-
Die
vorliegende Erfindung fällt
allgemein in das Gebiet der Molekularbiologie und Nucleinsäurechemie.
Genauer betrifft es Verfahren zur Amplifikation langer Nucleinsäureketten
durch die Polymerasekettenreaktion.
-
Die
Polymerasekettenreaktion (PCR), ein wirkungsvolles Werkzeug zur
Amplifikation von Nucleinsäuresequenzen,
ist in den U.S. Patenten Nr. 4,683,202; 4,683,195; 4,800,195 und
4,965,188 offenbart. In ihrer einfachsten Form ist die PCR ein in
vitro-Verfahren für
die enzymatische Synthese spezifischer DNA-Sequenzen, indem man
zwei Oligonucleotid-Primer benutzt, die an komplementäre Stränge hybridisieren
und die Regionen von Interesse in der Ziel-DNA flankieren. Eine
wiederholte Reihe von Reaktionsschritten, wie Denaturierung der
Matrize, Anlagerung der Primer sowie Verlängerung der angelagerten Primer
durch eine DNA-Polymerase, führt
schließlich
zu einer geometrischen Akkumulation eines spezifischen Fragmentes,
dessen Enden durch die 5'-Enden
der Primer definiert sind. Mit Hilfe der PCR lässt sich die selektive Anreicherung
einer spezifischen DNA-Sequenz um den Faktor 109 erreichen.
Das PCR-Verfahren ist auch in Saiki et al., 1985, Science 230:1350–1354, beschrieben.
-
Die
PCR findet breite Anwendung in der Molekularbiologie, molekularen
Evolutionsforschung, medizinischen Genetik, Bevölkerungsgenetik, forensischen
Biologie, der Kartierung des Genoms und bei Sequenzierungsprojekten.
Das heutige PCR-Verfahren findet jedoch seine Grenzen in der Größe der DNA-Region,
die zuverlässig
amplifiziert werden kann.
-
Versuche,
die Längenbegrenzung
der PCR aufzuheben, sind beschrieben bei Glukhov et al., 1991, Molek.
Biol. 25:1602–1610;
Kainz et al., 1992, Anal. Biochem. 202:46–49; Ohler und Rose, 1992,
PCR Meth. Applic. 2:51–59;
Ponce und Micol, 1992, Nucl. Acids Res. 20:623 und Rychlik et al.,
1990, Nucl. Acids Res. 18:6409–6412.
Obwohl Amplifikationen von 5–15
kb-Sequenzen erreicht wurden, ist die Ausbeute längerer Produkte doch sehr gering.
-
PCR-Verfahren
zur Amplifikation langer Nucleinsäuresquenzen würden die
Genom-Kartierung
und Sequenzierung ebenso erleichtern wie das molekulare Clonen durch
die Amplifikation von längerem
eingefügten
Material mit niedriger Kopienzahl. Ebenso würde dadurch eine Zusammenlagerung
von größeren rekombinanten
Konstrukten in der PCR-basierenden
Mutagenese möglich.
Es bleibt ein Bedarf für
Verfahren, die eine PCR-Amplifikation
von Zielen von mindestens 25 kb mit hoher Ausbeute ermöglichen.
-
Die
vorliegende Erfindung liefert verbesserte Verfahren und Reagenzien
für die
PCR-Amplifikation
langer DNA-Zielsequenzen.
-
Ein
Aspekt der Erfindung betrifft die Kombinationen thermostabiler DNA-Polymerasen,
die bei den Verfahren der vorliegenden Erfindung nützlich sind.
Die Kombinationen bestehen primär
aus der DNA-Polymerase von Thermus thermophilus, einer hochaktiven,
thermostabilen DNA-Polymerase, die keine 3'-zu 5'-Exonuclease-Aktivität zeigt, und zum anderen aus
einer DNA-Polymerse von Thermococcus litoralis, Pyrococcus-Art GB-D
oder Thermotoga maritima, alles thermostabile DNA-Polymerasen, die
eine 3'-zu 5'-Exonuclease-Aktivität zeigen.
-
Ein
weiterer Aspekt der Erfindung betrifft einen Puffer, der für die Amplifikation
langer Ziele nützlich
ist.
-
Ein
weiterer Aspekt der vorliegenden Erfindung betrifft die PCR-Amplifikationen
unter Verwendung spezifischer Kombinationen der vorstehend beschriebenen
thermostabilen Enzyme. Die Reaktionsbedingungen sind so festgelegt,
dass eine Amplifikation von Nucleinsäurezielsequenzen bis zu einer
Länge von
42 kb ermöglicht
wird.
-
Ein
weiterer Aspekt der Erfindung betrifft Kits, umfassend Reagenzien,
die zur Durchführung
der Verfahren der vorliegenden Erfindung nützlich sind. Diese Kits umfassen
eine Kombination thermostabiler DNA-Polymerasen wie vorstehend beschrieben,
und gegebenefalls weitere Reagenzien zur Amplifikation, die für die Verfahren
der vorliegenden Erfindung nützlich
sind.
-
Zum
besseren Verständnis
der Erfindung sind im folgenden einige Ausdrücke definiert.
-
Der
hier benutzte Ausdruck „Reaktionsgemisch
für die
Amplifikation" bezieht
sich auf eine wässrige Lösung, die
die verschiedenen Amplifikations-Reagenzien enthält, die zur Amplifikation einer
Ziel-Nucleinsäure
verwendet werden. Die Reagenzien beinhalten Primer, Enzyme, wässrige Puffer,
Salze, Ziel-Nucleinsäure und
Desoxynucleosidtriphosphate (konventionelle und unkonventionelle).
Je nach Kontext kann das Gemisch ein vollständiges oder ein unvollständiges Reaktionsgemisch
sein.
-
Die
hier benutzten Ausdrücke „Nucleinsäure" und „Oligonucleotid" beziehen sich auf
Primer, Sonden und Oligomerfragmente, die nachgewiesen werden sollen.
Sie gehören
zur Gattung der Polydesoxyribonucleotide (2-Desoxy-D-ribose enthaltend)
und der Polyribonucleotide (D-Ribose
enthaltend). Sie beziehen sich aber auch auf jede andere Art von
Polynucleotiden, die ein N-Glycosid einer Purin- oder Pyrimidinbase
oder eine modifizierte Purin- oder Pyrimidinbase ist. Mit den Ausdrücken „Nucleinsäure" und „Oligonucleotid" ist keine Längenunterscheidung
verbunden, die Ausdrücke
werden synonym verwandt, und beziehen sich nur auf die Primärstruktur
des Moleküls.
Daher umfassen diese Ausdrücke
sowohl doppelsträngige
als auch einzelsträngige
DNA, und ebenso doppelsträngige
und einzelsträngige
RNA.
-
Weil
Mononucleotide sich derartig zu Polynucleotiden verbinden, dass
die 5'-Phosphatgruppe
des Pentoseringes des einen Mononucleotids über eine Phophodiester-Bindung
in eine Richtung an den 3'-Sauerstoff
eines benachbarten Ringes bindet, bezeichnet man das Ende eines
Oligonucleotides als „5'-Ende", wenn die 5'-Phosphatgruppe nicht
mit dem 3'-Sauerstoff
des Pentoseringes eines Mononucleotids verbunden ist, und als 3'-Ende, wenn der 3'-Sauerstoff nicht mit einem 5'-Phosphat des Pentoseringes
eines folgenden Mononucleotids verbunden ist.
-
Die
exakte Größe eines
Oligonucleotids hängt
von vielen Faktoren ab, nicht zuletzt auch von der Funktion oder
der jeweiligen Verwendung des Oligonucleotids. Oligonucleotide können nach
verschiedenen geeigneten Verfahren hergestellt werden, z.B. durch
Clonierung und Restriktion geeigneter Sequenzen und durch direkte
chemische Synthese, z.B. durch das Phosphotriester-Verfahren von
Narang et al., 1979, Meth. Enzymol. 68:90–99, das Phosphodiester-Verfahren
von Brown et al., 1979, Meth. Enzymol. 68:109–151, das Diethylphospho ramidit-Verfahren
von Beaucage et al., 1981, Tetahedron Lett. 22:1859–1862; und
das Festphasenverfahren des U.S. Patents Nr. 4,458,066. Eine Übersicht
der Syntheseverfahren findet sich bei Goodchild, 1990, Bioconjugate
Chemistry 1 (3):165–187.
-
Der
hier benutzte Ausdruck „Hybridisierung" bezieht sich auf
die Bildung eines Doppelstranges aus zwei einzelsträngigen Nucleinsäuren durch
Paarung jeweiliger komplementärer
Basen. Hybridisierung kann sowohl zwischen zueinander komplementären Nucleinsäuresträngen stattfinden
als auch zwischen solchen, die kleinere, nicht zueinander passende
Regionen enthalten. Die Stabilität
eines Nucleinsäuredoppelstranges wird
durch die Schmelztemperatur oder „TM" angegeben. TM ist die Temperatur (unter definierter Ionenstärke und
definiertem pH-Wert), bei der 50% der Basenpaare dissoziiert sind.
Fachleute auf dem Gebiet der Nucleinsäuretechnologie können die
Doppelstrang-Stabilität
empirisch ermitteln, indem sie eine Reihe von Variablen berücksichtigen,
wie etwa die Länge
der Oligonucleotide, die Basenzusammensetzung und Sequenz der Oligonucleotide,
die Ionenstärke
und die Häufigkeit
nicht passender Basenpaare.
-
Bedingungen,
unter denen nur vollständig
komplementäre
Nucleinsäurestränge hybridisieren,
werden als „strenge
(stringente) Hybridisierungsbedingungen" bezeichnet. Strenge Hybridisierungsbedingungen
sind der Fachwelt gut bekannt (vgl. z.B. Sambrook et al., 1985,
Molecular Cloning – A
Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor,
New York). Allgemein werden strenge Bedingungen so ausgewählt, dass
sie bei einer Temperatur von etwa 5°C unter der Tm für die jeweilige
Sequenz bei definierter Ionenstärke
und definiertem pH-Wert liegen. Typische strenge Bedingungen sind
gegeben, wenn die Salzkonzentration mindestens etwa 0,2 molar ist,
der pH-Wert 7, und die Temperatur mindestens etwa 60°C beträgt. Eine Lockerung
der strengen Hybridisierungsbedingungen erlaubt die Tolerierung
von Fehlpaarungen innerhalb der Sequenz; der Grad der nicht verpaarten
Sequenzen kann durch eine entsprechende Einstellung der Hybridisierungsbedingungen
kontrolliert werden.
-
Zwei
einzelsträngige
Nucleinsäuren,
die bis auf kleinere Regionen, in denen keine Übereinstimmung vorliegt, komplementär zueinander
sind, werden als „im
wesentlichen komplementär" bezeichnet. Stabile
Doppelstränge
von im wesentlichen komplementären
Sequenzen erhält
man schon unter weniger strengen Hybridisierungsbedingungen. Fachleute
auf dem Gebiet der Nucleinsäuretechnologie
können
die Doppelstrang-Stabilität
empirisch ermitteln, in dem sie eine Reihe von Variablen berücksichtigen,
wie etwa die Länge und
Basenpaarkonzentration der Oligonucleotide, die Ionenstärke und
die Häufigkeit
nicht gepaarter Basenpaare.
-
Der
hier verwandte Ausdruck „Primer" bezieht sich auf
ein natürliches
oder synthetisch hergestelltes Oligonucleotid, das als Initiationspunkt
für eine
DNA-Synthese fungieren kann, unter Bedingungen, unter denen die
Synthese eines Primer-Verlängerungsproduktes
komplementär
zu einem Nucleinsäurestrang
induziert wird, d.h. in Anwesenheit der vier verschiedenen Nucleosidtriphosphate
und einem Reagenz für
die Polymerisation (DNA-Polymerase oder reverse Transcriptase) in
einem entsprechenden Puffer und bei einer geeigneten Temperatur.
Ein Primer ist vorzugsweise ein einsträngiges Oligodesoxyribonucleotid.
Die geeignete Länge eines
Primers hängt
von der Aufgabenstellung ab, typischerweise liegt sie im Bereich
von 15 bis 35 Nucleotiden. Kurze Primer-Moleküle brauchen generell kühlere Temperaturen,
um ausreichend stabile Hybrid-Komplexe mit der Matrize zu erzeugen.
-
Ein
Primer braucht nicht die genaue Sequenz der Matrize zu haben, muss
jedoch in genügendem Maße komplementär sein,
um mit der Matrize hybridisieren zu können. Primer können zusätzliche
Merkmale tragen, die ein Wiederauffinden oder eine Immobilisation
der Primer ermöglichen,
die aber zu keiner Beeinträchtigung
der Hauptaufgabe der Primer, nämlich
der Initiationspunkt einer DNA-Synthese zu sein, führen dürfen. Zum
Beispiel können
nichtkomplementäre
Sequenzen am Ende eines Primers lokalisiert sein, um Schnittstellen
für Restriktionsenzyme
bereitzustellen, die bei der Clonierung einer amplifizierten Sequenz
nützlich sind.
-
Die
hier benutzten Ausdrücke „stromaufwärts" und „stromabwärts" beziehen sich auf
die Lage der Primer-Bindungsstellen entlang der Zielsequenz. Der
stromaufwärts
liegende Primer hybridisiert mit dem nicht-codierenden Strang der
Zielsequenz und bildet daher das 5'-Ende der amplifizierten Sequenz, die
eine Subsequenz des codierenden Stranges der Zielsequenz ist. Entsprechend
hybridisiert der stromabwärts
liegende Primer mit dem codierenden Strang der Zielsequenz und bildet
daher das 5'-Ende
der amplifizierten Sequenz, die eine Subsequenz des nicht-codierenden
Stranges der Zielsequenz ist.
-
Die
hier benutzten Ausdrücke „Zielsequenz" und „Ziel-Nucleinsäuresequenz" beziehen sich auf
einen Bereich des Oligonucleotides, der amplifiziert oder nachgewiesen
werden soll, oder beides. Die Zielsequenz liegt zwischen den beiden
Primersequenzen, die für
die Amplifikation benutzt werden.
-
Der
hier benutzte Ausdruck „thermostabile
Nucleinsäure-Polymerase" bezieht sich auf
ein Enzym, das, z.B. verglichen mit Nucleotid-Polymerasen von E.
coli, relativ hitzestabil ist. Es katalysiert die Polymerisation
von Nucleosidtriphosphaten. Im allgemeinen startet das Enzym die
Synthese am 3'-Ende
des an die Zielsequenz angehefteten Primers und setzt sie in Richtung
5'-Ende entlang
der Matrize fort, bis die Synthese beendet ist.
-
Für die Verfahren
der vorliegenden Erfindung werden spezielle Kombinationen einer
DNA-Polymerase von
Thermus thermophilus (Tth) und einer DNA-Polymerase von Thermotoga
maritima (Tma), Pyrococcus-Art GB-D oder Thermococcus litoralis
(Tli) verwendet.
-
Die
hier benutzten Ausdrücke „ 3'-zu 5'-Nuclease-Aktivität" und „Korrekturlese-Aktivität" beziehen sich auf
die Aktivität
einer matrizenspezifischen Nucleinsäure-Polymerase, die nacheinander
Nucleotide vom 3'-Ende
eines Oligonucleotids entfernt.
-
Das
Maß für diejenige
Menge eines Enzyms, die benötigt
wird, um Nucleinsäure
mit einer bestimmten Rate zu synthetisieren, ist eine Einheit (Unit,
U). Die hier benannten Aktivitäts-Einheiten entsprechen
den Definitionen der jeweiligen Lieferanten der Polymerasen wie
nachstehend aufgeführt.
Da die Aktivitäten
unter unterschiedlichen spezifischen Bedingungen untersucht werden
können,
kann man die Aktivität
eines Enzymes nicht direkt mit der eines anderen Enzymes vergleichen.
-
Rekombinante
DNA-Polymerasen von Thermus thermophilus (rTth) und Thermatoga maritima
(UlTma) können über Perkin
Elmer, Norwalk, CT bezogen werden. Eine Einheit (Unit) von rTth-
oder UlTmaTM-DNA-Polymerase ist nach Definition
des Lieferanten Perkin Elmer diejenige Menge des Enzymes, die in 30
Minuten bei 74°C
10 nMol dNTP in säureunlösliches
Material einbaut, gemessen in einer 10-minütigen Inkubation in 50 μl Reaktionsgemisch
bestehend aus den folgenden Reagenzien:
200 μM je von
dATP, dGTP, dTTP
100 μM
[α-32p]-dCTP
(0.05 bis 0.1 Ci/mMol)
Aktivierte DNA von Lachsspermien
100
mM KCl
2,2 mM MgCl2
25 mM TAPS
[Natriumsalz der Tris-(hydroxymethyl)-methyl-amino-propansulfonsäure], pH
9,3 bei 25°C
1
mM beta-Mercaptoethanol
-
Rekombinante
DNA-Polymerasen von Thermococcus litoralis (VentR ® und
Pyrococcus-Art GB-D (Deep VentR ® können bezogen
werden über
New England Biolabs, Beverly, MA. Eine Einheit von VentR ® oder
Deep VentR ® ist
nach Definition des Lieferanten New England Biolabs diejenige Menge
des Enzyms, die in 30 Minuten bei 75°C 10nMol dNTP in säureunlösliches
Material einbaut. Das Reaktionsgemisch besteht dabei aus folgenden
Reagenzien:
200 μM
je von dNTP (dATP, dCTP, dGTP und 3H-dTTP)
0,2
mg/ml aktivierte DNA
10 mM KCl
10 mM (NH4)2SO4
20 mM Tris-HCl,
pH 8,8 bei 25°C
2
mM MgSO4
0,1 % Triton X-100
-
Konventionelle
Techniken aus der Molekularbiologie und der Mikrobiologie sowie
rekombinante DNA-Techniken, die zu den Arbeitsverfahren des Fachgebietes
gehören,
sind vollständig
in der Literatur dargestellt. Vgl. z.B. Sambrook, Fritsch und Maniatis,
Molecular Cloning: A Laboratory Manual, Second Edition (1989); Oligonucleotide
Synthesis (M.J. Gait, Herausgeber, 1984); Nucleic Acid Hybridisation
(B.D. Hames & S.J.
Higgins, Herausgeber, 1984); A Practical Guide to Molecular Cloning
(B. Perbal, 1984) und die Reihe Methods in Enzymology (Academic
Press, Inc.).
-
Die
vorliegende Erfindung liefert verbesserte Verfahren und Reagenzien
für die
PCR-Amplifikation
langer DNA-Zielsequenzen. Die PCR-Amplifikation für kurze
Nucleinsäure-Sequenzen ist der
Fachwelt gut bekannt und in den U.S. Patenten Nr. 4,683,195; 4,683,202
und 4,965,118 offenbart. Kommerzielle Lieferanten wie Perkin Elmer,
Norwalk, CT, verkau fen PCR-Reagenzien und veröffentlichen PCR-Protokolle.
Zum besseren Verständnis
der Vorteile, die die vorliegende Erfindung bietet, wird im folgenden
eine Zusammenfassung der PCR gegeben.
-
In
jedem Zyklus einer PCR-Amplifikation wird eine doppelsträngige Zielsequenz
denaturiert, Primer werden an jeden der beiden Stränge der
denaturierten Zielsequenz angelagert, um dann durch die Tätigkeit einer
DNA-Polymerase verlängert
zu werden. Typischerweise wird dieser Vorgang 25 bis 40 Mal wiederholt. Die
beiden Primer lagern sich an die gegenüberliegenden Enden der Ziel-Nucleinsäuresequenz,
und sind so ausgerichtet, dass das Verlängerungsprodukt eines jeden
Primers eine komplementäre
Kopie der Zielsequenz ist und nach Abspaltung von seinem Komplement
mit dem anderen Primer hybridisieren kann. Jeder Zyklus würde, wäre seine
Effektivität
100%, eine Verdoppelung der Zahl der Zielsequenzen bewirken.
-
Um
eine effiziente PCR-Amplifikation von langen Zielsequenzen zu erhalten,
müssen
mehrere Forderungen erfüllt
werden. Erstens müssen
die Zielsequenzen vollständig
denaturiert werden. Mit zunehmender Länge enthalten die Sequenzen
oft GC-reiche Abschnitte, die aufgrund ihrer relativ hohen Schmelztemperaturen
zu einer unvollständigen
Denaturierung neigen. Eine unvollständige Trennung der Stränge erlaubt
eine schnelle Renaturierung der Ziel-DNA, möglicherweise werden die Primer
an einer Anlagerung und Verlängerung
gehindert. Zweitens müssen
die Zeiten für
eine Verlängerung
ausreichend bemessen sein, um eine vollständige Strang-Synthese in jedem
PCR-Zyklus zu gewährleisten.
Drittens müssen
die langen Ziele vor einem Abbau während der Amplifikation geschützt werden.
Lange Ziele sind unter. PCR-Bedingungen
empfindlicher gegenüber
Abbau und Bruch der Stränge.
Die Unversehrtheit der Ausgangsmatrize und das Überleben der gebildeten Stränge während der
PCR sind daher wichtige Gesichtspunkte. Mit den Verfahren der vorliegenden Erfindung
sollen diese Forderungen für
lange PCR erfüllt
werden, ohne weder die Aktivität
der Polymerse noch die für
Einzelkopie-Gen-Amplifikationen von genomischer DNA notwendige Spezifität zu beeinträchtigen.
-
Verbesserung
der Zielstrang-Trennung, Ausdehnung der Verlängerungszeiten und Schutz der
Matrizen-DNA vor einem Abbau während
der Temperatur-Zyklen vergrößert die
maximale Amplifikationslänge
der Zielsequenzen ganz erheblich, sind aber nicht ausreichend, um
eine, effiziente Amplifikation der Zielsequenzen zu erzielen. Der
limitierende Faktor bei der Amplifikation langer Zielsequenzen im
Bereich von 23–42
kb ist die Genauigkeit der Nucleinsäuresynthese.
-
Der
falsche Einbau von Nucleotiden während
der Synthese der Primer-Verlängerungsprodukte
begrenzt die Länge
des Zieles, das erfolgreich amplifiziert werden kann. Die Auswirkungen
auf die Primer-Verlängerung
durch den Einbau einer 3'-Endbase,
die nicht mit der Matrize übereinstimmt,
wird in Huang et al., 1992, Nucl. Acids Res. 20:4567–4573 beschrieben.
Die Anwesenheit von falsch eingebauten Nucleotiden kann zu einer
vorzeitig abgebrochenen Strang-Synthese führen, was wiederum die Anzahl
der Matrizenstränge
für weitere
Runden der Amplifikation verringert, und damit die Gesamtausbeute
der Amplifikation langer Ziele. Selbst ein geringer Prozentsatz
falsch eingebauter Nucleotide kann für Sequenzen, die länger als
10 kb sind, kritisch werden.
-
Die
Genauigkeit bei der DNA-Synthese wird verbessert, wenn man eine
geringe Menge einer thermostabilen 3'-zu 5'-Exonuclease- oder „Korrekturlese"-Aktivität zusätzlich zur
DNA-Polymerase zum
Reaktionsgemisch dazu gibt. Die Korrekturlese-Aktivität verbessert
offensichtlich die Ausbeute an langen Produkten dadurch, dass falsch
eingebaute Nucleotide entfernt werden und der komplette Strang von
der vorherrschenden Polymeraseaktivität synthetisiert werden kann.
Ein wichtiger Aspekt der vorliegenden Erfindung bezieht sich auf
spezifische Gemische thermostabiler DNA-Polymerasen, die die maximale
amplifizierbare Länge
der Sequenz wesentlich erhöhen,
indem sie sowohl 3'-zu
5'-Exonuclease-
als auch Polymerase-Aktivität
bereitstellen.
-
Aktivität einer
korrekturlesenden Exonuclease konnte in der Tth-DNA-Polymerase nicht
nachgewiesen werden (Myers and Gelfand, 1991, Biochemistry 30:7661–7666),
ist aber sehr wohl enthalten in den DNA-Polymerasen von Thermococcus
litoralis (NEB Transcript (1991) Band 3, Nr. 1, XP000391405), Pyrococcus-Art GB-D
und Thermatoga maritima. Trotzdem ist eine Amplifikation langer
Ziele mit VentR ®-DNA-Polymerasen
allein nicht so effizient wie mit einer Tth-DNA-Polymerase, die
keine 3'-zu 5'-Exonuclease-Aktivität aufweist.
Die geringere Amplifikations-Effizienz beruht vermutlich, zumindest
zu einem Teil, auf einem Abbau der Primer und einer Abnahme der
Gesamtprozessierbarkeit, die von einer Konkurrenz zwischen der 3'-zu 5'-Exonuclease- und
der Polymerase-Aktivität
herrührt.
-
Die
relativen Anteile der Aktivitäten
von 3'-zu 5'-Exonuclease und
Polymerase können
durch das Mischen von DNA-Polymerasen kontrolliert werden. Mit der
Zugabe einer kleinen Menge einer sekundären Polymerase, die eine Korrekturlese-Aktivität besitzt,
wie etwa Tli-DNA-Polymerase,
zu einer aktiven primären
Polymerase, wie etwa Tth-DNA-Polymerase, kann der Vorteil einer
Korrekturlese-Aktivität
mit der großen
Aktivität der
DNA-Polymerase, die der primären
Polymerase eigen ist, verbunden werden.
-
Nahezu
alle Aspekte der PCR-Protokolle beeinflussen die Effizienz der Amplifikation
längerer
Zielsequenz-Moleküle.
Verlängerungszeiten,
Co-Lösungsmittel
und die Polymerasen (mit oder ohne 3'-zu 5'- Exonuclease-Aktivität) sind
die kritischsten Parameter, jedoch sind auch der pH-Wert, die Zusammensetzung
der Reaktionspuffer, die Salze (K+ und Mg2+) und die richtige Wahl der Primer wichtige
Variablen für
eine erfolgreiche Amplifikation langer Zielsequenzen. Die Auswirkungen
der einzelnen Komponenten einer PCR-Amplifikation auf die Effizienz
der Amplifikation langer Zielsequenzen wird im folgenden beschrieben.
-
Temperaturzyklus
-
Die
hier dargelegten Amplifikationsschritte verwenden einen zweistufigen
Temperatur-Zyklus, in dem die Reaktionstemperatur zwischen einer
hohen Temperatur, bei der die Ziel-Nucleinsäure denaturiert wird, und einer
niedrigeren Temperatur, bei der sich die Primer an die denaturierten
Ziel-Sequenzen anlagern, wechselt, und die Primer-Verlängerung
stattfindet. Zeitraum und Temperatur jedes einzelnen Schrittes in
jedem einzelnen Zyklus beeinflusst die Effizienz der Amplifikation.
-
Mit
steigender Denaturierungstemperatur steigt auch der Grad der Denaturierung
der Zielsequenz. Die Erhöhung
der Denaturierungstemperatur kann jedoch auch höhere Schadensraten nach sich
ziehen, etwa eine Depurinierung, die den Amplifikationserfolg schmälert, oder
einen erhöhten
Verlust der Polymerase-Aktivität.
Obwohl einerseits eine vollständige
Denaturierung der Ziel-Nucleinsäure-Sequenz
erreicht werden soll, müssen
andererseits gleichzeitig die Schäden an der Zielsequenz möglichst
gering gehalten werden. Folglich werden moderate Denaturierungstemperaturen
bevorzugt (z.B. etwa 94°C,
abhängig
vom GC-Gehalt), wobei die Vollständigkeit
der Denaturierung durch die Zugabe bestimmter Co-Lösungsmittel
verbesssert wird, wie im folgenden beschrieben.
-
Eine
relativ hohe Anlagerungstemperatur (z.B. etwa 68°C) reduziert die Hybridisierung
der Primer mit teilweise homologen Ziel-Sequenz-Abschnitten. Gleichzeitig
wird dadurch die Synthese von Produkten sekundärer Anlagerungsstellen minimiert.
Bei Amplifikationen mit Lambda-DNA-Zielsequenzen, wie in den Beispielen
beschrieben, ist mindestens eine Temperatur von 68°C über den
Zeitraum von 5–6
Minuten notwendig. Das Einfügen
eines stringenten Anlagerungsschrittes bei 70–75°C verbessert die Ausbeuten nicht
wesentlich. In ähnlicher
Weise haben auch komplexere Temperaturprofile mit Höchsttemperaturen
zur Anpassung möglicherweise
problematischer GC- oder AT-reicher Strangbereiche keine signifikanten
Verbesserungen gebracht.
-
Auch
der Verlängerungszeitraum,
der die vollständige
Strang-Synthese erlaubt, ist eine kritische Größe, wenn man lange Zielsequenzen erfolgreich amplifizieren
will. Für
die Amplifikation von Zielsequenzen, die länger als 20 kb sind, hat sich
eine Anlagerungs- und Verlängerungszeit
von mindestens 12 Minuten, aber nicht mehr als 22 Minuten in jedem
Zyklus als vorteilhaft erwiesen. Die Mindestverlängerungsdauer hängt von
anderen Faktoren ab, etwa der Konzentration der Co-Lösungsmittel,
wie im folgenden noch ausgeführt.
Bei Amplifikationsreaktionen, in denen man für den ersten Verlängerungszeitraum
eine Reaktionsdauer von etwa 12 Minuten wählt und diese Zeit in den folgenden
Zyklen um jeweils 15–20
Sekunden pro Zyklus verlängert,
erhält man
weniger unspezifische Produkte, als wenn man während der gesamten Amplifikation
jeweils eine Dauer von mehr als 15 Minuten ansetzt. Die von Perkin
Elmer, Norwalk, CT, vertriebene automatische Anlage zur Amplifikation
ermöglicht
auf einfache Weise, die jeweiligen Verlängerungszeiten während einer
Amplifikation zu erhöhen.
-
Reduzierung
der Amplifikation von nicht-spezifischen Zielsequenzen
-
Klassischerweise
werden die PCR-Reagentien vor dem ersten Denaturierungsschritt bei
Zimmertemperatur zusammengegeben. Bei der niedrigen, weniger stringenten
Temperatur können
die Primer sowohl untereinander als auch mit teilweise homologen
Abschnitten der Zielsequenz Bindungen eingehen. Diese unspezifischen
Primerbindungen können
zu Verlängerungsprodukten
führen,
die kurze Produkte ergeben, die sehr erfolgreich mit der eigentlichen
Zielsequenz konkurrieren und dadurch den Erfolg der gewünschten
Amplifikation langer Sequenzen erheblich mindern. Ein „Heiß-Start"- Verfahren minimiert
die Synthese von Primerverlängerungsprodukten
unspezifischer Primer-Hybridisierungen, durch Hemmung von Ver längerungsreaktionen, bis
die Reaktionstemperatur hoch genug ist, um solche unspezifischen
Bindungen nicht zustande kommen zu lassen. Da man davon ausgehen
kann, dass genomische Matrizen Sequenzen beinhalten, die teilweise
homolog mit Zielprimersequenzen sind, ist ein heißer Start
wichtig für
den Erfolg bei der Amplifikation langer Zielsequenzen.
-
Eine
Methode für
einen erfolgreichen Heiß-Start
beinhaltet das Zurückhalten
einer wesentlichen PCR-Reaktionskomponente, bis die Temperatur des
Amplifikationsgemisches 75–80°C erreicht
hat. Man kann zum Beispiel die DNA-Polymerase zurückhalten
oder Mg2+, das ein wesentlicher Katalysator
der DNA-Polymerase-Aktivität
ist. In einem Heiß-Start-Ansatz
werden die wesentlichen Komponenten per Hand zugegeben, nachdem
die Denaturierungstemperatur erreicht worden ist. Man kann die wesentlichen
Reaktionskomponenten auch zurückhalten,
indem man die Komponenten in einem Reaktionsgefäß zunächst durch eine hitzelabile Barriere
voneinander getrennt hält,
etwa durch Wachs, das bei Erreichen der Reaktionstemperatur schmilzt. Auf
diese Weise lässt
sich die Anzahl des wiederholten Öffnens des Reaktionsgefäßes minimieren,
wodurch die Möglichkeit
einer Verunreinigung gering gehalten wird.
-
Ein
anderer Heiß-Start-Ansatz,
der bei den Verfahren der vorliegenden Erfindung von Nutzen sein kann,
verwendet Uracil-N-glycosylase, die alle unspezifischen Produkte
abbaut, die vor Erreichen der eigentlichen Amplifikationsgemischtemperatur
gebildet werden (vgl. PCT-Patent-Veröffentlichung
Nr. WO 92/01814).
-
PCR-Reagenzien
-
In
einer PCR findet die Primer-Verlängerungsreaktion
statt, wenn das Primer-Matrizen-Gemisch
unter geeigneten Polymerisationsbedingungen mit einer DNA-Polymerase
inkubiert wird. Diese Bedingungen sind gegeben, wenn das Reaktionsgemisch
ein bivalentes Kation, ein monovalentes Kation, alle vier Desoxyribonucleotid-Triphosphate
(dNTPs) und einen Puffer enthält.
Co-Lösungsmittel,
die die Denaturierungsreaktion beeinflussen, können zum Reaktionsgemisch hinzugegeben
werden. Jeder dieser Komponenten beeinflusst die Effizienz der Verlängerungsreaktion
und wird im folgenden gesondert besprochen.
-
DNA-Polymerase
-
Die
Auswahl der Kombination von thermostabilen DNA-Polyrnerasen und
deren Konzentration ist von entscheidender Bedeutung, wenn die Länge oder
Komplexität
der Zielsequenz zunimmt. Mit der Kombination von Tth-DNA-Polymerase
und Tli-DNA-Polymerase erhält
man die beste Ausbeute bei der Amplifikation langer PCR-Produkte,
dabei sind Amplifikationen von Sequenzen von über 40 kb möglich.
-
Die
optimale Menge von DNA-Polymerase in einer PCR-Amplifikation hängt von
einer Reihe von Faktoren ab, einschließlich der Anzahl der Kopien
der Zielsequenzen in der Probe. Liegt eine große Anzahl von Kopien vor (107 Kopien der Zielsequenz), erhält man eine
höhere
Ausbeute bei einer Dosierung von 2–2,5 Einheiten (Units, U) Tth-DNA-Polymerase
pro 50 μl
Reaktionsgemisch. Eine weitere Erhöhung der Polymerase-Konzentration
führt zu
einem Anstieg der Amplifikation unspezifischer Zielmoleküle, die
sich in höheren
Hintergrundniveaus bemerkbar machen, wenn die amplifizierten Produkte
mittels Agarose-Gelelektrophorese nachgewiesen werden. Bei einer
geringen Anzahl von Kopien (104 Kopien der
Zielsequenz) erhält
man eine maximale Spezifizierung bei etwa 0,8–1 U Tth-DNA-Polymerase pro
50 μl Reaktionsgemisch.
Bei einer mittleren Kopienzahl von Zielsequenzen werden maximale
Ausbeuten bei mittleren Polymerasekonzentrationen erreicht. Die
optimale Polymerase-Konzentration
hängt auch
von der Konzentration des bivalenten Kations ab. Bei höheren Mg2+-Konzentrationen wurden die Polymerase-Konzentrationen
reduziert, um die Akkumulation unspezifischer Produkte zu minimieren.
-
Wurde
bei der PCR Tth-DNA-Polymerase allein verwendet, ergab sich bei
Proben mit einer großen Anzahl
von Kopien von Lambda-Phagen-DNA eine maximale Länge der Zielsequenz, die noch
amplifiziert wurde, von etwa 23 kb. In ähnlicher Weise war die maximale
amplifizierbare Länge
von Zielsequenzen bei einer geringen Anzahl von Kopien von Lambda-Phagen-DNA-Proben
auf etwa 10–12
kb beschränkt.
Dramatische Verbesserungen in der Länge der amplifizierbaren Zielsequenzen
ergaben sich durch die Zugabe einer geringen Menge von thermostabiler
3'-zu 5'-Exonuclease.
-
Wie
vorstehend beschrieben, findet man in der Tth-DNA-Polymerase keine
3'-zu 5'-Exonuclease-Aktivität. Die Aktivität des Korrekturlesens
ergibt sich erst bei der Kombination von Tth-DNA-Polymerase mit
einer geringen Menge thermostabiler DNA-Polymerase, die die Korrekturlese-Aktivität besitzt,
etwa DNA-Polymerasen von Thermococcus litoralis, Pyro coccus-Art
GB-D und Thermotoga maritima. Niedrige Konzentrationen einer dieser
DNA-Polymerasen
bewirken, dass längere
Zielsequenzen mit der PCR amplifiziert werden können als nur mit der Tth-DNA-Polymerase
allein. Eine Kombination von Tth- und Tli-DNA-Polymerasen hat sich als das verlässlichste
und wirkungsvollste Verfahren erwiesen.
-
Das
optimale Konzentrationsverhältnis
liegt bei 0,015–0,15
U Tli-DNA-Polymerase pro 2–2,5
U Tth-DNA-Polymerase für
Amplifikationen von Proben mit hoher Anzahl von Kopien (107 Kopien der Zielsequenz in 50 μl Reaktionsgemisch).
Für Amplifikationen
von Proben mit einer geringen Anzahl an Kopien (104 Kopien
der Zielsequenz in 50 μl
Reaktionsgemisch) beträgt
die optimale Konzentration etwa 0,015–0,15 U Tli-DNA-Polymerase
pro 0,8–1
U Tth-DNA-Polymerase.
Höhere
Konzentrationen von Tli-DNA-Polymerase reduzieren den Erfolg, vermutlich
durch Primerabbau.
-
Co-Lösungsmittel
-
Ein
Co-Lösungsmittel,
wie etwa Glycerin, ist eine kritische Reaktionskomponente für die effiziente
Amplifikation langer Zielsequenzen. Von einer Reihe von Co-Lösungsmitteln
wird berichtet, dass sie die PCR erleichtern, darunter Glycerin,
Dimethylsulfoxid (DMSO) Polyethylenglycol und Formamid. Ein Weg,
wodurch ein Co-Lösungsmittel
die Effizienz einer Amplifikation langer Zielsequenzen verbessern
kann, besteht in der Verbesserung der thermischen Stabilität der DNA-Polymerase.
Eine höhere
thermische Stabilität
verlangsamt den Verlust von DNA-Polymerase-Aktivität während der
wiederholten Denaturierungsschritte bei hohen Temperaturen.
-
Ein
anderer Effekt ist, dass ein Co-Lösungsmittel wesentlich die
Schmelz- und Strangtrennungstemperatur herabsetzen kann, wodurch
die Denaturierung der Matrize erleichtert wird und sich die Spezifität der Primer-Anlagerung
erhöht.
Die Schmelztemperatur kann zum Beispiel um 2,5–3°C verringert werden, wenn man
10% Glycerin zugibt. Auf diese Weise kann mit der Zugabe eines Co-Lösungsmittels
die Vollständigkeit der
Denaturierung der Zielsequenz erhöht werden, ohne dass die Denaturierungstemperatur
angehoben werden muss, was, wie vorstehend dargelegt, gleichzeitig
den Abbau der Zielsequenz-Moleküle
fördern
würde.
-
Ein
Standard-Tth-PCR-Puffer enthält
typischerweise 5% (v/v) Glycerin. Eine größere Menge Glycerin im Amplifikations-Reaktionsgemisch
kann die Amplifikation langer Zielsequenzen signifikant verbessern.
Signifikante Verbesserungen in der Ausbeute einer 9,4 kb-Zielsequenz
sind auf die Ergänzung
eines Standard-Tth-PCR-Puffers mit 5% (w/v) Glycerin zurückzuführen. Die
angegebenen Prozentzahlen berücksichtigen
nicht etwaige Glycerin-Zugaben durch die verschiedenen benutzten
Enzym-Stammlösungen.
-
DMSO,
vorzugsweise in einer Konzentration von etwa 5–6% (v/v), kann auch allein
verwendet werden. Kombinationen aus Glycerin und DMSO sind bei langen
Zielsequenzen allerdings wirkungsvoller. Bevorzugte Konzentrationskombinationen
beinhalten 5–14%
(w/v) Glycerin mit 0,5–5%
(v/v) DMSO. Zum Beispiel werden Amplifikationen von Lambda-Phagen-Zielsequenzen mit
25–34
kb Länge
verbessert, wenn man 1–3%
(v/v) DMSO mit 10% Glycerin zugibt oder 5% von beiden Co-Lösungsmitteln
verwendet. Amplifikationen von Lambda-Phagen-Zielsequenzen mit 35–42 kb Länge können mit
der Zugabe von 8–9%
Glycerin und 5% DMSO am erfolgreichsten durchgeführt werden. Zusätzlich werden
Zielsequenzen von bis zu 34 kb mit einer Kombination aus 3% DMSO
und 10% Glycerin in einer Verlängerungszeit
von 10 Minuten leicht amplifiziert. Bei einer Kombination von 1%
DMSO und 10% Glycerin war die Amplifikation auf 26 kb Länge der
Zielsequenz begrenzt. Eine bevorzugte Kombination besteht aus 10%
Glycerin und 2,25% DMSO.
-
Im
Unterschied zu Glycerin setzt DMSO die Thermostabilität der Polymerase
herab. Trotzdem ist die effektive Erniedrigung der Schmelz- und
Strangtrennungstemperatur um 5,5–6°C pro 10% DMSO wahrscheinlich
der dominierende Faktor für
die lange PCR. Die Zugabe von DMSO erhöht vermutlich auch die DNA-Stabilität durch
eine Herabsetzung der Depurinierungsrate und/oder der Rate der Kettenbrüche und
beschleunigt vermutlich die Strang-Renaturierung. Die Reduktion von Schmelz-
und Strangtrennungstemperaturen durch die Kombination von Glycerin
und DMSO stimmt generell mit der Gesamtreduktion überein,
die sich aus einer Summierung der Effekte der einzelnen Komponenten
allein ergeben würde.
Die Verbesserung der Ausbeuten durch die wirksame Senkung der Schmelz-
und Strangtrennungstemperaturen mit Hilfe der Zugabe eines Co-Lösungsmittels
wird, wie vorstehend besprochen, durch eine Erhöhung der Denaturierungs- oder
Anlagerungstemperatur während
der PCR nicht leicht verdoppelt.
-
Puffer
-
Der
pH-Wert eines Amplifikationsgemisches beeinflusst die Stabilität der Matrizen-DNA.
Eine Erhöhung
des pH-Wertes im Reaktionsgemisch kann den Abbau der Matrizen-DNA
während
der Temperaturzyklen vermindern. Obwohl PCR-Amplifikationsgemische
pH- gepuffert sind,
variiert der pH-Wert einer typischen PCR-Reaktion durch die Temperaturabhängigkeit
der Reaktionspuffer erheblich während
der Temperaturzyklen. Der normalerweise in einer PCR benutzte Puffer
ist Tris, das einen pKa-Wert von –0,031 pro °C aufweist. Die Schwankungen
des pH-Wertes während
der Temperaturzyklen können
durch die Verwendung eines Puffers mit einem kleinerem pKa-Wert
vermindert werden.
-
Zwei
geeignete Puffer sind Tris(hydroxymethyl)methylglycin (Tricin),
das einen pKa-Wert von –0,021 pro °C hat, und
N,N-Bis(hydroxyethyl)glycin (Bicin) mit einem pKa-Wert von –0,018 pro °C; beide
Werte gemessen bei 20°C
und einer Ionenstärke
von 0,1M (vgl. Good and Izawa, 1972, Meth. Enzymol. 24, Teil B: 53–68). Bei
Verwendung eines Tricin- oder Bicin-Puffers bleibt der pH-Wert bei den Reaktionsbedingungen
mit hoher Temperatur höher
als mit dem üblichen
Tris-Puffer, und die Schwankungen im pH-Wert aufgrund der Temperaturzyklen
sind geringer.
-
Optimale
Puffer und pH-Werte sind unter anderem abhängig von der verwendeten DNA-Polymerase. Benutzt
manTth-DNA-Polymerase, werden mit einem Puffer von 10–35 mM,
vorzugsweise 20–25
mM, Tricin bei einem pH-Wert von 8,5–8,7 (25°C) die verlässlichsten Ergebnisse erzielt.
Optimale Puffer-Bedingungen müssen
am besten empirisch für
die Amplifikation der spezifischen Zielsequenzen festgelegt werden.
-
Bivalentes Kation
-
Das
bevorzugte bivalente Kation für
eine DNA-Amplifikation ist Mg2+ Ohne eine
hinzugefügte
3'-zu 5'-Exonucleaseaktivität wird die
lange PCR am besten durch eine Gesamt- Mg2+-Konzentration von
1,72 mM vorangetrieben. In Anwesenheit einer Korrekturlese-Aktivität hingegen
werden die höchsten
Ausbeuten bei 0,9–1,3
mM Gesamtkonzentration Mg2+ erzielt. Bei
einigen Zielsequenzen kann man eine noch höhere Ausbeute erhalten, wenn
man die Mg2+-Konzentration bis auf 1,5 mM
erhöht
und gleichzeitig die Gesamt- Enzymkonzentration verringert, insbesondere
die der primären
Polymerasen (auf 1,25–2
U Tth-DNA-Polymerase). Bei einigen Zielsequenzen jedoch bewirkt
eine Reduzierung der Gesamt-Enzymkonzentration, durch die man ja
bei gleichzeitiger hoher Konzentration von Mg2+ die
Produktion unspezifischer Produkte vermindern wollte, eine insgesamt
geringere Ausbeute. Wie bei den nachstehend beschriebenen K+-Konzentarationen muss das Mg2+-Optimum
möglicherweise
für jedes
System empirisch eingestellt werden.
-
Monovalentes Kation
-
Das
bevorzugte monovalente Kation ist K+, in
Form von KOAc (K-acetat) oder KCl. Für die Amplifikation längerer Zielsequenzen
sind niedrigere K+-Konzentrationen von Vorteil.
Eine Reduzierung des unspezifischen Hintergrundes kann erreicht
werden, wenn K+ als KOAc statt als KCl bereitgestellt
wird. Allgemein gilt, K+-Konzentrationen,
die um 10–40%
reduziert sind, sind vorteilhafter für eine lange PCR als die Standardkonzentrationen
(100 mM KCl für
Tth-DNA-Polymerase). Bevorzugte Konzentrationen bei Verwendung der Tth-DNA-Polymerase sind 60–100 mM
KOAc, besonders 80–85
mM KOAc. Eine optimale Konzentration ist auch hier systemabhängig.
-
Für die Effizienz
der PCR-Amplifikationen mit Tricin- oder Bicin-Puffer ist unerheblich,
ob KCl oder KOAc als monovalentes Kation eingesetzt wird. Der Gebrauch
von einem Tricin/KOAc-Puffer erhöht
allerdings die Unempfindlichkeit der ablaufenden Reaktionen. Ein
Tricin/KOAc-Puffer hat eine etwas geringere Ionenstärke als
ein Tricin/KCl-Puffer, was dazu beitragen könnte, Sekundärstrukturen
in einer Matrize mit hohem G+C-Gehalt zu destabilisieren, was wiederum
die Vollständigkeit
der Denaturierung der Zielsequenz-Stränge verbessert.
-
Obgleich
KCl und KOAc die bevorzugten monovalenten Salze sind, können auch
andere monovalente Salze für
die Verfahren der hier vorliegende Erfindung nützlich sein, darunter NaCl,
(NH4)2SO4, K-Glutamat und NH4-Acetat.
-
Primer
-
Primer-Konzentrationen
müssen
möglicherweise
für jedes
System und die ungefähre
Anzahl von vorhandenen Matrizen-Kopien optimiert werden. Zum Beispiel
war bei den in den nachstehend angeführten Beispielen beschriebenen
Lambda-Phagen-Amplifikationen für
die Amplikation von Proben mit einer hohen Kopienzahl von Zielsequenzen
eine höhere
Primer-Konzentration
optimal als für
die Amplifikation von Proben, die eine geringere Anzahl von Zielsequenz-Kopien
enthielten. Für
die Reaktionen mit einer hohen Anzahl von Kopien (107 Kopien
der Zielsequenz) lag die optimale Konzentration bei 0,4–0,5 μM für jeden
Primer. Für
die Amplifikation weniger Kopien (104 Kopien
der Zielsequenz) waren 0,15–0,2 μM für jeden
Primer am effektivsten, wenn keine Korrekturlese-Aktivität vorhanden
war, und 0,2 μM
für jeden
Primer war am besten, wenn eine 3'-zu 5'-exonucleolytische Aktivität vorhanden
war. Bei Reaktionen mit mittlerer Anzahl an Kopien erwies sich eine Erhöhung der
Primer-Konzentrationen
auf über
0,2 μM für eine Erhöhung der
Ausbeute als mindestens ebenso effektiv wie eine Erhöhung der
DNA-Polymerase-Konzentrationen, wie bereits vorstehend diskutiert.
Die verbesserten PCR-Protokolle, die die Amplifikation von Nucleinsäure-Sequenzen
bis zu 42 kb Länge
ermöglichen,
sind in der untenstehenden Tabelle 1 zusammengefasst.
-
Tabelle 1
-
Optimale Bedingungen für lange
PCR
-
Temperaturprofil
-
- 25 bis 40 Amplifikationszyklen (abhängig von der Anzahl der Matrizen-Kopien)
Zwei-Temperaturen-Zyklus:
(a) Kurzer Denaturierungsschritt
(z.B. 94°C
für 10–15 Sekunden)
(b)
Langer Anlagerungs-/Verlängerungsschritt
(z.B. 68°C
für anfänglich 10–14 Minuten,
dann um 15–20
Sekunden pro Zyklus verlängert,
für mindestens
5–8 Zyklen)
Abschließendes
Halten auf 72°C
für mindestens
10 Minuten
-
Heiß-Start
-
Zurückhalten
von einem Reagenz (Mg2+, Enzym oder dNTPs),
bis alle Proben eine Temperatur von 75–80°C erreicht haben, am besten
durch eine Wachs-Trennschicht.
-
Primäre Polymerase
-
- 2,5 Einheiten Tth-DNA-Polymerase pro 50 μl für Matrizen mit hoher Anzahl
von Ausgangskopien (107 Kopien)
- 0,8-1 Einheiten) Tth-DNA-Polymerase pro 50 μl für Matrizen mit geringer Anzahl
von Ausgangskopien (104 Kopien)
- 3'- zu 5'-Exonuclease (für Matrizen
mit hoher oder geringer Anzahl von Kopien) 0,015–0,15 Einheiten Tli-DNA-Polymerase
pro 50 μl
-
Co-Lösungsmittel
-
- 5–14%
Glycerin mit 0,5–5%
DMSO
-
Puffer
-
- 20–25
mM Tricin oder Bicin, pH 8,5–8,7
-
Bivalentes Kation
-
- 0,9–1,5
mM Mg2+ gesamt, 0,2 mM Unterschiede können kritisch
wirken
-
Monovalentes Kation
-
-
Primer-Herstellung
-
- Entweder 20–23
Bp mit 50–60%
GC-Anteil oder längere
Sequenzen, um eine relativ hohe Anlagerungstemperatur zu ermöglichen.
-
Primer-Konzentration
-
- 0,4–0,5 μM für Matrizen
mit hoher Anzahl von Ausgangskopien (107 Kopien)
- 0,15–0,2 μM für Matrizen
mit geringer Anzahl von Ausgangskopien (104 Kopien)
-
dNTP- Konzentration
-
- 0,2 mM jeweils für
dATP, dCTP, dGTP, dTTP
-
Im
allgemeinen wird die Nucleinsäure
in der Probe DNA sein, in den meisten Fällen genomische DNA. Die vorliegende
Erfindung kann jedoch auch bei anderen Nucleinsäuren, etwa RNA oder clonierter
DNA angewendet werden, und die Nucleinsäure kann als Einzelstrang oder
als Doppelstrang in der Probe vorliegen und dennoch für die Zwecke
der vorliegenden Erfindung geeignet sein. Fachleute nehmen zur Kenntnis,
dass, wie auch immer die Nucleinsäure beschaffen ist, sie mit
geeigneten Modifikationen der vorgestellten Verfahren amplifziert
werden kann.
-
Aufgrund
der enormen Amplifikationsmöglichkeiten
mit der PCR können
schon geringe DNA-Verunreinigungen, etwa mit DNA aus einer hochkonzentrierten
DNA-Probe, von Positiv-Kontroll-Matrizen oder von vorausgegangenen
Amplifikationen, PCR-Produkte erzeugen, selbst wenn keine absichtlich
hinzugefügte
Matrizen-DNA in der Probe vorhanden ist. Wenn möglich sollten daher alle Reaktionsgemische
räumlich
getrennt von der PCR-Analyse und der Probenvorbereitung angesetzt
werden. Die Benutzung von gekennzeichneten oder Einmal-Gefäßen, Lösungen und
Pipetten (vorzugsweise Verdrängungspipetten)
für RNA/DNA-Präparation,
Reaktionsgemische und Probenanalyse minimieren eine gegenseitige
Verunreinigung. Vgl. auch Higuchi und Kwok, 1989, Nature 339:237–238; und
Kwok, und Orrego in Innis et al., Herausgeber, 1990 PCR Protocols:
A Guide to Methods and Applications, Academic Press, Inc., San Diego,
CA.
-
Enzymatische
Verfahren zur Verringerung des Problems einer PCR-Verunreinigung
durch die amplifizierte Nucleinsäure
aus vorhergehenden Reaktionen sind in der PCT-Patent-Veröffentlichung
Nr. WO 92/01814 und dem U.S. Patent Nr. 5,035,996 beschrieben. Die
Verfahren erlauben den enzymatischen Abbau jeder amplifizierter
DNA aus vorausgegangenen Reaktionen. PCR-Amplifikationen werden
in Gegenwart von dUTP anstatt dTTP durchgeführt. Das daraus resultierende
doppelsträngige
Amplifikationsprodukt, das Uracil enthält, wird von der Uracil-N-glycosylase
(UNG) angegriffen, während
eine normale Thyminenthaltende DNA nicht von UNG angegriffen wird.
Amplifikationsreaktionsgemische werden vor der Amplifikation mit
UNG behandelt, um alle Uracil-haltige DNA auszuschalten, die als
Ziel fungieren könnte.
Weil die einzige Quelle einer Uracil-haltigen DNA das amplifizierte
Produkt aus einer vorhergehenden Reaktion sein kann, verhindert
dieses Verfahren effektiv das Problem der Verunreinigung durch vorhergehende
Reaktionen (Carry-over). UNG wird durch Hitze zeitweise inaktiviert,
so dass die Denaturierungsschritte des Amplifikationsprozesses gleichzeitig auch
dazu dienen, die UNG zu inaktivieren. Neue Amplifikationsprodukte
werden demzufolge, obwohl sie Uracil einbauen, in einer UNG-inaktivierten
Umgebung gebildet und daher nicht abgebaut.
-
Die
Analyse der amplifizierten Produkte kann auf verschiedene Art und
Weise durchgeführt
werden, abhängig
von der jeweils gewünschten
Information. Die Nucleotidsequenz der amplifizierten Produkte kann durch
Standard-Techniken ermittelt. werden, wie sie bei Innis et al.,
1988, Proc. Natl. Acad. Sci. 85:9436–9440 beschrieben werden. Die
PCR-Amplifikationsprodukte können
direkt sequenziert werden (vgl. Saiki et al., 1988, Science 239:487–491) oder
indirekt, indem man sie zunächst
cloniert, und sie sich dann in einer geeigneten Wirtszelle replizieren
lässt.
-
Mit
den Verfahren für
Nachweis und Reinigung amplifizierter Nucleinsäure-Sequenzen ist die Fachwelt wohlvertraut
(vgl. Sambrook et al., 1989, siehe oben). Zur Reinigung der amplifizierten
Nucleinsäuren
können Verfahren
angewendet werden, die die Moleküle
nach ihrer Größe trennen,
zum Beispiel die Gelelektrophorese. Besonders die Agarose- und/oder
Acrylamid-Gelelektrophorese eignen sich zur Analyse amplifizierter
Produkte (vgl. Scharf et al., 1986, Science 233:1076–1078).
Für eine
bessere Größenauflösung können entweder die
Feldinversion-Gelelektrophorese oder die niedrigprozentige (0,3%)
Agarose-Gelelektrophorese angewendet werden, wie in den Beispielen
beschrieben.
-
Amplifizierte
Produkte können
durch direkte Anschauung ausgewertet werden, indem man die elektrophoretisch
nach Größe aufgetrennten
Produkte anfärbt,
zum Beispiel mit Ethidiumbromid. Alternativ kann man Oligonucleotid-Hybridisierungssonden
einsetzen, die kom plementär
zur Zielsequenz sind, um amplifizierte Produkte nachzuweisen. Unter
geeigneten Hybridisierungsbedingungen lagern sich die Sonden nur
an die Ziel-Nucleinsäuresequenzen
an. Die Anwesenheit hybridisierter Doppelstränge, die dann durch zahlreiche Verfahren
nachgewiesen werden können,
belegt die Anwesenheit amplifizierter Produkte. Um den Nachweis hybridisierter
Doppelstränge
aus Sonde und gesuchter Nucleinsäure-Sequenz
zu erleichtern, können
entweder die Primer oder die Sonden mit zusätzlichen Molekülen versehen
werden, etwa einem nachweisbaren Marker oder einem Molekül, das Primer
oder Sonde immobilisiert. In die Sonden eingebaute Marker zum Nachweis oder
zur Immobilisierung sollten die Hybridisierungseigenschaften der
Sonden allerdings nicht beeinträchtigen.
-
Zur
Kennzeichnung der Sonden eignen sich Marker, die sich mit spektroskopischen,
photochemischen, biochemischen, immunochemischen oder chemischen
Verfahren nachweisen lassen. Geeignete Marker sind zum Beispiel 32P, fluoreszierende Farbstoffe, elektronendichte
Reagenzien, Enzyme (wie üblicherweise
in ELISAs verwendet), Biotin oder Haptene oder Proteine, für die Antiseren
oder monoclonale Antikörper verfügbar sind.
Die Sonden können
auch an zusätzliche
Verbindungen gebunden werden, die die Sonde an einer festen Phase
fixieren.
-
Markierte
Sonden können
durch die oben beschriebenen Techniken zur Herstellung von Oligonucleotiden
synthetisiert und markiert werden. Zum Beispiel kann eine Sonde
am 5'-Ende mit 32P markiert werden, indem man sie mit 32P-ATP und einer Kinase inkubiert. Ein geeigneter
nicht-radioaktiver Marker für
SSO-Sonden ist Meerrettich-Peroxidase (HRP). Verfahren zur Herstellung
und zum Nachweis von Sonden mit diesem Marker werden in den U.S.
Patenten Nr. 4,914,210 und 4,962,029 beschrieben. Die Verwendung
von solchermaßen
markierten Sonden wird ebenfalls im U.S. Patent Nr. 4,789,630 beschrieben;
Saiki et al., 1988, N. Eng. J. Med. 319:537–541; und Bugawan et al.,1988,
Bio/Technology 6:943–947.
Geeignete Farbstoffe für
den Nachweis von HRP-markierten Sonden sind zum Beispiel Red leuco
dye und 3,3',5,5'-Tetramethylbenzidin (TMB).
-
Eine
zusätzliche
Verbindung, die in die Sonden eingebaut werden kann, um eine Immobilisierung
der Sonde zu erreichen, ist zum Beispiel ein langer Poly-dT „Schwanz", der durch Bestrahlung
auf einem Nylon-Untergrund fixiert werden kann; eine Technik, die
im einzelnen in der PCT-Patent-Veröffentlichung Nr. WO 89/11548
beschrieben ist.
-
Geeignete
Verfahren, Hybride zwischen Sonde und gesuchter Nucleinsäure-Sequenz
in einer Probe nachzuweisen, sind in der Fachwelt bekannt (Sambrook
et al., 1985, siehe oben). Sie umfassen zum Beispiel den Dot-Blot-
und den Revers-Dot-Blot-Test.
-
In
einem Dot-Blot-Format wird die unmarkierte amplifizierte Ziel-DNA
auf einem festen Untergrund, etwa einer Nylonmembran, fixiert. Der
Membran-Zielsequenz-Komplex wird mit einer markierten Sonde unter geeigneten
Hybridisierungsbedingungen inkubiert, nicht hybridisiertes Sondenmaterial
wird durch Waschen unter geeignet stringenten Bedingungen entfernt,
worauf die Membran auf die Anwesenheit von gebundenem Sondenmaterial
untersucht werden kann.
-
Eine
andere Untersuchung ist ein „umgekehrtes" Dot-Blot-Format,
in dem die amplifizierte Ziel-DNA markiert wird und die Sonden auf
einem festen Untergrund, etwa einer Nylon-Membran, fixiert sind. Die Ziel-DNA
wird üblicherweise
während
der Amplifikation durch den Einbau von markierten Primern markiert. Der
Membran-Sonden-Komplex und die markierte Probe werden unter geeigneten
Hybridisierungsbedingungen inkubiert, nichthybridisiertes Probenmaterial
wird durch Waschen unter geeignet stringenten Bedingungen enfernt,
worauf der Filter auf die Anwesenheit von gebundener Ziel-DNA untersucht
wird.
-
Alternativ
kann der Revers-Dot-Blot-Test auch durchgeführt werden, indem man einen
festen Träger mit
einer Vielzahl von Hybridisierungsstellen oder Vertiefungen benutzt.
Zum Beispiel ist eine Mikrotiter-Platte besonders sinnvoll für die in
großem
Umfang durchgeführten
klinischen Anwendungen der vorliegenden Verfahren. Ein Revers-Dot-Blot-Test
mit Verwendung einer Mikrotiter-Platte wird bei Loeffelholz et al.,
1992, in J. Clin. Microbiol. 30 (11):2847-2851 beschrieben. Sonden können durch
passive Bindung direkt an der Mikrotiter-Platte fixiert werden,
oder sie werden zunächst
an Rinderserumalbumin (BSA) gebunden, das sich an den Mikrotiter-Platten
anlagert.
-
Ein
weiteres geeignetes Untersuchungssystem ist im U.S. Patent Nr. 5,210,015
beschrieben, bei dem eine markierte Sonde während des PCR-Amplifikationsprozesses
zugegeben wird. Die Sonden sind so modifiziert, dass sie nicht als
Primer für
eine DNA-Synthese wirken können.
Jede Sonde, die während
jedes Synthese-Schrittes mit der Ziel-DNA hybridisiert, wird durch
die 5'-zu 3'-Exonuclease-Aktivität der DNA-Polymerase
abgebaut. Die Abbauprodukte der Sonde werden dann nachgewiesen.
Die Anwesenheit von Abbauprodukten der Sonde belegt, daß Hybridisierung
zwischen Sonde und Ziel-DNA stattgefunden hat.
-
Die
vorliegende Erfindung bezieht sich auch auf Kits, Einheiten mit
mehreren Behältern,
die hilfreiche Komponenten zur Anwendung der beschriebenen Verfahren
darstellen. Ein Kit enthält
eine Kombination der bevorzugten Polymerase-Enzyme in den Konzentrationsverhältnissen,
wie sie hier beschrieben sind. Zusätzliche Komponenten, die in
einem sinnvollen Kit zusammengestellt sein können, umfassen Primer für die PCR-Amplifikation
und Reagenzien für
die Durchführung
der PCR-Verfahrenn wie sie in der vorliegenden Erfindung beschrieben
werden.
-
Die
Fähigkeit,
Sequenzen von 10 bis 40 kb zu amplifizieren, findet zahlreiche Anwendungen
in Gebieten wie etwa Genomkartierung, Sequenzierung oder in der
Genetik. Kleine Lücken
in den Genkartierungen, die sich bisher als resistent gegenüber dem
molekularen Clonen erwiesen haben, können vielleicht über die Amplifikation
einer Sequenz, die von bekannten Sequenzen flankiert wird, zugänglich gemacht
werden. Die Amplifikation längerer
Zielsequenzen erlaubt außerdem
eine größere Flexibilität in der
Wahl der Primer, um problematische Sequenzen zu vermeiden, so wie
in dem Betaglobin-Gensystem, das nachstehend beschrieben ist. Von
der Verwendung längerer
Matrizen verspricht man sich durch die Abdeckung eines größeren Bereiches
bei jedem Sequenzierungsschritt auch eine Beschleunigung der Genomsequenzierung.
Ausgehend von bekannten Sequenzen können Amplifikationen nun längere Introns
umspannen, und vollständigere
Gensequenzen können
in einem Schritt amplifiziert werden. Die lange PCR vervollständigt daher
die Technologie für
eine schnelle, größere Abschnitte
umfassende Sequenzierung. Auch eine auf der PCR basierende Charakterisierung
und Diagnose von homozygoten und heterozygoten Trägern von
einer Reihe von medizinisch bedeutsamen Insertionen und Deletionen
mit einer Länge
von mehr als 4 kb wird möglich.
-
Die
hier vorgestellten Ergebnisse zeigen besonders die vielfältigen Möglichkeiten
bei der Anwendung dieser Verfahren für die Charakterisierung clonierter
Sequenzen. Die nachstehend beschriebenen J- und cro- Gen-Primer,
CF1018 (SEQ ID NO:23) und CF1019 (SEQ ID NO:24) sollten für nahezu
alle Insertionen, die mit Lambda-basierenden Vektoren cloniert sind,
nützlich
sein, für
Amplifikationen sowohl von Plaques als auch von isolierter DNA.
Die PCR-Produkte werden vollständig
durch Restriktionsabbau analysiert und sollten für die Se quenzierung geeignet
sein. Cosmid-Insertionen können
gleichfalls von Kolonien amplifizierbar sein. Die lange PCR wird
das molekulare Clonieren erleichtern, weil bereits wenige Kopien
amplifiziert werden können, und
auch die Zusammenlagerung längerer
rekombinanter Konstrukte in der PCR-basierenden Mutagenese wird
erleichtert.
-
Die
im folgenden angeführten
Beispiele in der vorliegenden Erfindung dienen lediglich der Erläuterung und
sollen nicht den Umfang der Erfindung begrenzen.
-
Beispiel 1
-
Material und Verfahren
-
Im
folgenden werden bevorzugte Verfahrensweisen und Reagenzien für die PCR-Amplifikation von langen
Lambda-Phagen- und menschlichen Betaglobin-Genkomplex-Sequenzen
beschrieben. Die Ergebnisse der Amplifikationen mit den folgenden
Verfahren sind in den darauffolgenden Beispielen beschrieben.
-
Ziel-Nucleinsäure-Sequenzen
-
Zwei
Matrizen-Nucleinsäure-Sequenzen
wurden zur Herstellung der nachstehend beschriebenen Amplifikations-Primer
benutzt: die Sequenz des Lambda-Phagen-Genoms (GenBank Zulassungsnummer M17233)
und die Sequenz des menschlichen Betaglobin-Genkomplexes (GenBank
Zulassungsnummer J00179). Lambda-Phagen- und menschliche DNA wurden
für die
nachstehend beschriebenen Amplifikationen verwendet.
-
Lambda-DNA
(1 ng/μl)
wurde von Perkin Elmer, Norwalk, CT bezogen. Aliquots (~100 ng)
von Lambda-DNA wurden einmal aufgetaut und dann bei 4°C gelagert.
Vollständige
genomische DNA aus der menschlichen Plazenta wurde von Sigma Chemicals,
St. Louis, MO bezogen. Alle Verdünnungen
von Matrizen-DNA wurden mit 10 mM Tris·Cl (pH 8 bei 25°C), 0,1 mM
EDTA angesetzt.
-
Eine
Bibliothek von menschlichen genomischen Clonen in Lambda-FIX II
wurde von Stratagene, La Jolla, CA bezogen, und wie vom Hersteller
empfohlen, auf Luria Broth-Agarplatten mit Top-Agarose weitergezüchtet. Per
Zufall ausgewählte
Plaques wurden mit Hilfe von sili konisierten Pasteurpipetten entnommen,
in 30 μl
25 mM Tris·Cl
(pH 8,3), 10 mM MgCl2 gegeben und bei 4°C gelagert.
Mengen von 1 μl
wurden jeweils für die
PCR verwendet.
-
Vollständige genomische
DNA einer lymphoblastoiden Zelllinie (KAS011B) wurde isoliert mit
Hilfe von 0,1 mg/ml Proteinase K und 0,5% SDS in 10 mM Tris·Cl (pH
8), 150 mM NaCl und 10 mM EDTA, über
Nacht bei 50°C.
Es folgte eine Extraktion mit Tris-gesättigtem Phenol (pH 8), eine
Ethanol-Fällung
mit NaOAc; die Probe wurde mit RNase A versetzt, dann mit Phenol-Chloroform
extrahiert und gegen 10 mM Tris·Cl (pH 8), 1 mM EDTA dialysiert.
-
Primer
-
Es
wurde ein Satz von Primern hergestellt, um die PCR-Amplifikation
von Lambda-Genom-Zielsequenzen
in der Größenordnung
von 1,5 bis 42,2 kb durchführen
zu können.
Es wurden stromaufwärts
liegende Primer hergestellt, um sie mit jedem der stromabwärts liegenden
Primer einsetzen zu können.
Im Ergebnis wurden eine Reihe von Zielsequenzen erhalten, die in
der Länge
um 1 bis 3 Kilobasen zunahmen.
-
Jeder
Primer des Satzes wurde so hergestellt, dass er in etwa die gleiche
optimale Anlagerungstemperatur (~68°C) besaß. Es wurden Primer-Sequenzen
zwischen 20 und 23 Basenpaaren Länge
ausgewählt, so
dass der Hybrid-Doppelstrang zwischen Primer und Zielsequenz eine
Zusammenstellung von insgesamt 12 G-C-Paarungen und 8–11 A-T-Paarungen
aufwies. Optimale Anlagerungstemperaturen wurden mit Hilfe des „Tp"-Algorithmus von
Wu et al., 1991, DNA Cell Biol. 10:233–238, ermittelt.
-
Ein
weiteres Primer-Paar, die J- und cro-Gen-Primer, wurden hergestellt,
um eine Amplifikation nahezu aller Insertionen, die mit Lambda-basierenden
Vektoren cloniert sind, zu ermöglichen.
Dies gilt sowohl für Plaques
als auch für
isolierte DNA, wie in Tabelle 2 dargestellt, siehe nachstehend.
-
In ähnlicher
Weise wurden Primer für
die Amplifikation von bestimmten Regionen des menschlichen Betaglobin-Gen-Komplexes
hergestellt. Die Primer wurden so ausgelegt, dass ein fixierter
stromabwärts
liegender Primer mit einer Reihe von stromaufwärts liegenden Primern benutzt
werden konnte, um Zielsequenzen von 7,5–22 kb zu amplifizieren. Die
Primer amplifizieren eine Zielsequenz, die sich stromaufwärts über das Deltaglobin-Gen
hinaus in das zweite Intron des A-Gammaglobin-Gens hinein erstreckt.
-
Die
Nucleotidsequenzen der Primer, die in den folgenden Beispielen benutzt
werden, werden (5'-zu
3') in der Tabelle
2, siehe nachstehend, dargestellt. Die Schmelztemperaturen (Tm)
wurden im wesentlichen berechnet wie bei Wetmur, 1991, Crit.Rev.Biochem.Mol.
Biol. 26: 227–259
angegeben. Die Schmelztemperaturen wurden unter folgenden Annahmen
berechnet: zwei freie Enden, 3,5 μg/ml
(~0,5 μM)
Primer, 80 mM Na
+ und 1,5 mM Mg
2+.
Die berechneten Schmelztemperaturen lagen zwischen 63–70°C. Die Zugabe
von 10% Glycerin senkt die Temperatur T
m um
2,5°C. Die
Nucleotidsequenzen der Primer wurden wegen möglicher sekundärer Bindungsstellen
innerhalb der Matrizen-DNA-Sequenzen und auf Inter- und Intra-Primer-Sequenz-Komplementierung
ausgewertet. Verwendet wurde dabei die Oligo 4,0 Software (National
Biosciences, Plymouth, MN).
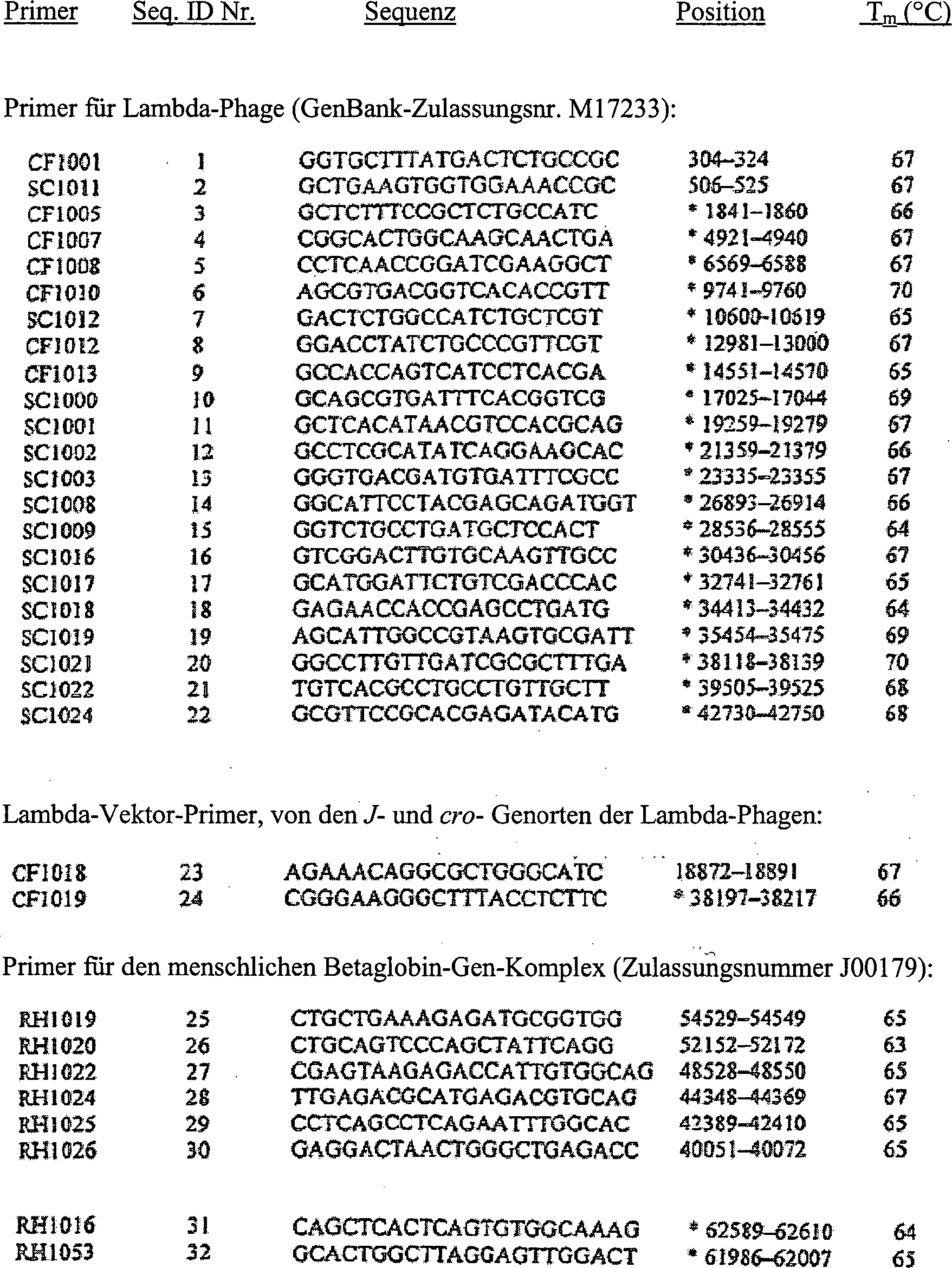
Tabelle
2 Primer
für die
Amplifikation
- *Stromabwärts liegende Primer komplementär zu den
aufgelisteten Positionsnummern
-
Primer
wurden mit dem Cyanoethoxyphosphoramidit-Verfahren (1μM Maßstab) auf
einem 394-DNA-Synthesizer (Applied Biosystems, Foster City, CA)
synthetisiert. Die Primer-Schutzgruppen
wurden entfernt und in 29% NH3/H2O vom Harz abgespalten, dann mit Sephadex
G25 (NAP-10-Säulen
von Pharmacia LKB, Piscataway, NJ) entsalzt. Die Ergebnisse jeder
Synthese wurden mit einer Polyacrylamid-Gelelektrophorese überprüft. Alle
Primer-Stammlösungen wurden
mit 10 mM Tris·Cl
(pH 8 bei 25°C),
0,1 mM EDTA angesetzt.
-
Thermostabile DNA-Polymerasen
-
Rekombinante
Tth-DNA-Polymerase (rTht) wurde von Perkin Elmer, Norwalk, CT bezogen.
Die Tli-DNA-Polymerase wird im U.S. Patent No. 5,210,036 beschrieben.
Die Tli-DNA-Polymerase
(VentR ® und DNA-Polymerase von
Pyrococcus-Art GB-D (Deep VentR ® wurden
von New England Biolabs, Beverly, MA bezogen. Die Tma-DNA-Polymerase
wird in der internationalen Patentveröffentlichung Nr. WO 92/03556
beschrieben und darin pTma12–3
genannt. Eine modifizierte DNA-Polymerase von Thermatoga maritima
ist bei Perkin Elmer, Norwalk, CT käuflich zu erwerben (UITmaTM).
-
Verdünnungen
(1/5 und 1/10) der VentR ®- und
Deep VentR ®-DNA-Polymerasen
werden vorzugsweise in Aufbewahrungspuffern nach den Angaben der
Hersteller hergestellt. In den folgenden Beispielen enthielten die
VentR ®-Verdünnungspuffer jedoch 1 mM EDTA
und 0,05% Tween 20 (Sigma Chemicals, St. Louis, MO) anstatt 0,1%
Triton X-100. Diese Modifizierung hatte keinen Einfluss auf die
Amplifikationsreaktionen. VentR ®-Polymerase-Lösungen wurden
wöchentlich
frisch angesetzt, Deep VentR -Polymerase wurde direkt vor dem Gebrauch
verdünnt.
Die Polymerase kann auch in dem Aufbewahrungspuffer der rTth-DNA-Polymerase
gelagert werden, der vom Hersteller mitgeliefert wird (100 mM KCl,
20 mM Tris-HCl, pH 8,0, 0,1 mM EDTA, 1 mM DTT, 0,5% Tween® 20,
50% (v/v) Glycerin).
-
Zusätzliche Puffer-Komponenten
-
Standard-Tth-Polymerase-Puffer
(5%(v/v) Glycerin, 10 mM Tris·Cl
(pH 8,3) 100 mM KCl, 0,75 mM EGTA, 0,05% Tween® 20)
für die
PCR wurde von Perkin Elmer, Norwalk, CT bezogen. Die Tricin-Puffer-Stammlösungen (Sigma
Chemicals, St. Louis, MO) mit 1,0 M wurden mit KOH auf ihren endgültigen pH-Wert
(bei 25°C)
eingestellt. Dimethylsulfoxid für
molekularbiologische Zwecke (DMSO) und Glycerin wurden von Sigma Chemicals,
St. Louis, MO beziehungsweise von J.T. Baker Chemicals, Phillipsburg,
NJ, bezogen. Kaliumacetat (KOAc) stammte ebenfalls von J.T. Baker
Chemicals. Der Beitrag von Glycerin aus dem Vorratspuffer für die Enzyme
(im allgemeinen 1%) wurde in keiner der Glycerin-Konzentrationen, die hier für die PCR-Puffer
angegeben wurden, berücksichtigt.
-
PCR-Methoden
-
Alle
Lambda-genomischen DNA-Amplifikationen wurden in einem GeneAmp® PCR
System 9600 Thermocycler unter Verwendung von MicroAmpTM-Gefäßen mit
Einzeldeckeln durchgeführt,
alles zu beziehen bei Perkin Elmer, Norwalk, CT. Reaktionsvolumina
waren entweder 50 oder 100 μl.
Die Konzentration von jeder dNTP betrug durchgehend 0,2 mM in allen
Reaktionen, die anderen Reaktionskomponenten wurden jedoch variiert,
wie im Text angegeben und in Tabelle 1 aufgelistet.
-
Zur
Minimierung der Amplifikation unspezifischer Sequenzen und der Bildung
von Primer-Dimeren wurden
manuelle „Heiß-Starts" durchgeführt, bei
denen das Mg2+ zurückgehalten wurde, bis die Proben
im Thermocycler für
etwa 90 Sekunden eine Temperatur von 75–80°C erreicht hatten. Das notwendige
Mg2+ wurde dann aus einer 25 mM Stammlösung (bei
Raumtemperatur) zugegeben. Nach der Zugabe von Mg2+ wurden die
Proben weitere 30–60
Sekunden inkubiert, insgesamt etwa 4–7 Minuten bei 75–80°C vor dem
ersten Denaturierungsschritt. Die Gesamtzeit schließt die Zeit
mit ein, die für
die Mg2+-Zugabe gebraucht wird, hängt also von
der Anzahl der Reaktionsgefäße ab. Eine
andere „Heiß-Start"-Vorgehensweise ist
in Beispiel 6 beschrieben.
-
Der
Thermocycler war für
ein Zwei-Stufen-Temperaturprofil programmiert. Jeder Amplifikations-Zyklus bestand
aus Denaturierung bei 94°C
für 10
Sekunden und anschließender
Anlagerungs-und Verlängerungsphase
bei 68°C
für 5–20 Minuten.
Der Denaturierungsschritt kann auch 15 Sekunden dauern. Für Anlagerungs-
und Verlängerungszeiten
von mehr als 12–14
Minuten wurde das Autoextensions-Programm des Thermocyclers benutzt,
um 15–20
Sekunden pro Zyklus hinzuzufügen,
bis zuletzt etwa 16–22
Minuten. Die Reaktionen erfolgten insgesamt in 25–40 Zyklen,
abhängig
von der Anzahl der Zielsequenz-Kopien zu Beginn, der Länge der
Zielsequenz und den Reaktionsbedingungen. Die meisten Versuchsansätze enthielten
einen ersten Inkubationsschritt bei 94°C für 10 Sekunden und einen letzten
Inkubationsschritt bei 72°C
für 10
Minuten.
-
Die
in den folgenden Beispielen beschriebenen Amplifikationen von menschlichen
Gen-Insertionen, die
in Lambda FIX II cloniert wurden, und von Regionen des menschlichen
Betaglobin-Genkomplexes wurden im wesentlichen so durchgeführt, wie
vorstehend beschrieben, jedoch mit den Modifikationen, die im folgenden genauer
beschrieben werden. Die spezifischen Bedingungen für die Amplifikation
von menschlichen genomischen Insertionen cloniert in Lambda-FIX
II aus Plaque-Suspensionen in 100 μl Reaktionsvolumina sind folgende:
25
mM Tricin (pH 8,7)
85 mM KOAc
12% (w/v) Glycerin
0,2
mM von jedem dNTP
0,4 μM
von jedem Primer
1,75 U Tth- Polymerase
0,02 U Tli-Polymerase
1,15
mM Mg(OAc)2
-
Es
wurde ein Heiß-Start
bei 80°C
mit einem Zwei-Stufen-Temperatur-Zyklus-Profil durchgeführt, wie vorstehend
beschrieben. Der Anlagerungs- und Verlängerungsschritt betrug anfangs
12 Minuten bei 68°C
und wurde um 15 Sekunden pro Zyklus verlängert, insgesamt über 32 Zyklen.
-
Die
genauen Bedingungen für
die Amplifikation einer Region des menschlichen Betaglobin-Genkomplexes von
37 ng der KAS011-DNA in 50 μl
Reaktionsvolumen waren folgende:
20 mM Tricin (pH 8,7)
85
mM KOAc
10% (w/v) Glycerin
2% (v/v) DMSO
0,2 mM von
jedem dNTP
0,2 μM
von jedem Primer
0,9 U Tth-Polymerase
0,02 U Tli-Polymerase
1,1
mM Mg(OAc)2
-
Es
wurde ein Heiß-Start
bei 78°C
mit einem Zwei-Stufen-Temperatur-Zyklus-Profil gewählt, wie
vorstehend beschrieben. Der Anlagerungs- und Verlängerungsschritt
betrug anfangs 12 Minuten bei 68°C über 12 Zyklen
und wurde dann um 15 Sekunden pro Zyklus über 24 Zyklen verlängert.
-
Eine
erhöhte
Ausbeute des amplifizierten Produktes kann durch die Zugabe von
bis zu 500 μg/ml nichtacetyliertem
BSA zur Amplifikationsreaktion erzielt werden.
-
Analyse der PCR-Produkte
-
Üblicherweise
werden 5–8 μl von jeder
PCR-Amplifikation mit Hilfe von standardisierten horizontalen Gels
untersucht. Sie bestehen aus 0,6% (w/v) SeaKem GTG-Agarose (FMC
BioProducts, Rockland, ME) in 1X TBE (89 mM Tris-Base, 89 mM Borsäure, 1 μM bis 2 mM
EDTA) oder 1X TAE (40 mM Tris-Acetat, 2 mM EDTA, pH 8–8,5) mit
0,5 μg/ml
Ethidiumbromid bei etwa 4–6
V/cm für
1,5–2
Stunden. Für
eine bessere Größenauflösung wurden
zwei Alternativen benutzt: Feld-Inversions-Gelelektrophorese und
0,3% Agarose-Gelelektrophorese.
-
Die
Feld-Inversions-Gelelektrophorese (FIGE) wurde in einem Hoefer-System
durchgeführt
(SuperSub Gel-Apparat, Switchback Pulse Controller und Netzteil,
alle von Hoefer, San Francisco, CA) mit einer Kühleinheit (2219 Multitemp II
von Pharmacia LKB). Zwischen 3 und 7 μl von jeder PCR-Amplifikation
wurden auf FIGE-Gelen aus 0,95% Agarose in 0,5x TBE (bei 1 μM EDTA) analysiert.
Die FIGE-Gele hatten einen Vorlauf von 15 Minuten bei 110 V, der
eigentliche Lauf ging über
22–25
Stunden bei 140–145
V und Pulszeiten von 0,65–1,95
oder 0,75–2
Sekunden (vorwärts:
rückwärts = 2,8:1
oder 3:1). Die Lauftemperaturen wurden auf 12–15°C geschätzt.
-
Alternativ
verwende man 2–5 μl auf 0,3%
Chromosomal Grade-Agarose (Bio-Rad, Richmond, CA) oder SeaKem GTG
oder Gold (FMC BioProducts, Rockland, ME) in 1X TAE. Das Gel wird
vor Entfernung des Kamms auf 4°C
gekühlt.
Nach Beladen mit 5–8 μl Probe erfolgt
der Lauf in 1XTAE mit 0,5% Ethidiumbromid bei 100 V für 2 Minuten,
dann entweder bei 1,5 V/cm für
6 Stunden oder 0,7 V/cm für
16 Stunden.
-
Die
Größe der amplifizierten
Produkte wurde durch einen Vergleich mit Molekulargewichtsmarkern
ermittelt, die in jedem Gel gleichzeitig mit der Probe mitliefen.
Die verwendeten Molekulargewichtsmarker waren Lambda/HindIII, entweder
von New England Biolabs oder Gibco BRL; Lambda/Mono Cut-Mix von
New England Biolabs und 1 kb-Leitern von Gibco BRL.
-
Für Restriktionsanalysen
wurden gleiche Mengen (10–16 μl) der PCR-Amplifikationsprodukte
von Lambda DNA-Amplifikationen vor der Elektrophorese mit BclI,
BssHII und MluI (New England Biolabs), oder mit BamHI, EcoRI und
HindIII (Gibco BRL) unter Verwendung der Hersteller-Puffer geschnitten.
Das Schneiden wurde innerhalb von 2,5–3 Stunden in 30–36 μl Reaktionsgemischen
durchgeführt.
Die Analyse der Proben erfolgte auf 0,6–8% Agarose-Gel. Gleiche Mengen
von Plaque-PCR-Proben (10–30 μl Aliquots)
wurden mit NotI (Stratagene) über
Nacht in 40 μl
Reaktionsgemischen geschnitten.
-
Beispiel 2
-
Amplifikation genomischer
Sequenzen des Lambda-Phagen
-
Die
Durchführung
der Amplifikationen erfolgte unter Verwendung von Zielsequenzen
mit einer hohen Anzahl von Ausgangskopien von Lambda-Phagen-DNA
in den Proben (107-108 Kopien
der Zielsequenz), wie im Beispiel 1 beschrieben, siehe oben. Zielsequenzen
von 1,5 bis 42,2 kb Länge
wurden innerhalb dieser ~50 kb-Sequenz (GenBank M17233) durch die
zahlreichen Paarungen der in Tabelle 2, siehe oben, aufgelisteten Primer
definiert.
-
Amplifizierte
Produkte wurden mit der Feldinversions-Gelelektrophorese (FIGE)
analysiert und mit einer Ethidiumbromid-Färbung sichtbar gemacht. Die
totalen Erträge
(pro 50 μl),
ermittelt durch den Vergleich mit einem Lambda/HindIII-Molekulargewichtsmarker,
lagen zwischen 0,7–1 μg eines 22,8
kb-Produktes und 0,2–0,3 μg eines 39
kb-Produktes. Eine 42,2 kb lange Zielsequenz, amplifiziert mit Hilfe
der Primer SC1011 (SEQ ID NO: 2) und SC1024 (SEQ ID NO: 22), wurde
mit geringerem Ertrag amplifiziert.
-
Beispiel 3
-
Lambda Clon-Amplifikation
von Plaques
-
Eine
wichtige Anwendung für
die Verfahren der vorliegenden Erfindung ist die Amplifikation von
Insertionen von Lambda-Klonen ohne vorherige arbeits- und zeitintensive
DNA-Isolierung.
Um die Nützlichkeit
der hier vorliegenden Verfahren für die Amplifikation solcher
Insertionen aufzeigen zu können,
wurden die Primer CF1018 (SEQ ID NO:23) und CF1019 (SEQ ID NO:24)
aus Sequenzen innerhalb der Lambda-Gene J und cro hergestellt (vgl.
Tabelle 2).
-
Die
Amplifikationen wurden wie in Beispiel 1 beschrieben durchgeführt, verwendet
wurden zufällig ausgewählte Plaques
aus der menschlichen Genbibliothek in Lambda FIX, in Beispiel 1
beschrieben. Die Amplifikationsprodukte wurden mit einer Gelelektrophorese
analysiert, nachdem sie mit NotI geschnitten worden waren, um die
Insertionen von den flankierenden Vektor-Sequenzen zu trennen. Die
Anwesenheit beider Vektorfragmente belegt, dass die gesamte Insertion
amplifiziert worden war.
-
Die
Größen der
amplifizierten Insertionen reichte von weniger als 10 kb bis größer als
20 kb. Schätzungen
des Herstellers besagen, dass Insertionen in der Größe zwischen
9 und 23 kb von diesem Lambda-Vektor transportiert werden können. Die
Größe der Insertionen
wurde relativ zu Molekulargewichtsmarkern in FIGE-Gelen gemessen.
-
Beispiel 4
-
Amplifikationen von Zielsequenzen
aus dem menschlichen Genom
-
Der
menschliche Betaglobin-Genkomplex wurde als ein Beispiel für Zielsequenzen
aus dem Genom ausgewählt,
die wahrscheinlich repetitive Sequenzen und homologe Abschnitte
an anderen Orten im Genom aufweisen. Die hergestellten Primer für den menschlichen
Betaglobin-Genkomplex sind in Tabelle 2 aufgelistet, siehe vorstehend.
Ein fixierter stromabwärts
liegender Primer wurde mit einer Reihe von stromaufwärts liegenden
Primern gepaart, die eine Region amplifizieren, die sich stromaufwärts über das
Deltaglobin-Gen hinweg in das zweite Intron eines A-Gammaglobin-Gens
hinein erstreckt. Es wurden Zielsequenzen von 13,5, 17,7, 19,6 und
22 kb aus 37 ng (104 Ausgangskopien) der
gesamten menschlichen Genom-DNA amplifiziert, wie in Beispiel 1
beschrieben. Je 12,5 μl
der amplifizierten Produkte wurden auf FIGE-Gele gegeben. Zum Vergleich
wurde ein Lambda/HindIII-Molekulargewichtsmarker
verwendet.
-
Zum
Vergleich wurden Zielsequenzen von 16,5, 18,8, 20,8 und 22,8 kb
von 0,05 pg (~103 Ausgangskopien) oder 0,5
pg (~104 Ausgangskopien) Lambda-Phagen-DNA
vor dem Hintergrund von ~3,7 ng bzw. 37 ng menschlicher genomischer
DNA aus der Plazenta unter gleichen Bedingungen amplifiziert. Amplifiziert
man eine Zielsequenz von einer niedrigen Anzahl von Ausgangskopien
zusätzlich
zu einer vorherigen Amplifikation mit einer hohen Anzahl von Ausgangskopien,
können
die Effekte einer Abnahme von Ausgangskopien getrennt werden von
den Effekten, die sich aus der Differenz der Zielsequenz ergeben.
-
Es
wurden Zielsequenzen vom Betaglobin-Genkomplex bis zu einer Länge von
22 kb amplifiziert. Die Betaglobin-Zielsequenzen wurden weniger
erfolgreich amplifiziert als Lambda-Sequenzen ähnlicher Länge, die auf einem Einzelkopie-Niveau
auf dem Hintergrund einer menschlichen Plazenta-DNA vorlagen, entweder in
der gleichen Gesamtkonzentration wie die Globin-Zielsequenz oder
in einer 10-fach niedrigeren Konzentration. Diese Unterschiede in
der Effizienz können
die relative Komplexität
der Sequenzen widerspiegeln, selbst wenn die Lambda-Zielsequenz
auch in einem Hintergrund menschlichen Genoms eingebettet war. Die
gestiegene Wahrscheinlichkeit, dass lange Zielsequenzen Abschnitte
enthalten, die ausreichend homolog sind, um als sekundäre Primer-Anlagerungsorte
zu wirken, und die Anwesenheit repetitiver Sequenzen im menschlichen
Genom erklären
möglicherweise,
warum Lambda-Zielsequenzen erfolgreicher amplifiziert werden können als
Betaglobin-Gen-Sequenzen vergleichbarer Länge.
-
Das
Problem der sekundären
Primer-Orte beeinflusste auch die Wahl von geeigneten Primern für die Amplifikation
der Betaglobin-Gen-Zielsequenzen. Es wurde der stromabwärts liegende
Primer RH1053 (SEQ ID NO: 32) ausgewählt, der 5' mit dem Betaglobin-Gen hybridisiert,
denn RH1016 (SEQ ID NO: 31), der innerhalb Exon 2 des Betaglobin-Gens
hybridisiert, hybridisiert bei Zielsequenzen, die länger als
14 kb sind, auch mit einem sekundären Anlagerungsort, was vielfältige Produkte
erzeugt. Der stromaufwärts
liegende Primer RH1020 (SEQ ID NO: 26) erzeugte vielfältige sekundäre Produkte,
wie auch der Gebrauch zweier anderer Primer (nicht aufgelistet)
innerhalb von 100 Basen von RH1020 (SEQ ID NO: 26). Alle drei liegen
innerhalb einer Alu-Wiederholungssequenz.
-
Ergebnisse
einer Amplifikation von Sequenzen bis zu 16 kb Länge vom menschlichen Neurofibromatose-1-Gen
deuten ebenso darauf hin, dass Verfahren zur Sicherstellung der
Primer-Spezifität von entscheidender
Wichtigkeit sind, um eine effiziente PCR-Amplifikation langer Zielsequenzen
durchführen
zu können.
-
Beispiel 5
-
DNA-Polymerase-Kombinationen
-
Um
die relative Effizienz verschiedener Kombinationen von DNA-Polymerasen
herauszufinden, wurden Amplifikationsreaktionen im wesentlichen
wie in Beispiel 1, siehe vorstehend, durchgeführt. Die verwendeten Primer
amplifizieren Zielsequenzen von 22,8, 26,4, 29,9 und 33,9 kb Länge. Die
miteinander verglichenen DNA-Polymerase-Kombinationen waren wie
folgt:
2,5 U rTth-DNA-Polymerase + 0,02 U VentR ®-DNA-Polymerase
2,5
U rTth-DNA-Polymerase + 0,06 U Deep VentR ®-DNA-Polymerase
3,15
U rTth-DNA-Polymerase + 0,5 U Tma-DNA-Polymerase
-
Alle
Reaktionen wurden in 50 μl
mit 107 Ausgangskopien von Lambda-DNA, 0,45 μM von jedem
Primer und 1,0–1,1
mM Mg(OAc)2 durchgeführt. Für die Amplifikationsreaktionen
galten die folgenden spezifischen Reaktionsbedingungen.
-
Reaktionen,
bei denen entweder rTth- und VentR ®-
oder rTth- und Deep VentR ®-DNA-Polymerasen verwendet
wurden, wurden in 20 mM Tricin (pH 8,7), 85 mM KOAc, 10% Glycerin
und 3% DMSO durchgeführt. Reaktionen,
bei denen rTth- und Tma-DNA-Polmerasen verwendet wurden, wurden
in 20 mM Tricin (pH 8,7), 85 mM KOAc, 10% Glycerin und 2,5% DMSO
durchgeführt.
-
Das
Temperaturzyklus-Profil war im wesentlichen wie in Beispiel 1, siehe
vorstehend, beschrieben. Für
die ersten 9 Zyklen wurde eine 13-minütige Verlängerungszeit gewählt. Die
Verlängerungszeit
wurde dann auf 13,5 Minuten ausgedehnt und bei jedem der folgenden
18 Zyklen um 20 Sekunden verlängert.
Aus jeder Reaktion wurden 7 μl
zusammen mit 150 ng des Lambda/HindIII-Molekulargewichtsmarker auf
ein Standard-Agarosegel gegeben.
-
Alle
Matrizen (bis zu 33,9 kb) wurden unter Benutzung von Kombinationen
von rTth-DNA-Polymerase mit
VentR ®-, Deep VentR – und Tma-DNA-Polymerase
amplifiziert. Die Kombination von 2,5 U rTth-DNA-Polymerase und
0,02 U VentR ®-DNA-Polymerase
amplifizierte alle Zielsequenzen mit größter Effizienz.
-
Beispiel 6
-
PCR-Amplifikations-Kit
-
Die
Reagenzien der Erfindung eignen sich für eine Verwendung in einem
Kit für
die Durchführung
der PCR-Amplifikation langer Zielsequenzen. Ein Kit enthält mindestens
ein DNA- Polymerase-Gemisch
wie hier beschrieben. Zusätzlich
können
Komponenten enthalten sein, die weitere für die Reaktionen verwendete
Reagenzien und Reaktionsbehälter,
wie nachstehend beschrieben, beinhalten.
-
Eine
bevorzugte DNA-Polymerase-Kombination, die sich für Zielsequenzen
sowohl mit einer hohen Anzahl von Ausgangskopien als auch mit einer
niedrigen Anzahl von Ausgangskopien eignet, besteht aus rTth- und
VentR -DNA-Polymerasen im Verhältnis
von 2 Einheiten (U) rTth-DNA-Polymerase zu 0,08 U VentR ®-DNA-Polymerase.
Wie nachstehend angeführt,
ist jedoch die bevorzugte Polymerase-Konzentration für die Amplifikation
einer hohen Anzahl von Ausgangskopien der Zielsequenz doppelt so
hoch wie die bevorzugte Konzentration für die Amplifikation von Zielsequenzen
mit einer niedrigen Anzahl von Ausgangskopien, das Verhältnis primärer und
sekundärer
Polymerasen zueinander bleibt aber gleich.
-
Ein
Reaktionspuffer, der in einen Kit aufgenommen werden kann, besteht
aus Tricin, KOAc, Glycerin und DMSO, etwa in den folgenden Konzentrationen:
25
mM Tricin (pH 8,7)
80 mM KOAc
10% (w/v) Glycerin
2,25%
(v/v) DMSO
-
Der
Ausdruck „etwa" bezieht sich auf
die Einstellung der Standard-Konzentration plus/minus einer 10%igen
Toleranz bei der Herstellung. Gewöhnlich kann der Reaktionspuffer
mit einer höheren
Konzentration gelagert werden, um dann vor dem Gebrauch verdünnt zu werden.
-
Die
Amplifikationen werden im wesentlichen unter VerwSETR der vorstehend
beschriebenen Kit-Komponenten durchgeführt, jedoch unter Verwendung
der im folgenden beschriebenen bevorzugten Reaktionsbedingungen.
Diese Reagenzien und Bedingungen sind sehr häufig benutzt worden und haben
sich als geeignet erwiesen, verlässliche
Amplifikationen langer Zielsequenzen zu ermöglichen.
-
Bevorzugte
Bedingungen für
eine Amplifikation von Zielsequenzen mit einer geringen Anzahl (2,0 × 104 Ausgangskopien) von Zielsequenzen (z.B.
aus dem menschlichen Genom) in 100 μl Reaktionsvolumen sind folgende:
25
mM Tricin (pH 8,7)
80 mM KOAc
10% (w/v) Glycerin
2,25%
(v/v) DMSO
0,2 mM von jedem dNTP
0,2 μM von jedem Primer
2 U
rTth-Polymerase
0,08 U VentR ®-Polymerase
1,1
mM Mg(OAc)2
-
Bevorzugte
Zyklus-Parameter für
die Amplifikation von Sequenzen mit geringer Anzahl von Ausgangskopien
(> 10 kb) sind folgende:
-
Bevorzugte
Bedingungen für
die Amplifikation von Zielsequenzen mit hoher Anzahl von Ausgangskopien
(2,0 × 107 Ausgangskopien), z.B. clonierte DNA, in
100 μl Reaktionsvolumen
sind folgende:
25 mM Tricin (pH 8,7)
80 mM KOAc
10%
(w/v) Glycerin
2,25% (v/v) DMSO
0,2 mM von jedem dNTP
0,4 μM von jedem
Primer
4 U rTth-Polymerase
0,16 U VentR ®-Polymerase
1,1
mM Mg(OAc)2
-
Bevorzugte
Zyklus-Parameter für
die Amplifikation von Zielsequenzen mit hoher Anzahl von Ausgangskopien
(> 10 kb) sind folgende:
-
Es
wird ein „Heiß-Start" durchgeführt, indem
die Reagenzien in den Reaktionsgefäßen mit Wachsperlen voneinander
getrennt werden (AmpliwaxTM PCR Gem 100,
entwickelt und hergestellt von Hoffmannn-La Roche und vertrieben
von Perkin Elmer, Norwalk, CT). Eine 40 μl Grundschicht aus den Reagenzien
Puffer (Tricin, KOAc, Glycerin und DMSO), Mg(OAc)2 und
den dNTPs wird ins Reaktionsgefäß gegeben. Über diese Grundschicht
wird eine Wachslage aus AmpliwaxTM PCR Gem
100 aufgebracht und das Ganze im Thermocycler zunächst 5 Minuten
bei 80°C,
dann 5 Minuten bei 25°C
inkubiert. Danach wird die oberste Schicht von 60 μl zugegeben,
enthaltend Puffer, die DNA-Polymerase-Kombination, die Primer und
die Zielsequenz-DNA.
-
Analysiert
werden die Proben, wie oben beschrieben, auf einem 0,6% Agarose-Gel
in 1X TAE und 0,5 μg/ml
EtBr für
1,5 Stunden bei 7 V/cm.