ES2759176T3 - Composiciones y procedimientos para el tratamiento del cáncer - Google Patents
Composiciones y procedimientos para el tratamiento del cáncer Download PDFInfo
- Publication number
- ES2759176T3 ES2759176T3 ES13178249T ES13178249T ES2759176T3 ES 2759176 T3 ES2759176 T3 ES 2759176T3 ES 13178249 T ES13178249 T ES 13178249T ES 13178249 T ES13178249 T ES 13178249T ES 2759176 T3 ES2759176 T3 ES 2759176T3
- Authority
- ES
- Spain
- Prior art keywords
- group
- antineoplastic agent
- protected
- cancer
- version
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 206010028980 Neoplasm Diseases 0.000 title claims description 155
- 201000011510 cancer Diseases 0.000 title claims description 100
- 238000011282 treatment Methods 0.000 title claims description 89
- 238000000034 method Methods 0.000 title description 131
- 239000000203 mixture Substances 0.000 title description 58
- 239000002246 antineoplastic agent Substances 0.000 claims abstract description 388
- 229940034982 antineoplastic agent Drugs 0.000 claims abstract description 356
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 129
- 239000001257 hydrogen Substances 0.000 claims abstract description 93
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 68
- 125000005842 heteroatom Chemical group 0.000 claims abstract description 64
- 230000000118 anti-neoplastic effect Effects 0.000 claims abstract description 9
- -1 duetoposide Chemical compound 0.000 claims description 163
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 75
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 claims description 69
- 210000004027 cell Anatomy 0.000 claims description 56
- 239000003814 drug Substances 0.000 claims description 55
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 31
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 28
- 229960004679 doxorubicin Drugs 0.000 claims description 27
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 claims description 22
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 claims description 21
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 claims description 20
- 229960005420 etoposide Drugs 0.000 claims description 20
- 125000003545 alkoxy group Chemical group 0.000 claims description 19
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 claims description 18
- 150000003839 salts Chemical class 0.000 claims description 18
- 229960002949 fluorouracil Drugs 0.000 claims description 17
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 claims description 16
- 229960004316 cisplatin Drugs 0.000 claims description 16
- 150000002431 hydrogen Chemical class 0.000 claims description 15
- 206010006187 Breast cancer Diseases 0.000 claims description 14
- 208000026310 Breast neoplasm Diseases 0.000 claims description 14
- 229960000975 daunorubicin Drugs 0.000 claims description 14
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 claims description 14
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 claims description 13
- 229960004528 vincristine Drugs 0.000 claims description 13
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 claims description 13
- 229940100198 alkylating agent Drugs 0.000 claims description 12
- 239000002168 alkylating agent Substances 0.000 claims description 12
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 claims description 12
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 claims description 12
- 210000001519 tissue Anatomy 0.000 claims description 12
- HVXBOLULGPECHP-WAYWQWQTSA-N Combretastatin A4 Chemical compound C1=C(O)C(OC)=CC=C1\C=C/C1=CC(OC)=C(OC)C(OC)=C1 HVXBOLULGPECHP-WAYWQWQTSA-N 0.000 claims description 11
- 229930012538 Paclitaxel Natural products 0.000 claims description 11
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 claims description 11
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 claims description 11
- 229960004397 cyclophosphamide Drugs 0.000 claims description 11
- 208000032839 leukemia Diseases 0.000 claims description 11
- 229960001592 paclitaxel Drugs 0.000 claims description 11
- 229960003048 vinblastine Drugs 0.000 claims description 11
- 229960004562 carboplatin Drugs 0.000 claims description 10
- 229960005537 combretastatin A-4 Drugs 0.000 claims description 10
- HVXBOLULGPECHP-UHFFFAOYSA-N combretastatin A4 Natural products C1=C(O)C(OC)=CC=C1C=CC1=CC(OC)=C(OC)C(OC)=C1 HVXBOLULGPECHP-UHFFFAOYSA-N 0.000 claims description 10
- 206010061289 metastatic neoplasm Diseases 0.000 claims description 10
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 claims description 9
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 claims description 9
- 229940123237 Taxane Drugs 0.000 claims description 9
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 claims description 9
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 claims description 9
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 claims description 9
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 claims description 9
- 108010006654 Bleomycin Proteins 0.000 claims description 8
- 108010092160 Dactinomycin Proteins 0.000 claims description 8
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 claims description 8
- 229960001904 epirubicin Drugs 0.000 claims description 8
- 208000020816 lung neoplasm Diseases 0.000 claims description 8
- 230000001394 metastastic effect Effects 0.000 claims description 8
- 229960001156 mitoxantrone Drugs 0.000 claims description 8
- 239000008194 pharmaceutical composition Substances 0.000 claims description 8
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 claims description 8
- 238000001959 radiotherapy Methods 0.000 claims description 8
- 229960000303 topotecan Drugs 0.000 claims description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 8
- 239000004037 angiogenesis inhibitor Substances 0.000 claims description 7
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 claims description 7
- 230000003211 malignant effect Effects 0.000 claims description 7
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 claims description 7
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 claims description 6
- 208000007766 Kaposi sarcoma Diseases 0.000 claims description 6
- 206010025323 Lymphomas Diseases 0.000 claims description 6
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 claims description 6
- ILTUTLWVTBBXNS-UHFFFAOYSA-N Tedanolide Natural products C1OC(=O)C(O)C(OC)C(C)C(=O)C(C)C(O)C(C)=CC(C)C(=O)CC(O)C(C)C(=O)C1C(O)C1(C)C(C(C)C=CC)O1 ILTUTLWVTBBXNS-UHFFFAOYSA-N 0.000 claims description 6
- VGQOVCHZGQWAOI-UHFFFAOYSA-N UNPD55612 Natural products N1C(O)C2CC(C=CC(N)=O)=CN2C(=O)C2=CC=C(C)C(O)=C12 VGQOVCHZGQWAOI-UHFFFAOYSA-N 0.000 claims description 6
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 claims description 6
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical class N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 claims description 6
- 229940127093 camptothecin Drugs 0.000 claims description 6
- 229960003668 docetaxel Drugs 0.000 claims description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 claims description 5
- 208000003174 Brain Neoplasms Diseases 0.000 claims description 5
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 claims description 5
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 5
- 206010039491 Sarcoma Diseases 0.000 claims description 5
- 229940122803 Vinca alkaloid Drugs 0.000 claims description 5
- 229960001561 bleomycin Drugs 0.000 claims description 5
- 229960000640 dactinomycin Drugs 0.000 claims description 5
- 229930013356 epothilone Natural products 0.000 claims description 5
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical class C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 claims description 5
- 229960000390 fludarabine Drugs 0.000 claims description 5
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 claims description 5
- 201000005962 mycosis fungoides Diseases 0.000 claims description 5
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 claims description 5
- 229960001278 teniposide Drugs 0.000 claims description 5
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 claims description 4
- YZBAXVICWUUHGG-UHFFFAOYSA-N 2-[[4-[2-[dimethyl(oxido)azaniumyl]ethylamino]-5,8-dihydroxy-9,10-dioxoanthracen-1-yl]amino]-n,n-dimethylethanamine oxide Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCC[N+](C)(C)[O-])=CC=C2NCC[N+](C)([O-])C YZBAXVICWUUHGG-UHFFFAOYSA-N 0.000 claims description 4
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 claims description 4
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 claims description 4
- 206010029260 Neuroblastoma Diseases 0.000 claims description 4
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 claims description 4
- HRHKSTOGXBBQCB-UHFFFAOYSA-N Porfiromycine Chemical compound O=C1C(N)=C(C)C(=O)C2=C1C(COC(N)=O)C1(OC)C3N(C)C3CN12 HRHKSTOGXBBQCB-UHFFFAOYSA-N 0.000 claims description 4
- 230000000340 anti-metabolite Effects 0.000 claims description 4
- 229940100197 antimetabolite Drugs 0.000 claims description 4
- 239000002256 antimetabolite Substances 0.000 claims description 4
- 229960002756 azacitidine Drugs 0.000 claims description 4
- 229930195731 calicheamicin Natural products 0.000 claims description 4
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 claims description 4
- 229960004630 chlorambucil Drugs 0.000 claims description 4
- 229960000684 cytarabine Drugs 0.000 claims description 4
- 208000005017 glioblastoma Diseases 0.000 claims description 4
- 206010020718 hyperplasia Diseases 0.000 claims description 4
- 229960001428 mercaptopurine Drugs 0.000 claims description 4
- 201000008968 osteosarcoma Diseases 0.000 claims description 4
- 210000000496 pancreas Anatomy 0.000 claims description 4
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 claims description 4
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 4
- 229910052697 platinum Inorganic materials 0.000 claims description 4
- 229960000624 procarbazine Drugs 0.000 claims description 4
- 201000000849 skin cancer Diseases 0.000 claims description 4
- 206010041823 squamous cell carcinoma Diseases 0.000 claims description 4
- PVYJZLYGTZKPJE-UHFFFAOYSA-N streptonigrin Chemical compound C=1C=C2C(=O)C(OC)=C(N)C(=O)C2=NC=1C(C=1N)=NC(C(O)=O)=C(C)C=1C1=CC=C(OC)C(OC)=C1O PVYJZLYGTZKPJE-UHFFFAOYSA-N 0.000 claims description 4
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 claims description 3
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 claims description 3
- WYXSYVWAUAUWLD-SHUUEZRQSA-N 6-azauridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=N1 WYXSYVWAUAUWLD-SHUUEZRQSA-N 0.000 claims description 3
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 claims description 3
- 201000010915 Glioblastoma multiforme Diseases 0.000 claims description 3
- 201000000582 Retinoblastoma Diseases 0.000 claims description 3
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 claims description 3
- 230000001154 acute effect Effects 0.000 claims description 3
- 208000009956 adenocarcinoma Diseases 0.000 claims description 3
- 235000013877 carbamide Nutrition 0.000 claims description 3
- 229960003901 dacarbazine Drugs 0.000 claims description 3
- 206010012818 diffuse large B-cell lymphoma Diseases 0.000 claims description 3
- 239000003085 diluting agent Substances 0.000 claims description 3
- 229960001842 estramustine Drugs 0.000 claims description 3
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 claims description 3
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 claims description 3
- 210000000265 leukocyte Anatomy 0.000 claims description 3
- 229960002340 pentostatin Drugs 0.000 claims description 3
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 claims description 3
- 229960004618 prednisone Drugs 0.000 claims description 3
- 210000000664 rectum Anatomy 0.000 claims description 3
- 201000006845 reticulosarcoma Diseases 0.000 claims description 3
- 208000029922 reticulum cell sarcoma Diseases 0.000 claims description 3
- 201000009410 rhabdomyosarcoma Diseases 0.000 claims description 3
- 210000002784 stomach Anatomy 0.000 claims description 3
- 229960001099 trimetrexate Drugs 0.000 claims description 3
- NOYPYLRCIDNJJB-UHFFFAOYSA-N trimetrexate Chemical compound COC1=C(OC)C(OC)=CC(NCC=2C(=C3C(N)=NC(N)=NC3=CC=2)C)=C1 NOYPYLRCIDNJJB-UHFFFAOYSA-N 0.000 claims description 3
- 150000003672 ureas Chemical class 0.000 claims description 3
- NNJPGOLRFBJNIW-HNNXBMFYSA-N (-)-demecolcine Chemical compound C1=C(OC)C(=O)C=C2[C@@H](NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-HNNXBMFYSA-N 0.000 claims description 2
- FLWWDYNPWOSLEO-HQVZTVAUSA-N (2s)-2-[[4-[1-(2-amino-4-oxo-1h-pteridin-6-yl)ethyl-methylamino]benzoyl]amino]pentanedioic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1C(C)N(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FLWWDYNPWOSLEO-HQVZTVAUSA-N 0.000 claims description 2
- CGMTUJFWROPELF-YPAAEMCBSA-N (3E,5S)-5-[(2S)-butan-2-yl]-3-(1-hydroxyethylidene)pyrrolidine-2,4-dione Chemical compound CC[C@H](C)[C@@H]1NC(=O)\C(=C(/C)O)C1=O CGMTUJFWROPELF-YPAAEMCBSA-N 0.000 claims description 2
- VRYALKFFQXWPIH-RANCGNPWSA-N (3r,4s,5r)-3,4,5,6-tetrahydroxy-2-tritiohexanal Chemical compound O=CC([3H])[C@@H](O)[C@H](O)[C@H](O)CO VRYALKFFQXWPIH-RANCGNPWSA-N 0.000 claims description 2
- AGNGYMCLFWQVGX-AGFFZDDWSA-N (e)-1-[(2s)-2-amino-2-carboxyethoxy]-2-diazonioethenolate Chemical compound OC(=O)[C@@H](N)CO\C([O-])=C\[N+]#N AGNGYMCLFWQVGX-AGFFZDDWSA-N 0.000 claims description 2
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 claims description 2
- QCXJFISCRQIYID-IAEPZHFASA-N 2-amino-1-n-[(3s,6s,7r,10s,16s)-3-[(2s)-butan-2-yl]-7,11,14-trimethyl-2,5,9,12,15-pentaoxo-10-propan-2-yl-8-oxa-1,4,11,14-tetrazabicyclo[14.3.0]nonadecan-6-yl]-4,6-dimethyl-3-oxo-9-n-[(3s,6s,7r,10s,16s)-7,11,14-trimethyl-2,5,9,12,15-pentaoxo-3,10-di(propa Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N=C2C(C(=O)N[C@@H]3C(=O)N[C@H](C(N4CCC[C@H]4C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]3C)=O)[C@@H](C)CC)=C(N)C(=O)C(C)=C2O2)C2=C(C)C=C1 QCXJFISCRQIYID-IAEPZHFASA-N 0.000 claims description 2
- ZGXJTSGNIOSYLO-UHFFFAOYSA-N 88755TAZ87 Chemical compound NCC(=O)CCC(O)=O ZGXJTSGNIOSYLO-UHFFFAOYSA-N 0.000 claims description 2
- HDZZVAMISRMYHH-UHFFFAOYSA-N 9beta-Ribofuranosyl-7-deazaadenin Natural products C1=CC=2C(N)=NC=NC=2N1C1OC(CO)C(O)C1O HDZZVAMISRMYHH-UHFFFAOYSA-N 0.000 claims description 2
- 208000003200 Adenoma Diseases 0.000 claims description 2
- 206010001233 Adenoma benign Diseases 0.000 claims description 2
- CEIZFXOZIQNICU-UHFFFAOYSA-N Alternaria alternata Crofton-weed toxin Natural products CCC(C)C1NC(=O)C(C(C)=O)=C1O CEIZFXOZIQNICU-UHFFFAOYSA-N 0.000 claims description 2
- 108010024976 Asparaginase Proteins 0.000 claims description 2
- 206010004146 Basal cell carcinoma Diseases 0.000 claims description 2
- VGGGPCQERPFHOB-MCIONIFRSA-N Bestatin Chemical compound CC(C)C[C@H](C(O)=O)NC(=O)[C@@H](O)[C@H](N)CC1=CC=CC=C1 VGGGPCQERPFHOB-MCIONIFRSA-N 0.000 claims description 2
- 206010005949 Bone cancer Diseases 0.000 claims description 2
- 208000018084 Bone neoplasm Diseases 0.000 claims description 2
- SHHKQEUPHAENFK-UHFFFAOYSA-N Carboquone Chemical compound O=C1C(C)=C(N2CC2)C(=O)C(C(COC(N)=O)OC)=C1N1CC1 SHHKQEUPHAENFK-UHFFFAOYSA-N 0.000 claims description 2
- 206010008263 Cervical dysplasia Diseases 0.000 claims description 2
- 206010061809 Cervix carcinoma stage 0 Diseases 0.000 claims description 2
- NNJPGOLRFBJNIW-UHFFFAOYSA-N Demecolcine Natural products C1=C(OC)C(=O)C=C2C(NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-UHFFFAOYSA-N 0.000 claims description 2
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 claims description 2
- 208000007569 Giant Cell Tumors Diseases 0.000 claims description 2
- 208000037147 Hypercalcaemia Diseases 0.000 claims description 2
- 102000006992 Interferon-alpha Human genes 0.000 claims description 2
- 108010047761 Interferon-alpha Proteins 0.000 claims description 2
- 102000003996 Interferon-beta Human genes 0.000 claims description 2
- 108090000467 Interferon-beta Proteins 0.000 claims description 2
- 102000008070 Interferon-gamma Human genes 0.000 claims description 2
- 108010074328 Interferon-gamma Proteins 0.000 claims description 2
- 102000000588 Interleukin-2 Human genes 0.000 claims description 2
- 108010002350 Interleukin-2 Proteins 0.000 claims description 2
- 239000005517 L01XE01 - Imatinib Substances 0.000 claims description 2
- 229920001491 Lentinan Polymers 0.000 claims description 2
- 208000007054 Medullary Carcinoma Diseases 0.000 claims description 2
- VFKZTMPDYBFSTM-KVTDHHQDSA-N Mitobronitol Chemical compound BrC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-KVTDHHQDSA-N 0.000 claims description 2
- KGTDRFCXGRULNK-UHFFFAOYSA-N Nogalamycin Natural products COC1C(OC)(C)C(OC)C(C)OC1OC1C2=C(O)C(C(=O)C3=C(O)C=C4C5(C)OC(C(C(C5O)N(C)C)O)OC4=C3C3=O)=C3C=C2C(C(=O)OC)C(C)(O)C1 KGTDRFCXGRULNK-UHFFFAOYSA-N 0.000 claims description 2
- 229930187135 Olivomycin Natural products 0.000 claims description 2
- 108010057150 Peplomycin Proteins 0.000 claims description 2
- 201000010208 Seminoma Diseases 0.000 claims description 2
- 208000000453 Skin Neoplasms Diseases 0.000 claims description 2
- 208000021712 Soft tissue sarcoma Diseases 0.000 claims description 2
- CGMTUJFWROPELF-UHFFFAOYSA-N Tenuazonic acid Natural products CCC(C)C1NC(=O)C(=C(C)/O)C1=O CGMTUJFWROPELF-UHFFFAOYSA-N 0.000 claims description 2
- FYAMXEPQQLNQDM-UHFFFAOYSA-N Tris(1-aziridinyl)phosphine oxide Chemical compound C1CN1P(N1CC1)(=O)N1CC1 FYAMXEPQQLNQDM-UHFFFAOYSA-N 0.000 claims description 2
- 208000025865 Ulcer Diseases 0.000 claims description 2
- IFJUINDAXYAPTO-UUBSBJJBSA-N [(8r,9s,13s,14s,17s)-17-[2-[4-[4-[bis(2-chloroethyl)amino]phenyl]butanoyloxy]acetyl]oxy-13-methyl-6,7,8,9,11,12,14,15,16,17-decahydrocyclopenta[a]phenanthren-3-yl] benzoate Chemical compound C([C@@H]1[C@@H](C2=CC=3)CC[C@]4([C@H]1CC[C@@H]4OC(=O)COC(=O)CCCC=1C=CC(=CC=1)N(CCCl)CCCl)C)CC2=CC=3OC(=O)C1=CC=CC=C1 IFJUINDAXYAPTO-UUBSBJJBSA-N 0.000 claims description 2
- 229950002684 aceglatone Drugs 0.000 claims description 2
- ZOZKYEHVNDEUCO-XUTVFYLZSA-N aceglatone Chemical compound O1C(=O)[C@H](OC(C)=O)[C@@H]2OC(=O)[C@@H](OC(=O)C)[C@@H]21 ZOZKYEHVNDEUCO-XUTVFYLZSA-N 0.000 claims description 2
- 208000017733 acquired polycythemia vera Diseases 0.000 claims description 2
- 229930183665 actinomycin Natural products 0.000 claims description 2
- 230000001919 adrenal effect Effects 0.000 claims description 2
- 229960000473 altretamine Drugs 0.000 claims description 2
- 229960002749 aminolevulinic acid Drugs 0.000 claims description 2
- BBDAGFIXKZCXAH-CCXZUQQUSA-N ancitabine Chemical compound N=C1C=CN2[C@@H]3O[C@H](CO)[C@@H](O)[C@@H]3OC2=N1 BBDAGFIXKZCXAH-CCXZUQQUSA-N 0.000 claims description 2
- 229950000242 ancitabine Drugs 0.000 claims description 2
- RGHILYZRVFRRNK-UHFFFAOYSA-N anthracene-1,2-dione Chemical class C1=CC=C2C=C(C(C(=O)C=C3)=O)C3=CC2=C1 RGHILYZRVFRRNK-UHFFFAOYSA-N 0.000 claims description 2
- 229950011321 azaserine Drugs 0.000 claims description 2
- 229950005567 benzodepa Drugs 0.000 claims description 2
- VFIUCBTYGKMLCM-UHFFFAOYSA-N benzyl n-[bis(aziridin-1-yl)phosphoryl]carbamate Chemical compound C=1C=CC=CC=1COC(=O)NP(=O)(N1CC1)N1CC1 VFIUCBTYGKMLCM-UHFFFAOYSA-N 0.000 claims description 2
- 229950008548 bisantrene Drugs 0.000 claims description 2
- 210000000621 bronchi Anatomy 0.000 claims description 2
- 229960002092 busulfan Drugs 0.000 claims description 2
- 108700002839 cactinomycin Proteins 0.000 claims description 2
- 229950009908 cactinomycin Drugs 0.000 claims description 2
- 229960002115 carboquone Drugs 0.000 claims description 2
- 208000002458 carcinoid tumor Diseases 0.000 claims description 2
- 201000001883 cholelithiasis Diseases 0.000 claims description 2
- ZYVSOIYQKUDENJ-WKSBCEQHSA-N chromomycin A3 Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@@H]1OC(C)=O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@@H](O)[C@H](O[C@@H]3O[C@@H](C)[C@H](OC(C)=O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@@H]1C[C@@H](O)[C@@H](OC)[C@@H](C)O1 ZYVSOIYQKUDENJ-WKSBCEQHSA-N 0.000 claims description 2
- 230000001684 chronic effect Effects 0.000 claims description 2
- 229960005052 demecolcine Drugs 0.000 claims description 2
- 108010067396 dornase alfa Proteins 0.000 claims description 2
- 239000003937 drug carrier Substances 0.000 claims description 2
- 210000000232 gallbladder Anatomy 0.000 claims description 2
- 208000001130 gallstones Diseases 0.000 claims description 2
- 235000011187 glycerol Nutrition 0.000 claims description 2
- 229930182470 glycoside Natural products 0.000 claims description 2
- 210000003128 head Anatomy 0.000 claims description 2
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 claims description 2
- 230000000148 hypercalcaemia Effects 0.000 claims description 2
- 208000030915 hypercalcemia disease Diseases 0.000 claims description 2
- 230000002390 hyperplastic effect Effects 0.000 claims description 2
- 229960002411 imatinib Drugs 0.000 claims description 2
- DBIGHPPNXATHOF-UHFFFAOYSA-N improsulfan Chemical compound CS(=O)(=O)OCCCNCCCOS(C)(=O)=O DBIGHPPNXATHOF-UHFFFAOYSA-N 0.000 claims description 2
- 229950008097 improsulfan Drugs 0.000 claims description 2
- 229960003130 interferon gamma Drugs 0.000 claims description 2
- 229960001388 interferon-beta Drugs 0.000 claims description 2
- 210000003734 kidney Anatomy 0.000 claims description 2
- 206010023841 laryngeal neoplasm Diseases 0.000 claims description 2
- 229940115286 lentinan Drugs 0.000 claims description 2
- 201000007270 liver cancer Diseases 0.000 claims description 2
- 208000014018 liver neoplasm Diseases 0.000 claims description 2
- WDRYRZXSPDWGEB-UHFFFAOYSA-N lonidamine Chemical compound C12=CC=CC=C2C(C(=O)O)=NN1CC1=CC=C(Cl)C=C1Cl WDRYRZXSPDWGEB-UHFFFAOYSA-N 0.000 claims description 2
- 229960003538 lonidamine Drugs 0.000 claims description 2
- 208000037841 lung tumor Diseases 0.000 claims description 2
- 230000000527 lymphocytic effect Effects 0.000 claims description 2
- 208000023356 medullary thyroid gland carcinoma Diseases 0.000 claims description 2
- 201000001441 melanoma Diseases 0.000 claims description 2
- QTFKTBRIGWJQQL-UHFFFAOYSA-N meturedepa Chemical compound C1C(C)(C)N1P(=O)(NC(=O)OCC)N1CC1(C)C QTFKTBRIGWJQQL-UHFFFAOYSA-N 0.000 claims description 2
- 229950009847 meturedepa Drugs 0.000 claims description 2
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 claims description 2
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 claims description 2
- 229960005485 mitobronitol Drugs 0.000 claims description 2
- 229960003539 mitoguazone Drugs 0.000 claims description 2
- MXWHMTNPTTVWDM-NXOFHUPFSA-N mitoguazone Chemical compound NC(N)=N\N=C(/C)\C=N\N=C(N)N MXWHMTNPTTVWDM-NXOFHUPFSA-N 0.000 claims description 2
- VFKZTMPDYBFSTM-GUCUJZIJSA-N mitolactol Chemical compound BrC[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-GUCUJZIJSA-N 0.000 claims description 2
- 229950010913 mitolactol Drugs 0.000 claims description 2
- CPTIBDHUFVHUJK-NZYDNVMFSA-N mitopodozide Chemical compound C1([C@@H]2C3=CC=4OCOC=4C=C3[C@H](O)[C@@H](CO)[C@@H]2C(=O)NNCC)=CC(OC)=C(OC)C(OC)=C1 CPTIBDHUFVHUJK-NZYDNVMFSA-N 0.000 claims description 2
- FOYWNSCCNCUEPU-UHFFFAOYSA-N mopidamol Chemical compound C12=NC(N(CCO)CCO)=NC=C2N=C(N(CCO)CCO)N=C1N1CCCCC1 FOYWNSCCNCUEPU-UHFFFAOYSA-N 0.000 claims description 2
- 229950010718 mopidamol Drugs 0.000 claims description 2
- 229960000951 mycophenolic acid Drugs 0.000 claims description 2
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 claims description 2
- 201000000050 myeloid neoplasm Diseases 0.000 claims description 2
- NJSMWLQOCQIOPE-OCHFTUDZSA-N n-[(e)-[10-[(e)-(4,5-dihydro-1h-imidazol-2-ylhydrazinylidene)methyl]anthracen-9-yl]methylideneamino]-4,5-dihydro-1h-imidazol-2-amine Chemical compound N1CCN=C1N\N=C\C(C1=CC=CC=C11)=C(C=CC=C2)C2=C1\C=N\NC1=NCCN1 NJSMWLQOCQIOPE-OCHFTUDZSA-N 0.000 claims description 2
- 210000003739 neck Anatomy 0.000 claims description 2
- QZGIWPZCWHMVQL-UIYAJPBUSA-N neocarzinostatin chromophore Chemical compound O1[C@H](C)[C@H](O)[C@H](O)[C@@H](NC)[C@H]1O[C@@H]1C/2=C/C#C[C@H]3O[C@@]3([C@@H]3OC(=O)OC3)C#CC\2=C[C@H]1OC(=O)C1=C(O)C=CC2=C(C)C=C(OC)C=C12 QZGIWPZCWHMVQL-UIYAJPBUSA-N 0.000 claims description 2
- 210000005036 nerve Anatomy 0.000 claims description 2
- 230000001537 neural effect Effects 0.000 claims description 2
- 210000002569 neuron Anatomy 0.000 claims description 2
- 229950008607 nitracrine Drugs 0.000 claims description 2
- YMVWGSQGCWCDGW-UHFFFAOYSA-N nitracrine Chemical compound C1=CC([N+]([O-])=O)=C2C(NCCCN(C)C)=C(C=CC=C3)C3=NC2=C1 YMVWGSQGCWCDGW-UHFFFAOYSA-N 0.000 claims description 2
- KGTDRFCXGRULNK-JYOBTZKQSA-N nogalamycin Chemical compound CO[C@@H]1[C@@](OC)(C)[C@@H](OC)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=C(O)C=C4[C@@]5(C)O[C@H]([C@H]([C@@H]([C@H]5O)N(C)C)O)OC4=C3C3=O)=C3C=C2[C@@H](C(=O)OC)[C@@](C)(O)C1 KGTDRFCXGRULNK-JYOBTZKQSA-N 0.000 claims description 2
- 229950009266 nogalamycin Drugs 0.000 claims description 2
- CZDBNBLGZNWKMC-MWQNXGTOSA-N olivomycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1)O[C@H]1O[C@@H](C)[C@H](O)[C@@H](OC2O[C@@H](C)[C@H](O)[C@@H](O)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@H](O)[C@H](OC)[C@H](C)O1 CZDBNBLGZNWKMC-MWQNXGTOSA-N 0.000 claims description 2
- 229950005848 olivomycin Drugs 0.000 claims description 2
- 230000000849 parathyroid Effects 0.000 claims description 2
- QIMGFXOHTOXMQP-GFAGFCTOSA-N peplomycin Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCCN[C@@H](C)C=1C=CC=CC=1)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C QIMGFXOHTOXMQP-GFAGFCTOSA-N 0.000 claims description 2
- 229950003180 peplomycin Drugs 0.000 claims description 2
- 208000028591 pheochromocytoma Diseases 0.000 claims description 2
- NJBFOOCLYDNZJN-UHFFFAOYSA-N pipobroman Chemical compound BrCCC(=O)N1CCN(C(=O)CCBr)CC1 NJBFOOCLYDNZJN-UHFFFAOYSA-N 0.000 claims description 2
- NUKCGLDCWQXYOQ-UHFFFAOYSA-N piposulfan Chemical compound CS(=O)(=O)OCCC(=O)N1CCN(C(=O)CCOS(C)(=O)=O)CC1 NUKCGLDCWQXYOQ-UHFFFAOYSA-N 0.000 claims description 2
- 229950001100 piposulfan Drugs 0.000 claims description 2
- 229960003171 plicamycin Drugs 0.000 claims description 2
- 208000037244 polycythemia vera Diseases 0.000 claims description 2
- 208000030266 primary brain neoplasm Diseases 0.000 claims description 2
- WOLQREOUPKZMEX-UHFFFAOYSA-N pteroyltriglutamic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(=O)NC(CCC(=O)NC(CCC(O)=O)C(O)=O)C(O)=O)C(O)=O)C=C1 WOLQREOUPKZMEX-UHFFFAOYSA-N 0.000 claims description 2
- 229940107568 pulmozyme Drugs 0.000 claims description 2
- BMKDZUISNHGIBY-UHFFFAOYSA-N razoxane Chemical compound C1C(=O)NC(=O)CN1C(C)CN1CC(=O)NC(=O)C1 BMKDZUISNHGIBY-UHFFFAOYSA-N 0.000 claims description 2
- 229960000460 razoxane Drugs 0.000 claims description 2
- 201000008261 skin carcinoma Diseases 0.000 claims description 2
- 206010040882 skin lesion Diseases 0.000 claims description 2
- 231100000444 skin lesion Toxicity 0.000 claims description 2
- 229960001052 streptozocin Drugs 0.000 claims description 2
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 claims description 2
- 229960001674 tegafur Drugs 0.000 claims description 2
- WFWLQNSHRPWKFK-ZCFIWIBFSA-N tegafur Chemical compound O=C1NC(=O)C(F)=CN1[C@@H]1OCCC1 WFWLQNSHRPWKFK-ZCFIWIBFSA-N 0.000 claims description 2
- 210000001685 thyroid gland Anatomy 0.000 claims description 2
- YFTWHEBLORWGNI-UHFFFAOYSA-N tiamiprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC(N)=NC2=C1NC=N2 YFTWHEBLORWGNI-UHFFFAOYSA-N 0.000 claims description 2
- 229950011457 tiamiprine Drugs 0.000 claims description 2
- 230000000699 topical effect Effects 0.000 claims description 2
- 229950001353 tretamine Drugs 0.000 claims description 2
- IUCJMVBFZDHPDX-UHFFFAOYSA-N tretamine Chemical compound C1CN1C1=NC(N2CC2)=NC(N2CC2)=N1 IUCJMVBFZDHPDX-UHFFFAOYSA-N 0.000 claims description 2
- HDZZVAMISRMYHH-LITAXDCLSA-N tubercidin Chemical compound C1=CC=2C(N)=NC=NC=2N1[C@@H]1O[C@@H](CO)[C@H](O)[C@H]1O HDZZVAMISRMYHH-LITAXDCLSA-N 0.000 claims description 2
- 229950009811 ubenimex Drugs 0.000 claims description 2
- SPDZFJLQFWSJGA-UHFFFAOYSA-N uredepa Chemical compound C1CN1P(=O)(NC(=O)OCC)N1CC1 SPDZFJLQFWSJGA-UHFFFAOYSA-N 0.000 claims description 2
- 229950006929 uredepa Drugs 0.000 claims description 2
- 229960005088 urethane Drugs 0.000 claims description 2
- 229950009268 zinostatin Drugs 0.000 claims description 2
- FBTUMDXHSRTGRV-ALTNURHMSA-N zorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(\C)=N\NC(=O)C=1C=CC=CC=1)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 FBTUMDXHSRTGRV-ALTNURHMSA-N 0.000 claims description 2
- 229960000641 zorubicin Drugs 0.000 claims description 2
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 claims 2
- 208000006168 Ewing Sarcoma Diseases 0.000 claims 1
- AHHFEZNOXOZZQA-ZEBDFXRSSA-N Ranimustine Chemical compound CO[C@H]1O[C@H](CNC(=O)N(CCCl)N=O)[C@@H](O)[C@H](O)[C@H]1O AHHFEZNOXOZZQA-ZEBDFXRSSA-N 0.000 claims 1
- VGQOVCHZGQWAOI-HYUHUPJXSA-N anthramycin Chemical compound N1[C@@H](O)[C@@H]2CC(\C=C\C(N)=O)=CN2C(=O)C2=CC=C(C)C(O)=C12 VGQOVCHZGQWAOI-HYUHUPJXSA-N 0.000 claims 1
- 108010047060 carzinophilin Proteins 0.000 claims 1
- 230000000968 intestinal effect Effects 0.000 claims 1
- 229960001420 nimustine Drugs 0.000 claims 1
- VFEDRRNHLBGPNN-UHFFFAOYSA-N nimustine Chemical compound CC1=NC=C(CNC(=O)N(CCCl)N=O)C(N)=N1 VFEDRRNHLBGPNN-UHFFFAOYSA-N 0.000 claims 1
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 claims 1
- 229960002185 ranimustine Drugs 0.000 claims 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 abstract description 35
- 125000004435 hydrogen atom Chemical group [H]* 0.000 abstract description 4
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 abstract 2
- 206010021143 Hypoxia Diseases 0.000 description 191
- 230000001146 hypoxic effect Effects 0.000 description 167
- 239000012190 activator Substances 0.000 description 131
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 128
- 150000001408 amides Chemical class 0.000 description 113
- 150000002148 esters Chemical class 0.000 description 86
- 150000001875 compounds Chemical class 0.000 description 84
- 229940124530 sulfonamide Drugs 0.000 description 74
- 229940002612 prodrug Drugs 0.000 description 73
- 239000000651 prodrug Substances 0.000 description 73
- 150000003456 sulfonamides Chemical group 0.000 description 72
- 125000000217 alkyl group Chemical group 0.000 description 61
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 56
- 229940079593 drug Drugs 0.000 description 54
- 150000001299 aldehydes Chemical class 0.000 description 51
- 125000004093 cyano group Chemical group *C#N 0.000 description 51
- 125000005843 halogen group Chemical group 0.000 description 51
- 150000003573 thiols Chemical class 0.000 description 51
- 150000003568 thioethers Chemical class 0.000 description 50
- 229910052799 carbon Inorganic materials 0.000 description 40
- 125000005647 linker group Chemical group 0.000 description 40
- YZEUHQHUFTYLPH-UHFFFAOYSA-N 2-nitroimidazole Chemical compound [O-][N+](=O)C1=NC=CN1 YZEUHQHUFTYLPH-UHFFFAOYSA-N 0.000 description 38
- 230000015572 biosynthetic process Effects 0.000 description 38
- 150000001734 carboxylic acid salts Chemical class 0.000 description 37
- 125000001153 fluoro group Chemical group F* 0.000 description 37
- 238000003786 synthesis reaction Methods 0.000 description 37
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 36
- 125000003118 aryl group Chemical group 0.000 description 35
- 239000003795 chemical substances by application Substances 0.000 description 35
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 33
- 125000003277 amino group Chemical group 0.000 description 28
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 26
- 230000009467 reduction Effects 0.000 description 25
- 238000006722 reduction reaction Methods 0.000 description 25
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 24
- 230000000694 effects Effects 0.000 description 23
- 230000007954 hypoxia Effects 0.000 description 23
- 229910052757 nitrogen Inorganic materials 0.000 description 21
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 20
- 238000006243 chemical reaction Methods 0.000 description 20
- 230000001472 cytotoxic effect Effects 0.000 description 20
- 125000000623 heterocyclic group Chemical group 0.000 description 20
- QAZLUNIWYYOJPC-UHFFFAOYSA-M sulfenamide Chemical compound [Cl-].COC1=C(C)C=[N+]2C3=NC4=CC=C(OC)C=C4N3SCC2=C1C QAZLUNIWYYOJPC-UHFFFAOYSA-M 0.000 description 20
- 150000003457 sulfones Chemical class 0.000 description 20
- 150000003459 sulfonic acid esters Chemical class 0.000 description 20
- 150000003462 sulfoxides Chemical class 0.000 description 20
- 229940127089 cytotoxic agent Drugs 0.000 description 19
- 239000000047 product Substances 0.000 description 19
- 239000003112 inhibitor Substances 0.000 description 18
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 18
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 17
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 17
- 238000002560 therapeutic procedure Methods 0.000 description 16
- 125000001072 heteroaryl group Chemical group 0.000 description 15
- 238000009472 formulation Methods 0.000 description 14
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 14
- 229910052760 oxygen Inorganic materials 0.000 description 14
- 239000000243 solution Substances 0.000 description 14
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 14
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 13
- 150000003857 carboxamides Chemical group 0.000 description 13
- 231100000433 cytotoxic Toxicity 0.000 description 13
- 239000000543 intermediate Substances 0.000 description 13
- 239000001301 oxygen Substances 0.000 description 13
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 12
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical compound N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 description 11
- 125000004036 acetal group Chemical group 0.000 description 11
- 125000003368 amide group Chemical group 0.000 description 11
- 229940041181 antineoplastic drug Drugs 0.000 description 11
- 125000004122 cyclic group Chemical group 0.000 description 11
- VQNATVDKACXKTF-XELLLNAOSA-N duocarmycin Chemical compound COC1=C(OC)C(OC)=C2NC(C(=O)N3C4=CC(=O)C5=C([C@@]64C[C@@H]6C3)C=C(N5)C(=O)OC)=CC2=C1 VQNATVDKACXKTF-XELLLNAOSA-N 0.000 description 11
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 11
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 11
- HAWSQZCWOQZXHI-FQEVSTJZSA-N 10-Hydroxycamptothecin Chemical compound C1=C(O)C=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 HAWSQZCWOQZXHI-FQEVSTJZSA-N 0.000 description 10
- 206010009944 Colon cancer Diseases 0.000 description 10
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 10
- 102000004190 Enzymes Human genes 0.000 description 10
- 108090000790 Enzymes Proteins 0.000 description 10
- VSNHCAURESNICA-UHFFFAOYSA-N Hydroxyurea Chemical compound NC(=O)NO VSNHCAURESNICA-UHFFFAOYSA-N 0.000 description 10
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 10
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 10
- 206010033128 Ovarian cancer Diseases 0.000 description 10
- 206010060862 Prostate cancer Diseases 0.000 description 10
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 10
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 10
- 125000002843 carboxylic acid group Chemical group 0.000 description 10
- 231100000599 cytotoxic agent Toxicity 0.000 description 10
- 229940088598 enzyme Drugs 0.000 description 10
- 235000019439 ethyl acetate Nutrition 0.000 description 10
- 238000003818 flash chromatography Methods 0.000 description 10
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 10
- 230000001988 toxicity Effects 0.000 description 10
- 231100000419 toxicity Toxicity 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- 229910019142 PO4 Inorganic materials 0.000 description 9
- 150000001412 amines Chemical class 0.000 description 9
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 9
- 239000003153 chemical reaction reagent Substances 0.000 description 9
- 238000002512 chemotherapy Methods 0.000 description 9
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 9
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 9
- 239000002254 cytotoxic agent Substances 0.000 description 9
- 229960005501 duocarmycin Drugs 0.000 description 9
- 229930184221 duocarmycin Natural products 0.000 description 9
- 125000001188 haloalkyl group Chemical group 0.000 description 9
- 125000002883 imidazolyl group Chemical group 0.000 description 9
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 9
- 125000000468 ketone group Chemical group 0.000 description 9
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 9
- 239000010452 phosphate Substances 0.000 description 9
- 239000007787 solid Substances 0.000 description 9
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 9
- 150000003536 tetrazoles Chemical class 0.000 description 9
- 229930194542 Keto Natural products 0.000 description 8
- 206010061535 Ovarian neoplasm Diseases 0.000 description 8
- 150000001241 acetals Chemical class 0.000 description 8
- 230000004913 activation Effects 0.000 description 8
- 125000003342 alkenyl group Chemical group 0.000 description 8
- 125000000304 alkynyl group Chemical group 0.000 description 8
- 230000015556 catabolic process Effects 0.000 description 8
- 238000006731 degradation reaction Methods 0.000 description 8
- 238000002474 experimental method Methods 0.000 description 8
- 231100000782 microtubule inhibitor Toxicity 0.000 description 8
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 8
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 8
- 230000001613 neoplastic effect Effects 0.000 description 8
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 8
- 230000001225 therapeutic effect Effects 0.000 description 8
- JXLYSJRDGCGARV-CFWMRBGOSA-N vinblastine Chemical compound C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-CFWMRBGOSA-N 0.000 description 8
- HAWSQZCWOQZXHI-UHFFFAOYSA-N CPT-OH Natural products C1=C(O)C=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 HAWSQZCWOQZXHI-UHFFFAOYSA-N 0.000 description 7
- 101100240526 Caenorhabditis elegans nhr-20 gene Proteins 0.000 description 7
- 238000005804 alkylation reaction Methods 0.000 description 7
- 229940045799 anthracyclines and related substance Drugs 0.000 description 7
- VGQOVCHZGQWAOI-YQRHFANHSA-N anthramycin Chemical compound N1[C@H](O)[C@@H]2CC(\C=C\C(N)=O)=CN2C(=O)C2=CC=C(C)C(O)=C12 VGQOVCHZGQWAOI-YQRHFANHSA-N 0.000 description 7
- 238000002648 combination therapy Methods 0.000 description 7
- 201000010099 disease Diseases 0.000 description 7
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 7
- 125000001033 ether group Chemical group 0.000 description 7
- 239000007789 gas Substances 0.000 description 7
- VLKZOEOYAKHREP-UHFFFAOYSA-N hexane Substances CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 7
- 239000002609 medium Substances 0.000 description 7
- 150000004957 nitroimidazoles Chemical class 0.000 description 7
- 235000013824 polyphenols Nutrition 0.000 description 7
- 239000011541 reaction mixture Substances 0.000 description 7
- 230000002829 reductive effect Effects 0.000 description 7
- 229910052702 rhenium Inorganic materials 0.000 description 7
- 229910052703 rhodium Inorganic materials 0.000 description 7
- 231100000331 toxic Toxicity 0.000 description 7
- 230000002588 toxic effect Effects 0.000 description 7
- 0 CCOC1OC(CC(C(COC2)C2C2c(cc3OC(C)=O)cc(*)c3OCC(*3C)=C*=C3[N+]([O-])=O)c3c2cc2OCOc2c3)C2OC(C)OC2C1O* Chemical compound CCOC1OC(CC(C(COC2)C2C2c(cc3OC(C)=O)cc(*)c3OCC(*3C)=C*=C3[N+]([O-])=O)c3c2cc2OCOc2c3)C2OC(C)OC2C1O* 0.000 description 6
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 6
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- OUUQCZGPVNCOIJ-UHFFFAOYSA-M Superoxide Chemical class [O-][O] OUUQCZGPVNCOIJ-UHFFFAOYSA-M 0.000 description 6
- 230000029936 alkylation Effects 0.000 description 6
- 230000008901 benefit Effects 0.000 description 6
- 150000002374 hemiaminals Chemical group 0.000 description 6
- 229960001330 hydroxycarbamide Drugs 0.000 description 6
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- 201000005202 lung cancer Diseases 0.000 description 6
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Substances [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 6
- 238000001356 surgical procedure Methods 0.000 description 6
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 6
- 108020004414 DNA Proteins 0.000 description 5
- 239000012448 Lithium borohydride Substances 0.000 description 5
- 229940122255 Microtubule inhibitor Drugs 0.000 description 5
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 208000029742 colonic neoplasm Diseases 0.000 description 5
- 230000006378 damage Effects 0.000 description 5
- 230000034994 death Effects 0.000 description 5
- 231100000517 death Toxicity 0.000 description 5
- 230000003111 delayed effect Effects 0.000 description 5
- 125000006575 electron-withdrawing group Chemical group 0.000 description 5
- 238000001727 in vivo Methods 0.000 description 5
- 238000002347 injection Methods 0.000 description 5
- 239000007924 injection Substances 0.000 description 5
- 230000007246 mechanism Effects 0.000 description 5
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 5
- 229960000485 methotrexate Drugs 0.000 description 5
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 5
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 125000003107 substituted aryl group Chemical group 0.000 description 5
- 229960003087 tioguanine Drugs 0.000 description 5
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 5
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 4
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 4
- WEAHRLBPCANXCN-UHFFFAOYSA-N Daunomycin Natural products CCC1(O)CC(OC2CC(N)C(O)C(C)O2)c3cc4C(=O)c5c(OC)cccc5C(=O)c4c(O)c3C1 WEAHRLBPCANXCN-UHFFFAOYSA-N 0.000 description 4
- 102000002177 Hypoxia-inducible factor-1 alpha Human genes 0.000 description 4
- 108050009527 Hypoxia-inducible factor-1 alpha Proteins 0.000 description 4
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 4
- 101710183280 Topoisomerase Proteins 0.000 description 4
- 125000002947 alkylene group Chemical group 0.000 description 4
- 239000003242 anti bacterial agent Substances 0.000 description 4
- 235000019445 benzyl alcohol Nutrition 0.000 description 4
- 208000035269 cancer or benign tumor Diseases 0.000 description 4
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 230000006872 improvement Effects 0.000 description 4
- 238000010253 intravenous injection Methods 0.000 description 4
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 4
- 239000003607 modifier Substances 0.000 description 4
- 125000002950 monocyclic group Chemical group 0.000 description 4
- 229930014626 natural product Natural products 0.000 description 4
- 231100000956 nontoxicity Toxicity 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 description 4
- 230000003389 potentiating effect Effects 0.000 description 4
- 125000006239 protecting group Chemical group 0.000 description 4
- 230000008707 rearrangement Effects 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 239000011550 stock solution Substances 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- 210000004881 tumor cell Anatomy 0.000 description 4
- 230000003442 weekly effect Effects 0.000 description 4
- WXSSUDRHAJTESR-UHFFFAOYSA-N (3-methyl-2-nitroimidazol-4-yl)methanol Chemical compound CN1C(CO)=CN=C1[N+]([O-])=O WXSSUDRHAJTESR-UHFFFAOYSA-N 0.000 description 3
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 3
- 208000036762 Acute promyelocytic leukaemia Diseases 0.000 description 3
- 229940122361 Bisphosphonate Drugs 0.000 description 3
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 3
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 3
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 3
- BRLQWZUYTZBJKN-UHFFFAOYSA-N Epichlorohydrin Chemical class ClCC1CO1 BRLQWZUYTZBJKN-UHFFFAOYSA-N 0.000 description 3
- 229940123414 Folate antagonist Drugs 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 208000034578 Multiple myelomas Diseases 0.000 description 3
- 102000004960 NAD(P)H dehydrogenase (quinone) Human genes 0.000 description 3
- 108020000284 NAD(P)H dehydrogenase (quinone) Proteins 0.000 description 3
- 229910003827 NRaRb Inorganic materials 0.000 description 3
- 208000033826 Promyelocytic Acute Leukemia Diseases 0.000 description 3
- 239000012979 RPMI medium Substances 0.000 description 3
- 108010077895 Sarcosine Proteins 0.000 description 3
- 206010041067 Small cell lung cancer Diseases 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 208000008383 Wilms tumor Diseases 0.000 description 3
- 230000002159 abnormal effect Effects 0.000 description 3
- 230000003213 activating effect Effects 0.000 description 3
- 238000011374 additional therapy Methods 0.000 description 3
- 229940088710 antibiotic agent Drugs 0.000 description 3
- 239000003886 aromatase inhibitor Substances 0.000 description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 3
- 150000004663 bisphosphonates Chemical class 0.000 description 3
- 201000008275 breast carcinoma Diseases 0.000 description 3
- 239000012267 brine Substances 0.000 description 3
- 231100000504 carcinogenesis Toxicity 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000009643 clonogenic assay Methods 0.000 description 3
- 231100000096 clonogenic assay Toxicity 0.000 description 3
- 210000001072 colon Anatomy 0.000 description 3
- 238000009096 combination chemotherapy Methods 0.000 description 3
- 230000008034 disappearance Effects 0.000 description 3
- 150000002170 ethers Chemical class 0.000 description 3
- 239000004052 folic acid antagonist Substances 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 229960001101 ifosfamide Drugs 0.000 description 3
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 3
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 3
- 239000007927 intramuscular injection Substances 0.000 description 3
- 238000010255 intramuscular injection Methods 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 229960001924 melphalan Drugs 0.000 description 3
- 229960004857 mitomycin Drugs 0.000 description 3
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 description 3
- 229940127073 nucleoside analogue Drugs 0.000 description 3
- 229960003552 other antineoplastic agent in atc Drugs 0.000 description 3
- 229940063179 platinol Drugs 0.000 description 3
- 150000003058 platinum compounds Chemical class 0.000 description 3
- 235000015320 potassium carbonate Nutrition 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 239000002599 prostaglandin synthase inhibitor Substances 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- 208000000587 small cell lung carcinoma Diseases 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- NVBFHJWHLNUMCV-UHFFFAOYSA-N sulfamide Chemical compound NS(N)(=O)=O NVBFHJWHLNUMCV-UHFFFAOYSA-N 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- DKPFODGZWDEEBT-QFIAKTPHSA-N taxane Chemical class C([C@]1(C)CCC[C@@H](C)[C@H]1C1)C[C@H]2[C@H](C)CC[C@@H]1C2(C)C DKPFODGZWDEEBT-QFIAKTPHSA-N 0.000 description 3
- 229960005267 tositumomab Drugs 0.000 description 3
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 3
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 3
- ZOCKGBMQLCSHFP-KQRAQHLDSA-N valrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC(OC)=C4C(=O)C=3C(O)=C21)(O)C(=O)COC(=O)CCCC)[C@H]1C[C@H](NC(=O)C(F)(F)F)[C@H](O)[C@H](C)O1 ZOCKGBMQLCSHFP-KQRAQHLDSA-N 0.000 description 3
- 239000003039 volatile agent Substances 0.000 description 3
- AADVCYNFEREWOS-UHFFFAOYSA-N (+)-DDM Natural products C=CC=CC(C)C(OC(N)=O)C(C)C(O)C(C)CC(C)=CC(C)C(O)C(C)C=CC(O)CC1OC(=O)C(C)C(O)C1C AADVCYNFEREWOS-UHFFFAOYSA-N 0.000 description 2
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 description 2
- IAKHMKGGTNLKSZ-INIZCTEOSA-N (S)-colchicine Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC IAKHMKGGTNLKSZ-INIZCTEOSA-N 0.000 description 2
- QFWCYNPOPKQOKV-UHFFFAOYSA-N 2-(2-amino-3-methoxyphenyl)chromen-4-one Chemical compound COC1=CC=CC(C=2OC3=CC=CC=C3C(=O)C=2)=C1N QFWCYNPOPKQOKV-UHFFFAOYSA-N 0.000 description 2
- BPBPYQWMFCTCNG-UHFFFAOYSA-N 2-(butan-2-yldisulfanyl)-1H-imidazole Chemical compound CCC(C)SSC1=NC=CN1 BPBPYQWMFCTCNG-UHFFFAOYSA-N 0.000 description 2
- FOAINFSTWSHMPH-UHFFFAOYSA-N 2-[ethyl(methyl)azaniumyl]acetate Chemical compound CCN(C)CC(O)=O FOAINFSTWSHMPH-UHFFFAOYSA-N 0.000 description 2
- CQOQDQWUFQDJMK-SSTWWWIQSA-N 2-methoxy-17beta-estradiol Chemical compound C([C@@H]12)C[C@]3(C)[C@@H](O)CC[C@H]3[C@@H]1CCC1=C2C=C(OC)C(O)=C1 CQOQDQWUFQDJMK-SSTWWWIQSA-N 0.000 description 2
- FTBBGQKRYUTLMP-UHFFFAOYSA-N 2-nitro-1h-pyrrole Chemical class [O-][N+](=O)C1=CC=CN1 FTBBGQKRYUTLMP-UHFFFAOYSA-N 0.000 description 2
- WUIABRMSWOKTOF-OYALTWQYSA-N 3-[[2-[2-[2-[[(2s,3r)-2-[[(2s,3s,4r)-4-[[(2s,3r)-2-[[6-amino-2-[(1s)-3-amino-1-[[(2s)-2,3-diamino-3-oxopropyl]amino]-3-oxopropyl]-5-methylpyrimidine-4-carbonyl]amino]-3-[(2r,3s,4s,5s,6s)-3-[(2r,3s,4s,5r,6r)-4-carbamoyloxy-3,5-dihydroxy-6-(hydroxymethyl)ox Chemical compound OS([O-])(=O)=O.N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C WUIABRMSWOKTOF-OYALTWQYSA-N 0.000 description 2
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 2
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 2
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 2
- 206010005003 Bladder cancer Diseases 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 2
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 2
- XCMYCICHMZBASJ-UHFFFAOYSA-N ClCCNP1OCCCN1 Chemical compound ClCCNP1OCCCN1 XCMYCICHMZBASJ-UHFFFAOYSA-N 0.000 description 2
- AADVCYNFEREWOS-OBRABYBLSA-N Discodermolide Chemical compound C=C\C=C/[C@H](C)[C@H](OC(N)=O)[C@@H](C)[C@H](O)[C@@H](C)C\C(C)=C/[C@H](C)[C@@H](O)[C@@H](C)\C=C/[C@@H](O)C[C@@H]1OC(=O)[C@H](C)[C@@H](O)[C@H]1C AADVCYNFEREWOS-OBRABYBLSA-N 0.000 description 2
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 2
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- 102000015696 Interleukins Human genes 0.000 description 2
- 108010063738 Interleukins Proteins 0.000 description 2
- 208000005125 Invasive Hydatidiform Mole Diseases 0.000 description 2
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 2
- 206010027452 Metastases to bone Diseases 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- 102000029749 Microtubule Human genes 0.000 description 2
- 108091022875 Microtubule Proteins 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 2
- 239000007832 Na2SO4 Substances 0.000 description 2
- 102000004316 Oxidoreductases Human genes 0.000 description 2
- 108090000854 Oxidoreductases Proteins 0.000 description 2
- 239000012661 PARP inhibitor Substances 0.000 description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 2
- SHGAZHPCJJPHSC-UHFFFAOYSA-N Panrexin Chemical compound OC(=O)C=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-UHFFFAOYSA-N 0.000 description 2
- 229940121906 Poly ADP ribose polymerase inhibitor Drugs 0.000 description 2
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 2
- HFVNWDWLWUCIHC-GUPDPFMOSA-N Prednimustine Chemical compound O=C([C@@]1(O)CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)[C@@H](O)C[C@@]21C)COC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 HFVNWDWLWUCIHC-GUPDPFMOSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- NAVMQTYZDKMPEU-UHFFFAOYSA-N Targretin Chemical compound CC1=CC(C(CCC2(C)C)(C)C)=C2C=C1C(=C)C1=CC=C(C(O)=O)C=C1 NAVMQTYZDKMPEU-UHFFFAOYSA-N 0.000 description 2
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 2
- 208000024313 Testicular Neoplasms Diseases 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- 239000000365 Topoisomerase I Inhibitor Substances 0.000 description 2
- 239000000317 Topoisomerase II Inhibitor Substances 0.000 description 2
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 2
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 2
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 2
- 241000863480 Vinca Species 0.000 description 2
- QTQAWLPCGQOSGP-VKNBREQOSA-N [(3r,5r,6s,7r,8e,10r,11r,12z,14e)-6-hydroxy-5,11,21-trimethoxy-3,7,9,15-tetramethyl-16,20,22-trioxo-17-azabicyclo[16.3.1]docosa-1(21),8,12,14,18-pentaen-10-yl] carbamate Chemical compound N1C(=O)\C(C)=C\C=C/[C@@H](OC)[C@H](OC(N)=O)\C(C)=C\[C@@H](C)[C@H](O)[C@H](OC)C[C@H](C)CC2=C(OC)C(=O)C=C1C2=O QTQAWLPCGQOSGP-VKNBREQOSA-N 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 230000001780 adrenocortical effect Effects 0.000 description 2
- 229940009456 adriamycin Drugs 0.000 description 2
- 125000003282 alkyl amino group Chemical group 0.000 description 2
- 230000002152 alkylating effect Effects 0.000 description 2
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 229940045719 antineoplastic alkylating agent nitrosoureas Drugs 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 229940046844 aromatase inhibitors Drugs 0.000 description 2
- GOLCXWYRSKYTSP-UHFFFAOYSA-N arsenic trioxide Inorganic materials O1[As]2O[As]1O2 GOLCXWYRSKYTSP-UHFFFAOYSA-N 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 229940120638 avastin Drugs 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 238000004166 bioassay Methods 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 150000001649 bromium compounds Chemical class 0.000 description 2
- 239000003560 cancer drug Substances 0.000 description 2
- 230000005907 cancer growth Effects 0.000 description 2
- CGRNNEJYAQQCOQ-UHFFFAOYSA-N carbamic acid;2-nitro-1h-imidazole Chemical compound NC(O)=O.[O-][N+](=O)C1=NC=CN1 CGRNNEJYAQQCOQ-UHFFFAOYSA-N 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 229940047495 celebrex Drugs 0.000 description 2
- 229960000590 celecoxib Drugs 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 238000007385 chemical modification Methods 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 229960002436 cladribine Drugs 0.000 description 2
- 230000003021 clonogenic effect Effects 0.000 description 2
- 238000011284 combination treatment Methods 0.000 description 2
- 229940125904 compound 1 Drugs 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 238000004132 cross linking Methods 0.000 description 2
- 230000003013 cytotoxicity Effects 0.000 description 2
- 231100000135 cytotoxicity Toxicity 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 230000008030 elimination Effects 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 229960004750 estramustine phosphate Drugs 0.000 description 2
- ADFOJJHRTBFFOF-RBRWEJTLSA-N estramustine phosphate Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)OP(O)(O)=O)[C@@H]4[C@@H]3CCC2=C1 ADFOJJHRTBFFOF-RBRWEJTLSA-N 0.000 description 2
- 239000002834 estrogen receptor modulator Substances 0.000 description 2
- LIQODXNTTZAGID-OCBXBXKTSA-N etoposide phosphate Chemical compound COC1=C(OP(O)(O)=O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 LIQODXNTTZAGID-OCBXBXKTSA-N 0.000 description 2
- 229960000752 etoposide phosphate Drugs 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 239000012091 fetal bovine serum Substances 0.000 description 2
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 2
- VVIAGPKUTFNRDU-ABLWVSNPSA-N folinic acid Chemical compound C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-ABLWVSNPSA-N 0.000 description 2
- 208000010749 gastric carcinoma Diseases 0.000 description 2
- 229940045109 genistein Drugs 0.000 description 2
- TZBJGXHYKVUXJN-UHFFFAOYSA-N genistein Natural products C1=CC(O)=CC=C1C1=COC2=CC(O)=CC(O)=C2C1=O TZBJGXHYKVUXJN-UHFFFAOYSA-N 0.000 description 2
- 235000006539 genistein Nutrition 0.000 description 2
- ZCOLJUOHXJRHDI-CMWLGVBASA-N genistein 7-O-beta-D-glucoside Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=C2C(=O)C(C=3C=CC(O)=CC=3)=COC2=C1 ZCOLJUOHXJRHDI-CMWLGVBASA-N 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 2
- 201000010536 head and neck cancer Diseases 0.000 description 2
- 208000014829 head and neck neoplasm Diseases 0.000 description 2
- 125000003875 hemiaminal group Chemical group 0.000 description 2
- 235000008216 herbs Nutrition 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 229940096120 hydrea Drugs 0.000 description 2
- 229960000908 idarubicin Drugs 0.000 description 2
- 150000002466 imines Chemical class 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- DKYWVDODHFEZIM-UHFFFAOYSA-N ketoprofen Chemical compound OC(=O)C(C)C1=CC=CC(C(=O)C=2C=CC=CC=2)=C1 DKYWVDODHFEZIM-UHFFFAOYSA-N 0.000 description 2
- 150000002596 lactones Chemical class 0.000 description 2
- 229960001691 leucovorin Drugs 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 2
- 230000000873 masking effect Effects 0.000 description 2
- 230000010534 mechanism of action Effects 0.000 description 2
- 229960004961 mechlorethamine Drugs 0.000 description 2
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical class ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- 210000004688 microtubule Anatomy 0.000 description 2
- HDZGCSFEDULWCS-UHFFFAOYSA-N monomethylhydrazine Chemical class CNN HDZGCSFEDULWCS-UHFFFAOYSA-N 0.000 description 2
- 239000002777 nucleoside Substances 0.000 description 2
- 150000003833 nucleoside derivatives Chemical class 0.000 description 2
- 238000011275 oncology therapy Methods 0.000 description 2
- 210000002997 osteoclast Anatomy 0.000 description 2
- 210000001672 ovary Anatomy 0.000 description 2
- 238000002638 palliative care Methods 0.000 description 2
- WRUUGTRCQOWXEG-UHFFFAOYSA-N pamidronate Chemical compound NCCC(O)(P(O)(O)=O)P(O)(O)=O WRUUGTRCQOWXEG-UHFFFAOYSA-N 0.000 description 2
- 201000002528 pancreatic cancer Diseases 0.000 description 2
- 208000008443 pancreatic carcinoma Diseases 0.000 description 2
- 239000000419 plant extract Substances 0.000 description 2
- 238000006116 polymerization reaction Methods 0.000 description 2
- 229920001282 polysaccharide Polymers 0.000 description 2
- 239000005017 polysaccharide Substances 0.000 description 2
- 150000004804 polysaccharides Chemical class 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 229960004694 prednimustine Drugs 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- AECPBJMOGBFQDN-YMYQVXQQSA-N radicicol Chemical compound C1CCCC(=O)C[C@H]2[C@H](Cl)C(=O)CC(=O)[C@H]2C(=O)O[C@H](C)C[C@H]2O[C@@H]21 AECPBJMOGBFQDN-YMYQVXQQSA-N 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 125000006413 ring segment Chemical group 0.000 description 2
- 229960004641 rituximab Drugs 0.000 description 2
- OHRURASPPZQGQM-GCCNXGTGSA-N romidepsin Chemical compound O1C(=O)[C@H](C(C)C)NC(=O)C(=C/C)/NC(=O)[C@H]2CSSCC\C=C\[C@@H]1CC(=O)N[C@H](C(C)C)C(=O)N2 OHRURASPPZQGQM-GCCNXGTGSA-N 0.000 description 2
- FSYKKLYZXJSNPZ-UHFFFAOYSA-N sarcosine Chemical compound C[NH2+]CC([O-])=O FSYKKLYZXJSNPZ-UHFFFAOYSA-N 0.000 description 2
- 238000005204 segregation Methods 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 125000005415 substituted alkoxy group Chemical group 0.000 description 2
- MLKXDPUZXIRXEP-MFOYZWKCSA-N sulindac Chemical compound CC1=C(CC(O)=O)C2=CC(F)=CC=C2\C1=C/C1=CC=C(S(C)=O)C=C1 MLKXDPUZXIRXEP-MFOYZWKCSA-N 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- AYUNIORJHRXIBJ-TXHRRWQRSA-N tanespimycin Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(NCC=C)C(=O)C=C1C2=O AYUNIORJHRXIBJ-TXHRRWQRSA-N 0.000 description 2
- UPSPUYADGBWSHF-UHFFFAOYSA-N tolmetin Chemical compound C1=CC(C)=CC=C1C(=O)C1=CC=C(CC(O)=O)N1C UPSPUYADGBWSHF-UHFFFAOYSA-N 0.000 description 2
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 230000004222 uncontrolled growth Effects 0.000 description 2
- 201000005112 urinary bladder cancer Diseases 0.000 description 2
- 229960000653 valrubicin Drugs 0.000 description 2
- 230000002792 vascular Effects 0.000 description 2
- 210000005166 vasculature Anatomy 0.000 description 2
- 229940053867 xeloda Drugs 0.000 description 2
- XRASPMIURGNCCH-UHFFFAOYSA-N zoledronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CN1C=CN=C1 XRASPMIURGNCCH-UHFFFAOYSA-N 0.000 description 2
- QCPDBEXGCHOIDE-UHFFFAOYSA-N (-)-6xi-Methyl-(2ar,4axi,8at,12bt,12ct)-2a,3,4,4a,5,6,7,8a,12b,12c-decahydro-5xi,12dxi-aethano-furo[4',3',2';4,10]anthra[9,1-bc]oxepin-2,9,12-trion Natural products CC1COC2C(C(C=CC3=O)=O)=C3C3C4C22CCC1C2CCC4C(=O)O3 QCPDBEXGCHOIDE-UHFFFAOYSA-N 0.000 description 1
- DOTUTEHJCHVMAB-UHFFFAOYSA-N (1-ethyl-4,5-dinitroimidazol-2-yl)methanol Chemical compound CCN1C(CO)=NC([N+]([O-])=O)=C1[N+]([O-])=O DOTUTEHJCHVMAB-UHFFFAOYSA-N 0.000 description 1
- JPNBVWIRDQVGAC-UHFFFAOYSA-N (2-nitrophenyl) hydrogen carbonate Chemical class OC(=O)OC1=CC=CC=C1[N+]([O-])=O JPNBVWIRDQVGAC-UHFFFAOYSA-N 0.000 description 1
- HBUBKKRHXORPQB-FJFJXFQQSA-N (2R,3S,4S,5R)-2-(6-amino-2-fluoro-9-purinyl)-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O HBUBKKRHXORPQB-FJFJXFQQSA-N 0.000 description 1
- RDJGLLICXDHJDY-NSHDSACASA-N (2s)-2-(3-phenoxyphenyl)propanoic acid Chemical compound OC(=O)[C@@H](C)C1=CC=CC(OC=2C=CC=CC=2)=C1 RDJGLLICXDHJDY-NSHDSACASA-N 0.000 description 1
- PSVUJBVBCOISSP-SPFKKGSWSA-N (2s,3r,4s,5s,6r)-2-bis(2-chloroethylamino)phosphoryloxy-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound OC[C@H]1O[C@@H](OP(=O)(NCCCl)NCCCl)[C@H](O)[C@@H](O)[C@@H]1O PSVUJBVBCOISSP-SPFKKGSWSA-N 0.000 description 1
- VRYALKFFQXWPIH-PBXRRBTRSA-N (3r,4s,5r)-3,4,5,6-tetrahydroxyhexanal Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)CC=O VRYALKFFQXWPIH-PBXRRBTRSA-N 0.000 description 1
- QHFKWIKCUHNXAU-UHFFFAOYSA-N (4-nitrophenyl) carbamate Chemical class NC(=O)OC1=CC=C([N+]([O-])=O)C=C1 QHFKWIKCUHNXAU-UHFFFAOYSA-N 0.000 description 1
- NQUUXKIFXGKMAF-UHFFFAOYSA-N (5-methyl-2-nitro-1h-imidazol-4-yl)methanol Chemical compound CC=1N=C([N+]([O-])=O)NC=1CO NQUUXKIFXGKMAF-UHFFFAOYSA-N 0.000 description 1
- FDZKMDGYTVIWIB-LCWDIZFYSA-N (6r,6as,8s)-3,8-dihydroxy-2,6-dimethoxy-5,6,6a,7,8,9-hexahydropyrrolo[2,1-c][1,4]benzodiazepin-11-one Chemical compound CO[C@H]1NC2=CC(O)=C(OC)C=C2C(=O)N2C[C@@H](O)C[C@@H]12 FDZKMDGYTVIWIB-LCWDIZFYSA-N 0.000 description 1
- NNYBQONXHNTVIJ-QGZVFWFLSA-N (R)-etodolac Chemical compound C1CO[C@](CC)(CC(O)=O)C2=C1C(C=CC=C1CC)=C1N2 NNYBQONXHNTVIJ-QGZVFWFLSA-N 0.000 description 1
- HJTAZXHBEBIQQX-UHFFFAOYSA-N 1,5-bis(chloromethyl)naphthalene Chemical compound C1=CC=C2C(CCl)=CC=CC2=C1CCl HJTAZXHBEBIQQX-UHFFFAOYSA-N 0.000 description 1
- ZOMATQMEHRJKLO-UHFFFAOYSA-N 1h-imidazol-2-ylmethanol Chemical class OCC1=NC=CN1 ZOMATQMEHRJKLO-UHFFFAOYSA-N 0.000 description 1
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 1
- XXGOYFOGYQNGJC-LBDSCFHVSA-N 2-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,2,4-triazine-3,5-dione;5-fluoro-1h-pyrimidine-2,4-dione Chemical compound FC1=CNC(=O)NC1=O.O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=N1 XXGOYFOGYQNGJC-LBDSCFHVSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- VEWCAFQJYMQLJB-UHFFFAOYSA-N 2-[ethyl(methyl)amino]acetic acid;hydrochloride Chemical compound Cl.CCN(C)CC(O)=O VEWCAFQJYMQLJB-UHFFFAOYSA-N 0.000 description 1
- 125000000022 2-aminoethyl group Chemical group [H]C([*])([H])C([H])([H])N([H])[H] 0.000 description 1
- KYVQCVBMLZMRKL-UHFFFAOYSA-N 2-ethoxy-5-(pyrrolidine-1-carbonylamino)benzenesulfonyl chloride Chemical compound C1=C(S(Cl)(=O)=O)C(OCC)=CC=C1NC(=O)N1CCCC1 KYVQCVBMLZMRKL-UHFFFAOYSA-N 0.000 description 1
- NXWTWYULZRDBSA-UHFFFAOYSA-N 2-fluoro-4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1F NXWTWYULZRDBSA-UHFFFAOYSA-N 0.000 description 1
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 1
- HUNDJKXYCIAYJT-UHFFFAOYSA-N 2-nitro-1,3-oxazole Chemical class [O-][N+](=O)C1=NC=CO1 HUNDJKXYCIAYJT-UHFFFAOYSA-N 0.000 description 1
- HQVAVYYAPZDXHV-UHFFFAOYSA-N 2-nitro-1h-imidazole;hydrobromide Chemical compound Br.[O-][N+](=O)C1=NC=CN1 HQVAVYYAPZDXHV-UHFFFAOYSA-N 0.000 description 1
- KLGQWSOYKYFBTR-UHFFFAOYSA-N 2-nitrobenzamide Chemical class NC(=O)C1=CC=CC=C1[N+]([O-])=O KLGQWSOYKYFBTR-UHFFFAOYSA-N 0.000 description 1
- SLAMLWHELXOEJZ-UHFFFAOYSA-N 2-nitrobenzoic acid Chemical compound OC(=O)C1=CC=CC=C1[N+]([O-])=O SLAMLWHELXOEJZ-UHFFFAOYSA-N 0.000 description 1
- FUBFWTUFPGFHOJ-UHFFFAOYSA-N 2-nitrofuran Chemical class [O-][N+](=O)C1=CC=CO1 FUBFWTUFPGFHOJ-UHFFFAOYSA-N 0.000 description 1
- IQUPABOKLQSFBK-UHFFFAOYSA-N 2-nitrophenol Chemical compound OC1=CC=CC=C1[N+]([O-])=O IQUPABOKLQSFBK-UHFFFAOYSA-N 0.000 description 1
- RVBUGGBMJDPOST-UHFFFAOYSA-N 2-thiobarbituric acid Chemical class O=C1CC(=O)NC(=S)N1 RVBUGGBMJDPOST-UHFFFAOYSA-N 0.000 description 1
- PRRZDZJYSJLDBS-UHFFFAOYSA-N 3-bromo-2-oxopropanoic acid Chemical compound OC(=O)C(=O)CBr PRRZDZJYSJLDBS-UHFFFAOYSA-N 0.000 description 1
- FYDIZCAZYUMWTJ-UHFFFAOYSA-N 3-nitro-2H-pyran-2-ol Chemical class [N+](=O)([O-])C=1C(OC=CC=1)O FYDIZCAZYUMWTJ-UHFFFAOYSA-N 0.000 description 1
- GIGCDIVNDFQKRA-LTCKWSDVSA-N 4-[(2s)-2-amino-2-carboxyethyl]-n,n-bis(2-chloroethyl)benzeneamine oxide;dihydrochloride Chemical compound Cl.Cl.OC(=O)[C@@H](N)CC1=CC=C([N+]([O-])(CCCl)CCCl)C=C1 GIGCDIVNDFQKRA-LTCKWSDVSA-N 0.000 description 1
- BQAPMRHAUAEUJV-UHFFFAOYSA-N 5-(bromomethyl)-1-methyl-2-nitroimidazole Chemical compound CN1C(CBr)=CN=C1[N+]([O-])=O BQAPMRHAUAEUJV-UHFFFAOYSA-N 0.000 description 1
- RSPZLRJUQLWMSD-UHFFFAOYSA-N 5-(chloromethoxymethyl)-2-nitro-1h-imidazole Chemical class [O-][N+](=O)C1=NC(COCCl)=CN1 RSPZLRJUQLWMSD-UHFFFAOYSA-N 0.000 description 1
- LXRCTUGTIGILID-UHFFFAOYSA-N 5-(chloromethyl)-2-nitro-1h-imidazole Chemical compound [O-][N+](=O)C1=NC(CCl)=CN1 LXRCTUGTIGILID-UHFFFAOYSA-N 0.000 description 1
- UKIWLFJYNMJPEG-UHFFFAOYSA-N 5-bromo-2h-isoquinolin-1-one Chemical compound C1=CC=C2C(O)=NC=CC2=C1Br UKIWLFJYNMJPEG-UHFFFAOYSA-N 0.000 description 1
- 150000004958 5-nitroimidazoles Chemical class 0.000 description 1
- VVIAGPKUTFNRDU-UHFFFAOYSA-N 6S-folinic acid Natural products C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-UHFFFAOYSA-N 0.000 description 1
- SHGAZHPCJJPHSC-ZVCIMWCZSA-N 9-cis-retinoic acid Chemical compound OC(=O)/C=C(\C)/C=C/C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-ZVCIMWCZSA-N 0.000 description 1
- 206010000830 Acute leukaemia Diseases 0.000 description 1
- 208000036832 Adenocarcinoma of ovary Diseases 0.000 description 1
- 201000004384 Alopecia Diseases 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 102400000068 Angiostatin Human genes 0.000 description 1
- 108010079709 Angiostatins Proteins 0.000 description 1
- 206010002660 Anoxia Diseases 0.000 description 1
- 241000976983 Anoxia Species 0.000 description 1
- 229940122815 Aromatase inhibitor Drugs 0.000 description 1
- DJHGAFSJWGLOIV-UHFFFAOYSA-L Arsenate2- Chemical compound O[As]([O-])([O-])=O DJHGAFSJWGLOIV-UHFFFAOYSA-L 0.000 description 1
- DJHGAFSJWGLOIV-UHFFFAOYSA-K Arsenate3- Chemical class [O-][As]([O-])([O-])=O DJHGAFSJWGLOIV-UHFFFAOYSA-K 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 206010003571 Astrocytoma Diseases 0.000 description 1
- 208000004736 B-Cell Leukemia Diseases 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 206010055113 Breast cancer metastatic Diseases 0.000 description 1
- IVFOEGDXOJDKMB-UHFFFAOYSA-N CC(OCc(cc1)ccc1OCc([n]1C)cnc1[NH+]([O-])O)=O Chemical compound CC(OCc(cc1)ccc1OCc([n]1C)cnc1[NH+]([O-])O)=O IVFOEGDXOJDKMB-UHFFFAOYSA-N 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 201000009030 Carcinoma Diseases 0.000 description 1
- 240000001829 Catharanthus roseus Species 0.000 description 1
- 229910052684 Cerium Inorganic materials 0.000 description 1
- FDZKMDGYTVIWIB-UHFFFAOYSA-N Chicamycin A Natural products COC1NC2=CC(O)=C(OC)C=C2C(=O)N2CC(O)CC12 FDZKMDGYTVIWIB-UHFFFAOYSA-N 0.000 description 1
- 208000006332 Choriocarcinoma Diseases 0.000 description 1
- 102000007644 Colony-Stimulating Factors Human genes 0.000 description 1
- 108010071942 Colony-Stimulating Factors Proteins 0.000 description 1
- 229940126062 Compound A Drugs 0.000 description 1
- 102100021906 Cyclin-O Human genes 0.000 description 1
- 229940122204 Cyclooxygenase inhibitor Drugs 0.000 description 1
- 102000004328 Cytochrome P-450 CYP3A Human genes 0.000 description 1
- 108010081668 Cytochrome P-450 CYP3A Proteins 0.000 description 1
- 101710112752 Cytotoxin Proteins 0.000 description 1
- 230000008265 DNA repair mechanism Effects 0.000 description 1
- 108010002156 Depsipeptides Proteins 0.000 description 1
- 101001031598 Dictyostelium discoideum Probable serine/threonine-protein kinase fhkC Proteins 0.000 description 1
- 102100031480 Dual specificity mitogen-activated protein kinase kinase 1 Human genes 0.000 description 1
- 101710146526 Dual specificity mitogen-activated protein kinase kinase 1 Proteins 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 102400001047 Endostatin Human genes 0.000 description 1
- 108010079505 Endostatins Proteins 0.000 description 1
- QXRSDHAAWVKZLJ-OXZHEXMSSA-N Epothilone B Natural products O=C1[C@H](C)[C@H](O)[C@@H](C)CCC[C@@]2(C)O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C QXRSDHAAWVKZLJ-OXZHEXMSSA-N 0.000 description 1
- XOZIUKBZLSUILX-SDMHVBBESA-N Epothilone D Natural products O=C1[C@H](C)[C@@H](O)[C@@H](C)CCC/C(/C)=C/C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C XOZIUKBZLSUILX-SDMHVBBESA-N 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- 102000018711 Facilitative Glucose Transport Proteins Human genes 0.000 description 1
- 201000006107 Familial adenomatous polyposis Diseases 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 1
- 201000003741 Gastrointestinal carcinoma Diseases 0.000 description 1
- 206010051066 Gastrointestinal stromal tumour Diseases 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 108091052347 Glucose transporter family Proteins 0.000 description 1
- 102000005720 Glutathione transferase Human genes 0.000 description 1
- 108010070675 Glutathione transferase Proteins 0.000 description 1
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Natural products NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- NYHBQMYGNKIUIF-UUOKFMHZSA-N Guanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O NYHBQMYGNKIUIF-UUOKFMHZSA-N 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 101710113864 Heat shock protein 90 Proteins 0.000 description 1
- 102100034051 Heat shock protein HSP 90-alpha Human genes 0.000 description 1
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 1
- 239000004705 High-molecular-weight polyethylene Substances 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 101000897441 Homo sapiens Cyclin-O Proteins 0.000 description 1
- 101001016865 Homo sapiens Heat shock protein HSP 90-alpha Proteins 0.000 description 1
- 101000582950 Homo sapiens Platelet factor 4 Proteins 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 208000035663 Invasive mole Diseases 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 229930045534 Me ester-Cyclohexaneundecanoic acid Natural products 0.000 description 1
- XOGTZOOQQBDUSI-UHFFFAOYSA-M Mesna Chemical compound [Na+].[O-]S(=O)(=O)CCS XOGTZOOQQBDUSI-UHFFFAOYSA-M 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 229930192392 Mitomycin Natural products 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- 108010045510 NADPH-Ferrihemoprotein Reductase Proteins 0.000 description 1
- 229910017711 NHRa Inorganic materials 0.000 description 1
- BLXXJMDCKKHMKV-UHFFFAOYSA-N Nabumetone Chemical compound C1=C(CCC(C)=O)C=CC2=CC(OC)=CC=C21 BLXXJMDCKKHMKV-UHFFFAOYSA-N 0.000 description 1
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 1
- 206010061309 Neoplasm progression Diseases 0.000 description 1
- IOVCWXUNBOPUCH-UHFFFAOYSA-M Nitrite anion Chemical compound [O-]N=O IOVCWXUNBOPUCH-UHFFFAOYSA-M 0.000 description 1
- 102000004459 Nitroreductase Human genes 0.000 description 1
- YJQPYGGHQPGBLI-UHFFFAOYSA-N Novobiocin Natural products O1C(C)(C)C(OC)C(OC(N)=O)C(O)C1OC1=CC=C(C(O)=C(NC(=O)C=2C=C(CC=C(C)C)C(O)=CC=2)C(=O)O2)C2=C1C YJQPYGGHQPGBLI-UHFFFAOYSA-N 0.000 description 1
- MSHZHSPISPJWHW-UHFFFAOYSA-N O-(chloroacetylcarbamoyl)fumagillol Chemical compound O1C(CC=C(C)C)C1(C)C1C(OC)C(OC(=O)NC(=O)CCl)CCC21CO2 MSHZHSPISPJWHW-UHFFFAOYSA-N 0.000 description 1
- 229940121682 Osteoclast inhibitor Drugs 0.000 description 1
- 206010061328 Ovarian epithelial cancer Diseases 0.000 description 1
- BELBBZDIHDAJOR-UHFFFAOYSA-N Phenolsulfonephthalein Chemical compound C1=CC(O)=CC=C1C1(C=2C=CC(O)=CC=2)C2=CC=CC=C2S(=O)(=O)O1 BELBBZDIHDAJOR-UHFFFAOYSA-N 0.000 description 1
- 101710179684 Poly [ADP-ribose] polymerase Proteins 0.000 description 1
- 102100023712 Poly [ADP-ribose] polymerase 1 Human genes 0.000 description 1
- 229920000776 Poly(Adenosine diphosphate-ribose) polymerase Polymers 0.000 description 1
- 208000006994 Precancerous Conditions Diseases 0.000 description 1
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 description 1
- CKIHZSGJPSDCNC-UHFFFAOYSA-N Quindoxin Chemical class C1=CC=C2N([O-])C=C[N+](=O)C2=C1 CKIHZSGJPSDCNC-UHFFFAOYSA-N 0.000 description 1
- 108091007187 Reductases Proteins 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 241000187081 Streptomyces peucetius Species 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 206010042971 T-cell lymphoma Diseases 0.000 description 1
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 206010057644 Testis cancer Diseases 0.000 description 1
- 102000002938 Thrombospondin Human genes 0.000 description 1
- 108060008245 Thrombospondin Proteins 0.000 description 1
- 208000033781 Thyroid carcinoma Diseases 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 206010047700 Vomiting Diseases 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 229960000583 acetic acid Drugs 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 238000005903 acid hydrolysis reaction Methods 0.000 description 1
- 238000010306 acid treatment Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- USZYSDMBJDPRIF-SVEJIMAYSA-N aclacinomycin A Chemical compound O([C@H]1[C@@H](O)C[C@@H](O[C@H]1C)O[C@H]1[C@H](C[C@@H](O[C@H]1C)O[C@H]1C[C@]([C@@H](C2=CC=3C(=O)C4=CC=CC(O)=C4C(=O)C=3C(O)=C21)C(=O)OC)(O)CC)N(C)C)[C@H]1CCC(=O)[C@H](C)O1 USZYSDMBJDPRIF-SVEJIMAYSA-N 0.000 description 1
- 229960004176 aclarubicin Drugs 0.000 description 1
- 231100000569 acute exposure Toxicity 0.000 description 1
- 238000009098 adjuvant therapy Methods 0.000 description 1
- 229940064305 adrucil Drugs 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 125000003172 aldehyde group Chemical group 0.000 description 1
- 229940060515 aleve Drugs 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229960001445 alitretinoin Drugs 0.000 description 1
- 229930013930 alkaloid Natural products 0.000 description 1
- 150000003797 alkaloid derivatives Chemical class 0.000 description 1
- 229940098174 alkeran Drugs 0.000 description 1
- PMMURAAUARKVCB-UHFFFAOYSA-N alpha-D-ara-dHexp Natural products OCC1OC(O)CC(O)C1O PMMURAAUARKVCB-UHFFFAOYSA-N 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XCPGHVQEEXUHNC-UHFFFAOYSA-N amsacrine Chemical compound COC1=CC(NS(C)(=O)=O)=CC=C1NC1=C(C=CC=C2)C2=NC2=CC=CC=C12 XCPGHVQEEXUHNC-UHFFFAOYSA-N 0.000 description 1
- 229960001220 amsacrine Drugs 0.000 description 1
- 206010002224 anaplastic astrocytoma Diseases 0.000 description 1
- 229960002932 anastrozole Drugs 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 1
- 230000007953 anoxia Effects 0.000 description 1
- 239000003817 anthracycline antibiotic agent Substances 0.000 description 1
- 230000001772 anti-angiogenic effect Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 239000000063 antileukemic agent Substances 0.000 description 1
- 239000003972 antineoplastic antibiotic Substances 0.000 description 1
- 229940045985 antineoplastic platinum compound Drugs 0.000 description 1
- 229940058965 antiprotozoal agent against amoebiasis and other protozoal diseases nitroimidazole derivative Drugs 0.000 description 1
- 229950006345 antramycin Drugs 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 229940078010 arimidex Drugs 0.000 description 1
- FZCSTZYAHCUGEM-UHFFFAOYSA-N aspergillomarasmine B Natural products OC(=O)CNC(C(O)=O)CNC(C(O)=O)CC(O)=O FZCSTZYAHCUGEM-UHFFFAOYSA-N 0.000 description 1
- 229940125717 barbiturate Drugs 0.000 description 1
- XFILPEOLDIKJHX-QYZOEREBSA-N batimastat Chemical compound C([C@@H](C(=O)NC)NC(=O)[C@H](CC(C)C)[C@H](CSC=1SC=CC=1)C(=O)NO)C1=CC=CC=C1 XFILPEOLDIKJHX-QYZOEREBSA-N 0.000 description 1
- 229950001858 batimastat Drugs 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 150000001556 benzimidazoles Chemical class 0.000 description 1
- ZYGHJZDHTFUPRJ-UHFFFAOYSA-N benzo-alpha-pyrone Natural products C1=CC=C2OC(=O)C=CC2=C1 ZYGHJZDHTFUPRJ-UHFFFAOYSA-N 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 229960002938 bexarotene Drugs 0.000 description 1
- 229940108502 bicnu Drugs 0.000 description 1
- 238000002306 biochemical method Methods 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 229960000074 biopharmaceutical Drugs 0.000 description 1
- 238000001815 biotherapy Methods 0.000 description 1
- HRQGCQVOJVTVLU-UHFFFAOYSA-N bis(chloromethyl) ether Chemical compound ClCOCCl HRQGCQVOJVTVLU-UHFFFAOYSA-N 0.000 description 1
- 125000006367 bivalent amino carbonyl group Chemical group [H]N([*:1])C([*:2])=O 0.000 description 1
- 238000004820 blood count Methods 0.000 description 1
- 230000036770 blood supply Effects 0.000 description 1
- 206010006007 bone sarcoma Diseases 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 125000005997 bromomethyl group Chemical group 0.000 description 1
- 208000003362 bronchogenic carcinoma Diseases 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- YDVJBLJCSLVMSY-UHFFFAOYSA-N carbamoyl cyanide Chemical compound NC(=O)C#N YDVJBLJCSLVMSY-UHFFFAOYSA-N 0.000 description 1
- 150000001721 carbon Chemical class 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 210000000845 cartilage Anatomy 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- GWXLDORMOJMVQZ-UHFFFAOYSA-N cerium Chemical compound [Ce] GWXLDORMOJMVQZ-UHFFFAOYSA-N 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- 208000029664 classic familial adenomatous polyposis Diseases 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229960001338 colchicine Drugs 0.000 description 1
- 230000005757 colony formation Effects 0.000 description 1
- 229940047120 colony stimulating factors Drugs 0.000 description 1
- 201000010989 colorectal carcinoma Diseases 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 150000004814 combretastatins Chemical class 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 239000012059 conventional drug carrier Substances 0.000 description 1
- 238000011443 conventional therapy Methods 0.000 description 1
- 229940088547 cosmegen Drugs 0.000 description 1
- 235000001671 coumarin Nutrition 0.000 description 1
- 150000004775 coumarins Chemical class 0.000 description 1
- 229940111134 coxibs Drugs 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 238000003235 crystal violet staining Methods 0.000 description 1
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 1
- 239000002619 cytotoxin Substances 0.000 description 1
- 229940070230 daypro Drugs 0.000 description 1
- 230000017858 demethylation Effects 0.000 description 1
- 238000010520 demethylation reaction Methods 0.000 description 1
- 229940070968 depocyt Drugs 0.000 description 1
- XOZIUKBZLSUILX-UHFFFAOYSA-N desoxyepothilone B Natural products O1C(=O)CC(O)C(C)(C)C(=O)C(C)C(O)C(C)CCCC(C)=CCC1C(C)=CC1=CSC(C)=N1 XOZIUKBZLSUILX-UHFFFAOYSA-N 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 229960001259 diclofenac Drugs 0.000 description 1
- DCOPUUMXTXDBNB-UHFFFAOYSA-N diclofenac Chemical compound OC(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl DCOPUUMXTXDBNB-UHFFFAOYSA-N 0.000 description 1
- KPHWPUGNDIVLNH-UHFFFAOYSA-M diclofenac sodium Chemical compound [Na+].[O-]C(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl KPHWPUGNDIVLNH-UHFFFAOYSA-M 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000002651 drug therapy Methods 0.000 description 1
- 229940087477 ellence Drugs 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- HKSZLNNOFSGOKW-UHFFFAOYSA-N ent-staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(C)O1 HKSZLNNOFSGOKW-UHFFFAOYSA-N 0.000 description 1
- HESCAJZNRMSMJG-HGYUPSKWSA-N epothilone A Natural products O=C1[C@H](C)[C@H](O)[C@H](C)CCC[C@H]2O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C HESCAJZNRMSMJG-HGYUPSKWSA-N 0.000 description 1
- QXRSDHAAWVKZLJ-PVYNADRNSA-N epothilone B Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 QXRSDHAAWVKZLJ-PVYNADRNSA-N 0.000 description 1
- XOZIUKBZLSUILX-GIQCAXHBSA-N epothilone D Chemical compound O1C(=O)C[C@H](O)C(C)(C)C(=O)[C@H](C)[C@@H](O)[C@@H](C)CCC\C(C)=C/C[C@H]1C(\C)=C\C1=CSC(C)=N1 XOZIUKBZLSUILX-GIQCAXHBSA-N 0.000 description 1
- 102000015694 estrogen receptors Human genes 0.000 description 1
- 108010038795 estrogen receptors Proteins 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 208000021045 exocrine pancreatic carcinoma Diseases 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 229940087476 femara Drugs 0.000 description 1
- RDJGLLICXDHJDY-UHFFFAOYSA-N fenoprofen Chemical compound OC(=O)C(C)C1=CC=CC(OC=2C=CC=CC=2)=C1 RDJGLLICXDHJDY-UHFFFAOYSA-N 0.000 description 1
- 229960001419 fenoprofen Drugs 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000009093 first-line therapy Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 229960000961 floxuridine Drugs 0.000 description 1
- 235000008191 folinic acid Nutrition 0.000 description 1
- 239000011672 folinic acid Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000006170 formylation reaction Methods 0.000 description 1
- QJIWQTQZVBIUTA-UHFFFAOYSA-N furan;1,3-thiazole Chemical compound C=1C=COC=1.C1=CSC=N1 QJIWQTQZVBIUTA-UHFFFAOYSA-N 0.000 description 1
- 201000008361 ganglioneuroma Diseases 0.000 description 1
- 238000002309 gasification Methods 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 201000011243 gastrointestinal stromal tumor Diseases 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 201000008822 gestational choriocarcinoma Diseases 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 229950011595 glufosfamide Drugs 0.000 description 1
- 229960002989 glutamic acid Drugs 0.000 description 1
- 230000002414 glycolytic effect Effects 0.000 description 1
- 208000024963 hair loss Diseases 0.000 description 1
- 230000003676 hair loss Effects 0.000 description 1
- 229940022353 herceptin Drugs 0.000 description 1
- 229940121372 histone deacetylase inhibitor Drugs 0.000 description 1
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 1
- 230000003054 hormonal effect Effects 0.000 description 1
- 102000052196 human PF4 Human genes 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 229940088013 hycamtin Drugs 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- QNXSIUBBGPHDDE-UHFFFAOYSA-N indan-1-one Chemical compound C1=CC=C2C(=O)CCC2=C1 QNXSIUBBGPHDDE-UHFFFAOYSA-N 0.000 description 1
- 229940089536 indocin Drugs 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- 150000002475 indoles Chemical class 0.000 description 1
- 229960000905 indomethacin Drugs 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000009830 intercalation Methods 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- 201000002313 intestinal cancer Diseases 0.000 description 1
- 210000004347 intestinal mucosa Anatomy 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- 229960000991 ketoprofen Drugs 0.000 description 1
- BWHLPLXXIDYSNW-UHFFFAOYSA-N ketorolac tromethamine Chemical compound OCC(N)(CO)CO.OC(=O)C1CCN2C1=CC=C2C(=O)C1=CC=CC=C1 BWHLPLXXIDYSNW-UHFFFAOYSA-N 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 210000002429 large intestine Anatomy 0.000 description 1
- 231100001231 less toxic Toxicity 0.000 description 1
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004324 lymphatic system Anatomy 0.000 description 1
- 208000025036 lymphosarcoma Diseases 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 238000009115 maintenance therapy Methods 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 229940087732 matulane Drugs 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- 231100000682 maximum tolerated dose Toxicity 0.000 description 1
- 206010027191 meningioma Diseases 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 229960004635 mesna Drugs 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 208000037819 metastatic cancer Diseases 0.000 description 1
- 208000011645 metastatic carcinoma Diseases 0.000 description 1
- 208000011575 metastatic malignant neoplasm Diseases 0.000 description 1
- YDCHPLOFQATIDS-UHFFFAOYSA-N methyl 2-bromoacetate Chemical compound COC(=O)CBr YDCHPLOFQATIDS-UHFFFAOYSA-N 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- ZDZOTLJHXYCWBA-BSEPLHNVSA-N molport-006-823-826 Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-BSEPLHNVSA-N 0.000 description 1
- VYGYNVZNSSTDLJ-HKCOAVLJSA-N monorden Natural products CC1CC2OC2C=C/C=C/C(=O)CC3C(C(=CC(=C3Cl)O)O)C(=O)O1 VYGYNVZNSSTDLJ-HKCOAVLJSA-N 0.000 description 1
- 229960004270 nabumetone Drugs 0.000 description 1
- 229940089466 nalfon Drugs 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 229940090008 naprosyn Drugs 0.000 description 1
- 229960002009 naproxen Drugs 0.000 description 1
- 229940109551 nipent Drugs 0.000 description 1
- OKXGHXHZNCJMSV-UHFFFAOYSA-N nitro phenyl carbonate Chemical compound [O-][N+](=O)OC(=O)OC1=CC=CC=C1 OKXGHXHZNCJMSV-UHFFFAOYSA-N 0.000 description 1
- LQNUZADURLCDLV-IDEBNGHGSA-N nitrobenzene Chemical group [O-][N+](=O)[13C]1=[13CH][13CH]=[13CH][13CH]=[13CH]1 LQNUZADURLCDLV-IDEBNGHGSA-N 0.000 description 1
- 150000005181 nitrobenzenes Chemical class 0.000 description 1
- 125000006502 nitrobenzyl group Chemical group 0.000 description 1
- 108020001162 nitroreductase Proteins 0.000 description 1
- OSTGTTZJOCZWJG-UHFFFAOYSA-N nitrosourea Chemical compound NC(=O)N=NO OSTGTTZJOCZWJG-UHFFFAOYSA-N 0.000 description 1
- 230000007959 normoxia Effects 0.000 description 1
- YJQPYGGHQPGBLI-KGSXXDOSSA-N novobiocin Chemical compound O1C(C)(C)[C@H](OC)[C@@H](OC(N)=O)[C@@H](O)[C@@H]1OC1=CC=C(C(O)=C(NC(=O)C=2C=C(CC=C(C)C)C(O)=CC=2)C(=O)O2)C2=C1C YJQPYGGHQPGBLI-KGSXXDOSSA-N 0.000 description 1
- 229960002950 novobiocin Drugs 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 230000000174 oncolytic effect Effects 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000002188 osteogenic effect Effects 0.000 description 1
- 229940127084 other anti-cancer agent Drugs 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 208000013371 ovarian adenocarcinoma Diseases 0.000 description 1
- 208000030339 ovarian transitional cell carcinoma Diseases 0.000 description 1
- 201000006588 ovary adenocarcinoma Diseases 0.000 description 1
- 201000001534 ovary transitional cell carcinoma Diseases 0.000 description 1
- OFPXSFXSNFPTHF-UHFFFAOYSA-N oxaprozin Chemical compound O1C(CCC(=O)O)=NC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 OFPXSFXSNFPTHF-UHFFFAOYSA-N 0.000 description 1
- 229940046231 pamidronate Drugs 0.000 description 1
- 229940096763 panretin Drugs 0.000 description 1
- 229960005489 paracetamol Drugs 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 229960003531 phenolsulfonphthalein Drugs 0.000 description 1
- 239000002953 phosphate buffered saline Substances 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 238000007747 plating Methods 0.000 description 1
- 150000003057 platinum Chemical class 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 231100000683 possible toxicity Toxicity 0.000 description 1
- 235000011181 potassium carbonates Nutrition 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 150000003138 primary alcohols Chemical class 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- LVTJOONKWUXEFR-FZRMHRINSA-N protoneodioscin Natural products O(C[C@@H](CC[C@]1(O)[C@H](C)[C@@H]2[C@]3(C)[C@H]([C@H]4[C@@H]([C@]5(C)C(=CC4)C[C@@H](O[C@@H]4[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@@H](O)[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@H](CO)O4)CC5)CC3)C[C@@H]2O1)C)[C@H]1[C@H](O)[C@H](O)[C@H](O)[C@@H](CO)O1 LVTJOONKWUXEFR-FZRMHRINSA-N 0.000 description 1
- 229940117820 purinethol Drugs 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 229940076788 pyruvate Drugs 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- 150000004053 quinones Chemical class 0.000 description 1
- 239000002534 radiation-sensitizing agent Substances 0.000 description 1
- 229930192524 radicicol Natural products 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 206010038038 rectal cancer Diseases 0.000 description 1
- 208000020615 rectal carcinoma Diseases 0.000 description 1
- 201000001275 rectum cancer Diseases 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 238000006479 redox reaction Methods 0.000 description 1
- 230000001850 reproductive effect Effects 0.000 description 1
- 150000004492 retinoid derivatives Chemical class 0.000 description 1
- 239000002342 ribonucleoside Substances 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- RZJQGNCSTQAWON-UHFFFAOYSA-N rofecoxib Chemical compound C1=CC(S(=O)(=O)C)=CC=C1C1=C(C=2C=CC=CC=2)C(=O)OC1 RZJQGNCSTQAWON-UHFFFAOYSA-N 0.000 description 1
- 229960003452 romidepsin Drugs 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000007423 screening assay Methods 0.000 description 1
- 150000003333 secondary alcohols Chemical class 0.000 description 1
- 125000000467 secondary amino group Chemical group [H]N([*:1])[*:2] 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- RAGFPHFDFVNLCG-INYQBOQCSA-N sibiromycin Chemical compound O[C@@H]1[C@@](O)(C)[C@@H](NC)[C@H](C)O[C@H]1OC(C(=C1O)C)=CC(C2=O)=C1N[C@H](O)[C@H]1N2C=C(\C=C\C)C1 RAGFPHFDFVNLCG-INYQBOQCSA-N 0.000 description 1
- RAGFPHFDFVNLCG-UHFFFAOYSA-N sibiromycin Natural products OC1C(O)(C)C(NC)C(C)OC1OC(C(=C1O)C)=CC(C2=O)=C1NC(O)C1N2C=C(C=CC)C1 RAGFPHFDFVNLCG-UHFFFAOYSA-N 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- MFBOGIVSZKQAPD-UHFFFAOYSA-M sodium butyrate Chemical compound [Na+].CCCC([O-])=O MFBOGIVSZKQAPD-UHFFFAOYSA-M 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 210000004872 soft tissue Anatomy 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000007480 spreading Effects 0.000 description 1
- 238000003892 spreading Methods 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- HKSZLNNOFSGOKW-FYTWVXJKSA-N staurosporine Chemical compound C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1[C@H]1C[C@@H](NC)[C@@H](OC)[C@]4(C)O1 HKSZLNNOFSGOKW-FYTWVXJKSA-N 0.000 description 1
- CGPUWJWCVCFERF-UHFFFAOYSA-N staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(OC)O1 CGPUWJWCVCFERF-UHFFFAOYSA-N 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 201000000498 stomach carcinoma Diseases 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 125000000565 sulfonamide group Chemical group 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid Substances OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- PFXVKGGZWQQTSE-UHFFFAOYSA-N sulfuryl dicyanide Chemical group N#CS(=O)(=O)C#N PFXVKGGZWQQTSE-UHFFFAOYSA-N 0.000 description 1
- 229960000894 sulindac Drugs 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 239000011885 synergistic combination Substances 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- FQZYTYWMLGAPFJ-OQKDUQJOSA-N tamoxifen citrate Chemical compound [H+].[H+].[H+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O.C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 FQZYTYWMLGAPFJ-OQKDUQJOSA-N 0.000 description 1
- 229940099419 targretin Drugs 0.000 description 1
- 229940061353 temodar Drugs 0.000 description 1
- 229960004964 temozolomide Drugs 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 125000001302 tertiary amino group Chemical group 0.000 description 1
- 230000002381 testicular Effects 0.000 description 1
- 201000003120 testicular cancer Diseases 0.000 description 1
- 229960003433 thalidomide Drugs 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 229960001196 thiotepa Drugs 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- 208000013077 thyroid gland carcinoma Diseases 0.000 description 1
- 229950002376 tirapazamine Drugs 0.000 description 1
- ORYDPOVDJJZGHQ-UHFFFAOYSA-N tirapazamine Chemical compound C1=CC=CC2=[N+]([O-])C(N)=N[N+]([O-])=C21 ORYDPOVDJJZGHQ-UHFFFAOYSA-N 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- 229960001017 tolmetin Drugs 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 229940019127 toradol Drugs 0.000 description 1
- 231100000820 toxicity test Toxicity 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 206010044412 transitional cell carcinoma Diseases 0.000 description 1
- 229960000575 trastuzumab Drugs 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 229960001727 tretinoin Drugs 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 229940086984 trisenox Drugs 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 230000005751 tumor progression Effects 0.000 description 1
- 229940072651 tylenol Drugs 0.000 description 1
- 150000004917 tyrosine kinase inhibitor derivatives Chemical class 0.000 description 1
- 229940054937 valstar Drugs 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- 229940063674 voltaren Drugs 0.000 description 1
- 230000008673 vomiting Effects 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 229960004276 zoledronic acid Drugs 0.000 description 1
- 229940002005 zometa Drugs 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4166—1,3-Diazoles having oxo groups directly attached to the heterocyclic ring, e.g. phenytoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/559—Redox delivery systems, e.g. dihydropyridine pyridinium salt redox systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6891—Pre-targeting systems involving an antibody for targeting specific cells
- A61K47/6899—Antibody-Directed Enzyme Prodrug Therapy [ADEPT]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82Y—SPECIFIC USES OR APPLICATIONS OF NANOSTRUCTURES; MEASUREMENT OR ANALYSIS OF NANOSTRUCTURES; MANUFACTURE OR TREATMENT OF NANOSTRUCTURES
- B82Y5/00—Nanobiotechnology or nanomedicine, e.g. protein engineering or drug delivery
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
Abstract
Un agente antineoplásico protegido de la fórmula Hip-N, en la que N es un agente antineoplásico, y un grupo hidroxilo protegible del antineoplásico está sustituido con Hip; Hip es una fracción que tiene la fórmula:**Fórmula** en la que R2 es hidrógeno; R3 es hidrógeno o alquilo C1-C6; R1 es alquilo C1-C6 o alcoxi C1-C6, opcionalmente sustituido con uno o más grupos que contienen heteroátomos; y R4 es hidrógeno, alquilo C1-C6 o alcoxi C1-C6, opcionalmente sustituido con uno o más grupos que contienen heteroátomos.
Description
DESCRIPCIÓN
Composiciones y procedimientos para el tratamiento del cáncer
Campo de la invención
En el presente documento se describen procedimientos, compuestos y composiciones útiles en el tratamiento del cáncer.
Antecedentes
El cáncer se refiere de forma general a una de un grupo de más de 100 enfermedades causadas por el crecimiento y la diseminación incontroladas de células anormales que pueden tomar la forma de tumores sólidos, de linfomas y de cánceres no sólidos como la leucemia. Al contrario que las células normales, que se reproducen hasta que se alcanza la maduración y después únicamente cuando es necesario para su sustitución, las células cancerosas crecen y se dividen indefinidamente, desplazando a las células próximas y finalmente diseminándose a otras partes del cuerpo, salvo que se detenga su progresión.
Una vez que las células cancerosas metastatizan al abandonar un tumor, viajarán a través del torrente sanguíneo o del sistema linfático a otras partes del cuerpo, en las que las células comienzan a multiplicarse y a desarrollarse en nuevos tumores. Este tipo de progresión del tumor hace que el cáncer sea peligrosamente mortal. Aunque se han producido grandes mejoras en el diagnóstico, el cuidado general del paciente, las técnicas quirúrgicas y las terapias coadyuvantes locales y sistémicas, la mayoría de las muertes por cáncer todavía son debidas a las metástasis y a otros cánceres que son resistentes a las terapias convencionales, incluyendo la radioterapia y la quimioterapia. La radioterapia normalmente sólo es eficaz para el tratamiento del cáncer en las fases tempranas e intermedias del cáncer, cuando el cáncer está localizado, y no es eficaz para la fase tardía de la enfermedad con metástasis. La quimioterapia puede ser eficaz en todas las fases de la enfermedad, pero pueden aparecer unos efectos secundarios graves, por ejemplo, vómitos, bajo recuento de glóbulos blancos, pérdida de cabello, pérdida de peso y otros efectos tóxicos, debidos a la radioterapia y a la quimioterapia. Debido a dichos efectos secundarios graves, muchos pacientes oncológicos no completan o no consiguen completar con éxito un régimen de tratamiento quimioterapéutico. Los efectos secundarios de la radiación y de los fármacos antineoplásicos pueden ser considerados como el resultado de una baja especificidad por el objetivo. Los fármacos antineoplásicos, administrados normalmente por vía intravenosa o más raramente por vía oral, circulan a través de la mayoría de los tejidos normales de los pacientes, así como de los tumores objetivo. Si el fármaco es tóxico para una célula normal, entonces esta circulación dará como resultado la muerte de las células normales, dando lugar a los efectos secundarios, y cuanto más tóxico sea el fármaco para las células normales, más graves serán los efectos secundarios. Debido a estos y otros problemas, algunos agentes quimioterapéuticos muy citotóxicos, unos agentes con unos valores de la CI50 nanomolares o subnanomolares frente a las células cancerosas, no se han desarrollado con éxito en fármacos aprobados.
Los profármacos se han investigado como un medio para reducir la toxicidad no deseada o algún otro atributo negativo de un fármaco sin una pérdida de la eficacia. Un profármaco es un fármaco que ha sido modificado químicamente para hacerlo inactivo pero que, después de su administración, es metabolizado o convertido de otro modo en el cuerpo en la forma activa del fármaco. Por ejemplo, en un esfuerzo por mejorar el direccionamiento del fármaco, se han desarrollado profármacos que son activados en unas condiciones hipóxicas. La hipoxia crea un entorno biorreductor y ciertos agentes anticancerosos han sido convertidos en profármacos que pueden ser activados en dichos entornos. Véanse las revisiones de Naylor y col., mayo de 2001, Mini. Rev. Med. 1 (1): 17-29, y de Denny, 2001, Eur. J. Med Chem. 36: 577-595. La “hipoxia” es un estado de bajo nivel de oxígeno; la mayoría de los tumores sólidos mayores de aproximadamente 1 mm de diámetro contienen regiones hipóxicas (véanse las referencias Coleman, 1988, J. Nat. Cane. Inst. 80: 310; y Vaupel y col., Cancer Res. 49: 6449).
Según crece un tumor, requiere un suministro de sangre, y por lo tanto, el crecimiento de una nueva vasculatura. La nueva vasculatura que da soporte al crecimiento del tumor a menudo está bastante desordenada, dejando unas porciones significativas del tumor poco vascularizadas y sometidas a un bloqueo vascular intermitente. La arquitectura vascular del tumor puede contribuir significativamente a la capacidad del cáncer para sobrevivir a la terapia farmacológica al menos de dos formas diferentes. En primer lugar, si el fármaco debe alcanzar el cáncer a través del torrente sanguíneo, entonces no tanto fármaco alcanzará las áreas poco vascularizadas hipóxicas del tumor. En segundo lugar, hasta el punto en el que el fármaco requiere oxígeno para ser eficaz, entonces el fármaco será menos eficaz en las regiones hipóxicas del tumor.
Sin embargo, por el contrario, el entorno hipóxico es propicio para los acontecimientos reductores que pueden usarse para la generación de derivados reducidos de una diversidad de grupos químicos (véase la referencia Workman y col., 1993, Cancer y Metast. Rev. 12: 73-82) y se han desarrollado compuestos profármacos biorreductores para explotar dichos entornos. Estos profármacos incluyen los antibióticos mitomicina C (MMC) y porfiromicina (POR), N-óxidos tales como la tirapazamina (TRZ; véase la referencia Zeeman y col., 1986, Inst. J.
Radiot. Oncol. Biol. Phys. 12: 1239), quinonas tales como la indoloquinona E09 (véase la referencia Bailey y col., 1992, Int. J. Radiot. Oncol. Biol. Phys. 22: 649), ciclopropamitosenos (EP-A-0868137) y un análogo N-óxido de amina terciaria de la mitoxantrona (AQ4N) que es activado por el citocromo P450 3A4 (véanse las referencias Patterson, 1993, Cancer Metast. Rev. 12: 119; y Patterson, 1994, Biochem. Pharm. Oncol. Res. 6: 533).
Otros compuestos profármacos activados biorreductoramente incluyen los derivados de nitroimidazol que han sido notificados como útiles en la radioterapia del cáncer como agentes radiosensibilizantes (véanse las publicaciones de patente EP312858 y WO91/11440) y potenciadores de agentes quimioterapéuticos (véase la patente de Estados Unidos n° 4.921.963). El nitroimidazol también ha sido conjugado con el agente antineoplásico PARP 5-bromoisoquinolinona (véase la referencia Parveen y col., julio de 1999, Bioorg. Med. Chem. Lett., 9: 2031-36). La propia fracción de nitroimidazol es, sin embargo, un poco citotóxica para las células normales, debido a que experimenta ciclos redox y genera superóxidos en unas condiciones oxigenadas.
Algunos compuestos profármacos activados biorreductoramente que incluyen un nitroimidazol unido a una diversidad de agentes antineoplásicos se han descrito en A 2-NITROIMIDAZOLE CARBAMATE PRODRUG OF 5-AMINO-1-(CHLOROMETHYL)-3-[5,6,7-TRIMETHOXYINDOL-2-YL)CARBONYL]-1,2-DIHYDRO-3H-BENZ[E]INDOLE(AMINO-SECO-CBI-TMI) FOR USE WITH ADEPT AND GDEPT, M. P. Hay y col., Bioorganic & Medicinal Chemistry Letters 9 (1999) 2237-2242 y en la publicación PCT WO 00/64864. En todos los profármacos descritos en estos documentos, el nitroimidazol está unido directamente a un conector de carbamato y el agente antineoplásico está protegido en un nitrógeno a través de un carbamato, o en un carbono a través de un enlace éster.
Por lo tanto, sigue habiendo una necesidad de proporcionar fármacos para el tratamiento del cáncer. Dichos fármacos serían especialmente beneficiosos si se dirigieran a las células cancerosas más eficazmente que los actuales fármacos y tuvieran menos efectos secundarios graves. Los compuestos y los procedimientos descritos actualmente ayudan a satisfacer esta necesidad.
Sumario
Las realizaciones de la presente solicitud se describen en las reivindicaciones. Las realizaciones que no están en estas reivindicaciones son únicamente con fines de referencia. La presente invención proporciona un agente antineoplásico protegido de la fórmula Hip-N, en la que N es un agente antineoplásico, y un grupo hidroxilo protegible del antineoplásico está sustituido con Hip; Hip es una fracción que tiene la fórmula:
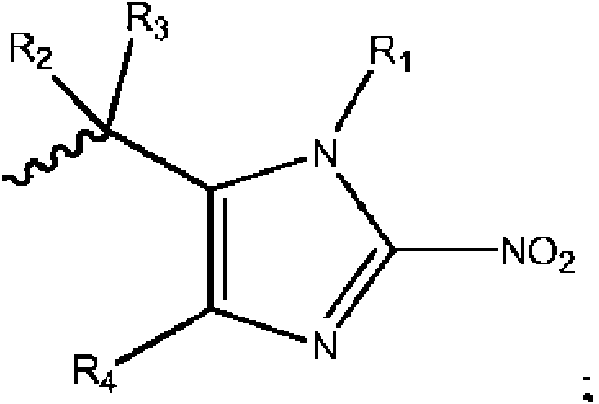
en la que
R2 es hidrógeno;
R3 es hidrógeno o alquilo C1-C6 ;
R1 es alquilo C1-C6 o alcoxi C1-C6 , opcionalmente sustituido con uno o más grupos que contienen heteroátomos; y
R4 es hidrógeno, alquilo C1-C6 o alcoxi C1-C6 , opcionalmente sustituido con uno o más grupos que contienen heteroátomos.
La presente invención proporciona adicionalmente dicho agente antineoplásico protegido o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento del cáncer.
Algunos ejemplos de agentes antineoplásicos que pueden usarse incluyen, pero no se limitan a, doxorrubicina, daunorrubicina, etopósido, duetopósido, combretastatina A-4, vinblastina, vincristina, camptotecina, topotecán, AQ4N, hidroxiurea, maitansinas, discodermólidos, epotilonas, taxanos, caliqueamicinas, tedanólidos, bleomicinas, caliqueamicinas, citarabina, epirrubicina, fludarabina, hidroxiureapentostatina, mitoxantrona, prednisona, docetaxel, paclitaxel, tedanólidos, tenipósido, vinca.
Los agentes antineoplásicos protegidos pueden usarse en el tratamiento del cáncer mediante la administración a un sujeto de una cantidad terapéuticamente eficaz de un agente antineoplásico protegido. Los agentes quimioterapéuticos que pueden usarse se describen con detalle en la sección de Descripción detallada.
Los cánceres que pueden ser tratados se describen con detalle en la sección de Descripción detallada, e incluyen
cáncer de pulmón, cáncer de pulmón no microcítico, cáncer de mama, cáncer de colon, cáncer de cabeza y cuello, cáncer de ovario, cáncer de páncreas y cáncer de próstata.
Descripción detallada
En esta descripción detallada se proporcionan en primer lugar unas definiciones útiles para la comprensión de los compuestos, de las composiciones y de los procedimientos descritos en esta patente. A continuación se proporciona una descripción de los compuestos profármacos que pueden usarse para el tratamiento del cáncer, seguido de las descripciones de los diversos componentes de estos compuestos, que incluyen (1) grupos activadores hipóxicos, (2) agentes antineoplásicos y (3) grupos conectores. Después se incluye una descripción de los procedimientos de tratamiento mediante el uso de los compuestos, incluyendo una descripción de los cánceres que pueden ser tratados, seguida de las descripciones de las formulaciones, los modos de administración, las dosis, etc. que pueden usarse con los compuestos y los procedimientos descritos en la patente. Después se describen los métodos de elaboración, seguido de las terapias de combinación, en las que los compuestos descritos en esta patente se usan junto con otros tratamientos, y finalmente se proporcionan ejemplos de los compuestos, las composiciones y los procedimientos descritos en esta patente.
Definiciones
Para facilitar la comprensión de los compuestos, las composiciones y los procedimientos descritos en esta patente, se proporcionan las siguientes definiciones y, salvo que se defina de otro modo, todos los términos técnicos y científicos usados en el presente documento tienen los significados que les atribuyen los expertos en los campos a los que pertenecen los compuestos y los procedimientos descritos en el presente documento.
Según se usa en el presente documento, “un/o” o “una” significa “al menos un/o/a” o “un/o/a o más”.
Según se usa en el presente documento “alquilo”, salvo que el contexto lo indique claramente de otro modo, incluye cualquier grupo alifático que contiene carbono e hidrógeno, incluyendo grupos alquilo de cadena lineal, de cadena ramificada, cíclicos y que contienen un carbociclo.
Según se usa en el presente documento, un “agente antineoplásico” o un “agente antitumoral” se refiere en general a cualquier agente usado en el tratamiento del cáncer. Dichos agentes pueden usarse solos o junto con otros compuestos y pueden aliviar, reducir, mejorar, prevenir o poner o mantener en un estado de remisión los síntomas clínicos o los marcadores diagnósticos asociados con un neoplasma, un tumor o un cáncer. Algunos agentes antineoplásicos incluyen, pero no se limitan a, agentes antiangiogénicos, agentes alquilantes, antimetabolitos, perturbadores de la polimerización de la microtubulina (por ejemplo, taxol), ciertos productos naturales, complejos de coordinación del platino, antracenedionas, ureas sustituidas, derivados de la metilhidrazina, supresores adrenocorticales, ciertas hormonas y antagonistas, polisacáridos antineoplásicos y ciertas hierbas medicinales u otros extractos vegetales.
Según se usa en el presente documento, un “tratamiento antineoplásico”, una “terapia para el cáncer” o un “tratamiento del cáncer”, se refieren a cualquier metodología para la mejora de los síntomas o el retraso de la progresión de un neoplasma, de un tumor o de un cáncer mediante la reducción del número o del crecimiento de las células cancerosas del cuerpo, normalmente (pero no se limita a) mediante la destrucción o la detención del crecimiento y de la división de las células cancerosas.
Según se usa en el presente documento, un “compuesto biorreductor” se refiere a un compuesto que acepta electrones en una reacción de oxidación-reducción.
Según se usa en el presente documento, “cáncer' se refiere a una de un grupo de más de 100 enfermedades causadas por el crecimiento y la diseminación incontrolados de células anormales que pueden tomar la forma de tumores sólidos, de linfomas y de cánceres no sólidos tales como la leucemia.
Según se usa en el presente documento, “maligna” se refiere a las células que tienen la capacidad de metastatizar, con una pérdida del control tanto del crecimiento como de la posición.
Según se usa en el presente documento, “neoplasma” (neoplasia) o “tumor” se refiere a un nuevo crecimiento anormal celular o tisular, que puede ser benigno o maligno.
Según se usa en el presente documento, un “profármaco” es un compuesto que, después de su administración, es metabolizado o convertido de otro modo en una forma activa o más activa con respecto a al menos una propiedad. Para producir un profármaco, puede modificarse químicamente un compuesto farmacéuticamente activo para hacerlo menos activo inactivo, pero la modificación química es tal que se genera una forma activa del compuesto mediante procesos metabólicos u otros procesos biológicos. Un profármaco puede tener, con respecto al fármaco, una estabilidad metabólica o unas características de transporte alteradas, menos efectos secundarios o una menor toxicidad o una mejora en el sabor, por ejemplo, (véase la referencia Nogrady, 1985, Medicinal Chemistry A
Biochemical Approach, Oxford University Press, Nueva York, páginas 388-392). Los profármacos también pueden ser preparados mediante el uso de compuestos que no son fármacos.
Agentes antineoplásicos protegidos
Los profármacos en particular descritos en esta patente que pueden usarse para el tratamiento del cáncer se denominan “agentes antineoplásicos protegidos”. En una versión, el agente antineoplásico protegido tiene la fórmula Hip-L-N o Hip-N, donde Hip es un activador hipóxico; L es un grupo conector; y N es un agente antineoplásico. Los diversos activadores hipóxicos, grupos conectores y agentes antineoplásicos que pueden usarse en estos agentes antineoplásicos protegidos se describen en las secciones del activador hipóxico, del grupo conector y del agente antineoplásico, a continuación.
En otra versión, el agente antineoplásico protegido es un agente antineoplásico en el que uno o más grupos hidroxilo y/o amino presentes en el agente antineoplásico están protegidos por un grupo activador hipóxico, Hip, que está unido directamente al grupo amino o hidroxilo, bien directamente o bien a través de un grupo conector, L. Los agentes antineoplásicos, los activadores hipóxicos y los grupos conectores que pueden usarse se describen en las secciones de los agentes antineoplásicos, del activador hipóxico y del grupo conector, a continuación. Cuando el grupo amino o hidroxilo del agente antineoplásico está protegido, el hidrógeno del grupo hidroxilo o una o más de las fracciones unidas al nitrógeno del grupo amino están sustituidas por un enlace con el grupo protector, es decir, por un enlace con el grupo Hip o con el grupo Hip-L. En una versión, la amina es un amina primaria y uno de los hidrógenos está sustituido por el grupo protector.
El siguiente esquema muestra la unión de un activador hipóxico a un oxígeno de un hidroxilo y un nitrógeno de una amina a través de un grupo conector, L.

Generalmente, puede protegerse cualquier número de grupos amino y cualquier número de hidroxilos de un agente antineoplásico. En una versión, se protege un grupo amino de un agente antineoplásico. En otra versión, se protege un hidroxilo de un agente antineoplásico. Si se protege más de un grupo hidroxilo y/o amino de un antineoplásico, los grupos protectores pueden ser iguales o pueden ser diferentes.
En una versión, el grupo protegido del agente antineoplásico es un grupo hidroxilo que está unido a un anillo aromático o heteroaromático del agente antineoplásico. El anillo aromático o heteroaromático puede estar sustituido o no sustituido, y puede estar condensado con uno o más anillos adicionales, que pueden ser aromáticos o no aromáticos y pueden contener cualquier número de heteroátomos en el anillo. En una versión, el hidroxilo protegido está unido a un anillo de fenilo del agente antineoplásico, y el anillo de fenilo puede estar sustituido o no sustituido y puede estar condensado con uno o más anillos adicionales, que pueden ser aromáticos o no aromáticos y pueden contener cualquier número de heteroátomos en el anillo.
En todas las versiones descritas en esta sección, la amina protegida o el hidroxilo protegido pueden estar unidos bien directamente a cualquier activador hipóxico o bien unidos a cualquier activador hipóxico a través de cualquier grupo conector descrito en la patente.
Naturaleza de los grupos en un agente antineoplásico que puede ser protegido : como se ha descrito más arriba, los grupos hidroxilo y amino de un agente antineoplásico pueden ser protegidos mediante su unión a un activador hipóxico, bien directamente o bien a través de un conector. Otros grupos que pueden ser protegidos
incluyen grupos basados en azufre, grupos aldehido y grupos ceto.
Los agentes antineoplásicos protegidos pueden liberar el agente antineoplásico o el agente antineoplásico modificado, incluyendo “supertoxinas" : dependiendo de si el agente antineoplásico está unido directamente al activador hipóxico y dependiendo de la naturaleza del grupo conector, si el agente antineoplásico está unido a través de un grupo conector al activador hipóxico, la molécula liberada tras la reducción del activador hipóxico es bien el agente antineoplásico o bien un agente antineoplásico modificado que incluye parte o la totalidad del grupo conector unido al agente antineoplásico. Como se usa en esta patente, un “agente antineoplásico modificado” se refiere a una especie que es liberada a partir de un agente antineoplásico protegido y que es diferente del propio agente antineoplásico. Por ejemplo, un agente antineoplásico protegido con la fórmula Hip-L-N puede producir un agente antineoplásico modificado de fórmula L-N después de la reducción del activador hipóxico. Cuando la reducción del activador hipóxico libera un agente antineoplásico modificado, el grupo conector unido al agente antineoplásico puede experimentar una reordenación o una degradación para producir bien el agente antineoplásico no modificado o bien algún otro agente antineoplásico modificado.
Algunos ejemplos generales de este aspecto de los agentes antineoplásicos protegidos se describen en la sección de grupos conectores.
El hecho de que la molécula liberada después de la reducción del activador hipóxico pueda ser diferente del agente antineoplásico que está siendo protegido, será apreciado por los expertos en la materia. Esto está ilustrado en los siguientes Ejemplos, uno de los cuales muestra que puede sintetizarse un agente antineoplásico protegido mediante la unión de doxorrubicina a un activador hipóxico para formar un agente antineoplásico protegido que libera un derivado de la doxorrubicina que contiene iminio que es bastante más tóxico que la doxorrubicina.
Los agentes antineoplásicos protegidos descritos en esta patente generalmente muestran una mayor eficacia y/o menos efectos secundarios que los compuestos previos. Por ejemplo, ciertos agentes antineoplásicos protegidos descritos en esta patente están conjugados con, o son activados por, unas condiciones hipóxicas para liberar unos agentes citotóxicos muy potentes, las “supertoxinas” con unos valores de la CI50 de menos de 100 nM frente a la mayoría de las líneas celulares cancerosas en el grupo de líneas celulares tumorales del NCI. Con respecto a la posible toxicidad de los agentes antineoplásicos protegidos, a pesar de que los agentes antineoplásicos protegidos todavía generan el superóxido que puede causar los indeseables efectos secundarios, esos efectos secundarios están muy reducidos con respecto a los efectos de los compuestos previos, debido, sobre una base molar, a que tiene que administrarse mucho menos agente antineoplásico protegido debido a la naturaleza muy citotóxica del agente antineoplásico liberado por el agente antineoplásico protegido. Generalmente (quizás con la excepción de los compuestos descritos en A2-NITROIMIDAZOLE CARBAMATE PRODRUG OF 5-AMINO-1-(CHLOROMETHYL)-3-[5,6,7-TRIMETHOXYINDOL-2-YL)CARBONYL]-1,2-DIHYDRO-3H-BENZ[E]INDOLE (AMINO-SECO-CBI-TMI) FOR USE WITH ADEPT AND GDEPT, M. P. Hay y col., Bioorganic & Medicinal Chemistry Letters 9 (1999) 2237-2242 y en la publicación PCT WO 00/64864.), los agentes antineoplásicos protegidos descritos en esta patente que liberan “supertoxinas” pueden usarse en unas dosis mucho menores que los profármacos de nitroimidazol conocidos hasta ahora. Estas dosis mucho menores producen menos superóxido (véase el análisis, a continuación) en un tejido normóxico.
Los agentes antineoplásicos protegidos pueden usarse para liberar una gran diversidad de agentes antineoplásicos según se describe en la sección de agentes antineoplásicos de esta patente.
Los agentes antineoplásicos protegidos pueden tener una toxicidad reducida : los agentes antineoplásicos protegidos, con respecto a los fármacos en los que son convertidos in vivo, pueden ser mucho menos tóxicos (al menos diez y hasta un millón de veces menos). La toxicidad reducida es el resultado de una modificación en el sitio de unión del conector L (como el caso en el que la activación de los agentes antineoplásicos protegidos libera el mismo agente citotóxico que se usó en la síntesis del fármaco) o de la generación de una fracción necesaria para la toxicidad mediante la eliminación del activador hipóxico (Hip). En cualquier caso, los agentes antineoplásicos protegidos se convierten en el correspondiente fármaco tóxico en los tejidos hipóxicos en virtud de la activación de la fracción del activador hipóxico, dando como resultado su eliminación y la concomitante liberación o generación del agente antineoplásico o de una versión modificada del agente antineoplásico.
Independientemente del agente antineoplásico, N, seleccionado para su incorporación en o su liberación por parte de los agentes antineoplásicos protegidos, en una versión, la fracción del conector y del activador hipóxico están unidas al agente antineoplásico, N, de una forma que enmascara o que reduce la actividad citotóxica del agente antineoplásico. Este efecto de enmascaramiento puede variar y puede depender de la actividad citotóxica del agente antineoplásico que se va a liberar. Normalmente, el agente antineoplásico protegido mostrará al menos aproximadamente 10 veces menos actividad citotóxica que el agente antineoplásico y puede manifestar hasta aproximadamente un millón de veces o más o menos actividad citotóxica. En una versión, la actividad citotóxica del agente antineoplásico protegido es de entre aproximadamente 100 veces y aproximadamente 10.000 veces menos que la actividad citotóxica del agente antineoplásico. Como un ejemplo, para un agente antineoplásico con una CI50 de 1 nM, la correspondiente CI50 del agente antineoplásico protegido puede ser de 1 microM o mayor.
En una versión, los agentes antineoplásicos protegidos descritos en esta patente incluyen como agentes antineoplásicos, N, cualquier agente que pueda ser unido a un activador hipóxico de una forma que produzca un agente antineoplásico protegido que sea entre al menos aproximadamente 10 veces y aproximadamente 1.000.000 veces, y normalmente entre aproximadamente 100 y aproximadamente 10.000 veces, menos activo como agente citotóxico que el agente antineoplásico o el agente antineoplásico modificado que es liberado desde el agente antineoplásico protegido en unas condiciones hipóxicas.
Posible mecanismo de acción de los agentes antineoplásicos protegidos: los nitroimidazoles se han usado previamente para formar profármacos para algunos supuestos agentes anticancerosos, incluyendo un inhibidor de la PARP (véase la referencia Parveen y col., 1999, Bioorganic y Medicinal Letters 9: 2031- 2036), una mostaza nitrogenada que fue activada, no liberada, por el nitroimidazol (véase la referencia Lee y col., 1998, Bioorganic y Medicinal Letters 8: 1741-1744) y los agentes descritos en A 2-NITROIMIDAZOLE CARBAMATE PRODRUG OF 5-AMINO-1-(CHLOROMETHYL)-3-[5,6,7-TRIMETHOXYINDOL-2-YL)CARBONYL]-1,2-DIHYDRO-3H-BENZ[E]INDOLE (AMINO-SECO-CBI-TMI) FOR USE WITH ADEPT AND GDEPT, M. P. Hay y col., Bioorganic & Medicinal Chemistry Letters 9 (1999) 2237-2242 y en la publicación PCT WO 00/64864. Se demostró que el inhibidor de la PARP era liberado químicamente, pero no se proporcionaron datos del cultivo celular. Se demostró que la mostaza nitrogenada quedaba activa en los datos del cultivo celular, y la selectividad entre toxicidad normóxica e hipóxica, aunque no se midió de forma precisa, se estableció como mayor de 7 veces en las células con unos mecanismos de reparación del ADN normales.
Los agentes antineoplásicos protegidos descritos en esta patente difieren de dichos profármacos conocidos en diversas formas que incluyen, pero no se limitan a, la naturaleza del agente antineoplásico liberado, la naturaleza de la conexión del activador hipóxico al agente antineoplásico, el mejor perfil de efectos secundarios, la presencia de más de una fracción activadora hipóxica o alguna combinación de estos atributos. Sin estar ligados a ninguna teoría, estas ventajas de los agentes antineoplásicos protegidos pueden apreciarse mejor con la comprensión de la farmacocinética de los profármacos activados por hipoxia en general y de los agentes antineoplásicos protegidos descritos en esta patente en particular.
En una versión, el agente antineoplásico protegido incluye un nitroimidazol como el activador hipóxico. El nitroimidazol es convertido, en ausencia de oxígeno, en una fracción que contiene un radical libre mediante una reductasa del citocromo P450. Si el nitroimidazol es apropiadamente unido covalentemente a otra fracción, una reducción adicional del radical libre desde el nitroimidazol puede dar lugar a la liberación de esa fracción. Sin embargo, en presencia de oxígeno, el radical libre reacciona con el oxígeno para formar superóxido y el nitroimidazol parental. El superóxido es una citotoxina, por lo que se cree que la producción de superóxido en tejidos normóxicos da lugar a efectos secundarios indeseados.
Ciertos profármacos que contienen nitroimidazol también pueden ser activados independientemente de la tensión de oxígeno por la diaforasa DT, que puede dar lugar a una activación en las células normóxicas, contribuyendo por lo tanto a los efectos secundarios indeseados. Esta vía de activación normóxica podría crear unos efectos secundarios significativos con un agente antineoplásico protegido en particular, sin embargo, se puede seleccionar otro agente antineoplásico protegido que contenga más de una fracción activada por hipoxia para reducir o eliminar dichos efectos secundarios.
Sin estar ligados a ninguna teoría, en el caso de los agentes antineoplásicos protegidos en los que el activador hipóxico es un nitroimidazol, el activador hipóxico es activado en unas condiciones hipóxicas a través del grupo nitro, que se reduce a una hidroxilamina o a una amina con la concomitante liberación de la porción de la molécula a la cual está unido el activador hipóxico. Este procedimiento de activación se muestra en el siguiente esquema. Aunque el esquema mostrado ilustra un enlace éter entre el activador hipóxico y el resto del agente antineoplásico protegido, se aplicarán unos mecanismos similares para los grupos acetal a los que está unido el activador hipóxico. Por ejemplo, para un agente antineoplásico protegido que contiene un nitroimidazol como el activador hipóxico, la reducción del activador hipóxico liberará al menos inicialmente un aldehído cuando el activador hipóxico esté unido a un grupo acetal y liberará, al menos inicialmente, un alcohol cuando el activador hipóxico esté unido a un grupo éter. Sin estar ligados a ninguna teoría, el siguiente esquema presenta un mecanismo para la liberación desde una versión de un activador hipóxico.

Activadores hipóxicos
Generalmente el activador hipóxico puede ser cualquier grupo que sea capaz de liberar el agente antineoplásico o una versión modificada del agente antineoplásico después de la reducción hipóxica del activador hipóxico. En una versión, el activador hipóxico es un grupo que es capaz de liberar el agente antineoplásico o una versión modificada del agente antineoplásico después de la reducción del activador hipóxico en unas condiciones hipóxicas, pero que no libera sustancialmente ningún agente antineoplásico ni ninguna versión modificada del agente antineoplásico en unas condiciones normóxicas.
Algunos ejemplos de activadores hipóxicos incluyen, pero no se limitan a, fracciones basadas en nitrobencenos deficientes en electrones, amidas del ácido nitrobenzoico deficientes en electrones, nitroazoles, nitroimidazoles, nitrotiofenos, nitrotiazoles, nitrooxazoles, nitrofuranos y nitropirroles en los que cada una de estas clases de fracciones puede estar sustituida o no sustituida. En una versión, el activador hipóxico es nitroimidazol sustituido. En una versión, el activador hipóxico es nitrobenceno sustituido. El experto en la materia comprenderá cómo sustituir estos y otros posibles grupos para proporcionar un potencial redox para el grupo en un intervalo capaz de experimentar una reducción en las condiciones hipóxicas descritas en esta patente. Por ejemplo, y según se describe con más detalle a continuación, una versión del activador hipóxico es un nitroimidazol que puede estar sustituido con una diversidad de grupos. En algunos ejemplos adicionales, el tiofeno, el furan tiazol y las fracciones pueden estar sustituidos con uno o más grupos donantes de electrones, que incluyen, pero no se limitan a, grupos metilo o metoxi o amino, para conseguir el intervalo deseado de potencial redox. En otro ejemplo, la fracción de nitropirrol puede requerir la sustitución del grupo aceptor de electrones que incluye, pero no se limita a, grupos ciano, carboxamida, -CF3 y sulfonamida, para conseguir el intervalo deseado de potencial redox. Generalmente, el experto en la materia comprenderá cómo modificar el potencial redox de un activador hipóxico mediante la sustitución de los grupos aceptores de electrones, de los grupos donantes de electrones o alguna combinación de dichos grupos. Como un ejemplo no limitante, pueden usarse grupos aceptores de electrones fuertes tales como ciano, sulfona, sulfonamida, carboxamida o -CF3 y el potencial redox puede ajustarse fino mediante el uso de grupos aceptores de electrones más suaves tales como -CH2 , -F, -Cl, -Br o mediante la adición de un separador de metileno entre el activador hipóxico y el grupo aceptor de electrones fuerte.
En el caso de que el activador hipóxico sea un nitroimidazol, en una versión el activador hipóxico tiene la fórmula

donde el enlace ondulado indica la posición de unión del activador hipóxico al grupo conector o al agente antineoplásico. En otra versión el activador hipóxico tiene la fórmula
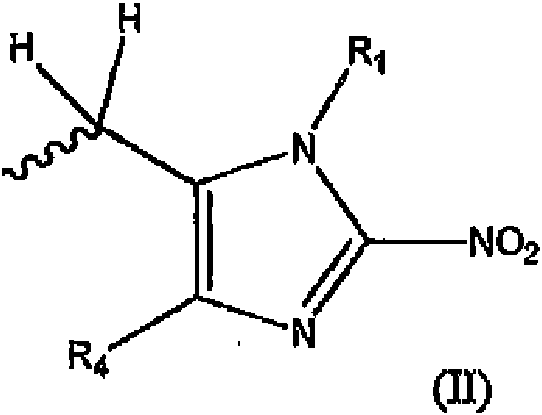
donde el enlace ondulado indica la posición de unión del activador hipóxico al grupo conector o al agente antineoplásico.
En una versión de los anteriores activadores hipóxicos (I & II), R2 es hidrógeno; R3 es -H o alquilo C1-C6 ; Ri se selecciona entre un alquilo C1-C6 o un alcoxi C1-C6 , donde el alquilo o el alcoxi está opcionalmente sustituido con uno o más grupos que contienen heteroátomos; y R4 se selecciona entre -H, alquilo C1-C6 o alcoxi C1-C6 , donde el alquilo o el alcoxi está opcionalmente sustituido con uno o más grupos que contienen heteroátomos.
En una versión de los anteriores activadores hipóxicos (I & II), R2 es hidrógeno; R3 es -H o alquilo C1-C6 ; R1 es alquilo C1-C6 , donde el alquilo está opcionalmente sustituido con uno o más grupos que contienen heteroátomos; y R4 se selecciona entre -H o alquilo C1-C6 , donde el alquilo está opcionalmente sustituido con uno o más grupos que contienen heteroátomos.
En una versión de los anteriores activadores hipóxicos (I & II), R2 es hidrógeno; R3 es -H o alquilo C1-C6 ; y R1 y R4 son cada uno independientemente -H o alquilo C1-C6 ; donde el alquilo está opcionalmente sustituido con uno o más grupos que contienen heteroátomos seleccionados entre hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster del ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; y con la condición de que R1 no sea hidrógeno.
En una versión, R2 es hidrógeno; R3 es -H o alquilo C1-C6 ; y R1 y R4 son cada uno independientemente un H, alquilo C1-C6 o alcoxi C1-C6 , estando el alquilo o el alcoxi opcionalmente sustituido con uno o más grupos seleccionados entre éter (-OR20), amino (-NH2), amino monosustituido (-NR20H), amino disustituido (-NR21R22), alquilamino C1-C5 cíclico, imidazolilo, alquilpiperazinilo C1-C6 , morfolino, tiol (-SH), tioéter -(SR20), tetrazol, ácido carboxílico (-COOH), éster (-COOR20), amida (-CONH2), amida monosustituida (-Co NHR20), amida disustituida (-CONR21R22), amida conectada por N (-NH2-C(=O)-R20), amida monosustituida conectada por N (-NHR21-C(=O)-R20), amida disustituida conectada por N (-n R21R22-S(=O)2-R20), sulfonamida conectada por N (-NH2-s (=O)2-R20), sulfonamida monosustituida conectada por N (-NHR21-S(=O)2-R20), sulfonamida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfoxi (-S(=O)2OH), sulfonato (S(=O)2OR20), sulfonilo (S(=O)2R20), sulfixi (S(=O)OH), sulfinato (S(=O)OR20), sulfinilo (S(=O)R20), fosfonooxi (OP(=O)(OH)2), fosfato (OP(=O)(OR20)2) y sulfonamida (-S(=O)2NH2 , -S(=O)2NHR21 o -S(=O)2NR21R22), donde R20, R21 y R22 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6 ; y con la condición de que R1 no sea hidrógeno.
En una versión de los anteriores activadores hipóxicos (I & II), R2 es hidrógeno; R3 es -H o alquilo C1-C3 ; y R1 y R4 son cada uno independientemente un -H o un alquilo seleccionado entre metilo, etilo, n-propilo, n-butilo, n-pentilo, tbutilo, ciclohexilo, ciclopentilo e isopropilo, donde el alquilo está opcionalmente sustituido con uno o más grupos que contienen heteroátomos; con la condición de que R1 no sea hidrógeno.
En una versión de los anteriores activadores hipóxicos (I & II), R2 es hidrógeno; R3 es -H o alquilo C1-C3 ; y R1 y R4 son cada uno independientemente -H o un alquilo seleccionado entre metilo, etilo, n-propilo, n-butilo, n-pentilo, tbutilo, ciclohexilo, ciclopentilo e isopropilo, donde el alquilo está opcionalmente sustituido con uno o más grupos que contienen heteroátomos seleccionados entre hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; con la condición de que R1 no sea hidrógeno.
En una versión de los anteriores activadores hipóxicos (I & II), R2 es hidrógeno; R3 es -H o alquilo C1-C3 ; y R1 y R4 son cada uno independientemente -H o un alquilo seleccionado entre metilo, etilo, n-propilo, n-butilo, n-pentilo, tbutilo, ciclohexilo, ciclopentilo e isopropilo, donde el alquilo está opcionalmente sustituido con uno o más grupos que contienen heteroátomos seleccionados entre éter (-OR20), amino (-NH2), amino monosustituido (-NR20H), amino disustituido (-NR21R22), alquilamino C1-C5 cíclico, imidazolilo, alquilpiperazinilo C1-6, morfolino, tiol (-SH), tioéter -(SR20), tetrazol, ácido carboxílico (-COOH), éster (-COOR20), amida (-CONH2), amida monosustituida (-CONHR20),
amida disustituida (-CONR21R22), amida conectada por N (-NH2-C(=O)-R20), amida monosustituida conectada por N (-NHR21-C(=O)-R20), amida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfonamida conectada por N (-NH2-S(=O)2-R20), sulfonamida monosustituida conectada por N (-NHR21-S(-O)2-R20), sulfonamida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfoxi (-S(=O)2OH), sulfonato (S(=O)2OR20), sulfonilo (S(=O)2R20), sulfixi (S(=O)OH), sulfinato (S(=O)OR20), sulfinilo (S(=O)R20), fosfonooxi (OP(=O)(OH)2), fosfato (OP(=O)(OR20)2) y sulfonamida (-S(=O)2NH2 , -S(=O)2NHR21 o -S(=O)2NR21R22), donde R20, R21 y R22 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6 ; con la condición de que R1 no sea hidrógeno.
En una versión de los anteriores activadores hipóxicos (I & II), R2 es hidrógeno; R3 es -H o alquilo C1-C3 ; y R1 y R4 se seleccionan cada uno independientemente entre metilo, etilo, n-propilo o n-butilo, cada alquilo opcionalmente sustituido con un grupo que contiene un heteroátomo, particularmente un grupo amino, un grupo ácido carboxílico o un grupo amida.
En una versión de los anteriores activadores hipóxicos (I & II), R2 es hidrógeno y R1 y R3 son cualquiera de las versiones descritas más arriba y R4 es alquilo C1-C3 opcionalmente sustituido con un grupo que contiene un heteroátomo, particularmente un grupo amino, un grupo ácido carboxílico o un grupo amida.
En una versión de los anteriores activadores hipóxicos (I & II), R2 es hidrógeno y R1 , R3 y R4 son cada uno independientemente etilo, n-propilo o n-butilo, cada uno opcionalmente sustituido con un grupo que contiene un heteroátomo, particularmente un grupo amino, un grupo ácido carboxílico o un grupo amida
En una versión de los anteriores activadores hipóxicos (I & II), R2 es hidrógeno; R3 es hidrógeno o alquilo C1-C3; R1 es metilo, acetato de metilo o etilo, n-propilo o n-butilo, cada uno opcionalmente sustituido con un grupo amino, un grupo ácido carboxílico o un grupo amida, y R4 es hidrógeno o alquilo C1-C3.
En una versión de los anteriores activadores hipóxicos (I & II), R2 es hidrógeno; R3 es hidrógeno o alquilo C1-C3; R1 es metilo, acetato de metilo o etilo, n-propilo o n-butilo, cada uno opcionalmente sustituido con un grupo amino, un grupo ácido carboxílico o un grupo amida, y R4 es hidrógeno.
En una versión de los anteriores activadores hipóxicos (I & II), R2 es hidrógeno, R1 es un grupo alquilo portador de un impedimento estérico, y R3 y R4 son cada uno independientemente -H o alquilo C1-C6 , opcionalmente sustituido con uno o más grupos que contienen heteroátomos. En una versión, R2 es hidrógeno, R1 es metilo y R3 y R4 son cada uno independientemente -H o alquilo C1-C6 , opcionalmente sustituido con uno o más grupos que contienen heteroátomos. En una versión, R2 es hidrógeno, R1 es acetato de metilo y R3 y R4 son cada uno independientemente -H o alquilo C1-C6 , opcionalmente sustituido con uno o más grupos que contienen heteroátomos.
En una versión de los anteriores activadores hipóxicos (I & II), R1, R3 y R4 son cualquiera de las versiones descritas más arriba y R2 se selecciona entre -H o alquilo C1-C6 , opcionalmente sustituido con uno o más grupos que contienen heteroátomos. En una versión, R1, R3 y R4 son cualquiera de las versiones descritas más arriba y R2 es -H, metilo, n-pentilo, t-butilo, ciclohexilo, ciclopentilo o isopropilo, todos opcionalmente sustituidos con uno o más grupos que contienen heteroátomos seleccionados entre hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano. En una versión, R1, R3 y R4 son cualquiera de las versiones descritas más arriba y R2 es H, alquilo C1-C6 o alcoxi C1-C6 , estando el alquilo o el alcoxi opcionalmente sustituido con uno o más grupos seleccionados entre éter (-OR20), amino (-NH2), amino monosustituido (-NR20H), amino disustituido (-NR21R22), alquilamino C1-C5 cíclico, imidazolilo, alquilpiperazinilo C1-6, morfolino, tiol (-SH), tioéter-(SR20), tetrazol, ácido carboxílico (-COOH), éster (-COOR20), amida (-CONH2), amida monosustituida (-CONHR20), amida disustituida (-CONR21R22), amida conectada por N (-NH2-c (=O)-R20), amida monosustituida conectada por N (-NHR21-C(=O)-R20), amida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfonamida conectada por N (-NH2-S(=O)2-R20), sulfonamida monosustituida conectada por N (-NHR21-S(=O)2-R20), sulfonamida disustituida conectada por N (-Nr21R22-S(=O)2-R20), sulfoxi (-S(=O)2OH), sulfonato (S(=O)2OR20), sulfonilo (S(=O)2R20), sulfixi (S(=O)OH), sulfinato (S(=O)OR20), sulfinilo (S(=O)R20), fosfonooxi (OP(=O)(OH)2), fosfato (OP(=O)(OR20)2) y sulfonamida (-S(=O)2NH2 , -S(=O)2 NHR21 o -S(=O)2NR21R22), donde R20, R21 y R22 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6.
En todas las anteriores versiones del activador hipóxico de nitroimidazol, para el R1 y el R4 el heteroátomo sustituyente en un grupo alquilo puede estar, en una versión, en la posición beta del grupo alquilo. Por ejemplo, pero no limitante, el etilo puede estar sustituido con metoxi en la posición beta para dar -(CH2)-CH2-O-CH3.
En el caso de que el activador hipóxico sea un nitroimidazol, en otra versión, el activador hipóxico tiene la fórmula

donde el enlace ondulado indica la posición de unión del activador hipóxico al grupo conector o al agente antineoplásico. En otra versión, el activador hipóxico tiene la fórmula
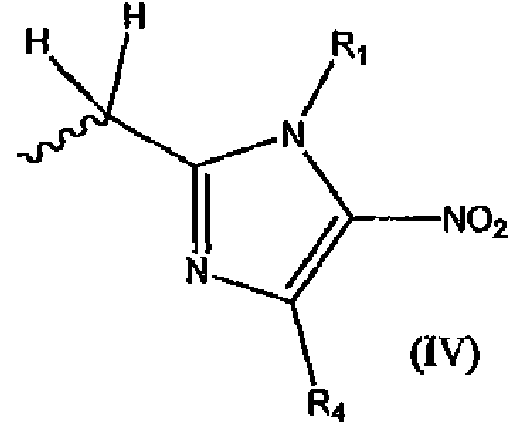
donde el enlace ondulado indica la posición de unión del activador hipóxico al grupo conector o al agente antineoplásico.
En una versión de los anteriores activadores hipóxicos (III & IV), R2 es hidrógeno; R3 es -H o alquilo C1-C6 ; R1 se selecciona entre un grupo aceptor de electrones, alquilo C1-C6 o alcoxi C1-C6 , donde el alquilo o el alcoxi está opcionalmente sustituido con uno o más grupos que contienen heteroátomos; y R4 se selecciona entre un grupo aceptor de electrones, -H, alquilo C1-C6 o alcoxi C1-C6 , donde el alquilo o el alcoxi está opcionalmente sustituido con uno o más grupos que contienen heteroátomos. En una versión del activador hipóxico descrito en este párrafo, al menos uno de R1 y R4 es un grupo aceptor de electrones.
En una versión de los anteriores activadores hipóxicos (III & IV), R2 es hidrógeno; R3 es -H o alquilo C1-C6 ; R1 se selecciona entre un grupo aceptor de electrones o alquilo C1-C6 , donde el alquilo está opcionalmente sustituido con uno o más grupos que contienen heteroátomos; y R4 se selecciona entre un grupo aceptor de electrones, -H o alquilo C1-C6 , donde el alquilo está opcionalmente sustituido con uno o más grupos que contienen heteroátomos. En una versión del activador hipóxico descrito en este párrafo, al menos uno de R1 y R4 es un grupo aceptor de electrones.
En una versión de los anteriores activadores hipóxicos (III & IV), R2 es hidrógeno; R3 es -H o alquilo C1-C6 ; y R1 y R4 son cada uno independientemente un grupo aceptor de electrones, -H o alquilo C1-C6 ; donde el alquilo está opcionalmente sustituido con uno o más grupos que contienen heteroátomos seleccionados entre hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; y donde el grupo aceptor de electrones se selecciona entre halo, ciano (-CN), haloalquilo, carboxamida, nitro, aldehído (-CHO), ceto (-COR20), alquenilo, alquinilo, amino cuaternario (-N+R20R21R22), éster (-COOR20), amida (-CONH2), amida monosustituida (-CONHR20), amida disustituida (-CONR21R22), amida conectada por N (-NH2-C(=O)-R20), amida monosustituida conectada por N (-NHR21-C(=O)-R20), amida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfonamida conectada por N (-NH2-S(=O)2-R20), sulfonamida monosustituida conectada por N (-Nh R21-S(=O)2-R20), sulfonamida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfoxi (-S(=O)2OH), sulfonato (S(=O)2OR20), sulfonilo (S(=O)2R20) y sulfonamida (-S(=O)2NH2 , - S(=O)2NHR21 o -S(=O)2NR21R22), donde R20, R21 y R22 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6 ; con la condición de que R1 no sea hidrógeno. En una versión del activador hipóxico descrito en este párrafo, al menos uno de R1 y R4 es un grupo aceptor de electrones.
En una versión de los anteriores activadores hipóxicos (III & IV), R2 es hidrógeno; R3 es -H o alquilo C1-C6 ; y R1 y R4 son cada uno independientemente un grupo aceptor de electrones, H, alquilo C1-C6 o alcoxi C1-C6 , estando el alquilo o el alcoxi opcionalmente sustituido con uno o más grupos seleccionados entre éter (-OR20), amino (-NH2), amino monosustituido (-NR20H), amino disustituido (-NR21R22), alquilamino C1-C5 cíclico, imidazolilo, alquilpiperazinilo C1-6, morfolino, tiol (-SH), tioéter -(SR20), tetrazol, ácido carboxílico (-COOH), éster (-COOR20), amida (-CONH2), amida monosustituida (-CONHR20), amida disustituida (-CONR21R22), amida conectada por N (-NH2-c (=O)-R20), amida
monosustituida conectada por N (-NHR21-C(=O)-R20), amida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfonamida conectada por N (-NH2-S(=O)2-R20), sulfonamida monosustituida conectada por N (-NHR21-S(=O)2-R20), sulfonamida disustituida conectada por N (-Nr21R22-S(=0)2-R20), sulfoxi (-S(=O)2OH), sulfonato (S(=O)2OR20), sulfonilo (S(=O)2R20), sulfixi (S(=O)OH), sulfinato (S(=O)OR20), sulfinilo (S(=O)R20), fosfonooxi (OP(=O)(OH)2), fosfato (OP(=O)(OR20)2) y sulfonamida (-S(=O)2NH2 , -S(=O)2NHR21 o -S(=O)2NR21R22), donde R20, R21 y seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6 ; donde el grupo aceptor de electrones se selecciona entre halo, ciano (-CN), haloalquilo, carboxamida, nitro, aldehído (-CHO), ceto (-COR20), alquenilo, alquinilo, amino cuaternario (-N+r20r21r22), éster (-COOR20), amida (-CONH2), amida monosustituida (-CONHR20), amida disustituida (-CONR21R22), amida conectada por N (-NH2-C(=O)-R20), amida monosustituida conectada por N (-NHR21-C(=O)-R20), amida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfonamida conectada por N (-NH2-S(=O)2-R20), sulfonamida monosustituida conectada por N (-NHR21-S(=O)2-R20), sulfonamida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfoxi (-S(=O)2OH), sulfonato (S(=O)2OR20), sulfonilo (S(=O)2R20) y sulfonamida (-S(=O)2NH2 , -S(=O)2NHR21 o -S(=O)2NR21R22), donde R20, R21 y R22 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6 ; con la condición de que R1 no sea hidrógeno. En una versión del activador hipóxico descrito en este párrafo, al menos uno de R1 y R4 es un grupo aceptor de electrones.
En una versión de los anteriores activadores hipóxicos (III & IV), R2 es hidrógeno; R3 es -H o alquilo C1-C3 ; y R1 y R4 son cada uno independientemente un grupo aceptor de electrones, -H o un alquilo seleccionado entre metilo, etilo, npropilo, n-butilo, n- pentilo, t-butilo, ciclohexilo, ciclopentilo e isopropilo, donde el alquilo está opcionalmente sustituido con uno o más grupos que contienen heteroátomos; donde el grupo aceptor de electrones se selecciona entre halo, ciano (-CN), haloalquilo, carboxamida, nitro, aldehído (-CHO), ceto (-COR20), alquenilo, alquinilo, amino cuaternario (-N+R20R21R22), éster (-COOR20), amida (-CONH2), amida monosustituida (-CONHR20), amida disustituida (-CONR21R22), amida conectada por N (-NH2-C(=O)-R20), amida monosustituida conectada por N (-NHR21-C(=O)-R20), amida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfonamida conectada por N (-NH2-S(=O)2-R20), sulfonamida monosustituida conectada por N (-NHR21-S(=O)2-R20), sulfonamida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfoxi (-S(=O)2OH), sulfonato (S(=O)2OR20), sulfonilo (S(=O)2R20) y sulfonamida (-S(=O)2NH2 , -S(=O)2NHR21 o -S(=O)2NR21R22), donde R20, R21 y R22 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6 ; con la condición de que R1 no sea hidrógeno. En una versión del activador hipóxico descrito en este párrafo, al menos uno de R1 y r4 es un grupo aceptor de electrones.
En una versión de los anteriores activadores hipóxicos (III & IV), R2 es hidrógeno; R3 es -H o alquilo C1-C3 ; y R1 y R4 son cada uno independientemente un grupo aceptor de electrones, -H o un alquilo seleccionado entre metilo, etilo, npropilo, n-butilo, n- pentilo, t-butilo, ciclohexilo, ciclopentilo e isopropilo, donde el alquilo está opcionalmente sustituido con uno o más grupos que contienen heteroátomos seleccionados entre hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; donde el grupo aceptor de electrones se selecciona entre halo, ciano (-CN), haloalquilo, carboxamida, nitro, aldehído (-CHO), ceto (-COR20), alquenilo, alquinilo, amino cuaternario (-N+R20R21R22), éster (-COOR20), amida (-CONH2), amida monosustituida (-CONHR20), amida disustituida (-c On R21R22), amida conectada por N (-NH2-C(=O)-R20), amida monosustituida conectada por N (-NHR21-C(=O)-R20), amida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfonamida conectada por N (-NH2-S(=O)2-R20), sulfonamida monosustituida conectada por N (-NHR21-S(=O)2-R20), sulfonamida disustituida conectada por N (-Nr21R22-S(=O)2-R20), sulfoxi (-S(=O)2OH), sulfonato (S(=O)2OR20), sulfonilo (S(=O)2R20) y sulfonamida (-S(=O)2NH2 , -S(=O)2NHR21 o -S(=O)2NR21R22), donde R20, R21 y R22 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6 ; con la condición de que R1 no sea hidrógeno. En una versión del activador hipóxico descrito en este párrafo, al menos uno de R1 y R4 es un grupo aceptor de electrones.
En una versión de los anteriores activadores hipóxicos (III & IV), R2 es hidrógeno; R3 es -H o alquilo C1-C3 ; y R1 y R4 son cada uno independientemente un grupo aceptor de electrones, -H o un alquilo seleccionado entre metilo, etilo, npropilo, n-butilo, n- pentilo, t-butilo, ciclohexilo, ciclopentilo e isopropilo, donde el alquilo está opcionalmente sustituido con uno o más grupos que contienen heteroátomos seleccionados entre éter (-OR20), amino (-NH2), amino monosustituido (-NR20H), amino disustituido (-NR21R22), alquilamino C1-C5 cíclico, imidazolilo, alquilpiperazinilo C1-C6, morfolino, tiol (-SH), tioéter -(SR20), tetrazol, ácido carboxílico (-COOH), éster (-COOR20), amida (-CONH2), amida monosustituida (-CONHR20), amida disustituida (-CONR21R22), amida conectada por N (-NH2-C(=O)-R20), amida monosustituida conectada por N (-NHR21-C(=O)-R20), amida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfonamida conectada por N (-NH2-S(=O)2-R20), sulfonamida monosustituida conectada por N (-NHR21-S(=O)2-R20), sulfonamida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfoxi (-S(=O)2OH), sulfonato (S(=O)2Or20), sulfonilo (S(=O)2R20), sulfixi (S(=O)OH), sulfinato (S(=O)OR20), sulfinilo (S(=O)R20), fosfonooxi (OP(=O)(OH)2), fosfato (OP(=O)(OR20)2) y sulfonamida (-S(=O)2NH2 , -S(=O)2NHR21 o -S(=O)2NR21R22), donde R20, R21 y seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6 ; donde el grupo aceptor de electrones se selecciona entre halo, ciano (-CN), haloalquilo, carboxamida, nitro, aldehído (-CHO), ceto (-COR20), alquenilo, alquinilo, amino cuaternario (N+R20R21R22), éster (-COOR20), amida (-CONH2), amida monosustituida (-CONHR20) amida disustituida (-CONR21R22), amida conectada por N (-NH2-C(=O)-R20), amida monosustituida conectada por N (-NHR21-C(=O)-R20), amida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfonamida conectada por N (-NH2-S(=O)2-R20), sulfonamida monosustituida conectada por N (-NHR21-S(=O)2-R20, sulfonamida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfoxi (-S(=O)2OH), sulfonato (S(=O)2OR20), sulfonilo (S(=O)2R20) y sulfonamida (-S(=O)2NH2 , -S(=O)2NHR21 o -S(=O)2NR21R22), donde R20, R21 y R22 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6 con la condición de que R1 no sea hidrógeno. En una versión del activador hipóxico descrito en este párrafo, al menos uno de R1 y R4 es un grupo aceptor de electrones.
En una versión de los anteriores activadores hipóxicos (III & IV), R2 es hidrógeno; R3 es -H o alquilo C1-C3 ; y R1 y R4 se seleccionan cada uno independientemente entre ciano, haloalquilo, carboxamida, metilo, etilo, n-propilo o n-butilo, cada uno opcionalmente sustituido con un grupo que contiene un heteroátomo, particularmente un grupo amino, un grupo ácido carboxílico o un grupo amida. En una versión del activador hipóxico descrito en este párrafo, al menos uno de R1 y R4 es un grupo aceptor de electrones.
En una versión de los anteriores activadores hipóxicos (III & IV), R2 es hidrógeno y R1 y R3 son cualquiera de las versiones descritas más arriba y R4 es alquilo C1-C3 opcionalmente sustituido con un grupo que contiene un heteroátomo, particularmente un grupo amino, un grupo ácido carboxílico o un grupo amida.
En una versión de los anteriores activadores hipóxicos (III & IV), R2 es hidrógeno y R1, R3 y R4 son cada uno independientemente etilo, n-propilo o n-butilo, cada uno opcionalmente sustituido con un grupo que contiene un heteroátomo, particularmente un grupo amino, un grupo ácido carboxílico o un grupo amida
En una versión de los anteriores activadores hipóxicos (III & IV), R2 es hidrógeno; R3 es hidrógeno o alquilo C1-C3 ; R1 es metilo, acetato de metilo o etilo, n-propilo o n-butilo, cada uno opcionalmente sustituido con un grupo amino, un grupo ácido carboxílico o un grupo amida y R4 es hidrógeno o alquilo C1-C3.
En una versión de los anteriores activadores hipóxicos (III & IV), R2 es hidrógeno; R3 es hidrógeno o alquilo C1-C3 ; R1 es metilo, acetato de metilo o etilo, n-propilo o n-butilo, cada uno opcionalmente sustituido con un grupo amino, un grupo ácido carboxílico o un grupo amida y R4 es hidrógeno.
En una versión de los anteriores activadores hipóxicos (III & IV), R2 es hidrógeno, R1 es un grupo alquilo portador de un impedimento estérico, y R3 y R4 son cada uno independientemente -H o alquilo C1-C6 , opcionalmente sustituido con uno o más grupos que contienen heteroátomos. En una versión, R2 es hidrógeno, R1 es metilo y R3 y R4 son cada uno independientemente -H o alquilo C1-C6 , opcionalmente sustituido con uno o más grupos que contienen heteroátomos. En una versión, R2 es hidrógeno, R1 es acetato de metilo y R3 y R4 son cada uno independientemente -H o alquilo C1-C6 , opcionalmente sustituido con uno o más grupos que contienen heteroátomos.
En otra versión de los activadores hipóxicos de nitroimidazol, el activador hipóxico se selecciona entre uno de los siguientes

o

En estas versiones del activador hipóxico de nitroimidazol, Ri, R2 , R3 y R4 son según se describe en cualquiera de las versiones anteriores.
En una versión, R1, R3 y R4 son cualquiera de las versiones descritas más arriba para cualquiera de los activadores hipóxicos, y R2 se selecciona entre -H o alquilo C1-C6 , opcionalmente sustituido con uno o más grupos que contienen heteroátomos. En una versión, R1, R3 y R4 son cualquiera de las versiones descritas más arriba, y R2 es -H, metilo, n-pentilo, t-butilo, ciclohexilo, ciclopentilo o isopropilo, todos opcionalmente sustituidos con uno o más grupos que contienen heteroátomos seleccionados entre hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano. En una versión, R1, R3 y R4 son cualquiera de las versiones descritas más arriba, y R2 es H, alquilo C1-C6 o alcoxi C1-C6 , estando el alquilo o el alcoxi opcionalmente sustituido con uno o más grupos seleccionados entre éter (-OR20), amino (-NH2), amino monosustituido (-NR20H), amino disustituido (-NR21R22), alquilamino C1-5 cíclico, imidazolilo, alquilpiperazinilo C1-6 , morfolino, tiol (-SH), tioéter -(SR20), tetrazol, ácido carboxílico (-COOH), éster (-COOR20), amida (-CONH2), amida monosustituida (-CONHR20), amida disustituida (-CONR21R22), amida conectada por N (-NH2-C(=O)-R20), amida monosustituida conectada por N (-NHR21-C(=O)-R20), amida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfonamida conectada por N (-NH2-S(=O)2-R20), sulfonamida monosustituida conectada por N (-NHR21-S(=O)2-R20), sulfonamida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfoxi (-S(=O)2OH), sulfonato (S(=O)2OR20), sulfonilo (S(=O)2R20), sulfixi (S(=O)OH), sulfinato (S(=O)OR20), sulfinilo (S(=O)R20), fosfonooxi (OP(=O)(OH)2), fosfato (OP(=O)(OR20)2) y sulfonamida (-S(=O)2NH2 , -S(=O)2NHR21 o -S(=O)2NR21R22), donde R20, R21 y R22 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6. En el caso de que el activador hipóxico sea un nitrobenceno, en una versión el activador hipóxico tiene la fórmula

donde R2 y R3 son según se han definido en cualquiera de las versiones anteriores, y R50, R51, R52 y R53 son independientemente un grupo aceptor de electrones, H, alquilo C1-C6 o alcoxi C1-C6 , estando dicho alquilo o alcoxi opcionalmente sustituido con uno o más grupos seleccionados entre éter (-OR20), amino (-NH2), amino monosustituido (-NR20H), amino disustituido (-NR21R22), alquilamino C1-5 cíclico, imidazolilo, alquilpiperazinilo C1-6 , morfolino, tiol (-SH), tioéter-(SR20), tetrazol, ácido carboxílico (-COOH), éster (-COOR20), amida (-CONH2), amida monosustituida (-CONHR20), amida disustituida (-CONR21R22), amida conectada por N (-NH2-c (=O)-R20), amida monosustituida conectada por N (-NHR21-C(=O)-R20), amida disustituida conectada por N (-NR21R22-S(=O)2-R20),
sulfonamida conectada por N (-NH2-S(=O)2-R20), sulfonamida monosustituida conectada por N (-NHR21-S(=O)2-R20), sulfonamida disustituida conectada por N (-Nr21R22-S(=0)2-R20), sulfoxi (-S(=O)2OH), sulfonato (S(=O)2OR20), sulfonilo (S(=O)2R20), sulfixi (S(=O)OH), sulfinato (S(=O)OR20), sulfinilo (S(=O)R20), fosfonooxi (OP(=O)(OH)2), fosfato (OP(=O)(OR20)2) y sulfonamida (-S(=O)2NH2 , -S(=O)2NHR21 o -S(=O)2NR21R22), donde R20, R21 y R22 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6 ; donde el grupo aceptor de electrones se selecciona entre halo, ciano (-CN), haloalquilo, carboxamida, nitro, aldehído (-CHO), ceto (-COR20), alquenilo, alquinilo, amino cuaternario (-N+r20r21r22), éster (-COOR20), amida (-CONH2), amida monosustituida (-CONHR20), amida disustituida (-CONR21R22), amida conectada por N (-NH2-C(=O)-R20), amida monosustituida conectada por N (-NHR21-C(=O)-R20), amida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfonamida conectada por N (-NH2-S(=O)2-R20), sulfonamida monosustituida conectada por N (-NHR21-S(=O)2-R20), sulfonamida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfoxi (-S(=O)2OH), sulfonato (S(=O)2OR20), sulfonilo (S(=O)2R20) y sulfonamida (-S(=O)2NH2 , -S(=O)2NHR21 o -S(=O)2NR21R22), donde R20, R21 y R22 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6.
En una versión, al menos uno de R50, R51, R52 y R53 es un grupo aceptor de electrones. En una versión, al menos uno de R50 y R52 es un grupo aceptor de electrones.
En una versión, uno o más de los carbonos del anillo de fenilo está sustituido con un heteroátomo. En una versión, uno de los átomos de carbono en el anillo si sustituido por un nitrógeno para dar un grupo piridilo.
En otra versión de los activadores hipóxicos de nitrobenceno, el activador hipóxico tiene la fórmula

En esta versión del nitrobenceno, R2 , R3 , R50, R51, R52 y R53 son según se describe en cualquiera de las versiones anteriores. En una versión, al menos uno de R50, R51, R52 y R53 es un grupo aceptor de electrones. En una versión, R52 es un grupo aceptor de electrones. En una versión, uno o más de los carbonos del anillo de fenilo está sustituido con un heteroátomo. En una versión, uno de los átomos de carbono en el anillo si sustituido por un nitrógeno para dar un grupo piridilo.
Grupo conector (L)
Si está presente en el agente antineoplásico protegido, el grupo conector, L, conecta el activador de la hipoxia con el agente antineoplásico. Esto es, el agente antineoplásico protegido tiene la fórmula Hip-L-N.
Generalmente, el grupo conector es un grupo que es susceptible de ser escindido a partir del activador hipóxico después de una reducción del activador hipóxico, produciendo un agente neoplásico modificado que es bien por sí mismo un agente neoplásico o bien a través de un reordenamiento, una degradación u otra modificación química produce un agente neoplásico. Algunos ejemplos específicos están incluidos en cualquier parte de esta patente de dichos reordenamientos y degradaciones de los agentes neoplásicos modificados resultantes de la escisión a partir del activador hipóxico.
En una versión, el grupo conector es un grupo de fórmula
/ u w X -------Y 'w w
donde X es un grupo éter o acetal, e Y es un grupo separador según se describe con más detalle a continuación. En la fórmula
/ v w x --------y o jw ^
el enlace ondulado en el grupo X muestra el punto de unión de X al activador hipóxico, y el enlace ondulado en el grupo Y muestra el punto de unión del grupo Y grupo al agente antineoplásico.
Grupo X
En una versión, X es uno de los siguientes grupos éter (X1) o acetal (X2)
«vvvr 0 'vru"u‘ 
X1 X2
donde la línea ondulada en el lado izquierdo del grupo X indica el punto de unión al activador hipóxico y la línea ondulada en el lado derecho del grupo X indica el punto de unión al grupo Y.
En una versión, R6 es un alquilo no sustituido o un alquilo sustituido con uno o más grupos que contienen heteroátomos; y R7 es hidrógeno, un alquilo no sustituido o un alquilo sustituido con uno o más grupos que contienen heteroátomos.
En una versión, R6 es un alquilo C1-C10 no sustituido o un alquilo C1-C10 sustituido con uno o más grupos que contienen heteroátomos, y R7 es hidrógeno, un alquilo C1-C10 no sustituido o un alquilo C1-C10 sustituido con uno o más grupos que contienen heteroátomos, donde tanto para R6 como para R7 el grupo que contiene un heteroátomo contiene uno o más de hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano. En una versión, R6 es un alquilo C1-C10 no sustituido.
En una versión, R6 es un alquilo C1-C3 no sustituido o un alquilo C1-C3 sustituido con uno o más grupos que contienen heteroátomos, y R7 es hidrógeno, un alquilo C1-C3 no sustituido o un alquilo C1-C3 sustituido con uno o más grupos que contienen heteroátomos, donde tanto para R6 como para R7 el grupo que contiene un heteroátomo contiene uno o más de hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano. En una versión, R6 es un alquilo C1-C3 no sustituido.
En una versión, R7 es hidrógeno y R6 es un alquilo C1-C3 no sustituido o un alquilo C1-C3 sustituido con uno o más grupos que contienen heteroátomos seleccionados entre hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano.
En una versión, R6 es un alquilo C1-C10 no sustituido y R7 es hidrógeno o un alquilo C1-C10 no sustituido.
En una versión, R6 es un alquilo C1-C3 no sustituido y R7 es hidrógeno o es un alquilo C1-C3 no sustituido.
En una versión, R6 es metilo y R7 es hidrógeno.
Grupo Y: generalmente Y puede ser cualquier grupo de forma que después de la reducción del activador hipóxico, el agente antineoplásico modificado liberado es citotóxico, o el agente antineoplásico modificado liberado experimenta una reordenación, una degradación u otra transformación química para producir un agente citotóxico. En una versión, Y es una cadena de -(CH2)n- con n = 1-4 no sustituida o una cadena de -(CH2)n- con n = 1-4 sustituida con uno o más grupos que contienen heteroátomos. En una versión, Y es una cadena de -(CH2)n- con n = 1-4 no sustituida o una cadena de -(CH2)n- con n = 1-4 sustituida con uno o más grupos que contienen heteroátomos seleccionados entre hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano.
En una versión, X es el grupo éter X1 e Y es -(CRcRd)- donde Rc y Rd son independientemente hidrógeno, alquilo C1-C3 no sustituido o alquilo C1-C3 sustituido con uno o más de hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano. En una versión, X e Y son
según se describe en la frase anterior, e Y está unido al agente antineoplásico a través del oxígeno de un grupo hidroxilo del agente antineoplásico; es decir, el agente antineoplásico protegido incluye un grupo formacetal y tiene la fórmula Hip-O-(CH2)-O-N', donde el agente antineoplásico N es N'-OH.
En una versión, X es el grupo éter X1 e Y es -(CH2)-. En una versión, X es el grupo éter X1, Y es -(CH2)- e Y está unido al agente antineoplásico a través del oxígeno de un grupo hidroxilo del agente antineoplásico; es decir, el agente antineoplásico protegido incluye un grupo formacetal y tiene la fórmula Hip-O-(CH2)-O-N', donde el agente antineoplásico N es N'-OH.
En una versión, X es el grupo acetal X2 e Y es una cadena de alquileno C3-C4 no sustituida o una cadena de alquileno C3-C4 sustituida con uno o más grupos que contienen heteroátomos. En una versión, X es el grupo acetal X2 e Y es una cadena de alquileno C3-C4 no sustituida o una cadena de alquileno C3-C4 sustituida con uno o más grupos que contienen heteroátomos seleccionados entre hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano. En una versión, X es el grupo acetal X2, Y es según se describe en cualquiera de las frases anteriores, e Y está unido al agente antineoplásico a través del nitrógeno de un grupo amino del agente antineoplásico; es decir, el agente antineoplásico protegido incluye tiene la fórmula Hip-O-(alquileno C3-C4 sustituido o no sustituido)-NR-N', donde el agente antineoplásico N es N'-NRR'.
Y es alquileno C 3 : en una versión, X es el grupo acetal X2 e Y es -(CReRf)-(CRgRh)-(CH2)-, donde Re, Rf, Rg y Rh son independientemente hidrógeno, alquilo C1-C3 no sustituido o alquilo C1-C3 sustituido con uno o más de hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano. En una versión, Re y Rf tomados en conjunto pueden formar un grupo =O; es decir, -(CReRf)- es -C(O)-. En una versión, Rg y Rh tomados en conjunto pueden formar un grupo =O; es decir, -(CRgRh)- es -C(O)-. En una versión, Re y Rf se seleccionan independientemente entre -H y -O-Ri, donde Ri es alquilo C1-C5 no sustituido. En una versión, Rg y Rh se seleccionan independientemente entre -H y -O-Ri, donde Ri es alquilo C1-C5 no sustituido. En una versión, X es el grupo acetal X2, Y es como en cualquiera de las versiones descritas en las frases anteriores del párrafo, e Y está unido al agente antineoplásico a través del nitrógeno de un grupo amino del agente antineoplásico; es decir, el agente antineoplásico protegido incluye tiene la fórmula Hip-O-(alquileno C3-C4 sustituido o no sustituido)-NR-N', donde el agente antineoplásico N es N'-NRR'.
Y es alquileno C 4 : en una versión, X es el grupo acetal X2 e Y es -(CReRf)-(CRgRh)-(CRjRk)-(CH2)-, donde Re, Rf, Rg, Rh, Rj y Rk son independientemente hidrógeno, alquilo C1-C3 no sustituido o alquilo C1-C3 sustituido con uno o más de hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano. En una versión, Re y Rf tomados en conjunto pueden formar un grupo =O; es decir, -(CReRf)- es -C(O)-. En una versión, Rg y Rh tomados en conjunto pueden formar un grupo =O; es decir, -(CRgRh)- es -C(O)-. En una versión, Rj y Rk tomados en conjunto pueden formar un grupo =O; es decir, -(CRjRk)- es -C(O)-. En una versión, Re y Rf se seleccionan independientemente entre -H y -O-Ri, donde Ri es -H o alquilo C1-C5 no sustituido. En una versión, Rg y Rh se seleccionan independientemente entre -H y -O-R', donde Ri es -H o alquilo C1-C5 no sustituido. En una versión, Rj y Rk se seleccionan independientemente entre -H y -O-Ri, donde Ri es -H o alquilo C1-C5 no sustituido. En una versión, X es el grupo acetal X2, Y es como en cualquiera de las versiones descritas en las frases anteriores del párrafo, e Y está unido al agente antineoplásico a través del nitrógeno de un grupo amino del agente antineoplásico; es decir, el agente antineoplásico protegido tiene la fórmula Hip-O-( alquileno C3-C4 sustituido o no sustituido)-NR-N', donde el agente antineoplásico N es N'-NRR'.
Y es alquileno C 4 sustituido con un heterogrupo de cadena: en una versión, X es el grupo acetal X2 e Y es -(CReRf)-Rm-(CRjRk)-(CH2)-, donde Re, Rf, Rg, Rh, Rj y Rk son según se han definido anteriormente más arriba, y Rm se selecciona entre -O-, -S-, -S(=O)2-, -S(=O)O- y -NR30-, donde R30 se selecciona entre -C(=O)R31, -C(=O)NR31R32, -H, alquilo C1-C10 o alquilo C1-C10 sustituido con uno o más grupos que contienen heteroátomos seleccionados entre hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano. Donde R31 y R32 se seleccionan independientemente entre alquilo C1-C10 o alquilo C1-C10 sustituido con uno o más grupos que contienen heteroátomos seleccionados entre hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano.
Y contiene un grupo de liberación retardada: en una versión, el grupo Y se elige de forma que el agente antineoplásico modificado liberado después de la reducción del activador hipóxico se convierte, después de la liberación, en el agente antineoplásico o en algún derivado activo del agente antineoplásico. En una versión, esta
degradación se produce mediante una degradación unimolecular con una semivida de segregación unimolecular de entre aproximadamente 0,01 segundos y aproximadamente 30 segundos. En una versión, esta degradación se produce mediante una degradación unimolecular con una semivida de segregación unimolecular de entre aproximadamente 0,5 segundos y aproximadamente 5 segundos.
En esta versión de liberación retardada de Y, el agente antineoplásico o algún derivado activo del agente antineoplásico es suministrado no solamente en el sitio de reducción hipóxica del agente antineoplásico protegido, sino también en el tejido circundante. Esto se produce debido a que el agente antineoplásico modificado liberado después de la reducción del activador hipóxico tiene tiempo para difundir al tejido circundante antes de ser degradado para liberar el agente antineoplásico o el derivado activo del agente antineoplásico.
En una versión de liberación retardada, Y tiene la fórmula
■A / w Rio— R-h— R-i2a-r'J
donde R11 es un grupo arilo o heteroarilo no sustituido o sustituido, R10 es un enlace o un grupo que une el grupo R11 al grupo X del conector, y R12 es un enlace o un grupo que une el grupo R11 al agente antineoplásico.
En una versión, X es el grupo éter e Y tiene la fórmula
/ w v Rio'— R-i i^— Ri2 'v v
con R10, R11 y R12 según se describe en cualquiera de las versiones de esta sección.
Generalmente, R11 puede ser cualquier grupo arilo o heteroarilo no sustituido o sustituido, y el grupo arilo o heteroarilo puede ser un grupo monocíclico o puede ser un sistema anular condensado.
Algunos ejemplos de sistemas anulares condensados que pueden usarse incluyen, pero no se limitan a, naftilo, quinolina o isoquinolina.
En una versión, el grupo arilo o heteroarilo no es un sistema anular aromático monocíclico o condensado 5 de miembros.
En una versión, el punto de unión del grupo arilo o heteroarilo a los grupos R10 y R12 puede ser generalmente cualquier átomo del anillo al que puede haber unido un grupo sustituyente y que permitirá la eliminación de los grupos R10 y R12. En una versión, el grupo arilo o heteroarilo es cualquier grupo que permita la eliminación en 1,6 o en 1,4.
En una versión, el grupo arilo o heteroarilo sustituido está sustituido con uno o más grupos seleccionados entre un grupo aceptor de electrones, sustituido o no sustituido, y un alcoxi sustituido o no sustituido.
En una versión, el grupo arilo o heteroarilo sustituido está sustituido con uno o más grupos seleccionados entre un grupo aceptor de electrones, alquilo C1-C6 no sustituido, alquilo C1-C6 sustituido, alcoxi C1-C6 no sustituido y alcoxi C1-C6 sustituido; donde el alquilo o el alcoxi sustituido están sustituidos con uno o más grupos seleccionados entre éter (-OR20), amino (-NH2), amino monosustituido (-NR20H), amino disustituido (-NR21R22), alquilamino C1-5 cíclico, imidazolilo, alquilpiperazinilo C1-6 , morfolino, tiol (-SH), tioéter -(SR20), tetrazol, ácido carboxílico (-COOH), éster (-COOR20), amida (-CONH2), amida monosustituida (-CONHR20), amida disustituida (-CONR21R22), amida conectada por N (-NH2-C(=O)-R20), amida monosustituida conectada por N (-NHR21-C(=O)-R20), amida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfonamida conectada por N (-NH2-S(=O)2-R20), sulfonamida monosustituida conectada por N (-Nh R21-S(=O)2-R20), sulfonamida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfoxi (-S(=O)2OH), sulfonato (S(=O)2OR20), sulfonilo (S(=O)2R20), sulfixi (S(=O)OH), sulfinato (S(=O)OR20), sulfinilo (S(=O)R20), fosfonooxi (OP(=O)(OH)2), fosfato (OP(=O)(OR20)2) y sulfonamida (-S(=O)2NH2 , -S(=O)2NHR21 o -S(=O)2NR21R22), donde R20, R21 y R22 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6 ; y donde el grupo aceptor de electrones se selecciona entre halo, ciano (-CN), haloalquilo, carboxamida, nitro, aldehído (-CHO), ceto (-COR20), alquenilo, alquinilo, amino cuaternario (-N+R20R21R22), éster (-COOR20), amida (-CONH2), amida monosustituida (-CONHR20), amida disustituida (-COn R21R22), amida conectada por N (-NH2-C(=O)-R20), amida monosustituida conectada por N (-NHR21-C(=O)-R20), amida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfonamida conectada por N (-NH2-S(=O)2-R20), sulfonamida monosustituida conectada por N (-NHR21-S(=O)2-R20), sulfonamida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfoxi (-S(=O)2OH), sulfonato (S(=O)2o R20), sulfonilo (S(=O)2R20) y sulfonamida (-S(=O)2NH2 , -S(=O)2NHR21 o -S(=O)2NR21 R22), donde R20, R21 y R22 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6.
En una versión, el grupo arilo o heteroarilo sustituido está sustituido con uno o más grupos seleccionados entre -F, Cl, -Br, -CN, -OCH3 , -NO2 , - NH2 , -NHR20, -NR20R21, -CH3 , - CF3 , -CHF2 , -CH2F, sulfamida (-S(=O)2NH2 , -S(=O)2NHR20, o -S(=O)2NR20R21), carboxamida (-C(=O)NH2 , -C(=O)NHR20 o -C(=O)NR20R21); donde R20 y R21 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6.
Algunos ejemplos de grupos heteroarilo monocíclicos que pueden usarse incluyen, pero no se limitan a, piridilo, piridazinilo y pirimidinilo. En todos los grupos heteroarilo monocíclicos, el punto de unión del grupo heteroarilo a los grupos R10 y R12 pueden estar generalmente en cualquier átomo del anillo al que pueda estar unido un grupo sustituyente y que permitirá la eliminación de los grupos R10 y R12.
En una versión, R11 es un arilo sin sustituir o sustituido, particularmente un fenilo no sustituido o sustituido. En una versión, R11 es un fenilo no sustituido o sustituido en el que los grupos R10 y R12 están en orto o en para entre sí; es decir, Y tiene la fórmula

En una versión, R13-R20 se seleccionan independientemente entre un grupo aceptor de electrones, alquilo C1-C6 no sustituido, alquilo C1-C6 sustituido, alcoxi C1-C6 no sustituido y alcoxi C1-C6 sustituido; donde el alquilo o el alcoxi sustituido están sustituidos con uno o más grupos seleccionados entre éter (-OR20), amino (-NH2), amino monosustituido (-NR20H), amino disustituido (-NR21R22), alquilamino C1-5 cíclico, imidazolilo, alquilpiperazinilo C1-6 , morfolino, tiol (-SH), tioéter-(SR20), tetrazol, ácido carboxílico (-COOH), éster (-COOR20), amida (-CONH2), amida monosustituida (-CONHR20), amida disustituida (-CONR21R22), amida conectada por N (-NH2-c (=O)-R20), amida monosustituida conectada por N (-NHR21-C(=O)-R20), amida disustituida conectada por N (-NR21R22-(=O)2-R20), sulfonamida conectada por N (-NH2-S(=O)2-R20), sulfonamida monosustituida conectada por N (-NHR21-S(=O)2-R20), sulfonamida disustituida conectada por N (-Nr21R22-S(=O)2-R20), sulfoxi (-S(=O)2OH), sulfonato (S(=O)2OR20), sulfonilo (S(=O)2R20), sulfixi (S(=O)OH), sulfinato (S(=O)OR20), sulfinilo (S(=O)R20), fosfonooxi (OP(=O)(OH)2), fosfato (OP(=O)(OR20)2) y sulfonamida (-S(=O)2NH2 , -S(=O)2NHR21 o -S(=O)2NR21R22), donde R20, R21 y R22 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6 ; y donde el grupo aceptor de electrones se selecciona entre halo, ciano (-CN), haloalquilo, carboxamida, nitro, aldehído (-CHO), ceto (-COR20), alquenilo, alquinilo, amino cuaternario (-N+R20r21r22), éster (-COOR20), amida (-CONH2), amida monosustituida (-CONHR20), amida disustituida (-CONR21R22), amida conectada por N (-NH2-C(=O)-R20), amida monosustituida conectada por N (-NHR21-C(=O)-R20), amida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfonamida conectada por N (-NH2-S(=O)2-R20), sulfonamida monosustituida conectada por N (-NHR21-S(=O)2-R20), sulfonamida disustituida conectada por N (-NR21R22-S(=O)2-R20), sulfoxi (-S(=O)2OH), sulfonato (S(=O)2OR20), sulfonilo (S(=O)2R20) y sulfonamida (-S(=O)2NH2 , -S(=O)2NHR21 o -S(=O)2NR21R22), donde R20, R21 y R22 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6.
En una versión, R13-R20 se seleccionan independientemente entre -F, -Cl, -Br, -CN, -OCH3 , - NO2 , -NH2 , -NHR20, -NR20R21, -CH3 , -CF3 , -CHF2 , -CH2F, sulfamida (-S(=O)2NH2 , - S(=O)2NHR20 o -S(=O)2NR20R21), carboxamida (-C(=O)NH2 , -C(=O)NHR20 o - C(=O)NR20R21); donde R20 y R21 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6.
En una versión, R13-R20 se seleccionan independientemente entre -F, -Cl, -Br, -CN, -OCH3 , - NO2 , -NH2 , -NHR20, -NR20R21, -CH3 , -CF3 , -CHF2 , -CH2F, sulfamida (-S(=O)2NH2 , - S(=O)2NHR20 o -S(=O)2NR2OR21), carboxamida (-C(=O)NH2 , -C(=O)NHR20 o - C(=O)NR20R21); donde R20 y R21 se seleccionan independientemente entre un grupo alquilo C1-C6 , un grupo heterocíclico C3-C20 o un grupo arilo C3-C20, preferentemente un grupo alquilo C1-C6 ; y alquilo C1-C10 o alquilo C1-C10 sustituido con uno o más grupos que contienen heteroátomos seleccionados entre hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano.
En una versión, el grupo R10 es un enlace y el grupo R11 está unido directamente al grupo conector X. En una versión, el grupo X es el éter X1, el grupo R10 es un enlace y R11 es un fenilo no sustituido o sustituido en el que los grupos R10 y R12 están en orto o en para entre sí; es decir, el grupo conector L tiene la fórmula

y donde R13-R20 son según se describe en cualquier versión anterior.
Grupo R12: R12 es generalmente cualquier grupo capaz de unir el grupo arilo o heteroarilo R11 a un grupo protegible del agente antineoplásico y que producirá el agente antineoplásico o un derivado citotóxico del agente antineoplásico después de la degradación del agente antineoplásico modificado que fue liberado después de la reducción del activador hipóxico.
En una versión, R12 tiene la fórmula -(CR40R41)-R42- donde R40 y R41 se seleccionan independientemente entre -H, alquilo C1-C10 o alquilo C1-C10 sustituido con uno o más grupos que contienen heteroátomos seleccionados entre hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; y R42 es un enlace o -OC(=O)-. En una versión, R12 tiene la fórmula -(CR40R41)-R42- donde R40 es -H y R41 se selecciona entre alquilo C1-C10 o alquilo C1-C10 sustituido con uno o más grupos que contienen heteroátomos seleccionados entre hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; y R42 es un enlace o -OC(=O)-. En una versión, R40, R41 y R42 son según se describe en cualquiera de las frases anteriores de este párrafo, que la condición de que R42 sea -OC(=O)-únicamente cuando el agente antineoplásico está unido al grupo conector a través del nitrógeno de una amina del agente antineoplásico. En una versión, R40 y R41 son hidrógeno.
En una versión, R12 es -CH2- y R12 está unido a un oxígeno del grupo hidroxilo del agente neoplásico. En una versión, R12 es -CH2- y R12 está unido a un nitrógeno del grupo amino del agente neoplásico En una versión, R12 es -CH2-O-C(=O)- y R12 está unido a un nitrógeno del grupo amino del agente neoplásico.
En una versión, R12 tiene la fórmula -(CR40R41)-CR43=CR44-R42- donde R40, R41 y R42 son como se ha descrito más arriba, y R43 y R44 se seleccionan independientemente entre -H, alquilo C1-C10 o alquilo C1-C10 sustituido con uno o más grupos que contienen heteroátomos seleccionados entre hidroxilo, éter, tiol, tioéter, éster sulfínico, sulfóxido, sulfona, ácido sulfónico, éster de ácido sulfónico, sulfenamida, sulfonamida, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano; preferentemente hidroxilo, éter, tiol, tioéter, ácido carboxílico, sal de un ácido carboxílico, éster, amida, aldehído, ceto, amino, halo y ciano. En una versión, R12 tiene la fórmula -(CR40R41)-CR43=CR44-R42- donde R40, R41 y R42 son como se ha descrito más arriba, y R43 y R44 son -H. Agentes antineoplásicos (N)
Generalmente, el agente antineoplásico, N, puede ser cualquier agente susceptible de ser protegido mediante el uso del activador hipóxico y de los grupos conectores descritos más arriba, y que genera un agente citotóxico antineoplásico o un agente antineoplásico modificado después de la liberación después de la reducción del activador hipóxico.
En una versión, N es un agente citotóxico que tiene una CI50 de menos de 100 microM y opcionalmente de menos de 1 microM, según se define mediante el ensayo de cribado del NCI como una CL50 después de un tratamiento con el fármaco durante 24 h de una línea celular sensible. En una versión, el agente citotóxico tiene una CI50 de menos de 10 nanomolar. En una versión, N es doxorrubicina. En una versión N es doxorrubicina unida al activador hipóxico a través de un grupo conector que es tal que la reducción del activador hipóxico libera la doxorrubicina modificada que tiene una CI50 en el intervalo nanomolar bajo.
En una versión, N se selecciona entre el grupo que consiste en maitansinas, enediyenos, discodermólidos, epotilonas, taxanos, caliqueamicinas y tedanólidos. En otra versión, N se selecciona entre el grupo que consiste enetopósido, vinblastina, vincristina, topotecán, 5-fluorouracilo, AQ4N e hidroxiurea.
Otros agentes antineoplásicos que pueden ser incorporados en o liberados desde los agentes antineoplásicos protegidos incluyen, pero no se limitan a, bleomicinas, caliqueamicinas, colchicina, ciclofosfamida, citarabina, dacarbazina, dactinomicina, daunorrubicina, discodermólidos, doxorrubicina y compuestos similares a la doxorrubicina tales como epirrubicina y derivados, enediyenos, epotilonas, etopósido, combretastatina A-4,
fludarabina, 5-fluorouracilo o profármacos de los mismos tales como Xeloda comercializado por Roche, hidroxiurea, hidroxiureapentostatina, maitansinas, 6-mercaptopurina, metotrexato, mitomicina, mitoxantrona, agentes que contienen platino que incluyen, pero no se limitan a, carboplatino y cisplatino, prednisona, procarbazina, taxanos que incluyen, pero no se limitan a, docetaxel y paclitaxel, tedanólidos, tenipósido, 6-tioguanina, topotecán y alcaloides de la vinca que incluyen, pero no se limitan a, vinblastina y vincristina.
Otros agentes antineoplásicos que pueden ser incorporados en o liberados desde los agentes antineoplásicos protegidos incluyen, pero no se limitan a, agentes antiangiogénicos, agentes alquilantes, antimetabolitos, perturbadores de la polimerización de la microtubulina (por ejemplo, taxol), ciertos productos naturales, complejos de coordinación con platino, antracenedionas, ureas sustituidas, derivados de metilhidrazina, supresores adrenocorticales, ciertas hormonas y antagonistas, polisacáridos antineoplásicos y ciertas hierbas medicinales u otros extractos vegetales.
En una versión, los agentes antineoplásicos que pueden ser protegidos mediante el uso de los activadores hipóxicos y de los grupos conectores descritos en el presente documento son la clase de antibióticos citotóxicos conocidos como antraciclinas. Las antraciclinas incluyen, pero no se limitan a, aclarrubicina, daunorrubicina, doxorrubicina, epirrubicina, idarrubicina, mitoxantrona, pirarrubicina y valrrubicina y cualquier análogo de las anteriores. Los análogos de la antraciclina son bien conocidos en la materia y están incluidos en la clase de las antraciclinas que pueden ser protegidas mediante el uso de los activadores hipóxicos y de los grupos conectores descritos en el presente documento.
Otros agentes antineoplásicos que pueden ser incorporados en o liberados desde los agentes antineoplásicos protegidos incluyen los análogos de cualquiera de los anteriores agentes descritos en esta sección. Con su conocimiento de la materia y las enseñanzas de esta patente, un experto en la materia comprenderá cómo identificar y sintetizar los análogos de los anteriores y cómo proteger los análogos de los anteriores para producir un agente antineoplásico protegido según se describe en esta patente.
Unión del agente antineoplásico al grupo conector o al activador hipóxico: el agente antineoplásico está unido al activador hipóxico (Hip) bien directamente o bien a través de un grupo conector (L); es decir, la estructura del agente antineoplásico protegido es Hip-N o Hip-L-N. Generalmente, el activador hipóxico o el grupo conector puede estar unido a cualquier fracción del agente antineoplásico susceptible de unión y que proporcione un profármaco del agente antineoplásico.
En una versión, el activador hipóxico o el grupo conector está unido a un grupo hidroxilo o amino del agente antineoplásico. En una versión, el hidrógeno del grupo hidroxilo y uno o más de los sustituyentes del grupo amino están sustituidos con un enlace a una fracción del activador hipóxico o del grupo conector. De esta forma, el agente antineoplásico puede estar unido al activador hipóxico o al grupo conector a través de una diversidad de grupos que incluyen, pero no se limitan a, éteres, carbamatos, carbonatos ésteres, acetales, amidas y aminas. En una versión, el activador hipóxico o el grupo conector está unido al agente antineoplásico a través de un éter, de una amina o de un carbamato.
El agente antineoplásico N puede estar representado como N'-Z, donde Z es -OH o -NRaRb y N' representa el resto del agente antineoplásico. Ra y Rb son tales que el - NRaRb es un grupo amino primario, secundario o terciario. Generalmente, -NRaRb es cualquier grupo de tipo amino capaz de unirse al activador hipóxico o al grupo conector mediante la sustitución de uno o de ambos Ra y Rb. En una versión, Z es -NH2. En una versión, Z en -NHRa.
En una versión, Z es un hidroxilo y el hidroxilo está unido a un grupo aromático del agente antineoplásico. En una versión, el grupo aromático al que está unido el hidroxilo es un fenilo o un fenilo sustituido. El fenilo sustituido puede ser parte de un sistema anular condensado.
En una versión, después de la protección mediante el activador hipóxico o el grupo conector, el agente antineoplásico está unido al activador hipóxico o al grupo conector de una de las siguientes formas


(éster)

En una versión, después de la protección mediante el activador hipóxico o un grupo conector, el agente antineoplásico es unido al activador hipóxico o al grupo conector a través bien del éter o bien del conector de carbamato, según se muestra más arriba.
Además de los nitrógenos de la amina, pueden protegerse otros nitrógenos de los agentes antineoplásicos mediante el uso de los activadores hipóxicos y de los grupos conectores descritos en esta patente. Algunos ejemplos de grupos con nitrógeno que pueden ser protegidos incluyen, pero no se limitan a, grupos amida, aminas heterocíclicas (que incluyen, pero no se limitan a, indoles, imidazoles y bencimidazoles) y el nitrógeno de una isoquinolina. En el caso de las amidas, en una versión, el grupo unido directamente al nitrógeno del agente antineoplásico no es un carbamato.
En una versión, el agente antineoplásico
Procedimientos de tratamiento mediante el uso de los agentes antineoplásicos protegidos
Los agentes antineoplásicos protegidos pueden usarse en los procedimientos para el tratamiento del cáncer. En dichos procedimientos, se administra una cantidad eficaz de un agente antineoplásico protegido al sujeto. Generalmente, el sujeto puede ser cualquier mamífero humano o no humano. El sujeto preferido que es un sujeto humano. Otros sujetos en particular incluyen, pero no se limitan a, primates no humanos, perros, gatos, animales de granja, caballos. En una versión, el agente antineoplásico protegido es administrado solo. En una versión, el agente antineoplásico protegido es administrado junto con uno o más agentes anticancerosos adicionales. En una versión, el agente antineoplásico protegido es administrado junto con un tratamiento terapéutico del cáncer que incluye, pero no se limita a, cirugía y radiación. El agente antineoplásico protegido normalmente ser administrado en una composición farmacéutica. Varias de las composiciones farmacéuticas que pueden usarse se describen en la sección de formulaciones de esta patente.
Los agentes antineoplásicos protegidos y sus composiciones farmacéuticas pueden usarse para el tratamiento de cualquier tipo de cáncer en un sujeto, particularmente en un sujeto humano. Los cánceres que pueden tratarse incluyen, pero no se limitan a, leucemia, cáncer de mama, cáncer de piel, cáncer óseo, cáncer de hígado, cáncer cerebral, cáncer de la laringe, de la vesícula biliar, del páncreas, del recto, de la paratiroides, de la tiroides, adrenal, del tejido neural, de cabeza y cuello, de estómago, de bronquios, de riñón, carcinoma de células basales, carcinoma epidermoide tanto del tipo ulcerativo como papilar, carcinoma de piel metastático, osteosarcoma, sarcoma de Ewing, reticulosarcoma, mieloma, tumor de células gigantes, tumor de pulmón microcítico, cálculos biliares, tumor de los islotes, tumor cerebral primario, tumores linfocíticos y granulocíticos agudos y crónicos, tumor de tricoleucocitos, adenoma, hiperplasia, carcinoma medular, feocromocitoma, neuronmas mucosales, ganglioneuromas intestinales, tumor hiperplásico del nervio corneal, tumor de constitución marfanoide, tumor de Wilm, seminoma, tumor leiomiomata, displasia cervical y carcinoma in situ, neuroblastoma, retinoblastoma, sarcoma del tejido blando, carcinoide maligno, lesión cutánea tópica, micosis fungoide, rabdomiosarcoma, sarcoma de Kaposi, sarcoma osteogénico y otros, hipercalcemia maligna, tumor de las células renales, policitermia vera, adenocarcinoma,
glioblastoma multiforme, leucemias, linternas, melanomas malignos y carcinomas epidermoides.
Los agentes antineoplásicos protegidos pueden usarse particularmente en el tratamiento de cánceres que contienen áreas significativas de tejido hipóxico. Dichos cánceres incluyen, pero no se limitan a, cáncer de pulmón, especialmente cáncer de pulmón no microcítico, cáncer de mama, cáncer de colon, cáncer de cabeza y cuello, cáncer de ovario, cáncer de páncreas y cáncer de próstata. Muchos de estos cánceres son analizados con fines ilustrativos a continuación. Los expertos en la materia apreciarán que la quimioterapia oncológica a menudo implica la administración simultánea o sucesiva de una diversidad de agentes anticancerosos y, según se analiza adicionalmente a continuación, los agentes antineoplásicos protegidos pueden usarse en terapias de combinación según se proporciona mediante los procedimientos descritos en el presente documento. Por lo tanto, en la descripción de los cánceres ilustrativos que contienen regiones hipóxicas susceptibles de un tratamiento con los agentes antineoplásicos protegidos, también se describen algunas terapias de combinación ilustrativas.
El cáncer de pulmón afecta a más de 100.000 varones y 50.000 mujeres en los estados Unidos, la mayoría de los cuales muere en el primer año del diagnóstico, haciendo que sea la principal causa de muerte por cáncer. Los protocolos actuales para el tratamiento del cáncer de pulmón implican la integración de la quimioterapia con o sin radioterapia o cirugía. Los agentes antineoplásicos protegidos, incluyendo aquellos que liberan los agentes quimioterapéuticos usados actualmente para el tratamiento de diversas formas del cáncer de pulmón, pueden usarse para el tratamiento del cáncer de pulmón, por ejemplo, mediante la sustitución de una forma no activada por hipoxia en la combinación, y pueden usarse otros agentes antineoplásicos protegidos en las terapias de combinación existentes. Se ha notificado una diversidad de regímenes quimioterapéuticos de combinación para el cáncer de pulmón microcítico, que incluyen las combinaciones que consisten en ciclofosfamida, doxorrubicina y vincristina (CAV); etopósido y cisplatino (VP-16); y ciclofosfamida, doxorrubicina y VP-16 (CAVP-16). Se han notificado unos beneficios de supervivencia modestos con el tratamiento con quimioterapias de combinación (etopósido más cisplatino) para el cáncer de pulmón no microcítico. Los agentes antineoplásicos protegidos descritos en esta patente pueden estar basados en cada uno de los agentes quimioterapéuticos enumerados más arriba.
Asimismo, diversos fármacos citotóxicos diferentes han producido una regresión al menos temporal del cáncer de ovario. Los fármacos más activos en el tratamiento del ovario han sido los agentes alquilantes, que incluyen ciclofosfamida, ifosfamida, melfalano, clorambucilo, tiotepa, cisplatino y carboplatino. Las terapias de combinación actuales para el cáncer de ovario incluyen cisplatino o carboplatino junto con ciclofosfamida a unos intervalos de entre 3 y 4 semanas durante entre seis y ocho ciclos. Los compuestos y los procedimientos descritos en el presente documento proporcionan formas de profármaco de cada uno de estos agentes y procedimientos para el tratamiento del cáncer de ovario en los que se usa un agente antineoplásico protegido según se describe en el presente documento en dichas combinaciones, bien para sustituir un agente o bien además del (los) agente(s) usado(s) actualmente.
El cáncer de próstata es la neoplasia más habitual en los hombres en los Estados Unidos y es la segunda causa más habitual de muerte por cáncer en los hombres por encima de la edad de 55, y se ha notificado que este cáncer consiste principalmente en tejido hipóxico. Se han notificado diversos protocolos de quimioterapia para su uso en la fase tardía de la enfermedad después de una recaída tras un tratamiento hormonal. Algunos agentes para el tratamiento del cáncer de próstata incluyen fosfato de estramustina, prednimustina y cisplatino, y las formas de profármaco de cada uno de estos agentes son proporcionadas por los compuestos y los procedimientos descritos en el presente documento, así como los procedimientos para el tratamiento del cáncer de próstata mediante el uso de dichos agentes. También se usa la quimioterapia de combinación para el tratamiento del cáncer de próstata, que incluye el tratamiento con fosfato de estramustina más prednimustina y cisplatino y 5-fluorouracilo, melfalano e hidroxiurea. Los compuestos y los procedimientos descritos en el presente documento proporcionan las formas de profármaco de cada uno de estos agentes y los procedimientos para el tratamiento del cáncer de próstata en los que se usa un agente antineoplásico protegido en dichas combinaciones, bien para sustituir un agente o bien además del (los) agente(s) usado(s) actualmente.
El cáncer de intestino grueso es la segunda causa más habitual de muerte por cáncer en los Estados Unidos, y asimismo es un cáncer caracterizado por regiones hipóxicas. Mientras que la quimioterapia en los pacientes con un cáncer colorrectal avanzado ha demostrado tener únicamente un beneficio mínimo, el 5-fluorouracilo es el tratamiento más eficaz para esta enfermedad. El 5-fluorouracilo es útil tanto solo como junto con otros fármacos, pero está asociado con únicamente una probabilidad de entre el 15 y el 20 por ciento de reducción en las masas tumorales medibles en un 50 por ciento o más. Por lo tanto, la forma de profármaco activada por hipoxia del 5-FU mediante el uso de los compuestos y de los procedimientos descritos en el presente documento y de los procedimientos para el tratamiento del cáncer de colon mediante el uso de ese profármaco, ofrece un beneficio terapéutico significativo y un potencial para satisfacer la necesidad no satisfecha de unos mejores procedimientos de tratamiento para esta enfermedad.
En una versión de los procedimientos de tratamiento, los agentes antineoplásicos protegidos pueden ser usados en varias metodologías conocidas en la terapia oncológica que incluyen, pero no se limitan a, “terapia con profármacos enzimáticos dirigidos a anticuerpos” (ADEPT), “terapia con profármacos enzimáticos dirigidos a virus (VDEPT),
“terapia con profármacos enzimáticos dirigidos a genes” (GDEPT) y “terapia con profármacos enzimáticos dirigidos a bacterias” (BDEPT). Los usos generales de los agentes antineoplásicos protegidos no están limitados a los anteriores procedimientos de tratamiento.
Formulaciones, modos de administración, dosis, etc.
Los agentes antineoplásicos protegidos normalmente estarán formulados en forma de formulaciones farmacéuticas para su administración a un sujeto. En esta sección se describen los modos de administración, las formulaciones y las dosis que pueden usarse cuando se tratan cánceres mediante el uso de los agentes antineoplásicos protegidos descritos en esta patente.
La administración de los agentes antineoplásicos protegidos para el tratamiento del cáncer puede ser efectuada mediante cualquier procedimiento que permita la administración de los profármacos en el sitio de acción, la región hipóxica de un tumor. Muchos fármacos oncológicos son administrados mediante una inyección intravenosa, y el agente antineoplásico protegido puede estar formulado para dicha administración, lo que incluye no solo formulaciones listas para su inyección sino también formulaciones liofilizadas o concentradas que deben ser rehidratadas o diluidas, respectivamente, antes de su inyección. Además de estas formulaciones, el agente antineoplásico protegido puede estar formulado para su administración por vía oral, por vía intraduodenal, mediante una inyección parenteral (incluyendo intravenosa, subcutánea, intramuscular, intravascular o infusión), por vía tópica y rectal. Los expertos en la materia reconocerán que el agente antineoplásico protegido puede ser activado por las bacterias del intestino. Si no se desea dicha activación, entonces el profesional puede emplear una vía de administración o una formulación que dé como resultado una absorción del agente antineoplásico protegido antes de su entrada en el intestino grueso o el colon. La vía de administración real y la correspondiente formulación de los agentes antineoplásicos protegidos dependerá del tipo de cáncer que se esté tratando, del agente antineoplásico protegido seleccionado para su administración, de la gravedad del cáncer y de la edad, el peso y el estado del paciente, entre otros factores.
De una forma similar, la cantidad de agente antineoplásico protegido administrada, y por lo tanto la cantidad de agente antineoplásico protegido contenida en la dosis administrada y en el producto que comprende esa dosis, dependerá del sujeto que se va a tratar, de la gravedad del cáncer, de la localización del cáncer, de la velocidad de administración, de la disposición del profármaco (por ejemplo, solubilidad y citotoxicidad), del agente citotóxico liberado por el agente antineoplásico protegido y del juicio del médico descriptor. Sin embargo, una dosis eficaz está normalmente en el intervalo de entre aproximadamente 0,001 y aproximadamente 100 mg por kg de peso corporal, preferentemente de entre aproximadamente 1 y aproximadamente 35 mg/kg/día, en dosis únicas o divididas. Era una persona de 70 kg, esto supondría entre aproximadamente 0,05 y aproximadamente 7 g/día, preferentemente entre aproximadamente 0,2 y aproximadamente 2,5 g/día. En algunos casos, los niveles de dosificación por debajo del límite inferior del intervalo mencionado anteriormente pueden ser más que adecuados, mientras que en otros casos pueden emplearse unas dosis todavía mayores sin causar ningún efecto secundario prejudicial; las dosis mayores también pueden dividirse en varias dosis pequeñas para su administración a lo largo del día.
Una formulación de un agente antineoplásico protegido puede estar, por ejemplo, en una forma adecuada para su administración oral en forma de un comprimido, de una cápsula, de un polvo para píldoras, de una formulación, de una solución y de una suspensión de liberación sostenida; para su inyección parenteral en forma de una solución, de una suspensión o de una emulsión estéril; para su administración tópica en forma de un ungüento o de una crema; y para su administración rectal en forma de un supositorio. Una formulación de un agente antineoplásico protegido puede estar en formas de dosificación unitarias adecuadas para una administración individual de las dosis precisas, y normalmente incluirán un portador o un excipiente farmacéutico convencional.
Algunos portadores farmacéuticos adecuados incluyen diluyentes inertes o agentes de relleno, agua y diversos disolventes orgánicos. Las composiciones farmacéuticas pueden contener, si se desea, ingredientes adicionales tales como saborizantes, aglutinantes, excipientes, y similares. Por lo tanto, para una administración oral, pueden emplearse comprimidos que contienen varios excipientes, tales como ácido cítrico, junto con varios disgregantes, tales como almidón, ácido algínico y ciertos silicatos complejos, y con agentes aglutinantes tales como sacarosa, gelatina y acacia. Adicionalmente, pueden usarse agentes lubricantes tales como estearato de magnesio, lauril sulfato de sodio y talco para la preparación de las formas en comprimidos de las formulaciones de los agentes antineoplásicos protegidos descritos en el presente documento. Pueden emplearse unas composiciones sólidas de un tipo similar en cápsulas de gelatina dura y blanda. Por lo tanto, algunos materiales preferidos incluyen lactosa o azúcar de leche y polietilenglicoles de alto peso molecular. Cuando se desean suspensiones acuosas o elixires para su administración oral, el profármaco de las mismas puede estar combinado con varios agentes edulcorantes o saborizantes, sustancias colorantes o pigmentos, y si se desea, agentes emulsionantes o agentes suspensores, junto con diluyentes tales como agua, etanol, propilenglicol, glicerina o combinaciones de los mismos.
Algunos ejemplos de formas de administración parenteral incluyen soluciones o suspensiones del profármaco activado por hipoxia en soluciones acuosas estériles, por ejemplo, propilenglicol acuoso o soluciones de dextrosa. Dichas formas de dosificación pueden estar adecuadamente tamponadas, si se desea.
Los procedimientos para la preparación de las diversas composiciones farmacéuticas con una cantidad específica de fármaco activo son conocidos, o serán evidentes, para los expertos en esta materia en vista de esta divulgación. Para algunos ejemplos, véase Remington's Pharmaceutical Sciences, Mack Publishing Company, Philadelphia, Pa., 17a Edición (1984).
Procedimientos de elaboración de los agentes antineoplásicos protegidos
Los agentes antineoplásicos protegidos descritos en esta patente pueden ser elaborados mediante una diversidad de procedimientos. Dados los procedimientos sintéticos descritos en los ejemplos, a continuación, y su conocimiento de la química medicinal sintética, un experto en la materia será capaz de sintetizar los agentes antineoplásicos protegidos de una forma directa.
Terapias de combinación
En una versión del procedimiento para el tratamiento del cáncer mediante el uso de los agentes antineoplásicos protegidos, se administra un agente antineoplásico protegido junto con una cantidad eficaz de uno o más agentes quimioterapéuticos, una cantidad eficaz de radioterapia, un procedimiento quirúrgico apropiado o cualquier combinación de dichas terapias adicionales.
Cuando un agente antineoplásico protegido se usa junto con una o más de las terapias adicionales, el agente antineoplásico protegido y a la terapia adicional pueden ser administrados al mismo tiempo o pueden ser administrados por separado. Por ejemplo, si un agente antineoplásico protegido es administrado con un agente quimioterapéutico adicional, los dos agentes pueden ser administrados simultáneamente o pueden ser administrados secuencialmente con algún tiempo entre las administraciones. Un experto en la materia comprenderá los procedimientos de administración de los agentes simultáneamente y secuencialmente y los posibles periodos de tiempo entre las administraciones.
Los agentes pueden ser administrados en la misma o en diferentes formulaciones, y pueden ser administrados a través de la misma o de diferentes vías.
Algunos agentes quimioterapéuticos que pueden usarse junto con los agentes antineoplásicos protegidos descritos en esta patente incluyen, pero no se limitan a, busulfano, improsulfano, piposulfano, benzodepa, carbocuona, 2-desoxi-D-glucosa, lonidamina, meturedepa, uredepa, altretamina, imatinib, trietilenmelamina, trietilenfosforamida, trietilentiofosforamida, trimetilolomelamina, clorambucilo, clornafazina, estramustina, ifosfamida, mecloretamina, clorhidrato de óxido de mecloretamina, melfalano, novembiquina, fenesterina, prednimustina, trofosfamida, mostaza de uracilo, carmustina, clorozotocina, fotemustina, nimustina, ranimustina, dacarbazina, manomustina, mitobronitol, mitolactol, pipobromano, aclacinomicinas, actinomicina F(1), antramicina, azaserina, bleomicina, cactinomicina, carubicina, carzinofilina, cromomicina, dactinomicina, daunorrubicina, daunomicina, 6-diazo-5-oxo-1-norleucina, ácido micofenólico, nogalamicina, olivomicina, peplomicina, plicamicina, porfiromicina, puromicina, estreptonigrina, estreptozocina, tubercidina, ubenimex, zinostatina, zorrubicina, denopterina, pteropterina, trimetrexato, fludarabina, 6-mercaptopurina, tiamiprina, tioguanina, ancitabina, azacitidina, 6-azauridina, carmofur, citarabina, didesoxiuridina, doxifluridina, enocitabina, floxuridina, 5-fluorouracilo, tegafur, L-asparraginasa, pulmozima, aceglatona, glicósido de aldofosfamida, ácido aminolevulínico, amsacrina, bestrabucilo, bisantreno, carboplatino, defofamida, demecolcina, diazicuona, elfornitina, acetato de eliptinio, etoglucida, flutamida, nitrato de galio, hidroxiurea, interferón alfa, interferón beta, interferón gamma, interleucina-2, lentinano, mitoguazona, mitoxantrona, mopidamol, nitracrina, pentostatina, fenamet, pirarrubicina, ácido podofilínico, 2-etil-hidrazida, procarbazina, razoxano, sizofirano, espirogermanio, paclitaxel, tamoxifeno, tenipósido, ácido tenuazonico, triazicuona, 2,2',2”-triclorotrietilamina, uretano, vinblastina, ciclofosfamida y vincristina. Otros agentes quimioterapéuticos que pueden usarse incluyen derivados de platino, que incluyen, pero no se limitan a, cisplatino, carboplatino y oxoplatino.
En una versión, los agentes antineoplásicos protegidos descritos en esta patente pueden usarse junto con un inhibidor de la antiangeogenisis que incluye, pero no se limita a, Avastin y algunos productos terapéuticos similares. En una versión de los procedimientos de tratamiento de combinación, se trata un sujeto con un inhibidor de la antiangeogenisis y posteriormente se trata con un agente antineoplásico protegido. En una versión de los procedimientos de tratamiento de combinación, un sujeto se trata con un inhibidor de la antiangeogenisis y posteriormente se trata con un agente antineoplásico protegido con otro agente quimioterapéutico, que incluye, pero no se limita a, cisplatino. En una versión de esta combinación de procedimientos de tratamiento mediante el uso de un inhibidor de la antiangeogenisis, el procedimiento se usa para el tratamiento del cáncer de mama.
En otra versión, un agente antineoplásico protegido es administrado con un agente antineoplásico que actúa bien directamente o bien indirectamente inhibiendo el factor inducible por hipoxia 1 alfa (HIF1a) o inhibiendo una proteína o una enzima, tales como un transportador de glucosa o el VEGF, cuya expresión o actividad está aumentada después del aumento de los niveles del HIF1a. Los inhibidores del HIF1a adecuados para su uso en esta versión de los procedimientos y de las composiciones descritas en el presente documento incluyen los inhibidores de la cinasa P13; LY294002; rapamicina; los inhibidores de la desacetilasa de histona tales como [(E)-(1S,4S,10S,21R)-7-[(Z)-etiliden]-4,21-diisopropil-2-oxa-12,13-ditia-5,8,20,23-tetraazabiciclo-[8,7,6]-tricos-16-eno-3,6,9,19,22-pentanona
(documento FR901228, depsipéptido); los inhibidores de la proteína de choque térmico 90 (Hsp90) tales como geldanamicina, 17-alilamino-geldanamicina (17-AAG) y otros análogos de la geldanamicina y radicicol y los derivados del radicicol tales como KF58333; genisteína; indanona; estaurosporina; los inhibidores de la cinasa de proteína 1 (MEK-1) tales como PD98059 (2'-amino-3'-metoxiflavona); PX-12 (disulfuro de 1-metilpropil 2-imidazolilo); pleurotina PX-478; 1,4-dióxidos de quinoxalina; butirato de sodio (NaB); nitropurrusiato de sodio (SNP) y otros donantes de NO; los inhibidores de los microtúbulos tales como novobiocina, panzem (2-metoxiestradiol o 2-ME2), vincristinas, taxanos, epotilonas, discodermólido y derivados de cualquiera de los anteriores; cumarinas; barbitúricos y los análogos del ácido tiobarbitúrico; camptotecinas; y el YC-1, un compuesto descrito en Biochem. Pharmacol., 15 de abril de 2001,61 (8): 947-954 y sus derivados.
En otra versión, un agente antineoplásico protegido es administrado con un agente antiangiogénico, que incluye, pero no se limita a, los agentes antiangiogénicos seleccionados entre el grupo que consiste en angiostatina, un agente que inhibe o que antagoniza de otro modo la acción del VEGF, batimastat, captoprilo, inhibidor derivado del cartílago, genisteína, endostatina, interleucina, lavendustina A, acetato de medroxipregesterona, factor de plaquetas humano recombinante 4, taxol, tecogalano, talidomida, trombospondina, TNP-470 y Avastin. Otros inhibidores de la angiogénesis útiles para los fines de las terapias de combinación proporcionados por los presentes procedimientos y composiciones descritos en el presente documento incluyen los inhibidores de la Cox-2 como celecoxib (Celebrex), diclofenaco (Voltaren), etodolaco (Lodine), fenoprofeno (Nalfon), indometacina (Indocin), ketoprofeno (Orudis, Oruvailo), ketoralaco (Toradol), oxaprozino (Daypro), nabumetona (Relaten), sulindaco (Clinorilo), tolmetina (Tolectin), rofecoxib (Vioxx), ibuprofeno (Advilo), naproxeno (Aleve, Naprosyn), ácido acetilsalicílico y paracetamol (Tylenol). Además, debido a que el ácido pirúvico juega un papel importante en la angiogénesis, pueden usarse miméticos del piruvato y de los inhibidores glicolíticos como los halopiruvatos, que incluyen el bromopiruvato, junto con un compuesto antiangiogénico y un agente antineoplásico protegido para el tratamiento del cáncer. En otra versión, un agente antineoplásico protegido es administrado con un agente antiangiogénico y otro agente antineoplásico que incluye, pero no se limita a, un agente citotóxico seleccionado entre el grupo que consiste en alquilantes, cisplatino, carboplatino y los inhibidores del ensamblaje de los microtúbulos, para el tratamiento del cáncer.
Además de la combinación de un agente antineoplásico protegido con los agentes descritos más arriba, los presentes procedimientos y composiciones descritos en el presente documento proporcionan una diversidad de combinaciones sinérgicas de un agente antineoplásico protegido y de otros fármacos antineoplásicos. Los expertos en la materia pueden determinar con facilidad los fármacos antineoplásicos que actúan “sinérgicamente” con un agente antineoplásico protegido según se describe en el presente documento. Por ejemplo, las referencias Vendetti, “Relevance of Transplantable Animal-Tumor Systems to the Selection of New Agents for Clinical Trial”, Pharmacological Basis of Cancer Chemotherapy, Williams y Wilkins, Baltimore, 1975, y Simpson Herren y col., 1985, “Evaluation of In Vivo Tumor Models for Predicting Clinical Activity for Anticancer Drugs”, Proc. Am. Assoc. Cancer Res. 26: 330, describen procedimientos para ayudar a determinar qué dos fármacos actúan sinérgicamente. Aunque la sinergia no es necesaria para el beneficio terapéutico según los procedimientos descritos en el presente documento, la sinergia puede mejorar el resultado terapéutico. Puede decirse que dos fármacos poseen sinergia terapéutica si un régimen de dosificación de combinación de los dos fármacos produce una destrucción de las células tumorales significativamente mejor que la suma de los agentes individuales a unas dosis toleradas óptimas o máximas. El “grado de sinergia” puede definirse como el log neto de la destrucción de las células tumorales con el régimen de combinación óptima menos el log neto de la destrucción de las células tumorales con la dosis óptima del agente individual más activo. Se considera que unas diferencias en la destrucción celular mayores de diez veces (un log) son concluyentemente indicativas de una sinergia terapéutica.
Cuando un agente antineoplásico protegido se usa con otro agente antineoplásico, un agente antineoplásico protegido se administrará, al menos en algunas versiones, antes del inicio de la terapia con el otro fármaco o fármacos, y la administración se continuará normalmente a lo largo del transcurso del tratamiento con el otro fármaco o fármacos. En algunas versiones, el fármaco administrado conjuntamente con un agente antineoplásico protegido ser administrado a una dosis más baja, y opcionalmente durante periodos de tiempos más largos de lo que sería el caso en ausencia de la administración de un agente antineoplásico protegido. Dichas terapias a una “dosis baja” pueden implicar, por ejemplo, la administración de un fármaco antineoplásico que incluye, pero no se limita a, paclitaxel, docetaxel, doxorrubicina, cisplatino o carboplatino, a una dosis menor que la aprobada y durante un periodo de tiempo más largo junto con un agente antineoplásico protegido administrado según los procedimientos descritos en el presente documento. Estos procedimientos pueden usarse para mejorar los resultados del paciente con respecto a las terapias realizadas actualmente mediante una destrucción más eficaz de las células cancerosas o una detención del crecimiento de las células cancerosas, así como la disminución de los efectos secundarios indeseados de la otra terapia. En otras versiones, el otro agente o agentes antineoplásicos serán administrados a los mismos niveles de dosificación usados cuando no se administra conjuntamente un agente antineoplásico protegido. Por lo tanto, cuando se emplea junto con un agente antineoplásico protegido, el (los) agente(s) antineoplásico(s) adicional(es) se administra(n) mediante el uso de cualquiera de las dosis habituales empleadas para esos agentes cuando se usan sin un agente antineoplásico protegido, o son menores que esas dosis habituales. La administración de un agente antineoplásico protegido según los procedimientos descritos en el presente documento puede facilitar por lo tanto al médico el tratamiento del cáncer con unos fármacos existentes (o aprobados posteriormente) a unas dosis más bajas (que las usadas actualmente), mejorando por lo tanto algunos o todos los efectos secundarios
tóxicos de dichos fármacos. La dosis exacta para un paciente dado varía entre paciente y paciente dependiendo de diversos factores que incluyen la combinación de fármacos empleada, la enfermedad en particular que se está tratando y el estado y los antecedentes clínicos del paciente, pero puede ser determinada únicamente mediante el uso de la pericia del artesano experto habitual en vista de las enseñanzas del presente documento.
Los regímenes de dosificación específicos de los agentes antineoplásicos conocidos y aprobados (es decir, la dosis eficaz recomendada) son conocidos por los médicos y se proporcionan, por ejemplo, en las descripciones de producto que se encuentran en el Physician's Desk Reference 2003, (Physicians' Desk Reference, 57a Ed) Medical Economics Company, Inc., Oradell, N.J y/o que están disponibles en la Federal Drug Administration. Algunos regímenes de dosificación ilustrativos para determinados fármacos antineoplásicos también se proporcionan a continuación.
Los fármacos para el cáncer pueden clasificarse de forma general como alquilantes, antraciclinas, antibióticos, inhibidores de la aromatasa, bisfosfonatos, inhibidores de la ciclooxigenasa, moderadores del receptor de estrógenos, antagonistas del folato, aresenatos inorgánicos, inhibidores de los microtúbulos, modificadores, nitrosoureas, análogos de nucleósido, inhibidores de los osteoclastos, compuestos que contienen platino, retinoides, inhibidores de la topoisomerasa 1, inhibidores de la topoisomerasa 2 e inhibidores de la cinasa de tirosina. Según los procedimientos descritos en el presente documento, un agente antineoplásico protegido puede ser administrado conjuntamente con cualquier fármaco antineoplásico de cualquiera de estas clases, o puede ser administrado antes o después del tratamiento con cualquiera de dichos fármacos o combinación de dichos fármacos. Además, un agente antineoplásico protegido puede ser administrado junto con una terapia biológica (por ejemplo, un tratamiento con interferones, interleucinas, factores estimulantes de colonias y anticuerpos monoclonales). Los productos biológicos usados para el tratamiento del cáncer son conocidos en la materia e incluyen, por ejemplo, trastuzumab (Herceptin), tositumomab y 1311 tositumomab (Bexxar), rituximab (Rituxan). En una versión, sin embargo, el fármaco antineoplásico administrado conjuntamente con un agente antineoplásico protegido no es un inhibidor de la topoisomerasa.
Algunos alquilantes útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, busulfano (Myleran, Busulfex), clorambucilo (Leuceran), ifosfamida (con o sin MESNA), ciclofosfamida (Cytoxan, Neosar), glufosfamida, melfalano, L-PAM (Alkeran), dacarbazina (DTIC-Dome) y temozolamida (Temodar). Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con un alquilante para el tratamiento del cáncer. En una versión, el cáncer es leucemia mielógena crónica, mieloma múltiple o astrocitoma anaplásico. Como un ejemplo, el compuesto 2-bis[(2-cloroetil)amino] tetrahidro-2H-1,3,2-oxazafosforina, 2-óxido, también conocido habitualmente como ciclofosfamida, es un alquilante usado en el tratamiento de linfomas malignos en las fases III y IV, del mieloma múltiple, de la leucemia, de micosis fungoides, del neuroblastoma, del adenocarcinoma de ovario, del retinoblastoma y del carcinoma de mama. La ciclofosfamida es administrada para una terapia de inducción en unas dosis de 1.500-1.800 mg/m2 que se administran por vía intravenosa en dosis divididas durante un periodo de entre tres y cinco días; para una terapia de mantenimiento, se administran 350-550 mg/m2 cada 7-10 días, o se administran 110-185 mg/m2 por vía intravenosa dos veces por semana. Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con ciclosfosfamida administrada a esas dosis o a unas dosis más bajas y/o durante una duración más larga de lo normal que para la administración de ciclosfosfamida sola.
Algunas antraciclinas útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, doxorrubicina (Adriamicina, Doxilo, Rubex), mitoxantrona (Novantrona), idarrubicina (Idamicina), valrrubicina (Valstar) y epirrubicina (Ellence). Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con una antraciclina para el tratamiento del cáncer. En una versión, el cáncer es una leucemia no linfocítica aguda, sarcoma de Kaposi, cáncer de próstata, cáncer de vejiga, carcinoma metastásico de ovario y cáncer de mama. Como un ejemplo, el compuesto (8S,10S)-10-[(3-amino-2,3,6-tridesoxi-.alfa.-L-lixo-hexopiranosil)oxi]-8-glicoloil-7,8,9,10-tetrahidro-6,8,11-trihidroxi-1-metoxi-5,12-naftacenediona, más conocido habitualmente como doxorrubicina, es un antibiótico de antraciclina citotóxico aislado a partir de cultivos de Streptomyces peucetius var. caesius. La doxorrubicina se ha usado con éxito para producir la regresión en afecciones neoplásicas diseminadas tales como leucemia linfoblástica aguda, leucemia mieloblástica aguda, tumor de Wilm, neuroblastoma, sarcomas de tejidos blandos y de hueso, carcinoma de mama, carcinoma de ovario, carcinoma de las células transicionales de la vejiga, carcinoma tiroideo, linfomas tanto del tipo Hodgkin como del tipo no Hodgkin, carcinoma broncogénico y carcinoma gástrico. La doxorrubicina se administra normalmente en una dosis en el intervalo de 30-75 mg/m2 en forma de una inyección intravenosa única administrada a intervalos de 21 días; una inyección intravenosa semanal a unas dosis de 20 mg/m2; o a unas dosis de 30 mg/m2 durante tres días sucesivos repetidos cada cuatro semanas. Según los procedimientos de los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente comenzando antes y continuando después de la administración de la doxorrubicina a dichas dosis (o a unas dosis más bajas).
Algunos antibióticos útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, dactinomicina, actinomicina D (Cosmegen), bleomicina (Blenoxano), daunorrubicina y daunomicina (Cerubidine, DanuoXome). Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con un antibiótico para el tratamiento del cáncer. En una versión, el cáncer
es un cáncer seleccionado entre el grupo que consiste en leucemia linfocítica aguda, otras leucemias y sarcoma de Kaposi.
Algunos inhibidores de la aromatasa útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, anastrozol (Arimidex) y letroazol (Femara). Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con un inhibidor de la aromatasa para el tratamiento del cáncer. En una versión, el cáncer es un cáncer de mama.
Algunos inhibidores de bisfosfonato útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, zoledronato (Zometa). Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con un inhibidor de bifosfonato para el tratamiento del cáncer. En una versión, el cáncer es un cáncer seleccionado entre el grupo que consiste en mieloma múltiple, metástasis óseas de tumores sólidos o cáncer de próstata.
Algunos inhibidores de la ciclooxigenasa útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, celecoxib (Celebrex). Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con un inhibidor de la ciclooxigenasa para el tratamiento del cáncer. En una versión, el cáncer es un cáncer de colon o un estado precanceroso conocido como poliposis adenomatosa familiar.
Algunos moduladores del receptor de estrógenos útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, tamoxifeno (Nolvadex) y fulvestrant (Faslodex). Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con un modulador del receptor de estrógenos para el tratamiento del cáncer. En una versión, el cáncer es un cáncer de mama o el tratamiento se administra para prevenir la aparición o la reaparición del cáncer de mama.
Algunos antagonistas del folato útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, metotrexato y tremetrexato. Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con un antagonista del folato para el tratamiento del cáncer. En una versión, el cáncer es un osteosarcoma. Como un ejemplo, el compuesto ácido N-[4-[[(2,4-diamino-6-pteridinil)metil metilamino]benzoil]-L-glutámico, conocido habitualmente como metotrexato, es un fármaco antifolato que se ha usado en el tratamiento del coriocarcinoma gestacional y en el tratamiento de pacientes con mola invasiva y mola hidatiforme. También es útil en el tratamiento de las fases avanzadas del linfoma maligno y en el tratamiento de los casos avanzados de micosis fungoides. El metotrexato se administra como sigue. Para el coriocarcinoma, se administran inyecciones intramusculares de unas dosis de entre 15 y 30 mg diariamente durante el transcurso de cinco días, dichas tandas se repiten según sea necesario con un periodo de reposo de una o más semanas interpuesto entre las tandas de terapia. Para las leucemias, se administran inyecciones intramusculares dos veces por semana en unas dosis de 30 mg/m2. Para la micosis fungoides, se administran inyecciones intramusculares semanales de unas dosis de 50 mg o, como alternativa, de 25 mg dos veces por semana. Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con metotrexato administrado a dichas dosis (o a unas dosis más bajas). La 5-metil-6-[[(3,4,5-trimetoxifenoil)-amino]metil]-2,4-quinazolindiamina (conocida habitualmente como trimetrexato) es otro fármaco antifolato que puede ser administrado conjuntamente con un agente antineoplásico protegido.
Algunos arsenatos inorgánicos útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, trióxido de arsénico (Trisenox). Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con un arsenato inorgánico para el tratamiento del cáncer. En una versión, el cáncer es una leucemia promielocítica aguda resistente (APL).
Algunos inhibidores de los microtúbulos (según se usa en el presente documento, un “inhibidor de los microtúbulos” es cualquier agente que interfiere en el ensamblaje o el desensamblaje de los microtúbulos) útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, vincristina (Oncovin), vinblastina (Velban), paclitaxel (Taxol, Paxene), vinorelbina (Navelbina), docetaxel (Taxotere), epotilona B o D o un derivado de cualquiera, y discodermolida o sus derivados. Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con un inhibidor de los microtúbulos para el tratamiento del cáncer. En una versión, el cáncer es un cáncer de ovario, un cáncer de mama, un cáncer de pulmón no microcítico, un sarcoma de Kaposi y un cáncer metastásico de origen mamario u ovárico. Como un ejemplo, el compuesto 22-oxo-vincaleucoblastina, también conocido habitualmente como vincristina, es un alcaloide que se obtiene a partir de la planta común vinca rosada (Vinca rosea, Linn.) y es útil en el tratamiento de la leucemia aguda. También se ha demostrado que es útil junto con otros agentes oncolíticos en el tratamiento de la enfermedad de Hodgkin, del linfosarcoma, del reticulosarcoma, del rabdomiosarcoma, del neuroblastoma y del tumor de Wilm. La vincristina se administra semanalmente a unas dosis intravenosas de 2 mg/m2 en niños y de 1,4 mg/m2 en adultos. Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con vincristina administrada a dichas dosis. En una versión, no se administra un agente antineoplásico protegido antes del tratamiento con un inhibidor de los microtúbulos, tales como un taxano, sino más bien, la administración de un agente antineoplásico protegido es administrada simultáneamente con o entre unos
pocos días y una semana después del inicio del tratamiento con un inhibidor de los microtúbulos.
Algunos modificadores útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, leucovorina (Wellcovorin), que se usa con otros fármacos tales como 5-fluorouracilo para el tratamiento del cáncer colorrectal. Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con un modificador y otro agente antineoplásico para el tratamiento del cáncer. En una versión, el cáncer es un cáncer de colon. En una versión, el modificador es un compuesto que aumenta la capacidad de una célula para captar la glucosa, que incluye, pero no se limita a, el compuesto N-hidroxiurea. Se ha notificado que la N-hidroxiurea mejora la capacidad de una célula para captar 2-desoxiglucosa (véase la referencia Smith y col., 1999, Cancer Letters 141: 85) y la administración de N-hidroxiurea a unos niveles que se notifica que aumentan la captación de un agente antineoplásico protegido o para el tratamiento leucemia junto con la administración de un agente antineoplásico protegido según se describe en el presente documento es una versión de los procedimientos terapéuticos proporcionados en el presente documento. En otra de dichas versiones, se administra un agente antineoplásico protegido conjuntamente con óxido nítrico o con un precursor de óxido nítrico, tal como un nitrito orgánico o un NONOato de espermina, para el tratamiento del cáncer, ya que estos últimos compuestos estimulan la captación de glucosa, y por lo tanto estimulan la captación de un agente antineoplásico protegido.
Algunas nitrosoureas útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, procarbazina (Matulane), lomustina, CCNU (CeeBU), carmustina (BCNU, BiCNU, Gliadel Wafer) y estramustina (Emcit). Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con una nitrosourea para el tratamiento del cáncer. En una versión, el cáncer es un cáncer de próstata o un glioblastoma, lo que incluye el glioblastoma multiforme recurrente.
Algunos análogos de nucleósido útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, mercaptopurina, 6-MP (Purinethol), fluorouracilo, 5-FU (Adrucilo), tioguanina, 6-TG (Tioguanina), hidroxiurea (Hydrea), citarabina (Cytosar-U, DepoCyt), floxuridina (FUDR), fludarabina (Fludara), pentostatina (Nipent), cladribina (Leustatina, 2-CdA), gemcitabina (Gemzar) y capecitabina (Xeloda). Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con un análogo de nucleósido para el tratamiento del cáncer. En una versión, el cáncer es una leucemia linfocítica B (CLL), una tricoleucemia, un adenocarcinoma del páncreas, un cáncer de mama metastásico, un cáncer de pulmón no microcítico o un carcinoma colorrectal metastásico. Como un ejemplo, el compuesto 5-fluoro-2,4(1H,3H)-pirimidinadiona, también conocido habitualmente como 5-fluorouracilo, es un antimetabolito análogo de nucleósido eficaz en el tratamiento paliativo del carcinoma de colon, de recto, de mama, de estómago y de páncreas en pacientes que se consideran incurables con cirugía u otros medios. El 5-fluorouracilo es administrado en una terapia inicial a unas dosis de 12 mg/m2 y administradas por vía intravenosa una vez al día durante 4 días sucesivos, no excediendo la dosis diaria los 800 mg. Si no se observa toxicidad en ningún momento durante el transcurso de la terapia, se administran 6 mg/kg por vía intravenosa los días 6°, 8°, 10° y 12°. No se administra ninguna terapia los días 5°, 7°, 9° ni 11°. En pacientes con poco riesgo o en aquellos que no están en un estado nutricional adecuado, se administra una dosis diaria de 6 mg/kg durante tres días, no excediendo la dosis diaria los 400 mg. Si no se observa toxicidad en ningún momento durante el tratamiento, pueden administrarse 3 mg/kg los días 5°, 7° y 9°. No se administra ninguna terapia los días 4°, 6° ni 8°. Una secuencia de inyecciones sobre cualquier programa constituye una tanda de terapia. Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con 5-FU administrado a dichas dosis dosis o con la forma de profármaco Xeloda con las dosis ajustadas correspondientemente. Como otro ejemplo, el compuesto 2-amino-1,7-dihidro-6H-purina- 6-tiona, también conocido habitualmente como 6-tioguanina, es un análogo de nucleósido eficaz en la terapia de las leucemias no pimfocíticas agudas. La 6-tioguanina se administra por vía oral a unas dosis de aproximadamente 2 mg/kg de peso corporal por día. La dosis diaria total puede ser administrada de una vez. Si después de cuatro semanas de dosis a este nivel no hay ninguna mejora, la dosis puede aumentarse con precaución hasta 3 mg/kg/día. Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con la 6-TG administrada a dichas dosis (o a unas dosis más bajas).
Algunos inhibidores de los osteoclastos útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, pamidronato (Aredia). Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con un inhibidor de los osteoclastos para el tratamiento del cáncer. En una versión, el cáncer son las metástasis óseas osteolíticas del cáncer de mama, y también se administra uno o más agentes anticancerosos adicionales conjuntamente con un agente antineoplásico protegido.
Algunos compuestos de platino útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, cisplatino (Platinol) y carboplatino (Paraplatin). Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con un compuesto de platino para el tratamiento del cáncer. En una versión, el cáncer es un cáncer testicular metastásico, un cáncer de ovario metastásico, un carcinoma de ovario y un cáncer de las células transicionales de la vejiga. Como un ejemplo, el compuesto cis-diaminodicloroplatino (II), conocido habitualmente como cisplatino, es útil en el tratamiento paliativo de tumores testiculares y ováricos metastásicos, y para el tratamiento del cáncer de las células transicionales de la
vejiga que no es abordable con cirugía ni con radioterapia. El cisplatino, cuando se usa para el cáncer de vejiga avanzado, es administrado en inyecciones intravenosas a unas dosis de 50-70 mg/m2 una vez cada entre tres y cuatro semanas. Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con cisplatino administrado a estas dosis (o a unas dosis más bajas). Pueden administrarse uno o más agentes anticancerosos adicionales conjuntamente con el compuesto de platino y un agente antineoplásico protegido. Como un ejemplo, puede administrarse Platinol, Blenoxano y Velbam conjuntamente con un agente antineoplásico protegido. Como otro ejemplo, puede administrarse Platinol y Adriamicina conjuntamente con un agente antineoplásico protegido.
Algunos retinoides útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, tretinoína, ATRA (Vesanoid), alitretinoína (Panretin) y bexaroteno (Targretin). Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con un retinoide para el tratamiento del cáncer. En una versión, el cáncer es un cáncer seleccionado entre el grupo que consiste en APL, sarcoma de Kaposi y linfoma de linfocitos T.
Algunos inhibidores de la topoisomerasa 1 útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, topotecán (Hycamtin) e irinotecán (Camptostar). Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con un inhibidor de la topoisomerasa 1 para el tratamiento del cáncer. En una versión, el cáncer es un carcinoma metastásico de ovario, de colon o de recto, o un cáncer de pulmón microcítico. Sin embargo, como se ha indicado más arriba, en una versión de los procedimientos descritos en el presente documento, la administración de un agente antineoplásico protegido puede preceder o seguir, o ambas, a la administración de un inhibidor de la topoisomerasa 1, pero no se administra junto con el mismo.
Algunos inhibidores de la topoisomerasa 2 útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, etopósido, vP-16 (Vepesid), tenipósido, VM-26 (Vumon) y fosfato de etopósido (Etopophos). Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con un inhibidor de la topoisomerasa 2 para el tratamiento del cáncer. En una versión, el cáncer es un cáncer seleccionado entre el grupo que consiste en tumores testiculares resistentes, la leucemia linfoblástica aguda resistente (ALL) y el cáncer de pulmón microcítico. Sin embargo, como se ha indicado más arriba, en una versión de los procedimientos descritos en el presente documento, la administración de un agente antineoplásico protegido puede preceder o seguir, o ambas, a la administración de un inhibidor de la topoisomerasa 2, pero no se administra junto con el mismo.
Algunos inhibidores de la cinasa de tirosina útiles en la práctica de los procedimientos descritos en el presente documento incluyen, pero no se limitan a, imatinib (Gleevec). Según los procedimientos descritos en el presente documento, se administra un agente antineoplásico protegido conjuntamente con un inhibidor de la cinasa de tirosina para el tratamiento del cáncer. En una versión, el cáncer es una CML o un tumor estromal gastrointestinal maligno metastásico o inoperable.
Por lo tanto, en el presente documento se describen procedimientos para el tratamiento del cáncer en los que se administra un agente antineoplásico protegido o una sal farmacéuticamente aceptable del mismo y uno o más agentes anticancerosos adicionales a un paciente. Algunas versiones específicas de dichos otros agentes anticancerosos incluyen, sin limitación, 5-metil-6-[[(3,4,5-trimetoxifenoil)amino]-metil]-2,4-quinazolinadiamina o una sal farmacéuticamente aceptable de la misma, (8S,10S)-10-(3-amino-2,3,6-trideoxi-alfa-L-lixo-hexopiranosil)oxi]-8-glicoloil-7,8,9,10-tetrahidro-6,8,11-trihidroxi-1-metoxi-5,12-naftacenediona o una sal farmacéuticamente aceptable de la misma; 5-fluoro-2,4(1H,3H)-pirimidinadiona o una sal farmacéuticamente aceptable de la misma; 2-amino-1,7-dihidro-6H-purina-6-tiona o una sal farmacéuticamente aceptable de la misma; 22-oxo-vincaleucoblastina o una sal farmacéuticamente aceptable de la misma; 2-bis[(2-cloroetil)amino]tetrahidro-2H-1,3,2-oxazafosforina, 2-óxido, o una sal farmacéuticamente aceptable del mismo; el ácido N-[4-[[(2,4-diamino-6-pteridinil)metil]-metilamino]benzoil]-L-glutámico, o una sal farmacéuticamente aceptable del mismo; o cis-diaminodicloroplatino (II). Los procedimientos descritos en el presente documento son aplicables de forma general a todos los cánceres, pero tienen un beneficio terapéutico particularmente significativo en el tratamiento de tumores sólidos, que se caracterizan por amplias regiones de tejido hipóxico. Algunos cánceres en particular que pueden ser tratados con los procedimientos descritos en el presente documento se analizan en la siguiente sección.
Ejemplos
Los siguientes ejemplos ilustran varios aspectos de los compuestos, de las composiciones y de los procedimientos descritos en esta patente. Estos ejemplos no pretenden limitar en modo alguno el ámbito de las reivindicaciones. Ejemplo 1
Síntesis de fracciones de un activador por hipoxia basado en nitroimidazol
Este ejemplo ilustra procedimientos para la síntesis de intermedios de una fracción de un activador hipóxico basado
en nitroimidazol útiles en los procedimientos descritos en el presente documento para la síntesis de los agentes antineoplásicos protegidos. Algunos procedimientos de síntesis adicionales se proporcionan en el Ejemplo 7. En la parte A, se proporciona un procedimiento ilustrativo para la síntesis de

N-1-metil-2-nitro-5-hidroximetil imidazol
a partir de clorhidrato de etil sarcosina. El (3-metil-2-nitro-3H-imidazol-4-il)-metanol es un ejemplo de lo que se indica en esta patente como un “alcohol primario de nitroimidazol”. Debido a que los agentes antineoplásicos protegidos que contienen este activador hipóxico pueden ser activados en algunas células incluso en unas condiciones normóxicas debido al ataque en el carbono primario del alcohol por parte de la glutatión-S-transferasa o a través de un mecanismo similar, y debido a que la sustitución adicional de este carbono puede reducir o eliminar dicha activación no deseada, también se describen en esta patente versiones de un alcohol secundario que se denominan “los alcoholes secundarios de nitroimidazol”. La síntesis de dichos alcoholes secundarios de nitroimidazol se presenta en la parte B de este ejemplo.
Parte A. Síntesis de un alcohol primario de nitroimidazol. El siguiente esquema proporciona un procedimiento para la síntesis del alcohol primario de nitroimidazol (el compuesto 2 del esquema) a partir de etil sarcosina. En este esquema, se convierte en primer lugar el clorhidrato de etilsarcosina en clorhidrato de etil-N-formil-C-formil sarcosina; un procedimiento adecuado para dicha conversión se describe en la referencia Jonas, 1949, J. Am. Chem. Soc. 71: 644. El último compuesto se convierte después en un compuesto denominado en este esquema “el éster de nitroimidazol” (el compuesto 1 del esquema); un procedimiento adecuado para dicha conversión se describe en la referencia Asato y col., 1972, J. Med. Chem. 15: 1086. Después, el éster de nitroimidazol se convierte en el alcohol primario de nitroimidazol; un procedimiento adecuado para dicha conversión se describe en Parveen y col., 1999, Bioorg. Med. Chem. Lett. 9: 2031




Todos los ejemplos mostrados a continuación tienen la fracción de nitroimidazol portadora de un grupo metilo en la posición 1. Este grupo metilo puede ser, como alternativa, un grupo alquilo portador de un impedimento estérico u otros grupos según se describe en esta patente. Dichos análogos serían útiles en la reducción de la actividad del grupo nitro en 2 frente a una reducción de dos electrones in vivo por parte de enzimas insensibles al oxígeno tales como la diaforasa DT. Mediante la reducción de la capacidad de los agentes antineoplásicos protegidos de ser activados por enzimas insensibles al oxígeno, se aumentará la selectividad hacia los tumores hipóxicos. Dichos análogos impedidos pueden ser sintetizados mediante el uso del esquema anterior y sustituyendo el éster de N metilglicina por un éster de N alquil glicina impedido tal como el éster de N neopentil glicina. Pueden contemplarse otros grupos impedidos que incluyen, pero no se limitan a, t-butilo, ciclohexilo, ciclopentilo, isopropilo o cualquier variantes sustituida con un heteroátomo.
Parte B. Síntesis de un alcohol secundario de nitroimidazol. Como se ha indicado más arriba, en esta sección se describen los alcoholes secundarios de nitroimidazol y los procedimientos para su síntesis. En tres procedimientos ilustrativos diferentes, según se muestra en los siguientes esquemas, se convierte bien el éster de nitroimidazol (el compuesto 1 en el esquema en la parte A y en los siguientes esquemas) o bien el alcohol primario de nitroimidazol (el compuesto 2 en el esquema en la parte A y en los siguientes esquemas) en el alcohol secundario de nitroimidazol mediante el uso bien de reactivos de cerio (segundo esquema, a continuación; véase Takeda y col., Organic Syntheses, volumen 76, página 228 y siguientes, y las referencias mencionadas en el mismo) o bien de reactivos de titanio (el primer y el tercer esquema, a continuación (véase Imwinkelried y col. Organic Syntheses, volumen CV 8, página 495 y siguientes, y las referencias mencionadas en el mismo).

Los alcoholes primarios y secundarios de nitroimidazol (el compuesto A, a continuación) pueden a su vez ser convertidos en el cloroformiato (el compuesto B, a continuación) y el clorometil éter (el compuesto C, a continuación), compuestos descritos en el presente documento, que se muestran a continuación.
R = H o Me

Los anteriores compuestos A, B y C, son útiles como intermedios en la síntesis de los agentes antineoplásicos protegidos activados por hipoxia, en los que sirven para bloquear el grupo hidroxi, amino y otros grupos del agente antineoplásico protegido.
Ejemplo 2
Etopósido
El siguiente es un ejemplo profético de un agente antineoplásico que contiene un grupo fenólico.
El fármaco antitumoral etopósido puede ser alquilado específicamente en su posición fenólica mediante el uso del reactivo 1-metil-2-nitro-5-bromometil-imidazol descrito en el Ejemplo 1 en unas condiciones suaves con una base moderada tal como carbonato de potasio en un disolvente anhidro tal como DMF. Alternativamente, la alquilación anterior puede ser efectuada en unas condicionas de Mitsunobu entre el metil-2-nitro-5-hidroximetil-imidazol y el etopósido mediante el uso de trifenil fosfina y de azodicarboxilato de isopropilo en DMF. Una reacción y unas condiciones experimentales análogas se proporcionan en Toki, et.al., Journal of Organic Chemistry, 2002, 67, 1866 1872, en el Ejemplo 1. La siguiente fórmula estructural presenta un ejemplo no limitante de dicha estructura.

Puede usarse un alquil éter fenólico de nitroimidazol portador de un alcohol bencílico en para o en orto para formar una conexión éter con el grupo fenólico del etopósido de una forma análoga a la descripción anterior. Puede acoplarse un alquil éter fenólico de nitroimidazol portador de un alcohol bencílico en para o en orto para formar un éter con el fenol del etopósido convirtiendo en primer lugar el alcohol bencílico en un bromuro y haciéndolo reaccionar después en las condiciones alquilantes habituales descritas más arriba, para efectuar el deseado enlace éter con el etopósido. Alternativamente, pueden aplicarse las condiciones de Mitsunobu descritas más arriba para formar el deseado éter. La siguiente fórmula estructural presenta un ejemplo no limitante de dicha estructura.
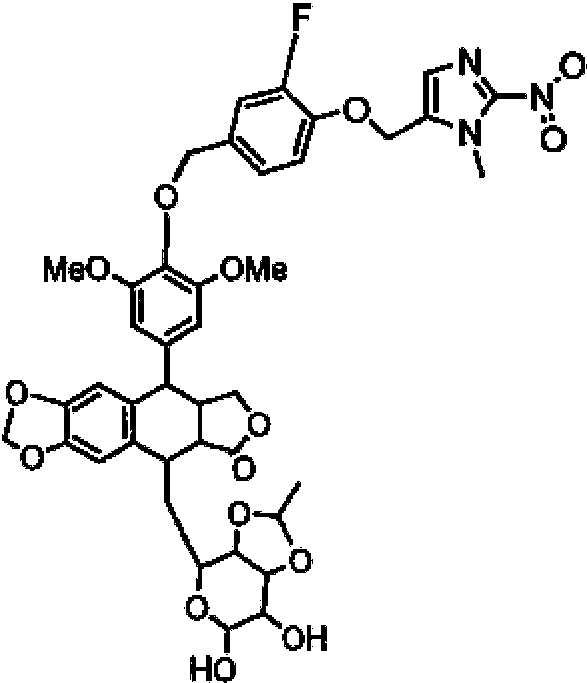
Ejemplo 3
Combretastatina A-4 y análogos
El siguiente es un ejemplo profético de un agente antineoplásico que contiene un grupo fenólico.
La combretastatina A-4 es un potente inhibidor de los microtúbulos y posee un único grupo fenólico que se cree que es esencial para la actividad citotóxica del compuesto. Existe un análogo de la combretastatina A-4 que difiere únicamente en la sustitución del grupo fenólico por un grupo amino, y es progresivamente más potente que la combretastatina A-4. Estos dos compuestos son modificados y transformados en inactivos, y después liberados hipóxicamente mediante el uso de los agentes antineoplásicos protegidos descritos en esta patente. (para una revisión de esta colección de análogos de la combretastatina, véase: Nam, N, Current Medicinal Chemistry, 2003, 10, 1697-1722). Muchos de estos análogos activos pueden ser protegidos como se describe a continuación de una forma análoga.
Las estructuras de la combretastatina A-4 y de su análogo amino se muestran a continuación (A-4: X = O, R1 = H y análogo amino: X = NH y R1 = H)

Pueden usarse los siguientes grupos protectores para proteger el grupo hidroxilo o amino de la combretastatina A-4 y de los análogos, donde el * indica el punto de unión.
Profármacos cuando X es O o NH

Profármaco cuando X es sólo NH

La conexión de la tercera construcción de nitroimidazol con la combretastatina A-4 parental es menos óptima, ya que el carbonato resultante puede ser inestable in vivo debido a las esterasas generales. La combretastatina A-4 de partida está disponible comercialmente. El análogo amino puede ser sintetizado según se notifica en Ohsumi, K, J. Med. Chem., 41 (16), 3022 -3032,1998. La síntesis de otro análogo puede encontrarse en las referencias descritas en Nam, N, Current Medicinal Chemistry, 2003,10, 1697-1722.
Ejemplo 4
La familia de la duocarmicina
El siguiente es un ejemplo profético de un agente antineoplásico que contiene un grupo fenólico.
La tecnología activada hipóxicamente descrita en esta patente también puede ser aplicada a la familia de la duocarmicina. Una conexión alquilo simple se describe en cualquier parte de esta patente y puede ser sintetizada de forma directa mediante los procedimientos descritos más arriba para el etopósido. La conexión del conector inferior al fenol de una duocarmicina puede ser sintetizada de forma directa como se ha descrito más arriba para el etopósido. Se han sintetizado muchos análogos de la duocarmicina y un experto en la materia comprenderá que el fenol libre en la posición indicada como O-R puede ser protegido mediante el uso de la tecnología del agente antineoplásico protegido descrita en esta patente, (véanse las referencias para las opciones de análogos: Denny, W, A, Current Medicinal Chemistry, 2001,533-44 y Searcey, M, Current Pharm. Discovery, 2002, 8, 1375-89.)

X puede ser una diversidad de grupos, que incluyen, pero no se limitan a, halo y sulfonolato y particularmente -Cl. Mediante el uso de los reactivos descritos más arriba para el etopósido, los profármacos activados hipóxicamente mostrados pueden ser sintetizados de forma directa, donde el asterisco es la conexión con el fenol o la amina de la duocarmicina.
Ejemplo 5
Barminomicina y análogos relacionados
El siguiente es un ejemplo profético de un agente antineoplásico que contiene un grupo hemiaminal.
La barminomicina es un producto natural relacionado con la daunorrubicina. Sirve como un análogo alquilante preactivado de la daunorrubicina al portar una funcionalidad hemiaminal que está en equilibrio tras la eliminación del agua para formar la imina activa (Moufarij, M. A., y col., Chemico-Biological Interactions, 138, 2001, 137-153). Esta funcionalidad añadida da como resultado una citotoxicidad que está aumentada en aproximadamente 1.000 veces con respecto a la daunorrubicina o a la doxorrubicina

Esta toxina ha sido el objeto de una solicitud de patente, la JP4187096, que la combina de una forma descrita vagamente con un anticuerpo que tiene un reconocimiento doble de la barminomicina y un antígeno específico La barminomicina puede ser modificada regioespecíficamente en el nitrógeno de la función hemiaminal con el carbonato de nitrofenilo derivado del 1-metil-2-nitro-5-hidroximetil imidazol para producir el producto mostrado más arriba, del que se muestra el R1 a continuación (estructura de la izquierda), siendo el asterisco la conexión con el nitrógeno. Alternativamente, puede hacerse reaccionar el nitrógeno del hemiaminal de la barminomicina con los carbonatos de nitrofenilo del éter alquil fenólico de nitroimidazol portador de un alcohol bencílico en para o en orto como se ha descrito previamente para producir el análogo de la barminomicina donde y el ejemplo de R1 se muestra a continuación (estructura de la derecha), siendo el asterisco la conexión con el nitrógeno.

Ejemplo 6
Antramicina y análogos relacionados
El siguiente es un ejemplo profético de un agente antineoplásico que contiene un grupo hemiaminal.
La antramicina es un producto antitumoral natural del que se han identificado muchos parientes naturales tales como tomaimcicina, sibiromicina, chicamicina A, neotramicina A y B y DC-81, entre otros. Se han sintetizado muchos análogos sintéticos sobre la base de estos productos naturales, y sus procedimientos de síntesis se han revisado bien en Kamal A., y col., Current Medicinal Chemistry-Anti-Cancer Agents, 2002, 2, 215-254. Estos agentes antineoplásicos comparten una importante característica común ya que en un disolvente acuoso existen en forma de un hemiaminal (R1 = H y R2 = H en la estructura mostrada a continuación para la estructura de la antramicina). Es esta fracción de hemiaminal, que se equilibra con la forma de imina, la que es capaz de reticular el N2 de una guanina del ADN después de la unión del fármaco en el surco menor del ADN. Este acontecimiento de reticulación es responsable de la toxicidad de estos compuestos y de su actividad antineoplásica.

El N del hemiaminal ha sido protegido como un profármaco en forma de un carbamato de paranitrofenilo por Thurston en un intento de crear una versión del profármaco que sea activada por una enzima nitrorreductasa de E Coli. Finalmente, esta enzima debía ser administrada en un tumor de una forma específica mediante su conjugación con un anticuerpo específico tumoral. Esta metodología se conoce de forma general como ADEPT. Un grupo nitrobencilo no es susceptible de ser reducido por las enzimas de los mamíferos dado su bajo potencial de reducción. No se realiza ninguna mención ni en la publicación periódica (Sagnou, M. J., Bioorganicand Medicinal Letters, 2000, 10, 2082) ni en la patente US 6608192 B1 ni en las presentaciones relacionadas, relativa al direccionamiento a las zonas hipóxicas de los tumores mediante el uso de un mecanismo de liberación específico hipóxico. Contemplamos la formación de profármacos de la serie de la antramicina mediante el uso de los mismos reactivos que los descritos más arriba para la barminomicina. Estas reacciones producirán profármacos de antramicina en los que R2 es H o un grupo alquilo tal como un metilo, dependiendo del pretratamiento con disolvente (agua, metanol u otro alcohol) de la antramicina y donde R1 se muestra a continuación de una forma análoga al ejemplo de la barminomicina, siendo el asterisco la unión al nitrógeno del hemiaminal.

Ejemplo 7
Síntesis del N-1-met¡l-2-nitro-5-h¡drox¡met¡l-¡m¡dazol
Se llevaron a cabo los siguientes procedimientos sintéticos para sintetizar el N-1-metil-2-nitro-5-hidroximetil-imidazol.

El éster: se sintetizó el N-1-metil-2-nitro -5-carboetoxiimidazol según el procedimiento descrito en la referencia, Asato G; Berkelhammer, G., J. Med. Chem. 1972, 15, 1086. En la etapa que implica una C-formilación, la base prescrita, KOtBU fue sustituida por NaH. El producto se caracterizó mediante una RMN H1
El alcohol: la reducción de la funcionalidad éster a un alcohol primario se llevó a cabo siguiendo una hidrólisis del éster al ácido. A una mezcla del ácido (49 mmol) en THF (1.500 ml) y trietilamina (71 mmol), enfriada a -10 C se añadió gota a gota cloroformiato de isobutilo (71 mmol) y se agitó durante 30 m. La temperatura se elevó hasta -5 C y se agitó durante 30 m. Después de la desaparición del material de partida, se añadió lentamente NaBH4 (263 mmol) a la mezcla de reacción seguido de la adición de agua (200 ml) durante 30 m. Después de 30 m adicionales, la mezcla de reacción se filtró con una capa de Na2SO4 anhidro, los volátiles se eliminaron con un rotavapor, y el residuo se purificó en una cromatografía en columna de gel de sílice mediante el uso de acetato de etilo para producir un sólido esencialmente puro que se recristalizó en EtOAc-hexanos para producir el producto puro. El producto se caracterizó mediante una CL-EM, mostrando un pico significativo en el peso molecular apropiado, y mediante una RMN H1
Síntesis del N-1-metil-5-nitro-2-hidroximetil-imidazol

A una mezcla del éster (disponible comercialmente) (2 g, 10 mmol), THF (100 ml) se añadió gota a gota LiBH4 (2 M en THF, 2 ml) a la temperatura ambiente. Después de la adición de la solución de LiBH4, la mezcla se agitó adicionalmente durante 20 h. Después de la desaparición del material de partida, se añadió una solución acuosa saturada de K2CO3 (1 ml) a la mezcla de reacción, después la mezcla se agitó durante 1 h, se añadió metanol (10 ml) a la mezcla de reacción. Una filtración y una purificación en columna ultrarrápida (eluyente EtOAc:Hexano = 1:1 (v/v)) dio el alcohol puro en forma de un sólido de color blanco (1 g, 65 %). El producto se caracterizó mediante una RMN H1
Ejemplo 8
Síntesis de los bromuros:
Los bromuros pueden ser preparados fácilmente a partir de los correspondientes alcoholes preparados según los Ejemplos 7 y 1 por medio de diversos procedimientos conocidos en la bibliografía. En uno de dichos procedimientos, se añadió una solución de N-1-metil-2-nitro-5- hidroximetil-imidazol (1 eq, 1,8 molar) y diisopropiletilamina (2 eq) en CH2CI2 anhidro a una solución de un complejo de PPh3-Br2 (comercial o en una solución preparada in situ, 3,5 molar) mientras se mantenía a 0 C y la mezcla de reacción se agitó a 0 C hasta que el análisis mediante una TLC mostró la completa desaparición del material de partida. Los volátiles se eliminaron a vacío y el residuo se purificó mediante una cromatografía en gel de sílice para producir el bromuro deseado, el reactivo B. El producto se caracterizó mediante una RMN H1.
El correspondiente derivado de 5-nitroimidazol se sintetizó de una forma similar a partir de N-1-etil-5-nitro-2-hidroximetil-nitroimidazol
Esquemas retrosintéticos generales para la síntesis de los Ejemplos 9, 10 y 11 Daunorrubicina protegida mediante un activador hipóxico unido a través de diversos grupos de liberación retardada
Estos ejemplos proporcionan los esquemas sintéticos para la síntesis de tres versiones del compuesto H en el siguiente esquema.

Ejemplo 9
Síntesis del Compuesto H con R1 es nitro, R2 es hidrógeno en el anterior esquema
1: síntesis del intermedio C, R1 es nitro, R2 es hidrógeno
En un matraz de fondo redondo de 100 ml se añadió una mezcla de A, Ri es nitro, R2 es hidrógeno, el reactivo disponible comercialmente (100 mg), B (110 mg), K2CO3 (200 mg) y acetona (anhidra, 1 ml). La mezcla se calentó a reflujo durante 4 h. Una vez terminada la reacción, los sólidos se eliminaron mediante una filtración a través del papel de filtro, y se obtuvo el producto puro C (145 mg) después de una cromatografía ultrarrápida (eluyente EtOAc:Hexano (50:50 (v/v))).
2: reducción de C: síntesis del intermedio D, R1 es nitro, R2 es hidrógeno::
En un matraz de fondo redondo de 100 ml se añadió una mezcla de C (145 mg), LiBH4 (2 M en THF, 1 ml) y THF anhidro (5 ml). La solución se agitó después a la temperatura ambiente durante 24 h. Una vez terminada la reacción, una purificación mediante una cromatografía ultrarrápida dio el alcohol puro D (87 mg).
3: síntesis del intermedio de carbonato F, R1 es nitro, R2 es hidrógeno
En un matraz de fondo redondo de 10 ml se añadió una mezcla de C, R1 es nitro, R2 es hidrógeno (87 mg), THF (2 ml), piridina (0,1 ml) y cloroformiato de p-nitrofenol (87 mg). La mezcla se agitó a la temperatura ambiente durante 5 h. La cromatografía ultrarrápida dio el producto puro (120 mg).
4: síntesis del Compuesto H, R1 es nitro, R2 es hidrógeno
En un matraz de fondo redondo de 25 ml se añadió una mezcla de F (20 mg), DMF (1 ml), la sal de HCI de la daunorrubicina (25 mg) y DIEA (diisopropiletilamina) (0,1 ml). La mezcla se agitó a la temperatura ambiente durante 4 h. Una vez terminada la reacción, la mezcla se vertió en 10 ml de diclorometano y se lavó con salmuera (3 x 5 ml). La cromatografía ultrarrápida dio el producto puro (30 mg).
Ensayo clonogénico
Ejemplo 10
Síntesis del Compuesto H con R1 es fluoro, R2 es fluoro
1: síntesis del intermedio C R1 es fluoro, R2 es fluoro
En un matraz de fondo redondo de 100 ml se añadió una mezcla de A, R1 es fluoro, R2 es fluoro (120 mg), B (100 mg), K2CO3 (310 mg) y acetona (anhidra, 2 ml). La mezcla se calentó a reflujo durante 4 h. Una vez terminada la reacción, la mezcla de reacción se diluyó con EtOAc (20 ml) y los sólidos se eliminaron mediante una filtración a través de papel de filtro y la solución orgánica se lavó con K2CO3 al 5 % (3 x 10 ml). La evaporación dio C, R1 es fluoro, R2 es fluoro en forma de un sólido de color blanco.
2: reducción de C R1 es fluoro, R2 es fluoro: síntesis del intermedio D R1 es fluoro, R2 es fluoro:
En un matraz de fondo redondo de 100 ml se añadió una mezcla de C- R1 es fluoro, R2 es fluoro (de la reacción anterior), LiBH4 (2 M en THF, 0,2 ml) y THF anhidro (10 ml). La solución se agitó después a la temperatura ambiente durante 24 h. Una vez terminada la reacción, una purificación mediante una cromatografía ultrarrápida dio el alcohol puro D, R1 es fluoro, R2 es fluoro.
3: síntesis del intermedio de carbonato F, R1 es fluoro, R2 es fluoro:
En un matraz de fondo redondo de 10 ml se añadió una mezcla de C, R1 es fluoro, R2 es fluoro (0,45 mmol), THF (2 ml), piridina (0,1 ml) y cloroformiato de p-nitrofenol (0,54 mg). La mezcla se agitó a la temperatura ambiente durante 5 h. La cromatografía ultrarrápida dio el producto puro.
4: síntesis del Compuesto H donde R1 es fluoro y R2 es fluoro
En un matraz de fondo redondo de 25 ml se añadió una mezcla de F, R1 es fluoro, R2 es fluoro (8,8 mg), DMF (1 ml), la sal de HCI de la daunorrubicina (10 mg) y DIEA (0,1 ml). La mezcla se agitó a la temperatura ambiente durante 4 h. Una vez terminada la reacción, la mezcla se vertió en 10 ml de diclorometano y se lavó con salmuera (3 x 5 ml). La cromatografía ultrarrápida dio el producto puro (6,6 mg).
Ejemplo 11
Síntesis del Compuesto H, R1 es fluoro, R? es hidrógeno:
1: síntesis del intermedio C, R1 es fluoro, R2 es hidrógeno
En un matraz de fondo redondo de 100 ml se añadió ácido 4-hidroxil-2-fluorobenzoico (1 g), metanol (10 ml) y ácido sulfúrico concentrado (al 98 %, 0,1 ml). La mezcla se calentó a reflujo durante 10 h. Una vez terminada la reacción, la mezcla se vertió en 100 ml de agua helada, y la filtración dio el producto puro A, R1 es fluoro, R2 es hidrógeno, (Ig) en forma de un sólido de color blanco.
En un matraz de fondo redondo de 100 ml se añadió una mezcla de A, R1 es fluoro, R2 es hidrógeno (100 mg), B (100 mg), K2CO3 (200 mg) y acetona (anhidra, 1 ml). La mezcla se calentó a reflujo durante 4 h. Una vez terminada la reacción, la mezcla de reacción se vertió en agua (10 ml) y se extrajo con EtOAc (3 x 15 ml). La solución orgánica combinada se lavó después con K2CO3 al 5 % (acuoso, 3 x 10 ml) para retirar el exceso de A y después se secó sobre Na2SO4. La evaporación dio C, R1 es fluoro, R2 es hidrógeno (130 mg) en forma de un sólido de color amarillo claro.
2: reducción de C, R1 es fluoro, R2 es hidrógeno: síntesis del intermedio D, R1 es fluoro, R2 es hidrógeno:
En un matraz de fondo redondo de 100 ml se añadió una mezcla de C, R1 es fluoro, R2 es hidrógeno (100 mg), LiBH4 (2 M en THF, 1 ml) y THF anhidro (5 ml). La solución se agitó después a la temperatura ambiente durante 24 h. Una vez terminada la reacción, una purificación mediante una cromatografía ultrarrápida dio el alcohol puro D, R1 es fluoro, R2 es hidrógeno (60 mg).
3: síntesis del intermedio de carbonato F, R1 es fluoro, R2 es hidrógeno:
En un matraz de fondo redondo de 10 ml se añadió una mezcla de C, R1 es fluoro, R2 es hidrógeno (10 mg), THF (2 ml), piridina (0,1 ml) y cloroformiato de p-nitrofenol (10 mg). La mezcla se agitó a la temperatura ambiente durante 5 h. La cromatografía ultrarrápida dio el producto puro (14 mg).
4: síntesis del Compuesto H donde R1 es fluoro y R2 es hidrógeno:
En un matraz de fondo redondo de 25 ml se añadió una mezcla de F, R1 es fluoro, R2 es hidrógeno (8,5 mg), DMF (1 ml), la sal de HCI de la daunorrubicina (10,7 mg) y DIEA (0,1 ml). La mezcla se agitó a la temperatura ambiente durante 2 h. Una vez terminada la reacción, la mezcla se vertió en 10 ml de diclorometano y se lavó con salmuera (3 x 5 ml). La cromatografía ultrarrápida dio el producto puro.
Los productos sintetizados en los Ejemplos 9, 10 y 11 se caracterizaron mediante una CL-EM con un pico principal del peso molecular apropiado
Ejemplo 12
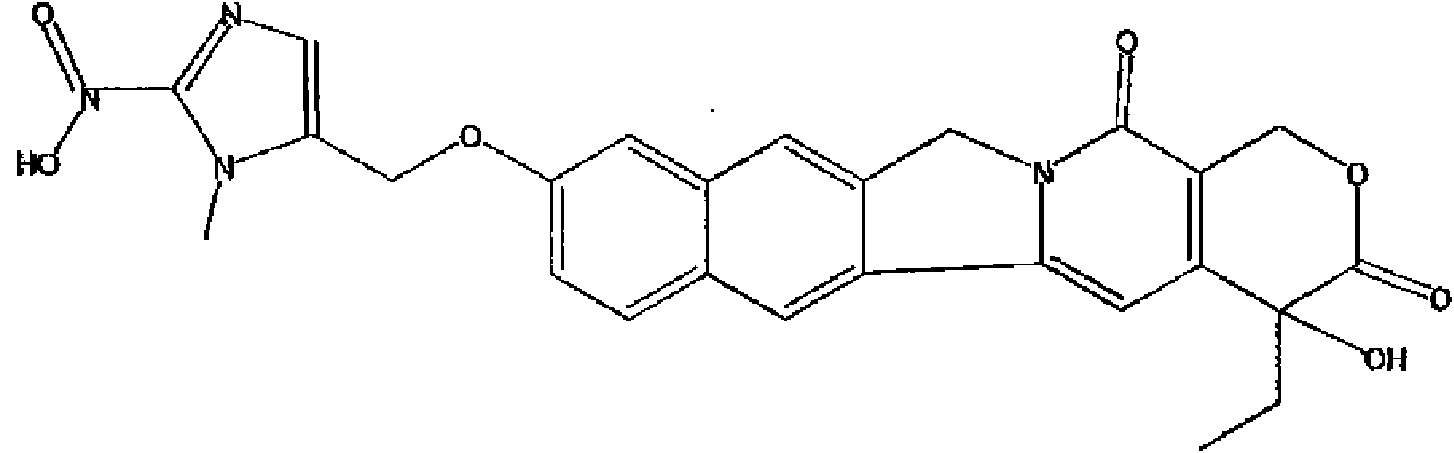
Alquilación de la 10-hidroxicamptotecina con N-1-metil-2-n¡tro-5-(bromomet¡l)-¡m¡dazol
La alquilación del grupo hidroxilo fenólico de la 10-hidroxicamptotecina se llevó a cabo en las condicionas convencionales usadas miles de veces en la bibliografía para alquilar la fracción fenólica de la 10-hidroxicamptotecina: se agitó una mezcla de 10-hidroxicamptotecina (2 eq, 0,1 milimolar) en DMF anhidra desclasificada, K2CO3 anhidro (3 eq) y el pertinente bromuro de nitroimidazol (1 eq) a la TA durante aproximadamente 16 h y los volátiles se eliminaron en un rotavapor. El residuo se purificó mediante una cromatografía en gel de sílice para producir los deseados éteres proporcionados en este Ejemplo 12 (5-nitro). Los compuestos se caracterizaron mediante una CL-EM mostrando un pico principal del peso molecular apropiado Ejemplo 13

Alquilación de la 10-hidroxicamptotecina con N-1-metil-5-nitro-2-(bromometil)-imidazol
La alquilación, la purificación y la caracterización se llevaron a cabo de una forma análoga a los procedimientos descritos en el anterior Ejemplo 12
Ejemplo 14
Pruebas biológicas
Se usaron los siguientes ensayos biológicos para la caracterización de varios de los agentes antineoplásicos protegidos.
A) Ensayo de toxicidad clonogénica
Introducción
Para determinar si un fármaco es un agente antineoplásico eficaz, usamos un ensayo clonogénico como una prueba rigurosa de la supervivencia celular. El ensayo clonogénico mide la capacidad reproductora de una célula superviviente mediante la determinación específica de si la célula puede crecer para formar una colonia de más de 50 células. En resumen, las células se tratan con un fármaco para una exposición aguda, y después se retira el fármaco. Las células se tripinizan para formar suspensiones celulares individuales y se ponen en placas unas cifras conocidas de células y se incuban hasta que se forman colonias. Se cuentan las colonias y la supervivencia de las células se calcula sobre la base del número de colonias formadas en los grupos tratados en comparación con el número de colonias formadas en los controles sin tratar. Con objeto de determinar si un fármaco es selectivamente tóxico en unas condiciones anóxicas, las células son expuestas al fármaco tanto al aire (normoxia) como completamente sin oxígeno (anoxia). Los términos aérobico y normóxico se usan de forma intercambiable.
Procedimientos experimentales
Se sembraron células humanas H460 de crecimiento exponencial (obtenidas a partir de la ATCC) en placas de vidrio con muescas de 60 mm a entre 2,5 y 5 x 105 células por placa, y se cultivaron en medio RPMI complementado con un 10 % de suero bovino fetal durante 2 días antes de iniciar el tratamiento. El día del experimento se prepararon soluciones madre de fármaco de concentraciones conocidas en medio completo y se añadieron 2 ml a cada placa. Las placas de vidrio se precintaron en recipientes herméticos de aluminio equipados con una válvula para controlar el flujo de gas. Para conseguir un equilibrio completo entre la fase gaseosa y la fase líquida se realizaron una serie de intercambios de gas en cada recipiente con agitación. Los recipientes se evacuaron y se gasificaron bien con una mezcla gaseosa anóxica certificada (95 % de nitrógeno y 5 % de dióxido de carbono) o bien con una mezcla gaseosa aeróbica (95 % de aire y 5 % de dióxido de carbono). Específicamente, cada recipiente se evacuó hasta menos de 26 pulgadas de mercurio y se mantuvo durante 15 segundos antes de gasificar a 20 psi y se mantuvo de nuevo durante 15 segundos. Después de una serie de cinco evacuaciones y gasificaciones, los recipientes se mantuvieron 5 minutos adicionales antes de la evacuación y el rellenado final de cada recipiente con la mezcla gaseosa deseada a 0,5 psi por encima de la presión atmosférica, las células se incubaron durante 2 horas a 37 °C. Al final del tratamiento, las placas se retiraron de cada recipiente y el fármaco se eliminó rápidamente de las células. Las placas se lavaron con solución salina tamponada con fosfato y una solución de tripsina-EDTA y después se tripsinizaron durante 5 minutos a 37 °C. Las células desprendidas se neutralizaron con medio más suero y se rotaron durante 5 min a 100 x g. Las células se resuspendieron aproximadamente a 1 x 106 células/ml y se diluyeron 10 veces para producir concentraciones madre para su colocación en placas. La concentración exacta de cada solución madre se determinó mediante el recuento con un contador de partículas Coulter Z2. Se colocaron en las placas unas cifras conocidas de células y se colocaron sin alterar en una estufa de incubación durante entre 7 y 10 días. Las colonias se fijaron y se tiñeron con una solución de etanol al 95 % con una tinción de violeta de cristal al 0,25 %. Se contaron las colonias de más de 50 células y se determinó la fracción superviviente.
Resultado
El control de daunorrubicina no mostró ninguna diferencia significativa en la toxicidad en el anterior ensayo
clonogénico (2 h de exposición al compuesto) entre las condiciones normóxicas (aire) y anóxicas. La CI 90 (90 % de inhibición de la formación de colonias) era de 0,5 micromolar, variando los datos entre 0,2 micromolar y 1 micromolar a lo largo de múltiples experimentos
El compuesto del Ejemplo 9 mostró en las mismas condiciones hipóxicas una CI 90 de 4 micromolar variando los datos entre 2 micromolar y 5 micromolar a lo largo de múltiples experimentos. Los datos normóxicos eran muy drásticos, sin observarse ninguna toxicidad a la concentración más alta de 20 micromolar, el límite de solubilidad en el medio celular para este ensayo. El compuesto del Ejemplo 9 mostró una notable ausencia de toxicidad en las condiciones normales con aire, lo que implica que el compuesto del Ejemplo 9 es un compuesto muy selectivo. El compuesto del Ejemplo 10 mostró en las mismas condiciones hipóxicas una CI 90 de 1 micromolar variando los datos entre 0,5 micromolar y 2 micromolar a lo largo de múltiples experimentos. Los datos normóxicos eran muy drásticos, sin observarse ninguna toxicidad a la concentración más alta de 10 micromolar, el límite de solubilidad en el medio celular para este ensayo. El compuesto del Ejemplo 10 mostró una notable ausencia de toxicidad en las condiciones normales con aire, lo que implica que el compuesto del Ejemplo 10 es un compuesto muy selectivo. El compuesto del Ejemplo 11 mostró en las mismas condiciones hipóxicas una CI 90 de 1 micromolar en un experimento. En unas condiciones normóxicas, la CI50 era de aproximadamente 5 micromolar en un experimento. El compuesto del Ejemplo 11 muestra por lo tanto una buena selectividad entre las condiciones anóxicas y normóxicas. Activación intracelular de los profármacos activados por hipoxia
Ensayo biológico
Introducción
Para determinar si un compuesto profármaco puede ser reducido mediante las reductasas celulares, y por lo tanto dar lugar a la liberación del fármaco activo, hemos ensayado el metabolismo del fármaco en el medio de las células expuestas al profármaco en unas condiciones aeróbicas o anóxicas.
Procedimientos experimentales
Se diluyeron soluciones madre de 10 hidroxi camptotecina y del compuesto descrito en el Ejemplo 12 (2 nitroimidazol 10 hidroxi camptotecina) hasta 30 uM en medio RPMI sin rojo fenol y se complementaron con HEPES 10 mM y suero bovino fetal al 10 %. Se recogieron células de crecimiento exponencia1H460 y se resuspendieron 1 x 107 células directamente en 1 ml de medio con fármaco. Las soluciones madre sólo de fármaco y las células con fármaco se colocaron en placas de vidrio con muescas de 60 mm y se expusieron a gases en anoxia o aeróbicos según se ha descrito previamente. Todos los grupos se incubaron durante 3 horas a 37 °C. Para finalizar el tratamiento y extraer el fármaco del medio, todos los grupos fueron transferidos a tubos de micrófuga y se rotaron 3 min a 15.000 x g para sedimentar las células. El sobrenadante se eliminó y se acidificó con ácido acético glacial (concentración final del 10 %). El fármaco se extrajo del medio RPMI mediante la adición de un volumen igual de acetato de etilo, la mezcla, la centrifugación y la eliminación de la fase orgánica superior. Todas las muestras se evaporaron a sequedad en un sistema con velocidad de rotación a vacío. Las muestras secas se almacenaron a -20 °C.

Interpretación
Como puede observarse según el siguiente informe analítico, el compuesto del Ejemplo 12 es activado para que libere la 10 hidroxi-camptotecina parental únicamente en unas condiciones anóxicas y en las presentes de las células H460. El grado de liberación (ensayo 4) es significativo, ya que más de la mitad del compuesto del Ejemplo 12 fue convertido limpiamente. El análisis mediante una CL-EM no mostró ningún otro pico significativo en las trazas de la CL a la longitud de onda selectiva de la 10 hidroxicamptotecina de 370 nm distinto a los cuantificados a
continuación. Todas las demás reacciones de control no mostraron ninguna liberación detectable de la 10 hidroxicamptotecina, incluyendo la incubación aeróbica con células H460 con el compuesto del Ejemplo 12. Estos resultados muestran un mecanismo de liberación muy selectivo (anóxico frente a hipóxico) para el compuesto del Ejemplo 12.
Informe analítico del ensayo celular del compuesto del Ejemplo 12

Ejemplo 15
Profármacos de doxorrubicina y compuestos relacionados
El siguiente es un ejemplo profético de agentes antineoplásicos protegidos según se describe en esta patente: este Ejemplo describe algunos agentes antineoplásicos protegidos ilustrativos que liberan derivados muy citotóxicos de doxorrubicina. Los expertos en la materia apreciarán que la doxorrubicina, la epirrubicina y la daunomicina y los numerosos análogos y derivados de sus compuestos que han sido y continúan siendo sintetizados, representan una clase de compuestos que pueden ser fácilmente convertidos en agentes antineoplásicos protegidos sobre la base de las enseñanzas del presente documento. Este ejemplo ilustra dichos agentes antineoplásicos protegidos que liberan compuestos muy citotóxicos en unas condiciones hipóxicas. Los derivados muy citotóxicos de la daunomicina se describen en la referencia Bakina y Farquhar, 1999, Anti-cancer Drug Design 14: 507. La Parte A de este Ejemplo ilustra unos agentes antineoplásicos protegidos que liberan dicho derivado muy citotóxico, denominado en el presente documento derivado de doxorrubicina, y que comprende una fracción activada por hipoxia. La Parte B de este Ejemplo ilustra un profármaco tal que comprende más de una de dichas fracciones. La Parte C de este Ejemplo ilustra un profármaco tal que libera un nuevo derivado dicatiónico.
Parte A. Profármaco derivado de la doxorrubicina activado por hipoxia. El único grupo amino libre de la doxorrubicina, de la daunomicina, de la epirrubicina y de los derivados de las mismas, que tienen sólo este único grupo amino libre, puede ser fácilmente modificado con un activador hipóxico, según se describe en el presente documento, para producir unos agentes antineoplásicos protegidos que tienen la siguiente estructura (DOX es doxorrubicina o uno de los compuestos relacionados mencionados previamente distintos al grupo amino libre):
En unas condiciones hipóxicas, la fracción activada por la hipoxia se elimina liberando el siguiente compuesto muy citotóxico mostrado como iminio DOX (o Super Dox), a continuación.

A continuación se muestra un esquema de otro profármaco ilustrativo de un análogo “super DOX” de los agentes antineoplásicos protegidos descritos en el presente documento.

Parte B. Profármaco derivado de la doxorrubicina activado por hipoxia que requiere la eliminación de múltiples fracciones activadas por hipoxia. Puede usarse la metodología descrita en la parte A de este ejemplo para generar agentes antineoplásicos protegidos que tengan dos o más fracciones activadas por hipoxia. Un agente antineoplásico ilustrativo protegido que tiene tres de dichas fracciones se muestra a continuación (X se define en la parte C).

Parte C. Profármaco derivado de diamino doxorrubicina activado por hipoxia. La doxorrubicina puede ser derivatizada según un procedimiento sintético según se describe en el presente documento con una fracción de aminoetilo para generar un derivado dicatiónico de doxorrubicina que es muy citotóxico y se une con más fuerza al ADN que la doxorrubicina. Un ejemplo de un agente antineoplásico protegido tiene la siguiente estructura (en la
siguiente estructura, X representa cualquiera de las diversas fracciones presentes en la doxorrubicina, en la daunomicina, en la epirrubicina y en sus derivados naturales y sintéticos):

Después de la liberación activada por la hipoxia, el compuesto se protona para convertir los carboxamatos en las correspondientes aminas. El profármaco se forma mediante la reacción de la doxorrubicina o de un derivado de la doxorrubicina con HC(O)CH2NHCO2R, en la que R se define como anteriormente, en primer lugar en presencia de NaB(OAc)3H y después en presencia de R'CO2CI, en la que R' es un alquilo inferior C1-6 y una base.
Ejemplo 16
Profármacos de etopósido
El siguiente es un ejemplo profético de agentes antineoplásicos protegidos según se describe en esta patente. El etopósido es un potente inhibidor de la topoisomerasa de ADN II no intercalante. Según los procedimientos sintéticos descritos en el presente documento, se pueden formar fácilmente agentes antineoplásicos protegidos que liberan etopósido en unas condiciones hipóxicas como sigue. La fracción activada por hipoxia está unida al etopósido en las posiciones hidroxi fenólicas a través de un conector éter.
Estos conectores pueden ser proporcionados por otros reactivos intermedios útiles según se describe en el presente documento, mostrados a continuación. Estos reactivos pueden ser preparados fácilmente a partir del correspondiente alcohol

A continuación se muestran dos profármacos ilustrativos, indicados como A y B a continuación, y un esquema para su formación a partir de etopósido.

Ejemplo 17
Profármacos de alcaloides de la vinca
El siguiente es un ejemplo profético de agentes antineoplásicos protegidos según se describe en esta patente. Los agentes antineoplásicos protegidos que liberan vinblastina (u otro alcaloide de la vinca) pueden ser preparados a partir de la vinblastina (o de otro alcaloide de la vinca) según se muestra en el siguiente esquema (el NH del indol puede ser unido selectivamente a la fracción activada por hipoxia por encima de la unión en los hidroxilos) mediante el uso de una metodología sintética sustancialmente similar a la descrita en el Ejemplo 16

Los agentes antineoplásicos protegidos que liberan vincristina se preparan de una forma análoga, partiendo de
vincristina.
Ejemplo 18
Profármacos de topotecán
El siguiente es un ejemplo profético de agentes antineoplásicos protegidos según se describe en esta patente. Los agentes antineoplásicos protegidos que liberan topotecán pueden ser preparados a partir de topotecán según se muestra en el siguiente esquema mediante el uso de una metodología sintética sustancialmente similar a la descrita en el Ejemplo 16.

Ejemplo 19
Profármacos de Taxanos
El siguiente es un ejemplo profético de agentes antineoplásicos protegidos según se describe en esta patente. Los agentes antineoplásicos protegidos que liberan paclitaxel, docetaxel u otro taxano pueden ser preparados a partir de paclitaxel (o de docetaxel o de otro taxano) mediante el uso de una metodología sintética sustancialmente similar a la descrita en el Ejemplo 16 para la preparación de compuestos con una estructura similar a la estructura mostrada a continuación.

Como se indica en la anterior estructura, dichos agentes antineoplásicos protegidos pueden tener una, dos, tres o cuatro fracciones activadas por hipoxia. En algunas versiones, estas fracciones activadas por hipoxia se seleccionan entre las que se muestran a continuación.

Ejemplo 21
Profármacos de 5-FU
El siguiente es un ejemplo profético de agentes antineoplásicos protegidos según se describe en esta patente. Los agentes antineoplásicos protegidos que liberan 5-fluorouracilo (5-FU) pueden ser preparados a partir de 5-FU mediante el uso de una metodología sintética sustancialmente similar a la descrita en el Ejemplo 16. Uno de dichos profármacos tiene la estructura que se muestra a continuación.

Otro agente antineoplásico protegido tiene dos fracciones activadas por hipoxia unidas, una a según se muestra en la anterior estructura, y la otra en el otro nitrógeno del anillo.
Ejemplo 21
Profármacos de agentes alquilantes
El siguiente es un ejemplo profético de agentes antineoplásicos protegidos según se describe en esta patente. Los agentes antineoplásicos protegidos que liberan un agente alquilante pueden ser preparados a partir de lo que se muestra en el siguiente esquema mediante el uso de una metodología sintética sustancialmente similar a la descrita en el Ejemplo 16. Se usan unos agentes antineoplásicos protegidos de ciclofosfamida para ilustrar este aspecto de los compuestos y de los procedimientos descritos en el presente documento. Estos agentes antineoplásicos protegidos pueden ser sintetizados según los procedimientos descritos en el presente documento mediante el uso del siguiente procedimiento sintético ilustrativo.

Ejemplo 22
Profármacos de hidroxiurea
El siguiente es un ejemplo profético de agentes antineoplásicos protegidos según se describe en esta patente. Los agentes antineoplásicos protegidos que liberan hidroxiurea (HU) pueden ser preparados a partir de hidroxiurea según se muestra en el siguiente esquema mediante el uso de una metodología sintética sustancialmente similar a la descrita en el Ejemplo 16. La HU (vendida como Hydrea por Bristol-Myers Squibb) es un fármaco antileucémico con un mecanismo de acción que se atribuye a la inhibición de la enzima sintética del ADN reductasa de ribonucleósido. También se ha demostrado que la HU es eficaz frente a tumores cerebrales (meningioma).

Q — grupo protector
Desprotección
X = Br, Ci, Y = NMe, Z = N
X = Br, Cl, Y = 0 ,Z = CH

Ejemplo 23
Profármacos de nitroimidazol-20-camptotecina
El siguiente es un ejemplo profético de agentes antineoplásicos protegidos según se describe en esta patente. A continuación se muestra un ejemplo representativo de un procedimiento para la preparación de unos agentes antineoplásicos protegidos de camptotecina ilustrativos.

e
( c o n c o n d ic ió n de que u n se a H) r r o i a r m a c o a c t iv a d o p o r h ip o x ia La fracción de N acetato de metilo del imidazol puede ser sintetizada mediante la desmetilación del derivado de hidroximetil imidazol con ácido yodhídrico y su realquilación con bromoacetato de metilo. Después de la formación del profármaco se elimina el éster de metoxi, opcionalmente con hidróxido de sodio, seguido de un tratamiento ácido para restaurar la lactona activa de la camptotecina.
Alternativamente, la lactona de la camptotecina puede ser hidrolizada hacia la forma abierta, y la fracción de alcohol derivatizada con clorometil nitroimidazol o un derivado de clorometoximetil nitroimidazol. No es necesario que este derivado de nitroimidazol porte un grupo ácido carboxílico fuera de la posición 1 del imidazol, pero puede hacerse estable frente a la reducción de dos electrones por parte de la diaforasa DT al tener un grupo alquilo impedido estéricamente, según se ha analizado más arriba. Esta reacción puede llevarse a cabo mediante la bis alquilación del ácido carboxílico libre y del alcohol seguida de una hidrólisis básica del éster, dejando un ácido libre para la solubilidad y enmascarando la fracción de nitroimidazol del profármaco la funcionalidad esencial de alcohol. La liberación del profármaco en unas condiciones hipóxicas in vivo dará como resultado la liberación del alcohol. La posterior ciclación con el ácido carboxílico en las condiciones ácidas del tumor generará una camptotecina activa. Dichos análogos de la camptotecina pueden ser preparados de una forma análoga mediante el uso de cualquiera de los numerosos análogos de la camptotecina conocidos en la materia como el fármaco que se va a proteger para producir un agente antineoplásico protegido.
Ejemplo 24
Profármacos de duocarmicina de nitroimidazol
El siguiente es un ejemplo profético de agentes antineoplásicos protegidos según se describe en esta patente.
Se conocen muchos análogos de la duocarmicina y sus profármacos (véase la patente de Estados Unidos n° 5.985.909, la publicación de patente PCT n° US02/17210 y la publicación de solicitud de patente de Estados Unidos n° 2003/0050331 A1). Ninguno de estos análogos o profármacos emplean un nitroimidazol activado para la liberación de la duocarmicina mediante el uso de una conexión estable de éter con el grupo fenólico de la duocarmicina. A continuación se muestra un ejemplo representativo de un agente antineoplásico protegido de duocarmicina de nitroimidazol, en el que el oxígeno fenólico está protegido en forma de un éter con el nitroimidazol, según se muestra. La síntesis comienza, en una versión, con el fenol libre de la duocarmicina y el derivado de nitroimidazol portador de un grupo bromometilo o clorometilo como el precursor para la conexión de éter. El Ri puede ser H, metilo o un alquilo inferior, R2 puede ser COOR, CN o NO2 , X puede ser Cl, Br, I o sulfonato, R3 y R4 pueden ser según se describe en el documento PCT/US02/17210. El grupo R5 del nitroimidazol puede ser metilo o un grupo alquilo impedido como se ha descrito más arriba.
Claims (14)
1. Un agente antineoplásico protegido de la fórmula Hip-N, en la que N es un agente antineoplásico, y un grupo hidroxilo protegible del antineoplásico está sustituido con Hip; Hip es una fracción que tiene la fórmula:

en la que
R2 es hidrógeno;
R3 es hidrógeno o alquilo C1-C6 ;
R1 es alquilo C1-C6 o alcoxi C1-C6 , opcionalmente sustituido con uno o más grupos que contienen heteroátomos; y R4 es hidrógeno, alquilo C1-C6 o alcoxi C1-C6 , opcionalmente sustituido con uno o más grupos que contienen heteroátomos.
2. El agente antineoplásico protegido según la reivindicación 1, en el que R1 es metilo; y R4 es hidrógeno o alquilo C1-C6 , opcionalmente sustituido con uno o más grupos que contienen heteroátomos.
3. El agente antineoplásico protegido según la reivindicación 1 o 2, en el que el agente antineoplásico N se selecciona entre el grupo que consiste en agentes antiangiogénicos, agentes alquilantes, antimetabolitos, complejos de coordinación con platino, antracenedionas y ureas sustituidas.
4. El agente antineoplásico protegido según la reivindicación 3, en el que el agente antineoplásico N es un agente alquilante.
5. El agente antineoplásico protegido según la reivindicación 3, en el que el agente antineoplásico N se selecciona entre el grupo que consiste en doxorrubicina, daunorrubicina, etopósido, duetopósido, combretastatina A-4, vinblastina, vincristina, camptotecina, topotecán, AQ4N, hidroxiurea, maitansinas, discodermólidos, epotilonas, taxanos, caliqueamicinas, tedanólidos, bleomicinas, citarabina, epirrubicina, fludarabina, hidroxiureapentostatina, mitoxantrona, prednisona, docetaxel, paclitaxel, tedanólidos, tenipósido y alcaloides de la vinca.
6. Un agente antineoplásico protegido según la reivindicación 1 que tiene la fórmula:

o una sal farmacéuticamente aceptable del mismo, en el que R3 es hidrógeno o alquilo C1-C6.
7. Un medicamento que comprende un agente antineoplásico protegido según una cualquiera de las reivindicaciones anteriores o una sal farmacéuticamente aceptable del mismo.
8. Una composición farmacéutica que comprende una cantidad terapéuticamente eficaz de un agente antineoplásico protegido según cualquiera de las reivindicaciones 1 a 6 o una sal farmacéuticamente aceptable del mismo y un excipiente, un portador y/o un diluyente farmacéuticamente aceptable.
9. Una composición farmacéutica según la reivindicación 8, en el que el diluyente es agua, etanol, propilenglicol, glicerina o combinaciones de los mismos.
10. Un agente antineoplásico protegido según cualquiera de las reivindicaciones 1 a 6 o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento del cáncer.
11. El agente antineoplásico protegido para su uso según la reivindicación 10, en el que el cáncer se selecciona entre el grupo que consiste en leucemia, cáncer de mama, cáncer de piel, cáncer óseo, cáncer de hígado, cáncer cerebral, cáncer de la laringe, de vesícula biliar, de páncreas, de recto, de paratiroides, de tiroides, adrenal, de tejido neural, de cabeza y cuello, de estómago, de bronquios, de riñones, carcinoma de células basales, carcinoma epidermoide tanto de tipo ulcerativo como de tipo papilar, carcinoma de piel metastásico, osteosarcoma, sarcoma de Ewing, reticulosarcoma, mieloma, tumor de células gigantes, tumor de pulmón microcítico, cálculos biliares, tumor de los islotes, tumor cerebral primario, tumores linfocíticos o granulocíticos agudos y crónicos, tumor de tricoleucocitos, adenoma, hiperplasia, carcinoma medular, feocromocitoma, neuronmas mucosales, ganglionauromas intestinales, tumor hiperplásico del nervio corneal, tumor de constitución marfanoide, tumor de Wilm, seminoma, tumor leiomiomata, displasia cervical o carcinoma in situ, neuroblastoma, retinoblastoma, sarcoma del tejido blando, carcinoide maligno, lesión cutánea tópica, micosis fungoide, rabdomiosarcoma, sarcoma de Kaposi, sarcoma osteogénico y otros, hipercalcemia maligna, tumor de las células renales, policitermia vera, adenocarcinoma, glioblastoma multiforme, leucemias, linfomas, melanomas malignos y carcinomas epidermoides.
12. El agente antineoplásico protegido para su uso según la reivindicación 10 u 11 junto con una cantidad eficaz de uno o más agentes anticancerosos, una cantidad eficaz de radioterapia, o cualquier combinación de los anteriores.
13. El agente antineoplásico protegido para su uso según la reivindicación 12, en el que el agente anticanceroso se selecciona entre el grupo que consiste en busulfano, improsulfano, piposulfano, benzodepa, carbocuona, 2-desoxi-D-glucosa, lonidamina, meturedepa, uredepa, altretamina, imatinib, trietilenmelamina, trietilenfosforamida, trietilentiofosforamida, trimetilolomelamina, clorambucilo, clornafazina, estramustina, ifosfamida, mecloretamina, clorhidrato de óxido de mecloretamina, melfalano, novembiquina, fenesterina, prednimustina, trofosfamida, mostaza de uracilo, carmustina, clorozotocina, fotemustina, nimustina, ranimustina, dacarbazina, manomustina, mitobronitol, mitolactol, pipobromano, aclacinomicinas, actinomicina F(1), antramicina, azaserina, bleomicina, cactinomicina, carubicinas, carzinofilina, cromomicina, dactinomicina, daunorrubicina, daunomicina, 6-diazo-5-oxo-1-norleucina, ácido micofenólico, nogalamicina, olivomicina, peplomicina, plicamicina, porfiromicina, puromicina, estreptonigrina, estreptozocina, tubercidina, ubenimex, zinostatina, zorrubicina, denopterina, pteropterina, trimetrexato, fludarabina, 6-mercaptopurina, tiamiprina, tioguanina, ancitabina, azacitidina, 6-azauridina, carmofur, citarabina, didesoxiuridina, doxifluridina, enocitabina, floxuridina, 5-fluorouracilo, tegafur, L-asparraginasa, pulmozima, aceglatona, glicósido de aldofosfamida, ácido aminolevulínico, amasacrina, bestrabucilo, bisantreno, carboplatino, defofamida, demecolcina, diazicuona, elformitina, acetato de eliptinio, etoglucida, flutamida, nitrato de galio, hidroxiurea, interferón alfa, interferón beta, interferón gamma, interleucina-2, lentinano, mitoguazona, mitoxantrona, mopidamol, nitracrina, pentostatina, fenamet, pirarrubicina, ácido podofilínico, 2-etilhidrazida, procarbazina, razoxano, sizofiran, espirogermanio, paclitaxel, tamoxifeno, tenipósido, ácido tenuazonico, triazicuona, 2,2',2”-triclorotrietilamina, uretano, vinblastina, ciclofosfamida, vincristina, cisplatino, carboplatino y oxoplatino.
14. El agente antineoplásico protegido para su uso según cualquiera de las reivindicaciones 10 a 13 en el tratamiento del cáncer en una cantidad de dosis de entre 1 y 35 mg/kg/día en dosis únicas o divididas.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US45884503P | 2003-03-28 | 2003-03-28 | |
| US46528103P | 2003-04-21 | 2003-04-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2759176T3 true ES2759176T3 (es) | 2020-05-07 |
Family
ID=33135116
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13178249T Expired - Lifetime ES2759176T3 (es) | 2003-03-28 | 2004-03-29 | Composiciones y procedimientos para el tratamiento del cáncer |
Country Status (14)
| Country | Link |
|---|---|
| US (2) | US7550496B2 (es) |
| EP (2) | EP1622608A4 (es) |
| JP (2) | JP2006521409A (es) |
| KR (2) | KR101198526B1 (es) |
| CN (2) | CN1791403A (es) |
| AU (1) | AU2004226338C1 (es) |
| CA (1) | CA2520000C (es) |
| ES (1) | ES2759176T3 (es) |
| IL (1) | IL170970A (es) |
| MX (1) | MXPA05010407A (es) |
| NO (1) | NO336266B1 (es) |
| NZ (1) | NZ543223A (es) |
| WO (1) | WO2004087075A2 (es) |
| ZA (1) | ZA200507752B (es) |
Families Citing this family (77)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ZA200507752B (en) * | 2003-03-28 | 2007-01-31 | Threshold Pharmaceuticals Inc | Compositions and methods for treating cancer |
| US7691838B2 (en) | 2003-05-30 | 2010-04-06 | Kosan Biosciences Incorporated | Method for treating diseases using HSP90-inhibiting agents in combination with antimitotics |
| DE50305909D1 (de) * | 2003-10-13 | 2007-01-18 | Salama Zoser B | Pharmazeutische Zusammensetzung umfassend Oxoplatin und dessen Salze |
| US20080132458A1 (en) * | 2004-03-10 | 2008-06-05 | Threshold Pharmaceuticals, Inc. | Hypoxia-Activated Anti-Cancer Agents |
| GB0421296D0 (en) * | 2004-09-24 | 2004-10-27 | Angiogene Pharm Ltd | Bioreductively-activated prodrugs |
| US7208611B2 (en) | 2005-02-23 | 2007-04-24 | Xenoport, Inc. | Platinum-containing compounds exhibiting cytostatic activity, synthesis and methods of use |
| JP2006282653A (ja) * | 2005-03-10 | 2006-10-19 | Kyoto Univ | 標的部位で選択的に活性化される新規化合物およびその利用 |
| JP5180824B2 (ja) | 2005-06-29 | 2013-04-10 | スレッシュホールド ファーマシューティカルズ, インコーポレイテッド | ホスホルアミデートアルキル化剤プロドラッグ |
| WO2008083101A1 (en) * | 2006-12-26 | 2008-07-10 | Threshold Pharmaceuticals, Inc. | Phosphoramidate alkylator prodrugs for the treatment of cancer |
| CN101765369A (zh) | 2007-03-19 | 2010-06-30 | 俄勒冈州由俄勒冈州立大学代表州高等教育委员会行使 | 曼尼希碱n-氧化物药物 |
| WO2008151253A1 (en) * | 2007-06-04 | 2008-12-11 | Threshold Pharmaceuticals, Inc. | Hypoxia activated prodrugs of antineoplastic agents |
| WO2009018163A1 (en) * | 2007-07-27 | 2009-02-05 | Threshold Pharmaceuticals, Inc. | Hypoxia activated prodrugs of anthracyclines |
| WO2009033165A1 (en) * | 2007-09-06 | 2009-03-12 | Threshold Pharmaceuticals, Inc. | Hypoxia activated prodrugs of bis-alkylating agents |
| US20090118031A1 (en) * | 2007-11-01 | 2009-05-07 | Qualizza Gregory K | Shaft Structure with Configurable Bending Profile |
| ES2884674T3 (es) | 2008-10-21 | 2021-12-10 | Immunogenesis Inc | Tratamiento del cáncer con el profármaco activado por hipoxia TH-302 en combinación con docetaxel o pemetrexed |
| WO2010073126A2 (en) * | 2008-12-22 | 2010-07-01 | The Governors Of The University Of Alberta | Compounds useful in delivering anti-neoplastic therapy and diagnostic imaging to hypoxic cells and methods of use thereof |
| CN101538256B (zh) * | 2009-04-28 | 2014-05-21 | 沈阳药科大学 | 3,4-二芳基呋喃-2,5-二酮类衍生物与3,4-二芳基-1h-吡咯-2,5-二酮类衍生物及其用途 |
| GB0907551D0 (en) | 2009-05-01 | 2009-06-10 | Univ Dundee | Treatment or prophylaxis of proliferative conditions |
| EP3311835B1 (en) | 2010-07-12 | 2021-03-24 | Threshold Pharmaceuticals Inc. | Administration of hypoxia activated prodrugs and antiangiogenic agents for the treatment of cancer |
| CN103327983A (zh) * | 2010-12-06 | 2013-09-25 | 梅里麦克制药股份有限公司 | 在使用包含蒽环类化疗剂的erbb2靶向免疫脂质体的治疗中防止心肌毒性的剂量和给药 |
| WO2012145684A1 (en) | 2011-04-22 | 2012-10-26 | The United States Of America, As Represented By The Secretary, Department Of Health & Human Services | Transient hypoxia inducers and their use |
| CN102260273B (zh) * | 2011-05-13 | 2015-04-22 | 兰州大学 | 脱氧鬼臼与5-氟尿嘧啶的拼合物及其制备与用途 |
| EP2793882A4 (en) | 2011-12-22 | 2015-04-29 | Threshold Pharmaceuticals Inc | ADMINISTRATION OF HYPOXIA ACTIVATED DRUGS IN COMBINATION WITH CHK1 INHIBITORS FOR THE TREATMENT OF CANCER |
| PT2797416T (pt) | 2011-12-28 | 2017-10-23 | Global Blood Therapeutics Inc | Compostos de benzaldeído substituído e métodos para a sua utilização no aumento da oxigenação de tecidos |
| ES2790358T3 (es) | 2011-12-28 | 2020-10-27 | Global Blood Therapeutics Inc | Compuestos de heteroaril aldehído sustituido y métodos para su uso en el aumento de la oxigenación tisular |
| CN104427984A (zh) | 2012-01-20 | 2015-03-18 | D·布朗 | 包括二脱水半乳糖醇和类似物的经取代的己糖醇类于治疗包括多形性胶质母细胞瘤和成神经管细胞瘤的肿瘤疾病和癌症干细胞的用途 |
| JP5676020B2 (ja) | 2012-02-13 | 2015-02-25 | 国立大学法人 筑波大学 | ニトロイミダゾールを用いたプロドラッグ |
| CN104204035B (zh) | 2012-03-30 | 2017-05-24 | 宇部兴产株式会社 | 具有封端基团的聚轮烷的制造方法 |
| WO2014058974A1 (en) * | 2012-10-10 | 2014-04-17 | Emory University | Methods of managing inflammation using glycolysis pathway inhibitors |
| US9278124B2 (en) | 2012-10-16 | 2016-03-08 | Halozyme, Inc. | Hypoxia and hyaluronan and markers thereof for diagnosis and monitoring of diseases and conditions and related methods |
| JP5922791B2 (ja) | 2012-10-29 | 2016-05-24 | 京セラ株式会社 | 弾性表面波センサ |
| JP2016501234A (ja) | 2012-12-03 | 2016-01-18 | メリマック ファーマスーティカルズ, インコーポレイテッドMerrimack Pharmaceuticals, Inc. | Her2陽性癌を処置するための併用療法 |
| EP2777694A1 (en) | 2013-03-14 | 2014-09-17 | Brij P. Giri | Hypoxia-Targeted Polymeric Micelles for Cancer Therapy and Imaging |
| US9604999B2 (en) | 2013-03-15 | 2017-03-28 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US10266551B2 (en) | 2013-03-15 | 2019-04-23 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| AP2015008718A0 (en) | 2013-03-15 | 2015-09-30 | Global Blood Therapeutics Inc | Compounds and uses thereof for the modulation of hemoglobin |
| CN105073728A (zh) | 2013-03-15 | 2015-11-18 | 全球血液疗法股份有限公司 | 化合物及其用于调节血红蛋白的用途 |
| WO2014150261A1 (en) | 2013-03-15 | 2014-09-25 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulaton of hemoglobin |
| US9422279B2 (en) | 2013-03-15 | 2016-08-23 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| MY180206A (en) | 2013-03-15 | 2020-11-25 | Global Blood Therapeutics Inc | Compounds and uses thereof for the modulation of hemoglobin |
| US20140274961A1 (en) | 2013-03-15 | 2014-09-18 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9802900B2 (en) | 2013-03-15 | 2017-10-31 | Global Blood Therapeutics, Inc. | Bicyclic heteroaryl compounds and uses thereof for the modulation of hemoglobin |
| US10100043B2 (en) | 2013-03-15 | 2018-10-16 | Global Blood Therapeutics, Inc. | Substituted aldehyde compounds and methods for their use in increasing tissue oxygenation |
| US9458139B2 (en) | 2013-03-15 | 2016-10-04 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US8952171B2 (en) | 2013-03-15 | 2015-02-10 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| JP2016519684A (ja) | 2013-04-08 | 2016-07-07 | デニス エム ブラウン | 準最適に投与された薬物療法の有効性を改善するための及び/又は副作用を低減するための方法および組成物 |
| WO2014181314A1 (en) | 2013-05-10 | 2014-11-13 | Department Of Biotechnology | Placental like alkaline phosphatase (plap) promoter mediated cell targeting |
| CN112472699A (zh) | 2013-07-26 | 2021-03-12 | 种族肿瘤学公司 | 改善比生群及衍生物的治疗益处的组合方法 |
| EP3024490A1 (en) | 2013-07-26 | 2016-06-01 | Threshold Pharmaceuticals, Inc. | Treatment of pancreatic cancer with a combination of a hypoxia-acti vated prodrug and a taxane |
| CN104434876B (zh) * | 2013-09-13 | 2018-04-27 | 布里吉·P·吉里 | 用于癌症疗法及成像的缺氧-标靶聚合微胞 |
| EA201992707A1 (ru) | 2013-11-18 | 2020-06-30 | Глобал Блад Терапьютикс, Инк. | Соединения и их применения для модуляции гемоглобина |
| DK3102208T3 (da) | 2014-02-07 | 2021-03-08 | Global Blood Therapeutics Inc | Krystallinsk polymorf af den frie base af 2-hydroxy-6-((2-(1-isopropyl-1h-pyrazol-5-yl)pyridin-3-yl)methoxy)benzaldehyd |
| CN106659765B (zh) | 2014-04-04 | 2021-08-13 | 德玛医药 | 二脱水半乳糖醇及其类似物或衍生物用于治疗非小细胞肺癌和卵巢癌的用途 |
| US10131683B2 (en) | 2014-07-17 | 2018-11-20 | Molecular Templates, Inc. | TH-302 solid forms and methods related thereto |
| AU2016229136B2 (en) | 2015-03-10 | 2018-08-09 | Obi Pharma, Inc. | DNA alkylating agents |
| MA41841A (fr) | 2015-03-30 | 2018-02-06 | Global Blood Therapeutics Inc | Composés aldéhyde pour le traitement de la fibrose pulmonaire, de l'hypoxie, et de maladies auto-immunes et des tissus conjonctifs |
| BR112017021167A2 (pt) | 2015-04-02 | 2018-07-03 | Obi Pharma, Inc. | composto, método de tratamento de câncer, e processo para preparar o composto de fórmula i |
| TWI772263B (zh) | 2015-06-24 | 2022-08-01 | 美商免疫原公司 | 含有dna烷化劑之氮丙啶 |
| BR112018005200A2 (pt) | 2015-09-16 | 2018-10-09 | Dfb Soria Llc | liberação de nanopartículas de fármaco e métodos de uso dos mesmos |
| CN105250325A (zh) * | 2015-10-28 | 2016-01-20 | 淄博齐鼎立专利信息咨询有限公司 | Demissine在制备治疗肾癌药物中的应用 |
| MA43373A (fr) | 2015-12-04 | 2018-10-10 | Global Blood Therapeutics Inc | Régimes posologiques pour 2-hydroxy-6-((2- (1-isopropyl-1h-pyrazol-5-yl)pyridin-3-yl)méthoxy)benzaldéhyde |
| AR108435A1 (es) | 2016-05-12 | 2018-08-22 | Global Blood Therapeutics Inc | Proceso para sintetizar 2-hidroxi-6-((2-(1-isopropil-1h-pirazol-5-il)-piridin-3-il)metoxi)benzaldehído |
| CN106554375B (zh) * | 2016-06-08 | 2019-10-18 | 浙江海正药业股份有限公司 | 一种蒽环类化合物、其制备方法及其用途 |
| TWI778983B (zh) | 2016-10-12 | 2022-10-01 | 美商全球血液治療公司 | 包含2-羥基-6-((2-(1-異丙基-1h-吡唑-5-基)吡啶-3-基)甲氧基)-苯甲醛之片劑 |
| ES2955884T3 (es) | 2017-03-15 | 2023-12-07 | Dfb Soria Llc | Terapia tópica para el tratamiento de malignidades de la piel con nanoparticulas de taxanos |
| CN106977501A (zh) * | 2017-03-20 | 2017-07-25 | 华东师范大学 | 一种基于2‑硝基咪唑‑1‑烷基醇的低氧激活前药 |
| CN107417672A (zh) * | 2017-04-12 | 2017-12-01 | 华东师范大学 | 一种基于2,2‑二甲基‑3‑(2‑硝基咪唑基)丙酸的低氧激活前药 |
| ES2935729T3 (es) * | 2017-09-14 | 2023-03-09 | Lankenau Inst Medical Res | Métodos y composiciones para el tratamiento del cáncer |
| CA3091027A1 (en) | 2018-02-02 | 2019-08-08 | Maverix Oncology, Inc. | Small molecule drug conjugates of gemcitabine monophosphate |
| JP7372252B2 (ja) | 2018-03-16 | 2023-10-31 | ディーエフビー ソリア リミテッド ライアビリティ カンパニー | タキサンのナノ粒子を用いる子宮頸部上皮内腫瘍(cin)および子宮頸癌の処置のための局所療法 |
| WO2020072377A1 (en) | 2018-10-01 | 2020-04-09 | Global Blood Therapeutics, Inc. | Modulators of hemoglobin for the treatment of sickle cell disease |
| KR20230023463A (ko) | 2021-08-10 | 2023-02-17 | 경북대학교 산학협력단 | 두경부암의 예후 예측을 위한 암 관련 섬유아세포의 바이오마커 |
| KR20240047452A (ko) | 2021-08-27 | 2024-04-12 | 아센타위츠 파마슈티컬즈 리미티드 | 동결건조 제형 용액과 동결건조 제형, 및 이의 방법과 용도 |
| WO2023025312A1 (zh) | 2021-08-27 | 2023-03-02 | 深圳艾欣达伟医药科技有限公司 | 使用th-302治疗parp抑制剂耐药的患者 |
| CN113968867A (zh) * | 2021-11-10 | 2022-01-25 | 聊城大学 | 一种喜树碱类前药及其用途 |
| CN114617876B (zh) * | 2022-01-28 | 2023-04-07 | 四川大学华西医院 | 一种抗肿瘤联合用药物 |
| CN116675620A (zh) * | 2022-02-22 | 2023-09-01 | 东南大学 | No供体化合物及其制备方法、药物组合物和应用 |
Family Cites Families (46)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3652579A (en) | 1969-06-26 | 1972-03-28 | Hoffmann La Roche | 1-methyl-2-substituted 5-nitroimidazoles |
| DE2229223C3 (de) | 1971-07-30 | 1975-12-18 | Gruppo Lepetit S.P.A., Mailand (Italien) | 2-Nitro-S-imidazol-Derivate und Verfahren zu deren Herstellung |
| US4921963A (en) * | 1987-04-13 | 1990-05-01 | British Columbia Cancer Foundation | Platinum complexes with one radiosensitizing ligand |
| JPH0819111B2 (ja) | 1987-10-22 | 1996-02-28 | ポーラ化成工業株式会社 | 2―ニトロイミダゾール誘導体及びこれを有効成分とする放射線増感剤 |
| US5190929A (en) | 1988-05-25 | 1993-03-02 | Research Corporation Technologies, Inc. | Cyclophosphamide analogs useful as anti-tumor agents |
| US4908356A (en) | 1988-05-25 | 1990-03-13 | Research Corporation Technologies, Inc. | Aldophosphamide derivatives useful as antitumor agents |
| US5403932A (en) | 1988-05-25 | 1995-04-04 | Research Corporation Technologies, Inc. | Phosphoramidates useful as antitumor agents |
| DE3835772A1 (de) | 1988-10-20 | 1990-04-26 | Deutsches Krebsforsch | Tumorhemmende saccharid-konjugate |
| JP2626727B2 (ja) | 1990-01-26 | 1997-07-02 | ポーラ化成工業株式会社 | 2―ニトロイミダゾール誘導体、その製造法及びこれを有効成分とする放射線増感剤 |
| US5233031A (en) * | 1991-09-23 | 1993-08-03 | University Of Rochester | Phosphoramidate analogs of 2'-deoxyuridine |
| WO1993008288A1 (en) | 1991-10-23 | 1993-04-29 | Cancer Research Campaign Technology Limited | Bacterial nitroreductase for the reduction of cb 1954 and analogues thereof to a cytotoxic form |
| US5750782A (en) | 1992-11-27 | 1998-05-12 | Cancer Research Campaign Technology Limited | Nitroaniline derivatives and their use as anti-tumour agents |
| US5306727A (en) | 1993-04-30 | 1994-04-26 | Research Corporation Technologies, Inc. | Phosphoramidates useful as antitumor agents |
| AU6859394A (en) | 1993-05-25 | 1994-12-20 | Auckland Uniservices Limited | Nitrobenzyl mustard quaternary salts and their use as hypoxia-selective cytotoxic agents |
| EP0648503B1 (en) | 1993-09-22 | 2000-07-26 | Hoechst Aktiengesellschaft | Pro-prodrugs, their production and use |
| JPH07309761A (ja) | 1994-05-20 | 1995-11-28 | Kyowa Hakko Kogyo Co Ltd | デュオカルマイシン誘導体の安定化法 |
| US5659061A (en) | 1995-04-20 | 1997-08-19 | Drug Innovation & Design, Inc. | Tumor protease activated prodrugs of phosphoramide mustard analogs with toxification and detoxification functionalities |
| GB9516943D0 (en) | 1995-08-18 | 1995-10-18 | Cancer Soc Auckland Div Nz Inc | Novel cyclopropylindoles and their secoprecursors,and their use as prodrugs |
| GB9526391D0 (en) | 1995-12-22 | 1996-02-21 | Diversey Equipment Technologie | Dispenser |
| US6218519B1 (en) | 1996-04-12 | 2001-04-17 | Pro-Neuron, Inc. | Compounds and methods for the selective treatment of cancer and bacterial infections |
| US6130237A (en) | 1996-09-12 | 2000-10-10 | Cancer Research Campaign Technology Limited | Condensed N-aclyindoles as antitumor agents |
| US6251933B1 (en) | 1996-12-13 | 2001-06-26 | The Cancer Research Campaign Technology Limited | Seco precursors of cyclopropylindolines and their use as prodrugs |
| DE19720312A1 (de) | 1997-05-15 | 1998-11-19 | Hoechst Ag | Zubereitung mit erhöhter in vivo Verträglichkeit |
| GB9818730D0 (en) | 1998-08-27 | 1998-10-21 | Univ Portsmouth | Collections of compounds |
| US6240925B1 (en) | 1999-03-23 | 2001-06-05 | Cynosure, Inc. | Photothermal vascular targeting with bioreductive agents |
| GB9909612D0 (en) | 1999-04-26 | 1999-06-23 | Cancer Res Campaign Tech | N-protected amines and their use as prodrugs |
| JP4629238B2 (ja) * | 1999-05-24 | 2011-02-09 | サザン・リサーチ・インスティテュート | イソホスホアミドマスタード・アナログ及びその使用 |
| AU5935600A (en) | 1999-07-14 | 2001-01-30 | Richard F. Borch | Phosphoramide compounds |
| DE60117247D1 (de) * | 2000-03-31 | 2006-04-20 | Purdue Research Foundation West Lafayette | Phosphoramidat-prodrugs |
| JP2002046334A (ja) | 2000-08-04 | 2002-02-12 | Riso Kagaku Corp | 孔版印刷装置及び該装置の印刷ドラム |
| EP1243276A1 (en) | 2001-03-23 | 2002-09-25 | Franciscus Marinus Hendrikus De Groot | Elongated and multiple spacers containing activatible prodrugs |
| US6506739B1 (en) | 2001-05-01 | 2003-01-14 | Telik, Inc. | Bis-(N,N'-bis-(2-haloethyl)amino)phosphoramidates as antitumor agents |
| KR20030033007A (ko) | 2001-05-31 | 2003-04-26 | 코울터 파머수티컬, 인코포레이티드 | 세포독소, 약물전구체, 링커 및 이에 유용한 안정화제 |
| FR2825926A1 (fr) * | 2001-06-14 | 2002-12-20 | Sod Conseils Rech Applic | Derives d'imidazoles modulant les canaux sodiques |
| US7091186B2 (en) | 2001-09-24 | 2006-08-15 | Seattle Genetics, Inc. | p-Amidobenzylethers in drug delivery agents |
| KR20030067275A (ko) | 2002-02-07 | 2003-08-14 | 주식회사 하이폭시 | 니트로이미다졸 및 위상 이성질화 효소 저해제를유효성분으로 포함하는 항암제 |
| JP4187096B2 (ja) | 2003-01-15 | 2008-11-26 | 独立行政法人科学技術振興機構 | 3次元フォトニック結晶製造方法 |
| GB0306907D0 (en) | 2003-03-26 | 2003-04-30 | Angiogene Pharm Ltd | Boireductively-activated prodrugs |
| GB0306908D0 (en) | 2003-03-26 | 2003-04-30 | Angiogene Pharm Ltd | Bioreductively activated stilbene prodrugs |
| ZA200507752B (en) * | 2003-03-28 | 2007-01-31 | Threshold Pharmaceuticals Inc | Compositions and methods for treating cancer |
| US6855695B2 (en) | 2003-06-13 | 2005-02-15 | Vion Pharmaceuticals, Inc. | Water-soluble SHPs as novel alkylating agents |
| CA2554463C (en) | 2004-02-06 | 2013-06-11 | Threshold Pharmaceuticals, Inc. | Anti-cancer therapies |
| WO2006057946A2 (en) | 2004-11-22 | 2006-06-01 | Threshold Pharmaceuticals, Inc. | Tubulin binding anti cancer agents and prodrugs thereof |
| WO2008011588A2 (en) | 2006-07-20 | 2008-01-24 | Threshold Pharmaceuticals, Inc. | Glycoconjugates of phosphoramidate alkylators for treatment of cancer |
| WO2008076826A1 (en) | 2006-12-13 | 2008-06-26 | Threshold Pharmaceuticals, Inc. | Pyrophosphoramide alkylators |
| WO2008083101A1 (en) | 2006-12-26 | 2008-07-10 | Threshold Pharmaceuticals, Inc. | Phosphoramidate alkylator prodrugs for the treatment of cancer |
-
2004
- 2004-03-28 ZA ZA200507752A patent/ZA200507752B/en unknown
- 2004-03-29 CA CA2520000A patent/CA2520000C/en not_active Expired - Lifetime
- 2004-03-29 AU AU2004226338A patent/AU2004226338C1/en not_active Expired
- 2004-03-29 US US10/549,545 patent/US7550496B2/en active Active
- 2004-03-29 NZ NZ543223A patent/NZ543223A/en not_active IP Right Cessation
- 2004-03-29 JP JP2006509466A patent/JP2006521409A/ja not_active Ceased
- 2004-03-29 EP EP04749522A patent/EP1622608A4/en not_active Withdrawn
- 2004-03-29 KR KR1020127002215A patent/KR101198526B1/ko active IP Right Grant
- 2004-03-29 MX MXPA05010407A patent/MXPA05010407A/es active IP Right Grant
- 2004-03-29 CN CNA2004800132768A patent/CN1791403A/zh active Pending
- 2004-03-29 WO PCT/US2004/009667 patent/WO2004087075A2/en active Application Filing
- 2004-03-29 EP EP13178249.2A patent/EP2671581B8/en not_active Expired - Lifetime
- 2004-03-29 ES ES13178249T patent/ES2759176T3/es not_active Expired - Lifetime
- 2004-03-29 KR KR1020057018367A patent/KR20050113670A/ko not_active IP Right Cessation
- 2004-03-29 CN CN2010105956028A patent/CN102161679A/zh active Pending
-
2005
- 2005-09-19 IL IL170970A patent/IL170970A/en active IP Right Grant
- 2005-10-26 NO NO20054988A patent/NO336266B1/no not_active IP Right Cessation
-
2009
- 2009-06-12 US US12/484,104 patent/US8299088B2/en active Active
-
2012
- 2012-01-26 JP JP2012014178A patent/JP5572644B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| KR20120035198A (ko) | 2012-04-13 |
| CA2520000A1 (en) | 2004-10-14 |
| NO20054988D0 (no) | 2005-10-26 |
| US7550496B2 (en) | 2009-06-23 |
| EP1622608A4 (en) | 2010-11-10 |
| MXPA05010407A (es) | 2006-05-31 |
| US20090312276A1 (en) | 2009-12-17 |
| IL170970A (en) | 2012-01-31 |
| JP5572644B2 (ja) | 2014-08-13 |
| NZ543223A (en) | 2008-11-28 |
| KR101198526B1 (ko) | 2012-11-06 |
| EP2671581A3 (en) | 2014-01-01 |
| EP1622608A2 (en) | 2006-02-08 |
| AU2004226338A1 (en) | 2004-10-14 |
| AU2004226338B2 (en) | 2011-05-19 |
| CA2520000C (en) | 2012-06-05 |
| CN102161679A (zh) | 2011-08-24 |
| EP2671581A2 (en) | 2013-12-11 |
| US20060258656A1 (en) | 2006-11-16 |
| US8299088B2 (en) | 2012-10-30 |
| EP2671581B8 (en) | 2019-11-13 |
| CN1791403A (zh) | 2006-06-21 |
| WO2004087075A2 (en) | 2004-10-14 |
| ZA200507752B (en) | 2007-01-31 |
| AU2004226338C1 (en) | 2012-01-12 |
| NO20054988L (no) | 2005-12-23 |
| EP2671581B1 (en) | 2019-10-02 |
| WO2004087075A3 (en) | 2005-03-24 |
| JP2012082227A (ja) | 2012-04-26 |
| JP2006521409A (ja) | 2006-09-21 |
| KR20050113670A (ko) | 2005-12-02 |
| NO336266B1 (no) | 2015-07-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2759176T3 (es) | Composiciones y procedimientos para el tratamiento del cáncer | |
| US9226932B2 (en) | Phosphoramidate alkylator prodrugs | |
| US20090042820A1 (en) | Tubulin Binding Anti Cancer Agents And Prodrugs Thereof | |
| US20080132458A1 (en) | Hypoxia-Activated Anti-Cancer Agents | |
| US20080214576A1 (en) | Topoisomerase Inhibitors and Prodrugs | |
| WO2007137196A2 (en) | Tubulin binding anti cancer compounds and prodrugs thereof |