-
Technisches
Gebiet der Erfindung
-
Diese
Erfindung betrifft die Gentherapie und insbesondere Materialien
und Verfahren, die zum Erzeugen hoher Titer rekombinanter AAV-Vektoren
zur Verwendung in gentherapeutischen Verfahren verwendet werden.
-
Hintergrund
der Erfindung
-
AAV-Vektoren
sind für
die Gentherapie nützlich,
aber ein großes
Hindernis war bisher das Unvermögen,
ausreichende Quantitäten
solcher rekombinanter Vektoren in Mengen herzustellen, die für die Anwendung
in der Humangentherapie klinisch nützlich wären. Dies ist ein besonderes
Problem für
in vivo Anwendungen, wie die direkte Abgabe an die Lunge.
-
Adeno-assoziierte
Virus- (AAV-) Vektoren gehören
zu einer kleinen Zahl rekombinanter Virusvektorsysteme, die sich
als in vivo Gentransfermittel nützlich
erwiesen haben (Überblick
bei Carter, 1992, Current Opinion in Biotechnology, 3:533:539; Muzyczka,
1992, Curr. Top. Microbiol. Immunol. 158:97-129) und daher potenziell
von großer
Bedeutung für
die Humangentherapie sind. AAV-Vektoren sind zur hochfrequenten,
stabilen DNA-Integration und -Expression in zahlreichen Zellen imstande,
einschließlich
zystischer Fibrose- (CF), bronchialer und nasaler Epithelzellen
(Flotte et al., 1992a, Am. J. Respir. Cell Mol. Biol., 7:349-356;
Egan et al., 1992, Nature, 358:581-584; Flotte et al., 1993a, J.
Biol. Chem., 268:3781-3790; Flotte et al., 1993b, Proc. Natl. Acad.
Sci. USA, in Druck), von menschlichem Knochenmark abgeleiteter Erythroleukämiezellen
(Walsh et al., 1992, Proc. Natl. Acad. Sci. USA, 89:7257-7261) und mehreren
anderen. AAV bedürfen
keiner aktiven Zellteilung für
eine stabile Expression, was ein deutlicher Vorteil gegenüber Retroviren
wäre, insbesondere
in Gewebe, wie dem menschlichen Atemwegepithel, wo die meisten Zellen
terminal differenziert und nicht teilend sind.
-
AAV
ist ein defektes Parvovirus, das nur in Zellen wächst, in welchen bestimmte
Funktionen durch ein co-infizierendes Helfervirus bereitgestellt
sind (siehe 1). Allgemeine Überblicke über das
AAV finden sich bei Carter, 1989, Handbook of Parvoviruses, Band
I, S. 169–228,
Berns, 1990, Virology, S. 1743–1764,
Raven Press (New York). Beispiele für co-infizierende Viren, die
Helferfunktionen für
das AAV-Wachstum und die Replikation bereitstellen, sind Adenoviren,
Herpesviren und in einigen Fällen
Pockenviren, wie Impfpocken. Die Eigenschaft der Helferfunktion
ist nicht bekannt, scheint aber eine gewisse indirekte Wirkung des
Helfervirus zu sein, das der Zelle die AAV-Replikation ermöglicht.
Dieses Konzept wird durch die Beobachtung unterstützt, dass
in bestimmen Fällen
eine AAV-Replikation bei einem geringen Maß an Effizienz in Abwesenheit
einer Helfervirus-Co-Infektion auftreten kann, wenn die Zellen mit
Mitteln behandelt werden, die entweder genotoxisch sind oder den
Zellzyklus aufbrechen.
-
Obwohl
AAV bis zu einem beschränkten
Maße in
Abwesenheit eines Helfervirus unter bestimmten unüblichen
Bedingungen replizieren können,
wie zuvor festgehalten wurde, ist das allgemeinere Ergebnis, dass die
Infektion von Zellen mit AAV in Abwesenheit von Helferfunktionen
zu einer Integration des AAV in das Wirtszellgenom führt. Das
integrierte AAV-Genom kann gewonnen und repliziert werden, um eine
Menge an infektiösen
AAV-Nachkommenpartikeln zu erhalten, wenn Zellen, die ein integriertes
AAV-Provirus enthalten mit einem Helfervirus, wie Adenovirus, superinfiziert
werden. Da die Integration von AAV ein effizientes Ereignis zu sein
scheint, legt dies nahe, dass AAV ein nützlicher Vektor zur Einführung von
Genen in Zellen für
eine stabile Expression für
Anwendungen wie der Humangentherapie sein könnte. Jüngere Ergebnisse (Kotin & Berns, 1989,
Virology, 170; 460–467;
Kotin et al., 1990, Proc. Natl. Acad. Sci. USA, 87:2211-2215; Samulski
et al., 1991, EMBO J. 10:3941-3950) haben nahegelegt, dass AAV eine
gewisse Präferenz
für eine
Integration an einer Stelle auf dem Humanchromosom 19 haben könnte, aber
das allgemeine Prinzip und der Mechanismus dieses Phänomens sind
noch nicht vollständig
geklärt.
-
AAV
hat einen sehr weiten Wirtsbereich, weder mit offensichtlicher Speziesnoch
Gewebespezifität, und
repliziert in nahezu jeder Zelllinie, die ihren Ursprung beim Menschen,
Affen oder Nagetier hat, vorausgesetzt, ein geeigneter Helfer ist
vorhanden. AAV ist überall
vorkommend und wurde von einer Vielzahl von Tierspezies isoliert,
einschließlich
der meisten Säugetier-
und mehrere Vogelspezies.
-
AAV
wird nicht mit der Ursache für
irgendeine Erkrankung in Zusammenhang gebracht. AAV ist kein transformierendes
oder onkogenes Virus. Die AAV-Integration
in Chromosome von Humanzelllinien bewirkt keine signifikante Veränderung
in den Wachstumseigenschaften oder morphologischen Eigenschaften
der Zellen. Diese Eigenschaften von AAV sind auch eine Empfehlung
für einen
möglicherweise
nützlichen
humangentherapeutischen Vektor, da die meisten der anderen viralen
Systeme, die für
diese Anwendung vorgeschlagen sind, wie Retroviren, Adenoviren,
Herpesviren oder Pockenviren, krankheitserregende Viren sind.
-
AAV-Partikel
bestehen aus einem Proteincapsid mit drei Capsidproteinen, VP1,
VP2 und VP3, die ein DNA-Genom einschließen. Das AAV-DNA-Genom ist
ein lineares, einzelsträngiges
DNA-Molekül
mit einem Molekulargewicht von etwa 1,5 × 106 Dalton
oder einer Länge
von etwa 4680 Nucleotiden. Stränge
jeder Komplementärrichtung, "Plus"- oder "Minus-" Stränge, sind
in einzelne Partikel verpackt, aber jedes Partikel hat nur ein DNA-Molekül. Gleiche
Anzahlen von AAV-Partikeln enthalten entweder einen Plus- oder einen
Minus-Strang. Jeder Strang ist gleichermaßen infektiös, und die Replikation erfolgt
durch Umkehr des elterlichen infizierenden Einzelstrangs zu einer
doppelsträngigen
Form und anschließende
Amplifizierung eines großen Pools
doppelsträngiger
Moleküle,
von welchen Nachkommen-Einzelstränge
abgelöst
und in Capside verpackt werden. Doppel- oder einzelsträngige Kopien
von AAV-Genomen, die in bakterielle Plasmide oder Phagemide eingesetzt
sind, sind infektiös,
wenn sie in Adenovirus-infizierte Zellen transfiziert werden, und
dies hat die Untersuchung der AAV-Genetik und die Entwicklung von
AAV-Vektoren ermöglicht.
Der Replikationszyklus von AAV ist in 1 dargestellt.
-
Das
AAV2-Genom hat eine Kopie der 145 Nucleotid langen ITR ("inverted terminal
repeat" – invertierte terminate
Wiederholung) jedes Endes einer einmaligen Sequenzregion mit einer
Länge von
etwa 4470 Nucleotiden (Srivastava et al., 1983, J. Virol., 45:555-564),
die zwei offene Hauptleserahmen für die rep- und cap-Gene enthält (Hermonat
et al., J. Virol. 51:329-339; Tratschin et al., 1984a, J. Virol.,
51:611-619). Die einmalige Region enthält drei Transkriptionspromotoren,
p5, p19 und p40 (Laughlin et al., 1979, Proc. Natl.. Acad. Sci.
USA, 76:5567-5571), die zur Expression der rep- und cap-Gene verwendet
werden. Die ITR-Sequenzen sind in cis erforderlich und sind ausreichend,
um einen funktionellen Replikationsursprung (ori) bereitzustellen, und sind
auch ausreichend, um Signale zu liefern, die für eine Integration in das Zellgenom
erforderlich sind, wie auch für
ein effizientes Ausschneiden und Gewinnen aus Wirtszellenchromosomen
oder aus rekombinanten Plasmiden. Zusätzlich wurde gezeigt, dass
die ITR direkt als Transkriptionspromotor in einem AAV-Vektor dienen
kann (Flotte et al., 1993, siehe oben).
-
Die
rep- und cap-Gene sind in trans erforderlich, um Funktionen für die Replikation
beziehungsweise Encapsidation eines viralen Genoms bereitzustellen.
Das rep-Gen wird von zwei Promotoren, p5 und
p19, exprimiert. Die Transkription von p5 ergibt eine ungespleißte 4,2 kb mRNA, die für ein Protein,
Rep78, codiert und eine gespleißte
3,9 kb mRNA, die für
ein Protein, Rep68, codiert. Die Transkription von p19 ergibt
eine ungespleißte
mRNA, die für
Rep52 codiert, und eine gespleißte
3,3 kb mRMA, die für
Rep40 codiert. Somit umfassen die vier Rep-Proteine alle eine gemeinsame
interne Regionsequenz, unterscheiden sich aber in Hinblick auf ihre
Amino- und Carboxyl-Endregionen. Nur Rep78 und Rep68 sind für eine AAV-Doppelstrang-DNA-Replikation
notwendig, aber Rep52 und Rep40 scheinen für die Nachkommen-Einzelstrang-DNA-Akkumulation erforderlich
zu sein. Mutationen in Rep78 und Rep68 sind phänotypisch Rep– ,
während
Mutationen, die nur Rep52 und Rep40 betreffen, Rep+,.
aber Ssd– sind.
Rep68 und Rep78 binden spezifisch an die Haarnadelstruktur der AAV-ITR
und besitzen mehrere Enzymaktivitäten, die zur Lösung der
Replikation an den AAV-Termini notwendig sind. Rep52 und Rep40 haben
keine dieser Eigenschaften.
-
Die
Rep-Proteine, vorwiegend Rep78 und Rep68 weisen mehrere pleiotrope
regulatorische Aktivitäten auf,
einschließlich
der positiven und negativen Regulation von AAV-Genen und der Expression
von einigen heterologen Promotoren, wie auch hemmende Wirkungen
auf das Zellwachstum (Tratschin et al., 1986, Mol. Cell Biol., 6:2884-2894;
Labow et al., 1987, Mol. Cell Biol., 7:1320-1325; Khleif et al.,
Virology, 181:738-741). Der AAV-p5-Promotor
ist durch Rep78 oder Rep68 negativ autoreguliert (Tratschin et al.,
1986, Mol. Cell Biol., 6:2884-2894). Wegen der hemmenden Wirkungen
der Expression von rep auf das Zellwachstum wurde eine konstitutive
Expression von rep in Zelllinien nicht leicht erreicht. Zum Beispiel
berichteten Mendelson et al. (1988, Virology, 166:154-165) von einem
sehr geringen Maß an
Expression einiger Rep-Proteine in bestimmten Zelllinien nach einer
stabilen Integration von AAV-Genomen.
-
Die
Proteine VP1, VP2 und VP3 haben alle eine gemeinsame überlappende
Sequenz, unterscheiden sich aber darin, dass VP1 und VP2 eine zusätzliche
aminoterminale Sequenz enthalten. Alle drei werden von demselben
cap-Gen-Leserahmen
codiert, der von einer gespleißten
2,3 kb mRNA exprimiert wird, die von dem p40-Promotor
transkribiert wird. VP2 und VP3 werden von derselben mRNA unter
Verwendung wechselnder Initiationscodons erzeugt. Für VP1 codiert
eine Minor-mRNA unter Verwendung einer 3'-Donorstelle, die 30 Nucleotide stromaufwärts von
dem 3'-Donor liegt,
der für
die Major-mRNA verwendet wird, die für VP2 und VP3 codiert. VP1,
VP2 und VP3 sind alle für
die Capsidproduktion notwendig. Mutationen, die alle drei Proteine (Cap–)
eliminieren, verhindern eine Ansammlung einer einzelsträngigen Nachkommen-AAV-DNA, während Mutationen
im VP1-Aminoterminus (Lip–, Inf–)
eine einzelsträngige
Produktion ermöglichen,
aber eine Zusammenfügung
stabiler infektiöser
Partikel vermeiden.
-
Die
genetische Analyse von AAV, die zuvor beschrieben wurde, beruht
auf der Mutationsanalyse von AAV-Genomen, die molekular zu bakteriellen
Plasmiden geklont wurden. In einer früheren Arbeit wurden molekulare
Klone infektiöser
Genome von AAV durch Einfügen
doppelsträngiger
Moleküle
von AAV in Plasmide durch Verfahren wie das GC-Tailing (Samulski
et al., 1982, Proc. Natl. Acad Sci. USA, 79:2077-2081), Hinzufügen synthetischer
Linker, die Restriktionsendonuclease enthalten (Laughlin et al.,
1983, Gene, 23:65-73) oder durch direkte, stumpfendige Ligation
(Senapathy & Carter,
1984, J. Biol. Chem., 259:4661-4666) konstruiert. Es wurde dann
gezeigt, dass die Transfektion solcher AAV-rekombinanter Plasmide
in Säugetierzellen,
die auch mit einem geeigneten Helfervirus, wie Adenovirus, infiziert
waren, zu einer Gewinnung und Exzision des AAV-Genoms frei von Plasmidsequenzen
und Replikation des gewonnenen Genoms und Erzeugung einer Ausbeute
von infektiösen
Nachkommen-AAV-Partikeln führte
(siehe 1). Dies schuf
die Basis für
die Durchführung
einer genetischen Analyse von AAV, wie oben zusammengefasst, und
ermöglichte
die Konstruktion von AAV-transduzierenden Vektoren.
-
Basierend
auf der genetischen Analyse wurden die allgemeinen Prinzipien der
AAV-Vektorkonstruktion definiert, wie kürzlich zusammengefasst (Carter,
1992, Current Opinions in Biotechnology, 3:533-539; Muzyczka, 1992,
Current Topics in Microbiology and Immunology, 158:97-129). AAV-Vektoren
werden in AAV rekombinanten Plasmiden durch Substituieren von Teilen
der AAV-Codierungssequenz durch Fremd-DNA konstruiert, um ein Vektorplasmid
zu erzeugen. In dem Vektorplasmid müssen die terminalen (ITR) Abschnitte
der AAV-Sequenz intakt bleiben, da diese Regionen in cis für mehrere
Funktionen notwendig sind, einschließlich der Exzision von dem
Plasmid nach der Transfektion, der Replikation des Vektorgenoms
und der Integration und Gewinnung aus einem Wirtszellengenom. Der
Vektor kann dann in ein AAV-Partikel verpackt werden, um ein AAV-transduzierendes
Virus durch Transfektion des Vektorplasmids in Zellen zu erzeugen,
die mit einem geeigneten Helfervirus, wie Adenovirus oder Herpesvirus,
infiziert sind. Um eine Replikation und Encapsidation des Vektorgenoms
in AAV-Partikel zu erreichen, muss das Vektorplasmid für alle AAV-Funktionen, die in trans
notwendig sind, nämlich
rep und cap, komplementiert werden, die bei der Konstruktion des
Vektorplasmids deletiert wurden.
-
Es
gibt wenigstens zwei wünschenswerte
Merkmale jedes AAV-Vektors, der zur Verwendung in der Humangentherapie
bestimmt ist. Erstens muss der transduzierende Vektor bei ausreichend
hohen Titern erzeugt werden, damit er als Abgabesystem brauchbar
ist. Dies ist besonders für
Gentherapiestratageme wichtig, die auf eine in vivo Abgabe des Vektors
gerichtet sind. Es ist wahrscheinlich, dass für viele gewünschte Anwendungen von AAV-Vektoren,
wie die Behandlung einer zystischen Fibrose durch direkte in vivo
Abgabe an den Atemweg, die erforderliche Dosis an transduzierendem
Vektor höher
als 1010 sein kann. Zweitens müssen die
Vektorzubereitungen frei von Wildtyp-AAV-Virus sein. Hohe Titer von AAV-Vektoren
sind aus mehreren Gründen
schwierig zu erreichen, die die bevorzugte Encapsidation von Wildtyp-AAV-Genomen
umfassen, wenn diese vorhanden sind oder durch Rekombination erzeugt
werden, sowie die Unfähigkeit,
ausreichende komplementierende Funktionen, wie rep oder cap zu erzeugen.
Geeignete Zelllinien, die solche komplementierenden Funktionen exprimieren,
wurden nicht erzeugt, zum Teil wegen mehrerer Hemmfunktionen des rep-Gens.
-
Die
ersten AAV-Vektoren, die beschrieben wurden, enthielten fremde Reportergene,
wie neo oder cat oder dhfr, die von AAV-Transkriptionspromotoren
oder einem SV40-Promotor exprimiert wurden (Tratschin et al., 1984b,
Mol. Cell. Biol., 4:2071-2081; Hermonat & Muzyczka, 1984, Proc. Natl. Acad.
Sci. USA, 81:6466-6470; Tratschin et al., 1985, Mol. Cell. Biol.,
5:3251-3260; McLaughlin et al., 1988, J. Virol., 62:1963-1973; Lebkowski
et al., 1988 Mol. Cell. Biol., 7:349-356). Diese Vektoren wurden
in AAV-transduzierende Partikel durch Co-Transfektion in Adenovirus-infizierte
Zellen gemeinsam mit einem zweiten Verpackungsplasmid verpackt,
das die AAV-rep- und -cap-Gene enthielt, die von den natürlichen
Wildtyp-AAV-Transkriptionspromotoren exprimiert werden. In einem
Versuch, eine Verpackung des Verpackungsplasmids zu vermeiden, das
die rep- und cap-Gene enthielt, die von den natürlichen Wildtyp-AAV-Transkriptionspromotoren exprimiert
werden. In einem Versuch, eine Verpackung des Verpackungsplasmids
in AAV-Partikel zu vermeiden, wurden mehrere Methoden angewandt.
In einigen Fällen
(Hermonat & Muzyczka,
1984; McLaughlin et al., 1988) war in dem Verpackungsplasmid eine
große
Region von Bakteriophage Lambda DNA in der AAV-Sequenz eingesetzt,
um ein übergroßes Genom
zu erzeugen, das nicht verpackt werden konnte. In anderen Fällen (Tratschin
et al., 1984b; Tratschin et al., 1985, Lebkowski et al., 1988) waren
bei dem Verpackungsplasmid die ITR-Regionen von AAV deletiert, so
dass es nicht exzisiert und repliziert und daher nicht verpackt
werden konnte. Mit allen diesen Methoden konnte die Erzeugung von
Partikeln, die Wildtyp-AAV-DNA enthielten, nicht vermieden werden
und es konnten keine effektiven hohen Titer von AAV-transduzierenden
Partikeln erzeugt werden. Tatsächlich
wurden Titer von nicht mehr als 104 ml von
Hermonat & Muzyczka,
1984, angegeben. Die Produktion von Wildtyp-AAV-Partikeln in diesen
Studien war wahrscheinlich auf die Gegenwart einer überlappenden
Homologie zwischen AAV-Sequenzen, die in dem Vektor und den Verpackungsplasmiden
vorhanden waren, zurückzuführen. Senapathy
und Carter (1984, J. Biol. Chem., 259:4661-4666) zeigten, dass das
Ausmaß der
Rekombination in einem solchen System etwa gleich dem Ausmaß an Sequenzüberlappung
ist. In einem Überblick über die
frühe Arbeit
(Carter 1989, Handbook of Parvoviruses, Band II, S. 247–284, CRC Press,
Boca Raton, FL) wurde nahegelegt, dass Titer von 106 pro
ml erhalten werden könnten,
aber dies beruhte auf den oben genannten Studien, in welchen große Mengen
an Wildtyp-AAV die Vektorzubereitung kontaminierten. Solche Vektorzubereitungen,
die Wildtyp-AAV enthalten, sind für die Humangentherapie nicht nützlich.
Ferner wiesen diese frühen
Vektoren geringe Transduktionseffizienzen auf und transduzierten
nicht mehr als 1 oder 2 % von Zellen in Kulturen verschiedener Humanzelllinien,
obwohl die Vektoren in Multiplizität von bis zu 50 000 Partikeln
pro Zelle zugeführt
wurden. Dies könnte
teilweise die Kontamination mit Wildtyp-AAV-Partikeln und die Gegenwart
des AAV-rep-Gens in dem Vektor reflektieren. Ferner zeigten Samulski et
al. (1989, J. Virol., 63:3822-3829), dass das Vorhandensein des
Wildtyp-AAV die Aus beute an verpacktem Vektor deutlich erhöhte. Somit
kann die Ausbeute an verpacktem Vektor in Verpackungssystemen, in
welchen die Produktion an Wildtyp-AAV beseitigt wird, tatsächlich geringer
sein. Dennoch ist es für
die Verwendung in jeder klinischen Anwendung am Menschen wichtig,
die Produktion an Wildtyp-AAV zu beseitigen.
-
Zusätzliche
Studien (McLaughlin et al., 1988; Lebkoswski et al., 1988) zur Erzeugung
von AAV-Vektoren, die das AAV-rep- oder -cap-Gen nicht enthalten,
stießen
weiterhin auf die Erzeugung von Wildtyp-AAV und erzeugten weiterhin
sehr geringe Transduktionsfrequenzen auf Humanzelllinien. Daher
berichteten McLaughlin et al., 1988, dass AAV-rep–cap–=Vektoren,
die das neo-Gen enthielten, das mit demselben Verpackungsplasmid
verpackt war, das früher
von Hermonat & Muzyczka
(1984) verwendet wurde, weiterhin Wildtyp-AAV enthielten. Folglich
war es nur möglich,
dieses Virus bei einer Multiplizität von 0,03 Partikeln pro Zelle (d.h.,
300 infektiösen
Einheiten pro 10 000 Zellen) zu verwenden, um Doppeltreffer mit
Vektor- und Wildtyp-Partikel zu vermeiden. Nachdem das Experiment
auf diese Weise, durch Infizieren von 32 000 Zellen mit 1000 infektiösen Einheiten,
durchgeführt
wurde, wurden durchschnittlich 800 Geneticin-resistente Kolonien
erhalten. Obwohl dies so interpretiert wurde, dass das Virus imstande
sei, eine Transduktionsfrequenz von 80 % zu erreichen, waren tatsächlich nur
2,5 % der Zellen transduziert. Somit war der effektiv nützliche
Titer dieses Vektors begrenzt. Ferner zeigt diese Studie nicht,
dass der tatsächliche
Titer der Vektorzubereitung höher
als jene war, die zuvor von Hermonat & Muzyczka (1984) erhalten worden
waren. Ebenso verpackten Lebkowski et al., 1988, AAV-Vektoren, die
weder ein rep- noch ein cap- Gen enthielten, und verwendeten ein
ori-Verpackungsplasmid pBalA, das mit jenem identisch ist, das früher von
Tratschin et al. (1984b, 1985) verwendet wurde, und berichteten
von Transduktionsfrequenzen, die ähnlich nieder waren, da für mehrere
Humanzelllinien nicht mehr als 1 % der Zellen zu einer Geneticinresistenz
transduziert werden konnten, auch nicht mit den am höchsten konzentrierten
Vektormaterialien. Lebkowski et al. (1988) gaben die tatsächlichen
Vektortiter nicht aussagekräftig
an, aber die biologischen Tests, die nicht mehr als 1 % Transduktionsfrequenz
zeigten, wenn 5 × 106 Zellen drei ml einer Vektorzubereitung
ausgesetzt wurden, lassen darauf schließen, dass der Titer geringer
als 2 × 104 war. Ebenso enthält das pBa1-Verpackungsplasmid überlappende
Homologie mit der ITR-Sequenz in dem Vektor und führt zur
Erzeugung von Wildtyp-AAV durch Rekombination.
-
Laface
et al. (1988) verwendete denselben Vektor wie jenen, der von Hermonat & Muzyczka (1984) verwendet
worden war, der auf dieselbe Weise hergestellt war, und erreichten
eine Transduktionsfrequenz von 1,5 % in murinen Knochenmarkkulturen,
wobei sich wieder ein sehr geringer Titer zeigte.
-
Samulski
et al. (1987, J. Virol., 61:3096-3101) konstruierten ein Plasmid,
das als pSub201 bezeichnet wurde, das ein intaktes AAV-Genom in
einem bakteriellen Plasmid ist, aber eine Deletion of 13 Nucleotiden
am Ende jeder ITR aufweist und somit weniger effizient als andere
AAV-Plasmide gewonnen und repliziert wird, die das gesamte AAV-Genom
enthalten. Samulski et al. (1989, J. Virol., 63:3822-3828) konstruierten
AAV-Vektoren auf der Basis von pSub201, die aber rep und cap deletiert
haben und entweder ein hyg- oder neo-Gen enthalten, das von einem
frühen
SV40 Genpromotor exprimiert wird. Sie verpackten diese Vektoren
durch Co-Transfektion mit einem Verpackungsplasmid, das als pAAV/Ad
bezeichnet wird, das aus der gesamten AAV-Nucleotidseugenz von Nucleotid
190 bis 4490 besteht, das an jedem Ende mit einer Kopie der Adenovirus-ITR
eingeschlossen ist. In diesem Verpackungsplasmid wurden die AAV-rep-
und cap-Gene von den natürlichen
AAV-Promotoren p5, p19 und
p40 exprimiert. Es wurde angenommen, dass
die Funktion der Adenovirus-ITR in pAAV/Ad den Expressionswert von
AAV-Capsidproteinen verstärkt.
Rep wird jedoch von seinem homologen Promotor exprimiert und wird
negativ reguliert und daher ist seine Expression begrenzt. Unter
Verwendung ihres Encapsidationssystems erzeugten Samulski et al.,
1989, AAV-Vektormaterialien, die im Wesentlichen frei von Wildtyp-AAV
waren, aber transduzierende Titer von nur 3 × 104 Hygromycin-resistenten
Einheiten pro ml Überstand
hatten. Wenn ein Wildtyp-AAV-Genom in dem Verpackungsplasmid verwendet
wurde, war der Titer der AAV-Vektorzubereitung auf 5 × 104 erhöht.
Der geringe Titer, der in diesem System erzeugt wurde, scheint somit
teilweise auf den Defekt in den ITR-Sequenzen des pSub201-Ausgangsplasmids
zurückzuführen zu
sein, das zur Vektorkonstruktion verwendet wurde, und teilweise
auf die begrenzende Expression von AAV-Genen von pAAV/Ad. In einem
Versuch, den Titer der AAVneo-Vektorzubereitung zu erhöhen, erzeugten
Samulski et al., 1989, Vektormaterialien durch Transfizieren, im
Bulk, von dreißig
10 cm Schalen mit 293-Zellen und Konzentrieren des Vektormaterials
durch Bandierung in CsCl. Dies erzeugte ein AAVneo Vektormaterial,
das insgesamt 108 Partikel enthielt, wie
durch einen DNA-Dot-Blot-Hybridisierungstest gemessen wurde. Wenn
dieses Vektormaterial in Multiplizitäten von bis zu 1000 Partikeln
pro Zelle verwendet wurde, wurde eine Transduktionsfrequenz von
70 erhalten. Dies legt nahe, dass das Partikel/Transduktionsverhältnis etwa
500 bis 1000 Partikel ist, da bei dem Verhältnis von einer transduzierenden
Einheit pro Zelle der erwartete Anteil von Zellen, die transduziert
sein sollten, gemäß der Poissonschen
Verteilung 63 % ist.
-
Obwohl
das System von Samulski et al., 1989, unter Verwendung des Vektorplasmids
pSub201 und des Verpackungsplasmids pAAV/Ad keine überlappende
AAV-Sequenzhomologie zwischen den zwei Plasmiden hatte, gibt es
eine überlappende
Homologie an den Xbal-Stellen und eine Rekombination dieser Stellen führt zur
Erzeugung eines vollständigen
Wildtyp-AAV. Das heißt,
obwohl keine überlappende
Homologie der AAV-Sequenz vorhanden ist, ist die vollständige AAV-Sequenz
in den zwei Plasmiden enthalten, und daher kann eine Rekombination
einen Wildtyp-AAV erzeugen, was unerwünscht ist. Dass diese Rekombinationsklasse
in AAV-Plasmiden auftritt, wurde von Senapathy & Carter (1984, J. Biol. Chem., 259:4661-4666)
gezeigt. Wegen der Probleme eines geringen Titers und der Möglichkeit,
Wildtyp-Rekombinante zu erzeugen, besitzt daher das System, das
von Samulski et al., 1989, beschrieben wurde, keine Nützlichkeit
für die
Humangentherapie.
-
Mehrere
andere Berichte haben AAV-Vektoren beschrieben. Srivastava et al.
(1989, Proc. Natl. Acad. Sci. USA, 86:8078-8082) beschrieben einen
AAV-Vektor auf der
Basis des pSub201-Plasmids von Samulski et al. (1987), in dem die
Kodierungssequenzen von AAV durch die Kodierungssequenzen eines
anderen Parvovirus, B19, ersetzt waren. Dieser Vektor wurde in AAV-Partikel
unter Verwendung des pAAV/Ad-Verpackungsplasmids verpackt und erzeugte
einen funktionalen Vektor, aber es wurden keine Titer angegeben.
Dieses System beruht auf pSub201 und hat daher den Mangel, der zuvor
für dieses
Plasmid beschrieben wurde. Zweitens enthielten sowohl der Vektor
als auch das Verpackungsplasmid überlappende
AAV-Sequenzen (die ITR-Regionen) und daher ist eine Rekombination,
die zu kontaminierendem Wildtyp-Virus führt, äußerst wahrscheinlich.
-
WO92/10574
betrifft die Hemmung von HIV. Dieses Dokument betrifft Pasmide mit
einem rep-Gen, das funktionsfähig
an den HIV-LTR-Promotor gebunden ist eine AAVITR-Sequenz enthält.
-
Chatterjee
et al. (1991, Vaccines 91, Cold Spring Harbor Laboratory Press,
S. 85–89),
Wong et al. (1991, Vaccines 91, Gold Spring Harbor Laboratory Press,
S. 183–189),
und Chatterjee et al. (1992, Science, 258:1485-1488) beschreiben
AAV-Vektoren, die zum Exprimieren von Antisense-RNA bestimmt sind,
die gegen infektiöse
Viren, wie HIV oder Herpes simplex Virus, gerichtet ist.
-
Die
Autoren gaben jedoch keine Titer für ihre AAV-Vektormaterialien
an. Ferner verpackten sie ihre Vektoren unter Verwendung eines Ori–-Verpackungsplasmids,
das jenem analog ist, das von Tratschin et al. (1984b, 1985) verwendet
wurde, das das Ba1A-Fragment des AAV-Genoms enthält, und daher enthielt ihr
Verpackungsplasmid AAV-Vektorsequenzen, die mit AAV-Sequenzen Homologie
aufweisen, die in ihren Vektorkonstrukten vorhanden waren. Dies
führt auch
zur Erzeugung von Wildtyp-AAV. So verwendeten Chatterjee et al.
und Wong et al. ein Verpackungssystem, von dem bekannt ist, dass
es nur einen geringen Titer ergibt, und das wegen der überlappenden
Homologie in den Vektor- und Verpackungssequenzen zur Erzeugung
von Wildtyp-AAV-Genomen
führen
kann.
-
Andere
Berichte haben die Verwendung von AAV-Vektoren zur Expression von
Genen in Humanlymphozyten (Muro-Cacho et al., 1992, J. Immunotherapy,
11:231-237) oder einer humanen, erythroiden Leukämiezelllinie (Walsh et al.,
1992, Proc. Natl. Acad. Sci. USA, 89:7257-7261) mit Vektoren, die
auf dem pSub201-Vektorplasmid basieren, und pAAV/Ad-Verpackungsplasmid
beschrieben. Auch hier wurden keine Titer von Vektormaterialien
angegeben und waren offensichtlich gering, da ein selektives Markergen
zur Identifizierung jener Zellen verwendet wurde, die erfolgreich
mit dem Vektor transduziert waren.
-
Es
wurde von der Transduktion humaner Luftweg-Epithelzellen, die in
vitro von einem Patienten mit zystischer Fibrose gezüchtet wurden,
mit einem AAV-Vektor
berichtet, der das selektive Markergen neo von dem AAV-p5-Promotor exprimiert (Flotte et al., 1992,
Am. H. Respir. Cell. Mol. Biol., 7:349-356). In dieser Studie wurde
der AAVneo-Vektor in AAV-Partikeln unter Verwendung des pAAV/Ad-Verpackungsplasmids
verpackt. Bis zu 70 % der Zellen in der Kultur konnten zu einer
Geneticinresistenz transduziert werden und das Partikel/Transduktionsverhältnis war ähnlich jenem,
das von Samulski et al.
-
(1989)
berichtet wurde. Somit war eine Multiplizität von bis zu mehreren hundert
Vektorpartikeln pro Zelle erforderlich, um eine Transduktion von
70 % der Zellen zu erreichen. Die Transduktion von humanen Luftweg-Epithelzellen
in einer in vitro Kultur unter Verwendung eines AAV-transduzierenden
Vektors, der das CFTR-Gen von dem AAV-ITR-Promotor exprimiert, zeigte,
dass die Zellen funktional hinsichtlich des elektrophysiologischen
Defekts in der Chloridkanalfunktion korrigiert werden konnten, der
in Zellen von Patienten mit zystischer Fibrose vorhanden ist (Egan
et al., Nature, 1992, 358:581-584; Flotte et al., J. Biol. Chem., 268:3781-3790).
-
Die
oben genannten Studien legen nahe, dass AAV-Vektoren eine mögliche Nützlichkeit
als Vektoren zur Behandlung von Erkrankungen des Menschen durch
Gentherapie haben könnten.
Die Fähigkeit,
ausreichende Mengen an AAV-Vektoren zu erzeugen, war jedoch eine
ernsthafte Beschränkung
in der Entwicklung einer Humangentherapie unter Verwendung von AAV-Vektoren.
Ein Aspekt dieser Einschränkung
hat auch zu dem bisherigen Fehlen von Studien geführt, die
AAV-Vektoren in in vitro Tiermodellen verwenden. Dies reflektiert
allgemein die Schwierigkeit, die mit der Erzeugung ausreichender
Mengen an AAV-Vektormaterialien mit ausreichend hohem Titer verbunden
ist, um bei der Analyse einer in vivo Abgabe und Genexpression nützlich zu
sein. Einer der einschränkenden
Faktoren für
die AAV-Gentherapie war die relative Ineffizienz der Vektorverpackungssysteme,
die verwendet wurden. Wegen des Mangels an Zelllinien, die die AAV-trans-komplementierenden
Funktionen exprimieren, wie rep und cap, wurde eine Verpackung von
AAV-Vektoren in Adenovirus-infizierten Zellen durch Co-Transfektion
eines Verpackungsplasmids und eines Vektorplasmids erreicht. Die
Effizienz dieses Verfahrens kann durch die Effizienz der Transfektion
jedes der Plasmidkonstrukte begrenzt sein, wie auch durch das Ausmaß der Expression
von Rep-Proteinen von den Verpackungsplasmiden, die bisher beschrieben
wurden. Jedes dieser Probleme scheint mit den biologischen Aktivitäten der AAV-Rep-Proteine
in Zusammenhang zu stehen. Zusätzlich,
wie zuvor festgehalten wurde, haben alle der zuvor beschriebenen
Verpackungssysteme die Fähigkeit,
Wildtyp-AAV durch Rekombination zu erzeugen.
-
Der
Mangel an Zelllinien, die stabil funktionales Rep exprimieren, reflektiert
offensichtlich eine zytotoxische oder zytostatische Funktion von
Rep, wie durch die Hemmung von Rep durch eine neo-resistenten Kolonieformation
gezeigt wird (Labow et al., 1987; Trempe et al., 1991). Dies scheint
auch mit der Neigung von Rep in Zusammenhang zu stehen, den immortalisierten
Phänotyp
in gezüchteten
Zellen umzukehren, wodurch die Produktion von Zelllinien, die funktionales
Rep stabil exprimieren, extrem schwierig wird. Es wurden mehrere
Versuche unternommen, Zelllinien zu erzeugen, die Rep exprimieren.
Mendelson et al. (1988, Virology, 166:154-165) berichteten, dass
sie in einer Zelllinie nach einer stabilen Transfektion von HeLa
oder 293-Zellen mit Plasmiden, die ein AAV-rep-Gen enthielten, eine
gewisse geringe Expression von AAV-Rep52-Protein erreichten, aber nicht von Rep78-
oder Rep68-Protein. Auf Grund des Fehlens von Rep78- und Rep68-Proteinen konnte
der Vektor in der Zelllinie nicht erzeugt werden. Eine andere Zelllinie
lieferte eine kaum erfassbare Menge an Rep78, die nicht funktional
war.
-
Vincent
et al. (1990, Vaccines 90, Cold Spring Harbor Laboratory Press,
S. 353-359) versuchten
Zelllinien herzustellen, die die AAV-rep- und -cap-Gene enthielten,
die von den normalen AAV-Promotoren exprimiert werden, aber diese
Versuche waren nicht erfolgreich, entweder weil die Vektoren mit
einem hundertfachen Überschuss
an Wildtyp-AAV-Partikeln kontaminiert waren, oder weil die Vektoren
bei nur sehr geringen Titern von weniger als 4 × 103 erzeugt
wurden.
-
In
einer anderen Methode konstruierten Lebkowski et al. (US Patent
5,173,414, erteilt am 22. Dez. 1992) Zelllinien, die AAV-Vektoren
in einem episomalen Plasmid enthielten. Diese Zelllinien konnten
dann mit Adenovirus infiziert und mit den trans-komplementierenden
AAV-Funktionen rep und cap transfiziert werden, um Zubereitungen
eines AAV-Vektors zu erzeugen. Es wird beansprucht, dass dies die
Herstellung von AVV-Materialien höherer Titer ermöglicht.
In den dargestellten Beispielen ist jedoch die einzige dargestellte
Information, die sich auf den Titer bezieht, dass eine Humanzelllinie,
K562, bei Effizienzen von nur 1 % oder weniger transduziert werden
konnte, was nicht auf eine Produktion hoher Titer eines AW-Vektors
hinweist. In diesem System wird der Vektor als Episomal (nichtintegriertes
Konstrukt) getragen und es wird behauptet, dass integrierte Kopien
des Vektors nicht bevorzugt sind.
-
Die
Methode zum Verpacken von AAV-Vektoren, die von Lebowski et al.,
1992, beschrieben ist, hat mehrere unerwünschte Aspekte. Erstens ist
es nicht wünschenswert,
den Vektor als nichtintegriertes, episomales Plasmid hoher Kopiezahl
in einer Zelllinie zu halten, da die Kopiezahl pro Zelle nicht streng
kontrolliert werden kann und episomale DNA eher eine Neuanordnung
erfährt,
die zur Herstellung defekter Vektoren führt. Zweitens muss in diesem
System der Vektor noch durch Infizieren der Zelllinie mit Adenovirus
und Einführen eines
Plasmids, das die AAV-rep- und -cap-Gene enthält, verpackt werden. Das Plasmid,
das von Lebkowski et al., 1992, verwendet wurde, war wieder pBa1,
das, wie zuvor festgehalten wurde, eine überlappende Homologie mit den
Vektor-ITR-Sequenzen hat und zur Erzeugung von Wildtyp-AAV führt. Drittens
wird in dem pBa1-Verpackungsplasmid, das von Lebkowski et al., 1988,
1992, verwendet wird, das rep-Gen von seinem p5-Promotor
exprimiert und ist daher negativ autoreguliert und daher ist wahrscheinlich,
dass die rep-Expression
begrenzt ist.
-
Das
Problem suboptimaler Werte der rep-Expression nach einer Plasmidtransfektion
kann mit einer anderen biologischen Aktivität dieser Proteine in Zusammenhang
stehen. Es gibt einen Hinweis (Tratschin et al., 1986, Mol. Cell.
Biol., 6:2884-2894), dass AAV-Rep-Proteine ihre eigene Expression
von dem AAV-p5-Promotor herabregulieren, der in allen
zuvor beschriebenen Verpackungskonstrukten verwendet wurde, wie pAAV/Ad
(Samulski et al., 1989) oder pBa1 (Lebkowski et al., 1988, 1992).
-
Kurzdarstellung
der Erfindung
-
Eine
der grundlegenden Herausforderungen für die Gentherapie war die Entwicklung
von Strategien zur Transduktion von Zellen und Geweben, die nicht
leicht ex vivo manipuliert werden können, oder die nicht aktiv
teilen. AAV-Vektoren
können
einen in vivo Gentransfer zum Beispiel im Atmungstrakt bewirken,
aber hohe Titer sind kritisch, so dass die Abgabe einer ausreichend
hohen Multiplizität
von Vektor in einem möglichst
geringen Volumen möglich
ist. Dadurch erhält
eine optimale Verpackungsmethodologie in der Bestimmung der Durchführbarkeit
einer Gentherapie auf AAV-Basis zentrale Bedeutung. Stabile AAV-Verpackungszelllinien
ohne Helfer waren unzuverlässig,
vorwiegend auf Grund der Aktivitäten
des Rep-Proteins, das seine eigene Expression herabreguliert und
die zellulare Immortalisierung umkehrt. Die Verfahren, die in dieser
Erfindung beschrieben sind, umgehen diese Probleme effektiv und
haben deutliche Verbesserungen in der Verpackungseffizienz ermöglicht.
-
Die
Verwendung eines HIV-LTR-Promotors zum Exprimieren hoher Werte von
AAV-Rep-Proteinen wurde an anderer Stelle berichtet (Antoni et al.,
1991), aber die Anwendung dieses Expressionssystems zur Verpackung
rekombinanter AAV-Vektoren ist eine neue Entwicklung.
-
Tatsächlich wurde
bisher nicht gezeigt, dass die Werte der Rep-Expression im Co-Transfektions-AAV-Vektor-Verpackungsprozess
begrenzend sind. Die Tatsache, dass eine zehnfache Erhöhung im
Verpackungstiter durch Erhöhen
der Rep-Expression erreicht wurde, liefert einen direkten Beweis,
dass die Rep-Werte
unter diesem Umstand begrenzend sind. Die Tatsache, dass eine pARtat-Co-Transfektion
die Verpackungseffizienz nicht weiter erhöhte, könnte entweder anzeigen, dass
(1) das Ausmaß der
Expression von dem HIV-Promotor in 293-Zellen selbst in Abwesenheit
von tat maximimiert war, oder (2), dass die Werte von Rep, die nur
mit pRS5 erreicht wurden, ausreichend waren um sicherzustellen,
dass die Rep-Expression für die
Verpackungseffizienz nicht mehr einschränkend war. Es wäre nun offensichtlich,
dass andere, Nicht-AAV-Promotoren
zur Erzeugung von Rep in Verpackungsplasmiden analog zu pRS5 verwendet
werden könnten.
-
Ebenso
ist das Phänomen
der Gewinnung integrierter rekombinanter AAV-Genome bekannt (Tratschin et al., 1985;
Flotte et al., 1993a), wurde aber noch nie zur Herstellung einer
vektorproduzierenden Zelllinie verwendet, wie hierin beschrieben
wurde.
-
Die
gesamte Verpackungseffizienz des pRS5-Vektor-Zellliniensystems war
wenigstens 104 Partikel pro Verpackungszelle,
was mehr als ausreichend ist, um eine Herstellung von rekombinanten
AAV-Vektorreagenzien klinischer Güte zu ermöglichen. Es wurde keine Wildtyp-AAV-Erzeugung
bei diesem Verfahren beobachtet, was ein zusätzlicher Vorteil gegenüber den
meisten Co-Transfektionsverfahren ist. Diese Verfahren machen die
Produktion von rekombinanten AAV-Vektoren klinischer Güte zur Verwendung
in der Gentherapie möglich.
-
Hierin
werden Verfahren und Konstrukte beschrieben, die die Herstellung
hoher Titer von AAV-Vektoren ohne Erzeugung von Wildtyp-AAV ermöglichen.
-
Daher
ist eine Ausführungsform
der Erfindung ein Verfahren zum Erzeugen hoher Titer rekombinanter AAV-Vektoren,
umfassend:
-
- (a) Bereitstellen von Zellen, die wenigstens
eine intakte Kopie eines stabil integrierten rekombinanten AAV-Vektors
enthalten, wobei der AAV-Vektor AAV invertierte terminale Wiederholungsregionen
(ITR-Regionen) und einen Transkriptionspromotor umfasst, der funktionsfähig an ein
Zielpolynucleotid gebunden ist, und wobei die Expression des rep-Gens
in diesen Zellen einschränkend
ist;
- (b) Bereitstellen eines AAV-Verpackungsplasmids, das die Expression
des Produktes des rep-Gens ermöglicht,
wobei in dem Plasmid das rep-Gen funktionsfähig an einen heterologen Promotor
gebunden ist und dem Verpackungsplasmid eine überlappende Homologie mit AAV-Sequenzen
in dem Vektor in der Zelle fehlt, die in (a) bereitgestellt wurde;
- (c) Einsetzen des AAV-Verpackungsplasmids in die Zelle und Inkubieren
der Zelle unter Bedingungen, die eine Replikation von AAV ermöglichen;
und
- (d) Isolieren rekombinanter AAV-Vektoren, die in Schritt (c)
erzeugt wurden.
-
In
dieser Ausführungsform
sind Verfahren enthalten, in welchen der Promotor in dem Verpackungsplasmid,
an den Rep funktionsfähig
gebunden ist, HIV-LTR ist, und jene Verfahren, in welchen das Verpackungsplasmid
pRS5 ist.
-
In
dieser Ausführungsform
sind auch Verfahren enthalten, in welchen das Zielpolynucleotid
für ein
Polypeptid codiert, das als CFTR ("cystic fibrosis transmembrane conductance
regulator") fungieren
kann.
-
Eine
weitere Ausführungsform
der Erfindung ist ein Verpackungssystem zum Erzeugen hoher Titer
rekombinanter AAV-Vektoren, umfassend:
-
- (a) Zellen, die wenigstens eine intakte Kopie
eines stabil integrierten rekombinanten AAV-Vektors enthalten, wobei
der AAV-Vektor AAV invertierte terminale Wiederholungsregionen (ITR-Regionen)
und einen Transkriptionspromotor umfasst, der funktionsfähig an ein
Zielpolynucleotid gebunden ist, und wobei die Expression des rep-Gens
in diesen Zellen einschränkend
ist; und
- (b) ein AAV-Verpackungsplasmid, das die Expression des Produktes
des rep-Gens ermöglicht,
wobei in dem Plasmid das rep-Gen funktionsfähig an einen heterologen Promotor
gebunden ist und dem Verpackungsplasmid eine überlappende Homologie mit AAV-Sequenzen
in dem Vektor in der Zelle, die in (a) bereitgestellt wird, fehlt.
-
Eine
weitere Ausführungsform
der Erfindung ist ein Verpackungsplasmid zur Verwendung in der Produktion
zum Erzeugen hoher Titer rekombinanter AAV-Vektoren, das die Expression von dem
Produkt des rep-Gens ermöglicht,
wobei das Plasmid aus einem rep-Gen besteht, das funktionsfähig an einen
heterologen Promotor gebunden ist.
-
Kurze Beschreibung
der Figuren
-
1 ist ein Diagramm des AAV-Lebenszyklus.
-
2 ist ein Schema, das die
Produktion des Verpackungsplasmids pRS5 und das Verhältnis von HIV1-LTR-Promotor
und der Codierungssequenz von AAV2-rep- und -cap-Genen zeigt.
-
3 ist eine Halbtonreproduktion
von Southern-Blots von Hirt-Extraktions-DNA-Proben, die gewinnbare intakte AAV-neo-Genome
zeigt, die in stabilen Zelllinien vorhanden sind.
-
4 ist eine Halbtonreproduktion
von Dot-Blot-Hybridisierungen, die den Nachweis von neo in Kontroll-
und verpackten AAV-neo-Genomen vergleicht.
-
5 ist eine Halbtonreproduktion
von Southern-Blots von Hirt-Extraktions-DNA-Proben, die die Gewinnung des AAV-CFTR-Vektors
(TRF42) aus vektorproduzierenden 293-Zell-Klonen zeigt.
-
6 ist eine Graphik des Prozentsatzes
von Lungenkarzinomzellen, die für
den CD44-Marker gefärbt
sind, wie durch Antikörperfärbung und
FACS-Analyse bestimmt wird.
-
Ausführliche Beschreibung der Erfindung
-
AAV-Vektoren
haben für
die Humangentherapie Relevanz, insbesondere für Erkrankungen wie die zystische
Fibrose und Sichelzellenanämie.
Die hierin beschriebene Erfindung stellt Methoden und Verfahren zur
Verwendung in der Produktion hoher Titer rekombinanter AAV-Vektoren
zur Verwendung in der Gentherapie bereit.
-
Bei
der Ausführung
der vorliegenden Erfindung werden, wenn nicht anders angegeben,
herkömmliche Techniken
der Molekularbiologie, Mikrobiologie, rekombinanten DNA und Immunologie
verwendet, die dem Fachmann bekannt sind. Solche Techniken sind
in der Literatur vollständig
erklärt.
Siehe z.B., Sambrook, Fritsch und Maniatis, Molecular Cloning: A
Laboratory Manual, zweite Ausgabe (1989), Oligonucleotide Synthesis
(M.J. Gait, Hrg., 1984), Animal Cell Culture (R.I. Freshney, Hrg.,
1987), die Reihe Methods in Enzymology (Academic Press, Inc.) Gene
Transfer Vectors for Mammalian Cells (J.M. Miller und M.P. Calos,
Hrg., 1987), Handbook of Experimental Immunology (D.M. Weir und
C.C. Blackwell, Hrg.), Current Protocols in Molecular Biolgy (F.M.
Ausubel, R. Brent, R.E. Kingston, D.D. Moore, J.G. Siedman, J.A.
Smith, und K. Struhl, Hrg., 1987) und Current Protocols in Immunology
(J.E. Coligan, A.M. Kruisbeek, D.H. Margulies, E.M. Shevach und
W. Strober, Hrg., 1991).
-
Die
Erzeugung hoher Titer rekombinanter AAV-Vektoren, die heterologe
Polynucleotide umfassen, die eine Transkription benötigen, wird
durch das folgende Verfahren ausgeführt.
-
In
dem Verfahren werden geklonte Zellen bereitgestellt, die ein geeignetes
AAV-Vektorplasmid enthalten. Das AAV-Vektorplasmid besteht aus den
AAV-ITR-Regionen
und einem Transkriptionspromotor, der funktionsfähig an ein Zielpolynucleotid
gebunden ist. Der Transkriptionspromotor, der an das Zielpolynucleotid
gebunden ist, ermöglicht
die Bildung von Transkripten, und enthält zum Beispiel Nicht-AAV-Promotoren,
wie auch AAV-Promotoren, wie p5, p19, p40, und AAV-ITR-Promotoren.
Die Transkriptions- und/oder Translationsprodukte des Zielpolynucleotids
sind vorzugsweise in der Gentherapie von Nutzen. Daher enthalten
Zielpolynucleotide Gene, die zur Gentherapie abgegeben werden, zum
Beispiel jene, die für
Untereinheitsketten von Hämoglobin, Enzymen,
Proteinen, wie den "cystic
fibrosis transmembrane conductane regulator (CFTR), und dergleichen codieren.
Zielpolynucleotide können
auch Polynucleotide sein, die, wenn sie transkribiert sind, eine
Aktivität wie
Anti- Sense-Moleküle, wie
Köder,
die an Transkriptions- oder Translationsfaktoren binden, wie Ribozyme und
dergleichen haben.
-
Eine
Vorraussetzung für
die geklonten Zellen, die für
das Verfahren bereitgestellt werden, ist, dass sie wenigstens eine
intakte Kopie des AAV-Vektorplasmids enthalten, die stabil in die
Zelle integriert ist und durch Infektion der transfizierten Zelle
mit einem Helfervirus, wie Adenovirus, gewonnen werden können, wenn
auch komplementäre
AAV-rep- oder rep- und cap-Funktionen vorhanden sind.
-
In
den in der Folge dargestellten Beispielen wurde ein AAVneo-Vektor
verwendet, wobei die anfängliche
Selektion der Zelllinie, die den Vektor enthält, durch Geneticinselektion
durchgeführt
wurde. Für
den Fachmann ist jedoch offensichtlich, dass zur Erzeugung von Zelllinien,
die einen Vektor enthalten, wie einen AAV-Vektor, der ein CFTR-Gen
enthält,
in dem kein selektiver Marker vorhanden ist, die Zellen gemeinsam
mit dem gewünschten
Vektorplasmid und einem zweiten Plasmid, das den selektiven Marker
enthält,
transfiziert werden können.
Nach der Selektion von Zellklonen auf der Basis des selektiven Markers
wäre es
offensichtlich, ein direktes Screening zu verwenden, um jene sofort
zu identifizieren, aus welchen ein Vektor mit hohem Titer gewonnen
werden kann. Ein Beispiel dafür
wird von Flotte et al. (1993a) berichtet, obwohl in diesem Beispiel
die Zellen durch Infektion mit AAV-Virus und Adenovirus gewonnen
wurden. Wie Flotte et al. (1993a) vermerkten, konnten auch produzierende
Zelllinien erhalten werden, die einen gewinnbaren AAV-Vektor enthielten,
der keinen selektierbaren Marker im Vektor aufwies. In diesem Beispiel
umfassten die AAV-Vektorplasmide Konstrukte, die die humane CFTRcDNA
enthielten, die funktionsfähig
an einen AAV-Promotor gebunden war, der aus der ITR bestand. Zelllinien,
die stabil integrierte Kopien dieser Vektoren enthielten, wurden
durch Co-Transfektion der Humanepithelzelllinie IB-3 mit dem AAV-CFTR-Vektorplasmid
und einem zweiten Plasmid, das den selektierbaren Macker neo enthielt,
abgeleitet. Nach der Selektion von Kolonien in Geneticin wurden einzelne
Klone erhalten, die den stabil integrierten Vektor enthielten, aus
dem der Vektor durch anschließende Infektion
mit Helfer-Adenovirus-
und Wildtyp-AAV-Partikeln gewonnen werden konnte. Dies ermöglicht eindeutig
die Erzeugung von Klonen mit stabil integrierten Kopien eines Vektors,
in welchen der Vektor selbst keinen selektierbaren Marker hat.
-
Das
Verfahren enthält
auch die Bereitstellung eines komplementierenden Verpackungsplasmids,
aus dem Rep- oder Rep- und Cap-Proteine von rep- oder rep- und cap-Genen exprimiert
werden können.
Dem Verpackungsplasmid fehlt überlappende
Homologie mit Sequenzen in dem Vektor zwischen den und einschließlich der
AAV-ITR-Sequenzen. Ferner kann die Kombination aus dem Verpackungsplasmid
und dem Vektor kein komplettes AAV-Genom ergeben. In dem Beispiel,
das unten angeführt
ist, fehlen die 120 Nucleotide langen Sequenzen um den AAV-p5-Promotor sowohl beim Verpackungsplasmid
als auch beim Vektor. Zusätzlich
wird in dem Verpackungsplasmid das rep-Gen nicht von dem AAV-p5-Promotor transkribiert, sondern ist vielmehr funktionsfähig an einen
heterologen Transkriptionspromotor gebunden, der durch die Expression
von rep nicht stark negativ autoreguliert ist.
-
In
dem bevorzugten Beispiel eines Verpackungsplasmids, wie durch pRS5
dargestellt, wurde HIV-LTR als heterologer Promotor verwendet, aber
es kann jeder heterologe Promotor, und vorzugsweise ein konstitutiver
oder induzierbarer Promotor, verwendet werden. HIV-LTR ist ein Beispiel
für beide
Arten von Promotor. Im allgemeinen ist dieser Promotor durch die
Wirkung des tat-Proteins
auf einen sehr hohen Expressionswert induzierbar. In dem hier gezeigten,
bevorzugten Beispiel zeigt der HIV-LTR-Promotor jedoch hohe Werte
einer konstitutiven Expression von rep, wenn er in 293-Zellen verwendet
wird. Dies ist darauf zurückzuführen, dass 293-Zellen
das Adenovirus-EIA-Genprodukt
exprimieren, von dem bekannt ist, dass es den HIV-LTR-Promotor transaktiviert.
Somit muss in 293-Zellen (die eine bevorzugte Zelllinie zur Etablierung
vektorhaltiger Zellen sind) die zusätzliche Transaktivierung des
HIV-LTR-Promotors in pRS5 nicht notwendig sein, um einen maximalen
Wert an funktionaler rep-Expression zu erhalten. Wenn vektorproduzierende
Zelllinien in anderen Zellen gebildet werden, dann könnte die
Transaktivierung von pRS5 durch Zugabe eines tat-Expressionsplasmids
wie pARtat (Antoni et al. 1991) wünschenswert sein. Solche Zelllinien
könnten
jede Humanzelllinie, wie HeLa, A549, KB, Detroit, WI38, oder jede
Zelllinie, in der geeignete Helferfunktionen exprimiert werden können, enthalten.
Wenn humanes Adenovirus als Helfer verwendet wird, könnte dieses
auch die gewünschte
Affenzelllinie, VERO, enthalten. Als Alternative, wenn Herpesviren
oder Pockenviren, wie Impfpocken oder Avipox, zur Bereitstellung
der Helferfunktion verwendet werden, kann jede geeignete Human-,
Nagetier- oder Affenzelllinie als vektorproduzierende Zelle ausreichend
sein.
-
Das
pRS5-Verpackungsplasmid dient als Modell sowohl für einen
induzierbaren als auch konstitutiven Promotor. Für den Fachmann wäre offensichtlich,
dass viele andere induzierbare oder konstitutive Promotoren in einem
Verpackungsplasmidkonstrukt verwendet werden könnten. Das primäre Merkmal
des Verpackungsplasmids ist, dass es den Wildtyp-AAV-p5-Promotor
nicht enthält
und daher durch rep nicht stark negativ autoreguliert ist. Beispiele
für andere
solche Promotoren wären
Mutationen des Wildtyp-p5-Promotors, die
eine Homologie mit dem Eltern-Wildtyp-Promotor entfernen oder negative
regulatorische Elemente dieses Promotors inaktivieren, wie die YYI-Region
des p5-Promotors.
Zu Beispielen für
induzierbare Promotoren zählen:
Metallioneninduzierbare Promotoren, wie Metallothioneinpromotor;
Steroidhormon-induzierbare Promotoren, wie der MMTV-Promotor; oder
der Wachstumshormonpromotor; Promotoren, die durch das Helfervirus,
wie Adenovirus, induzierbar wären;
frühe Gen-Promotoren,
die durch Adenovirus-EIA-Protein induzierbar sind, oder der Adenovirus-Major-Late-Promotor;
Herpesvirus-Promotor, der durch Herpesvirus-Proteine induzierbar
ist, wie VP16 oder 1CP4 oder Impfpocken- oder Pockenvirus-induzierbare
Promotoren oder Promotoren, die durch Pockenvirus-RNA-Polymerase
induzierbar sind, oder ein bakterieller Promotor, wie jener von
T7-Phage, der durch eine Pockenvirus-RNA-Polymerase induzierbar
wäre, oder
ein bakterieller Promotor, wie jener von T7-RNA-Polymerase.
-
Es
gibt viele starke konstitutive Promotoren, die zur Verwendung als
heterologer Promotor für
die rep-Expression in dem Verpackungsplasmid geeignet sind, einschließlich des
Adenovirus-Major-Late-Promotors, des Cytomegalovirus-Immediate-Early-Promotors,
des β-Wirkungspromotors,
oder des β-Globinpromotors.
Promotoren, die durch RNA-Polymerase III aktiviert werden, könnten auch
verwendet werden.
-
Die
Wirksamkeit des Verpackungsplasmids für die Komplementation der Rep- und Cap-Funktionen zur
Verpackung des AAV-Vektorplasmids kann unter Verwendung von AAV-Expressionsvektoren
getestet werden, denen die Rep- und/oder
Cap-Funktion fehlt, und die zusätzlich
einen Marker enthalten. Solche Expressionsvektoren sind in der Wissenschaft
bekannt und enthalten zum Beispiel das pAAVp5neo-Konstrukt
(Flotte et al., 1992). In den Beispielen wurde pAAVp5neo
als Vektorkonstrukt zum Testen jeder der beschriebenen Ver packungstechniken
verwendet, da es sowohl für
die Partikelzahl mit Hilfe des DNA-Blot-Dots (Samulski et al., 1989)
als auch mit Hilfe neo-transduzierender Titer titriert werden konnte.
-
Das
Verpackungsplasmid wird durch jede geeignete Technik, die in der
Wissenschaft bekannt ist, wie zum Beispiel Transfektion, Elektroporation
und dergleichen, in Zellen eingeführt, die das integrierte AAV-Vektorplasmid
enthalten. Nach dem Einführen
des Verpackungsplasmids werden die Zellen 3 bis 5 Tage unter Bedingungen
gezüchtet,
die eine Replikation von AAV ermöglichen,
Lysate werden hergestellt und die rekombinanten AAV-Vektorpartikel
werden durch Techniken, die in der Wissenschaft bekannt sind, gereinigt.
-
Die
in der Folge angeführten
Beispiele dienen als weitere Richtlinie für den praktizierenden Durchschnittsfachmann
und sind in keiner Weise als Einschränkung der Erfindung zu verstehen.
-
Das
Plasmid pRS5 in der E. coli DH5 Zelllinie (E. coli::pRS5 Stamm)
wurde am 9. November 1993 bei der American Type Culture Collection
(ATCC), 12301 Parklawn Dr., Rockville, Maryland 20852, hinterlegt
und erhielt die Zugangsnummer 69483. Die Hinterlegung erfolgte unter
den Bedingungen des Budapester Vertrags. Nach der Annahme und Erteilung
dieser Anmeldung als United States Patent werden alle Restriktionen hinsichtlich
der Verfügbarkeit
der Hinterlegung unwiderruflich aufgehoben; und der Zugriff auf
die genannten Hinterlegungen wird während der Anhängigkeit
der oben genannten Anmeldung für
jenen möglich
sein, der vom Bevollmächtigten
dazu unter 37 CFR § 1.14
und 35 USC § 1.22
bestimmt wird. Ferner werden die genannten Hinterlegungen für einen
Zeitraum von dreißig
(30) Jahren ab dem Tag der Hinterlegung, oder fünf (5) Jahren nach der letzten
Anfrage nach der Hinterlegung, oder über die geltend machbare Laufzeit
des US Patents, was auch immer länger
ist, aufrechterhalten. Die hierin erwähnten, hinterlegten Materialien
sollen nur der Einfachheit dienen und sind zur Ausführung der
vorliegenden Erfindung angesichts der vorliegenden Beschreibungen
nicht notwendig.
-
BEISPIELE
-
Beispiel 1
-
Verpackungsplasmid pRS5
-
Im
Verpackungsplasmid pRS5 ist der AAV-p5-Promotor
durch einen heterologen Promotor ersetzt, so dass die Expression
des rep-Genpolypeptids dessen eigene Synthese nicht negativ autoreguliert.
-
Das
Plasmid pRS5 wurde durch Ligieren des großen (5 kb) HindlIII bis Sphl
Fragments des zuvor beschrieben pHIVrep- (Antoni et al., J Virol.,
65:396-404) Plasmids (das den HIV-LTR-Promotor und rep-Gensequenzen,
einschließlich
der AAV-Nucleotide 263 bis 1886, flankiert von der pBR322 Plasmidsequenz,
enthält) mit
dem HindlII bis Sphl Fragment von einem Plasmid, das als pcap1 bezeichnet
wird (das das AAV-cap-Gen von den Nucleotiden 1886 bis 4491 ohne
AAV-ITR, wieder flankiert von pBR322-Sequenzen, enthält) konstruiert
(siehe 2). Nach dem
Ligieren dieser zwei Fragmente wurde ein Verpackungsplasmid, pRS5,
erzeugt, in dem das AAV-rep-Gen (Rep 68- und 78-Proteine) von dem HIV-LTR-Promotor transkribiert
ist, wobei die internen AAVp19- und p40-Promotoren die kleineren Rep-Produkte
(40 kD und 52 kD Rep-Proteine)
beziehungsweise die Cap-Proteine transkribieren. Somit enthält pRS5
die gesamte AAV-Codierungssequenz innerhalb der AAV-Nucleotidsequenz
von Nucleotid 263 bis 4491, die die rep-Codierungssequenz für Rep 78
und Rep 68 enthält,
die funktionsfähig
an den heterologen HIV-LTR-Promotor gebunden ist und Rep 52 und
Rep 40 von dem AAV-p19-Promotor und die
AAV-Capsidproteine
vom p40-Promotor exprimiert. Die Karte dieses Konstrukts ist in 1 dargestellt.
-
Das
pAAVp5neo-Konstrukt (Flotte et al., 1992)
wurde als Vektorkonstrukt zur Testung jeder der beschriebenen Verpackungstechniken
verwendet, da es sowohl für
die Partikelzahl mit Hilfe des DNA-Dot-Blots (Samulski et al., 1989)
als auch mit Hilfe neo-transduzierender Titer titriert werden konnte.
-
Beispiel 2
-
Zelllinien
-
Humane
293-31 Zellen (Graham et al., 1967, J. Gen. Virol., 36:59-72) wurden
in Eagle's Modified
Essential Medium mit 10 % fötalem
Kälberserum
bei 37 °C
in 5 % CO2 gezüchtet. Die 293-Zelllinie wurde
sowohl zum Verpacken von Vektorzubereitungen als auch für neo-Transduktionsexperimente
verwendet, um die neo-transduzierenden Titer zu verifizieren.
-
Beispiel 3
-
Verpacken von
AAV-Virionen mit einem HIV-LTR-Promotor-rep-Genplasmid
-
Das
pRS5-Konstrukt wurde zum Verpacken des pAAVp5neo-Vektorplasmids
durch Co-Transfektion in Adenovirus-Typ 5 (Ad5)-infizierte 293-Zellen
verwendet (Flotte et al., 1992). Insgesamt wurden zehn 10 cm Schalen,
die jeweils 2 × 106 293-Zellen (semikonfluent) enthielten,
mit 2 × 106 p.f.U. (Plage-bildenden Einheiten) Ad5
(m.o.i. ("multiplicity
of infection") =
1) 1 h vor der Transfektion mit jeweils 12,5 μg pRS5 und pAAVpSneo
infiziert. Dann wurden die Zellen 3 Tage bei 37 °C inkubiert und dann geerntet.
Die Zellen wurden danach abgeschabt und durch Zentrifugation bei
geringer Geschwindigkeit (4000 U/min × 10 min) gepoolt.
-
Das
Zellpellet wurde dann in 4 ml 10 mM Tris-HCl, pH 8,0, resuspendiert,
durch dreimaliges Gefrieren-Auftauen lysiert, mehrere Male durch
eine 25 g Nadel geschoben, um die Viskosität zu verringern, und mit Mikrokokken-Nuclease
(40 μl eines
300 μ/μl Stamms,
inkubiert bei 37 °C × 20 min,
und dann bei 4 °C über 10 min)
behandelt. Dann wurde CsCl auf eine Enddichte von 1,41 g/cc zugegeben.
Jedes Röhrchen
wurde mit 0,5 bis 1,0 ml Mineralöl
bedeckt und in einem Rotor mit frei ausschwingenden Bechern (SW50)
bei 35 000 U/min 12 h bei 4 °C
zentrifugiert.
-
Dann
wurden serielle 0,5 ml Fraktionen gesammelt und jede durch DNA-Dot-Blot-Hybridisierung
(Samulski et al., 1989) titriert, wobei zehnfache Verdünnungen
jeder Fraktion auf Nitrocellulosefilter geblottet und mit einer 32p-markierten DNA-Sonde, die aus einem 2,3
kb neo-Genfragment hergestellt wurde, unter Verwendung des Random-Priming-Kits
von Boehringer Mannheim hybridisiert. Die selektierten Fraktionen
wurden dann gegen Ringers Mineralsalzmedium, pH 7,4, dialysiert
und bei –20 °C zur Verwendung
in transduzierenden Titerexperimenten aufbewahrt.
-
Beispiel 4
-
Bestimmung transduzierender
Titer von AAV-neo Vektormaterialien
-
AAV-neo-Vektormaterialien,
die wie zuvor angeführt
hergestellt wurden, wurden in 293-31-Zellen durch Infizieren der
293-31-Zellen bei zehnfach zunehmenden Partikelmultiplizitäten, die
von 10 bis 1000 Partikel pro Zelle reichten, infiziert. In jedem
Fall wurden Zellen in Mikrotiter-Näpfchen mit 104 Zellen
pro Näpfchen geimpft,
und dann 2 h durch direkte Inokulation des Vektors in das Medium
infiziert. Die Zellen aus jedem Näpfchen wurden dann 24 h später trypsiniert
und auf 4 – 100
mm Schalen ausgestrichen. Drei Schalen jedes Satzes wurden dann
in G418 bei einer Dosis von 200 μg/ml
selektiert (aktiv, beginnend 48 h nach der ursprünglichen Infektion). Die vierte
Schale jedes Satzes wurde als Kontrolle der Ausstreicheffizienz
ohne G418 gezüchtet.
Die Zellen wurden 10 bis 14 Tage selektiert und dann mit Safranin-Rot
gefärbt.
Geneticin-resistente Kolonien wurden gezählt und eine Transduktionsfrequenz
durch Dividieren der mittleren Zahl von Genkolonien in jedem Satz
durch die Anzahl von Kolonien, die in der Ausstreicheffizienz-Kontrollplatte
erkennbar waren, bestimmt. In diesem Test wurde eine Transduktionseinheit
(t.u. – "transducing unit") dann als jenes
Impfmaterialvolumen pro Zelle definiert, das zum Transduzieren von
63% der Zellen zur Geneticinresistenz erforderlich war.
-
Beispiel 5
-
Verbesserung
der Co-Transfektion einer AAV-Vektorverpackung durch Verwendung
eines HIV-LTR-Protomor-Expressionsvektors
-
In
Tabelle 1 (Versuche 1 bis 3) sind die Ergebnisse von DNA-Blot-Dot-Titrationen
von AAV-neo-Partikelzubereitungen dargestellt, die durch die zuvor
etablierte pAAV/Ad-Co-Transfektionstechnik und eine parallele pRS5-Co-Transfektion erzeugt
wurden.
-
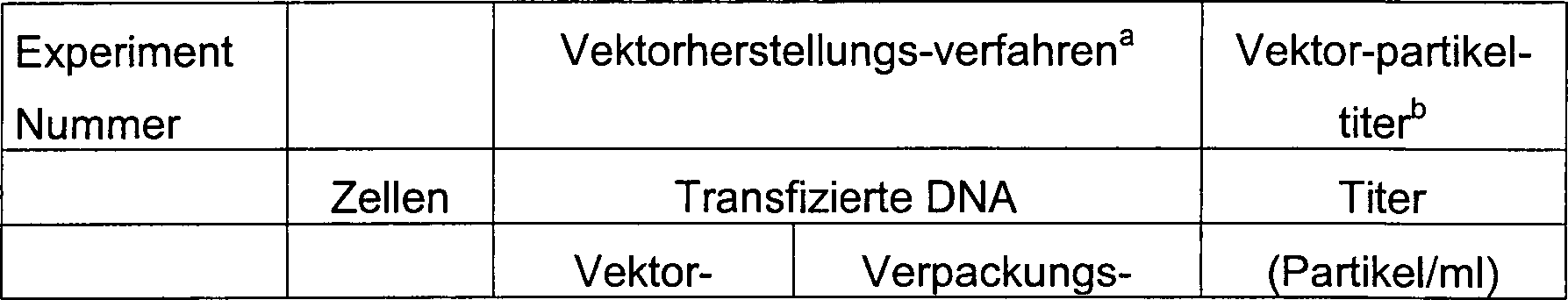
Tabelle
1. Partikeltiter von AAV-Vektorzubereitungen
-
-
Die
Ergebnisse in Tabelle 1 zeigen, dass Titer, die mit der pRS5-Konstruktion
erreicht wurden, das durch Co-Transfektion eingeführt wurde
(Experiment 4), ungefähr
fünf- bis
zehnmal höher
waren als jene, die unter Verwendung des pAAV/Ad-Verpackungsplasmids
erhalten wurden. Die Zugabe des HIV-trans-Transkriptionsaktivator- (tat-) Gens
durch Plasmid-Tri-Transfektion führte
zu einer sehr geringen zusätzlichen
Verpackungseffizienz. In dem Experiment, das in 3 dargestellt ist, gab es nur einen 30
% Anstieg im Partikeltiter der Fraktion mit Maximaltiter, von 3,0 × 1010 nur mit pRS5 auf 4,0 × 1010 mit
pRS5 + pARtat. Auf Grund dieser Ergebnisse schien es wahrscheinlich,
dass die Werte der rep- und cap-Expression in dieser Technik nicht
mehr einschränkend
waren.
-
Das
Experiment in 3 wurde
wie folgt durchgeführt.
-
Beispiel 6
-
Gewinnung intakter AAV-neo-Genome,
die in stabilen Zelllinien vorhanden sind
-
In
diesem Beispiel wurde eine Kultur aus 2 × 106 humanen
293-Zellen mit 5 Mikrogramm des AAV-Vektorplasmids pAAVp5neo (pSA206) transfiziert. Einzelne Kolonien
wurden unter Verwendung von 200 Mikrogramm aktivem G418 (Gibco-BRL)
pro ml Medium, beginnend 48 h nach der Transfektion, auf Geneticinresistenz
selektiert. Einzelne G-418-resistente Kolonien wurden mit sterilen
Klonierungszylindern isoliert und zu stabilen Zelllinien expandiert.
Von mehreren dieser Zelllinien wurden 2 × 106 Zellen
sowohl mit Adenovirus Typ 5 (moi = 2) als auch Wildtyp AAV2 (moi
= 2) infiziert. 48 Stunden später
wurde DNA mit geringem Molekulargewicht unter Verwendung des Hirtschen
High Salt-Detergent-Verfahrens selektiert und mittels 0,7% Agarosegel-Elektrophorese
und Southern-Blotting mit einer zufallsgeprimten 32p-markierten
neo-DNA-Sonde analysiert.
Die Ergebnisse der Southern-Blot-Hybridisierung der Hirt-extrahierten
DNA sind in 3 dargestellt.
-
Die
Figur zeigt, dass von 7 bis 8 einzelnen untersuchten Zelllinien
(Spur A bis G) die Vektorsequenz gewonnen und repliziert werden
konnte. Spur H zeigt ein Beispiel einer Geneticin-resistenten Zelllinie,
aus welcher der Vektor nicht gewonnen werden konnte.
-
Die
erwarteten intrazellularen replizierenden Spezies der Vektorgenome
(RFm für
monomere, doppelsträngige,
replizierende Form und RFd für
doppelsträngige,
dimere, replizierende Form) und die einzelsträngigen Nachkommen-Genome (SS) sind
angegeben. In wenigstens 6 der 8 Beispiele (Spüren A bis D und Spuren F und
G) wurden nicht neugeordnete Kopien des Vektors gewonnen.
-
Beispiel 7
-
Die Verwendung von Zelllinien
mit integriertem gewinnbarem AAV-Vektor verbessert die Verpackungseffizienz
-
Nach
der Verbesserung der Expression von AAV-rep-und-cap-Genen mit dem
pRS5-Konstrukt sollte die Effizienz der Verpackung durch die Herstellung
gleichförmiger
Zellpopulationen weiter verbessert werden, die integrierte, aber
gewinnbare Kopien des AAV-neo-Vektorgenoms enthielten. Die Kombination
einer pRS5-Transfektion von rep und cap mit der stabilen Zugabe
des pAAVp5neo-Vektors zu den Zelllinien
führte zu
einer signifikanten Verbesserung in der Verpackungseffizienz.
-
Zelllinien
wurden durch Transfizieren von AAVp5neo
durch Infizieren sowohl mit Wildtyp-AAV2 als auch Ad5 bei einer
moi von 5 für
jedes Virus hergestellt. Die Hirt-Extraktion wurde zur Isolierung
replizierender viraler DNA verwendet und diese DNA-Proben wurden
durch Elektrophorese und Southern-Blot-Hybridisierung unter Verwendung
einer 32p-markierten neo-Sonde analysiert.
Wie in 3 dargestellt,
waren gewinnbare AAV-neo-Rekombinanten in nahezu allen dieser Zelllinien
vorhanden. Die Muster der Banden enthielten, wie erwartet, Einzelstränge (unscharfe
Bande nahe dem Boden des Gels), doppelsträngiges Monomer mit einer Größe von 2,7
kb, Dimer mit 5,4 kg, größere multimere
Formen.
-
Zwei
Zelllinien, die auf gleiche Weise hergestellt und als neo4-6 und
neo4-9 bezeichnet wurden, wurden konstruiert und für anschließende Verpackungsexperimente
verwendet. Es wurde ein direkter Vergleich zwischen der Co-Transfektion von
pRS5 und pAAVp5neo in die 203-Zellen und
einer einzelnen Transfektion von pRS5 in entweder die neo4-6 oder
neo4-9 Zelllinie vorgenommen. Wie in Tabelle 1 dargestellt, erzeugte die
neo4-6 Zelllinie Titer von verpacktem AAV-neo, die 2,6 × 1011 betrugen.
-
Dieser
Partikeltiter stellt eine mehrfache Verbesserung gegenüber dem
293-Zellen-Co-Transfektionsverfahren
dar, und ergab die höchsten
Titer jeder Methode oder Kombination von Methoden, die verwendet wurden.
Der Gesamtpartikeltiter von nahezu 2,6 × 1011 Partikeln
wurde beginnend mit 2 × 107 Zellen erreicht und stellt somit eine Ausbeute
von 104 Partikeln pro Zelle dar.
-
Die
oben genannten Tests beruhen alle auf einer DNA/Dot-Blot-Technik,
die theoretisch auch Kopien von AAV-neo-Genomen erfassen könnte, die
nicht in AAV-Partikeln verpackt sind. Die Ergebnisse von zwei Arten
von Kontrollexperimenten schlossen diese Möglichkeit aus. Erstens wurden
AAVp5neo- und pRS5-Plasmide wie zuvor in
293-Zellen co-transfiziert, aber in Abwesenheit einer Ad5-Infektion.
Lysate dieser Zellen wurden wie jede der anderen Zubereitungen,
die oben genannt sind, behandelt. Wie in 4 dargestellt, zeigte die Kontroll-Dot-Blot-Hybridisierung,
dass die neo-Signale, verpackte AAV-neo-Genome reflektierten.
-
Die
DNA-Dot-Blot-Hybridisierung wurde wie folgt ausgeführt: Beginnend
mit 50 Mikrolitern jeder Fraktion wurden fünf zehnfache serielle Verdünnungen
in PBS hergestellt. Zur Lyse der Virionen wurde 1/10 Volumen (5 μl) 3N NaOH
zugegeben und die Mischung 1 h bei 65 °C inkubiert. Ein gleiches Volumen
(50 μl)
2M NH4Oac, pH 7,0, wurde zugegeben und das
gesamte 100 μl
Volumen wurde auf 0,45 μm
Nyonfilter mit einem Schleicher und Schuell Minifold I Mikroproben-Filtrationssammler übertragen.
Diese Filter wurden dann mit zufallsgeprimten, 32-p-markierten
neo-DNA-Sonden unter Standardbedingungen hybridisiert.
-
Serielle
zehnfache Verdünnungen
von Kontrollmaterialien, die wie beschrieben hergestellt worden
waren, wurden mit Verdünnungen
eines AAV-neo-Materials verglichen, das durch pRS5-Transfektion
der neo4-9-Zelllinie nach der Adenovirusinfektion hergestellt wurde
(dargestellt in Spalte A). Spalte B zeigt die pRS5-Transfektion
der neo4-9-Zelllinie ohne Adenovirusinfektion. Spalte C zeigt die
Adenovirusinfektion der neo4-6-Zelllinie ohne pRS5-Transfektion.
Spalte D zeigt die Adenovirusinfektion von pSA206-transfizierten 293-Zellen
ohne pRS5-Co-Transfektion. In keinem Fall ergab die übertragene
Zell- oder Plasmid-DNA ein signifikantes neo-Signal. Die Ergebnisse
von 4 zeigen, dass kein
erfassbares AAV-neo-Signal in der DNA/Dot-Blot-Hybridisierung von
diesem Experiment erfassbar war, was eindeutig darauf hinweist,
dass in diesen gereinigten Zubereitungen Plasmid-DNA nicht vorhanden
war.
-
Dann
wurde ein direkter Vergleich der transduzierenden Titer der AAV-neo-Vektormaterialien
durchgeführt,
die in dem dualen Plasmidtransfektionssystem oder durch Gewinnen
eines Vektors aus stabilen Linien erhalten worden waren. Wie Tabelle
2 zeigt, war die Transduktionsfrequenz, die mit einer äquivalenten
Anzahl von Vektorpartikeln erhalten wurde, die in jedem System erzeugt
wurden, ähnlich.
Dies zeigt, dass die Vektorpartikel, die durch Gewinnen des Vektors
aus stabilen Zelllinien erzeugt wurden, gleiche biologische Effizienz und
transduzierendes Potenzial aufwiesen wie jene, die in der dualen
Transfektion erzeugt wurden. Somit wiesen die Vektoren, die aus
Zelllinien gewonnen wurden, keine signifikanten biologischen Veränderungen
auf.
-
Tabelle
2. Biologische Äquivalenz
von AAV-neo-Vektorzubereitungen
a -
Die
oben genannten Ergebnisse zeigen, dass die kombinierten Modifizierungen,
die hier beschrieben sind, zu Erhöhungen bei Vektortitern von
etwa dem 50- bis 100-Fachen führen
können.
Ebenso kann dieses Verfahren im Vergleich zu den früher veröffentlichten
Berichten (z.B., Samulski et al., 1989) Vektorpartikel von wenigstens
2 × 1011 ergeben, was drei Größenordnungen höher ist,
als zuvor von einer gleichen Anzahl von Zellen erhalten wurde (d.h.,
humane 293-Zellen, die in insgesamt zehn 10 cm Zellkulturschalen
gezüchtet
wurden).
-
Beispiel 8
-
Verpackung von AAV-Vektoren,
die für
CFTR codieren
-
Ein
AAV-CFTR-Vektor, pTRF42, der die CFTR-cDNA enthält, die von einer AAV-ITR als
Promotor in Abwesenheit eines selektierbaren Markers exprimiert
wurde, wurde zur Erzeugung stabiler Vektorerzeugungslinien in der
293-Zelllinie durch
Co-Transfektion mit einem pSVneo-Plasmid verwendet. Dieser Vektor
konnte aus den stabilen Zelllinien gewonnen werden und der gewonnene
Vektor war intakt und nicht neugeordnet.
-
Die
Konstruktion von pTRF42 wurde beschrieben (Flotte et al., 1993a,
J. Biol. Chem., 268:3781-3790). Dieses Konstrukt enthält einen
AAV-CFTR-Vektor, der aus 145 Nucleotiden des AAV-5'-Endes (der ITR),
gefolgt von einem In-Frame-ATG-
(Met) Initiationskodon besteht, der direkt in die CFTR-Codierungssequenz
ab Aminosäure
1119 gelesen wird. Der Rest der CFTR-cDNA ist intakt bis zum nativen
Terminationskodon und bis zu Nucleotid 4629 der ursprünglichen
Sequenz. Darauf folgt ein synthetisches Polyadenylierungssignal
und dann AAV-Nucleotide 4490-4681 (3' ITR). Vier Mikrogramm dieses Vektors
wurden mit einem Mikrogramm des pSV2neo-Plasmids in 2 × 106 humane 293-Zellen co-transfiziert, die
dann mit 200 μg
aktivem G-418 48 h nach der Transfektion selektiert wurden. G-418-resistente
Klone wurden dann mit Klonierungszylindern isoliert und expandiert.
Die Klone wurden durch Gewinnung mit kombinierter Wildtyp-AAV2-
und Ad5-Infektion analysiert (jeweils moi = 2), wie bei den neo-Vektor-haltigen
Linien. Die DNA- (Hirt-) Extrakte mit geringem Molekulargewicht
wurden wieder durch 0,7 % Agarosegel-Elektrophorese und Southern-Blotting
mit einer zufallsgeprimten, 32pmarkierten
CFTR-cDNA-Sonde analysiert. Auch hier zeigte sich, dass die Zelllinien
gewinnbare, intakte Vektorsequenzen in den Größen hatten, die für die monomeren
und dimeren, replizierenden Formen (RF) vorhergesagt wurden, nämlich 4,6
beziehungsweise 9,2 kb. Dies bestätigte die Nützlichkeit der Methode, die
gewinnbare, vektorhaltige Zelllinien mit dem klinisch signifikanten
Beispiel eines AAV-CFTR-Vektors verwendete.
-
Die
zuvor beschriebenen Prinzipien und Lehren wurden zur Herstellung
zusätzlicher
Veranschaulichungsbeispiele der vorliegenden Erfindung verwendet,
von welchen einige in der Folge beschrieben sind, die die Nützlichkeit
der vorliegenden Erfindung näher
zeigen.
-
Beispiel 9
-
Integration
des lacZ-Gens in Lungenatemwegepithel
-
Die
in vivo Aktivität
von Vektoren, die durch die vorangehenden Methoden verpackt wurden,
wurde unter Verwendung des lacZ-Gens getestet, das für ein Enzym
mit β-Galactosidase-Aktivität codiert.
Der AAVp5lacZ-Vektor wurde durch Aufschluss
des pAAVp5neo-Konstrukts mit HindlII und
BamHI und Ligieren des großen
Fragments (das die AAV ITRs, die den AAVp5-Promotor
flankieren, und eine synthetische Polyadenylierungssequenz enthält) mit
einem HindlII- bis BamHI-Fragment von pSVBgal, das das E. coli lacZ-Gen
enthält,
hergestellt. Dieses anfängliche
Konstrukt wurde dann durch Aufschluss mit HindlII und Kpnl, Auffüllen ("Blunting") mit T4-Polymerase
(Boehringer Mannheim) und Religation des großen Fragments modifiziert.
Diese Endmanipulation ermöglichte
die Entfernung eines Sequenzzwischensegments, enthaltend vier ATG-Kodons
out-of-frame mit der lacZ-Codierungssequenz. Der Vektor wurde dann
unter Verwendung des pRS5-Plasmids wie zuvor beschrieben verpackt.
-
Aliquote
des verpackten AAVp5lacZ, die 1010 Partikel in 0,2 bis 0,5 ml enthielten,
wurden durch intraperitoneale Injektion an drei abgesetzte C57B1-Mäuse verabreicht.
Aliquote derselben Vehikellösung,
gegen die der Vektor dialysiert worden war, wurden in drei weitere
Mäuse injiziert,
die als Kontrolle dienten. Das Gewicht dieser Mäuse betrug jeweils etwa 30
gm, mit einer geschätzten
Gesamtkörperzellenanzahl
von 108 bis 109 Zellen.
Die durchschnittliche Vektordosis pro Zelle war daher zwischen 10
und 100 Partikeln pro Zelle, abhängig
vom Blutstrom. Vier Tage später
wurden die Tiere durch eine Pentobarbital-Überdosis getötet und
Proben der Bauchwand, Lunge, Leber, Milz, Nieren und Bauchspeicheldrüse entnommen
und in 2,5 % Glutaraldehyd fixiert.
-
Ein
in situ PVR-Test wurde an 5-Mikron, in Paraffin eingebetteten Gewebeschnitten
unter Verwendung vektorspezifischer Primer und eines nicht radioaktiven,
Digoxigenin-dUTP, Anti-Digoxigenin, alkalischen Phosphatase-Immunodetektionssystems
durchführt,
wie bereits beschrieben (Flotte et al., 1993). Es wurden Primer aus
der lacZ-Sequenz gewählt
(5'-Primer: 5'-ACAACTT-TAACGCCGTGCGCT-3'; 3'-Primer: 5'-TGCAGGAGCTCGTTATCGCTA-3'). Nach einem heißen Start
bei 82 °C
wurde eine 40-Zyklen PCR durchgeführt mit direkter Eingliederung
der Digoxidenin-markierten dUTP (Boehringer Mannheim) in die Rekationsprodukte.
Digoxigenin-markierte Nucleotide wurden dann mit einem mit alkalischer
Phosphatase bezeichnetem ("tagged") Anti-Digoxigenin-Antikörper (Genius
III Kit, Bohringer Mannheim) und einer immunohistochemischen Färbung (NBT,
X-phos) nachgewiesen.
-
Verschiedene
Gewebeschnitte für
injizierte Vektor- und Kontrollmäuse
wurden durch Lichtmikroskopie untersucht. Die Zellen, die Vektor-DNA
enthielten, wurden durch ein dunkelviolettbraunes Reaktionsprodukt angezeigt,
das den Nucleus überlagerte.
Die Färbung
wurde eindeutig in Schnitten von vektorinjizierten Mäusen beobachtet,
nicht aber in jenen von den Kontrollen. Vektor-DNA wurde in allen Zelltypen in den
Lungen erfasst, wie auch in den meisten Hepatozyten und pankreatischen
Azinuszellen. Nach der Absorption über die Lymphgefäße wären Vektorpartikel
in den venösen
Kreislauf eingetreten und auf den Lungenkreislauf getroffen, was
wahrscheinlich für
den sehr effizienten Gentransfer in der Lunge verantwortlich ist.
Sobald die Vektorpartikel durch den Lungen- und in den Körperkreislauf
gegangen sind, würden
sie wahrscheinlich äußerst effektiv
zu anderen Organen mit starkem Blutstrom verteilt werden.
-
Beispiel 10
-
Expression des
lacZ-Gens in vivo
-
Serielle
Schnitte derselben Gewebeproben, die für die in situ PCR-Erfassung
verwendet wurden, wurden auf β-Galactosidase-Aktivität mit X-gal-Reagens
(0,2 %, 37 °C,
16 h) gefärbt.
Schnitte von Mäusen,
denen AAVp5lacZ injiziert wurde, wurden
mit jenen verglichen, denen Vehikelkontrollen injiziert wurden.
In den Kontrolltieren war keine endogene β-Galactosidase-Aktivität in Schnitten,
die von der Lunge, Milz, Leber, Bauchspeicheldrüse, Niere oder dem Bauchfell
genommen wurden, nachweisbar.
-
Lungenschnitte
zeigten eine lacZ-Expression in den gesamten Segmenten des Atemwegepithels
von vektorinjizierten Tieren, nicht aber von Kontrollen. In einigen
der Atemwege waren > 75
% von 200 untersuchten Zellen positiv gefärbt. Etwa 25 % der Atemwege
zeigten dieses Maß an
positiver Färbung
(4 bis 6 untersuchte Atemwege pro Schnitt, ein bis zwei Schnitte
pro Tier), während
andere Regionen der Lunge geringere Expressionswerte hatten.
-
Milzschnitte
zeigten β-Galactosidase-Aktivität in nicht
lymphoiden Bereichen von vektorinjizierten Tieren, nicht aber in
Kontrollen. Lymphoide Follikel in der Milz waren im Wesentlichen
negativ. In Nicht-Epithelzellen in den Lungen oder in Zellen der
Leber, der Bauchwand und der Nieren von vektorbehandelten Tieren
wurde eine spärliche
Färbung
beobachtet, während
in der Bauchspeicheldrüse
keine Expression nachweisbar war.
-
Die
Verteilung von Vektor-DNA, wie durch einen in situ PCR-Test nachgewiesen,
wird mit der Verteilung der β-Galactosidase-Aktivität in Tabelle
3 wie folgt verglichen (n= 3): (--) keine Färbung, (+) X-gal gefärbte Zellen < 1 %, (++) 1 bis
25 % Färbung,
(+++) > 25 % Färbung, (nd)
nicht durchgeführt.
-
TABELLE
3 Gewebeverteilung
von lacZ-Gentransfer und Expression
-
Die
Expression der β-Galactosidase-Aktivität hängt daher
sowohl von der Verteilung des DNA-Vektors als auch der Gewebespezifität des Promotors
ab. Zum Beispiel ist der p5-Promotor in
Atemweg-Epithelzellen sehr aktiv, aber weitaus weniger aktiv in
anderen Zellen, die getestet wurden, wie jenen, die von der Bauchspeicheldrüse abgeleitet
wurden.
-
Beispiel 11
-
Integration und Expression
des CD44-Gens in Lungenkarzinomzellen
-
Der
pSAcd44-Vektor, der den Zellenoberflächenmarker CD44 von dem p5-Promotor
exprimiert, wurde direkt von dem pSA206- (AAVp5neo-)
Konstrukt subgeclont. Die Partikelzahlen wurden durch Dot-Blot-Analyse bestimmt.
Dieser Vektor und das pRS5-Verpackungsplasmid wurden in den Lungenkarzinomzelllinien
H209 und N82 co-transfektiert. Pro Aliquot wurden verschiedene Dosen
des Vektors in 4 × 104 Zellen getestet. 48 bis 72 Stunden nach
der Transfektion wurden die Zellen auf eine Zellenoberflächen-Genexpression
unter Verwendung eines Anti-CD44-Antikörpers fluoreszierend gefärbt. Die
Zellen wurden dann durch einen Fluoreszenz-aktivierten Zellzähler geleitet,
um den Anteil zu bestimmen, der positiv gefärbt war.
-
Ergebnisse
dieser Analyse sind in 6 dargestellt,
Virustiter sind in Einheiten von 104 angegeben; somit
stellt "1000" auf der horizontalen
Achse 250 Vektorpartikel pro Karzinomzelle dar. Die hohe Frequenz einer
CD44-Expression wurde bei Dosen von nur 100 Partikeln pro Zelle
erreicht.