WO2009018163A1 - Hypoxia activated prodrugs of anthracyclines - Google Patents
Hypoxia activated prodrugs of anthracyclines Download PDFInfo
- Publication number
- WO2009018163A1 WO2009018163A1 PCT/US2008/071228 US2008071228W WO2009018163A1 WO 2009018163 A1 WO2009018163 A1 WO 2009018163A1 US 2008071228 W US2008071228 W US 2008071228W WO 2009018163 A1 WO2009018163 A1 WO 2009018163A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- group
- alkylene
- heteroalkylene
- Prior art date
Links
- 0 C*1C([N+]([O-])=O)=NC=C1 Chemical compound C*1C([N+]([O-])=O)=NC=C1 0.000 description 4
- HBMINVNVQUDERA-UHFFFAOYSA-N C[n]1c([N+]([O-])=O)ncc1 Chemical compound C[n]1c([N+]([O-])=O)ncc1 HBMINVNVQUDERA-UHFFFAOYSA-N 0.000 description 2
- VOBFYAXUVBEUGM-UHFFFAOYSA-N Cc1ccc([N+]([O-])=O)[o]1 Chemical compound Cc1ccc([N+]([O-])=O)[o]1 VOBFYAXUVBEUGM-UHFFFAOYSA-N 0.000 description 2
- YZEUHQHUFTYLPH-UHFFFAOYSA-N [O-][N+](c1ncc[nH]1)=O Chemical compound [O-][N+](c1ncc[nH]1)=O YZEUHQHUFTYLPH-UHFFFAOYSA-N 0.000 description 2
- ZPTVNYMJQHSSEA-UHFFFAOYSA-N Cc(cc1)ccc1[N+]([O-])=O Chemical compound Cc(cc1)ccc1[N+]([O-])=O ZPTVNYMJQHSSEA-UHFFFAOYSA-N 0.000 description 1
- PLAZTCDQAHEYBI-UHFFFAOYSA-N Cc(cccc1)c1[N+]([O-])=O Chemical compound Cc(cccc1)c1[N+]([O-])=O PLAZTCDQAHEYBI-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/91—Nitro radicals
Definitions
- the present invention provides compositions and methods for the treatment of cancer, and generally relates to the fields of medicinal chemistry, medicine, pharmacology, molecular biology, and biology.
- Hypoxia activated prodrugs of anticancer agents are useful for tumor therapy.
- a HAP compound contains a bioreductive group, a linker, and an anticancer agent and is less cytotoxic than the corresponding anticancer agent under normoxic conditions or normoxia, such as those existing in a normal cell. Under hypoxia, however, the bioreductive group present in the HAP compound is reduced, and the cytotoxic anticancer agent is generated and/or released. In hypoxic regions such as those existing in solid tumors, a HAP compound generates and/or releases a cytotoxin and kills cancer cells selectively in and around the hypoxic tumor zone.
- HAP and/or bioreducible compounds are described, for example, in PCT Pat.
- HAP and/or bioreducible compounds currently, or previously tested, in the clinic include TH 302, AQ4N, and PR 104 (see Lalani et al, Clin. Cancer Res., 13:2216 (2007) and Patterson et al., Clin. Cancer Res., 13:3922 (2007)), each incorporated herein by reference, and WO 07/002931, supra). None of these compounds contain a 2-nitroimidazole moiety covalently bonded to an anticancer agent at the 1-N-position of the 2-nitroimidazole moiety.
- HAP compounds for the treatment of cancer, including HAP compounds with improved hypoxic compared to normoxic cytotoxicity.
- the present invention meets such needs.
- the present invention relates to hypoxia activated prodrug compounds, or HAP compounds, of anticancer agents comprising one or more bioreductive Hyp moieties covalently bonded to an anticancer agent, Q, via a linker moiety, L, wherein after hypoxic activation, the bioreduced Hyp moiety remains attached to the rest of the molecule, and wherein the anticancer agent, Q, is selected from the class of anthracycline anticancer agents, including but not limited to Daunorubicin, Doxorubicin, Epirubicin and Idarubicin:
- the present invention provides HAP compounds having structures of the formulas Hyp-L-Q and (Hyp-L) 2 -Q.
- Suitable Hyp moieties useful in the compounds of the present invention include, but are not limited to, nitro imidazoles, nitrofurans, nitrothiophenes, and nitrobenzenes.
- the nitroimidazole is a 2-nitroimidazole.
- the 2- nitroimidazole is covalently bonded to the anticancer agent via the L moiety wherein the L moiety is covalently bonded to the "1-N" position of the 2-nitroimidazole:
- Suitable L moieties useful in the compounds of the present invention include but are not limited to Ci-Ci 0 alkylene and Ci-Ci 0 heteroalkylene.
- the HAP compound of the present invention have structures of the formulas Hyp-L-Q and (Hyp-L) 2 -Q wherein each Hyp-L moiety is covalently bonded to the amino nitrogen atom in the anthracycline.
- Suitable L moieties useful in the present invention include Ci-C] 0 alkylene and Ci-Ci 0 heteroalkylene moieties.
- Other suitable L moieties useful in the present invention include Ci alkylene and Ci heteroalkylene moieties.
- Other suitable L moieties useful in the present invention include C 2 alkylene and C 2 heteroalkylene moieties.
- Other suitable L moieties useful in the present invention include C 3 alkylene and C 3 heteroalkylene moieties.
- suitable L moieties useful in the present invention include C 4 alkylene and C 4 heteroalkylene moieties. Other suitable L moieties useful in the present invention include C 5 alkylene and C 5 heteroalkylene moieties. Other suitable L moieties useful in the present invention include C 6 alkylene and C 6 heteroalkylene moieties. Other suitable L moieties useful in the present invention include C 7 alkylene and C 7 heteroalkylene moieties. Other suitable L moieties useful in the present invention include C 8 alkylene and C 8 heteroalkylene moieties. Other suitable L moieties useful in the present invention include C 9 alkylene and C 9 heteroalkylene moieties. Other suitable L moieties useful in the present invention include Ci 0 alkylene and Ci 0 heteroalkylene moieties.
- a suitable Hyp moiety useful in the present invention has a structure of the formula:
- Hyp moieties having a structure of formula:
- Suitable substituents include those substituents that increase the reactivity, DNA-, RNA-, or protein-cross linked lifetime, and/or tumor specific cytotoxicity of the HAP compounds of the present invention.
- the present invention provides a HAP compound having a structure of the formula:
- Ri is selected from the group consisting of hydrogen, CpC 6 alkyl, Ci-C 6 heteroalkyl
- L is selected from the group consisting of Ci-Ci 0 alkylene and Ci-Ci 0 heteroalkylene
- R 3 is selected from H and OH
- Hyp has a structure of the formula:
- Ri is hydrogen
- L is selected from Ci alkylene and Ci heteroalkylene moieties.
- L is selected from C 2 alkylene and C 2 heteroalkylene moieties.
- L is selected from C 3 alkylene and C 3 heteroalkylene moieties.
- L is selected from C 4 alkylene and C 4 heteroalkylene moieties.
- L is selected from C 5 alkylene and C 5 heteroalkylene moieties.
- L is selected from C 6 alkylene and C 6 heteroalkylene moieties.
- L is selected from C 7 alkylene and C 7 heteroalkylene moieties.
- L is selected from C 8 alkylene and C 8 heteroalkylene moieties. In certain embodiments, L is selected from C 9 alkylene and C 9 heteroalkylene moieties. In certain embodiments, L is selected from C 10 alkylene and Ci 0 heteroalkylene moieties.
- the anthracyclines useful in the HAP compounds of the present invention contain one or more amino groups, hi one embodiment, the amino group is a primary or secondary amino group, -NRi, as disclosed herein. A variety of functional groups are useful as the secondary group on the amino group in the anthracyclines useful in the present invention.
- the secondary group on the amino group, -NRi is selected from the group consisting of C]-C 6 alkyl, Ci-C 6 heteroalkyl, Ci-C 6 alkenyl, Ci-C 6 alkynyl, acyl, and heteroarylalkylenyloxycarbonyl.
- the present invention provides a HAP compound having a structure of the formula:
- R 3 is selected from H and OH, each L is independently selected from the group consisting of Ci-Ci 0 alkylene and Ci-Ci 0 heteroalkylene, and Hyp has a structure of the formula:
- the present invention provides compounds having a structure of the formula (Hyp-L)-Q-(CO 2 CR 4 R 5 -Brg) wherein Q is an anthracycline; L is selected from the group consisting of Ci-Ci 0 alkylene and Ci-Ci 0 heteroalkylene; Hyp is a moiety having a structure of the formula:
- each Of R 4 and R 5 is selected independently from the group consisting of hydrogen, Ci-C 6 alkyl Ci-C 6 heteroalkyl, C 3 -C 8 cycloalkyl, heterocyclyl, aryl and heteroaryl; Brg has a structure of the formula:
- R 6 is selected from the group consisting of hydrogen, Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl aryl, heteroaryl, and halo; and R 7 is Ci-C 6 alkyl, CpC 6 heteroalkyl, C 3 -C 8 cycloalkyl, heterocyclyl, aryl and heteroaryl; and the Hyp-L and CO 2 CR 4 R 5 -Brg , moieties are covalently bonded to an amino nitrogen atom in the anthracycline. In another embodiment, the Hyp-L and CO 2 CR 4 R 5 -Brg moieties are covalently bonded to the same amino nitrogen atom in the anthracycline.
- the anthracyclines useful in the present invention are selected from daunorubicin and doxorubicin.
- the present invention provides the HAP compounds of the present invention in substantially pure forms.
- the present invention provides methods of synthesizing HAP compounds of the present invention.
- the present invention provides pharmaceutically acceptable formulations each such formulation comprising a HAP compound of the present invention and pharmaceutically acceptable carriers, diluents, and/or excipients.
- the present invention provides a method of treating cancer and other hyperproliferative diseases comprising administering a therapeutically effective amount of a HAP compound of the present invention to a patient in need of such treatment.
- Section I provides useful definitions
- Section II describes the HAP compounds of the present invention and methods of their synthesis
- Section III describes therapies provided by the present invention
- Section IV provides illustrative methods for synthesizing and testing HAP compounds of the present invention. This detailed description is organized into sections only for the convenience of the reader, and disclosure found in any section is applicable to disclosure elsewhere in the specification.
- V/VOP refers to a position on a moiety which is covalently bonded to the rest of the molecule via a single bond.
- Acid salt refers to a compound of the present invention that contains relatively basic functionalities, to which a sufficient amount of an acid is added to form a salt.
- pharmaceutically acceptable acid addition salts include those derived from inorganic acids like hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric, hydriodic, or phosphorous acids and the like, as well as the salts derived from relatively nontoxic organic acids like acetic, propionic, isobutyric, malonic, benzoic, succinic, suberic, fumaric, mandelic, phthalic, benzenesulfonic, p-tolylsulfonic, citric, tartaric, methanesulfonic, and the like.
- salts of amino acids such as arginate and the like, and salts of organic acids like glucuronic or galactunoric acids and the like (see, e.g., Berge, S. M. et al ., “Pharmaceutical Salts,” Journal of Pharmaceutical Science, 66:1-19, 1977).
- alkenylene refers to a linear or branched, unsaturated, divalent, substituted or unsubstituted, hydrocarbon radical wherein the unsaturation is a carbon-carbon double bond and wherein there is at least one carbon atom covalently bonded to each side of the double bond.
- C 4 -C 6 alkenylene refers to a linear or branched, unsaturated, divalent, substituted or unsubstituted, hydrocarbon radical having 4-6 carbon atoms wherein the unsaturation is a carbon-carbon double bond and wherein there is at least one carbon atom covalently bonded to each side of the double bond.
- Alkoxy refers to a substituted or unsubstituted alkyl group covalently bonded to an oxygen atom.
- Ci-C 6 alkoxy refers to a substituted or unsubstituted alkyl group of 1-6 carbon atoms covalently bonded to an oxygen atom.
- a Ci-C 6 alkoxy group has the general structure -0-(Ci-C 6 ) alkyl.
- Ci-C 6 alkoxy groups include, for example, methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, sec-butoxy, tert-butoxy, n-pentoxy, 2-pentoxy, 3-pentoxy, isopentoxy, neopentoxy, hexoxy, 2-hexoxy, 3-hexoxy, and 3-methylpentoxy.
- Alkylenyloxy refers to a linear saturated divalent substituted or unsubstituted hydrocarbon radical or a branched saturated divalent hydrocarbon radical covalently bonded to an oxygen atom.
- a alkylenyloxy group has the general structure -0-(Ci-C 6 ) alkyl.
- CpC 6 alkoxy groups include, for example, methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, sec-butoxy, tert-butoxy, n-pentoxy, 2-pentoxy, 3-pentoxy, isopentoxy, neopentoxy, hexoxy, 2-hexoxy, 3-hexoxy, and 3-methylpentoxy.
- Alkylenyloxycarbonyl refers to an alkylenyloxy group covalently bonded to a carbonyl.
- Alkyl refers to a substituted or unsubstituted straight or branched chain alkyl group.
- Ci-C 6 alkyl refers to a substituted or unsubstituted straight or branched chain alkyl groups having 1-6 carbon atoms.
- Ci-C 6 alkyl groups include, for example, methyl, ethyl, propyl, isopropyl, n-butyl, sec-butyl, tert-butyl, pentyl, 2-pentyl, isopentyl, neopentyl, hexyl, 2-hexyl, 3-hexyl and 3-methylpentyl.
- a Ci-C 6 alkyl substituent may be covalently bonded to an atom within a molecule of interest via any chemically suitable portion of the C]-C 6 alkyl group.
- alkenyl by itself or as part of another substituent refers to a straight or branched chain, which may be mono or polyunsaturated, having the number of carbon atoms designated.
- ' ⁇ -Cgalkenyl means an alkenyl radical having from 2, 3, 4, 5 or
- substituted alkenyl has the same meaning with respect to alkenyl groups that substituted alkyl groups had with respect to unsubstituted alkyl groups.
- a substituted alkenyl group includes alkenyl groups in which a non-carbon or non-hydrogen atom is bonded to a carbon double bonded to another carbon and those in which one of the non-carbon or non- hydrogen atoms is bonded to a carbon not involved in a double bond to another carbon. Each site of unsaturation may be either cis or trans configuration about the double bond(s).
- Alkynyl by itself or as part of another substituent, means a straight or branched chain hydrocarbon radical, which may be mono- or polyunsaturated, having the number of carbon ato ms designated.
- C2-C6 alkynyl means an alkynyl radial having from 2 to 6 carbon atoms that is derived by the removal of one hydrogen atom from a single carbon atom of a parent alkane .
- Unsubstituted alkynyl refers to straight and branched chain groups such as those described with respect to unsubstituted alkyl groups as defined above, except that at least one triple bond exists between two carbon atoms. Examples include, but are not limited to ethynyl e.g. -C C(H), 1- propynyl e.g. -CC(CH 3 ), -
- substituted alkynyl has the same meaning with respect to alkynyl groups that substituted alkyl groups had with respect to unsubstituted alkyl groups.
- a substituted alkynyl group includes alkynyl groups in which a non-carbon or non-hydrogen atom is bonded to a carbon triple bonded to another carbon and those in which a non-carbon or non-hydrogen atom is bonded to a carbon not involved in a triple bond to another carbon.
- Alkylamino refers to a substituted or unsubstituted alkyl group covalently bonded to an -NH- moiety.
- C 1 -C 6 alkylamino refers to a substituted or unsubstituted alkyl group of 1-6 carbon atoms covalently bonded to an -NH- moiety.
- a Ci-C 6 alkylamino group has the general structure -NH-(Ci-C 6 ) alkyl.
- a di(Ci-C 6 ) alkylamino group has the general structure -N-[(Ci-C 6 ) alkyl] 2 .
- Ci-C 6 alkylamino groups include, for example, methylamino, ethylamino, propylamino and butylamino.
- Alkylene refers to a linear saturated divalent substituted or unsubstituted hydrocarbon radical or a branched saturated divalent hydrocarbon radical.
- Ci-Cio alkylene refers to a corresponding alkylene group having 1-10 carbon atoms.
- Ci-C 6 alkylene groups include, for example, methylene, ethylene, propylene, butylene, 2-methylpropylene, pentylene.
- Alkyl ether refers to a moiety with an oxygen atom and carbon atoms positioned such that at least one carbon atom is located on either side of the oxygen atom.
- C 2 -C 6 alkyl ether refers to a moiety with an oxygen atom and 2-6 carbon atoms positioned such that at least one carbon atom is located on either side of the oxygen atom.
- Alkynylene refers to a linear or branched, unsaturated, divalent, substituted or unsubstituted, hydrocarbon radical wherein the unsaturation is a carbon-carbon triple bond and wherein there is at least one carbon atom covalently bonded to each side of the triple bond.
- C 4 -C 6 alkynylene refers to a linear or branched, unsaturated, divalent, substituted or unsubstituted, hydrocarbon radical having 4-6 carbon atoms wherein the unsaturation is a carbon-carbon triple bond and wherein there is at least one carbon atom covalently bonded to each side of the triple bond.
- Amino refers to a monovalent radical -NR a R b or divalent radical -NR a -.
- alkylamino refers to the group -NR a R b where R a is alkyl and R b is H or alkyl.
- arylamino refers to the group -NR a R b where R a is aryl and R b is hydrogen, alkyl, aryl, or heterocyclyl.
- (alkyl)(aryl)amino refers to the group -NR a R b where R a is alkyl and R b is aryl.
- the alkyl portions can be the same or different and can also be combined to form a 3-7-membered ring with the nitrogen atom to which each is attached.
- a group represented as NR a R b is meant to include piperidinyl, pyrrolidinyl, morpholinyl, azetidinyl and the like.
- Anthracycline refers to anthracenedione anticancer agents and includes anthracycline analogs and anthracycline derivatives.
- Anthracyclines include, for example, aclarubicin, daunorubicin, doxorubicin, epirubicin, idarubicin, and pirarubicin.
- Anthracycline analogs are described, for example, in the references, Henry, Cancer Chemotherapy, A CS Symposium Series, 15-57 (1976); Nagy et al., Proc. Natl. Acad. Sci.
- Aryl refers to a substituted or unsubstituted cyclic moiety that includes one or more monocyclic or fused ring aromatic systems. Such moieties include any moiety that has one or more monocyclic or bicyclic fused ring aromatic systems, including but not limited to phenyl and naphthyl.
- Bioreductive group refers to a substituted or unsubstituted nitroaryl, nitroheteroaryl, indoloquinonyl, or a naphtoquinonyl moiety that can undergo reduction. Bioreductive groups are described for example in the U.S. Pat. Nos. 5,750,782; 5,780,585; 5,872,129; 6,251,933; 5,306,727; 5,403,932; 5,190,929; and 6,656,926; PCT Pat. Appl. Pub. Nos. WO 00/64864; 04/85361; 04/85421; 04/87075; 06/57946; and 07/02931; U.S. Pat. Appl. Pub.
- Cycloalkyl or “carbocycle” refers to, unless otherwise stated, cyclic versions of “alkyl”, “alkenyl” and “alkynyl” in which all ring atoms are carbon .
- Cycloalkyl or “carbocycle” refers to a mono- or polycyclic group.
- polycyclic refers herein to fused and non-fused alkyl cyclic structures.
- Cycloalkyl or “carbocycle” may form a bridged ring or a spiro ring.
- the cycloalkyl group may have one or more double or triple bond(s).
- cycloalkenyl refers to a cycloalkyl group that has at least one site of alkenyl unsaturation between the ring vertices.
- cycloalkynyl refers to a cycloalkyl group that has at least one site of alkynyl unsaturation between the ring vertices.
- cycloalkyl when used in combination with “alkyl”, as in C ⁇ .gcycloalky ⁇ .galkylene-, the cycloalkyl portion is meant to have the stated number of carbon atoms (e.g., from three to eight carbon atoms), while the alkylene portion has from one to eight carbon atoms. Typical cycloalkyl substituents have from 3 to 8 ring atoms. Examples of cycloalkyl include cyclopentyl, cyclohexyl, 1-cyclohexenyl, 3-cyclohexenyl, cycloheptyl, and the like.
- Halogen refers to by themselves or as part of another substituent, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom. Additionally, terms such as “haloalkyl,” are meant to include alkyl in which one or more hydrogen is substituted with halogen atoms which can be the same or different, in a number ranging from one up to the maximum number of halogens permitted e.g., for alkyl (2m'+l), where m' is the total number of carbon atoms in the alkyl group.
- haloCl-8alkyl is meant to include trifluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3-bromopropyl, and the like.
- perhaloalkyl means, unless otherwise stated, alkyl substituted with (2m'+l) halogen atoms, where m' is the total number of carbon atoms in the alkyl group.
- perhaloCl- ⁇ alkyl is meant to include trifluoromethyl, pentachloroethyl, 1,1,1- trifluoro-2-bromo-2-chloroethyl, and the like.
- haloalkoxy refers to an alkoxy radical substituted with one or more halogen atoms.
- Halide refers to the acid or anionic form of a halo group.
- Heteroalkyl means an alkyl radical as defined herein with one, two or three substituents independently selected from cyano, -ORw, -NRxRy, and -S(O)nRz (where n is an integer from 0 to 2 ), with the understanding that the point of attachment of the heteroalkyl radical is through a carbon atom of the heteroalkyl radical.
- Rw is hydrogen, alkyl, cycloalkyl, cycloalkyl-alkyl, aryl, araalkyl, alkoxycarbonyl, aryloxycarbonyl, carboxamido, or mono- or di-alkylcarbamoyl.
- Rx is hydrogen, alkyl, cycloalkyl, cycloalkyl-alkyl, aryl or araalkyl.
- Ry is hydrogen, alkyl, cycloalkyl, cycloalkyl-alkyl, aryl, araalkyl, alkoxycarbonyl, aryloxycarbonyl, carboxamido, mono- or di-alkylcarbamoyl or alkylsulfonyl.
- Rz is hydrogen (provided that n is 0), alkyl, cycloalkyl, cycloalkyl-alkyl, aryl, araalkyl, amino, mono- alkylamino, di-alkylamino, or hydroxyalkyl.
- Rw, Rx ,Ry, and Rz can be further substituted by amino, fluorine, alkylamino, di-alkylamino, OH or alkoxy.
- the prefix indicating the number of carbon atoms refers to the total number of carbon atoms in the portion of the heteroalkyl group exclusive of the cyano, -ORw, -NRxRy, or - S(O) n Rz portions.
- Heteroaryl refers to a substituted or unsubstituted monocyclic aromatic system having 5 or 6 ring atoms, or a fused ring bicyclic aromatic system having 8-20 atoms, in which the ring atoms are C, O, S, SO, SO 2 , or N, and at least one of the ring atoms is a heteroatom, i.e., O, S, SO, SO 2 , or N.
- Heteroaryl groups include, for example, acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothio-furanyl, benzothiophenyl, benzoxazolyl, benzothiazolyl, benzotriazolyl, benzotetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, NH-carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, dithiazinyl, furanyl, furazanyl, imidazolidinyl, imidazolinyl, imidazolyl, indazolyl, indolenyl, indolinyl, indolizinyl, indolyl, isobenzofuranyl, isochromanyl, isoindazolyl, isoindolinyl
- Heteroalkyl refers to an alkyl radical as defined herein with one, two or three substituents, independently selected from cyano, -ORw, -NRxRy, and -S(O)nRz (where n is an integer from 0 to 2 ), with the understanding that the point of attachment of the heteroalkyl radical is through a carbon atom of the heteroalkyl radical.
- Rw is hydrogen, alkyl, cycloalkyl, cycloalkyl-alkyl, aryl, araalkyl, alkoxycarbonyl, aryloxycarbonyl, carboxamido, or mono- or di-alkylcarbamoyl.
- Rx is hydrogen, alkyl, cycloalkyl, cycloalkyl- alkyl, aryl or araalkyl.
- Ry is hydrogen, alkyl, cycloalkyl, cycloalkyl-alkyl, aryl, araalkyl, alkoxycarbonyl, aryloxycarbonyl, carboxamido, mono- or di-alkylcarbamoyl or alkylsulfonyl.
- Rz is hydrogen (provided that n is 0), alkyl, cycloalkyl, cycloalkyl-alkyl, aryl, araalkyl, amino, mono-alkylamino, di-alkylamino, or hydroxyalkyl.
- Rw, Rx, Ry, and Rz can be further substituted by amino, fluorine, alkylamino, di-alkylamino, OH or alkoxy.
- the prefix indicating the number of carbon atoms refers to the total number of carbon atoms in the portion of the heteroalkyl group exclusive of the cyano, -ORw, -NRxRy, or -S(O) n Rz portions.
- Heterocyclyl refers to a monocyclic or fused ring multicyclic cycloalkyl group at least a portion of which is not aromatic and in which one or more of the carbon atoms in the ring system is replaced by a heteroatom selected from O, S, SO, SO 2 , P, or N.
- heterocyclyl groups include but are not limited to imidazolinyl, morpholinyl, piperidinyl, piperidin-2-onyl, piperazinyl, pyrrolidinyl, pyrrolidine-2-onyl, tetrahydrofuranyl, and tetrahydroimidazo [4,5-c] pyridinyl.
- Heteroalkylene refers to a Ci-C 6 alkylene as defined above wherein 1-3 carbon atoms in the hydrocarbon radical or a branched saturated divalent hydrocarbon radical is replaced with a heteroatom.
- Ci-Ci 0 heteroalkylene refers to a heteroalkylene group wherein 1 -5 carbon atoms in the hydrocarbon radical or a branched saturated divalent hydrocarbon radical is replaced with a heteroatom.
- Ci-C 6 heteroalkylene groups include, for example, -CH 2 CH 2 -O-CH 2 CH 2 - and -CH 2 CH 2 -S-CH 2 CH 2 -.
- Heteroarylalkylenyloxycarbonyl refers to an heteroarylalkylenyloxy group covalently bonded to a carbonyl.
- Leaving group refers to a moiety that can be replaced by a nucleophile. Leaving groups include, for example, halo and sulfonate.
- Nirate refers to the acid or anionic form of a nitro group.
- Niro refers to -NO 2 .
- “Pharmaceutically acceptable salts” refers to salts of the active compounds which are prepared with relatively nontoxic acids or bases, depending on the particular substituents found on the compounds described herein. When compounds of the present invention contain relatively acidic functionalities, base addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired base, either neat or in a suitable inert solvent. Examples of salts derived from pharmaceutically-acceptable inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic, manganous, potassium, sodium, zinc and the like.
- Salts derived from pharmaceutically-acceptable organic bases include salts of primary, secondary and tertiary amines, including substituted amines, cyclic amines, naturally-occurring amines and the like, such as arginine, betaine, caffeine, choline, N,N'-dibenzylethylenediamine, diethylamine, 2- diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N- ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like.
- acid addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired acid, either neat or in a suitable inert solvent.
- pharmaceutically acceptable acid addition salts include those derived from inorganic acids like hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric, hydriodic, or phosphorous acids and the like, as well as the salts derived from relatively nontoxic organic acids like acetic, propionic, isobutyric, malonic, benzoic, succinic, suberic, fumaric, mandelic, phthalic, benzenesulfonic, p- tolylsulfonic, citric, tartaric, methanesulfonic, and the like.
- salts of amino acids such as arginate and the like, and salts of organic acids like glucuronic or galactunoric acids and the like ⁇ see, e.g., Berge, S.M. et al ., "Pharmaceutical Salts," Journal of Pharmaceutical Science, 66:1-19, 1977).
- Certain specific compounds of the present invention contain both basic and acidic functionalities that allow the compounds to be converted into either base or acid addition salts.
- the neutral forms of the compounds may be regenerated by contacting the salt with a base or acid and isolating the parent compound in the conventional manner.
- the parent form of the compound differs from the various salt forms in certain physical properties, such as solubility in polar solvents, but otherwise the salts are equivalent to the parent form of the compound for the purposes of the present invention.
- Reducing agent refers to a compound which donates electrons in a reduction- oxidation reaction. In some embodiments the donation of electrons may be in the form of an atom or molecule, such as a hydrogen atom or molecule. Reducing agents include, for example, hydride reducing agents.
- Hydride reducing agents include, for example, NaBH 3 CN and NaBH(OAc) 3 Reducing agents are described, for example, in the references, or are prepared by methods known to those skilled in the art following procedures set forth in references such as Fieser and Fieser's Reagents for Organic Synthesis; Wiley & Sons: New York, 1967-2004, Volumes 1-22; Rodd's Chemistry of Carbon Compounds, Elsevier Science Publishers, 1989, Volumes 1-5 and Supplementals; and Organic Reactions, Wiley & Sons: New York, 2005, Volumes 1-6 J.
- C]-C 6 or Ci- 6 is meant to include all possible embodiments that have one fewer atom.
- Non-limiting examples include Ci -6 , C 2-6 , C 3-6 , C 4-6 , C 5-6 and the like.
- C 2 -Ci 0 or C 2-10 is meant to include all possible embodiments that have one fewer atom.
- Non- limiting examples include C 2- I 0 , C 3 -I 0 , C 4-I o, C 5-I o, C 6- I 0 , and the like.
- each of the terms herein is meant to include both “unsubstituted” and optionally “substituted” forms of the indicated radical, unless otherwise indicated.
- each radical is substituted with 0, 1, 2, 3, 4, or 5 substituents, unless otherwise indicated. Examples of substituents for each type of radical are provided below.
- Substituted refers to a group as defined herein in which one or more bonds to a carbon(s) or hydrogen(s) are replaced by a bond to non-hydrogen and non-carbon atom "substituents” such as, but not limited to, a halogen atom such as F, Cl, Br, and I; an oxygen atom in groups such as hydroxyl groups, alkoxy groups, aryloxy, and acyloxy groups; a sulfur atom in groups such as thiol groups, alkyl and aryl sulfide groups, sulfone groups, sulfonyl groups, and sulfoxide groups; a nitrogen atom in groups such as amino, alkylamines, dialkylamines, arylamines, alkylarylamines, diarylamines, alkoxyamino, hydroxyamino, acylamino, sulfonylamino, N-oxides, imides, and en
- Substituents also include groups in which one or more bonds to a carbon(s) or hydrogen(s) atom is replaced by a higher-order bond (e.g., a double- or triple-bond) to a heteroatom such as oxygen in oxo, acyl, amido, alkoxycarbonyl, aminocarbonyl, carboxyl, and ester groups; nitrogen in groups such as imines, oximes, hydrazones, and nitriles.
- a higher-order bond e.g., a double- or triple-bond
- nitrogen in groups such as imines, oximes, hydrazones, and nitriles.
- Substituents further include groups in which one or more bonds to a carbon(s) or hydrogen(s) atoms is replaced by a bond to a cycloalkyl, heterocyclyl, aryl, and heteroaryl groups.
- Representative “substituents” include, among others, groups in which one or more bonds to a carbon or hydrogen atom is/are replaced by one or more bonds to fluoro, chloro, or bromo group.
- Another representative “substituent” is the trifluoromethyl group and other groups that contain the trifluoromethyl group.
- substituted alkyl group contains a hydroxyl, alkoxy, or aryloxy group.
- substituted alkyl group contains a hydroxyl, alkoxy, or aryloxy group.
- substituted alkyl group includes alkyl groups that have an amine, or a substituted or unsubstituted alkylamine, dialkylamine, arylamine, (alkyl)(aryl)amine, diarylamine, heterocyclylamine, diheterocyclylamine, (alkyl)(heterocyclyl)amine, or (aryl)(heterocyclyl)amine group.
- substituted substituents include those in which one or more bonds to a carbon(s) or hydrogen(s) atoms is replaced by a bond to an alkyl, cycloalkyl, aryl, heteroaryl, or heterocyclyl group.
- the herein-defined groups may include prefixes and/or suffixes that are commonly used in the art to create additional well-recognized substituent groups.
- "alkylamino" refers to a group of the formula -NR a R b .
- R a and R b are each independently selected from H, alkyl, alkoxy, thioalkoxy, cycloalkyl, aryl, heteroaryl, or heterocyclyl or are optionally joined together with the atom(s) to which they are attached to form a cyclic group.
- R a and R b are attached to the same nitrogen atom, they can be combined with the nitrogen atom to form a 5-, 6- or 7-membered ring.
- -NR a R b is meant to include 1-pyrrolidinyl and 4-morpholinyl.
- R c , R d , R e and R f are each independently selected from alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, aryl, heteroaryl, heterocyclyl or alkylenearyl, as defined herein.
- a particular radical will have 0, 1, 2 or 3 substituents, with those groups having two or fewer substituents being preferred in the present invention. More preferably, a radical will be unsubstituted or monosubstituted. Most preferably, a radical will be unsubstituted.
- substituteduents refers to an atom or group, including, for example, amino, Ci-C 6 alkylamino or di(Ci-C 6 )alkylamino, Ci-C 6 alkoxy, C]-C 6 alkylthio, aryl, -COOH, -CONH 2 , cyano, ethenyl, ethynyl, halo, heteroaryl, hydroxy, mono- or di(Ci-C 6 )alkylcarboxamido, mono or di(Ci-C 6 )alkylsulfonamido, nitro, -OSO 2 -Ry, and -SO 2 NH 2 .
- Two of the "substituents" on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -T-C(O)-(CH 2 )q-U-, wherein T and U are independently -NH-, -0-, -CH2- or a single bond, and q is O, 1 or 2.
- two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -A-(CH2)r-B-, wherein A and B are independently -CH2-, -0-, -NH-, -S-, -S(O)-, -S(0)2-, -S(O) 2NR a - or a single bond, and r is 1, 2 or 3.
- One of the single bonds of the new ring so formed may optionally be replaced with a double bond.
- two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -(CH2)s-X-(CH2)t-, where s and t are independently integers of from 0 to 3, and X is -O-, -NR a -, -S-, -S(O)-, -S(O)2-, or -S(O) 2NR a -.
- the substituent R a in -NR a - and -S(O)2NR a - is selected from hydrogen or unsubstituted Cl-6alkyl. Otherwise, R' is as defined above.
- Sulfonate refers to a moiety having the structure of the formula -OSO 2 R y wherein R y is selected from the group consisting of Ci-C 6 alkyl, Ci-C 6 heteroalkyl, C 3 -Cg cycloalkyl, heterocyclyl, aryl, and heteroaryl.
- administering or "administration of a drug to a patient (and grammatical equivalents of this phrase) refers to direct administration, which may be administration to a patient by a medical professional or may be self-administration, and/or indirect administration, which may be the act of prescribing a drug.
- direct administration which may be administration to a patient by a medical professional or may be self-administration
- indirect administration which may be the act of prescribing a drug.
- a physician who instructs a patient to self-administer a drug and/or provides a patient with a prescription for a drug is administering the drug to the patient.
- Anoxia or "anoxic condition” refers to an oxygen level that is zero or immeasurably low.
- Hypoxia or “hypoxic condition” refers to an oxygen level lower than that observed under normoxia, and includes anoxia.
- Normaloxia or “normoxic condition” refers to an oxygen level observed in normally oxygenated cells and tissue, and in cells and tissue in contact with air.
- “Pharmaceutically acceptable carrier, excipient, or diluent” refers to a carrier, excipient, or diluent that is useful in preparing a pharmaceutical composition that is generally safe, non-toxic and neither biologically nor otherwise undesirable, and includes a carrier, excipient, or diluent that is acceptable for human pharmaceutical use as well as veterinary use.
- a “pharmaceutically acceptable carrier, excipient, or diluent” includes both one and more than one such carrier, excipient, or diluent.
- “Prodrug” refers to a compound that, after administration, is metabolized or otherwise converted to an active or more active form with respect to at least one property.
- a prodrug, relative to the drug it corresponds to, is modified chemically in a manner that renders it, relative to the drug, less active or inactive, but the chemical modification is such that the corresponding drug is generated by metabolic or other biological processes after the prodrug is administered.
- a prodrug may have, relative to the active drug, altered metabolic stability or transport characteristics, fewer side effects or lower toxicity, or improved flavor (for example, see the reference Nogrady, Medicinal Chemistry A Biochemical Approach, Oxford University Press, New York, 388-392 (1985), incorporated herein by reference).
- a prodrug does not have to be synthesized using the drug as an intermediate.
- Reduction of a symptom or symptoms refers to decreasing the severity or frequency of the symptom(s), or elimination of the symptom(s).
- Therapeutically effective amount of a substance refers to an amount of the substance that, when administered to a patient with cancer or another hyperproliferative disease, will have the intended therapeutic effect, e.g., alleviation, amelioration, palliation or elimination of one or more manifestations of cancer or another hyperproliferative disease in the patient.
- the full therapeutic effect does not necessarily occur by administration of one dose, and may occur only after administration of a series of doses.
- a therapeutically effective amount may be administered in one or more administrations.
- Treating or “treatment of a condition or patient refers to taking steps to obtain beneficial or desired results, including clinical results.
- beneficial or desired clinical results include, but are not limited to, alleviation or amelioration of one or more symptoms of cancer or another hyperproliferative disease; diminishn ⁇ ent of extent of disease; delay or slowing of disease progression; amelioration, palliation, or stabilization of the disease state, or other beneficial results.
- the present invention provides hypoxia activated prodrugs, or HAP compounds, of anticancer agents comprising one or more bioreductive Hyp moieties covalently bonded to the anticancer agent N, via a linker moiety L, wherein after hypoxic activation, the bioreduced Hyp moiety remains attached to the rest of the molecule, and the anticancer agent is selected from anthracyclines.
- the present invention provides a HAP compound having a structure of the formula selected from Hyp-L-Q and (Hyp-L) 2 -Q.
- Suitable Hyp moieties useful in the HAP compounds of the present invention include, but are not limited to, nitroimidazoles, nitrofurans, nitrothiophenes, and nitrobenzenes.
- the nitroimidazole is a 2-nitroimidazole.
- the 2-nitroimidazole is covalently bonded to the anticancer agent by the L moiety wherein the L moiety is covalently bonded to the "1-N" position of the 2-nitroimidazole.
- the imidazole moiety of the 2-nitroimidazole is suitably substituted.
- Suitable substituents include halo, Ci-C 6 alkyl, Ci-C 6 heteroalkyl, Ci-C 6 alkenyl, Cj-C 6 alkynyl, aryl, heteroaryl, and acyl.
- the imidazole moiety of the 2-nitroimidazole is suitably substituted at the 4-position of the imidazole moiety.
- each L is independently selected from the group consisting of Ci-Cio alkylene and Ci-Ci 0 heteroalkylene. In another embodiment, each L is independently selected from the group consisting Of C 2 -Ci 0 alkylene and C 2 -Ci 0 heteroalkylene. In another embodiment, each L is independently selected from the group consisting Of Ci-C 6 alkylene and Ci-C 6 heteroalkylene moieties. In another embodiment, each L is independently selected from the group consisting OfC 7 -Ci 0 alkylene C 7 -Ci 0 and heteroalkylene moieties.
- each L is independently selected from the group consisting of C 2 , C 3 , C 4 , C 5 C 6 , C 7 , C 8 , C 9 and Ci 0 alkylene and C 2 , C 3 , C 4 , C 5 C 6 , C 7 , C 8 , C 9 and C 10 heteroalkylene moieties, hi another embodiment, the L moiety is a C 5 alkylene moiety. In another embodiment, the L moiety is a C 5 heteroalkylene moiety.
- Other suitable L moieties useful in the HAP compounds of the present invention are described, for example, in U.S. Pat. Appl. Pub. Nos. US 2006/0258656 and U.S. Pat. Appl. No. 60/941,753 (each of which is incorporated herein by reference).
- the carbon atoms and/or heteroatoms within the Ci-Ci 0 alkylene and Ci-Ci 0 heteroalkylene L-moieties can be substituted with one or more substituents.
- Suitably substituted heteroatom moieties within a Ci-Ci 0 heteroalkylene L-moiety include -SO-, -SO 2 -, and -N(COR 2 )- wherein R 2 is selected from the group consisting of hydrogen, CpC 6 alkyl, Ci-C 6 heteroalkyl, C 3 -C 8 cycloalkyl, and heterocyclyl.
- the C 1 -Ci 0 heteroalkylene chain of the L-moiety suitably includes an ester, amide (-CONR 2 -), sulfonamide, urea, and a carbamate moiety wherein R 2 is defined as above.
- each L is independently selected from the group consisting of Ci-Ci 0 alkylene and C 1 -C 1O heteroalkylene moieties wherein adjacent carbon and/or heteroatoms are part of an aryl, a heteroaryl, a C 3 -C 8 cycloalkyl, and a heterocyclyl moiety.
- each L is independently selected from the group consisting OfC 4 -C 6 alkenylene and C 4 -C 6 alkynylene moieties wherein one or more carbon atoms in each of the C 4 -C 6 chain can be suitably substituted with one or more heteroatoms.
- the present invention provides HAP compounds having a structure of the formula Hyp-L-Q wherein L has a structure of the formula selected from the group consisting of: wherein Xi is selected from the group consisting of O, S, SO, SO 2 , CO, CF 2 , and -N(COR 2 )- wherein R 2 is defined as above.
- each L is independently selected from the group consisting of
- each L is independently selected from the group consisting of:
- R wherein is selected from the group consisting of an aryl, a heteroaryl, a C 3 -C 8 cycloalkyl, and a heterocyclyl moiety.
- the L moiety is covalently bonded to Hyp on the left side, and to Q on the right side of the L moieties, and wherein v/ww indicates the point of attachment of L to Hyp and Q.
- L has a structure of the formula -(CH 2 ) n - wherein n is 2-10. In embodiments, L has a structure of the formula -(CH 2 ) n - wherein n is independently selected from the group consisting of 2, 3, 4, 5, 6, 7, 8, 9, or 10. In another embodiment, L has a structure of the formula -(CH 2 ) 2 -(O-(CH 2 ) 2 -) m - wherein m is 1-2. In another embodiment, L is an ethyleneoxyethylene (-(CH 2 ) 2 -O-(CH 2 ) 2 -) moiety.
- the anthracycline or the anthracycline analog useful in the HAP compounds of the present invention contains one or more NHR 1 groups.
- a variety of functional groups are useful as R 1 in the anthracyclines useful in the present invention, hi one embodiment, R 1 is selected from the group consisting of hydrogen, C 1 -C 6 alkyl, Ci-C 6 heteroalkyl, Ci-C 6 alkenyl, Ci-C 6 alkynyl, and acyl.
- Anthracyclines suitable for use in the HAP compounds of the present invention include but are not limited to doxorubicin and daunorubicin.
- Other anthracyclines useful in the HAP compounds of the present invention are described for example in U.S. Pat. Appl. Pub. Nos. US 2007/0060534 and US 2006/0258656 and PCT Appl. Pub. No. WO 05/086951 (each of which is incorporated herein by reference).
- the present invention provides a HAP compound having a structures of the formula selected from Hyp-L-Q and (Hyp-L) 2 -Q wherein each Hyp-L moiety is covalently bonded to the NHRi -nitrogen atom of the anthracycline wherein Ri is defined as above.
- each L is independently selected from the group consisting Of Ci-C 6 alkylene and Ci-C 6 heteroalkylene
- Hyp is a moiety having a structure of the formula:
- the present invention provides a HAP compound having a structure of the formula:
- R 1 is selected from the group consisting of hydrogen, C 1 -C 6 alkyl, and Ci-C 6 heteroalkyl
- R 3 is selected from H and OH
- each L is independently selected from the group consisting of C]-Ci 0 alkylene and Ci-Ci 0 heteroalkylene
- Hyp has a structure of the formula:
- the present invention provides a HAP compound having a structure of the formula:
- R 3 is selected from H and OH, each L is independently selected from the group consisting of Ci -Ci 0 alkylene and Ci-Ci 0 heteroalkylene, and Hyp has a structure of the formula:
- each L is independently selected from the group consisting of the group consisting of -(CH 2 ) n - and -(CH 2 ) 2 -(O-(CH 2 ) 2 ) m - wherein n is 2-10 and m is 1-2.
- n is independently selected from the group consisting of 2, 3, 4, 5, 6, 7, 8, 9, or 10.
- Ri is hydrogen.
- the present invention provides a HAP compound having a structure of the formula (Hyp-L)-Q-(CO 2 CR 4 R 5 -Brg) wherein Q is an anthracycline, L is independently selected from the group consisting of Ci-Ci 0 alkylene and Ci-Ci 0 heteroalkylene, and Hyp is a moiety having a structure of the formula: each OfR 4 and R 5 is selected independently from the group consisting of hydrogen, Ci-C 6 alkyl Ci-C 6 heteroalkyl, C 3 -C 8 cycloalkyl, heterocyclyl, aryl and heteroaryl.
- Brg is a bioreductive moiety having a structure of the formula
- R 6 is selected from the group consisting of hydrogen, Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl aryl, heteroaryl, and halo and R 7 is Ci-C 6 alkyl Ci-C 6 heteroalkyl, C 3 -C 8 cycloalkyl, heterocyclyl, aryl and heteroaryl.
- R 6 is selected from the group consisting of hydrogen, Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl aryl, heteroaryl, and halo and R 7 is Ci-C 6 alkyl Ci-C 6 heteroalkyl, C 3 -C 8 cycloalkyl, heterocyclyl, aryl and heteroaryl.
- substituents include, but are not limited to, groups that as part of the compounds of the present invention enhance aqueous solubility of these compounds.
- substituents include electron withdrawing groups such as nitro, halo, cyano, haloalkyls, and the like. Certain other suitable substituents include electron donating substituents such as alkoxy, amino, alkylamino, dialkylamino, and the like.
- the anthracycline has a structure of the formula:
- each OfR 4 and R 5 is hydrogen, and Brg has a structure of the formula:
- each OfR 4 and R 5 is hydrogen.
- R 6 is hydrogen.
- each L is independently selected from the group consisting of C 5 alkylene and C 5 heteroalkylene.
- the present invention provides a HAP compounds having structure of the formula selected from:
- R 1 and -L-Hyp are as tabulated below.
- HAP compounds of the present invention kill cancer cells as described below.
- COMPOUND 1 undergoes reduction to a bis-hydroxylamine, and COMPOUND 2, or another HAP compound of the invention containing a single 2-nitroimidazole moiety, to a mono-hydroxylamine, that can alkylate DNA as shown below:
- the anthraquinone portion of daunorubicin can intercalate between DNA bases. Together, the intercalation and alkylation of the DNA hinders the replication of cancer cell DNA and kill cancer cells. Under normoxic conditions, such as those existing in a normal cell, the bis-hydroxylamine and mono-hydroxylamine alkylators are not formed, the DNA in those normal cells is not alkylated, and the normal cells are not killed or are killed to a lesser extent compared to hypoxic tumor cells.
- HAP compounds of the present invention can be synthesized according to the novel method described below and/or other methods known to one of skill in the art upon reading this disclosure.
- the present invention provides a method of synthesizing a HAP compound of the present invention having a structure of the formula Hyp-L-Q wherein Hyp is a bioreductive group, Q is an anticancer agent selected from anthracyclines, and L is a linker selected from C 2 -Ci 0 alkylene and C 2 -Ci 0 heteroalkylene, said method comprising reacting an anthracycline or an anthracycline analog containing one or more NHRi groups wherein R] is selected from the group consisting of hydrogen, Ci-C 6 alkyl, Ci-C 6 heteroalkyl, Ci -C 6 alkenyl, Ci-C 6 alkynyl, acyl, with a compound having a structure of the formula Hyp- Li-CHO wherein Li is selected from Ci -Cg alkylene and C1-C 9 heteroalkylene and a reducing agent to yield the compound having the structure of the formula Hyp-L-Q.
- Hyp is a bio
- Anthracycline are useful in the present synthetic methods as free bases and as ammonium salts.
- reacting an anthracycline or an anthracycline analog in the free base form according to the present methods yields a HAP compound having structure of the formula Hyp-L-Q.
- Ri is selected from Ci-C 6 alkyl and Ci-C 6 heteroalkyl.
- the anthracycline containing one or more NHRi groups is daunorubicin.
- the present invention provides a method of synthesizing a HAP compound having a structure of the formula:
- R 3 is selected from H and OH
- each L is independently selected from the group consisting Of C 2 -Ci 0 alkylene, and C 2 -Ci 0 heteroalkylene and Hyp has a structure of the formula:
- said method comprising reacting an anthracycline having a structure of the formula:
- Li is selected from -(CH 2 ) k - and -CH 2 -(O-(CH 2 ) 2 ) m - wherein k is 1-9 and m is 1-2.
- Li is selected from the group consisting Of -CH 2 -, -(CH 2 V, -(CH 2 ) 4 -, -CH 2 -O-(CH 2 ) 2 -, and -CH 2 -(O-(CH 2 ) 2 -) 2 -.
- the reducing agent is NaBH(OAc) 3 .
- the present invention provides a method of synthesizing a HAP compound of the present invention having a structure of the formula (Hyp-L) 2 -Q wherein Hyp is a bioreductive group, Q is an anticancer agent selected from anthracyclines, and L is a linker selected from C 2 -Ci O alkylene and C 2 -Ci 0 heteroalkylene, said method comprising reacting an anthracycline or an anthracycline analog containing one or more NH 2 groups with a compound having a structure of the formula Hyp-Li-CHO wherein Li is selected from C 1 -C 9 alkylene and C 1 -C 9 heteroalkylene and a reducing agent to yield the compound having a structure of the formula (Hyp-L) 2 -Q.
- Hyp is a bioreductive group
- Q is an anticancer agent selected from anthracyclines
- L is a linker selected from C 2 -Ci O alkylene and C
- reacting acid salts of anthracycline analogs containing one or more NH 2 groups according to the present methods yields a HAP compound having a structure of the formula (Hyp-L) 2 -Q.
- the anthracycline containing one or more NH 2 groups is selected from daunorubicin or doxorubicin.
- the present invention provides a method of synthesizing a HAP compound having a structure of the formula
- R 3 is selected from H and OH
- each L is independently selected from the group consisting OfC 2 -Ci 0 alkylene and C 2 -Ci 0 heteroalkylene
- Hyp has a structure of the formula:
- said method comprising reacting an ammonium salt of an anthracycline having a structure of the formula:
- Li is selected from -(CH 2 ) k - and -CH 2 -(O-(CH 2 ) 2 ) m - wherein k is 1-9 and m is 1-2.
- Li is selected from the group consisting of -CH 2 -, -(CH 2 ) 2 -, -(CH 2 ) 4 -, -CH 2 -O-(CH 2 ) 2 -, and -CH 2 -(O-(CH 2 ) 2 -) 2 -.
- the reducing agent is NaBH(OAc) 3 .
- Hyp moieties are useful in the synthetic methods of the present invention.
- the Hyp moiety is a substituted or an unsubstituted 2-nitroimidazole moiety.
- Substituted 2-nitroimidazoles useful in synthesizing the HAP compounds of the present invention can be synthesized according to methods adapted from PCT Pat. Pub. No. WO 07/02931, and U.S. Patent Application Nos. 60/941,753 and 60/970,364, and/or those known in the literature.
- the present invention provides methods of treating cancer and other hyperproliferative diseases comprising administering a therapeutically effective amount of a HAP compound of the present invention to a patient in need of such treatment.
- the HAP compound administered has a structure of the formula selected from Hyp-L-Q and (Hyp-L) 2 -Q, wherein Q is selected from anthracyclines, Hyp is a bioreductive group having a structure of the formula:
- the HAP compound administered includes, but is not limited to, COMPOUND 1, COMPOUND 2, COMPOUND 3, COMPOUND 4, COMPOUND 5, COMPOUND 6, COMPOUND 7, COMPOUND 8, COMPOUND 9, COMPOUND 10, COMPOUND 11, COMPOUND 12, COMPOUND 14, COMPOUND 15, COMPOUND 16 and COMPOUND 21.
- the therapeutically effective amount of the HAP compound is administered as a pharmaceutically acceptable formulation comprising a HAP compound of the present invention and pharmaceutically acceptable diluents or excipients.
- the therapeutically effective amount is administered in a daily dose.
- the therapeutically effective daily dose can be administered by employing suitable unit dose forms of the HAP compounds of the present invention.
- the daily dose is administered from once every day, once every two weeks, up to, once every month.
- the daily dose is administered parenterally or orally.
- a HAP compound of the present invention was safely administered at a daily dose of 10 mg/kg to mice once every week, for three weeks. Converting the mouse dose to human equivalent dose (HED) by dividing the mouse dose by 12.3 yields a HED of about 0.8 mg/kg, daily; thus in one embodiment of the present invention, COMPOUND 2 is administered to treat cancer in humans, by administering a daily dose of up to 0.8 mg/kg to a patient in need of such treatment. Lower effective daily doses can also be administered.
- HED human equivalent dose
- COMPOUND 2 can be administered at a daily dose of 5 mg/kg alone and in combination with another anticancer agent to xenograft tumor bearing mice.
- Another anthracycline such as doxorubicin is administered as control to mice bearing xenograft tumors, alone and in combination with the anticancer agent used with COMPOUND 2.
- the efficacy of COMPOUND 2 administration is determined by post-administration measurement of the mean and/or median tumor sizes and comparing them with the corresponding tumor sizes in the control groups.
- cancers can be treated according to the methods of the present invention by administering the HAP compounds the present invention.
- the cancer treated is selected from the group consisting of cancer of the adrenal gland, bone, brain, breast, bronchi, colon and/or rectum, gallbladder, head and neck, kidneys, larynx, liver, lung, neural tissue, pancreas, prostate, parathyroid, skin, stomach, and thyroid.
- the cancer treated is selected from the group consisting of acute and chronic lymphocytic and granulocytic tumors, adenocarcinoma, adenoma, basal cell carcinoma, cervical dysplasia and in situ carcinoma, Ewing's sarcoma, epidermoid carcinomas, giant cell tumor, glioblastoma multiforma, hairy-cell tumor, intestinal ganglioneuroma, hyperplastic corneal nerve tumor, islet cell carcinoma, Kaposi's sarcoma, leiomyoma, leukemias, lymphomas, malignant carcinoid, malignant melanomas, malignant hypercalcemia, marfanoid habitus tumor, medullary carcinoma, metastatic skin carcinoma, mucosal neuroma, myeloma, mycosis fungoides, neuroblastoma, osteo sarcoma, osteogenic and other sarcoma, ovarian tumor, pheochromocytoma, polycythemia ver
- the HAP compound of the present invention is administered for the treatment of cancer in combination with other anticancer agents or other anticancer therapies.
- Suitable anticancer therapies useful in accordance with the present methods include radiation therapy and surgery.
- Methods for treating cancer employing other hypoxia activated prodrugs are described, for example, in PCT Pat. Appl. Pub. Nos. WO 07/02931 and WO 06/57946 and U.S. Pat. Appl. Pub. No. US 2006/0258656 (each of which is incorporated herein by reference) and can be used for the treatment of cancer according to the present methods upon appropriate substitution of the other hypoxia activated prodrug compounds with the HAP compounds of the present invention.
- the present invention provides methods of treating non- cancer hyperproliferative diseases characterized by cellular hyperproliferation (e.g., an abnormally increased rate or amount of cellular proliferation) in accordance with the present methods.
- the hyperproliferative disease is selected from the group consisting of allergic angiitis and granulomatosis (Churg-Strauss disease), asbestosis, asthma, atrophic gastritis, benign prostatic hyperplasia, bullous pemphigoid, coeliac disease, chronic bronchitis and chronic obstructive airway disease, chronic sinusitis, Crohn's disease, demyelinating neuropathies, dermatomyositis, eczema, including atopic dermatitis, eustachean tube diseases, giant cell arteritis, graft rejection, hypersensitivity pneumonitis, hypersensitivity vasculitis (Henoch-Schonlein purpura), irritant dermatitis,
- the hyperproliferative disease treated is psoriasis, a disease characterized by the cellular hyperproliferation of keratinocytes which builds up on the skin to form elevated, scaly lesions.
- the hyperproliferative disease treated is multiple sclerosis, a disease characterized by progressive demyelination in the brain.
- the hyperproliferative diseases treated is rheumatoid arthritis, a multisystem chronic, relapsing, inflammatory disease that can lead to destruction and ankylosis of joints affected.
- a HAP compound of the present invention is administered to prevent a hyperproliferative disease resulting from cellular proliferation on a prosthesis implanted in a patient by coating the prosthesis with a composition containing a HAP compound of the present invention.
- Example IA describes the synthesis of COMPOUND 1, a HAP compound of the present invention having a structure of the formula (HyP-L) 2 -Q, wherein Hyp is an anthracycline containing an amino group, each L is a Ci-C 1O heteroalkylene moiety, and Hyp has a structure of the formula:
- COMPOUND 1 was synthesized according to a novel synthetic method of the present invention, starting from the ammonium salt of daunorubicin, daunorubicin hydrochloride, and a 2-nitroimidazole substituted at the 1-N-position with an aldehyde containing CpC 6 heteroalkylene moiety.
- OsO 4 (0.1 equivalent) was added to a mixture of l-N-allyloxyethyl-2-nitroimidazole (620 mg, 1 equivalent) in diethyl ether (Et 2 O, 10 mL) and water (10 mL) at room temperature (rt), followed by the addition OfNaIO 4 (1.68 g, 2.5 equivalent) over a period of 20 min.
- Example IB describes the synthesis of COMPOUND 2, a HAP compound of the present invention having a structure of the formula Hyp-L-Q wherein Hyp-L-Q wherein Hyp is an anthracycline containing an NH 2 group, L is a C 1 -Ci 0 heteroalkylene moiety, and Hyp has a structure of the formula:
- COMPOUND 2 was synthesized according to a novel synthetic method of the present invention described above, starting from daunorubicin and a 2-nitroimidazole substituted at the 1-N-position with an aldehyde containing Ci-C 6 heteroalkylene moiety.
- Triethylamine (TEA, 27 ⁇ L) was slowly added to a suspension of daunorubicin hydrochloride (102 mg) in dichloromethane (DCM, 5 mL) at O 0 C to yield daunorubicin that was used without further purification.
- DCM dichloromethane
- a solution of l-formylmethyloxyethyl-2-nitroimidazole (40 mg, synthesized as described in Example IA) in DCM (1 mL) was added to the daunorubicin containing mixture and stirred for 0.5 h at O 0 C, followed by the addition OfNaBH(OAc) 3 (127 mg). The mixture was allowed to come to rt and stirred for 2-3 h. Volatiles were removed under vacuo and the residue separated by column chromatography on silica gel employing DCM - DCM/MeOH (100:10) to obtain COMPOUND 2.
- COMPOUND 4 COMPOUND 5
- COMPOUND 8 COMPOUND 9 were synthesized according to the method described in Example IB starting from daunorubicin and upon suitable substitution of the corresponding starting aldehyde.
- COMPOUND 7 was synthesized according to the method described in Example IB starting from COMPOUND 2 and methoxyacetaldehyde.
- Daunorubicin aromatic ring 8.01 (IH, d); 7.80 (IH, t); 7.41 (IH, d); 2-Nitroimdazole ring: 7.18 (IH, s); 7.03 (IH, s).
- Daunorubicin aromatic ring 8.03 (IH, d); 7.79 (IH, t); 7.4 (IH, d); 2-Nitroimdazole ring: 7.15 (IH, s); 7.08 (IH, s).
- Example 2A(i) describes methods for determining cytotoxicities of HAP compounds of the present invention under hypoxia and normoxia by employing an AlamarBlue fluorescence intensity based detection of cell proliferation.
- H460 non-small cell lung cancer cells (10,000 - 15,000 cells/well/500 ⁇ L, ATCC HTB-177) were seeded in glass inserts on 24- well plates in RPMIl 640 medium supplemented with 10% FBS and 1% Penicillin/Streptomycin (Invitrogen Corporation, Carlsbad, CA).
- control group no test compound
- treatment groups in which the cells were kept in contact with the test compound at various concentrations for 2 h.
- the cells in the treatment groups were incubated for 2 hours with 6 different concentrations of a test compound, under hypoxia (5% CO 2 , 5% H 2 , 90% N 2 ) or normoxia (5% CO 2 , 95% air), media containing the test compound removed, fresh media added, and the cells incubated for 3 days.
- COMPOUND 2 a HAP compound of the present invention is about 400 times more cytotoxic under hypoxia than under normoxia. COMPOUND 2 also demonstrated an enhanced cytotoxicity under hypoxia compared to normoxia in a variety of other cell lines. In one embodiment of the present invention, COMPOUND 2 is administered to treat cancer by selectively killing hypoxic tumor cells and not killing or killing fewer of the normoxic, normal cells.
- cytotoxicities of the HAP compounds of the present invention were determined, under normoxia, in the MESSA-DX5 cell lines resistant to certain anticancer drugs and the results compared with the normoxic cytotoxicities determined in the corresponding non resistant, MESSA, cell line. Daunorubicin was used as a control compound.
- MESSA-DX5 cell lines are drug resistant due in part to the overexpression of the MDR-I protein efflux pump. The results are tabulated below.
- the present invention provides a method of treating drug resistant cancers by administering a therapeutically effective amount of a suitable HAP compound of the present invention to a patient in need of such treatment.
- This example describes testing the cytotoxicity of a HAP compound of the present invention employing the clonogenic survival method.
- Exponentially growing human H460 cells obtained from the ATCC
- RPMI medium supplemented with 10% fetal bovine serum for 2 days prior to initiating treatment with HAP compound of the present invention.
- HAP compound stocks of known concentrations were prepared in complete medium, and 2 mL of the desired stock added to each plate.
- the lid of the glass plate was removed and the plate shaken for 5 minutes on an orbital shaker.
- the plates were recovered and stored inside a glove-box.
- the glove-box was evacuated and gassed with either a certified anoxic gas mixture (95% nitrogen and 5% carbon dioxide) or with an aerobic (normoxic) gas mixture (95% air and 5% carbon dioxide). Cells were then incubated with the drug for 3 hours at 37°C.
- COMPOUND 2 was administered to mice and the in vivo pharmacokinetic parameters determined are tabulated below; daunorubicin was employed as a control compound. The data demonstrate that the pharmacokinetic properties of COMPOUND 2 are similar to that of the approved anticancer agent daunorubicin. In one embodiment of the present invention, thus, COMPOUND 2 is administered according to the present methods for the treatment of cancer.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Hypoxia activated prodrug compounds of anthracyclines are useful in the treatment of cancer and other hyperproliferative diseases. Certain compounds contain a 2-nitroimidazole moiety covalently bonded via the 1-N nitrogen atom to an alkylene and/or a heterolalyene linker; the alkylene and/or a heterolalyene linker is covalently bonded to the anthracycline.
Description
HYPOXIAACTIVATED PRODRUGS OF ANTHRACYCLINES
CROSS-REFERENCES TO RELATED APPLICATIONS [0001] This application claims priority to U.S. provisional application Serial No. 60/952,512, filed 27 July 2007 and U.S. provisional application Serial No. 60/972,162, filed 13 September 2007, each of which is incorporated herein by reference.
STATEMENT AS TO RIGHTS TO INVENTIONS MADE UNDER FEDERALLY SPONSORED RESEARCH OR DEVELOPMENT [0002] NOT APPLICABLE
REFERENCE TO A "SEQUENCE LISTING," A TABLE, OR A COMPUTER
PROGRAM LISTING APPENDIX SUBMITTED ON A COMPACT DISK. [0003] NOT APPLICABLE
FIELD OF THE INVENTION
[0004] The present invention provides compositions and methods for the treatment of cancer, and generally relates to the fields of medicinal chemistry, medicine, pharmacology, molecular biology, and biology.
BACKGROUND OF THE INVENTION
[0005] Hypoxia activated prodrugs of anticancer agents, or HAP compounds, are useful for tumor therapy. A HAP compound contains a bioreductive group, a linker, and an anticancer agent and is less cytotoxic than the corresponding anticancer agent under normoxic conditions or normoxia, such as those existing in a normal cell. Under hypoxia, however, the bioreductive group present in the HAP compound is reduced, and the cytotoxic anticancer agent is generated and/or released. In hypoxic regions such as those existing in solid tumors, a HAP compound generates and/or releases a cytotoxin and kills cancer cells selectively in and around the hypoxic tumor zone. HAP and/or bioreducible compounds are described, for example, in PCT Pat. Appl. Pub. Nos. WO 00/64864; 04/85361; 04/85421; 04/87075; 06/57946; and 07/02931.
[0006] HAP and/or bioreducible compounds currently, or previously tested, in the clinic include TH 302, AQ4N, and PR 104 (see Lalani et al, Clin. Cancer Res., 13:2216 (2007) and Patterson et al., Clin. Cancer Res., 13:3922 (2007)), each incorporated herein by reference, and WO 07/002931, supra). None of these compounds contain a 2-nitroimidazole moiety covalently bonded to an anticancer agent at the 1-N-position of the 2-nitroimidazole moiety.
[0007] Bioreducible compounds comprising an aromatic moiety tethered to the 1-N- position of a bioreductive group such as 2-nitroimidazole have been reported; however, none of these compounds were found to be clinically useful for the treatment of cancer (see Hodgkiss et al., J. Med. Chem., 34:2268-74 (1991); Papadopoulou et al., Int. J. Rad. Oncol, 42(4):775-79 (1998); Liu et al., Bioorg. Med. Chem., 4(9):2935-41 (2006); Papadopoulou et al., Bioorg. Med. Chem. Lett, 14(6):1523-25 (2004); and Buchko et al., Radiat. Res., 158(3):302-10 (2002)).
[0008] There remains a need for additional HAP compounds for the treatment of cancer, including HAP compounds with improved hypoxic compared to normoxic cytotoxicity. The present invention meets such needs.
BRIEF SUMMARY OF THE INVENTION
[0009] The present invention relates to hypoxia activated prodrug compounds, or HAP compounds, of anticancer agents comprising one or more bioreductive Hyp moieties covalently bonded to an anticancer agent, Q, via a linker moiety, L, wherein after hypoxic activation, the bioreduced Hyp moiety remains attached to the rest of the molecule, and wherein the anticancer agent, Q, is selected from the class of anthracycline anticancer agents, including but not limited to Daunorubicin, Doxorubicin, Epirubicin and Idarubicin:

Daunorubicin, Doxorubicin,

Epirubicin Idarubicin

Aclarubicin

Pirarubicin
Generally, the present invention provides HAP compounds having structures of the formulas Hyp-L-Q and (Hyp-L)2-Q.
[0010] Suitable Hyp moieties useful in the compounds of the present invention include, but are not limited to, nitro imidazoles, nitrofurans, nitrothiophenes, and nitrobenzenes. In one embodiment, the nitroimidazole is a 2-nitroimidazole. In one embodiment, the 2- nitroimidazole is covalently bonded to the anticancer agent via the L moiety wherein the L moiety is covalently bonded to the "1-N" position of the 2-nitroimidazole:

[0011] Suitable L moieties useful in the compounds of the present invention include but are not limited to Ci-Ci0 alkylene and Ci-Ci0 heteroalkylene.
[0012] The HAP compound of the present invention have structures of the formulas Hyp-L-Q and (Hyp-L)2-Q wherein each Hyp-L moiety is covalently bonded to the amino nitrogen atom in the anthracycline. Suitable L moieties useful in the present invention include Ci-C]0 alkylene and Ci-Ci0 heteroalkylene moieties. Other suitable L moieties useful in the present invention include Ci alkylene and Ci heteroalkylene moieties. Other suitable L moieties useful in the present invention include C2 alkylene and C2 heteroalkylene moieties. Other suitable L moieties useful in the present invention include C3 alkylene and C3 heteroalkylene moieties. Other suitable L moieties useful in the present invention include C4 alkylene and C4 heteroalkylene moieties. Other suitable L moieties useful in the present invention include C5 alkylene and C5 heteroalkylene moieties. Other suitable L moieties useful in the present invention include C6 alkylene and C6 heteroalkylene moieties. Other suitable L moieties useful in the present invention include C7 alkylene and C7 heteroalkylene moieties. Other suitable L moieties useful in the present invention include C8 alkylene and C8 heteroalkylene moieties. Other suitable L moieties useful in the present invention include C9 alkylene and C9 heteroalkylene moieties. Other suitable L moieties useful in the present invention include Ci0 alkylene and Ci0 heteroalkylene moieties. A suitable Hyp moiety useful in the present invention has a structure of the formula:

[0013] Other Hyp moieties having a structure of formula:

[0014] In one aspect, the present invention provides a HAP compound having a structure of the formula:


[0015] In another embodiment, Ri is hydrogen, hi certain embodiments, L is selected from Ci alkylene and Ci heteroalkylene moieties. In certain embodiments, L is selected from C2 alkylene and C2 heteroalkylene moieties. In certain embodiments, L is selected from C3 alkylene and C3 heteroalkylene moieties. In certain embodiments, L is selected from C4 alkylene and C4 heteroalkylene moieties. In certain embodiments, L is selected from C5 alkylene and C5 heteroalkylene moieties. In certain embodiments, L is selected from C6 alkylene and C6 heteroalkylene moieties. In certain embodiments, L is selected from C7 alkylene and C7 heteroalkylene moieties. In certain embodiments, L is selected from C8 alkylene and C8 heteroalkylene moieties. In certain embodiments, L is selected from C9 alkylene and C9 heteroalkylene moieties. In certain embodiments, L is selected from C10 alkylene and Ci0 heteroalkylene moieties. In certain embodiments, the anthracyclines useful in the HAP compounds of the present invention contain one or more amino groups, hi one embodiment, the amino group is a primary or secondary amino group, -NRi, as disclosed
herein. A variety of functional groups are useful as the secondary group on the amino group in the anthracyclines useful in the present invention. In one embodiment, the secondary group on the amino group, -NRi, is selected from the group consisting of C]-C6 alkyl, Ci-C6 heteroalkyl, Ci-C6 alkenyl, Ci-C6 alkynyl, acyl, and heteroarylalkylenyloxycarbonyl.
[0016] In another aspect, the present invention provides a HAP compound having a structure of the formula:


[0017] In another aspect, the present invention provides compounds having a structure of the formula (Hyp-L)-Q-(CO2CR4R5-Brg) wherein Q is an anthracycline; L is selected from the group consisting of Ci-Ci0 alkylene and Ci-Ci0 heteroalkylene; Hyp is a moiety having a structure of the formula:



[0018] Within this embodiment, R6 is selected from the group consisting of hydrogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl aryl, heteroaryl, and halo; and R7 is Ci-C6 alkyl, CpC6 heteroalkyl, C3-C8 cycloalkyl, heterocyclyl, aryl and heteroaryl; and the Hyp-L and CO2CR4R5-Brg , moieties are covalently bonded to an amino nitrogen atom in the anthracycline. In another embodiment, the Hyp-L and CO2CR4R5-Brg moieties are covalently bonded to the same amino nitrogen atom in the anthracycline.
[0019] In another embodiment, the anthracyclines useful in the present invention are selected from daunorubicin and doxorubicin.
[0020] In another embodiment, the present invention provides the HAP compounds of the present invention in substantially pure forms.
[0021] In another aspect, the present invention provides methods of synthesizing HAP compounds of the present invention.
[0022] In another aspect, the present invention provides pharmaceutically acceptable formulations each such formulation comprising a HAP compound of the present invention and pharmaceutically acceptable carriers, diluents, and/or excipients.
[0023] In another aspect, the present invention provides a method of treating cancer and other hyperproliferative diseases comprising administering a therapeutically effective amount of a HAP compound of the present invention to a patient in need of such treatment.
DETAILED DESCRIPTION OF THE INVENTION
[0024] This detailed description of the different aspects and embodiments of the present invention is organized as follows: Section I provides useful definitions; Section II describes the HAP compounds of the present invention and methods of their synthesis; Section III describes therapies provided by the present invention; and Section IV provides illustrative
methods for synthesizing and testing HAP compounds of the present invention. This detailed description is organized into sections only for the convenience of the reader, and disclosure found in any section is applicable to disclosure elsewhere in the specification.
I. Definitions
[0025] The following definitions are provided to assist the reader. Unless otherwise defined, all terms of art, notations, and other scientific or medical terms or terminology used herein are intended to have the meanings commonly understood by those of skill in the chemical and medical arts. In some cases, terms with commonly understood meanings are defined herein for clarity and/or for ready reference, and the inclusion of such definitions herein should not be construed as representing a substantial difference over the definition of the term as generally understood in the art.
[0026] "V/VOP" refers to a position on a moiety which is covalently bonded to the rest of the molecule via a single bond.
[0027] "Acid salt" refers to a compound of the present invention that contains relatively basic functionalities, to which a sufficient amount of an acid is added to form a salt. Examples of pharmaceutically acceptable acid addition salts include those derived from inorganic acids like hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric, hydriodic, or phosphorous acids and the like, as well as the salts derived from relatively nontoxic organic acids like acetic, propionic, isobutyric, malonic, benzoic, succinic, suberic, fumaric, mandelic, phthalic, benzenesulfonic, p-tolylsulfonic, citric, tartaric, methanesulfonic, and the like. Also included are salts of amino acids such as arginate and the like, and salts of organic acids like glucuronic or galactunoric acids and the like (see, e.g., Berge, S. M. et al ., "Pharmaceutical Salts," Journal of Pharmaceutical Science, 66:1-19, 1977).
[0028] "Alkenylene" refers to a linear or branched, unsaturated, divalent, substituted or unsubstituted, hydrocarbon radical wherein the unsaturation is a carbon-carbon double bond and wherein there is at least one carbon atom covalently bonded to each side of the double bond. "C4-C6 alkenylene" refers to a linear or branched, unsaturated, divalent, substituted or unsubstituted, hydrocarbon radical having 4-6 carbon atoms wherein the unsaturation is a carbon-carbon double bond and wherein there is at least one carbon atom covalently bonded to each side of the double bond.
[0029] "Alkoxy" refers to a substituted or unsubstituted alkyl group covalently bonded to an oxygen atom. "Ci-C6 alkoxy" refers to a substituted or unsubstituted alkyl group of 1-6 carbon atoms covalently bonded to an oxygen atom. In other words, a Ci-C6 alkoxy group has the general structure -0-(Ci-C6) alkyl. Ci-C6 alkoxy groups include, for example, methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, sec-butoxy, tert-butoxy, n-pentoxy, 2-pentoxy, 3-pentoxy, isopentoxy, neopentoxy, hexoxy, 2-hexoxy, 3-hexoxy, and 3-methylpentoxy.
[0030] "Alkylenyloxy" refers to a linear saturated divalent substituted or unsubstituted hydrocarbon radical or a branched saturated divalent hydrocarbon radical covalently bonded to an oxygen atom. In other words, a alkylenyloxy group has the general structure -0-(Ci-C6) alkyl. CpC6 alkoxy groups include, for example, methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, sec-butoxy, tert-butoxy, n-pentoxy, 2-pentoxy, 3-pentoxy, isopentoxy, neopentoxy, hexoxy, 2-hexoxy, 3-hexoxy, and 3-methylpentoxy.
[0031] "Alkylenyloxycarbonyl" refers to an alkylenyloxy group covalently bonded to a carbonyl. In other words, a alkylenylcarbonyl group has the general structure -C(=O)-O- alkyl-.
[0032] "Alkoxycarbonyl" refers to an alkoxy group covalently bonded to a carbonyl. In other words, a alkoxycarbonyl group has the general structure -C(=O)-O-alkyl. "Ci-C6 alkoxycarbonyl" refers to an (C]-C6) alkoxy group covalently bonded to a carbonyl. In other words, a Ci-C6 alkoxycarbonyl group has the general structure -C(=O)-O-(Ci-C6) alkyl.
[0033] "Alkyl" refers to a substituted or unsubstituted straight or branched chain alkyl group. "Ci-C6 alkyl" refers to a substituted or unsubstituted straight or branched chain alkyl groups having 1-6 carbon atoms. Ci-C6 alkyl groups include, for example, methyl, ethyl, propyl, isopropyl, n-butyl, sec-butyl, tert-butyl, pentyl, 2-pentyl, isopentyl, neopentyl, hexyl, 2-hexyl, 3-hexyl and 3-methylpentyl. A Ci-C6 alkyl substituent may be covalently bonded to an atom within a molecule of interest via any chemically suitable portion of the C]-C6 alkyl group.
[0034] "Alkenyl" by itself or as part of another substituent refers to a straight or branched chain, which may be mono or polyunsaturated, having the number of carbon atoms designated. For example, '^-Cgalkenyl" means an alkenyl radical having from 2, 3, 4, 5 or
6 atoms that is derived by the removal of one hydrogen atom from a single carbon atom of a
parent alkane. Examples include, but are not limited to vinyl, 2 propenyl i.e. - CH=C(H)(CH3), -CH=C(CH3)2, -C(CH3)=C(H)2, -C(CH3)=C(H)(CH3), C(CH2CH3)=CH2, butadienyl e.g. 2 (butadienyl), pentadienyl e.g. 2,4 pentadienyl and 3 (1,4 pentadienyl), and hexadienyl, among others, and higher homologs and stereoisomers thereof. The phrase "substituted alkenyl" has the same meaning with respect to alkenyl groups that substituted alkyl groups had with respect to unsubstituted alkyl groups. A substituted alkenyl group includes alkenyl groups in which a non-carbon or non-hydrogen atom is bonded to a carbon double bonded to another carbon and those in which one of the non-carbon or non- hydrogen atoms is bonded to a carbon not involved in a double bond to another carbon. Each site of unsaturation may be either cis or trans configuration about the double bond(s).
[0035] "Alkynyl" by itself or as part of another substituent, means a straight or branched chain hydrocarbon radical, which may be mono- or polyunsaturated, having the number of carbon ato ms designated. For example, "C2-C6 alkynyl" means an alkynyl radial having from 2 to 6 carbon atoms that is derived by the removal of one hydrogen atom from a single carbon atom of a parent alkane . "Unsubstituted alkynyl" refers to straight and branched chain groups such as those described with respect to unsubstituted alkyl groups as defined above, except that at least one triple bond exists between two carbon atoms. Examples include, but are not limited to ethynyl e.g. -C C(H), 1- propynyl e.g. -CC(CH3), -
CC(CH2CH3), -C(H2)CC(H), -C(H)2CC(CH3), and -C(H)2CC(CH2CH3) among others, and higher homologs and isomers thereof. The phrase "substituted alkynyl" has the same meaning with respect to alkynyl groups that substituted alkyl groups had with respect to unsubstituted alkyl groups. A substituted alkynyl group includes alkynyl groups in which a non-carbon or non-hydrogen atom is bonded to a carbon triple bonded to another carbon and those in which a non-carbon or non-hydrogen atom is bonded to a carbon not involved in a triple bond to another carbon.
[0036] "Alkylamino" refers to a substituted or unsubstituted alkyl group covalently bonded to an -NH- moiety. "C1-C6 alkylamino, " refers to a substituted or unsubstituted alkyl group of 1-6 carbon atoms covalently bonded to an -NH- moiety. In other words, a Ci-C6 alkylamino group has the general structure -NH-(Ci-C6) alkyl. Similarly a di(Ci-C6) alkylamino group has the general structure -N-[(Ci-C6) alkyl]2. Ci-C6 alkylamino groups include, for example, methylamino, ethylamino, propylamino and butylamino.
[0037] "Alkylene" refers to a linear saturated divalent substituted or unsubstituted hydrocarbon radical or a branched saturated divalent hydrocarbon radical. Similarly, Ci-Cio alkylene refers to a corresponding alkylene group having 1-10 carbon atoms. Ci-C6 alkylene groups include, for example, methylene, ethylene, propylene, butylene, 2-methylpropylene, pentylene.
[0038] "Alkyl ether" refers to a moiety with an oxygen atom and carbon atoms positioned such that at least one carbon atom is located on either side of the oxygen atom. "C2-C6 alkyl ether" refers to a moiety with an oxygen atom and 2-6 carbon atoms positioned such that at least one carbon atom is located on either side of the oxygen atom.
[0039] "Alkynylene" refers to a linear or branched, unsaturated, divalent, substituted or unsubstituted, hydrocarbon radical wherein the unsaturation is a carbon-carbon triple bond and wherein there is at least one carbon atom covalently bonded to each side of the triple bond. "C4-C6 alkynylene" refers to a linear or branched, unsaturated, divalent, substituted or unsubstituted, hydrocarbon radical having 4-6 carbon atoms wherein the unsaturation is a carbon-carbon triple bond and wherein there is at least one carbon atom covalently bonded to each side of the triple bond.
[0040] "Amino" refers to a monovalent radical -NRaRb or divalent radical -NRa-. The term "alkylamino" refers to the group -NRaRb where Ra is alkyl and Rb is H or alkyl. The term "arylamino" refers to the group -NRaRb where Ra is aryl and Rb is hydrogen, alkyl, aryl, or heterocyclyl. The term " (alkyl)(aryl)amino" refers to the group -NRaRb where Ra is alkyl and Rb is aryl. Additionally, for dialkylamino groups, the alkyl portions can be the same or different and can also be combined to form a 3-7-membered ring with the nitrogen atom to which each is attached. Accordingly, a group represented as NRaRb is meant to include piperidinyl, pyrrolidinyl, morpholinyl, azetidinyl and the like.
[0041] "Anthracycline" refers to anthracenedione anticancer agents and includes anthracycline analogs and anthracycline derivatives. Anthracyclines include, for example, aclarubicin, daunorubicin, doxorubicin, epirubicin, idarubicin, and pirarubicin. Anthracycline analogs are described, for example, in the references, Henry, Cancer Chemotherapy, A CS Symposium Series, 15-57 (1976); Nagy et al., Proc. Natl. Acad. Sci. USA, 93:2464-9 (1996); Bakina et al., Anti-Cancer Drug Design, 14:507-15 (1999); Perrin et al., Nucleic Acids Research, 1781 (1999); U.S. Patent App. No. 2007/0060534; U.S. Patent Nos. 4,301,277; 4,314,054; 4,464,529; 4,585,859; 4,591,637; 4,826,964; 5,843,903; and
6,184,374; 5,962,216; 5,196,522; 6,218,519; 6,433,150; PCT Pub. No. WO 98/13059; and Eur. Pat. No. EP 02/90744.
[0042] "Aryl" refers to a substituted or unsubstituted cyclic moiety that includes one or more monocyclic or fused ring aromatic systems. Such moieties include any moiety that has one or more monocyclic or bicyclic fused ring aromatic systems, including but not limited to phenyl and naphthyl.
[0043] "Bioreductive group" refers to a substituted or unsubstituted nitroaryl, nitroheteroaryl, indoloquinonyl, or a naphtoquinonyl moiety that can undergo reduction. Bioreductive groups are described for example in the U.S. Pat. Nos. 5,750,782; 5,780,585; 5,872,129; 6,251,933; 5,306,727; 5,403,932; 5,190,929; and 6,656,926; PCT Pat. Appl. Pub. Nos. WO 00/64864; 04/85361; 04/85421; 04/87075; 06/57946; and 07/02931; U.S. Pat. Appl. Pub. Nos. 2003/0008850; 2004/0254103; and 2005/0043244, and the references deGroot et al., Current Med. Chem., 8:1093-22 (2001); Borch et al., J Med. Chem., 43:2258-65 (2000); Borch et al., J. Med. Chem., 44:69-73 (2001); Borch et al., J. Med. Chem., 44:14-11 (2001); Hernick et al., J. Med. Chem., 45:3540-48 (2002); Hernick et al., J. Med. Chem., 46:148-54 (2003); Papot et al., Curr. Med. Chem., 2:155-85 (2002); Tercel et al., J. Med. Chem., 39:1084-94 (1996); and Tercel et al., J. Med. Chem., 44:3511-22 (2001) (each of which is incorporated herein by reference).
[0044] "Cycloalkyl" or "carbocycle", by themselves or in combination with other terms, refers to, unless otherwise stated, cyclic versions of "alkyl", "alkenyl" and "alkynyl" in which all ring atoms are carbon . "Cycloalkyl" or "carbocycle" refers to a mono- or polycyclic group. When used in connection with cycloalkyl substituents, the term "polycyclic" refers herein to fused and non-fused alkyl cyclic structures. "Cycloalkyl" or "carbocycle" may form a bridged ring or a spiro ring. The cycloalkyl group may have one or more double or triple bond(s). The term "cycloalkenyl" refers to a cycloalkyl group that has at least one site of alkenyl unsaturation between the ring vertices. The term "cycloalkynyl" refers to a cycloalkyl group that has at least one site of alkynyl unsaturation between the ring vertices. When "cycloalkyl" is used in combination with "alkyl", as in Cβ.gcycloalky^.galkylene-, the cycloalkyl portion is meant to have the stated number of carbon atoms (e.g., from three to eight carbon atoms), while the alkylene portion has from one to eight carbon atoms. Typical cycloalkyl substituents have from 3 to 8 ring atoms. Examples of cycloalkyl include cyclopentyl, cyclohexyl, 1-cyclohexenyl, 3-cyclohexenyl, cycloheptyl, and the like.
[0045] "Halogen" or halo" refers to by themselves or as part of another substituent, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom. Additionally, terms such as "haloalkyl," are meant to include alkyl in which one or more hydrogen is substituted with halogen atoms which can be the same or different, in a number ranging from one up to the maximum number of halogens permitted e.g., for alkyl (2m'+l), where m' is the total number of carbon atoms in the alkyl group. For example, the term "haloCl-8alkyl" is meant to include trifluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3-bromopropyl, and the like. The term "perhaloalkyl" means, unless otherwise stated, alkyl substituted with (2m'+l) halogen atoms, where m' is the total number of carbon atoms in the alkyl group. For example, the term "perhaloCl-βalkyl," is meant to include trifluoromethyl, pentachloroethyl, 1,1,1- trifluoro-2-bromo-2-chloroethyl, and the like. Additionally, the term "haloalkoxy" refers to an alkoxy radical substituted with one or more halogen atoms. "Halide" refers to the acid or anionic form of a halo group.
[0046] "Heteroalkyl" means an alkyl radical as defined herein with one, two or three substituents independently selected from cyano, -ORw, -NRxRy, and -S(O)nRz (where n is an integer from 0 to 2 ), with the understanding that the point of attachment of the heteroalkyl radical is through a carbon atom of the heteroalkyl radical. Rw is hydrogen, alkyl, cycloalkyl, cycloalkyl-alkyl, aryl, araalkyl, alkoxycarbonyl, aryloxycarbonyl, carboxamido, or mono- or di-alkylcarbamoyl. Rx is hydrogen, alkyl, cycloalkyl, cycloalkyl-alkyl, aryl or araalkyl. Ry is hydrogen, alkyl, cycloalkyl, cycloalkyl-alkyl, aryl, araalkyl, alkoxycarbonyl, aryloxycarbonyl, carboxamido, mono- or di-alkylcarbamoyl or alkylsulfonyl. Rz is hydrogen (provided that n is 0), alkyl, cycloalkyl, cycloalkyl-alkyl, aryl, araalkyl, amino, mono- alkylamino, di-alkylamino, or hydroxyalkyl. Representative examples include, for example, 2-hydroxyethyl, 2,3-dihydroxypropyl, 2-methoxyethyl, benzyloxymethyl, 2-cyanoethyl, and 2-methylsulfonyl-ethyl. For each of the above, Rw, Rx ,Ry, and Rz can be further substituted by amino, fluorine, alkylamino, di-alkylamino, OH or alkoxy. Additionally, the prefix indicating the number of carbon atoms (e.g., Ci-C10) refers to the total number of carbon atoms in the portion of the heteroalkyl group exclusive of the cyano, -ORw, -NRxRy, or - S(O)nRz portions.
[0047] "Heteroaryl" refers to a substituted or unsubstituted monocyclic aromatic system having 5 or 6 ring atoms, or a fused ring bicyclic aromatic system having 8-20 atoms, in which the ring atoms are C, O, S, SO, SO2, or N, and at least one of the ring atoms is a heteroatom, i.e., O, S, SO, SO2, or N. Heteroaryl groups include, for example, acridinyl,
azocinyl, benzimidazolyl, benzofuranyl, benzothio-furanyl, benzothiophenyl, benzoxazolyl, benzothiazolyl, benzotriazolyl, benzotetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, NH-carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, dithiazinyl, furanyl, furazanyl, imidazolidinyl, imidazolinyl, imidazolyl, indazolyl, indolenyl, indolinyl, indolizinyl, indolyl, isobenzofuranyl, isochromanyl, isoindazolyl, isoindolinyl, isoindolyl, isoquinolinyl, isothiazolyl, isoxazolyl, naphthyridinyl, octahydroisoquinolinyl, oxadiazolyl, oxazolidinyl, oxazolyl, oxazolidinyl, pyrimidinyl, phenanthridinyl, phenanthrolinyl, phenazinyl, phenothiazinyl, phenoxathiinyl, phenoxazinyl, phthalazinyl, piperazinyl, pteridinyl, purinyl, pyranyl, pyrazinyl, pyrazolidinyl, pyrazolinyl, pyrazolyl, pyridazinyl, pyridooxazolyl, pyridoimidazolyl, pyridothiazole, pyridinyl, pyridyl, pyrimidinyl, pyrrolyl, quinazolinyl, quinolinyl, quinoxalinyl, quinuclidinyl, tetrahydroisoquinolinyl, tetrahydroquinolinyl, tetrazolyl, thiadiazinyl, thiadiazolyl, thianthrenyl, thiazolyl, thienyl, thienothiazolyl, thienooxazolyl, thienoimidazolyl, thiophenyl, triazinyl and xanthenyl. Unless indicated otherwise, the arrangement of the heteroatoms within the ring may be any arrangement allowed by the bonding characteristics of the constituent ring atoms. "Heteroalkyl" refers to an alkyl radical as defined herein with one, two or three substituents, independently selected from cyano, -ORw, -NRxRy, and -S(O)nRz (where n is an integer from 0 to 2 ), with the understanding that the point of attachment of the heteroalkyl radical is through a carbon atom of the heteroalkyl radical. Rw is hydrogen, alkyl, cycloalkyl, cycloalkyl-alkyl, aryl, araalkyl, alkoxycarbonyl, aryloxycarbonyl, carboxamido, or mono- or di-alkylcarbamoyl. Rx is hydrogen, alkyl, cycloalkyl, cycloalkyl- alkyl, aryl or araalkyl. Ry is hydrogen, alkyl, cycloalkyl, cycloalkyl-alkyl, aryl, araalkyl, alkoxycarbonyl, aryloxycarbonyl, carboxamido, mono- or di-alkylcarbamoyl or alkylsulfonyl. Rz is hydrogen (provided that n is 0), alkyl, cycloalkyl, cycloalkyl-alkyl, aryl, araalkyl, amino, mono-alkylamino, di-alkylamino, or hydroxyalkyl. Representative examples include, for example, 2-hydroxyethyl, 2,3-dihydroxypropyl, 2-methoxyethyl, benzyloxymethyl, 2-cyanoethyl, and 2-methylsulfonyl-ethyl. For each of the above, Rw, Rx, Ry, and Rz can be further substituted by amino, fluorine, alkylamino, di-alkylamino, OH or alkoxy. Additionally, the prefix indicating the number of carbon atoms (e.g., Ci -Ci0) refers to the total number of carbon atoms in the portion of the heteroalkyl group exclusive of the cyano, -ORw, -NRxRy, or -S(O)nRz portions.
[0048] "Heterocyclyl" refers to a monocyclic or fused ring multicyclic cycloalkyl group at least a portion of which is not aromatic and in which one or more of the carbon atoms in the
ring system is replaced by a heteroatom selected from O, S, SO, SO2, P, or N. Examples of heterocyclyl groups include but are not limited to imidazolinyl, morpholinyl, piperidinyl, piperidin-2-onyl, piperazinyl, pyrrolidinyl, pyrrolidine-2-onyl, tetrahydrofuranyl, and tetrahydroimidazo [4,5-c] pyridinyl.
[0049] "Heteroalkylene" refers to a Ci-C6 alkylene as defined above wherein 1-3 carbon atoms in the hydrocarbon radical or a branched saturated divalent hydrocarbon radical is replaced with a heteroatom. Similarly, Ci-Ci0 heteroalkylene refers to a heteroalkylene group wherein 1 -5 carbon atoms in the hydrocarbon radical or a branched saturated divalent hydrocarbon radical is replaced with a heteroatom. Ci-C6 heteroalkylene groups include, for example, -CH2CH2-O-CH2CH2- and -CH2CH2-S-CH2CH2-.
[0050] "Heteroarylalkylenyloxycarbonyl" refers to an heteroarylalkylenyloxy group covalently bonded to a carbonyl. In other words, a heteroarylalkylenyloxycarbonyl group has the general structure -C(=O)-O-alkyl-heteroaryl.
[0051] "Leaving group" refers to a moiety that can be replaced by a nucleophile. Leaving groups include, for example, halo and sulfonate.
[0052] "Nitrate" refers to the acid or anionic form of a nitro group. [0053] "Nitro" refers to -NO2.
[0054] "Pharmaceutically acceptable salts" refers to salts of the active compounds which are prepared with relatively nontoxic acids or bases, depending on the particular substituents found on the compounds described herein. When compounds of the present invention contain relatively acidic functionalities, base addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired base, either neat or in a suitable inert solvent. Examples of salts derived from pharmaceutically-acceptable inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic, manganous, potassium, sodium, zinc and the like. Salts derived from pharmaceutically-acceptable organic bases include salts of primary, secondary and tertiary amines, including substituted amines, cyclic amines, naturally-occurring amines and the like, such as arginine, betaine, caffeine, choline, N,N'-dibenzylethylenediamine, diethylamine, 2- diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N- ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine
resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like. When compounds of the present invention contain relatively basic functionalities, acid addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired acid, either neat or in a suitable inert solvent. Examples of pharmaceutically acceptable acid addition salts include those derived from inorganic acids like hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric, hydriodic, or phosphorous acids and the like, as well as the salts derived from relatively nontoxic organic acids like acetic, propionic, isobutyric, malonic, benzoic, succinic, suberic, fumaric, mandelic, phthalic, benzenesulfonic, p- tolylsulfonic, citric, tartaric, methanesulfonic, and the like. Also included are salts of amino acids such as arginate and the like, and salts of organic acids like glucuronic or galactunoric acids and the like {see, e.g., Berge, S.M. et al ., "Pharmaceutical Salts," Journal of Pharmaceutical Science, 66:1-19, 1977). Certain specific compounds of the present invention contain both basic and acidic functionalities that allow the compounds to be converted into either base or acid addition salts.
[0055] The neutral forms of the compounds may be regenerated by contacting the salt with a base or acid and isolating the parent compound in the conventional manner. The parent form of the compound differs from the various salt forms in certain physical properties, such as solubility in polar solvents, but otherwise the salts are equivalent to the parent form of the compound for the purposes of the present invention.
[0056] "Reducing agent" refers to a compound which donates electrons in a reduction- oxidation reaction. In some embodiments the donation of electrons may be in the form of an atom or molecule, such as a hydrogen atom or molecule. Reducing agents include, for example, hydride reducing agents. Hydride reducing agents include, for example, NaBH3CN and NaBH(OAc)3 Reducing agents are described, for example, in the references, or are prepared by methods known to those skilled in the art following procedures set forth in references such as Fieser and Fieser's Reagents for Organic Synthesis; Wiley & Sons: New York, 1967-2004, Volumes 1-22; Rodd's Chemistry of Carbon Compounds, Elsevier Science Publishers, 1989, Volumes 1-5 and Supplementals; and Organic Reactions, Wiley & Sons: New York, 2005, Volumes 1-6 J.
[0057] In each of the above embodiments designating a number of atoms, e.g., "C]-C6 or Ci-6" is meant to include all possible embodiments that have one fewer atom. Non-limiting examples include Ci-6, C2-6, C3-6, C4-6, C5-6 and the like. C2-Ci0 or C2-10 is meant to include all possible embodiments that have one fewer atom. Non- limiting examples include C2-I0, C3-I0, C4-Io, C5-Io, C6-I0, and the like.
[0058] Each of the terms herein (e.g., "alkyl, " "heteroalkyl, " "aryl" and "heteroaryl") is meant to include both "unsubstituted" and optionally "substituted" forms of the indicated radical, unless otherwise indicated. Typically each radical is substituted with 0, 1, 2, 3, 4, or 5 substituents, unless otherwise indicated. Examples of substituents for each type of radical are provided below.
[0059] "Substituted" refers to a group as defined herein in which one or more bonds to a carbon(s) or hydrogen(s) are replaced by a bond to non-hydrogen and non-carbon atom "substituents" such as, but not limited to, a halogen atom such as F, Cl, Br, and I; an oxygen atom in groups such as hydroxyl groups, alkoxy groups, aryloxy, and acyloxy groups; a sulfur atom in groups such as thiol groups, alkyl and aryl sulfide groups, sulfone groups, sulfonyl groups, and sulfoxide groups; a nitrogen atom in groups such as amino, alkylamines, dialkylamines, arylamines, alkylarylamines, diarylamines, alkoxyamino, hydroxyamino, acylamino, sulfonylamino, N-oxides, imides, and enamines; and other heteroatoms in various other groups. "Substituents" also include groups in which one or more bonds to a carbon(s) or hydrogen(s) atom is replaced by a higher-order bond (e.g., a double- or triple-bond) to a heteroatom such as oxygen in oxo, acyl, amido, alkoxycarbonyl, aminocarbonyl, carboxyl, and ester groups; nitrogen in groups such as imines, oximes, hydrazones, and nitriles.
[0060] "Substituents" further include groups in which one or more bonds to a carbon(s) or hydrogen(s) atoms is replaced by a bond to a cycloalkyl, heterocyclyl, aryl, and heteroaryl groups. Representative "substituents" include, among others, groups in which one or more bonds to a carbon or hydrogen atom is/are replaced by one or more bonds to fluoro, chloro, or bromo group. Another representative "substituent" is the trifluoromethyl group and other groups that contain the trifluoromethyl group. Other representative "substituents" include those in which one or more bonds to a carbon or hydrogen atom is replaced by a bond to an oxygen atom such that the substituted alkyl group contains a hydroxyl, alkoxy, or aryloxy group. Other representative "substituents" include alkyl groups that have an amine, or a substituted or unsubstituted alkylamine, dialkylamine, arylamine, (alkyl)(aryl)amine,
diarylamine, heterocyclylamine, diheterocyclylamine, (alkyl)(heterocyclyl)amine, or (aryl)(heterocyclyl)amine group. Still other representative "substituents" include those in which one or more bonds to a carbon(s) or hydrogen(s) atoms is replaced by a bond to an alkyl, cycloalkyl, aryl, heteroaryl, or heterocyclyl group.
[0061] The herein-defined groups may include prefixes and/or suffixes that are commonly used in the art to create additional well-recognized substituent groups. As examples, "alkylamino" refers to a group of the formula -NRaRb. Unless stated otherwise, for the following groups containing Ra, Rb, Rc, Rd and Re: Ra and Rb are each independently selected from H, alkyl, alkoxy, thioalkoxy, cycloalkyl, aryl, heteroaryl, or heterocyclyl or are optionally joined together with the atom(s) to which they are attached to form a cyclic group. When Ra and Rb are attached to the same nitrogen atom, they can be combined with the nitrogen atom to form a 5-, 6- or 7-membered ring. For example, -NRaRb is meant to include 1-pyrrolidinyl and 4-morpholinyl.
[0062] Rc, Rd, Re and Rf, unless otherwise indicated, are each independently selected from alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, aryl, heteroaryl, heterocyclyl or alkylenearyl, as defined herein.
[0063] Typically, a particular radical will have 0, 1, 2 or 3 substituents, with those groups having two or fewer substituents being preferred in the present invention. More preferably, a radical will be unsubstituted or monosubstituted. Most preferably, a radical will be unsubstituted.
[0064] In some embodiments, "substituents" refers to an atom or group, including, for example, amino, Ci-C6alkylamino or di(Ci-C6)alkylamino, Ci-C6alkoxy, C]-C6alkylthio, aryl, -COOH, -CONH2, cyano, ethenyl, ethynyl, halo, heteroaryl, hydroxy, mono- or di(Ci-C6)alkylcarboxamido, mono or di(Ci-C6)alkylsulfonamido, nitro, -OSO2-Ry, and -SO2NH2.
[0065] "Substituents" for the alkyl and heteroalkyl radicals (as well as those groups referred to as alkylene, alkenyl, heteroalkylene, heteroalkenyl, alkynyl, cycloalkyl, heterocyclyl) can be a variety of groups selected from: -ORa, =0, =NRa, =N-0Ra, -NRaRb, -SRa, halogen, -SiRaRbRc, -OC(O)R3, -C(O)Ra, -CO2R3, -C0NRaRb, -OC(O)NRaRb, -NRbC(0)Ra, -NRa-C(0)NRbRc, -NRa-S02NRbRc, -NRbCO2Ra, -NH-C(NH2)=NH, -NRaC(NH2)=NH, -NH-C(NH2)=NRa, -S(O) Ra, -SO2Ra, -SO2NRaRb, -NRbSO2R, -CN and
-NO2, in a number ranging from zero to three, with those groups having zero, one or two substituents being particularly preferred.
[0066] In some embodiments, "substituents" for the alkyl and heteroalkyl radicals are selected from: -ORa, =0, -NRaRb, -SRa, halogen, -SiRaRbRc, -OC(O)R3, -C(O)Ra, -CO2R3, -CONRaRb, -OC(O)NRaRb, -NRbC(0)Ra, -NRbCO2R3, -NRa-SO2NRbRc, -S(O)R3, -SO2R3, -SO2NR3Rb, -NR0SO2R, -CN and -NO2, where Ra and Rb are as defined above. In some embodiments, substituents are selected from: -0Ra, =0, -NRaRb, halogen, -OC(O)Ra, -CO2R3, -C0NRaRb, -OC(O)NRaRb, -NRbC(0)Ra, -NRbC02Ra, -NRa-S02NRbRc, -SO2R3, -SO2NR3Rb, -NR11SO2R, -CN and -NO2.
[0067] Examples of substituted alkyl are: -(CH2)3NH2, -(CH2)3NH(CH3), -(CH2)3NH(CH3)2, -CH2C(OH2)CH2NH2, -CH2C(O)CH2NH2, -CH2S(=O)2CH3, -CH2OCH2NH2, -CO2H. Examples of substituents of substituted alkyl are: CH2OH, -OH, -OCH3, -OC2H5, -OCF3, -OC(=O)CH3, -OC(O)NH2, -OC(=O)N(CH3)2, -CN, -NO2, -C(O)CH3, -CO2H, -CO2CH3, -CONH2, -NH2, -N(CH3)2, -NHSO2CH3, -NHCOCH3, -NHC(O)OCH3, -NHSO2CH3, -SO2CH3, -SO2NH2, and halo.
[0068] Similarly, "substituents" for the aryl and heteroaryl groups are varied and are selected from: -halogen, -OR3, -OC(O) R3, -NR3Rb, -SR3, -R3, -CN, -N02, -CO2R3, -C0NRaRb, -C(O) Ra, -0C(0)NR3Rb, -NRbC(O) R3, -NRbC(0)2Ra, -NR3-C(0)NRbRc, -NH-C(NH2)=NH, -NR3C(NH2)=NH, -NH-C(NH2)=NR3, -S(O) R3, -S(O) 2 R3, -S(O)2NR'Rb, -N3, -CH(Ph)2, perfluoroCi-8alkoxy, and perfluoroCl-όalkyl, in a number ranging from zero to the total number of open valences on the aromatic ring system; and where Ra, Rb and Rc are independently selected from hydrogen, Ci-6alkyl and heteroalkyl, unsubstituted aryl and heteroaryl, (unsubstituted aryl)-Ci-8alkyl, and (unsubstituted aryl)oxy- Ci-6alkyl.
[0069] Two of the "substituents" on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -T-C(O)-(CH2)q-U-, wherein T and U are independently -NH-, -0-, -CH2- or a single bond, and q is O, 1 or 2. Alternatively, two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -A-(CH2)r-B-, wherein A and B are independently -CH2-, -0-, -NH-, -S-, -S(O)-, -S(0)2-, -S(O) 2NRa- or a single bond, and r is 1, 2 or 3. One of the single bonds of the new ring so formed may optionally be replaced with a double bond. Alternatively, two of the substituents on adjacent atoms of the aryl or heteroaryl ring may
optionally be replaced with a substituent of the formula -(CH2)s-X-(CH2)t-, where s and t are independently integers of from 0 to 3, and X is -O-, -NRa-, -S-, -S(O)-, -S(O)2-, or -S(O) 2NRa-. The substituent Ra in -NRa- and -S(O)2NRa- is selected from hydrogen or unsubstituted Cl-6alkyl. Otherwise, R' is as defined above.
[0070] Unless indicated otherwise, the nomenclature of substituents that are not explicitly defined herein are arrived at by naming the terminal portion of the functionality followed by the adjacent functionality toward the point of attachment. For example, the substituent "arylalkyloxycarbonyl" refers to the group (aryl)-(alkyl)-O-C(O)-.
[0071] "Sulfonate" refers to a moiety having the structure of the formula -OSO2Ry wherein Ry is selected from the group consisting of Ci-C6 alkyl, Ci-C6 heteroalkyl, C3-Cg cycloalkyl, heterocyclyl, aryl, and heteroaryl.
[0072] "Administering" or "administration of a drug to a patient (and grammatical equivalents of this phrase) refers to direct administration, which may be administration to a patient by a medical professional or may be self-administration, and/or indirect administration, which may be the act of prescribing a drug. For example, a physician who instructs a patient to self-administer a drug and/or provides a patient with a prescription for a drug is administering the drug to the patient.
[0073] "Anoxia" or "anoxic condition" refers to an oxygen level that is zero or immeasurably low.
[0074] "Hypoxia" or "hypoxic condition" refers to an oxygen level lower than that observed under normoxia, and includes anoxia.
[0075] "Normoxia" or "normoxic condition" refers to an oxygen level observed in normally oxygenated cells and tissue, and in cells and tissue in contact with air.
[0076] "Pharmaceutically acceptable carrier, excipient, or diluent" refers to a carrier, excipient, or diluent that is useful in preparing a pharmaceutical composition that is generally safe, non-toxic and neither biologically nor otherwise undesirable, and includes a carrier, excipient, or diluent that is acceptable for human pharmaceutical use as well as veterinary use. A "pharmaceutically acceptable carrier, excipient, or diluent" includes both one and more than one such carrier, excipient, or diluent.
[0077] "Prodrug" refers to a compound that, after administration, is metabolized or otherwise converted to an active or more active form with respect to at least one property. A prodrug, relative to the drug it corresponds to, is modified chemically in a manner that renders it, relative to the drug, less active or inactive, but the chemical modification is such that the corresponding drug is generated by metabolic or other biological processes after the prodrug is administered. A prodrug may have, relative to the active drug, altered metabolic stability or transport characteristics, fewer side effects or lower toxicity, or improved flavor (for example, see the reference Nogrady, Medicinal Chemistry A Biochemical Approach, Oxford University Press, New York, 388-392 (1985), incorporated herein by reference). A prodrug does not have to be synthesized using the drug as an intermediate.
[0078] "Reduction" of a symptom or symptoms (and grammatical equivalents of this phrase) refers to decreasing the severity or frequency of the symptom(s), or elimination of the symptom(s).
[0079] "Therapeutically effective amount" of a substance refers to an amount of the substance that, when administered to a patient with cancer or another hyperproliferative disease, will have the intended therapeutic effect, e.g., alleviation, amelioration, palliation or elimination of one or more manifestations of cancer or another hyperproliferative disease in the patient. The full therapeutic effect does not necessarily occur by administration of one dose, and may occur only after administration of a series of doses. Thus, a therapeutically effective amount may be administered in one or more administrations.
[0080] "Treating" or "treatment of a condition or patient refers to taking steps to obtain beneficial or desired results, including clinical results. For purposes of this invention, beneficial or desired clinical results include, but are not limited to, alleviation or amelioration of one or more symptoms of cancer or another hyperproliferative disease; diminishnαent of extent of disease; delay or slowing of disease progression; amelioration, palliation, or stabilization of the disease state, or other beneficial results.
II. Compounds and Synthetic Methods
[0081] In one aspect, the present invention provides hypoxia activated prodrugs, or HAP compounds, of anticancer agents comprising one or more bioreductive Hyp moieties covalently bonded to the anticancer agent N, via a linker moiety L, wherein after hypoxic activation, the bioreduced Hyp moiety remains attached to the rest of the molecule, and the anticancer agent is selected from anthracyclines. In one embodiment, the present invention
provides a HAP compound having a structure of the formula selected from Hyp-L-Q and (Hyp-L)2-Q.
[0082] Suitable Hyp moieties useful in the HAP compounds of the present invention include, but are not limited to, nitroimidazoles, nitrofurans, nitrothiophenes, and nitrobenzenes. In one embodiment, the nitroimidazole is a 2-nitroimidazole. In another embodiment, the 2-nitroimidazole is covalently bonded to the anticancer agent by the L moiety wherein the L moiety is covalently bonded to the "1-N" position of the 2-nitroimidazole. In another embodiment, the imidazole moiety of the 2-nitroimidazole is suitably substituted. Suitable substituents include halo, Ci-C6 alkyl, Ci-C6 heteroalkyl, Ci-C6 alkenyl, Cj-C6 alkynyl, aryl, heteroaryl, and acyl. hi another embodiment, the imidazole moiety of the 2-nitroimidazole is suitably substituted at the 4-position of the imidazole moiety.
[0083] In another embodiment, each L is independently selected from the group consisting of Ci-Cio alkylene and Ci-Ci0 heteroalkylene. In another embodiment, each L is independently selected from the group consisting Of C2-Ci0 alkylene and C2-Ci0 heteroalkylene. In another embodiment, each L is independently selected from the group consisting Of Ci-C6 alkylene and Ci-C6 heteroalkylene moieties. In another embodiment, each L is independently selected from the group consisting OfC7-Ci0 alkylene C7-Ci0 and heteroalkylene moieties. In another embodiment, each L is independently selected from the group consisting of C2, C3, C4, C5 C6, C7, C8, C9 and Ci0 alkylene and C2, C3, C4, C5 C6, C7, C8, C9 and C10 heteroalkylene moieties, hi another embodiment, the L moiety is a C5 alkylene moiety. In another embodiment, the L moiety is a C5 heteroalkylene moiety. Other suitable L moieties useful in the HAP compounds of the present invention are described, for example, in U.S. Pat. Appl. Pub. Nos. US 2006/0258656 and U.S. Pat. Appl. No. 60/941,753 (each of which is incorporated herein by reference).
[0084] In accordance with the present invention, the carbon atoms and/or heteroatoms within the Ci-Ci0 alkylene and Ci-Ci0 heteroalkylene L-moieties can be substituted with one or more substituents. Suitable substituents on a carbon atom include fluoro and double bonded oxygen atom (=0). Suitably substituted heteroatom moieties within a Ci-Ci0 heteroalkylene L-moiety include -SO-, -SO2-, and -N(COR2)- wherein R2 is selected from the group consisting of hydrogen, CpC6 alkyl, Ci-C6 heteroalkyl, C3-C8 cycloalkyl, and heterocyclyl. In another embodiment in accordance with the present invention, the C1-Ci0
heteroalkylene chain of the L-moiety suitably includes an ester, amide (-CONR2-), sulfonamide, urea, and a carbamate moiety wherein R2 is defined as above.
[0085] In certain other embodiments, each L is independently selected from the group consisting of Ci-Ci0 alkylene and C1-C1O heteroalkylene moieties wherein adjacent carbon and/or heteroatoms are part of an aryl, a heteroaryl, a C3-C8 cycloalkyl, and a heterocyclyl moiety.
[0086] In certain other embodiments, each L is independently selected from the group consisting OfC4-C6 alkenylene and C4-C6 alkynylene moieties wherein one or more carbon atoms in each of the C4-C6 chain can be suitably substituted with one or more heteroatoms.
[0087] In certain other embodiments, the present invention provides HAP compounds having a structure of the formula Hyp-L-Q wherein L has a structure of the formula selected from the group consisting of:
 wherein Xi is selected from the group consisting of O, S, SO, SO2, CO, CF2, and -N(COR2)- wherein R2 is defined as above. In another embodiment, each L is independently selected from the group consisting of
wherein Xi is selected from the group consisting of O, S, SO, SO2, CO, CF2, and -N(COR2)- wherein R2 is defined as above. In another embodiment, each L is independently selected from the group consisting of
-Xl,
wherein Xi is selected from the group consisting of -SO2- and -CO-. In certain other embodiments, each L is independently selected from the group consisting of:

R wherein is selected from the group consisting of an aryl, a heteroaryl, a C3-C8 cycloalkyl, and a heterocyclyl moiety. Within these embodiments, the L moiety is covalently bonded to Hyp on the left side, and to Q on the right side of the L moieties, and wherein v/ww indicates the point of attachment of L to Hyp and Q.
[0088] In another embodiment, L has a structure of the formula -(CH2)n- wherein n is 2-10. In embodiments, L has a structure of the formula -(CH2)n- wherein n is independently
selected from the group consisting of 2, 3, 4, 5, 6, 7, 8, 9, or 10. In another embodiment, L has a structure of the formula -(CH2)2-(O-(CH2)2-)m- wherein m is 1-2. In another embodiment, L is an ethyleneoxyethylene (-(CH2)2-O-(CH2)2-) moiety.
[0089] In another embodiment, the anthracycline or the anthracycline analog useful in the HAP compounds of the present invention contains one or more NHR1 groups. A variety of functional groups are useful as R1 in the anthracyclines useful in the present invention, hi one embodiment, R1 is selected from the group consisting of hydrogen, C1-C6 alkyl, Ci-C6 heteroalkyl, Ci-C6 alkenyl, Ci-C6 alkynyl, and acyl. Anthracyclines suitable for use in the HAP compounds of the present invention include but are not limited to doxorubicin and daunorubicin. Other anthracyclines useful in the HAP compounds of the present invention are described for example in U.S. Pat. Appl. Pub. Nos. US 2007/0060534 and US 2006/0258656 and PCT Appl. Pub. No. WO 05/086951 (each of which is incorporated herein by reference).
[0090] In another embodiment, the present invention provides a HAP compound having a structures of the formula selected from Hyp-L-Q and (Hyp-L)2-Q wherein each Hyp-L moiety is covalently bonded to the NHRi -nitrogen atom of the anthracycline wherein Ri is defined as above. In another embodiment, each L is independently selected from the group consisting Of Ci-C6 alkylene and Ci-C6 heteroalkylene, and Hyp is a moiety having a structure of the formula:

[0091] hi another aspect, the present invention provides a HAP compound having a structure of the formula:


[0092] In another aspects, the present invention provides a HAP compound having a structure of the formula:


[0093] In certain embodiments within these aspects, each L is independently selected from the group consisting of the group consisting of -(CH2)n- and -(CH2)2-(O-(CH2)2)m- wherein n is 2-10 and m is 1-2. In another embodiment, n is independently selected from the group consisting of 2, 3, 4, 5, 6, 7, 8, 9, or 10. In another embodiment, Ri is hydrogen.
[0094] In another aspect, the present invention provides a HAP compound having a structure of the formula (Hyp-L)-Q-(CO2CR4R5-Brg) wherein Q is an anthracycline, L is independently selected from the group consisting of Ci-Ci0 alkylene and Ci-Ci0 heteroalkylene, and Hyp is a moiety having a structure of the formula:
 each OfR4 and R5 is selected independently from the group consisting of hydrogen, Ci-C6 alkyl Ci-C6 heteroalkyl, C3-C8 cycloalkyl, heterocyclyl, aryl and heteroaryl.
each OfR4 and R5 is selected independently from the group consisting of hydrogen, Ci-C6 alkyl Ci-C6 heteroalkyl, C3-C8 cycloalkyl, heterocyclyl, aryl and heteroaryl.
[0095] Brg is a bioreductive moiety having a structure of the formula


wherein R6 is selected from the group consisting of hydrogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl aryl, heteroaryl, and halo and R7 is Ci-C6 alkyl Ci-C6 heteroalkyl, C3-C8 cycloalkyl, heterocyclyl, aryl and heteroaryl. One of skill in the art will appreciate upon reading this disclosure that each of the aryl and heteroaryl moieties shown above as part of the Hyp moieties can be further substituted in accordance with the present invention. Suitable substituents include, but are not limited to, groups that as part of the compounds of the present invention enhance aqueous solubility of these compounds. Other suitable substituents include electron withdrawing groups such as nitro, halo, cyano, haloalkyls, and the like. Certain other suitable substituents include electron donating substituents such as alkoxy, amino, alkylamino, dialkylamino, and the like.
[0096] In one embodiment, the anthracycline has a structure of the formula:
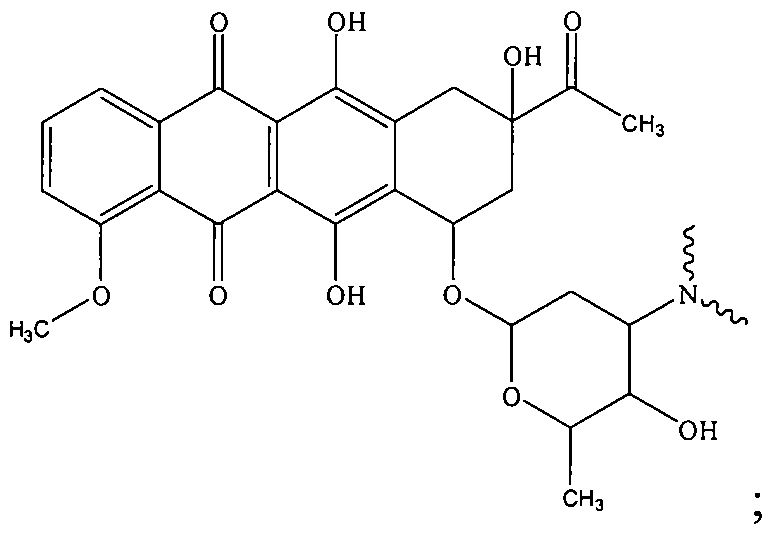

[0097] In another embodiment, each OfR4 and R5 is hydrogen. In another embodiment, R6 is hydrogen. In another embodiment, each L is independently selected from the group consisting of C5 alkylene and C5 heteroalkylene.
[0098] In another embodiment, the present invention provides a HAP compounds having structure of the formula selected from:



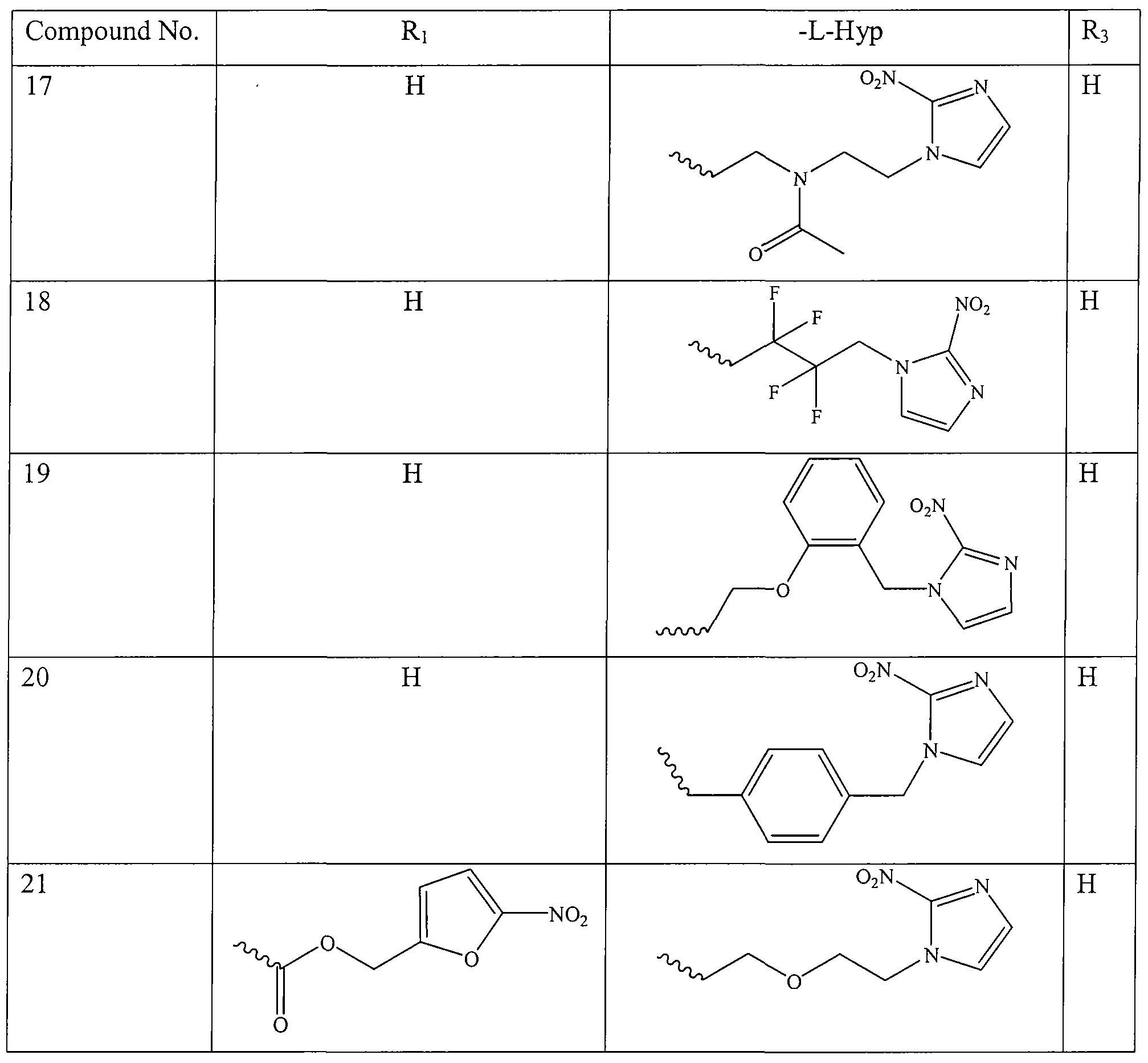
[0099] Without being bound by mechanism, HAP compounds of the present invention such as COMPOUND 1, COMPOUND 2, and the like, kill cancer cells as described below. In the hypoxic tumor zone, COMPOUND 1 undergoes reduction to a bis-hydroxylamine, and COMPOUND 2, or another HAP compound of the invention containing a single 2-nitroimidazole moiety, to a mono-hydroxylamine, that can alkylate DNA as shown below:

[0100] The anthraquinone portion of daunorubicin can intercalate between DNA bases. Together, the intercalation and alkylation of the DNA hinders the replication of cancer cell DNA and kill cancer cells. Under normoxic conditions, such as those existing in a normal cell, the bis-hydroxylamine and mono-hydroxylamine alkylators are not formed, the DNA in those normal cells is not alkylated, and the normal cells are not killed or are killed to a lesser extent compared to hypoxic tumor cells.
[0101] The HAP compounds of the present invention can be synthesized according to the novel method described below and/or other methods known to one of skill in the art upon reading this disclosure.
[0102] In another aspect, the present invention provides a method of synthesizing a HAP compound of the present invention having a structure of the formula Hyp-L-Q wherein Hyp is a bioreductive group, Q is an anticancer agent selected from anthracyclines, and L is a linker selected from C2-Ci0 alkylene and C2-Ci0 heteroalkylene, said method comprising reacting an anthracycline or an anthracycline analog containing one or more NHRi groups wherein R] is selected from the group consisting of hydrogen, Ci-C6 alkyl, Ci-C6 heteroalkyl, Ci -C6 alkenyl, Ci-C6 alkynyl, acyl, with a compound having a structure of the formula Hyp- Li-CHO wherein Li is selected from Ci -Cg alkylene and C1-C9 heteroalkylene and a reducing agent to yield the compound having the structure of the formula Hyp-L-Q. Anthracycline are
useful in the present synthetic methods as free bases and as ammonium salts. In one embodiment, reacting an anthracycline or an anthracycline analog in the free base form according to the present methods yields a HAP compound having structure of the formula Hyp-L-Q. In another embodiment, Ri is selected from Ci-C6 alkyl and Ci-C6 heteroalkyl. In another embodiment, the anthracycline containing one or more NHRi groups is daunorubicin.
[0103] In another aspect, the present invention provides a method of synthesizing a HAP compound having a structure of the formula:

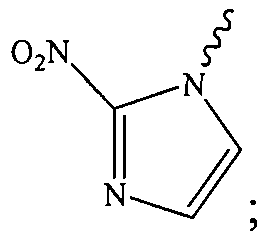

with a compound having a structure of the formula Hyp-Li-CHO, wherein Li is selected from C1-C9 alkylene and C1-C9 heteroalkylene, and a reducing agent to yield the compound having the structure of the formula:

[0104] In another embodiment, Li is selected from -(CH2)k- and -CH2-(O-(CH2)2)m- wherein k is 1-9 and m is 1-2. In another embodiment, Li is selected from the group consisting Of -CH2-, -(CH2V, -(CH2)4-, -CH2-O-(CH2)2-, and -CH2-(O-(CH2)2-)2-. In another embodiment, the reducing agent is NaBH(OAc)3.
[0105] In another aspect, the present invention provides a method of synthesizing a HAP compound of the present invention having a structure of the formula (Hyp-L)2-Q wherein Hyp is a bioreductive group, Q is an anticancer agent selected from anthracyclines, and L is a linker selected from C2-CiO alkylene and C2-Ci0 heteroalkylene, said method comprising reacting an anthracycline or an anthracycline analog containing one or more NH2 groups with a compound having a structure of the formula Hyp-Li-CHO wherein Li is selected from C1-C9 alkylene and C1-C9 heteroalkylene and a reducing agent to yield the compound having a structure of the formula (Hyp-L)2-Q. In one embodiment, reacting acid salts of anthracycline analogs containing one or more NH2 groups according to the present methods yields a HAP compound having a structure of the formula (Hyp-L)2-Q. In another embodiment, the anthracycline containing one or more NH2 groups is selected from daunorubicin or doxorubicin.
[0106] In another aspect, the present invention provides a method of synthesizing a HAP compound having a structure of the formula




[0107] In another embodiment, Li is selected from -(CH2)k- and -CH2-(O-(CH2)2)m- wherein k is 1-9 and m is 1-2. In another embodiment, Li is selected from the group consisting of -CH2-, -(CH2)2-, -(CH2)4-, -CH2-O-(CH2)2-, and -CH2-(O-(CH2)2-)2-. In another embodiment, the reducing agent is NaBH(OAc)3.
[0108] These, and other, compounds of the present invention are also synthesized by adapting methods known to one of skill in the art and upon reading this disclosure. Synthetic
methods thus adaptable are described, for example, in U.S. Patent Application No. 2008/0102026.
[0109] A variety of Hyp moieties are useful in the synthetic methods of the present invention. In one embodiment, the Hyp moiety is a substituted or an unsubstituted 2-nitroimidazole moiety. Substituted 2-nitroimidazoles useful in synthesizing the HAP compounds of the present invention can be synthesized according to methods adapted from PCT Pat. Pub. No. WO 07/02931, and U.S. Patent Application Nos. 60/941,753 and 60/970,364, and/or those known in the literature.
III. Therapies
[0110] In other aspects, the present invention provides methods of treating cancer and other hyperproliferative diseases comprising administering a therapeutically effective amount of a HAP compound of the present invention to a patient in need of such treatment. In one embodiment, the HAP compound administered has a structure of the formula selected from Hyp-L-Q and (Hyp-L)2-Q, wherein Q is selected from anthracyclines, Hyp is a bioreductive group having a structure of the formula:

[0111] In another embodiment, the therapeutically effective amount of the HAP compound is administered as a pharmaceutically acceptable formulation comprising a HAP compound of the present invention and pharmaceutically acceptable diluents or excipients.
[0112] In another embodiment, the therapeutically effective amount is administered in a daily dose. The therapeutically effective daily dose can be administered by employing suitable unit dose forms of the HAP compounds of the present invention. In another embodiment, the daily dose is administered from once every day, once every two weeks, up
to, once every month. In another embodiment, the daily dose is administered parenterally or orally.
[0113] A HAP compound of the present invention, COMPOUND 2, was safely administered at a daily dose of 10 mg/kg to mice once every week, for three weeks. Converting the mouse dose to human equivalent dose (HED) by dividing the mouse dose by 12.3 yields a HED of about 0.8 mg/kg, daily; thus in one embodiment of the present invention, COMPOUND 2 is administered to treat cancer in humans, by administering a daily dose of up to 0.8 mg/kg to a patient in need of such treatment. Lower effective daily doses can also be administered.
[0114] Lower effective daily doses of COMPOUND 2 useful for administration according to the present methods can be determined by in vivo efficacy studies. For example, COMPOUND 2 can be administered at a daily dose of 5 mg/kg alone and in combination with another anticancer agent to xenograft tumor bearing mice. Another anthracycline such as doxorubicin is administered as control to mice bearing xenograft tumors, alone and in combination with the anticancer agent used with COMPOUND 2. The efficacy of COMPOUND 2 administration is determined by post-administration measurement of the mean and/or median tumor sizes and comparing them with the corresponding tumor sizes in the control groups.
[0115] Various cancers can be treated according to the methods of the present invention by administering the HAP compounds the present invention. In certain embodiments, the cancer treated is selected from the group consisting of cancer of the adrenal gland, bone, brain, breast, bronchi, colon and/or rectum, gallbladder, head and neck, kidneys, larynx, liver, lung, neural tissue, pancreas, prostate, parathyroid, skin, stomach, and thyroid. In another embodiment, the cancer treated is selected from the group consisting of acute and chronic lymphocytic and granulocytic tumors, adenocarcinoma, adenoma, basal cell carcinoma, cervical dysplasia and in situ carcinoma, Ewing's sarcoma, epidermoid carcinomas, giant cell tumor, glioblastoma multiforma, hairy-cell tumor, intestinal ganglioneuroma, hyperplastic corneal nerve tumor, islet cell carcinoma, Kaposi's sarcoma, leiomyoma, leukemias, lymphomas, malignant carcinoid, malignant melanomas, malignant hypercalcemia, marfanoid habitus tumor, medullary carcinoma, metastatic skin carcinoma, mucosal neuroma, myeloma, mycosis fungoides, neuroblastoma, osteo sarcoma, osteogenic and other sarcoma, ovarian tumor, pheochromocytoma, polycythemia vera, primary brain tumor, small-cell lung
tumor, squamous cell carcinoma of both ulcerating and papillary type, hyperplasia, seminoma, soft tissue sarcoma, retinoblastoma, rhabdomyosarcoma, renal cell tumor, topical skin lesion, veticulum cell sarcoma, and Wilm's tumor.
[0116] In one embodiment, the HAP compound of the present invention is administered for the treatment of cancer in combination with other anticancer agents or other anticancer therapies. Suitable anticancer therapies useful in accordance with the present methods include radiation therapy and surgery. Methods for treating cancer employing other hypoxia activated prodrugs are described, for example, in PCT Pat. Appl. Pub. Nos. WO 07/02931 and WO 06/57946 and U.S. Pat. Appl. Pub. No. US 2006/0258656 (each of which is incorporated herein by reference) and can be used for the treatment of cancer according to the present methods upon appropriate substitution of the other hypoxia activated prodrug compounds with the HAP compounds of the present invention.
[0117] In certain embodiments, the present invention provides methods of treating non- cancer hyperproliferative diseases characterized by cellular hyperproliferation (e.g., an abnormally increased rate or amount of cellular proliferation) in accordance with the present methods. In certain embodiments, the hyperproliferative disease is selected from the group consisting of allergic angiitis and granulomatosis (Churg-Strauss disease), asbestosis, asthma, atrophic gastritis, benign prostatic hyperplasia, bullous pemphigoid, coeliac disease, chronic bronchitis and chronic obstructive airway disease, chronic sinusitis, Crohn's disease, demyelinating neuropathies, dermatomyositis, eczema, including atopic dermatitis, eustachean tube diseases, giant cell arteritis, graft rejection, hypersensitivity pneumonitis, hypersensitivity vasculitis (Henoch-Schonlein purpura), irritant dermatitis, inflammatory hemolytic anemia, inflammatory neutropenia, inflammatory bowel disease, Kawasaki's disease, multiple sclerosis, myocarditis, myositis, nasal polyps, nasolacrimal duct diseases, neoplastic vasculitis, pancreatitis, pemphigus vulgaris, primary glomerulonephritis, psoriasis, periodontal disease, polycystic kidney disease, polyarteritis nodosa, polyangitis overlap syndrome, primary sclerosing cholangitis, rheumatoid arthritis, serum sickness, surgical adhesions, stenosis or restenosis, scleritis, scleroderma, strictures of bile ducts, strictures (of duodenum, small bowel, and colon), silicosis and other forms of pneumoconiosis, type I diabetes, ulcerative colitis, ulcerative proctitis, vasculitis associated with connective tissue disorders, vasculitis associated with congenital deficiencies of the complement system, vasculitis of the central nervous system, and Wegener's granulomatosis.
[0118] In one embodiment, the hyperproliferative disease treated is psoriasis, a disease characterized by the cellular hyperproliferation of keratinocytes which builds up on the skin to form elevated, scaly lesions. In another embodiment, the hyperproliferative disease treated is multiple sclerosis, a disease characterized by progressive demyelination in the brain. In another embodiment, the hyperproliferative diseases treated is rheumatoid arthritis, a multisystem chronic, relapsing, inflammatory disease that can lead to destruction and ankylosis of joints affected. In another embodiment, a HAP compound of the present invention is administered to prevent a hyperproliferative disease resulting from cellular proliferation on a prosthesis implanted in a patient by coating the prosthesis with a composition containing a HAP compound of the present invention.
[0119] The invention, having been described in summary and in detail, is illustrated but not limited by the Examples below, which describe methods for synthesizing HAP compounds of the present invention and demonstrate various in vitro and in vivo properties of these compounds including cytotoxicity and pharmacokinetics.
IV. EXAMPLES
Example 1. Synthesis of HAP Compounds of the Present Invention A. Synthesis of COMPOUND 1
[0120] Example IA describes the synthesis of COMPOUND 1, a HAP compound of the present invention having a structure of the formula (HyP-L)2-Q, wherein Hyp is an anthracycline containing an amino group, each L is a Ci-C1O heteroalkylene moiety, and Hyp has a structure of the formula:

[0121] COMPOUND 1 was synthesized according to a novel synthetic method of the present invention, starting from the ammonium salt of daunorubicin, daunorubicin hydrochloride, and a 2-nitroimidazole substituted at the 1-N-position with an aldehyde containing CpC6 heteroalkylene moiety.
[0122] OsO4 (0.1 equivalent) was added to a mixture of l-N-allyloxyethyl-2-nitroimidazole (620 mg, 1 equivalent) in diethyl ether (Et2O, 10 mL) and water (10 mL) at room temperature (rt), followed by the addition OfNaIO4 (1.68 g, 2.5 equivalent) over a period of 20 min. The reaction mixture was stirred at rt for 16 h and extracted with ethyl acetate (EtOAc), the EtOAc portion was dried and concentrated to yield a residue that was separated by column chromatography on silica gel using EtOAc/Hexane (0-20%) as eluent to yield l-formylmethyloxyethyl-2-nitroimidazole that was used in the next step. A solution of l-formylemethyloxyethyl-2-nitroimidazole (70 mg) and daunorubicin hydrochloride (150 mg) in dichloromethane (DCM, 2 mL) was stirred at rt for 40 min, and cooled down to O0C, followed by the addition OfNaBH(OAc)3 (220 mg). The temperature of the reaction mixture was allowed to come to rt and stirred for 2 h. The reaction mixture was washed with water, the DCM portion was dried, and separated by thick layer chromatography on silica gel using MeOH/DCM (0-1%) as eluent to yield COMPOUND 1.
B. Synthesis of COMPOUND 2
[0123] Example IB describes the synthesis of COMPOUND 2, a HAP compound of the present invention having a structure of the formula Hyp-L-Q wherein Hyp-L-Q wherein Hyp is an anthracycline containing an NH2 group, L is a C1-Ci0 heteroalkylene moiety, and Hyp has a structure of the formula:

[0124] COMPOUND 2 was synthesized according to a novel synthetic method of the present invention described above, starting from daunorubicin and a 2-nitroimidazole substituted at the 1-N-position with an aldehyde containing Ci-C6 heteroalkylene moiety.
[0125] Triethylamine (TEA, 27 μL) was slowly added to a suspension of daunorubicin hydrochloride (102 mg) in dichloromethane (DCM, 5 mL) at O0C to yield daunorubicin that was used without further purification. After 10 minutes, a solution of l-formylmethyloxyethyl-2-nitroimidazole (40 mg, synthesized as described in Example IA) in DCM (1 mL) was added to the daunorubicin containing mixture and stirred for 0.5 h at O0C, followed by the addition OfNaBH(OAc)3 (127 mg). The mixture was allowed to come to rt and stirred for 2-3 h. Volatiles were removed under vacuo and the residue separated by
column chromatography on silica gel employing DCM - DCM/MeOH (100:10) to obtain COMPOUND 2.
C. Synthesis of Other Compounds of the Invention
[0126] COMPOUND 4, COMPOUND 5, COMPOUND 8, and COMPOUND 9 were synthesized according to the method described in Example IB starting from daunorubicin and upon suitable substitution of the corresponding starting aldehyde. COMPOUND 7 was synthesized according to the method described in Example IB starting from COMPOUND 2 and methoxyacetaldehyde.
D. 1H-NMR Data of Certain HAP Compounds of the Invention I) COMPOUND 1 δ (ppm CDC13) 14.1 (IH, s); 13.2 (IH, s); 8.01 (IH, d); 7.80 (IH, t); 7.41 (IH, d); 7.18 (IH, s); 7.03 (IH, s); 5.58 (IH, s); 5.31 (IH, s); 4.81 (lH,s); 4.58-4.50 (2H, m); 4.50-4.38 (2H, m); 4.10 (3H, s); 4.04 (IH, m); 3.57 (IH, s); 3.38 (2H, m); 3.30 (2H, m); 3.18 (IH, d); 2.95 (IH, d); 2.74 (3H, m); 2.42 (3H, s); 2.36 (IH, d); 2.13 (IH, dd); 2.03 (2H,s); 1.87 (2H, m); 1.72 (2H, bs); 1.40 (3H, d). Daunorubicin aromatic ring: 8.01 (IH, d); 7.80 (IH, t); 7.41 (IH, d); 2-Nitroimdazole ring: 7.18 (IH, s); 7.03 (IH, s).
2) COMPOUND 2 δ (ppm CDCl3) 14.1 (IH, s); 13.3 (IH, bs); 8.03 (IH, d); 7.79 (IH, t); 7.4 (IH, d); 7.15 (IH, s); 7.08 (IH, s); 5.54 (IH, s); 5.29 (IH, s); 4.56 (2H, m); 4.09 (4H, bs); 3.73 (2H, s); 3.67 (IH, s); 3.50 (2H, m); 3.20 (IH, d); 2.95 (2H, d); 2.78 (2H, m); 2.42 (3H, s); 2.36 (IH, d); 2.10 (IH, dd); 2.03 (2H,s); 1.77 (2H, m); 1.36 (3H, d). Daunorubicin aromatic ring: 8.03 (IH, d); 7.79 (IH, t); 7.4 (IH, d); 2-Nitroimdazole ring: 7.15 (IH, s); 7.08 (IH, s).
Example 2. Cytotoxicities of the HAP Compounds of the Present Invention A. Antiproliferation Assay Under Hypoxia and Normoxia
[0127] (i) Example 2A(i) describes methods for determining cytotoxicities of HAP compounds of the present invention under hypoxia and normoxia by employing an AlamarBlue fluorescence intensity based detection of cell proliferation. H460 non-small cell
lung cancer cells (10,000 - 15,000 cells/well/500 μL, ATCC HTB-177) were seeded in glass inserts on 24- well plates in RPMIl 640 medium supplemented with 10% FBS and 1% Penicillin/Streptomycin (Invitrogen Corporation, Carlsbad, CA). The cells were incubated overnight at 37°C in 5% CO2, 95% air and 100% relative humidity (these incubation conditions were used throughout the experiment unless otherwise mentioned) and divided into 2 groups: a "control group" (no test compound), and "treatment groups" (in which the cells were kept in contact with the test compound at various concentrations for 2 h).
[0128] The control fluorescence intensity, or Fo, proportional to the cell population of the control group at the beginning of the experiment, was determined following an AlamarBlue assay (λeX = 550 nm and λem = 590 nm). See also, Biosource International Inc., Tech Application Notes, Use of Alamar Blue in the Measurement of Cell Viability and Toxicity, Determining IC5o. The cells in the treatment groups were incubated for 2 hours with 6 different concentrations of a test compound, under hypoxia (5% CO2, 5% H2, 90% N2) or normoxia (5% CO2, 95% air), media containing the test compound removed, fresh media added, and the cells incubated for 3 days. The fluorescence intensities of the various treatment group cells incubated with different concentrations of the test compound and having different cell populations, and the control group cells at the end of the experiment (F1) having the highest cell population among all the groups, was determined following an AlamarBlue assay. The fluorescence intensities determined were background corrected by subtracting F0, and normalized by dividing with FrF0.
[0129] The background corrected and normalized fluorescence intensities of the control group after 3 days of incubation, and the various treatment groups after 3 days of incubation, were plotted against the corresponding concentrations of the test compound. The IC50 value for the test compound, i.e., the concentration of the test compound that stopped 50% of the cells from proliferating, was calculated based on a best-fit plot using an F test (GraphPad Prism4 software, San Diego, CA). Other cell lines such as colon (HT29) and prostate (PC3) were employed to test the antiproliferation activity of a HAP compound of the invention under hypoxia and normoxia. The results are tabulated below:


COMPOUND 3
[0130] The results demonstrate that COMPOUND 2, a HAP compound of the present invention is about 400 times more cytotoxic under hypoxia than under normoxia. COMPOUND 2 also demonstrated an enhanced cytotoxicity under hypoxia compared to normoxia in a variety of other cell lines. In one embodiment of the present invention, COMPOUND 2 is administered to treat cancer by selectively killing hypoxic tumor cells and not killing or killing fewer of the normoxic, normal cells.
[0131] (ii) Cytotoxitites of the HAP compounds of the present invention were also determined employing a method as described in Example 2A(i) but where the cells in the treatment groups were incubated for 4.5 hours with the HAP compounds. The results are tabulated below.


COMPOUND 6
B. Testing the Cytotoxicity of HAP Compounds in Drug-Resistant Cell Lines
[0132] The cytotoxicities of the HAP compounds of the present invention were determined, under normoxia, in the MESSA-DX5 cell lines resistant to certain anticancer drugs and the results compared with the normoxic cytotoxicities determined in the corresponding non resistant, MESSA, cell line. Daunorubicin was used as a control compound. MESSA-DX5 cell lines are drug resistant due in part to the overexpression of the MDR-I protein efflux pump. The results are tabulated below.

C. Clonogenic Survival Assay Under Normoxia and Hypoxia
[0134] This example describes testing the cytotoxicity of a HAP compound of the present invention employing the clonogenic survival method. Exponentially growing human H460 cells (obtained from the ATCC) were seeded into 60 mm notched glass plates at a density of between 2.5 and 5x105 cells per plate and grown in RPMI medium supplemented with 10% fetal bovine serum for 2 days prior to initiating treatment with HAP compound of the present invention. On the day of the test, HAP compound stocks of known concentrations were prepared in complete medium, and 2 mL of the desired stock added to each plate. To achieve complete equilibration between the surrounding gas phase and the liquid phase, the lid of the glass plate was removed and the plate shaken for 5 minutes on an orbital shaker. The plates were recovered and stored inside a glove-box. The glove-box was evacuated and gassed with either a certified anoxic gas mixture (95% nitrogen and 5% carbon dioxide) or with an aerobic (normoxic) gas mixture (95% air and 5% carbon dioxide). Cells were then incubated with the drug for 3 hours at 37°C.
[0135] At the end of treatment with the HAP compound, plates were removed from each vessel, and the prodrug was promptly removed from the cells. Plates were washed with phosphate buffered saline and a solution of trypsin-EDTA and then trypsinized for 5 minutes at 37°C. Detached cells were neutralized with medium plus serum and collected by centrifugation for 5 min at 100xg. Cells were resuspended at approximately 1x106 cells/mL and diluted 10-fold to yield stock concentrations for plating. The concentration of each stock was determined by counting with a Coulter Z2 particle counter. Known numbers of cells were plated, and the plates were placed in an incubator for 13 days. Colonies were fixed and stained with a solution of 95% ethanol and 0.25% crystal violet. Colonies having greater
than 50 cells were counted, and the surviving fraction was determined. Plating efficiencies (PEs) were determined by dividing the number of colonies by the actual number of cells plated. Surviving fractions were calculated by dividing the PEs for treated cells by the PEs of untreated cells. ICg0 values were determined by a best-fit method. COMPOUND 3 demonstrated IC90 values of 10 μM under normoxia and 0.1 μM under hypoxic conditions tested. The cytotoxicities of other HAP compounds of the present invention can be determined in a similar way in H460 and other cell lines.
Example 3. Demonstration of In Vivo Pharmacokinetic Stability of COMPOUND 3
[0136] COMPOUND 2 was administered to mice and the in vivo pharmacokinetic parameters determined are tabulated below; daunorubicin was employed as a control compound. The data demonstrate that the pharmacokinetic properties of COMPOUND 2 are similar to that of the approved anticancer agent daunorubicin. In one embodiment of the present invention, thus, COMPOUND 2 is administered according to the present methods for the treatment of cancer.
Pharmacokinetics of COMPOUND 3

* * *
[0137] While the present invention has been described with reference to the specific embodiments thereof, it should be understood by those skilled in the art that various changes can be made and equivalents can be substituted without departing from the scope of the invention. Pn addition, many modifications can be made to adapt a particular situation, material, composition of matter, process, process step or steps, to achieve the benefits provided by the present invention without departing from the scope of the present invention. All such modifications are intended to be within the scope of the claims appended hereto.
[0138] All publications and patent documents cited herein are incorporated herein by reference as if each such publication or document was specifically and individually indicated to be incorporated herein by reference. Citation of publications and patent documents is not intended as an indication that any such document is pertinent prior art, nor does it constitute any admission as to the contents or date of the same.
Claims
1. A compound having a structure of the formula selected from the group consisting of:
Hyp-L-Q and (Hyp-L)2-Q;
and pharmaceutically acceptable salts thereof;
wherein Q is an anthracycline, each L is independently selected from the group consisting of Ci-Ci0 alkylene and Ci-Ci0 heteroalkylene, Hyp is a moiety having a structure of the formula:

denotes the point of attachment to L.
2. The compound of claim 1, having the formula Hyp-L-Q, wherein Q has a structure of the formula:

wherein Ri is selected from the group consisting of hydrogen, Q-C6 alkyl, and Ci-C6 heteroalkyl R3 is selected from H and OH; and
~ denotes the point of attachment to L.
3. The compound of claim 1 having the formula (Hyp-L)2-Q, wherein the Q has a structure of the formula:

wherein R3 is selected from H and OH; and
~ denotes the point of attachment to L.
4. The compound of Claim 2, wherein L is selected from the group consisting of a C5 alkylene and a C5 heteroalkylene.
5. The compound of Claim 3, wherein each L is independently selected from the group consisting of a C5 alkylene and a C5 heteroalkylene.
6. The compound of claim 2, wherein said C1-Ci0 alkylene has a structure of the formula -(CH2)n-, wherein n is 2, 3, 4, 5, 6, 7, 8, 9 or 10, and wherein said C1-C10 heteroalkylene has a structure of the formula -(CH2)2-(O-(CH2)5-)m-, wherein m is 1 or 2.
7. The compound of claim 3 wherein said C1-C10 alkylene has a structure of the formula -(CH2)n-, wherein n is 2, 3, 4, 5, 6, 7, 8, 9 or 10, and wherein said Ci-C10 heteroalkylene moiety has a structure of the formula -(CH2)2-(O-(CH2)5-)m-, wherein m is 1 or 2.
8. The compound of claim 6, wherein R3 is H.
9. The compound of claim 6, wherein R3 is OH.
10. The compound of claim 7, wherein R3 is H.
11. The compound of claim 7, wherein R3 is OH.
12. The compound of claim 8, wherein L has a structure of formula -(CH2)2-(O-(CH2)5-)m-, wherein m is 1 or 2.
13. The compound of claim 12, having a structure of the formula:

14. The compound of any one of claims 1-3, wherein L is independently selected from the group consisting of C2alkylene, C3alkylene, C4alkylene, C5alkylene C6alkylene, C7alkylene, C8alkylene, Cgalkylene; C10alkylene; C2heteroalkylene, C3alkylene, C4heteroalkylene, C5heteroalkylene C6heteroalkylene, C7heteroalkylene, C8heteroalkylene, Cgheteroalkylene and Ci0heteroalkylene.
15. A compound having a structure of the formula (Hyp-L)-Q-
(CO2CR4R5-Brg), wherein Q is an anthracycline, L is selected from the group consisting of Ci-Cio alkylene and Ci-Ci0 heteroalkylene, and Hyp is a moiety having a structure of the formula:

each OfR4 and R5 is independently selected from the group consisting of hydrogen, Ci-C6 alkyl, Ci-C6 heteroalkyl, C3-C8 cycloalkyl, heterocyclyl, aryl and heteroaryl, Brg has a structure of the formula: 

R6 is selected from the group consisting of hydrogen, Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, aryl, heteroaryl, and halo, R7 is selected from the group consisting of Ci-C6 alkyl, Ci-C6 heteroalkyl, C3-C8 cycloalkyl, heterocyclyl, aryl and heteroaryl; and
~ denotes the point of attachment to the rest of the molecule.
16. The compound of claim 15, wherein Q has a structure of the formula:


~ denotes the point of attachment to L.
17. The compound of claim 16 wherein L is a C1-C1Q heteroalkylene.
18. The compound of claim 17 wherein the compound has a structure of the formula

19. A pharmaceutically acceptable formulation comprising a compound of claims 1-18 and a pharmaceutically acceptable carrier, excipient, or diluent.
20. A method of making a compound having a structure of the formula

wherein Ri is selected from the group consisting of hydrogen, C1-C6 alkyl, and Ci-C6 heteroalkyl, R3 is selected from H and OH, L is selected from the group consisting of C2-CiO alkyl ene and C2-Ci0 heteroalkylene, and Hyp has a structure of the formula:

the method comprising reacting an compound having a structure of the formula: 
with a compound having a structure of the formula: 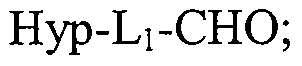
wherein L1 is selected from the group consisting of Ci-Cg alkylene and C1-C9 heteroalkylene, and a reducing agent to give the compound having a structure of the formula:

21. The method of claim 20 wherein the Li has a structure of the formula selected from the group consisting of -(CH2X- and -CH2-(O-(CH2)2)m-, wherein k is 1, 2, 3, 4, 5, 6, 7, 8 or 9 and m is 1-2 and the reducing agent is NaBH(OAc)3.
22. A method of synthesizing a compound having a structure of the formula 
wherein R3 is selected from H and OH, each L is independently selected from the group consisting Of C2-C1O alkylene and C2-C10 heteroalkylene; and Hyp has a structure of the formula:
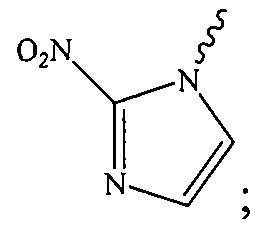
said method comprising reacting an acid salt of an anthracycline having a structure of the formula:

with a compound having a structure of the formula:
HyP-L1-CHO;
wherein L1 is selected from C2-Cg alkylene and C2-Cg heteroalkylene; and a reducing agent to give a compound having a structure of the formula:

23. The method of claim 22 wherein s the Li has a structure of the formula selected from the group consisting of -(CH2)k- and-CH2-(O-(CH2)2)m-, wherein k is 1 , 2, 3, 4, 5, 6, 7, 8 or 9 and m is 1-2 and the reducing agent is NaBH(OAc)3.
24. A method of treating cancer comprising administering a therapeutically effective amount of a compound of claims 1 - 17 to a patient in need of such treatment.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US95251207P | 2007-07-27 | 2007-07-27 | |
| US60/952,512 | 2007-07-27 | ||
| US97216207P | 2007-09-13 | 2007-09-13 | |
| US60/972,162 | 2007-09-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009018163A1 true WO2009018163A1 (en) | 2009-02-05 |
Family
ID=40304777
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/071228 WO2009018163A1 (en) | 2007-07-27 | 2008-07-25 | Hypoxia activated prodrugs of anthracyclines |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009018163A1 (en) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010048330A1 (en) | 2008-10-21 | 2010-04-29 | Threshold Pharmaceuticals, Inc. | Treatment of cancer using hypoxia activated prodrugs |
| WO2013122112A1 (en) | 2012-02-13 | 2013-08-22 | 国立大学法人筑波大学 | Prodrug using nitroimidazole |
| JP2016017059A (en) * | 2014-07-10 | 2016-02-01 | 国立大学法人 筑波大学 | Composition comprising antitumor prodrug |
| JP2018511612A (en) * | 2015-04-02 | 2018-04-26 | アセンタ ファーマシューティカルズ リミテッド | Nitrobenzyl derivatives of anticancer agents |
| US10364261B2 (en) | 2015-03-10 | 2019-07-30 | Obi Pharma, Inc. | DNA alkylating agents |
| US10409869B2 (en) | 2012-10-29 | 2019-09-10 | Obi Pharma, Inc. | (R)- and (S)-1-(3-(3-N,N-dimethylaminocarbonyl)phenoxyl-4-nitrophenyl)-1-ethyl-N,N'-bis (ethylene)phosphoramidate, compositions and methods for their use and preparation |
| US10654876B2 (en) | 2014-07-17 | 2020-05-19 | Molecular Templates, Inc. | TH-302 solid forms and methods related thereto |
| US10668047B2 (en) | 2015-06-24 | 2020-06-02 | Molecular Templates, Inc. | Aziridine containing DNA alkylating agents |
| WO2023025312A1 (en) | 2021-08-27 | 2023-03-02 | 深圳艾欣达伟医药科技有限公司 | Parp inhibitor-resistant patient treated with th-302 |
| WO2023025291A1 (en) | 2021-08-27 | 2023-03-02 | 深圳艾欣达伟医药科技有限公司 | Lyophilized formulation solution and lyophilized formulation, and method and use thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040191174A1 (en) * | 1991-10-29 | 2004-09-30 | Karen Linder | Rhenium and technetium complexes containing a hypoxia-localizing moiety |
| US20060258656A1 (en) * | 2003-03-28 | 2006-11-16 | Threshold Pharmaceuticals, Inc. | Compositions and methods for treating cancer |
| WO2007002931A2 (en) * | 2005-06-29 | 2007-01-04 | Threshold Pharmaceuticals, Inc. | Phosphoramidate alkylator prodrugs |
-
2008
- 2008-07-25 WO PCT/US2008/071228 patent/WO2009018163A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040191174A1 (en) * | 1991-10-29 | 2004-09-30 | Karen Linder | Rhenium and technetium complexes containing a hypoxia-localizing moiety |
| US20060258656A1 (en) * | 2003-03-28 | 2006-11-16 | Threshold Pharmaceuticals, Inc. | Compositions and methods for treating cancer |
| WO2007002931A2 (en) * | 2005-06-29 | 2007-01-04 | Threshold Pharmaceuticals, Inc. | Phosphoramidate alkylator prodrugs |
Non-Patent Citations (1)
| Title |
|---|
| NAGASAWA ET AL.: "Design of Hypoxia-targeting drugs as new cancer chemotherapeutics", BIOL. PHARM. BULL., vol. 29, no. 912, December 2006 (2006-12-01), pages 2335 - 2342, XP009085233 * |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010048330A1 (en) | 2008-10-21 | 2010-04-29 | Threshold Pharmaceuticals, Inc. | Treatment of cancer using hypoxia activated prodrugs |
| WO2013122112A1 (en) | 2012-02-13 | 2013-08-22 | 国立大学法人筑波大学 | Prodrug using nitroimidazole |
| JP5676020B2 (en) * | 2012-02-13 | 2015-02-25 | 国立大学法人 筑波大学 | Prodrug using nitroimidazole |
| US9655975B2 (en) | 2012-02-13 | 2017-05-23 | University Of Tsukuba | Prodrug using nitroimidazole |
| US10409869B2 (en) | 2012-10-29 | 2019-09-10 | Obi Pharma, Inc. | (R)- and (S)-1-(3-(3-N,N-dimethylaminocarbonyl)phenoxyl-4-nitrophenyl)-1-ethyl-N,N'-bis (ethylene)phosphoramidate, compositions and methods for their use and preparation |
| JP2016017059A (en) * | 2014-07-10 | 2016-02-01 | 国立大学法人 筑波大学 | Composition comprising antitumor prodrug |
| US10654876B2 (en) | 2014-07-17 | 2020-05-19 | Molecular Templates, Inc. | TH-302 solid forms and methods related thereto |
| US10766914B2 (en) | 2015-03-10 | 2020-09-08 | Obi Pharma, Inc. | DNA alkylating agents |
| US10364261B2 (en) | 2015-03-10 | 2019-07-30 | Obi Pharma, Inc. | DNA alkylating agents |
| JP2018511612A (en) * | 2015-04-02 | 2018-04-26 | アセンタ ファーマシューティカルズ リミテッド | Nitrobenzyl derivatives of anticancer agents |
| EP3277381A4 (en) * | 2015-04-02 | 2018-12-05 | Obi Pharma, Inc. | Nitrobenzyl derivatives of anti-cancer agents |
| AU2016244000B2 (en) * | 2015-04-02 | 2020-07-02 | Obi Pharma, Inc. | Nitrobenzyl derivatives of anti-cancer agents |
| CN108136214B (en) * | 2015-04-02 | 2020-08-28 | 深圳艾欣达伟医药科技有限公司 | Nitrobenzyl derivative anticancer agents |
| CN108136214A (en) * | 2015-04-02 | 2018-06-08 | 深圳艾衡昊医药科技有限公司 | Nitrobenzyl derivatives antitumor and anticancer agent |
| US10829437B2 (en) | 2015-04-02 | 2020-11-10 | Obi Pharma, Inc. | Nitrobenzyl derivatives of anti-cancer agents |
| CN112142692A (en) * | 2015-04-02 | 2020-12-29 | 深圳艾欣达伟医药科技有限公司 | Nitrobenzyl derivative anticancer agents |
| US11535585B2 (en) | 2015-04-02 | 2022-12-27 | Obi Pharma, Inc. | Nitrobenzyl derivatives of anti-cancer agents |
| US10668047B2 (en) | 2015-06-24 | 2020-06-02 | Molecular Templates, Inc. | Aziridine containing DNA alkylating agents |
| WO2023025312A1 (en) | 2021-08-27 | 2023-03-02 | 深圳艾欣达伟医药科技有限公司 | Parp inhibitor-resistant patient treated with th-302 |
| WO2023025291A1 (en) | 2021-08-27 | 2023-03-02 | 深圳艾欣达伟医药科技有限公司 | Lyophilized formulation solution and lyophilized formulation, and method and use thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009018163A1 (en) | Hypoxia activated prodrugs of anthracyclines | |
| WO2008151253A1 (en) | Hypoxia activated prodrugs of antineoplastic agents | |
| CA2613312C (en) | Phosphoramidate alkylator prodrugs | |
| SK284757B6 (en) | Arylsulfonanilide ureas, pharmaceutical composition containing them and use thereof | |
| CA2574032A1 (en) | Compounds and methods for the treatment of cancer | |
| CN106432014B (en) | Amido sulfur alcohol compound and preparation method thereof and its application in radiation protection | |
| FI71556B (en) | FOERFARANDE FOER FRAMSTAELLNING AV 5- (PARADEUTEROFENYL) -5-PHENYLHYDANTOIN VILKEN AER ETT CEEKEMEDEL ANVAENDBART MOT KONVULSIONER | |
| WO2009033165A1 (en) | Hypoxia activated prodrugs of bis-alkylating agents | |
| CN108299313B (en) | Compound and application thereof in pharmacy | |
| US20210309630A1 (en) | Allosteric inhibitor of wee1 kinase | |
| EP1558620B1 (en) | Ruthenium anticancer complexes | |
| WO2008076826A1 (en) | Pyrophosphoramide alkylators | |
| CN109761898B (en) | Double-target inhibitor and preparation method and application thereof | |
| US20210230105A1 (en) | Compounds with biguanidyl radical and uses thereof | |
| WO2013023274A1 (en) | Substituted 2-imidazolidinones and 2-imidazolones and their use in the treatment of cancer | |
| Datta et al. | Review on synthetic study of benzotriazole | |
| US5616563A (en) | Glutathione N-hydroxycarbamoyl thioesters and method of inhibiting neoplastic growth | |
| WO2012142698A1 (en) | Alkylurea derivatives active against cancer cells | |
| CN118320117B (en) | Molecular-based drug delivery system for multicellular organ targeting and in situ release | |
| WO2024089683A1 (en) | Anticancer drug conjugates | |
| Bolibrukh et al. | Synthesis of new thiosulfonate derivatives with quinone and quinoxaline fragments | |
| KR101484086B1 (en) | Novel emodin derivatives or pharmaceutically acceptable salt thereof, preparation method thereof and pharmaceutical composition for prevention or treatment of cancer disease containing the same as an active ingredient | |
| Karahan et al. | New Imidazolidindionedioximes and Their Pt (Ii) Complexes: Synthesis and Investigation of Their Antitumoral Activities on Breast Cancer Cells | |
| Mahmood | Aspects of the chemistry of 1, 4-naphthoquinones. An investigation of nucleophilic substitution reactions of alkylamines and hydroxyalyklamines on 1, 4 napthoquinones and the role of solvent on the position of substitution. | |
| CN102603635B (en) | Geldanamycin derivant and its production and use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08796653 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08796653 Country of ref document: EP Kind code of ref document: A1 |