WO2022202647A1 - ネガ型感光性樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス - Google Patents
ネガ型感光性樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス Download PDFInfo
- Publication number
- WO2022202647A1 WO2022202647A1 PCT/JP2022/012523 JP2022012523W WO2022202647A1 WO 2022202647 A1 WO2022202647 A1 WO 2022202647A1 JP 2022012523 W JP2022012523 W JP 2022012523W WO 2022202647 A1 WO2022202647 A1 WO 2022202647A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- formula
- resin composition
- acid
- Prior art date
Links
- 239000011342 resin composition Substances 0.000 title claims abstract description 252
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 59
- 239000004065 semiconductor Substances 0.000 title claims abstract description 18
- -1 nitrogen-containing cyclic compound Chemical class 0.000 claims abstract description 263
- 229920005989 resin Polymers 0.000 claims abstract description 205
- 239000011347 resin Substances 0.000 claims abstract description 205
- 239000003999 initiator Substances 0.000 claims abstract description 59
- 239000000203 mixture Substances 0.000 claims abstract description 50
- 125000004433 nitrogen atom Chemical group N* 0.000 claims abstract description 47
- 229940126062 Compound A Drugs 0.000 claims abstract description 42
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 claims abstract description 42
- 238000002835 absorbance Methods 0.000 claims abstract description 19
- 150000001875 compounds Chemical class 0.000 claims description 333
- 239000010410 layer Substances 0.000 claims description 141
- 239000002243 precursor Substances 0.000 claims description 108
- 238000010438 heat treatment Methods 0.000 claims description 83
- 229910052751 metal Inorganic materials 0.000 claims description 77
- 239000002184 metal Substances 0.000 claims description 77
- 239000000758 substrate Substances 0.000 claims description 41
- 239000003112 inhibitor Substances 0.000 claims description 24
- 238000006116 polymerization reaction Methods 0.000 claims description 24
- 239000003505 polymerization initiator Substances 0.000 claims description 23
- 230000000977 initiatory effect Effects 0.000 claims description 13
- 239000006087 Silane Coupling Agent Substances 0.000 claims description 10
- 238000010526 radical polymerization reaction Methods 0.000 claims description 10
- 239000011229 interlayer Substances 0.000 claims description 8
- 230000007423 decrease Effects 0.000 abstract description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 237
- 239000010408 film Substances 0.000 description 177
- 239000004642 Polyimide Substances 0.000 description 125
- 229920001721 polyimide Polymers 0.000 description 125
- 238000000034 method Methods 0.000 description 120
- 125000000962 organic group Chemical group 0.000 description 92
- 239000000047 product Substances 0.000 description 92
- 238000011161 development Methods 0.000 description 88
- 125000003118 aryl group Chemical group 0.000 description 83
- 125000000217 alkyl group Chemical group 0.000 description 79
- 125000002947 alkylene group Chemical group 0.000 description 77
- 125000001931 aliphatic group Chemical group 0.000 description 70
- 230000018109 developmental process Effects 0.000 description 68
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 68
- 239000003431 cross linking reagent Substances 0.000 description 67
- 239000004962 Polyamide-imide Substances 0.000 description 64
- 229920002312 polyamide-imide Polymers 0.000 description 64
- 150000003254 radicals Chemical class 0.000 description 64
- 239000002585 base Substances 0.000 description 51
- 229920000642 polymer Polymers 0.000 description 51
- 239000002253 acid Substances 0.000 description 48
- 229920002577 polybenzoxazole Polymers 0.000 description 44
- 239000002904 solvent Substances 0.000 description 41
- 125000004430 oxygen atom Chemical group O* 0.000 description 38
- 125000001424 substituent group Chemical group 0.000 description 37
- 230000008569 process Effects 0.000 description 34
- 229910052731 fluorine Inorganic materials 0.000 description 33
- 239000007787 solid Substances 0.000 description 33
- 239000000463 material Substances 0.000 description 32
- YEJRWHAVMIAJKC-UHFFFAOYSA-N 4-Butyrolactone Chemical compound O=C1CCCO1 YEJRWHAVMIAJKC-UHFFFAOYSA-N 0.000 description 30
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 30
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 30
- 238000006243 chemical reaction Methods 0.000 description 30
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 30
- 239000003960 organic solvent Substances 0.000 description 30
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 29
- 239000000126 substance Substances 0.000 description 29
- GTDPSWPPOUPBNX-UHFFFAOYSA-N ac1mqpva Chemical compound CC12C(=O)OC(=O)C1(C)C1(C)C2(C)C(=O)OC1=O GTDPSWPPOUPBNX-UHFFFAOYSA-N 0.000 description 28
- 150000004985 diamines Chemical class 0.000 description 28
- 125000001153 fluoro group Chemical group F* 0.000 description 28
- 229910052757 nitrogen Inorganic materials 0.000 description 28
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 27
- 238000000576 coating method Methods 0.000 description 27
- 125000004122 cyclic group Chemical group 0.000 description 27
- 125000005647 linker group Chemical group 0.000 description 27
- GZVHEAJQGPRDLQ-UHFFFAOYSA-N 6-phenyl-1,3,5-triazine-2,4-diamine Chemical compound NC1=NC(N)=NC(C=2C=CC=CC=2)=N1 GZVHEAJQGPRDLQ-UHFFFAOYSA-N 0.000 description 24
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 24
- 230000015572 biosynthetic process Effects 0.000 description 24
- 239000011248 coating agent Substances 0.000 description 24
- 239000007788 liquid Substances 0.000 description 24
- 239000000243 solution Substances 0.000 description 24
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 23
- 125000001183 hydrocarbyl group Chemical group 0.000 description 23
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 23
- 239000003795 chemical substances by application Substances 0.000 description 21
- 230000001976 improved effect Effects 0.000 description 21
- 125000003342 alkenyl group Chemical group 0.000 description 20
- 239000003963 antioxidant agent Substances 0.000 description 20
- 235000006708 antioxidants Nutrition 0.000 description 20
- BGTOWKSIORTVQH-UHFFFAOYSA-N cyclopentanone Chemical compound O=C1CCCC1 BGTOWKSIORTVQH-UHFFFAOYSA-N 0.000 description 20
- 235000019441 ethanol Nutrition 0.000 description 20
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 20
- 239000004094 surface-active agent Substances 0.000 description 20
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical group OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 19
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 19
- 125000001046 glycoluril group Chemical class [H]C12N(*)C(=O)N(*)C1([H])N(*)C(=O)N2* 0.000 description 19
- 238000003786 synthesis reaction Methods 0.000 description 19
- 125000004849 alkoxymethyl group Chemical group 0.000 description 18
- 150000002989 phenols Chemical class 0.000 description 18
- DQYSALLXMHVJAV-UHFFFAOYSA-M 3-heptyl-2-[(3-heptyl-4-methyl-1,3-thiazol-3-ium-2-yl)methylidene]-4-methyl-1,3-thiazole;iodide Chemical compound [I-].CCCCCCCN1C(C)=CS\C1=C\C1=[N+](CCCCCCC)C(C)=CS1 DQYSALLXMHVJAV-UHFFFAOYSA-M 0.000 description 17
- 239000000975 dye Substances 0.000 description 17
- 125000005843 halogen group Chemical group 0.000 description 17
- 150000003949 imides Chemical group 0.000 description 17
- 238000010521 absorption reaction Methods 0.000 description 16
- 230000004913 activation Effects 0.000 description 16
- 125000003545 alkoxy group Chemical group 0.000 description 16
- 125000003277 amino group Chemical group 0.000 description 16
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 16
- 125000006165 cyclic alkyl group Chemical group 0.000 description 16
- 230000000694 effects Effects 0.000 description 16
- 150000002148 esters Chemical class 0.000 description 16
- 238000011156 evaluation Methods 0.000 description 16
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 15
- 125000003710 aryl alkyl group Chemical group 0.000 description 15
- 230000000052 comparative effect Effects 0.000 description 15
- 238000001035 drying Methods 0.000 description 15
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 15
- ARXJGSRGQADJSQ-UHFFFAOYSA-N 1-methoxypropan-2-ol Chemical compound COCC(C)O ARXJGSRGQADJSQ-UHFFFAOYSA-N 0.000 description 14
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical group C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 14
- 239000007983 Tris buffer Substances 0.000 description 14
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 14
- 230000009471 action Effects 0.000 description 14
- 125000005042 acyloxymethyl group Chemical group 0.000 description 14
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 14
- 238000001723 curing Methods 0.000 description 13
- 125000000753 cycloalkyl group Chemical group 0.000 description 13
- 239000006185 dispersion Substances 0.000 description 13
- 238000001914 filtration Methods 0.000 description 13
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 13
- 230000005012 migration Effects 0.000 description 13
- 238000013508 migration Methods 0.000 description 13
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 13
- 229920001296 polysiloxane Polymers 0.000 description 13
- 125000006158 tetracarboxylic acid group Chemical group 0.000 description 13
- 239000010936 titanium Substances 0.000 description 13
- 229910052719 titanium Inorganic materials 0.000 description 13
- FVCSARBUZVPSQF-UHFFFAOYSA-N 5-(2,4-dioxooxolan-3-yl)-7-methyl-3a,4,5,7a-tetrahydro-2-benzofuran-1,3-dione Chemical compound C1C(C(OC2=O)=O)C2C(C)=CC1C1C(=O)COC1=O FVCSARBUZVPSQF-UHFFFAOYSA-N 0.000 description 12
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 12
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 12
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 12
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 12
- 125000000732 arylene group Chemical group 0.000 description 12
- LZCLXQDLBQLTDK-UHFFFAOYSA-N ethyl 2-hydroxypropanoate Chemical compound CCOC(=O)C(C)O LZCLXQDLBQLTDK-UHFFFAOYSA-N 0.000 description 12
- 238000005227 gel permeation chromatography Methods 0.000 description 12
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 11
- 125000004429 atom Chemical group 0.000 description 11
- 238000002156 mixing Methods 0.000 description 11
- LLHKCFNBLRBOGN-UHFFFAOYSA-N propylene glycol methyl ether acetate Chemical compound COCC(C)OC(C)=O LLHKCFNBLRBOGN-UHFFFAOYSA-N 0.000 description 11
- NQPJDJVGBDHCAD-UHFFFAOYSA-N 1,3-diazinan-2-one Chemical class OC1=NCCCN1 NQPJDJVGBDHCAD-UHFFFAOYSA-N 0.000 description 10
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 10
- 125000005529 alkyleneoxy group Chemical group 0.000 description 10
- 150000001408 amides Chemical class 0.000 description 10
- 230000003078 antioxidant effect Effects 0.000 description 10
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 10
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical compound C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 description 10
- 239000010949 copper Substances 0.000 description 10
- 125000003700 epoxy group Chemical group 0.000 description 10
- 241000894007 species Species 0.000 description 10
- 238000003860 storage Methods 0.000 description 10
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 10
- WGTYBPLFGIVFAS-UHFFFAOYSA-M tetramethylammonium hydroxide Chemical compound [OH-].C[N+](C)(C)C WGTYBPLFGIVFAS-UHFFFAOYSA-M 0.000 description 10
- VUMCUSHVMYIRMB-UHFFFAOYSA-N 1,3,5-tri(propan-2-yl)benzene Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1 VUMCUSHVMYIRMB-UHFFFAOYSA-N 0.000 description 9
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 9
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 9
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 9
- 102000012335 Plasminogen Activator Inhibitor 1 Human genes 0.000 description 9
- 108010022233 Plasminogen Activator Inhibitor 1 Proteins 0.000 description 9
- 239000002202 Polyethylene glycol Substances 0.000 description 9
- 239000004793 Polystyrene Substances 0.000 description 9
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical group CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 9
- 235000013877 carbamide Nutrition 0.000 description 9
- 229910052802 copper Inorganic materials 0.000 description 9
- 238000004132 cross linking Methods 0.000 description 9
- YAMHXTCMCPHKLN-UHFFFAOYSA-N imidazolidin-2-one Chemical class O=C1NCCN1 YAMHXTCMCPHKLN-UHFFFAOYSA-N 0.000 description 9
- 125000002950 monocyclic group Chemical group 0.000 description 9
- 229910052760 oxygen Inorganic materials 0.000 description 9
- 239000001301 oxygen Substances 0.000 description 9
- 229920001223 polyethylene glycol Polymers 0.000 description 9
- 229920002223 polystyrene Polymers 0.000 description 9
- 239000007870 radical polymerization initiator Substances 0.000 description 9
- 238000006798 ring closing metathesis reaction Methods 0.000 description 9
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 8
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 8
- 239000004593 Epoxy Substances 0.000 description 8
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical group OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 8
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 8
- 239000004698 Polyethylene Substances 0.000 description 8
- 239000006096 absorbing agent Substances 0.000 description 8
- 125000002252 acyl group Chemical group 0.000 description 8
- 125000005907 alkyl ester group Chemical group 0.000 description 8
- 239000007864 aqueous solution Substances 0.000 description 8
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 8
- 239000011737 fluorine Substances 0.000 description 8
- CATSNJVOTSVZJV-UHFFFAOYSA-N heptan-2-one Chemical compound CCCCCC(C)=O CATSNJVOTSVZJV-UHFFFAOYSA-N 0.000 description 8
- QQHJDPROMQRDLA-UHFFFAOYSA-N hexadecanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCCCC(O)=O QQHJDPROMQRDLA-UHFFFAOYSA-N 0.000 description 8
- ZSIAUFGUXNUGDI-UHFFFAOYSA-N hexan-1-ol Chemical compound CCCCCCO ZSIAUFGUXNUGDI-UHFFFAOYSA-N 0.000 description 8
- 238000003475 lamination Methods 0.000 description 8
- 229920000647 polyepoxide Polymers 0.000 description 8
- 125000006239 protecting group Chemical group 0.000 description 8
- 239000011541 reaction mixture Substances 0.000 description 8
- 239000007921 spray Substances 0.000 description 8
- 238000012719 thermal polymerization Methods 0.000 description 8
- TXBCBTDQIULDIA-UHFFFAOYSA-N 2-[[3-hydroxy-2,2-bis(hydroxymethyl)propoxy]methyl]-2-(hydroxymethyl)propane-1,3-diol Chemical compound OCC(CO)(CO)COCC(CO)(CO)CO TXBCBTDQIULDIA-UHFFFAOYSA-N 0.000 description 7
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 7
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 7
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 7
- 125000004450 alkenylene group Chemical group 0.000 description 7
- 230000008033 biological extinction Effects 0.000 description 7
- DKPFZGUDAPQIHT-UHFFFAOYSA-N butyl acetate Chemical compound CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 7
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 7
- 239000004202 carbamide Substances 0.000 description 7
- 239000012986 chain transfer agent Substances 0.000 description 7
- 239000007795 chemical reaction product Substances 0.000 description 7
- 125000004386 diacrylate group Chemical group 0.000 description 7
- 235000014113 dietary fatty acids Nutrition 0.000 description 7
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 7
- 239000003822 epoxy resin Substances 0.000 description 7
- 229930195729 fatty acid Natural products 0.000 description 7
- 239000000194 fatty acid Substances 0.000 description 7
- 125000000623 heterocyclic group Chemical group 0.000 description 7
- 150000002440 hydroxy compounds Chemical class 0.000 description 7
- 150000002739 metals Chemical class 0.000 description 7
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 7
- 125000002971 oxazolyl group Chemical group 0.000 description 7
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 description 7
- 239000002994 raw material Substances 0.000 description 7
- 229910052710 silicon Inorganic materials 0.000 description 7
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical group C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 6
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 6
- XLLIQLLCWZCATF-UHFFFAOYSA-N 2-methoxyethyl acetate Chemical compound COCCOC(C)=O XLLIQLLCWZCATF-UHFFFAOYSA-N 0.000 description 6
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 6
- OZJPLYNZGCXSJM-UHFFFAOYSA-N 5-valerolactone Chemical compound O=C1CCCCO1 OZJPLYNZGCXSJM-UHFFFAOYSA-N 0.000 description 6
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 6
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical group NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 6
- 239000004215 Carbon black (E152) Substances 0.000 description 6
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 6
- FFOPEPMHKILNIT-UHFFFAOYSA-N Isopropyl butyrate Chemical compound CCCC(=O)OC(C)C FFOPEPMHKILNIT-UHFFFAOYSA-N 0.000 description 6
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical compound CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 description 6
- 125000003647 acryloyl group Chemical group O=C([*])C([H])=C([H])[H] 0.000 description 6
- 239000003463 adsorbent Substances 0.000 description 6
- 229910052782 aluminium Inorganic materials 0.000 description 6
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 6
- 150000008064 anhydrides Chemical group 0.000 description 6
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 6
- XUPYJHCZDLZNFP-UHFFFAOYSA-N butyl butanoate Chemical compound CCCCOC(=O)CCC XUPYJHCZDLZNFP-UHFFFAOYSA-N 0.000 description 6
- 229910052799 carbon Inorganic materials 0.000 description 6
- 125000001142 dicarboxylic acid group Chemical group 0.000 description 6
- 150000002170 ethers Chemical class 0.000 description 6
- 229940116333 ethyl lactate Drugs 0.000 description 6
- 150000004665 fatty acids Chemical class 0.000 description 6
- NGAZZOYFWWSOGK-UHFFFAOYSA-N heptan-3-one Chemical compound CCCCC(=O)CC NGAZZOYFWWSOGK-UHFFFAOYSA-N 0.000 description 6
- 229930195733 hydrocarbon Natural products 0.000 description 6
- 238000007654 immersion Methods 0.000 description 6
- MLFHJEHSLIIPHL-UHFFFAOYSA-N isoamyl acetate Chemical compound CC(C)CCOC(C)=O MLFHJEHSLIIPHL-UHFFFAOYSA-N 0.000 description 6
- XMGQYMWWDOXHJM-UHFFFAOYSA-N limonene Chemical compound CC(=C)C1CCC(C)=CC1 XMGQYMWWDOXHJM-UHFFFAOYSA-N 0.000 description 6
- JDSHMPZPIAZGSV-UHFFFAOYSA-N melamine powder Natural products NC1=NC(N)=NC(N)=N1 JDSHMPZPIAZGSV-UHFFFAOYSA-N 0.000 description 6
- BDJRBEYXGGNYIS-UHFFFAOYSA-N nonanedioic acid Chemical compound OC(=O)CCCCCCCC(O)=O BDJRBEYXGGNYIS-UHFFFAOYSA-N 0.000 description 6
- 238000007363 ring formation reaction Methods 0.000 description 6
- 238000004528 spin coating Methods 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 150000003609 titanium compounds Chemical class 0.000 description 6
- 239000006097 ultraviolet radiation absorber Substances 0.000 description 6
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 6
- 235000012431 wafers Nutrition 0.000 description 6
- CDAWCLOXVUBKRW-UHFFFAOYSA-N 2-aminophenol Chemical compound NC1=CC=CC=C1O CDAWCLOXVUBKRW-UHFFFAOYSA-N 0.000 description 5
- HLBLWEWZXPIGSM-UHFFFAOYSA-N 4-Aminophenyl ether Chemical compound C1=CC(N)=CC=C1OC1=CC=C(N)C=C1 HLBLWEWZXPIGSM-UHFFFAOYSA-N 0.000 description 5
- QQGYZOYWNCKGEK-UHFFFAOYSA-N 5-[(1,3-dioxo-2-benzofuran-5-yl)oxy]-2-benzofuran-1,3-dione Chemical compound C1=C2C(=O)OC(=O)C2=CC(OC=2C=C3C(=O)OC(C3=CC=2)=O)=C1 QQGYZOYWNCKGEK-UHFFFAOYSA-N 0.000 description 5
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 5
- 229920000877 Melamine resin Polymers 0.000 description 5
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 5
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical compound CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 description 5
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 5
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 5
- 150000008065 acid anhydrides Chemical class 0.000 description 5
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 5
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 5
- 239000000654 additive Substances 0.000 description 5
- 150000001298 alcohols Chemical group 0.000 description 5
- 230000002744 anti-aggregatory effect Effects 0.000 description 5
- 125000004104 aryloxy group Chemical group 0.000 description 5
- 150000007514 bases Chemical class 0.000 description 5
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical class C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 5
- QRUDEWIWKLJBPS-UHFFFAOYSA-N benzotriazole Chemical compound C1=CC=C2N[N][N]C2=C1 QRUDEWIWKLJBPS-UHFFFAOYSA-N 0.000 description 5
- 239000012964 benzotriazole Substances 0.000 description 5
- 230000001588 bifunctional effect Effects 0.000 description 5
- PXKLMJQFEQBVLD-UHFFFAOYSA-N bisphenol F Chemical compound C1=CC(O)=CC=C1CC1=CC=C(O)C=C1 PXKLMJQFEQBVLD-UHFFFAOYSA-N 0.000 description 5
- 239000002981 blocking agent Substances 0.000 description 5
- 238000000354 decomposition reaction Methods 0.000 description 5
- 125000004427 diamine group Chemical group 0.000 description 5
- 150000001990 dicarboxylic acid derivatives Chemical class 0.000 description 5
- 150000005690 diesters Chemical class 0.000 description 5
- XXJWXESWEXIICW-UHFFFAOYSA-N diethylene glycol monoethyl ether Chemical compound CCOCCOCCO XXJWXESWEXIICW-UHFFFAOYSA-N 0.000 description 5
- USIUVYZYUHIAEV-UHFFFAOYSA-N diphenyl ether Chemical compound C=1C=CC=CC=1OC1=CC=CC=C1 USIUVYZYUHIAEV-UHFFFAOYSA-N 0.000 description 5
- 230000032050 esterification Effects 0.000 description 5
- 238000005886 esterification reaction Methods 0.000 description 5
- 238000005530 etching Methods 0.000 description 5
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 description 5
- 125000005842 heteroatom Chemical group 0.000 description 5
- 229910052739 hydrogen Inorganic materials 0.000 description 5
- 239000001257 hydrogen Substances 0.000 description 5
- 150000002576 ketones Chemical class 0.000 description 5
- 239000007769 metal material Substances 0.000 description 5
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 5
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 description 5
- 239000002245 particle Substances 0.000 description 5
- 239000011148 porous material Substances 0.000 description 5
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 5
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 5
- 150000003462 sulfoxides Chemical class 0.000 description 5
- 150000003536 tetrazoles Chemical group 0.000 description 5
- 238000012546 transfer Methods 0.000 description 5
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 description 4
- ZFPGARUNNKGOBB-UHFFFAOYSA-N 1-Ethyl-2-pyrrolidinone Chemical compound CCN1CCCC1=O ZFPGARUNNKGOBB-UHFFFAOYSA-N 0.000 description 4
- KBPLFHHGFOOTCA-UHFFFAOYSA-N 1-Octanol Chemical compound CCCCCCCCO KBPLFHHGFOOTCA-UHFFFAOYSA-N 0.000 description 4
- QFGCFKJIPBRJGM-UHFFFAOYSA-N 12-[(2-methylpropan-2-yl)oxy]-12-oxododecanoic acid Chemical compound CC(C)(C)OC(=O)CCCCCCCCCCC(O)=O QFGCFKJIPBRJGM-UHFFFAOYSA-N 0.000 description 4
- KJUGUADJHNHALS-UHFFFAOYSA-N 1H-tetrazole Chemical compound C=1N=NNN=1 KJUGUADJHNHALS-UHFFFAOYSA-N 0.000 description 4
- MSTZGVRUOMBULC-UHFFFAOYSA-N 2-amino-4-[2-(3-amino-4-hydroxyphenyl)-1,1,1,3,3,3-hexafluoropropan-2-yl]phenol Chemical compound C1=C(O)C(N)=CC(C(C=2C=C(N)C(O)=CC=2)(C(F)(F)F)C(F)(F)F)=C1 MSTZGVRUOMBULC-UHFFFAOYSA-N 0.000 description 4
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 4
- SVONRAPFKPVNKG-UHFFFAOYSA-N 2-ethoxyethyl acetate Chemical compound CCOCCOC(C)=O SVONRAPFKPVNKG-UHFFFAOYSA-N 0.000 description 4
- WXUAQHNMJWJLTG-UHFFFAOYSA-N 2-methylbutanedioic acid Chemical compound OC(=O)C(C)CC(O)=O WXUAQHNMJWJLTG-UHFFFAOYSA-N 0.000 description 4
- GPXCORHXFPYJEH-UHFFFAOYSA-N 3-[[3-aminopropyl(dimethyl)silyl]oxy-dimethylsilyl]propan-1-amine Chemical compound NCCC[Si](C)(C)O[Si](C)(C)CCCN GPXCORHXFPYJEH-UHFFFAOYSA-N 0.000 description 4
- XJMMNTGIMDZPMU-UHFFFAOYSA-N 3-methylglutaric acid Chemical compound OC(=O)CC(C)CC(O)=O XJMMNTGIMDZPMU-UHFFFAOYSA-N 0.000 description 4
- VVBLNCFGVYUYGU-UHFFFAOYSA-N 4,4'-Bis(dimethylamino)benzophenone Chemical compound C1=CC(N(C)C)=CC=C1C(=O)C1=CC=C(N(C)C)C=C1 VVBLNCFGVYUYGU-UHFFFAOYSA-N 0.000 description 4
- SGPJBCCVSFQQED-UHFFFAOYSA-N 4-[(4-aminophenyl)-[[dimethyl(trimethylsilyloxy)silyl]oxy-dimethylsilyl]oxy-methylsilyl]oxysilylaniline Chemical compound C=1C=C(N)C=CC=1[Si](C)(O[Si](C)(C)O[Si](C)(C)O[Si](C)(C)C)O[SiH2]C1=CC=C(N)C=C1 SGPJBCCVSFQQED-UHFFFAOYSA-N 0.000 description 4
- PLIKAWJENQZMHA-UHFFFAOYSA-N 4-aminophenol Chemical compound NC1=CC=C(O)C=C1 PLIKAWJENQZMHA-UHFFFAOYSA-N 0.000 description 4
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- PWLXTFFHCFWCGG-UHFFFAOYSA-N Heneicosanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCCCCCCCCC(O)=O PWLXTFFHCFWCGG-UHFFFAOYSA-N 0.000 description 4
- QCNWZROVPSVEJA-UHFFFAOYSA-N Heptadecanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCCCCC(O)=O QCNWZROVPSVEJA-UHFFFAOYSA-N 0.000 description 4
- 102100039856 Histone H1.1 Human genes 0.000 description 4
- 101001035402 Homo sapiens Histone H1.1 Proteins 0.000 description 4
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 4
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 4
- BTZVDPWKGXMQFW-UHFFFAOYSA-N Pentadecanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCCC(O)=O BTZVDPWKGXMQFW-UHFFFAOYSA-N 0.000 description 4
- KKEYFWRCBNTPAC-UHFFFAOYSA-N Terephthalic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-N 0.000 description 4
- QXGVRGZJILVMDF-UHFFFAOYSA-N Tetracosanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCCCCCCCCCCCC(O)=O QXGVRGZJILVMDF-UHFFFAOYSA-N 0.000 description 4
- 238000000862 absorption spectrum Methods 0.000 description 4
- 125000004423 acyloxy group Chemical group 0.000 description 4
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 4
- 125000004414 alkyl thio group Chemical group 0.000 description 4
- RWZYAGGXGHYGMB-UHFFFAOYSA-N anthranilic acid Chemical compound NC1=CC=CC=C1C(O)=O RWZYAGGXGHYGMB-UHFFFAOYSA-N 0.000 description 4
- 229910052786 argon Inorganic materials 0.000 description 4
- 125000005163 aryl sulfanyl group Chemical group 0.000 description 4
- 239000012965 benzophenone Substances 0.000 description 4
- 150000008366 benzophenones Chemical class 0.000 description 4
- 238000009835 boiling Methods 0.000 description 4
- 150000001721 carbon Chemical group 0.000 description 4
- 150000001244 carboxylic acid anhydrides Chemical class 0.000 description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 4
- 229910052801 chlorine Inorganic materials 0.000 description 4
- 238000006297 dehydration reaction Methods 0.000 description 4
- ISAOCJYIOMOJEB-UHFFFAOYSA-N desyl alcohol Natural products C=1C=CC=CC=1C(O)C(=O)C1=CC=CC=C1 ISAOCJYIOMOJEB-UHFFFAOYSA-N 0.000 description 4
- 150000001991 dicarboxylic acids Chemical class 0.000 description 4
- 229940075557 diethylene glycol monoethyl ether Drugs 0.000 description 4
- 239000000539 dimer Substances 0.000 description 4
- KZTYYGOKRVBIMI-UHFFFAOYSA-N diphenyl sulfone Chemical compound C=1C=CC=CC=1S(=O)(=O)C1=CC=CC=C1 KZTYYGOKRVBIMI-UHFFFAOYSA-N 0.000 description 4
- 238000009826 distribution Methods 0.000 description 4
- DGXRZJSPDXZJFG-UHFFFAOYSA-N docosanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCCCCCCCCCC(O)=O DGXRZJSPDXZJFG-UHFFFAOYSA-N 0.000 description 4
- 238000010894 electron beam technology Methods 0.000 description 4
- 125000006575 electron-withdrawing group Chemical group 0.000 description 4
- BHXIWUJLHYHGSJ-UHFFFAOYSA-N ethyl 3-ethoxypropanoate Chemical compound CCOCCC(=O)OCC BHXIWUJLHYHGSJ-UHFFFAOYSA-N 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 125000000524 functional group Chemical group 0.000 description 4
- 239000007789 gas Substances 0.000 description 4
- 235000011187 glycerol Nutrition 0.000 description 4
- 125000003055 glycidyl group Chemical group C(C1CO1)* 0.000 description 4
- JJWZFUFNJNGKAF-UHFFFAOYSA-N hexacosanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCCCCCCCCCCCCCC(O)=O JJWZFUFNJNGKAF-UHFFFAOYSA-N 0.000 description 4
- 229920001903 high density polyethylene Polymers 0.000 description 4
- 239000004700 high-density polyethylene Substances 0.000 description 4
- JJOJFIHJIRWASH-UHFFFAOYSA-N icosanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCCCCCCCC(O)=O JJOJFIHJIRWASH-UHFFFAOYSA-N 0.000 description 4
- 125000002883 imidazolyl group Chemical group 0.000 description 4
- 239000012535 impurity Substances 0.000 description 4
- 239000010954 inorganic particle Substances 0.000 description 4
- ZFSLODLOARCGLH-UHFFFAOYSA-N isocyanuric acid Chemical group OC1=NC(O)=NC(O)=N1 ZFSLODLOARCGLH-UHFFFAOYSA-N 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 4
- 229910052753 mercury Inorganic materials 0.000 description 4
- HSDFKDZBJMDHFF-UHFFFAOYSA-N methyl 3-ethoxypropanoate Chemical compound CCOCCC(=O)OC HSDFKDZBJMDHFF-UHFFFAOYSA-N 0.000 description 4
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 4
- WHTKRYWWSTYDNH-UHFFFAOYSA-N octacosanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCCCCCCCCCCCCCCCC(O)=O WHTKRYWWSTYDNH-UHFFFAOYSA-N 0.000 description 4
- 125000003566 oxetanyl group Chemical group 0.000 description 4
- VHDHONCVIHDOAO-UHFFFAOYSA-N pentacosanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCCCCCCCCCCCCC(O)=O VHDHONCVIHDOAO-UHFFFAOYSA-N 0.000 description 4
- 230000035699 permeability Effects 0.000 description 4
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 4
- WLJVNTCWHIRURA-UHFFFAOYSA-N pimelic acid Chemical compound OC(=O)CCCCCC(O)=O WLJVNTCWHIRURA-UHFFFAOYSA-N 0.000 description 4
- 238000009832 plasma treatment Methods 0.000 description 4
- 229920005575 poly(amic acid) Polymers 0.000 description 4
- 238000012545 processing Methods 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- CXMXRPHRNRROMY-UHFFFAOYSA-N sebacic acid Chemical compound OC(=O)CCCCCCCCC(O)=O CXMXRPHRNRROMY-UHFFFAOYSA-N 0.000 description 4
- 230000035945 sensitivity Effects 0.000 description 4
- 239000010703 silicon Substances 0.000 description 4
- 238000005507 spraying Methods 0.000 description 4
- 150000003512 tertiary amines Chemical class 0.000 description 4
- 150000000000 tetracarboxylic acids Chemical group 0.000 description 4
- HQHCYKULIHKCEB-UHFFFAOYSA-N tetradecanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCC(O)=O HQHCYKULIHKCEB-UHFFFAOYSA-N 0.000 description 4
- 125000003396 thiol group Chemical group [H]S* 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- 150000003852 triazoles Chemical group 0.000 description 4
- QJXPRGZXRIIGOP-UHFFFAOYSA-N tricosanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCCCCCCCCCCC(O)=O QJXPRGZXRIIGOP-UHFFFAOYSA-N 0.000 description 4
- HNAGHMKIPMKKBB-UHFFFAOYSA-N 1-benzylpyrrolidine-3-carboxamide Chemical compound C1C(C(=O)N)CCN1CC1=CC=CC=C1 HNAGHMKIPMKKBB-UHFFFAOYSA-N 0.000 description 3
- VLDPXPPHXDGHEW-UHFFFAOYSA-N 1-chloro-2-dichlorophosphoryloxybenzene Chemical compound ClC1=CC=CC=C1OP(Cl)(Cl)=O VLDPXPPHXDGHEW-UHFFFAOYSA-N 0.000 description 3
- LIPRQQHINVWJCH-UHFFFAOYSA-N 1-ethoxypropan-2-yl acetate Chemical compound CCOCC(C)OC(C)=O LIPRQQHINVWJCH-UHFFFAOYSA-N 0.000 description 3
- DMFAHCVITRDZQB-UHFFFAOYSA-N 1-propoxypropan-2-yl acetate Chemical compound CCCOCC(C)OC(C)=O DMFAHCVITRDZQB-UHFFFAOYSA-N 0.000 description 3
- HFZLSTDPRQSZCQ-UHFFFAOYSA-N 1-pyrrolidin-3-ylpyrrolidine Chemical compound C1CCCN1C1CNCC1 HFZLSTDPRQSZCQ-UHFFFAOYSA-N 0.000 description 3
- SBASXUCJHJRPEV-UHFFFAOYSA-N 2-(2-methoxyethoxy)ethanol Chemical compound COCCOCCO SBASXUCJHJRPEV-UHFFFAOYSA-N 0.000 description 3
- HCLJOFJIQIJXHS-UHFFFAOYSA-N 2-[2-[2-(2-prop-2-enoyloxyethoxy)ethoxy]ethoxy]ethyl prop-2-enoate Chemical compound C=CC(=O)OCCOCCOCCOCCOC(=O)C=C HCLJOFJIQIJXHS-UHFFFAOYSA-N 0.000 description 3
- LTHJXDSHSVNJKG-UHFFFAOYSA-N 2-[2-[2-[2-(2-methylprop-2-enoyloxy)ethoxy]ethoxy]ethoxy]ethyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCCOCCOCCOCCOC(=O)C(C)=C LTHJXDSHSVNJKG-UHFFFAOYSA-N 0.000 description 3
- HEQOJEGTZCTHCF-UHFFFAOYSA-N 2-amino-1-phenylethanone Chemical compound NCC(=O)C1=CC=CC=C1 HEQOJEGTZCTHCF-UHFFFAOYSA-N 0.000 description 3
- UHIDYCYNRPVZCK-UHFFFAOYSA-N 2-amino-4-[2-(3-amino-4-hydroxyphenyl)propan-2-yl]phenol Chemical compound C=1C=C(O)C(N)=CC=1C(C)(C)C1=CC=C(O)C(N)=C1 UHIDYCYNRPVZCK-UHFFFAOYSA-N 0.000 description 3
- ZDRNVPNSQJRIRN-UHFFFAOYSA-N 2-amino-5-[2-(4-amino-3-hydroxyphenyl)-1,1,1,3,3,3-hexafluoropropan-2-yl]phenol Chemical compound C1=C(O)C(N)=CC=C1C(C(F)(F)F)(C(F)(F)F)C1=CC=C(N)C(O)=C1 ZDRNVPNSQJRIRN-UHFFFAOYSA-N 0.000 description 3
- JDFAWEKPFLGRAK-UHFFFAOYSA-N 2-amino-5-[2-(4-amino-3-hydroxyphenyl)propan-2-yl]phenol Chemical compound C=1C=C(N)C(O)=CC=1C(C)(C)C1=CC=C(N)C(O)=C1 JDFAWEKPFLGRAK-UHFFFAOYSA-N 0.000 description 3
- ICPWFHKNYYRBSZ-UHFFFAOYSA-M 2-methoxypropanoate Chemical compound COC(C)C([O-])=O ICPWFHKNYYRBSZ-UHFFFAOYSA-M 0.000 description 3
- NVKGJHAQGWCWDI-UHFFFAOYSA-N 4-[4-amino-2-(trifluoromethyl)phenyl]-3-(trifluoromethyl)aniline Chemical group FC(F)(F)C1=CC(N)=CC=C1C1=CC=C(N)C=C1C(F)(F)F NVKGJHAQGWCWDI-UHFFFAOYSA-N 0.000 description 3
- LPEKGGXMPWTOCB-UHFFFAOYSA-N 8beta-(2,3-epoxy-2-methylbutyryloxy)-14-acetoxytithifolin Natural products COC(=O)C(C)O LPEKGGXMPWTOCB-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 3
- 102100027368 Histone H1.3 Human genes 0.000 description 3
- 101001009450 Homo sapiens Histone H1.3 Proteins 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- WOBHKFSMXKNTIM-UHFFFAOYSA-N Hydroxyethyl methacrylate Chemical compound CC(=C)C(=O)OCCO WOBHKFSMXKNTIM-UHFFFAOYSA-N 0.000 description 3
- WRQNANDWMGAFTP-UHFFFAOYSA-N Methylacetoacetic acid Chemical compound COC(=O)CC(C)=O WRQNANDWMGAFTP-UHFFFAOYSA-N 0.000 description 3
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical group C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 3
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 3
- DIQMPQMYFZXDAX-UHFFFAOYSA-N Pentyl formate Chemical compound CCCCCOC=O DIQMPQMYFZXDAX-UHFFFAOYSA-N 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 3
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical class C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 3
- 238000007259 addition reaction Methods 0.000 description 3
- 150000001335 aliphatic alkanes Chemical class 0.000 description 3
- 125000004644 alkyl sulfinyl group Chemical group 0.000 description 3
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 3
- 150000001412 amines Chemical group 0.000 description 3
- 239000002518 antifoaming agent Substances 0.000 description 3
- 150000004984 aromatic diamines Chemical class 0.000 description 3
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 3
- 125000005135 aryl sulfinyl group Chemical group 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 3
- ZYGHJZDHTFUPRJ-UHFFFAOYSA-N benzo-alpha-pyrone Natural products C1=CC=C2OC(=O)C=CC2=C1 ZYGHJZDHTFUPRJ-UHFFFAOYSA-N 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- RWCCWEUUXYIKHB-UHFFFAOYSA-N benzophenone Chemical compound C=1C=CC=CC=1C(=O)C1=CC=CC=C1 RWCCWEUUXYIKHB-UHFFFAOYSA-N 0.000 description 3
- 150000005130 benzoxazines Chemical class 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- WKDNYTOXBCRNPV-UHFFFAOYSA-N bpda Chemical compound C1=C2C(=O)OC(=O)C2=CC(C=2C=C3C(=O)OC(C3=CC=2)=O)=C1 WKDNYTOXBCRNPV-UHFFFAOYSA-N 0.000 description 3
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 3
- OBNCKNCVKJNDBV-UHFFFAOYSA-N butanoic acid ethyl ester Natural products CCCC(=O)OCC OBNCKNCVKJNDBV-UHFFFAOYSA-N 0.000 description 3
- IWPATTDMSUYMJV-UHFFFAOYSA-N butyl 2-methoxyacetate Chemical compound CCCCOC(=O)COC IWPATTDMSUYMJV-UHFFFAOYSA-N 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- 150000001733 carboxylic acid esters Chemical class 0.000 description 3
- 150000001735 carboxylic acids Chemical class 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 239000013522 chelant Substances 0.000 description 3
- 150000001805 chlorine compounds Chemical class 0.000 description 3
- 125000001309 chloro group Chemical group Cl* 0.000 description 3
- 229910052804 chromium Inorganic materials 0.000 description 3
- 239000011651 chromium Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 230000018044 dehydration Effects 0.000 description 3
- 238000000151 deposition Methods 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- 229940028356 diethylene glycol monobutyl ether Drugs 0.000 description 3
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 3
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical group [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 3
- ODQWQRRAPPTVAG-GZTJUZNOSA-N doxepin Chemical compound C1OC2=CC=CC=C2C(=C/CCN(C)C)/C2=CC=CC=C21 ODQWQRRAPPTVAG-GZTJUZNOSA-N 0.000 description 3
- 238000009713 electroplating Methods 0.000 description 3
- CKSRFHWWBKRUKA-UHFFFAOYSA-N ethyl 2-ethoxyacetate Chemical compound CCOCC(=O)OCC CKSRFHWWBKRUKA-UHFFFAOYSA-N 0.000 description 3
- WHRLOJCOIKOQGL-UHFFFAOYSA-N ethyl 2-methoxypropanoate Chemical compound CCOC(=O)C(C)OC WHRLOJCOIKOQGL-UHFFFAOYSA-N 0.000 description 3
- FJAKCEHATXBFJT-UHFFFAOYSA-N ethyl 2-oxobutanoate Chemical compound CCOC(=O)C(=O)CC FJAKCEHATXBFJT-UHFFFAOYSA-N 0.000 description 3
- XYIBRDXRRQCHLP-UHFFFAOYSA-N ethyl acetoacetate Chemical compound CCOC(=O)CC(C)=O XYIBRDXRRQCHLP-UHFFFAOYSA-N 0.000 description 3
- 229940093858 ethyl acetoacetate Drugs 0.000 description 3
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 3
- 239000010419 fine particle Substances 0.000 description 3
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 3
- XPFVYQJUAUNWIW-UHFFFAOYSA-N furfuryl alcohol Chemical compound OCC1=CC=CO1 XPFVYQJUAUNWIW-UHFFFAOYSA-N 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- VPVSTMAPERLKKM-UHFFFAOYSA-N glycoluril Chemical compound N1C(=O)NC2NC(=O)NC21 VPVSTMAPERLKKM-UHFFFAOYSA-N 0.000 description 3
- 230000002140 halogenating effect Effects 0.000 description 3
- 150000002430 hydrocarbons Chemical class 0.000 description 3
- 238000006358 imidation reaction Methods 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- 229910052742 iron Inorganic materials 0.000 description 3
- GJRQTCIYDGXPES-UHFFFAOYSA-N iso-butyl acetate Natural products CC(C)COC(C)=O GJRQTCIYDGXPES-UHFFFAOYSA-N 0.000 description 3
- 229940117955 isoamyl acetate Drugs 0.000 description 3
- FGKJLKRYENPLQH-UHFFFAOYSA-M isocaproate Chemical compound CC(C)CCC([O-])=O FGKJLKRYENPLQH-UHFFFAOYSA-M 0.000 description 3
- OQAGVSWESNCJJT-UHFFFAOYSA-N isovaleric acid methyl ester Natural products COC(=O)CC(C)C OQAGVSWESNCJJT-UHFFFAOYSA-N 0.000 description 3
- 235000001510 limonene Nutrition 0.000 description 3
- 229940087305 limonene Drugs 0.000 description 3
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 3
- PPFNAOBWGRMDLL-UHFFFAOYSA-N methyl 2-ethoxyacetate Chemical compound CCOCC(=O)OC PPFNAOBWGRMDLL-UHFFFAOYSA-N 0.000 description 3
- YVWPDYFVVMNWDT-UHFFFAOYSA-N methyl 2-ethoxypropanoate Chemical compound CCOC(C)C(=O)OC YVWPDYFVVMNWDT-UHFFFAOYSA-N 0.000 description 3
- AKWHOGIYEOZALP-UHFFFAOYSA-N methyl 2-methoxy-2-methylpropanoate Chemical compound COC(=O)C(C)(C)OC AKWHOGIYEOZALP-UHFFFAOYSA-N 0.000 description 3
- XPIWVCAMONZQCP-UHFFFAOYSA-N methyl 2-oxobutanoate Chemical compound CCC(=O)C(=O)OC XPIWVCAMONZQCP-UHFFFAOYSA-N 0.000 description 3
- 229940057867 methyl lactate Drugs 0.000 description 3
- CWKLZLBVOJRSOM-UHFFFAOYSA-N methyl pyruvate Chemical compound COC(=O)C(C)=O CWKLZLBVOJRSOM-UHFFFAOYSA-N 0.000 description 3
- 229910052759 nickel Inorganic materials 0.000 description 3
- 150000002923 oximes Chemical class 0.000 description 3
- JCGNDDUYTRNOFT-UHFFFAOYSA-N oxolane-2,4-dione Chemical compound O=C1COC(=O)C1 JCGNDDUYTRNOFT-UHFFFAOYSA-N 0.000 description 3
- NWVVVBRKAWDGAB-UHFFFAOYSA-N p-methoxyphenol Chemical compound COC1=CC=C(O)C=C1 NWVVVBRKAWDGAB-UHFFFAOYSA-N 0.000 description 3
- WXZMFSXDPGVJKK-UHFFFAOYSA-N pentaerythritol Chemical compound OCC(CO)(CO)CO WXZMFSXDPGVJKK-UHFFFAOYSA-N 0.000 description 3
- 238000000206 photolithography Methods 0.000 description 3
- 125000004193 piperazinyl group Chemical group 0.000 description 3
- 125000003386 piperidinyl group Chemical group 0.000 description 3
- 238000007747 plating Methods 0.000 description 3
- 229920001451 polypropylene glycol Polymers 0.000 description 3
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 3
- 239000004810 polytetrafluoroethylene Substances 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- CYIRLFJPTCUCJB-UHFFFAOYSA-N propyl 2-methoxypropanoate Chemical compound CCCOC(=O)C(C)OC CYIRLFJPTCUCJB-UHFFFAOYSA-N 0.000 description 3
- ILPVOWZUBFRIAX-UHFFFAOYSA-N propyl 2-oxopropanoate Chemical compound CCCOC(=O)C(C)=O ILPVOWZUBFRIAX-UHFFFAOYSA-N 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 3
- 230000001681 protective effect Effects 0.000 description 3
- 229960001860 salicylate Drugs 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 230000001629 suppression Effects 0.000 description 3
- 235000007586 terpenes Nutrition 0.000 description 3
- 150000003568 thioethers Chemical class 0.000 description 3
- 150000003573 thiols Chemical class 0.000 description 3
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 3
- QXJQHYBHAIHNGG-UHFFFAOYSA-N trimethylolethane Chemical compound OCC(C)(CO)CO QXJQHYBHAIHNGG-UHFFFAOYSA-N 0.000 description 3
- 239000008096 xylene Substances 0.000 description 3
- PAPBSGBWRJIAAV-UHFFFAOYSA-N ε-Caprolactone Chemical compound O=C1CCCCCO1 PAPBSGBWRJIAAV-UHFFFAOYSA-N 0.000 description 3
- WHIRALQRTSITMI-UJURSFKZSA-N (1s,5r)-6,8-dioxabicyclo[3.2.1]octan-4-one Chemical compound O1[C@@]2([H])OC[C@]1([H])CCC2=O WHIRALQRTSITMI-UJURSFKZSA-N 0.000 description 2
- QYGBYAQGBVHMDD-XQRVVYSFSA-N (z)-2-cyano-3-thiophen-2-ylprop-2-enoic acid Chemical compound OC(=O)C(\C#N)=C/C1=CC=CS1 QYGBYAQGBVHMDD-XQRVVYSFSA-N 0.000 description 2
- NOGBEXBVDOCGDB-NRFIWDAESA-L (z)-4-ethoxy-4-oxobut-2-en-2-olate;propan-2-olate;titanium(4+) Chemical compound [Ti+4].CC(C)[O-].CC(C)[O-].CCOC(=O)\C=C(\C)[O-].CCOC(=O)\C=C(\C)[O-] NOGBEXBVDOCGDB-NRFIWDAESA-L 0.000 description 2
- TYKCBTYOMAUNLH-MTOQALJVSA-J (z)-4-oxopent-2-en-2-olate;titanium(4+) Chemical class [Ti+4].C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O TYKCBTYOMAUNLH-MTOQALJVSA-J 0.000 description 2
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 2
- YXIWHUQXZSMYRE-UHFFFAOYSA-N 1,3-benzothiazole-2-thiol Chemical compound C1=CC=C2SC(S)=NC2=C1 YXIWHUQXZSMYRE-UHFFFAOYSA-N 0.000 description 2
- 125000001989 1,3-phenylene group Chemical group [H]C1=C([H])C([*:1])=C([H])C([*:2])=C1[H] 0.000 description 2
- 125000001140 1,4-phenylene group Chemical group [H]C1=C([H])C([*:2])=C([H])C([H])=C1[*:1] 0.000 description 2
- WVUYYXUATWMVIT-UHFFFAOYSA-N 1-bromo-4-ethoxybenzene Chemical compound CCOC1=CC=C(Br)C=C1 WVUYYXUATWMVIT-UHFFFAOYSA-N 0.000 description 2
- RTBFRGCFXZNCOE-UHFFFAOYSA-N 1-methylsulfonylpiperidin-4-one Chemical compound CS(=O)(=O)N1CCC(=O)CC1 RTBFRGCFXZNCOE-UHFFFAOYSA-N 0.000 description 2
- WJFKNYWRSNBZNX-UHFFFAOYSA-N 10H-phenothiazine Chemical compound C1=CC=C2NC3=CC=CC=C3SC2=C1 WJFKNYWRSNBZNX-UHFFFAOYSA-N 0.000 description 2
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 2
- CCUWGJDGLACFQT-UHFFFAOYSA-N 2,2,3,3,4,4-hexafluoropentanedioic acid Chemical compound OC(=O)C(F)(F)C(F)(F)C(F)(F)C(O)=O CCUWGJDGLACFQT-UHFFFAOYSA-N 0.000 description 2
- YUDUFRYTKFGQCL-UHFFFAOYSA-N 2,2,3,3-tetrafluorobutanedioic acid Chemical compound OC(=O)C(F)(F)C(F)(F)C(O)=O YUDUFRYTKFGQCL-UHFFFAOYSA-N 0.000 description 2
- KJDGGLOKWHMBOX-UHFFFAOYSA-N 2,2,3-trimethylbutanedioic acid Chemical compound OC(=O)C(C)C(C)(C)C(O)=O KJDGGLOKWHMBOX-UHFFFAOYSA-N 0.000 description 2
- CKUJONFLPBQEKM-UHFFFAOYSA-N 2,2,6,6-tetramethylheptanedioic acid Chemical compound OC(=O)C(C)(C)CCCC(C)(C)C(O)=O CKUJONFLPBQEKM-UHFFFAOYSA-N 0.000 description 2
- WUHHVDQBQZVSJV-UHFFFAOYSA-N 2,2-dibutylpropanedioic acid Chemical compound CCCCC(C(O)=O)(C(O)=O)CCCC WUHHVDQBQZVSJV-UHFFFAOYSA-N 0.000 description 2
- BTUDGPVTCYNYLK-UHFFFAOYSA-N 2,2-dimethylglutaric acid Chemical compound OC(=O)C(C)(C)CCC(O)=O BTUDGPVTCYNYLK-UHFFFAOYSA-N 0.000 description 2
- GOHPTLYPQCTZSE-UHFFFAOYSA-N 2,2-dimethylsuccinic acid Chemical compound OC(=O)C(C)(C)CC(O)=O GOHPTLYPQCTZSE-UHFFFAOYSA-N 0.000 description 2
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 2
- KLZYRCVPDWTZLH-UHFFFAOYSA-N 2,3-dimethylsuccinic acid Chemical compound OC(=O)C(C)C(C)C(O)=O KLZYRCVPDWTZLH-UHFFFAOYSA-N 0.000 description 2
- CPEXFJVZFNYXGU-UHFFFAOYSA-N 2,4,6-trihydroxybenzophenone Chemical compound OC1=CC(O)=CC(O)=C1C(=O)C1=CC=CC=C1 CPEXFJVZFNYXGU-UHFFFAOYSA-N 0.000 description 2
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 2
- INQDDHNZXOAFFD-UHFFFAOYSA-N 2-[2-(2-prop-2-enoyloxyethoxy)ethoxy]ethyl prop-2-enoate Chemical compound C=CC(=O)OCCOCCOCCOC(=O)C=C INQDDHNZXOAFFD-UHFFFAOYSA-N 0.000 description 2
- OJPDDQSCZGTACX-UHFFFAOYSA-N 2-[n-(2-hydroxyethyl)anilino]ethanol Chemical compound OCCN(CCO)C1=CC=CC=C1 OJPDDQSCZGTACX-UHFFFAOYSA-N 0.000 description 2
- KECOIASOKMSRFT-UHFFFAOYSA-N 2-amino-4-(3-amino-4-hydroxyphenyl)sulfonylphenol Chemical compound C1=C(O)C(N)=CC(S(=O)(=O)C=2C=C(N)C(O)=CC=2)=C1 KECOIASOKMSRFT-UHFFFAOYSA-N 0.000 description 2
- UZSDRHVOBLQYCX-UHFFFAOYSA-N 2-amino-6-hydroxybenzoic acid Chemical compound NC1=CC=CC(O)=C1C(O)=O UZSDRHVOBLQYCX-UHFFFAOYSA-N 0.000 description 2
- VRVRGVPWCUEOGV-UHFFFAOYSA-N 2-aminothiophenol Chemical compound NC1=CC=CC=C1S VRVRGVPWCUEOGV-UHFFFAOYSA-N 0.000 description 2
- POAOYUHQDCAZBD-UHFFFAOYSA-N 2-butoxyethanol Chemical compound CCCCOCCO POAOYUHQDCAZBD-UHFFFAOYSA-N 0.000 description 2
- VJKZIQFVKMUTID-UHFFFAOYSA-N 2-diazonio-5-sulfonaphthalen-1-olate Chemical compound N#[N+]C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1[O-] VJKZIQFVKMUTID-UHFFFAOYSA-N 0.000 description 2
- ALQPJHSFIXARGX-UHFFFAOYSA-N 2-ethynylaniline Chemical compound NC1=CC=CC=C1C#C ALQPJHSFIXARGX-UHFFFAOYSA-N 0.000 description 2
- SYUYTOYKQOAVDW-UHFFFAOYSA-N 2-nitrosonaphthalen-1-ol Chemical compound C1=CC=C2C(O)=C(N=O)C=CC2=C1 SYUYTOYKQOAVDW-UHFFFAOYSA-N 0.000 description 2
- WRMNZCZEMHIOCP-UHFFFAOYSA-N 2-phenylethanol Chemical compound OCCC1=CC=CC=C1 WRMNZCZEMHIOCP-UHFFFAOYSA-N 0.000 description 2
- DQEUFPARIOFOAI-UHFFFAOYSA-N 2-propan-2-ylpropanedioic acid Chemical compound CC(C)C(C(O)=O)C(O)=O DQEUFPARIOFOAI-UHFFFAOYSA-N 0.000 description 2
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical group C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 description 2
- CMLFRMDBDNHMRA-UHFFFAOYSA-N 2h-1,2-benzoxazine Chemical compound C1=CC=C2C=CNOC2=C1 CMLFRMDBDNHMRA-UHFFFAOYSA-N 0.000 description 2
- ULRPISSMEBPJLN-UHFFFAOYSA-N 2h-tetrazol-5-amine Chemical compound NC1=NN=NN1 ULRPISSMEBPJLN-UHFFFAOYSA-N 0.000 description 2
- DUHQIGLHYXLKAE-UHFFFAOYSA-N 3,3-dimethylglutaric acid Chemical compound OC(=O)CC(C)(C)CC(O)=O DUHQIGLHYXLKAE-UHFFFAOYSA-N 0.000 description 2
- AJHPGXZOIAYYDW-UHFFFAOYSA-N 3-(2-cyanophenyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound CC(C)(C)OC(=O)NC(C(O)=O)CC1=CC=CC=C1C#N AJHPGXZOIAYYDW-UHFFFAOYSA-N 0.000 description 2
- GIMFLOOKFCUUOQ-UHFFFAOYSA-N 3-amino-4-hydroxy-1,2-dihydropyrimidin-6-one Chemical compound NN1CN=C(O)C=C1O GIMFLOOKFCUUOQ-UHFFFAOYSA-N 0.000 description 2
- ZAJAQTYSTDTMCU-UHFFFAOYSA-N 3-aminobenzenesulfonic acid Chemical compound NC1=CC=CC(S(O)(=O)=O)=C1 ZAJAQTYSTDTMCU-UHFFFAOYSA-N 0.000 description 2
- KFFUEVDMVNIOHA-UHFFFAOYSA-N 3-aminobenzenethiol Chemical compound NC1=CC=CC(S)=C1 KFFUEVDMVNIOHA-UHFFFAOYSA-N 0.000 description 2
- CWLKGDAVCFYWJK-UHFFFAOYSA-N 3-aminophenol Chemical compound NC1=CC=CC(O)=C1 CWLKGDAVCFYWJK-UHFFFAOYSA-N 0.000 description 2
- 229940018563 3-aminophenol Drugs 0.000 description 2
- LMIOYAVXLAOXJI-UHFFFAOYSA-N 3-ethyl-3-[[4-[(3-ethyloxetan-3-yl)methoxymethyl]phenyl]methoxymethyl]oxetane Chemical compound C=1C=C(COCC2(CC)COC2)C=CC=1COCC1(CC)COC1 LMIOYAVXLAOXJI-UHFFFAOYSA-N 0.000 description 2
- XAWFHZMTJUGGEE-UHFFFAOYSA-N 3-ethyl-3-methylpentanedioic acid Chemical compound OC(=O)CC(C)(CC)CC(O)=O XAWFHZMTJUGGEE-UHFFFAOYSA-N 0.000 description 2
- NNKQLUVBPJEUOR-UHFFFAOYSA-N 3-ethynylaniline Chemical compound NC1=CC=CC(C#C)=C1 NNKQLUVBPJEUOR-UHFFFAOYSA-N 0.000 description 2
- UJBOOUHRTQVGRU-UHFFFAOYSA-N 3-methylcyclohexan-1-one Chemical compound CC1CCCC(=O)C1 UJBOOUHRTQVGRU-UHFFFAOYSA-N 0.000 description 2
- ZSSJQYMNCUJSBR-UHFFFAOYSA-N 4,5-dimethoxy-1,3-bis(methoxymethyl)imidazolidin-2-one Chemical compound COCN1C(OC)C(OC)N(COC)C1=O ZSSJQYMNCUJSBR-UHFFFAOYSA-N 0.000 description 2
- FWOLORXQTIGHFX-UHFFFAOYSA-N 4-(4-amino-2,3,5,6-tetrafluorophenyl)-2,3,5,6-tetrafluoroaniline Chemical group FC1=C(F)C(N)=C(F)C(F)=C1C1=C(F)C(F)=C(N)C(F)=C1F FWOLORXQTIGHFX-UHFFFAOYSA-N 0.000 description 2
- QYIMZXITLDTULQ-UHFFFAOYSA-N 4-(4-amino-2-methylphenyl)-3-methylaniline Chemical group CC1=CC(N)=CC=C1C1=CC=C(N)C=C1C QYIMZXITLDTULQ-UHFFFAOYSA-N 0.000 description 2
- LFEWXDOYPCWFHR-UHFFFAOYSA-N 4-(4-carboxybenzoyl)benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1C(=O)C1=CC=C(C(O)=O)C=C1 LFEWXDOYPCWFHR-UHFFFAOYSA-N 0.000 description 2
- WVDRSXGPQWNUBN-UHFFFAOYSA-N 4-(4-carboxyphenoxy)benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1OC1=CC=C(C(O)=O)C=C1 WVDRSXGPQWNUBN-UHFFFAOYSA-N 0.000 description 2
- SRKIZOFXPUNFBI-UHFFFAOYSA-N 4-[(4-hydroxy-3,5-dimethylphenyl)-(4-hydroxy-3-methoxyphenyl)methyl]-2,6-dimethylphenol Chemical compound C1=C(O)C(OC)=CC(C(C=2C=C(C)C(O)=C(C)C=2)C=2C=C(C)C(O)=C(C)C=2)=C1 SRKIZOFXPUNFBI-UHFFFAOYSA-N 0.000 description 2
- FVLYIHYVIMGFNV-UHFFFAOYSA-N 4-[[2-hydroxy-5-methyl-3-[(2,3,4-trihydroxyphenyl)methyl]phenyl]methyl]benzene-1,2,3-triol Chemical compound OC=1C(CC=2C(=C(O)C(O)=CC=2)O)=CC(C)=CC=1CC1=CC=C(O)C(O)=C1O FVLYIHYVIMGFNV-UHFFFAOYSA-N 0.000 description 2
- ALYNCZNDIQEVRV-PZFLKRBQSA-N 4-amino-3,5-ditritiobenzoic acid Chemical compound [3H]c1cc(cc([3H])c1N)C(O)=O ALYNCZNDIQEVRV-PZFLKRBQSA-N 0.000 description 2
- HVBSAKJJOYLTQU-UHFFFAOYSA-N 4-aminobenzenesulfonic acid Chemical compound NC1=CC=C(S(O)(=O)=O)C=C1 HVBSAKJJOYLTQU-UHFFFAOYSA-N 0.000 description 2
- WCDSVWRUXWCYFN-UHFFFAOYSA-N 4-aminobenzenethiol Chemical compound NC1=CC=C(S)C=C1 WCDSVWRUXWCYFN-UHFFFAOYSA-N 0.000 description 2
- ABJQKDJOYSQVFX-UHFFFAOYSA-N 4-aminonaphthalen-1-ol Chemical compound C1=CC=C2C(N)=CC=C(O)C2=C1 ABJQKDJOYSQVFX-UHFFFAOYSA-N 0.000 description 2
- WUBBRNOQWQTFEX-UHFFFAOYSA-N 4-aminosalicylic acid Chemical compound NC1=CC=C(C(O)=O)C(O)=C1 WUBBRNOQWQTFEX-UHFFFAOYSA-N 0.000 description 2
- JXYITCJMBRETQX-UHFFFAOYSA-N 4-ethynylaniline Chemical compound NC1=CC=C(C#C)C=C1 JXYITCJMBRETQX-UHFFFAOYSA-N 0.000 description 2
- WVYWICLMDOOCFB-UHFFFAOYSA-N 4-methyl-2-pentanol Chemical compound CC(C)CC(C)O WVYWICLMDOOCFB-UHFFFAOYSA-N 0.000 description 2
- CQMIJLIXKMKFQW-UHFFFAOYSA-N 4-phenylbenzene-1,2,3,5-tetracarboxylic acid Chemical compound OC(=O)C1=C(C(O)=O)C(C(=O)O)=CC(C(O)=O)=C1C1=CC=CC=C1 CQMIJLIXKMKFQW-UHFFFAOYSA-N 0.000 description 2
- NNJMFJSKMRYHSR-UHFFFAOYSA-N 4-phenylbenzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1C1=CC=CC=C1 NNJMFJSKMRYHSR-UHFFFAOYSA-N 0.000 description 2
- ZBIBQNVRTVLOHQ-UHFFFAOYSA-N 5-aminonaphthalen-1-ol Chemical compound C1=CC=C2C(N)=CC=CC2=C1O ZBIBQNVRTVLOHQ-UHFFFAOYSA-N 0.000 description 2
- FSBRKZMSECKELY-UHFFFAOYSA-N 5-aminonaphthalen-2-ol Chemical compound OC1=CC=C2C(N)=CC=CC2=C1 FSBRKZMSECKELY-UHFFFAOYSA-N 0.000 description 2
- FPKNJPIDCMAIDW-UHFFFAOYSA-N 5-aminonaphthalene-1-carboxylic acid Chemical compound C1=CC=C2C(N)=CC=CC2=C1C(O)=O FPKNJPIDCMAIDW-UHFFFAOYSA-N 0.000 description 2
- XVOBQVZYYPJXCK-UHFFFAOYSA-N 5-aminonaphthalene-2-carboxylic acid Chemical compound OC(=O)C1=CC=C2C(N)=CC=CC2=C1 XVOBQVZYYPJXCK-UHFFFAOYSA-N 0.000 description 2
- YDEUKNRKEYICTH-UHFFFAOYSA-N 5-aminoquinolin-8-ol Chemical compound C1=CC=C2C(N)=CC=C(O)C2=N1 YDEUKNRKEYICTH-UHFFFAOYSA-N 0.000 description 2
- GAYWCADKXYCKCG-UHFFFAOYSA-N 5-pyridin-3-yl-1,2-dihydro-1,2,4-triazole-3-thione Chemical compound N1NC(=S)N=C1C1=CC=CN=C1 GAYWCADKXYCKCG-UHFFFAOYSA-N 0.000 description 2
- QYFYIOWLBSPSDM-UHFFFAOYSA-N 6-aminonaphthalen-1-ol Chemical compound OC1=CC=CC2=CC(N)=CC=C21 QYFYIOWLBSPSDM-UHFFFAOYSA-N 0.000 description 2
- SERBLGFKBWPCJD-UHFFFAOYSA-N 6-aminonaphthalen-2-ol Chemical compound C1=C(O)C=CC2=CC(N)=CC=C21 SERBLGFKBWPCJD-UHFFFAOYSA-N 0.000 description 2
- CKSZZOAKPXZMAT-UHFFFAOYSA-N 6-aminonaphthalene-1-carboxylic acid Chemical compound OC(=O)C1=CC=CC2=CC(N)=CC=C21 CKSZZOAKPXZMAT-UHFFFAOYSA-N 0.000 description 2
- NZTPZUIIYNYZKT-UHFFFAOYSA-N 6-aminonaphthalene-2-carboxylic acid Chemical compound C1=C(C(O)=O)C=CC2=CC(N)=CC=C21 NZTPZUIIYNYZKT-UHFFFAOYSA-N 0.000 description 2
- FIHBHSQYSYVZQE-UHFFFAOYSA-N 6-prop-2-enoyloxyhexyl prop-2-enoate Chemical compound C=CC(=O)OCCCCCCOC(=O)C=C FIHBHSQYSYVZQE-UHFFFAOYSA-N 0.000 description 2
- ZYSOYLBBCYWEMB-UHFFFAOYSA-N 7-aminonaphthalen-1-ol Chemical compound C1=CC=C(O)C2=CC(N)=CC=C21 ZYSOYLBBCYWEMB-UHFFFAOYSA-N 0.000 description 2
- WSUYONLKFXZZRV-UHFFFAOYSA-N 7-aminonaphthalen-2-ol Chemical compound C1=CC(O)=CC2=CC(N)=CC=C21 WSUYONLKFXZZRV-UHFFFAOYSA-N 0.000 description 2
- BFBAHPDPLUEECU-UHFFFAOYSA-N 7-aminonaphthalene-1-carboxylic acid Chemical compound C1=CC=C(C(O)=O)C2=CC(N)=CC=C21 BFBAHPDPLUEECU-UHFFFAOYSA-N 0.000 description 2
- NBPYPKQPLKDTKB-UHFFFAOYSA-N 7-aminonaphthalene-2-carboxylic acid Chemical compound C1=CC(C(O)=O)=CC2=CC(N)=CC=C21 NBPYPKQPLKDTKB-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical group N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- 239000004925 Acrylic resin Substances 0.000 description 2
- 229920000178 Acrylic resin Polymers 0.000 description 2
- KLSJWNVTNUYHDU-UHFFFAOYSA-N Amitrole Chemical compound NC1=NC=NN1 KLSJWNVTNUYHDU-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- SDDLEVPIDBLVHC-UHFFFAOYSA-N Bisphenol Z Chemical compound C1=CC(O)=CC=C1C1(C=2C=CC(O)=CC=2)CCCCC1 SDDLEVPIDBLVHC-UHFFFAOYSA-N 0.000 description 2
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 2
- QEVGZEDELICMKH-UHFFFAOYSA-N Diglycolic acid Chemical compound OC(=O)COCC(O)=O QEVGZEDELICMKH-UHFFFAOYSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- XXRCUYVCPSWGCC-UHFFFAOYSA-N Ethyl pyruvate Chemical compound CCOC(=O)C(C)=O XXRCUYVCPSWGCC-UHFFFAOYSA-N 0.000 description 2
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 102100039855 Histone H1.2 Human genes 0.000 description 2
- 101001035375 Homo sapiens Histone H1.2 Proteins 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- VSNHCAURESNICA-UHFFFAOYSA-N Hydroxyurea Chemical class NC(=O)NO VSNHCAURESNICA-UHFFFAOYSA-N 0.000 description 2
- XNWPXDGRBWJIES-UHFFFAOYSA-N Maclurin Natural products OC1=CC(O)=CC(O)=C1C(=O)C1=CC=C(O)C(O)=C1 XNWPXDGRBWJIES-UHFFFAOYSA-N 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- IFAWYXIHOVRGHQ-UHFFFAOYSA-N Nonadecandioic acid Chemical compound OC(=O)CCCCCCCCCCCCCCCCCC(O)=O IFAWYXIHOVRGHQ-UHFFFAOYSA-N 0.000 description 2
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 2
- URLKBWYHVLBVBO-UHFFFAOYSA-N Para-Xylene Chemical group CC1=CC=C(C)C=C1 URLKBWYHVLBVBO-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- 206010034972 Photosensitivity reaction Diseases 0.000 description 2
- LGRFSURHDFAFJT-UHFFFAOYSA-N Phthalic anhydride Natural products C1=CC=C2C(=O)OC(=O)C2=C1 LGRFSURHDFAFJT-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 239000004952 Polyamide Substances 0.000 description 2
- 229920002582 Polyethylene Glycol 600 Polymers 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical group C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 2
- REFJWTPEDVJJIY-UHFFFAOYSA-N Quercetin Chemical compound C=1C(O)=CC(O)=C(C(C=2O)=O)C=1OC=2C1=CC=C(O)C(O)=C1 REFJWTPEDVJJIY-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 239000004115 Sodium Silicate Substances 0.000 description 2
- 235000000126 Styrax benzoin Nutrition 0.000 description 2
- 244000028419 Styrax benzoin Species 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 235000008411 Sumatra benzointree Nutrition 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- ZJCCRDAZUWHFQH-UHFFFAOYSA-N Trimethylolpropane Chemical compound CCC(CO)(CO)CO ZJCCRDAZUWHFQH-UHFFFAOYSA-N 0.000 description 2
- 229910021536 Zeolite Inorganic materials 0.000 description 2
- ULQMPOIOSDXIGC-UHFFFAOYSA-N [2,2-dimethyl-3-(2-methylprop-2-enoyloxy)propyl] 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCC(C)(C)COC(=O)C(C)=C ULQMPOIOSDXIGC-UHFFFAOYSA-N 0.000 description 2
- MPIAGWXWVAHQBB-UHFFFAOYSA-N [3-prop-2-enoyloxy-2-[[3-prop-2-enoyloxy-2,2-bis(prop-2-enoyloxymethyl)propoxy]methyl]-2-(prop-2-enoyloxymethyl)propyl] prop-2-enoate Chemical compound C=CC(=O)OCC(COC(=O)C=C)(COC(=O)C=C)COCC(COC(=O)C=C)(COC(=O)C=C)COC(=O)C=C MPIAGWXWVAHQBB-UHFFFAOYSA-N 0.000 description 2
- 125000004036 acetal group Chemical group 0.000 description 2
- 150000008062 acetophenones Chemical class 0.000 description 2
- 150000008360 acrylonitriles Chemical class 0.000 description 2
- ORILYTVJVMAKLC-UHFFFAOYSA-N adamantane Chemical compound C1C(C2)CC3CC1CC2C3 ORILYTVJVMAKLC-UHFFFAOYSA-N 0.000 description 2
- 239000001361 adipic acid Substances 0.000 description 2
- 235000011037 adipic acid Nutrition 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 125000005370 alkoxysilyl group Chemical group 0.000 description 2
- 125000005011 alkyl ether group Chemical group 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 125000003368 amide group Chemical group 0.000 description 2
- 229960004909 aminosalicylic acid Drugs 0.000 description 2
- JFCQEDHGNNZCLN-UHFFFAOYSA-N anhydrous glutaric acid Natural products OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 2
- 239000003945 anionic surfactant Substances 0.000 description 2
- 229940053200 antiepileptics fatty acid derivative Drugs 0.000 description 2
- 125000005018 aryl alkenyl group Chemical group 0.000 description 2
- 125000002102 aryl alkyloxo group Chemical group 0.000 description 2
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 2
- 229960002255 azelaic acid Drugs 0.000 description 2
- TZCXTZWJZNENPQ-UHFFFAOYSA-L barium sulfate Chemical compound [Ba+2].[O-]S([O-])(=O)=O TZCXTZWJZNENPQ-UHFFFAOYSA-L 0.000 description 2
- 229960002130 benzoin Drugs 0.000 description 2
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 2
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- SYEOWUNSTUDKGM-UHFFFAOYSA-N beta-methyladipic acid Natural products OC(=O)CC(C)CCC(O)=O SYEOWUNSTUDKGM-UHFFFAOYSA-N 0.000 description 2
- 230000005540 biological transmission Effects 0.000 description 2
- WXNRYSGJLQFHBR-UHFFFAOYSA-N bis(2,4-dihydroxyphenyl)methanone Chemical compound OC1=CC(O)=CC=C1C(=O)C1=CC=C(O)C=C1O WXNRYSGJLQFHBR-UHFFFAOYSA-N 0.000 description 2
- 125000001246 bromo group Chemical group Br* 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- YHWCPXVTRSHPNY-UHFFFAOYSA-N butan-1-olate;titanium(4+) Chemical compound [Ti+4].CCCC[O-].CCCC[O-].CCCC[O-].CCCC[O-] YHWCPXVTRSHPNY-UHFFFAOYSA-N 0.000 description 2
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 2
- JHIWVOJDXOSYLW-UHFFFAOYSA-N butyl 2,2-difluorocyclopropane-1-carboxylate Chemical compound CCCCOC(=O)C1CC1(F)F JHIWVOJDXOSYLW-UHFFFAOYSA-N 0.000 description 2
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 239000003093 cationic surfactant Substances 0.000 description 2
- BNWKXMCELVEAPW-UHFFFAOYSA-N chembl3305990 Chemical compound O=C1C(=[N+]=[N-])C=CC2=C1C=CC=C2S(=O)(=O)O BNWKXMCELVEAPW-UHFFFAOYSA-N 0.000 description 2
- 238000005229 chemical vapour deposition Methods 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 238000009833 condensation Methods 0.000 description 2
- 238000011109 contamination Methods 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- 238000003851 corona treatment Methods 0.000 description 2
- 235000001671 coumarin Nutrition 0.000 description 2
- 150000004775 coumarins Chemical class 0.000 description 2
- 125000004093 cyano group Chemical group *C#N 0.000 description 2
- 150000001923 cyclic compounds Chemical class 0.000 description 2
- 150000004292 cyclic ethers Chemical group 0.000 description 2
- PESYEWKSBIWTAK-UHFFFAOYSA-N cyclopenta-1,3-diene;titanium(2+) Chemical class [Ti+2].C=1C=C[CH-]C=1.C=1C=C[CH-]C=1 PESYEWKSBIWTAK-UHFFFAOYSA-N 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 238000007872 degassing Methods 0.000 description 2
- SWXVUIWOUIDPGS-UHFFFAOYSA-N diacetone alcohol Chemical compound CC(=O)CC(C)(C)O SWXVUIWOUIDPGS-UHFFFAOYSA-N 0.000 description 2
- 150000001988 diarylethenes Chemical class 0.000 description 2
- JQVDAXLFBXTEQA-UHFFFAOYSA-N dibutylamine Chemical compound CCCCNCCCC JQVDAXLFBXTEQA-UHFFFAOYSA-N 0.000 description 2
- OREAFAJWWJHCOT-UHFFFAOYSA-N dimethylmalonic acid Chemical compound OC(=O)C(C)(C)C(O)=O OREAFAJWWJHCOT-UHFFFAOYSA-N 0.000 description 2
- HNPSIPDUKPIQMN-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical compound O=[Si]=O.O=[Al]O[Al]=O HNPSIPDUKPIQMN-UHFFFAOYSA-N 0.000 description 2
- VFHVQBAGLAREND-UHFFFAOYSA-N diphenylphosphoryl-(2,4,6-trimethylphenyl)methanone Chemical compound CC1=CC(C)=CC(C)=C1C(=O)P(=O)(C=1C=CC=CC=1)C1=CC=CC=C1 VFHVQBAGLAREND-UHFFFAOYSA-N 0.000 description 2
- 238000010494 dissociation reaction Methods 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- UKMSUNONTOPOIO-UHFFFAOYSA-N docosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCCCC(O)=O UKMSUNONTOPOIO-UHFFFAOYSA-N 0.000 description 2
- SIGDBIXJUHHOIA-UHFFFAOYSA-N dotriacontanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC(O)=O SIGDBIXJUHHOIA-UHFFFAOYSA-N 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- JLEKJZUYWFJPMB-UHFFFAOYSA-N ethyl 2-methoxyacetate Chemical compound CCOC(=O)COC JLEKJZUYWFJPMB-UHFFFAOYSA-N 0.000 description 2
- IJUHLFUALMUWOM-UHFFFAOYSA-N ethyl 3-methoxypropanoate Chemical compound CCOC(=O)CCOC IJUHLFUALMUWOM-UHFFFAOYSA-N 0.000 description 2
- MSOLGAJLRIINNF-UHFFFAOYSA-N ethyl 7-(diethylamino)-2-oxochromene-3-carboxylate Chemical compound C1=C(N(CC)CC)C=C2OC(=O)C(C(=O)OCC)=CC2=C1 MSOLGAJLRIINNF-UHFFFAOYSA-N 0.000 description 2
- TVQGDYNRXLTQAP-UHFFFAOYSA-N ethyl heptanoate Chemical compound CCCCCCC(=O)OCC TVQGDYNRXLTQAP-UHFFFAOYSA-N 0.000 description 2
- SHZIWNPUGXLXDT-UHFFFAOYSA-N ethyl hexanoate Chemical compound CCCCCC(=O)OCC SHZIWNPUGXLXDT-UHFFFAOYSA-N 0.000 description 2
- FKRCODPIKNYEAC-UHFFFAOYSA-N ethyl propionate Chemical compound CCOC(=O)CC FKRCODPIKNYEAC-UHFFFAOYSA-N 0.000 description 2
- 229940117360 ethyl pyruvate Drugs 0.000 description 2
- UKFXDFUAPNAMPJ-UHFFFAOYSA-N ethylmalonic acid Chemical compound CCC(C(O)=O)C(O)=O UKFXDFUAPNAMPJ-UHFFFAOYSA-N 0.000 description 2
- 230000005281 excited state Effects 0.000 description 2
- LHGVFZTZFXWLCP-UHFFFAOYSA-N guaiacol Chemical compound COC1=CC=CC=C1O LHGVFZTZFXWLCP-UHFFFAOYSA-N 0.000 description 2
- 235000019382 gum benzoic Nutrition 0.000 description 2
- 229910052736 halogen Inorganic materials 0.000 description 2
- 230000020169 heat generation Effects 0.000 description 2
- 239000001307 helium Substances 0.000 description 2
- 229910052734 helium Inorganic materials 0.000 description 2
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 2
- LWGMOHWJLCZCLJ-UHFFFAOYSA-N hentriacontanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCCCCCCCCCCCCCCCCCCC(O)=O LWGMOHWJLCZCLJ-UHFFFAOYSA-N 0.000 description 2
- JPUCVAQYQIGVSJ-UHFFFAOYSA-N heptacosanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCCCCCCCCCCCCCCC(O)=O JPUCVAQYQIGVSJ-UHFFFAOYSA-N 0.000 description 2
- 125000001072 heteroaryl group Chemical group 0.000 description 2
- QNVRIHYSUZMSGM-UHFFFAOYSA-N hexan-2-ol Chemical compound CCCCC(C)O QNVRIHYSUZMSGM-UHFFFAOYSA-N 0.000 description 2
- AOGQPLXWSUTHQB-UHFFFAOYSA-N hexyl acetate Chemical compound CCCCCCOC(C)=O AOGQPLXWSUTHQB-UHFFFAOYSA-N 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- ZTVZLYBCZNMWCF-UHFFFAOYSA-N homocystine Chemical compound [O-]C(=O)C([NH3+])CCSSCCC([NH3+])C([O-])=O ZTVZLYBCZNMWCF-UHFFFAOYSA-N 0.000 description 2
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 239000011261 inert gas Substances 0.000 description 2
- 238000009413 insulation Methods 0.000 description 2
- 239000012948 isocyanate Substances 0.000 description 2
- IQPQWNKOIGAROB-UHFFFAOYSA-N isocyanate group Chemical group [N-]=C=O IQPQWNKOIGAROB-UHFFFAOYSA-N 0.000 description 2
- QQVIHTHCMHWDBS-UHFFFAOYSA-N isophthalic acid Chemical compound OC(=O)C1=CC=CC(C(O)=O)=C1 QQVIHTHCMHWDBS-UHFFFAOYSA-N 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 150000002596 lactones Chemical group 0.000 description 2
- 238000010030 laminating Methods 0.000 description 2
- HITOXZPZGPXYHY-UJURSFKZSA-N levoglucosenone Chemical compound O=C1C=C[C@H]2CO[C@@H]1O2 HITOXZPZGPXYHY-UJURSFKZSA-N 0.000 description 2
- HITOXZPZGPXYHY-UHFFFAOYSA-N levoglucosenone Natural products O=C1C=CC2COC1O2 HITOXZPZGPXYHY-UHFFFAOYSA-N 0.000 description 2
- PQXKHYXIUOZZFA-UHFFFAOYSA-M lithium fluoride Chemical compound [Li+].[F-] PQXKHYXIUOZZFA-UHFFFAOYSA-M 0.000 description 2
- HCZKYJDFEPMADG-TXEJJXNPSA-N masoprocol Chemical compound C([C@H](C)[C@H](C)CC=1C=C(O)C(O)=CC=1)C1=CC=C(O)C(O)=C1 HCZKYJDFEPMADG-TXEJJXNPSA-N 0.000 description 2
- KBOPZPXVLCULAV-UHFFFAOYSA-N mesalamine Chemical compound NC1=CC=C(O)C(C(O)=O)=C1 KBOPZPXVLCULAV-UHFFFAOYSA-N 0.000 description 2
- 229960004963 mesalazine Drugs 0.000 description 2
- 150000002734 metacrylic acid derivatives Chemical class 0.000 description 2
- 229910001507 metal halide Inorganic materials 0.000 description 2
- 150000005309 metal halides Chemical class 0.000 description 2
- BDJSOPWXYLFTNW-UHFFFAOYSA-N methyl 3-methoxypropanoate Chemical compound COCCC(=O)OC BDJSOPWXYLFTNW-UHFFFAOYSA-N 0.000 description 2
- 150000002762 monocarboxylic acid derivatives Chemical class 0.000 description 2
- LCEDQNDDFOCWGG-UHFFFAOYSA-N morpholine-4-carbaldehyde Chemical compound O=CN1CCOCC1 LCEDQNDDFOCWGG-UHFFFAOYSA-N 0.000 description 2
- QVEIBLDXZNGPHR-UHFFFAOYSA-N naphthalene-1,4-dione;diazide Chemical compound [N-]=[N+]=[N-].[N-]=[N+]=[N-].C1=CC=C2C(=O)C=CC(=O)C2=C1 QVEIBLDXZNGPHR-UHFFFAOYSA-N 0.000 description 2
- 125000004957 naphthylene group Chemical group 0.000 description 2
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 2
- BHZDBSUDGBEJDI-UHFFFAOYSA-N nonacosanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCCCCCCCCCCCCCCCCC(O)=O BHZDBSUDGBEJDI-UHFFFAOYSA-N 0.000 description 2
- 239000002736 nonionic surfactant Substances 0.000 description 2
- 229920003986 novolac Polymers 0.000 description 2
- RZJRJXONCZWCBN-UHFFFAOYSA-N octadecane Chemical compound CCCCCCCCCCCCCCCCCC RZJRJXONCZWCBN-UHFFFAOYSA-N 0.000 description 2
- BNJOQKFENDDGSC-UHFFFAOYSA-N octadecanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCCCCCC(O)=O BNJOQKFENDDGSC-UHFFFAOYSA-N 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 150000002921 oxetanes Chemical class 0.000 description 2
- MPQXHAGKBWFSNV-UHFFFAOYSA-N oxidophosphanium Chemical class [PH3]=O MPQXHAGKBWFSNV-UHFFFAOYSA-N 0.000 description 2
- 238000000059 patterning Methods 0.000 description 2
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 2
- 150000002978 peroxides Chemical class 0.000 description 2
- 229920001568 phenolic resin Polymers 0.000 description 2
- 239000005011 phenolic resin Substances 0.000 description 2
- 229950000688 phenothiazine Drugs 0.000 description 2
- ZQBAKBUEJOMQEX-UHFFFAOYSA-N phenyl salicylate Chemical compound OC1=CC=CC=C1C(=O)OC1=CC=CC=C1 ZQBAKBUEJOMQEX-UHFFFAOYSA-N 0.000 description 2
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 239000011574 phosphorus Substances 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 230000036211 photosensitivity Effects 0.000 description 2
- IEQIEDJGQAUEQZ-UHFFFAOYSA-N phthalocyanine Chemical class N1C(N=C2C3=CC=CC=C3C(N=C3C4=CC=CC=C4C(=N4)N3)=N2)=C(C=CC=C2)C2=C1N=C1C2=CC=CC=C2C4=N1 IEQIEDJGQAUEQZ-UHFFFAOYSA-N 0.000 description 2
- 238000005240 physical vapour deposition Methods 0.000 description 2
- 229920002647 polyamide Polymers 0.000 description 2
- 125000003367 polycyclic group Chemical group 0.000 description 2
- 229920000573 polyethylene Polymers 0.000 description 2
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical compound CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 2
- KIDHWZJUCRJVML-UHFFFAOYSA-N putrescine Chemical compound NCCCCN KIDHWZJUCRJVML-UHFFFAOYSA-N 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- WQGWDDDVZFFDIG-UHFFFAOYSA-N pyrogallol Chemical compound OC1=CC=CC(O)=C1O WQGWDDDVZFFDIG-UHFFFAOYSA-N 0.000 description 2
- 125000001422 pyrrolinyl group Chemical group 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 2
- 150000004053 quinones Chemical class 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 230000007261 regionalization Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 2
- 229930195734 saturated hydrocarbon Natural products 0.000 description 2
- 150000003335 secondary amines Chemical class 0.000 description 2
- 230000001235 sensitizing effect Effects 0.000 description 2
- NTHWMYGWWRZVTN-UHFFFAOYSA-N sodium silicate Chemical compound [Na+].[Na+].[O-][Si]([O-])=O NTHWMYGWWRZVTN-UHFFFAOYSA-N 0.000 description 2
- 229910052911 sodium silicate Inorganic materials 0.000 description 2
- TYFQFVWCELRYAO-UHFFFAOYSA-N suberic acid Chemical compound OC(=O)CCCCCCC(O)=O TYFQFVWCELRYAO-UHFFFAOYSA-N 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 150000005846 sugar alcohols Chemical class 0.000 description 2
- 229950000244 sulfanilic acid Drugs 0.000 description 2
- 150000003457 sulfones Chemical class 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- 150000008053 sultones Chemical group 0.000 description 2
- 238000004381 surface treatment Methods 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- VDZOOKBUILJEDG-UHFFFAOYSA-M tetrabutylammonium hydroxide Chemical compound [OH-].CCCC[N+](CCCC)(CCCC)CCCC VDZOOKBUILJEDG-UHFFFAOYSA-M 0.000 description 2
- ZUHZGEOKBKGPSW-UHFFFAOYSA-N tetraglyme Chemical compound COCCOCCOCCOCCOC ZUHZGEOKBKGPSW-UHFFFAOYSA-N 0.000 description 2
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 2
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical compound C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 2
- VXUYXOFXAQZZMF-UHFFFAOYSA-N titanium(IV) isopropoxide Chemical compound CC(C)O[Ti](OC(C)C)(OC(C)C)OC(C)C VXUYXOFXAQZZMF-UHFFFAOYSA-N 0.000 description 2
- LDHQCZJRKDOVOX-UHFFFAOYSA-N trans-crotonic acid Natural products CC=CC(O)=O LDHQCZJRKDOVOX-UHFFFAOYSA-N 0.000 description 2
- 238000002834 transmittance Methods 0.000 description 2
- KQTIIICEAUMSDG-UHFFFAOYSA-N tricarballylic acid Chemical compound OC(=O)CC(C(O)=O)CC(O)=O KQTIIICEAUMSDG-UHFFFAOYSA-N 0.000 description 2
- DXNCZXXFRKPEPY-UHFFFAOYSA-N tridecanedioic acid Chemical compound OC(=O)CCCCCCCCCCCC(O)=O DXNCZXXFRKPEPY-UHFFFAOYSA-N 0.000 description 2
- ZIBGPFATKBEMQZ-UHFFFAOYSA-N triethylene glycol Chemical compound OCCOCCOCCO ZIBGPFATKBEMQZ-UHFFFAOYSA-N 0.000 description 2
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- ARCGXLSVLAOJQL-UHFFFAOYSA-N trimellitic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C(C(O)=O)=C1 ARCGXLSVLAOJQL-UHFFFAOYSA-N 0.000 description 2
- DQZNLOXENNXVAD-UHFFFAOYSA-N trimethoxy-[2-(7-oxabicyclo[4.1.0]heptan-4-yl)ethyl]silane Chemical compound C1C(CC[Si](OC)(OC)OC)CCC2OC21 DQZNLOXENNXVAD-UHFFFAOYSA-N 0.000 description 2
- PYOKUURKVVELLB-UHFFFAOYSA-N trimethyl orthoformate Chemical compound COC(OC)OC PYOKUURKVVELLB-UHFFFAOYSA-N 0.000 description 2
- XFNJVJPLKCPIBV-UHFFFAOYSA-N trimethylenediamine Chemical compound NCCCN XFNJVJPLKCPIBV-UHFFFAOYSA-N 0.000 description 2
- 150000003672 ureas Chemical class 0.000 description 2
- 238000007740 vapor deposition Methods 0.000 description 2
- 239000010457 zeolite Substances 0.000 description 2
- QNODIIQQMGDSEF-UHFFFAOYSA-N (1-hydroxycyclohexyl)-phenylmethanone Chemical compound C=1C=CC=CC=1C(=O)C1(O)CCCCC1 QNODIIQQMGDSEF-UHFFFAOYSA-N 0.000 description 1
- XEDWWPGWIXPVRQ-UHFFFAOYSA-N (2,3,4-trihydroxyphenyl)-(3,4,5-trihydroxyphenyl)methanone Chemical class OC1=C(O)C(O)=CC=C1C(=O)C1=CC(O)=C(O)C(O)=C1 XEDWWPGWIXPVRQ-UHFFFAOYSA-N 0.000 description 1
- UCDVSEPOCIAPEM-UHFFFAOYSA-N (2,3,4-trihydroxyphenyl)-[2,4,5-tris(2,3,4-trihydroxybenzoyl)phenyl]methanone Chemical compound OC1=C(O)C(O)=CC=C1C(=O)C1=CC(C(=O)C=2C(=C(O)C(O)=CC=2)O)=C(C(=O)C=2C(=C(O)C(O)=CC=2)O)C=C1C(=O)C1=CC=C(O)C(O)=C1O UCDVSEPOCIAPEM-UHFFFAOYSA-N 0.000 description 1
- QWRVAXMLZCMVSL-UHFFFAOYSA-N (2,4,6-trihydroxyphenyl)-(3,4,5-trihydroxyphenyl)methanone Chemical compound OC1=CC(O)=CC(O)=C1C(=O)C1=CC(O)=C(O)C(O)=C1 QWRVAXMLZCMVSL-UHFFFAOYSA-N 0.000 description 1
- GBQZZLQKUYLGFT-UHFFFAOYSA-N (2,4-dihydroxyphenyl)-(2,3,4-trihydroxyphenyl)methanone Chemical compound OC1=CC(O)=CC=C1C(=O)C1=CC=C(O)C(O)=C1O GBQZZLQKUYLGFT-UHFFFAOYSA-N 0.000 description 1
- OKJFKPFBSPZTAH-UHFFFAOYSA-N (2,4-dihydroxyphenyl)-(4-hydroxyphenyl)methanone Chemical compound C1=CC(O)=CC=C1C(=O)C1=CC=C(O)C=C1O OKJFKPFBSPZTAH-UHFFFAOYSA-N 0.000 description 1
- AXIIFBJDJHDNGW-UHFFFAOYSA-N (2,5-dihydroxyphenyl)-(2,3,4-trihydroxyphenyl)methanone Chemical compound OC1=CC=C(O)C(C(=O)C=2C(=C(O)C(O)=CC=2)O)=C1 AXIIFBJDJHDNGW-UHFFFAOYSA-N 0.000 description 1
- ZODNDDPVCIAZIQ-UHFFFAOYSA-N (2-hydroxy-3-prop-2-enoyloxypropyl) 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCC(O)COC(=O)C=C ZODNDDPVCIAZIQ-UHFFFAOYSA-N 0.000 description 1
- XLBGWOVCPLFKCQ-UHFFFAOYSA-N (2-methylphenyl)-(2,3,4-trihydroxyphenyl)methanone Chemical compound CC1=CC=CC=C1C(=O)C1=CC=C(O)C(O)=C1O XLBGWOVCPLFKCQ-UHFFFAOYSA-N 0.000 description 1
- WYTZZXDRDKSJID-UHFFFAOYSA-N (3-aminopropyl)triethoxysilane Chemical compound CCO[Si](OCC)(OCC)CCCN WYTZZXDRDKSJID-UHFFFAOYSA-N 0.000 description 1
- UNMJLQGKEDTEKJ-UHFFFAOYSA-N (3-ethyloxetan-3-yl)methanol Chemical compound CCC1(CO)COC1 UNMJLQGKEDTEKJ-UHFFFAOYSA-N 0.000 description 1
- IQGIEMYBDGDBMR-UHFFFAOYSA-N (3-methyl-5-prop-2-enoyloxypentyl) prop-2-enoate Chemical compound C=CC(=O)OCCC(C)CCOC(=O)C=C IQGIEMYBDGDBMR-UHFFFAOYSA-N 0.000 description 1
- PGSHZSDDBWTHDU-UHFFFAOYSA-N (3-oxobutan-2-ylideneamino) 4-methylbenzenesulfonate Chemical compound CC(=O)C(C)=NOS(=O)(=O)C1=CC=C(C)C=C1 PGSHZSDDBWTHDU-UHFFFAOYSA-N 0.000 description 1
- MRUDPRFGMHTPFJ-UHFFFAOYSA-N (3-oxobutan-2-ylideneamino) acetate Chemical compound CC(=O)ON=C(C)C(C)=O MRUDPRFGMHTPFJ-UHFFFAOYSA-N 0.000 description 1
- SKCKRZWCOYHBEC-UHFFFAOYSA-N (3-oxobutan-2-ylideneamino) benzoate Chemical compound CC(=O)C(C)=NOC(=O)C1=CC=CC=C1 SKCKRZWCOYHBEC-UHFFFAOYSA-N 0.000 description 1
- JFFCVOSCPLKMLG-UHFFFAOYSA-N (3-oxobutan-2-ylideneamino) propanoate Chemical compound CCC(=O)ON=C(C)C(C)=O JFFCVOSCPLKMLG-UHFFFAOYSA-N 0.000 description 1
- HQLZSJJJXHJANW-UHFFFAOYSA-N (3-oxopentan-2-ylideneamino) acetate Chemical compound CCC(=O)C(C)=NOC(C)=O HQLZSJJJXHJANW-UHFFFAOYSA-N 0.000 description 1
- LTQBNYCMVZQRSD-UHFFFAOYSA-N (4-ethenylphenyl)-trimethoxysilane Chemical compound CO[Si](OC)(OC)C1=CC=C(C=C)C=C1 LTQBNYCMVZQRSD-UHFFFAOYSA-N 0.000 description 1
- ZRDYULMDEGRWRC-UHFFFAOYSA-N (4-hydroxyphenyl)-(2,3,4-trihydroxyphenyl)methanone Chemical compound C1=CC(O)=CC=C1C(=O)C1=CC=C(O)C(O)=C1O ZRDYULMDEGRWRC-UHFFFAOYSA-N 0.000 description 1
- CGCQHMFVCNWSOV-UHFFFAOYSA-N (4-morpholin-4-ylphenyl)-phenylmethanone Chemical compound C=1C=C(N2CCOCC2)C=CC=1C(=O)C1=CC=CC=C1 CGCQHMFVCNWSOV-UHFFFAOYSA-N 0.000 description 1
- VNFXPOAMRORRJJ-UHFFFAOYSA-N (4-octylphenyl) 2-hydroxybenzoate Chemical compound C1=CC(CCCCCCCC)=CC=C1OC(=O)C1=CC=CC=C1O VNFXPOAMRORRJJ-UHFFFAOYSA-N 0.000 description 1
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- WOGITNXCNOTRLK-VOTSOKGWSA-N (e)-3-phenylprop-2-enoyl chloride Chemical compound ClC(=O)\C=C\C1=CC=CC=C1 WOGITNXCNOTRLK-VOTSOKGWSA-N 0.000 description 1
- FFJCNSLCJOQHKM-CLFAGFIQSA-N (z)-1-[(z)-octadec-9-enoxy]octadec-9-ene Chemical compound CCCCCCCC\C=C/CCCCCCCCOCCCCCCCC\C=C/CCCCCCCC FFJCNSLCJOQHKM-CLFAGFIQSA-N 0.000 description 1
- YSIBYEBNVMDAPN-HJWRWDBZSA-N (z)-4-oxo-4-(3-triethoxysilylpropylamino)but-2-enoic acid Chemical compound CCO[Si](OCC)(OCC)CCCNC(=O)\C=C/C(O)=O YSIBYEBNVMDAPN-HJWRWDBZSA-N 0.000 description 1
- POILWHVDKZOXJZ-ARJAWSKDSA-M (z)-4-oxopent-2-en-2-olate Chemical compound C\C([O-])=C\C(C)=O POILWHVDKZOXJZ-ARJAWSKDSA-M 0.000 description 1
- OVSGBKZKXUMMHS-VGKOASNMSA-L (z)-4-oxopent-2-en-2-olate;propan-2-olate;titanium(4+) Chemical compound [Ti+4].CC(C)[O-].CC(C)[O-].C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O OVSGBKZKXUMMHS-VGKOASNMSA-L 0.000 description 1
- QKHRTQAMPODHIN-UHFFFAOYSA-N 1',1',3,3-tetramethyl-1,3'-spirobi[2h-indene]-4,4',5,5',6,6'-hexol Chemical compound C1=C(O)C(O)=C(O)C2=C1C(C)(C)CC21C(C=C(O)C(O)=C2O)=C2C(C)(C)C1 QKHRTQAMPODHIN-UHFFFAOYSA-N 0.000 description 1
- POFMQEVZKZVAPQ-UHFFFAOYSA-N 1,1,1',1'-tetramethyl-3,3'-spirobi[2h-indene]-5,5',6,6'-tetrol Chemical compound C12=CC(O)=C(O)C=C2C(C)(C)CC11C2=CC(O)=C(O)C=C2C(C)(C)C1 POFMQEVZKZVAPQ-UHFFFAOYSA-N 0.000 description 1
- ZOYHUCLRPNYEIY-UHFFFAOYSA-N 1,1,2,2-tetramethoxypropan-1-olate titanium(4+) Chemical compound COC(C([O-])(OC)OC)(C)OC.[Ti+4].COC(C([O-])(OC)OC)(C)OC.COC(C([O-])(OC)OC)(C)OC.COC(C([O-])(OC)OC)(C)OC ZOYHUCLRPNYEIY-UHFFFAOYSA-N 0.000 description 1
- AVQQQNCBBIEMEU-UHFFFAOYSA-N 1,1,3,3-tetramethylurea Chemical compound CN(C)C(=O)N(C)C AVQQQNCBBIEMEU-UHFFFAOYSA-N 0.000 description 1
- BWKAYBPLDRWMCJ-UHFFFAOYSA-N 1,1-diethoxy-n,n-dimethylmethanamine Chemical compound CCOC(N(C)C)OCC BWKAYBPLDRWMCJ-UHFFFAOYSA-N 0.000 description 1
- AUFHDXQTTXTQSC-UHFFFAOYSA-N 1,11-bis(2,3,4-trihydroxyphenyl)undecan-6-one Chemical compound OC1=C(O)C(O)=CC=C1CCCCCC(=O)CCCCCC1=CC=C(O)C(O)=C1O AUFHDXQTTXTQSC-UHFFFAOYSA-N 0.000 description 1
- BJTXPZYDJTYEBV-UHFFFAOYSA-N 1,13-bis(2,3,4-trihydroxyphenyl)tridecan-7-one Chemical compound OC1=C(O)C(O)=CC=C1CCCCCCC(=O)CCCCCCC1=CC=C(O)C(O)=C1O BJTXPZYDJTYEBV-UHFFFAOYSA-N 0.000 description 1
- VRKVWGGGHMMERE-UHFFFAOYSA-N 1,2-bis(methoxymethyl)benzene Chemical compound COCC1=CC=CC=C1COC VRKVWGGGHMMERE-UHFFFAOYSA-N 0.000 description 1
- VBYLUPVOJYUVQF-UHFFFAOYSA-N 1,2-dimethoxy-3,4-bis(methoxymethyl)benzene Chemical compound COCc1ccc(OC)c(OC)c1COC VBYLUPVOJYUVQF-UHFFFAOYSA-N 0.000 description 1
- LEEANUDEDHYDTG-UHFFFAOYSA-N 1,2-dimethoxypropane Chemical compound COCC(C)OC LEEANUDEDHYDTG-UHFFFAOYSA-N 0.000 description 1
- 125000002030 1,2-phenylene group Chemical group [H]C1=C([H])C([*:1])=C([*:2])C([H])=C1[H] 0.000 description 1
- JIHQDMXYYFUGFV-UHFFFAOYSA-N 1,3,5-triazine Chemical compound C1=NC=NC=N1 JIHQDMXYYFUGFV-UHFFFAOYSA-N 0.000 description 1
- XYXJKPCGSGVSBO-UHFFFAOYSA-N 1,3,5-tris[(4-tert-butyl-3-hydroxy-2,6-dimethylphenyl)methyl]-1,3,5-triazinane-2,4,6-trione Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C)=C1CN1C(=O)N(CC=2C(=C(O)C(=CC=2C)C(C)(C)C)C)C(=O)N(CC=2C(=C(O)C(=CC=2C)C(C)(C)C)C)C1=O XYXJKPCGSGVSBO-UHFFFAOYSA-N 0.000 description 1
- CYSGHNMQYZDMIA-UHFFFAOYSA-N 1,3-Dimethyl-2-imidazolidinon Chemical compound CN1CCN(C)C1=O CYSGHNMQYZDMIA-UHFFFAOYSA-N 0.000 description 1
- 125000000355 1,3-benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- XPJSWQIGULIVBS-UHFFFAOYSA-N 1,3-bis(2,3,4-trihydroxyphenyl)propane-1,3-dione Chemical compound OC1=C(O)C(O)=CC=C1C(=O)CC(=O)C1=CC=C(O)C(O)=C1O XPJSWQIGULIVBS-UHFFFAOYSA-N 0.000 description 1
- XKALZGSIEJZJCZ-UHFFFAOYSA-N 1,3-bis(methoxymethyl)urea Chemical compound COCNC(=O)NCOC XKALZGSIEJZJCZ-UHFFFAOYSA-N 0.000 description 1
- FJLAFBORFDOARE-UHFFFAOYSA-N 1,3-bis[4-(diethylamino)phenyl]prop-2-en-1-one Chemical compound C1=CC(N(CC)CC)=CC=C1C=CC(=O)C1=CC=C(N(CC)CC)C=C1 FJLAFBORFDOARE-UHFFFAOYSA-N 0.000 description 1
- YFMNMPLJIWVXER-UHFFFAOYSA-N 1,3-bis[4-(dimethylamino)phenyl]prop-2-en-1-one Chemical compound C1=CC(N(C)C)=CC=C1C=CC(=O)C1=CC=C(N(C)C)C=C1 YFMNMPLJIWVXER-UHFFFAOYSA-N 0.000 description 1
- OQZAQBGJENJMHT-UHFFFAOYSA-N 1,3-dibromo-5-methoxybenzene Chemical compound COC1=CC(Br)=CC(Br)=C1 OQZAQBGJENJMHT-UHFFFAOYSA-N 0.000 description 1
- KEQGZUUPPQEDPF-UHFFFAOYSA-N 1,3-dichloro-5,5-dimethylimidazolidine-2,4-dione Chemical compound CC1(C)N(Cl)C(=O)N(Cl)C1=O KEQGZUUPPQEDPF-UHFFFAOYSA-N 0.000 description 1
- YHMYGUUIMTVXNW-UHFFFAOYSA-N 1,3-dihydrobenzimidazole-2-thione Chemical compound C1=CC=C2NC(S)=NC2=C1 YHMYGUUIMTVXNW-UHFFFAOYSA-N 0.000 description 1
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 1
- WZCQRUWWHSTZEM-UHFFFAOYSA-N 1,3-phenylenediamine Chemical compound NC1=CC=CC(N)=C1 WZCQRUWWHSTZEM-UHFFFAOYSA-N 0.000 description 1
- CUJPFPXNDSIBPG-UHFFFAOYSA-N 1,3-propanediyl Chemical group [CH2]C[CH2] CUJPFPXNDSIBPG-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- 229940005561 1,4-benzoquinone Drugs 0.000 description 1
- FBMQNRKSAWNXBT-UHFFFAOYSA-N 1,4-diaminoanthracene-9,10-dione Chemical compound O=C1C2=CC=CC=C2C(=O)C2=C1C(N)=CC=C2N FBMQNRKSAWNXBT-UHFFFAOYSA-N 0.000 description 1
- CBCKQZAAMUWICA-UHFFFAOYSA-N 1,4-phenylenediamine Chemical compound NC1=CC=C(N)C=C1 CBCKQZAAMUWICA-UHFFFAOYSA-N 0.000 description 1
- ZNNRTWIXUUGIFI-UHFFFAOYSA-N 1,5-bis[4-(diethylamino)phenyl]penta-1,4-dien-3-one Chemical compound C1=CC(N(CC)CC)=CC=C1C=CC(=O)C=CC1=CC=C(N(CC)CC)C=C1 ZNNRTWIXUUGIFI-UHFFFAOYSA-N 0.000 description 1
- JWTSVUUPJIIXTO-UHFFFAOYSA-N 1,5-bis[4-(dimethylamino)phenyl]penta-1,4-dien-3-one Chemical compound C1=CC(N(C)C)=CC=C1C=CC(=O)C=CC1=CC=C(N(C)C)C=C1 JWTSVUUPJIIXTO-UHFFFAOYSA-N 0.000 description 1
- VWBVCOPVKXNMMZ-UHFFFAOYSA-N 1,5-diaminoanthracene-9,10-dione Chemical compound O=C1C2=C(N)C=CC=C2C(=O)C2=C1C=CC=C2N VWBVCOPVKXNMMZ-UHFFFAOYSA-N 0.000 description 1
- UWFRVQVNYNPBEF-UHFFFAOYSA-N 1-(2,4-dimethylphenyl)propan-1-one Chemical compound CCC(=O)C1=CC=C(C)C=C1C UWFRVQVNYNPBEF-UHFFFAOYSA-N 0.000 description 1
- GEWWCWZGHNIUBW-UHFFFAOYSA-N 1-(4-nitrophenyl)propan-2-one Chemical compound CC(=O)CC1=CC=C([N+]([O-])=O)C=C1 GEWWCWZGHNIUBW-UHFFFAOYSA-N 0.000 description 1
- HIYIGPNHCGAADM-UHFFFAOYSA-N 1-(methoxymethyl)-4-[4-(methoxymethyl)phenyl]-2,3-dimethylbenzene Chemical group CC=1C(=C(C=CC=1COC)C1=CC=C(C=C1)COC)C HIYIGPNHCGAADM-UHFFFAOYSA-N 0.000 description 1
- MODAACUAXYPNJH-UHFFFAOYSA-N 1-(methoxymethyl)-4-[4-(methoxymethyl)phenyl]benzene Chemical group C1=CC(COC)=CC=C1C1=CC=C(COC)C=C1 MODAACUAXYPNJH-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- GGZHVNZHFYCSEV-UHFFFAOYSA-N 1-Phenyl-5-mercaptotetrazole Chemical compound SC1=NN=NN1C1=CC=CC=C1 GGZHVNZHFYCSEV-UHFFFAOYSA-N 0.000 description 1
- HYLLZXPMJRMUHH-UHFFFAOYSA-N 1-[2-(2-methoxyethoxy)ethoxy]butane Chemical compound CCCCOCCOCCOC HYLLZXPMJRMUHH-UHFFFAOYSA-N 0.000 description 1
- RDSVPKGMYHANML-UHFFFAOYSA-N 1-[2-[1-(2-aminopropoxy)propan-2-yloxy]ethoxy]propan-2-amine Chemical compound CC(N)COCCOC(C)COCC(C)N RDSVPKGMYHANML-UHFFFAOYSA-N 0.000 description 1
- DSAUCRKFFBMTMD-UHFFFAOYSA-N 1-[5-[(5-acetyl-2,3,4-trihydroxyphenyl)methyl]-2,3,4-trihydroxyphenyl]ethanone Chemical compound OC1=C(O)C(C(=O)C)=CC(CC=2C(=C(O)C(O)=C(C(C)=O)C=2)O)=C1O DSAUCRKFFBMTMD-UHFFFAOYSA-N 0.000 description 1
- IANQTJSKSUMEQM-UHFFFAOYSA-N 1-benzofuran Chemical group C1=CC=C2OC=CC2=C1 IANQTJSKSUMEQM-UHFFFAOYSA-N 0.000 description 1
- JWYVGKFDLWWQJX-UHFFFAOYSA-N 1-ethenylazepan-2-one Chemical compound C=CN1CCCCCC1=O JWYVGKFDLWWQJX-UHFFFAOYSA-N 0.000 description 1
- RRQYJINTUHWNHW-UHFFFAOYSA-N 1-ethoxy-2-(2-ethoxyethoxy)ethane Chemical compound CCOCCOCCOCC RRQYJINTUHWNHW-UHFFFAOYSA-N 0.000 description 1
- JOLQKTGDSGKSKJ-UHFFFAOYSA-N 1-ethoxypropan-2-ol Chemical compound CCOCC(C)O JOLQKTGDSGKSKJ-UHFFFAOYSA-N 0.000 description 1
- KYWXRBNOYGGPIZ-UHFFFAOYSA-N 1-morpholin-4-ylethanone Chemical compound CC(=O)N1CCOCC1 KYWXRBNOYGGPIZ-UHFFFAOYSA-N 0.000 description 1
- KJCVRFUGPWSIIH-UHFFFAOYSA-N 1-naphthol Chemical compound C1=CC=C2C(O)=CC=CC2=C1 KJCVRFUGPWSIIH-UHFFFAOYSA-N 0.000 description 1
- YXAOOTNFFAQIPZ-UHFFFAOYSA-N 1-nitrosonaphthalen-2-ol Chemical compound C1=CC=CC2=C(N=O)C(O)=CC=C21 YXAOOTNFFAQIPZ-UHFFFAOYSA-N 0.000 description 1
- DKZRLCHWDNEKRH-UHFFFAOYSA-N 1-nonoxynonane Chemical compound CCCCCCCCCOCCCCCCCCC DKZRLCHWDNEKRH-UHFFFAOYSA-N 0.000 description 1
- WAPNOHKVXSQRPX-UHFFFAOYSA-N 1-phenylethanol Chemical compound CC(O)C1=CC=CC=C1 WAPNOHKVXSQRPX-UHFFFAOYSA-N 0.000 description 1
- TZMSYXZUNZXBOL-UHFFFAOYSA-N 10H-phenoxazine Chemical compound C1=CC=C2NC3=CC=CC=C3OC2=C1 TZMSYXZUNZXBOL-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- KGWYICAEPBCRBL-UHFFFAOYSA-N 1h-indene-1-carboxylic acid Chemical compound C1=CC=C2C(C(=O)O)C=CC2=C1 KGWYICAEPBCRBL-UHFFFAOYSA-N 0.000 description 1
- MVXVYAKCVDQRLW-UHFFFAOYSA-N 1h-pyrrolo[2,3-b]pyridine Chemical group C1=CN=C2NC=CC2=C1 MVXVYAKCVDQRLW-UHFFFAOYSA-N 0.000 description 1
- XIROXSOOOAZHLL-UHFFFAOYSA-N 2',3',4'-Trihydroxyacetophenone Chemical compound CC(=O)C1=CC=C(O)C(O)=C1O XIROXSOOOAZHLL-UHFFFAOYSA-N 0.000 description 1
- MEZZCSHVIGVWFI-UHFFFAOYSA-N 2,2'-Dihydroxy-4-methoxybenzophenone Chemical compound OC1=CC(OC)=CC=C1C(=O)C1=CC=CC=C1O MEZZCSHVIGVWFI-UHFFFAOYSA-N 0.000 description 1
- KGRVJHAUYBGFFP-UHFFFAOYSA-N 2,2'-Methylenebis(4-methyl-6-tert-butylphenol) Chemical compound CC(C)(C)C1=CC(C)=CC(CC=2C(=C(C=C(C)C=2)C(C)(C)C)O)=C1O KGRVJHAUYBGFFP-UHFFFAOYSA-N 0.000 description 1
- NOGFHTGYPKWWRX-UHFFFAOYSA-N 2,2,6,6-tetramethyloxan-4-one Chemical compound CC1(C)CC(=O)CC(C)(C)O1 NOGFHTGYPKWWRX-UHFFFAOYSA-N 0.000 description 1
- LTMRRSWNXVJMBA-UHFFFAOYSA-L 2,2-diethylpropanedioate Chemical compound CCC(CC)(C([O-])=O)C([O-])=O LTMRRSWNXVJMBA-UHFFFAOYSA-L 0.000 description 1
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 1
- HTQNYBBTZSBWKL-UHFFFAOYSA-N 2,3,4-trihydroxbenzophenone Chemical compound OC1=C(O)C(O)=CC=C1C(=O)C1=CC=CC=C1 HTQNYBBTZSBWKL-UHFFFAOYSA-N 0.000 description 1
- WCZNKVPCIFMXEQ-UHFFFAOYSA-N 2,3,5,6-tetramethylbenzene-1,4-diamine Chemical compound CC1=C(C)C(N)=C(C)C(C)=C1N WCZNKVPCIFMXEQ-UHFFFAOYSA-N 0.000 description 1
- YDYSEBSNAKCEQU-UHFFFAOYSA-N 2,3-diamino-n-phenylbenzamide Chemical compound NC1=CC=CC(C(=O)NC=2C=CC=CC=2)=C1N YDYSEBSNAKCEQU-UHFFFAOYSA-N 0.000 description 1
- KKTUQAYCCLMNOA-UHFFFAOYSA-N 2,3-diaminobenzoic acid Chemical compound NC1=CC=CC(C(O)=O)=C1N KKTUQAYCCLMNOA-UHFFFAOYSA-N 0.000 description 1
- ZVDSMYGTJDFNHN-UHFFFAOYSA-N 2,4,6-trimethylbenzene-1,3-diamine Chemical compound CC1=CC(C)=C(N)C(C)=C1N ZVDSMYGTJDFNHN-UHFFFAOYSA-N 0.000 description 1
- LXOFYPKXCSULTL-UHFFFAOYSA-N 2,4,7,9-tetramethyldec-5-yne-4,7-diol Chemical compound CC(C)CC(C)(O)C#CC(C)(O)CC(C)C LXOFYPKXCSULTL-UHFFFAOYSA-N 0.000 description 1
- ZXDDPOHVAMWLBH-UHFFFAOYSA-N 2,4-Dihydroxybenzophenone Chemical compound OC1=CC(O)=CC=C1C(=O)C1=CC=CC=C1 ZXDDPOHVAMWLBH-UHFFFAOYSA-N 0.000 description 1
- RWAOPZVGICHCOI-UHFFFAOYSA-N 2,4-diaminobenzene-1,3-diol Chemical compound NC1=CC=C(O)C(N)=C1O RWAOPZVGICHCOI-UHFFFAOYSA-N 0.000 description 1
- RLXBOUUYEFOFSW-UHFFFAOYSA-N 2,5-diaminobenzene-1,4-diol Chemical compound NC1=CC(O)=C(N)C=C1O RLXBOUUYEFOFSW-UHFFFAOYSA-N 0.000 description 1
- BWAPJIHJXDYDPW-UHFFFAOYSA-N 2,5-dimethyl-p-phenylenediamine Chemical compound CC1=CC(N)=C(C)C=C1N BWAPJIHJXDYDPW-UHFFFAOYSA-N 0.000 description 1
- QYXHDJJYVDLECA-UHFFFAOYSA-N 2,5-diphenylcyclohexa-2,5-diene-1,4-dione Chemical compound O=C1C=C(C=2C=CC=CC=2)C(=O)C=C1C1=CC=CC=C1 QYXHDJJYVDLECA-UHFFFAOYSA-N 0.000 description 1
- KUMMBDBTERQYCG-UHFFFAOYSA-N 2,6-bis(hydroxymethyl)-4-methylphenol Chemical compound CC1=CC(CO)=C(O)C(CO)=C1 KUMMBDBTERQYCG-UHFFFAOYSA-N 0.000 description 1
- DWKBMLWYFNGDCN-UHFFFAOYSA-N 2,6-bis(methoxymethyl)phenol Chemical compound COCC1=CC=CC(COC)=C1O DWKBMLWYFNGDCN-UHFFFAOYSA-N 0.000 description 1
- MAQOZOILPAMFSW-UHFFFAOYSA-N 2,6-bis[(2-hydroxy-5-methylphenyl)methyl]-4-methylphenol Chemical compound CC1=CC=C(O)C(CC=2C(=C(CC=3C(=CC=C(C)C=3)O)C=C(C)C=2)O)=C1 MAQOZOILPAMFSW-UHFFFAOYSA-N 0.000 description 1
- GOWFRAUGTCYVPS-UHFFFAOYSA-N 2,6-bis[(4-hydroxy-2,5-dimethylphenyl)methyl]-4-methylphenol Chemical compound OC=1C(CC=2C(=CC(O)=C(C)C=2)C)=CC(C)=CC=1CC1=CC(C)=C(O)C=C1C GOWFRAUGTCYVPS-UHFFFAOYSA-N 0.000 description 1
- CPIALDYHMPPRDJ-UHFFFAOYSA-N 2,6-bis[(4-hydroxy-3,5-dimethylphenyl)methyl]-4-methylphenol Chemical compound OC=1C(CC=2C=C(C)C(O)=C(C)C=2)=CC(C)=CC=1CC1=CC(C)=C(O)C(C)=C1 CPIALDYHMPPRDJ-UHFFFAOYSA-N 0.000 description 1
- FBJKAOHDILBGFG-UHFFFAOYSA-N 2,6-bis[(4-hydroxy-3,5-dimethylphenyl)methyl]cyclohexa-3,5-diene-1,1,3,4-tetrol Chemical compound CC1=C(O)C(C)=CC(CC2C(C(CC=3C=C(C)C(O)=C(C)C=3)=CC(O)=C2O)(O)O)=C1 FBJKAOHDILBGFG-UHFFFAOYSA-N 0.000 description 1
- QLTAOSCUNGRDIP-UHFFFAOYSA-N 2,6-bis[(4-hydroxy-3-methylphenyl)methyl]-4-methylphenol Chemical compound OC=1C(CC=2C=C(C)C(O)=CC=2)=CC(C)=CC=1CC1=CC=C(O)C(C)=C1 QLTAOSCUNGRDIP-UHFFFAOYSA-N 0.000 description 1
- HHEXUUDGIHLTSX-UHFFFAOYSA-N 2,6-bis[(4-hydroxy-3-methylphenyl)methyl]-4-phenylphenol Chemical compound C1=C(O)C(C)=CC(CC=2C(=C(CC=3C=C(C)C(O)=CC=3)C=C(C=2)C=2C=CC=CC=2)O)=C1 HHEXUUDGIHLTSX-UHFFFAOYSA-N 0.000 description 1
- CEHDSLDFUCVNKA-UHFFFAOYSA-N 2,6-bis[(4-hydroxyphenyl)methyl]-4-methylphenol Chemical compound OC=1C(CC=2C=CC(O)=CC=2)=CC(C)=CC=1CC1=CC=C(O)C=C1 CEHDSLDFUCVNKA-UHFFFAOYSA-N 0.000 description 1
- SFXPLYOSNTZJFW-UHFFFAOYSA-N 2,6-bis[[4-(diethylamino)phenyl]methylidene]-4-methylcyclohexan-1-one Chemical compound C1=CC(N(CC)CC)=CC=C1C=C(CC(C)C1)C(=O)C1=CC1=CC=C(N(CC)CC)C=C1 SFXPLYOSNTZJFW-UHFFFAOYSA-N 0.000 description 1
- LIFWPLOFXPGAJJ-UHFFFAOYSA-N 2,6-bis[[4-(diethylamino)phenyl]methylidene]cyclohexan-1-one Chemical compound C1=CC(N(CC)CC)=CC=C1C=C(CCC1)C(=O)C1=CC1=CC=C(N(CC)CC)C=C1 LIFWPLOFXPGAJJ-UHFFFAOYSA-N 0.000 description 1
- NXXYKOUNUYWIHA-UHFFFAOYSA-N 2,6-di-methyl phenol Natural products CC1=CC=CC(C)=C1O NXXYKOUNUYWIHA-UHFFFAOYSA-N 0.000 description 1
- DKCPKDPYUFEZCP-UHFFFAOYSA-N 2,6-di-tert-butylphenol Chemical compound CC(C)(C)C1=CC=CC(C(C)(C)C)=C1O DKCPKDPYUFEZCP-UHFFFAOYSA-N 0.000 description 1
- TYTPPOBYRKMHAV-UHFFFAOYSA-N 2,6-dimethylphenol Chemical compound CC1=CC=CC(C)=C1O.CC1=CC=CC(C)=C1O TYTPPOBYRKMHAV-UHFFFAOYSA-N 0.000 description 1
- XKWGWBPECLWUKL-UHFFFAOYSA-N 2-(1,3,6,8-tetratert-butylbenzo[d][1,3,2]benzodioxaphosphepin-10-yl)oxy-N,N-bis[2-(1,3,6,8-tetratert-butylbenzo[d][1,3,2]benzodioxaphosphepin-10-yl)oxyethyl]ethanamine Chemical compound C(C)(C)(C)C1=CC(=CC2=C1OP(OC1=C2C(=CC(=C1)C(C)(C)C)C(C)(C)C)C(C)(C)C)OCCN(CCOC1=CC2=C(OP(OC3=C2C(=CC(=C3)C(C)(C)C)C(C)(C)C)C(C)(C)C)C(=C1)C(C)(C)C)CCOC1=CC2=C(OP(OC3=C2C(=CC(=C3)C(C)(C)C)C(C)(C)C)C(C)(C)C)C(=C1)C(C)(C)C XKWGWBPECLWUKL-UHFFFAOYSA-N 0.000 description 1
- YAGPRJYCDKGWJR-UHFFFAOYSA-N 2-(2,4,8,10-tetratert-butylbenzo[d][1,3,2]benzodioxaphosphepin-6-yl)oxy-n,n-bis[2-(2,4,8,10-tetratert-butylbenzo[d][1,3,2]benzodioxaphosphepin-6-yl)oxyethyl]ethanamine Chemical compound O1C2=C(C(C)(C)C)C=C(C(C)(C)C)C=C2C2=CC(C(C)(C)C)=CC(C(C)(C)C)=C2OP1OCCN(CCOP1OC2=C(C=C(C=C2C=2C=C(C=C(C=2O1)C(C)(C)C)C(C)(C)C)C(C)(C)C)C(C)(C)C)CCOP(OC1=C(C=C(C=C11)C(C)(C)C)C(C)(C)C)OC2=C1C=C(C(C)(C)C)C=C2C(C)(C)C YAGPRJYCDKGWJR-UHFFFAOYSA-N 0.000 description 1
- STMDPCBYJCIZOD-UHFFFAOYSA-N 2-(2,4-dinitroanilino)-4-methylpentanoic acid Chemical compound CC(C)CC(C(O)=O)NC1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O STMDPCBYJCIZOD-UHFFFAOYSA-N 0.000 description 1
- VXQBJTKSVGFQOL-UHFFFAOYSA-N 2-(2-butoxyethoxy)ethyl acetate Chemical compound CCCCOCCOCCOC(C)=O VXQBJTKSVGFQOL-UHFFFAOYSA-N 0.000 description 1
- GZMAAYIALGURDQ-UHFFFAOYSA-N 2-(2-hexoxyethoxy)ethanol Chemical compound CCCCCCOCCOCCO GZMAAYIALGURDQ-UHFFFAOYSA-N 0.000 description 1
- PFTHQRBULKATPZ-UHFFFAOYSA-N 2-(3,4,5-trihydroxybenzoyl)oxyethyl 3,4,5-trihydroxybenzoate Chemical compound OC1=C(O)C(O)=CC(C(=O)OCCOC(=O)C=2C=C(O)C(O)=C(O)C=2)=C1 PFTHQRBULKATPZ-UHFFFAOYSA-N 0.000 description 1
- AZKQVMZEZRNXKG-UHFFFAOYSA-N 2-(3,5-dihydroxybenzoyl)oxyethyl 3,5-dihydroxybenzoate Chemical compound OC1=CC(O)=CC(C(=O)OCCOC(=O)C=2C=C(O)C=C(O)C=2)=C1 AZKQVMZEZRNXKG-UHFFFAOYSA-N 0.000 description 1
- KICYRZIVKKYRFS-UHFFFAOYSA-N 2-(3,5-dihydroxyphenyl)benzene-1,3,5-triol Chemical compound OC1=CC(O)=CC(C=2C(=CC(O)=CC=2O)O)=C1 KICYRZIVKKYRFS-UHFFFAOYSA-N 0.000 description 1
- JAHNSTQSQJOJLO-UHFFFAOYSA-N 2-(3-fluorophenyl)-1h-imidazole Chemical compound FC1=CC=CC(C=2NC=CN=2)=C1 JAHNSTQSQJOJLO-UHFFFAOYSA-N 0.000 description 1
- VOJRRQSAFQFFQU-UHFFFAOYSA-N 2-(3-triethoxysilylpropylcarbamoyl)benzoic acid Chemical compound CCO[Si](OCC)(OCC)CCCNC(=O)C1=CC=CC=C1C(O)=O VOJRRQSAFQFFQU-UHFFFAOYSA-N 0.000 description 1
- FXYQVUGCQHGGEX-UHFFFAOYSA-N 2-(5-chlorobenzotriazol-2-yl)-4-methyl-6-(2-methylpropyl)phenol Chemical compound CC(C)CC1=CC(C)=CC(N2N=C3C=C(Cl)C=CC3=N2)=C1O FXYQVUGCQHGGEX-UHFFFAOYSA-N 0.000 description 1
- PXMKSNKSDOKQGZ-UHFFFAOYSA-N 2-(5-chlorobenzotriazol-2-yl)-6-(2-methylbutan-2-yl)-4-(2-methylpropyl)phenol Chemical compound CCC(C)(C)C1=CC(CC(C)C)=CC(N2N=C3C=C(Cl)C=CC3=N2)=C1O PXMKSNKSDOKQGZ-UHFFFAOYSA-N 0.000 description 1
- ADFWZEGSIXNYRT-UHFFFAOYSA-N 2-(5-chlorobenzotriazol-2-yl)-6-(2-methylpropyl)-4-propylphenol Chemical compound CCCC1=CC(CC(C)C)=C(O)C(N2N=C3C=C(Cl)C=CC3=N2)=C1 ADFWZEGSIXNYRT-UHFFFAOYSA-N 0.000 description 1
- LHPPDQUVECZQSW-UHFFFAOYSA-N 2-(benzotriazol-2-yl)-4,6-ditert-butylphenol Chemical compound CC(C)(C)C1=CC(C(C)(C)C)=CC(N2N=C3C=CC=CC3=N2)=C1O LHPPDQUVECZQSW-UHFFFAOYSA-N 0.000 description 1
- FALRKNHUBBKYCC-UHFFFAOYSA-N 2-(chloromethyl)pyridine-3-carbonitrile Chemical compound ClCC1=NC=CC=C1C#N FALRKNHUBBKYCC-UHFFFAOYSA-N 0.000 description 1
- GJKGAPPUXSSCFI-UHFFFAOYSA-N 2-Hydroxy-4'-(2-hydroxyethoxy)-2-methylpropiophenone Chemical compound CC(C)(O)C(=O)C1=CC=C(OCCO)C=C1 GJKGAPPUXSSCFI-UHFFFAOYSA-N 0.000 description 1
- HDPLHDGYGLENEI-UHFFFAOYSA-N 2-[1-(oxiran-2-ylmethoxy)propan-2-yloxymethyl]oxirane Chemical compound C1OC1COC(C)COCC1CO1 HDPLHDGYGLENEI-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- AOBIOSPNXBMOAT-UHFFFAOYSA-N 2-[2-(oxiran-2-ylmethoxy)ethoxymethyl]oxirane Chemical compound C1OC1COCCOCC1CO1 AOBIOSPNXBMOAT-UHFFFAOYSA-N 0.000 description 1
- HWSSEYVMGDIFMH-UHFFFAOYSA-N 2-[2-[2-(2-methylprop-2-enoyloxy)ethoxy]ethoxy]ethyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCCOCCOCCOC(=O)C(C)=C HWSSEYVMGDIFMH-UHFFFAOYSA-N 0.000 description 1
- VIXBSRNUHGKKLM-UHFFFAOYSA-N 2-[3,5-bis(2-hydroxy-5-methylphenyl)phenyl]-4-methylphenol Chemical compound CC1=CC=C(O)C(C=2C=C(C=C(C=2)C=2C(=CC=C(C)C=2)O)C=2C(=CC=C(C)C=2)O)=C1 VIXBSRNUHGKKLM-UHFFFAOYSA-N 0.000 description 1
- SITYOOWCYAYOKL-UHFFFAOYSA-N 2-[4,6-bis(2,4-dimethylphenyl)-1,3,5-triazin-2-yl]-5-(3-dodecoxy-2-hydroxypropoxy)phenol Chemical compound OC1=CC(OCC(O)COCCCCCCCCCCCC)=CC=C1C1=NC(C=2C(=CC(C)=CC=2)C)=NC(C=2C(=CC(C)=CC=2)C)=N1 SITYOOWCYAYOKL-UHFFFAOYSA-N 0.000 description 1
- WPMUMRCRKFBYIH-UHFFFAOYSA-N 2-[4,6-bis(2-hydroxy-4-octoxyphenyl)-1,3,5-triazin-2-yl]-5-octoxyphenol Chemical compound OC1=CC(OCCCCCCCC)=CC=C1C1=NC(C=2C(=CC(OCCCCCCCC)=CC=2)O)=NC(C=2C(=CC(OCCCCCCCC)=CC=2)O)=N1 WPMUMRCRKFBYIH-UHFFFAOYSA-N 0.000 description 1
- KPXQQALZZHREIU-UHFFFAOYSA-N 2-[4,6-bis(2-hydroxyphenyl)triazin-5-yl]phenol Chemical class OC1=CC=CC=C1C1=NN=NC(C=2C(=CC=CC=2)O)=C1C1=CC=CC=C1O KPXQQALZZHREIU-UHFFFAOYSA-N 0.000 description 1
- SHKUUQIDMUMQQK-UHFFFAOYSA-N 2-[4-(oxiran-2-ylmethoxy)butoxymethyl]oxirane Chemical compound C1OC1COCCCCOCC1CO1 SHKUUQIDMUMQQK-UHFFFAOYSA-N 0.000 description 1
- YUZSJKBFHATJHV-UHFFFAOYSA-N 2-[4-[2-[4-(2-aminophenoxy)phenyl]-1,1,1,3,3,3-hexafluoropropan-2-yl]phenoxy]aniline Chemical compound NC1=CC=CC=C1OC1=CC=C(C(C=2C=CC(OC=3C(=CC=CC=3)N)=CC=2)(C(F)(F)F)C(F)(F)F)C=C1 YUZSJKBFHATJHV-UHFFFAOYSA-N 0.000 description 1
- FJYCAYKHNVQCJW-UHFFFAOYSA-N 2-[4-[4-(2-aminophenoxy)phenyl]sulfonylphenoxy]aniline Chemical compound NC1=CC=CC=C1OC1=CC=C(S(=O)(=O)C=2C=CC(OC=3C(=CC=CC=3)N)=CC=2)C=C1 FJYCAYKHNVQCJW-UHFFFAOYSA-N 0.000 description 1
- MSHXOTKVSBHKBZ-UHFFFAOYSA-N 2-[6-(2-hydroxyphenyl)triazin-4-yl]phenol Chemical class OC1=C(C=CC=C1)C1=CC(=NN=N1)C1=C(C=CC=C1)O MSHXOTKVSBHKBZ-UHFFFAOYSA-N 0.000 description 1
- WTYYGFLRBWMFRY-UHFFFAOYSA-N 2-[6-(oxiran-2-ylmethoxy)hexoxymethyl]oxirane Chemical compound C1OC1COCCCCCCOCC1CO1 WTYYGFLRBWMFRY-UHFFFAOYSA-N 0.000 description 1
- KUAUJXBLDYVELT-UHFFFAOYSA-N 2-[[2,2-dimethyl-3-(oxiran-2-ylmethoxy)propoxy]methyl]oxirane Chemical compound C1OC1COCC(C)(C)COCC1CO1 KUAUJXBLDYVELT-UHFFFAOYSA-N 0.000 description 1
- BXVXPASMKWYBFD-UHFFFAOYSA-N 2-[[2-hydroxy-3-(hydroxymethyl)-5-methylphenyl]methyl]-6-(hydroxymethyl)-4-methylphenol Chemical compound CC1=CC(CO)=C(O)C(CC=2C(=C(CO)C=C(C)C=2)O)=C1 BXVXPASMKWYBFD-UHFFFAOYSA-N 0.000 description 1
- VZKXQQSFNFJFSC-UHFFFAOYSA-N 2-[[2-hydroxy-3-[(2-hydroxy-3,5-dimethylphenyl)methyl]-5-methylphenyl]methyl]-4,6-dimethylphenol Chemical compound CC1=CC(C)=C(O)C(CC=2C(=C(CC=3C(=C(C)C=C(C)C=3)O)C=C(C)C=2)O)=C1 VZKXQQSFNFJFSC-UHFFFAOYSA-N 0.000 description 1
- OBWYKDNPBNLGON-UHFFFAOYSA-N 2-[[2-hydroxy-5-methyl-3-[(2,4,6-trihydroxyphenyl)methyl]phenyl]methyl]benzene-1,3,5-triol Chemical compound OC=1C(CC=2C(=CC(O)=CC=2O)O)=CC(C)=CC=1CC1=C(O)C=C(O)C=C1O OBWYKDNPBNLGON-UHFFFAOYSA-N 0.000 description 1
- YCKZAOPKIOWTEH-UHFFFAOYSA-N 2-[[4-(dimethylamino)phenyl]methylidene]-3h-inden-1-one Chemical compound C1=CC(N(C)C)=CC=C1C=C1C(=O)C2=CC=CC=C2C1 YCKZAOPKIOWTEH-UHFFFAOYSA-N 0.000 description 1
- QFPBHEMAYDQFHI-UHFFFAOYSA-N 2-[[6-hydroxy-3,5-dimethyl-3-[(2,4,6-trihydroxyphenyl)methyl]cyclohexa-1,5-dien-1-yl]methyl]benzene-1,3,5-triol Chemical compound C=1C(C)(CC=2C(=CC(O)=CC=2O)O)CC(C)=C(O)C=1CC1=C(O)C=C(O)C=C1O QFPBHEMAYDQFHI-UHFFFAOYSA-N 0.000 description 1
- VTYZTWSUUXQSPZ-UHFFFAOYSA-N 2-[bis(2-hydroxyethyl)amino]ethanol;propan-2-ol;titanium Chemical compound [Ti].CC(C)O.CC(C)O.OCCN(CCO)CCO.OCCN(CCO)CCO VTYZTWSUUXQSPZ-UHFFFAOYSA-N 0.000 description 1
- UTYHQSKRFPHMQQ-UHFFFAOYSA-N 2-amino-4-(3-amino-4-hydroxyphenoxy)phenol Chemical compound C1=C(O)C(N)=CC(OC=2C=C(N)C(O)=CC=2)=C1 UTYHQSKRFPHMQQ-UHFFFAOYSA-N 0.000 description 1
- KCFVSHSJPIVGCG-UHFFFAOYSA-N 2-amino-4-[(3-amino-4-hydroxyphenyl)methyl]phenol Chemical compound C1=C(O)C(N)=CC(CC=2C=C(N)C(O)=CC=2)=C1 KCFVSHSJPIVGCG-UHFFFAOYSA-N 0.000 description 1
- NPUFCLGAODQFLG-UHFFFAOYSA-N 2-amino-5-(4-aminophenyl)cyclohexa-2,4-diene-1,1-diol Chemical group C1C(O)(O)C(N)=CC=C1C1=CC=C(N)C=C1 NPUFCLGAODQFLG-UHFFFAOYSA-N 0.000 description 1
- RCYNJDVUURMJOZ-UHFFFAOYSA-N 2-amino-5-[(4-amino-3-hydroxyphenyl)methyl]phenol Chemical compound C1=C(O)C(N)=CC=C1CC1=CC=C(N)C(O)=C1 RCYNJDVUURMJOZ-UHFFFAOYSA-N 0.000 description 1
- ZMCHBSMFKQYNKA-UHFFFAOYSA-N 2-aminobenzenesulfonic acid Chemical compound NC1=CC=CC=C1S(O)(=O)=O ZMCHBSMFKQYNKA-UHFFFAOYSA-N 0.000 description 1
- MWGATWIBSKHFMR-UHFFFAOYSA-N 2-anilinoethanol Chemical compound OCCNC1=CC=CC=C1 MWGATWIBSKHFMR-UHFFFAOYSA-N 0.000 description 1
- UHFFVFAKEGKNAQ-UHFFFAOYSA-N 2-benzyl-2-(dimethylamino)-1-(4-morpholin-4-ylphenyl)butan-1-one Chemical compound C=1C=C(N2CCOCC2)C=CC=1C(=O)C(CC)(N(C)C)CC1=CC=CC=C1 UHFFVFAKEGKNAQ-UHFFFAOYSA-N 0.000 description 1
- SRGATTGYDONWOU-UHFFFAOYSA-N 2-cyclohexyl-5-methylphenol Chemical compound OC1=CC(C)=CC=C1C1CCCCC1 SRGATTGYDONWOU-UHFFFAOYSA-N 0.000 description 1
- ZVUNTIMPQCQCAQ-UHFFFAOYSA-N 2-dodecanoyloxyethyl dodecanoate Chemical compound CCCCCCCCCCCC(=O)OCCOC(=O)CCCCCCCCCCC ZVUNTIMPQCQCAQ-UHFFFAOYSA-N 0.000 description 1
- DORYOOVLRDMOQO-UHFFFAOYSA-N 2-dodecylbenzenesulfonic acid propan-2-olate titanium(4+) Chemical compound CC([O-])C.[Ti+4].C(CCCCCCCCCCC)C1=C(C=CC=C1)S(=O)(=O)O.C(CCCCCCCCCCC)C1=C(C=CC=C1)S(=O)(=O)O.C(CCCCCCCCCCC)C1=C(C=CC=C1)S(=O)(=O)O.CC([O-])C.CC([O-])C.CC([O-])C DORYOOVLRDMOQO-UHFFFAOYSA-N 0.000 description 1
- KKOHCQAVIJDYAF-UHFFFAOYSA-N 2-dodecylbenzenesulfonic acid;propan-2-ol;titanium Chemical compound [Ti].CC(C)O.CCCCCCCCCCCCC1=CC=CC=C1S(O)(=O)=O.CCCCCCCCCCCCC1=CC=CC=C1S(O)(=O)=O.CCCCCCCCCCCCC1=CC=CC=C1S(O)(=O)=O KKOHCQAVIJDYAF-UHFFFAOYSA-N 0.000 description 1
- 229940093475 2-ethoxyethanol Drugs 0.000 description 1
- WFSGQBNCVASPMW-UHFFFAOYSA-N 2-ethylhexanoyl chloride Chemical compound CCCCC(CC)C(Cl)=O WFSGQBNCVASPMW-UHFFFAOYSA-N 0.000 description 1
- NLGDWWCZQDIASO-UHFFFAOYSA-N 2-hydroxy-1-(7-oxabicyclo[4.1.0]hepta-1,3,5-trien-2-yl)-2-phenylethanone Chemical class OC(C(=O)c1cccc2Oc12)c1ccccc1 NLGDWWCZQDIASO-UHFFFAOYSA-N 0.000 description 1
- PCKZAVNWRLEHIP-UHFFFAOYSA-N 2-hydroxy-1-[4-[[4-(2-hydroxy-2-methylpropanoyl)phenyl]methyl]phenyl]-2-methylpropan-1-one Chemical compound C1=CC(C(=O)C(C)(O)C)=CC=C1CC1=CC=C(C(=O)C(C)(C)O)C=C1 PCKZAVNWRLEHIP-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- KXGFMDJXCMQABM-UHFFFAOYSA-N 2-methoxy-6-methylphenol Chemical compound [CH]OC1=CC=CC([CH])=C1O KXGFMDJXCMQABM-UHFFFAOYSA-N 0.000 description 1
- LWRBVKNFOYUCNP-UHFFFAOYSA-N 2-methyl-1-(4-methylsulfanylphenyl)-2-morpholin-4-ylpropan-1-one Chemical compound C1=CC(SC)=CC=C1C(=O)C(C)(C)N1CCOCC1 LWRBVKNFOYUCNP-UHFFFAOYSA-N 0.000 description 1
- RIWRBSMFKVOJMN-UHFFFAOYSA-N 2-methyl-1-phenylpropan-2-ol Chemical compound CC(C)(O)CC1=CC=CC=C1 RIWRBSMFKVOJMN-UHFFFAOYSA-N 0.000 description 1
- QTWJRLJHJPIABL-UHFFFAOYSA-N 2-methylphenol;3-methylphenol;4-methylphenol Chemical compound CC1=CC=C(O)C=C1.CC1=CC=CC(O)=C1.CC1=CC=CC=C1O QTWJRLJHJPIABL-UHFFFAOYSA-N 0.000 description 1
- SNOJPWLNAMAYSX-UHFFFAOYSA-N 2-methylpropan-1-ol;titanium Chemical compound [Ti].CC(C)CO.CC(C)CO.CC(C)CO.CC(C)CO SNOJPWLNAMAYSX-UHFFFAOYSA-N 0.000 description 1
- BNCADMBVWNPPIZ-UHFFFAOYSA-N 2-n,2-n,4-n,4-n,6-n,6-n-hexakis(methoxymethyl)-1,3,5-triazine-2,4,6-triamine Chemical compound COCN(COC)C1=NC(N(COC)COC)=NC(N(COC)COC)=N1 BNCADMBVWNPPIZ-UHFFFAOYSA-N 0.000 description 1
- JWAZRIHNYRIHIV-UHFFFAOYSA-N 2-naphthol Chemical compound C1=CC=CC2=CC(O)=CC=C21 JWAZRIHNYRIHIV-UHFFFAOYSA-N 0.000 description 1
- QCDWFXQBSFUVSP-UHFFFAOYSA-N 2-phenoxyethanol Chemical compound OCCOC1=CC=CC=C1 QCDWFXQBSFUVSP-UHFFFAOYSA-N 0.000 description 1
- XLLXMBCBJGATSP-UHFFFAOYSA-N 2-phenylethenol Chemical compound OC=CC1=CC=CC=C1 XLLXMBCBJGATSP-UHFFFAOYSA-N 0.000 description 1
- CUZKCNWZBXLAJX-UHFFFAOYSA-N 2-phenylmethoxyethanol Chemical compound OCCOCC1=CC=CC=C1 CUZKCNWZBXLAJX-UHFFFAOYSA-N 0.000 description 1
- WNYJRJRHKRZXEO-UHFFFAOYSA-N 2-propan-2-ylbenzene-1,4-diamine Chemical compound CC(C)C1=CC(N)=CC=C1N WNYJRJRHKRZXEO-UHFFFAOYSA-N 0.000 description 1
- HXIQYSLFEXIOAV-UHFFFAOYSA-N 2-tert-butyl-4-(5-tert-butyl-4-hydroxy-2-methylphenyl)sulfanyl-5-methylphenol Chemical compound CC1=CC(O)=C(C(C)(C)C)C=C1SC1=CC(C(C)(C)C)=C(O)C=C1C HXIQYSLFEXIOAV-UHFFFAOYSA-N 0.000 description 1
- IKEHOXWJQXIQAG-UHFFFAOYSA-N 2-tert-butyl-4-methylphenol Chemical compound CC1=CC=C(O)C(C(C)(C)C)=C1 IKEHOXWJQXIQAG-UHFFFAOYSA-N 0.000 description 1
- XBNVWXKPFORCRI-UHFFFAOYSA-N 2h-naphtho[2,3-f]quinolin-1-one Chemical class C1=CC=CC2=CC3=C4C(=O)CC=NC4=CC=C3C=C21 XBNVWXKPFORCRI-UHFFFAOYSA-N 0.000 description 1
- UAIUNKRWKOVEES-UHFFFAOYSA-N 3,3',5,5'-tetramethylbenzidine Chemical group CC1=C(N)C(C)=CC(C=2C=C(C)C(N)=C(C)C=2)=C1 UAIUNKRWKOVEES-UHFFFAOYSA-N 0.000 description 1
- JRBJSXQPQWSCCF-UHFFFAOYSA-N 3,3'-Dimethoxybenzidine Chemical group C1=C(N)C(OC)=CC(C=2C=C(OC)C(N)=CC=2)=C1 JRBJSXQPQWSCCF-UHFFFAOYSA-N 0.000 description 1
- NUIURNJTPRWVAP-UHFFFAOYSA-N 3,3'-Dimethylbenzidine Chemical group C1=C(N)C(C)=CC(C=2C=C(C)C(N)=CC=2)=C1 NUIURNJTPRWVAP-UHFFFAOYSA-N 0.000 description 1
- HSTOKWSFWGCZMH-UHFFFAOYSA-N 3,3'-diaminobenzidine Chemical group C1=C(N)C(N)=CC=C1C1=CC=C(N)C(N)=C1 HSTOKWSFWGCZMH-UHFFFAOYSA-N 0.000 description 1
- LFMCTLLZHYCABW-UHFFFAOYSA-N 3,3,3',3'-tetramethyl-1,1'-spirobi[2h-indene]-4,4',5,5',6,6'-hexol Chemical compound C12=CC(O)=C(O)C(O)=C2C(C)(C)CC11C(C=C(O)C(O)=C2O)=C2C(C)(C)C1 LFMCTLLZHYCABW-UHFFFAOYSA-N 0.000 description 1
- SDUDVCDCBOXLNZ-UHFFFAOYSA-N 3,3-bis(2,3,4-trihydroxyphenyl)-2-benzofuran-1-one Chemical compound OC1=C(O)C(O)=CC=C1C1(C=2C(=C(O)C(O)=CC=2)O)C2=CC=CC=C2C(=O)O1 SDUDVCDCBOXLNZ-UHFFFAOYSA-N 0.000 description 1
- QPFLFTVMWJJXKU-UHFFFAOYSA-N 3,3-bis(3,4-dihydroxyphenyl)-2-benzofuran-1-one Chemical compound C1=C(O)C(O)=CC=C1C1(C=2C=C(O)C(O)=CC=2)C2=CC=CC=C2C(=O)O1 QPFLFTVMWJJXKU-UHFFFAOYSA-N 0.000 description 1
- WEDOWOFLQFYLGV-UHFFFAOYSA-N 3,4-bis(hydroxymethyl)-2-methylphenol Chemical compound CC1=C(O)C=CC(CO)=C1CO WEDOWOFLQFYLGV-UHFFFAOYSA-N 0.000 description 1
- UHIZKBVOWLXJMK-UHFFFAOYSA-N 3,4-bis(methoxymethyl)-2-methylphenol Chemical compound COCC=1C(=C(C(=CC1)O)C)COC UHIZKBVOWLXJMK-UHFFFAOYSA-N 0.000 description 1
- UYMBCDOGDVGEFA-UHFFFAOYSA-N 3-(1h-indol-2-yl)-3h-2-benzofuran-1-one Chemical compound C12=CC=CC=C2C(=O)OC1C1=CC2=CC=CC=C2N1 UYMBCDOGDVGEFA-UHFFFAOYSA-N 0.000 description 1
- NDXGRHCEHPFUSU-UHFFFAOYSA-N 3-(3-aminophenyl)aniline Chemical group NC1=CC=CC(C=2C=C(N)C=CC=2)=C1 NDXGRHCEHPFUSU-UHFFFAOYSA-N 0.000 description 1
- LJGHYPLBDBRCRZ-UHFFFAOYSA-N 3-(3-aminophenyl)sulfonylaniline Chemical compound NC1=CC=CC(S(=O)(=O)C=2C=C(N)C=CC=2)=C1 LJGHYPLBDBRCRZ-UHFFFAOYSA-N 0.000 description 1
- ZADOWCXTUZWAKL-UHFFFAOYSA-N 3-(3-trimethoxysilylpropyl)oxolane-2,5-dione Chemical compound CO[Si](OC)(OC)CCCC1CC(=O)OC1=O ZADOWCXTUZWAKL-UHFFFAOYSA-N 0.000 description 1
- RNLHGQLZWXBQNY-UHFFFAOYSA-N 3-(aminomethyl)-3,5,5-trimethylcyclohexan-1-amine Chemical compound CC1(C)CC(N)CC(C)(CN)C1 RNLHGQLZWXBQNY-UHFFFAOYSA-N 0.000 description 1
- WWJCRUKUIQRCGP-UHFFFAOYSA-N 3-(dimethylamino)propyl 2-methylprop-2-enoate Chemical compound CN(C)CCCOC(=O)C(C)=C WWJCRUKUIQRCGP-UHFFFAOYSA-N 0.000 description 1
- BQGXXEGJJMEZMZ-UHFFFAOYSA-N 3-(n-ethyl-3-hydroxy-4-nitrosoanilino)propane-1-sulfonic acid Chemical compound OS(=O)(=O)CCCN(CC)C1=CC=C(N=O)C(O)=C1 BQGXXEGJJMEZMZ-UHFFFAOYSA-N 0.000 description 1
- PQFRTJPVZSPBFI-UHFFFAOYSA-N 3-(trifluoromethyl)benzene-1,2-diamine Chemical compound NC1=CC=CC(C(F)(F)F)=C1N PQFRTJPVZSPBFI-UHFFFAOYSA-N 0.000 description 1
- ZZYZUFNSKAIYCJ-UHFFFAOYSA-N 3-[(3-aminocyclohexyl)methyl]cyclohexan-1-amine Chemical compound C1C(N)CCCC1CC1CC(N)CCC1 ZZYZUFNSKAIYCJ-UHFFFAOYSA-N 0.000 description 1
- CKOFBUUFHALZGK-UHFFFAOYSA-N 3-[(3-aminophenyl)methyl]aniline Chemical compound NC1=CC=CC(CC=2C=C(N)C=CC=2)=C1 CKOFBUUFHALZGK-UHFFFAOYSA-N 0.000 description 1
- UCFMKTNJZCYBBJ-UHFFFAOYSA-N 3-[1-(2,3-dicarboxyphenyl)ethyl]phthalic acid Chemical compound C=1C=CC(C(O)=O)=C(C(O)=O)C=1C(C)C1=CC=CC(C(O)=O)=C1C(O)=O UCFMKTNJZCYBBJ-UHFFFAOYSA-N 0.000 description 1
- PAHZZOIHRHCHTH-UHFFFAOYSA-N 3-[2-(2,3-dicarboxyphenyl)propan-2-yl]phthalic acid Chemical compound C=1C=CC(C(O)=O)=C(C(O)=O)C=1C(C)(C)C1=CC=CC(C(O)=O)=C1C(O)=O PAHZZOIHRHCHTH-UHFFFAOYSA-N 0.000 description 1
- DKKYOQYISDAQER-UHFFFAOYSA-N 3-[3-(3-aminophenoxy)phenoxy]aniline Chemical compound NC1=CC=CC(OC=2C=C(OC=3C=C(N)C=CC=3)C=CC=2)=C1 DKKYOQYISDAQER-UHFFFAOYSA-N 0.000 description 1
- MFTFTIALAXXIMU-UHFFFAOYSA-N 3-[4-[2-[4-(3-aminophenoxy)phenyl]-1,1,1,3,3,3-hexafluoropropan-2-yl]phenoxy]aniline Chemical compound NC1=CC=CC(OC=2C=CC(=CC=2)C(C=2C=CC(OC=3C=C(N)C=CC=3)=CC=2)(C(F)(F)F)C(F)(F)F)=C1 MFTFTIALAXXIMU-UHFFFAOYSA-N 0.000 description 1
- WCXGOVYROJJXHA-UHFFFAOYSA-N 3-[4-[4-(3-aminophenoxy)phenyl]sulfonylphenoxy]aniline Chemical compound NC1=CC=CC(OC=2C=CC(=CC=2)S(=O)(=O)C=2C=CC(OC=3C=C(N)C=CC=3)=CC=2)=C1 WCXGOVYROJJXHA-UHFFFAOYSA-N 0.000 description 1
- DOYKFSOCSXVQAN-UHFFFAOYSA-N 3-[diethoxy(methyl)silyl]propyl 2-methylprop-2-enoate Chemical compound CCO[Si](C)(OCC)CCCOC(=O)C(C)=C DOYKFSOCSXVQAN-UHFFFAOYSA-N 0.000 description 1
- IKYAJDOSWUATPI-UHFFFAOYSA-N 3-[dimethoxy(methyl)silyl]propane-1-thiol Chemical compound CO[Si](C)(OC)CCCS IKYAJDOSWUATPI-UHFFFAOYSA-N 0.000 description 1
- LZMNXXQIQIHFGC-UHFFFAOYSA-N 3-[dimethoxy(methyl)silyl]propyl 2-methylprop-2-enoate Chemical compound CO[Si](C)(OC)CCCOC(=O)C(C)=C LZMNXXQIQIHFGC-UHFFFAOYSA-N 0.000 description 1
- MYKMRINTJPZLQB-UHFFFAOYSA-N 3-acetyl-7-(dimethylamino)chromen-2-one Chemical compound C1=C(C(C)=O)C(=O)OC2=CC(N(C)C)=CC=C21 MYKMRINTJPZLQB-UHFFFAOYSA-N 0.000 description 1
- LVYXPOCADCXMLP-UHFFFAOYSA-N 3-butoxy-n,n-dimethylpropanamide Chemical compound CCCCOCCC(=O)N(C)C LVYXPOCADCXMLP-UHFFFAOYSA-N 0.000 description 1
- KNWLMUXHNAQOJM-UHFFFAOYSA-N 3-ethyl-3-(3-ethylheptyl)oxetane Chemical compound CCCCC(CC)CCC1(CC)COC1 KNWLMUXHNAQOJM-UHFFFAOYSA-N 0.000 description 1
- FNYWFRSQRHGKJT-UHFFFAOYSA-N 3-ethyl-3-[(3-ethyloxetan-3-yl)methoxymethyl]oxetane Chemical compound C1OCC1(CC)COCC1(CC)COC1 FNYWFRSQRHGKJT-UHFFFAOYSA-N 0.000 description 1
- LBVMWHCOFMFPEG-UHFFFAOYSA-N 3-methoxy-n,n-dimethylpropanamide Chemical compound COCCC(=O)N(C)C LBVMWHCOFMFPEG-UHFFFAOYSA-N 0.000 description 1
- HAHWYTLUKCIGPH-UHFFFAOYSA-N 3-methylbutyl 2-(diethylamino)benzoate Chemical compound CCN(CC)C1=CC=CC=C1C(=O)OCCC(C)C HAHWYTLUKCIGPH-UHFFFAOYSA-N 0.000 description 1
- ARXVXVOLXMVYIT-UHFFFAOYSA-N 3-methylbutyl 2-(dimethylamino)benzoate Chemical compound CC(C)CCOC(=O)C1=CC=CC=C1N(C)C ARXVXVOLXMVYIT-UHFFFAOYSA-N 0.000 description 1
- VATRWWPJWVCZTA-UHFFFAOYSA-N 3-oxo-n-[2-(trifluoromethyl)phenyl]butanamide Chemical compound CC(=O)CC(=O)NC1=CC=CC=C1C(F)(F)F VATRWWPJWVCZTA-UHFFFAOYSA-N 0.000 description 1
- MECNWXGGNCJFQJ-UHFFFAOYSA-N 3-piperidin-1-ylpropane-1,2-diol Chemical compound OCC(O)CN1CCCCC1 MECNWXGGNCJFQJ-UHFFFAOYSA-N 0.000 description 1
- ATVJXMYDOSMEPO-UHFFFAOYSA-N 3-prop-2-enoxyprop-1-ene Chemical compound C=CCOCC=C ATVJXMYDOSMEPO-UHFFFAOYSA-N 0.000 description 1
- URDOJQUSEUXVRP-UHFFFAOYSA-N 3-triethoxysilylpropyl 2-methylprop-2-enoate Chemical compound CCO[Si](OCC)(OCC)CCCOC(=O)C(C)=C URDOJQUSEUXVRP-UHFFFAOYSA-N 0.000 description 1
- SJECZPVISLOESU-UHFFFAOYSA-N 3-trimethoxysilylpropan-1-amine Chemical compound CO[Si](OC)(OC)CCCN SJECZPVISLOESU-UHFFFAOYSA-N 0.000 description 1
- UUEWCQRISZBELL-UHFFFAOYSA-N 3-trimethoxysilylpropane-1-thiol Chemical compound CO[Si](OC)(OC)CCCS UUEWCQRISZBELL-UHFFFAOYSA-N 0.000 description 1
- XDLMVUHYZWKMMD-UHFFFAOYSA-N 3-trimethoxysilylpropyl 2-methylprop-2-enoate Chemical compound CO[Si](OC)(OC)CCCOC(=O)C(C)=C XDLMVUHYZWKMMD-UHFFFAOYSA-N 0.000 description 1
- KBQVDAIIQCXKPI-UHFFFAOYSA-N 3-trimethoxysilylpropyl prop-2-enoate Chemical compound CO[Si](OC)(OC)CCCOC(=O)C=C KBQVDAIIQCXKPI-UHFFFAOYSA-N 0.000 description 1
- TXFPEBPIARQUIG-UHFFFAOYSA-N 4'-hydroxyacetophenone Chemical compound CC(=O)C1=CC=C(O)C=C1 TXFPEBPIARQUIG-UHFFFAOYSA-N 0.000 description 1
- WECDUOXQLAIPQW-UHFFFAOYSA-N 4,4'-Methylene bis(2-methylaniline) Chemical compound C1=C(N)C(C)=CC(CC=2C=C(C)C(N)=CC=2)=C1 WECDUOXQLAIPQW-UHFFFAOYSA-N 0.000 description 1
- LATROIIBXZFCTH-UHFFFAOYSA-N 4,5-dihydroxy-1,3-bis(methoxymethyl)imidazolidin-2-one Chemical compound COCN1C(O)C(O)N(COC)C1=O LATROIIBXZFCTH-UHFFFAOYSA-N 0.000 description 1
- JEKBXKBRBLABQJ-UHFFFAOYSA-N 4,6-bis[(4-hydroxy-3,5-dimethylphenyl)methyl]benzene-1,2,3-triol Chemical compound CC1=C(O)C(C)=CC(CC=2C(=C(O)C(O)=C(CC=3C=C(C)C(O)=C(C)C=3)C=2)O)=C1 JEKBXKBRBLABQJ-UHFFFAOYSA-N 0.000 description 1
- DPYROBMRMXHROQ-UHFFFAOYSA-N 4,6-diaminobenzene-1,3-diol Chemical compound NC1=CC(N)=C(O)C=C1O DPYROBMRMXHROQ-UHFFFAOYSA-N 0.000 description 1
- FYYYKXFEKMGYLZ-UHFFFAOYSA-N 4-(1,3-dioxo-2-benzofuran-5-yl)-2-benzofuran-1,3-dione Chemical compound C=1C=C2C(=O)OC(=O)C2=CC=1C1=CC=CC2=C1C(=O)OC2=O FYYYKXFEKMGYLZ-UHFFFAOYSA-N 0.000 description 1
- QPINXWKZYKLBNB-UHFFFAOYSA-N 4-(2,3,4-trihydroxyphenyl)benzene-1,2,3-triol Chemical compound OC1=C(O)C(O)=CC=C1C1=CC=C(O)C(O)=C1O QPINXWKZYKLBNB-UHFFFAOYSA-N 0.000 description 1
- CFGDTWRKBRQUFB-UHFFFAOYSA-N 4-(2,4-dihydroxyphenyl)benzene-1,3-diol Chemical compound OC1=CC(O)=CC=C1C1=CC=C(O)C=C1O CFGDTWRKBRQUFB-UHFFFAOYSA-N 0.000 description 1
- ODPLCKKBVAJQEP-UHFFFAOYSA-N 4-(2-benzo[e][1,3]benzothiazol-2-ylethenyl)-n,n-dimethylaniline Chemical compound C1=CC(N(C)C)=CC=C1C=CC(S1)=NC2=C1C=CC1=CC=CC=C21 ODPLCKKBVAJQEP-UHFFFAOYSA-N 0.000 description 1
- RQBIGPMJQUKYAH-UHFFFAOYSA-N 4-(3,4-diaminophenoxy)benzene-1,2-diamine Chemical compound C1=C(N)C(N)=CC=C1OC1=CC=C(N)C(N)=C1 RQBIGPMJQUKYAH-UHFFFAOYSA-N 0.000 description 1
- LSJAPRRUOIMQSN-UHFFFAOYSA-N 4-(4-amino-2-fluorophenyl)-3-fluoroaniline Chemical group FC1=CC(N)=CC=C1C1=CC=C(N)C=C1F LSJAPRRUOIMQSN-UHFFFAOYSA-N 0.000 description 1
- QQWWWAQUMVHHQN-UHFFFAOYSA-N 4-(4-amino-4-phenylcyclohexa-1,5-dien-1-yl)aniline Chemical group C1=CC(N)=CC=C1C1=CCC(N)(C=2C=CC=CC=2)C=C1 QQWWWAQUMVHHQN-UHFFFAOYSA-N 0.000 description 1
- QNJWXYRVQVNUQC-UHFFFAOYSA-N 4-(4-aminophenyl)-3-(2-phenylphenyl)aniline Chemical group C1(=C(C=CC=C1)C1=C(C=CC(=C1)N)C1=CC=C(N)C=C1)C1=CC=CC=C1 QNJWXYRVQVNUQC-UHFFFAOYSA-N 0.000 description 1
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 1
- NBLFJUWXERDUEN-UHFFFAOYSA-N 4-[(2,3,4-trihydroxyphenyl)methyl]benzene-1,2,3-triol Chemical compound OC1=C(O)C(O)=CC=C1CC1=CC=C(O)C(O)=C1O NBLFJUWXERDUEN-UHFFFAOYSA-N 0.000 description 1
- FNFYXIMJKWENNK-UHFFFAOYSA-N 4-[(2,4-dihydroxyphenyl)methyl]benzene-1,3-diol Chemical compound OC1=CC(O)=CC=C1CC1=CC=C(O)C=C1O FNFYXIMJKWENNK-UHFFFAOYSA-N 0.000 description 1
- OMHOXRVODFQGCA-UHFFFAOYSA-N 4-[(4-amino-3,5-dimethylphenyl)methyl]-2,6-dimethylaniline Chemical compound CC1=C(N)C(C)=CC(CC=2C=C(C)C(N)=C(C)C=2)=C1 OMHOXRVODFQGCA-UHFFFAOYSA-N 0.000 description 1
- DZIHTWJGPDVSGE-UHFFFAOYSA-N 4-[(4-aminocyclohexyl)methyl]cyclohexan-1-amine Chemical compound C1CC(N)CCC1CC1CCC(N)CC1 DZIHTWJGPDVSGE-UHFFFAOYSA-N 0.000 description 1
- RSSGMIIGVQRGDS-UHFFFAOYSA-N 4-[(4-hydroxyphenyl)-phenylmethyl]phenol Chemical compound C1=CC(O)=CC=C1C(C=1C=CC(O)=CC=1)C1=CC=CC=C1 RSSGMIIGVQRGDS-UHFFFAOYSA-N 0.000 description 1
- BGQWVIOOEFRPOV-UHFFFAOYSA-N 4-[1,1,1,3,3,3-hexafluoro-2-[4-hydroxy-3,5-bis(hydroxymethyl)phenyl]propan-2-yl]-2,6-bis(hydroxymethyl)phenol Chemical compound OCC1=C(O)C(CO)=CC(C(C=2C=C(CO)C(O)=C(CO)C=2)(C(F)(F)F)C(F)(F)F)=C1 BGQWVIOOEFRPOV-UHFFFAOYSA-N 0.000 description 1
- IJJNNSUCZDJDLP-UHFFFAOYSA-N 4-[1-(3,4-dicarboxyphenyl)ethyl]phthalic acid Chemical compound C=1C=C(C(O)=O)C(C(O)=O)=CC=1C(C)C1=CC=C(C(O)=O)C(C(O)=O)=C1 IJJNNSUCZDJDLP-UHFFFAOYSA-N 0.000 description 1
- MMNLWVVVDFQANH-UHFFFAOYSA-N 4-[1-(4-hydroxy-3-methylphenyl)-1-phenylethyl]-2-methylphenol Chemical compound C1=C(O)C(C)=CC(C(C)(C=2C=CC=CC=2)C=2C=C(C)C(O)=CC=2)=C1 MMNLWVVVDFQANH-UHFFFAOYSA-N 0.000 description 1
- ASNOFHCTUSIHOM-UHFFFAOYSA-N 4-[10-(4-aminophenyl)anthracen-9-yl]aniline Chemical compound C1=CC(N)=CC=C1C(C1=CC=CC=C11)=C(C=CC=C2)C2=C1C1=CC=C(N)C=C1 ASNOFHCTUSIHOM-UHFFFAOYSA-N 0.000 description 1
- DYDBIHRVHOCPSZ-UHFFFAOYSA-N 4-[2,2,3-tris(4-hydroxyphenyl)propyl]phenol Chemical compound C1=CC(O)=CC=C1CC(C=1C=CC(O)=CC=1)(C=1C=CC(O)=CC=1)CC1=CC=C(O)C=C1 DYDBIHRVHOCPSZ-UHFFFAOYSA-N 0.000 description 1
- DQOPDYYQICTYEY-UHFFFAOYSA-N 4-[2-(1,3-benzoxazol-2-yl)ethenyl]-n,n-dimethylaniline Chemical compound C1=CC(N(C)C)=CC=C1C=CC1=NC2=CC=CC=C2O1 DQOPDYYQICTYEY-UHFFFAOYSA-N 0.000 description 1
- APXJLYIVOFARRM-UHFFFAOYSA-N 4-[2-(3,4-dicarboxyphenyl)-1,1,1,3,3,3-hexafluoropropan-2-yl]phthalic acid Chemical compound C1=C(C(O)=O)C(C(=O)O)=CC=C1C(C(F)(F)F)(C(F)(F)F)C1=CC=C(C(O)=O)C(C(O)=O)=C1 APXJLYIVOFARRM-UHFFFAOYSA-N 0.000 description 1
- GEYAGBVEAJGCFB-UHFFFAOYSA-N 4-[2-(3,4-dicarboxyphenyl)propan-2-yl]phthalic acid Chemical compound C=1C=C(C(O)=O)C(C(O)=O)=CC=1C(C)(C)C1=CC=C(C(O)=O)C(C(O)=O)=C1 GEYAGBVEAJGCFB-UHFFFAOYSA-N 0.000 description 1
- BEKFRNOZJSYWKZ-UHFFFAOYSA-N 4-[2-(4-aminophenyl)-1,1,1,3,3,3-hexafluoropropan-2-yl]aniline Chemical compound C1=CC(N)=CC=C1C(C(F)(F)F)(C(F)(F)F)C1=CC=C(N)C=C1 BEKFRNOZJSYWKZ-UHFFFAOYSA-N 0.000 description 1
- UHNUHZHQLCGZDA-UHFFFAOYSA-N 4-[2-(4-aminophenyl)ethyl]aniline Chemical compound C1=CC(N)=CC=C1CCC1=CC=C(N)C=C1 UHNUHZHQLCGZDA-UHFFFAOYSA-N 0.000 description 1
- ZYEDGEXYGKWJPB-UHFFFAOYSA-N 4-[2-(4-aminophenyl)propan-2-yl]aniline Chemical compound C=1C=C(N)C=CC=1C(C)(C)C1=CC=C(N)C=C1 ZYEDGEXYGKWJPB-UHFFFAOYSA-N 0.000 description 1
- VJMWNJWOAKHNDM-UHFFFAOYSA-N 4-[3,3,5-tris(4-hydroxyphenyl)pentyl]phenol Chemical compound C1=CC(O)=CC=C1CCC(C=1C=CC(O)=CC=1)(C=1C=CC(O)=CC=1)CCC1=CC=C(O)C=C1 VJMWNJWOAKHNDM-UHFFFAOYSA-N 0.000 description 1
- YUEUCKPWACQKRJ-UHFFFAOYSA-N 4-[3,5-bis(4-hydroxy-3,5-dimethylphenyl)phenyl]-2,6-dimethylphenol Chemical compound CC1=C(O)C(C)=CC(C=2C=C(C=C(C=2)C=2C=C(C)C(O)=C(C)C=2)C=2C=C(C)C(O)=C(C)C=2)=C1 YUEUCKPWACQKRJ-UHFFFAOYSA-N 0.000 description 1
- WUPRYUDHUFLKFL-UHFFFAOYSA-N 4-[3-(4-aminophenoxy)phenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC1=CC=CC(OC=2C=CC(N)=CC=2)=C1 WUPRYUDHUFLKFL-UHFFFAOYSA-N 0.000 description 1
- LTKJRDQWPMBSFN-UHFFFAOYSA-N 4-[3-(4-aminophenyl)-1,1,2,2,3,3-hexafluoropropyl]aniline Chemical compound C1=CC(N)=CC=C1C(F)(F)C(F)(F)C(F)(F)C1=CC=C(N)C=C1 LTKJRDQWPMBSFN-UHFFFAOYSA-N 0.000 description 1
- BOVVHULZWVFIOX-UHFFFAOYSA-N 4-[3-(4-aminophenyl)phenyl]aniline Chemical compound C1=CC(N)=CC=C1C1=CC=CC(C=2C=CC(N)=CC=2)=C1 BOVVHULZWVFIOX-UHFFFAOYSA-N 0.000 description 1
- JCRRFJIVUPSNTA-UHFFFAOYSA-N 4-[4-(4-aminophenoxy)phenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC(C=C1)=CC=C1OC1=CC=C(N)C=C1 JCRRFJIVUPSNTA-UHFFFAOYSA-N 0.000 description 1
- GACNZKUFHGKSEX-UHFFFAOYSA-N 4-[4-(4-aminophenyl)-1,1,2,2,3,3,4,4-octafluorobutyl]aniline Chemical compound C1=CC(N)=CC=C1C(F)(F)C(F)(F)C(F)(F)C(F)(F)C1=CC=C(N)C=C1 GACNZKUFHGKSEX-UHFFFAOYSA-N 0.000 description 1
- HBLYIUPUXAWDMA-UHFFFAOYSA-N 4-[4-[2-[4-(4-aminophenoxy)-3,5-bis(trifluoromethyl)phenyl]-1,1,1,3,3,3-hexafluoropropan-2-yl]-2,6-bis(trifluoromethyl)phenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC1=C(C(F)(F)F)C=C(C(C=2C=C(C(OC=3C=CC(N)=CC=3)=C(C=2)C(F)(F)F)C(F)(F)F)(C(F)(F)F)C(F)(F)F)C=C1C(F)(F)F HBLYIUPUXAWDMA-UHFFFAOYSA-N 0.000 description 1
- VCFYKCXKADGLPS-UHFFFAOYSA-N 4-[4-[2-[4-(4-aminophenoxy)-3,5-dimethylphenyl]-1,1,1,3,3,3-hexafluoropropan-2-yl]-2,6-dimethylphenoxy]aniline Chemical compound CC1=CC(C(C=2C=C(C)C(OC=3C=CC(N)=CC=3)=C(C)C=2)(C(F)(F)F)C(F)(F)F)=CC(C)=C1OC1=CC=C(N)C=C1 VCFYKCXKADGLPS-UHFFFAOYSA-N 0.000 description 1
- HHLMWQDRYZAENA-UHFFFAOYSA-N 4-[4-[2-[4-(4-aminophenoxy)phenyl]-1,1,1,3,3,3-hexafluoropropan-2-yl]phenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC1=CC=C(C(C=2C=CC(OC=3C=CC(N)=CC=3)=CC=2)(C(F)(F)F)C(F)(F)F)C=C1 HHLMWQDRYZAENA-UHFFFAOYSA-N 0.000 description 1
- KMKWGXGSGPYISJ-UHFFFAOYSA-N 4-[4-[2-[4-(4-aminophenoxy)phenyl]propan-2-yl]phenoxy]aniline Chemical compound C=1C=C(OC=2C=CC(N)=CC=2)C=CC=1C(C)(C)C(C=C1)=CC=C1OC1=CC=C(N)C=C1 KMKWGXGSGPYISJ-UHFFFAOYSA-N 0.000 description 1
- PDYQWKUIJVOAON-UHFFFAOYSA-N 4-[4-[2-[4-[4-amino-3-(trifluoromethyl)phenoxy]phenyl]-1,1,1,3,3,3-hexafluoropropan-2-yl]phenoxy]-2-(trifluoromethyl)aniline Chemical compound C1=C(C(F)(F)F)C(N)=CC=C1OC1=CC=C(C(C=2C=CC(OC=3C=C(C(N)=CC=3)C(F)(F)F)=CC=2)(C(F)(F)F)C(F)(F)F)C=C1 PDYQWKUIJVOAON-UHFFFAOYSA-N 0.000 description 1
- HYDATEKARGDBKU-UHFFFAOYSA-N 4-[4-[4-(4-aminophenoxy)phenyl]phenoxy]aniline Chemical group C1=CC(N)=CC=C1OC1=CC=C(C=2C=CC(OC=3C=CC(N)=CC=3)=CC=2)C=C1 HYDATEKARGDBKU-UHFFFAOYSA-N 0.000 description 1
- UTDAGHZGKXPRQI-UHFFFAOYSA-N 4-[4-[4-(4-aminophenoxy)phenyl]sulfonylphenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC1=CC=C(S(=O)(=O)C=2C=CC(OC=3C=CC(N)=CC=3)=CC=2)C=C1 UTDAGHZGKXPRQI-UHFFFAOYSA-N 0.000 description 1
- DPCDFSDBIWVMJC-UHFFFAOYSA-N 4-[4-[4-[4-amino-3-(trifluoromethyl)phenoxy]phenyl]phenoxy]-2-(trifluoromethyl)aniline Chemical group C1=C(C(F)(F)F)C(N)=CC=C1OC1=CC=C(C=2C=CC(OC=3C=C(C(N)=CC=3)C(F)(F)F)=CC=2)C=C1 DPCDFSDBIWVMJC-UHFFFAOYSA-N 0.000 description 1
- LACZRKUWKHQVKS-UHFFFAOYSA-N 4-[4-[4-amino-2-(trifluoromethyl)phenoxy]phenoxy]-3-(trifluoromethyl)aniline Chemical compound FC(F)(F)C1=CC(N)=CC=C1OC(C=C1)=CC=C1OC1=CC=C(N)C=C1C(F)(F)F LACZRKUWKHQVKS-UHFFFAOYSA-N 0.000 description 1
- JHIUAEPQGMOWHS-UHFFFAOYSA-N 4-[4-hydroxy-3,5-bis(methoxymethyl)phenyl]-2,6-bis(methoxymethyl)phenol Chemical compound COCC1=C(O)C(COC)=CC(C=2C=C(COC)C(O)=C(COC)C=2)=C1 JHIUAEPQGMOWHS-UHFFFAOYSA-N 0.000 description 1
- FPAKXKXRPMFMLR-UHFFFAOYSA-N 4-[5-(4-aminophenyl)-1,1,2,2,3,3,4,4,5,5-decafluoropentyl]aniline Chemical compound C1=CC(N)=CC=C1C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C1=CC=C(N)C=C1 FPAKXKXRPMFMLR-UHFFFAOYSA-N 0.000 description 1
- BOTFKTVSBXEXKF-UHFFFAOYSA-N 4-[7-(4-aminophenyl)-1,1,2,2,3,3,4,4,5,5,6,6,7,7-tetradecafluoroheptyl]aniline Chemical compound C1=CC(N)=CC=C1C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C1=CC=C(N)C=C1 BOTFKTVSBXEXKF-UHFFFAOYSA-N 0.000 description 1
- XBUUUMYXGIXUKW-UHFFFAOYSA-N 4-[[2-hydroxy-4,6-dimethyl-3-[(2,3,4-trihydroxyphenyl)methyl]phenyl]methyl]benzene-1,2,3-triol Chemical compound OC1=C(CC=2C(=C(O)C(O)=CC=2)O)C(C)=CC(C)=C1CC1=CC=C(O)C(O)=C1O XBUUUMYXGIXUKW-UHFFFAOYSA-N 0.000 description 1
- YGIJMKOMRCLGQA-UHFFFAOYSA-N 4-[[2-hydroxy-5-[[4-hydroxy-3,5-bis[(4-hydroxy-3,5-dimethylphenyl)methyl]phenyl]methyl]-3-[(4-hydroxy-3,5-dimethylphenyl)methyl]phenyl]methyl]-2,6-dimethylphenol Chemical compound CC1=C(O)C(C)=CC(CC=2C(=C(CC=3C=C(C)C(O)=C(C)C=3)C=C(CC=3C=C(CC=4C=C(C)C(O)=C(C)C=4)C(O)=C(CC=4C=C(C)C(O)=C(C)C=4)C=3)C=2)O)=C1 YGIJMKOMRCLGQA-UHFFFAOYSA-N 0.000 description 1
- BCKCGQKOVPLAAQ-UHFFFAOYSA-N 4-[[2-hydroxy-5-[[4-hydroxy-3-[(4-hydroxy-3,5-dimethylphenyl)methyl]-5-methylphenyl]methyl]-3-methylphenyl]methyl]-2,6-dimethylphenol Chemical compound CC1=C(O)C(C)=CC(CC=2C(=C(C)C=C(CC=3C=C(CC=4C=C(C)C(O)=C(C)C=4)C(O)=C(C)C=3)C=2)O)=C1 BCKCGQKOVPLAAQ-UHFFFAOYSA-N 0.000 description 1
- RMKPBDKDQDKFBH-UHFFFAOYSA-N 4-[[3,5-bis[(4-hydroxy-3,5-dimethylphenyl)sulfanylmethyl]-2,4,6-trimethylphenyl]methylsulfanyl]-2,6-dimethylphenol Chemical group CC1=C(O)C(C)=CC(SCC=2C(=C(CSC=3C=C(C)C(O)=C(C)C=3)C(C)=C(CSC=3C=C(C)C(O)=C(C)C=3)C=2C)C)=C1 RMKPBDKDQDKFBH-UHFFFAOYSA-N 0.000 description 1
- XUWFAPDKIXEUPR-UHFFFAOYSA-N 4-[[3-[[4-(diethylamino)phenyl]methylidene]cyclopentylidene]methyl]-n,n-diethylaniline Chemical compound C1=CC(N(CC)CC)=CC=C1C=C(CC1)CC1=CC1=CC=C(N(CC)CC)C=C1 XUWFAPDKIXEUPR-UHFFFAOYSA-N 0.000 description 1
- MCFMSTSGOOISJB-UHFFFAOYSA-N 4-[[4-hydroxy-2,5-dimethyl-3-[(2,3,4-trihydroxyphenyl)methyl]phenyl]methyl]benzene-1,2,3-triol Chemical compound CC=1C(CC=2C(=C(O)C(O)=CC=2)O)=C(O)C(C)=CC=1CC1=CC=C(O)C(O)=C1O MCFMSTSGOOISJB-UHFFFAOYSA-N 0.000 description 1
- OEHFFNCUDLHVHJ-UHFFFAOYSA-N 4-[[4-hydroxy-3,5-bis[(4-hydroxy-3-methylphenyl)methyl]phenyl]methyl]-2,6-bis[(4-hydroxy-3-methylphenyl)methyl]phenol Chemical compound C1=C(O)C(C)=CC(CC=2C(=C(CC=3C=C(C)C(O)=CC=3)C=C(CC=3C=C(CC=4C=C(C)C(O)=CC=4)C(O)=C(CC=4C=C(C)C(O)=CC=4)C=3)C=2)O)=C1 OEHFFNCUDLHVHJ-UHFFFAOYSA-N 0.000 description 1
- UGBCWMUGTYLMHK-UHFFFAOYSA-N 4-[[4-hydroxy-3,5-bis[(4-hydroxyphenyl)methyl]phenyl]methyl]-2,6-bis[(4-hydroxyphenyl)methyl]phenol Chemical compound C1=CC(O)=CC=C1CC1=CC(CC=2C=C(CC=3C=CC(O)=CC=3)C(O)=C(CC=3C=CC(O)=CC=3)C=2)=CC(CC=2C=CC(O)=CC=2)=C1O UGBCWMUGTYLMHK-UHFFFAOYSA-N 0.000 description 1
- UWFUILMHBCVIMK-UHFFFAOYSA-N 4-[bis(4-hydroxy-3,5-dimethylphenyl)methyl]benzene-1,2-diol Chemical compound CC1=C(O)C(C)=CC(C(C=2C=C(O)C(O)=CC=2)C=2C=C(C)C(O)=C(C)C=2)=C1 UWFUILMHBCVIMK-UHFFFAOYSA-N 0.000 description 1
- WFCQTAXSWSWIHS-UHFFFAOYSA-N 4-[bis(4-hydroxyphenyl)methyl]phenol Chemical compound C1=CC(O)=CC=C1C(C=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 WFCQTAXSWSWIHS-UHFFFAOYSA-N 0.000 description 1
- ACQVEWFMUBXEMR-UHFFFAOYSA-N 4-bromo-2-fluoro-6-nitrophenol Chemical compound OC1=C(F)C=C(Br)C=C1[N+]([O-])=O ACQVEWFMUBXEMR-UHFFFAOYSA-N 0.000 description 1
- QNPAQJRSQPGMSC-UHFFFAOYSA-N 4-cyclohexyl-2,6-bis[(4-hydroxyphenyl)methyl]phenol Chemical compound C1=CC(O)=CC=C1CC1=CC(C2CCCCC2)=CC(CC=2C=CC(O)=CC=2)=C1O QNPAQJRSQPGMSC-UHFFFAOYSA-N 0.000 description 1
- UZFMOKQJFYMBGY-UHFFFAOYSA-N 4-hydroxy-TEMPO Chemical compound CC1(C)CC(O)CC(C)(C)N1[O] UZFMOKQJFYMBGY-UHFFFAOYSA-N 0.000 description 1
- PRKPGWQEKNEVEU-UHFFFAOYSA-N 4-methyl-n-(3-triethoxysilylpropyl)pentan-2-imine Chemical compound CCO[Si](OCC)(OCC)CCCN=C(C)CC(C)C PRKPGWQEKNEVEU-UHFFFAOYSA-N 0.000 description 1
- XRBNDLYHPCVYGC-UHFFFAOYSA-N 4-phenylbenzene-1,2,3-triol Chemical compound OC1=C(O)C(O)=CC=C1C1=CC=CC=C1 XRBNDLYHPCVYGC-UHFFFAOYSA-N 0.000 description 1
- XESZUVZBAMCAEJ-UHFFFAOYSA-N 4-tert-butylcatechol Chemical compound CC(C)(C)C1=CC=C(O)C(O)=C1 XESZUVZBAMCAEJ-UHFFFAOYSA-N 0.000 description 1
- NSPMIYGKQJPBQR-UHFFFAOYSA-N 4H-1,2,4-triazole Chemical compound C=1N=CNN=1 NSPMIYGKQJPBQR-UHFFFAOYSA-N 0.000 description 1
- SGNZYJXNUURYCH-UHFFFAOYSA-N 5,6-dihydroxyindole Chemical compound C1=C(O)C(O)=CC2=C1NC=C2 SGNZYJXNUURYCH-UHFFFAOYSA-N 0.000 description 1
- VQVIHDPBMFABCQ-UHFFFAOYSA-N 5-(1,3-dioxo-2-benzofuran-5-carbonyl)-2-benzofuran-1,3-dione Chemical compound C1=C2C(=O)OC(=O)C2=CC(C(C=2C=C3C(=O)OC(=O)C3=CC=2)=O)=C1 VQVIHDPBMFABCQ-UHFFFAOYSA-N 0.000 description 1
- VZLUGGCFYPMLMI-UHFFFAOYSA-N 5-(3,5-dihydroxyphenyl)benzene-1,3-diol Chemical compound OC1=CC(O)=CC(C=2C=C(O)C=C(O)C=2)=C1 VZLUGGCFYPMLMI-UHFFFAOYSA-N 0.000 description 1
- UWSMKYBKUPAEJQ-UHFFFAOYSA-N 5-Chloro-2-(3,5-di-tert-butyl-2-hydroxyphenyl)-2H-benzotriazole Chemical compound CC(C)(C)C1=CC(C(C)(C)C)=CC(N2N=C3C=C(Cl)C=CC3=N2)=C1O UWSMKYBKUPAEJQ-UHFFFAOYSA-N 0.000 description 1
- ZHBXLZQQVCDGPA-UHFFFAOYSA-N 5-[(1,3-dioxo-2-benzofuran-5-yl)sulfonyl]-2-benzofuran-1,3-dione Chemical compound C1=C2C(=O)OC(=O)C2=CC(S(=O)(=O)C=2C=C3C(=O)OC(C3=CC=2)=O)=C1 ZHBXLZQQVCDGPA-UHFFFAOYSA-N 0.000 description 1
- LRSJXEJKRGBVHJ-UHFFFAOYSA-N 5-[4-(5-cyano-2,3,4-trihydroxybenzoyl)benzoyl]-2,3,4-trihydroxybenzonitrile Chemical compound OC1=C(O)C(C#N)=CC(C(=O)C=2C=CC(=CC=2)C(=O)C=2C(=C(O)C(O)=C(C#N)C=2)O)=C1O LRSJXEJKRGBVHJ-UHFFFAOYSA-N 0.000 description 1
- DRXGKQPTFWTTJW-UHFFFAOYSA-N 5-butoxy-2-[4-(4-butoxy-2-hydroxyphenyl)-6-(2,4-dibutoxyphenyl)-1,3,5-triazin-2-yl]phenol Chemical compound OC1=CC(OCCCC)=CC=C1C1=NC(C=2C(=CC(OCCCC)=CC=2)O)=NC(C=2C(=CC(OCCCC)=CC=2)OCCCC)=N1 DRXGKQPTFWTTJW-UHFFFAOYSA-N 0.000 description 1
- RZWRYPGAUIOOMK-UHFFFAOYSA-N 5-nitroso-8-quinolinol Chemical compound C1=CN=C2C(O)=CC=C(N=O)C2=C1 RZWRYPGAUIOOMK-UHFFFAOYSA-N 0.000 description 1
- IVQGJZCJWJVSJX-UHFFFAOYSA-N 5-phenylbenzene-1,2,3-triol Chemical compound OC1=C(O)C(O)=CC(C=2C=CC=CC=2)=C1 IVQGJZCJWJVSJX-UHFFFAOYSA-N 0.000 description 1
- SAPGBCWOQLHKKZ-UHFFFAOYSA-N 6-(2-methylprop-2-enoyloxy)hexyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCCCCCCOC(=O)C(C)=C SAPGBCWOQLHKKZ-UHFFFAOYSA-N 0.000 description 1
- NZUWGSJOJGLSCV-UHFFFAOYSA-N 6-[4-(2-hydroxy-3-methyl-4-propoxyphenyl)-6-(4-methylphenyl)-1,3,5-triazin-2-yl]-2-methyl-3-propoxyphenol Chemical compound CCCOc1ccc(c(O)c1C)-c1nc(nc(n1)-c1ccc(OCCC)c(C)c1O)-c1ccc(C)cc1 NZUWGSJOJGLSCV-UHFFFAOYSA-N 0.000 description 1
- SANIRTQDABNCHF-UHFFFAOYSA-N 7-(diethylamino)-3-[7-(diethylamino)-2-oxochromene-3-carbonyl]chromen-2-one Chemical compound C1=C(N(CC)CC)C=C2OC(=O)C(C(=O)C3=CC4=CC=C(C=C4OC3=O)N(CC)CC)=CC2=C1 SANIRTQDABNCHF-UHFFFAOYSA-N 0.000 description 1
- KNDQHSIWLOJIGP-UHFFFAOYSA-N 826-62-0 Chemical compound C1C2C3C(=O)OC(=O)C3C1C=C2 KNDQHSIWLOJIGP-UHFFFAOYSA-N 0.000 description 1
- GJCOSYZMQJWQCA-UHFFFAOYSA-N 9H-xanthene Chemical class C1=CC=C2CC3=CC=CC=C3OC2=C1 GJCOSYZMQJWQCA-UHFFFAOYSA-N 0.000 description 1
- SNCJAJRILVFXAE-UHFFFAOYSA-N 9h-fluorene-2,7-diamine Chemical compound NC1=CC=C2C3=CC=C(N)C=C3CC2=C1 SNCJAJRILVFXAE-UHFFFAOYSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 235000021357 Behenic acid Nutrition 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- KYNSBQPICQTCGU-UHFFFAOYSA-N Benzopyrane Chemical class C1=CC=C2C=CCOC2=C1 KYNSBQPICQTCGU-UHFFFAOYSA-N 0.000 description 1
- JMGZEFIQIZZSBH-UHFFFAOYSA-N Bioquercetin Natural products CC1OC(OCC(O)C2OC(OC3=C(Oc4cc(O)cc(O)c4C3=O)c5ccc(O)c(O)c5)C(O)C2O)C(O)C(O)C1O JMGZEFIQIZZSBH-UHFFFAOYSA-N 0.000 description 1
- FIPWRIJSWJWJAI-UHFFFAOYSA-N Butyl carbitol 6-propylpiperonyl ether Chemical compound C1=C(CCC)C(COCCOCCOCCCC)=CC2=C1OCO2 FIPWRIJSWJWJAI-UHFFFAOYSA-N 0.000 description 1
- XMTBDAXHYDJVCV-UHFFFAOYSA-N C(C(=O)C)(=O)O.C(C)(=O)OCC Chemical compound C(C(=O)C)(=O)O.C(C)(=O)OCC XMTBDAXHYDJVCV-UHFFFAOYSA-N 0.000 description 1
- ZGSCVOAWVNOCFV-UHFFFAOYSA-N C(C)(=O)OCC.C(C)OC(C(=O)O)(C)C Chemical compound C(C)(=O)OCC.C(C)OC(C(=O)O)(C)C ZGSCVOAWVNOCFV-UHFFFAOYSA-N 0.000 description 1
- VXOUILHMFVRYJI-UHFFFAOYSA-N CCCCOC(OCCCC)(OCCCC)CC(OCCCC)C(OCCCC)(OCCCC)NC1=NC(N)=NC(N)=N1 Chemical compound CCCCOC(OCCCC)(OCCCC)CC(OCCCC)C(OCCCC)(OCCCC)NC1=NC(N)=NC(N)=N1 VXOUILHMFVRYJI-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical class NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 239000004971 Cross linker Substances 0.000 description 1
- JPVYNHNXODAKFH-UHFFFAOYSA-N Cu2+ Chemical compound [Cu+2] JPVYNHNXODAKFH-UHFFFAOYSA-N 0.000 description 1
- FCKYPQBAHLOOJQ-UHFFFAOYSA-N Cyclohexane-1,2-diaminetetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)C1CCCCC1N(CC(O)=O)CC(O)=O FCKYPQBAHLOOJQ-UHFFFAOYSA-N 0.000 description 1
- 239000009261 D 400 Substances 0.000 description 1
- 239000004641 Diallyl-phthalate Substances 0.000 description 1
- ORAWFNKFUWGRJG-UHFFFAOYSA-N Docosanamide Chemical compound CCCCCCCCCCCCCCCCCCCCCC(N)=O ORAWFNKFUWGRJG-UHFFFAOYSA-N 0.000 description 1
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Natural products CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- GKQLYSROISKDLL-UHFFFAOYSA-N EEDQ Chemical compound C1=CC=C2N(C(=O)OCC)C(OCC)C=CC2=C1 GKQLYSROISKDLL-UHFFFAOYSA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- FPVVYTCTZKCSOJ-UHFFFAOYSA-N Ethylene glycol distearate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCCOC(=O)CCCCCCCCCCCCCCCCC FPVVYTCTZKCSOJ-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 102100022653 Histone H1.5 Human genes 0.000 description 1
- 101000899879 Homo sapiens Histone H1.5 Proteins 0.000 description 1
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 235000000177 Indigofera tinctoria Nutrition 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 1
- YXOLAZRVSSWPPT-UHFFFAOYSA-N Morin Chemical compound OC1=CC(O)=CC=C1C1=C(O)C(=O)C2=C(O)C=C(O)C=C2O1 YXOLAZRVSSWPPT-UHFFFAOYSA-N 0.000 description 1
- ZSXGLVDWWRXATF-UHFFFAOYSA-N N,N-dimethylformamide dimethyl acetal Chemical compound COC(OC)N(C)C ZSXGLVDWWRXATF-UHFFFAOYSA-N 0.000 description 1
- UBUCNCOMADRQHX-UHFFFAOYSA-N N-Nitrosodiphenylamine Chemical compound C=1C=CC=CC=1N(N=O)C1=CC=CC=C1 UBUCNCOMADRQHX-UHFFFAOYSA-N 0.000 description 1
- XQVWYOYUZDUNRW-UHFFFAOYSA-N N-Phenyl-1-naphthylamine Chemical compound C=1C=CC2=CC=CC=C2C=1NC1=CC=CC=C1 XQVWYOYUZDUNRW-UHFFFAOYSA-N 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- UTGIVGVJCOMGIV-UHFFFAOYSA-N NC(=O)N.C(CC)OC(=CC)OCCC Chemical compound NC(=O)N.C(CC)OC(=CC)OCCC UTGIVGVJCOMGIV-UHFFFAOYSA-N 0.000 description 1
- FELLDHNJLLNHEW-UHFFFAOYSA-N NC(=O)N.C(CC)OC(OCCC)C=C Chemical compound NC(=O)N.C(CC)OC(OCCC)C=C FELLDHNJLLNHEW-UHFFFAOYSA-N 0.000 description 1
- JEMJMXUPHOOGPY-UHFFFAOYSA-N NC(=O)N.C(CCC)OCC=C Chemical compound NC(=O)N.C(CCC)OCC=C JEMJMXUPHOOGPY-UHFFFAOYSA-N 0.000 description 1
- 229930192627 Naphthoquinone Natural products 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- HZYYJGBYLVDXGY-UHFFFAOYSA-N OC(COC1=CC(=C(C=C1)N1NC(=C(C(=N1)C1=C(C=C(C=C1)C)C)C1=C(C=CC=C1)O)C1=C(C=C(C=C1)C)C)O)COCCCCCCCCCCCCC Chemical class OC(COC1=CC(=C(C=C1)N1NC(=C(C(=N1)C1=C(C=C(C=C1)C)C)C1=C(C=CC=C1)O)C1=C(C=C(C=C1)C)C)O)COCCCCCCCCCCCCC HZYYJGBYLVDXGY-UHFFFAOYSA-N 0.000 description 1
- MAOLXURNLQQKOZ-UHFFFAOYSA-N OC1=C(C=CC(=C1)OCCC)C1=NC(=NC(=N1)C1=C(C=C(C=C1)OCCC)O)C1=C(C=C(C=C1)C)C.OC1=C(C=CC(=C1)O)C1=NC(=NC(=N1)C1=C(C=C(C=C1)C)C)C1=C(C=C(C=C1)C)C Chemical compound OC1=C(C=CC(=C1)OCCC)C1=NC(=NC(=N1)C1=C(C=C(C=C1)OCCC)O)C1=C(C=C(C=C1)C)C.OC1=C(C=CC(=C1)O)C1=NC(=NC(=N1)C1=C(C=C(C=C1)C)C)C1=C(C=C(C=C1)C)C MAOLXURNLQQKOZ-UHFFFAOYSA-N 0.000 description 1
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 1
- 239000004693 Polybenzimidazole Substances 0.000 description 1
- 229920000604 Polyethylene Glycol 200 Polymers 0.000 description 1
- 239000004721 Polyphenylene oxide Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- ZTHYODDOHIVTJV-UHFFFAOYSA-N Propyl gallate Chemical compound CCCOC(=O)C1=CC(O)=C(O)C(O)=C1 ZTHYODDOHIVTJV-UHFFFAOYSA-N 0.000 description 1
- 239000005700 Putrescine Substances 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- ZVOLCUVKHLEPEV-UHFFFAOYSA-N Quercetagetin Natural products C1=C(O)C(O)=CC=C1C1=C(O)C(=O)C2=C(O)C(O)=C(O)C=C2O1 ZVOLCUVKHLEPEV-UHFFFAOYSA-N 0.000 description 1
- HWTZYBCRDDUBJY-UHFFFAOYSA-N Rhynchosin Natural products C1=C(O)C(O)=CC=C1C1=C(O)C(=O)C2=CC(O)=C(O)C=C2O1 HWTZYBCRDDUBJY-UHFFFAOYSA-N 0.000 description 1
- 229910052581 Si3N4 Inorganic materials 0.000 description 1
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 1
- 229930183415 Suberin Natural products 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- QYTDEUPAUMOIOP-UHFFFAOYSA-N TEMPO Chemical compound CC1(C)CCCC(C)(C)N1[O] QYTDEUPAUMOIOP-UHFFFAOYSA-N 0.000 description 1
- UWHCKJMYHZGTIT-UHFFFAOYSA-N Tetraethylene glycol, Natural products OCCOCCOCCOCCO UWHCKJMYHZGTIT-UHFFFAOYSA-N 0.000 description 1
- 229920002359 Tetronic® Polymers 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical group C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical group C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- JADFUOUIMWDTFX-UHFFFAOYSA-N Triacontanedioic acid Chemical compound OC(=O)CCCCCCCCCCCCCCCCCCCCCCCCCCCCC(O)=O JADFUOUIMWDTFX-UHFFFAOYSA-N 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- QYKIQEUNHZKYBP-UHFFFAOYSA-N Vinyl ether Chemical compound C=COC=C QYKIQEUNHZKYBP-UHFFFAOYSA-N 0.000 description 1
- DPQRMIPRAHPPNE-UHFFFAOYSA-N [(1-oxo-1-phenylpropan-2-ylidene)amino] acetate Chemical compound CC(=O)ON=C(C)C(=O)C1=CC=CC=C1 DPQRMIPRAHPPNE-UHFFFAOYSA-N 0.000 description 1
- ZNZDJSGUWLGTLA-UHFFFAOYSA-N [(1-oxo-1-phenylpropan-2-ylidene)amino] benzoate Chemical compound C=1C=CC=CC=1C(=O)C(C)=NOC(=O)C1=CC=CC=C1 ZNZDJSGUWLGTLA-UHFFFAOYSA-N 0.000 description 1
- FLRQOWAOMJMSTP-JJTRIOAGSA-N [(2s)-2-[(2r)-3,4-dihydroxy-5-oxo-2h-furan-2-yl]-2-hydroxyethyl] (6z,9z,12z)-octadeca-6,9,12-trienoate Chemical compound CCCCC\C=C/C\C=C/C\C=C/CCCCC(=O)OC[C@H](O)[C@H]1OC(=O)C(O)=C1O FLRQOWAOMJMSTP-JJTRIOAGSA-N 0.000 description 1
- KIXNLSPBNAIKJU-UHFFFAOYSA-N [2,3-bis(2,3,4-trihydroxybenzoyl)phenyl]-(2,3,4-trihydroxyphenyl)methanone Chemical compound OC1=C(O)C(O)=CC=C1C(=O)C1=CC=CC(C(=O)C=2C(=C(O)C(O)=CC=2)O)=C1C(=O)C1=CC=C(O)C(O)=C1O KIXNLSPBNAIKJU-UHFFFAOYSA-N 0.000 description 1
- PICGUDVETMNGMN-UHFFFAOYSA-N [2,3-bis(hydroxymethyl)phenyl]-phenylmethanone Chemical compound OCc1cccc(C(=O)c2ccccc2)c1CO PICGUDVETMNGMN-UHFFFAOYSA-N 0.000 description 1
- WIMRPMLPJZLCAM-UHFFFAOYSA-N [2,3-bis(methoxymethyl)phenyl]-phenylmethanone Chemical compound COCc1cccc(C(=O)c2ccccc2)c1COC WIMRPMLPJZLCAM-UHFFFAOYSA-N 0.000 description 1
- QZKPXGFHHIFFTQ-UHFFFAOYSA-N [2-(2,3,4-trihydroxybenzoyl)phenyl]-(2,3,4-trihydroxyphenyl)methanone Chemical compound OC1=C(O)C(O)=CC=C1C(=O)C1=CC=CC=C1C(=O)C1=CC=C(O)C(O)=C1O QZKPXGFHHIFFTQ-UHFFFAOYSA-N 0.000 description 1
- RYSWVYFTSSTNMY-UHFFFAOYSA-N [2-(hydroxymethyl)-3,4-dimethoxyphenyl]methanol Chemical compound COC1=CC=C(CO)C(CO)=C1OC RYSWVYFTSSTNMY-UHFFFAOYSA-N 0.000 description 1
- UAGDZLRBMHVJNS-UHFFFAOYSA-N [2-(hydroxymethyl)phenyl] 2-(hydroxymethyl)benzoate Chemical compound OCc1ccccc1OC(=O)c1ccccc1CO UAGDZLRBMHVJNS-UHFFFAOYSA-N 0.000 description 1
- XMUZQOKACOLCSS-UHFFFAOYSA-N [2-(hydroxymethyl)phenyl]methanol Chemical compound OCC1=CC=CC=C1CO XMUZQOKACOLCSS-UHFFFAOYSA-N 0.000 description 1
- ZCWHTBJAXKQNAO-UHFFFAOYSA-N [2-(methoxymethyl)phenyl] 2-(methoxymethyl)benzoate Chemical compound COCc1ccccc1OC(=O)c1ccccc1COC ZCWHTBJAXKQNAO-UHFFFAOYSA-N 0.000 description 1
- YPCHGLDQZXOZFW-UHFFFAOYSA-N [2-[[4-methyl-3-[[3-prop-2-enoyloxy-2,2-bis(prop-2-enoyloxymethyl)propoxy]carbonylamino]phenyl]carbamoyloxymethyl]-3-prop-2-enoyloxy-2-(prop-2-enoyloxymethyl)propyl] prop-2-enoate Chemical compound CC1=CC=C(NC(=O)OCC(COC(=O)C=C)(COC(=O)C=C)COC(=O)C=C)C=C1NC(=O)OCC(COC(=O)C=C)(COC(=O)C=C)COC(=O)C=C YPCHGLDQZXOZFW-UHFFFAOYSA-N 0.000 description 1
- TWLPMXCINXVBJD-UHFFFAOYSA-N [3,4-bis(2,3,4-trihydroxybenzoyl)phenyl]-(2,3,4-trihydroxyphenyl)methanone Chemical compound OC1=C(O)C(O)=CC=C1C(=O)C(C=C1C(=O)C=2C(=C(O)C(O)=CC=2)O)=CC=C1C(=O)C1=CC=C(O)C(O)=C1O TWLPMXCINXVBJD-UHFFFAOYSA-N 0.000 description 1
- RHQMYFWTNGMTQQ-UHFFFAOYSA-N [3,5-bis(2,3,4-trihydroxybenzoyl)phenyl]-(2,3,4-trihydroxyphenyl)methanone Chemical compound OC1=C(O)C(O)=CC=C1C(=O)C1=CC(C(=O)C=2C(=C(O)C(O)=CC=2)O)=CC(C(=O)C=2C(=C(O)C(O)=CC=2)O)=C1 RHQMYFWTNGMTQQ-UHFFFAOYSA-N 0.000 description 1
- KTIARAWRWBOXEK-UHFFFAOYSA-N [3,5-bis(2,5-dihydroxybenzoyl)phenyl]-(2,5-dihydroxyphenyl)methanone Chemical compound OC1=CC=C(O)C(C(=O)C=2C=C(C=C(C=2)C(=O)C=2C(=CC=C(O)C=2)O)C(=O)C=2C(=CC=C(O)C=2)O)=C1 KTIARAWRWBOXEK-UHFFFAOYSA-N 0.000 description 1
- CPDIKOWXKCHINY-UHFFFAOYSA-N [3-(2,3,4-trihydroxybenzoyl)phenyl]-(2,3,4-trihydroxyphenyl)methanone Chemical compound OC1=C(O)C(O)=CC=C1C(=O)C1=CC=CC(C(=O)C=2C(=C(O)C(O)=CC=2)O)=C1 CPDIKOWXKCHINY-UHFFFAOYSA-N 0.000 description 1
- OUVCHDDCLKJYML-UHFFFAOYSA-N [3-(2,4,6-trihydroxybenzoyl)phenyl]-(2,4,6-trihydroxyphenyl)methanone Chemical compound OC1=CC(O)=CC(O)=C1C(=O)C1=CC=CC(C(=O)C=2C(=CC(O)=CC=2O)O)=C1 OUVCHDDCLKJYML-UHFFFAOYSA-N 0.000 description 1
- HIVQCJOGAHNXBO-UHFFFAOYSA-N [3-prop-2-enoyloxy-2-[[3-prop-2-enoyloxy-2,2-bis(prop-2-enoyloxymethyl)propoxy]methyl]-2-(prop-2-enoyloxymethyl)propyl] propanoate Chemical compound CCC(=O)OCC(COC(=O)C=C)(COC(=O)C=C)COCC(COC(=O)C=C)(COC(=O)C=C)COC(=O)C=C HIVQCJOGAHNXBO-UHFFFAOYSA-N 0.000 description 1
- RTTYWAHQPKZDFH-UHFFFAOYSA-N [4-(2,3,4-trihydroxy-5-methoxybenzoyl)phenyl]-(2,3,4-trihydroxy-5-methoxyphenyl)methanone Chemical compound OC1=C(O)C(OC)=CC(C(=O)C=2C=CC(=CC=2)C(=O)C=2C(=C(O)C(O)=C(OC)C=2)O)=C1O RTTYWAHQPKZDFH-UHFFFAOYSA-N 0.000 description 1
- ZBJXEKFLHASCMY-UHFFFAOYSA-N [4-(2,3,4-trihydroxy-5-methylbenzoyl)phenyl]-(2,3,4-trihydroxy-5-methylphenyl)methanone Chemical compound OC1=C(O)C(C)=CC(C(=O)C=2C=CC(=CC=2)C(=O)C=2C(=C(O)C(O)=C(C)C=2)O)=C1O ZBJXEKFLHASCMY-UHFFFAOYSA-N 0.000 description 1
- KWUMPOHPFZRMME-UHFFFAOYSA-N [4-(2,3,4-trihydroxy-5-nitrobenzoyl)phenyl]-(2,3,4-trihydroxy-5-nitrophenyl)methanone Chemical compound [O-][N+](=O)C1=C(O)C(O)=C(O)C(C(=O)C=2C=CC(=CC=2)C(=O)C=2C(=C(O)C(O)=C(C=2)[N+]([O-])=O)O)=C1 KWUMPOHPFZRMME-UHFFFAOYSA-N 0.000 description 1
- HPYQBBGJKRJCIY-UHFFFAOYSA-N [4-(2,3,4-trihydroxybenzoyl)phenyl]-(2,3,4-trihydroxyphenyl)methanone Chemical compound OC1=C(O)C(O)=CC=C1C(=O)C1=CC=C(C(=O)C=2C(=C(O)C(O)=CC=2)O)C=C1 HPYQBBGJKRJCIY-UHFFFAOYSA-N 0.000 description 1
- WPONNJIRMOCZTJ-UHFFFAOYSA-N [4-(2,4,6-trihydroxybenzoyl)phenyl]-(2,4,6-trihydroxyphenyl)methanone Chemical compound OC1=CC(O)=CC(O)=C1C(=O)C1=CC=C(C(=O)C=2C(=CC(O)=CC=2O)O)C=C1 WPONNJIRMOCZTJ-UHFFFAOYSA-N 0.000 description 1
- JFGQZILJTAQODG-UHFFFAOYSA-N [4-(3-bromo-2,5-dihydroxybenzoyl)phenyl]-(3-bromo-2,5-dihydroxyphenyl)methanone Chemical compound OC1=CC(Br)=C(O)C(C(=O)C=2C=CC(=CC=2)C(=O)C=2C(=C(Br)C=C(O)C=2)O)=C1 JFGQZILJTAQODG-UHFFFAOYSA-N 0.000 description 1
- OXIKYYJDTWKERT-UHFFFAOYSA-N [4-(aminomethyl)cyclohexyl]methanamine Chemical compound NCC1CCC(CN)CC1 OXIKYYJDTWKERT-UHFFFAOYSA-N 0.000 description 1
- KGUYZCLKIAMWTP-UHFFFAOYSA-N [4-(dimethylamino)phenyl]-(2-ethenylphenyl)methanone Chemical compound C1=CC(N(C)C)=CC=C1C(=O)C1=CC=CC=C1C=C KGUYZCLKIAMWTP-UHFFFAOYSA-N 0.000 description 1
- CNFQQUXCFJEREO-UHFFFAOYSA-N [4-[4-(hydroxymethyl)-2,3-dimethylphenyl]phenyl]methanol Chemical group CC=1C(=C(C=CC=1CO)C1=CC=C(C=C1)CO)C CNFQQUXCFJEREO-UHFFFAOYSA-N 0.000 description 1
- SFHGONLFTNHXDX-UHFFFAOYSA-N [4-[4-(hydroxymethyl)phenyl]phenyl]methanol Chemical group C1=CC(CO)=CC=C1C1=CC=C(CO)C=C1 SFHGONLFTNHXDX-UHFFFAOYSA-N 0.000 description 1
- VYGUBTIWNBFFMQ-UHFFFAOYSA-N [N+](#[C-])N1C(=O)NC=2NC(=O)NC2C1=O Chemical compound [N+](#[C-])N1C(=O)NC=2NC(=O)NC2C1=O VYGUBTIWNBFFMQ-UHFFFAOYSA-N 0.000 description 1
- XQAXKRLKNVVHSS-UHFFFAOYSA-J [Ti+4].Cc1cc([O-])c(C)c(C)c1C.Cc1cc([O-])c(C)c(C)c1C.Cc1cc([O-])c(C)c(C)c1C.Cc1cc([O-])c(C)c(C)c1C Chemical compound [Ti+4].Cc1cc([O-])c(C)c(C)c1C.Cc1cc([O-])c(C)c(C)c1C.Cc1cc([O-])c(C)c(C)c1C.Cc1cc([O-])c(C)c(C)c1C XQAXKRLKNVVHSS-UHFFFAOYSA-J 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 150000003869 acetamides Chemical class 0.000 description 1
- OHBRHBQMHLEELN-UHFFFAOYSA-N acetic acid;1-butoxybutane Chemical compound CC(O)=O.CCCCOCCCC OHBRHBQMHLEELN-UHFFFAOYSA-N 0.000 description 1
- NJYZCEFQAIUHSD-UHFFFAOYSA-N acetoguanamine Chemical compound CC1=NC(N)=NC(N)=N1 NJYZCEFQAIUHSD-UHFFFAOYSA-N 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- HFBMWMNUJJDEQZ-UHFFFAOYSA-N acryloyl chloride Chemical compound ClC(=O)C=C HFBMWMNUJJDEQZ-UHFFFAOYSA-N 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- MIBQYWIOHFTKHD-UHFFFAOYSA-N adamantane-1-carbonyl chloride Chemical compound C1C(C2)CC3CC2CC1(C(=O)Cl)C3 MIBQYWIOHFTKHD-UHFFFAOYSA-N 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 238000007754 air knife coating Methods 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 125000005103 alkyl silyl group Chemical group 0.000 description 1
- 229910045601 alloy Inorganic materials 0.000 description 1
- 239000000956 alloy Substances 0.000 description 1
- XPNGNIFUDRPBFJ-UHFFFAOYSA-N alpha-methylbenzylalcohol Natural products CC1=CC=CC=C1CO XPNGNIFUDRPBFJ-UHFFFAOYSA-N 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- MKSISPKJEMTIGI-LWTKGLMZSA-K aluminum (Z)-oxido-oxidoimino-phenylazanium Chemical compound [Al+3].[O-]\N=[N+](/[O-])c1ccccc1.[O-]\N=[N+](/[O-])c1ccccc1.[O-]\N=[N+](/[O-])c1ccccc1 MKSISPKJEMTIGI-LWTKGLMZSA-K 0.000 description 1
- MQPPCKJJFDNPHJ-UHFFFAOYSA-K aluminum;3-oxohexanoate Chemical compound [Al+3].CCCC(=O)CC([O-])=O.CCCC(=O)CC([O-])=O.CCCC(=O)CC([O-])=O MQPPCKJJFDNPHJ-UHFFFAOYSA-K 0.000 description 1
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 229910021417 amorphous silicon Inorganic materials 0.000 description 1
- 239000003957 anion exchange resin Substances 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 150000001454 anthracenes Chemical class 0.000 description 1
- 150000004056 anthraquinones Chemical class 0.000 description 1
- 125000005013 aryl ether group Chemical group 0.000 description 1
- PAQKRBLMHQTWAS-UHFFFAOYSA-N azane;n-hydroxy-n-naphthalen-1-ylnitrous amide Chemical compound N.C1=CC=C2C(N(N=O)O)=CC=CC2=C1 PAQKRBLMHQTWAS-UHFFFAOYSA-N 0.000 description 1
- NJVHCUNZAMFQNA-UHFFFAOYSA-N azane;n-hydroxy-n-phenylnitrous amide Chemical compound N.O=NN(O)C1=CC=CC=C1 NJVHCUNZAMFQNA-UHFFFAOYSA-N 0.000 description 1
- 125000000751 azo group Chemical group [*]N=N[*] 0.000 description 1
- DMLAVOWQYNRWNQ-UHFFFAOYSA-N azobenzene Chemical class C1=CC=CC=C1N=NC1=CC=CC=C1 DMLAVOWQYNRWNQ-UHFFFAOYSA-N 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 229940116226 behenic acid Drugs 0.000 description 1
- ZVSKZLHKADLHSD-UHFFFAOYSA-N benzanilide Chemical compound C=1C=CC=CC=1C(=O)NC1=CC=CC=C1 ZVSKZLHKADLHSD-UHFFFAOYSA-N 0.000 description 1
- GCAIEATUVJFSMC-UHFFFAOYSA-N benzene-1,2,3,4-tetracarboxylic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C(C(O)=O)=C1C(O)=O GCAIEATUVJFSMC-UHFFFAOYSA-N 0.000 description 1
- JGFLAAWSLCPCDY-UHFFFAOYSA-N benzene;cyclopenta-1,3-diene;iron Chemical class [Fe].C1C=CC=C1.C1=CC=CC=C1 JGFLAAWSLCPCDY-UHFFFAOYSA-N 0.000 description 1
- KXNQKOAQSGJCQU-UHFFFAOYSA-N benzo[e][1,3]benzothiazole Chemical compound C1=CC=C2C(N=CS3)=C3C=CC2=C1 KXNQKOAQSGJCQU-UHFFFAOYSA-N 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 1
- NEQRECSQVSRXLZ-UHFFFAOYSA-N benzyl 7-(dimethylamino)-2-oxochromene-3-carboxylate Chemical compound O=C1OC2=CC(N(C)C)=CC=C2C=C1C(=O)OCC1=CC=CC=C1 NEQRECSQVSRXLZ-UHFFFAOYSA-N 0.000 description 1
- JERAEGPJHSIFOW-UHFFFAOYSA-N benzyl(nitro)carbamic acid Chemical class OC(=O)N([N+]([O-])=O)CC1=CC=CC=C1 JERAEGPJHSIFOW-UHFFFAOYSA-N 0.000 description 1
- FKPSBYZGRQJIMO-UHFFFAOYSA-M benzyl(triethyl)azanium;hydroxide Chemical compound [OH-].CC[N+](CC)(CC)CC1=CC=CC=C1 FKPSBYZGRQJIMO-UHFFFAOYSA-M 0.000 description 1
- RRIWSQXXBIFKQM-UHFFFAOYSA-N benzylcarbamic acid Chemical class OC(=O)NCC1=CC=CC=C1 RRIWSQXXBIFKQM-UHFFFAOYSA-N 0.000 description 1
- 125000000649 benzylidene group Chemical group [H]C(=[*])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- NDKBVBUGCNGSJJ-UHFFFAOYSA-M benzyltrimethylammonium hydroxide Chemical compound [OH-].C[N+](C)(C)CC1=CC=CC=C1 NDKBVBUGCNGSJJ-UHFFFAOYSA-M 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 125000006267 biphenyl group Chemical group 0.000 description 1
- ZEFSGHVBJCEKAZ-UHFFFAOYSA-N bis(2,4-ditert-butyl-6-methylphenyl) ethyl phosphite Chemical compound CC=1C=C(C(C)(C)C)C=C(C(C)(C)C)C=1OP(OCC)OC1=C(C)C=C(C(C)(C)C)C=C1C(C)(C)C ZEFSGHVBJCEKAZ-UHFFFAOYSA-N 0.000 description 1
- PFYXSUNOLOJMDX-UHFFFAOYSA-N bis(2,5-dioxopyrrolidin-1-yl) carbonate Chemical compound O=C1CCC(=O)N1OC(=O)ON1C(=O)CCC1=O PFYXSUNOLOJMDX-UHFFFAOYSA-N 0.000 description 1
- SODJJEXAWOSSON-UHFFFAOYSA-N bis(2-hydroxy-4-methoxyphenyl)methanone Chemical compound OC1=CC(OC)=CC=C1C(=O)C1=CC=C(OC)C=C1O SODJJEXAWOSSON-UHFFFAOYSA-N 0.000 description 1
- RKTGAWJWCNLSFX-UHFFFAOYSA-M bis(2-hydroxyethyl)-dimethylazanium;hydroxide Chemical compound [OH-].OCC[N+](C)(C)CCO RKTGAWJWCNLSFX-UHFFFAOYSA-M 0.000 description 1
- AEWGGPYHSLODJJ-UHFFFAOYSA-N bis(3-amino-4-hydroxyphenyl)methanone Chemical compound C1=C(O)C(N)=CC(C(=O)C=2C=C(N)C(O)=CC=2)=C1 AEWGGPYHSLODJJ-UHFFFAOYSA-N 0.000 description 1
- TUQQUUXMCKXGDI-UHFFFAOYSA-N bis(3-aminophenyl)methanone Chemical compound NC1=CC=CC(C(=O)C=2C=C(N)C=CC=2)=C1 TUQQUUXMCKXGDI-UHFFFAOYSA-N 0.000 description 1
- QUDWYFHPNIMBFC-UHFFFAOYSA-N bis(prop-2-enyl) benzene-1,2-dicarboxylate Chemical compound C=CCOC(=O)C1=CC=CC=C1C(=O)OCC=C QUDWYFHPNIMBFC-UHFFFAOYSA-N 0.000 description 1
- HEYYMASVKJXRDV-UHFFFAOYSA-N bis[(3-ethyloxetan-3-yl)methyl] benzene-1,4-dicarboxylate Chemical compound C=1C=C(C(=O)OCC2(CC)COC2)C=CC=1C(=O)OCC1(CC)COC1 HEYYMASVKJXRDV-UHFFFAOYSA-N 0.000 description 1
- VYHBFRJRBHMIQZ-UHFFFAOYSA-N bis[4-(diethylamino)phenyl]methanone Chemical compound C1=CC(N(CC)CC)=CC=C1C(=O)C1=CC=C(N(CC)CC)C=C1 VYHBFRJRBHMIQZ-UHFFFAOYSA-N 0.000 description 1
- 150000001639 boron compounds Chemical class 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- OCWYEMOEOGEQAN-UHFFFAOYSA-N bumetrizole Chemical compound CC(C)(C)C1=CC(C)=CC(N2N=C3C=C(Cl)C=CC3=N2)=C1O OCWYEMOEOGEQAN-UHFFFAOYSA-N 0.000 description 1
- MTKOCRSQUPLVTD-UHFFFAOYSA-N butan-1-olate;titanium(2+) Chemical compound CCCCO[Ti]OCCCC MTKOCRSQUPLVTD-UHFFFAOYSA-N 0.000 description 1
- 125000006226 butoxyethyl group Chemical group 0.000 description 1
- IJRVQAXSAHHCNH-UHFFFAOYSA-M butyl(trimethyl)azanium;hydroxide Chemical compound [OH-].CCCC[N+](C)(C)C IJRVQAXSAHHCNH-UHFFFAOYSA-M 0.000 description 1
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 1
- 229930188620 butyrolactone Natural products 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- WUKWITHWXAAZEY-UHFFFAOYSA-L calcium difluoride Chemical compound [F-].[F-].[Ca+2] WUKWITHWXAAZEY-UHFFFAOYSA-L 0.000 description 1
- 229910001634 calcium fluoride Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- DKVNPHBNOWQYFE-UHFFFAOYSA-N carbamodithioic acid Chemical compound NC(S)=S DKVNPHBNOWQYFE-UHFFFAOYSA-N 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- YCIMNLLNPGFGHC-UHFFFAOYSA-N catechol Chemical compound OC1=CC=CC=C1O YCIMNLLNPGFGHC-UHFFFAOYSA-N 0.000 description 1
- 239000003729 cation exchange resin Substances 0.000 description 1
- 229910010293 ceramic material Inorganic materials 0.000 description 1
- ILGSNOOABSFVDL-UHFFFAOYSA-N cerium;n-hydroxy-n-phenylnitrous amide Chemical compound [Ce].O=NN(O)C1=CC=CC=C1 ILGSNOOABSFVDL-UHFFFAOYSA-N 0.000 description 1
- 239000012295 chemical reaction liquid Substances 0.000 description 1
- 238000005660 chlorination reaction Methods 0.000 description 1
- XTHPWXDJESJLNJ-UHFFFAOYSA-N chlorosulfonic acid Substances OS(Cl)(=O)=O XTHPWXDJESJLNJ-UHFFFAOYSA-N 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- 239000013065 commercial product Substances 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 239000013256 coordination polymer Substances 0.000 description 1
- 229910000365 copper sulfate Inorganic materials 0.000 description 1
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 1
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 1
- 238000005260 corrosion Methods 0.000 description 1
- 230000007797 corrosion Effects 0.000 description 1
- 229960000956 coumarin Drugs 0.000 description 1
- 239000007822 coupling agent Substances 0.000 description 1
- LDHQCZJRKDOVOX-NSCUHMNNSA-N crotonic acid Chemical compound C\C=C\C(O)=O LDHQCZJRKDOVOX-NSCUHMNNSA-N 0.000 description 1
- 238000007766 curtain coating Methods 0.000 description 1
- 125000002993 cycloalkylene group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- VKIRRGRTJUUZHS-UHFFFAOYSA-N cyclohexane-1,4-diamine Chemical compound NC1CCC(N)CC1 VKIRRGRTJUUZHS-UHFFFAOYSA-N 0.000 description 1
- RVOJTCZRIKWHDX-UHFFFAOYSA-N cyclohexanecarbonyl chloride Chemical compound ClC(=O)C1CCCCC1 RVOJTCZRIKWHDX-UHFFFAOYSA-N 0.000 description 1
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 1
- OQQXGCLLMDQESN-UHFFFAOYSA-N cyclohexylcarbamic acid Chemical group OC(=O)NC1CCCCC1 OQQXGCLLMDQESN-UHFFFAOYSA-N 0.000 description 1
- 125000004210 cyclohexylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- XCIXKGXIYUWCLL-UHFFFAOYSA-N cyclopentanol Chemical compound OC1CCCC1 XCIXKGXIYUWCLL-UHFFFAOYSA-N 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 229960002887 deanol Drugs 0.000 description 1
- 238000004042 decolorization Methods 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- VILAVOFMIJHSJA-UHFFFAOYSA-N dicarbon monoxide Chemical compound [C]=C=O VILAVOFMIJHSJA-UHFFFAOYSA-N 0.000 description 1
- 239000003989 dielectric material Substances 0.000 description 1
- OTARVPUIYXHRRB-UHFFFAOYSA-N diethoxy-methyl-[3-(oxiran-2-ylmethoxy)propyl]silane Chemical compound CCO[Si](C)(OCC)CCCOCC1CO1 OTARVPUIYXHRRB-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 229940019778 diethylene glycol diethyl ether Drugs 0.000 description 1
- PHDPNHJFOMABOA-UHFFFAOYSA-N difucol Chemical compound OC1=CC(O)=CC(O)=C1C1=C(O)C=C(O)C=C1O PHDPNHJFOMABOA-UHFFFAOYSA-N 0.000 description 1
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- WHGNXNCOTZPEEK-UHFFFAOYSA-N dimethoxy-methyl-[3-(oxiran-2-ylmethoxy)propyl]silane Chemical compound CO[Si](C)(OC)CCCOCC1CO1 WHGNXNCOTZPEEK-UHFFFAOYSA-N 0.000 description 1
- BEPAFCGSDWSTEL-UHFFFAOYSA-N dimethyl malonate Chemical compound COC(=O)CC(=O)OC BEPAFCGSDWSTEL-UHFFFAOYSA-N 0.000 description 1
- 239000012972 dimethylethanolamine Substances 0.000 description 1
- HTDKEJXHILZNPP-UHFFFAOYSA-N dioctyl hydrogen phosphate Chemical compound CCCCCCCCOP(O)(=O)OCCCCCCCC HTDKEJXHILZNPP-UHFFFAOYSA-N 0.000 description 1
- 238000003618 dip coating Methods 0.000 description 1
- CZZYITDELCSZES-UHFFFAOYSA-N diphenylmethane Chemical compound C=1C=CC=CC=1CC1=CC=CC=C1 CZZYITDELCSZES-UHFFFAOYSA-N 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 208000018459 dissociative disease Diseases 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 239000012990 dithiocarbamate Substances 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- MCPKSFINULVDNX-UHFFFAOYSA-N drometrizole Chemical compound CC1=CC=C(O)C(N2N=C3C=CC=CC3=N2)=C1 MCPKSFINULVDNX-UHFFFAOYSA-N 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- 238000002296 dynamic light scattering Methods 0.000 description 1
- 238000005485 electric heating Methods 0.000 description 1
- 238000007772 electroless plating Methods 0.000 description 1
- IVTMALDHFAHOGL-UHFFFAOYSA-N eriodictyol 7-O-rutinoside Natural products OC1C(O)C(O)C(C)OC1OCC1C(O)C(O)C(O)C(OC=2C=C3C(C(C(O)=C(O3)C=3C=C(O)C(O)=CC=3)=O)=C(O)C=2)O1 IVTMALDHFAHOGL-UHFFFAOYSA-N 0.000 description 1
- HCZKYJDFEPMADG-UHFFFAOYSA-N erythro-nordihydroguaiaretic acid Natural products C=1C=C(O)C(O)=CC=1CC(C)C(C)CC1=CC=C(O)C(O)=C1 HCZKYJDFEPMADG-UHFFFAOYSA-N 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- FWDBOZPQNFPOLF-UHFFFAOYSA-N ethenyl(triethoxy)silane Chemical compound CCO[Si](OCC)(OCC)C=C FWDBOZPQNFPOLF-UHFFFAOYSA-N 0.000 description 1
- NKSJNEHGWDZZQF-UHFFFAOYSA-N ethenyl(trimethoxy)silane Chemical compound CO[Si](OC)(OC)C=C NKSJNEHGWDZZQF-UHFFFAOYSA-N 0.000 description 1
- 125000005678 ethenylene group Chemical group [H]C([*:1])=C([H])[*:2] 0.000 description 1
- ZOOODBUHSVUZEM-UHFFFAOYSA-N ethoxymethanedithioic acid Chemical class CCOC(S)=S ZOOODBUHSVUZEM-UHFFFAOYSA-N 0.000 description 1
- IAJNXBNRYMEYAZ-UHFFFAOYSA-N ethyl 2-cyano-3,3-diphenylprop-2-enoate Chemical compound C=1C=CC=CC=1C(=C(C#N)C(=O)OCC)C1=CC=CC=C1 IAJNXBNRYMEYAZ-UHFFFAOYSA-N 0.000 description 1
- WNIHNYUROPJCLW-UHFFFAOYSA-N ethyl 2-ethoxy-2-methylpropanoate Chemical compound CCOC(=O)C(C)(C)OCC WNIHNYUROPJCLW-UHFFFAOYSA-N 0.000 description 1
- AYCOCJMEYVYNPM-UHFFFAOYSA-N ethyl 7-(dimethylamino)-2-oxochromene-3-carboxylate Chemical compound C1=C(N(C)C)C=C2OC(=O)C(C(=O)OCC)=CC2=C1 AYCOCJMEYVYNPM-UHFFFAOYSA-N 0.000 description 1
- YDMWUMUNUXUYKT-UHFFFAOYSA-N ethyl [(1-oxo-1-phenylpropan-2-ylidene)amino] carbonate Chemical compound CCOC(=O)ON=C(C)C(=O)C1=CC=CC=C1 YDMWUMUNUXUYKT-UHFFFAOYSA-N 0.000 description 1
- UHESRSKEBRADOO-UHFFFAOYSA-N ethyl carbamate;prop-2-enoic acid Chemical class OC(=O)C=C.CCOC(N)=O UHESRSKEBRADOO-UHFFFAOYSA-N 0.000 description 1
- KVFVBPYVNUCWJX-UHFFFAOYSA-M ethyl(trimethyl)azanium;hydroxide Chemical compound [OH-].CC[N+](C)(C)C KVFVBPYVNUCWJX-UHFFFAOYSA-M 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- STVZJERGLQHEKB-UHFFFAOYSA-N ethylene glycol dimethacrylate Chemical compound CC(=C)C(=O)OCCOC(=O)C(C)=C STVZJERGLQHEKB-UHFFFAOYSA-N 0.000 description 1
- 238000007765 extrusion coating Methods 0.000 description 1
- 239000003063 flame retardant Substances 0.000 description 1
- 229930002879 flavonoid pigment Natural products 0.000 description 1
- 150000004638 flavonoid pigments Chemical class 0.000 description 1
- FWQHNLCNFPYBCA-UHFFFAOYSA-N fluoran Chemical compound C12=CC=CC=C2OC2=CC=CC=C2C11OC(=O)C2=CC=CC=C21 FWQHNLCNFPYBCA-UHFFFAOYSA-N 0.000 description 1
- 150000003948 formamides Chemical class 0.000 description 1
- 238000013467 fragmentation Methods 0.000 description 1
- 238000006062 fragmentation reaction Methods 0.000 description 1
- PHLYOKFVXIVOJC-UHFFFAOYSA-N gallein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C(O)=C1OC1=C(O)C(O)=CC=C21 PHLYOKFVXIVOJC-UHFFFAOYSA-N 0.000 description 1
- 230000009477 glass transition Effects 0.000 description 1
- VOZRXNHHFUQHIL-UHFFFAOYSA-N glycidyl methacrylate Chemical compound CC(=C)C(=O)OCC1CO1 VOZRXNHHFUQHIL-UHFFFAOYSA-N 0.000 description 1
- 229940100608 glycol distearate Drugs 0.000 description 1
- 125000003827 glycol group Chemical group 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 238000007756 gravure coating Methods 0.000 description 1
- PKWIYNIDEDLDCJ-UHFFFAOYSA-N guanazole Chemical compound NC1=NNC(N)=N1 PKWIYNIDEDLDCJ-UHFFFAOYSA-N 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 230000026030 halogenation Effects 0.000 description 1
- 238000005658 halogenation reaction Methods 0.000 description 1
- 230000017525 heat dissipation Effects 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000002390 heteroarenes Chemical class 0.000 description 1
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 1
- ACCCMOQWYVYDOT-UHFFFAOYSA-N hexane-1,1-diol Chemical compound CCCCCC(O)O ACCCMOQWYVYDOT-UHFFFAOYSA-N 0.000 description 1
- NAQMVNRVTILPCV-UHFFFAOYSA-N hexane-1,6-diamine Chemical compound NCCCCCCN NAQMVNRVTILPCV-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000009775 high-speed stirring Methods 0.000 description 1
- 239000012456 homogeneous solution Substances 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 125000004464 hydroxyphenyl group Chemical group 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 150000007975 iminium salts Chemical class 0.000 description 1
- 229940097275 indigo Drugs 0.000 description 1
- COHYTHOBJLSHDF-UHFFFAOYSA-N indigo powder Chemical class N1C2=CC=CC=C2C(=O)C1=C1C(=O)C2=CC=CC=C2N1 COHYTHOBJLSHDF-UHFFFAOYSA-N 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000003402 intramolecular cyclocondensation reaction Methods 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 238000010884 ion-beam technique Methods 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- JEIPFZHSYJVQDO-UHFFFAOYSA-N iron(III) oxide Inorganic materials O=[Fe]O[Fe]=O JEIPFZHSYJVQDO-UHFFFAOYSA-N 0.000 description 1
- 230000001678 irradiating effect Effects 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- BHIWKHZACMWKOJ-UHFFFAOYSA-N isobutyric acid methyl ester Natural products COC(=O)C(C)C BHIWKHZACMWKOJ-UHFFFAOYSA-N 0.000 description 1
- LDHQCZJRKDOVOX-IHWYPQMZSA-N isocrotonic acid Chemical compound C\C=C/C(O)=O LDHQCZJRKDOVOX-IHWYPQMZSA-N 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 125000001261 isocyanato group Chemical group *N=C=O 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical group C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- MWDZOUNAPSSOEL-UHFFFAOYSA-N kaempferol Natural products OC1=C(C(=O)c2cc(O)cc(O)c2O1)c3ccc(O)cc3 MWDZOUNAPSSOEL-UHFFFAOYSA-N 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 239000004922 lacquer Substances 0.000 description 1
- 238000007561 laser diffraction method Methods 0.000 description 1
- QDLAGTHXVHQKRE-UHFFFAOYSA-N lichenxanthone Natural products COC1=CC(O)=C2C(=O)C3=C(C)C=C(OC)C=C3OC2=C1 QDLAGTHXVHQKRE-UHFFFAOYSA-N 0.000 description 1
- 238000013035 low temperature curing Methods 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 1
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 description 1
- 229960003951 masoprocol Drugs 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 150000002736 metal compounds Chemical class 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 125000005395 methacrylic acid group Chemical group 0.000 description 1
- VHRYZQNGTZXDNX-UHFFFAOYSA-N methacryloyl chloride Chemical compound CC(=C)C(Cl)=O VHRYZQNGTZXDNX-UHFFFAOYSA-N 0.000 description 1
- RBQRWNWVPQDTJJ-UHFFFAOYSA-N methacryloyloxyethyl isocyanate Chemical compound CC(=C)C(=O)OCCN=C=O RBQRWNWVPQDTJJ-UHFFFAOYSA-N 0.000 description 1
- 125000005394 methallyl group Chemical group 0.000 description 1
- ITNVWQNWHXEMNS-UHFFFAOYSA-N methanolate;titanium(4+) Chemical compound [Ti+4].[O-]C.[O-]C.[O-]C.[O-]C ITNVWQNWHXEMNS-UHFFFAOYSA-N 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- YDKNBNOOCSNPNS-UHFFFAOYSA-N methyl 1,3-benzoxazole-2-carboxylate Chemical compound C1=CC=C2OC(C(=O)OC)=NC2=C1 YDKNBNOOCSNPNS-UHFFFAOYSA-N 0.000 description 1
- LXEXJHKRLWTKGZ-UHFFFAOYSA-N methyl 7-(diethylamino)-2-oxochromene-3-carboxylate Chemical compound C1=C(C(=O)OC)C(=O)OC2=CC(N(CC)CC)=CC=C21 LXEXJHKRLWTKGZ-UHFFFAOYSA-N 0.000 description 1
- WJSJNBGIABLEGX-UHFFFAOYSA-M methyl(tripentyl)azanium;hydroxide Chemical compound [OH-].CCCCC[N+](C)(CCCCC)CCCCC WJSJNBGIABLEGX-UHFFFAOYSA-M 0.000 description 1
- LVHBHZANLOWSRM-UHFFFAOYSA-N methylenebutanedioic acid Natural products OC(=O)CC(=C)C(O)=O LVHBHZANLOWSRM-UHFFFAOYSA-N 0.000 description 1
- 238000004377 microelectronic Methods 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- CWQXQMHSOZUFJS-UHFFFAOYSA-N molybdenum disulfide Chemical compound S=[Mo]=S CWQXQMHSOZUFJS-UHFFFAOYSA-N 0.000 description 1
- JESXATFQYMPTNL-UHFFFAOYSA-N mono-hydroxyphenyl-ethylene Natural products OC1=CC=CC=C1C=C JESXATFQYMPTNL-UHFFFAOYSA-N 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- UXOUKMQIEVGVLY-UHFFFAOYSA-N morin Natural products OC1=CC(O)=CC(C2=C(C(=O)C3=C(O)C=C(O)C=C3O2)O)=C1 UXOUKMQIEVGVLY-UHFFFAOYSA-N 0.000 description 1
- 235000007708 morin Nutrition 0.000 description 1
- PHQOGHDTIVQXHL-UHFFFAOYSA-N n'-(3-trimethoxysilylpropyl)ethane-1,2-diamine Chemical compound CO[Si](OC)(OC)CCCNCCN PHQOGHDTIVQXHL-UHFFFAOYSA-N 0.000 description 1
- MQWFLKHKWJMCEN-UHFFFAOYSA-N n'-[3-[dimethoxy(methyl)silyl]propyl]ethane-1,2-diamine Chemical compound CO[Si](C)(OC)CCCNCCN MQWFLKHKWJMCEN-UHFFFAOYSA-N 0.000 description 1
- GXMIHVHJTLPVKL-UHFFFAOYSA-N n,n,2-trimethylpropanamide Chemical compound CC(C)C(=O)N(C)C GXMIHVHJTLPVKL-UHFFFAOYSA-N 0.000 description 1
- DKLYDESVXZKCFI-UHFFFAOYSA-N n,n-diphenylacetamide Chemical compound C=1C=CC=CC=1N(C(=O)C)C1=CC=CC=C1 DKLYDESVXZKCFI-UHFFFAOYSA-N 0.000 description 1
- YALAVAYMNJCEBU-UHFFFAOYSA-N n-(2-chloro-3-formylpyridin-4-yl)-2,2-dimethylpropanamide Chemical compound CC(C)(C)C(=O)NC1=CC=NC(Cl)=C1C=O YALAVAYMNJCEBU-UHFFFAOYSA-N 0.000 description 1
- UAOIEEWQVAXCFY-UHFFFAOYSA-N n-(3,4-dimethylphenyl)acetamide Chemical compound CC(=O)NC1=CC=C(C)C(C)=C1 UAOIEEWQVAXCFY-UHFFFAOYSA-N 0.000 description 1
- KBJFYLLAMSZSOG-UHFFFAOYSA-N n-(3-trimethoxysilylpropyl)aniline Chemical compound CO[Si](OC)(OC)CCCNC1=CC=CC=C1 KBJFYLLAMSZSOG-UHFFFAOYSA-N 0.000 description 1
- YUOMCRCBTBNCCS-JUKUECOXSA-N n-[(2s)-1-[[(5s)-5-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-6-hydroxyhexyl]amino]-1-oxo-3,3-diphenylpropan-2-yl]-6-methylpyridine-3-carboxamide Chemical compound N([C@H](C(=O)NCCCC[C@@H](CO)N(CC(C)C)S(=O)(=O)C=1C=CC(N)=CC=1)C(C=1C=CC=CC=1)C=1C=CC=CC=1)C(=O)C1=CC=C(C)N=C1 YUOMCRCBTBNCCS-JUKUECOXSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- PZYDAVFRVJXFHS-UHFFFAOYSA-N n-cyclohexyl-2-pyrrolidone Chemical compound O=C1CCCN1C1CCCCC1 PZYDAVFRVJXFHS-UHFFFAOYSA-N 0.000 description 1
- GNVRJGIVDSQCOP-UHFFFAOYSA-N n-ethyl-n-methylethanamine Chemical compound CCN(C)CC GNVRJGIVDSQCOP-UHFFFAOYSA-N 0.000 description 1
- LMTGCJANOQOGPI-UHFFFAOYSA-N n-methyl-n-phenylacetamide Chemical compound CC(=O)N(C)C1=CC=CC=C1 LMTGCJANOQOGPI-UHFFFAOYSA-N 0.000 description 1
- KADGVXXDDWDKBX-UHFFFAOYSA-N naphthalene-1,2,4,5-tetracarboxylic acid Chemical compound OC(=O)C1=CC=CC2=C(C(O)=O)C(C(=O)O)=CC(C(O)=O)=C21 KADGVXXDDWDKBX-UHFFFAOYSA-N 0.000 description 1
- HDLRSIQOZFLEPK-UHFFFAOYSA-N naphthalene-1,2,5,8-tetracarboxylic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C2=C(C(O)=O)C(C(=O)O)=CC=C21 HDLRSIQOZFLEPK-UHFFFAOYSA-N 0.000 description 1
- GNWCSWUWMHQEMD-UHFFFAOYSA-N naphthalene-1,2-dione diazide Chemical group [N-]=[N+]=[N-].[N-]=[N+]=[N-].C1=CC=C2C(=O)C(=O)C=CC2=C1 GNWCSWUWMHQEMD-UHFFFAOYSA-N 0.000 description 1
- DSCIZKMHZPGBNI-UHFFFAOYSA-N naphthalene-1,3,5,8-tetracarboxylic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C2=CC(C(=O)O)=CC(C(O)=O)=C21 DSCIZKMHZPGBNI-UHFFFAOYSA-N 0.000 description 1
- YPKJPFXVPWGYJL-UHFFFAOYSA-N naphthalene-1,4-dione;sulfuryl dichloride;diazide Chemical compound [N-]=[N+]=[N-].[N-]=[N+]=[N-].ClS(Cl)(=O)=O.C1=CC=C2C(=O)C=CC(=O)C2=C1 YPKJPFXVPWGYJL-UHFFFAOYSA-N 0.000 description 1
- KQSABULTKYLFEV-UHFFFAOYSA-N naphthalene-1,5-diamine Chemical compound C1=CC=C2C(N)=CC=CC2=C1N KQSABULTKYLFEV-UHFFFAOYSA-N 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- DOBFTMLCEYUAQC-UHFFFAOYSA-N naphthalene-2,3,6,7-tetracarboxylic acid Chemical compound OC(=O)C1=C(C(O)=O)C=C2C=C(C(O)=O)C(C(=O)O)=CC2=C1 DOBFTMLCEYUAQC-UHFFFAOYSA-N 0.000 description 1
- BTHGHFBUGBTINV-UHFFFAOYSA-N naphthalene-2,3,6-tricarboxylic acid Chemical compound C1=C(C(O)=O)C(C(O)=O)=CC2=CC(C(=O)O)=CC=C21 BTHGHFBUGBTINV-UHFFFAOYSA-N 0.000 description 1
- YTVNOVQHSGMMOV-UHFFFAOYSA-N naphthalenetetracarboxylic dianhydride Chemical compound C1=CC(C(=O)OC2=O)=C3C2=CC=C2C(=O)OC(=O)C1=C32 YTVNOVQHSGMMOV-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- SLCVBVWXLSEKPL-UHFFFAOYSA-N neopentyl glycol Chemical compound OCC(C)(C)CO SLCVBVWXLSEKPL-UHFFFAOYSA-N 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 150000002828 nitro derivatives Chemical class 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 150000002832 nitroso derivatives Chemical class 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000010606 normalization Methods 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- QUAMTGJKVDWJEQ-UHFFFAOYSA-N octabenzone Chemical compound OC1=CC(OCCCCCCCC)=CC=C1C(=O)C1=CC=CC=C1 QUAMTGJKVDWJEQ-UHFFFAOYSA-N 0.000 description 1
- 229940038384 octadecane Drugs 0.000 description 1
- WTBAHSZERDXKKZ-UHFFFAOYSA-N octadecanoyl chloride Chemical compound CCCCCCCCCCCCCCCCCC(Cl)=O WTBAHSZERDXKKZ-UHFFFAOYSA-N 0.000 description 1
- FMJSMJQBSVNSBF-UHFFFAOYSA-N octocrylene Chemical compound C=1C=CC=CC=1C(=C(C#N)C(=O)OCC(CC)CCCC)C1=CC=CC=C1 FMJSMJQBSVNSBF-UHFFFAOYSA-N 0.000 description 1
- 229920002114 octoxynol-9 Polymers 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000012788 optical film Substances 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 150000001451 organic peroxides Chemical class 0.000 description 1
- 125000005375 organosiloxane group Chemical group 0.000 description 1
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical group C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 description 1
- PFPYHYZFFJJQFD-UHFFFAOYSA-N oxalic anhydride Chemical compound O=C1OC1=O PFPYHYZFFJJQFD-UHFFFAOYSA-N 0.000 description 1
- 150000004893 oxazines Chemical class 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- DXGLGDHPHMLXJC-UHFFFAOYSA-N oxybenzone Chemical compound OC1=CC(OC)=CC=C1C(=O)C1=CC=CC=C1 DXGLGDHPHMLXJC-UHFFFAOYSA-N 0.000 description 1
- SOQBVABWOPYFQZ-UHFFFAOYSA-N oxygen(2-);titanium(4+) Chemical class [O-2].[O-2].[Ti+4] SOQBVABWOPYFQZ-UHFFFAOYSA-N 0.000 description 1
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 1
- ROTJZTYLACIJIG-UHFFFAOYSA-N pentane-1,3,5-tricarboxylic acid Chemical compound OC(=O)CCC(C(O)=O)CCC(O)=O ROTJZTYLACIJIG-UHFFFAOYSA-N 0.000 description 1
- 125000005010 perfluoroalkyl group Chemical group 0.000 description 1
- 239000013500 performance material Substances 0.000 description 1
- JGGWKXMPICYBKC-UHFFFAOYSA-N phenanthrene-1,8,9,10-tetracarboxylic acid Chemical compound C1=CC=C(C(O)=O)C2=C(C(O)=O)C(C(O)=O)=C3C(C(=O)O)=CC=CC3=C21 JGGWKXMPICYBKC-UHFFFAOYSA-N 0.000 description 1
- 239000002530 phenolic antioxidant Substances 0.000 description 1
- 229960005323 phenoxyethanol Drugs 0.000 description 1
- HLJDCFJLAODBJA-UHFFFAOYSA-N phenyl 2,3,4-trihydroxybenzoate Chemical compound OC1=C(O)C(O)=CC=C1C(=O)OC1=CC=CC=C1 HLJDCFJLAODBJA-UHFFFAOYSA-N 0.000 description 1
- HBZMQFJTPHSKNH-UHFFFAOYSA-N phenyl 3,4,5-trihydroxybenzoate Chemical compound OC1=C(O)C(O)=CC(C(=O)OC=2C=CC=CC=2)=C1 HBZMQFJTPHSKNH-UHFFFAOYSA-N 0.000 description 1
- 229960000969 phenyl salicylate Drugs 0.000 description 1
- DYFXGORUJGZJCA-UHFFFAOYSA-N phenylmethanediamine Chemical compound NC(N)C1=CC=CC=C1 DYFXGORUJGZJCA-UHFFFAOYSA-N 0.000 description 1
- 150000008301 phosphite esters Chemical group 0.000 description 1
- 150000003009 phosphonic acids Chemical group 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 238000000016 photochemical curing Methods 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- CLYVDMAATCIVBF-UHFFFAOYSA-N pigment red 224 Chemical compound C=12C3=CC=C(C(OC4=O)=O)C2=C4C=CC=1C1=CC=C2C(=O)OC(=O)C4=CC=C3C1=C42 CLYVDMAATCIVBF-UHFFFAOYSA-N 0.000 description 1
- 229960005235 piperonyl butoxide Drugs 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 229920001495 poly(sodium acrylate) polymer Polymers 0.000 description 1
- 229920002037 poly(vinyl butyral) polymer Polymers 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 229920001281 polyalkylene Polymers 0.000 description 1
- 229920001515 polyalkylene glycol Polymers 0.000 description 1
- 229920002480 polybenzimidazole Polymers 0.000 description 1
- 229910021420 polycrystalline silicon Inorganic materials 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920001225 polyester resin Polymers 0.000 description 1
- 239000004645 polyester resin Substances 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 229920000259 polyoxyethylene lauryl ether Polymers 0.000 description 1
- 229920005591 polysilicon Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 238000011085 pressure filtration Methods 0.000 description 1
- 150000003138 primary alcohols Chemical class 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 239000011164 primary particle Substances 0.000 description 1
- 238000007639 printing Methods 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- HKJYVRJHDIPMQB-UHFFFAOYSA-N propan-1-olate;titanium(4+) Chemical compound CCCO[Ti](OCCC)(OCCC)OCCC HKJYVRJHDIPMQB-UHFFFAOYSA-N 0.000 description 1
- CDQXHVDVGLVACE-UHFFFAOYSA-N propan-2-amine Chemical compound [CH2]C(C)N CDQXHVDVGLVACE-UHFFFAOYSA-N 0.000 description 1
- KVIKMJYUMZPZFU-UHFFFAOYSA-N propan-2-ol;titanium Chemical compound [Ti].CC(C)O.CC(C)O KVIKMJYUMZPZFU-UHFFFAOYSA-N 0.000 description 1
- OGHBATFHNDZKSO-UHFFFAOYSA-N propan-2-olate Chemical compound CC(C)[O-] OGHBATFHNDZKSO-UHFFFAOYSA-N 0.000 description 1
- RZWZRACFZGVKFM-UHFFFAOYSA-N propanoyl chloride Chemical compound CCC(Cl)=O RZWZRACFZGVKFM-UHFFFAOYSA-N 0.000 description 1
- WYVAMUWZEOHJOQ-UHFFFAOYSA-N propionic anhydride Chemical compound CCC(=O)OC(=O)CC WYVAMUWZEOHJOQ-UHFFFAOYSA-N 0.000 description 1
- 235000010388 propyl gallate Nutrition 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- AOHJOMMDDJHIJH-UHFFFAOYSA-N propylenediamine Chemical compound CC(N)CN AOHJOMMDDJHIJH-UHFFFAOYSA-N 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- MCSKRVKAXABJLX-UHFFFAOYSA-N pyrazolo[3,4-d]triazole Chemical compound N1=NN=C2N=NC=C21 MCSKRVKAXABJLX-UHFFFAOYSA-N 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical group C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- MIROPXUFDXCYLG-UHFFFAOYSA-N pyridine-2,5-diamine Chemical compound NC1=CC=C(N)N=C1 MIROPXUFDXCYLG-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 229940079877 pyrogallol Drugs 0.000 description 1
- GZTPJDLYPMPRDF-UHFFFAOYSA-N pyrrolo[3,2-c]pyrazole Chemical compound N1=NC2=CC=NC2=C1 GZTPJDLYPMPRDF-UHFFFAOYSA-N 0.000 description 1
- 239000010453 quartz Substances 0.000 description 1
- 235000005875 quercetin Nutrition 0.000 description 1
- 229960001285 quercetin Drugs 0.000 description 1
- FDRQPMVGJOQVTL-UHFFFAOYSA-N quercetin rutinoside Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC=2C(C3=C(O)C=C(O)C=C3OC=2C=2C=C(O)C(O)=CC=2)=O)O1 FDRQPMVGJOQVTL-UHFFFAOYSA-N 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 238000001226 reprecipitation Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229920003987 resole Polymers 0.000 description 1
- 230000027756 respiratory electron transport chain Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- 230000000630 rising effect Effects 0.000 description 1
- 238000007788 roughening Methods 0.000 description 1
- 235000005493 rutin Nutrition 0.000 description 1
- IKGXIBQEEMLURG-BKUODXTLSA-N rutin Chemical compound O[C@H]1[C@H](O)[C@@H](O)[C@H](C)O[C@@H]1OC[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](OC=2C(C3=C(O)C=C(O)C=C3OC=2C=2C=C(O)C(O)=CC=2)=O)O1 IKGXIBQEEMLURG-BKUODXTLSA-N 0.000 description 1
- ALABRVAAKCSLSC-UHFFFAOYSA-N rutin Natural products CC1OC(OCC2OC(O)C(O)C(O)C2O)C(O)C(O)C1OC3=C(Oc4cc(O)cc(O)c4C3=O)c5ccc(O)c(O)c5 ALABRVAAKCSLSC-UHFFFAOYSA-N 0.000 description 1
- 229960004555 rutoside Drugs 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000000790 scattering method Methods 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 239000003566 sealing material Substances 0.000 description 1
- 238000004062 sedimentation Methods 0.000 description 1
- AKLJIUURIIIOLU-UHFFFAOYSA-N silane 3-trimethoxysilylpropyl 2-methylprop-2-enoate Chemical compound [SiH4].CO[Si](OC)(OC)CCCOC(=O)C(C)=C AKLJIUURIIIOLU-UHFFFAOYSA-N 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- HQVNEWCFYHHQES-UHFFFAOYSA-N silicon nitride Chemical compound N12[Si]34N5[Si]62N3[Si]51N64 HQVNEWCFYHHQES-UHFFFAOYSA-N 0.000 description 1
- 229910052814 silicon oxide Inorganic materials 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000004332 silver Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 235000019795 sodium metasilicate Nutrition 0.000 description 1
- NNMHYFLPFNGQFZ-UHFFFAOYSA-M sodium polyacrylate Chemical compound [Na+].[O-]C(=O)C=C NNMHYFLPFNGQFZ-UHFFFAOYSA-M 0.000 description 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 1
- NMWCVZCSJHJYFW-UHFFFAOYSA-M sodium;3,5-dichloro-2-hydroxybenzenesulfonate Chemical compound [Na+].OC1=C(Cl)C=C(Cl)C=C1S([O-])(=O)=O NMWCVZCSJHJYFW-UHFFFAOYSA-M 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000004544 sputter deposition Methods 0.000 description 1
- 125000005504 styryl group Chemical group 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 229940014800 succinic anhydride Drugs 0.000 description 1
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 125000000565 sulfonamide group Chemical group 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 239000012756 surface treatment agent Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 150000003509 tertiary alcohols Chemical class 0.000 description 1
- 229940073455 tetraethylammonium hydroxide Drugs 0.000 description 1
- LRGJRHZIDJQFCL-UHFFFAOYSA-M tetraethylazanium;hydroxide Chemical compound [OH-].CC[N+](CC)(CC)CC LRGJRHZIDJQFCL-UHFFFAOYSA-M 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- DLNOIRAFPPLAPE-UHFFFAOYSA-L tetramethylazanium dihydroxide Chemical compound [OH-].[OH-].C[N+](C)(C)C.C[N+](C)(C)C DLNOIRAFPPLAPE-UHFFFAOYSA-L 0.000 description 1
- DCFYRBLFVWYBIJ-UHFFFAOYSA-M tetraoctylazanium;hydroxide Chemical compound [OH-].CCCCCCCC[N+](CCCCCCCC)(CCCCCCCC)CCCCCCCC DCFYRBLFVWYBIJ-UHFFFAOYSA-M 0.000 description 1
- JVOPCCBEQRRLOJ-UHFFFAOYSA-M tetrapentylazanium;hydroxide Chemical compound [OH-].CCCCC[N+](CCCCC)(CCCCC)CCCCC JVOPCCBEQRRLOJ-UHFFFAOYSA-M 0.000 description 1
- LPSKDVINWQNWFE-UHFFFAOYSA-M tetrapropylazanium;hydroxide Chemical compound [OH-].CCC[N+](CCC)(CCC)CCC LPSKDVINWQNWFE-UHFFFAOYSA-M 0.000 description 1
- 229920002803 thermoplastic polyurethane Polymers 0.000 description 1
- 239000010409 thin film Substances 0.000 description 1
- 125000005000 thioaryl group Chemical group 0.000 description 1
- 125000000101 thioether group Chemical group 0.000 description 1
- ANRHNWWPFJCPAZ-UHFFFAOYSA-M thionine Chemical compound [Cl-].C1=CC(N)=CC2=[S+]C3=CC(N)=CC=C3N=C21 ANRHNWWPFJCPAZ-UHFFFAOYSA-M 0.000 description 1
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea group Chemical group NC(=S)N UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 1
- 150000003585 thioureas Chemical class 0.000 description 1
- ZCUFMDLYAMJYST-UHFFFAOYSA-N thorium dioxide Chemical compound O=[Th]=O ZCUFMDLYAMJYST-UHFFFAOYSA-N 0.000 description 1
- 229910052718 tin Inorganic materials 0.000 description 1
- 239000011135 tin Substances 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- JMXKSZRRTHPKDL-UHFFFAOYSA-N titanium ethoxide Chemical compound [Ti+4].CC[O-].CC[O-].CC[O-].CC[O-] JMXKSZRRTHPKDL-UHFFFAOYSA-N 0.000 description 1
- 125000005424 tosyloxy group Chemical group S(=O)(=O)(C1=CC=C(C)C=C1)O* 0.000 description 1
- HTSABYAWKQAHBT-UHFFFAOYSA-N trans 3-methylcyclohexanol Natural products CC1CCCC(O)C1 HTSABYAWKQAHBT-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- NBXZNTLFQLUFES-UHFFFAOYSA-N triethoxy(propyl)silane Chemical compound CCC[Si](OCC)(OCC)OCC NBXZNTLFQLUFES-UHFFFAOYSA-N 0.000 description 1
- JXUKBNICSRJFAP-UHFFFAOYSA-N triethoxy-[3-(oxiran-2-ylmethoxy)propyl]silane Chemical compound CCO[Si](OCC)(OCC)CCCOCC1CO1 JXUKBNICSRJFAP-UHFFFAOYSA-N 0.000 description 1
- GKASDNZWUGIAMG-UHFFFAOYSA-N triethyl orthoformate Chemical compound CCOC(OCC)OCC GKASDNZWUGIAMG-UHFFFAOYSA-N 0.000 description 1
- JLGLQAWTXXGVEM-UHFFFAOYSA-N triethylene glycol monomethyl ether Chemical compound COCCOCCOCCO JLGLQAWTXXGVEM-UHFFFAOYSA-N 0.000 description 1
- YFNKIDBQEZZDLK-UHFFFAOYSA-N triglyme Chemical compound COCCOCCOCCOC YFNKIDBQEZZDLK-UHFFFAOYSA-N 0.000 description 1
- 125000004953 trihalomethyl group Chemical group 0.000 description 1
- SRPWOOOHEPICQU-UHFFFAOYSA-N trimellitic anhydride Chemical compound OC(=O)C1=CC=C2C(=O)OC(=O)C2=C1 SRPWOOOHEPICQU-UHFFFAOYSA-N 0.000 description 1
- NJMOHBDCGXJLNJ-UHFFFAOYSA-N trimellitic anhydride chloride Chemical compound ClC(=O)C1=CC=C2C(=O)OC(=O)C2=C1 NJMOHBDCGXJLNJ-UHFFFAOYSA-N 0.000 description 1
- HADKRTWCOYPCPH-UHFFFAOYSA-M trimethylphenylammonium hydroxide Chemical compound [OH-].C[N+](C)(C)C1=CC=CC=C1 HADKRTWCOYPCPH-UHFFFAOYSA-M 0.000 description 1
- AAAQKTZKLRYKHR-UHFFFAOYSA-N triphenylmethane Chemical compound C1=CC=CC=C1C(C=1C=CC=CC=1)C1=CC=CC=C1 AAAQKTZKLRYKHR-UHFFFAOYSA-N 0.000 description 1
- 150000004961 triphenylmethanes Chemical class 0.000 description 1
- GRPURDFRFHUDSP-UHFFFAOYSA-N tris(prop-2-enyl) benzene-1,2,4-tricarboxylate Chemical compound C=CCOC(=O)C1=CC=C(C(=O)OCC=C)C(C(=O)OCC=C)=C1 GRPURDFRFHUDSP-UHFFFAOYSA-N 0.000 description 1
- HIZCIEIDIFGZSS-UHFFFAOYSA-L trithiocarbonate Chemical compound [S-]C([S-])=S HIZCIEIDIFGZSS-UHFFFAOYSA-L 0.000 description 1
- 239000012989 trithiocarbonate Substances 0.000 description 1
- 229960004418 trolamine Drugs 0.000 description 1
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 239000010937 tungsten Substances 0.000 description 1
- 238000000870 ultraviolet spectroscopy Methods 0.000 description 1
- 125000002948 undecyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000003673 urethanes Chemical class 0.000 description 1
- 238000005292 vacuum distillation Methods 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- GPPXJZIENCGNKB-UHFFFAOYSA-N vanadium Chemical compound [V]#[V] GPPXJZIENCGNKB-UHFFFAOYSA-N 0.000 description 1
- ZTWTYVWXUKTLCP-UHFFFAOYSA-N vinylphosphonic acid Chemical class OP(O)(=O)C=C ZTWTYVWXUKTLCP-UHFFFAOYSA-N 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
Classifications
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/038—Macromolecular compounds which are rendered insoluble or differentially wettable
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/10—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
- C08G73/1067—Wholly aromatic polyimides, i.e. having both tetracarboxylic and diamino moieties aromatically bound
- C08G73/1071—Wholly aromatic polyimides containing oxygen in the form of ether bonds in the main chain
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/10—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
- C08G73/1003—Preparatory processes
- C08G73/1007—Preparatory processes from tetracarboxylic acids or derivatives and diamines
- C08G73/1025—Preparatory processes from tetracarboxylic acids or derivatives and diamines polymerised by radiations
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/22—Polybenzoxazoles
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/022—Quinonediazides
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/022—Quinonediazides
- G03F7/0226—Quinonediazides characterised by the non-macromolecular additives
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/022—Quinonediazides
- G03F7/023—Macromolecular quinonediazides; Macromolecular additives, e.g. binders
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/027—Non-macromolecular photopolymerisable compounds having carbon-to-carbon double bonds, e.g. ethylenic compounds
- G03F7/028—Non-macromolecular photopolymerisable compounds having carbon-to-carbon double bonds, e.g. ethylenic compounds with photosensitivity-increasing substances, e.g. photoinitiators
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/027—Non-macromolecular photopolymerisable compounds having carbon-to-carbon double bonds, e.g. ethylenic compounds
- G03F7/028—Non-macromolecular photopolymerisable compounds having carbon-to-carbon double bonds, e.g. ethylenic compounds with photosensitivity-increasing substances, e.g. photoinitiators
- G03F7/031—Organic compounds not covered by group G03F7/029
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/075—Silicon-containing compounds
- G03F7/0755—Non-macromolecular compounds containing Si-O, Si-C or Si-N bonds
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/20—Exposure; Apparatus therefor
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/26—Processing photosensitive materials; Apparatus therefor
- G03F7/30—Imagewise removal using liquid means
- G03F7/32—Liquid compositions therefor, e.g. developers
Definitions
- the present invention relates to a negative photosensitive resin composition, a cured product, a laminate, a method for producing a cured product, and a semiconductor device.
- Cyclized resins such as polyimide are used in a variety of applications due to their excellent heat resistance and insulating properties.
- the use is not particularly limited, but in the case of a semiconductor device for mounting, use as a material for an insulating film or a sealing material, or as a protective film can be mentioned. It is also used as a base film or coverlay for flexible substrates.
- the cyclized resin such as polyimide is used in the form of a negative photosensitive resin composition containing at least one of a cyclized resin such as polyimide and a precursor of the cyclized resin.
- a negative photosensitive resin composition is applied to a substrate, for example, by coating or the like to form a photosensitive film, and then, if necessary, exposure, development, heating, and the like are performed to form a cured product on the substrate. can be formed on.
- a precursor of the cyclized resin such as a polyimide precursor is cyclized, for example, by heating, and becomes a cyclized resin such as polyimide in the cured product.
- the negative photosensitive resin composition can be applied by a known coating method or the like, for example, the degree of freedom in designing the shape, size, application position, etc. when the negative photosensitive resin composition to be applied is applied. It can be said that it is excellent in adaptability in terms of manufacturing, such as high In addition to the high performance possessed by cyclized resins such as polyimide, from the viewpoint of such excellent applicability in production, the development of industrial applications of the negative photosensitive resin composition is increasingly expected.
- Patent Document 1 describes a negative photosensitive resin composition
- a negative photosensitive resin composition comprising (A) a polyamic acid ester having a specific structure, (B) a naphthoquinone diazide compound, and (C) a photopolymerization initiator as essential components. .
- Patent Document 2 discloses (A) a polyimide precursor which is a polyamic acid and/or a polyamic acid ester having a neutral functional group in a side chain which is a substitute for an acidic functional group or a basic functional group, polyhydroxy At least one resin selected from the group consisting of an amide polyoxazole precursor, polyaminoamide, polyamide, polyamideimide, polyimide, polybenzoxazole, polybenzimidazole, and polybenzthiazole: 100 parts by mass, and ( B) A resin composition containing 1 to 50 parts by mass of a compound having an aromatic sulfonate structure is described.
- the present invention provides a negative photosensitive resin composition which is excellent in the resolution of the obtained pattern and suppresses a decrease in film thickness due to development, a cured product obtained by curing the negative photosensitive resin composition, It aims at providing the laminated body containing the said hardened
- Condition i The composition further contains a nitrogen-containing cyclic compound as a component different from the resin, compound A, and photopolymerization initiator.
- Condition ii The resin includes a resin having a nitrogen-containing heterocyclic structure containing two or more nitrogen atoms as ring members.
- the compound A contains a naphthoquinone diazide compound.
- the negative photosensitive resin composition according to . ⁇ 5> Resin, a naphthoquinone diazide compound, and containing a photoinitiator, A negative photosensitive resin composition that satisfies at least one of the following conditions iii and iv.
- the composition further contains a nitrogen-containing cyclic compound as a component different from the resin, the naphthoquinone diazide compound and the photoinitiator.
- the resin includes a resin having a nitrogen-containing heterocyclic structure containing two or more nitrogen atoms as ring members.
- ⁇ 8> The negative photosensitive resin composition according to any one of ⁇ 1> to ⁇ 7>, further comprising a silane coupling agent.
- ⁇ 9> The negative photosensitive resin composition according to any one of ⁇ 1> to ⁇ 8>, further comprising a polymerization inhibitor.
- ⁇ 10> The negative photosensitive resin composition according to any one of ⁇ 1> to ⁇ 9>, further comprising a base generator.
- ⁇ 11> The negative photosensitive resin composition according to any one of ⁇ 1> to ⁇ 10>, wherein the photopolymerization initiator is a photoradical polymerization initiator.
- ⁇ 12> The negative photosensitive resin composition according to any one of ⁇ 1> to ⁇ 11>, which is used for forming an interlayer insulating film for a rewiring layer.
- ⁇ 13> A cured product obtained by curing the negative photosensitive resin composition according to any one of ⁇ 1> to ⁇ 12>.
- ⁇ 14> A laminate comprising two or more layers made of the cured product according to ⁇ 13> and a metal layer between any of the layers made of the cured product.
- a semiconductor device comprising the cured product according to ⁇ 13> or the laminate according to ⁇ 14>.
- a negative photosensitive resin composition that provides excellent pattern resolution and suppresses reduction in film thickness due to development, and a cured product obtained by curing the negative photosensitive resin composition.
- a product, a laminate containing the cured product, a method for producing the cured product, and a semiconductor device containing the cured product or the laminate are provided.
- a numerical range represented by the symbol "to” means a range including the numerical values before and after "to” as lower and upper limits, respectively.
- the term "process” is meant to include not only independent processes, but also processes that are indistinguishable from other processes as long as the desired effects of the process can be achieved.
- a description that does not describe substitution or unsubstituted includes a group (atomic group) having no substituent as well as a group (atomic group) having a substituent.
- alkyl group includes not only alkyl groups without substituents (unsubstituted alkyl groups) but also alkyl groups with substituents (substituted alkyl groups).
- exposure includes not only exposure using light but also exposure using particle beams such as electron beams and ion beams, unless otherwise specified.
- Light used for exposure includes actinic rays or radiation such as emission line spectra of mercury lamps, far ultraviolet rays represented by excimer lasers, extreme ultraviolet rays (EUV light), X-rays, and electron beams.
- (meth)acrylate means both or either of “acrylate” and “methacrylate”
- (meth)acrylic means both “acrylic” and “methacrylic”
- (meth)acryloyl means either or both of “acryloyl” and “methacryloyl”.
- Me in the structural formulas represents a methyl group
- Et represents an ethyl group
- Bu represents a butyl group
- Ph represents a phenyl group.
- total solid content refers to the total mass of all components of the composition excluding the solvent.
- the solid content concentration is the mass percentage of other components excluding the solvent with respect to the total mass of the composition.
- the weight average molecular weight (Mw) and number average molecular weight (Mn) are values measured using a gel permeation chromatography (GPC) method, unless otherwise specified, and are defined as polystyrene conversion values.
- the weight average molecular weight (Mw) and number average molecular weight (Mn) are, for example, HLC-8220GPC (manufactured by Tosoh Corporation), guard column HZ-L, TSKgel Super HZM-M, TSKgel It can be obtained by connecting Super HZ4000, TSKgel Super HZ3000, and TSKgel Super HZ2000 (manufactured by Tosoh Corporation) in series. Unless otherwise stated, their molecular weights were determined using THF (tetrahydrofuran) as an eluent.
- THF tetrahydrofuran
- NMP N-methyl-2-pyrrolidone
- detection in GPC measurement uses a UV ray (ultraviolet) wavelength detector of 254 nm.
- UV ray ultraviolet
- a third layer or element may be interposed between the reference layer and the other layer, and the reference layer and the other layer need not be in contact with each other.
- the direction in which the layers are stacked with respect to the base material is referred to as "upper", or when there is a resin composition layer, the direction from the base material to the resin composition layer is referred to as “upper”. and the opposite direction is called “down”.
- the composition may contain two or more compounds corresponding to each component contained in the composition.
- the content of each component in the composition means the total content of all compounds corresponding to that component.
- the temperature is 23° C.
- the pressure is 101,325 Pa (1 atm)
- the relative humidity is 50% RH, unless otherwise stated. Combinations of preferred aspects are more preferred aspects herein.
- the negative photosensitive resin composition in the first aspect of the present invention comprises a resin, a compound A whose absorbance at the exposure wavelength is reduced by exposure, and a photopolymerization initiator, and at least one of the following condition i and condition ii meet.
- Condition i The composition further contains a nitrogen-containing cyclic compound as a component different from the resin, compound A and photopolymerization initiator.
- Condition ii The resin includes a resin having a nitrogen-containing heterocyclic structure containing two or more nitrogen atoms as ring members.
- a negative photosensitive resin composition according to the second aspect of the present invention contains a resin, a naphthoquinone diazide compound, and a photopolymerization initiator, and satisfies at least one of condition iii and condition iv.
- Condition iii The composition further contains a nitrogen-containing cyclic compound as a component different from the resin, the naphthoquinone diazide compound and the photoinitiator.
- Condition iv The resin includes a resin having a nitrogen-containing heterocyclic structure containing two or more nitrogen atoms as ring members.
- the negative photosensitive resin composition of the present invention is preferably used for forming a photosensitive film subjected to exposure and development, and is used for forming a film subjected to exposure and development using a developer containing an organic solvent. More preferably used.
- INDUSTRIAL APPLICABILITY The resin composition of the present invention can be used, for example, to form an insulating film for semiconductor devices, an interlayer insulating film for rewiring layers, a stress buffer film, and the like, and can be used to form an interlayer insulating film for rewiring layers. preferable.
- the negative photosensitive resin composition of the present invention is used for forming a photosensitive film subjected to negative development.
- negative development refers to development in which non-exposed areas are removed by development in exposure and development
- positive development refers to development in which exposed areas are removed by development.
- the exposure method, the developer, and the development method include, for example, the exposure method described in the exposure step, the developer and the development method described in the development step in the description of the method for producing a cured product described later. is used.
- the resulting pattern has excellent resolution, and reduction in film thickness due to development is suppressed.
- the mechanism by which the above effects are obtained is unknown, it is presumed as follows.
- a photosensitive layer is formed using a negative photosensitive resin composition containing a resin and a photopolymerization initiator, and the photosensitive layer is exposed and developed to form a patterned cured product on a metal layer. It is done.
- the compound A a naphthoquinonediazide compound in the second aspect
- the above conditions i and ii in the second aspect , condition iii and condition iv
- the resolution of the pattern on the metal substrate is improved.
- the compound A (the naphthoquinone diazide compound in the second embodiment) absorbs the exposure light, so that the photopolymerization initiator can becomes less sensitive to light, and in regions where the amount of exposure is large, the compound A (in the second embodiment, the naphthoquinonediazide compound) is decolored (the absorbance of the exposure wavelength is reduced by exposure), thereby making the photopolymerization initiator It is presumed that this is because the photosensitivity of Furthermore, the negative photosensitive resin composition of the present invention contains a nitrogen-containing cyclic compound, or contains, as the resin, a resin having a nitrogen-containing heterocyclic structure containing two or more nitrogen atoms as ring members, or Including both.
- the negative photosensitive resin composition of the present invention contains a nitrogen-containing cyclic compound, a resin having a nitrogen-containing heterocyclic structure containing two or more nitrogen atoms as ring members, or both.
- the obtained cured product is also excellent in chemical resistance.
- the decomposition product for example, the decomposition product of the naphthoquinonediazide compound
- the cross-linking density is further increased, so the chemical resistance of the cured product is also improved. It is thought that That is, the negative photosensitive resin composition according to the second aspect of the present invention is a composition that gives a cured product having further excellent chemical resistance.
- the resin when the resin is a cyclized resin or a precursor thereof and the compound A is a naphthoquinonediazide compound, the resin and the decomposition product of the naphthoquinonediazide compound form a crosslinked structure, greatly improving chemical resistance.
- the resin and the decomposition product of the naphthoquinonediazide compound form a crosslinked structure, greatly improving chemical resistance.
- Patent Documents 1 and 2 negatives containing compound A, containing a resin having a nitrogen-containing heterocyclic structure containing two or more nitrogen atoms as a ring member as the resin, or containing both A type photosensitive resin composition is not described.
- the resin composition of the present invention contains a resin.
- the resin composition of the present invention preferably satisfies at least one of the following (1) and (2).
- the resin contained in the resin composition has a polymerizable group.
- the resin composition further contains a polymerizable compound described below.
- the resin is not particularly limited, and includes, for example, resins used in conventional pattern-forming compositions, but it preferably contains a cyclized resin or a precursor thereof (specific resin). It is more preferable to include
- the resin composition of the present invention contains, as the resin, a resin having a nitrogen-containing heterocyclic structure containing two or more nitrogen atoms as ring members. .
- the resin composition of the present invention contains, as the resin, a resin having a nitrogen-containing heterocyclic structure containing two or more nitrogen atoms as ring members. It's okay.
- a nitrogen-containing heterocyclic structure containing two or more nitrogen atoms as ring members may be monocyclic or polycyclic, preferably monocyclic or condensed, and may be a 5- or 6-membered ring.
- a monocyclic ring, or a condensed ring between five-membered rings, a condensed ring between six-membered rings, or a condensed ring between a five-membered ring and a six-membered ring is more preferable, and a monocyclic five-membered ring or a five-membered ring and a condensed 6-membered ring are more preferred.
- the nitrogen-containing heterocyclic structure is a double ring
- at least one single ring constituting the double ring contains two or more nitrogen atoms as ring members. That is, for example, a benzimidazole ring corresponds to a nitrogen-containing heterocyclic structure containing two or more nitrogen atoms as ring members in the present invention, while a 7-azaindole ring has two or more nitrogen atoms as ring members in the present invention. It shall not apply to nitrogen-containing heterocyclic structures containing atoms.
- the nitrogen-containing heterocyclic structure may have a plurality of nitrogen-containing heterocyclic structures containing two or more nitrogen atoms as ring members in the multicyclic structure, such as a purine ring structure.
- the above nitrogen-containing heterocyclic structure may contain a heteroatom other than a nitrogen atom as a ring member. is one.
- the heteroatom other than the nitrogen atom include an oxygen atom and a sulfur atom.
- the nitrogen-containing heterocyclic structure may be either an aliphatic ring or an aromatic ring, but is preferably an aromatic ring.
- the nitrogen-containing heterocycle preferably contains an imidazole skeleton, a triazole skeleton, or a tetrazole skeleton, and the imidazole ring, triazole ring, tetrazole ring, or A condensed ring of these rings and another ring is more preferable, and a benzimidazole ring, triazole ring, benzotriazole ring, tetrazole ring, or purine ring is even more preferable.
- the other ring is preferably an aromatic ring, more preferably a 5- or 6-membered aromatic ring, and more preferably a benzene ring.
- the nitrogen-containing heterocyclic structure is preferably present at the terminal of the specific resin, and is present at the main chain terminal of the specific resin. is more preferable.
- the main chain represents the relatively longest connecting chain in the resin molecule. At least one of the main chain ends of the specific resin preferably has the nitrogen-containing heterocyclic structure.
- the specific resin when the specific resin contains the nitrogen-containing heterocyclic structure, the specific resin preferably contains a structure represented by the following formula (1-1) or (2-1).
- the specific resin when the specific resin is polyimide, polyimide precursor, polyamideimide or polyamideimide precursor, it preferably contains a structure represented by the following formula (1-1).
- the specific resin when the specific resin is polybenzoxazole or a polybenzoxazole precursor, it preferably contains a structure represented by the following formula (2-1).
- L 1 is an n+1-valent linking group that does not contain a single bond or an imide ring structure
- R 1 is a nitrogen-containing heterocyclic structure containing two or more nitrogen atoms as ring members
- n is 1.
- L 2 is an m+1-valent linking group that does not contain a single bond or an oxazole ring structure
- R 2 is a nitrogen-containing heterocyclic structure containing two or more nitrogen atoms as ring members
- m is 1.
- Each of the above integers is represented, and * represents a binding site with another structure.
- RN represents a hydrogen atom or a monovalent substituent, preferably a hydrogen atom or a hydrocarbon group, more preferably a hydrogen atom, an alkyl group or an aromatic hydrocarbon group.
- the embodiment in which L 1 is a single bond is also one of the preferred embodiments of the present invention.
- R 1 in formula (1-1) are the same as the preferred embodiments of the nitrogen-containing heterocyclic structure described above.
- n is preferably an integer of 1 to 4, more preferably 1 or 2, and even more preferably 1.
- * represents a binding site with another structure.
- the embodiment in which two * are respectively bonded to the covalently bonded carbon atoms to form an imide ring structure containing the imide structure described in formula (1-1) is also one of the preferred embodiments of the present invention. is one.
- the imide ring structure for example, a five-membered ring structure is preferable.
- the embodiment in which L2 is a single bond is also one of the preferred embodiments of the present invention.
- R 2 in formula (2-1) are the same as the preferred embodiments of the nitrogen-containing heterocyclic structure described above.
- m is preferably an integer of 1 to 4, more preferably 1 or 2, and even more preferably 1.
- * represents a binding site with another structure.
- the embodiment in which two * are respectively bonded to the covalently bonded carbon atoms to form a ring structure containing a nitrogen atom and an oxygen atom described in formula (2-1) is also a preferred embodiment of the present invention.
- the specific resin preferably contains a structure represented by any one of the following formulas (1-2) to (1-3) as the structure containing the structure represented by formula (1-1).
- L 1 is an n+1 valent linking group that does not contain a single bond or an imide ring structure
- R 1 is a nitrogen-containing hetero ring containing two or more nitrogen atoms as ring members.
- n represents an integer of 1 or more
- * represents a bonding site with another structure.
- L 1 , R 1 and n are respectively synonymous with L 1 , R 1 and n in formula (1-1), and preferred embodiments are also the same. be.
- the specific resin preferably contains a structure represented by any one of the following formulas (2-2) to (2-3) as the structure containing the structure represented by formula (2-1).
- L 2 is an m+1 valent linking group that does not contain a single bond or an oxazole ring structure
- R 2 is a nitrogen-containing hetero ring containing two or more nitrogen atoms as ring members.
- m represents an integer of 1 or more
- * represents a binding site with another structure.
- L 2 , R 2 and m are respectively synonymous with L 2 , R 2 and m in formula (2-1), and preferred embodiments are also the same. be.
- the specific resin when the specific resin contains a structure represented by the above formula (1-1), the specific resin preferably also contains a structure represented by the following formula (1-4).
- a 1 represents an oxygen atom or —NH—
- R 115 represents a tetravalent organic group
- R 114 represents a hydrogen atom or a monovalent organic group
- L 1 represents An n+1 valent linking group that does not contain a single bond or an imide ring structure
- R 1 represents a nitrogen-containing heterocyclic structure containing two or more nitrogen atoms as a ring member
- n represents an integer of 1 or more
- * represents another Represents the binding site to the structure.
- a 1 , R 115 and R 114 have the same meanings as A 1 , R 115 and R 114 in formula (2) below, and preferred embodiments are also the same.
- L 1 , R 1 and n have the same definitions as L 1 , R 1 and n in formula (1-1) above, and preferred embodiments are also the same.
- the specific resin when the specific resin contains a structure represented by formula (1-4), the specific resin preferably contains a repeating unit represented by formula (2) described below. When the specific resin contains a repeating unit represented by formula (2) described later, A 1 , R 115 and R 114 in formula (1-4) are repeating units represented by formula (2) contained in the specific resin.
- the specific resin when the specific resin contains the structure represented by the above formula (1-1), the specific resin preferably also contains the structure represented by the following formula (1-5).
- R 132 represents a tetravalent organic group
- L 1 represents an n+1 valent linking group that does not contain a single bond or an imide ring structure
- R 1 represents two or more nitrogen atoms as ring members.
- n represents an integer of 1 or more
- * represents a binding site to another structure.
- R 132 has the same definition as R 132 in formula (4) below, and preferred embodiments are also the same.
- L 1 , R 1 and n have the same definitions as L 1 , R 1 and n in formula (1-1) above, and preferred embodiments are also the same.
- the specific resin when the specific resin contains a structure represented by formula (1-5), the specific resin preferably contains a repeating unit represented by formula (4) described below.
- R 132 in formula (1-5) is R in any of the repeating units represented by formula (4) contained in the specific resin The same structure as 132 is also preferred.
- * in formula (1-5) is also preferably a bonding site with R 131 in formula (4).
- the specific resin when the specific resin contains a structure represented by formula (2-1) above, the specific resin preferably also contains a structure represented by formula (2-4) below.
- R 122 represents a tetravalent organic group
- R 124 represents a hydrogen atom or a monovalent organic group
- L 2 is a single bond or an m+1 valent linkage that does not contain an oxazole ring structure
- R 2 represents a nitrogen-containing heterocyclic structure containing two or more nitrogen atoms as ring members
- m represents an integer of 1 or more
- * represents a bonding site with another structure.
- R 122 and R 124 have the same meanings as R 122 and R 124 in formula (3) below, and preferred embodiments are also the same.
- L 2 , R 2 and m have the same meanings as L 2 , R 2 and m in formula (2-1) above, and preferred embodiments are also the same.
- the specific resin when the specific resin contains a structure represented by formula (2-4), the specific resin preferably contains a repeating unit represented by formula (3) described below.
- R 122 and R 124 in formula (2-4) may be any of the repeating units represented by formula (3) contained in the specific resin. It is also preferred to have the same structure as R 122 and R 124 in (1).
- * in formula (2-4) is also preferably a bonding site with R 121 in formula (3).
- the specific resin when the specific resin contains a structure represented by formula (2-1) above, the specific resin preferably also contains a structure represented by formula (2-5) below.
- R 134 represents a tetravalent organic group
- L 2 is an m+1 valent linking group that does not contain a single bond or an oxazole ring structure
- R 2 is a nitrogen atom having two or more ring members.
- m represents an integer of 1 or more
- * represents a binding site with another structure.
- R 134 has the same definition as R 134 in formula (X) described below, and preferred embodiments are also the same.
- L 2 , R 2 and m have the same meanings as L 2 , R 2 and m in formula (1-1) above, and preferred embodiments are also the same.
- the specific resin preferably contains a repeating unit represented by formula (X) described below.
- R 134 in formula (2-5) is R in any of the repeating units represented by formula (X) contained in the specific resin The same structure as 134 is also preferred.
- * in formula (1-5) is also preferably a bonding site with R 133 in formula (X).
- R 113 and R 114 in formula (2) to be described later, R 123 and R 124 in formula (3) to be described later, R 113 in formula (PAI-2) to be described later, and the above nitrogen-containing heterocyclic structures may include
- the cyclized resin is preferably a resin containing an imide ring structure or an oxazole ring structure in its main chain structure.
- the main chain represents the relatively longest connecting chain in the resin molecule.
- cyclized resins include polyimide, polybenzoxazole, and polyamideimide.
- a precursor of a cyclized resin is a resin that undergoes a change in chemical structure by an external stimulus to become a cyclized resin, preferably a resin that undergoes a change in chemical structure by heat to become a cyclized resin.
- a resin that becomes a cyclized resin by forming a ring structure is more preferable.
- Precursors of the cyclized resin include polyimide precursors, polybenzoxazole precursors, polyamideimide precursors, and the like. That is, the resin composition of the present invention contains, as a specific resin, at least one selected from the group consisting of polyimides, polyimide precursors, polybenzoxazoles, polybenzoxazole precursors, polyamideimides, and polyamideimide precursors. It preferably contains a resin (specific resin).
- the resin composition of the present invention preferably contains polyimide or a polyimide precursor as the specific resin.
- the specific resin preferably has a polymerizable group, and more preferably contains a radically polymerizable group.
- the resin composition of the present invention preferably contains a radical polymerization initiator described later, and contains a radical polymerization initiator described later and a radical cross-linking agent described later. is more preferred. Further, if necessary, a sensitizer described later can be included. For example, a negative photosensitive film is formed from the resin composition of the present invention.
- polyimide precursor Although the type of the polyimide precursor used in the present invention is not particularly limited, it preferably contains a repeating unit represented by the following formula (2).
- a 1 and A 2 each independently represent an oxygen atom or -NH-
- R 111 represents a divalent organic group
- R 115 represents a tetravalent organic group
- R 113 and R 114 each independently represent a hydrogen atom or a monovalent organic group.
- a 1 and A 2 in formula (2) each independently represent an oxygen atom or —NH—, preferably an oxygen atom.
- R 111 in formula (2) represents a divalent organic group.
- divalent organic groups include groups containing linear or branched aliphatic groups, cyclic aliphatic groups and aromatic groups, linear or branched aliphatic groups having 2 to 20 carbon atoms, A cyclic aliphatic group having 3 to 20 carbon atoms, an aromatic group having 3 to 20 carbon atoms, or a group consisting of a combination thereof is preferable, and a group containing an aromatic group having 6 to 20 carbon atoms is more preferable.
- the hydrocarbon group in the chain may be substituted with a group containing a hetero atom, and in the cyclic aliphatic group and the aromatic group, the ring member hydrocarbon group is a hetero atom.
- may be substituted with a group containing Groups represented by -Ar- and -Ar-L-Ar- are exemplified as preferred embodiments of the present invention, and groups represented by -Ar-L-Ar- are particularly preferred.
- Ar is each independently an aromatic group
- L is a single bond, or an aliphatic hydrocarbon group having 1 to 10 carbon atoms which may be substituted with a fluorine atom, -O-, -CO -, -S-, -SO 2 - or -NHCO-, or a group consisting of a combination of two or more of the above. Preferred ranges for these are as described above.
- R 111 is preferably derived from a diamine.
- Diamines used in the production of polyimide precursors include linear or branched aliphatic, cyclic aliphatic or aromatic diamines. Only one type of diamine may be used, or two or more types may be used. Specifically, a linear or branched aliphatic group having 2 to 20 carbon atoms, a cyclic aliphatic group having 3 to 20 carbon atoms, an aromatic group having 3 to 20 carbon atoms, or a group consisting of a combination thereof is preferably a diamine containing, more preferably a diamine containing an aromatic group having 6 to 20 carbon atoms. In the straight-chain or branched aliphatic group, the hydrocarbon group in the chain may be substituted with a group containing a heteroatom. may be substituted with a group containing Examples of groups containing aromatic groups include:
- * represents a binding site with other structures.
- diamines include 1,2-diaminoethane, 1,2-diaminopropane, 1,3-diaminopropane, 1,4-diaminobutane or 1,6-diaminohexane; ,3-diaminocyclopentane, 1,2-, 1,3- or 1,4-diaminocyclohexane, 1,2-, 1,3- or 1,4-bis(aminomethyl)cyclohexane, bis-(4- aminocyclohexyl)methane, bis-(3-aminocyclohexyl)methane, 4,4′-diamino-3,3′-dimethylcyclohexylmethane and isophoronediamine; m- or p-phenylenediamine, diaminotoluene, 4,4′- or 3,3'-diaminobiphenyl, 4,4'-diaminodiphenyl ether, 3, 3,3
- diamines (DA-1) to (DA-18) described in paragraphs 0030 to 0031 of International Publication No. 2017/038598.
- diamines having two or more alkylene glycol units in the main chain described in paragraphs 0032 to 0034 of International Publication No. 2017/038598 are preferably used.
- R 111 is preferably represented by -Ar-L-Ar- from the viewpoint of the flexibility of the resulting organic film.
- Ar is each independently an aromatic group
- L is an aliphatic hydrocarbon group having 1 to 10 carbon atoms which may be substituted with a fluorine atom, -O-, -CO-, -S- , —SO 2 — or —NHCO—, or a group consisting of a combination of two or more of the above.
- Ar is preferably a phenylene group
- L is preferably an aliphatic hydrocarbon group having 1 or 2 carbon atoms which may be substituted with a fluorine atom, -O-, -CO-, -S- or -SO 2 - .
- the aliphatic hydrocarbon group here is preferably an alkylene group.
- R 111 is preferably a divalent organic group represented by the following formula (51) or (61).
- a divalent organic group represented by Formula (61) is more preferable.
- Equation (51) In formula (51), R 50 to R 57 are each independently a hydrogen atom, a fluorine atom or a monovalent organic group, and at least one of R 50 to R 57 is a fluorine atom, a methyl group or a trifluoro It is a methyl group, and each * independently represents a binding site to the nitrogen atom in formula (2).
- the monovalent organic groups represented by R 50 to R 57 include unsubstituted alkyl groups having 1 to 10 carbon atoms (preferably 1 to 6 carbon atoms), A fluorinated alkyl group and the like can be mentioned.
- R 58 and R 59 are each independently a fluorine atom, a methyl group, or a trifluoromethyl group, and * is each independently a binding site to the nitrogen atom in formula (2) show.
- Diamines that give the structure of formula (51) or (61) include 2,2′-dimethylbenzidine, 2,2′-bis(trifluoromethyl)-4,4′-diaminobiphenyl, 2,2′-bis (Fluoro)-4,4'-diaminobiphenyl, 4,4'-diaminooctafluorobiphenyl and the like. These may be used alone or in combination of two or more.
- R 115 in formula (2) represents a tetravalent organic group.
- a tetravalent organic group containing an aromatic ring is preferable, and a group represented by the following formula (5) or (6) is more preferable.
- each * independently represents a binding site to another structure.
- R 112 is a single bond or a divalent linking group, a single bond, or an aliphatic hydrocarbon group having 1 to 10 carbon atoms which may be substituted with a fluorine atom, —O—, -CO-, -S-, -SO 2 -, and -NHCO-, and preferably a group selected from a combination thereof, having 1 to 1 carbon atoms optionally substituted by a single bond or a fluorine atom 3 alkylene group, -O-, -CO-, -S- and -SO 2 -, and -CH 2 -, -C(CF 3 ) 2 -, -C( It is more preferably a divalent group selected from the group consisting of CH 3 ) 2 -, -O-, -CO-, -S- and -SO 2 -.
- R 115 specifically includes a tetracarboxylic acid residue remaining after removal of an anhydride group from a tetracarboxylic dianhydride.
- the polyimide precursor may contain only one type of tetracarboxylic dianhydride residue, or may contain two or more types thereof, as the structure corresponding to R115 .
- the tetracarboxylic dianhydride is preferably represented by the following formula (O).
- R 115 represents a tetravalent organic group.
- the preferred range of R 115 is synonymous with R 115 in formula (2), and the preferred range is also the same.
- tetracarboxylic dianhydrides include pyromellitic dianhydride (PMDA), 3,3′,4,4′-biphenyltetracarboxylic dianhydride, 3,3′,4,4′- Diphenyl sulfide tetracarboxylic dianhydride, 3,3′,4,4′-diphenylsulfonetetracarboxylic dianhydride, 3,3′,4,4′-benzophenonetetracarboxylic dianhydride, 3,3′ ,4,4′-diphenylmethanetetracarboxylic dianhydride, 2,2′,3,3′-diphenylmethanetetracarboxylic dianhydride, 2,3,3′,4′-biphenyltetracarboxylic dianhydride, 2,3,3′,4′-benzophenonetetracarboxylic dianhydride, 4,4′-oxydiphthalic dianhydride,
- tetracarboxylic dianhydrides (DAA-1) to (DAA-5) described in paragraph 0038 of WO 2017/038598 are also preferred examples.
- R 111 and R 115 has an OH group. More specifically, R 111 includes residues of bisaminophenol derivatives.
- R 113 and R 114 in formula (2) each independently represent a hydrogen atom or a monovalent organic group.
- the monovalent organic group preferably includes a linear or branched alkyl group, a cyclic alkyl group, an aromatic group, or a polyalkyleneoxy group.
- At least one of R 113 and R 114 preferably contains a polymerizable group, more preferably both contain a polymerizable group. It is also preferred that at least one of R 113 and R 114 contains two or more polymerizable groups.
- the polymerizable group is a group capable of undergoing a cross-linking reaction by the action of heat, radicals, or the like, and is preferably a radically polymerizable group.
- the polymerizable group examples include a group having an ethylenically unsaturated bond, an alkoxymethyl group, a hydroxymethyl group, an acyloxymethyl group, an epoxy group, an oxetanyl group, a benzoxazolyl group, a blocked isocyanate group, and an amino group. be done.
- a group having an ethylenically unsaturated bond is preferred.
- Groups having an ethylenically unsaturated bond include a vinyl group, an allyl group, an isoallyl group, a 2-methylallyl group, a group having an aromatic ring directly bonded to a vinyl group (e.g., a vinylphenyl group), and a (meth)acrylamide group.
- a (meth)acryloyloxy group a group represented by the following formula (III), and the like, and a group represented by the following formula (III) is preferable.
- R 200 represents a hydrogen atom, a methyl group, an ethyl group or a methylol group, preferably a hydrogen atom or a methyl group.
- * represents a binding site with another structure.
- R 201 represents an alkylene group having 2 to 12 carbon atoms, —CH 2 CH(OH)CH 2 —, a cycloalkylene group or a polyalkyleneoxy group.
- R 201 examples include ethylene, propylene, trimethylene, tetramethylene, pentamethylene, hexamethylene, octamethylene, alkylene groups such as dodecamethylene, 1,2-butanediyl, 1, 3-butanediyl group, —CH 2 CH(OH)CH 2 —, polyalkyleneoxy group, ethylene group, alkylene group such as propylene group, —CH 2 CH(OH)CH 2 —, cyclohexyl group, polyalkylene An oxy group is more preferred, and an alkylene group such as an ethylene group, a propylene group, or a polyalkyleneoxy group is even more preferred.
- alkylene groups such as dodecamethylene, 1,2-butanediyl, 1, 3-butanediyl group, —CH 2 CH(OH)CH 2 —, polyalkyleneoxy group, ethylene group, alkylene group such as propylene group, —CH 2 CH(OH)CH 2
- a polyalkyleneoxy group refers to a group in which two or more alkyleneoxy groups are directly bonded.
- the alkylene groups in the plurality of alkyleneoxy groups contained in the polyalkyleneoxy group may be the same or different.
- the arrangement of the alkyleneoxy groups in the polyalkyleneoxy group may be a random arrangement or a block arrangement. Alternatively, it may be arranged in a pattern such as an alternating pattern.
- the number of carbon atoms in the alkylene group (including the number of carbon atoms in the substituent when the alkylene group has a substituent) is preferably 2 or more, more preferably 2 to 10, and 2 to 6.
- the said alkylene group may have a substituent.
- Preferred substituents include alkyl groups, aryl groups, and halogen atoms.
- the number of alkyleneoxy groups contained in the polyalkyleneoxy group is preferably 2 to 20, more preferably 2 to 10, and even more preferably 2 to 6.
- a group to which an oxy group is bonded is preferable, a polyethyleneoxy group or a polypropyleneoxy group is more preferable, and a polyethyleneoxy group is still more preferable.
- the ethyleneoxy groups and the propyleneoxy groups may be arranged randomly, or may be arranged to form blocks. , may be arranged in a pattern such as alternately. Preferred embodiments of the number of repetitions of ethyleneoxy groups and the like in these groups are as described above.
- the polyimide precursor when R 113 is a hydrogen atom, or when R 114 is a hydrogen atom, the polyimide precursor may form a tertiary amine compound having an ethylenically unsaturated bond and a counter salt. good.
- tertiary amine compounds having ethylenically unsaturated bonds include N,N-dimethylaminopropyl methacrylate.
- the polyimide precursor preferably has a fluorine atom in its structure.
- the content of fluorine atoms in the polyimide precursor is preferably 10% by mass or more, and preferably 20% by mass or less.
- the polyimide precursor may be copolymerized with an aliphatic group having a siloxane structure.
- an aliphatic group having a siloxane structure there is an embodiment using bis(3-aminopropyl)tetramethyldisiloxane, bis(p-aminophenyl)octamethylpentasiloxane, or the like as the diamine.
- the repeating unit represented by formula (2) is preferably a repeating unit represented by formula (2-A). That is, at least one polyimide precursor used in the present invention is preferably a precursor having a repeating unit represented by formula (2-A). By including the repeating unit represented by the formula (2-A) in the polyimide precursor, it becomes possible to further widen the width of the exposure latitude.
- a 1 and A 2 represent an oxygen atom
- R 111 and R 112 each independently represent a divalent organic group
- R 113 and R 114 each independently represents a hydrogen atom or a monovalent organic group
- at least one of R 113 and R 114 is a group containing a polymerizable group, and both are preferably groups containing a polymerizable group.
- a 1 , A 2 , R 111 , R 113 and R 114 are each independently synonymous with A 1 , A 2 , R 111 , R 113 and R 114 in formula (2), and preferred ranges are also the same.
- R 112 has the same definition as R 112 in formula (5), and the preferred range is also the same.
- the polyimide precursor may contain one type of repeating unit represented by formula (2), but may contain two or more types. It may also contain structural isomers of the repeating unit represented by formula (2). It goes without saying that the polyimide precursor may also contain other types of repeating units in addition to the repeating units of formula (2) above.
- the content of the repeating unit represented by formula (2) is 50 mol% or more of the total repeating units.
- the total content is more preferably 70 mol % or more, still more preferably 90 mol % or more, and particularly preferably more than 90 mol %.
- the upper limit of the total content is not particularly limited, and all repeating units in the polyimide precursor excluding terminals may be repeating units represented by formula (2).
- the weight average molecular weight (Mw) of the polyimide precursor is preferably 5,000 to 100,000, more preferably 10,000 to 50,000, still more preferably 15,000 to 40,000. Also, the number average molecular weight (Mn) is preferably 2,000 to 40,000, more preferably 3,000 to 30,000, still more preferably 4,000 to 20,000.
- the polyimide precursor preferably has a molecular weight distribution of 1.5 or more, more preferably 1.8 or more, and even more preferably 2.0 or more. Although the upper limit of the polyimide precursor's molecular weight dispersity is not particularly defined, it is preferably 7.0 or less, more preferably 6.5 or less, and even more preferably 6.0 or less.
- the molecular weight dispersity is a value calculated by weight average molecular weight/number average molecular weight.
- the weight average molecular weight, number average molecular weight, and degree of dispersion of at least one polyimide precursor are preferably within the above ranges. It is also preferable that the weight-average molecular weight, the number-average molecular weight, and the degree of dispersion calculated from the plurality of types of polyimide precursors as one resin are within the ranges described above.
- the polyimide used in the present invention may be an alkali-soluble polyimide or a polyimide soluble in a developer containing an organic solvent as a main component.
- the alkali-soluble polyimide refers to a polyimide that dissolves at 23° C. by 0.1 g or more in 100 g of a 2.38% by mass tetramethylammonium aqueous solution, and from the viewpoint of pattern formation, 0.5 g or more. It is preferably a polyimide that dissolves, and more preferably a polyimide that dissolves 1.0 g or more. Although the upper limit of the dissolved amount is not particularly limited, it is preferably 100 g or less.
- the polyimide is preferably a polyimide having a plurality of imide structures in its main chain from the viewpoint of the film strength and insulating properties of the resulting organic film.
- the term "main chain” refers to the relatively longest linking chain in the molecule of the polymer compound that constitutes the resin, and the term “side chain” refers to the other linking chain.
- the polyimide preferably has a fluorine atom.
- a fluorine atom is preferably included in, for example, R132 in a repeating unit represented by formula (4) described later or R131 in a repeating unit represented by formula (4) described later. ) or R 131 in a repeating unit represented by formula (4) described later as a fluorinated alkyl group.
- the amount of fluorine atoms relative to the total mass of polyimide is preferably 5% by mass or more and preferably 20% by mass or less.
- the polyimide preferably has a silicon atom.
- a silicon atom for example, is preferably contained in R 131 in a repeating unit represented by formula (4) described later, and R 131 in a repeating unit represented by formula (4) described later is an organically modified (poly ) is more preferably contained as a siloxane structure.
- the silicon atom or the organically modified (poly)siloxane structure may be contained in the side chain of the polyimide, but is preferably contained in the main chain of the polyimide.
- the amount of silicon atoms relative to the total mass of polyimide is preferably 1% by mass or more, and more preferably 20% by mass or less.
- the polyimide preferably has an ethylenically unsaturated bond.
- the polyimide may have an ethylenically unsaturated bond at the end of its main chain or in a side chain, preferably in a side chain.
- the ethylenically unsaturated bond preferably has radical polymerizability.
- the ethylenically unsaturated bond is preferably contained in R 132 in a repeating unit represented by the formula (4) described later, or R 131 in a repeating unit represented by the formula (4) described later.
- the ethylenically unsaturated bond is preferably contained in R 131 in the repeating unit represented by formula (4) described later, and ethylene is contained in R 131 in the repeating unit represented by formula (4) described later It is more preferably included as a group having a polyunsaturated bond.
- the group having an ethylenically unsaturated bond includes a group having an optionally substituted vinyl group directly bonded to an aromatic ring such as a vinyl group, an allyl group, a vinylphenyl group, a (meth)acrylamide group, a (meth) Examples include an acryloyloxy group and a group represented by the following formula (IV).
- R 20 represents a hydrogen atom, a methyl group, an ethyl group or a methylol group, preferably a hydrogen atom or a methyl group.
- R 21 is an alkylene group having 2 to 12 carbon atoms, —O—CH 2 CH(OH)CH 2 —, —C( ⁇ O)O—, —O(C ⁇ O)NH— , a (poly)alkyleneoxy group having 2 to 30 carbon atoms (the number of carbon atoms in the alkylene group is preferably 2 to 12, more preferably 2 to 6, and particularly preferably 2 or 3; the number of repetitions is preferably 1 to 12, 1 to 6 are more preferable, and 1 to 3 are particularly preferable), or a group in which two or more of these are combined.
- the alkylene group having 2 to 12 carbon atoms may be a linear, branched, cyclic, or a combination of these alkylene groups.
- an alkylene group having 2 to 8 carbon atoms is preferable, and an alkylene group having 2 to 4 carbon atoms is more preferable.
- R 21 is preferably a group represented by any one of the following formulas (R1) to (R3), more preferably a group represented by formula (R1).
- L represents a single bond, an alkylene group having 2 to 12 carbon atoms, a (poly)alkyleneoxy group having 2 to 30 carbon atoms, or a group in which two or more of these are combined
- X represents an oxygen atom or a sulfur atom
- * represents a bonding site with another structure
- ⁇ represents a bonding site with the oxygen atom to which R 21 in formula (IV) bonds.
- a preferred embodiment of an alkylene group having 2 to 12 carbon atoms or a (poly)alkyleneoxy group having 2 to 30 carbon atoms in L is the above-mentioned R 21 having 2 to 12 carbon atoms. It is the same as the preferred embodiment of the 12 alkylene group or the (poly)alkyleneoxy group having 2 to 30 carbon atoms.
- X is preferably an oxygen atom.
- * has the same meaning as * in formula (IV), and preferred embodiments are also the same.
- the structure represented by formula (R1) is, for example, a polyimide having a hydroxy group such as a phenolic hydroxy group, and a compound having an isocyanato group and an ethylenically unsaturated bond (e.g., 2-isocyanatoethyl methacrylate, etc.). Obtained by reaction.
- the structure represented by formula (R2) can be obtained, for example, by reacting a polyimide having a carboxy group with a compound having a hydroxy group and an ethylenically unsaturated bond (eg, 2-hydroxyethyl methacrylate, etc.).
- the structure represented by formula (R3) can be obtained, for example, by reacting a polyimide having a hydroxy group such as a phenolic hydroxy group with a compound having a glycidyl group and an ethylenically unsaturated bond (e.g., glycidyl methacrylate, etc.) can get.
- a polyimide having a hydroxy group such as a phenolic hydroxy group
- a compound having a glycidyl group and an ethylenically unsaturated bond e.g., glycidyl methacrylate, etc.
- * represents a binding site with another structure, preferably a binding site with the main chain of polyimide.
- the amount of ethylenically unsaturated bonds relative to the total mass of the polyimide is preferably 0.0001-0.1 mol/g, more preferably 0.0005-0.05 mol/g.
- Polyimide may have a polymerizable group other than the group having an ethylenically unsaturated bond.
- Polymerizable groups other than groups having an ethylenically unsaturated bond include cyclic ether groups such as an epoxy group and an oxetanyl group, alkoxymethyl groups such as a methoxymethyl group, and methylol groups.
- a polymerizable group other than a group having an ethylenically unsaturated bond is preferably included, for example, in R 131 in a repeating unit represented by formula (4) described below.
- the amount of the polymerizable group other than the group having an ethylenically unsaturated bond with respect to the total mass of the polyimide is preferably 0.0001 to 0.1 mol / g, preferably 0.001 to 0.05 mol / g. more preferred.
- the acid value of polyimide is preferably 30 mgKOH/g or more, more preferably 50 mgKOH/g or more, and more preferably 70 mgKOH/g or more, from the viewpoint of improving developability. is more preferable. Also, the acid value is preferably 500 mgKOH/g or less, more preferably 400 mgKOH/g or less, and even more preferably 200 mgKOH/g or less. Further, when the polyimide is subjected to development using a developer containing an organic solvent as a main component (for example, "solvent development” described later), the acid value of the polyimide is preferably 1 to 35 mgKOH/g, and 2 to 30 mgKOH.
- the acid value is measured by a known method, for example, by the method described in JIS K 0070:1992.
- the acid group contained in the polyimide preferably has a pKa of 0 to 10, more preferably 3 to 8, from the viewpoint of both storage stability and developability.
- the pKa is expressed by the negative common logarithm pKa of the equilibrium constant Ka.
- pKa is a value calculated by ACD/ChemSketch (registered trademark).
- the values listed in the Chemical Society of Japan, Revised 5th Edition, Basics of Chemistry may be referred to.
- the acid group is a polyvalent acid such as phosphoric acid
- the pKa is the first dissociation constant.
- the polyimide preferably contains at least one selected from the group consisting of a carboxy group and a phenolic hydroxy group, more preferably a phenolic hydroxy group.
- the polyimide preferably has a phenolic hydroxy group from the viewpoint of making the development speed with an alkaline developer appropriate.
- the polyimide may have a phenolic hydroxy group at the end of the main chain or in the side chain.
- a phenolic hydroxy group is preferably contained in, for example, R 132 in a repeating unit represented by formula (4) described later or R 131 in a repeating unit represented by formula (4) described later.
- the amount of phenolic hydroxy groups relative to the total weight of the polyimide is preferably 0.1-30 mol/g, more preferably 1-20 mol/g.
- the polyimide used in the present invention is not particularly limited as long as it is a polymer compound having an imide structure, but it preferably contains a repeating unit represented by the following formula (4).
- R 131 represents a divalent organic group and R 132 represents a tetravalent organic group.
- the polymerizable group may be located on at least one of R 131 and R 132 , and the terminal of the polyimide as shown in the following formula (4-1) or (4-2) may be located in Formula (4-1)
- R 133 is a polymerizable group, and other groups are the same as in formula (4).
- Formula (4-2) At least one of R 134 and R 135 is a polymerizable group, and when it is not a polymerizable group, it is an organic group, and the other groups are as defined in formula (4).
- R 131 represents a divalent organic group.
- Examples of the divalent organic group are the same as those for R 111 in formula (2), and the preferred range is also the same.
- R 131 also includes a diamine residue remaining after removal of the amino group of the diamine.
- Diamines include aliphatic, cycloaliphatic or aromatic diamines.
- a specific example is the example of R 111 in formula (2) of the polyimide precursor.
- R 131 is preferably a diamine residue having at least two alkylene glycol units in its main chain from the viewpoint of more effectively suppressing warping during baking. More preferably, it is a diamine residue containing two or more ethylene glycol chains, propylene glycol chains, or both in one molecule, and more preferably the above diamine, which does not contain an aromatic ring. is.
- Diamines containing two or more ethylene glycol chains, propylene glycol chains, or both in one molecule include Jeffamine (registered trademark) KH-511, ED-600, ED-900, ED-2003, and EDR. -148, EDR-176, D-200, D-400, D-2000, D-4000 (trade names, manufactured by HUNTSMAN Co., Ltd.), 1-(2-(2-(2-aminopropoxy)ethoxy) propoxy)propan-2-amine, 1-(1-(1-(2-aminopropoxy)propan-2-yl)oxy)propan-2-amine, and the like.
- R 132 represents a tetravalent organic group.
- examples of the tetravalent organic group are the same as those for R 115 in formula (2), and the preferred range is also the same.
- four bonds of a tetravalent organic group exemplified as R 115 combine with four —C( ⁇ O)— moieties in the above formula (4) to form a condensed ring.
- R 132 includes, for example, a tetracarboxylic acid residue remaining after removal of an anhydride group from a tetracarboxylic dianhydride.
- a specific example is the example of R 115 in formula (2) of the polyimide precursor.
- R 132 is preferably an aromatic diamine residue having 1 to 4 aromatic rings.
- R 131 and R 132 has an OH group. More specifically, R 131 is 2,2-bis(3-hydroxy-4-aminophenyl)propane, 2,2-bis(3-hydroxy-4-aminophenyl)hexafluoropropane, 2,2- Bis(3-amino-4-hydroxyphenyl)propane, 2,2-bis(3-amino-4-hydroxyphenyl)hexafluoropropane, and the above (DA-1) to (DA-18) are preferred examples. and more preferred examples of R 132 are the above (DAA-1) to (DAA-5).
- the polyimide preferably has a fluorine atom in its structure.
- the content of fluorine atoms in the polyimide is preferably 10% by mass or more, and preferably 20% by mass or less.
- the polyimide may be copolymerized with an aliphatic group having a siloxane structure.
- the diamine component include bis(3-aminopropyl)tetramethyldisiloxane and bis(p-aminophenyl)octamethylpentasiloxane.
- the main chain end of the polyimide is blocked with a terminal blocking agent such as monoamine, acid anhydride, monocarboxylic acid, monoacid chloride compound, monoactive ester compound. preferably.
- monoamines examples include aniline, 2-ethynylaniline, 3-ethynylaniline, 4-ethynylaniline, 5-amino-8-hydroxyquinoline, 1-hydroxy-7 -aminonaphthalene, 1-hydroxy-6-aminonaphthalene, 1-hydroxy-5-aminonaphthalene, 1-hydroxy-4-aminonaphthalene, 2-hydroxy-7-aminonaphthalene, 2-hydroxy-6-aminonaphthalene, 2 -hydroxy-5-aminonaphthalene, 1-carboxy-7-aminonaphthalene, 1-carboxy-6-aminonaphthalene, 1-carboxy-5-aminonaphthalene, 2-carboxy-7-aminonaphthalene, 2-carboxy-6- Aminonaphthalene, 2-carboxy-5-aminonaphthalene, 2-aminobenzoic acid, 3-aminobenzoic acid, 4-aminobenzoic acid, 2-aminobenzoic acid
- the imidization rate (also referred to as "ring closure rate") of the polyimide is preferably 70% or more, more preferably 80% or more, from the viewpoint of the film strength, insulation properties, etc. of the resulting organic film. More preferably, it is 90% or more.
- the upper limit of the imidization rate is not particularly limited, and may be 100% or less.
- the imidization rate is measured, for example, by the method described below. The infrared absorption spectrum of the polyimide is measured, and the peak intensity P1 near 1377 cm ⁇ 1 , which is the absorption peak derived from the imide structure, is obtained. Next, after heat-treating the polyimide at 350° C.
- the polyimide may contain repeating units represented by the above formula (4) that all contain one type of R 131 or R 132 , and the above formula ( 4) may contain a repeating unit. Moreover, the polyimide may contain other types of repeating units in addition to the repeating units represented by the above formula (4). Other types of repeating units include, for example, repeating units represented by formula (2) above.
- polyimide for example, a method of reacting a tetracarboxylic dianhydride and a diamine (partially replaced with a monoamine terminal blocker) at a low temperature, a method of reacting a tetracarboxylic dianhydride (partially with an acid anhydride) at a low temperature a monoacid chloride compound or a monoactive ester compound) and a diamine, a diester is obtained by a tetracarboxylic dianhydride and an alcohol, and then a diamine (a part of which is a monoamine A method of reacting in the presence of a condensing agent) with a condensing agent, a diester is obtained by tetracarboxylic acid dianhydride and alcohol, then the remaining dicarboxylic acid is acid chloride, diamine (part of which is a monoamine Using a method such as a method of reacting with a terminal blocking agent) to obtain a polyimide precursor
- the weight average molecular weight (Mw) of the polyimide is preferably 5,000 to 100,000, more preferably 10,000 to 50,000, still more preferably 15,000 to 40,000. By setting the weight average molecular weight to 5,000 or more, the folding resistance of the cured film can be improved. A weight-average molecular weight of 15,000 or more is particularly preferable in order to obtain an organic film having excellent mechanical properties (e.g., elongation at break). Also, the number average molecular weight (Mn) of the polyimide is preferably 2,000 to 40,000, more preferably 3,000 to 30,000, still more preferably 4,000 to 20,000. The polyimide has a molecular weight distribution of preferably 1.5 or more, more preferably 1.8 or more, and even more preferably 2.0 or more.
- the upper limit of the polyimide molecular weight dispersion is not particularly defined, it is preferably 7.0 or less, more preferably 6.5 or less, and even more preferably 6.0 or less.
- the weight-average molecular weight, number-average molecular weight, and degree of dispersion of at least one type of polyimide are preferably within the above ranges. It is also preferable that the weight-average molecular weight, the number-average molecular weight, and the degree of dispersion calculated using the above plural kinds of polyimides as one resin are within the ranges described above.
- polybenzoxazole precursor Although the structure of the polybenzoxazole precursor used in the present invention is not particularly defined, it preferably contains a repeating unit represented by the following formula (3).
- R 121 represents a divalent organic group
- R 122 represents a tetravalent organic group
- R 123 and R 124 each independently represent a hydrogen atom or a monovalent organic group. show.
- R 123 and R 124 each have the same meaning as R 113 in formula (2), and the preferred ranges are also the same. That is, at least one is preferably a polymerizable group.
- R 121 represents a divalent organic group.
- the divalent organic group a group containing at least one of an aliphatic group and an aromatic group is preferred.
- the aliphatic group a linear aliphatic group is preferred.
- R 121 is preferably a dicarboxylic acid residue. Only one type of dicarboxylic acid residue may be used, or two or more types may be used.
- a dicarboxylic acid residue containing an aliphatic group and a dicarboxylic acid residue containing an aromatic group are preferable, and a dicarboxylic acid residue containing an aromatic group is more preferable.
- the dicarboxylic acid containing an aliphatic group is preferably a dicarboxylic acid containing a linear or branched (preferably linear) aliphatic group, a linear or branched (preferably linear) aliphatic group and two -COOH A dicarboxylic acid consisting of is more preferred.
- the number of carbon atoms in the linear or branched (preferably linear) aliphatic group is preferably 2 to 30, more preferably 2 to 25, even more preferably 3 to 20, and 4 to 15 is more preferred, and 5-10 is particularly preferred.
- the linear aliphatic group is preferably an alkylene group.
- Dicarboxylic acids containing linear aliphatic groups include malonic acid, dimethylmalonic acid, ethylmalonic acid, isopropylmalonic acid, di-n-butylmalonic acid, succinic acid, tetrafluorosuccinic acid, methylsuccinic acid, 2, 2-dimethylsuccinic acid, 2,3-dimethylsuccinic acid, dimethylmethylsuccinic acid, glutaric acid, hexafluoroglutaric acid, 2-methylglutaric acid, 3-methylglutaric acid, 2,2-dimethylglutaric acid, 3,3-dimethylglutaric acid, 3-ethyl-3-methylglutaric acid, adipic acid, octafluoroadipic acid, 3-methyladipic acid, pimelic acid, 2,2,6,6-tetramethylpimelic acid, suberin acid, dodecanedioic acid, azelaic acid, sebacic acid, hexadecanedi
- Z is a hydrocarbon group having 1 to 6 carbon atoms, and n is an integer of 1 to 6.
- the dicarboxylic acid containing an aromatic group the following dicarboxylic acid having an aromatic group is preferable, and the following dicarboxylic acid consisting of only a group having an aromatic group and two -COOH is more preferable.
- A is -CH 2 -, -O-, -S-, -SO 2 -, -CO-, -NHCO-, -C(CF 3 ) 2 -, and -C(CH 3 ) 2 - represents a divalent group selected from the group consisting of * independently represents a binding site to another structure.
- dicarboxylic acids containing aromatic groups include 4,4'-carbonyl dibenzoic acid, 4,4'-dicarboxydiphenyl ether, and terephthalic acid.
- R 122 represents a tetravalent organic group.
- the tetravalent organic group has the same meaning as R 115 in the above formula (2), and the preferred range is also the same.
- R 122 is also preferably a group derived from a bisaminophenol derivative.
- bisaminophenol derivatives having the following aromatic groups are preferred.
- X 1 represents -O-, -S-, -C(CF 3 ) 2 -, -CH 2 -, -SO 2 -, -NHCO-, and * and # respectively represent other structures and represents the binding site of R represents a hydrogen atom or a monovalent substituent, preferably a hydrogen atom or a hydrocarbon group, more preferably a hydrogen atom or an alkyl group.
- R 122 is also preferably a structure represented by the above formula.
- any two of the total four * and # are binding sites with the nitrogen atom to which R 122 in formula (3) binds, and Another two are preferably bonding sites with the oxygen atom to which R 122 in formula (3) is bonded, and two * are bonding sites with the oxygen atom to which R 122 in formula (3) is bonded. and two #s are binding sites to the nitrogen atom to which R 122 in formula (3) binds, or two * are binding sites to the nitrogen atom to which R 122 in formula (3) binds and two #s are more preferably a binding site to the oxygen atom to which R 122 in formula (3) binds, and two * are the oxygen to which R 122 in formula (3) binds. More preferably, it is a bonding site with an atom and two #s are bonding sites with a nitrogen atom to which R 122 in formula (3) is bonded.
- the bisaminophenol derivative is also preferably a compound represented by formula (As).
- R 1 is a hydrogen atom, alkylene, substituted alkylene, -O-, -S-, -SO 2 -, -CO-, -NHCO-, a single bond, or the following formula (A- It is an organic group selected from the group of sc).
- R2 is a hydrogen atom, an alkyl group, an alkoxy group, an acyloxy group, or a cyclic alkyl group, and may be the same or different.
- R3 is a hydrogen atom , a linear or branched alkyl group, an alkoxy group, an acyloxy group, or a cyclic alkyl group, and may be the same or different.
- R 1 is alkylene or substituted alkylene.
- alkylene and substituted alkylene for R 1 include linear or branched alkyl groups having 1 to 8 carbon atoms, among which —CH 2 — and —CH(CH 3 ) -, -C(CH 3 ) 2 - have sufficient solubility in solvents while maintaining the effect of high i-line transparency and high cyclization rate when cured at low temperature. It is more preferable in that a polybenzoxazole precursor having excellent properties can be obtained.
- the polybenzoxazole precursor may also contain other types of repeating units in addition to the repeating units of formula (3) above.
- the polybenzoxazole precursor preferably contains a diamine residue represented by the following formula (SL) as another type of repeating unit in that warping due to ring closure can be suppressed.
- Z has an a structure and a b structure
- R 1s is a hydrogen atom or a hydrocarbon group having 1 to 10 carbon atoms
- R 2s is a hydrocarbon group having 1 to 10 carbon atoms.
- At least one of R 3s , R 4s , R 5s and R 6s is an aromatic group, and the rest are hydrogen atoms or organic groups having 1 to 30 carbon atoms, which may be the same or different.
- Polymerization of a structure and b structure may be block polymerization or random polymerization.
- the mol % of the Z portion is 5 to 95 mol % for the a structure, 95 to 5 mol % for the b structure, and 100 mol % for a+b.
- preferred Z include those in which R 5s and R 6s in the b structure are phenyl groups.
- the molecular weight of the structure represented by formula (SL) is preferably 400 to 4,000, more preferably 500 to 3,000.
- tetracarboxylic acid residues include those of R 115 in formula (2).
- the weight average molecular weight (Mw) of the polybenzoxazole precursor is, for example, preferably 18,000 to 30,000, more preferably 20,000 to 29,000, still more preferably 22,000 to 28, 000. Also, the number average molecular weight (Mn) is preferably 7,200 to 14,000, more preferably 8,000 to 12,000, still more preferably 9,200 to 11,200.
- the molecular weight dispersity of the polybenzoxazole precursor is preferably 1.4 or more, more preferably 1.5 or more, and even more preferably 1.6 or more.
- the upper limit of the molecular weight dispersity of the polybenzoxazole precursor is not particularly defined, for example, it is preferably 2.6 or less, more preferably 2.5 or less, further preferably 2.4 or less, and 2.3 or less. is more preferable, and 2.2 or less is even more preferable.
- the resin composition contains a plurality of types of polybenzoxazole precursors as specific resins
- the weight-average molecular weight, number-average molecular weight, and degree of dispersion of at least one type of polybenzoxazole precursor are within the above ranges. preferable. It is also preferable that the weight-average molecular weight, the number-average molecular weight, and the degree of dispersion calculated from the plurality of types of polybenzoxazole precursors as one resin are within the ranges described above.
- Polybenzoxazole is not particularly limited as long as it is a polymer compound having a benzoxazole ring, but it is preferably a compound represented by the following formula (X), and a compound represented by the following formula (X) and more preferably a compound having a polymerizable group.
- a radically polymerizable group is preferred.
- R 133 represents a divalent organic group and R 134 represents a tetravalent organic group.
- the polymerizable group may be located at least one of R 133 and R 134 , and polybenzoxazole as shown in the following formula (X-1) or formula (X-2) may be located at the end of the Formula (X-1)
- at least one of R 135 and R 136 is a polymerizable group, and when it is not a polymerizable group, it is an organic group, and the other groups are as defined in formula (X).
- the polymerizable group is synonymous with the polymerizable group described in the polymerizable group possessed by the polyimide precursor above.
- R 133 represents a divalent organic group.
- Divalent organic groups include aliphatic groups and aromatic groups.
- a specific example is the example of R 121 in formula (3) of the polybenzoxazole precursor. Preferred examples thereof are the same as those of R121 .
- R 134 represents a tetravalent organic group.
- Tetravalent organic groups include examples of R 122 in the polybenzoxazole precursor formula (3). Moreover, the preferred examples thereof are the same as those of R122 .
- four bonds of a tetravalent organic group exemplified as R 122 combine with the nitrogen atom and oxygen atom in the above formula (X) to form a condensed ring.
- R 134 when R 134 is the following organic group, it forms the structure below. In the structures below, each * represents a bonding site with a nitrogen atom or an oxygen atom in formula (X).
- Polybenzoxazole preferably has an oxazole conversion rate of 85% or more, more preferably 90% or more.
- the upper limit is not particularly limited, and may be 100%.
- the oxazolization rate is measured, for example, by the method described below.
- the infrared absorption spectrum of polybenzoxazole is measured, and the peak intensity Q1 near 1650 cm ⁇ 1 , which is the absorption peak derived from the amide structure of the precursor, is obtained.
- the polybenzoxazole may contain repeating units of the above formula (X) that all contain one type of R 131 or R 132 , or may contain repeating units of the above formula (X) that contain two or more different types of R 131 or R 132 . ) repeating units.
- the polybenzoxazole may also contain other types of repeating units in addition to the repeating units of formula (X) above.
- Polybenzoxazole is obtained by, for example, reacting a bisaminophenol derivative with a dicarboxylic acid containing R 133 or a compound selected from dicarboxylic acid dichlorides and dicarboxylic acid derivatives of the above dicarboxylic acid to obtain a polybenzoxazole precursor. , which is obtained by oxazolating it using a known oxazolating reaction method.
- a dicarboxylic acid an active ester type dicarboxylic acid derivative obtained by pre-reacting 1-hydroxy-1,2,3-benzotriazole or the like may be used in order to increase the reaction yield.
- the weight average molecular weight (Mw) of polybenzoxazole is preferably from 5,000 to 70,000, more preferably from 8,000 to 50,000, even more preferably from 10,000 to 30,000.
- the weight average molecular weight is particularly preferably 20,000 or more.
- the weight average molecular weight of at least one kind of polybenzoxazole is within the above range.
- the number average molecular weight (Mn) of polybenzoxazole is preferably 7,200 to 14,000, more preferably 8,000 to 12,000, still more preferably 9,200 to 11,200. be.
- the polybenzoxazole has a molecular weight dispersity of preferably 1.4 or more, more preferably 1.5 or more, and even more preferably 1.6 or more.
- the upper limit of the polybenzoxazole molecular weight dispersity is not particularly defined, for example, it is preferably 2.6 or less, more preferably 2.5 or less, further preferably 2.4 or less, and even more preferably 2.3 or less.
- 2.2 or less is even more preferable.
- the weight average molecular weight, number average molecular weight, and degree of dispersion of at least one type of polybenzoxazole are preferably within the above ranges. It is also preferable that the weight-average molecular weight, the number-average molecular weight, and the degree of dispersion calculated from the plurality of types of polybenzoxazole as one resin are within the ranges described above.
- the polyamideimide precursor preferably contains a repeating unit represented by the following formula (PAI-2).
- R 117 represents a trivalent organic group
- R 111 represents a divalent organic group
- a 2 represents an oxygen atom or —NH—
- R 113 represents a hydrogen atom or a monovalent represents an organic group.
- R 117 is a linear or branched aliphatic group, a cyclic aliphatic group, an aromatic group, a heteroaromatic group, or two
- the above-linked groups are exemplified, straight-chain aliphatic groups having 2 to 20 carbon atoms, branched aliphatic groups having 3 to 20 carbon atoms, cyclic aliphatic groups having 3 to 20 carbon atoms, and 6 to 20 carbon atoms.
- alkylene group is preferably an alkylene group having 1 to 20 carbon atoms, more preferably an alkylene group having 1 to 10 carbon atoms, and even more preferably an alkylene group having 1 to 4 carbon atoms.
- halogenated alkylene group a halogenated alkylene group having 1 to 20 carbon atoms is preferable, a halogenated alkylene group having 1 to 10 carbon atoms is more preferable, and a halogenated alkylene group having 1 to 4 carbon atoms is more preferable.
- the halogen atom in the halogenated alkylene group includes a fluorine atom, a chlorine atom, a bromine atom, an iodine atom and the like, and a fluorine atom is preferable.
- the above halogenated alkylene group may have hydrogen atoms, or all of the hydrogen atoms may be substituted with halogen atoms, but it is preferred that all of the hydrogen atoms be substituted with halogen atoms.
- preferred halogenated alkylene groups include (ditrifluoromethyl)methylene groups and the like.
- the arylene group is preferably a phenylene group or a naphthylene group, more preferably a phenylene group, and still more preferably a 1,3-phenylene group or a 1,4-phenylene group.
- R 117 is preferably derived from a tricarboxylic acid compound in which at least one carboxy group may be halogenated. Chlorination is preferable as the halogenation.
- a compound having three carboxy groups is called a tricarboxylic acid compound. Two of the three carboxy groups of the tricarboxylic acid compound may be anhydrided.
- the optionally halogenated tricarboxylic acid compound used in the production of the polyamideimide precursor include branched aliphatic, cyclic aliphatic or aromatic tricarboxylic acid compounds. Only one of these tricarboxylic acid compounds may be used, or two or more thereof may be used.
- the tricarboxylic acid compound includes a linear aliphatic group having 2 to 20 carbon atoms, a branched aliphatic group having 3 to 20 carbon atoms, a cyclic aliphatic group having 3 to 20 carbon atoms, and a Tricarboxylic acid compounds containing 6 to 20 aromatic groups or groups in which two or more of these are combined via a single bond or a linking group are preferred, and aromatic groups having 6 to 20 carbon atoms or carbon atoms via a single bond or linking group are preferred. More preferred are tricarboxylic acid compounds containing groups in which two or more aromatic groups of numbers 6 to 20 are combined.
- tricarboxylic acid compounds include 1,2,3-propanetricarboxylic acid, 1,3,5-pentanetricarboxylic acid, citric acid, trimellitic acid, 2,3,6-naphthalenetricarboxylic acid, and phthalic acid.
- (or phthalic anhydride) and benzoic acid are a single bond, —O—, —CH 2 —, —C(CH 3 ) 2 —, —C(CF 3 ) 2 —, —SO 2 — or a phenylene group
- Linked compounds and the like are included.
- These compounds may be compounds in which two carboxy groups are anhydrided (e.g., trimellitic anhydride), or compounds in which at least one carboxy group is halogenated (e.g., trimellitic anhydride chloride). There may be.
- R 111 , A 2 and R 113 have the same meanings as R 111 , A 2 and R 113 in formula (2) above, and preferred embodiments are also the same.
- Polyamideimide precursors may further comprise other repeating units.
- Other repeating units include repeating units represented by the above formula (2) and repeating units represented by the following formula (PAI-1).
- R 116 represents a divalent organic group and R 111 represents a divalent organic group.
- R 116 is a linear or branched aliphatic group, a cyclic aliphatic group, an aromatic group, a heteroaromatic group, or two
- the above-linked groups are exemplified, straight-chain aliphatic groups having 2 to 20 carbon atoms, branched aliphatic groups having 3 to 20 carbon atoms, cyclic aliphatic groups having 3 to 20 carbon atoms, and 6 to 20 carbon atoms.
- alkylene group is preferably an alkylene group having 1 to 20 carbon atoms, more preferably an alkylene group having 1 to 10 carbon atoms, and even more preferably an alkylene group having 1 to 4 carbon atoms.
- halogenated alkylene group a halogenated alkylene group having 1 to 20 carbon atoms is preferable, a halogenated alkylene group having 1 to 10 carbon atoms is more preferable, and a halogenated alkylene group having 1 to 4 carbon atoms is more preferable.
- the halogen atom in the halogenated alkylene group includes a fluorine atom, a chlorine atom, a bromine atom, an iodine atom and the like, and a fluorine atom is preferable.
- the above halogenated alkylene group may have hydrogen atoms, or all of the hydrogen atoms may be substituted with halogen atoms, but it is preferred that all of the hydrogen atoms be substituted with halogen atoms.
- preferred halogenated alkylene groups include a (ditrifluoromethyl)methylene group and the like.
- the arylene group is preferably a phenylene group or a naphthylene group, more preferably a phenylene group, and still more preferably a 1,3-phenylene group or a 1,4-phenylene group.
- R 116 is preferably derived from a dicarboxylic acid compound or a dicarboxylic acid dihalide compound.
- a compound having two carboxy groups is called a dicarboxylic acid compound
- a compound having two halogenated carboxy groups is called a dicarboxylic acid dihalide compound.
- the carboxy group in the dicarboxylic acid dihalide compound may be halogenated, but is preferably chlorinated, for example. That is, the dicarboxylic acid dihalide compound is preferably a dicarboxylic acid dichloride compound.
- the optionally halogenated dicarboxylic acid compound or dicarboxylic acid dihalide compound used in the production of the polyamideimide precursor includes linear or branched aliphatic, cyclic aliphatic or aromatic dicarboxylic acid compounds or dicarboxylic acids. Examples include acid dihalide compounds. One of these dicarboxylic acid compounds or dicarboxylic acid dihalide compounds may be used, or two or more thereof may be used.
- the dicarboxylic acid compound or dicarboxylic acid dihalide compound includes a linear aliphatic group having 2 to 20 carbon atoms, a branched aliphatic group having 3 to 20 carbon atoms, and a cyclic aliphatic group having 3 to 20 carbon atoms.
- a dicarboxylic acid compound or dicarboxylic acid dihalide compound containing a group, an aromatic group having 6 to 20 carbon atoms, or a group in which two or more of these are combined via a single bond or a linking group is preferable, and an aromatic group having 6 to 20 carbon atoms.
- dicarboxylic acid compounds include malonic acid, dimethylmalonic acid, ethylmalonic acid, isopropylmalonic acid, di-n-butylmalonic acid, succinic acid, tetrafluorosuccinic acid, methylsuccinic acid, 2,2- dimethylsuccinic acid, 2,3-dimethylsuccinic acid, dimethylmethylsuccinic acid, glutaric acid, hexafluoroglutaric acid, 2-methylglutaric acid, 3-methylglutaric acid, 2,2-dimethylglutaric acid, 3, 3-dimethylglutaric acid, 3-ethyl-3-methylglutaric acid, adipic acid, octafluoroadipic acid, 3-methyladipic acid, pimelic acid, 2,2,6,6-tetramethylpimelic acid, suberic acid, dodecanedioic acid, azelaic acid, sebacic acid, hexadecanedioic acid,
- R 111 has the same definition as R 111 in formula (2) above, and preferred embodiments are also the same.
- the polyamideimide precursor preferably has a fluorine atom in its structure.
- the content of fluorine atoms in the polyamideimide precursor is preferably 10% by mass or more, and preferably 20% by mass or less.
- the polyamideimide precursor may be copolymerized with an aliphatic group having a siloxane structure.
- the diamine component bis(3-aminopropyl)tetramethyldisiloxane, bis(p-aminophenyl)octamethylpentasiloxane, etc. are used.
- a repeating unit represented by the formula (PAI-2), a repeating unit represented by the formula (PAI-1), and a repeating unit represented by the formula (2) An aspect in which the total content of units is 50 mol % or more of all repeating units is exemplified.
- the total content is more preferably 70 mol % or more, still more preferably 90 mol % or more, and particularly preferably more than 90 mol %.
- the upper limit of the total content is not particularly limited, and all repeating units in the polyamideimide precursor excluding the terminal are repeating units represented by formula (PAI-2), represented by formula (PAI-1). It may be either a repeating unit or a repeating unit represented by formula (2).
- the total content of repeating units represented by formula (PAI-2) and repeating units represented by formula (PAI-1) is An embodiment in which it is 50 mol % or more of all repeating units is mentioned.
- the total content is more preferably 70 mol % or more, still more preferably 90 mol % or more, and particularly preferably more than 90 mol %.
- the upper limit of the total content is not particularly limited, and all repeating units in the polyamideimide precursor excluding the terminal are repeating units represented by formula (PAI-2), or represented by formula (PAI-1) may be any of the repeating units provided.
- the weight average molecular weight (Mw) of the polyamideimide precursor is preferably 2,000 to 500,000, more preferably 5,000 to 100,000, still more preferably 10,000 to 50,000. .
- the number average molecular weight (Mn) is preferably 800 to 250,000, more preferably 2,000 to 50,000, still more preferably 4,000 to 25,000.
- the polyamidoimide precursor preferably has a molecular weight distribution of 1.5 or more, more preferably 1.8 or more, and even more preferably 2.0 or more.
- the upper limit of the molecular weight dispersity of the polyamideimide precursor is not particularly defined, it is preferably 7.0 or less, more preferably 6.5 or less, and even more preferably 6.0 or less.
- the weight average molecular weight, number average molecular weight, and degree of dispersion of at least one type of polyamideimide precursor are preferably within the above ranges. It is also preferable that the weight-average molecular weight, the number-average molecular weight, and the degree of dispersion calculated from the plurality of types of polyamideimide precursors as one resin are within the ranges described above.
- the polyamideimide used in the present invention may be an alkali-soluble polyamideimide or a polyamideimide soluble in a developer containing an organic solvent as a main component.
- the alkali-soluble polyamideimide refers to a polyamideimide that dissolves at 23° C. in an amount of 0.1 g or more in 100 g of a 2.38 mass % tetramethylammonium aqueous solution.
- a polyamideimide that dissolves 5 g or more is preferable, and a polyamideimide that dissolves 1.0 g or more is more preferable.
- the upper limit of the dissolved amount is not particularly limited, it is preferably 100 g or less.
- the polyamideimide is preferably a polyamideimide having a plurality of amide bonds and a plurality of imide structures in the main chain from the viewpoint of the film strength and insulating properties of the organic film to be obtained.
- the polyamideimide preferably has a fluorine atom.
- a fluorine atom is preferably contained in, for example, R 117 or R 111 in a repeating unit represented by formula (PAI-3) described later, and is preferably contained in a repeating unit represented by formula (PAI-3) described later It is more preferably contained in R 117 or R 111 as a fluorinated alkyl group.
- the amount of fluorine atoms is preferably 5% by mass or more and preferably 20% by mass or less with respect to the total mass of polyamideimide.
- the polyamideimide may have an ethylenically unsaturated bond.
- the polyamideimide may have an ethylenically unsaturated bond at the end of the main chain or in a side chain, preferably in the side chain.
- the ethylenically unsaturated bond preferably has radical polymerizability.
- the ethylenically unsaturated bond is preferably contained in R 117 or R 111 in the repeating unit represented by formula (PAI-3) described later, and the repeating unit represented by formula (PAI-3) described later.
- R 117 or R 111 It is more preferably contained as a group having an ethylenically unsaturated bond in R 117 or R 111 in .
- Preferred embodiments of the group having an ethylenically unsaturated bond are the same as the preferred embodiments of the group having an ethylenically unsaturated bond in the polyimide described above.
- the amount of ethylenically unsaturated bonds relative to the total mass of polyamideimide is preferably 0.0001 to 0.1 mol/g, more preferably 0.001 to 0.05 mol/g.
- Polyamideimide may have a polymerizable group other than the ethylenically unsaturated bond.
- the polymerizable groups other than the ethylenically unsaturated bond in the polyamideimide include the same groups as the above polymerizable groups other than the ethylenically unsaturated bond in the polyimide.
- a polymerizable group other than an ethylenically unsaturated bond is preferably included in R 111 in a repeating unit represented by formula (PAI-3) described later, for example.
- the amount of polymerizable groups other than ethylenically unsaturated bonds relative to the total mass of polyamideimide is preferably 0.05 to 10 mol/g, more preferably 0.1 to 5 mol/g.
- the acid value of the polyamideimide is preferably 30 mgKOH/g or more, more preferably 50 mgKOH/g or more, more preferably 70 mgKOH/g, from the viewpoint of improving developability. g or more is more preferable. Moreover, the acid value is preferably 500 mgKOH/g or less, more preferably 400 mgKOH/g or less, and even more preferably 200 mgKOH/g or less.
- the acid value of the polyamideimide is preferably 2 to 35 mgKOH/g, 3 ⁇ 30 mg KOH/g is more preferred, and 5 to 20 mg KOH/g is even more preferred.
- the acid value is measured by a known method, for example, by the method described in JIS K 0070:1992.
- the acid group contained in the polyamideimide the same groups as the acid group in the polyimide described above can be mentioned, and the preferred embodiments are also the same.
- the polyamideimide preferably has a phenolic hydroxy group.
- Polyamideimide may have a phenolic hydroxy group at the end of the main chain or in the side chain.
- a phenolic hydroxy group is preferably included in, for example, R 117 or R 111 in a repeating unit represented by formula (PAI-3) described later.
- the amount of phenolic hydroxy groups relative to the total mass of polyamideimide is preferably 0.1 to 30 mol/g, more preferably 1 to 20 mol/g.
- the polyamideimide used in the present invention is not particularly limited as long as it is a polymer compound having an imide structure and an amide bond, but it preferably contains a repeating unit represented by the following formula (PAI-3).
- R 111 and R 117 have the same definitions as R 111 and R 117 in formula (PAI-2), and preferred embodiments are also the same.
- the polymerizable group may be located at least one of R 111 and R 117 , or may be located at the end of the polyamideimide.
- the main chain end of the polyamideimide is blocked with a terminal blocker such as a monoamine, an acid anhydride, a monocarboxylic acid, a monoacid chloride compound, or a monoactive ester compound.
- a terminal blocker such as a monoamine, an acid anhydride, a monocarboxylic acid, a monoacid chloride compound, or a monoactive ester compound.
- Preferred aspects of the terminal blocker are the same as the preferred aspects of the terminal blocker in the polyimide described above.
- the imidization rate (also referred to as "ring closure rate") of polyamideimide is preferably 70% or more, more preferably 80% or more, from the viewpoint of the film strength, insulating properties, etc. of the resulting organic film. , more preferably 90% or more.
- the upper limit of the imidization rate is not particularly limited, and may be 100% or less.
- the imidization ratio is measured by the same method as for the polyimide ring closure ratio described above.
- Polyamideimide may contain repeating units represented by the above formula (PAI-3), all of which contain one type of R 111 or R 117 , and two or more different types of R 131 or R 132 . It may contain a repeating unit represented by the above formula (PAI-3). Moreover, the polyamideimide may contain other types of repeating units in addition to the repeating units represented by the above formula (PAI-3). Other types of repeating units include repeating units represented by the above formula (PAI-1) or formula (PAI-2).
- Polyamideimide is, for example, a method of obtaining a polyamideimide precursor by a known method and completely imidizing it using a known imidization reaction method, or stopping the imidization reaction in the middle and partially imidizing the imide structure and a method of partially introducing an imide structure by blending a completely imidized polymer with its polyamideimide precursor.
- the weight average molecular weight (Mw) of polyamideimide is preferably 5,000 to 70,000, more preferably 8,000 to 50,000, even more preferably 10,000 to 30,000. By setting the weight average molecular weight to 5,000 or more, the folding resistance of the cured film can be improved. In order to obtain an organic film having excellent mechanical properties, the weight average molecular weight is particularly preferably 20,000 or more. Further, the number average molecular weight (Mn) of the polyamideimide is preferably 800 to 250,000, more preferably 2,000 to 50,000, still more preferably 4,000 to 25,000. .
- the polyamidoimide has a molecular weight distribution of preferably 1.5 or more, more preferably 1.8 or more, and even more preferably 2.0 or more.
- the upper limit of the polyamidoimide molecular weight dispersity is not particularly defined, it is preferably 7.0 or less, more preferably 6.5 or less, and even more preferably 6.0 or less.
- the weight average molecular weight, number average molecular weight, and degree of dispersion of at least one type of polyamideimide are preferably within the above ranges. It is also preferable that the weight-average molecular weight, number-average molecular weight, and degree of dispersion calculated from the plurality of types of polyamideimide as one resin are within the ranges described above.
- Polyimide precursors and the like for example, a method of reacting a tetracarboxylic dianhydride and a diamine at a low temperature, a method of reacting a tetracarboxylic dianhydride and a diamine at a low temperature to obtain a polyamic acid, a condensing agent or an alkylating agent A method of esterification using a tetracarboxylic dianhydride and an alcohol to obtain a diester, followed by a reaction with a diamine in the presence of a condensing agent, a method of reacting a tetracarboxylic dianhydride and an alcohol to obtain a diester, After that, the remaining dicarboxylic acid can be acid-halogenated using a halogenating agent and reacted with a diamine.
- the method of obtaining a diester from a tetracarboxylic dianhydride and an alcohol, then acid-halogenating the remaining dicarboxylic acid with a halogenating agent, and reacting it with a diamine is more preferable.
- the condensing agent include dicyclohexylcarbodiimide, diisopropylcarbodiimide, 1-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline, 1,1-carbonyldioxy-di-1,2,3-benzotriazole, N, N'-disuccinimidyl carbonate, trifluoroacetic anhydride and the like can be mentioned.
- alkylating agent examples include N,N-dimethylformamide dimethyl acetal, N,N-dimethylformamide diethyl acetal, N,N-dialkylformamide dialkyl acetal, trimethyl orthoformate and triethyl orthoformate.
- halogenating agent examples include thionyl chloride, oxalyl chloride, phosphorus oxychloride and the like.
- organic solvent In the method for producing a polyimide precursor or the like, it is preferable to use an organic solvent in the reaction. One type of organic solvent may be used, or two or more types may be used.
- the organic solvent can be appropriately determined depending on the raw material, but pyridine, diethylene glycol dimethyl ether (diglyme), N-methylpyrrolidone, N-ethylpyrrolidone, ethyl propionate, dimethylacetamide, dimethylformamide, tetrahydrofuran, ⁇ -butyrolactone, and the like. is exemplified.
- a basic compound In the method for producing a polyimide precursor or the like, it is preferable to add a basic compound during the reaction.
- One type of basic compound may be used, or two or more types may be used.
- the basic compound can be appropriately determined depending on the raw material, but triethylamine, diisopropylethylamine, pyridine, 1,8-diazabicyclo[5.4.0]undec-7-ene, N,N-dimethyl-4-amino Pyridine and the like are exemplified.
- terminal blocking agents include monoalcohols, phenols, thiols, thiophenols, monoamines, and the like. It is more preferable to use monoalcohols, phenols and monoamines from the viewpoint of their properties.
- Preferred monoalcohol compounds include primary alcohols such as methanol, ethanol, propanol, butanol, hexanol, octanol, dodecinol, benzyl alcohol, 2-phenylethanol, 2-methoxyethanol, 2-chloromethanol and furfuryl alcohol, and isopropanol. , 2-butanol, cyclohexyl alcohol, cyclopentanol and 1-methoxy-2-propanol, and tertiary alcohols such as t-butyl alcohol and adamantane alcohol.
- Preferable phenolic compounds include phenols such as phenol, methoxyphenol, methylphenol, naphthalene-1-ol, naphthalene-2-ol, and hydroxystyrene.
- Preferred monoamine compounds include aniline, 2-ethynylaniline, 3-ethynylaniline, 4-ethynylaniline, 5-amino-8-hydroxyquinoline, 1-hydroxy-7-aminonaphthalene, 1-hydroxy-6- aminonaphthalene, 1-hydroxy-5-aminonaphthalene, 1-hydroxy-4-aminonaphthalene, 2-hydroxy-7-aminonaphthalene, 2-hydroxy-6-aminonaphthalene, 2-hydroxy-5-aminonaphthalene, 1- Carboxy-7-aminonaphthalene, 1-carboxy-6-aminonaphthalene, 1-carboxy-5-aminonaphthalene, 2-carboxy-7-aminonaphthalene, 2-carboxy-6-amin
- Preferred capping agents for amino groups are carboxylic acid anhydrides, carboxylic acid chlorides, carboxylic acid bromide, sulfonic acid chlorides, sulfonic anhydrides, sulfonic acid carboxylic acid anhydrides, etc., more preferably carboxylic acid anhydrides and carboxylic acid chlorides. preferable.
- Preferred carboxylic anhydride compounds include acetic anhydride, propionic anhydride, oxalic anhydride, succinic anhydride, maleic anhydride, phthalic anhydride, benzoic anhydride, 5-norbornene-2,3-dicarboxylic anhydride, and the like. are mentioned.
- Preferred compounds of carboxylic acid chlorides include acetyl chloride, acrylic acid chloride, propionyl chloride, methacrylic acid chloride, pivaloyl chloride, cyclohexanecarbonyl chloride, 2-ethylhexanoyl chloride, cinnamoyl chloride, and 1-adamantanecarbonyl chloride. , heptafluorobutyryl chloride, stearic acid chloride, benzoyl chloride, and the like.
- a step of depositing a solid may be included in the production of the polyimide precursor or the like. Specifically, after filtering off the water absorption by-products of the dehydration condensation agent coexisting in the reaction solution as necessary, water, aliphatic lower alcohol, or a poor solvent such as a mixture thereof, the obtained A polyimide precursor or the like can be obtained by adding a polymer component and depositing the polymer component, depositing it as a solid, and drying it. In order to improve the degree of purification, operations such as redissolution, reprecipitation, drying, etc. of the polyimide precursor may be repeated. Furthermore, a step of removing ionic impurities using an ion exchange resin may be included.
- the content of the specific resin in the resin composition of the present invention is preferably 20% by mass or more, more preferably 30% by mass or more, and 40% by mass or more with respect to the total solid content of the resin composition. is more preferable, and 50% by mass or more is even more preferable. Further, the content of the resin in the resin composition of the present invention is preferably 99.5% by mass or less, more preferably 99% by mass or less, more preferably 98% by mass, based on the total solid content of the resin composition. % or less, more preferably 97 mass % or less, and even more preferably 95 mass % or less.
- the resin composition of the present invention may contain only one type of specific resin, or may contain two or more types. When two or more types are included, the total amount is preferably within the above range.
- the resin composition of the present invention preferably contains at least two resins.
- the resin composition of the present invention may contain a total of two or more of the specific resin and other resins described later, or may contain two or more of the specific resins. It is preferable to include two or more kinds.
- the resin composition of the present invention contains two or more specific resins, for example, two or more polyimides that are polyimide precursors and have different dianhydride-derived structures (R 115 in the above formula (2)) It preferably contains a precursor.
- the resin composition of the present invention may contain other resins (hereinafter simply referred to as "other resins") different from the specific resins in addition to or instead of the specific resins described above.
- Other resins include phenolic resins, polyamides, epoxy resins, polysiloxanes, resins containing siloxane structures, (meth)acrylic resins, (meth)acrylamide resins, urethane resins, butyral resins, styryl resins, polyether resins, and polyester resins. etc.
- phenolic resins polyamides
- epoxy resins polysiloxanes
- resins containing siloxane structures resins containing siloxane structures
- (meth)acrylic resins (meth)acrylamide resins, urethane resins, butyral resins, styryl resins, polyether resins, and polyester resins.
- a (meth)acrylic resin by further adding a (meth)acrylic resin, a resin composition having excellent
- a high polymerizable group value having a weight average molecular weight of 20,000 or less for example, the molar amount of the polymerizable group in 1 g of the resin is 1 ⁇ 10 ⁇ 3 mol/g or more
- the coating properties of the resin composition and the solvent resistance of the pattern (cured product) can be improved. can.
- the content of the other resins is preferably 0.01% by mass or more, and 0.05% by mass or more, relative to the total solid content of the resin composition. More preferably, it is more preferably 1% by mass or more, even more preferably 2% by mass or more, even more preferably 5% by mass or more, and further preferably 10% by mass or more. More preferred.
- the content of other resins in the resin composition of the present invention is preferably 80% by mass or less, more preferably 75% by mass or less, based on the total solid content of the resin composition. It is more preferably 60% by mass or less, even more preferably 50% by mass or less.
- the content of other resins may be low.
- the content of the other resin is preferably 20% by mass or less, more preferably 15% by mass or less, and 10% by mass or less relative to the total solid content of the resin composition. is more preferable, 5% by mass or less is even more preferable, and 1% by mass or less is even more preferable.
- the lower limit of the content is not particularly limited as long as it is 0% by mass or more.
- the resin composition of the present invention may contain only one kind of other resin, or may contain two or more kinds thereof. When two or more types are included, the total amount is preferably within the above range.
- the negative photosensitive resin composition in the first aspect of the present invention contains a compound A that reduces absorbance at the exposure wavelength upon exposure.
- Whether or not a certain compound a contained in the negative photosensitive resin composition corresponds to compound A can be determined by the following method. .
- a solution of compound a having the same concentration as the concentration contained in the negative photosensitive resin composition is prepared, and the molar absorption coefficient of compound a at the wavelength of exposure light (mol ⁇ 1 L cm ⁇ 1 , “molar absorption ) is measured.
- the above measurements are performed quickly so that the effects of changes such as a decrease in the molar extinction coefficient of compound a are small.
- the solvent is used when the negative photosensitive resin composition contains a solvent, and N-methyl-2-pyrrolidone is used when the negative photosensitive resin composition does not contain a solvent.
- the solution of compound a is irradiated with exposure light.
- the exposure amount is 500 mJ as an integrated amount with respect to 1 mol of compound a.
- the molar absorption coefficient of compound a at the wavelength of the exposure light (mol ⁇ 1 ⁇ L ⁇ cm ⁇ 1 , also referred to as “molar absorption coefficient 2”) is measured. From the above molar extinction coefficient 1 and molar extinction coefficient 2, the attenuation rate (%) is calculated based on the following formula.
- Attenuation rate (%) 1 - molar extinction coefficient 2 / molar extinction coefficient 1 x 100
- the attenuation rate is preferably 10% or more, more preferably 20% or more.
- the lower limit of the attenuation rate is not particularly limited, and may be 0% or more.
- the wavelength of the exposure light may be a wavelength at which the photosensitive film is exposed.
- the wavelength of the exposure light is preferably a wavelength at which the photopolymerization initiator contained in the negative photosensitive resin composition is sensitive.
- the photoinitiator is sensitive to a certain wavelength means that the photoinitiator produces polymerization initiating species when exposed to light at a certain wavelength.
- the wavelength of the exposure light in relation to the light source, (1) semiconductor laser (wavelength: 830 nm, 532 nm, 488 nm, 405 nm, 375 nm, 355 nm etc.), (2) metal halide lamp, (3) high pressure mercury lamp, g-line (wavelength 436nm), h-line (wavelength 405nm), i-line (wavelength 365nm), broad (three wavelengths of g, h, i-line), (4) excimer laser, KrF excimer laser (wavelength 248nm), ArF excimer Laser (wavelength 193 nm), F2 excimer laser (wavelength 157 nm), (5) extreme ultraviolet; EUV (wavelength 13.6 nm), (6) electron beam, (7) YAG laser second harmonic 532 nm, third harmonic
- a wavelength at which the photopolymerization initiator is sensitive may be selected, but h-line (wavelength: 405 nm) or i-line (wavelength: 365 nm) is preferable, and i-line (wavelength: 365 nm) is more preferable.
- Compound A may be a compound that generates a radical polymerization initiating species upon exposure, but from the viewpoint of resolution and chemical resistance, it is preferably a compound that does not generate a radical polymerization initiating species upon exposure. Whether or not the compound A is a compound that generates a radical polymerization initiating species upon exposure is determined by the following method. A solution containing compound A at the same concentration as that contained in the negative photosensitive resin composition and a radical cross-linking agent is prepared. When the negative photosensitive resin composition contains a radical crosslinking agent, as the radical crosslinking agent in the solution, the same compound as the radical crosslinking agent contained in the negative photosensitive resin composition is used at the same concentration.
- the negative photosensitive resin composition does not contain a radical cross-linking agent
- methyl methacrylate is used at a concentration of 5 times that of compound A. After that, irradiation of exposure light is performed. The exposure amount is assumed to be 500 mJ as an integrated amount.
- the polymerization of the polymerizable compound is determined by high performance liquid chromatography, and if the ratio of the molar amount of the polymerizable compound polymerized to the total molar amount of the polymerizable compound is 10% or less, compound A is exposed. It is judged to be a compound that does not generate radical polymerization initiating species.
- the molar ratio is preferably 5% or less, more preferably 3% or less.
- the lower limit of the molar ratio is not particularly limited, and may be 0%.
- the wavelength of the exposure light may be a wavelength at which the photosensitive film is exposed.
- the wavelength of the exposure light is preferably a wavelength at which the photopolymerization initiator contained in the negative photosensitive resin composition is sensitive.
- the wavelength of the exposure light is preferably a wavelength at which the photopolymerization initiator contained in the negative photosensitive resin composition is sensitive.
- Compounds that generate radical polymerization initiation species upon exposure include the same compounds as the radical photopolymerization initiators described below.
- the composition contains a radical photopolymerization initiator as the compound A
- the compound A having the lowest polymerization initiation ability of the generated radical species is used as the photopolymerization initiator, and the others are used as the photopolymerization initiator.
- Examples of compounds that do not generate radical polymerization initiation species upon exposure include photoacid generators, photobase generators, and dyes whose absorption wavelength changes upon exposure.
- the compound A is preferably a naphthoquinonediazide compound or a dye whose absorbance changes upon exposure, and more preferably a naphthoquinonediazide compound.
- a combination of a photoacid generator or a photobase generator and a compound whose absorbance at the exposure wavelength decreases depending on the pH may be used.
- the negative photosensitive resin composition in the second aspect of the present invention contains a naphthoquinone diazide compound.
- the naphthoquinonediazide compound in the second aspect is synonymous with the naphthoquinonediazide compound included as a preferred aspect in the first aspect, and the preferred aspects thereof are also the same.
- the naphthoquinonediazide compound in the second aspect is preferably a compound whose absorbance at the exposure wavelength is reduced by exposure.
- the method of determining whether or not the compound is a compound whose absorbance at the exposure wavelength is reduced by exposure, and the preferred range of the decay rate are the same as those of the compound A in the first aspect described above.
- Naphthoquinone diazide compound examples include compounds that generate indenecarboxylic acid upon exposure to reduce the absorbance at the exposure wavelength, and compounds having a 1,2-naphthoquinonediazide structure are preferred.
- the naphthoquinonediazide compound is preferably a naphthoquinonediazide sulfonic acid ester of a hydroxy compound.
- the hydroxy compound compounds represented by any one of the following formulas (H1) to (H6) are preferable.
- R 1 and R 2 each independently represent a monovalent organic group
- R 3 and R 4 each independently represent a hydrogen atom or a monovalent organic group
- n1, n2, m1 and m2 are each independently an integer of 0-5, and at least one of m1 and m2 is an integer of 1-5.
- Z represents a tetravalent organic group
- L 1 , L 2 , L 3 and L 4 each independently represent a single bond or a divalent organic group
- R 5 , R 6 , R 7 and R 8 each independently represent a monovalent organic group
- n3, n4, n5 and n6 are each independently an integer of 0 to 3
- m3, m4, m5 and m6 are each independently 0 is an integer of ⁇ 2 and at least one of m3, m4, m5 and m6 is 1 or 2
- R 9 and R 10 each independently represent a hydrogen atom or a monovalent organic group
- L 5 each independently represents a divalent organic group
- n7 is an integer of 3 to 8. show.
- L6 represents a divalent organic group
- L7 and L8 each independently represent a divalent organic group containing an aliphatic tertiary or quaternary carbon.
- R 11 , R 12 , R 13 , R 14 , R 15 , R 16 , R 17 , R 18 , R 19 and R 20 are each independently a hydrogen atom, a halogen atom or a monovalent organic
- L 9 , L 10 and L 11 each independently represent a single bond or a divalent organic group
- m7, m8, m9 and m10 each independently represent an integer of 0 to 2, m7, At least one of m8, m9 and m10 is 1 or 2.
- R 42 , R 43 , R 44 and R 45 each independently represent a hydrogen atom or a monovalent organic group
- R 46 and R 47 each independently represent a monovalent organic group
- n16 and n17 each independently represent an integer of 0 to 4
- m11 and m12 each independently represent an integer of 0 to 4
- at least one of m11 and m12 is an integer of 1 to 4 be.
- R 1 and R 2 are each independently preferably a monovalent organic group having 1 to 60 carbon atoms, more preferably a monovalent organic group having 1 to 30 carbon atoms.
- Examples of monovalent organic groups for R 1 and R 2 include hydrocarbon groups which may have a substituent, such as aromatic hydrocarbon groups which may have a substituent such as a hydroxy group. mentioned.
- R 3 and R 4 are each independently preferably a monovalent organic group having 1 to 60 carbon atoms, more preferably a monovalent organic group having 1 to 30 carbon atoms. .
- Examples of monovalent organic groups for R 3 and R 4 include hydrocarbon groups which may have a substituent, such as hydrocarbon groups which may have a substituent such as a hydroxy group. .
- n1 and n2 are each independently preferably 0 or 1, more preferably 0.
- both m1 and m2 are preferably 1.
- the compound represented by formula (H1) is preferably a compound represented by any one of formulas (H1-1) to (H1-5).
- R 21 , R 22 and R 23 each independently represent a hydrogen atom or a monovalent organic group, preferably a hydrogen atom or a monovalent organic group having 1 to 20 carbon atoms, hydrogen An atom or a group represented by the following formula (R-1) is more preferable.
- R29 represents a hydrogen atom, an alkyl group or an alkoxy group
- n13 represents an integer of 0 to 2
- * represents a bonding site with another structure.
- n8, n9 and n10 each independently represents an integer of 0 to 2, preferably 0 or 1.
- R 24 represents a hydrogen atom or a monovalent organic group, preferably a hydrogen atom, an alkyl group having 1 to 20 carbon atoms or an alkoxy group having 1 to 20 carbon atoms.
- n14, n15 and n16 each independently represents an integer of 0 to 2;
- R30 represents a hydrogen atom or an alkyl group.
- R 25 , R 26 , R 27 and R 28 each independently represent a monovalent organic group and are represented by a hydrogen atom, an alkyl group or the above formula (R-1) It is preferably a group.
- n11, n12 and n13 each independently represent an integer of 0 to 2, preferably 0 or 1.
- H1-1 a compound represented by any one of the following formulas (H1-1-1) to (H1-1-4) is preferable.
- H1-2 a compound represented by the following formula (H1-2-1) or (H1-2-2) is preferable.
- H1-3 compounds represented by the following formulas (H1-3-1) to (H1-3-3) are preferable.
- Z is preferably a tetravalent group having 1 to 20 carbon atoms, more preferably a group represented by any one of the following formulas (Z-1) to (Z-4).
- * represents a binding site with another structure.
- L 1 , L 2 , L 3 and L 4 are each independently preferably a single bond or a methylene group.
- R 5 , R 6 , R 7 and R 8 are each independently preferably an organic group having 1 to 30 carbon atoms.
- n3, n4, n5 and n6 are each independently preferably an integer of 0 to 2, more preferably 0 or 1.
- m3, m4, m5 and m6 are each independently preferably 1 or 2, more preferably 1.
- compounds having the following structures are exemplified.
- R 9 and R 10 preferably each independently represent a hydrogen atom or a monovalent organic group having 1 to 20 carbon atoms.
- each L 5 is preferably independently a group represented by formula (L-1) below.
- R 30 represents a monovalent organic group having 1 to 20 carbon atoms
- n14 represents an integer of 1 to 5
- * represents a bonding site with another structure.
- n7 is preferably an integer of 4-6. Examples of the compound represented by formula (H3) include the following compounds. In the formula below, each n independently represents an integer of 0-9.
- L 7 and L 8 are each independently preferably a divalent organic group having 2 to 20 carbon atoms. Examples of the compound represented by formula (H4) include the following compounds.
- R 11 , R 12 , R 13 , R 14 , R 15 , R 16 , R 17 , R 18 , R 19 and R 20 are each independently a hydrogen atom, a halogen atom, an alkyl group, an alkenyl groups, alkoxy groups, allyl groups or acyl groups are preferred.
- R 31 and R 32 each independently represent a hydrogen atom, an alkyl group, an alkenyl group or an aryl group
- R 34 , R 35 , R 36 and R 37 each independently represents a hydrogen atom or an alkyl group
- n15 is an integer of 1 to 5
- R 38 , R 39 , R 40 and R 41 each independently represent a hydrogen atom or an alkyl group
- Compounds represented by formula (H5) include the following compounds.
- R 42 , R 43 , R 44 and R 45 each independently represent a hydrogen atom or a monovalent organic group, preferably a hydrogen atom or a monovalent organic group having 1 to 20 carbon atoms. , a hydrogen atom or an alkyl group having 1 to 20 carbon atoms is more preferable, and an alkyl group having 1 to 4 carbon atoms is more preferable.
- R 46 and R 47 are each independently preferably an alkyl group, an alkoxy group or an aryl group, more preferably an alkyl group.
- n16 and n17 are each independently preferably an integer of 0 to 2, more preferably 0 or 1.
- n16 and n17 are each independently preferably an integer of 1 to 3, more preferably 2 or 3. Examples of the compound represented by formula (H6) include the following compounds.
- hydroxy compounds include 2,3,4-trihydroxybenzophenone, 2,4,4'-trihydroxybenzophenone, 2,4,6-trihydroxybenzophenone, 2,3,4-trihydroxy-2'- methylbenzophenone, 2,3,4,4'-tetrahydroxybenzophenone, 2,2',4,4'-tetrahydroxybenzophenone, 2,4,6,3',4'-pentahydroxybenzophenone, 2,3, 4,2',4'-pentahydroxybenzophenone, 2,3,4,2',5'-pentahydroxybenzophenone, 2,4,6,3',4',5'-hexahydroxybenzophenone, 2,3 , 4,3′,4′,5′-hexahydroxybenzophenones and other polyhydroxybenzophenones, polyhydroxyphenyl alkyl ketones such as 2,3,4-trihydroxyacetophenone, 2,3,4-trihydroxyphenylpentyl ketone and 2,3,4-trihydroxyphenylhexyl ketone; Bis(2,
- polyhydroxybiphenyls bis(polyhydroxy)sulfides such as 4,4′-thiobis(1,3-dihydroxy)benzene, bis(polyhydroxyphenyl) ethers such as 2,2',4,4'-tetrahydroxydiphenyl ether, bis(polyhydroxyphenyl)sulfoxides such as 2,2′,4,4′-tetrahydroxydiphenylsulfoxide, bis(polyhydroxyphenyl)sulfones such as 2,2′,4,4′-diphenylsulfone, Tris(4-hydroxyphenyl)methane, 4,4′,4′′-trihydroxy-3,5,3′,5′-tetramethyltriphenylmethane, 4,4′,3′′,4′′-tetrahydroxy- 3,5,3′,5′-tetramethyltriphenylmethane, 4-[bis(3,5-dimethyl-4-hydroxyphenyl)methyl]-2-methoxy-phenol, 4,4′-(3,
- the naphthoquinonediazide sulfonic acid includes 6-diazo-5,6-dihydro-5-oxo-1-naphthalenesulfonic acid, 1,2-naphthoquinone-(2)-diazo-5-sulfonic acid and the like, and these may be mixed. may be used as
- the method for producing the naphthoquinonediazide sulfonate ester of a hydroxy compound is not particularly limited, but for example, naphthoquinone diazide sulfonic acid is converted to sulfonyl chloride with chlorosulfonic acid or thionyl chloride, and the obtained naphthoquinone diazide sulfonyl chloride is condensed with the hydroxy compound. It is obtained by reacting.
- a predetermined amount of a hydroxy compound and naphthoquinonediazidosulfonyl chloride are reacted in a solvent such as dioxane, acetone, or tetrahydrofuran in the presence of a basic catalyst such as triethylamine to effect esterification, and the resulting product is washed with water. , can be obtained by drying.
- the esterification rate of the naphthoquinonediazide sulfonic acid ester is not particularly limited, but is preferably 10% or more, more preferably 20% or more.
- the upper limit of the esterification rate is not particularly limited, and may be 100%.
- the esterification rate can be confirmed by 1 H-NMR or the like as the ratio of esterified groups among the hydroxy groups of the hydroxy compound.
- the dye whose absorbance changes with exposure is not particularly limited, and for example, known photochromic materials can be used.
- Leuco dyes diarylethene dyes, fulgides, spirooxazines, spiropyrans, or azobenzene compounds, etc. mentioned.
- leuco dyes include triarylmethane-based (eg, triphenylmethane-based), spiropyran-based, fluoran-based, diphenylmethane-based, rhodaminelactam-based, indolylphthalide-based, and leuco-auramine-based leuco dyes.
- triarylmethane-based leuco dyes are preferred.
- the leuco dye is a leuco dye that has a lactone ring, a sultine ring, or a sultone ring, and that the absorbance changes as the lactone ring, the sultine ring, or the sultone ring is opened or closed.
- the leuco dye a commercially available product such as Victoria Pure Blue-NAPS may be used.
- diarylethene compound for example, a diarylethene compound in which the above-mentioned absorbance changes by ring closure as described below is preferable.
- the conformation in the following formula is an example, and is not limited to this.
- the diarylethene compound is not particularly limited, and known compounds can be used. Examples include compounds described in paragraphs 0049 to 0056 of JP-A-2004-91638.
- the content of compound A with respect to the total solid content of the negative photosensitive resin composition in the first aspect of the present invention is not particularly limited, but is preferably 0.1 to 20% by mass, and 0.5 to 10% by mass. % by mass is more preferred, and 1 to 5% by mass is even more preferred.
- the content of the naphthoquinonediazide compound with respect to the total solid content of the negative photosensitive resin composition in the second aspect of the present invention is not particularly limited, but is preferably 0.1 to 20% by mass, and 0.5 to 0.5% by mass. It is more preferably 10% by mass, and even more preferably 1 to 5% by mass.
- the resin composition of the present invention contains a photopolymerization initiator.
- the photopolymerization initiator is preferably a photoradical polymerization initiator.
- the radical photopolymerization initiator is not particularly limited and can be appropriately selected from known radical photopolymerization initiators.
- a photoradical polymerization initiator having photosensitivity to light in the ultraviolet region to the visible region is preferred. It may also be an activator that produces an active radical by producing some action with a photoexcited sensitizer.
- the radical photopolymerization initiator contains at least one compound having a molar extinction coefficient of at least about 50 L ⁇ mol ⁇ 1 ⁇ cm ⁇ 1 within the wavelength range of about 240 to 800 nm (preferably 330 to 500 nm). is preferred.
- the molar extinction coefficient of a compound can be measured using known methods. For example, it is preferable to measure with an ultraviolet-visible spectrophotometer (Cary-5 spectrophotometer manufactured by Varian) using an ethyl acetate solvent at a concentration of 0.01 g/L.
- any known compound can be used as the photoradical polymerization initiator.
- halogenated hydrocarbon derivatives e.g., compounds having a triazine skeleton, compounds having an oxadiazole skeleton, compounds having a trihalomethyl group, etc.
- acylphosphine compounds such as acylphosphine oxide, hexaarylbiimidazole, oxime derivatives, etc.
- ketone compounds include compounds described in paragraph 0087 of JP-A-2015-087611, the content of which is incorporated herein.
- Kayacure-DETX-S manufactured by Nippon Kayaku Co., Ltd. is also suitably used.
- a hydroxyacetophenone compound, an aminoacetophenone compound, and an acylphosphine compound can be suitably used as the radical photopolymerization initiator. More specifically, for example, aminoacetophenone-based initiators described in JP-A-10-291969 and acylphosphine oxide-based initiators described in Japanese Patent No. 4225898 can be used. incorporated.
- ⁇ -hydroxyketone initiators include Omnirad 184, Omnirad 1173, Omnirad 2959, Omnirad 127 (manufactured by IGM Resins B.V.), IRGACURE 184 (IRGACURE is a registered trademark), DAROCUR 1173, IRGACURE 500, IRGACURE -2959 and IRGACURE 127 (trade names: both manufactured by BASF) can be used.
- ⁇ -aminoketone initiators examples include Omnirad 907, Omnirad 369, Omnirad 369E, Omnirad 379EG (manufactured by IGM Resins B.V.), IRGACURE 907, IRGACURE 369, and IRGACURE 379 (trade names: all BASF company) can be used.
- the compound described in JP-A-2009-191179 whose maximum absorption wavelength is matched to a wavelength light source such as 365 nm or 405 nm can also be used, the content of which is incorporated herein.
- Acylphosphine oxide initiators include 2,4,6-trimethylbenzoyl-diphenyl-phosphine oxide.
- Omnirad 819, Omnirad TPO (manufactured by IGM Resins B.V.), IRGACURE-819 and IRGACURE-TPO (trade names: all manufactured by BASF) can also be used.
- metallocene compounds examples include IRGACURE-784, IRGACURE-784EG (both manufactured by BASF) and Keycure VIS 813 (manufactured by King Brother Chem).
- the photoradical polymerization initiator is more preferably an oxime compound.
- an oxime compound By using an oxime compound, the exposure latitude can be improved more effectively.
- Oxime compounds are particularly preferred because they have a wide exposure latitude (exposure margin) and also act as photocuring accelerators.
- oxime compound examples include compounds described in JP-A-2001-233842, compounds described in JP-A-2000-080068, compounds described in JP-A-2006-342166, J. Am. C. S. Compounds described in Perkin II (1979, pp.1653-1660); C. S. Compounds described in Perkin II (1979, pp.156-162), compounds described in Journal of Photopolymer Science and Technology (1995, pp.202-232), compounds described in JP-A-2000-066385, Compounds described in JP-A-2004-534797, compounds described in JP-A-2017-019766, compounds described in Patent No.
- Preferred oxime compounds include, for example, compounds having the following structures, 3-(benzoyloxy(imino))butan-2-one, 3-(acetoxy(imino))butan-2-one, 3-(propionyloxy( imino))butan-2-one, 2-(acetoxy(imino))pentan-3-one, 2-(acetoxy(imino))-1-phenylpropan-1-one, 2-(benzoyloxy(imino)) -1-phenylpropan-1-one, 3-((4-toluenesulfonyloxy)(imino))butan-2-one, and 2-(ethoxycarbonyloxy(imino))-1-phenylpropan-1-one etc.
- an oxime compound an oxime-based radical photopolymerization initiator
- DFI-091 manufactured by Daito Chemix Co., Ltd.
- SpeedCure PDO manufactured by SARTOMER ARKEMA
- an oxime compound having the following structure can be used.
- An oxime compound having a fluorene ring can also be used as the photoradical polymerization initiator.
- Specific examples of the oxime compound having a fluorene ring include compounds described in JP-A-2014-137466 and compounds described in Japanese Patent No. 06636081, the contents of which are incorporated herein.
- an oxime compound having a skeleton in which at least one benzene ring of the carbazole ring is a naphthalene ring can also be used.
- Specific examples of such oxime compounds include compounds described in WO2013/083505, the contents of which are incorporated herein.
- oxime compound having a fluorine atom examples include compounds described in JP-A-2010-262028, compounds 24, 36-40 described in paragraph 0345 of JP-A-2014-500852, and JP-A-2013. and compound (C-3) described in paragraph 0101 of JP-A-164471, the contents of which are incorporated herein.
- An oxime compound having a nitro group can be used as the photopolymerization initiator.
- the oxime compound having a nitro group is also preferably a dimer.
- Specific examples of the oxime compound having a nitro group include the compounds described in paragraph numbers 0031 to 0047 of JP-A-2013-114249 and paragraph numbers 0008-0012 and 0070-0079 of JP-A-2014-137466; Included are compounds described in paragraphs 0007-0025 of Japanese Patent No. 4223071, the contents of which are incorporated herein.
- the oxime compound having a nitro group also includes ADEKA Arkles NCI-831 (manufactured by ADEKA Co., Ltd.).
- An oxime compound having a benzofuran skeleton can also be used as the photoradical polymerization initiator.
- Specific examples include OE-01 to OE-75 described in WO 2015/036910.
- an oxime compound in which a substituent having a hydroxy group is bonded to the carbazole skeleton can also be used.
- photoinitiators include compounds such as those described in WO2019/088055, the contents of which are incorporated herein.
- an oxime compound having an aromatic ring group Ar 2 OX1 in which an electron-withdrawing group is introduced into the aromatic ring (hereinafter also referred to as oxime compound OX) can be used.
- the electron-withdrawing group of the aromatic ring group Ar OX1 include an acyl group, a nitro group, a trifluoromethyl group, an alkylsulfinyl group, an arylsulfinyl group, an alkylsulfonyl group, an arylsulfonyl group, and a cyano group.
- a benzoyl group may have a substituent.
- substituents include halogen atoms, cyano groups, nitro groups, hydroxy groups, alkyl groups, alkoxy groups, aryl groups, aryloxy groups, heterocyclic groups, heterocyclic oxy groups, alkenyl groups, alkylsulfanyl groups, arylsulfanyl groups, It is preferably an acyl group or an amino group, more preferably an alkyl group, an alkoxy group, an aryl group, an aryloxy group, a heterocyclic oxy group, an alkylsulfanyl group, an arylsulfanyl group or an amino group.
- a sulfanyl group or an amino group is more preferred.
- the oxime compound OX is preferably at least one selected from the compounds represented by the formula (OX1) and the compounds represented by the formula (OX2), more preferably the compound represented by the formula (OX2). preferable.
- R X1 is an alkyl group, alkenyl group, alkoxy group, aryl group, aryloxy group, heterocyclic group, heterocyclicoxy group, alkylsulfanyl group, arylsulfanyl group, alkylsulfinyl group, arylsulfinyl group, alkylsulfonyl a group, an arylsulfonyl group, an acyl group, an acyloxy group, an amino group, a phosphinoyl group, a carbamoyl group or a sulfamoyl group
- R X2 is an alkyl group, alkenyl group, alkoxy group, aryl group, aryloxy group, heterocyclic group,
- R X12 is an electron-withdrawing group
- R X10 , R X11 , R X13 and R X14 are preferably hydrogen atoms.
- oxime compound OX examples include compounds described in paragraphs 0083 to 0105 of Japanese Patent No. 4600600, the contents of which are incorporated herein.
- oxime compounds having specific substituents shown in JP-A-2007-269779 and oxime compounds having a thioaryl group shown in JP-A-2009-191061. incorporated herein.
- photoradical polymerization initiators include trihalomethyltriazine compounds, benzyldimethylketal compounds, ⁇ -hydroxyketone compounds, ⁇ -aminoketone compounds, acylphosphine compounds, phosphine oxide compounds, metallocene compounds, oxime compounds, triaryl selected from the group consisting of imidazole dimers, onium salt compounds, benzothiazole compounds, benzophenone compounds, acetophenone compounds and derivatives thereof, cyclopentadiene-benzene-iron complexes and salts thereof, halomethyloxadiazole compounds, and 3-aryl-substituted coumarin compounds; are preferred.
- More preferred radical photopolymerization initiators are trihalomethyltriazine compounds, ⁇ -aminoketone compounds, acylphosphine compounds, phosphine oxide compounds, metallocene compounds, oxime compounds, triarylimidazole dimers, onium salt compounds, benzophenone compounds, and acetophenone compounds.
- At least one compound selected from the group consisting of trihalomethyltriazine compounds, ⁇ -aminoketone compounds, metallocene compounds, oxime compounds, triarylimidazole dimers, and benzophenone compounds is more preferred, and metallocene compounds or oxime compounds are even more preferred. .
- the photoradical polymerization initiator includes benzophenone, N,N'-tetraalkyl-4,4'-diaminobenzophenone such as N,N'-tetramethyl-4,4'-diaminobenzophenone (Michler's ketone), 2-benzyl -aromatic ketones such as 2-dimethylamino-1-(4-morpholinophenyl)-butanone-1,2-methyl-1-[4-(methylthio)phenyl]-2-morpholino-propanone-1, alkylanthraquinones, etc.
- benzophenone N,N'-tetraalkyl-4,4'-diaminobenzophenone
- 2-benzyl -aromatic ketones such as 2-dimethylamino-1-(4-morpholinophenyl)-butanone-1,2-methyl-1-[4-(methylthio)phenyl]-2-morpholino-propanone-1, alkylanthr
- benzoin ether compounds such as benzoin alkyl ether
- benzoin compounds such as benzoin and alkylbenzoin
- benzyl derivatives such as benzyl dimethyl ketal
- a compound represented by the following formula (I) can also be used.
- R 100 is an alkyl group having 1 to 20 carbon atoms, an alkyl group having 2 to 20 carbon atoms interrupted by one or more oxygen atoms, an alkoxy group having 1 to 12 carbon atoms, a phenyl group, Alternatively, an alkyl group having 1 to 20 carbon atoms, an alkoxy group having 1 to 12 carbon atoms, a halogen atom, a cyclopentyl group, a cyclohexyl group, an alkenyl group having 2 to 12 carbon atoms, a carbon number interrupted by one or more oxygen atoms a phenyl group or a biphenyl group substituted with at least one of an alkyl group having 2 to 18 carbon atoms and an alkyl group having 1 to 4 carbon atoms, and R I01 is a group represented by formula (II); It is the same group as R 100 , and each of R 102 to R 104 is independently an alkyl group having 1 to 12 carbon atoms
- R 105 to R 107 are the same as R 102 to R 104 in formula (I) above.
- radical photopolymerization initiator a difunctional or trifunctional or higher radical photopolymerization initiator may be used.
- a radical photopolymerization initiator two or more radicals are generated from one molecule of the radical photopolymerization initiator, so good sensitivity can be obtained.
- the crystallinity is lowered, the solubility in a solvent or the like is improved, and precipitation becomes difficult over time, and the stability over time of the resin composition can be improved.
- Specific examples of bifunctional or trifunctional or higher photoradical polymerization initiators include Japanese Patent Publication No. 2010-527339, Japanese Patent Publication No. 2011-524436, International Publication No.
- a photopolymerization initiator When a photopolymerization initiator is included, its content is preferably 0.1 to 30% by mass, more preferably 0.1 to 20% by mass, based on the total solid content of the resin composition of the present invention. , More preferably 0.5 to 15% by mass, still more preferably 1.0 to 10% by mass. Only one type of photopolymerization initiator may be contained, or two or more types may be contained. When two or more photopolymerization initiators are contained, the total amount is preferably within the above range. In addition, since the photopolymerization initiator may also function as a thermal polymerization initiator, the crosslinking by the photopolymerization initiator may be further advanced by heating with an oven, a hot plate, or the like.
- the resin composition may contain a sensitizer.
- a sensitizer absorbs specific actinic radiation and enters an electronically excited state.
- the sensitizer in an electronically excited state comes into contact with a thermal radical polymerization initiator, a photoradical polymerization initiator, or the like, and causes electron transfer, energy transfer, heat generation, or the like.
- the thermal radical polymerization initiator and the photoradical polymerization initiator undergo chemical changes and are decomposed to generate radicals, acids or bases.
- Usable sensitizers include benzophenones, Michler's ketones, coumarins, pyrazole azos, anilinoazos, triphenylmethanes, anthraquinones, anthracenes, anthrapyridones, benzylidenes, oxonols, and pyrazolotriazole azos. , pyridone azo, cyanine, phenothiazine, pyrrolopyrazole azomethine, xanthene, phthalocyanine, benzopyran, and indigo compounds.
- Sensitizers include, for example, Michler's ketone, 4,4'-bis(diethylamino)benzophenone, 2,5-bis(4'-diethylaminobenzal)cyclopentane, 2,6-bis(4'-diethylaminobenzal) Cyclohexanone, 2,6-bis(4'-diethylaminobenzal)-4-methylcyclohexanone, 4,4'-bis(dimethylamino)chalcone, 4,4'-bis(diethylamino)chalcone, p-dimethylaminocinnamyl denindanone, p-dimethylaminobenzylideneindanone, 2-(p-dimethylaminophenylbiphenylene)-benzothiazole, 2-(p-dimethylaminophenylvinylene)benzothiazole, 2-(p-dimethylaminophenylvinylene)iso naphthothiazole,
- the content of the sensitizer is preferably 0.01 to 20% by mass, preferably 0.1 to 15% by mass, based on the total solid content of the resin composition. more preferably 0.5 to 10% by mass.
- the sensitizers may be used singly or in combination of two or more.
- the resin composition of the present invention may contain a chain transfer agent.
- the chain transfer agent is defined, for example, in Kobunshi Jiten, 3rd edition (edited by Kobunshi Gakkai, 2005), pp. 683-684.
- Chain transfer agents include, for example, a group of compounds having —S—S—, —SO 2 —S—, —NO—, SH, PH, SiH, and GeH in the molecule, RAFT (Reversible Addition Fragmentation chain Transfer )
- Dithiobenzoate, trithiocarbonate, dithiocarbamate, xanthate compounds and the like having a thiocarbonylthio group used for polymerization are used. They can either donate hydrogen to less active radicals to generate radicals, or they can be oxidized and then deprotonated to generate radicals.
- thiol compounds can be preferably used.
- chain transfer agent can also use the compounds described in paragraphs 0152 to 0153 of International Publication No. 2015/199219, the contents of which are incorporated herein.
- the content of the chain transfer agent is preferably 0.01 to 20 parts by mass, preferably 0.01 to 20 parts by mass, based on 100 parts by mass of the total solid content of the resin composition of the present invention. 1 to 10 parts by mass is more preferable, and 0.5 to 5 parts by mass is even more preferable.
- One type of chain transfer agent may be used, or two or more types may be used. When two or more chain transfer agents are used, the total is preferably within the above range.
- the resin composition of the present invention further contains a nitrogen-containing cyclic compound as a component different from the resin, compound A and photopolymerization initiator.
- the resin composition of the present invention satisfies condition iii
- the resin composition of the present invention further contains a nitrogen-containing cyclic compound as a component different from the resin, the naphthoquinonediazide compound, and the photopolymerization initiator.
- the resin composition of the present invention satisfies condition ii or condition iv
- the resin composition of the present invention may further contain a nitrogen-containing cyclic compound.
- a nitrogen-containing cyclic compound is a compound having a ring structure containing a nitrogen atom as a ring member.
- the ring structure may be either an aliphatic ring structure or an aromatic ring structure, but is preferably an aromatic ring structure.
- the nitrogen-containing cyclic compound may be a compound having a ring structure containing one or more nitrogen atoms as ring members, but is preferably a compound having a ring structure containing two or more nitrogen atoms as ring members.
- the ring structure containing a nitrogen atom as a ring member is not particularly limited, but is pyrrole ring, furan ring, thiophene ring, imidazole ring, oxazole ring, thiazole ring, pyrazole ring, isoxazole ring, isothiazole ring, and tetrazole ring. , pyridine ring, pyridazine ring, pyrimidine ring, pyrazine ring, piperidine ring, piperazine ring, morpholine ring, 2H-pyran ring and 6H-pyran ring, triazine ring, isocyanurate ring and the like.
- These ring structures may form a multiple ring with another ring structure.
- a mode in which a ring structure such as a benzene ring and a condensed ring are formed such as a benzimidazole structure and a benzotriazole structure.
- triazole structures such as 1,2,4-triazole, benzotriazole, 3-amino-1,2,4-triazole, 3,5-diamino-1,2,4-triazole, 1H-tetrazole, 5
- a compound having a tetrazole structure such as -phenyltetrazole and 5-amino-tetrazole can be preferably used.
- nitrogen-containing cyclic compounds include, but are not limited to, the following compounds.
- the molecular weight of the nitrogen-containing cyclic compound is preferably 60-1000, more preferably 70-700.
- the content of the nitrogen-containing cyclic compound is 0.01 to 5.0% by mass relative to the total solid content of the resin composition of the present invention. is preferred, 0.05 to 2.0 mass % is more preferred, and 0.1 to 1.0 mass % is even more preferred.
- the content of the nitrogen-containing cyclic compound is preferably 0.5 to 200 parts by mass when the content of the compound A is 100 parts by mass. It is more preferably 0.8 to 100 parts by mass, even more preferably 0.1 to 40 parts by mass.
- the content of the nitrogen-containing cyclic compound is preferably 0.5 to 200 parts by mass when the content of the naphthoquinone diazide compound is 100 parts by mass. , more preferably 0.8 to 100 parts by mass, more preferably 0.1 to 40 parts by mass.
- the resin composition of the present invention may contain only one type of nitrogen-containing cyclic compound, or may contain two or more types. When two or more nitrogen-containing cyclic compounds are included, the total is preferably within the above range.
- the resin composition of the present invention preferably contains a polymerizable compound.
- Polymerizable compounds include radical cross-linking agents or other cross-linking agents. Although the polymerizable compounds exemplified below include nitrogen-containing cyclic compounds, these are not included in the above-mentioned "nitrogen-containing cyclic compounds”.
- the resin composition of the present invention preferably contains a radical cross-linking agent.
- a radical cross-linking agent is a compound having a radically polymerizable group.
- the radically polymerizable group a group containing an ethylenically unsaturated bond is preferred.
- Examples of the group containing an ethylenically unsaturated bond include groups containing an ethylenically unsaturated bond such as a vinyl group, an allyl group, a vinylphenyl group, a (meth)acryloyl group, a maleimide group, and a (meth)acrylamide group.
- the group containing an ethylenically unsaturated bond is preferably a (meth)acryloyl group, a (meth)acrylamide group, or a vinylphenyl group, and more preferably a (meth)acryloyl group from the viewpoint of reactivity.
- the radical cross-linking agent is preferably a compound having one or more ethylenically unsaturated bonds, and more preferably a compound having two or more.
- the radical cross-linking agent may have 3 or more ethylenically unsaturated bonds.
- the compound having two or more ethylenically unsaturated bonds is preferably a compound having 2 to 15 ethylenically unsaturated bonds, more preferably a compound having 2 to 10 ethylenically unsaturated bonds, and 2 to 6.
- the resin composition of the present invention contains a compound having two ethylenically unsaturated bonds and a compound having three or more ethylenically unsaturated bonds. It is also preferred to include
- the molecular weight of the radical cross-linking agent is preferably 2,000 or less, more preferably 1,500 or less, and even more preferably 900 or less.
- the lower limit of the molecular weight of the radical cross-linking agent is preferably 100 or more.
- radical cross-linking agent examples include unsaturated carboxylic acids (eg, acrylic acid, methacrylic acid, itaconic acid, crotonic acid, isocrotonic acid, maleic acid, etc.), their esters, and amides. They are esters of saturated carboxylic acids and polyhydric alcohol compounds, and amides of unsaturated carboxylic acids and polyhydric amine compounds.
- addition reaction products of unsaturated carboxylic acid esters or amides having a nucleophilic substituent such as a hydroxy group, an amino group, or a sulfanyl group with monofunctional or polyfunctional isocyanates or epoxies, or monofunctional or polyfunctional is also preferably used.
- addition reaction products of unsaturated carboxylic acid esters or amides having electrophilic substituents such as isocyanate groups and epoxy groups with monofunctional or polyfunctional alcohols, amines, and thiols, and halogeno groups
- substitution reaction products of unsaturated carboxylic acid esters or amides having a leaving substituent such as a tosyloxy group and monofunctional or polyfunctional alcohols, amines, and thiols.
- paragraphs 0113 to 0122 of JP-A-2016-027357 can be referred to, and the contents thereof are incorporated herein.
- the radical cross-linking agent is preferably a compound having a boiling point of 100°C or higher under normal pressure.
- examples include polyethylene glycol di(meth)acrylate, trimethylolethane tri(meth)acrylate, neopentyl glycol di(meth)acrylate, pentaerythritol tri(meth)acrylate, pentaerythritol tetra(meth)acrylate, dipentaerythritol penta(meth)acrylate, dipentaerythritol hexa(meth)acrylate, hexanediol di(meth)acrylate, trimethylolpropane tri(acryloyloxypropyl)ether, tri(acryloyloxyethyl)isocyanurate, glycerin, trimethylolethane, etc.
- polyfunctional (meth)acrylate obtained by reacting polyfunctional carboxylic acid with a compound having a cyclic ether group such as glycidyl (meth)acrylate and an ethylenically unsaturated bond can also be used.
- JP-A-2010-160418, JP-A-2010-129825, JP-A-4364216, etc. have a fluorene ring and an ethylenically unsaturated bond. It is also possible to use compounds having two or more groups and cardo resins.
- JP-B-46-043946 JP-B-01-040337, JP-B-01-040336, and JP-A-02-025493.
- vinyl phosphonic acid compounds and the like can also be mentioned.
- Compounds containing perfluoroalkyl groups described in JP-A-61-022048 can also be used.
- the journal of the Japan Adhesive Association vol. 20, No. 7, pp. 300-308 (1984) as photopolymerizable monomers and oligomers can also be used.
- dipentaerythritol triacrylate (commercially available as KAYARAD D-330 (manufactured by Nippon Kayaku Co., Ltd.)), dipentaerythritol tetraacrylate (commercially available as KAYARAD D-320 (Nippon Kayaku ( Ltd.), A-TMMT (manufactured by Shin-Nakamura Chemical Co., Ltd.), dipentaerythritol penta(meth)acrylate (commercially available as KAYARAD D-310 (manufactured by Nippon Kayaku Co., Ltd.)), dipenta Erythritol hexa(meth)acrylate (commercially available products are KAYARAD DPHA (manufactured by Nippon Kayaku Co., Ltd.) and A-DPH (manufactured by Shin-Nakamura Chemical Co., Ltd.)), and their (meth)acryloyl groups are ethylene glycol,
- radical cross-linking agents examples include SR-494, a tetrafunctional acrylate having four ethyleneoxy chains, manufactured by Sartomer, SR-209, a bifunctional methacrylate having four ethyleneoxy chains, manufactured by Sartomer. 231, 239, Nippon Kayaku Co., Ltd.
- DPCA-60 a hexafunctional acrylate having 6 pentyleneoxy chains, TPA-330, a trifunctional acrylate having 3 isobutyleneoxy chains, urethane oligomer UAS-10 , UAB-140 (manufactured by Nippon Paper Industries), NK Ester M-40G, NK Ester 4G, NK Ester M-9300, NK Ester A-9300, UA-7200 (manufactured by Shin-Nakamura Chemical Co., Ltd.), DPHA-40H (Japan Kayaku Co., Ltd.), UA-306H, UA-306T, UA-306I, AH-600, T-600, AI-600 (manufactured by Kyoeisha Chemical Co., Ltd.), Blenmer PME400 (manufactured by NOF Corporation) etc.
- radical cross-linking agents examples include urethane acrylates such as those described in JP-B-48-041708, JP-A-51-037193, JP-B-02-032293, JP-B-02-016765, Urethane compounds having an ethylene oxide skeleton described in JP-B-58-049860, JP-B-56-017654, JP-B-62-039417 and JP-B-62-039418 are also suitable.
- compounds having an amino structure or a sulfide structure in the molecule described in JP-A-63-277653, JP-A-63-260909, and JP-A-01-105238 are used. can also
- the radical cross-linking agent may be a radical cross-linking agent having an acid group such as a carboxy group or a phosphoric acid group.
- a radical cross-linking agent having an acid group is preferably an ester of an aliphatic polyhydroxy compound and an unsaturated carboxylic acid. is more preferable.
- the aliphatic polyhydroxy compound is pentaerythritol or dipentaerythritol is a compound.
- Examples of commercially available products include polybasic acid-modified acrylic oligomers manufactured by Toagosei Co., Ltd. such as M-510 and M-520.
- the acid value of the radical cross-linking agent having an acid group is preferably 0.1-300 mgKOH/g, particularly preferably 1-100 mgKOH/g. If the acid value of the radical cross-linking agent is within the above range, the handleability in production is excellent, and furthermore the developability is excellent. Moreover, the polymerizability is good. The acid value is measured according to JIS K 0070:1992.
- the resin composition preferably uses a bifunctional methacrylate or acrylate.
- Specific compounds include triethylene glycol diacrylate, triethylene glycol dimethacrylate, tetraethylene glycol dimethacrylate, tetraethylene glycol diacrylate, PEG (polyethylene glycol) 200 diacrylate, PEG200 dimethacrylate, PEG600 diacrylate, and PEG600 diacrylate.
- PEG200 diacrylate is a polyethylene glycol diacrylate having a polyethylene glycol chain formula weight of about 200.
- a monofunctional radical cross-linking agent can be preferably used as the radical cross-linking agent from the viewpoint of suppressing warpage associated with the elastic modulus control of the pattern (cured product).
- Monofunctional radical cross-linking agents include n-butyl (meth)acrylate, 2-ethylhexyl (meth)acrylate, 2-hydroxyethyl (meth)acrylate, butoxyethyl (meth)acrylate, carbitol (meth)acrylate, cyclohexyl (meth)acrylate, ) acrylate, benzyl (meth) acrylate, phenoxyethyl (meth) acrylate, N-methylol (meth) acrylamide, glycidyl (meth) acrylate, polyethylene glycol mono (meth) acrylate, polypropylene glycol mono (meth) acrylate, etc.
- N-vinyl compounds such as N-vinylpyrrolidone and N-vinylcaprolactam
- allyl glycidyl ether are preferably used.
- the monofunctional radical cross-linking agent a compound having a boiling point of 100° C. or higher under normal pressure is also preferable in order to suppress volatilization before exposure.
- Other di- or higher functional radical cross-linking agents include allyl compounds such as diallyl phthalate and triallyl trimellitate.
- a radical cross-linking agent When a radical cross-linking agent is contained, its content is preferably more than 0% by mass and 60% by mass or less with respect to the total solid content of the resin composition of the present invention. More preferably, the lower limit is 5% by mass or more. The upper limit is more preferably 50% by mass or less, and even more preferably 30% by mass or less.
- a single radical cross-linking agent may be used alone, or two or more may be used in combination. When two or more are used in combination, the total amount is preferably within the above range.
- the resin composition of the present invention contains another cross-linking agent different from the radical cross-linking agent described above.
- the other cross-linking agent refers to a cross-linking agent other than the radical cross-linking agent described above, and other compounds or reaction products thereof in the composition are formed by exposure of the photoacid generator or the photobase generator.
- the compound has a plurality of groups in the molecule that promote the reaction to form a covalent bond between and forms a covalent bond with other compounds or reaction products thereof in the composition
- Compounds having a plurality of groups in the molecule, the reaction of which is promoted by the action of an acid or base are preferred.
- the acid or base is preferably an acid or base generated from a photoacid generator or a photobase generator in the exposure step.
- a compound having at least one group selected from the group consisting of acyloxymethyl group, methylol group and alkoxymethyl group is preferable, and selected from the group consisting of acyloxymethyl group, methylol group and alkoxymethyl group. More preferred is a compound having a structure in which at least one group is directly bonded to a nitrogen atom.
- cross-linking agents include, for example, an amino group-containing compound such as melamine, glycoluril, urea, alkylene urea, and benzoguanamine, which is reacted with formaldehyde or formaldehyde and alcohol, and the hydrogen atom of the amino group is converted to an acyloxymethyl group, methylol group, or A compound having a structure substituted with an alkoxymethyl group can be mentioned.
- the method for producing these compounds is not particularly limited as long as they have the same structure as the compounds produced by the above methods. Oligomers formed by self-condensation of methylol groups of these compounds may also be used.
- a melamine-based crosslinking agent is a melamine-based crosslinking agent
- a glycoluril, urea or alkyleneurea-based crosslinking agent is a urea-based crosslinking agent
- an alkyleneurea-based crosslinking agent is an alkyleneurea-based crosslinking agent.
- a cross-linking agent using benzoguanamine is called a benzoguanamine-based cross-linking agent.
- the resin composition of the present invention preferably contains at least one compound selected from the group consisting of urea-based cross-linking agents and melamine-based cross-linking agents. More preferably, it contains at least one compound selected from the group consisting of agents.
- an alkoxymethyl group or an acyloxymethyl group is directly substituted on the nitrogen atom of an aromatic group or the following urea structure, or on a triazine.
- the alkoxymethyl group or acyloxymethyl group of the above compound preferably has 2 to 5 carbon atoms, preferably 2 or 3 carbon atoms, and more preferably 2 carbon atoms.
- the total number of alkoxymethyl groups and acyloxymethyl groups in the above compound is preferably 1-10, more preferably 2-8, and particularly preferably 3-6.
- the molecular weight of the compound is preferably 1500 or less, preferably 180-1200.
- R 100 represents an alkyl group or an acyl group.
- R 101 and R 102 each independently represent a monovalent organic group and may combine with each other to form a ring.
- Examples of compounds in which an alkoxymethyl group or an acyloxymethyl group is directly substituted by an aromatic group include compounds represented by the following general formula.
- X represents a single bond or a divalent organic group
- each R 104 independently represents an alkyl group or an acyl group
- R 103 represents a hydrogen atom, an alkyl group, an alkenyl group, an aryl group, an aralkyl group , or a group that decomposes under the action of an acid to produce an alkali-soluble group (e.g., a group that leaves under the action of an acid, a group represented by —C(R 4 ) 2 COOR 5 (R 4 is independently It represents a hydrogen atom or an alkyl group having 1 to 4 carbon atoms, and R 5 represents a group that leaves under the action of an acid.)).
- R 105 each independently represents an alkyl group or alkenyl group, a, b and c are each independently 1 to 3, d is 0 to 4, e is 0 to 3, f is 0 to 3 , a+d is 5 or less, b+e is 4 or less, and c+f is 4 or less.
- R 5 in the group represented by —C(R 4 ) 2 COOR 5 a group that is decomposed by the action of an acid to produce an alkali-soluble group, a group that is eliminated by the action of an acid, and —C(R 36 )(R 37 )(R 38 ), —C(R 36 )(R 37 )(OR 39 ), —C(R 01 )(R 02 )(OR 39 ), and the like.
- R 36 to R 39 each independently represent an alkyl group, cycloalkyl group, aryl group, aralkyl group or alkenyl group.
- R 36 and R 37 may combine with each other to form a ring.
- alkyl group an alkyl group having 1 to 10 carbon atoms is preferable, and an alkyl group having 1 to 5 carbon atoms is more preferable.
- the alkyl group may be linear or branched.
- a cycloalkyl group having 3 to 12 carbon atoms is preferable, and a cycloalkyl group having 3 to 8 carbon atoms is more preferable.
- the cycloalkyl group may have a monocyclic structure or a polycyclic structure such as a condensed ring.
- the aryl group is preferably an aromatic hydrocarbon group having 6 to 30 carbon atoms, more preferably a phenyl group.
- an aralkyl group having 7 to 20 carbon atoms is preferable, and an aralkyl group having 7 to 16 carbon atoms is more preferable.
- the aralkyl group is intended to be an aryl group substituted with an alkyl group, and preferred embodiments of these alkyl and aryl groups are the same as the preferred embodiments of the alkyl and aryl groups described above.
- the alkenyl group is preferably an alkenyl group having 3 to 20 carbon atoms, more preferably an alkenyl group having 3 to 16 carbon atoms. Moreover, these groups may further have a known substituent within the range in which the effects of the present invention can be obtained.
- R 01 and R 02 each independently represent a hydrogen atom, an alkyl group, a cycloalkyl group, an aryl group, an aralkyl group or an alkenyl group.
- the group that is decomposed by the action of an acid to form an alkali-soluble group or the group that is eliminated by the action of an acid is preferably a tertiary alkyl ester group, an acetal group, a cumyl ester group, an enol ester group, or the like. More preferred are tertiary alkyl ester groups and acetal groups.
- compounds having an alkoxymethyl group include the following structures.
- Examples of the compound having an acyloxymethyl group include compounds obtained by changing the alkoxymethyl group of the following compounds to an acyloxymethyl group.
- Compounds having an alkoxymethyl group or acyloxymethyl in the molecule include, but are not limited to, the following compounds.
- the compound containing at least one of an alkoxymethyl group and an acyloxymethyl group a commercially available one or a compound synthesized by a known method may be used. From the viewpoint of heat resistance, compounds in which an alkoxymethyl group or acyloxymethyl group is directly substituted on an aromatic ring or a triazine ring are preferred.
- melamine-based cross-linking agents include hexamethoxymethylmelamine, hexaethoxymethylmelamine, hexapropoxymethylmelamine, and hexabutoxybutylmelamine.
- urea-based cross-linking agents include monohydroxymethylated glycoluril, dihydroxymethylated glycoluril, trihydroxymethylated glycoluril, tetrahydroxymethylated glycoluril, monomethoxymethylated glycoluril, and dimethoxymethylated glycol.
- Uril trimethoxymethylated glycoluril, tetramethoxymethylated glycoluril, monoethoxymethylated glycoluril, diethoxymethylated glycoluril, triethoxymethylated glycoluril, tetraethoxymethylated glycoluril, monopropoxymethylated glycoluril , dipropoxymethylated glycoluril, tripropoxymethylated glycoluril, tetrapropoxymethylated glycoluril, monobutoxymethylated glycoluril, dibutoxymethylated glycoluril, tributoxymethylated glycoluril, or tetrabutoxymethylated glycoluril glycoluril-based crosslinkers such as uril; urea-based cross-linking agents such as bismethoxymethylurea, bisethoxymethylurea, bispropoxymethylurea, and bisbutoxymethylurea; monohydroxymethylated ethyleneurea or dihydroxymethylated ethyleneurea, monomethoxymethylated ethyleneurea, dimethoxymethylated
- benzoguanamine-based cross-linking agents include monohydroxymethylated benzoguanamine, dihydroxymethylated benzoguanamine, trihydroxymethylated benzoguanamine, tetrahydroxymethylated benzoguanamine, monomethoxymethylated benzoguanamine, dimethoxymethylated benzoguanamine, and trimethoxymethylated benzoguanamine.
- tetramethoxymethylated benzoguanamine monoethoxymethylated benzoguanamine, diethoxymethylated benzoguanamine, triethoxymethylated benzoguanamine, tetraethoxymethylated benzoguanamine, monopropoxymethylated benzoguanamine, dipropoxymethylated benzoguanamine, tripropoxymethylated benzoguanamine, tetra propoxymethylated benzoguanamine, monobutoxymethylated benzoguanamine, dibutoxymethylated benzoguanamine, tributoxymethylated benzoguanamine, tetrabutoxymethylated benzoguanamine, and the like.
- the compound having at least one group selected from the group consisting of a methylol group and an alkoxymethyl group includes at least one group selected from the group consisting of a methylol group and an alkoxymethyl group on an aromatic ring (preferably a benzene ring).
- Compounds to which a seed group is directly attached are also preferably used. Specific examples of such compounds include benzenedimethanol, bis(hydroxymethyl)cresol, bis(hydroxymethyl)dimethoxybenzene, bis(hydroxymethyl)diphenyl ether, bis(hydroxymethyl)benzophenone, hydroxymethylphenyl hydroxymethylbenzoate.
- suitable commercial products include 46DMOC, 46DMOEP (manufactured by Asahi Organic Chemicals Industry Co., Ltd.), DML-PC, DML-PEP, DML-OC, and DML-OEP.
- DML-34X DML-PTBP, DML-PCHP, DML-OCHP, DML-PFP, DML-PSBP, DML-POP, DML-MBOC, DML-MBPC, DML-MTrisPC, DML-BisOC-Z, DML-BisOCHP -Z, DML-BPC, DMLBisOC-P, DMOM-PC, DMOM-PTBP, DMOM-MBPC, TriML-P, TriML-35XL, TML-HQ, TML-BP, TML-pp-BPF, TML-BPE, TML -BPA, TML-BPAF, TML-BPAP, TMOM-BP, TMOM-BPE, TMOM-BPA, TMOM-BPAF, TMOM-BPAP, HML-TPPHBA, HML-TPHAP, HMOM-TPPHBA, HMOM-TPHAP (Honshu Chemical Industry Co., Ltd.), Nikalac (registered
- the resin composition of the present invention preferably contains at least one compound selected from the group consisting of epoxy compounds, oxetane compounds, and benzoxazine compounds as another cross-linking agent.
- Epoxy compound (compound having an epoxy group) -
- the epoxy compound is preferably a compound having two or more epoxy groups in one molecule.
- the epoxy group undergoes a cross-linking reaction at 200° C. or less and does not undergo a dehydration reaction resulting from the cross-linking, so film shrinkage does not easily occur. Therefore, containing an epoxy compound is effective for low-temperature curing and suppression of warpage of the resin composition of the present invention.
- the epoxy compound preferably contains a polyethylene oxide group.
- the polyethylene oxide group means that the number of repeating units of ethylene oxide is 2 or more, and the number of repeating units is preferably 2-15.
- epoxy compounds include bisphenol A type epoxy resin; bisphenol F type epoxy resin; propylene glycol diglycidyl ether, neopentyl glycol diglycidyl ether, ethylene glycol diglycidyl ether, butylene glycol diglycidyl ether, hexamethylene glycol diglycidyl ether.
- alkylene glycol type epoxy resins such as trimethylolpropane triglycidyl ether or polyhydric alcohol hydrocarbon type epoxy resins
- polyalkylene glycol type epoxy resins such as polypropylene glycol diglycidyl ether
- epoxy groups such as polymethyl (glycidyloxypropyl) siloxane Examples include, but are not limited to, containing silicones and the like.
- Epiclon (registered trademark) 850-S Epiclon (registered trademark) HP-4032, Epiclon (registered trademark) HP-7200, Epiclon (registered trademark) HP-820, Epiclon (registered trademark) HP-4700, Epiclon (registered trademark) HP-4770, Epiclon (registered trademark) EXA-830LVP, Epiclon (registered trademark) EXA-8183, Epiclon (registered trademark) EXA-8169, Epiclon (registered trademark) N-660, Epiclon (registered trademark) N-665-EXP-S, Epiclon (registered trademark) N-740 (trade name, manufactured by DIC Corporation), Ricaresin (registered trademark) BEO-20E, Jamaicaresin (registered trademark) BEO-60E, Ricaresin (registered trademark) ) HBE-100, Ricaresin (registered trademark) DME-100, Ricaresin (registered trademark)
- n is an integer of 1-5 and m is an integer of 1-20.
- n 1 to 2 and m is 3 to 7 from the viewpoint of achieving both heat resistance and elongation improvement.
- oxetane compound compound having an oxetanyl group
- the oxetane compounds include compounds having two or more oxetane rings in one molecule, 3-ethyl-3-hydroxymethyloxetane, 1,4-bis ⁇ [(3-ethyl-3-oxetanyl)methoxy]methyl ⁇ benzene, 3-ethyl-3-(2-ethylhexylmethyl)oxetane, 1,4-benzenedicarboxylic acid-bis[(3-ethyl-3-oxetanyl)methyl]ester and the like can be mentioned.
- Aron oxetane series manufactured by Toagosei Co., Ltd. eg, OXT-121, OXT-221
- OXT-121, OXT-221 can be suitably used, and these can be used alone or in combination of two or more. good.
- a benzoxazine compound (compound having a benzoxazolyl group)-
- a benzoxazine compound is preferable because it is a cross-linking reaction derived from a ring-opening addition reaction, so that degassing does not occur during curing, and thermal shrinkage is reduced to suppress warping.
- benzoxazine compounds include Pd-type benzoxazine, Fa-type benzoxazine (these are trade names, manufactured by Shikoku Kasei Kogyo Co., Ltd.), benzoxazine adducts of polyhydroxystyrene resins, phenol novolac-type dihydrobenzoxazines, oxazine compounds. These may be used alone or in combination of two or more.
- the content of the other cross-linking agent is preferably 0.1 to 30% by mass, more preferably 0.1 to 20% by mass, more preferably 0.1 to 20% by mass, based on the total solid content of the resin composition of the present invention. It is more preferably 5 to 15% by mass, particularly preferably 1.0 to 10% by mass.
- Other cross-linking agents may be contained alone, or may be contained in two or more. When two or more other cross-linking agents are contained, the total is preferably within the above range.
- the resin composition of the present invention may contain a base generator.
- the base generator is a compound capable of generating a base by physical or chemical action.
- Preferred base generators for the resin composition of the present invention include thermal base generators and photobase generators.
- the resin composition when the resin composition contains a cyclized resin precursor, the resin composition preferably contains a base generator.
- the base generator may be an ionic base generator or a non-ionic base generator.
- bases generated from base generators include secondary amines and tertiary amines. There are no particular restrictions on the base generator used in the present invention, and known base generators can be used. Examples of known base generators include carbamoyloxime compounds, carbamoylhydroxylamine compounds, carbamic acid compounds, formamide compounds, acetamide compounds, carbamate compounds, benzylcarbamate compounds, nitrobenzylcarbamate compounds, sulfonamide compounds, imidazole derivative compounds, and amine imides.
- nonionic base generator examples include compounds represented by formula (B1), formula (B2), or formula (B3). Although the base generators exemplified below include nitrogen-containing cyclic compounds, these are not included in the above-mentioned "nitrogen-containing cyclic compounds".
- Rb 1 , Rb 2 and Rb 3 are each independently an organic group having no tertiary amine structure, a halogen atom or a hydrogen atom. However, Rb 1 and Rb 2 are not hydrogen atoms at the same time. Also, none of Rb 1 , Rb 2 and Rb 3 has a carboxy group.
- the tertiary amine structure refers to a structure in which all three bonds of a trivalent nitrogen atom are covalently bonded to a hydrocarbon-based carbon atom. Therefore, when the bonded carbon atom is a carbon atom forming a carbonyl group, that is, when forming an amide group together with the nitrogen atom, this is not the case.
- Rb 1 , Rb 2 and Rb 3 preferably contains a cyclic structure, and more preferably at least two of them contain a cyclic structure.
- the cyclic structure may be either a single ring or a condensed ring, preferably a single ring or a condensed ring in which two single rings are condensed.
- the monocyclic ring is preferably a 5- or 6-membered ring, more preferably a 6-membered ring.
- the monocyclic ring is preferably a cyclohexane ring and a benzene ring, more preferably a cyclohexane ring.
- Rb 1 and Rb 2 are a hydrogen atom, an alkyl group (preferably 1 to 24 carbon atoms, more preferably 2 to 18 carbon atoms, even more preferably 3 to 12 carbon atoms), an alkenyl group (preferably 2 to 24 carbon atoms). , more preferably 2 to 18, more preferably 3 to 12), an aryl group (preferably 6 to 22 carbon atoms, more preferably 6 to 18, even more preferably 6 to 10), or an arylalkyl group (7 carbon atoms to 25 are preferred, 7 to 19 are more preferred, and 7 to 12 are even more preferred). These groups may have substituents to the extent that the effects of the present invention are exhibited. Rb 1 and Rb 2 may combine with each other to form a ring.
- the ring to be formed is preferably a 4- to 7-membered nitrogen-containing heterocyclic ring.
- Rb 1 and Rb 2 are particularly linear, branched or cyclic alkyl groups (having preferably 1 to 24 carbon atoms, more preferably 2 to 18 carbon atoms, and still more preferably 3 to 12 carbon atoms) which may have a substituent.
- Rb 3 is an alkyl group (preferably 1 to 24 carbon atoms, more preferably 2 to 18 carbon atoms, still more preferably 3 to 12 carbon atoms), an aryl group (preferably 6 to 22 carbon atoms, more preferably 6 to 18 carbon atoms, 6 to 10 are more preferred), alkenyl groups (preferably 2 to 24 carbon atoms, more preferably 2 to 12, more preferably 2 to 6), arylalkyl groups (preferably 7 to 23 carbon atoms, more preferably 7 to 19 preferably 7 to 12), arylalkenyl groups (preferably 8 to 24 carbon atoms, more preferably 8 to 20, more preferably 8 to 16), alkoxyl groups (preferably 1 to 24 carbon atoms, 2 to 18 is more preferred, and 3 to 12 are even more preferred), an aryloxy group (preferably 6 to 22 carbon atoms, more preferably 6 to 18, and even more preferably 6 to 12), or an arylalkyloxy group (preferably 7 to 12 carbon atoms).
- an aryl group preferably
- Rb 3 may further have a substituent as long as the effects of the present invention are exhibited.
- the compound represented by Formula (B1) is preferably a compound represented by Formula (B1-1) or Formula (B1-2) below.
- Rb 11 and Rb 12 and Rb 31 and Rb 32 are respectively the same as Rb 1 and Rb 2 in formula (B1).
- Rb 13 is an alkyl group (preferably 1 to 24 carbon atoms, more preferably 2 to 18 carbon atoms, still more preferably 3 to 12 carbon atoms), an alkenyl group (preferably 2 to 24 carbon atoms, more preferably 2 to 18 carbon atoms, 3 to 12 is more preferred), an aryl group (preferably 6 to 22 carbon atoms, more preferably 6 to 18, more preferably 6 to 12), an arylalkyl group (preferably 7 to 23 carbon atoms, more preferably 7 to 19, 7 to 12 are more preferable), and may have a substituent within the range in which the effects of the present invention are exhibited.
- Rb 13 is preferably an arylalkyl group.
- Rb 33 and Rb 34 each independently represents a hydrogen atom, an alkyl group (preferably 1 to 12 carbon atoms, more preferably 1 to 8 carbon atoms, and even more preferably 1 to 3 carbon atoms), an alkenyl group (preferably 2 to 12 carbon atoms , more preferably 2 to 8, more preferably 2 to 3), an aryl group (preferably 6 to 22 carbon atoms, more preferably 6 to 18, more preferably 6 to 10), an arylalkyl group (7 to 23 is preferred, 7 to 19 are more preferred, and 7 to 11 are even more preferred), and a hydrogen atom is preferred.
- an alkyl group preferably 1 to 12 carbon atoms, more preferably 1 to 8 carbon atoms, and even more preferably 1 to 3 carbon atoms
- an alkenyl group preferably 2 to 12 carbon atoms , more preferably 2 to 8, more preferably 2 to 3
- an aryl group preferably 6 to 22 carbon atoms, more preferably 6 to 18, more preferably 6 to 10
- Rb 35 is an alkyl group (preferably 1 to 24 carbon atoms, more preferably 1 to 12 carbon atoms, still more preferably 3 to 8 carbon atoms), an alkenyl group (preferably 2 to 12 carbon atoms, more preferably 2 to 10 carbon atoms, 3 to 8 is more preferred), an aryl group (preferably 6 to 22 carbon atoms, more preferably 6 to 18, even more preferably 6 to 12), an arylalkyl group (preferably 7 to 23 carbon atoms, more preferably 7 to 19 , 7 to 12 are more preferred), and aryl groups are preferred.
- the compound represented by formula (B1-1) is also preferably the compound represented by formula (B1-1a).
- Rb 11 and Rb 12 have the same definitions as Rb 11 and Rb 12 in formula (B1-1).
- Rb 15 and Rb 16 are hydrogen atoms, alkyl groups (preferably 1 to 12 carbon atoms, more preferably 1 to 6, even more preferably 1 to 3), alkenyl groups (preferably 2 to 12 carbon atoms, 2 to 6 more preferably 2 to 3), an aryl group (preferably 6 to 22 carbon atoms, more preferably 6 to 18, even more preferably 6 to 10), an arylalkyl group (preferably 7 to 23 carbon atoms, 7 to 19 are more preferred, and 7 to 11 are even more preferred), and a hydrogen atom or a methyl group is preferred.
- Rb 17 is an alkyl group (preferably 1 to 24 carbon atoms, more preferably 1 to 12 carbon atoms, still more preferably 3 to 8 carbon atoms), an alkenyl group (preferably 2 to 12 carbon atoms, more preferably 2 to 10 carbon atoms, 3 to 8 is more preferred), an aryl group (preferably 6 to 22 carbon atoms, more preferably 6 to 18, more preferably 6 to 12), an arylalkyl group (preferably 7 to 23 carbon atoms, more preferably 7 to 19, 7 to 12 are more preferable), and aryl groups are particularly preferable.
- an alkyl group preferably 1 to 24 carbon atoms, more preferably 1 to 12 carbon atoms, still more preferably 3 to 8 carbon atoms
- an alkenyl group preferably 2 to 12 carbon atoms, more preferably 2 to 10 carbon atoms, 3 to 8 is more preferred
- an aryl group preferably 6 to 22 carbon atoms, more preferably 6 to 18, more preferably 6 to 12
- L is a divalent hydrocarbon group having a saturated hydrocarbon group on a connecting chain route connecting adjacent oxygen atoms and carbon atoms, wherein the number of atoms on the connecting chain route is represents a hydrocarbon group of 3 or more.
- RN1 and RN2 each independently represent a monovalent organic group.
- the term “connected chain” refers to the shortest (minimum number of atoms) of atomic chains on a path connecting two atoms or groups of atoms to be connected.
- L is composed of a phenylene ethylene group, has an ethylene group as a saturated hydrocarbon group
- the linking chain is composed of four carbon atoms, and on the route of the linking chain
- the number of atoms of (that is, the number of atoms constituting the linked chain, hereinafter also referred to as "linked chain length" or "linked chain length”) is 4.
- the number of carbon atoms in L (including carbon atoms other than carbon atoms in the connecting chain) in formula (B3) is preferably 3-24.
- the upper limit is more preferably 12 or less, still more preferably 10 or less, and particularly preferably 8 or less. More preferably, the lower limit is 4 or more.
- the upper limit of the linking chain length of L is preferably 12 or less, more preferably 8 or less, further preferably 6 or less, and 5 The following are particularly preferred.
- the linking chain length of L is preferably 4 or 5, most preferably 4.
- Specific preferred compounds of the base generator include, for example, compounds described in paragraph numbers 0102 to 0168 of WO2020/066416, and compounds described in paragraph numbers 0143 to 0177 of WO2018/038002. mentioned.
- the base generator preferably contains a compound represented by the following formula (N1).
- R N1 and R N2 each independently represent a monovalent organic group
- R C1 represents a hydrogen atom or a protecting group
- L represents a divalent linking group
- L is a divalent linking group, preferably a divalent organic group.
- the linking chain length of the linking group is preferably 1 or more, more preferably 2 or more.
- the upper limit is preferably 12 or less, more preferably 8 or less, and even more preferably 5 or less.
- the linking chain length is the number of atoms present in the atomic arrangement that provides the shortest path between two carbonyl groups in the formula.
- R 1 N1 and R 2 N2 each independently represent a monovalent organic group (preferably 1 to 24 carbon atoms, more preferably 2 to 18 carbon atoms, more preferably 3 to 12 carbon atoms), and a hydrocarbon group ( preferably 1 to 24 carbon atoms, more preferably 1 to 12 carbon atoms, more preferably 1 to 10 carbon atoms), specifically, an aliphatic hydrocarbon group (preferably 1 to 24 carbon atoms, 1 to 12 is more preferable, 1 to 10 is more preferable) or an aromatic hydrocarbon group (preferably 6 to 22 carbon atoms, more preferably 6 to 18, more preferably 6 to 10), and an aliphatic hydrocarbon groups are preferred.
- a monovalent organic group preferably 1 to 24 carbon atoms, more preferably 2 to 18 carbon atoms, more preferably 3 to 12 carbon atoms
- a hydrocarbon group preferably 1 to 24 carbon atoms, more preferably 1 to 12 carbon atoms, more preferably 1 to 10 carbon atoms
- an aliphatic hydrocarbon group
- an aliphatic hydrocarbon group as R N1 and R N2 because the generated base has high basicity.
- the aliphatic hydrocarbon group and the aromatic hydrocarbon group may have a substituent, and the aliphatic hydrocarbon group and the aromatic hydrocarbon group are in the aliphatic hydrocarbon chain or in the aromatic ring, You may have an oxygen atom in the substituent.
- an aspect in which the aliphatic hydrocarbon group has an oxygen atom in the hydrocarbon chain is exemplified.
- Aliphatic hydrocarbon groups constituting R N1 and R N2 include linear or branched chain alkyl groups, cyclic alkyl groups, groups related to combinations of chain alkyl groups and cyclic alkyl groups, and oxygen atoms in the chains.
- Alkyl groups having The linear or branched chain alkyl group preferably has 1 to 24 carbon atoms, more preferably 2 to 18 carbon atoms, and still more preferably 3 to 12 carbon atoms.
- Linear or branched chain alkyl groups are, for example, methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, isopropyl group, isobutyl group, secondary butyl group, tertiary butyl group, isopentyl group, neopentyl group, tertiary pentyl group, isohexyl group and the like.
- the cyclic alkyl group preferably has 3 to 12 carbon atoms, more preferably 3 to 6 carbon atoms.
- Cyclic alkyl groups include, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and cyclooctyl groups.
- Groups associated with a combination of a chain alkyl group and a cyclic alkyl group preferably have 4 to 24 carbon atoms, more preferably 4 to 18 carbon atoms, and even more preferably 4 to 12 carbon atoms.
- Groups related to combinations of chain alkyl groups and cyclic alkyl groups include, for example, a cyclohexylmethyl group, a cyclohexylethyl group, a cyclohexylpropyl group, a methylcyclohexylmethyl group, and an ethylcyclohexylethyl group.
- the alkyl group having an oxygen atom in the chain preferably has 2 to 12 carbon atoms, more preferably 2 to 6 carbon atoms, and still more preferably 2 to 4 carbon atoms.
- An alkyl group having an oxygen atom in the chain may be chain or cyclic, and may be linear or branched.
- R 3 N1 and R 3 N2 are preferably alkyl groups having 5 to 12 carbon atoms, from the viewpoint of raising the boiling point of the decomposition base described below.
- a group having a cyclic alkyl group or an alkyl group having 1 to 8 carbon atoms is preferable.
- RN1 and RN2 may be linked to each other to form a ring structure.
- the chain may have an oxygen atom or the like.
- the cyclic structure formed by R N1 and R N2 may be a monocyclic ring or a condensed ring, but is preferably a monocyclic ring.
- the cyclic structure to be formed is preferably a 5- or 6-membered ring containing a nitrogen atom in formula (N1), such as pyrrole ring, imidazole ring, pyrazole ring, pyrroline ring, pyrrolidine ring, imidazolidine ring, A pyrazolidine ring, a piperidine ring, a piperazine ring, a morpholine ring and the like can be mentioned, and a pyrroline ring, a pyrrolidine ring, a piperidine ring, a piperazine ring and a morpholine ring are preferably mentioned.
- N1 nitrogen atom in formula (N1)
- R C1 represents a hydrogen atom or a protecting group, preferably a hydrogen atom.
- the protective group is preferably a protective group that is decomposed by the action of an acid or a base, and preferably includes a protective group that is decomposed by an acid.
- protecting groups include chain or cyclic alkyl groups or chain or cyclic alkyl groups having an oxygen atom in the chain.
- Chain or cyclic alkyl groups include methyl group, ethyl group, isopropyl group, tert-butyl group, cyclohexyl group and the like.
- the chain alkyl group having an oxygen atom in the chain specifically includes an alkyloxyalkyl group, more specifically a methyloxymethyl (MOM) group, an ethyloxyethyl (EE) group, and the like. mentioned.
- Cyclic alkyl groups having an oxygen atom in the chain include epoxy group, glycidyl group, oxetanyl group, tetrahydrofuranyl group, tetrahydropyranyl (THP) group and the like.
- the divalent linking group constituting L is not particularly defined, but is preferably a hydrocarbon group, more preferably an aliphatic hydrocarbon group.
- the hydrocarbon group may have substituents and may have atoms of types other than carbon atoms in the hydrocarbon chain. More specifically, it is preferably a divalent hydrocarbon linking group which may have an oxygen atom in the chain, and a divalent aliphatic hydrocarbon which may have an oxygen atom in the chain group, a divalent aromatic hydrocarbon group, or a group related to a combination of a divalent aliphatic hydrocarbon group which may have an oxygen atom in the chain and a divalent aromatic hydrocarbon group, A divalent aliphatic hydrocarbon group which may have an oxygen atom in the chain is more preferred.
- the divalent hydrocarbon linking group preferably has 1 to 24 carbon atoms, more preferably 2 to 12 carbon atoms, and even more preferably 2 to 6 carbon atoms.
- the divalent aliphatic hydrocarbon group preferably has 1 to 12 carbon atoms, more preferably 2 to 6 carbon atoms, and still more preferably 2 to 4 carbon atoms.
- the divalent aromatic hydrocarbon group preferably has 6 to 22 carbon atoms, more preferably 6 to 18 carbon atoms, and even more preferably 6 to 10 carbon atoms.
- a group related to a combination of a divalent aliphatic hydrocarbon group and a divalent aromatic hydrocarbon group preferably has 7 to 22 carbon atoms, more preferably 7 to 18, and 7 to 10 is more preferred.
- linking group L examples include a linear or branched chain alkylene group, a cyclic alkylene group, a group related to a combination of a chain alkylene group and a cyclic alkylene group, and an alkylene group having an oxygen atom in the chain.
- a linear or branched alkenylene group, a cyclic alkenylene group, an arylene group and an arylenealkylene group are preferred.
- the linear or branched chain alkylene group preferably has 1 to 12 carbon atoms, more preferably 2 to 6 carbon atoms, and still more preferably 2 to 4 carbon atoms.
- the cyclic alkylene group preferably has 3 to 12 carbon atoms, more preferably 3 to 6 carbon atoms.
- the group associated with the combination of a chain alkylene group and a cyclic alkylene group preferably has 4 to 24 carbon atoms, more preferably 4 to 12 carbon atoms, and even more preferably 4 to 6 carbon atoms.
- An alkylene group having an oxygen atom in the chain may be chain or cyclic, and may be linear or branched.
- the alkylene group having an oxygen atom in the chain preferably has 1 to 12 carbon atoms, more preferably 1 to 6 carbon atoms, and still more preferably 1 to 3 carbon atoms.
- the linear or branched chain alkenylene group preferably has 2 to 12 carbon atoms, more preferably 2 to 6 carbon atoms, and still more preferably 2 to 3 carbon atoms.
- the linear or branched chain alkenylene group preferably has 1 to 10 C ⁇ C bonds, more preferably 1 to 6, even more preferably 1 to 3.
- the cyclic alkenylene group preferably has 3 to 12 carbon atoms, more preferably 3 to 6 carbon atoms.
- the number of C ⁇ C bonds in the cyclic alkenylene group is preferably 1-6, more preferably 1-4, even more preferably 1-2.
- the arylene group preferably has 6 to 22 carbon atoms, more preferably 6 to 18 carbon atoms, and even more preferably 6 to 10 carbon atoms.
- the arylene alkylene group preferably has 7 to 23 carbon atoms, more preferably 7 to 19 carbon atoms, and even more preferably 7 to 11 carbon atoms.
- a chain alkylene group, a cyclic alkylene group, an alkylene group having an oxygen atom in the chain, a chain alkenylene group, an arylene group, and an arylene alkylene group are preferable, and a 1,2-ethylene group and a propanediyl group (especially 1, 3-propanediyl group), cyclohexanediyl group (especially 1,2-cyclohexanediyl group), vinylene group (especially cis-vinylene group), phenylene group (1,2-phenylene group), phenylenemethylene group (especially 1,2-phenylene methylene group) and ethyleneoxyethylene group (especially 1,2-ethyleneoxy-1,2-ethylene group) are more preferable.
- base generators include the following, but the present invention should not be construed as being limited thereto.
- the molecular weight of the nonionic base generator is preferably 800 or less, more preferably 600 or less, and even more preferably 500 or less.
- the lower limit is preferably 100 or more, more preferably 200 or more, and even more preferably 300 or more.
- Specific preferred compounds of the ionic base generator include, for example, compounds described in paragraphs 0148 to 0163 of International Publication No. 2018/038002.
- ammonium salts include the following compounds, but the present invention is not limited thereto.
- iminium salts include the following compounds, but the present invention is not limited thereto.
- the content of the base generator is preferably 0.1 to 50 parts by mass with respect to 100 parts by mass of the resin in the resin composition of the present invention.
- the lower limit is more preferably 0.3 parts by mass or more, and even more preferably 0.5 parts by mass or more.
- the upper limit is more preferably 30 parts by mass or less, still more preferably 20 parts by mass or less, even more preferably 10 parts by mass or less, and may be 5 parts by mass or less, or may be 4 parts by mass or less.
- One or two or more base generators can be used. When two or more are used, the total amount is preferably within the above range.
- the resin composition of the present invention preferably contains a solvent. Any known solvent can be used as the solvent.
- the solvent is preferably an organic solvent.
- Organic solvents include compounds such as esters, ethers, ketones, cyclic hydrocarbons, sulfoxides, amides, ureas, and alcohols.
- Esters such as ethyl acetate, n-butyl acetate, isobutyl acetate, hexyl acetate, amyl formate, isoamyl acetate, butyl propionate, isopropyl butyrate, ethyl butyrate, butyl butyrate, methyl lactate, ethyl lactate, ⁇ -butyrolactone , ⁇ -caprolactone, ⁇ -valerolactone, alkyl alkyloxyacetates (e.g. methyl alkyloxyacetate, ethyl alkyloxyacetate, butyl alkyloxyacetate (e.g.
- 3-alkyloxypropionic acid alkyl esters e.g., methyl 3-alkyloxypropionate, ethyl 3-alkyloxypropionate, etc.
- 2-alkyloxypropionate alkyl esters e.g., methyl 2-alkyloxypropionate, ethyl 2-alkyloxypropionate, 2-alkyl propyl oxypropionate (e.g., methyl 2-methoxypropionate, ethyl 2-methoxypropionate, propyl 2-methoxypropionate, methyl 2-ethoxypropionate, ethyl 2-ethoxypropionate)
- 2-alkyloxy- Methyl 2-methylpropionate and ethyl 2-alkyloxy-2-methylpropionate e.g., methyl 2-methoxy-2-methylpropionate, ethyl 2-ethoxy-2-methylpropionate, etc.
- ethers include ethylene glycol dimethyl ether, diethylene glycol dimethyl ether, diethylene glycol diethyl ether, diethylene glycol ethyl methyl ether, diethylene glycol butyl methyl ether, triethylene glycol dimethyl ether, tetraethylene glycol dimethyl ether, tetrahydrofuran, ethylene glycol monomethyl ether, ethylene glycol monoethyl ether, Methyl cellosolve acetate, ethyl cellosolve acetate, diethylene glycol monomethyl ether, diethylene glycol monoethyl ether, diethylene glycol monobutyl ether, propylene glycol monomethyl ether, propylene glycol dimethyl ether, propylene glycol dimethyl ether, propylene glycol monomethyl ether acetate, propylene glycol monoethyl ether acetate, ethylene glycol monobutyl ether, ethylene glycol Preferred examples include monobutyl ether acetate
- Suitable ketones include, for example, methyl ethyl ketone, cyclohexanone, cyclopentanone, 2-heptanone, 3-heptanone, 3-methylcyclohexanone, levoglucosenone, dihydrolevoglucosenone and the like.
- Suitable examples of cyclic hydrocarbons include aromatic hydrocarbons such as toluene, xylene and anisole, and cyclic terpenes such as limonene.
- Suitable sulfoxides include, for example, dimethyl sulfoxide.
- Suitable ureas include N,N,N',N'-tetramethylurea, 1,3-dimethyl-2-imidazolidinone, and the like.
- Alcohols such as methanol, ethanol, 1-propanol, 2-propanol, 1-butanol, 1-pentanol, 1-hexanol, benzyl alcohol, ethylene glycol monomethyl ether, 1-methoxy-2-propanol, 2-ethoxyethanol, Diethylene glycol monoethyl ether, diethylene glycol monohexyl ether, triethylene glycol monomethyl ether, propylene glycol monoethyl ether, propylene glycol monomethyl ether, polyethylene glycol monomethyl ether, polypropylene glycol, tetraethylene glycol, ethylene glycol monobutyl ether, ethylene glycol monobenzyl ether, ethylene glycol monophenyl ether, methylphenyl carbinol, n-amyl alcohol, methyl amyl alcohol, diacetone alcohol and the like.
- a combination of dimethyl sulfoxide and ⁇ -butyrolactone or a combination of N-methyl-2-pyrrolidone and ethyl lactate is particularly
- the content of the solvent is preferably an amount such that the total solid concentration of the resin composition of the present invention is 5 to 80% by mass, more preferably 5 to 75% by mass. More preferably, the amount is from 10 to 70% by mass, and even more preferably from 20 to 70% by mass.
- the solvent content may be adjusted according to the desired thickness of the coating and the method of application.
- the resin composition of the present invention may contain only one type of solvent, or may contain two or more types. When two or more solvents are contained, the total is preferably within the above range.
- the resin composition of the present invention preferably contains a metal adhesion improver for improving adhesion to metal materials used for electrodes, wiring, and the like.
- metal adhesion improvers include alkoxysilyl group-containing silane coupling agents, aluminum-based adhesion aids, titanium-based adhesion aids, compounds having a sulfonamide structure and compounds having a thiourea structure, phosphoric acid derivative compounds, and ⁇ -ketoesters. compounds, amino compounds, and the like.
- the resin composition of the present invention preferably contains a silane coupling agent.
- cyclic compounds containing nitrogen among the metal adhesion improvers exemplified below these are not included in the above-mentioned "nitrogen-containing cyclic compounds”.
- silane coupling agent examples include compounds described in paragraph 0167 of WO 2015/199219, compounds described in paragraphs 0062 to 0073 of JP 2014-191002, and paragraphs of WO 2011/080992.
- Compounds described in 0063-0071, compounds described in paragraphs 0060-0061 of JP-A-2014-191252, compounds described in paragraphs 0045-0052 of JP-A-2014-041264, International Publication No. 2014/097594 Compounds described in paragraph 0055, compounds described in paragraphs 0067 to 0078 of JP-A-2018-173573, the contents of which are incorporated herein.
- silane coupling agents include, for example, vinyltrimethoxysilane, vinyltriethoxysilane, 2-(3,4-epoxycyclohexyl)ethyltrimethoxysilane, 3-glycidoxypropylmethyldimethoxysilane, 3-glycid.
- xypropyltrimethoxysilane 3-glycidoxypropylmethyldiethoxysilane, 3-glycidoxypropyltriethoxysilane, p-styryltrimethoxysilane, 3-methacryloxypropylmethyldimethoxysilane, 3-methacryloxypropyltrimethoxysilane Silane, 3-methacryloxypropylmethyldiethoxysilane, 3-methacryloxypropyltriethoxysilane, 3-acryloxypropyltrimethoxysilane, N-2-(aminoethyl)-3-aminopropylmethyldimethoxysilane, N-2 -(aminoethyl)-3-aminopropyltrimethoxysilane, 3-aminopropyltrimethoxysilane, 3-aminopropyltrimethoxysilane, 3-aminopropyltrimethoxysilane, 3-aminopropyltrimeth
- Aluminum-based adhesion aids include aluminum tris(ethylacetoacetate), aluminum tris(acetylacetonate), ethylacetoacetate aluminum diisopropylate, and the like.
- the content of the metal adhesion improver is preferably 0.01 to 30 parts by mass, more preferably 0.1 to 10 parts by mass, and still more preferably 0.01 to 30 parts by mass with respect to 100 parts by mass of the specific resin. It is in the range of 5 to 5 parts by mass. When it is at least the above lower limit value, the adhesiveness between the pattern and the metal layer is improved, and when it is at most the above upper limit value, the heat resistance and mechanical properties of the pattern are improved.
- One type of metal adhesion improver may be used, or two or more types may be used. When two or more types are used, the total is preferably within the above range.
- the resin composition of the present invention preferably further contains a migration inhibitor.
- a migration inhibitor By containing the migration inhibitor, it becomes possible to effectively suppress the migration of metal ions derived from the metal layer (metal wiring) into the film.
- the migration inhibitor is not particularly limited, but includes thioureas, compounds having a sulfanyl group, hindered phenol compounds, salicylic acid derivative compounds, and hydrazide derivative compounds.
- an ion trapping agent that traps anions such as halogen ions can be used.
- Other migration inhibitors include rust inhibitors described in paragraph 0094 of JP-A-2013-015701, compounds described in paragraphs 0073 to 0076 of JP-A-2009-283711, and JP-A-2011-059656.
- the compound described in paragraph 0052, the compound described in paragraphs 0114, 0116 and 0118 of JP-A-2012-194520, the compound described in paragraph 0166 of WO 2015/199219, etc. can be used, and these The contents are incorporated herein.
- the compounds included in the nitrogen-containing cyclic compounds described above are not included in the migration inhibitor.
- the content of the migration inhibitor is preferably 0.01 to 5.0% by mass with respect to the total solid content of the resin composition of the present invention. , more preferably 0.05 to 2.0% by mass, and even more preferably 0.1 to 1.0% by mass.
- migration inhibitor Only one type of migration inhibitor may be used, or two or more types may be used. When two or more migration inhibitors are used, the total is preferably within the above range.
- the resin composition of the present invention preferably contains a polymerization inhibitor.
- Polymerization inhibitors include phenol compounds, quinone compounds, amino compounds, N-oxyl free radical compound compounds, nitro compounds, nitroso compounds, heteroaromatic compounds, metal compounds and the like.
- Specific compounds of polymerization inhibitors include p-hydroquinone, o-hydroquinone, o-methoxyphenol, p-methoxyphenol, di-tert-butyl-p-cresol, pyrogallol, p-tert-butylcatechol, 1, 4-benzoquinone, diphenyl-p-benzoquinone, 4,4′-thiobis(3-methyl-6-tert-butylphenol), 2,2′-methylenebis(4-methyl-6-tert-butylphenol), N-nitrosophenyl hydroxylamine cerium salt, N-nitroso-N-phenylhydroxyamine aluminum salt, N-nitrosodiphenylamine, N-phenylnaphthylamine, ethylenediaminetetraacetic acid, 1,2-cyclohexanediaminetetraacetic acid, glycol etherdiaminetetraacetic acid, 2, 6-di-tert-butyl-4-methylphenol,
- the content of the polymerization inhibitor is preferably 0.01 to 20% by mass with respect to the total solid content of the resin composition of the present invention. It is more preferably 0.02 to 15% by mass, and even more preferably 0.05 to 10% by mass.
- polymerization inhibitor Only one type of polymerization inhibitor may be used, or two or more types may be used. When two or more polymerization inhibitors are used, the total is preferably within the above range.
- the resin composition of the present invention may optionally contain various additives, such as surfactants, higher fatty acid derivatives, thermal polymerization initiators, inorganic particles, ultraviolet absorbers, as long as the effects of the present invention can be obtained.
- additives such as surfactants, higher fatty acid derivatives, thermal polymerization initiators, inorganic particles, ultraviolet absorbers, as long as the effects of the present invention can be obtained.
- Organic titanium compounds, antioxidants, anti-agglomerating agents, phenolic compounds, other polymer compounds, plasticizers and other auxiliaries (for example, antifoaming agents, flame retardants, etc.) can be blended. Properties such as film physical properties can be adjusted by appropriately containing these components. These components are described, for example, from paragraph number 0183 of JP-A-2012-003225 (paragraph number 0237 of corresponding US Patent Application Publication No.
- the total blending amount is preferably 3% by mass or less of the solid content of the resin composition of the present invention.
- surfactant various surfactants such as fluorine-based surfactants, silicone-based surfactants, and hydrocarbon-based surfactants can be used.
- the surfactant may be a nonionic surfactant, a cationic surfactant, or an anionic surfactant.
- the liquid properties (especially fluidity) when prepared as a coating liquid are further improved, and the uniformity of coating thickness and liquid saving are further improved. can do. That is, when a film is formed using a coating liquid to which a composition containing a surfactant is applied, the interfacial tension between the surface to be coated and the coating liquid is reduced, and the wettability to the surface to be coated is improved. , the coatability to the surface to be coated is improved. Therefore, it is possible to more preferably form a film having a uniform thickness with little unevenness in thickness.
- fluorosurfactants include Megafac F171, F172, F173, F176, F177, F141, F142, F143, F144, R30, F437, F475, F479, F482, F554, F780, RS-72-K (manufactured by DIC Corporation), Florado FC430, FC431, FC171, Novec FC4430, FC4432 (manufactured by 3M Japan Ltd.), Surflon S-382, SC-101, SC-103, SC-104, SC-105, SC1068, SC-381, SC-383, S393, KH-40 (Asahi Glass Co., Ltd.
- Fluorinated surfactants compounds described in paragraphs 0015 to 0158 of JP-A-2015-117327, compounds described in paragraphs 0117-0132 of JP-A-2011-132503 can also be used, the contents of which are incorporated herein.
- a block polymer can also be used as the fluorosurfactant, and specific examples thereof include compounds described in JP-A-2011-89090, the contents of which are incorporated herein.
- the fluorosurfactant has a repeating unit derived from a (meth)acrylate compound having a fluorine atom and 2 or more (preferably 5 or more) alkyleneoxy groups (preferably ethyleneoxy groups and propyleneoxy groups) (meta)
- a fluorine-containing polymer compound containing a repeating unit derived from an acrylate compound can also be preferably used, and the following compounds are also exemplified as fluorine-based surfactants used in the present invention.
- the weight average molecular weight of the above compound is preferably 3,000 to 50,000, more preferably 5,000 to 30,000.
- a fluorine-containing polymer having an ethylenically unsaturated group in a side chain can also be used as a fluorine-based surfactant. Specific examples include compounds described in paragraphs 0050 to 0090 and paragraphs 0289 to 0295 of JP-A-2010-164965, the contents of which are incorporated herein.
- Commercially available products include Megafac RS-101, RS-102 and RS-718K manufactured by DIC Corporation.
- the fluorine content in the fluorine-based surfactant is preferably 3-40% by mass, more preferably 5-30% by mass, and particularly preferably 7-25% by mass.
- a fluorosurfactant having a fluorine content within this range is effective in terms of uniformity of the thickness of the coating film and saving liquid, and has good solubility in the composition.
- silicone-based surfactants examples include Toray Silicone DC3PA, Toray Silicone SH7PA, Toray Silicone DC11PA, Toray Silicone SH21PA, Toray Silicone SH28PA, Toray Silicone SH29PA, Toray Silicone SH30PA, and Toray Silicone SH8400 (the above, Toray Dow Corning Co., Ltd.
- TSF-4440, TSF-4300, TSF-4445, TSF-4460, TSF-4452 manufactured by Momentive Performance Materials
- KP341, KF6001, KF6002 manufactured by Shin-Etsu Silicone Co., Ltd.
- BYK307, BYK323, and BYK330 manufactured by BYK-Chemie Co., Ltd.
- Hydrocarbon surfactants include, for example, Pionin A-76, Nucalgen FS-3PG, Pionin B-709, Pionin B-811-N, Pionin D-1004, Pionin D-3104, Pionin D-3605, Pionin D-6112, Pionin D-2104-D, Pionin D-212, Pionin D-931, Pionin D-941, Pionin D-951, Pionin E-5310, Pionin P-1050-B, Pionin P-1028-P, Pionin P-4050-T and the like (manufactured by Takemoto Oil & Fat Co., Ltd.), and the like.
- Nonionic surfactants include glycerol, trimethylolpropane, trimethylolethane and their ethoxylates and propoxylates (e.g., glycerol propoxylate, glycerol ethoxylate, etc.), polyoxyethylene lauryl ether, polyoxyethylene stearyl ether, Examples include polyoxyethylene oleyl ether, polyoxyethylene octylphenyl ether, polyoxyethylene nonylphenyl ether, polyethylene glycol dilaurate, polyethylene glycol distearate, and sorbitan fatty acid ester.
- cationic surfactants include organosiloxane polymer KP341 (manufactured by Shin-Etsu Chemical Co., Ltd.), (meth)acrylic acid-based (co)polymer Polyflow No. 75, No. 77, No. 90, No. 95 (manufactured by Kyoeisha Chemical Co., Ltd.), W001 (manufactured by Yusho Co., Ltd.), and the like.
- anionic surfactants include W004, W005, W017 (manufactured by Yusho Co., Ltd.), and Sandet BL (manufactured by Sanyo Kasei Co., Ltd.).
- the surfactant content is preferably 0.001 to 2.0% by mass, more preferably 0.005 to 1.0% by mass, based on the total solid content of the composition.
- the resin composition of the present invention is added with a higher fatty acid derivative such as behenic acid or behenic acid amide, and the resin composition of the present invention is dried in the process of drying after coating. may be unevenly distributed on the surface of the
- the content of the higher fatty acid derivative is preferably 0.1 to 10% by mass relative to the total solid content of the resin composition of the present invention. Only one type of higher fatty acid derivative may be used, or two or more types thereof may be used. When two or more higher fatty acid derivatives are used, the total is preferably within the above range.
- the resin composition of the present invention may contain a thermal polymerization initiator, particularly a thermal radical polymerization initiator.
- a thermal radical polymerization initiator is a compound that generates radicals by thermal energy and initiates or accelerates the polymerization reaction of a polymerizable compound. By adding a thermal radical polymerization initiator, the polymerization reaction of the resin and the polymerizable compound can be advanced, so that the solvent resistance can be further improved.
- the photopolymerization initiator described above may also have a function of initiating polymerization by heat, and may be added as a thermal polymerization initiator.
- thermal radical polymerization initiators include compounds described in paragraphs 0074 to 0118 of JP-A-2008-063554, the contents of which are incorporated herein.
- thermal polymerization initiator When a thermal polymerization initiator is included, its content is preferably 0.1 to 30% by mass, more preferably 0.1 to 20% by mass, based on the total solid content of the resin composition of the present invention. , more preferably 0.5 to 15% by mass.
- One type of thermal polymerization initiator may be contained, or two or more types may be contained. When two or more thermal polymerization initiators are contained, the total amount is preferably within the above range.
- the resin composition of the present invention may contain inorganic fine particles.
- inorganic particles include calcium carbonate, calcium phosphate, silica, kaolin, talc, titanium dioxide, alumina, barium sulfate, calcium fluoride, lithium fluoride, zeolite, molybdenum sulfide, and glass.
- the average particle diameter of the inorganic particles is preferably 0.01 to 2.0 ⁇ m, more preferably 0.02 to 1.5 ⁇ m, still more preferably 0.03 to 1.0 ⁇ m, and 0.04 to 0.5 ⁇ m. Especially preferred.
- the average particle size of the fine particles is the primary particle size and the volume average particle size.
- the volume average particle size can be measured by a dynamic light scattering method using Nanotrac WAVE II EX-150 (manufactured by Nikkiso Co., Ltd.). If the above measurement is difficult, the centrifugal sedimentation light transmission method, X-ray transmission method, or laser diffraction/scattering method can be used.
- the composition of the present invention may contain an ultraviolet absorber.
- an ultraviolet absorber As the ultraviolet absorber, salicylate-based, benzophenone-based, benzotriazole-based, substituted acrylonitrile-based, and triazine-based ultraviolet absorbers can be used.
- the cyclic compound containing nitrogen also exists in the ultraviolet absorber illustrated below, these shall not be included in the above-mentioned "nitrogen-containing cyclic compound.”
- Examples of salicylate-based UV absorbers include phenyl salicylate, p-octylphenyl salicylate, pt-butylphenyl salicylate, and the like.
- benzophenone-based UV absorbers examples include 2,2'-dihydroxy-4- Methoxybenzophenone, 2,2'-dihydroxy-4,4'-dimethoxybenzophenone, 2,2',4,4'-tetrahydroxybenzophenone, 2-hydroxy-4-methoxybenzophenone, 2,4-dihydroxybenzophenone, 2- and hydroxy-4-octoxybenzophenone.
- benzotriazole-based UV absorbers examples include 2-(2'-hydroxy-3',5'-di-tert-butylphenyl)-5-chlorobenzotriazole, 2-(2'-hydroxy-3 '-tert-butyl-5'-methylphenyl)-5-chlorobenzotriazole, 2-(2'-hydroxy-3'-tert-amyl-5'-isobutylphenyl)-5-chlorobenzotriazole, 2-( 2'-hydroxy-3'-isobutyl-5'-methylphenyl)-5-chlorobenzotriazole, 2-(2'-hydroxy-3'-isobutyl-5'-propylphenyl)-5-chlorobenzotriazole, 2 -(2'-hydroxy-3',5'-di-tert-butylphenyl)benzotriazole, 2-(2'-hydroxy-5'-methylphenyl)benzotriazole, 2-[2'-hydroxy-5' -(1,
- Examples of substituted acrylonitrile UV absorbers include ethyl 2-cyano-3,3-diphenylacrylate and 2-ethylhexyl 2-cyano-3,3-diphenylacrylate.
- examples of triazine-based UV absorbers include 2-[4-[(2-hydroxy-3-dodecyloxypropyl)oxy]-2-hydroxyphenyl]-4,6-bis(2,4-dimethylphenyl )-1,3,5-triazine, 2-[4-[(2-hydroxy-3-tridecyloxypropyl)oxy]-2-hydroxyphenyl]-4,6-bis(2,4-dimethylphenyl) -mono(hydroxyphenyl)triazine compounds such as 1,3,5-triazine, 2-(2,4-dihydroxyphenyl)-4,6-bis(2,4-dimethylphenyl)-1,3,5-triazine 2,4-bis(2-hydroxy-4-propyloxyphenyl)-6-(
- the above various ultraviolet absorbers may be used singly or in combination of two or more.
- the composition of the present invention may or may not contain an ultraviolet absorber, but when it does, the content of the ultraviolet absorber is 0.001% by mass with respect to the total solid mass of the composition of the present invention. It is preferably at least 1% by mass, more preferably at least 0.01% by mass and not more than 0.1% by mass.
- the resin composition of this embodiment may contain an organic titanium compound. By including the organic titanium compound in the resin composition, it is possible to form a resin layer having excellent chemical resistance even when cured at a low temperature.
- Organotitanium compounds that can be used include those in which organic groups are attached to titanium atoms through covalent or ionic bonds. Specific examples of organotitanium compounds are shown below in I) to VII): I) Titanium chelate compound: Among them, a titanium chelate compound having two or more alkoxy groups is more preferable because the storage stability of the resin composition is good and a good curing pattern can be obtained.
- titanium bis(triethanolamine) diisopropoxide titanium di(n-butoxide) bis(2,4-pentanedionate), titanium diisopropoxide bis(2,4-pentanedionate ), titanium diisopropoxide bis(tetramethylheptanedionate), titanium diisopropoxide bis(ethylacetoacetate), and the like.
- Tetraalkoxytitanium compounds for example titanium tetra(n-butoxide), titanium tetraethoxide, titanium tetra(2-ethylhexoxide), titanium tetraisobutoxide, titanium tetraisopropoxide, titanium tetramethoxide , titanium tetramethoxypropoxide, titanium tetramethylphenoxide, titanium tetra(n-nonyloxide), titanium tetra(n-propoxide), titanium tetrastearyloxide, titanium tetrakis[bis ⁇ 2,2-(allyloxymethyl) butoxide ⁇ ] and the like.
- Titanocene compounds for example, pentamethylcyclopentadienyltitanium trimethoxide, bis( ⁇ 5-2,4-cyclopentadien-1-yl)bis(2,6-difluorophenyl)titanium, bis( ⁇ 5-2, 4-cyclopentadien-1-yl)bis(2,6-difluoro-3-(1H-pyrrol-1-yl)phenyl)titanium and the like.
- Monoalkoxy titanium compounds for example, titanium tris(dioctylphosphate) isopropoxide, titanium tris(dodecylbenzenesulfonate) isopropoxide, and the like.
- Titanium oxide compounds for example, titanium oxide bis(pentanedionate), titanium oxide bis(tetramethylheptanedionate), phthalocyanine titanium oxide and the like.
- the organotitanium compound at least one compound selected from the group consisting of I) titanium chelate compounds, II) tetraalkoxytitanium compounds, and III) titanocene compounds provides better chemical resistance. It is preferable from the viewpoint of performance.
- titanium diisopropoxide bis(ethylacetoacetate), titanium tetra(n-butoxide) and bis( ⁇ 5-2,4-cyclopentadien-1-yl)bis(2,6-difluoro-3-(1H) -pyrrol-1-yl)phenyl)titanium is preferred.
- the blending amount is preferably 0.05 to 10 parts by mass, more preferably 0.1 to 2 parts by mass, per 100 parts by mass of the specific resin.
- the amount is 0.05 parts by mass or more, the resulting cured pattern exhibits good heat resistance and chemical resistance more effectively. Excellent.
- compositions of the present invention may contain antioxidants.
- an antioxidant By containing an antioxidant as an additive, it is possible to improve the elongation properties of the cured film and the adhesion to metal materials.
- Antioxidants include phenol compounds, phosphite ester compounds, thioether compounds and the like. Any phenolic compound known as a phenolic antioxidant can be used as the phenolic compound.
- Preferred phenolic compounds include hindered phenolic compounds.
- a compound having a substituent at a site adjacent to the phenolic hydroxy group (ortho position) is preferred.
- a substituted or unsubstituted alkyl group having 1 to 22 carbon atoms is preferable as the above substituent.
- the antioxidant is also preferably a compound having a phenol group and a phosphite ester group in the same molecule.
- Phosphorus-based antioxidants can also be suitably used as antioxidants.
- a phosphorus antioxidant tris[2-[[2,4,8,10-tetrakis(1,1-dimethylethyl)dibenzo[d,f][1,3,2]dioxaphosphepin-6 -yl]oxy]ethyl]amine, tris[2-[(4,6,9,11-tetra-tert-butyldibenzo[d,f][1,3,2]dioxaphosphepin-2-yl ) oxy]ethyl]amine, ethyl bis(2,4-di-tert-butyl-6-methylphenyl) phosphite, and the like.
- antioxidants examples include Adekastab AO-20, Adekastab AO-30, Adekastab AO-40, Adekastab AO-50, Adekastab AO-50F, Adekastab AO-60, Adekastab AO-60G, Adekastab AO-80. , ADEKA STAB AO-330 (manufactured by ADEKA Corporation) and the like.
- compounds described in paragraphs 0023 to 0048 of Japanese Patent No. 6268967 can also be used, the contents of which are incorporated herein.
- the composition of the present invention may also contain latent antioxidants, if desired.
- the latent antioxidant is a compound in which the site functioning as an antioxidant is protected with a protective group, and is heated at 100 to 250°C, or heated at 80 to 200°C in the presence of an acid/base catalyst.
- a compound that functions as an antioxidant by removing the protective group by the reaction is exemplified.
- latent antioxidants include compounds described in WO 2014/021023, WO 2017/030005, and JP 2017-008219, the contents of which are incorporated herein.
- Commercially available latent antioxidants include ADEKA Arkles GPA-5001 (manufactured by ADEKA Co., Ltd.).
- Examples of preferred antioxidants include 2,2-thiobis(4-methyl-6-t-butylphenol), 2,6-di-t-butylphenol and compounds of formula (3).
- R 5 represents a hydrogen atom or an alkyl group having 2 or more carbon atoms (preferably 2 to 10 carbon atoms), and R 6 represents alkylene having 2 or more carbon atoms (preferably 2 to 10 carbon atoms). represents a group.
- R 7 represents a monovalent to tetravalent organic group containing at least one of an alkylene group having 2 or more carbon atoms (preferably 2 to 10 carbon atoms), an oxygen atom and a nitrogen atom.
- k represents an integer of 1 to 4;
- the compound represented by formula (3) suppresses oxidative deterioration of the aliphatic groups and phenolic hydroxy groups possessed by the resin.
- metal oxidation can be suppressed by the antirust action on the metal material.
- R7 includes an alkyl group, a cycloalkyl group, an alkoxy group, an alkyl ether group, an alkylsilyl group, an alkoxysilyl group, an aryl group, an aryl ether group, a carboxyl group, a carbonyl group, an allyl group, a vinyl group, a heterocyclic group, - O--, --NH--, --NHNH--, combinations thereof, and the like, which may further have a substituent.
- Examples of compounds represented by general formula (3) include the following, but are not limited to the structures below.
- the amount of antioxidant to be added is preferably 0.1 to 10 parts by mass, more preferably 0.5 to 5 parts by mass, per 100 parts by mass of the specific resin.
- the addition amount 0.1 parts by mass or more By making the addition amount 0.1 parts by mass or more, the effect of improving elongation characteristics and adhesion to metal materials can be easily obtained even in a high-temperature and high-humidity environment.
- the interaction with the agent improves the sensitivity of the resin composition.
- Only one kind of antioxidant may be used, or two or more kinds thereof may be used. When two or more kinds are used, it is preferable that the total amount thereof is within the above range.
- the resin composition of the present embodiment may contain an anti-aggregation agent as necessary.
- Anti-aggregating agents include sodium polyacrylate and the like.
- the aggregation inhibitor may be used alone or in combination of two or more.
- the composition of the present invention may or may not contain an anti-aggregating agent, but when it is included, the content of the anti-aggregating agent is 0.01% by mass relative to the total solid mass of the composition of the present invention. It is preferably at least 10% by mass, more preferably at least 0.02% by mass and not more than 5% by mass.
- the resin composition of the present embodiment may contain a phenolic compound as necessary.
- phenolic compounds include Bis-Z, BisP-EZ, TekP-4HBPA, TrisP-HAP, TrisP-PA, BisOCHP-Z, BisP-MZ, BisP-PZ, BisP-IPZ, BisOCP-IPZ, BisP-CP, BisRS-2P, BisRS-3P, BisP-OCHP, methylenetris-FR-CR, BisRS-26X (these are trade names, manufactured by Honshu Chemical Industry Co., Ltd.), BIP-PC, BIR-PC, BIR-PTBP, BIR -BIPC-F (these are trade names, manufactured by Asahi Organic Chemicals Industry Co., Ltd.) and the like.
- one type of phenolic compound may be used alone, or two or more types may be used in combination.
- the composition of the present invention may or may not contain a phenolic compound, but if it does, the content of the phenolic compound is 0.01% by mass relative to the total solid mass of the composition of the present invention. It is preferably at least 30% by mass, more preferably at least 0.02% by mass and not more than 20% by mass.
- Other polymer compounds include siloxane resins, (meth)acrylic polymers obtained by copolymerizing (meth)acrylic acid, novolac resins, resole resins, polyhydroxystyrene resins, and copolymers thereof.
- Other polymer compounds may be modified products into which cross-linking groups such as methylol groups, alkoxymethyl groups and epoxy groups have been introduced.
- composition of the present invention may or may not contain other polymer compounds, but when it does, the content of the other polymer compound is 0 relative to the total solid mass of the composition of the present invention. It is preferably 0.01% by mass or more and 30% by mass or less, and more preferably 0.02% by mass or more and 20% by mass or less.
- the viscosity of the resin composition of the present invention can be adjusted by adjusting the solid content concentration of the resin composition. From the viewpoint of coating film thickness, it is preferably 20 mm 2 /s to 12,000 mm 2 /s, more preferably 30 mm 2 /s to 10,000 mm 2 /s, and even more preferably 50 mm 2 /s to 8,000 mm 2 /s. . If it is the said range, it will become easy to obtain a coating film with high uniformity.
- the water content of the resin composition of the present invention is preferably less than 2.0% by mass, more preferably less than 1.5% by mass, and even more preferably less than 1.0% by mass. If it is less than 2.0%, the storage stability of the resin composition is improved. Methods for maintaining the moisture content include adjusting the humidity in the storage conditions and reducing the porosity of the storage container during storage.
- the metal content of the resin composition of the present invention is preferably less than 5 mass ppm (parts per million), more preferably less than 1 mass ppm, and even more preferably less than 0.5 mass ppm.
- metals include sodium, potassium, magnesium, calcium, iron, copper, chromium, and nickel, but metals contained as complexes of organic compounds and metals are excluded. When multiple metals are included, the total of these metals is preferably within the above range.
- a raw material having a low metal content is selected as a raw material constituting the resin composition of the present invention.
- the raw material constituting the product is filtered through a filter, or the inside of the apparatus is lined with polytetrafluoroethylene or the like to perform distillation under conditions in which contamination is suppressed as much as possible.
- the resin composition of the present invention preferably has a halogen atom content of less than 500 ppm by mass, more preferably less than 300 ppm by mass, and less than 200 ppm by mass from the viewpoint of wiring corrosion. is more preferred.
- those present in the form of halogen ions are preferably less than 5 ppm by mass, more preferably less than 1 ppm by mass, and even more preferably less than 0.5 ppm by mass.
- Halogen atoms include chlorine and bromine atoms. It is preferable that the total amount of chlorine atoms and bromine atoms or chlorine ions and bromine ions is within the above ranges.
- ion exchange treatment and the like are preferably mentioned.
- a conventionally known container can be used as the container for the resin composition of the present invention.
- the inner wall of the container is a multi-layer bottle composed of 6 types and 6 layers of resin, and 6 types of resin are used. It is also preferred to use bottles with a seven-layer structure. Examples of such a container include the container described in JP-A-2015-123351.
- a cured product of this resin composition By curing the resin composition of the present invention, a cured product of this resin composition can be obtained.
- the cured product of the present invention is a cured product obtained by curing the resin composition of the present invention. Curing of the resin composition is preferably by heating, and the heating temperature is more preferably in the range of 120°C to 400°C, more preferably in the range of 140°C to 380°C, and 170°C. It is particularly preferred to be in the range of -350°C.
- the form of the cured product of the resin composition is not particularly limited, and can be selected from film-like, rod-like, spherical, pellet-like, etc. according to the application.
- this cured product is preferably in the form of a film.
- pattern processing of the resin composition can be used to form protective films on walls, form via holes for conduction, adjust impedance, capacitance or internal stress, and impart heat dissipation functions. You can also choose the shape.
- the film thickness of the cured product (film made of the cured product) is preferably 0.5 ⁇ m or more and 150 ⁇ m or less.
- the shrinkage ratio when the resin composition of the present invention is cured is preferably 50% or less, more preferably 45% or less, and even more preferably 40% or less.
- the imidization reaction rate of the cured product of the resin composition of the present invention is preferably 70% or higher, more preferably 80% or higher, and even more preferably 90% or higher. If it is 70% or more, a cured product having excellent mechanical properties may be obtained.
- the elongation at break of the cured product of the resin composition of the present invention is preferably 30% or more, more preferably 40% or more, and even more preferably 50% or more.
- the glass transition temperature (Tg) of the cured product of the resin composition of the present invention is preferably 180° C. or higher, more preferably 210° C. or higher, and even more preferably 230° C. or higher.
- the resin composition of the present invention can be prepared by mixing the components described above.
- the mixing method is not particularly limited, and conventionally known methods can be used. Mixing can be performed by mixing with a stirring blade, mixing with a ball mill, mixing by rotating the tank itself, or the like.
- the temperature during mixing is preferably 10-30°C, more preferably 15-25°C.
- the filter pore size is, for example, 5 ⁇ m or less, preferably 1 ⁇ m or less, more preferably 0.5 ⁇ m or less, and even more preferably 0.1 ⁇ m or less.
- the material of the filter is preferably polytetrafluoroethylene, polyethylene or nylon. HDPE (high density polyethylene) is more preferable when the material of the filter is polyethylene.
- a filter that has been pre-washed with an organic solvent may be used. In the filter filtration step, multiple types of filters may be connected in series or in parallel for use.
- filters with different pore sizes or materials may be used in combination.
- a connection mode for example, a mode in which an HDPE filter with a pore size of 1 ⁇ m is connected in series as a first stage and an HDPE filter with a pore size of 0.2 ⁇ m as a second stage are connected in series.
- various materials may be filtered multiple times.
- circulation filtration may be used.
- you may filter by pressurizing.
- the pressure to be applied is, for example, 0.01 MPa or more and 1.0 MPa or less, preferably 0.03 MPa or more and 0.9 MPa or less, and more preferably 0.05 MPa or more and 0.7 MPa or less.
- impurities may be removed using an adsorbent.
- You may combine filter filtration and the impurity removal process using an adsorbent.
- a known adsorbent can be used as the adsorbent. Examples thereof include inorganic adsorbents such as silica gel and zeolite, and organic adsorbents such as activated carbon.
- the resin composition filled in the bottle may be subjected to a degassing step under reduced pressure.
- the method for producing a cured product of the present invention includes a film forming step of applying a resin composition on a substrate to form a film, an exposure step of selectively exposing the film, and a film exposed by the exposure step. It is preferable to include a developing step of developing using a developer to form a pattern.
- the method for producing a cured product of the present invention includes the film forming step, the exposing step, the developing step, and a heating step of heating the pattern obtained by the developing step, and after development of exposing the pattern obtained by the developing step. It is particularly preferred to include at least one of the exposure steps.
- the manufacturing method of the present invention preferably includes the film forming step and the step of heating the film. Details of each step will be described below.
- the resin composition of the present invention can be used in a film-forming step in which a film is formed by applying it onto a substrate.
- the method for producing a cured product of the present invention preferably includes a film forming step of applying the resin composition onto a substrate to form a film.
- the type of base material can be appropriately determined according to the application, and includes semiconductor manufacturing base materials such as silicon, silicon nitride, polysilicon, silicon oxide, and amorphous silicon, quartz, glass, optical films, ceramic materials, vapor deposition films, Magnetic films, reflective films, metal substrates such as Ni, Cu, Cr, and Fe (for example, substrates formed from metals, and substrates having metal layers formed by plating, vapor deposition, etc.) ), paper, SOG (Spin On Glass), TFT (Thin Film Transistor) array substrates, mold substrates, plasma display panel (PDP) electrode plates, etc., and are not particularly limited.
- semiconductor manufacturing base materials such as silicon, silicon nitride, polysilicon, silicon oxide, and amorphous silicon, quartz, glass, optical films, ceramic materials, vapor deposition films, Magnetic films, reflective films, metal substrates such as Ni, Cu, Cr, and Fe (for example, substrates formed from metals, and substrates having metal layers formed by plating, vapor deposition, etc.
- a semiconductor fabrication substrate is particularly preferable, and a silicon substrate, a Cu substrate and a mold substrate are more preferable.
- these substrates may be provided with a layer such as an adhesion layer or an oxide layer made of hexamethyldisilazane (HMDS) or the like on the surface.
- HMDS hexamethyldisilazane
- the shape of the substrate is not particularly limited, and may be circular or rectangular.
- the diameter is, for example, 100 to 450 mm, preferably 200 to 450 mm.
- the short side length is, for example, 100 to 1000 mm, preferably 200 to 700 mm.
- the base material for example, a plate-like base material (substrate), preferably a panel-like base material (substrate) is used.
- the resin layer or metal layer serves as the base material.
- Specific means to be applied include dip coating, air knife coating, curtain coating, wire bar coating, gravure coating, extrusion coating, spray coating, spin coating, slit coating, An inkjet method and the like are exemplified. From the viewpoint of uniformity of film thickness, spin coating, slit coating, spray coating, or inkjet method is more preferable, and spin coating from the viewpoint of uniformity of film thickness and productivity. and slit coating methods are preferred.
- a film having a desired thickness can be obtained by adjusting the solid content concentration and application conditions of the resin composition according to the method.
- the coating method can be appropriately selected depending on the shape of the substrate. Spin coating, spray coating, ink jet method, etc.
- slit coating and spray coating are preferable for rectangular substrates.
- method, inkjet method, and the like are preferred.
- spin coating for example, it can be applied at a rotation speed of 500 to 3,500 rpm for about 10 seconds to 3 minutes.
- a method of transferring a coating film, which is formed on a temporary support in advance by the above application method, onto a base material can also be applied.
- the transfer method the manufacturing methods described in paragraphs 0023 and 0036 to 0051 of JP-A-2006-023696 and paragraphs 0096-0108 of JP-A-2006-047592 can also be suitably used in the present invention.
- a step of removing excess film at the edge of the substrate may be performed.
- processes include edge bead rinsing (EBR), back rinsing, and the like.
- EBR edge bead rinsing
- a pre-wetting process may also be employed in which the substrate is coated with various solvents before the resin composition is applied to the substrate to improve the wettability of the substrate, and then the resin composition is applied.
- the film may be subjected to a step of drying the formed film (layer) to remove the solvent (drying step) after the film forming step (layer forming step). That is, the method for producing a cured product of the present invention may include a drying step of drying the film formed by the film forming step. Moreover, the drying step is preferably performed after the film formation step and before the exposure step.
- the drying temperature of the film in the drying step is preferably 50 to 150°C, more preferably 70 to 130°C, even more preferably 90 to 110°C. Moreover, you may dry by pressure reduction.
- the drying time is exemplified from 30 seconds to 20 minutes, preferably from 1 minute to 10 minutes, more preferably from 2 minutes to 7 minutes.
- the film may be subjected to an exposure step that selectively exposes the film. That is, the method for producing a cured product of the present invention may include an exposure step of selectively exposing the film formed in the film forming step. Selectively exposing means exposing a portion of the film. Also, by selectively exposing, the film is formed with exposed regions (exposed portions) and non-exposed regions (non-exposed portions). The amount of exposure is not particularly defined as long as the resin composition of the present invention can be cured . is more preferred.
- the exposure wavelength can be appropriately determined in the range of 190-1,000 nm, preferably 240-550 nm.
- the exposure wavelength is as follows: (1) semiconductor laser (wavelength 830 nm, 532 nm, 488 nm, 405 nm, 375 nm, 355 nm etc.), (2) metal halide lamp, (3) high pressure mercury lamp, g-line (wavelength 436 nm), h-line (wavelength 405 nm), i-line (wavelength 365 nm), broad (three wavelengths of g, h, i-line), (4) excimer laser, KrF excimer laser (wavelength 248 nm), ArF excimer laser (wavelength 193 nm) ), F2 excimer laser (wavelength 157 nm), (5) extreme ultraviolet; EUV (wavelength 13.6 nm), (6) electron beam, (7) YAG laser second harmonic 532 nm, third harmonic 355 nm, etc.
- the resin composition of the present invention exposure with a high-pressure mercury lamp is particularly preferred, and exposure with i-line is particularly preferred. Thereby, particularly high exposure sensitivity can be obtained.
- the method of exposure is not particularly limited as long as at least a part of the film made of the resin composition of the present invention is exposed. mentioned.
- the film may be subjected to a step of heating after exposure (post-exposure heating step). That is, the method for producing a cured product of the present invention may include a post-exposure heating step of heating the exposed film in the exposure step.
- the post-exposure heating step can be performed after the exposure step and before the development step.
- the heating temperature in the post-exposure heating step is preferably 50°C to 140°C, more preferably 60°C to 120°C.
- the heating time in the post-exposure heating step is preferably 30 seconds to 300 minutes, more preferably 1 minute to 10 minutes.
- the heating rate in the post-exposure heating step is preferably 1 to 12° C./min, more preferably 2 to 10° C./min, still more preferably 3 to 10° C./min, from the temperature at the start of heating to the maximum heating temperature. Also, the rate of temperature increase may be appropriately changed during heating.
- the heating means in the post-exposure heating step is not particularly limited, and known hot plates, ovens, infrared heaters and the like can be used. It is also preferable to perform the heating in an atmosphere of low oxygen concentration by flowing an inert gas such as nitrogen, helium, or argon.
- the film after exposure may be subjected to a development step in which the film is developed using a developer to form a pattern.
- the method for producing a cured product of the present invention may include a development step of developing a film exposed in the exposure step with a developer to form a pattern. By performing development, one of the exposed and non-exposed portions of the film is removed to form a pattern.
- development in which the unexposed portion of the film is removed by the development process is called negative development
- development in which the exposed portion of the film is removed by the development process is called positive development.
- Examples of the developer used in the development step include a developer containing an alkaline aqueous solution or an organic solvent.
- basic compounds that the alkaline aqueous solution may contain include inorganic alkalis, primary amines, secondary amines, tertiary amines, and quaternary ammonium salts.
- TMAH tetramethylammonium hydroxide hydroxide
- potassium hydroxide sodium carbonate, sodium hydroxide, sodium silicate, sodium metasilicate, ammonia, ethylamine, n-propylamine, diethylamine, di-n-butylamine, triethylamine, methyldiethylamine, dimethylethanolamine, triethanolamine, tetraethylammonium hydroxide, tetrapropylammonium hydroxide, tetrabutylammonium hydroxide, tetrapentylammonium hydroxide, tetrahexylammonium hydroxide, tetraoctylammonium hydroxide, ethyltrimethylammonium Hydroxide, butyltrimethylammonium hydroxide, methyltriamylammonium hydroxide, dibutyldipentylammonium hydroxide, dimethylbis(2-hydroxyethyl)
- the content of the basic compound in the developer is preferably 0.01 to 10% by mass, more preferably 0.1 to 5% by mass, more preferably 0.3 to 3% by mass, based on the total mass of the developer. is more preferred.
- the organic solvent may be an ester such as ethyl acetate, n-butyl acetate, amyl formate, isoamyl acetate, isobutyl acetate, butyl propionate, isopropyl butyrate, ethyl butyrate, butyl butyrate, Methyl lactate, ethyl lactate, ⁇ -butyrolactone, ⁇ -caprolactone, ⁇ -valerolactone, alkyl alkyloxyacetate (e.g. methyl alkyloxyacetate, ethyl alkyloxyacetate, butyl alkyloxyacetate (e.g.
- 3-alkyloxypropionate alkyl esters e.g., methyl 3-alkyloxypropionate, ethyl 3-alkyloxypropionate, etc. (e.g., 3-methoxy methyl propionate, ethyl 3-methoxypropionate, methyl 3-ethoxypropionate, ethyl 3-ethoxypropionate, etc.
- 2-alkyloxypropionate alkyl esters e.g.
- methyl 2-alkyloxypropionate, 2- ethyl alkyloxypropionate, propyl 2-alkyloxypropionate, etc. e.g., methyl 2-methoxypropionate, ethyl 2-methoxypropionate, propyl 2-methoxypropionate, methyl 2-ethoxypropionate, 2-ethoxypropionic acid ethyl
- methyl 2-alkyloxy-2-methylpropionate and ethyl 2-alkyloxy-2-methylpropionate e.g.
- ethers such as diethylene glycol dimethyl ether, tetrahydrofuran, Ethylene glycol monomethyl ether, ethylene glycol monoethyl ether, methyl cellosolve acetate, ethyl cellosolve acetate, diethylene glycol monomethyl ether, diethylene glycol monoethyl ether, diethylene glycol monobutyl ether, propylene glycol monomethyl ether (PGME), propylene glycol monomethyl ether acetate (PGMEA), propylene Glycol monoethyl ether acetate
- the organic solvent can be used singly or in combination of two or more.
- a developer containing at least one selected from the group consisting of cyclopentanone, ⁇ -butyrolactone, dimethylsulfoxide, N-methyl-2-pyrrolidone, and cyclohexanone is particularly preferred, and cyclopentanone and ⁇ -butyrolactone. and dimethylsulfoxide is more preferred, and a developer containing cyclopentanone is most preferred.
- the content of the organic solvent relative to the total weight of the developer is preferably 50% by mass or more, more preferably 70% by mass or more, and 80% by mass or more. is more preferable, and 90% by mass or more is particularly preferable. Moreover, the content may be 100% by mass.
- the developer may further contain other components.
- Other components include, for example, known surfactants and known antifoaming agents.
- the method of supplying the developer is not particularly limited as long as the desired pattern can be formed, and a method of immersing the substrate on which the film is formed in the developer, and supplying the developer to the film formed on the substrate using a nozzle.
- the type of nozzle is not particularly limited, and straight nozzles, shower nozzles, spray nozzles and the like can be mentioned. From the viewpoint of permeability of the developer, removability of the non-image area, and efficiency in production, a method of supplying the developer with a straight nozzle or a method of continuously supplying the developer with a spray nozzle is preferable.
- the method of supplying with a spray nozzle is more preferable.
- the substrate is spun to remove the developer from the substrate.
- a step of removing from above may be employed, and this step may be repeated multiple times.
- the method of supplying the developer in the development process includes a process in which the developer is continuously supplied to the base material, a process in which the developer is kept substantially stationary on the base material, and a process in which the developer exceeds the developer on the base material.
- a process of vibrating with sound waves or the like and a process of combining them can be employed.
- the development time is preferably 10 seconds to 10 minutes, more preferably 20 seconds to 5 minutes.
- the temperature of the developer during development is not particularly limited, but is preferably 10 to 45°C, more preferably 18 to 30°C.
- the pattern may be washed (rinsed) with a rinse.
- a method of supplying the rinse liquid before the developer in contact with the pattern is completely dried may be employed.
- Rinse liquid When the developer is an alkaline aqueous solution, water, for example, can be used as the rinse.
- the developer is a developer containing an organic solvent, for example, a solvent different from the solvent contained in the developer (for example, water, an organic solvent different from the organic solvent contained in the developer) is used as the rinse liquid. be able to.
- the organic solvent includes esters such as ethyl acetate, n-butyl acetate, amyl formate, isoamyl acetate, isobutyl acetate, butyl propionate, isopropyl butyrate, ethyl butyrate, and butyl butyrate. , methyl lactate, ethyl lactate, ⁇ -butyrolactone, ⁇ -caprolactone, ⁇ -valerolactone, alkyl alkyloxyacetates (e.g. methyl alkyloxyacetate, ethyl alkyloxyacetate, butyl alkyloxyacetate (e.g.
- 3-alkyloxypropionate alkyl esters e.g., methyl 3-alkyloxypropionate, ethyl 3-alkyloxypropionate, etc. (e.g., 3- methyl methoxypropionate, ethyl 3-methoxypropionate, methyl 3-ethoxypropionate, ethyl 3-ethoxypropionate, etc.
- 2-alkyloxypropionate alkyl esters e.g.
- methyl 2-alkyloxypropionate 2 -ethyl alkyloxypropionate, propyl 2-alkyloxypropionate, etc. (e.g., methyl 2-methoxypropionate, ethyl 2-methoxypropionate, propyl 2-methoxypropionate, methyl 2-ethoxypropionate, 2-ethoxypropionate ethyl acid)), methyl 2-alkyloxy-2-methylpropionate and ethyl 2-alkyloxy-2-methylpropionate (e.g.
- ethers such as diethylene glycol dimethyl ether, tetrahydrofuran , ethylene glycol monomethyl ether, ethylene glycol monoethyl ether, methyl cellosolve acetate, ethyl cellosolve acetate, diethylene glycol monomethyl ether, diethylene glycol monoethyl ether, diethylene glycol monobutyl ether, propylene glycol monomethyl ether (PGME), propylene glycol monomethyl ether acetate (PGMEA), Propylene glycol monoethyl ether a
- the organic solvent can be used singly or in combination of two or more.
- the organic solvent can be used singly or in combination of two or more.
- cyclopentanone, ⁇ -butyrolactone, dimethylsulfoxide, N-methylpyrrolidone, cyclohexanone, PGMEA and PGME are particularly preferred, cyclopentanone, ⁇ -butyrolactone, dimethylsulfoxide, PGMEA and PGME are more preferred, and cyclohexanone and PGMEA are more preferred. More preferred.
- the rinse liquid contains an organic solvent
- the rinse liquid is preferably 50% by mass or more of the organic solvent, more preferably 70% by mass or more of the organic solvent, and 90% by mass or more of the organic solvent. is more preferred. Further, 100% by mass of the rinse liquid may be an organic solvent.
- the rinse solution may further contain other components.
- Other components include, for example, known surfactants and known antifoaming agents.
- the method of supplying the rinse solution is not particularly limited as long as the desired pattern can be formed.
- From the viewpoint of the permeability of the rinse liquid, the removability of the non-image areas, and the efficiency in manufacturing there are methods of supplying the rinse liquid using a shower nozzle, a straight nozzle, a spray nozzle, etc., and a continuous supply method using a spray nozzle is preferable.
- the method of supplying the rinsing liquid with a spray nozzle is more preferable.
- the rinsing step is preferably a step of supplying the rinse liquid to the film after exposure through a straight nozzle or a step of continuously supplying the same, and more preferably a step of supplying the rinse liquid through a spray nozzle.
- the method of supplying the rinse liquid in the rinse step includes a process in which the rinse liquid is continuously supplied to the base material, a process in which the rinse liquid is kept substantially stationary on the base material, and a process in which the rinse liquid is kept on the base material in a substantially stationary state. A process of vibrating with sound waves or the like and a process of combining them can be employed.
- the rinse time is preferably 10 seconds to 10 minutes, more preferably 20 seconds to 5 minutes.
- the temperature of the rinsing liquid during rinsing is not particularly specified, but is preferably 10 to 45°C, more preferably 18 to 30°C.
- the pattern obtained by the development step may be subjected to a heating step of heating the pattern obtained by the development. That is, the method for producing a cured product of the present invention may include a heating step of heating the pattern obtained by the developing step. Moreover, the method for producing a cured product of the present invention may include a heating step of heating a pattern obtained by another method without performing the developing step or a film obtained by the film forming step. In the heating step, a resin such as a polyimide precursor is cyclized into a resin such as polyimide.
- the heating temperature (maximum heating temperature) in the heating step is preferably 50 to 450°C, more preferably 150 to 350°C, still more preferably 150 to 250°C, even more preferably 160 to 250°C, particularly 160 to 230°C. preferable.
- the heating step is preferably a step of promoting the cyclization reaction of the polyimide precursor in the pattern by the action of the base generated from the base generator by heating.
- Heating in the heating step is preferably carried out at a temperature rising rate of 1 to 12° C./min from the temperature at the start of heating to the maximum heating temperature.
- the rate of temperature increase is more preferably 2 to 10°C/min, still more preferably 3 to 10°C/min.
- By setting the temperature increase rate to 1°C/min or more it is possible to prevent excessive volatilization of the acid or solvent while ensuring productivity.
- the residual stress of the object can be relaxed.
- the temperature at the start of heating is preferably 20°C to 150°C, more preferably 20°C to 130°C, and even more preferably 25°C to 120°C.
- the temperature at the start of heating refers to the temperature at which the process of heating up to the maximum heating temperature is started.
- the temperature of the film (layer) after drying is, for example, the boiling point of the solvent contained in the resin composition of the present invention.
- the heating time (heating time at the highest heating temperature) is preferably 5 to 360 minutes, more preferably 10 to 300 minutes, even more preferably 15 to 240 minutes.
- the heating temperature is preferably 30° C. or higher, more preferably 80° C. or higher, further preferably 100° C. or higher, from the viewpoint of adhesion between layers. 120° C. or higher is particularly preferred.
- the upper limit of the heating temperature is preferably 350° C. or lower, more preferably 250° C. or lower, and even more preferably 240° C. or lower.
- Heating may be done in stages. As an example, the temperature is raised from 25° C. to 120° C. at 3° C./min, held at 120° C. for 60 minutes, heated from 120° C. to 180° C. at 2° C./min, and held at 180° C. for 120 minutes. , may be performed. It is also preferable to carry out the treatment while irradiating ultraviolet rays as described in US Pat. No. 9,159,547. Such a pretreatment process can improve the properties of the film.
- the pretreatment step is preferably performed for a short time of about 10 seconds to 2 hours, more preferably 15 seconds to 30 minutes.
- the pretreatment may be performed in two or more steps.
- the first pretreatment step may be performed in the range of 100 to 150°C, and then the second pretreatment step may be performed in the range of 150 to 200°C. good. Further, cooling may be performed after heating, and the cooling rate in this case is preferably 1 to 5°C/min.
- the heating step is preferably carried out in an atmosphere of low oxygen concentration, such as by flowing an inert gas such as nitrogen, helium or argon, or under reduced pressure, in order to prevent decomposition of the specific resin.
- the oxygen concentration is preferably 50 ppm (volume ratio) or less, more preferably 20 ppm (volume ratio) or less.
- a heating means in the heating step is not particularly limited, and examples thereof include a hot plate, an infrared furnace, an electric heating oven, a hot air oven, an infrared oven and the like.
- the pattern obtained by the development step (the pattern after rinsing when the rinsing step is performed) is subjected to a post-development exposure step of exposing the pattern after the development step instead of or in addition to the heating step.
- the method for producing a cured product of the present invention may include a post-development exposure step of exposing the pattern obtained by the development step.
- the method for producing a cured product of the present invention may include a heating step and a post-development exposure step, or may include only one of the heating step and the post-development exposure step.
- a reaction in which cyclization of the polyimide precursor or the like proceeds by exposure of the photobase generator can be promoted.
- the post-development exposure step at least part of the pattern obtained in the development step may be exposed, but it is preferable that the entire pattern be exposed.
- the exposure amount in the post-development exposure step is preferably 50 to 20,000 mJ/cm 2 , more preferably 100 to 15,000 mJ/cm 2 in terms of exposure energy at the wavelength to which the photosensitive compound is sensitive. preferable.
- the post-development exposure step can be performed using, for example, the light source used in the exposure step described above, and broadband light is preferably used.
- the pattern obtained by the development step may be subjected to a metal layer forming step of forming a metal layer on the pattern. That is, the method for producing a cured product of the present invention includes a metal layer forming step of forming a metal layer on the pattern obtained by the developing step (preferably subjected to at least one of the heating step and the post-development exposure step). is preferred.
- the metal layer is not particularly limited, and existing metal species can be used. Examples include copper, aluminum, nickel, vanadium, titanium, chromium, cobalt, gold, tungsten, tin, silver, and alloys containing these metals. copper and aluminum are more preferred, and copper is even more preferred.
- the method of forming the metal layer is not particularly limited, and existing methods can be applied. For example, use the methods described in JP-A-2007-157879, JP-A-2001-521288, JP-A-2004-214501, JP-A-2004-101850, US Pat. can do.
- photolithography, PVD (Physical Vapor Deposition), CVD (Chemical Vapor Deposition), lift-off, electroplating, electroless plating, etching, printing, and a combination thereof can be considered. More specifically, a patterning method combining sputtering, photolithography and etching, and a patterning method combining photolithography and electroplating can be used.
- a preferred embodiment of plating is electroplating using a copper sulfate or copper cyanide plating solution.
- the thickness of the metal layer is preferably 0.01 to 50 ⁇ m, more preferably 1 to 10 ⁇ m, at the thickest part.
- Fields to which the cured product of the present invention can be applied include insulating films for electronic devices, interlayer insulating films for rewiring layers, and stress buffer films.
- pattern formation by etching of a sealing film, a substrate material (a base film or coverlay of a flexible printed circuit board, an interlayer insulating film), or an insulating film for mounting purposes as described above can be used.
- the method for producing the cured product of the present invention or the cured product of the present invention can also be used for the production of plates such as offset plates or screen plates, for etching molded parts, for protective lacquers and dielectrics in electronics, especially microelectronics. It can also be used for the production of layers and the like.
- the laminate of the present invention refers to a structure having a plurality of layers made of the cured product of the present invention.
- the laminate of the present invention is a laminate containing two or more layers made of a cured product, and may be a laminate in which three or more layers are laminated. Of the two or more layers of the cured product contained in the laminate, at least one is a layer made of the cured product of the present invention, and the shrinkage of the cured product, or the deformation of the cured product due to the shrinkage, etc. From the viewpoint of suppression, it is also preferable that all the layers made of the cured product contained in the laminate are layers made of the cured product of the present invention.
- the method for producing the laminate of the present invention preferably includes the method for producing the cured product of the present invention, and more preferably includes repeating the method for producing the laminate of the present invention multiple times.
- the laminate of the present invention includes two or more layers made of the cured material and a metal layer between any of the layers made of the cured material.
- the metal layer is preferably formed by the metal layer forming step. That is, it is preferable that the method for producing the laminate of the present invention further includes a metal layer forming step of forming a metal layer on the layer made of the cured product between the production methods for the cured product that are performed multiple times. Preferred aspects of the metal layer forming step are as described above.
- the laminate for example, a laminate containing at least a layer structure in which three layers of a layer made of the first cured product, a metal layer, and a layer made of the second cured product are laminated in this order is preferable. be done.
- both the layer comprising the first cured product and the layer comprising the second cured product are layers comprising the cured product of the present invention.
- the resin composition of the present invention used for forming the layer comprising the first cured product and the resin composition of the present invention used for forming the layer comprising the second cured product have the same composition. It may be a product or a composition having a different composition.
- the metal layer in the laminate of the present invention is preferably used as a metal wiring such as a rewiring layer.
- the method for manufacturing the laminate of the present invention includes a lamination step.
- the lamination step means that the surface of the pattern (resin layer) or metal layer is again subjected to (a) film formation step (layer formation step), (b) exposure step, (c) development step, (d) heating step and development It is a series of steps including performing at least one of the post-exposure steps in this order. However, at least one of (a) the film forming step and (d) the heating step and the post-development exposure step may be repeated. Moreover, after at least one of the (d) heating step and the post-development exposure step, (e) a metal layer forming step may be included. Needless to say, the lamination step may further include the drying step and the like as appropriate.
- a surface activation treatment process may be further performed.
- a plasma treatment is exemplified as the surface activation treatment. Details of the surface activation treatment will be described later.
- the lamination step is preferably performed 2 to 20 times, more preferably 2 to 9 times.
- a configuration of 2 to 20 resin layers such as resin layer/metal layer/resin layer/metal layer/resin layer/metal layer is preferable, and a configuration of 2 to 9 layers is more preferable.
- Each of the layers described above may have the same composition, shape, film thickness, etc., or may differ from each other.
- a cured product (resin layer) of the resin composition of the present invention so as to cover the metal layer after providing the metal layer.
- the film forming step, (b) the exposure step, (c) the developing step, (d) at least one of the heating step and the post-development exposure step, and (e) the metal layer forming step are repeated in this order.
- the film forming step, (d) at least one of the heating step and the post-development exposure step, and (e) the metal layer forming step are repeated in this order.
- the method for producing a laminate of the present invention preferably includes a surface activation treatment step of subjecting at least part of the metal layer and the resin composition layer to surface activation treatment.
- the surface activation treatment step is usually performed after the metal layer formation step, but after the development step (preferably after at least one of the heating step and the post-development exposure step), the resin composition layer is subjected to surface activation treatment.
- the metal layer forming step may be performed.
- the surface activation treatment may be performed only on at least part of the metal layer, may be performed only on at least part of the resin composition layer after exposure, or may be performed on the metal layer and the resin composition layer after exposure. Both may be done at least partially, respectively.
- the surface activation treatment is preferably performed on at least part of the metal layer, and it is preferable to perform the surface activation treatment on part or all of the area of the metal layer on which the resin composition layer is formed.
- the surface of the metal layer By subjecting the surface of the metal layer to the surface activation treatment in this manner, the adhesiveness to the resin composition layer (film) provided on the surface can be improved.
- the resin composition layer when the resin composition layer is cured, such as in the case of negative development, it is less likely to be damaged by surface treatment, and the adhesion is likely to be improved.
- Specific examples of the surface activation treatment include plasma treatment of various source gases (oxygen, hydrogen, argon, nitrogen, nitrogen/hydrogen mixed gas, argon/oxygen mixed gas, etc.), corona discharge treatment, and CF 4 /O 2 . , NF 3 /O 2 , SF 6 , NF 3 , NF 3 /O 2 etching treatment, surface treatment by ultraviolet (UV) ozone method, immersion in hydrochloric acid aqueous solution to remove the oxide film, and then amino groups and thiol groups.
- UV ultraviolet
- the treatment is selected from immersion treatment in an organic surface treatment agent containing at least one compound and mechanical surface roughening treatment using a brush.
- Plasma treatment is preferred, and oxygen plasma treatment using oxygen as a raw material gas is particularly preferred.
- the energy is preferably 500-200,000 J/m 2 , more preferably 1000-100,000 J/m 2 , most preferably 10,000-50,000 J/m 2 .
- the present invention also discloses a semiconductor device comprising the cured product of the present invention or the laminate of the present invention.
- the present invention also discloses a method for producing a semiconductor device including the method for producing the cured product of the present invention or the method for producing the laminate of the present invention.
- Specific examples of a semiconductor device using the resin composition of the present invention for forming an interlayer insulating film for a rewiring layer can refer to the description of paragraphs 0213 to 0218 of JP-A-2016-027357 and the description of FIG. The contents of which are incorporated herein.
- Polymer PE was prepared by dissolving 0.600 g (3.00 mmol) of oxydianiline (hereinafter abbreviated as “ODA”) in 8.50 ml of N,N-dimethylacetamide (DMAc) with 4,4′-biphenyltetracarboxylic acid. 0.883 g (3.00 mmol) of acid dianhydride (hereinafter abbreviated as "BPDA”) was added at once and stirred for 12 hours at room temperature to obtain a highly viscous transparent polymer PE solution.
- the intrinsic viscosity of polymer PE in DMAc was 0.73 dL/g at a concentration of 0.5 g/dL at 30°C.
- DMAc was purified by vacuum distillation, and ODA and BPDA were recrystallized from tetrahydrofuran (THF) and acetic anhydride, respectively.
- THF tetrahydrofuran
- Mw weight average molecular weight
- the resulting reaction solution was added to 3 liters of ethyl alcohol to form a precipitate consisting of a crude polymer.
- the resulting crude polymer was separated by filtration and dissolved in 1.5 liters of tetrahydrofuran to obtain a crude polymer solution.
- the resulting crude polymer solution was dropped into 28 liters of water to precipitate the polymer, and the obtained precipitate was filtered and dried in vacuum to obtain a powdery polymer PA-1.
- Mw weight average molecular weight
- Polymer PB-1 was obtained by carrying out the reaction in the same manner as described in . When the molecular weight of polymer PB-1 was measured by gel permeation chromatography (converted to standard polystyrene), the weight average molecular weight (Mw) was 22,000.
- the polyimide precursor was then precipitated in 5 liters of water and the water-polyimide precursor mixture was stirred at a speed of 5000 rpm for 15 minutes.
- the polyimide precursor was collected by filtration, put into 4 liters of water again, stirred for another 30 minutes, and filtered again.
- the resulting polyimide precursor was then dried under reduced pressure at 45° C. for 3 days to obtain polymer PA-2.
- Mw weight average molecular weight
- Synthesis of polymer PB-2> In Synthesis Example 4, 19.0 g (64.5 mmol) of 3,3'4,4'-biphenyl tetra- Except for using a carboxylic acid dianhydride, the reaction was carried out in the same manner as in Synthesis Example 4 to obtain a polymer PB-1.
- Mw weight average molecular weight
- the separable flask was cooled in a water bath at 15-20°C.
- the time required for dropping was 40 minutes, and the maximum temperature of the reaction solution was 30°C.
- 30.8 g (0.2 mol) of 1,2-cyclohexyldicarboxylic anhydride was added to the reaction solution and left to stir at room temperature for 15 hours to remove 99% of all amine terminal groups on the polymer chain. It was capped with a carboxycyclohexylamide group.
- the reaction rate at this time can be easily calculated by tracking the remaining amount of the charged 1,2-cyclohexyldicarboxylic anhydride by high performance liquid chromatography (HPLC).
- Examples and Comparative Examples> the components shown in the table below were mixed to obtain each negative photosensitive resin composition.
- the components shown in the table below were mixed to obtain each comparative composition.
- the content (blended amount) of each component described in the table other than the solvent is the amount (parts by mass ).
- the content (mixture amount) of the solvent is such that the solid content concentration of the composition is the value of "solid content concentration [mass%]" in the table, and the content of each solvent relative to the total mass of the solvent is It was made to be the amount (parts by mass) described in the "Addition amount” column.
- the obtained negative photosensitive resin composition and the comparative composition were subjected to pressure filtration using a polytetrafluoroethylene filter having a pore width of 0.45 ⁇ m.
- the description of "-" indicates that the composition does not contain the corresponding component.
- A-1 to A-4 and A-6 to A-8 are compounds that do not generate radical polymerization initiating species upon exposure, and A-5 is a compound that generates radical polymerization initiating species upon exposure.
- CA-1 is not a compound whose absorbance at the exposure wavelength is reduced by exposure, and does not correspond to compound A.
- F-1 Tetraethylene glycol dimethacrylate
- F-2 Light ester BP-6EM (manufactured by Kyoeisha Chemical Co., Ltd.)
- F-3 Light ester NP (manufactured by Kyoeisha Chemical Co., Ltd.)
- F-4 Light ester 1.6HX (manufactured by Kyoeisha Chemical Co., Ltd.)
- F-5 KAYARAD DPHA (manufactured by Nippon Kayaku Co., Ltd.)
- ⁇ F-6 Daicel Celoxide CEL2081 (manufactured by Daicel Co., Ltd.)
- the prepared negative photosensitive resin composition or comparative composition was applied onto a copper wafer by spin coating.
- the wafer is dried on a hot plate at the temperature and time shown in the table, and the thickness shown in the "Coating film thickness” column of the table on the copper wafer is a curable resin composition with a uniform thickness.
- a layer was formed.
- the curable resin composition layer on the copper substrate is exposed using a stepper using a photomask with contact holes of 5 ⁇ m to 20 ⁇ m in increments of 1 ⁇ m. It was exposed at a wavelength of 365 nm.
- the exposure amount was changed in increments of 50 mJ/cm 2 within the range of 250 to 650 mJ/cm 2 .
- a direct exposure device ADTEC DE-6UH III
- the exposure wavelength was set to 405 nm, and the exposure amount was varied in the range of 250 to 650 mJ/cm 2 in increments of 50 mJ/cm 2 .
- the wafer was subjected to heat treatment after exposure at the indicated temperature/time.
- the exposed curable resin composition layer was developed with cyclopentanone for 40 seconds and then rinsed with PGMEA (propylene glycol monomethyl ether acetate).
- the line width of the smallest contact hole pattern size was defined as the "minimum hole diameter" and evaluated according to the following evaluation criteria. It can be said that the smaller the minimum hole diameter, the better the resolution.
- the measurement limit is 5 ⁇ m.
- the evaluation results are shown in the "Resolution" column in the table. A or B is preferred, and A is more preferred. -Evaluation criteria- When the coating film thickness is 5 ⁇ m: A: The minimum hole diameter was 5 ⁇ m.
- B The minimum hole diameter was 5 ⁇ m or more and less than 7 ⁇ m.
- C The minimum hole diameter was 7 ⁇ m or more and less than 10 ⁇ m.
- D The minimum hole diameter was 10 ⁇ m or more.
- A The minimum hole diameter was less than 10 ⁇ m.
- B The minimum hole diameter was 10 ⁇ m or more and less than 15 ⁇ m.
- C The minimum hole diameter was 15 ⁇ m or more and less than 20 ⁇ m.
- D The minimum hole diameter was 20 ⁇ m or more.
- the post-development residual film ratio at an exposure amount capable of forming the minimum hole diameter was calculated by measuring the film thickness before and after development and calculating the ratio of the post-development film thickness to the pre-development film thickness. Evaluation was performed according to the following evaluation criteria from the calculated value of the residual film ratio, and the evaluation results were described in the column of "Remaining film ratio after development" in the table. The higher the residual film ratio, the better the result. A or B is preferred, and A is more preferred.
- Remaining film ratio (%) film thickness after development/film thickness before development x 100 -Evaluation criteria- A: The residual film rate was 90% or more. B: The residual film rate was 85% or more and less than 90%. C: The residual film rate was 80% or more and less than 85%. D: The residual film rate was less than 80%.
- a curable resin composition layer was formed on a silicon wafer by the same method as the formation of the curable resin composition layer in the evaluation of resolution described above. Thereafter, in each example or comparative example, the entire surface of the curable resin composition layer was exposed to light without using a photomask to obtain a resin film.
- the exposure method was the same as in the evaluation of resolution described above, except that no photomask was used. The exposure amount was 500 mJ/cm 2 . In evaluating the chemical resistance, the post-exposure heating step was not performed.
- the resin film obtained in each example or comparative example was heated at a rate of 10° C./min in a nitrogen atmosphere, and the temperature and time shown in the columns of "temperature” and “time” under “curing conditions” in the table were obtained. The temperature was maintained at the specified temperature and time to form a cured film.
- the obtained cured film was immersed in the following chemicals under the following conditions, and the dissolution rate was calculated.
- DMSO dimethyl sulfoxide
- TMAH tetramethylammonium hydroxide
- Remaining film ratio (%) film thickness after immersion / film thickness before immersion ⁇ 100
- the obtained value of the residual film ratio was evaluated according to the following evaluation criteria, and the evaluation results were described in the "Chemical resistance" column. It can be said that the higher the residual film ratio, the better the chemical resistance. A or B is preferred, and A is more preferred.
- C The residual film rate was 55% or more and less than 70%.
- D The residual film rate was less than 55%.
- the comparative composition according to Comparative Example 1 contains a nitrogen-containing cyclic compound but does not contain compound A. In such an example, it can be seen that the resolution is inferior.
- a comparative composition according to Comparative Example 2 contains CA-1 in place of compound A and a nitrogen-containing cyclic compound. In such an example, it can be seen that the residual film ratio after development is inferior.
- the comparative compositions according to Comparative Examples 3-5 do not contain a nitrogen-containing cyclic compound. It can be seen that in such an example, both the resolution and the post-development residual film ratio are inferior.
- Example 101 The resin composition used in Example 1 was applied in a layer by spin coating to the surface of the thin copper layer of the resin substrate having the thin copper layer formed on the surface, and dried at 100° C. for 5 minutes to obtain a film thickness. After forming a photosensitive film of 20 ⁇ m, it was exposed using a stepper (NSR1505 i6 manufactured by Nikon Corporation). Exposure was performed at a wavelength of 365 nm through a mask (a binary mask with a 1:1 line-and-space pattern and a line width of 10 ⁇ m). After the above exposure, the film was developed with cyclohexanone for 2 minutes and rinsed with PGMEA for 30 seconds to obtain a layer pattern.
- NSR1505 i6 manufactured by Nikon Corporation
- the temperature was raised at a rate of 10° C./min, reaching 230° C., and then maintained at 230° C. for 180 minutes to form an interlayer insulating film for rewiring layers.
- This interlayer insulating film for rewiring layer was excellent in insulating properties.
- a semiconductor device was manufactured using these interlayer insulating films for rewiring layers, it was confirmed that the device operated without any problem.
Abstract
Description
このようなネガ型感光性樹脂組成物を、例えば塗布等により基材に適用して感光膜を形成し、その後、必要に応じて露光、現像、加熱等を行うことにより、硬化物を基材上に形成することができる。
ポリイミド前駆体等の上記環化樹脂の前駆体は、例えば加熱により環化され、硬化物中でポリイミド等の環化樹脂となる。
ネガ型感光性樹脂組成物は、公知の塗布方法等により適用可能であるため、例えば、適用されるネガ型感光性樹脂組成物の適用時の形状、大きさ、適用位置等の設計の自由度が高いなど、製造上の適応性に優れるといえる。ポリイミド等の環化樹脂が有する高い性能に加え、このような製造上の適応性に優れる観点から、上述のネガ型感光性樹脂組成物の産業上の応用展開がますます期待されている。
特許文献2には、(A)酸性官能基又は塩基性官能基の置換体である中性官能基を側鎖中に有し、ポリアミド酸及び/又はポリアミド酸エステルであるポリイミド前駆体、ポリヒドロキシアミドであるポリオキサゾール前駆体、ポリアミノアミド、ポリアミド、ポリアミドイミド、ポリイミド、ポリベンゾオキサゾール、ポリベンゾイミダゾール、並びにポリベンズチアゾールから成る群から選択される少なくとも1種である樹脂:100質量部、並びに(B)芳香族スルホン酸エステル構造を有する化合物:1~50質量部を含む、樹脂組成物が記載されている。
<1> 樹脂、
露光によりその露光波長の吸光度が小さくなる化合物A、及び、
光重合開始剤を含み、
下記条件i及び条件iiの少なくとも一方を満たす
ネガ型感光性樹脂組成物。
条件i:組成物が、上記樹脂、上記化合物A及び上記光重合開始剤とは異なる成分として含窒素環状化合物を更に含む。
条件ii:上記樹脂として、環員として2以上の窒素原子を含む含窒素ヘテロ環構造を有する樹脂を含む。
<2> 上記化合物Aが、露光によりラジカル重合開始種を発生しない化合物である、<1>に記載のネガ型感光性樹脂組成物。
<3> 上記化合物Aが、ナフトキノンジアジド化合物を含む、<1>又は<2>に記載のネガ型感光性樹脂組成物。
<4> 上記化合物Aの含有量を100質量部とした場合の、上記含窒素環状化合物の含有量が、0.5~200質量部である、<1>~<3>のいずれか1つに記載のネガ型感光性樹脂組成物。
<5> 樹脂、
ナフトキノンジアジド化合物、及び、
光重合開始剤を含み、
下記条件iii及び条件ivの少なくとも一方を満たす
ネガ型感光性樹脂組成物。
条件iii:組成物が、上記樹脂、上記ナフトキノンジアジド化合物及び上記光重合開始とは異なる成分として含窒素環状化合物を更に含む。
条件iv:上記樹脂として、環員として2以上の窒素原子を含む含窒素ヘテロ環構造を有する樹脂を含む。
<6> 上記樹脂が環化樹脂又はその前駆体である、<1>~<5>のいずれか1つに記載のネガ型感光性樹脂組成物。
<7> 重合性化合物を更に含む、<1>~<6>のいずれか1つに記載のネガ型感光性樹脂組成物。
<8> シランカップリング剤を更に含む、<1>~<7>のいずれか1つに記載のネガ型感光性樹脂組成物。
<9> 重合禁止剤を更に含む、<1>~<8>のいずれか1つに記載のネガ型感光性樹脂組成物。
<10> 塩基発生剤を更に含む、<1>~<9>のいずれか1つに記載のネガ型感光性樹脂組成物。
<11> 上記光重合開始剤が、光ラジカル重合開始剤である、<1>~<10>のいずれか1つに記載のネガ型感光性樹脂組成物。
<12> 再配線層用層間絶縁膜の形成に用いられる、<1>~<11>のいずれか1つに記載のネガ型感光性樹脂組成物。
<13> <1>~<12>のいずれか1つに記載のネガ型感光性樹脂組成物を硬化してなる硬化物。
<14> <13>に記載の硬化物からなる層を2層以上含み、上記硬化物からなる層同士のいずれかの間に金属層を含む積層体。
<15> <1>~<12>のいずれか1つに記載のネガ型感光性樹脂組成物を基板に適用して膜を形成する膜形成工程、上記膜を選択的に露光する露光工程、及び、露光工程により露光された膜を現像液を用いて現像してパターンを形成する現像工程を含む、硬化物の製造方法。
<16> 上記膜を50~450℃で加熱する加熱工程を含む、<15>に記載の硬化物の製造方法。
<17> <13>に記載の硬化物又は<14>に記載の積層体を含む、半導体デバイス。
本明細書において「~」という記号を用いて表される数値範囲は、「~」の前後に記載される数値をそれぞれ下限値及び上限値として含む範囲を意味する。
本明細書において「工程」との語は、独立した工程だけではなく、その工程の所期の作用が達成できる限りにおいて、他の工程と明確に区別できない工程も含む意味である。
本明細書における基(原子団)の表記において、置換及び無置換を記していない表記は、置換基を有しない基(原子団)と共に置換基を有する基(原子団)をも包含する。例えば、「アルキル基」とは、置換基を有しないアルキル基(無置換アルキル基)のみならず、置換基を有するアルキル基(置換アルキル基)をも包含する。
本明細書において「露光」とは、特に断らない限り、光を用いた露光のみならず、電子線、イオンビーム等の粒子線を用いた露光も含む。また、露光に用いられる光としては、水銀灯の輝線スペクトル、エキシマレーザーに代表される遠紫外線、極紫外線(EUV光)、X線、電子線等の活性光線又は放射線が挙げられる。
本明細書において、「(メタ)アクリレート」は、「アクリレート」及び「メタクリレート」の両方、又は、いずれかを意味し、「(メタ)アクリル」は、「アクリル」及び「メタクリル」の両方、又は、いずれかを意味し、「(メタ)アクリロイル」は、「アクリロイル」及び「メタクリロイル」の両方、又は、いずれかを意味する。
本明細書において、構造式中のMeはメチル基を表し、Etはエチル基を表し、Buはブチル基を表し、Phはフェニル基を表す。
本明細書において、全固形分とは、組成物の全成分から溶剤を除いた成分の総質量をいう。また本明細書において、固形分濃度とは、組成物の総質量に対する、溶剤を除く他の成分の質量百分率である。
本明細書において、重量平均分子量(Mw)及び数平均分子量(Mn)は、特に述べない限り、ゲル浸透クロマトグラフィ(GPC)法を用いて測定した値であり、ポリスチレン換算値として定義される。本明細書において、重量平均分子量(Mw)及び数平均分子量(Mn)は、例えば、HLC-8220GPC(東ソー(株)製)を用い、カラムとしてガードカラムHZ-L、TSKgel Super HZM-M、TSKgel Super HZ4000、TSKgel Super HZ3000、及び、TSKgel Super HZ2000(以上、東ソー(株)製)を直列に連結して用いることによって求めることができる。それらの分子量は特に述べない限り、溶離液としてTHF(テトラヒドロフラン)を用いて測定したものとする。ただし、溶解性が低い場合など、溶離液としてTHFが適していない場合にはNMP(N-メチル-2-ピロリドン)を用いることもできる。また、GPC測定における検出は特に述べない限り、UV線(紫外線)の波長254nm検出器を使用したものとする。
本明細書において、積層体を構成する各層の位置関係について、「上」又は「下」と記載したときには、注目している複数の層のうち基準となる層の上側又は下側に他の層があればよい。すなわち、基準となる層と上記他の層の間に、更に第3の層や要素が介在していてもよく、基準となる層と上記他の層は接している必要はない。また、特に断らない限り、基材に対し層が積み重なっていく方向を「上」と称し、又は、樹脂組成物層がある場合には、基材から樹脂組成物層へ向かう方向を「上」と称し、その反対方向を「下」と称する。なお、このような上下方向の設定は、本明細書中における便宜のためであり、実際の態様においては、本明細書における「上」方向は、鉛直上向きと異なることもありうる。
本明細書において、特段の記載がない限り、組成物は、組成物に含まれる各成分として、その成分に該当する2種以上の化合物を含んでもよい。また、特段の記載がない限り、組成物における各成分の含有量とは、その成分に該当する全ての化合物の合計含有量を意味する。
本明細書において、特に述べない限り、温度は23℃、気圧は101,325Pa(1気圧)、相対湿度は50%RHである。
本明細書において、好ましい態様の組み合わせは、より好ましい態様である。
本発明の第1の態様におけるネガ型感光性樹脂組成物は、樹脂、露光によりその露光波長の吸光度が小さくなる化合物A、及び、光重合開始剤を含み、下記条件i及び条件iiの少なくとも一方を満たす。
条件i:組成物が、上記樹脂、上記化合物A及び上記光重合開始剤とは異なる成分として含窒素環状化合物を更に含む。
条件ii:上記樹脂として、環員として2以上の窒素原子を含む含窒素ヘテロ環構造を有する樹脂を含む。
本発明の第2の態様におけるネガ型感光性樹脂組成物は、樹脂、ナフトキノンジアジド化合物、及び、光重合開始剤を含み、条件iii及び条件ivの少なくとも一方を満たす。
条件iii:組成物が、上記樹脂、上記ナフトキノンジアジド化合物及び上記光重合開始とは異なる成分として含窒素環状化合物を更に含む。
条件iv:上記樹脂として、環員として2以上の窒素原子を含む含窒素ヘテロ環構造を有する樹脂を含む。
以下、単に「ネガ型感光性樹脂組成物」又は「樹脂組成物」と記載した場合には、第1の態様におけるネガ型感光性樹脂組成物、及び、第2の態様におけるネガ型感光性樹脂組成物の両方を含むものとする。
本発明の樹脂組成物は、例えば、半導体デバイスの絶縁膜、再配線層用層間絶縁膜、ストレスバッファ膜等の形成に用いることができ、再配線層用層間絶縁膜の形成に用いられることが好ましい。
また、本発明のネガ型感光性樹脂組成物は、ネガ型現像に供される感光膜の形成に用いられる。
本発明において、ネガ型現像とは、露光及び現像において、現像により非露光部が除去される現像をいい、ポジ型現像とは、現像により露光部が除去される現像をいう。
上記露光の方法、上記現像液、及び、上記現像の方法としては、例えば、後述する硬化物の製造方法の説明における露光工程において説明された露光方法、現像工程において説明された現像液及び現像方法が使用される。
上記効果が得られるメカニズムは不明であるが、下記のように推測される。
ここで、本発明においては、露光により露光波長の吸光度が小さくなる化合物A(第2の態様においては、ナフトキノンジアジド化合物)を含み、かつ、上述の条件i及び条件ii(第2の態様においては、条件iii及び条件iv)の少なくとも一方を満たすことにより、金属基板上でのパターンの解像性が向上することを見出した。
これは、化合物A(第2の態様においては、ナフトキノンジアジド化合物)が露光光を吸収することにより、露光量が小さい領域(例えば、実際に露光したい領域の周辺の領域)においては光重合開始剤が感光しにくくなり、また、露光量が大きい領域においては、化合物A(第2の態様においては、ナフトキノンジアジド化合物)の消色(露光により露光波長の吸光度が小さくなること)により光重合開始剤が感光しやすくなるためであると推測される。
更に、本発明のネガ型感光性樹脂組成物は、含窒素環状化合物を含むか、上記樹脂として、環員として2以上の窒素原子を含む含窒素ヘテロ環構造を有する樹脂を含むか、又は、その両方を含む。これにより、金属層から感光層への金属のマイグレーションが抑制され、未露光部に現像残渣を生じにくくなると考えられる。結果として、上述の化合物A(第2の態様においては、ナフトキノンジアジド化合物)の消色による効果がさらに際立ち、露光部と未露光部の溶解コントラストが拡大してパターンの解像性に優れ、かつ、現像による膜厚の減少が抑制されると推測される。
また、本発明のネガ型感光性樹脂組成物は含窒素環状化合物か、上記樹脂として、環員として2以上の窒素原子を含む含窒素ヘテロ環構造を有する樹脂を含むか、又は、その両方を含むため、金属層から感光層への金属のマイグレーションが抑制され、露光部の重合が金属により妨げられることが抑制され、硬化が十分に進行しやすいと考えられる。そのため、得られる硬化物の耐薬品性にも優れると考えられる。
更に、化合物Aが消色してできた分解物(例えば、ナフトキノンジアジド化合物の分解物)が樹脂等の架橋にも組み込まれ、より架橋密度が増大することから、硬化物の耐薬品性も向上すると考えられる。すなわち、本発明の第2の態様に係るネガ型感光性樹脂組成物は、得られる硬化物の耐薬品性に更に優れた組成物である。
特に、樹脂が環化樹脂又はその前駆体であり、かつ、化合物Aがナフトキノンジアジド化合物である場合に、樹脂とナフトキノンジアジド化合物の分解物とが架橋構造を形成し、耐薬品性が大きく向上する場合がある。
本発明の樹脂組成物は、樹脂を含む。
本発明において、本発明の樹脂組成物は、下記(1)及び(2)の少なくとも一方を満たすことが好ましい。
(1)樹脂組成物に含まれる樹脂が重合性基を有する
(2)樹脂組成物が後述する重合性化合物を更に含む。
樹脂としては、特に限定されず、例えば従来のパターン形成用組成物において用いられる樹脂が挙げられるが、環化樹脂又はその前駆体(特定樹脂)を含むことが好ましく、環化樹脂の前駆体を含むことがより好ましい。
本発明の樹脂組成物が上述の条件i又は条件iiiを満たす場合、本発明の樹脂組成物は、上記樹脂として、環員として2以上の窒素原子を含む含窒素ヘテロ環構造を有する樹脂を含んでもよい。
ただし、含窒素ヘテロ環構造が複環である場合、複環を構成する少なくとも1つの単環において2以上の窒素原子を環員として含む。すなわち、例えば、ベンゾイミダゾール環は本発明でいう環員として2以上の窒素原子を含む含窒素ヘテロ環構造に該当するが、7-アザインドール環は、本発明でいう環員として2以上の窒素原子を含む含窒素ヘテロ環構造には該当しないものとする。
また、上記含窒素ヘテロ環構造は、プリン環構造のように、環員として2以上の窒素原子を含む含窒素ヘテロ環構造を複環構造内に複数有していてもよい。
上記含窒素ヘテロ環構造は、窒素原子以外のヘテロ原子を環員として含んでもよいが、含窒素ヘテロ環構造が窒素原子以外のヘテロ原子を環員として含まない態様も、本発明の好ましい態様の一つである。上記窒素原子以外のヘテロ原子としては、酸素原子、硫黄原子等が挙げられる。
上記含窒素ヘテロ環構造は、脂肪族環であっても芳香族環であってもよいが、芳香族環であることが好ましい。
上記他の環としては、芳香環が好ましく、5員環又は6員環の芳香環がより好ましく、ベンゼン環がより好ましい。
本発明において、主鎖とは、樹脂分子中で相対的に最も長い結合鎖を表す。
特定樹脂の主鎖末端のうち、少なくとも1つが上記含窒素ヘテロ環構造を有することが好ましい。
特に、特定樹脂がポリイミド、ポリイミド前駆体、ポリアミドイミド又はポリアミドイミド前駆体のいずれかである場合には、下記式(1-1)で表される構造を含むことが好ましい。
また、特定樹脂がポリベンゾオキサゾール、又は、ポリベンゾオキサゾール前駆体である場合には、下記式(2-1)で表される構造を含むことが好ましい。
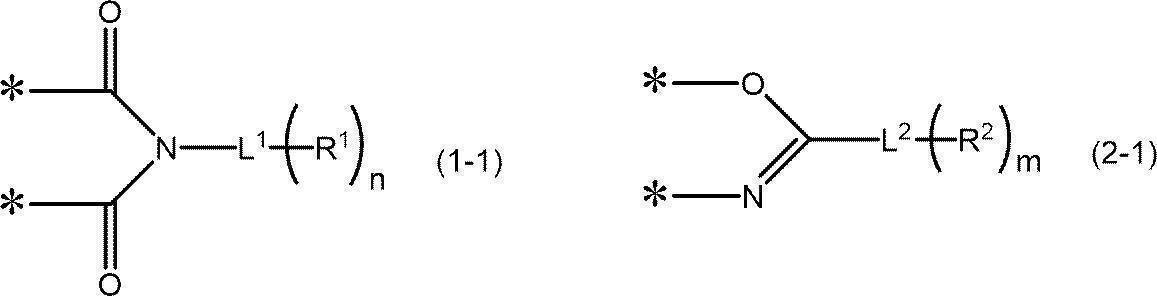
式(1-1)中、L1は単結合又はイミド環構造を含まないn+1価の連結基を、R1は環員として2以上の窒素原子を含む含窒素ヘテロ環構造を、nは1以上の整数をそれぞれ表し、*は他の構造との結合部位を表す。
式(2-1)中、L2は単結合又はオキサゾール環構造を含まないm+1価の連結基を、R2は環員として2以上の窒素原子を含む含窒素ヘテロ環構造を、mは1以上の整数をそれぞれ表し、*は他の構造との結合部位を表す。
上記RNは水素原子又は1価の置換基を表し、水素原子又は炭化水素基であることが好ましく、水素原子、アルキル基又は芳香族炭化水素基であることがより好ましい。
また、L1が単結合である態様も、本発明の好ましい態様の1つである。
また、L2が単結合である態様も、本発明の好ましい態様の1つである。

式(1-2)又は式(1-3)中、L1は単結合又はイミド環構造を含まないn+1価の連結基を、R1は環員として2以上の窒素原子を含む含窒素ヘテロ環構造を、nは1以上の整数をそれぞれ表し、*は他の構造との結合部位を表す。
式(1-2)又は式(1-3)中、L1、R1及びnはそれぞれ、式(1-1)中のL1、R1及びnと同義であり、好ましい態様も同様である。

式(2-2)又は式(2-3)中、L2は単結合又はオキサゾール環構造を含まないm+1価の連結基を、R2は環員として2以上の窒素原子を含む含窒素ヘテロ環構造を、mは1以上の整数をそれぞれ表し、*は他の構造との結合部位を表す。
式(2-2)又は式(2-3)中、L2、R2及びmはそれぞれ、式(2-1)中のL2、R2及びmと同義であり、好ましい態様も同様である。
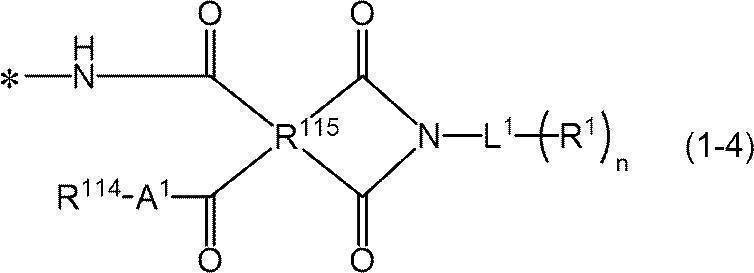
式(1-4)中、A1は酸素原子又は-NH-を表し、R115は、4価の有機基を表し、R114は、水素原子又は1価の有機基を表し、L1は単結合又はイミド環構造を含まないn+1価の連結基を、R1は環員として2以上の窒素原子を含む含窒素ヘテロ環構造を、nは1以上の整数をそれぞれ表し、*は他の構造との結合部位を表す。
式(1-4)中、A1、R115及びR114は、後述の式(2)におけるA1、R115及びR114と同義であり、好ましい態様も同様である。
式(1-4)中、L1、R1及びnは、上述の式(1-1)におけるL1、R1及びnと同義であり、好ましい態様も同様である。
また、特定樹脂が式(1-4)で表される構造を含む場合、特定樹脂は、後述の式(2)で表される繰返し単位を含むことが好ましい。
特定樹脂が後述の式(2)で表される繰返し単位を含む場合、式(1-4)におけるA1、R115及びR114は、特定樹脂に含まれる式(2)で表される繰返し単位のいずれかにおけるA1、R115及びR114と同一の構造であることも好ましい。
また、特定樹脂が後述の式(2)で表される繰返し単位を含む場合、式(1-4)における*は、式(2)中のR111との結合部位であることも好ましい。
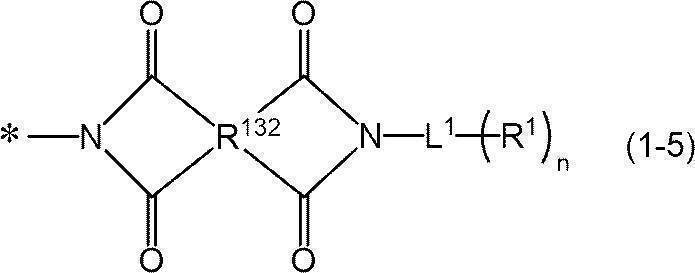
式(1-5)中、R132は、4価の有機基を表し、L1は単結合又はイミド環構造を含まないn+1価の連結基を、R1は環員として2以上の窒素原子を含む含窒素ヘテロ環構造を、nは1以上の整数をそれぞれ表し、*は他の構造との結合部位を表す。
式(1-5)中、R132は、後述の式(4)におけるR132と同義であり、好ましい態様も同様である。
式(1-5)中、L1、R1及びnは、上述の式(1-1)におけるL1、R1及びnと同義であり、好ましい態様も同様である。
また、特定樹脂が式(1-5)で表される構造を含む場合、特定樹脂は、後述の式(4)で表される繰返し単位を含むことが好ましい。
特定樹脂が後述の式(4)で表される繰返し単位を含む場合、式(1-5)におけるR132は、特定樹脂に含まれる式(4)で表される繰返し単位のいずれかにおけるR132と同一の構造であることも好ましい。
また、特定樹脂が後述の式(4)で表される繰返し単位を含む場合、式(1-5)における*は、式(4)中のR131との結合部位であることも好ましい。

式(2-4)中、R122は、4価の有機基を表し、R124は水素原子又は1価の有機基を表し、L2は単結合又はオキサゾール環構造を含まないm+1価の連結基を、R2は環員として2以上の窒素原子を含む含窒素ヘテロ環構造を、mは1以上の整数をそれぞれ表し、*は他の構造との結合部位を表す。
式(2-4)中、R122及びR124は、後述の式(3)におけるR122及びR124と同義であり、好ましい態様も同様である。
式(2-4)中、L2、R2及びmは、上述の式(2-1)におけるL2、R2及びmと同義であり、好ましい態様も同様である。
また、特定樹脂が式(2-4)で表される構造を含む場合、特定樹脂は、後述の式(3)で表される繰返し単位を含むことが好ましい。
特定樹脂が後述の式(3)で表される繰返し単位を含む場合、式(2-4)におけるR122及びR124は、特定樹脂に含まれる式(3)で表される繰返し単位のいずれかにおけるR122及びR124と同一の構造であることも好ましい。
また、特定樹脂が後述の式(3)で表される繰返し単位を含む場合、式(2-4)における*は、式(3)中のR121との結合部位であることも好ましい。

式(2-5)中、R134は、4価の有機基を表し、L2は単結合又はオキサゾール環構造を含まないm+1価の連結基を、R2は環員として2以上の窒素原子を含む含窒素ヘテロ環構造を、mは1以上の整数をそれぞれ表し、*は他の構造との結合部位を表す。
式(2-5)中、R134は、後述の式(X)におけるR134と同義であり、好ましい態様も同様である。
式(2-5)中、L2、R2及びmは、上述の式(1-1)におけるL2、R2及びmと同義であり、好ましい態様も同様である。
また、特定樹脂が式(2-5)で表される構造を含む場合、特定樹脂は、後述の式(X)で表される繰返し単位を含むことが好ましい。
特定樹脂が後述の式(X)で表される繰返し単位を含む場合、式(2-5)におけるR134は、特定樹脂に含まれる式(X)で表される繰返し単位のいずれかにおけるR134と同一の構造であることも好ましい。
また、特定樹脂が後述の式(X)で表される繰返し単位を含む場合、式(1-5)における*は、式(X)中のR133との結合部位であることも好ましい。

本発明において、主鎖とは、樹脂分子中で相対的に最も長い結合鎖を表す。
環化樹脂としては、ポリイミド、ポリベンゾオキサゾール、ポリアミドイミド等が挙げられる。
環化樹脂の前駆体とは、外部刺激により化学構造の変化を生じて環化樹脂となる樹脂をいい、熱により化学構造の変化を生じて環化樹脂となる樹脂が好ましく、熱により閉環反応を生じて環構造が形成されることにより環化樹脂となる樹脂がより好ましい。
環化樹脂の前駆体としては、ポリイミド前駆体、ポリベンゾオキサゾール前駆体、ポリアミドイミド前駆体等が挙げられる。
すなわち、本発明の樹脂組成物は、特定樹脂として、ポリイミド、ポリイミド前駆体、ポリベンゾオキサゾール、ポリベンゾオキサゾール前駆体、ポリアミドイミド、及び、ポリアミドイミド前駆体よりなる群から選ばれた少なくとも1種の樹脂(特定樹脂)を含むことが好ましい。
本発明の樹脂組成物は、特定樹脂として、ポリイミド又はポリイミド前駆体を含むことが好ましい。
また、特定樹脂は重合性基を有することが好ましく、ラジカル重合性基を含むことがより好ましい。
特定樹脂がラジカル重合性基を有する場合、本発明の樹脂組成物は、後述のラジカル重合開始剤を含むことが好ましく、後述のラジカル重合開始剤を含み、かつ、後述のラジカル架橋剤を含むことがより好ましい。さらに必要に応じて、後述の増感剤を含むことができる。このような本発明の樹脂組成物からは、例えば、ネガ型感光膜が形成される。
本発明で用いるポリイミド前駆体は、その種類等特に定めるものではないが、下記式(2)で表される繰返し単位を含むことが好ましい。

式(2)中、A1及びA2は、それぞれ独立に、酸素原子又は-NH-を表し、R111は、2価の有機基を表し、R115は、4価の有機基を表し、R113及びR114は、それぞれ独立に、水素原子又は1価の有機基を表す。
式(2)におけるR111は、2価の有機基を表す。2価の有機基としては、直鎖又は分岐の脂肪族基、環状の脂肪族基及び芳香族基を含む基が例示され、炭素数2~20の直鎖又は分岐の脂肪族基、炭素数3~20の環状の脂肪族基、炭素数3~20の芳香族基、又は、これらの組み合わせからなる基が好ましく、炭素数6~20の芳香族基を含む基がより好ましい。上記直鎖又は分岐の脂肪族基は鎖中の炭化水素基がヘテロ原子を含む基で置換されていてもよく、上記環状の脂肪族基および芳香族基は環員の炭化水素基がヘテロ原子を含む基で置換されていてもよい。本発明の好ましい実施形態として、-Ar-および-Ar-L-Ar-で表される基であることが例示され、特に好ましくは-Ar-L-Ar-で表される基である。但し、Arは、それぞれ独立に、芳香族基であり、Lは、単結合、又は、フッ素原子で置換されていてもよい炭素数1~10の脂肪族炭化水素基、-O-、-CO-、-S-、-SO2-若しくは-NHCO-、あるいは、上記の2つ以上の組み合わせからなる基である。これらの好ましい範囲は、上述のとおりである。
具体的には、炭素数2~20の直鎖又は分岐の脂肪族基、炭素数3~20の環状の脂肪族基、炭素数3~20の芳香族基、又は、これらの組み合わせからなる基を含むジアミンであることが好ましく、炭素数6~20の芳香族基を含むジアミンであることがより好ましい。上記直鎖又は分岐の脂肪族基は鎖中の炭化水素基がヘテロ原子を含む基で置換されていてもよく上記環状の脂肪族基および芳香族基は環員の炭化水素基がヘテロ原子を含む基で置換されていてもよい。芳香族基を含む基の例としては、下記が挙げられる。

式中、Aは単結合又は2価の連結基を表し、単結合、又は、フッ素原子で置換されていてもよい炭素数1~10の脂肪族炭化水素基、-O-、-C(=O)-、-S-、-SO2-、-NHCO-、又は、これらの組み合わせから選択される基であることが好ましく、単結合、又は、フッ素原子で置換されていてもよい炭素数1~3のアルキレン基、-O-、-C(=O)-、-S-、若しくは、-SO2-から選択される基であることがより好ましく、-CH2-、-O-、-S-、-SO2-、-C(CF3)2-、又は、-C(CH3)2-であることが更に好ましい。
式中、*は他の構造との結合部位を表す。
式(51)

式(51)中、R50~R57は、それぞれ独立に、水素原子、フッ素原子又は1価の有機基であり、R50~R57の少なくとも1つは、フッ素原子、メチル基又はトリフルオロメチル基であり、*はそれぞれ独立に、式(2)中の窒素原子との結合部位を表す。
R50~R57の1価の有機基としては、炭素数1~10(好ましくは炭素数1~6)の無置換のアルキル基、炭素数1~10(好ましくは炭素数1~6)のフッ化アルキル基等が挙げられる。

式(61)中、R58及びR59は、それぞれ独立に、フッ素原子、メチル基、又はトリフルオロメチル基であり、*はそれぞれ独立に、式(2)中の窒素原子との結合部位を表す。
式(51)又は(61)の構造を与えるジアミンとしては、2,2’-ジメチルベンジジン、2,2’-ビス(トリフルオロメチル)-4,4’-ジアミノビフェニル、2,2’-ビス(フルオロ)-4,4’-ジアミノビフェニル、4,4’-ジアミノオクタフルオロビフェニル等が挙げられる。これらは1種で又は2種以上を組み合わせて用いてもよい。
式(5)又は式(6)中、*はそれぞれ独立に、他の構造との結合部位を表す。

式(5)中、R112は単結合又は2価の連結基であり、単結合、又は、フッ素原子で置換されていてもよい炭素数1~10の脂肪族炭化水素基、-O-、-CO-、-S-、-SO2-、及び-NHCO-、ならびに、これらの組み合わせから選択される基であることが好ましく、単結合、フッ素原子で置換されていてもよい炭素数1~3のアルキレン基、-O-、-CO-、-S-及び-SO2-から選択される基であることがより好ましく、-CH2-、-C(CF3)2-、-C(CH3)2-、-O-、-CO-、-S-及び-SO2-からなる群から選択される2価の基であることが更に好ましい。
テトラカルボン酸二無水物は、下記式(O)で表されることが好ましい。

式(O)中、R115は、4価の有機基を表す。R115の好ましい範囲は式(2)におけるR115と同義であり、好ましい範囲も同様である。
エチレン性不飽和結合を有する基としては、ビニル基、アリル基、イソアリル基、2-メチルアリル基、ビニル基と直接結合した芳香環を有する基(例えば、ビニルフェニル基など)、(メタ)アクリルアミド基、(メタ)アクリロイルオキシ基、下記式(III)で表される基などが挙げられ、下記式(III)で表される基が好ましい。

式(III)において、*は他の構造との結合部位を表す。
式(III)において、R201は、炭素数2~12のアルキレン基、-CH2CH(OH)CH2-、シクロアルキレン基又はポリアルキレンオキシ基を表す。
好適なR201の例は、エチレン基、プロピレン基、トリメチレン基、テトラメチレン基、ペンタメチレン基、ヘキサメチレン基、オクタメチレン基、ドデカメチレン基等のアルキレン基、1,2-ブタンジイル基、1,3-ブタンジイル基、-CH2CH(OH)CH2-、ポリアルキレンオキシ基が挙げられ、エチレン基、プロピレン基等のアルキレン基、-CH2CH(OH)CH2-、シクロヘキシル基、ポリアルキレンオキシ基がより好ましく、エチレン基、プロピレン基等のアルキレン基、又はポリアルキレンオキシ基が更に好ましい。
本発明において、ポリアルキレンオキシ基とは、アルキレンオキシ基が2以上直接結合した基をいう。ポリアルキレンオキシ基に含まれる複数のアルキレンオキシ基におけるアルキレン基は、それぞれ同一であっても異なっていてもよい。
ポリアルキレンオキシ基が、アルキレン基が異なる複数種のアルキレンオキシ基を含む場合、ポリアルキレンオキシ基におけるアルキレンオキシ基の配列は、ランダムな配列であってもよいし、ブロックを有する配列であってもよいし、交互等のパターンを有する配列であってもよい。
上記アルキレン基の炭素数(アルキレン基が置換基を有する場合、置換基の炭素数を含む)は、2以上であることが好ましく、2~10であることがより好ましく、2~6であることがより好ましく、2~5であることが更に好ましく、2~4であることが一層好ましく、2又は3であることが特に好ましく、2であることが最も好ましい。
また、上記アルキレン基は、置換基を有していてもよい。好ましい置換基としては、アルキル基、アリール基、ハロゲン原子等が挙げられる。
また、ポリアルキレンオキシ基に含まれるアルキレンオキシ基の数(ポリアルキレンオキシ基の繰返し数)は、2~20が好ましく、2~10がより好ましく、2~6が更に好ましい。
ポリアルキレンオキシ基としては、溶剤溶解性及び耐溶剤性の観点からは、ポリエチレンオキシ基、ポリプロピレンオキシ基、ポリトリメチレンオキシ基、ポリテトラメチレンオキシ基、又は、複数のエチレンオキシ基と複数のプロピレンオキシ基とが結合した基が好ましく、ポリエチレンオキシ基又はポリプロピレンオキシ基がより好ましく、ポリエチレンオキシ基が更に好ましい。上記複数のエチレンオキシ基と複数のプロピレンオキシ基とが結合した基において、エチレンオキシ基とプロピレンオキシ基とはランダムに配列していてもよいし、ブロックを形成して配列していてもよいし、交互等のパターン状に配列していてもよい。これらの基におけるエチレンオキシ基等の繰返し数の好ましい態様は上述の通りである。
式(2-A)

式(2-A)中、A1及びA2は、酸素原子を表し、R111及びR112は、それぞれ独立に、2価の有機基を表し、R113及びR114は、それぞれ独立に、水素原子又は1価の有機基を表し、R113及びR114の少なくとも一方は、重合性基を含む基であり、両方が重合性基を含む基であることが好ましい。
R112は、式(5)におけるR112と同義であり、好ましい範囲も同様である。
上記ポリイミド前駆体の分子量の分散度は、1.5以上が好ましく、1.8以上がより好ましく、2.0以上であることが更に好ましい。ポリイミド前駆体の分子量の分散度の上限値は特に定めるものではないが、例えば、7.0以下が好ましく、6.5以下がより好ましく、6.0以下が更に好ましい。
本明細書において、分子量の分散度とは、重量平均分子量/数平均分子量により算出される値である。
また、樹脂組成物が特定樹脂として複数種のポリイミド前駆体を含む場合、少なくとも1種のポリイミド前駆体の重量平均分子量、数平均分子量、及び、分散度が上記範囲であることが好ましい。また、上記複数種のポリイミド前駆体を1つの樹脂として算出した重量平均分子量、数平均分子量、及び、分散度が、それぞれ、上記範囲内であることも好ましい。
本発明に用いられるポリイミドは、アルカリ可溶性ポリイミドであってもよく、有機溶剤を主成分とする現像液に対して可溶なポリイミドであってもよい。
本明細書において、アルカリ可溶性ポリイミドとは、100gの2.38質量%テトラメチルアンモニウム水溶液に対し、23℃で0.1g以上溶解するポリイミドをいい、パターン形成性の観点からは、0.5g以上溶解するポリイミドであることが好ましく、1.0g以上溶解するポリイミドであることが更に好ましい。上記溶解量の上限は特に限定されないが、100g以下であることが好ましい。
また、ポリイミドは、得られる有機膜の膜強度及び絶縁性の観点からは、複数個のイミド構造を主鎖に有するポリイミドであることが好ましい。
本明細書において、「主鎖」とは、樹脂を構成する高分子化合物の分子中で相対的に最も長い結合鎖をいい、「側鎖」とはそれ以外の結合鎖をいう。
得られる有機膜の膜強度の観点からは、ポリイミドは、フッ素原子を有することも好ましい。
フッ素原子は、例えば、後述する式(4)で表される繰返し単位におけるR132、又は、後述する式(4)で表される繰返し単位におけるR131に含まれることが好ましく、後述する式(4)で表される繰返し単位におけるR132、又は、後述する式(4)で表される繰返し単位におけるR131にフッ化アルキル基として含まれることがより好ましい。
ポリイミドの全質量に対するフッ素原子の量は、5質量%以上が好ましく、また、20質量%以下が好ましい。
得られる有機膜の膜強度の観点からは、ポリイミドは、ケイ素原子を有することも好ましい。
ケイ素原子は、例えば、後述する式(4)で表される繰返し単位におけるR131に含まれることが好ましく、後述する式(4)で表される繰返し単位におけるR131に後述する有機変性(ポリ)シロキサン構造として含まれることがより好ましい。
また、上記ケイ素原子又は上記有機変性(ポリ)シロキサン構造はポリイミドの側鎖に含まれていてもよいが、ポリイミドの主鎖に含まれることが好ましい。
ポリイミドの全質量に対するケイ素原子の量は、1質量%以上が好ましく、20質量%以下がより好ましい。
得られる有機膜の膜強度の観点からは、ポリイミドは、エチレン性不飽和結合を有することが好ましい。
ポリイミドは、エチレン性不飽和結合を主鎖末端に有していてもよいし、側鎖に有していてもよいが、側鎖に有することが好ましい。
上記エチレン性不飽和結合は、ラジカル重合性を有することが好ましい。
エチレン性不飽和結合は、後述する式(4)で表される繰返し単位におけるR132、又は、後述する式(4)で表される繰返し単位におけるR131に含まれることが好ましく、後述する式(4)で表される繰返し単位におけるR132、又は、後述する式(4)で表される繰返し単位におけるR131にエチレン性不飽和結合を有する基として含まれることがより好ましい。
これらの中でも、エチレン性不飽和結合は、後述する式(4)で表される繰返し単位におけるR131に含まれることが好ましく、後述する式(4)で表される繰返し単位におけるR131にエチレン性不飽和結合を有する基として含まれることがより好ましい。
エチレン性不飽和結合を有する基としては、ビニル基、アリル基、ビニルフェニル基等の芳香環に直接結合した、置換されていてもよいビニル基を有する基、(メタ)アクリルアミド基、(メタ)アクリロイルオキシ基、下記式(IV)で表される基などが挙げられる。

また、上記炭素数2~12のアルキレン基としては、直鎖状、分岐鎖状、環状又はこれらの組み合わせにより表されるアルキレン基のいずれであってもよい。
上記炭素数2~12のアルキレン基としては、炭素数2~8のアルキレン基が好ましく、炭素数2~4のアルキレン基がより好ましい。

式(R1)~(R3)中、Lは単結合、又は、炭素数2~12のアルキレン基、炭素数2~30の(ポリ)アルキレンオキシ基若しくはこれらを2以上結合した基を表し、Xは酸素原子又は硫黄原子を表し、*は他の構造との結合部位を表し、●は式(IV)中のR21が結合する酸素原子との結合部位を表す。
式(R1)~(R3)中、Lにおける炭素数2~12のアルキレン基、又は、炭素数2~30の(ポリ)アルキレンオキシ基の好ましい態様は、上述のR21における、炭素数2~12のアルキレン基、又は、炭素数2~30の(ポリ)アルキレンオキシ基の好ましい態様と同様である。
式(R1)中、Xは酸素原子であることが好ましい。
式(R1)~(R3)中、*は式(IV)中の*と同義であり、好ましい態様も同様である。
式(R1)で表される構造は、例えば、フェノール性ヒドロキシ基等のヒドロキシ基を有するポリイミドと、イソシアナト基及びエチレン性不飽和結合を有する化合物(例えば、2-イソシアナトエチルメタクリレート等)とを反応することにより得られる。
式(R2)で表される構造は、例えば、カルボキシ基を有するポリイミドと、ヒドロキシ基及びエチレン性不飽和結合を有する化合物(例えば、2-ヒドロキシエチルメタクリレート等)とを反応することにより得られる。
式(R3)で表される構造は、例えば、フェノール性ヒドロキシ基等のヒドロキシ基を有するポリイミドと、グリシジル基及びエチレン性不飽和結合を有する化合物(例えば、グリシジルメタクリレート等)とを反応することにより得られる。
ポリイミドは、エチレン性不飽和結合を有する基以外の重合性基を有していてもよい。
エチレン性不飽和結合を有する基以外の重合性基としては、エポキシ基、オキセタニル基等の環状エーテル基、メトキシメチル基等のアルコキシメチル基、メチロール基等が挙げられる。
エチレン性不飽和結合を有する基以外の重合性基は、例えば、後述する式(4)で表される繰返し単位におけるR131に含まれることが好ましい。
ポリイミドの全質量に対するエチレン性不飽和結合を有する基以外の重合性基の量は、0.0001~0.1mol/gであることが好ましく、0.001~0.05mol/gであることがより好ましい。
ポリイミドがアルカリ現像に供される場合、現像性を向上する観点からは、ポリイミドの酸価は、30mgKOH/g以上であることが好ましく、50mgKOH/g以上であることがより好ましく、70mgKOH/g以上であることが更に好ましい。
また、上記酸価は500mgKOH/g以下であることが好ましく、400mgKOH/g以下であることがより好ましく、200mgKOH/g以下であることが更に好ましい。
また、ポリイミドが有機溶剤を主成分とする現像液を用いた現像(例えば、後述する「溶剤現像」)に供される場合、ポリイミドの酸価は、1~35mgKOH/gが好ましく、2~30mgKOH/gがより好ましく、5~20mgKOH/gが更に好ましい。
上記酸価は、公知の方法により測定され、例えば、JIS K 0070:1992に記載の方法により測定される。
また、ポリイミドに含まれる酸基としては、保存安定性及び現像性の両立の観点から、pKaが0~10である酸基が好ましく、3~8である酸基がより好ましい。
pKaとは、酸から水素イオンが放出される解離反応を考え、その平衡定数Kaをその負の常用対数pKaによって表したものである。本明細書において、pKaは、特に断らない限り、ACD/ChemSketch(登録商標)による計算値とする。又は、日本化学会編「改定5版 化学便覧 基礎編」に掲載の値を参照してもよい。
また、酸基が例えばリン酸等の多価の酸である場合、上記pKaは第一解離定数である。
このような酸基として、ポリイミドは、カルボキシ基、及び、フェノール性ヒドロキシ基よりなる群から選ばれた少なくとも1種を含むことが好ましく、フェノール性ヒドロキシ基を含むことがより好ましい。
アルカリ現像液による現像速度を適切なものとする観点からは、ポリイミドは、フェノール性ヒドロキシ基を有することが好ましい。
ポリイミドは、フェノール性ヒドロキシ基を主鎖末端に有してもよいし、側鎖に有してもよい。
フェノール性ヒドロキシ基は、例えば、後述する式(4)で表される繰返し単位におけるR132、又は、後述する式(4)で表される繰返し単位におけるR131に含まれることが好ましい。
ポリイミドの全質量に対するフェノール性ヒドロキシ基の量は、0.1~30mol/gであることが好ましく、1~20mol/gであることがより好ましい。

式(4)中、R131は、2価の有機基を表し、R132は、4価の有機基を表す。
重合性基を有する場合、重合性基は、R131及びR132の少なくとも一方に位置していてもよいし、下記式(4-1)又は式(4-2)に示すようにポリイミドの末端に位置していてもよい。
式(4-1)

式(4-1)中、R133は重合性基であり、他の基は式(4)と同義である。
式(4-2)

R134及びR135の少なくとも一方は重合性基であり、重合性基でない場合は有機基であり、他の基は式(4)と同義である。
R131は、2価の有機基を表す。2価の有機基としては、式(2)におけるR111と同様のものが例示され、好ましい範囲も同様である。
また、R131としては、ジアミンのアミノ基の除去後に残存するジアミン残基が挙げられる。ジアミンとしては、脂肪族、環式脂肪族又は芳香族ジアミンなどが挙げられる。具体的な例としては、ポリイミド前駆体の式(2)中のR111の例が挙げられる。
例えば、R115として例示される4価の有機基の4つの結合子が、上記式(4)中の4つの-C(=O)-の部分と結合して縮合環を形成する。
ポリイミドのイミド化率(「閉環率」ともいう)は、得られる有機膜の膜強度、絶縁性等の観点からは、70%以上であることが好ましく、80%以上であることがより好ましく、90%以上であることがより好ましい。
上記イミド化率の上限は特に限定されず、100%以下であればよい。
上記イミド化率は、例えば下記方法により測定される。
ポリイミドの赤外吸収スペクトルを測定し、イミド構造由来の吸収ピークである1377cm-1付近のピーク強度P1を求める。次に、そのポリイミドを350℃で1時間熱処理した後、再度、赤外吸収スペクトルを測定し、1377cm-1付近のピーク強度P2を求める。得られたピーク強度P1、P2を用い、下記式に基づいて、ポリイミドのイミド化率を求めることができる。
イミド化率(%)=(ピーク強度P1/ピーク強度P2)×100
また、ポリイミドの数平均分子量(Mn)は、好ましくは2,000~40,000であり、より好ましくは3,000~30,000であり、更に好ましくは4,000~20,000である。
上記ポリイミドの分子量の分散度は、1.5以上が好ましく、1.8以上がより好ましく、2.0以上であることが更に好ましい。ポリイミドの分子量の分散度の上限値は特に定めるものではないが、例えば、7.0以下が好ましく、6.5以下がより好ましく、6.0以下が更に好ましい。
また、樹脂組成物が特定樹脂として複数種のポリイミドを含む場合、少なくとも1種のポリイミドの重量平均分子量、数平均分子量、及び、分散度が上記範囲であることが好ましい。また、上記複数種のポリイミドを1つの樹脂として算出した重量平均分子量、数平均分子量、及び、分散度が、それぞれ、上記範囲内であることも好ましい。
本発明で用いるポリベンゾオキサゾール前駆体は、その構造等について特に定めるものではないが、好ましくは下記式(3)で表される繰返し単位を含む。

式(3)中、R121は、2価の有機基を表し、R122は、4価の有機基を表し、R123及びR124は、それぞれ独立に、水素原子又は1価の有機基を表す。
式(3)において、R121は、2価の有機基を表す。2価の有機基としては、脂肪族基及び芳香族基の少なくとも一方を含む基が好ましい。脂肪族基としては、直鎖の脂肪族基が好ましい。R121は、ジカルボン酸残基が好ましい。ジカルボン酸残基は、1種のみ用いてもよいし、2種以上用いてもよい。
脂肪族基を含むジカルボン酸としては、直鎖又は分岐(好ましくは直鎖)の脂肪族基を含むジカルボン酸が好ましく、直鎖又は分岐(好ましくは直鎖)の脂肪族基と2つの-COOHからなるジカルボン酸がより好ましい。直鎖又は分岐(好ましくは直鎖)の脂肪族基の炭素数は、2~30であることが好ましく、2~25であることがより好ましく、3~20であることが更に好ましく、4~15であることが一層好ましく、5~10であることが特に好ましい。直鎖の脂肪族基はアルキレン基であることが好ましい。
直鎖の脂肪族基を含むジカルボン酸としては、マロン酸、ジメチルマロン酸、エチルマロン酸、イソプロピルマロン酸、ジ-n-ブチルマロン酸、スクシン酸、テトラフルオロスクシン酸、メチルスクシン酸、2,2-ジメチルスクシン酸、2,3-ジメチルスクシン酸、ジメチルメチルスクシン酸、グルタル酸、ヘキサフルオログルタル酸、2-メチルグルタル酸、3-メチルグルタル酸、2,2-ジメチルグルタル酸、3,3-ジメチルグルタル酸、3-エチル-3-メチルグルタル酸、アジピン酸、オクタフルオロアジピン酸、3-メチルアジピン酸、ピメリン酸、2,2,6,6-テトラメチルピメリン酸、スベリン酸、ドデカフルオロスベリン酸、アゼライン酸、セバシン酸、ヘキサデカフルオロセバシン酸、1,9-ノナン二酸、ドデカン二酸、トリデカン二酸、テトラデカン二酸、ペンタデカン二酸、ヘキサデカン二酸、ヘプタデカン二酸、オクタデカン二酸、ノナデカン二酸、エイコサン二酸、ヘンエイコサン二酸、ドコサン二酸、トリコサン二酸、テトラコサン二酸、ペンタコサン二酸、ヘキサコサン二酸、ヘプタコサン二酸、オクタコサン二酸、ノナコサン二酸、トリアコンタン二酸、ヘントリアコンタン二酸、ドトリアコンタン二酸、ジグリコール酸、更に下記式で表されるジカルボン酸等が挙げられる。

(式中、Zは炭素数1~6の炭化水素基であり、nは1~6の整数である。)

式中、Aは-CH2-、-O-、-S-、-SO2-、-CO-、-NHCO-、-C(CF3)2-、及び、-C(CH3)2-からなる群から選択される2価の基を表し、*はそれぞれ独立に、他の構造との結合部位を表す。
R122は、また、ビスアミノフェノール誘導体由来の基であることが好ましく、ビスアミノフェノール誘導体由来の基としては、例えば、3,3’-ジアミノ-4,4’-ジヒドロキシビフェニル、4,4’-ジアミノ-3,3’-ジヒドロキシビフェニル、3,3’-ジアミノ-4,4’-ジヒドロキシジフェニルスルホン、4,4’-ジアミノ-3,3’-ジヒドロキシジフェニルスルホン、ビス-(3-アミノ-4-ヒドロキシフェニル)メタン、2,2-ビス(3-アミノ-4-ヒドロキシフェニル)プロパン、2,2-ビス-(3-アミノ-4-ヒドロキシフェニル)ヘキサフルオロプロパン、2,2-ビス-(4-アミノ-3-ヒドロキシフェニル)ヘキサフルオロプロパン、ビス-(4-アミノ-3-ヒドロキシフェニル)メタン、2,2-ビス-(4-アミノ-3-ヒドロキシフェニル)プロパン、4,4’-ジアミノ-3,3’-ジヒドロキシベンゾフェノン、3,3’-ジアミノ-4,4’-ジヒドロキシベンゾフェノン、4,4’-ジアミノ-3,3’-ジヒドロキシジフェニルエーテル、3,3’-ジアミノ-4,4’-ジヒドロキシジフェニルエーテル、1,4-ジアミノ-2,5-ジヒドロキシベンゼン、1,3-ジアミノ-2,4-ジヒドロキシベンゼン、1,3-ジアミノ-4,6-ジヒドロキシベンゼンなどが挙げられる。これらのビスアミノフェノールは、単独にて、あるいは混合して使用してもよい。
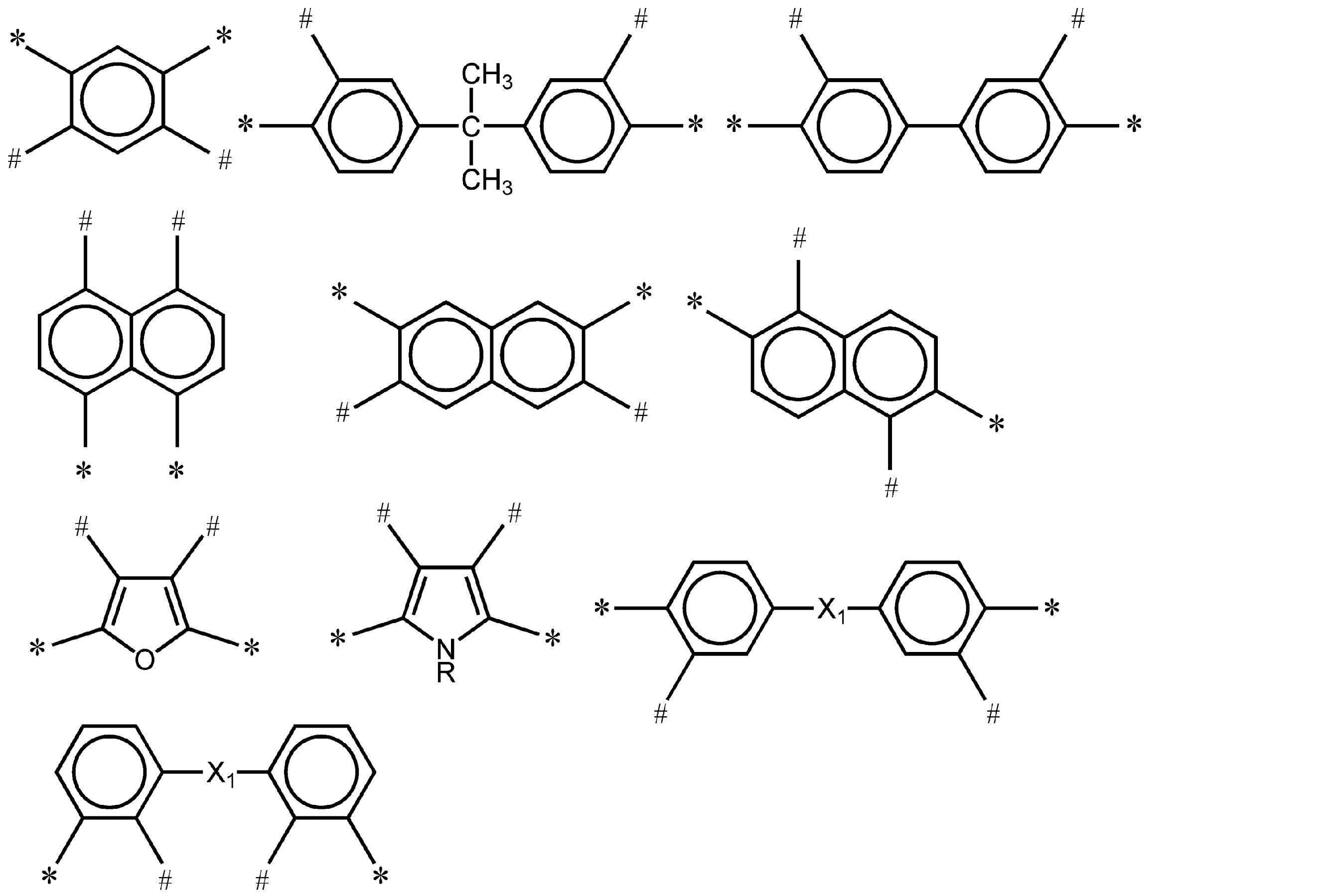
式中、X1は、-O-、-S-、-C(CF3)2-、-CH2-、-SO2-、-NHCO-を表し、*及び#はそれぞれ、他の構造との結合部位を表す。Rは水素原子又は1価の置換基を表し、水素原子又は炭化水素基が好ましく、水素原子又はアルキル基がより好ましい。また、R122は、上記式により表される構造であることも好ましい。R122が、上記式により表される構造である場合、計4つの*及び#のうち、いずれか2つが式(3)中のR122が結合する窒素原子との結合部位であり、かつ、別の2つが式(3)中のR122が結合する酸素原子との結合部位であることが好ましく、2つの*が式(3)中のR122が結合する酸素原子との結合部位であり、かつ、2つの#が式(3)中のR122が結合する窒素原子との結合部位であるか、又は、2つの*が式(3)中のR122が結合する窒素原子との結合部位であり、かつ、2つの#が式(3)中のR122が結合する酸素原子との結合部位であることがより好ましく、2つの*が式(3)中のR122が結合する酸素原子との結合部位であり、かつ、2つの#が式(3)中のR122が結合する窒素原子との結合部位であることが更に好ましい。

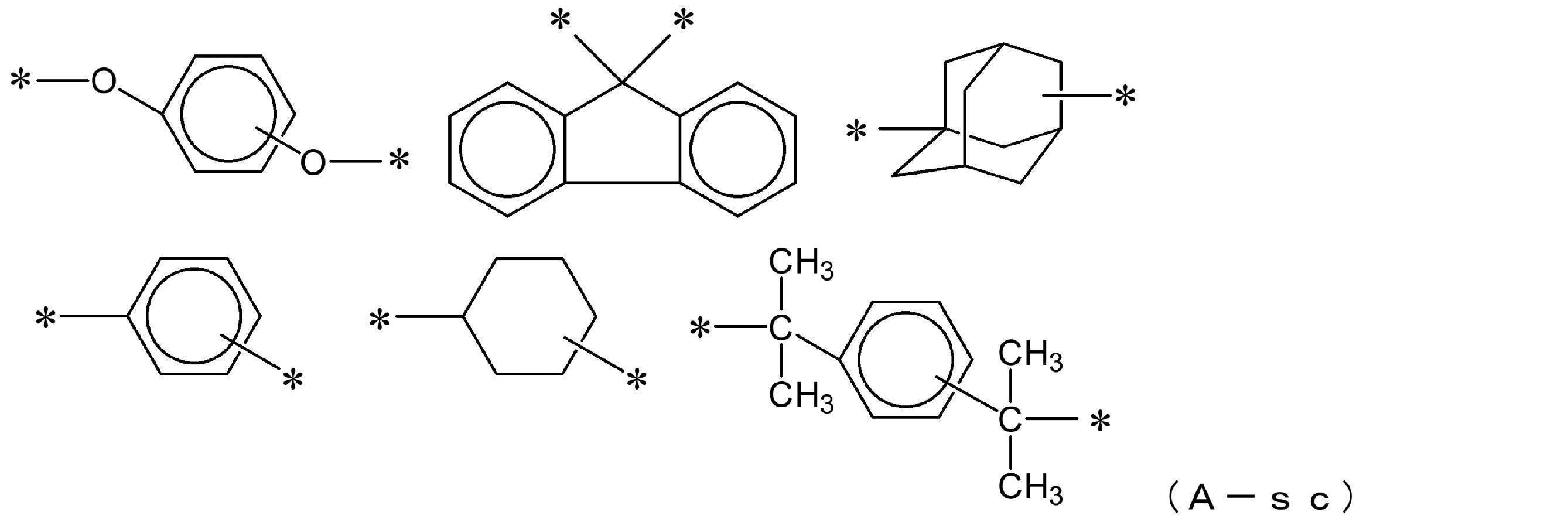
ポリベンゾオキサゾール前駆体は、閉環に伴う反りの発生を抑制できる点で、下記式(SL)で表されるジアミン残基を他の種類の繰返し単位として含むことが好ましい。

式(SL)中、Zは、a構造とb構造を有し、R1sは、水素原子又は炭素数1~10の炭化水素基であり、R2sは炭素数1~10の炭化水素基であり、R3s、R4s、R5s、R6sのうち少なくとも1つは芳香族基で、残りは水素原子又は炭素数1~30の有機基で、それぞれ同一でも異なっていてもよい。a構造及びb構造の重合は、ブロック重合でもランダム重合でもよい。Z部分のモル%は、a構造は5~95モル%、b構造は95~5モル%であり、a+bは100モル%である。
上記ポリベンゾオキサゾール前駆体の分子量の分散度は、1.4以上であることが好ましく、1.5以上がより好ましく、1.6以上であることが更に好ましい。ポリベンゾオキサゾール前駆体の分子量の分散度の上限値は特に定めるものではないが、例えば、2.6以下が好ましく、2.5以下がより好ましく、2.4以下が更に好ましく、2.3以下が一層好ましく、2.2以下がより一層好ましい。
また、樹脂組成物が特定樹脂として複数種のポリベンゾオキサゾール前駆体を含む場合、少なくとも1種のポリベンゾオキサゾール前駆体の重量平均分子量、数平均分子量、及び、分散度が上記範囲であることが好ましい。また、上記複数種のポリベンゾオキサゾール前駆体を1つの樹脂として算出した重量平均分子量、数平均分子量、及び、分散度が、それぞれ、上記範囲内であることも好ましい。
ポリベンゾオキサゾールとしては、ベンゾオキサゾール環を有する高分子化合物であれば、特に限定はないが、下記式(X)で表される化合物であることが好ましく、下記式(X)で表される化合物であって、重合性基を有する化合物であることがより好ましい。上記重合性基としては、ラジカル重合性基が好ましい。

式(X)中、R133は、2価の有機基を表し、R134は、4価の有機基を表す。
重合性基を有する場合、重合性基は、R133及びR134の少なくとも一方に位置していてもよいし、下記式(X-1)又は式(X-2)に示すようにポリベンゾオキサゾールの末端に位置していてもよい。
式(X-1)
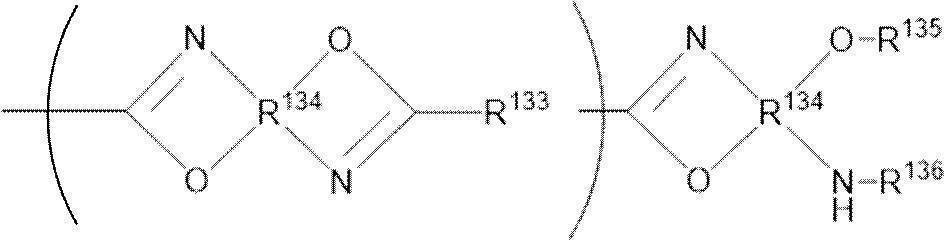
式(X-1)中、R135及びR136の少なくとも一方は、重合性基であり、重合性基でない場合は有機基であり、他の基は式(X)と同義である。
式(X-2)

式(X-2)中、R137は重合性基であり、他は置換基であり、他の基は式(X)と同義である。
例えば、R122として例示される4価の有機基の4つの結合子が、上記式(X)中の窒素原子、酸素原子と結合して縮合環を形成する。例えば、R134が、下記有機基である場合、下記構造を形成する。下記構造中、*はそれぞれ、式(X)中の窒素原子又は酸素原子との結合部位を表す。

上記オキサゾール化率は、例えば下記方法により測定される。
ポリベンゾオキサゾールの赤外吸収スペクトルを測定し、前駆体のアミド構造に由来する吸収ピークである1650cm-1付近のピーク強度Q1を求める。次に、1490cm-1付近に見られる芳香環の吸収強度で規格化する。そのポリベンゾオキサゾールを350℃で1時間熱処理した後、再度、赤外吸収スペクトルを測定し、1650cm-1付近のピーク強度Q2を求め、1490cm-1付近に見られる芳香環の吸収強度で規格化する。得られたピーク強度Q1、Q2の規格値を用い、下記式に基づいて、ポリベンゾオキサゾールのオキサゾール化率を求めることができる。
オキサゾール化率(%)=(ピーク強度Q1の規格値/ピーク強度Q2の規格値)×100
なお、ジカルボン酸の場合には反応収率等を高めるため、1-ヒドロキシ-1,2,3-ベンゾトリアゾール等を予め反応させた活性エステル型のジカルボン酸誘導体を用いてもよい。
また、ポリベンゾオキサゾールの数平均分子量(Mn)は、好ましくは7,200~14,000であり、より好ましくは8,000~12,000であり、更に好ましくは9,200~11,200である。
上記ポリベンゾオキサゾールの分子量の分散度は、1.4以上であることが好ましく、1.5以上がより好ましく、1.6以上であることが更に好ましい。ポリベンゾオキサゾールの分子量の分散度の上限値は特に定めるものではないが、例えば、2.6以下が好ましく、2.5以下がより好ましく、2.4以下が更に好ましく、2.3以下が一層好ましく、2.2以下がより一層好ましい。
また、樹脂組成物が特定樹脂として複数種のポリベンゾオキサゾールを含む場合、少なくとも1種のポリベンゾオキサゾールの重量平均分子量、数平均分子量、及び、分散度が上記範囲であることが好ましい。また、上記複数種のポリベンゾオキサゾールを1つの樹脂として算出した重量平均分子量、数平均分子量、及び、分散度が、それぞれ、上記範囲内であることも好ましい。
ポリアミドイミド前駆体は、下記式(PAI-2)で表される繰返し単位を含むことが好ましい。

式(PAI-2)中、R117は3価の有機基を表し、R111は2価の有機基を表し、A2は酸素原子又は-NH-を表し、R113は水素原子又は1価の有機基を表す。
上記連結基としては、-O-、-S-、-C(=O)-、-S(=O)2-、アルキレン基、ハロゲン化アルキレン基、アリーレン基、又はこれらを2以上結合した連結基が好ましく、-O-、-S-、アルキレン基、ハロゲン化アルキレン基、アリーレン基、又はこれらを2以上結合した連結基がより好ましい。
上記アルキレン基としては、炭素数1~20のアルキレン基が好ましく、炭素数1~10のアルキレン基がより好ましく、炭素数1~4のアルキレン基が更に好ましい。
上記ハロゲン化アルキレン基としては、炭素数1~20のハロゲン化アルキレン基が好ましく、炭素数1~10のハロゲン化アルキレン基がより好ましく、炭素数1~4のハロゲン化アルキレン基がより好ましい。また、上記ハロゲン化アルキレン基におけるハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられ、フッ素原子が好ましい。上記ハロゲン化アルキレン基は、水素原子を有していても、水素原子の全てがハロゲン原子で置換されていてもよいが、水素原子の全てがハロゲン原子で置換されていることが好ましい。好ましいハロゲン化アルキレン基の例としては、(ジトリフルオロメチル)メチレン基等が挙げられる。
上記アリーレン基としては、フェニレン基又はナフチレン基が好ましく、フェニレン基がより好ましく、1,3-フェニレン基又は1,4-フェニレン基が更に好ましい。
本発明において、カルボキシ基を3つ有する化合物をトリカルボン酸化合物という。
上記トリカルボン酸化合物の3つのカルボキシ基のうち2つのカルボキシ基は酸無水物化されていてもよい。
ポリアミドイミド前駆体の製造に用いられるハロゲン化されていてもよいトリカルボン酸化合物としては、分岐鎖状の脂肪族、環状の脂肪族又は芳香族のトリカルボン酸化合物などが挙げられる。
これらのトリカルボン酸化合物は、1種のみ用いてもよいし、2種以上用いてもよい。
これらの化合物は、2つのカルボキシ基が無水物化した化合物(例えば、トリメリット酸無水物)であってもよいし、少なくとも1つのカルボキシ基がハロゲン化した化合物(例えば、無水トリメリット酸クロリド)であってもよい。
他の繰返し単位としては、上述の式(2)で表される繰返し単位、下記式(PAI-1)で表される繰返し単位等が挙げられる。

式(PAI-1)中、R116は、直鎖状又は分岐鎖状の脂肪族基、環状の脂肪族基、及び芳香族基、複素芳香族基、又は単結合若しくは連結基によりこれらを2以上連結した基が例示され、炭素数2~20の直鎖の脂肪族基、炭素数3~20の分岐の脂肪族基、炭素数3~20の環状の脂肪族基、炭素数6~20の芳香族基、又は、単結合若しくは連結基によりこれらを2以上組み合わせた基が好ましく、炭素数6~20の芳香族基、又は、単結合若しくは連結基により炭素数6~20の芳香族基を2以上組み合わせた基がより好ましい。
上記連結基としては、-O-、-S-、-C(=O)-、-S(=O)2-、アルキレン基、ハロゲン化アルキレン基、アリーレン基、又はこれらを2以上結合した連結基が好ましく、-O-、-S-、アルキレン基、ハロゲン化アルキレン基、アリーレン基、又はこれらを2以上結合した連結基がより好ましい。
上記アルキレン基としては、炭素数1~20のアルキレン基が好ましく、炭素数1~10のアルキレン基がより好ましく、炭素数1~4のアルキレン基が更に好ましい。
上記ハロゲン化アルキレン基としては、炭素数1~20のハロゲン化アルキレン基が好ましく、炭素数1~10のハロゲン化アルキレン基がより好ましく、炭素数1~4のハロゲン化アルキレン基がより好ましい。また、上記ハロゲン化アルキレン基におけるハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられ、フッ素原子が好ましい。上記ハロゲン化アルキレン基は、水素原子を有していても、水素原子の全てがハロゲン原子で置換されていてもよいが、水素原子の全てがハロゲン原子で置換されていることが好ましい。好ましいハロゲン化アルキレン基の例としては、(ジトリフルオロメチル)メチレン基等が挙げられる。
上記アリーレン基としては、フェニレン基又はナフチレン基が好ましく、フェニレン基がより好ましく、1,3-フェニレン基又は1,4-フェニレン基が更に好ましい。
本発明において、カルボキシ基を2つ有する化合物をジカルボン酸化合物、ハロゲン化されたカルボキシ基を2つ有する化合物をジカルボン酸ジハライド化合物という。
ジカルボン酸ジハライド化合物におけるカルボキシ基は、ハロゲン化されていればよいが、例えば、塩素化されていることが好ましい。すなわち、ジカルボン酸ジハライド化合物は、ジカルボン酸ジクロリド化合物であることが好ましい。
ポリアミドイミド前駆体の製造に用いられるハロゲン化されていてもよいジカルボン酸化合物又はジカルボン酸ジハライド化合物としては、直鎖状又は分岐鎖状の脂肪族、環状の脂肪族又は芳香族ジカルボン酸化合物又はジカルボン酸ジハライド化合物などが挙げられる。
これらのジカルボン酸化合物又はジカルボン酸ジハライド化合物は、1種のみ用いてもよいし、2種以上用いてもよい。
ジカルボン酸ジハライド化合物の具体例としては、上記ジカルボン酸化合物の具体例における2つのカルボキシ基をハロゲン化した構造の化合物が挙げられる。
また、本発明におけるポリアミドイミド前駆体の別の一実施形態として、式(PAI-2)で表される繰返し単位、及び、式(PAI-1)で表される繰返し単位の合計含有量が、全繰返し単位の50モル%以上である態様が挙げられる。上記合計含有量は、70モル%以上であることがより好ましく、90モル%以上であることが更に好ましく、90モル%超であることが特に好ましい。上記合計含有量の上限は、特に限定されず、末端を除くポリアミドイミド前駆体における全ての繰返し単位が、式(PAI-2)で表される繰返し単位、又は、式(PAI-1)で表される繰返し単位のいずれかであってもよい。
ポリアミドイミド前駆体の分子量の分散度は、1.5以上が好ましく、1.8以上がより好ましく、2.0以上であることが更に好ましい。ポリアミドイミド前駆体の分子量の分散度の上限値は特に定めるものではないが、例えば、7.0以下が好ましく、6.5以下がより好ましく、6.0以下が更に好ましい。 また、樹脂組成物が特定樹脂として複数種のポリアミドイミド前駆体を含む場合、少なくとも1種のポリアミドイミド前駆体の重量平均分子量、数平均分子量、及び、分散度が上記範囲であることが好ましい。また、上記複数種のポリアミドイミド前駆体を1つの樹脂として算出した重量平均分子量、数平均分子量、及び、分散度が、それぞれ、上記範囲内であることも好ましい。
本発明に用いられるポリアミドイミドは、アルカリ可溶性ポリアミドイミドであってもよく、有機溶剤を主成分とする現像液に対して可溶なポリアミドイミドであってもよい。
本明細書において、アルカリ可溶性ポリアミドイミドとは、100gの2.38質量%テトラメチルアンモニウム水溶液に対し、23℃で0.1g以上溶解するポリアミドイミドをいい、パターン形成性の観点からは、0.5g以上溶解するポリアミドイミドであることが好ましく、1.0g以上溶解するポリアミドイミドであることが更に好ましい。上記溶解量の上限は特に限定されないが、100g以下であることが好ましい。
また、ポリアミドイミドは、得られる有機膜の膜強度及び絶縁性の観点からは、複数個のアミド結合及び複数個のイミド構造を主鎖に有するポリアミドイミドであることが好ましい。
得られる有機膜の膜強度の観点からは、ポリアミドイミドは、フッ素原子を有することが好ましい。
フッ素原子は、例えば、後述する式(PAI-3)で表される繰返し単位におけるR117、又は、R111に含まれることが好ましく、後述する式(PAI-3)で表される繰返し単位におけるR117、又は、R111にフッ化アルキル基として含まれることがより好ましい。
ポリアミドイミドの全質量に対するフッ素原子の量は、5質量%以上が好ましく、また、20質量%以下が好ましい。
得られる有機膜の膜強度の観点からは、ポリアミドイミドは、エチレン性不飽和結合を有してもよい。
ポリアミドイミドは、エチレン性不飽和結合を主鎖末端に有していてもよいし、側鎖に有していてもよいが、側鎖に有することが好ましい。
上記エチレン性不飽和結合は、ラジカル重合性を有することが好ましい。
エチレン性不飽和結合は、後述する式(PAI-3)で表される繰返し単位におけるR117、又は、R111に含まれることが好ましく、後述する式(PAI-3)で表される繰返し単位におけるR117、又は、R111にエチレン性不飽和結合を有する基として含まれることがより好ましい。
エチレン性不飽和結合を有する基の好ましい態様は、上述のポリイミドにおけるエチレン性不飽和結合を有する基の好ましい態様と同様である。
ポリアミドイミドは、エチレン性不飽和結合以外の重合性基を有していてもよい。
ポリアミドイミドにおけるエチレン性不飽和結合以外の重合性基としては、上述のポリイミドにおけるエチレン性不飽和結合以外の重合性基と同様の基が挙げられる。
エチレン性不飽和結合以外の重合性基は、例えば、後述する式(PAI-3)で表される繰返し単位におけるR111に含まれることが好ましい。
ポリアミドイミドの全質量に対するエチレン性不飽和結合以外の重合性基の量は、0.05~10mol/gであることが好ましく、0.1~5mol/gであることがより好ましい。
ポリアミドイミドがアルカリ現像に供される場合、現像性を向上する観点からは、ポリアミドイミドの酸価は、30mgKOH/g以上であることが好ましく、50mgKOH/g以上であることがより好ましく、70mgKOH/g以上であることが更に好ましい。
また、上記酸価は500mgKOH/g以下であることが好ましく、400mgKOH/g以下であることがより好ましく、200mgKOH/g以下であることが更に好ましい。
また、ポリアミドイミドが有機溶剤を主成分とする現像液を用いた現像(例えば、後述する「溶剤現像」)に供される場合、ポリアミドイミドの酸価は、2~35mgKOH/gが好ましく、3~30mgKOH/gがより好ましく、5~20mgKOH/gが更に好ましい。
上記酸価は、公知の方法により測定され、例えば、JIS K 0070:1992に記載の方法により測定される。
また、ポリアミドイミドに含まれる酸基としては、上述のポリイミドにおける酸基と同様の基が挙げられ、好ましい態様も同様である。
アルカリ現像液による現像速度を適切なものとする観点からは、ポリアミドイミドは、フェノール性ヒドロキシ基を有することが好ましい。
ポリアミドイミドは、フェノール性ヒドロキシ基を主鎖末端に有してもよいし、側鎖に有してもよい。
フェノール性ヒドロキシ基は、例えば、後述する式(PAI-3)で表される繰返し単位におけるR117、又は、R111に含まれることが好ましい。
ポリアミドイミドの全質量に対するフェノール性ヒドロキシ基の量は、0.1~30mol/gであることが好ましく、1~20mol/gであることがより好ましい。

式(PAI-3)中、R111及びR117はそれぞれ、式(PAI-2)中のR111及びR117と同義であり、好ましい態様も同様である。
重合性基を有する場合、重合性基は、R111及びR117の少なくとも一方に位置していてもよいし、ポリアミドイミドの末端に位置していてもよい。
ポリアミドイミドのイミド化率(「閉環率」ともいう)は、得られる有機膜の膜強度、絶縁性等の観点からは、70%以上であることが好ましく、80%以上であることがより好ましく、90%以上であることがより好ましい。
上記イミド化率の上限は特に限定されず、100%以下であればよい。
上記イミド化率は、上述のポリイミドの閉環率と同様の方法により測定される。
また、ポリアミドイミドの数平均分子量(Mn)は、好ましくは800~250,000であり、より好ましくは、2,000~50,000であり、更に好ましくは、4,000~25,000である。
ポリアミドイミドの分子量の分散度は、1.5以上が好ましく、1.8以上がより好ましく、2.0以上であることが更に好ましい。ポリアミドイミドの分子量の分散度の上限値は特に定めるものではないが、例えば、7.0以下が好ましく、6.5以下がより好ましく、6.0以下が更に好ましい。
また、樹脂組成物が特定樹脂として複数種のポリアミドイミドを含む場合、少なくとも1種のポリアミドイミドの重量平均分子量、数平均分子量、及び、分散度が上記範囲であることが好ましい。また、上記複数種のポリアミドイミドを1つの樹脂として算出した重量平均分子量、数平均分子量、及び、分散度が、それぞれ、上記範囲内であることも好ましい。
ポリイミド前駆体等は、例えば、低温中でテトラカルボン酸二無水物とジアミンを反応させる方法、低温中でテトラカルボン酸二無水物とジアミンを反応させてポリアミック酸を得、縮合剤又はアルキル化剤を用いてエステル化する方法、テトラカルボン酸二無水物とアルコールとによりジエステルを得て、その後ジアミンと縮合剤の存在下で反応させる方法、テトラカルボン酸二無水物とアルコールとによりジエステルを得、その後残りのジカルボン酸をハロゲン化剤を用いて酸ハロゲン化し、ジアミンと反応させる方法、などの方法を利用して得ることができる。上記製造方法のうち、テトラカルボン酸二無水物とアルコールとによりジエステルを得、その後残りのジカルボン酸をハロゲン化剤を用いて酸ハロゲン化し、ジアミンと反応させる方法がより好ましい。
上記縮合剤としては、例えばジシクロヘキシルカルボジイミド、ジイソプロピルカルボジイミド、1-エトキシカルボニル-2-エトキシ-1,2-ジヒドロキノリン、1,1-カルボニルジオキシ-ジ-1,2,3-ベンゾトリアゾール、N,N’-ジスクシンイミジルカーボネート、無水トリフルオロ酢酸等が挙げられる。
上記アルキル化剤としては、N,N-ジメチルホルムアミドジメチルアセタール、N,N-ジメチルホルムアミドジエチルアセタール、N,N-ジアルキルホルムアミドジアルキルアセタール、オルトギ酸トリメチル、オルトギ酸トリエチル等が挙げられる。
上記ハロゲン化剤としては、塩化チオニル、塩化オキサリル、オキシ塩化リン等が挙げられる。
ポリイミド前駆体等の製造方法では、反応に際し、有機溶剤を用いることが好ましい。有機溶剤は1種でもよいし、2種以上でもよい。
有機溶剤としては、原料に応じて適宜定めることができるが、ピリジン、ジエチレングリコールジメチルエーテル(ジグリム)、N-メチルピロリドン、N-エチルピロリドン、プロピオン酸エチル、ジメチルアセトアミド、ジメチルホルムアミド、テトラヒドロフラン、γ-ブチロラクトン等が例示される。
ポリイミド前駆体等の製造方法では、反応に際し、塩基性化合物を添加することが好ましい。塩基性化合物は1種でもよいし、2種以上でもよい。
塩基性化合物は、原料に応じて適宜定めることができるが、トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン、N,N-ジメチル-4-アミノピリジン等が例示される。
ポリイミド前駆体等の製造方法に際し、保存安定性をより向上させるため、ポリイミド前駆体等の樹脂末端に残存するカルボン酸無水物、酸無水物誘導体、或いは、アミノ基を封止することが好ましい。樹脂末端に残存するカルボン酸無水物、及び酸無水物誘導体を封止する際、末端封止剤としては、モノアルコール、フェノール、チオール、チオフェノール、モノアミン等が挙げられ、反応性、膜の安定性から、モノアルコール、フェノール類やモノアミンを用いることがより好ましい。モノアルコールの好ましい化合物としては、メタノール、エタノール、プロパノール、ブタノール、ヘキサノール、オクタノール、ドデシノール、ベンジルアルコール、2-フェニルエタノール、2-メトキシエタノール、2-クロロメタノール、フルフリルアルコール等の1級アルコール、イソプロパノール、2-ブタノール、シクロヘキシルアルコール、シクロペンタノール、1-メトキシ-2-プロパノール等の2級アルコール、t-ブチルアルコール、アダマンタンアルコール等の3級アルコールが挙げられる。フェノール類の好ましい化合物としては、フェノール、メトキシフェノール、メチルフェノール、ナフタレン-1-オール、ナフタレン-2-オール、ヒドロキシスチレン等のフェノール類などが挙げられる。また、モノアミンの好ましい化合物としては、アニリン、2-エチニルアニリン、3-エチニルアニリン、4-エチニルアニリン、5-アミノ-8-ヒドロキシキノリン、1-ヒドロキシ-7-アミノナフタレン、1-ヒドロキシ-6-アミノナフタレン、1-ヒドロキシ-5-アミノナフタレン、1-ヒドロキシ-4-アミノナフタレン、2-ヒドロキシ-7-アミノナフタレン、2-ヒドロキシ-6-アミノナフタレン、2-ヒドロキシ-5-アミノナフタレン、1-カルボキシ-7-アミノナフタレン、1-カルボキシ-6-アミノナフタレン、1-カルボキシ-5-アミノナフタレン、2-カルボキシ-7-アミノナフタレン、2-カルボキシ-6-アミノナフタレン、2-カルボキシ-5-アミノナフタレン、2-アミノ安息香酸、3-アミノ安息香酸、4-アミノ安息香酸、4-アミノサリチル酸、5-アミノサリチル酸、6-アミノサリチル酸、2-アミノベンゼンスルホン酸、3-アミノベンゼンスルホン酸、4-アミノベンゼンスルホン酸、3-アミノ-4,6-ジヒドロキシピリミジン、2-アミノフェノール、3-アミノフェノール、4-アミノフェノール、2-アミノチオフェノール、3-アミノチオフェノール、4-アミノチオフェノールなどが挙げられる。これらを2種以上用いてもよく、複数の末端封止剤を反応させることにより、複数の異なる末端基を導入してもよい。
また、樹脂末端のアミノ基を封止する際、アミノ基と反応可能な官能基を有する化合物で封止することが可能である。アミノ基に対する好ましい封止剤は、カルボン酸無水物、カルボン酸クロリド、カルボン酸ブロミド、スルホン酸クロリド、無水スルホン酸、スルホン酸カルボン酸無水物などが好ましく、カルボン酸無水物、カルボン酸クロリドがより好ましい。カルボン酸無水物の好ましい化合物としては、無水酢酸、無水プロピオン酸、無水シュウ酸、無水コハク酸、無水マレイン酸、無水フタル酸、無水安息香酸、5-ノルボルネン-2,3-ジカルボン酸無水物などが挙げられる。また、カルボン酸クロリドの好ましい化合物としては、塩化アセチル、アクリル酸クロリド、プロピオニルクロリド、メタクリル酸クロリド、ピバロイルクロリド、シクロヘキサンカルボニルクロリド、2-エチルヘキサノイルクロリド、シンナモイルクロリド、1-アダマンタンカルボニルクロリド、ヘプタフルオロブチリルクロリド、ステアリン酸クロリド、ベンゾイルクロリド、などが挙げられる。
ポリイミド前駆体等の製造に際し、固体を析出する工程を含んでいてもよい。具体的には、反応液中に共存している脱水縮合剤の吸水副生物を必要に応じて濾別した後、水、脂肪族低級アルコール、又はその混合液等の貧溶媒に、得られた重合体成分を投入し、重合体成分を析出させることで、固体として析出させ、乾燥させることでポリイミド前駆体等を得ることができる。精製度を向上させるために、ポリイミド前駆体等を再溶解、再沈析出、乾燥等の操作を繰返してもよい。さらに、イオン交換樹脂を用いてイオン性不純物を除去する工程を含んでいてもよい。
本発明の樹脂組成物における特定樹脂の含有量は、樹脂組成物の全固形分に対し20質量%以上であることが好ましく、30質量%以上であることがより好ましく、40質量%以上であることが更に好ましく、50質量%以上であることが一層好ましい。また、本発明の樹脂組成物における樹脂の含有量は、樹脂組成物の全固形分に対し、99.5質量%以下であることが好ましく、99質量%以下であることがより好ましく、98質量%以下であることが更に好ましく、97質量%以下であることが一層好ましく、95質量%以下であることがより一層好ましい。
本発明の樹脂組成物は、特定樹脂を1種のみ含んでいてもよいし、2種以上含んでいてもよい。2種以上含む場合、合計量が上記範囲となることが好ましい。
具体的には、本発明の樹脂組成物は、特定樹脂と、後述する他の樹脂とを合計で2種以上含んでもよいし、特定樹脂を2種以上含んでいてもよいが、特定樹脂を2種以上含むことが好ましい。
本発明の樹脂組成物が特定樹脂を2種以上含む場合、例えば、ポリイミド前駆体であって、二無水物由来の構造(上述の式(2)でいうR115)が異なる2種以上のポリイミド前駆体を含むことが好ましい。
本発明の樹脂組成物は、上述した特定樹脂に加えて、又は、上述の特定樹脂に代えて、特定樹脂とは異なる他の樹脂(以下、単に「他の樹脂」ともいう)を含んでもよい。
他の樹脂としては、フェノール樹脂、ポリアミド、エポキシ樹脂、ポリシロキサン、シロキサン構造を含む樹脂、(メタ)アクリル樹脂、(メタ)アクリルアミド樹脂、ウレタン樹脂、ブチラール樹脂、スチリル樹脂、ポリエーテル樹脂、ポリエステル樹脂等が挙げられる。
例えば、(メタ)アクリル樹脂を更に加えることにより、塗布性に優れた樹脂組成物が得られ、また、耐溶剤性に優れたパターン(硬化物)が得られる。
例えば、後述する重合性化合物に代えて、又は、後述する重合性化合物に加えて、重量平均分子量が20,000以下の重合性基価の高い(例えば、樹脂1gにおける重合性基の含有モル量が1×10-3モル/g以上である)(メタ)アクリル樹脂を樹脂組成物に添加することにより、樹脂組成物の塗布性、パターン(硬化物)の耐溶剤性等を向上させることができる。
また、本発明の樹脂組成物における、他の樹脂の含有量は、樹脂組成物の全固形分に対し、80質量%以下であることが好ましく、75質量%以下であることがより好ましく、70質量%以下であることが更に好ましく、60質量%以下であることが一層好ましく、50質量%以下であることがより一層好ましい。
また、本発明の樹脂組成物の好ましい一態様として、他の樹脂の含有量が低含有量である態様とすることもできる。上記態様において、他の樹脂の含有量は、樹脂組成物の全固形分に対し、20質量%以下であることが好ましく、15質量%以下であることがより好ましく、10質量%以下であることが更に好ましく、5質量%以下であることが一層好ましく、1質量%以下であることがより一層好ましい。上記含有量の下限は特に限定されず、0質量%以上であればよい。
本発明の樹脂組成物は、他の樹脂を1種のみ含んでいてもよいし、2種以上含んでいてもよい。2種以上含む場合、合計量が上記範囲となることが好ましい。
本発明の第1の態様におけるネガ型感光性樹脂組成物は、露光によりその露光波長の吸光度が小さくなる化合物Aを含む。
まず、ネガ型感光性樹脂組成物に含まれる濃度と同濃度の化合物aの溶液を調製し、露光光の波長における化合物aのモル吸光係数(mol-1・L・cm-1、「モル吸光係数1」ともいう。)を測定する。上記測定は、化合物aのモル吸光係数の低下などの変化の影響が小さくなるよう手早く行う。上記溶液における溶剤は、ネガ型感光性樹脂組成物が溶剤を含む場合はその溶剤を、ネガ型感光性樹脂組成物が溶剤を含まない場合はN-メチル-2-ピロリドンを用いる。
次に、上記化合物aの溶液に対して露光光の照射を行う。露光量は1モルの化合物aに対して積算量として500mJとする。
その後、露光後の上記化合物aの溶液を用い、露光光の波長における化合物aのモル吸光係数(mol-1・L・cm-1、「モル吸光係数2」ともいう。)を測定する。
上記モル吸光係数1及びモル吸光係数2から、下記式に基づいて減衰率(%)を算出し、減衰率(%)が5%以上である場合に、化合物aは露光によりその露光波長の吸光度が小さくなる化合物(すなわち、化合物A)であると判断する。
減衰率(%)=1-モル吸光係数2/モル吸光係数1×100
上記減衰率は、10%以上であることが好ましく、20%以上であることがより好ましい。また、上記減衰率の下限は特に限定されず、0%以上であればよい。
上記露光光の波長としては、ネガ型感光性樹脂組成物を感光膜の形成に用いる場合にはその感光膜が露光される波長であればよい。
また、上記露光光の波長としては、ネガ型感光性樹脂組成物に含まれる光重合開始剤が感度を有する波長であることが好ましい。光重合開始剤がある波長に対して感度を有するとは、光重合開始剤をある波長において露光した際に重合開始種を生じることをいう。
上記露光光の波長としては、光源との関係でいうと、(1)半導体レーザー(波長 830nm、532nm、488nm、405nm、375nm、355nm etc.)、(2)メタルハライドランプ、(3)高圧水銀灯、g線(波長 436nm)、h線(波長 405nm)、i線(波長 365nm)、ブロード(g,h,i線の3波長)、(4)エキシマレーザー、KrFエキシマレーザー(波長 248nm)、ArFエキシマレーザー(波長 193nm)、F2エキシマレーザー(波長 157nm)、(5)極端紫外線;EUV(波長 13.6nm)、(6)電子線、(7)YAGレーザーの第二高調波532nm、第三高調波355nm等が挙げられる。
露光光の波長は、例えば光重合開始剤が感度を有する波長を選択すればよいが、h線(波長 405nm)又はi線(波長 365nm)が好ましく、i線(波長 365nm)がより好ましい。
化合物Aが露光によりラジカル重合開始種を発生する化合物であるか否かは、下記の方法により判定される。
ネガ型感光性樹脂組成物に含まれる濃度と同濃度の化合物A、及び、ラジカル架橋剤を含む溶液を調製する。ネガ型感光性樹脂組成物がラジカル架橋剤を含む場合、上記溶液中のラジカル架橋剤としては、ネガ型感光性樹脂組成物に含まれるラジカル架橋剤と同一の化合物を同濃度で使用する。ネガ型感光性樹脂組成物がラジカル架橋剤を含まない場合、メタクリル酸メチルを化合物Aの5倍の濃度で使用する。
その後、露光光の照射を行う。露光量は積算量として500mJとする。
露光後に、例えば高速液体クロマトグラフィにより重合性化合物の重合を判断し、重合性化合物の全モル量に対して重合した重合性化合物のモル量の割合が10%以下である場合に、化合物Aが露光によりラジカル重合開始種を発生しない化合物であると判定する。
上記モル量の割合は5%以下であることが好ましく、3%以下であることがより好ましい。また、上記モル量の割合の下限は特に限定されず、0%であってもよい。
上記露光光の波長としては、ネガ型感光性樹脂組成物を感光膜の形成に用いる場合にはその感光膜が露光される波長であればよい。
また、上記露光光の波長としては、ネガ型感光性樹脂組成物に含まれる光重合開始剤が感度を有する波長であることが好ましい。
また、上記露光光の波長としては、ネガ型感光性樹脂組成物に含まれる光重合開始剤が感度を有する波長であることが好ましい。
露光によりラジカル重合開始種を発生しない化合物としては、光酸発生剤、光塩基発生剤、その他、露光により吸収波長が変化する色素等が挙げられる。
これらの中でも、化合物Aとしては、ナフトキノンジアジド化合物、又は、露光により吸光度が変化する色素であることが好ましく、ナフトキノンジアジド化合物であることがより好ましい。
また、化合物Aとして、例えば、光酸発生剤又は光塩基発生剤と、pHにより露光波長の吸光度が小さくなる化合物とを組み合わせて用いることも考えられる。
第2の態様におけるナフトキノンジアジド化合物は、第1の態様において好ましい態様として含まれるナフトキノンジアジド化合物と同義であり、その好ましい態様も同様である。
第2の態様におけるナフトキノンジアジド化合物は、露光によりその露光波長の吸光度が小さくなる化合物であることが好ましい。露光によりその露光波長の吸光度が小さくなる化合物であるか否かの判定方法、及び、減衰率の好ましい範囲は、上述の第1の態様における化合物Aにおける判定方法及び減衰率と同様である。
ナフトキノンジアジド化合物としては、露光によりインデンカルボン酸を生じてその露光波長の吸光度が小さくなる化合物が挙げられ、1,2-ナフトキノンジアジド構造を有する化合物が好ましい。
ナフトキノンジアジド化合物としては、ヒドロキシ化合物のナフトキノンジアジドスルホン酸エステルであることが好ましい。
上記ヒドロキシ化合物としては、下記式(H1)~(H6)のいずれかで表される化合物が好ましい。

式(H2)中、Zは4価の有機基を表し、L1、L2、L3及びL4はそれぞれ独立に、単結合又は2価の有機基を表し、R5、R6、R7及びR8はそれぞれ独立に、1価の有機基を表し、n3、n4、n5及びn6はそれぞれ独立に、0~3の整数であり、m3、m4、m5及びm6はそれぞれ独立に、0~2の整数であり、m3、m4、m5及びm6のうち少なくとも1つは1又は2である。
式(H3)中、R9及びR10はそれぞれ独立に、水素原子又は1価の有機基を表し、L5はそれぞれ独立に、2価の有機基を表し、n7は3~8の整数を表す。
式(H4)中、L6は2価の有機基を表し、L7及びL8はそれぞれ独立に、脂肪族の3級又は4級炭素を含む2価の有機基を表す。
式(H5)中、R11、R12、R13、R14、R15、R16、R17、R18、R19及びR20はそれぞれ独立に、水素原子、ハロゲン原子又は1価の有機基を表し、L9、L10およびL11はそれぞれ独立に、単結合又は2価の有機基を表し、m7、m8、m9、m10はそれぞれ独立に、0~2の整数を表し、m7、m8、m9、m10のうち少なくとも1つは1又は2である。
式(H6)中、R42、R43、R44、及びR45はそれぞれ独立に、水素原子又は1価の有機基を表し、R46、及びR47はそれぞれ独立に、1価の有機基を表し、n16及びn17はそれぞれ独立に、0~4の整数を表し、m11及びm12はそれぞれ独立に、0~4の整数を表し、m11及びm12のうち少なくとも1つは1~4の整数である。
式(H1)中、R3及びR4はそれぞれ独立に、炭素数1~60の1価の有機基であることが好ましく、炭素数1~30の1価の有機基であることがより好ましい。R3及びR4における1価の有機基としては、置換基を有してもよい炭化水素基が挙げられ、例えば、ヒドロキシ基等の置換基を有してもよい炭化水素基等が挙げられる。
式(H1)中、n1及びn2はそれぞれ独立に、0又は1であることが好ましく、0であることがより好ましい。
式(H1)中、m1及びm2はいずれも1であることが好ましい。


式(R-1)中、R29は水素原子、アルキル基又はアルコキシ基を表し、n13は0~2の整数を表し、*は他の構造との結合部位を表す。
(H1-1)中、n8、n9及びn10はそれぞれ独立に、0~2の整数を表し、0又は1が好ましい。
式(H1-3)中、n11、n12及びn13はそれぞれ独立に、0~2の整数を表し、0又は1が好ましい。
式(H1-2)で表される化合物としては下記式(H1-2-1)または(H1-2-2)で表される化合物が好ましい。
式(H1-3)で表される化合物としては下記式(H1-3-1)~式(H1-3-3)で表される化合物が好ましい。


式(H2)中、L1、L2、L3及びL4はそれぞれ独立に、単結合又はメチレン基であることが好ましい。
式(H2)中、R5、R6、R7及びR8はそれぞれ独立に、炭素数1~30の有機基が好ましい。
式(H2)中、n3、n4、n5及びn6はそれぞれ独立に、0~2の整数であることが好ましく、0又は1であることがより好ましい。
式(H2)中、m3、m4、m5及びm6はそれぞれ独立に、1又は2であることが好ましく、1であることがより好ましい。
式(H2)で表される化合物としては、下記構造の化合物が例示される。
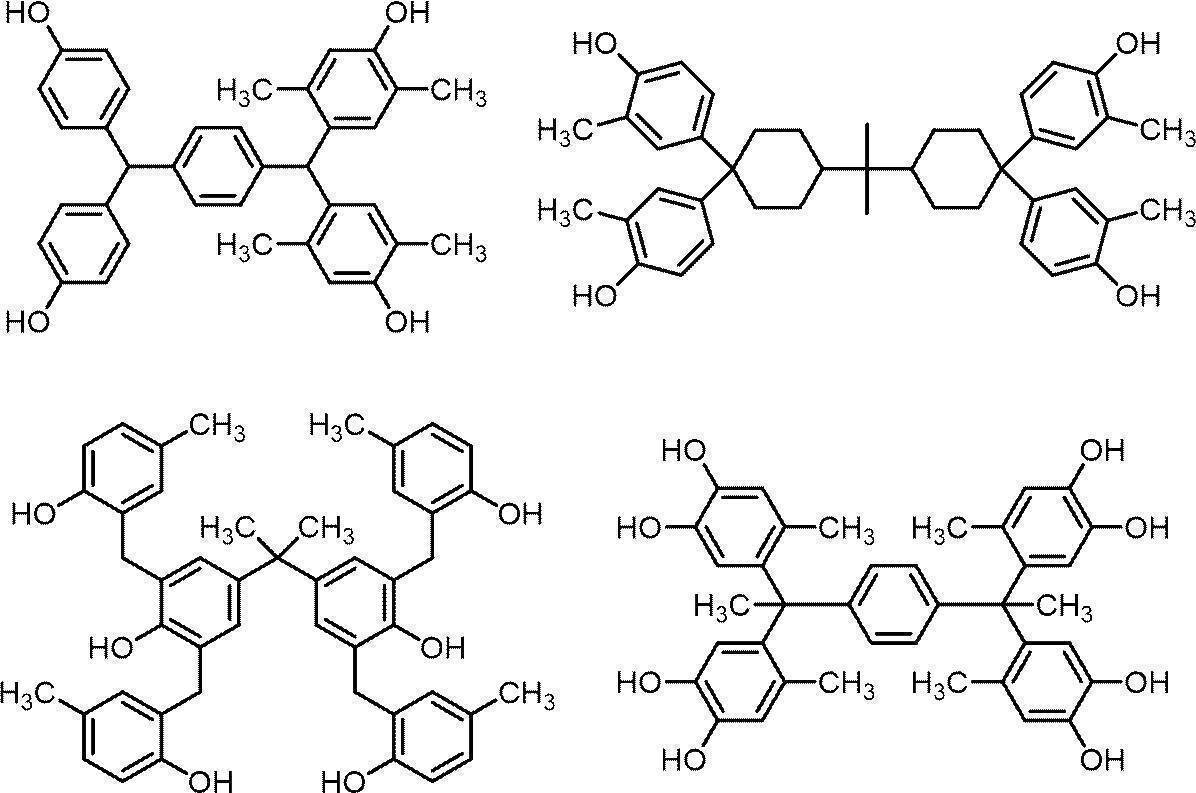
式(H3)中、L5はそれぞれ独立に、下記式(L-1)で表される基であることが好ましい。

式(L-1)中、R30は炭素数1~20の1価の有機基を表し、n14は1~5の整数を表し、*は他の構造との結合部位を表す。
式(H3)中、n7は4~6の整数であることが好ましい。
式(H3)で表される化合物としては、下記化合物が挙げられる。下記式中、nはそれぞれ独立に、0~9の整数を表す。

式(H4)中、L7及びL8はそれぞれ独立に、炭素数2~20の2価の有機基であることが好ましい。
式(H4)で表される化合物としては、下記化合物が挙げられる。

式(H5)中、L9、L10およびL11はそれぞれ独立に、単結合、-O-、-S-、-S(=O)2-、-C(=O)-、-C(=O)O-、シクロペンチリデン、シクロヘキシリデン、フェニレンまたは炭素数1~20の2価の有機基が好ましく、下記式(L-2)~式(L-4)のいずれかで表される基であることがより好ましい。
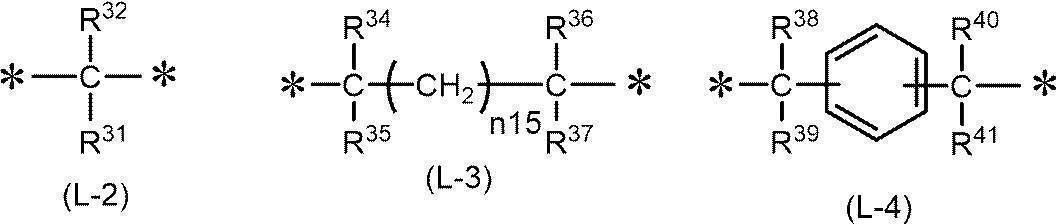
式(L-2)~式(L-4)中、R31及びR32はそれぞれ独立に、水素原子、アルキル基、アルケニル基又はアリール基を表し、R34、R35、R36及びR37はそれぞれ独立に、水素原子又はアルキル基を表し、n15は、1~5の整数であり、R38、R39、R40及びR41はそれぞれ独立に、水素原子又はアルキル基を表し、*は他の構造との結合部位を表す。
式(H5)で表される化合物としては、下記化合物が挙げられる。

式(H6)中、R46、及びR47はそれぞれ独立に、アルキル基、アルコキシ基又はアリール基が好ましく、アルキル基がより好ましい。
式(H6)中、n16及びn17はそれぞれ独立に、0~2の整数が好ましく、0又は1がより好ましい。
式(H6)中、n16及びn17はそれぞれ独立に、1~3の整数が好ましく、2又は3がより好ましい。
式(H6)で表される化合物としては、下記化合物が挙げられる。
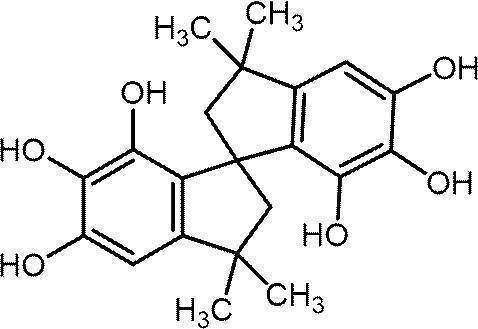
2,3,4-トリヒドロキシアセトフェノン、2,3,4-トリヒドロキシフェニルペンチルケトン、2,3,4-トリヒドロキシフェニルヘキシルケトン等のポリヒドロキシフェニルアルキルケトン類、
ビス(2,4-ジヒドロキシフェニル)メタン、ビス(2,3,4-トリヒドロキシフェニル)メタン、ビス(2,4-ジヒドロキシフェニル)プロパン-1、ビス(2,3,4-トリヒドロキシフェニル)プロパン-1、ノルジヒドログアイアレチン酸、1,1-ビス(4-ヒドロキシフェニル)シクロヘキサン等のビス((ポリ)ヒドロキシフェニル)アルカン類、
3,4,5-トリヒドロキシ安息香酸プロピル、2,3,4-トリヒドロキシ安息香酸フェニル、3,4,5-トリヒドロキシ安息香酸フェニル等のポリヒドロキシ安息香酸エステル類、
ビス(2,3,4-トリヒドロキシベンゾイル)メタン、ビス(3-アセチル-4,5,6-トリヒドロキシフェニル)-メタン、ビス(2,3,4-トリヒドロキシベンゾイル)ベンゼン、ビス(2,4,6-トリヒドロキシベンゾイル)ベンゼン等のビス(ポリヒドロキシベンゾイル)アルカン又はビス(ポリヒドロキシベンゾイル)アリール類、
エチレングリコール-ジ(3,5-ジヒドロキシベンゾエート)、エチレングリコール-ジ(3,4,5-トリヒドロキシベンゾエート)等のアルキレン-ジ(ポリヒドロキシベンゾエート)類、
2,3,4-ビフェニルトリオール、3,4,5-ビフェニルトリオール、3,5,3′,5′-ビフェニルテトロール、2,4,2′,4′-ビフェニルテトロール、2,4,6,3′,5′-ビフェニルペントール、2,4,6,2′,4′,6′-ビフェニルヘキソール、2,3,4,2′,3′,4′-ビフェニルヘキソール等のポリヒドロキシビフェニル類、
4,4′-チオビス(1,3-ジヒドロキシ)ベンゼン等のビス(ポリヒドロキシ)スルフィド類、
2,2′,4,4′-テトラヒドロキシジフェニルエーテル等のビス(ポリヒドロキシフェニル)エーテル類、
2,2′,4,4′-テトラヒドロキシジフェニルスルフォキシド等のビス(ポリヒドロキシフェニル)スルフォキシド類、
2,2′,4,4′-ジフェニルスルフォン等のビス(ポリヒドロキシフェニル)スルフォン類、
トリス(4-ヒドロキシフェニル)メタン、4,4′,4″-トリヒドロキシ-3,5,3′,5′-テトラメチルトリフェニルメタン、4,4′,3″,4″-テトラヒドロキシ-3,5,3′,5′-テトラメチルトリフェニルメタン、4-[ビス(3,5-ジメチル-4-ヒドロキシフェニル)メチル]-2-メトキシ-フェノール、4,4′-(3,4-ジオール-ベンジリデン)ビス[2,6-ジメチルフェノール]、4,4′-[(2-ヒドロキシ-フェニル)メチレン]ビス[2-シクロヘキシル-5-メチルフェノール、4,4′,2″,3″,4″-ペンタヒドロキシ-3,5,3′,5′-テトラメチルトリフェニルメタン、2,3,4,2′,3′,4′-ヘキサヒドロキシ-5,5′-ジアセチルトリフェニルメタン、2,3,4,2′,3′,4′,3″,4″-オクタヒドロキシ-5,5′-ジアセチルトリフェニルメタン、2,4,6,2′,4′,6′-ヘキサヒドロキシ-5,5′-ジプロピオニルトリフェニルメタン等のポリヒドロキシトリフェニルメタン類、4,4′-(フェニルメチレン)ビスフェノール、4,4′-(1-フェニル-エチリデン)ビス[2-メチルフェノール]、4,4′,4″-エチリデン-トリスフェノール等のポリヒドロキシトリフェニルエタン類、
3,3,3′,3′-テトラメチル-1,1′-スピロビ-インダン-5,6,5′,6′-テトロール、3,3,3′,3′-テトラメチル-1,1′-スピロビ-インダン-5,6,7,5′,6′,7′-ヘキソオール、3,3,3′,3′-テトラメチル-1,1′-スピロビ-インダン-4,5,6,4′,5′,6′-ヘキソオール、3,3,3′,3′-テトラメチル-1,1′-スピロビ-インダン-4,5,6,5′,6′,7′-ヘキソオール等のポリヒドロキシスピロビーインダン類、2,4,4-トリメチル-2′,4′,7′-トリヒドロキシフラバン、等のポリヒドロキシフラバン類、
3,3-ビス(3,4-ジヒドロキシフェニル)フタリド、3,3-ビス(2,3,4-トリヒドロキシフェニル)フタリド、3′,4′,5′,6′-テトラヒドロキシスピロ[フタリド-3,9′-キサンテン]等のポリヒドロキシフタリド類、モリン、ケルセチン、ルチン等のフラボノ色素類、
α,α′,α″-トリス(4-ヒドロキシフェニル)1,3,5-トリイソプロピルベンゼン、α,α′,α″-トリス(3,5-ジメチル-4-ヒドロキシフェニル)1,3,5-トリイソプロピルベンゼン、α,α′,α″-トリス(3,5-ジエチル-4-ヒドロキシフェニル)1,3,5-トリイソプロピルベンゼン、α,α′,α″-トリス(3,5-ジn-プロピル-4-ヒドロキシフェニル)1,3,5-トリイソプロピルベンゼン、α,α′,α″-トリス(3,5-ジイソプロピル-4-ヒドロキシフェニル)1,3,5-トリイソプロピルベンゼン、α,α′,α″-トリス(3,5-ジn-ブチル-4-ヒドロキシフェニル)1,3,5-トリイソプロピルベンゼン、α,α′,α″-トリス(3-メチル-4-ヒドロキシフェニル)1,3,5-トリイソプロピルベンゼン、α,α′,α″-トリス(3-メトキシ-4-ヒドロキシフェニル)1,3,5-トリイソプロピルベンゼン、α,α′,α″-トリス(2,4-ジヒドロキシフェニル)1,3,5-トリイソプロピルベンゼン、1,3,5-トリス(3,5-ジメチル-4-ヒドロキシフェニル)ベンゼン、1,3,5-トリス(5-メチル-2-ヒドロキシフェニル)ベンゼン、2,4,6-トリス(3,5-ジメチル-4-ヒドロキシフェニルチオメチル)メシチレン、1-[α-メチル-α-(4′-ヒドロキシフェニル)エチル]-4-[α,α’-ビス(4″-ヒドロキシフェニル)エチル]ベンゼン、1-[α-メチル(4′-ヒドロキシフェニル)エチル]-3-[α,α’-ビス(4″-ヒドロキシフェニル)エチル]ベンゼン、1-[α-メチル-α-(3′,5′-ジメチル-4′-ヒドロキシフェニル)エチル]-4-[α,α′-ビス(3″,5″-ジメチル-4″-ヒドロキシフェニル)エチル]ベンゼン、1-[α-メチル(3′-メチル-4′-ヒドロキシフェニル)エチル]-4-[α′,α′-ビス(3″-メチル-4″-ヒドロキシフェニル)エチル]ベンゼン、1-[α-メチル-α-(3′-メトキシ-4′-ヒドロキシフェニル)エチル]-4-[α′,α′-ビス(3″-メトキシ-4″-ヒドロキシフェニル)エチル]ベンゼン、1-[α-メチル-α-(2′,4′-ジヒドロキシフェニル)エチル]-4-[α′,α′-ビス(4″-ヒドロキシフェニル)エチル]ベンゼン、1-[α-メチル(2′,4′-ジヒドロキシフェニル)エチル]-3-[α′,α′-ビス(4″-ヒドロキシフェニル)エチル]ベンゼン等の特開平4-253058に記載のポリヒドロキシ化合物、α,α,α′,α′,α″,α″-ヘキサキス-(4-ヒドロキシフェニル)-1,3,5-トリエチルベンゼン等の特開平5-224410号に記載のポリヒドロキシ化合物、1,2,2,3-テトラ(p-ヒドロキシフェニル)プロパン、1,3,3,5-テトラ(p-ヒドロキシフェニル)ペンタン等の特開平5-303200号、EP-530148に記載のポリ(ヒドロキシフェニル)アルカン類、
p-ビス(2,3,4-トリヒドロキシベンゾイル)ベンゼン、p-ビス(2,4,6-トリヒドロキシベンゾイル)ベンゼン、m-ビス(2,3,4-トリヒドロキシベンゾイル)ベンゼン、m-ビス(2,4,6-トリヒドロキシベンゾイル)ベンゼン、p-ビス(2,5-ジヒドロキシ-3-ブロムベンゾイル)ベンゼン、p-ビス(2,3,4-トリヒドロキシ-5-メチルベンゾイル)ベンゼン、p-ビス(2,3,4-トリヒドロキシ-5-メトキシベンゾイル)ベンゼン、p-ビス(2,3,4-トリヒドロキシ-5-ニトロベンゾイル)ベンゼン、p-ビス(2,3,4-トリヒドロキシ-5-シアノベンゾイル)ベンゼン、1,3,5-トリス(2,5-ジヒドロキシベンゾイル)ベンゼン、1,3,5-トリス(2,3,4-トリヒドロキシベンゾイル)ベンゼン、1,2,3-トリス(2,3,4-トリヒドロキシベンゾイル)ベンゼン、1,2,4-トリス(2,3,4-トリヒドロキシベンゾイル)ベンゼン、1,2,4,5-テトラキス(2,3,4-トリヒドロキシベンゾイル)ベンゼン、α,α′-ビス(2,3,4-トリヒドロキシベンゾイル)p-キシレン、α,α′,α′-トリス(2,3,4-トリヒドロキシベンゾイル)メシレン、
2,6-ビス-(2-ヒドロキシ-3,5-ジメチルベンジル)-p-クレゾール、2,6-ビス-(2-ヒドロキシ-5′-メチルベンジル)-p-クレゾール、2,6-ビス-(2,4,6-トリヒドロキシベンジル)-p-クレゾール、2,6-ビス-(2,3,4-トリヒドロキシベンジル)-p-クレゾール、2,6-ビス(2,3,4-トリヒドロキシベンジル)-3,5-ジメチル-フェノール、4,6-ビス-(4-ヒドロキシ-3,5-ジメチルベンジル)-ピロガロール、2,6-ビス-(4-ヒドロキシ-3,5-ジメチルベンジル)-1,3,4-トリヒドロキシ-フェノール、4,6-ビス-(2,4,6-トリヒドロキシベンジル)-2,4-ジメチル-フェノール、4,6-ビス-(2,3,4-トリヒドロキシベンジル)-2,5-ジメチル-フェノール、2,6-ビス-(4-ヒドロキシベンジル)-p-クレゾール、2,6-ビス(4-ヒドロキシベンジル)-4-シクロヘキシルフェノール、2,6-ビス(4-ヒドロキシ-3-メチルベンジル)-p-クレゾール、2,6-ビス(4-ヒドロキシ-3,5-ジメチルベンジル)-p-クレゾール、2,6-ビス(4-ヒドロキシ-2,5-ジメチルベンジル)-p-クレゾール、2,6-ビス(4-ヒドロキシ-3-メチルベンジル)-4-フェニル-フェノール、2,2′,6,6′-テトラキス[(4-ヒドロキシフェニル)メチル]-4,4′-メチレンジフェノール、2,2′,6,6′-テトラキス[(4-ヒドロキシ-3,5-ジメチルフェニル)メチル]-4,4′-メチレンジフェノール、2,2′,6,6′-テトラキス[(4-ヒドロキシ-3-メチルフェニル)メチル]-4,4′-メチレンジフェノール、2,2′-ビス[(4-ヒドロキシ-3,5-ジメチルフェニル)メチル]6,6′-ジメチル-4,4′-メチレンジフェノール、2,2’,3,3’-テトラヒドロ-3,3,3’,3’-テトラメチル-1,1’-スピロビ(1H-インデン)-5,5’,6,6’,7,7’ヘキサノール、ビス(4-ヒドロキシ-3,5-ジメチルフェニル)-(4-ヒドロキシ-3-メトキシフェニル)メタン等を挙げることができる。
また、ノボラック樹脂等フェノール樹脂の低核体を用いる事もできる。
例えば、ヒドロキシ化合物とナフトキノンジアジドスルホニルクロリドの所定量をジオキサン、アセトン、又はテトラヒドロフラン等の溶媒中において、トリエチルアミン等の塩基性触媒の存在下で反応させてエステル化を行い、得られた生成物を水洗、乾燥することにより得ることができる。
上記エステル化率は、ヒドロキシ化合物が有するヒドロキシ基のうち、エステル化された基の割合として、1H-NMR等により確認することができる。
露光により吸光度が変化する色素としては、特に限定されず、例えば公知のフォトクロミック材料を使用することができるが、ロイコ色素、ジアリールエテン色素、フルギド類、スピロオキサジン類、スピロピラン類、あるいはアゾベンゼン系化合物等が挙げられる。
ロイコ色素としては、例えば、トリアリールメタン系(例えばトリフェニルメタン系)、スピロピラン系、フルオラン系、ジフェニルメタン系、ローダミンラクタム系、インドリルフタリド系、ロイコオーラミン系等のロイコ色素が挙げられる。これらの中でも、トリアリールメタン系のロイコ色素が好ましい。
ロイコ色素としては、市販品を使用してもよく、ビクトリアピュアブル-NAPS等が挙げられる。

本発明の第1の態様におけるネガ型感光性樹脂組成物の全固形分に対する化合物Aの含有量は、特に限定されないが、0.1~20質量%であることが好ましく、0.5~10質量%であることがより好ましく、1~5質量%であることが更に好ましい。
本発明の第2の態様におけるネガ型感光性樹脂組成物の全固形分に対するナフトキノンジアジド化合物の含有量は、特に限定されないが、0.1~20質量%であることが好ましく、0.5~10質量%であることがより好ましく、1~5質量%であることが更に好ましい。
本発明の樹脂組成物は、光重合開始剤を含む。
光重合開始剤は、光ラジカル重合開始剤であることが好ましい。光ラジカル重合開始剤としては、特に制限はなく、公知の光ラジカル重合開始剤の中から適宜選択することができる。例えば、紫外線領域から可視領域の光線に対して感光性を有する光ラジカル重合開始剤が好ましい。また、光励起された増感剤と何らかの作用を生じ、活性ラジカルを生成する活性剤であってもよい。



式中、RX1は、アルキル基、アルケニル基、アルコキシ基、アリール基、アリールオキシ基、複素環基、複素環オキシ基、アルキルスルファニル基、アリールスルファニル基、アルキルスルフィニル基、アリールスルフィニル基、アルキルスルホニル基、アリールスルホニル基、アシル基、アシルオキシ基、アミノ基、ホスフィノイル基、カルバモイル基またはスルファモイル基を表し、
RX2は、アルキル基、アルケニル基、アルコキシ基、アリール基、アリールオキシ基、複素環基、複素環オキシ基、アルキルスルファニル基、アリールスルファニル基、アルキルスルフィニル基、アリールスルフィニル基、アルキルスルホニル基、アリールスルホニル基、アシルオキシ基またはアミノ基を表し、
RX3~RX14は、それぞれ独立して水素原子または置換基を表す;
ただし、RX10~RX14のうち少なくとも一つは、電子求引性基である。


なお、光重合開始剤は熱重合開始剤としても機能する場合があるため、オーブンやホットプレート等の加熱によって光重合開始剤による架橋を更に進行させられる場合がある。
樹脂組成物は、増感剤を含んでいてもよい。増感剤は、特定の活性放射線を吸収して電子励起状態となる。電子励起状態となった増感剤は、熱ラジカル重合開始剤、光ラジカル重合開始剤などと接触して、電子移動、エネルギー移動、発熱などの作用が生じる。これにより、熱ラジカル重合開始剤、光ラジカル重合開始剤は化学変化を起こして分解し、ラジカル、酸又は塩基を生成する。
使用可能な増感剤として、ベンゾフェノン系、ミヒラーズケトン系、クマリン系、ピラゾールアゾ系、アニリノアゾ系、トリフェニルメタン系、アントラキノン系、アントラセン系、アントラピリドン系、ベンジリデン系、オキソノール系、ピラゾロトリアゾールアゾ系、ピリドンアゾ系、シアニン系、フェノチアジン系、ピロロピラゾールアゾメチン系、キサンテン系、フタロシアニン系、ベンゾピラン系、インジゴ系等の化合物を使用することができる。
増感剤としては、例えば、ミヒラーズケトン、4,4’-ビス(ジエチルアミノ)ベンゾフェノン、2,5-ビス(4’-ジエチルアミノベンザル)シクロペンタン、2,6-ビス(4’-ジエチルアミノベンザル)シクロヘキサノン、2,6-ビス(4’-ジエチルアミノベンザル)-4-メチルシクロヘキサノン、4,4’-ビス(ジメチルアミノ)カルコン、4,4’-ビス(ジエチルアミノ)カルコン、p-ジメチルアミノシンナミリデンインダノン、p-ジメチルアミノベンジリデンインダノン、2-(p-ジメチルアミノフェニルビフェニレン)-ベンゾチアゾール、2-(p-ジメチルアミノフェニルビニレン)ベンゾチアゾール、2-(p-ジメチルアミノフェニルビニレン)イソナフトチアゾール、1,3-ビス(4’-ジメチルアミノベンザル)アセトン、1,3-ビス(4’-ジエチルアミノベンザル)アセトン、3,3’-カルボニル-ビス(7-ジエチルアミノクマリン)、3-アセチル-7-ジメチルアミノクマリン、3-エトキシカルボニル-7-ジメチルアミノクマリン、3-ベンジロキシカルボニル-7-ジメチルアミノクマリン、3-メトキシカルボニル-7-ジエチルアミノクマリン、3-エトキシカルボニル-7-ジエチルアミノクマリン(7-(ジエチルアミノ)クマリン-3-カルボン酸エチル)、N-フェニル-N’-エチルエタノールアミン、N-フェニルジエタノールアミン、N-p-トリルジエタノールアミン、N-フェニルエタノールアミン、4-モルホリノベンゾフェノン、ジメチルアミノ安息香酸イソアミル、ジエチルアミノ安息香酸イソアミル、2-メルカプトベンズイミダゾール、1-フェニル-5-メルカプトテトラゾール、2-メルカプトベンゾチアゾール、2-(p-ジメチルアミノスチリル)ベンズオキサゾール、2-(p-ジメチルアミノスチリル)ベンゾチアゾール、2-(p-ジメチルアミノスチリル)ナフト(1,2-d)チアゾール、2-(p-ジメチルアミノベンゾイル)スチレン、ジフェニルアセトアミド、ベンズアニリド、N-メチルアセトアニリド、3‘,4’-ジメチルアセトアニリド等が挙げられる。
また、他の増感色素を用いてもよい。
増感色素の詳細については、特開2016-027357号公報の段落0161~0163の記載を参酌でき、この内容は本明細書に組み込まれる。
本発明の樹脂組成物は、連鎖移動剤を含有してもよい。連鎖移動剤は、例えば高分子辞典第三版(高分子学会編、2005年)683-684頁に定義されている。連鎖移動剤としては、例えば、分子内に-S-S-、-SO2-S-、-N-O-、SH、PH、SiH、及びGeHを有する化合物群、RAFT(Reversible Addition Fragmentation chain Transfer)重合に用いられるチオカルボニルチオ基を有するジチオベンゾアート、トリチオカルボナート、ジチオカルバマート、キサンタート化合物等が用いられる。これらは、低活性のラジカルに水素を供与して、ラジカルを生成するか、若しくは、酸化された後、脱プロトンすることによりラジカルを生成しうる。特に、チオール化合物を好ましく用いることができる。
本発明の樹脂組成物が条件iを満たす場合、本発明の樹脂組成物は、上記樹脂、上記化合物A及び上記光重合開始剤とは異なる成分として含窒素環状化合物を更に含む。
本発明の樹脂組成物が条件iiiを満たす場合、本発明の樹脂組成物は、上記樹脂、上記ナフトキノンジアジド化合物及び上記光重合開始とは異なる成分として含窒素環状化合物を更に含む。
本発明の樹脂組成物が条件ii又は条件ivを満たす場合、本発明の樹脂組成物は含窒素環状化合物を更に含んでもよい。
含窒素環状化合物は、窒素原子を環員として含む環構造を有する化合物である。上記環構造は、脂肪族環構造であっても芳香族環構造であってもよいが、芳香族環構造であることが好ましい。
また、含窒素環状化合物は、環員として窒素原子を1つ以上含む環構造を有する化合物であればよいが、環員として窒素原子を2つ以上含む環構造を有する化合物であることが好ましい。
上記窒素原子を環員として含む環構造としては、特に制限はないが、ピロール環、フラン環、チオフェン環、イミダゾール環、オキサゾール環、チアゾール環、ピラゾール環、イソオキサゾール環、イソチアゾール環、テトラゾール環、ピリジン環、ピリダジン環、ピリミジン環、ピラジン環、ピペリジン環、ピペラジン環、モルホリン環、2H-ピラン環及び6H-ピラン環、トリアジン環、イソシアヌレート環等が挙げられる。
これらの環構造は、他の環構造と複環を形成していてもよい。例えば、ベンゾイミダゾール構造、ベンゾトリアゾール構造等のように、ベンゼン環等の環構造と縮合環を形成する態様が挙げられる。
これらの中でも、1,2,4-トリアゾール、ベンゾトリアゾール、3-アミノ-1,2,4-トリアゾール、3,5-ジアミノ-1,2,4-トリアゾール等のトリアゾール構造、1H-テトラゾール、5-フェニルテトラゾール、5-アミノ-テトラゾール等のテトラゾール構造を有する化合物が好ましく使用できる。

本発明の第1の態様における樹脂組成物における、上記化合物Aの含有量を100質量部とした場合の上記含窒素環状化合物の含有量は、0.5~200質量部であることが好ましく、0.8~100質量部であることがより好ましく、0.1~40質量部であることが更に好ましい。
本発明の第2の態様における樹脂組成物における、上記ナフトキノンジアジド化合物の含有量を100質量部とした場合の上記含窒素環状化合物の含有量は、0.5~200質量部であることが好ましく、0.8~100質量部であることがより好ましく、0.1~40質量部であることが更に好ましい。
本発明の樹脂組成物は含窒素環状化合物を1種のみ含んでもよいし、2種以上含んでよい。含窒素環状化合物を2種以上含む場合は、その合計が上記範囲であることが好ましい。
本発明の樹脂組成物は、重合性化合物を含むことが好ましい。
重合性化合物としては、ラジカル架橋剤、又は、他の架橋剤が挙げられる。なお、以下に例示した重合性化合物の中には窒素を含む環状化合物も存在するが、これらは上述の「含窒素環状化合物」には含まれないものとする。
本発明の樹脂組成物は、ラジカル架橋剤を含むことが好ましい。
ラジカル架橋剤は、ラジカル重合性基を有する化合物である。ラジカル重合性基としては、エチレン性不飽和結合を含む基が好ましい。上記エチレン性不飽和結合を含む基としては、ビニル基、アリル基、ビニルフェニル基、(メタ)アクリロイル基、マレイミド基、(メタ)アクリルアミド基などのエチレン性不飽和結合を有する基が挙げられる。
これらの中でも、上記エチレン性不飽和結合を含む基としては、(メタ)アクリロイル基、(メタ)アクリルアミド基、ビニルフェニル基が好ましく、反応性の観点からは、(メタ)アクリロイル基がより好ましい。
上記エチレン性不飽和結合を2個以上有する化合物としては、エチレン性不飽和結合を2~15個有する化合物が好ましく、エチレン性不飽和結合を2~10個有する化合物がより好ましく、2~6個有する化合物が更に好ましい。
また、得られるパターン(硬化物)の膜強度の観点からは、本発明の樹脂組成物は、エチレン性不飽和結合を2個有する化合物と、上記エチレン性不飽和結合を3個以上有する化合物とを含むことも好ましい。
具体的な化合物としては、トリエチレングリコールジアクリレート、トリエチレングリコールジメタクリレート、テトラエチレングリコールジメタクリレート、テトラエチレングリコールジアクリレート、PEG(ポリエチレングリコール)200ジアクリレート、PEG200ジメタクリレート、PEG600ジアクリレート、PEG600ジメタクリレート、ポリテトラエチレングリコールジアクリレート、ポリテトラエチレングリコールジメタクリレート、ネオペンチルグリコールジアクリレート、ネオペンチルグリコールジメタクリレート、3-メチル-1,5-ペンタンジオールジアクリレート、1,6-ヘキサンジオールジアクリレート、1,6ヘキサンジオールジメタクリレート、ジメチロール-トリシクロデカンジアクリレート、ジメチロール-トリシクロデカンジメタクリレート、ビスフェノールAのEO(エチレンオキシド)付加物ジアクリレート、ビスフェノールAのEO付加物ジメタクリレート、ビスフェノールAのPO(プロピレンオキシド)付加物ジアクリレート、ビスフェノールAのPO付加物ジメタクリレート、2-ヒドロキシー3-アクリロイロキシプロピルメタクリレート、イソシアヌル酸EO変性ジアクリレート、イソシアヌル酸変性ジメタクリレート、その他ウレタン結合を有する2官能アクリレート、ウレタン結合を有する2官能メタクリレートを使用することができる。これらは必要に応じ、2種以上を混合し使用することができる。
なお、例えばPEG200ジアクリレートとは、ポリエチレングリコールジアクリレートであって、ポリエチレングリコール鎖の式量が200程度のものをいう。
本発明の樹脂組成物は、パターン(硬化物)の弾性率制御に伴う反り抑制の観点から、ラジカル架橋剤として、単官能ラジカル架橋剤を好ましく用いることができる。単官能ラジカル架橋剤としては、n-ブチル(メタ)アクリレート、2-エチルヘキシル(メタ)アクリレート、2-ヒドロキシエチル(メタ)アクリレート、ブトキシエチル(メタ)アクリレート、カルビトール(メタ)アクリレート、シクロヘキシル(メタ)アクリレート、ベンジル(メタ)アクリレート、フェノキシエチル(メタ)アクリレート、N-メチロール(メタ)アクリルアミド、グリシジル(メタ)アクリレート、ポリエチレングリコールモノ(メタ)アクリレート、ポリプロピレングリコールモノ(メタ)アクリレート等の(メタ)アクリル酸誘導体、N-ビニルピロリドン、N-ビニルカプロラクタム等のN-ビニル化合物類、アリルグリシジルエーテル等が好ましく用いられる。単官能ラジカル架橋剤としては、露光前の揮発を抑制するため、常圧下で100℃以上の沸点を持つ化合物も好ましい。
その他、2官能以上のラジカル架橋剤としては、ジアリルフタレート、トリアリルトリメリテート等のアリル化合物類が挙げられる。
本発明の樹脂組成物は、上述したラジカル架橋剤とは異なる、他の架橋剤を含むことも好ましい。
本発明において、他の架橋剤とは、上述したラジカル架橋剤以外の架橋剤をいい、光酸発生剤、又は、光塩基発生剤の感光により、組成物中の他の化合物又はその反応生成物との間で共有結合を形成する反応が促進される基を分子内に複数個有する化合物であることが好ましく、組成物中の他の化合物又はその反応生成物との間で共有結合を形成する反応が酸又は塩基の作用によって促進される基を分子内に複数個有する化合物が好ましい。
上記酸又は塩基は、露光工程において、光酸発生剤又は光塩基発生剤から発生する酸又は塩基であることが好ましい。
他の架橋剤としては、アシルオキシメチル基、メチロール基及びアルコキシメチル基よりなる群から選ばれた少なくとも1種の基を有する化合物が好ましく、アシルオキシメチル基、メチロール基及びアルコキシメチル基よりなる群から選ばれた少なくとも1種の基が窒素原子に直接結合した構造を有する化合物がより好ましい。
他の架橋剤としては、例えば、メラミン、グリコールウリル、尿素、アルキレン尿素、ベンゾグアナミンなどのアミノ基含有化合物にホルムアルデヒド又はホルムアルデヒドとアルコールを反応させ、上記アミノ基の水素原子をアシルオキシメチル基、メチロール基又はアルコキシメチル基で置換した構造を有する化合物が挙げられる。これらの化合物の製造方法は特に限定されず、上記方法により製造された化合物と同様の構造を有する化合物であればよい。また、これらの化合物のメチロール基同士が自己縮合してなるオリゴマーであってもよい。
上記のアミノ基含有化合物として、メラミンを用いた架橋剤をメラミン系架橋剤、グリコールウリル、尿素又はアルキレン尿素を用いた架橋剤を尿素系架橋剤、アルキレン尿素を用いた架橋剤をアルキレン尿素系架橋剤、ベンゾグアナミンを用いた架橋剤をベンゾグアナミン系架橋剤という。
これらの中でも、本発明の樹脂組成物は、尿素系架橋剤及びメラミン系架橋剤よりなる群から選ばれた少なくとも1種の化合物を含むことが好ましく、後述するグリコールウリル系架橋剤及びメラミン系架橋剤よりなる群から選ばれた少なくとも1種の化合物を含むことがより好ましい。
上記化合物が有するアルコキシメチル基又はアシルオキシメチル基は、炭素数2~5が好ましく、炭素数2又は3が好ましく、炭素数2がより好ましい。
上記化合物が有するアルコキシメチル基及びアシルオキシメチル基の総数は1~10が好ましく、より好ましくは2~8、特に好ましくは3~6である。
上記化合物の分子量は好ましくは1500以下であり、180~1200が好ましい。

R101及びR102は、それぞれ独立に、一価の有機基を表し、互いに結合して環を形成してもよい。
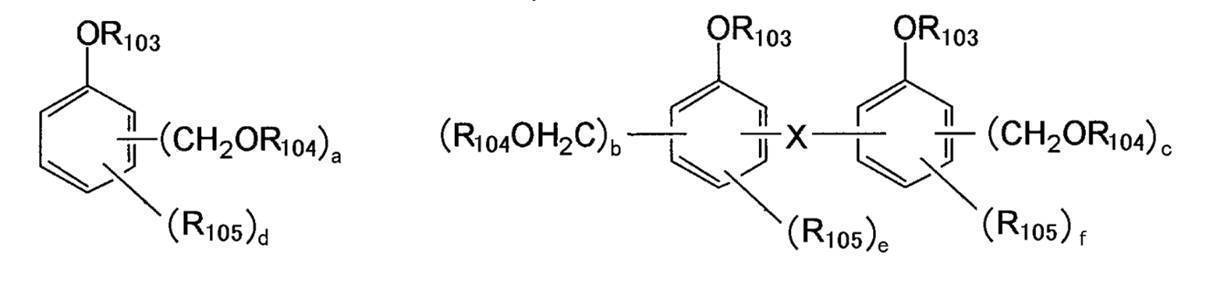
R105は各々独立にアルキル基又はアルケニル基を示し、a、b及びcは各々独立に1~3であり、dは0~4であり、eは0~3であり、fは0~3であり、a+dは5以下であり、b+eは4以下であり、c+fは4以下である。
酸の作用により分解し、アルカリ可溶性基を生じる基、酸の作用により脱離する基、-C(R4)2COOR5で表される基におけるR5については、例えば、-C(R36)(R37)(R38)、-C(R36)(R37)(OR39)、-C(R01)(R02)(OR39)等を挙げることができる。
式中、R36~R39は、各々独立に、アルキル基、シクロアルキル基、アリール基、アラルキル基又はアルケニル基を表す。R36とR37とは、互いに結合して環を形成してもよい。
上記アルキル基としては、炭素数1~10のアルキル基が好ましく、炭素数1~5のアルキル基がより好ましい。
上記アルキル基は、直鎖状、分岐鎖状のいずれであってもよい。
上記シクロアルキル基としては、炭素数3~12のシクロアルキル基が好ましく、炭素数3~8のシクロアルキル基がより好ましい。
上記シクロアルキル基は単環構造であってもよいし、縮合環等の多環構造であってもよい。
上記アリール基は炭素数6~30の芳香族炭化水素基であることが好ましく、フェニル基であることがより好ましい。
上記アラルキル基としては、炭素数7~20のアラルキル基が好ましく、炭素数7~16のアラルキル基がより好ましい。
上記アラルキル基はアルキル基により置換されたアリール基を意図しており、これらのアルキル基及びアリール基の好ましい態様は、上述のアルキル基及びアリール基の好ましい態様と同様である。
上記アルケニル基は炭素数3~20のアルケニル基が好ましく、炭素数3~16のアルケニル基がより好ましい。
また、これらの基は本発明の効果が得られる範囲内で、公知の置換基を更に有していてもよい。

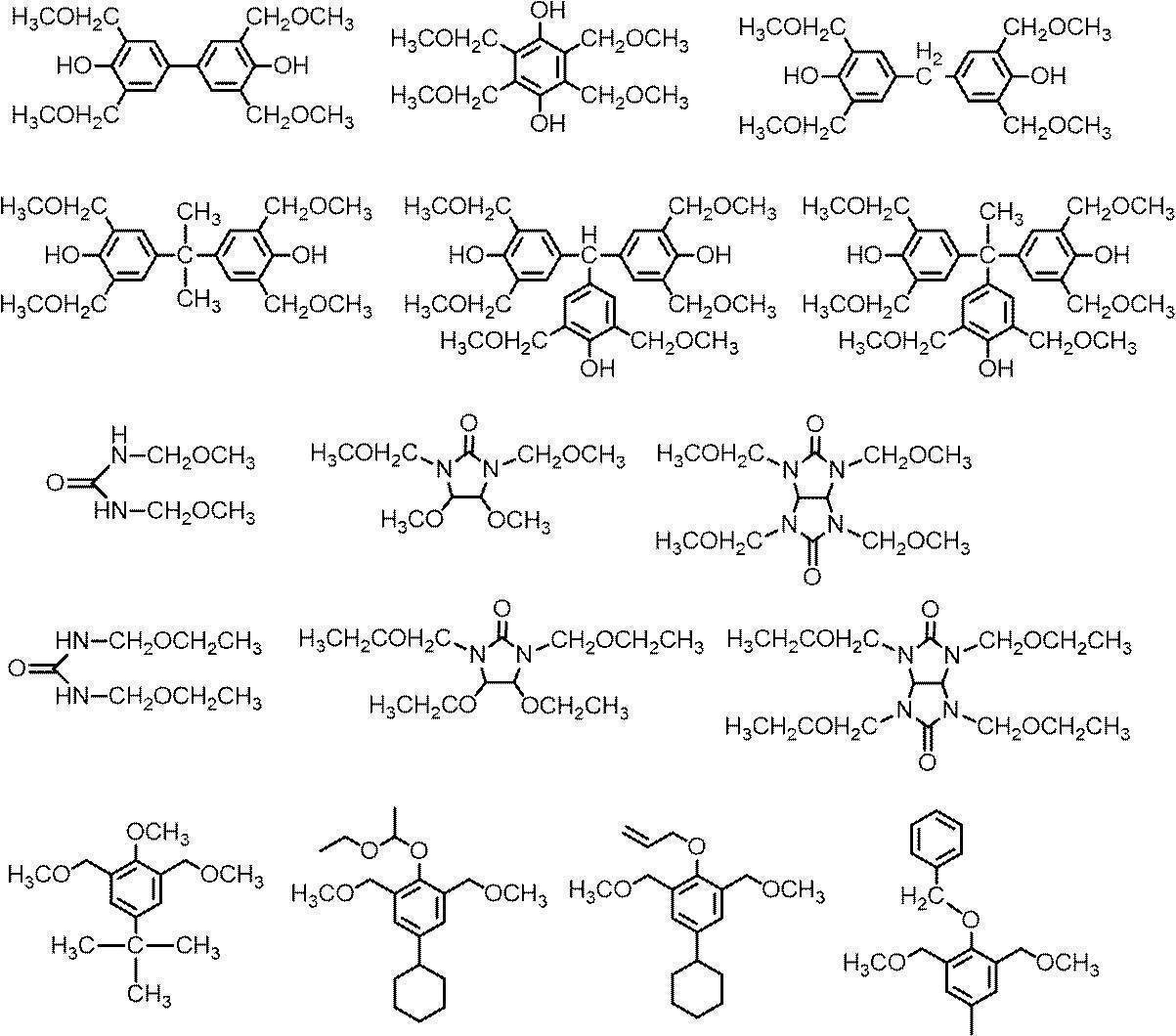
耐熱性の観点で、アルコキシメチル基又はアシルオキシメチル基が、直接芳香環やトリアジン環上に置換した化合物が好ましい。
ビスメトキシメチル尿素、ビスエトキシメチル尿素、ビスプロポキシメチル尿素、ビスブトキシメチル尿素等の尿素系架橋剤、
モノヒドロキシメチル化エチレン尿素又はジヒドロキシメチル化エチレン尿素、モノメトキシメチル化エチレン尿素、ジメトキシメチル化エチレン尿素、モノエトキシメチル化エチレン尿素、ジエトキシメチル化エチレン尿素、モノプロポキシメチル化エチレン尿素、ジプロポキシメチル化エチレン尿素、モノブトキシメチル化エチレン尿素、又は、ジブトキシメチル化エチレン尿素などのエチレン尿素系架橋剤、
モノヒドロキシメチル化プロピレン尿素、ジヒドロキシメチル化プロピレン尿素、モノメトキシメチル化プロピレン尿素、ジメトキシメチル化プロピレン尿素、モノジエトキシメチル化プロピレン尿素、ジエトキシメチル化プロピレン尿素、モノプロポキシメチル化プロピレン尿素、ジプロポキシメチル化プロピレン尿素、モノブトキシメチル化プロピレン尿素、又は、ジブトキシメチル化プロピレン尿素などのプロピレン尿素系架橋剤、
1,3-ジ(メトキシメチル)4,5-ジヒドロキシ-2-イミダゾリジノン、1,3-ジ(メトキシメチル)-4,5-ジメトキシ-2-イミダゾリジノンなどが挙げられる。
このような化合物の具体例としては、ベンゼンジメタノール、ビス(ヒドロキシメチル)クレゾール、ビス(ヒドロキシメチル)ジメトキシベンゼン、ビス(ヒドロキシメチル)ジフェニルエーテル、ビス(ヒドロキシメチル)ベンゾフェノン、ヒドロキシメチル安息香酸ヒドロキシメチルフェニル、ビス(ヒドロキシメチル)ビフェニル、ジメチルビス(ヒドロキシメチル)ビフェニル、ビス(メトキシメチル)ベンゼン、ビス(メトキシメチル)クレゾール、ビス(メトキシメチル)ジメトキシベンゼン、ビス(メトキシメチル)ジフェニルエーテル、ビス(メトキシメチル)ベンゾフェノン、メトキシメチル安息香酸メトキシメチルフェニル、ビス(メトキシメチル)ビフェニル、ジメチルビス(メトキシメチル)ビフェニル、4,4’,4’’-エチリデントリス[2,6-ビス(メトキシメチル)フェノール]、5,5’-[2,2,2‐トリフルオロ‐1‐(トリフルオロメチル)エチリデン]ビス[2‐ヒドロキシ‐1,3‐ベンゼンジメタノール]、3,3’,5,5’-テトラキス(メトキシメチル)-1,1’-ビフェニル-4,4’-ジオール等が挙げられる。
エポキシ化合物としては、一分子中にエポキシ基を2以上有する化合物であることが好ましい。エポキシ基は、200℃以下で架橋反応し、かつ、架橋に由来する脱水反応が起こらないため膜収縮が起きにくい。このため、エポキシ化合物を含有することは、本発明の樹脂組成物の低温硬化及び反りの抑制に効果的である。

オキセタン化合物としては、一分子中にオキセタン環を2つ以上有する化合物、3-エチル-3-ヒドロキシメチルオキセタン、1,4-ビス{[(3-エチル-3-オキセタニル)メトキシ]メチル}ベンゼン、3-エチル-3-(2-エチルヘキシルメチル)オキセタン、1,4-ベンゼンジカルボン酸-ビス[(3-エチル-3-オキセタニル)メチル]エステル等を挙げることができる。具体的な例としては、東亞合成(株)製のアロンオキセタンシリーズ(例えば、OXT-121、OXT-221)が好適に使用することができ、これらは単独で、又は2種以上混合してもよい。
ベンゾオキサジン化合物は、開環付加反応に由来する架橋反応のため、硬化時に脱ガスが発生せず、更に熱収縮を小さくして反りの発生が抑えられることから好ましい。
本発明の樹脂組成物は、塩基発生剤を含んでもよい。ここで、塩基発生剤とは、物理的または化学的な作用によって塩基を発生することができる化合物である。本発明の樹脂組成物にとって好ましい塩基発生剤としては、熱塩基発生剤および光塩基発生剤が挙げられる。
特に、樹脂組成物が環化樹脂の前駆体を含む場合、樹脂組成物は塩基発生剤を含むことが好ましい。樹脂組成物が熱塩基発生剤を含有することによって、例えば加熱により前駆体の環化反応を促進でき、硬化物の機械特性や耐薬品性が良好なものとなり、例えば半導体パッケージ中に含まれる再配線層用層間絶縁膜としての性能が良好となる。
塩基発生剤としては、イオン型塩基発生剤でもよく、非イオン型塩基発生剤でもよい。塩基発生剤から発生する塩基としては、例えば、2級アミン、3級アミンが挙げられる。
本発明に係る塩基発生剤について特に制限はなく、公知の塩基発生剤を用いることができる。公知の塩基発生剤としては、例えば、カルバモイルオキシム化合物、カルバモイルヒドロキシルアミン化合物、カルバミン酸化合物、ホルムアミド化合物、アセトアミド化合物、カルバメート化合物、ベンジルカルバメート化合物、ニトロベンジルカルバメート化合物、スルホンアミド化合物、イミダゾール誘導体化合物、アミンイミド化合物、ピリジン誘導体化合物、α-アミノアセトフェノン誘導体化合物、4級アンモニウム塩誘導体化合物、ピリジニウム塩、α-ラクトン環誘導体化合物、アミンイミド化合物、フタルイミド誘導体化合物、アシルオキシイミノ化合物、などを用いることができる。
非イオン型塩基発生剤の具体的な化合物としては、式(B1)、式(B2)、又は式(B3)で表される化合物が挙げられる。
なお、以下に例示した塩基発生剤の中には窒素を含む環状化合物も存在するが、これらは上述の「含窒素環状化合物」には含まれないものとする。
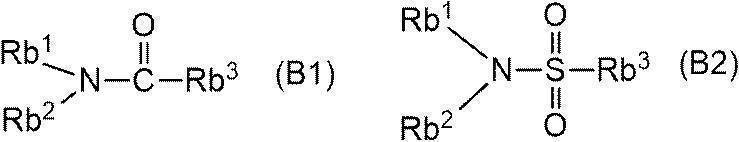
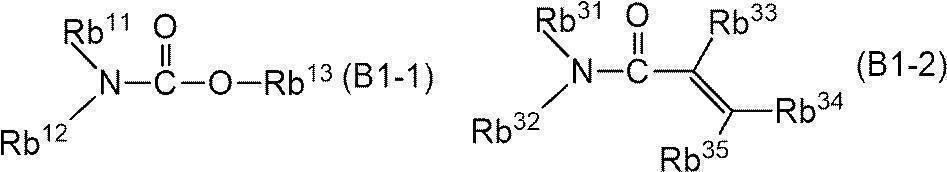
Rb13はアルキル基(炭素数1~24が好ましく、2~18がより好ましく、3~12が更に好ましい)、アルケニル基(炭素数2~24が好ましく、2~18がより好ましく、3~12が更に好ましい)、アリール基(炭素数6~22が好ましく、6~18がより好ましく、6~12が更に好ましい)、アリールアルキル基(炭素数7~23が好ましく、7~19がより好ましく、7~12が更に好ましい)であり、本発明の効果を奏する範囲で置換基を有していてもよい。中でも、Rb13はアリールアルキル基が好ましい。

Rb15及びRb16は水素原子、アルキル基(炭素数1~12が好ましく、1~6がより好ましく、1~3が更に好ましい)、アルケニル基(炭素数2~12が好ましく、2~6がより好ましく、2~3が更に好ましい)、アリール基(炭素数6~22が好ましく、6~18がより好ましく、6~10が更に好ましい)、アリールアルキル基(炭素数7~23が好ましく、7~19がより好ましく、7~11が更に好ましい)であり、水素原子又はメチル基が好ましい。
Rb17はアルキル基(炭素数1~24が好ましく、1~12がより好ましく、3~8が更に好ましい)、アルケニル基(炭素数2~12が好ましく、2~10がより好ましく、3~8が更に好ましい)、アリール基(炭素数6~22が好ましく、6~18がより好ましく、6~12が更に好ましい)、アリールアルキル基(炭素数7~23が好ましく、7~19がより好ましく、7~12が更に好ましい)であり、中でもアリール基が好ましい。

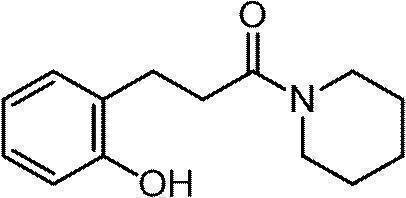

環状アルキル基は、炭素数3~12のものが好ましく、3~6がより好ましい。環状アルキル基は、例えば、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、シクロオクチル基等が挙げられる。
鎖状アルキル基と環状アルキル基の組合せに係る基は、炭素数4~24のものが好ましく、4~18がより好ましく、4~12がさらに好ましい。鎖状アルキル基と環状アルキル基の組合せに係る基は、例えば、シクロヘキシルメチル基、シクロヘキシルエチル基、シクロヘキシルプロピル基、メチルシクロヘキシルメチル基、エチルシクロヘキシルエチル基等が挙げられる。
酸素原子を鎖中に有するアルキル基は、炭素数2~12のものが好ましく、2~6がより好ましく、2~4がさらに好ましい。酸素原子を鎖中に有するアルキル基は、鎖状でも環状でもよく、直鎖でも分岐でもよい。
なかでも、後述する分解生成塩基の沸点を高める観点で、RN1およびRN2は炭素数5~12のアルキル基が好ましい。ただし、金属(例えば銅)の層と積層する際の密着性を重視する処方においては、環状のアルキル基を有する基や炭素数1~8のアルキル基であることが好ましい。
2価の炭化水素連結基は、炭素数1~24のものが好ましく、2~12がより好ましく、2~6がさらに好ましい。2価の脂肪族炭化水素基は、炭素数1~12のものが好ましく、2~6がより好ましく、2~4がさらに好ましい。2価の芳香族炭化水素基は、炭素数6~22のものが好ましく、6~18がより好ましく、6~10がさらに好ましい。2価の脂肪族炭化水素基と2価の芳香族炭化水素基の組み合わせに係る基(例えば、アリーレンアルキル基)は、炭素数7~22のものが好ましく、7~18がより好ましく、7~10がさらに好ましい。
直鎖または分岐の鎖状アルキレン基は、炭素数1~12のものが好ましく、2~6がより好ましく、2~4がさらに好ましい。
環状アルキレン基は、炭素数3~12のものが好ましく、3~6がより好ましい。
鎖状アルキレン基と環状アルキレン基の組み合わせに係る基は、炭素数4~24のものが好ましく、4~12がより好ましく、4~6がさらに好ましい。
酸素原子を鎖中に有するアルキレン基は、鎖状でも環状でもよく、直鎖でも分岐でもよい。酸素原子を鎖中に有するアルキレン基は、炭素数1~12のものが好ましく、1~6がより好ましく、1~3がさらに好ましい。
環状のアルケニレン基は、炭素数3~12のものが好ましく、3~6がより好ましい。環状のアルケニレン基は、C=C結合の数は1~6が好ましく、1~4がより好ましく、1~2がさらに好ましい。
アリーレン基は、炭素数6~22のものが好ましく、6~18がより好ましく、6~10がさらに好ましい。
アリーレンアルキレン基は、炭素数7~23のものが好ましく、7~19がより好ましく、7~11がさらに好ましい。
中でも、鎖状アルキレン基、環状アルキレン基、酸素原子を鎖中に有するアルキレン基、鎖状のアルケニレン基、アリーレン基、アリーレンアルキレン基が好ましく、1,2-エチレン基、プロパンジイル基(特に1,3-プロパンジイル基)、シクロヘキサンジイル基(特に1,2-シクロヘキサンジイル基)、ビニレン基(特にシスビニレン基)、フェニレン基(1,2-フェニレン基)、フェニレンメチレン基(特に1,2-フェニレンメチレン基)、エチレンオキシエチレン基(特に1,2-エチレンオキシ-1,2-エチレン基)がより好ましい。



塩基発生剤は、1種又は2種以上を用いることができる。2種以上を用いる場合は、合計量が上記範囲であることが好ましい。
本発明の樹脂組成物は、溶剤を含むことが好ましい。
溶剤は、公知の溶剤を任意に使用できる。溶剤は有機溶剤が好ましい。有機溶剤としては、エステル類、エーテル類、ケトン類、環状炭化水素類、スルホキシド類、アミド類、ウレア類、アルコール類などの化合物が挙げられる。
本発明の樹脂組成物は、電極や配線などに用いられる金属材料との接着性を向上させるための金属接着性改良剤を含んでいることが好ましい。金属接着性改良剤としては、アルコキシシリル基を有するシランカップリング剤、アルミニウム系接着助剤、チタン系接着助剤、スルホンアミド構造を有する化合物及びチオウレア構造を有する化合物、リン酸誘導体化合物、βケトエステル化合物、アミノ化合物等が挙げられる。
これらの中でも、本発明の樹脂組成物は、シランカップリング剤を含むことが好ましい。
なお、以下に例示した金属密着性改良剤の中には窒素を含む環状化合物も存在するが、これらは上述の「含窒素環状化合物」には含まれないものとする。
シランカップリング剤としては、例えば、国際公開第2015/199219号の段落0167に記載の化合物、特開2014-191002号公報の段落0062~0073に記載の化合物、国際公開第2011/080992号の段落0063~0071に記載の化合物、特開2014-191252号公報の段落0060~0061に記載の化合物、特開2014-041264号公報の段落0045~0052に記載の化合物、国際公開第2014/097594号の段落0055に記載の化合物、特開2018-173573の段落0067~0078に記載の化合物が挙げられ、これらの内容は本明細書に組み込まれる。また、特開2011-128358号公報の段落0050~0058に記載のように異なる2種以上のシランカップリング剤を用いることも好ましい。また、シランカップリング剤は、下記化合物を用いることも好ましい。以下の式中、Meはメチル基を、Etはエチル基を表す。

アルミニウム系接着助剤としては、例えば、アルミニウムトリス(エチルアセトアセテート)、アルミニウムトリス(アセチルアセトネート)、エチルアセトアセテートアルミニウムジイソプロピレート等を挙げることができる。
本発明の樹脂組成物は、マイグレーション抑制剤を更に含むことが好ましい。マイグレーション抑制剤を含むことにより、金属層(金属配線)由来の金属イオンが膜内へ移動することを効果的に抑制可能となる。
ただし、これらのうち、上述の含窒素環状化合物に含まれる化合物は、マイグレーション抑制剤には含まれないものとする。
本発明の樹脂組成物は、重合禁止剤を含むことが好ましい。重合禁止剤としてはフェノール系化合物、キノン系化合物、アミノ系化合物、N-オキシルフリーラジカル化合物系化合物、ニトロ系化合物、ニトロソ系化合物、ヘテロ芳香環系化合物、金属化合物などが挙げられる。
本発明の樹脂組成物は、本発明の効果が得られる範囲で、必要に応じて、各種の添加物、例えば、界面活性剤、高級脂肪酸誘導体、熱重合開始剤、無機粒子、紫外線吸収剤、有機チタン化合物、酸化防止剤、凝集防止剤、フェノール系化合物、他の高分子化合物、可塑剤及びその他の助剤類(例えば、消泡剤、難燃剤など)等を配合することができる。これらの成分を適宜含有させることにより、膜物性などの性質を調整することができる。これらの成分は、例えば、特開2012-003225号公報の段落番号0183以降(対応する米国特許出願公開第2013/0034812号明細書の段落番号0237)の記載、特開2008-250074号公報の段落番号0101~0104、0107~0109等の記載を参酌でき、これらの内容は本明細書に組み込まれる。これらの添加剤を配合する場合、その合計配合量は本発明の樹脂組成物の固形分の3質量%以下とすることが好ましい。
界面活性剤としては、フッ素系界面活性剤、シリコーン系界面活性剤、炭化水素系界面活性剤などの各種界面活性剤を使用できる。界面活性剤はノニオン型界面活性剤であってもよく、カチオン型界面活性剤であってもよく、アニオン型界面活性剤であってもよい。
フッ素系界面活性剤は、フッ素原子を有する(メタ)アクリレート化合物に由来する繰り返し単位と、アルキレンオキシ基(好ましくはエチレンオキシ基、プロピレンオキシ基)を2以上(好ましくは5以上)有する(メタ)アクリレート化合物に由来する繰り返し単位と、を含む含フッ素高分子化合物も好ましく用いることができ、下記化合物も本発明で用いられるフッ素系界面活性剤として例示される。

フッ素系界面活性剤は、エチレン性不飽和基を側鎖に有する含フッ素重合体をフッ素系界面活性剤として用いることもできる。具体例としては、特開2010-164965号公報の段落0050~0090および段落0289~0295に記載された化合物が挙げられ、この内容は本明細書に組み込まれる。また、市販品としては、例えばDIC(株)製のメガファックRS-101、RS-102、RS-718K等が挙げられる。
界面活性剤の含有量は、組成物の全固形分に対して、0.001~2.0質量%が好ましく、0.005~1.0質量%がより好ましい。
本発明の樹脂組成物は、酸素に起因する重合阻害を防止するために、ベヘン酸やベヘン酸アミドのような高級脂肪酸誘導体を添加して、塗布後の乾燥の過程で本発明の樹脂組成物の表面に偏在させてもよい。
本発明の樹脂組成物は、熱重合開始剤を含んでもよく、特に熱ラジカル重合開始剤を含んでもよい。熱ラジカル重合開始剤は、熱のエネルギーによってラジカルを発生し、重合性を有する化合物の重合反応を開始又は促進させる化合物である。熱ラジカル重合開始剤を添加することによって樹脂及び重合性化合物の重合反応を進行させることもできるので、より耐溶剤性を向上できる。また、上述した光重合開始剤も熱により重合を開始する機能を有する場合があり、熱重合開始剤として添加することができる場合がある。
本発明の樹脂組成物は、無機微粒子を含んでもよい。無機粒子として、具体的には、炭酸カルシウム、リン酸カルシウム、シリカ、カオリン、タルク、二酸化チタン、アルミナ、硫酸バリウム、フッ化カルシウム、フッ化リチウム、ゼオライト、硫化モリブデン、ガラス等を含むことができる。
微粒子の上記平均粒子径は、一次粒子径であり、また体積平均粒子径である。体積平均粒子径は、Nanotrac WAVE II EX-150(日機装社製)による動的光散乱法で測定できる。
上記測定が困難である場合は、遠心沈降光透過法、X線透過法、レーザー回折・散乱法で測定することもできる。
本発明の組成物は、紫外線吸収剤を含んでいてもよい。紫外線吸収剤としては、サリシレート系、ベンゾフェノン系、ベンゾトリアゾール系、置換アクリロニトリル系、トリアジン系などの紫外線吸収剤を使用することができる。
なお、以下に例示した紫外線吸収剤の中には窒素を含む環状化合物も存在するが、これらは上述の「含窒素環状化合物」には含まれないものとする。
サリシレート系紫外線吸収剤の例としては、フェニルサリシレート、p-オクチルフェニルサリシレート、p-t-ブチルフェニルサリシレートなどが挙げられ、ベンゾフェノン系紫外線吸収剤の例としては、2,2’-ジヒドロキシ-4-メトキシベンゾフェノン、2,2’-ジヒドロキシ-4,4’-ジメトキシベンゾフェノン、2,2’,4,4’-テトラヒドロキシベンゾフェノン、2-ヒドロキシ-4-メトキシベンゾフェノン、2,4-ジヒドロキシベンゾフェノン、2-ヒドロキシ-4-オクトキシベンゾフェノンなどが挙げられる。また、ベンゾトリアゾール系紫外線吸収剤の例としては、2-(2’-ヒドロキシ-3’,5’-ジ-tert-ブチルフェニル)-5-クロロベンゾトリアゾール、2-(2’-ヒドロキシ-3’-tert-ブチル-5’-メチルフェニル)-5-クロロベンゾトリアゾール、2-(2’-ヒドロキシ-3’-tert-アミル-5’-イソブチルフェニル)-5-クロロベンゾトリアゾール、2-(2’-ヒドロキシ-3’-イソブチル-5’-メチルフェニル)-5-クロロベンゾトリアゾール、2-(2’-ヒドロキシ-3’-イソブチル-5’-プロピルフェニル)-5-クロロベンゾトリアゾール、2-(2’-ヒドロキシ-3’,5’-ジ-tert-ブチルフェニル)ベンゾトリアゾール、2-(2’-ヒドロキシ-5’-メチルフェニル)ベンゾトリアゾール、2-[2’-ヒドロキシ-5’-(1,1,3,3-テトラメチル)フェニル]ベンゾトリアゾールなどが挙げられる。
本発明の組成物は、紫外線吸収剤を含んでも含まなくてもよいが、含む場合、紫外線吸収剤の含有量は、本発明の組成物の全固形分質量に対して、0.001質量%以上1質量%以下であることが好ましく、0.01質量%以上0.1質量%以下であることがより好ましい。
本実施形態の樹脂組成物は、有機チタン化合物を含有してもよい。樹脂組成物が有機チタン化合物を含有することにより、低温で硬化した場合であっても耐薬品性に優れる樹脂層を形成できる。
有機チタン化合物の具体例を、以下のI)~VII)に示す:
I)チタンキレート化合物:中でも、樹脂組成物の保存安定性がよく、良好な硬化パターンが得られることから、アルコキシ基を2個以上有するチタンキレート化合物がより好ましい。具体的な例は、チタニウムビス(トリエタノールアミン)ジイソプロポキサイド、チタニウムジ(n-ブトキサイド)ビス(2,4-ペンタンジオネート)、チタニウムジイソプロポキサイドビス(2,4-ペンタンジオネート)、チタニウムジイソプロポキサイドビス(テトラメチルヘプタンジオネート)、チタニウムジイソプロポキサイドビス(エチルアセトアセテート)等である。
II)テトラアルコキシチタン化合物:例えば、チタニウムテトラ(n-ブトキサイド)、チタニウムテトラエトキサイド、チタニウムテトラ(2-エチルヘキソキサイド)、チタニウムテトライソブトキサイド、チタニウムテトライソプロポキサイド、チタニウムテトラメトキサイド、チタニウムテトラメトキシプロポキサイド、チタニウムテトラメチルフェノキサイド、チタニウムテトラ(n-ノニロキサイド)、チタニウムテトラ(n-プロポキサイド)、チタニウムテトラステアリロキサイド、チタニウムテトラキス[ビス{2,2-(アリロキシメチル)ブトキサイド}]等である。
III)チタノセン化合物:例えば、ペンタメチルシクロペンタジエニルチタニウムトリメトキサイド、ビス(η5-2,4-シクロペンタジエン-1-イル)ビス(2,6-ジフルオロフェニル)チタニウム、ビス(η5-2,4-シクロペンタジエン-1-イル)ビス(2,6-ジフルオロ-3-(1H-ピロール-1-イル)フェニル)チタニウム等である。
IV)モノアルコキシチタン化合物:例えば、チタニウムトリス(ジオクチルホスフェート)イソプロポキサイド、チタニウムトリス(ドデシルベンゼンスルホネート)イソプロポキサイド等である。
V)チタニウムオキサイド化合物:例えば、チタニウムオキサイドビス(ペンタンジオネート)、チタニウムオキサイドビス(テトラメチルヘプタンジオネート)、フタロシアニンチタニウムオキサイド等である。
VI)チタニウムテトラアセチルアセトネート化合物:例えば、チタニウムテトラアセチルアセトネート等である。
VII)チタネートカップリング剤:例えば、イソプロピルトリドデシルベンゼンスルホニルチタネート等である。
本発明の組成物は、酸化防止剤を含んでいてもよい。添加剤として酸化防止剤を含有することで、硬化後の膜の伸度特性や、金属材料との密着性を向上させることができる。酸化防止剤としては、フェノール化合物、亜リン酸エステル化合物、チオエーテル化合物などが挙げられる。フェノール化合物としては、フェノール系酸化防止剤として知られる任意のフェノール化合物を使用することができる。好ましいフェノール化合物としては、ヒンダードフェノール化合物が挙げられる。フェノール性ヒドロキシ基に隣接する部位(オルト位)に置換基を有する化合物が好ましい。上述の置換基としては炭素数1~22の置換又は無置換のアルキル基が好ましい。また、酸化防止剤は、同一分子内にフェノール基と亜リン酸エステル基を有する化合物も好ましい。また、酸化防止剤は、リン系酸化防止剤も好適に使用することができる。リン系酸化防止剤としてはトリス[2-[[2,4,8,10-テトラキス(1,1-ジメチルエチル)ジベンゾ[d,f][1,3,2]ジオキサホスフェピン-6-イル]オキシ]エチル]アミン、トリス[2-[(4,6,9,11-テトラ-tert-ブチルジベンゾ[d,f][1,3,2]ジオキサホスフェピン-2-イル)オキシ]エチル]アミン、亜リン酸エチルビス(2,4-ジ-tert-ブチル-6-メチルフェニル)などが挙げられる。酸化防止剤の市販品としては、例えば、アデカスタブ AO-20、アデカスタブ AO-30、アデカスタブ AO-40、アデカスタブ AO-50、アデカスタブ AO-50F、アデカスタブ AO-60、アデカスタブ AO-60G、アデカスタブ AO-80、アデカスタブ AO-330(以上、(株)ADEKA製)などが挙げられる。また、酸化防止剤は、特許第6268967号公報の段落番号0023~0048に記載された化合物を使用することもでき、この内容は本明細書に組み込まれる。また、本発明の組成物は、必要に応じて、潜在酸化防止剤を含有してもよい。潜在酸化防止剤としては、酸化防止剤として機能する部位が保護基で保護された化合物であって、100~250℃で加熱するか、又は酸/塩基触媒存在下で80~200℃で加熱することにより保護基が脱離して酸化防止剤として機能する化合物が挙げられる。潜在酸化防止剤としては、国際公開第2014/021023号、国際公開第2017/030005号、特開2017-008219号公報に記載された化合物が挙げられ、この内容は本明細書に組み込まれる。潜在酸化防止剤の市販品としては、アデカアークルズGPA-5001((株)ADEKA製)等が挙げられる。
好ましい酸化防止剤の例としては、2,2-チオビス(4-メチル-6-t-ブチルフェノール)、2,6-ジ-t-ブチルフェノールおよび式(3)で表される化合物が挙げられる。





本実施形態の樹脂組成物は、必要に応じて凝集防止剤を含有してもよい。凝集防止剤としては、ポリアクリル酸ナトリウム等が挙げられる。
本発明の組成物は、凝集防止剤を含んでも含まなくてもよいが、含む場合、凝集防止剤の含有量は、本発明の組成物の全固形分質量に対して、0.01質量%以上10質量%以下であることが好ましく、0.02質量%以上5質量%以下であることがより好ましい。
本実施形態の樹脂組成物は、必要に応じてフェノール系化合物を含有してもよい。フェノール系化合物としては、Bis-Z、BisP-EZ、TekP-4HBPA、TrisP-HAP、TrisP-PA、BisOCHP-Z、BisP-MZ、BisP-PZ、BisP-IPZ、BisOCP-IPZ、BisP-CP、BisRS-2P、BisRS-3P、BisP-OCHP、メチレントリス-FR-CR、BisRS-26X(以上、商品名、本州化学工業(株)製)、BIP-PC、BIR-PC、BIR-PTBP、BIR-BIPC-F(以上、商品名、旭有機材工業(株)製)等が挙げられる。
本発明の組成物は、フェノール系化合物を含んでも含まなくてもよいが、含む場合、フェノール系化合物の含有量は、本発明の組成物の全固形分質量に対して、0.01質量%以上30質量%以下であることが好ましく、0.02質量%以上20質量%以下であることがより好ましい。
他の高分子化合物としては、シロキサン樹脂、(メタ)アクリル酸を共重合した(メタ)アクリルポリマー、ノボラック樹脂、レゾール樹脂、ポリヒドロキシスチレン樹脂およびそれらの共重合体などが挙げられる。他の高分子化合物はメチロール基、アルコキシメチル基、エポキシ基などの架橋基が導入された変性体であってもよい。
本発明の組成物は、他の高分子化合物を含んでも含まなくてもよいが、含む場合、他の高分子化合物の含有量は、本発明の組成物の全固形分質量に対して、0.01質量%以上30質量%以下であることが好ましく、0.02質量%以上20質量%以下であることがより好ましい。
本発明の樹脂組成物の粘度は、樹脂組成物の固形分濃度により調整できる。塗布膜厚の観点から、20mm2/s~12,000mm2/sが好ましく、30mm2/s~10,000mm2/sがより好ましく、50mm2/s~8,000mm2/sが更に好ましい。上記範囲であれば、均一性の高い塗布膜を得ることが容易になる。20mm2/s以上、更には、1,000mm2/s以上であれば、例えば再配線用絶縁膜として必要とされる膜厚で塗布することが容易であり、12,000mm2/s以下であれば、塗布面状に優れた塗膜が得られる。
本発明の樹脂組成物の含水率は、2.0質量%未満であることが好ましく、1.5質量%未満であることがより好ましく、1.0質量%未満であることが更に好ましい。2.0%未満であれば、樹脂組成物の保存安定性が向上する。
水分の含有量を維持する方法としては、保管条件における湿度の調整、保管時の収容容器の空隙率低減などが挙げられる。
ハロゲン原子の含有量を調節する方法としては、イオン交換処理などが好ましく挙げられる。
本発明の樹脂組成物を硬化することにより、この樹脂組成物の硬化物を得ることができる。
本発明の硬化物は、本発明の樹脂組成物を硬化してなる硬化物である。
樹脂組成物の硬化は加熱によるものであることが好ましく、加熱温度が120℃~400℃の範囲内であることがより好ましく、140℃~380℃の範囲内にあることが更に好ましく、170℃~350℃の範囲内にあることが特に好ましい。樹脂組成物の硬化物の形態は、特に限定されず、フィルム状、棒状、球状、ペレット状など、用途に合わせて選択することができる。本発明において、この硬化物は、フィルム状であることが好ましい。また、樹脂組成物のパターン加工によって、壁面への保護膜の形成、導通のためのビアホール形成、インピーダンスや静電容量あるいは内部応力の調整、放熱機能付与など、用途にあわせて、この硬化物の形状を選択することもできる。この硬化物(硬化物からなる膜)の膜厚は、0.5μm以上150μm以下であることが好ましい。
本発明の樹脂組成物を硬化した際の収縮率は、50%以下が好ましく、45%以下がより好ましく、40%以下が更に好ましい。ここで、収縮率は、樹脂組成物の硬化前後の体積変化の百分率を指し、下記の式より算出することができる。
収縮率[%]=100-(硬化後の体積÷硬化前の体積)×100
本発明の樹脂組成物の硬化物のイミド化反応率は、70%以上が好ましく、80%以上がより好ましく、90%以上が更に好ましい。70%以上であれば、機械特性に優れた硬化物となる場合がある。
本発明の樹脂組成物の硬化物の破断伸びは、30%以上が好ましく、40%以上がより好ましく、50%以上が更に好ましい。
本発明の樹脂組成物の硬化物のガラス転移温度(Tg)は、180℃以上であることが好ましく、210℃以上であることがより好ましく、230℃以上であることがさらに好ましい。
本発明の樹脂組成物は、上記各成分を混合して調製することができる。混合方法は特に限定はなく、従来公知の方法で行うことができる。
混合は撹拌羽による混合、ボールミルによる混合、タンク自身を回転させる混合などを採用することができる。
混合中の温度は10~30℃が好ましく、15~25℃がより好ましい。
フィルターを用いたろ過の他、吸着材を用いた不純物の除去処理を行ってもよい。フィルターろ過と吸着材を用いた不純物除去処理とを組み合わせてもよい。吸着材としては、公知の吸着材を用いることができる。例えば、シリカゲル、ゼオライトなどの無機系吸着材、活性炭などの有機系吸着材が挙げられる。
更にフィルターを用いたろ過後、ボトルに充填した樹脂組成物を減圧下に置き、脱気する工程を施しても良い。
本発明の硬化物の製造方法は、樹脂組成物を基材上に適用して膜を形成する膜形成工程、上記膜を選択的に露光する露光工程、及び、露光工程により露光された膜を現像液を用いて現像してパターンを形成する現像工程を含むことが好ましい。
本発明の硬化物の製造方法は、上記膜形成工程、上記露光工程、上記現像工程、並びに、現像工程により得られたパターンを加熱する加熱工程及び現像工程により得られたパターンを露光する現像後露光工程の少なくとも一方を含むことが特に好ましい。
また、本発明の製造方法は、上記膜形成工程、及び、上記膜を加熱する工程を含むことも好ましい。
以下、各工程の詳細について説明する。
本発明の樹脂組成物は、基材上に適用して膜を形成する膜形成工程に用いることができる。
本発明の硬化物の製造方法は、樹脂組成物を基材上に適用して膜を形成する膜形成工程を含むことが好ましい。
基材の種類は、用途に応じて適宜定めることができるが、シリコン、窒化シリコン、ポリシリコン、酸化シリコン、アモルファスシリコンなどの半導体作製基材、石英、ガラス、光学フィルム、セラミック材料、蒸着膜、磁性膜、反射膜、Ni、Cu、Cr、Feなどの金属基材(例えば、金属から形成された基材、及び、金属層が例えばめっきや蒸着等により形成された基材のいずれであってもよい)、紙、SOG(Spin On Glass)、TFT(薄膜トランジスタ)アレイ基材、モールド基材、プラズマディスプレイパネル(PDP)の電極板などが挙げられ、特に制約されない。本発明では、特に、半導体作製基材が好ましく、シリコン基材、Cu基材およびモールド基材がより好ましい。
また、これらの基材にはヘキサメチルジシラザン(HMDS)等による密着層や酸化層などの層が表面に設けられていてもよい。
また、基材の形状は特に限定されず、円形状であってもよく、矩形状であってもよい。
基材のサイズとしては、円形状であれば、例えば直径が100~450mmであり、好ましくは200~450mmである。矩形状であれば、例えば短辺の長さが100~1000mmであり、好ましくは200~700mmである。
また、基材としては、例えば板状、好ましくはパネル状の基材(基板)が用いられる。
また、あらかじめ仮支持体上に上記付与方法によって付与して形成した塗膜を、基材上に転写する方法を適用することもできる。
転写方法に関しては特開2006-023696号公報の段落0023、0036~0051や、特開2006-047592号公報の段落0096~0108に記載の作製方法を本発明においても好適に用いることができる。
また、基材の端部において余分な膜の除去を行なう工程を行なってもよい。このような工程の例には、エッジビードリンス(EBR)、バックリンスなどが挙げられる。
また樹脂組成物を基材に塗布する前に基材を種々の溶剤を塗布し、基材の濡れ性を向上させた後に樹脂組成物を塗布するプリウェット工程を採用しても良い。
上記膜は、膜形成工程(層形成工程)の後に、溶剤を除去するために形成された膜(層)を乾燥する工程(乾燥工程)に供されてもよい。
すなわち、本発明の硬化物の製造方法は、膜形成工程により形成された膜を乾燥する乾燥工程を含んでもよい。
また、上記乾燥工程は膜形成工程の後、露光工程の前に行われることが好ましい。
乾燥工程における膜の乾燥温度は50~150℃であることが好ましく、70℃~130℃がより好ましく、90℃~110℃が更に好ましい。また、減圧により乾燥を行っても良い。乾燥時間としては、30秒~20分が例示され、1分~10分が好ましく、2分~7分がより好ましい。
上記膜は、膜を選択的に露光する露光工程に供されてもよい。
すなわち、本発明の硬化物の製造方法は、膜形成工程により形成された膜を選択的に露光する露光工程を含んでもよい。
選択的に露光するとは、膜の一部を露光することを意味している。また、選択的に露光することにより、膜には露光された領域(露光部)と露光されていない領域(非露光部)が形成される。
露光量は、本発明の樹脂組成物を硬化できる限り特に定めるものではないが、例えば、波長365nmでの露光エネルギー換算で50~10,000mJ/cm2が好ましく、200~8,000mJ/cm2がより好ましい。
また、露光の方式は特に限定されず、本発明の樹脂組成物からなる膜の少なくとも一部が露光される方式であればよいが、フォトマスクを使用した露光、レーザーダイレクトイメージング法による露光等が挙げられる。
上記膜は、露光後に加熱する工程(露光後加熱工程)に供されてもよい。
すなわち、本発明の硬化物の製造方法は、露光工程により露光された膜を加熱する露光後加熱工程を含んでもよい。
露光後加熱工程は、露光工程後、現像工程前に行うことができる。
露光後加熱工程における加熱温度は、50℃~140℃であることが好ましく、60℃~120℃であることがより好ましい。
露光後加熱工程における加熱時間は、30秒間~300分間が好ましく、1分間~10分間がより好ましい。
露光後加熱工程における昇温速度は、加熱開始時の温度から最高加熱温度まで1~12℃/分が好ましく、2~10℃/分がより好ましく、3~10℃/分が更に好ましい。
また、昇温速度は加熱途中で適宜変更してもよい。
露光後加熱工程における加熱手段としては、特に限定されず、公知のホットプレート、オーブン、赤外線ヒーター等を用いることができる。
また、加熱に際し、窒素、ヘリウム、アルゴンなどの不活性ガスを流す等により、低酸素濃度の雰囲気で行うことも好ましい。
露光後の上記膜は、現像液を用いて現像してパターンを形成する現像工程に供されてもよい。
すなわち、本発明の硬化物の製造方法は、露光工程により露光された膜を現像液を用いて現像してパターンを形成する現像工程を含んでもよい。 現像を行うことにより、膜の露光部及び非露光部のうち一方が除去され、パターンが形成される。
ここで、膜の非露光部が現像工程により除去される現像をネガ型現像といい、膜の露光部が現像工程により除去される現像をポジ型現像という。
現像工程において用いられる現像液としては、アルカリ水溶液、又は、有機溶剤を含む現像液が挙げられる。
他の成分としては、例えば、公知の界面活性剤や公知の消泡剤等が挙げられる。
現像液の供給方法は、所望のパターンを形成できれば特に制限は無く、膜が形成された基材を現像液に浸漬する方法、基材上に形成された膜にノズルを用いて現像液を供給するパドル現像、または、現像液を連続供給する方法がある。ノズルの種類は特に制限は無く、ストレートノズル、シャワーノズル、スプレーノズル等が挙げられる。
現像液の浸透性、非画像部の除去性、製造上の効率の観点から、現像液をストレートノズルで供給する方法、又はスプレーノズルにて連続供給する方法が好ましく、画像部への現像液の浸透性の観点からは、スプレーノズルで供給する方法がより好ましい。
また、現像液をストレートノズルにて連続供給後、基材をスピンし現像液を基材上から除去し、スピン乾燥後に再度ストレートノズルにて連続供給後、基材をスピンし現像液を基材上から除去する工程を採用してもよく、この工程を複数回繰り返しても良い。
また現像工程における現像液の供給方法としては、現像液が連続的に基材に供給され続ける工程、基材上で現像液が略静止状態で保たれる工程、基材上で現像液を超音波等で振動させる工程及びそれらを組み合わせた工程などが採用可能である。
現像液がアルカリ水溶液である場合、リンス液としては、例えば水を用いることができる。現像液が有機溶剤を含む現像液である場合、リンス液としては、例えば、現像液に含まれる溶剤とは異なる溶剤(例えば、水、現像液に含まれる有機溶剤とは異なる有機溶剤)を用いることができる。
他の成分としては、例えば、公知の界面活性剤や公知の消泡剤等が挙げられる。
リンス液の供給方法は、所望のパターンを形成できれば特に制限は無く、基材をリンス液に浸漬する方法、基材上でのパドル現像、基材にリンス液をシャワーで供給する方法、基材上にストレートノズル等の手段により現像液を連続供給する方法がある。
リンス液の浸透性、非画像部の除去性、製造上の効率の観点から、リンス液をシャワーノズル、ストレートノズル、スプレーノズルなどで供給する方法があり、スプレーノズルにて連続供給する方法が好ましく、画像部へのリンス液の浸透性の観点からは、スプレーノズルで供給する方法がより好ましい。ノズルの種類は特に制限は無く、ストレートノズル、シャワーノズル、スプレーノズル等が挙げられる。
すなわち、リンス工程は、リンス液を上記露光後の膜に対してストレートノズルにより供給、又は、連続供給する工程であることが好ましく、リンス液をスプレーノズルにより供給する工程であることがより好ましい。
またリンス工程におけるリンス液の供給方法としては、リンス液が連続的に基材に供給され続ける工程、基材上でリンス液が略静止状態で保たれる工程、基材上でリンス液を超音波等で振動させる工程及びそれらを組み合わせた工程などが採用可能である。
現像工程により得られたパターン(リンス工程を行う場合は、リンス後のパターン)は、上記現像により得られたパターンを加熱する加熱工程に供されてもよい。
すなわち、本発明の硬化物の製造方法は、現像工程により得られたパターンを加熱する加熱工程を含んでもよい。
また、本発明の硬化物の製造方法は、現像工程を行わずに他の方法で得られたパターン、又は、膜形成工程により得られた膜を加熱する加熱工程を含んでもよい。
加熱工程において、ポリイミド前駆体等の樹脂は環化してポリイミド等の樹脂となる。
また、特定樹脂、又は特定樹脂以外の架橋剤における未反応の架橋性基の架橋なども進行する。
加熱工程における加熱温度(最高加熱温度)としては、50~450℃が好ましく、150~350℃がより好ましく、150~250℃が更に好ましく、160~250℃が一層好ましく、160~230℃が特に好ましい。
加えて、急速加熱可能なオーブンの場合、加熱開始時の温度から最高加熱温度まで1~8℃/秒の昇温速度で行うことが好ましく、2~7℃/秒がより好ましく、3~6℃/秒が更に好ましい。
上記加熱温度の上限は、350℃以下であることが好ましく、250℃以下であることがより好ましく、240℃以下であることが更に好ましい。
更に、加熱後冷却してもよく、この場合の冷却速度としては、1~5℃/分であることが好ましい。
加熱工程における加熱手段としては、特に限定されないが、例えばホットプレート、赤外炉、電熱式オーブン、熱風式オーブン、赤外線オーブンなどが挙げられる。
現像工程により得られた(リンス工程を行う場合は、リンス後のパターン)は、上記加熱工程に代えて、又は、上記加熱工程に加えて、現像工程後のパターンを露光する現像後露光工程に供されてもよい。
すなわち、本発明の硬化物の製造方法は、現像工程により得られたパターンを露光する現像後露光工程を含んでもよい。本発明の硬化物の製造方法は、加熱工程及び現像後露光工程を含んでもよいし、加熱工程及び現像後露光工程の一方のみを含んでもよい。
現像後露光工程においては、例えば、光塩基発生剤の感光によってポリイミド前駆体等の環化が進行する反応などを促進することができる。
現像後露光工程においては、現像工程において得られたパターンの少なくとも一部が露光されればよいが、上記パターンの全部が露光されることが好ましい。
現像後露光工程における露光量は、感光性化合物が感度を有する波長における露光エネルギー換算で、50~20,000mJ/cm2であることが好ましく、100~15,000mJ/cm2であることがより好ましい。
現像後露光工程は、例えば、上述の露光工程における光源を用いて行うことができ、ブロードバンド光を用いることが好ましい。
現像工程により得られたパターン(加熱工程及び現像後露光工程の少なくとも一方に供されたものが好ましい)は、パターン上に金属層を形成する金属層形成工程に供されてもよい。
すなわち、本発明の硬化物の製造方法は、現像工程により得られたパターン(加熱工程及び現像後露光工程少なくとも一方に供されたものが好ましい)上に金属層を形成する金属層形成工程を含むことが好ましい。
本発明の硬化物の製造方法、又は、本発明の硬化物の適用可能な分野としては、電子デバイスの絶縁膜、再配線層用層間絶縁膜、ストレスバッファ膜などが挙げられる。そのほか、封止フィルム、基板材料(フレキシブルプリント基板のベースフィルムやカバーレイ、層間絶縁膜)、又は上記のような実装用途の絶縁膜をエッチングでパターン形成することなどが挙げられる。これらの用途については、例えば、サイエンス&テクノロジー(株)「ポリイミドの高機能化と応用技術」2008年4月、柿本雅明/監修、CMCテクニカルライブラリー「ポリイミド材料の基礎と開発」2011年11月発行、日本ポリイミド・芳香族系高分子研究会/編「最新ポリイミド 基礎と応用」エヌ・ティー・エス,2010年8月等を参照することができる。
本発明の積層体とは、本発明の硬化物からなる層を複数層有する構造体をいう。
本発明の積層体は、硬化物からなる層を2層以上含む積層体であり、3層以上積層した積層体としてもよい。
上記積層体に含まれる2層以上の上記硬化物からなる層のうち、少なくとも1つが本発明の硬化物からなる層であり、硬化物の収縮、又は、上記収縮に伴う硬化物の変形等を抑制する観点からは、上記積層体に含まれる全ての硬化物からなる層が本発明の硬化物からなる層であることも好ましい。
すなわち、本発明の積層体の製造方法は、複数回行われる硬化物の製造方法の間に、硬化物からなる層上に金属層を形成する金属層形成工程を更に含むことが好ましい。金属層形成工程の好ましい態様は上述の通りである。
上記積層体としては、例えば、第一の硬化物からなる層、金属層、第二の硬化物からなる層の3つの層がこの順に積層された層構造を少なくとも含む積層体が好ましいものとして挙げられる。
上記第一の硬化物からなる層及び上記第二の硬化物からなる層は、いずれも本発明の硬化物からなる層であることが好ましい。上記第一の硬化物からなる層の形成に用いられる本発明の樹脂組成物と、上記第二の硬化物からなる層の形成に用いられる本発明の樹脂組成物とは、組成が同一の組成物であってもよいし、組成が異なる組成物であってもよい。本発明の積層体における金属層は、再配線層などの金属配線として好ましく用いられる。
本発明の積層体の製造方法は、積層工程を含むことが好ましい。
積層工程とは、パターン(樹脂層)又は金属層の表面に、再度、(a)膜形成工程(層形成工程)、(b)露光工程、(c)現像工程、(d)加熱工程及び現像後露光工程の少なくとも一方を、この順に行うことを含む一連の工程である。ただし、(a)の膜形成工程および(d)加熱工程及び現像後露光工程の少なくとも一方を繰り返す態様であってもよい。また、(d)加熱工程及び現像後露光工程の少なくとも一方の後には(e)金属層形成工程を含んでもよい。積層工程には、更に、上記乾燥工程等を適宜含んでいてもよいことは言うまでもない。
例えば、樹脂層/金属層/樹脂層/金属層/樹脂層/金属層のように、樹脂層を2層以上20層以下とする構成が好ましく、2層以上9層以下とする構成が更に好ましい。
上記各層はそれぞれ、組成、形状、膜厚等が同一であってもよいし、異なっていてもよい。
本発明の積層体の製造方法は、上記金属層および樹脂組成物層の少なくとも一部を表面活性化処理する、表面活性化処理工程を含むことが好ましい。
表面活性化処理工程は、通常、金属層形成工程の後に行うが、上記現像工程の後(好ましくは、加熱工程及び現像後露光工程の少なくとも一方の後)、樹脂組成物層に表面活性化処理工程を行ってから、金属層形成工程を行ってもよい。
表面活性化処理は、金属層の少なくとも一部のみに行ってもよいし、露光後の樹脂組成物層の少なくとも一部のみに行ってもよいし、金属層および露光後の樹脂組成物層の両方について、それぞれ、少なくとも一部に行ってもよい。表面活性化処理は、金属層の少なくとも一部について行うことが好ましく、金属層のうち、表面に樹脂組成物層を形成する領域の一部または全部に表面活性化処理を行うことが好ましい。このように、金属層の表面に表面活性化処理を行うことにより、その表面に設けられる樹脂組成物層(膜)との密着性を向上させることができる。
また、表面活性化処理は、露光後の樹脂組成物層(樹脂層)の一部または全部についても行うことが好ましい。このように、樹脂組成物層の表面に表面活性化処理を行うことにより、表面活性化処理した表面に設けられる金属層や樹脂層との密着性を向上させることができる。特にネガ型現像を行う場合など、樹脂組成物層が硬化されている場合には、表面処理によるダメージを受けにくく、密着性が向上しやすい。
表面活性化処理としては、具体的には、各種原料ガス(酸素、水素、アルゴン、窒素、窒素/水素混合ガス、アルゴン/酸素混合ガスなど)のプラズマ処理、コロナ放電処理、CF4/O2、NF3/O2、SF6、NF3、NF3/O2によるエッチング処理、紫外線(UV)オゾン法による表面処理、塩酸水溶液に浸漬して酸化皮膜を除去した後にアミノ基とチオール基を少なくとも一種有する化合物を含む有機表面処理剤への浸漬処理、ブラシを用いた機械的な粗面化処理から選択され、プラズマ処理が好ましく、特に原料ガスに酸素を用いた酸素プラズマ処理が好ましい。コロナ放電処理の場合、エネルギーは、500~200,000J/m2が好ましく、1000~100,000J/m2がより好ましく、10,000~50,000J/m2が最も好ましい。
本発明は、本発明の硬化物、又は、本発明の積層体を含む半導体デバイスも開示する。
本発明は、本発明の硬化物の製造方法、又は、本発明の積層体の製造方法を含む半導体デバイスの製造方法も開示する。本発明の樹脂組成物を再配線層用層間絶縁膜の形成に用いた半導体デバイスの具体例としては、特開2016-027357号公報の段落0213~0218の記載及び図1の記載を参酌でき、これらの内容は本明細書に組み込まれる。
ポリマーPEは、オキシジアニリン(以下、「ODA」と略記する)0.600g(3.00mmol)のN,N-ジメチルアセトアミド(DMAc)8.50ml溶液中に、4,4'-ビフェニルテトラカルボン酸二無水物(以下、「BPDA」と略記する)0.883g(3.00mmol)を一度に加え、室温環境下12時間撹拌せしめることにより、粘性の高い透明溶液のポリマーPE溶液を得た。DMAc中のポリマーPEの固有粘度は、30℃、0.5g/dLの濃度で0.73dL/gであった。なお、DMAcは、減圧蒸留により精製したものを、ODA及びBPDAは、それぞれテトラヒドロフラン(THF)、無水酢酸から再結晶したものを用いた。ポリマーPEの分子量をゲルパーミエーションクロマトグラフィー(標準ポリスチレン換算)で測定したところ、重量平均分子量(Mw)は12,000であった。
4,4’-オキシジフタル酸二無水物(ODPA)155.1gをセパラブルフラスコに入れ、2-ヒドロキシエチルメタクリレート(HEMA)131.2gとγ-ブチロラクトン400mlを入れて室温下で撹拌し、撹拌しながらピリジン81.5gを加えて反応混合物を得た。反応による発熱の終了後に室温まで放冷し、16時間放置した。
次に、氷冷下において、ジシクロヘキシルカルボジイミド(DCC)206.3gをγ-ブチロラクトン180mlに溶解した溶液を撹拌しながら40分かけて反応混合物に加え、続いて4,4’-ジアミノジフェニルエーテル(DADPE)93.0gをγ-ブチロラクトン350mlに懸濁したものを撹拌しながら60分かけて加えた。更に室温で2時間撹拌した後、エチルアルコール30mlを加えて1時間撹拌し、次に、γ-ブチロラクトン400mlを加えた。反応混合物に生じた沈殿物をろ過により取り除き、反応液を得た。
得られた反応液を3リットルのエチルアルコールに加えて粗ポリマーからなる沈殿物を生成した。生成した粗ポリマーを濾別し、テトラヒドロフラン1.5リットルに溶解して粗ポリマー溶液を得た。得られた粗ポリマー溶液を28リットルの水に滴下してポリマーを沈殿させ、得られた沈殿物を濾別した後、真空乾燥して粉末状のポリマーPA-1を得た。ポリマーPA-1の分子量をゲルパーミエーションクロマトグラフィー(標準ポリスチレン換算)で測定したところ、重量平均分子量(Mw)は20,000であった。
上記合成例2における4,4’-オキシジフタル酸二無水物155.1gに代えて、3,3’4,4’-ビフェニルテトラカルボン酸二無水物147.1gを用いた以外は、合成例2に記載の方法と同様にして反応を行い、ポリマーPB-1を得た。ポリマーPB-1の分子量をゲルパーミエーションクロマトグラフィー(標準ポリスチレン換算)で測定したところ、重量平均分子量(Mw)は22,000であった。
20.0g(64.5ミリモル)の4,4’-オキシジフタル酸二無水物(140°Cで12時間乾燥)と、17.12g(131.58ミリモル)の2-ヒドロキシエチルメタクリレート(HEMA)とを、50mlのN-メチルピロリドンに懸濁させ、モレキュラーシーブで乾燥させた。懸濁液を100℃で3時間加熱した。加熱を開始してから数分後に透明な溶液が得られた。反応混合物を室温に冷却し、21.43g(270.9ミリモル)のピリジン及び90mlのN-メチルピロリドンを加えた。次いで、反応混合物を-10℃に冷却し、温度を-10±4℃に保ちながら16.12g(135.5ミリモル)のSOCl2を10分かけて加えた。SOCl2を加えている間、粘度が増加した。50mlのN-メチルピロリドンで希釈した後、反応混合物を室温で2時間撹拌した。次いで、100mlのN-メチルピロリドンに11.75g(58.7ミリモル)の4,4’-ジアミノジフェニルエーテルを溶解させた溶液を、20~23℃で20分かけて反応混合物に滴下した。次いで、反応混合物を室温で1晩撹拌した。次いで、5リットルの水の中でポリイミド前駆体を沈殿させ、水-ポリイミド前駆体混合物を5000rpmの速度で15分間撹拌した。ポリイミド前駆体を濾取し、再度4リットルの水に投入してさらに30分間撹拌し再び濾過した。次いで、得られたポリイミド前駆体を減圧下で、45℃で3日間乾燥し、ポリマーPA-2を得た。ポリマーPA-2の分子量をゲルパーミエーションクロマトグラフィー(標準ポリスチレン換算)で測定したところ、重量平均分子量(Mw)は20,000であった。
合成例4において、20.0g(64.5ミリモル)の4,4’-オキシジフタル酸二無水物に代えて、19.0g(64.5ミリモル)の3,3’4,4’-ビフェニルテトラカルボン酸二無水物を用いた以外は、合成例4に記載の方法と同様にして反応を行い、ポリマーPB-1を得た。ポリマーPB-2の分子量をゲルパーミエーションクロマトグラフィー(標準ポリスチレン換算)で測定したところ、重量平均分子量(Mw)は21,000であった。
セパラブルフラスコ中で、2,2-ビス(3-アミノ-4-ヒドロキシフェニル)-ヘキサフルオロプロパン 183.1g(0.5モル)、N,N-ジメチルアセトアミド(DMAc) 640.9g、ピリジン 63.3g(0.8モル)を室温(25℃)で混合撹拌し、均一溶液とした。これに、4,4’-ジフェニルエーテルジカルボニルクロリド 118.0g(0.4モル)をジエチレングリコールジメチルエーテル(DMDG) 354gに溶解したものを滴下ロートより滴下した。この際、セパラブルフラスコは15~20℃の水浴で冷却した。滴下に要した時間は40分、反応液温は最大で30℃であった。
滴下終了から3時間後、反応液に1,2-シクロヘキシルジカルボン酸無水物 30.8g(0.2mol)を添加し、室温で15時間撹拌放置し、ポリマー鎖の全アミン末端基の99%をカルボキシシクロヘキシルアミド基で封止した。この際の反応率は投入した1,2-シクロヘキシルジカルボン酸無水物の残量を高速液体クロマトグラフィー(HPLC)で追跡することにより容易に算出することができる。その後上記反応液を2Lの水に高速撹拌下で滴下し重合体を分散析出させ、これを回収し、適宜水洗、脱水の後に真空乾燥を施し、ゲルパーミエーションクロマトグラフィー(GPC)法で測定した重量平均分子量9,000(ポリスチレン換算)の粗ポリベンゾオキサゾール前駆体を得た。
上記で得られた粗ポリベンゾオキサゾール前駆体をγ-ブチロラクトン(GBL)に再溶解した後、これを陽イオン交換樹脂及び陰イオン交換樹脂にて処理し、それにより得られた溶液をイオン交換水中に投入後、析出したポリマーを濾別、水洗、真空乾燥することにより精製されたポリベンゾオキサゾール前駆体(ポリマーPC)を得た。ポリマーPB-2の分子量をゲルパーミエーションクロマトグラフィー(標準ポリスチレン換算)で測定したところ、重量平均分子量(Mw)は18,000であった。
<合成例7:ポリマーPDの合成>
H Seino et al., J. Polym. Sci., Part A: Polym. Chem., 36, 2261 (1998)のPFAAの合成方法と同様の方法により、ピロメリット酸二無水物、3,3’4,4’-ビフェニルテトラカルボン酸二無水物、及び、2,2’-ビス(トリフルオロメチル)ベンジジンから、ポリマーPDを合成した。ポリマーPDの分子量をゲルパーミエーションクロマトグラフィー(標準ポリスチレン換算)で測定したところ、重量平均分子量(Mw)は15,000であった。
各実施例において、それぞれ、下記表に記載の成分を混合し、各ネガ型感光性樹脂組成物を得た。また、各比較例において、それぞれ、下記表に記載の成分を混合し、各比較用組成物を得た。
具体的には、溶剤以外の表に記載の各成分の含有量(配合量)は、表の「固形分中の濃度[質量部]」の「添加量」の欄に記載の量(質量部)とした。
溶剤の含有量(配合量)は、組成物の固形分濃度が表中の「固形分濃度[質量%]」の値となるようにし、溶剤の全質量に対する各溶剤の含有量は、表中の「添加量」の欄に記載の量(質量部)となるようにした。
得られたネガ型感光性樹脂組成物及び比較用組成物を、細孔の幅が0.45μmのポリテトラフルオロエチレン製フィルターを用いて加圧ろ過した。
また、表中、「-」の記載は該当する成分を組成物が含有していないことを示している。




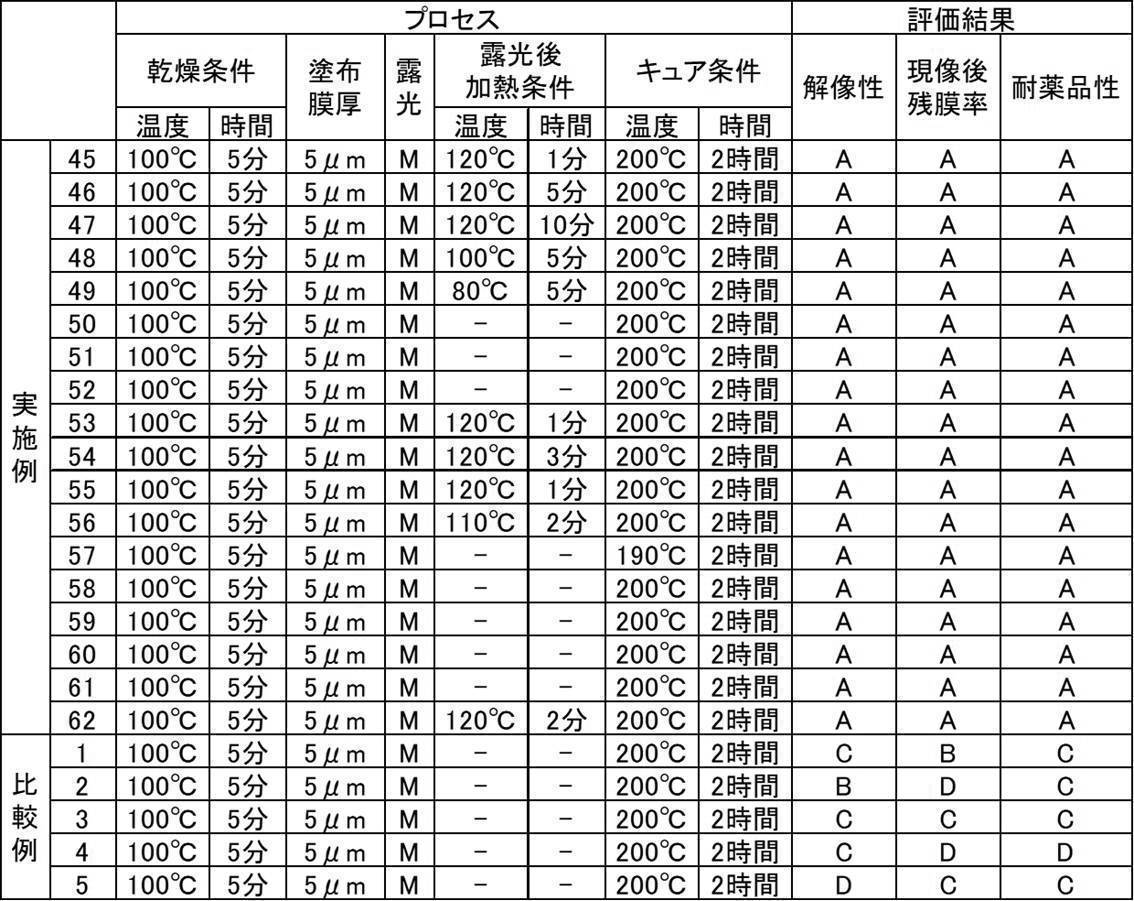
・樹脂PA-1、PA-2、PB-1、PB-2、PC、PD、PE:上記合成例により得られた樹脂
・A-1:2,6-ビス(2,3,4-トリヒドロキシベンジル)-p-クレゾールと6-ジアゾ5,6-ジヒドロ-5-オキソ-1-ナフタレンスルホン酸とのエステル
・A-2:2,2’,3,3’-テトラヒドロ-3,3,3’,3’-テトラメチル-1,1’-スピロビ(1H-インデン)-5,5’,6,6’,7,7’ヘキサノールと1,2-ナフトキノン-(2)-ジアゾ-5-スルホン酸とのエステル
・A-3:ビス(4-ヒドロキシ-3,5-ジメチルフェニル)-(4-ヒドロキシ-3-メトキシフェニル)メタンと6-ジアゾ-5,6-ジヒドロ-5-オキソナフタレン-1-スルホン酸とのエステル
・A-4:ビクトリアピュアブル-NAPS(ロイコ色素、保土谷化学工業(株)製)
・A-5:下記オキシム化合物
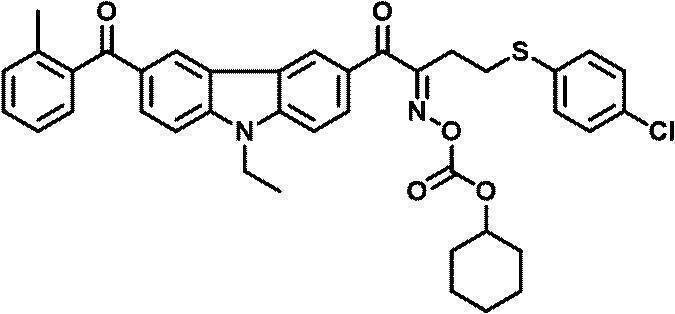
・A-6:下記ジアリールエテン色素

・A-7:4-[ビス(4-ヒドロキシ-3,5-ジメチルフェニル)メチル]1,2-ベンゼンジオールと6-ジアゾ-5,6-ジヒドロ-5-オキソナフタレン-1-スルホン酸とのエステル
・A-8:4,4’-(メンタン-1,8-ジイル)ビス[2-(4-ヒドロキシベンジル)-6-メチルフェノールと6-ジアゾ-5-オキソ-5,6-ジヒドロナフタレン-1-スルホン酸とのエステル
・CA-1:下記クマリン色素

上記化合物中、A-1~A-8は露光によりその露光波長の吸光度が小さくなる化合物であり、化合物Aに該当する。
また、A-1~A-4、A-6~A-8は露光時にラジカル重合開始種を発生しない化合物であり、A-5は露光時にラジカル重合開始種を発生する化合物である。
CA-1は露光によりその露光波長の吸光度が小さくなる化合物ではなく、化合物Aに該当しない。
・I-1:1-フェニル-1,2-プロパンジオン-2-(O-エトキシカルボニル)-オキシム(極大吸収波長:365nm)
・I-2:NCI 831(極大吸収波長:365nm) ((株)ADEKA製)
・I-3:Irgacure OXE01(極大吸収波長:365nm) (BASF社製)
・I-4:Irgacure OXE02(極大吸収波長:365nm) (BASF社製)
・I-5:Irgacure 784(極大吸収波長:405nm) (BASF社製)
・N-1:1H-テトラゾール
・N-2:5-アミノテトラゾール
・N-3:3-アミノトリアゾール
・N-4:ベンゾトリアゾール
・N-5:下記化合物

・F-1:テトラエチレングリコールジメタクリレート
・F-2:ライトエステル BP-6EM (共栄社化学(株)製)
・F-3:ライトエステルNP (共栄社化学(株)製)
・F-4:ライトエステル1.6HX (共栄社化学(株)製)
・F-5:KAYARAD DPHA(日本化薬(株)製)
・F-6:ダイセル セロキサイド CEL2081 ((株)ダイセル製)
・G-1:N-[3-(トリエトキシシリル)プロピル]フタルアミド酸
・G-2:N-[3-(トリエトキシシリル)プロピル]マレインアミド酸
・G-3:3-メタクリロキシプロピルトリメトキシシラン
・G-4:2-(3,4-エポキシシクロヘキシル)エチルトリメトキシシラン
・G-5:下記化合物

・H-1:p-ベンゾキノン
・H-2:4-メトキシフェノール
・H-3:2-ニトロソ-1-ナフト-ル
・K-1:N-cyclohexyl-N,N-dimethyl-N-phenacylammonium Maleate

・K-2:下記化合物
・K-3:下記化合物

・K-4:下記化合物

・M-1:7-ジエチルアミノ-3-エトキシカルボニルクマリン
・M-2:N-フェニルジエタノールアミン
・S-1:N-メチルピロリドン
・S-2:乳酸エチル
・S-3:γ-ブチロラクトン
・S-4:ジメチルスルホキシド
・S-5:N,N-ジメチルアセトアミド
〔解像性評価〕
各実施例又は比較例において、調製したネガ型感光性樹脂組成物又は比較用組成物を、銅ウエハ上にスピンコート法により塗布した。上記ウエハをホットプレート上で、表に記載の温度及び時間で乾燥し、銅ウエハ上に表の「塗布膜厚」の欄に記載の厚さであって、均一な厚さの硬化性樹脂組成物層を形成した。
「露光」の欄に「M」と記載した例においては、銅基板上の硬化性樹脂組成物層を、ステッパーを用い、5μm~20μmまで1μm刻みのコンタクトホールのフォトマスクを使用して、露光波長365nmで露光した。露光量は250~650mJ/cm2の範囲で50mJ/cm2刻みで変化させた。
「露光」の欄に「D」と記載した例においては、ダイレクト露光装置(アドテック DE-6UH III)を用いて、5μm~20μmまで1μm刻みのコンタクトホールになるようなレーザーダイレクトイメージング露光を行った。露光波長は405nmとし、露光量は250~650mJ/cm2の範囲で50mJ/cm2刻みで変化させた。
表の「露光後加熱工程」の欄に数値の記載のある実施例に関しては、記載の温度/時間で露光後にウェハの加熱処理を実施した。加熱はホットプレートで大気下にて実施した。
露光した硬化性樹脂組成物層を、シクロペンタノンで40秒間現像した後に、PGMEA(プロピレングリコールモノメチルエーテルアセテート)でリンスした。
現像後に得られたパターンにおいて、コンタクトホールパターンサイズが最小であるものの線幅を「最小ホール径」として、下記評価基準に従って評価した。最小ホール径が小さければ小さいほど解像性に優れるといえ、例えば、その後のめっき工程で形成される金属配線幅を微細化できることを表すため、好ましい結果となる。測定限界は5μmである。評価結果は、表中の「解像性」の欄に記載した。A又はBが好ましく、Aがより好ましい。
-評価基準-
塗布膜厚が5μmの場合:
A:最小ホール径が5μmであった。
B:最小ホール径が5μm以上7μm未満であった。
C:最小ホール径が7μm以上10μm未満であった。
D:最小ホール径が10μm以上であった。
塗布膜厚が13μmの場合:
A:最小ホール径が10μm未満であった。
B:最小ホール径が10μm以上15μm未満であった。
C:最小ホール径が15μm以上20μm未満であった。
D:最小ホール径が20μm以上であった。
上記解像性評価において、最小ホール径を形成できる露光量における現像後の残膜率を、現像前後の膜厚を測定し、現像前の膜厚に対する現像後の膜厚の割合として算出した。算出された残膜率の値から下記評価基準に従って評価を行い、評価結果は表の「現像後残膜率」の欄に記載した。残膜率が高いほど好ましい結果となる。A又はBが好ましく、Aがより好ましい。
残膜率(%)=現像後の膜厚/現像前の膜厚×100
-評価基準-
A:残膜率が90%以上であった。
B:残膜率が85%以上90%未満であった。
C:残膜率が80%以上85%未満であった。
D:残膜率が80%未満であった。
各実施例又は比較例において、上述の解像性評価における硬化性樹脂組成物層の形成と同様の方法により、シリコンウェハ上に硬化性樹脂組成物層を形成した。
その後、各実施例又は比較例において、フォトマスクを使用せずに硬化性樹脂組成物層の全面に対して露光を行い、樹脂膜を得た。
露光方法は、フォトマスクを使用しなかった以外は、上述の解像性評価と同様の方法とした。露光量は500mJ/cm2とした。
耐薬品性の評価においては、露光後加熱工程は行わなかった。
次いで、各実施例又は比較例において得られた樹脂膜を、窒素雰囲気下で、10℃/分の昇温速度で昇温し、表の「キュア条件」の「温度」及び時間の欄に示した温度及び時間でその温度を維持し、硬化膜を形成した。
得られた硬化膜を下記の薬品に下記の条件で浸漬し、溶解速度を算定した。
薬品:ジメチルスルホキシド(DMSO)と25質量%のテトラメチルアンモニウムヒドロキシド(TMAH)水溶液の90:10(質量比)の混合物
評価条件:上記硬化膜を上記薬品に75℃で15分間浸漬して浸漬前後の硬化膜の膜厚を比較し、浸漬後の残膜率を算出した。
残膜率(%)=浸漬後の膜厚/浸漬前の膜厚×100
得られた残膜率の値について、下記評価基準に従って評価し、評価結果を「耐薬品性」の欄に記載した。残膜率が大きいほど、耐薬品性に優れるといえる。A又はBが好ましく、Aがより好ましい。
-評価基準-
A:残膜率が75%以上であった。
B:残膜率が70%以上75%未満であった。
C:残膜率が55%以上70%未満であった。
D:残膜率が55%未満であった。
比較例1に係る比較用組成物は、含窒素環状化合物を含むが化合物Aを含まない。このような例においては、解像性に劣ることがわかる。
比較例2に係る比較用組成物は、化合物Aの代わりにCA-1を含み、かつ、含窒素環状化合物を含む。このような例においては、現像後残膜率に劣ることがわかる。
比較例3~5に係る比較用組成物は、含窒素環状化合物を含まない。このような例においては、解像性、現像後残膜率のいずれにも劣ることがわかる。
実施例1において使用した樹脂組成物を、表面に銅薄層が形成された樹脂基材の銅薄層の表面にスピンコート法により層状に適用して、100℃で5分間乾燥し、膜厚20μmの感光膜を形成した後、ステッパー((株)ニコン製、NSR1505 i6)を用いて露光した。露光はマスク(パターンが1:1ラインアンドスペースであり、線幅が10μmであるバイナリマスク)を介して、波長365nmで行った。上記露光後、シクロヘキサノンで2分間現像し、PGMEAで30秒間リンスし、層のパターンを得た。
次いで、窒素雰囲気下で、10℃/分の昇温速度で昇温し、230℃に達した後、230℃で180分間維持して、再配線層用層間絶縁膜を形成した。この再配線層用層間絶縁膜は、絶縁性に優れていた。
また、これらの再配線層用層間絶縁膜を使用して半導体デバイスを製造したところ、問題なく動作することを確認した。
Claims (17)
- 樹脂、
露光によりその露光波長の吸光度が小さくなる化合物A、及び、
光重合開始剤を含み、
下記条件i及び条件iiの少なくとも一方を満たす
ネガ型感光性樹脂組成物。
条件i:組成物が、前記樹脂、前記化合物A及び前記光重合開始剤とは異なる成分として含窒素環状化合物を更に含む。
条件ii:前記樹脂として、環員として2以上の窒素原子を含む含窒素ヘテロ環構造を有する樹脂を含む。 - 前記化合物Aが、露光によりラジカル重合開始種を発生しない化合物である、請求項1に記載のネガ型感光性樹脂組成物。
- 前記化合物Aが、ナフトキノンジアジド化合物を含む、請求項1又は2に記載のネガ型感光性樹脂組成物。
- 前記化合物Aの含有量を100質量部とした場合の、前記含窒素環状化合物の含有量が、0.5~200質量部である、請求項1~3のいずれか1項に記載のネガ型感光性樹脂組成物。
- 樹脂、
ナフトキノンジアジド化合物、及び、
光重合開始剤を含み、
下記条件iii及び条件ivの少なくとも一方を満たす
ネガ型感光性樹脂組成物。
条件iii:組成物が、前記樹脂、前記ナフトキノンジアジド化合物及び前記光重合開始とは異なる成分として含窒素環状化合物を更に含む。
条件iv:前記樹脂として、環員として2以上の窒素原子を含む含窒素ヘテロ環構造を有する樹脂を含む。 - 前記樹脂が環化樹脂又はその前駆体である、請求項1~5のいずれか1項に記載のネガ型感光性樹脂組成物。
- 重合性化合物を更に含む、請求項1~6のいずれか1項に記載のネガ型感光性樹脂組成物。
- シランカップリング剤を更に含む、請求項1~7のいずれか1項に記載のネガ型感光性樹脂組成物。
- 重合禁止剤を更に含む、請求項1~8のいずれか1項に記載のネガ型感光性樹脂組成物。
- 塩基発生剤を更に含む、請求項1~9のいずれか1項に記載のネガ型感光性樹脂組成物。
- 前記光重合開始剤が、光ラジカル重合開始剤である、請求項1~10のいずれか1項に記載のネガ型感光性樹脂組成物。
- 再配線層用層間絶縁膜の形成に用いられる、請求項1~11のいずれか1項に記載のネガ型感光性樹脂組成物。
- 請求項1~12のいずれか1項に記載のネガ型感光性樹脂組成物を硬化してなる硬化物。
- 請求項13に記載の硬化物からなる層を2層以上含み、前記硬化物からなる層同士のいずれかの間に金属層を含む積層体。
- 請求項1~12のいずれか1項に記載のネガ型感光性樹脂組成物を基板に適用して膜を形成する膜形成工程、前記膜を選択的に露光する露光工程、及び、露光工程により露光された膜を現像液を用いて現像してパターンを形成する現像工程を含む、硬化物の製造方法。
- 前記膜を50~450℃で加熱する加熱工程を含む、請求項15に記載の硬化物の製造方法。
- 請求項13に記載の硬化物又は請求項14に記載の積層体を含む、半導体デバイス。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202280023407.9A CN117099045A (zh) | 2021-03-22 | 2022-03-18 | 负型感光性树脂组合物、固化物、层叠体、固化物的制造方法及半导体器件 |
| EP22775435.5A EP4317287A1 (en) | 2021-03-22 | 2022-03-18 | Negative photosensitive resin composition, cured product, laminate, method for producing cured product, and semiconductor device |
| JP2023509118A JPWO2022202647A1 (ja) | 2021-03-22 | 2022-03-18 | |
| KR1020237032232A KR20230148224A (ko) | 2021-03-22 | 2022-03-18 | 네거티브형 감광성 수지 조성물, 경화물, 적층체, 경화물의 제조 방법, 및, 반도체 디바이스 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021046917 | 2021-03-22 | ||
| JP2021-046917 | 2021-03-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022202647A1 true WO2022202647A1 (ja) | 2022-09-29 |
Family
ID=83395794
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/012523 WO2022202647A1 (ja) | 2021-03-22 | 2022-03-18 | ネガ型感光性樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP4317287A1 (ja) |
| JP (1) | JPWO2022202647A1 (ja) |
| KR (1) | KR20230148224A (ja) |
| CN (1) | CN117099045A (ja) |
| TW (1) | TW202248755A (ja) |
| WO (1) | WO2022202647A1 (ja) |
Citations (119)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4643946B1 (ja) | 1967-11-09 | 1971-12-27 | ||
| JPS4864183A (ja) | 1971-12-09 | 1973-09-05 | ||
| JPS4841708B1 (ja) | 1970-01-13 | 1973-12-07 | ||
| JPS4943191B1 (ja) | 1969-07-11 | 1974-11-19 | ||
| JPS506034B1 (ja) | 1970-08-11 | 1975-03-10 | ||
| JPS5137193A (ja) | 1974-09-25 | 1976-03-29 | Toyo Boseki | |
| JPS5230490B2 (ja) | 1972-03-21 | 1977-08-09 | ||
| JPS5617654B2 (ja) | 1970-12-28 | 1981-04-23 | ||
| JPS5849860B2 (ja) | 1973-12-07 | 1983-11-07 | ヘキスト アクチェンゲゼルシャフト | コウジユウゴウセイフクシヤザイリヨウ |
| JPS6122048A (ja) | 1984-06-08 | 1986-01-30 | ヘキスト・アクチエンゲゼルシヤフト | 重合可能な化合物、その製法、およびこれを含有する放射線感性複写層 |
| JPS6239417B2 (ja) | 1978-05-20 | 1987-08-22 | Hoechst Ag | |
| JPS6239418B2 (ja) | 1978-05-20 | 1987-08-22 | Hoechst Ag | |
| JPS63260909A (ja) | 1987-03-28 | 1988-10-27 | ヘキスト・アクチエンゲゼルシヤフト | 光重合性混合物及びこの混合物から製造される記録材料 |
| JPS63277653A (ja) | 1987-03-28 | 1988-11-15 | ヘキスト・アクチエンゲゼルシヤフト | 重合可能な化合物、これを含有する放射線重合可能な混合物及び放射線重合可能な記録材料 |
| JPH01105238A (ja) | 1987-03-28 | 1989-04-21 | Hoechst Ag | 光重合可能な混合物および光重合可能な記録材料 |
| JPH0140337B2 (ja) | 1979-12-29 | 1989-08-28 | Hoechst Ag | |
| JPH0140336B2 (ja) | 1979-12-29 | 1989-08-28 | Hoechst Ag | |
| JPH0225493A (ja) | 1988-05-21 | 1990-01-26 | Hoechst Ag | アルケニルホスホン酸エステルおよびアルケニルホスフイン酸エルテル、その製法並びに当該化合物を含有する放射線重合性混合物および記録材料 |
| JPH0216765B2 (ja) | 1980-09-29 | 1990-04-18 | Hoechst Ag | |
| JPH0232293B2 (ja) | 1980-12-22 | 1990-07-19 | Hoechst Ag | |
| JPH04253058A (ja) | 1991-01-29 | 1992-09-08 | Fuji Photo Film Co Ltd | ポジ型フオトレジスト組成物 |
| EP0530148A1 (de) | 1991-08-30 | 1993-03-03 | OCG Microelectronic Materials Inc. | Positiv-Fotoresists mit erhöhtem Auflösungsvermögen und geringer Kristallisationsneigung, sowie neue Tetra(hydroxyphenyl)alkane |
| JPH05224410A (ja) | 1992-02-12 | 1993-09-03 | Fuji Photo Film Co Ltd | ポジ型フオトレジスト組成物 |
| JPH05303200A (ja) | 1992-04-27 | 1993-11-16 | Fuji Photo Film Co Ltd | ポジ型フオトレジスト組成物 |
| JPH06130664A (ja) | 1992-10-22 | 1994-05-13 | Sumitomo Bakelite Co Ltd | ネガ型感光性樹脂組成物 |
| JPH1062986A (ja) | 1996-08-21 | 1998-03-06 | Fuji Photo Film Co Ltd | 感放射線性着色組成物 |
| JPH10291969A (ja) | 1996-12-06 | 1998-11-04 | Ciba Specialty Chem Holding Inc | 新規α−アミノアセトフェノン光開始剤 |
| JP2000070025A (ja) | 1998-09-01 | 2000-03-07 | Seigo Nakamura | 傘 |
| JP2000080068A (ja) | 1998-06-26 | 2000-03-21 | Ciba Specialty Chem Holding Inc | 新規o―アシルオキシム光開始剤 |
| JP2001233842A (ja) | 1999-12-15 | 2001-08-28 | Ciba Specialty Chem Holding Inc | オキシムエステルの光開始剤 |
| JP2001521288A (ja) | 1997-10-20 | 2001-11-06 | フリップ・チップ・テクノロジーズ・エルエルシー | チップスケールパッケージ及びその形成方法 |
| JP2004091638A (ja) | 2002-08-30 | 2004-03-25 | Kingo Uchida | フォトクロミック材料 |
| JP2004101850A (ja) | 2002-09-09 | 2004-04-02 | Sumitomo Bakelite Co Ltd | 感光性有機無機複合材料およびそれを用いた半導体装置 |
| JP2004214501A (ja) | 2003-01-07 | 2004-07-29 | Sony Corp | ウエハーレベル・チップサイズ・パッケージおよびその製造方法 |
| JP2004254595A (ja) | 2003-02-26 | 2004-09-16 | Hiroshima Pref Gov | マンノシルエリスリトールリピッドの製造方法 |
| JP2006023696A (ja) | 2004-06-07 | 2006-01-26 | Fuji Photo Film Co Ltd | 着色感光性樹脂組成物、着色感光性樹脂組成物の塗布膜、感光性樹脂転写材料、感光性樹脂層の形成方法、カラーフィルター、カラーフィルターの製造方法、及び液晶表示装置。 |
| JP2006047592A (ja) | 2004-08-03 | 2006-02-16 | Fuji Photo Film Co Ltd | 遮光膜付基板、エレクトロルミネッセンス表示装置用遮光膜付基板、及び該遮光膜付基板を用いたエレクトロルミネッセンス表示装置 |
| JP2006251009A (ja) * | 2005-03-08 | 2006-09-21 | Chisso Corp | 感光性組成物およびそれを用いた表示素子 |
| JP2006342166A (ja) | 2001-06-11 | 2006-12-21 | Ciba Specialty Chem Holding Inc | 組み合わされた構造を有するオキシムエステルの光開始剤 |
| JP2007157879A (ja) | 2005-12-02 | 2007-06-21 | Sony Corp | 半導体装置及びその製造方法、並びに半導体ウェーハ |
| JP2007269779A (ja) | 2006-02-24 | 2007-10-18 | Fujifilm Corp | オキシム誘導体、光重合性組成物、カラーフィルタおよびその製造方法 |
| JP2008063554A (ja) | 2006-08-11 | 2008-03-21 | Fujifilm Corp | 分解性樹脂組成物、パターン形成材料およびパターン形成方法 |
| JP2008250074A (ja) | 2007-03-30 | 2008-10-16 | Fujifilm Corp | 感光性樹脂組成物、感光性フィルム、感光性積層体、永久パターン形成方法、及びプリント基板 |
| JP2008292970A (ja) | 2006-09-27 | 2008-12-04 | Fujifilm Corp | 化合物及びその互変異性体、金属錯体化合物、感光性着色硬化性組成物、カラーフィルタ、及びその製造方法 |
| JP4223071B2 (ja) | 2006-12-27 | 2009-02-12 | 株式会社Adeka | オキシムエステル化合物及び該化合物を含有する光重合開始剤 |
| JP4225898B2 (ja) | 2001-08-21 | 2009-02-18 | チバ ホールディング インコーポレーテッド | 深色モノ−及びビス−アシルホスフィンオキシド及びスルフィド並びに光開始剤としてのこれらの使用 |
| JP2009191061A (ja) | 2007-08-27 | 2009-08-27 | Fujifilm Corp | 新規化合物、光重合性組成物、カラーフィルタ用光重合性組成物、カラーフィルタ、及びその製造方法、固体撮像素子、並びに、平版印刷版原版 |
| JP2009191179A (ja) | 2008-02-15 | 2009-08-27 | Toyo Ink Mfg Co Ltd | 光重合開始剤、重合性組成物、および重合物の製造方法。 |
| JP4364216B2 (ja) | 2005-12-30 | 2009-11-11 | チェイル インダストリーズ インコーポレイテッド | 感光性樹脂組成物及びこれを用いたブラックマトリックス |
| JP2009283711A (ja) | 2008-05-22 | 2009-12-03 | Hitachi Chemical Dupont Microsystems Ltd | 半導体装置及びその製造方法、感光性樹脂組成物並びに電子部品 |
| JP2010129825A (ja) | 2008-11-28 | 2010-06-10 | Sumitomo Chemical Co Ltd | 有機エレクトロルミネッセンス素子およびその製造方法 |
| JP2010160418A (ja) | 2009-01-09 | 2010-07-22 | Hitachi Chem Co Ltd | 感光性樹脂組成物,並びにこれを用いた感光性エレメント,ソルダーレジスト及びプリント配線板 |
| JP2010164965A (ja) | 2008-12-19 | 2010-07-29 | Mitsubishi Chemicals Corp | カラーフィルタ画素形成用組成物、カラーフィルタ、液晶表示装置及び有機elディスプレイ |
| JP2010527339A (ja) | 2007-05-11 | 2010-08-12 | ビーエーエスエフ ソシエタス・ヨーロピア | オキシムエステル光重合開始剤 |
| JP2010262028A (ja) | 2009-04-30 | 2010-11-18 | Nippon Steel Chem Co Ltd | ブラックマトリックス用感光性樹脂組成物 |
| JP4600600B1 (ja) | 2009-06-17 | 2010-12-15 | 東洋インキ製造株式会社 | 新規オキシムエステル化合物およびそれを含んでなるラジカル重合開始剤および重合性組成物およびそれを用いたネガ型レジストおよびそれを用いた画像パターン形成方法 |
| US7888181B2 (en) | 2008-09-22 | 2011-02-15 | Stats Chippac, Ltd. | Method of forming a wafer level package with RDL interconnection over encapsulant between bump and semiconductor die |
| JP2011059656A (ja) | 2009-06-04 | 2011-03-24 | Asahi Kasei E-Materials Corp | ネガ型感光性樹脂組成物、硬化レリーフパターン形成・製造方法、並びに半導体装置 |
| JP2011089090A (ja) | 2009-10-26 | 2011-05-06 | Toray Ind Inc | ポリエステル樹脂組成物、その製造方法、およびフィルム |
| JP2011128358A (ja) | 2009-12-17 | 2011-06-30 | Hitachi Chemical Dupont Microsystems Ltd | ポジ型感光性樹脂組成物、それを用いた硬化膜及び電子部品 |
| WO2011080992A1 (ja) | 2009-12-28 | 2011-07-07 | 東レ株式会社 | ポジ型感光性樹脂組成物 |
| JP2011132503A (ja) | 2009-11-25 | 2011-07-07 | Sumitomo Chemical Co Ltd | 樹脂組成物及び表示装置 |
| JP2011524436A (ja) | 2008-06-06 | 2011-09-01 | ビーエーエスエフ ソシエタス・ヨーロピア | 光開始剤混合物 |
| JP2012003225A (ja) | 2010-01-27 | 2012-01-05 | Fujifilm Corp | ソルダーレジスト用重合性組成物及びソルダーレジストパターンの形成方法 |
| JP2012014052A (ja) | 2010-07-02 | 2012-01-19 | Fujifilm Corp | 着色感光性樹脂組成物、カラーフィルタ、カラーフィルタの製造方法、及び液晶表示装置 |
| JP2012194520A (ja) | 2010-08-05 | 2012-10-11 | Asahi Kasei E-Materials Corp | 感光性樹脂組成物、硬化レリーフパターンの製造方法及び半導体装置 |
| JP2013015701A (ja) | 2011-07-05 | 2013-01-24 | Hitachi Chemical Dupont Microsystems Ltd | 感光性樹脂組成物、該樹脂組成物を用いたパターン硬化膜の製造方法及び電子部品 |
| JP2013072935A (ja) | 2011-09-27 | 2013-04-22 | Toray Ind Inc | ポジ型感光性樹脂組成物 |
| JP2013114249A (ja) | 2011-12-01 | 2013-06-10 | Toppan Printing Co Ltd | 黒色感光性樹脂組成物およびカラーフィルタ |
| WO2013083505A1 (en) | 2011-12-07 | 2013-06-13 | Basf Se | Oxime ester photoinitiators |
| JP2013522445A (ja) | 2010-03-22 | 2013-06-13 | ヘンケル コーポレイション | マクロ光開始剤およびそれらの硬化性組成物 |
| JP2013164471A (ja) | 2012-02-09 | 2013-08-22 | Jsr Corp | 硬化性樹脂組成物、表示素子用硬化膜、表示素子用硬化膜の形成方法及び表示素子 |
| WO2013167515A1 (en) | 2012-05-09 | 2013-11-14 | Basf Se | Oxime ester photoinitiators |
| JP2013256506A (ja) | 2006-10-24 | 2013-12-26 | Sumitomo Bakelite Co Ltd | ビス(アミノフェノール)誘導体及びその製造方法、並びにポリアミド樹脂類、ポジ型感光性樹脂組成物、保護膜、層間絶縁膜、半導体装置及び表示素子 |
| JP2014500852A (ja) | 2010-10-05 | 2014-01-16 | ビーエーエスエフ ソシエタス・ヨーロピア | ベンゾカルバゾール化合物のオキシムエステル誘導体ならびに前記誘導体の光重合性の組成物における光開始剤としての使用 |
| WO2014021023A1 (ja) | 2012-07-31 | 2014-02-06 | 株式会社Adeka | 潜在性添加剤及び該添加剤を含有する組成物 |
| JP2014041264A (ja) | 2012-08-22 | 2014-03-06 | Sumitomo Bakelite Co Ltd | 感光性樹脂組成物、硬化膜、保護膜、絶縁膜、およびそれを用いた半導体装置、表示体装置 |
| WO2014097594A1 (ja) | 2012-12-21 | 2014-06-26 | 日立化成デュポンマイクロシステムズ株式会社 | ポリイミド前駆体樹脂組成物 |
| JP2014130173A (ja) | 2012-12-27 | 2014-07-10 | Fujifilm Corp | カラーフィルタ用組成物、赤外線透過フィルタ及びその製造方法、並びに赤外線センサー |
| JP2014137466A (ja) | 2013-01-16 | 2014-07-28 | Jsr Corp | 感放射線性着色組成物、着色硬化膜及び表示素子 |
| JP2014186186A (ja) | 2013-03-25 | 2014-10-02 | Toray Ind Inc | 耐熱性樹脂及びその前駆体組成物 |
| JP2014191002A (ja) | 2013-03-26 | 2014-10-06 | Toray Ind Inc | ポジ型感光性樹脂組成物、それを用いた硬化パターンの製造方法、それから得られるレリーフパターンおよびそれを有する発光素子 |
| JP2014191252A (ja) | 2013-03-28 | 2014-10-06 | Sumitomo Bakelite Co Ltd | 感光性樹脂組成物、硬化膜、保護膜、半導体装置および表示体装置 |
| WO2015004565A1 (en) | 2013-07-08 | 2015-01-15 | Basf Se | Oxime ester photoinitiators |
| JP2015034964A (ja) | 2013-02-28 | 2015-02-19 | 富士フイルム株式会社 | 透明樹脂層形成用組成物、透明樹脂層、固体撮像素子およびオプトエレクトロニクスデバイス |
| WO2015036910A1 (en) | 2013-09-10 | 2015-03-19 | Basf Se | Oxime ester photoinitiators |
| JP2015087611A (ja) | 2013-10-31 | 2015-05-07 | 富士フイルム株式会社 | 積層体、有機半導体製造用キットおよび有機半導体製造用レジスト組成物 |
| JP2015117327A (ja) | 2013-12-19 | 2015-06-25 | Dic株式会社 | 界面活性剤組成物、コーティング組成物及びレジスト組成物 |
| JP2015123351A (ja) | 2013-12-27 | 2015-07-06 | コダマ樹脂工業株式会社 | 耐薬品性吹込み成形積層容器 |
| JP2015127817A (ja) | 2009-04-14 | 2015-07-09 | 日立化成デュポンマイクロシステムズ株式会社 | 感光性樹脂組成物及びこれを用いた回路形成用基板 |
| WO2015125469A1 (ja) | 2014-02-19 | 2015-08-27 | 日立化成デュポンマイクロシステムズ株式会社 | 樹脂組成物、それによって形成される硬化膜及びパターン硬化膜、及びそれらの製造方法 |
| WO2015152153A1 (ja) | 2014-04-04 | 2015-10-08 | 株式会社Adeka | オキシムエステル化合物及び該化合物を含有する光重合開始剤 |
| US9159547B2 (en) | 2013-09-17 | 2015-10-13 | Deca Technologies Inc. | Two step method of rapid curing a semiconductor polymer layer |
| JP2015187211A (ja) | 2014-03-26 | 2015-10-29 | 富士フイルム株式会社 | 着色組成物、硬化膜、カラーフィルタ、パターン形成方法、カラーフィルタの製造方法、固体撮像素子、および、画像表示装置 |
| US9177926B2 (en) | 2011-12-30 | 2015-11-03 | Deca Technologies Inc | Semiconductor device and method comprising thickened redistribution layers |
| WO2015199219A1 (ja) | 2014-06-27 | 2015-12-30 | 富士フイルム株式会社 | 熱塩基発生剤、熱硬化性樹脂組成物、硬化膜、硬化膜の製造方法および半導体デバイス |
| JP2016027357A (ja) | 2014-03-27 | 2016-02-18 | 富士フイルム株式会社 | 感光性樹脂組成物、硬化膜、硬化膜の製造方法および半導体デバイス |
| WO2016034963A1 (en) | 2014-09-04 | 2016-03-10 | Basf Se | Polycyclic photoinitiators |
| JP2017008219A (ja) | 2015-06-23 | 2017-01-12 | 株式会社Adeka | 組成物 |
| WO2017030005A1 (ja) | 2015-08-17 | 2017-02-23 | 株式会社Adeka | 組成物 |
| WO2017033680A1 (ja) | 2015-08-26 | 2017-03-02 | パナソニックヘルスケアホールディングス株式会社 | 超低温フリーザ |
| WO2017038598A1 (ja) | 2015-08-28 | 2017-03-09 | 富士フイルム株式会社 | 硬化膜の製造方法、再配線層用層間絶縁膜の製造方法、および、半導体デバイスの製造方法 |
| JP2017523465A (ja) | 2014-07-15 | 2017-08-17 | 常州強力電子新材料股▲ふん▼有限公司Cahngzhou Tronly New Electronic Materials Co.,Ltd. | オキシムエステル類光開始剤含有感光性組成物及びその使用 |
| JP2017151342A (ja) | 2016-02-26 | 2017-08-31 | 東洋インキScホールディングス株式会社 | 感光性着色組成物およびカラーフィルタ |
| JP2017167399A (ja) | 2016-03-17 | 2017-09-21 | 株式会社Dnpファインケミカル | カラーフィルタ用感光性着色樹脂組成物、カラーフィルタ、表示装置 |
| WO2017164127A1 (ja) | 2016-03-25 | 2017-09-28 | 東レ株式会社 | 着色樹脂組成物、カラーフィルタ基板、および液晶表示装置 |
| JP2017198865A (ja) | 2016-04-27 | 2017-11-02 | 東京応化工業株式会社 | 感光性組成物 |
| WO2018038002A1 (ja) | 2016-08-25 | 2018-03-01 | 富士フイルム株式会社 | 積層体の製造方法および電子デバイスの製造方法 |
| JP6301489B2 (ja) | 2014-03-18 | 2018-03-28 | 常州強力電子新材料股▲ふん▼有限公司Cahngzhou Tronly New Electronic Materials Co.,Ltd. | ニトロ基含有ビスオキシムエステル系光重合開始剤及びその合成製造方法と応用 |
| WO2018110179A1 (ja) | 2016-12-13 | 2018-06-21 | 日油株式会社 | ペルオキシシンナメート誘導体、該化合物を含有する重合性組成物 |
| JP2018173573A (ja) | 2017-03-31 | 2018-11-08 | 日立化成デュポンマイクロシステムズ株式会社 | 感光性樹脂組成物、パターン硬化物の製造方法、硬化物、層間絶縁膜、カバーコート層、表面保護膜、及び電子部品 |
| WO2018221177A1 (ja) | 2017-06-01 | 2018-12-06 | 日油株式会社 | トリアジンペルオキシド誘導体、該化合物を含有する重合性組成物 |
| JP2019044030A (ja) | 2017-08-31 | 2019-03-22 | 学校法人東京理科大学 | ペルオキシエステル基を有するチオキサントン誘導体を含有する重合性組成物およびその硬化物、当該硬化物の製造方法 |
| JP2019043864A (ja) | 2017-08-31 | 2019-03-22 | 学校法人東京理科大学 | ペルオキシエステル基を有するベンゾフェノン誘導体、該化合物を含有する重合性組成物およびその硬化物、当該硬化物の製造方法 |
| WO2019088055A1 (ja) | 2017-10-30 | 2019-05-09 | 株式会社Adeka | 化合物、組成物、硬化物及び硬化物の製造方法 |
| JP2019167313A (ja) | 2018-03-26 | 2019-10-03 | 日油株式会社 | ペルオキシシンナメート誘導体、該化合物を含有する重合性組成物およびその硬化物、並びに当該硬化物の製造方法 |
| JP2019206689A (ja) | 2018-05-29 | 2019-12-05 | 旭化成株式会社 | 樹脂組成物、及び硬化膜の製造方法 |
| WO2020066416A1 (ja) | 2018-09-28 | 2020-04-02 | 富士フイルム株式会社 | 感光性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、および半導体デバイス |
| WO2021039782A1 (ja) * | 2019-08-26 | 2021-03-04 | 富士フイルム株式会社 | 硬化性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、半導体デバイス、樹脂、及び、樹脂の製造方法 |
-
2022
- 2022-03-17 TW TW111109738A patent/TW202248755A/zh unknown
- 2022-03-18 EP EP22775435.5A patent/EP4317287A1/en active Pending
- 2022-03-18 CN CN202280023407.9A patent/CN117099045A/zh active Pending
- 2022-03-18 KR KR1020237032232A patent/KR20230148224A/ko unknown
- 2022-03-18 JP JP2023509118A patent/JPWO2022202647A1/ja active Pending
- 2022-03-18 WO PCT/JP2022/012523 patent/WO2022202647A1/ja active Application Filing
Patent Citations (124)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4643946B1 (ja) | 1967-11-09 | 1971-12-27 | ||
| JPS4943191B1 (ja) | 1969-07-11 | 1974-11-19 | ||
| JPS4841708B1 (ja) | 1970-01-13 | 1973-12-07 | ||
| JPS506034B1 (ja) | 1970-08-11 | 1975-03-10 | ||
| JPS5617654B2 (ja) | 1970-12-28 | 1981-04-23 | ||
| JPS4864183A (ja) | 1971-12-09 | 1973-09-05 | ||
| JPS5230490B2 (ja) | 1972-03-21 | 1977-08-09 | ||
| JPS5849860B2 (ja) | 1973-12-07 | 1983-11-07 | ヘキスト アクチェンゲゼルシャフト | コウジユウゴウセイフクシヤザイリヨウ |
| JPS5137193A (ja) | 1974-09-25 | 1976-03-29 | Toyo Boseki | |
| JPS6239417B2 (ja) | 1978-05-20 | 1987-08-22 | Hoechst Ag | |
| JPS6239418B2 (ja) | 1978-05-20 | 1987-08-22 | Hoechst Ag | |
| JPH0140336B2 (ja) | 1979-12-29 | 1989-08-28 | Hoechst Ag | |
| JPH0140337B2 (ja) | 1979-12-29 | 1989-08-28 | Hoechst Ag | |
| JPH0216765B2 (ja) | 1980-09-29 | 1990-04-18 | Hoechst Ag | |
| JPH0232293B2 (ja) | 1980-12-22 | 1990-07-19 | Hoechst Ag | |
| JPS6122048A (ja) | 1984-06-08 | 1986-01-30 | ヘキスト・アクチエンゲゼルシヤフト | 重合可能な化合物、その製法、およびこれを含有する放射線感性複写層 |
| JPH01105238A (ja) | 1987-03-28 | 1989-04-21 | Hoechst Ag | 光重合可能な混合物および光重合可能な記録材料 |
| JPS63260909A (ja) | 1987-03-28 | 1988-10-27 | ヘキスト・アクチエンゲゼルシヤフト | 光重合性混合物及びこの混合物から製造される記録材料 |
| JPS63277653A (ja) | 1987-03-28 | 1988-11-15 | ヘキスト・アクチエンゲゼルシヤフト | 重合可能な化合物、これを含有する放射線重合可能な混合物及び放射線重合可能な記録材料 |
| JPH0225493A (ja) | 1988-05-21 | 1990-01-26 | Hoechst Ag | アルケニルホスホン酸エステルおよびアルケニルホスフイン酸エルテル、その製法並びに当該化合物を含有する放射線重合性混合物および記録材料 |
| JPH04253058A (ja) | 1991-01-29 | 1992-09-08 | Fuji Photo Film Co Ltd | ポジ型フオトレジスト組成物 |
| EP0530148A1 (de) | 1991-08-30 | 1993-03-03 | OCG Microelectronic Materials Inc. | Positiv-Fotoresists mit erhöhtem Auflösungsvermögen und geringer Kristallisationsneigung, sowie neue Tetra(hydroxyphenyl)alkane |
| JPH05224410A (ja) | 1992-02-12 | 1993-09-03 | Fuji Photo Film Co Ltd | ポジ型フオトレジスト組成物 |
| JPH05303200A (ja) | 1992-04-27 | 1993-11-16 | Fuji Photo Film Co Ltd | ポジ型フオトレジスト組成物 |
| JPH06130664A (ja) | 1992-10-22 | 1994-05-13 | Sumitomo Bakelite Co Ltd | ネガ型感光性樹脂組成物 |
| JPH1062986A (ja) | 1996-08-21 | 1998-03-06 | Fuji Photo Film Co Ltd | 感放射線性着色組成物 |
| JPH10291969A (ja) | 1996-12-06 | 1998-11-04 | Ciba Specialty Chem Holding Inc | 新規α−アミノアセトフェノン光開始剤 |
| JP2001521288A (ja) | 1997-10-20 | 2001-11-06 | フリップ・チップ・テクノロジーズ・エルエルシー | チップスケールパッケージ及びその形成方法 |
| JP2000080068A (ja) | 1998-06-26 | 2000-03-21 | Ciba Specialty Chem Holding Inc | 新規o―アシルオキシム光開始剤 |
| JP2000070025A (ja) | 1998-09-01 | 2000-03-07 | Seigo Nakamura | 傘 |
| JP2001233842A (ja) | 1999-12-15 | 2001-08-28 | Ciba Specialty Chem Holding Inc | オキシムエステルの光開始剤 |
| JP2006342166A (ja) | 2001-06-11 | 2006-12-21 | Ciba Specialty Chem Holding Inc | 組み合わされた構造を有するオキシムエステルの光開始剤 |
| JP4225898B2 (ja) | 2001-08-21 | 2009-02-18 | チバ ホールディング インコーポレーテッド | 深色モノ−及びビス−アシルホスフィンオキシド及びスルフィド並びに光開始剤としてのこれらの使用 |
| JP2004091638A (ja) | 2002-08-30 | 2004-03-25 | Kingo Uchida | フォトクロミック材料 |
| JP2004101850A (ja) | 2002-09-09 | 2004-04-02 | Sumitomo Bakelite Co Ltd | 感光性有機無機複合材料およびそれを用いた半導体装置 |
| JP2004214501A (ja) | 2003-01-07 | 2004-07-29 | Sony Corp | ウエハーレベル・チップサイズ・パッケージおよびその製造方法 |
| JP2004254595A (ja) | 2003-02-26 | 2004-09-16 | Hiroshima Pref Gov | マンノシルエリスリトールリピッドの製造方法 |
| JP2006023696A (ja) | 2004-06-07 | 2006-01-26 | Fuji Photo Film Co Ltd | 着色感光性樹脂組成物、着色感光性樹脂組成物の塗布膜、感光性樹脂転写材料、感光性樹脂層の形成方法、カラーフィルター、カラーフィルターの製造方法、及び液晶表示装置。 |
| JP2006047592A (ja) | 2004-08-03 | 2006-02-16 | Fuji Photo Film Co Ltd | 遮光膜付基板、エレクトロルミネッセンス表示装置用遮光膜付基板、及び該遮光膜付基板を用いたエレクトロルミネッセンス表示装置 |
| JP2006251009A (ja) * | 2005-03-08 | 2006-09-21 | Chisso Corp | 感光性組成物およびそれを用いた表示素子 |
| JP2007157879A (ja) | 2005-12-02 | 2007-06-21 | Sony Corp | 半導体装置及びその製造方法、並びに半導体ウェーハ |
| JP4364216B2 (ja) | 2005-12-30 | 2009-11-11 | チェイル インダストリーズ インコーポレイテッド | 感光性樹脂組成物及びこれを用いたブラックマトリックス |
| JP2007269779A (ja) | 2006-02-24 | 2007-10-18 | Fujifilm Corp | オキシム誘導体、光重合性組成物、カラーフィルタおよびその製造方法 |
| JP2008063554A (ja) | 2006-08-11 | 2008-03-21 | Fujifilm Corp | 分解性樹脂組成物、パターン形成材料およびパターン形成方法 |
| JP2008292970A (ja) | 2006-09-27 | 2008-12-04 | Fujifilm Corp | 化合物及びその互変異性体、金属錯体化合物、感光性着色硬化性組成物、カラーフィルタ、及びその製造方法 |
| JP2013256506A (ja) | 2006-10-24 | 2013-12-26 | Sumitomo Bakelite Co Ltd | ビス(アミノフェノール)誘導体及びその製造方法、並びにポリアミド樹脂類、ポジ型感光性樹脂組成物、保護膜、層間絶縁膜、半導体装置及び表示素子 |
| JP4223071B2 (ja) | 2006-12-27 | 2009-02-12 | 株式会社Adeka | オキシムエステル化合物及び該化合物を含有する光重合開始剤 |
| JP2008250074A (ja) | 2007-03-30 | 2008-10-16 | Fujifilm Corp | 感光性樹脂組成物、感光性フィルム、感光性積層体、永久パターン形成方法、及びプリント基板 |
| JP2010527339A (ja) | 2007-05-11 | 2010-08-12 | ビーエーエスエフ ソシエタス・ヨーロピア | オキシムエステル光重合開始剤 |
| JP2009191061A (ja) | 2007-08-27 | 2009-08-27 | Fujifilm Corp | 新規化合物、光重合性組成物、カラーフィルタ用光重合性組成物、カラーフィルタ、及びその製造方法、固体撮像素子、並びに、平版印刷版原版 |
| JP2009191179A (ja) | 2008-02-15 | 2009-08-27 | Toyo Ink Mfg Co Ltd | 光重合開始剤、重合性組成物、および重合物の製造方法。 |
| JP2009283711A (ja) | 2008-05-22 | 2009-12-03 | Hitachi Chemical Dupont Microsystems Ltd | 半導体装置及びその製造方法、感光性樹脂組成物並びに電子部品 |
| JP2011524436A (ja) | 2008-06-06 | 2011-09-01 | ビーエーエスエフ ソシエタス・ヨーロピア | 光開始剤混合物 |
| US7888181B2 (en) | 2008-09-22 | 2011-02-15 | Stats Chippac, Ltd. | Method of forming a wafer level package with RDL interconnection over encapsulant between bump and semiconductor die |
| JP2010129825A (ja) | 2008-11-28 | 2010-06-10 | Sumitomo Chemical Co Ltd | 有機エレクトロルミネッセンス素子およびその製造方法 |
| JP2010164965A (ja) | 2008-12-19 | 2010-07-29 | Mitsubishi Chemicals Corp | カラーフィルタ画素形成用組成物、カラーフィルタ、液晶表示装置及び有機elディスプレイ |
| JP2010160418A (ja) | 2009-01-09 | 2010-07-22 | Hitachi Chem Co Ltd | 感光性樹脂組成物,並びにこれを用いた感光性エレメント,ソルダーレジスト及びプリント配線板 |
| JP2015127817A (ja) | 2009-04-14 | 2015-07-09 | 日立化成デュポンマイクロシステムズ株式会社 | 感光性樹脂組成物及びこれを用いた回路形成用基板 |
| JP2010262028A (ja) | 2009-04-30 | 2010-11-18 | Nippon Steel Chem Co Ltd | ブラックマトリックス用感光性樹脂組成物 |
| JP2011059656A (ja) | 2009-06-04 | 2011-03-24 | Asahi Kasei E-Materials Corp | ネガ型感光性樹脂組成物、硬化レリーフパターン形成・製造方法、並びに半導体装置 |
| JP4600600B1 (ja) | 2009-06-17 | 2010-12-15 | 東洋インキ製造株式会社 | 新規オキシムエステル化合物およびそれを含んでなるラジカル重合開始剤および重合性組成物およびそれを用いたネガ型レジストおよびそれを用いた画像パターン形成方法 |
| JP2011089090A (ja) | 2009-10-26 | 2011-05-06 | Toray Ind Inc | ポリエステル樹脂組成物、その製造方法、およびフィルム |
| JP2011132503A (ja) | 2009-11-25 | 2011-07-07 | Sumitomo Chemical Co Ltd | 樹脂組成物及び表示装置 |
| JP2011128358A (ja) | 2009-12-17 | 2011-06-30 | Hitachi Chemical Dupont Microsystems Ltd | ポジ型感光性樹脂組成物、それを用いた硬化膜及び電子部品 |
| WO2011080992A1 (ja) | 2009-12-28 | 2011-07-07 | 東レ株式会社 | ポジ型感光性樹脂組成物 |
| JP2012003225A (ja) | 2010-01-27 | 2012-01-05 | Fujifilm Corp | ソルダーレジスト用重合性組成物及びソルダーレジストパターンの形成方法 |
| US20130034812A1 (en) | 2010-01-27 | 2013-02-07 | Fujifilm Corporation | Polymerizable composition for solder resist, and solder resist pattern formation method |
| JP2013522445A (ja) | 2010-03-22 | 2013-06-13 | ヘンケル コーポレイション | マクロ光開始剤およびそれらの硬化性組成物 |
| JP2012014052A (ja) | 2010-07-02 | 2012-01-19 | Fujifilm Corp | 着色感光性樹脂組成物、カラーフィルタ、カラーフィルタの製造方法、及び液晶表示装置 |
| JP2012194520A (ja) | 2010-08-05 | 2012-10-11 | Asahi Kasei E-Materials Corp | 感光性樹脂組成物、硬化レリーフパターンの製造方法及び半導体装置 |
| JP2014500852A (ja) | 2010-10-05 | 2014-01-16 | ビーエーエスエフ ソシエタス・ヨーロピア | ベンゾカルバゾール化合物のオキシムエステル誘導体ならびに前記誘導体の光重合性の組成物における光開始剤としての使用 |
| JP2013015701A (ja) | 2011-07-05 | 2013-01-24 | Hitachi Chemical Dupont Microsystems Ltd | 感光性樹脂組成物、該樹脂組成物を用いたパターン硬化膜の製造方法及び電子部品 |
| JP2013072935A (ja) | 2011-09-27 | 2013-04-22 | Toray Ind Inc | ポジ型感光性樹脂組成物 |
| JP2013114249A (ja) | 2011-12-01 | 2013-06-10 | Toppan Printing Co Ltd | 黒色感光性樹脂組成物およびカラーフィルタ |
| WO2013083505A1 (en) | 2011-12-07 | 2013-06-13 | Basf Se | Oxime ester photoinitiators |
| US9177926B2 (en) | 2011-12-30 | 2015-11-03 | Deca Technologies Inc | Semiconductor device and method comprising thickened redistribution layers |
| JP2013164471A (ja) | 2012-02-09 | 2013-08-22 | Jsr Corp | 硬化性樹脂組成物、表示素子用硬化膜、表示素子用硬化膜の形成方法及び表示素子 |
| JP2017019766A (ja) | 2012-05-09 | 2017-01-26 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | オキシムエステル光開始剤 |
| WO2013167515A1 (en) | 2012-05-09 | 2013-11-14 | Basf Se | Oxime ester photoinitiators |
| WO2014021023A1 (ja) | 2012-07-31 | 2014-02-06 | 株式会社Adeka | 潜在性添加剤及び該添加剤を含有する組成物 |
| JP2014041264A (ja) | 2012-08-22 | 2014-03-06 | Sumitomo Bakelite Co Ltd | 感光性樹脂組成物、硬化膜、保護膜、絶縁膜、およびそれを用いた半導体装置、表示体装置 |
| WO2014097594A1 (ja) | 2012-12-21 | 2014-06-26 | 日立化成デュポンマイクロシステムズ株式会社 | ポリイミド前駆体樹脂組成物 |
| JP2014130173A (ja) | 2012-12-27 | 2014-07-10 | Fujifilm Corp | カラーフィルタ用組成物、赤外線透過フィルタ及びその製造方法、並びに赤外線センサー |
| JP2014137466A (ja) | 2013-01-16 | 2014-07-28 | Jsr Corp | 感放射線性着色組成物、着色硬化膜及び表示素子 |
| JP6065596B2 (ja) | 2013-01-16 | 2017-01-25 | Jsr株式会社 | 感放射線性着色組成物、着色硬化膜及び表示素子 |
| JP2015034964A (ja) | 2013-02-28 | 2015-02-19 | 富士フイルム株式会社 | 透明樹脂層形成用組成物、透明樹脂層、固体撮像素子およびオプトエレクトロニクスデバイス |
| JP2014186186A (ja) | 2013-03-25 | 2014-10-02 | Toray Ind Inc | 耐熱性樹脂及びその前駆体組成物 |
| JP2014191002A (ja) | 2013-03-26 | 2014-10-06 | Toray Ind Inc | ポジ型感光性樹脂組成物、それを用いた硬化パターンの製造方法、それから得られるレリーフパターンおよびそれを有する発光素子 |
| JP2014191252A (ja) | 2013-03-28 | 2014-10-06 | Sumitomo Bakelite Co Ltd | 感光性樹脂組成物、硬化膜、保護膜、半導体装置および表示体装置 |
| JP6469669B2 (ja) | 2013-07-08 | 2019-02-13 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | オキシムエステル光開始剤 |
| JP2016532675A (ja) | 2013-07-08 | 2016-10-20 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | オキシムエステル光開始剤 |
| WO2015004565A1 (en) | 2013-07-08 | 2015-01-15 | Basf Se | Oxime ester photoinitiators |
| WO2015036910A1 (en) | 2013-09-10 | 2015-03-19 | Basf Se | Oxime ester photoinitiators |
| US9159547B2 (en) | 2013-09-17 | 2015-10-13 | Deca Technologies Inc. | Two step method of rapid curing a semiconductor polymer layer |
| JP2015087611A (ja) | 2013-10-31 | 2015-05-07 | 富士フイルム株式会社 | 積層体、有機半導体製造用キットおよび有機半導体製造用レジスト組成物 |
| JP2015117327A (ja) | 2013-12-19 | 2015-06-25 | Dic株式会社 | 界面活性剤組成物、コーティング組成物及びレジスト組成物 |
| JP2015123351A (ja) | 2013-12-27 | 2015-07-06 | コダマ樹脂工業株式会社 | 耐薬品性吹込み成形積層容器 |
| WO2015125469A1 (ja) | 2014-02-19 | 2015-08-27 | 日立化成デュポンマイクロシステムズ株式会社 | 樹脂組成物、それによって形成される硬化膜及びパターン硬化膜、及びそれらの製造方法 |
| JP6301489B2 (ja) | 2014-03-18 | 2018-03-28 | 常州強力電子新材料股▲ふん▼有限公司Cahngzhou Tronly New Electronic Materials Co.,Ltd. | ニトロ基含有ビスオキシムエステル系光重合開始剤及びその合成製造方法と応用 |
| JP2015187211A (ja) | 2014-03-26 | 2015-10-29 | 富士フイルム株式会社 | 着色組成物、硬化膜、カラーフィルタ、パターン形成方法、カラーフィルタの製造方法、固体撮像素子、および、画像表示装置 |
| JP2016027357A (ja) | 2014-03-27 | 2016-02-18 | 富士フイルム株式会社 | 感光性樹脂組成物、硬化膜、硬化膜の製造方法および半導体デバイス |
| WO2015152153A1 (ja) | 2014-04-04 | 2015-10-08 | 株式会社Adeka | オキシムエステル化合物及び該化合物を含有する光重合開始剤 |
| WO2015199219A1 (ja) | 2014-06-27 | 2015-12-30 | 富士フイルム株式会社 | 熱塩基発生剤、熱硬化性樹脂組成物、硬化膜、硬化膜の製造方法および半導体デバイス |
| JP2017523465A (ja) | 2014-07-15 | 2017-08-17 | 常州強力電子新材料股▲ふん▼有限公司Cahngzhou Tronly New Electronic Materials Co.,Ltd. | オキシムエステル類光開始剤含有感光性組成物及びその使用 |
| WO2016034963A1 (en) | 2014-09-04 | 2016-03-10 | Basf Se | Polycyclic photoinitiators |
| JP2017008219A (ja) | 2015-06-23 | 2017-01-12 | 株式会社Adeka | 組成物 |
| WO2017030005A1 (ja) | 2015-08-17 | 2017-02-23 | 株式会社Adeka | 組成物 |
| WO2017033680A1 (ja) | 2015-08-26 | 2017-03-02 | パナソニックヘルスケアホールディングス株式会社 | 超低温フリーザ |
| WO2017038598A1 (ja) | 2015-08-28 | 2017-03-09 | 富士フイルム株式会社 | 硬化膜の製造方法、再配線層用層間絶縁膜の製造方法、および、半導体デバイスの製造方法 |
| JP2017151342A (ja) | 2016-02-26 | 2017-08-31 | 東洋インキScホールディングス株式会社 | 感光性着色組成物およびカラーフィルタ |
| JP2017167399A (ja) | 2016-03-17 | 2017-09-21 | 株式会社Dnpファインケミカル | カラーフィルタ用感光性着色樹脂組成物、カラーフィルタ、表示装置 |
| WO2017164127A1 (ja) | 2016-03-25 | 2017-09-28 | 東レ株式会社 | 着色樹脂組成物、カラーフィルタ基板、および液晶表示装置 |
| JP2017198865A (ja) | 2016-04-27 | 2017-11-02 | 東京応化工業株式会社 | 感光性組成物 |
| WO2018038002A1 (ja) | 2016-08-25 | 2018-03-01 | 富士フイルム株式会社 | 積層体の製造方法および電子デバイスの製造方法 |
| WO2018110179A1 (ja) | 2016-12-13 | 2018-06-21 | 日油株式会社 | ペルオキシシンナメート誘導体、該化合物を含有する重合性組成物 |
| JP2018173573A (ja) | 2017-03-31 | 2018-11-08 | 日立化成デュポンマイクロシステムズ株式会社 | 感光性樹脂組成物、パターン硬化物の製造方法、硬化物、層間絶縁膜、カバーコート層、表面保護膜、及び電子部品 |
| WO2018221177A1 (ja) | 2017-06-01 | 2018-12-06 | 日油株式会社 | トリアジンペルオキシド誘導体、該化合物を含有する重合性組成物 |
| JP2019044030A (ja) | 2017-08-31 | 2019-03-22 | 学校法人東京理科大学 | ペルオキシエステル基を有するチオキサントン誘導体を含有する重合性組成物およびその硬化物、当該硬化物の製造方法 |
| JP2019043864A (ja) | 2017-08-31 | 2019-03-22 | 学校法人東京理科大学 | ペルオキシエステル基を有するベンゾフェノン誘導体、該化合物を含有する重合性組成物およびその硬化物、当該硬化物の製造方法 |
| WO2019088055A1 (ja) | 2017-10-30 | 2019-05-09 | 株式会社Adeka | 化合物、組成物、硬化物及び硬化物の製造方法 |
| JP2019167313A (ja) | 2018-03-26 | 2019-10-03 | 日油株式会社 | ペルオキシシンナメート誘導体、該化合物を含有する重合性組成物およびその硬化物、並びに当該硬化物の製造方法 |
| JP2019206689A (ja) | 2018-05-29 | 2019-12-05 | 旭化成株式会社 | 樹脂組成物、及び硬化膜の製造方法 |
| WO2020066416A1 (ja) | 2018-09-28 | 2020-04-02 | 富士フイルム株式会社 | 感光性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、および半導体デバイス |
| WO2021039782A1 (ja) * | 2019-08-26 | 2021-03-04 | 富士フイルム株式会社 | 硬化性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、半導体デバイス、樹脂、及び、樹脂の製造方法 |
Non-Patent Citations (8)
| Title |
|---|
| "Basics and development of polyimide materials", November 2011, CMC TECHNICAL LIBRARY |
| "High functionality and applied technology of polyimide", April 2008, SCIENCE & TECHNOLOGY CO. |
| "Latest Polyimide Basics and Applications", August 2010, NTS INC. |
| "The Society of Polymer Science", 2005, article "Polymer Dictionary", pages: 683 - 684 |
| H SEINO ET AL., J. POLYM. SCI., PART A: POLYM. CHEM., vol. 36, 1998, pages 2261 |
| J. C. S. PERKIN II, 1979, pages 1653 - 1660 |
| JOURNAL OF PHOTOPOLYMER SCIENCE AND TECHNOLOGY, 1995, pages 202 - 232 |
| JOURNAL OF THE ADHESION SOCIETY OF JAPAN, vol. 20, no. 7, 1984, pages 300 - 308 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN117099045A (zh) | 2023-11-21 |
| TW202248755A (zh) | 2022-12-16 |
| JPWO2022202647A1 (ja) | 2022-09-29 |
| KR20230148224A (ko) | 2023-10-24 |
| EP4317287A1 (en) | 2024-02-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7259141B1 (ja) | 硬化物の製造方法、積層体の製造方法、及び、半導体デバイスの製造方法、並びに、処理液 | |
| WO2022070730A1 (ja) | 硬化物の製造方法、積層体の製造方法、及び、半導体デバイスの製造方法 | |
| WO2022145356A1 (ja) | 樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス | |
| WO2022045060A1 (ja) | 硬化性樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス | |
| WO2022045124A1 (ja) | 硬化性樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス | |
| WO2022210532A1 (ja) | 樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス、並びに、ポリイミド前駆体及びその製造方法 | |
| WO2022145355A1 (ja) | 樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス | |
| JP7367053B2 (ja) | パターン形成方法、光硬化性樹脂組成物、積層体の製造方法、及び、電子デバイスの製造方法 | |
| WO2022145136A1 (ja) | 樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス、並びに、化合物 | |
| WO2022050041A1 (ja) | 硬化物の製造方法、積層体の製造方法、及び、電子デバイスの製造方法 | |
| WO2022050278A1 (ja) | 硬化性樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス、並びに、光塩基発生剤 | |
| WO2021085072A1 (ja) | パターン形成方法、感光性樹脂組成物、積層体の製造方法、及び、半導体デバイスの製造方法 | |
| WO2022202647A1 (ja) | ネガ型感光性樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス | |
| WO2023032475A1 (ja) | 硬化物の製造方法、積層体の製造方法、及び、半導体デバイスの製造方法、並びに、処理液、及び、樹脂組成物 | |
| WO2022176869A1 (ja) | 永久膜の製造方法、積層体の製造方法、及び、半導体デバイスの製造方法 | |
| JP7425088B2 (ja) | 硬化性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、半導体デバイス、及び、樹脂 | |
| WO2023032821A1 (ja) | 樹脂組成物、硬化物、積層体、硬化物の製造方法、積層体の製造方法、半導体デバイスの製造方法、及び、半導体デバイス | |
| WO2022210226A1 (ja) | 樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス、並びに、化合物 | |
| WO2022210225A1 (ja) | 樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス | |
| WO2023032820A1 (ja) | 樹脂組成物、硬化物、積層体、硬化物の製造方法、積層体の製造方法、半導体デバイスの製造方法、及び、半導体デバイス、並びに、化合物 | |
| WO2022210324A1 (ja) | 樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス、並びに、塩基発生剤 | |
| WO2022172996A1 (ja) | 樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス、並びに、塩基発生剤 | |
| WO2023026892A1 (ja) | 樹脂組成物、硬化物、積層体、硬化物の製造方法、積層体の製造方法、半導体デバイスの製造方法、及び、半導体デバイス、並びに、塩基発生剤 | |
| WO2022224838A1 (ja) | 樹脂組成物、硬化物、積層体、硬化物の製造方法、半導体デバイス、及び、樹脂 | |
| WO2022210466A1 (ja) | 樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス、並びに、ポリイミド前駆体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22775435 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023509118 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 20237032232 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020237032232 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202280023407.9 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022775435 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2022775435 Country of ref document: EP Effective date: 20231023 |