KR20200067930A - 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 - Google Patents
폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 Download PDFInfo
- Publication number
- KR20200067930A KR20200067930A KR1020207015960A KR20207015960A KR20200067930A KR 20200067930 A KR20200067930 A KR 20200067930A KR 1020207015960 A KR1020207015960 A KR 1020207015960A KR 20207015960 A KR20207015960 A KR 20207015960A KR 20200067930 A KR20200067930 A KR 20200067930A
- Authority
- KR
- South Korea
- Prior art keywords
- mmol
- compound
- reaction mixture
- concentrated
- mixture
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/14—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/498—Pyrazines or piperazines ortho- and peri-condensed with carbocyclic ring systems, e.g. quinoxaline, phenazine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/529—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim forming part of bridged ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5365—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/537—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines spiro-condensed or forming part of bridged ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/18—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/22—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed systems contains four or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/18—Bridged systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Abstract
인간 면역결핍 바이러스 (HIV) 감염의 치료에 사용하기 위한 화합물이 개시되어 있다. 화합물은 하기 화학식 I을 갖는다 (그의 입체이성질체 및 제약상 허용되는 염을 포함). 이러한 화합물의 제조 및 사용과 연관된 방법, 뿐만 아니라 이러한 화합물을 포함하는 제약 조성물이 또한 개시되어 있다.
<화학식 I>

상기 식에서 R1, X, W, Y1, Y2, Z1, 및 Z4는 본원에 정의된 바와 같다.
<화학식 I>
상기 식에서 R1, X, W, Y1, Y2, Z1, 및 Z4는 본원에 정의된 바와 같다.
Description
관련 출원에 대한 상호 참조
본원은 35 U.S.C. §119(e) 하에 2012년 12월 21일에 출원된 미국 특허 가출원 번호 61/745,375, 2013년 3월 15일에 출원된 미국 특허 가출원 번호 61/788,397, 및 2013년 7월 12일에 출원된 미국 특허 가출원 번호 61/845,803을 우선권 주장한다. 상기 출원은 그 전문이 본원에 참조로 포함된다.
인간 면역결핍 바이러스 (HIV) 감염의 치료를 위한 화합물, 조성물 및 방법이 개시되어 있다. 특히, 신규 폴리시클릭 카르바모일피리돈 화합물 및 그의 제조 방법 및 치료제 또는 예방제로서의 용도가 개시되어 있다.
인간 면역결핍 바이러스 감염 및 관련 질환은 전세계적으로 주요 공중 보건 문제이다. 제1형 인간 면역결핍 바이러스 (HIV-1)는 바이러스 복제에 요구되는 3종의 효소: 역전사효소, 프로테아제 및 인테그라제를 코딩한다. 역전사효소 및 프로테아제를 표적화하는 약물이 널리 사용되고 유효성을 나타낼지라도, 특히 조합되어 사용되는 경우에, 독성 및 내성 균주의 발생은 그의 유용성을 제한하였다 (Palella, et al. N. Engl. J Med. (1998) 338:853-860; Richman, D. D. Nature (2001) 410:995-1001).
프레그난 X 수용체 (PXR)는 소분자의 대사 및 신체로부터의 제거에 관여하는 효소의 주요 조절자 중 하나인 핵 수용체이다. PXR의 활성화는 대사 효소, 예컨대 시토크롬 P450 3A4 (CYP3A4) 뿐만 아니라 수송에 관여하는 효소, 예컨대 간 및 장 내의 OATP2를 상향-조절하거나 또는 그의 생산을 유도하는 것으로 공지되어 있다 (Endocrine Reviews (2002) 23(5):687-702). 한 약물이 PXR의 활성화에 의해 이들 및 다른 효소의 상향-조절을 유발하는 경우에, 이는 상향-조절된 효소에 감수성인 공-투여된 약물의 흡수 및/또는 노출을 감소시킬 수 있다. 이러한 유형의 약물-약물 상호작용의 위험을 최소화하기 위해, PXR 활성화를 최소화하는 것이 바람직하다. 또한, PXR이 많은 상이한 부류의 분자에 의해 활성화되는 것으로 공지되어 있다 (Endocrine Reviews (2002) 23(5):687-702). 따라서, 다른 약물과 공-투여될 약물의 경우에, PXR 활성화에 대해 시험하고 이러한 활성화를 최소화하는 것이 중요하다.
수송체는 약물의 약동학, 안전성 및 효능 프로파일에서 역할을 하는 것으로 확인되고, 특정 약물-약물 상호작용은 수송체에 의해 매개된다. 문헌 [Giacomini KM, et al. "Membrane transporters in drug development," Nat.Rev Drug Discov. 9: 215-236, 2010; Zhang L, et al. "Transporter-Mediated Drug-Drug Interactions," Clin. Pharm. Ther. 89(4):481-484 (2011)]을 참조한다. 한 수송체, 유기 양이온 수송체 2 (OCT2; SLC22A2)는 수송체 중 용질 운반체 (SLC) 슈퍼-패밀리의 구성원이고, 주로 신장 근위 세관의 기저측 막 상에 위치한다. OCT2는 정단 발현된 다중약물 및 독소 배출 (MATE) 수송체 1 및 2-K와 협력하여 신장에서 주요 양이온 분비 경로를 형성하는 것으로 여겨지고, 크레아티닌을 비롯한 내인성 화합물 및 메트포르민을 비롯한 생체이물을 수송하는 것으로 밝혀졌다. 따라서 OCT2의 억제는 증가된 수준의 혈청 크레아티닌 및 다른 OCT2 기질의 증가된 수준에 대한 가능성으로 이어질 수 있다. 약물의 OCT2 억제를 시험하고 이를 감소시키는 것이 또한 중요하다.
항레트로바이러스 요법의 목표는 HIV 감염된 환자에서 바이러스 억제를 달성하는 것이다. 미국 보건복지부에 의해 확립된 치료 가이드라인에는 바이러스 억제의 달성이 조합 요법의 사용, 즉 적어도 2종 이상의 약물 부류로부터의 여러 약물의 사용을 필요로 하는 것으로 제공되어 있다. (Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. Available at http://aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Section accessed March 14, 2013.) 또한, HIV 감염된 환자의 치료에 관한 판단은 환자가 다른 의학적 상태에 대한 치료를 요구하는 경우에 복잡해진다 (Id. at E-12). HIV를 억제할 뿐만 아니라 환자가 겪을 수 있는 다른 상태를 치료하기 위해 표준 치료가 다중의 상이한 약물의 사용을 요구하기 때문에, 약물 상호작용에 대한 가능성은 약물 요법의 선택을 위한 기준이다. 따라서, 감소된 약물 상호작용 가능성을 갖는 항레트로바이러스 요법에 대한 필요성이 존재한다.
따라서, 다른 약물과 공-투여되는 경우에 HIV의 복제를 억제하고 PXR 활성화를 최소화하는 신규 작용제에 대한 필요성이 존재한다.
본 발명은 항바이러스 활성을 갖는, 그의 입체이성질체 및 제약상 허용되는 염을 비롯한 신규 폴리시클릭 카르바모일피리돈 화합물, 및 HIV 감염의 치료에서 이러한 화합물의 용도에 관한 것이다. 본 발명의 화합물은 HIV 인테그라제의 활성을 억제하는데 사용될 수 있고, HIV 복제를 감소시키는데 사용될 수 있다.
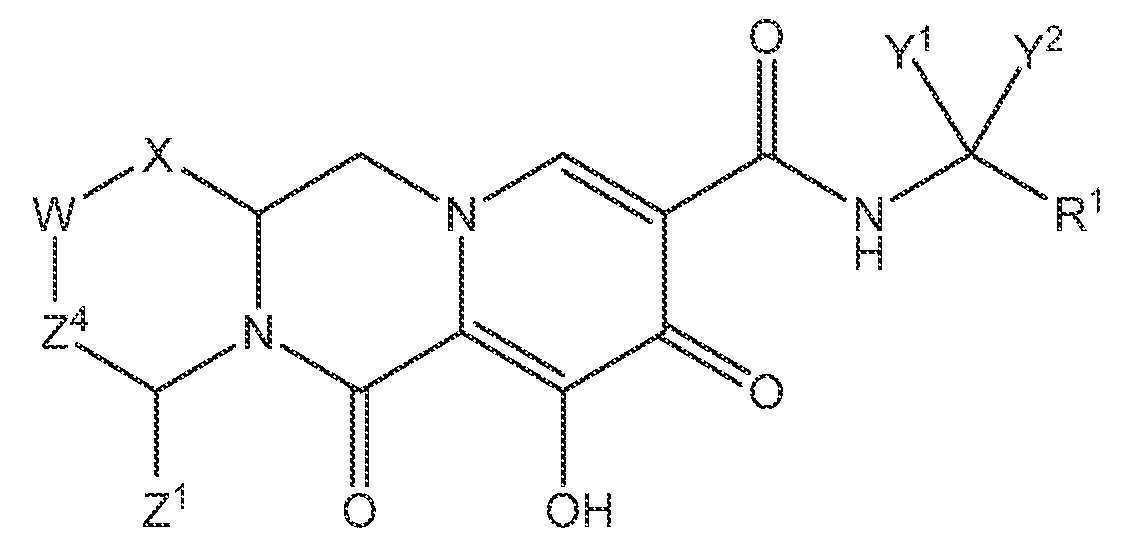
본 발명의 한 실시양태에서, 하기 화학식 I을 갖는 화합물 또는 그의 입체이성질체 또는 제약상 허용되는 염이 제공된다.
<화학식 I>
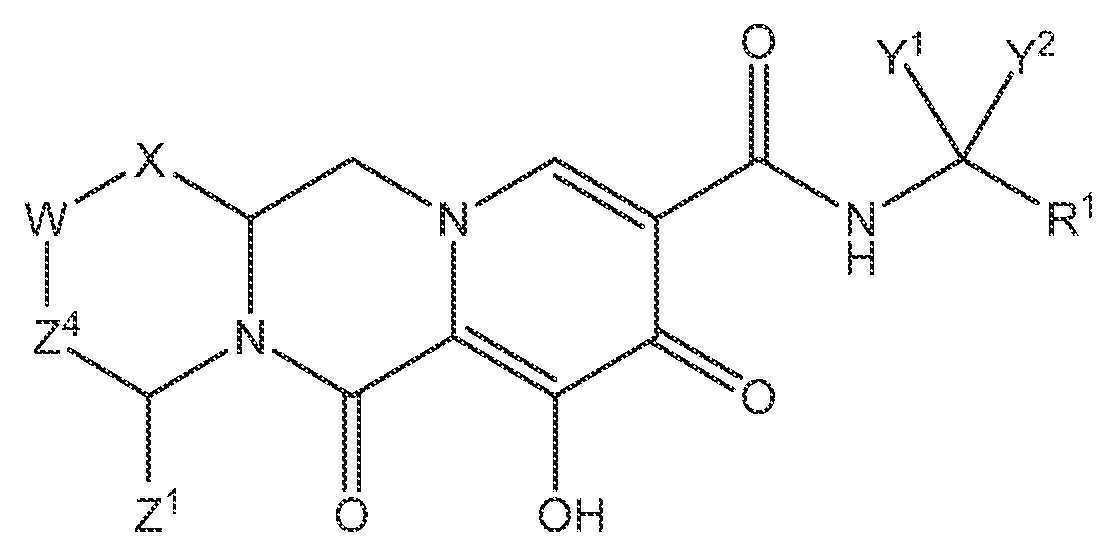
상기 식에서:
X는 -O- 또는 -NZ3- 또는 -CHZ3-이고;
W는 -CHZ2-이고;
Z1, Z2 및 Z3은 각각, 독립적으로, 수소 또는 C1-3알킬이거나, 또는 여기서 Z1 및 Z2 또는 Z1 및 Z3은 함께 -L-을 형성하고, 여기서 L은 -C(Ra)2-, -C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2C(Ra)2-, 또는 -C(Ra)2C(Ra)2C(Ra)2C(Ra)2-이고, 여기서 Z1 및 Z2 또는 Z1 및 Z3 중 적어도 하나는 함께 -L-을 형성하고;
Z4는 결합, -CH2-, 또는 -CH2CH2-이고;
Y1 및 Y2는 각각, 독립적으로, 수소, C1-3알킬 또는 C1-3할로알킬이고;
R1은 1 내지 3개의 할로겐으로 치환된 페닐이고;
각각의 Ra는 독립적으로, 수소, 할로, 히드록실 또는 C1-4알킬이다.
본 발명의 또 다른 실시양태에서, 하기 화학식 I을 갖는 화합물 또는 그의 입체이성질체 또는 제약상 허용되는 염이 제공된다.
<화학식 I>

상기 식에서:
X는 -O- 또는 -NZ3- 또는 -CHZ3-이고;
W는 -O- 또는 -NZ2- 또는 -CHZ2-이고;
Z1, Z2 및 Z3은 각각, 독립적으로, 수소 또는 C1-3알킬이거나, 또는 여기서 Z1 및 Z2 또는 Z1 및 Z3은 함께 -L-을 형성하고, 여기서 L은 -C(Ra)2-, -C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2C(Ra)2C(Ra)2-, -C(Ra)2OC(Ra)2-, -C(Ra)2NRaC(Ra)2-, -C(Ra)2SC(Ra)2-, -C(Ra)2S(O)C(Ra)2-, -C(Ra)2SO2C(Ra)2-, -C(Ra)2OC(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2OC(Ra)2-, -C(Ra)2NRaC(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2NRaC(Ra)2-, -C(Ra)2SC(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2SC(Ra)2-, -C(Ra)2S(O)C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2S(O)C(Ra)2-, -C(Ra)2SO2C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2SO2C(Ra)2-, -C(Ra)2SO2NRaC(Ra)2- 또는 -C(Ra)2NRaSO2C(Ra)2-이고;
Z4는 결합 또는 -CH2-, -CH2CH2-, -CH2CH2CH2-, -CH2OCH2-, -CH2NRaCH2-, -CH2SCH2-, -CH2S(O)CH2- 또는 -CH2SO2CH2-이고;
Y1 및 Y2는 각각, 독립적으로, 수소, C1-3알킬 또는 C1-3할로알킬이거나, 또는 Y1 및 Y2는 이들이 부착되어 있는 탄소 원자와 함께 3 내지 6개의 고리 원자를 갖는 카르보시클릭 고리 또는 3 내지 6개의 고리 원자를 갖는 헤테로시클릭 고리를 형성하고, 여기서 카르보시클릭 또는 헤테로시클릭 고리는 1개 이상의 Ra로 임의로 치환되고;
R1은 임의로 치환된 아릴 또는 임의로 치환된 헤테로아릴이고;
각각의 Ra는 독립적으로, 수소, 할로, 히드록실 또는 C1-4알킬이거나, 또는 여기서 2개의 Ra 기는 이들이 부착되어 있는 탄소 원자와 함께 =O를 형성하고,
여기서 하기 중 적어도 하나이다: (i) Z1 및 Z2 또는 Z1 및 Z3은 함께 -L-을 형성하거나; 또는 (ii) Y1 및 Y2는 이들이 부착되어 있는 탄소 원자와 함께 3 내지 6개의 고리 원자를 갖는 카르보시클릭 고리 또는 3 내지 6개의 고리 원자를 갖는 헤테로시클릭 고리를 형성한다.
또 다른 실시양태에서, 화학식 I을 갖는 화합물 또는 그의 입체이성질체 또는 제약상 허용되는 염, 및 제약상 허용되는 담체, 희석제 또는 부형제를 포함하는 제약 조성물이 제공된다.
본 발명은 또한 HIV 감염에 걸리거나 또는 걸릴 위험이 있는 인간에서 HIV 감염을 치료하기 위한 상기 기재된 바와 같은 제약 조성물의 용도를 제공한다.
또 다른 실시양태에서, 요법에서 화학식 I을 갖는 화합물을 사용하는 방법이 제공된다. 특히, 포유동물 (예를 들어, 인간)에게 화학식 I을 갖는 화합물 또는 그의 입체이성질체 또는 제약상 허용되는 염, 및 제약상 허용되는 담체, 희석제 또는 부형제를 투여하는 것을 포함하는, 포유동물에서 HIV 바이러스의 증식을 치료하거나, AIDS를 치료하거나, 또는 AIDS 또는 ARC 증상의 개시를 지연시키는 방법이 제공된다.
또 다른 실시양태에서, HIV 감염에 걸리거나 또는 걸릴 위험이 있는 인간에서 HIV 감염을 치료하기 위한, 본원에 기재된 바와 같은 화학식 I의 화합물 또는 그의 제약상 허용되는 염의 용도가 개시되어 있다.
또 다른 실시양태에서, HIV 감염에 걸리거나 또는 걸릴 위험이 있는 인간에서 HIV 감염을 치료하기 위한 의약의 제조를 위한, 본원에 기재된 바와 같은 화학식 I의 화합물 또는 그의 제약상 허용되는 염의 용도가 개시되어 있다.
또 다른 실시양태에서, HIV 감염을 치료하는데 효과적인 조성물; 및 상기 조성물이 HIV에 의한 감염을 치료하는데 사용될 수 있음을 나타내는 표지를 포함하는 포장 재료를 포함하는 제조품이 개시되어 있다. 예시적인 조성물은 본 발명에 따른 화학식 I의 화합물 또는 그의 제약상 허용되는 염을 포함한다.
또 다른 실시양태에서, HIV의 증식을 억제하는 방법이 개시되어 있다. 상기 방법은 바이러스를 HIV의 증식을 억제하는 조건 하에 유효량의 화학식 I의 화합물 또는 그의 염에 노출시키는 것을 포함한다.
또 다른 실시양태에서, HIV 인테그라제 효소의 활성을 억제하기 위한 화학식 I의 화합물의 용도가 개시되어 있다.
또 다른 실시양태에서, HIV의 복제를 억제하기 위한 화학식 I의 화합물 또는 그의 염의 용도가 개시되어 있다.
다른 실시양태, 목적, 특징 및 이점이 하기 실시양태의 상세한 설명에 기재될 것이고, 부분적으로는 그러한 설명으로부터 명백할 것이거나, 또는 청구된 본 발명의 실시에 의해 파악할 수 있다. 이들 목적 및 이점은 기재된 설명 및 그의 청구범위에서 특히 언급되어 있는 방법 및 조성물에 의해 실현되고 이루어질 것이다. 상기 발명의 내용은 그것이 본원에 개시된 실시양태 중 일부의 간략하고 일반적인 개요로서 간주된다는 이해 하에 기재되었고, 단지 독자의 이익 및 편의를 위해 제공되었으며, 첨부된 특허청구범위가 합법적으로 청구하는 범위나 그와 등가인 범주를 임의의 방식으로 제한하고자 의도되는 것은 아니다.
본 발명의 화합물은 HIV 인테그라제의 활성을 억제하는데 사용될 수 있고, HIV 복제를 감소시키는데 사용될 수 있다.
하기 설명에는 본 발명의 다양한 실시양태의 충분한 이해를 제공하기 위한 특정의 구체적 세부사항이 기재되어 있다. 그러나, 통상의 기술자는 본 발명이 이들 세부사항없이 실시될 수 있다는 것을 이해할 것이다. 하기의 여러 실시양태의 설명은 본 개시내용이 청구되는 주제의 예시로서 간주된다는 이해 하에 기재되었고, 첨부된 특허청구범위를 예시된 구체적 실시양태로 제한하고자 의도되는 것은 아니다. 본 개시내용 전반에 걸쳐 사용된 표제는 단지 편의상 제공되고, 특허청구범위를 임의의 방식으로 제한하는 것으로 해석되어서는 안된다. 임의의 표제 하에 예시된 실시양태는 임의의 다른 표제 하에 예시된 실시양태와 조합될 수 있다.
정의
문맥상 달리 요구되지 않는 한, 본 명세서 및 특허청구범위의 전반에 걸쳐 용어 "포함하다" 및 그의 변형, 예컨대, "포함한다" 및 "포함하는"은 개방된 포괄적인 의미로, "포함하나 이에 제한되지는 않는"으로 해석되어야 한다.
본 명세서 전반에 걸쳐 "한 실시양태" 또는 "하나의 실시양태"에 대한 언급은 그 실시양태와 관련하여 기재된 특정한 특징, 구조 또는 특성이 본 발명의 적어도 하나의 실시양태에 포함된다는 것을 의미한다. 따라서, 본 명세서 전반에 걸쳐 다양한 위치에서 어구 "한 실시양태에서" 또는 "하나의 실시양태에서"의 출현은 반드시 동일한 실시양태를 모두 지칭하는 것은 아니다. 또한, 특정한 특징, 구조, 또는 특성은 하나 이상의 실시양태에서 임의의 적합한 방식으로 조합될 수 있다.
문맥상 달리 요구되지 않는 한, 본 명세서 전반에 걸쳐 "화학식 I의 화합물" 또는 "화학식 I의 화합물들"에 대한 언급은 예를 들어 화학식 II-A, II-B, II-C, III-A, III-B, III-C, III-D, III-E, III-F, III-G, III-H, IV-AA, IV-AB, IV-AC, IV-AD, IV-AE, IV-AF, IV-AG, IV-AH, IV-BA, IV-BB, IV-BC, IV-BD, IV-BE, IV-BF, IV-BG, 및 IV-BH의 화합물, 뿐만 아니라 본원에 개시된 구체적 화합물을 비롯한, 화학식 I의 모든 실시양태를 지칭한다.
"아미노"는 -NH2 라디칼을 지칭한다.
"시아노"는 -CN 라디칼을 지칭한다.
"히드록시" 또는 "히드록실"은 -OH 라디칼을 지칭한다.
"이미노"는 =NH 치환기를 지칭한다.
"니트로"는 -NO2 라디칼을 지칭한다.
"옥소"는 =O 치환기를 지칭한다.
"티옥소"는 =S 치환기를 지칭한다.
"알킬"은 탄소 및 수소 원자만으로 이루어지고, 포화 또는 불포화이고 (즉 1개 이상의 이중 및/또는 삼중 결합을 함유함), 1 내지 12개의 탄소 원자 (C1-C12 알킬), 바람직하게는 1 내지 8개의 탄소 원자 (C1-C8 알킬) 또는 1 내지 6개의 탄소 원자 (C1-C6 알킬)를 갖고, 단일 결합에 의해 분자의 나머지에 부착되는 직쇄형 또는 분지형 탄화수소 쇄 라디칼, 예를 들어 메틸, 에틸, n-프로필, 1-메틸에틸 (이소-프로필), n-부틸, n-펜틸, 1,1-디메틸에틸 (t-부틸), 3-메틸헥실, 2-메틸헥실, 에테닐, 프로프-1-에닐, 부트-1-에닐, 펜트-1-에닐, 펜타-1,4-디에닐, 에티닐, 프로피닐, 부티닐, 펜티닐, 헥시닐 등을 지칭한다. 명세서에서 특별히 달리 언급되지 않는 한, 알킬 기는 임의로 치환될 수 있다.
"알킬렌" 또는 "알킬렌 쇄"는 탄소 및 수소만으로 이루어지고, 포화 또는 불포화이고 (즉 1개 이상의 이중 및/또는 삼중 결합을 함유함), 1 내지 12개의 탄소 원자를 갖는, 분자의 나머지가 라디칼 기에 연결된 직쇄형 또는 분지형 2가 탄화수소 쇄, 예를 들어 메틸렌, 에틸렌, 프로필렌, n-부틸렌, 에테닐렌, 프로페닐렌, n-부테닐렌, 프로피닐렌, n-부티닐렌 등을 지칭한다. 알킬렌 쇄는 단일 또는 이중 결합을 통해 분자의 나머지에 부착되고, 단일 또는 이중 결합을 통해 라디칼 기에 부착된다. 분자의 나머지 및 라디칼 기에 대한 알킬렌 쇄의 부착 지점은 쇄 내 1개의 탄소 또는 임의의 2개의 탄소를 통할 수 있다. 명세서에서 특별히 달리 언급되지 않는 한, 알킬렌 쇄는 임의로 치환될 수 있다.
"알콕시"는 화학식 -ORA의 라디칼을 지칭하며, 여기서 RA는 1 내지 12개의 탄소 원자를 함유하는 상기 정의된 바와 같은 알킬 라디칼이다. 명세서에서 특별히 달리 언급되지 않는 한, 알콕시 기는 임의로 치환될 수 있다.
"알킬아미노"는 화학식 -NHRA 또는 -NRARA의 라디칼을 지칭하며, 여기서 각각의 RA는 독립적으로 1 내지 12개의 탄소 원자를 함유하는 상기 정의된 바와 같은 알킬 라디칼이다. 명세서에서 특별히 달리 언급되지 않는 한, 알킬아미노 기는 임의로 치환될 수 있다.
"티오알킬"은 화학식 -SRA의 라디칼을 지칭하며, 여기서 RA는 1 내지 12개의 탄소 원자를 함유하는 상기 정의된 바와 같은 알킬 라디칼이다. 명세서에서 특별히 달리 언급되지 않는 한, 티오알킬 기는 임의로 치환될 수 있다.
"아릴"은 수소 및 6 내지 18개의 탄소 원자를 포함하는 모노시클릭 탄화수소 고리계 라디칼을 지칭한다. 아릴 라디칼은 벤젠으로부터 유래된 아릴 라디칼을 포함하나, 이에 제한되지는 않는다. 명세서에서 특별히 달리 언급되지 않는 한, 용어 "아릴" 또는 접두어 "아르-" (예컨대 "아르알킬"에서)는 임의로 치환된 아릴 라디칼을 포함하는 것으로 의도된다.
"아르알킬"은 화학식 -RB-RC의 라디칼을 지칭하며, 여기서 RB는 상기 정의된 바와 같은 알킬렌 쇄이고 RC는 상기 정의된 바와 같은 1개 이상의 아릴 라디칼, 예를 들어 벤질이다. 명세서에서 특별히 달리 언급되지 않는 한, 아르알킬 기는 임의로 치환될 수 있다.
"시클로알킬" 또는 "카르보시클릭 고리"는 탄소 및 수소 원자만으로 이루어지고, 3 내지 15개의 탄소 원자, 바람직하게는 3 내지 10개의 탄소 원자를 갖고, 포화 또는 불포화이고, 단일 결합에 의해 분자의 나머지에 부착되는 안정한 비-방향족 모노시클릭 탄화수소 라디칼을 지칭한다. 모노시클릭 라디칼은 예를 들어 시클로프로필, 시클로부틸, 시클로펜틸, 시클로헥실, 시클로헵틸 및 시클로옥틸을 포함한다. 명세서에서 특별히 달리 언급되지 않는 한, 시클로알킬 기는 임의로 치환될 수 있다.
"시클로알킬알킬"은 화학식 -RBRD의 라디칼을 지칭하며, 여기서 RB는 상기 정의된 바와 같은 알킬렌 쇄이고 RD는 상기 정의된 바와 같은 시클로알킬 라디칼이다. 명세서에서 특별히 달리 언급되지 않는 한, 시클로알킬알킬 기는 임의로 치환될 수 있다.
"할로" 또는 "할로겐"은 브로모, 클로로, 플루오로 또는 아이오도를 지칭한다.
"할로알킬"은 상기 정의된 바와 같은 1개 이상의 할로 라디칼에 의해 치환된 상기 정의된 바와 같은 알킬 라디칼, 예를 들어 트리플루오로메틸, 디플루오로메틸, 트리클로로메틸, 2,2,2-트리플루오로에틸, 1,2-디플루오로에틸, 3-브로모-2-플루오로프로필, 1,2-디브로모에틸 등을 지칭한다. 명세서에서 특별히 달리 언급되지 않는 한, 할로알킬 기는 임의로 치환될 수 있다.
"헤테로시클릴" 또는 "헤테로시클릭 고리"는 2 내지 12개의 탄소 원자, 및 질소, 산소 및 황으로 이루어진 군으로부터 선택된 1 내지 6개의 헤테로원자로 이루어진 안정한 3- 내지 18-원 비-방향족 고리 라디칼을 지칭한다. 본원에 개시된 실시양태에서, 헤테로시클릴 라디칼은 모노시클릭 고리계이고; 헤테로시클릴 라디칼은 부분 또는 완전 포화일 수 있다. 이러한 헤테로시클릴 라디칼의 예는 디옥솔라닐, 티에닐, [1,3]디티아닐, 이미다졸리닐, 이미다졸리디닐, 이소티아졸리디닐, 이속사졸리디닐, 모르폴리닐, 2-옥소피페라지닐, 2-옥소피페리디닐, 2-옥소피롤리디닐, 옥사졸리디닐, 피페리디닐, 피페라지닐, 4-피페리도닐, 피롤리디닐, 피라졸리디닐, 티아졸리디닐, 테트라히드로푸릴, 트리티아닐, 테트라히드로피라닐, 티오모르폴리닐, 티아모르폴리닐, 1-옥소-티오모르폴리닐 및 1,1-디옥소-티오모르폴리닐을 포함하나, 이에 제한되지는 않는다. 명세서에서 특별히 달리 언급되지 않는 한, 헤테로시클릴 기는 임의로 치환될 수 있다.
"N-헤테로시클릴"은 적어도 1개의 질소를 함유하는 상기 정의된 바와 같은 헤테로시클릴 라디칼을 지칭하고, 분자의 나머지에 대한 헤테로시클릴 라디칼의 부착 지점은 헤테로시클릴 라디칼 내의 질소 원자를 통한다. 명세서에서 특별히 달리 언급되지 않는 한, N-헤테로시클릴 기는 임의로 치환될 수 있다.
"헤테로시클릴알킬"은 화학식 -RBRE의 라디칼을 지칭하며, 여기서 RB는 상기 정의된 바와 같은 알킬렌 쇄이고 RE는 상기 정의된 바와 같은 헤테로시클릴 라디칼이고, 헤테로시클릴이 질소-함유 헤테로시클릴인 경우에, 헤테로시클릴은 질소 원자에서 알킬 라디칼에 부착될 수 있다. 명세서에서 특별히 달리 언급되지 않는 한, 헤테로시클릴알킬 기는 임의로 치환될 수 있다.
"헤테로아릴"은 수소 원자, 1 내지 13개의 탄소 원자, 질소, 산소 및 황으로 이루어진 군으로부터 선택된 1 내지 6개의 헤테로원자를 포함하는 5- 내지 14-원 모노시클릭 고리계 라디칼을 지칭한다. 예는 아제피닐, 푸라닐, 푸라노닐, 이소티아졸릴, 이미다졸릴, 이속사졸릴, 옥사디아졸릴, 2-옥소아제피닐, 옥사졸릴, 옥시라닐, 1-옥시도피리디닐, 1-옥시도피리미디닐, 1-옥시도피라지닐, 1-옥시도피리다지닐, 피라졸릴, 피리디닐, 피라지닐, 피리미디닐, 피리다지닐, 티아졸릴, 티아디아졸릴, 트리아졸릴, 테트라졸릴, 트리아지닐, 티오페닐 및 티에닐을 포함하나, 이에 제한되지는 않는다. 명세서에서 특별히 달리 언급되지 않는 한, 헤테로아릴 기는 임의로 치환될 수 있다.
"N-헤테로아릴"은 적어도 1개의 질소를 함유하는 상기 정의된 바와 같은 헤테로아릴 라디칼을 지칭하고, 분자의 나머지에 대한 헤테로아릴 라디칼의 부착 지점은 헤테로아릴 라디칼 내의 질소 원자를 통한다. 명세서에서 특별히 달리 언급되지 않는 한, N-헤테로아릴 기는 임의로 치환될 수 있다.
"헤테로아릴알킬"은 화학식 -RBRF의 라디칼을 지칭하며, 여기서 RB는 상기 정의된 바와 같은 알킬렌 쇄이고 RF는 상기 정의된 바와 같은 헤테로아릴 라디칼이다. 명세서에서 특별히 달리 언급되지 않는 한, 헤테로아릴알킬 기는 임의로 치환될 수 있다. 본원에 사용된 용어 "치환된"은 적어도 1개의 수소 원자가 비-수소 원자, 예컨대 이에 제한되지는 않는 할로겐 원자, 예컨대 F, Cl, Br, 및 I; 히드록실 기, 알콕시 기 및 에스테르 기와 같은 기 내 산소 원자; 티올 기, 티오알킬 기, 술폰 기, 술포닐 기 및 술폭시드 기와 같은 기 내 황 원자; 아민, 아미드, 알킬아민, 디알킬아민, 아릴아민, 알킬아릴아민, 디아릴아민, N-옥시드, 이미드 및 엔아민과 같은 기 내 질소 원자; 트리알킬실릴 기, 디알킬아릴실릴 기, 알킬디아릴실릴 기 및 트리아릴실릴 기와 같은 기 내 규소 원자; 및 다양한 다른 기 내 다른 헤테로원자에 대한 결합에 의해 대체된 임의의 상기 기 (즉 알킬, 알킬렌, 알콕시, 알킬아미노, 티오알킬, 아릴, 아르알킬, 시클로알킬, 시클로알킬알킬, 할로알킬, 헤테로시클릴, N-헤테로시클릴, 헤테로시클릴알킬, 헤테로아릴, N-헤테로아릴 및/또는 헤테로아릴알킬)를 의미한다. "치환된"은 또한 1개 이상의 수소 원자가 헤테로원자, 예컨대 옥소, 카르보닐, 카르복실 및 에스테르 기 내 산소; 및 이민, 옥심, 히드라존 및 니트릴과 같은 기 내 질소에 대한 보다 높은 차수의 결합 (예를 들어, 이중- 또는 삼중-결합)에 의해 대체된 임의의 상기 기를 의미한다. 예를 들어, "치환된"은 1개 이상의 수소 원자가 -NRGRH, -NRGC(=O)RH, -NRGC(=O)NRGRH, -NRGC(=O)ORH, -NRGC(=NRG)NRGRH, -NRGSO2RH, -OC(=O)NRGRH, -ORG, -SRG, -SORG, -SO2RG, -OSO2RG, -SO2ORG, =NSO2RG, 및 -SO2NRGRH로 대체된 임의의 상기 기를 포함한다. "치환된"은 또한 1개 이상의 수소 원자가 -C(=O)RG, -C(=O)ORG, -C(=O)NRGRH, -CH2SO2RG, -CH2SO2NRGRH로 대체된 임의의 상기 기를 의미한다. 상기에서, RG 및 RH는 동일하거나 상이하고, 독립적으로 수소, 알킬, 알콕시, 알킬아미노, 티오알킬, 아릴, 아르알킬, 시클로알킬, 시클로알킬알킬, 할로알킬, 헤테로시클릴, N-헤테로시클릴, 헤테로시클릴알킬, 헤테로아릴, N-헤테로아릴 및/또는 헤테로아릴알킬이다. "치환된"은 또한 1개 이상의 수소 원자가 아미노, 시아노, 히드록실, 이미노, 니트로, 옥소, 티옥소, 할로, 알킬, 알콕시, 알킬아미노, 티오알킬, 아릴, 아르알킬, 시클로알킬, 시클로알킬알킬, 할로알킬, 헤테로시클릴, N-헤테로시클릴, 헤테로시클릴알킬, 헤테로아릴, N-헤테로아릴 및/또는 헤테로아릴알킬 기에 대한 결합에 의해 대체된 임의의 상기 기를 의미한다. 또한, 각각의 상기 치환기는 1개 이상의 상기 치환기에 의해 또한 임의로 치환될 수 있다.
본원에 사용된 용어 "보호기"는 합성 절차 동안 목적하지 않은 반응에 대해, 제한 없이 히드록실 및 아미노 기를 포함하는 반응성 기를 보호하기 위한 것으로 관련 기술분야에 공지되어 있는 불안정성 화학적 모이어티를 지칭한다. 보호기로 보호된 히드록실 및 아미노 기는 본원에서 각각 "보호된 히드록실 기" 및 "보호된 아미노 기"로 지칭된다. 보호기는 전형적으로, 다른 반응성 부위에서의 반응 동안 부위를 보호하기 위해 선택적 및/또는 직교적으로 사용되고, 이어서 비보호된 기는 남겨두고 그대로 제거될 수 있거나 추가의 반응을 위해 사용될 수 있다. 관련 기술분야에 공지된 바와 같은 보호기는 일반적으로 문헌 [Greene and Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons, New York (1999)]에 기재되어 있다. 일반적으로, 기는 보호되거나 또는 적절한 시점에 그의 최종 기로의 전환을 위해 모 분자의 다른 영역을 변형시키는 반응에 대해 불활성일 전구체로서 제공된다. 추가의 대표적인 보호기 또는 전구체 기는 문헌 [Agrawal, et al., Protocols for Oligonucleotide Conjugates, Eds, Humana Press; New Jersey, 1994; Vol. 26 pp. 1-72]에 논의되어 있다. "히드록실 보호기"의 예는 t-부틸, t-부톡시메틸, 메톡시메틸, 테트라히드로피라닐, 1-에톡시에틸, 1-(2-클로로에톡시)에틸, 2-트리메틸실릴에틸, p-클로로페닐, 2,4-디니트로페닐, 벤질, 2,6-디클로로벤질, 디페닐메틸, p-니트로벤질, 트리페닐메틸, 트리메틸실릴, 트리에틸실릴, t-부틸디메틸실릴, t-부틸디페닐실릴 (TBDPS), 트리페닐실릴, 벤조일포르메이트, 아세테이트, 클로로아세테이트, 트리클로로아세테이트, 트리플루오로아세테이트, 피발로에이트, 벤조에이트, p-페닐벤조에이트, 9-플루오레닐메틸 카르보네이트, 메실레이트 및 토실레이트를 포함하나, 이에 제한되지는 않는다. "아미노 보호기"의 예는 카르바메이트-보호기, 예컨대 2-트리메틸실릴에톡시카르보닐 (Teoc), 1-메틸-1-(4-비페닐릴)에톡시카르보닐 (Bpoc), t-부톡시카르보닐 (BOC), 알릴옥시카르보닐 (Alloc), 9-플루오레닐메틸옥시카르보닐 (Fmoc) 및 벤질옥시카르보닐 (Cbz); 아미드 보호기, 예컨대 포르밀, 아세틸, 트리할로아세틸, 벤조일 및 니트로페닐아세틸; 술폰아미드-보호기, 예컨대 2-니트로벤젠술포닐; 및 이민 및 시클릭 이미드 보호기, 예컨대 프탈이미도 및 디티아숙시노일을 포함하나, 이에 제한되지는 않는다.
본원에 개시된 본 발명은 또한 상이한 원자 질량 또는 질량수를 갖는 원자에 의해 대체된 1개 이상의 원자를 가짐으로써 동위원소-표지된, 화학식 I의 모든 제약상 허용되는 화합물을 포괄하는 것으로 의도된다. 개시된 화합물로 혼입될 수 있는 동위원소의 예는 수소, 탄소, 질소, 산소, 인, 플루오린, 염소 및 아이오딘의 동위원소, 예컨대 각각 2H, 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 31P, 32P, 35S, 18F, 36Cl, 123I, 및 125I를 포함한다. 이들 방사성표지된 화합물은, 예를 들어 작용 부위 또는 방식, 또는 약리학상 중요한 작용 부위에 대한 결합 친화도를 특성화함으로써 화합물의 유효성을 결정 또는 측정하는 것을 돕는데 유용할 수 있다. 특정 동위원소-표지된 화학식 I의 화합물, 예를 들어 방사성 동위원소가 혼입된 것은 약물 및/또는 기질 조직 분포 연구에 유용하다. 방사성 동위원소 삼중수소, 즉 3H, 및 탄소-14, 즉 14C가 혼입의 용이성 및 용이한 검출 수단의 측면에서 이러한 목적에 특히 유용하다.
보다 무거운 동위원소, 예컨대 중수소, 즉 2H로의 치환은 더 큰 대사 안정성으로 인한 특정의 치료 이점을 제공할 수 있다. 예를 들어, 생체내 반감기가 증가할 수 있거나 투여량 요건이 감소될 수 있다. 따라서, 보다 무거운 동위원소는 일부 상황에서 바람직할 수 있다.
양전자 방출 동위원소, 예컨대 11C, 18F, 15O 및 13N으로의 치환은 기질 수용체 점유율을 조사하기 위한 양전자 방출 단층촬영 (PET) 연구에 유용할 수 있다. 화학식 I의 동위원소-표지된 화합물은 일반적으로 통상의 기술자에게 공지된 통상의 기술에 의해 제조될 수 있거나, 또는 기존에 사용되던 비-표지된 시약 대신 적절한 동위원소-표지된 시약을 사용하여 하기 기재된 바와 같은 실시예에 기재된 것과 유사한 방법에 의해 제조될 수 있다.
본원에 개시된 본 발명은 또한 개시된 화합물의 생체내 대사 산물을 포괄하는 것으로 의도된다. 이러한 산물은, 주로 효소적 과정으로 인한, 예를 들어 투여된 화합물의 산화, 환원, 가수분해, 아미드화, 에스테르화 등으로부터 생성될 수 있다. 따라서, 본 발명은 본 발명의 화합물을 그의 대사 산물이 생성되기에 충분한 시간 기간 동안 포유동물에게 투여하는 것을 포함하는 방법에 의해 생성된 화합물을 포함한다. 이러한 산물은 전형적으로 방사성표지된 본 발명의 화합물을 검출가능한 용량으로 동물, 예컨대 래트, 마우스, 기니 피그, 원숭이 또는 인간에게 투여하고, 충분한시간 동안 대사가 일어나게 하고, 뇨, 혈액 또는 다른 생물학적 샘플로부터 그의 전환 산물을 단리하는 것에 의해 확인된다.
"안정한 화합물" 및 "안정한 구조"는 반응 혼합물로부터 유용한 정도의 순도로의 단리를 견디고 효과적인 치료제로 제제화되기에 충분히 강건한 화합물을 나타내는 것으로 의도된다.
"포유동물"은 인간, 및 실험 동물 및 가정용 애완 동물과 같은 가축 동물 (예를 들어, 고양이, 개, 돼지, 소, 양, 염소, 말, 토끼) 및 야생 동물 등과 같은 비-가축 동물 둘 다를 포함한다.
"임의적인" 또는 "임의로"는 이후에 기재되는 상황의 사건이 일어날 수 있거나 또는 일어나지 않을 수 있고, 상기 기재가 상기 사건 또는 상황이 일어난 경우 및 일어나지 않은 경우를 포함함을 의미한다. 예를 들어, "임의로 치환된 아릴"은 아릴 라디칼이 치환될 수 있거나 또는 치환되지 않을 수 있고, 상기 기재가 치환된 아릴 라디칼 및 어떠한 치환도 갖지 않는 아릴 라디칼을 둘 다 포함함을 의미한다.
"제약상 허용되는 담체, 희석제 또는 부형제"는 미국 식품 의약품국에 의해 인간 또는 가축 동물에서의 사용에 대해 허용가능한 것으로 승인된, 임의의 아주반트, 담체, 부형제, 활택제, 감미제, 희석제, 보존제, 염료/착색제, 향미 증진제, 계면활성제, 습윤제, 분산제, 현탁화제, 안정화제, 등장화제, 용매 또는 유화제를 제한 없이 포함한다.
"제약상 허용되는 염"은 제약상 허용되고 모 화합물의 목적하는 약리학적 활성을 보유하는 (또는 보유하는 형태로 전환될 수 있는) 화합물의 염을 지칭한다. 본원에 개시된 화합물의 "제약상 허용되는 염"의 예는 적절한 염기, 예컨대 알칼리 금속 (예를 들어, 나트륨), 알칼리 토금속 (예를 들어, 마그네슘), 암모늄 및 NX4 + (여기서 X는 C1-C4 알킬임)로부터 유래된 염을 포함한다. 질소 원자 또는 아미노 기의 제약상 허용되는 염은 예를 들어 유기 카르복실산, 예컨대 아세트산, 벤조산, 캄포르술폰산, 시트르산, 글루코헵톤산, 글루콘산, 락트산, 푸마르산, 타르타르산, 말레산, 말론산, 말산, 만델산, 이세티온산, 락토비온산, 숙신산, 2-나프탈렌술폰산, 올레산, 팔미트산, 프로피온산, 스테아르산 및 트리메틸아세트산; 유기 술폰산, 예컨대 메탄술폰산, 에탄술폰산, 벤젠술폰산 및 p-톨루엔술폰산; 및 무기 산, 예컨대 염산, 브로민화수소산, 황산, 질산, 인산 및 술팜산의 염을 포함한다. 히드록시 기의 화합물의 제약상 허용되는 염은 적합한 양이온, 예컨대 Na+ 및 NX4 + (여기서 X는 독립적으로 H 또는 C1-C4 알킬 기로부터 선택됨)와 조합된 상기 화합물의 음이온을 포함한다. 제약상 허용되는 염은 또한 모 화합물 내에 존재하는 산성 양성자가 금속 이온, 예를 들어 알칼리 금속 이온, 알칼리 토류 이온 또는 알루미늄 이온에 의해 대체되거나; 또는 유기 염기, 예컨대 디에탄올아민, 트리에탄올아민, N-메틸글루카민 등과 배위될 때 형성되는 염을 포함한다. 또한, 본 정의에는 암모늄 및 치환되거나 4급화된 암모늄 염이 포함된다. 제약상 허용되는 염의 대표적인 비제한적 목록은 문헌 [S.M. Berge et al., J. Pharma Sci., 66(1), 1-19 (1977), 및 Remington: The Science and Practice of Pharmacy, R. Hendrickson, ed., 21st edition, Lippincott, Williams & Wilkins, Philadelphia, PA, (2005), at p. 732, Table 38-5]에서 찾아볼 수 있고, 이는 둘 다 본원에 참조로 포함된다.
치료 용도를 위해, 본원에 개시된 화합물의 활성 성분의 염은 전형적으로 제약상 허용될 것이고, 즉 생리학상 허용되는 산 또는 염기로부터 유래된 염일 것이다. 그러나, 제약상 허용되지 않는 산 또는 염기의 염은 또한 예를 들어 화학식 I의 화합물 또는 본 발명의 또 다른 화합물의 제조 또는 정제에서 용도를 발견할 수 있다. 모든 염은, 생리학상 허용되는 산 또는 염기로부터 유래된 것인지에 관계없이, 본 발명의 범위 내에 포함된다.
금속 염은 전형적으로 금속 수산화물을 본 발명의 화합물과 반응시킴으로써 제조된다. 이러한 방식으로 제조된 금속 염의 예는 Li+, Na+, 및 K+를 함유하는 염이다. 덜 가용성인 금속 염은 적합한 금속 화합물의 첨가에 의해 보다 가용성인 염의 용액으로부터 침전될 수 있다.
또한, 염은 염기성 중심, 전형적으로 아민에의 특정 유기 및 무기 산, 예를 들어 HCl, HBr, H2SO4, H3PO4 또는 유기 술폰산의 산 첨가로부터 형성될 수 있다. 마지막으로, 본원의 조성물은 본원에 개시된 화합물을 그의 비-이온화 형태 뿐만 아니라 쯔비터이온 형태로, 그리고 수화물에서와 같이 화학량론적 양의 물과의 조합물로 포함하는 것으로 이해되어야 한다.
종종 결정화는 본 발명의 화합물의 용매화물을 생성한다. 본원에 사용된 용어 "용매화물"은 본 발명의 화합물의 1개 이상의 분자를 1개 이상의 용매 분자와 함께 포함하는 응집체를 지칭한다. 용매는 물일 수 있고, 이 경우에 용매화물은 수화물일 수 있다. 대안적으로, 용매는 유기 용매일 수 있다. 따라서, 본 발명의 화합물은 1수화물, 2수화물, 반수화물, 1.5수화물, 3수화물, 4수화물 등을 비롯한 수화물 뿐만 아니라 상응하는 용매화 형태로 존재할 수 있다. 본 발명의 화합물은 진성 용매화물일 수 있는 반면에, 다른 경우에 본 발명의 화합물은 단지 외래의 물을 보유할 수 있거나 또는 물에 더하여 몇몇 외래 용매의 혼합물일 수 있다.
"제약 조성물"은 본 발명의 화합물, 및 포유동물, 예를 들어 인간에게 생물학적으로 활성인 화합물을 전달하기 위해 관련 기술분야에서 일반적으로 허용되는 매질의 제제를 지칭한다. 이러한 매질은 모든 제약상 허용되는 담체, 희석제 또는 부형제를 포함한다.
"유효량" 또는 "치료 유효량"은 그를 필요로 하는 환자에게 투여된 경우에 화합물이 유용성을 갖는 질환-상태, 병태 또는 장애를 위한 치료를 달성하기에 충분한 본 발명에 따른 화합물의 양을 지칭한다. 이러한 양은 연구원 또는 임상의가 추구하는 조직계 또는 환자의 생물학적 또는 의학적 반응을 도출하는데 충분할 것이다. 치료 유효량을 구성하는 본 발명에 따른 화합물의 양은 화합물 및 그의 생물학적 활성, 투여를 위해 사용되는 조성물, 투여 시간, 투여 경로, 화합물의 배설 속도, 치료 지속기간, 치료할 질환-상태 또는 장애의 유형 및 그의 중증도, 본 발명의 화합물과 조합되어 또는 동시에 사용되는 약물, 및 환자의 연령, 체중, 전반적 건강, 성별 및 식이와 같은 인자에 따라 달라질 것이다. 이러한 치료 유효량은 그 자신의 지식, 최신 기술, 및 본 개시내용과 관련이 있는 통상의 기술자에 의해 통상적으로 결정될 수 있다.
본원에 사용된 용어 "치료"는 HIV 감염의 증상을 완화 또는 제거하고/거나 환자에서 바이러스 로드를 감소시키기 위한 본 발명에 따른 화합물 또는 조성물의 투여를 의미하는 것으로 의도된다. 용어 "치료"는 또한 바이러스 노출-후이지만 질환의 증상의 출현 전 및/또는 혈액에서 바이러스의 검출 전 개체에의, 질환의 증상의 출현을 방지하고/거나 바이러스가 혈액에서 검출가능한 수준에 이르는 것을 방지하기 위한 본 발명에 따른 화합물 또는 조성물의 투여, 및 출산 전 모체 및 생후 초일 내 소아에게 투여함으로써 모체에서 아기로의 HIV의 주산기 감염을 방지하기 위한 본 발명에 따른 화합물 또는 조성물의 투여를 포괄한다.
본원에 사용된 용어 "항바이러스제"는 인간에서 바이러스의 형성 및/또는 복제에 필요한 숙주 또는 바이러스 메카니즘을 방해하는 작용제를 포함하나 이에 제한되지는 않는, 인간에서 바이러스의 형성 및/또는 복제를 억제하는데 효과적인 작용제 (화합물 또는 생물학적)를 의미하는 것으로 의도된다.
본원에 사용된 용어 "HIV 복제의 억제제"는 시험관내, 생체외 또는 생체내에 관계없이 숙주 세포에서 HIV의 복제 능력을 감소시키거나 제거할 수 있는 작용제를 의미하는 것으로 의도된다.
본 발명의 화합물 또는 그의 제약상 허용되는 염은 1개 이상의 비대칭 중심을 함유할 수 있고, 따라서 절대 입체화학의 관점에서 (R)- 또는 (S)-로서, 또는 아미노산의 경우에 (D)- 또는 (L)-로서 정의될 수 있는 거울상이성질체, 부분입체이성질체 및 기타 입체이성질체 형태를 생성할 수 있다. 본 발명은 모든 이러한 가능한 이성질체, 뿐만 아니라 그의 라세미 및 광학적으로 순수한 형태를 포함하는 것으로 의도된다. 광학 활성 (+) 및 (-), (R)- 및 (S)-, 또는 (D)- 및 (L)- 이성질체는 키랄 합성단위체 또는 키랄 시약을 사용하여 제조될 수 있거나, 또는 통상의 기술, 예를 들어 크로마토그래피 및 분별 결정화를 사용하여 분할될 수 있다. 개별 거울상이성질체의 제조/단리를 위한 통상의 기술은 적합한 광학적으로 순수한 전구체로부터의 키랄 합성, 또는 예를 들어 키랄 고압 액체 크로마토그래피 (HPLC)를 사용한 라세미체 (또는 염 또는 유도체의 라세미체)의 분할을 포함한다. 본원에 기재된 화합물이 올레핀계 이중 결합 또는 다른 기하학적 비대칭 중심을 함유하는 경우에, 달리 명시되지 않는 한, 상기 화합물은 E 및 Z 기하 이성질체를 둘 다 포함하는 것으로 의도된다. 마찬가지로, 모든 호변이성질체 형태가 또한 포함되는 것으로 의도된다.
"입체이성질체"는 동일한 결합에 의해 결합된 동일한 원자로 이루어져 있으나 상호교환될 수 없는 상이한 3차원 구조를 갖는 화합물을 지칭한다. 본 발명은 다양한 입체이성질체 및 그의 혼합물을 고려하고, 분자들이 서로 중첩될 수 없는 거울상인 2종의 입체이성질체를 지칭하는 "거울상이성질체"를 포함한다.
"호변이성질체"는 분자의 한 원자로부터 동일한 분자의 또 다른 원자로의 양성자 이동을 지칭한다. 본 발명은 임의의 상기 화합물의 호변이성질체를 포함한다.
"전구약물"은 경구 전달에 대한 생물학적 장벽을 극복한 후 모 약물을 효율적으로 유리하도록 화학적으로 설계된 화합물을 지칭한다. 특정 실시양태에서, 본 발명은 화학식 I의 화합물의 전구약물을 포함한다.
화합물
상기 기재된 바와 같이, 본 발명의 한 실시양태에서, 항바이러스 활성을 갖는 하기 화학식 I을 갖는 화합물 또는 그의 입체이성질체 또는 제약상 허용되는 염이 제공된다.
<화학식 I>

상기 식에서:
X는 -O- 또는 -NZ3- 또는 -CHZ3-이고;
W는 -CHZ2-이고;
Z1, Z2 및 Z3은 각각, 독립적으로, 수소 또는 C1-3알킬이거나, 또는 여기서 Z1 및 Z2 또는 Z1 및 Z3은 함께 -L-을 형성하고, 여기서 L은 -C(Ra)2-, -C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2C(Ra)2-, 또는 -C(Ra)2C(Ra)2C(Ra)2C(Ra)2-이고, 여기서 Z1 및 Z2 또는 Z1 및 Z3 중 적어도 하나는 함께 -L-을 형성하고;
Z4는 결합, -CH2-, 또는 -CH2CH2-이고;
Y1 및 Y2는 각각, 독립적으로, 수소, C1-3알킬 또는 C1-3할로알킬이고;
R1은 1 내지 3개의 할로겐으로 치환된 페닐이고;
각각의 Ra는 독립적으로, 수소, 할로, 히드록실 또는 C1-4알킬이다.
또 다른 실시양태에서, 하기 화학식 II-A를 갖는 화합물이 제공된다.
<화학식 II-A>
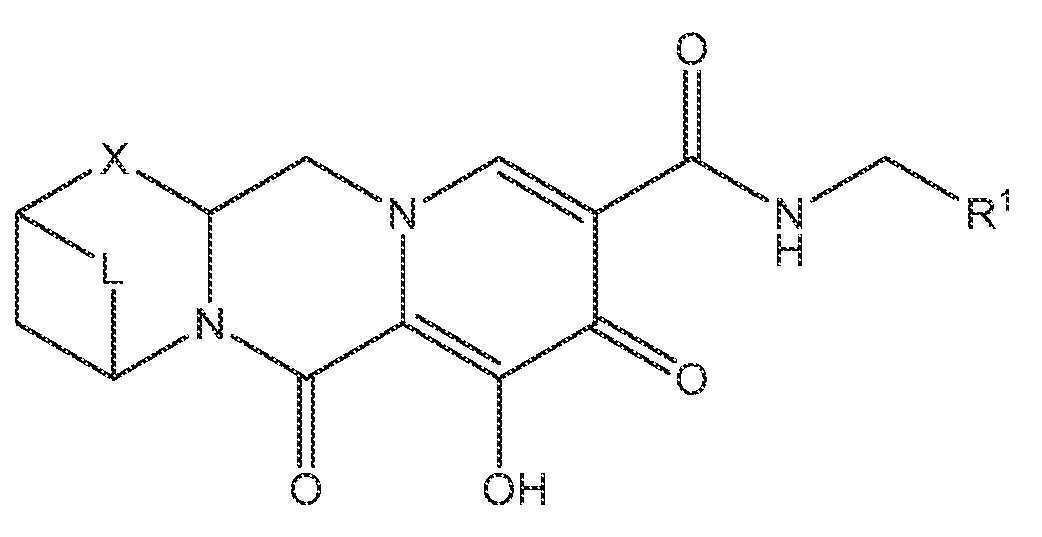
또 다른 실시양태에서, 하기 화학식 II-B를 갖는 화합물이 제공된다.
<화학식 II-B>

또 다른 실시양태에서, 하기 화학식 II-C를 갖는 화합물이 제공된다.
<화학식 II-C>

또 다른 실시양태에서, L은 -C(Ra)2-이다. 추가 실시양태에서, L은 -C(Ra)2C(Ra)2-이다. 추가 실시양태에서, L은 -C(Ra)2C(Ra)2C(Ra)2-이다. 추가 실시양태에서, 각각의 Ra는 수소이다. 추가 실시양태에서, 1개의 Ra는 메틸이고, 각각의 나머지 Ra는 수소이다. 추가 실시양태에서, 1개의 Ra는 할로겐이고, 각각의 나머지 Ra는 수소이다. 추가 실시양태에서, 2개의 Ra는 할로겐이고, 각각의 나머지 Ra는 수소이다. 추가 실시양태에서, 1개의 Ra는 할로겐이고, 각각의 나머지 Ra는 수소이다.
또 다른 실시양태에서, X는 -O-이다. 또 다른 실시양태에서, X는 -NZ3-이다. 또 다른 실시양태에서, X는 -NH-이다. 또 다른 실시양태에서, X는 -CHZ3-이고, Z1 및 Z3은 함께 -L-을 형성한다. 추가 실시양태에서, Z2는 수소이다. 또 다른 실시양태에서, X는 -CH2-이다.
또 다른 실시양태에서, Z4는 결합 또는 -CH2-이다. 또 다른 실시양태에서, Z4는 -CH2-이다. 또 다른 실시양태에서, Z4는 결합이다.
또 다른 실시양태에서, Y1 및 Y2는 각각 독립적으로 수소, 메틸 또는 트리플루오로메틸이다.
또 다른 실시양태에서, R1은 1개의 할로겐으로 치환된다. 추가 실시양태에서, R1은 4-플루오로페닐 또는 2-플루오로페닐이다.
또 다른 실시양태에서, R1은 2개의 할로겐으로 치환된다. 추가 실시양태에서, R1은 2,4-디플루오로페닐, 2,3-디플루오로페닐, 2,6-디플루오로페닐, 3-플루오로-4-클로로페닐, 3,4-디플루오로페닐, 2-플루오로-4-클로로페닐 또는 3,5-디플루오로페닐이다. 추가 실시양태에서, R1은 2,4-디플루오로페닐이다.
또 다른 실시양태에서, R1은 3개의 할로겐으로 치환된다. 추가 실시양태에서, R1은 2,4,6-트리플루오로페닐 또는 2,3,4-트리플루오로페닐이다. 추가 실시양태에서, R1은 2,4,6-트리플루오로페닐이다.
한 실시양태에서, 상기 기재된 바와 같은 화학식 I, II-A, II-B, 또는 II-C 중 어느 하나의 화합물 또는 그의 입체이성질체 또는 제약상 허용되는 염, 및 제약상 허용되는 담체, 희석제 또는 부형제를 포함하는 제약 조성물이 제공된다.
HIV 감염에 걸리거나 또는 걸릴 위험이 있는 인간에게 치료 유효량의 상기 기재된 바와 같은 화학식 I, II-A, II-B, 또는 II-C 중 어느 하나의 화합물 또는 그의 제약 조성물을 투여하는 것에 의해 상기 인간에서 HIV 감염을 치료하는 방법을 포함하는 또 다른 실시양태가 제공된다. HIV 감염에 걸리거나 또는 걸릴 위험이 있는 인간에게 치료 유효량의 상기 기재된 바와 같은 화학식 I, II-A, II-B, 또는 II-C 중 어느 하나의 화합물 또는 그의 제약 조성물을 투여하는 것에 의해 상기 인간에서 HIV 감염을 치료 또는 예방하는 방법을 포함하는 또 다른 실시양태가 제공된다.
또 다른 실시양태에서, HIV 감염에 걸리거나 또는 걸릴 위험이 있는 인간에서 HIV 감염을 치료하기 위한, 상기 기재된 바와 같은 화학식 I, II-A, II-B, 또는 II-C 중 어느 하나의 화합물 또는 그의 제약 조성물의 용도가 제공된다. 또 다른 실시양태에서, HIV 감염에 걸리거나 또는 걸릴 위험이 있는 인간에서 HIV 감염을 치료 또는 예방하기 위한, 상기 기재된 바와 같은 화학식 I, II-A, II-B, 또는 II-C 중 어느 하나의 화합물 또는 그의 제약 조성물의 용도가 제공된다.
또 다른 실시양태에서, 상기 기재된 바와 같은 화학식 I, II-A, II-B, 또는 II-C 중 어느 하나의 화합물 또는 그의 제약 조성물의 의료 요법에서의 용도가 제공된다.
또 다른 실시양태에서, HIV 감염의 치유적 치료에 사용하기 위한, 상기 기재된 바와 같은 화학식 I, II-A, II-B, 또는 II-C 중 어느 하나의 화합물 또는 그의 제약 조성물의 용도가 제공된다. 또 다른 실시양태에서, HIV 감염의 예방적 또는 치유적 치료에 사용하기 위한, 상기 기재된 바와 같은 화학식 I, II-A, II-B, 또는 II-C 중 어느 하나의 화합물 또는 그의 제약 조성물의 용도가 제공된다.
상기 추가로 기재된 바와 같이, 본 발명의 또 다른 실시양태에서, 항바이러스 활성을 갖는 하기 화학식 I을 갖는 화합물 또는 그의 입체이성질체 또는 제약상 허용되는 염이 제공된다.
<화학식 I>

상기 식에서:
X는 -O- 또는 -NZ3- 또는 -CHZ3-이고;
W는 -O- 또는 -NZ2- 또는 -CHZ2-이고;
Z1, Z2 및 Z3은 각각, 독립적으로, 수소, C1-3알킬 또는 C1-3할로알킬이거나, 또는 여기서 Z1 및 Z2 또는 Z1 및 Z3은 함께 -L-을 형성하고, 여기서 L은 -C(Ra)2-, -C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2C(Ra)2C(Ra)2-, -C(Ra)2OC(Ra)2-, -C(Ra)2NRaC(Ra)2-, -C(Ra)2SC(Ra)2-, -C(Ra)2S(O)C(Ra)2-, -C(Ra)2SO2C(Ra)2-, -C(Ra)2OC(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2OC(Ra)2-, -C(Ra)2NRaC(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2NRaC(Ra)2-, -C(Ra)2SC(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2SC(Ra)2-, -C(Ra)2S(O)C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2S(O)C(Ra)2-, -C(Ra)2SO2C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2SO2C(Ra)2-, -C(Ra)2SO2NRaC(Ra)2- 또는 -C(Ra)2NRaSO2C(Ra)2-이고;
Z4는 결합 또는 -CH2-, -CH2CH2-, -CH2CH2CH2-, -CH2OCH2-, -CH2NRaCH2-, -CH2SCH2-, -CH2S(O)CH2- 또는 -CH2SO2CH2-이고;
Y1 및 Y2는 각각, 독립적으로, 수소 또는 C1-3알킬이거나, 또는 Y1 및 Y2는 이들이 부착되어 있는 탄소 원자와 함께 3 내지 6개의 고리 원자를 갖는 카르보시클릭 고리 또는 3 내지 6개의 고리 원자를 갖는 헤테로시클릭 고리를 형성하고, 여기서 카르보시클릭 또는 헤테로시클릭 고리는 1개 이상의 Ra로 임의로 치환되고;
R1은 임의로 치환된 아릴 또는 임의로 치환된 헤테로아릴이고;
각각의 Ra는, 독립적으로, 수소, 할로, 히드록실 또는 C1-4알킬이거나, 또는 여기서 2개의 Ra 기는 이들이 부착되어 있는 탄소 원자와 함께 =O를 형성하고,
여기서 하기 중 적어도 하나이다: (i) Z1 및 Z2 또는 Z1 및 Z3은 함께 -L-을 형성하거나; 또는 (ii) Y1 및 Y2는 이들이 부착되어 있는 탄소 원자와 함께 3 내지 6개의 고리 원자를 갖는 카르보시클릭 고리 또는 3 내지 6개의 고리 원자를 갖는 헤테로시클릭 고리를 형성한다.
또 다른 실시양태에서, W는 -CHZ2-이다-.
또 다른 실시양태에서, Z1 및 Z2 또는 Z1 및 Z3은 함께 -L-을 형성한다.
또 다른 실시양태에서, 하기 화학식 II-A, II-B 또는 II-C 중 하나를 갖는 화합물이 제공된다.
<화학식 II-A>
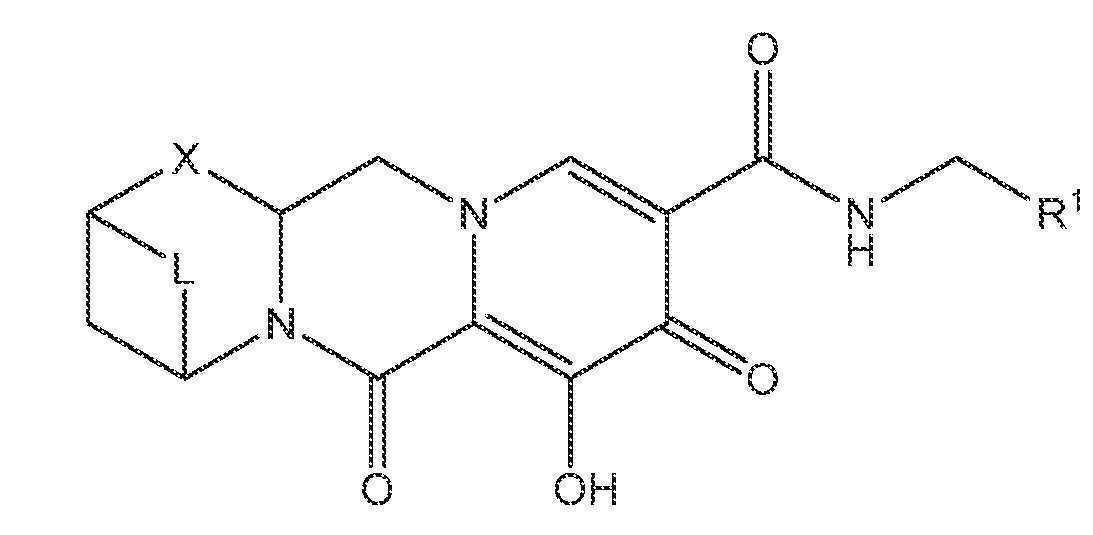
<화학식 II-B>
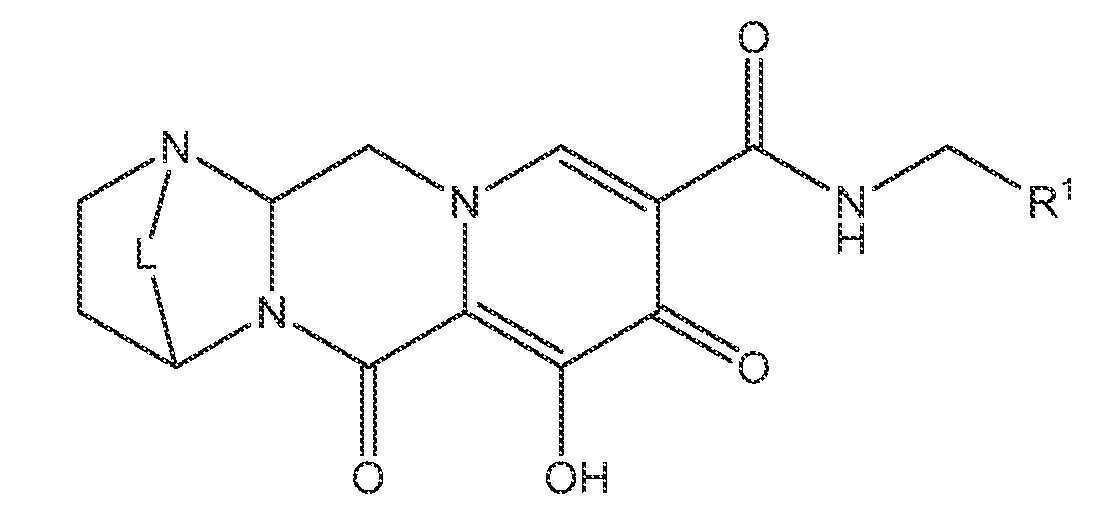
<화학식 II-C>

상기 식에서 L은 -C(Ra)2-, -C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2C(Ra)2C(Ra)2-, -C(Ra)2OC(Ra)2-, -C(Ra)2NRaC(Ra)2-, -C(Ra)2SC(Ra)2-, -C(Ra)2S(O)C(Ra)2-, -C(Ra)2SO2C(Ra)2-, -C(Ra)2OC(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2OC(Ra)2-, -C(Ra)2NRaC(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2NRaC(Ra)2-, -C(Ra)2SC(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2SC(Ra)2-, -C(Ra)2S(O)C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2S(O)C(Ra)2-, -C(Ra)2SO2C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2SO2C(Ra)2-, -C(Ra)2SO2NRaC(Ra)2- 또는 -C(Ra)2NRaSO2C(Ra)2-이다.
또 다른 실시양태에서, Y1 및 Y2는 이들이 부착되어 있는 탄소 원자와 함께 3 내지 6개의 고리 원자를 갖는 카르보시클릭 고리 또는 3 내지 6개의 고리 원자를 갖는 헤테로시클릭 고리를 형성한다.
또 다른 실시양태에서, 하기 화학식 III-A, III-B, III-C 또는 III-D 중 하나를 갖는 화합물이 제공된다.
<화학식 III-A>

<화학식 III-B>
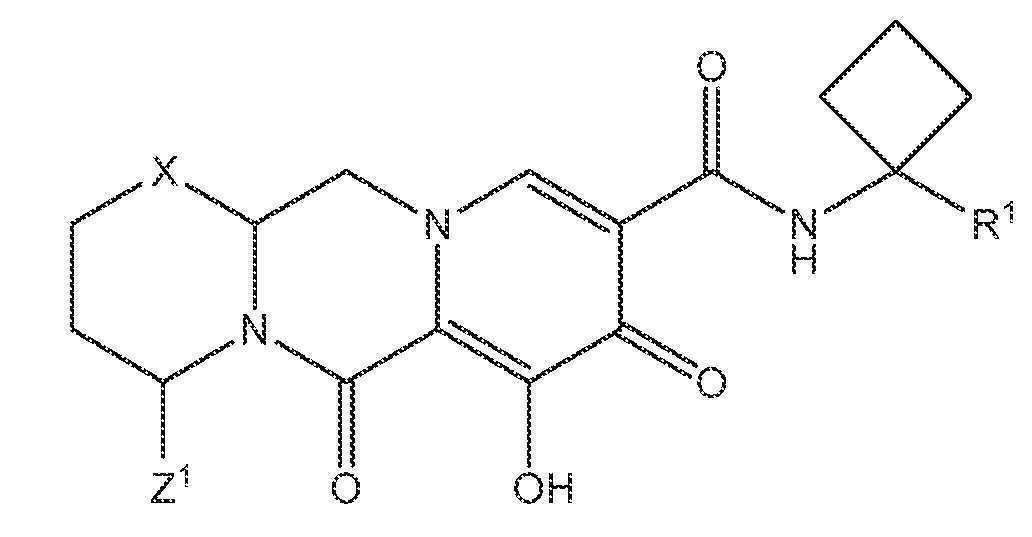
<화학식 III-C>

<화학식 III-D>
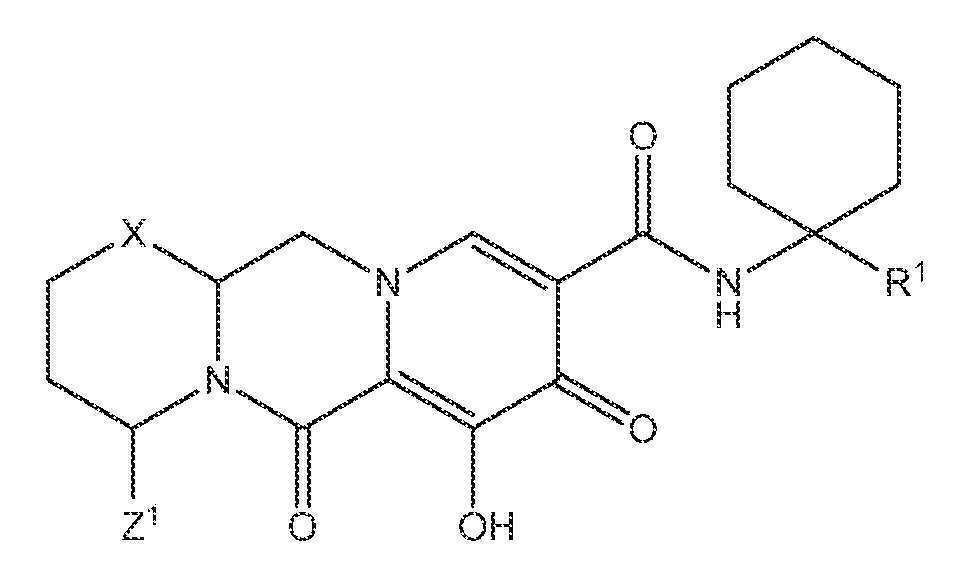
상기 식에서 Z1 및 Z3은 각각, 독립적으로, 수소 또는 C1-3알킬이다.
또 다른 실시양태에서, 하기 화학식 III-E, III-F, III-G 또는 III-H 중 하나를 갖는 화합물이 제공된다.
<화학식 III-E>
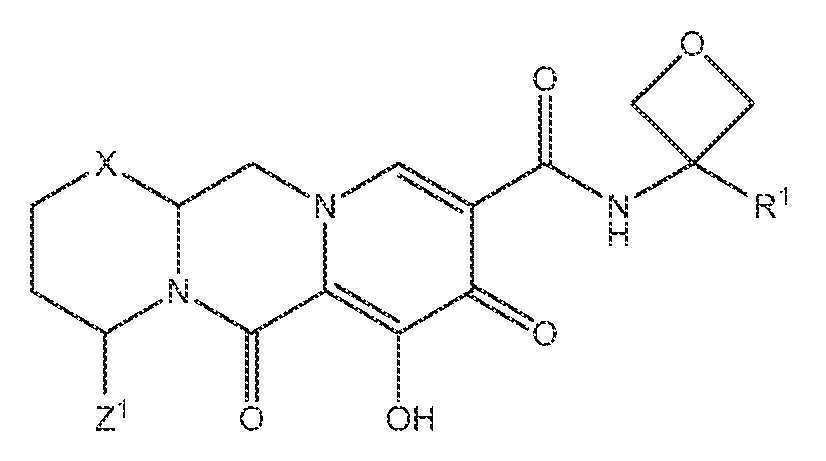
<화학식 III-F>
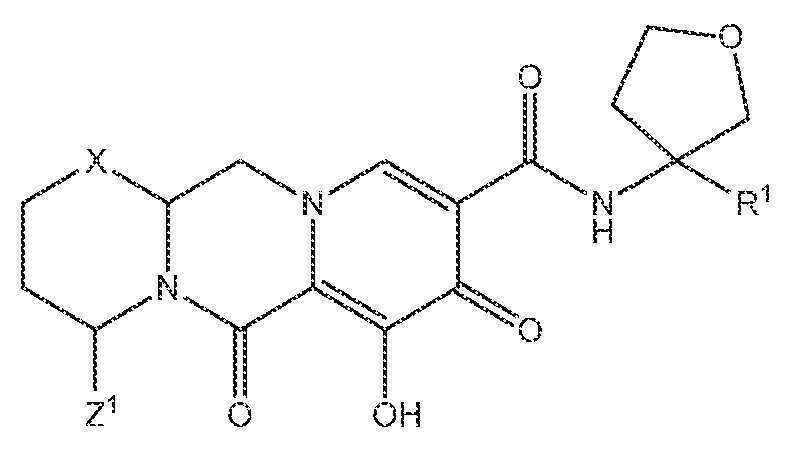
<화학식 III-G>

<화학식 III-H>

상기 식에서 Z1 및 Z3은 각각, 독립적으로, 수소 또는 C1-3알킬이다.
또 다른 실시양태에서, (i) Z1 및 Z2 또는 Z1 및 Z3은 함께 -L-을 형성하고, 또한 (ii) Y1 및 Y2는 이들이 부착되어 있는 탄소 원자와 함께 3 내지 6개의 고리 원자를 갖는 카르보시클릭 고리 또는 3 내지 6개의 고리 원자를 갖는 헤테로시클릭 고리를 형성한다.
또 다른 실시양태에서, 하기 화학식 IV-AA, IV-AB, IV-AC, IV-AD, IV-AE, IV-AF, IV-AG 또는 IV-AH 중 하나를 갖는 화합물이 제공된다.
<화학식 IV-AA>

<화학식 IV-AB>

<화학식 IV-AC>
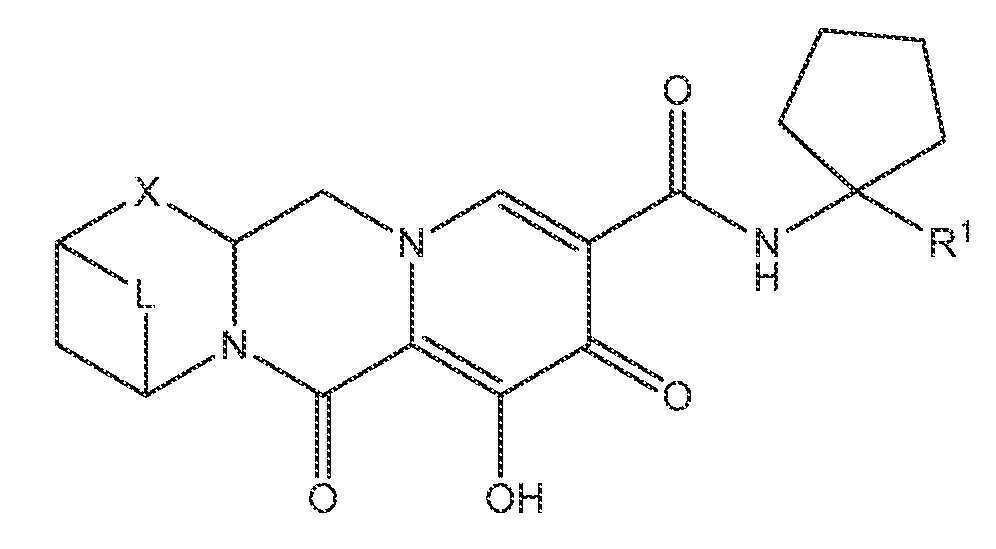
<화학식 IV-AD>
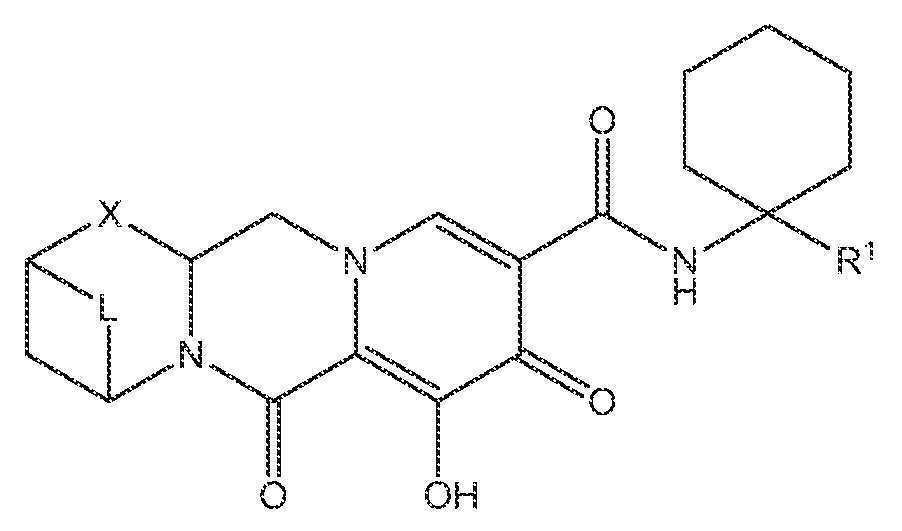
<화학식 IV-AE>

<화학식 IV-AF>
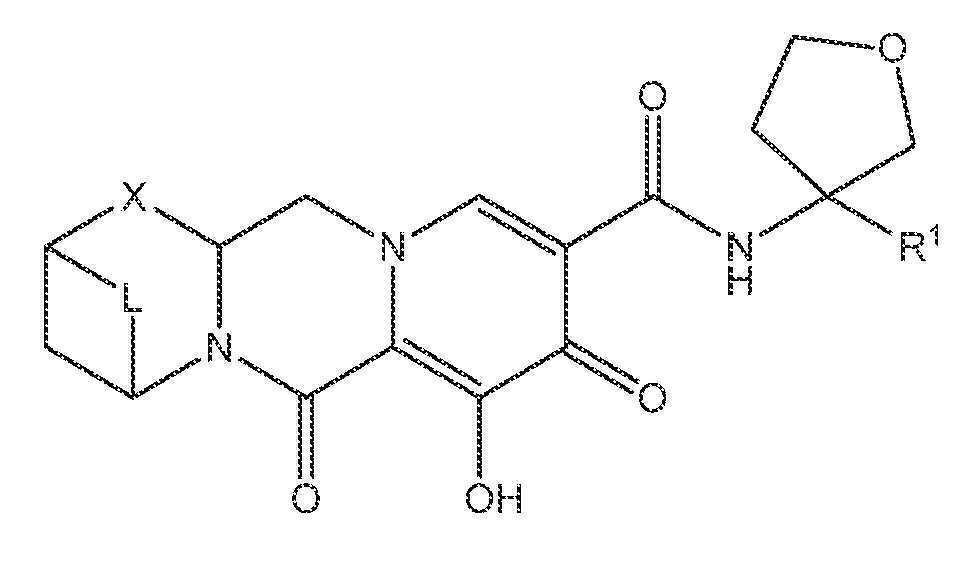
<화학식 IV-AG>
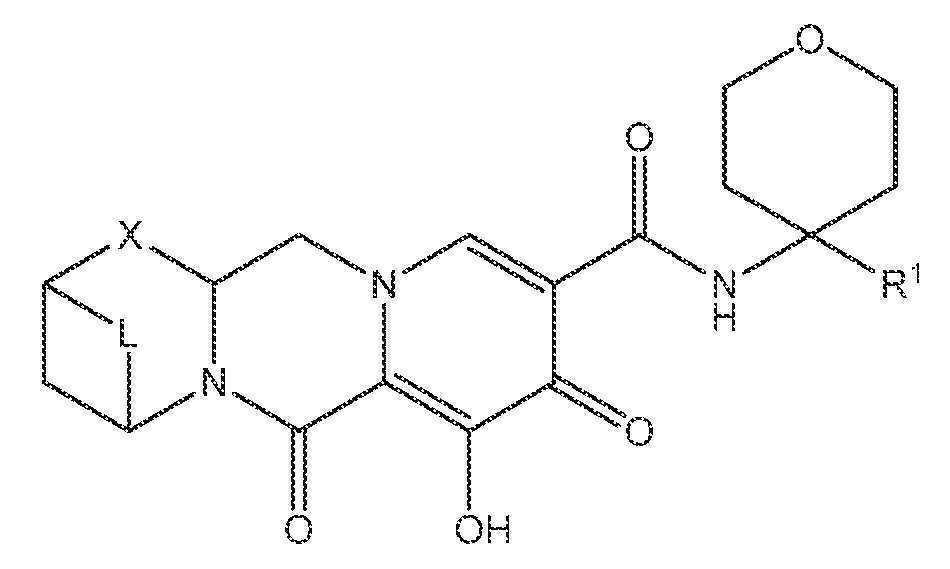
<화학식 IV-AH>
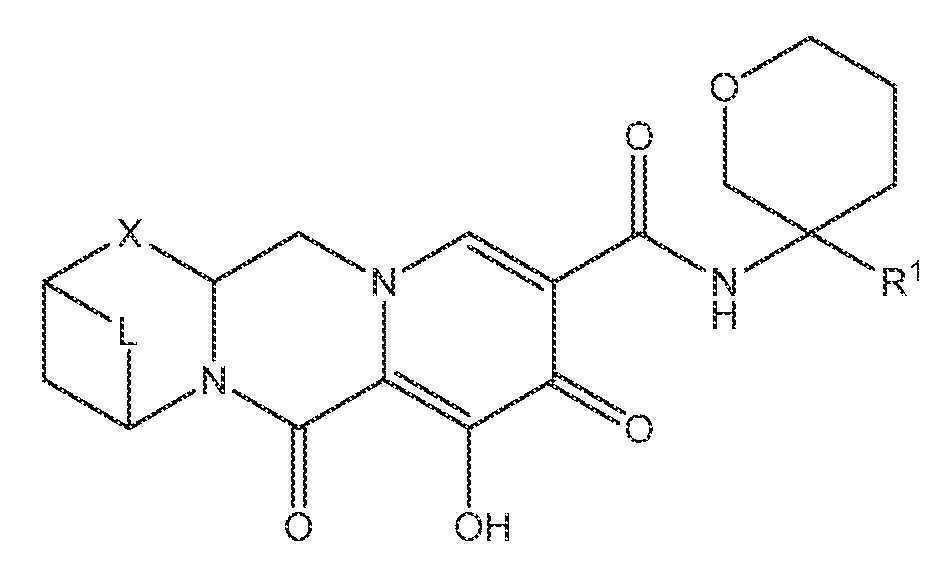
상기 식에서 L은 -C(Ra)2-, -C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2C(Ra)2C(Ra)2-, -C(Ra)2OC(Ra)2-, -C(Ra)2NRaC(Ra)2-, -C(Ra)2SC(Ra)2-, -C(Ra)2S(O)C(Ra)2-, -C(Ra)2SO2C(Ra)2-, -C(Ra)2OC(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2OC(Ra)2-, -C(Ra)2NRaC(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2NRaC(Ra)2-, -C(Ra)2SC(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2SC(Ra)2-, -C(Ra)2S(O)C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2S(O)C(Ra)2-, -C(Ra)2SO2C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2SO2C(Ra)2-, -C(Ra)2SO2NRaC(Ra)2- 또는 -C(Ra)2NRaSO2C(Ra)2-이다.
또 다른 실시양태에서, 하기 화학식 IV-BA, IV-BB, IV-BC, IV-BD, IV-BE, IV-BF, IV-BG 또는 IV-BH 중 하나를 갖는 화합물이 제공된다.
<화학식 IV-BA>

<화학식 IV-BB>

<화학식 IV-BC>

<화학식 IV-BD>

<화학식 IV-BE>
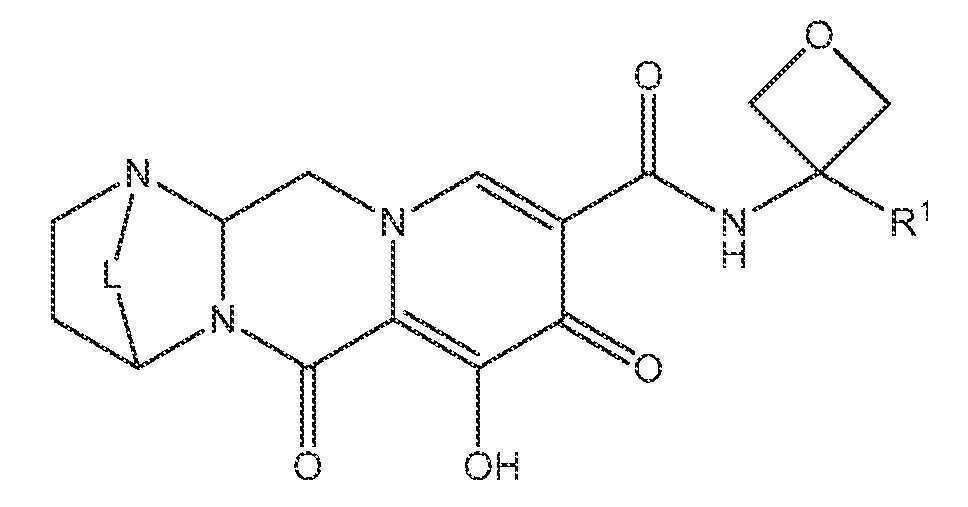
<화학식 IV-BF>
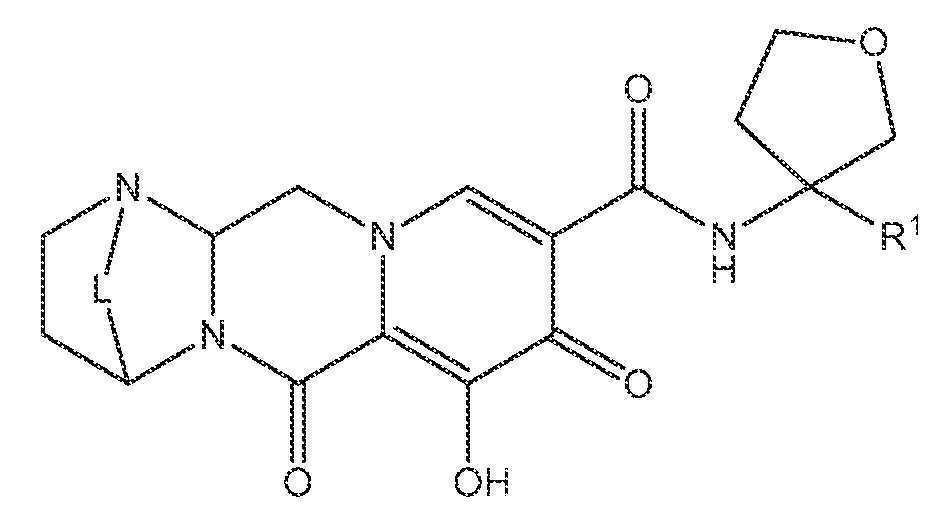
<화학식 IV-BG>

<화학식 IV-BH>
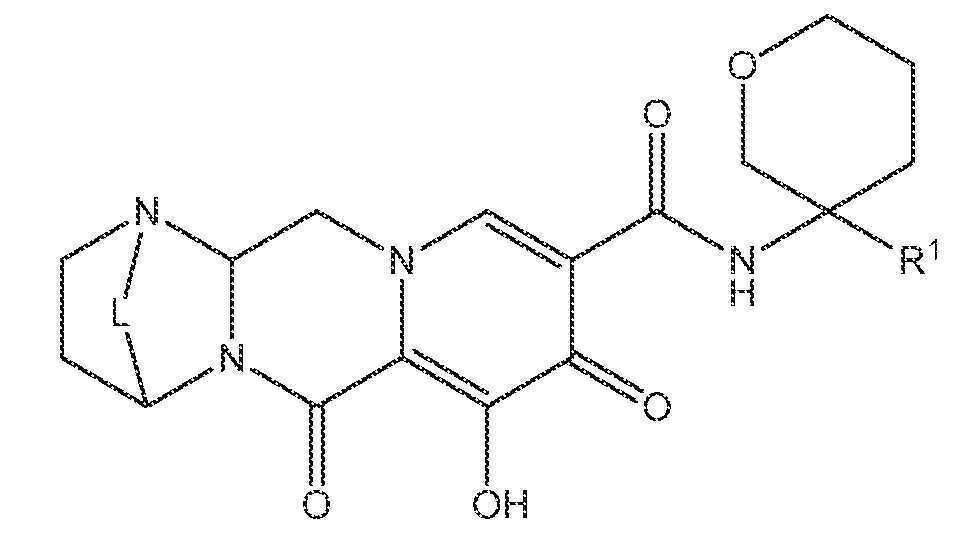
상기 식에서 L은 -C(Ra)2-, -C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2C(Ra)2C(Ra)2-, -C(Ra)2OC(Ra)2-, -C(Ra)2NRaC(Ra)2-, -C(Ra)2SC(Ra)2-, -C(Ra)2S(O)C(Ra)2-, -C(Ra)2SO2C(Ra)2-, -C(Ra)2OC(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2OC(Ra)2-, -C(Ra)2NRaC(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2NRaC(Ra)2-, -C(Ra)2SC(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2SC(Ra)2-, -C(Ra)2S(O)C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2S(O)C(Ra)2-, -C(Ra)2SO2C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2SO2C(Ra)2-, -C(Ra)2SO2NRaC(Ra)2- 또는 -C(Ra)2NRaSO2C(Ra)2-이다.
또 다른 실시양태에서, L은 -C(Ra)2-, -C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2C(Ra)2-, 또는 -C(Ra)2C(Ra)2C(Ra)2C(Ra)2-이다. 추가 실시양태에서, L은 -C(Ra)2-이다. 추가 실시양태에서, L은 -C(Ra)2C(Ra)2-이다. 추가 실시양태에서, L은 -C(Ra)2C(Ra)2C(Ra)2-이다. 추가 실시양태에서, 각각의 Ra는 수소이다. 추가 실시양태에서, 1개의 Ra는 메틸이고, 각각의 나머지 Ra는 수소이다. 추가 실시양태에서, 1개의 Ra는 할로겐이고, 각각의 나머지 Ra는 수소이다. 추가 실시양태에서, 2개의 Ra는 할로겐이고, 각각의 나머지 Ra는 수소이다. 추가 실시양태에서, 1개의 Ra는 할로겐이고, 각각의 나머지 Ra는 수소이다.
또 다른 실시양태에서, L은 -C(Ra)2OC(Ra)2-, -C(Ra)2NRaC(Ra)2-, -C(Ra)2SC(Ra)2-, -C(Ra)2S(O)C(Ra)2-, 또는 -C(Ra)2SO2C(Ra)2-이다. 추가 실시양태에서, L은 -C(Ra)2OC(Ra)2-이다. 추가 실시양태에서, 각각의 Ra는 수소이다. 추가 실시양태에서, 1개의 Ra는 메틸이고, 각각의 나머지 Ra는 수소이다. 추가 실시양태에서, 1개의 Ra는 할로겐이고, 각각의 나머지 Ra는 수소이다. 추가 실시양태에서, 2개의 Ra는 할로겐이고, 각각의 나머지 Ra는 수소이다. 추가 실시양태에서, 1개의 Ra는 할로겐이고, 각각의 나머지 Ra는 수소이다.
또 다른 실시양태에서, X는 -O-이다. 추가 실시양태에서, Z2는 수소이다. 또 다른 실시양태에서, X는 -NZ3-이다. 또 다른 실시양태에서, X는 -NH-이다. 또 다른 실시양태에서, X는 -CHZ3-이다. 또 다른 실시양태에서, X는 -CH2-이다.
또 다른 실시양태에서, Z4는 결합 또는 -CH2-이다. 또 다른 실시양태에서, Z4는 -CH2-이다. 또 다른 실시양태에서, Z4는 결합이다.
또 다른 실시양태에서, Y1 및 Y2는 각각 독립적으로 수소, 메틸 또는 트리플루오로메틸이다.
또 다른 실시양태에서, R1은 1개의 할로겐으로 치환된다. 추가 실시양태에서, R1은 4-플루오로페닐 또는 2-플루오로페닐이다.
또 다른 실시양태에서, R1은 페닐이다. 또 다른 실시양태에서, R1은 피리디닐이다.
또 다른 실시양태에서, R1은 적어도 1개의 할로겐으로 치환된다.
또 다른 실시양태에서, R1은 1개의 할로겐으로 치환된다. 추가 실시양태에서, R1은 4-플루오로페닐 또는 2-플루오로페닐이다.
또 다른 실시양태에서, R1은 2개의 할로겐으로 치환된다. 추가 실시양태에서, R1은 2,4-디플루오로페닐, 2,3-디플루오로페닐, 2,6-디플루오로페닐, 3-플루오로-4-클로로페닐, 3,4-디플루오로페닐, 2-플루오로-4-클로로페닐 또는 3,5-디플루오로페닐이다. 추가 실시양태에서, R1은 2,4-디플루오로페닐이다.
또 다른 실시양태에서, R1은 3개의 할로겐으로 치환된다. 추가 실시양태에서, R1은 2,4,6-트리플루오로페닐 또는 2,3,4-트리플루오로페닐이다. 추가 실시양태에서, R1은 2,4,6-트리플루오로페닐이다.
또 다른 실시양태에서, R1은 3-트리플루오로메틸-4-플루오로페닐 또는 2-시클로프로폭시-4-플루오로페닐이다.
한 실시양태에서, 상기 기재된 바와 같은 화학식 I, II-A, II-B, II-C, III-A, III-B, III-C, III-D, III-E, III-F, III-G, III-H, IV-AA, IV-AB, IV-AC, IV-AD, IV-AE, IV-AF, IV-AG, IV-AH, IV-BA, IV-BB, IV-BC, IV-BD, IV-BE, IV-BF, IV-BG, 및 IV-BH 중 어느 하나의 화합물 또는 그의 입체이성질체 또는 제약상 허용되는 염, 및 제약상 허용되는 담체, 희석제 또는 부형제를 포함하는 제약 조성물이 제공된다.
HIV 감염에 걸리거나 또는 걸릴 위험이 있는 인간에게 치료 유효량의 상기 기재된 바와 같은 화학식 I, II-A, II-B, II-C, III-A, III-B, III-C, III-D, III-E, III-F, III-G, III-H, IV-AA, IV-AB, IV-AC, IV-AD, IV-AE, IV-AF, IV-AG, IV-AH, IV-BA, IV-BB, IV-BC, IV-BD, IV-BE, IV-BF, IV-BG, 및 IV-BH 중 어느 하나의 화합물 또는 그의 제약 조성물을 투여하는 것에 의해 상기 인간에서 HIV 감염을 치료하는 방법을 포함하는 또 다른 실시양태가 제공된다. HIV 감염에 걸리거나 또는 걸릴 위험이 있는 인간에게 치료 유효량의 상기 기재된 바와 같은 화학식 I, II-A, II-B, II-C, III-A, III-B, III-C, III-D, III-E, III-F, III-G, III-H, IV-AA, IV-AB, IV-AC, IV-AD, IV-AE, IV-AF, IV-AG, IV-AH, IV-BA, IV-BB, IV-BC, IV-BD, IV-BE, IV-BF, IV-BG, 및 IV-BH 중 어느 하나의 화합물 또는 그의 제약 조성물을 투여하는 것에 의해 상기 인간에서 HIV 감염을 치료 또는 예방하는 방법을 포함하는 또 다른 실시양태가 제공된다.
또 다른 실시양태에서, HIV 감염에 걸리거나 또는 걸릴 위험이 있는 인간에서 HIV 감염을 치료하기 위한, 상기 기재된 바와 같은 화학식 I, II-A, II-B, II-C, III-A, III-B, III-C, III-D, III-E, III-F, III-G, III-H, IV-AA, IV-AB, IV-AC, IV-AD, IV-AE, IV-AF, IV-AG, IV-AH, IV-BA, IV-BB, IV-BC, IV-BD, IV-BE, IV-BF, IV-BG, 및 IV-BH 중 어느 하나의 화합물 또는 그의 제약 조성물의 용도가 제공된다. 또 다른 실시양태에서, HIV 감염에 걸리거나 또는 걸릴 위험이 있는 인간에서 HIV 감염을 치료 또는 예방하기 위한, 상기 기재된 바와 같은 화학식 I, II-A, II-B, II-C, III-A, III-B, III-C, III-D, III-E, III-F, III-G, III-H, IV-AA, IV-AB, IV-AC, IV-AD, IV-AE, IV-AF, IV-AG, IV-AH, IV-BA, IV-BB, IV-BC, IV-BD, IV-BE, IV-BF, IV-BG, 및 IV-BH 중 어느 하나의 화합물 또는 그의 제약 조성물의 용도가 제공된다.
또 다른 실시양태에서, 상기 기재된 바와 같은 화학식 I, II-A, II-B, II-C, III-A, III-B, III-C, III-D, III-E, III-F, III-G, III-H, IV-AA, IV-AB, IV-AC, IV-AD, IV-AE, IV-AF, IV-AG, IV-AH, IV-BA, IV-BB, IV-BC, IV-BD, IV-BE, IV-BF, IV-BG, 및 IV-BH 중 어느 하나의 화합물 또는 그의 제약 조성물의 의료 요법에서의 용도가 제공된다.
또 다른 실시양태에서, HIV 감염의 치유적 치료에 사용하기 위한, 상기 기재된 바와 같은 화학식 I, II-A, II-B, II-C, III-A, III-B, III-C, III-D, III-E, III-F, III-G, III-H, IV-AA, IV-AB, IV-AC, IV-AD, IV-AE, IV-AF, IV-AG, IV-AH, IV-BA, IV-BB, IV-BC, IV-BD, IV-BE, IV-BF, IV-BG, 및 IV-BH 중 어느 하나의 화합물 또는 그의 제약 조성물의 용도가 제공된다. 또 다른 실시양태에서, HIV 감염의 예방적 또는 치유적 치료에 사용하기 위한, 상기 기재된 바와 같은 화학식 I, II-A, II-B, II-C, III-A, III-B, III-C, III-D, III-E, III-F, III-G, III-H, IV-AA, IV-AB, IV-AC, IV-AD, IV-AE, IV-AF, IV-AG, IV-AH, IV-BA, IV-BB, IV-BC, IV-BD, IV-BE, IV-BF, IV-BG, 및 IV-BH 중 어느 하나의 화합물 또는 그의 제약 조성물의 용도가 제공된다.
상기 기재된 바와 같은 화학식 I, II-A, II-B, II-C, III-A, III-B, III-C, III-D, III-E, III-F, III-G, III-H, IV-AA, IV-AB, IV-AC, IV-AD, IV-AE, IV-AF, IV-AG, IV-AH, IV-BA, IV-BB, IV-BC, IV-BD, IV-BE, IV-BF, IV-BG, 및 IV-BH의 화합물 중 임의의 실시양태, 및 상기 기재된 바와 같은 화학식 I, II-A, II-B, II-C, III-A, III-B, III-C, III-D, III-E, III-F, III-G, III-H, IV-AA, IV-AB, IV-AC, IV-AD, IV-AE, IV-AF, IV-AG, IV-AH, IV-BA, IV-BB, IV-BC, IV-BD, IV-BE, IV-BF, IV-BG, 및 IV-BH의 화합물에서 R1, Ra, X, W, Y1, Y2, L, Z1, Z2, Z3, 또는 Z4 기에 대해 본원에 기재된 임의의 구체적 치환기는 독립적으로 화학식 I, II-A, II-B, II-C, III-A, III-B, III-C, III-D, III-E, III-F, III-G, III-H, IV-AA, IV-AB, IV-AC, IV-AD, IV-AE, IV-AF, IV-AG, IV-AH, IV-BA, IV-BB, IV-BC, IV-BD, IV-BE, IV-BF, IV-BG, 및 IV-BH의 화합물 중 다른 실시양태 및/또는 치환기와 조합되어 구체적으로 상기 기재되지 않은 본 발명의 실시양태를 형성할 수 있음을 이해한다. 또한, 치환기 목록이 특정한 실시양태 및/또는 청구항에서 임의의 특정한 R1, Ra, X, W, Y1, Y2, L, Z1, Z2, Z3, 또는 Z4에 대해 열거된 경우에, 각각의 개별 치환기는 특정한 실시양태 및/또는 청구항에서 삭제될 수 있고, 나머지 치환기 목록이 본 발명이 범위 내에 있는 것으로 간주될 것임을 이해한다.
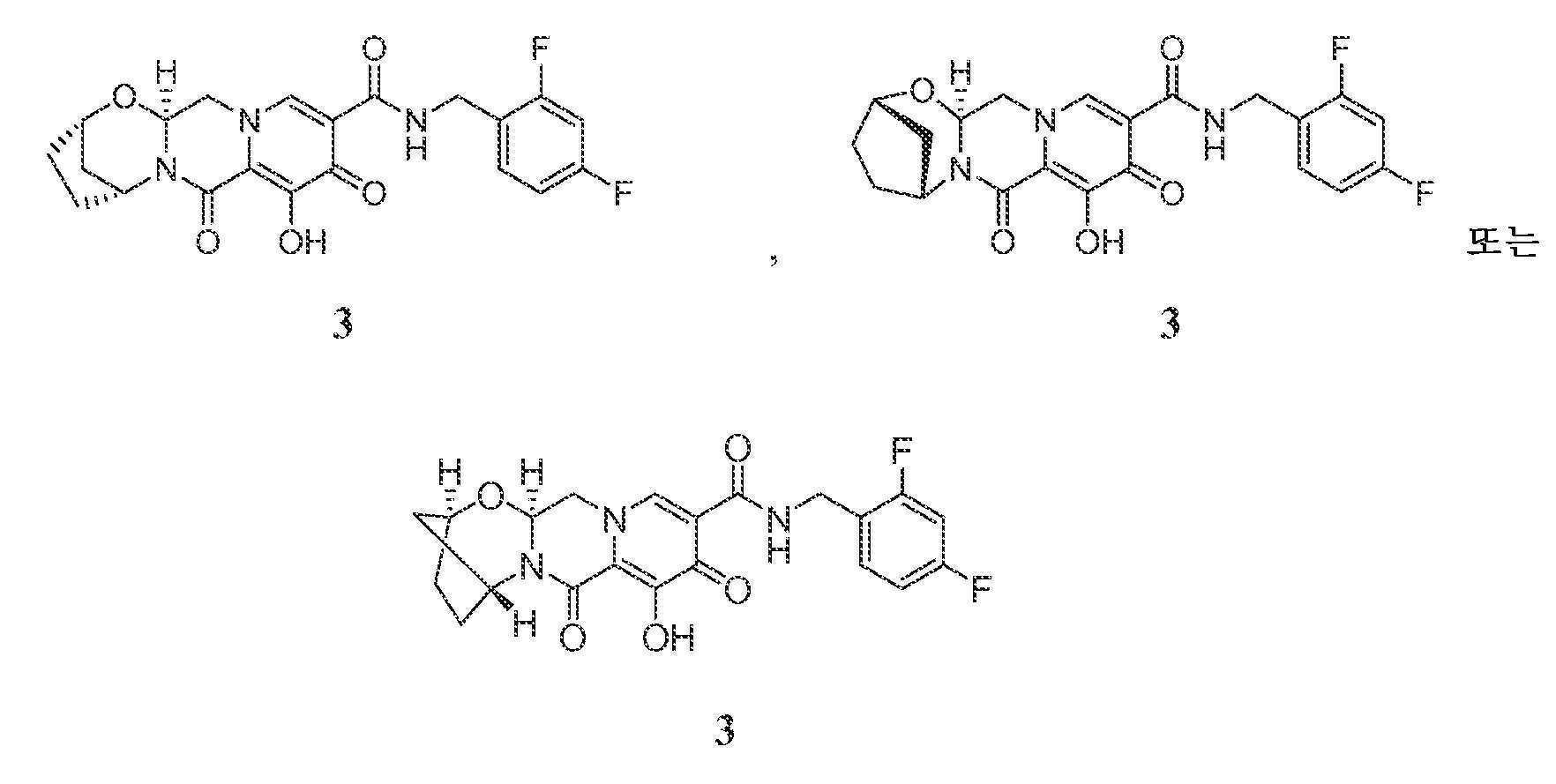
통상의 기술자가 인식할 것과 같이, Z1 및 Z2 또는 Z1 및 Z3이 함께 -L-을 형성한 화학식 I, II-A, II-B, II-C, IV-AA, IV-AB, IV-AC, IV-AD, IV-AE, IV-AF, IV-AG, IV-AH, IV-BA, IV-BB, IV-BC, IV-BD, IV-BE, IV-BF, IV-BG, 및 IV-BH의 화합물은 여러 상이한 방식으로 제시될 수 있다. 예를 들어, 실시예 3의 화합물 3은 하기와 같이 제시될 수 있다:
제약 조성물
투여 목적을 위해, 특정 실시양태에서, 본원에 기재된 화합물은 미가공 화학물질로서 투여되거나 또는 제약 조성물로서 제제화된다. 본원에 개시된 제약 조성물은 화학식 I의 화합물, 및 제약상 허용되는 담체, 희석제 또는 부형제 중 하나 이상을 포함한다. 화학식 I의 화합물은 조성물 중에 특정한 관심 질환 또는 상태를 치료하는데 효과적인 양으로 존재한다. 화학식 I의 화합물의 활성은 예를 들어 하기 실시예에 기재된 바와 같이 통상의 기술자에 의해 결정될 수 있다. 적절한 농도 및 투여량은 통상의 기술자에 의해 용이하게 결정될 수 있다. 특정 실시양태에서, 화학식 I의 화합물은 제약 조성물 중에 약 25 mg 내지 약 500 mg의 양으로 존재한다. 특정 실시양태에서, 화학식 I의 화합물은 제약 조성물 중에 약 100 mg 내지 약 300 mg의 양으로 존재한다. 특정 실시양태에서, 화학식 I의 화합물은 제약 조성물 중에 약 25 mg, 50 mg, 100 mg, 200 mg, 300 mg, 400 mg 또는 약 500 mg의 양으로 존재한다.
순수한 형태로 또는 적절한 제약 조성물로의 본 발명의 화합물 또는 그의 제약상 허용되는 염의 투여는 유사한 유용성을 제공하기 위한 작용제의 허용되는 임의의 투여 방식을 통해 이루어진다. 본 발명의 제약 조성물은 본 발명의 화합물을 적절한 제약상 허용되는 담체, 희석제 또는 부형제와 조합함으로써 제조되고, 구체적 실시양태에서 고체, 반-고체, 액체 또는 기체상 형태의 제제로, 예컨대 정제, 캡슐, 분말, 과립, 연고, 용액, 좌제, 주사, 흡입제, 겔, 마이크로구체 및 에어로졸로 제제화된다. 이러한 제약 조성물의 예시적인 투여 경로는 제한 없이 경구, 국소, 경피, 흡입, 비경구, 설하, 협측, 직장, 질, 및 비강내를 포함한다. 본 발명의 제약 조성물은 환자에게 조성물의 투여 시 내부에 함유되어 있는 활성 성분이 생체이용가능하도록 제제화된다. 대상체 또는 환자에게 투여될 조성물은 하나 이상의 투여 단위 형태를 취하며, 예를 들어 정제는 단일 투여 단위일 수 있고, 에어로졸 형태의 본 발명의 화합물의 용기는 다수의 투여 단위를 보유할 수 있다. 이러한 투여 형태를 제조하는 실제 방법은 통상의 기술자에게 공지되어 있거나 또는 그에게 자명할 것이며; 예를 들어 문헌 [Remington: The Science and Practice of Pharmacy, 20th Edition (Philadelphia College of Pharmacy and Science, 2000)]을 참조한다. 투여될 조성물은 어떠한 경우라도 본원에 기재된 교시에 따라 관심 질환 또는 상태의 치료를 위한 치료 유효량의 본 발명의 화합물 또는 그의 제약상 허용되는 염을 함유할 것이다.
본원에 개시된 제약 조성물은 제약 업계에 널리 공지되어 있는 방법론에 의해 제조된다. 예를 들어, 특정 실시양태에서, 주사에 의해 투여되도록 의도되는 제약 조성물은 본 발명의 화합물을 멸균, 증류수와 조합하여 용액을 형성함으로서 제조된다. 일부 실시양태에서, 균질 용액 또는 현탁액의 형성을 용이하게 하기 위해 계면활성제가 첨가된다. 계면활성제는 본 발명의 화합물과 비공유적으로 상호작용하여 수성 전달 시스템 중 화합물의 용해 또는 균질 현탁을 용이하게 하는 화합물이다.
본 발명의 화합물 또는 그의 제약상 허용되는 염은 치료 유효량으로 투여되며, 이는 사용된 구체적 화합물의 활성; 화합물의 대사 안정성 및 작용 기간; 환자의 연령, 체중, 전반적 건강, 성별 및 식이; 투여 방식 및 시간; 배설 속도; 약물 조합; 특정한 장애 또는 상태의 중증도; 및 대상체에서 진행중인 요법을 비롯한 다양한 인자에 따라 달라질 것이다.
조합 요법
한 실시양태에서, HIV 감염에 걸리거나 또는 걸릴 위험이 있는 인간에게 치료 유효량의 본원에 개시된 화합물 또는 그의 제약상 허용되는 염을 치료 유효량의 하나 이상의 추가의 치료제와 조합하여 투여하는 것을 포함하는, 상기 인간에서 HIV 감염을 치료 또는 예방하는 방법이 제공된다.
한 실시양태에서, 본원에 개시된 화합물 또는 그의 제약상 허용되는 염을 하나 이상의 추가의 치료제 및 제약상 허용되는 담체, 희석제 또는 부형제와 조합하여 포함하는 제약 조성물이 제공된다.
한 실시양태에서, 본원에 개시된 화합물 또는 그의 제약상 허용되는 염을 하나 이상의 추가의 치료제와 조합하여 포함하는 조합 제약 작용제가 제공된다.
상기 실시양태에서, 추가의 치료제는 항-HIV 작용제일 수 있다. 예를 들어, 일부 실시양태에서, 추가의 치료제는 HIV 프로테아제 억제제, 역전사효소의 HIV 비-뉴클레오시드 억제제, 역전사효소의 HIV 뉴클레오시드 억제제, 역전사효소의 HIV 뉴클레오티드 억제제, HIV 인테그라제 억제제, HIV 비-촉매 부위 (또는 알로스테릭) 인테그라제 억제제, 진입 억제제 (예를 들어, CCR5 억제제, gp41 억제제 (즉, 융합 억제제)과 CD4 부착 억제제), CXCR4 억제제, gp120 억제제, G6PD 및 NADH-옥시다제 억제제, HIV 캡시드를 표적으로 하는 화합물 ("캡시드 억제제"; 예를 들어, 캡시드 중합 억제제 또는 캡시드 교란 화합물, 예컨대 WO 2013/006738 (길리아드 사이언시스(Gilead Sciences)), US 2013/0165489 (펜실베니아 대학교(University of Pennsylvania)), 및 WO 2013/006792 (파마 리소시스(Pharma Resources))에 개시되어 있는 것, 약동학적 인핸서 및 HIV를 치료하기 위한 다른 약물, 및 그의 조합으로 이루어진 군으로부터 선택된다. 추가 실시양태에서, 추가의 치료제는 하기 중 하나 이상으로부터 선택된다:
(1) 암프레나비르, 아타자나비르, 포삼프레나비르, 인디나비르, 로피나비르, 리토나비르, 넬피나비르, 사퀴나비르, 티프라나비르, 브레카나비르, 다루나비르, TMC-126, TMC-114, 모제나비르 (DMP-450), JE-2147 (AG1776), L-756423, RO0334649, KNI-272, DPC-681, DPC-684, GW640385X, DG17, PPL-100, DG35 및 AG 1859로 이루어진 군으로부터 선택되는 HIV 프로테아제 억제제;
(2) 카프라비린, 에미비린, 델라비리딘, 에파비렌즈, 네비라핀, (+) 칼라놀리드 A, 에트라비린, GW5634, DPC-083, DPC-961, DPC-963, MIV-150, TMC-120, 릴피비린, BILR 355 BS, VRX 840773, 레르시비린 (UK-453061), RDEA806, KM023 및 MK-1439로 이루어진 군으로부터 선택되는 역전사효소의 HIV 비-뉴클레오시드 또는 비-뉴클레오티드 억제제;
(3) 지도부딘, 엠트리시타빈, 디다노신, 스타부딘, 잘시타빈, 라미부딘, 아바카비르, 암독소비르, 엘부시타빈, 알로부딘, MIV-210, ±-FTC, D-d4FC, 엠트리시타빈, 포스파지드, 포지부딘 티독실, 아프리시티빈 (AVX754), KP-1461, GS-9131 (길리아드 사이언시스) 및 포살부딘 티독실 (종전 HDP 99.0003)로 이루어진 군으로부터 선택되는 역전사효소의 HIV 뉴클레오시드 억제제;
(4) 테노포비르, 테노포비르 디소프록실 푸마레이트, 테노포비르 알라페나미드 푸마레이트 (길리아드 사이언시스), GS-7340 (길리아드 사이언시스), GS-9148 (길리아드 사이언시스), 아데포비르, 아데포비르 디피복실, CMX-001 (키메릭스) 또는 CMX-157 (키메릭스)로 이루어진 군으로부터 선택되는 역전사효소의 HIV 뉴클레오티드 억제제;
(5) 쿠르쿠민, 쿠르쿠민의 유도체, 키코르산, 키코르산의 유도체, 3,5-디카페오일퀸산, 3,5-디카페오일퀸산의 유도체, 아우린트리카르복실산, 아우린트리카르복실산의 유도체, 카페인산 페네틸 에스테르, 카페인산 페네틸 에스테르의 유도체, 티르포스틴, 티르포스틴의 유도체, 퀘르세틴, 퀘르세틴의 유도체, S-1360, AR-177, L-870812 및 L-870810, 랄테그라비르, BMS-538158, GSK364735C, BMS-707035, MK-2048, BA 011, 엘비테그라비르, 돌루테그라비르 및 GSK-744로 이루어진 군으로부터 선택되는 HIV 인테그라제 억제제;
(6) BI-224436, CX0516, CX05045, CX14442, 각각 그 전문이 본원에 참조로 포함되는 WO 2009/062285 (베링거 잉겔하임(Boehringer Ingelheim)), WO 2010/130034 (베링거 잉겔하임), WO 2013/159064 (길리아드 사이언시스), WO 2012/145728 (길리아드 사이언시스), WO 2012/003497 (길리아드 사이언시스), WO 2012/003498 (길리아드 사이언시스)에 개시되어 있는 화합물을 포함하나 이에 제한되지는 않는, HIV 비-촉매 부위, 또는 알로스테릭, 인테그라제 억제제 (NCINI);
(7) 엔푸비르티드, 시푸비르티드, 알부비르티드, FB006M 및 TRI-1144로 이루어진 군으로부터 선택되는 gp41 억제제;
(8) CXCR4 억제제 AMD-070;
(9) 진입 억제제 SP01A;
(10) gp120 억제제 BMS-488043;
(11) G6PD 및 NADH-옥시다제 억제제 이뮤니틴;
(12) 아플라비록, 비크리비록, 마라비록, 세니크리비록, PRO-140, INCB15050, PF-232798 (화이자(Pfizer)) 및 CCR5mAb004로 이루어진 군으로부터 선택되는 CCR5 억제제;
(13) 이발리주맙 (TMB-355) 및 BMS-068 (BMS-663068)로 이루어진 군으로부터 선택되는 CD4 부착 억제제;
(14) 코비시스타트 및 SPI-452로 이루어진 군으로부터 선택되는 약동학적 인핸서;
(15) BAS-100, SPI-452, REP 9, SP-01A, TNX-355, DES6, ODN-93, ODN-112, VGV-1, PA-457 (베비리마트), HRG214, VGX-410, KD-247, AMZ 0026, CYT 99007A-221 HIV, DEBIO-025, BAY 50-4798, MDX010 (이필리무맙), PBS 119, ALG 889, 및 PA-1050040 (PA-040)으로 이루어진 군으로부터 선택되는 HIV를 치료하기 위한 다른 약물,
및 그의 조합.
특정 실시양태에서, 본원에 개시된 화합물 또는 그의 제약상 허용되는 염은 2, 3, 4종 또는 그 초과의 추가의 치료제와 조합된다. 특정 실시양태에서, 본원에 개시된 화합물 또는 그의 제약상 허용되는 염은 2종의 추가의 치료제와 조합된다. 다른 실시양태에서, 본원에 개시된 화합물 또는 그의 제약상 허용되는 염은 3종의 추가의 치료제와 조합된다. 추가 실시양태에서, 본원에 개시된 화합물 또는 그의 제약상 허용되는 염은 4종의 추가의 치료제와 조합된다. 2, 3, 4종 또는 그 초과의 추가의 치료제는 동일한 부류의 치료제로부터 선택된 상이한 치료제일 수 있거나, 또는 상이한 부류의 치료제로부터 선택될 수 있다. 구체적 실시양태에서, 본원에 개시된 화합물 또는 그의 제약상 허용되는 염은 역전사효소의 HIV 뉴클레오티드 억제제 및 역전사효소의 HIV 비-뉴클레오시드 억제제와 조합된다. 또 다른 구체적 실시양태에서, 본원에 개시된 화합물 또는 그의 제약상 허용되는 염은 역전사효소의 HIV 뉴클레오티드 억제제 및 HIV 프로테아제 억제 화합물과 조합된다. 추가 실시양태에서, 본원에 개시된 화합물 또는 그의 제약상 허용되는 염은 역전사효소의 HIV 뉴클레오티드 억제제, 역전사효소의 HIV 비-뉴클레오시드 억제제 및 HIV 프로테아제 억제 화합물과 조합된다. 추가 실시양태에서, 본원에 개시된 화합물 또는 그의 제약상 허용되는 염은 역전사효소의 HIV 뉴클레오티드 억제제, 역전사효소의 HIV 비-뉴클레오시드 억제제 및 약동학적 인핸서와 조합된다.
특정 실시양태에서, 본원에 개시된 화합물이 상기 기재된 바와 같은 하나 이상의 추가의 치료제와 조합되는 경우에, 조성물의 성분은 동시 또는 순차 요법으로서 투여된다. 순차적으로 투여되는 경우에, 조합물은 2회 이상의 투여로 투여될 수 있다.
특정 실시양태에서, 본원에 개시된 화합물은 하나 이상의 추가의 치료제와, 환자에게 동시 투여를 위한 단위 투여 형태로, 예를 들어 경구 투여를 위한 고체 투여 형태로서 조합된다.
특정 실시양태에서, 본원에 개시된 화합물은 하나 이상의 추가의 치료제와 함께 투여된다. 본원에 개시된 화합물의 하나 이상의 추가의 치료제와의 공-투여는 일반적으로, 치료 유효량의 본원에 개시된 화합물 및 하나 이상의 추가의 치료제가 환자의 신체 내에 둘 다 존재하도록 하는, 본원에 개시된 화합물과 하나 이상의 추가의 치료제의 동시 또는 순차적 투여로 지칭된다.
공-투여는 하나 이상의 추가의 치료제의 단위 투여량의 투여 전 또는 후의 본원에 개시된 화합물의 단위 투여량의 투여, 예를 들어 하나 이상의 추가의 치료제의 투여 수초, 수분 또는 수시간 내의 본원에 개시된 화합물의 투여를 포함한다. 예를 들어, 일부 실시양태에서, 본원에 개시된 화합물의 단위 용량이 먼저 투여되고, 이어서 수초 또는 수분 내에 하나 이상의 추가의 치료제의 단위 용량이 투여된다. 대안적으로, 다른 실시양태에서, 하나 이상의 추가의 치료제의 단위 용량이 먼저 투여되고, 이어서 수초 또는 수분 내에 본원에 개시된 화합물의 단위 용량이 투여된다. 일부 실시양태에서, 본원에 개시된 화합물의 단위 용량이 먼저 투여되고, 이어서 수시간의 기간 후 (예를 들어, 1-12시간) 하나 이상의 추가의 치료제의 단위 용량이 투여된다. 다른 실시양태에서, 하나 이상의 추가의 치료제의 단위 용량이 먼저 투여되고, 이어서 수시간의 기간 후 (예를 들어, 1-12시간), 본원에 개시된 화합물의 단위 용량이 투여된다.
하기 실시예는 본 발명의 화합물, 즉 하기 화학식 I의 화합물의 다양한 제조 방법을 예시한다.
<화학식 I>
상기 식에서 R1, X, W, Y1, Y2, Z1, Z2, 또는 Z4는 상기 정의된 바와 같다. 통상의 기술자는 이들 화합물을 유사한 방법 또는 통상의 기술자에게 공지되어 있는 다른 방법과의 조합에 의해 제조할 수 있음을 이해한다. 또한, 통상의 기술자는 하기 기재된 것과 유사한 방식으로, 적절한 출발 물질을 사용하고 필요에 따라 합성 파라미터를 변형시킴으로써 하기에 구체적으로 예시되지 않은 다른 화학식 I의 화합물을 제조할 수 있을 것임을 이해한다. 일반적으로, 출발 성분은 시그마 알드리치(Sigma Aldrich), 랭커스터 신테시스, 인크.(Lancaster Synthesis, Inc.), 메이브리지(Maybridge), 매트릭스 사이언티픽(Matrix Scientific), TCI, 및 플루오로켐 USA(Fluorochem USA) 등과 같은 공급원으로부터 입수할 수 있거나, 또는 통상의 기술자에게 공지되어 있는 공급원에 따라 합성할 수 있거나 (예를 들어, 문헌 [Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5th edition (Wiley, December 2000)] 참조), 또는 본원에 기재된 바와 같이 제조할 수 있다.
하기 실시예는 제한이 아닌 예시의 목적으로 제공된다.
실시예
일반적 합성 반응식
반응식 1-3은 본 발명의 추가 실시양태로서 제공되며, 화학식 I을 갖는 화합물을 제조하는데 사용되었고 화학식 I을 갖는 추가의 화합물을 제조하는데 사용될 수 있는 일반적 방법을 예시한다.
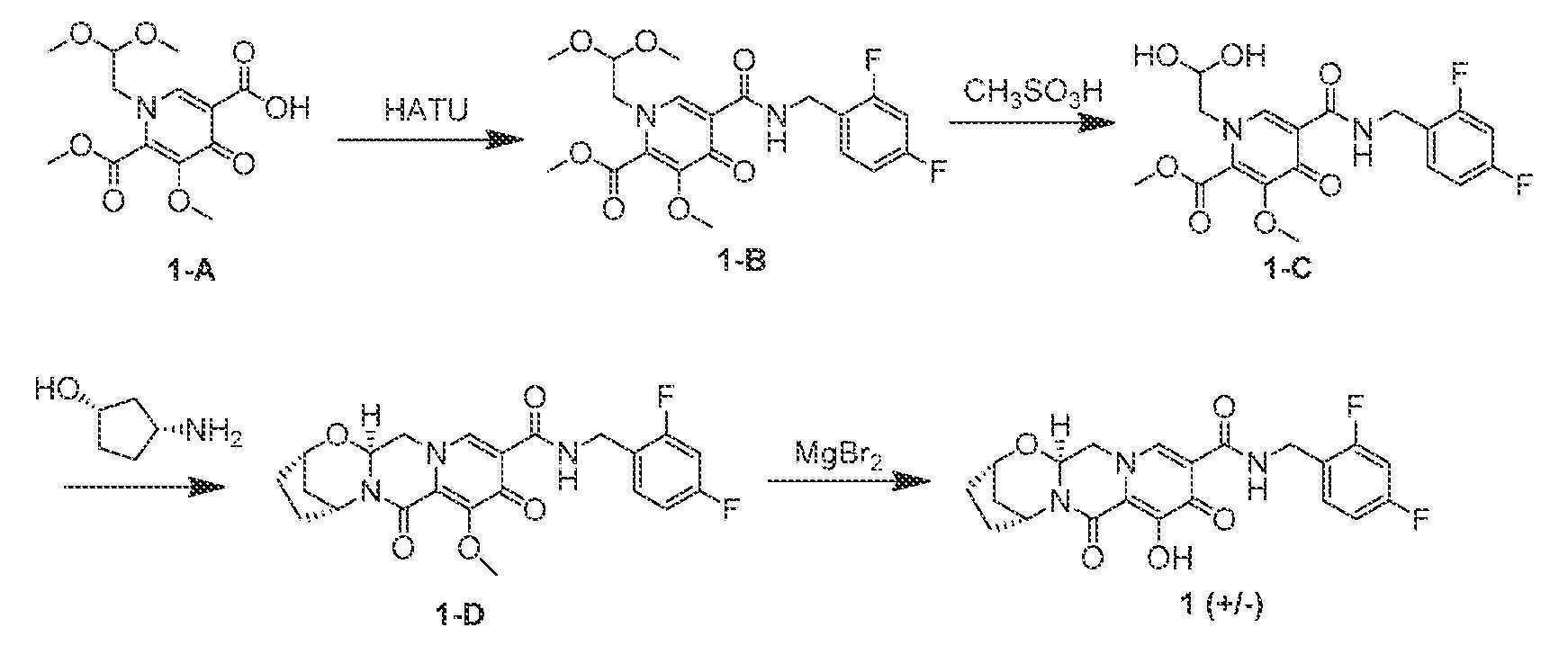
<반응식 1>

A1은 적절한 아민 및 커플링 시약, 예컨대 HATU 또는 EDCI에 의해 아미드 A2로 전환될 수 있다. A2는 강산, 예컨대 메탄술폰산에 의해 A3으로 전환될 수 있다. A3은 적절한 시클릭 디아민 또는 시클릭 아미노알콜과의 가열에 이어 브로민화마그네슘과 같은 시약에 의한 메틸 탈보호에 의해 A5 또는 A4로 전환될 수 있다.
대안적으로, A1은 강산, 예컨대 메탄술폰산에 의한 처리에 의해 A6으로 전환될 수 있다. A6은 적절한 시클릭 디아민 또는 시클릭 아미노알콜과 축합된 후 브로민화마그네슘과 같은 시약에 의해 메틸 탈보호되어 각각 A7 또는 A8을 형성할 수 있다. A7 또는 A8은 적절한 아민 및 커플링 시약, 예컨대 HATU 또는 EDCI에 의한 처리에 이어 브로민화마그네슘과 같은 시약에 의한 메틸 탈보호에 의해 아미드 A5 및 A4로 전환될 수 있다.
<반응식 2>

B1 (WO2012/018065에 기재된 바와 같음)은 디아민과 환류 조건 하에 축합되어 B2를 생성한다. B2는 가수분해되고 아민과 아미드-형성 방법에 의해 커플링되어 벤질 보호기의 제거 시 생성물 B3이 수득된다.
대표적 화합물
실시예 1
화합물 1의 제조
N-(2,4-디플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

단계 1
WO2011/119566 A1에 기재된 바와 같이 제조된, 1-(2,2-디메톡시에틸)-5-메톡시-6-(메톡시카르보닐)-4-옥소-1,4-디히드로피리딘-3-카르복실산 (1-A, 0.300 g, 0.95 mmol)을 건조 톨루엔으로부터 1회 증발시키고, 아세토니트릴 (4 mL) 중에 현탁시키고, N,N-디이소프로필에틸아민 (DIPEA) (0.329 mL, 1.90 mmol), 2,4-디플루오로벤질아민 (0.125 mL, 1.05 mmol) 및 HATU (0.433 g, 1.14 mmol)로 처리하였다. 반응 혼합물을 10분 동안 교반하고, 농축시켰다. 잔류물을 실리카 겔 상에서 플래쉬 크로마토그래피 (10에서 60% 에틸 아세테이트:디클로로메탄)에 의해 정제하여 화합물 메틸 5-(2,4-디플루오로벤질카르바모일)-1-(2,2-디메톡시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트, 1-B를 수득하였다.

단계 2
아세토니트릴 (0.9 mL) 및 아세트산 (0.1 mL) 중 메틸 5-(2,4-디플루오로벤질카르바모일)-1-(2,2-디메톡시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트 (1-B, 0.106 g, 0.24 mmol)를 메탄술폰산 (0.005 mL, 0.072 mmol)으로 처리하고, 황색 마개로 밀봉하고, 70℃로 가열하였다. 16시간 후, 혼합물을 냉각시켜 메틸 5-(2,4-디플루오로벤질카르바모일)-1-(2,2-디히드록시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트, 1-C의 조 용액을 수득하였다.
단계 3 및 4
메틸 5-(2,4-디플루오로벤질카르바모일)-1-(2,2-디히드록시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트 (1-C, 이전 단계로부터의 조 혼합물 0.65 mL, 0.17 mmol)를 아세토니트릴 (0.65 mL) 및 시스-3-아미노시클로펜탄올 (0.06 mL)로 처리하였다. 반응 혼합물을 밀봉하고, 90℃로 가열하였다. 30분 후, 반응 혼합물을 냉각시키고, 브로민화마그네슘 (0.063 g, 0.34 mmol)을 첨가하였다. 혼합물을 재밀봉하고, 50℃로 가열하였다. 10분 후, 반응 혼합물을 디클로로메탄과 염산 (0.2 M 수성) 사이에 분배하였다. 유기 층을 제거하고, 수성 층을 디클로르메탄으로 다시 추출하였다. 합한 유기 층을 황산나트륨 상에서 건조시키고, 여과하고, 농축시켰다. 정제용 HPLC 정제 (30-70% 아세토니트릴:물, 0.1% TFA)로 화합물 1을 라세미 혼합물로서 수득하였다.

실시예 2 및 3
화합물 2 및 3의 제조
(2R,5S,13aR)-N-(2,4-디플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드 (2) 및 (2S,5R,13aS)-N-(2,4-디플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드 (3)

화합물 1 (16 mg)을 키랄팩 AS-H를 사용하는 키랄 HPLC에 의해 용리액으로서 100% 에탄올로 분리하여 화합물 2 및 3을 거울상이성질체적으로 풍부한 형태로 수득하였다. 화합물 2의 경우: LCMS-ESI+ (m/z): [M+H]+ 계산치, C21H20F2N3O5: 432.14; 실측치: 432.2, 키랄 HPLC 체류 시간 = 4.50분 (키랄팩 AS-H, 150 x 4.6 mm, 1 mL/분 EtOH). 화합물 3의 경우: LCMS-ESI+ (m/z): [M+H]+ 계산치, C21H20F2N3O5: 432.14; 실측치: 432.2, 키랄 HPLC 체류 시간 = 6.84분 (키랄팩 AS-H, 150 x 4.6 mm, 1 mL/분 EtOH).

대안적으로, 화합물 3을 다음과 같이 제조하였다:
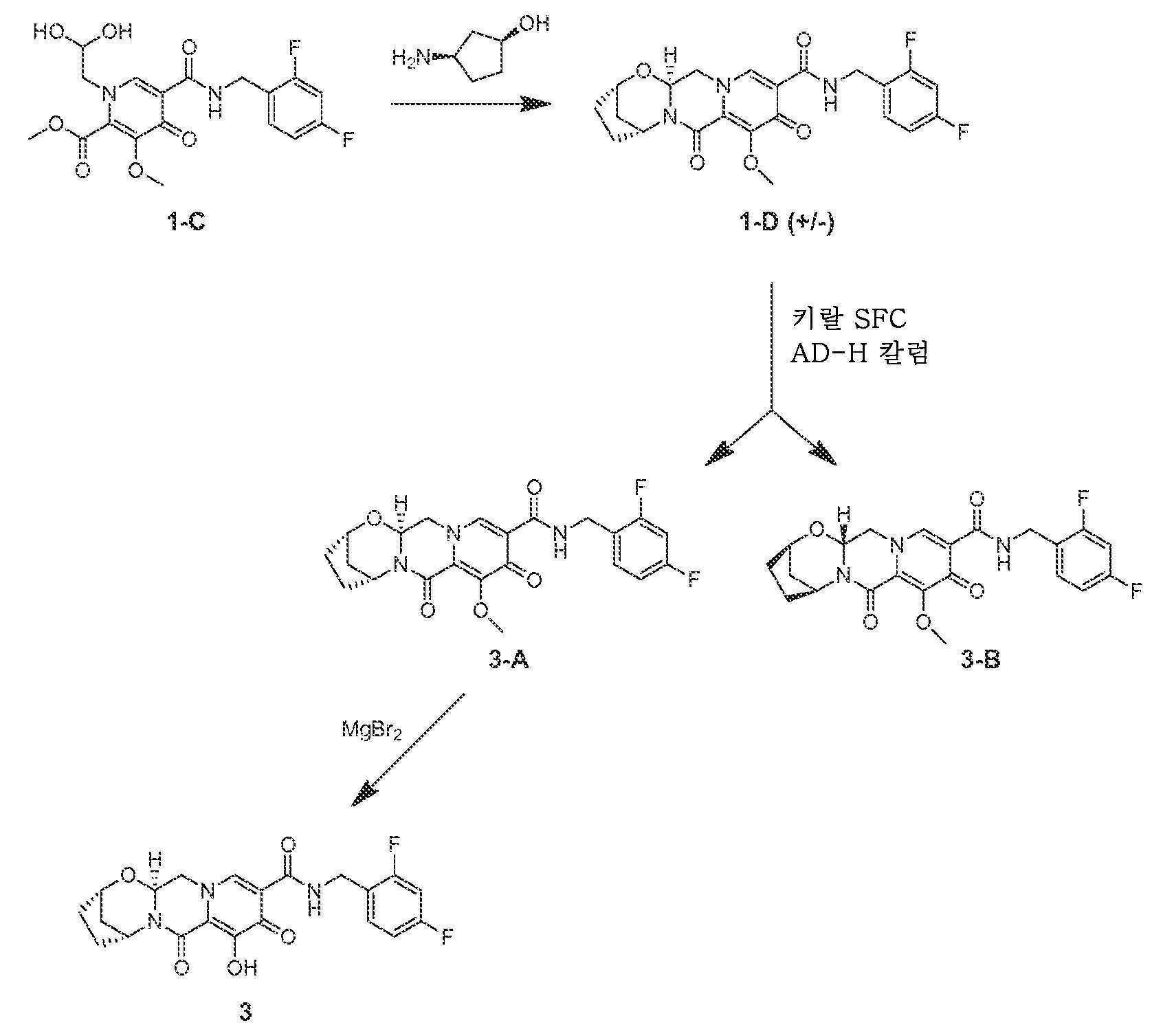
메틸 5-(2,4-디플루오로벤질카르바모일)-1-(2,2-디히드록시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트 (1-C, 0.026 mL 메탄술폰산을 함유하는 9:1 아세토니트릴:아세트산 5 mL 중 1.2 mmol)를 아세토니트릴 (5.0 mL) 및 시스-3-아미노시클로펜탄올 (0.24 g, 2.4 mmol)로 처리하였다. 반응 혼합물을 밀봉하고, 90℃로 가열하였다. 30분 후, 반응 혼합물을 냉각시키고, 탄산칼륨 (0.332 g, 2.4 mmol)으로 처리하고, 밀봉하고, 90℃로 재가열하였다. 15분 후, 혼합물을 냉각시키고, 디클로르메탄과 염산 (0.2 M 수성) 사이에 분배하였다. 유기 층을 제거하고, 수용액을 디클로로메탄으로 다시 추출하였다. 합한 유기 층을 황산나트륨 (무수) 상에서 건조시키고, 여과하고, 농축시켰다. 잔류물을 플래쉬 크로마토그래피 (디클로로메탄 중 0-8% 에탄올 (11% 포화 수성 수산화암모늄 함유))에 의해 정제하여 중간체 1-D를 수득하였다.
중간체 1-D (270 mg)를 50 mm 키랄팩 AD-H 칼럼 상에서 키랄 SFC에 의해 용리액으로서 초임계 이산화탄소 중 50% (1:1 메탄올:아세토니트릴)를 사용하여 분리하여 중간체 3-A (제1 용리 피크) 및 3-B (제2 용리 피크)를 거울상이성질체풍부화 형태로 수득하였다. 3-A의 경우: LCMS-ESI+ (m/z): [M+H]+ 계산치, C22H22F2N3O5: 446.15; 실측치: 446.2. 3-B의 경우: LCMS-ESI+ (m/z): [M+H]+ 계산치, C22H22F2N3O5: 446.15; 실측치: 446.2.
아세토니트릴 (5 mL) 중 중간체 3-A (0.110 g, 0.247 mmol)를 브로민화마그네슘 (0.091 g, 0.494 mmol)으로 조금씩 처리하고, 밀봉하고, 50℃로 가열하였다. 10분 후, 혼합물을 냉각시키고, 디클로로메탄과 염산 (0.2 M 수성) 사이에 분배하였다. 유기 층을 분리하고, 수층을 디클로로메탄으로 다시 추출하였다. 합한 유기 층을 황산나트륨 상에서 건조시키고, 여과하고, 농축시켰다. 정제용 HPLC 정제 (30-70% 아세토니트릴:물, 0.1% TFA)로 화합물 3을 거울상이성질체풍부화 형태로 수득하였다. 키랄 HPLC 체류 시간 = 6.51분 (키랄팩 AS-H, 150 x 4.6 mm, 1 mL/분 EtOH).

실시예 4
화합물 4의 제조
(1S,4R)-N-(2,4-디플루오로벤질)-7-히드록시-6,8-디옥소-3,4,6,8,12,12a-헥사히드로-2H-1,4-메타노피리도[1',2':4,5]피라지노[1,2-a]피리미딘-9-카르복스아미드

메틸 5-(2,4-디플루오로벤질카르바모일)-1-(2,2-디히드록시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트 (1-C, 0.002 mL 메탄술폰산을 함유하는 9:1 아세토니트릴:아세트산 0.53 mL 중 0.12 mmol)를 아세토니트릴에 이어서 (R)-피롤리딘-3-아민 (0.032 mL, 0.36 mmol)으로 처리하였다. 반응 혼합물을 마개를 막고, 90℃로 5.5시간 동안 가열하였다. 냉각시킨 후, 혼합물을 디클로로메탄과 중탄산나트륨 (1M 수성) 사이에 분배하였다. 유기 층을 분리하고, 수층을 에틸 아세테이트로 다시 추출하였다. 합한 유기 층을 황산나트륨 (무수) 상에서 건조시키고, 여과하고, 농축시켰다. 잔류물을 아세토니트릴 (1 mL) 중에 용해시키고, 브로민화마그네슘 (0.022 g, 0.12 mmol)으로 처리하고, 마개를 막고, 50℃로 10분 동안 가열하였다. 냉각시킨 후, 혼합물을 디클로로메탄과 염화암모늄 (포화) 사이에 분배하였다. 유기 층을 분리하고, 수층을 디클로로메탄으로 다시 추출하였다. 수성 층을 HCl (수성)을 사용하여 pH = 1로 조정하고, 디클로로메탄을 사용하여 다시 추출하였다. 수용액을 NaOH (수성)을 사용하여 pH = 3으로 조정하고, 디클로로메탄을 사용하여 다시 추출하였다. 합한 유기 층을 황산나트륨 상에서 건조시키고, 여과하고, 농축시켰다. 정제용 HPLC 정제 (10-55% 아세토니트릴:물, 0.1% TFA)로 화합물 4를 수득하였다.

실시예 5
화합물 5의 제조
(4R,12aS)-N-(1-(2,4-디플루오로페닐)시클로프로필)-7-히드록시-4-메틸-6,8-디옥소-3,4,6,8,12,12a-헥사히드로-2H-[1,3]옥사지노[3,2-d]피리도[1,2-a]피라진-9-카르복스아미드
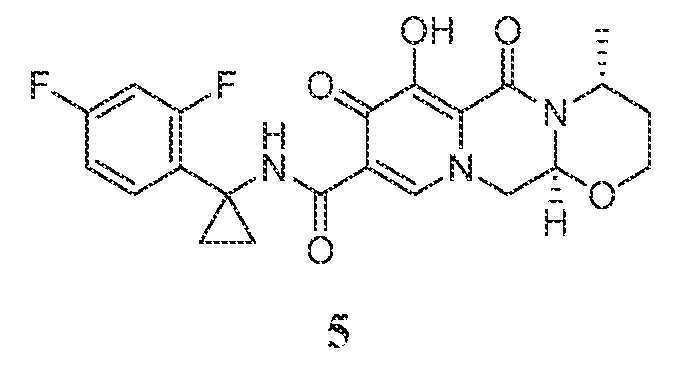

단계 1
(4R,12aS)-7-메톡시-4-메틸-6,8-디옥소-3,4,6,8,12,12a-헥사히드로-2H-[1,3]옥사지노[3,2-d]피리도[1,2-a]피라진-9-카르복실산 (중간체 5-A)을 WO2011/119566에 기재된 바와 같이 (3S,11aR)-6-메톡시-3-메틸-5,7-디옥소-2,3,5,7,11,11a-헥사히드로옥사졸로[3,2-d]피리도[1,2-a]피라진-8-카르복실산과 유사한 방식으로 (S)-2-아미노프로판-1-올을 (R)-3-아미노부탄-1-올로 치환하여 제조하였다. WO2011/119566은 그 전문이 본원에 참조로 포함된다. CH2Cl2 (2 mL) 중 중간체 5-A (24.8 mg, 0.080 mmol), 1-(2,4-디플루오로페닐)시클로프로판아민 HCl 염 (5-B, 21.9 mg, 0.107 mmol), 및 HATU (48 mg, 0.126 mmol)의 현탁액을 주위 온도에서 교반하면서 N,N-디이소프로필에틸아민 (DIPEA) (0.1 mL, 0.574 mmol)을 첨가하였다. 30분 후, 반응 혼합물을 에틸 아세테이트로 희석한 후 10% 수성 시트르산 용액 (x1) 및 포화 수성 NaHCO3 용액 (x1)으로 세척하였다. 수성 분획을 에틸 아세테이트 (x1)로 추출한 후, 유기 분획을 합하고, 건조 (MgSO4)시키고, 농축시켰다. 잔류물을 콤비플래쉬 (12 g 칼럼)에 의해 헥산, 에틸 아세테이트, 및 에틸 아세테이트 중 20% 메탄올을 사용하여 정제하여 (4R,12aS)-N-(1-(2,4-디플루오로페닐)시클로프로필)-7-메톡시-4-메틸-6,8-디옥소-3,4,6,8,12,12a-헥사히드로-2H-[1,3]옥사지노[3,2-d]피리도[1,2-a]피라진-9-카르복스아미드, 중간체 5-C를 수득하였다.
단계 2
아세토니트릴 (2 mL) 중 중간체 5-C (39 mg, 0.080 mmol) 및 브로민화마그네슘 (42 mg, 0.2282 mmol)의 현탁액을 50℃에서 교반하였다. 1시간 후, 1 N HCl (2 mL)을 첨가하고 반응 혼합물을 0℃ 조에서 교반하였다. 생성된 혼합물을 물 (~20 mL)로 희석한 후, 생성물을 디클로로메탄 (x3)으로 추출하고, 합한 추출물을 건조 (MgSO4)시키고, 농축시켰다. 잔류물을 정제용 HPLC에 의해 정제하여 (4R,12aS)-N-(1-(2,4-디플루오로페닐)시클로프로필)-7-히드록시-4-메틸-6,8-디옥소-3,4,6,8,12,12a-헥사히드로-2H-[1,3]옥사지노[3,2-d]피리도[1,2-a]피라진-9-카르복스아미드, 화합물 5를 TFA 염으로서 수득하였다.

실시예 6
화합물 6의 제조
(1R,4S)-N-(2,4-디플루오로벤질)-7-히드록시-6,8-디옥소-3,4,6,8,12,12a-헥사히드로-2H-1,4-메타노피리도[1',2':4,5]피라지노[1,2-a]피리미딘-9-카르복스아미드

메틸 5-(2,4-디플루오로벤질카르바모일)-1-(2,2-디히드록시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트 (1-C, 0.100 g, 0.243 mmol), (S)-피롤리딘-3-아민 (0.043 mL, 0.485 mmol) 및 탄산칼륨 (0.067 g, 0.485 mmol)을 아세토니트릴 (1.9 mL) 및 아세트산 (0.1 mL) 중에 현탁시키고, 90℃로 1.5시간 동안 가열하였다. 냉각시킨 후, 혼합물을 브로민화마그네슘 (0.090 g)으로 처리하고, 50℃로 30분 동안 가열하였다. 냉각시킨 후, 혼합물을 디클로로메탄과 0.2 M HCl 사이에 분배하였다. 유기 층을 분리하고, 수층을 디클로로메탄으로 다시 추출하였다. 합한 유기 층을 황산나트륨 (무수) 상에서 건조시키고, 여과하고, 농축시켰다. 정제용 HPLC 정제 (25-50% 아세토니트릴:물, 0.1% TFA)로 화합물 6을 수득하였다.

실시예 7
화합물 7의 제조
(2S,6R)-N-(2,4-디플루오로벤질)-9-히드록시-8,10-디옥소-3,4,5,6,8,10,14,14a-옥타히드로-2H-2,6-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사조신-11-카르복스아미드

메틸 5-(2,4-디플루오로벤질카르바모일)-1-(2,2-디히드록시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트 (1-C, 0.050 g, 0.121 mmol), (1S,3R)-3-아미노시클로헥산올 (0.028 g, 0.243 mmol) 및 탄산칼륨 (0.034 g, 0.243 mmol)을 아세토니트릴 (0.95 mL) 중에 현탁시키고, 90℃로 0.5시간 동안 가열하였다. 냉각시킨 후, 아세트산 (0.050 mL)을 첨가하고, 혼합물을 90℃로 2시간 동안 재가열하였다. 냉각시킨 후, 혼합물을 브로민화마그네슘 (0.044 g)으로 처리하고, 50℃로 1시간 동안 가열하였다. 냉각시킨 후, 제2 부분의 브로민화마그네슘 (0.044 g)을 첨가하고, 혼합물을 50℃로 15분 동안 재가열하였다. 냉각시킨 후, 혼합물을 디클로로메탄과 0.2 M HCl 사이에 분배하였다. 유기 층을 분리하고, 수층을 디클로로메탄으로 다시 추출하였다. 합한 유기 층을 황산나트륨 (무수) 상에서 건조시키고, 여과하고, 농축시켰다. 정제용 HPLC 정제 (40-80% 아세토니트릴:물, 0.1% TFA)로 화합물 7을 수득하였다.

실시예 8
화합물 8의 제조
(2R,6S)-N-(2,4-디플루오로벤질)-9-히드록시-8,10-디옥소-3,4,5,6,8,10,14,14a-옥타히드로-2H-2,6-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사조신-11-카르복스아미드
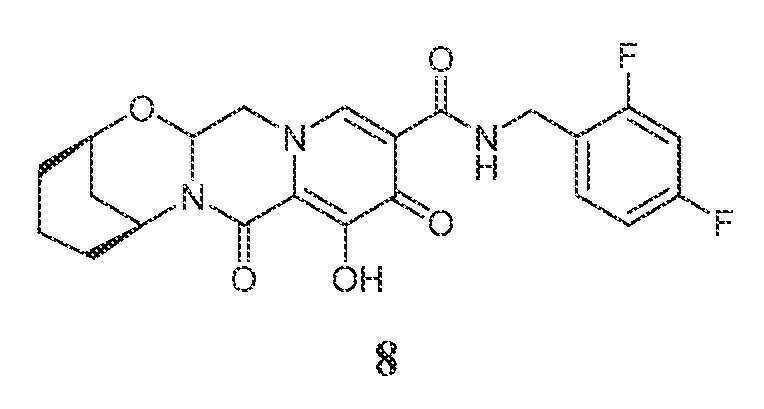
화합물 8을 화합물 7과 유사한 방식으로 (1S,3R)-3-아미노시클로헥산올 대신 (1R,3S)-3-아미노시클로헥산올을 사용하여 제조하였다.

실시예 9 및 10
화합물 9 및 10의 제조
(2S,5R,13aS)-N-((R)-1-(4-플루오로페닐)에틸)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드 9 및 (2R,5S,13aR)-N-((R)-1-(4-플루오로페닐)에틸)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드 10
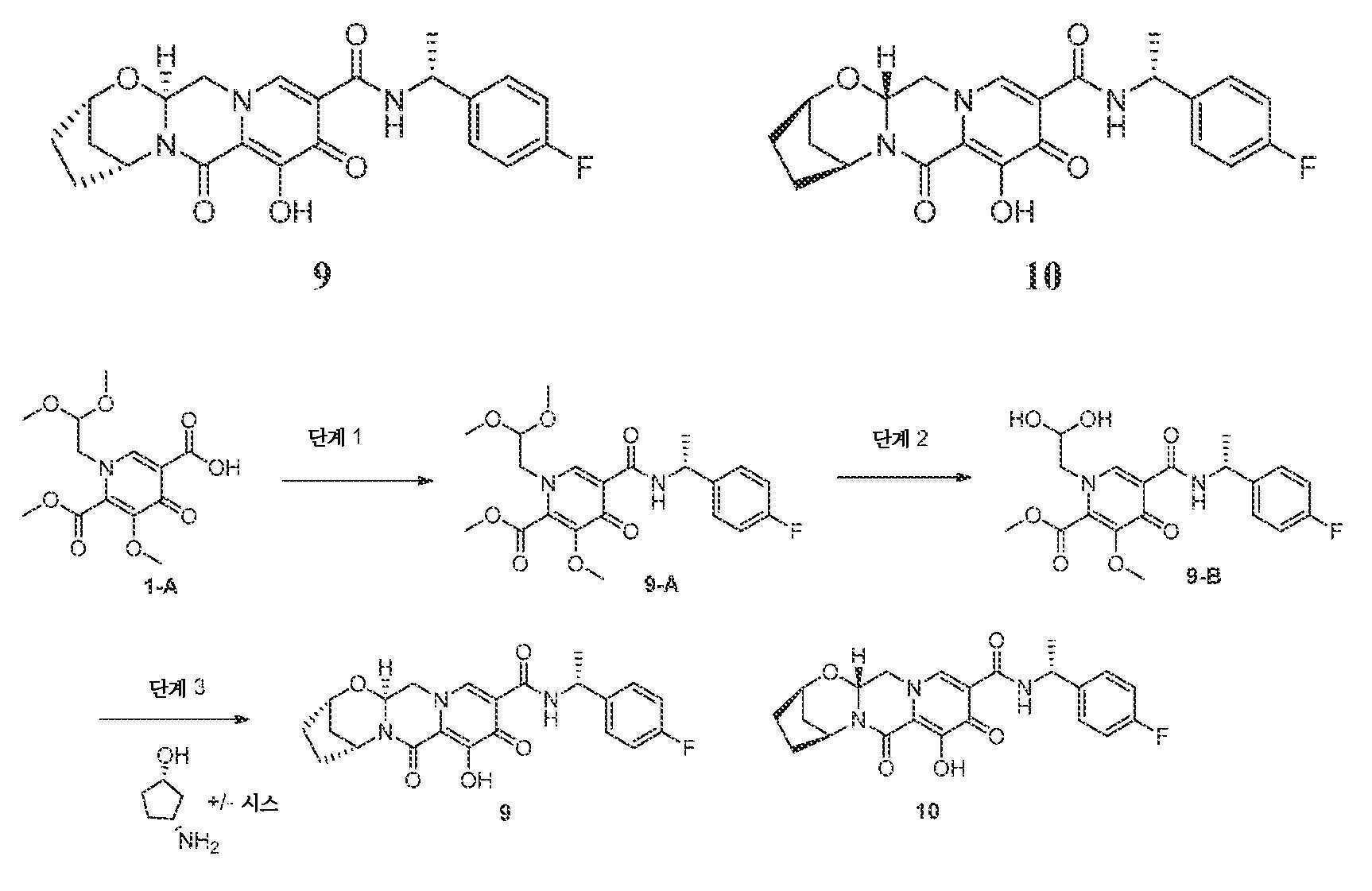
단계 1
1-(2,2-디메톡시에틸)-5-메톡시-6-(메톡시카르보닐)-4-옥소-1,4-디히드로피리딘-3-카르복실산 (1-A, 0.500 g, 1.59 mmol)을 아세토니트릴 (6 mL) 중에 현탁시키고, N,N-디이소프로필에틸아민 (DIPEA) (0.550 mL, 3.17 mmol), (R)-1-(4-플루오로페닐)에탄아민 (0.242 mg, 1.74 mmol) 및 HATU (0.661 g, 1.74 mmol)로 처리하였다. 반응 혼합물을 2시간 동안 교반하고, 에틸 아세테이트와 물 사이에 분배하였다. 유기 층을 분리하고, HCl (10% 수성), 중탄산나트륨 (1M 수성)으로 세척하고, 황산나트륨 상에서 건조시키고, 여과하고, 농축시켜 조 (R)-메틸 1-(2,2-디메톡시에틸)-5-(1-(4-플루오로페닐)에틸카르바모일)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트를 수득하였으며, 이를 후속 단계에 정제 없이 사용하였다:
단계 2
(R)-메틸 1-(2,2-디메톡시에틸)-5-(1-(4-플루오로페닐)에틸카르바모일)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트를 아세토니트릴 (5.7 mL) 및 아세트산 (0.6 mL) 중에 현탁시키고, 메탄 술폰산 (0.031 mL, 0.477 mmol)으로 처리하였다. 혼합물을 마개를 막고, 75℃로 가열하였다. 7시간 후, 혼합물을 냉각시키고, 후속 단계에 정제 없이 사용하였다:
단계 3
(R)-메틸 1-(2,2-디히드록시에틸)-5-(1-(4-플루오로페닐)에틸카르바모일)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트 (단계 2로부터의 조 혼합물 3.6 mL, 0.8 mmol)를 아세토니트릴 (3.6 mL)로 희석하고, 시스-3-아미노시클로펜탄올, HCl 염 (0.219 g, 1.6 mmol) 및 탄산칼륨 (0.276 g, 2.0 mmol)으로 처리하였다. 혼합물을 마개를 막고, 90℃로 가열하였다. 20분 후, 반응 혼합물을 냉각시키고, 디클로로메탄과 HCl (0.2 M 수성) 사이에 분배하였다. 층을 분리하고, 수성 층을 디클로로메탄으로 다시 추출하였다. 합한 유기 층을 소량의 아세토니트릴로 처리하고, 황산나트륨 상에서 건조시키고, 여과하고, 농축시켰다.
잔류물을 아세토니트릴 (4 mL) 중에 현탁시키고, 브로민화마그네슘 (0.177 g)으로 처리하였다. 혼합물을 마개를 막고, 50℃로 가열하였다. 10분 후, 반응 혼합물을 냉각시키고, 디클로로메탄과 HCl (0.2 M 수성) 사이에 분배하였다. 층을 분리하고, 수성 층을 디클로르메탄으로 다시 추출하였다. 합한 유기 층을 황산나트륨 상에서 건조시키고, 여과하고, 농축시켰다. 잔류물을 실리카 겔 상에서 플래쉬 크로마토그래피 (0-8% 에탄올:DCM)에 의해 정제하여 목적하는 9 및 10의 부분입체이성질체 혼합물을 수득하였다.
혼합물을 키랄팩 AD-H를 사용하는 키랄 HPLC에 의해 용리액으로서 100% 에탄올로 분리하여 화합물 9 및 10을 거울상이성질체적으로 풍부한 형태로 수득하였다:
화합물 9의 경우: LCMS-ESI+ (m/z): [M+H]+ 계산치, C22H23FN3O5: 428.16; 실측치: 428.1. 키랄 HPLC 체류 시간 = 10.177분 (키랄팩 AD-H, 150 x 4.6 mm, 1 mL/분 EtOH).

화합물 10의 경우: LCMS-ESI+ (m/z): [M+H]+ 계산치, C22H23FN3O5: 428.16; 실측치: 428.1. 키랄 HPLC 체류 시간 = 14.061분 (키랄팩 AD-H, 150 x 4.6 mm, 1 mL/분 EtOH).

실시예 11
화합물 11의 제조
(2S,5R,13aS)-N-((R)-1-(2,4-디플루오로페닐)에틸)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

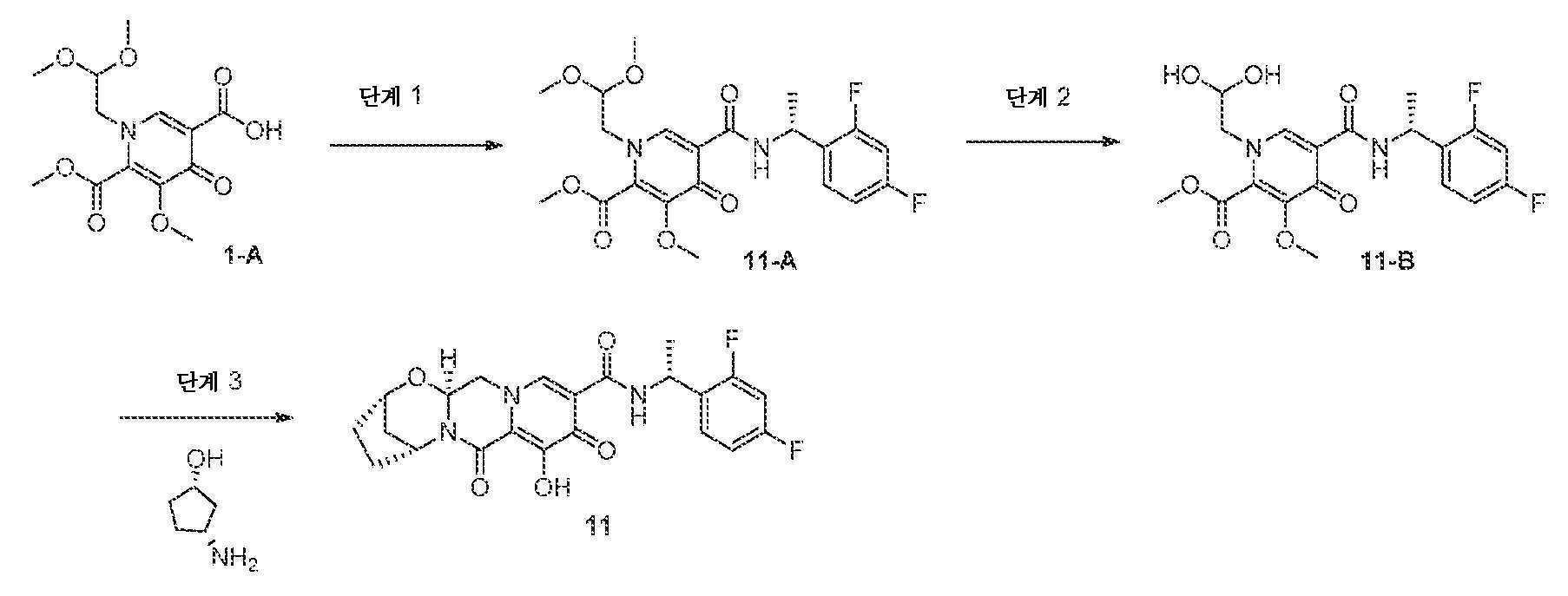
단계 1
1-(2,2-디메톡시에틸)-5-메톡시-6-(메톡시카르보닐)-4-옥소-1,4-디히드로피리딘-3-카르복실산 (1-A, 0.315 g, 1.00 mmol)을 아세토니트릴 (4 mL) 중에 현탁시키고, N,N-디이소프로필에틸아민 (DIPEA) (0.348 mL, 2.00 mmol), (R)-1-(2,4-디플루오로페닐)에탄아민 HCl 염 (0.213 mg, 1.10 mmol) 및 HATU (0.418 g, 1.10 mmol)로 처리하였다. 반응 혼합물을 1시간 동안 교반하고, 디클로로메탄과 HCl (10% 수성) 사이에 분배하였다. 유기 층을 분리하고, 중탄산나트륨 (1M 수성)으로 세척하고, 황산나트륨 상에서 건조시키고, 여과하고, 농축시켜 조 (R)-메틸 5-(1-(2,4-디플루오로페닐)에틸카르바모일)-1-(2,2-디메톡시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트를 수득하였고, 이를 후속 단계에 정제 없이 사용하였다.
단계 2
(R)-메틸 5-(1-(2,4-디플루오로페닐)에틸카르바모일)-1-(2,2-디메톡시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트를 아세토니트릴 (3.6 mL) 및 아세트산 (0.4 mL) 중에 현탁시키고, 메탄 술폰산 (0.020 mL)으로 처리하였다. 혼합물을 마개를 막고, 75℃로 가열하였다. 16시간 후, 조 혼합물을 냉각시키고, 후속 단계에 정제 없이 사용하였다.
단계 3
(R)-메틸 5-(1-(2,4-디플루오로페닐)에틸카르바모일)-1-(2,2-디히드록시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트 (단계 2로부터의 조 혼합물의 절반, 대략 0.5 mmol)를 아세토니트릴 (2.5 mL)로 희석하고, (1S,3R)-3-아미노시클로펜탄올 (0.110 g, 1.09 mmol) 및 탄산칼륨 (0.069 g, 0.50 mmol)으로 처리하였다. 혼합물을 마개를 막고, 90℃로 가열하였다. 15분 후, 반응 혼합물을 냉각시키고, 브로민화마그네슘 (0.184 g)을 첨가하였다. 반응 혼합물을 50℃로 가열하였다. 10분 후, 혼합물을 냉각시키고, 추가의 브로민화마그네슘 부분 (0.184 g)으로 처리하였다. 반응 혼합물을 50℃로 재가열하고, 10분 동안 교반하였다. 냉각시킨 후, 혼합물을 디클로로메탄과 HCl (0.2 M 수성) 사이에 분배하였다. 층을 분리하고, 수성 층을 디클로로메탄으로 다시 추출하였다. 합한 유기 층을 황산나트륨 상에서 건조시키고, 여과하고, 농축시켰다. 정제용 HPLC 정제 (30-60% 아세토니트릴:물, 0.1% TFA)로 목적 화합물 11을 수득하였다.

실시예 12
화합물 12의 제조
(2R,5S,13aR)-N-((R)-1-(2,4-디플루오로페닐)에틸)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

화합물 12를 화합물 11과 유사한 방식으로 (1S,3R)-3-아미노시클로펜탄올 대신 (1R,3S)-3-아미노시클로펜탄올을 사용하여 제조하였다.

실시예 13
화합물 13의 제조
(2S,5R,13aS)-N-((S)-1-(2,4-디플루오로페닐)에틸)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

화합물 13을 화합물 11과 유사한 방식으로 (R)-1-(2,4-디플루오로페닐)에탄아민 대신 (S)-1-(2,4-디플루오로페닐)에탄아민을 사용하고 단지 단일 부분의 브로민화마그네슘 (0.184 g)을 사용하여 제조하였다.

실시예 14
화합물 14의 제조
(2R,5S,13aR)-N-((S)-1-(2,4-디플루오로페닐)에틸)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드
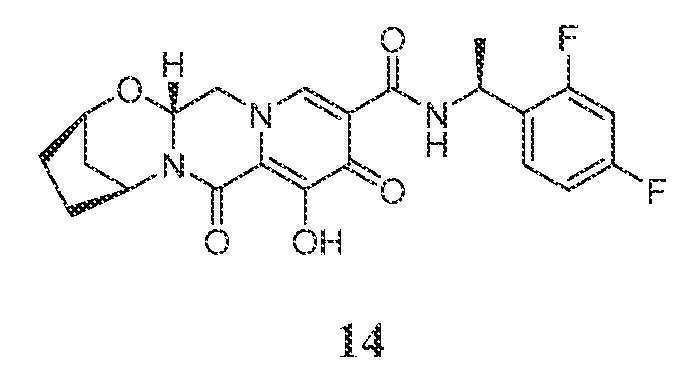
화합물 14를 화합물 11과 유사한 방식으로 (R)-1-(2,4-디플루오로페닐)에탄아민 대신 (S)-1-(2,4-디플루오로페닐)에탄아민을 사용하고 (1S,3R)-3-아미노시클로펜탄올 대신 (1R,3S)-3-아미노시클로펜탄올을 사용하여 제조하였다.

실시예 15
화합물 15의 제조
(2S,5R,13aS)-N-(4-플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드


단계 1
아세토니트릴 (36 mL) 및 아세트산 (4 mL) 중에 현탁시킨 1-(2,2-디메톡시에틸)-5-메톡시-6-(메톡시카르보닐)-4-옥소-1,4-디히드로피리딘-3-카르복실산 (1-A, 3.15 g, 10.0 mmol)을 메탄 술폰산 (0.195 mL)으로 처리하였다. 혼합물을 75℃로 가열하였다. 7시간 후, 조 혼합물을 냉각시키고, -10℃에서 3일 동안 보관하였다. 조 혼합물을 75℃로 2시간 동안 재가열하고, 냉각시키고, 후속 단계에 정제 없이 사용하였다.
단계 2
조 1-(2,2-디히드록시에틸)-5-메톡시-6-(메톡시카르보닐)-4-옥소-1,4-디히드로피리딘-3-카르복실산 (단계 1로부터의 조 혼합물의 16.8 mL, 대략 4 mmol)을 (1S,3R)-3-아미노시클로펜탄올 (0.809 g, 8 mmol)과 합하고, 아세토니트릴 (16.8 mL)로 희석하고, 탄산칼륨 (0.553 g, 4 mmol)으로 처리하였다. 반응 혼합물을 85℃로 가열하고, 15분 동안 교반하고, 주위 온도로 냉각시키고, 추가로 16시간 동안 교반하였다. HCl (50 mL, 0.2M 수성)을 첨가하고, 투명한 황색 용액을 디클로로메탄으로 3회 추출하였다. 합한 유기부를 황산나트륨 상에서 건조시키고, 여과하고, 황색 고체로 농축시켰다. 이 조 물질을 디클로로메탄/헥산으로부터 침전시켜 목적 중간체 15-B를 담베이지색 분말로서 수득하였다.
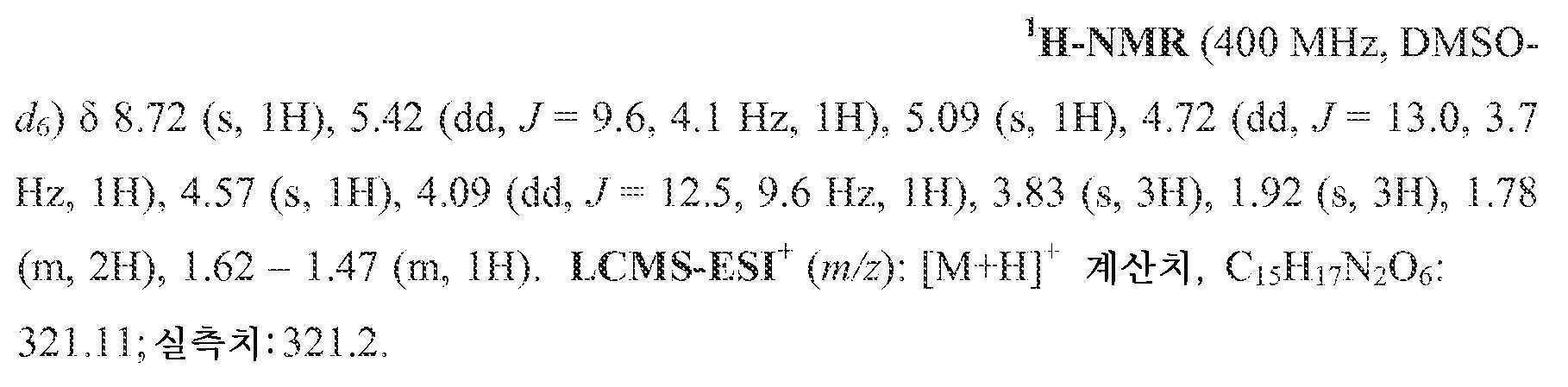
단계 3
중간체 15-B (0.040 g, 0.125 mmol) 및 (4-플루오로페닐)메탄아민 (0.017 g, 0.137 mmol)을 아세토니트릴 (1 mL) 중에 현탁시키고, N,N-디이소프로필에틸아민 (DIPEA) (0.033 mL, 0.187 mmol) 및 HATU (0.052 g, 0.137 mmol)로 처리하였다. 30분 동안 교반한 후, 반응 혼합물을 브로민화마그네슘 (0.046 g, 0.25 mmol)으로 처리하고, 50℃로 가열하였다. 10분 후, 반응 혼합물을 냉각시키고, HCl (2 mL, 10% 수성)로 처리하였다. 몇분 후, 침전물을 여과하고, HCl (10% 수성) 및 물로 세척하였다. 침전물의 정제용 HPLC 정제 (20-65% 아세토니트릴:물, 0.1% TFA)로 목적 화합물 15를 수득하였다.

실시예 16
화합물 16의 제조
(2S,5R,13aS)-N-(2,3-디플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드
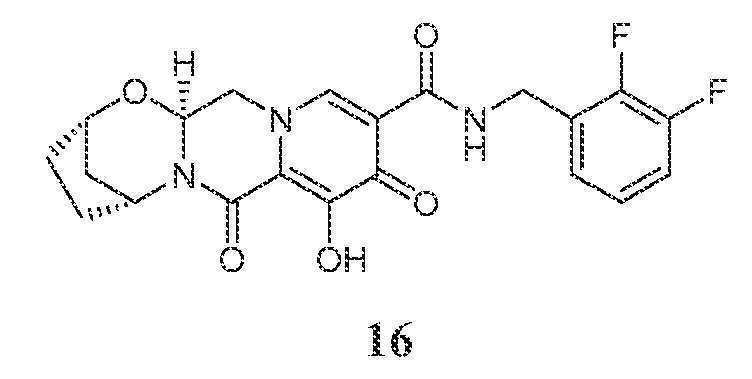
화합물 16을 화합물 15와 유사한 방식으로 (4-플루오로페닐)메탄아민 대신 (2,3-디플루오로페닐)메탄아민을 사용하여 제조하였다.

실시예 17
화합물 17의 제조
(2S,5R,13aS)-N-(4-클로로-2-플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

화합물 17을 화합물 15와 유사한 방식으로 (4-플루오로페닐)메탄아민 대신 (4-클로로-2-플루오로페닐)메탄아민을 사용하여 제조하였다.

실시예 18
화합물 18의 제조
(2S,5R,13aS)-N-(3,4-디플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드
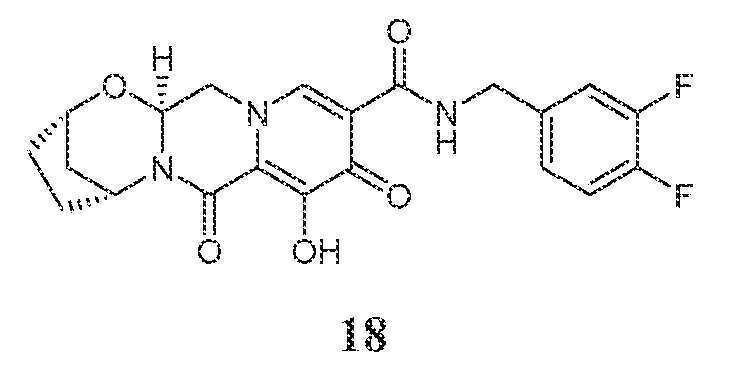
화합물 18을 화합물 15와 유사한 방식으로 (4-플루오로페닐)메탄아민 대신 (3,4-디플루오로페닐)메탄아민을 사용하여 제조하였다.

실시예 19
화합물 19의 제조
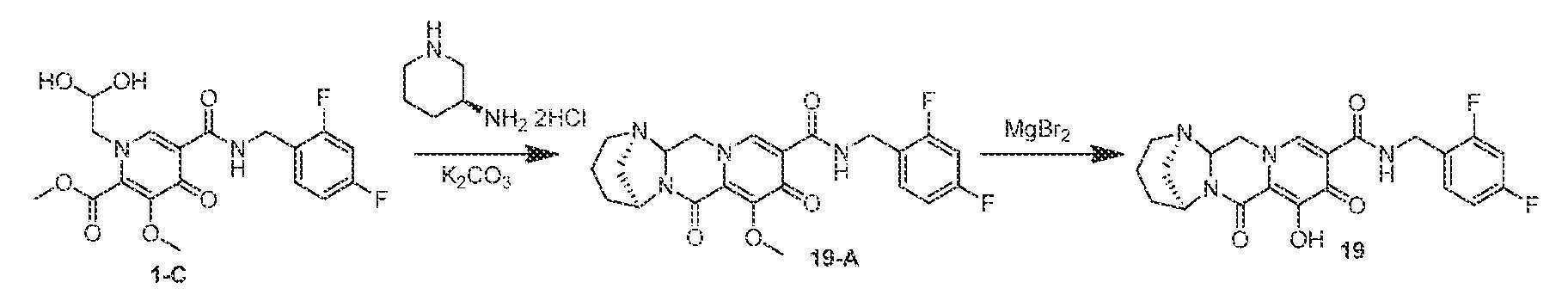
(1R,5S)-N-(2,4-디플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-1,5-메타노피리도[1',2':4,5]피라지노[1,2-a][1,3]디아제핀-10-카르복스아미드

단계 1 및 2
메틸 5-(2,4-디플루오로벤질카르바모일)-1-(2,2-디히드록시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트 (1-C, 97.5 mg, 0.236 mmol)를 아세토니트릴 (1.9 mL), 아세트산 (0.1 mL), 탄산칼륨 (145 mg, 1.05 mmol), 및 (S)-피페리딘-3-아민 디히드로클로라이드 (82 mg, 0.472 mmol)로 처리하였다. 반응 혼합물을 밀봉하고, 90℃로 가열하였다. 60분 후, 반응 혼합물을 냉각시키고, 염수와 디클로로메탄 사이에 분배하였다. 수성 상을 디클로로메탄으로 3회 추출하고, 합한 유기 상을 합하고, MgSO4 상에서 건조시키고, 여과하고, 농축시켰다. 조 생성물을 아세토니트릴 (2 mL) 중에 용해시키고, 브로민화마그네슘 (89.1 mg, 0.48 mmol)을 첨가하였다. 혼합물을 재밀봉하고, 50℃로 가열하였다. 90분 후, 반응 혼합물을 0.2M HCl(수성) ~5 mL로 켄칭하고, pH를 ~10으로 조정하고, 염수로 희석하고, DCM으로 3회 추출하였다. HPLC 정제 (아세토니트릴:물, 0.1% TFA)로 화합물 19를 수득하였다.

실시예 20
화합물 20의 제조
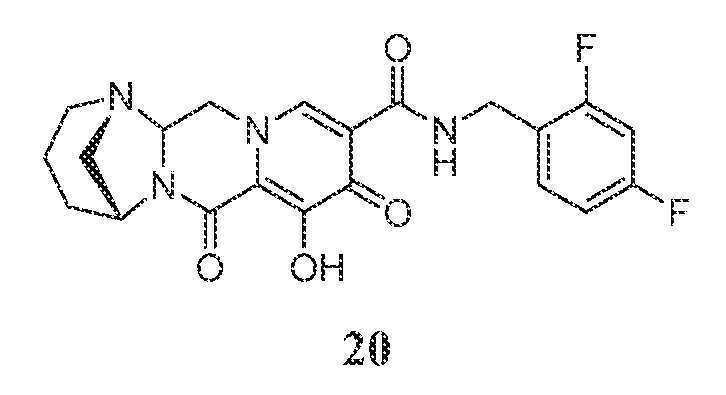
(1S,5R)-N-(2,4-디플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-1,5-메타노피리도[1',2':4,5]피라지노[1,2-a][1,3]디아제핀-10-카르복스아미드

단계 1 및 2
메틸 5-(2,4-디플루오로벤질카르바모일)-1-(2,2-디히드록시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트 (1-C, 103.3 mg, 0.25 mmol)를 아세토니트릴 (1.9 mL), 아세트산 (0.1 mL), 탄산칼륨 (159.8 mg, 1.16 mmol), 및 (R)-피페리딘-3-아민 디히드로클로라이드 (90 mg, 0.52 mmol)로 처리하였다. 반응 혼합물을 밀봉하고, 90℃로 가열하였다. 40분 후, 반응 혼합물을 냉각시키고, 염수와 디클로로메탄 사이에 분배하였다. 수성 상을 디클로로메탄으로 3회 추출하고, 합한 유기 상을 합하고, MgSO4 상에서 건조시키고, 여과하고, 농축시켰다. 조 생성물을 아세토니트릴 (2 mL) 중에 용해시키고, 브로민화마그네슘 (96.5 mg, 0.52 mmol)을 첨가하였다. 혼합물을 재밀봉하고, 50℃로 가열하였다. 80분 후, 반응 혼합물을 0.2M HCl (수성) ~5 mL로 켄칭하고, pH를 ~10로 조정하고, 염수로 희석하고, DCM으로 3회 추출하였다. HPLC 정제 (아세토니트릴:물, 0.1% TFA)로 화합물 20을 수득하였다.

실시예 21
화합물 21의 제조
(2S,5R,13aS)-N-((S)-1-(4-플루오로페닐)에틸)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드


단계 1 및 2
(S)-메틸 1-(2,2-디히드록시에틸)-5-(1-(4-플루오로페닐)에틸카르바모일)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트 (21-A, 1 mL, 19:1 아세토니트릴:아세트산 중 0.23 M 용액, 실시예 9로부터 (R)-메틸 1-(2,2-디히드록시에틸)-5-(1-(4-플루오로페닐)에틸카르바모일)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트 9-A에 따라 (R)-1-(4-플루오로페닐)에탄아민 대신 (S)-1-(4-플루오로페닐)에탄아민을 사용하여 제조됨)를 (1S,3R)-3-아미노시클로펜탄올 (62 mg, 0.61 mmol) 및 탄산칼륨 (34 mg, 0.25 mmol)으로 처리하였다. 반응 혼합물을 밀봉하고, 90℃로 가열하였다. 60분 후, 반응 혼합물을 냉각시키고, 염수와 디클로로메탄 사이에 분배하였다. 수성 상을 디클로로메탄으로 3회 추출하고, 합한 유기 상을 합하고, MgSO4 상에서 건조시키고, 여과하고, 농축시켰다. 조 생성물을 아세토니트릴 (2 mL) 중에 용해시키고, 브로민화마그네슘 (74 mg, 0.4 mmol)을 첨가하였다. 혼합물을 재밀봉하고, 50℃로 가열하였다. 100분 후, 반응 혼합물을 0.2M HCl (수성)로 켄칭하고, 염수로 희석하고, DCM으로 3회 추출하였다. HPLC 정제 (아세토니트릴:물, 0.1% TFA)로 화합물 21을 수득하였다.

실시예 22
화합물 22의 제조
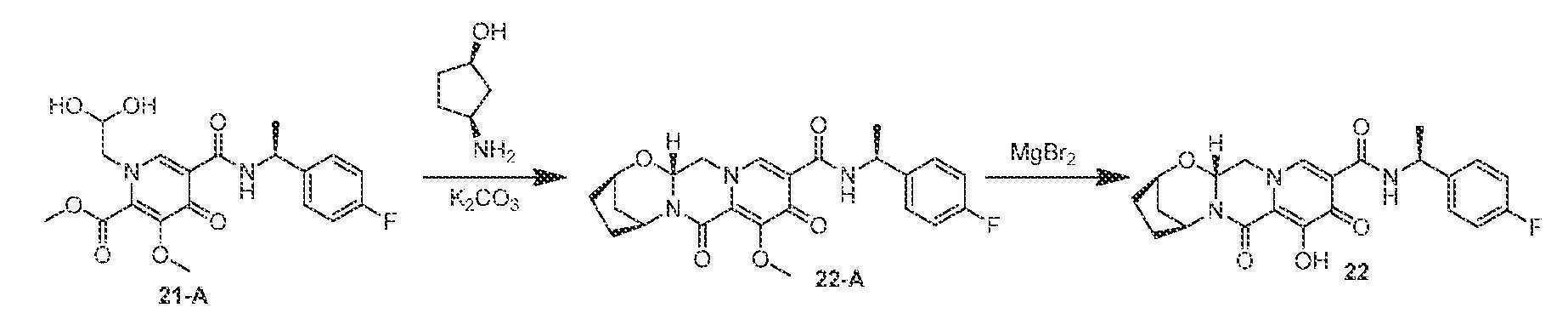
(2R,5S,13aR)-N-((S)-1-(4-플루오로페닐)에틸)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

단계 1 및 2
(S)-메틸 1-(2,2-디히드록시에틸)-5-(1-(4-플루오로페닐)에틸카르바모일)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트 (21-A, 1 mL, 19:1 아세토니트릴:아세트산 중 0.23 M 용액)을 (1R,3S)-3-아미노시클로펜탄올 (52 mg, 0.51 mmol) 및 탄산칼륨 (31 mg, 0.22 mmol)으로 처리하였다. 반응 혼합물을 밀봉하고, 90℃로 가열하였다. 60분 후, 반응 혼합물을 냉각시키고, 염수와 디클로로메탄 사이에 분배하였다. 수성 상을 디클로로메탄으로 3회 추출하고, 합한 유기 상을 합하고, MgSO4 상에서 건조시키고, 여과하고, 농축시켰다. 조 생성물을 아세토니트릴 (2 mL) 중에 용해시키고, 브로민화마그네슘 (91 mg, 0.49 mmol)을 첨가하였다. 혼합물을 재밀봉하고, 50℃로 가열하였다. 100분 후, 반응 혼합물을 0.2M HCl(수성)로 켄칭하고, 염수로 희석하고, DCM으로 3회 추출하였다. HPLC 정제 (아세토니트릴:물, 0.1% TFA)로 화합물 22를 수득하였다.

실시예 23
화합물 23의 제조
(2S,5R,13aS)-N-(2-플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드


단계 1 및 2
15-B (41 mg, 0.13 mmol)를 아세토니트릴 (1 mL), (2-플루오로페닐)메탄아민 (17 mg, 0.14 mmol), HATU (67 mg, 0.18 mmol), 및 N,N-디이소프로필에틸아민 (DIPEA) (24 mg, 0.19 mmol)으로 처리하였다. 반응 혼합물을 실온에서 1시간 동안 교반하고, 브로민화마그네슘 (47 mg, 0.26 mmol)을 첨가하였다. 혼합물을 밀봉하고, 50℃로 가열하였다. 60분 후, 반응 혼합물을 0.2M HCl (수성)로 켄칭하고, 염수로 희석하고, DCM으로 3회 추출하였다. HPLC 정제 (아세토니트릴:물, 0.1% TFA)로 화합물 23을 수득하였다.

실시예 24
화합물 24의 제조
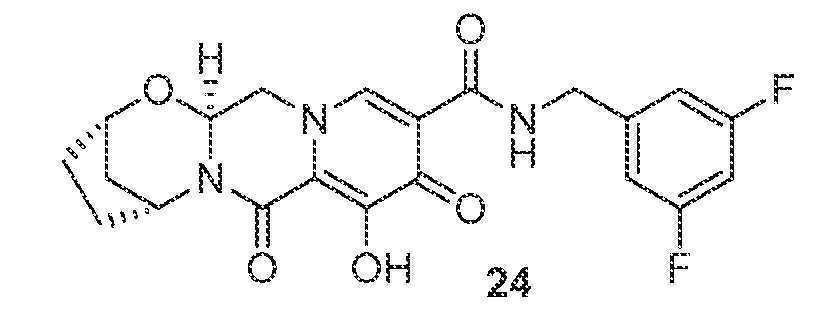
(2S,5R,13aS)-N-(3,5-디플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

단계 1 및 2
15-B (44 mg, 0.14 mmol)를 아세토니트릴 (1 mL), (3,5-디플루오로페닐)메탄아민 (32 mg, 0.23 mmol), HATU (54 mg, 0.14 mmol), 및 N,N-디이소프로필에틸아민 (37 mg, 0.29 mmol)으로 처리하였다. 반응 혼합물을 실온에서 1시간 동안 교반하고, 브로민화마그네슘 (57 mg, 0.31 mmol)을 첨가하였다. 혼합물을 밀봉하고, 50℃로 가열하였다. 60분 후, 반응 혼합물을 0.2M HCl (수성)로 켄칭하고, 염수로 희석하고, DCM으로 3회 추출하였다. HPLC 정제 (아세토니트릴:물, 0.1% TFA)로 화합물 24를 수득하였다.

실시예 25
화합물 25의 제조
(2S,5R,13aS)-N-(4-플루오로-3-(트리플루오로메틸)벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

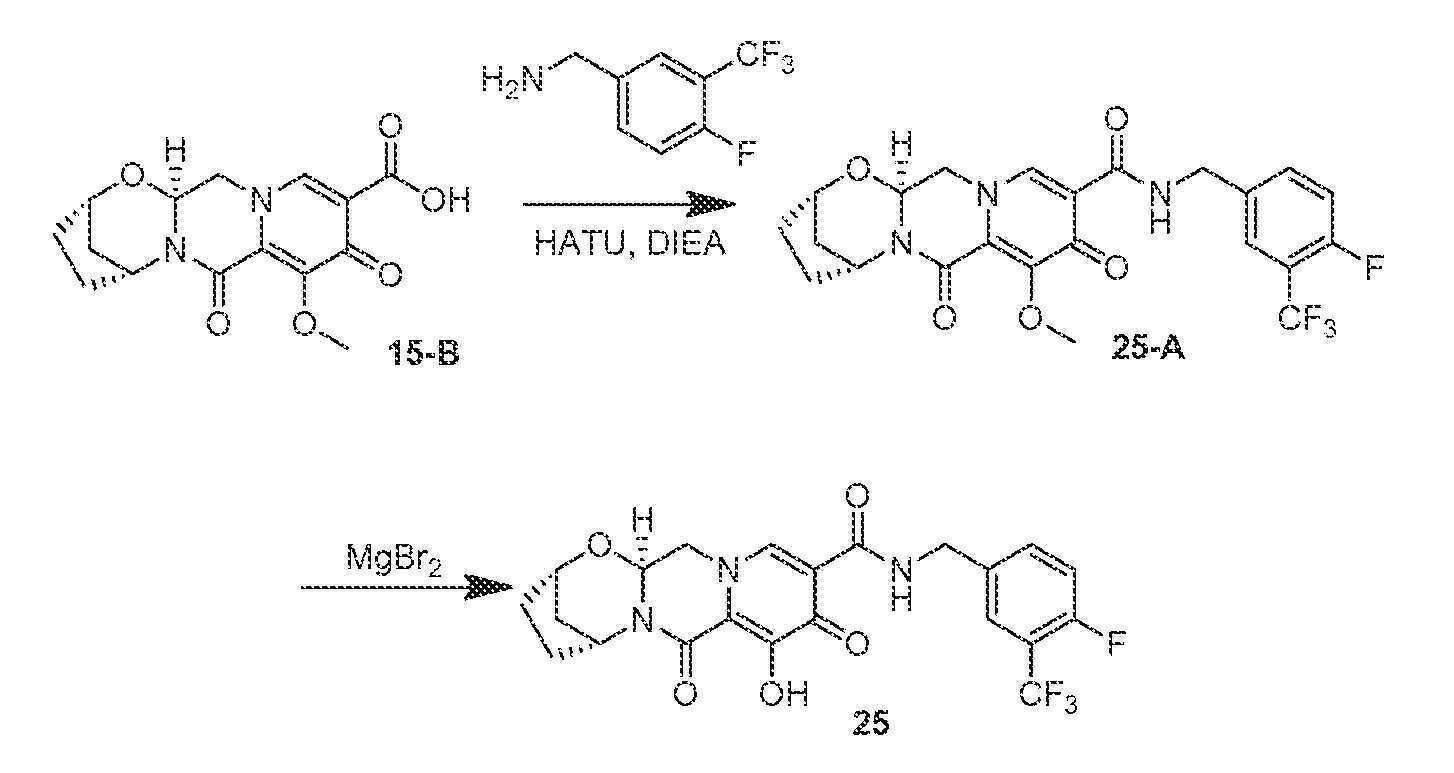
단계 1 및 2
15-B (43 mg, 0.13 mmol)를 아세토니트릴 (1 mL), (4-플루오로-3-(트리플루오로메틸)페닐)메탄아민 (29 mg, 0.15 mmol), HATU (62 mg, 0.16 mmol), 및 N,N-디이소프로필에틸아민 (26 mg, 0.20 mmol)으로 처리하였다. 반응 혼합물을 실온에서 1시간 동안 교반하고, 브로민화마그네슘 (62 mg, 0.34 mmol)을 첨가하였다. 혼합물을 밀봉하고, 50℃로 가열하였다. 60분 후, 반응 혼합물을 0.2M HCl(수성)로 켄칭하고, 염수로 희석하고, DCM으로 3회 추출하였다. HPLC 정제 (아세토니트릴:물, 0.1% TFA)로 화합물 25를 수득하였다.

실시예 26
화합물 26의 제조
(2S,5R,13aS)-N-(4-클로로-3-플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드


단계 1 및 2
15-B (41 mg, 0.13 mmol)를 아세토니트릴 (1 mL), (4-클로로-3-플루오로페닐)메탄아민 (40 mg, 0.25 mmol), HATU (60 mg, 0.16 mmol), 및 N,N-디이소프로필에틸아민 (28 mg, 0.22 mmol)으로 처리하였다. 반응 혼합물을 실온에서 1시간 동안 교반하고, 브로민화마그네슘 (48 mg, 0.26 mmol)을 첨가하였다. 혼합물을 밀봉하고, 50℃로 가열하였다. 60분 후, 반응 혼합물을 0.2M HCl (수성)로 켄칭하고, 염수로 희석하고, DCM으로 3회 추출하였다. HPLC 정제 (아세토니트릴:물, 0.1% TFA)로 화합물 26을 수득하였다.

실시예 27
화합물 27의 제조
(2S,5R)-N-(1-(2,4-디플루오로페닐)시클로프로필)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드
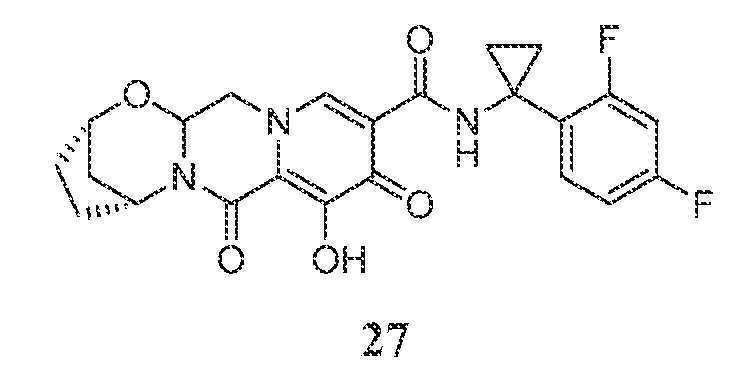

단계 1
CH2Cl2 (20 mL) 중 화합물 1-A (1.004 g, 3.19 mmol), 아민 27-A (688 mg, 3.35 mmol), 및 HATU (1.453 g 3.82 mmol)의 현탁액을 0℃ 조에서 교반하면서 N,N-디이소프로필에틸아민 (DIPEA) (2 mL, 11.48 mmol)을 첨가하였다. 0℃에서 1시간 후, 반응 혼합물을 시럽으로 농축시키고, 에틸 아세테이트로 희석하고, 물 (x 2)로 세척하였다. 수성 분획을 에틸 아세테이트 (x 1)로 추출한 후, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 콤비플래쉬 (120 g 칼럼)에 의해 용리액으로서 헥산-에틸 아세테이트를 사용하여 정제하였다. 주요 피크를 합하고, 농축시켜 생성물 27-B 1.082 g (73%)을 수득하였다. 부차 피크를 합한 후, 농축시키고, 농축된 잔류물을 CH2Cl2 중에 용해시키고, 일부 불용성 물질을 여과하였다. 여과물을 농축시켜 추가의 생성물 27-B 361 mg (24%)을 수득하였다.
단계 2 및 3
화합물 27-B (81 mg, 0.174 mmol)를 실온에서 아세토니트릴 (22 mL), AcOH (2 mL), 및 메탄술폰산 (0.14 mL, 2.16 mmol)의 혼합물 (1 mL) 중에 용해시키고, 생성된 용액을 65℃에서 20시간 동안 교반하였다.
생성된 용액을 실온으로 냉각시킨 후, 아미노알콜 27-D (50 mg, 라세미, 0.363 mmol), K2CO3 (50 mg, 0.362 mmol), 및 아세토니트릴 (2 mL)을 용액에 첨가하였다. 생성된 혼합물을 65℃ 조에서 1시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시킨 후, 이것을 1 N HCl (~2 mL)로 산성화시키고, 물 (~8 mL)로 희석하고, CH2Cl2 (x 3)로 추출하였다. 합한 추출물을 건조 (Na2SO4)시키고, 농축시키고, 콤비플래쉬에 의해 정제하여 화합물 27-E 67 mg (82%)을 수득하였다.

단계 4
MeCN (3 mL) 중 화합물 27-E (67 mg, 0.142 mmol) 및 MgBr2 (66 mg, 0.358 mmol)의 혼합물을 50℃에서 30분 동안 교반하고, 0℃로 냉각시킨 후, 1 N HCl (3 mL)로 처리하였다. 혼합물을 물 (~30 mL)로 희석한 후, 생성물을 CH2Cl2 (x 3)로 추출하고, 합한 추출물을 건조 (Na2SO4)시키고, 농축시켰다. 생성물을 정제용 HPLC에 의해 정제하고, 동결건조시켜 생성물 27을 트리플루오로아세트산과의 1:1 혼합물로서 수득하였다.

실시예 28
화합물 28의 제조
(2S,6R)-N-(1-(2,4-디플루오로페닐)시클로프로필)-9-히드록시-8,10-디옥소-3,4,5,6,8,10,14,14a-옥타히드로-2H-2,6-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사조신-11-카르복스아미드

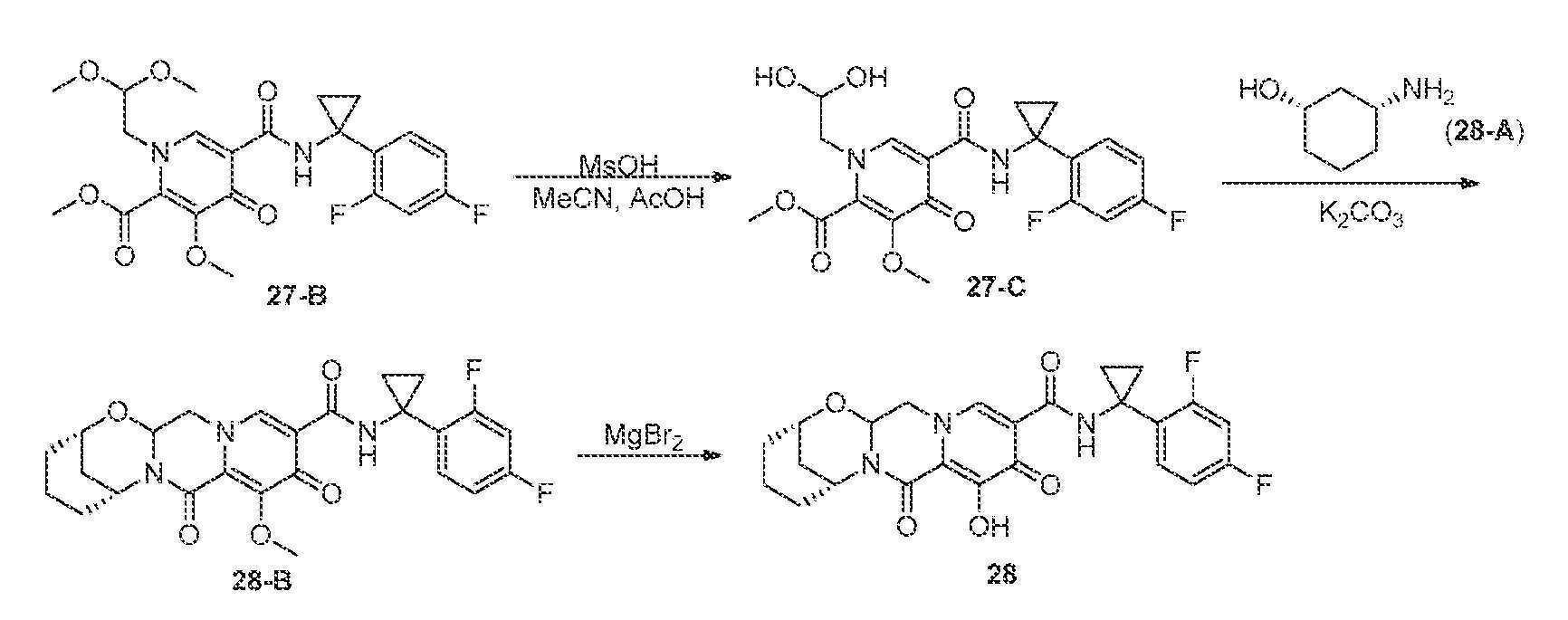
단계 1 및 2
화합물 27-B (87 mg, 0.187 mmol)를 실온에서 아세토니트릴 (22 mL), AcOH (2 mL), 및 메탄술폰산 (0.14 mL, 2.16 mmol)의 혼합물 (2 mL) 중에 용해시키고, 생성된 용액을 65℃에서 20시간 동안 교반하였다.
생성된 용액을 실온으로 냉각시킨 후, 아미노알콜 28-A (44 mg, 라세미, 0.382 mmol) 및 아세토니트릴 (2 mL)을 용액에 첨가하였다. 생성된 혼합물을 65℃ 조에서 30분 동안 교반한 후, K2CO3 (41 mg, 0.297 mmol)을 첨가하고, 혼합물을 65℃에서 21시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시키고, 이것을 1 N HCl (~2 mL)로 산성화시키고, 물 (~8 mL)로 희석하고, CH2Cl2 (x 3)로 추출하였다. 합한 추출물을 건조 (Na2SO4)시키고, 농축시키고, 정제용 HPLC에 의해 정제하고, 생성물을 함유하는 분획을 동결건조시켰다. 잔류물을 에틸 아세테이트 중에 용해시킨 후, 용액을 포화 NaHCO3 (x 1)으로 세척하고, 건조 (Na2SO4)시키고, 농축시켜 화합물 28-B를 트리플루오로아세트산과의 1:1 혼합물로서 18 mg (20%) 수득하였다.

단계 3
화합물 28-B (18 mg, 0.037 mmol)를 화합물 27-E 합성의 단계 4에 기재된 바와 같이 MgBr2로 처리하여 화합물 28을 수득하였다.

실시예 29
화합물 29의 제조
(2R,6S)-N-(1-(2,4-디플루오로페닐)시클로프로필)-9-히드록시-8,10-디옥소-3,4,5,6,8,10,14,14a-옥타히드로-2H-2,6-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사조신-11-카르복스아미드
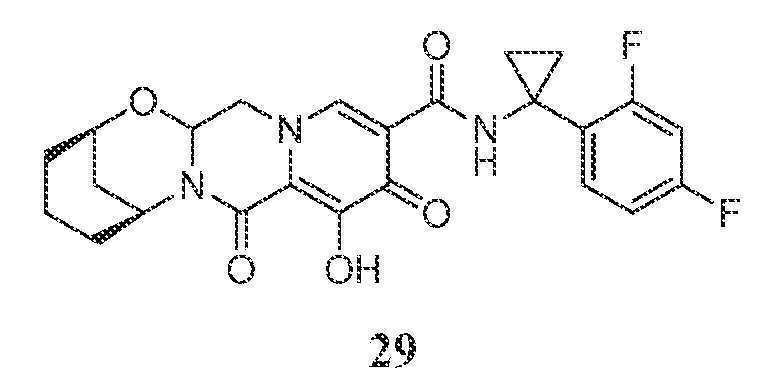
단계 1 및 2
화합물 29-B (13 mg, 14%)를 화합물 27-B (87 mg, 0.187 mmol) 및 아미노알콜 29-A (45 mg, 0.391 mmol)로부터 화합물 28-B 합성의 단계 1에 기재된 것과 유사한 방식으로 제조하였다.

단계 3
화합물 29를 화합물 29-B로부터 화합물 16 합성의 단계 2에 기재된 것과 유사한 방식으로 제조하였다.

실시예 30
화합물 30의 제조
(2S,5R,13aS)-N-(1-(2,4-디플루오로페닐)시클로프로필)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드


단계 1 및 2
화합물 27-B (150 mg, 0.322 mmol)를 실온에서 아세토니트릴 (2 mL), AcOH (0.2 mL), 및 메탄술폰산 (0.007 mL, 0.108 mmol) 중에 용해시키고, 생성된 용액을 65℃에서 20시간 동안 교반하였다. 생성된 용액을 실온으로 냉각시킨 후, 아미노알콜 30-A (72.1 mg, 키랄, 0.713 mmol), K2CO3 (89.4 mg, 0.647 mmol), 및 아세토니트릴 (2 mL)을 용액에 첨가하였다. 생성된 혼합물을 65℃ 조에서 0.5시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시킨 후, 이것을 1 N HCl (~3 mL)로 산성화시키고, 물 (~12 mL)로 희석하고, CH2Cl2 (x 3)로 추출하였다. 합한 추출물을 건조 (Na2SO4)시키고, 농축시키고, 콤비플래쉬에 의해 정제하여 화합물 30-B 128 mg (84%)을 수득하였다.

단계 3
MeCN (5 mL) 중 화합물 30-B (128 mg, 0.272 mmol) 및 MgBr2 (130 mg, 0.706 mmol)의 혼합물을 50℃에서 30분 동안 교반하고, 0℃로 냉각시킨 후, 1 N HCl (4 mL)로 처리하였다. 혼합물을 물로 희석한 후, 생성물을 CH2Cl2 (x 3)로 추출하고, 합한 추출물을 건조 (Na2SO4)시키고, 농축시켰다. 생성물을 콤비플래쉬에 의해 정제하여 생성물 30을 수득하였다.

실시예 31
화합물 31의 제조
(2R,5S)-N-(1-(2,4-디플루오로페닐)시클로프로필)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드


단계 1 및 2
화합물 31-B (123 mg, 81%)를 화합물 27-B (150 mg, 0. 322 mmol) 및 아미노알콜 31-A (70.3 mg, 0.695 mmol)로부터 화합물 30-B 합성의 단계 1 및 2에 기재된 것과 유사한 방식으로 제조하였다.

단계 3
화합물 31을 화합물 31-B로부터 화합물 30 합성의 단계 3에 기재된 것과 유사한 방식으로 제조하였다.

실시예 32
화합물 32의 제조
(2S,5R)-N-(1-(2,4-디플루오로페닐)시클로부틸)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드


CH2Cl2 (2 mL) 중 화합물 32-A (22.2 mg, 0.069 mmol), 화합물 32-B (18.7 mg, 0.102 mmol), 및 HATU (43 mg, 0.113 mmol)의 용액을 실온에서 교반하면서 N,N-디이소프로필에틸아민 (DIPEA) (0.075 mL, 0.431 mmol)을 첨가하였다. 30분 후, 반응 혼합물을 에틸 아세테이트로 희석하고, 물 (x 2)로 세척하였다. 수성 분획을 EA (x 1)로 추출한 후, 유기 분획을 합하고, 건조시키고, 농축시키고, 진공 하에 건조시켰다.
MeCN (2 mL) 중 상기 조 생성물 및 MgBr2 (35 mg, 0.190 mmol)의 혼합물을 50℃ 조에서 1시간 동안 교반하고, 0℃로 냉각시킨 후, 1 N HCl (~ 1 mL)로 처리하였다. 생성된 용액을 물로 희석하고, CH2Cl2 (x 3)로 추출하였다. 합한 추출물을 건조 (Na2SO4)시키고, 농축시켰다. 생성물을 정제용 HPLC에 의해 정제하고, 동결건조시켜 화합물 32를 수득하였다.

실시예 33
화합물 33의 제조
(2S,5R)-N-(1-(2,4-디플루오로페닐)시클로펜틸)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

화합물 33을 화합물 32-A 및 화합물 33-A로부터 화합물 32의 합성에 기재된 바와 같이 수득하였다.

실시예 34
화합물 34의 제조
(2S,5R)-N-(1-(2,4-디플루오로페닐)시클로헥실)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

화합물 34를 화합물 32-A 및 화합물 34-A로부터 화합물 32의 합성에 기재된 바와 같이 수득하였다.

실시예 35
화합물 35의 제조
(2S,5R)-N-(4-(2,4-디플루오로페닐)테트라히드로-2H-피란-4-일)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

화합물 35를 화합물 32-A 및 화합물 35-A로부터 화합물 32의 합성에 기재된 바와 같이 수득하였다.

실시예 36
화합물 36의 제조
(2S,5R)-N-((S)-1-(2,4-디플루오로페닐)-2,2,2-트리플루오로에틸)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드


화합물 36을 화합물 32-A 및 화합물 36-A로부터 화합물 32의 합성에 기재된 바와 같이 수득하였다.

실시예 37
화합물 37의 제조
(3S,11aR)-N-(1-(2,4-디플루오로페닐)시클로프로필)-6-히드록시-3-메틸-5,7-디옥소-2,3,5,7,11,11a-헥사히드로옥사졸로[3,2-a]피리도[1,2-d]피라진-8-카르복스아미드
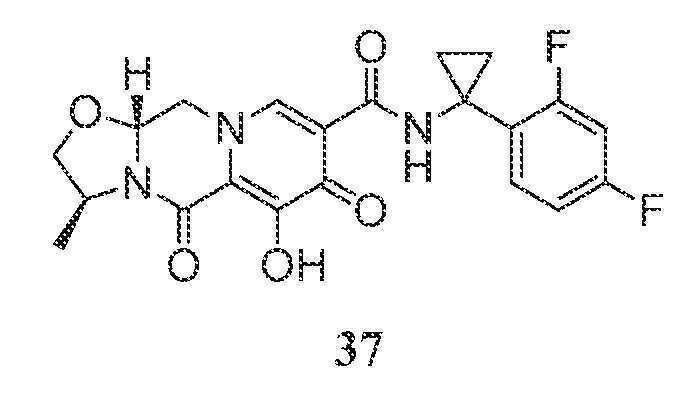
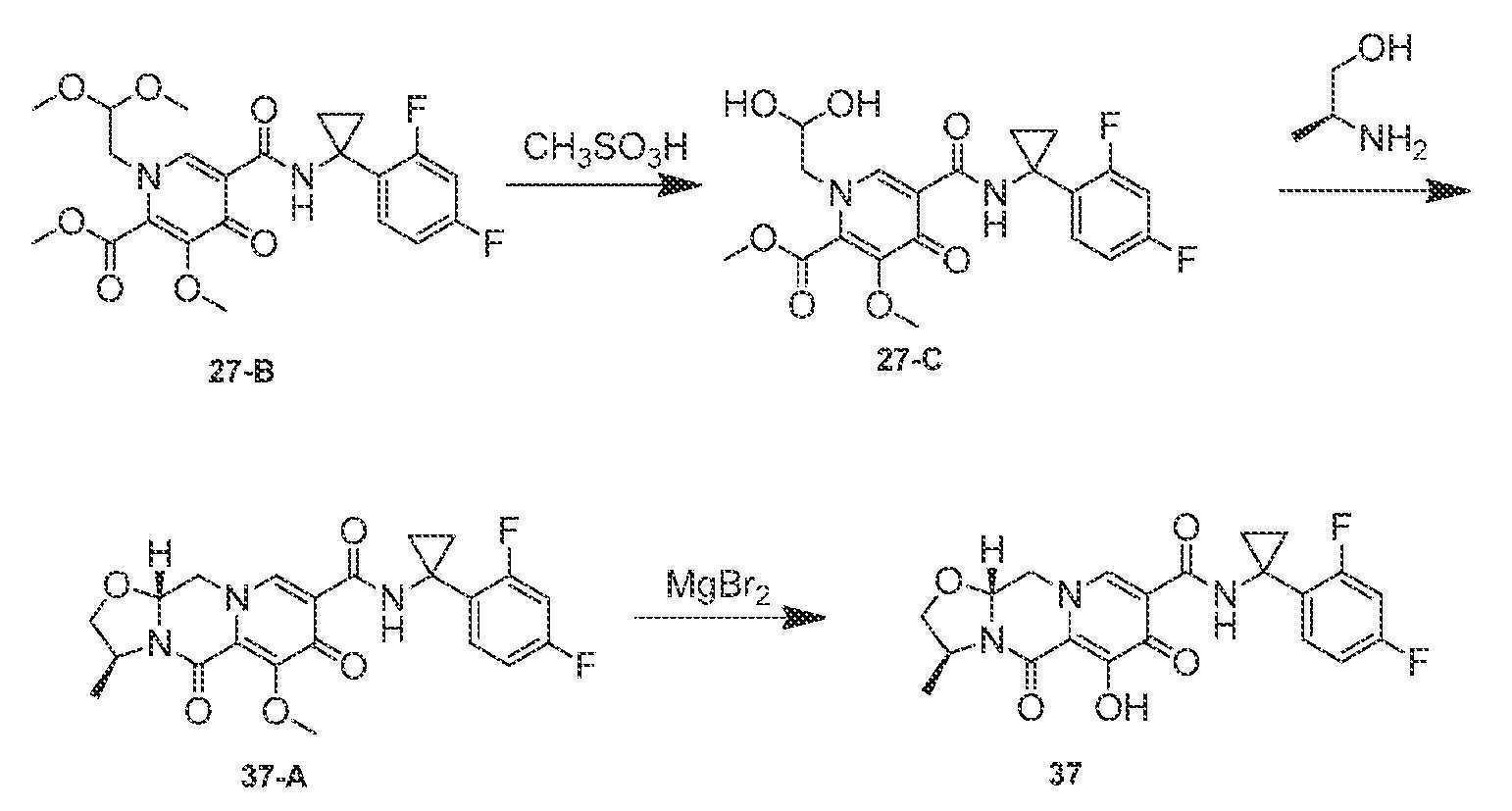
단계 1
아세토니트릴 (1.5 mL) 및 아세트산 (0.2 mL) 중 메틸 5-(1-(2,4-디플루오로페닐)시클로프로필카르바모일)-1-(2,2-디메톡시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트 (27-B, 0.150 g, 0.32 mmol)를 메탄술폰산 (0.05 mL)으로 처리하고, 황색 마개로 밀봉하고, 70℃로 가열하였다. 16시간 후, 혼합물을 냉각시켜 메틸 5-(1-(2,4-디플루오로페닐)시클로프로필카르바모일)-1-(2,2-디히드록시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트의 조 용액 27-C를 수득하였다.
단계 2 및 3
메틸 5-(1-(2,4-디플루오로페닐)시클로프로필카르바모일)-1-(2,2-디히드록시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트 (27-C, 0.32 mmol, 이전 단계로부터의 조 혼합물)을 아세토니트릴 (1.5 mL) 및 아세트산 (0.2 mL) 중에 용해시켰다. (S)-2-아미노프로판-1-올 (0.048 g, 0.64 mmol) 및 K2CO3 (0.088 g, 0.64 mmol)을 반응 혼합물에 첨가하였다. 반응 혼합물을 밀봉하고, 70℃로 가열하였다. 3시간 후, 반응 혼합물을 냉각시키고, 브로민화마그네슘 (0.081 g, 0.44 mmol)을 첨가하였다. 혼합물을 재밀봉하고, 50℃로 가열하였다. 10분 후, 반응 혼합물을 0℃로 냉각시키고, 1 N 염산 (0.5 mL)을 첨가하였다. 이어서, 반응 혼합물을 MeOH (2 mL)로 희석하였다. 여과한 후, 조 물질을 정제용 HPLC (30-70% 아세토니트릴:물, 0.1% TFA)에 의해 정제하여 화합물 37을 TFA 염으로서 수득하였다.

실시예 38
화합물 38의 제조
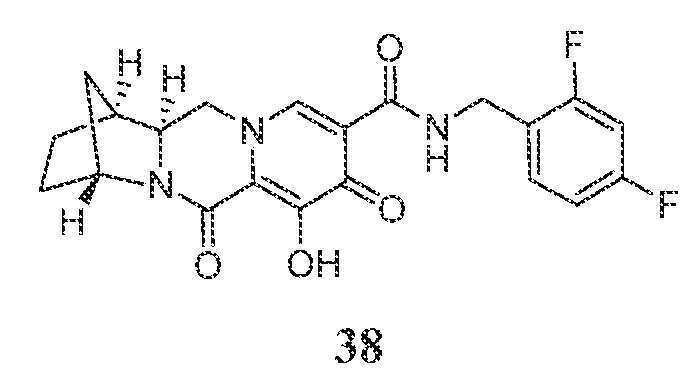
(1S,4R,12aR)-N-(2,4-디플루오로벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
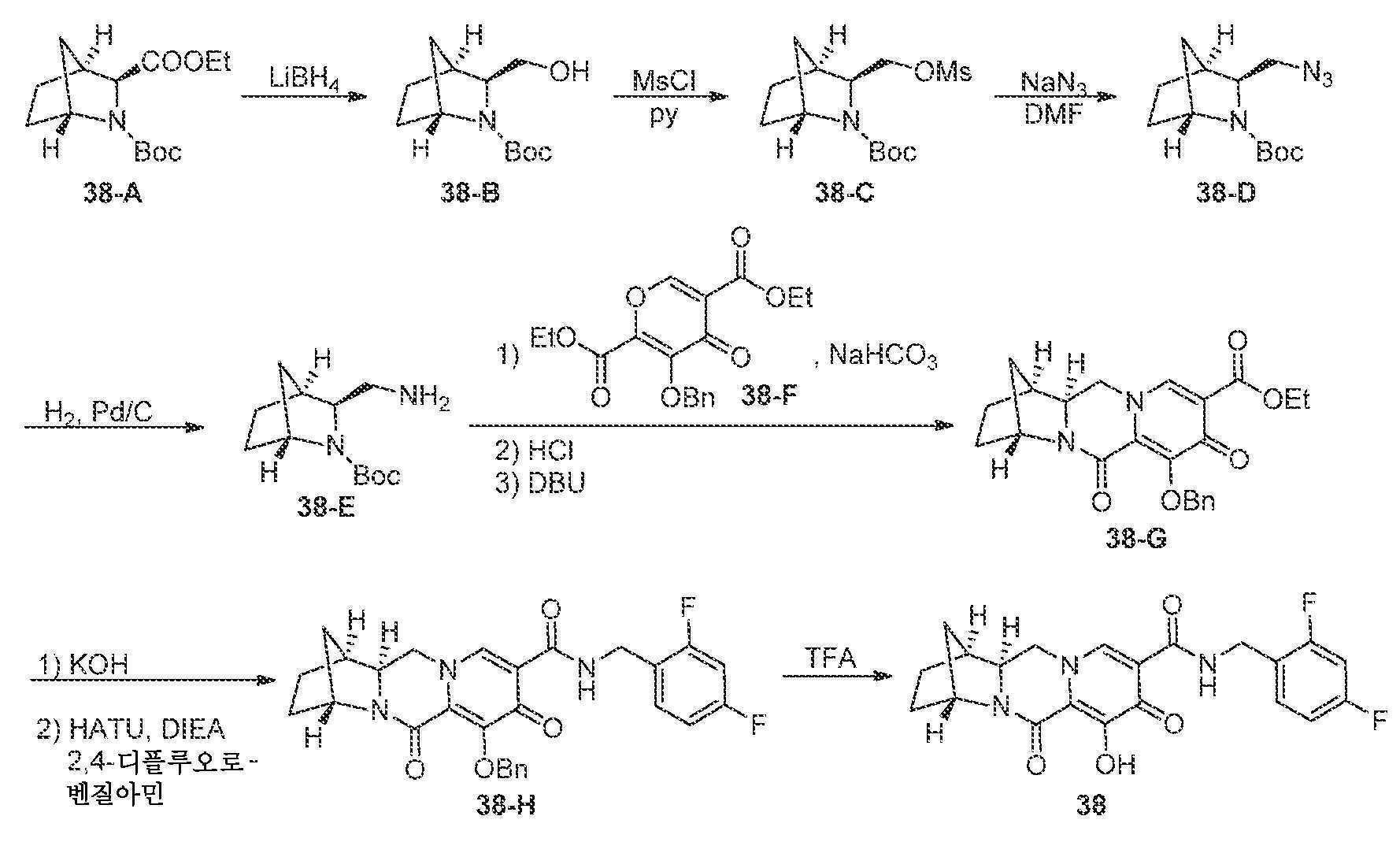
단계 1
THF (10 mL) 중 화합물 38-A (1562 mg, 5.799 mmol) (WO 97/05139의 실시예 41b 참조)의 용액을 -78℃에서 교반하면서 THF 중 2.0 M LiBH4 (3.2 mL)를 첨가하고, 생성된 혼합물을 실온에서 교반하였다. 3시간 후, 추가의 THF 중 2.0 M LiBH4 (3.2 mL)를 첨가하고, 용액을 실온에서 17.5시간 동안 교반하였다. 반응 혼합물을 에틸 아세테이트로 희석하고 물을 천천히 첨가한 후, 2개의 상을 분리하고, 분리된 수성 분획을 에틸 아세테이트 (x 1)로 추출하였다. 2개의 유기 분획을 물 (x 1)로 세척하고, 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 콤비플래쉬 (40 g 칼럼)에 의해 용리액으로서 헥산 - 에틸 아세테이트를 사용하여 정제하여 화합물 38-B를 수득하였다.

단계 2
CH2Cl2 (6 mL) 중 화합물 38-B (589 mg, 2.591 mmol) 및 NEt3 (0.47 mL, 3.369 mmol)의 용액을 0℃에서 교반하면서 MsCl (0.22 mL, 2.842 mmol)을 첨가하였다. 실온에서 1시간 후, 혼합물을 에틸 아세테이트로 희석하고, 물 (x 2)로 세척하였다. 수성 분획을 에틸 아세테이트 (x 1)로 추출하고, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 콤비 플래쉬 (40 g 칼럼)에 의해 용리액으로서 헥산 - 에틸 아세테이트를 사용하여 정제하여 화합물 38-C를 수득하였다.

단계 3
DMF (5 mL) 중 화합물 38-C (769 mg, 2.518 mmol)의 용액에 아지드화나트륨 (819 mg, 12.6 mmol)을 첨가하였다. 반응 혼합물을 50℃에서 15시간, 80℃에서 5시간, 및 100℃에서 19시간 동안 교반하였다. 반응 혼합물을 5% LiCl 용액으로 희석하고, 생성물을 에틸 아세테이트 (x 2)로 추출하였다. 유기 분획을 물 (x 1)로 세척한 후, 2개의 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 콤비플래쉬 (40 g 칼럼)에 의해 용리액으로서 헥산 - 에틸 아세테이트를 사용하여 정제하여 화합물 38-D를 수득하였다.

단계 4
에틸 아세테이트 (10 mL) 및 EtOH (10 mL) 중 화합물 38-D (507 mg, 2.009 mmol)의 용액에 10% Pd/C (52 mg)를 첨가하였다. 반응 혼합물을 H2 분위기 하에 1.5시간 동안 교반하였다. 혼합물을 셀라이트를 통해 여과하고, 여과물을 농축시켜 조 화합물 38-E를 수득하였다.
단계 5
물 (3 mL) 및 EtOH (3 mL) 중 조 화합물 38-E (206 mg, 0.910 mmol), 화합물 38-F (330 mg, 0.953 mmol), 및 NaHCO3 (154 mg, 1.833 mmol)의 혼합물을 실온에서 20시간 동안 교반하였다. 반응 혼합물을 물로 희석하고 에틸 아세테이트 (x 2)로 추출한 후, 추출물을 물 (x 1)로 세척하고, 합하고, 건조 (Na2SO4)시키고, 농축시켜 조 피리딘 생성물을 수득하였다.
조 잔류물 (388 mg)을 CH2Cl2 (4 mL) 및 디옥산 중 4 N HCl (4 mL) 중에 용해시켰다. 1.5시간 후, 추가의 디옥산 중 4 N HCl (4 mL)을 첨가하고, 실온에서 1시간 동안 교반하였다. 혼합물을 농축 건조시키고, 톨루엔 (x 1)과 공증발시키고, 진공 하에 30분 동안 건조시켰다.
톨루엔 (10 mL) 중 조 잔류물 및 1,8-디아자비시클로운데스-7-엔 (DBU) (1.06 mL, 7.088 mmol)을 110℃ 조에서 교반하였다. 30분 후, 혼합물을 농축시키고, 잔류물을 콤비플래쉬 (40 g 칼럼)에 의해 용리액으로서 에틸 아세테이트 - 20% MeOH/에틸 아세테이트를 사용하여 정제하여 화합물 38-G를 수득하였다.

단계 6
THF (3 mL) 및 MeOH (3 mL) 중 화합물 38-G (232 mg, 0.568 mmol)의 혼합물을 실온에서 교반하면서 1 N KOH (3 mL)를 첨가하였다. 1시간 후, 반응 혼합물을 1 N HCl (~3.1 mL)로 중화시키고, 농축시키고, 잔류물을 톨루엔 (x 3)과 함께 농축시켰다. 잔류물을 진공 하에 30분 동안 건조시킨 후, CH2Cl2 (4 mL) 및 DMF (4 mL) 중 조 잔류물, 2,4-디플루오로벤질아민 (86 mg, 0.601 mmol) 및 HATU (266 mg, 0.700 mmol)의 현탁액을 0℃에서 교반하면서 N,N-디이소프로필에틸아민 (DIPEA) (0.7 mL, 4.019 mmol)을 첨가하였다. 45분 후, 추가의 2,4-디플루오로벤질아민 (86 mg, 0.559 mmol), HATU (266 mg, 0.700 mmol), 및 N,N-디이소프로필에틸아민 (DIPEA) (0.7 mL, 4.019 mmol)을 실온에서 첨가하였다. 1.25시간 후, 혼합물을 농축시켜 대부분의 CH2Cl2를 제거하고, 에틸 아세테이트로 희석하고, 5% LiCl (x 2)로 세척하였다. 수성 분획을 에틸 아세테이트 (x 1)로 추출한 후, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 콤비플래쉬 (40 g 칼럼)에 의해 용리액으로서 에틸 아세테이트 -20%MeOH/에틸 아세테이트를 사용하여 정제하여 화합물 38-H를 수득하였다.

단계 7
화합물 38-H (240 mg, 0.475 mmol)를 TFA (3 mL) 중에 실온에서 30분 동안 용해시키고, 용액을 농축시켰다. 잔류물을 콤비플래쉬 (40 g 칼럼)에 의해 용리액으로서 CH2Cl2-CH2Cl2 중 20% MeOH를 사용하여 정제하였다. 수집된 생성물 분획을 농축시킨 후, 잔류물을 MeCN (~2 mL) 중에서 0℃에서 15분 동안 연화처리하고, 고체를 여과하고, MeCN으로 세척하였다. 수집된 고체를 진공 하에 건조시켜 화합물 38을 수득하였다.
여과물을 농축시키고, 잔류물을 MeCN (~1 mL) 및 물 (~1 mL) 중에 가열하여 용해시켰다. 용액을 천천히 실온으로 냉각시킨 다음 빙조에서 15분 동안 냉각시켰다. 고체를 여과하고, MeCN으로 세척하고, 진공 하에 건조시켜 추가의 화합물 38을 수득하였다.

실시예 39 및 40
화합물 39 및 40의 제조
(2R,3S,5R,13aS)-N-(2,4-디플루오로벤질)-8-히드록시-3-메틸-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드 39 및 (2S,3R,5S,13aR)-N-(2,4-디플루오로벤질)-8-히드록시-3-메틸-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드 40


단계 1
제1구리 시아나이드 (290 mg, 3.27 mmol)를 3.3 mL THF 중에 현탁시키고, -78℃로 냉각시켰다. 디에틸 에테르 중 MeLi의 1.6M 용액 (4.1 mL, 6.56 mmol)을 적가하고, 반응 용액을 실온으로 2시간의 기간에 걸쳐 가온되도록 하고, -78℃로 재냉각시켰다. tert-부틸 (1R,3R,5S)-6-옥사비시클로[3.1.0]헥산-3-일카르바메이트 (330 mg, 1.66 mmol)를 3.3 mL THF에 이어서 삼플루오린화붕소 디에틸 에테레이트 (0.25 mL, 1.99 mmol)에 적가하고, 30분에 걸쳐 -30℃로 가온되도록 하고, -35℃ 내지 -25℃에서 1시간 동안 교반하였다. 이어서, 반응 용액을 실온으로 가온하고, 포화 NH3(수성)/NH4(수성)의 혼합물로 켄칭하고, EtOAc로 추출하고, 염수로 세척하고, MgSO4 상에서 건조시키고, 여과하고, 농축시키고, SGC (0-10% EtOH/DCM)에 의해 정제하여 라세미 tert-부틸 (1S,3S,4S)-3-히드록시-4-메틸시클로펜틸카르바메이트를 수득하였다.

단계 2
3 mL HCl/디옥산 (4M, 12 mmol)을 3 mL 디옥산 중 라세미 tert-부틸 (1S,3S,4S)-3-히드록시-4-메틸시클로펜틸카르바메이트 (182 mg, 0.85 mmol)의 용액에 첨가하였다. 반응 혼합물을 실온에서 2시간 동안 교반하고, 농축시키고, 톨루엔으로 2회 체이싱하여 라세미 (1S,2S,4S)-4-아미노-2-메틸시클로펜탄올을 수득하였다.
단계 3
메틸 5-(2,4-디플루오로벤질카르바모일)-1-(2,2-디히드록시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트 (1-C, 310 mg, 0.75 mmol), 라세미 (1S,2S,4S)-4-아미노-2-메틸시클로펜탄올 (115 mg, 0.76 mmol), 및 탄산칼륨 (232 mg, 1.68 mmol)을 3.8 mL 아세토니트릴/0.2 mL 아세트산에 녹이고, 90℃에서 2시간 동안 교반한 후, 반응 혼합물을 DCM과 염수 사이에 분배하고, 수성 상을 DCM으로 추출하고, 합한 유기 상을 MgSO4 상에서 건조시키고, 여과하고, 농축시키고, SGC (0-10% EtOH/DCM)에 의해 정제하여 중간체 39-A를 수득하였다.
단계 4
중간체 39-A (190 mg)를 룩스(Lux) 셀룰로스-2 칼럼 상에서 키랄 정제용 HPLC에 의해 용리액으로서 9:1 ACN:MeOH를 사용하여 분리하여 중간체 39-B (제1 용리 피크) 및 40-A (제2 용리 피크)를 거울상이성질체풍부화 형태로 수득하였다. 중간체 39-B의 경우: (X선 결정학에 의해 절대 입체화학이 확인됨), 키랄 HPLC 체류 시간 = 3.98분 (룩스 셀룰로스-2 IC, 150 x 4.6 mm, 2 mL/분 9:1 ACN:MeOH). 중간체 40-A의 경우: (X선 결정학에 의해 절대 입체화학이 확인됨), 키랄 HPLC 체류 시간 = 6.35분 (룩스 셀룰로스-2 IC, 150 x 4.6 mm, 2 mL/분 9:1 ACN:MeOH).
단계 5a
브로민화마그네슘 (68 mg, 0.37 mmol)을 2 mL 아세토니트릴 중 중간체 39-B (83 mg, 0.18 mmol)의 용액에 첨가하였다. 반응 혼합물을 50℃에서 1시간 동안 교반하고, 10% 수성 HCl로 산성화시키고, 수성 및 디클로로메탄 사이에 분배하고, 수성 상을 디클로로메탄으로 추출하였다. 합한 유기 상을 MgSO4 상에서 건조시키고, 여과하고, 농축시키고, 실리카 겔 크로마토그래피 (0-10% EtOH/DCM)에 의해 정제하여 화합물 39를 수득하였다.

단계 5b
브로민화마그네슘 (59 mg, 0.32 mmol)을 2 mL 아세토니트릴 중 중간체 40-A (70 mg, 0.15 mmol)의 용액에 첨가하였다. 반응 혼합물을 50℃에서 1시간 동안 교반하고, 10% 수성 HCl로 산성화시키고, 수성 및 디클로로메탄 사이에 분배하고, 수성 상을 디클로로메탄으로 추출하였다. 합한 유기 상을 MgSO4 상에서 건조시키고, 여과하고, 농축시키고, 실리카 겔 크로마토그래피 (0-10% EtOH/DCM)에 의해 정제하여 화합물 40을 수득하였다.

실시예 41
화합물 41의 제조
(1R,4S,12aR)-7-히드록시-6,8-디옥소-N-(2,4,6-트리플루오로벤질)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드


단계 1
THF (14 mL) 중 41-A (2020 mg, 7.463 mmol) (38-A와 동일한 방법으로 제조함)의 용액을 0℃에서 교반하면서 THF 중 2.0 M LiBH4 (7.5 mL, 15 mmol)를 첨가하였다. 생성된 혼합물을 실온에서 21시간 동안 교반한 후, 0℃에서 냉각시키고 EA로 희석한 후, 물을 천천히 첨가하여 켄칭하였다. 2개의 상을 분리한 후, 수성 분획을 EA (x 1)로 추출하고, 2개의 유기 분획을 물 (x 1)로 세척하고, 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 콤비플래쉬 (120 g 칼럼)에 의해 용리액으로서 헥산 - EA를 사용하여 정제하여 41-B를 수득하였다.
단계 2
100-mL 둥근 바닥 플라스크에 DCM (20 mL) 중 반응물 41-B (1.6 g, 7.05 mmol) 및 트리에틸아민 (0.94 g, 9.3 mmol)을 채웠다. 메탄술포닐 클로라이드 (0.91 g, 8.0 mmol)를 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 실온에서 3시간 동안 교반하였다. 혼합물을 EA (100 mL)로 희석하고, 물 (2x)로 세척하였다. 수성 분획을 EA (1x)로 추출하고, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 콤비 플래쉬 (120 g 칼럼, 카트리지 사용)에 의해 용리액으로서 헥산 - EA를 사용하여 정제하여 41-C를 수득하였다.
단계 3
100-mL 둥근 바닥 플라스크에 DMF (10 mL) 중 반응물 41-C (2.1 g, 6.9 mmol) 및 아지드화나트륨 (2.3 g, 34.5 mmol)을 채웠다. 이어서, 반응 혼합물을 100℃에서 밤새 교반하였다. 혼합물을 EA (100 mL)로 희석하고, 물 (2x)로 세척하였다. 수성 분획을 EA (1x)로 추출하고, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 콤비 플래쉬 (120 g 칼럼, 카트리지 사용)에 의해 용리액으로서 헥산 - EA를 사용하여 정제하여 41-D를 수득하였다.
단계 4
EA (20 mL) 및 EtOH (20 mL) 중 반응물 41-D (1.3 g)의 용액 (N2로 퍼징함)에 Pd/C (130 mg)를 첨가하였다. 혼합물을 H2 하에 3시간 동안 교반하였다. 혼합물을 셀라이트를 통해 여과하고, 여과물을 농축시켜 화합물 41-E를 수득하였다.
단계 5
100-mL 둥근 바닥 플라스크에 에탄올 (20 mL) 중 반응물 41-E (1.05 g, 4.62 mmol) 및 반응물 38-F (1.6 g, 4.62 mmol)를 채웠다. 물 (20 mL) 중 중탄산나트륨 (0.77 g, 9.2 mmol)을 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 실온에서 밤새 교반하였다. 혼합물을 EA (100 mL)로 희석하고, 물 (2x)로 세척하였다. 수성 분획을 EA (1x)로 추출하고, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 조 생성물 (2.4 g)을 후속 단계에 추가 정제 없이 사용하였다. LCMS-ESI+ (m/z): [M+H]+ 계산치, C18H19F2N2O7: 556; 실측치: 556.
100-mL 둥근 바닥 플라스크에 4 N HCl /디옥산 중 이전 반응으로부터의 조 생성물 (24.7 mL)을 채웠다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하였다. 농축시킨 후, 톨루엔 (30 mL) 중 중간체 (2.1 g) 및 DBU (3.27 g, 21.5 mmol)를 1시간 동안 교반하면서 110℃로 가열하였다. 농축시킨 후, 잔류물을 콤비플래쉬 (120 g 칼럼)에 의해 용리액으로서 헥산 - 에틸 아세테이트를 사용하여 정제하여 41-F를 수득하였다.
단계 6
100-mL 둥근 바닥 플라스크에 THF (5 mL) 및 MeOH (5 mL) 중 반응물 41-F (0.5 g, 1.22 mmol)를 채웠다. 1 N KOH (3.7 mL)를 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하였다. 1 N HCl (3.7 mL)을 첨가하여 반응 혼합물을 산성화시키고, 농축시켜 대부분의 유기 용매를 제거하고, EtOAc (2 X)로 추출하였다. 유기 층을 합하고, 건조 (Na2SO4)시키고, 농축시켜 화합물 41-G를 수득하였다.
단계 7
100-mL 둥근 바닥 플라스크에 반응물 41-G (0.14 g, 0.37 mmol), (2,4,6-트리플루오로페닐)메탄아민 (0.12 g, 0.73 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.24 g, 1.84 mmol) 및 HATU (0.28 g, 0.74 mmol)를 채우고, DCM (5 mL) 중에 용해시켰다. 반응 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 EA (100 mL)로 희석하고, 포화 NaHCO3 (2x), 포화 NH4Cl (2x)로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 헥산-EtOAc를 사용하여 정제하여 화합물 41-H를 수득하였다.
단계 8
50-mL 둥근 바닥 플라스크에 TFA (2 mL) 중 반응물 41-H (0.13 g, 0.25 mmol)를 채웠다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 EtOAc-MeOH를 사용하여 정제하여 화합물 41을 수득하였다.

실시예 42
화합물 42의 제조
(2R,5S,13aR)-8-히드록시-7,9-디옥소-N-(2,4,6-트리플루오로벤질)-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드


단계 1
아세토니트릴 (36 mL) 및 아세트산 (4 mL) 중 1-(2,2-디메톡시에틸)-5-메톡시-6-(메톡시카르보닐)-4-옥소-1,4-디히드로피리딘-3-카르복실산 (3.15 g, 10 mmol)을 메탄술폰산 (0.195 mL, 3 mmol)으로 처리하고, 75℃ 조에 두었다. 반응 혼합물을 7시간 동안 교반하고, 냉각시키고, -10℃에서 3일 동안 보관하고, 추가로 2시간 동안 75℃로 재가열하였다. 이 물질을 냉각시키고 조 상태로 후속 단계를 수행하였다.
단계 2
단계 1로부터의 조 반응 혼합물 (20 mL, 4.9 mmol)을 (1R,3S)-3-아미노시클로펜탄올 (0.809 g, 8 mmol)이 담긴 플라스크로 옮겼다. 혼합물을 아세토니트릴 (16.8 mL)로 희석하고, 탄산칼륨 (0.553 g, 4 mmol)으로 처리하고, 85℃로 가열하였다. 2시간 후, 반응 혼합물을 주위 온도로 냉각시키고, 밤새 교반하였다. 0.2M HCl (50 mL)을 첨가하고, 투명한 황색 용액을 디클로로메탄 (2x150 mL)으로 추출하였다. 합한 유기 층을 황산나트륨 상에서 건조시키고, 여과하고, 밝은 오렌지색 고체 1.49 g으로 농축시켰다. 디클로르메탄:헥산으로부터 재결정화하여 목적 중간체 42A를 수득하였다:
단계 3
중간체 42-A (0.225 g, 0.702 mmol) 및 (2,4,6-트리플루오로페닐)메탄아민 (0.125 g, 0.773 mmol)을 아세토니트릴 (4 mL) 중에 현탁시키고, N,N-디이소프로필에틸아민 (DIPEA) (0.183 mmol, 1.05 mmol)으로 처리하였다. 이 현탁액에 (디메틸아미노)-N,N-디메틸(3H-[1,2,3]트리아졸로[4,5-b]피리딘-3-일옥시)메탄이미늄 헥사플루오로포스페이트 (HATU, 0.294 g, 0.774 mmol)를 첨가하였다. 1.5시간 후, 조 반응 혼합물을 후속 단계에 사용하였다.
단계 4
이전 단계의 조 반응 혼합물에 MgBr2 (0.258 g, 1.40 mmol)를 첨가하였다. 반응 혼합물을 50℃에서 10분 동안 교반하고, 10% 수성 HCl로 산성화시키고, 디클로로메탄으로 2회 추출하였다. 합한 유기 상을 MgSO4 상에서 건조시키고, 여과하고, 농축시키고, 실리카 겔 크로마토그래피 (EtOH/디클로르메탄)에 이어서 HPLC (ACN/H2O, 0.1% TFA 개질제 함유)를 사용하여 정제하여 화합물 42를 수득하였다:

실시예 43
화합물 43의 제조
(12aR)-N-((R)-1-(2,4-디플루오로페닐)에틸)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드

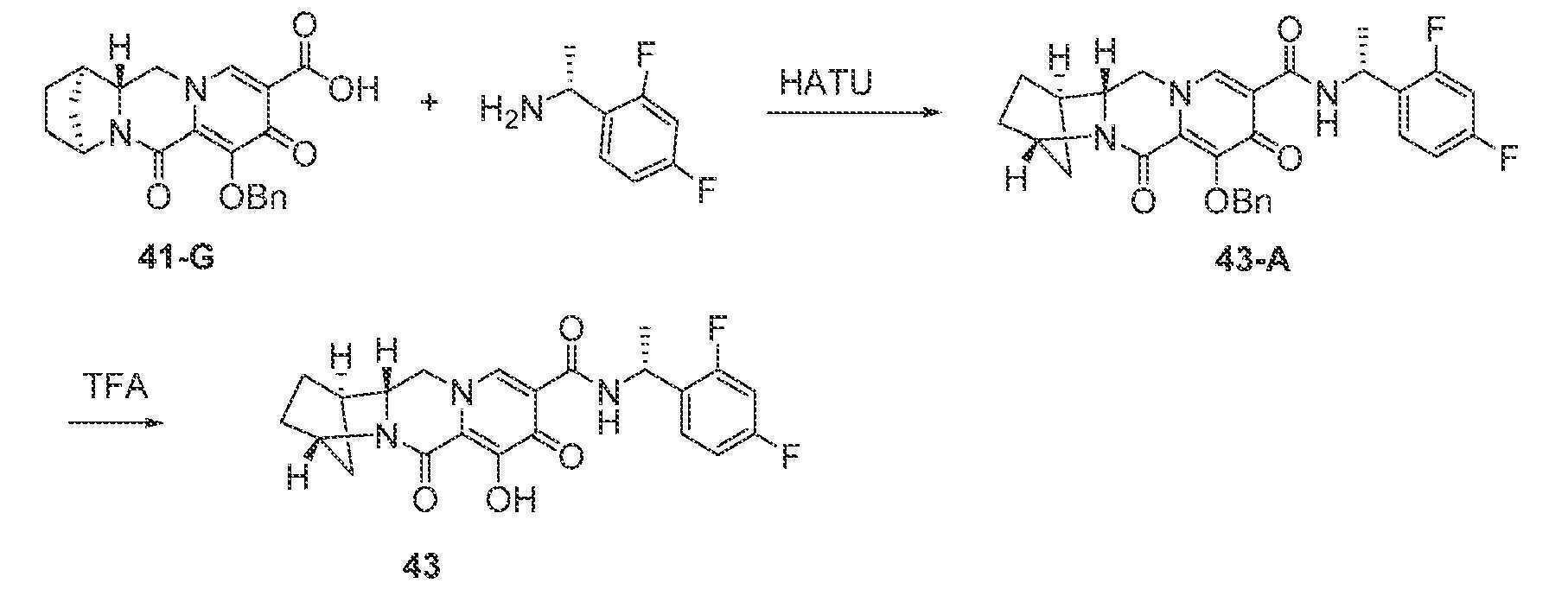
단계 1
100-mL 둥근 바닥 플라스크에 반응물 41-G (0.14 g, 0.37 mmol), (R)-1-(2,4-디플루오로페닐)에탄아민 (0.12 g, 0.74 mmol), N,N-디이소프로필에틸아민 (0.24 g, 1.84 mmol) 및 HATU (0.28 g, 0.74 mmol)를 채우고, DCM (5 mL) 중에 용해시켰다. 반응 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 EA (100 mL)로 희석하고, 포화 NaHCO3 (2x), 포화 NH4Cl (2x)로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 헥산-EtOAc를 사용하여 정제하여 화합물 43-A를 수득하였다.
단계 2
50-mL 둥근 바닥 플라스크에 TFA (2 mL) 중 반응물 43-A (0.14 g, 0.27 mmol)를 채웠다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 EtOAc-MeOH를 사용하여 정제하여 화합물 43을 수득하였다.

실시예 44
화합물 44의 제조
(13aS)-8-히드록시-7,9-디옥소-N-(2,3,4-트리플루오로벤질)-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

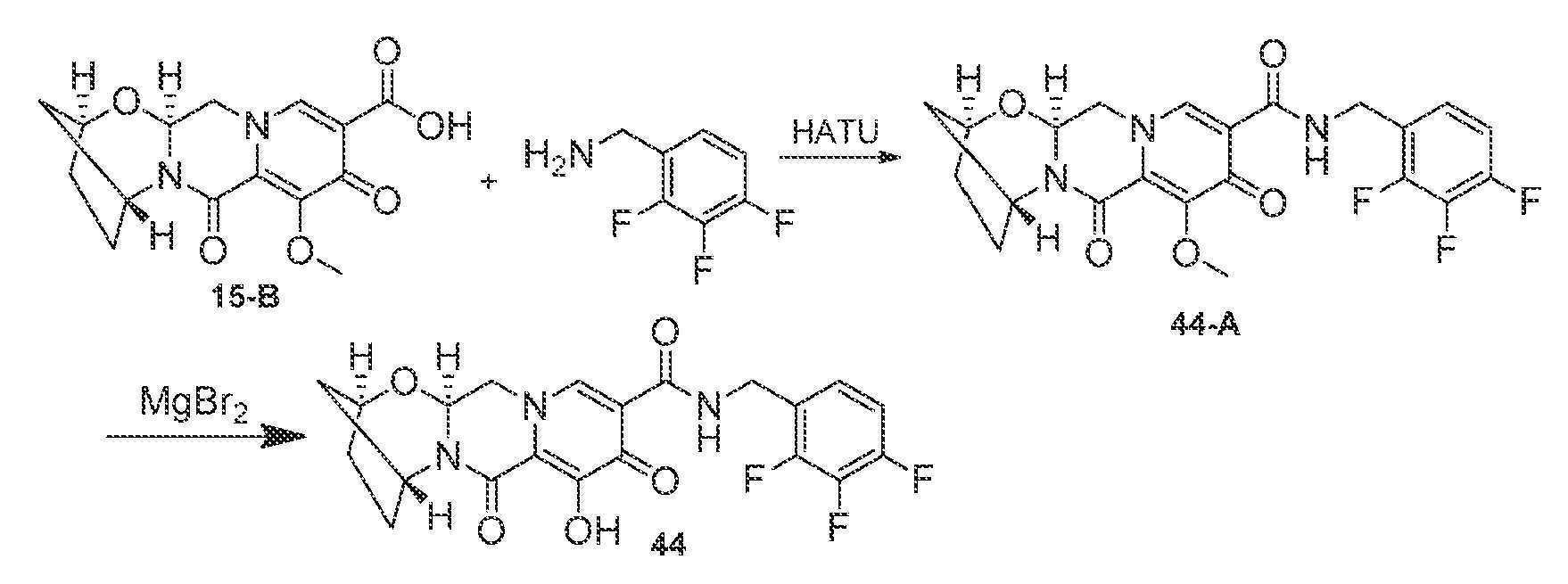
단계 1
화합물 15-B (40 mg, 0.12 mmol)를 1 mL 아세토니트릴에 녹이고, 2,3,4-트리플루오로벤질아민 (29 mg, 0.18 mmol), HATU (53 mg, 0.14 mmol), N,N-디이소프로필에틸아민 (DIPEA) (20 mg, 0.16 mmol)으로 처리하고, 실온에서 2시간 동안 교반한 후, LCMS 분석하여 화합물 15-B의 완전한 소모 및 중간체 44-A의 형성을 밝혀내었다. 반응 혼합물로 후속 단계를 수행하였다.
단계 2
이전 단계의 조 반응 용액에 MgBr2 (63 mg, 0.34 mmol)를 첨가하였다. 반응 혼합물을 50℃에서 1시간 동안 교반하고, 10% 수성 HCl로 산성화시키고, 수성 및 디클로로메탄 사이에 분배하고, 수성 상을 디클로로메탄으로 추출하였다. 합한 유기 상을 MgSO4 상에서 건조시키고, 여과하고, 농축시키고, HPLC (ACN/H2O, 0.1% TFA 개질제 함유)에 의해 화합물 44로 정제하였다.

실시예 45
화합물 45의 제조
(13aS)-8-히드록시-7,9-디옥소-N-(2,4,6-트리플루오로벤질)-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

단계 1
화합물 15-B (38 mg, 0.12 mmol)를 1 mL 아세토니트릴에 녹이고, 2,4,6-트리플루오로벤질아민 (34 mg, 0.21 mmol), HATU (50 mg, 0.13 mmol), N,N-디이소프로필에틸아민 (DIPEA) (23 mg, 0.18 mmol)으로 처리하고, 실온에서 2시간 동안 교반한 후, LCMS 분석하여 화합물 15-B의 완전한 소모 및 중간체 45-A의 형성을 밝혀내었다. 반응 혼합물로 후속 단계를 수행하였다.
단계 2
이전 단계의 조 반응 용액에 MgBr2 (55 mg, 0.30 mmol)를 첨가하였다. 반응 혼합물을 50℃에서 1시간 동안 교반하고, 10% 수성 HCl로 산성화시키고, 수성 및 디클로로메탄 사이에 분배하고, 수성 상을 디클로로메탄으로 추출하였다. 합한 유기 상을 MgSO4 상에서 건조시키고, 여과하고, 농축시키고, HPLC (ACN/H2O, 0.1% TFA 개질제 함유)에 의해 정제하여 화합물 45를 수득하였다.

실시예 46
화합물 46의 제조
(13aS)-N-(2,6-디플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드


단계 1
화합물 15-B (38 mg, 0.12 mmol)를 1 mL 아세토니트릴에 녹이고, 2,6-디플루오로벤질아민 (19 mg, 0.14 mmol), HATU (56 mg, 0.15 mmol), N,N-디이소프로필에틸아민 (DIPEA) (20 mg, 0.15 mmol)으로 처리하고, 실온에서 90분 동안 교반한 후, LCMS 분석하여 화합물 A의 완전한 소모 및 중간체 46-A의 형성을 밝혀내었다. 반응 혼합물로 후속 단계를 수행하였다.
단계 2
이전 단계의 조 반응 용액에 MgBr2 (50 mg, 0.27 mmol)를 첨가하였다. 반응 혼합물을 50℃에서 1시간 동안 교반하고, 10% 수성 HCl로 산성화시키고, 수성 및 디클로로메탄 사이에 분배하고, 수성 상을 디클로로메탄으로 추출하였다. 합한 유기 상을 MgSO4 상에서 건조시키고, 여과하고, 농축시키고, HPLC (ACN/H2O, 0.1% TFA 개질제 함유)에 의해 정제하여 화합물 46을 수득하였다.

실시예 47
화합물 47의 제조
(1R,4S,12aR)-N-(2,4-디플루오로벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드

단계 1
조 산 41-G (0.45 g, 1.18 mmol), 2,4-디플루오로벤질아민 (0.35 g, 2.44 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.79 g, 6.11 mmol) 및 HATU (0.93 g, 2.44 mmol)를 DCM (10 mL) 중에 용해시켰다. 반응 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 EA (100 mL)로 희석하고, 포화 NaHCO3 (2x), 포화 NH4Cl (2x)로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 헥산-EtOAc를 사용하여 정제하여 화합물 47-A를 수득하였다.
단계 2
50-mL 둥근 바닥 플라스크에 TFA (6 mL) 중 반응물 47-A (0.5 g, 0.99 mmol)를 채웠다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 EtOAc-MeOH를 사용하여 정제하여 화합물 47을 수득하였다.

실시예 48
화합물 48의 제조
(1S,4R,12aS)-N-(2,4-디플루오로벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
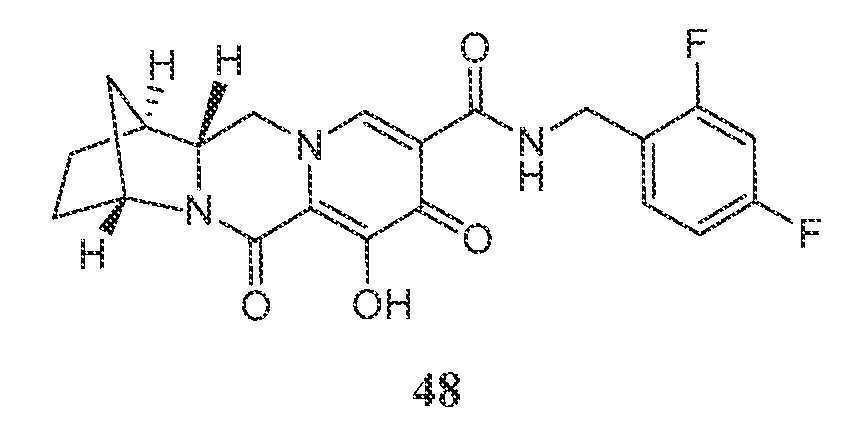

48-B를 실시예 55에서 55-H와 유사하게 55-A를 48-A로 치환하여 제조하였다. 화합물 48은 실시예 38에서 화합물 38에 대해 기재된 바와 같이 38-B를 48-B로 치환하여 제조하여 화합물 48을 수득하였다.

실시예 49
화합물 49의 제조
(2S,5R,13aS)-8-히드록시-7,9-디옥소-N-((3-(트리플루오로메틸)피리딘-2-일)메틸)-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드
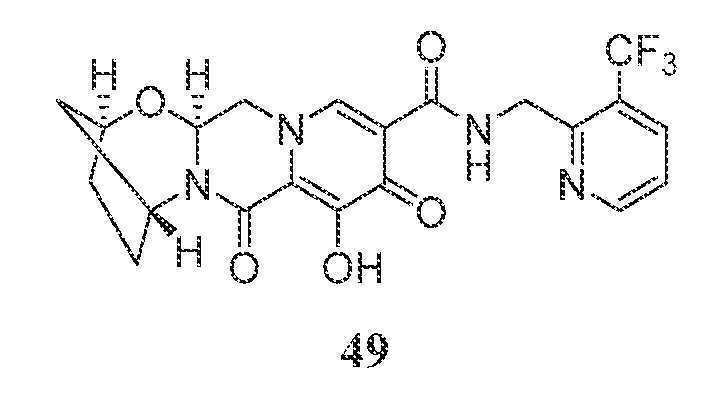

단계 1
화합물 15-B (44 mg, 0.14 mmol)를 1 mL 아세토니트릴에 녹이고, (3-(트리플루오로메틸)피리딘-2-일)메탄아민 (38 mg, 0.18 mmol, HCl 염), HATU (69 mg, 0.18 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.07 mL, 0.40 mmol)으로 처리하고, 실온에서 1시간 동안 교반한 후, LCMS 분석하여 화합물 15-B의 완전한 소모 및 중간체 49-A의 형성을 밝혀내었다. 반응 혼합물로 후속 단계를 수행하였다.
단계 2
이전 단계의 조 반응 용액에 MgBr2 (51 mg, 0.28 mmol)를 첨가하였다. 반응 혼합물을 50℃에서 90분 동안 교반하고, 10% 수성 HCl로 산성화시키고, 수성 및 디클로로메탄 사이에 분배하고, 수성 상을 디클로로메탄으로 추출하였다. 합한 유기 상을 MgSO4 상에서 건조시키고, 여과하고, 농축시키고, 메탄올에 이어 디에틸 에테르로 연화처리하여 화합물 49를 수득하였다.

실시예 50 및 51
화합물 50 및 51의 제조
N-(2,4-디플루오로벤질)-9-히드록시-8,10-디옥소-2,3,5,6,8,10,14,14a-옥타히드로-2,6-메타노피리도[1',2':4,5]피라지노[2,1-b][1,6,3]디옥사조신-11-카르복스아미드 50 및 51


단계 1
메틸 5-(2,4-디플루오로벤질카르바모일)-1-(2,2-디히드록시에틸)-3-메톡시-4-옥소-1,4-디히드로피리딘-2-카르복실레이트 (1-C, 392 mg, 0.95 mmol) (실시예 87), 라세미 시스-5-아미노테트라히드로-2H-피란-3-올 (WO 2012/145569 (Bennett, B. L. et al.), 2012년 4월 20일 출원) (112 mg, 0.95 mmol), 및 탄산칼륨 (134 mg, 0.97 mmol)을 3.8 mL 아세토니트릴/0.2 mL 아세트산에 녹이고, 90℃에서 90분 동안 교반한 후, 반응 혼합물을 DCM과 염수 사이에 분배하고, 수성 상을 DCM으로 추출하고, 합한 유기 상을 MgSO4 상에서 건조시키고, 여과하고, 농축시키고, SGC (0-10% EtOH/DCM)에 의해 정제하여 중간체 50-A를 수득하였다.
단계 2
중간체 50-A (40 mg)를 키랄팩 IC 칼럼 상에서 키랄 SFC에 의해 용리액으로서 초임계 이산화탄소 중 10% DMF를 사용하여 분리하여 중간체 50-B (제1 용리 피크) 및 51-A (제2 용리 피크)를 거울상이성질체풍부화 형태로 수득하였다. 중간체 50-B의 경우: (미지의 절대 입체화학), 키랄 HPLC 체류 시간 = 11.48분 (키랄팩 IC, 150 x 4.6 mm, 1 mL/분 MeOH). 중간체 51-A의 경우: (미지의 절대 입체화학), 키랄 HPLC 체류 시간 = 14.35분 (키랄팩 IC, 150 x 4.6 mm, 1 mL/분 MeOH).
단계 3a
브로민화마그네슘 (12 mg, 0.06 mmol)을 1 mL 아세토니트릴 중 중간체 50-B (10.5 mg, 0.02 mmol, 미지의 절대 입체화학)의 용액에 첨가하였다. 반응 혼합물을 50℃에서 1시간 동안 교반하고, 10% 수성 HCl로 산성화시키고, 수성 및 디클로로메탄 사이에 분배하고, 수성 상을 디클로로메탄으로 추출하였다. 합한 유기 상을 MgSO4 상에서 건조시키고, 여과하고, 농축시키고, HPLC (ACN/H2O, 0.1% TFA 개질제 함유)에 의해 정제하여 화합물 50을 수득하였다.

단계 3b
브로민화마그네슘 (13 mg, 0.07 mmol)을 1 mL 아세토니트릴 중 중간체 51-A (13.2 mg, 0.03 mmol, 미지의 절대 입체화학)의 용액에 첨가하였다. 반응 혼합물을 50℃에서 1시간 동안 교반하고, 10% 수성 HCl로 산성화시키고, 수성 및 디클로로메탄 사이에 분배하고, 수성 상을 디클로로메탄으로 추출하였다. 합한 유기 상을 MgSO4 상에서 건조시키고, 여과하고, 농축시키고, HPLC (ACN/H2O, 0.1% TFA 개질제 함유)에 의해 정제하여 화합물 51을 수득하였다.

실시예 52
화합물 52의 제조
(2S,5R,13aS)-N-(2-시클로프로폭시-4-플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

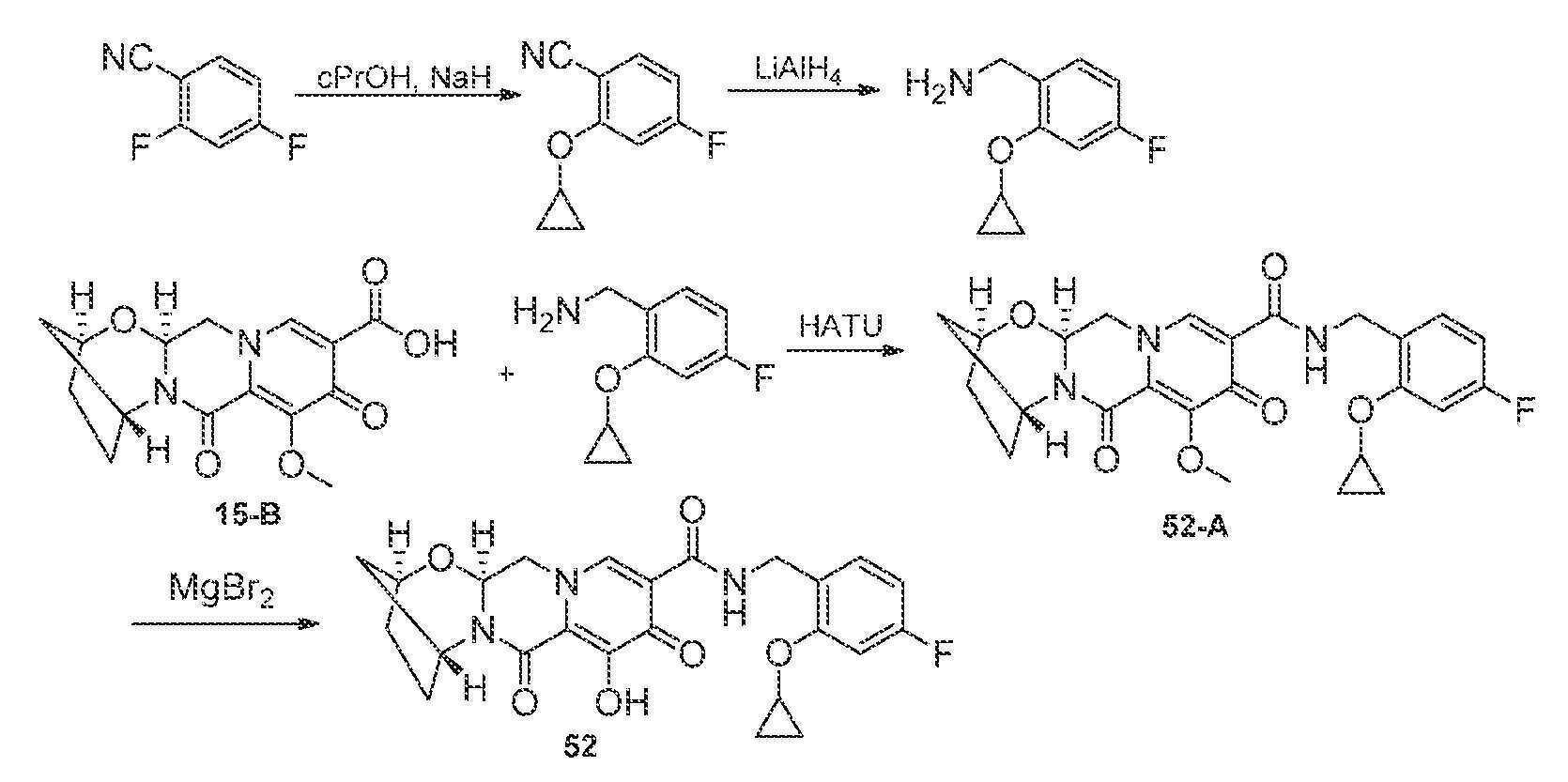
단계 1
20 mL 디옥산 중 시클로프로판올 (1.9 g, 29 mmol)의 용액을 80 mL 디옥산 중 수소화나트륨의 0℃ 용액 (광유 중 60% 분산액, 1.04 g, 26 mmol)에 적가하였다. 반응 혼합물이 실온으로 가온되도록 하고, 2,4-디플루오로벤조니트릴 (3.48 g, 25 mmol)을 조금씩 첨가하고, 반응 온도를 95℃로 상승시켰다. 18시간 동안 교반한 후 반응 용액을 실온으로 냉각시키고, 에틸 아세테이트로 희석하고, 물로 2회 및 염수로 2회 세척하고, MgSO4 상에서 건조시키고, 여과하고, 실리카 겔 상에서 농축시켰다. 실리카 겔 크로마토그래피 (0-10% EtOAc/헥산)에 의해 정제하여 2-시클로프로폭시-4-플루오로벤조니트릴을 수득하였다.
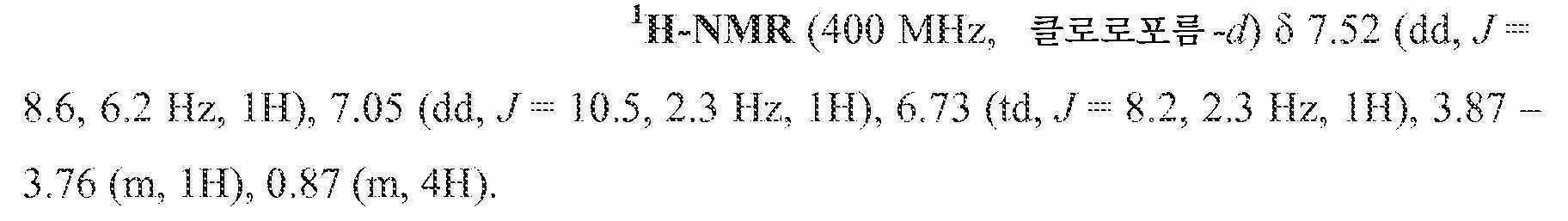
단계 2
THF 중 수소화알루미늄리튬의 0℃ 현탁액 (1M, 15 mL, 15 mmol)에 14 mL 디에틸 에테르 중 2-시클로프로폭시-4-플루오로벤조니트릴을 적가하였다. 반응 용액을 3시간 동안 교반하고, 실온으로 서서히 가온하고, 0℃로 재냉각시키고, 추가의 THF 중 수소화알루미늄리튬 8 mL (1M, 8 mmol)를 첨가하고, 추가로 90분 동안 교반하였다. 반응물을 0.9 mL 물, 0.9 mL 15% NaOH(수성), 및 2.7 mL 물의 순차적 첨가에 의해 켄칭하였다. 반응물을 디에틸 에테르로 헹구면서 셀라이트를 통해 여과하고, MgSO4 상에서 건조시키고, 농축시켜 충분한 순도의 2-시클로프로폭시-4-플루오로벤질아민을 수득하였고, 조 물질로서 수행하였다.
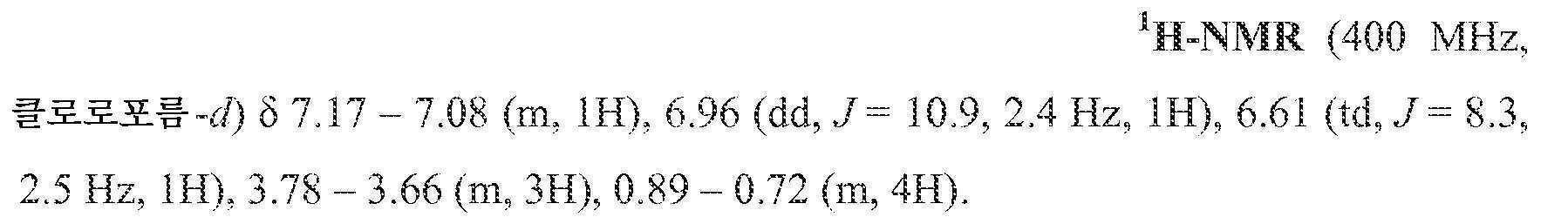
단계 3
화합물 15-B (46 mg, 0.14 mmol)를 1 mL 아세토니트릴에 녹이고, 2-시클로프로폭시-4-플루오로벤질아민 (32 mg, 0.18 mmol), HATU (62 mg, 0.16 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.04 mL, 0.22 mmol)으로 처리하고, 실온에서 2시간 동안 교반한 후, LCMS 분석하여 화합물 15-B의 완전한 소모 및 중간체 52-A의 형성을 밝혀내었다. 반응 혼합물로 후속 단계를 수행하였다.
단계 4
이전 단계의 조 반응 용액에 MgBr2 (56 mg, 0.30 mmol)를 첨가하였다. 반응 혼합물을 50℃에서 90분 동안 교반하고, 10% 수성 HCl로 산성화시키고, 수성 및 디클로로메탄 사이에 분배하고, 수성 상을 디클로로메탄으로 추출하였다. 합한 유기 상을 MgSO4 상에서 건조시키고, 여과하고, 농축시키고, HPLC (ACN/H2O, 0.1% TFA 개질제 함유)에 의해 정제하여 화합물 52를 수득하였다.

실시예 53
화합물 53의 제조
(2R,5S,13aR)-N-(2-시클로프로폭시-4-플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드


단계 1
화합물 42-A (46 mg, 0.14 mmol)를 1 mL 아세토니트릴에 녹이고, 2-시클로프로폭시-4-플루오로벤질아민 (33 mg, 0.18 mmol), HATU (61 mg, 0.16 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.04 mL, 0.24 mmol)으로 처리하고, 실온에서 2시간 동안 교반한 후, LCMS 분석하여 화합물 42-A의 완전한 소모 및 중간체 53-A의 형성을 밝혀내었다. 반응 혼합물로 후속 단계를 수행하였다.
단계 2
이전 단계의 조 반응 용액에 MgBr2 (55 mg, 0.30 mmol)를 첨가하였다. 반응 혼합물을 50℃에서 90분 동안 교반하고, 10% 수성 HCl로 산성화시키고, 수성 및 디클로로메탄 사이에 분배하고, 수성 상을 디클로로메탄으로 추출하였다. 합한 유기 상을 MgSO4 상에서 건조시키고, 여과하고, 농축시키고, HPLC (ACN/H2O, 0.1% TFA 개질제 함유)에 의해 정제하여 화합물 53을 수득하였다.

실시예 54
화합물 54의 제조
(2R,5S)-N-((S)-1-(2,4-디플루오로페닐)-2,2,2-트리플루오로에틸)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드
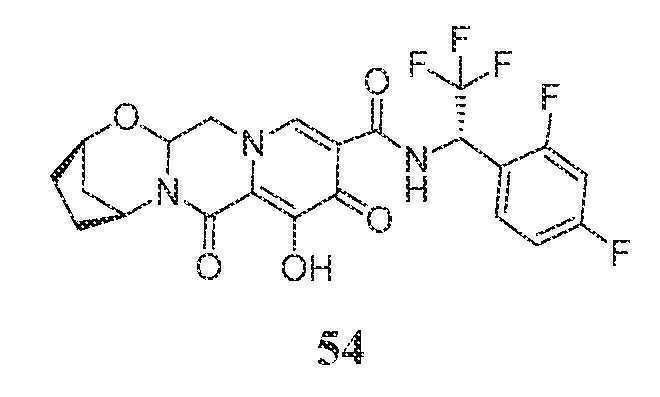
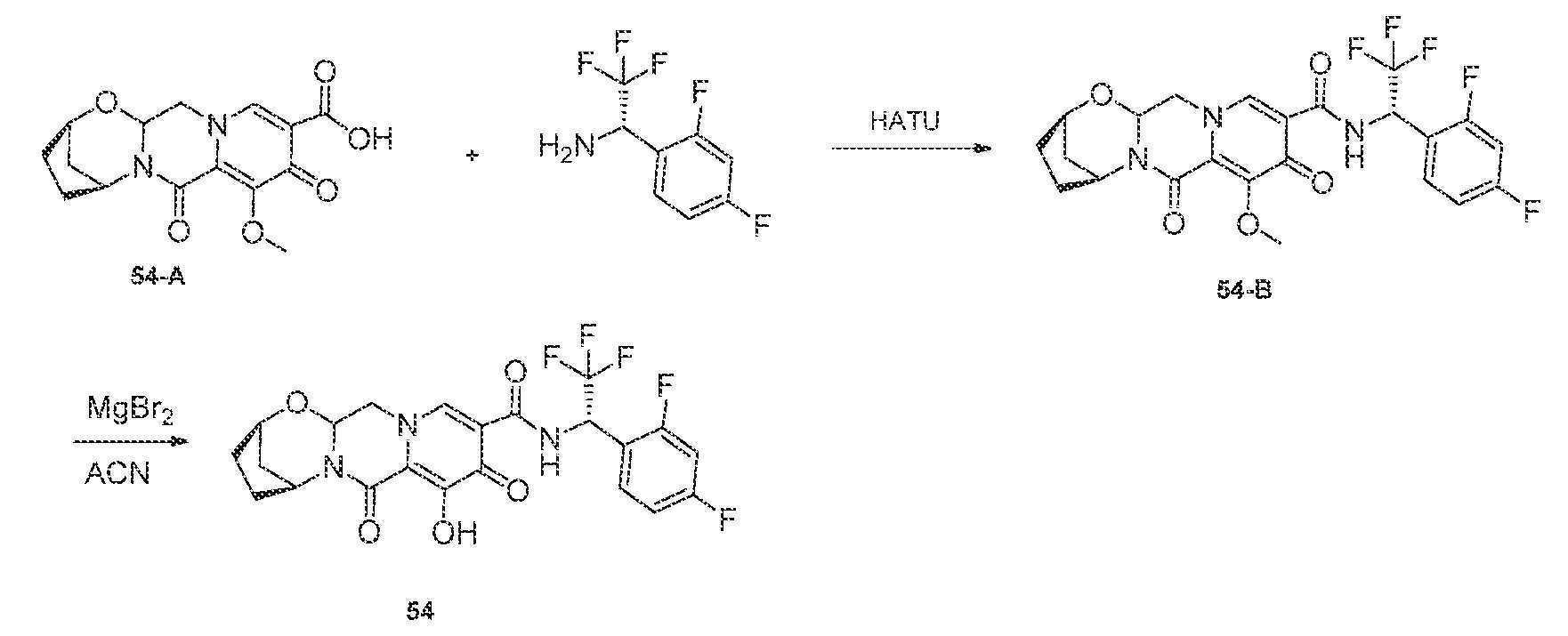
단계 1
50-mL 둥근 바닥 플라스크에 DCM (2 ml) 중 반응물 54-A (0.02 g, 0.06 mmol), (S)-1-(2,4-디플루오로페닐)-2,2,2-트리플루오로에탄아민 (0.019 g, 0.09 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.048 g, 0.38 mmol) 및 HATU (0.036 g, 0.09 mmol)를 채웠다. 반응 혼합물을 실온에서 1시간 동안 교반하였다. 반응 혼합물을 농축시키고, EtOAc (50 mL) 중에 재용해시키고, 포화 NaHCO3 (2x), 포화 NH4Cl로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 헥산-EtOAc를 사용하여 정제하여 54-B를 수득하였다.
단계 2
50-mL 둥근 바닥 플라스크에 아세토니트릴 (2 mL) 중 반응물 54-B (0.03 g, 0.058 mmol) 및 브로민화마그네슘 (0.03 g, 0.15mmol)을 채웠다. 반응 혼합물을 50℃로 가열하였다. 10분 후, 반응 혼합물을 0℃로 냉각시키고, 1 N 염산 (0.5 mL)을 첨가하였다. 이어서, 반응 혼합물을 MeOH (2 mL)로 희석하였다. 여과한 후, 조 물질을 정제용 HPLC 정제 (30-70% 아세토니트릴:물, 0.1% TFA)에 의해 정제하여 화합물 54를 TFA 염으로서 수득하였다.

실시예 55
화합물 55의 제조
(1R,4S,12aS)-7-히드록시-6,8-디옥소-N-(2,4,6-트리플루오로벤질)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
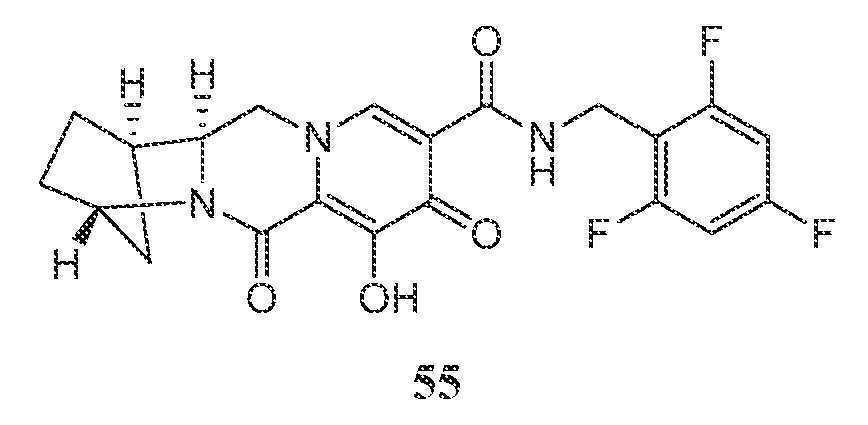

단계 1
H2 분위기 하의 EtOH (400 mL) 중 화합물 55-A (40.60 g, 150 mmol) 및 Pd(OH)2/C (12 g)의 혼합물을 실온에서 밤새 교반하였다. 반응 혼합물을 여과하고, HCl/EtOH (400 ml)로 처리하였다. 혼합물을 실온에서 2시간 동안 교반하였다. 반응 혼합물을 농축시켜 화합물 55-B를 수득하였으며, 이를 후속 단계에 정제 없이 사용하였다.
단계 2
CH3CN (1 L) 중 화합물 55-B (92.25 g, 0.45 mol) 및 K2CO3 (186.30 g, 1.35 mol)의 용액에 0℃에서 벤질 브로마이드 (76.50 g, 0.45 mol)를 첨가하였다. 혼합물을 실온에서 밤새 교반하였다. 반응 혼합물을 여과하고, 농축시키고, 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하여 화합물 55-C를 수득하였다.
단계 3
THF (400 mL) 중 디이소프로필아민 (50 g, 0.50 mol)의 혼합물에 n-BuLi (200 mL, 0.50 mol)를 -78℃에서 N2 분위기 하에 첨가하였다. 0.5시간 후, 반응 혼합물을 20℃로 가온하고, 0.5시간 동안 교반하였다. 혼합물을 -78℃로 냉각시키고, THF (600 mL) 중 화합물 55-C (64.75 g, 0.25 mol)의 용액을 N2 분위기 하에 첨가하였다. 혼합물을 4시간 동안 교반하고, 포화 NH4Cl 용액으로 켄칭하였다. 혼합물을 EtOAc로 추출하고, 유기 층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하여 화합물 55-D를 수득하였다.
단계 4
4N HCl (1.30 L) 중 화합물 55-D (129.50 g 0.50 mol)의 혼합물을 4시간 동안 환류하였다. 혼합물을 농축시켰다. 잔류물을 HPLC에 의해 정제하여 화합물 55-E를 수득하였다.
단계 5
H2 분위기 하의 EtOH (400 mL) 중 화합물 55-E (47 g, 176 mmol) 및 Pd(OH)2/C (9 g)의 혼합물을 실온에서 밤새 교반하였다. 반응 혼합물을 농축시켜 화합물 55-F를 수득하였으며, 이를 후속 단계에 정제 없이 사용하였다.

단계 6
디옥산 (120 mL) 중 화합물 55-F (29.20 g, 165 mmol) 및 2N NaOH 용액 (330 mL, 0.66 mol)의 혼합물에 0℃에서 Boc2O (39.60 g, 181 mmol)를 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하였다. 혼합물을 3N HCl을 사용하여 pH=5~6으로 조정하고, DCM으로 추출하였다. 유기 층을 Na2SO4 상에서 건조시키고, 여과하고, 농축시켜 55-G를 수득하였다.

단계 7
0℃로 냉각시킨 THF (10 mL) 중 화합물 55-G (500 mg, 2.07 mmol)의 혼합물에 BH3-DMS THF 착물 (THF 중 2N, 8.23 mmol, 4.1 mL)을 천천히 첨가하였다. 기체가 발생하였다. 내부 온도를 모니터링하여 어떠한 주요 발열도 없음을 보장하였다. 반응물을 밤새 실온으로 가온되도록 하였다. LC/MS에 의하면 일부 출발 물질이 남아있었고, 추가의 2 mL BH3-DMS THF 착물을 첨가하고, 혼합물을 추가로 3시간 동안 교반한 다음, 반응물을 0℃로 냉각시키고, 메탄올로 천천히 켄칭하였다 (기체 발생). 내부 온도를 모니터링하여 발열을 25℃ 미만으로 보장하였다. 혼합물을 농축시킨 다음, 실리카 겔 크로마토그래피 (20-40% EtOAc/헥산)에 의해 정제하여 55-H를 수득하였다.
단계 8
화합물 55를 실시예 41에 기재된 바와 같이, 41-B를 55-H로 치환하여 제조하여 화합물 55를 수득하였다.

실시예 56
화합물 56의 제조
(1R,2S,4R,12aR)-2-플루오로-7-히드록시-6,8-디옥소-N-(2,4,6-트리플루오로벤질)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드


단계 1
테트라히드로푸란 (65 ml) 중 56-A (5 g, 19.43 mmol)의 용액을 빙조에서 냉각시키면서 0.5 M 9-보라비시클로[3.3.1]노난 (48.58 ml)을 적가하였다. 반응 혼합물을 실온으로 가온하였다. 18시간 후, 반응물을 0℃로 냉각시키고, 2M 수산화나트륨 (34 ml) 및 과산화수소 (9.34 ml, 97.15 mmol)의 혼합물을 적가하였다. 0℃에서 2시간 후, 반응물을 실온으로 가온하고, 1시간 동안 교반하였다. 혼합물을 EtOAc로 희석하고, 물로 세척하였다. 수성 분획을 EtOAc로 추출하고, 합한 유기 분획을 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 실리카 칼럼 크로마토그래피 (50-70% EtOAc/헥산)에 의해 정제하여 56-B (3.05 g, 57%)를 수득하였다.
단계 2
N,N-디메틸포름아미드 (12 ml) 중 56-B (1.45 g, 5.27 mmol)의 용액에 tert-부틸클로로디페닐실란 (1.51 ml, 5.79 mmol) 및 이미다졸 (1.08 g, 15.8 mmol)을 첨가하였다. 18시간 후, 혼합물을 물로 희석하고, EtOAc (2x)로 추출하고, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 실리카 칼럼 크로마토그래피 (10-20% EtOAc/헥산)에 의해 정제하여 56-C (2.6 g, 96.1%)를 수득하였다.
단계 3
EtOH (26 mL) 및 아세트산 (3 mL) 중 56-C (3.27 g, 6.36 mmol)의 용액에 10% PdOH/C (0.52 g, 3.7 mmol)를 첨가하고, 현탁액을 파르 장치에서 50 atm에서 20시간 동안 진탕시켰다. 셀라이트를 통해 여과한 후, 케이크를 EtOH로 세척하고, 여과물을 진공 하에 농축시켰다. 잔류물을 에탄올 (26 ml) 및 아세트산 (3 ml, 52.4 mmol) 중에 용해시키고, 10% PdOH/C (0.52 g, 3.7 mmol)로 처리하고, 파르 장치에서 50 atm에서 20시간 동안 진탕시켰다. 셀라이트를 통해 여과하고, 케이크를 EtOH로 세척하고, 여과물을 진공 하에 농축 건조시켜 조 탈보호된 생성물 (2.07g, 79.4%)을 수득하였다.
THF (20 ml) 중 조 잔류물 (2 g, 4.88 mmol) 및 디-tert-부틸 디카르보네이트 97% (2.14 g, 9.79 mmol)에 N,N-디이소프로필에틸아민 (DIPEA) (2.14 ml, 12.27 mmol)을 첨가하였다. 20시간 후, 반응 혼합물을 물로 희석하고, EtOAC (2x)로 추출하고, 2개의 유기 분획을 물로 세척하고, 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 실리카 칼럼 크로마토그래피 (10-20% EtOAc/헥산)에 의해 정제하여 56-D (2.13 g, 86.14%)를 수득하였다.
단계 4
THF (20 ml) 중 56-D (2.07 g, 4.06 mmol)의 용액을 빙조에서 교반하면서 THF 중 2.0 M LiBH4 (4.07 ml)를 첨가하고, 생성된 혼합물을 실온에서 18시간 동안 교반하였다. 그 후, 반응 혼합물을 에틸 아세테이트로 희석하고, 천천히 물로 처리하였다. 2개의 상을 분리하고, 수성 분획을 에틸 아세테이트로 다시 추출하였다. 2개의 유기 분획을 물로 세척하고, 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 실리카 칼럼 크로마토그래피 (20-40% EOAc/헥산)에 의해 정제하여 56-E (1.59 g, 81.3%)를 수득하였다.
단계 5
THF (90 ml) 중 56-E (1.58 g, 3.28 mmol), 프탈이미드 (0.79 g, 5.38 mmol) 및 트리페닐포스핀 (1.93 g, 7.37 mmol)의 혼합물을 빙조에서 냉각시켰다. 디이소프로필 아조디카르복실레이트, 95% (1.46 ml, 7.42 mmol)를 첨가하였다. 이어서, 혼합물을 실온으로 가온하고, 20시간 동안 교반하였다. 그 후, 반응 혼합물을 농축시키고, 잔류물을 에테르 중에 용해시키고, 빙조에서 냉각시키고, 1.5시간 동안 교반하였다. 고체를 여과하고, 여과물을 농축시켰다. 잔류물을 실리카 칼럼 크로마토그래피 (10-30% EtOAc/헥산)에 의해 정제하여 보호된 아미노 화합물 (1.86 g, 92.8%)을 수득하였다.
에탄올 (19 ml) 중 보호된 아미노 화합물 56-F (1.85 g, 3.03 mmol) 및 히드라진 수화물 (0.6 ml, 12.39 mmol)의 용액을 70℃에서 2시간 동안 교반하였다. 반응 혼합물을 빙조에서 냉각시키고, 에테르 (10 ml)를 첨가하고, 혼합물을 30분 동안 교반하였다. 형성된 고체를 여과하고, 여과물을 진공 하에 농축 건조시켰다.
단계 6
물 (15 mL) 및 EtOH (15 mL) 중 조 아미노 화합물 56-F (991 mg, 2.06 mmol), 화합물 38-F (실시예 38) (714 mg, 2.06 mmol) 및 NaHCO3 (347 mg, 4.12 mmol)의 혼합물을 20시간 동안 교반하였다. 반응 혼합물을 진공 하에 농축시키고, 잔류물을 물과 EtOAc 사이에 분배하였다. 수성 층을 EtOAc로 재추출하고, 합한 유기 층을 건조 (Na2SO4)시키고, 농축시켰다. 잔류물 (1.5 g)을 CH2Cl2 (5 mL) 중에 용해시키고, 디옥산 중 4N HCl (18.6 mL)을 첨가하였다. 1.5시간 후, 혼합물을 농축 건조시키고, 톨루엔과 공증발시키고, 진공 하에 건조시켰다.
톨루엔 (25 ml) 중 조 잔류물 (1.38 g) 및 DBU (1.4 ml, 9.38 mmol)를 110℃에서 교반하였다. 35분 후, 혼합물을 농축시키고, 잔류물을 실리카 칼럼 크로마토그래피 (5-15% MeOH/EtOAc)에 의해 정제하여 56-G (450 mg, 72.3%)를 수득하였다.
단계 7
MeOH (14 ml) 및 THF (14 ml) 중 56-G (890 mg, 1.34 mmol)의 혼합물을 실온에서 교반하면서 1M KOH (7.09 ml)를 첨가하였다. 30분 후, 반응 혼합물을 1N HCl로 중화시키고, EtOAc (2x)로 추출하고, 합한 유기 추출물을 건조 (Na2SO4)시키고, 농축시켰다.
디클로로메탄 (5 ml) 중 조 잔류물 (850 mg), 2,4,6-트리플루오로벤질아민 (248 mg, 1.54 mmol) 및 HATU (662 mg, 1.74 mmol)의 현탁액을 실온에서 교반하면서 N,N-디이소프로필에틸아민 (DIPEA) (1.63 ml, 9.37 mmol)을 첨가하였다. 1시간 후, 추가의 2,4,6-디플루오로벤질아민 (32 mg, 0.2 mmol), HATU (153 mg, 0.4 mmol) 및 N,N-디이소프로필에틸아민 (DIPEA) (0.12 ml, 0.67 mmol)을 첨가하였다. 30분 후, 혼합물을 물로 희석하고, EtOAc (3x)로 추출하고, 합한 유기 상을 건조 (Na2SO4)시키고, 농축시키고, 잔류물을 실리카 칼럼 크로마토그래피 (50-75% EtOAc/헥산)에 의해 정제하여 56-H (919 mg, 88.23%)를 수득하였다.
단계 8
THF (5 ml) 중 56-H (915 mg, 1.18 mmol)의 용액을 빙조에서 교반하면서 THF 중 1.0 M 테트라부틸암모늄 플루오라이드 (1.18 ml)를 적가하였다. 생성된 혼합물을 실온에서 30분 동안 교반하였다. 반응 혼합물을 진공 하에 농축시키고, 잔류물을 EtOAc로 희석하고, 물로 세척하고, 건조시키고 (Na2SO4), 농축시키고, 잔류물을 실리카 칼럼 크로마토그래피 (50-75% EtOAc/헥산에 이어서 5% MeOH/EtOAc)에 의해 정제하였다. 생성된 물질 (248 mg, 0.46 mmol)을 -78℃로 냉각시킨 디클로로메탄 (2 ml) 중에 용해시키면서 디에틸아미노황 트리플루오라이드 (0.07 mL, 0.55 mmol)를 적가하고, 반응물을 실온으로 가온하고, 1시간 동안 교반하였다. 반응물을 빙조에서 냉각시키고, 포화 NaHCO3으로 켄칭하고, 2개의 상을 분리하고, 분리된 수성 분획을 CH2Cl2로 추출하였다. 2개의 유기 분획을 합하고, 건조시키고 (Na2SO4), 농축시켰다. 잔류물을 실리카 칼럼 크로마토그래피 (1% MeOH/EtOAc)에 의해 정제하여 56-J (75 mg) (LCMS-ESI+ (m/z): [M+H]+ 계산치, C28H23F4N3O4: 541.49; 실측치: 542.320) 및 56-I (30 mg) (LCMS-ESI+ (m/z): [M+H]+ 계산치, C28H22F3N3O4: 521.49; 실측치: 522.05)를 수득하였다.
단계 9
화합물 56-J (75 mg, 139 mmol)를 TFA (1 mL) 중에 용해시키고, 실온에서 10분 동안 교반하고, 용액을 농축시켰다. 잔류물을 역상 HPLC (제미니(Gemini), 15에서 43% ACN/H2O + 0.1% TFA)에 의해 정제하여 화합물 56을 수득하였다.

실시예 57
화합물 57의 제조
(1R,4R,12aR)-2,2-디플루오로-7-히드록시-6,8-디옥소-N-(2,4,6-트리플루오로벤질)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
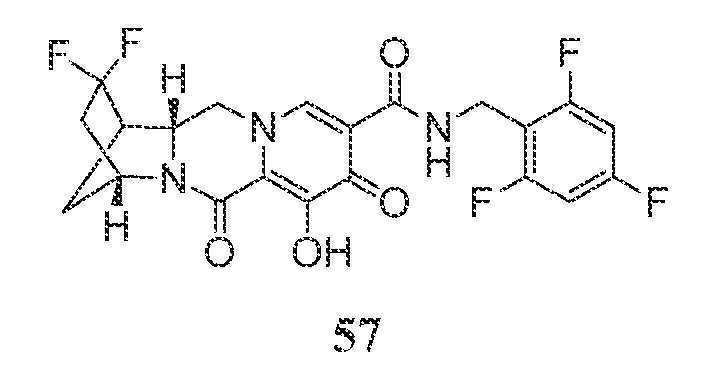
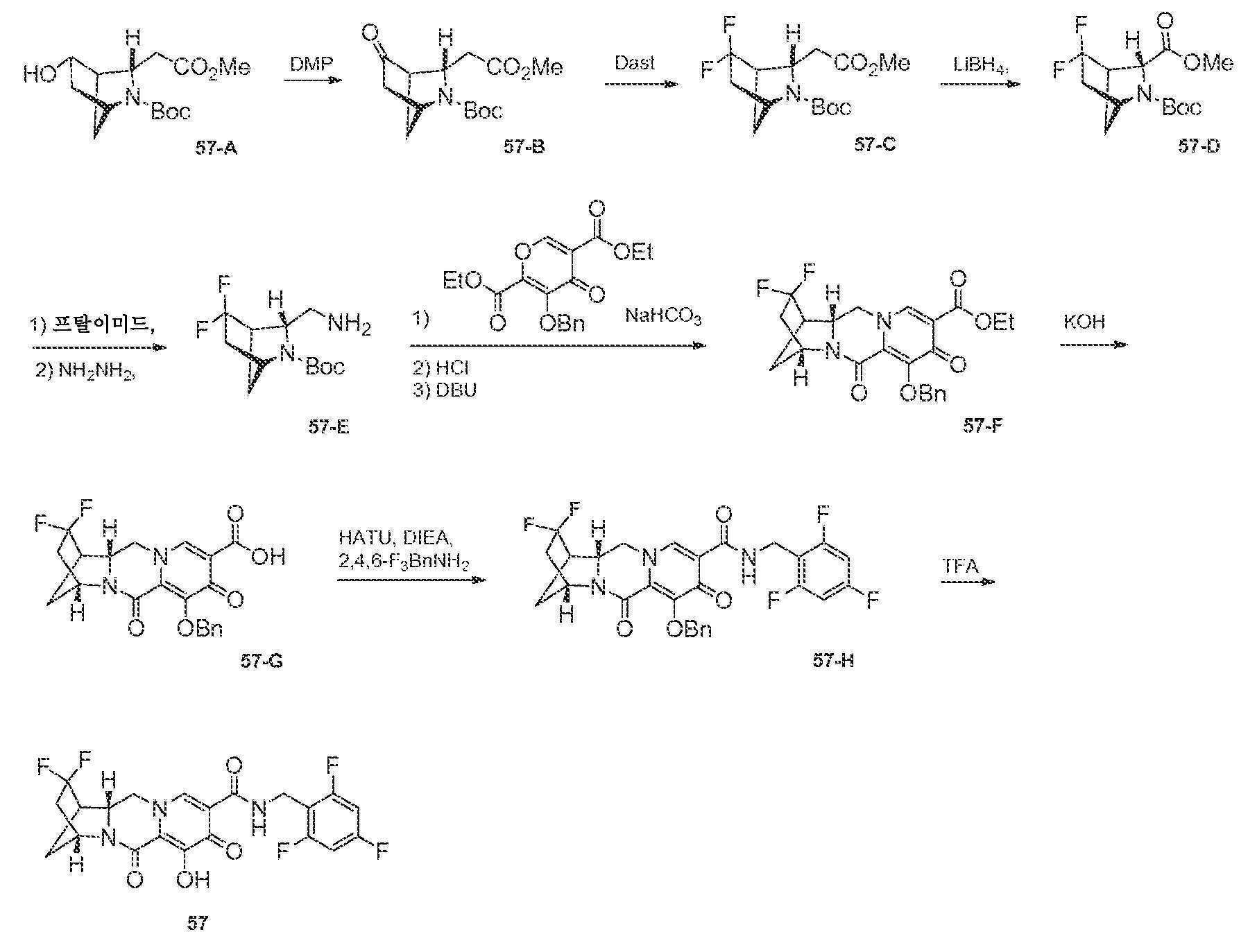
단계 1
디클로로메탄 (30 ml) 중 57-A (1.45 g, 5.34 mmol)의 용액을 빙조에서 냉각시키면서 데스 마르틴 퍼아이오디난 (4.53 g, 10.69 mmol)을 여러 부분으로 첨가하고, 반응물을 실온에서 18시간 동안 교반하였다. 반응물을 물의 첨가에 의해 켄칭하고, 침전물을 여과하고, Na2S2O3의 포화 용액을 첨가하였다. 2상 용액으로 변화될 때까지 혼합물을 교반한 다음, 포화 NaHCO3을 첨가하고, 수성 층을 CH2Cl2로 추출하였다. 합한 유기 분획을 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 실리카 칼럼 크로마토그래피 (30-50% EtOAc/헥산)에 의해 정제하여 57-B (1.13 g, 78.2%)를 수득하였다.
단계 2
디클로로메탄 (10 ml) 중 57-B (0.5 g, 1.86 mmol)의 용액을 -78℃로 냉각시키면서 디에틸아미노황 트리플루오라이드 (0.52 mL, 3.91 mmol)를 적가하고, 반응물을 실온으로 가온하고, 18시간 동안 교반하였다. 반응물을 빙조에서 냉각시키고, 포화 NaHCO3으로 켄칭하고, 2개의 상을 분리하고, 분리된 수성 분획을 CH2Cl2로 추출하였다. 2개의 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 실리카 칼럼 크로마토그래피 (20-50% EtOAc/헥산)에 의해 정제하여 57-C (518 mg, 95.39%)를 수득하였다.

단계 3
THF (10 ml) 중 57-C (935 mg, 3.21 mmol)의 용액을 빙조에서 교반하면서 THF 중 2.0 M LiBH4 (3.22 ml)를 첨가하고, 생성된 혼합물을 실온에서 18시간 동안 교반하였다. 그 후, 반응 혼합물을 에틸 아세테이트로 희석하고, 물을 천천히 첨가하였다. 2개의 상을 분리하고, 분리된 수성 분획을 에틸 아세테이트로 추출하였다. 2개의 유기 분획을 물로 세척하고, 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 실리카 칼럼 크로마토그래피 (20-40% EtOAc/헥산)에 의해 정제하여 57-D (724 mg, 85.67%)를 수득하였다.
단계 4
THF (45 ml) 중 57-D (720 mg, 2.74 mmol), 프탈이미드 (402 mg, 2.73 mmol) 및 트리페닐포스핀 (1.61 g, 6.15 mmol)의 혼합물을 빙조에서 냉각시켰다. 디이소프로필 아조디카르복실레이트, 95% (1.22 ml, 6.19 mmol)를 첨가하였다. 이어서, 혼합물을 실온으로 가온하고, 20시간 동안 교반하였다. 그 후, 반응 혼합물을 농축시키고, 잔류물을 에테르 중에 용해시키고, 빙조에서 냉각시키고, 1.5시간 동안 교반하였다. 고체를 여과한 후, 여과물을 농축시켰다. 잔류물을 실리카 칼럼 크로마토그래피 (40-60% EtOAc/헥산)에 의해 정제하여 프탈이미드 부가물 (1.07 g, 99.7%)을 수득하였다.
에탄올 (10 ml) 중 프탈이미드 부가물 (1.07 g, 2.73 mmol) 및 히드라진 수화물 (0.54 mL, 11.15 mmol)의 용액을 70℃에서 2시간 동안 교반하였다. 반응 혼합물을 빙조에서 냉각시키고, 에테르 (10 ml)를 첨가하였다. 혼합물을 30분 동안 교반하였다. 형성된 고체를 여과하고, 여과물을 진공 하에 농축 건조시켜 조 57-E를 수득하였다.
단계 5
물 (15 mL) 및 EtOH (15 mL) 중 조 57-E (709 mg, 2.7 mmol), 화합물 38-F (실시예 38) (936 mg, 2.7 mmol) 및 NaHCO3 (454 mg, 5.41 mmol)의 혼합물을 20시간 동안 교반하였다. 반응 혼합물을 진공 하에 농축시키고, 잔류물을 물과 EtOAc 사이에 분배하였다. 수성 층을 EtOAc로 재추출하고, 합한 유기 층을 건조 (Na2SO4)시키고, 농축시켰다. 잔류물 (1.5 g)을 CH2Cl2 (7 mL) 중에 용해시키고, 디옥산 중 4N HCl (26.9 mL)을 첨가하였다. 1.5시간 후, 혼합물을 농축 건조시키고, 톨루엔과 공증발시키고, 진공 하에 건조시켰다. 톨루엔 (25 ml) 중 조 잔류물 (1.3 g) 및 DBU (2 ml, 13.4 mmol)를 110℃에서 교반하였다. 35분 후, 혼합물을 농축시키고, 잔류물을 실리카 칼럼 크로마토그래피 (5-15% MeOH/EtOAc)에 의해 정제하여 57-F (426 mg, 36.17%)를 수득하였다.
단계 6
MeOH (7 ml) 및 THF (7 ml) 중 화합물 57-F (426 mg, 0.96 mmol)의 혼합물을 실온에서 교반하면서 1M KOH (5.06 ml)를 첨가하였다. 30분 후, 반응 혼합물을 1N HCl로 중화시키고, EtOAc (2x)로 추출하고, 합한 유기 추출물을 건조 (Na2SO4)시키고, 조 57-G로 농축시켰다.
단계 7
디클로로메탄 (3 ml) 중 조 잔류물 57-G (189 mg), 2,4,6-트리플루오로벤질아민 (95 mg, 0.59 mmol) 및 HATU (276 mg, 0.73 mmol)의 현탁액을 실온에서 교반하면서 N,N-디이소프로필에틸아민 (DIPEA) (0.59 ml, 3.4 mmol)을 첨가하였다. 1시간 후, 혼합물을 물로 희석하고, EtOAc (3x)로 추출하였다. 합한 유기 상을 건조 (Na2SO4)시키고, 57-H로 농축시켰다.
단계 8
화합물 57-H (150 mg, 0.27 mmol)를 TFA (2 mL) 중에 용해시키고, 실온에서 10분 동안 교반하고, 용액을 농축시켰다. 잔류물을 역상 HPLC (제미니, 15에서 60% ACN/H2O + 0.1% TFA)에 의해 정제하여 화합물 57 (85 mg, 67.5%)을 수득하였다.

실시예 58
화합물 58의 제조
(1R,4R,12aR)-N-(3-클로로-2,4-디플루오로벤질)-2,2-디플루오로-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
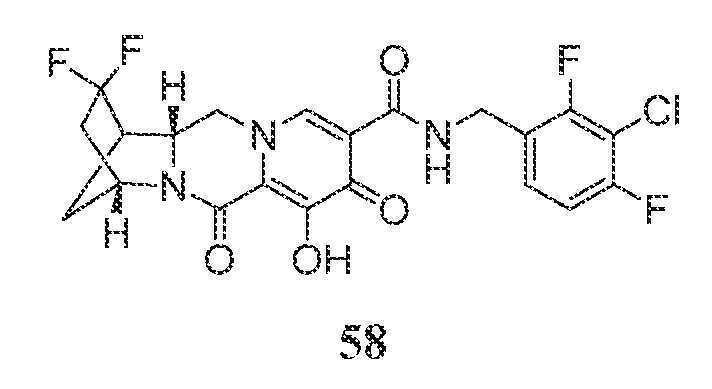

단계 1
디클로로메탄 (3 ml) 중 조 잔류물 57-G (120 mg), 3-클로로,2,4-디플루오로벤질아민 (67 mg, 0.38 mmol) 및 HATU (175 mg, 0.46 mmol)의 현탁액을 실온에서 교반하면서 N,N-디이소프로필에틸아민 (DIPEA) (0.38 ml, 0.28 mmol)을 첨가하였다. 1시간 후, 혼합물을 물로 희석하고, EtOAc (3x)로 추출하고, 합한 유기 상을 건조 (Na2SO4)시키고, 농축시켜 58-A를 수득하였다.
단계 2
화합물 58-A (166 mg)를 TFA (2 mL) 중에 용해시키고, 실온에서 10분 동안 교반하고, 용액을 농축시켰다. 잔류물을 역상 HPLC (제미니, 15에서 70% ACN/H2O + 0.1% TFA)에 의해 정제하여 화합물 57 (60 mg, 42.8%)을 수득하였다.

실시예 59
화합물 59의 제조
(1R,2R,4R,12aR)-2-플루오로-7-히드록시-6,8-디옥소-N-(2,4,6-트리플루오로벤질)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
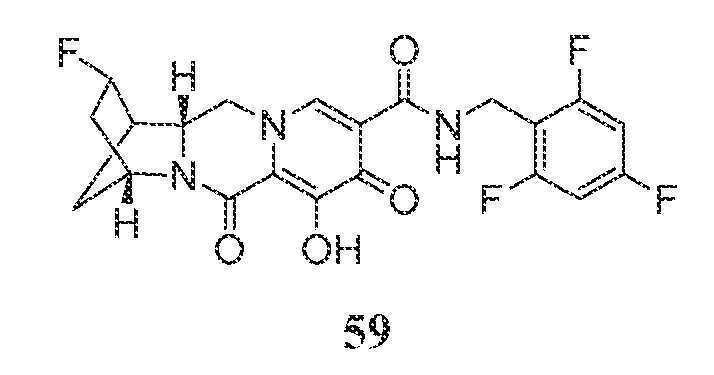
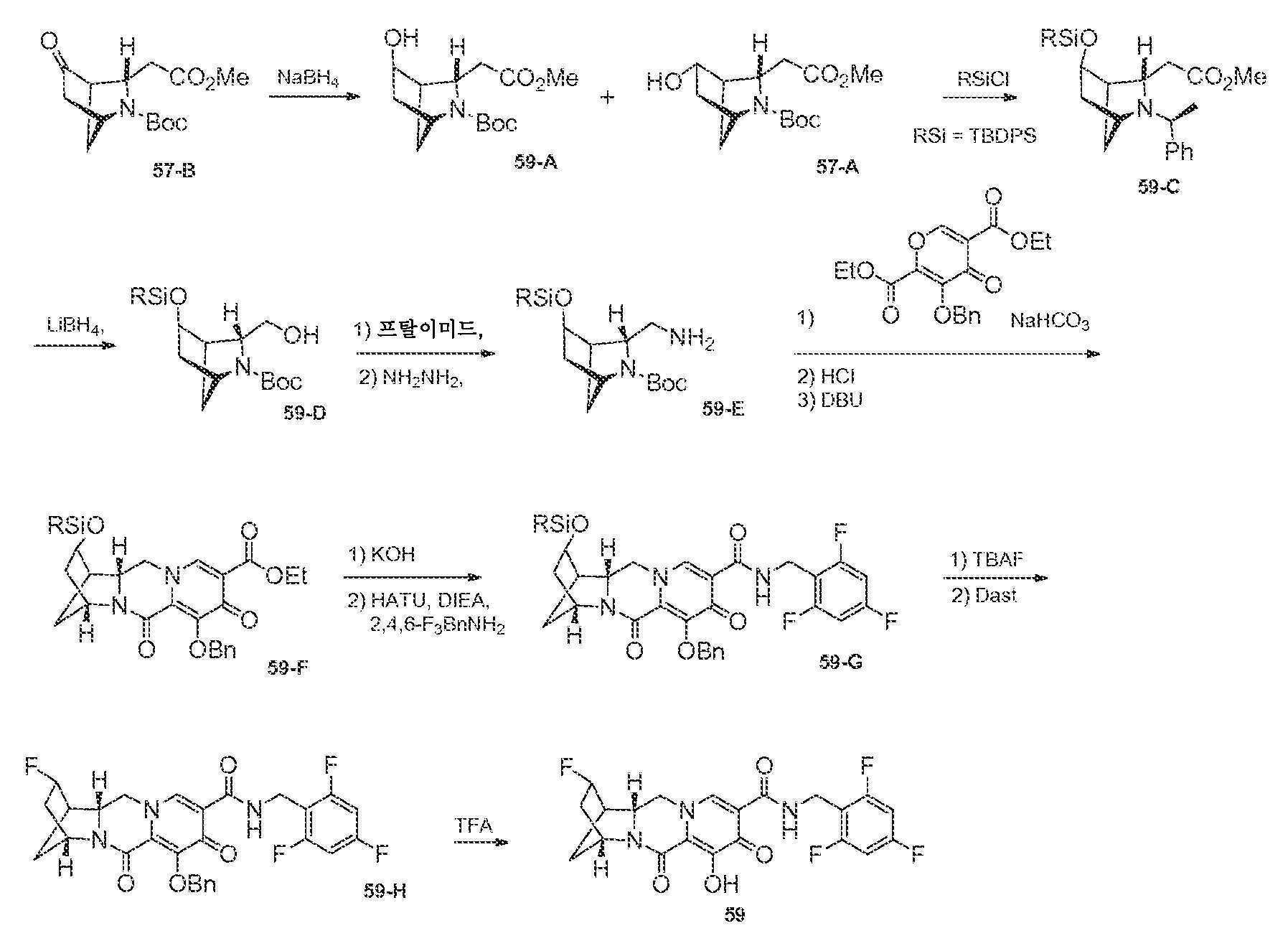
단계 1
메탄올 (35 mL) 중 57-B (1.9 g, 7.06 mmol)의 용액을 0℃에서 교반하면서 수소화붕소나트륨 (667 mg, 17.64 mmol)을 조금씩 첨가하고, 생성된 혼합물을 실온에서 30분 동안 교반하였다. 반응 혼합물을 빙조에서 냉각시키고, 물을 첨가하여 켄칭하고, 농축시켰다. 잔류물을 물과 EtOAc 사이에 분배하였다. 수성 층을 EtOAc로 재추출하고, 합한 유기 층을 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 실리카 칼럼 크로마토그래피 (30-60% EtOAc/헥산)에 의해 정제하여 59-A (1.49 g)를 수득하였다.

단계 2
N,N-디메틸포름아미드 (5 ml) 중 59-A (686 mg, 2.53 mmol)의 용액에 tert-부틸클로로디페닐실란 (0.723 mL, 2.78 mmol) 및 이미다졸 (516 mg, 7.56 mmol)을 첨가하였다. 18시간 후, 혼합물을 물로 희석하고, EtOAc (2x)로 추출하고, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 실리카 칼럼 크로마토그래피 (10-20% EtOAc/헥산)에 의해 정제하여 59-C를 수득하였다.
단계 3
THF (13 ml) 중 59-C (1.23 g, 2.41 mmol)의 용액을 빙조에서 교반하면서 THF 중 2.0 M LiBH4 (2.42mL, 4.84 mmol)를 첨가하고, 생성된 혼합물을 실온에서 18시간 동안 교반하였다. 반응 혼합물을 에틸 아세테이트로 희석한 후, 물을 천천히 첨가하고, 2개의 상을 분리하고, 분리된 수성 분획을 에틸 아세테이트로 추출하였다. 2개의 유기 분획을 물로 세척하고, 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 실리카 칼럼 크로마토그래피 (20-40% EtOAc/헥산)에 의해 정제하여 59-D를 수득하였다.
단계 4
THF (50 ml) 중 59-D (963 mg, 2.0 mmol), 프탈이미드 (482 mg, 3.28 mmol) 및 트리페닐포스핀 (1.18 g, 4.49 mmol)의 혼합물을 빙조에서 냉각시켰다. 디이소프로필 아조디카르복실레이트, 95% (0.89 mL, 4.52 mmol)를 첨가하였다. 이어서, 혼합물을 실온으로 가온하고, 20시간 동안 교반하였다. 그 후, 반응 혼합물을 농축시키고, 잔류물을 에테르 중에 용해시키고, 빙조에서 냉각시키고, 1.5시간 동안 교반하였다. 그 후, 고체를 여과하고, 여과물을 농축시켰다. 잔류물을 실리카 칼럼 크로마토그래피 (10-30% EtOAc/헥산)에 의해 정제하여 프탈이미드 부가물을 수득하였다.
에탄올 (12 ml) 중 프탈이미드 부가물 (1.2 g, 1.97 mmol) 및 히드라진 수화물 (0.4 ml, 8.03 mmol)의 용액을 70℃에서 2시간 동안 교반하였다. 반응 혼합물을 빙조에서 냉각시키고, 에테르 (10 ml)를 첨가하고, 혼합물을 30분 동안 교반하였다. 형성된 고체를 여과하고, 여과물을 진공 하에 농축 건조시켜 59-E를 수득하였다.
단계 5
물 (12 mL) 및 EtOH (12 mL) 중 조 59-E (770 mg, 1.60 mmol), 화합물 38-F (실시예 38) (555 mg, 1.60 mmol) 및 NaHCO3 (269 mg, 3.20 mmol)의 혼합물을 20시간 동안 교반하였다. 반응 혼합물을 진공 하에 농축시키고, 잔류물을 물과 EtOAc 사이에 분배하였다. 수성 층을 EtOAc로 재추출하고, 합한 유기 층을 건조 (Na2SO4)시키고, 농축시켰다.
잔류물 (1.29 g)을 CH2Cl2 (4 mL) 중에 용해시키고, 디옥산 중 4N HCl (15.6 mL)을 첨가하였다. 1.5시간 후, 혼합물을 농축 건조시키고, 톨루엔과 공증발시키고, 진공 하에 건조시켰다.
톨루엔 (20 ml) 중 조 잔류물 (1.09 mg) 및 DBU (1.17 ml, 7.8 mmol)를 110℃에서 교반하였다. 35분 후, 혼합물을 농축시키고, 잔류물을 실리카 칼럼 크로마토그래피 (5-15% MeOH/EtOAc)에 의해 정제하여 59-F를 수득하였다.
단계 6
MeOH (10 ml) 및 THF (10 ml) 중 59-F (680 mg, 1.03 mmol)의 혼합물을 실온에서 교반하면서 1M KOH (5.42 ml)를 첨가하였다. 30분 후, 반응 혼합물을 1N HCl로 중화시키고, EtOAc (2x)로 추출하고, 합한 유기 추출물을 건조 (Na2SO4)시키고, 농축시켰다.
디클로로메탄 (6 ml) 중 조 잔류물 (650 mg), 2,4,6-트리플루오로벤질아민 (214 mg, 1.33 mmol) 및 HATU (623 mg, 1.64 mmol)의 현탁액을 실온에서 교반하면서 N,N-디이소프로필에틸아민 (DIPEA) (1.34 ml, 7.68 mmol)을 첨가하였다. 2시간 후, 혼합물을 물로 희석하고, EtOAc (3x)로 추출하고, 합한 유기 상을 건조 (Na2SO4)시키고, 농축시키고, 잔류물을 실리카 칼럼 크로마토그래피 (50-75% EtOAc/헥산)에 의해 정제하여 59-G를 수득하였다.
단계 7
THF (10 ml) 중 59-G (648 mg, 0.83 mmol)의 용액을 빙조에서 교반하면서 THF 중 1.0 M 테트라부틸암모늄 플루오라이드 (0.83 ml)를 적가하고, 생성된 혼합물을 실온에서 30분 동안 교반하였다. 추가의 THF 중 1.0 M 테트라부틸암모늄 플루오라이드 (0.1 ml)를 적가하였다. 30분 후, 반응 혼합물을 진공 하에 농축시키고, 잔류물을 EtOAc로 희석하고, 물로 세척하고, 건조 (Na2SO4)시키고, 농축시키고, 잔류물을 실리카 칼럼 크로마토그래피 (5% MeOH/EtOAc)에 의해 정제하였다. 디클로로메탄 (3 ml) 중 잔류물 (290 mg, 0.54 mmol)의 용액을 -78℃로 냉각시키면서 디에틸아미노황 트리플루오라이드 (0.09 mL, 0.65 mmol)를 적가하고, 반응물을 실온으로 가온하고, 2.5시간 동안 교반하였다. 반응물을 빙조에서 냉각시키고, 포화 NaHCO3으로 켄칭하고, 2개의 상을 분리하고, 분리된 수성 분획을 CH2Cl2로 추출하였다. 2개의 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 실리카 칼럼 크로마토그래피 (1% MeOH/EtOAc)에 의해 정제하여 59-H를 수득하였다.
단계 8
화합물 59-H (103 mg, 0.19 mmol)를 TFA (1.4 mL) 중에 실온에서 15분 동안 용해시키고, 용액을 농축시켰다. 잔류물을 DMF 중에 현탁시키고, 여과하고, 침전된 생성물을 물로 세척하고, 진공 하에 건조시켜 화합물 59를 수득하였다.

실시예 60
화합물 60의 제조
(1R,4S,12aR)-N-(2,3-디클로로벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드

단계 1
MeOH (100 mL) 중 디메틸 3-메톡시-4-옥소-4H-피란-2,5-디카르복실레이트 (5.5 g, 23 mmol)의 용액에 41-E (실시예 41) (5 g, 22 mmol) 및 중탄산나트륨 (3.6 g, 43 mmol)을 첨가하였다. 용액을 실온에서 1.5시간 동안 교반하였다. 4M HCl (디옥산 중, 55 mL, 221 mmol)을 첨가하고, 용액을 50℃로 2시간 동안 가열하였다. 반응물을 실온으로 냉각시키고, 진공 하에 농축시켰다. 생성된 오일을 중탄산나트륨 중에 용해시키고, EtOAc로 세척하였다. 이어서, 수성 층을 CH2Cl2 (4x)로 추출하였다. 합한 CH2Cl2 추출물을 Na2SO4 상에서 건조시키고, 농축시켜 60-A를 수득하였다.
단계 2
MeOH (12 mL) 및 THF (23 mL) 중 60-A (3.7 g, 11.6 mmol)의 현탁액에 수성 KOH (2M, 15.7 mL, 31.4 mmol)를 첨가하였다. 생성된 용액을 실온에서 10분 동안 교반하였다. 휘발성 물질을 진공 하에 제거하고, 생성된 수성 층을 1N HCl로 산성화시켰다. 생성된 백색 고체를 여과하고, 물로 세척하고, 진공 하에 건조시켜 60-B를 수득하였다.

단계 3
CH2Cl2 (3.5 mL) 중 60-B (0.10 g, 0.33 mmol)의 용액에 (2,3-디클로로페닐)메탄아민 (0.12 g, 0.70 mmol), HATU (0.25 g, 0.66 mmol), 및 N,N-디이소프로필에틸아민 (DIPEA) (0.29 mL, 1.64 mmol)을 첨가하였다. LC/MS에 의해 완료된 것으로 판단될 때까지 생성된 용액을 실온에서 교반하였다. 반응 혼합물을 CH2Cl2로 희석하고, 1N HCl로 세척하였다. 수성 층을 CH2Cl2로 역추출하고, 합한 유기 층을 Na2SO4 상에서 건조시키고, 진공 하에 농축시켰다. 조 물질을 뜨거운 DMF 중에 용해시키고, 냉각 시 침전되도록 하였다. 여과하여 60-C를 수득하였다.
단계 4
아세토니트릴 (4.5 mL) 중 60-C (0.11 g, 0.24 mmol)의 슬러리에 브로민화마그네슘 (0.089 g, 0.48 mmol)을 첨가하였다. 반응 혼합물을 45℃로 2.5시간 동안 가열한 다음, 실온으로 냉각시켰다. 슬러리를 CH2Cl2로 희석하고, 1N HCl 및 염수로 세척하였다. 수성 층을 CH2Cl2 (2x)로 역추출하고, 합한 유기 층을 Na2SO4 상에서 건조시키고, 진공 하에 농축시켰다. 조 고체를 메탄올로 연화처리하고, 여과하여 60을 수득하였다.

실시예 61
화합물 61의 제조
(1R,4S,12aS)-N-(3-클로로-2,4-디플루오로벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드

61을 실시예 60과 유사하게, 41-E를 (1S,3S,4R)-tert-부틸 3-(아미노메틸)-2-아자비시클로[2.2.1]헵탄-2-카르복실레이트 (실시예 55에서 제조됨)로, (2,3-디클로로페닐)메탄아민을 (3-클로로-2,4-디플루오로페닐)메탄아민으로 치환하여 제조하였다.

실시예 62
화합물 62의 제조
(2R,5S,13aR)-N-(4-플루오로-2-(트리플루오로메틸)벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

화합물 62를 화합물 42와 유사한 방식으로 (2,4,6-트리플루오로페닐페닐)메탄아민 대신 (4-플루오로-2-(트리플루오로메틸)페닐)메탄아민을 사용하여 제조하였다.

실시예 63
화합물 63의 제조
(2R,5S,13aR)-N-(2-클로로-4-플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

화합물 63을 화합물 42와 유사한 방식으로 (2,4,6-트리플루오로페닐페닐)메탄아민 대신 (2-클로로-4-플루오로페닐)메탄아민을 사용하여 제조하였다.

실시예 64
화합물 64의 제조
(2R,5S,13aR)-8-히드록시-7,9-디옥소-N-(2,4,5-트리플루오로벤질)-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

화합물 64를 화합물 42와 유사한 방식으로 (2,4,6-트리플루오로페닐페닐)메탄아민 대신 (2,4,5-트리플루오로페닐)메탄아민을 사용하여 제조하였다.

실시예 65
화합물 65의 제조
(2R,5S,13aR)-N-(5-클로로-2,4-디플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

화합물 65를 화합물 42와 유사한 방식으로 (2,4,6-트리플루오로페닐페닐)메탄아민 대신 (5-클로로-2,4-디플루오로페닐)메탄아민을 사용하여 제조하였다.

실시예 66
화합물 66의 제조
(1R,4S,12aR)-N-(3,4-디플루오로벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드

화합물 66을 화합물 60과 유사한 방식으로 (2,3-디클로로페닐)메탄아민 대신 (3,4-디플루오로페닐)메탄아민을 사용하여 제조하였다.

실시예 67
화합물 67의 제조
(1R,4S,12aR)-N-(4-플루오로-2-(트리플루오로메틸)벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
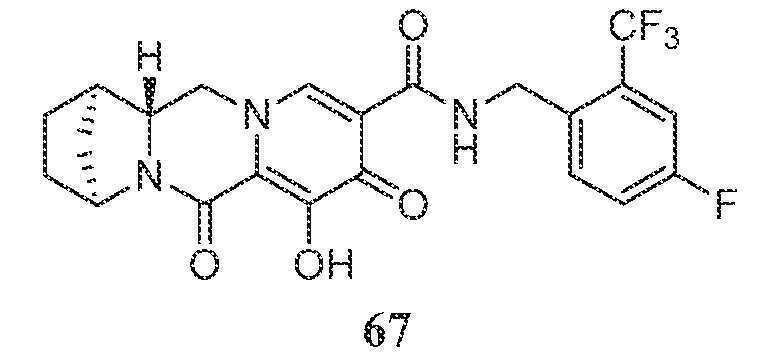
화합물 67을 화합물 60과 유사한 방식으로 (2,3-디클로로페닐)메탄아민 대신 (4-플루오로-2-(트리플루오로메틸)페닐)메탄아민을 사용하여 제조하였다.

실시예 68
화합물 68의 제조
(1R,4S,12aR)-N-(2-클로로-4-플루오로벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
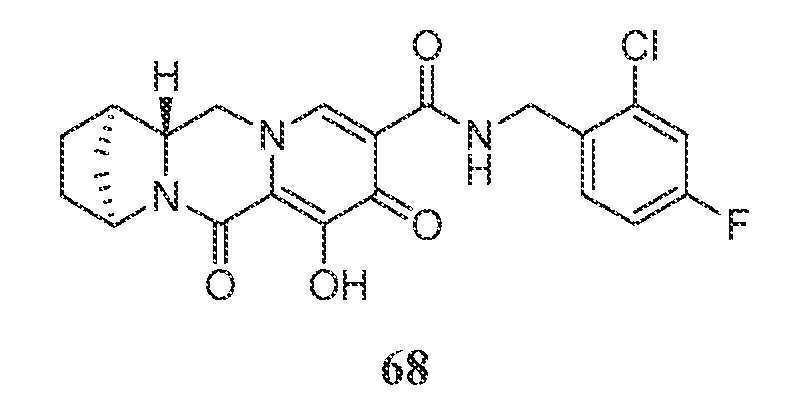
화합물 68을 화합물 60과 유사한 방식으로 (2,3-디클로로페닐)메탄아민 대신 (2-클로로-4-플루오로페닐)메탄아민을 사용하여 제조하였다.

실시예 69
화합물 69의 제조
(1R,4S,12aR)-N-(3-클로로-2,4-디플루오로벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
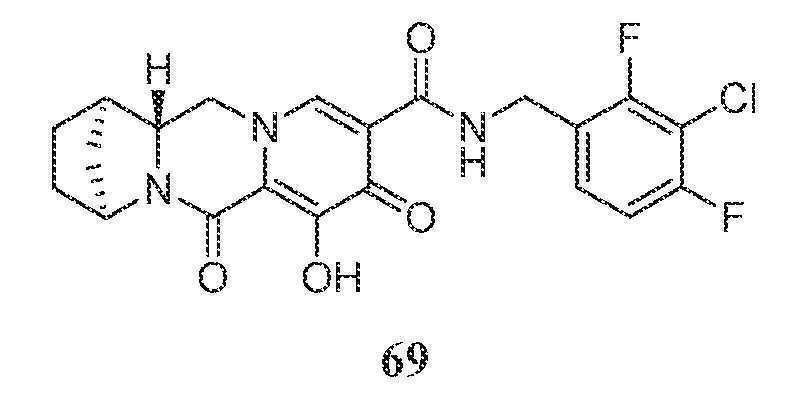
화합물 69를 화합물 60과 유사한 방식으로 (2,3-디클로로페닐)메탄아민 대신 (3-클로로-2,4-디플루오로페닐)메탄아민을 사용하여 제조하였다.

실시예 70
화합물 70의 제조
(1R,4S,12aR)-N-(2-플루오로-3-메틸벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드

화합물 70을 화합물 60과 유사한 방식으로 (2,3-디클로로페닐)메탄아민 대신 (2-플루오로-3-메틸페닐)메탄아민을 사용하여 제조하였다.

실시예 71
화합물 71의 제조
(1R,4S,12aR)-N-(3,6-디클로로-2-플루오로벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
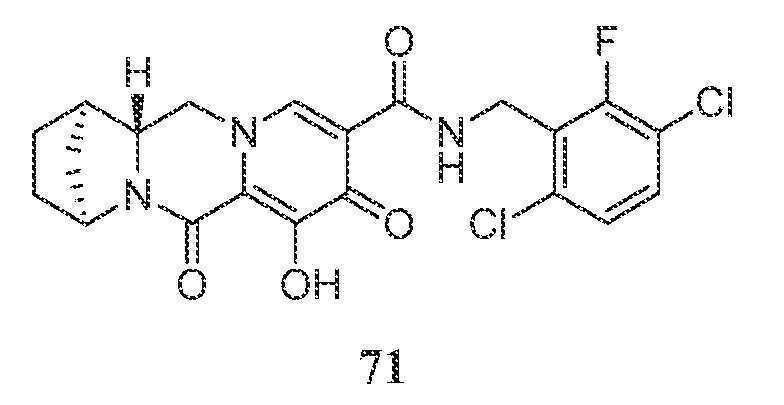
화합물 71을 화합물 60과 유사한 방식으로 (2,3-디클로로페닐)메탄아민 대신 (3,6-디클로로-2-플루오로페닐)메탄아민을 사용하여 제조하였다.

실시예 72
화합물 72의 제조
(1R,4S,12aR)-N-(3-클로로벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드

화합물 72를 화합물 60과 유사한 방식으로 (2,3-디클로로페닐)메탄아민 대신 (3-클로로페닐)메탄아민을 사용하여 제조하였다.

실시예 73
화합물 73의 제조
(1R,4S,12aR)-N-(3-클로로-2,6-디플루오로벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
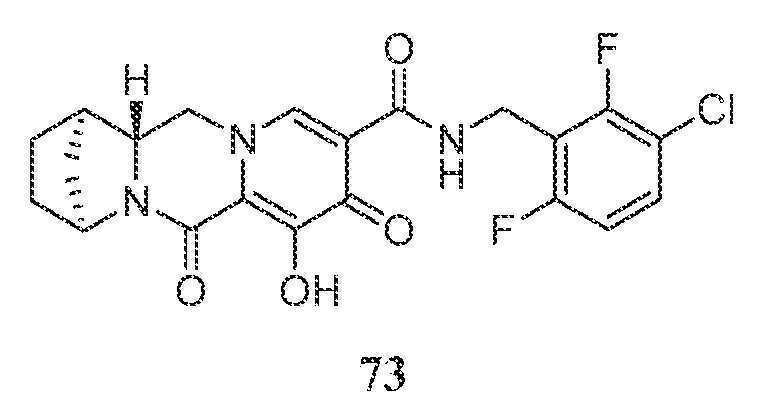
화합물 73을 화합물 60과 유사한 방식으로 (2,3-디클로로페닐)메탄아민 대신 (3-클로로-2,6-디플루오로페닐)메탄아민을 사용하여 제조하였다.

실시예 74
화합물 74의 제조
(1R,4S,12aR)-N-(2-플루오로-3-(트리플루오로메틸)벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드

화합물 74를 화합물 60과 유사한 방식으로 (2,3-디클로로페닐)메탄아민 대신 (2-플루오로-3-(트리플루오로메틸)페닐)메탄아민을 사용하여 제조하였다.

실시예 75
화합물 75의 제조
(1R,4S,12aR)-N-(3-클로로-4-플루오로벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
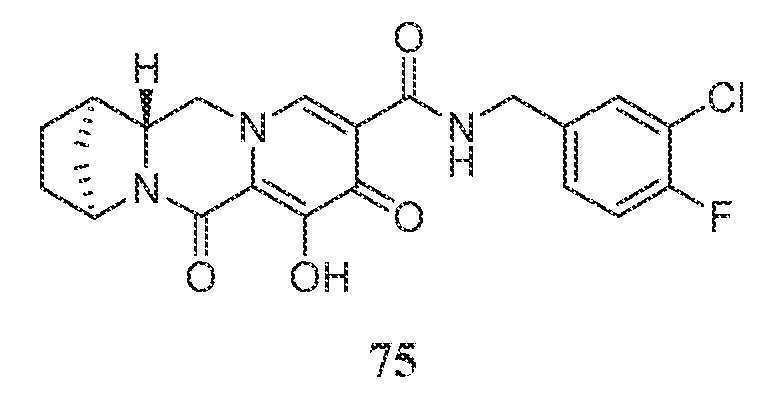
화합물 75를 화합물 60과 유사한 방식으로 (2,3-디클로로페닐)메탄아민 대신 (3-클로로-4-플루오로페닐)메탄아민을 사용하여 제조하였다.

실시예 76
화합물 76의 제조
(1R,4S,12aR)-N-((3,5-디플루오로피리딘-2-일)메틸)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드

화합물 76을 화합물 60과 유사한 방식으로 (2,3-디클로로페닐)메탄아민 대신 (3,5-디플루오로피리딘-2-일)메탄아민을 사용하여 제조하였다.

실시예 77
화합물 77의 제조
(1R,4S,12aR)-7-히드록시-6,8-디옥소-N-((R)-1-(2,4,6-트리플루오로페닐)에틸)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드

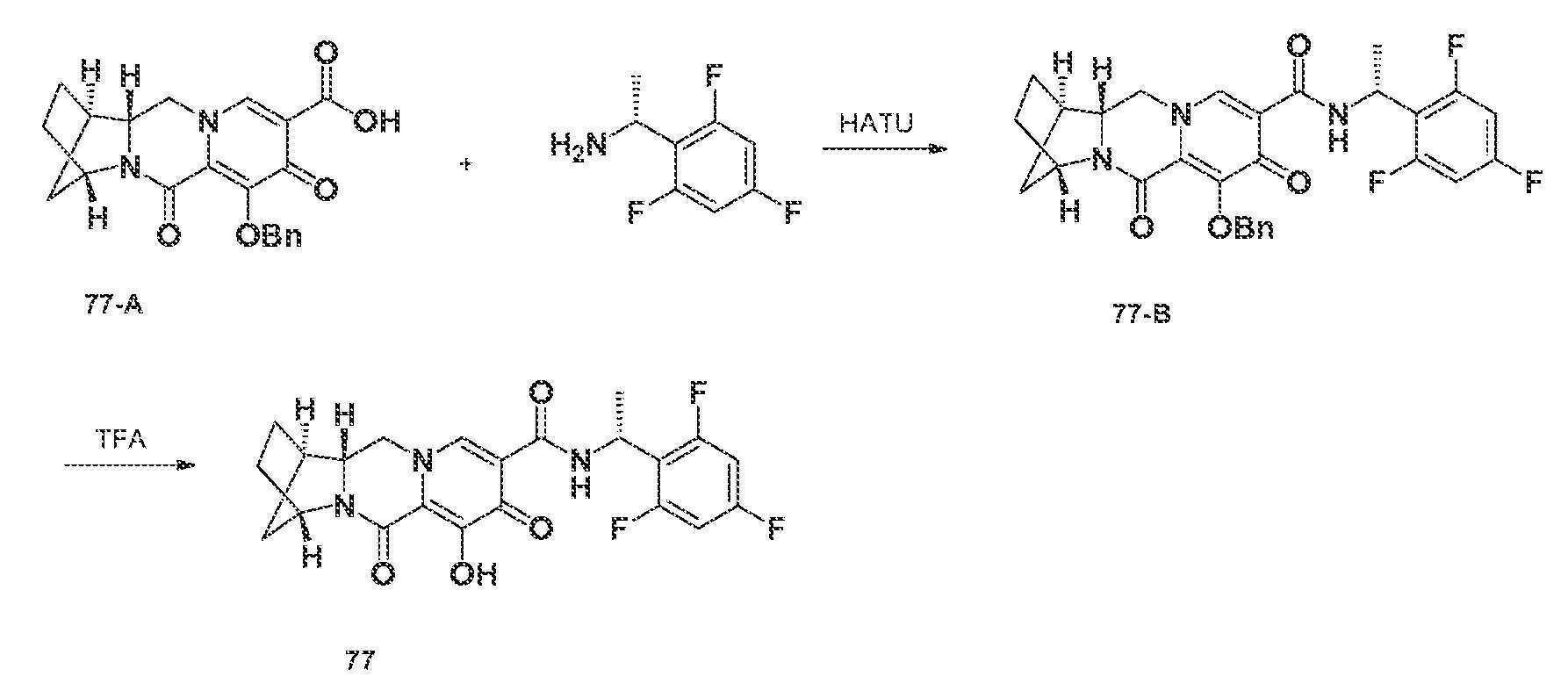
단계 1
50-mL 둥근 바닥 플라스크에 DCM (10 ml) 중 77-A (0.15 g, 0.39 mmol), (R)-1-(2,4,6-트리플루오로페닐)에탄아민 (0.14 g, 0.78 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.25 g, 1.97 mmol) 및 HATU (0.29 g, 0.79 mmol)를 채웠다. 반응 혼합물을 실온에서 1시간 동안 교반하였다. 반응 혼합물을 농축시키고, EtOAc (50 mL) 중에 재용해시키고, 포화 NaHCO3 (2x), 포화 NH4Cl로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 헥산-EtOAc를 사용하여 정제하여 77-B를 백색 고체로서 수득하였다.
단계 2
50-mL 둥근 바닥 플라스크에 TFA (2 mL) 중 77-B (0.20 g, 0.37 mmol)를 채웠다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 용액을 농축시키고, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 EtOAc-EtOAc 중 20% MeOH를 사용하여 정제하여 화합물 77을 수득하였다.

실시예 78
화합물 78의 제조
(2R,13aR)-8-히드록시-7,9-디옥소-N-((R)-1-(2,4,6-트리플루오로페닐)에틸)-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드
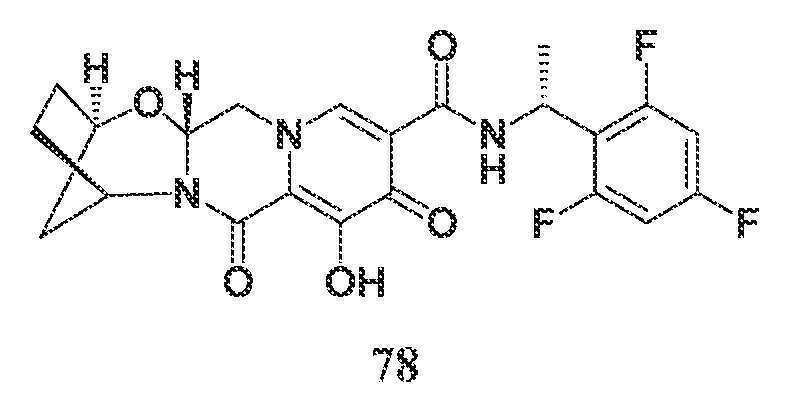

단계 1
50-mL 둥근 바닥 플라스크에 DCM (10 ml) 중 78-A (0.30 g, 0.94 mmol), (R)-1-(2,4,6-트리플루오로페닐) 에탄아민 (0.39 g, 1.87 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.61 g, 4.87 mmol) 및 HATU (0.71 g, 1.87 mmol)를 채웠다. 반응 혼합물을 실온에서 1시간 동안 교반하였다. 반응 혼합물을 농축시키고, EtOAc (50 mL) 중에 재용해시키고, 포화 NaHCO3 (2x), 포화 NH4Cl로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 헥산-EtOAc를 사용하여 정제하여 78-B를 백색 고체로서 수득하였다.
단계 2
50-mL 둥근 바닥 플라스크에 아세토니트릴 (5 mL) 중 78-B (0.4 g, 0.84 mmol) 및 브로민화마그네슘 (0.4 g, 2.2 mmol)을 채웠다. 반응 혼합물을 50℃로 가열하였다. 10분 후, 반응 혼합물을 0℃로 냉각시키고, 1 N 염산 (4 mL)을 첨가하였다. 추가의 물 (~ 5 mL)을 첨가하고, 고체를 여과하고, 물로 세척하고, 건조시켜 화합물 78을 수득하였다.

실시예 79
화합물 79의 제조
(1R,4S,12aR)-7-히드록시-6,8-디옥소-N-(2,4,5-트리플루오로벤질)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드


단계 1
50-mL 둥근 바닥 플라스크에 DCM (10 ml) 중 79-A (0.12 g, 0.32 mmol), (2,4,5-트리플루오로페닐)메탄아민 (0.10 g, 0.63 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.20 g, 1.58 mmol) 및 HATU (0.24 g, 0.63 mmol)를 채웠다. 반응 혼합물을 실온에서 1시간 동안 교반하였다. 반응 혼합물을 농축시키고, EtOAc (50 mL) 중에 재용해시키고, 포화 NaHCO3 (2x), 포화 NH4Cl로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 헥산-EtOAc를 사용하여 정제하여 79-B를 백색 고체로서 수득하였다.
단계 2
50-mL 둥근 바닥 플라스크에 TFA (2 mL) 중 79-B (0.15 g, 0.29 mmol)를 채웠다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 용액을 농축시키고, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 EtOAc-EtOAc 중 20% MeOH를 사용하여 정제하여 화합물 79를 수득하였다.

실시예 80
화합물 80의 제조
(1R,4S,12aR)-N-(5-클로로-2,4-디플루오로벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드


단계 1
50-mL 둥근 바닥 플라스크에 DCM (10 ml) 중 80-A (0.12 g, 0.32 mmol), (5-클로로-2,4-디플루오로페닐)메탄아민 (0.11 g, 0.63 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.20 g, 1.58 mmol) 및 HATU (0.24 g, 0.63 mmol)를 채웠다. 반응 혼합물을 실온에서 1시간 동안 교반하였다. 반응 혼합물을 농축시키고, EtOAc (50 mL) 중에 재용해시키고, 포화 NaHCO3 (2x), 포화 NH4Cl로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 헥산-EtOAc를 사용하여 정제하여 80-B를 백색 고체로서 수득하였다.
단계 2
50-mL 둥근 바닥 플라스크에 TFA (2 mL) 중 80-B (0.14 g, 0.26 mmol)를 채웠다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 용액을 농축시키고, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 EtOAc-EtOAc 중 20% MeOH를 사용하여 정제하여 화합물 80을 수득하였다.

실시예 81
화합물 81의 제조
(1R,3S,4S,12aS)-3-플루오로-7-히드록시-6,8-디옥소-N-(2,4,6-트리플루오로벤질)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
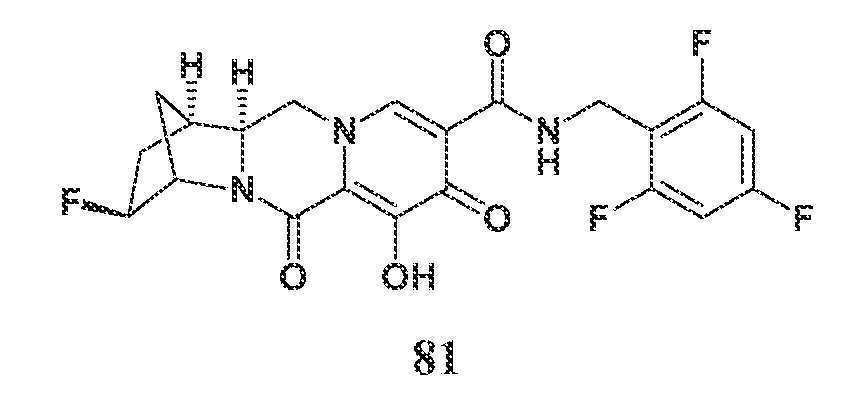

단계 1
100-mL 둥근 바닥 플라스크에 DCM (10 mL) 중 81-A (1.0 g, 3.7 mmol)를 채웠다. 반응 혼합물을 0℃로 냉각시켰다. 디에틸아미노황 트리플루오라이드 (DAST) (0.58 mL, 4.1 mmol)를 천천히 첨가하였다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하였다. 혼합물을 0℃로 다시 냉각시켰다. 포화 NaHCO3 (5 mL)을 적가하여 반응을 켄칭하였다. 이어서, 반응 혼합물을 EtOAc (100 mL)로 희석하고, 포화 NaHCO3, 염수로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 81-B를 수득하였다.
단계 2
100-mL 둥근 바닥 플라스크에 THF (10 mL) 중 81-B (0.8 g, 3.0 mmol)를 채웠다. 반응 혼합물을 -78℃에서 교반하였다. THF 중 2.0 M LiBH4 (3.2 mL, 6.4 mmol)를 천천히 첨가하였다. 이어서, 반응 혼합물을 가온하고, 실온에서 3시간 동안 교반하였다. 이어서, 반응 혼합물을 EtOAc (100 mL)로 희석하고, 물로 천천히 처리하였다 (H2 발생). 2개의 상을 분리한 후, 수성 분획을 EtOAc로 추출하고, 2개의 유기 분획을 합하고, 물로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 81-C를 수득하였다.
단계 3
100-mL 둥근 바닥 플라스크에 THF (15 mL) 중 81-C (0.57 g, 2.3 mmol), 트리페닐포스핀 (1.3 g, 5.1 mmol) 및 프탈이미드 (0.55 g, 3.7 mmol)를 채웠다. 이어서, 반응 혼합물을 교반하면서 0℃로 냉각시켰다. 디이소프로필 아조디카르복실레이트 (DIAD) (1.0 mL, 5.1 mmol)를 반응 혼합물에 천천히 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하였다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 81-D를 수득하였다.
단계 4
EtOH (40 mL) 중 81-D (0.8 g, 2.1 mmol)의 용액에 히드라진 1수화물 (0.6 mL)을 첨가하였다. 반응 혼합물을 3시간 동안 교반하면서 70℃로 가열하였다. 여과하여 고체를 제거한 후, 여과물을 농축시켜 81-E를 수득하였다.
단계 5
100-mL 둥근 바닥 플라스크에 에탄올 (7 mL) 중 81-E (0.49 g, 2.0 mmol) 및 81-F (0.7 g, 2.0 mmol)를 채웠다. 물 (7 mL) 중 중탄산나트륨 (0.34 g, 4.0 mmol)을 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 실온에서 밤새 교반하였다. 혼합물을 EtOAc (50 mL)로 희석하고, 물 (2 x)로 세척하였다. 수성 분획을 EtOAc (1 x)로 추출하고, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 조 81-G를 후속 단계에 추가 정제 없이 사용하였다.
단계 6
100-mL 둥근 바닥 플라스크에 4 N HCl /디옥산 (11 mL) 중 81-G (1.1 g, 1.9 mmol)를 채웠다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하였다. 농축시킨 후, 1.0 g 중간체를 수득하였다. 중간체 및 DBU (1.3 g, 8.8 mmol)를 톨루엔 (10 mL) 중에 용해시켰다. 반응 혼합물을 1시간 동안 교반하면서 110℃로 가열하였다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 81-H를 수득하였다.
단계 7
100-mL 둥근 바닥 플라스크에 THF (5 mL) 및 MeOH (5 mL) 중 81-H (0.56 g, 1.4 mmol)를 채웠다. 1 N KOH (4 mL)를 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하였다. 반응 혼합물을 1 N HCl (4 mL)의 첨가에 의해 산성화시켰다. 농축시킨 후, 잔류물을 톨루엔 (3 x)과 공증발시켰다. 조 산의 절반, 2,4,6-트리플루오로벤질아민 (0.2 g, 1.3 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.41 g, 3.1 mmol) 및 HATU (0.48 g, 1.25 mmol)를 DMF (10 mL) 중에 용해시켰다. 반응 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 EtOAc (100 mL)로 희석하고, 포화 NaHCO3 (2x), 포화 NH4Cl (2x)로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 헥산-EtOAc를 사용하여 정제하여 81-I를 수득하였다.
단계 8
50-mL 둥근 바닥 플라스크에 TFA (3 mL) 중 81-I (0.31 g, 0.58 mmol)를 채웠다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 EtOAc-MeOH를 사용하여 정제하여 화합물 81을 수득하였다.

실시예 82
화합물 82의 제조
(1S,3R,4R,12aR)-3-플루오로-7-히드록시-6,8-디옥소-N-(2,4,6-트리플루오로벤질)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드

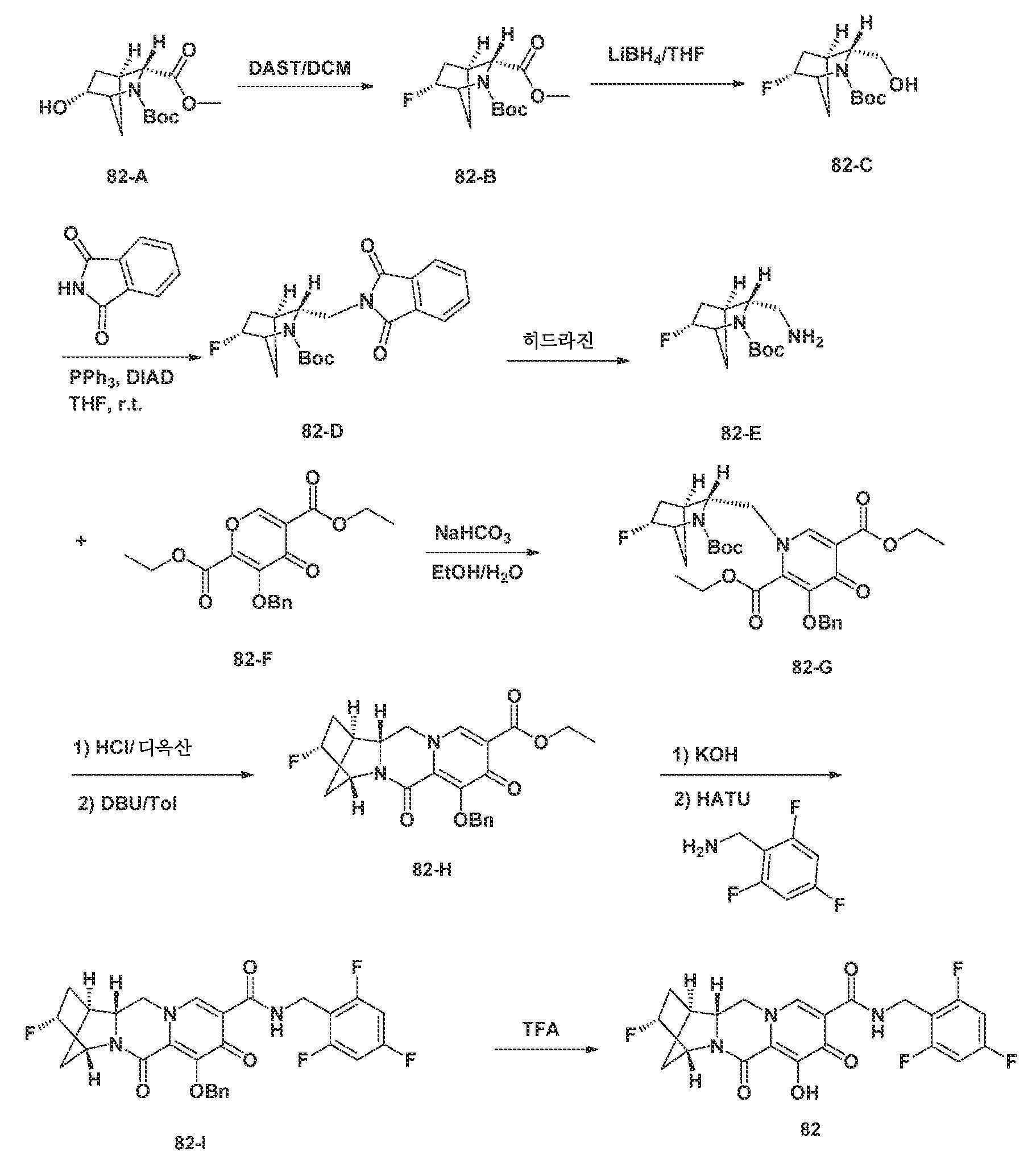
단계 1
100-mL 둥근 바닥 플라스크에 DCM (6 mL) 중 82-A (0.6 g, 2.1 mmol)를 채웠다. 반응 혼합물을 0℃로 냉각시켰다. DAST (0.35 mL, 3.0 mmol)를 천천히 첨가하였다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하였다. 혼합물을 0℃로 다시 냉각시켰다. 포화 NaHCO3 (5 mL)을 적가하여 반응을 켄칭하였다. 이어서, 반응 혼합물을 EtOAc (100 mL)로 희석하고, 포화 NaHCO3, 염수로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 82-B를 수득하였다.
단계 2
100-mL 둥근 바닥 플라스크에 THF (10 mL) 중 82-B (0.4 g, 1.5 mmol)를 채웠다. 반응 혼합물을 -78℃에서 교반하였다. THF 중 2.0 M LiBH4 (1.6 mL, 3.2 mmol)를 천천히 첨가하였다. 이어서, 반응 혼합물을 가온하고, 실온에서 3시간 동안 교반하였다. 이어서, 반응 혼합물을 EtOAc (100 mL)로 희석하고, 물을 천천히 첨가하였다 (H2 발생). 2개의 상을 분리한 후, 수성 분획을 EtOAc로 추출하고, 2개의 유기 분획을 합하고, 물로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 82-C를 수득하였다.
단계 3
100-mL 둥근 바닥 플라스크에 THF (10 mL) 중 82-C (0.25 g, 1.0 mmol), 트리페닐포스핀 (0.59 g, 2.2 mmol) 및 프탈이미드 (0.24 g, 1.6 mmol)를 채웠다. 이어서, 반응 혼합물을 교반하면서 0℃로 냉각시켰다. DIAD (0.44 mL, 2.2 mmol)를 반응 혼합물에 천천히 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하였다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 82-D를 수득하였다.
단계 4
EtOH (20 mL) 중 82-D (0.35 g, 0.9 mmol)의 용액에 히드라진 1수화물 (0.3 mL)을 첨가하였다. 반응 혼합물을 3시간 동안 교반하면서 70℃로 가열하였다. 여과하여 고체를 제거한 후, 여과물을 농축시켜 82-E를 수득하였다.
단계 5
100-mL 둥근 바닥 플라스크에 에탄올 (7 mL) 중 82-E (0.21 g, 0.87 mmol) 및 82-F (0.3 g, 0.87 mmol)를 채웠다. 물 (7 mL) 중 중탄산나트륨 (0.15 g, 1.7 mmol)을 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 실온에서 밤새 교반하였다. 혼합물을 EtOAc (50 mL)로 희석하고, 물 (2 x)로 세척하였다. 수성 분획을 EtOAc로 추출하고, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 조 82-G를 후속 단계에 추가 정제 없이 사용하였다.
단계 6
100-mL 둥근 바닥 플라스크에 4 N HCl /디옥산 (5 mL) 중 82-G (0.49 g, 0.86 mmol)를 채웠다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하였다. 농축시킨 후, 0.4 g 중간체를 수득하였다. 중간체 및 DBU (0.6 g, 4.0 mmol)를 톨루엔 (10 mL) 중에 용해시켰다. 반응 혼합물을 1시간 동안 교반하면서 110℃로 가열하였다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 82-H를 수득하였다.
단계 7
100-mL 둥근 바닥 플라스크에 THF (5 mL) 및 MeOH (5 mL) 중 82-H (0.2 g, 0.49 mmol)를 채웠다. 1 N KOH (1.5 mL)를 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하였다. 반응 혼합물을 1 N HCl (1.5 mL)의 첨가에 의해 산성화시켰다. 농축시킨 후, 잔류물을 톨루엔 (3 x)과 공증발시켰다. 조 산, 2,4,6-트리플루오로벤질아민 (0.15 g, 0.95 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.31 g, 2.4 mmol) 및 HATU (0.36 g, 0.95 mmol)를 DCM (10 mL) 중에 용해시켰다. 반응 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 EtOAc (100 mL)로 희석하고, 포화 NaHCO3 (2x), 포화 NH4Cl (2x)로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 헥산-EtOAc를 사용하여 정제하여 82-I를 수득하였다.
단계 8
50-mL 둥근 바닥 플라스크에 TFA (3 mL) 중 82-I (0.22 g, 0.41 mmol)를 채웠다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 EtOAc-MeOH를 사용하여 정제하여 화합물 82를 수득하였다.
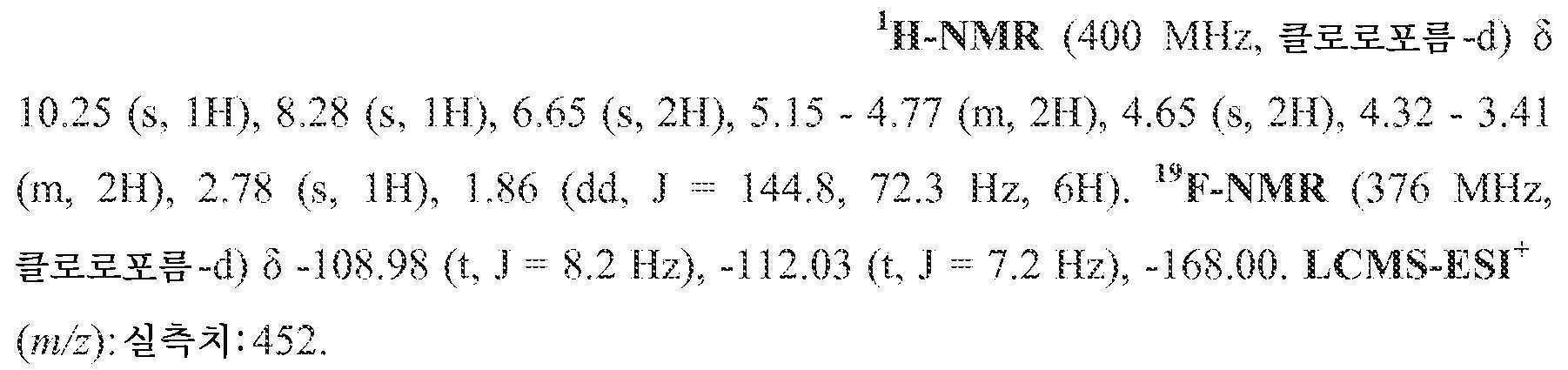
실시예 83
화합물 83의 제조
(1S,4R,12aS)-3,3-디플루오로-7-히드록시-6,8-디옥소-N-(2,4,6-트리플루오로벤질)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
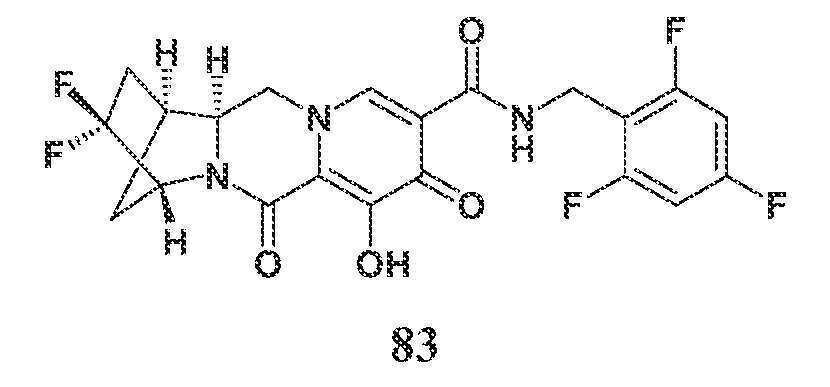

단계 1
100-mL 둥근 바닥 플라스크에 DCM (20 mL) 중 83-A (1.0 g, 3.7 mmol)를 채웠다. 반응 혼합물을 0℃로 냉각시켰다. 데스-마르틴 퍼아이오디난 (1.8 g, 4.2 mmol)을 천천히 첨가하였다. 이어서, 반응 혼합물을 실온에서 3시간 동안 교반하였다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 83-B를 수득하였다.
단계 2
100-mL 둥근 바닥 플라스크에 DCM (15 mL) 중 83-B (0.85 g, 3.2 mmol)를 채웠다. 반응 혼합물을 0℃로 냉각시켰다. DAST (1.5 mL, 11.3 mmol)를 천천히 첨가하였다. 이어서, 반응 혼합물을 실온에서 밤새 교반하였다. 혼합물을 0℃로 다시 냉각시켰다. 포화 NaHCO3 (5 mL)을 적가하여 반응을 켄칭하였다. 이어서, 반응 혼합물을 EtOAc (100 mL)로 희석하고, 포화 NaHCO3, 염수로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 83-C를 수득하였다.
단계 3
100-mL 둥근 바닥 플라스크에 THF (6 mL) 중 83-C (0.44 g, 1.5 mmol)를 채웠다. 반응 혼합물을 -78℃에서 교반하였다. THF 중 2.0 M LiBH4 (1.6 mL, 3.2 mmol)를 천천히 첨가하였다. 이어서, 반응 혼합물을 가온하고, 실온에서 3시간 동안 교반하였다. 이어서, 반응 혼합물을 EtOAc (100 mL)로 희석하고, 물을 천천히 첨가하였다 (H2 발생). 2개의 상을 분리한 후, 수성 분획을 EtOAc로 추출하고, 2개의 유기 분획을 합하고, 물로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 83-D를 수득하였다.
단계 4
100-mL 둥근 바닥 플라스크에 THF (10 mL) 중 83-D (0.17 g, 0.65 mmol), 트리페닐포스핀 (0.37 g, 1.4 mmol) 및 프탈이미드 (0.15 g, 1.0 mmol)를 채웠다. 이어서, 반응 혼합물을 교반하면서 0℃로 냉각시켰다. DIAD (0.28 mL, 1.4 mmol)를 반응 혼합물에 천천히 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하였다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 83-E를 수득하였다.
단계 5
EtOH (20 mL) 중 83-E (0.25 g, 0.64 mmol)의 용액에 히드라진 1수화물 (0.3 mL)을 첨가하였다. 반응 혼합물을 3시간 동안 교반하면서 70℃로 가열하였다. 여과하여 고체를 제거한 후, 여과물을 농축시켜 83-F를 수득하였다.
단계 6
100-mL 둥근 바닥 플라스크에 에탄올 (7 mL) 중 83-F (0.18 g, 0.69 mmol) 및 83-G (0.324g, 0.69 mmol)를 채웠다. 물 (7 mL) 중 중탄산나트륨 (0.12 g, 1.4 mmol)을 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 실온에서 밤새 교반하였다. 혼합물을 EtOAc (50 mL)로 희석하고, 물로 세척하였다. 수성 분획을 EtOAc로 추출하고, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 조 83-H를 후속 단계에 추가 정제 없이 사용하였다.
단계 7
100-mL 둥근 바닥 플라스크에 4 N HCl /디옥산 (3.8 mL) 중 83-H (0.4 g, 0.68 mmol)를 채웠다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하였다. 농축시킨 후, 0.35 g 중간체를 수득하였다. 중간체 및 DBU (0.51 g, 3.3 mmol)를 톨루엔 (10 mL) 중에 용해시켰다. 반응 혼합물을 1시간 동안 교반하면서 110℃로 가열하였다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 83-I를 수득하였다.
단계 8
100-mL 둥근 바닥 플라스크에 THF (5 mL) 및 MeOH (5 mL) 중 83-I (0.2 g, 0.47 mmol)를 채웠다. 1 N KOH (1.4 mL)를 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하였다. 반응 혼합물을 1 N HCl (1.4 mL)의 첨가에 의해 산성화시켰다. 농축시킨 후, 잔류물을 톨루엔 (3 x)과 공증발시켰다. 조 산, 2,4,6-트리플루오로벤질아민 (0.14 g, 0.91 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.29 g, 2.2 mmol) 및 HATU (0.35 g, 0.91 mmol)를 DCM (10 mL) 중에 용해시켰다. 반응 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 EtOAc (100 mL)로 희석하고, 포화 NaHCO3 (2x), 포화 NH4Cl (2x)로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 헥산-EtOAc를 사용하여 정제하여 83-J를 수득하였다.
단계 9
50-mL rbf에 TFA (3 mL) 중 83-J (0.18 g, 0.32 mmol)를 채웠다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 EtOAc-MeOH를 사용하여 정제하여 화합물 83을 백색 고체로서 수득하였다.

실시예 84
화합물 84의 제조
(1S,2R,4S,12aR)-7-히드록시-2-메틸-6,8-디옥소-N-(2,4,6-트리플루오로벤질)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
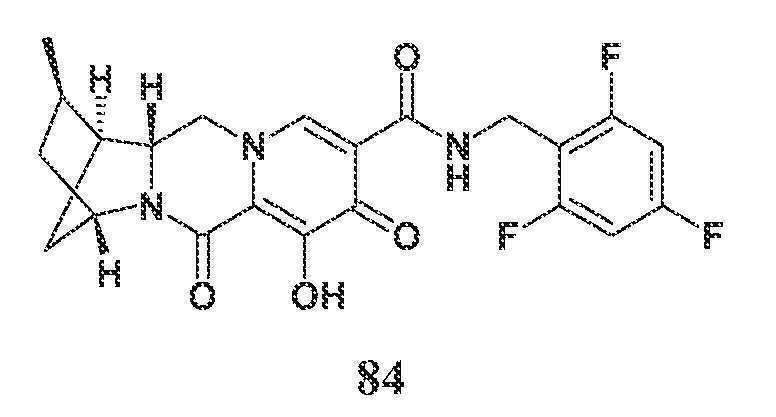
단계 1
100-mL 둥근 바닥 플라스크에 DCM (20 mL) 중 84-A (1.6 g, 5.9 mmol)를 채웠다. 반응 혼합물을 0℃로 냉각시켰다. 데스-마르틴 퍼아이오디난 (4.9 g, 11.7 mmol)을 천천히 첨가하였다. 이어서, 반응 혼합물을 실온에서 3시간 동안 교반하였다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 84-B를 수득하였다.
단계 2
100-mL 둥근 바닥 플라스크에 THF (30 mL) 중 84-B (1.3 g, 4.8 mmol)를 채웠다. 반응 혼합물을 0℃로 냉각시켰다. 테베(Tebbe) 시약 (톨루엔 중 0.5 M, 19.4 mL, 9.7 mmol)을 천천히 첨가하였다. 이어서, 반응 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 0℃로 다시 냉각시켰다. 포화 NaHCO3 (5 mL)을 적가하여 반응을 켄칭하였다. 반응 혼합물을 실온에서 추가로 15분 동안 교반하고, 셀라이트를 통해 여과하였다. 여과된 케이크를 DCM (2 x)으로 세척하였다. 합한 여과물을 진공 하에 농축시키고, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 84-C를 수득하였다.
단계 3
EtOH (20 mL) 중 84-C (0.9 g, 3.4 mmol)의 용액 (N2로 퍼징함)에 Pd/C (0.18 g)를 첨가하였다. 혼합물을 H2 하에 3시간 동안 교반하였다. 혼합물을 셀라이트를 통해 여과하고, 여과물을 농축시켜 84-D를 수득하였다.
단계 4
100-mL 둥근 바닥 플라스크에 THF (6 mL) 중 84-D (0.9 g, 3.3 mmol)를 채웠다. 반응 혼합물을 -78℃에서 교반하였다. THF 중 2.0 M LiBH4 (13.2 mL, 26.4 mmol)를 천천히 첨가하였다. 이어서, 반응 혼합물을 가온하고, 실온에서 3시간 동안 교반하였다. 이어서, 반응 혼합물을 EtOAc (100 mL)로 희석하고, 물을 천천히 첨가하였다 (H2 발생). 2개의 상을 분리한 후, 수성 분획을 EtOAc로 추출하고, 2개의 유기 분획을 합하고, 물로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 84-E를 수득하였다.
단계 5
100-mL 둥근 바닥 플라스크에 THF (15 mL) 중 84-E (0.4 g, 1.66 mmol), 트리페닐포스핀 (0.96 g, 3.6 mmol) 및 프탈이미드 (0.39 g, 2.7 mmol)를 채웠다. 이어서, 반응 혼합물을 교반하면서 0℃로 냉각시켰다. DIAD (0.7 mL, 3.6 mmol)를 반응 혼합물에 천천히 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하였다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 84-F를 수득하였다.
단계 6
EtOH (20 mL) 중 84-F (0.55 g, 1.5 mmol)의 용액에 히드라진 1수화물 (0.3 mL)을 첨가하였다. 반응 혼합물을 3시간 동안 교반하면서 70℃로 가열하였다. 여과하여 고체를 제거한 후, 여과물을 농축시켜 84-G를 수득하였다.
단계 7
100-mL 둥근 바닥 플라스크에 에탄올 (10 mL) 중 84-G (0.35 g, 1.4 mmol) 및 84-H (0.5g, 1.4 mmol)를 채웠다. 물 (10 mL) 중 중탄산나트륨 (0.24 g, 2.8 mmol)을 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 실온에서 밤새 교반하였다. 혼합물을 EtOAc (50 mL)로 희석하고, 물 (2 x)로 세척하였다. 수성 분획을 EtOAc로 추출하고, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 조 84-I를 후속 단계에 추가 정제 없이 사용하였다.
단계 8
100-mL rbf에 4 N HCl /디옥산 (8.2 mL) 중 84-I (0.84 g, 1.4 mmol)를 채웠다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하였다. 농축시킨 후, 0.74 g 중간체를 수득하였다. 중간체 및 DBU (1.1 g, 7.2 mmol)를 톨루엔 (10 mL) 중에 용해시켰다. 반응 혼합물을 1시간 동안 교반하면서 110℃로 가열하였다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 84-J를 수득하였다.
단계 9
100-mL 둥근 바닥 플라스크에 THF (5 mL) 및 MeOH (5 mL) 중 84-J (0.4 g, 0.98 mmol)를 채웠다. 1 N KOH (3.0 mL)를 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하였다. 반응 혼합물을 1 N HCl (3.0 mL)의 첨가에 의해 산성화시켰다. 농축시킨 후, 잔류물을 톨루엔 (3 x)과 공증발시켰다. 조 산, 2,4,6-트리플루오로벤질아민 (0.32 g, 1.96 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.63 g, 4.9 mmol) 및 HATU (0.74 g, 1.9 mmol)를 DCM (10 mL) 중에 용해시켰다. 반응 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 EtOAc (100 mL)로 희석하고, 포화 NaHCO3 (2x), 포화 NH4Cl (2x)로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 헥산-EtOAc를 사용하여 정제하여 84-K를 수득하였다.
단계 10
50-mL 둥근 바닥 플라스크에 TFA (6 mL) 중 84-K (0.5 g, 0.93 mmol)를 채웠다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 EtOAc-MeOH를 사용하여 정제하여 화합물 84를 수득하였다.

실시예 85
화합물 85의 제조
(6aS,7R,11S)-1-히드록시-2,13-디옥소-N-(2,4,6-트리플루오로벤질)-6,6a,7,8,9,10,11,13-옥타히드로-2H-7,11-메타노피리도[1',2':4,5]피라지노[1,2-a]아제핀-3-카르복스아미드


단계 1
DMSO (6 mL) 및 물 (0.75 mL) 중 85-A (1100 mg, 3.855 mmol)의 용액을 실온에서 교반하면서 N-아이오도숙신이미드 (885 mg, 3.934 mmol)를 첨가하였다. 2시간 후, 추가의 N-아이오도숙신이미드 (88 mg, 0.391 mmol)를 첨가하고, 생성된 혼합물을 실온에서 1.5시간 동안 교반하였다. 암갈색 반응 혼합물을 EtOAc로 희석하고, 10% 수성 Na2S2O3 용액 및 수성 NaHCO3 용액 (~1:4 혼합물)에 이어서 물 (약간의 염수 포함)로 세척하였다. 수성 분획을 EtOAc로 추출한 후, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 85-B를 수득하였다.

단계 2
톨루엔 (15 mL) 중 85-B (993 mg, 2.313 mmol), AIBN (305 mg, 1.857 mmol), 및 트리부틸주석 히드라이드 (1392 mg, 4.799 mmol)의 용액을 100℃에서 교반하였다. 2시간 후, 반응 혼합물을 실온으로 냉각시키고, EtOAc로 희석하고, 물 및 염수로 세척하였다. 수성 분획을 EtOAc로 추출한 후, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 85-C를 수득하였다.

단계 3
EtOH (25 mL) 및 디옥산 중 4 N HCl (0.9 mL) 중 85-C (725 mg, 2.39 mmol) 및 20% Pd(OH)2/C (351 mg)의 혼합물을 H2 분위기 하에 교반하였다. 2시간 후, 반응 혼합물을 여과하고, 여과물을 농축시켰다. LCMS-ESI+ (m/z): [M+H]+ 계산치, C10H18NO3: 200.13; 실측치: 200.1. 잔류물을 톨루엔 (x 2)과 공증발시킨 후, THF (15 mL) 중 잔류물 및 Boc2O (720 mg, 3.299 mmol)을 실온에서 교반하면서 N,N-디이소프로필에틸아민 (DIPEA) (1.2 mL, 6.889 mmol)을 첨가하였다. 1시간 후, 반응 혼합물을 물로 희석하고, EtOAc (x 2)로 추출하였다. 유기 추출물을 물로 세척한 후, 합한 추출물을 건조 (Na2SO4)시키고 농축시켰다. 잔류물을 플래쉬에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 85-D를 수득하였고, 이는 회전이성질체의 혼합물인 것으로 보였다.

단계 4
THF (5 mL) 중 85-D (568 mg, 1.897 mmol) 및 피리딘 (0.25 mL, 3.091 mmol)의 용액에 0℃에서 페닐 클로로티오노포르메이트 (0.3 mL, 2.169 mmol)를 첨가하였고, 불용성 물질이 신속하게 생성되었다. 0℃에서 ~30분 후, 추가의 피리딘 (0.3 mL, 3.709 mmol) 및 페닐 클로로티오노포르메이트 (0.3 mL, 2.169 mmol)를 첨가하였다. 0℃에서 1.5시간 및 실온에서 1시간 후, 혼합물을 농축시키고, 잔류물을 EtOAc 및 물 중에 용해시켰다. 2개의 층을 분리한 후, 유기 분획을 ~0.1 N HCl, 포화 수성 NaHCO3, 및 염수로 세척하였다. 수성 분획을 EtOAc로 추출한 후, 합한 유기 분획을 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 EtOAc/헥산을 사용하여 정제하여 85-E를 수득하였다.

단계 5
톨루엔 (8 mL) 중 85-E (602 mg, 1.382 mmol), AIBN (182 mg, 1.108 mmol), 및 트리부틸주석 히드라이드 (608 mg, 2.096 mmol)의 혼합물을 100℃에서 교반하였다. 1시간 후, 반응 혼합물을 농축시키고, 잔류물을 EtOAc 중에 용해시킨 후, 물 및 염수로 세척하였다. 수성 분획을 EtOAc로 추출한 후, 합한 유기 분획을 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 EtOAc/헥산을 사용하여 정제하여 85-F를 수득하였고, 이는 회전이성질체의 혼합물인 것으로 보였다.

단계 6
85-F (420 mg)를 재정제하고, THF 중 정제된 85-F (3 mL)를 0℃에서 교반하면서 THF 중 2.0 M LiBH4 (1.5 mL)를 첨가하였다. 5분 후, 혼합물을 실온에서 17시간 동안 교반하고, 추가의 THF 중 2.0 M LiBH4 (1.5 mL)를 실온에서 첨가하였다. 실온에서 23시간 후, 추가의 THF 중 2.0 M LiBH4 (3 mL)를 첨가하고, 생성된 혼합물을 ~72시간 동안 교반하였다. 반응 혼합물을 0℃에서 교반하면서 물을 천천히 첨가하고 추가로 물로 희석한 후, 생성물을 EtOAc (x 2)로 추출하였다. 추출물을 물로 세척하고, 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 85-G를 수득하였다.

단계 7
THF (10 mL) 중 85-G (198 mg, 0.820 mmol), 프탈이미드 (200 mg, 1.359 mmol), 및 PPh3 (488 mg, 1.861 mmol)의 용액을 0℃ 조에서 교반하면서 DIAD (0.36 mL, 1.828 mmol)를 첨가하였다. 0℃에서 30분 후, 혼합물을 실온에서 17시간 동안 교반하였다. 반응 혼합물을 농축시키고, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산-EtOAc를 사용하여 85-H로 정제하였고, 이는 회전이성질체의 혼합물인 것으로 보였다.

단계 8
EtOH (12 mL) 중 85-H (270 mg, 0.729 mmol)의 용액에 실온에서 히드라진 수화물 (0.145 mL, 3.083 mmol)을 첨가하고, 생성된 용액을 70℃에서 교반하였다. 1.5시간 후, 혼합물을 0℃로 냉각시키고, 에테르 (30 mL)로 희석한 후, 1시간 동안 0℃에서 교반하였다. 혼합물을 여과하고, 여과물을 농축시켰다. 잔류물을 CH2Cl2 중에 용해시키고, 여과하여 약간의 불용성 물질을 제거하였다. 생성된 여과물을 농축시켰다. 잔류물을 물 (3 mL) 및 EtOH (3 mL) 중 85-I (257 mg, 0.742 mmol) 및 NaHCO3 (131 mg, 1.559 mmol)과 합하고, 실온에서 교반하였다. 1시간 후, 혼합물을 물로 희석하고, EtOAc (x 2)로 추출하였다. 추출물을 물로 세척한 후, 유기 추출물을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. CH2Cl2 중 잔류물의 용액 (2 mL)에 디옥산 중 4N HCl (6 mL)을 첨가하였다. 실온에서 1.5시간 후, 용액을 농축시키고, 톨루엔과 공증발시켰다. 톨루엔 (5 mL) 중 잔류물 및 DBU (0.6 mL, 4.012 mmol)의 혼합물을 100℃ 조에서 교반하였다. 1시간 후, 추가의 DBU (0.3 mL, 2.006 mmol)를 첨가하고, 혼합물을 100℃에서 추가로 1시간 동안 교반하였다. 혼합물을 농축시킨 후, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 EtOAc - 20% MeOH/EtOAc를 사용하여 정제하여 85-J를 수득하였다.

단계 9
THF (4 mL) 및 MeOH (4 mL) 중 85-J (214 mg, 0.507 mmol)의 혼합물을 실온에서 교반하면서 1 N KOH (1.1 mL)를 첨가하였다. 30분 후, 반응 혼합물을 ~1 mL로 농축시키고, 1 N HCl (~1.2 mL)로 산성화시키고, 염수로 희석한 후, CH2Cl2 (20 mL x 2)로 추출하였다. 합한 추출물을 건조 (Na2SO4)시키고, 농축시켜 조 산을 수득하였다.
CH2Cl2 (6 mL) 중 조 산 (199 mg, 0.505 mmol), 2,4,6-트리플루오로벤질 아민 (130 mg, 0.807 mmol), 및 HATU (304 mg, 0.800 mmol)의 혼합물을 실온에서 교반하면서 N,N-디이소프로필에틸아민 (DIPEA) (0.62 mL, 3.559 mmol)을 첨가하였다. 30분 후, 반응 혼합물을 농축시키고, 잔류물을 EtOAc 중에 용해시키고, 포화 수성 NH4Cl (x 2), 포화 수성 NaHCO3 (x 2), 및 염수로 세척하였다. 수성 분획을 EtOAc로 추출한 후, 2개의 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 플래쉬에 의해 용리액으로서 EtOAc-20%MeOH/EA를 사용하여 정제하여 85-K를 수득하였다.

단계 10
85-K (187 mg, 0.348 mmol)를 실온에서 트리플루오로아세트산 (3 mL) 중에 용해시키고, 실온에서 교반하였다. 1시간 후, 용액을 농축시키고, 잔류물을 CH2Cl2 중에 용해시켰다. 용액을 0.1 N HCl로 세척한 후, 수성 분획을 CH2Cl2 (x 2)로 추출하였다. 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 CH2Cl2-CH2Cl2 중 20% MeOH를 사용하여 정제하여 화합물 85 150 mg (96%)을 수득하였다. 화합물 85를 추가로 메탄올 (10 mL)로부터의 재결정화에 의해 정제하여 화합물 85를 수득하였다.

실시예 86
화합물 86의 제조
(1R,3S,4R,12aS)-7-히드록시-3-메틸-6,8-디옥소-N-(2,4,6-트리플루오로벤질)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드

단계 1
DMSO (52 mL) 및 물 (6.5 mL) 중 86-A (10.160 g, 39.48 mmol)의 용액을 실온에서 교반하면서 N-아이오도숙신이미드 (8.888 g, 39.50 mmol)를 첨가하였다. 30분 후, 암갈색 반응 혼합물을 EtOAc로 희석하고, 포화 수성 NaHCO3 용액, 10% 수성 Na2S2O3 용액, 및 염수로 세척하였다. 수성 분획을 EtOAc로 추출한 후, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 86-B를 백색 고체로서 수득하였다.

단계 2
톨루엔 (150 mL) 중 86-B (12.468 g, 31.07 mmol), 아조비스이소부티로니트릴 (AIBN) (4.082 g, 24.86 mmol), 및 트리부틸주석 히드라이드 (18.047 g, 62.22 mmol)의 용액을 100℃에서 교반하였다. 30분 후, 반응 혼합물을 실온으로 냉각시키고, EtOAc로 희석하고, 물 및 염수로 세척하였다. 수성 분획을 EtOAc로 추출한 후, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 플래쉬 크로마토그래피 2회에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 86-C를 수득하였다.

단계 3
EtOH (100 mL) 및 디옥산 중 4 N HCl (5.7 mL) 중 86-C (4.187 g, 15.21 mmol) 및 20% Pd(OH)2/C (1.022 g)의 혼합물을 H2 분위기 하에 교반하였다. 1.5시간 후, 반응 혼합물을 여과하고, 여과물을 농축시켰다. 잔류물을 톨루엔과 공증발시킨 후, 잔류물을 후속 단계에 사용하였다. LCMS-ESI+ (m/z): [M+H]+ 계산치, C8H14NO3: 172.1; 실측치: 172.1.
잔류물을 톨루엔과 공증발시킨 후, THF (45 mL) 중 잔류물 및 Boc2O (5.712 g, 26.17 mmol)를 실온에서 교반하면서 N,N-디이소프로필에틸아민 (DIPEA) (8 mL, 45.93 mmol)을 첨가하였다. 30분 후, 반응 혼합물을 물로 희석하고, EtOAc (x 2)로 추출하였다. 유기 추출물을 물로 세척한 후, 합한 추출물을 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 86-D를 수득하였다. 1H NMR 스펙트럼은 회전이성질체의 혼합물을 시사하였다.

단계 4
CH2Cl2 (35 mL) 중 86-D (1659 mg, 6.115 mmol)의 용액을 0℃ 조에서 교반하면서 데스-마르틴 퍼아이오디난 (5.183 g, 12.22 mmol)을 여러 부분으로 첨가하였다. 5분 후, 혼합물을 실온에서 교반하였다. 2시간 후, 반응 혼합물을 빙조에서 냉각시키고, 물로 켄칭하고, 여과하였다. 여과물을 포화 NaHCO3으로 세척하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 86-E를 수득하였다. 1H NMR 스펙트럼은 2종의 회전이성질체를 시사하였다.

단계 5
THF (12 mL) 중 86-E (528 mg, 1.961mmol)의 용액을 0℃에서 교반하면서 톨루엔 중 테베 시약 0.5 M 용액 (7.9 mL, 3.95 mmol)을 적가하였다. 첨가한 후, 갈색 용액을 실온으로 천천히 가온되도록 하고, 실온에서 2.5시간 동안 교반하였다. 반응 혼합물을 0℃ 조에서 교반하면서 포화 수성 NaHCO3 용액의 첨가에 의해 반응물을 조심스럽게 켄칭하였다. 혼합물을 CH2Cl2로 희석하고 실온에서 15분 동안 교반한 후, 생성된 혼합물을 셀라이트 패드를 통해 여과하고, 필터 케이크를 CH2Cl2로 세척하였다. 여과물 중 2개의 분획을 분리한 후, 수성 분획을 CH2Cl2로 추출하고, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산 - EtOAc를 사용하여 정제하여 86-F를 수득하였다. 1H NMR 스펙트럼은 2종의 회전이성질체를 시사하였다.

단계 6
EtOH (5 mL) 중 86-F (333 mg, 1.246 mmol) 및 20% Pd(OH)2/C (53 mg)의 혼합물을 H2 분위기 하에 교반하였다. 30분 후, 혼합물을 여과하고, 여과물을 농축시켜 86-G를 수득하였다. 1H NMR 스펙트럼은 2종의 회전이성질체를 시사하였다.

단계 7
THF (5 mL) 중 86-G (336 mg, 1.482 mmol)의 용액을 0℃에서 교반하면서 THF 중 2.0 M LiBH4 (1.5 mL)를 첨가하였다. 5분 후, 혼합물을 실온에서 교반하였다. 2시간 후, 추가의 THF 중 2.0 M LiBH4 (1.5 mL)를 첨가하였다. 실온에서 21시간 후, 추가의 THF 중 2.0 M LiBH4 (3 mL)를 첨가하였다. 실온에서 3시간 후, 용액을 35℃에서 18시간 동안 가열하였다. 반응 혼합물을 0℃로 냉각시키고, 물로 조심스럽게 켄칭하였다. 혼합물을 EtOAc (x 2)로 추출한 후, 2개의 유기 분획을 물로 세척하고, 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 플래쉬 크로마토그래피에 의해 헥산 - EtOAc를 사용하여 정제하여 86-H를 수득하였다.

단계 8
THF (10 mL) 중 86-H (218 mg, 0.903 mmol), 프탈이미드 (218 mg, 1.482 mmol), 및 PPh3 (535 mg, 2.040 mmol)의 용액을 0℃ 조에서 교반하면서 DIAD (0.40 mL, 2.032 mmol)를 첨가하였다. 0℃에서 10분 후, 혼합물을 실온에서 19시간 동안 교반하였다. 반응 혼합물을 농축시키고, 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 헥산-EtOAc를 사용하여 정제하여 86-I를 수득하였다. 1H NMR은 2종의 회전이성질체를 시사하였다.

단계 9
EtOH (12 mL) 중 86-I (319 mg, 0.861 mmol)의 용액에 실온에서 히드라진 수화물 (0.17 mL, 3.494 mmol)을 첨가하고, 생성된 용액을 70℃ 조에서 교반하였다. 1.5시간 후, 혼합물을 0℃로 냉각시키고, 에테르 (25 mL)로 희석한 후, 1시간 동안 0℃에서 교반하였다. 혼합물을 여과하고, 여과물을 농축시켰다. 잔류물을 CH2Cl2 중에 용해시키고, 여과하여 약간의 불용성 물질을 제거하였다. 생성된 여과물을 농축시켜 조 아민을 수득하였다.
조 아민을 톨루엔과 공증발시킨 후, 물 (3 mL) 및 EtOH (3 mL) 중 조 아민, 85-I (300 mg, 0.866 mmol), 및 NaHCO3 (150 mg, 1.845 mmol)의 혼합물을 실온에서 교반하였다. 2시간 후, 혼합물을 물로 희석하고, EtOAc (x 2)로 추출하였다. 추출물을 물로 세척한 후, 유기 추출물을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. CH2Cl2 중 잔류물의 용액 (2 mL)에 디옥산 중 4 N HCl (6 mL)을 첨가하였다. 실온에서 1.5시간 후, 용액을 농축시키고, 톨루엔과 공증발시켰다. 톨루엔 (6 mL) 중 잔류물 및 DBU (0.65 mL, 4.347 mmol)의 혼합물을 100℃에서 교반하였다. 1시간 후, 추가의 DBU (0.65 mL, 4.347 mmol)를 첨가하고, 혼합물을 100℃에서 교반하였다. 추가의 DBU (0.65 mL, 4.347 mmol)를 1시간 후에 첨가하고, 혼합물을 100℃에서 추가로 2.5시간 동안 교반하였다. 혼합물을 CH2Cl2로 희석하고, 1 N HCl 3 mL를 함유하는 물로 세척하였다. 유기 분획을 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 EtOAc-20% MeOH/EtOAc를 사용하여 정제하여 86-J를 수득하였다.

단계 10
THF (2 mL) 및 EtOH (2 mL) 중 86-J (83 mg, 0.196 mmol)의 혼합물을 실온에서 교반하면서 1 N KOH (0.4 mL)를 첨가하였다. 30분 후, 반응 혼합물을 물로 희석하고, CH2Cl2로 세척하였다. 수성 분획을 1 N HCl (0.45 mL)로 산성화시킨 후, 생성물을 CH2Cl2 (x 2)로 추출하였다. 합한 추출물을 건조 (Na2SO4)시키고, 농축시켜 조 산을 수득하였다. LCMS-ESI+ (m/z): [M +H]+ 계산치, C22H23N2O5: 395.2; 실측치: 395.2.
CH2Cl2 (3 mL) 중 조 산 (69 mg, 0.175 mmol), 2,4,6-트리플루오로벤질 아민 (42 mg, 0.261 mmol), 및 HATU (106 mg, 0.279 mmol)의 혼합물을 실온에서 교반하면서 N,N-디이소프로필에틸아민 (DIPEA) (0.25 mL, 1.435 mmol)을 첨가하였다. 30분 후, 반응 혼합물을 농축시키고, 잔류물을 EtOAc 중에 용해시키고, 포화 수성 NH4Cl (x 2), 포화 수성 NaHCO3 (x 2), 및 염수로 세척하였다. 수성 분획을 EtOAc로 추출한 후, 2개의 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 EtOAc-20%MeOH/EtOAc를 사용하여 정제하여 86-K를 수득하였다.

단계 11
86-K (61 mg, 0.113 mmol)를 트리플루오로아세트산 (2 mL) 중에 용해시키고, 실온에서 교반하였다. 1시간 후, 용액을 농축시키고, 잔류물을 CH2Cl2 중에 용해시켰다. 용액을 0.1 N HCl로 세척한 후, 수성 분획을 CH2Cl2 (x 2)로 추출하였다. 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 플래쉬 크로마토그래피에 의해 용리액으로서 CH2Cl2-CH2Cl2 중 20% MeOH를 사용하여 정제하여 화합물 86을 수득하였다.

실시예 87
시스-5-아미노테트라히드로-2H-피란-3-올의 제조

단계 1
20 mL 메탄올 중 벤질 (5-옥소테트라히드로-2H-피란-3-일)카르바메이트 (740 mg, 3.0 mmol) 및 염화세륨 (III) 7수화물 (1.12 g, 3.0 mmol)의 용액을 0℃로 냉각시키고, 이어서 수소화붕소나트륨 (120 mg, 3.2 mmol)을 조금씩 첨가하였다. 반응 혼합물을 0℃에서 45분 동안 교반되도록 한 다음, 1 mL 아세톤을 천천히 첨가하여 켄칭하고, 이어서 실온에서 3시간 교반하였다. 반응 혼합물을 물과 디클로로메탄 사이에 분배하고, 수성 상을 디클로로메탄에 이어서 2-부탄올로 추출하였다. 합한 유기 상을 황산마그네슘 상에서 건조시키고, 여과하고, 농축시키고, 잔류물을 플래쉬 크로마토그래피 (0-100% EtOAc/헥산)에 의해 정제하여 목적 시스-벤질 ((3R,5S)-5-히드록시테트라히드로-2H-피란-3-일)카르바메이트를 수득하였다.

목적하지 않은 트랜스-이성질체도 또한 단리되었다.
단계 2
5 mL 1:1 DCM:EtOH 중 시스-벤질 ((3R,5S)-5-히드록시테트라히드로-2H-피란-3-일)카르바메이트 (290 mg, 1.16 mmol)의 용액에 10wt% Pd/C (255 mg)를 첨가하였다. 이 혼합물을 풍선 압력 수소 하에 18시간 동안 교반하고, 에탄올로 헹구면서 셀라이트를 통한 여과에 의해 팔라듐을 제거하였다. 여과물의 농축 시, 시스-5-아미노테트라히드로-2H-피란-3-올이 수득되었고, 이를 조 물질로서 수행하였다.
실시예 88
화합물 88의 제조
(2R,5S,13aR)-N-(3-클로로-2-플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드
화합물 88을 화합물 15와 유사한 방식으로 (4-플루오로페닐)메탄아민 대신 (3-클로로-2-플루오로페닐)메탄아민을 사용하여 제조하였다.

실시예 89
화합물 89의 제조
(2R,5S,13aR)-N-(2,5-디플루오로벤질)-8-히드록시-7,9-디옥소-2,3,4,5,7,9,13,13a-옥타히드로-2,5-메타노피리도[1',2':4,5]피라지노[2,1-b][1,3]옥사제핀-10-카르복스아미드

화합물 89를 화합물 15와 유사한 방식으로 (4-플루오로페닐)메탄아민 대신 (2,5-디플루오로페닐)메탄아민을 사용하여 제조하였다.

실시예 90
화합물 90의 제조
(1R,4S,12aR)-N-(3-클로로-2-플루오로벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드

화합물 90을 화합물 41과 유사한 방식으로 (2,4,6-트리플루오로페닐)메탄아민 대신 (3-클로로-2-플루오로페닐)메탄아민을 사용하여 제조하였다.

실시예 91
화합물 91의 제조
(1R,4S,12aR)-7-히드록시-6,8-디옥소-N-(2,3,4-트리플루오로벤질)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
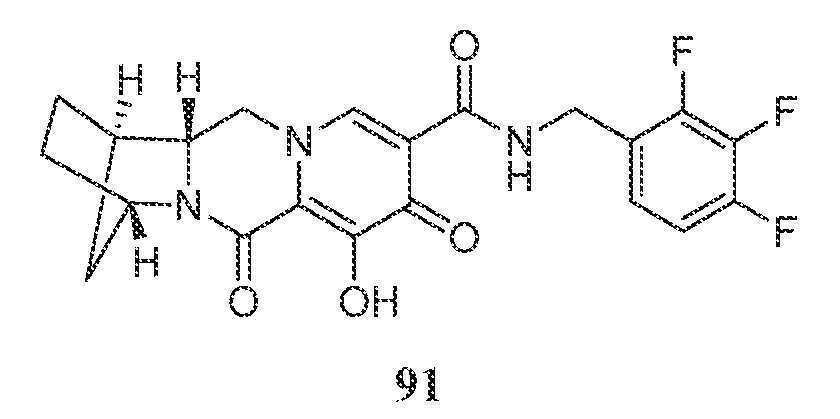
화합물 91을 화합물 41과 유사한 방식으로 (2,4,6-트리플루오로페닐)메탄아민 대신 (2,3,4-트리플루오로페닐)메탄아민을 사용하여 제조하였다.

실시예 92
화합물 92의 제조
(1R,4S,12aR)-N-(4-클로로-2-플루오로벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드

화합물 92를 화합물 41과 유사한 방식으로 (2,4,6-트리플루오로페닐)메탄아민 대신 (4-클로로-2-플루오로페닐)메탄아민을 사용하여 제조하였다.

실시예 93
화합물 93의 제조
(1R,4S,12aR)-N-(2-클로로-4,6-디플루오로벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드

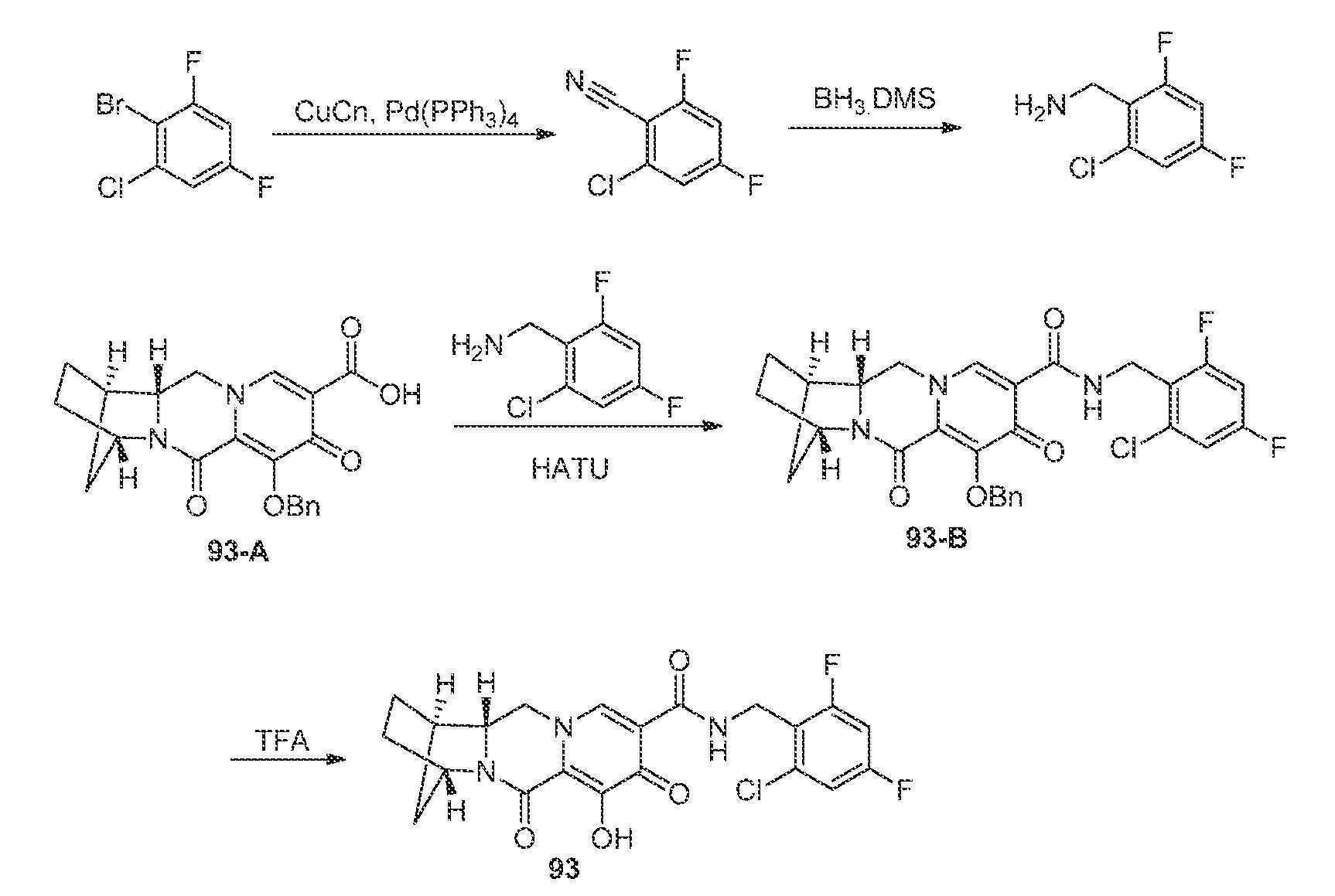
단계 1
5 mL 마이크로웨이브 바이알에 2-브로모-1-클로로-3,5-디플루오로벤젠 (540 mg, 2.4 mmol), 제1구리 시아나이드 (436 mg, 4.87 mmol), 테트라키스(트리페닐포스핀)팔라듐 (63 mg, 0.05 mmol)을 채우고, 밀봉하고, 배기하고/질소로 재충전하였다. 여기에 5 mL 탈기된 DMF를 첨가하였다. 밀봉된 용기를 110℃에서 18시간 동안 가열하고, 에틸 아세테이트로 희석하고, 9:1 NH4OH:NH4Cl(수성) 2회, 5% LiCl(수성) 2회, 및 염수로 순차적으로 세척하였다. 이어서, 유기 상을 황산마그네슘 상에서 건조시키고, 여과하고, 농축시켰다. 조 잔류물을 플래쉬 크로마토그래피 (100% 헥산)에 의해 정제하여 2-클로로-4,6-디플루오로벤조니트릴을 수득하였다.

단계 2
2.4 mL THF 중 2-클로로-4,6-디플루오로벤조니트릴 (210 mg, 1.2 mmol)의 용액에 THF 중 보란-DMS의 2M 용액 (0.6 mL)을 첨가하였다. 이 반응 혼합물을 환류 온도에서 18시간 동안 교반되도록 하여 모든 용매가 상실되게 하였다. 잔류물을 3 mL THF 중에 재용해시키고, 0℃로 냉각시키고, HCl(수성)의 6M 용액을 조심스럽게 첨가하고, 혼합물을 30분 동안 환류 온도로 되돌렸다. 반응 혼합물을 다시 한번 0℃로 냉각시키고, 4M NaOH(수성)로 처리하였다. 수성 상을 DCM으로 추출하고, 합한 유기 상을 황산마그네슘 상에서 건조시키고, 여과하고, 농축시켰다. 조 잔류물을 플래쉬 크로마토그래피 (0-10% MeOH/DCM)에 의해 정제하여 (2-클로로-4,6-디플루오로페닐)메탄아민을 수득하였다.

단계 3 및 4
1 mL 디클로로메탄 중 93-A (74 mg, 0.11 mmol), (2-클로로-4,6-디플루오로페닐)메탄아민 (48.5 mg, 0.27 mmol), HATU (100 mg, 0.26 mmol), 및 N,N-디이소프로필에틸아민 (0.1 mL, 0.57 mmol)의 용액을 실온에서 1시간 동안 교반하였고, 그 시점에 LCMS에 의해 93-A의 완전한 소멸 및 93-B의 형성을 관찰하였다. TFA (0.65 M)를 첨가하고, 혼합물을 실온에서 1시간 동안 교반하고, 1 mL DMF을 첨가하였다. 이어서, 반응 혼합물을 농축시키고, 정제용 HPLC (ACN/H2O + 0.1% TFA)에 의해 정제하여 화합물 93을 수득하였다.

실시예 94
화합물 94의 제조
(1R,4S,12aR)-N-벤질-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
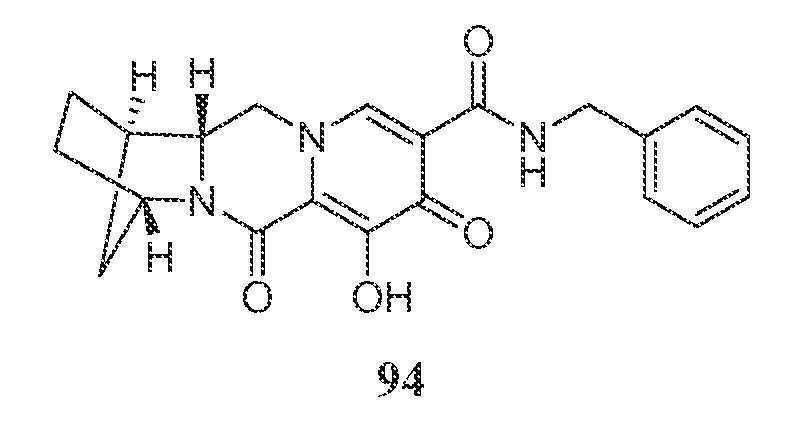
화합물 94를 화합물 41과 유사한 방식으로 (2,4,6-트리플루오로페닐)메탄아민 대신 페닐메탄아민을 사용하여 제조하였다.

실시예 95
키랄 tert-부틸 3-((1,3-디옥소이소인돌린-2-일)메틸)-2-아자비시클로[2.1.1]헥산-2-카르복실레이트 95-A 및 95-B의 제조

단계 1
9 mL THF 중 라세미 tert-부틸 3-(히드록시메틸)-2-아자비시클로[2.1.1]헥산-2-카르복실레이트 (285 mg, 1.34 mmol), 트리페닐포스핀 (425 mg, 1.62 mmol), 및 프탈이미드 (240 mg, 1.62 mmol)의 0℃ 용액에 1 ml THF 중 디이소프로필 아조디카르복실레이트 (0.35 mL, 1.8 mmol)의 용액을 적가하였다. 반응 혼합물을 실온으로 가온하고, 90분 동안 교반하고, 실리카 상에서 농축시키고, 플래쉬 크로마토그래피 (0-25% EtOAc/헥산)에 의해 정제하여 tert-부틸 3-((1,3-디옥소이소인돌린-2-일)메틸)-2-아자비시클로[2.1.1]헥산-2-카르복실레이트를 라세미 혼합물로서 수득하였다.
단계 2
라세미 tert-부틸 3-((1,3-디옥소이소인돌린-2-일)메틸)-2-아자비시클로[2.1.1]헥산-2-카르복실레이트 (655 mg, 1.91 mmol)를 룩스 셀룰로스-2 칼럼 상에서 키랄 HPLC에 의해 아세토니트릴 용리액을 사용하여 분리하여 키랄 95-A (제1 용리 피크) 및 95-B (제2 용리 피크)를 거울상이성질체풍부화 형태로 수득하였다. 95-A의 경우: 144 mg, 98%ee (미지의 절대 입체화학). 95-B의 경우: 242 mg, 49%ee (미지의 절대 입체화학).
실시예 96
화합물 96의 제조
(1R,3R,11aS)-6-히드록시-5,7-디옥소-N-(2,4,6-트리플루오로벤질)-2,3,5,7,11,11a-헥사히드로-1H-1,3-메타노피리도[1,2-a]피롤로[1,2-d]피라진-8-카르복스아미드
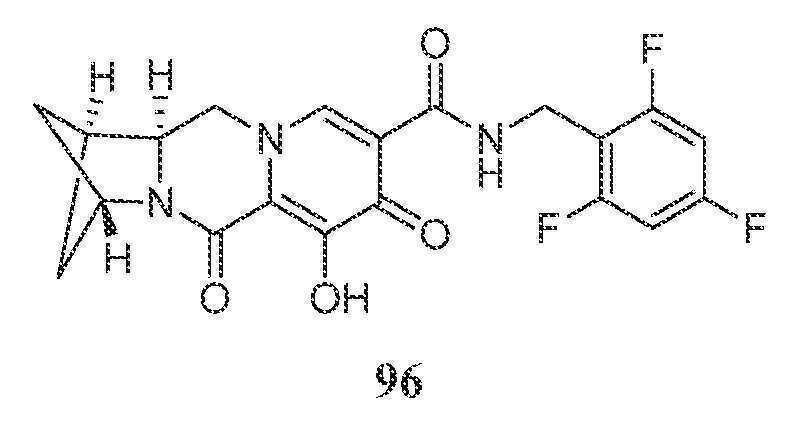
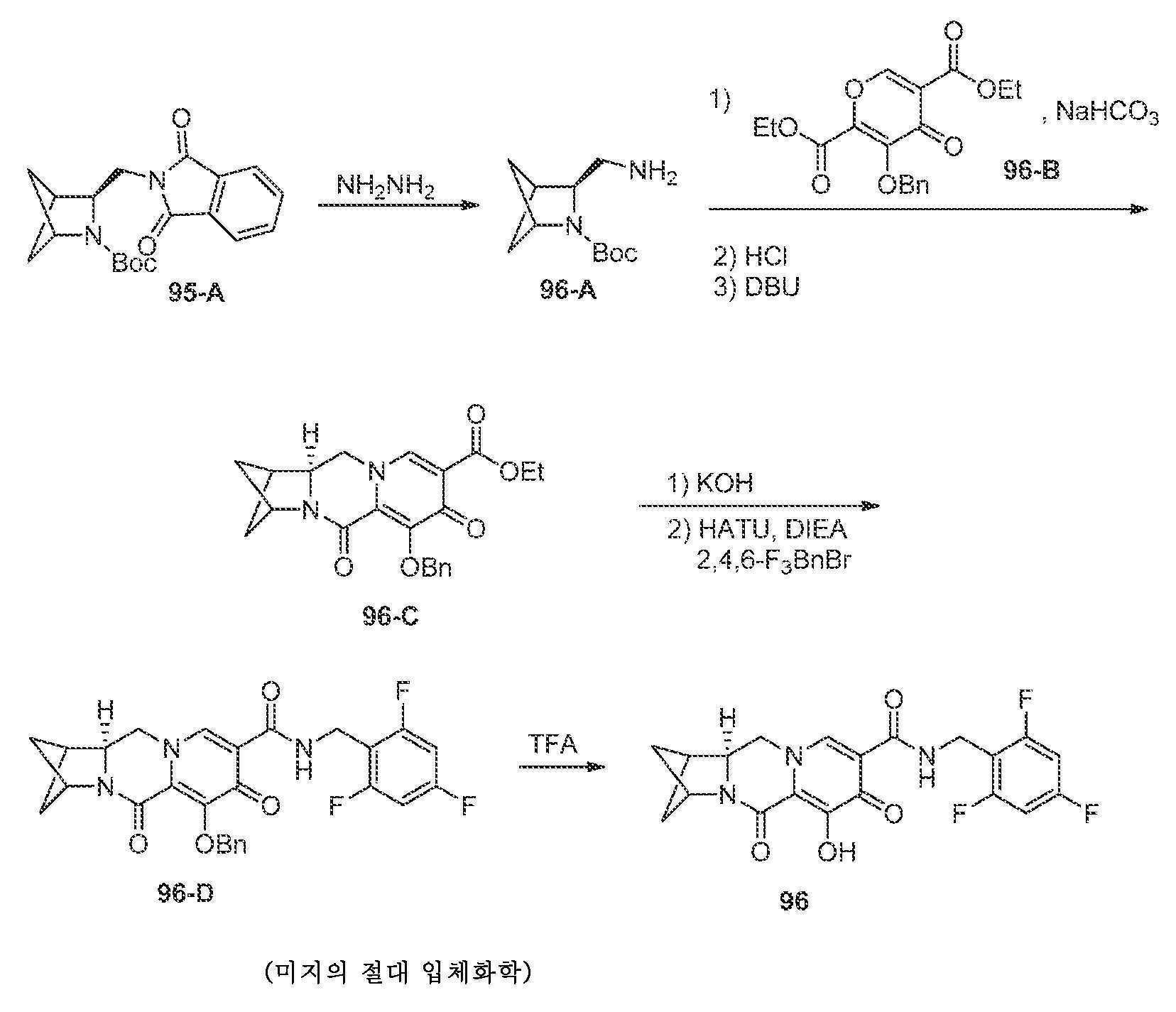
단계 1
9 mL 에탄올 중 중간체 95-A (141 mg, 0.41 mmol, 98% ee, 미지의 절대 입체화학)의 용액에 히드라진 수화물 (0.5 mL, 10.3 mmol)을 첨가하고, 70℃에서 18시간 동안 교반하여 미지의 절대 입체화학의 96-A를 수득하였다. 고체를 여과에 의해 제거하고, 여과물을 농축시키고, 조 물질로서 수행하였다.
단계 2
2 mL 물 및 2 mL 에탄올 중 조 96-A (0.41 mmol 추정치), 96-B (430 mg, 1.25 mmol), 및 중탄산나트륨 (69 mg, 0.82 mmol)의 혼합물을 실온에서 18시간 동안 교반한 후, 반응 혼합물을 물로 희석하고, 에틸 아세테이트로 3회 추출하였다. 합한 유기 상을 황산마그네슘 상에서 건조시키고, 여과하고, 농축시켰다. 조 잔류물 (222 mg)을 1.5 mL DCM 중에 용해시키고, 디옥산 중 4N HCl (4 mL)을 첨가하고, 실온에서 90분 동안 교반하였다. 혼합물을 농축 건조시키고, 톨루엔과 공증발시켰다. 6 mL 메탄올 중 조 잔류물 및 DBU (0.3 mL, 2.0 mmol)를 50℃에서 90분 동안 교반하였다. 이어서, 반응 혼합물을 실리카 겔 상에서 농축시키고, 플래쉬 크로마토그래피 (0-10% MeOH/DCM)에 의해 정제하여 96-C를 수득하였다.
단계 3
96-C (112 mg, 0.28 mmol), 1M 수성 수산화칼륨 (1 mL), 4 mL 메탄올, 및 4 mL THF의 혼합물을 실온에서 3시간 동안 교반하고, 혼합물을 디클로로메탄으로 희석하고, 1M 수성 염화수소의 첨가에 의해 산성화시키고, 유기 상을 디클로로메탄으로 추출하였다. 합한 유기부를 건조시키고, 여과하고, 톨루엔으로부터 농축시켰다. 진공 하에 건조시킨 후, 잔류물을 1.5 mL DCM 중에 현탁시키고, 트리플루오로벤질아민 (62 mg, 0.38 mmol), HATU (220 mg, 0.58 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.15 mL, 0.86 mmol)을 첨가하였다. 이 반응 혼합물을 실온에서 2시간 동안 교반하여 96-D를 수득하였고, 조 물질로서 수행하였다.
단계 4
트리플루오로아세트산 (1.7 mL, 22.2 mmol)을 이전 단계로부터의 96-D를 함유하는 조 반응 용액에 첨가하고, 반응 혼합물을 실온에서 90분 동안 교반되도록 하였다. 이어서, DMF 1 mL를 첨가하고, 반응 혼합물을 ~1 mL로 농축시키고, 여과하고, 정제용 HPLC (ACN/물 + 0.1% TFA)에 의해 정제하여 화합물 96 (미지의 절대 입체화학)을 수득하였다.

실시예 97
화합물 97의 제조
(1S,3S,11aR)-6-히드록시-5,7-디옥소-N-(2,4,6-트리플루오로벤질)-2,3,5,7,11,11a-헥사히드로-1H-1,3-메타노피리도[1,2-a]피롤로[1,2-d]피라진-8-카르복스아미드

화합물 97 (49% ee, 미지의 절대 입체화학)을 화합물 96과 유사한 방식으로 거울상이성질체적으로 대향인 중간체 95-A 대신 중간체 95-B (49% ee, 미지의 절대 입체화학)를 사용하여 제조하였다.

실시예 98
화합물 98의 제조
(1S,4R,12aR)-3,3-디플루오로-7-히드록시-6,8-디옥소-N-(2,4,6-트리플루오로벤질)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
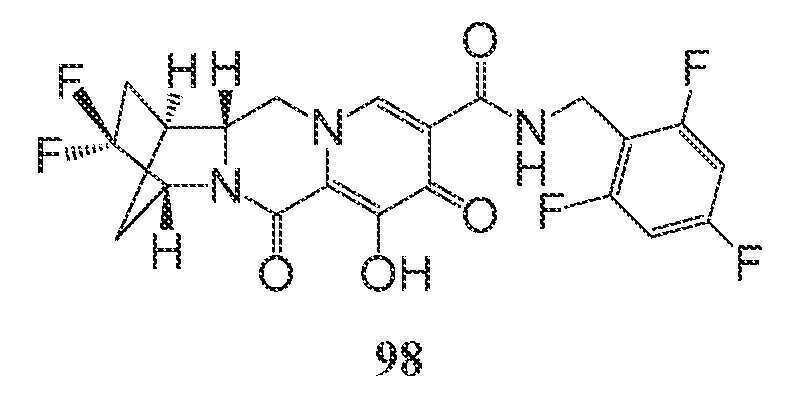

단계 1
98-A (0.5g, 1.87mmol)를 DCM (20 mL) 중에 용해시키고, 질소 하에 0℃로 냉각시켰다. 데스-마르틴 퍼아이오디난 (1.59 g, 3.74 mmol)을 천천히 첨가하였다. 혼합물을 실온에서 2시간 동안 교반하고, Na2S2O3/NaHCO3 (7:1) 포화 수용액 (160 mL)으로 켄칭하고, 2개의 층이 분리될 때까지 격렬히 교반하였다. 조 생성물을 DCM으로 2회 추출하였다. 합한 유기 층을 황산나트륨 상에서 건조시키고, 농축시켰다. 조 생성물을 실리카 겔 상에서 플래쉬 크로마토그래피에 의해 0-20%MeOH/DCM을 사용하여 정제하여 98-B를 수득하였다.

단계 2
DCM (15 mL) 중 98-B (504 mg, 1.87mmol)의 용액을 0℃에서 교반하였다. DAST (1ml)를 반응 혼합물에 적가하였다. 실온에서 밤새 교반한 후, 반응 혼합물을 0℃로 다시 냉각시켰다. 포화 NaHCO3 (10 mL)을 천천히 첨가하였다. 혼합물을 DCM으로 2회 추출하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 잔류물을 플래쉬 크로마토그래피 0-50% EtOAc/헥산에 의해 정제하여 98-C를 수득하였다.

단계 3
THF (20 mL) 중 98-C (476 mg, 1.634mmol)를 0℃에서 교반하면서 THF 중 2.0 M LiBH4 (2.4 mL, 4.8mmol)를 첨가하였다. 혼합물을 실온으로 가온하고, 4시간 동안 교반하였다. 반응 혼합물을 얼음으로 켄칭하고, EtOAc 및 포화 NH4Cl로 희석하였다 (약간의 H2 발생). 2개의 상을 분리한 후, 유기 분획을 염수로 세척하고, 건조 (Na2SO4)시키고, 농축시켰다. 조 생성물 98-D를 후속 단계에 그대로 사용하였다.
단계 4
THF (10 mL) 중 98-D (1.634mmol), 프탈이미드 (0.36 g, 2.45mmol), 및 PPh3 (0.855 g, 3.26mmol)을 0℃ 조에서 교반하면서 DIAD (0.642 mL, 3.26mmol)를 첨가하였다. 첨가한 후, 혼합물을 0℃에서 30분 동안 교반한 다음, 실온에서 16시간 동안 교반하였다. 이것을 EtOAc, 및 포화 NH4Cl로 희석하였다. 5분 동안 교반한 후, 고체를 여과하고, 2개의 상을 분리하였다. 유기 상을 염수로 세척하고, 건조 (Na2SO4)시키고, 농축시켰다. 조 생성물을 플래쉬 크로마토그래피에 의해 용리액으로서 0-50%EA/Hex를 사용하여 정제하여 98-E를 수득하였다. 1H NMR은 2종의 회전이성질체의 혼합물을 시사하였다.

단계 5
EtOH (10mL) 중 98-E (696 mg, 1.774mmol)의 용액에 실온에서 히드라진 수화물 (1mL)을 첨가하고, 생성된 용액을 실온에서 2시간 동안 교반하였다. 혼합물을 에틸 에테르 (30 mL)로 희석하고, 0℃에서 60분 동안 교반한 후 여과하였다. 여과물을 농축시키고, 잔류물을 CH2Cl2 중에 용해시키고, 여과하였다. 여과물을 농축시키고, 실리카 겔 상에서 플래쉬 크로마토그래피에 의해 0-20% MeOH (0.2% TEA) /DCM을 사용하여 정제하여 98-F를 수득하였다.

단계 6, 7 및 8
물 (5 mL) 및 EtOH (5 mL) 중 98-G (375.8 mg, 1.55 mmol), 98-E (370 mg, 1.41 mmol), 및 NaHCO3 (261 mg, 3.10 mmol)의 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 염수로 희석하고, EtOAc (x 2)로 추출하였다. 추출물을 합하고, 건조시키고 (Na2SO4), 농축시키고, 진공 하에 건조시켜 조 A를 수득하였다. LCMS-ESI+ (m/z): [M+H]+ 591.59. CH2Cl2 (5 mL) 중 조 A (1.38mmol)에 디옥산 중 4 N HCl (5 mL)을 첨가하였다. 실온에서 2시간 후, 혼합물을 농축 건조시켰다. 이를 톨루엔과 1회 공증발시키고, 진공 하에 건조시켜 조 B를 수득하였다. 무수 MeOH (15 mL) 중 B (1.38mmol + 0.442mmol) 및 DBU (3 mL, 11mmol)를 50℃ 조에서 40분 동안 교반하였다. 혼합물을 농축시켰다. 잔류물을 플래쉬 크로마토그래피 (80 g 칼럼)에 의해 용리액으로서 0 - 20% MeOH/DCM을 사용하여 정제하여 98-H를 수득하였다.

단계 9,10 및 11
나머지 단계를 실시예 41과 유사한 절차를 사용하여 수행하여 목적 화합물 98을 수득하였다.

실시예 99
화합물 99의 제조
(1R,3S,4R,12aR)-7-히드록시-3-메틸-6,8-디옥소-N-(2,4,6-트리플루오로벤질)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
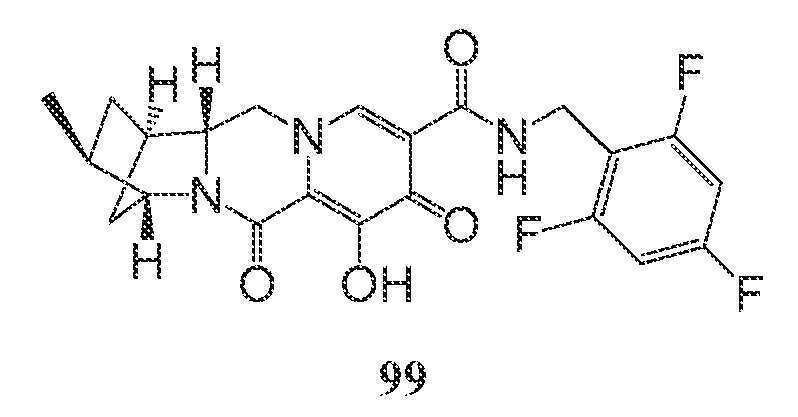
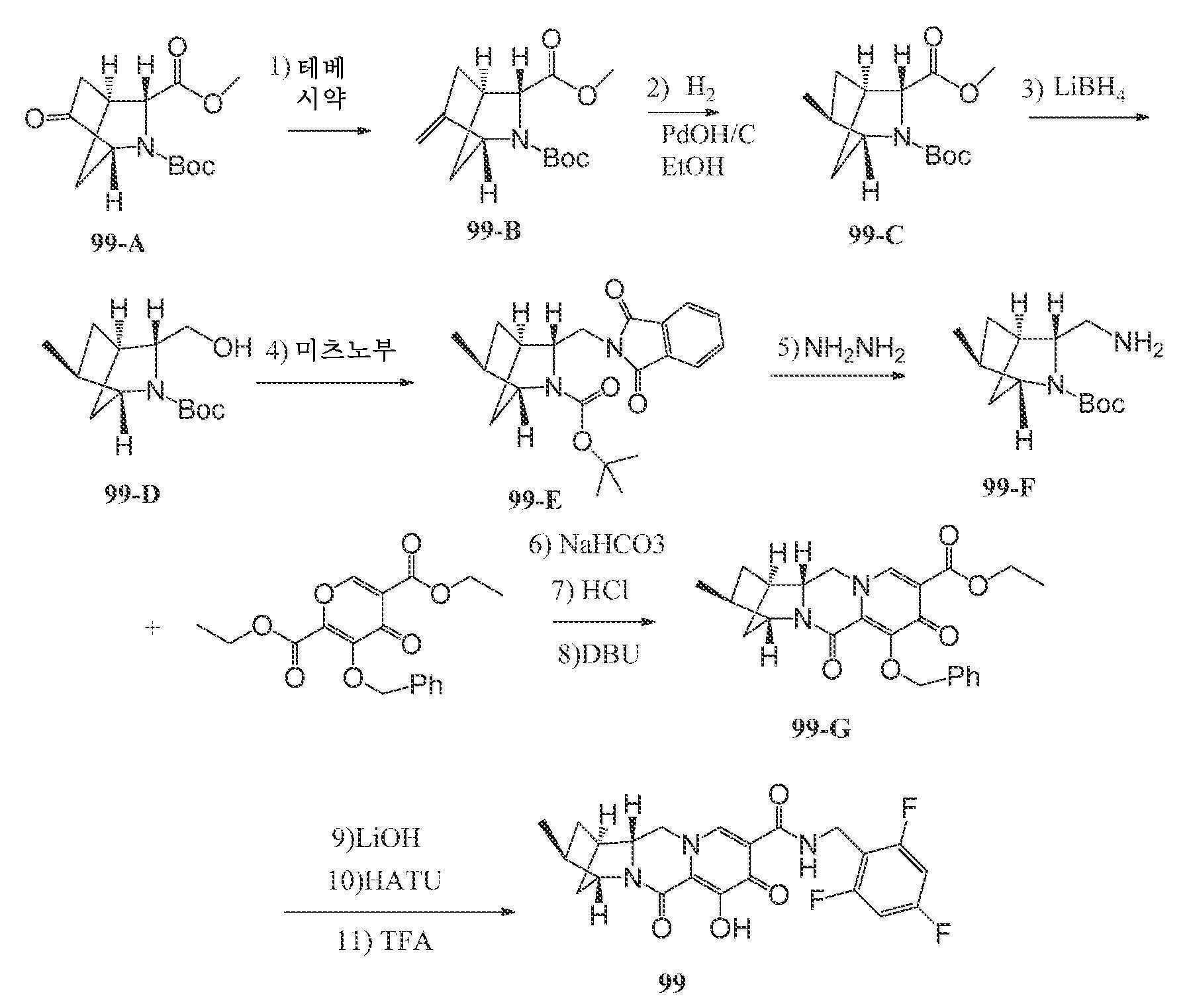
단계 1
THF (20 mL) 중 99-A (1 g, 3.71 mmol)의 교반 용액에 테베 시약 용액 (톨루엔 중 0.5 M, 14.85mL, 7.42 mmol)을 0℃에서 적가하였다. 첨가한 후, 갈색 용액을 실온으로 천천히 가온되도록 하고, 실온에서 2시간 동안 교반하였다. 반응물을 0℃에서의 포화 NaHCO3 용액의 첨가에 의해 조심스럽게 켄칭하고, 혼합물을 실온에서 15분 동안 교반하였다. 혼합물을 셀라이트를 통해 여과하고, 필터 케이크를 에테르 및 DCM (1:1)으로 2회 세척하였다. 층을 분리한 후, 유기부를 합하고, 진공 하에 농축시키고, 잔류물을 실리카 겔 칼럼 상에서 칼럼 크로마토그래피에 의해 0-50% EtOAc/헥산을 사용하여 정제하여 99-B를 수득하였다.

단계 2
EtOH (50 mL) 중 99-B (675 mg, 2.506 mmol) 및 20% Pd(OH)2/C (500 mg)의 혼합물을 H2 분위기 하에 교반하였다. 혼합물을 셀라이트를 통해 여과하고, 여과물을 농축시켜 99-C를 수득하였다.

단계 3
THF (20 mL) 중 99-C (670 mg, 2.488 mmol)를 0℃에서 교반하면서 THF 중 2.0 M LiBH4 (3.7mL, 7.46 mmol)를 첨가하였다. 혼합물을 실온으로 가온하고, 4시간 동안 교반하였다. 반응 혼합물을 얼음으로 켄칭하고, EtOAc 및 포화 NH4Cl로 희석하였다 (약간의 H2 발생). 2개의 상을 분리한 후, 유기 분획을 염수로 세척하고, 건조 (Na2SO4)시키고, 농축시켰다. 조 알콜 99-D를 후속 단계에 그대로 사용하였다.
단계 4 및 5
단계 4 및 5를 실시예 41에서의 절차와 유사한 절차를 사용하여 수행하여 99-F를 수득하였다.
단계 6, 7 및 8
단계 6, 7 및 8을 실시예 41의 절차와 유사한 절차를 사용하여 수행하여 99-G를 수득하였다.
단계 9, 10 및 11
나머지 단계를 실시예 41과 유사한 절차를 사용하여 수행하여 화합물 99를 수득하였다.

실시예 100
화합물 100의 제조
(1R,4R,12aS)-N-(2,4-디플루오로벤질)-7-히드록시-6,8-디옥소-1,2,3,4,6,8,12,12a-옥타히드로-1,4-에타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
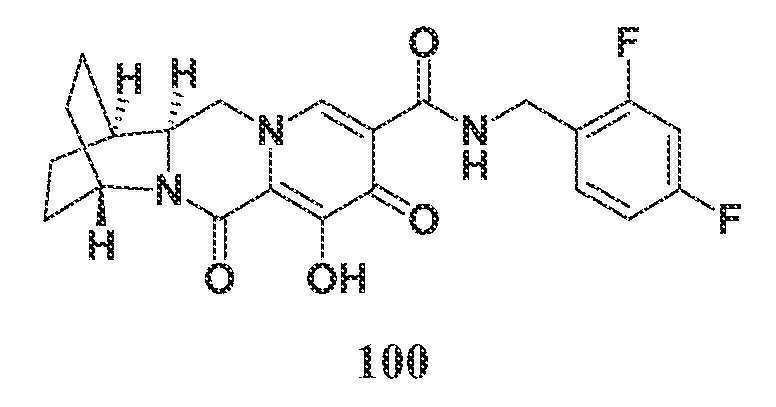

단계 1
100-mL rbf에 THF (20 mL) 중 100-A (2.0 g, 7.8 mmol)를 채웠다. 반응 혼합물을 0℃로 냉각시켰다. 보란 디메틸 술피드 (THF 중 2 N, 17.6 mL)를 천천히 첨가하였다. 이어서, 반응 혼합물을 실온에서 밤새 교반하였다. 반응 혼합물을 0℃로 다시 냉각시켰다. 메탄올 (8 mL)을 적가하여 반응을 켄칭하였다. 농축시킨 후, 잔류물을 콤비 플래쉬 (40 g 칼럼, 카트리지 사용)에 의해 용리액으로서 헥산 - EA를 사용하여 정제하여 100-B를 수득하였다.
단계 2
100-mL rbf에 THF (30 mL) 중 100-B (1.8 g, 7.4 mmol), 트리페닐포스핀 (4.3 g, 16.2 mmol) 및 프탈이미드 (1.8 g, 12.2 mmol)를 채웠다. 이어서, 반응 혼합물을 교반하면서 0℃로 냉각시켰다. DIAD (3.2 mL, 16.2 mmol)를 반응 혼합물에 천천히 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하였다. 농축시킨 후, 잔류물을 콤비 플래쉬 (80 g 칼럼, 카트리지 사용)에 의해 용리액으로서 헥산 - EA를 사용하여 정제하여 100-C를 수득하였다.
단계 3
EtOH (50 mL) 중 100-C (2.5 g, 6.8 mmol)의 용액에 히드라진 1수화물 (1.7 mL)을 첨가하였다. 반응 혼합물을 3시간 동안 교반하면서 70℃로 가열하였다. 여과하여 고체를 제거한 후, 여과물을 농축시켜 100-D를 수득하였다.
단계 4
100-mL rbf에 에탄올 (30 mL) 중 100-D (1.6 g, 6.7 mmol) 및 100-E (2.3 g, 6.7 mmol)를 채웠다. 물 (30 mL) 중 중탄산나트륨 (1.2 g, 1.4 mmol)을 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 실온에서 밤새 교반하였다. 혼합물을 EA (200 mL)로 희석하고, 물 (2 x)로 세척하였다. 수성 분획을 EA (1 x)로 추출하고, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 조 100-F를 후속 단계에 추가 정제 없이 사용하였다.
단계 5
100-mL rbf에 4 N HCl /디옥산 (38 mL) 중 100-F (3.7 g, 6.5 mmol)를 채웠다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하였다. 농축시킨 후, 3.2 g 중간체를 수득하였다. 중간체 및 DBU (5.1 g, 33.8 mmol)를 톨루엔 (100 mL) 중에 용해시켰다. 반응 혼합물을 1시간 동안 교반하면서 110℃로 가열하였다. 농축시킨 후, 잔류물을 콤비 플래쉬 (80 g 칼럼, 카트리지 사용)에 의해 용리액으로서 헥산 - EA를 사용하여 정제하여 100-G를 수득하였다.
단계 6
100-mL rbf에 THF (20 mL) 및 MeOH (20 mL) 중 100-G (2.0 g, 4.7 mmol)를 채웠다. 1 N KOH (18.9 mL)를 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하였다. 반응 혼합물을 1 N HCl (18.9 mL)의 첨가에 의해 산성화시켰다. 농축시킨 후, 잔류물을 톨루엔 (3 x)과 공증발시켰다. 조 산 (0.28 g, 0.72 mmol), 2,4-디플루오로벤질아민 (0.2 g, 1.44 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.47 g, 3.6 mmol) 및 HATU (0.55 g, 1.44 mmol)를 DCM (20 mL) 중에 용해시켰다. 반응 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 EA (100 mL)로 희석하고, 포화 NaHCO3 (2x), 포화 NH4Cl (2x)로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 헥산-EtOAc를 사용하여 정제하여 100-H를 수득하였다.
단계 7
50-mL rbf에 TFA (5 mL) 중 100-H (0.36 g, 0.69 mmol)를 채웠다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 EtOAc-MeOH를 사용하여 정제하여 화합물 100을 수득하였다.

실시예 101
화합물 101의 제조
(1R,4R,12aS)-7-히드록시-6,8-디옥소-N-(2,4,6-트리플루오로벤질)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-에타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드
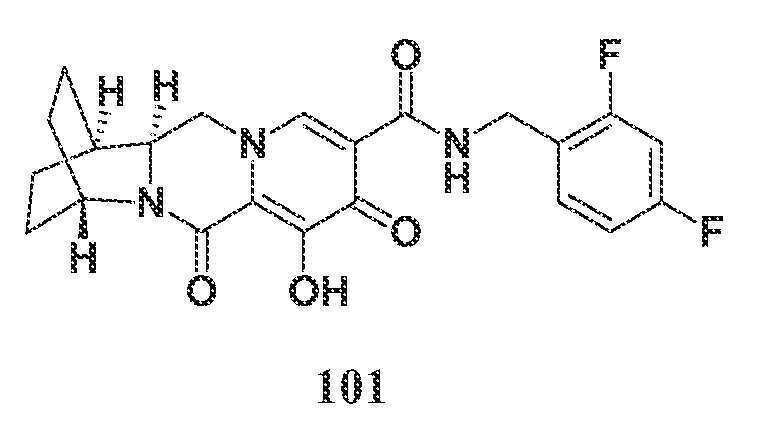

단계 1
100-mL rbf에 THF (2 mL) 및 MeOH (2 mL) 중 101-A (0.3 g, 0.72 mmol)를 채웠다. 1 N KOH (2.1 mL)를 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하였다. 반응 혼합물을 1 N HCl (2.1 mL)의 첨가에 의해 산성화시켰다. 농축시킨 후, 잔류물을 톨루엔 (3 x)과 공증발시켰다. 조 산 (0.72 mmol), 2, 4, 6-트리플루오로벤질아민 (0.23 g, 1.44 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.47 g, 3.6 mmol) 및 HATU (0.55 g, 1.44 mmol)를 DCM (20 mL) 중에 용해시켰다. 반응 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 EA (100 mL)로 희석하고, 포화 NaHCO3 (2x), 포화 NH4Cl (2x)로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 헥산-EtOAc를 사용하여 정제하여 101-B를 수득하였다.
단계 2
50-mL rbf에 TFA (5 mL) 중 101-B (0.36 g, 0.67 mmol)를 채웠다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 EtOAc-MeOH를 사용하여 정제하여 화합물 101을 수득하였다.

실시예 102
화합물 102의 제조
(1S,4S,12aR)-7-히드록시-6,8-디옥소-N-(2,4,6-트리플루오로벤질)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-에타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드

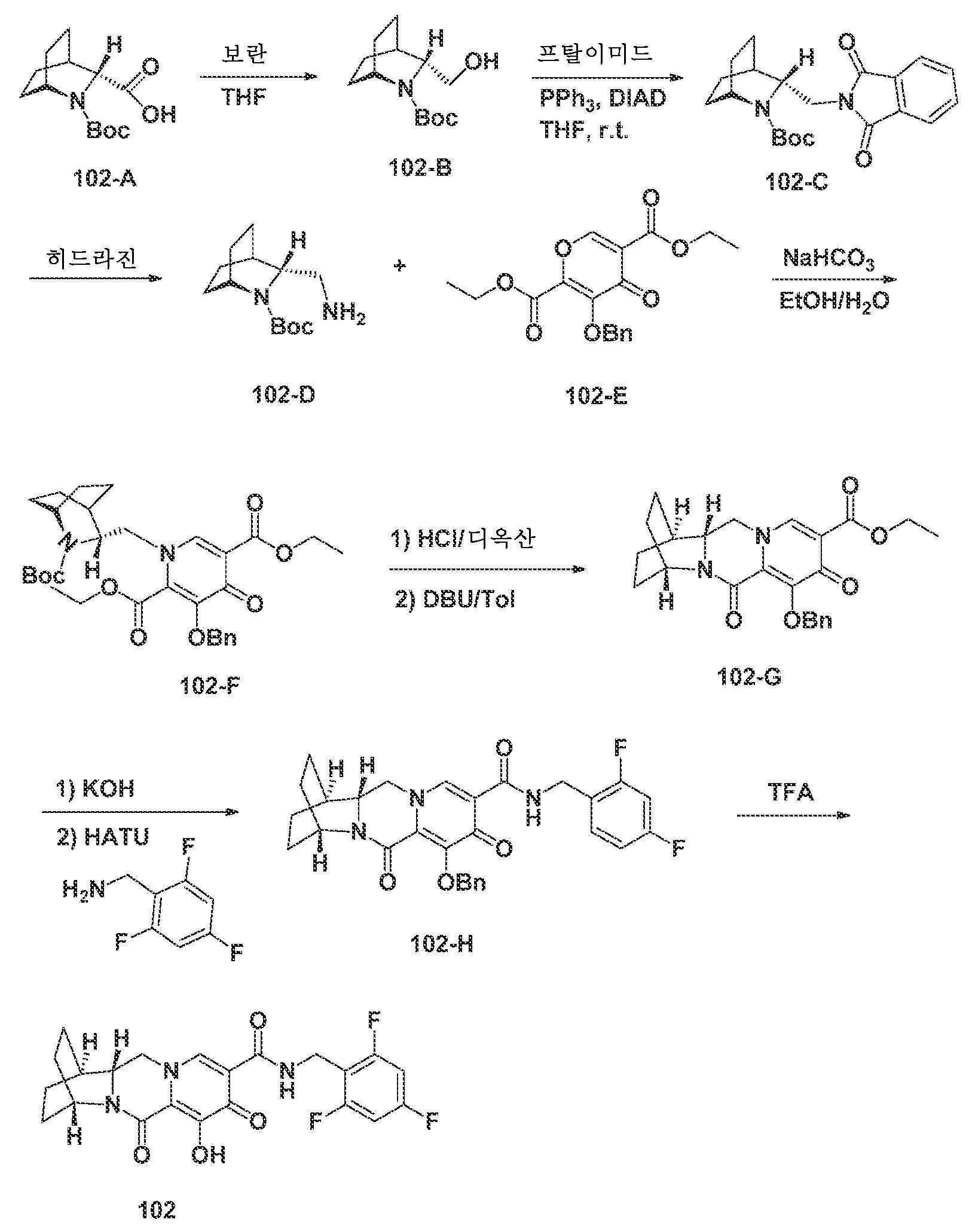
단계 1
100-mL rbf에 THF (20 mL) 중 102-A (2.0 g, 7.8 mmol)를 채웠다. 반응 혼합물을 0℃로 냉각시켰다. 보란 디메틸 술피드 (THF 중 2 N, 17.6 mL)를 천천히 첨가하였다. 이어서, 반응 혼합물을 실온에서 밤새 교반하였다. 반응 혼합물을 0℃로 다시 냉각시켰다. 메탄올 (8 mL)을 적가하여 반응을 켄칭하였다. 농축시킨 후, 잔류물을 콤비 플래쉬 (40 g 칼럼, 카트리지 사용)에 의해 용리액으로서 헥산 - EA를 사용하여 정제하여 102-B를 수득하였다.
단계 2
100-mL rbf에 THF (30 mL) 중 102-B (1.8 g, 7.4 mmol), 트리페닐포스핀 (4.3 g, 16.2 mmol) 및 프탈이미드 (1.8 g, 12.2 mmol)를 채웠다. 이어서, 반응 혼합물을 교반하면서 0℃로 냉각시켰다. DIAD (3.2 mL, 16.2 mmol)를 반응 혼합물에 천천히 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하였다. 농축시킨 후, 잔류물을 콤비 플래쉬 (80 g 칼럼, 카트리지 사용)에 의해 용리액으로서 헥산 - EA를 사용하여 정제하여 102-C를 수득하였다.
단계 3
EtOH (50 mL) 중 102-C (2.5 g, 6.8 mmol)의 용액에 히드라진 1수화물 (1.7 mL)을 첨가하였다. 반응 혼합물을 3시간 동안 교반하면서 70℃로 가열하였다. 여과하여 고체를 제거한 후, 여과물을 농축시켜 102-D를 수득하였다.
단계 4
100-mL rbf에 에탄올 (30 mL) 중 102-D (1.6 g, 6.7 mmol) 및 102-E (2.3 g, 6.7 mmol)를 채웠다. 물 (30 mL) 중 중탄산나트륨 (1.2 g, 1.4 mmol)을 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 실온에서 밤새 교반하였다. 혼합물을 EA (200 mL)로 희석하고, 물 (2 x)로 세척하였다. 수성 분획을 EA (1 x)로 추출하고, 유기 분획을 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 조 102-F를 후속 단계에 추가 정제 없이 사용하였다.
단계 5
100-mL rbf에 4 N HCl /디옥산 (38 mL) 중 102-F (3.7 g, 6.5 mmol)를 채웠다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하였다. 농축시킨 후, 3.2 g 중간체를 수득하였다. 중간체 및 DBU (5.1 g, 33.8 mmol)를 톨루엔 (100 mL) 중에 용해시켰다. 반응 혼합물을 1시간 동안 교반하면서 110℃로 가열하였다. 농축시킨 후, 잔류물을 콤비 플래쉬 (80 g 칼럼, 카트리지 사용)에 의해 용리액으로서 헥산 - EA를 사용하여 정제하여 102-G를 수득하였다.
단계 6
100-mL rbf에 THF (2 mL) 및 MeOH (2 mL) 중 102-G (0.3 g, 0.72 mmol)를 채웠다. 1 N KOH (2.1 mL)를 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하였다. 반응 혼합물을 1 N HCl (2.1 mL)의 첨가에 의해 산성화시켰다. 농축시킨 후, 잔류물을 톨루엔 (3x)과 공증발시켰다. 조 산 (0.72 mmol), 2, 4, 6-트리플루오로벤질아민 (0.23 g, 1.44 mmol), N,N-디이소프로필에틸아민 (DIPEA) (0.47 g, 3.6 mmol) 및 HATU (0.55 g, 1.44 mmol)를 DCM (20 mL) 중에 용해시켰다. 반응 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 EA (100 mL)로 희석하고, 포화 NaHCO3 (2x), 포화 NH4Cl (2x)로 세척하고, Na2SO4 상에서 건조시켰다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 헥산-EtOAc를 사용하여 정제하여 102-H를 수득하였다.
단계 7
50-mL rbf에 TFA (5 mL) 중 102-H (0.36 g, 0.67 mmol)를 채웠다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 농축시킨 후, 조 물질을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 EtOAc-MeOH를 사용하여 정제하여 화합물 102를 수득하였다.

실시예 103
화합물 103의 제조
(1R,4R,12aR)-2,3-디플루오로-7-히드록시-6,8-디옥소-N-(2,4,6-트리플루오로벤질)-1,2,3,4,6,8,12,12a-옥타히드로-1,4-메타노디피리도[1,2-a:1',2'-d]피라진-9-카르복스아미드

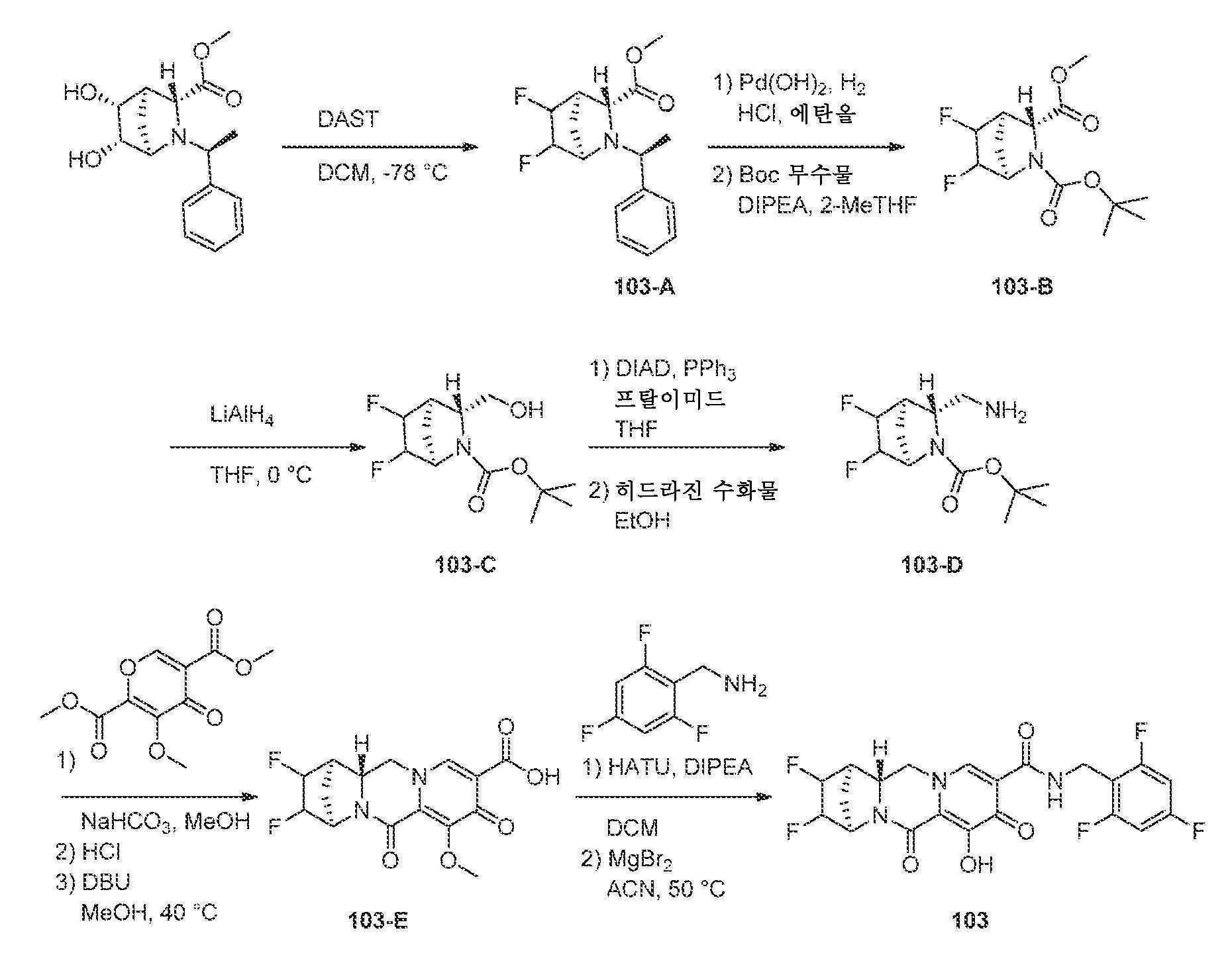
단계 1
DCM (27mL) 중 (1R,3R,4R,5R,6S)-메틸 5,6-디히드록시-2-((S)-1-페닐에틸)-2-아자비시클로[2.2.1]헵탄-3-카르복실레이트 (2.0g, 6.9mmol)의 용액을 드라이 아이스/아세톤 조에서 -78℃로 냉각시켰다. 이 용액에 플라스틱 말단의 피펫을 통해 DAST (2.18 ml, 16.48 mmol)를 첨가하였다. 용액을 -78℃에서 30분 동안 교반한 후, 조에서 꺼내고, 천천히 실온으로 가온되도록 하고, 실온에서 1시간 동안 교반하였다. 교반 중인 포화 중탄산나트륨 용액 (150mL)에 플라스틱 말단의 피펫을 통해 반응 혼합물을 천천히 첨가하여 반응을 켄칭시켰다. 층을 분리하고, 수성 층을 디클로로메탄으로 역추출하였다. 합한 유기 층을 황산마그네슘 상에서 건조시키고, 여과하고, 진공 하에 농축시켰다. 조 생성물을 실리카 겔 크로마토그래피 (7-28% 에틸 아세테이트/헥산)에 의해 정제하여 103-A를 수득하였다.

단계 2 및 3
에탄올 (36.01 ml) 및 1.25M HCl-에탄올 (4.09 ml) 중 103-A (0.96 g, 3.24 mmol)의 용액에 20% PdOH/C (1.14 g, 1.62 mmol)를 첨가하고, 현탁액을 수소 분위기 하에 22시간 동안 교반하였다. 셀라이트를 통해 여과한 후, 케이크를 EtOH로 세척하고, 여과물을 진공 하에 농축 건조시켜, 후속 단계를 위한 3.24mmol로 추정되는 조 탈보호 생성물을 수득하였다.
2-메틸테트라히드로푸란 (32.43 ml) 중 조 잔류물 (0.62 g, 3.24 mmol) 및 디-tert-부틸 디카르보네이트 (1.06 g, 4.86 mmol)에 N,N-디이소프로필에틸아민 (0.56 ml, 0 mol)을 첨가하였다. 완료된 후, 반응 혼합물을 물로 희석하고, EtOAC (2x)로 추출하고, 유기 분획을 물로 세척하고, 합하고, 건조 (Na2SO4)시키고, 농축시켰다. 잔류물을 실리카 칼럼 크로마토그래피 (0-55% EtOAc/헥산)에 의해 정제하여 103-B를 수득하였다.

단계 4
THF (15 ml) 중 103-B (0.68 g, 2.33 mmol)의 용액을 빙조에서 교반하면서 THF 중 1.0 M LiBH4 (4.65 ml)를 첨가하고, 생성된 혼합물을 0℃에서 30분 동안 교반하였고, 이 때 TLC에 의하면 완료된 것으로 나타났다. 반응 혼합물을 물 (0.3 mL)에 이어서 NaOH (~15%, 3.5M, 0.3 mL)로 조심스럽게 처리하고, 최종적으로 추가의 물 (0.9 mL)로 처리하였다. 혼합물을 실온에서 15분 동안 교반하고, 형성된 ppt를 여과하고, 디에틸 에테르로 세척하고, 상청액을 농축시켜 103-C를 수득하였다.
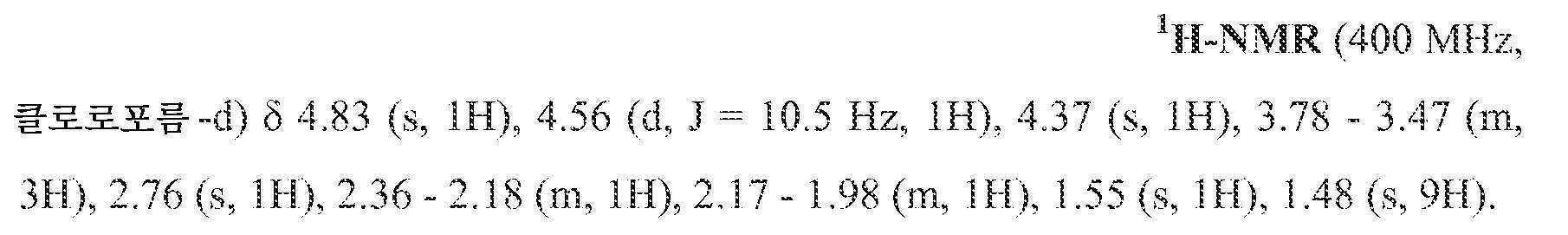
단계 5 및 6
THF (11 ml) 중 103-C (0.59 g, 2.25 mmol), 프탈이미드 (0.53 g, 3.6 mmol) 및 트리페닐포스핀 (1.3 g, 4.95 mmol)의 혼합물을 빙조에서 냉각시켰다. 디이소프로필 아조디카르복실레이트 (0.97 ml, 4.95 mmol)를 첨가하였다. 이어서, 혼합물을 실온으로 가온하고, 14시간 동안 교반한 다음 진공 하에 농축시켰다. 잔류물을 에테르 중에 용해시키고, 1시간 동안 교반한 다음, 고체를 여과하고, 여과물을 농축시켰다. 잔류물을 실리카 칼럼 크로마토그래피 (10-31-91% EtOAc/헥산)에 의해 정제하여 보호된 아미노 화합물 (2.25mmol의 생성물로 추정됨)을 수득하였다.
에탄올 (22 ml) 중 보호된 아미노 화합물 (0.88 g, 2.25 mmol) 및 히드라진 수화물 (0.46 ml, 9.52 mmol)의 용액을 60℃에서 2시간 동안 교반하였다. 반응 혼합물을 빙조에서 냉각시키고, 에테르 (10 ml)를 첨가하고, 혼합물을 30분 동안 교반하였다. 형성된 고체를 여과하고, 여과물을 진공 하에 농축 건조시켜 103-D를 수득하였다.

단계 7, 8 및 9
화합물 103을 화합물 60과 유사한 방식으로 41-E 대신 103-D를 사용하고 (2,3-디클로로페닐)메탄아민 대신 (2,4,6-트리플루오로페닐)메탄아민을 사용하여 제조하엿다. 단일 부분입체이성질체가 생성되었다. 플루오린의 입체화학은 미지이다.

항바이러스 검정
실시예 104
MT4 세포에서의 항바이러스 검정
MT4 세포를 사용한 항바이러스 검정을 위해, DMSO 중 189X 시험 농도의 3배 연속 희석된 화합물 0.4 μL를 384-웰 검정 플레이트 (10개 농도)의 각 웰 내 세포 성장 배지 (RPMI 1640, 10% FBS, 1% 페니실린/스트렙토마이신(Streptomycine), 1% L-글루타민, 1% HEPES) 40 μL에 4중으로 첨가하였다.
2 x 106개 MT4 세포의 1 mL 분취액을 세포 성장 배지 (모의-감염) 또는 HIV-IIIb 농축 ABI 원액의 새로운 1:250 희석물 (MT4 세포의 경우 0.004 m.o.i.) 25 μL (MT4)로 37℃에서 각각 1 및 3시간 동안 사전-감염시켰다. 감염 및 비감염된 세포를 세포 성장 배지 중에서 희석하고, 2000개 (MT4의 경우) 세포 35 μL를 검정 플레이트의 각 웰에 첨가하였다.
이어서, 검정 플레이트를 37℃ 인큐베이터에서 인큐베이션하였다. 인큐베이션 5일 후, 2X 농축 셀타이터-글로(CellTiter-Glo)™ 시약 (카탈로그 # G7573, 프로메가 바이오사이언시스, 인크.(Promega Biosciences, Inc.), 위스콘신주 매디슨) 25 μL를 검정 플레이트의 각 웰에 첨가하였다. 2-3분 동안 실온에서 인큐베이션하여 세포 용해를 수행한 다음, 엔비전(Envision) 판독기 (퍼킨엘머(PerkinElmer))를 사용하여 화학발광을 판독하였다.
본 발명의 화합물은 하기 표 1에 도시된 바와 같이 본 검정에서 항바이러스 활성을 입증하였다. 따라서, 본 발명의 화합물은 HIV 바이러스의 증식을 치료하거나, AIDS를 치료하거나, 또는 AIDS 또는 ARC 증상의 개시를 지연시키는데 유용할 수 있다.
<표 1>
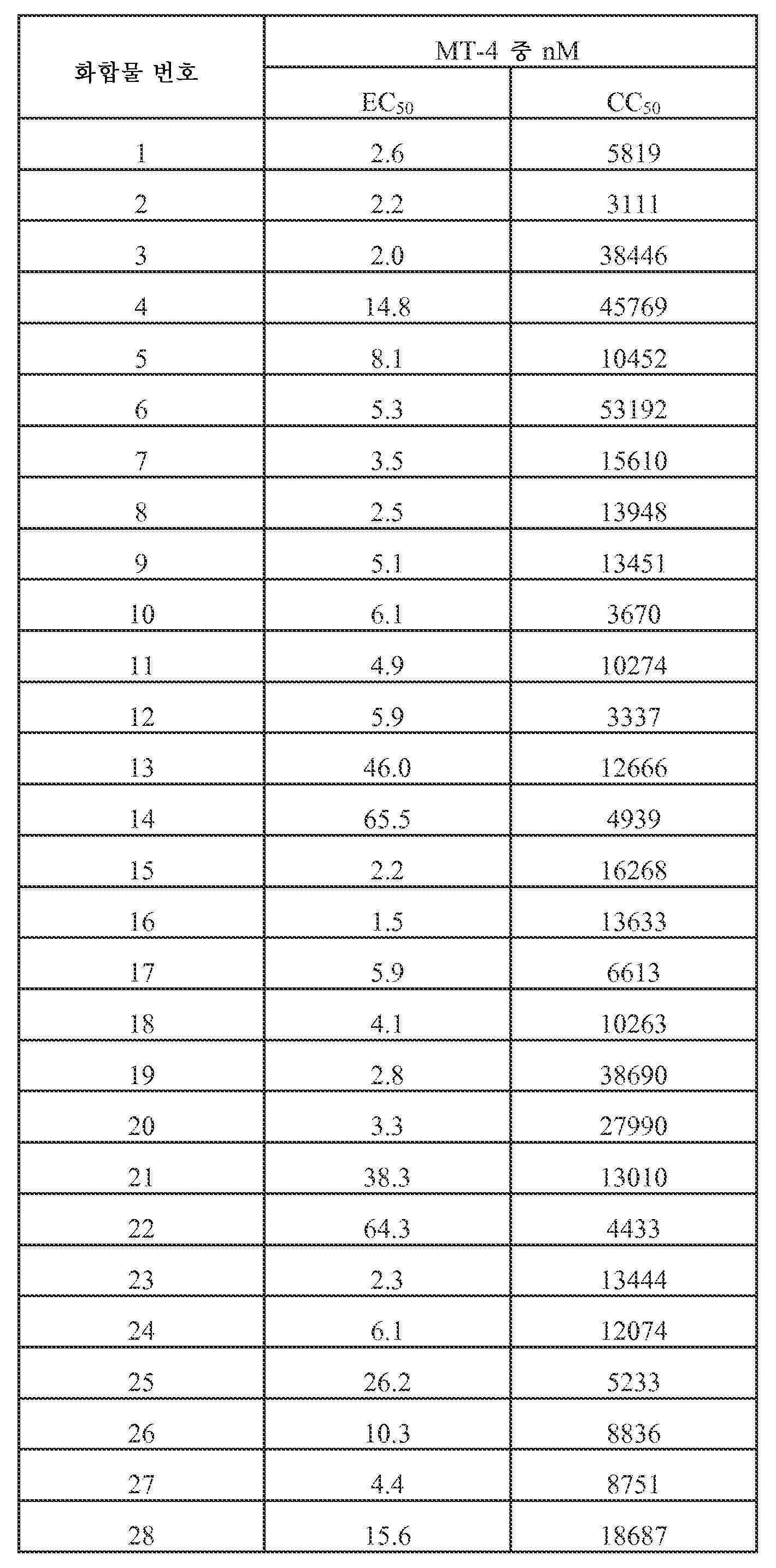
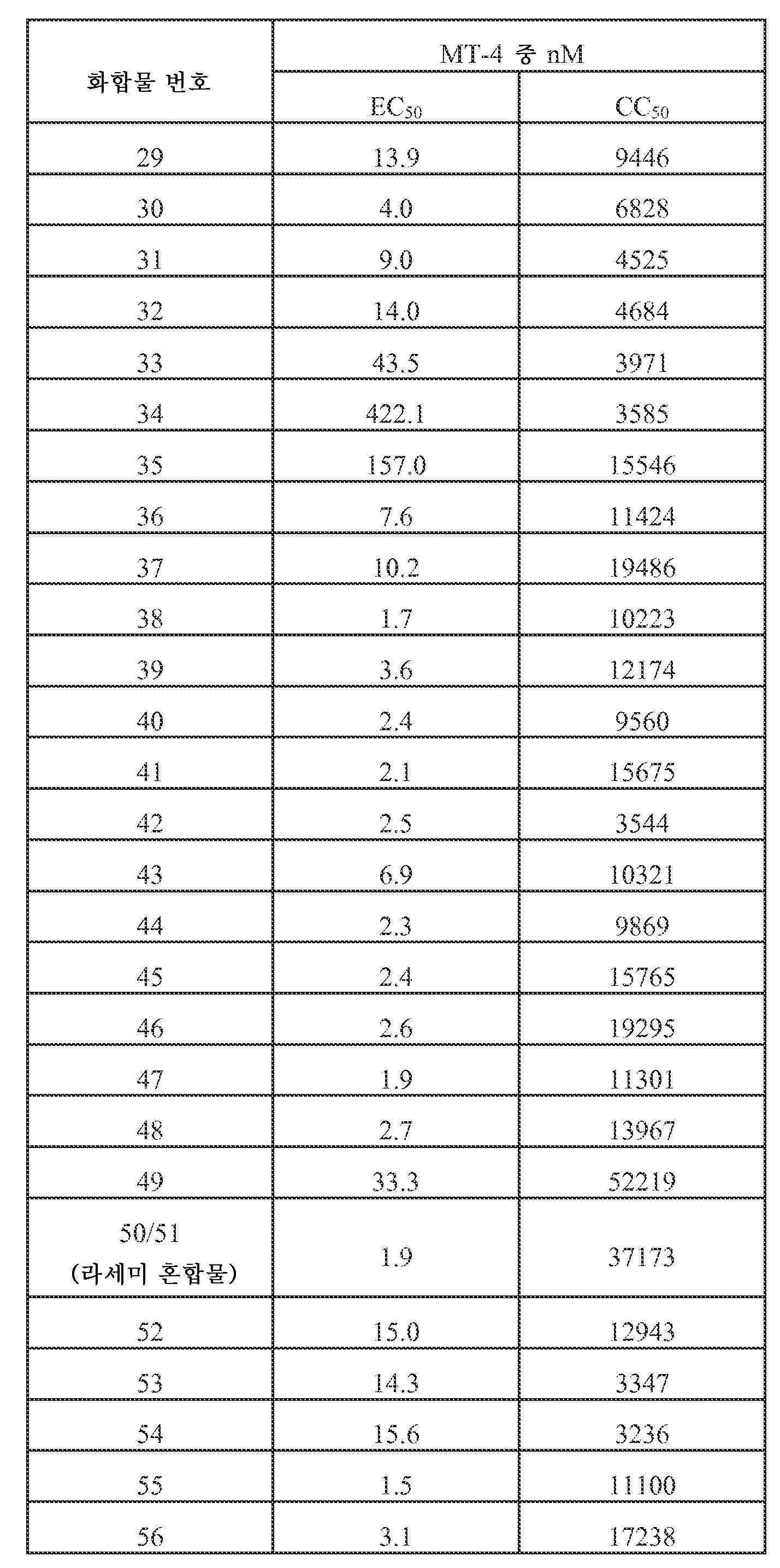

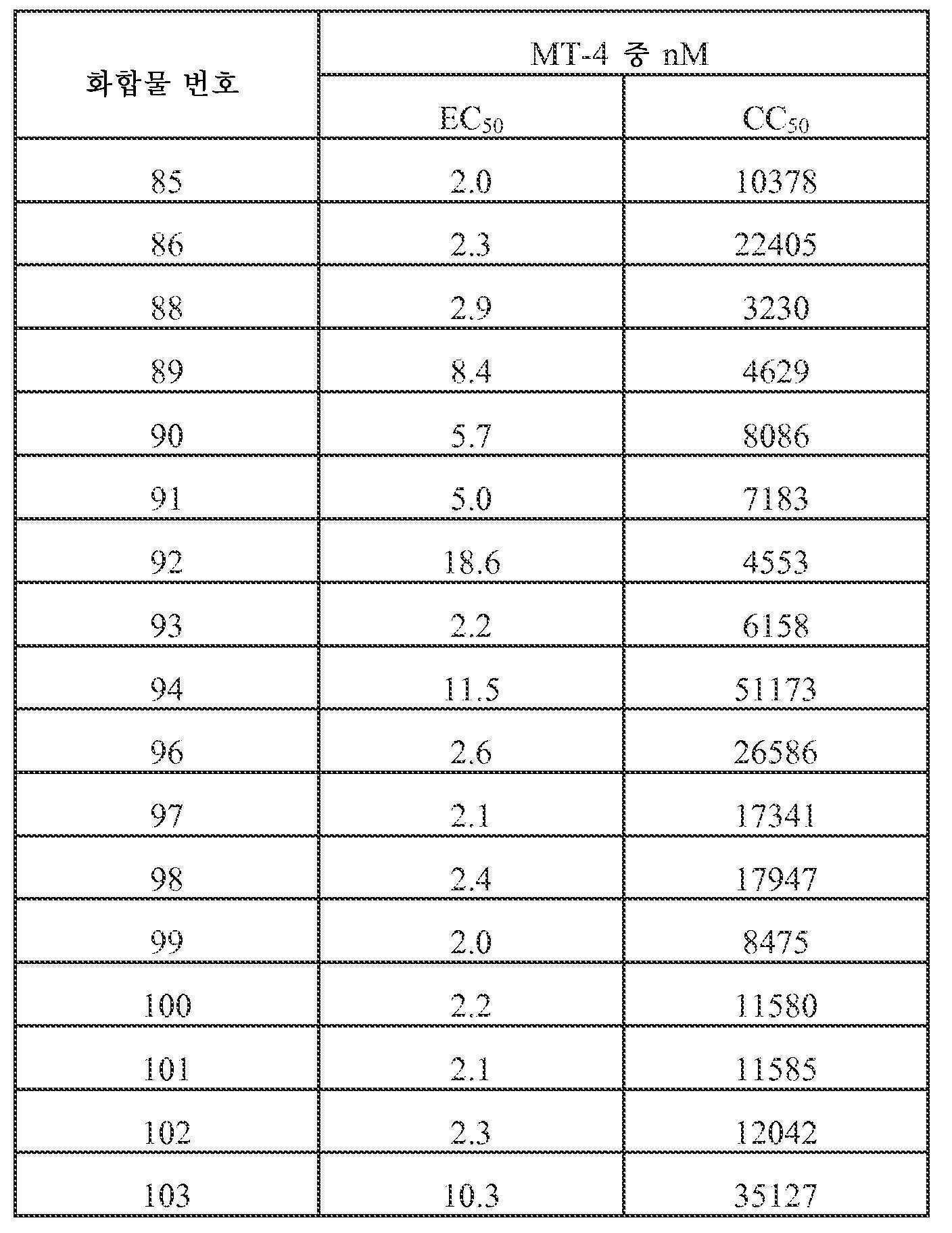
실시예 105
인간 PXR 활성화 검정
루시페라제 리포터 유전자 검정. 안정하게 형질전환된 종양 세포주 (DPX2)를 96-웰 마이크로타이터 플레이트 상에 플레이팅하였다. DPX2 세포는 인간 PXR 유전자 (NR1I2), 및 인간 CYP3A4 유전자에서 확인되는 2개의 프로모터, 즉 XREM 및 PXRE에 연결된 루시페라제 리포터 유전자를 보유한다. 세포를 각 화합물의 6종의 농도 (0.15 ~ 50 μM)로 처리하고, 24시간 동안 인큐베이션하였다. 생존 세포의 수를 결정하고, 리포터 유전자 활성을 평가하였다. 양성 대조군: 6종의 농도 (0.1 ~ 20 μM)에서의 리팜피신. 10 또는 20 μM RIF에 의한 최대 배수 유도에 관한 %Emax를 DMSO 배경에 대해 조정된 하기 방정식에 따라 시험 화합물에 대해 계산하였다: %Emax = (배수 유도 - 1)/(RIF에 의한 최대 배수 유도 - 1) x 100%.
<표 2>

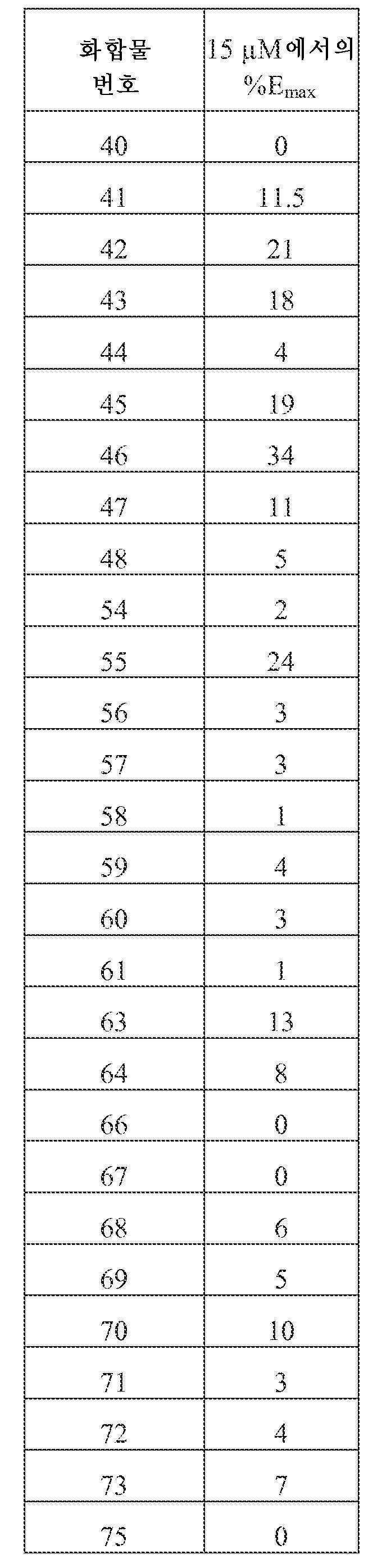
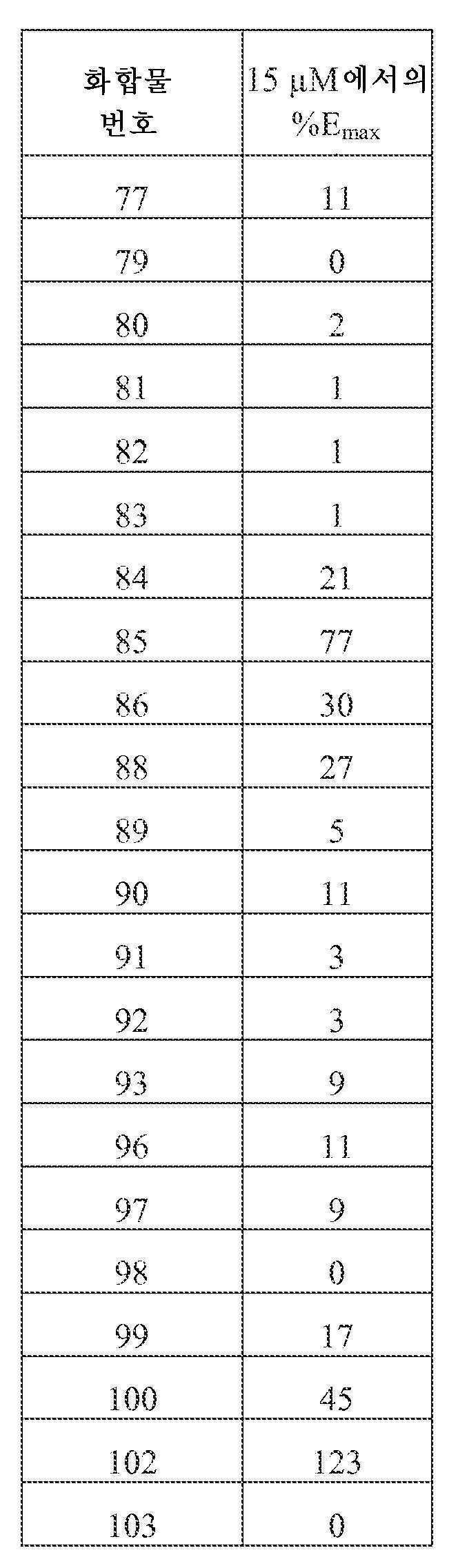
실시예 106
OCT2 억제 검정
시험 화합물에 의한 모델 기질 14C-테트라에틸암모늄 (TEA)의 OCT2 매개 흡수의 용량 의존적 억제를 야생형 및 OCT2-형질감염된 MDCKII 세포에서 0.014 μM 내지 10 μM의 7종의 농도에서 연구하였다.
MDCKII 세포를 37℃, 90% 습도 및 5% CO2로 설정한 인큐베이터 내에서 1% Pen/Strep, 10% 태아 소 혈청, 및 0.25 mg/mL 히그로마이신 B를 함유하는 최소 필수 배지 (MEM) 중에 유지시켰다. 검정 24시간 전, 5 mM 부티르산나트륨을 함유하는 배지를 플라스크 내 MDCKII 세포에 첨가하고, 세포를 80-90% 전면생장률로 성장시켰다. 검정 당일에, 세포를 트립신처리하고, 크렙스-헨셀라이트 완충제(Krebs-Henseleit Buffer, KHB), pH 7.4 중에 5 x 106 백만개 세포/mL로 재현탁시켰다. 세포를 검정 플레이트 내에서 15분 동안 예비인큐베이션한 후 시험 화합물 또는 기질을 첨가하였다.
시험 화합물은 DMSO 중에서 연속 희석한 다음, 야생형 또는 OCT2-형질감염된 세포를 함유하는 0.4 mL KHB 완충제 내에 스파이킹하고 (2 μL), 10분 동안 인큐베이션하였다. KHB 완충제 중 100 μM 14C-TEA 0.1 mL의 첨가에 의해 검정을 개시하였다 (혼합 후 20 μM 최종 농도). TEA의 농도는 Km에 기초하였다. 인큐베이션 10분 후, 빙냉 1X PBS 완충제 0.5 mL의 첨가에 의해 검정 혼합물을 켄칭하였다. 이어서 샘플을 1000 rpm에서 5분 동안 원심분리하고, 상청액을 제거하였다. 세척 단계를 빙냉 PBS로 4회 반복하였다. 마지막으로, 세포 펠릿을 0.2N NaOH로 용해하고, 실온에서 적어도 30분 동안 두어 완전한 용해를 보장하였다. 이어서 샘플을 액체 섬광 계수기 상에서 계수하고, dpm 계수를 사용하여 하기 계산을 수행하였다. % 억제는 하기와 같이 계산하였다: % 억제 = [1 - {[OCT2]i-[WT]ni} / {[OCT2]ni-[WT]ni}]*100, 상기 식에서 각각 [OCT2]i는 OCT2 세포에 대한 시험 화합물의 존재 하의 dpm 계수를 나타내고, [OCT2]ni는 OCT2 세포에 대한 시험 화합물의 부재 하의 dpm 계수를 나타내고, [WT]ni는 야생형 세포에 대한 시험 화합물의 부재 하의 dpm 계수를 나타낸다.
<표 3>
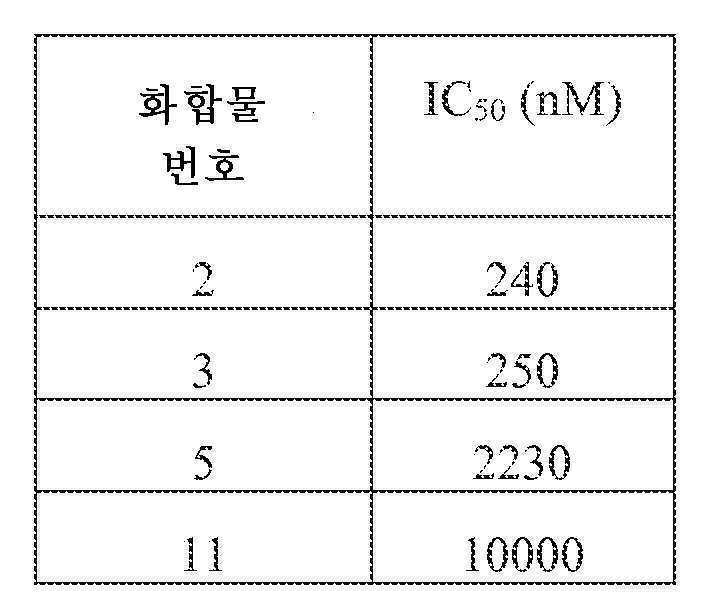
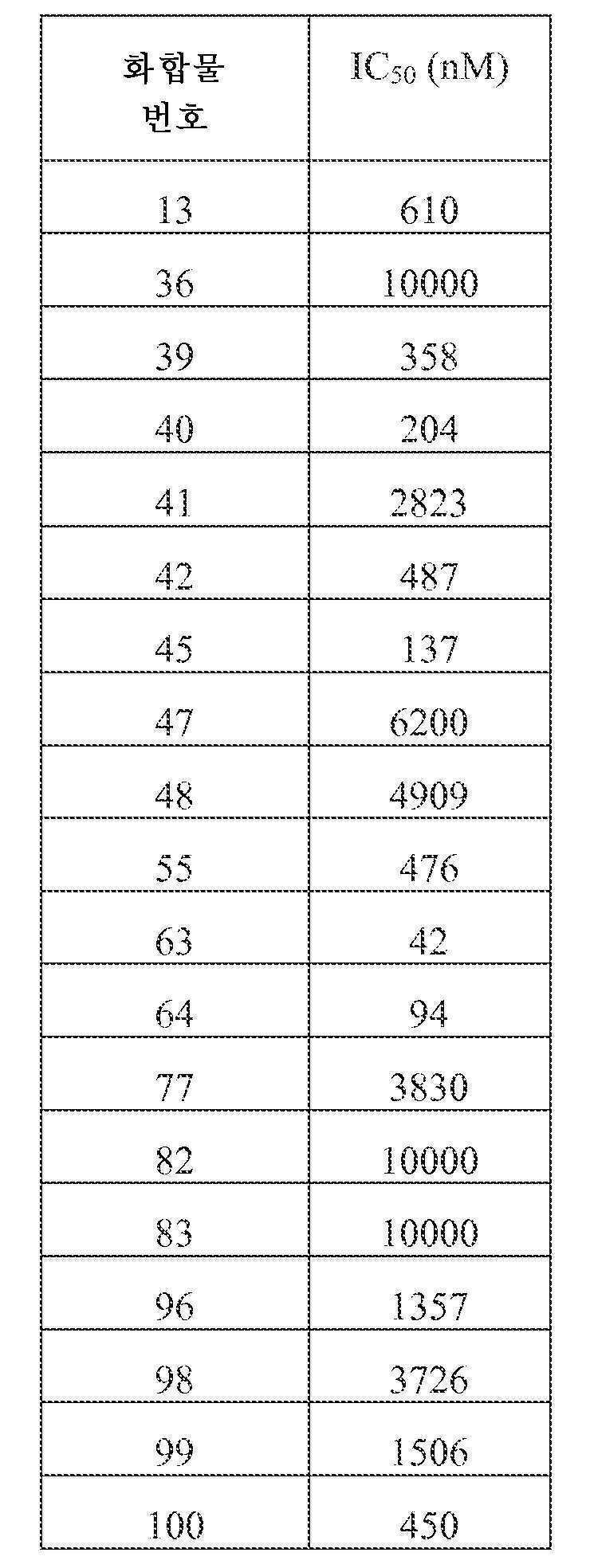
표 1, 2 및 3의 데이터는 각각의 화합물에 대한 각각의 검정의 시간에 걸친 평균을 나타낸다. 특정 화합물의 경우에, 프로젝트 기간에 걸쳐 다중 검정을 수행하였다. 따라서, 표 1, 2 및 3에 보고된 데이터는 선행 문서에서 보고된 데이터, 뿐만 아니라 개입 기간에 수행된 검정으로부터의 데이터도 포함한다.
실시예 107
비글 개에게 경구 또는 정맥내 투여 후 약동학적 분석
약동학적 분석을 다양한 시험 화합물에 대해, 비글 개에게 정맥내 또는 경구 투여 후 수행하였다.
정맥내로 투여된 화합물의 약동학적 분석을 위해, 시험 화합물을 0.1 mg/mL의 IV 주입을 위해 5% 에탄올, 55% PEG 300, 및 40% 물 중에 제제화하였다. 경구로 투여된 화합물의 약동학적 분석을 위해, 시험 화합물을 1 mg/kg의 Di수 중 0.1% 트윈(Tween) 20, 0.5% HPMC LV100의 수성 현탁액으로서 제제화하였다.
각 투여 군은 3마리의 수컷, 비-나이브 순종 비글 개로 이루어졌다. 투여 시, 동물은 10 내지 13 kg으로 칭량되었다. 동물을 용량 투여 전 밤새 및 투여 후 최대 4시간 금식시켰다. 정맥내 투여의 연구를 위해, 시험 물품을 정맥내 주입에 의해 30분에 걸쳐 동물에게 투여하였다. 주입 속도는 각 동물의 체중에 따라 0.5 mg/kg의 용량이 전달되도록 조정하였다. 경구 투여의 연구를 위해, 시험 물품을 각 동물의 체중에 따라 1 mg/kg의 용량이 전달되도록 투여하였다.
정맥내로 투여된 화합물의 약동학적 분석을 위해, 일련의 정맥 혈액 샘플 (각각 대략 1 mL)을 각각의 동물로부터 투여 후 0, 0.250, 0.483, 0.583, 0.750, 1.00, 1.50, 2.00, 4.00, 8.00, 12.0, 및 24.0시간에 채취하였다. 혈액 샘플을 항응고제로서 EDTA-K2가 담긴 바큐테이너(Vacutainer)™ 튜브에 수집하고, 즉시 혈장용 습윤 얼음 상 계류 원심분리에 두었다. LC/MS/MS 방법을 사용하여 혈장 내 시험 화합물의 농도를 측정하였다. 각 혈장 샘플의 100 μL 분취액을 깨끗한 96 웰 플레이트에 첨가하고, 차가운 아세토니트릴/내부 표준 용액 (ACN)/(ISTD) 400 μL를 첨가하였다. 단백질 침전 후, 상청액의 110 μL 분취액을 깨끗한 96-웰 플레이트로 옮기고, 물 300 μL로 희석하였다. 상기 용액의 25 μL 분취액을 하이퍼실 골드(Hypersil Gold) C18 HPLC 칼럼 (50 X 3.0 mm, 5 μm; 써모-하이퍼실(Thermo-Hypersil) 파트 # 25105-053030)을 사용하는 TSQ 퀀텀 울트라(Quantum Ultra) LC/MS/MS 시스템에 주사하였다. 용리 및 분리를 위해 애질런트(Agilent) 1200 시리즈 이원 펌프 (P/N G1312A 빈(P/N G1312A Bin) 펌프)를 사용하고, 샘플 주사를 위해 HTS 팔(HTS Pal) 오토샘플러 (립 테크놀로지스(LEAP Technologies), 노스캐롤라이나주 카버러)를 사용하였다. TSQ 퀀텀 울트라 삼중 사중극자 질량 분광계를 선택적 반응 모니터링 모드로 사용하였다 (써모 피니간(Thermo Finnigan), 캘리포니아주 산호세). 액체 크로마토그래피는 2개의 이동상을 사용하여 수행하였다: 이동상 A는 pH 3.0으로 2.5 mM 포름산암모늄 수용액 중 1% 아세토니트릴을 함유하였고, 이동상 B는 pH 4.6으로 10 mM 포름산암모늄 중 90% 아세토니트릴을 함유하였다. 혈장 농도-시간 데이터에 대해 비-구획화 약동학적 분석을 수행하였다. 결과 데이터를 하기 표 4의 처음 3개의 열에 제시한다. 표 4에서, CL은 클리어런스를 지칭하며, 이는 약물이 혈장으로부터 제거되는 속도를 특성화한다. 약물의 클리어런스가 낮을수록 신체 내 배설 반감기는 더 길다. Vss는 분포의 정상 상태 부피를 지칭하고, 약물이 얼마나 잘 조직에 분포되었는지를 나타낸다. Vss가 클수록, 신체 내 배설 반감기는 더 길다. MRT는 평균 체류 시간을 지칭하며, 이는 분자가 신체 내에서 존재하는 평균 시간의 측정치이다.
경구로 투여된 화합물의 약동학적 분석을 위해, 일련의 정맥 혈액 샘플 (각각 대략 0.3 mL)을 각각의 동물로부터 투여 후 0, 0.25, 0.50, 1.0, 2.0, 4.0, 6.0, 8.0, 12.0 및 24.0시간의 시점에 채취하였다. 혈액 샘플을 상기 기재된 정맥내 연구와 유사한 방식으로 수집하고, 준비하고, 분석하였다. 혈장 농도-시간 데이터에 대해 비-구획화 약동학적 분석을 수행하였다. 결과 데이터를 하기 표 4의 마지막 3개의 열에 제시한다. 표 4에서, F (%)는 경구 생체이용률을 지칭한다. Cmax는 투여 후 화합물의 최대 혈장 농도를 지칭한다. AUC는 곡선하 면적을 지칭하고, 이는 표시된 화합물의 전체 혈장 노출의 측정치이다.
<표 4>
본 명세서에서 언급되는 모든 미국 특허, 미국 특허 출원 공개, 미국 특허 출원, 외국 특허, 외국 특허 출원 및 비-특허 간행물은 본 발명의 설명과 비일치하지 않는 정도로 그 전문이 본원에 참조로 포함된다.
상기로부터, 본 발명의 구체적 실시양태가 예시 목적을 위해 본원에 기재되었지만, 본 발명의 취지 및 범위를 벗어나지 않으면서 다양한 변형이 이루어질 수 있음을 인지할 것이다. 따라서, 본 발명은 첨부된 특허청구범위에 의하는 것을 제외하고는 제한되지 않는다.
Claims (1)
- 하기 화학식 I을 갖는 화합물 또는 그의 입체이성질체 또는 제약상 허용되는 염의 용도.
<화학식 I>

상기 식에서:
X는 -O- 또는 -NZ3- 또는 -CHZ3-이고;
W는 -CHZ2-이고;
Z1, Z2 및 Z3은 각각, 독립적으로, 수소 또는 C1-3알킬이거나, 또는 여기서 Z1 및 Z2 또는 Z1 및 Z3은 함께 -L-을 형성하고, 여기서 L은 -C(Ra)2-, -C(Ra)2C(Ra)2-, -C(Ra)2C(Ra)2C(Ra)2-, 또는 -C(Ra)2C(Ra)2C(Ra)2C(Ra)2-이고, 여기서 Z1 및 Z2 또는 Z1 및 Z3 중 적어도 하나는 함께 -L-을 형성하고;
Z4는 결합, -CH2-, 또는 -CH2CH2-이고;
Y1 및 Y2는 각각, 독립적으로, 수소, C1-3알킬 또는 C1-3할로알킬이고;
R1은 1 내지 3개의 할로겐으로 치환된 페닐이고;
각각의 Ra는 독립적으로, 수소, 할로, 히드록실 또는 C1-4알킬이다.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020217012463A KR102351912B1 (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261745375P | 2012-12-21 | 2012-12-21 | |
| US61/745,375 | 2012-12-21 | ||
| US201361788397P | 2013-03-15 | 2013-03-15 | |
| US61/788,397 | 2013-03-15 | ||
| US201361845803P | 2013-07-12 | 2013-07-12 | |
| US61/845,803 | 2013-07-12 | ||
| PCT/US2013/076367 WO2014100323A1 (en) | 2012-12-21 | 2013-12-19 | Polycyclic-carbamoylpyridone compounds and their pharmaceutical use |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020197031864A Division KR102120875B1 (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020217012463A Division KR102351912B1 (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR20200067930A true KR20200067930A (ko) | 2020-06-12 |
| KR102246963B1 KR102246963B1 (ko) | 2021-04-30 |
Family
ID=49917297
Family Applications (8)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020217012463A KR102351912B1 (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
| KR1020227001078A KR102406288B1 (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
| KR1020227018650A KR102527797B1 (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
| KR1020177016697A KR102040007B1 (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
| KR1020237014240A KR20230061576A (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
| KR1020157019194A KR101770048B1 (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
| KR1020197031864A KR102120875B1 (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
| KR1020207015960A KR102246963B1 (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
Family Applications Before (7)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020217012463A KR102351912B1 (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
| KR1020227001078A KR102406288B1 (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
| KR1020227018650A KR102527797B1 (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
| KR1020177016697A KR102040007B1 (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
| KR1020237014240A KR20230061576A (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
| KR1020157019194A KR101770048B1 (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
| KR1020197031864A KR102120875B1 (ko) | 2012-12-21 | 2013-12-19 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
Country Status (41)
Families Citing this family (151)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9540343B2 (en) | 2011-07-06 | 2017-01-10 | Gilead Sciences, Inc. | Compounds for the treatment of HIV |
| EP3243826B1 (en) | 2012-04-26 | 2019-10-30 | Bristol-Myers Squibb Company | Imidazothiadiazole and imidazopyrazine derivatives as protease activated receptor 4 (par4) inhibitors for treating platelet aggregation |
| TWI568722B (zh) | 2012-06-15 | 2017-02-01 | 葛蘭馬克製藥公司 | 作爲mPGES-1抑制劑之三唑酮化合物 |
| US9216996B2 (en) * | 2012-12-21 | 2015-12-22 | Gilead Sciences, Inc. | Substituted 2,3,4,5,7,9,13,13a-octahydropyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepines and methods for treating viral infections |
| UA111305C2 (uk) | 2012-12-21 | 2016-04-11 | Пфайзер Інк. | Конденсовані лактами арилу та гетероарилу |
| JP6437452B2 (ja) | 2013-01-14 | 2018-12-12 | インサイト・ホールディングス・コーポレイションIncyte Holdings Corporation | Pimキナーゼ阻害剤として有用な二環式芳香族カルボキサミド化合物 |
| ME03780B (me) | 2013-01-15 | 2021-04-20 | Incyte Holdings Corp | Jedinjenja tiazolkarboksamida i piridinkarboksamida korisna kao inhibitori pim kinaze |
| JP6411491B2 (ja) * | 2013-07-12 | 2018-10-24 | ギリアード サイエンシス インコーポレーテッド | 多環式カルバモイルピリドン化合物およびhiv感染症を処置するためのその使用 |
| NO2865735T3 (ko) | 2013-07-12 | 2018-07-21 | ||
| WO2015027124A1 (en) | 2013-08-23 | 2015-02-26 | Incyte Corporation | Furo- and thieno-pyridine carboxamide compounds useful as pim kinase inhibitors |
| WO2015177537A1 (en) * | 2014-05-20 | 2015-11-26 | Cipla Limited | Process for preparing polycyclic carbamoyl pyridone derivatives and intermediates thereof |
| CR20160574A (es) | 2014-06-17 | 2017-02-23 | Pfizer | Compuestos de dihidroisoquinolinona sustituida |
| TWI744723B (zh) | 2014-06-20 | 2021-11-01 | 美商基利科學股份有限公司 | 多環型胺甲醯基吡啶酮化合物之合成 |
| TW201613936A (en) | 2014-06-20 | 2016-04-16 | Gilead Sciences Inc | Crystalline forms of(2R,5S,13aR)-8-hydroxy-7,9-dioxo-n-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide |
| NO2717902T3 (ko) | 2014-06-20 | 2018-06-23 | ||
| US9822124B2 (en) | 2014-07-14 | 2017-11-21 | Incyte Corporation | Bicyclic heteroaromatic carboxamide compounds useful as Pim kinase inhibitors |
| US9580418B2 (en) | 2014-07-14 | 2017-02-28 | Incyte Corporation | Bicyclic aromatic carboxamide compounds useful as Pim kinase inhibitors |
| EP3196201B1 (en) * | 2014-08-22 | 2021-04-28 | Shionogi & Co., Ltd. | Polycyclic pyridone derivative having integrase-inhibiting activity |
| US20160067255A1 (en) | 2014-09-04 | 2016-03-10 | Gilead Sciences, Inc. | Methods of treating or preventing hiv in patients using a combination of tenofovir alafenamide and dolutegravir |
| WO2016090545A1 (en) * | 2014-12-09 | 2016-06-16 | Merck Sharp & Dohme Corp. | Spirocyclic heterocycle compounds useful as hiv integrate inhibitors |
| TWI738321B (zh) * | 2014-12-23 | 2021-09-01 | 美商基利科學股份有限公司 | 多環胺甲醯基吡啶酮化合物及其醫藥用途 |
| MX2017008520A (es) | 2014-12-24 | 2018-03-01 | Gilead Sciences Inc | Compuestos de pirimidina fusionada para el tratamiento del virus de inmunodeficiencia humana (vih). |
| TW202237569A (zh) | 2014-12-24 | 2022-10-01 | 美商基利科學股份有限公司 | 喹唑啉化合物 |
| JP6356919B2 (ja) | 2014-12-24 | 2018-07-11 | ギリアード サイエンシーズ, インコーポレイテッド | Hivの処置のためのイソキノリン化合物 |
| DK3236972T3 (en) | 2014-12-26 | 2021-10-04 | Univ Emory | Antivirale N4-hydroxycytidin-derivativer |
| MD3097102T2 (ro) | 2015-03-04 | 2018-02-28 | Gilead Sciences Inc | Compuşi de 4,6-diamino-pirido[3,2-D]pirimidină modulatori ai receptorilor de tip Toll |
| CN107531727B (zh) * | 2015-04-02 | 2019-11-29 | 吉利德科学公司 | 多环氨甲酰基吡啶酮化合物及其药物用途 |
| CN107709321A (zh) * | 2015-04-28 | 2018-02-16 | 盐野义制药株式会社 | 经取代的多环性吡啶酮衍生物及其前药 |
| US9540347B2 (en) | 2015-05-29 | 2017-01-10 | Incyte Corporation | Pyridineamine compounds useful as Pim kinase inhibitors |
| US20210292327A1 (en) | 2015-08-26 | 2021-09-23 | Gilead Sciences, Inc. | Deuterated toll-like receptor modulators |
| AR105967A1 (es) | 2015-09-09 | 2017-11-29 | Incyte Corp | Sales de un inhibidor de pim quinasa |
| EP3356355B1 (en) | 2015-09-30 | 2020-05-13 | Gilead Sciences, Inc. | Compounds and combinations for the treatment of hiv |
| TW201718546A (zh) | 2015-10-02 | 2017-06-01 | 英塞特公司 | 適用作pim激酶抑制劑之雜環化合物 |
| SI3346995T1 (sl) * | 2015-11-09 | 2019-11-29 | Gilead Sciences Inc | Terapevtski sestavek za zdravljenje virusa človeške imunske pomanjkljivosti |
| SG11201804787UA (en) | 2015-12-15 | 2018-07-30 | Gilead Sciences Inc | Human immunodeficiency virus neutralizing antibodies |
| WO2017197055A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | Heterocyclic degronimers for target protein degradation |
| WO2017197046A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | C3-carbon linked glutarimide degronimers for target protein degradation |
| CN109562113A (zh) | 2016-05-10 | 2019-04-02 | C4医药公司 | 用于靶蛋白降解的螺环降解决定子体 |
| WO2018005328A1 (en) * | 2016-06-27 | 2018-01-04 | Concert Pharmaceuticals, Inc. | Deuterated bictegravir |
| HUE063811T2 (hu) | 2016-08-19 | 2024-02-28 | Gilead Sciences Inc | HIV-vírus-fertõzés megelõzõ vagy terápiás kezelésére alkalmas terápiás vegyületek |
| WO2018042331A1 (en) | 2016-08-31 | 2018-03-08 | Glaxosmithkline Intellectual Property (No.2) Limited | Combinations and uses and treatments thereof |
| WO2018042332A1 (en) | 2016-08-31 | 2018-03-08 | Glaxosmithkline Intellectual Property (No.2) Limited | Combinations and uses and treatments thereof |
| WO2018045150A1 (en) | 2016-09-02 | 2018-03-08 | Gilead Sciences, Inc. | 4,6-diamino-pyrido[3,2-d]pyrimidine derivaties as toll like receptor modulators |
| PT3507276T (pt) | 2016-09-02 | 2022-01-11 | Gilead Sciences Inc | Compostos moduladores do recetor de tipo toll |
| WO2018051250A1 (en) | 2016-09-14 | 2018-03-22 | Viiv Healthcare Company | Combination comprising tenofovir alafenamide, bictegravir and 3tc |
| EP3518935A1 (en) | 2016-09-27 | 2019-08-07 | Gilead Sciences, Inc. | Therapeutic compositions for treatment of human immunodeficiency virus |
| US10800772B2 (en) | 2016-09-28 | 2020-10-13 | Gilead Sciences, Inc. | Therapeutic compounds |
| EP3532478B1 (en) | 2016-10-27 | 2021-05-26 | Gilead Sciences, Inc. | Crystalline form of darunavir free base |
| JOP20190130A1 (ar) | 2016-12-02 | 2019-06-02 | Merck Sharp & Dohme | مركبات حلقية غير متجانسة رباعية الحلقات مفيدة كمثبطات إنزيم مدمج لفيروس نقص المناعة البشرية (hiv) |
| CN108250215B (zh) * | 2016-12-28 | 2022-04-19 | 华创合成制药股份有限公司 | 一种新型抗hiv药物及其制备方法和用途 |
| AR110768A1 (es) | 2017-01-31 | 2019-05-02 | Gilead Sciences Inc | Formas cristalinas de tenofovir alafenamida |
| JOP20180009A1 (ar) | 2017-02-06 | 2019-01-30 | Gilead Sciences Inc | مركبات مثبط فيروس hiv |
| CN106860464A (zh) * | 2017-02-16 | 2017-06-20 | 江苏艾迪药业有限公司 | 用于联合抗病毒治疗的药物组合物及应用 |
| CN106860414B (zh) * | 2017-02-16 | 2019-12-24 | 江苏艾迪药业股份有限公司 | 一种用于抗hiv的复方制剂及其制备方法与应用 |
| WO2018229798A1 (en) | 2017-06-13 | 2018-12-20 | Cipla Limited | Process for the preparation of bictegravir and intermediate thereof |
| CN110831965B (zh) | 2017-06-21 | 2023-03-07 | 吉利德科学公司 | 靶向hiv gp120和cd3的多特异性抗体 |
| SI3661937T1 (sl) | 2017-08-01 | 2021-11-30 | Gilead Sciences, Inc. | Kristalinične oblike etil((S)-((((2R,5R)-5-(6-amino-9H-purin-9-IL)-4- fluoro-2,5-dihidrofuran-2-IL)oksi)metil)(fenoksi)fosforil)-L-alaninata (GS-9131) za zdravljenje virusnih okužb |
| WO2019030626A1 (en) | 2017-08-09 | 2019-02-14 | Viiv Healthcare Company | COMBINATIONS, USES AND CORRESPONDING TREATMENTS |
| WO2019030625A1 (en) | 2017-08-09 | 2019-02-14 | Viiv Healthcare Company | COMBINATIONS, USES AND CORRESPONDING TREATMENTS |
| TW202024061A (zh) | 2017-08-17 | 2020-07-01 | 美商基利科學股份有限公司 | Hiv蛋白質膜抑制劑之固體形式 |
| AR112412A1 (es) | 2017-08-17 | 2019-10-23 | Gilead Sciences Inc | Formas de sal de colina de un inhibidor de la cápside del vih |
| US11286262B2 (en) | 2017-10-06 | 2022-03-29 | Shionogi & Co., Ltd. | Stereoselective process for preparing substituted polycyclic pyridone derivatives |
| JOP20180092A1 (ar) | 2017-10-13 | 2019-04-13 | Gilead Sciences Inc | مثبطات hiv بروتياز |
| WO2019084020A1 (en) | 2017-10-24 | 2019-05-02 | Gilead Sciences, Inc. | METHODS OF TREATING PATIENTS CO-INFECTED BY A VIRUS AND TUBERCULOSIS |
| SG11202004403QA (en) | 2017-12-07 | 2020-06-29 | Univ Emory | N4-hydroxycytidine and derivatives and anti-viral uses related thereto |
| US10596161B2 (en) | 2017-12-08 | 2020-03-24 | Incyte Corporation | Low dose combination therapy for treatment of myeloproliferative neoplasms |
| AU2018392212B9 (en) | 2017-12-20 | 2021-03-18 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3' cyclic dinucleotides with phosphonate bond activating the STING adaptor protein |
| US10966999B2 (en) | 2017-12-20 | 2021-04-06 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| AU2019209594B2 (en) * | 2018-01-19 | 2021-09-23 | Gilead Sciences, Inc. | Metabolites of bictegravir |
| AU2019218837A1 (en) | 2018-02-09 | 2020-09-24 | Sandoz Ag | Crystalline form of bictegravir sodium |
| KR102587510B1 (ko) | 2018-02-15 | 2023-10-11 | 길리애드 사이언시즈, 인코포레이티드 | 피리딘 유도체 및 hiv 감염을 치료하기 위한 그의 용도 |
| WO2019159199A1 (en) | 2018-02-16 | 2019-08-22 | Cipla Limited | Continues flow process for the preparation of active pharmaceutical ingredients - polycyclic carbamoyl pyridone derivatives and intermediates thereof |
| WO2019161280A1 (en) | 2018-02-16 | 2019-08-22 | Gilead Sciences, Inc. | Methods and intermediates for preparing a therapeutic compound useful in the treatment of retroviridae viral infection |
| TW202005654A (zh) | 2018-04-06 | 2020-02-01 | 捷克科學院有機化學與生物化學研究所 | 2,2,─環二核苷酸 |
| TWI818007B (zh) | 2018-04-06 | 2023-10-11 | 捷克科學院有機化學與生物化學研究所 | 2'3'-環二核苷酸 |
| WO2019193543A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides |
| CN109020911B (zh) * | 2018-04-16 | 2022-06-28 | 常州制药厂有限公司 | 用于制备bictegravir的中间体及其制备方法 |
| CA3098274A1 (en) | 2018-04-26 | 2019-10-31 | Mylan Laboratories Limited | Polymorphic forms of bictegravir and its sodium salt |
| TW202014193A (zh) | 2018-05-03 | 2020-04-16 | 捷克科學院有機化學與生物化學研究所 | 包含碳環核苷酸之2’3’-環二核苷酸 |
| CN110526930B (zh) * | 2018-05-23 | 2022-06-03 | 莫云芬 | 抗hiv病毒的含硫多环-羟基吡啶酮甲酰胺类似物及其应用 |
| SG11202011386QA (en) | 2018-05-31 | 2020-12-30 | Shionogi & Co | Polycyclic pyridone derivative |
| JP7328962B2 (ja) | 2018-05-31 | 2023-08-17 | 塩野義製薬株式会社 | 多環性カルバモイルピリドン誘導体 |
| WO2020004443A1 (ja) | 2018-06-27 | 2020-01-02 | 国立大学法人北海道大学 | 多環性カルバモイルピリドン誘導体を含有するアレナウイルス増殖阻害剤 |
| WO2020003151A1 (en) | 2018-06-28 | 2020-01-02 | Honour Lab Limited | Process for the preparation of sodium (2r,5s,13ar)-7,9-dioxo-10-((2,4,6-trifluorobenzyl)carbamoyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1', 2':4,5]pyrazino[2,1-b] [1,3] oxazepin-8-olate and its polymorphic form |
| SG11202012043RA (en) | 2018-07-03 | 2021-01-28 | Gilead Sciences Inc | Antibodies that target hiv gp120 and methods of use |
| US11634431B2 (en) | 2018-07-12 | 2023-04-25 | Laurus Labs Limited | Process for purification of protected polycyclic carbamoylpyridone derivatives |
| CA3216031A1 (en) | 2018-07-16 | 2020-01-23 | Gilead Sciences, Inc. | Capsid inhibitors for the treatment of hiv |
| TWI766172B (zh) | 2018-07-30 | 2022-06-01 | 美商基利科學股份有限公司 | 抗hiv化合物 |
| EP3852761A1 (en) | 2018-09-19 | 2021-07-28 | Gilead Sciences, Inc. | Integrase inhibitors for the prevention of hiv |
| WO2020072656A1 (en) | 2018-10-03 | 2020-04-09 | Gilead Sciences, Inc. | Imidozopyrimidine derivatives |
| BR112021007622A2 (pt) * | 2018-10-22 | 2021-07-27 | Board Of Regents Of The University Of Nebraska | pró-fármacos antivirais e nanoformulações destes |
| CA3117556A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds as hpk1 inhibitors |
| EP3873608A1 (en) | 2018-10-31 | 2021-09-08 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds having hpk1 inhibitory activity |
| EP3653629A1 (en) * | 2018-11-16 | 2020-05-20 | Sandoz AG | Acid addition salts of an integrase strand transfer inhibitor |
| US20220144851A1 (en) | 2019-02-07 | 2022-05-12 | Cipla Limited | Novel polymorphs of integrase inhibitor |
| WO2020176505A1 (en) | 2019-02-25 | 2020-09-03 | Gilead Sciences, Inc. | Protein kinase c agonists |
| WO2020176510A1 (en) | 2019-02-25 | 2020-09-03 | Gilead Sciences, Inc. | Protein kinase c agonists |
| CN113543851A (zh) | 2019-03-07 | 2021-10-22 | 捷克共和国有机化学与生物化学研究所 | 2’3’-环二核苷酸及其前药 |
| JP7350872B2 (ja) | 2019-03-07 | 2023-09-26 | インスティチュート オブ オーガニック ケミストリー アンド バイオケミストリー エーエスシーアール,ヴイ.ヴイ.アイ. | 3’3’-環状ジヌクレオチドおよびそのプロドラッグ |
| WO2020178768A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| US11084832B2 (en) * | 2019-03-22 | 2021-08-10 | Gilead Sciences, Inc. | Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical use |
| US20200347036A1 (en) | 2019-04-17 | 2020-11-05 | Gilead Sciences, Inc. | Solid forms of an hiv protease inhibitor |
| TW202210480A (zh) | 2019-04-17 | 2022-03-16 | 美商基利科學股份有限公司 | 類鐸受體調節劑之固體形式 |
| TWI751517B (zh) | 2019-04-17 | 2022-01-01 | 美商基利科學股份有限公司 | 類鐸受體調節劑之固體形式 |
| TW202104210A (zh) | 2019-04-17 | 2021-02-01 | 美商基利科學股份有限公司 | Hiv蛋白酶抑制劑 |
| TWI762925B (zh) | 2019-05-21 | 2022-05-01 | 美商基利科學股份有限公司 | 鑑別對使用gp120 v3聚醣導向之抗體的治療敏感之hiv病患的方法 |
| CN110229174A (zh) * | 2019-05-22 | 2019-09-13 | 博诺康源(北京)药业科技有限公司 | Bictegravir原料药基因毒性杂质的合成方法 |
| TWI826690B (zh) | 2019-05-23 | 2023-12-21 | 美商基利科學股份有限公司 | 經取代之烯吲哚酮化物及其用途 |
| RU2717101C1 (ru) * | 2019-06-03 | 2020-03-18 | Андрей Александрович Иващенко | Анелированные 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-a]пиразин-7-карбоксамиды - ингибиторы интегразы ВИЧ, способы их получения и применения |
| CN110263404B (zh) * | 2019-06-12 | 2023-03-21 | 江苏大学 | 一种基于dpm模型的一体化预制泵站沉积特性的计算方法 |
| US20220305115A1 (en) | 2019-06-18 | 2022-09-29 | Janssen Sciences Ireland Unlimited Company | Combination of hepatitis b virus (hbv) vaccines and pyridopyrimidine derivatives |
| EP3993797A4 (en) * | 2019-06-18 | 2023-10-25 | Laurus Labs Limited | METHOD AND POLYMORPHOLIC FORMS OF BICTEGRAVIR AND PHARMACEUTICALLY ACCEPTABLE SALTS OR CO-CRYSTALS THEREOF |
| US20200398978A1 (en) | 2019-06-20 | 2020-12-24 | Bell Helicopter Textron Inc. | Low-drag rotor blade extension |
| EP3990476A1 (en) | 2019-06-25 | 2022-05-04 | Gilead Sciences, Inc. | Flt3l-fc fusion proteins and methods of use |
| US11248005B2 (en) | 2019-07-08 | 2022-02-15 | Lupin Limited | Process for preparation of intermediates used for the synthesis of HIV integrase inhibitor |
| US11795210B2 (en) | 2019-07-16 | 2023-10-24 | Gilead Sciences, Inc. | HIV vaccines and methods of making and using |
| US20220257619A1 (en) | 2019-07-18 | 2022-08-18 | Gilead Sciences, Inc. | Long-acting formulations of tenofovir alafenamide |
| WO2021034804A1 (en) | 2019-08-19 | 2021-02-25 | Gilead Sciences, Inc. | Pharmaceutical formulations of tenofovir alafenamide |
| CN110698473B (zh) * | 2019-10-08 | 2020-12-18 | 浙江大学 | 哌嗪酮并羟基吡啶酮-5-羧基类化合物及制备和应用 |
| KR20220106165A (ko) | 2019-11-26 | 2022-07-28 | 길리애드 사이언시즈, 인코포레이티드 | Hiv의 예방을 위한 캡시드 억제제 |
| US20230059640A1 (en) | 2019-11-28 | 2023-02-23 | Shionogi & Co., Ltd. | Prophylactic and therapeutic pharmaceutical agent for hiv infectious diseases characterized by comprising combination of integrase inhibitor and anti-hiv agent |
| US20230058677A1 (en) * | 2019-11-28 | 2023-02-23 | Shionogi & Co., Ltd. | Polycyclic pyridopyrazine derivative |
| PE20230376A1 (es) | 2019-12-24 | 2023-03-06 | Carna Biosciences Inc | Compuestos moduladores de la diacilglicerol quinasa |
| CR20220418A (es) | 2020-02-24 | 2022-10-10 | Gilead Sciences Inc | Compuestos tetracíclicos para el tratamiento de infecciones por vih |
| EP4121437A1 (en) | 2020-03-20 | 2023-01-25 | Gilead Sciences, Inc. | Prodrugs of 4'-c-substituted-2-halo-2'-deoxyadenosine nucleosides and methods of making and using the same |
| WO2021236944A1 (en) | 2020-05-21 | 2021-11-25 | Gilead Sciences, Inc. | Pharmaceutical compositions comprising bictegravir |
| CN113698420A (zh) * | 2020-05-22 | 2021-11-26 | 上海迪赛诺生物医药有限公司 | 比克替拉韦钠的新晶型及其制备方法 |
| EP4172157A1 (en) | 2020-06-25 | 2023-05-03 | Gilead Sciences, Inc. | Capsid inhibitors for the treatment of hiv |
| EP4192474A1 (en) | 2020-08-07 | 2023-06-14 | Gilead Sciences, Inc. | Prodrugs of phosphonamide nucleotide analogues and their pharmaceutical use |
| CN116390924A (zh) | 2020-09-30 | 2023-07-04 | 吉利德科学公司 | 桥连三环氨基甲酰基吡啶酮化合物及其用途 |
| TWI815194B (zh) | 2020-10-22 | 2023-09-11 | 美商基利科學股份有限公司 | 介白素2-Fc融合蛋白及使用方法 |
| TW202227445A (zh) * | 2020-10-30 | 2022-07-16 | 大陸商上海拓界生物醫藥科技有限公司 | 抑制基因缺陷的hiv病毒的用途 |
| WO2022103758A1 (en) | 2020-11-11 | 2022-05-19 | Gilead Sciences, Inc. | METHODS OF IDENTIFYING HIV PATIENTS SENSITIVE TO THERAPY WITH gp120 CD4 BINDING SITE-DIRECTED ANTIBODIES |
| HUE064467T2 (hu) | 2021-01-19 | 2024-03-28 | Gilead Sciences Inc | Szubsztituált piridotriazin-származékok és alkalmazásuk |
| TW202313094A (zh) | 2021-05-18 | 2023-04-01 | 美商基利科學股份有限公司 | 使用FLT3L—Fc融合蛋白之方法 |
| EP4359415A1 (en) | 2021-06-23 | 2024-05-01 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| US11932634B2 (en) | 2021-06-23 | 2024-03-19 | Gilead Sciences, Inc. | Diacylglycerol kinase modulating compounds |
| KR20240005901A (ko) | 2021-06-23 | 2024-01-12 | 길리애드 사이언시즈, 인코포레이티드 | 디아실글리세롤 키나제 조절 화합물 |
| CN117377671A (zh) | 2021-06-23 | 2024-01-09 | 吉利德科学公司 | 二酰基甘油激酶调节化合物 |
| WO2023006087A1 (zh) * | 2021-07-30 | 2023-02-02 | 南京明德新药研发有限公司 | 大环吡啶酮类化合物及其应用 |
| TW202337439A (zh) | 2021-12-03 | 2023-10-01 | 美商基利科學股份有限公司 | 用於hiv病毒感染之治療性化合物 |
| US20230203071A1 (en) | 2021-12-03 | 2023-06-29 | Zhimin Du | Therapeutic compounds for hiv virus infection |
| WO2023102523A1 (en) | 2021-12-03 | 2023-06-08 | Gilead Sciences, Inc. | Therapeutic compounds for hiv virus infection |
| CN114605437A (zh) * | 2022-04-01 | 2022-06-10 | 遵义医科大学 | 连续一锅法制备三个替拉韦药物的合成工艺 |
| TW202400165A (zh) | 2022-04-06 | 2024-01-01 | 美商基利科學股份有限公司 | 橋聯三環胺甲醯基吡啶酮化合物及其用途 |
| WO2023248240A1 (en) | 2022-06-21 | 2023-12-28 | Mylan Laboratories Limited | Polymorphic forms of bictegravir sodium |
| WO2024006982A1 (en) | 2022-07-01 | 2024-01-04 | Gilead Sciences, Inc. | Therapeutic compounds useful for the prophylactic or therapeutic treatment of an hiv virus infection |
| WO2024015741A1 (en) | 2022-07-12 | 2024-01-18 | Gilead Sciences, Inc. | Hiv immunogenic polypeptides and vaccines and uses thereof |
| US20240083984A1 (en) | 2022-08-26 | 2024-03-14 | Gilead Sciences, Inc. | Dosing and scheduling regimen for broadly neutralizing antibodies |
| WO2024061257A1 (zh) * | 2022-09-20 | 2024-03-28 | 吉斯凯(苏州)制药有限公司 | 多环氮杂环酮类化合物及其应用 |
| WO2024076915A1 (en) | 2022-10-04 | 2024-04-11 | Gilead Sciences, Inc. | 4'-thionucleoside analogues and their pharmaceutical use |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20050092755A (ko) * | 2003-01-14 | 2005-09-22 | 길리애드 사이언시즈, 인코포레이티드 | 복합 항바이러스 치료를 위한 조성물 및 방법 |
| WO2006116764A1 (en) * | 2005-04-28 | 2006-11-02 | Smithkline Beecham Corporation | Polycyclic carbamoylpyridone derivative having hiv integrase inhibitory activity |
| KR20070011588A (ko) * | 2004-05-17 | 2007-01-24 | 티보텍 파마슈티칼즈 리미티드 | 치환된 1-페닐-1,5-디히드로-피리도-[3,2-b]인돌-2-온 및다른 hiv 억제제의 배합물 |
| EP2412709A1 (en) * | 2009-03-26 | 2012-02-01 | Shionogi & Co., Ltd. | Process for producing pyrone and pyridone derivatives |
| KR20170029578A (ko) * | 2014-07-11 | 2017-03-15 | 길리애드 사이언시즈, 인코포레이티드 | Hiv의 치료를 위한 톨-유사 수용체의 조정제 |
Family Cites Families (129)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE788516A (fr) | 1971-09-10 | 1973-03-07 | Lonza Ag | Procede de fabrication d'esters alcoxyacetylacetiques |
| GB1528382A (en) | 1974-12-26 | 1978-10-11 | Teijin Ltd | Cyclopentene diols and acyl esters thereof and processes for their preparation |
| DE2658401A1 (de) | 1976-12-23 | 1978-07-06 | Merck Patent Gmbh | Cyclopentan-1-amine, verfahren zu ihrer herstellung und diese verbindungen enthaltende mittel |
| US4575694A (en) | 1984-03-05 | 1986-03-11 | Allied Corporation | Coaxial connector |
| DE3900735A1 (de) | 1989-01-12 | 1990-07-26 | Hoechst Ag | Neue mehrfunktionelle (alpha)-diazo-(beta)-ketoester, verfahren zu ihrer herstellung und deren verwendung |
| US6703396B1 (en) | 1990-02-01 | 2004-03-09 | Emory University | Method of resolution and antiviral activity of 1,3-oxathiolane nuclesoside enantiomers |
| US5204466A (en) | 1990-02-01 | 1993-04-20 | Emory University | Method and compositions for the synthesis of bch-189 and related compounds |
| US5914331A (en) | 1990-02-01 | 1999-06-22 | Emory University | Antiviral activity and resolution of 2-hydroxymethyl-5-(5-fluorocytosin-1-yl)-1,3-oxathiolane |
| DE4014649A1 (de) | 1990-05-08 | 1991-11-14 | Hoechst Ag | Neue mehrfunktionelle verbindungen mit (alpha)-diazo-ss-ketoester- und sulfonsaeureester-einheiten, verfahren zu ihrer herstellung und deren verwendung |
| GB9301000D0 (en) | 1993-01-20 | 1993-03-10 | Glaxo Group Ltd | Chemical compounds |
| US5922695A (en) | 1996-07-26 | 1999-07-13 | Gilead Sciences, Inc. | Antiviral phosphonomethyoxy nucleotide analogs having increased oral bioavarilability |
| SE9702772D0 (sv) | 1997-07-22 | 1997-07-22 | Astra Pharma Prod | Novel compounds |
| US5935946A (en) | 1997-07-25 | 1999-08-10 | Gilead Sciences, Inc. | Nucleotide analog composition and synthesis method |
| AU1403099A (en) | 1997-11-14 | 1999-06-07 | Merck & Co., Inc. | Alpha-1a adrenergic receptor antagonists |
| US6479531B1 (en) | 1998-11-09 | 2002-11-12 | James Black Foundation Limited | Gastrin and cholecystokinin receptor ligands |
| GB2345058A (en) | 1998-12-01 | 2000-06-28 | Cerebrus Pharm Ltd | Hydroxypyridone compounds useful in the treatment of oxidative damage to the central nervous system |
| CZ20012160A3 (cs) | 1998-12-25 | 2001-10-17 | Shionogi & Co., Ltd. | Heteroaromatické deriváty s inhibiční aktivitou proti HIV integráze |
| WO2001095905A1 (en) | 2000-06-14 | 2001-12-20 | Shionogi & Co., Ltd. | Inhibitor for enzyme having two divalent metal ions as active centers |
| AU1532802A (en) * | 2000-10-12 | 2002-04-22 | Merck & Co Inc | Aza- and polyaza-naphthalenyl-carboxamides useful as hiv integrase inhibitors |
| CN100491349C (zh) | 2001-08-10 | 2009-05-27 | 盐野义制药株式会社 | 抗病毒药 |
| MY169670A (en) | 2003-09-03 | 2019-05-08 | Tibotec Pharm Ltd | Combinations of a pyrimidine containing nnrti with rt inhibitors |
| WO2003030897A1 (en) | 2001-10-03 | 2003-04-17 | Ucb, S.A. | Pyrrolidinone derivatives |
| PL212914B1 (pl) | 2001-10-26 | 2012-12-31 | Angeletti P Ist Richerche Bio | N-podstawione hydroksypirymidynonokarboksyamidy jako inhibitory integrazy HIV |
| US7109186B2 (en) | 2002-07-09 | 2006-09-19 | Bristol-Myers Squibb Company | HIV integrase inhibitors |
| JP2006506352A (ja) | 2002-09-11 | 2006-02-23 | メルク エンド カムパニー インコーポレーテッド | Hivインテグラーゼ阻害剤として有用なジヒドロキシピリドピラジン−1,6−ジオン化合物 |
| DK1564210T5 (da) | 2002-11-20 | 2010-05-03 | Japan Tobacco Inc | 4-Oxoquinolinforbindelser og anvendelse deraf som HIV-integraseinhibitorer |
| TW200510425A (en) | 2003-08-13 | 2005-03-16 | Japan Tobacco Inc | Nitrogen-containing fused ring compound and use thereof as HIV integrase inhibitor |
| TW200528440A (en) | 2003-10-31 | 2005-09-01 | Fujisawa Pharmaceutical Co | 2-cyanopyrrolidinecarboxamide compound |
| CN101014571A (zh) | 2004-01-30 | 2007-08-08 | 默克公司 | 用作hiv整合酶抑制剂的n-苄基-3,4-二羟基吡啶-2-羧酰胺类和n-苄基-2,3-二羟基吡啶-4-羧酰胺类化合物 |
| CA2555176A1 (en) | 2004-02-11 | 2005-08-25 | Smithkline Beecham Corporation | Hiv integrase inhibitors |
| CN101014574A (zh) | 2004-03-09 | 2007-08-08 | 默克公司 | Hiv整合酶抑制剂 |
| EP1755586A2 (en) | 2004-04-29 | 2007-02-28 | The Regents of the University of California | Hydroxypyridinone, hydroxypyridinethione, pyrone, and thiopyrone metalloprotein inhibitors |
| WO2005110399A2 (en) | 2004-04-29 | 2005-11-24 | The Regents Of The University Of California | Zinc-binding groups for metalloprotein inhibitors |
| WO2005110414A2 (en) | 2004-05-07 | 2005-11-24 | Merck & Co., Inc. | Hiv integrase inhibitors |
| US7273859B2 (en) | 2004-05-12 | 2007-09-25 | Bristol-Myers Squibb Company | HIV integrase inhibitors: cyclic pyrimidinone compounds |
| MY134672A (en) | 2004-05-20 | 2007-12-31 | Japan Tobacco Inc | Stable crystal of 4-oxoquinoline compound |
| US7531554B2 (en) | 2004-05-20 | 2009-05-12 | Japan Tobacco Inc. | 4-oxoquinoline compound and use thereof as HIV integrase inhibitor |
| US8633219B2 (en) | 2004-05-21 | 2014-01-21 | Japan Tobacco Inc. | Combination therapy |
| US7550463B2 (en) | 2004-09-15 | 2009-06-23 | Shionogi & Co., Ltd. | Carbamoylpyridone derivatives having inhibitory activity against HIV integrase |
| JP2006118669A (ja) | 2004-10-25 | 2006-05-11 | Sanoh Industrial Co Ltd | 樹脂チューブ |
| EP1973906A1 (en) | 2004-12-23 | 2008-10-01 | Virochem Pharma Inc. | Hydroxydihydropyridopy razine-1,8-diones and methods for inhibiting hiv integrase |
| ATE516026T1 (de) | 2005-02-21 | 2011-07-15 | Shionogi & Co | Bicyclisches carbamoylpyridonderivat mit hiv- integrase-hemmender wirkung |
| WO2006125048A2 (en) | 2005-05-16 | 2006-11-23 | Gilead Sciences, Inc. | Hiv-integrase inhibitor compounds |
| TWI471145B (zh) | 2005-06-13 | 2015-02-01 | Bristol Myers Squibb & Gilead Sciences Llc | 單一式藥學劑量型 |
| EP1906971A2 (en) | 2005-07-27 | 2008-04-09 | Gilead Sciences, Inc. | Antiviral compounds |
| CA2626956A1 (en) | 2005-10-27 | 2007-05-03 | Shionogi & Co., Ltd. | Polycyclic carbamoylpyridone derivative having hiv integrase inhibitory activity |
| CA2635468C (en) | 2005-12-30 | 2016-08-09 | Gilead Sciences, Inc. | Methods for improving the pharmacokinetics of hiv integrase inhibitors |
| US7601844B2 (en) | 2006-01-27 | 2009-10-13 | Bristol-Myers Squibb Company | Piperidinyl derivatives as modulators of chemokine receptor activity |
| EP2452682A1 (en) | 2006-02-01 | 2012-05-16 | Japan Tobacco, Inc. | Use of 6-(3-chloro-2-fluorobenzyl)-1-[(2s)-1-hydroxy-3-methylbutan-2-yl]-7-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid or salt thereof for treating retrovirus infection |
| IN2014CN00532A (ko) | 2006-03-06 | 2015-04-03 | Japan Tobacco Inc | |
| AU2007223260C1 (en) | 2006-03-06 | 2011-02-03 | Japan Tobacco Inc. | Method for producing 4-oxoquinoline compound |
| US7893055B2 (en) | 2006-06-28 | 2011-02-22 | Bristol-Myers Squibb Company | HIV integrase inhibitors |
| EP2046328A4 (en) | 2006-07-19 | 2009-10-28 | Univ Georgia Res Found | PYRIDINONE DIKETOIC ACIDS: INHIBITORS OF HIV REPLICATION IN COMBINATION THERAPY |
| EP2644587A3 (en) | 2006-09-12 | 2013-10-23 | Gilead Sciences, Inc. | Process and intermediates for preparing integrase inhibitors |
| AU2007313293A1 (en) | 2006-10-18 | 2008-04-24 | Merck & Co., Inc. | HIV integrase inhibitors |
| US9891239B2 (en) | 2007-02-23 | 2018-02-13 | Gilead Sciences, Inc. | Modulators of pharmacokinetic properties of therapeutics |
| US20080280945A1 (en) | 2007-05-09 | 2008-11-13 | Sachin Lohani | Crystalline forms of an HIV integrase inhibitor |
| SG182229A1 (en) | 2007-06-29 | 2012-07-30 | Gilead Sciences Inc | Therapeutic compositions and the use thereof |
| US20090093482A1 (en) | 2007-06-29 | 2009-04-09 | Gilead Sciences, Inc. | Therapeutic compositions and methods |
| US20090012103A1 (en) | 2007-07-05 | 2009-01-08 | Matthew Abelman | Substituted heterocyclic compounds |
| AU2008282273A1 (en) | 2007-07-31 | 2009-02-05 | Limerick Biopharma, Inc. | Pyrone analog compositions and methods |
| AR068403A1 (es) | 2007-09-11 | 2009-11-18 | Gilead Sciences Inc | Proceso e intermediarios para la preparacion de inhibidores de integrasa |
| JP5285709B2 (ja) | 2007-11-16 | 2013-09-11 | ギリアード サイエンシス インコーポレーテッド | ヒト免疫不全ウイルスの複製阻害薬 |
| GB0803019D0 (en) | 2008-02-19 | 2008-03-26 | Btg Int Ltd | Fluorinated compounds |
| US8129398B2 (en) | 2008-03-19 | 2012-03-06 | Bristol-Myers Squibb Company | HIV integrase inhibitors |
| WO2010011813A1 (en) | 2008-07-23 | 2010-01-28 | Alkermes, Inc. | Complex of trospium and pharmaceutical compositions thereof |
| EP2465858B1 (en) * | 2008-07-25 | 2013-12-25 | VIIV Healthcare Company | Process for preparing a pyrido[1,2-a]pyrrolo[1',2':3,4]imidazo[1,2-d]pyrazine-8-carboxamide derivative |
| WO2010011815A1 (en) | 2008-07-25 | 2010-01-28 | Smithkline Beecham Corporation | Chemical compounds |
| WO2010011819A1 (en) | 2008-07-25 | 2010-01-28 | Smithkline Beecham Corporation | Chemical compounds |
| WO2010011818A1 (en) * | 2008-07-25 | 2010-01-28 | Smithkline Beecham Corporation | Chemical compounds |
| CN102149385B (zh) | 2008-07-25 | 2015-04-01 | 盐野义制药株式会社 | 化合物 |
| EP2320908B9 (en) | 2008-07-25 | 2014-06-18 | VIIV Healthcare Company | Dolutegravir Prodrugs |
| KR101847887B1 (ko) | 2008-12-11 | 2018-04-11 | 시오노기세야쿠 가부시키가이샤 | 카르바모일피리돈 hiv 인테그라제 억제제 및 중간체의 합성 |
| JP5572168B2 (ja) | 2008-12-11 | 2014-08-13 | ビーブ・ヘルスケア・カンパニー | カルバモイルピリドンhivインテグラーゼ阻害剤の製造方法および中間体 |
| ES2550101T3 (es) | 2008-12-17 | 2015-11-04 | Merck Patent Gmbh | Inhibidores de proteína quinasa de benzonaftiridinona tricíclica modificada con anillo C y su uso |
| SG173544A1 (en) | 2009-02-06 | 2011-09-29 | Gilead Sciences Inc | Tablets for combination therapy |
| WO2010110231A1 (ja) | 2009-03-26 | 2010-09-30 | 塩野義製薬株式会社 | 置換された3-ヒドロキシ-4-ピリドン誘導体 |
| WO2010147068A1 (ja) | 2009-06-15 | 2010-12-23 | 塩野義製薬株式会社 | 置換された多環性カルバモイルピリドン誘導体 |
| WO2011028044A2 (ko) | 2009-09-02 | 2011-03-10 | 이화여자대학교 산학협력단 | 피라졸 유도체, 이의 제조방법 및 이를 포함하는 골다공증 예방 및 치료용 조성물 |
| MX367937B (es) | 2010-01-27 | 2019-09-12 | Viiv Healthcare Co | Combinaciones de compuestos que comprenden inhibidores de la integrasa de vih y otros agentes terapéuticos. |
| SG183484A1 (en) | 2010-02-26 | 2012-09-27 | Japan Tobacco Inc | 1,3,4,8-tetrahydro-2h-pyrido[1,2-a]pyrazine derivative and use of same as hiv integrase inhibitor |
| TWI582097B (zh) | 2010-03-23 | 2017-05-11 | Viiv醫療保健公司 | 製備胺甲醯吡啶酮衍生物及中間體之方法 |
| WO2011139637A1 (en) | 2010-05-03 | 2011-11-10 | Philadelphia Health & Education Corporation | Small-molecule modulators of hiv-1 capsid stability and methods thereof |
| US9102614B2 (en) | 2010-07-02 | 2015-08-11 | Gilead Sciences, Inc. | Naphth-2-ylacetic acid derivatives to treat AIDS |
| AU2011274323B2 (en) | 2010-07-02 | 2015-08-06 | Gilead Sciences, Inc. | 2 -quinolinyl- acetic acid derivatives as HIV antiviral compounds |
| WO2012009009A2 (en) | 2010-07-14 | 2012-01-19 | Addex Pharma S.A. | Novel 2-amino-4-pyrazolyl-thiazole derivatives and their use as allosteric modulators of metabotropic glutamate receptors |
| BR112013002461A2 (pt) | 2010-08-05 | 2016-05-24 | Shionogi & Co | método para produzir um composto, e, cristal de um composto |
| TR201810736T4 (tr) | 2010-09-24 | 2018-08-27 | Shionogi & Co | Substitüe polisiklik karbamoil piridon derivatı ön-ilacı. |
| PE20170521A1 (es) | 2010-11-19 | 2017-05-27 | Gilead Sciences Inc | Combinacion farmaceutica que comprende rilpivirina hcl, tenofovir disoproxil fumarato y emtricitabina |
| WO2012106534A2 (en) | 2011-02-02 | 2012-08-09 | The Regents Of The University Of California | Hiv integrase inhibitors |
| AP2015008931A0 (en) | 2011-04-21 | 2015-12-31 | Gilead Sciences Inc | Benzothiazole compounds and their pharmaceutical use |
| US20140213553A1 (en) | 2011-05-03 | 2014-07-31 | Concert Pharmaceuticals Inc. | Carbamoylpyridone derivatives |
| US9328075B2 (en) | 2011-05-05 | 2016-05-03 | St. Jude Children's Research Hospital | Pyrimidinone compounds and methods for treating influenza |
| US9121496B2 (en) | 2011-06-29 | 2015-09-01 | Arvinmeritor Technology, Llc | Drive axle system and a method of control |
| US9540343B2 (en) | 2011-07-06 | 2017-01-10 | Gilead Sciences, Inc. | Compounds for the treatment of HIV |
| CN103826616B (zh) | 2011-07-07 | 2016-08-10 | 爱尔兰詹森科学公司 | 地瑞纳韦组合配制品 |
| CN102863512B (zh) | 2011-07-07 | 2016-04-20 | 上海泓博智源医药技术有限公司 | 抗病毒化合物 |
| US9206197B2 (en) | 2011-09-14 | 2015-12-08 | Mapi Pharma Ltd. | Amorphous form of dolutegravir |
| EP2774928B1 (en) | 2011-10-12 | 2017-08-30 | Shionogi & Co., Ltd. | Polycyclic pyridone derivative having integrase-inhibiting activity |
| ES2650915T3 (es) | 2011-12-12 | 2018-01-23 | Bayer Intellectual Property Gmbh | Imidazopiridazinas amino-sustituidas |
| WO2013103738A1 (en) | 2012-01-04 | 2013-07-11 | Gilead Sciences, Inc. | Napthalene acetic acid derivatives against hiv infection |
| WO2014008636A1 (en) | 2012-07-11 | 2014-01-16 | Merck Sharp & Dohme Corp. | Macrocyclic compounds as hiv integrase inhibitors |
| US20150166520A1 (en) | 2012-07-20 | 2015-06-18 | Merck Sharp & Dohme Corp. | Amido-substituted pyrimidinone derivatives useful for the treatment of hiv infection |
| EP2877469A4 (en) | 2012-07-25 | 2016-04-06 | Merck Sharp & Dohme | SUBSTITUTED NAPHTHYRIDINEDIONE DERIVATIVES AS INHIBITORS OF HIV INTEGRASE |
| CN104520275B (zh) | 2012-08-03 | 2016-11-02 | 吉利德科学公司 | 用于制备整合酶抑制剂的方法和中间产物 |
| US9657009B2 (en) | 2012-11-08 | 2017-05-23 | Bristol-Myers Squibb Company | Heteroaryl substituted pyridyl compounds useful as kinase modulators |
| CA2893959A1 (en) | 2012-12-14 | 2014-06-19 | Shenshen Cai | Pharmaceutical compositions |
| EP2931730B1 (en) | 2012-12-17 | 2019-08-07 | Merck Sharp & Dohme Corp. | 4-pyridinonetriazine derivatives as hiv integrase inhibitors |
| US20140221355A1 (en) | 2012-12-21 | 2014-08-07 | Gilead Sciences, Inc. | Polycyclic-carbamoylpyridone compounds and their pharmaceutical use |
| US9216996B2 (en) * | 2012-12-21 | 2015-12-22 | Gilead Sciences, Inc. | Substituted 2,3,4,5,7,9,13,13a-octahydropyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepines and methods for treating viral infections |
| US20160000721A1 (en) | 2012-12-21 | 2016-01-07 | Merck Sharp & Dohme Corp. | Gastro-retentive formulations |
| JP6320039B2 (ja) | 2012-12-27 | 2018-05-09 | 日本たばこ産業株式会社 | 置換されたスピロピリド[1,2−a]ピラジン誘導体及びそのHIVインテグラーゼ阻害剤としての医薬用途 |
| WO2014172188A2 (en) | 2013-04-16 | 2014-10-23 | Merck Sharp & Dohme Corp. | 4-pyridone derivative compounds and uses thereof as hiv integrase inhibitors |
| WO2014200880A1 (en) | 2013-06-13 | 2014-12-18 | Merck Sharp & Dohme Corp. | Fused tricyclic heterocyclic compounds as hiv integrase inhibitors |
| JP6411491B2 (ja) * | 2013-07-12 | 2018-10-24 | ギリアード サイエンシス インコーポレーテッド | 多環式カルバモイルピリドン化合物およびhiv感染症を処置するためのその使用 |
| NO2865735T3 (ko) | 2013-07-12 | 2018-07-21 | ||
| WO2015039348A1 (en) | 2013-09-23 | 2015-03-26 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds useful as hiv integrase inhibitors |
| MD4794B1 (ro) | 2013-09-27 | 2022-02-28 | Merck Sharp & Dohme Corp | Derivaţi de chinolizină substituiţi utili ca inhibitori de integrază HIV |
| WO2015089847A1 (en) | 2013-12-20 | 2015-06-25 | Merck Sharp & Dohme Corp. | Spirocyclic heterocycle compounds useful as hiv integrase inhibitors |
| EP3096763B1 (en) | 2014-01-21 | 2019-12-25 | Laurus Labs Limited | Novel process for the preparation of dolutegravir and pharmaceutically acceptable salts thereof |
| WO2015138933A1 (en) | 2014-03-13 | 2015-09-17 | Assia Chemical Industries Ltd. | Solid state forms of dolutegravir sodium |
| NO2717902T3 (ko) | 2014-06-20 | 2018-06-23 | ||
| TWI744723B (zh) | 2014-06-20 | 2021-11-01 | 美商基利科學股份有限公司 | 多環型胺甲醯基吡啶酮化合物之合成 |
| TW201613936A (en) | 2014-06-20 | 2016-04-16 | Gilead Sciences Inc | Crystalline forms of(2R,5S,13aR)-8-hydroxy-7,9-dioxo-n-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide |
| TWI738321B (zh) | 2014-12-23 | 2021-09-01 | 美商基利科學股份有限公司 | 多環胺甲醯基吡啶酮化合物及其醫藥用途 |
| CN107531727B (zh) | 2015-04-02 | 2019-11-29 | 吉利德科学公司 | 多环氨甲酰基吡啶酮化合物及其药物用途 |
| CA2921336A1 (en) | 2015-06-30 | 2016-12-30 | Gilead Sciences, Inc. | Pharmaceutical formulations |
| US20170000807A1 (en) | 2015-06-30 | 2017-01-05 | Gilead Sciences, Inc. | Pharmaceutical formulations |
| SI3346995T1 (sl) | 2015-11-09 | 2019-11-29 | Gilead Sciences Inc | Terapevtski sestavek za zdravljenje virusa človeške imunske pomanjkljivosti |
-
2013
- 2013-12-19 US US14/133,858 patent/US9216996B2/en active Active
- 2013-12-19 HU HUE15201519A patent/HUE046559T2/hu unknown
- 2013-12-19 PL PL19185121.1T patent/PL3608325T3/pl unknown
- 2013-12-19 PL PL15201519T patent/PL3067358T3/pl unknown
- 2013-12-19 KR KR1020217012463A patent/KR102351912B1/ko active IP Right Grant
- 2013-12-19 TW TW112109785A patent/TWI830624B/zh active
- 2013-12-19 MD MDA20170067A patent/MD4754C1/ro active IP Right Grant
- 2013-12-19 EA EA201591027A patent/EA030003B1/ru active Protection Beyond IP Right Term
- 2013-12-19 NZ NZ709260A patent/NZ709260A/en unknown
- 2013-12-19 KR KR1020227001078A patent/KR102406288B1/ko active IP Right Grant
- 2013-12-19 MY MYPI2016001880A patent/MY191741A/en unknown
- 2013-12-19 LT LTEP19185121.1T patent/LT3608325T/lt unknown
- 2013-12-19 PH PH12016500389A patent/PH12016500389A1/en unknown
- 2013-12-19 PT PT152015194T patent/PT3067358T/pt unknown
- 2013-12-19 KR KR1020227018650A patent/KR102527797B1/ko active IP Right Grant
- 2013-12-19 UY UY35213A patent/UY35213A/es not_active Application Discontinuation
- 2013-12-19 ME MEP-2016-93A patent/ME02400B/me unknown
- 2013-12-19 DK DK19185121.1T patent/DK3608325T3/da active
- 2013-12-19 SI SI201332004T patent/SI3608325T1/sl unknown
- 2013-12-19 ES ES15201519T patent/ES2753548T3/es active Active
- 2013-12-19 TW TW110146626A patent/TWI815245B/zh active
- 2013-12-19 EP EP15201519.4A patent/EP3067358B1/en active Active
- 2013-12-19 CR CR20170279A patent/CR20170279A/es unknown
- 2013-12-19 MY MYPI2015001580A patent/MY164352A/en unknown
- 2013-12-19 RS RS20160407A patent/RS54873B1/sr unknown
- 2013-12-19 CN CN202310322212.0A patent/CN116640140A/zh active Pending
- 2013-12-19 CA CA3012242A patent/CA3012242C/en active Active
- 2013-12-19 DK DK13815937.1T patent/DK2822954T3/en active
- 2013-12-19 HU HUE19185121A patent/HUE059688T2/hu unknown
- 2013-12-19 MX MX2015008009A patent/MX344879B/es active IP Right Grant
- 2013-12-19 ES ES13815937.1T patent/ES2577283T3/es active Active
- 2013-12-19 KR KR1020177016697A patent/KR102040007B1/ko active IP Right Grant
- 2013-12-19 UA UAA201506209A patent/UA114351C2/uk unknown
- 2013-12-19 HU HUE13815937A patent/HUE028284T2/en unknown
- 2013-12-19 EA EA201890236A patent/EA037633B1/ru unknown
- 2013-12-19 KR KR1020237014240A patent/KR20230061576A/ko not_active Application Discontinuation
- 2013-12-19 DK DK15201519T patent/DK3067358T3/da active
- 2013-12-19 EP EP13815937.1A patent/EP2822954B1/en active Active
- 2013-12-19 AR ARP130104930A patent/AR094197A1/es active IP Right Grant
- 2013-12-19 AU AU2013361401A patent/AU2013361401C1/en active Active
- 2013-12-19 PH PH12019501848A patent/PH12019501848A1/en unknown
- 2013-12-19 PT PT191851211T patent/PT3608325T/pt unknown
- 2013-12-19 PL PL13815937.1T patent/PL2822954T3/pl unknown
- 2013-12-19 CN CN201711080644.6A patent/CN107674086B/zh active Active
- 2013-12-19 SI SI201330197A patent/SI2822954T1/sl unknown
- 2013-12-19 CN CN202010188107.9A patent/CN111303152B/zh active Active
- 2013-12-19 TW TW102147242A patent/TWI642669B/zh active
- 2013-12-19 LT LT15201519T patent/LT3067358T/lt unknown
- 2013-12-19 SI SI201331609T patent/SI3067358T1/sl unknown
- 2013-12-19 EP EP22178867.2A patent/EP4122935A1/en active Pending
- 2013-12-19 KR KR1020157019194A patent/KR101770048B1/ko active IP Right Grant
- 2013-12-19 TW TW109111537A patent/TWI752457B/zh active
- 2013-12-19 PE PE2015001036A patent/PE20151499A1/es active IP Right Grant
- 2013-12-19 WO PCT/US2013/076367 patent/WO2014100323A1/en active Application Filing
- 2013-12-19 CA CA3131094A patent/CA3131094A1/en active Pending
- 2013-12-19 MX MX2017000362A patent/MX357940B/es unknown
- 2013-12-19 NZ NZ718708A patent/NZ718708A/en unknown
- 2013-12-19 TW TW107127529A patent/TWI693224B/zh active
- 2013-12-19 MD MDA20200093A patent/MD4841B1/ro unknown
- 2013-12-19 CA CA2893843A patent/CA2893843C/en active Active
- 2013-12-19 ES ES19185121T patent/ES2926068T3/es active Active
- 2013-12-19 KR KR1020197031864A patent/KR102120875B1/ko active IP Right Grant
- 2013-12-19 EP EP19185121.1A patent/EP3608325B1/en active Active
- 2013-12-19 PT PT138159371T patent/PT2822954E/pt unknown
- 2013-12-19 PE PE2016000694A patent/PE20170528A1/es unknown
- 2013-12-19 JP JP2015549669A patent/JP6028105B2/ja active Active
- 2013-12-19 UA UAA201612380A patent/UA123572C2/uk unknown
- 2013-12-19 HR HRP20220899TT patent/HRP20220899T1/hr unknown
- 2013-12-19 SG SG11201504857SA patent/SG11201504857SA/en unknown
- 2013-12-19 AP AP2015008510A patent/AP2015008510A0/xx unknown
- 2013-12-19 BR BR122015029881-4A patent/BR122015029881B1/pt active IP Right Grant
- 2013-12-19 MD MDA20150064A patent/MD4736C1/ro active IP Right Grant
- 2013-12-19 BR BR112015014714-3A patent/BR112015014714B1/pt active IP Right Grant
- 2013-12-19 CN CN201380073134.XA patent/CN104995198B/zh active Active
- 2013-12-19 KR KR1020207015960A patent/KR102246963B1/ko active IP Right Grant
-
2015
- 2015-06-10 IL IL239316A patent/IL239316A/en active IP Right Grant
- 2015-06-18 CL CL2015001756A patent/CL2015001756A1/es unknown
- 2015-06-18 MX MX2022013596A patent/MX2022013596A/es unknown
- 2015-06-18 MX MX2020003055A patent/MX2020003055A/es unknown
- 2015-06-22 HK HK15105909.2A patent/HK1205124A1/xx unknown
- 2015-06-22 PH PH12015501445A patent/PH12015501445A1/en unknown
- 2015-07-17 CR CR20150380A patent/CR20150380A/es unknown
- 2015-07-31 US US14/815,504 patent/US9663528B2/en active Active
- 2015-10-26 ZA ZA2015/07997A patent/ZA201507997B/en unknown
-
2016
- 2016-04-11 CL CL2016000837A patent/CL2016000837A1/es unknown
- 2016-04-21 HK HK18110106.0A patent/HK1250711A1/zh unknown
- 2016-04-21 HK HK16104629.3A patent/HK1216643A1/zh unknown
- 2016-05-20 HR HRP20160544TT patent/HRP20160544T1/hr unknown
- 2016-05-23 IL IL245780A patent/IL245780B/en active IP Right Grant
- 2016-05-30 CY CY20161100472T patent/CY1117570T1/el unknown
- 2016-06-01 SM SM201600157T patent/SMT201600157B/it unknown
- 2016-06-21 JP JP2016122743A patent/JP6411409B2/ja active Active
- 2016-08-17 IL IL247311A patent/IL247311A0/en unknown
- 2016-11-11 US US15/349,934 patent/US9732092B2/en active Active
- 2016-11-24 AU AU2016262722A patent/AU2016262722B2/en active Active
-
2017
- 2017-05-11 CL CL2017001191A patent/CL2017001191A1/es unknown
- 2017-05-18 US US15/599,151 patent/US10035809B2/en active Active
-
2018
- 2018-06-25 US US16/017,371 patent/US10689399B2/en active Active
- 2018-08-17 LU LU00083C patent/LUC00083I2/en unknown
- 2018-08-21 LT LTPA2018511C patent/LTC2822954I2/lt unknown
- 2018-08-22 NL NL300947C patent/NL300947I2/nl unknown
- 2018-08-23 CY CY2018022C patent/CY2018022I2/el unknown
- 2018-08-23 HU HUS1800035C patent/HUS1800035I1/hu unknown
- 2018-09-19 AU AU2018232957A patent/AU2018232957B2/en active Active
- 2018-09-26 JP JP2018179844A patent/JP6571256B2/ja active Active
-
2019
- 2019-02-22 ZA ZA2019/03516A patent/ZA201903516B/en unknown
- 2019-08-07 JP JP2019145317A patent/JP6933690B2/ja active Active
- 2019-10-29 HR HRP20191957TT patent/HRP20191957T1/hr unknown
- 2019-11-05 CY CY20191101151T patent/CY1122246T1/el unknown
-
2020
- 2020-05-01 AU AU2020202914A patent/AU2020202914B2/en active Active
- 2020-05-06 US US16/868,119 patent/US11548901B2/en active Active
- 2020-05-28 IL IL274988A patent/IL274988A/en unknown
-
2021
- 2021-08-19 JP JP2021134172A patent/JP7301918B2/ja active Active
- 2021-08-23 AU AU2021221427A patent/AU2021221427B2/en active Active
-
2023
- 2023-06-21 JP JP2023101636A patent/JP2023134483A/ja active Pending
- 2023-12-30 AU AU2023286038A patent/AU2023286038A1/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20050092755A (ko) * | 2003-01-14 | 2005-09-22 | 길리애드 사이언시즈, 인코포레이티드 | 복합 항바이러스 치료를 위한 조성물 및 방법 |
| KR100860136B1 (ko) * | 2003-01-14 | 2008-09-25 | 길리애드 사이언시즈, 인코포레이티드 | 복합 항바이러스 치료를 위한 조성물 및 방법 |
| KR20070011588A (ko) * | 2004-05-17 | 2007-01-24 | 티보텍 파마슈티칼즈 리미티드 | 치환된 1-페닐-1,5-디히드로-피리도-[3,2-b]인돌-2-온 및다른 hiv 억제제의 배합물 |
| WO2006116764A1 (en) * | 2005-04-28 | 2006-11-02 | Smithkline Beecham Corporation | Polycyclic carbamoylpyridone derivative having hiv integrase inhibitory activity |
| EP2412709A1 (en) * | 2009-03-26 | 2012-02-01 | Shionogi & Co., Ltd. | Process for producing pyrone and pyridone derivatives |
| KR20170029578A (ko) * | 2014-07-11 | 2017-03-15 | 길리애드 사이언시즈, 인코포레이티드 | Hiv의 치료를 위한 톨-유사 수용체의 조정제 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR102120875B1 (ko) | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A107 | Divisional application of patent | ||
| E902 | Notification of reason for refusal | ||
| E701 | Decision to grant or registration of patent right | ||
| A107 | Divisional application of patent | ||
| GRNT | Written decision to grant |