WO2015145968A1 - トナーおよびトナーの製造方法 - Google Patents
トナーおよびトナーの製造方法 Download PDFInfo
- Publication number
- WO2015145968A1 WO2015145968A1 PCT/JP2015/000957 JP2015000957W WO2015145968A1 WO 2015145968 A1 WO2015145968 A1 WO 2015145968A1 JP 2015000957 W JP2015000957 W JP 2015000957W WO 2015145968 A1 WO2015145968 A1 WO 2015145968A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- toner
- resin
- particles
- group
- parts
- Prior art date
Links
Images



Classifications
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/097—Plasticisers; Charge controlling agents
- G03G9/09733—Organic compounds
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/0802—Preparation methods
- G03G9/0804—Preparation methods whereby the components are brought together in a liquid dispersing medium
- G03G9/0806—Preparation methods whereby the components are brought together in a liquid dispersing medium whereby chemical synthesis of at least one of the toner components takes place
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/0802—Preparation methods
- G03G9/081—Preparation methods by mixing the toner components in a liquefied state; melt kneading; reactive mixing
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/0825—Developers with toner particles characterised by their structure; characterised by non-homogenuous distribution of components
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/087—Binders for toner particles
- G03G9/08784—Macromolecular material not specially provided for in a single one of groups G03G9/08702 - G03G9/08775
- G03G9/08795—Macromolecular material not specially provided for in a single one of groups G03G9/08702 - G03G9/08775 characterised by their chemical properties, e.g. acidity, molecular weight, sensitivity to reactants
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/087—Binders for toner particles
- G03G9/08784—Macromolecular material not specially provided for in a single one of groups G03G9/08702 - G03G9/08775
- G03G9/08797—Macromolecular material not specially provided for in a single one of groups G03G9/08702 - G03G9/08775 characterised by their physical properties, e.g. viscosity, solubility, melting temperature, softening temperature, glass transition temperature
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/097—Plasticisers; Charge controlling agents
- G03G9/09708—Inorganic compounds
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/097—Plasticisers; Charge controlling agents
- G03G9/09733—Organic compounds
- G03G9/0975—Organic compounds anionic
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/097—Plasticisers; Charge controlling agents
- G03G9/09783—Organo-metallic compounds
Definitions
- the present invention relates to a toner for developing an electrostatic charge image used in image forming methods such as electrophotography and electrostatic printing and a method of producing the toner.
- Patent Document 1 proposes a toner containing a charge control resin having a benzyloxysalicylic acid structure. According to this, it has favorable chargeability, and even when stored under high temperature and high humidity, it is possible to suppress the decrease in fluidity and to stabilize the charging performance.
- Patent Document 2 proposes a toner having a resin having a power generation function inside the toner and having resin particles having a charge dissipation function fixed to the toner surface.
- the charge density on the toner surface is kept appropriately low, electrostatic aggregation is suppressed even if the external additive is embedded after printing on a large number of sheets, image defects are less likely to occur, and durability is improved in a normal environment. doing.
- the stability and durability of the charging performance under severe environments are not sufficient.
- the durability after printing on a large number of sheets is insufficient in a high temperature and high humidity environment, and the image stability is not satisfactory.
- toner with stable chargeability, sufficient durability, and high image stability in a high-speed, one-component development system, and a method for producing the same. It is to do.
- the present invention is a toner having toner particles obtained by fixing resin particles on the surface of toner base particles containing a binder resin, a colorant and a release agent,
- the present invention relates to a toner characterized in that the resin particles contain a resin having an ionic functional group and a pKa (acid dissociation constant) of 7.0 or more and 9.0 or less.
- the present invention is also a method for producing a toner having toner particles obtained by fixing resin particles on the surface of toner base particles containing a binder resin, a colorant and a release agent, (I) forming, in an aqueous medium, particles of a polymerizable monomer composition containing a polymerizable monomer and a colorant; (Ii) polymerizing the polymerizable monomer contained in the particles of the polymerizable monomer composition to obtain a dispersion B containing the toner base particles; (Iii) adding the resin particles to the dispersion B to obtain a dispersion C; (Iv) heating the dispersion C above the glass transition temperature (Tg) of the toner base particles to fix the resin particles on the surface of the toner base particles to obtain toner particles in this order
- the present invention relates to a method for producing a toner, wherein the resin particles contain a resin having an ionic functional group and a pKa (acid dissociation constant) of
- the chargeability is stable not only in a normal environment but also in a severe environment, and the durability is excellent, and the image formation is performed at high speed in a one-component development system. Also, it is possible to provide a toner capable of obtaining excellent image stability.
- FIG. 2 is an enlarged view of a developing unit of the electrophotographic apparatus.
- FIG. 1 is a cross-sectional view of an electrophotographic apparatus using the image forming method of the present invention.
- a resin having a pKa (acid dissociation constant) having an ionic functional group on the surface of a toner base particle containing a binder resin, a coloring agent, and a releasing agent is 7.0 or more and 9.0 or less
- the present invention relates to a toner having toner particles to which resin particles containing resin A) are fixed.
- Such toners have stable chargeability not only in a normal environment but also in severe environments, and are excellent in durability, and are cases where image formation is performed at high speed in a one-component development system. Also, it has the excellent property that excellent image stability can be obtained.
- the resin having a pKa (acid dissociation constant) having an ionic functional group of 7.0 or more and 9.0 or less exhibits excellent charging performance in a high humidity environment, so that point will be described.
- a resin which has an ionic functional group what has functional groups, such as a sulfonic acid and carboxylic acid, is used in many cases.
- functional groups such as a sulfonic acid and carboxylic acid
- the amount of charge may decrease due to the influence.
- pKa (acid dissociation constant) is 7.0 or more and 9.0 or less, the hygroscopicity of the resin can be reduced, and the reduction of the charge amount in a high humidity environment can be suppressed.
- pKa acid dissociation constant
- the amount of adsorbed water increases, and the chargeability decreases under high humidity.
- the pKa acid dissociation constant
- the chargeability is low and a sufficient charge amount can not be expressed.
- Any resin having an ionic functional group may be used as long as it satisfies the above pKa (acid dissociation constant).
- a resin having a hydroxyl group bonded to an aromatic ring and a resin having a carboxyl group bonded to an aromatic ring are exemplified.
- the resin A is more preferably a resin having a monovalent group a represented by the following formula (1) as a molecular structure.
- R 1 represents a hydroxy group, a carboxy group, an alkyl group having 1 to 18 carbon atoms, or an alkoxy group having 1 to 18 carbon atoms
- R 2 represents a hydrogen atom or a hydroxy group Represents an alkyl group having 1 to 18 carbon atoms, or an alkoxy group having 1 to 18 carbon atoms
- g represents an integer of 1 to 3
- h represents an integer of 0 to 3 h is 2 Or when it is 3, h R 1 may be the same or different.
- Examples of the alkyl group in R 1 and R 2 include methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, s-butyl group, t-butyl group and the like, and examples of the alkoxy group And examples thereof include a methoxy group, an ethoxy group and a propoxy group.
- a more preferable structure of the monovalent group a represented by the formula (1) is that R 1 represents an alkyl group having 1 to 18 carbon atoms, or an alkoxy group having 1 to 18 carbon atoms, and R 2 represents Represents a hydrogen atom, g represents an integer of 1 or more and 3 or less, h represents an integer of 0 or more and 3 or less, and when h is 2 or 3, h R 1 s are identical or different May be
- the main chain structure of the resin A is not particularly limited.
- vinyl polymers, polyester polymers, polyamide polymers, polyurethane polymers, polyether polymers and the like can be mentioned.
- the polymer of the hybrid type which these 2 or more types were combined is also mentioned.
- polyester polymers or vinyl polymers are preferable. More preferably, it is a vinyl polymer having a monovalent group a represented by the formula (1) as a partial structure represented by the following formula (2).
- R 3 represents a hydroxy group, a carboxy group, an alkyl group having 1 to 18 carbon atoms, or an alkoxy group having 1 to 18 carbon atoms
- R 4 represents a hydrogen atom, a hydroxy group, an alkyl group having 1 to 18 carbon atoms, or an alkoxy group having 1 to 18 carbon atoms
- R 5 represents a hydrogen atom or a methyl group
- i represents an integer of 1 or more and 3 or less
- j represents an integer of 0 or more and 3 or less
- R 3 can be independently selected.
- Examples of the alkyl group in R 3 and R 4 include methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, s-butyl group, t-butyl group and the like, and examples of alkoxy group And the like, and examples thereof include a methoxy group, an ethoxy group and a propoxy group.
- the weight average molecular weight calculated by gel permeation chromatography is 1000 or more and 100000 or less. Within this range, the strength and chargeability of the resin particles can be maintained in a well-balanced manner. In order to make a weight average molecular weight into the said range, it is controllable by changing conditions, such as the amount of reagents at the time of manufacturing resin A, reaction temperature, solvent concentration. Further, resin A having a desired molecular weight can be obtained by separation and separation by GPC.
- content of monovalent group a shown by Formula (1) per 1 g of resin A is 50 to 1000 micromol.
- amount By setting the amount to 50 ⁇ mol or more, good chargeability and durability can be exhibited. Moreover, charge up can be suppressed by setting it as 1000 micromol or less.
- charge control resin those having an acidic polar group such as sulfonic acid or carboxylic acid are often used. Such resins tend to adsorb moisture, and under high temperature and humidity conditions, the charging performance may not be exhibited due to the influence.
- Resin particles containing the resin A having a monovalent group a represented by the formula (1) exhibit charging performance.
- the part corresponding to the polar group is constituted by a monovalent group a represented by the formula (1).
- the monovalent group a represented by the formula (1) has a large pKa (acid dissociation constant) as compared with the structure of the polar part used in a general charge control resin. Therefore, it is considered that the pKa of the resin A is also increased, and the influence of water adsorption is reduced.
- the pKa of the resin A is more preferably 7.0 or more and 8.0 or less. If it is 7.0 or more, the effect of reducing the environmental difference of the chargeability by water adsorption becomes high. By setting it as 8.0 or less, a favorable charge amount can be exhibited. Although the method of determining pKa will be described later, it can be determined from the neutralization titration result.
- a manufacturing method of resin A It does not specifically limit as a manufacturing method of resin A, It can manufacture by a well-known method.
- a vinyl-based polymer for example, a polymerizable monomer M (following formula (3)) having a monovalent group a represented by the formula (1) and a vinyl-based monomer are exemplified. It is a method of copolymerizing using a polymerization initiator.
- R 6 represents a hydroxy group, a carboxy group, an alkyl group having 1 to 18 carbon atoms, or an alkoxy group having 1 to 18 carbon atoms
- R 7 represents a hydrogen atom, a hydroxy group, an alkyl group having 1 to 18 carbon atoms, or an alkoxy group having 1 to 18 carbon atoms
- R 8 represents a hydrogen atom or a methyl group
- k represents an integer of 1 or more and 3 or less
- l represents an integer of 0 or more and 3 or less
- R 6 can be independently selected.
- the vinyl monomer to be copolymerized with the polymerizable monomer M is not particularly limited.
- styrene such as styrene, o-methylstyrene, m-methylstyrene, p-methylstyrene, ⁇ -methylstyrene and derivatives thereof; ethylene unsaturated monoolefins such as ethylene, propylene, butylene and isobutylene; chloride Vinyl, vinylidene chloride, vinyl bromide, vinyl halides such as vinyl fluoride; vinyl ester acids such as vinyl acetate, vinyl propionate, vinyl benzoate; n-butyl acrylate, such as 2-ethylhexyl acrylate Acrylic acid esters; Methacrylic acid esters in which the acrylic acid esters are converted to methacrylic acid; methacrylic acid amino esters such as dimethylaminoethyl methacrylate, diethylaminoethyl me
- a polymerization initiator which can be used when copolymerizing the above-mentioned polymerizable monomer component
- various things such as a peroxide system polymerization initiator and an azo system polymerization initiator
- peroxide type polymerization initiator which can be used, peroxy ester, peroxy dicarbonate, dialkyl peroxide, peroxy ketal, ketone peroxide, hydroperoxide, diacyl peroxide is mentioned as an organic type.
- examples of the inorganic type include persulfates and hydrogen peroxide.
- Peroxy esters such as butyrate, t-butylperoxyisopropyl monocarbonate, t-butylperoxy 2-ethylhexyl monocarbonate, etc .
- diacyl peroxides such as benzoyl peroxide
- peroxy dicarbonates such as diisopropyl peroxy dicarbonate
- 1 Peroxyketals such as 1-di-t-hexylperoxycyclohexane
- dialkyl peroxides such as di-t-butyl peroxide
- others such as t-butyl peroxy allyl monocarbonate etc.
- azo polymerization initiator which can be used, 2,2'-azobis- (2,4-dimethylvaleronitrile), 2,2'-azobisisobutyronitrile, 1,1'-azobis (cyclohexane- 1-carbonitrile), 2,2'-azobis-4-methoxy-2,4-dimethylvaleronitrile, azobisisobutyronitrile, dimethyl-2,2'-azobis (2-methyl propionate), etc. It is illustrated.
- polymerization initiators 2 or more types can also be used simultaneously as needed. It is preferable that the usage-amount of the polymerization initiator used in this case is 0.100 mass part or more and 20.0 mass parts or less with respect to 100 mass parts of polymerizable monomers.
- the polymerization method any method such as solution polymerization, suspension polymerization, emulsion polymerization, dispersion polymerization, precipitation polymerization, bulk polymerization can be used, and it is not particularly limited.
- resin A which has monovalent group a shown by Formula (1) is a polyester resin
- various well-known manufacturing methods can be utilized. For example, I) A method of converting into a monovalent group a represented by the formula (1) by an organic reaction using a reaction residue of a carboxy group or a hydroxy group contained in a polyester structure; II) A method of producing a polyester using a polyhydric alcohol or polyhydric carboxylic acid having a monovalent group a represented by the formula (1) as a substituent; III) A method of previously introducing a functional group capable of easily introducing a monovalent group a represented by the formula (1) as a substituent into a polyhydric alcohol or polyhydric carboxylic acid; Etc.
- IV) A method of hybridizing a polyester resin containing a monovalent group a represented by the formula (1) as a substituent with a vinyl monomer; V) A method of converting a carboxy group into a structure represented by the formula (1) by an organic reaction after polymerization using a monomer having a carboxy group such as acrylic acid or methacrylic acid as a vinyl monomer; VI) A method of hybridizing a polyester resin using a polymerizable monomer M having a structure a represented by the formula (3); Etc.
- Known methods can be used as a method of hybridizing a polyester resin with a vinyl monomer, and it is effective as a method of IV). Specifically, a method of vinyl-modifying a polyester with a peroxide-based initiator, a method of graft-modifying a polyester resin having an unsaturated group to prepare a hybrid resin, and the like can be mentioned.
- the carboxy group present in the resin is converted to the amino group to the monovalent group a represented by the formula (1) And the like, and the like.
- the polymerizable monomer M represented by the above-mentioned Formula (3) can be used.
- the weight average molecular weight of the resin A As a method of adjusting the weight average molecular weight of the resin A, known methods can be used. Specifically, in the case of a polyester resin, it can be arbitrarily adjusted by adjusting the preparation ratio of the acid component and the alcohol component and the polymerization time. In addition to the adjustment of the molecular weight of the polyester component in the hybrid resin, the adjustment of the weight average molecular weight of the polymer is also possible by the adjustment of the molecular weight of the vinyl modification unit. Specifically, it can be arbitrarily adjusted by adjusting the amount of radical initiator, polymerization temperature and the like in the reaction step of vinyl modification. As a vinyl monomer which can be used for hybridization of a polyester resin by this invention, the vinyl type monomer mentioned above can be used.
- the content of the monovalent group a represented by the formula (1) based on the total mass of the resin A can be determined by the method described below. First, the acid value of the resin A is quantified by titrating the resin A according to the method described later, and the amount of carboxy group derived from the monovalent group a represented by the formula (1) that the resin A has is calculated. Then, based on this, the content ( ⁇ mol) of the monovalent group a represented by the formula (1) per 1 g of the resin A can be calculated.
- NMR NMR can be measured, the molar ratio of each component can be calculated from the integral value derived from the characteristic chemical shift value of each monomer component, and the content ( ⁇ mol) can be calculated based thereon.
- the toner of the present invention is a toner in which resin particles containing resin A are fixed.
- the resin particles may be produced by any method, and may be in the form of powder or dispersed in any medium, but those dispersed in an aqueous medium are preferably used.
- those produced by known methods such as emulsion polymerization, soap-free emulsion polymerization and phase inversion emulsification can be used.
- the phase inversion emulsification method is particularly preferable because it does not require an emulsifier or a dispersion stabilizer and resin particles having a smaller particle diameter can be easily obtained.
- a resin having a self-dispersing property or a resin capable of exhibiting a self-dispersing property by neutralization is used.
- self-dispersion in an aqueous medium is exhibited in a resin having a hydrophilic group in the molecule.
- good self-dispersion is exhibited in a resin having a polyether group or an ionic group.
- the resin A since a carboxy group exists in the monovalent group a, self-dispersion is thereby expressed. By neutralizing this carboxy group, the hydrophilicity is increased and self-dispersion in an aqueous medium becomes possible.
- phase inversion emulsification method it is possible to obtain a stable aqueous dispersion of resin particles without substantially using an emulsifier or a dispersion stabilizer.
- the toner of the present invention since embedding of the external additive can be suppressed even in a high temperature and high humidity environment, the image stability (so-called durability) can be maintained even after printing on a large number of sheets.
- a resin having a monovalent group a represented by the formula (1) is used, particularly remarkable effects can be obtained.
- the mechanism is not clear but is considered as follows.
- the monovalent group a represented by the formula (1) has a structure in which the salicylic acid moiety is bonded via a benzyloxy group, so the flexibility is high, and the resin a has a structure which protrudes from the main chain.
- the carboxy group of this salicylic acid is considered to be oriented to the outermost surface because of its high polarity. Therefore, the resin particle containing the resin A having the monovalent group a is disposed at the outermost surface and in close proximity to the salicylic acid site, so that a network of hydrogen bonds is formed and the surface strength of the resin particle is increased. I think that is. Since the concentration is higher than when resin A is added as a component of the toner base particle in advance, a dense hydrogen bond is formed, and it is considered that a higher effect is exhibited.
- the fixed amount of the resin particles is preferably 0.1 parts by mass or more and less than 5.0 parts by mass with respect to 100 parts by mass of the toner base particles.
- the content is preferably 0.1 parts by mass or more and less than 5.0 parts by mass with respect to 100 parts by mass of the toner base particles.
- the resin particles may be embedded in the toner base particles by mechanical impact in order to be sufficiently fixed after being attached to the surface of the toner base particles.
- the resin particles may be fixed by heating to a temperature higher than the glass transition temperature (Tg) of the resin particles and smoothing.
- the toner particles contain at least one metal element selected from magnesium, calcium, barium and aluminum because the durability is further excellent.
- the present invention is that the carboxy group on the toner base particle surface and the carboxy group on the resin particle are bonded to the same metal element and the resin particle is firmly fixed to the toner base particle surface. The person is thinking.
- the toner base particles contain a resin having a carboxy group.
- the resin having a carboxy group those similar to the resins usable for the binder resin described later are suitably used. It is preferable that a carboxy group is introduced into these resins so that the acid value of the resin is 5.0 mg KOH / g or more and 30.0 mg KOH / g or less. When the acid value is in this range, the carboxy group is effectively oriented on the surface of the toner base particle, and the introduction of the metal element is promoted.
- the metal element is preferably contained in an amount of 10 ppm to 1000 ppm based on the total mass of the toner particles. Furthermore, it is preferable that it is 20 ppm or more and 200 ppm or less with respect to the total mass of the toner particles. Furthermore, it is particularly preferable that the content is 50 ppm or more and 200 ppm or less with respect to the total mass of the toner particles. When the content of the metal element is in this range, the chargeability and durability under high temperature and high humidity conditions are particularly excellent. The method of quantifying the content of the metal element will be described later.
- any method may be used to contain the metal element in the toner, a method in which resin particles are fixed to the surface of toner base particles in an aqueous medium is preferable.
- the resin particles are fixed at a pH higher than pKa, the carboxy groups on the surface of the resin particles are likely to dissociate, thereby promoting bonding with the metal element and promoting the content of the metal element.
- binder resin used in the toner of the present invention there are no particular restrictions on the binder resin used in the toner of the present invention.
- the following can be illustrated.
- the following are preferably used on the toner characteristics.
- polyester resin a polyester resin which is usually produced using a polyhydric alcohol and a carboxylic acid, or a carboxylic acid anhydride, or a carboxylic acid ester as a raw material monomer can be used.
- the same polyhydric alcohol component and polyhydric carboxylic acid component as the polyester resin described above can be used.
- polyester resins obtained by condensation polymerization of the components listed below are particularly preferable.
- a diol component a bisphenol derivative.
- a carboxylic acid comprising a divalent or higher carboxylic acid or an acid anhydride thereof; fumaric acid, maleic acid, maleic anhydride, phthalic acid, terephthalic acid, trimellitic acid, lower alkyl esters such as pyromellitic acid component.
- the toner of the present invention can also be used as a magnetic toner, in which case the following magnetic materials are used.
- Iron oxides such as magnetite, maghemite and ferrite, or iron oxides including other metal oxides; metals such as Fe, Co, Ni, or these metals and Al, Co, Cu, Pb, Mg, Ni, Sn, Alloys with metals such as Zn, Sb, Ca, Mn, Se, Ti, and mixtures thereof.
- Iron trioxide Fe 3 O 4
- ferric oxide ⁇ -Fe 2 O 3
- iron zinc oxide ZnFe 2 O 4
- iron copper oxide CuFe 2 O 4
- iron neodymium oxide NdFe 2 O
- barium iron oxide BaFe 12 O 19
- magnesium iron oxide MgFe 2 O 4
- iron manganese oxide MnFe 2 O 4
- the magnetic materials described above are used alone or in combination of two or more. Particularly suitable magnetic materials are fine powders of iron trioxide or ⁇ -iron trioxide.
- the average particle diameter of these magnetic substances is preferably 0.1 ⁇ m to 2 ⁇ m, and more preferably 0.1 ⁇ m to 0.3 ⁇ m.
- the magnetic properties at 795.8 kA / m (10 k Oe) application have a coercivity (Hc) of 1.6 kA / m to 12 kA / m (20 to 150 Oe) and a saturation magnetization ( ⁇ s) of 5 Am 2 / It is more than kg and 200 Am 2 / kg or less. Preferably, it is 50 Am 2 / kg or more and 100 Am 2 / kg or less. Residual magnetization (.sigma.r) is, 2Am 2 / kg or more 20 Am 2 / kg or less being preferred.
- the magnetic material is preferably 10.0 parts by mass or more and 200 parts by mass or less, and more preferably 20.0 parts by mass or more and 150 parts by mass or less with respect to 100 parts by mass of the binder resin.
- colorants for use as nonmagnetic toners known colorants such as various dyes and pigments conventionally known can be used.
- color pigments for cyan include copper phthalocyanine compounds and derivatives thereof, anthraquinone compounds, basic dye lake compounds and the like. Specifically, C.I. I. Pigment blue 1, 7, 15, 15: 1, 15: 2, 15: 3, 15: 4, 60, 62, 66 and the like.
- color pigments for yellow include compounds represented by condensation azo compounds, isoindolinone compounds, anthraquinone compounds, azo metal complexes, methine compounds and allylamide compounds. Specifically, C.I. I. Pigment yellow 1, 2, 3, 4, 5, 6, 7, 10, 11, 12, 13, 14, 15, 16, 17, 23, 62, 65, 73, 74, 83, 93, 94, 95, 97, 109, 110, 111, 120, 127, 128, 129, 147, 151, 154, 155, 168, 174, 175, 176, 180, 181, 185; I. Bat Yellow 1, 3, 20 can be mentioned.
- black colorant carbon black, aniline black, acetylene black, titanium black, and those toned in black using the yellow / magenta / cyan colorants described above can be used.
- the toner of the present invention may contain a release agent.
- aliphatic hydrocarbon wax such as low molecular weight polyethylene, low molecular weight polypropylene, microcrystalline wax, paraffin wax; oxide of aliphatic hydrocarbon wax such as oxidized polyethylene wax; aliphatic hydrocarbon wax Block copolymers of a fatty acid ester such as carnauba wax, sazole wax, montanic acid ester wax, and deoxidized fatty acid ester such as deacidified carnauba wax partially or entirely, behenic acid Examples thereof include partially esterified fatty acids such as monoglycerides and polyhydric alcohols; and methyl ester compounds having a hydroxy group obtained by hydrogenating vegetable oils and fats.
- the molecular weight distribution of the releasing agent is preferably such that the main peak is in the range of molecular weight 400 or more and 2400 or less, and more preferably in the range of 430 or more and 2000 or less. By this, it is possible to impart preferable thermal characteristics to the toner.
- the addition amount of the release agent is preferably 2.50 parts by mass or more and 40.0 parts by mass or less, and is 3.00 parts by mass or more and 15.0 parts by mass or less with respect to 100 parts by mass of the binder resin. It is more preferable that
- the toner particles in the present invention may be produced by any method as long as they are obtained by fixing resin particles on the surface of toner base particles containing a binder resin, a colorant and a releasing agent. Is preferably a method of production by a suspension polymerization method.
- the toner particles using the suspension polymerization method preferred in the present invention are produced through the following steps (i) to (iv).
- the resin particles are characterized by containing a resin having an ionic functional group and having a pKa (acid dissociation constant) of 7.0 or more and 9.0 or less.
- a polymerizable monomer composition containing a polymerizable monomer and a colorant is added to an aqueous medium to form particles of the polymerizable monomer composition in the aqueous medium.
- a colorant is added to the polymerizable monomer which is the main constituent material of the toner particles, and these are uniformly dissolved using a homogenizer such as a homogenizer, ball mill, colloid mill, or ultrasonic dispersion machine.
- a dispersed polymerizable monomer composition is prepared.
- a polyfunctional monomer if necessary, a chain transfer agent, a wax as a releasing agent, a charge control agent, a plasticizer, and a dispersing agent
- a polyfunctional monomer if necessary, a chain transfer agent, a wax as a releasing agent, a charge control agent, a plasticizer, and a dispersing agent
- a wax as a releasing agent
- a charge control agent if necessary, a wax as a releasing agent, a charge control agent, a plasticizer, and a dispersing agent
- a dispersing agent if necessary, a polyfunctional monomer, a chain transfer agent, a wax as a releasing agent, a charge control agent, a plasticizer, and a dispersing agent
- Such additives can be added as appropriate.
- the above polymerizable monomer composition is introduced into an aqueous medium prepared in advance, and suspended by using a high-speed disperser such as a high-speed stirrer or an ultrasonic disperser to perform granulation.
- a high-speed disperser such as a high-speed stirrer or an ultrasonic disperser to perform granulation.
- the aqueous medium contains a dispersion stabilizer from the viewpoint of uniformly adhering the resin particles and enhancing the adhesion between the toner base particles and the resin particles.
- calcium phosphate compound, aluminum phosphate compound, magnesium phosphate compound, calcium hydroxide compound, aluminum hydroxide compound, magnesium hydroxide compound, calcium carbonate compound, aluminum carbonate compound, and magnesium carbonate compound It is particularly preferred that it is at least one selected.
- it is possible to control the particle size of toner base particles.
- the metal element derived from the dispersion stabilizer is present on the surface of the toner base particle, it is considered that the toner base particle and the resin particle are bonded via the metal element, and the adhesion strength between the toner base particle and the resin particle is increased.
- the polymerization initiator may be mixed with other additives when preparing the polymerizable monomer composition, or may be mixed into the polymerizable monomer composition just before suspending in the aqueous medium. Good. Moreover, it can also be added in the state melt
- particles of the polymerizable monomer composition are formed in the aqueous medium.
- the suspension after the step (i) is heated to 50 ° C. or more and 90 ° C. or less, and the particles of the polymerizable monomer composition in the suspension maintain the particle state
- the polymerization reaction is carried out with stirring so as not to cause floating or settling of particles.
- the polymerization initiator is easily decomposed by heating to generate free radicals (radicals).
- the generated radical is added to the unsaturated bond of the polymerizable monomer to newly generate an adduct radical.
- the generated adduct radical is further added to the unsaturated bond of the polymerizable monomer.
- the polymerization reaction proceeds to form toner base particles containing the above-mentioned polymerizable monomer as a main constituent material, whereby a dispersion B containing toner base particles is obtained. If necessary, this may be followed by a distillation step to remove the remaining polymerizable monomer.
- step (iii) resin particles are added to the dispersion B, and the resin particles are applied to the surface of the toner base particles to obtain a dispersion C.
- the resin fine particles exhibit a negative property. Therefore, when the toner base particle surface is in a positive state, resin particles can be applied to the toner base particle surface.
- a method for making the surface of the toner base particle positive a method in which a cationic surfactant is contained in the toner base particle, a method in which a dispersion stabilizer of a metal salt is adsorbed on the surface of the toner base particle, etc. are suitably used. .
- the method using a toner in which the dispersion stabilizer is adsorbed on the surface of the toner base particles is particularly preferable.
- the resin particles having the same polarity as the toner base particles with respect to the dispersion stabilizer are added to the dispersion liquid B in a state of being dispersed in an aqueous medium.
- the resin particles can be densely and uniformly adhered to the toner base particles in a state in which the dispersion stabilizer is adsorbed on the surface.
- a preferable addition rate is 0.1 parts by mass / minute or more and 5.0 parts by mass / minute or less as solid content of resin particles with respect to 100 parts by mass of solid content of dispersion liquid B containing toner mother particles.
- the temperature at which the resin particles are added to the dispersion B may be a temperature at which the resin particles do not cause single aggregation.
- the resin particles may be added in a state in which the dispersion B is kept at a temperature above the Tg of the toner base particles in advance.
- the average particle size of the resin particles is preferably in the range of 5 nm or more and 200 nm or less, as a value of median diameter determined by particle size distribution measurement by a laser scattering method. More preferably, it is used in the range of 20 nm or more and 130 nm or less.
- the average particle size is less than 5 nm, sufficient durability may not be obtained. In addition, when the average particle size exceeds 200 nm, non-uniform adhesion may occur.
- the ratio (D50 / D10) of the volume-based median diameter (D50) of the resin particles to the particle diameter (D10) at which the cumulative number of particles in the volume distribution is 10% is 1.0 or more and 3.0 or less preferable.
- the ratio (D90 / D50) of the volume-based median diameter (D50) of the resin particles to the particle diameter (D90) at which the cumulative number of particles in the volume distribution is 90% is 1.0 or more and 3.0 or less Is preferred. Being within these ranges means that the particle size distribution of the resin particles is uniform, and as a result, the resin particles to be fixed have little variation among toners, and stable performance can be obtained. More preferably, the resin particles contain a resin A having a monovalent group a represented by the above formula (1).
- step (iv) the dispersion C is heated to a temperature equal to or higher than the glass transition temperature (Tg) of toner base particles to obtain toner particles.
- Tg glass transition temperature
- the resin particles adhere to the dispersion stabilizer adsorbed to the toner base particles. Thereafter, the dispersion energy is transferred to the surface of the non-adsorbed toner matrix particles upon contact with the stirring energy, and comes into contact. At this time, when heating is performed at or above Tg of the toner base particles, the surface of the toner base particles is softened, and thus the contacted resin particles are fixed to the surface of the toner base particles. When the heating time is longer than the Tg of the toner base particles, the fixation becomes strong, and the adhesion between the toner base particles and the resin particles can be enhanced.
- the resin particles fill the exposed portion of the toner base particle surface and are present more than that, the resin particles are kept adhering on the dispersion stabilizer by heating at a temperature higher than the Tg of the resin particles, and Smoothing can be advanced to improve adhesion. By sufficiently enhancing the adhesion, good durability can be realized.
- a dispersion stabilizer may be additionally added to suppress aggregation and further improve production stability. Also, a small amount of surfactant can be added.
- the dispersion stabilizer is removed at a temperature lower than the Tg of the resin particles. Thereafter, it is filtered, washed and dried by a known method to obtain toner particles.
- the toner particles may be added with a fluidity improver as an external additive.
- Fluidity improvers include fine powders of vinylidene fluoride fine powder, fluorine-based resin powder such as polytetrafluoroethylene fine powder; fine powder of silica by wet process, fine powder of silica such as fine powder of silica by dry process, Treated silica fine powder surface-treated with treating agents such as silane coupling agent, titanium coupling agent, silicone oil; titanium oxide fine powder; alumina fine powder, treated titanium oxide fine powder, treated alumina oxide fine powder .
- the flowability improver has a specific surface area of at least 30.0 m 2 / g, preferably at least 50.0 m 2 / g, as measured by the BET method using nitrogen adsorption, and gives good results.
- the addition amount of the flowability improving agent is preferably 0.010 parts by mass or more and 8.0 parts by mass or less, and more preferably 0.10 parts by mass or more and 4.0 parts by mass or less with respect to 100 parts by mass of toner particles. .
- the toner preferably has a weight average particle diameter (D4) of 3.0 ⁇ m or more and 15.0 ⁇ m or less, and is 4.0 ⁇ m or more and 12.0 ⁇ m or less, in order to faithfully develop smaller latent image dots. Is more preferred. Moreover, it is preferable that ratio (D4 / D1) of the weight average particle diameter (D4) with respect to a number average particle diameter (D1) is less than 1.40.
- the toner of the present invention can be mixed with a magnetic carrier and used as a two-component developer.
- a magnetic carrier metal particles such as surface oxidized or unoxidized iron, lithium, calcium, magnesium, nickel, copper, zinc, cobalt, manganese, chromium, rare earths, their alloy particles, oxide particles and ferrite are micronized Can be used.
- a coated carrier in which the surface of the magnetic carrier core is coated with a resin.
- a coating method a method in which a coating solution prepared by dissolving or suspending a coating material such as a resin in a solvent is adhered to the surface of a magnetic carrier core, and a method in which a magnetic carrier core and a coating material are mixed with powder Be
- the coating material for the magnetic carrier core examples include silicone resins, polyester resins, styrene resins, acrylic resins, polyamides, polyvinyl butyral, and amino acrylate resins. These are used singly or in combination.
- the treatment amount of the coating material is preferably 0.10% by mass or more and 30% by mass or less, and more preferably 0.50% by mass or more and 20% by mass or less with respect to the carrier core particles.
- the average particle diameter of the magnetic carrier is preferably 10.0 ⁇ m or more and 100 ⁇ m or less, and more preferably 20.0 ⁇ m or more and 70.0 ⁇ m or less in terms of 50% particle diameter (D50) based on volume.
- the mixing ratio is preferably 2.0% by mass or more and 15% by mass or less, more preferably 4.0% by mass or more and 13% by mass or less as the toner concentration in the developer It is below.
- the glass transition temperature (Tg) of the toner and the resin particles can be determined, for example, using a differential scanning calorimeter (Q1000) manufactured by TA Instruments, in the following manner.
- the temperature rise rate is 2 ° C./min and the modulation amplitude ⁇ 0 at a measurement temperature range of 20 ° C. to 150 ° C. in a nitrogen atmosphere. Measure at 6 ° C and 1 frequency / minute.
- the weight average particle diameter (D4) and the number average particle diameter (D1) of the toner are calculated as follows.
- a precise particle size distribution measuring apparatus “Coulter Counter Multisizer 3” (registered trademark, manufactured by Beckman Coulter, Inc.) having a 100 ⁇ m aperture tube and using a pore electrical resistance method is used.
- the setting of measurement conditions and the analysis of measurement data use the attached special software "Beckman Coulter Multisizer 3 Version 3.51" (manufactured by Beckman Coulter, Inc.). The measurement is performed with 25,000 channels of effective measurement channels.
- electrolytic aqueous solution used for the measurement a solution in which special grade sodium chloride is dissolved in ion exchange water so that the concentration is 1% by mass, for example, “ISOTON II” (manufactured by Beckman Coulter, Inc.) can be used.
- the bin interval is set to logarithmic particle size
- the particle size bin is set to 256 particle size bin
- the particle size range is set to 2 ⁇ m to 60 ⁇ m.
- the specific measurement method is as follows. (1) Put 200 mL of the aqueous electrolytic solution in a 250 mL round bottom beaker made of Multisizer 3 exclusively for use in a glass, set it on a sample stand, and stir the stirrer rod counterclockwise at 24 rotations / second. Then, dirt and air bubbles in the aperture tube are removed by the "aperture flush" function of special software. (2) Put 30 mL of the electrolytic aqueous solution into a 100 mL flat bottom beaker made of glass.
- contaminone N nonionic surfactant, anionic surfactant, 10% by weight aqueous solution of neutral detergent for pH 7 precision measuring instrument cleaning consisting of organic builders as a dispersing agent, Wako Pure Chemical Industries, Ltd. 3.
- contaminone N nonionic surfactant, anionic surfactant, 10% by weight aqueous solution of neutral detergent for pH 7 precision measuring instrument cleaning consisting of organic builders as a dispersing agent, Wako Pure Chemical Industries, Ltd. 3.
- Two oscillators with an oscillation frequency of 50 kHz are built in with 180 degrees of phase shift, and an ultrasonic dispersion device “Ultrasonic Dispersion System Tetra 150” (manufactured by Nikkaki Bios Co., Ltd.) having an electrical output of 120 W is prepared.
- ultrasonic dispersion processing is continued for another 60 seconds.
- the water temperature of the water tank is appropriately adjusted so as to be 10 ° C. or more and 40 ° C. or less.
- the aqueous electrolyte solution of (5) in which the toner is dispersed using a pipette is dropped to adjust the measurement concentration to 5%. Then, measurement is performed until the number of measurement particles reaches 50,000.
- the measurement data is analyzed by the dedicated software attached to the device to calculate the weight average particle diameter (D4) and the number average particle diameter (D1).
- the “average diameter” on the “analysis / volume statistical value (arithmetic mean)” screen when the graph / volume% is set in the dedicated software is the weight average particle diameter (D4). Further, the “average diameter” on the “Analysis / number statistics (arithmetic average)” screen when the graph / number% is set by the dedicated software is the number average particle diameter (D1).
- the volume-based median diameter (D50) of the resin particles is calculated by measuring the particle diameter using Dynamic Light Scattering (DLS), using Zetasizer Nano-ZS (manufactured by MALVERN).
- Measurement mode Particle size Material: Polystyrene latex (RI: 1.59, Absorption: 0.01) Dispersant: Water (Temperature: 25 ° C, Viscosity: 0.8872 cP, RI: 1.330) Temperature: 25.0 ° C Cell: Clear disposable zeta cell Measurement duration: Automatic
- the sample is prepared by diluting with water so as to be 0.50% by mass, and filled in a disposable capillary cell (DTS 1060), and the cell is loaded into the cell holder of the apparatus.
- DTS 1060 disposable capillary cell
- D50 is calculated based on volume-based particle size distribution data obtained by converting the light intensity distribution obtained from DLS measurement by Mie theory.
- the acid value is the number of mg of potassium hydroxide necessary to neutralize the acid contained in 1 g of the sample.
- the acid value in the present invention is measured according to JIS K 0070-1992, and specifically, it is measured according to the following procedure.
- the titration is performed using a 0.1 mol / L potassium hydroxide ethyl alcohol solution (manufactured by Kishida Chemical Co., Ltd.).
- the factor of the potassium hydroxide ethyl alcohol solution can be determined using a potentiometric titration apparatus (a potentiometric titration measurement apparatus AT-510 manufactured by Kyoto Denshi Kogyo Co., Ltd.).
- 100 mL of 0.100 mol / L hydrochloric acid is taken in a 250 mL tall beaker, titrated with the potassium hydroxide ethyl alcohol solution, and determined from the amount of the potassium hydroxide ethyl alcohol solution required for neutralization.
- the 0.100 mol / L hydrochloric acid used is prepared according to JIS K 8001-1998.
- Titration device Potentiometric titration device AT-510 (manufactured by Kyoto Denshi Kogyo Co., Ltd.)
- Electrode Composite glass electrode double junction type (manufactured by Kyoto Denshi Kogyo Co., Ltd.)
- Control software for titrator AT-WIN Titration analysis software: Tview The titration parameters and control parameters at the time of titration are performed as follows.
- the acid value is calculated by substituting the obtained result into the following equation.
- A [(C ⁇ B) ⁇ f ⁇ 5.611] / S (Wherein, A: acid value (mg KOH / g), B: addition amount of potassium hydroxide ethyl alcohol solution of blank test (mL), C: addition amount of potassium hydroxide ethyl alcohol solution of this test (mL), f: Factor of potassium hydroxide solution, S: sample (g))
- ⁇ PKa> 0.100 g of the measurement sample is precisely weighed in a 250 mL tall beaker, 150 mL of THF is added, and dissolved over 30 minutes. Put a pH electrode into this solution and read the pH of the sample THF solution. Thereafter, 10 ⁇ L each of 0.1 mol / L potassium hydroxide ethyl alcohol solution (manufactured by Kishida Chemical Co., Ltd.) is added, and pH is read and titration is performed each time. Add a 0.1 mol / L potassium hydroxide ethyl alcohol solution until the pH becomes 10 or more and the pH does not change even when 30 ⁇ L is added.
- the pH is plotted against the addition amount of 0.1 mol / L potassium hydroxide ethyl alcohol solution to obtain a titration curve. From the obtained titration curve, the point where the slope of pH change is the largest is taken as the neutralization point.
- pKa is determined as follows. The pH at half the amount of the 0.1 mol / L potassium hydroxide ethyl alcohol solution required until the neutralization point is read from the titration curve, and the value of pH read is taken as pKa.
- ⁇ NMR> The content of the monovalent group a contained in the resin A is determined using nuclear magnetic resonance spectroscopy (1 H-NMR) [400 MHz, CDCl 3 , room temperature (25 ° C.)].
- the molar ratio of each monomer component is determined from the integral value of the spectrum obtained, and based on this, the mol% of the monovalent group a contained in the resin A is calculated. Then, the number of moles of the group a per 1 g of the resin A is calculated.
- the molecular weight of the resin A is calculated by gel permeation chromatography (GPC) in terms of polystyrene.
- GPC gel permeation chromatography
- the column elution rate also depends on the amount of the sulfonic acid group, so the accurate molecular weight and molecular weight distribution can not be measured. Therefore, it is necessary to prepare a sample capped with a sulfonic acid group in advance.
- Methyl esterification is preferable for capping, and commercially available methyl esterification agents can be used. Specifically, a method of treating with trimethylsilyldiazomethane can be mentioned.
- the measurement of molecular weight by GPC is performed as follows.
- the above resin is added to THF (tetrahydrofuran), and the solution left at room temperature for 24 hours is filtered with a solvent resistant membrane filter “Misholy Disc” (manufactured by Tosoh Corporation) having a pore diameter of 0.2 ⁇ m to obtain a sample solution, Measure under the following conditions.
- the amount of THF is adjusted so that the concentration of the resin is 0.8% by mass.
- a basic solvent such as DMF.
- HLC8120 GPC (detector: RI) (made by Tosoh Corporation) Column: 7 series of Shodex KF-801, 802, 803, 804, 805, 806, 807 (made by Showa Denko) Eluent: Tetrahydrofuran (THF) Flow rate: 1.0 mL / min Oven temperature: 40.0 ° C Sample injection amount: In calculating the molecular weight of a 0.10 mL sample, a molecular weight calibration curve prepared using the standard polystyrene resin column listed below is used.
- TSK Standard Polystyrene F-850, F-450, F-288, F-128, F-80, F-40, F-20, F-10, F manufactured by Toso -4, F-2, F-1, A-5000, A-2500, A-1000, A-500 ".
- the amount of dispersion stabilizer contained in the toner particles is quantified by fluorescent X-ray when an inorganic dispersant is used. Measurement of fluorescent X-rays conforms to JIS K 0119-1969, and specifically, it is as follows.
- the measurement equipment includes a wavelength dispersive fluorescent X-ray analyzer “Axios” (manufactured by PANalytical), and a dedicated software “SuperQ ver. 4.0F” (made by PANalytical) for setting measurement conditions and analyzing measured data. Use).
- Rh is used as the anode of the X-ray tube
- the measurement atmosphere is vacuum
- the measurement diameter is 27 mm
- the measurement time is 10 seconds.
- a proportional counter (PC) is used.
- SC scintillation counter
- toner particles As a measurement sample, 4 g of toner particles are placed in a dedicated press aluminum ring and flattened, and the tablet molding and compression machine “BRE-32” (manufactured by Maekawa Test Instruments Co., Ltd.) is used. It is pressed for a second, and a pellet molded to a thickness of 2 mm and a diameter of 39 mm is used.
- BRE-32 tablet molding and compression machine
- the measurement is performed under the above conditions, the element is identified based on the peak position of the obtained X-ray, and the concentration is calculated from the counting rate (unit: cps) which is the number of X-ray photons per unit time.
- the metal element is quantified using this measurement result and a calibration curve prepared in advance for the metal element to be quantified.
- Step 1 100 g of 2,5-dihydroxybenzoic acid and 1441 g of 80% sulfuric acid were heated and mixed at 50 ° C. To this dispersion, 144 g of tert-butyl alcohol was added and stirred at 50 ° C. for 30 minutes. Thereafter, 144 g of tert-butyl alcohol was added to this dispersion and stirred for 30 minutes three times.
- the reaction solution was cooled to room temperature and poured slowly into 1 kg of ice water. The precipitate was filtered, washed with water and then washed with hexane. The precipitate was dissolved in 200 mL of methanol and reprecipitated in 3.6 L of water. After filtration, it was dried at 80 ° C. to obtain 74.9 g of a salicylic acid intermediate represented by the following formula (4).
- a salicylic acid intermediate was obtained in the same manner as in the synthesis of the polymerizable monomer M-1 (Step 1) except that 144 g of tert-butyl alcohol was changed to 253 g of 2-octanol.
- a polymerizable monomer M-5 of the following formula (9) was obtained by the same method as the synthesis (step 2) of the polymerizable monomer M-1 except that 32 g of the salicylic acid intermediate obtained here was used.
- reaction solution was cooled and dropped into 1 L of methanol to obtain a precipitate.
- the obtained precipitate was dissolved in 120 mL of THF and then added dropwise to 1.80 L of methanol to precipitate a white precipitate, which was filtered and dried at 90 ° C. under reduced pressure to obtain 57.6 g of resin A-1 .
- the NMR and acid value of the obtained resin A-1 were measured to confirm the content of the component derived from the polymer monomer M-1.
- the polymerization reaction was carried out for 10 hours while maintaining the above temperature, and after cooling, the reaction solution was dropped into hexane for reprecipitation purification, filtered and dried to obtain a polar resin F.
- the obtained polar resin F had a carboxyl group-derived acid value of 0 mg KOH / g.
- Example 1 (Preparation of toner base particles) 850.0 parts of 0.1 mol / L Na 3 PO 4 aqueous solution is added to a container equipped with a high-speed stirring device Claire Mix (manufactured by M. Technique), the rotation speed is adjusted to 15000 rpm, and the temperature is raised to 60 ° C. did. 68.0 parts of a 1.0 mol / L-CaCl 2 aqueous solution is added thereto to prepare an aqueous medium containing a fine sparingly water-soluble dispersant Ca 3 (PO 4 ) 2 and after stirring for 30 minutes, the pH is 6 Adjusted to .0.
- the mixed solution was heated to a temperature of 60 ° C. and then stirred at 9000 r / min with a TK type homomixer (manufactured by Tokushu Kika Kogyo) to dissolve and disperse.
- a TK type homomixer manufactured by Tokushu Kika Kogyo
- a polymerizable monomer composition Into this was dissolved 10.0 parts of a polymerization initiator 2,2'-azobis (2,4-dimethylvaleronitrile) to prepare a polymerizable monomer composition.
- the polymerizable monomer composition was charged into the aqueous medium, and granulated at a temperature of 60 ° C. for 15 minutes while being rotated at 15000 rpm.
- a small amount of the obtained dispersion liquid of toner base particles is extracted, and after adjusting pH to 1.0 by adding 10% hydrochloric acid and stirring for 2 hours, it is filtered, sufficiently washed with ion exchanged water and then dried.
- the glass transition temperature Tg was measured. The Tg was 50.3 ° C.
- a toner 1 was obtained by mixing 2.0 parts of hydrophobic silica fine powder with 100.0 parts of the toner particles 1 with a Henschel mixer (manufactured by Mitsui Miike) at 3000 rpm for 15 minutes.
- the hydrophobic silica fine powder was treated with dimethyl silicone oil (20% by mass) as a fluidity improver, and the primary particles having a number average diameter of 10 nm and a BET specific surface area of 170 m 2 / g were used. .
- Example 2 to Example 27 The same procedure as in Example 1 was carried out except that the pH adjustment, the type and number of addition parts of the aqueous dispersion of resin particles, the addition temperature of the aqueous dispersion of resin particles, and the heating temperature were changed as shown in Table 4. Toners 2 to 27 were obtained.
- Example 28 The resin A-1 was freeze-crushed to obtain a freeze-crushed product of the resin A-1.
- Example 2 In the same manner as in Example 1, a dispersion of toner base particles was obtained. The pH of the obtained dispersion of toner base particles was adjusted to 1.5. The mixture was stirred for 2 hours as it was, and after repeating filtration and washing with water three times, the solid content was recovered and dried in a vacuum dryer at 30 ° C. for 1 day.
- a toner 28 was obtained by mixing 2.0 parts of hydrophobic silica fine powder with 100.0 parts of the toner particles 28 with a Henschel mixer (manufactured by Mitsui Miike) at 3000 rpm for 15 minutes.
- the hydrophobic silica fine powder was treated with dimethyl silicone oil (20% by mass) as a fluidity improver, and the primary particles having a number average diameter of 10 nm and a BET specific surface area of 170 m 2 / g were used. .
- Example 29 The aqueous dispersion of resin particles E-1 was dried to obtain a dried product of resin particles E-1.
- the dried product of the resin particle E-1 obtained was freeze-crushed to obtain a freeze-crushed product of the resin particle E-1.
- Toner 29 was obtained in the same manner as in Example 28, except that the freeze-pulverized product of resin A-1 was changed to the freeze-pulverized product of resin particle E-1 obtained.
- Example 30 The process until preparation of toner base particles was performed in the same manner as in Example 1 to obtain a toner base particle dispersion.
- toner particles 30 (heating temperature) using a heating oil bath, and stirring was continued for 1 hour.
- the dispersion was then cooled to 20 ° C., adjusted to pH 1.5 with 10% hydrochloric acid (acid-treated pH), stirred for 2 hours (acid-treated time), filtered, and thoroughly washed with ion-exchanged water , Dried and classified to obtain toner particles 30.
- the toner particles 30 were obtained by externally adding hydrophobic silica fine powder in the same manner as in Example 1, and toner 30 was obtained.
- Example 31 Dispersion of toner mother particles was carried out in the same manner as in Example 1 except that the 1.0 mol / L CaCl 2 aqueous solution in (Preparation of toner mother particles) was changed to a 1.0 mol / L MgCl 2 aqueous solution. I got a liquid.
- Example 32 Dispersion of toner mother particles is performed in the same manner as in Example 1 except that the 1.0 mol / L CaCl 2 aqueous solution in (Preparation of toner mother particles) in Example 1 is changed to a 1.0 mol / L BaCl 2 aqueous solution. I got a liquid.
- Example 33 (Preparation of toner base particles) Dispersion of toner mother particles was carried out in the same manner as in Example 1 except that the 1.0 mol / L CaCl 2 aqueous solution in (Preparation of toner mother particles) in Example 1 was changed to a 0.7 mol / L AlCl 3 aqueous solution. I got a liquid.
- Example 34 An aqueous solution prepared by dissolving 13.2 parts of calcium chloride in 250 parts of ion-exchanged water was placed in a container equipped with a high-speed stirrer Creamix (manufactured by M. Technique), and the rotational speed was adjusted to 18,000 rpm. An aqueous solution prepared by dissolving 4.8 parts of sodium hydroxide in 50 parts of ion-exchanged water is gradually added thereto with stirring to prepare a calcium hydroxide colloid (a poorly water-soluble metal hydroxide colloid) dispersion. , PH was adjusted to 6.0.
- the mixed solution was heated to a temperature of 60 ° C. and then stirred at 9000 r / min with a TK type homomixer (manufactured by Tokushu Kika Kogyo) to dissolve and disperse.
- a TK type homomixer manufactured by Tokushu Kika Kogyo
- a polymerizable monomer composition Into this was dissolved 10.0 parts of a polymerization initiator 2,2'-azobis (2,4-dimethylvaleronitrile) to prepare a polymerizable monomer composition.
- the polymerizable monomer composition was charged into the aqueous medium, and granulated at a temperature of 60 ° C. for 15 minutes while being rotated at 18,000 rpm.
- Example 35 Toner 35 was obtained in the same manner as in Example 34 except that 13.2 parts of calcium chloride in Example 34 was changed to 11.3 parts of magnesium chloride.
- Example 36 Toner 36 was obtained in the same manner as in Example 34 except that 13.2 parts of calcium chloride in Example 34 was changed to 24.7 parts of barium chloride.
- Example 37 Toner 37 was obtained in the same manner as in Example 34 except that 13.2 parts of calcium chloride in Example 34 was changed to 10.5 parts of aluminum chloride.
- Example 38 An aqueous solution of 12.6 parts of sodium carbonate dissolved in 250 parts of ion-exchanged water was placed in a container equipped with a high-speed stirrer Creamix (manufactured by M. Technics Co., Ltd.), and the rotational speed was adjusted to 18,000 rpm. An aqueous solution prepared by dissolving 13.2 parts of calcium chloride in 50 parts of ion-exchanged water was added at once with stirring and stirred for 30 minutes, and then the pH was adjusted to 6.0.
- a high-speed stirrer Creamix manufactured by M. Technics Co., Ltd.
- the mixed solution was heated to a temperature of 60 ° C. and then stirred at 9000 r / min with a TK type homomixer (manufactured by Tokushu Kika Kogyo) to dissolve and disperse.
- a TK type homomixer manufactured by Tokushu Kika Kogyo
- a polymerizable monomer composition Into this was dissolved 10.0 parts of a polymerization initiator 2,2'-azobis (2,4-dimethylvaleronitrile) to prepare a polymerizable monomer composition.
- the polymerizable monomer composition was charged into the aqueous medium, and granulated at a temperature of 60 ° C. for 15 minutes while being rotated at 18,000 rpm.
- the mixture was transferred to a propeller stirrer and reacted at a temperature of 70 ° C. for 5 hours while stirring at 100 rpm, and then the temperature was raised to 80 ° C., and reaction was further performed for 5 hours.
- Example 39 Toner 39 was obtained in the same manner as in Example 38 except that 13.2 parts of calcium chloride in Example 38 was changed to 11.3 parts of magnesium chloride.
- Toner 40 was obtained in the same manner as in Example 38 except that 13.2 parts of calcium chloride in Example 38 was changed to 24.7 parts of barium chloride.
- Toner 41 was obtained in the same manner as in Example 38 except that 13.2 parts of calcium chloride in Example 38 was changed to 10.5 parts of aluminum chloride.
- Toners 42 to 52 were obtained in the same manner as in Example 30 except that the fixing pH, the acid treatment pH, and the acid treatment time in the fixing step of Example 30 were changed as shown in Table 5.
- Example 53 A toner 53 was obtained in the same manner as in Example 30 except that the saturated polyester resin in Example 30 was changed to a polar resin F.
- Example 54 Toner particles 54 were obtained in the same manner as in Example 1 except that the acid treatment temperature in Example 1 was changed to 65 ° C. The obtained toner particles were extremely coarse and no subsequent toner formation and evaluation were conducted.
- Example 55 The toner was manufactured by the dissolution suspension method according to the following procedure.
- the aqueous medium and the solution were adjusted according to the following procedure to prepare a toner.
- a mixture of 660.0 parts of water and 25.0 parts of a 48.5 mass% aqueous solution of sodium dodecyl diphenyl ether disulfonate is mixed and stirred, and the aqueous medium is stirred at 10,000 rpm using a TK homomixer (manufactured by Tokushu Kika Kogyo) Prepared.
- aqueous medium 150.0 parts is placed in a container, stirred at a rotation number of 12000 rpm using a TK type homomixer (manufactured by Tokushu Kika Kogyo Co., Ltd.), 100 parts of the above solution is added thereto, and mixed for 10 minutes.
- the emulsion slurry was prepared.
- the obtained filtrate was re-dispersed with ion exchanged water, 10% hydrochloric acid was added to adjust the pH to 1.0, and the mixture was stirred for 2 hours. Thereafter, the resultant is filtered, sufficiently washed with ion exchange water, dried and classified to obtain toner particles 55.
- the toner particles 55 were obtained by externally adding hydrophobic silica fine powder in the same manner as in Example 1 to obtain toner 55.
- Comparative Example 1 A toner 56 was obtained in the same manner as in Example 1 except that the aqueous dispersion of the resin particles E-1 was changed to the aqueous dispersion of the resin particles E-18.
- Comparative Example 2 A toner 57 was obtained in the same manner as in Example 1 except that the aqueous dispersion of the resin particles E-1 was changed to the aqueous dispersion of the resin particles E-19.
- Comparative Example 3 (Preparation of toner base particles) 850.0 parts of 0.1 mol / L Na 3 PO 4 aqueous solution is added to a container equipped with a high-speed stirring device Claire Mix (manufactured by M. Technique), the rotation speed is adjusted to 15000 rpm, and the temperature is raised to 60 ° C. did. 68.0 parts of a 1.0 mol / L-CaCl 2 aqueous solution is added thereto to prepare an aqueous medium containing a fine sparingly water-soluble dispersant Ca 3 (PO 4 ) 2 and after stirring for 30 minutes, the pH is 6 Adjusted to .0.
- the mixed solution was heated to a temperature of 60 ° C. and then stirred at 9000 r / min with a TK type homomixer (manufactured by Tokushu Kika Kogyo) to dissolve and disperse.
- a TK type homomixer manufactured by Tokushu Kika Kogyo
- a polymerizable monomer composition Into this was dissolved 10.0 parts of a polymerization initiator 2,2'-azobis (2,4-dimethylvaleronitrile) to prepare a polymerizable monomer composition.
- the polymerizable monomer composition was charged into the aqueous medium, and granulated at a temperature of 60 ° C. for 15 minutes while being rotated at 15000 rpm.
- a toner 58 is obtained by mixing 2.0 parts of hydrophobic silica fine powder with 100.0 parts of the toner particles 58 with a Henschel mixer (manufactured by Mitsui Miike) at 3000 rpm for 15 minutes.
- the hydrophobic silica fine powder was treated with dimethyl silicone oil (20% by mass) as a fluidity improver, and the primary particles having a number average diameter of 10 nm and a BET specific surface area of 170 m 2 / g were used. .
- the metal element content was quantified by performing fluorescent X-ray measurement.
- the thing below detection limit is made into ND, and the result is shown in Table 6.
- the toners 1 to 53 and the toners 55 to 58 were subjected to performance evaluation according to the following method. The results are shown in Table 7.
- Magnetic carrier F813-300 (made by Powder Tech) 276 g and evaluation toner 24 g are put into a 500 mL plastic bottle with a lid, and a shaker (YS-LD: made by Yayoi Co., Ltd.), 4 reciprocations per second Shake for 1 minute.
- the toner and the two-component developer were evaluated as follows.
- the potential of the electrometer 9 at this time is V (volt).
- 8 is a capacitor, and its capacity is C ( ⁇ F).
- the weight of the entire measurement container after suction is weighed to obtain W2 (g).
- the developing device shown in FIG. 2 was attached to the unit 104 a of FIG. 3 under the environment of high temperature and high humidity (temperature 30 ° C., humidity 85% RH).
- the process speed was 200 mm / s in the cyan single color mode.
- the solid image (image printing rate 4%) is continuously printed on the transfer paper so that the loading amount of the toner amount is 0.40 mg / cm 2, and the image density of the first, 4000, and 8000 sheets images And fog was measured.
- the results are shown in Table 7.
- the toner 2, the toner 30 to the toner 53, and the toner 55 to the toner 58 were also evaluated in the following mode.
- the developing device shown in FIG. 2 was attached to the unit 104 a of FIG. 3 under the environment of high temperature and high humidity (temperature 30 ° C., humidity 85% RH).
- the process speed was 200 mm / s in the cyan single color mode.
- the results are shown in Table 8.
- the reflectance (%) of the non-image area of the printed out image was measured with “REFLECTOMETER MODEL TC-6DS” (manufactured by Tokyo Denshoku Co., Ltd.). The obtained reflectance was evaluated using the value (%) which deducted from the reflectance (%) of the unused printout paper (standard paper) similarly measured. As the numerical value is smaller, the image fogging is suppressed.
Landscapes
- Physics & Mathematics (AREA)
- General Physics & Mathematics (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Chemical & Material Sciences (AREA)
- Inorganic Chemistry (AREA)
- Developing Agents For Electrophotography (AREA)
Abstract
Description
該樹脂粒子が、イオン性官能基を有するpKa(酸解離定数)が7.0以上9.0以下である樹脂を含有することを特徴とするトナーに関する。
(i)重合性単量体および着色剤を含有する重合性単量体組成物の粒子を水系媒体中で形成する工程と、
(ii)該重合性単量体組成物の該粒子に含有される該重合性単量体を重合させて、該トナー母粒子を含有する分散液Bを得る工程と、
(iii)該分散液Bに該樹脂粒子を添加して、分散液Cを得る工程と、
(iv)該分散液Cを該トナー母粒子のガラス転移温度(Tg)以上に加熱して、該トナー母粒子の表面に該樹脂粒子を固着させて、トナー粒子を得る工程と
をこの順に有し、
該樹脂粒子が、イオン性官能基を有するpKa(酸解離定数)が7.0以上9.0以下である樹脂を含有することを特徴とするトナーの製造方法に関する。
このようなトナーは、通常の環境のみならず、苛酷な環境下においても、帯電性が安定しており、また、耐久性にすぐれ、一成分現像システムにおいて高速で画像形成を行う場合であっても、優れた画像安定性が得られるという優れた特性を有する。
上記イオン性官能基を有するpKa(酸解離定数)が7.0以上9.0以下である樹脂は、高湿環境で優れた帯電性能を発揮するため、その点について説明する。
一般的にイオン性官能基を有する樹脂としては、スルホン酸やカルボン酸などの官能基を有したものが多く用いられている。しかしこのような樹脂は水分を吸着し易く、高温高湿下ではその影響で帯電量が低下する場合がある。しかしpKa(酸解離定数)が7.0以上9.0以下であれば、樹脂の吸湿性を低減し、高湿環境での帯電量の低下を抑制できる。
pKa(酸解離定数)が7.0未満の場合、水分吸着量が増え、高湿下で帯電性が低下してしまう。pKa(酸解離定数)が9.0を超える場合、帯電能力が低く十分な帯電量を発現することができない。
pKa(酸解離定数)の求め方は後述するが、中和滴定結果から求めることができる。
イオン性官能基を有する樹脂としては、上記pKa(酸解離定数)を満たすものであればどのようなものでも構わない。例えば、芳香環に結合した水酸基を有する樹脂や、芳香環に結合したカルボキシル基を有する樹脂が例示される。

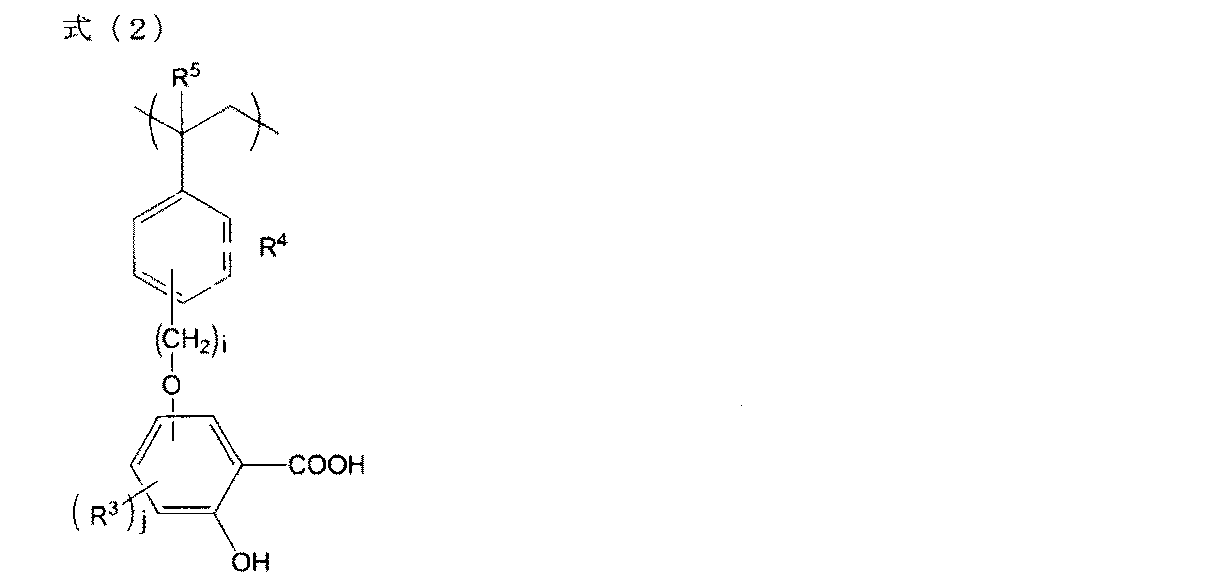
R4は、水素原子、ヒドロキシ基、炭素数1以上18以下のアルキル基、または、炭素数1以上18以下のアルコキシ基を表し、
R5は、水素原子またはメチル基を表し、
iは、1以上3以下の整数を表し、jは0以上3以下の整数を表し、jが2または3の場合、R3はそれぞれ独立して選択できる。)

R7は、水素原子、ヒドロキシ基、炭素数1以上18以下のアルキル基、または、炭素数1以上18以下のアルコキシ基を表し、
R8は、水素原子またはメチル基を表し、
kは1以上3以下の整数を表し、lは0以上3以下の整数を表し、lが2または3の場合、R6はそれぞれ独立して選択できる。)

I)ポリエステル構造に含まれるカルボキシ基やヒドロキシ基の反応残基を利用して、有機反応により、式(1)で示される1価の基aに変換する方法;
II)式(1)で示される1価の基aを置換基として有する多価アルコールまたは多価カルボン酸を用いてポリエステルを作製する方法;
III)多価アルコールまたは多価カルボン酸に、式(1)で示される1価の基aを置換基として導入させやすい官能基をあらかじめ導入しておく方法;
等が挙げられる。
IV)式(1)で示される1価の基aを置換基として含有するポリエステル樹脂をビニル単量体によりハイブリッド化する方法;
V)ビニル単量体としてアクリル酸、メタクリル酸等のカルボキシ基を有するものを用いて重合した後に、そのカルボキシ基を有機反応により、式(1)で示される構造に変換する方法;
VI)式(3)で示される構造aを有する重合性単量体Mを用いてポリエステル樹脂をハイブリッド化する方法;
等が挙げられる。
まず、樹脂Aを後述の方法により滴定することにより、樹脂Aの酸価を定量し、樹脂Aが有する式(1)で示される1価の基aに由来するカルボキシ基の量を算出する。そして、これを基に樹脂A1g当りの式(1)で示される1価の基aの含有量(μmol)を算出することができる。なお、樹脂Aが式(1)で示される1価の基a以外の部位にカルボキシ基を有している場合は、樹脂Aを作製する際に式(1)で示される1価の基aを付加反応させる直前の化合物(例えばポリエステル樹脂)の酸価をあらかじめ測定しておく。式(1)で示される1価の基aの付加量は、付加反応後の樹脂Aの酸価との差で算出することができる。
(ii)該重合性単量体組成物の該粒子に含有される該重合性単量体を重合させて、該トナー母粒子を含有する分散液Bを得る工程と、
(iii)該分散液Bに該樹脂粒子を添加して、分散液Cを得る工程と、
(iv)該分散液Cを該トナー母粒子のガラス転移温度(Tg)以上に加熱して、該トナー母粒子の表面に該樹脂粒子を固着させて、トナー粒子を得る工程と
をこの順に有し、
該樹脂粒子が、イオン性官能基を有するpKa(酸解離定数)が7.0以上9.0以下である樹脂を含有することを特徴とする。
樹脂粒子としては、上記式(1)で示される1価の基aを有する樹脂Aを含有することがさらに好ましい。
二成分系現像剤を調製する場合、その混合比率は、現像剤中のトナー濃度として2.0質量%以上15質量%以下であることが好ましく、より好ましくは4.0質量%以上13質量%以下である。
トナー及び樹脂粒子のガラス転移温度(Tg)は、例えば、TAインスツルメント社製の示差走査熱量計(Q1000)を用い、以下のようにして求めることができる。
トナーの重量平均粒径(D4)および個数平均粒径(D1)は、以下のようにして算出する。測定装置としては、100μmのアパーチャーチューブを備えた細孔電気抵抗法による精密粒度分布測定装置「コールター・カウンター Multisizer 3」(登録商標、ベックマン・コールター社製)を用いる。測定条件の設定及び測定データの解析は、付属の専用ソフト「ベックマン・コールター Multisizer 3 Version3.51」(ベックマン・コールター社製)を用いる。尚、測定は実効測定チャンネル数2万5千チャンネルで行う。
(1)Multisizer 3専用のガラス製250mL丸底ビーカーに前記電解水溶液200mLを入れ、サンプルスタンドにセットし、スターラーロッドの撹拌を反時計回りで24回転/秒にて行う。そして、専用ソフトの「アパーチャーのフラッシュ」機能により、アパーチャーチューブ内の汚れと気泡を除去しておく。
(2)ガラス製の100mL平底ビーカーに前記電解水溶液30mLを入れる。この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業社製)をイオン交換水で3質量倍に希釈した希釈液を0.3mL加える。
(3)発振周波数50kHzの発振器2個を位相を180度ずらした状態で内蔵し、電気的出力120Wの超音波分散器「Ultrasonic Dispersion System Tetra150」(日科機バイオス社製)を準備する。超音波分散器の水槽内に3.3Lのイオン交換水を入れ、この水槽中にコンタミノンNを2mL添加する。
(4)前記(2)のビーカーを前記超音波分散器のビーカー固定穴にセットし、超音波分散器を作動させる。そして、ビーカー内の電解水溶液の液面の共振状態が最大となるようにビーカーの高さ位置を調整する。
(5)前記(4)のビーカー内の電解水溶液に超音波を照射した状態で、トナー10mgを少量ずつ前記電解水溶液に添加し、分散させる。そして、さらに60秒間超音波分散処理を継続する。尚、超音波分散にあたっては、水槽の水温が10℃以上40℃以下となる様に適宜調節する。
(6)サンプルスタンド内に設置した前記(1)の丸底ビーカーに、ピペットを用いてトナーを分散した前記(5)の電解質水溶液を滴下し、測定濃度が5%となるように調整する。そして、測定粒子数が50000個になるまで測定を行う。
(7)測定データを装置付属の前記専用ソフトにて解析を行い、重量平均粒径(D4)および個数平均粒径(D1)を算出する。尚、前記専用ソフトでグラフ/体積%と設定したときの、「分析/体積統計値(算術平均)」画面の「平均径」が重量平均粒径(D4)である。また、前記専用ソフトでグラフ/個数%と設定したときの、「分析/個数統計値(算術平均)」画面の「平均径」が個数平均粒径(D1)である。
樹脂粒子の体積基準のメジアン径(D50)は、ゼータサイザーNano-ZS(MALVERN社製)を用い、動的光散乱法(DLS:Dynamic Light Scattering)で粒子径を測定することにより算出する。
測定モード:粒子径
Material:Polystyrene latex(RI:1.59、Absorption:0.01)
Dispersant:Water(Temperature:25℃、Viscosity:0.8872cP、RI:1.330)
Temperature:25.0℃
Cell:Clear disposable zeta cell
Measurement duration:Automatic
酸価は試料1gに含まれる酸を中和するために必要な水酸化カリウムのmg数である。本発明における酸価は、JIS K 0070-1992に準じて測定されるが、具体的には、以下の手順に従って測定する。
滴定装置:電位差滴定装置AT-510(京都電子工業株式会社製)
電極:複合ガラス電極ダブルジャンクション型(京都電子工業株式会社製)
滴定装置用制御ソフトウェア:AT-WIN
滴定解析ソフト:Tview
滴定時における滴定パラメーター並びに制御パラメーターは下記のように行う。
滴定パラメーター
滴定モード:ブランク滴定
滴定様式:全量滴定
最大滴定量:20mL
滴定前の待ち時間:30秒
滴定方向:自動
制御パラメーラー
終点判断電位:30dE
終点判断電位値:50dE/dmL
終点検出判断:設定しない
制御速度モード:標準
ゲイン:1
データ採取電位:4mV
データ採取滴定量:0.1mL
測定サンプル0.100gを250mLのトールビーカーに精秤し、トルエン/エチルアルコール(3:1)の混合溶液150mLを加え、1時間かけて溶解する。前記電位差滴定装置を用い、前記水酸化カリウムエチルアルコール溶液を用いて滴定する。
試料を用いない(すなわちトルエン/エチルアルコール(3:1)の混合溶液のみとする)以外は、上記操作と同様の滴定を行う。
A=[(C-B)×f×5.611]/S
(式中、A:酸価(mgKOH/g)、B:空試験の水酸化カリウムエチルアルコール溶液の添加量(mL)、C:本試験の水酸化カリウムエチルアルコール溶液の添加量(mL)、f:水酸化カリウム溶液のファクター、S:試料(g)である。)
測定サンプル0.100gを250mLのトールビーカーに精秤し、THF150mLを加え、30分かけて溶解する。この溶液にpH電極を入れ、サンプルのTHF溶液のpHを読み取る。その後、0.1モル/L水酸化カリウムエチルアルコール溶液(キシダ化学社製)を10μLずつ添加し、その都度pHを読み取り滴定を行う。pHが10以上となり、30μL添加してもpHの変化がなくなるまで0.1モル/L水酸化カリウムエチルアルコール溶液を加える。得られた結果から0.1モル/L水酸化カリウムエチルアルコール溶液添加量に対するpHをプロットし、滴定曲線を得る。得られた滴定曲線からpH変化の傾きが一番大きいところを中和点とする。pKaは次のようにして求める。中和点までに必要とした0.1モル/L水酸化カリウムエチルアルコール溶液量の半分量でのpHを滴定曲線から読み取り、読み取ったpHの値をpKaとする。
樹脂Aに含まれる1価の基aの含有量は核磁気共鳴分光分析(1H-NMR)[400MHz、CDCl3、室温(25℃)]を用いて行う。
測定装置:FT NMR装置 JNM-EX400(日本電子社製)
測定周波数:400MHz
パルス条件:5.0μs
周波数範囲:10500Hz
積算回数:64回
樹脂Aの分子量はゲルパーミュエーションクロマトグラフィー(GPC)によって、ポリスチレン換算で算出される。スルホン酸基を有する重合体は、カラム溶出速度がスルホン酸基の量にも依存してしまうため、正確な分子量及び分子量分布を測定したことにはならない。そのため、予めスルホン酸基をキャッピングした試料を用意する必要がある。キャッピングにはメチルエステル化が好ましく、市販のメチルエステル化剤が使用できる。具体的には、トリメチルシリルジアゾメタンで処理する方法が挙げられる。
装置:HLC8120 GPC(検出器:RI)(東ソー社製)
カラム:Shodex KF-801、802、803、804、805、806、807の7連(昭和電工社製)
溶離液:テトラヒドロフラン(THF)
流速:1.0mL/min
オーブン温度:40.0℃
試料注入量:0.10mL試料の分子量の算出にあたっては、以下に列挙する標準ポリスチレン樹脂カラムを用いて作成した分子量校正曲線を使用する。具体的には、東ソ-社製の商品名「TSKスタンダード ポリスチレン F-850、F-450、F-288、F-128、F-80、F-40、F-20、F-10、F-4、F-2、F-1、A-5000、A-2500、A-1000、A-500」である。
各元素の蛍光X線の測定は、JIS K 0119-1969に準ずるが、具体的には以下の通りである。
(工程1)
2,5-ジヒドロキシ安息香酸100gと80%硫酸1441gとを50℃に加熱混合した。この分散液にtert-ブチルアルコール144gを加えて50℃で30分間撹拌した。その後、この分散液にtert-ブチルアルコール144gを加え30分間撹拌する操作を3回行った。反応液を室温まで冷却し、氷水1kgにゆっくり注いだ。析出物を濾過、水洗し、その後、ヘキサン洗浄した。この析出物をメタノール200mLに溶解させ、水3.6Lに再沈殿させた。濾過後、80℃にて乾燥することで下記式(4)に示すサリチル酸中間体74.9gを得た。

得られたサリチル酸中間体25.0gをメタノール150mLに溶解させ、炭酸カリウム36.9gを加えて65℃に加熱した。この反応液に4-(クロロメチル)スチレン18.7gとメタノール100mLの混合液を滴下し、65℃にて3時間反応させた。反応液を冷却後、濾過し、濾液を濃縮して粗生成物を得た。粗生成物をpH=2の水1.5Lに分散させ、酢酸エチルを加えて抽出した。その後、水洗し、硫酸マグネシウムで乾燥させ、減圧下、酢酸エチルを留去して析出物を得た。析出物をヘキサン洗浄後、トルエンと酢酸エチルにて再結晶することで精製し、下記式(5)に示す重合性単量体M-1を20.1g得た。
式(4)のサリチル酸中間体を2,4-ジヒドロキシ安息香酸18gに変更する以外は、重合性単量体M-1の合成(工程2)と同じ方法で、下記式(6)の重合性単量体M-2を得た。

式(4)のサリチル酸中間体を2,3-ジヒドロキシ安息香酸18g に変更する以外は、重合性単量体M-1の合成(工程2)と同じ方法で、下記式(7)の重合性単量体M-3を得た。
式(4)のサリチル酸中間体を2,6-ジヒドロキシ安息香酸18gに変更する以外は、重合性単量体M-1の合成(工程2)と同じ方法で、下記式(8)の重合性単量体M-4を得た。

tert-ブチルアルコール144gを2-オクタノール253gに変更する以外は、重合性単量体M-1の合成(工程1)と同じ方法で、サリチル酸中間体を得た。ここで得られるサリチル酸中間体32gを用いる以外は、重合性単量体M-1の合成(工程2)と同じ方法で、下記式(9)の重合性単量体M-5を得た。
式(4)のサリチル酸中間体を2,5-ジヒドロキシ-3-メトキシ安息香酸22gに変更する以外は、重合性単量体M-1の合成(工程2)と同じ方法で、下記式(10)の重合性単量体M-6を得た。

式(5)に示す重合性単量体M-1 9.0g、2-エチルヘキシルアクリレート 15.2g、スチレン 45.8gをDMF42.0mLに溶解させ、窒素バブリングをしながら1時間撹拌した後、110℃まで加熱した。この反応液に、開始剤としてtert-ブチルパーオキシイソプロピルモノカルボネート(日本油脂株式会社製、商品名パーブチルI)2.1gとトルエン42mLの混合液を滴下した。更に110℃にて4時間反応した。その後、冷却しメタノール1Lに滴下し、析出物を得た。得られた析出物をTHF120mLに溶解後、メタノール1.80Lに滴下し、白色析出物を析出させ、濾過し、減圧下90℃にて乾燥させることで、樹脂A-1を57.6g得た。得られた樹脂A-1のNMRと酸価を測定し、重合体単量体M-1に由来する成分の含有量を確認した。
原料の仕込み量を表2のように変更する以外は樹脂A-1の合成例と同様にして樹脂A-2~樹脂A-17を得た。
撹拌機、コンデンサー、温度計、窒素導入管を付した反応容器にキシレン200部を仕込み、窒素気流下で還流した。単量体として、
2-アクリルアミド-2-メチルプロパンスルホン酸 6.0部
スチレン 72.0部
2-エチルヘキシルアクリレート 18.0部
を混合し、前記反応容器に撹拌しながら滴下し10時間保持した。その後、蒸留を行って溶剤を留去し、減圧下40℃で乾燥し樹脂D-1を得た。得られた樹脂D-1のNMRと酸価を測定し、式(1)で示される1価の基aの含有量を確認した。
撹拌機、コンデンサー、温度計、窒素導入管を付した反応容器にキシレン200部を仕込み、窒素気流下で還流した。
5-ビニルサリチル酸 9.0部
スチレン 75.0部
2-エチルヘキシルアクリレート 16.0部
ジメチル-2,2’-アゾビス(2-メチルプロピオネート) 5.0部
を混合し、前記反応容器に撹拌しながら滴下し10時間保持した。その後、蒸留を行って溶剤を留去し、減圧下40℃で乾燥し樹脂D-2を得た。得られた樹脂D-2のNMRと酸価を測定し、式(1)で示される1価の基aの含有量に由来する成分の含有量を確認した。

撹拌機、コンデンサー、温度計、窒素導入管を備えた反応容器に、メチルエチルケトン200.0部を仕込み、樹脂A-1を100.0部加えて溶解した。次いで、1.0モル/L水酸化カリウム水溶液をゆっくり加え、10分間撹拌を行った後、イオン交換水500.0部をゆっくり滴下し、乳化させた。得られた乳化物を減圧蒸留して脱溶剤し、イオン交換水を加えて樹脂濃度が20%になるように調製することで、樹脂粒子E-1の水分散体を得た。得られた樹脂粒子Aの水分散体の物性値を表3に示す。
樹脂A-1と、1.0モル/L水酸化カリウム水溶液の量を表3に示すように変更した以外は、樹脂粒子E-1の製造例と同様にして樹脂粒子E-2~樹脂粒子E-19の水分散体を得た。得られた樹脂粒子E-1~樹脂粒子E-19の水分散体の物性値を表3に示す。

撹拌機、コンデンサー、温度計、窒素導入管を備えた反応容器にメチルエチルケトン100.0部を仕込み、窒素雰囲気下、温度80℃に昇温した。次いで、以下の単量体からなる混合物に重合開始剤としてt-ブチルパーオキシ-2-エチルヘキサノエート3.0部を添加し、撹拌しながら2時間かけて滴下した。
スチレン 72.2部
n-ブチルアクリレート(アクリル酸n-ブチル) 14.0部
2-ヒドロキシエチルメタクリレート 4.0部
(トナー母粒子の作製)
高速撹拌装置クレアミックス(エム・テクニック社製)を備えた容器中に0.1mol/L-Na3PO4水溶液850.0部を添加し、回転数を15000rpmに調整し、60℃に加温した。ここに1.0mol/L-CaCl2水溶液68.0部を添加し、微細な難水溶性分散剤Ca3(PO4)2を含む水系媒体を調製し、30分間撹拌した後、pHを6.0に調整した。
・スチレン 70.0部
・n-ブチルアクリレート 30.0部
・飽和ポリエステル樹脂 3.0部
(テレフタル酸-プロピレンオキサイド変性ビスフェノールA共重合体、酸価13mgKOH/g、Mw14500)
・C.I.ピグメントブルー15:3 6.5部
・エステルワックス 12.0部
(主成分C21H43COOC22H45、融点72.5℃)
還流冷却管、撹拌機、温度計を備えた反応容器に上記トナー母粒子の分散液500.0部(固形分100.0部)を入れ、撹拌しながら、炭酸ナトリウム水溶液を加え、pHを8.5に調整した(固着pH)。ここに、樹脂粒子E-1の水分散体15.0部(固形分3.0部)を22℃で(添加温度)緩やかに添加し、200回転/分で15分間撹拌を行った。次いで、加熱用オイルバスを用いて樹脂粒子が付着したトナー母粒子の分散液の温度を80℃(加熱温度)に保持し、1時間撹拌を続けた。その後分散液を20℃まで冷却した後(酸処理温度)、10%塩酸を加えてpHを1.0に調整して2時間撹拌し、ろ過し、イオン交換水で洗浄した(酸処理)。得られたろ物をイオン交換水で再分散し、10%塩酸を加えてpHを1.0に調整し、2時間撹拌してろ過した。ろ物を再分散し、酸処理する操作を3回繰り返した。その後、乾燥および分級してトナー粒子1を得た。
実施例1において、pH調整、樹脂粒子の水分散体の種類及び添加部数、樹脂粒子の水分散体の添加温度、加熱温度を表4に示すように変更した以外は、実施例1と同様にしてトナー2~27を得た。

樹脂A-1を凍結粉砕して、樹脂A-1の凍結粉砕品を得た。
乾燥させたトナー母粒子100.0部に樹脂A-1の凍結粉砕品を3.0部添加し、乾式粒子複合化装置(ホソカワミクロン製 ノビルタNOB-130)に投入した。処理温度30℃、回転式処理ブレード90m/sec.の条件にて固着を行い、トナー粒子28を得た。
樹脂粒子E-1の水分散体を乾燥させ、樹脂粒子E-1の乾燥品を得た。得られた樹脂粒子E-1の乾燥品を凍結粉砕して樹脂粒子E-1の凍結粉砕品を得た。
トナー母粒子の作製までは実施例1と同様に行い、トナー母粒子分散液を得た。
還流冷却管、撹拌機、温度計を備えた反応容器に上記トナー母粒子の分散液500.0部(固形分100.0部)を入れ、撹拌しながら、炭酸ナトリウム水溶液を加え、pHを9.0に調整した(固着pH)。ここに、樹脂粒子E-2の水分散体15.0部(固形分3.0部)を22℃で(添加温度)緩やかに添加し、200回転/分で15分間撹拌を行った。次いで、加熱用オイルバスを用いて樹脂粒子が付着したトナー母粒子の分散液の温度を80℃(加熱温度)に保持し、1時間撹拌を続けた。その後分散液を20℃まで冷却した後、10%塩酸でpHを1.5に調整し(酸処理pH)2時間撹拌(酸処理時間)した後、ろ過し、イオン交換水で十分に洗浄し、乾燥および分級してトナー粒子30を得た。
実施例1の(トナー母粒子の作製)における1.0mol/L-CaCl2水溶液を1.0mol/L-MgCl2水溶液に変更した以外は、実施例1と同様に行い、トナー母粒子の分散液を得た。
以降は、実施例30と同様にして、トナー31を得た。
実施例1の(トナー母粒子の作製)における1.0mol/L-CaCl2水溶液を1.0mol/L-BaCl2水溶液に変更した以外は、実施例1と同様に行い、トナー母粒子の分散液を得た。
以降は、実施例30と同様にして、トナー32を得た。
(トナー母粒子の作製)
実施例1の(トナー母粒子の作製)における1.0mol/L-CaCl2水溶液を0.7mol/L-AlCl3水溶液に変更した以外は、実施例1と同様に行い、トナー母粒子の分散液を得た。
以降は、実施例30と同様にして、トナー33を得た。
高速撹拌装置クレアミックス(エム・テクニック社製)を備えた容器中に、塩化カルシウム13.2部をイオン交換水250部に溶解した水溶液を入れ回転数を18000rpmに調整した。ここに、イオン交換水50部に水酸化ナトリウム4.8部を溶解した水溶液を撹拌しながら徐々に添加して、水酸化カルシウムコロイド(難水溶性の金属水酸化物コロイド)分散液を調製し、pHを6.0に調整した。
・スチレン 70.0部
・n-ブチルアクリレート 30.0部
・飽和ポリエステル樹脂 3.0部
(テレフタル酸-プロピレンオキサイド変性ビスフェノールA共重合体、酸価13mgKOH/g、Mw14500)
・C.I.ピグメントブルー15:3 6.5部
・エステルワックス 12.0部
(主成分C21H43COOC22H45、融点72.5℃)
以降は、実施例30と同様にしてトナー34を得た。
実施例34の塩化カルシウム13.2部を塩化マグネシウム11.3部に変更した以外は、実施例34と同様にしてトナー35を得た。
実施例34の塩化カルシウム13.2部を塩化バリウム24.7部に変更した以外は、実施例34と同様にしてトナー36を得た。
実施例34の塩化カルシウム13.2部を塩化アルミニウム10.5部に変更した以外は、実施例34と同様にしてトナー37を得た。
高速撹拌装置クレアミックス(エム・テクニック社製)を備えた容器中に、炭酸ナトリウム12.6部をイオン交換水250部に溶解した水溶液を入れ回転数を18000rpmに調整した。ここに、イオン交換水50部に塩化カルシウム13.2部を溶解した水溶液を撹拌しながら一気に添加して、30分間撹拌した後、pHを6.0に調整した。
・スチレン 70.0部
・n-ブチルアクリレート 30.0部
・飽和ポリエステル樹脂 3.0部
(テレフタル酸-プロピレンオキサイド変性ビスフェノールA共重合体、酸価13mgKOH/g、Mw14500)
・C.I.ピグメントブルー15:3 6.5部
・エステルワックス 12.0部
(主成分C21H43COOC22H45、融点72.5℃)
以降は、実施例30と同様にしてトナー38を得た。
実施例38の塩化カルシウム13.2部を塩化マグネシウム11.3部に変更した以外は実施例38と同様にしてトナー39を得た。
実施例38の塩化カルシウム13.2部を塩化バリウム24.7部に変更した以外は実施例38と同様にしてトナー40を得た。
実施例38の塩化カルシウム13.2部を塩化アルミニウム10.5部に変更した以外は実施例38と同様にしてトナー41を得た。
実施例30の樹脂粒子の固着工程での固着pH、酸処理pH、及び酸処理時間を表5のように変更した以外は実施例30と同様にしてトナー42~トナー52を得た。

実施例30の飽和ポリエステル樹脂を極性樹脂Fに変更した以外は実施例30と同様にしてトナー53を得た。
実施例1の酸処理温度を65℃に変更した以外は実施例1と同様にし、トナー粒子54を得た。得られたトナー粒子は著しく粗粒化しており、その後のトナー化と評価は実施しなかった。
下記の手順に従って、溶解懸濁法によってトナーを製造した。
・スチレン-n-ブチルアクリレート共重合体(共重合比:スチレン/n-ブチルアクリレート=75/25、Mp=17000) 100.0部
・飽和ポリエステル樹脂 3.0部
(テレフタル酸-プロピレンオキサイド変性ビスフェノールA共重合体、酸価13mgKOH/g、Mw14500)
・C.I.ピグメントブルー15:3 6.5部
・最大吸熱ピークのピーク温度が77℃の炭化水素ワックス(HNP-51,日本精蝋社製) 9.0部
樹脂粒子E-1の水分散体を樹脂粒子E-18の水分散体に変更する以外は実施例1と同様にしてトナー56を得た。
樹脂粒子E-1の水分散体を樹脂粒子E-19の水分散体に変更する以外は実施例1と同様にしてトナー57を得た。
(トナー母粒子の作製)
高速撹拌装置クレアミックス(エム・テクニック社製)を備えた容器中に0.1mol/L-Na3PO4水溶液850.0部を添加し、回転数を15000rpmに調整し、60℃に加温した。ここに1.0mol/L-CaCl2水溶液68.0部を添加し、微細な難水溶性分散剤Ca3(PO4)2を含む水系媒体を調製し、30分間撹拌した後、pHを6.0に調整した。
・スチレン 70.0部
・n-ブチルアクリレート 30.0部
・飽和ポリエステル樹脂 3.0部
(テレフタル酸-プロピレンオキサイド変性ビスフェノールA共重合体、酸価13mgKOH/g、Mw14500)
・樹脂A-1 3.0部
・C.I.ピグメントブルー15:3 6.5部
・エステルワックス 12.0部
(主成分C21H43COOC22H45、融点72.5℃)
次に、イオン交換水を200.0部添加して、還流管を取り外し、蒸留装置を取り付けた。容器内の温度が100℃の蒸留を5時間行った。蒸留留分は700.0部であった。30℃まで冷却し、重合体スラリーを得た。得られた重合体スラリーのpHを1.0に調製した。そのまま2時間撹拌し、ろ過と水による洗浄を3回繰り返した後に固形分を回収し、30℃の減圧乾燥機で1日間乾燥し、トナー粒子58を得た。
下記のように二成分現像剤を作製した。
帯電量の測定は二成分現像剤30gを分取し、高温高湿環境(30℃/85%RH)で5昼夜放置し、その後50mLの絶縁性のプラスチック容器に入れ、200回/分の速度で3分間振とうさせ、図1の装置を用いて測定した。
図1に示す底に500メッシュ(目開き25μm)のスクリーン3のある金属製の測定容器2に摩擦帯電量を測定しようとする二成分現像剤を0.500g入れ金属製のフタ4をする。このときの測定容器2全体の重量を秤りWl(g)とする。次に、吸引機1(測定容器2と接する部分は少なくとも絶縁体)において、吸引口7から吸引し風量調節弁6を調整して真空計5の圧力を250mmAqとする。この状態で充分、好ましくは2分間吸引を行いトナーを吸引除去する。
摩擦帯電量(mC/kg)=(C×V)/(W1-W2)
二成分現像剤の放置環境が、低温低湿環境(10℃/15%RH)である以外は前述の高温高湿下でのトナー帯電量の評価に記載の方法と同様にトナー帯電量の測定を行った。評価は低温低湿時と高温高湿時の帯電量の比(低温低湿下の帯電量/高温高湿下の帯電量)の絶対値を計算した。
二成分現像剤30gを分取し、高温高湿環境(30℃/85%RH)で5昼夜放置し、その後50mLの絶縁性のプラスチック容器に入れた。200回/分の速度で30秒間振とうさせ、図1の装置を用いて帯電量を測定した。高温高湿下でのトナー帯電量の評価にて求めた摩擦帯電量を飽和帯電量とし、立ち上がり(%)を下記式により算出した。
立ち上がり(%)=
{180回振とう時の帯電量(mC/kg)/飽和帯電量(mC/kg)}×100
図2に示す一成分接触現像システムの現像装置の改造機(Satera LBP5300;キヤノン製)において、現像剤容器にトナーを70g充填する。なお、転写紙としてはXerox4200(ゼロックス社製、75g/m2紙)を用いた。
ベタ部分の画像濃度により評価した。尚、画像濃度の測定には「マクベス反射濃度計 RD918」(マクベス社製)を用いて、原稿濃度が0.00の白下地部分のプリントアウト画像に対する相対濃度を測定した。
プリントアウトした画像の非画像部の反射率(%)を「REFLECTOMETER MODEL TC-6DS」(東京電色社製)で測定した。得られた反射率を、同様にして測定した未使用のプリントアウト用紙(標準紙)の反射率(%)から差し引いた数値(%)を用いて評価した。数値が小さい程、画像カブリが抑制されていることになる。
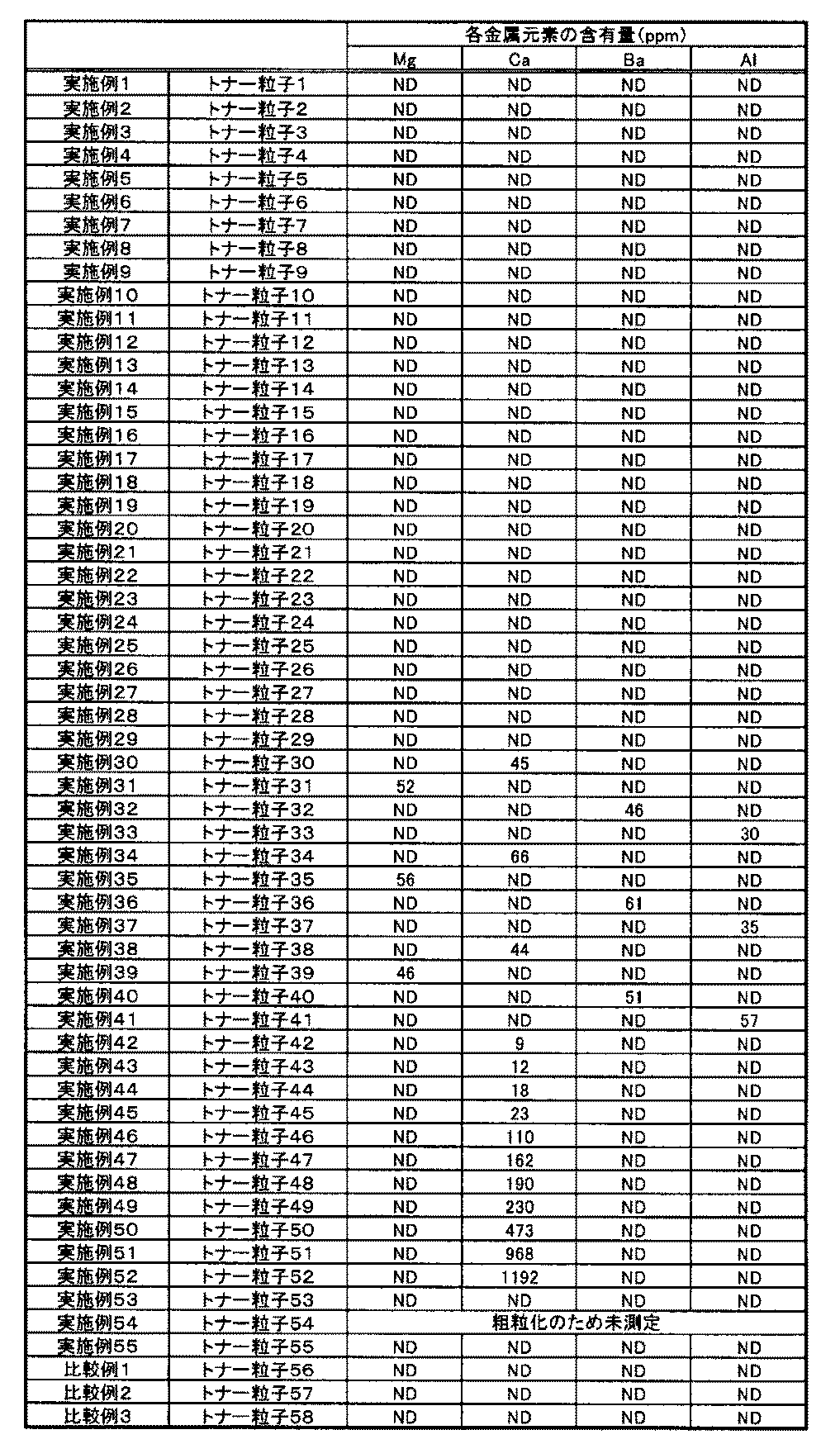

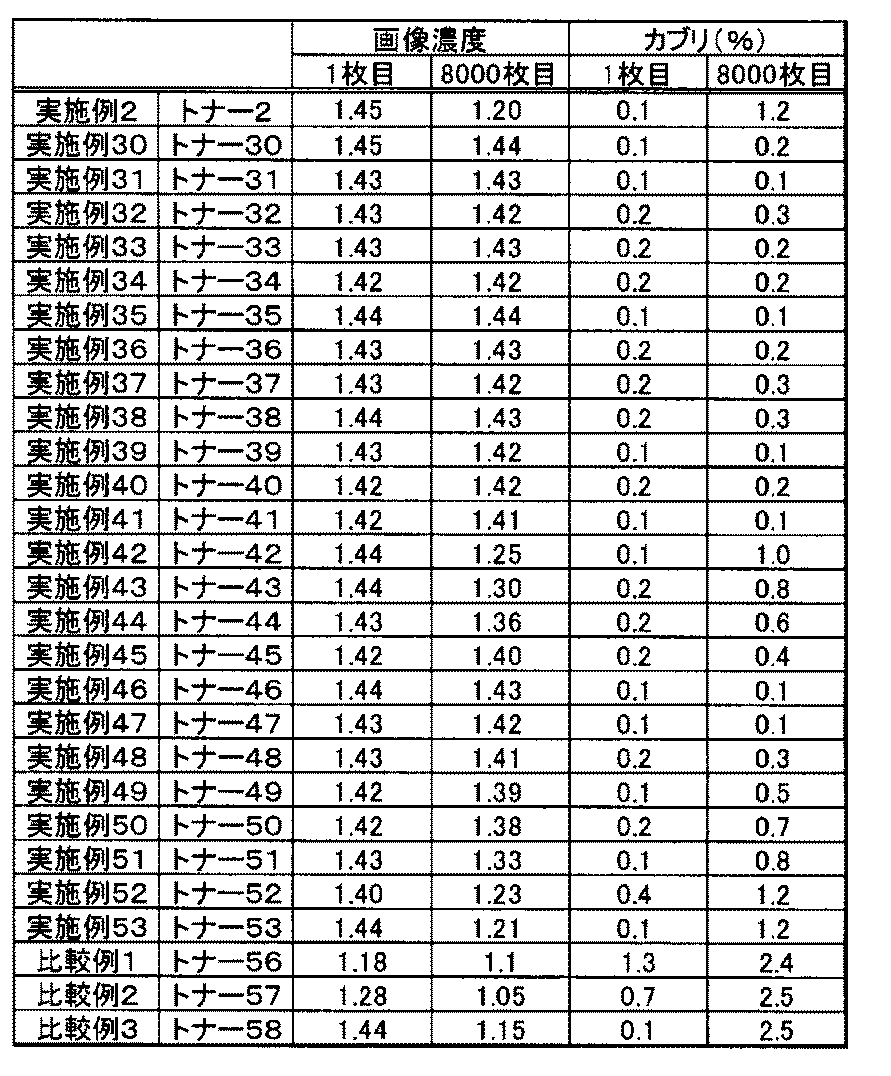
Claims (12)
- 結着樹脂、着色剤および離型剤を含有するトナー母粒子の表面に、樹脂粒子を固着させることにより得られたトナー粒子を有するトナーであって、
前記樹脂粒子が、樹脂Aを含有し、
前記該樹脂Aが、イオン性官能基を有し、且つpKa(酸解離定数)が7.0以上9.0以下である、
ことを特徴とするトナー。 - 前記樹脂Aが、下記式(1)で示される1価の基aを有することを特徴とする請求項1に記載のトナー。
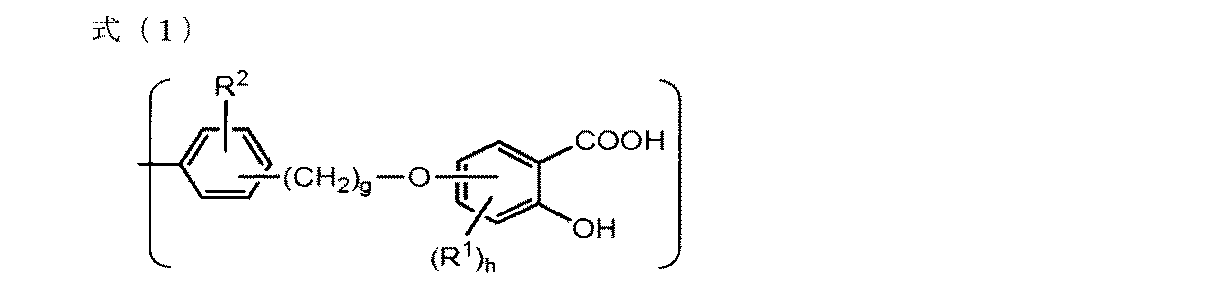
- 前記基aの含有量が、前記樹脂A1g当り、50μmol以上1000μmol以下である請求項2に記載のトナー。
- 前記トナー粒子が、マグネシウム、カルシウム、バリウムおよびアルミニウムからなる群より選択される少なくとも1種の金属元素を含有する請求項1乃至3のいずれか1項に記載のトナー。
- 前記トナー粒子に含有される前記金属元素の含有量が、前記トナー粒子の全質量に対して10ppm以上1000ppm以下である請求項4に記載のトナー。
- 前記トナー粒子に含有される前記金属元素の含有量が、前記トナー粒子の全質量に対して20ppm以上200ppm以下である請求項4に記載のトナー。
- 前記トナー母粒子が、カルボキシ基を有する樹脂を含有する請求項1乃至6のいずれか1項に記載のトナー。
- 結着樹脂、着色剤および離型剤を含有するトナー母粒子の表面に、樹脂粒子が固着されたトナー粒子の製造方法であって、
(i)重合性単量体および着色剤を含有する重合性単量体組成物の粒子を水系媒体中で形成する工程と、
(ii)該重合性単量体組成物の該粒子に含有される該重合性単量体を重合させて、該トナー母粒子を含有する分散液Bを得る工程と、
(iii)該分散液Bに該樹脂粒子を添加して、分散液Cを得る工程と、
(iv)該分散液Cを該トナー母粒子のガラス転移温度(Tg)以上に加熱して、該トナー母粒子の表面に該樹脂粒子を固着させて、トナー粒子を得る工程と
をこの順に有し、
該樹脂粒子が、樹脂Aを含有し、
該樹脂Aが、イオン性官能基を有し、且つpKa(酸解離定数)が7.0以上9.0以下である、ことを特徴とするトナー粒子の製造方法。 - 該樹脂Aが、下記式(1)で示される1価の基aを有することを特徴とする請求項8に記載のトナー粒子の製造方法。

- 前記水系媒体が、無機分散安定剤を含有する請求項8または9に記載のトナー粒子の製造方法。
- 前記無機分散安定剤が、リン酸カルシウム化合物、リン酸アルミニウム化合物、リン酸マグネシウム化合物、水酸化カルシウム化合物、水酸化アルミニウム化合物、水酸化マグネシウム化合物、炭酸カルシウム化合物、炭酸アルミニウム化合物、および炭酸マグネシウム化合物からなる群より選択される少なくとも1種である請求項8乃至10のいずれか1項に記載のトナー粒子の製造方法。
- 前記製造方法が、前記トナー粒子を得る工程の後に、前記トナー母粒子のガラス転移温度(Tg)以下の温度で前記無機分散安定剤を除去する工程を有する請求項8乃至11のいずれか1項に記載のトナー粒子の製造方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP15767896.2A EP3125044B1 (en) | 2014-03-27 | 2015-02-25 | Toner and process for producing toner |
| BR112016017396A BR112016017396A2 (pt) | 2014-03-27 | 2015-02-25 | Toner e método para produzir toner |
| RU2016141933A RU2016141933A (ru) | 2014-03-27 | 2015-02-25 | Тонер и способ изготовления тонера |
| CN201580016480.3A CN106133613A (zh) | 2014-03-27 | 2015-02-25 | 调色剂和调色剂的制造方法 |
| US14/745,345 US9829820B2 (en) | 2014-03-27 | 2015-06-19 | Toner and method for producing toner |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014-067127 | 2014-03-27 | ||
| JP2014067127 | 2014-03-27 | ||
| JP2014-199726 | 2014-09-30 | ||
| JP2014199726 | 2014-09-30 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/745,345 Continuation US9829820B2 (en) | 2014-03-27 | 2015-06-19 | Toner and method for producing toner |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015145968A1 true WO2015145968A1 (ja) | 2015-10-01 |
Family
ID=54194524
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/000957 WO2015145968A1 (ja) | 2014-03-27 | 2015-02-25 | トナーおよびトナーの製造方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US9829820B2 (ja) |
| EP (1) | EP3125044B1 (ja) |
| JP (1) | JP2016066048A (ja) |
| CN (1) | CN106133613A (ja) |
| BR (1) | BR112016017396A2 (ja) |
| RU (1) | RU2016141933A (ja) |
| WO (1) | WO2015145968A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016200814A (ja) * | 2015-04-08 | 2016-12-01 | キヤノン株式会社 | トナー |
| JP2016218370A (ja) * | 2015-05-25 | 2016-12-22 | キヤノン株式会社 | 画像形成方法 |
| JP2017102236A (ja) * | 2015-12-01 | 2017-06-08 | キヤノン株式会社 | トナー粒子の製造方法 |
| US9921501B2 (en) | 2016-03-18 | 2018-03-20 | Canon Kabushiki Kaisha | Toner and process for producing toner |
Families Citing this family (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9952523B2 (en) | 2015-02-25 | 2018-04-24 | Canon Kabushiki Kaisha | Toner and toner production method |
| US9733583B2 (en) | 2015-04-08 | 2017-08-15 | Canon Kabushiki Kaisha | Toner |
| JP6797660B2 (ja) | 2016-01-08 | 2020-12-09 | キヤノン株式会社 | トナーの製造方法 |
| JP6659143B2 (ja) * | 2016-01-08 | 2020-03-04 | キヤノン株式会社 | トナー粒子の製造方法 |
| DE102017101171B4 (de) | 2016-01-28 | 2021-07-22 | Canon Kabushiki Kaisha | Toner |
| US9897932B2 (en) | 2016-02-04 | 2018-02-20 | Canon Kabushiki Kaisha | Toner |
| JP6855289B2 (ja) * | 2016-03-18 | 2021-04-07 | キヤノン株式会社 | トナー及びトナーの製造方法 |
| JP6808542B2 (ja) | 2016-03-18 | 2021-01-06 | キヤノン株式会社 | トナー及びトナーの製造方法 |
| JP6849370B2 (ja) * | 2016-10-03 | 2021-03-24 | キヤノン株式会社 | トナー粒子の製造方法 |
| JP2018060009A (ja) * | 2016-10-04 | 2018-04-12 | キヤノン株式会社 | トナー粒子の製造方法 |
| JP6873649B2 (ja) * | 2016-10-04 | 2021-05-19 | キヤノン株式会社 | トナー粒子の製造方法 |
| US10338487B2 (en) | 2017-05-15 | 2019-07-02 | Canon Kabushiki Kaisha | Toner |
| JP6887868B2 (ja) | 2017-05-15 | 2021-06-16 | キヤノン株式会社 | トナー |
| US10503090B2 (en) | 2017-05-15 | 2019-12-10 | Canon Kabushiki Kaisha | Toner |
| US10345726B2 (en) | 2017-05-15 | 2019-07-09 | Canon Kabushiki Kaisha | Method of manufacturing toner |
| US10551758B2 (en) | 2017-05-15 | 2020-02-04 | Canon Kabushiki Kaisha | Toner |
| US10310396B2 (en) | 2017-05-15 | 2019-06-04 | Canon Kabushiki Kaisha | Method of producing toner |
| US10353308B2 (en) | 2017-05-15 | 2019-07-16 | Canon Kabushiki Kaisha | Toner |
| CN107329380A (zh) * | 2017-06-26 | 2017-11-07 | 安徽天兴凯顿信息科技有限公司 | 一种用于复印机色调剂树脂及青色色调剂制备方法 |
| JP7146403B2 (ja) | 2018-01-26 | 2022-10-04 | キヤノン株式会社 | トナー |
| JP7267750B2 (ja) | 2018-01-26 | 2023-05-02 | キヤノン株式会社 | トナー |
| JP2019128516A (ja) | 2018-01-26 | 2019-08-01 | キヤノン株式会社 | トナー |
| US10635011B2 (en) | 2018-04-27 | 2020-04-28 | Canon Kabushiki Kaisha | Toner |
| JP7210222B2 (ja) | 2018-10-19 | 2023-01-23 | キヤノン株式会社 | トナー |
| JP7443047B2 (ja) | 2018-12-28 | 2024-03-05 | キヤノン株式会社 | トナー |
| JP7391658B2 (ja) | 2018-12-28 | 2023-12-05 | キヤノン株式会社 | トナー |
| EP3674802B1 (en) | 2018-12-28 | 2022-05-18 | Canon Kabushiki Kaisha | Toner and toner manufacturing method |
| JP7267740B2 (ja) | 2018-12-28 | 2023-05-02 | キヤノン株式会社 | トナー |
| JP7286314B2 (ja) | 2018-12-28 | 2023-06-05 | キヤノン株式会社 | トナー |
| JP7412903B2 (ja) * | 2019-06-11 | 2024-01-15 | キヤノン株式会社 | トナーの製造方法 |
| JP7309481B2 (ja) | 2019-07-02 | 2023-07-18 | キヤノン株式会社 | トナー |
| JP7336293B2 (ja) | 2019-07-25 | 2023-08-31 | キヤノン株式会社 | トナー |
| JP7328048B2 (ja) | 2019-07-25 | 2023-08-16 | キヤノン株式会社 | トナー |
| JP7321810B2 (ja) | 2019-07-25 | 2023-08-07 | キヤノン株式会社 | トナー |
| JP2021165835A (ja) * | 2020-04-06 | 2021-10-14 | キヤノン株式会社 | トナー及びトナーの製造方法 |
| JP7512089B2 (ja) * | 2020-06-01 | 2024-07-08 | キヤノン株式会社 | トナー |
| JP7458915B2 (ja) | 2020-06-25 | 2024-04-01 | キヤノン株式会社 | トナー |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06130726A (ja) * | 1992-10-19 | 1994-05-13 | Ricoh Co Ltd | 静電荷現像用トナー |
| JP2007086567A (ja) * | 2005-09-26 | 2007-04-05 | Kyocera Chemical Corp | 画像形成方法、トナー及びトナーの製造方法 |
| JP2011075934A (ja) * | 2009-09-30 | 2011-04-14 | Nippon Zeon Co Ltd | 重合トナーの製造方法 |
| JP2011215311A (ja) * | 2010-03-31 | 2011-10-27 | Mitsubishi Chemicals Corp | 静電荷像現像用トナーの製造方法 |
| WO2012157713A1 (ja) * | 2011-05-18 | 2012-11-22 | オリヱント化学工業株式会社 | 荷電制御樹脂及び荷電制御樹脂の製造方法 |
| JP2012256042A (ja) * | 2011-05-18 | 2012-12-27 | Canon Inc | トナー |
| JP2013120210A (ja) * | 2011-12-06 | 2013-06-17 | Morimura Chemicals Ltd | 静電像現像トナーおよびその製造方法 |
| JP2014067021A (ja) * | 2012-09-06 | 2014-04-17 | Mitsubishi Chemicals Corp | 静電荷像現像用トナー |
Family Cites Families (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE69025416T2 (de) | 1989-03-29 | 1996-08-01 | Bando Chemical Ind | Entwickler für die Elektrophotographie und deren Herstellungsverfahren |
| JPH02256071A (ja) | 1989-03-29 | 1990-10-16 | Bando Chem Ind Ltd | 静電潜像現像用トナーの製造方法 |
| US5066559A (en) | 1990-01-22 | 1991-11-19 | Minnesota Mining And Manufacturing Company | Liquid electrophotographic toner |
| JP4835628B2 (ja) * | 2000-06-19 | 2011-12-14 | 三菱化学株式会社 | 静電荷像現像用トナー及びその製造方法 |
| JP2004176036A (ja) | 2002-04-26 | 2004-06-24 | Canon Inc | 重合性化合物、高分子化合物、それらを用いた組成物及び記録方法並びに記録装置 |
| JP4708731B2 (ja) | 2003-05-27 | 2011-06-22 | キヤノン株式会社 | 液体組成物のセット、それを用いた液体付与方法および液体付与装置 |
| JP4324120B2 (ja) * | 2005-02-18 | 2009-09-02 | キヤノン株式会社 | 磁性トナー |
| JP2008070671A (ja) * | 2006-09-15 | 2008-03-27 | Ricoh Co Ltd | 画像形成方法並びに装置およびプロセスカートリッジ |
| KR101261106B1 (ko) | 2008-02-25 | 2013-05-06 | 캐논 가부시끼가이샤 | 토너 |
| JP5400758B2 (ja) | 2008-02-25 | 2014-01-29 | キヤノン株式会社 | トナー |
| JP2009244494A (ja) * | 2008-03-31 | 2009-10-22 | Brother Ind Ltd | 負帯電トナーの製造方法 |
| JP5371407B2 (ja) * | 2008-12-16 | 2013-12-18 | キヤノン株式会社 | トナーおよびトナーの製造方法 |
| JP2010276754A (ja) * | 2009-05-27 | 2010-12-09 | Konica Minolta Business Technologies Inc | トナーの製造方法、トナー |
| JP5630601B2 (ja) * | 2009-06-04 | 2014-11-26 | 戸田工業株式会社 | 電子写真現像剤用磁性キャリア及びその製造方法、並びに二成分系現像剤 |
| JP5444890B2 (ja) * | 2009-06-30 | 2014-03-19 | 日本ゼオン株式会社 | 重合トナーの製造方法 |
| JP5541673B2 (ja) * | 2009-12-28 | 2014-07-09 | キヤノン株式会社 | トナー |
| JP5773752B2 (ja) * | 2010-06-11 | 2015-09-02 | キヤノン株式会社 | トナー及びトナーの製造方法 |
| JP5765132B2 (ja) * | 2010-12-06 | 2015-08-19 | 株式会社リコー | 静電荷像現像用トナーと該トナーを用いる現像剤、及び画像形成装置、並びにプロセスカートリッジ |
| CN103459436B (zh) * | 2011-03-30 | 2015-01-21 | 佳能株式会社 | 可聚合单体、高分子化合物、包含高分子化合物的电荷控制剂以及包含电荷控制剂的显影剂承载构件和调色剂 |
| US8609312B2 (en) | 2011-05-18 | 2013-12-17 | Canon Kabushiki Kaisha | Toner |
| US8574801B2 (en) | 2011-05-18 | 2013-11-05 | Canon Kabushiki Kaisha | Toner |
| CN103547969B (zh) * | 2011-05-18 | 2016-06-22 | 佳能株式会社 | 调色剂 |
| US9098002B2 (en) * | 2011-05-18 | 2015-08-04 | Canon Kabushiki Kaisha | Toner |
| KR20130028661A (ko) | 2011-09-09 | 2013-03-19 | 캐논 가부시끼가이샤 | 토너 |
| JP2013061384A (ja) * | 2011-09-12 | 2013-04-04 | Konica Minolta Business Technologies Inc | トナー粒子の製造方法 |
| JP6031347B2 (ja) | 2011-12-27 | 2016-11-24 | 花王株式会社 | 電子写真用トナーの製造方法 |
| JP5681147B2 (ja) * | 2012-06-20 | 2015-03-04 | 京セラドキュメントソリューションズ株式会社 | 磁性1成分現像用トナー |
| JP2014032401A (ja) * | 2012-07-12 | 2014-02-20 | Canon Inc | トナーの製造方法 |
| US9423708B2 (en) | 2014-03-27 | 2016-08-23 | Canon Kabushiki Kaisha | Method for producing toner particle |
-
2015
- 2015-02-25 WO PCT/JP2015/000957 patent/WO2015145968A1/ja active Application Filing
- 2015-02-25 CN CN201580016480.3A patent/CN106133613A/zh active Pending
- 2015-02-25 EP EP15767896.2A patent/EP3125044B1/en active Active
- 2015-02-25 BR BR112016017396A patent/BR112016017396A2/pt not_active Application Discontinuation
- 2015-02-25 RU RU2016141933A patent/RU2016141933A/ru unknown
- 2015-03-27 JP JP2015065489A patent/JP2016066048A/ja active Pending
- 2015-06-19 US US14/745,345 patent/US9829820B2/en active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06130726A (ja) * | 1992-10-19 | 1994-05-13 | Ricoh Co Ltd | 静電荷現像用トナー |
| JP2007086567A (ja) * | 2005-09-26 | 2007-04-05 | Kyocera Chemical Corp | 画像形成方法、トナー及びトナーの製造方法 |
| JP2011075934A (ja) * | 2009-09-30 | 2011-04-14 | Nippon Zeon Co Ltd | 重合トナーの製造方法 |
| JP2011215311A (ja) * | 2010-03-31 | 2011-10-27 | Mitsubishi Chemicals Corp | 静電荷像現像用トナーの製造方法 |
| WO2012157713A1 (ja) * | 2011-05-18 | 2012-11-22 | オリヱント化学工業株式会社 | 荷電制御樹脂及び荷電制御樹脂の製造方法 |
| JP2012256042A (ja) * | 2011-05-18 | 2012-12-27 | Canon Inc | トナー |
| JP2013120210A (ja) * | 2011-12-06 | 2013-06-17 | Morimura Chemicals Ltd | 静電像現像トナーおよびその製造方法 |
| JP2014067021A (ja) * | 2012-09-06 | 2014-04-17 | Mitsubishi Chemicals Corp | 静電荷像現像用トナー |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3125044A4 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016200814A (ja) * | 2015-04-08 | 2016-12-01 | キヤノン株式会社 | トナー |
| JP2016218370A (ja) * | 2015-05-25 | 2016-12-22 | キヤノン株式会社 | 画像形成方法 |
| JP2017102236A (ja) * | 2015-12-01 | 2017-06-08 | キヤノン株式会社 | トナー粒子の製造方法 |
| US9921501B2 (en) | 2016-03-18 | 2018-03-20 | Canon Kabushiki Kaisha | Toner and process for producing toner |
Also Published As
| Publication number | Publication date |
|---|---|
| CN106133613A (zh) | 2016-11-16 |
| RU2016141933A (ru) | 2018-04-28 |
| US20150286157A1 (en) | 2015-10-08 |
| BR112016017396A2 (pt) | 2017-08-08 |
| EP3125044A1 (en) | 2017-02-01 |
| US9829820B2 (en) | 2017-11-28 |
| EP3125044A4 (en) | 2017-09-06 |
| RU2016141933A3 (ja) | 2018-04-28 |
| JP2016066048A (ja) | 2016-04-28 |
| EP3125044B1 (en) | 2020-05-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2015145968A1 (ja) | トナーおよびトナーの製造方法 | |
| US9423708B2 (en) | Method for producing toner particle | |
| JP6351296B2 (ja) | トナー | |
| JP2018010288A (ja) | トナー、該トナーを備えた現像装置及び画像形成装置 | |
| JP2018010286A (ja) | トナー、現像装置及び画像形成装置 | |
| JP2011154351A (ja) | トナー | |
| KR20120088839A (ko) | 토너용 수지, 및 토너 | |
| JP5800450B2 (ja) | トナー | |
| JP2017102399A (ja) | トナー及びトナーの製造方法 | |
| US9556300B2 (en) | Method for manufacturing resin particles and method for manufacturing toner particles | |
| JP2018173499A (ja) | トナー | |
| JP5455746B2 (ja) | トナー粒子の製造方法及びイエロートナー | |
| JP2014032401A (ja) | トナーの製造方法 | |
| JP2018004879A (ja) | トナー、及び該トナーを備えた現像装置 | |
| JP5995672B2 (ja) | トナー | |
| JP5995669B2 (ja) | トナー | |
| JP5419586B2 (ja) | トナー | |
| JP6541481B2 (ja) | トナー及びその製造方法 | |
| JP5294890B2 (ja) | トナー | |
| JP6576187B2 (ja) | トナー | |
| JP5414339B2 (ja) | トナー及び該トナーの製造方法 | |
| JP2017062366A (ja) | 画像形成方法 | |
| US20240219855A1 (en) | Toner for electrostatic charge image development and method for manufacturing toner for electrostatic charge image development | |
| JP6120701B2 (ja) | トナーの製造方法 | |
| JP2017062443A (ja) | トナー |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15767896 Country of ref document: EP Kind code of ref document: A1 |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015767896 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015767896 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112016017396 Country of ref document: BR |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2016141933 Country of ref document: RU Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 112016017396 Country of ref document: BR Kind code of ref document: A2 Effective date: 20160727 |