KR101100064B1 - 나프록센의 니트로옥시유도체 제조방법 - Google Patents
나프록센의 니트로옥시유도체 제조방법 Download PDFInfo
- Publication number
- KR101100064B1 KR101100064B1 KR1020057003509A KR20057003509A KR101100064B1 KR 101100064 B1 KR101100064 B1 KR 101100064B1 KR 1020057003509 A KR1020057003509 A KR 1020057003509A KR 20057003509 A KR20057003509 A KR 20057003509A KR 101100064 B1 KR101100064 B1 KR 101100064B1
- Authority
- KR
- South Korea
- Prior art keywords
- compound
- formula
- reaction
- hydrogen
- naproxen
- Prior art date
Links
- 0 C[*@]([C@]1*)C1=*N Chemical compound C[*@]([C@]1*)C1=*N 0.000 description 3
- RXYPXQSKLGGKOL-UHFFFAOYSA-N CN1CCN(C)CC1 Chemical compound CN1CCN(C)CC1 RXYPXQSKLGGKOL-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C269/00—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C269/06—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups by reactions not involving the formation of carbamate groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C201/00—Preparation of esters of nitric or nitrous acid or of compounds containing nitro or nitroso groups bound to a carbon skeleton
- C07C201/02—Preparation of esters of nitric acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C203/00—Esters of nitric or nitrous acid
- C07C203/02—Esters of nitric acid
- C07C203/04—Esters of nitric acid having nitrate groups bound to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/26—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atom of at least one of the carbamate groups bound to a carbon atom of a six-membered aromatic ring
- C07C271/28—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atom of at least one of the carbamate groups bound to a carbon atom of a six-membered aromatic ring to a carbon atom of a non-condensed six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C303/00—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides
- C07C303/26—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfonic acids
- C07C303/28—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfonic acids by reaction of hydroxy compounds with sulfonic acids or derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/10—Preparation of carboxylic acid esters by reacting carboxylic acids or symmetrical anhydrides with ester groups or with a carbon-halogen bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/66—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety
- C07C69/73—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of unsaturated acids
- C07C69/734—Ethers
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
본 발명은 화학식 (B)의 화합물을 화학식 (C)의 화합물과 반응시키는 것을 포함하여, 화학식 (A)의 화합물을 제조하는 방법에 관한 것이다:

R-COOZ (B)

상기 식에서,
R은 나프록센 또는 브로모나프록센의 기이며,
R1-R12는 수소, 또는 알킬 그룹이고,
m, n, o, q, r 및 s는 각각 독립적으로 0 내지 6의 정수이고, p는 0 또는 1이고,
X는 O, S, SO, SO2, NR13 또는 PR13, 또는 아릴, 헤테로아릴 그룹이고,
Z는 수소, 또는 Li+, Na+, K+, Ca++, Mg++, 테트라알킬암모늄 및 테트라알킬포스포늄으로부터 선택된 양이온이고;
Y는 적합한 이탈기이다.
2-(S)-(5-브로모-6-메톡시-2-나프틸)-프로파노산, 4-(니트로옥시)부틸 에스테르, 나프록센, 브로모나프록센
Description
본 발명은 나프록센 (2-(S)-(6-메톡시-2-나프틸)-프로파노산) 또는 브로모나프록센 (2-(S)-(5-브로모-6-메톡시-2-나프틸)-프로파노산) [Tetrahedron 1989, 45권, 페이지 4243-4252]의 니트로옥시알킬에스테르의 제조방법에 관한 것이다.
(2-(S)-(6-메톡시-2-나프틸)-프로파노산)의 항염증성 활성이 시판되고 있는 제품 (나프록센)인 S 거울상 이성질체에 기인한다는 것은 선행 기술에서 잘 알려졌다.
WO 01/10814는 (2-(S)-(6-메톡시-2-나프틸)-프로피오닐 클로라이드를 메틸렌 클로라이드 중에서 탄산칼륨의 존재 하에 4-니트로옥시부탄-1-올과 반응시킴으로써 2-(S)-(6-메톡시-2-나프틸)-프로피오산의 니트로옥시부틸에스테르를 제조하는 방법을 개시한다. 획득된 에스테르는 97% 이상의 거울상 이성질체 과잉률 (e.e.)을 가진다. 이 방법은 몇몇 부산물이 형성된다는 단점이 있으며, 실제로 순수한 형태의 니트로옥시알킬 알콜 및 높은 화학적 및 거울상 이성질체 순도의 2-아릴프로파노일 할라이드를 획득하는 것은 매우 어렵다. 더욱이, 예를들어 4-니트로옥시부탄-1-올은 용액 내에서만 안정하며, 순수한 물질로 분리될 수 없다.
본 발명은 출발 나프록센 또는 브로모나프록센의 것만큼 높은 거울상 이성질체 과잉률을 가진 나프록센 또는 브로모나프록센의 니트로옥시알킬에스테르의 새로운 제조방법을 제공하며, 여기에서는 불순물 및 부산물이 본질적으로 무시할 만한 양으로 존재한다. 그러므로, 거울상 이성질체적으로 순수한 나프록센을 출발물질로 하여, 거울상 이성질체적으로 순수한 에스테르가 얻어진다. 이것은 특별히 중요한데, 왜냐하면: ⅰ) 대부분의 나프록센의 니트로옥시알킬 에스테르는 낮은 융점이거나 액체 물질이고, 따라서 획득된 조 에스테르의 e.e.는 통상적인 물리적 방법에 의해서 향상될 수 없고. ⅱ) 고려 중인 분자들에서 에스테르 이외의 기능기의 부재는 정제를 어렵게 만들기 때문이다.
본 발명의 또 다른 이점은 출발 화합물이 안정하다는 것이다. 본 발명의 방법은 출발물질로서 나프록센의 염 및 알킬 쇄 내에 치환기로서 이탈기를 갖는 니트로옥시 알킬 유도체를 사용한다.
나프록센 염은 암모늄 또는 알칼리 금속염으로서 사용된다. 나트륨염은 화학적 및 거울상 이성질체적으로 안정하며, 대규모로 상업적으로 이용가능하지 않으며 화학적으로 불안정하고 라세미화되기 쉬운 2-(S)-(6-메톡시-2-나프틸)-프로파노일 클로라이드 (나프록센 클로라이드)를 대신하여 상업적으로 이용가능하다.
또한, 니트로옥시 알킬 유도체는 상응하는 니트로옥시알킬 알콜에 비해서 더 안정하다. 따라서, 본 발명의 방법에 포함되는 두 가지 시약은 모두 선행기술에 기록된 것들에 비해서 훨씬 더 안정하다.
나프록센 염에 의한 치환반응에서 방법 선택성의 수반된 상실과 경쟁하는 것으로 예상되는 니트로옥시 알킬 유도체상의 두 가지 치환기, 즉 니트로옥시 및 이탈기의 존재로 인하여, 본 발명의 방법에서 관찰되는 높은 선택성은 예상되지 않았었다. 본 발명의 또 다른 이점은 출발 화합물이 안정하다는 것이다. 본 발명의 방법은 출발물질로서 종래 방법의 산 클로라이드 대신에, 나프록센 염, 특히 안정하고 상업적으로 이용가능한 산물인 나트륨염을 사용한다.
브로모나프록센 니트로옥시알킬에스테르는 그 자체가 생물학적으로 활성이고, 통상적인 방법에 의해서 상응하는 나프록센 에스테르로 전환될 수 있다.
본 발명은 i) 화학식 (B)의 화합물을 화학식 (C)의 화합물과 반응시키고, ii) 임의로 R'가 Br인 화학식 (A)의 화합물을 R'가 수소인 화학식 (A)의 화합물로 전환시키는 것을 포함하여, 화학식 (A)의 화합물을 제조하는 방법에 관한 것이다.

R-COOZ (B)

상기 식에서,
R은 다음과 같고

R'는 수소 원자 또는 Br이며,
R1-R12는 동일하거나 다르고, 독립적으로 수소, 또는 임의로 아릴에 의해서 치환된 직선형 또는 가지형 C1-C6 알킬이며,
m, n, o, q, r 및 s는 각각 독립적으로 0 내지 6의 정수이고, p는 0 또는 1이고,
X는 O, S, SO, SO2, NR13 또는 PR13이고, 여기서 R13는 수소 또는 C1-C6 알킬이거나, X는
- 사이클로알킬렌 환 내에 5 내지 7 개의 탄소 원자를 갖는 사이클로알킬렌 (여기서, 환은 최종적으로 곁사슬 T로 치환되고, T는 1 내지 10 개의 탄소 원자를 가진 직선형 또는 가지형 알킬, 바람직하게는 CH3이다);
- 임의로 하나 이상의 할로겐 원자, 1 내지 4 개의 탄소 원자를 함유하는 직선형 또는 가지형 알킬기, 또는 직선형 또는 가지형 C1-C3 퍼플루오로알킬로 치환된 아릴렌;
- 다음으로부터 선택된 5 또는 6 원 포화, 불포화, 또는 방향족 헤테로사이클릭 환으로 구성된 그룹으로부터 선택되며

(여기서, 결합이 정의되지 않은 위치를 갖는 경우에 이들은 환 내의 어떤 가능한 위치에나 존재할 수 있음을 의미한다);
Z는 수소, 또는 Li+, Na+, K+, Ca++, Mg++, 암모늄, 트리알킬암모늄, 테트라알킬암모늄 및 테트라알킬포스포늄으로부터 선택된 양이온이고;
삭제
삭제
삭제
삭제
삭제
Y는
삭제
- 할로겐 원자
- -BF4, -SbF6, FSO3-, ClO4-, RASO3- (여기서 RA는 하나 이상의 할로겐 원자에 의해서 임의로 치환된 직선형 또는 가지형 C1-C6 알킬, 또는 C1-C6 알킬아릴이다);
- RBCOO- (여기서 RB는 하나 이상의 할로겐 원자 또는 NO2기에 의해서 임의로 치환된 아릴, 직선형 또는 가지형 C1-C6 알킬, 또는 질소, 산소, 황 또는 인으로부터 선택된 동일하거나 다른 하나 이상의 이종원자들을 함유하는 C4-C10 헤테로아릴이다);
- 하나 이상의 할로겐 원자 또는 NO2기들로 임의로 치환된 아릴옥시, 또는 헤테로아릴옥시로부터 선택된다.
삭제
바람직하게는 본 발명은
치환기 R1-R12가 동일하거나 다르며, 독립적으로 수소 또는 직선형 또는 가지형 C1-C3 알킬이고,
m, n, o, p, q, r 및 s 들은 상기 정의된 것과 같으며,
X는 O, S 또는


가장 바람직하게는 본 발명은 R1-R4 및 R7-R10은 수소이고, m, n, q, r은 1이며, o 및 s는 0이고, p는 0 또는 1이고, X는 O 또는 S인 화학식 (A)의 화합물을 제조하는 방법에 관한 것이다.
화학식(C)의 화합물에서, 바람직하게는 Y는 Br, Cl, I, -BF4, ClO4-, -SbF6, FSO3-, CF3SO3-, C2F5SO3-, C3F7SO3-, C4F9SO3-, p-CH3C6H4SO3-로 구성된 그룹으로부터 선택된다.
화학식(B)의 화합물과 화학식(C)의 화합물 사이의 반응은 아세톤, 테트라하이드로퓨란, 디메틸포름아마이드, N-메틸피롤리돈, 설포란 및 아세토니트릴로부터 선택된 유기용매 중에서 수행될 수 있다.
대신으로, 반응은 오늄염, 예를 들어, 테트라알킬암모늄 및 테트라알킬포스포늄 염과 같은 상전이 촉매의 존재 하에, 톨루엔, 클로로벤젠, 니트로벤젠, tert-부틸-메틸에테르로부터 선택된 비양성자성 쌍극 용매 및 수용액을 포함하는 이상성계 내에서 수행될 수 있으며, 여기서 유기용액은 (C)를 함유하고 수용액은 (B)의 알칼리 금속염을 함유한다.
반응은 0℃ 내지 100℃ 사이의 온도에서 2-0.5의 (B)/(C) 몰비로 수행된다.
카르복실산 염은 별도로 제조될 수 있거나, "원위치에서", 예를 들어, 삼급 아민의 화학량적 양의 존재 하에서 (B) 및 (C) 사이 반응을 실행하거나, 상기 아민을 과량으로 사용하여 생성될 수 있다.
화학식 (C)의 화합물은 이하에 제시된 화학식 (D)의 화합물을, 예를 들어, 설포니트릭 혼합물 등으로부터 선택된 니트로화제로 니트로화함으로써 제조될 수 있다:

여기서 M는 OH이고,
Y, X, m, n, o, p, q, r, s 및 R1-R12는 상기 언급된 의미를 가진다.
대신으로, 화학식 (C)의 화합물은 화학식 (E)의 화합물을, 예를 들어, 알칼리 금속 질산염, 4급 암모늄 질산염, 4급 포스포늄염 및 AgNO3, Zn(NO)2ㆍ6H2O로부터 선택된 니트로화제와 반응시킴으로써 획득될 수 있다.:

여기서:
Y, X, m, n, o, p, q, r, s 및 R1-R12는 상기 언급된 의미를 가진다.
대신으로, 화학식 (C)의 화합물은 화학식 (F)의 화합물을, W가 OH인 경우에는 알킬 및 아릴 설포닐클로라이드, 트리플루오로메탄설폰산 무수물, 및 W가 할로겐인 경우에는 AgSbF6, AgBF4, AgClO4, CF3SO3Ag, AgSO3CH3, CH3C6H4SO3Ag로부터 선택된 화합물과 반응시킴으로써 획득될 수 있다:

여기서 W는 OH 또는 할로겐이다.
화합물 (D)의 니트로화는 유기용매, 일반적으로 아세톤, 테트라하이드로퓨란, 디메틸포름아미드, N-메틸피롤리돈, 설포란, 아세토니트릴, 메틸렌 클로라이드 등으로부터 선택된 용매 중에서, 전이 금속염으로부터 선택된 니트로화제, 또는 M이 OH일 때는 설포니트릭 혼합물과 같이 질산을 기본으로 하는 니트로화 시스템을 사용하여 수행된다.
(D)/니트로화제 몰비는 2 내지 0.5이고, 특히 1.5 내지 0.5이며, 니트로화는 0℃ 내지 100℃, 바람직하게는 15℃ 내지 80℃의 온도에서 수행된다.
반응 생성물 (C)는 분리될 수 있거나, 그의 용액을 (A)를 제공하기 위한 기질 (B)와의 반응에 그 자체로 사용할 수 있다.
화합물 (E)의 니트로화는 유기용매 중에서, 일반적으로 아세톤, 테트라하이드로퓨란, 디메틸포름아미드, N-메틸피롤리돈, 설포란, 아세토니트릴, 메틸렌 클로라이드 등으로부터 선택된 용매 중에서, 알칼리 금속 질산염, 오늄염 질산염, 예를 들어, 테트라알킬암모늄, 테트라알킬-포스포늄 또는 트리알킬암모늄 질산염 등과 같은 친핵성 니트로화제를 사용하여 수행될 수 있다.
반응은 0℃ 내지 100℃, 특히 15℃ 내지 80℃의 온도에서 20 내지 2, 바람직하게는 8 내지 1의 (E)/니트로화제 몰비로 수행된다.
반응 생성물 (C)는 분리될 수 있거나, 그의 용액을 (A)를 제공하기 위한 기질 (B)와의 반응에 그 자체로 사용할 수 있다.
(F)로부터 화합물 (C)를 획득하기 위한 반응은 일반적으로 아세톤, 테트라하이드로퓨란, 디메틸포름아미드, N-메틸피롤리돈, 설포란, 아세토니트릴, 메틸렌 클로라이드 등으로 구성된 그룹으로부터 선택된 유기용매 중에서 은, 아연, 수은의 염으로부터 선택된 전이 금속염을 사용하여 수행될 수 있거나, W가 OH인 경우에 반응은 메탄설포닐 클로라이드 등과 같은 산 클로라이드를 사용하거나, 트리플루오로메탄설폰산 무수물과 같은 적합한 무수물을 사용하여 수행될 수 있다.
반응은 -20℃ 내지 100℃, 특히 -20℃ 내지 60℃ 범위의 온도에서 2 내지 0.5, 바람직하게는 1.5 내지 0.5의 화합물 (F)/시약 몰비로 수행된다.
반응 생성물 (C)는 분리될 수 있거나, 그의 용액을 (A)를 제공하기 위한 기질 (B)와의 반응에 그 자체로 사용할 수 있다.
문헌 [Chem. Pharm. Bull., 1993, 41, 1040]에 따르는 4-니트로옥시부틸 브로마이드의 제조
질산 (90%, 0.8 mol)을 0℃로 유지되는 황산 (0.8 mol) 중에 교반 하에서 적가한 다음에, 혼합물을 0℃에서 80 분간 교반하였다. 그렇게 얻어지고 0℃에서 유지된 용액에, 교반 하에서 4-브로모부탄올을 적가하고 (0.4 mol), 혼합물을 같은 온도에서 추가로 210 분간 다시 교반하였다. 그 후, 용액을 물-얼음 혼합물에 붓고, 디에틸 에테르로 두 번 추출하였다.
에테르 추출물을 함께 합하고, 중탄산나트륨 포화 용액으로 세척하였다. 용매를 진공 하에서 증발시켜 황색 오일을 수득하였다 (수율 : 84.8%).
실시예 1
4-니트로옥시부틸 p-톨루엔설포네이트의 제조
질소 대기 하의 교반 하에서 0℃로 유지되는 피리딘 (50 ㎖) 중의 4-브로모부탄올 (5.0 g, 33 mmol)의 용액에 토실 클로라이드 (6.8 g, 36 mmol)를 첨가하였다. 생성된 용액을 추가로 20 분 동안 교반 하에 유지시킨 다음에 -18℃에서 밤새 저장하였다. 반응 혼합물은 물/얼음 혼합물 (약 400 ㎖)에 붓고, 에틸 에테르 (500 ㎖)로 추출하였다. 유기상을 6 N 염산 (500 ㎖)으로 세척하고, 황산나트륨 상에서 건조시켰다. 진공 하에서 용매를 증발시켜 오일상 잔류물 (7 g)을 제공하였다. 실온에서 질소 하에 교반하면서 유지되는 아세토니트릴 (50 ㎖) 중의 오일상 잔류물 (7 g, 23 mmol)의 용액에 질산은 (7.8 g, 46 mmol)을 첨가하였다. 대략 15 분 후에, 황색의 불용성 생성물의 형성이 관찰되었다. 불균질 혼합물을 밤새 교반 하에서 유지시켰다. 불용성 물질을 여과에 의해 제거하고, 용액을 물 (200 ㎖)에 붓고, 에틸 에테르(2×250 ㎖)로 추출하였다. 유기 추출물을 합하여 황산나트륨 상에서 건조시켰다. 진공 하에서 용매를 증발시켜 오일상 잔류물을 수득하였다 (5 g).
용출제로 헥산/에틸 에테르 혼합물을 사용하는 실리카겔 (100 g) 상에서의 잔류물의 크로마토그래피로 m.p. 38-40℃ 및 HPLC에 의해 결정된 것으로 98% 보다 큰 순도를 갖는 표제 생성물(3 g)을 제공하였다.
FTIR (고체 KBr, cm -1) : 2966, 1626, 1355, 1281, 1177, 1097, 959, 876, 815, 663, 553.
300 MHz 1H NMR (CDCl3) 델타 1.77 (m, 4H); 2.35 (s, 3H); 4.06 (m, 2H); 4.38 (m, 2H); 4.38 (m, 2H); 7.36 (2H); 7.7 (2H).
실시예 2
2-(S)-(6-메톡시-2-나프틸)-프로파노산, 4-(니트로옥시)부틸 에스테르의 합성
KHCO3 (5.22 g, 52 mmol)를 질소 하에서 DMF (200 ㎖) 중의 2-(S)-(6-메톡시-2-나프틸)-프로파노산 (나프록센) (키랄 HPLC에 의해 결정된 것으로 99 e.e.) (10.0 g, 43 mmol)의 용액에 첨가하였다.
불균질 혼합물을 50-60℃까지 가열하고, 이 온도에서 질소 하 및 자기 교반 하에서 90 분간 유지시켰다. 반응 혼합물을 실온으로 냉각시켰다. 요오드화 칼륨 (2.14 g, 12.9 mmol) 및 4-브로모부틸니트레이트 (14.48 g, 73 mmol)를 상기 혼합물에 첨가하고, 반응 혼합물을 질소 하에 실온에서 25 시간 동안 교반하였다. 물 (200 ㎖)을 반응 혼합물에 5 분에 걸쳐 적가하였다. 혼합물을 t-BuOMe (200 ㎖)로 추출하고, 유기상을 NaCl 10% 수용액 (2×200 ㎖)으로 세척하고, Na2SO4 상에서 건조시켰다. 진공 하에서 용매를 증발시켜 오일상 잔류물 (17.3 g)을 제공하였다. 잔류물을 실리카겔 상에서 크로마토그래피 (용출제 헥산/에틸 아세테이트)하여 황색 오일상 화합물로서 2-(S)-(6-메톡시-2-나프틸)-프로파노산, 4-(니트로옥시)부틸 에스테르를 제공하였다 (10.8 g, 73% 수율, HPLC에 의해 결정된 것으로 99%보다 큰 e.e.).
생성물은 표준 샘플과 비교하여 확인되었다.
실시예 3
2-(S)-(6-메톡시-2-나프틸)-프로파노산, 4-(니트로옥시)부틸 에스테르의 합성
KHCO3 (5.22 g, 52 mmol)를 질소 하에서 DMF (200 ㎖) 중의 2-(S)-(6-메톡시-2-나프틸)-프로파노산 (나프록센) (키랄 HPLC에 의해 결정된 것으로 99 e.e.) (10.0 g, 43 mmol)의 용액에 첨가하였다.
불균질 혼합물을 50-60℃까지 가열하고, 이 온도에서 질소 하 및 자기 교반 하에서 90 분 동안 유지시켰다. 반응 혼합물을 실온으로 냉각시켰다. 4-(니트로옥시)부틸-4-메틸벤젠설포네이트 (21.1 g, 73 mmol)를 상기 혼합물에 첨가하고, 반응 혼합물을 질소 하에 실온에서 25 시간 동안 교반하였다. 통상적인 수성 후처리에 이은 반응 조생성물의 실리카겔 상에서의 크로마토그래피 (용출제 헥산/에틸 아세테이트)에 의해 2-(S)-(6-메톡시-2-나프틸)-프로파노산, 4-(니트로옥시)부틸 에스테르를 제공하였다 (10.4 g, 70% 수율, HPLC에 의해 결정된 것으로 99 %보다 큰 e.e.).
실시예 4
2-(S)-(+)-(5-브로모-6-메톡시-2-나프틸)-프로파노산, 4-(니트로옥시)부틸 에스테르의 합성
DMF (120 ㎖) 중의 트리에틸아민 (5.25 g, 52 mmol), 2-(S)-(5-브로모-6-메톡시-2-나프틸)-프로파노산 (브로모-나프록센) (13.3 g, 43 mmol; e.e. 99%) 및 4-브로모부틸니트레이트 (43 mmol)의 혼합물을 질소 하에 25℃에서 2일간 교반하였다.
진공 하에서 DMF를 제거하고, 이어서 통상적인 수성 후처리에 의해 반응 조생성물을 제공하였다. 잔류물을 실리카겔 상에서 크로마토그래피 (용출제 헥산/에틸 아세테이트)하여 2-(S)-(5-브로모-6-메톡시-2-나프틸)-프로파노산, 4-(니트로옥시)부틸 에스테르를 수득하였다 (11.9 g, 65% 수율, HPLC에 의해 결정된 것으로 99 %보다 큰 e.e.).
생성물은 분광법에 의해 확인되었다.
Claims (9)
- 화학식 (B)의 화합물을 화학식 (C)의 화합물과 반응시키는 것을 포함하는, 화학식 (A)의 화합물을 제조하는 방법:(A)
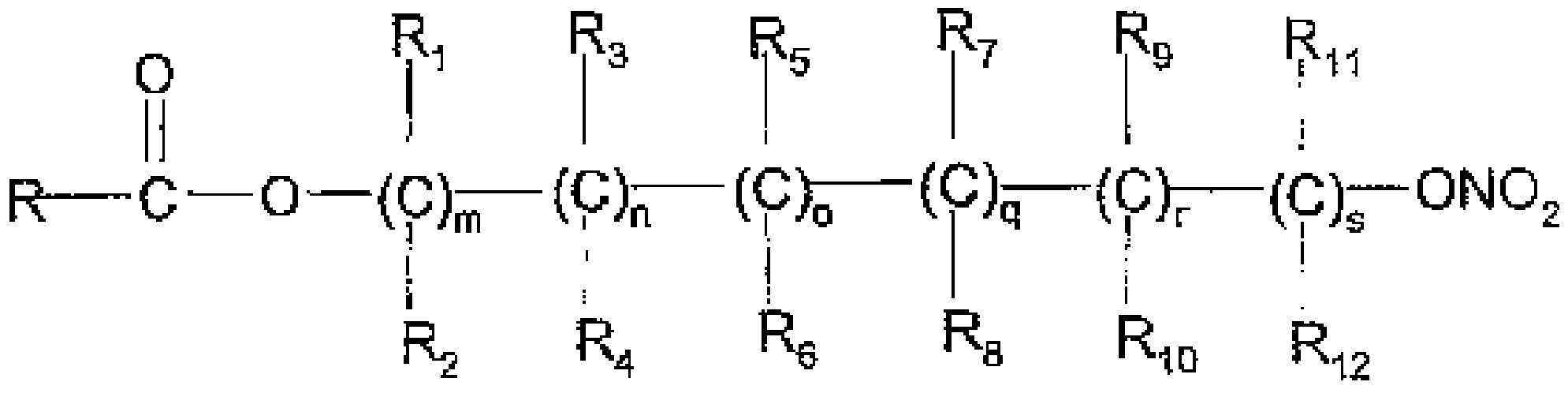 R-COOZ (B)(C)
R-COOZ (B)(C) 상기 식에서:R은 다음과 같고,
상기 식에서:R은 다음과 같고, R'는 수소 원자 또는 Br이며,R1-R12는 수소이며,m, n, o, q, r 및 s는 각각 독립적으로 0 내지 6의 정수이고, 여기에서 m, n, o, q, r 및 s의 합은 4이고,Z는 수소이며,Y는 Br 또는 CH3C6H4SO3-이다.
R'는 수소 원자 또는 Br이며,R1-R12는 수소이며,m, n, o, q, r 및 s는 각각 독립적으로 0 내지 6의 정수이고, 여기에서 m, n, o, q, r 및 s의 합은 4이고,Z는 수소이며,Y는 Br 또는 CH3C6H4SO3-이다. - 제1항에 있어서, 생성된 R'가 Br인 화학식 (A)의 화합물을 R'가 수소인 화학식 (A)의 화합물로 전환시키는 것을 더 포함하는, 화학식 (A)의 화합물을 제조하는 방법.
- 제1항에 있어서, 반응이 아세톤, 테트라하이드로퓨란, 디메틸포름아마이드, N-메틸피롤리돈, 설포란 및 아세토니트릴로부터 선택된 유기용매 중에서 수행되는 화학식 (A)의 화합물의 제조방법.
- 제1항에 있어서, 반응이 상전이 촉매의 존재 하에 톨루엔, 클로로벤젠, 니트로벤젠, tert-부틸메틸에테르로부터 선택된 비양성자성 쌍극 용매 및 수용액을 포함하는 이상성계 중에서 수행되며, 여기에서 유기 용액은 (C)를 함유하고, 수용액은 (B)의 알칼리금속염을 함유하는 화학식 (A)의 화합물의 제조방법.
- 제1항, 제3항 및 제4항 중 어느 한 항에 있어서, 반응이 0℃ 내지 100℃사이의 온도에서 수행되는 화학식 (A)의 화합물의 제조방법.
- 제1항, 제3항 및 제4항 중 어느 한 항에 있어서, 화학식 (B) 및 (C)의 화합물을 2-0.5의 (B)/(C) 몰비로 반응시키는 화학식 (A)의 화합물의 제조방법.
- 삭제
- 삭제
- 삭제
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ITMI2002A001861 | 2002-08-29 | ||
| IT001861A ITMI20021861A1 (it) | 2002-08-29 | 2002-08-29 | Procedimento di sintesi di nitrossialchilesteri di acidi carbossilici, intermedi impiegabili in detto procedimento e loro preparazione. |
| PCT/EP2003/008698 WO2004020384A1 (en) | 2002-08-29 | 2003-08-06 | Process for preparing nitrooxyderivatives of naproxen |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR20050057057A KR20050057057A (ko) | 2005-06-16 |
| KR101100064B1 true KR101100064B1 (ko) | 2011-12-29 |
Family
ID=31972201
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020057003509A KR101100064B1 (ko) | 2002-08-29 | 2003-08-06 | 나프록센의 니트로옥시유도체 제조방법 |
Country Status (20)
| Country | Link |
|---|---|
| US (3) | US7723382B2 (ko) |
| EP (2) | EP1532098B1 (ko) |
| JP (2) | JP2005536559A (ko) |
| KR (1) | KR101100064B1 (ko) |
| CN (1) | CN1326830C (ko) |
| AT (1) | ATE449060T1 (ko) |
| AU (2) | AU2003266966B2 (ko) |
| CA (1) | CA2497187C (ko) |
| CY (1) | CY1109769T1 (ko) |
| DE (1) | DE60330152D1 (ko) |
| DK (1) | DK1532098T3 (ko) |
| ES (1) | ES2335656T3 (ko) |
| IL (1) | IL166587A0 (ko) |
| IT (1) | ITMI20021861A1 (ko) |
| NZ (1) | NZ537993A (ko) |
| PT (1) | PT1532098E (ko) |
| RU (1) | RU2315035C2 (ko) |
| SI (1) | SI1532098T1 (ko) |
| WO (2) | WO2004020385A1 (ko) |
| ZA (1) | ZA200500890B (ko) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7163958B2 (en) | 2002-07-03 | 2007-01-16 | Nitromed Inc. | Nitrosated nonsteroidal antiinflammatory compounds, compositions and methods of use |
| ITMI20021861A1 (it) * | 2002-08-29 | 2004-02-29 | Nicox Sa | Procedimento di sintesi di nitrossialchilesteri di acidi carbossilici, intermedi impiegabili in detto procedimento e loro preparazione. |
| US7745496B2 (en) * | 2004-11-25 | 2010-06-29 | Nicox S.A. | Process for preparing halogenoalkylnitrates |
| WO2009149053A2 (en) * | 2008-06-02 | 2009-12-10 | Dr. Reddy's Laboratories Ltd. | Naproxcinod process and solid dispersion |
| ITRM20080325A1 (it) * | 2008-06-20 | 2009-12-21 | Nicox Sa | Metodo per purificare 4-(nitroosi)butil(2s)-2-(6-metossi-2-naftil) propanoato |
| US8062653B2 (en) | 2009-02-18 | 2011-11-22 | Bezwada Biomedical, Llc | Controlled release of nitric oxide and drugs from functionalized macromers and oligomers |
| CN101885684B (zh) * | 2010-07-01 | 2013-10-30 | 南京中医药大学 | 带有一氧化氮供体的芳香酸前体药物及其制备方法和其应用 |
| TWI417276B (zh) * | 2011-03-01 | 2013-12-01 | Everlight Chem Ind Corp | 2-(s)-(6-甲氧基-2-萘基)-丙酸之酸酐衍生物、其製法暨及其用途 |
| WO2014111957A1 (en) | 2013-01-21 | 2014-07-24 | Apparao Satyam | Nitric oxide releasing prodrugs of therapeutic agents |
| MX2021004581A (es) * | 2018-10-25 | 2021-06-15 | Dsm Ip Assets Bv | Composiciones acuosas que comprenden omega nitrooxi-1-alcanoles. |
| CN111333560B (zh) * | 2020-04-13 | 2024-01-23 | 合肥工业大学 | 一种制备螺环β-内酰胺的方法 |
| CN112321420A (zh) * | 2020-11-03 | 2021-02-05 | 浙江海翔川南药业有限公司 | 一种萘普生杂质及其制备方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995030641A1 (en) | 1994-05-10 | 1995-11-16 | Nicox S.A. | Nitro compounds and their compositions having anti-inflammatory, analgesic and anti-thrombotic acitivities |
| WO2001010814A1 (en) | 1999-08-04 | 2001-02-15 | Nicox S.A. | Process for the preparation of naproxene nitroxyalkylesters |
Family Cites Families (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5279359A (en) * | 1975-12-26 | 1977-07-04 | Hitachi Ltd | Cleaning material arrestor |
| US5057427A (en) * | 1988-04-07 | 1991-10-15 | Sepracor, Inc. | Method for resolution of stereoisomers |
| JPH05279359A (ja) * | 1992-02-06 | 1993-10-26 | Kawaken Fine Chem Co Ltd | アポビンカミン酸エステルの製造方法 |
| JPH05247015A (ja) * | 1992-03-05 | 1993-09-24 | Nippon Iyakuhin Kogyo Kk | 新規ピペラジン誘導体 |
| SI0722434T1 (en) * | 1993-10-06 | 1998-12-31 | Nicox S.A. | Nitric esters having anti-inflammatory and/or analgesic activity and process for their preparation |
| IT1276071B1 (it) * | 1995-10-31 | 1997-10-24 | Nicox Ltd | Compositi ad attivita' anti-infiammatoria |
| JP2000516235A (ja) * | 1996-08-16 | 2000-12-05 | ハンドフォース インベストメンツ リミテッド | 非潰瘍誘発性の鎮痛/抗炎症クロニキシン誘導体 |
| IT1288123B1 (it) * | 1996-09-04 | 1998-09-10 | Nicox Sa | Uso di nitroderivati per l'incontinenza urinaria |
| FR2757159B1 (fr) * | 1996-12-12 | 1999-12-17 | Hoechst Marion Roussel Inc | Nouveaux derives nitres analgesiques, anti-inflammatoires et anti-thrombotiques, leur procede de preparation, leur application comme medicaments |
| IT1311924B1 (it) * | 1999-04-13 | 2002-03-20 | Nicox Sa | Composti farmaceutici. |
| IT1314184B1 (it) * | 1999-08-12 | 2002-12-06 | Nicox Sa | Composizioni farmaceutiche per la terapia di condizioni di stressossidativo |
| US6429223B1 (en) * | 2000-06-23 | 2002-08-06 | Medinox, Inc. | Modified forms of pharmacologically active agents and uses therefor |
| ITMI20010985A1 (it) * | 2001-05-15 | 2002-11-15 | Nicox Sa | Farmaci per il morbo di alzheimer |
| ITMI20011308A1 (it) * | 2001-06-21 | 2002-12-21 | Nicox Sa | Farmaci per il dolore cronico |
| ITMI20011307A1 (it) * | 2001-06-21 | 2002-12-21 | Nicox Sa | Farmaci per l'epilessia |
| WO2003045896A1 (en) * | 2001-11-27 | 2003-06-05 | Astrazeneca Ab | New process |
| US7220749B2 (en) * | 2002-06-11 | 2007-05-22 | Nitromed, Inc. | Nitrosated and/or nitrosylated cyclooxygenase-2 selective inhibitors, compositions and methods of use |
| US7163958B2 (en) * | 2002-07-03 | 2007-01-16 | Nitromed Inc. | Nitrosated nonsteroidal antiinflammatory compounds, compositions and methods of use |
| ITMI20021861A1 (it) * | 2002-08-29 | 2004-02-29 | Nicox Sa | Procedimento di sintesi di nitrossialchilesteri di acidi carbossilici, intermedi impiegabili in detto procedimento e loro preparazione. |
| US7199947B2 (en) * | 2004-12-28 | 2007-04-03 | Motorola Inc. | Miniature digital camera lens |
-
2002
- 2002-08-29 IT IT001861A patent/ITMI20021861A1/it unknown
-
2003
- 2003-08-06 CA CA2497187A patent/CA2497187C/en not_active Expired - Fee Related
- 2003-08-06 JP JP2004532055A patent/JP2005536559A/ja not_active Ceased
- 2003-08-06 DK DK03747879.9T patent/DK1532098T3/da active
- 2003-08-06 NZ NZ537993A patent/NZ537993A/en not_active IP Right Cessation
- 2003-08-06 JP JP2004532054A patent/JP4528123B2/ja not_active Expired - Fee Related
- 2003-08-06 KR KR1020057003509A patent/KR101100064B1/ko not_active IP Right Cessation
- 2003-08-06 ES ES03747879T patent/ES2335656T3/es not_active Expired - Lifetime
- 2003-08-06 CN CNB038206056A patent/CN1326830C/zh not_active Expired - Fee Related
- 2003-08-06 AU AU2003266966A patent/AU2003266966B2/en not_active Ceased
- 2003-08-06 EP EP03747879A patent/EP1532098B1/en not_active Expired - Lifetime
- 2003-08-06 RU RU2005104419/04A patent/RU2315035C2/ru not_active IP Right Cessation
- 2003-08-06 WO PCT/EP2003/008700 patent/WO2004020385A1/en active Application Filing
- 2003-08-06 WO PCT/EP2003/008698 patent/WO2004020384A1/en active Application Filing
- 2003-08-06 AT AT03747879T patent/ATE449060T1/de active
- 2003-08-06 DE DE60330152T patent/DE60330152D1/de not_active Expired - Lifetime
- 2003-08-06 EP EP03790866A patent/EP1537070A1/en not_active Withdrawn
- 2003-08-06 SI SI200331743T patent/SI1532098T1/sl unknown
- 2003-08-06 PT PT03747879T patent/PT1532098E/pt unknown
- 2003-08-06 US US10/522,986 patent/US7723382B2/en not_active Expired - Fee Related
- 2003-08-06 US US10/523,722 patent/US7199258B2/en not_active Expired - Fee Related
- 2003-08-06 AU AU2003266261A patent/AU2003266261A1/en not_active Abandoned
-
2005
- 2005-01-31 IL IL16658705A patent/IL166587A0/xx not_active IP Right Cessation
- 2005-01-31 ZA ZA200500890A patent/ZA200500890B/xx unknown
-
2010
- 2010-02-10 CY CY20101100132T patent/CY1109769T1/el unknown
- 2010-03-30 US US12/750,381 patent/US20100217028A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995030641A1 (en) | 1994-05-10 | 1995-11-16 | Nicox S.A. | Nitro compounds and their compositions having anti-inflammatory, analgesic and anti-thrombotic acitivities |
| WO2001010814A1 (en) | 1999-08-04 | 2001-02-15 | Nicox S.A. | Process for the preparation of naproxene nitroxyalkylesters |
Non-Patent Citations (1)
| Title |
|---|
| J.Med.Chem., 1993, Vol.36, pp.815-819 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6013828A (en) | Processes and intermediates useful to make antifolates | |
| KR101100064B1 (ko) | 나프록센의 니트로옥시유도체 제조방법 | |
| US6262262B1 (en) | Processes and intermediates useful to make antifolates | |
| US6696591B1 (en) | Process for obtaining (nitroxymethyl)phenyl esters of salicylic acid derivatives | |
| EP0905128B1 (en) | Processes and intermediates useful to make antifolates | |
| JPS6087251A (ja) | 置換ベンズアミド誘導体の製造法 | |
| US4009172A (en) | 2,3,3A,6,7,7A-Hexahydro-thieno[3,2-b]pyridin-(4H)5-ones | |
| KR100514819B1 (ko) | 키랄 글리시딜 유도체의 제조방법 | |
| US6090168A (en) | Processes and intermediates useful to make antifolates | |
| EP0183797B1 (en) | 5-halo-4h-1,3-dioxin-4-one compounds, process for their preparation and their use in the preparation of alpha-halo-acetoacetic esters | |
| JP2006176531A (ja) | 光学活性ベンズイミダゾール誘導体の光学異性体 | |
| JP4318803B2 (ja) | ビス(パーフルオロアルキルスルホニル)メタンの製造方法 | |
| KR0179320B1 (ko) | (±)-2-(톨릴술피닐)-시클로펜타데칸-1-온 및 그의 제법 | |
| JP4686466B2 (ja) | 環状N−置換α−イミノカルボン酸の製法 | |
| HU203316B (en) | Process for producing mercapto-benzoic acid derivatives | |
| KR910003635B1 (ko) | 2-(2-나프틸옥시)프로피온아닐리드 유도체의 제조방법 | |
| KR100201582B1 (ko) | 제초성 피리미딜옥시벤조산 옥심에스테르 유도체의제조방법 | |
| JP2558491B2 (ja) | α,βー不飽和カルボニル化合物のオルガノチオメチル化方法 | |
| JPS62126162A (ja) | 2,3,5,6−テトラフルオロ−4−メルカプトベンゾニトリルおよびその製造方法 | |
| KR20050056157A (ko) | 광학적으로 순수한 펜에틸 아민 유도체의 제조방법 | |
| JPH0316339B2 (ko) | ||
| JPH0316338B2 (ko) | ||
| KR19990027763A (ko) | ο-(클로로메틸)벤조산 에스테르 유도체의 제조방법 | |
| JPH06145131A (ja) | シアノアリールオキシアミン類の製造方法 | |
| JPH03123769A (ja) | ビスメチルスルホノキシメチルエーテルの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A201 | Request for examination | ||
| E902 | Notification of reason for refusal | ||
| E90F | Notification of reason for final refusal | ||
| E701 | Decision to grant or registration of patent right | ||
| GRNT | Written decision to grant | ||
| LAPS | Lapse due to unpaid annual fee |