Anatomy 0.000 description 1
- 201000008815 extraosseous osteosarcoma Diseases 0.000 description 1
- 201000001155 extrinsic allergic alveolitis Diseases 0.000 description 1
- 206010016165 failure to thrive Diseases 0.000 description 1
- 229940087861 faslodex Drugs 0.000 description 1
- 229940087476 femara Drugs 0.000 description 1
- RDJGLLICXDHJDY-UHFFFAOYSA-N fenoprofen Chemical compound OC(=O)C(C)C1=CC=CC(OC=2C=CC=CC=2)=C1 RDJGLLICXDHJDY-UHFFFAOYSA-N 0.000 description 1
- 229960001419 fenoprofen Drugs 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 230000004992 fission Effects 0.000 description 1
- 229960005304 fludarabine phosphate Drugs 0.000 description 1
- 235000008191 folinic acid Nutrition 0.000 description 1
- 239000011672 folinic acid Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- YAKWPXVTIGTRJH-UHFFFAOYSA-N fotemustine Chemical compound CCOP(=O)(OCC)C(C)NC(=O)N(CCCl)N=O YAKWPXVTIGTRJH-UHFFFAOYSA-N 0.000 description 1
- 229960004783 fotemustine Drugs 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 150000002256 galaktoses Chemical class 0.000 description 1
- 201000010175 gallbladder cancer Diseases 0.000 description 1
- 201000007487 gallbladder carcinoma Diseases 0.000 description 1
- 229940044658 gallium nitrate Drugs 0.000 description 1
- 201000005649 gangliocytoma Diseases 0.000 description 1
- 201000011243 gastrointestinal stromal tumor Diseases 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 229940020967 gemzar Drugs 0.000 description 1
- 238000010363 gene targeting Methods 0.000 description 1
- 238000010914 gene-directed enzyme pro-drug therapy Methods 0.000 description 1
- 210000004907 gland Anatomy 0.000 description 1
- 229940080856 gleevec Drugs 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 150000002303 glucose derivatives Chemical class 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 210000002768 hair cell Anatomy 0.000 description 1
- 201000009277 hairy cell leukemia Diseases 0.000 description 1
- 235000015220 hamburgers Nutrition 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- 150000002373 hemiacetals Chemical class 0.000 description 1
- 208000007475 hemolytic anemia Diseases 0.000 description 1
- 125000004404 heteroalkyl group Chemical group 0.000 description 1
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 229940121372 histone deacetylase inhibitor Drugs 0.000 description 1
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 102000052196 human PF4 Human genes 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 229940042795 hydrazides for tuberculosis treatment Drugs 0.000 description 1
- 229940096120 hydrea Drugs 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- 229960001330 hydroxycarbamide Drugs 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 230000000148 hypercalcaemia Effects 0.000 description 1
- 208000030915 hypercalcemia disease Diseases 0.000 description 1
- 230000000222 hyperoxic effect Effects 0.000 description 1
- 208000022098 hypersensitivity pneumonitis Diseases 0.000 description 1
- 201000006362 hypersensitivity vasculitis Diseases 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 229940099279 idamycin Drugs 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- 208000015446 immunoglobulin a vasculitis Diseases 0.000 description 1
- DBIGHPPNXATHOF-UHFFFAOYSA-N improsulfan Chemical compound CS(=O)(=O)OCCCNCCCOS(C)(=O)=O DBIGHPPNXATHOF-UHFFFAOYSA-N 0.000 description 1
- 229950008097 improsulfan Drugs 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- SNWQUNCRDLUDEX-UHFFFAOYSA-N inden-1-one Chemical compound C1=CC=C2C(=O)C=CC2=C1 SNWQUNCRDLUDEX-UHFFFAOYSA-N 0.000 description 1
- 229940089536 indocin Drugs 0.000 description 1
- 150000002475 indoles Chemical class 0.000 description 1
- HOBCFUWDNJPFHB-UHFFFAOYSA-N indolizine Chemical compound C1=CC=CN2C=CC=C21 HOBCFUWDNJPFHB-UHFFFAOYSA-N 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 229960000905 indomethacin Drugs 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 238000007689 inspection Methods 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 229960003130 interferon gamma Drugs 0.000 description 1
- 229960001388 interferon-beta Drugs 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 238000003402 intramolecular cyclocondensation reaction Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- XMBWDFGMSWQBCA-YPZZEJLDSA-N iodane Chemical compound [125IH] XMBWDFGMSWQBCA-YPZZEJLDSA-N 0.000 description 1
- 229940044173 iodine-125 Drugs 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 208000001875 irritant dermatitis Diseases 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 210000002510 keratinocyte Anatomy 0.000 description 1
- 229960000991 ketoprofen Drugs 0.000 description 1
- OZWKMVRBQXNZKK-UHFFFAOYSA-N ketorolac Chemical compound OC(=O)C1CCN2C1=CC=C2C(=O)C1=CC=CC=C1 OZWKMVRBQXNZKK-UHFFFAOYSA-N 0.000 description 1
- BWHLPLXXIDYSNW-UHFFFAOYSA-N ketorolac tromethamine Chemical compound OCC(N)(CO)CO.OC(=O)C1CCN2C1=CC=C2C(=O)C1=CC=CC=C1 BWHLPLXXIDYSNW-UHFFFAOYSA-N 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 210000002429 large intestine Anatomy 0.000 description 1
- 201000011061 large intestine cancer Diseases 0.000 description 1
- 150000002605 large molecules Chemical class 0.000 description 1
- 201000010260 leiomyoma Diseases 0.000 description 1
- 229940115286 lentinan Drugs 0.000 description 1
- 229960003881 letrozole Drugs 0.000 description 1
- 229940063725 leukeran Drugs 0.000 description 1
- 238000013332 literature search Methods 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 208000037841 lung tumor Diseases 0.000 description 1
- 230000001926 lymphatic effect Effects 0.000 description 1
- 230000000527 lymphocytic effect Effects 0.000 description 1
- 208000025036 lymphosarcoma Diseases 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 208000026037 malignant tumor of neck Diseases 0.000 description 1
- 210000005075 mammary gland Anatomy 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- MQXVYODZCMMZEM-ZYUZMQFOSA-N mannomustine Chemical compound ClCCNC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CNCCCl MQXVYODZCMMZEM-ZYUZMQFOSA-N 0.000 description 1
- 229950008612 mannomustine Drugs 0.000 description 1
- 150000002703 mannose derivatives Chemical class 0.000 description 1
- 241001515942 marmosets Species 0.000 description 1
- 230000000873 masking effect Effects 0.000 description 1
- 229940087732 matulane Drugs 0.000 description 1
- 229960004616 medroxyprogesterone Drugs 0.000 description 1
- PSGAAPLEWMOORI-PEINSRQWSA-N medroxyprogesterone acetate Chemical compound C([C@@]12C)CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2CC[C@]2(C)[C@@](OC(C)=O)(C(C)=O)CC[C@H]21 PSGAAPLEWMOORI-PEINSRQWSA-N 0.000 description 1
- 208000023356 medullary thyroid gland carcinoma Diseases 0.000 description 1
- 229960004635 mesna Drugs 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- GXHMMDRXHUIUMN-UHFFFAOYSA-N methanesulfonic acid Chemical compound CS(O)(=O)=O.CS(O)(=O)=O GXHMMDRXHUIUMN-UHFFFAOYSA-N 0.000 description 1
- 125000004458 methylaminocarbonyl group Chemical group [H]N(C(*)=O)C([H])([H])[H] 0.000 description 1
- HRHKSTOGXBBQCB-VFWICMBZSA-N methylmitomycin Chemical compound O=C1C(N)=C(C)C(=O)C2=C1[C@@H](COC(N)=O)[C@@]1(OC)[C@H]3N(C)[C@H]3CN12 HRHKSTOGXBBQCB-VFWICMBZSA-N 0.000 description 1
- QTFKTBRIGWJQQL-UHFFFAOYSA-N meturedepa Chemical compound C1C(C)(C)N1P(=O)(NC(=O)OCC)N1CC1(C)C QTFKTBRIGWJQQL-UHFFFAOYSA-N 0.000 description 1
- 229950009847 meturedepa Drugs 0.000 description 1
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 1
- 230000033607 mismatch repair Effects 0.000 description 1
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 1
- 229960005485 mitobronitol Drugs 0.000 description 1
- 229960003539 mitoguazone Drugs 0.000 description 1
- MXWHMTNPTTVWDM-NXOFHUPFSA-N mitoguazone Chemical compound NC(N)=N\N=C(/C)\C=N\N=C(N)N MXWHMTNPTTVWDM-NXOFHUPFSA-N 0.000 description 1
- 229950010913 mitolactol Drugs 0.000 description 1
- VFKZTMPDYBFSTM-GUCUJZIJSA-N mitolactol Chemical compound BrC[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-GUCUJZIJSA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000004264 monolayer culture Methods 0.000 description 1
- HDZGCSFEDULWCS-UHFFFAOYSA-N monomethylhydrazine Chemical class CNN HDZGCSFEDULWCS-UHFFFAOYSA-N 0.000 description 1
- VYGYNVZNSSTDLJ-HKCOAVLJSA-N monorden Natural products CC1CC2OC2C=C/C=C/C(=O)CC3C(C(=CC(=C3Cl)O)O)C(=O)O1 VYGYNVZNSSTDLJ-HKCOAVLJSA-N 0.000 description 1
- FOYWNSCCNCUEPU-UHFFFAOYSA-N mopidamol Chemical compound C12=NC(N(CCO)CCO)=NC=C2N=C(N(CCO)CCO)N=C1N1CCCCC1 FOYWNSCCNCUEPU-UHFFFAOYSA-N 0.000 description 1
- 229950010718 mopidamol Drugs 0.000 description 1
- 208000001725 mucocutaneous lymph node syndrome Diseases 0.000 description 1
- 235000010460 mustard Nutrition 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 description 1
- 229960000951 mycophenolic acid Drugs 0.000 description 1
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 1
- YKYONYBAUNKHLG-UHFFFAOYSA-N n-Propyl acetate Natural products CCCOC(C)=O YKYONYBAUNKHLG-UHFFFAOYSA-N 0.000 description 1
- NJSMWLQOCQIOPE-OCHFTUDZSA-N n-[(e)-[10-[(e)-(4,5-dihydro-1h-imidazol-2-ylhydrazinylidene)methyl]anthracen-9-yl]methylideneamino]-4,5-dihydro-1h-imidazol-2-amine Chemical compound N1CCN=C1N\N=C\C(C1=CC=CC=C11)=C(C=CC=C2)C2=C1\C=N\NC1=NCCN1 NJSMWLQOCQIOPE-OCHFTUDZSA-N 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229960004270 nabumetone Drugs 0.000 description 1
- 229940089466 nalfon Drugs 0.000 description 1
- 229940090008 naprosyn Drugs 0.000 description 1
- 229960002009 naproxen Drugs 0.000 description 1
- 208000016366 nasal cavity polyp Diseases 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 229940086322 navelbine Drugs 0.000 description 1
- 230000017074 necrotic cell death Effects 0.000 description 1
- 230000001338 necrotic effect Effects 0.000 description 1
- QZGIWPZCWHMVQL-UIYAJPBUSA-N neocarzinostatin chromophore Chemical compound O1[C@H](C)[C@H](O)[C@H](O)[C@@H](NC)[C@H]1O[C@@H]1C/2=C/C#C[C@H]3O[C@@]3([C@@H]3OC(=O)OC3)C#CC\2=C[C@H]1OC(=O)C1=C(O)C=CC2=C(C)C=C(OC)C=C12 QZGIWPZCWHMVQL-UIYAJPBUSA-N 0.000 description 1
- 231100000417 nephrotoxicity Toxicity 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 230000003448 neutrophilic effect Effects 0.000 description 1
- VFEDRRNHLBGPNN-UHFFFAOYSA-N nimustine Chemical compound CC1=NC=C(CNC(=O)N(CCCl)N=O)C(N)=N1 VFEDRRNHLBGPNN-UHFFFAOYSA-N 0.000 description 1
- 229960001420 nimustine Drugs 0.000 description 1
- 229940109551 nipent Drugs 0.000 description 1
- 229960005130 niridazole Drugs 0.000 description 1
- YMVWGSQGCWCDGW-UHFFFAOYSA-N nitracrine Chemical compound C1=CC([N+]([O-])=O)=C2C(NCCCN(C)C)=C(C=CC=C3)C3=NC2=C1 YMVWGSQGCWCDGW-UHFFFAOYSA-N 0.000 description 1
- 229950008607 nitracrine Drugs 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 239000002840 nitric oxide donor Substances 0.000 description 1
- 108020001162 nitroreductase Proteins 0.000 description 1
- 125000005245 nitryl group Chemical group [N+](=O)([O-])* 0.000 description 1
- KGTDRFCXGRULNK-JYOBTZKQSA-N nogalamycin Chemical compound CO[C@@H]1[C@@](OC)(C)[C@@H](OC)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=C(O)C=C4[C@@]5(C)O[C@H]([C@H]([C@@H]([C@H]5O)N(C)C)O)OC4=C3C3=O)=C3C=C2[C@@H](C(=O)OC)[C@@](C)(O)C1 KGTDRFCXGRULNK-JYOBTZKQSA-N 0.000 description 1
- 229950009266 nogalamycin Drugs 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 230000000474 nursing effect Effects 0.000 description 1
- 235000003715 nutritional status Nutrition 0.000 description 1
- 230000000414 obstructive effect Effects 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- CZDBNBLGZNWKMC-MWQNXGTOSA-N olivomycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1)O[C@H]1O[C@@H](C)[C@H](O)[C@@H](OC2O[C@@H](C)[C@H](O)[C@@H](O)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@H](O)[C@H](OC)[C@H](C)O1 CZDBNBLGZNWKMC-MWQNXGTOSA-N 0.000 description 1
- 230000000174 oncolytic effect Effects 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- OFPXSFXSNFPTHF-UHFFFAOYSA-N oxaprozin Chemical compound O1C(CCC(=O)O)=NC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 OFPXSFXSNFPTHF-UHFFFAOYSA-N 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 229960003978 pamidronic acid Drugs 0.000 description 1
- 201000002094 pancreatic adenocarcinoma Diseases 0.000 description 1
- 229940096763 panretin Drugs 0.000 description 1
- 201000003913 parathyroid carcinoma Diseases 0.000 description 1
- 208000017954 parathyroid gland carcinoma Diseases 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- QIMGFXOHTOXMQP-GFAGFCTOSA-N peplomycin Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCCN[C@@H](C)C=1C=CC=CC=1)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C QIMGFXOHTOXMQP-GFAGFCTOSA-N 0.000 description 1
- 229950003180 peplomycin Drugs 0.000 description 1
- 230000000505 pernicious effect Effects 0.000 description 1
- 239000002831 pharmacologic agent Substances 0.000 description 1
- 230000006611 pharmacological activation Effects 0.000 description 1
- 125000004351 phenylcyclohexyl group Chemical group C1(=CC=CC=C1)C1(CCCCC1)* 0.000 description 1
- 208000028591 pheochromocytoma Diseases 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 150000003014 phosphoric acid esters Chemical class 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- FAIAAWCVCHQXDN-UHFFFAOYSA-N phosphorus trichloride Chemical compound ClP(Cl)Cl FAIAAWCVCHQXDN-UHFFFAOYSA-N 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- NJBFOOCLYDNZJN-UHFFFAOYSA-N pipobroman Chemical compound BrCCC(=O)N1CCN(C(=O)CCBr)CC1 NJBFOOCLYDNZJN-UHFFFAOYSA-N 0.000 description 1
- 229960000952 pipobroman Drugs 0.000 description 1
- NUKCGLDCWQXYOQ-UHFFFAOYSA-N piposulfan Chemical compound CS(=O)(=O)OCCC(=O)N1CCN(C(=O)CCOS(C)(=O)=O)CC1 NUKCGLDCWQXYOQ-UHFFFAOYSA-N 0.000 description 1
- 229950001100 piposulfan Drugs 0.000 description 1
- 229960001221 pirarubicin Drugs 0.000 description 1
- 150000003057 platinum Chemical class 0.000 description 1
- 229960003171 plicamycin Drugs 0.000 description 1
- 206010035653 pneumoconiosis Diseases 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 201000006292 polyarteritis nodosa Diseases 0.000 description 1
- 208000030761 polycystic kidney disease Diseases 0.000 description 1
- 208000037244 polycythemia vera Diseases 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 150000004804 polysaccharides Chemical class 0.000 description 1
- 229950004406 porfiromycin Drugs 0.000 description 1
- 230000002980 postoperative effect Effects 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 1
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 1
- 229910000105 potassium hydride Inorganic materials 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 208000030266 primary brain neoplasm Diseases 0.000 description 1
- 201000000742 primary sclerosing cholangitis Diseases 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 229940090181 propyl acetate Drugs 0.000 description 1
- 201000001514 prostate carcinoma Diseases 0.000 description 1
- 210000000064 prostate epithelial cell Anatomy 0.000 description 1
- 229940107568 pulmozyme Drugs 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 229950010131 puromycin Drugs 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- 150000004728 pyruvic acid derivatives Chemical class 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 229930192524 radicicol Natural products 0.000 description 1
- 230000003439 radiotherapeutic effect Effects 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- BMKDZUISNHGIBY-UHFFFAOYSA-N razoxane Chemical compound C1C(=O)NC(=O)CN1C(C)CN1CC(=O)NC(=O)C1 BMKDZUISNHGIBY-UHFFFAOYSA-N 0.000 description 1
- 229960000460 razoxane Drugs 0.000 description 1
- 230000036647 reaction Effects 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 238000005057 refrigeration Methods 0.000 description 1
- 229940087462 relafen Drugs 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 238000009774 resonance method Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 235000020945 retinal Nutrition 0.000 description 1
- 239000011604 retinal Substances 0.000 description 1
- 229930002330 retinoic acid Natural products 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- RZJQGNCSTQAWON-UHFFFAOYSA-N rofecoxib Chemical compound C1=CC(S(=O)(=O)C)=CC=C1C1=C(C=2C=CC=CC=2)C(=O)OC1 RZJQGNCSTQAWON-UHFFFAOYSA-N 0.000 description 1
- 229960003452 romidepsin Drugs 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 208000010157 sclerosing cholangitis Diseases 0.000 description 1
- 238000011519 second-line treatment Methods 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 238000003375 selectivity assay Methods 0.000 description 1
- 230000008313 sensitization Effects 0.000 description 1
- 206010040400 serum sickness Diseases 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- 229950001403 sizofiran Drugs 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- MFBOGIVSZKQAPD-UHFFFAOYSA-M sodium butyrate Chemical compound [Na+].CCCC([O-])=O MFBOGIVSZKQAPD-UHFFFAOYSA-M 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 229940063675 spermine Drugs 0.000 description 1
- 229950006315 spirogermanium Drugs 0.000 description 1
- 208000022159 squamous carcinoma in situ Diseases 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- CGPUWJWCVCFERF-UHFFFAOYSA-N staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(OC)O1 CGPUWJWCVCFERF-UHFFFAOYSA-N 0.000 description 1
- 230000036262 stenosis Effects 0.000 description 1
- 208000037804 stenosis Diseases 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- PVYJZLYGTZKPJE-UHFFFAOYSA-N streptonigrin Chemical compound C=1C=C2C(=O)C(OC)=C(N)C(=O)C2=NC=1C(C=1N)=NC(C(O)=O)=C(C)C=1C1=CC=C(OC)C(OC)=C1O PVYJZLYGTZKPJE-UHFFFAOYSA-N 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 229960005137 succinic acid Drugs 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- FDDDEECHVMSUSB-UHFFFAOYSA-N sulfanilamide Chemical compound NC1=CC=C(S(N)(=O)=O)C=C1 FDDDEECHVMSUSB-UHFFFAOYSA-N 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 238000005987 sulfurization reaction Methods 0.000 description 1
- MLKXDPUZXIRXEP-MFOYZWKCSA-N sulindac Chemical compound CC1=C(CC(O)=O)C2=CC(F)=CC=C2\C1=C/C1=CC=C(S(C)=O)C=C1 MLKXDPUZXIRXEP-MFOYZWKCSA-N 0.000 description 1
- 230000003319 supportive effect Effects 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 239000011885 synergistic combination Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- FQZYTYWMLGAPFJ-OQKDUQJOSA-N tamoxifen citrate Chemical compound [H+].[H+].[H+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O.C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 FQZYTYWMLGAPFJ-OQKDUQJOSA-N 0.000 description 1
- 229940099419 targretin Drugs 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229940063683 taxotere Drugs 0.000 description 1
- 229960001674 tegafur Drugs 0.000 description 1
- WFWLQNSHRPWKFK-ZCFIWIBFSA-N tegafur Chemical compound O=C1NC(=O)C(F)=CN1[C@@H]1OCCC1 WFWLQNSHRPWKFK-ZCFIWIBFSA-N 0.000 description 1
- 229940061353 temodar Drugs 0.000 description 1
- 206010043207 temporal arteritis Diseases 0.000 description 1
- ISIJQEHRDSCQIU-UHFFFAOYSA-N tert-butyl 2,7-diazaspiro[4.5]decane-7-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCCC11CNCC1 ISIJQEHRDSCQIU-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229960003433 thalidomide Drugs 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 238000004809 thin layer chromatography Methods 0.000 description 1
- 125000004149 thio group Chemical group *S* 0.000 description 1
- 150000003568 thioethers Chemical class 0.000 description 1
- YFTWHEBLORWGNI-UHFFFAOYSA-N tiamiprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC(N)=NC2=C1NC=N2 YFTWHEBLORWGNI-UHFFFAOYSA-N 0.000 description 1
- 230000008467 tissue growth Effects 0.000 description 1
- 235000015149 toffees Nutrition 0.000 description 1
- 229960001017 tolmetin Drugs 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- 229940019127 toradol Drugs 0.000 description 1
- 231100000167 toxic agent Toxicity 0.000 description 1
- 239000003440 toxic substance Substances 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 125000005270 trialkylamine group Chemical group 0.000 description 1
- PXSOHRWMIRDKMP-UHFFFAOYSA-N triaziquone Chemical compound O=C1C(N2CC2)=C(N2CC2)C(=O)C=C1N1CC1 PXSOHRWMIRDKMP-UHFFFAOYSA-N 0.000 description 1
- 229960004560 triaziquone Drugs 0.000 description 1
- OVCXRBARSPBVMC-UHFFFAOYSA-N triazolopyridine Chemical compound C=1N2C(C(C)C)=NN=C2C=CC=1C=1OC=NC=1C1=CC=C(F)C=C1 OVCXRBARSPBVMC-UHFFFAOYSA-N 0.000 description 1
- RXJKFRMDXUJTEX-UHFFFAOYSA-N triethylphosphine Chemical compound CCP(CC)CC RXJKFRMDXUJTEX-UHFFFAOYSA-N 0.000 description 1
- KCTAHLRCZMOTKM-UHFFFAOYSA-N tripropylphosphane Chemical compound CCCP(CCC)CCC KCTAHLRCZMOTKM-UHFFFAOYSA-N 0.000 description 1
- 229940086984 trisenox Drugs 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- UMKFEPPTGMDVMI-UHFFFAOYSA-N trofosfamide Chemical compound ClCCN(CCCl)P1(=O)OCCCN1CCCl UMKFEPPTGMDVMI-UHFFFAOYSA-N 0.000 description 1
- 229960000875 trofosfamide Drugs 0.000 description 1
- HDZZVAMISRMYHH-LITAXDCLSA-N tubercidin Chemical compound C1=CC=2C(N)=NC=NC=2N1[C@@H]1O[C@@H](CO)[C@H](O)[C@H]1O HDZZVAMISRMYHH-LITAXDCLSA-N 0.000 description 1
- 229940072651 tylenol Drugs 0.000 description 1
- 229950009811 ubenimex Drugs 0.000 description 1
- 231100000397 ulcer Toxicity 0.000 description 1
- 230000004222 uncontrolled growth Effects 0.000 description 1
- 229960001055 uracil mustard Drugs 0.000 description 1
- SPDZFJLQFWSJGA-UHFFFAOYSA-N uredepa Chemical compound C1CN1P(=O)(NC(=O)OCC)N1CC1 SPDZFJLQFWSJGA-UHFFFAOYSA-N 0.000 description 1
- 229950006929 uredepa Drugs 0.000 description 1
- 208000010570 urinary bladder carcinoma Diseases 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 208000022625 uterine cervix carcinoma in situ Diseases 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 229940054937 valstar Drugs 0.000 description 1
- 230000004862 vasculogenesis Effects 0.000 description 1
- 229940065658 vidaza Drugs 0.000 description 1
- JXLYSJRDGCGARV-CFWMRBGOSA-N vinblastine Chemical compound C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-CFWMRBGOSA-N 0.000 description 1
- CILBMBUYJCWATM-PYGJLNRPSA-N vinorelbine ditartrate Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O.OC(=O)[C@H](O)[C@@H](O)C(O)=O.C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC CILBMBUYJCWATM-PYGJLNRPSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- 229940053867 xeloda Drugs 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229950009268 zinostatin Drugs 0.000 description 1
- 229940002005 zometa Drugs 0.000 description 1
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/22—Amides of acids of phosphorus
- C07F9/24—Esteramides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/337—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having four-membered rings, e.g. taxol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/513—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim having oxo groups directly attached to the heterocyclic ring, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/683—Diesters of a phosphorus acid with two hydroxy compounds, e.g. phosphatidylinositols
- A61K31/685—Diesters of a phosphorus acid with two hydroxy compounds, e.g. phosphatidylinositols one of the hydroxy compounds having nitrogen atoms, e.g. phosphatidylserine, lecithin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/645—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having two nitrogen atoms as the only ring hetero atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/645—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having two nitrogen atoms as the only ring hetero atoms
- C07F9/6503—Five-membered rings
- C07F9/6506—Five-membered rings having the nitrogen atoms in positions 1 and 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6536—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having nitrogen and sulfur atoms with or without oxygen atoms, as the only ring hetero atoms
- C07F9/6539—Five-membered rings
- C07F9/65392—Five-membered rings containing two nitrogen atoms
- C07F9/65397—Five-membered rings containing two nitrogen atoms having the two nitrogen atoms in positions 1 and 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6564—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K33/00—Medicinal preparations containing inorganic active ingredients
- A61K33/24—Heavy metals; Compounds thereof
- A61K33/243—Platinum; Compounds thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Diabetes (AREA)
- Immunology (AREA)
- Hematology (AREA)
- Pulmonology (AREA)
- Rheumatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Urology & Nephrology (AREA)
- Physical Education & Sports Medicine (AREA)
- Neurology (AREA)
- Dermatology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pain & Pain Management (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Transplantation (AREA)
- Neurosurgery (AREA)
- Obesity (AREA)
- Biomedical Technology (AREA)
- Cardiology (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
当氨基磷酸酯烷化剂前体药物单独施用或与其它一种或多种抗肿瘤药联合施用时,氨基磷酸酯烷化剂前体药物可被用来治疗癌症。
Description
相关申请的交叉参考
[0001]本申请要求2005年6月29日提交的美国临时专利申请第60/695,755号的权益,将其引入本文作为参考。
发明背景
发明领域
[0002]本发明提供含有氨基磷酸酯烷化剂前体药物的组合物和使用氨基磷酸酯烷化剂前体药物治疗癌症和其它增生性疾病的方法。一般而言,本发明涉及化学、生物学、分子生物学、药理学和医学领域。
相关技术描述
[0003]用于癌症化学疗法的烷基化剂(“烷化剂(alkylator)”或“芥子(mustard)”)包括多种多样的化学药品类,所述化学药品具有在生理条件下烷化生物重要大分子(biologically vital macromolecule)如DNA的能力(参见Hardman等,The Pharmacological Basis of Therapeutics,2001,1389-1399,McGraw-Hill,NewYork,USA)。据推测,在烷化剂的抗肿瘤活性中,DNA烷化是重要的机制。化学治疗的烷化剂作为强亲电体,例如通过形成相邻杂原子稳定的鎓中间体如氮丙啶或氮丙啶鎓阳离子而起作用。
[0004]用于癌症疗法的氨基磷酸酯基烷化剂,例如环磷酰胺和异环磷酰胺是化学疗法烷化剂的重要亚类。环磷酰胺和异环磷酰胺都在肝脏被活化,并且释放的活性烷化剂烷化肿瘤细胞内的亲核部分如DNA,以便起着化学治疗剂的作用。如果活性烷化剂被释放离开肿瘤,则健康非癌细胞的生物分子的DNA和其它亲核部分如磷酸酯、氨基、巯基、羟基、羧基和咪唑并基团可能被烷化。健康细胞的这类烷化可以导致患者的不期望的毒性事件(toxic event)(参见Hardman等,同上)。

环磷酰胺 和 异环磷酰胺
[0005]仍需要能用来治疗癌症或其它增生性疾病的新型氨基磷酸酯基烷化剂,优选地为对正常细胞具有更低毒性的化合物。本发明满足这些需要并且提供新的氨基磷酸酯烷化剂前体药物,以及使用它们进行治疗的方法,如在下面的部分概述的。
发明简述
[0006]在一方面,本发明提供低氧活化的氨基磷酸酯烷化剂前体药物和合成它们的方法。本发明的氨基磷酸酯烷化剂前体药物可具有分子式ALK-T,其中ALK是氨基磷酸酯烷化剂,T是L-Z3,其中L是连接体,Z3是生物还原基团。
[0007]在一个方面,本发明提供了分子式(I)的氨基磷酸酯烷化剂前体药物:

其中
[0008]Y1是O、S、NR6、或NSO2R6,其中每一R6独立为C1-C6烷基、C1-C6杂烷基、芳基或杂芳基;
[0009]Y2是O、S、NR6、NCOR6或NSO2R6;
[0010]R1-R5的每一个独立地是氢、羟基、氨基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;或者R1-R5的任意两个一起形成C3-C10杂环;或者R1-R5的每一个独立为触发物T,其中T是L-Z3;
[0011]L选自
-[C(Z1)2-Y3]v-[C(=O)-O]q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]z-[-C(Z1)=C(Z1)]g-Z3和
-[C(Z1)2-Y3]v-(S(=O)2)q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]z-[C(Z1)=C(Z1)]g-Z3;其中每一个z、v、q、u和g独立地为0或者1。
[0012]Y3是O、S或NR7,其中每一R7独立地是氢、羟基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;
[0013]Y4是O、S或者-NR7-C(=O)-O-;
[0014]每一Z1独立为氢、卤素、C1-C6烷基、C1-C6杂烷基、芳基、杂芳基、C3-C8环烷基、杂环基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;
[0015]Z2是C1-C6亚烷基、C1-C6杂亚烷基,
其中每一X1独立为N或CR8,每一R8独立地是氢、卤素、硝基、氰基、CO2H、C1-C6烷基、C1-C6杂烷基、C1-C6环烷基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、CON(R7)2、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基:
[0016]X2是NR7、S或O;和
[0017]Z3选自:



[0018]条件是,在分子式(I)中:
[0019](i)R1-R5的至少两个选自2-卤烷基、2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;
[0020](ii)R1-R5的至少一个选自2-卤烷基、2-C1-C6烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;并且NR2R3和NR4R5中至少一个是 或者
或者
[0021](iii)NR2R3和NR4R5都合起来是以及
[0022]提供其单独的异构体或异构体的外消旋或非外消旋混合物、生物电子等排物、药效团、药学上可接受的盐、溶剂化物、水合物或前体药物。
[0023]在一实施方式中,Z3是在氧化还原反应中可接受一个或多个电子的生物还原基团。
[0024]在相关的实施方式中,本发明提供在处于低氧的细胞中具有50μM到0.01nm IC50或GI50的氨基磷酸酯烷化剂前体药物。在相关的实施方式中,本发明提供在相应的常氧的细胞中具有毒性低至百万倍、10,000倍和1000倍的低氧细胞毒性(hypoxic cytotoxicity)的氨基磷酸酯烷化剂前体药物。在相关的实施方式中,通过抗增殖试验并且使用含氧量低和常氧的细胞中的化合物的相对IC50值,测量细胞毒性。在相关的实施方式中,通过产克隆试验(clonogenic assay)并且使用含氧量低和常氧的细胞中的化合物的相对C10、C50或C90值,测量细胞毒性。
[0025]在另一相关的实施方式中,本发明提供在处于低氧的细胞中具有50μM到0.01nM IC50值的氨基磷酸酯烷化剂前体药物。在另一相关的实施方式中,本发明提供在相应的常氧的细胞中毒性低至5000倍的氨基磷酸酯烷化剂前体药物,如通过含氧量低和常氧的细胞中的相对IC50值进行测量。在另一相关的实施方式中,本发明提供氨基磷酸酯烷化剂前体药物,其在处于低氧的细胞中具有50μM到0.01nM的IC50,并且在相应的常氧的细胞中其毒性低至1000倍,如通过在含氧量低和常氧的细胞中的相对IC50值进行测量。
[0026]在相关的实施方式中,本发明的氨基磷酸酯烷化剂前体药物具有0.1nM到50μM的低氧细胞毒性(hypoxic cytotoxicity)和10到100,000的低氧细胞毒性比率(hypoxia cytotoxicity ratio)HCR,其通过常氧和含氧量低的细胞毒性的比率进行测量,并且其在本申请的后面进行更详细的定义。在相关的实施方式中,本发明的氨基磷酸酯烷化剂前体药物具有0.1nM到50μM的低氧细胞毒性和25到100,000的HCR。在另一相关的实施方式中,本发明的氨基磷酸酯烷化剂前体药物具有0.1nM到5μM的低氧细胞毒性和50到10,000的HCR。
[0027]在一方面,本发明提供用于治疗癌症和其它增生性疾病的新的氨基磷酸酯烷化剂。
[0028]在一方面,本发明提供包含本发明的氨基磷酸酯烷化剂前体药物和药学上可接受的赋形剂、载体或稀释剂的药物制剂。
[0029]在一方面,本发明提供治疗癌症和其它增生性疾病的方法,包括将治疗有效量的本发明的氨基磷酸酯烷化剂前体药物或者已知的氨基磷酸酯烷化剂前体药物施用给需要该治疗的患者。在一实施方式中,治疗的癌症对第一线、第二线或第三线治疗具有抗性,或者其是复发癌症。在另一实施方式,治疗的癌症是转移癌。在另一实施方式,本发明的氨基磷酸酯烷化剂前体药物或者已知的氨基磷酸酯烷化剂前体药物与至少另外的抗癌药剂(anticancer agent)联合施用。
附图简述
[0030]图1说明化合物25(50mg/kg)对H460异种移植小鼠模型中肿瘤生长的影响。
[0031]图2说明化合物25(100mg/kg)对H460异种移植小鼠模型中肿瘤生长的影响。
[0032]图3说明化合物25(150mg/kg)与CDDP联合给药对H460异种移植小鼠模型中肿瘤生长的影响。
[0033]图4说明化合物25与CDDP联合给药对H460异种移植小鼠模型中肿瘤生长的影响。
[0034]图5、6和7说明化合物与吉西他滨(Gemcitabine)联合给药对H460异种移植小鼠模型中肿瘤生长的影响。
发明详述
[0035]本发明不同的方面和实施方式的详细描述被组织如下:第I部分提供有用的定义;第II部分描述本发明的化合物和制备它们的方法;第III部分描述使用本发明的化合物单独或联合处理、治疗、施用和配制的方法;和第IV部分提供本发明化合物的合成方法和生物试验的实施例。该详细的描述被组织成各部分,仅仅是为了读者的方便,在任何部分的公开内容可用于本发明的任意方面。
I.定义
[0036]下面的定义被提供用来帮助读者。除非另有其它定义,本文应用的所有的技术术语、符号和其它科学或医学词语或术语,意图具有化学和医学领域的技术人员普遍理解的含义。在一些情况下,为了清楚和/或为了容易参考,具有普遍理解含义的术语在本文中被定义,在本文中加入这些定义不应被解释为表示与本领域通常理解的术语定义具有实质上的差异。
[0037]如本文所用,“一个(a)”或“一个(an)”指“至少一个(at least one)”或“一个或更多(one or more)”。
[0038]“烷基”指直链饱和单价烃基或支链饱和单价烃基,其具有词缀表示的碳原子数目。如在本公开内容所使用的,词缀(C1-Cqq)、Q1-qq或Q1-Cqq具有相同的含义,其中qq是从2-20的整数。例如(C1-C8)烷基、C1-8烷基或C1-C8烷基包括甲基、乙基、正丙基、2-丙基、正丁基、2-丁基、叔丁基、戊基和类似烷基。对于本文的每一定义(例如烷基、烯基、烷氧基、芳烷氧基(araalkyloxy)),当不含有表示烷基部分主链碳原子数的词缀时,其基团或部分将具有六个或更少的主链碳原子。任选地,(C1-C6)烷基可以任选地进一步用取代基取代,所述取代基包括,例如,氘(“D”)、羟基、氨基、单(C1-C6)烷基氨基或二(C1-C6)烷基氨基、卤素、C2-C6烯基醚、氰基、硝基、乙烯基、乙炔基、C1-C6烷氧基、C1-C6烷硫基、-COOH、-CONH2、单-(C1-C6)烷基-酰胺基或二-(C1-C6)烷基-酰胺基、-SO2NH2、-OSO2-(C1-C6)烷基、单(C1-C6)烷基磺酰氨基或二(C1-C6)烷基磺酰氨基、芳基、杂芳基、烷基磺酰氧基、杂烷基磺酰氧基、芳基磺酰氧基或杂芳基磺酰氧基。
[0039]“烯基”指直链单价烃基或支链单价烃基,其具有词缀表示的碳原子数目并且含有至少一个双键,但是不超过三个双键。例如(C2-C6)烯基包括乙烯基、丙烯基、1,3-丁二烯基和类似烯基。任选地,链烯基可以进一步用取代基取代,所述取代基包括,例如,氘(“D”)、羟基、氨基、单(C1-C6)烷基氨基或二(C1-C6)烷基氨基、卤素、C2-C6烯基醚、氰基、硝基、乙烯基、乙炔基、C1-C6烷氧基、C1-C6烷硫基、-COOH、-CONH2、单-(C1-C6)烷基-酰胺基或二-(C1-C6)烷基-酰胺基、-SO2NH2、-OSO2-(C1-C6)烷基、单(C1-C6)烷基磺酰氨基或二(C1-C6)烷基磺酰氨基、芳基、杂芳基、烷基或杂烷基磺酰氧基、以及芳基或杂芳基磺酰氧基。
[0040]“烷化剂”指通过与大分子上的亲核体的亲电反应可形成共价烷基键连接到大分子的反应部分。“氨基磷酸酯烷化剂(Phosphoramidate alkylator)”指存在氮丙啶或氮丙啶鎓亲电体或通过分子内环化产生氮丙啶或氮丙啶鎓亲电体的烷化剂。
[0041]“亚烷基”指具有一到十二个碳原子的直链饱和二价烃基或具有一到十二个碳原子的支链饱和二价烃基,任选地可以进一步用取代基取代,所述取代基包括,例如,氘(“D”)、羟基、氨基、单(C1-C6)烷基氨基或二(C1-C6)烷基氨基、卤素、C2-C6烯基醚、氰基、硝基、乙烯基、乙炔基、C1-C6烷氧基、C1-C6烷硫基、-COOH、-CONH2、单-(C1-C6)烷基-酰胺基或二-(C1-C6)烷基-酰胺基、-SO2NH2、-OSO2-(C1-C6)烷基、单(C1-C6)烷基磺酰氨基或二(C1-C6)烷基磺酰氨基、芳基、杂芳基、烷基或杂烷基磺酰氧基、以及芳基或杂芳基磺酰氧基。例如,亚烷基包括亚甲基、亚乙基、亚丙基、2-甲基-亚丙基、亚戊基、亚己基和类似亚烷基。
[0042]“杂亚烷基”基本上具有前面给出的亚烷基的含义,除了在亚烷基的双基中可以存在一个或多个杂原子(即氧、硫、氮和/或磷)。例如,杂亚烷基包括-CH2OCH2O-、-CH2CH2OCH2CH2-、-CH2CH2N(CH3)CH2CH2-、-CH2CH2SCH2CH2-和类似物。
[0043]“芳基”是指6到10个环原子的单价单环或双环芳族烃基,其被一个到八个,优选一个、两个、三个、四个或五个取代基独立取代,所述取代基选自氘(“D”)、烷基、环烷基、环烷基烷基、卤素、硝基、氰基、羟基、烷氧基、氨基、酰氨基、单烷基氨基、二烷基氨基、卤代烷基、卤代烷氧基、杂烷基、COR(其中R是氢、烷基、环烷基、环烷基烷基、苯基或苯基烷基)、-(CR′R")n-COOR(其中n是0至5的整数,R′和R"独立地为氢或烷基,R是氢、烷基、环烷基、环烷基烷基、苯基或苯基烷基)或-(CR′R")n-CONRxRy(其中n是0至5的整数,R′和R"独立地为氢或烷基,Rx和Ry独立地选自氢、烷基、环烷基、环烷基烷基、苯基或苯基烷基)。在一实施方式中,Rx和Ry一起为环烷基或杂环基。更具体地,术语芳基包括但不限于,苯基、联苯基、1-萘基和2-萘基,以及它们的取代形式。
[0044]“环烷基”是指具有3至7个环碳的单价环状烃基。环烷基可以具有一个或多个双键,任选地也可独立地用一个、两个、三个或四个取代基取代,所述取代基选自烷基、任选取代的苯基或-C(O)Rz(其中Rz为氢、烷基、卤代烷基、氨基、单烷基氨基、二烷基氨基、羟基、烷氧基或任选取代的苯基)。更具体地,术语环烷基包括,例如,环丙基、环己基、环已烯基、苯基环已基、4-羧基环已基、2-酰胺基-环已烯基、2-二甲基氨基羰基-环已基和类似物。
[0045]“杂烷基(Heteroalkyl)”是指如本文定义的烷基,其带有一个、两个或三个取代基,所述取代基独立地选自氰基、-ORw、-NRxRy和-S(O)PRZ(其中p是0至2的整数),可以理解,杂烷基的连接点是通过所述杂烷基的碳原子。Rw是氢、烷基、环烷基、环烷基-烷基、芳基、芳烷基、烷氧基羰基、芳氧基羰基、酰胺基、或单烷基氨基甲酰基或二烷基氨基甲酰基。Rx是氢、烷基、环烷基、环烷基-烷基、芳基或芳烷基(araalkyl)。Ry是氢、烷基、环烷基、环烷基-烷基、芳基、芳烷基、烷氧基羰基、芳氧基羰基、酰胺基、单-烷基甲酰基或二-烷基甲酰基或烷基磺酰基。Rz是氢(条件是n为0)、烷基、环烷基、环烷基-烷基、芳基、芳烷基、氨基、单烷基氨基、二烷基氨基或羟烷基。代表性实例包括,例如,2-羟乙基、2,3-二羟基丙基、2-甲氧基乙基、苄氧基甲基、2-氰乙基和2-甲基磺酰基-乙基。对于上述的每一个,Rw、Rx、Ry和Rz可以进一步被氨基、卤素、氟、烷基氨基、二烷基氨基、OH或烷氧基取代。另外,表示碳原子数目的词缀(例如C1-C10)是指杂烷基中除氰基、-ORW、-NRxRy或-S(O)PRZ部分之外的部分中的碳原子总数。
[0046]在一实施方式中,Rx和Ry一起为环烷基或杂环基。
[0047]“杂芳基”是指具有5至12个环原子的单价单环、双环或三环基,具有至少一个含有一个、两个或三个选自N、O或S的环杂原子的芳环,剩余的环原子为C,可以理解,杂芳基的连接点是在芳环上。杂芳环任选地被一个到八个,优选一个、两个、三个或四个取代基独立取代,所述取代基选自烷基、环烷基、环烷基-烷基、卤素、硝基、氰基、羟基、烷氧基、氨基、酰氨基、单烷基氨基、二烷基氨基、卤代烷基、卤代烷氧基、杂烷基、-COR(其中R是氢、烷基、苯基或苯基烷基)、-(CR′R")n-COOR(其中n是0至5的整数,R′和R"独立地为氢或烷基,R是氢、烷基、环烷基、环烷基-烷基、苯基或苯基烷基)或-(CR′R")n-CONRxRy(其中n是0至5的整数,R′和R"独立地为氢或烷基,Rx和Ry彼此独立地选自氢、烷基、环烷基、环烷基-烷基、苯基或苯基烷基)。在一实施方式中,Rx和Ry一起为环烷基或杂环基。更具体地,术语杂芳基包括但不限于吡啶基、呋喃基、噻吩基、噻唑基、异噻唑基、三唑基、咪唑基、异噁唑基、吡咯基、吡唑基、哒嗪基、嘧啶基、苯并呋喃基、四氢化苯并呋喃基、异苯并呋喃基、苯并噻唑基、苯并异噻唑基、苯并三唑基、吲哚基、异吲哚基、苯并噁唑基、喹啉基、四氢喹啉基、异喹啉基、苯并咪唑基、苯并异噁唑基或苯并噻吩基、吲唑基、吡咯并嘧啶基(pyrrolopyrymidinyl)、中氮茚基(indolizinyl)、吡唑并吡啶基、三唑并吡啶基、吡唑并嘧啶基、三唑并嘧啶基、吡咯并三嗪基、吡唑并三嗪基、三唑并三嗪基、吡唑并四嗪基、六吖茚基和七吖茚基、以及它们的衍生物。除非另外指明,杂原子在环中的排列可以是组成环原子的键合特性所允许的任何排列。
[0048]“杂环基”或“环杂烷基”是指具有3至8个环原子的饱和或不饱和非芳族环状基团,其中1至4个环原子是选自O、NR(其中R为氢、烷基、环烷基、环烷基烷基、苯基或苯基烷基)、P(=O)ORw或S(O)p(其中p是0至2的整数)的杂原子,其余的环原子是C,其中1个或2个C原子可以任选地被羰基取代。杂环基环可以任选地独立地用1、2、3或4个取代基取代,所述取代基选自烷基、芳基、芳烷基、杂芳基、杂芳烷基、环烷基、环烷基烷基、卤素、硝基、氰基、羟基、烷氧基、氨基、单烷基氨基、二烷基氨基、卤代烷基、卤代烷氧基、-COR(其中R是氢、烷基、环烷基、环烷基烷基、苯基或苯基烷基)、-(CR′R")n-COOR(n是0至5的整数,R′和R"独立地为氢或烷基,R是氢、烷基、环烷基、环烷基烷基、苯基或苯基烷基)、或-(CR′R")n-CONRxRy(其中n是0至5的整数,R′和R"独立地为氢或烷基,Rx和Ry彼此独立地为氢、烷基、环烷基、环烷基烷基、苯基或苯基烷基)。更具体地,术语杂环基包括但不限于吡啶基、四氢吡喃基、N-甲基哌啶-3-基、N-甲基吡咯烷-3-基、2-吡咯烷酮-1-基、呋喃基、喹啉基、噻吩基、苯并噻吩基、吡咯烷基、哌啶基、吗啉基、吡咯烷基、四氢呋喃基、四氢硫代呋喃基、1,1-二氧代-六氢-1Δ6-硫代吡喃-4-基、四氢咪唑并[4,5-c]吡啶基、咪唑啉基、哌嗪基和哌啶-2-酮基和它们的衍生物。表示碳原子数目的词缀(例如C3-C10)是指环杂烷基或杂环基部分中除杂原子数目之外的碳原子总数。
[0049]“C1-C6酰基”指-CO-(C1-C6烷基),其中术语烷基定义如上。
[0050]“C1-C6杂酰基”指-CO-(C1-C6杂烷基),其中术语杂烷基定义如上。
[0051]“芳酰基”指-CO-芳基,其中术语芳基定义如上。
[0052]“杂芳酰基”指-CO-杂芳基,其中术语杂芳基定义如上。
[0053]“Rsul磺酰氧基(Rsulsulfonyloxy)”指Rsu1-S(=O2)-O-,其包括烷基磺酰氧基、杂烷基磺酰氧基、环烷基磺酰氧基、杂环烷基磺酰氧基、芳基磺酰氧基和杂芳基磺酰氧基,其中Rsul分别是烷基、杂烷基、环烷基、杂环基、芳基和杂芳基,其中烷基、杂烷基、环烷基、杂环基、芳基和杂芳基定义如上。烷基磺酰氧基的实例包括Me-S(=O)2-O-、Et-S(=O)2-O-、CF3-S(=O)2-O-和类似物,芳基磺酰氧基的实例包括 和类似物。烷基磺酰氧基、杂烷基磺酰氧基、环烷基磺酰氧基、杂环烷基磺酰氧基、芳基磺酰氧基和杂芳基磺酰氧基基团在氨基磷酸酯烷化剂中可以是离去基团,在细胞中可被核酸如DNA或RNA和蛋白质的咪唑、羧酸酯或硫醇所取代,这引起烷基化以及细胞死亡。各种Rsul磺酰氧基基团与核酸、蛋白质或水的反应速率可根据例如Rsul部分的吸电子性质和空间体积进行调控,并且这些反应可提供氨基磷酸酯烷化剂和其前体药物,特别地,它们在肿瘤的普通和氧含量低的区域对肿瘤比对健康细胞具有更强的毒性。
和类似物。烷基磺酰氧基、杂烷基磺酰氧基、环烷基磺酰氧基、杂环烷基磺酰氧基、芳基磺酰氧基和杂芳基磺酰氧基基团在氨基磷酸酯烷化剂中可以是离去基团,在细胞中可被核酸如DNA或RNA和蛋白质的咪唑、羧酸酯或硫醇所取代,这引起烷基化以及细胞死亡。各种Rsul磺酰氧基基团与核酸、蛋白质或水的反应速率可根据例如Rsul部分的吸电子性质和空间体积进行调控,并且这些反应可提供氨基磷酸酯烷化剂和其前体药物,特别地,它们在肿瘤的普通和氧含量低的区域对肿瘤比对健康细胞具有更强的毒性。
[0054]“取代基”与在上述每一基团的定义中具体描述的取代基一起,指选自下列的取代基:氘、-卤素、-OR′、-NR′R"、-SR′、-SiR′R"R′"、-OC(O)R′、-C(O)R′、-CO2R′、-CONR′R"、-OC(O)NR′R"、-NR"C(O)R′、-NR′-、C(O)NR"R′"、-NR"C(O)2R′、-NH-C(NH2)=NH、-NR′C(NH2)=NH、-NH-C(NH2)=NR′、-S(O)R′、-S(O)2R′、-S(O)2NR′R"、-NR′S(O)2R"、-CN和-NO2、-R′、-N3、全氟(C1-C4)烷氧基和全氟(C1-C4)烷基,其数目在零到基团上开放化合价总数之间;和其中R′、R"和R′"独立地选自氢、C1-8烷基、C3-6环烷基、C2-8烯基、C2-8炔基、未取代的芳基和杂芳基、(未取代芳基)-C1-4烷基、和未取代烷氧基-C1-4烷基、用1-3个卤素取代的芳基、未取代的C1-8烷基、C1-8烷氧基或C1-8硫代烷氧基、或者未取代的芳基-C1-4烷基。当R′和R"被连接到同一氮原子,它们可与氮原子结合形成3-、4-、5-、6-或7-元环。例如-NR′R"指包括1-吡咯烷基和4-吗啉基。其它适合的取代基包括每一个通过1-4个碳原子的亚烷基范围连接到环原子的上述芳基取代基。芳基或杂芳基环的相邻原子上的取代基中的两个可任选被具有式-T2-C(O)-(CH2)q-U3-的取代基所取代,其中T2和U3独立地为-NH-、-O-、-CH2-或单键,并且q是从0到2的整数。可选地,芳基或杂芳基环的相邻原子上的取代基中的两个可任选被具有式-A-(CH2)r-B-的取代基所取代,其中A和B独立地为-CH2-、-O-、-NH-、-S-、-S(O)-、-S(O)2-、-S(O)2NR′-或单键,并且r是从1到3的整数。如此形成的新环的单键中的一个可任选地被双键所取代。可选地,芳基或杂芳基环的相邻原子上的取代基中的两个可任选被具有式-(CH2)s-X5-(CH2)t-的取代基所取代,其中s和t独立地为从0到3的整数,并且X5是-O-、-NR′-、-S-、-S(O)-、-S(O)2-或-S(O)2NR′-。在-NR′-和-S(O)2NR′中的取代基R′选自氢或未取代的C1-6烷基。
[0055]本发明的某些化合物拥有不对称碳原子(光学中心)或双键;外消旋物、非对映体、几何异构体、结构异构体(regioisomer)和单独的异构体(例如分离的对映体)都拟被包括在本发明的范围之内。本发明的化合物在组成这些化合物的一个或多个原子上也可包括非天然部分的原子同位素。例如,化合物可用放射性同位素放射标记,所述放射性同位素例如氚(3H)、碘-125(125I)或碳-14(14C)。本发明的化合物的所有同位素的变体——无论是不是具有放射性,都拟被包括在本发明的范围之内。
[0056]术语“药学上可接受的盐”是指包括用相对无毒性的酸或碱——这取决于在本文描述的化合物上的具体取代基,制备的活性化合物的盐。当本发明的化合物含有相对酸性官能度时,通过将这些化合物的中性形式与足够量的需要的碱相接触可得到碱性加成的盐,所述的碱是纯的或者在适合的惰性溶剂中。源自药学上可接受的无机碱的盐的实例包括:铝、铵、钙、铜、铁、亚铁、锂、锰、镁、亚锰、钾、钠、锌和类似物。源自药学上可接受的有机碱的盐包括下述物质的盐:伯胺、仲胺和叔胺,这包括取代的胺、环胺、天然存在的胺和类似胺,例如精氨酸、甜菜碱、咖啡碱、胆碱、N,N′-二卞基乙二胺、二乙胺、2-二乙氨基乙醇、2-二甲氨基乙醇、乙醇胺、乙二胺、N-乙基吗啉、N-乙基哌啶、葡糖胺(glucamine)、氨基葡糖(glucosamine)、组氨酸、哈胺、异丙胺、赖氨酸、甲基葡糖胺、吗啉、哌嗪、哌啶(piperadine)、聚胺树脂、普鲁卡因、嘌呤、可可碱、三乙胺、三甲胺、三丙胺、三羟甲基甲胺(tromethamine)和类似物。当本发明的化合物含有相对碱性官能度时,通过将这些化合物的中性形式与足够量的需要的酸相接触可得到酸性加成的盐,所述的酸是纯的或者在适合的惰性溶剂中。药学上可接受的酸加成的盐的实例包括源自无机酸的盐,所述无机酸如盐酸、氢溴酸、硝酸、碳酸、一氢碳酸、磷酸、一氢磷酸、二氢磷酸、硫酸、一氢硫酸、氢碘酸、或亚磷酸和类似物,以及源自相对无毒性的有机酸的盐,所述有机酸如乙酸、丙酸、异丁酸、马来酸、安息香酸、琥珀酸、辛二酸、反丁烯二酸、扁桃酸、邻苯二甲酸、苯磺酸、对-甲基磺酸(p-tolysulfonic acid)、柠檬酸、酒石酸、甲磺酸(methanesulfonic acid)和类似物。也包括氨基酸如精氨酸和类似物的盐,以及有机酸如葡糖醛酸或半乳糖醛酸和类似物的盐(参见如Berge,S.M.等,"Pharmaceutical Salts",Journal ofPharmaceutical Science,1977,66,1-19)。本发明某些具体的化合物同时含有碱性和酸性官能度,这样允许化合物被转化为碱或酸加成的盐。
[0057]化合物的中性形式可通过将盐与碱或酸接触并且用传统的方式分离母体化合物而再产生。化合物的母体形式在某些物理性质上与各种盐形式不同,例如在极性溶剂中的溶解度,但是在其它方面,对于本发明,盐与化合物的母体形式是相同的。
[0058]本发明的某些化合物可以以非溶剂化形式以及溶剂化形式存在,所述溶剂化形式包括水合形式。通常而言,溶剂化形式与非溶剂化形式相同,并拟被包括在本发明的范围内。本发明的某些化合物可以以多晶型或非晶型形式存在。通常而言,所有的物理形式对于本发明考虑的用途而言是相同的,并拟被包括在本发明的范围内。
[0059]如本文使用的,“葡萄糖类似物”包括单-、二-和三糖。葡萄糖类似物包括含有氨基葡糖、N-乙酰基-氨基葡糖的糖;果糖;甘露糖和甘露糖衍生物;葡萄糖和葡萄糖衍生物,包括但不限于2-脱氧葡萄糖(2-DG)、N-乙酰基-2-氨基-2-脱氧葡萄糖、3-氨基-3-脱氧-葡萄糖、2-氨基-2-脱氧-葡萄糖;和半乳糖和半乳糖衍生物,包括但不限于D-2-脱氧-D-半乳糖、D-4-氨基-4-脱氧-半乳糖和D-2-氨基-2-脱氧-半乳糖。因此,葡萄糖类似物可以与葡萄糖或衍生物如DG和氨基葡糖不同,因为其是其差向异构体。另外,葡萄糖类似物可以是任意前述化合物的氟化衍生物。而且,任意前述化合物的环中的氧可用选自硫、砜和类似物的等排物所取代。例如葡萄糖类似物可以是5-硫代-D-葡萄糖或其衍生物。
[0060]波浪线 指将一个基团或部分连接到另一个基团或部分的位置。例如
指将一个基团或部分连接到另一个基团或部分的位置。例如 和
和 表示硫代基团是连接另一个基团或部分的位置。
表示硫代基团是连接另一个基团或部分的位置。
[0061]术语CO、C(O)、-CO-在本文可以互相交换使用。术语CO2和COO在本文可以互相交换使用。术语SO2、S(O)2在本文可以互相交换使用。术语SO和S(=O)在本文可以互相交换使用。术语PO和P(=O)在本文可以互相交换使用。
[0062]如本文使用的,化学部分如分子、基团或原子的“生物等排物”指具有相似大小和空间位置的一对电子对或多对电子对的另外的化学部分。生物等排物和生物等排性是公知的用于预测化合物生物活性的工具,这基于如此假设:具有相似大小、形状和电子密度的化合物可具有相似的生物活性。已知的生物等排取代包括,例如-F、-OH、-NH2、-Cl和CH3的相互交换;-Br和-i-C3H7的互相交换;-I和-t-C4H9的互相交换;-O-、-S-、-NH-、-CH2-和-Se-的互相交换;-N=、-CH=和-P=(在环状或非环状部分)的互相交换;苯基和吡啶基的互相交换;-C=C-和-S-(例如苯和噻吩)的互相交换;芳族氮(Rar-N(Rar)-Rar)与不饱和碳(Rar-C(=Rar)-Rar)的互相交换;以及-CO-、-SO-和-SO2-的互相交换。这些实例不限于生物等排等价物的范围,本领域的技术人员将能鉴定本领域其它已知的生物等排取代。参见,例如Patani等,1996,Chem.Rev.96:3147-76;和Burger,1991,A Prog.Drug Res.37:287-371。
[0063]已知分子的结合能力或功能的合适的定量预测可基于分子中少量的原子或官能团的空间排列进行。如本文使用的,这种排列被称为“药效团”,在分子中的一个或多个药效团被鉴定后,该信息可被用来鉴定含有相同或相似药效团的其它分子。该方法对医学化学领域的普通技术人员是公知的,并且作为在本申请中描述的结构信息鉴别氨基磷酸酯烷化剂前体药物和氨基磷酸酯烷化剂的药效团。进行药效团相关搜索可用的程序的实例是来自Chemical Computing Group的3D药效团搜索程序(参见http://www.chemcomp.com/fdept/prodinfo.htm)。
[0064]“任选的”或“任选地”指其后描述的事件或情况可以发生,但不是必须发生,并且指该描述包括事件或情况发生的情形和事件或情况不发生的情形。例如,“任选地用烷基基团单或双取代的杂环基团”指,烷基可以但不是必须存在,并且该描述包括杂环基团被用烷基基团单或双取代的情形和杂环基团没有被用烷基基团取代的情形。
[0065]取代基或变化的组合是允许的,仅仅如果这类组合产生稳定或化学可行的化合物。稳定的化合物或化学可行的化合物是当被保持在4℃或更低温度下,在不存在湿气或其它化学反应条件时,化学结构可在至少一周内基本上不发生改变的化合物。
[0066]如本文使用的,“前体药物”是这样的化合物,其在给药后被代谢或通过其它方式转变为活性形式,或比前体药物本身在至少一个生物性质上更有活性的形式。为了产生前体药物,药学活性化合物(或其适当的前体)可以被化学修饰,以便修饰形式具有较低活性或失活,但是该化学修饰在某些生物条件下可被有效逆转,使得化合物的药学活性形式通过代谢或其它生物过程产生。相对于该药物而言,前体药物可以具有,例如改变的代谢稳定性或转运特性、较少的副作用或较低的毒性、或改善的味道(参见文献Nogrady,1985,Medicinal Chemistry ABiochemical Approach,Oxford University Press,New York,pages 388-392)。前体药物也可使用如此的化合物制备,这些化合物不是药物但是其在某些生物条件下活化后,产生药学活性化合物。如本文使用的氨基磷酸酯烷化剂前体药物是在活化后释放活性氨基磷酸酯烷化剂的前体药物。
[0067]如本文使用的“细胞毒剂”是对细胞产生毒性影响的试剂或化合物。如本文使用的,“细胞抑制剂”是抑制或压制细胞生长和增殖的试剂。
[0068]如本文使用的“低氧的细胞”是居留在低氧的体内环境中的细胞,例如在低氧的肿瘤区或在体外的细胞。如本文使用的“常氧的细胞”是居留在常氧的体内或体外环境中的细胞。如本文使用的化合物或试剂的“低氧细胞毒性”是其对低氧细胞的细胞毒性。如本文使用的化合物或试剂的“常氧细胞毒性”是其对常氧细胞的细胞毒性。
[0069]如本文使用的“生物还原基团”指在氧化还原反应中接受电子的基团。生物还原基团是下列基团:(1)可被还原,即,能接受电子、氢和/或氢负离子的基团;(2)可在体内和/或体外被还原;(3)可在低氧下在体内和/或体外被还原;(4)可通过DT-心肌黄酶、硫醇或通过光化学或电化学方法在体内和/或体外被还原;或(5)可通过生物方法例如酶水解、代谢等被除去和/或切割。
[0070]例如,以及如下面更详细的描述,一种生物还原基团是可用多种基团取代的硝基咪唑。生物还原基团的其它实例包括但不限于基于缺电子硝基苯(electron deficient nitrobenzene)、缺电子硝基苯甲酸酰胺、硝基吡咯(nitroazole)、硝基咪唑、硝基噻吩、硝基噻唑、硝基噁唑、硝基呋喃和硝基吡咯(nitropyrrole),其中可任选取代这些类部分的每一个,这样生物还原基团的氧化还原电位处于如此范围内——在该范围内在肿瘤的低氧条件下,基团可通过DT-心肌黄酶和/或硫醇进行还原。根据本文的公开内容,本领域的技术人员将理解如何取代这些和其它的生物还原基团,以提供具有处于所述范围内氧化还原电位的生物还原基团。
[0071]一般而言,本领域技术人员可通过修饰基团以含有吸电子基团、给电子基团或这类基团的组合,来“调整”生物还原基团的氧化还原电位。例如,硝基噻吩、硝基呋喃和硝基噻唑基团可用一种或多种给电子基团取代以达到需要的氧化还原电位,所述给电子基团包括但不限于甲基、甲氧基或氨基基团。在另一实例中,可用吸电子基团取代硝基吡咯部分以达到需要的氧化还原电位,所述吸电子基团包括但不限于氰基、酰胺、-CF3和氨磺酰基团。为了这个目标,可以使用强吸电子基团例如氰基、砜、氨磺酰、酰胺、或-CF3和轻度吸电子基团例如-CH2-卤素,其中卤素是-F、-Cl或-Br。
[0072]如本文使用的,“抗瘤药”、“抗肿瘤药”或“抗癌药”指用于治疗癌症的任何药剂。这类药剂可单独使用或者与其它化合物联合使用,它们可缓解、减轻、改善、抑制或者置于或保持在与瘤、肿瘤或癌症相关的临床症状或诊断标记的缓解状态。抗瘤剂包括但不限于抗血管生成药、烷基化剂(alkylating agent)或烷化剂(alkylator)、抗代谢物、某些天然产物、铂配位络合物(platinum coordinationcomplexe)、蒽醌、取代脲、甲基肼衍生物、皮质激素抑制剂、某些激素和拮抗剂、抗癌多糖、化学保护剂以及某些草本或其它植物提取液。
[0073]如本文使用的,“癌症”指异常细胞的不受控制的生长和扩散所导致的一类100多种疾病中的一种,其可以采取实体瘤、淋巴瘤和非实体瘤如白血病的形式。
[0074]如本文使用的,“恶性癌症”指具有转移能力、并且失去了生长和位置控制的癌细胞或者癌症。
[0075]如本文使用的,“瘤”(瘤形成)或“肿瘤”指异常的新细胞或组织生长,其可以是良性的或者恶性的。
[0076]如本文使用的,“治疗”疾病或患者是指采取步骤获得有益或所需的结果,包括临床结果。对于本发明,有益或所需的临床结果包括但不限于癌症或其它增生性疾病的一个或多个症状的缓解或改善,疾病程度减轻,疾病进展延迟或减慢,疾病状态改善、减轻或稳定,和如下所述的其它有益结果。
[0077]如本文使用的,一个症状或多个症状的“减轻(reduction)”(和此词语的语法等价词)是指降低症状(一种或多种)的严重程度或频率,或消除症状(一种或多种)。
[0078]如本文使用的,“给药”或向对象“给药”(和此词语的语法等价词)可以包括直接给药,包括自给药,和/或间接给药,包括开药物处方的行为。例如,如本文使用的,指导患者自给药和/或为患者提供药物处方的医生是向患者给药。
[0079]如本文使用的,药物的“治疗有效量”是这样的药物量,当给予患有癌症的对象时,其将具有预期的治疗效果,例如,对象的癌症的一个或多个表现的缓解、改善、减轻或消除。全部治疗效应不必通过给予一个剂量而出现,可以仅在给予一系列剂量之后出现。因此,治疗有效量可以在一次或多次给药中给予。
[0080]如本文使用的,药物的“预防有效量”是这样的药物量,当给予对象时,其将具有预期的预防效果,例如,防止或延迟疾病或症状的发生(或再发)或降低疾病或症状的发生(或再发)的可能性。全部预防效应不必通过给予一个剂量而出现,可以仅在给予一系列剂量之后出现。因此,预防有效量可以在一次或多次给药中给予。
[0081]如本文使用的,“第二线”治疗指如此治疗,其对一次化学疗法方案(regimen)或“第一线”化学疗法没有应答的癌症给于治疗。“第三线”治疗指如此治疗,当初始治疗——第一线治疗和接下来的治疗——第二线治疗不起作用或者不再起作用时,给与癌症的治疗。
[0082]如本文使用的“LogP”指物质亲脂性的测量,亲脂性基于辛醇和水之间物质的分配而确定。
IIa.化合物
[0083]大多数药物调节的癌症治疗——包括基于氨基磷酸酯烷化剂的治疗,依赖于被称为细胞毒剂的毒素,其对分裂细胞靶向具有选择性,所述细胞靶向例如其复制的DNA、微管和多种生长因子和生长因子受体。这些药物是有效的,因为通常癌细胞比正常细胞分裂更频繁。然而,这些药物不可避免地几乎不能杀死患者的所有癌细胞。一个原因是癌细胞可突变并且发展出药物抗性。另一个原因是不是所有的癌细胞都比正常细胞分裂更频繁,缓慢分裂的癌细胞可能如同正常细胞对这样的细胞毒剂一样不敏感,或者甚至比正常细胞对细胞毒剂更不敏感。
[0084]有些癌细胞存在于血管化较差的实体瘤中,不能产生细胞分裂所需要的能量,并且分裂缓慢。当肿瘤生长时,它需要血液供应,因而需要生长出新的血管。支持肿瘤生长的新血管通常是混乱的;这使肿瘤的重要区域血管化较差,并且甚至使血管化区域受到间歇式阻塞。肿瘤中这些血管化较差的和阻塞的区域的含氧量变低——与相应的正常组织相比它们具有较低的氧浓度或较低的氧分压,它们中的细胞表现出较慢的分裂速率。因而,仅有10%的实体瘤的中值氧浓度(median oxygen concentration)落入了正常范围40-60mm Hg内,而有50%的实体瘤的中值氧浓度低于10mm Hg。
[0085]肿瘤的低氧区域代表癌细胞转移和癌细胞对抗治疗的重要来源(参见,例如De Jaeger等,Br J Cancer.2001,84(9):1280-5和Rofstad等,Br J Cancer.1999,80(11):1697-707)。那么,不值得奇怪的是,较低肿瘤氧水平与对治疗反应不足、增加的转移和较差的存活率相关。环磷酰胺和异环磷酰胺的活化和作用的机制可举例说明这些药剂如何不可能特异性靶向该困难,以杀死低氧区域的肿瘤。
[0086]环磷酰胺和异环磷酰胺都是前体药物并且可在肝脏中经由中间体被氧化激活,以产生活性氨基磷酸酯烷化剂,分别为烷化剂1(环磷酰胺芥子)和烷化剂2(异环磷酰胺芥子)(参见下面)。电中性半缩醛1和2可以具有数分钟的半衰期,并且可渗透进或渗透出细胞。相反,阴离子烷化剂1和2具有小得多的细胞膜渗透能力,并且一旦在细胞外形成,不能有效地通过烷化细胞DNA而杀死细胞。
[0087]当氨基磷酸酯烷化剂到达肿瘤,它们通常杀死快速生长、良好血管化、常氧、肿瘤外部区域的细胞。然而,这些氨基磷酸酯烷化剂在渗透入血管化更差、较缓慢生长、逐渐低氧的内部肿瘤区域方面并且在杀死其内的肿瘤细胞方面不是那么有效。在任意的这些活化烷化剂到达肿瘤之前,它们可与健康细胞反应并且导致毒性和/或细胞死亡。

[0088]虽然低氧肿瘤很难治疗,但低氧肿瘤区可以产生各种化学基团的还原衍生物(参见文献Workman等,1993,Cancer and Metast.Rev.12:73-82),并且细胞毒素的前体药物可以被开发以利用这种生物还原环境(PCT申请US04/009667和US 05/08161;PCT/US2005/041959和PCT/US2005/042095,都属于Matteucci等)。通过一起使用生物还原基团(Z3)和烷化剂,可构建这类低氧还原(或低氧活化)前体药物。生物还原基团可用作共价结合或连接到氨基磷酸酯烷化剂的触发物(Trigger)部分的一部分。
[0089]本发明的化合物通常被描述为氨基磷酸酯烷化剂前体药物。一般而言,本发明的氨基磷酸酯烷化剂前体药物具有下列结构:
Alk-触发物
其中Alk是氨基磷酸酯烷化剂,触发物T具有结构L-Z3,其中连接体L被结合到生物还原基团Z3。在一实施方式中,触发物T是低氧激活触发物。
[0090]氨基磷酸酯烷化剂衍生物在下列参看文献中被报道:Borch等,J.Med.Chem.2000,43:2258-65;2001,44:69-73;2001,44:74-7;Hernick等J.Med.Chem.2002,45:3540-8;Hernick等,J.Med.Chem.2003,46:148-54;美国专利第4,908,356;5,306,727;5,403,932;5,190,929;5,472,956;和6,656,926号;美国专利申请公开第2003/0008850号;和Papot等,Curr.Med.Chem.,2002,2,155-85,它们分离的化合物在其中被公开,这不是本发明的主题。在一些实施方式中,本发明的氨基磷酸酯烷化剂前体药物具有一种或多种下列特性:(i)在低氧的组织中,更高的低氧毒性或更低的IC50或IC90值,(ii)更低的含氧正常的细胞毒性,以及(iii)更低的毒副作用情况或这些特性的一些组合。在一些实施方式中,本发明的氨基磷酸酯烷化剂前体药物与已知的氨基磷酸酯烷化剂衍生物不同,在于:(i)释放的氨基磷酸酯烷化剂的性质,(ii)连接体(L)和/或生物还原基团Z3的性质,(iii)一种以上生物还原基团部分的存在,或这些特性的一些组合,(iv)通过更大的HCR值测定的增加的低氧选择性细胞毒性,(v)增加的水溶性,(vi)增加的对肝脏微粒降解的稳定性和/或(vii)提供有效的氨基磷酸酯烷化剂前体药物,所述前体药物是非手性的,并且避免在体内代谢中特异性的对映体。
[0091]为了理解为什么本发明的前体药物化合物代表对已知抗癌氨基磷酸酯烷化剂衍生物的明显进步,了解肿瘤生物学特别是在低氧条件下的肿瘤生物学,了解本文提供的前体药物的药物动力学和药效动力学是特别有帮助的。
[0092]为了有效的肿瘤治疗,低氧活化前体药物对于健康的常氧的细胞比对低氧肿瘤细胞具有少得多的毒性。在一些实施方式中,本发明的低氧活化前体药物对于常氧的细胞比对于低氧细胞具有更少的活性和更低的毒性。当这类本发明的前体药物遇倒实体瘤组织内的低氧、还原环境时,生物还原基团的还原引起氨基磷酸酯烷化剂或活性细胞毒素的分解。在肿瘤区内释放氨基磷酸酯烷化剂,并且可更容易地渗入实体瘤的低氧区。这些氨基磷酸酯烷化剂可在很难到达的实体瘤的低氧区杀死细胞,同时减少了非癌健康细胞的死亡和对患者的毒副作用。因此,本发明提供低氧活化前体药物,对比于低氧、肿瘤细胞,其对健康、常氧的细胞具有低得多的毒性。
[0093]在某些实施方式中,本发明的氨基磷酸酯烷化剂前体药物使用含有硝基的芳族或吲哚醌部分作为触发物T中的生物还原基团。在低氧肿瘤中,硝基基团被还原为羟氨基或氨基基团,电子对从氨基或羟氨基流动穿过触发物T的共轭π电子体系释放氨基磷酸酯烷化剂。在另一实施方式中,在低氧肿瘤中,吲哚醌被还原为吲哚氢醌,电子对从氢醌流动穿过触发物T释放氨基磷酸酯烷化剂。释放的氨基磷酸酯烷化剂在低氧肿瘤中和/或在低氧肿瘤附近杀死细胞。
[0094]许多酶可负责触发物中生物还原基团Z3的还原。例如,细胞色素P450还原酶可在第一步分别将生物还原基团中的硝基或醌部分还原成NO2(-)或半醌自由基阴离子。与常氧的组织相比,低氧肿瘤区可具有更高浓度的还原酶。在常氧的请况下,如在良好血管化的健康组织中,在存在氧的情况下,形成的NO2(-)或半醌自由基阴离子可与氧反应以回复到生物还原基团,并且最终不会产生或释放氨基磷酸酯烷化剂。共价结合到NO2(-)或半醌自由基阴离子的芳基或杂芳基部分调控自由基阴离子的氧敏感性。
[0095]生物还原基团的氧敏感性可部分地根据生物还原基团的还原电位而变化。因此,例如,一生物还原基团在具有1%氧的低氧肿瘤区可被还原,另一生物还原基团在具有0.1%氧的低氧肿瘤区被还原,而又一生物还原基团在具有0.01%氧的低氧肿瘤区被还原。
[0096]当生物还原基团如此容易被还原以致细胞色素P450还原酶或其它还原剂(“还原剂”)在健康常氧的组织中,在存在氧的情况下可将其还原时,生物还原基团失去了一些或所有其低氧特异性。如果生物还原基团中的NO2(-)或半醌自由基阴离子不与氧反应或者与氧反应缓慢,自由基阴离子本身可释放氨基磷酸酯烷化剂,或者其可被进一步还原并且释放氨基磷酸酯烷化剂,这引起对健康常氧的细胞和组织的毒性。本发明的新的氨基磷酸酯烷化剂前体药物对低氧癌细胞和组织比对健康常氧的细胞和组织的毒性更大。
[0097]将生物还原基团Z3还原的容易或困难性可通过生物还原基团的还原电位进行量度,并且其受连接体(L)和氨基磷酸酯烷化剂(Alk-H)影响。例如,与将生物还原基团共价结合到富含电子的连接体或富含电子的氨基磷酸酯烷化剂时相比,将生物还原基团连接到吸电子连接体或吸电子氨基磷酸酯烷化剂可使生物还原基团更容易被还原。
[0098]触发物T可被氧化、水解或硫化,并且可以以低氧非选择性(non-senselective)方式释放氨基磷酸酯烷化剂。TelcytaTM,一种临床的氨基磷酸酯烷化剂前体药物,可在不存在低氧的情况下,通过谷胱甘肽转移酶的作用释放活性毒素(参见,例如,“Methods of Treatment”部分的氨基磷酸酯烷化剂1f)。关于氨基磷酸酯烷化剂的释放,连接体和/或氨基磷酸酯烷化剂的化学性质可影响前体药物的氧化、水解或硫解的稳定性。在本发明的一实施方式中,低氧活化的氨基磷酸酯烷化剂前体药物在低氧非特异性、氧化、水解或硫解中,不释放氨基磷酸酯烷化剂。
[0099]根据本发明,在氨基磷酸酯烷化剂前体药物中合适地使用的触发物T可被用来“调整”前体药物的药物动力学,而不会改变细胞毒性性质。例如,抗癌药的大体积分布确保前体药物被组织很快吸收。根据本发明,在一实施方式中,氨基磷酸酯烷化剂前体药物分布的体积,可通过使用含有在生理条件下可形成铵阳离子的氨基基团的触发物T进行调节。在一实施方式中,含有季铵基团的触发物T可产生本发明的前体药物化合物,其具有大体积分布同时避免可能的内体截留。在另一实施方式,含有羰基官能度的触发物T将作为阴离子羧酸酯阴离子形式存在。正常健康组织外部的细胞外空间的CO2(-)不容易穿过正常细胞膜。肿瘤细胞外空间的更低pH可将CO2(-)转化为不带电荷的“CO2H”形式,这允许前体药物通过肿瘤细胞膜。
[0100]含有羟基、氨基、巯基和/或羰基基团的氨基磷酸酯烷化剂可被转化为前体药物,这是通过将触发物T共价结合到一个或多个这些官能团上完成的。在从氨基磷酸酯烷化剂转化为前体药物的过程中,氨基磷酸酯烷化剂中的羟基基团可被转化为例如醚或缩醛;氨基被转化为烷基氨基、氨基甲酸酯或酰胺;羰基基团转化为酯;以及巯基团转化为硫醚或硫代酰基,如下面在合成方法和实验部分更详细描述的。这些转化可产生前体药物,与相应的氨基磷酸酯烷化剂相比,其具有更低的极性或更高的亲脂性。非极性氨基磷酸酯烷化剂前体药物不容易溶于含水药物载体或稀释液中。可溶性增强子基团如CO2H、氨基、烷基氨基、二烷基氨基和羟基可被使用在触发物T中,以调整前体药物的溶解度,并且克服在制备氨基磷酸酯烷化剂前体药物的含水制剂中遇到的任何问题。
[0101]本发明的氨基磷酸酯烷化剂可具有一个或多个N-(2-卤烷基)或N-(2-卤乙基)和/或一个或多个共价结合到P=O部分的氮丙啶 部分,如下文所示。在阴离子氨基磷酸酯烷化剂部分释放后,氮丙啶或氮丙啶鎓类形成,其可烷化DNA(参见实施例部分,实施例36)。取决于R2和R3取代基的吸电子特性,氮丙啶鎓形成动力学可变化。例如,如下面的反应顺序所示,当NR2R3部分从NH2改变为
部分,如下文所示。在阴离子氨基磷酸酯烷化剂部分释放后,氮丙啶或氮丙啶鎓类形成,其可烷化DNA(参见实施例部分,实施例36)。取决于R2和R3取代基的吸电子特性,氮丙啶鎓形成动力学可变化。例如,如下面的反应顺序所示,当NR2R3部分从NH2改变为 时,烷化的速度可增加(参见Engle等,J.Med.Chem.,1987,25:1347-57)。氮原子上的取代基可改变氨基磷酸酯烷化剂的几何形状、P=O部分中的该氮原子上的孤对电子的离域作用、用于氮丙啶鎓或随后的氮丙啶形成的氮孤对电子的有效性、以及氨基磷酸酯烷化剂前体药物和氨基磷酸酯烷化剂的水溶性。
时,烷化的速度可增加(参见Engle等,J.Med.Chem.,1987,25:1347-57)。氮原子上的取代基可改变氨基磷酸酯烷化剂的几何形状、P=O部分中的该氮原子上的孤对电子的离域作用、用于氮丙啶鎓或随后的氮丙啶形成的氮孤对电子的有效性、以及氨基磷酸酯烷化剂前体药物和氨基磷酸酯烷化剂的水溶性。

[0102]本发明部分源于如此发现:使用2-硝基咪唑-生物还原基团的氨基磷酸酯烷化剂前体药物表现了出人意料高的低氧细胞毒性,低常氧毒性和高HCR和提高的溶解度。例如,在处于低氧的细胞中用0.05μM的IC50的抗增殖细胞毒性试验中,化合物24和25在低氧细胞中的毒性分别为常氧细胞中的400到1000倍(参见实施例部分)。在抗增殖细胞毒性试验中,含有异环磷酰胺芥子或异环磷酰胺芥子类似物和具有下列分子式的氨基磷酸酯烷化剂前体药物,出人意料地在低氧细胞比常氧的细胞中毒性更高,和/或与已知的具有2-硝基噻吩-5-基、2-硝基呋喃-5-基或5-硝基咪唑基、生物还原基团(Z3)和N,N’(四-2-氯乙基(choloroethyl))氨基磷酸酯芥子或环磷酰胺芥子,或者以吲哚醌基团作为Z3和具有异环磷酰胺芥子的氨基磷酸酯烷化剂衍生物的HCR值相比,拥有出人意料高的HCR值(参见,例如,Borch等,J.Med.Chem.中化合物P4,P14-17,P19,和P21-22,和美国专利第6,656,926号,它们都在前面提到),所述分子式为:Z3-CH2-O-P(=O)(NHCH2CH2X4)2、Z3-CH2-O-P(=O)(NHCH(R9)CH2X4)2和Z3-CH(Z2)-O-P(=O)(NHCH(R9)CH2X4)2;其中Z2是甲基;R9是氢、甲基或异丙基;Z3是1-N-甲基-2-硝基咪唑-5-基、2-硝基噻吩-5-基或2-硝基呋喃-5-基;并且每一个X4是Cl或Br。
[0103]在一个方面,本发明提供了具有分子式(I)的氨基磷酸酯烷化剂前体药物:

其中
[0104]Y1是O、S、NR6或NSO2R6,其中每一R6独立为C1-C6烷基、C1-C6杂烷基、芳基或杂芳基;
[0105]Y2是O、S、NR6、NCOR6或NSO2R6;
[0106]R1-R5的每一个是氢、羟基、氨基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;或者R1-R5的任意两个一起形成C3-C10杂环;或者R1-R5的每一个独立为触发物T,其中触发物是L-Z3;
[0107]L选自
-[C(Z1)2-Y3]v-[C(=O)-O]q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]z-[-C(Z1)=C(Z1)]g-;和
-[C(Z1)2-Y3]v-(S(=O)2)q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]2-[C(Z1)=C(Z1)]g-;
其中每一个z、v、q、u和g独立为0或者1;
[0108]Y3是S、O或NR7,其中每一R7独立地是氢、羟基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;
[0109]Y4是O、S或者-NR7-C(=O)-O-;
[0110]每一Z1独立为氢、卤素、C1-C6烷基、C1-C6杂烷基、芳基、杂芳基、C3-C8环烷基、杂环基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;
[0111]Z2是C1-C6亚烷基、C1-C6杂亚烷基,
[0112]其中每一X1独立为N或CR8,其中R8独立地是氢、卤素、OH、OP(=O)(OH)2、硝基、氰基、CO2H、C1-C6烷基、C1-C6杂烷基、C1-C6环烷基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、CON(R7)2、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;
[0113]X2是NR7、S或O;和
[0114]Z3是选自下面的生物还原基团:

和 条件是,在分子式(I)中:
条件是,在分子式(I)中:
[0115](i)R1-R5的至少两个选自2-卤烷基、2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;
[0116](ii)R1-R5的至少一个选自2-卤烷基、2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;并且NR2R3和NR4R5的至少一个是 或者
或者
[0117](iii)NR2R3和NR4R5都是 以及
以及
[0118]提供其单独的异构体或异构体的外消旋或非外消旋混合物、生物电子等排物、药效团、药学上可接受的盐、溶剂化物、水合物或前体药物。
[0119]在一实施方式中,z是1。
[0120]在一实施方式中,R2-R5不是相同的。
[0121]在一实施方式中,R2-R5的任意一个是
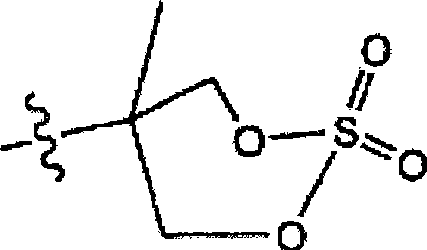

[0122]在一实施方式中,本发明提供每一个使用两个氨基磷酸酯烷化剂的低氧活化氨基磷酸酯烷化剂前体药物。在一实施方式中,本发明的氨基磷酸酯烷化剂前体药物使用1-N-烷基-2-硝基咪唑-5-基部分或1-N-甲基-2-硝基咪唑-5-基部分作为生物还原基团或Z3。在一实施方式中,本发明的氨基磷酸酯烷化剂前体药物使用2-硝基呋喃部分作为生物还原基团或Z3。
[0123]在一实施方式中,本发明不包括下列化合物:



其中Ra是H、Br(P14)、NMe2(P15)、CN(P16)或CONH2(P17),





其中R是H、Me或烯丙基;
[0124]3-(5-甲氧基-1-甲基-4,7-吲哚醌基)-甲基二[N-甲基-N-(2-溴乙基)]二氨基磷酸酯(P27),
[0125]3-(5-甲氧基-1-甲基-4,7-吲哚醌基)甲基N,N-二(2-溴乙基)-二氨基磷酸酯(P28),
[0126]2-(5-甲氧基-1-甲基-4,7-吲哚醌基)甲基二[N-甲基-N-(2-溴乙基)]二氨基磷酸酯(P29),
[0127]2-(5-甲氧基-1-甲基-4,7-吲哚醌基)甲基N,N-二(2-氯乙基)二氨基磷酸酯(P30),
[0128]2-(5-甲氧基-1-甲基-4,7-吲哚醌基)甲基N,N-二(2-溴乙基)二氨基磷酸酯(P31),
[0129]3-(5-甲氧基-1-甲基-4,7-吲哚醌基)甲基N,N-二(2-溴乙基)二氨基磷酸酯(P32),
[0130]2-(5-甲氧基-1-甲基-4,7-吲哚醌基)甲基二[N-甲基-N-(2-溴乙基)]二氨基磷酸酯(P33),
[0131]2-(5-甲氧基-1-甲基-4,7-吲哚醌基)甲基N,N-二(2-氯乙基)二氨基磷酸酯(P34),和
[0132]2-(5-甲氧基-1-甲基-4,7-吲哚醌基)甲基N,N-二(2-溴乙基)二氨基磷酸酯(P35)。
[0133]在相关实施方式中,本发明提供分子式(I)的化合物,条件是:
[0134](i)R1-R5的至少一个选自2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基,以及
[0135]R1-R5的至少一个选自2-卤烷基、2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;或者
[0136](ii)R1-R5的至少一个选自2-卤烷基、2-C1-C6烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;并且NR2R3和NR4R5的至少一个是或者
[0137](iii)NR2R3和NR4R5每一个是
[0138]在另一相关的实施方式中,本发明提供分子式(I)的化合物,条件是分子式(I)不包括R2和R3一起形成吗啉环或者R4和R5一起形成吗啉环。
[0139]在一实施方式中,本发明不包括具有下列结构的化合物:

其中,Z1是氢或C1-C6烷基。
[0140]在一实施方式中,本发明提供化合物,其中触发物T是:
[C(Z1)2-Y3]-(C(=O)-O)-[C(Z1)2-Z2-Y4]-[C(Z1)2]z-[C(Z1)=C(Z1)]-Z3;
[C(Z1)2-Y3]-[C(Z1)2-Z2-Y4]-[C(Z1)2]z-[C(Z1)=C(Z1)]-Z3;
[C(Z1)2-Y3]-[C(Z1)2]z-[C(Z1)=C(Z1)]-Z3;
[C(Z1)2-Y3-[C(Z1)2]z-Z3;
[C(Z1)2-Y3]-(C(=O)-O)-[C(Z1)2]z-[C(z1)=C(Z1)]-Z3;
[C(z1)2-Y3]-(C(=O)-O)-[C(z1)2]z-Z3;
[C(Z1)2-Y3]-(C(=O)-O)-[C(Z1)2]z-[C(z1)=C(Z1)]-Z3;
[C(Z1)2-Z2-Y4]-[C(Z1)2]z-[C(Z1)=C(Z1)]-Z3;
-[C(Z1)2]z-[C(Z1)=C(Z1)]-Z3;

[0141]在另外的实施方式中,Z3是:

[0142]在一实施方式中,每一-C(Z1)2-是:-CH2-,-CHMe-,-CH(CN)-,-CH(CO2H)-,-CH(CONH2)-,-CH(CF3)-,-CH(CHF2)-,-C(Me)2-,-C(Et)2-,-CH(CH2NMe2)-,-CH(CH2NMe2)-,-C(CH2NMe2)2-,或-C(CH2CO2H)2-。
[0143]在一实施方式中,-C(Z1)2-Y3是:-CH2-O-,-CH2-S-,-CH2-NMe,-CH2-NH-,CH(Me)-O-,CH(Me)-S-;-CH(Me)-NMe-,-CH(Me)-NH-;-CMe2-NMe-,-CMe2-NMe-,或-CMe2-NMe-。
[0144]在一实施方式中,-Z2-Y4合起来是:



[0145]在一实施方式中,-[C(Z1)=C(Z1)]-是:-CH=CH-,-C(CN)=CH-,-CH=C(CN)-,-C(Ar)=CH-,-CH=CAr-,-C(COAr)=CH-,-CH=C(COAr)-,-C(COR12)=CH-或-CH=C(COR12)-,其中Ar是芳基,其任选被多达5个选自下列的取代基所取代:OH,OMe,CF3,O-CHF2,OCF3,NO2,CN,卤素,卤甲基,二卤甲基,三卤甲基,羟甲基,CO2H,CONH2,CONMe2,和CONHMe;并且R12独立为氢,C1-C6烷基,C1-C6杂烷基,C3-C8环烷基,或杂环。
[0146]在另一实施方式中,触发物是:

在另一实施方式中,触发物是

其中每一Z1独立为H或C1-C6烷基。
[0147]在另一实施方式中,触发物是

其中每一Z1为H或C1-C6烷基,R8是H、OH或-OP(=O)(OH)2。
[0148]在一实施方式中,本发明提供分子式(II)和分子式(III)的化合物:


其中每一个R2-R5独立地选自氢、羟基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基和杂芳基;或者R2-R5的任意两个一起形成C3-C10杂环;每一个Y1独立地是S或O;以及每个触发物T如在式(I)中所定义;
[0149]条件是在分子式(II)或(III)中:
[0150](i)R1-R5的至少二个选自2-卤烷基、2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;或
[0151](ii)R1-R5的至少一个选自2-卤烷基、2-C1-C6烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;并且NR2R3和NR4R5的至少一个是 或者
或者
[0152](iii)NR2R3和NR4R5每一个是 以及
以及
[0153]提供其单独的异构体或异构体的外消旋或非外消旋混合物、生物电子等排物、药效团、药学上可接受的盐、溶剂化物、水合物或前体药物。
[0154]在一实施方式中,本发明提供分子式(II)的化合物,其中触发物T是-CH2-Z3、-CH(Z1)-Z3或-C(Z1)2-Z3,其中Z1是C1-烷基和Z3是:

条件是在分子式(II)中:
[0155](i)R2和R3的一个是H,并且R4和R5的一个是H;
[0156](ii)R2和R3的一个是C1-烷基,并且R4和R5的一个是C1-烷基;或者
[0157](iii)R2-R5的至少一个是氢、氨基、C3-C8环烷基、杂环基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基、C1-C6酰基、C1-C6杂酰基、或芳酰基或杂芳酰基。
[0158]在一实施方式中,本发明提供式(II)的化合物,其中Z3是选自下列的生物还原基团:

条件是在分子式(I)中:
[0159](i)R1-R5的至少一个选自2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基,以及
[0160]R1-R5的至少一个选自2-卤烷基、2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;或者
[0161](ii)R1-R5的至少一个选自2-卤烷基、2-C1-C6烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;并且NR2R3和NR4R5的至少一个是 或者
或者
[0162](iii)NR2R3和NR4R5每一个是
[0163]在一方面,本发明提供分子式(I)的氨基磷酸酯烷化剂前体药物:

其中
[0164]R1是-[C(Z1)2-Y3]v-[C(=O)-O]q-[C(Z1)2-Z2-Y4]u-Z3或-[C(Z1)2-Y3]v-(S(=O)2)q-[C(Z1)2-Z2-Y4]u-Z3,其中每一个v、q和u独立为0或者1;Z3是葡萄糖或其类似物,条件是其不包括参考文件Wiessler等的美国专利第5,622,936号中描述的氨基磷酸酯烷化剂的葡萄糖偶联物(conjugate);
[0165]R2-R5的每一个独立是氢、羟基、氨基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基和杂芳基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;或者R1-R5的任意两个一起形成C3-C10杂环;
[0166]条件是在分子式(I)中:
[0167](i)R2-R5的至少两个选自2-卤烷基、2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;
[0168](ii)R2-R5的至少一个选自2-卤烷基、2-C1-C6烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;并且NR2R3和NR4R5至少一个是 或者
或者
[0169](iii)NR2R3和NR4R5每一个是以及
[0170]提供其单独的异构体或异构体的外消旋或非外消旋混合物、生物电子等排物、药效团、药学上可接受的盐、溶剂化物、水合物或前体药物。
[0171]在另一实施方式中,本发明提供化合物:

[0172]在另一实施方式中,本发明提供化合物:
和
其中X4和Z3如前面定义。
[0173]在另一实施方式中,本发明提供化合物:

在一实施方式中,R6是-(N-CH2CH2X4)2。
[0174]在另一实施方式中,本发明提供化合物:


其中R2-R5如在分子式(II)中所定义。
[0175]下列的方案举例说明将氨基磷酸酯烷化剂前体药物低氧还原以产生相应的氨基磷酸酯烷化剂。
[0176]在另一实施方式中,本发明提供化合物:


其中,R2-R5如在分子式(II)所定义。
[0177]下列的方案举例说明将氨基磷酸酯烷化剂前体药物低氧还原以产生相应的氨基磷酸酯烷化剂。

[0178]在一实施方式中,本发明提供分子式(IV)-(VII)的化合物:

和
其中每一R9独立为氢、氘、芳基、杂芳基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C1-C6酰基、C1-C6杂酰基、芳酰基、杂芳酰基、C1-C6烷氧基羰基、C1-C6烷基氨基羰基、二C1-C6烷基氨基羰基或C1-C6烷氧基;或者两个R9基团一起形成杂环;每一R10是氢、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、芳酰基或杂芳酰基或者两个R10基团一起形成杂环;
[0179]R11独立为氢、氘、芳基、杂芳基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C1-C6酰基、C1-C6杂酰基、芳酰基、杂芳酰基、C1-C6烷氧基羰基、C1-C6烷基氨基羰基、二C1-C6烷基氨基羰基或C1-C6烷氧基;或者两个R9基团一起形成杂环,条件是,当R11是C1-C6烷基或C1-C6杂烷基时,那么R11不包或者两个R11基团一起形成杂环;
[0180]X4是Cl、Br、烷基磺酰氧基、杂烷基磺酰氧基、芳基磺酰氧基或杂烷基磺酰氧基;和
[0181]触发物T是
[C(Z1)2-Y3]v-(C(=O)-O)q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]z-[C(Z1)=C(Z1)]g-Z3。
[0182]在相关实施方式中,在分子式(IV)-(VII)中,每一R9独立为氢、氘、C1-C3烷基、C1-C6杂烷基、C3-C6环烷基、杂环基、芳基或杂芳基。在另一实施方式中,每一R9独立为氢、氘或C1-C6烷基。在另一相关实施方式中,每一R9独立为甲基、乙基、丙基、异丙基、异丁基、叔丁基或环丙基。
[0183]在一实施方式中,本发明提供分子式(IV)的化合物,其中一个R10是-(CH2)e-嵌入剂(Intercalator),其中嵌入剂是能嵌入到核酸碱基对之间的芳族或杂芳族部分。
[0184]在另一实施方式时,本发明提供化合物:
其中X4和R10如在分子式(IV)中所定义。
[0185]在另一实施方式中,本发明提供化合物:

[0186]在一实施方式中,本发明提供分子式(VIII)的化合物:

其中每一R9独立为氢、甲基、乙基、丙基、异丙基或环丙基;并且N(R10)2选自NH2,NHMe,NMe2,Net2,
 NHOMe和NHOH。
NHOMe和NHOH。
[0187]在一实施方式中,本发明提供分子式(IX)的化合物:
其中每一R9独立为氢、甲基、乙基、丙基、异丙基或环丙基。
[0188]在一实施方式中,本发明提供分子式(X)的化合物:

其中每一R9独立为氢、甲基、乙基、丙基、异丙基或环丙基;并且每一R11独立为氢、甲基、乙基、丙基、异丙基、苄基、取代的甲基、环丙基、甲氧基和羟基;或者两个R11一起形成杂环。
[0189]在一实施方式中,本发明提供分子式(X-A)、(X-B)和(X-C)的化合物:



其中X2和X4如分子式(I)中所定义,并且R10和R11如分子式(IV)、(VI)和(VII)中所定义。
[0190]在一实施方式中,本发明提供分子式(XI)-(XV)的化合物:

其中每一R11独立为氢、甲基或取代的甲基、苄基、异丙基、丙基、环丙基、甲氧基和羟基;并且X1、X2和Z3如上所定义;并且X4是Cl、Br、烷基磺酰氧基、杂烷基磺酰氧基、环烷基磺酰氧基、杂环烷基磺酰氧基、芳基磺酰氧基或杂芳基磺酰氧基。在一实施方式中,在分子式(XII)、(XIV)和(XV)的化合物中,当X4是Cl或Br时,那么R11不包括异丙基。在一实施方式中,分子式(X)的化合物不包括其中Z3是下述的化合物:


[0191]在一实施方式中,本发明提供分子式(XII)、(XIV)或(XV)的化合物,其中每一R11是氢。分子式XII、XIV或XV的化合物的实例包括化合物5、7、8、9、10、13、14、15、19、23、24、25、26、32、34和36。在一实施方式中,本发明提供分子式XII、XIV或XV的氨基磷酸酯烷化剂前体药物,其中R11不包括丙基或异丙基。在另一实施方式中,本发明不包括下列化合物:

[0192]在一实施方式中,本发明提供氨基磷酸酯烷化剂前体药物,其中R11是C3-C8环烷基。在另一实施方式中,环烷基是环丙烷基。一般而言,对于在细胞中氧化代谢蛋白质,环丙基可能比烷基更稳定,特别在肝脏中,与已知的氨基磷酸酯烷化剂前体药物相比,本发明的前体药物化合物提供药物动力学改善的氨基磷酸酯烷化剂前体药物。
[0193]在一实施方式中,本发明提供分子式(XVI)的化合物:

其中K是C1-C6亚烷基或C1-C6杂亚烷基。在一实施方式中,K是(C(R12)2)e、CH2CH2(-X6-CH2CH2)f、或CH2(-X6-CH2)f,其中e是1-10,f是0-3,X6是O、S或NR12,其中每一R12如上独立定义。
[0194]在一实施方式中,本发明提供分子式(XVI)-(XVIII)的化合物

其中e是0-4,X4是Cl或Br、烷基磺酰氧基、杂烷基磺酰氧基、芳基磺酰氧基或杂芳基磺酰氧基;X6是O、S或NR12,其中R12如上所定义。
[0195]在一实施方式中,本发明提供分子式(XIX)的化合物:

其中e是0-4,X4是Cl或Br、烷基磺酰氧基、杂烷基磺酰氧基、芳基磺酰氧基或杂芳基磺酰氧基。在相关实施方式中,本发明提供分子式(XIX)的化合物,其中e是1。参见实施例部分的本文所述分子式化合物的实例。
[0196]在一实施方式中,本发明提供分子式(XX)的化合物:

其中Rg是葡萄糖或葡萄糖类似物;其中e是0-4,X4是Cl、Br、烷基磺酰氧基、杂烷基磺酰氧基、芳基磺酰氧基或杂芳基磺酰氧基。如本文所使用的,葡萄糖类似物包括单、二和三糖。在相关实施方式中,本发明提供分子式XX的化合物,其中e是1。
[0197]在一实施方式中,本发明提供下列化合物:



其中X4是Cl、Br或烷基磺酰氧基。
[0198]在一实施方式中,本发明提供下列化合物:


其中R9和X4如在分子式VI中所定义。
[0199]在一实施方式中,本发明提供分子式(XXI)的化合物:

其中Y1是S或O;并且触发物T如在分子式(I)中所定义。
[0200]在另一实施方式,本发明提供肟-氨基磷酸酯烷化剂偶联物:

[0201]在一实施方式中,这类肟-氨基磷酸酯烷化剂偶联物可被酶促水解以产生

[0202]在另一方面,本发明提供分子式(XXII)的化合物:
其中
[0203]R1-R5、Y1和Y2如在分子式(I)中所定义;
[0204]每一R1-R5和R1 *-R5 *独立选自氢、羟基、C1-C6烷基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基;或者R2和R2 *一起形成杂环;或者每一R1-R5和R1 *-R5 *独立为选自下列的触发物T:
-[C(Z1)2-Y3]v-[C(=O)-O]q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]z-[-C(Z1)=C(Z1)]g-Z3和
-[C(Z1)2-Y3]v-(S(=O)2)q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]2-[C(Z1)=C(Z1)]g-Z3-;
条件是在分子式(XXII)中:
[0205](i)R1-R5和R1 *-R5 *的至少两个是2-卤烷基、2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基或2-杂烷基磺酰氧基烷基;或者
[0206](ii)R1-R5和R1 *-R5 *的至少一个是2-卤烷基、2-C1-C6烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基或2-杂烷基磺酰氧基烷基;并且NR2R3和NR2 *R3 *中至少一个是 或者
或者
[0207](iii)NR2R3和NR2 *R3 *每一个是以及
提供其单独的异构体或异构体的外消旋或非外消旋混合物、生物电子等排物、药效团、药学上可接受的盐、溶剂化物、水合物或前体药物。
[0208]每一个Z独立为C、S或P;
[0209]每一个t独立为1或2;
[0210]每一个r独立为0或1;
[0211]K选自C1-C6亚烷基、C1-C6杂亚烷基、亚芳基或杂亚芳基、(C(R9)2)n、和(Y5-(C(R9)2)m-Y4-(C(R9)2)m-Y6)n,其中n是1-8;
[0212]每一个m独立为1-4;
[0213]每一R9独立为C1-C6烷基或杂烷基,或者当共价结合到同一碳原子或相邻碳原子时,一起为环烷基或杂环基;和
[0214]每一Y4、Y5和Y6独立为O、S、NR7或键;条件是Y4、Y5和Y6的一个必须为O、S或NR7。
[0215]在另一方面,本发明提供分子式(XXIII)的化合物:

其中
[0216]R1-R5、Y1和Y2如在分子式(I)中所定义;
[0217]每一R1-R5和R1 *-R5 *独立选自氢、羟基、C1-C6烷基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基;或者R2和R2 *一起形成杂环;或者每一R1 *-R5 *独立为选自下列的触发物T:
-[C(Z1)2-Y3]v-[C(=O)-O]q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]z-[-C(Z1)=C(Z1)]g-Z3和
-[C(Z1)2-Y3]v-(S(=O)2)q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]2-[C(Z1)=C(Z1)]g-Z3-;
条件是在分子式(XXIII)中:
[0218](i)R2-R5和R2 *-R5 *的至少两个选自2-卤烷基、2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基或2-杂烷基磺酰氧基烷基;
[0219](ii)R2-R5和R2 *-R5 *的至少一个是2-卤烷基、2-C1-C6烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基或2-杂烷基磺酰氧基烷基;并且NR2R3和NR2 *R3 *中一个是 或者
或者
[0220](iii)NR2R3和NR2 *R3 *都是 或NR4R5和NR4 *R5 *都是
或NR4R5和NR4 *R5 *都是 以及
以及
[0221]提供其单独的异构体或异构体的外消旋或非外消旋混合物、生物电子等排物、药效团、药学上可接受的盐、溶剂化物、水合物或前体药物。
[0222]L2是

其中X如上所定义。
[0223]在另一实施方式中,本发明提供分子式(XXIV)的化合物:

其中R2、R3、R4、R2 *、R3 *、R4 *、Z、K和触发物如在分子式(XXII)中所定义。
[0224]在另一实施方式中,本发明提供具有式(XXV)或(XXVI)结构的分子式(XXIV)的化合物:
[0225]在另一实施方式中,本发明提供分子式(XXVI)的化合物:

其中X1、X2、X4和e如在分子式(XXV)中所定义。
[0226]在另一实施方式中,本发明提供分子式(XXVII)的化合物:

其中R2-R5、r、k、Y1和触发物T如在分子式(XXIV)中所定义。
[0227]在一实施方式中,本发明提供下列分子式的化合物:

其中T是L-Z3;
L1是CH2,CHMe,C(Me)2,CH2OCH2,(CH2)3,CH2S(CH2)2,CH2S(CH2)3,
[0228]在另一实施方式中,本发明提供选自下列的Z3:



[0229]在另一实施方式中,本发明提供具有下列分子式的部分:

其选自:



[0230]在另一实施方式中,本发明提供选自下列的T:

和

其中每一Z1、R7和R8如上所定义。在本实施方式中,Z1是氢、甲基或乙基;R7是甲基、三氟乙基、乙基、丙基和环己基;以及R8是OH或OP(=O)(OH)2。在本实施方式中,

其中每一R9是氢或C1-C6烷基,每一X4是卤素或RsulS(=O)2O-。在另一实施方式中,R9是氢、甲基、乙基、异丙基或异丁基;X4是氯、溴或者甲磺酰氧基。
[0231]在另一实施方式中,本发明提供下列分子式的化合物:

其中T(触发物)如上所定义,或者更具体而言,T是L-Z3,其中L是CH2、CHMe、CMe2、

并且Z3是



[0232]在一方面,本发明提供下列分子式的氘化氨基磷酸酯烷化剂和氘化氨基磷酸酯烷化剂前体药物

其中X4是卤素或RsulS(=O)2O。在另一实施方式,X4是Cl或Br。这类氘化氨基磷酸酯烷化剂和它们的前体药物与其非氘化或氢化的类似物例如化合物25、36和类似物,对于低氧肿瘤组织具有相等的细胞毒性。然而,与其相应的氨基磷酸酯烷化剂和/或氨基磷酸酯烷化剂前体药物相比,通过核磁共振方法,这类氘化类似物在体内例如在血浆中的存在可以被更有效地确定,而且这类氘化类似物可用于确定氨基磷酸酯烷化剂和/或氨基磷酸酯烷化剂前体药物的药物动力学或药效动力学特性。氨基磷酸酯烷化剂和/或氨基磷酸酯烷化剂前体药物的药物动力学和/或药效动力学信息被用于确定剂量、给药频率和相似的给药相关参数。八氘化化合物25和八氘化异环磷酰胺烷化剂的合成在实施例部分进行描述。
[0233]在另一组的实施方式中,本发明提供了对实施例中化合物的单独的和选择性的分组。本发明的化合物的实例包括:






[0234]在一实施方式中,氨基磷酸酯烷化剂前体药物包含

作为Z3,并且表现出对低氧肿瘤的特异性毒性,而对健康常氧的组织具有少得多的毒性。
[0235]在一实施方式中,本发明提供新的氨基磷酸酯烷化剂前体药物,其在生物还原后,释放相应的新的或已知的氨基磷酸酯烷化剂

其中,X4如在分子式(I)中所定义,R9、R10和R11如在分子式(IV)-(VII)中所定义,并提供它们的离子化形式。在相关的实施方式中,X4是Cl、Br、甲磺酰氧基、苯磺酰氧基或对-甲苯磺酰氧基。
[0236]在一实施方式中,本发明提供新的氨基磷酸酯烷化剂前体药物,其在生物还原后,释放相应的新的或已知的氨基磷酸酯烷化剂,和它们的离子化形式;

其中N(R10)2选自NH2,NHMe,NMe2,NEt2,

[0237]当用于癌症治疗时,抗癌药环磷酰胺代谢为1d(R10是氢)并且异环磷酰胺代谢为1e(每一R11是氢)。葡膦酰胺(Glufosfamide),其在临床癌症治疗中正被评估,释放分子式1e的烷化剂(每一个R11是氢,参见Wiessler等,美国专利第5,622,936号;PCT申请第US05/03370号,其名称为"Anti Cancer Therapies",美国专利申请第60/638995号,其名称为"Glufosfamide Combination Therapy"和2005年5月11日提交的名称为"Glufosfamide Combination Therapy"的代理案卷第021305-005900US号)。在临床癌症治疗中正被评估的TelcytaTM释放1f(Rosen等,Clin Cancer Res.2004,10(11):3689-98)。
[0238]已知的氨基磷酸酯烷化剂前体药物例如异环磷酰胺和环磷酰胺,代谢产生细胞毒性的副产物(by product)例如丙烯醛和氯乙醛,其引起不期望的患者副作用例如出血膀胱炎、昏迷或死亡。在一实施方式中,本发明提供氨基磷酸酯烷化剂前体药物,与通过异环磷酰胺和/或环磷酰胺代谢产生的那些相比,其每次治疗在代谢后产生更少的毒性副产物。在一实施方式中,本发明的氨基磷酸酯烷化剂前体药物不通过体内代谢产生丙烯醛。由本发明的前体药物的代谢产生的副产物的实例包括氯-、溴-、烷基磺酰氧基-、杂烷基磺酰氧基-、芳基磺酰氧基-或杂芳基磺酰氧基-乙醛,(对于来自异环磷酰胺的氯乙醛的代谢产物,参见上面提到的Hardman等的参考文献的第1396页)。在另一实施方式中,本发明提供氨基磷酸酯烷化剂前体药物,其在氧化代谢后产生5-95%这么多的氯乙醛或者与上面定义的每次治疗中异环磷酰胺代谢产生的氯乙醛相等。
[0239]在还原Z3后形成的氨基磷酸酯烷化剂衍生物可与被保护的氨基磷酸酯烷化剂和氨基磷酸酯烷化剂前体药物不同,并且被称为修饰的氨基磷酸酯烷化剂前体药物。例如,在还原生物还原基团(Z3)后,氨基磷酸酯烷化剂前体药物可产生修饰的氨基磷酸酯烷化剂前体药物Alk-触发物mod。当生物还原基团还原形成修饰的氨基磷酸酯烷化剂前体药物时,结合到氨基磷酸酯烷化剂的连接体(L)可进行降解,产生氨基磷酸酯烷化剂或一些其它的修饰的氨基磷酸酯烷化剂前体药物。
[0240]在一实施方式中,本发明提供通过引入上述的连接体(L)而在低氧组织中活化后表现出旁观者效应的化合物。在一实施方式中,该旁观者效应使本发明的修饰的氨基磷酸酯烷化剂扩散或渗入进低氧不足以活化本发明前体药物化合物的肿瘤区,但是驻留在可活化这些前体药物的低氧肿瘤区附近。
[0241]在触发物T中的生物还原基团(Z3)还原后,修饰为Z3-mod,产生修饰的氨基磷酸酯烷化剂前体药物如氨基磷酸酯烷化剂-TM或Alk-TM偶联物。在一实施方式中,TM选自:
[C(Z1)2-Y3]-(C(=O)-O)-[C(Z1)2-Z2-Y4]-[C(Z1)2]z-[C(Z1)=C(Z1)]-Z3-mod;
[C(Z1)2-Y3]-[C(Z1)2-Z2-Y4]-[C(Z1)2]z-[C(Z1)=C(Z1)]-Z3-mod;
[C(Z1)2-Y3]-[C(Z1)2]z-[C(Z0=C(Z1)]-Z3-mod;
[C(Z1)2-Y3]-[C(Z1)2]z-Z3-mod;
[C(Z1)2-Y3]-(C(=O)-O)-[C(Z1)2]z-[C(Z1)=C(Z1)]-Z3-mod;
[C(Z1)2-Y3]-(C(=O)-O)-[C(Z1)2]z-Z3-mod;
[C(Z1)2-Y3]-(C(=O)-O)-[C(Z1)2]z-[C(Z1)=C(Z1)]-Z3-mod;
[C(Z1)2-Z2-Y4]-[C(Z1)2]z-[C(Z1)=C(Z1)]-Z3-mod;和
-[C(Z1)2]z-[C(Z1)=C(Z1)]-Z3-mod其中,Z3-mod是被生物还原或其它方式还原或修饰的Z3。
[0242]在另一实施方式中,TM选自:
[C(Z1)2-Y3]-(C(=O)-O)-[C(Z1)2-Z2-Y4]-H;[C(Z1)2-Y3]-[C(Z1)2-Z2-Y4]-H;和[C(Z1)2-Y3]-H;
[0243]在一实施方式中,触发物T包括具有下列分子式的连接体(L):
-[C(Z1)2-Y3]-(C(=O)-O)-[C(Z1)2-Z2-Y4]-[C(Z1)2]z-[C(Z1)=C(Z1)]-;
-[C(Z1)2-Y3]-[C(Z1)2-Z2-Y4]-[C(Z1)2]z-[C(Z1)=C(Z1)]-;
-[C(Z1)2-Y3]-[C(Z1)2]z-[C(Z1)=C(Z1)]-;-[C(Z1)2-Y3]-[C(Z1)2]z-;
-[C(Z1)2-Y3]-(C(=O)-O)-[C(Z1)2]z-[C(Z1)=C(Z1)]-;-[C(Z1)2-Y3]-(C(=O)-O)-C(Z1)2-;
-[C(Z1)2-Y3]-(C(=O)-O)-[C(Z1)2]z-[C(Z1)=C(Z1)]-;和
-[C(Z1)2-Z2-Y4]-[C(Z1)2]z-[C(Z1)=C(Z1)]-;-[C(Z1)2]z-[C(Z1)=C(Z1)]-和-[C(Z1)2]z-。
[0244]在一实施方式中,本发明提供这样的触发物T,在生物还原后,其被修饰为触发物Mod或TM,并且在0.1秒以内从TM分离氨基磷酸酯烷化剂。在另一实施方式中,在0.01到0.10秒之内从TM分离氨基磷酸酯烷化剂。在另一实施方式中,在0.1到1.0秒之内从TM分离氨基磷酸酯烷化剂。在另一实施方式中,在1.0到10.0秒之内从TM分离氨基磷酸酯烷化剂。在另一实施方式中,在10.0到100.0秒之内从TM分离氨基磷酸酯烷化剂。
[0245]在相关实施方式中,在活化或还原之后,氨基磷酸酯烷化剂前体药物产生具有修饰的触发物T(TM)的前体药物,其然后从活化或还原位置,释放20到500μm的氨基磷酸酯烷化剂;或者从活化或还原位置,释放20到100μm的氨基磷酸酯烷化剂。使用细胞球状体和多层细胞实验(这类实验例如,参见Kyle等,CancerRes.2004,64(17):6304-9and West等,Cancer Chemother.Pharmacol,1987,20(2):109-14),可以测量本发明的氨基磷酸酯烷化剂前体药物的旁观者效应,并且如在实施例35与37更详细描述。将肿瘤细胞在培养物中培养成多细胞球状体,以产生实体瘤的肿瘤微环境体外模型,其含有低氧区和对有限营养和废物增加的环境压力应答的休眠细胞群。当细胞聚集体继续分裂和向外生长时,这些球状体具有形成氧和营养物梯度的独特性质。在存活边缘(viable rim)达到大约150μm大小后,低氧区形成,这使该区域中的细胞进入休眠状态并且最终细胞死亡。由于死亡的细胞,坏死核(necrotic core)形成。球状体可被分为4个不同的区,以模拟低氧活化前体药物的有效性:1)外层的需氧和活化分裂区;2)中间低氧区;3)细胞不处于周期中的低氧区;4)和含有死亡细胞和细胞残渣的坏死核。药物的响应将依赖于许多因素;化合物渗入球状体最深区域的能力。通过硝基还原酶,低氧活化前体药物(HAP)的活化;已被活化的细胞内活化药物的反应性;和活化的药物从被活化的位置离开和杀死附近细胞的能力(旁观者效应)。因此,可在多种不同的水平上,进行化合物有效性的评估。单独化合物的效果可对于单层培养物中的细胞相对于完整球状体进行比较。HAP可被用作单一治疗。球状体的低氧部分可通过改变平衡气体的O2浓度而调整,并且因此改变需氧和低氧区的比例。HAP可与其它化学治疗剂联合,这些化学治疗剂或者仅靶向外层需氧细胞或者能靶向整个球状体。通过知道低氧部分和每一单一治疗的预期细胞杀死可预测预期的细胞杀死。
[0246]在一实施方式中,本发明提供氨基磷酸酯烷化剂前体药物,其在活化例如生物还原后,释放具有半衰期在0.1秒以下的氨基磷酸酯烷化剂;半衰期在0.01到0.1秒之间的氨基磷酸酯烷化剂、半衰期在0.1到1.0秒之间的氨基磷酸酯烷化剂、半衰期在1.0到10.0秒之间的氨基磷酸酯烷化剂和半衰期在10.0到100.0秒之间的氨基磷酸酯烷化剂。
[0247]抗癌药可结合到脉管系统周围的组织和/或具有高分子量,这阻碍了扩散并且不能以治疗有效浓度到达低氧肿瘤区,该低氧肿瘤区离脉管系统可达150-200μm。在一实施方式中,本发明提供氨基磷酸酯烷化剂前体药物,其能到达远离脉管系统的低氧癌细胞。一些确定旁观者效应的方法在实施例35和37被更详细描述。用于低氧活化前体药物的氨基磷酸酯烷化剂在有效杀死肿瘤细胞方面起重要作用。例如,对于低氧活化氨基磷酸酯烷化剂前体药物,氨基磷酸酯烷化剂的细胞毒性和其细胞内烷化速率以及前体药物和氨基磷酸酯烷化剂对细胞膜的渗透性影响氨基磷酸酯烷化剂前体药物的低氧选择性和低氧细胞毒性。
[0248]在一实施方式中,本发明提供氨基磷酸酯烷化剂前体药物,其比在体内形成的相应的氨基磷酸酯烷化剂安全(安全至少10倍以及多达1,000,000倍)。在一实施方式中,提高的安全性是由于对触发物T的连接部位的修饰(氨基磷酸酯烷化剂前体药物的活化释放烷化剂/细胞毒剂)。在任一情形中,氨基磷酸酯烷化剂前体药物在低氧组织中依靠生物还原基团(Z3)的活化或还原被转化为相应的烷化剂,这导致其去除以及同时或随后释放或产生氨基磷酸酯烷化剂。
[0249]在一实施方式中,以掩蔽或降低氨基磷酸酯烷化剂细胞毒性的方式,触发物T被共价结合到氨基磷酸酯烷化剂。这种掩蔽效应可不同并且依赖于氨基磷酸酯烷化剂的细胞毒性活性。典型地,氨基磷酸酯烷化剂前体药物比相应的氨基磷酸酯烷化剂将显示出至少小大约10倍的细胞毒性活性,并且可以显示出小大约至100万倍或更少的细胞毒性活性。在一情况下,氨基磷酸酯烷化剂前体药物的细胞毒性活性比相应的氨基磷酸酯烷化剂的细胞毒性活性小大约100倍到大约10,000倍。作为一实例,对于具有1nM的IC50、IC90或LC50的氨基磷酸酯烷化剂,相应的氨基磷酸酯烷化剂前体药物的IC50、IC90或LC50可以是1μM或更大。
[0250]在一情况下,本文提供的化合物作为氨基磷酸酯烷化剂前体药物包括任何氨基磷酸酯烷化剂,该氨基磷酸酯烷化剂可以以产生氨基磷酸酯烷化剂前体药物的方式连接到触发物T,所述氨基磷酸酯烷化剂前体药物作为细胞毒剂比相应的氨基磷酸酯烷化剂或修饰的氨基磷酸酯烷化剂活性小至少约10倍到约1,000,000倍,一般地约100倍到约10,000倍,所述氨基磷酸酯烷化剂在低氧条件下从所述化合物释放。
[0251]为了确定在缺氧或低氧条件下氨基磷酸酯烷化剂前体药物是否是选择性地有活性的,将细胞在或者有空气(常氧)或者没有氧气(缺氧)或者有很少氧气(低氧)的情况下暴露于药物。本领域技术人员将认识到,在抗增殖实验中测量的氨基磷酸酯烷化剂前体药物的细胞毒性通过IC50表示;用产克隆存活试验测量的氨基磷酸酯烷化剂前体药物的细胞毒性表示为IC10或LC10、IC90获LC90、或者IC99或LC99。例如,通过在常氧和低氧中确定的IC50、IC90、LC50、LC90或LC99测量的细胞毒性的比率被称为低氧细胞毒性比(HCR),并且可以作为本发明的前体药物的低氧选择细胞毒性的量度。氨基磷酸酯烷化剂前体药物的HCR越大,低氧细胞选择毒性越高,相对于健康、常氧的细胞,前体药物的低氧肿瘤杀死能力越大。基于IC99或LC99确定的HCR比基于IC90或LC90确定的HCR大。
[0252]在相关实施方式中,本发明的氨基磷酸酯烷化剂前体药物具有0.1nM到50μM的低氧细胞毒性和10到100,000的HCR。在相关实施方式中,本发明的氨基磷酸酯烷化剂前体药物具有0.1nM到50μM的低氧细胞毒性和25到100,000的HCR(参见实施例部分)。在另一相关实施方式中,本发明的氨基磷酸酯烷化剂前体药物具有0.1nM到5μM的低氧细胞毒性和50到10,000的HCR,例如在实施例29、30和31中描述的化合物。
[0253]在一实施方式中,本发明提供具有比相应常氧毒性多5到1,000,000倍的低氧毒性的氨基磷酸酯烷化剂前体药物。在另一实施方式中,本发明提供具有比相应常氧毒性多10到10,000倍的低氧毒性的氨基磷酸酯烷化剂前体药物。在另一实施方式中,本发明提供具有比相应常氧毒性多25到5,000倍的低氧毒性的氨基磷酸酯烷化剂前体药物。
[0254]肿瘤具有氧浓度梯度,其可从脉管系统附近的组织中的10%变化为大约150μM远的组织中的0.5%,并且在进一步远离脉管系统并且靠近坏死核的组织中更低。在一实施方式中,本发明提供这样的氨基磷酸酯烷化剂前体药物,其在多种氧浓度下,可产生比相应前体药物的毒性大5-1,000,00、10-10,00和25-5,000倍的氨基磷酸酯烷化剂。在一实施方式中,本发明提供这样的氨基磷酸酯烷化剂前体药物,其在大约0.5-0.6%的氧浓度下,可产生比相应前体药物的毒性大5-1,000,00、10-10,00和25-5,000倍的氨基磷酸酯烷化剂。
[0255]本发明的氨基磷酸酯烷化剂前体药物的logP可测量前体药物的亲脂性或亲水性。在一实施方式中,本发明提供具有小于0的logP的氨基磷酸酯烷化剂前体药物。这类氨基磷酸酯烷化剂前体药物可以是亲水性的,例如具有分子式XV的前体药物,其中每一R11是H,并且它们容易被配制成用于静脉内注射(i.v.)或腹膜内注射(i.p.)的含水制剂。这类前体药物的另一实施例是化合物24、25和36。
[0256]在一实施方式中,本发明提供具有logP在0以上的氨基磷酸酯烷化剂前体药物。在一实施方式中,本发明提供具有logP在0和4之间的氨基磷酸酯烷化剂前体药物,例如分子式XIV、XX和XV所示例的那些,其中每一R11是甲基或环丙基,并且被施用于患者可通过细胞膜渗透到癌细胞内部。另一实例是具有logP在0和5、6、7或16之间的前体药物。(对于本发明的氨基磷酸酯烷化剂前体药物测量的logP,参见实施例部分)。
IIb.合成方法
[0257]本发明部分源于如此发现:不能通过在适当的溶剂中将 1-N-甲基-2-硝基咪唑-5-甲醇和正-丁基锂反应而分离的化合物36,通过采用Mitsunobu型反应容易被合成,其中通过加入三苯膦和二异丙基偶氮二羧酸酯,将1-N-甲基-2-硝基咪唑-5-甲醇活化,并且与反应以产生化合物36。
1-N-甲基-2-硝基咪唑-5-甲醇和正-丁基锂反应而分离的化合物36,通过采用Mitsunobu型反应容易被合成,其中通过加入三苯膦和二异丙基偶氮二羧酸酯,将1-N-甲基-2-硝基咪唑-5-甲醇活化,并且与反应以产生化合物36。
[0258]因此,在一方面,本发明提供合成氨基磷酸酯化合物的方法,包括将氨基磷酸或二氨基磷酸与醇反应产生氨基磷酸酯。在另一方面,本发明提供合成本发明的新的氨基磷酸酯烷化剂前体药物化合物和已知的那些氨基磷酸酯烷化剂前体药物化合物的方法。在一实施方式中,本发明提供合成氨基磷酸酯烷化剂前体药物的方法,包括将新的或已知的氨基磷酸酯烷化剂、触发物-OH、三取代膦和二烷基偶氮二羧酸酯反应,产生新的或已知的氨基磷酸酯烷化剂前体药物。在该方法的一实施方式中,在第一步,触发物-OH与三取代膦和二烷基偶氮二羧酸酯反应产生中间体,在第二步,将氨基磷酸酯烷化剂加入到从第一步得到的中间体,以产生产物。这类Mitsunobu型反应特别适合于合成新的或已知的氨基磷酸酯烷化剂前体药物或衍生物,Alk-触发物,其中触发物是L-Z3,其中Z3是

Alk是

其中,R9如上所定义。
[0259]在一实施方式中,本发明提供合成氨基磷酸酯烷化剂前体药物的方法,包括将每一新的或已知的氨基磷酸酯烷化剂:

与触发物-OH、三取代膦和二烷基偶氮二羧酸酯反应,分别产生:
其中,X4、R5、R7和R8如在分子式(I)中所定义。
[0260]在一实施方式中,本发明提供合成具有下列分子式的化合物的方法,

包括使(a)、(b)和(c)反应。
(a)具有下列分子式的新的或已知的氨基磷酸酯烷化剂:
其中,R2-R5如分子式(I)中所定义,条件是
(i)R2-R5的至少两个选自2-卤烷基、2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;
(ii)R1-R5的至少一个选自2-卤烷基、2-C1-C6烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;并且NR2R3和NR4R5的至少一个是或者
(iii)NR2R3和NR4R5都合起来是
(b)触发物-OH,其中触发物如在分子式(I)中所定义,三取代磷酸氢,和
(c)二烷基偶氮二羧酸酯,以产生具有下列分子式的化合物

[0261]在一实施方式中,具有下列分子式的化合物

选自下列


[0262]在另一实施方式,具有分子式的下列基团

选自:




[0263]在另一实施方式中,反应包括溶剂如THF、二噁烷、C1-C6烷基乙酸酯、氯仿、二氯甲烷、乙腈和类似物。在另一实施方式中,三取代膦中每一取代基独立选自C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、芳基、杂芳基和C1-C6烷氧基取代基。在另一实施方式中,触发物T是



其中X1、X2、Z1和Z2如在分子式(I)中所定义。
[0264]在另一实施方式中,本发明提供合成氨基磷酸酯烷化剂前体药物的方法,包括
[0265](i)在选自THF、二噁烷、二氯甲烷、氯仿、乙酸乙酯、乙酸丙酯、乙酸丁酯或乙腈的溶剂中,使下述反应:具有下列分子式的化合物

其中每一个R11独立为氢、环丙基、甲基、乙基、苄基、或甲氧基;每一个R9独立为氢、甲基、乙基、丙基或环丙基;并且X4是卤素、甲基磺酰氧基、苯基磺酰氧基、4-甲基苯基磺酰氧基和4-卤苯基磺酰氧基;
(ii)选自三苯膦、三丁基膦和三丁基亚磷酸酯的三取代膦;和
(iii)二乙基或二异内基偶氮二羧酸酯;
产生下列分子式的产物:

[0266]在另一实施方式中,本发明提供合成下列分子式的化合物的方法:

包括下列步骤:
(i)在非质子溶剂中,将触发物-OH——其中触发物如分子式(I)所定义、三取代膦和二烷基偶氮二羧酸酯反应,产生中间体(i);
(ii)将从步骤(i)得到的中间体(i)与下列分子式的化合物反应

其中,每一R9、R11和X4如分子式(I)所定义,产生下列分子式的化合物:
[0267]在另一实施方式中,三取代膦是P(R12)3,其中每一R12是H、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、芳基或杂芳基。在另一实施方式中,三取代膦是聚合物支载的(polymer supported)三取代膦。在另一实施方式中,三取代膦是三苯膦、三丁基膦、三丙基膦、三乙基膦或三甲基膦。在另一实施方式中,三取代膦是聚合物支载的三苯膦。聚合物支载的三取代膦是商业上可获得的,例如从Varian Inc.of Palo Alto,California。在另一实施方式中,本发明提供合成其中每一个R11是氢的化合物的方法。在另一实施方式中,本发明提供合成下列化合物的方法:

[0268]在另一实施方式中,本发明提供制造化合物的方法,其中触发物选自

和Z3是



[0269]在一实施方式中,本发明提供合成氨基磷酸酯烷化剂前体药物的方法,包括下列步骤:
[0270](a)用N-2-卤乙基-N-(R13)铵盐回流POCl3,产生二氯氨基磷酸酯中间体,其中R13是氢、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、芳基、杂芳基;
[0271](b)将步骤(a)中的二氯氨基磷酸酯中间体与N-2-卤乙基-N-(R13)铵盐和碱在溶剂中反应,产生一氯氨基磷酸酯中间体,其中R13是H、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、芳基、杂芳基;和
[0272](c)将步骤(b)中获得的一氯氨基磷酸酯中间体与触发物-OH和碱在溶剂中反应,产生氨基磷酸酯烷化剂前体药物。
[0273]在一实施方式中,步骤(a)中的二氯氨基磷酸酯中间体从剩下的反应混合物中分离,然后将它进行步骤(b)的反应。在另一实施方式中,通过首先在真空中除去过量的POCl3,然后在减压下蒸馏二氯氨基磷酸酯,进行分离。
[0274]在一实施方式中,通过在硅胶上的急骤柱层析从剩下的反应混合物中分离步骤(c)中的氨基磷酸酯烷化剂前体药物。在一实施方式中,在步骤(b)中使用的碱是叔胺。在步骤(b)中使用的适合的叔胺包括三烷基胺如三乙胺或二异丙基乙胺。在一实施方式中,在步骤(b)中使用的溶剂是四氢呋喃(THF)或二噁烷。
[0275]在一实施方式中,通过在硅胶上的急骤柱层析从剩下的反应混合物中分离步骤(b)中的一氯氨基磷酸酯中间体,然后将它进行步骤(c)的反应。在一实施方式中,可用于步骤(c)中的碱是六烷基二硅氮化锂(lithium hexaalkyldisilazide)、六烷基二硅氮化钠或六烷基二硅氮化钾;氢化钠或氢化钾;或者二异丙基酰胺锂(lithium diisopropylamide)。在一实施方式中,在步骤(c)中使用的溶剂是二甲氧基乙烷、二甘醇二甲醚、二乙醚或THF。
[0276]在一实施方式中,本发明提供合成氨基磷酸酯烷化剂前体药物的方法,包括下列步骤:
[0277](a)在溶剂中反应大约每个1当量的POCl3、触发物-OH和碱,产生二氯氨基磷酸酯中间体;和
[0278](b)将步骤(a)中的二氯氨基磷酸酯中间体与N-2-卤乙基-N-(R13)铵盐和碱在溶剂中反应,产生氨基磷酸酯烷化剂前体药物,其中R13是氢、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、芳基、杂芳基。
[0279]在一实施方式中,在0℃以下进行步骤(a)和步骤(b)。在另一实施方式中,在高于步骤(a)的温度20-100℃以上的温度进行步骤(b)。
[0280]在另一实施方式中,本发明提供合成如下所示的本发明的杂环氨基磷酸酯烷化剂前体药物的方法:

其中,X4=Br或Cl;e=1-3
[0281]在一实施方式中,本发明提供合成氨基磷酸酯烷化剂前体药物的方法,包括下列步骤:
[0282](a)使PCl3与N,N-二(2-卤乙基)铵盐和碱在溶剂中反应,产生一氯磷酰胺衍生物;
[0283](b)将一氯磷酰胺衍生物与触发物-OH反应,产生中间物;和
[0284](c)氧化步骤(b)中的中间体,产生氨基磷酸酯烷化剂前体药物。
[0285]在一实施方式中,在步骤(b)中使用的碱是三乙胺。在另一实施方式中,在步骤(c)中使用的溶剂是二甲氧基乙烷、二甘醇二甲醚或C1-C6烷基乙酸酯。在另一实施方式,在步骤(c)中的触发物-OH是

[0286]多种1-N-烷基-2-氨基咪唑-5-羧酸酯可按照下面示意性的描述进行合成:

[0287]可还原1-N-烷基-2-氨基咪唑-5-羧酸酯,以产生本发明中使用的1-N-烷基-2-氨基-5羟基甲基咪唑衍生物,作为生物还原基团Z3。
[0288]在下面的实施例部分进一步详细地提供合成方法。
[0289]生物还原基团和氨基磷酸酯烷化剂前体药物的合成和本发明的方法可从下列的参考文献中改变得到:Matteucci等,PCT申请公开第WO 04/009667号,和名称为"Hypoxia Activated anti-Cancer Agents"的低氧活化前体药物美国专利申请;deGroot等,2001,Current Med.Chem.8:1093-1122;Denny等,美国专利第5,750,782;5,780,585;5,872,129;和6,251,933号;Davis等,PCT申请公开第WO 04/85421和WO04/85361号;以及Lin等,美国专利申请公开第2004/254103和2005/043244号,以及Borch等(如前所述)。
[0290]在下面的“实施例”部分进一步详细地提供合成本发明的氨基磷酸酯烷化剂前体药物的方法的实施例。
IIIa.治疗方法
[0291]在一实施方式中,本发明提供通过施用本发明的氨基磷酸酯烷化剂前体药物或者已知的氨基磷酸酯烷化剂前体药物给患者,治疗需要治疗的患者的癌症的方法。已知的氨基磷酸酯烷化剂在前面所述的Borch等的参考文献中提供。在一实施方式中,在根据本发明提供的方法治疗癌症中使用的氨基磷酸酯烷化剂前体药物具有选自(I)-(XXVII)的分子式。在一实施方式中,在根据本发明提供的方法治疗癌症中使用的氨基磷酸酯烷化剂前体药物选自在实施例部分举例的化合物。
[0292]用烷化剂治疗癌症可导致形成对这些烷化剂具有抗性的癌症。与在缓慢生长的低氧癌症区的癌细胞相比,烷化剂可以杀死在更快速分裂或含更高氧的癌症区的癌细胞。缓慢生长的低氧癌症区的癌细胞在烷化剂治疗后仍存活,并且可以产生对这类烷化剂具有抗性的细胞。鸟嘌呤-O6-烷基转移酶、谷胱甘肽、谷胱甘肽转移酶、核苷酸切除修复通路和/或错配修复蛋白的增强的活性,以及主动运输的药物例如氮芥和苯丙氨酸氮芥降低的渗透性,被推测对癌症的烷化剂抗性负责(例如,参见Hardman等,第1393和1433页,如前所述)。
[0293]本发明的前体药物对于治疗对其它疗法具有抗性的癌症是有效的。在低氧癌症区缓慢分裂的癌细胞作为抗性癌细胞和株系(strain)的来源,其被本发明的前体药物杀死。在一实施方式中,通过单独施用或者与其它抗癌剂一起联合施用本发明的化合物,本发明提供治疗对一种或多种烷化剂的治疗具有抗性的癌症的方法。在一实施方式中,本发明的氨基磷酸酯烷化剂前体药物与基本不具有肾毒性的药物一起联合施用。在一实施方式中,氨基磷酸酯烷化剂前体药物与卡铂一起施用。
[0294]在一实施方式中,本发明提供与已知烷化剂不具有交叉抗性的氨基磷酸酯烷化剂前体药物。在另一实施方式中,本发明提供与下列烷化剂不具有交叉抗性的氨基磷酸酯烷化剂前体药物:环磷酰胺、异环磷酰胺、葡磷酰胺、氮芥、苯丙氨酸氮芥、苯丁酸氮芥、达卡巴嗪、替莫唑胺、卡莫司汀、链脲霉素、苯达莫司汀、白消安、噻替哌、顺铂、卡铂和奥沙利铂。
[0295]在一实施方式中,通过单独或者与其它抗癌剂一起联合施用本发明的化合物作为第一线治疗,本发明提供治疗癌症的方法。在另一实施方式中,通过单独或者与其它抗癌剂一起联合施用本发明的化合物作为第一线治疗,本发明提供治疗转移癌的方法。在一实施方式中,通过单独或者与其它抗癌剂一起联合施用本发明的化合物作为第二线治疗,本发明提供治疗癌症的方法。在一实施方式中,通过单独或者与其它抗癌剂一起联合施用本发明的化合物作为第三线治疗,本发明提供治疗癌症的方法。在一实施方式中,通过在用手术和/或放射疗法的在先治疗后单独或者与其它抗癌剂一起联合施用本发明的化合物,本发明提供治疗癌症的方法。在一实施方式中,通过单独或者与其它抗癌剂一起联合施用本发明的化合物,本发明提供治疗癌症的方法,所述癌症在先前的化学疗法、手术、放疗或它们任意的组合后已经复发。
[0296]在本发明提供的治疗癌症的方法中,将有效量的氨基磷酸酯烷化剂前体药物施用给对象。一般而言,对象可以是任何人或非人哺乳动物。优选的对象是人对象。其它具体的对象包括但不限于,非人灵长类、狗、猫、农场动物和马。在一情况下,氨基磷酸酯烷化剂前体药物被单独施用。在一种情况下,氨基磷酸酯烷化剂前体药物与一种或多种另外的抗癌剂联合使用。在一情况下,与癌症治疗一起,施用氨基磷酸酯烷化剂前体药物,所述癌症治疗包括但不限于手术和放疗。一般地,氨基磷酸酯烷化剂前体药物将以药学组合物施用。可被使用的多种药学组合物,在下文的制剂部分进行描述。
[0297]氨基磷酸酯烷化剂前体药物和它们的药学组合物可被用来治疗对象特别是人对象的任意类型癌症。可被治疗的癌症包括但不限于白血病、乳癌、皮肤癌、骨癌、肝癌、脑癌、喉癌、胆囊癌、胰腺癌、直肠癌、甲状旁腺癌、甲状腺癌、肾上腺癌、神经组织癌、头部和颈部癌、胃癌、支气管癌、肾癌,基底细胞癌,溃疡状和乳头状两种类型的鳞状细胞癌,转移性皮肤癌,骨肉瘤,尤因氏肉瘤(Ewing′s Sarcoma),网状细胞(veticulum cell)肉瘤,骨髓瘤,巨细胞瘤,小细胞肺肿瘤,胆石,胰岛细胞癌,原发性脑瘤,急性和慢性淋巴细胞瘤和粒细胞瘤,毛细胞瘤,腺瘤,增生,髓样癌,嗜铬细胞瘤,粘膜神经瘤,肠内神经节细胞瘤,增生性角膜神经瘤,类马伐氏症候群瘤(marfanoid habitus tumor),维尔母斯肿瘤,精原细胞瘤,平滑肌瘤,子宫颈异常和原位癌,成神经细胞瘤,视网膜母细胞瘤,软组织肉瘤,恶性类癌瘤,局部皮肤病变,蕈样肉芽肿病,横纹肌肉瘤,卡波西肉瘤,成骨和其它肉瘤,恶性高钙血症,肾细胞瘤,真性红细胞增多症,腺癌,多形性成胶质细胞瘤,白血病,淋巴瘤,恶性黑素瘤,和表皮样癌。
[0298]氨基磷酸酯烷化剂前体药物可具体被用来治疗含有明显低氧组织的癌症。这类癌症包括但不限于肺癌特别是非小细胞肺癌、乳腺癌、结肠癌、头部和颈部癌、卵巢癌、胰腺癌和前列腺癌。可用本发明的氨基磷酸酯烷化剂前体药物治疗的癌症类型的实例在下面的参考文献中提供,它们中的每一个被全部引入本文作为参考:Tidmarsh等,2005年12月22日提交的PCT专利申请第PCT/US2005/047314号和名称为"Glufosfamide combination therapy"的PCT专利申请,其律师案卷号为021305-005900PC;以及美国专利申请第60/760,599和60/719,787号和PCT专利申请公开第WO 2005/076888号。这些癌症中的几个在下面为阐明的目的进行评述。本领域技术人员将明白,癌症化学疗法通常涉及同时或连续施用多种抗癌剂,并且如下面进一步讨论的,氨基磷酸酯烷化剂前体药物可被用于联合治疗,如本文方法所描述的。因此,在含有适于用氨基磷酸酯烷化剂前体药物治疗的低氧区的示例性癌症的描述中,也描述了联合治疗的实例。
[0299]在美国,肺癌影响100,000以上男性和50,000以上女性,他们中的大多数在诊断的一年内死去,这使得肺癌成为癌症死亡的主要因素。目前治疗肺癌的方法涉及在进行或不进行放疗或手术的情况下结合化学疗法。氨基磷酸酯烷化剂前体药物可被用作单一药剂或者与现有的联合治疗组合。多种联合化学治疗方法已被报道用于小细胞肺癌,包括由环磷酰胺、阿霉素和长春花新碱(CAV)组成的联合;依托泊甙苷和顺铂(VP-16)组成的联合;和环磷酰胺、阿霉素和VP-16(CAVP-16)组成的联合。对于非小细胞肺癌,从联合化疗(依托泊甙苷和顺铂)得到的适度的存活益处(modest survival benefit)已被报道。
[0300]同样地,几种不同的细胞毒性药已经产生卵巢癌至少暂时的退化。在治疗卵巢癌中最具活性的药物是烷化剂,包括环磷酰胺、异环磷酰胺、苯丙氨酸氮芥、苯丁酸氮芥、噻替哌、顺铂和卡铂。目前用于卵巢癌的联合治疗包括顺铂或卡铂与环磷酰胺结合,以三到四周为间隔,进行六到八个周期。本文描述的化合物和方法提供治疗卵巢癌的前体药物形式和方法,其中本文描述的氨基磷酸酯烷化剂前体药物被用作单一药剂或用于现有的这种联合治疗,或者取代或加入目前使用的药剂(一种或多种)。
[0301]在美国,前列腺癌是男性中最常见的恶性肿瘤,它是55岁以上男性的癌症死亡中第二最常见的因素,这种癌症已被报道主要由低氧组织构成。几种化学疗法已被报道用于在激素治疗之后复发后的后期阶段疾病。用于治疗前列腺癌的药剂包括烷化剂磷酸雌二醇氮芥、泼尼莫司汀和顺铂。联合化学治疗也被用来治疗前列腺癌,包括用磷酸雌二醇氮芥加泼尼莫司汀和顺铂的治疗,以及用5-氟尿嘧啶、苯丙氨酸氮芥和烃脲的治疗。本发明提供治疗前列腺癌的方法,其中本发明的氨基磷酸酯烷化剂前体药物被用于这类联合,或者取代目前使用的药剂或者加入目前使用的药剂(一种或多种)。
[0302]在美国,大肠癌症是癌症死亡的第二最常见因素,并且同样是以低氧区为特征的癌症。虽然患有晚期直肠癌的患者进行化学治疗已证明仅仅具有有限的益处(marginal benefit),但5-氟尿嘧啶对于这种疾病是最有效的治疗。5-氟尿嘧啶可单独使用或与其它药物联合使用,但是其与将可测量的肿瘤质量降低50%或更多的仅为15%到20%的可能性相关。使用5-氟尿嘧啶(5-FU)与本文描述的化合物和方法以及使用前体药物治疗结肠癌的方法相联合,可提供明显的治疗益处和潜力,因为其满足了更好治疗这种疾病的方法所需的未满足的要求。
[0303]在一种治疗方法的情况中,氨基磷酸酯烷化剂前体药物可用于多种已知的癌症治疗方法,包括但不限于“抗体导向酶前体药疗法(antibody directedenzyme prodrug therapy,ADEPT)”、“病毒导向酶前体药疗法(VDEPT)”、“基因导向酶前体药疗法(GDEPT)”和“细菌导向酶前体药疗法(BDEPT)”。氨基磷酸酯烷化剂前体药物总的应用不限于前述治疗方法。
[0304]在另一方面,本发明提供治疗非癌增生性疾病的方法,该疾病的特征为细胞增殖(例如异常增加的细胞增殖速率或数量)。在一实施方式中,根据本发明治疗的增生性疾病选自过敏性脉管炎和肉芽肿病(丘-施二氏疾病)、石棉沉着病、哮喘、萎缩性胃炎、良性前列腺增生、大疱性类天疱疮、乳糜泻、慢性支气管炎和慢性阻塞性呼吸道疾病、慢性鼻窦炎、克罗恩病、脱髓鞘神经性病、皮肌炎、包括特应性皮炎的湿疹、咽鼓管疾病(eustachean tube disease)、巨细胞动脉炎、移植排斥、超敏感性肺炎、高敏感性脉管炎(亨诺-许兰二氏紫癜)、刺激性皮炎、炎性溶血性贫血、炎性嗜中性粒细胞减少、炎性肠疾病、川崎氏病、多硬化症、心肌炎、肌炎、鼻息肉、鼻泪管疾病、新增性血管炎、胰腺炎、寻常天疱疹、原发性肾小球性肾炎、牛皮癣、牙周病、多囊肾疾病、结节性多动脉炎、多脉管炎重叠综合症、原发性硬化性胆管炎、类风湿性关节炎、血清病、手术后粘连(surgicaladhesion)、狭窄或再狭窄、巩膜炎、硬皮病、胆管狭窄、(十二指肠、小肠和结肠)狭窄、矽肺和其它形式尘肺病、I型糖尿病、溃疡性结肠炎、溃疡性直肠炎、与结缔组织紊乱相关的血管炎、与补体系统先天性缺陷相关的血管炎、中枢神经系统血管炎和韦格纳肉芽肿病。
[0305]在本发明的一些实施方式中,施用本发明的化合物治疗选自下列的增生性疾病:牛皮癣、多硬化症、类风湿性关节炎、再狭窄和良性前列腺增生。在一实施方式中,被治疗的增生性疾病是牛皮癣,一种以角质化细胞的细胞增殖为特征的疾病,其在皮肤上聚集形成突出的鳞状损伤。在另一实施方式中,被治疗的增生性疾病是多硬化症,一种以在脑中逐步脱髓鞘为特征的疾病。在另一实施方式中,被治疗的增生性疾病是类风湿性关节炎,一种多系统慢性、复发性炎性疾病,其可导致受影响关节的破坏和强直(ankyiosis)。在另一实施方式中,通过用含有本发明化合物的组合物涂敷假体施用本发明的化合物,以防止由于在植入对象的假体上细胞增殖导致的增生性疾病。在另一实施方式中,被治疗的增生性疾病是良性前列腺增生,一种其中前列腺上皮细胞生长异常并且因此阻断尿流动的疾病。
IIIb.制剂、给药方式、剂量
[0306]氨基磷酸酯烷化剂前体药物一般被配制为用于施用给对象的药物制剂。在这部分描述的是当使用本文描述的氨基磷酸酯烷化剂前体药物治疗癌症时,可被使用的给药方式、制剂和剂量。
[0307]可通过任何可将药物输送到作用位置——肿瘤低氧区的方法,实现氨基磷酸酯烷化剂前体药物的给药以治疗癌症。通过静脉内注射,施用多种癌症药物,氨基磷酸酯烷化剂前体药物可被配制成如此制剂,不但包括随时注射(ready-for-injection)制剂,而且包括冻干或浓缩的制剂,冻干或浓缩的制剂在注射之前,必须先分别再水化或稀释。除了这些制剂,可配制氨基磷酸酯烷化剂前体药物,用于经口途径、十二指肠内途径、肠胃外注射(包括静脉内、皮下、肌肉内、血管内或灌流)、外用和直肠途径给药。本领域技术人员将认识到,氨基磷酸酯烷化剂前体药物在消化道内可被细菌活化。如果这种活化是不需要的,那么从业者可使用这样的给药途经或制剂——其导致氨基磷酸酯烷化剂前体药物在进入大肠或结肠之前被吸收。氨基磷酸酯烷化剂前体药物的实际给药途经和相应的剂型将取决于被治疗的癌症类型、选择用于给药的氨基磷酸酯烷化剂前体药物、癌症的严重性和患者的年龄、体重和状况,以及其它因素。
[0308]氨基磷酸酯烷化剂前体药物施用的量,以及因此包含在施用的药剂中的氨基磷酸酯烷化剂前体药物的量和包含药剂的产物的量,将取决于被治疗的对象、癌症的严重性、癌症的位置、给药速率、前体药物的特性(例如分子量、溶解度以及低氧和常氧的细胞毒性)、氨基磷酸酯烷化剂前体药物释放的细胞毒剂和处方医师的判断。
[0309]在一实施方式中,本发明提供治疗患者癌症的方法,其中有效剂量一般在每千克体重大约0.001到大约0.1g的范围,或者以单剂量或分次剂量为大约1.0到大约35mg/kg/天。对于70kg的人,这总计为大约0.05到大约7g/天,大约0.2到大约2.5g/天。在一些情况下,在前述范围的下限以下的给药水平可能远远足够,而在另一些情况下,可以使用更大的剂量,而没有引起任何有害的副作用;更大的剂量可被分成几个较小的剂量来整日给药,这通过灌流一小时或者持续使用经外周插入的中心导管(PICC管线)和便携式静脉注射袋和泵来实现。
[0310]在一实施方式中,本发明治疗癌症和其它增生性疾病的化合物的有效剂量以单剂量或分次剂量在大约0.1到大约35mg/kg/天;大约0.5到大约20mg/kg/天;大约0.5到大约15mg/kg/天;大约0.5到大约10mg/kg/天;大约0.5到大约8mg/kg/天;和大约1到大约5mg/kg/天的范围。在一实施方式中,本发明治疗癌症和其它增生性疾病的化合物的有效剂量以单剂量或分次剂量在大约2到大约8mg/kg/天;大约2到大约4mg/kg/天;和大约2mg/kg/天的范围。在一实施方式中,本发明治疗癌症和其它增生性疾病的化合物的有效剂量以单剂量或分次剂量在大约0.25到大约2.5mg/kg/天;大约0.25到大约1mg/kg/天;和大约0.25到大约0.5mg/kg/天的范围。在一实施方式中,或者作为单一疗法(只用本发明的化合物),或者与其它护理治疗标准联合(组合),通过每日静脉注射,给予该剂量。在一实施方式中,本发明治疗癌症和其它增生性疾病的化合物的有效剂量在如先前描述的范围内,每周给药一次。
[0311]在一实施方式中,间歇性(更少的频率)施用更大的剂量;在两周内每隔三天一次,施用大约3到大约20mg/kg的剂量;大约6到大约10mg/kg的剂量;或8mg/kg的剂量。在另一实施方式中,在四周内每周一次,施用大约5到大约30mg/kg的剂量;大约10到大约15mg/kg的剂量;或者大约12.5mg/kg的氨基磷酸酯烷化剂前体药物。在一实施方式中,施用大约0.5到大约8mg/kg/天范围内的剂量5天,两周为一周期。
[0312]在另一实施方式中,对于治疗人类患者,氨基磷酸酯烷化剂前体药物的最大日剂量不大于500mg/kg患者体重,因此氨基磷酸酯烷化剂前体药物的每日给药剂量在每千克患者体重大约1mg氨基磷酸酯烷化剂前体药物到每千克患者体重大约500mg氨基磷酸酯烷化剂前体药物的范围内。在一实施方式中,氨基磷酸酯烷化剂前体药物的每日给药剂量在待被治疗的患者体重的大约5mg/kg到大约500mg/kg的范围内。在另一实施方式中,氨基磷酸酯烷化剂前体药物的日剂量的治疗有效量为待被治疗的患者体重的大约10mg/kg到大约250mg/kg。在另一实施方式中,氨基磷酸酯烷化剂前体药物的治疗有效量为待被治疗的患者体重的大约25mg/kg到大约150mg/kg。在另一实施方式中,氨基磷酸酯烷化剂前体药物的治疗有效量为待被治疗的患者体重的大约25mg/kg到大约50mg/kg。在另一实施方式中,氨基磷酸酯烷化剂前体药物的治疗有效量为待被治疗的患者体重的大约1.25mg/kg到大约12.5mg/kg。
[0313]通过和根据对于人类以及任何其它哺乳动物的研究,也可提供关于给药的指导。可将为动物确定的治疗有效量转化为如下表所述的相应的人的相当剂量(HED):
| 动物 | 人的相当剂量(HED)转化因子a |
| 小鼠 | 12.3 |
| 仓鼠 | 7.4 |
| 大鼠 | 6.2 |
| 雪貂 | 5.3 |
| 豚鼠 | 4.6 |
| 微型猪(Micro-pig) | 1.4 |
| 小型猪(Mini-pig) | 1.1 |
| 兔 | 3.1 |
| 狗 | 1.8 |
| 猴b | 3.1 |
| 狨 | 6.2 |
| 松鼠猴 | 5.3 |
| 狒狒 | 1.8 |
a为了将以mg/kg为单位的动物剂量转化为以mg/kg为单位的HED(假设60kg人),将动物剂量除以HED转化因子。对于没有列出的物种或者在标准范围以外的重量,人的相当剂量(HED)可从下面的公式计算:HED=以mg/kg为单位的动物剂量×(kg为单位的动物体重/kg为单位的人体重)0.33。
b例如猕猴、恒河猴或桩尾猴。
[0314]为了得到治疗效果,氨基磷酸酯烷化剂前体药物的治疗有效日剂量通常多次施用给患者。在一实施方式中,氨基磷酸酯烷化剂前体药物在一段时期内按日施用。一般而言,将至少连续三天进行日给药。在相关实施方式中,至少连续5天、至少连续7天或至少连续10天给药。根据医师选择的剂量、制剂和给药途径以及患者的方便程度,全部日剂量可每天一次给药,或者,日剂量也可在一天的进程中分为多次较小的剂量给药(包括泵输注或静脉内给药)。例如,可将剂量分为较小的两个剂量,每日给药两次,或分为较小的三个剂量,每日给药三次。对癌症治疗领域技术人员显而易见的是,如本文所采用,"日"给药并不限于每天给药一次,而是可包括多次给药。
[0315]也可采用除连续地日给药之外的其它给药方案。每隔一天(qod)给药一次是特别方便的,在某些情况下采取每三日给药一次,或每星期给药一次可能是合适的,但在任何一种情况下,都在一段时间内进行氨基磷酸酯烷化剂前体药物的重复给药。例如,无论给药是按日(如上所述,包括多次的日剂量)、每隔一天或次数更少,在一种实施方式中,氨基磷酸酯烷化剂前体药物每星期至少2天给药,持续至少二、三、四、五个或至少六个连续的星期,或者可选地,在六个月的期间内进行至少两、三、四、五个或至少六个星期,或者可选地,在十二个月的期间内进行至少两、三、四、五个或至少六个星期。在一个实施方式中,氨基磷酸酯烷化剂前体药物每星期至少3天给药,持续至少二、三、四、五个或至少六个连续的星期,或者可选地,在六个月期间内持续至少两、三、四、五个或至少六个星期,或者可选地,在十二个月期间内持续至少两、三、四、五个或至少六个星期。在一种实施方式中,氨基磷酸酯烷化剂前体药物每月至少10天给药,任选为每月至少20天,持续至少一个月或至少两、三、四、五个或至少六个连续月,或者可选地,在六个月期间内持续至少一、二、三、四、五个或至少六个月。
[0316]在一实施方式中,治疗有效剂量的给药持续进行多天,典型地为至少三个连续日,通常为至少五到十个连续日,或一星期,或几个星期或更长。因而,患者可按照本方法进行几天、一星期、一个月、两个月、三个月、六个月、或一年或更长时间的氨基磷酸酯烷化剂前体药物给药。
[0317]与其它抗癌剂的给药方案相一致,氨基磷酸酯烷化剂前体药物可以进行多“轮”给药。例如,在一些实施方式中,氨基磷酸酯烷化剂前体药物的给药可为每日一次,持续至少三到十,或至少五到十个连续日,并且该三到十日或五到十日的治疗可重复一次、两次或三次或更多次,有时候在多日治疗的每次之间存在长达一到几星期的无治疗(用氨基磷酸酯烷化剂前体药物)期间。类似地,在一些实施方式中,氨基磷酸酯烷化剂前体药物每隔一天进行二到十次给药,更经常为三到十次给药,或五到十次给药,并且该二、三或五到十次每隔一天(qod)给药可以重复一次、两次或三次或更多次,在多日给药的每次之间存在长达一个到多个星期的无治疗(用氨基磷酸酯烷化剂前体药物)期间。其它的多轮给药计划对于受到本公开指导的熟练医师来说是显而易见的。
[0318]一方面,"给予治疗有效剂量或治疗有效方案的氨基磷酸酯烷化剂前体药物"是指(i)在所述的范围内给予氨基磷酸酯烷化剂前体药物(例如,1mg-1g氨基磷酸酯烷化剂前体药物/kg患者体重,典型为25-150mg氨基磷酸酯烷化剂前体药物/kg患者体重),在规定的时期内持续规定的最少天数,其中氨基磷酸酯烷化剂前体药物的给药对患者的癌症具有疗效。示例性的氨基磷酸酯烷化剂前体药物的治疗有效剂量方案包括本文所述的那些方案,如氨基磷酸酯烷化剂前体药物给药进行3个连续日,5个连续日,7个连续日,10个连续日,每周至少3天,每周至少3天并持续一个月,每月至少10天,以及每月至少20天。
[0319]在根据本发明优化氨基磷酸酯烷化剂前体药物治疗方案中,可对氨基磷酸酯烷化剂前体药物给药的剂量和频率进行选择,从而在治疗期间的血浆浓度曲线(AUC)下获得最大的持续面积。如AUC所测定的,理论上最佳的剂量给药方案使肿瘤细胞最大程度地暴露于氨基磷酸酯烷化剂前体药物,同时将任何单次给药的最大血浆浓度(Cmax)降至最低。较高的Cmax将会带来毒性,而AUC将决定疗效。正如其它癌症治疗药物的领域中所理解的一样,如果观察到毒性或者为了方便患者,可以暂时停止采用氨基磷酸酯烷化剂前体药物的治疗,这并不偏离本发明的范围,然后再重新开始。
[0320]在一实施方式中,用于治疗癌症的本发明氨基磷酸酯烷化剂前体药物的药物动力学可确定剂量、给药方法和用氨基磷酸酯烷化剂前体药物治疗的癌症的种类。在一实施方式中,本发明的氨基磷酸酯烷化剂前体药物可具有1到300分钟之间的体内半衰期。在一实施方式中,本发明的化合物可具有3到10分钟之间的体内半衰期。在一实施方式中,本发明的化合物可具有10到30分钟之间的体内半衰期。氨基磷酸酯烷化剂前体药物的短半衰期可能要求治疗的灌流时间长于具有更长半衰期的氨基磷酸酯烷化剂前体药物所需要的灌流时间。氨基磷酸酯烷化剂前体药物的短半衰期可能增加对该前体药物的最大耐受剂量(MTD)。
[0321]在另一实施方式中,本发明提供这样的氨基磷酸酯烷化剂前体药物——当与小鼠肝脏微粒(如果可行,用人的实例和数据更新)蛋白培养30分钟时,其保持多达20%未改变。在另一实施方式中,本发明提供这样的氨基磷酸酯烷化剂前体药物——当与小鼠肝脏微粒蛋白培养30分钟时,其保持20%-80%未改变。在另一实施方式中,本发明提供这样的氨基磷酸酯烷化剂前体药物——当与小鼠肝脏微粒蛋白培养30分钟时,其保持80%以上未改变。在另一实施方式中,本发明的氨基磷酸酯烷化剂前体药物示例——当与小鼠肝脏微粒蛋白培养30分钟时,其保持80%以上未改变,包括1、25和36。本发明的前体药物的MLM稳定性越高,其治疗有效剂量和患者不期望的副作用越低。
[0322]在相关实施方式中,本发明的氨基磷酸酯烷化剂前体药物的生物还原基团,在低氧肿瘤区还原/活化后,形成氨基磷酸酯烷化剂-TM偶联物。在转移疾病的情况下,氨基磷酸酯烷化剂-TM偶联物可扩散并且到达肿瘤的其它部分或其它肿瘤。多个药物动力学参数如在稳定状态下的分布体积(Vss)、清除率(clearance,CL)、曲线下面积(AUC)、小鼠肝脏微粒稳定性(MLM稳定性)、血浆稳定性和本发明的氨基磷酸酯烷化剂前体药物的Cmax被测量并在实施例部分列出(也参见Hardman等,如前所述)。
[0323]在再治疗方案中,可调整剂量以反映患者对先前治疗的耐受性。在任何情况下,当在重复给药期间观察到毒性时,如观察到严重的症状,可暂时停止给药。当带有毒性的第一器官不再含有显著浓度的氨基磷酸酯烷化剂前体药物或其释放的氨基磷酸酯烷化剂(可通过症状的停止而间接地测定或确定)时,可在此时终止给药的暂停期(药物假期)。因此,间歇给药期间不仅可用具体的天数来定义,也可通过基于症状和氨基磷酸酯烷化剂前体药物或其释放的氨基磷酸酯烷化剂的正常器官清除率的药物假期来具体化。
[0324]例如,氨基磷酸酯烷化剂前体药物的制剂可以是适合经口给药的形式,如片剂、胶囊、药粉(pill powder)、持续释放剂型、溶液和悬浮液;适合肠胃外注射的形式,如无菌溶液、悬浮液或乳浊液;适合外用给药的形式,如软膏或乳脂;和适合直肠给药的形式,如栓剂。氨基磷酸酯烷化剂前体药物的制剂可以是适合精确剂量单次给药的单位剂量形式,其一般包括传统的药物载体或赋形剂。
[0325]适合的药物载体包括惰性稀释剂或填料、水和多种有机溶剂。如果需要,药物组合物可含有另外的成分如调味剂、粘合剂、赋形剂和类似物。因此对于经口给药,含有多种赋形剂如柠檬酸的片剂可与多种崩解剂如淀粉、褐藻酸和某些复合硅酸酯,以及与粘合剂如蔗糖、明胶和阿拉伯胶一起使用。另外,润滑剂如硬脂酸镁、十二烷基硫酸钠和滑石可被用来制备本文描述的氨基磷酸酯烷化剂前体药物的片剂形式的制剂。相似类型的固体组合物可被用于软和硬填充明胶胶囊。因此,优选的材料包括乳糖或奶糖和高分子量聚乙二醇。当需要水悬浮液或酏剂用于经口给药时,本文的前体药物可与多种增甜剂或调味剂、着色物质或染料,以及如果需要的话,乳化剂或悬浮剂组合,与稀释剂如水、乙醇、丙二醇、甘油或它们的组合结合。
[0326]示例性肠胃外给药形式包括无菌水溶液中的氨基磷酸酯烷化剂前体药物的溶液或悬浮液,所述无菌水溶液例如含水聚乙二醇、丙二醇或右旋糖溶液。如果需要,这类剂型可被适当缓冲。
[0327]根据本公开内容,制备多种含有具体量活性药物的药学组合物方法对本领域技术人员是已知的,或者是显而易见的。例如,参见Remington′sPharmaceutical Sciences,Mack Publishing Company,Philadelphia,Pa.,第17版(1984)。
[0328]使用本发明氨基磷酸酯烷化剂前体药物治疗癌症的方法在杀死生长在肿瘤低氧区最难杀死的癌细胞是有效的。一旦在低氧区释放,氨基磷酸酯烷化剂前体药物可从低氧细胞扩散并杀死含有快速分裂细胞增长群的邻近区域的癌细胞。低氧区作为药物工厂在肿瘤内产生用于杀死相邻常氧癌细胞的烷化剂,在肿瘤中产生相对于正常组织更高浓度的氨基磷酸酯烷化剂。前体药物用于在肿瘤内产生氨基磷酸酯烷化剂,可降低由于正常细胞毒性引起的毒副作用。在肿瘤常氧区域中的癌细胞被破坏后,低氧区可变成常氧区并开始分裂。如此,这类细胞可以被从本发明的氨基磷酸酯烷化剂前体药物或已知的那些氨基磷酸酯烷化剂前体药物产生的氨基磷酸酯烷化剂杀死,或者被与氨基磷酸酯烷化剂前体药物一起施用的其它抗癌药或细胞毒素杀死,如下面部分描述。
IIIc.联合治疗
[0329]根据本发明的方法,氨基磷酸酯烷化剂前体药物可与其它抗癌药("抗癌剂")联合进行共同给药。不拟受限于任何具体的机理或效果,这类共同给药在某些情况下可以提供优于已知癌症疗法的数个优势中的一个或多个,如,例如,共同施用氨基磷酸酯烷化剂前体药物和抗癌剂对诱导癌细胞死亡具有协同效应。共同给药提供了较单独施用抗癌剂更好的治疗结果,例如,癌症的一个或多个症状的更大程度的缓解或减轻,疾病程度的减轻,疾病进展的延迟或减慢,疾病状态的改善、缓解或稳定,部分或完全好转,存活延长或其它有益的治疗结果。
[0330]氨基磷酸酯烷化剂前体药物的共同给药增加了癌细胞对抗癌剂的敏感性,使得可以将更低剂量的抗癌剂施用给患者,或者允许抗癌剂用于治疗在其它情况下对该抗癌剂有抗性的细胞或治疗在其它情况下难以治疗的细胞。已知的抗癌剂一般靶向于常氧区中的快速分裂细胞,而本发明的氨基磷酸酯烷化剂前体药物靶向在肿瘤区域中不能被单独的抗癌药有效杀死的低氧细胞。
[0331]如本文所用,当氨基磷酸酯烷化剂前体药物与另一抗癌剂(本文也称为“药剂(agent)”)作为相同疗程的一部分而给予时,氨基磷酸酯烷化剂前体药物与该药剂被“共同给予”。在一实施方式中,在给予药剂(即,开始其它癌症疗法)之前,首先给予氨基磷酸酯烷化剂前体药物,并且在给予药剂的过程(即,其它疗法的过程)中,持续应用氨基磷酸酯烷化剂前体药物进行治疗。在另一实施方式中,在其它癌症疗法开始或完成之后,给予氨基磷酸酯烷化剂前体药物。在其它实施方式中,氨基磷酸酯烷化剂前体药物随其它癌症疗法开始的同时被首先给予。例如,参见在实施例部分描述的联合治疗。
[0332]在一实施方式中,在给予药剂之前,首先给予氨基磷酸酯烷化剂前体药物,并且在停止给予药剂后,继续应用氨基磷酸酯烷化剂前体药物进行治疗。在一实施方式中,在给予药剂之前,首先给予氨基磷酸酯烷化剂前体药物,并且在给予药剂期间的一部分期间,继续应用氨基磷酸酯烷化剂前体药物进行治疗。对于某些药物,例如某些拓扑异构酶抑制剂,可以在给予该第二药物之前,开始和完成氨基磷酸酯烷化剂前体药物的给予。
[0333]在存在氧的情况下,还原Z3而形成的自由基阴离子与氧反应,产生超氧化物和Z3。超氧化物是细胞毒素并且超氧化物在常氧组织中的产生可引起不期望的副作用。在一实施方式中,本发明提供了与化学保护试剂(chemoprotective agent)或化学保护剂(chemoprotectant)联合给予的氨基磷酸酯烷化剂前体药物。化学保护剂保护健康组织免遭抗癌药的毒性作用。在一实施方式中,化学保护剂是硫醇或二硫化物。在一实施方式中,化学保护剂可以还原超氧化物。在另一实施方式中,化学保护剂可以和产生于氨基磷酸酯烷化剂前体药物的“Michael受体”反应并且防止“Michael受体”与蛋白质和核酸反应(参见下面)。

[0334]现今的抗癌药治疗通常涉及多轮或“多个周期”地给予抗癌药(一种或多种)。在给予氨基磷酸酯烷化剂前体药物的上下文中,每个给药周期(以及全部的完整周期组)可以被视为给予第二药物。氨基磷酸酯烷化剂前体药物可以在用其它药剂的多个治疗周期中的任意周期或所有周期中给予;一般地,氨基磷酸酯烷化剂前体药物在每一周期中,每日给予,持续至少2天或更多天。在本发明的一个方面,根据每轮重复的时间表,将氨基磷酸酯烷化剂前体药物与药剂共同给予。
[0335]在应用氨基磷酸酯烷化剂前体药物治疗癌症的方法的一个方案中,氨基磷酸酯烷化剂前体药物与有效量的一种或多种化疗药、有效量的放疗、适当的手术过程或这些额外治疗的任何组合联合给予。
[0336]当氨基磷酸酯烷化剂前体药物与一种或多种另外的治疗联合使用时,氨基磷酸酯烷化剂前体药物和另外的治疗可以同时给予或者可以分开给予。例如,如果氨基磷酸酯烷化剂前体药物与另外的化疗药一起给予时,两种药可以同时给予或者可以相继给予,在给药之间相隔一段时间。本领域技术人员将理解同时和相继给予药物的方法和给药之间的可能时间段。例如,参见在实施例部分描述的联合治疗。
[0337]药剂可以作为相同或不同的制剂给予,并且可以通过相同或不同的途径给予。
[0338]可以和本发明的氨基磷酸酯烷化剂前体药物联合应用的化疗剂包括但不限于白消安、英丙舒凡、哌泊舒凡、苯佐替派、卡波醌、2-脱氧-D-葡萄糖、氯尼达明及其类似物(refrence apps)、葡磷酰胺、吉母赛他宾(gemcitibine)、埃罗替尼(erlotinib)、美妥替哌、乌瑞替派、六甲基蜜胺、伊马替尼、三亚乙基蜜胺、三乙撑磷酰胺、三亚乙基硫代磷酰胺、三羟甲基蜜胺(trimethylolomelamine)、苯丁酸氮芥、萘氮芥、雌莫司汀、异环磷酰胺、吉非替尼、氮芥、氮芥氧化物盐酸(mechlorethamine oxide hydrochloride)、苯丙氨酸氮芥、novembichin、苯芥胆固醇、泼尼莫司汀、曲磷胺、尿嘧啶氮芥、卡莫司汀、氯脲菌素、福莫司汀、尼莫司汀、雷莫司汀、达卡巴嗪、甘露莫司汀、二溴甘露醇、二溴卫矛醇、哌泊溴烷、阿克拉霉素、放线菌素F(1)、安曲霉素、偶氮丝氨酸、博来霉素、放线菌素C、卡柔比星、嗜癌素、色霉素、放线菌素D、柔红霉素、柔毛霉素、6-叠氮-5-氧代-1-正亮氨酸、麦考酚酸、诺拉霉素、橄榄霉素、培洛霉素、普卡霉素、泊非霉素、嘌罗霉素、绛色霉素、链脲霉素、杀结核菌素、乌苯美司、净司他丁、佐柔比星、二甲叶酸、丁喋翼素、三甲曲沙、氟达拉滨、6-巯基嘌呤、硫咪嘌呤、硫鸟嘌呤、安西他滨、阿扎胞苷、6-氮尿苷、卡莫氟、阿糖胞苷、双脱氧尿苷、去氧氟尿苷、依诺他滨、氟尿苷、5-氟尿嘧啶、替加氟、L-天冬酰胺酶、阿法链道酶(pulmozyme)、醋葡醛内酯、醛磷酰胺苷(aldophosphamide glycoside)、氨基乙酰丙酸、安吖啶、bestrabucil、比生群、卡铂、defofamide、地美可辛、地吖醌、elfornithine、依利醋铵、依托格鲁、氟他氨、硝酸镓、羟基脲、干扰素α、干扰素β、干扰素γ、白介素-2、香菇多糖、米托胍腙、米托蒽醌、莫哌达醇、尼西吖啶(nitracrine)、喷司他丁、phenamet、吡柔比星、依托泊甙酸(podophyllinicacid)、2-乙基酰肼、丙卡巴肼、雷佐生、西佐喃、锗螺胺、紫杉醇、他莫昔芬、erlotonib、替尼泊甙、细交链孢菌酮酸、三亚胺醌、2,2′,2"-三氯三乙胺、尿烷(urethan)、长春花碱、环磷酰胺和长春新碱。可以应用的其它化疗药包括铂衍生物,包括但不限于顺铂、卡铂和oxoplatin。
[0339]在一个方案中,本文描述的氨基磷酸酯烷化剂前体药物可以和抗血管生成(antiangeogenisis)抑制剂联合应用,所述血管生成抑制剂包括但不限于阿瓦斯丁(Avastin)和类似的治疗剂。在联合治疗方法的一个方案中,对象用血管生成抑制剂来治疗,随后应用氨基磷酸酯烷化剂前体药物来治疗。在联合治疗方法的一个方案中,对象用血管生成抑制剂来治疗,随后应用氨基磷酸酯烷化剂前体药物和其它化疗剂来治疗,其它化疗剂包括但不限于顺铂和卡铂。在应用血管生成抑制剂治疗的这些联合方法的一个方案中,应用该方法来治疗乳腺癌。
[0340]在另一实施方式中,氨基磷酸酯烷化剂前体药物与抗癌药一起给予,所述抗癌药直接或间接起作用来抑制表皮生长因子或EGFR受体。适于与本发明氨基磷酸酯烷化剂前体药物共同给予的EGFR抑制剂包括吉非替尼和erlotonib。
[0341]在另一方案中,氨基磷酸酯烷化剂前体药物与抗癌药一起给予,所述抗癌药直接或间接起作用来抑制低氧诱导因子1α(HIF1α)或抑制蛋白质或酶例如葡萄糖转运蛋白或VEGF,其表达或活性随着HIF1α水平增加而增加。适合用于本文所述方法和组合物的这一方案的HIF1α抑制剂包括P13激酶抑制剂;LY294002;雷帕霉素;组蛋白脱乙酰酶抑制剂例如[(E)-(1S,4S,10S,21R)-7-[(Z)-亚乙基]-4,21-二异丙基-2-氧杂-12,13-二硫代-5,8,20,23-四氮杂双环-[8,7,6]-二十三-16-烯-3,6,9,19,22-戊酮(FR901228,depsipeptide);热休克蛋白90(Hsp90)抑制剂例如格尔德霉素、17-烯丙基氨基-格尔德霉素(17-AAG)和其它格尔德霉素类似物,和根赤壳菌素和根赤壳菌素衍生物例如KF58333;金转停(genistein);茚满酮;星形孢菌素(staurosporin);蛋白激酶-1(MEK-1)抑制剂例如PD98059(2′-氨基-3′-甲氧基黄酮);PX-12(1-甲基丙基2-咪唑二硫化物);北风菌素PX-478;喹噁啉1,4-二氧化物;丁酸钠(NaB);硝普钠(SNP)和其它NO供体;微管抑制剂例如新生霉素、panzem(2-甲氧雌二醇或2-ME2)、长春新碱、紫杉烷、埃坡霉素、discodermolide和前述任何物质的衍生物;香豆素;巴比妥酸盐和硫代巴比妥酸类似物;喜树碱;和YC-1,描述于Biochem.Pharmacol.,15 Apr 2001,61(8):947-954中的化合物,所述文献并入本文作为参考,及其衍生物。
[0342]在另一方案中,氨基磷酸酯烷化剂前体药物与抗血管生成药一起给予,所述抗血管生成药包括但不限于选自下列的抗血管生成药:血管抑素(angiostatin),一种抑制或者以其它方式拮抗VEGF作用的药剂、巴马司他、卡托普利、软骨来源的抑制剂、genistein、内皮他丁、白介素、lavendustin A、甲羟孕酮乙酸酯、重组人血小板因子4、紫杉醇、tecogalan、沙利度胺、血小板反应素、TNP-470和阿瓦斯汀(Avastin)。用于由本文所述的方法和组合物提供的联合治疗的其它有用的血管生成抑制剂包括Cox-2抑制剂如celecoxib(Celebrex)、双氯芬酸(扶他林)、依托度酸(Lodine)、非诺洛芬(Nalfon)、吲哚美辛(Indocin)、酮洛芬(Orudis、Oruvail)、ketoralac(Toradol)、奥沙普秦(Daypro)、萘丁美酮(Relafen)、舒林酸(Clinoril)、托美丁(Tolectin)、rofecoxib(Vioxx)、布洛芬(Advil)、萘普生(Aleve、Naprosyn)、阿司匹林和乙酰氨基酚(Tylenol)。
[0343]此外,由于丙酮酸在血管生成中起重要作用,丙酮酸盐模拟物和糖酵解抑制物如卤代丙酮酸盐,包括溴丙酮酸盐,可以和抗血管生成化合物以及氨基磷酸酯烷化剂前体药物联合应用以治疗癌症。在另一方案中,氨基磷酸酯烷化剂前体药物与抗血管生成药和另一抗癌药一起给予以治疗癌症,另一抗癌药包括但不限于选自烷化剂、顺铂、卡铂和微管装配抑制剂的细胞毒性剂。
[0344]除了氨基磷酸酯烷化剂前体药物与上述药剂的联合之外,本文所述的方法和组合物还提供了氨基磷酸酯烷化剂前体药物与其它抗癌药的多种协同联合。本领域技术人员可以容易地确定与本文所述的氨基磷酸酯烷化剂前体药物“协同地”起作用的抗癌药。例如,参考文献Vendetti,"Relevance of TransplantableAnimal-Tumor Systems to the Selection of New Agents for Clinical Trial,"Pharmacological Basis of Cancer Chemotherapy,Williams and Wilkins,Baltimore,1975和Simpson Herren等,1985,"Evaluation of In Vivo Tumor Models for PredictingClinical Activity for Anticancer Drugs,"proc.Am.Assoc.Cancer Res.26:330,每一文献并入本文作为参考,其描述了帮助确定两种药物是否协同起作用的方法。
[0345]虽然根据本文所述的方法,协同对于治疗益处而言并不是必须的,但在一实施方式中,本发明提供了治疗癌症的方法,其中在氨基磷酸酯烷化剂前体药物与另一种抗癌药之间存在协同作用。如果两种药剂的联合给药方案产生比最优或最大耐受剂量的单一药剂的总和明显更好的肿瘤细胞杀伤作用,则两种药物可以称作具有治疗协同作用。“协同度(degree of synergy)”可以定义为,最优的联合方案引起的肿瘤细胞杀伤的净对数减去最优剂量的最具活性单一药剂引起的肿瘤细胞杀伤的净对数。10倍(1个对数)以上的细胞杀伤差异被结论性地认为表明了治疗协同作用。
[0346]当氨基磷酸酯烷化剂前体药物与另一抗癌药应用时,在至少一些实施方式中,氨基磷酸酯烷化剂前体药物将在其它一种或多种药物治疗开始之前给予,并且给药一般将在其它一种或多种药物的整个疗程中持续。在一些实施方式中,与氨基磷酸酯烷化剂前体药物共同给予的药物将以低剂量输送,并且任选地,与不给予氨基磷酸酯烷化剂前体药物的情况相比,持续更长的时期。这种“低剂量”治疗可以包括,例如,与根据本文所述方法给予的氨基磷酸酯烷化剂前体药物一起,给予低于批准剂量和持续更长时期的抗癌药,所述抗癌药包括但不限于紫杉醇、多烯紫杉醇、阿霉素、顺铂或卡铂。
[0347]由于更有效地杀死癌细胞或终止癌细胞生长以及减少了其它疗法的不期望副作用,相对于目前实施的疗法,这些方法可以用于改善患者的结局。当与氨基磷酸酯烷化剂前体药物联合使用时,其它抗癌药(一种或多种)可应用这些药剂所用的标准剂量(即,未使用氨基磷酸酯烷化剂前体药物时应用的剂量)或低于这些标准剂量的剂量,进行给药。
[0348]因此,根据本文所述方法给予氨基磷酸酯烷化剂前体药物使得医生可以用更低剂量(比目前应用的)的现有(或者以后批准的)药物治疗癌症,从而缓解这些药物的一些或全部毒性副作用。对特定患者的确切剂量随患者而变化,这取决于许多因素,包括所使用的药物联合、待治疗的具体疾病和患者的状况以及过去病史,但是可以仅仅由本领域普通技术人员根据本文的教导就可以确定。
[0349]已知的和批准的化疗药或抗肿瘤药的具体给药方案(即,推荐的有效剂量)是医生已知的,并且在例如,Physician′s Desk Reference 2003,(Physicians′DeskReference,第57版)Medical Economics Company,Inc.,Oradell,N.J的产品描述中给出,和/或可以从美国食品药品管理局(Federal Drug Administration)得到。一些抗癌药的示例性给药方案也被提供如下。
[0350]癌症药物一般可以分为烷化剂、蒽环类、抗生素、芳香酶抑制剂、二膦酸酯、环加氧酶抑制剂、雌激素受体调节剂、叶酸拮抗剂、无机砷酸盐、微管抑制剂、修饰剂、亚硝基脲、核苷类似物、破骨细胞抑制剂、含铂化合物、视黄醛、拓扑异构酶1抑制剂、拓扑异构酶2抑制剂和酪氨酸激酶抑制剂。根据本文所述方法,氨基磷酸酯烷化剂前体药物可以和来自任意这些类型的任何抗癌药共同给予,或者可以在用任何这些药物或这些药物的组合治疗之前或之后给予。此外,氨基磷酸酯烷化剂前体药物可以和生物疗法联合给予(例如,用干扰素、白介素、集落刺激因子和单克隆抗体进行治疗)。用于治疗癌症的生物学试剂是本领域已知的,包括,例如,曲妥珠单抗(Herceptin)、托西莫单抗和131I托西莫单抗(Bexxar)、利妥昔单抗(Rituxan)。
[0351]用于实施本文所述方法的烷化剂包括但不限于白消安(Myleran,Busulfex)、苯丁酸氮芥(Leukeran)、异环磷酰胺(带有或不带有MESNA)、环磷酰胺(Cytoxan,Neosar)、葡磷酰胺、苯丙氨酸氮芥、L-PAM(Alkeran)、达卡巴嗪(DTIC-Dome)和替莫唑胺(Temodar)。根据本文所述方法,氨基磷酸酯烷化剂前体药物与烷化剂共同给予以治疗癌症。在一个方案中,癌症是慢性骨髓性白血病、多发性骨髓瘤或多形性成胶质细胞瘤。
[0352]在一实施方式中,本发明提供了治疗可以通过给予烷化剂治疗的癌症的方法,这是通过给予本发明的氨基磷酸酯烷化剂前体药物,或单独给予或者与至少另一种烷化剂或其前体药物联合给予来进行的。烷化剂如,例如,环磷酰胺、异环磷酰胺、葡磷酰胺、氮芥、苯丙氨酸氮芥、苯丁酸氮芥、达卡巴嗪、替莫唑胺、卡莫司汀、链脲霉素、苯达莫司汀、白消安、噻替哌、顺铂、卡铂和奥沙利铂,以及单独应用这些烷化剂中的任一种或与其它抗癌药或化学保护剂联合应用来治疗的癌症类型描述于例如Hardman等的参考文献(如前所述)中。
[0353]在一实施方式中,本发明提供了治疗癌症的方法,其通过共同给予氨基磷酸酯烷化剂前体药物和至少烷化剂环磷酰胺,来治疗III阶段和IV阶段的恶性淋巴瘤、多发性骨髓瘤、白血病、蕈样霉菌病、神经细胞瘤、卵巢腺癌、视网膜母细胞瘤以及乳腺癌。对于诱导疗法,环磷酰胺的给药剂量为1500-1800mg/m2,其以分次剂量进行静脉内给药,时间为3—5日;对于维持疗法,每7-10日给药350-550mg/m2,或者一周两次静脉内给药110-185mg/m2。根据本文描述的方法,氨基磷酸酯烷化剂前体药物与环磷酰胺一起共同给药,其中与环磷酰胺单独给药的标准量相比,环磷酰胺按照这样的剂量或更少的剂量给药,和/或给药持续时间更长。
[0354]在一实施方式中,本发明提供了治疗癌症的方法,这通过给予本发明的氨基磷酸酯烷化剂前体药物和使用至少烷化剂氮芥的癌症治疗方案一起来完成。例如,氮芥被用于与化疗方案MOPP(氮芥、长春新碱(长春花新碱))、丙卡巴肼(procarbazine)和泼尼松)联合使用于患有霍杰金病的患者,并且通过在每一疗程的28天循环的第一天和第八天,以6mg/m2的剂量静脉内快速给药方式进行给予。
[0355]在一实施方式中,本发明提供了治疗癌症的方法,这通过给予本发明的氨基磷酸酯烷化剂前体药物和使用至少烷化剂异环磷酰胺的癌症治疗方案来完成。异环磷酰胺被用来治疗儿童和成人肉瘤、颈和肺癌,以及与其它药物联合来治疗睾丸癌。异环磷酰胺被用作ICE(异环磷酰胺、卡铂和依托泊甙)和RICE(利妥昔单抗和ICE)疗法的一部分,用于治疗淋巴瘤(参见Hardman等,如前所述)。
[0356]在一实施方式中,本发明提供了治疗癌症的方法,其通过给予本发明氨基磷酸酯烷化剂前体药物和应用至少烷化剂葡磷酰胺的癌症治疗方案来完成。葡磷酰胺在临床中用于治疗胰腺癌或盐酸吉他西宾(Gemzar)抗性胰腺癌。葡磷酰胺可以用于治疗乳腺癌、霍杰金病、胃肠道癌症,或者作为GCE(葡磷酰胺、卡铂和依托泊甙)或RGCE(利妥昔单抗和GCE)疗法的一部分用于治疗淋巴瘤。(Tidmarsh等,2005年12月22日提交的PCT专利申请第PCT/US2005/047314号,以及名称为"Glufosfamide combination therapy"的PCT专利申请,律师案卷号021305-005900PC;和美国专利申请第60/760,599和60/719,787号,以及PCT专利公开第WO2005/076888号,引入这些文献的全部作为参考)。
[0357]在一实施方式中,本发明提供了治疗癌症的方法,通过给予本发明氨基磷酸酯烷化剂前体药物和应用至少一种选自吖丙啶和甲基蜜胺的烷化剂的癌症治疗方案来完成。在另一实施方式中,吖丙啶是三亚乙基蜜胺或噻替哌(Thiotepa)。
[0358]噻替哌可被用来治疗乳腺癌、卵巢癌和膀胱癌、恶性淋巴癌、支气管肺癌和维尔姆斯肿瘤。噻替哌以高剂量与使用环磷酰胺的化疗联合应用,用于患有用自体骨移植治疗的难治愈恶性肿瘤的患者,而且治疗多种癌症,包括膀胱癌、卵巢癌、乳腺癌、肺癌、脑癌和淋巴癌(参见International Agency for Research onCancer Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Humans,1975,9:286,Lyon,France;International Agency for Research on Cancer Monographson the Evaluation of Carcinogenic Risk of Chemicals to Humans,1990,50:415,Lyon,France;和MEDLINEplus,2003,Drug Information:Thiotepa,National Library ofMedicine)。在第一轮治疗失败后,甲基蜜胺——六甲蜜胺,被用来治疗晚期卵巢癌。
[0359]在一实施方式中,本发明提供了治疗癌症的方法,通过给予本发明氨基磷酸酯烷化剂前体药物和应用至少烷化剂苯丙氨酸氮芥、苯丁酸氮芥或苯达莫司汀的癌症治疗方案来完成。苯丙氨酸氮芥被用来治疗多发性骨髓瘤,并可被经口给药。苯丁酸氮芥被用来治疗慢性淋巴细胞白血病和原发性巨球蛋白血症(macroblobulinemia)。苯达莫司汀是Salmedix Inc.开发的,可被用来治疗血液恶性肿瘤,如,例如非霍杰金淋巴瘤、慢性淋巴细胞白血病和多发性骨髓瘤。
[0360]在一实施方式中,本发明提供了治疗癌症的方法,通过给予本发明氨基磷酸酯烷化剂前体药物和应用至少烷化剂白消安的癌症治疗方案来完成。白消安被用来治疗慢性粒细胞性白血病和慢性骨髓性白血病。高剂量的白消安可与环磷酰胺联合使用,在骨髓移植前治疗患有急性骨髓性白血病的患者。
[0361]在一实施方式中,本发明提供了治疗癌症的方法,通过给予本发明氨基磷酸酯烷化剂前体药物和应用至少亚硝基脲烷化剂的癌症治疗方案来完成。在另一实施方式中,亚硝基脲烷化剂是卡莫司汀。卡莫司汀可被用来治疗霍杰金病、淋巴瘤、骨髓瘤、恶性星形细胞瘤、脑转移瘤、黑素瘤和胃肠肿瘤。在另一实施方式中,亚硝基脲是用来治疗胰岛细胞癌的链脲霉素。
[0362]在一实施方式中,本发明提供了治疗癌症的方法,通过给予本发明氨基磷酸酯烷化剂前体药物和至少应用三氮烯烷化剂的癌症治疗方案来完成。在一实施方式中,三氮烯烷化剂是达卡巴嗪。达卡巴嗪被用来治疗恶性黑素瘤、霍杰金病和成人肉瘤。在另一实施方式中,三氮烯烷化剂是替莫唑胺。替莫唑胺可被用来治疗恶性神经胶质瘤。
[0363]在一实施方式中,本发明提供了治疗癌症的方法,通过给予本发明氨基磷酸酯烷化剂前体药物和应用至少铂配位配合物烷化剂的癌症治疗方案来完成。在一实施方式中,铂配位配合物烷化剂是顺铂。顺铂可用于治疗膀胱癌、头和颈癌、子宫内膜癌、小细胞肺癌以及一些儿童肿瘤。顺铂单独或与环磷酰胺联合用于治疗晚期卵巢癌。顺铂与博来霉素、依托泊甙和长春花碱的联合化疗用于治疗晚期睾丸癌,与紫杉醇、环磷酰胺或阿霉素之一的联合化疗用于治疗卵巢癌。
[0364]可用于实施本文所述方法的蒽环类包括但不限于阿霉素(Adriamycin,Doxil,Rubex)、米托蒽醌(Novantrone)、伊达比星(Idamycin)、戊柔比星(valrubicin)(Valstar)和表阿霉素(Ellence)。根据本文所述方法,氨基磷酸酯烷化剂前体药物与蒽环类共同给予以治疗癌症。在一个方案中,癌症是急性非淋巴细胞白血病、卡波西肉瘤、前列腺癌、膀胱癌、卵巢转移癌和乳腺癌。
[0365]作为一个实例,化合物(8S,10S)-10-[(3-氨基-2,3,6-三脱氧-α-L-来苏-己吡喃糖基)氧代]-8-乙醇酰-7,8,9,10-四氢-6,8,11-三羟基-1-甲氧基-5,12-并四苯二酮,更通常称作阿霉素(doxorubicin),是分离自波塞链霉菌表灰变种(Streptomycespeucetiusv ar.caesius)培养物的细胞毒性蒽环类抗生素。阿霉素(多柔比星)已经被成功用于在播散性瘤性疾病中产生退化,如急性淋巴细胞白血病、急性粒细胞白血病、威尔姆斯瘤、神经母细胞瘤、软组织肉瘤和骨肉瘤、乳腺癌、卵巢癌、膀胱移行细胞癌(transitional cell bladder carcinoma)、甲状腺癌、霍杰金和非霍杰金型淋巴瘤、支气管癌和胃癌。阿霉素一般以30-75mg/m2范围的剂量给药,以21天间隔静脉注射一次给予;以20mg/m2的剂量每周静脉注射;或以30mg/m2的剂量在三个连续日每日给予,每四周重复进行。根据本文所述方法,在给予这样剂量(或更低剂量)的阿霉素之前开始共同给予氨基磷酸酯烷化剂前体药物,并且在所述给予之后继续共同给予。用于实施本文所述方法的环状蒽环类细胞毒素前体药物由文献Matteuci等,PCT专利申请号US05/008161提供。
[0366]用于实施本文所述方法的抗生素包括但不限于放线菌素D、放线菌素(Cosmegen)、博来霉素(Blenoxane)、柔红霉素和道诺霉素(Cerubidine,DanuoXome)。根据本文所述方法,氨基磷酸酯烷化剂前体药物与抗生素共同给予以治疗癌症。在一个方案中,癌症是选自急性淋巴细胞白血病、其它白血病和卡波西肉瘤的癌症。
[0367]用于实施本文所述方法的芳香酶抑制剂包括但不限于阿那曲唑(Arimidex)和来曲唑(letroazole)(Femara)。根据本文所述方法,氨基磷酸酯烷化剂前体药物与芳香酶抑制剂共同给予以治疗癌症。在一个方案中,癌症是乳腺癌。
[0368]用于实施本文所述方法的二磷酸盐抑制剂包括但不限于唑来磷酸(Zoledronate)(Zometa)。根据本文所述方法,氨基磷酸酯烷化剂前体药物与二磷酸盐抑制剂共同给予以治疗癌症。在一个方案中,癌症是选自多发性骨髓瘤、实体瘤转移的骨癌或前列腺癌的癌症。
[0369]用于实施本文所述方法的环加氧酶抑制剂包括但不限于塞来考昔(西乐葆(Celebrex))。根据本文所述方法,氨基磷酸酯烷化剂前体药物与环加氧酶抑制剂共同给予以治疗癌症。在一个方案中,癌症是结肠癌或称为家族性腺瘤性息肉病的癌前状态。
[0370]用于实施本文所述方法的雌激素受体调节剂包括但不限于他莫西芬(Nolvadex)和氟维司群(fulvestrant)(Faslodex)。根据本文所述方法,氨基磷酸酯烷化剂前体药物与雌激素受体调节剂共同给予以治疗癌症。在一个方案中,癌症是乳腺癌,或治疗被给予用来防止乳腺癌的发生或复发。
[0371]用于实施本文所述方法的叶酸拮抗剂包括但不限于氨甲喋呤和三甲氧蝶呤(tremetrexate)。根据本文所述方法,氨基磷酸酯烷化剂前体药物与叶酸拮抗剂共同给予以治疗癌症。在一个方案中,癌症是骨肉瘤。
[0372]作为一个实例,化合物N-[4-[[(2,4-二氨基-6-蝶啶基)甲基甲氨基]苯甲酰基]-L-谷氨酸,通常称为氨甲喋呤,是已用于治疗妊娠性绒癌和治疗患有破坏性绒毛膜腺瘤和水泡样胎块的患者的抗叶酸药。在晚期恶性淋巴瘤的治疗中和在蕈样肉芽肿病晚期情况的治疗中,它也是有用的。氨甲喋呤被如下给予。对于绒毛膜癌,每日肌肉内注射15至30mg的剂量,持续5天疗程,按照需要重复这一疗程,疗程间插入的休息期间是1周或更多周。对于白血病,给予每周两次肌肉内注射,剂量是30mg/m2。对于蕈样肉芽肿病,肌肉内注射50mg剂量,每周一次,或者可选地,给予25mg剂量,每周两次。根据本文所述方法,氨基磷酸酯烷化剂前体药物与以此类剂量(或更低剂量)给予的氨甲喋呤共同给予。5-甲基-6-[[(3,4,5-三甲氧基苯基)-氨基]甲基]-2,4-喹唑啉二胺(通常称为三甲曲沙、三甲氧蝶呤)是能够与氨基磷酸酯烷化剂前体药物共同给予的另一抗叶酸药。
[0373]可用于实施本文所述方法的无机砷酸盐包括但不限于三氧化二砷(Trisenox)。根据本文所述方法,氨基磷酸酯烷化剂前体药物与无机砷酸盐共同给予以治疗癌症。在一个方案中,癌症是难治性急性前髓细胞性白血病(APL)。
[0374]可用于实施本文所述方法的微管抑制剂(如本文所用,“微管抑制剂”是干扰微管的装配或分解的任何药剂)包括但不限于长春新碱(Oncovin)、长春花碱(Velban)、紫杉醇(paclitaxel)(Taxol,Paxene)、长春瑞宾(vinorelbine)(Navelbine)、多烯紫杉醇(docetaxel)(Taxotere)、埃坡霉素B或D或其任一种的衍生物,以及discodermolide或其衍生物。可被用于本发明实践的微管蛋白结合抗癌药和其前体药物在如下参考文献中提供:Matteucci等,PCT专利申请第PCT/US2005/042095号;名称为"Tubulin Binding Anti Cancer Agents and Prodrugs Thereof”的美国专利申请(律师案卷号021305-008500US、021305-008400US和021305-004520US)。根据本文所述方法,氨基磷酸酯烷化剂前体药物与微管抑制剂共同给予以治疗癌症。在一个方案中,癌症是卵巢癌、乳腺癌、非小细胞肺癌、卡波西肉瘤和乳腺或卵巢器官的转移癌。作为一个实例,化合物22-氧代-长春花碱,通常也称为长春新碱,是获自普通长春花植物(夹竹桃科植物长春花(Vinca rosea,Linn.))的生物碱,在治疗急性白血病中是有用的。也已表明,其与其它溶瘤细胞药联合,在治疗霍杰金病、淋巴肉瘤、网状组织细胞肉瘤、横纹肌肉瘤、神经母细胞瘤和威尔姆斯瘤中是有用的。对于儿童,长春新碱每周静脉给予2mg/m2剂量,对于成人给予1.4mg/m2。根据本文所述方法,氨基磷酸酯烷化剂前体药物与以此类剂量给予的长春新碱共同给予。在一个方案中,在用微管抑制剂如紫杉烷治疗之前,不给予氨基磷酸酯烷化剂前体药物,而是在用微管抑制剂的治疗开始的同时或在其后数日至一周内,给予氨基磷酸酯烷化剂前体药物。
[0375]可用于实施本文所述方法的修饰剂包括但不限于亚叶酸(Wellcovorin),其与其它药物如5-氟尿嘧啶一起应用以治疗结肠直肠癌。根据本文所述方法,氨基磷酸酯烷化剂前体药物与修饰剂和另一抗癌药共同给予以治疗癌症。在一个方案中,癌症是结肠癌。在一个方案中,修饰剂是增强细胞摄取葡萄糖的能力的化合物,包括但不限于化合物N-羟基脲。已经报道,N-羟基脲增强细胞摄取2-脱氧葡萄糖的能力(参见文献Smith等,1999,Cancer Letters 141:85,其并入本文作为参考),以据报道可增强2-脱氧葡萄糖摄取或治疗白血病的水平给予N-羟基脲并连同给予2-脱氧葡萄糖和如本文描述的氨基磷酸酯烷化剂前体药物是本文提供的治疗方法的一个方案。在另一个这样的方案中,氨基磷酸酯烷化剂前体药物与一氧化一氮或一氧化一氮前体如有机亚硝酸盐或精胺二醇二氮烯鎓(spermineNONOate)共同给予,以治疗癌症,因为后者化合物刺激葡萄糖的摄取。
[0376]可用于实施本文所述方法的亚硝基脲包括但不限于丙卡巴肼(Matulane)、罗莫司丁(lomustine)、CCNU(CeeBU)、卡莫司汀(BCNU,BiCNU,Gliadel Wafer)和雌莫司汀(Emcyt)。根据本文所述方法,氨基磷酸酯烷化剂前体药物与亚硝基脲共同给予以治疗癌症。在一个方案中,癌症是前列腺癌或胶质母细胞瘤,包括复发性多形性成胶质母细胞瘤。
[0377]可用于实施本文所述方法的核苷类似物包括但不限于巯基嘌呤、6-MP(Purinethol)、氟尿嘧啶、5-FU(Adrucil)、硫鸟嘌呤、6-TG(Thioguanine)、羟基脲(Hydrea)、阿糖胞苷(Cytosar-U,DepoCyt)、氟尿苷(FUDR)、氟达拉滨(fludarabine)(Fludara)、氮杂胞苷(azacytidine)(Vidaza)、喷司他丁(pentostatin)(Nipent)、克拉屈滨(Leustatin,2-CdA)、吉西他滨(Gemzar)和卡培他滨(Xeloda)。根据本文所述方法,氨基磷酸酯烷化剂前体药物与核苷类似物共同给予以治疗癌症。在一个方案中,癌症是B细胞淋巴细胞白血病(CLL)、毛细胞白血病、胰腺腺癌、转移性乳腺癌、非小细胞肺癌或转移结肠直肠癌。作为一个实例,化合物5-氟-2,4(1H,3H)-嘧啶二酮,通常也称为5-氟尿嘧啶,是在被认为通过手术或其它方式不可治愈的患者的结肠癌、直肠癌、乳腺癌、胃癌和胰腺癌的姑息疗法(palliative management)中有效的抗代谢物核苷类似物。5-氟尿嘧啶在初始治疗中的给予剂量是12mg/m2,每日一次静脉内给予,持续4个连续日,每日剂量不超过800mg。如果在治疗期间的任何时间没有观察到毒性,则在第6天、第8天、第10天和第12天静脉给予6mg/kg。在第5天、第7天、第9天或第11天不给予治疗。对于风险大的患者或不处于足够营养状态的那些患者,给予6mg/kg的日剂量,持续3日,日剂量不超过400mg。如果在治疗期间的任何时间没有观察到毒性,可以在第5天、第7天和第9天给予3mg/kg。在第4天、第6天或第8天不给予治疗。按任一时间表的注射顺序构成了疗程。根据本文所述方法,氨基磷酸酯烷化剂前体药物与以此类剂量给予的5-FU共同给予,或与相应调整剂量的前体药物形式Xeloda共同给予。作为另一实例,化合物2-氨基-1,7-二氢-6H-嘌呤-6-硫酮,一般也称为6-硫鸟嘌呤,是在治疗急性非淋巴细胞白血病中有效的核苷类似物。6-硫鸟嘌呤以每日约2mg/kg体重的剂量经口给予。日总剂量可以一次给予。如果在此水平剂量4周后没有改善,剂量可以谨慎增加至3mg/kg/日。根据本文所述方法,氨基磷酸酯烷化剂前体药物与以此类剂量(或更低剂量)给予的6-TG共同给予。
[0378]可用于实施本文所述方法的破骨细胞抑制剂包括但不限于帕米膦酸(Aredia)。根据本文所述方法,氨基磷酸酯烷化剂前体药物与破骨细胞抑制剂共同给予,以治疗癌症。在一个方案中,癌症是乳腺癌破骨性骨转移,并且一种或多种其它抗癌药也与氨基磷酸酯烷化剂前体药物共同给予。
[0350]可用于实施本文所述方法的铂化合物包括但不限于顺铂(Platinol)和卡铂(Paraplatin)。根据本文所述方法,氨基磷酸酯烷化剂前体药物与铂化合物共同给予,以治疗癌症。在一个方案中,癌症是转移睾丸癌、转移卵巢癌、卵巢癌和膀胱移行细胞癌。作为一个实例,化合物顺-二胺二氯铂(II),通常称为顺铂,在转移睾丸肿瘤和转移卵巢肿瘤的姑息疗法中是有用的,并且可用于治疗手术或放疗无法改善的膀胱移行细胞癌。顺铂在用于晚期膀胱癌时,每3至4周一次静脉注射给药,剂量是50-70mg/m2。根据本文所述方法,氨基磷酸酯烷化剂前体药物与以这些剂量(或更低剂量)给予的顺铂共同给予。一种或多种其它抗癌药可以与顺铂化合物和氨基磷酸酯烷化剂前体药物共同给予。作为一个实例,Platinol、Blenoxane和Velbam可以与氨基磷酸酯烷化剂前体药物共同给予。作为另一个例子,顺铂和阿霉素(adriamycin)可以与氨基磷酸酯烷化剂前体药物共同给予。
[0351]可用于实施本文所述方法的视黄素包括但不限于维甲酸、ATRA(Vesanoid)、9-顺式视黄酸(alitretinoin)(Panretin)和贝沙罗汀(bexarotene)(Targretin)。根据本文所述方法,氨基磷酸酯烷化剂前体药物与视黄素共同给予,以治疗癌症。在一个方案中,癌症是选自APL、卡波西肉瘤和T细胞淋巴瘤的癌症。
[0352]可用于本文所述方法的实践中的拓扑异构酶1抑制剂包括但不限于托泊替康(topotecan)(Hycamtin)和伊立替康(irinotecan)(Camptostar)。根据本文所述的方法,氨基磷酸酯烷化剂前体药物与拓扑异构酶1抑制剂共同给予,以治疗癌症。可用于本发明所述方法的实践中的拓扑异构酶抑制剂及其前体药物提供于文献Matteucci等,美国专利申请PCT/US2005/041959中。在一个方案中,癌症是卵巢、结肠或直肠转移癌,或小细胞肺癌。然而,如上所指出,在本文所述方法的一个方案中,氨基磷酸酯烷化剂前体药物的给予是在给予拓扑异构酶1抑制剂之前或之后或两者给予,但不是与其同时给予。
[0353]可用于本文所述方法的实践中的拓扑异构酶2抑制剂包括但不限于依托泊甙(etoposide)、VP-16(Vepesid)、替尼泊苷(teniposide)、VM-26(Vumon)和依托泊甙磷酸盐(Etopophos)。根据本文所述的方法,氨基磷酸酯烷化剂前体药物与拓扑异构酶2抑制剂共同给予,以治疗癌症。在一个方案中,癌症是选自下列的癌症:难治性睾丸肿瘤、难治性急性淋巴细胞白血病(ALL)和小细胞肺癌。然而,如上所指,在本文所述方法的一个方案中,氨基磷酸酯烷化剂前体药物的给予是在给予拓扑异构酶2抑制剂之前或之后,或在之前和之后,但是不与其同时给予。
[0354]可用于本文所述方法的实践中的酪氨酸激酶抑制剂包括但不限于伊马替尼(imatinib)(Gleevec)。根据本文所述的方法,氨基磷酸酯烷化剂前体药物与酪氨酸激酶抑制剂共同给予,以治疗癌症。在一个方案中,癌症是CML或转移的或不可切除的恶性胃肠道基质肿瘤。
[0355]可用于本发明实践中的氯尼达明(lonidamine)类似物提供于文献Matteucci等美国专利申请第11/346632、60/764,427、60/764,438号和名称为"Heterocyclic Lonidamine Analogs"申请(律师案卷号021305-007220US、021305-007900US)和PCT公开第WO 2006/015191、WO 2006/015263和WO2006/01007 A2号。
[0356]因此,治疗癌症的方法在本文中被描述,其中氨基磷酸酯烷化剂前体药物或其药学上可接受的盐和一种或多种其它抗癌药被给予患者。这些其它抗癌药的具体方案包括但不限于:5-甲基-6-[[(3,4,5-三甲氧基苯基)氨基]-甲基]-2,4-喹唑啉二胺或其药学上可接受的盐;(8S,10S)-10-(3-氨基-2,3,6-三脱氧-α-L-来苏-已吡喃糖基)氧代]-8-乙醇酰基-7,8,9,10-四氢-6,8,11-三羟基-1-甲氧基-5,12-并四苯二酮或其药学上可接受的盐;5-氟-2,4(1H,3H)-嘧啶二酮或其药学上可接受的盐;2-氨基-1,7-二氢-6H-嘌呤-6-硫酮或其药学上可接受的盐;22-氧代-长春花碱或其药学上可接受的盐;2-二[(2-氯乙基)氨基]四氢-2H-1,3,2-氧氮杂膦(oxazaphosphorine),2-氧化物或其药学上可接受的盐;N-[4-[[(2,4-二氨基-6-蝶啶基)甲基]-甲氨基]苯甲酰基]-L-谷氨酸或其药学上可接受的盐;或顺二氨二氯-铂(II)。
IV.实施例
[0357]在下面的实施例中,对文字所表示的化合物的任何提及是对在相应反应方案中接着该文字显示的结构,或是该文字上方的结构的提及。
合成
[0358]合成本发明氨基磷酸酯烷化剂前体药物的方法在IIb部分提供。当可获得时,用来合成本发明氨基磷酸酯烷化剂前体药物的原材料从商业制造商购买,例如,如Sigma-Aldrich Co.1-N-甲基-2-硝基咪唑-5-甲醇从Syngene,India购买。非商业可获得的原材料经标准文献方法合成。这类方法可由文献搜索工具如American Chemical Society的SciFinder或可从MDL Software获得的Beilstein进行确定。
[0359]与湿敏化合物如,例如POCl3和PCl3以及它们的一氯或二氯衍生物的反应使用无水溶剂并在氮气或氩气中进行。从反应混合物中分离产物使用装置(work-up)进行,当需要时,接着进行真空蒸馏、结晶、柱层析或制备型厚层层析(preparative thick layer chromatography)。化合物柱层析的适当洗脱液可通过阅读本公开内容而确定,和/或通过薄层层析确定化合物的Rf以及选择使需要的化合物与不需要的化合物中分离的溶剂而确定。具体洗脱液的选择可取决于化合物的极性性质、其它精细(closely)洗脱化合物的存在、使用的固定相的类型如硅胶或矾土和用于通过固定相洗脱溶剂的压力量以及其它因素。在实践中,溶剂的不同组合可被用来分离同一化合物。
[0360]通过标准的分析技术例如TCL、NMR光谱和LC-MS分析分离的化合物的纯度,分离的化合物被贮藏于冷冻室或冰箱,以避免湿气、光或空气。氨基磷酸酯烷化剂前体药物化合物的储存溶液在DMSO中制备,并且存于冰箱中。
实施例1
合成化合物23
[0361]在-78℃下,向5-硝呋乙醇(5-nitrofurfuryl alcohol)(200mg,1.4mmol)的THF(10ml)溶液中,加入一份POCl3,然后逐滴加入三乙胺(TEA,0.22ml,1.54mmol)。在1小时内,将温度升高到-30℃,然后加入2-盐酸氯乙胺,再加入TEA(1ml、7mmol)。在将温度升高到室温后(rt)后,继续反应一个小时以上,用水淬火反应混合物,并且分离有机层。用DCM提取水层,并且干燥和浓缩混合的有机溶液。通过急骤柱层析分离化合物23,并且用LC/MS和NMR光谱分析化合物23,其是纯的。
实施例2
合成化合物5

[0362]将N-甲基-2-氯乙基氯化铵(10gm)的POCl3(40ml)悬浮液回流(135℃)过夜。在真空中除去过量POCl3后,在真空中将产物5i蒸馏出,其为浅黄色油,通过1H和31P NMR光谱分析其是纯的。
[0363]在-78℃下,向5i(1gm,4.75mmol)和N-甲基-2-氯乙基氯化铵(0.62gm,4.75mmol)的THF溶液中,缓慢加入二异丙基乙胺(DIEA,1.65ml,9.5mmol),并将反应混合物加热到室温。在室温下搅拌1小时后,用乙酸乙酯将反应混合物稀释,并且用盐水洗涤反应混合物。通过MgSO4干燥有机层,并且将其浓缩以产生剩余物,通过急骤柱层析分离该剩余物,以产生油状化合物5ii。
[0364]在-78℃下,向N-甲基-2-硝基咪唑-5-甲醇(0.5g,3.2mmol)的二甲氧基乙烷(DME)溶液中,加入二(三甲基甲硅烷基)酰胺锂(lithium bis(trimethylsilyl)amide)(1M THF溶液,3.2mmol,3.2ml)。5分钟后,加入5ii(2.9mmol,770mg),并且将反应混合物温热至-20℃,用乙酸乙酯将反应混合物稀释,并且用盐水洗涤反应混合物。通过MgSO4干燥有机层,并且将其浓缩。通过用DCM中6-12%甲醇的急骤层析纯化,产生5。
[0365]使用制备化合物5所用的过程合成化合物8和16。

实施例3
合成化合物35

[0366]向乙醇胺(6.03mL,100mmol)和K2CO3(13.8g,100mmol)的DMF(38mL)溶液中,在室温下,逐滴加入对甲苯磺酰氯溶液(19g,100mmol),并且将反应混合物加热到120℃(浴温)。将K2CO3(27.6g,200mmol)加入到反应混合物,然后逐滴加入1,3-二溴丙烷(10g,50mmol)。再加热2小时后,将反应冷却至室温,倒入水(250mL)中,并且用乙酸乙酯提取。用Na2SO4干燥有机层,并且将其浓缩产生化合物35a,其为黄色油状物,在下个反应中使用。
[0367]蒸馏化合物35a的含水HBr(48%,50ml)溶液,以除去水部分(大约20ml),并且回流反应混合物40小时。通过蒸馏除去另外的水部分(5ml),并且回流反应混合物(4小时)。将反应混合物冷却至室温,用水(20mL)稀释,并且通过才里特垫(celite pad)过滤。将滤液浓缩至干燥,以产生剩余物,将该剩余物与乙醇共蒸发3次,然后加入大量丙酮,过滤白色固体产物35b,用丙酮洗涤2遍,并且将其用于下面提供的磷酸化中。
[0368]将化合物35b(1g)的POCl3(14mL)悬浮液在130℃加热大约14小时,在130℃(浴温)下,真空中除去过量POCl3。通过在硅胶上使用10-80%ETOAc/己烷进行柱层析纯化剩余物,产生产物35c,使用柱层析分离硅胶和10-80%丙酮/甲苯作为洗脱液,使用与实施例2提供的相同的过程,采用柱层析分离硅胶并以10-80%丙酮/甲苯为洗脱液,将其转化为本发明的化合物35。
实施例4
合成化合物7
[0369]使用N-环丙基-2-氯乙基氯化铵,如下面提供的方法制备化合物7

[0370]向环丙胺(25g)的干燥THF(30ml)溶液中,在超过35分钟的时间逐滴加入2-溴乙醇(17.6g,0.141mol)在30ml THF中的溶液。在室温下搅拌反应混合物1小时,并且在50℃下,加热75分钟。冷却后,浓缩反应混合物产生橙色油状物,将其中加入氢氧化钠(7g)的水(50ml)溶液。搅拌反应混合物10分钟,并且用乙酸乙酯(75ml)提取4次。将混合的有机层干燥(MgSO4)并且蒸发,以得到橙色油状剩余物。在53-56℃真空(1mm Hg)下,蒸馏该剩余物,产生透明、无色液体的中间体醇(5.94g,42%产率),使用LC/MS和1H NMR分析其是纯的。
[0371]向中间体醇(3.7g,36.6mmol)的干燥THF(30ml)溶液中,加入HCl的二噁烷(4.0M,18.3ml,73.2mmol)溶液。将反应混合物冷却到0℃,通过注射器加入SOCl2(6.50g,54.9mmol)。将反应混合物回流(6h)、冷却并且浓缩,产生剩余物。将剩余物与干燥醚(100ml)研磨,过滤并且在真空中除去可挥发的剩余物,产生7i(5.42g,95%产率),通过1H NMR分析其是纯的。
[0372]将7i(3.00g,19.2mmol)加入到POCl3(15ml)中,在氮气下回流7.5小时。浓缩反应混合物,并且在真空中通过短程蒸馏设备(short path distillation apparatus)蒸馏所形成的油状物,产生透明、浅黄色油状物7ii(3.6g,79%产率),使用1H NMR分析其是纯的。
[0373]将7ii(0.50g,2.11mmol)和N-环丙基-2-氯乙胺盐酸(0.33g,2.11mmol)在氩气下,在干燥THF中组合。将反应混合物冷却至-78℃,并且通过注射器缓慢加入DIEA(0.545g,4.22mmol),缓慢暖至室温,搅拌1.5小时,并且浓缩,得到橙色油状剩余物。通过二氧化硅上使用乙酸乙酯中0-50%己烷进行急骤层析,分离剩余物,以给出315mg(理论上为47%)浅黄色油状物,通过MS分析其为7iii。
[0374]将N-甲基-2-硝基咪唑-5-甲醇(76.8mg,0.489mmol),在氩气下部分溶于干燥THF(2ml)中。将反应混合物冷却至-78℃,加入二(三甲基甲硅烷基)酰胺锂的THF溶液中(1.6M,0.306ml,0.489mmol)。15分钟后,加入7iii(172mg,0.538mmol)在2ml THF中的溶液。15分钟后将反应混合物缓慢温至室温,搅拌2小时,倒入25ml水中并且用乙酸乙酯(30ml)萃取3次。通过MgSO4干燥合并的有机层,并且将其浓缩,以得到黄色油状剩余物。通过用DCM中0-10%甲醇急骤层析分离该剩余物,产生为黄色油状物的化合物7(110mg,51%产率),通过LC-MS和1H NMR分析其是纯的。
实施例5
合成化合物6和15

[0375]向二(2-氯乙基)氯化铵(1.43g,8.01mmol)的二氯甲烷(DCM)悬浮液中,在室温下加入三氯化磷(0.32ml,3.64mmol),然后,加入TEA(3.05ml,21.84mmol)。在室温下搅拌反应混合物30分钟,然后加入DME中的N-甲基-2-硝基咪唑基甲醇(0.474g,3.31mmol)。搅拌0.5小时后,将反应混合物冷却至-20℃,加入叔丁基氢过氧化物(癸烷中0.7ml,3.82mmol,5.5M)。在超过一小时的时间内将反应混合物加热至室温,并且倒入10%含水HCl中。分离有机层,用DCM提取水层。通过MgSO4干燥混合的有机溶液,并且将其浓缩,得到剩余物,用DCM中6-12%甲醇急骤层析纯化该剩余物,产生6。
[0376]使用上面描述的合成化合物6的方法合成化合物15。

实施例6
合成化合物23、26和36

[0377]向N-甲基-2-硝基咪唑-5-甲醇(180mg,1.14mmol)、三苯膦(300mg,1.14mmol)和异磷酰胺芥子(1c,127mg,0.57mmol)的THF(10ml)溶液中,在室温下逐滴加入二异丙基偶氮二羧酸酯(diisopropyl azodicarboxylate,DIAD,0.22ml,1.14mmol)。两小时后,浓缩反应混合物,通过使用甲苯中30-100%丙酮,急骤层析分离剩余物,产生化合物36。
[0378]使用实施例6的方法,合成化合物23和26。
实施例7
合成化合物1

[0379]将N-甲基-2-硝基咪唑-5-甲醇(50mg,0.318mmol),在氩气下溶于干燥THF(2ml)。将溶液冷却至-78℃,通过注射器加入二(三甲基甲硅烷基)酰胺锂溶液(在甲苯中1M,0.35ml,0.35mmol)。5分钟后,加入二(氯乙基)氨基磷酸二氯化物(91mg,0.35mmol)的THF(2ml)溶液。在78℃下搅拌30分钟后,使用NaCl/冰浴将温度降至-20℃,用无水氨鼓泡反应混合物5分钟。用氮气清洗反应混合物,温至室温,倒入25ml水中并且用乙酸乙酯(4×25ml)萃取。(MgSO4)干燥混合的有机层,并且将其浓缩,得到浅黄色油状物,通过在硅胶上用二氯甲烷中的0-10%甲醇急骤层析分离该浅黄色油状物,产生油状的化合物1(32mg,28%产率),其立刻(onstanding)固化,并且通过LC/MS和1H NMR分析其是纯的。
实施例8
合成化合物25、26
[0380]在10℃,向2-溴乙基溴化铵(19.4g)的DCM(90mL)溶液中,加入POCl3(2.3mL)的DCM(4mL)溶液,然后,加入TEA(14.1mL)的DCM(25mL)溶液。将反应混合物过滤,将滤液浓缩至原来体积的约30%,并过滤。用DCM(3×25mL)洗涤剩余物,并且将混合的DCM部分浓缩,产生固体,将THF(6mL)和水(8mL)的混合物加入到该固体中。在旋转蒸发器(rotary evaporator)中除去THF,将所形成的溶液在冰箱中冷却过夜。过滤得到的沉淀物,用水(10mL)和醚(30mL)洗涤,然后在真空中干燥以产生2.1g

[0381]异磷酰胺芥子
可通过使用实施例8提供的方法合成,其中用2-氯乙基氯化铵取代2-溴乙基溴化铵。合成异磷酰胺芥子的方法已有描述(参见,如Wiessler等,如前所述)。
[0382]使用实施例6提供的方法和适当的触发物-OH,将氨基磷酸酯烷化剂毒素:

转化为化合物24和25。
实施例9
合成化合物37-105
[0383]使用描述用于上面的25或36合成的Mitsunobu型偶联,使用适当取代的触发物-OH和异环磷酰胺芥子类似物,合成下面的化合物37-105。例如,对于合成化合物40、81、83、87、89、95、96、100和104,使用的异环磷酰胺芥子类似物是HOP(=O)(NHCH2CH2Cl)2;在化合物50、53、55、56、58-65、68-71、73-75、77-80、82、84-86、88、90-92、94、97-99、101-103和105中,使用的异环磷酰胺芥子类似物是HOP(=O)(NHCH2CH2Br)2;在化合物37、39、52、54和93中,使用的异环磷酰胺芥子类似物是HOP(=O)(NHCHMeCH2Cl)2的R对映体;在化合物38、41、51和57中,使用的异环磷酰胺芥子类似物是HOP(=O)(NHCHMeCH2Cl)2的S对映体;在化合物43-45和49中,使用的异环磷酰胺芥子类似物是HOP(=O)(NHCH(CHMe2)CH2Cl)2的R对映体;以及在化合物46-48中,使用的异环磷酰胺芥子类似物是HOP(=O)(NHCH(CHMe2)CH2Cl)2的S对映体。
[0384]在合成化合物37-105中使用多种触发物-OH化合物,包括下列触发物-OH化合物:1-N-甲基-2-硝基咪唑-5-甲醇、1-N-甲基-5-硝基咪唑-2-甲醇、5-硝基呋喃-2-甲醇、5-硝基噻吩-2-甲醇;



[0385]下列化合物根据实施例6中描述的方法。


实施例10-26描述了在合成本发明的氨基磷酸酯烷化剂前体药物中使用的多种触发物-OH化合物的合成。
实施例10
合成化合物52i

[0386]化合物52ii(100mg,0.48mmol)、52iii(73mg,0.48mmol)和KOAc(190mg,1.92mmol)的DMF(5ml)溶液被脱气3次,并且在室温下,氩气气氛中加入PdCl2(dppf)(36mg,0.048mmol)。将反应混合物在60℃加热两小时,用乙酸乙酯(EA)将反应混合物稀释,并且用盐水洗涤反应混合物。干燥有机层,并进行浓缩,在硅胶上使用EA/Hex(0-80%)作为洗脱液通过柱层析分离剩余物,以产生52i。
[0387]以相似的方式,如下面示意描述的,制备化合物55i、63i、59i、65i和68i:

实施例11

[0388]在室温下,向化合物68ii(100mg,0.31mmol)和3-氨基-1-丙醇(0.047g,0.62mmol)的THF(2.5ml)溶液中,加入DIEA(0.162ml,0.93mmol)。将反应混合物搅拌过夜,并且浓缩产生剩余物,在硅胶上使用EA/Hex(0-80%)作为洗脱液通过柱层析分离该剩余物,产生化合物68i。
[0389]相似地制造化合物69i,如下面方案描述。

实施例12
[0390]在室温下,向化合物70ii(100mg,0.87mmol)和化合物70iii(112mg,0.87mmol)的丙酮(8ml)溶液中,加入K2CO3(78.6mg,0.87mmol)。将反应混合物在60℃下加热,并搅拌1小时,过滤并且浓缩,产生剩余物,在硅胶上使用(EA/Hex)0-60%通过柱层析分离该剩余物,产生化合物70i。
[0391]类似地制造化合物51i,如下面方案描述。

实施例13
(HAP触发物-检查编号(check #s))

[0392]化合物59ii(200mg,0.96mmol)和59iii(127mg,0.96mmol)的DMF(3ml)溶液被脱气3次,并且在室温下,氩气气氛中向其加入PdCl2(dppf)(50mg,0.07mmol),然后加入CuI(8.5mg,0.043mmol)和TEA(0.27ml,1.92mmol),并且将反应混合物在60℃加热两小时。用乙酸乙酯(EA)将反应混合物稀释,并且用盐水洗涤反应混合物,分离、干燥有机层,并进行浓缩以产生剩余物,在硅胶上使用EA\Hex(0-70%)作为洗脱液,通过柱层析分离该剩余物,产生化合物58i。
实施例14

[0393]在-20℃,向67i(472mg,2.69mmol)的DCM(20ml)悬浮液中,加入苯基二氯磷酸酯(0.2ml,1.34mmol),然后逐滴加入TEA(0.75ml,5.38mmol)并进行搅拌。将反应混合物温至室温,在室温下搅拌1小时,倒入盐水中,分离有机层,并用DCM萃提水层。用MgSO4干燥混合的有机层,并进行浓缩。在硅胶上使用EA/Hex(10-100%)作为洗脱液,通过柱层析分离剩余物,以产生化合物67ii。向化合物67ii(42mg)的EtOH(5ml)的溶液中加入氧化铂(IV)(20mg),将反应混合物脱气,并在氢气中强力搅拌0.5小时。用MeOH稀释反应混合物,通过针筒过滤器过滤,滤液在真空中浓缩,并与甲苯一起蒸发以产生化合物67iii。应用如合成化合物36描述的Mitsunobu型反应,将化合物67iii与1-N-甲基-2-硝基咪唑-5-甲醇反应。
实施例15
合成化合物106和107

[0394]在-78℃下,向5-硝呋乙醇(200mg,1.4mmol)的THF(10ml)溶液中,加入POCl3(0.13ml,1.4mmol),然后逐滴加入三乙胺(TEA,0.216ml,1.54mmol)。在1小时内,将反应温度升高到-10℃,然后加入2-(苯基磺酰基)乙胺盐酸(832mg,3.5mmol),然后加入TEA(1ml、7mmol)。将反应升高到室温,搅拌一小时,用水淬火,并且分离有机层。用DCM萃提水层两遍,并且干燥和浓缩混合的有机层,产生剩余物,在硅胶上使用丙酮\甲苯(30%到100%)作为洗脱液,通过柱层析分离该剩余物,产生产物106。使用相似的方法合成化合物107:

[0395]下面示出的化合物108-112:

使用在实施例3中合成化合物35所描述的方法,并用 取代
取代 而进行合成。
而进行合成。
实施例16
合成化合物113-117

[0396]根据在实施例7中描述的方法合成化合物113,如这里所述。在-78℃下,向113ii(181mg,1.16mmol)的THF(8mL)溶液中,逐滴加入LiN(TMS)2(1.2mL,1MTHF溶液,1.2mmol),然后加入1i。将反应混合物暖至-20℃,用NH3鼓泡反应混合物5分钟。将水(20mL)加入到反应混合物中,并用EA(30mL)萃取反应混合物3次。干燥混合的有机层,并且将其浓缩,以产生剩余物,在硅胶上用丙酮\甲苯(30-100%)柱层析,分离该剩余物,产生化合物113。
[0397]根据对于化合物13描述的方法,并用合适的触发物-OH作为原料代替 合成化合物114-117。
合成化合物114-117。

实施例17
合成八氘化异环磷酰胺和化合物64(八氘化的化合物25)

[0398]在0℃下,将48%HBr(60mL)逐滴加入到d4-乙醇胺。在室温下,搅拌反应混合物1小时,然后进行温和回流和缓慢蒸馏,在2小时内收集16mL液体,直到155℃(油浴)。用60mL、48%的HBr置换两次,然后继续蒸馏另外5个小时。收集90mL液体。所形成的溶液在165℃下加热2小时,并在真空中蒸发。残留物从无水乙醇(10mL)-乙酸乙酯(30mL)重结晶为11.3g d4-2-溴乙胺氢溴化物(化合物64i)。在-20℃下,氩气中将化合物64i(19.5mmol,1.0当量)逐滴加入d4-2-溴乙胺氢溴化物(40.0mmol,2.05当量)在干燥DCM(100mL)中的悬浮液,然后在-20℃下逐滴加入TEA(81.9mmol,4.2当量)。在-20℃下搅拌反应混合物0.5小时,并在室温下搅拌2小时,倒入水中,并用DCM(30mL)萃取2遍。用盐水洗涤混合的有机层,通过Na2SO4干燥该有机层,并且在减压下将其浓缩以产生剩余物,在硅胶上使用己烷/EA(100:70(v/v))作为洗脱液,通过柱层析,分离该剩余物,产生7.0g化合物64ii。将PtO2(0.7g)加入到化合物64ii(7.0g)的MeOH(160mL)溶液中,将反应混合物脱气,并且与H2交换三次,在室温下,H2中搅拌3小时,并且用MeOH稀释直到反应混合物中的白色固体溶解。过滤稀释的反应混合物,在减压下将滤液浓缩,产生剩余物,用无水醚洗涤该剩余物2次,产生2.9g化合物64iii。在0℃下,氩气中向化合物64iii(1.92g,1.0当量)、1-N-甲基-2-硝基咪唑甲醇(1.01g,1.1当量)和PPh3(2.39g,1.5当量)的THF(20mL)悬浮液中,加入DIAD(1.76ml,1.5当量)。搅拌反应混合物2小时,同时将反应混合物从0℃加热到室温,然后在真空中除去挥发性物质,产生剩余物。在硅胶上使用丙酮/甲苯(100:70(v/v))作为洗脱液,通过急骤层析,分离该剩余物,产生1.35g化合物64。
实施例18
合成化合物2i
错误!不能编辑域代码产生目标。
2ii 2iii 2i
[0399]根据参考文献Cavalleri等,J.Het.Chem.,1972,9:979合成乙烯基衍生物2iii,并且如下进行羟汞化。将Hg(OAc)2(208mg,0.653mmol)溶于水(0.7mL)和THF(0.7mL),然后加入化合物2iii(100mg,0.653mmol)。在室温下,搅拌反应混合物1.5小时,将NaBH4(25mg)分次(in portions)加入,在搅拌15分钟后,将反应混合物倒入水中,用EA萃取,干燥EA层,并且进行浓缩以产生剩余物,使用EA/己烷(0-100%)作为洗脱液,通过硅胶柱层析,分离该剩余物,产生化合物2i(16mg)。
实施例19
合成化合物94i
错误!不能编辑域代码产生目标。
94iii 94ii 94i
[0400]在0℃下,将94iii(7.1g)的Ac2O(9.7mL)溶液逐滴加入发烟硝酸(1.5mL)的AcOH(12ml)溶液中。将反应混合物加热至室温,搅拌1小时,将发烟硝酸(1mL)逐滴加入,并且搅拌1.5小时。将反应混合物倒入水中,用EA萃取,干燥EA层,并且进行浓缩以产生剩余物,使用EA/己烷(0-100%)作为洗脱液,通过硅胶柱层析,分离该剩余物,产生化合物94ii。在0℃下,将化合物94ii(600mg,1.77mmol)悬浮于甲醇(10ml)中,然后在超过5分钟时间将NaBH4(141mg)分次加入反应混合物。每小时3次一次性加入NaBH4(100mg),将反应混合物搅拌3.5小时,将反应混合物倒入水中,用EA萃取,干燥EA层,并且进行浓缩以产生剩余物,使用EA/己烷(0-100%)作为洗脱液,通过硅胶柱层析,分离该剩余物,产生化合物94i(289mg),其为黄色固体。
实施例20
合成化合物96i
错误!不能编辑域代码产生目标。
96ii 96iii 96i
[0401]在140℃下,搅拌A(1.4g)、CuCN(0.56g)和DMF(25mL)的混合物35分钟,并且在300mL碎冰上搅拌10分钟。然后过滤反应混合物,并且使用己烷:EA(1:0到2:3)作为洗脱液,通过柱层析分离剩余物,产生黄色油状化合物96iii(617mg)。将化合物96iii转化为醇96i,并且通过柱层析分离,然后使用与化合物94iii所用的相似方法,使用THF代替MeOH作为反应中溶剂。
实施例21
合成化合物99i
错误!不能编辑域代码产生目标。
99ii 99iii 99i
[0402]将99ii(500mg)、PdCl2(PPh3)2(208mg)和CuI(56.4mg)的混合物悬浮于TEA(15mL)中,将反应混合物脱气,并且每一次用Ar吹洗6次。将丙炔鼓泡通过反应混合物15分钟,然后在丙炔气氛下50℃浴中继续反应2小时。将反应混合物倒入EA中,将其过滤,浓缩滤液以产生残留物,使用EA/己烷(0-100%)为洗脱液,通过硅胶柱层析分离该残留物,产生化合物99i(286mg)。
实施例22
合成1-N-甲基-2-氨基咪唑-5-羧酸乙酯

[0403]将甲酸乙酯(500mL)加入到1L圆底烧瓶中含有的肌氨酸甲酯盐酸(82g,585.7mmol,在反应前磨成粉末)中。在冰水浴中,将反应混合物冷却,搅拌,将气体出口连接到烧瓶,在2小时期间将NaH(60%油状悬浮液,54g,1.35mol)缓慢加入,并且在室温下搅拌大约14小时。使用旋转蒸发器除去挥发物,产生剩余物,将其与己烷(500mL)一起研磨两次,产生粘性浅棕色糊状物,将该糊状物溶解在乙醇(400mL)和浓HCl(50mL)中,并在110℃下搅拌1.5小时。在反应混合物冷却后,过滤出白色沉淀物,并用2×25mL乙醇洗涤残留物。蒸发滤液,产生深棕色油状物,向其中加入10%含水HOAc、H2NCN(45g,1.07mol)和乙酸钠(88g,1.07mol)。在90-100℃下,将反应混合物搅拌1.5小时,产生透明溶液,将溶液冷却,使用浓HCl将其pH调节到1,使用旋转蒸发器在45℃以下的温度,将所形成的溶液浓缩至原来体积的1/5。通过加入K2CO3,将浓缩的反应混合物仔细中和到8-9的pH,并且用EA(5 x 200mL,然后3 x 50mL)萃取。通过MgSO4干燥混合的乙酸乙酯层,过滤,并且除去挥发物以产生48g 1-N-甲基-2-氨基咪唑-5-羧酸乙酯。
实施例23
合成1-N-甲基-2-氨基咪唑-5-羧酸乙酯

[0404]将甲酸乙酯(850mL)加入到肌氨酸甲酯盐酸盐(205g,1.46mol,在反应前磨成粉末)、碳酸钾(205g,1.48mo1)和EtOH(800mL)中,在室温下搅拌整夜,并且过滤。在旋转蒸发器中,浓缩滤液,在此期间,剩余物分成两层。分离上层,并用EA萃取下层。通过MgSO4干燥混合的EA层和上层,过滤,并浓缩,产生185g(81%)N-甲酰肌氨酸甲酯,其被用于下面的反应。在1小时内,分几部分小心将NaH(60%油状悬浮液,16.0g,0.4mol)加入到在冰水浴中冷却的N-甲酰肌氨酸甲酯(50g,0.34mol)和甲酸乙酯(160mL)的混合物中。搅拌反应混合物,并将温度升至室温,继续搅拌过夜。将反应混合物与己烷(每次100mL)一起研磨两次,将剩余物溶解在EtOH(100mL)和浓HCl(60mL)中,在110℃下搅拌反应混合物。1小时后,将反应混合物冷却,过滤,用EtOH洗涤剩余物,将滤液浓缩,产生深棕色油状物。将该油状物加入到10%HOAc的水(200mL)溶液、NH2CN(35g)和乙酸钠(90g)中,在95℃下搅拌。1小时后,在旋转蒸发器中将反应混合物浓缩至原来体积的1/3,并且通过加入碳酸钠将其pH调节到大约9。然后用EA(8x100mL)萃取反应混合物,干燥组合的EA层,过滤,并且浓缩以产生剩余物,通过重结晶将该剩余物纯化,产生1-N-甲基-2-氨基咪唑-5-羧酸乙酯(“氨基酯”)。
实施例24
合成1-N-甲基-2-硝基咪唑-5-羧酸乙酯
[0405]将氨基酯(36.94g,0.218mol)在200ml乙酸中的溶液逐滴加入到在冰水浴中冷却的亚硝酸钠(100g,1.449mol)和水(300ml)的溶液中,并且搅拌。测量为大约-5-10℃的反应混合物的温度,被升高到室温,并且搅拌反应混合物过夜。用DCM(3×150mL)萃取反应混合物。干燥并蒸发混合的DCM层,以产生微红的剩余物,在硅胶上使用EA/己烷(30%)作为洗脱液,通过柱层析分离该剩余物,产生为浅棕色固体(27g,产率62%)的1-N-甲基-2-硝基咪唑-5-羧酸乙酯(“硝基酯”)。
[0406]在实施例24中描述的并使用含水乙酸的方法是对使用大约7%硫酸(v/v)的方法的改进,用于从氨基酯形成重氮鎓离子。使用含水硫酸,反应体积变大,这引起难以有效搅拌反应混合物。例如,包含150g氨基酯的反应要求大约12L的反应混合体积。粘性硝基酯作为产物在含水硫酸中形成,并且中断了对反应混合物的搅拌。
实施例25
合成1-N-甲基-2-硝基咪唑-5-羧酸

[0407]在室温下,搅拌硝基酯(39.2g,196.9rnmol)在1N NaOH(600mL)和水(200mL)中的悬浮液大约20小时,得到透明浅棕色溶液。通过加入浓HCl将反应混合物的pH调节到大约1,并且用EA(5x150mL)萃取反应混合物。通过MgSO4干燥并浓缩混合的乙酸乙酯层,产生为浅棕色固体(32.2g,产率95%)的1-N-甲基-2-硝基咪唑-5-羧酸(“硝基酸”)。
实施例26
合成1-N-甲基-2-硝基咪唑-5-羧酸

[0408]搅拌硝基酸(30.82g,180.23mmol)和三乙胺(140mL,285mmol)在无水THF(360mL)中的混合物,同时在干冰-乙腈浴(温度<-20℃)中冷却该反应混合物。在10分钟的期间,将氯甲酸异丁酯(37.8mL,288mmol)逐滴加入该冷却的反应混合物中,并且搅拌1小时,然后加入氢硼化钠(36g,947mmol)和在1小时期间逐滴加入水,同时保持温度在大约0℃或以下。将反应混合物温至0℃。过滤掉固体并且用THF洗涤。蒸发组合的THF部分,产生为橙色固体(25g)的1-N-甲基-2-硝基咪唑-5-甲醇,将它们从乙酸乙酯重结晶。
实施例27
合成化合物119
[0409]在-78℃下,向1-N-甲基-2-硝基咪唑-5-甲醇(50mg,0.32mmol)在DME中的悬浮液,加入LiN(TMS)2,伴随强力搅拌。10分钟后,将化合物119i(67mg,0.32mmol)加入,并将反应混合物温至室温。1小时后,浓缩反应混合物,并且通过硅胶上层析(0-100%丙酮\甲苯)分离剩余物,产生化合物119。

实施例28A-28V
[0410]通过使用相应取代的氨基磷酸酯和羟基取代的触发物(触发物-OH),根据上面的实施例1-27中描述的方法,合成化合物134到155。
实施例29A
[0411]下列化合物的溶解度在下面列出:
| 化合物 | 溶解度(室温下盐水中) |
| 10 | 10mg/mL |
| 25 | 15mg/mL |
| 73 | 10mg/mL |
| 155 | <1mg/mL |
实施例29B
抗增殖分析
[0412]为了确定氨基磷酸酯烷化剂前体药物对细胞增殖的效应,在基于多孔Alamar Blue的分析中检测这些化合物的抗增殖活性。比较测试化合物存在和不存在时的细胞生长,这通过荧光平板读数器在激发波长550nm和发射波长590nm处测定(参见Biosource International Inc.,Tech Application Notes,Use of Alamar Bluein the measurement of Cell Viability and Toxicity,Determining IC50)。用20,000细胞/孔/500μl培养基,检测下列细胞系:NCI-H460细胞(ATCC HTB-177,RPMI培养基(Gibco Products,Invitrogen Corporation,Carlsbad,CA))、HT29细胞(ATCC HTB-38,RPMI培养基(Gibco))、MES-SA细胞(ATCC CRL-1976,McCoy′s 5a培养基(ATCC))、MES-SA/Dx5细胞((ATCC CRL-1977),McCoy′s5a培养基(ATCC))、ACHN细胞(ATCC CRL-1611,极限必需培养基,Eagle(ATCC))、PC3细胞(ATCCCRL-1435,Ham′s F12K培养基(ATCC))。将这些细胞在位于24孔平板中的每一孔中的玻璃插入物上接种,其密度和培养基如上规定,接种一天后进行化合物测试。24小时后,将这些平板分成2组——缺氧组和空气组。将测试化合物以从100、30、10、3、1、0.3、0.1、0.03到0.01μM的浓度加入到治疗组的每一个孔(200μl体积)中。所有测试化合物在完全培养基中连续稀释,在每个孔中最终的DMSO浓度小于或等于1%。缺氧治疗组中的细胞在Bactron II厌氧培养室中培养2小时。在空气治疗组中的细胞在标准组织培养培养箱中培养2小时。两小时后,用测试化合物治疗,将测试化合物从每一孔中除去,用500μl培养基洗涤细胞,并且在500μl新鲜培养基中培养三天。3天后,用10%Abamar Blue染色细胞2小时,然后测量细胞增殖能力(如前面提到的),计算测试化合物的50%生长抑制浓度(GI50(本文也称为IC50)),并将它们列在下面的表X中。
表X:IC50值(μM)
| 化合物 | H460缺氧/空气 | HT29缺氧/空气 | MES-SA缺氧/空气 | MES-SA/Dx5缺氧/空气 | ACHN缺氧/空气 | PC3缺氧/空气 |
| P2 | 44/>100 | |||||
| 0.4/72 | 50/>100 | 1/>100 | ||||
| 23 | 0.04/5 | 7.5/- | ||||
| 23 | 0.1/14 | |||||
| 154 | 0.9/2 | |||||
| 139 | 16/100 | |||||
| 140 | 5/65 | |||||
| 2 | 8/>100 | |||||
| 5 | 0.05/6 | 10/>100 | ||||
| 22 | 0.7/16 | |||||
| 3 | >20/>100 | |||||
| 142 | >40/>100 | |||||
| 4 | 40/>100 | |||||
| 143 | 4.5/3.5 | |||||
| 6 | 0.7/>100 | 22/>100 | 5/>100 | |||
| 144 | 7/>100 | |||||
| 145 | >100/>100 | |||||
| 147 | >100/>100 | |||||
| 7 | 0.14/25 | 7/>100 | 0.59/83 | |||
| 11 | 5.2/>100 | |||||
| 12 | 1.7/>100 | |||||
| 9 | >10/>100 | |||||
| 8 | 0.013/0.6 | |||||
| 36 | 0.88/>100 | 55/>100 | 5/>100 | 7.5/>100 | ||
| 149 | 50/>100 | |||||
| 15 | 0.08/1 | |||||
| 16 | 1.6/>100 |
| 17 | 1/9 | |||||
| 18 | 3.4/9 | |||||
| 14 | 8.5/>100 | |||||
| 150 | -/21 | |||||
| 25 | 0.15/86 | 16/>100 | 0.9/>100 | 0.3/>100 | 0.2/62 | 0.6/>100 |
| 26 | 0.1/35 | |||||
| 10 | 100/>100 | |||||
| 31 | 83/>100 | |||||
| 24 | 0.01/4 | 0.1/2 | 0.1/0.8 | |||
| 27 | 25/100 | |||||
| 28 | 0.15/20 | |||||
| 32 | 50/100 | |||||
| 33 | 8/46 | |||||
| 34 | 0.01/1.8 | 0.8/57 | 0.13/10 | |||
| 35 | 0.075/50 | |||||
| 35 | 0.05/55 | 1.8/>100 | 1/100 | |||
| 74 | >100/>100 | |||||
| 75 | 9/26 | |||||
| 76 | 9/>100 | |||||
| 77 | 1.6/5.5 | |||||
| 78 | >100/>100 | |||||
| 79 | 3.5/3.5 | |||||
| 118 | >100/>100 | |||||
| 80 | >100/>100 | |||||
| 81 | 0.05/0.3 | |||||
| 82 | 0.03/0.02 | |||||
| 83 | 0.3/>100 | |||||
| 84 | 0.003/40 | |||||
| 85 | 0.7/100 | |||||
| 86 | 1.4/3 | |||||
| 119 | 0.3/>100 | |||||
| 37 | 0.36/>100 | |||||
| 87 | 22/>100 | |||||
| 88 | 0.03/0.53 | |||||
| 89 | 0.33/3.7 | |||||
| 90 | 0.01/3.4 | |||||
| 38 | 0.33/>100 | |||||
| 106 | 0.09/3.5 | |||||
| 107 | 0.06/2.8 | |||||
| 108 | 1.1/>100 | |||||
| 109 | 0.3/13 | |||||
| 110 | 0.3/21 |
| 39 | 0.2/5 | |||||
| 91 | -/>100 | |||||
| 92 | >100/>100 | |||||
| 41 | -/7 | |||||
| 42 | 0.5/9 | |||||
| 93 | 0.1/3.8 | |||||
| 94 | 0.3/2 | |||||
| 95 | -/2.7 | |||||
| 96 | 0.1/0.1 | |||||
| 120 | 0.3/50 | |||||
| 121 | 0.04/1 | |||||
| 122 | 0.04/1.3 | |||||
| 43 | 2/60 | |||||
| 44 | 3/100 | |||||
| 45 | 6/>100 | |||||
| 46 | 5/>100 | |||||
| 47 | 4/>100 | |||||
| 48 | -/>100 | |||||
| 97 | 0.01/0.1 | |||||
| 49 | -/>100 | |||||
| 50 | 0.1/3 | |||||
| 98 | 0.1/2 | |||||
| 51 | 3/7 | |||||
| 52 | 15/20 | |||||
| 53 | 3/10 | |||||
| 99 | 0.1/1 | |||||
| 100 | 0.5/35 | |||||
| 54 | 1/60 | |||||
| 55 | 5/12 | |||||
| 56 | 0.5/10 | |||||
| 123 | 100/>100 | |||||
| 57 | 14/100 | |||||
| 124 | -/0 | |||||
| 125 | -/100 | |||||
| 126 | -/0 | |||||
| 111 | 50/100 | |||||
| 58 | 5/10 | |||||
| 59 | 2/6 | |||||
| 60 | 15/15 | |||||
| 61 | 0.3/4 | |||||
| 62 | 2/45 | |||||
| 63 | 1/8 | |||||
| 127 | 0.02/5 | |||||
| 128 | 0.02/10 | |||||
| 112 | 70/>100 | |||||
| 103 | 0.02/2 | |||||
| 113 | 1/100 |
| 65 | 25/75 | |||||
| 114 | 1/80 | |||||
| 129 | 1/100 | |||||
| 115 | 0.5/5 | |||||
| 116 | 0.5/15 | |||||
| 130 | 0.7/20 | |||||
| 66 | 0.3/100 | |||||
| 67 | 48/>100 | |||||
| 68 | 100/>100 | |||||
| 69 | 71/>100 | |||||
| 70 | 2/65 | |||||
| 117 | 8/70 | |||||
| 71 | 0.1/0.1 | |||||
| 72 | 0.5/12 | |||||
| 131 | >100/>100 | |||||
| 132 | 3/3 | |||||
| 133 | 22/>100 | |||||
| 104 | 0.4/12 | |||||
| 105 | <0.1/1 |
实施例30
抗增殖分析-氧依赖性
[0413]为了确定氨基磷酸酯烷化剂前体药物的氧依赖性,如前所述(参见实施例29),在基于多孔Alamar Blue的分析中检测这些化合物的抗增殖活性。在测试前一天,将NCI-H460细胞(ATCC HTB-177,RPMI培养基(Gibco))或HT29细胞(ATCC HTB-38,RPMI培养基(Gibco))以20,000细胞/孔/500μl培养基,接种于位于24孔平板中的玻璃插入物中。将细胞在Bactron II厌氧培养室中培养2小时,该厌氧培养室用期望氧浓度的气体冲洗,期望氧浓度从缺氧、0.1%、0.3%、0.6%、1%、10%氧气到空气。计算的IC50值(μM)列在下面的表Y1(H460细胞)或表Y2(HT29细胞)中。
表Y1:H460细胞中的IC50值(μM)
| 化合物 | N2 | 0.1%O2 | 0.3%O2 | 0.6%O2 | 1%O2 | 10%O2 | 空气 |
| 1 | 0.3 | 10 | 7 | 50 | 100 | ||
| 23 | 0.05 | 5 | 6 | 5 | 5 |
| 5 | 0.03 | 1 | 1 | 10 | 5 | 40 | |
| 36 | 1 | 30 | 60 | 60 | >100 | >100 | |
| 16 | 0.3 | 10 | 10 | 100 | >100 | ||
| 25 | 0.1 | 1 | 3 | 5 | 10 | 25 | 55 |
| 26 | 0.3 | 3 | 6 | 5 | 10 | 40 | |
| 10 | >100 | >100 | >100 | >100 | >100 | ||
| 24 | 0.007 | 0.85 | >1 | ||||
| 34 | 0.01 | 1 | 5 | ||||
| 35 | 0.05 | 6 | 5 | 40 | 50 | ||
| 84 | 0 | 3 | 40 | ||||
| 119 | 0.3 | >100 | >100 | ||||
| 37 | 0.5 | 25 | >100 | ||||
| 88 | 0.03 | 0.5 | 0.2 | 0.5 | |||
| 38 | 0.4 | 45 | >100 | ||||
| 106 | 0.1 | 0.7 | 4 | ||||
| 108 | 1 | >100 | >100 | ||||
| 109 | 0.3 | 10 | 15 | ||||
| 110 | 0.3 | 3 | 25 | ||||
| 44 | 45 | >100 | |||||
| 46 | 50 | 100 | |||||
| 47 | 60 | 100 | |||||
| 97 | 0.006 | 0.01 | 0.02 | ||||
| 49 | 100 | 100 | |||||
| 50 | 3 | 3 | |||||
| 98 | 0.5 | 2 | |||||
| 51 | 7 | 7 | |||||
| 52 | 10 | 20 | |||||
| 53 | 5 | 10 | |||||
| 99 | 0.5 | 1 | |||||
| 100 | 0.5 | 10 | 35 | ||||
| 54 | 1 | 30 | 60 |
| 55 | 5 | 8 | 12 | ||||
| 56 | 0.5 | 8 | 10 | ||||
| 123 | >100 | >100 | >100 | ||||
| 61 | 0.3 | 4 | 4 | ||||
| 62 | 2 | 30 | 45 | ||||
| 63 | 1 | 15 | 8 | ||||
| 127 | 0.02 | 1 | 5 | ||||
| 128 | 0.02 | 1 | 10 | ||||
| 113 | 1 | >100 | >100 | ||||
| 114 | 1 | 5 | 80 | ||||
| 66 | 0.3 | 20 | 100 | ||||
| 70 | 2 | 30 | 65 |
表Y2:HT29细胞中的IC50值(μM)
| 化合物 | N2 | 0.1%O2 | 0.3%O2 | 0.6%O2 | 1%O2 | 10%O2 | 空气 |
| 25 | 2 | 25 | >100 |
实施例31
产克隆分析.氧依赖性
[04141为了确定氨基磷酸酯烷化剂前体药物的氧依赖性,进行产克隆存活分析。在化合物测试前2天,将细胞铺于60mm玻璃皿中(在5mL培养基中每个皿中5×105个细胞)。检测下列细胞系:NCI-H460细胞(ATCC HTB-177,RPMI培养基(Gibco))、HT29细胞(ATCC HTB-38,RPMI培养基(Gibco))、PC3细胞(ATCCCRL.1435,Ham's F12K培养基(ATCC))。在测试之前即刻在完全培养基中制造测试化合物的溶液,并且直接加入到细胞中(2mL体积)。通过将玻璃皿暴露于Bactron II厌氧培养室中或铝容器中(参见实施例33)两小时,实现缺氧或低氧(在200ppm O2以下)。对于厌氧培养室,通过在实验之前用预先标定的气体冲洗厌氧培养室,实现在200ppm和空气之间的期望水平的加氧作用。对于铝容器,在37℃水浴中,在摇动平台上,通过将预先加热的、气密的铝固定装置(ji劫中的玻璃皿暴露于一系列5次迅速抽空(rapid evacuation),并且用95%氮气和5%二氧化碳冲洗(对照样品也进行冲洗),实现缺氧或低氧。在第五次抽空和冲洗后,摇动平台(其具有水浴和固定装置)5分钟,然后施行再一次抽空和冲洗,将固定装置转移到37℃培养箱的摇床上,进行剩余的1到2小时的药物暴露。通过改变抽空程度和次数,达到200ppm和空气之间的加氧水平。使用特别修改的便于监控气相和液相的铝固定装置中的氧电极(Anima,Phoenixville,PA),检测培养基和气相中的氧浓度。在暴露于药物之后,从培养箱或铝容器移走玻璃皿,并且通过用培养基冲洗将药物从细胞洗掉。然后胰酶消化细胞,并将细胞铺板,以在塑料皮氏培养皿中进行产克隆存活。10到14天后,用结晶紫(在95%乙醇中0.25%)对培养皿染色,计数含有超过50个细胞的群落(参见实施例33)。计算测试化合物的90%生长抑制浓度(IC90,90%杀死,10%存活),并将它们列在下面的表Y3中。
表Y3:IC90值(μM)
| 化合物〔细胞系〕 | N2 | 0.1%O2 | 0.6%O2 | 空气 |
| 23(H460) | 0.3 | 0.6 | 5 | |
| 25(H460) | 0.1 | 0.4 | 5 | 30 |
| 25(HT29) | 0.2 | 3 | 40 | |
| 25(PC3) | 0.3 | 50 | ||
| 24(H460) | 0.07 | 0.25 | 14 | |
| 35(H460) | 0.5 | 3 | 30 | |
| 37(H460) | 0.2 | 5 | 90 | |
| 70(H460) | 2 | 8 | 20 |
实施例32
电化学
[0415]为了确定氨基磷酸酯烷化剂前体药物的电化学特性和还原电位,由Bioanalytical Systems,Inc.产生这些化合物的循环伏安图(cyclic voltammogram)。用玻璃化碳黑(3.0mm直径)工作电极、Ag/AgCl参比电极和铂线辅助电极,进行所有的实验。在加入9mL磷酸缓冲盐溶液(PBS)之后,将化合物溶于1mL甲醇中,制造在0.5至1.5mM之间的最终药物浓度。将溶液加入到电化学细胞小瓶中,并且用氩气喷射该溶液5分钟,以除去大部分的氧。在100mV/sec和10,000mV/sec扫描速率下,在玻璃化碳黑工作电极处,进行循环伏安法。以CGME汞电极(SMDE模式中的CGME,150μm内径毛细管,体积为8液滴(size8drop)),进行一次测试试验,但是在汞和玻璃化碳黑伏安图之间只观察到很小差异,所以没有进一步使用汞电极。化合物的单电子或多电子还原电位以每一扫描速率产生,并且在下面的表中列出。
表:还原电位(mV)
| 化合物 | 100mV/sec | 10,000mV/scc |
| 1 | -596 | -638 |
| 5 | -606 | -634 |
| 36 | -609 | -634 |
| 25 | -594 | -626 |
| 24 | -568 | -636 |
| 34 | -584 | -663 |
| 78 | -704 | -746 |
| 82 | -428,-610 | -414,-769 |
| 88 | -559 | -629 |
| 108 | -614 | -593 |
| 103 | -638,-769,-875 | -756 |
| 2-NO2-咪唑 | -634 | -693 |
| 5-NO2-呋喃 | -487 | -638 |
| 4-NO2-苯 | -712,-1106 | -735,-1268 |
实施例33
产克隆存活分析
[0416]在如下的分析中检测本发明的氨基磷酸酯烷化剂前体药物。将指数生长的人H460细胞(丛ATCC获得)以每板2.5至5×105之间的密度接种于60mm具缺刻的玻璃板中,并且在开始药物治疗之前,将细胞在补充有10%胎牛血清的RPMI培养基中生长2天。在检测那天,在完全培养基中制备已知浓度的药物原液,并且每块平板加入2ml需要的原液。为了达到在周围气相和液相之间的完全平衡,除去玻璃平板的盖子并且将平板在轨道摇床上摇动5分钟。复原平板,并且将其储存于无菌隔离罩(或手套箱)内。将无菌隔离罩抽空,并且用标定的缺氧气体混合物(95%氮气和5%二氧化碳)或者用需氧(常氧)气体混合物(95%空气和5%二氧化碳)进行气体处理。然后在37℃下用药物培养细胞2小时。
[0417]在前体药物治疗的最后,将平板从每个容器中移出,并将前体药物从细胞迅速除去。用磷酸缓冲盐溶液和胰蛋白酶-EDTA溶液洗涤平板,然后在37℃下胰酶消化5分钟。用培养基和血清中和脱离的细胞,并且通过100×g下离心5分钟进行收集。以大约1×106细胞/ml将细胞再悬浮,并且将其稀释10倍以产生用于铺板的原液浓度。每个原液的浓度通过用Coulter Z2颗粒计数器计数,进行确定。将已知数量的细胞铺板,并且将平板放置于培养箱7天到10天之间。用95%乙醇和0.25%结晶紫的溶液固定并染色集落(colony)。计数大于50个细胞的集落,确定存活分数。
[0418]基于HT29和细胞的产克隆分析以与上述和实施例31中描述的相同的方法进行。
[0419]在低氧和常氧中,通过如下方法确定化合物细胞毒性(表1A和1B):使用H460和HT29细胞系的产克隆分析,如在实施例31和33中提供的,并且表达为单位为μM的IC90;使用H460,HT29,HCT1 16和DX-5细胞系,通过修改Hay等,J.Med.Chem.,2003,46:169-82描述的多孔分析,进行抗增殖分析,并且表达为单位为μM的IC50(参见实施例29)。在常氧和低氧中确定的IC50或IC90的比率被称为低氧细胞毒性比(HCR),并且可作为本发明前体药物的低氧选择性细胞毒性的量度。
表1A

表1B

P=增殖;C=产克隆;H=低氧;N=常氧
实施例34
化合物25对细胞周期分布的影响
[0420]以1.0×106细胞/3ml培养基的密度,将细胞(H60、PC3和HT29)接种于每个60mm的皿中。附着24小时后,在常氧(空气)或低氧(氮气)下,将细胞暴露于指定浓度的化合物25,暴露2小时。将细胞洗涤二次,并且在新鲜培养基中另外温育22小时。胰酶消化细胞,离心,并且在-20℃下,在75%乙醇中固定至少24小时。使用Guava细胞周期试剂(Guava,Hayward,CA),通过流式细胞仪(Guava,Hayward,CA)确定细胞周期分布。数据显示,在多种人癌细胞系中,化合物25诱导细胞周期以氧-依赖性和浓度-依赖性的方式停止。
H460细胞
| μM | G0/GI | S | G2/M | |
| 0 | 空气 | 56 | 12 | 30 |
| 氮气 | 59 | 11 | 26 | |
| 0.005 | 空气 | 38 | 18 | 42 |
| 氮气 | 50 | 12 | 38 | |
| 0.05 | 空气 | 58 | 11 | 28 |
| 氮气 | 30 | 7 | 59 | |
| 0.5 | 空气 | 58 | 11 | 28 |
| 氮气 | 2.3 | 31 | 40 | |
| 5 | 空气 | 42 | 6 | 59 |
| 氮气 | 47 | 15 | 17 | |
| 50 | 空气 | 14 | 19 | 65 |
| 氮气 | 33 | 14 | 11 |
PC3细胞
| μM | G0/G1 | S | G2/M | |
| 0 | 空气 | 54 | 13 | 33 |
| 氮气 | 60 | 12 | 28 | |
| 0.0005 | 空气 | 55 | 12 | 32 |
| 氮气 | 59 | 10 | 31 | |
| 0.005 | 空气 | 52 | 13 | 34 |
| 氮气 | 56 | 11 | 32 | |
| 0.05 | 空气 | 55 | 12 | 33 |
| 氮气 | 43 | 12 | 44 | |
| 0.5 | 空气 | 55 | 13 | 32 |
| 氮气 | 21 | 33 | 46 | |
| 5 | 空气 | 55 | 12 | 32 |
| 氮气 | 35 | 38 | 26 |
HT29细胞
| μM | G0/G1 | S | G2/M | |
| 0 | 空气 | 50 | 14 | 36 |
| 氮气 | 47 | 13 | 39 | |
| 0.005 | 空气 | 52 | 12 | 35 |
| 氮气 | 46 | 14 | 40 | |
| 0.05 | 空气 | 50 | 15 | 35 |
| 氮气 | 37 | 11 | 52 | |
| 0.5 | 空气 | 48 | 14 | 37 |
| 氮气 | 8 | 8 | 84 | |
| 5 | 空气 | 47 | 13 | 39 |
| 氮气 | 14 | 50 | 36 | |
| 5 | 空气 | |||
| 氮气 |
实施例35
球状体模型
[0421]两种人癌细胞系被用于这些球状体研究,以确定低氧活化氨基磷酸酯烷化剂前体药物的功效。将HT29结直肠腺癌(colorectal adenocarcinoma)(结肠癌)细胞以10,000细胞/mL直接接种入125ml旋转瓶中,在补充有10%FBS和抗生素的RPMI培养基中生长。当这些细胞分裂时,它们彼此吸附,形成球状体。将H460肺癌细胞接种于非粘附表面包被的瓶中以形成小的细胞球,该小的细胞球可接种于旋转瓶。为了开始H460细胞接种,将150cm2组织培养瓶用1%琼脂包被,然后每瓶加入10,000细胞,使其在补充有10%FBS和抗生素的RPMI培养基中生长3到5天,然后接种入旋转培养瓶。对于两种细胞系,在球状体可以用眼睛看到后,每天更换生长培养基。
[0422]为了确定完整球状体中低氧区的形态和位置,制备所有球状体进行组织学研究。对于冷冻的部分,在磷酸缓冲盐溶液(PBS)中洗涤完整球状体,并且将其嵌入OCT,在干冰/2-甲基丁烷溶液中迅速冷冻,然后储藏于-80℃。对于包埋于石蜡的部分,将完整球状体固定在新鲜制备的4%的低聚甲醛PBS溶液中,接下来进行包埋和切片。
[0423]为了评估氨基磷酸酯烷化剂前体药物渗透到内部低氧癌细胞、活化、释放氨基磷酸酯烷化剂和杀死那些内部癌细胞的能力,测量暴露于药物2小时的球状体的产克隆存活率。
[0424]将球状体放置于新的生长培养基中,并且在开始实验之前温育至少一小时。通过一系列限定大小的无菌筛孔滤器(mesh filter)过滤球状体培养物,分离在500至600μM之间的球状体。将10至20之间的球状体放置于硅化具缺刻的60mm Pyrex皿上,在具有需要浓度检测化合物的3mL培养基中。将这些皿放置于密封的铝容器中,并且进行一系列抽空,以及用含有5%CO2和限定量O2(0%O2、3%O2、10%O2或空气)的标定气体进行气体处理。将球状体培养在摇动水浴中2小时,以确保溶解O2在溶液中的平衡和溶液中球状体的完整性。将检测化合物除去,并且洗涤球状体,然后用胰蛋白酶完全消化。因为坏死核含有细胞碎片,需要用DNase I处理以产生均匀的单个细胞悬液。以106/mL将细胞重悬浮,并且铺板以测量产克隆存活率。
[0425]在氮气、0.6%O2或空气下,在单层细胞中进行最初的剂量反应实验,以确定从氨基磷酸酯烷化剂前体药物释放的氨基磷酸酯烷化剂的适当的剂量范围和氧依赖性。产克隆存活率是终点,并且通过IC90或C90值(杀死90%细胞并且产生10%存活所需要的抑制浓度)总结数据。柔红霉素和顺铂——它们中的每一个以不同程度渗透入球状体,被用来杀死球状体外部的需氧癌细胞。由于其对细胞很高的亲和性,柔红霉素被用来渗透入多层细胞球状体的外层,而以适合仅杀死外部需要癌细胞的剂量使用顺铂。作为由于高反应性和低渗透性而杀死低氧下单层培养物中细胞但是不杀死多层细胞培养物中细胞的生物还原药物对照,替拉扎明(Tirapazamine)被用于基于单层的实验中和球状体中,如下面对于暴露2小时的H460细胞所列出。
暴露的H460细胞作为单层或球状体的IC90值
[0426]在球状体中,检测一系列氨基磷酸酯烷化剂前体药物,以确定它们渗透入位于内部的低氧癌细胞、活化和杀死低氧细胞的能力。结果在下面列出。
将H460细胞作为单层或球状体暴露于氨基磷酸酯前体药物2小时的IC90值

[0427]化合物25功效的相似结果在HT29球状体中示出,如下面列出的:
[0428]将氨基磷酸酯烷化剂前体药物与顺铂或柔红霉素同时组合,将球状体暴露于该组合2小时,然后测量产克隆存活率。结果在下面列出:
| 化合物 | IC50(μM) |
| 柔红霉素 | 17 |
| 化合物25 | 9 |
| 柔红霉素+化合物25 | 2.3 |
| 化合物 | IC50(μM) | IC99(μM) |
| 顺铂 | 14 | |
| 化合物25 | 12 | |
| 顺铂+化合物25 | 2.3 | 5.4 |
[0429]氨基磷酸酯烷化剂前体药物显示了渗透入球状体中内部细胞和单独以及与其它靶向需氧癌细胞的药剂联合以杀死低氧癌细胞的能力。
实施例36
抗增殖分析-DNA突变体修复细胞
[0430]从ATCC得到特异性DNA修复途径的中国仓鼠卵细胞突变体。用2,500或3,000细胞/孔/500μL补充有10%胎牛血清和抗生素的Dulbecco′s Modified Eagle培养基(Gibco),检测下面的细胞系:AA8细胞(ATCC CRL-1859)、EM9细胞(ATCCCRL-1861)、UV41细胞(ATCC CRL-1860)、UV135细胞(ATCC CRL-1867)、IRS1SF细胞。最初用抗增殖分析筛选所有细胞系,用产克隆分析(如先前描述)重新检测表现敏感性的那些细胞系,以证实增殖结果。在低氧或需氧条件下,将细胞暴露于选定剂量的本发明氨基磷酸酯烷化剂前体药物中2小时,除去检验化合物,并分析细胞。下表列出了细胞系、突变途径和特异性基因缺陷:
| 细胞系 | 突变体途径 | 基因缺陷 |
| AA8 | 没有(野生型) | (没有) |
| EM9 | 碱基切除修复 | XRCC1 |
| UV135 | 核苷酸切除修复 | XPG |
| UV41 | 核苷酸切除修复和同源重组 | XPF |
| IrslSF | 同源重组 | XRCC3 |
[0431]下表列出在缺氧或需氧条件下将各种细胞系暴露于化合物25和36的影响,如通过IC50测量的增殖所分析。
| 化合物 | AA8(缺氧/空气) | EM9(缺氧/空气) | UV41(缺氧/空气) | UV135(缺氧/空气) | IRS1SF(缺氧/空气) |
| 36 | 2/>100 | 4/>100 | 0.03/20 | 2/>100 | 0.3/59 |
| 25 | 8/>100 | 7/>100 | 0.2/95 | 6/>100 | 2/>100 |
[0432]下表列出在缺氧或需氧条件下将选择的细胞暴露于化合物25的产克隆存活率的IC90值。

[0433]低氧下仅在同源重组中有缺陷的细胞系对化合物敏感。因为UV41涉及核苷酸切除修复途径以及同源重组修复途径,化合物25也可能产生明显数量的一元加成物。然而,也涉及核苷酸切除修复的UV135对化合物25不敏感。化合物25产生的主要损伤是DNA链间交联。用产克隆分析,在UV41和irslSF细胞中证实了这些结果。需氧条件下的暴露产生与低氧下所见的相同的敏感谱,这表明需氧细胞毒性也由DNA链间交联形成所引起。在突变细胞系,化合物36表现了相似的敏感模式(pattern of sensitivity),这表明化合物36也产生DNA链间交联。
实施例37
多层细胞培养物分析
[0434]该实施例表示化合物25对使用多层细胞培养物(MCC)的组织渗透的影响,并且评估任何旁观者效应。用氧合培养基(20%O2和5%O2)或低氧培养基(大约0%O2)培养MCC,并且从一侧(暴露的表面,常氧侧)暴露检测化合物,而另一侧被暂时隔离(远侧,低氧侧)。当MCC在20%O2或5%O2的培养基中培养时,从暴露于培养基的表面到培养物的远侧表面的氧梯度形成。最远50μm的组织耗空了氧。在用5%O2处理的培养基中O2的耗空程度比用20%O2处理的培养基中O2的耗空程度大;用5%O2培养最接近地反应了体内环境。用0%O2培养基培养MCC模拟了灌注有限的低氧,其中肿瘤血管完全耗空了氧,检测化合物必须渗透极大的距离到达所有细胞。因此,如果活化药物的结合起着限制其渗透的作用,这种情况对药物渗透具有更大的障碍。
[0435]在用检测化合物培养之前和用检测化合物培养期间,使用用0、5或20%O2气体处理的培养基进行基于MCC的实验45分钟。在固体支持体上将HCT116细胞生长为150μm的厚度,培养物的一侧被夹紧,以形成扩散有限的低氧。在0%O2、5%O2或20%O2下,将培养物暴露于检测化合物1小时,通过测量BrdU并入的抑制,评估功效。在新鲜培养基中,在20%O2下将培养物再培养1小时,从设备移走培养物,将培养物回到正常生长培养箱——这里培养基流过MCC的两侧。在BrdUrd标记和接下来的冷冻切片之前,将培养物培育24小时。使用免疫组织化学染色、显微镜成像和计算机图像分析分析,分析在MCC上暴露侧和远侧上的BrdUrd标记,以评估化合物25对细胞增殖的影响。
[0436]当在20%O2下,将培养物暴露于化合物25的分次剂量(graded dose)时,与暴露侧(常氧)相比,远侧(低氧)需要的化合物少5倍,以产生相当的结果,这显示了化合物25的渗透和低氧活化。当在含有5%O2的更加生理相关的条件下,将MCC暴露于检测化合物时,化合物25抑制BrdU并入低氧侧比抑制BrdU并入常氧侧有效10倍。在5%和20%O2下,培养物的常氧侧通过暴露于化合物25,受相同的影响。
[0437]在5%O2下化合物25对培养物的低氧侧比在0%O2下化合物25对培养物的低氧侧更有效。在5%O2下,培养物常氧侧与低氧侧的比较表明,化合物25更有效地渗透穿过氧合相对完好的组织。化合物25能杀死位于距功能性血管大约150μm处的低氧细胞。相对于暴露于5%O2条件,在0%O2条件下观察到将低氧侧暴露于化合物25,减少大约3倍。仅在最高的浓度,观察到旁观者效应。
[0438]下表列出暴露于化合物25的分次剂量的影响,如IC50(抑制50%的BrdU并入的浓度)所测量。
| 侧 | 0%O2(μM) | 5%O2(μM) | 20%O2(μM) |
| 低氧 | ~1.1 | 0.7 | 2.6 |
| 常氧 | ~1.7 | 8.0 | >10 |
实施例38
人和小鼠微粒蛋白对化合物25的代谢
[0439]使用含有细胞色素P450酶的人(HLM)、大鼠(RLM)和小鼠(MLM)肝脏微粒蛋白,施行氨基磷酸酯烷化剂前体药物(化合物25)的代谢能力的体外评估。通过在水:甲醇盐桥溶液中将DMSO原液溶液稀释100倍、将微粒蛋白(1mg/mL)加入PBS/MgCl2中和通过加入NADPH溶液引发的酶促反应,制备化合物25(500μL,5μM)的溶液。在加入NADPH溶液后0、10、20和30分钟,吸取50μL反应混合物,用乙腈沉淀蛋白质,并通过反相LC-MS/MS分析透明上清液中的化合物25量。硝苯吡啶和睾丸素被用作阳性对照。第一研究对比RLM和MLM(表1),第二研究对比HLM和RLM(表2A和2B)。
表1

表2A

表2B

实施例39
氨基磷酸酯烷化剂前体药物的体内药物动力学
[0440]氨基磷酸酯烷化剂前体药物的各种血浆药物动力学参数在CD-1 小鼠中确定,除了在下面表3中注明的。
表3
| 药物(mg/kg) | 剂量(mg/kg) | 途径 | 制剂 | Tmax(min) | Cmax(μg/mL) | AUC(μg-h/mL) | 半衰期(min) |
| 23 | 50 | i.p. | 25%PEG/75%盐水 | 5.00 | 8.50 | 210 | 9.60 |
| 23 | 50 | i.v. | 25%PEG/75%盐水 | 5.00 | 7.80 | 136 | 8.87 |
| 36 | 50 | i.p. | 25%PEG/75%盐水 | 15.0 | 44.0 | 1439 | 11.0 |
| 36 | 50 | i.v. | 25%PEG/75%盐水 | 5.00 | 27.7 | 646 | 14.1 |
| 1a | 50 | i.p. | 25%PEG/75%盐水 | 15.0 | 29.9 | - | - |
| 1a | 50 | i.v. | 25%PEG/75%盐水 | 5.00 | 35.0 | 12.5 | 15.3 |
| 5 | 50 | i.p. | 25%PEG/75%盐水 | 5.00 | 3.00 | 57.4 | 2.56 |
| 5 | 50 | i.v. | 25%PEG/75%盐水 | 5.00 | 16.9 | 270 | 4.67 |
| 37 | 20 | i.p. | 聚乙烯篦麻油:乙醇:盐水(1:2:7) | 2.00 | 12.6 | 196 | 23.2 |
| 37 | 20 | i.v. | 聚乙烯篦麻油:乙醇:盐水(1:2:7) | 2.00 | 15.0 | 172 | 9.00 |
| 85 | 25 | i.p. | 10%PEG | 5.00 | 3.93 | 89.1 | l0.0 |
| 128 | 25 | i.p. | 10%PEG | 5.00 | 3.85 | 102 | 8.31 |
| 24 | 50 | i.p. | 盐水 | 5.00 | 7.60 | 64.0 | 4.10 |
aBalb/c小鼠
实施例40
化合物25的体内药物动力学
[0441]化合物25的多种血浆或肿瘤药物动力学参数在CD-1小鼠中确定,除了在下面表4中注明的。
表4
| 剂量(mg/kg) | 途径 | 制剂 | Tmax(min) | Cmax(μg/mL) | AUC(μg-h/mL) | 半衰期(min) | Fc(%) |
| 150a | i.p. | 盐水 | 5.00 | 90.1 | 1239 | 58.7 | - |
| 150a,b | i.p. | 盐水 | 15.0 | 3.38 | 307 | ND | - |
| 100 | p.o. | 盐水 | 15.0 | 15.8 | 784 | 95.2 | - |
| 50 | i.p. | 30%PEG/70%盐水 | 5.00 | 22.9 | 438 | 11.0 | - |
| 50 | i.v. | 30%PEG/70%盐水 | 2.0 | 27.5 | 325 | 6.7 | - |
| 50 | i.p. | 30%PEG/70%盐水 | 15.0 | 9.2 | - | - | - |
| 50 | i.p. | 30%PEG/170%盐水 | 2.0 | 27.5 | 177 | 10.1 | - |
| 50 | i.p. | 盐水 | 5.00 | 38.5 | 635 | 7.91 | - |
| 50 | p.o. | 盐水 | 15.0 | 0.93 | 40.4 | 25.7 | 13.6 |
| 25 | i.p. | 10%PEG | 45.0 | 6.33 | 247 | 4.43 |
a具有H460肿瘤的裸鼠
b肿瘤PK
c生物有效度
实施例41
细胞色素450抑制化合物25的代谢
[0442]含有100μL溶液的8个反应孔——所述溶液含有pH 7.4的50mM磷酸钾、2.6mM NADP+、6.6mM葡萄糖-6-磷酸、0.8U/mL葡萄糖-6-磷酸脱氢酶和检测化合物(例如化合物25)的1:3连续稀释液——和适当阳性对照抑制剂(例如对于CYP1A2用呋拉茶碱,对于CYP2C9用磺胺苯吡唑、对于CYPC219用N-苄基尼凡诺,对于CYP2D6用奎尼丁和对于CYP3A4用酮康唑)的1:3连续稀释液的8个孔被一起制备。检测化合物的浓度范围从0.0229μM到200μM。通过加入100μL预热的酶/底物溶液开始反应。在0时刻点,通过加入50mL、10%的甲酸(对于2C19,用400mL乙腈)水溶液到100mL协同因子溶液中以使酶失活,然后加入100mL酶/底物溶液,制备对照反应。也制备不含抑制剂的对照反应。在37℃下,适当温育后,通过加入50mL、10%甲酸水溶液(对于2C19,用400mL乙腈)中止反应。制备反应并且使用HPLC/MS/MS分析反应的探针底物(对于CYP1A2为非那西丁,对于CYP2C9为双氯芬酸、对于CYPC219为(S)-美芬妥英,对于CYP2D6为右美沙芬和对于CYP3A4为咪达唑仑、睾丸素和硝苯地平)的代谢形式。每一个分析进行两遍。IC50值的总结在下面列出。
表5

NI=没有检测到明显抑制
实施例42
在小鼠、大鼠、狗和人肝细胞中形成的化合物25的潜在代谢物的确定
[0443]10μM浓度的化合物25与小鼠、大鼠、狗、猴和人冻存肝细胞一起温育。通过用乙腈淬灭,反应在0(预培育)、30、60和120分钟停止,然后离心,并且通过高效液体色谱仪(HPLC)结合串联质谱仪(LC/MS/MS)进行分析。通过进行从100到520amu的全扫描,鉴别潜在的代谢物。接下来收集可能代谢物的产物离子谱,并且与母体化合物的产物离子谱对比,以确定每一可能代谢物是否与化合物25相关。通过对比获得的每一时间点的峰高度,监控随着时间母体化合物(化合物25)的消失和可能代谢物的出现。
实施例43
大鼠、狗和猴中化合物25和其代谢物(一种或多种)的体内药物动力学确定
[0444]在Sprague Dawley大鼠中,单一静脉内施用5、20、50和100mg/kg化合物25后,确定化合物25和其代谢物(一种或多种)的药物动力学参数。也将在毕尔格猎犬和猕猴(cynomologus monkey)中,单一静脉内施用20mg/kg化合物25之后,确定化合物25和其代谢物(一种或多种)的药物动力学。通过LC/MS/MS方法,确定血浆中化合物25和其代谢物(一种或多种)的浓度,并且计算平均的药物动力学参数。
实施例44
大鼠中的质量平衡研究
[0445]将14C-化合物25作为单一静脉内药剂,施用给正常和胆汁插管的Sprague-Dawley大鼠。在规定时间收集血浆、尿和大便,通过液体闪烁计数(LSC)确定总的放射性浓度。
实施例45
定量的全身放射自显影
[0446]将14C-化合物25的单一静脉内药剂,施用给Sprague-Dawley大鼠。在规定时间,在每一时间点使一只大鼠安乐死。离心血液以得到血浆,并且分析血液和血浆的放射性浓度。将冷冻大鼠尸体包埋于2%CMC中,冷冻成块,并且在Leica CM 3600冷冻切片机中切割成40μm片。将收集的切片冷冻干燥,与14C放射自显影标准一起安装和暴露于感光成像平板上,14C放射自显影标准用于接下来的图像分析软件校准。使用Molecular Dynamics Storm 820或860扫描暴露的屏幕(screen)。通过图像分析,测量含有脂肪(棕色和白色)、肾上腺、血液、脑(大脑、小脑、髓质)骨、骨髓、盲肠和内含物、附睾、食道、眼球(葡萄膜、房水、晶状体)、哈氏腺、心脏、肾脏(皮层、髓质、乳突和完整切片)、大肠和内含物、肝脏、肺、颌下淋巴结、胰脏、垂体、前列腺、唾液腺、精囊、骨骼肌、皮肤、胃(和内含物)、小肠(和内含物)、脾脏、脊髓、气管、甲状腺和膀胱(和内含物)的选择组织中的放射性的浓度。对于每一动物,得到放射自发光图片和数字图像。
实施例46
化合物25的血浆蛋白结合
[0447]使用超滤,确定化合物25在小鼠、大鼠、狗、猴和人血浆中的蛋白结合。通过将以3种浓度掺加有化合物25的血浆等分加入Centrifree 仪器,一种三份,进行超滤。然后,将所有血浆样品平衡到37℃。在37℃下,以2500xg,离心Centrifree仪器30分钟。将超滤液的75μL等分液掺加入I.S.(氘化化合物25),并且用LC/MS/MS分析。使用用于校正曲线的人超滤液标准分析和定量超滤液。
仪器,一种三份,进行超滤。然后,将所有血浆样品平衡到37℃。在37℃下,以2500xg,离心Centrifree仪器30分钟。将超滤液的75μL等分液掺加入I.S.(氘化化合物25),并且用LC/MS/MS分析。使用用于校正曲线的人超滤液标准分析和定量超滤液。
实施例47
[0448]实施例47使用HT-29人结肠癌异种移植小鼠模型,表明了本发明的化合物治疗癌症的有效性。
[0449]使7-8周大的雌性CB17/SCID小鼠(购自Charles River,Cambridge,MA)适应至少3天,并在无病原体的条件下进行处理。从American Type CultureCollectjon得到人结肠癌细胞系HT-29。将细胞系在补充10%胎牛血清的RPMI1640培养基中培养。将细胞保持在37℃含有5%CO2的培养箱中。从培养物收获HT-29细胞,并且以3×106细胞/动物接种于腹膜皮下空间。当肿瘤长到100mm3的平均体积(第8天)时,给予10只小鼠的每组如下物质3周:仅给载体(组1,盐水和PEG(每只10mL/kg));以20、60或200mg/kg日剂量(分别为组2、组3和组4)仅给化合物36(溶解在30%环式糊精的PBS中);和在给予10mg/kg 5FU(盐水中)2-3小时后,以20、60和200mg/kg日剂量(分别为组5、组6和组7)给予化合物36,并且与仅以10mg/kg接受5FU的组(组8)进行比较,如在下面列出的。
[0450]每周记录每只鼠的体重两次。通过使用数字测径器,每周两次外表上测量肿瘤的二维,监控每个异种移植的生长。通过下面的方程确定肿瘤体积(V):V=(L×W2)/2,其中L是异种移植物的长度,W是异种移植物的宽度。每周测量两次肿瘤体积。
[0451]以20、60和200mg/kg/天施用化合物36,与单独施用载体相比,每种剂量都减少了肿瘤生长。与施用载体相比,施用化合物36和5FU组合,导致对肿瘤生长的更大的和剂量相关的抑制。另外,60和200mg/kg化合物36的组合减少了肿瘤生长,其减少程度大于单独给予5FU。

[0452]与这些抗肿瘤效果相关,存在一定程度的体重减轻和偶然的死亡率,特别是在那些用高剂量化合物36治疗的组中,但是在其它组中也存在。总之,化合物36显示出抑制肿瘤生长的不同速率。
实施例48
[0453]实施例48使用NCI H460人结肠癌异种移植物小鼠模型,表明本发明的化合物治疗癌症的有效性。
[0454]使7-8周大的雌性CB17/SCID小鼠(购自Charles River,Cambridge,MA)适应至少3天,然后在无病原体的条件下进行处理。从American Type CultureCollection得到人结肠癌细胞系NCI H460。根据实施例47中描述的过程,培养和收获细胞系,并且以1×106细胞/动物接种于腹膜皮下空间。当肿瘤长到100mm3的平均体积(第8天)时,每组小鼠给药3周,如下表列出:化合物25(10%PEG中2.5mg/ml,给药途径—腹膜内注射)和紫杉醇(5%EtOH、5%Cremophor(聚乙烯蓖麻油)和90%盐水中1mg/ml;给药途径—在施用化合物25后2小时静脉内注射)。如在上面实施例47中的描述,测量体重和肿瘤体积。
治疗方案
组1a和1b,n=5
q1d/qd=每天;q2d=每第二天;q7d=每第7天
[0455]当载体治疗的小鼠已经达到946mm3的体积,基于第29天测量的肿瘤体积的结果在表X2中呈现。加入接受盐水或不治疗的5只小鼠的组,以便表示任何载体的影响,但是不用于这些分析的比较。

[0456]结果表明,给药化合物25的所有三个方案提供相似的肿瘤生长抑制程度,而且联合治疗特别是每天给药的联合治疗提供了额外的益处。每一联合治疗与一定程度的体重减轻相关,但是没有大到足以引起任何死亡。总之,结果表明,化合物25在肺癌的这个模型中是有效的,并且比由标准化疗剂紫杉醇所提供的,提供了额外的益处。
[0457]使用小鼠向HED转化,对于治疗癌症,特别是肺癌,以大约2到大约8mg/kg/天的治疗有效剂量,单独或与TaxolTM联合,施用化合物25,其中,与更低的剂量相比,对于更高的剂量,日剂量可以以降低的给药频率施用。
实施例49
[0458]实施例49描述了本发明的化合物治疗癌症的有效性,如使用H460非小肺癌异种移植小鼠模型所表明。使7-8周大的雌性CB17/SCID小鼠(购自CharlesRiver,Cambridge,MA)适应至少3天,然后在无病原体的条件下进行处理。从AmericanType Culture Collection得到人非小肺癌细胞系NCI H460。如上面实施例47中描述的,培养和收获细胞系,并且以3×106细胞/动物接种于腹膜皮下空间。当肿瘤长到100mm3的平均体积(第8天)时,每组小鼠(每组10只)给药3周,如下表列出:化合物25(10%PEG中2.5mg/ml,施用途径—腹膜内注射);化合物24(10%PEG中0.3、0.1mg/ml,施用途径—腹膜内注射)和紫杉醇(5%EtOH、5%Cremophor和90%盐水中1mg/ml;在施用检测化合物后2小时,静脉内注射施用)。
治疗方法
| 组 | 小鼠数量 | 治疗 | 剂量(mg/kg) | 方案 |
| 1 | 10 | 载体* | NA | (q1d×5d)/周×3周 |
| 2 | 8 | 紫杉醇 | 10 | (q2d×3)/周×2周 |
| 3 | 8 | 化合物24 | 3 | (q1d×5d)/周×3周 |
| 4 | 9 | 化合物24紫杉醇 | 110 | (q1d×5d)/周×3周(q2d×3)/周×2周 |
| 5 | 8 | 化合物24 | 3 | (q1d×5d)/周×3周 |
| 紫杉醇 | 10 | (q2d×3)/周×2周 | ||
| 6 | 8 | 化合物25紫杉醇 | 2510 | (q1d×5d)/周×3周(q2d×3)/周×2周 |
*-50%PEG
[0459]如在上面实施例47中的描述,测量体重和肿瘤体积。第27天测量的肿瘤生长抑制的结果在下面列出。在第27天进行对比,因为那是载体组测量的最后一天而且那些动物已死亡。

[0460]这些结果表明,3mg/kg的化合物24和25mg/kg的化合物25的日剂量抑制肿瘤生长,并且化合物25单独治疗和与紫杉醇联合治疗都具有稍多的益处。这些效果伴随有轻微的体重减轻,特别在化合物25+紫杉醇组。
[0461]使用小鼠向HED转化,对于治疗癌症,特别是肺癌,以2mg/kg/天的治疗有效剂量,单独或与TaxolTM联合,施用化合物25,以及对于治疗癌症,特别是肺癌,以0.25mg/kg/天的治疗有效剂量单独或与TaxolTM联合施用化合物24。
实施例50
[0462]实施例50描述本发明的化合物治疗癌症的有效性,如使用HT-29人结肠癌异种移植物小鼠模型所表明。使7-8周大的雌性CB17/SCID小鼠(购自CharlesRiver,Cambridge,MA)适应至少3天,然后在无病原体的条件下进行处理。从American Type Culture Collection得到人结肠癌细胞系HT29。如上面实施例47中描述的,培养和收获细胞系,并且以3×106细胞/动物接种于腹膜皮下空间。当肿瘤长到100mm3的平均体积(第8天)时,每组小鼠(每组10只)如下表列出的给药3周:在计划联合治疗的那天,在施用5-FU或顺铂(CDDP;盐水中)之前2小时,施用化合物24(10%PEG中),施用途径—腹膜内注射;仅给予5FU(在盐水中),或者仅施用CDDP。
治疗方案
| 组 | 小鼠数量 | 检测项目 | 剂量(mg/kg) | 途径,方案 |
| 1* | 8 | 盐水 | 10ml/kg | iv,q3d×4 |
| 2* | 8 | 5-FU | 50 | iv,q3d×4 |
| 3* | 4 | 没有治疗 | N/A | N/A |
| 4** | 9 | 载体(盐水) | 10ml/kg | ip,(q1d×5)/周×3周 |
| 5** | 9 | 5-FU | 50 | iv,q3d×4 |
| 6** | 9 | CDDP | 5 | iv,一次 |
| 7** | 9 | 化合物24 | 3 | ip,(q1d×5)/周×3周 |
| 8** | 9 | 化合物24 | 6 | ip,(q1d×5)/周×3周 |
| 9** | 9 | 化合物245-FU | 650 | ip,(q1d×5)/周×3周iv,q3d×4 |
| 10** | 9 | 化合物24CDDP | 35 | ip,(q1d×5)/周×3周iv,一次 |
| 11** | 9 | 化合物24CDDP | 65 | ip,(q1d×5)/周×3周iv,一次 |
| 12** | 8 | 没有治疗 | N/A | N/A |
*-肿瘤位于侧腹;**-肿瘤位于腹膜;q3d=每第三天
[0463]在对照组,将肿瘤移植入两个位置,作为位置对对照组肿瘤生长影响的单独研究的一部分。这些结果对该研究的解释没有影响,并且将所有治疗与机体同一部位带有肿瘤的载体组进行比较。如在上面实施例47中的描述,测量体重和肿瘤体积。第25天测量的肿瘤生长抑制在下面列出,第25时载体肿瘤已经达到最大的尺寸,并且该组里的动物已死亡。

[0464]这些结果表明,作为单一治疗的化合物24导致稍高于40%的肿瘤生长抑制,而组合的化合物24与CDDP或5FU联合施用提供了大约50-70%的生长抑制。根据该实施例,最治疗有效的联合是化合物24和5FU的联合。对肿瘤生长的影响与治疗期间小鼠体重较小的下降相关;然而在治疗结束时,小鼠恢复了失去的重量。
[0465]使用小鼠向HED转化,对于治疗癌症,特别是肺癌,以大约0.25到大约0.50mg/kg/天的治疗有效剂量,单独或与5FU或CDDP联合施用化合物24。
实施例51
[0466],实施例51描述本发明的化合物治疗癌症的有效性,如使用H460非小肺癌异种移植物小鼠模型所表明。使7-8周大的雌性CB17/SCID小鼠(购自CharlesRiver,Cambridge,MA)适应至少3天,然后在无病原体的条件下进行处理。从American Type Culture Collection得到人非小肺癌细胞系NCI H460。如上面实施例47中描述的,培养和收获细胞系,并且以3×106细胞/动物接种于腹膜皮下空间。当肿瘤长到100mm3的平均体积时,开始治疗,其中10只小鼠的组接受载体(组1);3或6mg/kg的CDDP(分别为组2和组3,一次静脉内注射);盐水中的50mg/kg的化合物25,每周5次持续两周(组4);100mg/kg的化合物25,每三天一次,进行5次(组5),或者每一剂量的化合物25与3或6mg/kg的CDPP的联合(分别为组6和组7)。接受50mg/kg化合物25的组的结果在图1中阐明。图2示出100mg/kg化合物25的相似结果。
[0467]使用化合物25的盐水配制形式进行的这些结果表明,与使用50mg/kg日剂量的那些结果相比,使用更低频率给药的50mg/kg和100mg/kg的日剂量,明显地剂量相关地,肿瘤体积减小并且肿瘤生长延迟增加。这些数据也表明,在该模型中,两种给药方案都增加了CDDP的效果。
[0468]使用小鼠向HED转化,对于治疗癌症,特别是肺癌,以大约4到大约8mg/kg/天的治疗有效剂量,单独或与5FU或CDDP联合施用化合物25,其中,与更低的剂量相比,对于更高的剂量,日剂量可以以降低的给药频率施用。
实施例52
[0469]实施例52描述了本发明的化合物治疗癌症的有效性,如使用HT-29人结肠癌异种移植物小鼠模型所表明。使7-8周大的雌性CB17/SCID小鼠(购自Charles River,Cambridge,MA)适应至少3天,然后在无病原体的条件下进行处理。从American Type Culture Collection得到人结肠癌细胞系HT29。如上面实施例47中描述的,培养和收获细胞系,并且以3×106细胞/动物接种于腹膜皮下空间。当肿瘤长到100mm3的平均体积(第8天)时,每组小鼠(每组10只)如下表列出给药3周:在计划联合治疗的那天,在施用CDDP之前2小时,施用盐水中的化合物25,施用途径—腹膜内注射;和CDDP(盐水中,静脉内注射)。
治疗方案
| 组(n=9) | 检测项目 | 剂量(mg/kg) | 给药方案 |
| 1 | 盐水 | 10ml/mg | (qd×5)/周×2周 |
| 2 | CDDP | 5 | 一次 |
| 3 | 化合物25 | 50 | (qd×5)/周×2周 |
| 4 | 化合物25 | 100 | (q2d×3)/周×2周 |
| 5 | 化合物25 | 100 | Q3d×5 |
| 6 | 化合物25 | 100 | Q7d×2 |
| 7 | 化合物25 | 50 | (qd×5)/周×2周 |
| CDDP | 5 | 一次 | |
| 8 | 化合物25CDDP | 1005 | (q2d×3)/周×2周一次 |
| 9 | 化合物25CDDP | 1005 | Q3d×5一次 |
| 10 | 化合物25CDDP | 1005 | Q7d×2一次 |
[0470]如在上面实施例47中的描述,测量体重和肿瘤体积。数据基于第25天的肿瘤体积,此时载体组的肿瘤已经达到足够使小鼠死亡的大小。肿瘤生长抑制的结果在下面列出。

[0471]结果表明,以50mg/kg/天和100mg/kg/天,多种剂量方案,单一治疗施用在盐水中配制的化合物25,导致在该结肠癌模型中抑制肿瘤生长,并且化合物25和CDPP的治疗联合加强了化合物25治疗该模型中结肠癌的有效性。这些效果伴随有轻微的体重减轻,在联合组中更是如此;在治疗结束后,小鼠恢复了失去的重量。
[0472]使用小鼠向HED转化,对于治疗癌症,特别是肺癌,以大约4到大约8mg/kg/天的治疗有效剂量,单独或与CDDP联合施用化合物25,其中,与更低的剂量相比,对于更高的剂量,日剂量可以以降低的给药频率施用。
实施例53
[0473]实施例53描述了本发明的化合物治疗癌症的有效性,如使用H460非小肺癌异种移植小鼠模型所表明。使7-8周大的雌性CB17/SCID小鼠(购自CharlesRiver,Cambridge,MA)适应至少3天,然后在无病原体的条件下进行处理。从American Type Culture Collection得到人非小肺癌细胞系NCI H460。如上面实施例47中描述的,培养和收获细胞系,并且以3×106细胞/动物接种于腹膜皮下空间。当肿瘤长到100mm3的平均体积时,开始治疗,其中10只小鼠的组接受载体(组1);6mg/kg的CDDP(一次静脉内注射,组2);盐水中150mg/kg的化合物25,每周一次持续两周(组3,腹膜内注射);或者两种药剂联合(组4)。
[0474]图3所示的结果表明,每周150mg/kg的化合物25比仅给予CDDP提供更大的肿瘤生长减小,并且两种药剂的联合导致增加的益处。这些结果也表明,在两周的给药期间肿瘤体积不变化,这表示完全抑制了肿瘤的生长。这些数据表明,以一周一次150mg/kg/天施用化合物25作为单一治疗,是前述实施例(实施例47-52)中描述的所有给药方案中最有效的。观察到很小的体重改变,这表明该给药方案减小毒性。
[0475]使用小鼠向HED转化,对于治疗癌症,特别是非小细胞肺癌,以大约12mg/kg/天的治疗有效剂量,单独或与CDDP联合施用化合物25,任选以每周一次的频率给药。
实施例54
[0476]实施例54描述在H460异种移植物小鼠模型中,经腹膜内快速注射或腹膜内灌流,化合物25单独或与顺铂联合的有效性。使6周大的雌性Nu-Foxnlnu纯合nu/nu小鼠(购自Charles River,Cambridge,MA)适应至少3天,然后在无病原体的条件下进行处理。从AmericanType Culture Collection得到人结肠癌细胞系HT29。如上面实施例47中描述的,培养和收获细胞系,并且以3×106细胞/动物接种于腹膜皮下空间。当肿瘤长到100mm3的平均体积(第8天)时,每组小鼠(每组10只)如下表列出给药3周:在计划联合治疗的那天,在施用CDDP之前2小时,施用化合物25(配制为15mg/ml盐水溶液,施用方式——腹膜内注射);和盐水中CDDP,静脉内注射。
治疗方案
| 组 | 检测项目 | 剂量(mg/kg) | 方案 | 剂量浓度:剂量体积 |
| 1 | 盐水 | 10 | Q7d×2 | 0mg/ml:10mL/kg |
| 2 | CDDP | 6 | Q7d×2 | 0.6mg/ml:10mL/kg |
| 3 | 化合物25 | 150 | Q7d×2 | 15mg/ml:10mL/kg |
| 4 | 化合物25CDDP | 1506 | q7d×2q7d×2 | 15mg/ml:10mL/kg0.6mg/ml:10m[/kg |
| 5 | 化合物25 | 150 | Q7d×2 | 10mg/ml:15mL/kg |
| 6 | 盐水 | 0.2ml | 200μl-每周×2* | 0mg/ml:1μL/小时 |
| 7 | 化合物25 | 15mg/ml | 200μl-每周×2* | 15mg/ml:1μL/小时 |
*-Alzet泵,200μl,每周×2(在一星期末,重新植入新的泵)
[0477]如在上面实施例47中的描述,测量体重和肿瘤体积。结果表示在图4中。数据表明,当持续单独或与CDDP联合应用化合物25有效时,间歇给药,如每周一次给药,在治疗某些癌症例如非小细胞肺癌中可提供更大的治疗益处。
实施例55
化合物25和吉西他滨联合治疗
[0478]将化合物25和吉西他滨联合施用给裸鼠,所述裸鼠携带衍生自MiPaca2型人胰腺癌细胞的肿瘤。MiaPaca-2肿瘤是高度浸润性的、快速生长的肿瘤,其导致未治疗的动物在20-30天内死亡。用红色荧光蛋白基因转染肿瘤细胞。通过腹膜内注射给药,对小鼠施用载体对照药剂、吉西他滨、化合物25、化合物24或吉西他滨/化合物25联合或吉西他滨/化合物24,如在下面列出的(8只鼠/组)。将化合物24和25配制于盐水中,由Threshold Pharmaceuticals,Inc.作为干粉提供。吉西他滨从商业获得,并且根据制造商的指导,新鲜制备。
治疗方案
*qd=每天;qw=每周
[0479]每周作肿瘤图像1次,直到研究结束,此时获得打开的身体图像(openbody image),以证实效果。在组1中,肿瘤快速生长(图5),并且到30天产生100%致命性(图6)。
[0480]组3和组4对肿瘤体积产生较小的影响,对存活率也产生很小的影响。组2明显减小了肿瘤体积并且延长了存活。组6提供了肿瘤大小微小的减小,但是对存活率没有另外的影响。相反地,组5明显表明了减小的肿瘤生长,并且相比于组2,明显延长了存活。在治疗后,组5中八分之五的肿瘤迅速退化(regress),并且在短期内没有发出荧光(图7)。
[0481]直到实验结束,这些肿瘤中的四个保持零荧光,认为肿瘤己被治愈。在组2中,没有肿瘤被认为已治愈。这些结果表明,与基于护理的(with the standof care)吉西他滨单一治疗相比,用化合物25和吉西他滨联合治疗在该癌症模型的治疗中更有益处。这些结果表明,用30mg/kg/天的化合物25和吉西他滨联合给药的动物中的肿瘤减小明显比用吉西他滨作为单一药剂治疗的动物的肿瘤减小更大。
[0842]使用小鼠向HED转化,对于治疗癌症,特别是胰腺癌,以大约2.5mg/kg/天的治疗有效剂量,单独或与吉西他滨联合施用化合物25。
实施例36
[0483]已经知道,治疗人疾病包括癌症的有效分子,在剂量接近达到有益效果必需的剂量或者有时比达到有益效果必需的剂量多得多时,可能是有毒性的。为了确定给予这样的化合物的适当剂量和途径,需要了解其毒性。常规地,确定毒性剂量的最初方法涉及使用啮齿类动物如小鼠来提供初步数据,其可能支持在更大的动物和人中进行的相似研究的设计。检测化合物(化合物24、25和36)在小鼠中检测,作为确定在更大的动物中使用的初步实验。以高达300mg/kg作为单一药剂的剂量检测化合物25,发现化合物25引起肾毒性例如肾小管坏死和蛋白溢入尿。也观察到白细胞短暂的减少。然而,在更低剂量(100和200mg/kg)时,注意到很小的毒性。选择的这些剂量表示了可用于更大动物如大鼠和狗的大概的剂量,目的在于证实这类毒性存在以及预测人的肾功能是否应该进行测定。
[0484]虽然本发明已参照具体实施方式被详细描述,但本领域技术人员将知道,修改和改进在本发明的范围和精神内,如所附权利要求书中所述。本文引用的所有出版物和专利文献(专利、公布的专利申请和未公布的专利申请)并入本文作为参考,如同每一这样的出版物或文献被特别地和具体地指明并入本文作为参考。对出版物和专利文献的引用并不意图承认,任何这种文献是有关的现有技术,也不构成对其内容或其日期的任何承认。本发明现在已通过书面说明和实施例的方式被描述,本领域技术人员将知道,本发明可以以多种实施方式实践,并且前面的描述和实施例是为了阐明的目的,而不是对所附权利要求的限制。
Claims (42)
1.分子式(I)的化合物:

其单独的异构体或者异构体的外消旋或非外消旋混合物、生物电子等排物、药效团、药学上可接受的盐、溶剂化物、水合物或前体药物;
其中
Y1是O、S、NR6或NSO2R6
Y2是O、S、NR6、NCOR6或NSO2R6,其中每一R6独立为(C1-C6)烷基、(C1-C6)杂烷基、芳基或杂芳基;
R1-R5的每一个独立地是氢、羟基、氨基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;或者R1-R5的任意两个一起形成C3-C10杂环;或者R1-R5的每一个独立为具有式L-Z3的触发物T;
L选自
-[C(Z1)2-Y3]v-[C(=O)-O]q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]z-[-C(Z1)=C(Z1)]g-;和
-[C(Z1)2-Y3]v-(S(=O)2)q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]z-[C(Z1)=C(Z1)]g;其中每一个
z、v、q、u和g独立为0或者1;
Y3是S、O或NR7,其中每一R7独立地是氢、羟基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;
Y4是O、S或者-NR7-C(=O)-O-;
每一Z1和Z1独立为氢、卤素、C1-C6烷基、C1-C6杂烷基、芳基、杂芳基、C3-C8环烷基、杂环基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;
Z2是C1-C6亚烷基、C1-C6杂亚烷基,
其中每一X1独立为N或CR8,其中R8独立地是氢、卤素、硝基、氰基、CHF2、CF3、CO2H、氨基、C1-C6烷基、C1-C6杂烷基、C1-C6环烷基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、CON(R7)2、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;
X2是NR7、S或O;和
Z3是具有选自下面的分子式的生物还原基团:



条件是在分子式(I)中:
(ia)R1-R5的至少两个选自2-卤烷基、2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;
(ib)R1-R5的至少一个选自2-卤烷基、2-C1-C6烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;并且NR2R3或NR4R5至少一个是 或者
或者
(ic)NR2R3和NR4R5都是 和
和
(ii)R1-R5的至少一个是所述触发物T;及
(iii)不包括具有选自下列的分子式的化合物:


其中Ra是H、Br、NMe2、CN或CONH2,

其中R是H、Me或烯丙基;
3-(5-甲氧基-1-甲基-4,7-吲哚醌基)-甲基二[N-甲基-N-(2-溴乙基)]二氨基磷酸酯,
3-(5-甲氧基-1-甲基-4,7-吲哚醌基)甲基N,N-二(2-溴乙基)-二氨基磷酸酯,
2-(5-甲氧基-1-甲基-4,7-吲哚醌基)甲基二[N-甲基-N-(2-溴乙基)]二氨基磷酸酯,
2-(5-甲氧基-1-甲基-4,7-吲哚醌基)甲基N,N-二(2-氯乙基)二氨基磷酸酯,
2-(5-甲氧基-1-甲基-4,7-吲哚醌基)甲基N,N-二(2-溴乙基)二氨基磷酸酯,
3-(5-甲氧基-1-甲基-4,7-吲哚醌基)甲基N,N-二(2-溴乙基)二氨基磷酸酯,
2-(5-甲氧基-1-甲基-4,7-吲哚醌基)甲基二[N-甲基-N-(2-溴乙基)]二氨基磷酸酯,
2-(5-甲氧基-1-甲基-4,7-吲哚醌基)甲基N,N-二(2-氯乙基)二氨基磷酸酯,和
2-(5-甲氧基-1-甲基-4,7-吲哚醌基)甲基N,N-二(2-溴乙基)二氨基磷酸酯。
2.权利要求1所述的化合物,其中Y1和Y2是O。
3.前述权利要求的任一项所述的化合物,其中R1是T。
4.权利要求1所述的化合物,其具有分子式(II)或(III):


和其单独的异构体或异构体的外消旋或非外消旋混合物、生物电子等排物、药效团、药学上可接受的盐、溶剂化物、水合物或前体药物,
其中R2-R5的每一个独立是氢、羟基、氨基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;或者R2-R5的任意两个一起形成C3-C10杂环;每一Y1独立为S或O;
条件是在分子式(II)或(III)中:
(i)R2-R5的至少二个选自2-卤烷基、2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;
(ii)R2-R5的至少一个选自2-卤烷基、2-C1-C6烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;并且NR2R3和NR4R5的一个是 或者
或者
(iii)NR2R3和NR4R5都合起来是
5.权利要求4所述的化合物,其中L选自:
[C(Z1)2-Y3]-(C(=O)-O)-[C(Z1)2-Z2-Y4]-[C(Z1)2]z-[C(Z1)=C(Z1)]-;
[C(Z1)2-Y3]-[C(Z1)2-Z2-Y4]-[C(Z1)2]z-[C(Z1)=C(Z1)]-;
[C(Z1)2-Y3]-[C(Z1)2]z-[C(Z1)=C(Z1)]-;
[C(Z1)2-Y3]-[C(Z1)2]z-;
[C(Z1)2-Y3]-(C(=O)-O)-[C(Z1)2]z-[C(Z1)=C(Z1)]-;
[C(Z1)2-Y3]-(C(=O)-O)-[C(Z1)2]z-;
[C(z1)2-Y3]-(C(=O)-O)-[C(Z1)2]z-[C(z1)=C(Z1)]-;
[C(Z1)2-Z2-Y4]-[C(Z1)2]z-[C(Z1)=C(Z1)]-;
-[C(Z1)2]z-[C(Z1)=C(Z1)]-;
和-[C(Z1)2]z-。
6.权利要求5所述的化合物,其中L是-[C(Z1)2]z-。
7.权利要求5所述的化合物,其中每一-C(Z1)2-独立地是:-CH2-、-CHMe-、-CH(CN)-、-CH(CO2H)-、-CH(CONH2)-、-CH(CF3)-、-CH(CHF2)-、-C(Me)2-、-C(Et)2-、-CH(CH2NMe2)-、-CH(CH2NMe2)-、-C(CH2NMe2)2-或-C(CH2CO2H)2-。
8.权利要求5所述的化合物,其中-C(Z1)2-Y3-是:-CH2-O-、-CH2-S-、-CH2-NMe、-CH2-NH-、CH(Me)-O-、CH(Me)-S-、-CH(Me)-NMe-、-CH(Me)-NH-、-CMe2-NMe-、-CMe2-NMe-或-CMe2-NMe-。
9.权利要求5所述的化合物,其中-Z2-Y4-合起来选自:


10.权利要求5所述的化合物,其中-[C(Z1)=C(Z1)]-是:-CH=CH-、-C(CN)=CH-、-CH=C(CN)-、-C(Ar)=CH-、-CH=CAr-、-C(COAr)=CH-、-CH=C(COAr)-、-C(COR12)=CH-或-CH=C(COR12)-,其中Ar是芳基和R12独立为氢、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基或杂环基。
11.权利要求5所述的化合物,其中所述Z3选自:

12.权利要求4所述的化合物,具有分子式(IV)、(V)、(VI)或(VII):

和
其中每一R9独立为氢、芳基、杂芳基、C1-C4烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C3-C8环烷基、杂环基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基、C1-C6烷氧基羰基、C1-C6烷基氨基羰基、C1-C6二烷基氨基羰基或C1-C6烷氧基;
每一R10是氢、C1-C6烷基或杂烷基、C3-C8环烷基、杂环基、C1-C6烷氧基、芳基、杂芳基或者两个R10一起形成杂环;
每一R11独立为氢、芳基、杂芳基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基或C1-C6烷氧基;或者两个R11一起形成杂环;条件是当每一R11独立地是C1-C6烷基、C1-C6杂烷基时则R11不包括
X4是Cl、Br、烷基磺酰氧基、杂烷基磺酰氧基、芳基磺酰氧基或杂烷基磺酰氧基。
13.权利要求12所述的化合物,具有分子式(IV),其中每一R9独立为氢、甲基、乙基、丙基、异丙基或环丙基;并且N(R10)2选自NH2、NHMe、NMe2、NEt2、 NHOMe和NHOH。
NHOMe和NHOH。
14.权利要求12所述的化合物,具有分子式(V),其中每一R9独立为氢、甲基、乙基、丙基、异丙基或环丙基。
15.权利要求12所述的化合物,具有分子式(VI),其中每一R9独立为氢、甲基、乙基、丙基、异丙基或环丙基,并且R11为氢或甲基。
16.权利要求12所述的化合物,具有分子式(VII),其中每一R9独立为氢、甲基、乙基、丙基、异丙基或环丙基;并且每一R11独立为氢、甲基、乙基、丙基、异丙基、苄基、取代的甲基、环丙基、甲氧基和羟基;或者两个R11一起形成杂环。
17.权利要求1所述的化合物,其中T具有分子式:
18.权利要求1所述的化合物,其中T具有分子式:

其中每一Z1独立为H或C1-C6烷基。
19.权利要求1所述的化合物,其中T具有分子式:

其中每一Z1为氢或C1-C6烷基,R8是H、OH或-OP(=O)(OH)2。
20.权利要求12所述的化合物,具有分子式(VIII)、(IX)或(X):


21.权利要求12所述的化合物,具有分子式(XI)、(XII)、(XIII)、(XIV)或(XV):


其中每一R11独立为氢、甲基或取代的甲基、苄基、异丙基、环丙基、甲氧基和羟基;并且X4是Cl、Br、烷基磺酰氧基、杂烷基磺酰氧基、环烷基磺酰氧基、杂环烷基磺酰氧基、芳基磺酰氧基或杂芳基磺酰氧基;条件是:
(i)在(XII)、(XIV)或(XV)中,当X4是Cl或Br时,则R11不包括异丙基,和
(ii)分子式(XVIII)不包括其中Z3是下列的化合物:


22.权利要求15和权利要求21所述的化合物,具有分子式XII、XIV或XV,其中每一R11是氢。
23.权利要求4所述的化合物,具有分子式(XIX):
其中K是C1-C6亚烷基或C1-C6杂亚烷基。
24.权利要求23所述的化合物,其中K是(C(R12)2)e、CH2CH2(-X6-CH2CH2)f、或CH2(-X6-CH2)f,其中e是1-10,f是0-3,X6是O、S或NR12,其中每一R12独立为氢、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基或杂环基。
25.权利要求21所述的化合物,具有分子式(XVII)或(XVIII):

其中e是0-4,X4是Cl或Br、烷基磺酰氧基、杂烷基磺酰氧基、芳基磺酰氧基或杂芳基磺酰氧基。
26.权利要求25所述的化合物,具有分子式:

其中e是0-4,X4是Cl、Br、烷基磺酰氧基、杂烷基磺酰氧基、芳基磺酰氧基或杂芳基磺酰氧基。
27.权利要求4所述的化合物,具有分子式(XXII):

28.权利要求4所述的化合物,具有分子式(II),其中触发物T是-CH2-Z3或-CH(Z1)-Z3,其中Z1是C1-烷基,并且Z3是:

条件是在分子式(II)中:
(i)R2和R3的一个是H,并且R4和R5的一个是H;
(ii)R2和R3的一个是C1-烷基,并且R4和R5的一个是C1-烷基;或者
(iii)R2-R5的至少一个是羟基、氨基、C3-C8环烷基、杂环基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基、C1-C6酰基、C1-C6杂酰基、或芳酰基或杂芳酰基。
29.权利要求4所述的化合物,具有分子式(II),其中Z3是选自下列的生物还原基团:
或
条件是在分子式(I)中:
(i)R1-R5的至少一个选自2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基以及
R1-R5的至少一个选自2-卤烷基、2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;或者
(ii)R1-R5的至少一个选自2-卤烷基、2-C1-C6烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;并且NR2R3和NR4R5的至少一个是 或者
或者
(iii)NR2R3和NR4R5都合起来是
30.具有下列分子式的化合物:

和其单独的异构体或异构体的外消旋或非外消旋混合物、生物电子等排物、药效团、药学上可接受的盐、溶剂化物、水合物或前体药物,
其中Y1是O、S、NR6或NSO2R6;
Y2是O、S、NR6或NSO2R6,其中每一R6独立为(C1-C6)烷基、C1-C6杂烷基、芳基或杂芳基;
R1-R4和R1 *-R4 *的每一个独立选自氢、羟基、氨基、C1-C6烷基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;或者R1-R4或R1 *-R4 *的任意两个一起形成杂环;或者R1-R4和R1 *-R4 *的每一个独立为选自下列的触发物T:
-[C(Z1)2-Y3]v-[C(=O)-O]q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]z-[-C(Z1)=C(Z1)]g-Z3和
-[C(Z1)2-Y3]v-(S(=O)2)q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]z-[C(Z1)=C(Z1)]g-Z3;
其中每一个z、v、q、u和g独立为0或者1;
Y3是S、O或NR7,其中每一R7独立地是氢、羟基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基;
Y4是O、S或者-NR7-C(=O)-O-;
每一Z1和Z1独立为氢、卤素、C1-C6烷基、C1-C6杂烷基、芳基、杂芳基、C3-C8环烷基、杂环基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;
Z2是
其中每一X1独立为N或CR8,其中R8独立地是氢、卤素、硝基、氰基、CHF2、CF3、CO2H、C1-C6烷基、C1-C6杂烷基、C1-C6环烷基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基和CON(R7)2;
X2是NR7、S或O;和
Z3是具有下面分子式的生物还原基团:



每一个Z独立为C、S或P;每一个t独立为1或2;每一个r独立为0或1;
K是C1-C6亚烷基、C1-C6杂亚烷基、亚芳基或杂亚芳基;
条件是在分子式(XXI)中:
(i)R1-R4的至少一个和R1 *-R4 *的至少一个选自2-卤烷基、2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;
(ii)R1-R4和R1 *-R4 *的至少一个选自2-卤烷基、2-C1-C6烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;并且NR2R3和NR2 *R3 *的一个是 或者
或者
(iii)NR2R3和NR2 *R3 *都合起来是
31.分子式(XXI)的化合物:

和其单独的异构体或异构体的外消旋或非外消旋混合物、生物电子等排物、药效团、药学上可接受的盐、溶剂化物、水合物或前体药物,
其中,Y1是O、S、NR6或NSO2R6;
Y2是O、S、NR6或NSO2R6,其中每一R6独立为C1-C6烷基、C1-C6杂烷基、芳基或杂芳基;
R2-R5和R2 *-R5 *的每一个独立选自氢、羟基、C1-C6烷基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基;或者R2 *-R5 *的任意两个一起形成杂环;或者R2 *-R5 *的每一个独立为选自下列的触发物T:
-[C(Z1)2-Y3]v-[C(=O)-O]q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]z-[-C(Z1)=C(Z1)]g-Z3和-[C(Z1)2-Y3]v-(S(=O)2)q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]z-[C(Z1)=C(Z1)]g-Z3;其中每一个z、v、q、u和g独立为0或者1;
Y3是S、O或NR7,其中每一R7独立地是氢、羟基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基:
Y4是O、S或者-NR7-C(=O)-O-;
每一Z1和Z1独立为氢、卤素、C1-C6烷基、C1-C6杂烷基、芳基、杂芳基、C3-C8环烷基、杂环基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;
Z2是

其中每一X1独立为N或CR8,其中R8独立地是氢、卤素、硝基、氰基、CHF2、CF3、CO2H、C1-C6烷基、C1-C6杂烷基、C1-C6环烷基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基和CON(R7)2;
X2是NR7、S或O;和
Z3是具有下面分子式的生物还原基团:

或
L2是

其中Y4如上定义;
条件是在分子式(XXII)中:
(i)R2-R5的至少一个和R2 *-R5 *的至少一个选自2-卤烷基、2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;
(ii)R2-R5和R2 *-R5 *的至少一个选自2-卤烷基、2-C1-C6烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基和2-杂烷基磺酰氧基烷基;并且NR2R3和NR2 *R3 *的至少一个是 或者
或者
(iii)NR2R3和NR2 *R3 *都合起来是
32.分子式(XXVI)的化合物:

和其单独的异构体或异构体的外消旋或非外消旋混合物、生物电子等排物、药效团、药学上可接受的盐、溶剂化物、水合物或前体药物,
其中Y1是O、S、NR6或NSO2R6;
Y2是O、S、NR6或NSO2R6,其中每一R6独立为(C1-C6)烷基、C1-C6杂烷基、芳基或杂芳基;
R2-R5和R2 *-R5 *的每一个独立选自氢、羟基、C1-C6烷基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基;或者R2-R5或R2 *-R5 *的任意两个起形成杂环;
每个触发物T选自:
-[C(Z1)2-Y3]v-[C(=O)-O]q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]z-[-C(Z1)=C(Z1)]g-Z3和
-[C(Z1)2-Y3]v-(S(=O)2)q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]z-[C(Z1)=C(Z1)]g-Z3;
其中每一个z、v、q、u和g独立为0或者1;
Y3是S、O或NR7,其中每一R7独立地是氢、羟基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基;
Y4是O、S或者-NR7-C(=O)-O-;
每一Z1和Z1独立为氢、卤素、C1-C6烷基、C1-C6杂烷基、芳基、杂芳基、C3-C8环烷基、杂环基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;
Z2是

其中每一X1独立为N或CR8,其中R8独立地是氢、卤素、硝基、氰基、CHF2、CF3、CO2H、C1-C6烷基、C1-C6杂烷基、C1-C6环烷基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基和CON(R7)2;
X2是NR7、S或O;和
Z3是具有下面分子式的生物还原基团:



每一个Z独立为C、S或P;每一个t独立为1或2;每一个r独立为0或1;
K是C1-C6亚烷基、C1-C6杂亚烷基、亚芳基或杂亚芳基;
条件是在分子式(XXI)中:
(i)R1-R4和R1-R4 *的至少两个选自2-卤烷基、2-烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基或2-杂烷基磺酰氧基烷基;
(ii)R1-R4和R1-R4 *的至少一个选自2-卤烷基、2-C1-C6烷基磺酰氧基烷基、2-杂烷基磺酰氧基烷基、2-芳基磺酰氧基烷基或2-杂烷基磺酰氧基烷基;并且NR2R3和NR2 *R3 *的一个是或者
(iii)NR2R3和NR2 *R3 *都合起来是
33.权利要求1或权利要求15所述的化合物,其中触发物T选自:

34.权利要求33所述的化合物,其中Z1是氢、甲基或乙基;R7是甲基、三氟乙基、乙基、丙基和环己基;以及R8是OH或OP(=O)(OH)2;R9是氢或C1-C6烷基,并且每一X4是卤素或Rsu1S(=O)2O-。
35.权利要求34所述的化合物,其中R9是氢、甲基、乙基、异丙基或异丁基;并且X4是氯、溴或者甲磺酰氧基。
36.权利要求33所述的化合物,具有下列分子式:

其中T是L-Z3;
L是CH2、CHMe、CMe2、

和Z3是



37.权利要求1所述的化合物,具有选自下列的分子式:


38.权利要求1所述的化合物,具有分子式:

39.权利要求1所述的化合物,具有分子式:

40.药物制剂,其含有权利要求1-39任一项所述的化合物和药学可接受的赋形剂、载体或稀释剂。
41.治疗癌症的方法,包括将权利要求40所述的药物制剂施用给需要治疗的患者。
42.合成分子式Alk-触发物的化合物的方法,包括使Alk-H与触发物-OH、三取代膦和二烷基偶氮二羧酸酯反应,以产生分子式Alk-触发物的化合物,
其中Alk是:



其中每一R9独立为氢、芳基、杂芳基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基、C1-C6烷氧基羰基、C1-C6烷基氨基羰基、C1-C6二烷基氨基羰基或C1-C6烷氧基;
每一R10是氢、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C1-C6烷氧基、芳基、杂芳基或者两个R10一起形成杂环;
每一R11独立为氢、芳基、杂芳基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基或C1-C6烷氧基;或者两个R11一起形成杂环;条件是当每一R11独立是C1-C6烷基、C1-C6杂烷基时,则R11不包括和
X4是Cl、Br、烷基磺酰氧基、杂烷基磺酰氧基、芳基磺酰氧基或杂烷基磺酰氧基;
其中触发物是
[C(Z1)2-Y3]v-(C(=O)-O)q-[C(Z1)2-Z2-Y4]u-[C(Z1)2]z-[C(Z1)=C(Z1)]g-Z3;
其中每一个z、v、q、u和g独立为0或者1;
Y3是S、O或NR7,其中每一R7独立地是氢、羟基、C1-C6烷基、C1-C6杂烷基、C3-C8环烷基、杂环基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、杂芳基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;
Y4是O、S或者-NR7-C(=O)-O-;
每一Z1和Z1独立为氢、卤素、C1-C6烷基、C1-C6杂烷基、芳基、杂芳基、C3-C8环烷基、杂环基、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;
Z2是

其中每一X1独立为N或CR8,其中R8独立地是氢、卤素、硝基、氰基、CHF2、CF3、CO2H、氨基、C1-C6烷基、C1-C6杂烷基、C1-C6环烷基、C1-C6烷氧基、C1-C6烷基氨基、C1-C6二烷基氨基、芳基、CON(R7)2、C1-C6酰基、C1-C6杂酰基、芳酰基或杂芳酰基;
X2是NR7、S或O;和
Z3是具有下面分子式的生物还原基团:



Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201210251557.3A CN102746336B (zh) | 2005-06-29 | 2006-06-29 | 氨基磷酸酯烷化剂前体药物 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US69575505P | 2005-06-29 | 2005-06-29 | |
| US60/695,755 | 2005-06-29 | ||
| PCT/US2006/025881 WO2007002931A2 (en) | 2005-06-29 | 2006-06-29 | Phosphoramidate alkylator prodrugs |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201210251557.3A Division CN102746336B (zh) | 2005-06-29 | 2006-06-29 | 氨基磷酸酯烷化剂前体药物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN101501054A true CN101501054A (zh) | 2009-08-05 |
| CN101501054B CN101501054B (zh) | 2012-09-05 |
Family
ID=37596083
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201210251557.3A Active CN102746336B (zh) | 2005-06-29 | 2006-06-29 | 氨基磷酸酯烷化剂前体药物 |
| CN2006800300828A Active CN101501054B (zh) | 2005-06-29 | 2006-06-29 | 氨基磷酸酯烷化剂前体药物 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201210251557.3A Active CN102746336B (zh) | 2005-06-29 | 2006-06-29 | 氨基磷酸酯烷化剂前体药物 |
Country Status (24)
| Country | Link |
|---|---|
| US (4) | US8003625B2 (zh) |
| EP (2) | EP2336141B1 (zh) |
| JP (2) | JP5180824B2 (zh) |
| KR (1) | KR101198571B1 (zh) |
| CN (2) | CN102746336B (zh) |
| AU (2) | AU2006263433B8 (zh) |
| BR (1) | BRPI0612845A8 (zh) |
| CA (1) | CA2613312C (zh) |
| CY (1) | CY1113250T1 (zh) |
| DK (1) | DK1896040T3 (zh) |
| ES (2) | ES2579235T3 (zh) |
| HK (1) | HK1113754A1 (zh) |
| HR (1) | HRP20120803T1 (zh) |
| IL (1) | IL188236A (zh) |
| NO (1) | NO334420B1 (zh) |
| NZ (1) | NZ565378A (zh) |
| PL (1) | PL1896040T3 (zh) |
| PT (1) | PT1896040E (zh) |
| RS (1) | RS52505B (zh) |
| RU (1) | RU2414475C2 (zh) |
| SI (1) | SI1896040T1 (zh) |
| TW (1) | TWI384989B (zh) |
| WO (1) | WO2007002931A2 (zh) |
| ZA (1) | ZA200800314B (zh) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102850397A (zh) * | 2011-06-29 | 2013-01-02 | 北京大学 | 多靶点抗肿瘤化合物及其制备方法和应用 |
| CN102924507A (zh) * | 2011-11-10 | 2013-02-13 | 安徽四维药业有限公司 | 一种抗肿瘤化合物及其制备方法与应用、药物组合物 |
| CN104583224A (zh) * | 2012-07-03 | 2015-04-29 | 百时美施贵宝公司 | 制备用于治疗病毒感染的核苷化合物的富含非对映体的氨基磷酸酯衍生物的方法 |
| WO2015067215A1 (zh) * | 2013-11-07 | 2015-05-14 | 四川恒康发展有限责任公司 | 一种抗肿瘤前药及其激活剂、组合物和应用 |
| CN105037427A (zh) * | 2015-06-22 | 2015-11-11 | 石家庄学院 | 丹参酮iia亚乙基亚胺磷酸酯衍生物及其制备方法与应用 |
| CN105814032A (zh) * | 2013-10-10 | 2016-07-27 | 默克专利有限公司 | 通过Cα-取代的N-烷基-甘氨酸酯衍生物合成1-烷基-2-氨基-咪唑-5-羧酸酯 |
| CN107530556A (zh) * | 2015-03-10 | 2018-01-02 | 深圳艾衡昊医药科技有限公司 | Dna烷化剂 |
| CN109219610A (zh) * | 2016-04-04 | 2019-01-15 | 新泽西州立拉特格斯大学 | 拓扑异构酶毒物 |
| CN109705187A (zh) * | 2019-01-29 | 2019-05-03 | 石家庄学院 | 一种雷公藤红素衍生物及其制备方法与应用 |
| CN110746459A (zh) * | 2018-07-24 | 2020-02-04 | 上海喀露蓝科技有限公司 | 一种艾伏磷酰胺的制备方法 |
| WO2021083315A1 (zh) * | 2019-11-01 | 2021-05-06 | 深圳艾欣达伟医药科技有限公司 | 口服给药的固体剂型药物 |
| WO2023025291A1 (zh) | 2021-08-27 | 2023-03-02 | 深圳艾欣达伟医药科技有限公司 | 冻干制剂溶液及冻干制剂、方法和用途 |
| WO2024179467A1 (zh) * | 2023-02-27 | 2024-09-06 | 深圳艾欣达伟医药科技有限公司 | 溶液、冻干制剂、冻干制剂单位包装、注射液及注射液配制方法 |
Families Citing this family (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ565378A (en) | 2005-06-29 | 2011-03-31 | Threshold Pharmaceuticals Inc | Phosphoramidate alkylator prodrugs |
| JP5357049B2 (ja) * | 2006-12-26 | 2013-12-04 | スレッシュオールド ファーマシューティカルズ, インコーポレイテッド | 癌の治療のためのフォスフォルアミデートアルキル化剤プロドラッグ |
| US8765690B2 (en) | 2007-04-05 | 2014-07-01 | Threshold Pharmaceuticals, Inc. | Treatment of cancer with glufosfamide in patients not receiving insulin therapy |
| WO2009018163A1 (en) * | 2007-07-27 | 2009-02-05 | Threshold Pharmaceuticals, Inc. | Hypoxia activated prodrugs of anthracyclines |
| UA104579C2 (uk) | 2007-12-10 | 2014-02-25 | Байокрист Фармасьютикалз, Инк. | Спосіб лікування раку кровотворної системи із застосуванням фородезину у комбінації з ритуксимабом |
| CN102256966B (zh) | 2008-10-17 | 2016-02-10 | 白头生物医学研究所 | 可溶性mTOR复合物和其调节剂 |
| ES2884674T3 (es) | 2008-10-21 | 2021-12-10 | Immunogenesis Inc | Tratamiento del cáncer con el profármaco activado por hipoxia TH-302 en combinación con docetaxel o pemetrexed |
| RU2013102398A (ru) * | 2010-06-28 | 2014-08-10 | Тресхолд Фармасьютикалз, Инк. | Лечение рака крови |
| MX2012014428A (es) * | 2010-07-12 | 2013-03-05 | Threshold Pharmaceuticals Inc | Administracion de profarmacos activados por hipoxia y de agentes antiangiogenicos para el tratamiento de cancer. |
| CN103458896B (zh) * | 2011-04-01 | 2016-02-10 | 施瑞修德制药公司 | 用于治疗癌症的方法 |
| EP2696858A4 (en) * | 2011-04-15 | 2014-09-03 | Threshold Pharmaceuticals Inc | UNIT DOSE FOR ORAL ADMINISTRATION |
| US9402820B2 (en) * | 2011-04-22 | 2016-08-02 | The United States Of America As Represented By The Secretary, Department Of Health And Human Services | Use of pyruvate or succinate to enhance the efficacy of a hypoxia activated prodrug for the treatment of tumors |
| EP2793899A4 (en) * | 2011-12-22 | 2015-06-17 | Threshold Pharmaceuticals Inc | HYPOXIA ACTIVATED DRUGS AND MTOR INHIBITORS FOR TREATING CANCER |
| US9254299B2 (en) | 2011-12-22 | 2016-02-09 | Threshold Pharmaceuticals, Inc. | Administration of hypoxia activated prodrugs in combination with Chk1 inhibitors for treating cancer |
| US20160045522A1 (en) * | 2012-02-21 | 2016-02-18 | Threshold Pharmaceuticals, Inc. | Treatment of Cancer |
| WO2013154778A1 (en) | 2012-04-11 | 2013-10-17 | Dana-Farber Cancer Institute, Inc. | Host targeted inhibitors of dengue virus and other viruses |
| WO2014062856A1 (en) | 2012-10-16 | 2014-04-24 | Halozyme, Inc. | Hypoxia and hyaluronan and markers thereof for diagnosis and monitoring of diseases and conditions and related methods |
| US10000483B2 (en) | 2012-10-19 | 2018-06-19 | Dana-Farber Cancer Institute, Inc. | Bone marrow on X chromosome kinase (BMX) inhibitors and uses thereof |
| WO2014069063A1 (ja) | 2012-10-29 | 2014-05-08 | 京セラ株式会社 | 弾性表面波センサ |
| US20160158253A1 (en) | 2013-07-26 | 2016-06-09 | Threshold Pharmaceuticals, Inc. | Treatment of pancreatic cancer with a combination of a hypoxia-activated prodrug and a taxane |
| WO2015025283A2 (en) | 2013-08-20 | 2015-02-26 | Stichting Maastricht Radiation Oncology "Maastro-Clinic" | Dual action carbonic anhydrase inhibitors |
| US10071109B2 (en) | 2013-11-06 | 2018-09-11 | Molecular Templates, Inc. | Predictive biomarker for hypoxia-activated prodrug therapy |
| EP3105238A4 (en) | 2014-02-13 | 2017-11-08 | Ligand Pharmaceuticals, Inc. | Prodrug compounds and their uses |
| US10131683B2 (en) | 2014-07-17 | 2018-11-20 | Molecular Templates, Inc. | TH-302 solid forms and methods related thereto |
| CN108136214B (zh) | 2015-04-02 | 2020-08-28 | 深圳艾欣达伟医药科技有限公司 | 硝基苄基衍生物抗癌试剂 |
| PL412787A1 (pl) | 2015-06-22 | 2017-01-02 | Magdalena Król | Oparty na makrofagach celowany system dostarczania związków związanych z ferrytyną |
| ES2872533T3 (es) * | 2015-06-24 | 2021-11-02 | Immunogenesis Inc | Agentes alquilantes de ADN que contienen aziridina |
| CA2990696C (en) * | 2015-11-16 | 2024-01-02 | Obi Pharma, Inc. | (r)- and (s)-1-(3-(3-n,n-dimethylaminocarbonyl)phenoxyl-4-nitrophenyl)-1-ethyl-n,n'-bis (ethylene)phosphoramidate, compositions and methods for their use and preparation |
| EP3565806B1 (en) | 2017-01-06 | 2022-03-02 | Rivus Pharmaceuticals, Inc. | Novel phenyl derivatives |
| CN111788196A (zh) | 2018-01-09 | 2020-10-16 | 配体药物公司 | 缩醛化合物及其治疗用途 |
| CN112533918A (zh) | 2018-06-13 | 2021-03-19 | 安菲斯塔治疗有限责任公司 | 用于靶向Rpn11的双功能分子 |
| CA3103205A1 (en) | 2018-06-13 | 2019-12-19 | Amphista Therapeutics Ltd | Bifunctional molecules for targeting uchl5 |
| WO2019238886A1 (en) | 2018-06-13 | 2019-12-19 | University Of Dundee | Bifunctional molecules for targeting usp14 |
| EP3650046A1 (en) | 2018-11-08 | 2020-05-13 | Cellis AG | Mesenchymal stem cell based targeted active ingredient delivery system |
| US20230174624A1 (en) | 2020-03-18 | 2023-06-08 | Cellis Ag | Ferritin variants with increased stability, complexation ability and transferrin receptor affinity |
| WO2022195092A1 (en) | 2021-03-18 | 2022-09-22 | Cellis Ag | Ferritin variants with increased stability and complexation ability |
| CN118076359A (zh) | 2021-08-27 | 2024-05-24 | 深圳艾欣达伟医药科技有限公司 | 使用th-302治疗parp抑制剂耐药的患者 |
| WO2024056413A1 (en) | 2022-09-13 | 2024-03-21 | Cellis Ag | Isolated targeted delivery system for the treatment of glioma |
| WO2024112663A1 (en) | 2022-11-21 | 2024-05-30 | Rivus Pharmaceuticals, Inc. | Deuterium enriched phenyl derivatives for treating mitochondriarelated disorders or conditions |
| WO2024133541A1 (en) | 2022-12-20 | 2024-06-27 | Cellis Sp. Z O.O. [Ltd.] | Isolated targeted delivery system for the treatment of ovarian cancer |
Family Cites Families (87)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3652579A (en) | 1969-06-26 | 1972-03-28 | Hoffmann La Roche | 1-methyl-2-substituted 5-nitroimidazoles |
| DE2229248C3 (de) | 1971-07-30 | 1980-03-27 | Gruppo Lepetit S.P.A., Mailand (Italien) | 5-Iminomethyl-2-nitroimidazoI-Derivate und Verfahren zu deren Herstellung |
| US4921963A (en) | 1987-04-13 | 1990-05-01 | British Columbia Cancer Foundation | Platinum complexes with one radiosensitizing ligand |
| JPH0819111B2 (ja) | 1987-10-22 | 1996-02-28 | ポーラ化成工業株式会社 | 2―ニトロイミダゾール誘導体及びこれを有効成分とする放射線増感剤 |
| US5190929A (en) | 1988-05-25 | 1993-03-02 | Research Corporation Technologies, Inc. | Cyclophosphamide analogs useful as anti-tumor agents |
| US4908356A (en) | 1988-05-25 | 1990-03-13 | Research Corporation Technologies, Inc. | Aldophosphamide derivatives useful as antitumor agents |
| US5403932A (en) | 1988-05-25 | 1995-04-04 | Research Corporation Technologies, Inc. | Phosphoramidates useful as antitumor agents |
| DE3835772A1 (de) * | 1988-10-20 | 1990-04-26 | Deutsches Krebsforsch | Tumorhemmende saccharid-konjugate |
| JP2626727B2 (ja) | 1990-01-26 | 1997-07-02 | ポーラ化成工業株式会社 | 2―ニトロイミダゾール誘導体、その製造法及びこれを有効成分とする放射線増感剤 |
| HU9201539D0 (en) | 1990-09-11 | 1992-08-28 | Kortec Ag | Method and device for gasifying gasifiable materials and/or transforming gas as well as heat exchanger of high temperature for executing said method |
| US5233031A (en) | 1991-09-23 | 1993-08-03 | University Of Rochester | Phosphoramidate analogs of 2'-deoxyuridine |
| US5633158A (en) | 1991-10-23 | 1997-05-27 | Cancer Research Campaign Technology Limited | Bacterial nitroreductase for the reduction of CB 1954 and analogues thereof to a cytotoxic form |
| US5750782A (en) | 1992-11-27 | 1998-05-12 | Cancer Research Campaign Technology Limited | Nitroaniline derivatives and their use as anti-tumour agents |
| US5306727A (en) | 1993-04-30 | 1994-04-26 | Research Corporation Technologies, Inc. | Phosphoramidates useful as antitumor agents |
| JPH08510469A (ja) | 1993-05-25 | 1996-11-05 | オークランド ユニサービシーズ リミテッド | ニトロベンジルマスタード四級塩およびその低酸素選択性細胞毒剤としての利用 |
| EP0648503B1 (en) | 1993-09-22 | 2000-07-26 | Hoechst Aktiengesellschaft | Pro-prodrugs, their production and use |
| JPH07309761A (ja) * | 1994-05-20 | 1995-11-28 | Kyowa Hakko Kogyo Co Ltd | デュオカルマイシン誘導体の安定化法 |
| US5659061A (en) * | 1995-04-20 | 1997-08-19 | Drug Innovation & Design, Inc. | Tumor protease activated prodrugs of phosphoramide mustard analogs with toxification and detoxification functionalities |
| GB9516943D0 (en) | 1995-08-18 | 1995-10-18 | Cancer Soc Auckland Div Nz Inc | Novel cyclopropylindoles and their secoprecursors,and their use as prodrugs |
| US6218519B1 (en) | 1996-04-12 | 2001-04-17 | Pro-Neuron, Inc. | Compounds and methods for the selective treatment of cancer and bacterial infections |
| US5728271A (en) | 1996-05-20 | 1998-03-17 | Rti Resource Transforms International Ltd. | Energy efficient liquefaction of biomaterials by thermolysis |
| JP4371433B2 (ja) | 1996-05-20 | 2009-11-25 | ダイナモーティブ・エナジー・システムズ・コーポレイション | 熱分解によるバイオマテリアルのエネルギ効率的な液化 |
| US6130237A (en) | 1996-09-12 | 2000-10-10 | Cancer Research Campaign Technology Limited | Condensed N-aclyindoles as antitumor agents |
| US6251933B1 (en) | 1996-12-13 | 2001-06-26 | The Cancer Research Campaign Technology Limited | Seco precursors of cyclopropylindolines and their use as prodrugs |
| DE19720312A1 (de) | 1997-05-15 | 1998-11-19 | Hoechst Ag | Zubereitung mit erhöhter in vivo Verträglichkeit |
| US6017948A (en) | 1998-10-30 | 2000-01-25 | Supergen, Inc. | Water-miscible pharmaceutical compositions |
| US7914994B2 (en) | 1998-12-24 | 2011-03-29 | Cepheid | Method for separating an analyte from a sample |
| US6240925B1 (en) | 1999-03-23 | 2001-06-05 | Cynosure, Inc. | Photothermal vascular targeting with bioreductive agents |
| GB9909612D0 (en) | 1999-04-26 | 1999-06-23 | Cancer Res Campaign Tech | N-protected amines and their use as prodrugs |
| WO2000071134A1 (en) | 1999-05-24 | 2000-11-30 | Southern Research Institute | Isophosphoramide mustard analogs and use thereof |
| CA2377232A1 (en) | 1999-06-11 | 2000-12-21 | Henceforth Hibernia, Inc. | Prophylactic, therapeutic and industrial antioxidant compositions enhanced with stabilized atomic hydrogen/free electrons and methods to prepare and use such compositions |
| AU5935600A (en) | 1999-07-14 | 2001-01-30 | Richard F. Borch | Phosphoramide compounds |
| JP2004538240A (ja) | 2000-03-31 | 2004-12-24 | パーデュー・リサーチ・ファウンデーション | ホスホラミデートおよびそのための方法 |
| WO2002076499A2 (en) | 2001-03-23 | 2002-10-03 | Aphton Corporation | Combination treatment of pancreatic cancer |
| EP1243276A1 (en) | 2001-03-23 | 2002-09-25 | Franciscus Marinus Hendrikus De Groot | Elongated and multiple spacers containing activatible prodrugs |
| US6506739B1 (en) * | 2001-05-01 | 2003-01-14 | Telik, Inc. | Bis-(N,N'-bis-(2-haloethyl)amino)phosphoramidates as antitumor agents |
| AU2002303929B9 (en) | 2001-05-31 | 2007-01-25 | E. R. Squibb & Sons, L.L.C. | Cytotoxins, prodrugs, linkers and stabilizers useful therefor |
| FR2825926A1 (fr) | 2001-06-14 | 2002-12-20 | Sod Conseils Rech Applic | Derives d'imidazoles modulant les canaux sodiques |
| US7091186B2 (en) | 2001-09-24 | 2006-08-15 | Seattle Genetics, Inc. | p-Amidobenzylethers in drug delivery agents |
| US20040009667A1 (en) | 2002-02-07 | 2004-01-15 | Etsuo Iijima | Etching method |
| KR20030067275A (ko) | 2002-02-07 | 2003-08-14 | 주식회사 하이폭시 | 니트로이미다졸 및 위상 이성질화 효소 저해제를유효성분으로 포함하는 항암제 |
| HUE026218T2 (en) | 2002-02-21 | 2016-05-30 | Inst Virology | MN / CA IX-Specific Monoclonal Antibodies and Methods for MN / CA IX Deficient Mice |
| JP4227771B2 (ja) | 2002-07-17 | 2009-02-18 | 三菱重工業株式会社 | バイオマスのガス化方法 |
| US6833390B2 (en) | 2002-07-22 | 2004-12-21 | Bayer Polymers Llc | Process for preparing closed-cell water-blown rigid polyurethane foams having improved mechanical properties |
| DE10237931A1 (de) | 2002-08-14 | 2004-02-26 | Endress + Hauser Gmbh + Co. Kg | Vorrichtung zur Überwachung eines vorbestimmten Füllstands eines Messmediums in einem Behälter |
| EA009337B1 (ru) | 2003-01-09 | 2007-12-28 | Пфайзер Инк. | Трициклические соединения, представляющие собой ингибиторы протеинкиназ, для увеличения эффективности противоопухолевых агентов и лучевой терапии |
| GB0306908D0 (en) | 2003-03-26 | 2003-04-30 | Angiogene Pharm Ltd | Bioreductively activated stilbene prodrugs |
| GB0306907D0 (en) | 2003-03-26 | 2003-04-30 | Angiogene Pharm Ltd | Boireductively-activated prodrugs |
| ZA200507752B (en) | 2003-03-28 | 2007-01-31 | Threshold Pharmaceuticals Inc | Compositions and methods for treating cancer |
| US6855695B2 (en) | 2003-06-13 | 2005-02-15 | Vion Pharmaceuticals, Inc. | Water-soluble SHPs as novel alkylating agents |
| US7340059B2 (en) | 2003-06-17 | 2008-03-04 | Intel Corporation | Programmable scrambler and De-scrambler for digital telephony equipment |
| JP2005041733A (ja) | 2003-07-28 | 2005-02-17 | National Institute Of Advanced Industrial & Technology | バイオマスによる水素製造法 |
| WO2005076888A2 (en) | 2004-02-06 | 2005-08-25 | Threshold Pharmaceuticals, Inc. | Anti-cancer therapies |
| US7432106B2 (en) | 2004-03-24 | 2008-10-07 | Applied Biosystems Inc. | Liquid processing device including gas trap, and system and method |
| JP4426621B2 (ja) | 2004-05-04 | 2010-03-03 | バイエル ヘルスケア エルエルシー | Mn/ca ix/ca9及び腎臓癌の予後診断 |
| IL162713A (en) | 2004-06-24 | 2011-04-28 | Desalitech Ltd | Apparatus and methods for continuous desalination in closed circuit without containers |
| WO2006015263A2 (en) | 2004-07-29 | 2006-02-09 | Threshold Pharmaceuticals, Inc. | Lonidamine analogs |
| WO2006015191A2 (en) | 2004-07-29 | 2006-02-09 | Threshold Pharmaceuticals, Inc. | Multicyclic lonidamine analogs |
| EP1819338A4 (en) | 2004-11-22 | 2009-11-04 | Threshold Pharmaceuticals Inc | ANTICANCER TUBULIN-BINDING AGENTS AND THEIR PRODUCTS |
| US20070117784A1 (en) | 2005-03-04 | 2007-05-24 | Novacea, Inc. | Treatment of hyperproliferative diseases with anthraquinones |
| NZ565378A (en) | 2005-06-29 | 2011-03-31 | Threshold Pharmaceuticals Inc | Phosphoramidate alkylator prodrugs |
| US20090136521A1 (en) | 2005-10-03 | 2009-05-28 | Genetix Pharmaceuticals , Inc. | Method for Selectively Depleting Hypoxic Cells |
| WO2008011588A2 (en) | 2006-07-20 | 2008-01-24 | Threshold Pharmaceuticals, Inc. | Glycoconjugates of phosphoramidate alkylators for treatment of cancer |
| WO2008033041A1 (en) | 2006-09-11 | 2008-03-20 | Auckland Uniservices Limited | Cancer treatment |
| JP2008069017A (ja) | 2006-09-12 | 2008-03-27 | Matsushita Electric Ind Co Ltd | 水素製造方法 |
| US7807454B2 (en) | 2006-10-18 | 2010-10-05 | The Regents Of The University Of California | Microfluidic magnetophoretic device and methods for using the same |
| WO2008076826A1 (en) | 2006-12-13 | 2008-06-26 | Threshold Pharmaceuticals, Inc. | Pyrophosphoramide alkylators |
| JP5357049B2 (ja) | 2006-12-26 | 2013-12-04 | スレッシュオールド ファーマシューティカルズ, インコーポレイテッド | 癌の治療のためのフォスフォルアミデートアルキル化剤プロドラッグ |
| JP5174411B2 (ja) | 2007-09-28 | 2013-04-03 | 独立行政法人石油天然ガス・金属鉱物資源機構 | 管式リフォーマーの有効熱利用方法 |
| US8216607B2 (en) | 2008-03-17 | 2012-07-10 | Roger Williams Hospital | Combination of ceramide and gemcitabine for inducing cell death and uses thereof in treating cancer |
| CN102026634B (zh) | 2008-04-10 | 2014-01-22 | 弗吉尼亚州立邦联大学 | 诱导肿瘤缺氧以治疗癌症 |
| CA2725754C (en) | 2008-06-11 | 2017-05-23 | Hazel Joan Dyke | Diazacarbazoles and methods of use |
| JP4665021B2 (ja) | 2008-09-03 | 2011-04-06 | 三菱重工業株式会社 | バイオマスのガス化方法 |
| ES2884674T3 (es) | 2008-10-21 | 2021-12-10 | Immunogenesis Inc | Tratamiento del cáncer con el profármaco activado por hipoxia TH-302 en combinación con docetaxel o pemetrexed |
| WO2010108091A2 (en) | 2009-03-20 | 2010-09-23 | Pbs Biotech, Inc. | Automatable aseptic sample withdrawal system |
| WO2010129622A1 (en) | 2009-05-04 | 2010-11-11 | Macusight, Inc. | Mtor pathway inhibitors for treating ocular disorders |
| JP2013510180A (ja) | 2009-11-06 | 2013-03-21 | インフィニティ ファーマスーティカルズ、インク. | ヘッジホッグ経路阻害剤の経口製剤 |
| JP5659536B2 (ja) | 2010-03-31 | 2015-01-28 | 新日鐵住金株式会社 | タール含有ガスの改質用触媒及びその製造方法、並びにタール含有ガスの改質方法 |
| RU2013102398A (ru) | 2010-06-28 | 2014-08-10 | Тресхолд Фармасьютикалз, Инк. | Лечение рака крови |
| MX2012014428A (es) | 2010-07-12 | 2013-03-05 | Threshold Pharmaceuticals Inc | Administracion de profarmacos activados por hipoxia y de agentes antiangiogenicos para el tratamiento de cancer. |
| US10357577B2 (en) | 2010-07-16 | 2019-07-23 | Auckland Uniservices Limited | Bacterial nitroreductase enzymes and methods relating thereto |
| CN103458896B (zh) | 2011-04-01 | 2016-02-10 | 施瑞修德制药公司 | 用于治疗癌症的方法 |
| EP2696858A4 (en) | 2011-04-15 | 2014-09-03 | Threshold Pharmaceuticals Inc | UNIT DOSE FOR ORAL ADMINISTRATION |
| JP5805989B2 (ja) | 2011-04-26 | 2015-11-10 | 大塚電子株式会社 | 電気泳動移動度測定用セル並びにそれを用いた測定装置及び測定方法 |
| JP5995873B2 (ja) | 2011-12-20 | 2016-09-21 | 学校法人長崎総合科学大学 | 合成ガスの生成方法及び製造装置、並びに、液体燃料の合成方法及び合成装置 |
| EP2793899A4 (en) | 2011-12-22 | 2015-06-17 | Threshold Pharmaceuticals Inc | HYPOXIA ACTIVATED DRUGS AND MTOR INHIBITORS FOR TREATING CANCER |
| WO2013116385A1 (en) | 2012-01-31 | 2013-08-08 | Threshold Pharmaceuticals, Inc. | Predictive biomarker for hypoxia-activated prodrug therapy |
-
2006
- 2006-06-29 NZ NZ565378A patent/NZ565378A/en not_active IP Right Cessation
- 2006-06-29 WO PCT/US2006/025881 patent/WO2007002931A2/en active Application Filing
- 2006-06-29 PL PL06786161T patent/PL1896040T3/pl unknown
- 2006-06-29 CN CN201210251557.3A patent/CN102746336B/zh active Active
- 2006-06-29 CN CN2006800300828A patent/CN101501054B/zh active Active
- 2006-06-29 KR KR1020087002436A patent/KR101198571B1/ko active IP Right Grant
- 2006-06-29 EP EP11157963.7A patent/EP2336141B1/en active Active
- 2006-06-29 TW TW095123590A patent/TWI384989B/zh active
- 2006-06-29 EP EP06786161A patent/EP1896040B1/en active Active
- 2006-06-29 BR BRPI0612845A patent/BRPI0612845A8/pt active Search and Examination
- 2006-06-29 JP JP2008519666A patent/JP5180824B2/ja active Active
- 2006-06-29 SI SI200631455T patent/SI1896040T1/sl unknown
- 2006-06-29 AU AU2006263433A patent/AU2006263433B8/en active Active
- 2006-06-29 RS RS20120449A patent/RS52505B/en unknown
- 2006-06-29 CA CA2613312A patent/CA2613312C/en active Active
- 2006-06-29 US US11/993,822 patent/US8003625B2/en active Active
- 2006-06-29 PT PT06786161T patent/PT1896040E/pt unknown
- 2006-06-29 ES ES11157963.7T patent/ES2579235T3/es active Active
- 2006-06-29 DK DK06786161.7T patent/DK1896040T3/da active
- 2006-06-29 ZA ZA200800314A patent/ZA200800314B/xx unknown
- 2006-06-29 RU RU2008103195/04A patent/RU2414475C2/ru not_active IP Right Cessation
- 2006-06-29 ES ES06786161T patent/ES2389532T3/es active Active
-
2007
- 2007-12-19 IL IL188236A patent/IL188236A/en active IP Right Grant
-
2008
- 2008-01-23 NO NO20080442A patent/NO334420B1/no not_active IP Right Cessation
- 2008-08-26 HK HK08109465.9A patent/HK1113754A1/xx unknown
-
2011
- 2011-05-05 AU AU2011202075A patent/AU2011202075B8/en active Active
- 2011-06-17 US US13/163,303 patent/US8507464B2/en active Active
-
2012
- 2012-05-16 JP JP2012112647A patent/JP5781007B2/ja active Active
- 2012-10-05 HR HRP20120803TT patent/HRP20120803T1/hr unknown
- 2012-10-23 CY CY20121100999T patent/CY1113250T1/el unknown
-
2013
- 2013-07-12 US US13/941,261 patent/US8664204B2/en active Active
- 2013-12-10 US US14/102,213 patent/US9226932B2/en active Active
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102850397B (zh) * | 2011-06-29 | 2015-02-18 | 北京大学 | 多靶点抗肿瘤化合物及其制备方法和应用 |
| CN102850397A (zh) * | 2011-06-29 | 2013-01-02 | 北京大学 | 多靶点抗肿瘤化合物及其制备方法和应用 |
| CN102924507A (zh) * | 2011-11-10 | 2013-02-13 | 安徽四维药业有限公司 | 一种抗肿瘤化合物及其制备方法与应用、药物组合物 |
| CN104583224A (zh) * | 2012-07-03 | 2015-04-29 | 百时美施贵宝公司 | 制备用于治疗病毒感染的核苷化合物的富含非对映体的氨基磷酸酯衍生物的方法 |
| CN105814032A (zh) * | 2013-10-10 | 2016-07-27 | 默克专利有限公司 | 通过Cα-取代的N-烷基-甘氨酸酯衍生物合成1-烷基-2-氨基-咪唑-5-羧酸酯 |
| WO2015067215A1 (zh) * | 2013-11-07 | 2015-05-14 | 四川恒康发展有限责任公司 | 一种抗肿瘤前药及其激活剂、组合物和应用 |
| CN104628772A (zh) * | 2013-11-07 | 2015-05-20 | 四川恒康发展有限责任公司 | 一种抗肿瘤前药及其激活剂、组合物和应用 |
| CN107530556A (zh) * | 2015-03-10 | 2018-01-02 | 深圳艾衡昊医药科技有限公司 | Dna烷化剂 |
| CN107530556B (zh) * | 2015-03-10 | 2020-04-10 | 深圳艾欣达伟医药科技有限公司 | Dna烷化剂 |
| CN105037427A (zh) * | 2015-06-22 | 2015-11-11 | 石家庄学院 | 丹参酮iia亚乙基亚胺磷酸酯衍生物及其制备方法与应用 |
| CN109219610A (zh) * | 2016-04-04 | 2019-01-15 | 新泽西州立拉特格斯大学 | 拓扑异构酶毒物 |
| CN110746459A (zh) * | 2018-07-24 | 2020-02-04 | 上海喀露蓝科技有限公司 | 一种艾伏磷酰胺的制备方法 |
| CN109705187A (zh) * | 2019-01-29 | 2019-05-03 | 石家庄学院 | 一种雷公藤红素衍生物及其制备方法与应用 |
| WO2021083315A1 (zh) * | 2019-11-01 | 2021-05-06 | 深圳艾欣达伟医药科技有限公司 | 口服给药的固体剂型药物 |
| CN112755001A (zh) * | 2019-11-01 | 2021-05-07 | 深圳艾欣达伟医药科技有限公司 | 口服给药的固体剂型药物 |
| CN112755001B (zh) * | 2019-11-01 | 2022-04-12 | 深圳艾欣达伟医药科技有限公司 | 口服给药的固体剂型药物 |
| WO2023025291A1 (zh) | 2021-08-27 | 2023-03-02 | 深圳艾欣达伟医药科技有限公司 | 冻干制剂溶液及冻干制剂、方法和用途 |
| WO2024179467A1 (zh) * | 2023-02-27 | 2024-09-06 | 深圳艾欣达伟医药科技有限公司 | 溶液、冻干制剂、冻干制剂单位包装、注射液及注射液配制方法 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101501054B (zh) | 氨基磷酸酯烷化剂前体药物 | |
| JP6798890B2 (ja) | グルタミナーゼ阻害剤との併用療法 | |
| Weiss et al. | Palladium-mediated dealkylation of N-propargyl-floxuridine as a bioorthogonal oxygen-independent prodrug strategy | |
| Babichev et al. | PI3K/AKT/mTOR inhibition in combination with doxorubicin is an effective therapy for leiomyosarcoma | |
| US20040192656A1 (en) | Methods and compositions for the treatment of human and animal cancers | |
| CN110087649A (zh) | 用辛伐他汀和化疗药物与七甲川花菁染料偶联体提高肿瘤对激素拮抗剂和药物的敏感性 | |
| CN102161679A (zh) | 治疗癌症的组合物和方法 | |
| US11248016B2 (en) | Glycolipids and pharmaceutical compositions thereof for use in therapy | |
| US20080214576A1 (en) | Topoisomerase Inhibitors and Prodrugs | |
| CN105246483A (zh) | 用于γ-谷氨酰循环调节的方法和组合物 | |
| WO2019107341A1 (ja) | オゾンナノバブルの抗がん剤 | |
| WO2014205179A1 (en) | Ribonucleotide reductase inhibitors sensitize tumor cells to dna damaging agents | |
| Chen et al. | Targeted Cancer Therapies, from Small Molecules to Antibodies |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CP03 | Change of name, title or address | ||
| CP03 | Change of name, title or address |
Address after: Texas, USA Patentee after: Molecular Template Co.,Ltd. Address before: California, USA Patentee before: Threshold Pharmaceuticals, Inc. |
|
| TR01 | Transfer of patent right | ||
| TR01 | Transfer of patent right |
Effective date of registration: 20231114 Address after: Texas, USA Patentee after: Immune Response Co.,Ltd. Address before: Texas, USA Patentee before: Molecular Template Co.,Ltd. |