WO2020209239A1 - 前駆体、前駆体の製造方法、正極材、正極材の製造方法、および、リチウムイオン二次電池 - Google Patents
前駆体、前駆体の製造方法、正極材、正極材の製造方法、および、リチウムイオン二次電池 Download PDFInfo
- Publication number
- WO2020209239A1 WO2020209239A1 PCT/JP2020/015606 JP2020015606W WO2020209239A1 WO 2020209239 A1 WO2020209239 A1 WO 2020209239A1 JP 2020015606 W JP2020015606 W JP 2020015606W WO 2020209239 A1 WO2020209239 A1 WO 2020209239A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- positive electrode
- electrode material
- precursor
- nickel
- manganese
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G53/00—Compounds of nickel
- C01G53/006—Compounds containing, besides nickel, two or more other elements, with the exception of oxygen or hydrogen
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G53/00—Compounds of nickel
- C01G53/40—Nickelates
- C01G53/42—Nickelates containing alkali metals, e.g. LiNiO2
- C01G53/44—Nickelates containing alkali metals, e.g. LiNiO2 containing manganese
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G53/00—Compounds of nickel
- C01G53/40—Nickelates
- C01G53/42—Nickelates containing alkali metals, e.g. LiNiO2
- C01G53/44—Nickelates containing alkali metals, e.g. LiNiO2 containing manganese
- C01G53/50—Nickelates containing alkali metals, e.g. LiNiO2 containing manganese of the type [MnO2]n-, e.g. Li(NixMn1-x)O2, Li(MyNixMn1-x-y)O2
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
- H01M10/0525—Rocking-chair batteries, i.e. batteries with lithium insertion or intercalation in both electrodes; Lithium-ion batteries
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/04—Processes of manufacture in general
- H01M4/0471—Processes of manufacture in general involving thermal treatment, e.g. firing, sintering, backing particulate active material, thermal decomposition, pyrolysis
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/50—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese
- H01M4/505—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese of mixed oxides or hydroxides containing manganese for inserting or intercalating light metals, e.g. LiMn2O4 or LiMn2OxFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/52—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron
- H01M4/525—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron of mixed oxides or hydroxides containing iron, cobalt or nickel for inserting or intercalating light metals, e.g. LiNiO2, LiCoO2 or LiCoOxFy
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/70—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data
- C01P2002/72—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data by d-values or two theta-values, e.g. as X-ray diagram
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/70—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data
- C01P2002/74—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data by peak-intensities or a ratio thereof only
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/80—Crystal-structural characteristics defined by measured data other than those specified in group C01P2002/70
- C01P2002/85—Crystal-structural characteristics defined by measured data other than those specified in group C01P2002/70 by XPS, EDX or EDAX data
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/51—Particles with a specific particle size distribution
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/62—Submicrometer sized, i.e. from 0.1-1 micrometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/40—Electric properties
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M2004/021—Physical characteristics, e.g. porosity, surface area
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M2004/026—Electrodes composed of, or comprising, active material characterised by the polarity
- H01M2004/028—Positive electrodes
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Definitions
- the present invention relates to a precursor, a method for producing a precursor, a positive electrode material, a method for producing a positive electrode material, and a lithium ion secondary battery.
- Lithium cobalt oxide is widely used as a positive electrode material (positive electrode active material) for a lithium ion secondary battery.
- cobalt contained in lithium cobalt oxide is a rare metal with an annual production of only about 20,000 tons. Therefore, from the viewpoint of resource quantity and cost, a positive electrode material replacing lithium cobalt oxide is required. Therefore, conventionally, a lithium-containing nickel-manganese composite oxide has been proposed as a cobalt-free positive electrode material (Patent Document 1).
- a conventional lithium ion secondary battery using a lithium-containing nickel-manganese composite oxide as a positive electrode material may have insufficient discharge capacity and cycle characteristics.
- a precursor of a positive electrode material used in a lithium ion secondary battery which is at least one selected from the group consisting of nickel-manganese composite hydroxide and nickel-manganese composite oxide, and contains nickel and manganese.
- [3] The precursor according to the above [1] or [2], wherein the amount of mass loss when heated from room temperature to 1050 ° C. in an air atmosphere is 16% by mass or more.
- the [001] / [101] peak ratio which is the peak intensity ratio between the peak intensity in the [001] direction and the peak intensity in the [101] direction in X-ray diffraction, is 14 or less.
- the peak intensity in the [001] direction is the maximum peak intensity in the range where the diffraction angle 2 ⁇ is 17 to 21 °
- the peak in the [101] direction is the maximum in the range where the diffraction angle 2 ⁇ is 30 to 40 °. Peak intensity.
- the ratio of the ammonium-equivalent content of the ammonium source to the sum of the nickel-equivalent content of the nickel source and the manganese-equivalent content of the manganese source is 0 in terms of molar ratio.
- a positive electrode material used for a lithium ion secondary battery which is a lithium-containing nickel-manganese composite oxide, contains lithium, nickel, and manganese, and is a precursor according to any one of the above [1] to [4].
- Positive electrode material obtained by using the body is a positive electrode material used for a lithium ion secondary battery, which is a lithium-containing nickel-manganese composite oxide containing lithium, nickel and manganese, and has a content of a composite oxide represented by the formula Li 2 MnO 3.
- a method for producing a positive electrode material which comprises mixing and calcining the obtained mixture to obtain a calcined product.
- the ratio of the lithium-equivalent content of the lithium-containing compound to the sum of the nickel-equivalent content of the precursor and the manganese-equivalent content of the precursor is more than 1.03 in molar ratio.
- a lithium ion secondary battery having excellent discharge capacity and cycle characteristics can be obtained.
- the precursor of the present invention is a precursor of a positive electrode material used in a lithium ion secondary battery, and is at least one selected from the group consisting of nickel-manganese composite hydroxide and nickel-manganese composite oxide, and is nickel and manganese.
- the ratio of the nickel content to the total of the nickel content and the manganese content is 0.45 or more and 0.60 or less in terms of molar ratio, and the average valence of manganese is less than 4.0. It is a precursor.
- a positive electrode material lithium-containing nickel-manganese composite hydroxide described later is obtained.
- the lithium ion secondary battery using the obtained positive electrode material is excellent in discharge capacity and cycle characteristics. The reason is presumed as follows.
- a positive electrode material lithium-containing nickel-manganese composite oxide
- a precursor containing manganese having a high valence for example, Mn 4+
- lithium (Li + ) and the precursor manganese (Mn 4+ ) are produced. Due to the large repulsive force with, lithium cannot be uniformly diffused into the precursor. Therefore, the discharge capacity and the cycle characteristics are lowered.
- the precursor of the present invention has a low average valence of manganese of less than 4.0.
- the repulsive force between lithium and manganese in the precursor is relatively small (lithium and the precursor are likely to react), and lithium is uniform even inside the precursor. Easy to spread to. Therefore, it is excellent in discharge capacity and cycle characteristics.
- the precursor of the present invention contains nickel (Ni) and manganese (Mn).
- Ni nickel
- Mn manganese
- the ratio of the nickel content to the total of the nickel content and the manganese content (hereinafter, may be referred to as “Ni / (Ni + Mn)”) is 0.45 or more in terms of molar ratio. It is 0.60 or less, preferably 0.48 or more and 0.55 or less. That is, the precursors of the present invention contain substantially the same proportions of nickel and manganese.
- the precursor of the present invention preferably contains substantially no cobalt (Co) from the viewpoint of resource quantity and cost.
- the cobalt content in the precursor of the present invention is preferably 0.1% by mass or less, more preferably 0.01% by mass or less, and further preferably 0.001% by mass or less. It is particularly preferred that the precursor of the present invention does not contain cobalt (does not contain any cobalt).
- composition of the precursor is determined by inductively coupled plasma (ICP) emission spectroscopy.
- the average valence of manganese in the precursor of the present invention is less than 4.0, preferably 3.8 or less, more preferably 3.5 or less, because of better discharge capacity and cycle characteristics. It is preferable, and 3.2 or less is more preferable.
- the average valence of manganese in the precursor of the present invention is, for example, 2.5 or more, preferably 2.7 or more, and more preferably 2.9 or more.
- the average valence of manganese (Mn) is determined by X-ray photoelectron spectroscopy (XPS). Specifically, using an XPS device (QuanteraSXM, manufactured by ULVAC-PHI), a narrow scan analysis of the precursor is performed under the following conditions, and a photoelectron spectrum (also referred to as "narrow spectrum") of a 3s orbital (Mn3s) of manganese is performed. ). The exchange split width ( ⁇ E) of the obtained narrow spectrum is measured. Next, MnO (valence: 2), Mn 2 O 3 (valence: 3) and MnO 2 (valence: 4) are used as standard substances, and ⁇ E of each standard substance is measured in the same manner.
- XPS X-ray photoelectron spectroscopy
- the exchange split width ( ⁇ E) of Mn3s of the precursor of the present invention is preferably 4.9 eV or more, more preferably 5.0 eV or more, because the average valence of manganese (less than 4.0) described above can be easily obtained. ..
- the ⁇ E of Mn3s of the precursor of the present invention is preferably 5.7 eV or less, more preferably 5.5 eV or less.
- the precursor of the present invention preferably has a small average particle size (also referred to as "primary particle size") of the primary particles.
- the average particle size of the primary particles in the precursor of the present invention is preferably less than 0.6 ⁇ m, more preferably 0.1 ⁇ m or less.
- the lower limit is not particularly limited, but is, for example, 0.01 ⁇ m or more, preferably 0.03 ⁇ m or more.
- the primary particle size of the precursor (average particle size of the primary particles) is determined as follows. First, the precursor is observed using a scanning electron microscope (SEM) to obtain an SEM image. From the obtained SEM image, 200 or more primary particles are randomly extracted. Using image analysis software, the diameter equivalent to the projected area circle of the extracted primary particles (the diameter of the circle having the same area as the area of the particles on the SEM image) is obtained. The number average diameter of the obtained diameters is defined as the average particle diameter of the primary particles.
- SEM scanning electron microscope
- the precursor of the present invention is, for example, spherical secondary particles formed by aggregating a plurality of primary particles.
- the shape of the primary particles include a plate shape, a needle shape, a spherical shape, a rectangular parallelepiped shape, and the like, and among them, a plate shape is preferable.
- the precursor of the present invention has a large amount of mass loss (also simply referred to as “mass loss” in this paragraph) when heated from room temperature to 1050 ° C. in an air atmosphere.
- a precursor having a large amount of mass loss means that the hydroxide is high.
- the positive electrode material is produced using a precursor containing a large amount of hydroxide, the discharge capacity and cycle characteristics are more excellent.
- the mass reduction amount of the precursor of the present invention is preferably 16% by mass or more, more preferably 17% by mass or more, and further preferably 19% by mass or more.
- the upper limit is not particularly limited, but the mass reduction amount is, for example, 25% by mass or less, preferably 22% by mass or less.
- the amount of mass loss is determined by the ignition loss measurement described below. First, 1 g of a sample (precursor) placed in a crucible is heated to 1050 ° C. using an electric furnace, and then naturally cooled. The mass of the naturally cooled sample is then measured. The amount of mass loss is determined from the difference between the mass of the sample before heating and the mass of the sample after heating.
- the precursor of the present invention preferably has a small [001] / [101] peak ratio, which is a peak intensity ratio between the peak intensity in the [001] direction and the peak intensity in the [101] direction in X-ray diffraction.
- the peak intensity in the [001] direction is the maximum peak intensity in the range where the diffraction angle 2 ⁇ is in the range of 17 to 21 °.
- the peak in the [101] direction is the maximum peak intensity in the range where the diffraction angle 2 ⁇ is in the range of 30 to 40 °.
- the [001] / [101] peak ratio may be simply referred to as a “peak ratio”.
- Precursors with a small peak ratio [001] / [101] tend to be highly amorphous. Although the details are not clear, it is considered that nickel is unlikely to be replaced with lithium sites in the positive electrode material produced using the amorphous precursor. As a result, the discharge capacity and cycle characteristics are more excellent. At this time, it is considered that the peak ratio has an appropriate range, not only that the crystallinity is high.
- the [001] / [101] peak ratio of the precursor of the present invention is preferably 14 or less, more preferably 10 or less, further preferably 4.5 or less, particularly preferably 4 or less, and 3 or less. Most preferred.
- the lower limit is not particularly limited, but the [001] / [101] peak ratio is, for example, 1 or more.
- the method for producing a precursor of the present invention is the above-mentioned method for producing a precursor of the present invention, in which a nickel source, a manganese source, an ammonium source, and an alkaline aqueous solution are used in a reaction vessel having a pH of 9 or more and 12 or less. It is a method of introducing into a liquid to obtain a precipitate (at least one selected from the group consisting of nickel-manganese composite hydroxide and nickel-manganese composite oxide). The resulting precipitate (more specifically, for example, the precipitate separated from the reaction vessel solution and dried) is the precursor of the present invention described above.
- the method for producing the precursor of the present invention is the so-called coprecipitation method.
- the coprecipitation method nickel and manganese can be uniformly dispersed at the atomic level.
- the ratio of the ammonium-equivalent content of the ammonium source to the sum of the nickel-equivalent content of the nickel source and the manganese-equivalent content of the manganese source in the raw material aqueous solution (hereinafter referred to as "NH 4 / (Ni + Mn)").
- the molar ratio is preferably more than 0 and 1 or less, more preferably 0.1 or more and 0.8 or less, and further preferably 0.2 or more and 0.6 or less.
- the ratio (Ni / Mn) of the nickel-equivalent content of the nickel source to the manganese-equivalent content of the manganese source (Ni / Mn) in the raw material aqueous solution is preferably 1/1 in terms of molar ratio.
- the pH of the raw material aqueous solution is preferably 6 or less, more preferably 5.5 or less, and even more preferably 5 or less.
- the lower limit is not particularly limited, but the pH of the raw material aqueous solution is, for example, 3 or more, preferably 4 or more.
- nickel source examples include nickel salts such as nickel sulfate, nickel carbonate, nickel nitrate, nickel acetate, and nickel chloride, and nickel sulfate (NiSO 4 ) is preferable.
- manganese source examples include manganese salts such as manganese sulfate, manganese carbonate, manganese nitrate, manganese acetate, and manganese chloride, and manganese sulfate (MnSO 4 ) is preferable.
- ammonium source examples include ammonium salts such as ammonium sulfate, ammonium chloride, ammonium nitrate and ammonium carbonate, and ammonium sulfate ((NH 4 ) 2 SO 4 ) is preferable.
- ammonium salts such as ammonium sulfate, ammonium chloride, ammonium nitrate and ammonium carbonate, and ammonium sulfate ((NH 4 ) 2 SO 4 ) is preferable.
- the nickel source, the manganese source, and the ammonium source it is preferable that an aqueous solution is used. In each aqueous solution, the concentrations (contents) of the nickel source, the manganese source, and the ammonium source are preferably adjusted so as to have the above-mentioned molar ratios.
- a sodium hydroxide (NaOH) aqueous solution is preferable.
- the reaction tank liquid is the content liquid of the reaction tank, and as described above, the pH is 9 or more and 12 or less.
- the reaction tank liquid is prepared by adding, for example, pure water to an alkaline aqueous solution such as an aqueous sodium hydroxide solution. When obtaining the precipitate, it is preferable to stir the reaction tank liquid using a stirring rod or the like.
- the temperature of the reaction vessel liquid is preferably 30 ° C. or higher and 60 ° C. or lower, and more preferably 35 ° C. or higher and 45 ° C. or lower.
- the element A source may be further introduced into the reaction vessel liquid.
- the element A source is preferably contained in the raw material aqueous solution.
- the element A source include salts of element A such as sulfates, carbonates, nitrates, and acetates of element A. The amount of the element A source is appropriately adjusted according to the desired composition.
- the precipitate obtained by the coprecipitation method is separated from the reaction tank liquid (solid-liquid separation), washed with water, and then dried.
- the temperature at which the precipitate is dried is preferably low because oxidation of the precipitate due to the dehydration reaction is suppressed and the above-mentioned valence of manganese can be easily obtained.
- the drying temperature is preferably 100 ° C. or lower, more preferably 90 ° C. or lower, further preferably 80 ° C. or lower, particularly preferably 70 ° C. or lower, and most preferably 60 ° C. or lower.
- the lower limit is not particularly limited, but the drying temperature is, for example, 30 ° C. or higher, preferably 40 ° C. or higher.
- the atmosphere for drying the precipitate is preferably a non-oxidizing atmosphere because it suppresses oxidation of the precipitate and tends to reduce the valence of manganese.
- the non-oxidizing atmosphere include a non-oxidizing atmosphere having an oxygen concentration of 10% by volume or less, and a specific example thereof is a vacuum atmosphere (for example, 0.1 MPa or less).
- the time (drying time) for drying the precipitate is more preferably 5 hours or more.
- the positive electrode material of the present invention is also called a positive electrode active material.
- the positive electrode material (first aspect) of the present invention is a positive electrode material used for a lithium ion secondary battery, which is a lithium-containing nickel-manganese composite oxide, contains lithium, nickel, and manganese, and is a precursor of the present invention described above. It is a positive electrode material obtained by using a body.
- the lithium ion secondary battery using the positive electrode material obtained by using the precursor of the present invention is excellent in discharge capacity and cycle characteristics.
- the positive electrode material (second aspect) of the present invention is a positive electrode material used for a lithium ion secondary battery, which is a lithium-containing nickel-manganese composite oxide, contains lithium, nickel, and manganese, and is represented by the formula Li 2 MnO 3 . It is a positive electrode material in which the content of the represented composite oxide is more than 0% by mass and 20% by mass or less.
- the lithium-containing nickel-manganese composite oxide is preferably hexagonal.
- the “composite oxide represented by the formula Li 2 MnO 3 ” is also referred to as a “second phase composite oxide” or simply a “second phase”.
- a lithium ion secondary battery using a positive electrode material having a second phase content of more than 0% by mass and 20% by mass or less is excellent in discharge capacity and cycle characteristics.
- the content of the second phase is preferably 2% by mass or more and 19% by mass or less, and more preferably 3% by mass or more and 17% by mass or less because the discharge capacity and the cycle characteristics are more excellent.
- the content of the second phase in the positive electrode material is determined as follows. First, an X-ray diffraction (XRD) pattern of the positive electrode material is obtained under the following conditions. Next, the obtained XRD pattern is Rietveld analyzed using RIETAN-FP (profile: extended pseudo-Voigt function) and pattern fitting is performed. In this way, the content of the second phase is determined.
- XRD X-ray diffraction
- the composite oxide of the second phase is preferably a monoclinic composite oxide. More specifically, in the positive electrode material (second aspect) of the present invention, the hexagonal composite oxide is mixed with the monoclinic composite oxide as a heterogeneous phase (second phase composite oxide). Is more preferable.
- the positive electrode material (lithium-containing nickel-manganese composite oxide) of the present invention contains lithium (Li), nickel (Ni) and manganese (Mn).
- the atoms of each other undergo a disproportionation reaction.
- the change in the valence of Ni which has a large free energy change ⁇ G before and after charging and discharging and has a high voltage, can be used for the charging / discharging reaction, and a lithium ion secondary battery having a high voltage and a high capacity can be obtained.
- the crystal structure is stable even in the charged state, the obtained lithium ion secondary battery has excellent cycle characteristics.
- the positive electrode material of the present invention further comprises aluminum (Al), silicon (Si), titanium (Ti), zirconium (Zr), calcium (Ca), potassium (K), barium (Ba), strontium (Sr) and sulfur. It may contain at least one element A selected from the group consisting of (S).
- the positive electrode material (lithium-containing nickel-manganese composite oxide) of the present invention preferably contains a composite oxide represented by the following formula (1).
- (1) Li a Ni x Mn 1-x A y O 2
- a is a number more than 0.95 and less than 1.10
- x is a number of 0.45 or more and 0.60 or less
- y is a number of 0 or more and 0.020 or less
- A Is at least one selected from the group consisting of Al, Si, Ti, Zr, Ca, K, Ba, Sr and S.
- the composition of the formula (1) is a composition containing the second phase (composite oxide represented by the formula Li 2 MnO 3 ).
- the composition of the positive electrode material is determined by inductively coupled plasma (ICP) emission spectroscopy.
- the positive electrode material of the present invention utilizes the disproportionation reaction caused by mutual atoms due to the combination of manganese and nickel. In order to allow this reaction to proceed sufficiently, it is preferable that the manganese atom and the nickel atom are adjacent to each other in the positive electrode material and are uniformly distributed. Therefore, the positive electrode material of the present invention has a median value of 0.85 or more and 1.20 or less and a half width in the relative frequency distribution of the molar ratio (Mn / Ni) between the manganese content and the nickel content. Is preferably 0.90 or less. The median value is more preferably 0.90 or more and 1.10 or less.
- the half width is more preferably 0.80 or less. As a result, the obtained lithium ion secondary battery has a high voltage and is more excellent in discharge capacity and cycle characteristics.
- the lower limit of the half width is not particularly limited, and the smaller the half width is, the more preferable.
- the median and full width at half maximum of the relative frequency distribution of the molar ratio (Mn / Ni) of the positive electrode material is determined by observation using a transmission electron microscope (TEM) and energy dispersive X-ray analysis (EDX). Specifically, it is as follows. First, the powder of the positive electrode material is embedded in a resin, and then sliced using a focused ion beam processing device to obtain a sample for TEM observation. The obtained sample is observed using TEM (JEM-F200, manufactured by JEOL Ltd.) (observation condition: accelerating voltage 200 kV) to obtain a HAADF-STEM image.
- TEM transmission electron microscope
- EDX energy dispersive X-ray analysis
- the HAADF-STEM image is morphologically observed, EDX is performed using the device (Dual SDD, manufactured by JEOL) attached to the TEM (analysis condition: accelerating voltage 200 kV), and element mapping is performed (map resolution: 1.96 nm / pixel). ). From the obtained element mapping result, only the positive electrode material portion is extracted, and a simple quantitative calculation is performed for each pixel so that the total content (molar amount) of Mn and Ni is 100%.
- the molar ratio (Mn / Ni) is determined by dividing the Mn content by the Ni content.
- the molar ratio (Mn / Ni) ratio is taken at 0.01 pitch on the horizontal axis, and the relative frequency is taken on the vertical axis to create a relative frequency distribution. Obtain the median and full width at half maximum for the created relative frequency distribution.
- the positive electrode material (lithium-containing nickel-manganese composite oxide) of the present invention has a mass increase when left in an air atmosphere at a temperature of 25 ° C. and a humidity of 60% for 240 hours (also simply referred to as “mass increase” in this paragraph). ) Is less.
- the temperature "25 ° C” means “25 ⁇ 3 ° C”
- the humidity "60%” means "60 ⁇ 5%”.
- the small amount of mass increase when the positive electrode material is left in the air atmosphere means that the deterioration due to the reaction with moisture and carbon dioxide in the air atmosphere is small.
- the positive electrode material having a small amount of mass increase suppresses deterioration in the air atmosphere and is excellent in handleability. It also has an excellent effect of suppressing decomposition of the electrolytic solution.
- the smaller the amount of mass increase when the positive electrode material is left in the air atmosphere the smaller the amount of lithium extracted from the positive electrode material by the reaction with water and carbon dioxide. In this case, the decrease in charge / discharge capacity due to the extraction of lithium from the positive electrode material is suppressed.
- the mass increase amount of the positive electrode material of the present invention is preferably 0.75% by mass or less, more preferably 0.70% by mass or less, further preferably 0.60% by mass or less, and 0.50% by mass.
- the amount of mass increase is calculated as follows. First, a predetermined amount of the sample (positive electrode material) is weighed and placed in a sample bottle. Next, the sample bottle containing the sample is stored in a constant temperature and humidity chamber maintained at a temperature of 25 ° C. and a humidity of 60% in an air atmosphere, and left for 240 hours. The amount of mass increase is obtained from the difference between the mass of the sample before standing and the mass of the sample after leaving.
- the method for producing a positive electrode material of the present invention is the above-mentioned method for producing a positive electrode material of the present invention, in which the above-mentioned precursor of the present invention and a lithium-containing compound are mixed and the obtained mixture is fired.
- This is a method for producing a positive electrode material for obtaining a fired product.
- the fired product lithium-containing nickel-manganese composite oxide
- Li / (Ni + Mn) the ratio of the lithium-equivalent content of the lithium-containing compound to the total of the nickel-equivalent content of the precursor and the manganese-equivalent content of the precursor.
- Li / (Ni + Mn) the ratio of the lithium-equivalent content of the lithium-containing compound to the total of the nickel-equivalent content of the precursor and the manganese-equivalent content of the precursor.
- ) Is preferably more than 1.03 and less than 1.10, and more preferably 1.04 or more and 1.08 or less in terms of molar ratio.
- Li / (Ni + Mn) is within this range, the content of the second phase tends to be an appropriate amount in the obtained positive electrode material (second aspect) of the present invention.
- lithium-containing compound examples include lithium hydroxide and lithium carbonate, and among them, lithium hydroxide is preferable because the reaction temperature is low.
- a compound containing the element A (hereinafter, also referred to as “A-containing compound”) may be further mixed with the mixture.
- A-containing compound include, but are not limited to, hydroxides, oxides, chlorides, salts (for example, sulfates, carbonates, nitrates, etc.) of the element A.
- the mixing amount of the A-containing compound is appropriately adjusted according to the desired composition.
- the mixture obtained by mixing is calcined to obtain a calcined product.
- the mixture is tentatively fired at a temperature of 400 ° C. or higher and 700 ° C. or lower, and then main fired at a temperature of 800 ° C. or higher and 1000 ° C. or lower.
- the content of the second phase tends to be an appropriate amount in the obtained positive electrode material (second aspect) of the present invention.
- the temperature of the main firing is more preferably 900 ° C. or higher and 1000 ° C. or lower, and further preferably 925 ° C. or higher and 975 ° C. or lower, because the content of the second phase tends to be more appropriate.
- the atmosphere of the tentative firing is not particularly limited, and examples thereof include an oxidizing atmosphere (for example, an atmospheric atmosphere) and a non-oxidizing atmosphere. Among them, a non-oxidizing atmosphere is preferable. Examples of the non-oxidizing atmosphere include a non-oxidizing atmosphere having an oxygen concentration of 10% by volume or less (specifically, for example, a nitrogen atmosphere). By calcination in a non-oxidizing atmosphere, the formation of nickel and / or manganese oxides with low activity is suppressed, and lithium is likely to diffuse uniformly in the calcined product.
- the atmosphere of the main firing is not particularly limited, and examples thereof include an oxidizing atmosphere (for example, an atmospheric atmosphere) or a non-oxidizing atmosphere.
- the firing time is not particularly limited.
- the firing time of the tentative firing is preferably 6 hours or more and 48 hours or less, and more preferably 12 hours or more and 36 hours or less.
- the firing time of this firing is preferably 5 hours or more and 30 r or less, and more preferably 10 hours or more and 25 hours or less.
- the fired product is preferably washed with water.
- the fired product washed with water is then appropriately dried to become a positive electrode material.
- excess lithium that has not entered the inside of the obtained positive electrode material is washed away. Therefore, the amount of residual lithium in the charged state is reduced, and the amount of mass increase described above is reduced.
- the lithium ion secondary battery of the present invention includes a positive electrode containing the positive electrode material of the present invention described above, a negative electrode, and an ionic conduction medium that is interposed between the positive electrode and the negative electrode to conduct lithium ions. It is a secondary battery.
- the lithium ion secondary battery of the present invention is excellent in discharge capacity and cycle characteristics.
- the ionic conduction medium is, for example, an electrolyte such as a non-aqueous electrolyte solution.
- the lithium ion secondary battery of the present invention may further include a separator.
- a conventionally known configuration of a lithium ion secondary battery can be adopted except that the positive electrode material of the present invention is used.
- Precursor No. 1 >> 0.4 mol / L nickel sulfate (NiSO 4 ) aqueous solution, 0.4 mol / L manganese sulfate (MnSO 4 ) aqueous solution, 0.2 mol / L ammonium sulfate ((NH 4 ) 2 SO 4 ) aqueous solution was mixed to obtain an aqueous raw material solution.
- the molar ratio (NH 4 / (Ni + Mn)) in the raw material aqueous solution was 0.25.
- the pH of the raw material aqueous solution was 4.6. 1 L of pure water and an aqueous sodium hydroxide solution were added to the reaction vessel to obtain a reaction vessel liquid adjusted to pH 11.
- the raw material aqueous solution was added to the reaction vessel solution at a rate of 150 mL / h.
- an alkaline aqueous solution (10% by mass sodium hydroxide aqueous solution) was charged into the reaction tank liquid while controlling the pH of the reaction tank liquid to be 11.
- a precipitate was obtained.
- the temperature of the reaction tank liquid was controlled to 40 ° C. while stirring the reaction tank liquid at 450 rpm with a stirring rod.
- the precipitate was filtered and washed with water. Then, the precipitate was dried at 50 ° C. for 10 hours in a vacuum atmosphere using a vacuum dryer.
- the precursor No. I got 1.
- Precursor No. 3 Ammonium sulfate aqueous solution was not mixed with the raw material aqueous solution. The pH of this raw material aqueous solution was 6.5. The aqueous raw material solution and 0.06 mol / L aqueous ammonia were charged into the reaction vessel solution at a rate of 150 mL / h, respectively. During the addition of the raw material aqueous solution and the ammonia water, an alkaline aqueous solution (10% by mass sodium hydroxide aqueous solution) was added to the reaction tank liquid while controlling the pH of the reaction tank liquid to be 11. Thus, a precipitate was obtained. Other than these points, the precursor No. Precursor No. 1 in the same manner as in 1. I got 3.
- Precursor No. 4 The dry atmosphere of the precipitate was changed to the air atmosphere. The drying temperature of the precipitate was set to 110 ° C. Other than these points, the precursor No. Precursor No. 1 in the same manner as in 1. I got 4.
- Precursor No. 5 The dry atmosphere of the precipitate was changed to the air atmosphere. The drying temperature of the precipitate was set to 110 ° C. Other than these points, the precursor No. In the same manner as in No. 3, the precursor No. I got 5.
- Positive electrode material No. 1 >> Precursor No. 1 and lithium hydroxide were mixed to obtain a mixture. The molar ratio (Li / (Ni + Mn)) at the time of mixing was 1.05. The obtained mixture was calcined to obtain a calcined product. More specifically, the mixture was calcined at 650 ° C. for 24 hours in a nitrogen atmosphere and then fired for 15 hours at 950 ° C. in an air atmosphere. The obtained calcined product was crushed using a mortar. The fired product was not washed with water. In this way, the positive electrode material No. I got 1.
- Positive electrode material No. 2-Positive material No. 5 >> Precursor No. 2-Precursor No. 5 was used. Other than this point, the positive electrode material No. In the same manner as in No. 1, the positive electrode material No. 2-Positive material No. I got 5.
- Positive electrode material No. 6-Positive electrode material No. 10 >> Al-containing compound (aluminum nitrate), Ti-containing compound (titanium nitrate), Zr-containing compound (zirconium nitrate), K-containing compound (potassium nitrate), and Ba-containing compound (barium nitrate) were further mixed with the mixture, respectively. Other than this point, the positive electrode material No. In the same manner as in No. 1, the positive electrode material No. 6-Positive electrode material No. I got 10.
- N-Methyl-2pyrrolidone was added to the positive electrode material (90% by mass), acetylene black (5% by mass) and polyvinylidene fluoride (5% by mass) and kneaded to obtain a mixture.
- the obtained mixture was applied to an aluminum current collector to a thickness of 320 ⁇ m to form a coating film.
- the coating film and the laminated body of the aluminum current collector were pressurized using a roll press having a gap set to 80 ⁇ m.
- a disk having a diameter of 14 mm was punched out from the pressurized laminate. The punched disk was vacuum dried at 150 ° C. for 15 hours. The disk after vacuum drying was used as the positive electrode.
- a lithium metal sheet was used as the negative electrode.
- a polypropylene porous membrane (Cellguard # 2400) was used as the separator. 1 mol of LiPF 6 was dissolved in 1 L of a mixed solution having a volume ratio (EC / DMC) of ethylene carbonate (EC) and dimethyl carbonate (DMC) of 1/1 to obtain a non-aqueous electrolyte solution.
- a lithium ion secondary battery (test cell) was prepared in a glove box substituted with argon.
- the current value was set to a constant value of 0.2C, and the voltage was charged and discharged in the range of 2.75 to 4.4V to determine the discharge capacity [mAh / g].
- Precursor No. 1 having an average valence of Mn of less than 4.0.
- 1-Precursor No. Positive electrode material No. 3 produced using No. 3.
- 1-Positive electrode material No. 3 and positive electrode material No. 6-Positive electrode material No. No. 10 is a precursor No. 10 having an average valence of Mn of 4.0 or more.
- 4-Precursor No. Positive electrode material No. 5 produced using No. 4 to positive electrode material No. The discharge capacity and cycle characteristics were superior to those of 5.
- Positive electrode material No. 11 >> Precursor No. 1 and lithium hydroxide were mixed to obtain a mixture. The molar ratio (Li / (Ni + Mn)) at the time of mixing was 1.05. The obtained mixture was calcined to obtain a calcined product. More specifically, the mixture was calcined at 650 ° C. for 24 hours in a nitrogen atmosphere and then fired for 5 hours at 1000 ° C. in an air atmosphere. The obtained calcined product was crushed using a mortar. The fired product was not washed with water. In this way, the positive electrode material No. I got 11.
- Positive electrode material No. 12 >> The firing temperature in the main firing was 900 ° C., and the firing time was 15 hours. The points other than these are the positive electrode material No. In the same manner as in No. 11, the positive electrode material No. I got twelve.
- Positive electrode material No. 13 >> The firing temperature in the main firing was 950 ° C., and the firing time was 10 hours. The points other than these are the positive electrode material No. In the same manner as in No. 11, the positive electrode material No. I got 13.
- Positive electrode material No. 14 >> The firing temperature in the main firing was 950 ° C., and the firing time was 15 hours. The fired product was washed with water and then dried. The points other than these are the positive electrode material No. In the same manner as in No. 11, the positive electrode material No. I got 14.
- Positive electrode material No. 15 The S-containing compound (lithium sulfate) was further mixed with the mixture. The firing temperature in the main firing was 950 ° C., and the firing time was 15 hours. The points other than these are the positive electrode material No. In the same manner as in No. 11, the positive electrode material No. I got 15.
- Positive electrode material No. 16 >> The molar ratio (Li / (Ni + Mn)) at the time of mixing was set to 1.20. The firing temperature in the main firing was 950 ° C., and the firing time was 10 hours. The points other than these are the positive electrode material No. In the same manner as in No. 11, the positive electrode material No. I got 16.
- Positive electrode material No. 17 >> The firing temperature in the main firing was 780 ° C., and the firing time was 15 hours. The points other than these are the positive electrode material No. In the same manner as in No. 11, the positive electrode material No. I got 17.
- Positive electrode material No. 18 >> The firing temperature in the main firing was 1080 ° C., and the firing time was 15 hours. The points other than these are the positive electrode material No. In the same manner as in No. 11, the positive electrode material No. I got 18.
- the positive electrode material No. 1 in which the content of the second phase (Li 2 MnO 3 ) is more than 0% by mass and 20% by mass or less.
- Reference numeral 15 denotes a positive electrode material No. 15 in which the content of the second phase (Li 2 MnO 3 ) is 0% by mass or more than 20% by mass.
- 16-Positive electrode material No. The discharge capacity and cycle characteristics were superior to those of 18.
Landscapes
- Chemical & Material Sciences (AREA)
- Inorganic Chemistry (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Manufacturing & Machinery (AREA)
- Materials Engineering (AREA)
- Battery Electrode And Active Subsutance (AREA)
- Inorganic Compounds Of Heavy Metals (AREA)
Abstract
Description
しかし、コバルト酸リチウムに含まれるコバルトは、年産量が20,000t程度しかないレアメタルである。このため、資源量やコスト面の観点から、コバルト酸リチウムに代わる正極材が求められている。
そこで、従来、コバルトを含有しない正極材として、リチウム含有ニッケルマンガン複合酸化物が提案されている(特許文献1)。
また、本発明は、放電容量およびサイクル特性に優れたリチウムイオン二次電池が得られる正極材およびその製造方法を提供することを目的とする。
更に、本発明は、放電容量およびサイクル特性に優れたリチウムイオン二次電池を提供することを目的とする。
[1]リチウムイオン二次電池に用いる正極材の前駆体であって、ニッケルマンガン複合水酸化物およびニッケルマンガン複合酸化物からなる群から選ばれる少なくとも1種であり、ニッケルおよびマンガンを含有し、ニッケル含有量とマンガン含有量との合計に対するニッケル含有量の比が、モル比で、0.45以上0.60以下であり、マンガンの平均価数が4.0未満である、前駆体。
[2]一次粒子の平均粒子径が0.6μm未満である、上記[1]に記載の前駆体。
[3]大気雰囲気下で室温から1050℃まで加熱したときの質量減少量が16質量%以上である、上記[1]または[2]に記載の前駆体。
[4]X線回折における[001]方向のピーク強度と[101]方向のピーク強度とのピーク強度比である[001]/[101]ピーク比が、14以下である、上記[1]~[3]のいずれかに記載の前駆体。
ただし、上記[001]方向のピーク強度は、回折角2θが17~21°の範囲における最大ピーク強度であり、上記[101]方向のピークは、回折角2θが30~40°の範囲における最大ピーク強度である。
[5]上記[1]~[4]のいずれかに記載の前駆体を製造する方法であって、ニッケル源、マンガン源、アンモニウム源、および、アルカリ性水溶液を、pHが9以上12以下である反応槽液に導入して、沈殿物を得る、前駆体の製造方法。
[6]上記ニッケル源、上記マンガン源、および、上記アンモニウム源を含有する水溶液を原料水溶液とし、上記原料水溶液と上記アルカリ性水溶液とを上記反応槽液に導入して、上記沈殿物を得る、上記[5]に記載の前駆体の製造方法。
[7]上記原料水溶液において、上記ニッケル源のニッケル換算の含有量と上記マンガン源のマンガン換算の含有量との合計に対する、上記アンモニウム源のアンモニウム換算の含有量の比が、モル比で、0超1以下である、上記[6]に記載の前駆体の製造方法。
[8]上記原料水溶液のpHが6以下である、上記[6]または[7]に記載の前駆体の製造方法。
[9]上記沈殿物を、100℃以下の温度で乾燥する、上記[6]~[8]のいずれかに記載の前駆体の製造方法。
[10]上記沈殿物を、非酸化性雰囲気下で乾燥する、上記[6]~[9]のいずれかに記載の前駆体の製造方法。
[11]リチウムイオン二次電池に用いる正極材であって、リチウム含有ニッケルマンガン複合酸化物であり、リチウム、ニッケルおよびマンガンを含有し、上記[1]~[4]のいずれかに記載の前駆体を用いて得られる、正極材。
[12]リチウムイオン二次電池に用いる正極材であって、リチウム含有ニッケルマンガン複合酸化物であり、リチウム、ニッケルおよびマンガンを含有し、式Li2MnO3で表わされる複合酸化物の含有量が、0質量%超20質量%以下である、正極材。
[13]更に、アルミニウム、ケイ素、チタン、ジルコニウム、カルシウム、カリウム、バリウム、ストロンチウムおよび硫黄からなる群から選ばれる少なくとも1種の元素Aを含有する、上記[11]または[12]に記載の正極材。
[14]マンガン含有量とニッケル含有量とのモル比の相対度数分布において、中央値が0.85以上1.20以下であり、かつ、半値幅が0.90以下である、上記[11]~[13]のいずれかに記載の正極材。
[15]温度25℃、湿度60%の大気雰囲気下に240時間放置したときの質量増加量が0.75質量%以下である、上記[11]~[14]のいずれかに記載の正極材。
[16]上記[11]~[15]のいずれかに記載の正極材を製造する方法であって、上記[1]~[4]のいずれかに記載の前駆体と、リチウム含有化合物とを混合し、得られた混合物を焼成して焼成物を得る、正極材の製造方法。
[17]上記前駆体のニッケル換算の含有量と上記前駆体のマンガン換算の含有量との合計に対する、上記リチウム含有化合物のリチウム換算の含有量の比が、モル比で、1.03超1.10未満である、上記[16]に記載の正極材の製造方法。
[18]上記混合物を、400℃以上700℃以下の温度で仮焼成し、その後、800℃以上1000℃以下の温度で本焼成して、上記焼成物を得る、上記[16]または[17]に記載の正極材の製造方法。
[19]上記焼成物を水洗する、上記[16]~[18]のいずれかに記載の正極材の製造方法。
[20]上記[11]~[15]のいずれかに記載の正極材を含有する正極と、負極と、上記正極と上記負極との間に介在してリチウムイオンを伝導するイオン伝導媒体と、を備えるリチウムイオン二次電池。
本発明の前駆体は、リチウムイオン二次電池に用いる正極材の前駆体であって、ニッケルマンガン複合水酸化物およびニッケルマンガン複合酸化物からなる群から選ばれる少なくとも1種であり、ニッケルおよびマンガンを含有し、ニッケル含有量とマンガン含有量との合計に対するニッケル含有量の比が、モル比で、0.45以上0.60以下であり、マンガンの平均価数が4.0未満である、前駆体である。
これに対して、本発明の前駆体は、マンガンの平均価数が4.0未満と低い。このような前駆体を用いて正極材を製造する場合、リチウムと前駆体のマンガンとの反発力が相対的に小さく(リチウムと前駆体とが反応しやすく)、リチウムが前駆体の内部まで均一に拡散しやすい。このため、放電容量およびサイクル特性に優れる。
本発明の前駆体は、ニッケル(Ni)およびマンガン(Mn)を含有する。
本発明の前駆体において、ニッケル含有量とマンガン含有量との合計に対するニッケル含有量の比(以下、「Ni/(Ni+Mn)」と表記する場合がある)は、モル比で、0.45以上0.60以下であり、0.48以上0.55以下が好ましい。すなわち、本発明の前駆体は、実質的に同比率のニッケルおよびマンガンを含む。
上述したように、本発明の前駆体におけるマンガンの平均価数は、4.0未満であり、放電容量およびサイクル特性がより優れるという理由から、3.8以下が好ましく、3.5以下がより好ましく、3.2以下が更に好ましい。
一方、本発明の前駆体におけるマンガンの平均価数は、例えば、2.5以上であり、2.7以上が好ましく、2.9以上がより好ましい。
具体的には、XPS装置(QuanteraSXM、ULVAC-PHI社製)を用いて、下記条件で、前駆体をナロースキャン分析して、マンガンの3s軌道(Mn3s)の光電子スペクトル(「ナロースペクトル」ともいう)を得る。得られたナロースペクトルの交換分裂幅(ΔE)を計測する。
次に、標準物質として、MnO(価数:2)、Mn2O3(価数:3)およびMnO2(価数:4)を用い、各標準物質のΔEを同様に計測する。
マンガンは、その価数に応じて、3s軌道のナロースペクトルの交換分裂幅(ΔE)が変化することが知られている。
各標準物質のΔEから検量線を作成する。作成した検量線と、前駆体のΔEとから、前駆体のMnの価数を求める。
前駆体ごとにΔEの計測は3回行ない、3回の平均値を、各前駆体のMn平均価数とする。
線源:X線モノクロAl-Kα
電圧:15kV
出力:25kW
ビーム径:100μmφ
・分析領域:100μmφ
・ナロースキャン分析条件
Mn3s Pass Energy:55eV
Step Size:0.1eV
一方、本発明の前駆体のMn3sのΔEは、5.7eV以下が好ましく、5.5eV以下がより好ましい。
本発明の前駆体は、一次粒子の平均粒子径(「一次粒子径」ともいう)が小さい方が好ましい。一次粒子径が小さい前駆体を用いて正極材を製造することにより、リチウムが前駆体の内部を移動する距離が短く、リチウムを前駆体の内部に均一に拡散しやすくなり、放電容量およびサイクル特性がより優れる。
具体的には、本発明の前駆体における一次粒子の平均粒子径は、0.6μm未満が好ましく、0.1μm以下がより好ましい。下限は特に限定されないが、例えば、0.01μm以上であり、0.03μm以上が好ましい。
まず、前駆体を、走査型電子顕微鏡(SEM)を用いて観察して、SEM画像を得る。得られたSEM画像から、無作為に200個以上の一次粒子を抽出する。画像解析ソフトを用いて、抽出した一次粒子の投影面積円相当径(SEM画像上の粒子の面積と同一の面積の円の直径)を得る。得られた径の個数平均径を、一次粒子の平均粒子径とする。
本発明の前駆体は、大気雰囲気下で室温から1050℃まで加熱したときの質量減少量(本段落において、単に「質量減少量」ともいう)が多い方が好ましい。
質量減少量が多い前駆体(ニッケルマンガン複合水酸化物およびニッケルマンガン複合酸化物からなる群から選ばれる少なくとも1種)は、水酸化物が多いことを意味する。水酸化物が多い前駆体を用いて正極材を製造する場合、放電容量およびサイクル特性などがより優れる。これは、詳細は不明であるが、前駆体にヒドロキシ基が複雑に配置されることで、得られる正極材(リチウム含有ニッケルマンガン複合酸化物)の結晶構造が適度に整えられるためと考えられる。
具体的には、本発明の前駆体の質量減少量は、16質量%以上が好ましく、17質量%以上がより好ましく、19質量%以上が更に好ましい。
上限は、特に限定されないが、質量減少量は、例えば、25質量%以下であり、22質量%以下が好ましい。
質量減少量は、以下に説明する強熱減量測定により求める。
まず、ルツボに入れた試料(前駆体)1gを、電気炉を用いて、1050℃まで加熱し、その後、自然冷却する。次いで、自然冷却した試料の質量を測定する。加熱前の試料の質量と加熱後の試料の質量との差から、質量減少量を求める。
本発明の前駆体は、X線回折における[001]方向のピーク強度と[101]方向のピーク強度とのピーク強度比である[001]/[101]ピーク比が小さいことが好ましい。[001]方向のピーク強度は、回折角2θが17~21°の範囲における最大ピーク強度である。[101]方向のピークは、回折角2θが30~40°の範囲における最大ピーク強度である。以下、[001]/[101]ピーク比を、単に「ピーク比」と呼ぶ場合がある。
[001]/[101]ピーク比が小さい前駆体は、非晶質性が高い傾向にある。詳細は明らかではないが、非晶質な前駆体を用いて製造した正極材は、リチウムサイトにニッケルが置換しにくいと考えられる。その結果、放電容量およびサイクル特性などがより優れる。このとき、単に非結晶性が高いことのみが要求されるのではなく、ピーク比に適度な範囲があると考えられる。
具体的には、本発明の前駆体の[001]/[101]ピーク比は、14以下が好ましく、10以下がより好ましく、4.5以下が更に好ましく、4以下が特に好ましく、3以下が最も好ましい。
下限は、特に限定されないが、[001]/[101]ピーク比は、例えば、1以上である。
X線回折装置(X線源:CuKα、管電圧:40kV、管電流:40mA)を用いて、前駆体のX線回折(XRD)パターンを得て、[001]方向のピーク強度と、[101]方向のピーク強度とからピーク強度比([001]/[101]ピーク比)を求める。
次に、本発明の前駆体の製造方法を説明する。
本発明の前駆体の製造方法は、上述した本発明の前駆体を製造する方法であって、ニッケル源、マンガン源、アンモニウム源、および、アルカリ性水溶液を、pHが9以上12以下である反応槽液に導入して、沈殿物(ニッケルマンガン複合水酸化物およびニッケルマンガン複合酸化物からなる群から選ばれる少なくとも1種)を得る方法である。
得られる沈殿物(より詳細には、例えば、沈殿物を、反応槽液からろ別し乾燥したもの)が、上述した本発明の前駆体となる。
本発明の前駆体の製造方法は、いわゆる共沈法である。共沈法を用いることにより、ニッケルとマンガンとを原子レベルで均一に分散できる。
本発明における共沈法では、ニッケル源、マンガン源、および、アンモニウム源を含有する水溶液を原料水溶液とし、この原料水溶液とアルカリ性水溶液とを反応槽液に導入して、沈殿物を得ることが好ましい。
すなわち、ニッケル源およびマンガン源とアンモニウム源とをそれぞれ別々に反応槽液に導入するのではなく、ニッケル源およびマンガン源とアンモニウム源とを予め混合したものを、反応槽液に導入することが好ましい。
これにより、反応槽液において、生成した沈殿物にアンモニウムが作用することが回避され、一次粒子の必要以上の成長が抑制されやすい。
また、原料水溶液において、アンモニウム(NH4 +)がニッケルイオンおよびマンガンイオンに配位して安定化することも、一次粒子の成長を抑制できる理由の1つと考えられる。
マンガン源としては、例えば、硫酸マンガン、炭酸マンガン、硝酸マンガン、酢酸マンガン、塩化マンガンなどのマンガン塩が挙げられ、硫酸マンガン(MnSO4)が好ましい。
アンモニウム源としては、例えば、硫酸アンモニウム、塩化アンモニウム、硝酸アンモニウム、炭酸アンモニウムなどのアンモニウム塩が挙げられ、硫酸アンモニウム((NH4)2SO4)が好ましい。
ニッケル源、マンガン源およびアンモニウム源は、それぞれ、水溶液の態様が用いられることが好ましい。
各水溶液において、ニッケル源、マンガン源およびアンモニウム源の濃度(含有量)は、それぞれ、上述したモル比となるように調整されることが好ましい。
沈殿物を得る際に、攪拌棒などを用いて、反応槽液を攪拌することが好ましい。
反応槽液の温度は、30℃以上60℃以下が好ましく、35℃以上45℃以下がより好ましい。
元素A源としては、例えば、元素Aの硫酸塩、炭酸塩、硝酸塩、酢酸塩などの元素Aの塩が挙げられる。
元素A源の量は、所望する組成に応じて、適宜調整される。
共沈法により得られた沈殿物は、反応槽液からろ別(固液分離)し、水洗した後、乾燥することが好ましい。
具体的には、乾燥温度は、100℃以下が好ましく、90℃以下がより好ましく、80℃以下が更に好ましく、70℃以下が特に好ましく、60℃以下が最も好ましい。
下限は特に限定されないが、乾燥温度は、例えば、30℃以上であり、40℃以上が好ましい。
次に、本発明の正極材を説明する。正極材は、正極活物質とも呼ばれる。
本発明の正極材(第1態様)は、リチウムイオン二次電池に用いる正極材であって、リチウム含有ニッケルマンガン複合酸化物であり、リチウム、ニッケルおよびマンガンを含有し、上述した本発明の前駆体を用いて得られる、正極材である。
本発明の前駆体を用いて得られた正極材を用いたリチウムイオン二次電池は、放電容量およびサイクル特性に優れる。
本発明の正極材(第2態様)は、リチウムイオン二次電池に用いる正極材であって、リチウム含有ニッケルマンガン複合酸化物であり、リチウム、ニッケルおよびマンガンを含有し、式Li2MnO3で表わされる複合酸化物の含有量が、0質量%超20質量%以下である、正極材である。ここで、リチウム含有ニッケルマンガン複合酸化物は、六方晶であることが好ましい。
以下、「式Li2MnO3で表わされる複合酸化物」を、「第2相の複合酸化物」または単に「第2相」ともいう。
第2相の含有量が0質量%超20質量%以下である正極材を用いたリチウムイオン二次電池は、放電容量およびサイクル特性に優れる。
放電容量およびサイクル特性がより優れるという理由から、第2相の含有量は、2質量%以上19質量%以下が好ましく、3質量%以上17質量%以下がより好ましい。
まず、下記条件にて、正極材のX線回折(XRD)パターンを得る。次いで、得られたXRDパターンについて、RIETAN-FP(プロファイル:拡張擬フォークト関数)を用いてリートベルト解析し、パターンフィッティングする。こうして、第2相の含有量を求める。
・装置名:デバイ・シェラー型回折計BL5S2(あいちシンクロトロン光センター)
・X線波長:0.7Å
・検出器:二次元半導体検出器ピラタス
・測定時間:10分/1試料
・試料:リンデマンガラスキャピラリー(0.3mmφ)に試料を充填
・測定方法:透過法
・測定温度:室温
より詳細には、本発明の正極材(第2態様)においては、六方晶の複合酸化物に、異相として単斜晶の複合酸化物(第2相の複合酸化物)が混合していることがより好ましい。
本発明の正極材(リチウム含有ニッケルマンガン複合酸化物)は、リチウム(Li)、ニッケル(Ni)およびマンガン(Mn)を含有する。
ニッケルとマンガンとを組み合わせることにより、相互の原子が不均化反応を起こす。これにより、充放電前後の自由エネルギー変化ΔGが大きく高電圧なNiの価数変化を、充放電反応に利用でき、高電圧かつ高容量のリチウムイオン二次電池が得られる。また、充電状態でも安定な結晶構造となるため、得られるリチウムイオン二次電池は、サイクル特性に優れる。
(1)LiaNixMn1-xAyO2
式(1)中、aは0.95超1.10未満の数であり、xは0.45以上0.60以下の数であり、yは0以上0.020以下の数であり、AはAl、Si、Ti、Zr、Ca、K、Ba、SrおよびSからなる群から選ばれる少なくとも1種である。
上述したように、本発明の正極材は、マンガンとニッケルとの組み合わせにより相互の原子が起こす不均化反応を利用している。この反応を十分に進行させるためには、マンガン原子とニッケル原子とが正極材中で隣接し、均一に分布していることが好ましい。
このため、本発明の正極材は、マンガン含有量とニッケル含有量とのモル比(Mn/Ni)の相対度数分布において、中央値が0.85以上1.20以下であり、かつ、半値幅が0.90以下であることが好ましい。中央値は0.90以上1.10以下がより好ましい。半値幅は0.80以下がより好ましい。
これにより、得られるリチウムイオン二次電池は、高電圧となり、放電容量およびサイクル特性がより優れる。
半値幅の下限は特に限定されず、半値幅は小さいほど好ましい。
まず、正極材の粉末を樹脂に包埋し、その後、集束イオンビーム加工装置を用いて薄片化することにより、TEM観察用の試料を得る。
得られた試料について、TEM(JEM-F200、JEOL社製)を用いて、観察し(観察条件:加速電圧200kV)、HAADF-STEM像を得る。
HAADF-STEM像について、形態観察し、TEMに付属する装置(Dual SDD、JEOL製)を用いてEDXを行ない(分析条件:加速電圧200kV)、元素マッピングする(マップの分解能:1.96nm/ピクセル)。
得られた元素マッピング結果から、正極材部分のみを抜き出し、MnおよびNiの含有量(モル量)の合計が100%となるように、各ピクセルにおいて、簡易定量計算する。 Mn含有量をNi含有量で除して、モル比(Mn/Ni)を求める。横軸にモル比(Mn/Ni)比を0.01ピッチでとり、縦軸に相対度数をとり、相対度数分布を作成する。作成した相対度数分布について、中央値および半値幅を求める。
本発明の正極材(リチウム含有ニッケルマンガン複合酸化物)は、温度25℃、湿度60%の大気雰囲気下に240時間放置したときの質量増加量(本段落において、単に「質量増加量」ともいう)が少ない方が好ましい。
なお、ここで、温度の「25℃」は「25±3℃」を意味し、湿度の「60%」は「60±5%」を意味する。
正極材を大気雰囲気下に放置すると、大気雰囲気中の水分および二酸化炭素と、正極材中のリチウムまたは正極材の表面に残留しているリチウム含有化合物とが反応して、正極材の質量が増加する場合がある。
正極材を大気雰囲気下に放置したときの質量増加量が少ないことは、大気雰囲気中の水分および二酸化炭素との反応による劣化が少ないことを意味する。質量増加量が少ない正極材は、大気雰囲気中での劣化が抑制され、ハンドリング性に優れる。また、電解液の分解を抑制する効果にも優れる。
また、正極材を大気雰囲気下に放置したときの質量増加量が少ないほど、水分および二酸化炭素との反応によって正極材中から引き抜かれるリチウムの量が少ない。この場合、正極材中からリチウムが引き抜かれることによる充放電容量の低下が抑制される。
具体的には、本発明の正極材の質量増加量は、0.75質量%以下が好ましく、0.70質量%以下がより好ましく、0.60質量%以下が更に好ましく、0.50質量%以下が特に好ましい。
質量増加量は、次のように求める。まず、試料(正極材)を所定量秤量してサンプル瓶に入れる。次いで、試料を入れたサンプル瓶を、大気雰囲気下で、温度25℃、湿度60%に保持した恒温恒湿槽に保管し、240時間放置する。放置前の試料の質量と放置後の試料の質量との差から、質量増加量を求める。
次に、本発明の正極材の製造方法を説明する。
本発明の正極材の製造方法は、上述した本発明の正極材を製造する方法であって、上述した本発明の前駆体と、リチウム含有化合物とを混合し、得られた混合物を焼成して焼成物を得る、正極材の製造方法である。
焼成物(リチウム含有ニッケルマンガン複合酸化物)が、適宜解砕等がなされた後に、上述した本発明の正極材となる。
本発明の前駆体と、リチウム含有化合物とを混合し、混合物を得る。
このとき、前駆体のニッケル換算の含有量と前駆体のマンガン換算の含有量との合計に対する、リチウム含有化合物のリチウム換算の含有量の比(以下、「Li/(Ni+Mn)」と表記する場合がある)は、モル比で、1.03超1.10未満が好ましく、1.04以上1.08以下がより好ましい。
Li/(Ni+Mn)がこの範囲内であれば、得られる本発明の正極材(第2態様)において、第2相の含有量が適量になりやすい。
A含有化合物としては、例えば、元素Aの水酸化物、酸化物、塩化物、塩(例えば、硫酸塩、炭酸塩、硝酸塩など)などが挙げられるが、これらに限定されない。
A含有化合物の混合量は、所望する組成に応じて、適宜調整される。
混合により得られた混合物を焼成して、焼成物を得る。
このとき、混合物を、400℃以上700℃以下の温度で仮焼成し、その後、800℃以上1000℃以下の温度で本焼成することが好ましい。
このような焼成条件であれば、得られる本発明の正極材(第2態様)において、第2相の含有量が適量になりやすい。
第2相の含有量がより適量になりやすいという理由から、本焼成の温度は、900℃以上1000℃以下がより好ましく、925℃以上975℃以下が更に好ましい。
本焼成の雰囲気は、特に限定されず、例えば、酸化性雰囲気(例えば、大気雰囲気など)または非酸化性雰囲気が挙げられる。
仮焼成の焼成時間は、6h以上48h以下が好ましく、12h以上36h以下がより好ましい。
本焼成の焼成時間は、5h以上30r以下が好ましく、10h以上25h以下がより好ましい。
焼成物は、水洗することが好ましい。水洗した焼成物は、その後、適宜乾燥して、正極材となる。水洗することにより、得られる正極材は、内部に入り込んでいない余分なリチウムが洗い流される。このため、充電状態での残留リチウム量が低減し、上述した質量増加量が少なくなる。
水洗および乾燥の後、更に、200℃以上800℃以下で焼成することが好ましい。
本発明のリチウムイオン二次電池は、上述した本発明の正極材を含有する正極と、負極と、正極と負極との間に介在してリチウムイオンを伝導するイオン伝導媒体と、を備えるリチウムイオン二次電池である。
本発明のリチウムイオン二次電池は、放電容量およびサイクル特性に優れる。
イオン伝導媒体は、例えば、非水電解液などの電解質である。
本発明のリチウムイオン二次電池は、更に、セパレータを備えていてもよい。
そのほか、本発明の正極材を用いること以外は、従来公知のリチウムイオン二次電池の構成を採用できる。
以下のようにして、前駆体No.1~前駆体No.5を製造した。
0.4モル/Lの硫酸ニッケル(NiSO4)水溶液と、0.4モル/Lの硫酸マンガン(MnSO4)水溶液と、0.2モル/Lの硫酸アンモニウム((NH4)2SO4)水溶液とを混合して、原料水溶液を得た。原料水溶液におけるモル比(NH4/(Ni+Mn))は0.25であった。原料水溶液のpHは4.6であった。
反応槽に、1Lの純水と水酸化ナトリウム水溶液とを加えて、pH11に調整した反応槽液を得た。
反応槽液に、原料水溶液を150mL/hの速度で投入した。原料水溶液の投入中、アルカリ性水溶液(10質量%の水酸化ナトリウム水溶液)を、反応槽液のpHが11になるように制御しながら、反応槽液に投入した。こうして、沈殿物を得た。このとき、攪拌棒によって反応槽液を450rpmで攪拌しつつ、反応槽液の温度を40℃に制御した。
沈殿物を、ろ別し、水洗した。その後、沈殿物を、真空乾燥機を用いて、真空雰囲気下にて、50℃で10h乾燥した。
こうして、前駆体No.1を得た。
沈殿物の乾燥雰囲気を大気雰囲気にした。この点以外は、前駆体No.1と同様にして、前駆体No.2を得た。
原料水溶液に硫酸アンモニウム水溶液を混合しなかった。この原料水溶液のpHは6.5であった。この原料水溶液と、0.06モル/Lのアンモニア水とをそれぞれ150mL/hの速度で反応槽液に投入した。原料水溶液およびアンモニア水の投入中、アルカリ性水溶液(10質量%の水酸化ナトリウム水溶液)を、反応槽液のpHが11になるように制御しながら、反応槽液に投入した。こうして、沈殿物を得た。
これらの点以外は、前駆体No.1と同様にして、前駆体No.3を得た。
沈殿物の乾燥雰囲気を大気雰囲気にした。沈殿物の乾燥温度を110℃にした。
これらの点以外は、前駆体No.1と同様にして、前駆体No.4を得た。
沈殿物の乾燥雰囲気を大気雰囲気にした。沈殿物の乾燥温度を110℃にした。
これらの点以外は、前駆体No.3と同様にして、前駆体No.5を得た。
得られた前駆体No.1~前駆体No.5について、それぞれ、モル比(Ni/(Ni+Mn))、Mn3sの交換分裂幅(ΔE)、Mnの平均価数、一次粒子径、質量減少量(大気雰囲気下で室温から1050℃まで加熱したときの質量減少量)、および、[001]/[101]ピーク比を求めた。結果を下記表1に示す。

得られた前駆体No.1~前駆体No.5を用いて、以下のようにして、正極材No.1~正極材No.10を製造した。
前駆体No.1と、水酸化リチウムとを混合して混合物を得た。混合の際のモル比(Li/(Ni+Mn))は1.05とした。得られた混合物を焼成して焼成物を得た。より詳細には、混合物を、窒素雰囲気下、650℃で24h仮焼成し、その後、大気雰囲気下、950℃で15h本焼成した。得られた焼成物を、乳鉢を用いて解砕した。焼成物は水洗しなかった。こうして、正極材No.1を得た。
それぞれ前駆体No.2~前駆体No.5を用いた。
この点以外は、正極材No.1と同様にして、それぞれ、正極材No.2~正極材No.5を得た。
混合物に、それぞれ、Al含有化合物(硝酸アルミニウム)、Ti含有化合物(硝酸チタン)、Zr含有化合物(硝酸ジルコニウム)、K含有化合物(硝酸カリウム)、および、Ba含有化合物(硝酸バリウム)を更に混合した。
この点以外は、正極材No.1と同様にして、それぞれ、正極材No.6~正極材No.10を得た。
得られた正極材No.1~正極材No.10について、組成、質量増加量(温度25℃、湿度60%の大気雰囲気下に240時間放置したときの質量増加量)、放電容量およびサイクル特性を求めた。結果を下記表2に示す。
放電容量およびサイクル特性は、以下のように求めた(後述する第2態様においても同様)。
正極材(90質量%)、アセチレンブラック(5質量%)およびポリフッ化ビニリデン(5質量%)に、N-メチル-2ピロリドンを添加し混練して、混合物を得た。得られた混合物を、アルミニウム集電体に、厚さ320μmで塗布し、塗膜を形成した。塗膜およびアルミニウム集電体の積層体を、隙間を80μmに設定したロールプレスを用いて加圧した。加圧した積層体から、直径14mmの円板を打ち抜いた。打ち抜いた円板を、150℃で15h真空乾燥した。真空乾燥後の円板を正極とした。
負極として、リチウム金属シートを用いた。セパレータとして、ポリプロピレン製の多孔質膜(セルガード#2400)を用いた。
エチレンカーボネート(EC)とジメチルカーボネート(DMC)との体積比(EC/DMC)が1/1である混合溶液1Lに、1モルのLiPF6を溶解させて、非水電解液を得た。
これらの正極、負極、セパレータおよび非水電解液を用いて、アルゴンで置換したグローブボックス内で、リチウムイオン二次電池(試験セル)を作製した。作製した試験セルを用いて、電流値を0.2Cの一定値とし、かつ、電圧を2.75~4.4Vの範囲で、充放電して、放電容量[mAh/g]を求めた。
上述した充放電を、電流値を0.5Cとして、40回(40サイクル)繰り返した。得られた放電容量[mAh/g]から、下記式を用いて、サイクル特性を計算した。
サイクル特性[%]=(第40サイクルの放電容量/第1サイクルの放電容量)×100

上記表1および表2に示すように、Mnの平均価数が4.0未満である前駆体No.1~前駆体No.3を用いて製造した正極材No.1~正極材No.3および正極材No.6~正極材No.10は、Mnの平均価数が4.0以上である前駆体No.4~前駆体No.5を用いて製造した正極材No.4~正極材No.5よりも、放電容量およびサイクル特性が優れていた。
得られた前駆体No.1を用いて、以下のようにして、正極材No.11~正極材No.18を製造した。
前駆体No.1と、水酸化リチウムとを混合して混合物を得た。混合の際のモル比(Li/(Ni+Mn))は1.05とした。得られた混合物を焼成して焼成物を得た。より詳細には、混合物を、窒素雰囲気下、650℃で24h仮焼成し、その後、大気雰囲気下、1000℃で5h本焼成した。得られた焼成物を、乳鉢を用いて解砕した。焼成物は水洗しなかった。こうして、正極材No.11を得た。
本焼成における焼成温度を900℃とし、焼成時間を15hとした。
これら以外の点は、正極材No.11と同様にして、正極材No.12を得た。
本焼成における焼成温度を950℃とし、焼成時間を10hとした。
これら以外の点は、正極材No.11と同様にして、正極材No.13を得た。
本焼成における焼成温度を950℃とし、焼成時間を15hとした。
焼成物を水洗し、その後、乾燥した。
これら以外の点は、正極材No.11と同様にして、正極材No.14を得た。
混合物に、S含有化合物(硫酸リチウム)を更に混合した。
本焼成における焼成温度を950℃とし、焼成時間を15hとした。
これら以外の点は、正極材No.11と同様にして、正極材No.15を得た。
混合の際のモル比(Li/(Ni+Mn))を1.20とした。
本焼成における焼成温度を950℃とし、焼成時間を10hとした。
これら以外の点は、正極材No.11と同様にして、正極材No.16を得た。
本焼成における焼成温度を780℃とし、焼成時間を15hとした。
これら以外の点は、正極材No.11と同様にして、正極材No.17を得た。
本焼成における焼成温度を1080℃とし、焼成時間を15hとした。
これら以外の点は、正極材No.11と同様にして、正極材No.18を得た。
得られた正極材No.11~正極材No.18について、組成、第2相(Li2MnO3)の含有量、質量増加量(温度25℃、湿度60%の大気雰囲気下に240時間放置したときの質量増加量)、放電容量およびサイクル特性を求めた。結果を下記表3に示す。
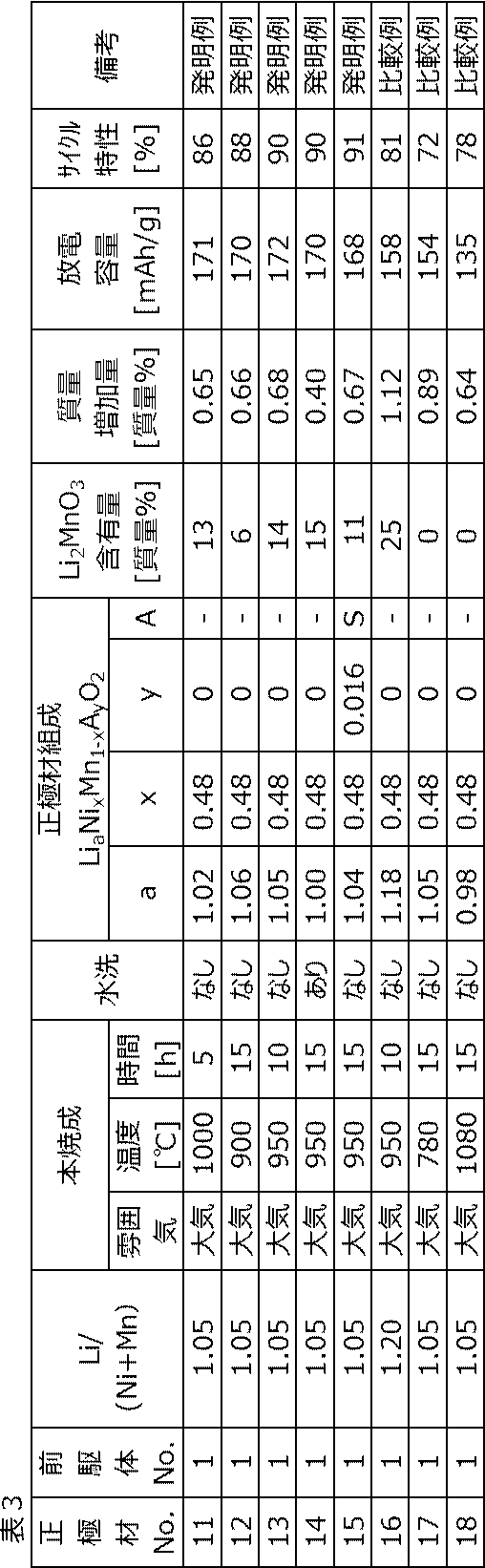
上記表3に示すように、第2相(Li2MnO3)の含有量が0質量%超20質量%以下である正極材No.11~正極材No.15は、第2相(Li2MnO3)の含有量が0質量%または20質量%超である正極材No.16~正極材No.18よりも、放電容量およびサイクル特性が優れていた。
正極材No.1~正極材No.3および正極材No.11について、上述した方法に従って、モル比(Mn/Ni)の相対度数分布の中央値および半値幅を求めた。放電容量およびサイクル特性の結果と併せて、下記表4に示す。
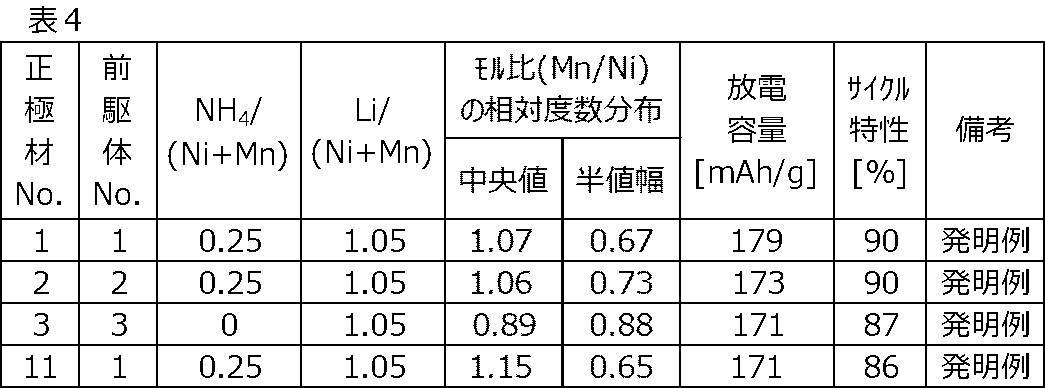
このとき、中央値が0.90以上1.10以下である正極材No.1および正極材No.2は、これを満たさない正極材No.3および正極材No.11と比較して、放電容量およびサイクル特性がより優れていた。
Claims (20)
- リチウムイオン二次電池に用いる正極材の前駆体であって、
ニッケルマンガン複合水酸化物およびニッケルマンガン複合酸化物からなる群から選ばれる少なくとも1種であり、
ニッケルおよびマンガンを含有し、
ニッケル含有量とマンガン含有量との合計に対するニッケル含有量の比が、モル比で、0.45以上0.60以下であり、
マンガンの平均価数が4.0未満である、前駆体。 - 一次粒子の平均粒子径が0.6μm未満である、請求項1に記載の前駆体。
- 大気雰囲気下で室温から1050℃まで加熱したときの質量減少量が16質量%以上である、請求項1または2に記載の前駆体。
- X線回折における[001]方向のピーク強度と[101]方向のピーク強度とのピーク強度比である[001]/[101]ピーク比が、14以下である、請求項1~3のいずれか1項に記載の前駆体。
ただし、前記[001]方向のピーク強度は、回折角2θが17~21°の範囲における最大ピーク強度であり、前記[101]方向のピークは、回折角2θが30~40°の範囲における最大ピーク強度である。 - 請求項1~4のいずれか1項に記載の前駆体を製造する方法であって、
ニッケル源、マンガン源、アンモニウム源、および、アルカリ性水溶液を、pHが9以上12以下である反応槽液に導入して、沈殿物を得る、前駆体の製造方法。 - 前記ニッケル源、前記マンガン源、および、前記アンモニウム源を含有する水溶液を原料水溶液とし、
前記原料水溶液と前記アルカリ性水溶液とを前記反応槽液に導入して、前記沈殿物を得る、請求項5に記載の前駆体の製造方法。 - 前記原料水溶液において、
前記ニッケル源のニッケル換算の含有量と前記マンガン源のマンガン換算の含有量との合計に対する、前記アンモニウム源のアンモニウム換算の含有量の比が、モル比で、0超1以下である、請求項6に記載の前駆体の製造方法。 - 前記原料水溶液のpHが6以下である、請求項6または7に記載の前駆体の製造方法。
- 前記沈殿物を、100℃以下の温度で乾燥する、請求項6~8のいずれか1項に記載の前駆体の製造方法。
- 前記沈殿物を、非酸化性雰囲気下で乾燥する、請求項6~9のいずれか1項に記載の前駆体の製造方法。
- リチウムイオン二次電池に用いる正極材であって、
リチウム含有ニッケルマンガン複合酸化物であり、
リチウム、ニッケルおよびマンガンを含有し、
請求項1~4のいずれか1項に記載の前駆体を用いて得られる、正極材。 - リチウムイオン二次電池に用いる正極材であって、
リチウム含有ニッケルマンガン複合酸化物であり、
リチウム、ニッケルおよびマンガンを含有し、
式Li2MnO3で表わされる複合酸化物の含有量が、0質量%超20質量%以下である、正極材。 - 更に、アルミニウム、ケイ素、チタン、ジルコニウム、カルシウム、カリウム、バリウム、ストロンチウムおよび硫黄からなる群から選ばれる少なくとも1種の元素Aを含有する、請求項11または12に記載の正極材。
- マンガン含有量とニッケル含有量とのモル比の相対度数分布において、中央値が0.85以上1.20以下であり、かつ、半値幅が0.90以下である、請求項11~13のいずれか1項に記載の正極材。
- 温度25℃、湿度60%の大気雰囲気下に240時間放置したときの質量増加量が0.75質量%以下である、請求項11~14のいずれか1項に記載の正極材。
- 請求項11~15のいずれか1項に記載の正極材を製造する方法であって、
請求項1~4のいずれか1項に記載の前駆体と、リチウム含有化合物とを混合し、得られた混合物を焼成して焼成物を得る、正極材の製造方法。 - 前記前駆体のニッケル換算の含有量と前記前駆体のマンガン換算の含有量との合計に対する、前記リチウム含有化合物のリチウム換算の含有量の比が、モル比で、1.03超1.10未満である、請求項16に記載の正極材の製造方法。
- 前記混合物を、400℃以上700℃以下の温度で仮焼成し、その後、800℃以上1000℃以下の温度で本焼成して、前記焼成物を得る、請求項16または17に記載の正極材の製造方法。
- 前記焼成物を水洗する、請求項16~18のいずれか1項に記載の正極材の製造方法。
- 請求項11~15のいずれか1項に記載の正極材を含有する正極と、負極と、前記正極と前記負極との間に介在してリチウムイオンを伝導するイオン伝導媒体と、を備えるリチウムイオン二次電池。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202080027397.7A CN113646929A (zh) | 2019-04-11 | 2020-04-07 | 前体、前体的制造方法、正极材料、正极材料的制造方法和锂离子二次电池 |
| EP20788108.7A EP3955347A4 (en) | 2019-04-11 | 2020-04-07 | PRECURSOR, METHOD OF MAKING A PRECURSOR, POSITIVE ELECTRODE MATERIAL, METHOD OF MAKING A POSITIVE ELECTRODE MATERIAL, AND LITHIUM-ION SECONDARY BATTERY |
| JP2020542028A JP7160930B2 (ja) | 2019-04-11 | 2020-04-07 | 前駆体、前駆体の製造方法、正極材、正極材の製造方法、および、リチウムイオン二次電池 |
| US17/602,422 US20220173391A1 (en) | 2019-04-11 | 2020-04-07 | Precursor, method for manufacturing precursor, positive electrode material, method for manufacturing positive electrode material, and lithium-ion secondary cell |
| CA3136583A CA3136583A1 (en) | 2019-04-11 | 2020-04-07 | Precursor, method for manufacturing precursor, positive electrode material, method for manufacturing positive electrode material, and lithium-ion secondary cell |
| KR1020217031999A KR20210134752A (ko) | 2019-04-11 | 2020-04-07 | 전구체, 전구체의 제조 방법, 정극재, 정극재의 제조 방법, 및 리튬이온 2 차 전지 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019075282 | 2019-04-11 | ||
| JP2019-075282 | 2019-04-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020209239A1 true WO2020209239A1 (ja) | 2020-10-15 |
Family
ID=72751279
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/015606 WO2020209239A1 (ja) | 2019-04-11 | 2020-04-07 | 前駆体、前駆体の製造方法、正極材、正極材の製造方法、および、リチウムイオン二次電池 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US20220173391A1 (ja) |
| EP (1) | EP3955347A4 (ja) |
| JP (1) | JP7160930B2 (ja) |
| KR (1) | KR20210134752A (ja) |
| CN (1) | CN113646929A (ja) |
| CA (1) | CA3136583A1 (ja) |
| TW (1) | TWI795640B (ja) |
| WO (1) | WO2020209239A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021256526A1 (ja) * | 2020-06-17 | 2021-12-23 | Basf戸田バッテリーマテリアルズ合同会社 | 非水電解質二次電池用正極活物質の製造方法 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116490469A (zh) * | 2020-11-30 | 2023-07-25 | 松下知识产权经营株式会社 | 非水电解质二次电池用正极活性物质及非水电解质二次电池 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002042813A (ja) | 2000-07-27 | 2002-02-08 | Matsushita Electric Ind Co Ltd | 正極活物質およびこれを含む非水電解質二次電池 |
| JP2003017049A (ja) * | 2001-06-27 | 2003-01-17 | Toyota Central Res & Dev Lab Inc | リチウム二次電池正極活物質用リチウム遷移金属複合酸化物およびその製造方法 |
| JP2005097087A (ja) * | 2003-07-18 | 2005-04-14 | Tosoh Corp | 新規なリチウム・ニッケル・マンガン複合酸化物およびその製造方法 |
| JP2014099295A (ja) * | 2012-11-13 | 2014-05-29 | Nippon Chemicon Corp | リチウムイオン二次電池用電極材料、この電極材料の製造方法、及びリチウムイオン二次電池 |
| WO2018169004A1 (ja) * | 2017-03-16 | 2018-09-20 | 国立研究開発法人産業技術総合研究所 | ニッケルマンガン系複合酸化物及びその製造方法 |
Family Cites Families (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004002141A (ja) * | 2002-03-07 | 2004-01-08 | Tosoh Corp | リチウム−ニッケル−マンガン複合酸化物とその製造方法及びそれを用いるリチウムイオン二次電池 |
| CN100381365C (zh) * | 2003-04-17 | 2008-04-16 | 清美化学股份有限公司 | 含锂-镍-钴-锰复合氧化物及锂二次电池用正极活性物质用原料和它们的制造方法 |
| EP1699099A4 (en) * | 2003-11-06 | 2008-12-24 | Panasonic Corp | ALKALIBATTERY AND MATERIAL FOR THE POSITIVE ELECTRODE OF AN ALKALIBATTERY |
| US8080340B2 (en) * | 2004-09-03 | 2011-12-20 | Uchicago Argonne, Llc | Manganese oxide composite electrodes for lithium batteries |
| JP5348510B2 (ja) * | 2009-09-04 | 2013-11-20 | トヨタ自動車株式会社 | リチウム二次電池用正極活物質およびその利用 |
| WO2011052607A1 (ja) * | 2009-10-29 | 2011-05-05 | Agcセイミケミカル株式会社 | リチウムイオン二次電池用正極材料の製造方法 |
| JP5973128B2 (ja) * | 2010-11-29 | 2016-08-23 | 住友金属鉱山株式会社 | 非水系電解質二次電池用正極活物質およびその製造方法、ならびに該正極活物質を用いた非水系電解質二次電池 |
| JP4915488B1 (ja) * | 2011-03-28 | 2012-04-11 | 住友金属鉱山株式会社 | ニッケルマンガン複合水酸化物粒子とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、および非水系電解質二次電池 |
| WO2012164752A1 (ja) * | 2011-05-30 | 2012-12-06 | 住友金属鉱山株式会社 | 非水系二次電池用正極活物質とその製造方法、ならびに該正極活物質を用いた非水系電解質二次電池 |
| US20170050864A1 (en) * | 2011-05-30 | 2017-02-23 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for nonaqueous secondary batteries, method for producing same, and nonaqueous electrolyte secondary battery using positive electrode active material |
| KR101958038B1 (ko) * | 2011-06-24 | 2019-03-13 | 스미또모 가가꾸 가부시끼가이샤 | 리튬 이온 이차 전지용 정극 활물질의 제조 방법 |
| TWI636613B (zh) * | 2013-07-18 | 2018-09-21 | 日商東曹股份有限公司 | 鎳-錳系複合氧氫氧化物及其製造方法、以及其用途 |
| JP6244713B2 (ja) * | 2013-07-24 | 2017-12-13 | 住友金属鉱山株式会社 | 非水電解質二次電池用正極活物質の製造方法 |
| JP6237331B2 (ja) * | 2014-02-27 | 2017-11-29 | 住友金属鉱山株式会社 | 非水電解質二次電池用正極活物質の前駆体とその製造方法、及び非水電解質二次電池用正極活物質とその製造方法 |
| JP6587804B2 (ja) * | 2015-01-23 | 2019-10-09 | 住友化学株式会社 | 正極活物質、リチウムイオン二次電池用正極およびリチウムイオン二次電池 |
| JP6347227B2 (ja) * | 2015-04-28 | 2018-06-27 | 住友金属鉱山株式会社 | マンガンニッケルチタン複合水酸化物粒子とその製造方法、および、非水系電解質二次電池用正極活物質の製造方法 |
| JP6929793B2 (ja) * | 2016-02-03 | 2021-09-01 | 住友化学株式会社 | 正極活物質、リチウムイオン二次電池用正極およびリチウムイオン二次電池 |
| JP6292250B2 (ja) * | 2016-05-16 | 2018-03-14 | 住友金属鉱山株式会社 | 非水系電解質二次電池用正極活物質およびその製造方法、ならびに該正極活物質を用いた非水系電解質二次電池 |
| KR102373218B1 (ko) * | 2016-05-24 | 2022-03-10 | 스미또모 가가꾸 가부시끼가이샤 | 정극 활물질, 그 제조 방법 및 리튬 이온 이차 전지용 정극 |
| JP6583359B2 (ja) * | 2017-07-27 | 2019-10-02 | 住友金属鉱山株式会社 | ニッケルコバルトマンガン複合水酸化物 |
| TWI651272B (zh) * | 2017-09-07 | 2019-02-21 | 明志科技大學 | 一種富鋰-鋰鎳錳氧化物陰極複合材料的製備方法及其用途 |
-
2020
- 2020-04-07 JP JP2020542028A patent/JP7160930B2/ja active Active
- 2020-04-07 KR KR1020217031999A patent/KR20210134752A/ko not_active Application Discontinuation
- 2020-04-07 CA CA3136583A patent/CA3136583A1/en active Pending
- 2020-04-07 US US17/602,422 patent/US20220173391A1/en active Pending
- 2020-04-07 CN CN202080027397.7A patent/CN113646929A/zh active Pending
- 2020-04-07 WO PCT/JP2020/015606 patent/WO2020209239A1/ja unknown
- 2020-04-07 EP EP20788108.7A patent/EP3955347A4/en active Pending
- 2020-04-10 TW TW109112216A patent/TWI795640B/zh active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002042813A (ja) | 2000-07-27 | 2002-02-08 | Matsushita Electric Ind Co Ltd | 正極活物質およびこれを含む非水電解質二次電池 |
| JP2003017049A (ja) * | 2001-06-27 | 2003-01-17 | Toyota Central Res & Dev Lab Inc | リチウム二次電池正極活物質用リチウム遷移金属複合酸化物およびその製造方法 |
| JP2005097087A (ja) * | 2003-07-18 | 2005-04-14 | Tosoh Corp | 新規なリチウム・ニッケル・マンガン複合酸化物およびその製造方法 |
| JP2014099295A (ja) * | 2012-11-13 | 2014-05-29 | Nippon Chemicon Corp | リチウムイオン二次電池用電極材料、この電極材料の製造方法、及びリチウムイオン二次電池 |
| WO2018169004A1 (ja) * | 2017-03-16 | 2018-09-20 | 国立研究開発法人産業技術総合研究所 | ニッケルマンガン系複合酸化物及びその製造方法 |
Non-Patent Citations (2)
| Title |
|---|
| ELECTROCHEMICAL SOCIETY BATTERY TECHNICAL COMMITTEE: "DENCHI HANDBOOK [Battery Handbook]", 30 November 2009, THE ELECTROCHEMICAL SOCIETY OF JAPAN, JP, ISBN: 978-4-274-20805-8, article OHMSHA LTD.: "Passage", pages: 450 - 457, XP009524164 * |
| See also references of EP3955347A4 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021256526A1 (ja) * | 2020-06-17 | 2021-12-23 | Basf戸田バッテリーマテリアルズ合同会社 | 非水電解質二次電池用正極活物質の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2020209239A1 (ja) | 2021-05-06 |
| CN113646929A (zh) | 2021-11-12 |
| EP3955347A4 (en) | 2022-06-08 |
| TWI795640B (zh) | 2023-03-11 |
| EP3955347A1 (en) | 2022-02-16 |
| CA3136583A1 (en) | 2020-10-15 |
| US20220173391A1 (en) | 2022-06-02 |
| JP7160930B2 (ja) | 2022-10-25 |
| KR20210134752A (ko) | 2021-11-10 |
| TW202039371A (zh) | 2020-11-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Li et al. | Retarded phase transition by fluorine doping in Li-rich layered Li1. 2Mn0. 54Ni0. 13Co0. 13O2 cathode material | |
| US20190221838A1 (en) | Positive electrode active material containing lithium composite oxide and covering material and battery | |
| US9564634B2 (en) | Positive electrode active substance particles and process for producing the same, and non-aqueous electrolyte secondary battery | |
| CN110121481A (zh) | 镍锰复合氢氧化物和其制造方法、非水系电解质二次电池用正极活性物质和其制造方法以及非水系电解质二次电池 | |
| US11239463B2 (en) | Process for producing cathode active material, cathode active material, positive electrode, and lithium ion secondary battery | |
| JP7160930B2 (ja) | 前駆体、前駆体の製造方法、正極材、正極材の製造方法、および、リチウムイオン二次電池 | |
| KR20180105144A (ko) | 정극 활물질, 리튬 이온 이차 전지용 정극 및 리튬 이온 이차 전지 | |
| JP7415336B2 (ja) | リチウムイオン二次電池用正極活物質前駆体、リチウムイオン二次電池用正極活物質、リチウムイオン二次電池用正極活物質前駆体の製造方法、リチウムイオン二次電池用正極活物質の製造方法、リチウムイオン二次電池 | |
| JP7414056B2 (ja) | リチウムイオン二次電池用正極活物質、リチウムイオン二次電池用正極活物質の製造方法、リチウムイオン二次電池 | |
| JP7363885B2 (ja) | リチウムイオン二次電池用正極活物質、リチウムイオン二次電池用正極活物質の製造方法、リチウムイオン二次電池 | |
| JP2018160323A (ja) | 非水系電解質二次電池用正極活物質とその前駆体、及びそれらの製造方法 | |
| US20230163294A1 (en) | Lithium-metal composite oxide, positive electrode active material for lithium secondary battery, positive electrode for lithium secondary battery, and lithium secondary battery | |
| Yamashita et al. | Hydrothermal synthesis and electrochemical properties of Li2FexMnxCo1− 2xSiO4/C cathode materials for lithium-ion batteries | |
| JP7441999B1 (ja) | リチウム金属複合酸化物、リチウム二次電池用正極活物質、リチウム二次電池用正極、および、リチウム二次電池 | |
| JP7148685B1 (ja) | リチウム二次電池用正極活物質、リチウム二次電池用正極及びリチウム二次電池 | |
| JP7416897B1 (ja) | リチウム金属複合酸化物、リチウム二次電池用正極活物質、リチウム二次電池用正極、及びリチウム二次電池 | |
| CN114400317B (zh) | 一种正极材料及其制备方法、锂离子电池 | |
| JP7441998B1 (ja) | リチウム金属複合酸化物、リチウム二次電池用正極活物質、リチウム二次電池用正極、および、リチウム二次電池 | |
| JP7454642B1 (ja) | リチウム金属複合酸化物、リチウム二次電池用正極活物質、リチウム二次電池用正極及びリチウム二次電池 | |
| JP7359911B1 (ja) | 前駆体及びリチウム二次電池用正極活物質の製造方法 | |
| JP7412485B1 (ja) | 金属複合水酸化物粒子及びリチウム二次電池用正極活物質の製造方法 | |
| WO2023106274A1 (ja) | リチウム二次電池用正極活物質、リチウム二次電池用正極及びリチウム二次電池 | |
| JP7395944B2 (ja) | リチウムイオン二次電池用正極活物質前駆体、リチウムイオン二次電池用正極活物質、リチウムイオン二次電池用正極活物質前駆体の製造方法、リチウムイオン二次電池用正極活物質の製造方法、リチウムイオン二次電池 | |
| WO2024014551A1 (ja) | 金属複合化合物、金属複合化合物の製造方法、及びリチウム金属複合酸化物の製造方法 | |
| WO2023249013A1 (ja) | リチウム二次電池用正極活物質、リチウム二次電池用正極、リチウム二次電池、およびリチウム二次電池用正極活物質の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2020542028 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20788108 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20217031999 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 3136583 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2020788108 Country of ref document: EP Effective date: 20211111 |