JP5240988B2 - イソブテン及び第3級ブタノールの製造方法 - Google Patents
イソブテン及び第3級ブタノールの製造方法 Download PDFInfo
- Publication number
- JP5240988B2 JP5240988B2 JP2007542611A JP2007542611A JP5240988B2 JP 5240988 B2 JP5240988 B2 JP 5240988B2 JP 2007542611 A JP2007542611 A JP 2007542611A JP 2007542611 A JP2007542611 A JP 2007542611A JP 5240988 B2 JP5240988 B2 JP 5240988B2
- Authority
- JP
- Japan
- Prior art keywords
- catalyst
- reaction
- isobutene
- tba
- producing
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 title claims description 188
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical compound CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 title claims description 182
- 238000000034 method Methods 0.000 title claims description 63
- 230000008569 process Effects 0.000 title claims description 11
- 239000003054 catalyst Substances 0.000 claims description 167
- 238000006243 chemical reaction Methods 0.000 claims description 116
- 238000004519 manufacturing process Methods 0.000 claims description 76
- 239000011148 porous material Substances 0.000 claims description 42
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 claims description 41
- 239000007864 aqueous solution Substances 0.000 claims description 31
- 239000002994 raw material Substances 0.000 claims description 31
- 239000000203 mixture Substances 0.000 claims description 30
- 229930195733 hydrocarbon Natural products 0.000 claims description 27
- 150000002430 hydrocarbons Chemical class 0.000 claims description 27
- 238000006297 dehydration reaction Methods 0.000 claims description 25
- 239000004215 Carbon black (E152) Substances 0.000 claims description 23
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 23
- 239000011964 heteropoly acid Substances 0.000 claims description 22
- VXNZUUAINFGPBY-UHFFFAOYSA-N 1-Butene Chemical compound CCC=C VXNZUUAINFGPBY-UHFFFAOYSA-N 0.000 claims description 18
- 230000000887 hydrating effect Effects 0.000 claims description 8
- 229910004298 SiO 2 Inorganic materials 0.000 claims description 7
- 239000012535 impurity Substances 0.000 description 54
- 230000000694 effects Effects 0.000 description 33
- 238000000926 separation method Methods 0.000 description 31
- 239000002253 acid Substances 0.000 description 29
- 238000004821 distillation Methods 0.000 description 26
- 239000012071 phase Substances 0.000 description 23
- 230000007423 decrease Effects 0.000 description 22
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 20
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 20
- 239000003463 adsorbent Substances 0.000 description 20
- 229910052799 carbon Inorganic materials 0.000 description 20
- 238000004064 recycling Methods 0.000 description 20
- 238000010924 continuous production Methods 0.000 description 19
- 239000000463 material Substances 0.000 description 19
- 239000000243 solution Substances 0.000 description 19
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 18
- 239000003456 ion exchange resin Substances 0.000 description 18
- 229920003303 ion-exchange polymer Polymers 0.000 description 18
- 238000000151 deposition Methods 0.000 description 16
- 239000000126 substance Substances 0.000 description 16
- 230000008021 deposition Effects 0.000 description 15
- 239000011734 sodium Substances 0.000 description 15
- 239000007800 oxidant agent Substances 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- 238000006703 hydration reaction Methods 0.000 description 12
- 239000007789 gas Substances 0.000 description 11
- XEEYBQQBJWHFJM-UHFFFAOYSA-N iron Substances [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 11
- 230000007797 corrosion Effects 0.000 description 10
- 238000005260 corrosion Methods 0.000 description 10
- 230000006866 deterioration Effects 0.000 description 10
- 230000001965 increasing effect Effects 0.000 description 10
- 230000002378 acidificating effect Effects 0.000 description 9
- 150000001336 alkenes Chemical class 0.000 description 9
- 230000000052 comparative effect Effects 0.000 description 9
- 238000002474 experimental method Methods 0.000 description 8
- 230000006870 function Effects 0.000 description 8
- 229910052742 iron Inorganic materials 0.000 description 8
- 229910021645 metal ion Inorganic materials 0.000 description 8
- 238000003860 storage Methods 0.000 description 8
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 7
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 7
- 238000004458 analytical method Methods 0.000 description 7
- 230000003197 catalytic effect Effects 0.000 description 7
- -1 iron ions Chemical class 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- 230000007774 longterm Effects 0.000 description 7
- PXHVJJICTQNCMI-UHFFFAOYSA-N nickel Substances [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 7
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 7
- 230000001590 oxidative effect Effects 0.000 description 7
- 239000000047 product Substances 0.000 description 7
- 239000011973 solid acid Substances 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 6
- 238000000465 moulding Methods 0.000 description 6
- 230000009257 reactivity Effects 0.000 description 6
- 238000001179 sorption measurement Methods 0.000 description 6
- 150000001450 anions Chemical class 0.000 description 5
- 239000003729 cation exchange resin Substances 0.000 description 5
- 239000011651 chromium Substances 0.000 description 5
- 229910052750 molybdenum Inorganic materials 0.000 description 5
- 229910052759 nickel Inorganic materials 0.000 description 5
- 239000003921 oil Substances 0.000 description 5
- DHRLEVQXOMLTIM-UHFFFAOYSA-N phosphoric acid;trioxomolybdenum Chemical compound O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.OP(O)(O)=O DHRLEVQXOMLTIM-UHFFFAOYSA-N 0.000 description 5
- 229910001220 stainless steel Inorganic materials 0.000 description 5
- 239000010935 stainless steel Substances 0.000 description 5
- FXNDIJDIPNCZQJ-UHFFFAOYSA-N 2,4,4-trimethylpent-1-ene Chemical compound CC(=C)CC(C)(C)C FXNDIJDIPNCZQJ-UHFFFAOYSA-N 0.000 description 4
- 239000003513 alkali Substances 0.000 description 4
- 150000001875 compounds Chemical class 0.000 description 4
- 230000018044 dehydration Effects 0.000 description 4
- 238000005187 foaming Methods 0.000 description 4
- 230000035484 reaction time Effects 0.000 description 4
- 239000002699 waste material Substances 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 3
- 230000009102 absorption Effects 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 238000000862 absorption spectrum Methods 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 238000009835 boiling Methods 0.000 description 3
- 239000006227 byproduct Substances 0.000 description 3
- 239000007795 chemical reaction product Substances 0.000 description 3
- 229910052804 chromium Inorganic materials 0.000 description 3
- 238000009826 distribution Methods 0.000 description 3
- 238000011156 evaluation Methods 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 229910052739 hydrogen Inorganic materials 0.000 description 3
- 229910052500 inorganic mineral Inorganic materials 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 239000011707 mineral Substances 0.000 description 3
- 239000011733 molybdenum Substances 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 150000002894 organic compounds Chemical class 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 239000011347 resin Substances 0.000 description 3
- 229920005989 resin Polymers 0.000 description 3
- CGFYHILWFSGVJS-UHFFFAOYSA-N silicic acid;trioxotungsten Chemical compound O[Si](O)(O)O.O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1.O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1.O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1.O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1 CGFYHILWFSGVJS-UHFFFAOYSA-N 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 3
- 239000010457 zeolite Substances 0.000 description 3
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 2
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 2
- KAKZBPTYRLMSJV-UHFFFAOYSA-N Butadiene Chemical compound C=CC=C KAKZBPTYRLMSJV-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical compound CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 description 2
- MCMNRKCIXSYSNV-UHFFFAOYSA-N ZrO2 Inorganic materials O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 description 2
- 239000003377 acid catalyst Substances 0.000 description 2
- 239000002518 antifoaming agent Substances 0.000 description 2
- 239000011324 bead Substances 0.000 description 2
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 238000005336 cracking Methods 0.000 description 2
- 238000000354 decomposition reaction Methods 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 238000005469 granulation Methods 0.000 description 2
- 230000003179 granulation Effects 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 230000036571 hydration Effects 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 238000009776 industrial production Methods 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 238000005342 ion exchange Methods 0.000 description 2
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 2
- 239000007791 liquid phase Substances 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000003472 neutralizing effect Effects 0.000 description 2
- 229910017604 nitric acid Inorganic materials 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- IYDGMDWEHDFVQI-UHFFFAOYSA-N phosphoric acid;trioxotungsten Chemical compound O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.OP(O)(O)=O IYDGMDWEHDFVQI-UHFFFAOYSA-N 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 238000011084 recovery Methods 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 238000007086 side reaction Methods 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 239000000057 synthetic resin Substances 0.000 description 2
- 229920003002 synthetic resin Polymers 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- CHRJZRDFSQHIFI-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;styrene Chemical compound C=CC1=CC=CC=C1.C=CC1=CC=CC=C1C=C CHRJZRDFSQHIFI-UHFFFAOYSA-N 0.000 description 1
- 229910016341 Al2O3 ZrO2 Inorganic materials 0.000 description 1
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 229910001369 Brass Inorganic materials 0.000 description 1
- 229910000975 Carbon steel Inorganic materials 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 229920001661 Chitosan Polymers 0.000 description 1
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 239000006087 Silane Coupling Agent Substances 0.000 description 1
- 229910002800 Si–O–Al Inorganic materials 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- 229910021536 Zeolite Inorganic materials 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 150000004703 alkoxides Chemical class 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 229910045601 alloy Inorganic materials 0.000 description 1
- 239000000956 alloy Substances 0.000 description 1
- 150000004645 aluminates Chemical class 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- 239000001099 ammonium carbonate Substances 0.000 description 1
- 235000012501 ammonium carbonate Nutrition 0.000 description 1
- 229910000963 austenitic stainless steel Inorganic materials 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 239000010951 brass Substances 0.000 description 1
- 238000001354 calcination Methods 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 239000010962 carbon steel Substances 0.000 description 1
- 238000003763 carbonization Methods 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 229940023913 cation exchange resins Drugs 0.000 description 1
- 239000003518 caustics Substances 0.000 description 1
- 239000013522 chelant Substances 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000002826 coolant Substances 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 229910052593 corundum Inorganic materials 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- HNPSIPDUKPIQMN-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical compound O=[Si]=O.O=[Al]O[Al]=O HNPSIPDUKPIQMN-UHFFFAOYSA-N 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 238000006056 electrooxidation reaction Methods 0.000 description 1
- 238000000921 elemental analysis Methods 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000001125 extrusion Methods 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 239000000295 fuel oil Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 229910000856 hastalloy Inorganic materials 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- QOSATHPSBFQAML-UHFFFAOYSA-N hydrogen peroxide;hydrate Chemical compound O.OO QOSATHPSBFQAML-UHFFFAOYSA-N 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 238000004898 kneading Methods 0.000 description 1
- MRELNEQAGSRDBK-UHFFFAOYSA-N lanthanum oxide Inorganic materials [O-2].[O-2].[O-2].[La+3].[La+3] MRELNEQAGSRDBK-UHFFFAOYSA-N 0.000 description 1
- 239000008204 material by function Substances 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 239000007769 metal material Substances 0.000 description 1
- MEFBJEMVZONFCJ-UHFFFAOYSA-N molybdate Chemical compound [O-][Mo]([O-])(=O)=O MEFBJEMVZONFCJ-UHFFFAOYSA-N 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 238000006864 oxidative decomposition reaction Methods 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 238000005192 partition Methods 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000005096 rolling process Methods 0.000 description 1
- 238000005204 segregation Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 150000003440 styrenes Chemical class 0.000 description 1
- 125000001174 sulfone group Chemical group 0.000 description 1
- 125000000542 sulfonic acid group Chemical group 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 235000011149 sulphuric acid Nutrition 0.000 description 1
- 229920005613 synthetic organic polymer Polymers 0.000 description 1
- 238000005979 thermal decomposition reaction Methods 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 238000006276 transfer reaction Methods 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229910001845 yogo sapphire Inorganic materials 0.000 description 1
Images



Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J19/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J19/18—Stationary reactors having moving elements inside
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C1/00—Preparation of hydrocarbons from one or more compounds, none of them being a hydrocarbon
- C07C1/20—Preparation of hydrocarbons from one or more compounds, none of them being a hydrocarbon starting from organic compounds containing only oxygen atoms as heteroatoms
- C07C1/24—Preparation of hydrocarbons from one or more compounds, none of them being a hydrocarbon starting from organic compounds containing only oxygen atoms as heteroatoms by elimination of water
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D3/00—Distillation or related exchange processes in which liquids are contacted with gaseous media, e.g. stripping
- B01D3/009—Distillation or related exchange processes in which liquids are contacted with gaseous media, e.g. stripping in combination with chemical reactions
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J19/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J19/0053—Details of the reactor
- B01J19/0066—Stirrers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/02—Boron or aluminium; Oxides or hydroxides thereof
- B01J21/04—Alumina
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/02—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the alkali- or alkaline earth metals or beryllium
- B01J23/04—Alkali metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J27/00—Catalysts comprising the elements or compounds of halogens, sulfur, selenium, tellurium, phosphorus or nitrogen; Catalysts comprising carbon compounds
- B01J27/14—Phosphorus; Compounds thereof
- B01J27/186—Phosphorus; Compounds thereof with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J27/188—Phosphorus; Compounds thereof with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium with chromium, molybdenum, tungsten or polonium
-
- B01J35/60—
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C11/00—Aliphatic unsaturated hydrocarbons
- C07C11/02—Alkenes
- C07C11/08—Alkenes with four carbon atoms
- C07C11/09—Isobutene
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/03—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by addition of hydroxy groups to unsaturated carbon-to-carbon bonds, e.g. with the aid of H2O2
- C07C29/04—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by addition of hydroxy groups to unsaturated carbon-to-carbon bonds, e.g. with the aid of H2O2 by hydration of carbon-to-carbon double bonds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2219/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J2219/18—Details relating to the spatial orientation of the reactor
- B01J2219/185—Details relating to the spatial orientation of the reactor vertical
-
- B01J35/615—
-
- B01J35/633—
-
- B01J35/66—
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2521/00—Catalysts comprising the elements, oxides or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium or hafnium
- C07C2521/02—Boron or aluminium; Oxides or hydroxides thereof
- C07C2521/04—Alumina
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/02—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the alkali- or alkaline earth metals or beryllium
- C07C2523/04—Alkali metals
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Description
〔1〕 第3級ブタノールを原料として、イソブテンを製造する方法であって、
Na含有量がNa2Oに換算して0.1〜0.6重量%の範囲であり、Si含有量がSiO2に換算して0.4重量%以下であり、且つ、比表面積が200〜600m2/gであるアルミナ触媒を用いて、200〜450℃の反応温度にて、気相下、脱水反応を行う工程を含むことを特徴とするイソブテンの製造方法、
〔2〕 前記アルミナ触媒が、触媒中の全細孔容積が0.1〜0.5cc/gの範囲にあり、且つ、細孔半径70Å以上の細孔が有する細孔容積が全細孔容積の60%以上を占める範囲にあることを特徴とする〔1〕に記載のイソブテンの製造方法。
〔3〕 前記第3級ブタノールが、イソブテン及びn−ブテンを含む炭化水素混合物から、水及びヘテロポリ酸触媒を含む触媒水溶液を用いて、選択的にイソブテンを水和して、第3級ブタノールを製造する方法によって製造されることを特徴とする〔1〕又は〔2〕に記載のイソブテンの製造方法、
を提供する。
〔4〕 イソブテン及びn−ブテンを含む炭化水素混合物から、水及び触媒を含む触媒水溶液を用いて、選択的にイソブテンを水和して、第3級ブタノールを連続的に製造する方法であって、
前記炭化水素混合物、前記触媒及び第3級ブタノールのうち少なくとも1種を循環するリサイクル系を用いて、前記少なくとも1種をリサイクルさせる工程と、
該リサイクル系から蓄積性不純物の一部を除去する工程と、
を含むことを特徴とする第3級ブタノールの連続的製造方法、
〔5〕 前記蓄積性不純物を除去する工程において、多孔質吸着剤及び/又はイオン交換樹脂を用いることを特徴とする〔4〕記載の製造方法、
〔6〕 前記多孔質吸着剤の細孔半径が、0.5〜500nmの範囲であることを特徴とする〔4〕又は〔5〕に記載の製造方法、
〔7〕 前記触媒が、Hammettの酸度関数Hoで、−5.6≧Hoで表される強酸又は強酸塩であることを特徴とする〔4〕ないし〔6〕のうち何れか一項に記載の製造方法、
〔8〕 前記触媒が、ヘテロポリ酸であることを特徴とする〔4〕ないし〔7〕のうち何れか一項に記載の製造方法、
〔9〕 前記ヘテロポリ酸が、リンモリブデン酸、リンモリブドバナジン酸、リンタングステン酸、リンタングストバナジン酸、ケイタングステン酸及びそれらの塩からなる群から選ばれ、それぞれ単独で、又は2種類以上のヘテロポリ酸を混合して用いることを特徴とする〔8〕に記載の製造方法、
〔10〕 前記イオン交換樹脂が、陽イオン交換樹脂であることを特徴とする〔5〕に記載の製造方法、
〔11〕 前記触媒水溶液及び前記炭化水素混合物が、向流となるように供給しながら反応させる工程を含むことを特徴とする〔4〕ないし〔10〕のうち何れか一項に記載の製造方法、
〔12〕 酸化剤を、連続的又は不連続的に添加する工程をさらに含むことを特徴とする〔4〕ないし〔11〕のうち何れか一項に記載の製造方法、
を提供する。
〔13〕 少なくとも1つの攪拌槽型反応器と、前記攪拌槽型反応器に接続した蒸留塔と、前記攪拌槽型反応器及び/又は前記蒸留塔と接続し、不純物を除去する分離器と、前記分離器と、前記攪拌槽型反応器及び/又は前記蒸留塔とを連通させるボトム液配管と、を備える、第3級ブタノールの連続製造装置、
〔14〕 前記攪拌槽型反応器が、触媒分離器を備えることを特徴とする〔13〕に記載の第3級ブタノールの連続製造装置、
〔15〕 前記分離器が、前記触媒分離器と連通していることを特徴とする〔14〕に記載の第3級ブタノールの連続製造装置、
〔16〕 前記分離器が、吸着剤カラム及び/又はイオン交換樹脂カラムを備えることを特徴とする〔13〕ないし〔15〕に記載の第3級ブタノールの連続製造装置、
〔17〕 前記攪拌槽型反応器が、触媒水溶液と原料炭化水素混合物が向流となるように供給しながら反応させる向流型反応器であることを特徴とする〔13〕ないし〔16〕のうちいずれか一項に記載の第3級ブタノールの連続製造装置、
〔18〕 前記連続製造装置において、触媒が接触する部位の材質が、少なくともCrを17〜21%、Niを8〜14%含み、Cが0.10%以下であるステンレス鋼から構成されることを特徴とする〔13〕ないし〔17〕のうち何れか一項に記載の第3級ブタノールの連続製造装置、
〔19〕 前記触媒が、ヘテロポリ酸であることを特徴とする〔18〕に記載の第3級ブタノールの連続製造装置、
を提供する。
(触媒のリサイクル系において)
まず、触媒であるヘテロポリ酸は、その陰イオンの直径が約1nmであり、一般的な鉱酸である、硝酸、塩酸、硫酸に比べ大きな陰イオンである。このことは、他の鉱酸に比べプロトンとしての酸強度を高める結果となる。イソブテンの水和反応において同じプロトン濃度において硝酸などの一般的な鉱酸に比べ、高い反応活性を示す。また、大きな陰イオンとイソブテンとの相互作用がn−ブテンより強いと推定され、高い選択水和反応特性を引き出すと考えられている。さらに、例えば、100℃程度の反応温度領域では安定であり、熱劣化のあるイオン交換樹脂に比して、寿命の観点からも有利である。したがって、触媒としてはイオン交換樹脂に比べ優れている。しかしながら、近年、年間オーダーの長時間に渡り連続的に反応を継続していると、緩やかではあるが反応活性が低下していくという問題が発生した。本発明者らはその原因や劣化を詳細に検討した結果、本発明のTBAの製造方法では、以下のような解決策を見出した。
また、原料であるイソブテン及びn−ブテンを含む炭化水素混合物におけるブテン類は、ビニル基という反応性の官能基を有することから何らかの重合反応等によって高沸点の蓄積性有機化合物の生成が推定される。
蒸留塔での分離性能悪化現象では、蓄積性不純物の濃度が高くなると悪くなるなどの関係も実験的にも検証できた。イソブテンの水和反応生成物であるTBAは、触媒水溶液中にある範囲濃度で含まれると、特開2000−44502号に示されているように、蒸留塔において起こる発泡現象を消泡する機能を有することが見出されている。アルコール類は発泡現象を抑制する消泡剤として用いられるため、本反応での目的生成物であるTBAも消泡剤として機能すると考えられる。
以下、実施例および比較例によって本発明を具体的に説明するが、これらは本発明の範囲を何等限定するものではない。なお、本発明のイソブテン製造方法の実施例において採用した測定方法は以下の通りである。
(1)アルミナ触媒中のNa含有量、Si含有量の測定
標準試料を用いた蛍光X線分析法にて、リガク製RIX1000を用いて分析を実施した。
(2)アルミナ触媒の比表面積の測定
窒素吸着法にて、島津製Gemini2360を用いて比表面積を測定した。
(3)アルミナ触媒の細孔容積、細孔分布の測定
水銀圧入法にて、QUANTACHROME製Pore Master GT33を用いて細孔容積、細孔分布を測定した。
Na含有量がNa2Oに換算して0.25重量%、Si含有量がSiO2に換算して0.06重量%、比表面積が402m2/g、全細孔容積が0.21cc/g、細孔半径70Å以上の細孔が有する細孔容積が全細孔容積の78%であり、直径2〜5mmの球状に成型されたアルミナ触媒(触媒A)を、外部に電気炉を有する内径10mm、長さ30cmのSUS316製の縦型管状反応管に10ml充填し、電気炉の温度を360℃に設定した。イソブテン及びn−ブテンを含む炭化水素混合物から、水及びヘテロポリ酸触媒を含む触媒水溶液を用いて、選択的にイソブテンを水和して製造されたTBAの85重量%水溶液を100℃に予熱して20ml/Hr(LHSV=2.0hr−1)で反応器塔頂部からフィードし、反応管内の圧力が4Kg/cm2Gになるようにして反応を行った。反応管下部より排出される気液混合物を液相部と気相部に分離した。実験開始後、一定時間経過したところの気相部の反応生成物をガスクロマトグラフィーで分析し、反応成績を求めた。5時間経過したところTBA転化率95%、イソブテン選択率99%、収率94%であった。800時間経過したところTBA転化率94%、イソブテン選択率99%、収率93%であった。
物性値が異なる各種アルミナ触媒(触媒B、C、D、E、F、G)を触媒として、実施例1と全く同一の反応条件下で性能評価を行った。その結果を表1に示す。主な副生物はジイソブテンとsec−ブタノールであった。
表1からイソブテンの収率、特に800時間後の収率の高い触媒は、触媒A、Bであることが分かる。
実施例1で用いた反応管に触媒Aを10ml充填し、電気炉の温度を250℃に設定した。実施例1で用いたTBAの85重量%水溶液を100℃に予熱して5ml/Hr(LHSV=0.5hr−1)で反応器塔頂部からフィードし、反応管内の圧力が4Kg/cm2Gになるようにして反応を行った。結果を表2に示した。
実施例1で用いた反応管に触媒Aを10ml充填し、電気炉の温度を400℃に設定した。実施例1で用いたTBAの85重量%水溶液を100℃に予熱して10ml/Hr(LHSV=1.0hr−1)で反応器塔頂部からフィードし、反応管内の圧力が4Kg/cm2Gになるようにして反応を行った。結果を表2に示した。
実施例2、3の電気炉の温度を150℃および500℃に設定し、その他の条件は全て同様にして反応を行った。結果を表2に示した。
外部にオイル加熱槽を有する内径25.4mm、長さ280cmのSUS316製の縦型管状反応管に触媒Aを600ml充填し、電気炉の温度を350℃に設定した。実施例1で用いたTBAの85重量%水溶液を100℃に予熱して600ml/Hr(LHSV=1.5hr−1)でフィードし、反応管内の圧力を4Kg/cm2Gに設定して反応を行った。上記条件における触媒寿命テストを実施し、通算反応時間が1800時間と9000時間における反応結果を表3に示す。
原料であるTBA水溶液を変更した以外は実施例1と全く同一の触媒および反応条件下で性能評価を行った。比較例8ではプロピレンオキサイドを製造する際に副生するTBAを用い、比較例9ではイソブテン及びn−ブテンを含む炭化水素混合物からイオン交換樹脂にて酢酸存在下にて水和されたTBAを用い、比較例10ではイソブテン及びn−ブテンを含む炭化水素混合物からイオン交換樹脂にて水和されたTBAを用いた。比較のため、実施例1で用いたTBAの組成と反応の結果を併せて、通算反応時間が800時間における結果を表4に示した。
〔参考実験1〕蓄積性不純物(界面活性能の原因物質)の解析:
約50重量%のリンモリブデン酸を含み約1年間連続反応に用いた触媒水溶液1000gとビーズ状吸着剤(三菱化学製、セパビーズSP207:修飾スチレン系、比表面積:630m2/g、最高頻度細孔直径:21nm)250gを約60℃で3時間かき混ぜながら接触させた。その後、吸着剤をロ別し、洗浄液が無色透明になるまで、蒸留水で水洗した。吸着剤を風乾したのち、1000mlのアセトン溶液に浸漬し約1時間放置するとアセトン溶液は茶褐色に着色した。茶褐色に着色したアセトン溶液を、ロータリーエバポレーターを用いて減圧下、60℃でアセトンを除去すると茶褐色の固体状物質が得られた。茶褐色の固体状物質の元素分析を行った結果、リン、モリブデン、炭素、窒素および水素から構成される物質であった。また、その物質の赤外吸収スペクトルを測定し、スペクトル図を得た。図1は、本発明で得られた蓄積性不純物の赤外吸収スペクトルを示す。この赤外吸収スメクトルには、ケギン型ヘテロポリ酸に特徴的な1062cm-1、960cm-1、880cm-1、808cm-1の吸収、2970cm-1のC−H伸縮振動等、有機物化合物由来の吸収が見られ、アルキル基とアニオンを持つも化合物が蓄積性不純物の一つとして確認できた。
新しい試薬から調製したリンモリブデン酸50重量%の水溶液50mlとイソブテンを43重量%含有するラフネ−ト50mlを耐圧のガラス容器に採取し、激しく3分間振り混ぜた後に静止して界面が形成される時間を測定した。5回繰り返した結果、2相に分離し界面形成までの時間は平均6秒であった。
新しい試薬から調製したリンモリブデン酸50重量%の水溶液50mlに参考実験1で得た蓄積性不純物を0.5g加え、イソブテンを43重量%含有するラフネ−ト50mlを耐圧のガラス容器に採取し、激しく3分間振リ混ぜた後に静止して界面が形成される時間を測定した。5回繰り返した結果、2相に分離し界面形成までの時間は平均120秒であり、界面が不明確であった。
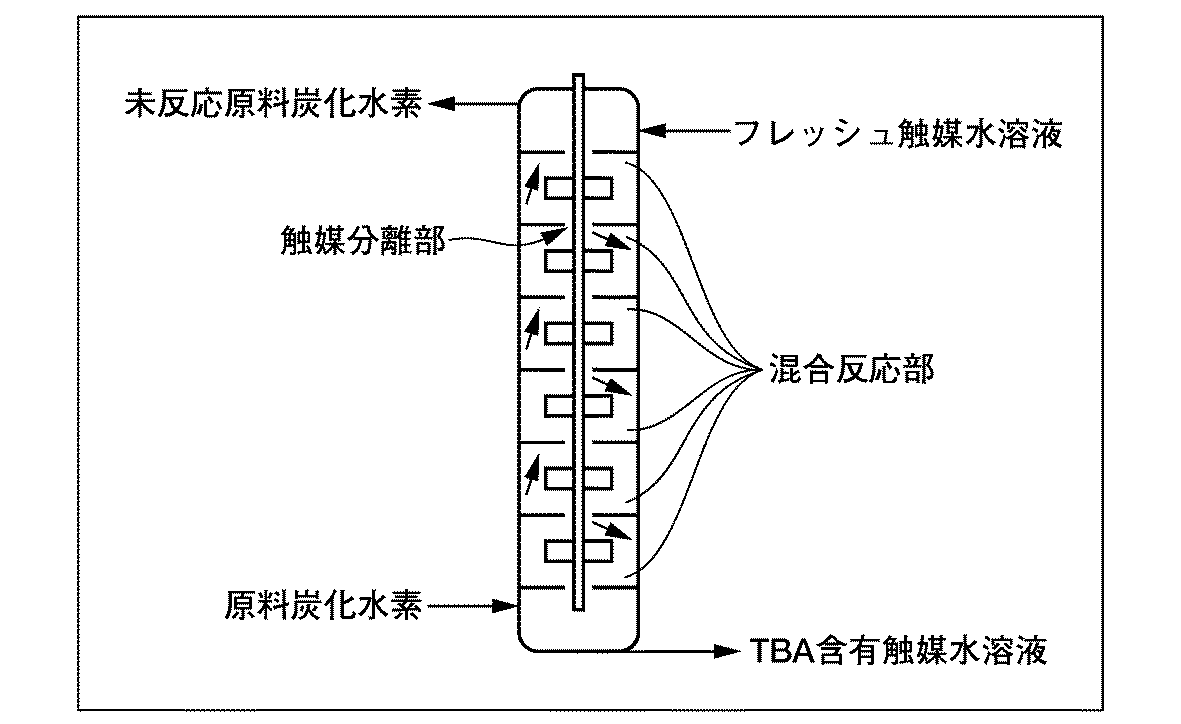
図2は、本発明によるTBAの製造方法を実施するための一つの態様としての連続製造装置を例示する。本発明における連続製造装置は、攪拌槽型反応器及び配管等部位全てがSUS304(Cr18.0%、Ni8.0%C:0.08%以下)で構成され、反応容積が10Lの攪拌式反応槽と、2Lの容積の触媒分離部とから構成される攪拌槽型反応器を3つ備える。なお、図2では、3つの攪拌槽型反応器を例示するが、本発明の連続製造装置は攪拌型反応器を3つ備えるものに限定されず、少なくとも一つの攪拌槽型反応器を備える。各反応器に設置した触媒分離器で分離した各炭化水素相(オイル相)と触媒水溶液(水相)を分離し、原料である炭化水素と触媒水溶液が向流となるフローで反応させ、最終反応槽の触媒分離器で分離した生成TBAを含む触媒水溶液を理論段数20段の充填塔式蒸留塔に供給し、蒸留塔の上部からTBAを分離し、ボトム温度が70℃になるように減圧に管理し、ボトムから触媒水溶液を回収する操作を行った。また、ボトム液配管ラインには、三菱化学社製、酸型陽イオン交換樹脂(SK1B)を500ミリリットル充填したカラム及び吸着剤として三菱化学社製合成樹脂系(セパビーズ:P207、最高頻度細孔直径:21nm、充填量500ミリリットル)を設置し、ボトム液量の約1/10の液量を循環させ、不純物金属イオン、蓄積性不純物を連続的に吸着除去しながら触媒水溶液を攪拌槽型反応器へ供給した(図2参照)。触媒として50重量%の新しいリンモリブデン酸(H3PMo12O40)溶液(Hammett酸度関数Ho−5.6≧Ho)を調製し、図2に例示する全ての反応器へ50L/hで供給し、炭化水素原料として、43重量%のイソブテンを含むラフネート1を20L/hで向流となるように供給した。反応温度は、各反応器の攪拌式反応槽が70℃になるように管理した。さらに反応開始から、48時間経過ごとに、蒸留塔ボトム配管に10wt%の過酸化水素水100mlを1時間かけて供給する間欠操作を行ない、約2000時間の連続運転を行った。反応初期のイソブテンの転化率95%、水を除いたTBAの選択率は99%を示し、2000時間経過時点でもほぼ同等の反応成績を維持することができた。また、触媒分離器における触媒分離状態の悪化による未反応炭化水素への触媒流出、及び蒸留塔の差圧上昇を伴う運転性の悪化も全く発生しなかった。このときの触媒水溶液50mlと新しい吸着材セパビーズ30mlと接触させ蓄積不純物量を吸着させた。その後参考例1と同様な操作で吸着材を水洗して分離後、アセトンで吸着剤から蓄積性不純物を溶解させ、アセトンを蒸発させ残渣として不純物の量を求めたところ、触媒水溶液中の蓄積性不純物量は2.1wt%であった。
実施例6のボトム液配管ラインの酸型陽イオン交換樹脂カラム及び吸着剤カラムを設置しない以外は実施例6と同様の操作で連続反応を行った。反応開始から100時間は転化率95%を維持したが、約150時間経過ごろから蒸留塔の差圧の上昇がみられ、さらに未反応炭化水素に若干の濁りが発生した。未反応炭化水素を蒸発させ残渣を蛍光X線で分析したところモリブデンが検出された。触媒分離部の分離性能低下及び蒸留塔の分離悪化により反応継続が困難になったためを停止した。反応停止後、触媒水溶液を抜き出し、参考実験2の触媒分離性能をテストした。5回繰り返した結果、2相に分離した界面形成までの時間は平均160秒であり、界面も不明確であった。このときの触媒水溶液50mlと新しい吸着材セパビーズ30mlと接触させ蓄積不純物量を吸着させた。その後参考例と同様な操作で蓄積性不純物の量を求めたところ、触媒水溶液中の蓄積性不純物量は11.5wt%であった。
過酸化水素による酸化処理を行わなかった以外は、実施例6と同様の操作で反応を行った。反応初期約10時間目のイソブテンの転化率は95%であった。反応運転は安定に維持され長時間運転が可能であった。しかし、約500時間以降緩やかに活性の低下が認められ、約1000時間目のイソブテン転化率は94.2%、2000時間経過時点では93.6%であり、長期反応では活性低下が認められた。
攪拌器の材質をSUS304よりも一般的には、さらに耐酸腐食性に優れるとされるハステロイB(Ni67%、Mo28%、Fe5%)に変更し、イオン交換樹脂を設置しない以外は実施例6と同様操作で行った。反応初期約10時間目のイソブテンの転化率は95%であった。反応運転は安定に維持され長時間運転が可能であった。しかし、約300時間以降緩やかに活性の低下が認められ、約1000時間目のイソブテン転化率は94.5%、2000時間経過時点では94.1%であり、長期反応では活性低下が認められた。反応停止後触媒水溶液を分析した結果、鉄イオンが800ppmと約700ppmの増加し、一般的な耐腐食性から予想した結果となることを確認した。
実施例6の反応器3基を図3に示したような構造の混合反応部8室、各混合反応部の容積15Lの向流型反応器1基に変更した以外は同様で反応を行った。イソブテンの転化率は97.5%と高い値が得られることから、向流に触媒と原料を供給する方法及び向流型反応器の高い反応性が確認できた。
触媒をリンタングステン酸(H3PW12O40)に変更し、プロトン濃度を同じに設定した以外は、実施例6と同様の操作で500時間の反応を行った。反応10時間後のイソブテンの転化率は95.2%であり500時間経過も転化率は95.2%、運転状の問題も全くなかった。
触媒をリンバナドモリブデン酸(H4PVMo11O40)に変更し、プロトン濃度を同じに設定した以外は、実施例6と同様の操作で500時間の反応を行った。反応10時間後のイソブテンの転化率は94.7%であり500時間経過も転化率は94.8%、運転状の問題も全くなかった。
触媒をケイタングステン酸(H4SiW12O40)に変更し、プロトン濃度を同じに設定した以外は、実施例6と同様の操作で500時間の反応を行った。反応10時間後のイソブテンの転化率は95.3%であり500時間経過も転化率は95.2%、運転状の問題も全くなかった。
攪拌器の材質をSUS304からモリブデンをさらに少量含むSUS316(Cr:18.0%、Ni:10.0%、C:0.08%以下、Mo:2.0%)に変更し、イオン交換樹脂を設置しない以外は実施例6と同様操作で行った。反応初期約10時間目のイソブテンの転化率は95.2%であった。反応運転は安定に維持され長時間運転が可能で、約300時間以降も変化は見られず約1000時間目のイソブテン転化率は95.1%、2000時間経過時点では95.2%であり、長期反応でも活性低下は認められなかった。反応停止後触媒水溶液を分析した結果、鉄イオンは98ppmと初期から大きな変化は認められなかった。
Claims (3)
- 第3級ブタノールを原料として、イソブテンを製造する方法であって、
Na含有量がNa2Oに換算して0.1〜0.6重量%の範囲であり、Si含有量がSiO2に換算して0.4重量%以下であり、且つ、比表面積が200〜600m2/gであるアルミナ触媒を用いて、200〜450℃の反応温度にて、気相下、脱水反応を行う工程、
を含むことを特徴とするイソブテンの製造方法。 - 前記アルミナ触媒が、触媒中の全細孔容積が0.1〜0.5cc/gの範囲にあり、且つ、細孔半径70Å以上の細孔が有する細孔容積が全細孔容積の60%以上を占める範囲にあることを特徴とする請求項1に記載のイソブテンの製造方法。
- 前記第3級ブタノールが、イソブテン及びn−ブテンを含む炭化水素混合物から、水及びヘテロポリ酸触媒を含む触媒水溶液を用いて、選択的にイソブテンを水和して、第3級ブタノールを製造する方法によって製造されることを特徴とする請求項1又は2に記載のイソブテンの製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007542611A JP5240988B2 (ja) | 2005-11-01 | 2006-10-25 | イソブテン及び第3級ブタノールの製造方法 |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005317904 | 2005-11-01 | ||
| JP2005317904 | 2005-11-01 | ||
| JP2005373789 | 2005-12-27 | ||
| JP2005373789 | 2005-12-27 | ||
| JP2007542611A JP5240988B2 (ja) | 2005-11-01 | 2006-10-25 | イソブテン及び第3級ブタノールの製造方法 |
| PCT/JP2006/321215 WO2007052505A1 (ja) | 2005-11-01 | 2006-10-25 | イソブテン及び第3級ブタノールの製造方法 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012197535A Division JP5582621B2 (ja) | 2005-11-01 | 2012-09-07 | イソブテン及び第3級ブタノールの製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2007052505A1 JPWO2007052505A1 (ja) | 2009-04-30 |
| JP5240988B2 true JP5240988B2 (ja) | 2013-07-17 |
Family
ID=38005657
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007542611A Active JP5240988B2 (ja) | 2005-11-01 | 2006-10-25 | イソブテン及び第3級ブタノールの製造方法 |
| JP2012197535A Active JP5582621B2 (ja) | 2005-11-01 | 2012-09-07 | イソブテン及び第3級ブタノールの製造方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012197535A Active JP5582621B2 (ja) | 2005-11-01 | 2012-09-07 | イソブテン及び第3級ブタノールの製造方法 |
Country Status (8)
| Country | Link |
|---|---|
| US (3) | US8637716B2 (ja) |
| EP (2) | EP1944282B1 (ja) |
| JP (2) | JP5240988B2 (ja) |
| KR (1) | KR100969616B1 (ja) |
| CN (2) | CN101300211B (ja) |
| SG (1) | SG158189A1 (ja) |
| TW (3) | TWI483922B (ja) |
| WO (1) | WO2007052505A1 (ja) |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5017173B2 (ja) * | 2007-05-16 | 2012-09-05 | 三菱レイヨン株式会社 | α−アシロキシアクリル酸および/またはそのエステルを製造するための触媒およびα−アシロキシアクリル酸および/またはそのエステルの製造方法 |
| JP5410052B2 (ja) * | 2008-09-03 | 2014-02-05 | 東ソー株式会社 | 硫酸鉄の生成抑制方法 |
| JP5461442B2 (ja) * | 2009-02-03 | 2014-04-02 | 株式会社日本触媒 | フィルターの再生方法 |
| DE102009026585A1 (de) * | 2009-05-29 | 2010-12-02 | Evonik Oxeno Gmbh | Herstellung von 3-Methylbut-1-en durch Dehydratisierung von 3-Methylbutan-1-ol |
| US9522854B2 (en) | 2009-07-29 | 2016-12-20 | The United States Of America As Represented By The Secretary Of The Navy | Process and apparatus for the selective dimerization of terpenes and poly-alpha-olefins with a single-stage reactor and a single-stage fractionation system |
| US8785702B2 (en) | 2009-07-29 | 2014-07-22 | The United States Of America As Represented By The Secretary Of The Navy | Turbine and diesel fuels and methods for making the same |
| US9242226B2 (en) * | 2009-07-29 | 2016-01-26 | The Government Of The United States Of America As Represented By The Secretary Of The Navy | Process for the dehydration of aqueous bio-derived terminal alcohols to terminal alkenes |
| US9649626B2 (en) | 2009-07-29 | 2017-05-16 | The United States Of America As Represented By The Secretary Of The Navy | Process for the dehydration of aqueous bio-derived terminal alcohols to terminal alkenes |
| US8912373B2 (en) | 2009-07-29 | 2014-12-16 | The United States Of America As Represented By The Secretary Of The Navy | Process for the dehydration of aqueous bio-derived terminal alcohols to terminal alkenes |
| US8969636B2 (en) | 2009-07-29 | 2015-03-03 | The United States Of America As Represented By The Secretary Of The Navy | Homogeneous metallocene ziegler-natta catalysts for the oligomerization of olefins in aliphatic-hydrocarbon solvents |
| RU2012122587A (ru) * | 2009-11-03 | 2013-12-10 | Басф Се | Способ обращения с водными растворами метансульфокислоты |
| EP2578559A1 (en) | 2011-10-07 | 2013-04-10 | Metabolic Explorer | Process for producing isobutene from isobutylamine |
| WO2014092849A1 (en) * | 2012-12-14 | 2014-06-19 | Washington State University | Process and catalyst for conversion of acetic acid to isobutene |
| WO2014204509A1 (en) * | 2013-06-18 | 2014-12-24 | Washington State University | Process and catalyst for conversion of acetic acid to isobutene and propylene |
| SG11201607927WA (en) | 2014-05-07 | 2016-11-29 | Mitsubishi Rayon Co | Method for producing isobutylene from isobutanol |
| US10792642B2 (en) | 2014-12-03 | 2020-10-06 | China Petroleum & Chemical Corporation | Catalyst and preparation method thereof, and method for preparing isobutylene by applying the same |
| KR20170035621A (ko) * | 2015-09-23 | 2017-03-31 | 롯데케미칼 주식회사 | 터트-부탄올로부터 이소부틸렌의 제조 방법 |
| CN105732326A (zh) * | 2016-03-18 | 2016-07-06 | 安徽三联泵业股份有限公司 | 一种利用树脂处理正丁醇生产废液的方法 |
| KR102080381B1 (ko) * | 2018-01-09 | 2020-02-21 | 한화토탈 주식회사 | 알루미늄과 코발트를 중심원소로 하는 헤테로폴리산 촉매, 그 제조방법 및 상기 촉매를 이용하여 n-부텐의 수화반응으로부터 2-부탄올을 제조하는 방법 |
| JP7020623B2 (ja) * | 2018-03-27 | 2022-02-16 | 株式会社クラレ | イソブテンの製造方法 |
| FR3084267B1 (fr) * | 2018-07-25 | 2021-10-08 | Axens | Alumine a acidite et structure de porosite optimales |
| CN109096026B (zh) * | 2018-08-28 | 2021-07-06 | 宁波昊德化学工业股份有限公司 | 一种异丁烯的生产方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5562031A (en) * | 1978-10-31 | 1980-05-10 | Asahi Chem Ind Co Ltd | Preparation of tertiary butanol from mixed butylene |
| JPS6341431A (ja) * | 1986-08-06 | 1988-02-22 | Mitsubishi Rayon Co Ltd | イソブチレンの製造法 |
| JPH04300840A (ja) * | 1991-03-29 | 1992-10-23 | Mitsui Petrochem Ind Ltd | 低級オレフィン類の製造方法 |
| JPH08143493A (ja) * | 1994-11-15 | 1996-06-04 | Mitsui Toatsu Chem Inc | オレフィンの接触水和方法 |
| JP2000044497A (ja) * | 1998-07-30 | 2000-02-15 | Asahi Chem Ind Co Ltd | イソブチレンを分離したオレフィン混合物の処理方法 |
Family Cites Families (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3665048A (en) * | 1970-01-14 | 1972-05-23 | Atlantic Richfield Co | Process to produce high purity isobutylene |
| JPS5527045B1 (ja) * | 1971-03-31 | 1980-07-17 | ||
| JPS5124894Y2 (ja) | 1971-06-16 | 1976-06-25 | ||
| JPS4841918A (ja) * | 1971-10-04 | 1973-06-19 | ||
| JPS54135710A (en) * | 1978-04-10 | 1979-10-22 | Nippon Oil Co Ltd | Preparation of isobutene by decomposition of tertiary butanol |
| JPS5839134B2 (ja) | 1978-06-08 | 1983-08-27 | 旭化成株式会社 | 混合ブチレンよりタ−シヤリ−ブタノ−ルの製造方法 |
| DE2922545C2 (de) * | 1978-06-08 | 1982-06-03 | Asahi Kasei Kogyo K.K., Osaka | Verfahren zur Herstellung von tert.-Butanol |
| JPS557213A (en) | 1978-06-30 | 1980-01-19 | Asahi Chem Ind Co Ltd | Separation of isobutylene from mixed butylene |
| JPS5551028A (en) | 1978-10-11 | 1980-04-14 | Asahi Chem Ind Co Ltd | Preparation of tertiary butanol from mixed butylene |
| JPS6051451B2 (ja) | 1978-11-06 | 1985-11-14 | 三菱レイヨン株式会社 | 第3級ブチルアルコ−ルの製造法 |
| JPS562855A (en) | 1979-06-19 | 1981-01-13 | Satake Eng Co Ltd | Device for humidifying cereal grain |
| JPS5610124A (en) | 1979-07-05 | 1981-02-02 | Sumitomo Chem Co Ltd | Preparation of tert-butyl alcohol |
| JPS56166134A (en) * | 1980-05-27 | 1981-12-21 | Mitsui Toatsu Chem Inc | Preparation of alcohol |
| FR2492809A1 (fr) * | 1980-10-29 | 1982-04-30 | Inst Francais Du Petrole | Procede d'obtention d'une olefine par decomposition de l'ether correspondant |
| JPS5839806A (ja) | 1981-09-04 | 1983-03-08 | Hitachi Ltd | 油圧サ−ボ弁 |
| DE3151446A1 (de) | 1981-12-24 | 1983-07-14 | Chemische Werke Hüls AG, 4370 Marl | Verfahren zur herstellung von hochreinem isobuten durch dehydratisierung von tertiaer-butanol |
| DE3628008C1 (ja) * | 1986-08-19 | 1987-11-05 | Deutsche Texaco Ag, 2000 Hamburg, De | |
| JP2585594B2 (ja) * | 1987-05-11 | 1997-02-26 | ペガサスミシン製造 株式会社 | ミシンの糸調子装置 |
| ATE141579T1 (de) * | 1991-02-04 | 1996-09-15 | Mitsui Petrochemical Ind | Verfahren zur herstellung von propylene |
| US5191143A (en) * | 1992-01-10 | 1993-03-02 | Texaco Chemical Company | Preparation of isobutylene |
| EP0579153B1 (en) * | 1992-07-13 | 1998-04-08 | Tosoh Corporation | Process for producing tertiary alcohols |
| US5716895A (en) * | 1993-04-01 | 1998-02-10 | Nippon Kayaku Kabushiki Kaisha | Process for regeneration of catalysts |
| US5756604A (en) * | 1995-08-31 | 1998-05-26 | Hodogaya Chemical Co., Ltd. | Process for producing polyether, and process for recycling and reusing herteropolyacid |
| CN1067972C (zh) | 1996-06-27 | 2001-07-04 | 中国石化齐鲁石油化工公司 | 一种由混合碳四或抽余碳四中异丁烯制叔丁醇的方法 |
| JPH11193255A (ja) | 1997-12-26 | 1999-07-21 | Mitsubishi Rayon Co Ltd | 第3級ブチルアルコールの製造方法 |
| JP2000034242A (ja) * | 1998-07-16 | 2000-02-02 | Asahi Chem Ind Co Ltd | 第3級ブタノールの製造方法 |
| JP4174103B2 (ja) | 1998-07-30 | 2008-10-29 | キヤノン株式会社 | インクジェット記録装置およびインクジェット記録方法 |
| JP4197771B2 (ja) | 1998-07-30 | 2008-12-17 | 旭化成ケミカルズ株式会社 | オレフィン混合物より第3級ブチルアルコールの回収方法 |
| CN1221506C (zh) | 2002-12-31 | 2005-10-05 | 中国石化集团齐鲁石油化工公司 | 一种制备叔丁醇的方法 |
| DE10338581A1 (de) * | 2003-08-22 | 2005-03-17 | Oxeno Olefinchemie Gmbh | Verfahren zur Erzeugung von tert.-Butanol |
| DE102004030943B4 (de) * | 2004-06-26 | 2013-10-02 | Evonik Oxeno Gmbh | Verfahren zur Herstellung von tert.-Butanol aus Isobuten-haltigen Kohlenwasserstoffgemischen |
-
2006
- 2006-10-25 WO PCT/JP2006/321215 patent/WO2007052505A1/ja active Application Filing
- 2006-10-25 JP JP2007542611A patent/JP5240988B2/ja active Active
- 2006-10-25 US US12/091,016 patent/US8637716B2/en active Active
- 2006-10-25 SG SG200908714-9A patent/SG158189A1/en unknown
- 2006-10-25 CN CN2006800408194A patent/CN101300211B/zh active Active
- 2006-10-25 KR KR20087009030A patent/KR100969616B1/ko active IP Right Grant
- 2006-10-25 EP EP06822192.8A patent/EP1944282B1/en active Active
- 2006-10-25 EP EP10006919.4A patent/EP2266939B1/en active Active
- 2006-10-25 CN CN201110301139.6A patent/CN102516030B/zh active Active
- 2006-11-01 TW TW098119272A patent/TWI483922B/zh not_active IP Right Cessation
- 2006-11-01 TW TW095140458A patent/TW200728249A/zh not_active IP Right Cessation
- 2006-11-01 TW TW103144662A patent/TWI537240B/zh not_active IP Right Cessation
-
2012
- 2012-09-07 JP JP2012197535A patent/JP5582621B2/ja active Active
-
2013
- 2013-12-20 US US14/136,404 patent/US9145342B2/en active Active
-
2015
- 2015-08-26 US US14/836,669 patent/US9919283B2/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5562031A (en) * | 1978-10-31 | 1980-05-10 | Asahi Chem Ind Co Ltd | Preparation of tertiary butanol from mixed butylene |
| JPS6341431A (ja) * | 1986-08-06 | 1988-02-22 | Mitsubishi Rayon Co Ltd | イソブチレンの製造法 |
| JPH04300840A (ja) * | 1991-03-29 | 1992-10-23 | Mitsui Petrochem Ind Ltd | 低級オレフィン類の製造方法 |
| JPH08143493A (ja) * | 1994-11-15 | 1996-06-04 | Mitsui Toatsu Chem Inc | オレフィンの接触水和方法 |
| JP2000044497A (ja) * | 1998-07-30 | 2000-02-15 | Asahi Chem Ind Co Ltd | イソブチレンを分離したオレフィン混合物の処理方法 |
Non-Patent Citations (1)
| Title |
|---|
| JPN6012035874; SWECKER, J. L. and DATYE, A. K.: JOURNAL OF CATALYSIS 121, 1990, p.196-201 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102516030A (zh) | 2012-06-27 |
| SG158189A1 (en) | 2010-01-29 |
| TW200940486A (en) | 2009-10-01 |
| KR20080046735A (ko) | 2008-05-27 |
| CN101300211A (zh) | 2008-11-05 |
| EP2266939A1 (en) | 2010-12-29 |
| JP5582621B2 (ja) | 2014-09-03 |
| US20090124835A1 (en) | 2009-05-14 |
| EP1944282A1 (en) | 2008-07-16 |
| EP2266939B1 (en) | 2016-09-28 |
| TW201514139A (zh) | 2015-04-16 |
| US20150360198A1 (en) | 2015-12-17 |
| CN102516030B (zh) | 2016-05-04 |
| US20140107387A1 (en) | 2014-04-17 |
| US9919283B2 (en) | 2018-03-20 |
| US8637716B2 (en) | 2014-01-28 |
| JP2013006864A (ja) | 2013-01-10 |
| JPWO2007052505A1 (ja) | 2009-04-30 |
| TWI483922B (zh) | 2015-05-11 |
| TWI322797B (ja) | 2010-04-01 |
| US9145342B2 (en) | 2015-09-29 |
| TW200728249A (en) | 2007-08-01 |
| EP1944282A4 (en) | 2009-05-06 |
| CN101300211B (zh) | 2012-10-17 |
| EP1944282B1 (en) | 2016-06-29 |
| TWI537240B (zh) | 2016-06-11 |
| KR100969616B1 (ko) | 2010-07-14 |
| WO2007052505A1 (ja) | 2007-05-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5240988B2 (ja) | イソブテン及び第3級ブタノールの製造方法 | |
| BRPI0607726B1 (pt) | Processo para fabricar acroleína | |
| JP2009500383A (ja) | 混合アルコールの脱水のための反応性蒸留 | |
| CN102614916A (zh) | 用于异丁烷与丁烯烷基化的强酸性氟化树脂催化剂的制备方法 | |
| JP5890849B2 (ja) | グリコールエーテルを利用した高純度のイソブテンの製造方法 | |
| WO2013008279A1 (en) | Process for preparing catalyst used in production of acrolein and/or acrylic acid and process for preparing acrolein and/or acrylic acid by dehydration reaction of glycerin | |
| WO2010071011A1 (ja) | 酢酸エステルの製造方法 | |
| JP5336239B2 (ja) | オレフィン二量体の製造方法、オレフィン二量体 | |
| US5183947A (en) | One step synthesis of methyl t-butyl ether from t-butanol using fluorophosphoric acid-modified clay catalysts | |
| RU2786385C1 (ru) | Способ очистки гликолей от примесей | |
| JPH069472A (ja) | ハロゲン化酸で改質された粘土触媒を使用するアルキル第三級アルキルエーテルの合成方法 | |
| JPH07258133A (ja) | 粗アセトンからのイソプロピル−tert−ブチルエーテルの製造方法 | |
| JP5290937B2 (ja) | アルコールの製造方法 | |
| JPH07316084A (ja) | アルキルスルホン酸で改質された酸化物触媒を用いるアルキル第三級アルキルエーテルの合成方法 | |
| JP2004148177A (ja) | 低級脂肪酸エステル製造用触媒、及び低級脂肪酸エステルの製造法 | |
| RU2400467C1 (ru) | Способ получения диизопропилового эфира | |
| GB2623213A (en) | Method for producing alcohol | |
| Phalak | Reaction engineering studies in ion exchange resin catalyzed esterification reactions | |
| CN109232156A (zh) | 一种制备异戊烯的方法 | |
| Ahmadpour et al. | LIQUID PHASE ALKYLATION OF BENZENE WITH 1-DECENE OVER SILICA SUPPORTED H14 [NaP5W30O110] AS A GREEN AND REUSABLE CATALYST | |
| KR20140070197A (ko) | 크롬산화물이 고분산된 중형 기공성 실리카 촉매의 제조방법 및 이로써 제조된 촉매를 이용한 프로필렌의 제조방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20090811 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120710 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120907 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120921 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20130327 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20130401 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20160412 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 Ref document number: 5240988 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313111 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| S531 | Written request for registration of change of domicile |
Free format text: JAPANESE INTERMEDIATE CODE: R313531 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |