JP2017535271A - ガイドrnaのペアを使用したターゲティングによる遺伝子改変の方法及び組成物 - Google Patents
ガイドrnaのペアを使用したターゲティングによる遺伝子改変の方法及び組成物 Download PDFInfo
- Publication number
- JP2017535271A JP2017535271A JP2017527267A JP2017527267A JP2017535271A JP 2017535271 A JP2017535271 A JP 2017535271A JP 2017527267 A JP2017527267 A JP 2017527267A JP 2017527267 A JP2017527267 A JP 2017527267A JP 2017535271 A JP2017535271 A JP 2017535271A
- Authority
- JP
- Japan
- Prior art keywords
- crispr rna
- sequence
- cell
- rna recognition
- nucleic acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 238000000034 method Methods 0.000 title claims abstract description 384
- 230000008685 targeting Effects 0.000 title claims abstract description 276
- 108020005004 Guide RNA Proteins 0.000 title claims abstract description 167
- 239000000203 mixture Substances 0.000 title abstract description 22
- 238000012239 gene modification Methods 0.000 title description 24
- 230000005017 genetic modification Effects 0.000 title description 22
- 235000013617 genetically modified food Nutrition 0.000 title description 22
- 108090000623 proteins and genes Proteins 0.000 claims abstract description 465
- 102000004169 proteins and genes Human genes 0.000 claims abstract description 285
- 108700028369 Alleles Proteins 0.000 claims abstract description 230
- 241000282414 Homo sapiens Species 0.000 claims abstract description 134
- 230000004075 alteration Effects 0.000 claims abstract description 24
- 230000003834 intracellular effect Effects 0.000 claims abstract description 19
- 108091079001 CRISPR RNA Proteins 0.000 claims description 558
- 210000004027 cell Anatomy 0.000 claims description 557
- 150000007523 nucleic acids Chemical class 0.000 claims description 327
- 102000039446 nucleic acids Human genes 0.000 claims description 279
- 108020004707 nucleic acids Proteins 0.000 claims description 279
- 108020004414 DNA Proteins 0.000 claims description 249
- 102000053602 DNA Human genes 0.000 claims description 224
- 210000000349 chromosome Anatomy 0.000 claims description 179
- 239000002773 nucleotide Substances 0.000 claims description 156
- 125000003729 nucleotide group Chemical group 0.000 claims description 156
- 239000013598 vector Substances 0.000 claims description 128
- 108091028113 Trans-activating crRNA Proteins 0.000 claims description 116
- 230000014509 gene expression Effects 0.000 claims description 116
- 239000000523 sample Substances 0.000 claims description 116
- 238000012217 deletion Methods 0.000 claims description 104
- 230000037430 deletion Effects 0.000 claims description 104
- 230000004048 modification Effects 0.000 claims description 99
- 238000012986 modification Methods 0.000 claims description 99
- 230000000694 effects Effects 0.000 claims description 80
- 210000001161 mammalian embryo Anatomy 0.000 claims description 69
- 108091033409 CRISPR Proteins 0.000 claims description 67
- 230000005782 double-strand break Effects 0.000 claims description 63
- 230000035772 mutation Effects 0.000 claims description 60
- 238000006243 chemical reaction Methods 0.000 claims description 54
- 238000003780 insertion Methods 0.000 claims description 52
- 230000037431 insertion Effects 0.000 claims description 52
- 101710163270 Nuclease Proteins 0.000 claims description 47
- 230000027455 binding Effects 0.000 claims description 42
- 238000005215 recombination Methods 0.000 claims description 39
- 230000006798 recombination Effects 0.000 claims description 39
- 230000006780 non-homologous end joining Effects 0.000 claims description 38
- 108010008532 Deoxyribonuclease I Proteins 0.000 claims description 37
- 102000007260 Deoxyribonuclease I Human genes 0.000 claims description 37
- 210000003527 eukaryotic cell Anatomy 0.000 claims description 27
- 241000283984 Rodentia Species 0.000 claims description 21
- 230000001965 increasing effect Effects 0.000 claims description 21
- 210000005260 human cell Anatomy 0.000 claims description 20
- 210000004962 mammalian cell Anatomy 0.000 claims description 20
- 108091032973 (ribonucleotides)n+m Proteins 0.000 claims description 16
- 230000001939 inductive effect Effects 0.000 claims description 16
- 201000010099 disease Diseases 0.000 claims description 15
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 15
- 108020004999 messenger RNA Proteins 0.000 claims description 15
- 230000007423 decrease Effects 0.000 claims description 12
- 102000003960 Ligases Human genes 0.000 claims description 10
- 108090000364 Ligases Proteins 0.000 claims description 10
- 210000000130 stem cell Anatomy 0.000 claims description 9
- 230000034431 double-strand break repair via homologous recombination Effects 0.000 claims description 8
- 210000004504 adult stem cell Anatomy 0.000 claims description 7
- 210000002230 centromere Anatomy 0.000 claims description 7
- 238000004519 manufacturing process Methods 0.000 claims description 7
- 230000002441 reversible effect Effects 0.000 claims description 7
- 108020004682 Single-Stranded DNA Proteins 0.000 claims description 6
- 210000001840 diploid cell Anatomy 0.000 claims description 6
- 241000282412 Homo Species 0.000 claims description 4
- 206010059866 Drug resistance Diseases 0.000 claims description 3
- 108010064218 Poly (ADP-Ribose) Polymerase-1 Proteins 0.000 claims 1
- 102000015087 Poly (ADP-Ribose) Polymerase-1 Human genes 0.000 claims 1
- 235000018102 proteins Nutrition 0.000 description 249
- 238000003776 cleavage reaction Methods 0.000 description 114
- 230000007017 scission Effects 0.000 description 114
- 241000699666 Mus <mouse, genus> Species 0.000 description 99
- 229920002477 rna polymer Polymers 0.000 description 60
- 238000003556 assay Methods 0.000 description 53
- 238000002744 homologous recombination Methods 0.000 description 47
- 230000006801 homologous recombination Effects 0.000 description 47
- 241000700159 Rattus Species 0.000 description 46
- 108091028043 Nucleic acid sequence Proteins 0.000 description 44
- 230000000295 complement effect Effects 0.000 description 38
- 241001465754 Metazoa Species 0.000 description 31
- 102000040430 polynucleotide Human genes 0.000 description 27
- 108091033319 polynucleotide Proteins 0.000 description 27
- 239000002157 polynucleotide Substances 0.000 description 27
- 230000001105 regulatory effect Effects 0.000 description 25
- 210000002257 embryonic structure Anatomy 0.000 description 23
- 108091060290 Chromatid Proteins 0.000 description 20
- 210000004756 chromatid Anatomy 0.000 description 20
- 238000009396 hybridization Methods 0.000 description 19
- 230000001404 mediated effect Effects 0.000 description 17
- 210000001519 tissue Anatomy 0.000 description 17
- 102100021386 Trans-acting T-cell-specific transcription factor GATA-3 Human genes 0.000 description 16
- 238000006467 substitution reaction Methods 0.000 description 16
- 108010091086 Recombinases Proteins 0.000 description 14
- 239000003795 chemical substances by application Substances 0.000 description 14
- 230000006870 function Effects 0.000 description 14
- 230000007246 mechanism Effects 0.000 description 14
- 230000008439 repair process Effects 0.000 description 14
- 238000012216 screening Methods 0.000 description 14
- 230000002068 genetic effect Effects 0.000 description 13
- 108090000765 processed proteins & peptides Proteins 0.000 description 13
- 230000010076 replication Effects 0.000 description 13
- 230000014759 maintenance of location Effects 0.000 description 12
- -1 Csm2 Proteins 0.000 description 11
- 210000000287 oocyte Anatomy 0.000 description 11
- 229920001184 polypeptide Polymers 0.000 description 11
- 102000004196 processed proteins & peptides Human genes 0.000 description 11
- 238000002474 experimental method Methods 0.000 description 10
- 230000010354 integration Effects 0.000 description 10
- 230000013011 mating Effects 0.000 description 10
- 108020004705 Codon Proteins 0.000 description 9
- 241000699800 Cricetinae Species 0.000 description 9
- 108010077850 Nuclear Localization Signals Proteins 0.000 description 9
- 102000018120 Recombinases Human genes 0.000 description 9
- 239000003814 drug Substances 0.000 description 9
- 230000008569 process Effects 0.000 description 9
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 8
- 241000699670 Mus sp. Species 0.000 description 8
- 235000001014 amino acid Nutrition 0.000 description 8
- 238000010586 diagram Methods 0.000 description 8
- 239000012634 fragment Substances 0.000 description 8
- 210000004940 nucleus Anatomy 0.000 description 8
- 238000003753 real-time PCR Methods 0.000 description 8
- 241000124008 Mammalia Species 0.000 description 7
- 208000009869 Neu-Laxova syndrome Diseases 0.000 description 7
- 108091005461 Nucleic proteins Proteins 0.000 description 7
- 108700008625 Reporter Genes Proteins 0.000 description 7
- 210000004436 artificial bacterial chromosome Anatomy 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 229940079593 drug Drugs 0.000 description 7
- 239000003112 inhibitor Substances 0.000 description 7
- 239000003550 marker Substances 0.000 description 7
- 230000000394 mitotic effect Effects 0.000 description 7
- 210000000472 morula Anatomy 0.000 description 7
- 230000005783 single-strand break Effects 0.000 description 7
- 108091035539 telomere Proteins 0.000 description 7
- 210000003411 telomere Anatomy 0.000 description 7
- 102000055501 telomere Human genes 0.000 description 7
- 238000013518 transcription Methods 0.000 description 7
- 230000035897 transcription Effects 0.000 description 7
- 238000001890 transfection Methods 0.000 description 7
- 108091026890 Coding region Proteins 0.000 description 6
- 108091027544 Subgenomic mRNA Proteins 0.000 description 6
- 125000003275 alpha amino acid group Chemical group 0.000 description 6
- 210000001106 artificial yeast chromosome Anatomy 0.000 description 6
- 210000002459 blastocyst Anatomy 0.000 description 6
- 210000000805 cytoplasm Anatomy 0.000 description 6
- 230000004927 fusion Effects 0.000 description 6
- 230000001976 improved effect Effects 0.000 description 6
- 238000000338 in vitro Methods 0.000 description 6
- 238000000520 microinjection Methods 0.000 description 6
- 238000012546 transfer Methods 0.000 description 6
- 101150113197 CMAH gene Proteins 0.000 description 5
- 101100300807 Drosophila melanogaster spn-A gene Proteins 0.000 description 5
- 108091029865 Exogenous DNA Proteins 0.000 description 5
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 5
- 102000004144 Green Fluorescent Proteins Human genes 0.000 description 5
- 238000002944 PCR assay Methods 0.000 description 5
- 241001494479 Pecora Species 0.000 description 5
- 229940024606 amino acid Drugs 0.000 description 5
- 150000001413 amino acids Chemical class 0.000 description 5
- 210000004102 animal cell Anatomy 0.000 description 5
- 230000002759 chromosomal effect Effects 0.000 description 5
- 210000004602 germ cell Anatomy 0.000 description 5
- 230000037361 pathway Effects 0.000 description 5
- 108010054624 red fluorescent protein Proteins 0.000 description 5
- 230000002829 reductive effect Effects 0.000 description 5
- 238000011160 research Methods 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- 241000271566 Aves Species 0.000 description 4
- 241000283690 Bos taurus Species 0.000 description 4
- 101100220616 Caenorhabditis elegans chk-2 gene Proteins 0.000 description 4
- 101100440312 Mus musculus C5 gene Proteins 0.000 description 4
- 101100113907 Mus musculus Clspn gene Proteins 0.000 description 4
- 241000282898 Sus scrofa Species 0.000 description 4
- 102000040945 Transcription factor Human genes 0.000 description 4
- 108091023040 Transcription factor Proteins 0.000 description 4
- 101100113908 Xenopus laevis clspn gene Proteins 0.000 description 4
- 230000002159 abnormal effect Effects 0.000 description 4
- 125000000539 amino acid group Chemical group 0.000 description 4
- 238000000137 annealing Methods 0.000 description 4
- 108091005948 blue fluorescent proteins Proteins 0.000 description 4
- 239000002131 composite material Substances 0.000 description 4
- 238000007796 conventional method Methods 0.000 description 4
- 230000006378 damage Effects 0.000 description 4
- 210000002950 fibroblast Anatomy 0.000 description 4
- 108091006047 fluorescent proteins Proteins 0.000 description 4
- 102000034287 fluorescent proteins Human genes 0.000 description 4
- 238000012224 gene deletion Methods 0.000 description 4
- 239000005090 green fluorescent protein Substances 0.000 description 4
- 230000012447 hatching Effects 0.000 description 4
- 238000010348 incorporation Methods 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- 108091070501 miRNA Proteins 0.000 description 4
- 239000002679 microRNA Substances 0.000 description 4
- 230000035699 permeability Effects 0.000 description 4
- 230000001737 promoting effect Effects 0.000 description 4
- 230000007026 protein scission Effects 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 239000004055 small Interfering RNA Substances 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 230000009261 transgenic effect Effects 0.000 description 4
- 108091005957 yellow fluorescent proteins Proteins 0.000 description 4
- 210000004340 zona pellucida Anatomy 0.000 description 4
- 241000282472 Canis lupus familiaris Species 0.000 description 3
- 241000283707 Capra Species 0.000 description 3
- 241000282994 Cervidae Species 0.000 description 3
- 108050008316 DNA endonuclease RBBP8 Proteins 0.000 description 3
- 102100039524 DNA endonuclease RBBP8 Human genes 0.000 description 3
- 230000033616 DNA repair Effects 0.000 description 3
- 102100039116 DNA repair protein RAD50 Human genes 0.000 description 3
- 102100034484 DNA repair protein RAD51 homolog 3 Human genes 0.000 description 3
- 102100033996 Double-strand break repair protein MRE11 Human genes 0.000 description 3
- 241000282326 Felis catus Species 0.000 description 3
- 241000287828 Gallus gallus Species 0.000 description 3
- 101000743929 Homo sapiens DNA repair protein RAD50 Proteins 0.000 description 3
- 101001132271 Homo sapiens DNA repair protein RAD51 homolog 3 Proteins 0.000 description 3
- 101000591400 Homo sapiens Double-strand break repair protein MRE11 Proteins 0.000 description 3
- 101001128138 Homo sapiens NACHT, LRR and PYD domains-containing protein 2 Proteins 0.000 description 3
- 101000981336 Homo sapiens Nibrin Proteins 0.000 description 3
- 102000006496 Immunoglobulin Heavy Chains Human genes 0.000 description 3
- 108010019476 Immunoglobulin Heavy Chains Proteins 0.000 description 3
- 101150090152 Lig1 gene Proteins 0.000 description 3
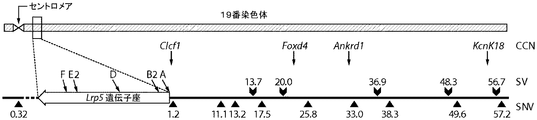
- 101100344029 Mus musculus Lrp5 gene Proteins 0.000 description 3
- 101100155034 Mus musculus Ubap2 gene Proteins 0.000 description 3
- 101100385413 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) csm-3 gene Proteins 0.000 description 3
- 101100355599 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) mus-11 gene Proteins 0.000 description 3
- 102100024403 Nibrin Human genes 0.000 description 3
- 101710179684 Poly [ADP-ribose] polymerase Proteins 0.000 description 3
- 102100023712 Poly [ADP-ribose] polymerase 1 Human genes 0.000 description 3
- 229920000776 Poly(Adenosine diphosphate-ribose) polymerase Polymers 0.000 description 3
- 102000053062 Rad52 DNA Repair and Recombination Human genes 0.000 description 3
- 108700031762 Rad52 DNA Repair and Recombination Proteins 0.000 description 3
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 3
- 241000193996 Streptococcus pyogenes Species 0.000 description 3
- 239000004098 Tetracycline Substances 0.000 description 3
- 101150092793 Trpa1 gene Proteins 0.000 description 3
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 3
- 241000700605 Viruses Species 0.000 description 3
- 102000002258 X-ray Repair Cross Complementing Protein 1 Human genes 0.000 description 3
- 108010000443 X-ray Repair Cross Complementing Protein 1 Proteins 0.000 description 3
- 239000012190 activator Substances 0.000 description 3
- 238000007792 addition Methods 0.000 description 3
- 235000004279 alanine Nutrition 0.000 description 3
- 125000003295 alanine group Chemical group N[C@@H](C)C(=O)* 0.000 description 3
- 230000000692 anti-sense effect Effects 0.000 description 3
- 230000008827 biological function Effects 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 230000025084 cell cycle arrest Effects 0.000 description 3
- 210000000170 cell membrane Anatomy 0.000 description 3
- 235000013330 chicken meat Nutrition 0.000 description 3
- 238000005520 cutting process Methods 0.000 description 3
- 108010082025 cyan fluorescent protein Proteins 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- 210000001900 endoderm Anatomy 0.000 description 3
- 239000003623 enhancer Substances 0.000 description 3
- 108010021843 fluorescent protein 583 Proteins 0.000 description 3
- 108020001507 fusion proteins Proteins 0.000 description 3
- 102000037865 fusion proteins Human genes 0.000 description 3
- 238000010363 gene targeting Methods 0.000 description 3
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 3
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 3
- 210000000688 human artificial chromosome Anatomy 0.000 description 3
- 230000002779 inactivation Effects 0.000 description 3
- 230000003993 interaction Effects 0.000 description 3
- 244000144972 livestock Species 0.000 description 3
- 210000003470 mitochondria Anatomy 0.000 description 3
- 239000000178 monomer Substances 0.000 description 3
- 230000036961 partial effect Effects 0.000 description 3
- 239000013612 plasmid Substances 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- 229910052594 sapphire Inorganic materials 0.000 description 3
- 239000010980 sapphire Substances 0.000 description 3
- 230000035945 sensitivity Effects 0.000 description 3
- 210000001082 somatic cell Anatomy 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 229960002180 tetracycline Drugs 0.000 description 3
- 229930101283 tetracycline Natural products 0.000 description 3
- 235000019364 tetracycline Nutrition 0.000 description 3
- 150000003522 tetracyclines Chemical class 0.000 description 3
- 230000005945 translocation Effects 0.000 description 3
- 238000011144 upstream manufacturing Methods 0.000 description 3
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 2
- YMHOBZXQZVXHBM-UHFFFAOYSA-N 2,5-dimethoxy-4-bromophenethylamine Chemical compound COC1=CC(CCN)=C(OC)C=C1Br YMHOBZXQZVXHBM-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical compound O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 2
- 102000006822 Agouti Signaling Protein Human genes 0.000 description 2
- 108010072151 Agouti Signaling Protein Proteins 0.000 description 2
- 108700020463 BRCA1 Proteins 0.000 description 2
- 102000036365 BRCA1 Human genes 0.000 description 2
- 101150072950 BRCA1 gene Proteins 0.000 description 2
- 102000052609 BRCA2 Human genes 0.000 description 2
- 108700020462 BRCA2 Proteins 0.000 description 2
- 102100026189 Beta-galactosidase Human genes 0.000 description 2
- 101710201279 Biotin carboxyl carrier protein Proteins 0.000 description 2
- 101150008921 Brca2 gene Proteins 0.000 description 2
- 108010077544 Chromatin Proteins 0.000 description 2
- 108700010070 Codon Usage Proteins 0.000 description 2
- 241000484025 Cuniculus Species 0.000 description 2
- 102220605874 Cytosolic arginine sensor for mTORC1 subunit 2_D10A_mutation Human genes 0.000 description 2
- 108010061914 DNA polymerase mu Proteins 0.000 description 2
- 102100027828 DNA repair protein XRCC4 Human genes 0.000 description 2
- 102100029764 DNA-directed DNA/RNA polymerase mu Human genes 0.000 description 2
- 108010053770 Deoxyribonucleases Proteins 0.000 description 2
- 102000016911 Deoxyribonucleases Human genes 0.000 description 2
- 108700024394 Exon Proteins 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- 230000010337 G2 phase Effects 0.000 description 2
- 108010070675 Glutathione transferase Proteins 0.000 description 2
- 101710154606 Hemagglutinin Proteins 0.000 description 2
- 102100029100 Hematopoietic prostaglandin D synthase Human genes 0.000 description 2
- 108091027305 Heteroduplex Proteins 0.000 description 2
- 108010033040 Histones Proteins 0.000 description 2
- 102000006947 Histones Human genes 0.000 description 2
- 101100440311 Homo sapiens C5 gene Proteins 0.000 description 2
- 101000649315 Homo sapiens DNA repair protein XRCC4 Proteins 0.000 description 2
- 101000904868 Homo sapiens Transcriptional regulator ATRX Proteins 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- 108010025815 Kanamycin Kinase Proteins 0.000 description 2
- 241000186660 Lactobacillus Species 0.000 description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 2
- 239000004472 Lysine Substances 0.000 description 2
- 241000282560 Macaca mulatta Species 0.000 description 2
- 108060004795 Methyltransferase Proteins 0.000 description 2
- 102000007474 Multiprotein Complexes Human genes 0.000 description 2
- 108010085220 Multiprotein Complexes Proteins 0.000 description 2
- 101100038118 Mus musculus Ror1 gene Proteins 0.000 description 2
- 101100482469 Mus musculus Trpa1 gene Proteins 0.000 description 2
- YJDAOHJWLUNFLX-UHFFFAOYSA-N NU 1025 Chemical compound C1=CC=C2C(=O)NC(C)=NC2=C1O YJDAOHJWLUNFLX-UHFFFAOYSA-N 0.000 description 2
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- 101710093908 Outer capsid protein VP4 Proteins 0.000 description 2
- 101710135467 Outer capsid protein sigma-1 Proteins 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- 101710176177 Protein A56 Proteins 0.000 description 2
- 102000001195 RAD51 Human genes 0.000 description 2
- 101150006234 RAD52 gene Proteins 0.000 description 2
- 230000004570 RNA-binding Effects 0.000 description 2
- 108010068097 Rad51 Recombinase Proteins 0.000 description 2
- 101100523530 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) RAD55 gene Proteins 0.000 description 2
- 108091027967 Small hairpin RNA Proteins 0.000 description 2
- 108020004459 Small interfering RNA Proteins 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 238000002105 Southern blotting Methods 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- 102000002933 Thioredoxin Human genes 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- 102100023931 Transcriptional regulator ATRX Human genes 0.000 description 2
- 108700019146 Transgenes Proteins 0.000 description 2
- 241000545067 Venus Species 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 230000001580 bacterial effect Effects 0.000 description 2
- 108010005774 beta-Galactosidase Proteins 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 230000022131 cell cycle Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 2
- 102000021178 chitin binding proteins Human genes 0.000 description 2
- 108091011157 chitin binding proteins Proteins 0.000 description 2
- 210000003763 chloroplast Anatomy 0.000 description 2
- 210000003483 chromatin Anatomy 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 210000003981 ectoderm Anatomy 0.000 description 2
- 238000004520 electroporation Methods 0.000 description 2
- 210000001671 embryonic stem cell Anatomy 0.000 description 2
- 210000002889 endothelial cell Anatomy 0.000 description 2
- 230000007159 enucleation Effects 0.000 description 2
- 230000007613 environmental effect Effects 0.000 description 2
- 231100000221 frame shift mutation induction Toxicity 0.000 description 2
- 230000037433 frameshift Effects 0.000 description 2
- 102000054766 genetic haplotypes Human genes 0.000 description 2
- 210000001654 germ layer Anatomy 0.000 description 2
- 239000000185 hemagglutinin Substances 0.000 description 2
- 238000011577 humanized mouse model Methods 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000005304 joining Methods 0.000 description 2
- 210000003292 kidney cell Anatomy 0.000 description 2
- 229940039696 lactobacillus Drugs 0.000 description 2
- 238000012423 maintenance Methods 0.000 description 2
- 241001515942 marmosets Species 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 230000033607 mismatch repair Effects 0.000 description 2
- 238000010369 molecular cloning Methods 0.000 description 2
- 210000005087 mononuclear cell Anatomy 0.000 description 2
- 210000002569 neuron Anatomy 0.000 description 2
- 238000005457 optimization Methods 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 210000001672 ovary Anatomy 0.000 description 2
- 230000008775 paternal effect Effects 0.000 description 2
- RDOWQLZANAYVLL-UHFFFAOYSA-N phenanthridine Chemical compound C1=CC=C2C3=CC=CC=C3C=NC2=C1 RDOWQLZANAYVLL-UHFFFAOYSA-N 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- 210000001778 pluripotent stem cell Anatomy 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 150000004892 pyridazines Chemical class 0.000 description 2
- 238000012207 quantitative assay Methods 0.000 description 2
- 230000007057 regulation of double-strand break repair via nonhomologous end joining Effects 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 108091008146 restriction endonucleases Proteins 0.000 description 2
- 230000035939 shock Effects 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- 239000000243 solution Substances 0.000 description 2
- 238000012453 sprague-dawley rat model Methods 0.000 description 2
- 238000010809 targeting technique Methods 0.000 description 2
- ZFXYFBGIUFBOJW-UHFFFAOYSA-N theophylline Chemical compound O=C1N(C)C(=O)N(C)C2=C1NC=N2 ZFXYFBGIUFBOJW-UHFFFAOYSA-N 0.000 description 2
- 108060008226 thioredoxin Proteins 0.000 description 2
- 229940094937 thioredoxin Drugs 0.000 description 2
- 108091006107 transcriptional repressors Proteins 0.000 description 2
- 238000003151 transfection method Methods 0.000 description 2
- 238000013519 translation Methods 0.000 description 2
- 210000004291 uterus Anatomy 0.000 description 2
- 210000005253 yeast cell Anatomy 0.000 description 2
- SGKRLCUYIXIAHR-AKNGSSGZSA-N (4s,4ar,5s,5ar,6r,12ar)-4-(dimethylamino)-1,5,10,11,12a-pentahydroxy-6-methyl-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1=CC=C2[C@H](C)[C@@H]([C@H](O)[C@@H]3[C@](C(O)=C(C(N)=O)C(=O)[C@H]3N(C)C)(O)C3=O)C3=C(O)C2=C1O SGKRLCUYIXIAHR-AKNGSSGZSA-N 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- AVRPFRMDMNDIDH-UHFFFAOYSA-N 1h-quinazolin-2-one Chemical compound C1=CC=CC2=NC(O)=NC=C21 AVRPFRMDMNDIDH-UHFFFAOYSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- NEAQRZUHTPSBBM-UHFFFAOYSA-N 2-hydroxy-3,3-dimethyl-7-nitro-4h-isoquinolin-1-one Chemical class C1=C([N+]([O-])=O)C=C2C(=O)N(O)C(C)(C)CC2=C1 NEAQRZUHTPSBBM-UHFFFAOYSA-N 0.000 description 1
- 108020005345 3' Untranslated Regions Proteins 0.000 description 1
- MOVBBVMDHIRCTG-LJQANCHMSA-N 4-[(3s)-1-azabicyclo[2.2.2]oct-3-ylamino]-3-(1h-benzimidazol-2-yl)-6-chloroquinolin-2(1h)-one Chemical compound C([N@](CC1)C2)C[C@@H]1[C@@H]2NC1=C(C=2NC3=CC=CC=C3N=2)C(=O)NC2=CC=C(Cl)C=C21 MOVBBVMDHIRCTG-LJQANCHMSA-N 0.000 description 1
- MDOJTZQKHMAPBK-UHFFFAOYSA-N 4-iodo-3-nitrobenzamide Chemical compound NC(=O)C1=CC=C(I)C([N+]([O-])=O)=C1 MDOJTZQKHMAPBK-UHFFFAOYSA-N 0.000 description 1
- NEEVCWPRIZJJRJ-LWRDCAMISA-N 5-(benzylideneamino)-6-[(e)-benzylideneamino]-2-sulfanylidene-1h-pyrimidin-4-one Chemical group C=1C=CC=CC=1C=NC=1C(=O)NC(=S)NC=1\N=C\C1=CC=CC=C1 NEEVCWPRIZJJRJ-LWRDCAMISA-N 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- 241001136782 Alca Species 0.000 description 1
- 102000007698 Alcohol dehydrogenase Human genes 0.000 description 1
- 108010021809 Alcohol dehydrogenase Proteins 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- 108091093088 Amplicon Proteins 0.000 description 1
- 241000272517 Anseriformes Species 0.000 description 1
- 241000620196 Arthrospira maxima Species 0.000 description 1
- 102000002804 Ataxia Telangiectasia Mutated Proteins Human genes 0.000 description 1
- 108010004586 Ataxia Telangiectasia Mutated Proteins Proteins 0.000 description 1
- 241000906059 Bacillus pseudomycoides Species 0.000 description 1
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 1
- 241000283726 Bison Species 0.000 description 1
- 108010045123 Blasticidin-S deaminase Proteins 0.000 description 1
- 101150095530 CDS1 gene Proteins 0.000 description 1
- 101150018129 CSF2 gene Proteins 0.000 description 1
- 101150069031 CSN2 gene Proteins 0.000 description 1
- 101100257372 Caenorhabditis elegans sox-3 gene Proteins 0.000 description 1
- 102000000584 Calmodulin Human genes 0.000 description 1
- 108010041952 Calmodulin Proteins 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 241000579895 Chlorostilbon Species 0.000 description 1
- 108091005960 Citrine Proteins 0.000 description 1
- 102100040484 Claspin Human genes 0.000 description 1
- 101710117926 Claspin Proteins 0.000 description 1
- 241000193163 Clostridioides difficile Species 0.000 description 1
- 241000193155 Clostridium botulinum Species 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- 102100032182 Crooked neck-like protein 1 Human genes 0.000 description 1
- 101150074775 Csf1 gene Proteins 0.000 description 1
- 102100022928 DNA repair protein RAD51 homolog 1 Human genes 0.000 description 1
- 101710136590 DNA repair protein RAD51 homolog 1 Proteins 0.000 description 1
- 230000004543 DNA replication Effects 0.000 description 1
- 230000007018 DNA scission Effects 0.000 description 1
- 230000004568 DNA-binding Effects 0.000 description 1
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 1
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 102000004533 Endonucleases Human genes 0.000 description 1
- 108010042407 Endonucleases Proteins 0.000 description 1
- 102100030013 Endoribonuclease Human genes 0.000 description 1
- 101710199605 Endoribonuclease Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 101001091269 Escherichia coli Hygromycin-B 4-O-kinase Proteins 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- 241000206602 Eukaryota Species 0.000 description 1
- 108091092566 Extrachromosomal DNA Proteins 0.000 description 1
- 230000010190 G1 phase Effects 0.000 description 1
- 101150106478 GPS1 gene Proteins 0.000 description 1
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 1
- KOSRFJWDECSPRO-WDSKDSINSA-N Glu-Glu Chemical compound OC(=O)CC[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(O)=O KOSRFJWDECSPRO-WDSKDSINSA-N 0.000 description 1
- 241000700721 Hepatitis B virus Species 0.000 description 1
- 102000008157 Histone Demethylases Human genes 0.000 description 1
- 108010074870 Histone Demethylases Proteins 0.000 description 1
- 102000011787 Histone Methyltransferases Human genes 0.000 description 1
- 108010036115 Histone Methyltransferases Proteins 0.000 description 1
- 102000003893 Histone acetyltransferases Human genes 0.000 description 1
- 108090000246 Histone acetyltransferases Proteins 0.000 description 1
- 102000003964 Histone deacetylase Human genes 0.000 description 1
- 108090000353 Histone deacetylase Proteins 0.000 description 1
- 241001272567 Hominoidea Species 0.000 description 1
- 101000921063 Homo sapiens Crooked neck-like protein 1 Proteins 0.000 description 1
- 101000956431 Homo sapiens Cytokine receptor-like factor 1 Proteins 0.000 description 1
- 101000744174 Homo sapiens DNA-3-methyladenine glycosylase Proteins 0.000 description 1
- 101000882584 Homo sapiens Estrogen receptor Proteins 0.000 description 1
- 101001103039 Homo sapiens Inactive tyrosine-protein kinase transmembrane receptor ROR1 Proteins 0.000 description 1
- 101001046587 Homo sapiens Krueppel-like factor 1 Proteins 0.000 description 1
- 101001139146 Homo sapiens Krueppel-like factor 2 Proteins 0.000 description 1
- 101001139134 Homo sapiens Krueppel-like factor 4 Proteins 0.000 description 1
- 101001139130 Homo sapiens Krueppel-like factor 5 Proteins 0.000 description 1
- 101001043594 Homo sapiens Low-density lipoprotein receptor-related protein 5 Proteins 0.000 description 1
- 101000984042 Homo sapiens Protein lin-28 homolog A Proteins 0.000 description 1
- 101000777293 Homo sapiens Serine/threonine-protein kinase Chk1 Proteins 0.000 description 1
- 101000777277 Homo sapiens Serine/threonine-protein kinase Chk2 Proteins 0.000 description 1
- 101100482468 Homo sapiens TRPA1 gene Proteins 0.000 description 1
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 1
- 108091092195 Intron Proteins 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- 102100022248 Krueppel-like factor 1 Human genes 0.000 description 1
- 102100020675 Krueppel-like factor 2 Human genes 0.000 description 1
- 102100020677 Krueppel-like factor 4 Human genes 0.000 description 1
- 102100020680 Krueppel-like factor 5 Human genes 0.000 description 1
- 102000015335 Ku Autoantigen Human genes 0.000 description 1
- 108010025026 Ku Autoantigen Proteins 0.000 description 1
- 241000713666 Lentivirus Species 0.000 description 1
- 101710152191 Low molecular weight antigen MTB12 Proteins 0.000 description 1
- 108060001084 Luciferase Proteins 0.000 description 1
- 239000005089 Luciferase Substances 0.000 description 1
- 101710175625 Maltose/maltodextrin-binding periplasmic protein Proteins 0.000 description 1
- 241000206589 Marinobacter Species 0.000 description 1
- 102100025169 Max-binding protein MNT Human genes 0.000 description 1
- 102000016397 Methyltransferase Human genes 0.000 description 1
- 240000000249 Morus alba Species 0.000 description 1
- 235000008708 Morus alba Nutrition 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 241000711408 Murine respirovirus Species 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 101100219625 Mus musculus Casd1 gene Proteins 0.000 description 1
- 101100355584 Mus musculus Rad51 gene Proteins 0.000 description 1
- 101100310657 Mus musculus Sox1 gene Proteins 0.000 description 1
- 101100257376 Mus musculus Sox3 gene Proteins 0.000 description 1
- 241000282339 Mustela Species 0.000 description 1
- 241000282341 Mustela putorius furo Species 0.000 description 1
- 108091057508 Myc family Proteins 0.000 description 1
- 102100038895 Myc proto-oncogene protein Human genes 0.000 description 1
- 101710135898 Myc proto-oncogene protein Proteins 0.000 description 1
- 101150030193 Nanp gene Proteins 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 241001515112 Nitrosococcus watsonii Species 0.000 description 1
- 241001223105 Nodularia spumigena Species 0.000 description 1
- 108020004485 Nonsense Codon Proteins 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 108700026244 Open Reading Frames Proteins 0.000 description 1
- 239000012661 PARP inhibitor Substances 0.000 description 1
- 108010088535 Pep-1 peptide Proteins 0.000 description 1
- 241000286209 Phasianidae Species 0.000 description 1
- 102000011755 Phosphoglycerate Kinase Human genes 0.000 description 1
- 241000512220 Polaromonas Species 0.000 description 1
- 229940121906 Poly ADP ribose polymerase inhibitor Drugs 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229920002873 Polyethylenimine Polymers 0.000 description 1
- 101710149951 Protein Tat Proteins 0.000 description 1
- 102100025460 Protein lin-28 homolog A Human genes 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 102000014450 RNA Polymerase III Human genes 0.000 description 1
- 108010078067 RNA Polymerase III Proteins 0.000 description 1
- 241000700157 Rattus norvegicus Species 0.000 description 1
- 101001023863 Rattus norvegicus Glucocorticoid receptor Proteins 0.000 description 1
- 101100247004 Rattus norvegicus Qsox1 gene Proteins 0.000 description 1
- 101100047461 Rattus norvegicus Trpm8 gene Proteins 0.000 description 1
- 102000006382 Ribonucleases Human genes 0.000 description 1
- 108010083644 Ribonucleases Proteins 0.000 description 1
- 108091028664 Ribonucleotide Proteins 0.000 description 1
- 108020004422 Riboswitch Proteins 0.000 description 1
- 101150086694 SLC22A3 gene Proteins 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- 101710113029 Serine/threonine-protein kinase Proteins 0.000 description 1
- 102100031081 Serine/threonine-protein kinase Chk1 Human genes 0.000 description 1
- 102100031075 Serine/threonine-protein kinase Chk2 Human genes 0.000 description 1
- 101150117538 Set2 gene Proteins 0.000 description 1
- 241000700584 Simplexvirus Species 0.000 description 1
- 101150001847 Sox15 gene Proteins 0.000 description 1
- 241000194022 Streptococcus sp. Species 0.000 description 1
- 241000194020 Streptococcus thermophilus Species 0.000 description 1
- 101001091268 Streptomyces hygroscopicus Hygromycin-B 7''-O-kinase Proteins 0.000 description 1
- 241000203587 Streptosporangium roseum Species 0.000 description 1
- 241000271567 Struthioniformes Species 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 101710137500 T7 RNA polymerase Proteins 0.000 description 1
- 101710192266 Tegument protein VP22 Proteins 0.000 description 1
- 101001099217 Thermotoga maritima (strain ATCC 43589 / DSM 3109 / JCM 10099 / NBRC 100826 / MSB8) Triosephosphate isomerase Proteins 0.000 description 1
- 102000006601 Thymidine Kinase Human genes 0.000 description 1
- 108020004440 Thymidine kinase Proteins 0.000 description 1
- 101710150448 Transcriptional regulator Myc Proteins 0.000 description 1
- 108091023045 Untranslated Region Proteins 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 210000001766 X chromosome Anatomy 0.000 description 1
- 210000002593 Y chromosome Anatomy 0.000 description 1
- 210000001015 abdomen Anatomy 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- 108020002494 acetyltransferase Proteins 0.000 description 1
- 102000005421 acetyltransferase Human genes 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000001464 adherent effect Effects 0.000 description 1
- 210000001789 adipocyte Anatomy 0.000 description 1
- KOSRFJWDECSPRO-UHFFFAOYSA-N alpha-L-glutamyl-L-glutamic acid Natural products OC(=O)CCC(N)C(=O)NC(CCC(O)=O)C(O)=O KOSRFJWDECSPRO-UHFFFAOYSA-N 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-L aspartate group Chemical group N[C@@H](CC(=O)[O-])C(=O)[O-] CKLJMWTZIZZHCS-REOHCLBHSA-L 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 238000002869 basic local alignment search tool Methods 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 210000001172 blastoderm Anatomy 0.000 description 1
- 210000002449 bone cell Anatomy 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 244000309466 calf Species 0.000 description 1
- 210000004413 cardiac myocyte Anatomy 0.000 description 1
- 101150055766 cat gene Proteins 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000012820 cell cycle checkpoint Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 210000003855 cell nucleus Anatomy 0.000 description 1
- 210000004671 cell-free system Anatomy 0.000 description 1
- 230000030570 cellular localization Effects 0.000 description 1
- 230000010094 cellular senescence Effects 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- OTAFHZMPRISVEM-UHFFFAOYSA-N chromone Chemical compound C1=CC=C2C(=O)C=COC2=C1 OTAFHZMPRISVEM-UHFFFAOYSA-N 0.000 description 1
- 239000013611 chromosomal DNA Substances 0.000 description 1
- 230000008711 chromosomal rearrangement Effects 0.000 description 1
- 230000005770 chromosome separation Effects 0.000 description 1
- 239000011035 citrine Substances 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 239000003184 complementary RNA Substances 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 101150055601 cops2 gene Proteins 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 230000001186 cumulative effect Effects 0.000 description 1
- 235000013365 dairy product Nutrition 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 239000000412 dendrimer Substances 0.000 description 1
- 229920000736 dendritic polymer Polymers 0.000 description 1
- 239000005547 deoxyribonucleotide Substances 0.000 description 1
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 238000006471 dimerization reaction Methods 0.000 description 1
- 229960003722 doxycycline Drugs 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 108010057988 ecdysone receptor Proteins 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 101150097231 eg gene Proteins 0.000 description 1
- 239000010976 emerald Substances 0.000 description 1
- 229910052876 emerald Inorganic materials 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 230000004049 epigenetic modification Effects 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- 230000004720 fertilization Effects 0.000 description 1
- 238000001917 fluorescence detection Methods 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 238000002509 fluorescent in situ hybridization Methods 0.000 description 1
- 229920001002 functional polymer Polymers 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 238000003209 gene knockout Methods 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 230000004077 genetic alteration Effects 0.000 description 1
- 231100000118 genetic alteration Toxicity 0.000 description 1
- 230000007614 genetic variation Effects 0.000 description 1
- 238000010362 genome editing Methods 0.000 description 1
- 108010055341 glutamyl-glutamic acid Proteins 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 210000003494 hepatocyte Anatomy 0.000 description 1
- 102000055426 human LRP5 Human genes 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 238000007901 in situ hybridization Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- 230000006882 induction of apoptosis Effects 0.000 description 1
- 229950002133 iniparib Drugs 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- PXZQEOJJUGGUIB-UHFFFAOYSA-N isoindolin-1-one Chemical compound C1=CC=C2C(=O)NCC2=C1 PXZQEOJJUGGUIB-UHFFFAOYSA-N 0.000 description 1
- VDBNYAPERZTOOF-UHFFFAOYSA-N isoquinolin-1(2H)-one Chemical compound C1=CC=C2C(=O)NC=CC2=C1 VDBNYAPERZTOOF-UHFFFAOYSA-N 0.000 description 1
- 210000002510 keratinocyte Anatomy 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 238000011813 knockout mouse model Methods 0.000 description 1
- 108091008792 l-Myc Proteins 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 230000008774 maternal effect Effects 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 230000021121 meiosis Effects 0.000 description 1
- 210000002752 melanocyte Anatomy 0.000 description 1
- 210000003716 mesoderm Anatomy 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 230000025608 mitochondrion localization Effects 0.000 description 1
- 230000011278 mitosis Effects 0.000 description 1
- 108091005601 modified peptides Proteins 0.000 description 1
- 238000003032 molecular docking Methods 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 210000000663 muscle cell Anatomy 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 108091008800 n-Myc Proteins 0.000 description 1
- WGLUNLJVYNJMBU-UHFFFAOYSA-N n-[2-(dimethylamino)ethyl]-n-[(3-methyl-1-oxo-2,4-dihydroisoquinolin-3-yl)methyl]decanamide Chemical compound C1=CC=C2C(=O)NC(CN(CCN(C)C)C(=O)CCCCCCCCC)(C)CC2=C1 WGLUNLJVYNJMBU-UHFFFAOYSA-N 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 210000004498 neuroglial cell Anatomy 0.000 description 1
- 229960003966 nicotinamide Drugs 0.000 description 1
- 235000005152 nicotinamide Nutrition 0.000 description 1
- 239000011570 nicotinamide Substances 0.000 description 1
- 230000037434 nonsense mutation Effects 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- FAQDUNYVKQKNLD-UHFFFAOYSA-N olaparib Chemical compound FC1=CC=C(CC2=C3[CH]C=CC=C3C(=O)N=N2)C=C1C(=O)N(CC1)CCN1C(=O)C1CC1 FAQDUNYVKQKNLD-UHFFFAOYSA-N 0.000 description 1
- 229960000572 olaparib Drugs 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 210000003463 organelle Anatomy 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 210000003101 oviduct Anatomy 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- RZFVLEJOHSLEFR-UHFFFAOYSA-N phenanthridone Chemical compound C1=CC=C2C(O)=NC3=CC=CC=C3C2=C1 RZFVLEJOHSLEFR-UHFFFAOYSA-N 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- IJAPPYDYQCXOEF-UHFFFAOYSA-N phthalazin-1(2H)-one Chemical compound C1=CC=C2C(=O)NN=CC2=C1 IJAPPYDYQCXOEF-UHFFFAOYSA-N 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000008488 polyadenylation Effects 0.000 description 1
- 108010011110 polyarginine Proteins 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 235000020004 porter Nutrition 0.000 description 1
- 230000032029 positive regulation of DNA repair Effects 0.000 description 1
- 230000029279 positive regulation of transcription, DNA-dependent Effects 0.000 description 1
- 230000004481 post-translational protein modification Effects 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 210000001236 prokaryotic cell Anatomy 0.000 description 1
- 230000004853 protein function Effects 0.000 description 1
- 230000004850 protein–protein interaction Effects 0.000 description 1
- 108010045647 puromycin N-acetyltransferase Proteins 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 102000005912 ran GTP Binding Protein Human genes 0.000 description 1
- 102000037983 regulatory factors Human genes 0.000 description 1
- 108091008025 regulatory factors Proteins 0.000 description 1
- 230000008263 repair mechanism Effects 0.000 description 1
- 230000008672 reprogramming Effects 0.000 description 1
- 238000002271 resection Methods 0.000 description 1
- 230000028617 response to DNA damage stimulus Effects 0.000 description 1
- 239000002336 ribonucleotide Substances 0.000 description 1
- 125000002652 ribonucleotide group Chemical group 0.000 description 1
- 230000001568 sexual effect Effects 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 238000002741 site-directed mutagenesis Methods 0.000 description 1
- 210000004683 skeletal myoblast Anatomy 0.000 description 1
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 238000003153 stable transfection Methods 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 230000010741 sumoylation Effects 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 238000010381 tandem affinity purification Methods 0.000 description 1
- 230000002123 temporal effect Effects 0.000 description 1
- 101150024821 tetO gene Proteins 0.000 description 1
- 229960000278 theophylline Drugs 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 229940126585 therapeutic drug Drugs 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- 108091006106 transcriptional activators Proteins 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 238000003146 transient transfection Methods 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 230000034512 ubiquitination Effects 0.000 description 1
- 238000010798 ubiquitination Methods 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 230000009107 upstream regulation Effects 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 229940075420 xanthine Drugs 0.000 description 1
Images





























Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/90—Stable introduction of foreign DNA into chromosome
- C12N15/902—Stable introduction of foreign DNA into chromosome using homologous recombination
- C12N15/907—Stable introduction of foreign DNA into chromosome using homologous recombination in mammalian cells
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K67/00—Rearing or breeding animals, not otherwise provided for; New or modified breeds of animals
- A01K67/027—New or modified breeds of vertebrates
- A01K67/0275—Genetically modified vertebrates, e.g. transgenic
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/102—Mutagenizing nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/8509—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells for producing genetically modified animals, e.g. transgenic
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases RNAses, DNAses
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6813—Hybridisation assays
- C12Q1/6827—Hybridisation assays for detection of mutation or polymorphism
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6888—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for detection or identification of organisms
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2217/00—Genetically modified animals
- A01K2217/07—Animals genetically altered by homologous recombination
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2227/00—Animals characterised by species
- A01K2227/10—Mammal
- A01K2227/105—Murine
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPRs]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2537/00—Reactions characterised by the reaction format or use of a specific feature
- C12Q2537/10—Reactions characterised by the reaction format or use of a specific feature the purpose or use of
- C12Q2537/16—Assays for determining copy number or wherein the copy number is of special importance
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Biomedical Technology (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Plant Pathology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Analytical Chemistry (AREA)
- Environmental Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Animal Husbandry (AREA)
- Biodiversity & Conservation Biology (AREA)
- Animal Behavior & Ethology (AREA)
- Cell Biology (AREA)
- Mycology (AREA)
- Immunology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
Abstract
Description
本出願は、2014年11月12日出願の米国特許出願第62/083,005号、2015年6月19日出願の米国特許出願第62/182,314号、及び2015年8月28日出願の米国特許出願第62/211,421号の利益を主張するものであり、これらの出願のそれぞれをその全容にわたってすべての目的で本明細書に参照により援用するものである。
472225SEQLIST.txtのファイルに記載の配列表は32.7kbであり、2015年11月20日に作成されたものであり、本明細書に参照によって援用する。
本明細書で互換的に使用される「タンパク質」、「ポリペプチド」、及び「ペプチド」なる用語には、コード及び非コードアミノ酸、並びに化学的又は生化学的に修飾又は誘導体化されたアミノ酸を含む、任意の長さのアミノ酸のポリマー形態が含まれる。これらの用語には、修飾されたペプチド主鎖を有するポリペプチドなどの修飾されたポリマーも含まれる。
(発明を実施するための形態)
細胞内のゲノムを改変するための方法及び組成物が提供される。本方法及び組成物は、1つのゲノム標的遺伝子座内の異なる部位をターゲティングする2種類のガイドRNA(gRNA)を使用したCRISPR/Casシステムを用いるものである。例えば、本方法及び組成物は、2種類のガイドRNA(gRNA)を使用したCRISPR/Casシステムを用いることによって、1つのゲノム標的遺伝子座内の異なる部位に二本鎖切断のペアを形成することができる。また、本方法及び組成物は、2種類のガイドRNA(gRNA)を使用したCRISPR/Casシステムを用いることによって、1つのゲノム標的遺伝子座内の異なる部位に一本鎖切断のペアを形成することもできる。特定の方法では、2種類以上のガイドRNA(例えば3種類又は4種類)を使用することで例えば1つのゲノム標的遺伝子座内の異なる部位に2箇所以上の一本鎖切断又は二本鎖切断を形成することができる。
本明細書に開示される方法及び組成物は、CRISPR(Clustered Regularly Interspersed Short Palindromic Repeats)/CRISPR関連(Cas)システム、又はかかるシステムの構成要素を利用して細胞内のゲノムを改変することが可能である。CRISPR/Casシステムは、Cas遺伝子の発現に関与するか又はその活性を誘導する転写産物及び他のエレメントを含む。CRISPR/Casシステムは、I型、II型、又はIII型のシステムであってよい。本明細書に開示される方法及び組成物は、CRISPR複合体(Casタンパク質と複合体を形成したガイドRNA(gRNA)からなる)を核酸の部位特異的な切断に利用することによってCRISPR/Casシステムを用いるものである。
Casタンパク質は一般的に少なくとも1つのRNA認識又は結合ドメインを有している。そのようなドメインは、ガイドRNA(gRNA、下記により詳しく説明する)と相互作用することができる。Casタンパク質は、ヌクレアーゼドメイン(例えば、DNase又はRNaseドメイン)、DNA結合ドメイン、ヘリカーゼドメイン、タンパク質間相互作用ドメイン、二量化ドメイン、及び他のドメインを含んでもよい。ヌクレアーゼドメインは、核酸切断のための触媒活性を有する。切断は、核酸分子の共有結合の切断を含む。切断は、平滑末端又は付着末端を生じさせることができ、一本鎖又は二本鎖でありうる。
「ガイドRNA」又は「gRNA」はCasタンパク質と結合して、Casタンパク質を標的DNA内の特定の位置に標的化するRNA分子を含む。ガイドRNAは、2つの部分、すなわち「DNAターゲティングセグメント」と「タンパク質結合セグメント」とを含むことができる。「セグメント」には、RNA内のヌクレオチドの連続的区間のような、分子のセグメント、部分、又は領域が含まれる。特定のgRNAは、2個の別々のRNA分子、すなわち「アクチベーターRNA」と「ターゲッターRNA」とを含む。他のgRNAは、「単鎖分子gRNA」、「単鎖ガイドRNA」、又は「sgRNA」とも呼ばれる場合もある単鎖RNA分子(単鎖RNAポリヌクレオチド)である。例えば、それぞれの全容をすべての目的で本明細書に参照により援用するところの国際公開第2013/176772A1号、同第2014/065596A1号、同第2014/089290A1号、同第2014/093622A2号、同第2014/099750A2号、同第2013142578A1号、及び同第2014/131833A1号を参照されたい。「ガイドRNA」及び「gRNA」なる用語には、二分子gRNA及び一分子gRNAの両方が含まれる。
「CRISPR RNA認識配列」なる用語は、gRNAのDNAターゲティングセグメントが結合する標的DNA内に存在する核酸配列を含むが、ただし、結合のための充分な条件が存在することを条件とする。例えば、CRISPR RNA認識配列は、ガイドRNAがそれに対する相補性を有するように設計される配列を含み、CRISPR RNA認識配列とDNAターゲティング配列との間のハイブリダイゼーションによってCRISPR複合体の形成が促進される。ハイブリダイゼーションが生じてCRISPR複合体の形成を促進するだけの充分な相補性があれば、完全な相補性は必ずしも必要ではない。CRISPR RNA認識配列は、下記により詳細に説明するCasタンパク質の切断部位も含む。CRISPR RNA認識配列は、例えば、細胞の核若しくは細胞質又はミトコンドリア若しくは葉緑体などの細胞の小器官内に存在しうる任意のポリヌクレオチドを含むことができる。
本明細書に開示される方法及び組成物では、核酸インサート及びホモロジーアームを含むターゲティングベクターを使用して細胞内のゲノムを改変することもできる。かかる方法では、核酸インサートは、相同組換え事象を介してホモロジーアームによって決定されるゲノム標的遺伝子座に組み込まれる。本明細書で提供される方法は、相同組換え事象と組み合わせてヌクレアーゼ剤(例えばCasタンパク質)を利用することができる。かかる方法では、ヌクレアーゼ剤によってヌクレアーゼ切断部位に形成されるニック又は二本鎖切断を相同組換えと組み合わせて利用することで、ゲノム標的遺伝子座への核酸インサートのターゲティングによる挿入を促進することができる。
(1)核酸インサート
本明細書に開示される方法では1以上の別々の核酸インサートを使用することができ、これらの核酸インサートは、別々のターゲティングベクターにより、又は同じターゲティングベクター上で細胞に導入することができる。核酸インサートは、ゲノム標的遺伝子座に組み込むためのDNAのセグメントを含む。標的遺伝子座への核酸インサートの組み込みは、標的遺伝子座への対象となる核酸配列の付加、標的遺伝子座における対象となる核酸配列の欠失、及び/又は標的遺伝子座における対象となる核酸配列の置換を生じることができる。
標的ゲノム遺伝子座に核酸インサートを導入するためにターゲティングベクターを用いることができるが、このようなターゲティングベクターは核酸インサート及び核酸インサートに隣接したホモロジーアームを含む。ターゲティングベクターは直鎖状又は環状であってよく、一本鎖又は二本鎖であってもよい。ターゲティングベクターは、デオキシリボ核酸(DNA)又はリボ核酸(RNA)であってよい。参照を容易にするため、本明細書ではホモロジーアームを、5’及び3’(すなわち、上流及び下流)ホモロジーアームと呼ぶ。この呼び方は、ターゲティングベクター内の核酸インサートに対するホモロジーアームの相対位置に関する。5’及び3’ホモロジーアームは、本明細書においてそれぞれ「5’」標的配列及び「3’」標的配列と呼ばれる標的遺伝子座内の領域に対応している。特定のターゲティングベクターは、5’及び3’ホモロジーアームを有するが核酸インサートは有さない。そのようなターゲティングベクターは、核酸インサートを挿入することなく5’標的配列と3’標的配列との間の配列を欠失させるように機能することができる。
一部のターゲティングベクターは、「ラージターゲティングベクター」又は「LTVEC」であり、これには、細胞内で相同組み換えを行うことを目的とした他の手法で一般的に使用される核酸配列よりも大きな核酸配列に相当し、このような大きな核酸配列から誘導されたホモロジーアームを含むターゲティングベクターが含まれる。LTVECには、細胞内で相同組み換えを行うことを目的とした他の手法で一般的に使用される核酸配列よりも大きな核酸配列を有する核酸インサートを含むターゲティングベクターも含まれる。例えば、LTVECは、従来のプラスミド系ターゲティングベクターではそれらのサイズ制限のために対処することができない大きな遺伝子座の改変を可能にする。例えば、標的遺伝子座は、ヌクレアーゼ剤(例えば、Casタンパク質)によって誘導されるニック若しくは二本鎖切断が存在しなければ、従来法を用いてターゲティングできないか、又は不正確にターゲティングされるだけであるか若しくは著しく低い効率でしかターゲティングできない細胞内の遺伝子座であってよい(すなわち5’及び3’ホモロジーアームがこの遺伝子座に相当してよい)。
1細胞期胚に使用するためのターゲティングベクターは、長さが5kb以下であり、デオキシリボ核酸(DNA)又はリボ核酸(RNA)であってよく、一本鎖又は二本鎖であってよく、環状又は直鎖状であってよい。1細胞期胚に使用するための代表的なターゲティングベクターは、約50ヌクレオチド〜約5kbの長さである。例えば、1細胞期胚に使用するためのターゲティングベクターは、約50〜約100、約100〜約200、約200〜約300、約300〜約400、約400〜約500、約500〜約600、約600〜約700、約700〜約800、約800〜約900、又は約900〜約1,000ヌクレオチドの長さであってよい。あるいは、1細胞期胚に使用するためのターゲティングベクターは、約1kb〜約1.5kb、約1.5kb〜約2kb、約2kb〜約2.5kb、約2.5kb〜約3kb、約3kb〜約3.5kb、約3.5kb〜約4kb、約4kb〜約4.5kb、又は約4.5kb〜約5kbの長さであってもよい。あるいは、1細胞期胚に使用するためのターゲティングベクターは、例えば、5kb、4.5kb、4kb、3.5kb、3kb、2.5kb、2kb、1.5kb、1kb、900ヌクレオチド、800ヌクレオチド、700ヌクレオチド、600ヌクレオチド、500ヌクレオチド、400ヌクレオチド、300ヌクレオチド、200ヌクレオチド、100ヌクレオチド、又は50ヌクレオチド以下の長さであってもよい。一本鎖DNAのドナーの場合では、代表的なターゲティングベクターは、約60ヌクレオチド〜約200ヌクレオチド(例えば、約60ヌクレオチド〜約80ヌクレオチド、約80ヌクレオチド〜約100ヌクレオチド、約100ヌクレオチド〜約120ヌクレオチド、約120ヌクレオチド〜約140ヌクレオチド、約140ヌクレオチド〜約160ヌクレオチド、約160ヌクレオチド〜約180ヌクレオチド、又は約180ヌクレオチド〜約200ヌクレオチド)であってよい。
本明細書に述べられる様々な核酸配列は、プロモーターと機能的に連結することができる。そのようなプロモーターは、例えば、多能性ラットの細胞、真核細胞、哺乳動物細胞、非ヒト哺乳動物細胞、ヒト細胞、げっ歯類細胞、マウス細胞、又はハムスター細胞中で活性なものであってよい。1細胞期胚において活性であるプロモーターを使用することもできる。プロモーターは例えば、構成的に活性であるプロモーター、条件的プロモーター、誘導性プロモーター、時間的に制限されたプロモーター(例えば発生的に調節されるプロモーター)、又は空間的に制限されたプロモーター(例えば細胞特異的又は組織特異的プロモーター)とすることができる。プロモーターの例は、例えば国際公開第2013/176772号に見ることができる。尚、その全容を参照により本明細書に援用する。
A.ゲノムを改変する方法
2種類のガイドRNAを使用して1つのゲノム標的遺伝子座内の異なる領域をターゲティングすることによって細胞内のゲノムを改変するための様々な方法が提供される。2種類以上(例えば3種類のガイドRNA又は4種類のガイドRNA)のガイドRNAを使用して1つのゲノム標的遺伝子座内の異なる領域をターゲティングする方法も提供される。これらの方法は、インビトロ、エクスビボ、又はインビボで行うことができる。かかる方法は、両対立遺伝子の遺伝子改変の形成を促進し、ゲノム破壊又は他のターゲティングによる改変、例えばゲノム内の核酸配列の欠失と外因性の核酸配列による置換を同時に行うことを含みうる。
本明細書では、細胞内のゲノムに両対立遺伝子改変を行うか、又は細胞内のゲノムに対する両対立遺伝子改変を促進するか若しくはその頻度を高めるための方法が提供される。かかる方法によって、例えばゲノムを破壊して、後に組換えを生じるゲノムDNAの2つの配列間の大きなゲノムDNAの部分を除去することができる。かかる方法によって、核酸インサートを挿入するか、又は大きなゲノムDNAの部分を欠失させて核酸インサートで置換することもできる。
特定の方法では、改変しようとするゲノムは第1の対立遺伝子についてヘテロ接合型である細胞内にあり、この遺伝子を第1の対立遺伝子についてホモ接合型となるように改変する。ヘテロ接合型なる用語には、ゲノムが、1個以上の対応する染色体遺伝子座に異なる対立遺伝子(例えば相同染色体上の対応する遺伝子座に異なる対立遺伝子)を有する場合含まれる。ホモ接合型なる用語には、ゲノムが、対応する染色体遺伝子座(例えば対応する相同染色体上の)に同じ対立遺伝子を有する場合が含まれる。特定のこのような方法では、ホモ接合は、細胞が第1の対立遺伝子をドナー配列として用いて遺伝子変換などの相同組換えによって対応する第2の対立遺伝子の二本鎖切断を修復することによって得られる。一般的に、遺伝子変換の範囲は数百塩基対に限定されている。例えば、Kasparek & Humphrey(2011)Seminars in Cell & Dev.Biol.22:886〜897を参照されたい。尚、その全容をすべての目的で参照により本明細書に援用する。しかしながら、1個の遺伝子座内の異なる切断部位での切断を誘導するガイドRNAのペアを用いることにより、より大きな範囲にわたった遺伝子変換能を促進及び向上させることができる。
遺伝子改変された非ヒト動物を、本明細書に開示される様々な方法を用いて作成することができる。特定の場合では、遺伝子改変された非ヒト動物を作成する方法は、(1)上記に述べた方法を用いて多能性細胞のゲノムを改変することと、(2)前記遺伝子改変された多能性細胞を選択することと、(3)前記遺伝子改変された多能性細胞を宿主胚に導入することと、(4)前記遺伝子改変された多能性細胞を有する前記宿主胚を代理母に移植することと、を含む。前記遺伝子改変された多能性細胞から子孫細胞が作成される。このドナー細胞を例えば胚盤胞期又は前桑実胚期(すなわち4細胞期又は8細胞期胚)などの任意の段階の宿主胚に導入することができる。前記遺伝子改変を生殖細胞系に伝えることが可能な子孫細胞が作成される。多能性細胞は、本明細書の他の箇所で述べるようなES細胞(例えばマウスES細胞又はラットES細胞)であってよい。例えば、米国特許第7,294,754号を参照されたい。尚、その全容をすべての目的で参照により本明細書に援用する。
本明細書に開示される方法によって改変されたゲノム又はゲノム標的遺伝子座は、細胞内のDNAの任意のセグメント又は領域を有することができる。ゲノム又はゲノム標的遺伝子座は、細胞に天然に存在するものでもよく、細胞のゲノムに組み込まれた異種又は外因性のDNAのセグメントであってもよく、又はそれらの組み合わせであってもよい。そのような異種又は外因性のDNAのセグメントは、導入遺伝子、発現カセット、選択マーカーをコードしたポリヌクレオチド、又はゲノムDNAの異種若しくは外因性領域を含むことができる。
特定の方法では、上記のゲノムを接触させることは、1以上のCasタンパク質、1以上のCRISPR RNA、及び1以上のtracrRNAを細胞内に導入することを含む。このような導入は任意の手段によって行うことができ、成分のうちの1つ以上(例えば成分のうちの2つ、又は成分のすべて)を任意の組み合わせで同時又は順次に細胞内に導入することができる。
本明細書では、細胞内に核酸を導入することを可能とする様々な組成物及び方法を提供する。特定の場合では、核酸を導入するために用いられるシステムは、特定のゲノム遺伝子座におけるターゲティングによる組み込みを可能とする。かかるシステムは様々な要素を使用するものであり、説明の便宜上、「ターゲティングによるゲノム組み込みシステム」なる用語には、一般的に、導入事象に必要とされるすべての要素(例えば、ヌクレアーゼ剤、ヌクレアーゼ切断部位、挿入DNAポリヌクレオチド、ターゲティングベクター、標的ゲノム遺伝子座、及び対象とするポリヌクレオチドのうちの1つ以上)が含まれるものである。
組換えとは、2個のポリヌクレオチド間での遺伝情報の交換のあらゆるプロセスを含むものであり、あらゆる機構によって生じうる。二本鎖切断(DSB)に応じた組換えは、非相同末端結合(NHEJ)及び相同組換え(HR)という2つの保存されたDNA修復経路で主として生じる。Kasparek & Humphrey(2011)Seminars in Cell & Dev.Biol.22:886〜897を参照されたい。尚、その全容をすべての目的で参照により本明細書に援用する。NHEJは、相同性鋳型を必要とせずに、切断末端同士を互いに直接連結することによる核酸の二重鎖切断の修復を含む。NHEJによる不連続配列の連結は、二重鎖切断部位の近くに欠失、挿入、又は転位をしばしば生じうる。組換えは、相同組換え型修復(HDR)又は相同組換え(HR)によっても生じうる。HDR又はHRは、ヌクレオチド配列の相同性を必要としうる核酸修復の形を含み、「標的」分子(すなわち二本鎖切断が生じた分子)の修復の鋳型とするために「ドナー」分子を利用し、ドナーから標的への遺伝情報の転移を生じる。いかなる特定の理論に束縛されることも望むものではないが、このような移動は、切断が生じた標的とドナーとの間で形成されるヘテロ二重鎖DNAのミスマッチ修復、及び/又は標的の一部になることとなる遺伝情報を再合成するためにドナーが使用される合成依存性鎖アニーリング、及び/又は関連するプロセスに関与してもよい。場合によっては、ドナーポリヌクレオチド、ドナーポリヌクレオチドの部分、ドナーポリヌクレオチドの複製、又はドナーポリヌクレオチドの複製の部分は、標的DNA内に組み込まれる。
本明細書で提供される様々な組成物及び方法では、例えば動物由来の細胞などの細胞を用いる。かかる細胞は非ヒト動物に由来するものでよい。かかる細胞は、例えば真菌細胞(例えば酵母)、植物細胞、動物細胞、哺乳動物細胞、及びヒト細胞などの真核細胞であってよい。哺乳動物細胞は、例えば非ヒト哺乳動物細胞、ヒト細胞、げっ歯類細胞、ラット細胞、マウス細胞、ハムスター細胞、線維芽細胞、又はCHO細胞であってよい。真核細胞は、全能性細胞、例えば非ヒト多能性細胞(例えばマウス胚性幹(ES)細胞若しくはラットES細胞)若しくはヒト多能性細胞などの多能性細胞、又は非多能性細胞であってよい。全能性細胞としては、あらゆる細胞型を生じることができる未分化細胞が挙げられ、多能性細胞としては、1以上の分化した細胞型に発生する能力を有する未分化細胞が挙げられる。かかる多能性及び/又は全能性細胞は、例えば胚性幹(ES)細胞又は人工多能性幹(iPS)細胞のようなES様細胞であってもよい。胚性幹細胞としては、胚への導入後に発生中の胚のあらゆる組織に寄与することが可能である胚由来の全能性又は多能性細胞が挙げられる。ES細胞は胚盤胞の内部細胞塊から誘導することができ、脊椎動物の3つの胚葉(内胚葉、外胚葉、及び中胚葉)のいずれの細胞にも分化することが可能である。
上記の方法の一部のものは、改変されたゲノムを有する細胞を同定することを更に含む。様々な方法を用いて、欠失又は挿入などのターゲティングによる改変を有する細胞を同定することができる。そのような方法は、標的遺伝子座(例えば第1のCRISPR RNA認識配列と第2のCRISPR RNAとの間)にターゲティングによる改変を有する1個の細胞を同定することを含む。スクリーニングを行って改変されたゲノム遺伝子座を有するこうした細胞を同定することができる。
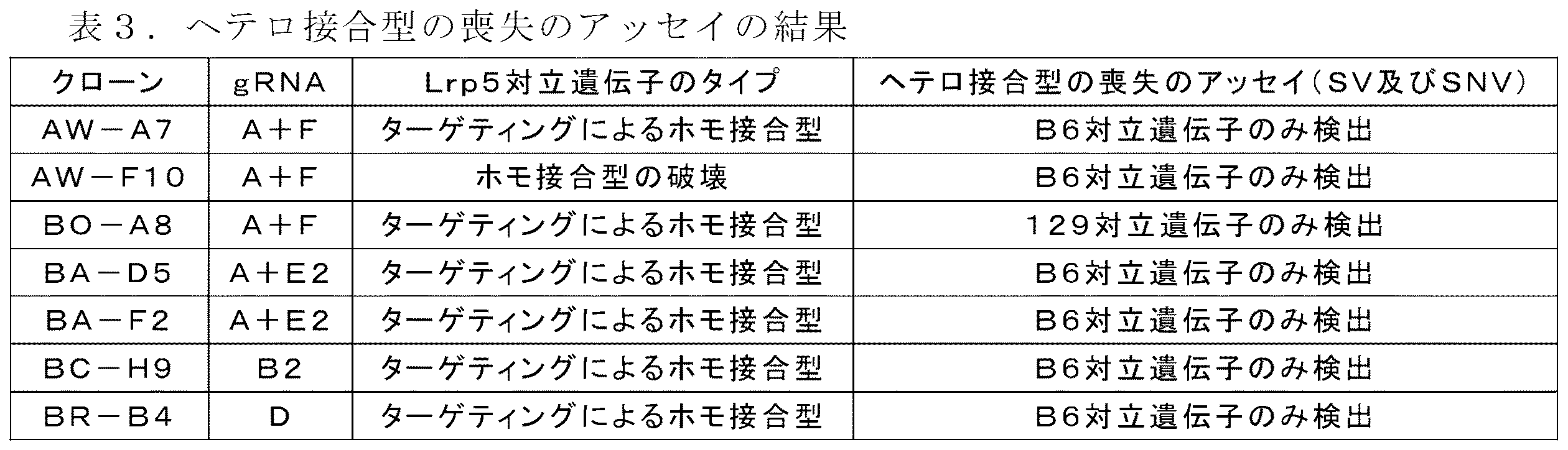
材料及び方法
ES細胞の培養、スクリーニング、及び電気穿孔法
ここに述べる各実験は、本発明者らによるC57BL6NTac/129S6SvEvF1ハイブリッドXY ES細胞株であるVGF1を使用して行った(Poueymirou et al.(2007)Nat.Biotechnol.25:91〜99;Valenzuela et al.(2003)Nat.Biotechnol.21:652〜659)。ES細胞をこれまでに記載されているようにして培養した(Matise et al.(2000)in Joyner,A.L.ed.Gene Targeting:a practical approach,pp.100〜132,Oxford University Press,New York)。
Lrp5又は他の標的化遺伝子の欠失させた部分の内側50bp、100bp、500bp、又は1kbの位置の周辺の約200bpのDNA(上流及び下流の両方)を、CRISPR設計ツール(crispr.mit.edu)に入力して可能なgRNA配列を取得した。次いで、可能性のあるgRNA配列を、内因性のDNAのみが切断され、LTVECのヒト化インサートは切断されないようにフィルタリングした。
sgRNAを、シームレスなRNA発現を行うために77bpのスカフォールドと融合したpMB_sgRNA(U6プロモーター)内のBsmbI部位に二重鎖オリゴ(IDT社)としてクローニングするか、又はジーンコピア社(GeneCopoeia)より確認済みの発現プラスミドとして購入した(LRP5ガイドA、B、B2、E2、E、及びF)。社内で作成したプラスミドをPCR及びサンガー法により配列決定して確認した。
ES細胞からDNAを精製し、ターゲティングベクター、及びCas9を発現するプラスミド、及び複数のガイドRNA(gRNA)の1つを発現するプラスミド又は異なるgRNAの組み合わせを発現する2種類のプラスミドを電気穿孔法で導入したES細胞からクローンを誘導した。対立遺伝子改変(すなわち対立遺伝子喪失又は対立遺伝子獲得)定量PCRアッセイにより、マウスの標的遺伝子座のターゲティングによる欠失及びターゲティングベクターの挿入を有するか、又はCas9/gRNAにより誘導された欠失を有するものとして同定されたクローンをフォローアップとしての従来のPCRアッセイを行うために選択した。
gRNAのそれぞれの組み合わせについて2つのPCRアッセイを設計した。第1のPCRは、異なるgRNAの組み合わせのCRISPR RNA認識配列間の破壊を検出するための欠失アッセイとした。第2のPCRは、5’アッセイであり、2つのPCRアッセイを含むものとした。最初のものはヒト化された対立遺伝子についての5’ヒトアッセイであり、マウス/ヒト接合部にわたって設計した。第2のものは、内因性のマウス対立遺伝子についての5’マウスアッセイであり、ターゲティングによる5’欠失接合部にわたって設計した。
タカラバイオ株式会社(TaKaRa)製LA Taq DNA ポリメラーゼ(カタログ番号RR002M)を使用してES細胞のDNA鋳型を増幅した。各PCRアッセイ反応ミックス及び水のネガティブコントロールで反応を行った。アッセイ混合物は以下を含むものとした。すなわち、ES細胞DNA鋳型0.005mL;1X LA PCRバッファーII(Mg2+添加);0.01mM dNTPミックス;0.0075mMフォワードオリゴ(それぞれに);0.0075mMリバースオリゴ(それぞれに);5000単位/mL LA Taqポリメラーゼ;及びddH2Oを加えて0.025mLとした。
白色のコロニーを0.025mg/mLのカナマイシンを含んだLB中に接種し、37℃で振盪しながら一晩インキュベートした。各コロニーは、アッセイした産物の集団からの1つのアンプリコンを表す。キアジェン社(QUIAGEN)製plasmid miniprepキット(カタログ番号12123)を使用して各細菌培養物からDNAを抽出した。インサートのDNA配列を、0.002mL TOPOクローニングしたPCR、1xPCRxエンハンサー溶液(10xストック)(カタログ番号X11495−017)、0.0075mMオリゴ(M13F又はM13R)、及び0.015mLとなるまでddH2Oを含んだシークエンシング反応ミックス中で決定した。
シークエンシングの結果から、不確定配列及びpCR4−TOPOベクター配列をトリミングし、PCRインサート配列を単離した。配列決定したフラグメントを基準配列とアラインして変異を分析した。
破壊したポジティブクローンからのPCR産物を製造者の指示にしたがってpCR4−TOPOベクター(インビトロジェン社(Invitrogen)カタログ番号K4575−02)にクローニングし、次いでOne Shot Top10細胞に化学的に形質導入し、0.060mg/mLのX−gal及び0.025mg/mLのカナマイシンアガープレートに播種した。キアジェン社(QUIAGEN)製plasmid miniprepキット(カタログ番号12123)を使用して細菌培養物からDNAを抽出した。次いでインサートのシークエンシングの結果を予想した破壊基準配列とアラインしてインデル変異を分析した。Cas9は、gRNAによって認識された配列内にPAMから3塩基対の位置で切断することが予想された。予想された切断内の配列を基準配列から欠失させ、残りを用いて結果とアラインした。
TaqMan(登録商標)対立遺伝子識別反応液は、ゲノムDNA、それぞれの多型に対する特異的プローブ/プライマー、及びTaqMan(登録商標)遺伝子発現PCRマスターミックスを含む0.008mlのものとした。プローブは、ライフ・テクノロジーズ社(Life Technologies)(サーモ社(Thermo))より、プライマーはIDT社より注文した。129対立遺伝子に対するプローブをVIC色素で標識し、B6対立遺伝子に対するプローブをFAM色素で標識した。各TaqMan(登録商標)対立遺伝子アッセイは、384ウェルプレートで4重に行い、アプライド・バイオシステムズ社(Applied BioSystems)製ViiA 7プラットフォーム上で行った。SNV PCRサイクリングプログラムは以下とした。すなわち、95℃で10分間の後、95℃で15秒、60℃で60秒、及び60℃で30秒を40サイクル。反応の分析及び結果の評価は、ViiA7ソフトウェアv1.1を使用して行った。
選択されたES細胞クローンは、セルライン・ジェネティクス社(Cell Line Genetics)(ウィスコンシン州マディソン)、又はバン・アンデル・インスティテュート社(Van Andel Institute)(ミシガン州グランドラピッズ)により、各社の標準的手順によって蛍光in situハイブリダイゼーション(FISH)を用いて分析を行った。本発明者らは、マウス及びヒトBACを2色分析用のプローブとして提供した。
げっ歯類遺伝子の全体又は一部の正確な1工程での欠失及び場合によりそのヒトホモログの全体又は一部による同時置換を行うため、本発明者らは電気穿孔法によりげっ歯類ES細胞に以下の核酸分子を導入した。すなわち、(1)LTVEC、(2)Cas9エンドヌクレアーゼをコードしたプラスミド又はmRNA、及び(3)1以上のCRISPR単鎖ガイドRNA(gRNA)をコードした1以上のプラスミド又はgRNA自体。各実験でLTVECは直鎖化した。一部の実験では、LTVECは、げっ歯類遺伝子を欠失させてヒト遺伝子を挿入する相同組換え事象を導くように設計されたげっ歯類のホモロジーアームを隣接させた遺伝子産物(タンパク質又はRNA)をコードしたヒト遺伝子の全体又は一部を含むものとした。他の実験では、LTVECは、Ch25h遺伝子座のような別の遺伝子座をターゲティングするように設計した。いずれの場合も、LTVECは、抗生物質(例えばG418)に対する耐性を与える酵素(例えばネオマイシンホスホトランスフェラーゼ)の発現を誘導する薬剤選択カセットも有していた。
一群の実験において、LTVECは、マウスLrp5(低密度リポタンパク質受容体関連タンパク質5)遺伝子の、エクトドメインをコードする部分の68kbの欠失を生じ、同時にヒトLRP5遺伝子(図1)からの相同配列の91kbのフラグメントで置換するように設計した。LTVECは、欠失させようとするマウスLrp5遺伝子の68kbの配列に隣接したマウスLrp5遺伝子座の部分から誘導された7kb及び33kbのゲノムDNAを含むホモロジーアームを隣接させたヒトLRP5遺伝子の91kbのフラグメントを含むものとした。別々の実験において、このLrp5をヒト化するLTVECを、Cas9をコードしたプラスミド、及び欠失の標的とされるマウスLrp5遺伝子の領域内に二本鎖切断を形成するように設計された8種類のgRNA(A、B、B2、C、D、E2、E、F)のうちの1つをコードした第2のプラスミドと組み合わせた。各gRNAは、ヒトLRP5遺伝子の挿入される部分内のどの配列も認識しないように設計した。他の実験では、LTVEC、及びCas9をコードしたプラスミドを、欠失の標的とされるマウスLrp5遺伝子の領域内の異なる部位を標的とする2つの異なるgRNAをコードしたプラスミドと組み合わせた。
別の実験群では、LTVECは、補体成分5(C5又はHc(溶血性補体))のマウス遺伝子の76kbの欠失と同時に相同なヒトC5遺伝子の97kbのフラグメントによる置換を行うように設計した(図6)。標的遺伝子座は、C5(Hc)遺伝子の終止コドンまでのエクソン2を含むものとした。LTVECは、欠失させようとするマウスC5(Hc)遺伝子の76kbの配列に隣接したマウスC5(Hc)遺伝子座の部分から誘導された35kb及び31kbのゲノムDNAを含むホモロジーアームを隣接させたヒトC5遺伝子の97kbのフラグメントを含むものとした。別々の実験において、このC5(Hc)をヒト化するLTVECを、Cas9をコードしたプラスミド、及び欠失の標的とされるマウスC5(Hc)遺伝子の領域内に二本鎖切断を形成するように設計された6種類のgRNA(A、B、C、D、E、及びE2、表1を参照)のうちの1つをコードした第2のプラスミドと組み合わせた。各gRNAは、ヒトC5遺伝子の挿入される部分内のどの配列も認識しないように設計した。他の実験では、LTVEC、及びCas9をコードしたプラスミドを、欠失の標的とされるマウスC5(Hc)遺伝子の領域内の異なる部位を標的とする2つの異なるgRNAをコードしたプラスミドと組み合わせた。特定の実験では、C5(Hc)をヒト化するLTVECの代わりにCh25hを標的とするコントロールLTVECを使用した。コントロールLTVECは、Ch25h(約1kb)のコード配列全体を欠失させてCh25h遺伝子座内にプロマイシン及びネオマイシン選択カセットを挿入するように設計されたものであり、C5(Hc)遺伝子座における相同組換えについてターゲティングされなかった薬剤耐性クローンを選択するための手段として用いた。
別の実験群では、LTVECは、マウスRor1(チロシンタンパク質キナーゼ膜貫通受容体ROR1)遺伝子の110kbの欠失と同時に相同なヒトROR1遺伝子の134kbのフラグメントによる置換を行うように設計した(図8)。LTVECは、欠失させようとするマウスRor1遺伝子の110kbの配列に隣接したマウスRor1遺伝子座の部分から誘導された41.8kb及び96.4kbのゲノムDNAを含むホモロジーアームを隣接させたヒトROR1遺伝子の134kbのフラグメントを含むものとした。別々の実験において、このRor1をヒト化するLTVECを、Cas9をコードしたプラスミド、及び欠失の標的とされるマウスRor1遺伝子の領域内に二本鎖切断を形成するように設計された6種類のgRNA(A、B、C、D、E、及びF、表1を参照)のうちの1つをコードした第2のプラスミドと組み合わせた。各gRNAは、ヒトROR1遺伝子の挿入される部分内のどの配列も認識しないように設計した。他の実験では、LTVEC、及びCas9をコードしたプラスミドを、欠失の標的とされるRor1遺伝子内の異なる部位を標的とする2つの異なるgRNAをコードしたプラスミドと組み合わせた。
別の実験群では、LTVECは、マウスTrpa1(一過性受容体電位カチオンチャンネル、サブファミリーA、メンバー1)遺伝子の45.3kbの欠失と同時に相同なヒトTRPA1遺伝子の54.5kbのフラグメントによる置換を行うように設計した(図9)。LTVECは、欠失させようとするマウスTrpa1遺伝子の45.3kbの配列に隣接したマウスTrpa1遺伝子座の部分から誘導された41.0kb及び58.0kbのゲノムDNAを含むホモロジーアームを隣接させたヒトTRPA1遺伝子の54.5kbのフラグメントを含むものとした。別々の実験において、このTrpa1をヒト化するLTVECを、Cas9をコードしたプラスミド、及び欠失の標的とされるマウスTrpa1遺伝子の領域内に二本鎖切断を形成するように設計された8種類のgRNA(A、A2、B、C、D、E2、E、及びF、表1を参照)のうちの1つをコードした第2のプラスミドと組み合わせた。各gRNAは、ヒトTRPA1遺伝子の挿入される部分内のどの配列も認識しないように設計した。他の実験では、LTVEC、及びCas9をコードしたプラスミドを、欠失の標的とされるTrpa1遺伝子内の異なる部位を標的とする2つの異なるgRNAをコードしたプラスミドと組み合わせた。
gRNAの組み合わせによって誘導される大きな欠失の対立遺伝子構造
2種類のgRNAによって導かれるCas9切断事象により誘導される大きな欠失を有するクローンに更なる配列分析を行った(表7を参照)。これらの大きな欠失は、gRNAを、Lrp5 LTVEC又はほぼ30Mb離れたCh25h遺伝子を標的としたLTVECと組み合わせた場合にLrp5遺伝子座における大きな欠失がほぼ同じ頻度で得られたことから、同じ遺伝子座におけるLTVEC誘導型の相同組換えとは独立しているようであった。大きな欠失の特性評価を行うため、本発明者らは、4つのヒト化からの37個のクローン(15個のヘミ接合型のクローン及び22個の両対立遺伝子の大きな欠損を有するクローン)で欠失をスパンするようなPCRを行い、個々のPCR産物のクローンをシークエンシングした。配列では、38kb〜109kbの範囲の大きな欠失が確認された。これらのES細胞クローンのうちの2つ(Lrp5クローンAW−A8及びBP−D3)は、予想されるCas9切断部位の間に完全に修復された正確な欠失(68.2kb)を有しており、1つのクローン(HcクローンP−B12)は、38.1kbの欠失以外に1塩基対の挿入を有していた。ES細胞クローンのうち27個は、非相同性末端結合(NHEJ)による不正確な修復と符合する、Cas9切断部位を超えて延びる欠失を有していた。残りの7つのES細胞クローンは明らかなNHEJ誘導欠失と挿入の組み合わせを有しており(例えばLrp5クローンBP−F6及びHcクローンO−E4)、そのうち4つは200bpよりも大きい挿入を有しており、その由来するゲノム遺伝子座にマッピングすることができた(データは示さず)。Lrp5クローンBO−E9の210bpの挿入は、セントロメア方向にgRNA Fの標的部位の約2600bp外側に位置する同じ配列(+19番染色体、3589138〜3589347)に対して反転した向きであった。この配列は、Lrp5 LTVECの長い3’ホモロジーアーム内に存在していた。Lrp5クローンBP−F6及びBP−G7は、Lrp5 gRNA A及びFを、Cas9、及びLrp5からテロメア方向に30Mb離れたCh25h遺伝子を標的としたLTVECと組み合わせた実験から誘導された。クローンBP−F6は、ベクター主鎖の一部と同じ103bpのフラグメントがCh25h近傍の配列と同じでかつLTVECの長腕にも存在する163bpのフラグメント(+19番染色体、34478136〜34478298)と連結されたもので構成されていたことからCh25h LTVECの一方の末端から誘導されたものと考えられる266bpの挿入を有していた。このフラグメントを、内因性の染色体配列に対して反転した向きで欠失に挿入した。HcクローンO−E4は、gRNA Aの認識部位から約3.1kb離れた欠失配列内に見られる同じ配列に対して反転した254bpの挿入を有していた。HcクローンS−D5の1304bpの挿入は、2つのフラグメント、すなわち、予想されるgRNA E2に誘導されるCas9切断部位から約1.4kb離れた欠失配列内に見られる同じ配列と同じ向きの1238bpの部分と、gRNA E2切断部位の25bp外側の同じ配列の反転された向きの複製である第2の66bpの部分とから構成されていた。
両対立遺伝子の大きな欠失を有する22個のES細胞クローンのうちの21個が1つの固有の配列のみを有しており(表7)、これらがホモ接合型の対立遺伝子であることを示した。HcクローンS−A11では、12個のPCRクローンのうち、11個で同じ配列が見られた。異なる配列を有する1個のクローンは、2個の異なる欠失対立遺伝子を示唆している可能性があるが、本発明者らは、Hcヘミ接合型クローンのうちの2つ、N−D11及びO−F12でも同じ結果を見出した。複数のクローンにおける異なるホモ接合型の欠失対立遺伝子は、これらが、一方の染色体上の欠失が相同染色体上のCas9切断の相同組換え修復の鋳型として機能する遺伝子変換機構によって生じた可能性を示唆した。本発明者らは、VGF1 ES細胞株の129S6SvEvTac(129)とC57BL/6NTac(B6)とのF1ハイブリッド組成(Poueymirou et al.(2007)Nat.Biotechnol.25:91〜99;Valenzuela et al.(2003)Nat.Biotechnol.21:652〜659)を利用して、これらの細胞株間で19番染色体上のLrp5遺伝子座及び2番染色体上のHc遺伝子座(図示せず)の周辺の構造変異体(SV)及び1ヌクレオチド変異体(SNV)(下記で用いる5つのSVアッセイ及び10個のSNVアッセイについては図5を参照)におけるヘテロ接合性の喪失として遺伝子変換についてアッセイを行った(Lefebvre et al.(2001)Nat.Genet.27:257〜258)。いずれのヘミ接合性の喪失も、染色体全体が喪失した結果でないことを確認するため、129株とB6株との間で同じであった部位で染色体コピー数(CCN)アッセイを行った。Lrp5がヒト化又は欠失された対立遺伝子については、19番染色体のLrp5からテロメア方向に1.2Mb離れた点から長腕の末端までに位置する複数のSV及びSNVについてアッセイを行った(図5)。Lrp5の位置がセントロメアに近いために、この遺伝子のセントロメア側にはSVは見つからず、SNVが1個だけ見つかった。Hcについては、2番染色体上のこの遺伝子の両側で複数のSV及びSNVについてアッセイすることができた(図示せず)。Lrp5クローンのうちの6つについての結果を図10A〜E及び11A〜Cに示す。
ターゲティング効率に対するLTVECのホモロジーアームのサイズの影響を調べるため、補体成分5(C5又はHc(溶血性補体))のマウス遺伝子の76kbの欠失と同時に相同なヒトC5遺伝子の97kbのフラグメントによる置換を行うように設計された2種類のLTVECを比較した(図13)。標的遺伝子座は、C5(Hc)遺伝子の終止コドンまでのエクソン2を含むものとした。第1のLTVECは、欠失させようとするマウスC5(Hc)遺伝子の76kbの配列に隣接したマウスC5(Hc)遺伝子座の部分から誘導された35kb及び31kbのゲノムDNAを含むホモロジーアームを隣接させたヒトC5遺伝子の97kbのフラグメントを含むものとした(図13でLTVECとして示されたターゲティングベクターを参照)。第2のLTVECは、欠失させようとするマウスC5(Hc)遺伝子の76kbの配列に隣接したマウスC5(Hc)遺伝子座の部分から誘導された各5kbのゲノムDNAを含むホモロジーアームを隣接させたヒトC5遺伝子の97kbのフラグメントを含むものとした(図13でsTVECとして示されたターゲティングベクターを参照)。
CRISPR RNAの認識配列間及び切断部位間の距離をより短くすることがターゲティング効率に与える影響を調べるため、シチジン一リン酸−N−アセチルノイラミン酸ヒドロキシラーゼ(Cmah)のマウス遺伝子の18.2kbの欠失と同時にlacZレポーター及びヒグロマイシン耐性選択カセットを含んだインサートによる置換を行うように設計されたLTVECを設計した。このLTVECを、互いに間隔の近い配列を標的とした2種類のgRNAとともに使用した(図14)。標的遺伝子座は、Cmah遺伝子の最初の5個のエクソンを含むものとした。LTVECは、欠失させようとするマウスCmah遺伝子の18.2kbの配列に隣接したマウスCmah遺伝子座の部分から誘導された120kb及び57kbのゲノムDNAを含むホモロジーアームを隣接させた8.8kbのlazZ−hygrインサートを含むものとした。このLTVECを、Cas9及びターゲティングにより欠失させようとするマウスCmah遺伝子の領域の5’末端の近くに二本鎖切断を形成するように設計された2種類のgRNA(A及びB)をコードしたプラスミドと組み合わせた。これら2種類のgRNAは、欠失させようとする配列の5’末端のATGに近くの、互いに間隔の近い配列を標的としたものであり、標的切断部位は27bp離れていた(図15を参照)。2種類のgRNAにより導かれたCas9による切断によって、平滑末端を有する27bpの切り取られた配列が生じる。LTVEC単独をコントロールとして使用した。
1細胞期胚においてターゲティングによる大きな欠失を行うため、エクトドメインをコードするマウスLrp5(低密度リポタンパク質受容体関連タンパク質5)の部分の68kbの欠失と、場合により、2個の60ヌクレオチドのホモロジーアームを隣接させた4ヌクレオチドのインサートを含む一本鎖DNAドナー配列(124ヌクレオチドの長さ)を使用して4ヌクレオチドの挿入による置換とが同時に行われるように実験を設計した。4ヌクレオチドのインサートは、標的遺伝子座に挿入されて制限酵素部位を形成した。別々の実験において、Cas9タンパク質をタンパク質の形態で細胞質注入(CI)により導入するか、又はCas9をmRNAの形態で前核注入(PNI)又は電気穿孔法(EP)により導入した。Cas9は、ターゲティングにより欠失させようとするマウスLrp5遺伝子の領域内に二本鎖切断を形成するように設計された2種類のgRNA(A+F)と、また、場合により相同組換えドナーと組み合わせた。gRNAはRNAの形態で注入した。この後、得られた1対立遺伝子及び両対立遺伝子突然変異の頻度を評価した。
標準的な「対立遺伝子の改変(modification-of-allele)」(MOA)スクリーニング手法(例えば図17Aを参照)は、各試料の4つの生物学的複製物のCt値の平均をすべての試料のCt値の中央値と比較することによってTaqMan(登録商標)コピー数を決定するものである。「対立遺伝子の喪失(loss-of-allele)」では、TaqMan(登録商標)プローブを、ターゲティングにより欠失させようとする標的ゲノム遺伝子座の領域の上流(mTU)及び下流(mTD)領域に対して用いる。「対立遺伝子の獲得(gain-of-allele)」では、TaqMan(登録商標)プローブをネオマイシン耐性カセットに対して用いる。しかしながら、これらのプローブは、核酸インサートの任意の領域に対して設計することができる。二倍体のヘテロ接合型にターゲティングされたクローンでは、mTU、mTD、及びNeoプローブのそれぞれのTaqMan(登録商標)コピー数は1となる。二倍体のホモ接合型にターゲティングされたクローンでは、mTU及びmTDのそれぞれのTaqMan(登録商標)コピー数は0となり、NeoのTaqMan(登録商標)コピー数は2となる。同様に、二倍体のターゲティングされないクローンでは、mTU及びmTDのそれぞれのTaqMan(登録商標)コピー数は2となり、NeoのTaqMan(登録商標)コピー数は0となる。二倍体のヘテロ接合型に破壊されたクローンでは、mTU及びmTDのTaqMan(登録商標)コピー数は1となり、Neoのコピー数は0となる。二倍体のホモ接合型に破壊されたクローンでは、mTU、mTD、及びNeoプローブのそれぞれのTaqMan(登録商標)コピー数は0となる。
改変マウス免疫グロブリン重鎖インサートの約900kbの領域の正確な1工程の欠失と同時にloxP部位を隣接させたPgk−Neoインサート(ネオマイシンホスホトランスフェラーゼ遺伝子と機能的に連結されたホスホグリセリン酸キナーゼIプロモーター)による置換を行うため、本発明者らは以下の核酸分子を電気穿孔法によってマウスES細胞に導入した。すなわち、(1)LTVEC、(2)Cas9エンドヌクレアーゼをコードしたプラスミド、及び(3)4種類のCRISPR単鎖ガイドRNA(gRNA)をコードした1以上のプラスミド。各実験でLTVECは直鎖化した。改変の標的となる遺伝子は、可変領域遺伝子のセグメント(VH、DH、TH)を相当するヒトの遺伝子セグメントで置換したマウス免疫グロブリン重鎖遺伝子座の約900kbの領域とした。LTVECは、標的遺伝子座の約900kbの領域を欠失させ、ネオマイシンホスホトランスフェラーゼの発現を誘導してG418に対する耐性を付与する薬剤耐性カセットを挿入する相同組換え事象を誘導するように設計された19kbの5’ホモロジーアームと13kbの3’ホモロジーアームとを隣接させた約2kbの長さを有するPgk−Neoインサートを含むものとした。
Claims (81)
- 細胞内のゲノムに両対立遺伝子改変を行うための方法であって、前記ゲノムを、
(a)第1のCasタンパク質、
(b)ゲノム標的遺伝子座内の第1のCRISPR RNA認識配列にハイブリダイズする第1のCRISPR RNA、
(c)前記ゲノム標的遺伝子座内の第2のCRISPR RNA認識配列にハイブリダイズする第2のCRISPR RNA、
(d)tracrRNA、及び
(e)前記細胞が1細胞期胚である場合にターゲティングベクターの長さは5kb以下であるものとして、5’標的配列にハイブリダイズする5’ホモロジーアームと3’標的配列にハイブリダイズする3’ホモロジーアームとを隣接させた核酸インサートを含むターゲティングベクター、と接触させることを含み、
前記ゲノムが、前記ゲノム標的遺伝子座を含む第1及び第2の相同染色体のペアを含み、
前記第1のCasタンパク質が、前記第1及び第2のCRISPR RNA認識配列の少なくとも一方を切断して前記第1及び第2の相同染色体の少なくとも一方に少なくとも1つの二本鎖切断を生ずる、方法。 - 前記改変されたゲノムを有する細胞を同定することを更に含む、請求項1に記載の方法。
- 前記核酸インサートが、第1の標的配列にハイブリダイズする第1のホモロジーアームに隣接した選択カセットを含み
前記第1のホモロジーアームが前記5’ホモロジーアームであり、かつ前記第1の標的配列が前記5’標的配列であるか、又は前記第1のホモロジーアームが前記3’ホモロジーアームであり、かつ前記第1の標的配列が前記3’標的配列であり、
前記同定することが、
(a)前記細胞からDNAを得ることと、
(b)前記細胞の前記DNAを、前記第1の標的配列内に結合するプローブ、前記核酸インサート内に結合するプローブ、及び既知のコピー数を有する参照遺伝子内に結合するプローブに曝露することであって、各プローブは、結合する際に検出可能なシグナルを発生することと、
(c)前記プローブのそれぞれの結合からのシグナルを検出することと、
(d)前記参照遺伝子プローブからのシグナルを前記第1の標的配列プローブからのシグナルと比較して前記第1の標的配列のコピー数を決定し、前記参照遺伝子プローブからのシグナルを前記核酸インサートプローブからのシグナルと比較して前記核酸インサートのコピー数を決定することと、を含み、
前記核酸インサートのコピー数が1又は2であり、前記第1の標的配列のコピー数が2である場合には、前記ゲノム標的遺伝子座への前記核酸インサートのターゲティングによる挿入を示し、
前記核酸インサートのコピー数が1以上であり、前記第1の標的配列のコピー数が3以上である場合には、前記ゲノム標的遺伝子座以外のゲノム遺伝子座への前記核酸インサートのランダムな挿入を示す、請求項2に記載の方法。 - 前記第1のCasタンパク質が、前記第1及び第2の相同染色体のそれぞれの前記第1及び第2のCRISPR RNA認識配列の少なくとも一方を切断して前記第1及び第2の相同染色体のそれぞれに少なくとも1つの二本鎖切断を生ずる、請求項1〜3のいずれかに記載の方法。
- 前記第1のCasタンパク質が、前記第1及び第2の相同染色体の少なくとも一方の前記第1及び第2のCRISPR RNA認識配列を切断して前記第1及び第2の相同染色体の少なくとも一方に少なくとも2つの二本鎖切断を生ずる、請求項1〜4のいずれかに記載の方法。
- 前記ゲノムを、
(f)前記ゲノム標的遺伝子座内の第3のCRISPR RNA認識配列にハイブリダイズする第3のCRISPR RNA、及び
(g)前記ゲノム標的遺伝子座内の第4のCRISPR RNA認識配列にハイブリダイズする第4のCRISPR RNAと接触させることを更に含む、請求項1〜5のいずれかに記載の方法。 - (a)前記第1のCRISPR RNA認識配列と前記第3のCRISPR RNA認識配列とが、約25bp〜約50bp、約50bp〜約100bp、約100bp〜約150bp、約150bp〜約200bp、約200bp〜約250bp、約250bp〜約300bp、約300bp〜約350bp、約350bp〜約400bp、約400bp〜約450bp、約450bp〜約500bp、約500bp〜約600bp、約600bp〜約700bp、約700bp〜約800bp、約800bp〜約900bp、約900bp〜約1kb、約1kb〜約2kb、約2kb〜約3kb、約3kb〜約4kb、約4kb〜約5kb、約5kb〜約6kb、約6kb〜約7kb、約7kb〜約8kb、約8kb〜約9kb、約9kb〜約10kb、約10kb〜約20kb、約20kb〜約30kb、約30kb〜約40kb、約40kb〜約50kb、約50kb〜約60kb、約60kb〜約70kb、約70kb〜約80kb、約80kb〜約90kb、又は約90kb〜約100kbだけ離れており、かつ/又は
(b)前記第2のCRISPR RNA認識配列と前記第4のCRISPR RNA認識配列とが、約25bp〜約50bp、約50bp〜約100bp、約100bp〜約150bp、約150bp〜約200bp、約200bp〜約250bp、約250bp〜約300bp、約300bp〜約350bp、約350bp〜約400bp、約400bp〜約450bp、約450bp〜約500bp、約500bp〜約600bp、約600bp〜約700bp、約700bp〜約800bp、約800bp〜約900bp、約900bp〜約1kb、約1kb〜約2kb、約2kb〜約3kb、約3kb〜約4kb、約4kb〜約5kb、約5kb〜約6kb、約6kb〜約7kb、約7kb〜約8kb、約8kb〜約9kb、約9kb〜約10kb、約10kb〜約20kb、約20kb〜約30kb、約30kb〜約40kb、約40kb〜約50kb、約50kb〜約60kb、約60kb〜約70kb、約70kb〜約80kb、約80kb〜約90kb、又は約90kb〜約100kbだけ離れている、請求項6に記載の方法。 - 前記第1及び第3のCRISPR RNA認識配列がCRISPR RNA認識配列の第1のペアであり、前記第2及び第4のCRISPR RNA認識配列がCRISPR RNA認識配列の第2のペアであり、前記第1のペアと前記第2のペアとが、約25bp〜約50bp、約50bp〜約100bp、約100bp〜約150bp、約150bp〜約200bp、約200bp〜約250bp、約250bp〜約300bp、約300bp〜約350bp、約350bp〜約400bp、約400bp〜約450bp、約450bp〜約500bp、約500bp〜約600bp、約600bp〜約700bp、約700bp〜約800bp、約800bp〜約900bp、約900bp〜約1kb、約1kb〜約5kb、約5kb〜約10kb、約10kb〜約20kb、約20kb〜約40kb、約40kb〜約60kb、約60kb〜約80kb、約80kb〜約100kb、約100kb〜約150kb、約150kb〜約200kb、約200kb〜約300kb、約300kb〜約400kb、約400kb〜約500kb、約500kb〜約1Mb、約1Mb〜約1.5Mb、約1.5Mb〜約2Mb、約2Mb〜約2.5Mb、約2.5Mb〜約3Mb、約3Mb〜約4Mb、約4Mb〜約5Mb、約5Mb〜約10Mb、約10Mb〜約20Mb、約20Mb〜約30Mb、約30Mb〜約40Mb、約40Mb〜約50Mb、約50Mb〜約60Mb、約60Mb〜約70Mb、約70Mb〜約80Mb、約80Mb〜約90Mb、又は約90Mb〜約100Mbだけ離れている、請求項6又は7に記載の方法。
- 前記第1のCasタンパク質が、前記第1、第2、第3、及び第4のCRISPR RNA認識配列のうちの少なくとも2つを切断して前記第1及び第2の相同染色体の少なくとも一方に少なくとも2つの二本鎖切断を生ずる、請求項8に記載の方法。
- 前記第1のCasタンパク質が、前記第1、第2、第3、及び第4のCRISPR RNA認識配列のうちの少なくとも2つを切断して前記第1及び第2の相同染色体の両方に少なくとも2つの二本鎖切断を生ずる、請求項9に記載の方法。
- 前記ゲノムを前記第1及び第2のCRISPR RNAの両方と接触させることにより、前記ゲノムを前記第1のCRISPR RNA又は第2のCRISPR RNAのいずれか単独と接触させる場合と比較して両対立遺伝子改変の効率が高くなる、請求項1〜10のいずれかに記載の方法。
- 前記核酸インサートが前記5’標的配列と前記3’標的配列との間に挿入される、請求項1〜11のいずれかに記載の方法。
- 前記細胞が二倍体であり、前記両対立遺伝子改変によって、前記ゲノム標的遺伝子座にホモ接合性、複合ヘテロ接合性、又はヘミ接合性が生じる、請求項1〜12のいずれかに記載の方法。
- 前記両対立遺伝子改変が、前記第1の相同染色体の前記第1のCRISPR RNA認識配列と前記第2のCRISPR RNA認識配列との間の欠失を含む、請求項1〜13のいずれかに記載の方法。
- 前記両対立遺伝子改変が、前記第1及び第2の相同染色体の両方の前記第1のCRISPR RNA認識配列と前記第2のCRISPR RNA認識配列との間の前記欠失を含む、請求項14に記載の方法。
- 前記両対立遺伝子改変が、前記第1及び第2の相同染色体の両方の前記5’標的配列と前記3’標的配列との間への前記核酸インサートの挿入を更に含む、請求項15に記載の方法。
- 前記両対立遺伝子改変が、
(a)前記第1及び第2の相同染色体の両方の前記第1のCRISPR RNA認識配列と前記第2のCRISPR RNA認識配列との間の前記欠失、及び前記第2の相同染色体ではなく、前記第1の相同染色体の前記5’標的配列と前記3’標的配列との間への前記核酸インサートの挿入、
(b)前記第1の相同染色体の前記第1のCRISPR RNA認識配列と前記第2のCRISPR RNA認識配列との間の前記欠失、及び前記第2の相同染色体の前記第1のCRISPR RNA認識配列と前記第2のCRISPR RNA認識配列との間の遺伝子座の破壊、
(c)前記第1の相同染色体の前記第1のCRISPR RNA認識配列と前記第2のCRISPR RNA認識配列との間の前記欠失、前記第1の相同染色体の前記5’標的配列と前記3’標的配列との間への前記核酸インサートの挿入、及び前記第2の相同染色体の前記5’標的配列と前記3’標的配列との間の遺伝子座の破壊、又は、
(d)前記第1の相同染色体の前記第1のCRISPR RNA認識配列と前記第2のCRISPR RNA認識配列との間の前記欠失、及び前記第1の相同染色体の前記5’標的配列と前記3’標的配列との間への前記核酸インサートの挿入を含み、前記核酸インサート配列が、前記欠失される配列とホモロガスであるか又はオルソロガスである、請求項14に記載の方法。 - (a)前記第1及び第2のCRISPR RNA認識配列がそれぞれ、前記5’及び3’標的配列の両方から、少なくとも50bp、少なくとも100bp、少なくとも200bp、少なくとも300bp、少なくとも400bp、少なくとも500bp、少なくとも600bp、少なくとも700bp、少なくとも800bp、少なくとも900bp、少なくとも1kb、少なくとも2kb、少なくとも3kb、少なくとも4kb、少なくとも5kb、少なくとも6kb、少なくとも7kb、少なくとも8kb、少なくとも9kb、少なくとも10kb、少なくとも20kb、少なくとも30kb、少なくとも40kb、少なくとも50kb、少なくとも60kb、少なくとも70kb、少なくとも80kb、少なくとも90kb、若しくは少なくとも100kbに位置しているか、
(b)前記第1及び第2のCRISPR RNA認識配列がそれぞれ、前記5’及び3’標的配列の両方から、50bpより遠く、100bpより遠く、200bpより遠く、300bpより遠く、400bpより遠く、500bpより遠く、600bpより遠く、700bpより遠く、800bpより遠く、900bpより遠く、1kbより遠く、2kbより遠く、3kbより遠く、4kbより遠く、5kbより遠く、6kbより遠く、7kbより遠く、8kbより遠く、9kbより遠く、10kbより遠く、20kbより遠く、30kbより遠く、40kbより遠く、50kbより遠く、60kbより遠く、70kbより遠く、80kbより遠く、90kbより遠く、若しくは100kbより遠くに位置しているか、又は、
(c)前記第1及び第2のCRISPR RNA認識配列がそれぞれ、前記5’及び3’標的配列の両方から、約50bp〜約100bp、約200bp〜約300bp、約300bp〜約400bp、約400bp〜約500bp、約500bp〜約600bp、約600bp〜約700bp、約700bp〜約800bp、約800bp〜約900bp、約900bp〜約1kb、約1kb〜約2kb、約2kb〜約3kb、約3kb〜約4kb、約4kb〜約5kb、約5kb〜約10kb、約10kb〜約20kb、約20kb〜約30kb、約30kb〜約40kb、約40kb〜約50kb、若しくは約50kb〜約100kbに位置している、請求項1〜17のいずれかに記載の方法。 - (a)前記第1のCRISPR RNA認識配列と前記第2のCRISPR RNA認識配列とが、約1kb〜約5kb、約5kb〜約10kb、約10kb〜約20kb、約20kb〜約40kb、約40kb〜約60kb、約60kb〜約80kb、約80kb〜約100kb、約100kb〜約150kb、約150kb〜約200kb、約200kb〜約300kb、約300kb〜約400kb、約400kb〜約500kb、約500kb〜約1Mb、約1Mb〜約1.5Mb、約1.5Mb〜約2Mb、約2Mb〜約2.5Mb、若しくは約2.5Mb〜約3Mbだけ離れているか、
(b)前記第1のCRISPR RNA認識配列と前記第2のCRISPR RNA認識配列とが、少なくとも1kb、少なくとも2kb、少なくとも3kb、少なくとも4kb、少なくとも5kb、少なくとも10kb、少なくとも20kb、少なくとも30kb、少なくとも40kb、少なくとも50kb、少なくとも60kb、少なくとも70kb、少なくとも80kb、少なくとも90kb、少なくとも100kb、少なくとも110kb、少なくとも120kb、少なくとも130kb、少なくとも140kb、少なくとも150kb、少なくとも160kb、少なくとも170kb、少なくとも180kb、少なくとも190kb、少なくとも200kb、少なくとも250kb、少なくとも300kb、少なくとも350kb、少なくとも400kb、少なくとも450kb、若しくは少なくとも500kbだけ離れているか、
(c)前記第1のCRISPR RNA認識配列と前記第2のCRISPR RNA認識配列とが、約25bp〜約50bp、約50bp〜約100bp、約100bp〜約150bp、約150bp〜約200bp、約200bp〜約250bp、約250bp〜約300bp、約300bp〜約350bp、約350bp〜約400bp、約400bp〜約450bp、約450bp〜約500bp、約500bp〜約600bp、約600bp〜約700bp、約700bp〜約800bp、約800bp〜約900bp、若しくは約900bp〜約1kbだけ離れているか、又は、
(d)前記第1のCRISPR RNA認識配列と前記第2のCRISPR RNA認識配列とが、25bp未満、50bp未満、100bp未満、150bp未満、200bp未満、250bp未満、300bp未満、350bp未満、400bp未満、450bp未満、500bp未満、600bp未満、700bp未満、800bp未満、900bp未満、1kb未満、2kb未満、3kb未満、4kb未満、5kb未満、若しくは10kb未満だけ離れている、請求項1〜18のいずれかに記載の方法。 - (a)前記欠失される核酸が、約5kb〜約10kb、約10kb〜約20kb、約20kb〜約40kb、約40kb〜約60kb、約60kb〜約80kb、約80kb〜約100kb、約100kb〜約150kb、約150kb〜約200kb、約200kb〜約300kb、約300kb〜約400kb、約400kb〜約500kb、約500kb〜約1Mb、約1Mb〜約1.5Mb、約1.5Mb〜約2Mb、約2Mb〜約2.5Mb、若しくは約2.5Mb〜約3Mbであるか、又は、
(b)前記欠失される核酸が、少なくとも20kb、少なくとも30kb、少なくとも40kb、少なくとも50kb、少なくとも60kb、少なくとも70kb、少なくとも80kb、少なくとも90kb、少なくとも100kb、少なくとも110kb、少なくとも120kb、少なくとも130kb、少なくとも140kb、少なくとも150kb、少なくとも160kb、少なくとも170kb、少なくとも180kb、少なくとも190kb、少なくとも200kb、少なくとも250kb、少なくとも300kb、少なくとも350kb、少なくとも400kb、少なくとも450kb、若しくは少なくとも500kbであるか、又は、
(c)前記欠失される核酸が、少なくとも550kb、少なくとも600kb、少なくとも650kb、少なくとも700kb、少なくとも750kb、少なくとも800kb、少なくとも850kb、少なくとも900kb、少なくとも950kb、少なくとも1Mb、少なくとも1.5Mb、若しくは少なくとも2Mbである、請求項14〜19のいずれか一項に記載の方法。 - 前記5’及び3’標的配列が、前記ゲノム標的遺伝子座内にある、請求項1〜20のいずれかに記載の方法。
- 前記ターゲティングベクターが直鎖状の形態である、請求項1〜21のいずれかに記載の方法。
- 前記ターゲティングベクターが一本鎖又は二本鎖である、請求項1〜22のいずれかに記載の方法。
- 前記細胞が真核細胞である、請求項1〜23のいずれかに記載の方法。
- 前記真核細胞が、哺乳動物細胞、ヒト細胞、非ヒト細胞、げっ歯類細胞、マウス細胞、ラット細胞、多能性細胞、非多能性細胞、非ヒト多能性細胞、ヒト多能性細胞、げっ歯類多能性細胞、マウス多能性細胞、ラット多能性細胞、マウス胚性幹(ES)細胞、ラットES細胞、ヒトES細胞、ヒト成人幹細胞、発生的に制限されたヒト前駆細胞、ヒト人工多能性幹(iPS)細胞、又は1細胞期胚である、請求項24に記載の方法。
- 前記細胞が1細胞期胚であり、
(a)前記ターゲティングベクターの長さが約50ヌクレオチド〜約5kbであるか、又は、
(b)前記ターゲティングベクターが一本鎖DNAであり、その長さが約60〜約200ヌクレオチドである、請求項25に記載の方法。 - 前記細胞が1細胞期胚ではなく、
(a)前記ターゲティングベクターが、少なくとも10kbのラージターゲティングベクター(LTVEC)であるか、又は、
(b)前記ターゲティングベクターがラージターゲティングベクター(LTVEC)であり、前記LTVECの前記5’及び3’ホモロジーアームの合計が少なくとも10kbである、請求項25に記載の方法。 - (a)前記LTVECが、約50kb〜約300kb、約50kb〜約75kb、約75kb〜約100kb、約100kb〜約125kb、約125kb〜約150kb、約150kb〜約175kb、約175kb〜約200kb、約200kb〜約225kb、約225kb〜約250kb、約250kb〜約275kb、若しくは約275kb〜約300kbであるか、又は、
(b)前記LTVECの前記5’及び3’ホモロジーアームの合計が、約10kb〜約20kb、約20kb〜約30kb、約30kb〜約40kb、約40kb〜約50kb、約50kb〜約60kb、約60kb〜約70kb、約70kb〜約80kb、約80kb〜約90kb、約90kb〜約100kb、約100kb〜約110kb、約110kb〜約120kb、約120kb〜約130kb、約130kb〜約140kb、約140kb〜約150kb、約150kb〜約160kb、約160kb〜約170kb、約170kb〜約180kb、約180kb〜約190kb、又は約190kb〜約200kbである、請求項27に記載の方法。 - 前記細胞が1細胞期胚ではなく、
前記ターゲティングベクターがラージターゲティングベクター(LTVEC)であり、前記5’及び3’ホモロジーアームの合計が少なくとも10kbであり、
前記第1及び第2のCRISPR RNA認識配列がそれぞれ、前記5’及び3’標的配列の両方から、200bpより遠く、300bpより遠く、400bpより遠く、500bpより遠く、600bpより遠く、700bpより遠く、800bpより遠く、900bpより遠く、1kbより遠く、2kbより遠く、3kbより遠く、4kbより遠く、5kbより遠く、6kbより遠く、7kbより遠く、8kbより遠く、9kbより遠く、10kbより遠く、20kbより遠く、30kbより遠く、40kbより遠く、50kbより遠く、60kbより遠く、70kbより遠く、80kbより遠く、90kbより遠く、若しくは100kbより遠くに位置しており、
前記第1のCasタンパク質が、前記第1及び第2の相同染色体の少なくとも一方の前記第1及び第2のCRISPR RNA認識配列を切断して前記第1及び第2の相同染色体の少なくとも一方に少なくとも2つの二本鎖切断を生じ、
前記両対立遺伝子改変が、前記第1の相同染色体の前記第1のCRISPR RNA認識配列と前記第2のCRISPR RNA認識配列との間の前記欠失、及び前記第1の相同染色体の前記5’標的配列と前記3’標的配列との間への前記核酸インサートの挿入を含み、前記核酸インサート配列が、前記欠失される配列とホモロガスであるか又はオルソロガスである、請求項1〜3のいずれか一項に記載の方法。 - 第1の対立遺伝子についてヘテロ接合型である細胞内のゲノムを改変するための方法であって、前記ゲノムを、
(a)第1のCasタンパク質、
(b)tracrRNA、及び
(c)第1の非対立遺伝子特異的CRISPR RNA認識配列にハイブリダイズする第1のCRISPR RNA、と接触させることを含み、前記第1の対立遺伝子が第1の相同染色体上にあり、前記CRISPR RNA認識配列が、第2の相同染色体上の、前記第1の対立遺伝子に対応する遺伝子座に対してセントロメア側にあり、
前記第1のCasタンパク質が前記第1のCRISPR RNA認識配列を切断して二本鎖切断を生じ、前記細胞が前記第1の対立遺伝子についてホモ接合型となるように改変される、方法。 - 前記ゲノムを、第2の相同染色体上の、前記第1の対立遺伝子に対応する遺伝子座に対してセントロメア側の第2の非対立遺伝子特異的CRISPR RNA認識配列にハイブリダイズする第2のCRISPR RNAと接触させることを更に含み、
前記第1のCasタンパク質が、前記第1及び第2のCRISPR RNA認識配列の少なくとも一方を切断して少なくとも1つの二本鎖切断を生ずる、請求項30に記載の方法。 - 前記第1のCasタンパク質が、前記第1のCRISPR RNA認識配列及び前記第2のCRISPR RNA認識配列を切断する、請求項31に記載の方法。
- ヘテロ接合性の喪失が前記二本鎖切断のテロメア側で生じる、請求項30〜32のいずれか一項に記載の方法。
- 前記第1及び第2のCRISPR RNA認識配列が、前記第2の相同染色体上に位置し、前記第1の相同染色体上には位置していない、請求項31〜33のいずれか一項に記載の方法。
- 前記第1のCRISPR RNA認識部位が、セントロメアから約100bp〜約1kb、約1kb〜約10kb、約10kb〜約100kb、約100kb〜約1Mb、約1Mb〜約10Mb、約10Mb〜約20Mb、約20Mb〜約30Mb、約30Mb〜約40Mb、約40Mb〜約50Mb、約50Mb〜約60Mb、約60Mb〜約70Mb、約70Mb〜約80Mb、約80Mb〜約90Mb、又は約90Mb〜約100Mbにある、請求項30〜34のいずれか一項に記載の方法。
- 前記第1の対立遺伝子が、前記第1のCRISPR RNA認識配列から約100bp〜約1kb、約1kb〜約10kb、約10kb〜約100kb、約100kb〜約1Mb、約1Mb〜約10Mb、約10Mb〜約20Mb、約20Mb〜約30Mb、約30Mb〜約40Mb、約40Mb〜約50Mb、約50Mb〜約60Mb、約60Mb〜約70Mb、約70Mb〜約80Mb、約80Mb〜約90Mb、又は約90Mb〜約100Mbにある、請求項30〜35のいずれか一項に記載の方法。
- ヘテロ接合性の喪失によって置換される前記第2の相同染色体の領域が、約100bp〜約1kb、約1kb〜約10kb、約10kb〜約100kb、約100kb〜約1Mb、約1Mb〜約10Mb、約10Mb〜約20Mb、約20Mb〜約30Mb、約30Mb〜約40Mb、約40Mb〜約50Mb、約50Mb〜約60Mb、約60Mb〜約70Mb、約70Mb〜約80Mb、約80Mb〜約90Mb、又は約90Mb〜約100Mbである、請求項30〜36のいずれか一項に記載の方法。
- (a)前記第1の対立遺伝子が突然変異を有するか、又は、
(b)前記第1の対立遺伝子が野生型対立遺伝子であり、前記第2の相同染色体上の対応する遺伝子座が突然変異を有する、請求項30〜37のいずれか一項に記載の方法。 - 前記第1の対立遺伝子が突然変異を有し、前記突然変異がターゲティングによる改変である、請求項38に記載の方法。
- 第1の対立遺伝子についてヘテロ接合型である細胞内のゲノムを改変するための方法であって、前記ゲノムを、
(a)第1のCasタンパク質、
(b)tracrRNA、
(c)第2の対立遺伝子内の第1のCRISPR RNA認識配列にハイブリダイズする第1のCRISPR RNAであって、前記第1の対立遺伝子が第1の相同染色体上にあり、前記第2の対立遺伝子が第2の相同染色体上の対応する遺伝子座にある、第1のCRISPR RNA、及び
(d)前記第2の対立遺伝子内の第2のCRISPR RNA認識配列にハイブリダイズする第2のCRISPR RNA、と接触させることを含み、
前記第1のCasタンパク質が前記第1及び第2のCRISPR RNA認識配列の少なくとも一方を切断することで、少なくとも1つの二本鎖切断及び組換えを起こす末端配列を生じ、前記組換えが前記第1の対立遺伝子と前記第2の対立遺伝子との間で起こることで、前記第1の対立遺伝子についてホモ接合型である改変されたゲノムを生じる、方法。 - 前記第1のCasタンパク質が、前記第1のCRISPR RNA認識配列及び前記第2のCRISPR RNA認識配列を切断する、請求項40に記載の方法。
- 前記第1及び第2のCRISPR RNA認識配列が前記第2の対立遺伝子内に位置し、前記第1の対立遺伝子内には位置していない、請求項40又は41に記載の方法。
- (a)前記第1のCRISPR RNA認識配列と前記第2のCRISPR RNA認識配列とが、約1kb〜約5kb、約5kb〜約10kb、約10kb〜約20kb、約20kb〜約40kb、約40kb〜約60kb、約60kb〜約80kb、約80kb〜約100kb、約100kb〜約150kb、約150kb〜約200kb、約200kb〜約300kb、約300kb〜約400kb、約400kb〜約500kb、約500kb〜約1Mb、約1Mb〜約1.5Mb、約1.5Mb〜約2Mb、約2Mb〜約2.5Mb、若しくは約2.5Mb〜約3Mbだけ離れているか、又は、
(b)前記第1のCRISPR RNA認識配列と前記第2のCRISPR RNA認識配列とが、少なくとも1kb、少なくとも2kb、少なくとも3kb、少なくとも4kb、少なくとも5kb、少なくとも10kb、少なくとも20kb、少なくとも30kb、少なくとも40kb、少なくとも50kb、少なくとも60kb、少なくとも70kb、少なくとも80kb、少なくとも90kb、少なくとも100kb、少なくとも110kb、少なくとも120kb、少なくとも130kb、少なくとも140kb、少なくとも150kb、少なくとも160kb、少なくとも170kb、少なくとも180kb、少なくとも190kb、少なくとも200kb、少なくとも250kb、少なくとも300kb、少なくとも350kb、少なくとも400kb、少なくとも450kb、若しくは少なくとも500kbだけ離れている、請求項40〜42のいずれか一項に記載の方法。 - (a)前記第1の対立遺伝子と前記第2の対立遺伝子との間の配列の相違が、約100bp〜約200bp、約200bp〜約400bp、約400bp〜約600bp、約600bp〜約800bp、約800bp〜約1kb、約1kb〜約2kb、約2kb〜約3kb、約4kb〜約5kb、約5kb〜約10kb、約10kb〜約20kb、約20kb〜約40kb、約40kb〜約60kb、約60kb〜約80kb、約80kb〜約100kb、約100kb〜約150kb、約150kb〜約200kb、約200kb〜約300kb、約300kb〜約400kb、約400kb〜約500kb、約500kb〜約1Mb、約1Mb〜約1.5Mb、約1.5Mb〜約2Mb、約2Mb〜約2.5Mb、若しくは約2.5Mb〜約3Mbにわたるか、又は、
(b)前記第1の対立遺伝子と前記第2の対立遺伝子との間の配列の相違が、少なくとも100bp、少なくとも200bp、少なくとも300bp、少なくとも400bp、少なくとも500bp、少なくとも600bp、少なくとも700bp、少なくとも800bp、少なくとも800bp、少なくとも1kb、少なくとも2kb、少なくとも3kb、少なくとも4kb、少なくとも5kb、少なくとも6kb、少なくとも7kb、少なくとも8kb、少なくとも9kb、少なくとも10kb、20kb、少なくとも30kb、少なくとも40kb、少なくとも50kb、少なくとも60kb、少なくとも70kb、少なくとも80kb、少なくとも90kb、少なくとも100kb、少なくとも110kb、少なくとも120kb、少なくとも130kb、少なくとも140kb、少なくとも150kb、少なくとも160kb、少なくとも170kb、少なくとも180kb、少なくとも190kb、少なくとも200kb、少なくとも250kb、少なくとも300kb、少なくとも350kb、少なくとも400kb、少なくとも450kb、又は少なくとも500kbにわたる、請求項40〜43のいずれか一項に記載の方法。 - (a)前記第1の対立遺伝子がターゲティングによる改変を有し、前記第2の対立遺伝子が野生型対立遺伝子であるか、又は、
(b)前記第1の対立遺伝子が野生型対立遺伝子であり、前記第2の対立遺伝子が疾患原因突然変異を有する、請求項40〜44のいずれか一項に記載の方法。 - 前記組換えが、遺伝子変換又はヘテロ接合性の喪失(LOH)を含む、請求項40〜45のいずれか一項に記載の方法。
- 前記第1の対立遺伝子についてホモ接合型である細胞を同定することを更に含む、請求項30〜46のいずれか一項に記載の方法。
- 前記Casタンパク質と前記第1のCRISPR RNAとが天然に一緒に存在していない、請求項30〜47のいずれか一項に記載の方法。
- 前記細胞が真核細胞である、請求項30〜48のいずれか一項に記載の方法。
- 前記真核細胞が、哺乳動物細胞、ヒト細胞、非ヒト細胞、げっ歯類細胞、マウス細胞、ラット細胞、多能性細胞、非多能性細胞、非ヒト多能性細胞、ヒト多能性細胞、げっ歯類多能性細胞、マウス多能性細胞、ラット多能性細胞、マウス胚性幹(ES)細胞、ラットES細胞、ヒトES細胞、ヒト成人幹細胞、発生的に制限されたヒト前駆細胞、ヒト人工多能性幹(iPS)細胞、又は1細胞期胚である、請求項49に記載の方法。
- 前記第1のCasタンパク質がCas9である、請求項1〜50のいずれかに記載の方法。
- 前記第1のCasタンパク質が、二本鎖DNAの両方の鎖に対してヌクレアーゼ活性を有する、請求項1〜51のいずれかに記載の方法。
- 前記第1のCasタンパク質がニッカーゼである、請求項1〜51のいずれか一項に記載の方法。
- 前記第1のCasタンパク質がニッカーゼであり、前記方法が、前記ゲノムを、
(f)ニッカーゼである第2のCasタンパク質、
(g)第3のCRISPR RNA認識配列にハイブリダイズする第3のCRISPR RNA、及び
(h)第4のCRISPR RNA認識配列にハイブリダイズする第4のCRISPR RNA、と接触させることを更に含み、
前記第1のCasタンパク質が、前記第1のCRISPR RNA認識配列内及び前記第2のCRISPR RNA認識配列内のゲノムDNAの第1の鎖を切断し、前記第2のCasタンパク質が、前記第3のCRISPR RNA認識配列内及び前記第4のCRISPR RNA認識配列内のゲノムDNAの第2の鎖を切断する、請求項1〜39及び請求項41〜53のいずれか一項に記載の方法。 - (a)前記第1のCRISPR RNAと前記tracrRNAとが互いに第1のガイドRNA(gRNA)として融合され、かつ/若しくは前記第2のCRISPR RNAと前記tracrRNAとが互いに第2のgRNAとして融合されているか、又は、
(b)前記第1のCRISPR RNAと前記tracrRNAとが別々のRNA分子であり、かつ/若しくは前記第2のCRISPR RNAと前記tracrRNAとが別々のRNA分子である、請求項1〜29及び請求項31〜54のいずれか一項に記載の方法。 - 前記接触させることが、前記第1のCasタンパク質、前記第1及び第2のCRISPR RNA、及び前記tracrRNAを前記細胞に導入することを含む、請求項1〜55のいずれかに記載の方法。
- (a)前記第1のCasタンパク質が、タンパク質、前記第1のCasタンパク質をコードしたメッセンジャーRNA(mRNA)、若しくは前記第1のCasタンパク質をコードしたDNAの形態で前記細胞に導入され、
(b)前記第1のCRISPR RNAが、RNAの形態で、若しくは前記第1のCRISPR RNAをコードしたDNAの形態で前記細胞に導入され、
(c)前記第2のCRISPR RNAが、RNAの形態で、若しくは前記第2のCRISPR RNAをコードしたDNAの形態で前記細胞に導入され、かつ/又は
(d)前記tracrRNAが、RNAの形態で、若しくは前記tracrRNAをコードしたDNAの形態で前記細胞に導入される、請求項56に記載の方法。 - 前記第1のCasタンパク質、前記第1のCRISPR RNA、及び前記tracrRNAが、第1のタンパク質/RNA複合体として前記細胞に導入され、かつ/又は前記第1のCasタンパク質、前記第2のCRISPR RNA、及び前記tracrRNAが、第2のタンパク質/RNA複合体として前記細胞に導入される、請求項57に記載の方法。
- (a)前記第1のCasタンパク質をコードした前記DNAが、第1の発現コンストラクト内の第1のプロモーターと機能的に連結され、
(b)前記第1のCRISPR RNAをコードした前記DNAが、第2の発現コンストラクト内の第2のプロモーターと機能的に連結され、
(c)前記第2のCRISPR RNAをコードした前記DNAが、第3の発現コンストラクト内の第3のプロモーターと機能的に連結され、かつ/又は
(d)前記tracrRNAをコードした前記DNAが、第4の発現コンストラクト内の第4のプロモーターと機能的に連結されており、
前記第1、第2、第3、及び第4のプロモーターが前記細胞内で活性である、請求項57に記載の方法。 - 前記第1、第2、第3、及び/又は第4の発現コンストラクトが、1個の核酸分子の構成要素である、請求項59に記載の方法。
- (a)前記第1のCasタンパク質をコードした前記DNAが、第1の発現コンストラクト内の第1のプロモーターと機能的に連結され、
(b)前記第1のCRISPR RNAをコードした前記DNAと前記tracrRNAをコードした前記DNAとが、第1のガイドRNA(gRNA)をコードするDNAに互いに融合されるとともに第2の発現コンストラクト内の第2のプロモーターと機能的に連結され、かつ/又は、
(c)前記第2のCRISPR RNAをコードした前記DNAと前記tracrRNAをコードした前記DNAとが、第2のgRNAをコードするDNAに互いに融合されるとともに第3の発現コンストラクト内の第3のプロモーターと機能的に連結されており、
前記第1、第2、及び第3のプロモーターが前記細胞内で活性である、請求項57に記載の方法。 - 前記第1、第2、及び/又は第3の発現コンストラクトが、1個の核酸分子の構成要素である、請求項61に記載の方法。
- 前記細胞が、非相同性末端結合(NHEJ)を低下させるか、かつ/又は遺伝子変換若しくは相同組換え型修復(HDR)を増大させるように改変されている、請求項1〜62のいずれかに記載の方法。
- 前記細胞が、DNA−PK、PARP1、及びリガーゼIVのうちの1つ以上の発現又は活性を低下させるように改変されている、請求項63に記載の方法。
- 前記発現又は活性の低下が、誘導性、可逆性、時間特異的、及び/又は空間特異的である、請求項64に記載の方法。
- F0世代の非ヒト動物を作成するための方法であって、
(a)請求項1〜25及び請求項27〜65のいずれか一項に記載の方法によって作成された非ヒトES細胞を非ヒト宿主胚に導入することと、
(b)前記非ヒト宿主胚を代理母に懐胎させることと、を含み、
前記代理母が、前記両対立遺伝子改変を有する前記F0世代の非ヒト動物を作る、方法。 - F0世代の非ヒト動物を作成するための方法であって、請求項1〜26及び請求項30〜65のいずれか一項に記載の方法によって作成された遺伝子改変された1細胞期胚を代理母に移植することを含み、
前記代理母が、前記両対立遺伝子改変を有する前記F0世代の非ヒト動物を作る、方法。 - 前記非ヒト動物がマウス又はラットである、請求項66又は67に記載の方法。
- 1細胞期胚ではない二倍体細胞の標的ゲノム遺伝子座への核酸インサートのターゲティングによる挿入を同定するための方法であって、
(a)前記細胞からDNAを得ることであって、前記細胞が、第1の標的配列にハイブリダイズする第1のホモロジーアームと第2の標的配列にハイブリダイズする第2のホモロジーアームとを隣接させた前記核酸インサートを含むラージターゲティングベクター(LTVEC)と接触させられ、前記核酸インサートが、前記第1のホモロジーアームに隣接した選択カセットを含むことと、
(b)前記細胞の前記DNAを、前記第1の標的配列内に結合するプローブ、前記核酸インサート内に結合するプローブ、及び既知のコピー数を有する参照遺伝子内に結合するプローブに曝露することであって、各プローブは、結合する際に検出可能なシグナルを発生することと、
(c)前記プローブのそれぞれの結合からのシグナルを検出することと、
(d)前記参照遺伝子プローブからのシグナルを前記第1の標的配列プローブからのシグナルと比較して前記第1の標的配列のコピー数を決定し、前記参照遺伝子プローブからのシグナルを前記核酸インサートプローブからのシグナルと比較して前記核酸インサートのコピー数を決定することと、を含み、
前記核酸インサートのコピー数が1又は2であり、前記第1の標的配列のコピー数が2である場合には、前記標的ゲノム遺伝子座への前記核酸インサートのターゲティングによる挿入を示し、
前記核酸インサートのコピー数が1以上であり、前記第1の標的配列のコピー数が3以上である場合には、前記標的ゲノム遺伝子座以外のゲノム遺伝子座への前記核酸インサートのランダムな挿入を示す、方法。 - 前記第1の標的配列プローブの結合からのシグナルを用いて前記第1の標的配列の閾サイクル(Ct)値を決定し、前記参照遺伝子プローブの結合からのシグナルを用いて前記参照遺伝子の閾サイクル(Ct)値を決定し、前記第1の標的配列のCt値と前記参照遺伝子のCt値とを比較することによって前記第1の標的配列のコピー数を決定し、
前記核酸インサートプローブの結合からのシグナルを用いて前記核酸インサートの閾サイクル(Ct)値を決定し、前記第1の標的配列のCt値と前記参照遺伝子のCt値とを比較することによって前記核酸インサートのコピー数を決定する、請求項69に記載の方法。 - 前記選択カセットが薬剤耐性遺伝子を含む、請求項69又は70に記載の方法。
- 前記核酸インサートが、少なくとも5kb、少なくとも10kb、少なくとも20kb、少なくとも30kb、少なくとも40kb、少なくとも50kb、少なくとも60kb、少なくとも70kb、少なくとも80kb、少なくとも90kb、少なくとも100kb、少なくとも150kb、少なくとも200kb、少なくとも250kb、少なくとも300kb、少なくとも350kb、少なくとも400kb、少なくとも450kb、又は少なくとも500kbである、請求項69〜71のいずれか一項に記載の方法。
- 前記プローブが前記第1の標的配列及び前記選択カセット内で結合する配列間の距離が、100ヌクレオチド、200ヌクレオチド、300ヌクレオチド、400ヌクレオチド、500ヌクレオチド、600ヌクレオチド、700ヌクレオチド、800ヌクレオチド、900ヌクレオチド、1kb、1.5kb、2kb、2.5kb、3kb、3.5kb、4kb、4.5kb、又は5kb以下である、請求項69〜72のいずれか一項に記載の方法。
- 前記第2の標的配列のコピー数を決定することを更に含む、請求項69〜73のいずれか一項に記載の方法。
- 前記工程(b)が、前記細胞の前記DNAを前記第2の標的配列に結合するプローブに曝露することを更に含み、
前記工程(c)が、前記第2の標的配列プローブの結合からのシグナルを検出することを更に含み、
前記工程(d)が、前記参照遺伝子プローブからのシグナルを前記第2の標的配列プローブからのシグナルと比較して前記第2の標的配列のコピー数を決定することを更に含む、請求項74に記載の方法。 - 前記核酸インサート内の1つ以上の更なる配列の前記コピー数を決定することを更に含む、請求項69〜75のいずれか一項に記載の方法。
- 前記工程(b)が、前記細胞の前記DNAを前記核酸インサートに結合する1つ以上の更なるプローブに曝露することを更に含み、
前記工程(c)が、前記1つ以上の更なるプローブの結合からのシグナルを検出することを更に含み、
前記工程(d)が、前記参照遺伝子プローブからのシグナルを、前記1つ以上の更なる核酸インサートプローブからのシグナルと比較して前記核酸インサート内の前記1つ以上の更なる配列のコピー数を決定することを更に含む、請求項76に記載の方法。 - 前記核酸インサート内の前記1つ以上の更なる配列が、前記第2の標的配列に隣接した配列を含む、請求項76又は77に記載の方法。
- 前記LTVECが、前記標的ゲノム遺伝子座から内因性配列を欠失させるように設計されているか、又は
前記細胞が、Casタンパク質、標的ゲノム遺伝子座内の第1のCRISPR RNA認識配列にハイブリダイズする第1のCRISPR RNA、前記標的ゲノム遺伝子座内の第2のCRISPR RNA認識配列にハイブリダイズする第2のCRISPR RNA、及びtracrRNAと更に接触させられている、請求項69〜78のいずれか一項に記載の方法。 - 前記方法が、標的ゲノム遺伝子座における前記内因性配列のコピー数を決定することを更に含む、請求項79に記載の方法。
- 前記工程(b)が、前記細胞の前記DNAを前記標的ゲノム遺伝子座における前記内因性配列に結合するプローブに曝露することを更に含み、
前記工程(c)が、前記内因性配列プローブの結合からのシグナルを検出することを更に含み、
前記工程(d)が、前記参照遺伝子プローブからのシグナルを前記内因性配列プローブからのシグナルと比較して前記内因性配列のコピー数を決定することを更に含む、請求項80に記載の方法。
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462083005P | 2014-11-21 | 2014-11-21 | |
| US62/083,005 | 2014-11-21 | ||
| US201562182314P | 2015-06-19 | 2015-06-19 | |
| US62/182,314 | 2015-06-19 | ||
| US201562211421P | 2015-08-28 | 2015-08-28 | |
| US62/211,421 | 2015-08-28 | ||
| PCT/US2015/062023 WO2016081923A2 (en) | 2014-11-21 | 2015-11-20 | METHODS AND COMPOSITIONS FOR TARGETED GENETIC MODIFICATION USING PAIRED GUIDE RNAs |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2020112438A Division JP7101211B2 (ja) | 2014-11-21 | 2020-06-30 | ガイドrnaのペアを使用したターゲティングによる遺伝子改変の方法及び組成物 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2017535271A true JP2017535271A (ja) | 2017-11-30 |
| JP2017535271A5 JP2017535271A5 (ja) | 2018-12-20 |
| JP6727199B2 JP6727199B2 (ja) | 2020-07-22 |
Family
ID=54771219
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2017527267A Active JP6727199B2 (ja) | 2014-11-21 | 2015-11-20 | ガイドrnaのペアを使用したターゲティングによる遺伝子改変の方法及び組成物 |
| JP2020112438A Active JP7101211B2 (ja) | 2014-11-21 | 2020-06-30 | ガイドrnaのペアを使用したターゲティングによる遺伝子改変の方法及び組成物 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2020112438A Active JP7101211B2 (ja) | 2014-11-21 | 2020-06-30 | ガイドrnaのペアを使用したターゲティングによる遺伝子改変の方法及び組成物 |
Country Status (24)
| Country | Link |
|---|---|
| US (4) | US10457960B2 (ja) |
| EP (2) | EP3221457B1 (ja) |
| JP (2) | JP6727199B2 (ja) |
| KR (3) | KR102683423B1 (ja) |
| CN (2) | CN113444747A (ja) |
| AU (2) | AU2015349692B2 (ja) |
| BR (1) | BR112017010547A2 (ja) |
| CA (2) | CA3176380A1 (ja) |
| CY (1) | CY1121738T1 (ja) |
| DK (1) | DK3221457T3 (ja) |
| ES (1) | ES2731437T3 (ja) |
| HR (1) | HRP20190949T1 (ja) |
| HU (1) | HUE044907T2 (ja) |
| IL (3) | IL283585B2 (ja) |
| LT (1) | LT3221457T (ja) |
| MX (2) | MX2017006670A (ja) |
| NZ (1) | NZ731962A (ja) |
| PL (1) | PL3221457T3 (ja) |
| PT (1) | PT3221457T (ja) |
| RS (1) | RS58893B1 (ja) |
| RU (2) | RU2734770C2 (ja) |
| SG (2) | SG11201703747RA (ja) |
| SI (1) | SI3221457T1 (ja) |
| WO (1) | WO2016081923A2 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020050294A1 (ja) * | 2018-09-05 | 2020-03-12 | 学校法人慶應義塾 | 相同組換え効率上昇剤及びその使用 |
| JP6867635B1 (ja) * | 2020-04-06 | 2021-04-28 | 株式会社Logomix | ゲノム改変方法及びゲノム改変キット |
| JP2021511038A (ja) * | 2018-01-17 | 2021-05-06 | バーテックス ファーマシューティカルズ インコーポレイテッドVertex Pharmaceuticals Incorporated | Dna−pk阻害剤 |
| WO2023054573A1 (ja) * | 2021-09-30 | 2023-04-06 | 国立大学法人大阪大学 | 相同染色体の一方に特異的なdna欠失を有する細胞を製造する方法 |
| US12005127B2 (en) | 2018-01-17 | 2024-06-11 | Vertex Pharmaceuticals Incorporated | DNA-PK inhibitors |
| US12121524B2 (en) | 2019-01-16 | 2024-10-22 | Vertex Pharmaceuticals Incorporated | DNA-PK inhibitors |
Families Citing this family (110)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6596541B2 (en) | 2000-10-31 | 2003-07-22 | Regeneron Pharmaceuticals, Inc. | Methods of modifying eukaryotic cells |
| US20050144655A1 (en) | 2000-10-31 | 2005-06-30 | Economides Aris N. | Methods of modifying eukaryotic cells |
| EP2395016A3 (en) | 2003-05-30 | 2012-12-19 | Merus B.V. | Design and use of paired variable regions of specific binding molecules |
| US20100069614A1 (en) | 2008-06-27 | 2010-03-18 | Merus B.V. | Antibody producing non-human mammals |
| CA2853829C (en) | 2011-07-22 | 2023-09-26 | President And Fellows Of Harvard College | Evaluation and improvement of nuclease cleavage specificity |
| WO2013163628A2 (en) | 2012-04-27 | 2013-10-31 | Duke University | Genetic correction of mutated genes |
| JP6475172B2 (ja) | 2013-02-20 | 2019-02-27 | リジェネロン・ファーマシューティカルズ・インコーポレイテッドRegeneron Pharmaceuticals, Inc. | ラットの遺伝子組換え |
| RS62263B1 (sr) | 2013-04-16 | 2021-09-30 | Regeneron Pharma | Ciljana modifikacija genoma pacova |
| US20150044192A1 (en) | 2013-08-09 | 2015-02-12 | President And Fellows Of Harvard College | Methods for identifying a target site of a cas9 nuclease |
| US9359599B2 (en) | 2013-08-22 | 2016-06-07 | President And Fellows Of Harvard College | Engineered transcription activator-like effector (TALE) domains and uses thereof |
| US9228207B2 (en) | 2013-09-06 | 2016-01-05 | President And Fellows Of Harvard College | Switchable gRNAs comprising aptamers |
| US9388430B2 (en) | 2013-09-06 | 2016-07-12 | President And Fellows Of Harvard College | Cas9-recombinase fusion proteins and uses thereof |
| US9737604B2 (en) | 2013-09-06 | 2017-08-22 | President And Fellows Of Harvard College | Use of cationic lipids to deliver CAS9 |
| CA2930015A1 (en) | 2013-11-07 | 2015-05-14 | Editas Medicine, Inc. | Crispr-related methods and compositions with governing grnas |
| RU2725520C2 (ru) | 2013-12-11 | 2020-07-02 | Регенерон Фармасьютикалс, Инк. | Способы и композиции для направленной модификации генома |
| US11053481B2 (en) | 2013-12-12 | 2021-07-06 | President And Fellows Of Harvard College | Fusions of Cas9 domains and nucleic acid-editing domains |
| EP3708671A1 (en) | 2014-06-06 | 2020-09-16 | Regeneron Pharmaceuticals, Inc. | Methods and compositions for modifying a targeted locus |
| US9902971B2 (en) | 2014-06-26 | 2018-02-27 | Regeneron Pharmaceuticals, Inc. | Methods for producing a mouse XY embryonic (ES) cell line capable of producing a fertile XY female mouse in an F0 generation |
| US10077453B2 (en) | 2014-07-30 | 2018-09-18 | President And Fellows Of Harvard College | CAS9 proteins including ligand-dependent inteins |
| ES2741387T3 (es) | 2014-10-15 | 2020-02-10 | Regeneron Pharma | Métodos y composiciones para generar o mantener células pluripotentes |
| CA2963820A1 (en) | 2014-11-07 | 2016-05-12 | Editas Medicine, Inc. | Methods for improving crispr/cas-mediated genome-editing |
| SI3221457T1 (sl) | 2014-11-21 | 2019-08-30 | Regeneron Pharmaceuticals, Inc. | Postopki in sestavki za ciljno genetsko modifikacijo z uporabo vodilnih RNK v parih |
| WO2016100819A1 (en) | 2014-12-19 | 2016-06-23 | Regeneron Pharmaceuticals, Inc. | Methods and compositions for targeted genetic modification through single-step multiple targeting |
| EP3294896A1 (en) | 2015-05-11 | 2018-03-21 | Editas Medicine, Inc. | Optimized crispr/cas9 systems and methods for gene editing in stem cells |
| US10285388B2 (en) | 2015-05-29 | 2019-05-14 | Regeneron Pharmaceuticals, Inc. | Non-human animals having a disruption in a C9ORF72 locus |
| JP7396783B2 (ja) | 2015-06-09 | 2023-12-12 | エディタス・メディシン、インコーポレイテッド | 移植を改善するためのcrispr/cas関連方法および組成物 |
| CN108291218B (zh) | 2015-07-15 | 2022-08-19 | 新泽西鲁特格斯州立大学 | 核酸酶非依赖性靶向基因编辑平台及其用途 |
| US11667911B2 (en) | 2015-09-24 | 2023-06-06 | Editas Medicine, Inc. | Use of exonucleases to improve CRISPR/CAS-mediated genome editing |
| WO2017066497A2 (en) | 2015-10-13 | 2017-04-20 | Duke University | Genome engineering with type i crispr systems in eukaryotic cells |
| IL294014B2 (en) | 2015-10-23 | 2024-07-01 | Harvard College | Nucleobase editors and their uses |
| EP3370513A1 (en) * | 2015-11-06 | 2018-09-12 | The Jackson Laboratory | Large genomic dna knock-in and uses thereof |
| CA3011359A1 (en) | 2016-01-13 | 2017-07-20 | Regeneron Pharmaceuticals, Inc. | Rodents having an engineered heavy chain diversity region |
| US11666666B2 (en) * | 2016-02-11 | 2023-06-06 | The Regents Of The University Of California | Methods and compositions for modifying a mutant dystrophin gene in a cell's genome |
| US11597924B2 (en) | 2016-03-25 | 2023-03-07 | Editas Medicine, Inc. | Genome editing systems comprising repair-modulating enzyme molecules and methods of their use |
| EP4047092A1 (en) | 2016-04-13 | 2022-08-24 | Editas Medicine, Inc. | Cas9 fusion molecules, gene editing systems, and methods of use thereof |
| AU2017268458B2 (en) * | 2016-05-20 | 2022-07-21 | Regeneron Pharmaceuticals, Inc. | Methods for breaking immunological tolerance using multiple guide RNAS |
| US20210010022A1 (en) * | 2016-05-27 | 2021-01-14 | Cambridge Enterprise Limited | Novel nucleic acid construct |
| US20190323038A1 (en) * | 2016-06-17 | 2019-10-24 | Montana State Univesity | Bidirectional targeting for genome editing |
| WO2017220527A1 (en) * | 2016-06-20 | 2017-12-28 | Glycotope Gmbh | Means and methods for modifying multiple alleles |
| EP3476865B1 (en) * | 2016-06-28 | 2023-09-13 | Biocytogen Pharmaceuticals (Beijing) Co., Ltd. | Method for constructing pd-1 gene-modified humanized animal model and use thereof |
| US11124805B2 (en) | 2016-07-13 | 2021-09-21 | Vertex Pharmaceuticals Incorporated | Methods, compositions and kits for increasing genome editing efficiency |
| CN109563505A (zh) | 2016-07-28 | 2019-04-02 | 帝斯曼知识产权资产管理有限公司 | 用于真核细胞的组装系统 |
| AU2017302657A1 (en) | 2016-07-29 | 2019-02-14 | Regeneron Pharmaceuticals, Inc. | Mice comprising mutations resulting in expression of c-truncated fibrillin-1 |
| IL308426A (en) | 2016-08-03 | 2024-01-01 | Harvard College | Adenosine nuclear base editors and their uses |
| US11661590B2 (en) | 2016-08-09 | 2023-05-30 | President And Fellows Of Harvard College | Programmable CAS9-recombinase fusion proteins and uses thereof |
| US11542509B2 (en) | 2016-08-24 | 2023-01-03 | President And Fellows Of Harvard College | Incorporation of unnatural amino acids into proteins using base editing |
| CA3038548A1 (en) | 2016-09-30 | 2018-04-05 | Regeneron Pharmaceuticals, Inc. | Non-human animals having a hexanucleotide repeat expansion in a c9orf72 locus |
| SG11201903089RA (en) | 2016-10-14 | 2019-05-30 | Harvard College | Aav delivery of nucleobase editors |
| MX2019005256A (es) | 2016-11-04 | 2019-08-05 | Regeneron Pharma | Animales no humanos que tienen un locus de la cadena ligera lambda de inmunoglobulina modificado geneticamente. |
| CN106520831B (zh) * | 2016-11-18 | 2020-05-26 | 青岛市畜牧兽医研究所 | 一种哺乳动物基因组修饰方法 |
| WO2018119359A1 (en) | 2016-12-23 | 2018-06-28 | President And Fellows Of Harvard College | Editing of ccr5 receptor gene to protect against hiv infection |
| US12065666B2 (en) | 2017-01-05 | 2024-08-20 | Rutgers, The State University Of New Jersey | Targeted gene editing platform independent of DNA double strand break and uses thereof |
| EP4095263A1 (en) | 2017-01-06 | 2022-11-30 | Editas Medicine, Inc. | Methods of assessing nuclease cleavage |
| CN110446781A (zh) | 2017-02-15 | 2019-11-12 | 蓝鸟生物公司 | 供体修复模板多重基因组编辑 |
| US11898179B2 (en) | 2017-03-09 | 2024-02-13 | President And Fellows Of Harvard College | Suppression of pain by gene editing |
| EP3592777A1 (en) | 2017-03-10 | 2020-01-15 | President and Fellows of Harvard College | Cytosine to guanine base editor |
| JP7191388B2 (ja) | 2017-03-23 | 2022-12-19 | プレジデント アンド フェローズ オブ ハーバード カレッジ | 核酸によってプログラム可能なdna結合蛋白質を含む核酸塩基編集因子 |
| WO2018201086A1 (en) | 2017-04-28 | 2018-11-01 | Editas Medicine, Inc. | Methods and systems for analyzing guide rna molecules |
| US11560566B2 (en) | 2017-05-12 | 2023-01-24 | President And Fellows Of Harvard College | Aptazyme-embedded guide RNAs for use with CRISPR-Cas9 in genome editing and transcriptional activation |
| KR20200010311A (ko) * | 2017-05-16 | 2020-01-30 | 제네로스 바이오파마 리미티드 | 알츠하이머병의 치료를 위한 조성물 및 방법 |
| WO2018227114A1 (en) | 2017-06-09 | 2018-12-13 | Editas Medicine, Inc. | Engineered cas9 nucleases |
| SG11201911886PA (en) | 2017-06-27 | 2020-01-30 | Regeneron Pharma | Non-human animals comprising a humanized asgr1 locus |
| US11866726B2 (en) | 2017-07-14 | 2024-01-09 | Editas Medicine, Inc. | Systems and methods for targeted integration and genome editing and detection thereof using integrated priming sites |
| CN111801345A (zh) | 2017-07-28 | 2020-10-20 | 哈佛大学的校长及成员们 | 使用噬菌体辅助连续进化(pace)的进化碱基编辑器的方法和组合物 |
| US11021719B2 (en) | 2017-07-31 | 2021-06-01 | Regeneron Pharmaceuticals, Inc. | Methods and compositions for assessing CRISPER/Cas-mediated disruption or excision and CRISPR/Cas-induced recombination with an exogenous donor nucleic acid in vivo |
| CA3067872A1 (en) | 2017-07-31 | 2019-02-07 | Regeneron Pharmaceuticals, Inc. | Cas-transgenic mouse embryonic stem cells and mice and uses thereof |
| US11319532B2 (en) | 2017-08-30 | 2022-05-03 | President And Fellows Of Harvard College | High efficiency base editors comprising Gam |
| BR112020003609A2 (pt) | 2017-09-29 | 2020-09-01 | Regeneron Pharmaceuticals, Inc. | sistema e método para formar uma emulsão |
| WO2019072241A1 (en) | 2017-10-13 | 2019-04-18 | Beijing Biocytogen Co., Ltd | NON-HUMAN ANIMAL GENETICALLY MODIFIED WITH PD-1 HUMAN OR CHIMERIC |
| CN111757937A (zh) | 2017-10-16 | 2020-10-09 | 布罗德研究所股份有限公司 | 腺苷碱基编辑器的用途 |
| US20190141966A1 (en) | 2017-11-10 | 2019-05-16 | Regeneron Pharmaceuticals, Inc. | Non-Human Animals Comprising SLC30A8 Mutation And Methods Of Use |
| KR102709884B1 (ko) | 2017-11-30 | 2024-09-26 | 리제너론 파마슈티칼스 인코포레이티드 | 인간화된 trkb 유전자좌를 포함하는 비인간 동물 |
| JP7430636B2 (ja) | 2017-12-05 | 2024-02-13 | リジェネロン・ファーマシューティカルズ・インコーポレイテッド | 操作された免疫グロブリンラムダ軽鎖を有する非ヒト動物及びその使用 |
| WO2019148166A1 (en) * | 2018-01-29 | 2019-08-01 | Massachusetts Institute Of Technology | Methods of enhancing chromosomal homologous recombination |
| JP7334178B2 (ja) | 2018-03-19 | 2023-08-28 | リジェネロン・ファーマシューティカルズ・インコーポレイテッド | CRISPR/Cas系を使用した動物での転写モジュレーション |
| US20210071202A1 (en) | 2018-03-29 | 2021-03-11 | Jichi Medical University | Genome editing method, composition, cell, cell preparation, and method for producing cell preparation |
| WO2019213183A1 (en) * | 2018-04-30 | 2019-11-07 | The Trustees Of The University Of Pennsylvania | In utero crispr-mediated therapeutic editing of genes |
| US20210230637A1 (en) * | 2018-05-08 | 2021-07-29 | Osaka University | Method for producing homozygous cells |
| KR20210045360A (ko) | 2018-05-16 | 2021-04-26 | 신테고 코포레이션 | 가이드 rna 설계 및 사용을 위한 방법 및 시스템 |
| IL305557A (en) | 2018-06-14 | 2023-10-01 | Regeneron Pharma | Non-human animals with transgenic DH–DH rearrangement capacity, and their uses |
| BR112021003380A2 (pt) * | 2018-08-28 | 2021-05-18 | Flagship Pioneering Innovations Vi, Llc | métodos e composições para modulação de um genoma |
| US11845958B2 (en) * | 2018-09-05 | 2023-12-19 | Wisconsin Alumni Research Foundation | Genetically modified genes and cells, and methods of using same for silencing virus gene expression |
| CA3112612C (en) | 2018-09-13 | 2024-02-27 | Regeneron Pharmaceuticals, Inc. | Complement factor h gene knockout rat as a model of c3 glomerulopathy |
| WO2020067004A1 (ja) * | 2018-09-25 | 2020-04-02 | 公益財団法人微生物化学研究会 | 新規ウイルスベクターおよびその製造方法と使用方法 |
| CA3120799A1 (en) | 2018-12-20 | 2020-06-25 | Regeneron Pharmaceuticals, Inc. | Nuclease-mediated repeat expansion |
| WO2020191243A1 (en) | 2019-03-19 | 2020-09-24 | The Broad Institute, Inc. | Methods and compositions for editing nucleotide sequences |
| SG11202108454RA (en) | 2019-04-04 | 2021-09-29 | Regeneron Pharma | Non-human animals comprising a humanized coagulation factor 12 locus |
| JP2022534867A (ja) | 2019-06-04 | 2022-08-04 | リジェネロン・ファーマシューティカルズ・インコーポレイテッド | ベータスリップ変異を有するヒト化ttr遺伝子座を含む非ヒト動物と使用方法 |
| MA56117A (fr) | 2019-06-05 | 2022-04-13 | Regeneron Pharma | Animaux non humains ayant un répertoire de chaînes légères lambda limité exprimé à partir du locus kappa et leurs utilisations |
| CN113939595A (zh) | 2019-06-07 | 2022-01-14 | 瑞泽恩制药公司 | 包括人源化白蛋白基因座的非人动物 |
| WO2021108363A1 (en) | 2019-11-25 | 2021-06-03 | Regeneron Pharmaceuticals, Inc. | Crispr/cas-mediated upregulation of humanized ttr allele |
| EP4096396A1 (en) | 2020-01-28 | 2022-12-07 | Regeneron Pharmaceuticals, Inc. | Non-human animals comprising a humanized pnpla3 locus and methods of use |
| WO2021158883A1 (en) | 2020-02-07 | 2021-08-12 | Regeneron Pharmaceuticals, Inc. | Non-human animals comprising a humanized klkb1 locus and methods of use |
| US20210292754A1 (en) * | 2020-03-16 | 2021-09-23 | Pairwise Plants Services, Inc. | Natural guide architectures and methods of making and using the same |
| US20230102342A1 (en) | 2020-03-23 | 2023-03-30 | Regeneron Pharmaceuticals, Inc. | Non-human animals comprising a humanized ttr locus comprising a v30m mutation and methods of use |
| WO2021222327A1 (en) * | 2020-04-27 | 2021-11-04 | Duke University | A high-throughput screening method to discover optimal grna pairs for crispr-mediated exon deletion |
| DE112021002672T5 (de) | 2020-05-08 | 2023-04-13 | President And Fellows Of Harvard College | Vefahren und zusammensetzungen zum gleichzeitigen editieren beider stränge einer doppelsträngigen nukleotid-zielsequenz |
| EP4171215A2 (en) | 2020-06-26 | 2023-05-03 | Regeneron Pharmaceuticals, Inc. | Non-human animals comprising a humanized ace2 locus |
| CN112481297A (zh) * | 2020-11-02 | 2021-03-12 | 深圳先进技术研究院 | 一种制造染色体结构变异的方法 |
| CN112382339B (zh) * | 2020-11-17 | 2024-08-13 | 中国人民解放军军事科学院军事医学研究院 | 一种识别合子型基因激活zga基因的方法及装置 |
| US20240317849A1 (en) | 2020-12-23 | 2024-09-26 | Regeneron Pharmaceuticals, Inc. | Nucleic acids encoding anchor modified antibodies and uses thereof |
| WO2022251179A1 (en) * | 2021-05-26 | 2022-12-01 | Inscripta, Inc. | Crispr editing in diploid genomes |
| CN113832189B (zh) * | 2021-09-06 | 2022-10-21 | 中国人民解放军军事科学院军事医学研究院 | 一种用于敲除猪免疫球蛋白重链IGHG区的gRNA及其应用 |
| CN118251491A (zh) | 2021-10-28 | 2024-06-25 | 瑞泽恩制药公司 | 用于敲除C5的CRISPR/Cas相关方法及组合物 |
| KR20240117571A (ko) | 2021-12-08 | 2024-08-01 | 리제너론 파마슈티칼스 인코포레이티드 | 돌연변이 마이오실린 질환 모델 및 이의 용도 |
| WO2023122506A1 (en) | 2021-12-20 | 2023-06-29 | Regeneron Pharmaceuticals, Inc. | Non-human animals comprising humanized ace2 and tmprss loci |
| WO2023150798A1 (en) | 2022-02-07 | 2023-08-10 | Regeneron Pharmaceuticals, Inc. | Compositions and methods for defining optimal treatment timeframes in lysosomal disease |
| US20230257432A1 (en) | 2022-02-11 | 2023-08-17 | Regeneron Pharmaceuticals, Inc. | Compositions and methods for screening 4r tau targeting agents |
| WO2023250511A2 (en) | 2022-06-24 | 2023-12-28 | Tune Therapeutics, Inc. | Compositions, systems, and methods for reducing low-density lipoprotein through targeted gene repression |
| US20240224964A9 (en) | 2022-09-29 | 2024-07-11 | Regeneron Pharmaceuticals, Inc. | Correction of hepatosteatosis in humanized liver animals through restoration of il6/il6r/gp130 signaling in human hepatocytes |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004536556A (ja) * | 2000-10-31 | 2004-12-09 | リジェネロン・ファーマシューティカルズ・インコーポレイテッド | 真核生物細胞を改変する方法 |
| WO2014034412A1 (ja) * | 2012-08-31 | 2014-03-06 | 京セラメディカル株式会社 | 人工心肺ポンプ用駆動装置 |
| WO2014104878A1 (en) * | 2012-12-27 | 2014-07-03 | Keygene N.V. | Method for removing genetic linkage in a plant |
Family Cites Families (178)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4516228A (en) | 1983-08-25 | 1985-05-07 | Mobil Oil Corporation | Acoustic well logging device for detecting compressional and shear waves |
| US5830729A (en) | 1996-04-18 | 1998-11-03 | Institut Pasteur | I Sce I-induced gene replacement and gene conversion in embryonic stem cells |
| AU8587598A (en) | 1997-07-26 | 1999-02-16 | Wisconsin Alumni Research Foundation | Trans-species nuclear transfer |
| JP2002533130A (ja) | 1998-12-31 | 2002-10-08 | ザ ジェイ. デビッド グラッドストーン インスティテューツ | Hiv共受容体を発現するトランスジェニック齧歯類動物および齧歯類細胞株 |
| US6503717B2 (en) | 1999-12-06 | 2003-01-07 | Sangamo Biosciences, Inc. | Methods of using randomized libraries of zinc finger proteins for the identification of gene function |
| JP2002535995A (ja) | 1999-02-03 | 2002-10-29 | ザ チルドレンズ メディカル センター コーポレイション | 染色体標的部位での二本鎖dna切断の誘導を含む遺伝子修復 |
| CA2410516A1 (en) | 2000-05-31 | 2001-12-06 | Chiron Corporation | Compositions and methods for treating neoplastic disease using chemotherapy and radiation sensitizers |
| US20050144655A1 (en) | 2000-10-31 | 2005-06-30 | Economides Aris N. | Methods of modifying eukaryotic cells |
| US6596541B2 (en) | 2000-10-31 | 2003-07-22 | Regeneron Pharmaceuticals, Inc. | Methods of modifying eukaryotic cells |
| US7105348B2 (en) | 2000-10-31 | 2006-09-12 | Regeneron Pharmaceuticals, Inc. | Methods of modifying eukaryotic cells |
| AUPR451401A0 (en) | 2001-04-20 | 2001-05-24 | Monash University | A method of nuclear transfer |
| AU2003251286B2 (en) | 2002-01-23 | 2007-08-16 | The University Of Utah Research Foundation | Targeted chromosomal mutagenesis using zinc finger nucleases |
| US7612250B2 (en) | 2002-07-29 | 2009-11-03 | Trustees Of Tufts College | Nuclear transfer embryo formation method |
| US20030175968A1 (en) | 2002-10-30 | 2003-09-18 | Golic Kent G. | Gene targeting method |
| EP1587545A2 (en) | 2003-01-13 | 2005-10-26 | Mahendra S. Rao | Persistent expression of candidate molecule in proliferating stem and progenitor cells for delivery of therapeutic products |
| SG150548A1 (en) | 2003-12-01 | 2009-03-30 | Kudos Pharm Ltd | Dna damage repair inhibitors for treatment of cancer |
| ES2463476T3 (es) | 2004-10-19 | 2014-05-28 | Regeneron Pharmaceuticals, Inc. | Método para generar un ratón homocigótico para una modificación genética |
| FR2879622B1 (fr) | 2004-12-17 | 2008-02-01 | Agronomique Inst Nat Rech | Procede in vitro de production d'ovocytes ou d'oeufs presentant une modification genomique ciblee |
| GB0615327D0 (en) | 2006-03-30 | 2006-09-13 | Univ Edinburgh | Culture medium containing kinase inhibitors and uses thereof |
| EP2505058A1 (en) | 2006-03-31 | 2012-10-03 | Medarex, Inc. | Transgenic animals expressing chimeric antibodies for use in preparing human antibodies |
| CN101117633B (zh) | 2006-08-03 | 2011-07-20 | 上海交通大学附属儿童医院 | 一种细胞核移植方法 |
| US7771967B2 (en) | 2006-12-22 | 2010-08-10 | The J. David Gladstone Institutes | Nucleic acid encoding apolipoprotein E-I3 |
| CN101679977B (zh) | 2007-04-26 | 2013-05-01 | 桑格摩生物科学股份有限公司 | 向ppp1r12c基因座的靶向整合 |
| AU2008259939B2 (en) | 2007-06-01 | 2014-03-13 | Open Monoclonal Technology, Inc. | Compositions and methods for inhibiting endogenous immunoglobulin genes and producing transgenic human idiotype antibodies |
| KR20160015400A (ko) | 2008-08-22 | 2016-02-12 | 상가모 바이오사이언스 인코포레이티드 | 표적화된 단일가닥 분할 및 표적화된 통합을 위한 방법 및 조성물 |
| CN102625655B (zh) | 2008-12-04 | 2016-07-06 | 桑格摩生物科学股份有限公司 | 使用锌指核酸酶在大鼠中进行基因组编辑 |
| US20110041195A1 (en) * | 2009-08-11 | 2011-02-17 | Sangamo Biosciences, Inc. | Organisms homozygous for targeted modification |
| US8518392B2 (en) | 2009-08-14 | 2013-08-27 | Regeneron Pharmaceuticals, Inc. | Promoter-regulated differentiation-dependent self-deleting cassette |
| CA2779337A1 (en) | 2009-10-28 | 2011-05-05 | Helmholtz Zentrum Muenchen Deutsches Forschungszentrum Fuer Gesundheit U Nd Umwelt (Gmbh) | Homologous recombination in the oocyte |
| AU2010319894B2 (en) | 2009-10-29 | 2015-03-05 | Regeneron Pharmaceuticals, Inc. | Multifunctional alleles |
| US20120315670A1 (en) | 2009-11-02 | 2012-12-13 | Gen9, Inc. | Compositions and Methods for the Regulation of Multiple Genes of Interest in a Cell |
| WO2011068103A1 (ja) | 2009-12-01 | 2011-06-09 | 独立行政法人国立がん研究センター | ラット胚性幹細胞を用いたキメララットの作製法 |
| ES2583060T3 (es) | 2009-12-21 | 2016-09-16 | Keygene N.V. | Técnicas mejoradas para la transfección de protoplastos |
| UA113493C2 (xx) | 2010-01-22 | 2017-02-10 | Спосіб видалення ділянки днк в рослині | |
| JP2013518602A (ja) | 2010-02-09 | 2013-05-23 | サンガモ バイオサイエンシーズ, インコーポレイテッド | 部分的に一本鎖のドナー分子による標的化ゲノム改変 |
| EP3156062A1 (en) | 2010-05-17 | 2017-04-19 | Sangamo BioSciences, Inc. | Novel dna-binding proteins and uses thereof |
| GB201009732D0 (en) | 2010-06-10 | 2010-07-21 | Gene Bridges Gmbh | Direct cloning |
| CN104711218B (zh) | 2010-06-11 | 2018-09-25 | 瑞泽恩制药公司 | 由xy es细胞制备能育的xy雌性动物 |
| EP2596011B1 (en) | 2010-07-21 | 2018-10-03 | Sangamo Therapeutics, Inc. | Methods and compositions for modification of a hla locus |
| WO2012018726A1 (en) | 2010-08-02 | 2012-02-09 | Cellectis Sa | Method for increasing double-strand break-induced gene targeting |
| WO2012129198A1 (en) | 2011-03-23 | 2012-09-27 | Transposagen Biopharmaceuticals, Inc. | Genetically modified rat models for obesity and diabetes |
| WO2012168307A2 (en) | 2011-06-07 | 2012-12-13 | Helmholtz Zentrum München - Deutsches Forschungszentrum für Gesundheit und Umwelt (GmbH) | Improved recombination efficiency by inhibition of nhej dna repair |
| PL3424947T3 (pl) | 2011-10-28 | 2021-06-14 | Regeneron Pharmaceuticals, Inc. | Myszy z genetycznie zmodyfikowanym receptorem komórek t |
| US9637739B2 (en) | 2012-03-20 | 2017-05-02 | Vilnius University | RNA-directed DNA cleavage by the Cas9-crRNA complex |
| WO2013141680A1 (en) | 2012-03-20 | 2013-09-26 | Vilnius University | RNA-DIRECTED DNA CLEAVAGE BY THE Cas9-crRNA COMPLEX |
| ES2683071T3 (es) | 2012-04-25 | 2018-09-24 | Regeneron Pharmaceuticals, Inc. | Direccionamiento mediado por nucleasas con grandes vectores de direccionamiento |
| CN104471067B (zh) | 2012-05-07 | 2020-08-14 | 桑格摩生物治疗股份有限公司 | 用于核酸酶介导的转基因靶向整合的方法和组合物 |
| DK2800811T3 (en) | 2012-05-25 | 2017-07-17 | Univ Vienna | METHODS AND COMPOSITIONS FOR RNA DIRECTIVE TARGET DNA MODIFICATION AND FOR RNA DIRECTIVE MODULATION OF TRANSCRIPTION |
| BR112014031080A2 (pt) | 2012-06-12 | 2018-05-08 | Genentech Inc | métodos e composições de geração de alelos knock-out condicionais. |
| JP2015527889A (ja) | 2012-07-25 | 2015-09-24 | ザ ブロード インスティテュート, インコーポレイテッド | 誘導可能なdna結合タンパク質およびゲノム撹乱ツール、ならびにそれらの適用 |
| SG11201503059XA (en) | 2012-10-23 | 2015-06-29 | Toolgen Inc | Composition for cleaving a target dna comprising a guide rna specific for the target dna and cas protein-encoding nucleic acid or cas protein, and use thereof |
| KR102243092B1 (ko) | 2012-12-06 | 2021-04-22 | 시그마-알드리치 컴퍼니., 엘엘씨 | Crispr-기초된 유전체 변형과 조절 |
| WO2014093655A2 (en) | 2012-12-12 | 2014-06-19 | The Broad Institute, Inc. | Engineering and optimization of systems, methods and compositions for sequence manipulation with functional domains |
| US8697359B1 (en) | 2012-12-12 | 2014-04-15 | The Broad Institute, Inc. | CRISPR-Cas systems and methods for altering expression of gene products |
| EP4286402A3 (en) | 2012-12-12 | 2024-02-14 | The Broad Institute, Inc. | Crispr-cas component systems, methods and compositions for sequence manipulation |
| US20140310830A1 (en) * | 2012-12-12 | 2014-10-16 | Feng Zhang | CRISPR-Cas Nickase Systems, Methods And Compositions For Sequence Manipulation in Eukaryotes |
| SG11201504523UA (en) | 2012-12-12 | 2015-07-30 | Broad Inst Inc | Delivery, engineering and optimization of systems, methods and compositions for sequence manipulation and therapeutic applications |
| DK3064585T3 (da) | 2012-12-12 | 2020-04-27 | Broad Inst Inc | Konstruering og optimering af forbedrede systemer, fremgangsmåder og enzymsammensætninger til sekvensmanipulation |
| CN105121641A (zh) | 2012-12-17 | 2015-12-02 | 哈佛大学校长及研究员协会 | Rna-引导的人类基因组工程化 |
| WO2014127287A1 (en) | 2013-02-14 | 2014-08-21 | Massachusetts Institute Of Technology | Method for in vivo tergated mutagenesis |
| JP6475172B2 (ja) | 2013-02-20 | 2019-02-27 | リジェネロン・ファーマシューティカルズ・インコーポレイテッドRegeneron Pharmaceuticals, Inc. | ラットの遺伝子組換え |
| US10227610B2 (en) | 2013-02-25 | 2019-03-12 | Sangamo Therapeutics, Inc. | Methods and compositions for enhancing nuclease-mediated gene disruption |
| EP2922393B2 (en) | 2013-02-27 | 2022-12-28 | Helmholtz Zentrum München - Deutsches Forschungszentrum für Gesundheit und Umwelt (GmbH) | Gene editing in the oocyte by cas9 nucleases |
| US10612043B2 (en) * | 2013-03-09 | 2020-04-07 | Agilent Technologies, Inc. | Methods of in vivo engineering of large sequences using multiple CRISPR/cas selections of recombineering events |
| EP2971167B1 (en) | 2013-03-14 | 2019-07-31 | Caribou Biosciences, Inc. | Compositions and methods of nucleic acid-targeting nucleic acids |
| US20140273230A1 (en) | 2013-03-15 | 2014-09-18 | Sigma-Aldrich Co., Llc | Crispr-based genome modification and regulation |
| US9234213B2 (en) | 2013-03-15 | 2016-01-12 | System Biosciences, Llc | Compositions and methods directed to CRISPR/Cas genomic engineering systems |
| WO2014153470A2 (en) | 2013-03-21 | 2014-09-25 | Sangamo Biosciences, Inc. | Targeted disruption of t cell receptor genes using engineered zinc finger protein nucleases |
| WO2014165825A2 (en) | 2013-04-04 | 2014-10-09 | President And Fellows Of Harvard College | Therapeutic uses of genome editing with crispr/cas systems |
| US20160186208A1 (en) | 2013-04-16 | 2016-06-30 | Whitehead Institute For Biomedical Research | Methods of Mutating, Modifying or Modulating Nucleic Acid in a Cell or Nonhuman Mammal |
| RS62263B1 (sr) | 2013-04-16 | 2021-09-30 | Regeneron Pharma | Ciljana modifikacija genoma pacova |
| EP2986709A4 (en) | 2013-04-16 | 2017-03-15 | University Of Washington Through Its Center For Commercialization | Activating an alternative pathway for homology-directed repair to stimulate targeted gene correction and genome engineering |
| EP2796558A1 (en) | 2013-04-23 | 2014-10-29 | Rheinische Friedrich-Wilhelms-Universität Bonn | Improved gene targeting and nucleic acid carrier molecule, in particular for use in plants |
| AU2014262867B2 (en) | 2013-05-10 | 2019-12-05 | Sangamo Therapeutics, Inc. | Delivery methods and compositions for nuclease-mediated genome engineering |
| CA2913865C (en) | 2013-05-29 | 2022-07-19 | Cellectis | A method for producing precise dna cleavage using cas9 nickase activity |
| US20140359795A1 (en) | 2013-05-31 | 2014-12-04 | Recombinetics, Inc. | Genetic techniques for making animals with sortable sperm |
| WO2014201015A2 (en) | 2013-06-11 | 2014-12-18 | The Regents Of The University Of California | Methods and compositions for target dna modification |
| CA2915845A1 (en) | 2013-06-17 | 2014-12-24 | The Broad Institute, Inc. | Delivery, engineering and optimization of systems, methods and compositions for targeting and modeling diseases and disorders of post mitotic cells |
| EP4245853A3 (en) | 2013-06-17 | 2023-10-18 | The Broad Institute, Inc. | Optimized crispr-cas double nickase systems, methods and compositions for sequence manipulation |
| CN106062197A (zh) | 2013-06-17 | 2016-10-26 | 布罗德研究所有限公司 | 用于序列操纵的串联指导系统、方法和组合物的递送、工程化和优化 |
| KR20160056869A (ko) | 2013-06-17 | 2016-05-20 | 더 브로드 인스티튜트, 인코퍼레이티드 | 바이러스 구성성분을 사용하여 장애 및 질환을 표적화하기 위한 crispr-cas 시스템 및 조성물의 전달, 용도 및 치료 적용 |
| WO2014204723A1 (en) | 2013-06-17 | 2014-12-24 | The Broad Institute Inc. | Oncogenic models based on delivery and use of the crispr-cas systems, vectors and compositions |
| CA2915842C (en) | 2013-06-17 | 2022-11-29 | The Broad Institute, Inc. | Delivery and use of the crispr-cas systems, vectors and compositions for hepatic targeting and therapy |
| BR112015031639A2 (pt) | 2013-06-19 | 2019-09-03 | Sigma Aldrich Co Llc | integração alvo |
| US10011850B2 (en) | 2013-06-21 | 2018-07-03 | The General Hospital Corporation | Using RNA-guided FokI Nucleases (RFNs) to increase specificity for RNA-Guided Genome Editing |
| EP3019595A4 (en) | 2013-07-09 | 2016-11-30 | THERAPEUTIC USES OF A GENERIC CHANGE WITH CRISPR / CAS SYSTEMS | |
| US11060083B2 (en) | 2013-07-19 | 2021-07-13 | Larix Bioscience Llc | Methods and compositions for producing double allele knock outs |
| CN105392885B (zh) | 2013-07-19 | 2020-11-03 | 赖瑞克斯生物科技公司 | 用于产生双等位基因敲除的方法和组合物 |
| JP2016525004A (ja) | 2013-07-24 | 2016-08-22 | メディック アクティフ フェアトリープス ゲーエムベーハー | 食材細断装置 |
| US10563225B2 (en) | 2013-07-26 | 2020-02-18 | President And Fellows Of Harvard College | Genome engineering |
| MX2016002306A (es) * | 2013-08-22 | 2016-07-08 | Du Pont | Promotor u6 de polimerasa iii de soja y metodos de uso. |
| WO2015031775A1 (en) | 2013-08-29 | 2015-03-05 | Temple University Of The Commonwealth System Of Higher Education | Methods and compositions for rna-guided treatment of hiv infection |
| DE202014010413U1 (de) | 2013-09-18 | 2015-12-08 | Kymab Limited | Zellen und Organismen |
| WO2015048690A1 (en) | 2013-09-27 | 2015-04-02 | The Regents Of The University Of California | Optimized small guide rnas and methods of use |
| WO2015048577A2 (en) | 2013-09-27 | 2015-04-02 | Editas Medicine, Inc. | Crispr-related methods and compositions |
| WO2015052231A2 (en) | 2013-10-08 | 2015-04-16 | Technical University Of Denmark | Multiplex editing system |
| ES2881473T3 (es) | 2013-10-17 | 2021-11-29 | Sangamo Therapeutics Inc | Métodos de suministro y composiciones para la modificación por ingeniería genética del genoma mediada por nucleasas |
| WO2015068785A1 (ja) | 2013-11-06 | 2015-05-14 | 国立大学法人広島大学 | 核酸挿入用ベクター |
| CA2930015A1 (en) | 2013-11-07 | 2015-05-14 | Editas Medicine, Inc. | Crispr-related methods and compositions with governing grnas |
| US10787684B2 (en) | 2013-11-19 | 2020-09-29 | President And Fellows Of Harvard College | Large gene excision and insertion |
| JP2016538001A (ja) | 2013-11-28 | 2016-12-08 | ホライズン・ジェノミクス・ゲーエムベーハー | 体細胞半数体ヒト細胞株 |
| RU2725520C2 (ru) | 2013-12-11 | 2020-07-02 | Регенерон Фармасьютикалс, Инк. | Способы и композиции для направленной модификации генома |
| CN106536729A (zh) | 2013-12-12 | 2017-03-22 | 布罗德研究所有限公司 | 使用粒子递送组分靶向障碍和疾病的crispr‑cas系统和组合物的递送、用途和治疗应用 |
| KR20160097327A (ko) | 2013-12-12 | 2016-08-17 | 더 브로드 인스티튜트, 인코퍼레이티드 | 유전자 산물, 구조 정보 및 유도성 모듈형 cas 효소의 발현의 변경을 위한 crispr-cas 시스템 및 방법 |
| AU2014361784A1 (en) | 2013-12-12 | 2016-06-23 | Massachusetts Institute Of Technology | Delivery, use and therapeutic applications of the CRISPR-Cas systems and compositions for HBV and viral diseases and disorders |
| WO2015089462A1 (en) | 2013-12-12 | 2015-06-18 | The Broad Institute Inc. | Delivery, use and therapeutic applications of the crispr-cas systems and compositions for genome editing |
| CA2932472A1 (en) | 2013-12-12 | 2015-06-18 | Massachusetts Institute Of Technology | Compositions and methods of use of crispr-cas systems in nucleotide repeat disorders |
| EP4219699A1 (en) | 2013-12-12 | 2023-08-02 | The Broad Institute, Inc. | Engineering of systems, methods and optimized guide compositions with new architectures for sequence manipulation |
| KR102274445B1 (ko) | 2013-12-19 | 2021-07-08 | 아미리스 인코퍼레이티드 | 게놈 삽입을 위한 방법 |
| JP6747974B2 (ja) | 2014-01-08 | 2020-08-26 | プレジデント アンド フェローズ オブ ハーバード カレッジ | Rna誘導型遺伝子ドライブ |
| JP6479024B2 (ja) | 2014-01-24 | 2019-03-06 | ザ チルドレンズ メディカル センター コーポレーション | 抗体親和性の最適化のための高スループットマウスモデル |
| WO2015117041A1 (en) | 2014-01-30 | 2015-08-06 | Nair Ramesh B | Gene modification-mediated methods and compositions for generating dominant traits in eukaryotic systems |
| US10233456B2 (en) | 2014-01-30 | 2019-03-19 | The Board Of Trustees Of The University Of Arkansas | Method, vectors, cells, seeds and kits for stacking genes into a single genomic site |
| EP3114227B1 (en) | 2014-03-05 | 2021-07-21 | Editas Medicine, Inc. | Crispr/cas-related methods and compositions for treating usher syndrome and retinitis pigmentosa |
| WO2015138510A1 (en) | 2014-03-10 | 2015-09-17 | Editas Medicine., Inc. | Crispr/cas-related methods and compositions for treating leber's congenital amaurosis 10 (lca10) |
| EP3116533B1 (en) | 2014-03-12 | 2020-08-12 | Precision Biosciences, Inc. | Dystrophin gene exon deletion using engineered nucleases |
| IL247679B1 (en) | 2014-03-14 | 2024-07-01 | Cibus Europe Bv | Methods and compositions for increasing the efficiency of targeted gene modification through the use of oligonucleotide-mediated gene repair |
| EP3122877B1 (en) | 2014-03-26 | 2023-03-22 | University of Maryland, College Park | Targeted genome editing in zygotes of domestic large animals |
| GB201406968D0 (en) | 2014-04-17 | 2014-06-04 | Green Biologics Ltd | Deletion mutants |
| WO2015163733A1 (en) | 2014-04-24 | 2015-10-29 | Institute For Basic Science | A method of selecting a nuclease target sequence for gene knockout based on microhomology |
| GB201407852D0 (en) | 2014-05-02 | 2014-06-18 | Iontas Ltd | Preparation of libraries od protein variants expressed in eukaryotic cells and use for selecting binding molecules |
| US20170088819A1 (en) | 2014-05-16 | 2017-03-30 | Vrije Universiteit Brussel | Genetic correction of myotonic dystrophy type 1 |
| JP2017518372A (ja) | 2014-05-30 | 2017-07-06 | ザ ボード オブ トラスティーズ オブ ザ レランド スタンフォード ジュニア ユニバーシティー | 潜伏性ウイルス感染用の処置剤を送達するための組成物および方法 |
| EP3708671A1 (en) | 2014-06-06 | 2020-09-16 | Regeneron Pharmaceuticals, Inc. | Methods and compositions for modifying a targeted locus |
| AU2015277369B2 (en) | 2014-06-16 | 2021-08-19 | The Johns Hopkins University | Compositions and methods for the expression of CRISPR guide RNAs using the H1 promoter |
| US9902971B2 (en) | 2014-06-26 | 2018-02-27 | Regeneron Pharmaceuticals, Inc. | Methods for producing a mouse XY embryonic (ES) cell line capable of producing a fertile XY female mouse in an F0 generation |
| KR20170032406A (ko) | 2014-07-15 | 2017-03-22 | 주노 쎄러퓨티크스 인코퍼레이티드 | 입양 세포 치료를 위한 조작된 세포 |
| US9944933B2 (en) | 2014-07-17 | 2018-04-17 | Georgia Tech Research Corporation | Aptamer-guided gene targeting |
| WO2016011428A1 (en) | 2014-07-17 | 2016-01-21 | University Of Pittsburgh - Of The Commonwealth System Of Higher Education | Methods of treating cells containing fusion genes |
| ES2730378T3 (es) | 2014-08-27 | 2019-11-11 | Caribou Biosciences Inc | Procedimientos para incrementar la eficiencia de la modificación mediada por Cas9 |
| EP3188763B1 (en) | 2014-09-02 | 2020-05-13 | The Regents of The University of California | Methods and compositions for rna-directed target dna modification |
| WO2016049163A2 (en) | 2014-09-24 | 2016-03-31 | The Broad Institute Inc. | Use and production of chd8+/- transgenic animals with behavioral phenotypes characteristic of autism spectrum disorder |
| WO2016049024A2 (en) | 2014-09-24 | 2016-03-31 | The Broad Institute Inc. | Delivery, use and therapeutic applications of the crispr-cas systems and compositions for modeling competition of multiple cancer mutations in vivo |
| WO2016049258A2 (en) | 2014-09-25 | 2016-03-31 | The Broad Institute Inc. | Functional screening with optimized functional crispr-cas systems |
| AU2015323973A1 (en) | 2014-09-29 | 2017-04-20 | The Jackson Laboratory | High efficiency, high throughput generation of genetically modified mammals by electroporation |
| AU2015324935B2 (en) | 2014-10-01 | 2021-04-08 | The General Hospital Corporation | Methods for increasing efficiency of nuclease-induced homology-directed repair |
| EP3204496A1 (en) | 2014-10-10 | 2017-08-16 | Editas Medicine, Inc. | Compositions and methods for promoting homology directed repair |
| WO2016061073A1 (en) | 2014-10-14 | 2016-04-21 | Memorial Sloan-Kettering Cancer Center | Composition and method for in vivo engineering of chromosomal rearrangements |
| ES2741387T3 (es) | 2014-10-15 | 2020-02-10 | Regeneron Pharma | Métodos y composiciones para generar o mantener células pluripotentes |
| WO2016061481A1 (en) | 2014-10-17 | 2016-04-21 | The Penn State Research Foundation | Methods and compositions for multiplex rna guided genome editing and other rna technologies |
| US20170306306A1 (en) | 2014-10-24 | 2017-10-26 | Life Technologies Corporation | Compositions and Methods for Enhancing Homologous Recombination |
| WO2016073955A2 (en) | 2014-11-06 | 2016-05-12 | President And Fellows Of Harvard College | Cells lacking b2m surface expression and methods for allogeneic administration of such cells |
| CA2963820A1 (en) | 2014-11-07 | 2016-05-12 | Editas Medicine, Inc. | Methods for improving crispr/cas-mediated genome-editing |
| SI3221457T1 (sl) | 2014-11-21 | 2019-08-30 | Regeneron Pharmaceuticals, Inc. | Postopki in sestavki za ciljno genetsko modifikacijo z uporabo vodilnih RNK v parih |
| KR20170081268A (ko) | 2014-11-27 | 2017-07-11 | 단지거 이노베이션즈 엘티디. | 게놈 편집용 핵산 구조체 |
| US20170266320A1 (en) | 2014-12-01 | 2017-09-21 | President And Fellows Of Harvard College | RNA-Guided Systems for In Vivo Gene Editing |
| WO2016094880A1 (en) | 2014-12-12 | 2016-06-16 | The Broad Institute Inc. | Delivery, use and therapeutic applications of crispr systems and compositions for genome editing as to hematopoietic stem cells (hscs) |
| EP3230452A1 (en) | 2014-12-12 | 2017-10-18 | The Broad Institute Inc. | Dead guides for crispr transcription factors |
| WO2016094874A1 (en) | 2014-12-12 | 2016-06-16 | The Broad Institute Inc. | Escorted and functionalized guides for crispr-cas systems |
| WO2016097751A1 (en) | 2014-12-18 | 2016-06-23 | The University Of Bath | Method of cas9 mediated genome engineering |
| WO2016100819A1 (en) | 2014-12-19 | 2016-06-23 | Regeneron Pharmaceuticals, Inc. | Methods and compositions for targeted genetic modification through single-step multiple targeting |
| EP3239298A4 (en) | 2014-12-26 | 2018-06-13 | Riken | Gene knockout method |
| WO2016112242A1 (en) | 2015-01-08 | 2016-07-14 | President And Fellows Of Harvard College | Split cas9 proteins |
| EP3242938B1 (en) | 2015-01-09 | 2020-01-08 | Bio-Rad Laboratories, Inc. | Detection of genome editing |
| CN107429263A (zh) | 2015-01-15 | 2017-12-01 | 斯坦福大学托管董事会 | 调控基因组编辑的方法 |
| WO2016130697A1 (en) | 2015-02-11 | 2016-08-18 | Memorial Sloan Kettering Cancer Center | Methods and kits for generating vectors that co-express multiple target molecules |
| GB2535532B (en) | 2015-02-23 | 2021-05-12 | Knorr Bremse Systeme Fuer Nutzfahrzeuge Gmbh | Brake valve arrangement |
| EP3262171A2 (en) | 2015-02-23 | 2018-01-03 | Crispr Therapeutics AG | Materials and methods for treatment of hemoglobinopathies |
| US20180200387A1 (en) | 2015-02-23 | 2018-07-19 | Crispr Therapeutics Ag | Materials and methods for treatment of human genetic diseases including hemoglobinopathies |
| US11261466B2 (en) | 2015-03-02 | 2022-03-01 | Sinai Health System | Homologous recombination factors |
| GB201504223D0 (en) | 2015-03-12 | 2015-04-29 | Genome Res Ltd | Biallelic genetic modification |
| WO2016154579A2 (en) | 2015-03-26 | 2016-09-29 | Editas Medicine, Inc. | Crispr/cas-mediated gene conversion |
| MX2017012407A (es) | 2015-03-27 | 2018-03-07 | Harvard College | Celulas t modificadas y métodos para hacer y usar las mismas. |
| EP3277816B1 (en) | 2015-04-01 | 2020-06-17 | Editas Medicine, Inc. | Crispr/cas-related methods and compositions for treating duchenne muscular dystrophy and becker muscular dystrophy |
| US10155938B2 (en) | 2015-04-14 | 2018-12-18 | City Of Hope | Coexpression of CAS9 and TREX2 for targeted mutagenesis |
| US20180346879A1 (en) | 2015-04-22 | 2018-12-06 | Genea Ip Holdings Pty Limited | Generation of muscle-lineage clels from stem cells |
| EP3289081B1 (en) | 2015-04-27 | 2019-03-27 | Genethon | Compositions and methods for the treatment of nucleotide repeat expansion disorders |
| WO2016176690A2 (en) | 2015-04-30 | 2016-11-03 | The Trustees Of Columbia University In The City Of New York | Gene therapy for autosomal dominant diseases |
| CN107849547B (zh) | 2015-05-16 | 2022-04-19 | 建新公司 | 深内含子突变的基因编辑 |
| US9790490B2 (en) | 2015-06-18 | 2017-10-17 | The Broad Institute Inc. | CRISPR enzymes and systems |
| KR20180054871A (ko) | 2015-10-08 | 2018-05-24 | 프레지던트 앤드 펠로우즈 오브 하바드 칼리지 | 다중화 게놈 편집 |
| AU2016341041A1 (en) | 2015-10-20 | 2018-03-15 | Pioneer Hi-Bred International, Inc. | Methods and compositions for marker-free genome modification |
| EP3370513A1 (en) | 2015-11-06 | 2018-09-12 | The Jackson Laboratory | Large genomic dna knock-in and uses thereof |
| US11597924B2 (en) | 2016-03-25 | 2023-03-07 | Editas Medicine, Inc. | Genome editing systems comprising repair-modulating enzyme molecules and methods of their use |
| WO2017173004A1 (en) | 2016-03-30 | 2017-10-05 | Mikuni Takayasu | A method for in vivo precise genome editing |
| EP3443080B1 (en) | 2016-04-13 | 2021-08-11 | University of Massachusetts | Repairing compound heterozygous recessive mutations by allele exchange |
| AU2017268458B2 (en) | 2016-05-20 | 2022-07-21 | Regeneron Pharmaceuticals, Inc. | Methods for breaking immunological tolerance using multiple guide RNAS |
| WO2019148166A1 (en) | 2018-01-29 | 2019-08-01 | Massachusetts Institute Of Technology | Methods of enhancing chromosomal homologous recombination |
-
2015
- 2015-11-20 SI SI201530759T patent/SI3221457T1/sl unknown
- 2015-11-20 JP JP2017527267A patent/JP6727199B2/ja active Active
- 2015-11-20 PT PT15804289T patent/PT3221457T/pt unknown
- 2015-11-20 BR BR112017010547A patent/BR112017010547A2/pt not_active Application Discontinuation
- 2015-11-20 IL IL283585A patent/IL283585B2/en unknown
- 2015-11-20 KR KR1020237015378A patent/KR102683423B1/ko active IP Right Grant
- 2015-11-20 KR KR1020227021859A patent/KR102531016B1/ko active IP Right Grant
- 2015-11-20 IL IL301900A patent/IL301900A/en unknown
- 2015-11-20 PL PL15804289T patent/PL3221457T3/pl unknown
- 2015-11-20 KR KR1020177016481A patent/KR102415093B1/ko active IP Right Grant
- 2015-11-20 LT LTEP15804289.5T patent/LT3221457T/lt unknown
- 2015-11-20 NZ NZ731962A patent/NZ731962A/en unknown
- 2015-11-20 CA CA3176380A patent/CA3176380A1/en active Pending
- 2015-11-20 EP EP15804289.5A patent/EP3221457B1/en active Active
- 2015-11-20 EP EP19161085.6A patent/EP3521437A1/en active Pending
- 2015-11-20 RU RU2017121367A patent/RU2734770C2/ru active
- 2015-11-20 CN CN202110716022.8A patent/CN113444747A/zh active Pending
- 2015-11-20 AU AU2015349692A patent/AU2015349692B2/en active Active
- 2015-11-20 ES ES15804289T patent/ES2731437T3/es active Active
- 2015-11-20 DK DK15804289.5T patent/DK3221457T3/da active
- 2015-11-20 HU HUE15804289 patent/HUE044907T2/hu unknown
- 2015-11-20 CN CN201580063428.3A patent/CN107208078B/zh active Active
- 2015-11-20 SG SG11201703747RA patent/SG11201703747RA/en unknown
- 2015-11-20 CA CA2968440A patent/CA2968440A1/en active Pending
- 2015-11-20 SG SG10201913829YA patent/SG10201913829YA/en unknown
- 2015-11-20 WO PCT/US2015/062023 patent/WO2016081923A2/en active Application Filing
- 2015-11-20 US US14/948,221 patent/US10457960B2/en active Active
- 2015-11-20 MX MX2017006670A patent/MX2017006670A/es unknown
- 2015-11-20 RS RS20190760A patent/RS58893B1/sr unknown
- 2015-11-20 RU RU2020134412A patent/RU2020134412A/ru unknown
-
2017
- 2017-05-09 IL IL252181A patent/IL252181B/en active IP Right Grant
- 2017-05-19 MX MX2022000378A patent/MX2022000378A/es unknown
-
2019
- 2019-05-24 HR HRP20190949TT patent/HRP20190949T1/hr unknown
- 2019-06-20 CY CY20191100640T patent/CY1121738T1/el unknown
- 2019-09-16 US US16/572,124 patent/US11697828B2/en active Active
- 2019-09-16 US US16/572,137 patent/US20200002731A1/en not_active Abandoned
-
2020
- 2020-06-30 JP JP2020112438A patent/JP7101211B2/ja active Active
-
2021
- 2021-12-22 AU AU2021290301A patent/AU2021290301B2/en active Active
-
2023
- 2023-05-15 US US18/317,465 patent/US20230332185A1/en active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004536556A (ja) * | 2000-10-31 | 2004-12-09 | リジェネロン・ファーマシューティカルズ・インコーポレイテッド | 真核生物細胞を改変する方法 |
| WO2014034412A1 (ja) * | 2012-08-31 | 2014-03-06 | 京セラメディカル株式会社 | 人工心肺ポンプ用駆動装置 |
| WO2014104878A1 (en) * | 2012-12-27 | 2014-07-03 | Keygene N.V. | Method for removing genetic linkage in a plant |
Non-Patent Citations (5)
| Title |
|---|
| BIOCHEM. BIOPHYS. RES. COMMUN., vol. 445, JPN6019051417, January 2014 (2014-01-01), pages 791 - 794, ISSN: 0004273248 * |
| FEBS J., vol. 281, JPN6019051407, April 2014 (2014-04-01), pages 1717 - 1725, ISSN: 0004185209 * |
| GENETICS, vol. 196, JPN6019051414, April 2014 (2014-04-01), pages 961 - 971, ISSN: 0004273247 * |
| NAT. COMMUN., vol. 5, JPN6019051420, June 2014 (2014-06-01), pages 4240, ISSN: 0004273249 * |
| NUCL. ACID. RES., vol. 43, no. 3, JPN6019051412, 20 November 2014 (2014-11-20), pages 21, ISSN: 0004273246 * |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2021511038A (ja) * | 2018-01-17 | 2021-05-06 | バーテックス ファーマシューティカルズ インコーポレイテッドVertex Pharmaceuticals Incorporated | Dna−pk阻害剤 |
| JP7466448B2 (ja) | 2018-01-17 | 2024-04-12 | バーテックス ファーマシューティカルズ インコーポレイテッド | Dna-pk阻害剤 |
| US12005127B2 (en) | 2018-01-17 | 2024-06-11 | Vertex Pharmaceuticals Incorporated | DNA-PK inhibitors |
| WO2020050294A1 (ja) * | 2018-09-05 | 2020-03-12 | 学校法人慶應義塾 | 相同組換え効率上昇剤及びその使用 |
| US12121524B2 (en) | 2019-01-16 | 2024-10-22 | Vertex Pharmaceuticals Incorporated | DNA-PK inhibitors |
| JP6867635B1 (ja) * | 2020-04-06 | 2021-04-28 | 株式会社Logomix | ゲノム改変方法及びゲノム改変キット |
| JP2021164443A (ja) * | 2020-04-06 | 2021-10-14 | 株式会社Logomix | ゲノム改変方法及びゲノム改変キット |
| WO2023054573A1 (ja) * | 2021-09-30 | 2023-04-06 | 国立大学法人大阪大学 | 相同染色体の一方に特異的なdna欠失を有する細胞を製造する方法 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7101211B2 (ja) | ガイドrnaのペアを使用したターゲティングによる遺伝子改変の方法及び組成物 | |
| JP7095066B2 (ja) | 単一ステップの複数標的化を通じた標的化された遺伝子修飾のための方法及び組成物 | |
| DK3161128T3 (en) | METHODS AND COMPOSITIONS FOR TARGETED GENTICAL MODIFICATIONS AND PROCEDURES FOR USE THEREOF |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20181112 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20181112 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20191227 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200327 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20200601 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20200630 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6727199 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |