JP5638515B2 - ピロール化合物 - Google Patents
ピロール化合物 Download PDFInfo
- Publication number
- JP5638515B2 JP5638515B2 JP2011508745A JP2011508745A JP5638515B2 JP 5638515 B2 JP5638515 B2 JP 5638515B2 JP 2011508745 A JP2011508745 A JP 2011508745A JP 2011508745 A JP2011508745 A JP 2011508745A JP 5638515 B2 JP5638515 B2 JP 5638515B2
- Authority
- JP
- Japan
- Prior art keywords
- compound
- fluoropyridin
- ethyl acetate
- fluoro
- sulfonyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CCC(C[C@](C**C(C)C)CC1)[C@]1C(C)(C)C Chemical compound CCC(C[C@](C**C(C)C)CC1)[C@]1C(C)(C)C 0.000 description 3
Images


Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pyrrole Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Description
消化性潰瘍および逆流性食道炎等の治療を目的に、胃酸の分泌を抑制するオメプラゾールに代表されるプロトンポンプ阻害剤が広く臨床現場で使用されている。しかしながら、既存のプロトンポンプ阻害剤には効果、副作用の点で問題点が存在する。すなわち、既存のプロトンポンプ阻害剤は酸性条件下で不安定であることから腸溶製剤として処方されることが多く、その場合、作用の発現までに数時間を要し、更に連投で最大薬効を示すまでに約5日を要する。また、既存のプロトンポンプ阻害剤は代謝酵素多型に基づく治療効果のバラツキやジアゼパム等の薬剤との薬物間相互作用が懸念され、改良が望まれている。プロトンポンプ阻害作用を有するピロ−ル化合物として、特許文献1には式

〔1〕式(I)
〔2〕Aが、式(A−1)[式中、R1およびR3は、ともに水素原子を示し、R2は、ハロゲン原子、ハロゲンで置換されていてもよいC1−6アルキル基、またはハロゲンで置換されていてもよいC1−6アルコキシ基を示す。]で表される上記〔1〕記載の化合物またはその塩、
〔3〕R2が、C1−6アルキル基、またはC1−6アルコキシ基である上記〔2〕記載の化合物またはその塩、
〔4〕1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N-メチルメタンアミンまたはその塩、
〔5〕1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−2−イル)スルホニル]−1H-ピロール−3−イル}−N-メチルメタンアミンまたはその塩、
〔6〕1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メトキシピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
〔7〕1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
〔8〕1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(6−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
〔9〕上記〔1〕記載の化合物またはその塩のプロドラッグ、
〔10〕上記〔1〕記載の化合物もしくはその塩またはそれらのプロドラッグを含有してなる医薬、
〔11〕酸分泌抑制剤である上記〔10〕記載の医薬、
〔12〕カリウムイオン競合型アシッドブロッカー(Potassium−Competitive Acid Blocker)である上記〔10〕記載の医薬、
〔13〕消化性潰瘍、ゾリンジャー・エリソン(Zollinger−Ellison)症候群、胃炎、逆流性食道炎、症候性胃食道逆流症(Symptomatic Gastroesophageal Reflux Disease(Symptomatic GERD))、Barrett食道、機能性ディスペプシア(Functional Dyspepsia)、胃癌、胃MALTリンパ腫、非ステロイド系抗炎症剤に起因する潰瘍あるいは手術後ストレスによる胃酸過多ならびに潰瘍の治療もしくは予防剤;または消化性潰瘍、急性ストレス潰瘍、出血性胃炎あるいは侵襲ストレスによる上部消化管出血の抑制剤である上記〔10〕記載の医薬、
〔14〕哺乳動物に対して、上記〔1〕記載の化合物もしくはそれらの塩またはそのプロドラッグの有効量を投与することを特徴とする、消化性潰瘍、ゾリンジャー・エリソン(Zollinger−Ellison)症候群、胃炎、逆流性食道炎、症候性胃食道逆流症(Symptomatic Gastroesophageal Reflux Disease(Symptomatic GERD))、Barrett食道、機能性ディスペプシア(Functional Dyspepsia)、胃癌、胃MALTリンパ腫、非ステロイド系抗炎症剤に起因する潰瘍あるいは手術後ストレスによる胃酸過多ならびに潰瘍の治療または予防方法;または消化性潰瘍、急性ストレス潰瘍、出血性胃炎あるいは侵襲ストレスによる上部消化管出血の抑制方法、
〔15〕消化性潰瘍、ゾリンジャー・エリソン(Zollinger−Ellison)症候群、胃炎、逆流性食道炎、症候性胃食道逆流症(Symptomatic Gastroesophageal Reflux Disease(Symptomatic GERD))、Barrett食道、機能性ディスペプシア(Functional Dyspepsia)、胃癌、胃MALTリンパ腫、非ステロイド系抗炎症剤に起因する潰瘍あるいは手術後ストレスによる胃酸過多ならびに潰瘍の治療もしくは予防剤;または消化性潰瘍、急性ストレス潰瘍、出血性胃炎あるいは侵襲ストレスによる上部消化管出血の抑制剤を製造するための上記〔1〕記載の化合物もしくはその塩またはそれらのプロドラッグの使用、および
〔16〕Aが、少なくとも1個の置換基を有するピリジル基、式(A−1)または式(A−2)[式中、R1およびR3のいずれか一方が、ハロゲン原子、ハロゲンで置換されていてもよいC1−6アルキル基、またはハロゲンで置換されていてもよいC1−6アルコキシ基を示し、他方が、水素原子、ハロゲン原子、ハロゲンで置換されていてもよいC1−6アルキル基、またはハロゲンで置換されていてもよいC1−6アルコキシ基を示し、R2が、水素原子、ハロゲン原子、ハロゲンで置換されていてもよいC1−6アルキル基、またはハロゲンで置換されていてもよいC1−6アルコキシ基を示し、R4およびR6は、それぞれ水素原子、ハロゲン原子、またはハロゲンで置換されていてもよいC1−6アルキル基を示し、R5は、水素原子、ハロゲン原子、ハロゲンで置換されていてもよいC1−6アルキル基、またはハロゲンで置換されていてもよいC1−6アルコキシ基を示し、R7は、水素原子、またはハロゲンで置換されていてもよいC1−6アルキル基を示す]で表される上記〔1〕化合物またはその塩、
に関する。
本明細書中、「ハロゲン原子」、「ハロゲン」としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子が挙げられる。式(I)中、Aは少なくとも1個の置換基を有するピリジル基を示す。Aで示される「少なくとも1個の置換基を有するピリジル基」としては、式
R1として、特に好ましくは、水素原子またはC1−6アルキル基である。R2として、特に好ましくは、水素原子、C1−6アルキル基またはC1−6アルコキシ基である。R3として、特に好ましくは、水素原子、ハロゲン原子、またはC1−6アルコキシ基である。R4として、特に好ましくは、水素原子またはC1−6アルキル基である。R5として、特に好ましくは、水素原子、ハロゲン原子またはC1−6アルキル基である。R6として、特に好ましくは、水素原子またはC1−6アルキル基である。R7として、特に好ましくは、水素原子またはC1−6アルキル基である。
式(I)中、Aは、以下の態様として分類することができる。
(i)Aが、式(A−1)であり、式中、R1およびR3は、ともに水素原子を示し、R2は、ハロゲン原子、ハロゲンで置換されていてもよいC1−6アルキル基、またはハロゲンで置換されていてもよいC1−6アルコキシ基を示す。
(ii)Aが、少なくとも1個の置換基を有するピリジル基、式(A−1)または式(A−2)であり、式中、R1およびR3のいずれか一方が、ハロゲン原子、ハロゲンで置換されていてもよいC1−6アルキル基、またはハロゲンで置換されていてもよいC1−6アルコキシ基を示し、他方が、水素原子、ハロゲン原子、ハロゲンで置換されていてもよいC1−6アルキル基、またはハロゲンで置換されていてもよいC1−6アルコキシ基を示し、R2が、水素原子、ハロゲン原子、ハロゲンで置換されていてもよいC1−6アルキル基、またはハロゲンで置換されていてもよいC1−6アルコキシ基を示し、R4およびR6は、それぞれ水素原子、ハロゲン原子、またはハロゲンで置換されていてもよいC1−6アルキル基を示し、R5は、水素原子、ハロゲン原子、ハロゲンで置換されていてもよいC1−6アルキル基、またはハロゲンで置換されていてもよいC1−6アルコキシ基を示し、R7は、水素原子、またはハロゲンで置換されていてもよいC1−6アルキル基を示す)を示す。
Aで示される「少なくとも1個の置換基を有するピリジル基」として、好ましくは、式
化合物(I)の特に好ましい別の態様として、下記式(Ia−1)、式(Ia−2)、式(Ib)または式(Ic)で表される化合物またはその塩が挙げられる。
式(Ib)のR5は、水素原子、ハロゲン原子、ハロゲンで置換されていてもよいC1−6アルキル基、またはハロゲンで置換されていてもよいC1−6アルコキシ基を示し、R5として好ましくは、水素原子、ハロゲン原子、またはハロゲンで置換されていてもよいC1−6アルキル基であり、特に好ましくは、水素原子、ハロゲン原子、またはC1−6アルキル基である。
式(Ic)のR7は、水素原子、またはハロゲンで置換されていてもよいC1−6アルキル基を示し、R7として好ましくは、水素原子またはハロゲンで置換されていてもよいC1−6アルキル基、特に好ましくは、水素原子またはC1−6アルキル基である。
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(3−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メトキシピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(6−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(2−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−メトキシピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{1−[(5−クロロピリジン−3−イル)スルホニル]−4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{4−フルオロ−1−[(5−フルオロ−6−メチルピリジン−2−イル)スルホニル]−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{4−フルオロ−1−[(5−フルオロ−4−メチルピリジン−2−イル)スルホニル]−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{4−フルオロ−1−[(5−フルオロ−4−メトキシピリジン−2−イル)スルホニル]−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−メトキシピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{4−フルオロ−1−[(5−フルオロ−6−メチルピリジン−3−イル)スルホニル]−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{1−[(4,6−ジメチルピリジン−2−イル)スルホニル]−4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{1−[(5−クロロピリジン−2−イル)スルホニル]−4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{1−[(5,6−ジメチルピリジン−2−イル)スルホニル]−4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{1−[(4,5−ジメチルピリジン−2−イル)スルホニル]−4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、および
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩。
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N-メチルメタンアミンまたはその塩、
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−2−イル)スルホニル]−1H-ピロール−3−イル}−N-メチルメタンアミンまたはその塩、
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4-メトキシピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩、および
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(6−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩。
化合物(VII)は化合物(V)と式(VI)
(i)プロトンポンプ阻害薬、例、オメプラゾール(omeprazole)、エソメプラゾール(esomeprazole)、パントプラゾール(pantoprazole)、ラベプラゾール(rabeprazole)、テナトプラゾール(tenatoprazole)、イラプラゾール(ilaprazole)およびランソプラゾール(lansoprazole);
(ii)経口制酸合剤、例、Maalox、AludroxおよびGaviscon;
(iii)粘膜保護剤、例、ポラプレジンク(polaprezinc)、エカベトナトリウム(ecabe sodium)、レバミピド(rebamipide)、テプレノン(teprenone)、セトラキサート(cetraxate)、スクラルファート(sucralfate)、クロロピリン銅(chloropylline−copper)およびプラウノトール(plaunotol);
(iv)抗胃剤、例、抗ガストリンワクチン、イトリグルミド(itriglumide)およびZ−360;
(v)5−HT3アンタゴニスト、例、ドラセトロン(dolasetron)、パロノセトロン(palonosetron)、アロセトロン(alosetron)、アザセトロン(azasetron)、ラモセトロン(ramosetron)、ミトラザピン(mitrazapine)、グラニセトロン(granisetron)、トロピセトロン(tropisetron)、E−3620、オンダンセトロン(ondansetron)およびインジセトロン(indisetron);
(vi)5−HT4アゴニスト、例、テガセロド(tegaserod)、モサプリド(mosapride)、シニタプリド(cinitapride)およびオキシトリプタン(oxtriptane);
(vii)緩下剤、例、Trifyba、Fybogel、Konsyl、Isogel、Regulan、CelevacおよびNormacol;
(viii)GABABアゴニスト、例、バクロフェン(baclofen)およびAZD−3355;
(ix)GABABアンタゴニスト、例、GAS−360およびSGS−742;
(x)カルシウムチャネルブロッカー、例、アラニジピン(aranidipine)、ラシジピン(lacidipine)、ファロジピン(falodipine)、アゼルニジピン(azelnidipine)、クリニジピン(clinidipine)、ロメリジン(lomerizine)、ジルチアゼム(diltiazem)、ガロパミル(gallopamil)、エフォニジピン(efonidipine)、ニソルピジン(nisoldipine)、アムロジピン(amlodipine)、レルカニジピン(lercanidipine)、ベバントロール(bevantolol)、ニカルジピン(nicardipine)、イスラジピン(isradipine)、ベニジピン(benidipine)、ベラパミル(verapamil)、ニトレンジピン(nitrendipine)、バルニジピン(barnidipine)、プロパフェノン(propafenone)、マニジピン(manidipine)、ベプリジル(bepridil)、ニフェジピン(nifedipine)、ニルバジピン(nilvadipine)、ニモジピン(nimodipine)およびファスジル(fasudil);
(xi)ドーパミンアンタゴニスト、例、メトクロプラミド(metoclopramide)、ドンペリドン(domperidone)およびレボスルピリド(levosulpiride);
(xii)タキキニン(NK)アンタゴニスト、特に、NK−3、NK−2およびNK−1アンタゴニスト、例、ネパズタント(nepadutant)、サレズタント(saredutant)、タルネタント(talnetant)、(αR,9R)−7−[3,5−ビス(トリフルオロメチル)ベンジル]−8,9,10,11−テトラヒドロ−9−メチル−5−(4−メチルフェニル)−7H−[1,4]ジアゾシノ[2,1−g][1,7]ナフチリジン−6, 13−ジオン(TAK−637)、5−[(2R,3S)−2−[(1R)−1−[3,5−ビス(トリフルオロメチル)フェニル]エトキシ−3−(4−フルオロフェニル)−4−モルホリニル]メチル]−1,2−ジヒドロ−3H−1,2,4−トリアゾール−3−オン(MK−869)、ラネピタント(lanepitant)、ダピタント(dapitant)および(2S,3S)−3−[[2−メトキシ−5−(トリフルオロメトキシ)フェニル]メチルアミノ]−2−フェニル−ピペリジン;
(xiii)一酸化窒素シンターゼ阻害薬、例、GW−274150、ティラルギニン(tilarginine)、P54、グアニジオエチルジスルフィド(guanidioethyldisulfide)およびニトロフルビプロフェン(nitroflurbiprofen);
(xiv)バニロイドレセプター1アンタゴニスト、例、AMG−517およびGW−705498;
(xv)グレリンアゴニスト、例、カプロモレリン(capromorelin)およびTZP−101;
(xvi)AchE阻害剤、例、Z−338およびKW−5092。
s:シングレット(singlet)、d:ダブレット(doublet)、dd:ダブルダブレット(double doublet)、ddd:トリプルダブレット(triple doublet)、dt:ダブルトリプレット(double triplet)、t:トリプレット(triplet)、q:カルテット(quartet)、dq:ダブルカルテット(double quartet)、m:マルチプレット(multiplet)、br:ブロード(broad)、brs:ブロードシングレット(broad singlet)、J:カップリング定数(coupling constant)、Hz:ヘルツ(Hertz)。
(2−オキソエチル)カルバミン酸 tert−ブチル
(2−ヒドロキシエチル)カルバミン酸 tert−ブチルの(10.0g)のジメチルスルホキシド(50mL)とトリエチルアミン(12.3g)の混合溶液に三酸化硫黄ピリジン錯体(15.0g)を氷冷下で加え、1時間攪拌した。反応液を室温にてさらに3時間攪拌した後に、1mol/L塩酸を加え、酢酸エチルで抽出した。分離した水層を酢酸エチルで再度抽出した。合わせた有機層を飽和食塩水で洗浄し、硫酸マグネシウムで乾燥後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=17:3→13:7)で精製することにより表題化合物を淡黄色油状物として得た(収量6.50g、収率66%)。
1H−NMR(CDCl3)δ:1.46(9H,s),4.08(2H,d,J=4.5Hz),5.19(1H,brs),9.66(1H,s).
4−[(tert−ブトキシカルボニル)アミノ]−2,2−ジフルオロ−3−{[(4−メチルフェニル)スルホニル]オキシ}ブタン酸エチル
亜鉛粉末(23.0g)を0.1mol/L塩酸、エタノール、ジエチルエーテルで洗浄した後に、減圧下で乾燥させた。アルゴン雰囲気下、洗浄した亜鉛粉末のテトラヒドロフラン(300mL)懸濁液に(2−オキソエチル)カルバミン酸 tert−ブチル(35.0g)のテトラヒドロフラン(50mL)溶液を加え、さらに氷冷下でブロモジフルオロ酢酸エチル(75.9g)を徐々に滴下した後、15分間攪拌した。反応液に、1mol/L塩酸を加え、酢酸エチルで抽出した。分離した水層を酢酸エチルで再度抽出した。合わせた有機層を、飽和炭酸水素ナトリウム水溶液、水、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後に、減圧濃縮した。残留物をテトラヒドロフラン(30mL)とピリジン(40mL)混合溶液に溶解し、トリエチルアミン(19mL)、4−ジメチルアミノピリジン(3.35g)と塩化 4−メチルベンゼンスルホニル(39.2g)を室温で加え、2時間攪拌した。反応液を減圧濃縮し、残留物を酢酸エチルに溶解し、1mol/L塩酸で二度洗浄した。分離した水層を酢酸エチルで再度抽出した。合わせた有機層を飽和炭酸水素ナトリウム水溶液、水、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=4:1)で精製することにより表題化合物を淡黄色油状物として得た(収量44.8g、収率46%)。
1H−NMR(CDCl3)δ:1.33(3H,t,J=7.2Hz),1.46(9H,s),2.46(3H,s),3.26−3.43(1H,m),3.71(1H,brs),4.28(2H,q,J=7.1Hz),4.77(1H,brs),5.08−5.24(1H,m),7.35(2H,d,J=8.1Hz),7.80(2H,d,J=8.1Hz).
3,3−ジフルオロ−4−{[(4−メチルフェニル)スルホニル]オキシ}−2−オキソピロリジン−1−カルボン酸 tert−ブチル
4−[(tert−ブトキシカルボニル)アミノ]−2,2−ジフルオロ−3−{[(4−メチルフェニル)スルホニル]オキシ}ブタン酸エチル(44.8g)の酢酸エチル(50mL)溶液に4mol/L塩化水素−酢酸エチル溶液(100mL)を加え3時間攪拌した。反応液を減圧濃縮し、残留物を2回トルエンで共沸した。得られた混合物をアセトニトリル(20mL)に溶解し、トリエチルアミン(15.6g)を加え、3時間攪拌した。さらに二炭酸ジ−tert−ブチル(33.6g)と4−ジメチルアミノピリジン(3.76g)を室温で加え1時間攪拌した。反応液を減圧濃縮し、残留物を酢酸エチルに溶解し、1mol/L塩酸で洗浄した。分離した水層を酢酸エチルで再度抽出した。合わせた有機層を飽和炭酸水素ナトリウム水溶液、水、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=2:1)で精製することにより表題化合物を淡黄色油状物として得た(収量32.0g、収率80%)。
1H−NMR(CDCl3)δ:1.55(9H,s),2.48(3H,s),3.81−3.91(1H,m),4.09−4.18(1H,m),4.94−5.06(1H,m),7.40(2H,d,J=8.1Hz),7.83(2H,d,J=8.1Hz).
4−メチルベンゼンスルホン酸 4,4−ジフルオロ−5−(2−フルオロピリジン−3−イル)−3,4−ジヒドロ−2H−ピロール−3−イル
ジイソプロピルアミン(8.76g)のテトラヒドロフラン(230mL)溶液に1.6mol/L n−ブチルリチウムヘキサン溶液(51mL)を−78℃で加え1時間攪拌した。この溶液に、2−フルオロピリジン(11.2g)を滴下し2時間攪拌した。生じた淡黄色の懸濁液に3,3−ジフルオロ−4−{[(4−メチルフェニル)スルホニル]オキシ}−2−オキソピロリジン−1−カルボン酸 tert−ブチル(22.6g)のテトラヒドロフラン(50mL)溶液をゆっくりと滴下し、さらに1時間攪拌した。反応混合物に水を加えた後、室温へと温度を上げ、減圧濃縮した。残留物を、酢酸エチルで希釈し、水で洗浄した。分離した水層を再度酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥させた後、減圧濃縮した。得られた混合物をジクロロメタン(30mL)に溶解し、トリフルオロ酢酸(100mL)を氷冷下で滴下し、徐々に室温まで昇温し4時間攪拌した。反応液を減圧濃縮した後、酢酸エチルで希釈し、中性になるまで飽和炭酸水素ナトリウム水溶液を加えた。分離した水層を酢酸エチルで再度抽出した。合わせた有機層を飽和食塩水で洗浄し、硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=9:1→3:1)で精製することにより表題化合物を無色固体として得た(収量10.9g、収率51%)。
1H−NMR(CDCl3)δ:2.48(3H,s),4.17−4.28(1H,m),4.42−4.54(1H,m),5.06−5.13(1H,m),7.31(1H,ddd,J=7.6,4.9,1.9Hz),7.39(2H,d,J=7.9Hz),7.85(2H,d,J=8.3Hz),8.22−8.31(1H,m),8.34−8.39(1H,m).
2−フルオロ−3−(3−フルオロ−1H−ピロール−2−イル)ピリジン
4−メチルベンゼンスルホン酸 4,4−ジフルオロ−5−(2−フルオロピリジン−3−イル)−3,4−ジヒドロ−2H−ピロール−3−イル(18.0g)のテトラヒドロフラン(180mL)溶液に水素化ホウ素ナトリウム(3.68g)を氷冷下で加え、さらにメタノール(90mL)を加え、3時間攪拌した。反応混合物を減圧濃縮した後に、残留物を酢酸エチルで希釈し、水で洗浄した。分離した水層を酢酸エチルで再度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後に減圧濃縮し、4−メチルベンゼンスルホン酸 4,4−ジフルオロ−5−(2−フルオロピリジン−3−イル)ピロリジン−3−イルを得た。水素化ナトリウム(9.74g)のテトラヒドロフラン(100mL)懸濁液に4−メチルベンゼンスルホン酸 4,4−ジフルオロ−5−(2−フルオロピリジン−3−イル)ピロリジン−3−イルのテトラヒドロフラン(100mL)溶液を氷冷下で滴下し、さらに15−クラウン−5(32.2g)を加え、3時間攪拌した。反応液に飽和塩化アンモニウム水溶液を加えた後に、減圧濃縮した。残留物を酢酸エチルで希釈し、1mol/L塩酸で洗浄した。分離した水層を再度酢酸エチルで抽出した。合わせた有機層を飽和炭酸水素ナトリウム水溶液、水、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後に減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=9:1→3:1)で精製することにより表題化合物を無色固体として得た(収量6.35g、収率72%)。
1H−NMR(CDCl3)δ:6.10(1H,t,J=2.9Hz),6.69(1H,dt,J=4.6,3.4Hz),7.20−7.30(1H,m),8.00(1H,dt,J=4.7,1.7Hz),8.25(1H,ddd,J=10.3,7.8,1.9Hz),8.69(1H,brs).
2−フルオロ−3−{3−フルオロ−1−[トリス(1−メチルエチル)シリル]−1H−ピロール−2−イル}ピリジン
水素化ナトリウム(3.32g)のテトラヒドロフラン(70mL)懸濁液に2−フルオロ−3−(3−フルオロ−1H−ピロール−2−イル)ピリジン(5.98g)のテトラヒドロフラン(30mL)溶液を氷冷下で滴下し30分間攪拌した。さらに15−クラウン−5(18.3g)とトリフルオロメタンスルホン酸トリス(1−メチルエチル)シリル(25.4g)を加え、1時間攪拌した。減圧下で溶媒を半量留去した後に水を加え、酢酸エチルで抽出した。分離した水層を酢酸エチルで再度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後に、減圧濃縮した。残留物を塩基性シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン→ヘキサン−酢酸エチル=19:1)で精製することにより表題化合物を淡黄色油状物として得た(収量10.9g、収率98%)。
1H−NMR(CDCl3)δ:1.04(18H,d,J=7.0Hz),1.09−1.19(3H,m),6.17(1H,dd,J=3.2,1.5Hz),6.70(1H,dd,J=4.8,3.3Hz),7.21(1H,ddd,J=7.3,4.9,1.7Hz),7.78(1H,ddd,J=9.3,7.3,2.1Hz).
4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−カルバルデヒド
アルゴン雰囲気下でN,N−ジメチルホルムアミド(717mg)のジクロロメタン(20mL)溶液に塩化オキザリル(1.13g)を氷冷下で加え、10分間攪拌した。得られた懸濁液に2−フルオロ−3−{3−フルオロ−1−[トリス(1−メチルエチル)シリル]−1H−ピロール−2−イル}ピリジン(1.50g)のジクロロメタン(5mL)溶液を加え還流条件下で10時間攪拌した。反応混合物を氷冷下で冷却した後に1mol/L水酸化ナトリウム水溶液(30mL)を加え15分間攪拌した。溶媒を減圧下で半量濃縮し、残留物に酢酸エチルを加えて分液した。分離した水層を酢酸エチルで再度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮した。残留固体をジイソプロピルエーテル(30mL)で洗浄し吸引濾過することにより、表題化合物を無色固体として得た(収量726mg、収率78%)。
1H−NMR(CDCl3)δ:7.29−7.40(2H,m),8.11(1H,dt,J=4.8,1.6Hz),8.29(1H,ddd,J=10.0,7.9,1.9Hz),9.22(1H,brs),9.90(1H,s).
{[4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル]メチル}メチルカルバミン酸 tert−ブチル
4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−カルバルデヒド(261mg)のテトラヒドロフラン(1mL)−メタノール(2mL)溶液に40%メチルアミンメタノール溶液(4mL)を室温で加え、20分間攪拌した。反応混合物に水素化ホウ素ナトリウム(142mg)を加え1時間攪拌した。反応混合物を減圧濃縮した後に、水(4mL)と酢酸エチル(4mL)を加えた。得られた混合物に室温で二炭酸ジ−tert−ブチル(410mg)を加え、1時間攪拌した。反応混合物を酢酸エチルと水層に分離し、分離した水層を再度酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=9:1→3:1)で精製することにより表題化合物を無色固体として得た(収量347mg、収率86%)。1H−NMR(CDCl3)δ:1.49(9H,s),2.88(3H,s),4.31(2H,s),6.46−6.94(1H,m),7.15−7.32(1H,m),8.00(1H,dt,J=4.7,1.7Hz),8.23(1H,ddd,J=10.2,7.9,1.9Hz),8.66(1H,brs).
2−(ベンジルスルファニル)−3−メチルピリジン
水素化ナトリウム(60%油性、1.44g)のテトラヒドロフラン(45mL)懸濁液に室温でフェニルメタンチオール(465mg)を滴下して15分間攪拌した。反応液に2−ブロモ−3−メチルピリジン(2.0g)を加え、60℃で1時間半攪拌した。反応液を水で希釈した後、減圧濃縮した。残った水層を酢酸エチルで2度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン→ヘキサン−酢酸エチル=97:3)で精製することにより表題化合物を灰色油状物として得た(収量1.79g、収率72%)。
1H−NMR(CDCl3)δ: 2.23(3H,s),4.49(2H,s),6.93(1H,dd,J=7.6,4.9Hz),7.19−7.35(5H,m),7.39−7.48(1H,m),8.32(1H,dd,J=4.9,1.1Hz).
塩化 3−メチルピリジン−2−スルホニル
2−(ベンジルスルファニル)−3−メチルピリジン(1.79g)の酢酸(16mL)−水(8mL)溶液に室温でN−クロロコハク酸イミド(3.33g)を加え2時間攪拌した。反応液を減圧濃縮した後に、飽和炭酸水素ナトリウム水溶液を加え酢酸エチルで2度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=9:1→3:2)で精製することにより粗表題化合物を淡黄色油状物として得た(収量153mg)。
1H−NMR(CDCl3)δ:2.78(3H,s),7.57(1H,dd,J=7.9,4.5Hz),7.82(1H,ddd,J=7.7,1.5,0.8Hz),8.61(1H,dd,J=4.5,1.1Hz).
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(3−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル
水素化ナトリウム(60%油性、20mg)のテトラヒドロフラン(2mL)懸濁液に室温で、{[4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル]メチル}メチルカルバミン酸 tert−ブチル(161mg)、15−クラウン−5(110mg)、粗塩化 3−メチルピリジン−2−スルホニル(153mg)のテトラヒドロフラン(1.5mL)溶液を室温で加え、72時間攪拌した。反応液を水で希釈し、酢酸エチルで抽出した。分離した水層を再度酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=9:1→11:9)で精製することにより表題化合物を無色油状物として得た(収量113mg、収率47%)。
1H−NMR(CDCl3)δ:1.47(9H,s),2.43(3H,s),2.90(3H,s),4.32(2H,brs),7.20(1H,ddd,J=7.4,5.0,1.7Hz),7.29(1H,d,J=5.7Hz),7.36(1H,dd,J=7.8,4.6Hz),7.61(1H,dd,J=7.8,0.8Hz),7.76−7.85(1H,m),8.19(1H,ddd,J=4.9,2.0,1.0Hz),8.29(1H,dd,J=4.5,0.9Hz).
2−(ベンジルスルファニル)−4−メチルピリジン
水素化ナトリウム(60%油性、512mg)のテトラヒドロフラン(45mL)懸濁液に室温でフェニルメタンチオール(1.52g)を滴下し、さらに反応液に2−ブロモ−4−メチルピリジン(2.0g)を加え、60℃で72時間攪拌した。反応液を水で希釈し酢酸エチルで2度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン→ヘキサン−酢酸エチル=24:1)で精製することにより表題化合物を褐色油状物として得た(収量1.40g、収率56%)。
1H−NMR(CDCl3)δ:2.26(3H,s),4.43(2H,s),6.82(1H,d,J=5.1Hz),6.99(1H,s),7.17−7.32(3H,m),7.35−7.44(2H,m),8.31(1H,d,J=5.1Hz).
フッ化 4−メチルピリジン−2−スルホニル
2−(ベンジルスルファニル)−4−メチルピリジン(1.40g)の酢酸(10mL)−水(5mL)溶液に氷冷下でN−クロロコハク酸イミド(3.48g)を加え、徐々に室温に戻した後4時間攪拌した。さらに反応液にフッ化カリウム(379mg)を室温で加え18時間攪拌した。反応液を減圧濃縮した後に、酢酸エチルで希釈し飽和炭酸水素ナトリウム水溶液で洗浄した。分離した水層を酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=4:1→1:1)で精製することにより粗表題化合物を淡黄色油状物として得た(収量343mg、収率30%)。
1H−NMR(CDCl3)δ:2.54(3H,s),7.50(1H,dt,J=4.9,0.7Hz),7.95(1H,d,J=0.8Hz),8.69(1H,d,J=4.9Hz).
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル
水素化ナトリウム(60%油性、60mg)のテトラヒドロフラン懸濁液(3mL)に室温で、{[4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル]メチル}メチルカルバミン酸 tert−ブチル(323mg)、15−クラウン−5(330mg)、フッ化 4−メチルピリジン−2−スルホニル(343mg)を室温で加え、41時間攪拌した。反応液を水で希釈し、酢酸エチルで抽出した。分離した水層を再度酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=4:1→1:1)で精製することにより表題化合物を淡黄色油状物として得た(収量333mg、収率70%)。
1H−NMR(CDCl3)δ:1.47(9H,s),2.38(3H,s),2.86(3H,s),4.27(2H,brs),7.27−7.34(3H,m),7.36(1H,s),7.87(1H,ddd,J=9.2,7.5,1.9Hz),8.26(1H,d,J=3.8Hz),8.45(1H,d,J=4.9Hz).
2−(ベンジルスルファニル)−5−フルオロピリジン
水素化ナトリウム(60%油性、440mg)のテトラヒドロフラン(40mL)懸濁液に室温でフェニルメタンチオール(1.37g)を滴下し、さらに反応液に2−ブロモ−5−フルオロピリジン(1.76g)を加え、60℃で5時間攪拌した。反応液を水で希釈し減圧濃縮した。残った水層を酢酸エチルで2度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン→ヘキサン−酢酸エチル=97:3)で精製することにより粗表題化合物を褐色油状物として得た(収量244mg)。
フッ化 5−フルオロピリジン−2−スルホニル
粗2−(ベンジルスルファニル)−5−フルオロピリジン(244mg)の酢酸(3mL)−水(1.5mL)溶液に氷冷下でN−クロロコハク酸イミド(594mg)を加え、徐々に室温に戻した後2時間攪拌した。さらに反応液にフッ化カリウム(65mg)を室温で加え1時間攪拌した。反応液を減圧濃縮した後に、水で希釈し酢酸エチルで抽出した。分離した水層を酢酸エチルで再度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=9:1→3:1)で精製することにより粗表題化合物を無色固体として得た(収量69mg、収率35%)。
1H−NMR(CDCl3)δ:7.75(1H,ddd,J=8.7,7.4,2.7Hz),8.20(1H,dd,J=8.8,4.1Hz),8.66(1H,d,J=2.8Hz).
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル
水素化ナトリウム(60%油性、40mg)のテトラヒドロフラン(2.5mL)懸濁液に室温で、{[4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル]メチル}メチルカルバミン酸 tert−ブチル(162mg)、15−クラウン−5(220mg)、フッ化 5−フルオロピリジン−2−スルホニル(120mg)を室温で加え、28時間攪拌した。反応液を水で希釈し、酢酸エチルで抽出した。分離した水層を再度酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=9:1→7:3)で精製することにより表題化合物を淡黄色油状物として得た(収量69mg、収率29%)。
1H−NMR(CDCl3)δ:1.48(9H,s),2.88(3H,s),4.27(2H,brs),7.24−7.34(2H,m),7.52(1H,ddd,J=8.7,7.5,2.8Hz),7.68(1H,dd,J=8.7,4.1Hz),7.85(1H,ddd,J=9.2,7.4,2.0Hz),8.27(1H,ddd,J=4.8,1.8,0.9Hz),8.45(1H,d,J=2.6Hz).
2−(ベンジルスルファニル)−4−メトキシピリジン
2−クロロ−4−メトキシピリジン(786mg)のトルエン(10mL)溶液にフェニルメタンチオール(683mg)、N,N−ジイソプロピルエチルアミン(1.56g)、トリス(ジベンジリデンアセトン)二パラジウム(0)(202mg)、4,5−ビス(ジフェニルホスフィノ)−9,9−ジメチルキサンテン(256mg)を加え、アルゴン雰囲気下80℃で26時間攪拌した。反応液をシリカゲルで濾過し、濾液を減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン→ヘキサン−酢酸エチル=19:1)で精製することにより表題化合物を橙色油状物として得た(収量454mg、収率38%)。
1H−NMR(CDCl3)δ:3.79(3H,s),4.43(2H,s),6.57(1H,dd,J=5.9,2.5Hz),6.68(1H,d,J=2.3Hz),7.19−7.34(3H,m),7.36−7.44(2H,m),8.27(1H,d,J=5.7Hz).
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メトキシピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル
2−(ベンジルスルファニル)−4−メトキシピリジン(453mg)の酢酸(4mL)−水(2mL)溶液に氷冷下でN−クロロコハク酸イミド(1.10g)を加え、徐々に室温に戻した後に5時間攪拌した。反応液を減圧濃縮した後に、水で希釈し酢酸エチルで2度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=3:1→1:1)で精製することにより粗塩化 4−メトキシピリジン−2−スルホニルを淡黄色油状物として得た。次に、水素化ナトリウム(60%油性、30mg)のテトラヒドロフラン(2.5mL)懸濁液に室温で、{[4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル]メチル}メチルカルバミン酸 tert−ブチル(162mg)、15−クラウン−5(165mg)、上記で得られた粗塩化 4−メトキシピリジン−2−スルホニルのテトラヒドロフラン(2mL)溶液を室温で加え、18時間攪拌した。反応液を水で希釈し、酢酸エチルで抽出した。分離した水層を再度酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=17:3→1:1)で精製することにより表題化合物を無色油状物として得た(収量96mg、2工程収率9%)。
1H−NMR(CDCl3)δ:1.47(9H,s),2.87(3H,s),3.84(3H,s),4.27(2H,brs),6.94(1H,dd,J=5.6,2.4Hz),7.07(1H,d,J=2.4Hz),7.28(1H,dd,J=5.3,2.1Hz),7.31(1H,d,J=5.7Hz),7.87(1H,ddd,J=9.2,7.5,1.8Hz),8.26(1H,d,J=4.7Hz),8.39(1H,d,J=5.7Hz).
3−(ベンジルスルファニル)−5−フルオロピリジン
3−ブロモ−5−フルオロピリジン(522mg)のトルエン(5mL)溶液にフェニルメタンチオール(370mg)、N,N−ジイソプロピルエチルアミン(831mg)、トリス(ジベンジリデンアセトン)二パラジウム(0)(108mg)、4,5−ビス(ジフェニルホスフィノ)−9,9−ジメチルキサンテン(138mg)を加え、アルゴン雰囲気下80℃で2時間攪拌した。反応液を水で希釈し、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、シリカゲルで濾過し、濾液を減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン→ヘキサン−酢酸エチル=10:1)で精製することにより表題化合物を橙色油状物として得た(収量587mg、収率90%)。
1H−NMR(CDCl3)δ:4.13(2H,s),7.23−7.33(6H,m),8.25−8.26(1H,m),8.30−8.31(1H,m).
塩化 5−フルオロピリジン−3−スルホニル
3−(ベンジルスルファニル)−5−フルオロピリジン(573mg)の酢酸(7.5mL)−水(2.5mL)溶液に室温でN−クロロコハク酸イミド(1.40g)を加え1時間半攪拌した。反応液を減圧濃縮した後に、水で希釈し酢酸エチルで2度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をトルエンで共沸した後にシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=9:1→7:3)で精製することにより表題化合物を無色油状物として得た(収量376mg、収率74%)。
1H−NMR(CDCl3)δ:8.04(1H,ddd,J=7.0,2.7,2.0Hz),8.85(1H,d,J=2.6Hz),9.10(1H,dd,J=1.1,0.8Hz).
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル
水素化ナトリウム(60%油性、20mg)のテトラヒドロフラン(2mL)懸濁液に室温で、{[4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル]メチル}メチルカルバミン酸 tert−ブチル(162mg)、15−クラウン−5(132mg)、塩化 5−フルオロピリジン−3−スルホニル(127mg)のテトラヒドロフラン(1mL)溶液を室温で加え、1時間攪拌した。反応液を水で希釈し、酢酸エチルで抽出した。分離した水層を再度酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物を塩基性シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=9:1→7:3)で精製することにより表題化合物を無色油状物として得た(収量224mg、収率93%)。
1H−NMR(CDCl3)δ:1.48(9H,s),2.88(3H,s),4.27(2H,s),7.28−7.36(2H,m),7.38(1H,d,J=7.2Hz),7.73−7.86(1H,m),8.34(1H,d,J=4.2Hz),8.46(1H,s),8.69(1H,d,J=2.7Hz).
3−(ベンジルスルファニル)−4−メチルピリジン
3−ブロモ−4−メチルピリジン(1.0g)のトルエン(12mL)溶液にフェニルメタンチオール(794mg)、N,N−ジイソプロピルエチルアミン(1.65g)、トリス(ジベンジリデンアセトン)二パラジウム(0)(213mg)、4,5−ビス(ジフェニルホスフィノ)−9,9−ジメチルキサンテン(269mg)を加え、アルゴン雰囲気下80℃で1時間半攪拌した。反応液をシリカゲルで濾過し、濾液を減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=19:1→3:1→1:1)で精製することにより表題化合物を黄色油状物として得た(収量740mg、収率59%)。
1H−NMR(CDCl3)δ:2.27(3H,s),4.07(2H,s),7.06(1H,d,J=4.9Hz),7.14−7.35(5H,m),8.30(1H,d,J=5.3Hz),8.45(1H,s).
塩化 4−メチルピリジン−3−スルホニル
3−(ベンジルスルファニル)−4−メチルピリジン(740mg)の酢酸(9mL)−水(3mL)溶液に室温でN−クロロコハク酸イミド(1.84g)を加え2時間攪拌した。反応液を減圧濃縮した後に、水で希釈し酢酸エチルで2度抽出した。合わせた有機層を飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をトルエンで共沸した後にシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=9:1→3:1)で精製することにより粗表題化合物を無色油状物として得た(収量676mg)。
1H−NMR(CDCl3)δ:2.82(3H,s),7.34−7.44(1H,m),8.77(1H,d,J=4.9Hz),9.19(1H,s).
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル
水素化ナトリウム(60%油性、24mg)のテトラヒドロフラン(2mL)懸濁液に室温で、{[4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル]メチル}メチルカルバミン酸 tert−ブチル(161mg)、15−クラウン−5(132mg)、粗塩化 4−メチルピリジン−3−スルホニル(125mg)のテトラヒドロフラン(1mL)溶液を室温で加え、1時間攪拌した。反応液を水で希釈し、酢酸エチルで抽出した。分離した水層を再度酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=17:3→1:1)で精製することにより表題化合物を淡黄色油状物として得た(収量127mg、収率53%)。
1H−NMR(CDCl3)δ:1.49(9H,s),2.36(3H,s),2.92(3H,s),4.32(2H,s),7.19(1H,d,J=5.1Hz),7.23−7.31(1H,m),7.41(1H,brs),7.82(1H,dt,J=8.3,1.9Hz),8.18−8.26(2H,m),8.58(1H,d,J=5.1Hz).
3−(ベンジルスルファニル)−5−メチルピリジン
3−ブロモ−5−メチルピリジン(888mg)のトルエン(10mL)溶液にフェニルメタンチオール(705mg)、N,N−ジイソプロピルエチルアミン(1.47g)、トリス(ジベンジリデンアセトン)二パラジウム(0)(189mg)、4,5−ビス(ジフェニルホスフィノ)−9,9−ジメチルキサンテン(239mg)を加え、アルゴン雰囲気下80℃で1時間半攪拌した。反応液をシリカゲルで濾過し、濾液を減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=19:1→17:3)で精製することにより表題化合物を黄色油状物として得た(収量1.06g、収率95%)。
1H−NMR(CDCl3)δ:2.26(3H,d,J=0.8Hz),4.09(2H,s),7.20−7.33(5H,m),7.37(1H,dt,J=2.1,0.8Hz),8.25(1H,d,J=1.3Hz),8.33(1H,d,J=2.1Hz).
塩化 5−メチルピリジン−3−スルホニル
3−(ベンジルスルファニル)−5−メチルピリジン(1.06g)の酢酸(15mL)−水(5mL)溶液に室温でN−クロロコハク酸イミド(2.63g)を加え2時間攪拌した。反応液を減圧濃縮した後に、水で希釈し酢酸エチルで2度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=19:1→17:3)で精製することにより表題化合物を無色油状物として得た(収量700mg、収率74%)。
1H−NMR(CDCl3)δ:2.52(3H,s),7.96−8.22(1H,m),8.78(1H,d,J=1.5Hz),9.06(1H,d,J=2.3Hz).
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル
水素化ナトリウム(60%油性、24mg)のテトラヒドロフラン(3mL)懸濁液に室温で、{[4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル]メチル}メチルカルバミン酸 tert−ブチル(323mg)、15−クラウン−5(264mg)、塩化 4−メチルピリジン−3−スルホニル(249mg)のテトラヒドロフラン(2mL)溶液を室温で加え、1時間攪拌した。反応液を水で希釈し、酢酸エチルで抽出した。分離した水層を再度酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=9:1→1:1)で精製することにより表題化合物を淡黄色油状物として得た(収量370mg、収率77%)。
1H−NMR(CDCl3)δ:1.48(9H,s),2.35(3H,d,J=0.4Hz),2.86(3H,s),4.26(2H,brs),7.26(1H,s),7.32(1H,ddd,J=7.3,5.2,1.5Hz),7.38(1H,brs),7.76−7.90(1H,m),8.25−8.34(1H,m),8.46(1H,d,J=2.1Hz),8.63(1H,d,J=1.5Hz).
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(6−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル
水素化ナトリウム(60%油性、31mg)のテトラヒドロフラン(3mL)懸濁液に室温で、{[4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル]メチル}メチルカルバミン酸 tert−ブチル(100mg)、15−クラウン−5(170mg)、塩化 6−メチルピリジン−3−スルホニル塩酸塩(91mg)を室温で加え、1時間半攪拌した。反応液を水で希釈し、酢酸エチルで抽出した。分離した水層を再度酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=17:3→1:1)で精製することにより表題化合物を淡黄色油状物として得た(収量123mg、収率83%)。
1H−NMR(CDCl3)δ:1.48(9H,s),2.62(3H,s),2.86(3H,s),4.26(2H,s),7.20(1H,d,J=8.0Hz),7.27−7.34(2H,m),7.51(1H,dd,J=8.0,1.9Hz),7.76−7.86(1H,m),8.27−8.36(1H,m),8.50(1H,d,J=2.3Hz).
3−(ベンジルスルファニル)−2−メチルピリジン
3−ブロモ−2−メチルピリジン(1.0g)のトルエン(12mL)溶液にフェニルメタンチオール(794mg)、N,N−ジイソプロピルエチルアミン(1.65g)、トリス(ジベンジリデンアセトン)二パラジウム(0)(213mg)、4,5−ビス(ジフェニルホスフィノ)−9,9−ジメチルキサンテン(269mg)を加え、アルゴン雰囲気下80℃で4時間攪拌した。反応液をシリカゲルで濾過し、濾液を減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=9:1→3:1)で精製することにより表題化合物を黄色固体として得た(収量742mg、収率59%)。
1H−NMR(CDCl3)δ:2.56(3H,s),4.08(2H,s),7.03(1H,dd,J=7.6,5.0Hz),7.21−7.34(5H,m),7.48(1H,dd,J=7.8,1.6Hz),8.30(1H,dd,J=4.8,1.6Hz).
塩化 2−メチルピリジン−3−スルホニル
3−(ベンジルスルファニル)−2−メチルピリジン(731mg)の酢酸(9mL)−水(3mL)溶液に室温でN−クロロコハク酸イミド(1.81g)を加え4時間攪拌した。反応液を減圧濃縮した後に、水で希釈し酢酸エチルで2度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=19:1→7:3)で精製することにより表題化合物を無色油状物として得た(収量175mg、収率27%)。
1H−NMR(CDCl3)δ:3.03(3H,s),7.40(1H,dd,J=8.1,4.7Hz),8.33(1H,dd,J=8.1,1.7Hz),8.80(1H,dd,J=4.7,1.7Hz).
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(2−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル
水素化ナトリウム(60%油性、34mg)のテトラヒドロフラン(2mL)懸濁液に室温で、{[4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル]メチル}メチルカルバミン酸 tert−ブチル(226mg)、15−クラウン−5(185mg)、塩化 2−メチルピリジン−3−スルホニル(174mg)のテトラヒドロフラン(1mL)溶液を室温で加え、18時間攪拌した。反応液を水で希釈し、酢酸エチルで抽出した。分離した水層を再度酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=17:3→1:1)で精製することにより表題化合物を黄色油状物として得た(収量288mg、収率86%)。
1H−NMR(CDCl3)δ:1.49(9H,s),2.61(3H,s),2.92(3H,s),4.32(2H,s),7.03(1H,dd,J=8.1,4.7Hz),7.21−7.26(1H,m),7.34(1H,dd,J=8.1,1.7Hz),7.42(1H,brs),7.79(1H,ddd,J=9.2,7.3,2.1Hz),8.19−8.26(1H,m),8.63(1H,dd,J=4.9,1.5Hz).
2−(ベンジルスルファニル)−5−メトキシピリジン
2−ブロモ−5−メトキシピリジン(1.13g)のトルエン(15mL)溶液にフェニルメタンチオール(820mg)、N,N−ジイソプロピルエチルアミン(1.71g)、トリス(ジベンジリデンアセトン)二パラジウム(0)(220mg)、4,5−ビス(ジフェニルホスフィノ)−9,9−ジメチルキサンテン(278mg)を加え、アルゴン雰囲気下80℃で3時間攪拌した。反応液をシリカゲルで濾過し、濾液を減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=49:1→19:1)で精製することにより表題化合物を黄色油状物として得た(収量1.47g、収率定量的)。
1H−NMR(CDCl3)δ:3.83(3H,s),4.37(2H,s),6.99−7.10(2H,m),7.19−7.31(3H,m),7.33−7.40(2H,m),8.21(1H,dd,J=2.6,0.9Hz).
塩化 5−メトキシピリジン−2−スルホニル
2−(ベンジルスルファニル)−5−メトキシピリジン(1.47g)の酢酸(9mL)−水(3mL)溶液に室温でN−クロロコハク酸イミド(3.20g)を加え2時間攪拌した。反応液を減圧濃縮した後に、水で希釈し酢酸エチルで2度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=19:1→17:3)で精製することにより表題化合物を無色固体として得た(収量984mg、収率79%)。
1H−NMR(CDCl3)δ:4.00(3H,s),7.38(1H,dd,J=8.9,2.8Hz),8.08(1H,d,J=8.7Hz),8.43(1H,d,J=2.8Hz).
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−メトキシピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル
水素化ナトリウム(60%油性、168mg)のテトラヒドロフラン(10mL)懸濁液に室温で、{[4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル]メチル}メチルカルバミン酸 tert−ブチル(970mg)、15−クラウン−5(925mg)、塩化 5−メトキシピリジン−2−スルホニル(984mg)のテトラヒドロフラン(15mL)溶液を室温で加え、30分間攪拌した。反応液を半量まで減圧濃縮した後に、水で希釈し、酢酸エチルで抽出した。分離した水層を再度酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=17:3→1:1)で精製することにより表題化合物を黄色油状物として得た(収量1.38g、収率93%)。
1H−NMR(CDCl3)δ:1.47(9H,s),2.87(3H,s),3.91(3H,s),4.26(2H,brs),7.16(1H,dd,J=8.8,2.9Hz),7.24−7.30(1H,m),7.32(1H,d,J=5.5Hz),7.52(1H,d,J=8.9Hz),7.87(1H,ddd,J=9.2,7.4,2.1Hz),8.23(1H,d,J=2.4Hz),8.26(1H,ddd,J=4.9,1.9,0.9Hz).
トリフルオロメタンスルホン酸 5−クロロピリジン−3−イル
5−クロロピリジン−3−オール(1.30g)のテトラヒドロフラン(50mL)溶液にトリエチルアミン(1.21g)、N−フェニルビス(トリフルオロメタンスルホンイミド)(3.93g)を室温で加え、20分間攪拌した。反応液を減圧濃縮し、残留物を酢酸エチルで希釈した後に1mol/L塩酸で洗浄した。分離した水層を酢酸エチルで再度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=99:1→19:1)で精製することにより表題化合物を無色油状物として得た(収量1.73g、収率66%)。
1H−NMR(CDCl3)δ:7.69(1H,t,J=2.3Hz),8.52(1H,d,J=2.3Hz),8.64(1H,d,J=1.9Hz).
3−(ベンジルスルファニル)−5−クロロピリジン
トリフルオロメタンスルホン酸 5−クロロピリジン−3−イル(1.73g)のトルエン(15mL)溶液にフェニルメタンチオール(861mg)、N,N−ジイソプロピルエチルアミン(1.88g)、トリス(ジベンジリデンアセトン)二パラジウム(0)(121mg)、4,5−ビス(ジフェニルホスフィノ)−9,9−ジメチルキサンテン(153mg)を加え、アルゴン雰囲気下80℃で3時間攪拌した。反応液をシリカゲルで濾過し、濾液を減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン→ヘキサン−酢酸エチル=19:1)で精製することにより表題化合物を黄色油状物として得た(収量1.63g、収率定量的)。
1H−NMR(CDCl3)δ:4.12(2H,s),7.21−7.36(5H,m),7.53(1H,t,J=2.1Hz),8.36(2H,d,J=1.9Hz).
塩化 5−クロロピリジン−3−スルホニル
3−(ベンジルスルファニル)−5−クロロピリジン(1.63g)の酢酸(9mL)−水(3mL)溶液に室温でN−クロロコハク酸イミド(3.53g)を加え2時間攪拌した。反応液を減圧濃縮した後に、水で希釈し酢酸エチルで2度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をトルエンで共沸した後にシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=49:1→9:1)で精製することにより表題化合物を無色油状物として得た(収量1.26g、収率90%)。
1H−NMR(CDCl3)δ:8.29(1H,t,J=2.2Hz),8.91(1H,d,J=2.2Hz),9.12(1H,d,J=1.9Hz).
({1−[(5−クロロピリジン−3−イル)スルホニル]−4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル
水素化ナトリウム(60%油性、52mg)のテトラヒドロフラン(3mL)懸濁液に室温で、{[4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル]メチル}メチルカルバミン酸 tert−ブチル(323mg)、15−クラウン−5(286mg)、塩化 5−クロロピリジン−3−スルホニル(318mg)のテトラヒドロフラン(2mL)溶液を室温で加え、20分間攪拌した。反応液を水で希釈し、酢酸エチルで抽出した。分離した水層を再度酢酸エチルで抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=9:1→7:3)で精製することにより表題化合物を無色油状物として得た(収量391mg、収率78%)。
1H−NMR(CDCl3)δ:1.48(9H,s),2.88(3H,s),4.27(2H,s),7.26(1H,s),7.33(1H,ddd,J=7.3,5.2,1.5Hz),7.61(1H,t,J=2.1Hz),7.80(1H,ddd,J=9.2,7.5,1.9Hz),8.26−8.38(1H,m),8.50(1H,d,J=1.9Hz),8.76(1H,d,J=2.3Hz).
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(3−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン フマル酸塩
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(3−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル(107mg)の酢酸エチル(2mL)、2−プロパノール(1mL)溶液に4mol/L塩化水素−酢酸エチル溶液(3mL)を加え、混合物を室温で1時間攪拌した。反応混合物を減圧濃縮し、残留物を飽和炭酸水素ナトリウム水溶液で希釈し、酢酸エチルで抽出した。分離した水層を酢酸エチルで再度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物を塩基性シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=4:1→1:1)で精製することにより1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(3−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンを淡黄色油状物として得た(収量45mg、収率54%)。得られた1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(3−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンの酢酸エチル(2mL)溶液をフマル酸(14mg)のエタノール(2mL)溶液に滴下し減圧濃縮した。残留物を酢酸エチル−エタノールから再結晶することにより、表題化合物を白色固体として得た(収量51mg、収率88%)。
1H−NMR(DMSO−d6)δ:2.35(3H,s),2.38(3H,s),3.73(2H,s),6.53(2H,s),7.32−7.39(1H,m),7.48(1H,d,J=5.7Hz),7.63(1H,dd,J=7.8,4.4Hz),7.74−7.83(1H,m),7.93(1H,d,J=7.6Hz),8.27(1H,d,J=4.2Hz),8.41(1H,d,J=4.5Hz),3H未検出.
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン 塩酸塩
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル(333mg)の酢酸エチル(2mL)、2−プロパノール(1mL)溶液に4mol/L塩化水素−酢酸エチル溶液(3mL)を加え、混合物を室温で1時間攪拌した。反応混合物を減圧濃縮し、残留物を酢酸エチル−エタノールから再結晶することにより、表題化合物を白色固体として得た(収量191mg、収率66%)。
1H−NMR(DMSO−d6)δ:2.37(3H,s),2.56(3H,s),4.05(2H,s),7.45(1H,ddd,J=7.3,5.0,1.7Hz),7.54(1H,s),7.59−7.66(1H,m),7.77−7.90(2H,m),8.33−8.40(1H,m),8.55(1H,d,J=4.9Hz),9.11(2H,brs).
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン 塩酸塩
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル(69mg)の酢酸エチル(1.5mL)、2−プロパノール(1mL)溶液に4mol/L塩化水素−酢酸エチル溶液(2mL)を加え、混合物を室温で2時間半攪拌した。反応混合物を減圧濃縮し、残留物を酢酸エチル−エタノールから再結晶することにより、表題化合物を白色固体として得た(収量31mg、収率51%)。
1H−NMR(DMSO−d6)δ:2.58(3H,s),4.07(2H,s),7.41−7.49(1H,m),7.80(1H,d,J=5.5Hz),7.82−7.91(2H,m),8.05(1H,dt,J=8.6,2.8Hz),8.36(1H,ddd,J=4.9,1.9,0.9Hz),8.78(1H,d,J=2.8Hz),8.97(2H,brs).
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メトキシピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン 塩酸塩
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メトキシピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル(94mg)の酢酸エチル(2mL)、および2−プロパノール(1mL)溶液に4mol/L塩化水素−酢酸エチル溶液(2mL)を加え、混合物を室温で4時間攪拌した。反応混合物を減圧濃縮し、残留物を酢酸エチル−エタノールから再結晶することにより、表題化合物を白色固体として得た(収量65mg、収率79%)。
1H−NMR(DMSO−d6)δ:2.57(3H,s),3.87(3H,s),4.06(2H,s),7.17(1H,d,J=2.3Hz),7.33(1H,dd,J=5.7,2.7Hz),7.46(1H,ddd,J=6.9,5.2,1.5Hz),7.80(1H,d,J=5.7Hz),7.89(1H,ddd,J=9.3,7.6,1.7Hz),8.30−8.39(1H,m),8.51(1H,d,J=5.7Hz),9.01(2H,brs).
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン 塩酸塩
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル(224mg)の酢酸エチル(2mL)、2−プロパノール(1mL)溶液に4N塩化水素−酢酸エチル溶液(3mL)を加え、混合物を室温で3時間攪拌した。反応混合物を減圧濃縮し、残留物を酢酸エチル−エタノールから再結晶することにより、表題化合物を白色固体として得た(収量127mg、収率65%)。
1H−NMR(DMSO−d6)δ:2.58(3H,s),4.05(2H,s),7.46−7.55(1H,m),7.87−7.97(2H,m),8.03(1H,dt,J=7.6,2.3Hz),8.42(1H,d,J=4.2Hz),8.49(1H,s),9.04(1H,d,J=2.3Hz),9.09(2H,brs).
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン フマル酸塩
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル(127mg)の酢酸エチル(2mL)、2−プロパノール(0.5mL)溶液に4N塩化水素−酢酸エチル溶液(2mL)を加え、混合物を室温で2時間攪拌した。反応混合物を減圧濃縮し、残留物を飽和炭酸水素ナトリウム水溶液で希釈し、酢酸エチルで抽出した。分離した水層を酢酸エチルで再度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮することにより1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンを淡黄色油状物として得た(収量97mg、収率97%)。得られた1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンの酢酸エチル(2mL)溶液をフマル酸(30mg)のエタノール(2mL)溶液に滴下し減圧濃縮した。残留物をエタノールから再結晶することにより、表題化合物を白色固体として得た(収量103mg、収率81%)。
1H−NMR(DMSO−d6)δ:2.33(3H,s),2.40(3H,s),3.76(2H,s),6.53(2H,s),7.43(1H,ddd,J=7.3,5.1,1.8Hz),7.52(1H,d,J=5.1Hz),7.72(1H,d,J=5.7Hz),7.85(1H,ddd,J=9.5,7.4,1.9Hz),8.14(1H,s),8.32(1H,ddd,J=4.9,1.9,0.9Hz),8.70(1H,d,J=5.1Hz),3H未検出.
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン フマル酸塩
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル(370mg)の酢酸エチル(2mL)、2−プロパノール(1mL)溶液に4N塩化水素−酢酸エチル溶液(3mL)を加え、混合物を室温で3時間攪拌した。反応混合物を減圧濃縮し、残留物を飽和炭酸水素ナトリウム水溶液で希釈し、酢酸エチルで抽出した。分離した水層を酢酸エチルで再度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮することにより1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンを淡黄色油状物として得た(収量256mg、収率88%)。得られた1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンの酢酸エチル(2mL)溶液をフマル酸(78mg)のエタノール(2mL)溶液に滴下し減圧濃縮した。残留物をエタノール−水から再結晶することにより、表題化合物を白色固体として得た(収量288mg、収率87%)。
1H−NMR(DMSO−d6)δ:2.33(3H,s),2.35(3H,s),3.70(2H,s),6.54(2H,s),7.50(1H,ddd,J=7.3,5.1,1.9Hz),7.63−7.71(2H,m),7.90(1H,ddd,J=9.6,7.5,2.0Hz),8.36−8.41(1H,m),8.42(1H,d,J=2.3Hz),8.76(1H,d,J=1.3Hz),3H未検出.
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(6−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン 0.5フマル酸塩
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(6−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル(123mg)の酢酸エチル(2mL)、2−プロパノール(1mL)溶液に4mol/L塩化水素−酢酸エチル溶液(3mL)を加え、混合物を室温で3時間攪拌した。反応混合物を減圧濃縮し、残留物を飽和炭酸水素ナトリウム水溶液で希釈し、酢酸エチルで抽出した。分離した水層を酢酸エチルで再度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物を塩基性シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=1:1)で精製することにより1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(6−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンを無色油状物として得た(収量88mg、収率91%)。得られた1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(6−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンの酢酸エチル(2mL)溶液をフマル酸(27mg)のエタノール(2mL)溶液に滴下し減圧濃縮した。残留物をエタノール−水から再結晶することにより、表題化合物を白色固体として得た(収量78mg、収率77%)。
1H−NMR(DMSO−d6)δ:2.30(3H,s),2.56(3H,s),3.61(2H,s),6.51(1H,s),7.45−7.53(2H,m),7.60(1H,d,J=5.7Hz),7.79(1H,dd,J=8.3,2.3Hz),7.91(1H,ddd,J=9.6,7.5,1.9Hz),8.35−8.40(1H,m),8.48(1H,d,J=2.3Hz),2H未検出.
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(2−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン フマル酸塩
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(2−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル(288mg)の酢酸エチル(2mL)、2−プロパノール(1mL)溶液に4mol/L塩化水素−酢酸エチル溶液(3mL)を加え、混合物を室温で3時間攪拌した。反応混合物を減圧濃縮し、残留物を飽和炭酸水素ナトリウム水溶液で希釈し、酢酸エチルで抽出した。分離した水層を酢酸エチルで再度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮することにより1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(2−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンを無色油状物として得た(収量220mg、収率97%)。得られた1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(2−メチルピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンの酢酸エチル(3mL)溶液をフマル酸(67mg)のエタノール(3mL)溶液に滴下し減圧濃縮した。残留物をエタノールから再結晶することにより、表題化合物を白色固体として得た(収量253mg、収率88%)。
1H−NMR(DMSO−d6)δ:2.41(3H,s),2.49(3H,s),3.77(2H,s),6.53(2H,s),7.31(1H,dd,J=8.1,4.7Hz),7.42(1H,ddd,J=7.2,5.1,1.7Hz),7.47(1H,dd,J=8.3,1.5Hz),7.71(1H,d,J=5.7Hz),7.84(1H,ddd,J=9.6,7.5,1.9Hz),8.28−8.34(1H,m),8.73(1H,dd,J=4.7,1.7Hz),3H未検出.
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−メトキシピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン 塩酸塩
({4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−メトキシピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル(1.38g)の酢酸エチル(6mL)、2−プロパノール(3mL)溶液に4mol/L塩化水素−酢酸エチル溶液(9mL)を加え、混合物を室温で1時間半攪拌した。反応混合物を減圧濃縮し、残留物をエタノール−水から再結晶することにより、表題化合物を白色固体として得た(収量1.06g、収率88%)。
1H−NMR(DMSO−d6)δ:2.54(3H,s),3.91(3H,s),4.03(2H,s),7.38−7.46(1H,m),7.51−7.58(1H,m),7.62−7.70(1H,m),7.75−7.87(2H,m),8.33(1H,dt,J=4.7,0.8Hz),8.36(1H,d,J=3.0Hz),9.20(2H,brs).
1−{1−[(5−クロロピリジン−3−イル)スルホニル]−4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミン 塩酸塩
({1−[(5−クロロピリジン−3−イル)スルホニル]−4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}メチル)メチルカルバミン酸 tert−ブチル(391mg)の酢酸エチル(2mL)、2−プロパノール(1mL)溶液に4mol/L塩化水素−酢酸エチル溶液(3mL)を加え、混合物を室温で1時間半攪拌した。反応混合物を減圧濃縮し、残留物をエタノールから再結晶することにより、表題化合物を白色固体として得た(収量298mg、収率95%)。
1H−NMR(DMSO−d6)δ:2.57(3H,s),4.05(2H,s),7.52(1H,ddd,J=7.3,5.1,1.9Hz),7.93(1H,ddd,J=9.6,7.5,2.0Hz),8.01(1H,d,J=5.5Hz),8.11(1H,t,J=2.2Hz),8.43(1H,ddd,J=4.9,1.8,0.8Hz),8.57(1H,d,J=2.1Hz),9.05(1H,d,J=2.1Hz),9.33(2H,brs).
1−{4−フルオロ−1−[(5−フルオロ−6−メチルピリジン−2−イル)スルホニル]−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミン
6−ブロモ−3−フルオロ−2−メチルピリジンを用いて参考例15、参考例16、参考例17および実施例3と同様の方法で合成する。
1−{4−フルオロ−1−[(5−フルオロ−4−メチルピリジン−2−イル)スルホニル]−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミン
2−ブロモ−5−フルオロ−4−メチルピリジンを用いて参考例15、参考例16、参考例17および実施例3と同様の方法で合成する。
1−{4−フルオロ−1−[(5−フルオロ−4−メトキシピリジン−2−イル)スルホニル]−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミン
4−tert−ブトキシ−2,5−ジフルオロピリジンよりtert−ブチル基を除去、メチル化して2,5−ジフルオロ−4−メトキシピリジンを合成し、参考例15、参考例16、参考例17および実施例3と同様の方法で合成する。
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−メトキシピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン
3−ブロモ−5−メトキシピリジンを用いて参考例20、参考例21、参考例22および実施例5と同様の方法で合成する。
1−{4−フルオロ−1−[(5−フルオロ−6−メチルピリジン−3−イル)スルホニル]−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミン
5−クロロ−3−フルオロ−2−ヨードピリジンよりボロン酸カップリング反応を用いて5−クロロ−3−フルオロ−2−メチルピリジンを合成し、参考例20、参考例21、参考例22および実施例5と同様の方法で合成する。
1−{1−[(4,6−ジメチルピリジン−2−イル)スルホニル]−4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミン
2−ブロモ−4,6−ジメチルピリジンを用いて参考例15、参考例16、参考例17および実施例3と同様の方法で合成する。
1−{1−[(5−クロロピリジン−2−イル)スルホニル]−4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミン
塩化 5−クロロピリジン−2−スルホニルを用いて、参考例17および実施例3と同様の方法で合成する。
1−{1−[(5,6−ジメチルピリジン−2−イル)スルホニル]−4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミン
6−ブロモ−2,3−ジメチルピリジンを用いて、参考例15、参考例16、参考例17および実施例3と同様の方法で合成する。
1−{1−[(4,5−ジメチルピリジン−2−イル)スルホニル]−4−フルオロ−5−(2−フルオロピリジン−3−イル)−1H−ピロール−3−イル}−N−メチルメタンアミン
2−ブロモ−4,5−ジメチルピリジンを用いて、参考例15、参考例16、参考例17および実施例3と同様の方法で合成する。
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン 塩酸塩(751mg)を飽和重曹水に溶解し、酢酸エチルで二度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮することにより表題化合物を淡黄色油状物として得た(収量647mg、収率95%)。
1H−NMR(CDCl3)δ:2.38(3H,s),2.45(3H,s),3.64(2H,s),7.23−7.30(2H,m),7.33(1H,d,J=5.7Hz),7.36(1H,s),7.88(1H,ddd,J=9.3,7.4,1.9Hz),8.22−8.29(1H,m),8.45(1H,d,J=4.5Hz),1H未検出.
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン 塩酸塩(780mg)を飽和重曹水に溶解し、酢酸エチルで二度抽出した。合わせた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮した。残留物を塩基性シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル=3:1→3:7)で精製することにより表題化合物を淡黄色油状物として得た(収量619mg、収率87%)。
1H−NMR(CDCl3)δ:2.46(3H,s),3.65(2H,s),7.28−7.36(2H,m),7.41(1H,dt,J=7.3,2.2Hz),7.80(1H,ddd,J=9.2,7.4,2.0Hz),8.28−8.39(1H,m),8.48(1H,s),8.68(1H,d,J=2.6Hz),1H未検出.
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン フマル酸塩
フマル酸(58mg)のエタノール(2mL)溶液に1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン(189mg)の酢酸エチル(2mL)溶液を加え、混合物を減圧濃縮した。残留物をエタノールから再結晶することにより、表題化合物を白色固体として得た(収量224mg、収率91%)。
1H−NMR(DMSO−d6)δ:2.35−2.40(6H,m),3.73(2H,s),6.53(2H,s),7.44(1H,ddd,J=7.3,5.1,1.8Hz),7.49−7.55(2H,m),7.59(1H,d,J=4.9Hz),7.86(1H,ddd,J=9.5,7.4,1.9Hz),8.27−8.39(1H,m),8.54(1H,d,J=4.9Hz),3H未検出.
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン コハク酸塩
コハク酸(59mg)のエタノール(2mL)溶液に1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン(189mg)の酢酸エチル(2mL)溶液を加え、混合物を減圧濃縮した。残留物をエタノール−水から再結晶することにより、表題化合物を白色固体として得た(収量232mg、収率93%)。
1H−NMR(DMSO−d6)δ:2.34(3H,s),2.36(4H,s),2.37(3H,s),3.66(2H,s),7.39−7.49(2H,m),7.52(1H,s),7.55−7.63(1H,m),7.86(1H,ddd,J=9.5,7.4,1.9Hz),8.34(1H,ddd,J=4.9,1.9,0.9Hz),8.54(1H,d,J=4.9Hz),3H未検出.
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン 0.5フマル酸塩
フマル酸(36mg)のエタノール(2mL)溶液に1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン(120mg)の酢酸エチル(2mL)溶液を加え、混合物を減圧濃縮した。残留物をエタノールから再結晶することにより、表題化合物を白色固体として得た(収量113mg、収率82%)。
1H−NMR(DMSO−d6)δ:2.32(3H,s),3.63(2H,s),6.52(1H,s),7.50(1H,ddd,J=7.3,5.1,1.9Hz),7.67(1H,d,J=5.7Hz),7.94(1H,ddd,J=9.6,7.5,2.0Hz),7.98−8.05(1H,m),8.31−8.42(1H,m),8.48(1H,s),9.00(1H,d,J=2.8Hz),2H未検出.
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン コハク酸塩
コハク酸(46mg)のエタノール(4mL)溶液を1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン(150mg)に加え、混合物を減圧濃縮した。残留物をエタノールから再結晶することにより、表題化合物を白色固体として得た(収量167mg、収率85%)。
1H−NMR(DMSO−d6)δ:2.33(3H,s),2.38(4H,s),3.64(2H,s),7.50(1H,ddd,J=7.2,5.0,1.9Hz),7.66(1H,d,J=5.7Hz),7.94(1H,ddd,J=9.6,7.5,2.0Hz),7.98−8.03(1H,m),8.34−8.42(1H,m),8.44−8.53(1H,m),9.01(1H,d,J=2.6Hz),3H未検出.
1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン L−酒石酸塩
L−酒石酸(59mg)のエタノール(4mL)溶液を1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(5−フルオロピリジン−3−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミン(150mg)に加え、混合物を減圧濃縮した。残留物をエタノールから再結晶することにより、表題化合物を白色固体として得た(収量184mg、収率88%)。
1H−NMR(DMSO−d6)δ:2.44(3H,s),3.81(2H,s),4.00(2H,s),7.51(1H,ddd,J=7.3,5.2,1.9Hz),7.75(1H,d,J=5.7Hz),7.93(1H,ddd,J=9.5,7.4,1.9Hz),8.00(1H,dt,J=7.8,2.3Hz),8.38−8.43(1H,m),8.49(1H,s),9.02(1H,d,J=2.6Hz),5H未検出.
プロトン・カリウム−アデノシントリホスファターゼ(H+,K+−ATPase)阻害活性試験
ウォールマーク(Wallmark)らの方法[Biochim.Biophys.Acta,728,31(1983年)]に準じて胃粘膜ミクロソーム画分をブタの胃から調製した。まず、胃体部を摘出後、水道水で洗浄した後、3mol/L食塩水に浸し、ペーパータオルで粘膜表面を拭いた。胃粘膜を剥離し、細切後、1mmol/L EDTAおよび10mmol/Lトリス塩酸を含む0.25mol/L蔗糖液(pH6.8)中でポリトロン(キネマティカ)を用いてホモジナイズした。得られたホモジネートを20,000×gで30分間遠心分離後、上清を100,000×gで90分間遠心分離した。沈殿部を0.25mol/L蔗糖液で懸濁後、7.5%フィコールを含む0.25mol/L蔗糖液上に重層し、100,000×gで5時間遠心分離した。両層の界面部画分を回収し、0.25mol/L蔗糖液で遠心洗浄を行った。得られたミクロソーム画分はプロトン、カリウム−アデノシントリホスファターゼ標品として用いた。
Advanced Chemistry Developmet,Inc.のPhyschem Batch(Ver.10)を用いてpKa値を計算により求めた。その結果を表4に示す。
ATP含量試験
ヒト肝ガン由来細胞株HepG2(ATCC No.HB−8065)は、10%ウシ胎児血清(fetal bovine serum;FBS;TRACE SCIENTIFIC LTD.)、1mmol/Lピルビン酸ナトリウム(Invitrogen)、2mmol/L L−グルタミン(Invitrogen)、50IU/mLペニシリン(Invitrogen)、50μg/mLストレプトマイシン(Invitrogen)を含むDulbecco’s modified Eagle medium(DMEM;Invitrogen)培地を用いて5%CO2、37℃で維持・継代した。被検試薬は、DMSOにて10mMに調製し、さらに、0.5%FBS、1mmol/Lピルビン酸ナトリウム、2mmol/L L−グルタミン、50IU/mLペニシリン、50μg/mLストレプトマイシンを含むDMEM培地を用いてDMSOの最終濃度が0.1%となるように希釈した。96ウェル白色プレート(Costar)上でHepG2(2x104cells/well)を被検試薬と共に5%CO2、37℃で培養した。1日培養後、細胞内のATP含量をATPLiteTM(PerkinElmer Life Sciences)を用いて測定した。その結果をコントロール(薬物無添加)に対する相対値(%)として表4に示す(n≧3,平均値±SD)。
Caspase−3/7活性試験
試験例3と同様の方法で1日培養した細胞内のCaspase−3/7活性をCaspase−Glo 3/7Assay(Promega)を用いて測定した。その結果をスタウロスポリンを曝露させた時のCaspase−3/7活性の最大値を100%、被検試薬無添加時の活性を0%とした各被検試薬の相対的な活性(%)として表4に示す(n≧3,平均値±SD)。
麻酔ラット胃灌流モデルにおける灌流液pH測定
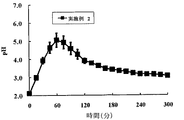
8週齢のJcl:SD雄性ラットを約24時間絶食後、実験に用いた。被験薬物はDMAA:PEG400=1:1液にて溶解し、1mL/kgの投与量になるように調製した。ウレタン(1.2 g/kg、i.p.)麻酔下に十二指腸および前胃部から胃内にカニューレを挿入し、食道部を結紮して生理食塩液(0.5 mL/min)で胃灌流を行った。灌流液は微量流通形ガラス電極(6961−15Cおよび2461A−15T,HORIBA)を用いて連続的にpHの測定を行った。ヒスタミン2塩酸塩8 mg/kg/hを1時間以上静脈内持続投与してpHが安定した後、被験薬物を静脈内投与した。被験薬物投与後5時間後まで灌流液pHの測定を行った。その結果を図1、2、3に示す。
Claims (9)
- 式(I):
[式中、R2は、ハロゲン原子、ハロゲンで置換されていてもよいC1−6アルキル基、またはハロゲンで置換されていてもよいC1−6アルコキシ基を示す。]で表される化合物またはその塩。 - R2が、C1−6アルキル基、またはC1−6アルコキシ基である請求項1記載の化合物またはその塩。
- 1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メチルピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N-メチルメタンアミンまたはその塩。
- 1−{4−フルオロ−5−(2−フルオロピリジン−3−イル)−1−[(4−メトキシピリジン−2−イル)スルホニル]−1H−ピロール−3−イル}−N−メチルメタンアミンまたはその塩。
- 請求項1記載の化合物またはその塩を含有してなる医薬。
- 酸分泌抑制剤である請求項5記載の医薬。
- カリウムイオン競合型アシッドブロッカー(Potassium−Competitive Acid Blocker)である請求項5記載の医薬。
- 消化性潰瘍、ゾリンジャー・エリソン(Zollinger−Ellison)症候群、胃炎、逆流性食道炎、症候性胃食道逆流症(Symptomatic Gastroesophageal Reflux Disease(Symptomatic GERD))、Barrett食道、機能性ディスペプシア(Functional Dyspepsia)、胃癌、胃MALTリンパ腫、非ステロイド系抗炎症剤に起因する潰瘍あるいは手術後ストレスによる胃酸過多ならびに潰瘍の治療もしくは予防剤;または消化性潰瘍、急性ストレス潰瘍、出血性胃炎あるいは侵襲ストレスによる上部消化管出血の抑制剤である請求項5記載の医薬。
- 消化性潰瘍、ゾリンジャー・エリソン(Zollinger−Ellison)症候群、胃炎、逆流性食道炎、症候性胃食道逆流症(Symptomatic Gastroesophageal Reflux Disease(Symptomatic GERD))、Barrett食道、機能性ディスペプシア(Functional Dyspepsia)、胃癌、胃MALTリンパ腫、非ステロイド系抗炎症剤に起因する潰瘍あるいは手術後ストレスによる胃酸過多ならびに潰瘍の治療もしくは予防剤;または消化性潰瘍、急性ストレス潰瘍、出血性胃炎あるいは侵襲ストレスによる上部消化管出血の抑制剤を製造するための請求項1記載の化合物またはその塩の使用。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011508745A JP5638515B2 (ja) | 2008-08-27 | 2009-08-26 | ピロール化合物 |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008218851 | 2008-08-27 | ||
| JP2008218851 | 2008-08-27 | ||
| JP2008269099 | 2008-10-17 | ||
| JP2008269099 | 2008-10-17 | ||
| JP2011508745A JP5638515B2 (ja) | 2008-08-27 | 2009-08-26 | ピロール化合物 |
| PCT/JP2009/065279 WO2010024451A1 (en) | 2008-08-27 | 2009-08-26 | Pyrrole compounds |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2012501296A JP2012501296A (ja) | 2012-01-19 |
| JP2012501296A5 JP2012501296A5 (ja) | 2012-09-13 |
| JP5638515B2 true JP5638515B2 (ja) | 2014-12-10 |
Family
ID=41508823
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011508745A Active JP5638515B2 (ja) | 2008-08-27 | 2009-08-26 | ピロール化合物 |
Country Status (39)
| Country | Link |
|---|---|
| US (3) | US8592597B2 (ja) |
| EP (1) | EP2318390B1 (ja) |
| JP (1) | JP5638515B2 (ja) |
| KR (1) | KR101629181B1 (ja) |
| CN (1) | CN102197033B (ja) |
| AR (1) | AR073136A1 (ja) |
| AU (1) | AU2009284867B2 (ja) |
| BR (1) | BRPI0917705A2 (ja) |
| CA (1) | CA2735162C (ja) |
| CL (1) | CL2011000402A1 (ja) |
| CO (1) | CO6321166A2 (ja) |
| CR (1) | CR20110169A (ja) |
| CY (1) | CY1114452T1 (ja) |
| DK (1) | DK2318390T3 (ja) |
| DO (1) | DOP2011000061A (ja) |
| EA (1) | EA019741B1 (ja) |
| EC (1) | ECSP11010912A (ja) |
| ES (1) | ES2423289T3 (ja) |
| GE (1) | GEP20135786B (ja) |
| HK (1) | HK1157756A1 (ja) |
| HR (1) | HRP20130653T1 (ja) |
| IL (1) | IL211335A (ja) |
| JO (1) | JO2871B1 (ja) |
| MA (1) | MA32647B1 (ja) |
| ME (1) | ME01598B (ja) |
| MX (1) | MX2011001955A (ja) |
| MY (1) | MY152008A (ja) |
| NZ (1) | NZ591793A (ja) |
| PE (2) | PE20110300A1 (ja) |
| PL (1) | PL2318390T3 (ja) |
| PT (1) | PT2318390E (ja) |
| RS (1) | RS52872B (ja) |
| SI (1) | SI2318390T1 (ja) |
| SM (1) | SMT201300085B (ja) |
| TW (1) | TWI473798B (ja) |
| UA (1) | UA105185C2 (ja) |
| UY (1) | UY32074A (ja) |
| WO (1) | WO2010024451A1 (ja) |
| ZA (1) | ZA201101774B (ja) |
Families Citing this family (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AR073136A1 (es) * | 2008-08-27 | 2010-10-13 | Takeda Pharmaceutical | Compuestos de pirrol |
| US20110229879A1 (en) * | 2010-03-19 | 2011-09-22 | University Of Rochester | Methods and compositions for nuclear staining |
| CN102452979B (zh) * | 2010-11-03 | 2015-04-01 | 天津药明康德新药开发有限公司 | 5-氟-3-吡啶磺酰氯的合成方法 |
| CN102863371B (zh) * | 2011-07-06 | 2016-04-13 | 中国科学院上海有机化学研究所 | 氟代二氢吡咯或氟代吡咯 |
| US10039813B2 (en) | 2012-02-07 | 2018-08-07 | Massachusetts Institute Of Technology | Use of antagonists of ghrelin or ghrelin receptor to prevent or treat stress-sensitive psychiatric illness |
| NZ703528A (en) | 2012-06-27 | 2016-08-26 | Takeda Pharmaceutical | Liquid preparations of amines and organic acids stabilized by salts |
| EP2963019B1 (en) | 2013-02-28 | 2020-08-12 | Takeda Pharmaceutical Company Limited | Method for producing pyridine-3-sulfonyl chloride |
| US9724396B2 (en) | 2013-03-15 | 2017-08-08 | Massachusetts Institute Of Technology | Use of antagonists of growth hormone or growth hormone receptor to prevent or treat stress-sensitive psychiatric illness |
| KR102084185B1 (ko) * | 2013-08-29 | 2020-03-04 | 주식회사 대웅제약 | 테트라히드로사이클로펜타피롤 유도체 및 이의 제조방법 |
| CN105367550A (zh) * | 2014-08-11 | 2016-03-02 | 江苏柯菲平医药股份有限公司 | 四氢环戊二烯并[c]吡咯类衍生物、其制备方法及其在医药上的应用 |
| UY36547A (es) | 2015-02-05 | 2016-06-01 | Bayer Cropscience Ag | Derivados heterocíclicos condensados bicíclicos sustituidos por 2-(het)arilo como pesticidas |
| WO2016138099A1 (en) | 2015-02-24 | 2016-09-01 | Massachusetts Institute Of Technology | Use of ghrelin or functional ghrelin receptor agonists to prevent and treat stress-sensitive psychiatric illness |
| TWI710379B (zh) | 2015-07-30 | 2020-11-21 | 日商武田藥品工業股份有限公司 | 錠劑 |
| US10667517B2 (en) | 2015-08-07 | 2020-06-02 | Bayer Cropscience Aktiengesellschaft | 2-(Het)aryl-substituted fused heterocycle derivatives as pesticides |
| CN106430006B (zh) * | 2016-08-18 | 2018-06-12 | 国网湖南省电力公司带电作业中心 | 配电线路移动式绝缘登高作业装置及方法 |
| CN108203430A (zh) * | 2016-12-16 | 2018-06-26 | 天地人和生物科技有限公司 | 一种新型可逆性质子泵抑制剂及其制备方法和用途 |
| PE20200442A1 (es) | 2017-07-10 | 2020-02-28 | Takeda Pharmaceuticals Co | Preparacion que comprende vonoprazan |
| CN113527536B (zh) * | 2020-04-21 | 2024-03-22 | 杭州德柯医疗科技有限公司 | 一种含氟多糖高分子化合物及其制备方法 |
| KR20210156235A (ko) * | 2020-06-17 | 2021-12-24 | 일동제약(주) | 신규한 산 분비 억제제 및 이의 용도 |
| EP4074317A1 (en) | 2021-04-14 | 2022-10-19 | Bayer AG | Phosphorus derivatives as novel sos1 inhibitors |
| CN113620930B (zh) * | 2021-07-12 | 2022-08-16 | 南京烁慧医药科技有限公司 | 一种含磺酰胺结构的化合物及其制备方法和应用、一种药物组合物及应用 |
| WO2023113474A1 (ko) * | 2021-12-15 | 2023-06-22 | 일동제약(주) | 1-설포닐 피롤 유도체의 신규 염, 이의 제조 방법 및 이를 포함하는 약학 조성물 |
| WO2023113458A1 (en) * | 2021-12-15 | 2023-06-22 | Ildong Pharmaceutical Co., Ltd. | Novel salt of 1-sulfonyl pyrrole derivative, preparation method thereof and pharmaceutical composition comprising thereof |
| KR20230102353A (ko) * | 2021-12-30 | 2023-07-07 | 주식회사 대웅제약 | 삼중음성유방암의 예방 또는 치료용 약학적 조성물 |
Family Cites Families (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0259085B1 (en) | 1986-08-29 | 1991-08-21 | Pfizer Inc. | 2-guanidino-4-arylthiazoles for treatment of peptic ulcers |
| US5128366A (en) | 1990-07-05 | 1992-07-07 | Shinogi & Co., Ltd. | Pyrrole derivatives |
| US5331006A (en) * | 1990-08-31 | 1994-07-19 | Warner-Lambert Company | Amino acid analogs as CCK antagonists |
| WO1992004025A1 (en) | 1990-08-31 | 1992-03-19 | Warner-Lambert Company | Amino acid analogs as cck antagonists |
| HU219131B (hu) | 1991-10-18 | 2001-02-28 | Monsanto Co. | Módszer és fungicid készítmény növények torsgombabetegségének gátlására és a hatóanyagok |
| MX9205392A (es) | 1991-10-29 | 1993-04-01 | Du Pont | Triazolcarbozamidas herbicidas y procedimiento para su obtencion. |
| JPH06135961A (ja) | 1992-10-23 | 1994-05-17 | Nippon Iyakuhin Kogyo Kk | 新規ジフェニルピロリルフラン誘導体 |
| US5286742A (en) | 1992-11-03 | 1994-02-15 | American Cyanamid Company | Pyrrole thiocarboxamide insecticidal and acaricidal agents |
| US5480902A (en) | 1993-08-31 | 1996-01-02 | American Cyanamid Company | Thienylpyrrole fungicidal agents |
| JPH08119936A (ja) | 1994-10-18 | 1996-05-14 | Fujisawa Pharmaceut Co Ltd | 複素環式誘導体 |
| JPH0930967A (ja) | 1995-07-17 | 1997-02-04 | Chugai Pharmaceut Co Ltd | 抗潰瘍剤 |
| RU2221782C2 (ru) | 1996-08-28 | 2004-01-20 | Дзе Проктер Энд Гэмбл Компани | Замещенные циклические аминовые ингибиторы металлопротеаз |
| SK86699A3 (en) | 1996-12-23 | 2000-11-07 | Du Pont Pharm Co | Nitrogen containing heteroaromatics as factor xa inhibitors |
| JPH11209344A (ja) | 1998-01-26 | 1999-08-03 | Kyowa Hakko Kogyo Co Ltd | 含窒素複素環化合物 |
| GB9817548D0 (en) * | 1998-08-12 | 1998-10-07 | Novartis Ag | Organic compounds |
| US7105564B1 (en) | 1999-03-10 | 2006-09-12 | Shionogi & Co., Ltd. | Pharmaceutical composition comprising a dual antagonist against PGD2/TXA2 receptors having a [2.2.1] or [3.1.1] bicyclic skeleton |
| TW575561B (en) | 1999-03-25 | 2004-02-11 | Hoffmann La Roche | 1-arenesulfonyl-2-aryl-pyrrolidine and piperidine derivatives |
| AU3783100A (en) | 1999-06-14 | 2000-12-21 | Dow Agrosciences Llc | Substituted triazoles, imidazoles and pyrazoles as herbicides |
| DE60024120T2 (de) * | 1999-08-26 | 2006-07-27 | Aventis Pharmaceuticals Inc. | Substituierte (aminoiminomethyl oder aminomethyl) dihydrobenzofurane und benozopyrane |
| US6911468B2 (en) | 2000-05-22 | 2005-06-28 | Takeda Chemical Industries, Ltd. | Tyrosine phosphatase inhibitors |
| US6589978B2 (en) | 2000-06-30 | 2003-07-08 | Hoffman-La Roche Inc. | 1-sulfonyl pyrrolidine derivatives |
| GB0016453D0 (en) | 2000-07-04 | 2000-08-23 | Hoffmann La Roche | Pyrrole derivatives |
| CN1582281A (zh) | 2001-10-01 | 2005-02-16 | 大正制药株式会社 | Mch受体拮抗剂 |
| MXPA04004464A (es) | 2001-11-08 | 2004-08-11 | Upjohn Co | Compuestos de heteroarilo sustituido con azabiciclo para el tratamiento de enfermedades. |
| AU2002342909A1 (en) | 2001-11-22 | 2003-06-10 | Ciba Specialty Chemicals Holding Inc. | Pyrrole synthesis |
| US20050101657A1 (en) | 2001-12-28 | 2005-05-12 | Takeda Chemical Industries Ltd. | Androgen receptor antagonists |
| AU2003216274A1 (en) | 2002-02-11 | 2003-09-04 | Neurocrine Biosciences, Inc. | Pyrrole derivatives as ligands of melanocortin receptors |
| AU2003209114A1 (en) * | 2002-02-14 | 2003-09-04 | Wyeth | Pyrrolylalkylidene-hydrazinecarboximidamide derivatives as 5-hydroxytryptamine-6 ligands |
| TW200306191A (en) | 2002-02-22 | 2003-11-16 | Teijin Ltd | Pyrrolopyrimidine derivatives |
| BR0311707A (pt) | 2002-06-13 | 2005-03-15 | Du Pont | Composto, composição e método de controle de pelo menos uma praga invertebrada |
| CA2495216A1 (en) | 2002-08-12 | 2004-02-19 | Sugen, Inc. | 3-pyrrolyl-pyridopyrazoles and 3-pyrrolyl-indazoles as novel kinase inhibitors |
| JP2004315511A (ja) | 2003-03-31 | 2004-11-11 | Taisho Pharmaceut Co Ltd | Mch受容体アンタゴニスト |
| EP1628957B1 (en) | 2003-05-26 | 2010-09-29 | Takeda Pharmaceutical Company Limited | Sulfopyrroles |
| JP5173192B2 (ja) | 2004-09-30 | 2013-03-27 | 武田薬品工業株式会社 | プロトンポンプ阻害薬 |
| EP1655284A1 (en) | 2004-10-26 | 2006-05-10 | Aponetics AG | 2-Phenylsulfopyrroles |
| RS52473B9 (sr) * | 2005-08-30 | 2019-07-31 | Takeda Pharmaceuticals Co | 1-heterociklilsulfonil, 2-aminometil, 5-(hetero-)aril supstituisani derivati 1-h-pirola kao inhibitori sekrecije kiseline |
| US8512761B2 (en) * | 2006-01-27 | 2013-08-20 | Yale University | Fast acting inhibitor of gastric acid secretion |
| US7994205B2 (en) * | 2006-03-31 | 2011-08-09 | Takeda Pharmaceutical Company Limited | Aryl-or heteroaryl-sulfonyl compounds as acid secretion inhibitors |
| US8933105B2 (en) * | 2007-02-28 | 2015-01-13 | Takeda Pharmaceutical Company Limited | Pyrrole compounds |
| KR101558011B1 (ko) | 2007-09-28 | 2015-10-06 | 다케다 야쿠힌 고교 가부시키가이샤 | 양성자 펌프 억제제로서의 5-원 이종원자고리 화합물 |
| US20090318429A1 (en) * | 2008-04-28 | 2009-12-24 | Institute For Oneworld Health | Compounds, Compositions and Methods Comprising Heteroaromatic Derivatives |
| CL2009001203A1 (es) | 2008-05-16 | 2009-10-23 | Takeda San Diego Inc | Compuestos derivados de indazol y pirazol sustituidos; composicion farmaceutica de dichos compuestos; kit farmaceutico; y su uso como activadores de la glucoquinasa para tratar enfermedades metabolicas tales como hiperglicemia, diabetes, dislipidemias, obesidad, sindrome metabolico x y enfermedades cardiovasculares. |
| AR073136A1 (es) * | 2008-08-27 | 2010-10-13 | Takeda Pharmaceutical | Compuestos de pirrol |
-
2009
- 2009-08-26 AR ARP090103278A patent/AR073136A1/es unknown
- 2009-08-26 RS RS20130319A patent/RS52872B/en unknown
- 2009-08-26 TW TW98128638A patent/TWI473798B/zh not_active IP Right Cessation
- 2009-08-26 PL PL09788053T patent/PL2318390T3/pl unknown
- 2009-08-26 UA UAA201103600A patent/UA105185C2/uk unknown
- 2009-08-26 AU AU2009284867A patent/AU2009284867B2/en not_active Ceased
- 2009-08-26 WO PCT/JP2009/065279 patent/WO2010024451A1/en active Application Filing
- 2009-08-26 US US13/061,078 patent/US8592597B2/en active Active
- 2009-08-26 ES ES09788053T patent/ES2423289T3/es active Active
- 2009-08-26 MY MYPI20110861 patent/MY152008A/en unknown
- 2009-08-26 EP EP09788053.8A patent/EP2318390B1/en active Active
- 2009-08-26 PT PT97880538T patent/PT2318390E/pt unknown
- 2009-08-26 PE PE2011000194A patent/PE20110300A1/es not_active Application Discontinuation
- 2009-08-26 JP JP2011508745A patent/JP5638515B2/ja active Active
- 2009-08-26 PE PE2013002359A patent/PE20141174A1/es not_active Application Discontinuation
- 2009-08-26 DK DK09788053.8T patent/DK2318390T3/da active
- 2009-08-26 UY UY0001032074A patent/UY32074A/es not_active Application Discontinuation
- 2009-08-26 CA CA2735162A patent/CA2735162C/en not_active Expired - Fee Related
- 2009-08-26 CN CN200980142371.0A patent/CN102197033B/zh active Active
- 2009-08-26 ME MEP-2013-90A patent/ME01598B/me unknown
- 2009-08-26 JO JO2009322A patent/JO2871B1/en active
- 2009-08-26 KR KR1020117006779A patent/KR101629181B1/ko active IP Right Grant
- 2009-08-26 BR BRPI0917705A patent/BRPI0917705A2/pt not_active IP Right Cessation
- 2009-08-26 US US12/547,778 patent/US8969387B2/en active Active
- 2009-08-26 MX MX2011001955A patent/MX2011001955A/es active IP Right Grant
- 2009-08-26 EA EA201170384A patent/EA019741B1/ru not_active IP Right Cessation
- 2009-08-26 GE GEAP200912153A patent/GEP20135786B/en unknown
- 2009-08-26 SI SI200930654T patent/SI2318390T1/sl unknown
- 2009-08-26 NZ NZ591793A patent/NZ591793A/xx not_active IP Right Cessation
-
2011
- 2011-02-21 IL IL211335A patent/IL211335A/en not_active IP Right Cessation
- 2011-02-24 CL CL2011000402A patent/CL2011000402A1/es unknown
- 2011-02-25 DO DO2011000061A patent/DOP2011000061A/es unknown
- 2011-03-08 ZA ZA2011/01774A patent/ZA201101774B/en unknown
- 2011-03-18 MA MA33715A patent/MA32647B1/fr unknown
- 2011-03-25 CO CO11036583A patent/CO6321166A2/es active IP Right Grant
- 2011-03-25 CR CR20110169A patent/CR20110169A/es unknown
- 2011-03-25 EC EC2011010912A patent/ECSP11010912A/es unknown
- 2011-11-04 HK HK11111932.5A patent/HK1157756A1/xx not_active IP Right Cessation
-
2013
- 2013-07-10 HR HRP20130653AT patent/HRP20130653T1/hr unknown
- 2013-07-26 CY CY20131100636T patent/CY1114452T1/el unknown
- 2013-07-26 SM SM201300085T patent/SMT201300085B/xx unknown
- 2013-10-01 US US14/043,018 patent/US8993598B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5638515B2 (ja) | ピロール化合物 | |
| JP5687293B2 (ja) | 酸分泌抑制薬 | |
| EP2114917B1 (en) | Pyrrole compounds | |
| EP3162798B1 (en) | 5-membered heterocyclic compound | |
| WO2007114338A1 (ja) | 酸分泌抑制薬 | |
| JP5552481B2 (ja) | ピラゾール化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120726 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20120726 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140128 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140319 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20140924 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20141022 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5638515 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |