WO2017002925A1 - ヒドロクロロフルオロオレフィンの製造方法、および2,3,3,3-テトラフルオロプロペンの製造方法 - Google Patents
ヒドロクロロフルオロオレフィンの製造方法、および2,3,3,3-テトラフルオロプロペンの製造方法 Download PDFInfo
- Publication number
- WO2017002925A1 WO2017002925A1 PCT/JP2016/069464 JP2016069464W WO2017002925A1 WO 2017002925 A1 WO2017002925 A1 WO 2017002925A1 JP 2016069464 W JP2016069464 W JP 2016069464W WO 2017002925 A1 WO2017002925 A1 WO 2017002925A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hcfo
- formula
- producing
- raw material
- metal
- Prior art date
Links
- 0 *C(C(F)(F)F)=CCl Chemical compound *C(C(F)(F)F)=CCl 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/35—Preparation of halogenated hydrocarbons by reactions not affecting the number of carbon or of halogen atoms in the reaction
- C07C17/358—Preparation of halogenated hydrocarbons by reactions not affecting the number of carbon or of halogen atoms in the reaction by isomerisation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/02—Boron or aluminium; Oxides or hydroxides thereof
- B01J21/04—Alumina
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/06—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of zinc, cadmium or mercury
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/24—Chromium, molybdenum or tungsten
- B01J23/26—Chromium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/40—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals of the platinum group metals
- B01J23/44—Palladium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J27/00—Catalysts comprising the elements or compounds of halogens, sulfur, selenium, tellurium, phosphorus or nitrogen; Catalysts comprising carbon compounds
- B01J27/06—Halogens; Compounds thereof
- B01J27/08—Halides
- B01J27/122—Halides of copper
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J27/00—Catalysts comprising the elements or compounds of halogens, sulfur, selenium, tellurium, phosphorus or nitrogen; Catalysts comprising carbon compounds
- B01J27/06—Halogens; Compounds thereof
- B01J27/125—Halogens; Compounds thereof with scandium, yttrium, aluminium, gallium, indium or thallium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J27/00—Catalysts comprising the elements or compounds of halogens, sulfur, selenium, tellurium, phosphorus or nitrogen; Catalysts comprising carbon compounds
- B01J27/06—Halogens; Compounds thereof
- B01J27/128—Halogens; Compounds thereof with iron group metals or platinum group metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/08—Heat treatment
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/23—Preparation of halogenated hydrocarbons by dehalogenation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C21/00—Acyclic unsaturated compounds containing halogen atoms
- C07C21/02—Acyclic unsaturated compounds containing halogen atoms containing carbon-to-carbon double bonds
- C07C21/18—Acyclic unsaturated compounds containing halogen atoms containing carbon-to-carbon double bonds containing fluorine
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/06—Silicon, titanium, zirconium or hafnium; Oxides or hydroxides thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
Definitions
- the present invention relates to a method for producing hydrochlorofluoroolefin and a method for producing 2,3,3,3-tetrafluoropropene.
- (Z) -1-chloro-2,3,3,3-tetrafluoropropene (HCFO-1224yd (Z)) and (Z) -1,2-dichloro-3,3,3-trifluoropropene (HCFO—) 1223xd (Z)) is useful as a foaming agent, a solvent, a cleaning agent, a refrigerant, a working fluid, a propellant, a fluororesin raw material, and the like for rigid polyurethane foam.
- Patent Document 1 discloses a potassium chloride catalyst in which 1,2-dichloro-2,3,3,3-tetrafluoropropane (HCFC-234ba) is supported on carbon in a gas phase. Describes a method for obtaining HCFO-1224yd by dehydrochlorination reaction by contacting with.
- HCFC-234ba 1-chloro-2,2,3,3,3-pentafluoropropane
- Patent Document 2 also describes that 2,3,3,3-tetrafluoropropene (HFO-1234yf) is obtained by reducing the obtained HCFO-1224yd with hydrogen in the presence of a catalyst.
- Patent Document 3 carbon is loaded with a source compound gas containing at least one of 1,1-dichloro-2,3,3,3-tetrafluoropropene (CFO-1214ya) and HCFO-1224yf and hydrogen. Describes a method of reacting in the presence of a palladium catalyst to obtain HFO-1234yf. Patent Document 3 also describes that CFO-1214ya is reduced by hydrogen to produce HCFO-1224yd.
- the present invention has been made to solve the above-mentioned problems, and is an industrially advantageous and efficient method for producing an Z form by isomerizing an E form of HCFO-1224yd or HCFO-1223xd.
- the purpose is to provide.
- Another object of the present invention is to provide a method for producing HFO-1234yf capable of efficiently producing HFO-1234yf while suppressing the production of by-products.
- the method for producing hydrochlorofluoroolefin of the present invention isomerizes a compound represented by the following formula (1) contained in the raw material composition under the condition that the compound represented by the following formula (1) is isomerized. It is made to react and the compound represented by following formula (2) is manufactured.
- X is a fluorine atom or a chlorine atom, and X in the formula (1) and formula (2) is the same.
- the method for producing 2,3,3,3-tetrafluoropropene of the present invention comprises the step of obtaining (Z) -1-chloro-2,3,3,3-tetrafluoropropene by the above-mentioned method for producing hydrofluoroolefin. And (Z) -1-chloro-2,3,3,3-tetrafluoropropene obtained in the above step is reduced with hydrogen to obtain 2,3,3,3-tetrafluoropropene. .
- the Z form can be produced by isomerizing the E form of HCFO-1224yd or HCFO-1223xd by an industrially advantageous and efficient method. Further, according to the method for producing HFO-1234yf of the present invention, it is possible to efficiently produce HFO-1234yf while suppressing the formation of by-products.
- HCFO (1) a compound represented by the following formula (1)
- HCFO (2) is the Z form.
- X is a fluorine atom or a chlorine atom, and X in the above formulas (1) and (2) is the same.
- X in HCFO (1) used as a starting material in the isomerization reaction represented by the above formula (3) is a fluorine (F) atom or a chlorine (Cl) atom.
- HCFO (1) is HCFO-1224yd (E) when X is an F atom, and HCFO-1223xd (E) when X is a Cl atom.
- HCFO-1224yd (E) and HCFO-1223xd (E) can both be produced by known methods.
- the raw material composition may be composed only of HCFO (1), or may be composed of compounds other than HCFO (1) and HCFO (1).
- HCFO (2) which is a Z body of HCFO (1) is mentioned.
- the compound other than HCFO (1) may be an impurity such as a by-product generated in addition to HCFO (1) when HCFO (1) is produced as a raw material for producing HCFO (1).
- a by-product generated from the impurity can be removed by a known means such as distillation.
- HCFO-1224yd (E) in which X is an F atom in HCFO (1) is obtained by supporting a mixed gas of CFO-1214ya and hydrogen on activated carbon. It can be produced by supplying it to a catalyst layer made of palladium or the like and hydrogenating CFO-1214ya.
- CFO-1214ya used in this method can be produced by a known method.
- HCFO-1224yd is usually obtained as a mixture of Z-form and E-form.
- a mixture of the Z form and the E form of HCFO-1224yd may be used as a raw material composition as it is, or the Z form and the E form are separated by a known method such as distillation. Then, it may be prepared to a desired mixing ratio and used as a raw material composition.
- HCFO-1224yd (E) is produced by using CFO-1214ya as a production raw material, and 1-chloro-1,2,2,3,3,3-hexafluoropropane (HCFC-) produced as a by-product in the production process.
- HCFO-1223xd is usually obtained as a mixture of Z-form and E-form.
- the mixture of Z form and E form of HCFO-1223xd may be used as a raw material composition as it is, or the Z form and E form may be separated by a known method such as distillation. You may adjust to a desired mixing ratio and use it as a raw material composition.
- HCFO-1223xd (E) includes HCFC-233da as a raw material for production, 1-chloro-3,3,3-trifluoropropyne by-produced in the production process, (E) -1-chloro- 3,3,3-trifluoropropene (HCFO-1233zd (E)), (Z) -1-chloro-3,3,3-trifluoropropene (HCFO-1233zd (Z)), 1,1-dichloro- As a raw material composition in which 3,3,3-trifluoropropene (HCFO-1223za), the target products HCFO-1223xd (Z), HCFO-1224yd (Z), HCFO-1224yd (E) and the like are mixed as impurities
- the isomerization reaction may be performed.
- HCFO (1) and HCFO (2) are present in a predetermined ratio.
- the equilibrium ratio of HCFO (1) (E form) and HCFO (2) (Z form) in the equilibrium state of this isomerization is a molar ratio represented by HCFO (1) / HCFO (2) at 150 ° C. and atmospheric pressure. The inventors confirmed that the ratio was 1.5 / 98.5 or more and 4/96 or less.
- HCFO (1) is subjected to isomerization conditions (1), thereby increasing the reaction rate of the isomerization reaction and rapidly isomerizing HCFO (1) to HCFO (2).
- HCFO (2) is efficiently produced.
- HCFO (1) / HCFO (2) in the raw material composition for example, within the range of the preferable equilibrium ratio, the conversion rate from HCFO (1) to HCFO (2) can be improved. it can.
- HCFO (1) / HCFO (2) in the raw material composition is preferably 5/95 or more in molar ratio, / 70 or more is more preferable. It is more preferable to use only HCFO (1) among HCFO (1) and HCFO (2) as a raw material composition.
- the raw material composition having a small HCFO (1) / HCFO (2) has an apparent conversion rate from HCFO (1) to HCFO (2) as compared with a case where HCFO (1) / HCFO (2) is large.
- the above isomerization reaction, HCFO (1) and HCFO (2) obtained by the isomerization reaction, and reisomerization of HCFO (1) obtained by distillation separation By repeating the above, HCFO (2) can be obtained from HCFO (1) in an industrially advantageous and efficient manner.
- HCFO (2) It is also possible to convert HCFO (2) to HCFO (1) by adjusting the isomerization conditions to cause the reverse reaction of the isomerization reaction shown in the above formula (3).
- the isomerization reaction is performed under the condition that HCFO (2) is isomerized to HCFO (1) (hereinafter also referred to as “isomerization condition (2)”).
- Examples of the isomerization condition (2) include a case where HCFO (1) / HCFO (2) in the raw material composition subjected to the isomerization equilibrium reaction is smaller than the equilibrium ratio in the isomerization equilibrium state. It is done. From the viewpoint of improving the conversion rate from HCFO (2) to HCFO (1), HCFO (1) / HCFO (2) in the raw material composition is preferably less than 5/95 in molar ratio. More preferably, it is less than 5 / 98.5. More preferably, only HCFO (2) is used as the raw material composition.
- HCFO (1) is obtained by isomerization reaction of HCFO (2)
- the isomerization reaction from HCFO (2) to HCFO (1) HCFO (1) obtained by isomerization reaction
- HCFO (1) obtained by isomerization reaction By repeating HCFO (2) distillation separation and HCFO (2) re-isomerization obtained by distillation separation, HCFO (1) can be obtained from HCFO (2) in an industrially advantageous and efficient manner. .
- the isomerization equilibrium is formed by contacting HCFO (1) with a metal catalyst in a reactor, contacting HCFO (1) with a radical generator in the reactor, HCFO ( The method etc. which heat 1) can be used.
- HCFO The method etc. which heat 1
- These methods are suitable as an industrial method for producing HCFO (2) by isomerizing HCFO (1) because an equilibrium of isomerization can be quickly formed.
- the reactor for the isomerization reaction of HCFO (1) is not particularly limited as long as it can withstand the temperature and pressure in the reactor described later.
- a cylindrical vertical reactor can be used.
- As a material of the reactor glass, iron, nickel, iron, an alloy mainly containing nickel, or the like is used. Further, the reactor may be provided with an electric heater for heating the inside of the reactor.
- the isomerization reaction represented by the above formula (3) can be performed by either a batch method or a continuous flow method.
- the HCFO production method of the present invention is preferably a continuous method in terms of production efficiency.
- the isomerization reaction in the HCFO production method of the present invention is usually carried out in a gas phase state of the raw material composition containing HCFO (1).
- the raw material composition is preferably preheated and introduced into the reactor.
- the preheating temperature of the raw material composition is preferably 20 ° C. or higher and 300 ° C. or lower, more preferably 50 ° C. or higher and 250 ° C. or lower from the viewpoint of vaporizing the raw material composition and improving the reactivity.
- the manufacturing method of HCFO of this invention demonstrates the reaction conditions in the case of making a raw material composition perform a isomerization reaction by a continuous type in the gaseous-phase state, it is not limited to this.
- a dilution gas is used together with the raw material composition in terms of suppressing side reactions, ease of supply of starting materials to the reactor, and adjusting the flow rate. It is preferable to supply to the reactor. Moreover, when performing the isomerization reaction shown by said Formula (3) in presence of the metal catalyst mentioned later, there also exists an advantage of improving durability of a metal catalyst by using dilution gas.
- Examples of the dilution gas include nitrogen, carbon dioxide gas, rare gas (such as helium), and organic compounds that are inert in the isomerization reaction.
- examples of inert organic compounds include saturated hydrocarbons such as methane, ethane, propane, butane, pentane, and hexane, trifluoromethane (CHF 3 , HFC-23), difluoromethane (CH 2 F 2 , HFC-32), Pentafluoroethane (CF 3 —CHF 2 , HFC-125), tetrafluoroethane (CF 3 —CFH 2 , HFC-134a), trifluoroethane (CF 3 —CH 3 , HFC-143a), difluoroethane (CF 2 H Fluorocarbons such as —CH 3 , HFC-152a) and tetrafluoropropane (CF 3 —CFH—CH 3 , HFC-254eb).
- the amount of the dilution gas is not particularly limited, but specifically, it is preferably 1 mol% or more and 10000 mol% or less, more preferably 10 mol% or more and 1000 mol based on HCFO (1) supplied to the reactor. % Or less, more preferably 30 mol% or more and 500 mol% or less.
- the raw material composition and the dilution gas may be mixed after preheating to the above temperature, and then supplied to the reactor, or may be mixed and preheated to the above temperature and then supplied to the reactor.
- the isomerization reaction can be performed by bringing HCFO (1) into contact with the metal catalyst in the reactor.
- a metal catalyst is accommodated in a reactor, a reaction part is formed, and HCFO (1) is passed through the reaction part.
- the metal catalyst may be accommodated in either a fixed bed type or a fluidized bed type.
- a fixed bed type either a horizontal fixed bed type or a vertical fixed bed type may be used, but when a mixed gas composed of multiple components is generated by an isomerization reaction, due to a difference in specific gravity. Since it is easy to prevent the concentration distribution of each component from occurring depending on the location, a vertical fixed bed type is preferable.
- the metal catalyst used for the isomerization reaction represented by the above formula (3) has a catalytic action for the isomerization reaction represented by the above formula (3).
- the metal catalyst include at least one substance selected from the group consisting of simple metals, metal oxides, and metal halides.
- the metal catalyst one kind of substance may be used alone, or two or more kinds of substances may be used in combination.
- the metal catalyst is preferably a metal oxide or a metal halide because HCFO (1) can be efficiently isomerized to HCFO (2).
- the metal constituting the metal catalyst examples include at least one element selected from the group consisting of transition metal elements, Group 12 metal elements of the periodic table, and Group 13 metal elements.
- the metals constituting the metal catalyst are Group 4 metal elements, Group 6 metal elements, Group 8 metal elements, Group 9 metal elements, Group 10 metal elements, Group 11 metal elements, At least one element selected from the group consisting of Group 12 metal elements and Group 13 metal elements is preferred.
- Group 4 metal elements, Group 6 metal elements, Group 8 metal elements, Group 10 metal elements, Group 11 More preferred is at least one element selected from the group consisting of group metal elements, group 12 metal elements, and group 13 metal elements.
- the metal constituting the metal catalyst titanium, zirconium, hafnium, chromium, iron, ruthenium, cobalt, rhodium, nickel, palladium, platinum, copper, zinc, aluminum are preferable, zirconium, chromium, iron, nickel, palladium, copper Zinc and aluminum are more preferable.
- the metal catalyst is a single metal
- the single metal may be one of the above metals or an alloy of two or more metals.
- the metal oxide may be an oxide of one of the above metals or a composite oxide of two or more metals.
- the metal halide may be a single halide of the above-described metal or a composite halide of two or more metals.
- metal catalysts include iron, cobalt, nickel, palladium, chromium oxide (chromia), aluminum oxide (alumina), zinc oxide, iron fluoride, aluminum fluoride, aluminum chloride, chromium fluoride, chromium chloride, etc. Is mentioned.
- metal catalysts at least one selected from the group consisting of aluminum oxide (alumina) and chromium oxide (chromia) is easy in obtaining and capable of efficiently isomerizing HCFO (1) to HCFO (2). Substances are preferred.
- the metal catalyst may be supported on a carrier.
- the carrier include an alumina carrier, a zirconia carrier, a silica carrier, a silica alumina carrier, a carbon carrier represented by activated carbon, a barium sulfate carrier, and a calcium carbonate carrier.
- the activated carbon include activated carbon prepared from raw materials such as wood, charcoal, fruit glass, coconut shell, peat, lignite and coal.
- the metal catalyst is preferably activated in advance from the viewpoint of improving the reactivity.
- the activation treatment method include a method in which a metal catalyst is brought into contact with an activation treatment agent with heating or without heating.
- an activation treatment agent for example, hydrogen fluoride, hydrogen chloride, fluorine-containing hydrocarbon, or the like can be used.
- an activation processing agent 1 type may be used independently and 2 or more types may be used together. Among them, it is preferable to use a fluorinated hydrocarbon as the activation treatment agent.
- fluorine-containing hydrocarbon used as the activation treatment agent examples include trichlorofluoromethane (CFC-11), dichlorodifluoromethane (CFC-12), chlorotrifluoromethane (CFC-13), and dichlorofluoromethane (HCFC-21). Chlorodifluoromethane (HCFC-22), trifluoromethane (HFC-23), tetrafluoroethylene (FO-1114) and the like are preferable. Further, HCFO-1224yd (E) or HCFO-1224yd (Z) as a raw material can be used as an activation treatment agent.
- a reactivation treatment can be performed on the metal catalyst. That is, in the isomerization reaction, when the activity of the metal catalyst decreases and the conversion rate to the target HCFO (2) decreases (when it is difficult to form an isomerization equilibrium), the metal catalyst is re-started. It is preferable to perform an activation treatment. Thereby, the activity of the metal catalyst can be regenerated and the metal catalyst can be reused.
- a reactivation treatment method there is a method in which a metal catalyst is brought into contact with a treatment agent for reactivation treatment (reactivation treatment agent) under heating or non-heating as in the activation treatment before use. Can be mentioned.
- oxygen, hydrogen fluoride, hydrogen chloride, chlorine-containing, fluorine-containing hydrocarbon, or the like can be used as the reactivation treatment agent.
- Chlorine-containing or fluorine-containing hydrocarbons include carbon tetrachloride, chloroform, dichloromethane (HCC-30), chloromethane, vinyl chloride, CFC-11, CFC-12, CFC-13, CFC-21, HCFC-22, HFC -23, FO-1114, HCFO-1224yd (E / Z), and the like.
- an inert gas such as nitrogen, argon, or helium can be used to dilute the reactivation treatment agent from the viewpoint of suppressing side reactions and improving the durability of the metal catalyst.
- the metal catalyst may be activated before being accommodated in the reactor.
- the activation treatment is preferably performed in the state accommodated in the reactor. Therefore, it is preferable to perform the activation treatment by introducing the activation treatment agent into the reactor containing the metal catalyst.
- the activation treatment agent may be introduced into the reactor at room temperature, but from the viewpoint of improving reactivity, it is preferable to adjust the temperature by heating or the like when introduced into the reactor.
- the temperature in the reactor is preferably heated to 50 ° C. or higher and 400 ° C. or lower.
- the HCFO (1) introduced into the reactor is brought into contact with the metal catalyst in the reactor.
- the reaction pressure for example, when pressurization is required for the purpose of shortening the reaction time, the pressurization condition is 1.0 MPa or less, the internal pressure in the reactor is 1.0 MPa or less at normal pressure.
- the contact temperature (reaction temperature) of HCFO (1) and the metal catalyst is 0 ° C. or higher and 500 ° C. or lower, preferably 50 ° C. or higher and 500 ° C. or lower, more preferably 50 ° C. or higher and 350 ° C. or lower, as the temperature in the reactor. 100 degreeC or more and 250 degrees C or less are more preferable, and 150 degreeC or more and 250 degrees C or less are the most preferable. If the reaction temperature is too low, it is difficult to form the isomerization equilibrium, so that the conversion rate of HCFO (1) to HCFO (2) decreases. On the other hand, when the reaction temperature is too high, byproducts are generated due to decomposition of HCFO (1) and the conversion rate to HCFO (2) decreases.
- the contact time (reaction time) of HCFO (1) and the metal catalyst in the reactor is preferably from 0.1 second to 1000 seconds, and more preferably from 1 second to 100 seconds.
- the contact time corresponds to the residence time of HCFO (1) in the reactor, and can be controlled by adjusting the supply amount (flow rate) of HCFO (1) to the reactor.
- Examples of the method of bringing HCFO (1) into contact with the radical generator include a method in which HCFO (1) is brought into contact with a radical generator activated by heat or light in a reactor.
- a method of bringing HCFO (1) and the activated radical generator into contact with each other in the reactor a method in which the radical generator is activated in advance and then supplied to the reactor, a mixture of the radical generator and HCFO (1) is used. Is introduced into the reactor, and the radical generator is activated in the reactor.
- the HCFO (1) and the radical generator may be supplied to the reactor either first or simultaneously. That is, during the supply of either one of HCFO (1) and the radical generator, even if the other is not supplied into the reactor, the HCFO (1) or the radical generator that has been supplied is being retained. Then, components to be supplied later are supplied, and the radical generator is appropriately activated.
- HCFO (1) and the activated radical generator are brought into contact with each other for a predetermined time in the reactor. That's fine.
- a mixture of HCFO (1) and radical generator is supplied to the reactor, and the radical activator in the reactor. Is preferably activated.
- the radical generator may be activated with either heat or light, or may be activated in combination with both, but industrially, it is preferably activated only with heat,
- a method of supplying a mixture of HCFO (1) and a radical generator to a heated reactor, applying thermal energy to the mixture in the reactor, and activating the radical generator with heat is preferable.
- the radical generator is activated by heat or light to generate radicals.
- a radical is a chemical species such as an atom, molecule, or ion that has an unpaired electron. It can be a radical cation with a positive charge, a radical anion with a negative charge, a radical with a neutral charge, a biradical, a carbene, etc. Including. Specific examples of the radical include a fluorine radical, chlorine radical, bromine radical, iodine radical, oxygen radical, hydrogen radical, hydroxy radical, nitroxy radical, nitrogen radical, alkyl radical, difluorocarbene or carbon radical.
- the radical generator for generating radicals as described above is not particularly limited as long as it generates radicals by applying external energy such as heat or light.
- the radical generator is preferably one that easily generates radicals in the reaction system.
- halogen gas such as chlorine and bromine, halogenated hydrocarbon, etc., air, oxygen, ozone, hydrogen peroxide Etc.
- Halogenated hydrocarbons include a part of hydrogen atoms bonded to carbon atoms in alkanes such as methane, ethane, propane, butane, pentane and hexane, and alkenes such as ethene, propene, butene, pentene and hexene, or Halogenated hydrocarbons that are all substituted with fluorine, chlorine, bromine or iodine atoms, wherein the halogenated hydrocarbons contain at least one atom of fluorine, chlorine, bromine or iodine atoms.
- the halogenated hydrocarbon serving as the radical generator does not include the starting material or the compound serving as the target material in the HCFO production method of the present invention, that is, HCFO-1224yd and HCFO-1223xd.
- radical cleavage of a compound having 4 or more fluorine atoms may not occur easily. In such a case, it is preferable to appropriately optimize radical generation conditions such as temperature.
- a radical generator may be used individually by 1 type, and may use 2 or more types together.
- radical generators oxygen, air, and chlorine are preferable because they are inexpensive and easily available.
- Chlorine is suitable as a radical generator because it easily generates radicals, but is very corrosive.
- chlorine is used as the radical generator, the product after completion of the isomerization reaction must be washed with a basic aqueous solution to which a reducing agent is added to remove chlorine.
- halogen or halogenated hydrocarbon is used as a radical generator, a small amount of halide, that is, halides of HCFO (1) and HCFO (2) are generated as by-products, and the target product (HCFO (2 )) May be difficult to purify.
- air and oxygen have the advantage that they can be easily separated from the product. Therefore, it is more preferable to use air or oxygen as the radical generator.
- the amount of the radical generator supplied to the reactor is very small. This is because the generation of radicals is chained. Moreover, addition of an excessive radical generator not only wastes auxiliary materials, but also causes a load on the separation process between the target substance or starting material after the reaction and the radical generator. Even in the case of oxygen or air, which can be easily separated from the target substance and starting material, for example, if the amount of air added increases, the ability of the coagulation process or distillation process decreases.
- chlorine is added excessively as a radical generator, a compound in which chlorine is added to the double bond of HCFO (1) and HCFO (2) (chlorine adduct) is generated as described above. Since this chlorine adduct is hydrochlorofluorocarbon (HCFC) which is a global warming and ozone depleting substance, it is preferable that the amount of by-product of the chlorine adduct is small.
- HCFC hydrochlorofluorocarbon
- the amount of HCFO (1) and radical generator is preferably 0.0001 / 99.9999 or more and 10/90 or less in terms of a molar ratio represented by radical generator / HCFO (1). More preferably, it is 0.0001 / 99.9999 or more and 0.1 / 99.95 or less. Further, the amount of HCFO (1) and radical generator is such that, in the present embodiment in which HCFO (1) is reacted in a gas phase state, when the radical generator is supplied in the form of a gas, the radical generator / The volume ratio shown by HCFO (1) should just be the range similar to the said molar ratio. In the present embodiment in which the isomerization reaction is performed by a continuous method, the supply amounts of HCFO (1) and the radical generator are shown as supply amounts per unit time.
- the reaction when the radical generator is activated by heat, the reaction is preferably performed at normal pressure or a slight pressure of 0.2 MPa or less as in the case of using a metal catalyst.
- the reaction temperature if the reaction temperature is too low, activation of the radical generator becomes insufficient, and the conversion rate of HCFO (1) to HCFO (2) decreases.
- the reaction temperature when the reaction temperature is too high, byproducts are generated due to decomposition of HCFO (1) and the conversion rate to HCFO (2) decreases. Therefore, the reaction temperature (contact temperature between the HCFO (1) and the radical generator) is preferably 100 ° C. or higher and 800 ° C. or lower, and more preferably 200 ° C. or higher and 600 ° C. or lower.
- the contact time (reaction time) between HCFO (1) and the radical generator in the reactor is preferably 0.01 seconds to 1000 seconds, more preferably 0.05 seconds to 100 seconds.
- the contact time corresponds to the residence time of the HCFO (1) and the radical generator in the reactor, and is controlled by adjusting the supply amount (flow rate) of the HCFO (1) and the radical generator to the reactor. it can.
- the radical generator when the radical generator is activated by light, the radical generator may be irradiated with light.
- Specific examples of the irradiation light include ultraviolet rays and visible rays including light having a wavelength of 200 nm to 400 nm.
- examples of the light source capable of performing such light irradiation include a high pressure mercury lamp, a low pressure mercury lamp, and a metal halide lamp.
- the light irradiation method is not particularly limited as long as it can sufficiently activate the radical activator present in the reaction system throughout the reaction time.
- HCFO (1) and the radical activator are mixed in advance and reacted.
- Examples include a method of inserting a light source equipped with a jacket made of a material into the gas of the component and irradiating the component with light from the inside of the component.
- the jacket is preferably a jacket having a cooling means depending on the reaction temperature.
- the isomerization reaction can be performed by heating HCFO (1) in a reactor.
- HCFO (1) can be supplied into a reactor heated by a heating furnace such as an electric furnace.
- the reaction pressure at this time is preferably carried out at normal pressure or slight pressurization of 0.2 MPa or less. If the reaction temperature is too low, it is difficult to form the isomerization equilibrium, so that the conversion rate of HCFO (1) to HCFO (2) decreases.
- the heating temperature is preferably 400 ° C. or higher and 1000 ° C. or lower, and more preferably 500 ° C. or higher and 900 ° C. or lower.
- the residence time (reaction time) of HCFO (1) in the reactor is preferably 0.001 seconds to 1000 seconds, and more preferably 0.01 seconds to 100 seconds.
- the conversion rate to HCFO (2) can be improved by increasing the reaction temperature within the above-mentioned preferable range or increasing the reaction time within the above-mentioned preferable range.
- the target substance HCFO (2) can be obtained as the outlet gas of the reactor.
- the outlet gas may contain by-products generated from impurities or the like contained in the raw material composition or generated by decomposition of HCFO (1). These by-products in the outlet gas can be removed to the extent desired by known means such as distillation.
- HCFO (1) and HCFO (2) in the outlet gas are, for example, when HCFO (1) is HCFO-1224yd (E), the boiling point of HCFO (1) (E form) is 17 ° C., HCFO (2) Since there is a difference in boiling point such that the boiling point of (Z-form) is 15 ° C., it can be separated by a normal distillation method. Therefore, the outlet gas obtained above is subjected to acid washing, alkali washing, dehydration with an adsorbent such as synthetic zeolite, removal of by-products, and distillation, if necessary, so that high purity HCFO (2) and HCFO are obtained. (1) can be obtained respectively.
- the outlet gas containing HCFO (1) and HCFO (2) which has been washed as described above, is supplied to a distillation column for distillation, and a distillation containing HCFO (2) as a main component from the top of the column. From the bottom of the tower, the bottoms containing HCFO (1) can be obtained.
- HCFO (1) is highly purified and HCFO (1) / HCFO (2) is greater than the equilibrium ratio. Become. For this reason, the HCFO (1) in the bottom product can be converted to HCFO (2) by subjecting the bottom product to isomerization conditions in the HCFO production method of the present invention as a raw material composition. Is possible.
- HCFO (1) / HCFO (2) in the distillate obtained by distillation is smaller than the equilibrium ratio
- the isomerization conditions in the method for producing HCFO of the present invention using the distillate as a raw material composition HCFO (2) in the distillate can be converted to HCFO (1).
- the HCFO can be efficiently performed.
- HCFO (1) can be obtained from (2).
- HCFO (1) When HCFO (1) is obtained from HCFO (2), a method for obtaining HCFO (2) from HCFO (1), specifically, a method using a metal catalyst, a method using a radical generator, heating,
- the conditions such as HCFO (1) / HCFO (2), reaction temperature, reaction time, etc. in the raw material composition may be adjusted so as to be the isomerization condition (2).
- HCFO (2) can be converted to HCFO (1) by making HCFO (1) / HCFO (2) in the raw material composition smaller than the equilibrium ratio in the equilibrium reaction of isomerization. it can.
- HCFO (2) (Z form) can be produced by isomerizing HCFO (1) (E form) by an industrially advantageous and efficient method. Further, if HCFO (1) / HCFO (2) in the raw material composition is made smaller than the equilibrium ratio, HCFO (2) (Z-form) is isomerized by an industrially advantageous and efficient method, and HCFO ( 1) (E body) can be manufactured.
- the working medium for heat cycle of the present invention contains HCFO-1224yd (Z) obtained by isomerizing HCFO-1224yd (E) in which X is an F atom in the above formula (1) by the above HCFO production method. To do.
- the content of HCFO-1224yd (Z) with respect to 100% by mass of the working medium for heat cycle is preferably 10% by mass or more, more preferably 20% by mass to 100% by mass, and even more preferably 40% by mass to 100% by mass.
- 60 mass% or more and 100 mass% or less are more preferable, and 90 mass% or more and 100 mass% or less are the most preferable.
- the working medium for heat cycle of the present invention may further contain an optional component other than HCFO-1224yd (Z), which is usually contained in the working medium for heat cycle, in addition to HCFO-1224yd (Z).
- Optional components include hydrofluorocarbons (HFC) such as HFC-32, difluoroethane, trifluoroethane, tetrafluoroethane, HFC-125, pentafluoropropane, hexafluoropropane, heptafluoropropane, pentafluorobutane, heptafluorocyclopentane, etc.
- HFC hydrofluorocarbons
- HFO-1234yf 1,2-difluoroethylene (HFO-1132), 2-fluoropropene (HFO-1261yf), 1,1,2-trifluoropropene (HFO-1243yc), 1,2,3,3; 3-pentafluoropropene (HFO-1225ye), 1,3,3,3-tetrafluoropropene (HFO-1234ze), 3,3,3-trifluoropropene (HFO-1243zf), 1,1,1,4 , 4,4-Hexaful Hydrofluoroolefins (HFO) such as 2-butene (HFO-1336mzz); 1-chloro-2,2-difluoroethylene (HCFO-1122), 1,2-dichlorofluoroethylene (HCFO-1121), 1- Chloro-2-fluoroethylene (HCFO-1131), 2-chloro-3,3,3-trifluoropropene (HCFO-1233xf), 1-chloro-3,3,3-tetrafluoropropene (HCFO-1233
- the working medium for the heat cycle of the present invention includes mineral oil refrigerating machine oil such as naphthenic refrigerating machine oil and paraffinic refrigerating machine oil in addition to stabilizers such as an oxidation resistance improving agent, a heat resistance improving agent, and a metal deactivator And mixed with refrigeration oils such as ester refrigeration oils, ether refrigeration oils, polyglycol refrigeration oils, hydrocarbon refrigeration oils, and other synthetic oil refrigeration oils. Can be applied.
- a heat cycle system using a heat exchanger such as a condenser or an evaporator is used without particular limitation.
- a heat cycle system for example, a refrigeration cycle
- a gaseous working medium is compressed by a compressor, cooled by a condenser to produce a high-pressure liquid, the pressure is reduced by an expansion valve, and vaporized by vaporizing at a low temperature by an evaporator. It has a mechanism that takes heat away with heat.
- the heat cycle system to which the working medium for heat cycle of the present invention is applied is preferably a centrifugal refrigerator that is a kind of the air conditioner.
- the centrifugal refrigerator is preferably a low-pressure centrifugal refrigerator of the low-pressure type and the high-pressure type.
- the low-pressure type is, for example, a working medium not subject to the application of the high-pressure gas safety method, that is, “a liquefied gas having a pressure of 0.2 MPa or more at a normal temperature and having a pressure of 0.2 MPa or more.
- HCFO-1224yd (Z) has a low global warming potential because it has a carbon-carbon double bond in the molecule. Moreover, there are many halogen ratios which suppress combustibility in a molecule
- the method for producing HFFO-1234yf of the present invention comprises a step of obtaining HCFO-1224yd (Z) by isomerizing HCFO-1224yd (E) wherein X in the formula (1) is an F atom by the HCFO production method.
- HCFO-1224yd (Z) obtained in the step of obtaining HCFO-1224yd (Z) is reacted with hydrogen and reduced to obtain HFO-1234yf.
- HCFO-1224yd (Z) and hydrogen generate HFO-1234yf by the reaction shown in the following formula (4).
- the hydrogen reduction reaction of HCFO-1224yd (Z) is preferably carried out in a gas phase in the presence of a palladium catalyst supported on activated carbon.
- a catalyst layer is formed by filling a palladium catalyst supported on activated carbon in a reactor formed of a material such as glass, iron, nickel, or an alloy containing these as a main component.
- HCFO-1224yd (Z) and hydrogen can be supplied to the catalyst layer.
- the palladium catalyst is not limited to palladium alone but may be a palladium alloy. Further, the palladium catalyst may be a mixture catalyst of palladium and another metal, or a composite catalyst in which palladium and another metal are separately supported on a carrier. When the palladium catalyst is a palladium alloy, examples of the palladium catalyst include a palladium / platinum alloy catalyst and a palladium / rhodium alloy catalyst.
- Examples of the activated carbon include those prepared using wood, charcoal, fruit husk, coconut husk, peat, lignite, coal, etc. as raw materials, those obtained from plant materials are preferred over mineral materials, and coconut shell activated carbon is particularly preferred. preferable.
- Examples of the shape of the activated carbon include formed charcoal having a length of about 2 mm or more and 5 mm or less, crushed coal of about 4 mesh or more and 50 mesh or less, granular charcoal, and the like. Of these, crushed charcoal of 4 mesh or more and 20 mesh or less, or formed coal is preferable.
- the packing density of the palladium-supported activated carbon in the catalyst layer is preferably 0.5 g / cm 3 or more and 1 g / cm 3 or less, and more preferably 0.6 g / cm 3 or more and 0.8 g / cm 3 or less.
- the ratio of HCFO-1224yd (Z) to hydrogen introduced into the catalyst layer is 0.7 or less in terms of the ratio (H 2 / Cl) of the number of moles of chlorine atoms to the number of moles of hydrogen in HCFO-1224yd (Z).
- H 2 / Cl is preferably 0.6 or less, and more preferably 0.5 or less, from the viewpoint of reducing the by-product of HFC-254eb.
- H 2 / Cl is preferably 0.1 or more, more preferably 0.2 or more, from the viewpoint of the yield of HFO-1234yf.
- the temperature of the catalyst layer is preferably 50 ° C. or higher, more preferably 60 ° C. or higher, and particularly preferably 80 ° C. or higher from the viewpoint of reactivity.
- the reaction pressure is preferably normal pressure from the viewpoint of handleability.
- Examples 1 to 20 and Examples 24 to 25 are Examples, and Examples 21 to 23 are Reference Examples.
- Catalyst Preparation Example 1 In a tube reactor made of stainless steel (SUS316) having an inner diameter of 23.4 mm and a height of 400 mm equipped with an electric furnace, a chromium magnesium composite oxide catalyst (Cr 2 O 3 : 98 mass%, MgO: 2 mass%, AG ⁇ 23, Sakai Chemical) was charged, and the temperature was raised to 200 ° C. while flowing nitrogen (N 2 ) gas. The temperature was maintained until no water flow out from the outlet of the reactor, and the catalyst was dried.
- SUS316 stainless steel
- N 2 nitrogen
- HCFC-22 was passed through the reactor together with N 2 gas, and when the hot spot due to the activation of the packed catalyst reached the outlet end of the reactor, the reactor temperature was raised to 250 ° C., This state was maintained for 8 hours, and the catalyst was activated to obtain catalyst 1.
- Catalyst preparation example 2 A catalyst 2 was obtained in the same manner as in Catalyst Preparation Example 1 except that the reactor was filled with an alumina catalyst (N612N, JGC catalyst conversion) instead of the chromium magnesium composite oxide catalyst.
- Catalyst Preparation Example 3 A catalyst 3 was obtained in the same manner as in Catalyst Preparation Example 1 except that the reactor was charged with an aluminum fluoride catalyst (reagent deer grade, Kanto Chemical) instead of the chromium magnesium composite oxide catalyst.
- an aluminum fluoride catalyst (reagent deer grade, Kanto Chemical) instead of the chromium magnesium composite oxide catalyst.
- Catalyst Preparation Example 4 Prepared in the same manner as in Catalyst Preparation Example 1 except that the reactor was filled with a zirconium-zinc composite oxide catalyst (ZrO 2 : 95% by mass, ZnO: 5% by mass, N Chemcat) instead of the chromium magnesium composite oxide catalyst. And catalyst 4 was obtained.
- a zirconium-zinc composite oxide catalyst ZrO 2 : 95% by mass, ZnO: 5% by mass, N Chemcat
- Catalyst Preparation Example 5 Instead of the chromium magnesium composite oxide catalyst in the reactor, a catalyst in which 0.5% palladium is supported on carbon (the ratio of palladium to the total amount of the catalyst is 0.5 mass%, manufactured by NV Chemcat, 0.5% A catalyst 5 was obtained in the same manner as in Catalyst Preparation Example 1 except that Pd / C) was charged.
- the temperature was raised to 200 ° C. while flowing. The temperature was maintained until no water flow out from the outlet of the reactor, and the catalyst was dried. After drying of the catalyst, HCFC-22 was passed through the reactor together with N 2 gas, and when the hot spot due to the activation of the packed catalyst reached the outlet end of the reactor, the reactor temperature was raised to 250 ° C., This state was maintained for 8 hours, and the catalyst was activated to obtain catalyst 6.
- Catalyst Preparation Example 7 A catalyst 7 was obtained in the same manner as in Catalyst Preparation Example 6 except that a 17% by mass nickel chloride (II) aqueous solution was used instead of the 25% by mass iron (II) chloride aqueous solution.
- Catalyst Preparation Example 8 A catalyst 8 was obtained in the same manner as in Catalyst Preparation Example 6 except that a 25 mass% copper (II) chloride aqueous solution was used instead of the 25 mass% iron (II) chloride aqueous solution.
- HCFO-1224yd was produced by the same method as described in Patent Document 3.
- a tube reactor made of stainless steel (SUS316) palladium-supported activated carbon in which palladium was supported on activated carbon was filled to form a catalyst layer.
- the hydrogen (H 2 ) / CFO-1214ya molar ratio 1/1 mixed with CFO-1214ya and hydrogen mixed in the reactor.
- Gas was supplied.
- the mixed gas was supplied into the reactor so that the residence time of the mixed gas of CFO-1214ya and hydrogen in the catalyst layer was 25 seconds.
- the outlet gas obtained from the reactor was passed through a 10% by weight aqueous potassium hydroxide (KOH) solution to remove acidic components, and then passed through a dehydration tower packed with synthetic zeolite (Molecular Sieves 4A). And dehydrated.
- KOH potassium hydroxide
- the outlet gas after dehydration was trapped in a cylinder cooled with dry ice. Using this trapped outlet gas as a sample, it is supplied to the bottom of a distillation column having about 30 theoretical plates, and distillation is performed by batch distillation at an operating pressure of 0.02 MPa (gauge pressure).
- a distillate containing 79.1% -1224yd (Z) and 19.3% HCFO-1224yd (E) (hereinafter, this distillate is referred to as “raw material composition A”) was obtained.
- Example 1 A tube reactor (hereinafter also referred to as a reactor) made of stainless steel (SUS316) having an inner diameter of 23.4 mm and a height of 400 mm was charged with 81.5 mL of catalyst 1.
- the raw material composition A was mixed at a rate of 27.9 NmL / min and nitrogen gas (N 2 ) as a diluent gas at a rate of 55.7 NmL / min, and preheated to 50 ° C. in a heating furnace. Thereafter, the preheated raw material composition A and nitrogen gas mixed gas were passed through a reactor maintained at 50 ° C. by a heating furnace at almost atmospheric pressure.
- the composition of the outlet gas was that the area percentage by GC (GC area%) was HCFO-1224yd (Z) of 88.2 (GC area) and HCFO-1224yd (E) was 11.1% (GC area%). Further, the reaction time (the residence time of the raw material composition A in the reactor) was 49.5 seconds.
- Examples 2--7 Except that the temperature in the reactor (also referred to as reaction temperature), the flow rate of nitrogen gas (also referred to as nitrogen flow rate), and the flow rate of raw material composition A (also referred to as raw material flow rate) were changed to the conditions shown in Table 1. The same operation as 1 was performed. The reactor exit gas was collected and analyzed by GC. The GC analysis results are shown in Table 1.
- Example 8-12 The same operation as in Example 1 was performed except that the metal catalyst was changed to catalyst 2 and the reaction temperature, nitrogen flow rate, and raw material flow rate were changed to the conditions shown in Table 1.
- the reactor exit gas was collected and analyzed by GC. The GC analysis results are shown in Table 1.
- Examples 13 to 18 As shown in Table 2, 81.5 mL of catalyst 3 to 8 was charged in the same tube reactor as in Example 1.
- the raw material composition A was mixed at a rate of 19.0 NmL / min and nitrogen gas (N 2 ) at a rate of 38.1 NmL / min and preheated to 50 ° C.
- N 2 nitrogen gas
- a preheated raw material composition A and nitrogen mixed gas was passed through the reactor under almost atmospheric pressure.
- the outlet gas immediately after flowing through the reactor was passed through an aqueous solution of potassium hydroxide (KOH) having a concentration of 10% by mass to remove acidic components (acid cleaning), and then dehydrated with synthetic zeolite (Molecular Sieves 4A).
- KOH potassium hydroxide
- the water was passed through a tower for dehydration, and the outlet gas after dehydration was collected in a cylinder cooled with dry ice.
- the collected outlet gas was collected and its composition was analyzed by GC.
- the GC analysis results are shown in Table
- HCFO-1224yd (E) can be converted to HCFO-1224yd (Z) with a very high yield (conversion rate).
- HCFO-1223xd (Synthesis Example 2: Synthesis of HCFO-1223xd) HCFO-1223xd was produced in the same manner as described in International Publication No. 2014/046250. Specifically, HCFC-233da was dropped into a heated potassium hydroxide aqueous solution to cause the reaction. By separating the reaction liquid after the reaction into an aqueous phase and an oil phase, 7194 g of a reaction crude liquid containing 90.3% HCFO-1223xd (Z) and 5.7% HCFO-1223xd (E) as an oil phase. Got.
- the obtained reaction crude liquid was purified by distillation, and a bottom liquid containing 17.1% HCFO-1223xd (Z) and 79.6% HCFO-1223xd (E) (hereinafter, this bottom liquid was referred to as “raw composition”. B)), and a distillate containing 99.8% HCFO-1223xd (Z) and 0.2% HCFO-1223xd (E) (hereinafter, this distillate is referred to as “Raw Material Composition C”). 3597 g of ".”
- Example 19 In Example 1, the same operation as in Example 1 was performed except that the raw material composition B was used instead of the raw material composition A, and the reaction temperature, nitrogen flow rate, and raw material flow rate were changed as shown in Table 3.
- the reactor outlet gas was collected as in Example 1, and the composition of the collected outlet gas was analyzed by GC. The GC analysis results are shown in Table 3.
- HCFO-1223xd (E) can be converted to HCFO-1223xd (Z) with a very high yield (conversion rate).
- Example 21 In Example 1, the same operation as in Example 1 was performed except that the raw material composition C was used instead of the raw material composition A, and the reaction temperature, the nitrogen flow rate, and the raw material flow rate were changed as shown in Table 4, respectively.
- the reactor outlet gas was collected as in Example 1, and the composition of the collected outlet gas was analyzed by GC. The GC analysis results are shown in Table 4.
- HCFO-1223xd (Z) is changed to HCFO-1223xd (E) by making HCFO-1223xd (E) / HCFO-1223xd (Z) in the raw material composition smaller than the equilibrium ratio. It can be seen that it can be converted.
- Example 24 Using the same tube reactor as in Example 1 (SUS316 tube reactor having an inner diameter of 23.4 mm and a height of 400 mm), the temperature of the heating furnace was set to 400 ° C., and the reactor was heated.
- the mixture was mixed and preheated to 200 ° C., then, supplied to the reactor heated to the above temperature, and allowed to flow under almost atmospheric pressure.
- the outlet gas immediately after flowing through the reactor was passed through a 10% by weight aqueous KOH solution to remove acidic components (acid cleaning), and then passed through a dehydration tower packed with synthetic zeolite (Molecular Sieves 4A).
- the dehydrated outlet gas was trapped in a cylinder cooled with dry ice.
- the trapped outlet gas was collected and its composition was analyzed by GC. The results of GC analysis are shown in Table 5.
- HCFO-1224yd (E) can be converted to HCFO-1224yd (Z) with a very high yield (conversion rate).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Physics & Mathematics (AREA)
- Thermal Sciences (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Catalysts (AREA)
Abstract
HCFO-1224ydまたはHCFO-1223xdのE体を異性化させて、そのZ体を製造する工業的に有利かつ効率的な方法の提供。 【解決手段】特定のHCFO(E体)が異性化される条件下で、原料組成物に含まれる前記HCFO(E体)を異性化反応させて、HCFO(Z体)を製造するHCFOの製造方法。
Description
本発明は、ヒドロクロロフルオロオレフィンの製造方法、および2,3,3,3-テトラフルオロプロペンの製造方法に関する。
(Z)-1-クロロ-2,3,3,3-テトラフルオロプロペン(HCFO-1224yd(Z))および(Z)-1,2-ジクロロ-3,3,3-トリフルオロプロペン(HCFO-1223xd(Z))は、硬質ポリウレタンフォームの発泡剤、溶剤、洗浄剤、冷媒、作動流体、噴射剤、フッ素樹脂の原料等として有用である。
本明細書において、ハロゲン化炭化水素については、化合物名の後の括弧内にその化合物の略称を記し、必要に応じて化合物名に代えてその略称を用いる。また、分子内に二重結合を有し、E体とZ体が存在する化合物については、E体とZ体をそれぞれ化合物の略称の末尾に(E)、(Z)と表記して示す。なお、化合物名の略称の末尾に(E)、(Z)の表記がないものは、E体および/またはZ体を示す。
HCFO-1224ydの製造方法として、特許文献1には、1,2-ジクロロ-2,3,3,3-テトラフルオロプロパン(HCFC-234ba)を気相状態で、カーボンに担持された塩化カリウム触媒に接触させることで脱塩化水素反応させ、HCFO-1224ydを得る方法が記載されている。また、特許文献2には、1-クロロ-2,2,3,3,3-ペンタフルオロプロパン(HCFC-235cb)を水酸化カリウム等の塩基を用いて脱フッ化水素反応させ、HCFO-1224ydを得る方法が記載されている。特許文献2には、触媒の存在下、得られたHCFO-1224ydを水素で還元することで、2,3,3,3-テトラフルオロプロペン(HFO-1234yf)を得ることも記載されている。
また、特許文献3には、1,1-ジクロロ-2,3,3,3-テトラフルオロプロペン(CFO-1214ya)およびHCFO-1224yfの少なくとも一方を含む原料化合物ガスと水素を、カーボンに担持させたパラジウム触媒の存在下で反応させて、HFO-1234yfを得る方法が記載されている。特許文献3には、CFO-1214yaが水素還元されて、HCFO-1224ydが生成することも記載されている。
しかしながら、これらの方法ではHCFO-1224ydが、通常、E体とZ体の混合物として得られ、一方の幾何異性体のみを利用する場合には不都合である。そのため、工業的に有利かつ効率的な方法で、HCFO-1224yd(E)とHCFO-1224yd(Z)のうちいずれか一方を選択的に製造する方法が求められている。
また、HCFO-1223xdについても同様に、E体とZ体のうちいずれか一方を選択的に製造する工業的に有利かつ効率的な方法が求められている。
本発明は、上記した課題を解決するためになされたものであって、HCFO-1224ydまたはHCFO-1223xdのE体を異性化させて、そのZ体を製造する工業的に有利かつ効率的な方法を提供することを目的とする。また、本発明は、副生物の生成を抑制してHFO-1234yfを効率よく製造することのできるHFO-1234yfの製造方法を提供することを目的とする。
本発明のヒドロクロロフルオロオレフィンの製造方法は、下記式(1)で表される化合物が異性化される条件下で、原料組成物に含まれる下記式(1)で表される化合物を異性化反応させて、下記式(2)で表される化合物を製造することを特徴とする。


(前記式(1)および式(2)中、Xはフッ素原子または塩素原子であり、前記式(1)および式(2)におけるXは同一である。)
本発明の2,3,3,3-テトラフルオロプロペンの製造方法は、上記ヒドロフルオロオレフィンの製造方法により(Z)-1-クロロ-2,3,3,3-テトラフルオロプロペンを得る工程と、前記工程で得られた前記(Z)-1-クロロ-2,3,3,3-テトラフルオロプロペンを水素により還元して2,3,3,3-テトラフルオロプロペンを得る工程とを有する。
本発明のHCFOの製造方法によれば、工業的に有利かつ効率的な方法でHCFO-1224ydまたはHCFO-1223xdのE体を異性化して、そのZ体を製造することができる。また、本発明のHFO-1234yfの製造方法によれば、副生物の生成を抑制してHFO-1234yfを効率よく製造することができる。
以下、本発明の実施形態を詳細に説明する。
(HCFOの製造方法)
本発明のヒドロクロロフルオロオレフィン(HCFO)の製造方法は、下記式(3)で示すように、下記式(1)で表される化合物(以下「HCFO(1)」という。)が異性化される条件下で、HCFO(1)を含有する原料組成物(以下、原料組成物ともいう。)に含まれるHCFO(1)を異性化反応させて、下記式(2)(以下「HCFO(2)」という。)で表される化合物を製造する。HCFO(1)は、上記HCFOのE体であり、HCFO(2)はZ体である。
本発明のヒドロクロロフルオロオレフィン(HCFO)の製造方法は、下記式(3)で示すように、下記式(1)で表される化合物(以下「HCFO(1)」という。)が異性化される条件下で、HCFO(1)を含有する原料組成物(以下、原料組成物ともいう。)に含まれるHCFO(1)を異性化反応させて、下記式(2)(以下「HCFO(2)」という。)で表される化合物を製造する。HCFO(1)は、上記HCFOのE体であり、HCFO(2)はZ体である。


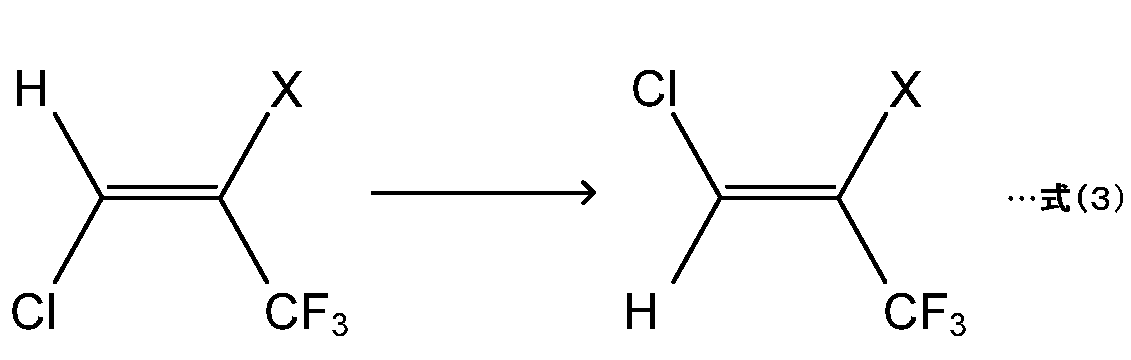
上記式(1)および式(2)中、Xはフッ素原子または塩素原子であり、上記式(1)および式(2)におけるXは同一である。
上記式(3)で示す異性化反応(以下単に「異性化反応」ともいう。)において出発物質として用いるHCFO(1)の有するXは、フッ素(F)原子または塩素(Cl)原子である。HCFO(1)は、XがF原子である場合には、HCFO-1224yd(E)であり、XがCl原子である場合には、HCFO-1223xd(E)である。これら、HCFO-1224yd(E)およびHCFO-1223xd(E)は、いずれも公知の方法で製造可能である。
また、原料組成物は、HCFO(1)のみから構成されてもよく、HCFO(1)とHCFO(1)以外の化合物から構成されてもよい。原料組成物中に含まれるHCFO(1)以外の化合物としては、HCFO(1)のZ体であるHCFO(2)が挙げられる。その他、HCFO(1)以外の化合物は、HCFO(1)の製造原料、HCFO(1)を製造する際にHCFO(1)以外に生成する副生物等の不純物であってもよい。なお、上記不純物を含有する原料組成物を用いて異性化反応させた場合、不純物から生成する副生物は、蒸留等の既知の手段により除去することが可能である。
出発物質として用いるHCFO(1)の製造方法として、具体的に、HCFO(1)においてXがF原子であるHCFO-1224yd(E)は、CFO-1214yaと水素の混合ガスを、活性炭に担持させたパラジウム等の触媒層に供給して、CFO-1214yaを水素還元することにより製造することができる。この方法に用いられるCFO-1214yaは公知の方法で製造可能である。
なお、上記方法では、通常、HCFO-1224ydはZ体およびE体の混合物として得られる。本発明のHCFOの製造方法では、上記したように、HCFO-1224ydのZ体およびE体の混合物をそのまま原料組成物として用いてもよいし、Z体およびE体を蒸留等公知の方法で分離して所望の混合比に調製して原料組成物として用いてもよい。さらに、HCFO-1224yd(E)は、これに製造原料であるCFO-1214yaや、製造工程で副生する、1-クロロ-1,2,2,3,3,3-ヘキサフルオロプロパン(HCFC-226ca)、1-クロロ-1,1,2,2,3,3-ヘキサフルオロプロパン(HCFC-226cb)、1-クロロ-2,2,3,3,3-テトラフルオロプロペン(HCFO-1224xe)、1,1,1,2,2-ペンタフルオロプロパン(HFC-254eb)、(E)-1,3,3,3-テトラフルオロプロペン(HFO-1234ze(E))、(Z)-1,3,3,3-テトラフルオロプロペン(HFO-1234ze(Z))、1,1-ジクロロ-2,3,3,3-テトラフルオロプロペン(CFO-1214ya)、1,3-ジクロロ-1,2,3,3-テトラフルオロプロペン(CFO-1214yb)、1,2-ジクロロ-1,3,3,3-テトラフルオロプロペン(CFO-1214xb)、1-クロロ-2,3,3,3-テトラフルオロプロパン(HCFC-244eb)、2,2-ジクロロ-1,1,3,3,3-ペンタフルオロプロパン(HCFC-225aa)、1,2-ジクロロ-1,2,3,3,3-ペンタフルオロプロパン(HCFC-225ba)、1,2-ジクロロ-1,1,2,3,3-ペンタフルオロプロパン(HCFC-225bb)、1,1-ジクロロ-2,2,3,3,3-ペンタフルオロプロパン(HCFC-225ca)、1,3-ジクロロ-1,1,2,2,3-ペンタフルオロプロパン(HCFC-225cb)、1,1-ジクロロ-1,2,2,3,3-ペンタフルオロプロパン(HCFC-225cc)、HCFO-1223xd(E)、HCFO-1223xd(Z)、目的物であるHCFO-1224yd(Z)等が不純物として混合された原料組成物として、異性化反応させてもよい。
HCFO(1)においてXがCl原子であるHCFO-1223xd(E)は、1,1,2-トリクロロ-3,3,3-トリフルオロプロパン(HCFC-233da)を、相間移動触媒の存在下で水酸化カリウム等の塩基と反応させる方法等によって得られる。
上記方法では、通常、HCFO-1223xdはZ体およびE体の混合物として得られる。本発明のHCFOの製造方法においては、このHCFO-1223xdのZ体およびE体の混合物をそのまま原料組成物として用いてもよいし、Z体およびE体を蒸留等の公知の方法で分離して所望の混合比に調製して原料組成物として用いてもよい。さらに、HCFO-1223xd(E)は、これに製造原料であるHCFC-233da等や、製造工程で副生する1-クロロ-3,3,3-トリフルオロプロピン、(E)-1-クロロ-3,3,3-トリフルオロプロペン(HCFO-1233zd(E))、(Z)-1-クロロ-3,3,3-トリフルオロプロペン(HCFO-1233zd(Z))、1,1-ジクロロ-3,3,3-トリフルオロプロペン(HCFO-1223za)、目的物であるHCFO-1223xd(Z)、HCFO-1224yd(Z)、HCFO-1224yd(E)等が不純物として混合された原料組成物として、異性化反応させてもよい。
(異性化の条件)
ここで、上記式(3)に示されるHCFO(1)のHCFO(2)への異性化反応が行われる条件では、通常、HCFO(2)のHCFO(1)への異性化反応が同時に行われる。すなわち、HCFO(1)およびHCFO(2)間の異性化反応は平衡反応である。本発明のHCFOの製造方法は、該異性化の平衡反応において、HCFO(1)がHCFO(2)に異性化される条件(以下「異性化条件(1)」ともいう。)で異性化反応を行うものである。
ここで、上記式(3)に示されるHCFO(1)のHCFO(2)への異性化反応が行われる条件では、通常、HCFO(2)のHCFO(1)への異性化反応が同時に行われる。すなわち、HCFO(1)およびHCFO(2)間の異性化反応は平衡反応である。本発明のHCFOの製造方法は、該異性化の平衡反応において、HCFO(1)がHCFO(2)に異性化される条件(以下「異性化条件(1)」ともいう。)で異性化反応を行うものである。
なお、異性化の平衡状態において、HCFO(1)とHCFO(2)は所定の比率で存在する。この異性化の平衡状態におけるHCFO(1)(E体)およびHCFO(2)(Z体)の平衡比は、150℃、大気圧において、HCFO(1)/HCFO(2)で示されるモル比で、1.5/98.5以上4/96以下であることを発明者らは確認した。
本発明のHCFOの製造方法は、HCFO(1)を異性化条件(1)に供することで、上記異性化反応の反応速度を上げ、HCFO(1)を迅速にHCFO(2)に異性化させることで、HCFO(2)を効率よく製造するものである。
また、HCFO(1)とHCFO(2)を含む原料組成物が異性化の平衡反応に供される場合、原料組成物中のHCFO(1)とHCFO(2)の比(以下「HCFO(1)とHCFO(2)の比」を「HCFO(1)/HCFO(2)」という。)が、異性化の平衡状態におけるHCFO(1)/HCFO(2)(以下、異性化の平衡状態におけるHCFO(1)/HCFO(2)を「平衡比」という。)よりも大きいとき、異性化条件(1)が満たされるため、HCFO(1)を異性化させてHCFO(2)に変換することができる。
また、原料組成物中のHCFO(1)/HCFO(2)を調節することで、例えば上記好ましい平衡比の範囲内で、HCFO(1)からHCFO(2)への変換率を向上させることができる。HCFO(1)からHCFO(2)への変換率を向上させる点から、原料組成物中のHCFO(1)/HCFO(2)は、モル比で、5/95以上であることが好ましく、30/70以上であることがより好ましい。原料組成物としてHCFO(1)およびHCFO(2)のうちHCFO(1)のみを用いることがさらに好ましい。
なお、HCFO(1)/HCFO(2)の小さい原料組成物は、HCFO(1)/HCFO(2)が大きい場合に比べて、HCFO(1)からHCFO(2)への見かけ上の変換率は小さくなるが、例えば、後述するように、上記異性化反応と、異性化反応で得られるHCFO(1)およびHCFO(2)の蒸留分離、蒸留分離により得られるHCFO(1)の再異性化を繰り返すことで、工業的に有利かつ効率的な方法でHCFO(1)からHCFO(2)を得ることができる。
また、異性化条件を調節することにより、上記式(3)に示す異性化反応の逆反応を生起させて、HCFO(2)をHCFO(1)に変換することも可能である。この場合、HCFO(2)がHCFO(1)に異性化される条件(以下「異性化条件(2)」ともいう。)で異性化反応を行う。
異性化条件(2)としては、例えば、異性化の平衡反応に供される原料組成物中のHCFO(1)/HCFO(2)が、異性化の平衡状態における平衡比よりも小さい場合が挙げられる。HCFO(2)からHCFO(1)への変換率を向上させる点から、原料組成物中のHCFO(1)/HCFO(2)は、モル比で、5/95未満であることが好ましく、1.5/98.5未満であることがより好ましい。原料組成物としてHCFO(2)のみを用いることがさらに好ましい。HCFO(2)を異性化反応させてHCFO(1)を得る場合にも、上記同様、HCFO(2)からHCFO(1)への異性化反応と、異性化反応で得られるHCFO(1)およびHCFO(2)の蒸留分離、蒸留分離により得られるHCFO(2)の再異性化を繰り返すことで、工業的に有利かつ効率的な方法でHCFO(2)からHCFO(1)を得ることができる。
上記異性化の平衡を形成させる方法として具体的には、反応器内でHCFO(1)と金属触媒を接触させる方法、反応器内でHCFO(1)とラジカル発生剤を接触させる方法、HCFO(1)を加熱する方法等を用いることができる。これらの方法によれば異性化の平衡を速やかに形成させることができるため、上記方法は、HCFO(1)を異性化させてHCFO(2)を製造する工業的な方法として適している。
(反応器)
HCFO(1)を異性化反応させる反応器としては、後述する反応器内の温度および圧力に耐えるものであれば、特に限定されず、例えば、円筒状の縦型反応器を用いることができる。反応器の材質としては、ガラス、鉄、ニッケル、鉄またはニッケルを主成分とする合金等が用いられる。また、反応器は、反応器内を加熱する電気ヒータ等を備えていてもよい。
HCFO(1)を異性化反応させる反応器としては、後述する反応器内の温度および圧力に耐えるものであれば、特に限定されず、例えば、円筒状の縦型反応器を用いることができる。反応器の材質としては、ガラス、鉄、ニッケル、鉄またはニッケルを主成分とする合金等が用いられる。また、反応器は、反応器内を加熱する電気ヒータ等を備えていてもよい。
(異性化反応)
上記式(3)で示す異性化反応は、バッチ式、連続流通式のどちらの方法でも可能である。本発明のHCFOの製造方法は、製造効率の点で連続式の方法であるのが好ましい。
上記式(3)で示す異性化反応は、バッチ式、連続流通式のどちらの方法でも可能である。本発明のHCFOの製造方法は、製造効率の点で連続式の方法であるのが好ましい。
本発明のHCFOの製造方法における異性化反応は、通常HCFO(1)を含む原料組成物が気相の状態で行われる。原料組成物は、予熱して反応器に導入することが好ましい。この際の原料組成物の予熱温度は、原料組成物を気化させ、反応性を向上させる点から、20℃以上300℃以下であることが好ましく、50℃以上250℃以下であることがより好ましい。以下、本発明のHCFOの製造方法を、原料組成物が気相の状態で、連続式で異性化反応を行わせる場合の反応条件について説明するが、これに限定されない。
異性化反応を原料組成物が気相の状態で行う場合、副反応の抑制、出発物質の反応器への供給のしやすさ、流量の調整などの点から、原料組成物とともに、希釈ガスを反応器に供給することが好ましい。また、上記式(3)で示す異性化反応を後述する金属触媒の存在下で行う場合、希釈ガスを用いることで金属触媒の耐久性を向上させるという利点もある。
希釈ガスとしては、窒素、二酸化炭素ガス、希ガス(ヘリウムなど)、上記異性化反応において不活性な有機化合物等が挙げられる。不活性な有機化合物としては、メタン、エタン、プロパン、ブタン、ペンタン、ヘキサン等の飽和炭化水素や、トリフルオロメタン(CHF3、HFC-23)、ジフルオロメタン(CH2F2、HFC-32)、ペンタフルオロエタン(CF3-CHF2、HFC-125)、テトラフルオロエタン(CF3-CFH2、HFC-134a)、トリフルオロエタン(CF3-CH3、HFC-143a)、ジフルオロエタン(CF2H-CH3、HFC-152a)、テトラフルオロプロパン(CF3-CFH-CH3、HFC-254eb)等のフルオロ炭化水素が挙げられる。希釈ガスの量は、特に制限されないが、具体的には、反応器に供給されるHCFO(1)に対して、好ましくは1モル%以上10000モル%以下、より好ましくは10モル%以上1000モル%以下、さらに好ましくは30モル%以上500モル%以下である。
原料組成物と希釈ガスは、反応性を向上させる点から、上記した好ましい温度に予熱してから反応器に導入することが好ましい。原料組成物と希釈ガスは、それぞれ上記温度に予熱してから混合し、その後反応器に供給してもよいし、混合してから上記温度に予熱し、その後反応器に供給してもよい。
(金属触媒との接触による異性化)
HCFO(1)を金属触媒と接触させることでHCFO(1)を異性化反応させる場合、異性化反応は、反応器内でHCFO(1)と金属触媒を接触させて行うことができる。具体的に、例えば、反応器内に金属触媒を収容し、反応部を形成して、この反応部にHCFO(1)を通流させることで行う。この場合、金属触媒は、固定床型または流動床型のいずれの形式で収容されていてもよい。また、固定床型である場合には、水平固定床型または垂直固定床型のいずれであってもよいが、異性化反応によって多成分から構成される混合ガスが生じた際に、比重差により場所によって各成分の濃度分布が生じることを防ぎやすいことから、垂直固定床型であることが好ましい。
HCFO(1)を金属触媒と接触させることでHCFO(1)を異性化反応させる場合、異性化反応は、反応器内でHCFO(1)と金属触媒を接触させて行うことができる。具体的に、例えば、反応器内に金属触媒を収容し、反応部を形成して、この反応部にHCFO(1)を通流させることで行う。この場合、金属触媒は、固定床型または流動床型のいずれの形式で収容されていてもよい。また、固定床型である場合には、水平固定床型または垂直固定床型のいずれであってもよいが、異性化反応によって多成分から構成される混合ガスが生じた際に、比重差により場所によって各成分の濃度分布が生じることを防ぎやすいことから、垂直固定床型であることが好ましい。
上記式(3)で示す異性化反応に用いられる金属触媒は、上記式(3)で示す異性化反応に対して触媒作用を有する。金属触媒としては、例えば、金属単体、金属酸化物、金属ハロゲン化物からなる群から選ばれる少なくとも1種の物質が挙げられる。金属触媒は、1種の物質を単独で用いてもよく、2種以上の物質を併用してもよい。
これらの中でも、HCFO(1)を効率よくHCFO(2)に異性化できることから、金属触媒は金属酸化物または金属ハロゲン化物であることが好ましい。
金属触媒を構成する金属としては、遷移金属元素、周期表の第12族金属元素、第13族金属元素からなる群から選ばれる少なくとも1種の元素が挙げられる。中でも、金属触媒を構成する金属は、周期表の第4族金属元素、第6族金属元素、第8族金属元素、第9族金属元素、第10族金属元素、第11族金属元素、第12族金属元素、第13族金属元素からなる群から選ばれる少なくとも1種の元素が好ましく、第4族金属元素、第6族金属元素、第8族金属元素、第10族金属元素、第11族金属元素、第12族金属元素、第13族金属元素からなる群から選ばれる少なくとも1種の元素がより好ましい。
金属触媒を構成する金属としては、チタン、ジルコニウム、ハフニウム、クロム、鉄、ルテニウム、コバルト、ロジウム、ニッケル、パラジウム、プラチナ、銅、亜鉛、アルミニウムが好ましく、ジルコニウム、クロム、鉄、ニッケル、パラジウム、銅、亜鉛、アルミニウムがより好ましい。金属触媒が金属単体である場合、金属単体は、上記した金属の1種であってもよく、2種以上の金属の合金であってもよい。金属触媒が金属酸化物である場合、金属酸化物は、上記した金属の1種の酸化物であってもよく、2種以上の金属の複合酸化物であってもよい。金属触媒が金属ハロゲン化物である場合、金属ハロゲン化物は、上記した金属の1種のハロゲン化物であってもよく、2種以上の金属の複合ハロゲン化物であってもよい。
金属触媒としては、具体的に、鉄、コバルト、ニッケル、パラジウム、酸化クロム(クロミア)、酸化アルミニウム(アルミナ)、酸化亜鉛、フッ化鉄、フッ化アルミニウム、塩化アルミニウム、フッ化クロム、塩化クロムなどが挙げられる。これらの金属触媒の中でも、入手が容易でHCFO(1)を効率よくHCFO(2)に異性化できる点で、酸化アルミニウム(アルミナ)および酸化クロム(クロミア)からなる群から選ばれる1種以上の物質が好ましい。
また、金属触媒は、担体に担持されていてもよい。担体としては、例えば、アルミナ担体、ジルコニア担体、シリカ担体、シリカアルミナ担体、活性炭に代表されるカーボン担体、硫酸バリウム担体、炭酸カルシウム担体などが挙げられる。活性炭としては、例えば、木材、木炭、果実ガラ、ヤシガラ、泥炭、亜炭、石炭などの原料から調製した活性炭などが挙げられる。
また、金属触媒は、反応性向上の観点から、あらかじめ活性化処理されていることが好ましい。活性化処理の方法としては、加熱下または非加熱下で金属触媒を活性化処理剤と接触させる方法が挙げられる。活性化処理剤としては、例えば、フッ化水素、塩化水素、含フッ素炭化水素などを用いることができる。活性化処理剤としては1種を単独で用いてもよく、2種以上を併用してもよい。中でも、活性化処理剤としては、含フッ素炭化水素を用いることが好ましい。活性化処理剤として用いる含フッ素炭化水素としては、例えば、トリクロロフルオロメタン(CFC-11)、ジクロロジフルオロメタン(CFC-12)、クロロトリフルオロメタン(CFC-13)、ジクロロフルオロメタン(HCFC-21)、クロロジフルオロメタン(HCFC-22)、トリフルオロメタン(HFC-23)、テトラフルオロエチレン(FO-1114)等が好適である。また、原料であるHCFO-1224yd(E)またはHCFO-1224yd(Z)を活性化処理剤に用いることもできる。
また、金属触媒に対しては、このような反応前の活性化処理の他に、再活性化処理を行うことができる。すなわち、異性化反応において、金属触媒の活性が落ち、目的物であるHCFO(2)への変換率が低下したとき(異性化の平衡が形成されにくくなったとき)には、金属触媒を再活性化処理することが好ましい。これにより、金属触媒の活性を再生させて金属触媒を再利用することができる。再活性化処理の方法としては、使用前の活性化処理と同様に、加熱下または非加熱下で金属触媒を再活性化処理のための処理剤(再活性化処理剤)と接触させる方法が挙げられる。再活性化処理剤としては、酸素、フッ化水素、塩化水素、含塩素または含フッ素炭化水素等を用いることができる。含塩素または含フッ素炭化水素としては、四塩化炭素、クロロホルム、ジクロロメタン(HCC-30)、クロロメタン、塩化ビニル、CFC-11、CFC-12、CFC-13、CFC-21、HCFC-22、HFC-23、FO-1114、HCFO-1224yd(E/Z)等を挙げることができる。また、再活性化処理において、副反応の抑制および金属触媒の耐久性向上等の点から、再活性化処理剤を希釈するために窒素、アルゴン、ヘリウム等の不活性ガスを用いることができる。
金属触媒は、反応器に収容される前に活性化処理が行われていてもよいが、操作が簡便で作業効率が良いため、反応器に収容した状態で活性化処理を行うことが好ましい。そのため、金属触媒を収容した反応器に活性化処理剤を導入して活性化処理を行うことが好ましい。活性化処理剤は、常温のまま反応器に導入してもよいが、反応性を向上させる観点から、反応器に導入する際に加熱等により温度調節を行うことが好ましい。
また、活性化処理の効率を高めるために、反応器内を加熱した状態で活性化処理をすることが好ましい。その場合、反応器内の温度は50℃以上400℃以下に加熱することが好ましい。
このようにして、反応器に導入したHCFO(1)は、反応器内で金属触媒と接触される。このとき、反応圧力については、例えば、反応時間の短縮等の目的で加圧を必要とする場合には、1.0MPa以下の加圧条件、反応器内の内圧で常圧以上1.0MPa以下の圧力条件とすることが可能であるが、工業的な実施のしやすさの点から、常圧、もしくは0.2MPa以下の微加圧で反応を行うことが好ましい。
HCFO(1)と金属触媒の接触温度(反応温度)は、反応器内の温度として0℃以上500℃以下であり、50℃以上500℃以下が好ましく、50℃以上350℃以下がより好ましく、100℃以上250℃以下がさらに好ましく、150℃以上250℃以下が最も好ましい。反応温度が低すぎると、上記異性化の平衡を形成し難いため、HCFO(1)のHCFO(2)への変換率が低下する。一方、反応温度が高すぎると、HCFO(1)が分解する等により副生物が生成してHCFO(2)への変換率が低下する。また、反応器内でのHCFO(1)と金属触媒の接触時間(反応時間)は、0.1秒以上1000秒以下が好ましく、1秒以上100秒以下がより好ましい。なお、接触時間は、HCFO(1)の反応器内での滞留時間に相当し、HCFO(1)の反応器への供給量(流量)を調節することで制御できる。
(ラジカル発生剤との接触による異性化)
HCFO(1)とラジカル発生剤を接触させる方法としては、HCFO(1)と熱または光によって活性化させたラジカル発生剤とを反応器内で接触させる方法が挙げられる。
HCFO(1)とラジカル発生剤を接触させる方法としては、HCFO(1)と熱または光によって活性化させたラジカル発生剤とを反応器内で接触させる方法が挙げられる。
HCFO(1)と活性化させたラジカル発生剤を反応器内で接触させる方法としては、ラジカル発生剤を予め活性化してから反応器へ供給する方法、ラジカル発生剤とHCFO(1)との混合物を反応器に導入して、反応器内でラジカル発生剤を活性化する方法が挙げられる。いずれの方法でも、HCFO(1)とラジカル発生剤の反応器への供給はいずれが先であってもよく、あるいは同時であってもよい。すなわち、HCFO(1)とラジカル発生剤のいずれか一方の供給の際に、反応器内に他方が供給されていない場合でも、先に供給されたHCFO(1)またはラジカル発生剤の滞留中に、後から供給される成分が供給され、さらに、ラジカル発生剤が適宜活性化されて、最終的に、HCFO(1)と活性化されたラジカル発生剤とが反応器内で所定の時間接触すればよい。ただし、HCFO(1)と活性化されたラジカル発生剤を効率的に接触させるためには、HCFO(1)とラジカル発生剤を混合した混合物を反応器へ供給し、反応器内でラジカル活性剤を活性化させることが好ましい。
また、ラジカル発生剤は、熱または光のいずれか一方で活性化させてもよく、両者を併用して活性化させてもよいが、工業的には、熱のみで活性化させることが好ましく、加熱した反応器にHCFO(1)とラジカル発生剤の混合物を供給し、反応器内で当該混合物に熱エネルギーを付与して、熱によりラジカル発生剤を活性化させる方法が簡便で好ましい。
ラジカル発生剤は、熱または光によって活性化されてラジカルを発生するものである。ラジカルは、不対電子を有する原子、分子、あるいはイオン等の化学種であり、化学種の電荷がプラスのラジカルカチオン、電荷がマイナスのラジカルアニオン、電荷が中性のラジカル、ビラジカル、カルベン等を含む。ラジカルとして、具体的には、フッ素ラジカル、塩素ラジカル、臭素ラジカル、ヨウ素ラジカル、酸素ラジカル、水素ラジカル、ヒドロキシラジカル、ニトロキシラジカル、窒素ラジカル、アルキルラジカル、ジフルオロカルベンまたは炭素ラジカル等が挙げられる。
上記したようなラジカルを発生させるラジカル発生剤としては、熱や光等の外部エネルギーの付与によって、ラジカルを発生するものであれば特に限定されない。ラジカル発生剤として、具体的には、反応系内に容易にラジカルを発生させるものが好ましく、例えば、塩素、臭素等のハロゲンガス、またはハロゲン化炭化水素等、空気、酸素、オゾン、過酸化水素等が挙げられる。ハロゲン化炭化水素としては、メタン、エタン、プロパン、ブタン、ペンタン、ヘキサン等のアルカン類や、エテン、プロペン、ブテン、ペンテン、ヘキセン等のアルケン類中の炭素原子に結合した水素原子の一部または全部をフッ素、塩素、臭素またはヨウ素原子で置換したハロゲン化炭化水素であって、当該ハロゲン化炭化水素はフッ素、塩素、臭素またはヨウ素原子の少なくとも1つの原子を含むものである。なお、ラジカル発生剤となるハロゲン化炭化水素として、本発明のHCFOの製造方法における出発物質または目的物質となる化合物、すなわち、HCFO-1224ydおよびHCFO-1223xdは含まれない。また、フッ素原子数が4以上の化合物はラジカル解裂が起こりにくい場合があり、その場合は、温度等、ラジカル発生条件の最適化を適宜行うことが好ましい。ラジカル発生剤は1種を単独で使用してもよく、2種以上を併用してもよい。
ハロゲン化炭化水素の具体例としては、CH3Cl、CH2Cl2、CHCl3、CCl4、CH3CH2Cl、CH3CCl3、CH2ClCH2Cl、CH2=CCl2、CHCl=CCl2、CCl2=CCl2、CHCl2CHCl2、CCl3CH2Cl、CH3CH2CH2Cl、CH3CHClCH3、CH3CHClCH2Cl、CH3Br、CH2Br2、CHBr3、CBr4、CH3CH2Br、CH3CBr3、CH2BrCH2Br、CH2=CBr2、CHBr=CBr2、CBr2=CBr2、CHBr2CHBr2、CBr3CH2Br、CH3CH2CH2Br、CH3CHBrCH3、CH3CHBrCH2Br、CH3I、CH2I2、CHI3、CH3CH2I、CH3CI3、CH2ICH2I、CH2=CI2、CHI=CI2、CI2=CI2、CHI2CHI2、CI3CH2I、CH3CH2CH2I、CH3CHICH3、CH3CHICH2I、CF2HCl、CF3I、CF2I2、CF3Br、CF2Br2等が挙げられる。
ラジカル発生剤として、上記したなかでは、安価で入手のし易さから、酸素、空気および塩素が好ましい。塩素はラジカルを発生させやすい点でラジカル発生剤としては好適であるが、非常に腐食性が高い。また、ラジカル発生剤として塩素を用いた場合、異性化反応終了後の生成物を、還元剤の添加された塩基性水溶液等で洗浄し、塩素を除去する必要がある。また、ラジカル発生剤として、ハロゲンや、ハロゲン化炭化水素を用いると、微量のハロゲン化物、すなわち、HCFO(1)およびHCFO(2)のハロゲン化物が副生物として生成し、目的物(HCFO(2))の精製が困難な場合がある。
これに対して、空気および酸素は生成物との分離が容易であるという利点がある。そのため、ラジカル発生剤として、空気または酸素を使用することがより好ましい。
反応器に供給されるラジカル発生剤の量は、微量であることが好ましい。ラジカルの生成が連鎖するためである。また、過剰のラジカル発生剤の添加は、副資材の無駄となるだけでなく、反応後の目的物質あるいは出発物質と、ラジカル発生剤との分離工程の負荷を生じる。目的物質および出発物質との分離が容易な、酸素または空気の場合であっても、例えば空気の添加量が多くなると凝集工程や蒸留工程の能力が低下する。また、ラジカル発生剤として塩素を過剰に添加すると、上記したように、HCFO(1)およびHCFO(2)の二重結合に塩素が付加した化合物(塩素付加体)が生成する。この塩素付加体は、地球温暖化、オゾン層破壊物質であるヒドロクロロフルオロカーボン(HCFC)であるため、塩素付加体の副生は少ない方が好ましい。
上記した点から、HCFO(1)とラジカル発生剤の量は、ラジカル発生剤/HCFO(1)で示されるモル比で、0.0001/99.9999以上10/90以下であることが好ましく、0.0001/99.9999以上0.1/99.95以下であることがより好ましい。また、HCFO(1)とラジカル発生剤の量は、HCFO(1)が気相の状態で反応を行わせる本実施形態において、ラジカル発生剤が気体で供給される場合には、ラジカル発生剤/HCFO(1)で示される体積比が上記モル比同様の範囲であればよい。なお、上記異性化反応を連続式の方法で行う本実施形態では、HCFO(1)とラジカル発生剤の供給量は、単位時間当たりの供給量で示すものとする。
上記異性化反応において、ラジカル発生剤を熱により活性化させる場合、反応圧力は、金属触媒を用いる場合と同様、常圧もしくは0.2MPa以下の微加圧で反応を行うことが好ましい。また、反応温度が低すぎると、ラジカル発生剤の活性化が不充分となるため、HCFO(1)のHCFO(2)への変換率が低下する。一方、反応温度が高すぎると、HCFO(1)が分解する等により副生物が生成してHCFO(2)への変換率が低下する。そのため、反応温度(HCFO(1)とラジカル発生剤の接触温度)は、100℃以上800℃以下が好ましく、200℃以上600℃以下がより好ましい。反応器内でのHCFO(1)とラジカル発生剤の接触時間(反応時間)は、0.01秒以上1000秒以下が好ましく、0.05秒以上100秒以下がより好ましい。なお、接触時間は、HCFO(1)とラジカル発生剤の反応器内での滞留時間に相当し、HCFO(1)とラジカル発生剤の反応器への供給量(流量)を調節することで制御できる。
また、ラジカル発生剤を光によって活性化させる場合、ラジカル発生剤に対して光を照射すればよい。照射する光として具体的には、波長200nm以上400nm以下の光を含む紫外線や可視光線などが挙げられる。上記式(3)で示す異性化反応において、このような光照射を行うことが可能な光源としては、例えば、高圧水銀ランプ、低圧水銀ランプ、メタルハライドランプ等を挙げることができる。
光照射の方法としては、反応時間を通じて反応系に存在するラジカル活性剤を十分に活性化させ得る方法であれば特に制限されないが、例えば、HCFO(1)とラジカル活性剤を予め混合して反応器に供給する場合、少なくとも上記異性化反応に必要な波長の光を透過し、反応系に存在する成分(HCFO(1)、HCFO(2)およびラジカル発生剤等)に不活性で、耐食性の材料で構成されたジャケットを装着した光源を当該成分のガス中に挿入し、成分内部から成分に対して光を照射する等の方法が挙げられる。また、光源が熱を発生する場合には、反応温度によっては、上記ジャケットは冷却手段を有するジャケットであることが好ましい。
(加熱による異性化)
上記異性化反応を、HCFO(1)を加熱する方法によって行う場合、上記異性化反応は、反応器内でHCFO(1)を加熱して行うことができる。具体的に、例えば、電気炉等の加熱炉により加熱された反応器内に、HCFO(1)を供給して行うことができる。この際の反応圧力は、金属触媒を用いる場合と同様、常圧もしくは0.2MPa以下の微加圧で反応を行うことが好ましい。反応温度が低すぎると、上記異性化の平衡を形成し難いため、HCFO(1)のHCFO(2)への変換率が低下する。一方、反応温度が高すぎると、HCFO(1)が分解する等により副生物が生成してHCFO(2)への変換率が低下する。そのため、加熱温度(反応温度)は、400℃以上1000℃以下が好ましく、500℃以上900℃以下がより好ましい。反応器内でのHCFO(1)の滞留時間(反応時間)は、0.001秒以上1000秒以下が好ましく、0.01秒以上100秒以下がより好ましい。反応温度を上記した好ましい範囲内で高くする、または反応時間を上記した好ましい範囲内で長くすることで、HCFO(2)への変換率を向上させることができる。
上記異性化反応を、HCFO(1)を加熱する方法によって行う場合、上記異性化反応は、反応器内でHCFO(1)を加熱して行うことができる。具体的に、例えば、電気炉等の加熱炉により加熱された反応器内に、HCFO(1)を供給して行うことができる。この際の反応圧力は、金属触媒を用いる場合と同様、常圧もしくは0.2MPa以下の微加圧で反応を行うことが好ましい。反応温度が低すぎると、上記異性化の平衡を形成し難いため、HCFO(1)のHCFO(2)への変換率が低下する。一方、反応温度が高すぎると、HCFO(1)が分解する等により副生物が生成してHCFO(2)への変換率が低下する。そのため、加熱温度(反応温度)は、400℃以上1000℃以下が好ましく、500℃以上900℃以下がより好ましい。反応器内でのHCFO(1)の滞留時間(反応時間)は、0.001秒以上1000秒以下が好ましく、0.01秒以上100秒以下がより好ましい。反応温度を上記した好ましい範囲内で高くする、または反応時間を上記した好ましい範囲内で長くすることで、HCFO(2)への変換率を向上させることができる。
(出口ガス)
本発明のHCFOの製造方法においては、目的物質であるHCFO(2)を、上記反応器の出口ガスとして得ることができる。出口ガス中には、原料組成物に含まれる不純物等から生成したり、HCFO(1)が分解等したりして生成した副生物が含有されることがある。出口ガス中のこれらの副生物は、蒸留等の既知の手段で望まれる程度に除去することができる。
本発明のHCFOの製造方法においては、目的物質であるHCFO(2)を、上記反応器の出口ガスとして得ることができる。出口ガス中には、原料組成物に含まれる不純物等から生成したり、HCFO(1)が分解等したりして生成した副生物が含有されることがある。出口ガス中のこれらの副生物は、蒸留等の既知の手段で望まれる程度に除去することができる。
また、上記異性化反応では、上述したように、異性化される条件において異性化の平衡状態が形成されるため、反応条件(異性化条件)を好適なものに調整したとしても、出口ガス中には、目的物質であるHCFO(2)に加えて、出発物質であるHCFO(1)が含まれる。
出口ガス中のHCFO(1)およびHCFO(2)は、例えば、HCFO(1)がHCFO-1224yd(E)である場合、HCFO(1)(E体)の沸点が17℃、HCFO(2)(Z体)の沸点が15℃であるように、沸点に差があるため、通常の蒸留方法によって分離することが可能である。したがって、上記で得られる出口ガスを、必要に応じて酸洗浄、アルカリ洗浄、合成ゼオライトなどの吸着剤による脱水、副生物の除去を行い、蒸留することで、高純度のHCFO(2)およびHCFO(1)をそれぞれ得ることができる。具体的には、上記で洗浄等を行った、HCFO(1)とHCFO(2)を含む出口ガスを、蒸留塔に供給して蒸留し、塔頂からHCFO(2)を主成分とする留出物を、塔底からHCFO(1)を含む缶出物をそれぞれ得ることができる。
このように、蒸留によって得られた留出物および缶出物のうち、缶出物については、HCFO(1)が高純度化され、HCFO(1)/HCFO(2)が平衡比よりも大きくなる。このため、缶出物をさらに、原料組成物として本発明のHCFOの製造方法における異性化される条件に供することで、缶出物中のHCFO(1)をHCFO(2)へ変換させることが可能である。
また、蒸留により得られる留出物中のHCFO(1)/HCFO(2)が平衡比よりも小さい場合、留出物を原料組成物として、本発明のHCFOの製造方法における異性化される条件に供することで、留出物中のHCFO(2)をHCFO(1)へ変換させることが可能である。このように、異性化反応と、異性化反応で得られるHCFO(1)およびHCFO(2)の蒸留分離、蒸留分離により得られるHCFO(1)の再異性化を繰り返すことで、効率的にHCFO(2)からHCFO(1)を得ることができる。
なお、HCFO(2)からHCFO(1)を得る場合には、上記HCFO(1)からHCFO(2)を得る方法、具体的には、金属触媒を用いる方法、ラジカル発生剤を用いる方法、加熱による方法において、原料組成物におけるHCFO(1)/HCFO(2)、反応温度、反応時間等の条件を、異性化条件(2)となるように、調整すればよい。特に、上記したように原料組成物におけるHCFO(1)/HCFO(2)を、異性化の平衡反応における平衡比よりも小さくすることで、HCFO(2)をHCFO(1)に変換することができる。
以上、本発明のHCFOの製造方法によれば、工業的に有利かつ効率的な方法でHCFO(1)(E体)を異性化して、HCFO(2)(Z体)を製造することができる。また、原料組成物中のHCFO(1)/HCFO(2)を平衡比よりも小さくすれば、工業的に有利かつ効率的な方法でHCFO(2)(Z体)を異性化して、HCFO(1)(E体)を製造することができる。
(熱サイクル用作動媒体)
本発明の熱サイクル用作動媒体は、上記HCFOの製造方法によって、上記式(1)においてXがF原子であるHCFO-1224yd(E)を異性化して得られたHCFO-1224yd(Z)を含有する。熱サイクル用作動媒体の100質量%に対するHCFO-1224yd(Z)の含有量は、10質量%以上が好ましく、20質量%以上100質量%以下がより好ましく、40質量%以上100質量%以下が一層好ましく、60質量%以上100質量%以下がさらに好ましく、90質量%以上100質量%以下が最も好ましい。
本発明の熱サイクル用作動媒体は、上記HCFOの製造方法によって、上記式(1)においてXがF原子であるHCFO-1224yd(E)を異性化して得られたHCFO-1224yd(Z)を含有する。熱サイクル用作動媒体の100質量%に対するHCFO-1224yd(Z)の含有量は、10質量%以上が好ましく、20質量%以上100質量%以下がより好ましく、40質量%以上100質量%以下が一層好ましく、60質量%以上100質量%以下がさらに好ましく、90質量%以上100質量%以下が最も好ましい。
本発明の熱サイクル用作動媒体は、HCFO-1224yd(Z)にさらに、通常熱サイクル用作動媒体が含有するHCFO-1224yd(Z)以外の任意成分を含有していてもよい。任意成分としては、HFC-32、ジフルオロエタン、トリフルオロエタン、テトラフルオロエタン、HFC-125、ペンタフルオロプロパン、ヘキサフルオロプロパン、ヘプタフルオロプロパン、ペンタフルオロブタン、ヘプタフルオロシクロペンタン等のヒドロフルオロカーボン(HFC);HFO-1234yf、1,2-ジフルオロエチレン(HFO-1132)、2-フルオロプロペン(HFO-1261yf)、1,1,2-トリフルオロプロペン(HFO-1243yc)、1,2,3,3,3-ペンタフルオロプロペン(HFO-1225ye)、1,3,3,3-テトラフルオロプロペン(HFO-1234ze)、3,3,3-トリフルオロプロペン(HFO-1243zf)、1,1,1,4,4,4-ヘキサフルオロ-2-ブテン(HFO-1336mzz)等のヒドロフルオロオレフィン(HFO);1-クロロ-2,2-ジフルオロエチレン(HCFO-1122)、1,2-ジクロロフルオロエチレン(HCFO-1121)、1-クロロ-2-フルオロエチレン(HCFO-1131)、2-クロロ-3,3,3-トリフルオロプロペン(HCFO-1233xf)、1-クロロ-3,3,3-テトラフルオロプロペン(HCFO-1233zd)、HCFO-1224yd(E)等のHCFO-1224yd(Z)以外のHCFO;二酸化炭素、炭化水素等が挙げられる。
本発明の熱サイクル用作動媒体は、耐酸化性向上剤、耐熱性向上剤、金属不活性剤等の安定剤に加えて、さらに、ナフテン系冷凍機油、パラフィン系冷凍機油等の鉱油系冷凍機油や、エステル系冷凍機油、エーテル系冷凍機油、ポリグリコール系冷凍機油、炭化水素系冷凍機油等の合成油系冷凍機油などの冷凍機油と混合されて、熱サイクルシステム用組成物として熱サイクルシステムに適用することができる。
本発明の熱サイクル用作動媒体が適用される熱サイクルシステムとしては、凝縮器や蒸発器等の熱交換器による熱サイクルシステムが特に制限なく用いられる。熱サイクルシステム、例えば、冷凍サイクルにおいては、気体の作動媒体を圧縮機で圧縮し、凝縮器で冷却して圧力の高い液体をつくり、膨張弁で圧力を下げ、蒸発器で低温気化させて気化熱で熱を奪う機構を有する。
熱サイクルシステムとしては、冷凍・冷蔵機器、空調機器、発電システム、熱輸送装置および二次冷却機等を特に限定せず採用することができる。本発明の熱サイクル用作動媒体の適用される熱サイクルシステムとしては、上記空調機器の一種である、遠心式冷凍機であることが好ましい。遠心式冷凍機としては、低圧型、高圧型のうち、低圧型の遠心式冷凍機であることが好ましい。なお、低圧型とは、例えば、高圧ガス保安法の適用を受けない作動媒体、すなわち、「常用の温度において、圧力0.2MPa以上となる液化ガスで現にその圧力が0.2MPa以上であるもの、または圧力が0.2MPa以上となる場合の温度が35℃以下である液化ガス」に該当しない作動媒体を用いた遠心式冷凍機をいう。
HCFO-1224yd(Z)は、分子内に炭素-炭素二重結合を有するため地球温暖化係数が低い。また、分子内に燃焼性を抑えるハロゲンの割合が多い。そのため、本発明の熱サイクル用作動媒体によれば、燃焼性が抑えられ、地球温暖化への影響が少なく、優れたサイクル性能を得られる熱サイクル用作動媒体とすることができる。特に、熱サイクル用作動媒体の充填量が多い遠心式冷凍機に適用することで、地球温暖化への影響が少なく、安全性が高く、優れたサイクル性能を得られる熱サイクルシステムを得ることができる。
(HFO-1234yfの製造方法)
本発明のHFO-1234yfの製造方法は、上記HCFOの製造方法によって、上記式(1)のXがF原子であるHCFO-1224yd(E)を異性化してHCFO-1224yd(Z)を得る工程と、HCFO-1224yd(Z)を得る工程で得られたHCFO-1224yd(Z)を水素と反応させて還元することでHFO-1234yfを得る工程とを有する。この水素還元反応において、HCFO-1224yd(Z)と水素は、下記式(4)に示す反応により、HFO-1234yfを生成する。
本発明のHFO-1234yfの製造方法は、上記HCFOの製造方法によって、上記式(1)のXがF原子であるHCFO-1224yd(E)を異性化してHCFO-1224yd(Z)を得る工程と、HCFO-1224yd(Z)を得る工程で得られたHCFO-1224yd(Z)を水素と反応させて還元することでHFO-1234yfを得る工程とを有する。この水素還元反応において、HCFO-1224yd(Z)と水素は、下記式(4)に示す反応により、HFO-1234yfを生成する。
CF3CF=CHCl + H2 → CF3CF=CH2+ HCl
・・・(4)
・・・(4)
本発明のHFO-1234yfの製造方法において、HCFO-1224yd(Z)の水素還元反応は、活性炭に担持されたパラジウム触媒の存在下、気相によって行うことが好ましい。上記水素還元反応は、例えば、ガラス、鉄、ニッケル、またはこれらを主成分とする合金等の材質で形成された反応器内に、活性炭に担持されたパラジウム触媒を充填して触媒層を形成し、当該触媒層に、HCFO-1224yd(Z)と水素を供給して行うことができる。
パラジウム触媒としてはパラジウム単体のみならず、パラジウム合金であってもよい。また、パラジウム触媒は、パラジウムと他の金属との混合物やパラジウムと他の金属とを担体に別々に担持させた複合触媒であってもよい。パラジウム触媒がパラジウム合金の場合、パラジウム触媒としては、パラジウム/白金合金触媒やパラジウム/ロジウム合金触媒などが挙げられる。
活性炭としては、木材、木炭、果実殻、ヤシ殻、泥炭、亜炭、石炭等を原料として調製したものが挙げられ、鉱物質原料よりも植物原料から得られたものが好ましく、ヤシ殻活性炭が特に好ましい。活性炭の形状としては、長さ2mm以上5mm以下程度の成形炭、4メッシュ以上50メッシュ以下程度の破砕炭、粒状炭等が挙げられる。なかでも、4メッシュ以上20メッシュ以下の破砕炭、または成形炭が好ましい。
触媒層におけるパラジウム担持活性炭の充填密度は、0.5g/cm3以上1g/cm3以下が好ましく、0.6g/cm3以上0.8g/cm3以下がより好ましい。触媒層に導入するHCFO-1224yd(Z)と水素の割合は、HCFO-1224yd(Z)中の塩素原子のモル数と水素のモル数との比(H2/Cl)で0.7以下が好ましい。H2/Clは、HFC-254ebの副生を低減する点で、0.6以下が好ましく、0.5以下がより好ましい。また、H2/Clは、HFO-1234yfの収率の点から、0.1以上が好ましく、0.2以上がより好ましい。
触媒層の温度は、気相反応である場合、反応性の点から、50℃以上が好ましく、60℃以上がより好ましく、80℃以上が特に好ましい。反応圧力は、取り扱い性の点から、常圧が好ましい。
本発明のHFO-1234yfの製造方法によれば、副生物の生成を抑制してHFO-1234yfを効率よく製造することができる。
次に、実施例について説明するが、本発明はこれらに限定されない。例1~20、例24~25は実施例、例21~23は参考例である。
(触媒調製例1)
電気炉を備えた内径23.4mm、高さ400mmのステンレス鋼(SUS316)製のチューブ型反応器に、クロムマグネシウム複合酸化物触媒(Cr2O3:98質量%、MgO:2質量%、AG-23、堺化学)を81.5mL充填し、窒素(N2)ガスを流しながら200℃まで昇温した。反応器の出口から水の流出が見られなくなるまで温度を維持し触媒を乾燥した。触媒の乾燥終了後、N2ガスと共にHCFC-22を反応器に通流させ、充填した触媒の活性化によるホットスポットが反応器の出口端に達した所で反応器温度を250℃に上げ、その状態を8時間保ち、触媒の活性化処理を行い、触媒1を得た。
電気炉を備えた内径23.4mm、高さ400mmのステンレス鋼(SUS316)製のチューブ型反応器に、クロムマグネシウム複合酸化物触媒(Cr2O3:98質量%、MgO:2質量%、AG-23、堺化学)を81.5mL充填し、窒素(N2)ガスを流しながら200℃まで昇温した。反応器の出口から水の流出が見られなくなるまで温度を維持し触媒を乾燥した。触媒の乾燥終了後、N2ガスと共にHCFC-22を反応器に通流させ、充填した触媒の活性化によるホットスポットが反応器の出口端に達した所で反応器温度を250℃に上げ、その状態を8時間保ち、触媒の活性化処理を行い、触媒1を得た。
(触媒調製例2)
反応器に、クロムマグネシウム複合酸化物触媒の代わりにアルミナ触媒(N612N、日揮触媒化成)を充填した以外は触媒調製例1と同様に調製を行い、触媒2を得た。
反応器に、クロムマグネシウム複合酸化物触媒の代わりにアルミナ触媒(N612N、日揮触媒化成)を充填した以外は触媒調製例1と同様に調製を行い、触媒2を得た。
(触媒調製例3)
反応器に、クロムマグネシウム複合酸化物触媒の代わりにフッ化アルミニウム触媒(試薬鹿特級、関東化学)を充填した以外は触媒調製例1と同様に調製を行い、触媒3を得た。
反応器に、クロムマグネシウム複合酸化物触媒の代わりにフッ化アルミニウム触媒(試薬鹿特級、関東化学)を充填した以外は触媒調製例1と同様に調製を行い、触媒3を得た。
(触媒調製例4)
反応器に、クロムマグネシウム複合酸化物触媒の代わりにジルコニウム亜鉛複合酸化物触媒(ZrO2:95質量%、ZnO:5質量%、エヌイーケムキャット)を充填した以外は触媒調製例1と同様に調製を行い、触媒4を得た。
反応器に、クロムマグネシウム複合酸化物触媒の代わりにジルコニウム亜鉛複合酸化物触媒(ZrO2:95質量%、ZnO:5質量%、エヌイーケムキャット)を充填した以外は触媒調製例1と同様に調製を行い、触媒4を得た。
(触媒調製例5)
反応器に、クロムマグネシウム複合酸化物触媒の代わりに、カーボンに0.5%パラジウムを担持した触媒(触媒の全体量に対するパラジウムの割合が0.5質量%、エヌイーケムキャット社製、0.5%Pd/C)を充填した以外は触媒調製例1と同様に調製を行い、触媒5を得た。
反応器に、クロムマグネシウム複合酸化物触媒の代わりに、カーボンに0.5%パラジウムを担持した触媒(触媒の全体量に対するパラジウムの割合が0.5質量%、エヌイーケムキャット社製、0.5%Pd/C)を充填した以外は触媒調製例1と同様に調製を行い、触媒5を得た。
(触媒調製例6)
25質量%塩化鉄(II)水溶液を調製し、円柱状の活性炭(日本エンバイロケミカルズ、現大阪ガスケミカル社製、粒状白鷺G2X)200mLを浸漬させ、3時間保持した。濾過した活性炭を減圧下、90℃で乾燥させて、塩化鉄(II)担持活性炭を得た。電気炉を備えた内径23.4mm、高さ400mmのステンレス鋼(SUS316)製のチューブ型反応器に、上記で得られた塩化鉄(II)担持活性炭を81.5mL充填し、N2ガスを流しながら200℃まで昇温した。反応器の出口から水の流出が見られなくなるまで温度を維持し触媒を乾燥した。触媒の乾燥終了後、N2ガスと共にHCFC-22を反応器に通流させ、充填した触媒の活性化によるホットスポットが反応器の出口端に達した所で反応器温度を250℃に上げ、その状態を8時間保ち、触媒の活性化処理を行い、触媒6を得た。
25質量%塩化鉄(II)水溶液を調製し、円柱状の活性炭(日本エンバイロケミカルズ、現大阪ガスケミカル社製、粒状白鷺G2X)200mLを浸漬させ、3時間保持した。濾過した活性炭を減圧下、90℃で乾燥させて、塩化鉄(II)担持活性炭を得た。電気炉を備えた内径23.4mm、高さ400mmのステンレス鋼(SUS316)製のチューブ型反応器に、上記で得られた塩化鉄(II)担持活性炭を81.5mL充填し、N2ガスを流しながら200℃まで昇温した。反応器の出口から水の流出が見られなくなるまで温度を維持し触媒を乾燥した。触媒の乾燥終了後、N2ガスと共にHCFC-22を反応器に通流させ、充填した触媒の活性化によるホットスポットが反応器の出口端に達した所で反応器温度を250℃に上げ、その状態を8時間保ち、触媒の活性化処理を行い、触媒6を得た。
(触媒調製例7)
25質量%塩化鉄(II)水溶液に代えて、17質量%塩化ニッケル(II)水溶液を用いた以外は、触媒調製例6と同様に調製を行い、触媒7を得た。
25質量%塩化鉄(II)水溶液に代えて、17質量%塩化ニッケル(II)水溶液を用いた以外は、触媒調製例6と同様に調製を行い、触媒7を得た。
(触媒調製例8)
25質量%塩化鉄(II)水溶液に代えて、25質量%塩化銅(II)水溶液を用いた以外は、触媒調製例6と同様に調製を行い、触媒8を得た。
25質量%塩化鉄(II)水溶液に代えて、25質量%塩化銅(II)水溶液を用いた以外は、触媒調製例6と同様に調製を行い、触媒8を得た。
(合成例1:HCFO-1224ydの製造)
特許文献3に記載の方法と同様の方法でHCFO-1224ydを製造した。ステンレス鋼(SUS316)製のチューブ型反応器内に、活性炭にパラジウムを担持させたパラジウム担持活性炭を充填して、触媒層を形成した。その後、反応器を、加熱炉によって80℃に保持した状態で、反応器内に、水素(H2)/CFO-1214yaモル比=1/1になる割合で混合したCFO-1214yaと水素の混合ガスを供給した。この際、CFO-1214yaと水素の混合ガスの触媒層における滞留時間が25秒になるように上記混合ガスを反応器内に供給した。
特許文献3に記載の方法と同様の方法でHCFO-1224ydを製造した。ステンレス鋼(SUS316)製のチューブ型反応器内に、活性炭にパラジウムを担持させたパラジウム担持活性炭を充填して、触媒層を形成した。その後、反応器を、加熱炉によって80℃に保持した状態で、反応器内に、水素(H2)/CFO-1214yaモル比=1/1になる割合で混合したCFO-1214yaと水素の混合ガスを供給した。この際、CFO-1214yaと水素の混合ガスの触媒層における滞留時間が25秒になるように上記混合ガスを反応器内に供給した。
上記反応器から得られた出口ガスを濃度10質量%の水酸化カリウム(KOH)水溶液に通流させて酸性成分を除去した後、合成ゼオライト(モレキュラーシーブス4A)を充填した脱水塔に通流させて脱水した。脱水後の出口ガスを、ドライアイスで冷却されたシリンダー内に捕捉した。この捕捉された出口ガスを試料として、理論段数約30段の蒸留塔の塔底に供給し、運転圧力0.02MPa(ゲージ圧)で、バッチ蒸留による蒸留を行い、蒸留塔の塔頂からHCFO-1224yd(Z)を79.1%、HCFO-1224yd(E)を19.3%含む留出液(以下、本留出液を「原料組成物A」という。)を得た。
(例1)
内径23.4mm、高さ400mmのステンレス鋼(SUS316)製のチューブ型反応器(以下、反応器ともいう。)に触媒1の81.5mLを充填した。原料組成物Aを27.9NmL/min、希釈ガスとして窒素ガス(N2)を55.7NmL/minの割合で混合して、加熱炉によって50℃に予熱した。その後、予熱した原料組成物Aと窒素ガスの混合ガスを、加熱炉によって50℃に保持した反応器に、ほぼ大気圧下で通流させた。反応器からの出口ガスを捕集し、GC(ガスクロマトグラフィー)によって分析したところ、出口ガスの組成は、GCによる面積百分率(GC area%)でHCFO-1224yd(Z)が88.2(GC area%)で、HCFO-1224yd(E)が11.1%(GC area%)であった。また、反応時間(原料組成物Aの反応器内の滞留時間)は49.5秒であった。
内径23.4mm、高さ400mmのステンレス鋼(SUS316)製のチューブ型反応器(以下、反応器ともいう。)に触媒1の81.5mLを充填した。原料組成物Aを27.9NmL/min、希釈ガスとして窒素ガス(N2)を55.7NmL/minの割合で混合して、加熱炉によって50℃に予熱した。その後、予熱した原料組成物Aと窒素ガスの混合ガスを、加熱炉によって50℃に保持した反応器に、ほぼ大気圧下で通流させた。反応器からの出口ガスを捕集し、GC(ガスクロマトグラフィー)によって分析したところ、出口ガスの組成は、GCによる面積百分率(GC area%)でHCFO-1224yd(Z)が88.2(GC area%)で、HCFO-1224yd(E)が11.1%(GC area%)であった。また、反応時間(原料組成物Aの反応器内の滞留時間)は49.5秒であった。
(例2~7)
反応器内の温度(反応温度ともいう。)、窒素ガスの流量(窒素流量ともいう。)、原料組成物Aの流量(原料流量ともいう。)を、表1の条件に変更した以外は例1と同様の操作を行った。反応器の出口ガスを捕集し、GCによって分析した。GC分析結果を表1に示す。
反応器内の温度(反応温度ともいう。)、窒素ガスの流量(窒素流量ともいう。)、原料組成物Aの流量(原料流量ともいう。)を、表1の条件に変更した以外は例1と同様の操作を行った。反応器の出口ガスを捕集し、GCによって分析した。GC分析結果を表1に示す。
(例8~12)
金属触媒を触媒2に変更し、反応温度、窒素流量、原料流量を表1の条件に変更した以外は例1と同様の操作を行った。反応器の出口ガスを捕集し、GCによって分析した。GC分析結果を表1に示す。
金属触媒を触媒2に変更し、反応温度、窒素流量、原料流量を表1の条件に変更した以外は例1と同様の操作を行った。反応器の出口ガスを捕集し、GCによって分析した。GC分析結果を表1に示す。

(例13~18)
例1と同様のチューブ型反応器に、表2に示す通り、触媒3~8をそれぞれ81.5mL充填した。原料組成物Aを19.0NmL/min、窒素ガス(N2)を38.1NmL/minの割合で混合して50℃に予熱した。反応器を加熱炉によって200℃に保持した状態で、予熱された原料組成物Aと窒素の混合ガスを反応器にほぼ大気圧下で流通させた。反応器を流通した直後の出口ガスを、濃度10質量%の水酸化カリウム(KOH)水溶液に通流させて酸性成分を除去した(酸洗浄)後、合成ゼオライト(モレキュラーシーブス4A)を充填した脱水塔に通流させて脱水し、脱水後の出口ガスをドライアイスで冷却したシリンダー中に捕集した。捕集した出口ガスを分取し、その組成をGCによって分析した。GC分析結果を表2に示す。
例1と同様のチューブ型反応器に、表2に示す通り、触媒3~8をそれぞれ81.5mL充填した。原料組成物Aを19.0NmL/min、窒素ガス(N2)を38.1NmL/minの割合で混合して50℃に予熱した。反応器を加熱炉によって200℃に保持した状態で、予熱された原料組成物Aと窒素の混合ガスを反応器にほぼ大気圧下で流通させた。反応器を流通した直後の出口ガスを、濃度10質量%の水酸化カリウム(KOH)水溶液に通流させて酸性成分を除去した(酸洗浄)後、合成ゼオライト(モレキュラーシーブス4A)を充填した脱水塔に通流させて脱水し、脱水後の出口ガスをドライアイスで冷却したシリンダー中に捕集した。捕集した出口ガスを分取し、その組成をGCによって分析した。GC分析結果を表2に示す。

表1、2に示されるように、HCFO-1224yd(E)を、非常に高い収率(変換率)でHCFO-1224yd(Z)に変換することができることがわかる。
(合成例2:HCFO-1223xdの合成)
国際公開第2014/046250号に記載の方法と同様の方法で、HCFO-1223xdを製造した。具体的には、加熱した水酸化カリウム水溶液中にHCFC-233daを滴下して反応させた。反応後の反応液を水相と油相に分離することで、油相として、HCFO-1223xd(Z)を90.3%、HCFO-1223xd(E)を5.7%含む反応粗液の7194gを得た。得られた反応粗液を蒸留精製し、HCFO-1223xd(Z)を17.1%、HCFO-1223xd(E)を79.6%含む缶出液(以下、本缶出液を「原料組成物B」ともいう。)を54g、およびHCFO-1223xd(Z)を99.8%、HCFO-1223xd(E)を0.2%含む留出液(以下、本留出液を「原料組成物C」ともいう。)を3597g得た。
国際公開第2014/046250号に記載の方法と同様の方法で、HCFO-1223xdを製造した。具体的には、加熱した水酸化カリウム水溶液中にHCFC-233daを滴下して反応させた。反応後の反応液を水相と油相に分離することで、油相として、HCFO-1223xd(Z)を90.3%、HCFO-1223xd(E)を5.7%含む反応粗液の7194gを得た。得られた反応粗液を蒸留精製し、HCFO-1223xd(Z)を17.1%、HCFO-1223xd(E)を79.6%含む缶出液(以下、本缶出液を「原料組成物B」ともいう。)を54g、およびHCFO-1223xd(Z)を99.8%、HCFO-1223xd(E)を0.2%含む留出液(以下、本留出液を「原料組成物C」ともいう。)を3597g得た。
(例19、20)
例1において、原料組成物Aに代えて原料組成物Bを用い、反応温度、窒素流量、原料流量を、表3に示すように変更した以外は例1と同様の操作を行った。例1と同様に反応器の出口ガスを捕集し、捕集された出口ガスの組成をGCによって分析した。GC分析結果を表3に示す。
例1において、原料組成物Aに代えて原料組成物Bを用い、反応温度、窒素流量、原料流量を、表3に示すように変更した以外は例1と同様の操作を行った。例1と同様に反応器の出口ガスを捕集し、捕集された出口ガスの組成をGCによって分析した。GC分析結果を表3に示す。

表3に示されるように、HCFO-1223xd(E)を非常に高い収率(変換率)でHCFO-1223xd(Z)に変換することができることがわかる。
(例21~23)
例1において、原料組成物Aに代えて原料組成物Cを用い、反応温度、窒素流量、原料流量、それぞれ表4に示すように変更した以外は例1と同様の操作を行った。例1と同様に反応器の出口ガスを捕集し、捕集された出口ガスの組成をGCによって分析した。GC分析結果を表4に示す。
例1において、原料組成物Aに代えて原料組成物Cを用い、反応温度、窒素流量、原料流量、それぞれ表4に示すように変更した以外は例1と同様の操作を行った。例1と同様に反応器の出口ガスを捕集し、捕集された出口ガスの組成をGCによって分析した。GC分析結果を表4に示す。

表4に示されるように、原料組成物中のHCFO-1223xd(E)/HCFO-1223xd(Z)を平衡比よりも小さくすることで、HCFO-1223xd(Z)をHCFO-1223xd(E)に変換することができることがわかる。
(例24)
例1と同様のチューブ型反応器(内径23.4mm、高さ400mmのSUS316製のチューブ型反応器)を用い、加熱炉の温度を400℃に設定して反応器を加熱した。原料組成物Aを442.6NmL/min、ラジカル発生剤として、塩素を5.2NmL/minの割合(原料組成物A中のHCFO-1224yd(E)/塩素モル比=85.1/1)で混合して200℃に予熱し、その後、上記温度に加熱された反応器に供給し、ほぼ大気圧下で流通させた。反応器を流通した直後の出口ガスを、濃度10質量%のKOH水溶液に通流させて酸性成分を除去した(酸洗浄)後、合成ゼオライト(モレキュラーシーブス4A)を充填した脱水塔に通流させて脱水し、脱水後の出口ガスをドライアイスで冷却したシリンダー中に捕捉した。捕捉した出口ガスを分取し、その組成をGCによって分析した。GC分析結果を表5に示す。
例1と同様のチューブ型反応器(内径23.4mm、高さ400mmのSUS316製のチューブ型反応器)を用い、加熱炉の温度を400℃に設定して反応器を加熱した。原料組成物Aを442.6NmL/min、ラジカル発生剤として、塩素を5.2NmL/minの割合(原料組成物A中のHCFO-1224yd(E)/塩素モル比=85.1/1)で混合して200℃に予熱し、その後、上記温度に加熱された反応器に供給し、ほぼ大気圧下で流通させた。反応器を流通した直後の出口ガスを、濃度10質量%のKOH水溶液に通流させて酸性成分を除去した(酸洗浄)後、合成ゼオライト(モレキュラーシーブス4A)を充填した脱水塔に通流させて脱水し、脱水後の出口ガスをドライアイスで冷却したシリンダー中に捕捉した。捕捉した出口ガスを分取し、その組成をGCによって分析した。GC分析結果を表5に示す。
(例25)
例24において、ラジカル発生剤の種類を塩素から空気に代えた(原料組成物A中のHCFO-1224yd(E)/空気(体積比)=85.1/1)以外は例24と同様の操作を行った。シリンダー中に捕捉した出口ガスを分取し、その組成をGCによって分析した。GC分析結果を表5に示す。
例24において、ラジカル発生剤の種類を塩素から空気に代えた(原料組成物A中のHCFO-1224yd(E)/空気(体積比)=85.1/1)以外は例24と同様の操作を行った。シリンダー中に捕捉した出口ガスを分取し、その組成をGCによって分析した。GC分析結果を表5に示す。

表5に示されるように、HCFO-1224yd(E)を非常に高い収率(変換率)でHCFO-1224yd(Z)に変換することができることがわかる。
Claims (13)
- 下記式(1)で表される化合物が異性化される条件下で、原料組成物に含まれる下記式(1)で表される化合物を異性化反応させて、下記式(2)で表される化合物を製造することを特徴とするヒドロクロロフルオロオレフィンの製造方法。


- 前記式(1)で表される化合物を金属触媒と接触させることで、前記式(1)で表される化合物を異性化反応させる請求項1記載のヒドロクロロフルオロオレフィンの製造方法。
- 前記金属触媒が、金属単体、金属酸化物、金属ハロゲン化物からなる群から選ばれる少なくとも1種の物質である、請求項2記載のヒドロクロロフルオロオレフィンの製造方法。
- 前記金属触媒を構成する金属が、第4族金属元素、第6族金属元素、第8族金属元素、第9族金属元素、第10族金属元素、第11族金属元素、第12族金属元素、第13族金属元素からなる群から選ばれる少なくとも1種の元素である、請求項2または3記載のヒドロクロロフルオロオレフィンの製造方法。
- 前記金属触媒は、金属酸化物である請求項2~4のいずれか1項記載のヒドロクロロフルオロオレフィンの製造方法。
- 前記金属触媒は、酸化アルミニウムおよび酸化クロムからなる群から選ばれる1種以上の物質である、請求項2~5のいずれか1項記載のヒドロクロロフルオロオレフィンの製造方法。
- 前記原料組成物中に前記式(2)で表される化合物が含まれ、前記原料組成物中の前記式(1)で表される化合物と前記原料組成物中の前記式(2)で表される化合物とのモル比(前記原料組成物中の前記式(1)で表される化合物のモル数/前記原料組成物中の前記式(2)で表される化合物のモル数)が5/95以上である、請求項1~6のいずれか1項記載のヒドロクロロフルオロオレフィンの製造方法。
- 前記式(1)で表される化合物を気相で異性化反応させる、請求項1~7のいずれか1項に記載のヒドロクロロフルオロオレフィンの製造方法。
- 前記式(1)で表される化合物と前記金属触媒との接触温度は50℃以上500℃以下である、請求項2~8のいずれか1項に記載のヒドロクロロフルオロオレフィンの製造方法。
- 前記式(1)で表される化合物と前記金属触媒との接触時間は0.1秒以上1000秒以下である、請求項2~9のいずれか1項に記載のヒドロクロロフルオロオレフィンの製造方法。
- 前記原料組成物は前記式(1)で表される化合物として少なくとも(E)-1-クロロ-2,3,3,3-テトラフルオロプロペンを含み、前記(E)-1-クロロ-2,3,3,3-テトラフルオロプロペンが異性化される条件下で前記(E)-1-クロロ-2,3,3,3-テトラフルオロプロペンを異性化反応させて、前記(2)で表される化合物として(Z)-1-クロロ-2,3,3,3-テトラフルオロプロペンを製造する、請求項1~10のいずれか1項に記載のヒドロクロロフルオロオレフィンの製造方法。
- 前記原料組成物は前記式(2)で表される化合物として少なくとも(Z)-1-クロロ-2,3,3,3-テトラフルオロプロペンを含む、請求項11に記載のヒドロクロロフルオロオレフィンの製造方法。
- 請求項11または12に記載のヒドロクロロフルオロオレフィンの製造方法により(Z)-1-クロロ-2,3,3,3-テトラフルオロプロペンを得る工程と、前記工程で得られた前記(Z)-1-クロロ-2,3,3,3-テトラフルオロプロペンを水素により還元して2,3,3,3-テトラフルオロプロペンを得る工程とを有することを特徴とする2,3,3,3-テトラフルオロプロペンの製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP16818033.9A EP3318548A4 (en) | 2015-06-30 | 2016-06-30 | PREPARATION OF HYDROCHLOROFLUOROLEFINE AND METHOD OF MANUFACTURING 2,3,3,3-TETRAFLUORPROPES |
| JP2017526435A JP7101478B2 (ja) | 2015-06-30 | 2016-06-30 | ヒドロクロロフルオロオレフィンの製造方法、および2,3,3,3-テトラフルオロプロペンの製造方法 |
| CN201680037956.6A CN107709278B (zh) | 2015-06-30 | 2016-06-30 | 氢氯氟烯烃的制造方法以及2,3,3,3-四氟丙烯的制造方法 |
| US15/856,999 US10442744B2 (en) | 2015-06-30 | 2017-12-28 | Method of producing hydrochlorofluoroolefin and method of producing 2,3,3,3-tetrafluoropropene |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015130833 | 2015-06-30 | ||
| JP2015-130833 | 2015-06-30 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/856,999 Continuation US10442744B2 (en) | 2015-06-30 | 2017-12-28 | Method of producing hydrochlorofluoroolefin and method of producing 2,3,3,3-tetrafluoropropene |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017002925A1 true WO2017002925A1 (ja) | 2017-01-05 |
Family
ID=57608385
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/069464 WO2017002925A1 (ja) | 2015-06-30 | 2016-06-30 | ヒドロクロロフルオロオレフィンの製造方法、および2,3,3,3-テトラフルオロプロペンの製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US10442744B2 (ja) |
| EP (1) | EP3318548A4 (ja) |
| JP (3) | JP7101478B2 (ja) |
| CN (1) | CN107709278B (ja) |
| WO (1) | WO2017002925A1 (ja) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018047972A1 (ja) * | 2016-09-12 | 2018-03-15 | 旭硝子株式会社 | 1-クロロ-2,3,3-トリフルオロプロペンの製造方法 |
| WO2019036049A1 (en) * | 2017-08-18 | 2019-02-21 | The Chemours Company, Fc, Llc | COMPOSITIONS AND USES OF Z-1-CHLORO-2,3,3,3-TETRAFLUOROPROP-1-ENE |
| WO2019088035A1 (ja) * | 2017-11-01 | 2019-05-09 | Agc株式会社 | 硬質発泡合成樹脂の製造方法 |
| WO2019123759A1 (ja) * | 2017-12-22 | 2019-06-27 | Agc株式会社 | 溶剤組成物、洗浄方法、塗膜形成用組成物、塗膜付き基材の製造方法、エアゾール組成物、リンス組成物、部材の洗浄方法および部材の洗浄装置 |
| US20190390878A1 (en) * | 2017-08-10 | 2019-12-26 | Mitsubishi Heavy Industries Thermal Systems, Ltd. | Heat pump and method for designing the same |
| WO2020008865A1 (ja) * | 2018-07-06 | 2020-01-09 | セントラル硝子株式会社 | 1,2-ジクロロ-3,3,3-トリフルオロプロペンの製造方法 |
| CN110869461A (zh) * | 2017-07-26 | 2020-03-06 | Agc株式会社 | 共沸或类共沸组合物、热循环用工作介质和热循环系统 |
| JP2021059498A (ja) * | 2019-10-03 | 2021-04-15 | セントラル硝子株式会社 | 不飽和クロロフルオロカーボンの製造方法 |
| JP7502615B2 (ja) | 2020-07-01 | 2024-06-19 | セントラル硝子株式会社 | Z-1,2-ジクロロ-3,3,3-トリフルオロプロペンを製造する方法 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3492547B1 (en) * | 2016-07-29 | 2023-10-18 | Agc Inc. | Working fluid for heat cycle |
| JP6753434B2 (ja) * | 2018-06-13 | 2020-09-09 | ダイキン工業株式会社 | ジフルオロエチレンの製造方法 |
| JP6870704B2 (ja) * | 2019-06-10 | 2021-05-12 | ダイキン工業株式会社 | 1−ハロ−2−フルオロエチレンの製造方法 |
| EP4119531A1 (en) * | 2019-07-03 | 2023-01-18 | The Chemours Company FC, LLC | Compositions and methods for synthesis of 2,3-dichloro-1,1,1,2-tetrafluoropropane and 2,3,3,3-tetrafluoropropene |
| WO2024071127A1 (ja) * | 2022-09-28 | 2024-04-04 | ダイキン工業株式会社 | ジフルオロエチレンの製造方法 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010523635A (ja) * | 2007-04-11 | 2010-07-15 | イネオス、フラウアー、ホールディングス、リミテッド | (ヒドロハロ)フルオロアルケンを異性化する方法 |
| JP2010529111A (ja) * | 2007-09-11 | 2010-08-26 | ダイキン工業株式会社 | 2,3,3,3−テトラフルオロプロペンの製造方法 |
| JP2010536777A (ja) * | 2007-08-16 | 2010-12-02 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー | アルミニウム触媒を用いた1,2,3,3,3−ペンタフルオロプロペンのe異性体とz異性体間での触媒異性化 |
| JP2012509324A (ja) * | 2008-11-19 | 2012-04-19 | アーケマ・インコーポレイテッド | ヒドロクロロフルオロオレフィンを製造するための方法 |
| JP2012512160A (ja) * | 2008-12-12 | 2012-05-31 | ハネウェル・インターナショナル・インコーポレーテッド | 1−クロロ−3,3,3−トリフルオロプロペンの異性化 |
| JP2013180964A (ja) * | 2012-03-01 | 2013-09-12 | Asahi Glass Co Ltd | 2,3,3,3−テトラフルオロプロペンの製造方法 |
| WO2014046251A1 (ja) * | 2012-09-21 | 2014-03-27 | セントラル硝子株式会社 | 1,2-ジクロロ-3,3,3-トリフルオロプロペンの製造方法 |
| JP2014513673A (ja) * | 2011-02-21 | 2014-06-05 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー | ヒドロクロロフルオロカーボンの触媒接触脱塩化水素化 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6226239A (ja) * | 1985-05-28 | 1987-02-04 | イ−・アイ・デユポン・デ・ニモアス・アンド・カンパニ− | フルオロオレフインの接触ハロゲン交換法 |
| US8987535B2 (en) | 2008-11-19 | 2015-03-24 | Arkema Inc. | Process for the manufacture of hydrochlorofluoroolefins |
| US8987534B2 (en) | 2008-11-19 | 2015-03-24 | Arkema Inc. | Process for the manufacture of hydrochlorofluoroolefins |
| TW201219111A (en) | 2010-06-23 | 2012-05-16 | Asahi Glass Co Ltd | Process for producing 2, 3, 3, 3-tetrafluoropropene |
| GB2519572B (en) * | 2013-10-25 | 2015-12-30 | Mexichem Amanco Holding Sa | Process for isomerising (hydro)(halo)fluoroalkenes |
-
2016
- 2016-06-30 EP EP16818033.9A patent/EP3318548A4/en active Pending
- 2016-06-30 WO PCT/JP2016/069464 patent/WO2017002925A1/ja active Application Filing
- 2016-06-30 JP JP2017526435A patent/JP7101478B2/ja active Active
- 2016-06-30 CN CN201680037956.6A patent/CN107709278B/zh active Active
-
2017
- 2017-12-28 US US15/856,999 patent/US10442744B2/en not_active Expired - Fee Related
-
2020
- 2020-11-25 JP JP2020195457A patent/JP2021028340A/ja not_active Ceased
-
2022
- 2022-03-28 JP JP2022052007A patent/JP2022076023A/ja not_active Withdrawn
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010523635A (ja) * | 2007-04-11 | 2010-07-15 | イネオス、フラウアー、ホールディングス、リミテッド | (ヒドロハロ)フルオロアルケンを異性化する方法 |
| JP2010536777A (ja) * | 2007-08-16 | 2010-12-02 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー | アルミニウム触媒を用いた1,2,3,3,3−ペンタフルオロプロペンのe異性体とz異性体間での触媒異性化 |
| JP2010529111A (ja) * | 2007-09-11 | 2010-08-26 | ダイキン工業株式会社 | 2,3,3,3−テトラフルオロプロペンの製造方法 |
| JP2012509324A (ja) * | 2008-11-19 | 2012-04-19 | アーケマ・インコーポレイテッド | ヒドロクロロフルオロオレフィンを製造するための方法 |
| JP2012512160A (ja) * | 2008-12-12 | 2012-05-31 | ハネウェル・インターナショナル・インコーポレーテッド | 1−クロロ−3,3,3−トリフルオロプロペンの異性化 |
| JP2014513673A (ja) * | 2011-02-21 | 2014-06-05 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー | ヒドロクロロフルオロカーボンの触媒接触脱塩化水素化 |
| JP2013180964A (ja) * | 2012-03-01 | 2013-09-12 | Asahi Glass Co Ltd | 2,3,3,3−テトラフルオロプロペンの製造方法 |
| WO2014046251A1 (ja) * | 2012-09-21 | 2014-03-27 | セントラル硝子株式会社 | 1,2-ジクロロ-3,3,3-トリフルオロプロペンの製造方法 |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018047972A1 (ja) * | 2016-09-12 | 2018-03-15 | 旭硝子株式会社 | 1-クロロ-2,3,3-トリフルオロプロペンの製造方法 |
| CN110869461A (zh) * | 2017-07-26 | 2020-03-06 | Agc株式会社 | 共沸或类共沸组合物、热循环用工作介质和热循环系统 |
| US11441821B2 (en) * | 2017-08-10 | 2022-09-13 | Mitsubishi Heavy Industries Thermal System, Ltd. | Heat pump and method for designing the same |
| US20190390878A1 (en) * | 2017-08-10 | 2019-12-26 | Mitsubishi Heavy Industries Thermal Systems, Ltd. | Heat pump and method for designing the same |
| JP2020531633A (ja) * | 2017-08-18 | 2020-11-05 | ザ ケマーズ カンパニー エフシー リミテッド ライアビリティ カンパニー | Z−1−クロロ−2,3,3,3−テトラフルオロプロパ−1−エンの組成物及び使用 |
| US11679291B2 (en) | 2017-08-18 | 2023-06-20 | The Chemours Company Fc, Llc | Compositions and uses of Z-1-chloro-2,3,3,3-tetrafluoroprop-1-ene |
| WO2019036049A1 (en) * | 2017-08-18 | 2019-02-21 | The Chemours Company, Fc, Llc | COMPOSITIONS AND USES OF Z-1-CHLORO-2,3,3,3-TETRAFLUOROPROP-1-ENE |
| WO2019088035A1 (ja) * | 2017-11-01 | 2019-05-09 | Agc株式会社 | 硬質発泡合成樹脂の製造方法 |
| CN111263780A (zh) * | 2017-11-01 | 2020-06-09 | Agc株式会社 | 硬质发泡合成树脂的制造方法 |
| CN111263780B (zh) * | 2017-11-01 | 2022-04-05 | Agc株式会社 | 硬质发泡合成树脂的制造方法 |
| WO2019123759A1 (ja) * | 2017-12-22 | 2019-06-27 | Agc株式会社 | 溶剤組成物、洗浄方法、塗膜形成用組成物、塗膜付き基材の製造方法、エアゾール組成物、リンス組成物、部材の洗浄方法および部材の洗浄装置 |
| JPWO2020008865A1 (ja) * | 2018-07-06 | 2021-07-08 | セントラル硝子株式会社 | 1,2−ジクロロ−3,3,3−トリフルオロプロペンの製造方法 |
| WO2020008865A1 (ja) * | 2018-07-06 | 2020-01-09 | セントラル硝子株式会社 | 1,2-ジクロロ-3,3,3-トリフルオロプロペンの製造方法 |
| JP7315856B2 (ja) | 2018-07-06 | 2023-07-27 | セントラル硝子株式会社 | 1,2-ジクロロ-3,3,3-トリフルオロプロペンの製造方法 |
| JP2021059498A (ja) * | 2019-10-03 | 2021-04-15 | セントラル硝子株式会社 | 不飽和クロロフルオロカーボンの製造方法 |
| JP7502615B2 (ja) | 2020-07-01 | 2024-06-19 | セントラル硝子株式会社 | Z-1,2-ジクロロ-3,3,3-トリフルオロプロペンを製造する方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN107709278A (zh) | 2018-02-16 |
| EP3318548A4 (en) | 2019-02-06 |
| US20180134639A1 (en) | 2018-05-17 |
| JP2021028340A (ja) | 2021-02-25 |
| US10442744B2 (en) | 2019-10-15 |
| EP3318548A1 (en) | 2018-05-09 |
| CN107709278B (zh) | 2020-12-18 |
| JP2022076023A (ja) | 2022-05-18 |
| JPWO2017002925A1 (ja) | 2018-04-19 |
| JP7101478B2 (ja) | 2022-07-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7101478B2 (ja) | ヒドロクロロフルオロオレフィンの製造方法、および2,3,3,3-テトラフルオロプロペンの製造方法 | |
| JP6885447B2 (ja) | トリフルオロエチレンを含む流体の精製方法、およびトリフルオロエチレンの製造方法 | |
| US10072194B2 (en) | Working fluid for heat cycle | |
| JP6379391B2 (ja) | トリフルオロエチレンを含む組成物 | |
| JP6650523B2 (ja) | 1−クロロ−3,3,3−トリフルオロプロピレン、2,3,3,3−テトラフルオロプロピレン及び1,3,3,3−テトラフルオロプロピレンの同時生産方法 | |
| EP1943203B1 (en) | Method for producing fluorinated organic compounds | |
| CN101801894B (zh) | 通过金属氟化物催化剂制备含卤素和氢的烯烃 | |
| JP2015229767A (ja) | 熱サイクル用作動媒体 | |
| JP2011500695A (ja) | フッ素化されたオレフィンの合成のための方法 | |
| JP2014503494A (ja) | 2,3,3,3−テトラフルオロプロペンの製造法 | |
| JP2013523882A (ja) | テトラフルオロオレフィンを製造するための方法 | |
| JP6152476B2 (ja) | 2,3,3,3−テトラフルオロプロペンの製造方法 | |
| JP2024144738A (ja) | 2,3-ジクロロ-1,1,1,2-テトラフルオロプロパン及び2,3,3,3-テトラフルオロプロペンの合成のための組成物及び方法 | |
| WO2018047972A1 (ja) | 1-クロロ-2,3,3-トリフルオロプロペンの製造方法 | |
| KR20080066853A (ko) | 플루오르화 유기 화합물의 제조 방법 | |
| JP2016023145A (ja) | トリフルオロエチレンの精製方法 | |
| JP6549553B2 (ja) | フッ素化オレフィンの製造プロセス |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16818033 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017526435 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016818033 Country of ref document: EP |