CN103841964A - 提供药物立即释放的抗破碎片剂 - Google Patents
提供药物立即释放的抗破碎片剂 Download PDFInfo
- Publication number
- CN103841964A CN103841964A CN201280038092.1A CN201280038092A CN103841964A CN 103841964 A CN103841964 A CN 103841964A CN 201280038092 A CN201280038092 A CN 201280038092A CN 103841964 A CN103841964 A CN 103841964A
- Authority
- CN
- China
- Prior art keywords
- tablet
- granule
- pharmacologically active
- chemical compounds
- host material
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images





Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
- A61K9/2077—Tablets comprising drug-containing microparticles in a substantial amount of supporting matrix; Multiparticulate tablets
- A61K9/2081—Tablets comprising drug-containing microparticles in a substantial amount of supporting matrix; Multiparticulate tablets with microcapsules or coated microparticles according to A61K9/50
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
- A61K31/137—Arylalkylamines, e.g. amphetamine, epinephrine, salbutamol, ephedrine or methadone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
- A61K31/138—Aryloxyalkylamines, e.g. propranolol, tamoxifen, phenoxybenzamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
Abstract
本发明涉及一种抗破碎片剂,这种片剂包含超过片剂总重量的三分之一的量的基质材料;和少于片剂总重量的三分之二的量的多个包衣颗粒;其中所述颗粒包含药理学活性化合物和生理学上可接受的聚合物,优选地为聚环氧烷;并形成基质材料中的非连续相;其优选地按照欧洲药典,在体外条件下提供药理学活性化合物的立即释放。
Description
发明领域
本发明涉及包含基质材料和多个包衣颗粒的抗破碎片剂(tamper-resistant tablets),所述包衣颗粒包含药理学活性化合物并形成基质材料中的非连续相,优选地在体外条件下提供药理学活性化合物的立即释放。
发明背景
大量的药理活性物质具有被滥用或误用的可能性,即它们可被用于产生与其打算的用途不一致的作用。因此,例如在控制严重至极其严重的疼痛方面呈现良好疗效的阿片样物质,经常被滥用以引起与沉醉相似的欣快状态。具体地讲,具有精神效应的活性物质因此被滥用。
为了能够滥用,相应的剂型比如片剂或胶囊剂被粉碎,例如被滥用者磨碎,优选地使用水性液体自如此得到的粉末提取活性物质,并在任选地通过棉絮或纤维素填絮过滤后,非肠道特别是静脉内给予生成的溶液。这种类型的剂量与对于滥用者期望的结果即刺激(kick)的口服滥用相比较,导致活性物质的甚至更快的扩散。如果鼻腔给予粉末状剂型即吸入,也可达到这种刺激或这些陶醉样欣快状态。
已经出现了为避免药物滥用的各种观念。
已提出以一种方式在剂型中加入厌恶剂(aversive agents)和/或拮抗剂,以使得当剂型被篡用时,它们仅产生其厌恶和/或拮抗作用。然而,这样的厌恶剂的存在大体上是不可取的,因此存在着提供足够的抗破碎而不依赖于厌恶剂和/或拮抗剂的需求。
防止滥用的另一个概念依赖于药用剂型的机械性能,特别是增加的断裂强度(抗破碎性)。这样的药用剂型的主要优点是通过常规手段碾碎,特别是粉碎,例如以研钵研磨或通过锤子破碎是不可能的,或者至少基本上受到阻碍。因此,防碍了潜在的滥用者通常可得到的剂型的粉化过程或者至少使得这种粉化过程更加麻烦,而粉化过程对滥用是必须的。
这样的药用剂型可用于避免其中含有的药理学活性化合物的药物滥用,因为它们不能通过常规手段被粉碎,因此不能以粉末状形式即鼻腔给予。这些药用剂型的机械性能,特别是高断裂强度使得它们抗破碎。在这样的抗破碎药用剂型的情况下,其可参照例如WO
2005/016313、WO 2005/016314、WO 2005/ 063214、WO
2005/102286、WO 2006/002883、WO 2006/002884、WO
2006/002886、WO 2006/082097、WO 2006/082099和WO2009/092601。
这些确保抗滥用的剂型以具有滥用可能性的活性物质的控制释放,优选地以延迟释放(retarded
release)为区别特征。然而,活性物质的快速释放对于众多治疗应用,例如使用具有滥用可能性的活性物质缓解疼痛是必要的。
WO
2010/140007公开了包含熔融挤出的未包衣颗粒的剂型,这种未包衣颗粒包含药物,其中所述熔融挤出的颗粒作为基质中的非连续相存在。所述剂型提供药物的延长释放。
WO
2008/107149公开了含有一种或多种具有滥用可能性的活性物质、至少一种合成或天然的聚合物和至少一种崩解剂的阻碍滥用的多颗粒剂型,所述片剂的单个颗粒具有至少500 N的断裂强度,和45分钟后至少75%的活性物质释放。示例性的胶囊剂提供药理学活性化合物的快速释放。
US
2010/0092553和US 2007/224129 A1公开了其组成和结构使得有可能避免误用的固体多颗粒口服药用形式。所述微粒具有确保药物的改良释放的极厚涂层,并同时赋予所包衣的微粒抗破碎性,以避免误用。
WO
2008/033523公开了一种可包含颗粒的药用组合物,这种颗粒可至少包含一种易被滥用的活性药用成分。这种颗粒含有醇溶性和醇不溶性的和至少部分水溶性的材料两者。两种材料在醇和水的存在下成为粒状。颗粒也可在颗粒上包含呈现破抗碎性的包衣。使用醇基溶剂实施颗粒上的材料沉积。
然而,胶囊剂的性能在每一方面,例如关于崩解时间、患者依从性(例如可吞咽性)和易于制造方面不能令人满意。此外,胶囊剂通常含有胶质,因此有引起牛海绵状脑病(BSE或TSE)的风险。就涉及抗破碎剂型而言,胶囊剂是不利的,因为它们一般地可易于打开,从而释放以粉末状或颗粒形式存在的成分而不需要任何机械撞击。如果在胶囊中含有不同类型的组分,例如除了不含药物的颗粒外还有含有药物的颗粒,潜在的滥用者也许能够在视觉上区分不同类型的完好的未破坏组分(例如依据其颜色、大小或其它宏观性质)而容许手工分离。
然而,这些抗破碎剂型的性质在每个方面都不能令人满意。需要具有破抗碎性和尽可能快地释放(立即释放)药理学活性化合物的抗破碎剂型,即应在约30-45分钟或更早时显示达到85%-100%的释放逐渐增加。所述剂型应有利地具有可易于口服的形状、大小和重量。当然,所述剂型也应易于以成本有效的方式制备。当试图篡用剂型以制备适合于通过静脉内给予滥用的制剂时,可通过注射器与剩余部分分开的剂型的液体部分应尽可能地少,例如应含有不多于20
wt.-%的起初在剂型中含有的药理学活性化合物。
然而,功能性包衣的微粒或颗粒的制备关于通过在有机喷涂程序中应用薄膜包衣过度生产的努力方面不能令人满意,这种喷涂程序需要防止蒸汽爆炸的广泛措施。从环境和毒理学的观点来看,有机溶剂的使用是更加不合需要的。此外,应用功能性薄膜包衣通常需要高度努力以确保功能性屏障的完整性,并因此为产生高制备成本的生产步骤。
依据本发明的一个目的是提供抗破碎的药用剂型,其提供药理学活性化合物的快速释放,并且其与先前技术的抗破碎药用剂型相比较具有优势。
该目的已经通过本专利权利要求书实现。
发明概述
本发明涉及优选地用于口服给予的抗破碎片剂,其包含
(i) 超过片剂总重量的三分之一的量的基质材料;和
(ii) 少于片剂总重量的三分之二的量的多个包衣颗粒;其中所述颗粒包含药理学活性化合物和生理学上可接受的聚合物,优选聚环氧烷;并形成基质材料中的非连续相;
其优选地按照欧洲药典,在体外条件下提供药理学活性化合物的立即释放。
已经令人惊讶地发现,抗破碎剂型的体外释放曲线可通过在基质材料中嵌入含有药理学活性化合物的颗粒,并增加基质材料与包衣颗粒的相对重量比来加速。
进一步地,已经令人惊讶地发现,任选地以预先压实的或预先成颗粒形式存在的基质材料的混合物,可与包衣颗粒混合,并随后被压实成片剂,这样的片剂依次呈现优良的,即加速的崩解时间和体外释放特性。
更进一步地,已经令人惊讶地发现,口服剂型可被设计在抗破碎性、崩解时间和药物释放、载药量、加工性能(尤其是成片性(tablettability))和患者依从性之间提供最佳的折中。
特别是,已经令人惊讶地发现,当提供具有包衣,优选地具有包含水溶性聚合物的包衣材料的颗粒时,崩解和药物释放可被加速。已经出乎意料地发现,所述包衣的溶解不另外分别阻碍崩解和药物释放,但是引起其显著的加速。
发明详述
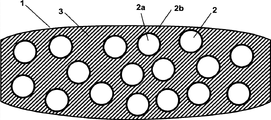
图1图解说明依据本发明的片剂的优选实施方案。
图2图解说明依据本发明的片剂的另一个优选的实施方案。
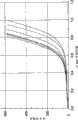
图3显示具有不同组成和颗粒大小的依据本发明的不同片剂的体外释放曲线。
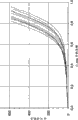
图4显示具有不同组成的依据本发明的不同片剂的体外释放曲线。
图5说明当经受断裂强度试验时,依据本发明的片剂中含有的颗粒的行为,特别是其可变形性。
图6说明当经受断裂强度试验时常规颗粒的行为。
图7显示通过测量常规颗粒的机械性能得到的距离-力-图表。
图8显示通过测量依据本发明的颗粒的机械性能得到的距离-力-图表。
图9显示通过测量依据本发明的颗粒的机械性能得到的距离-力-图表。
本文使用的术语“片剂”指包含药理学活性化合物,并且实际上给予患者或由患者摄取的药用实体。其可在其制备中被压制或模制,并且其可具有几乎任何大小、形状、重量和颜色。大多数片剂打算被整个吞咽,因此优选的依据本发明的片剂被设计用于口服给予。然而,备选地片剂可溶解于口中、咀嚼、或在吞咽之前溶于液体中,并且一些片剂可置于体腔中。因此,依据本发明的片剂可备选地适合于颊、舌、直肠或阴道给予。植入也是可能的。
依据本发明的片剂优选地可被视为MUPS制剂(多单元颗粒系统)。在一个优选的实施方案中,依据本发明的片剂为单片的。在另一个优选的实施方案中,依据本发明的片剂不为单片的。在这方面,单片的优选地意指片剂由没有连接或接缝的材料形成或组成,或者由单一的单元组成或构成。
优选地,依据本发明的片剂含有其与胶囊剂具有相对高密度相比较的致密压实单元中的所有成分。
依据本发明的片剂包含具有不同形态学和性质的亚单元,即含有药物的颗粒和基质材料,其中包衣颗粒形成基质材料中的非连续相。包衣颗粒一般地具有不同于基质材料的机械性能的机械性能。优选地,包衣颗粒具有比基质材料更高的机械强度。依据本发明的片剂中的包衣颗粒可通过常规手段例如固态核磁共振光谱学、光栅电子显微镜、太赫光谱等目测。
依据本发明的片剂的一个优点是相同的颗粒可与不同量的基质材料混合,从而产生不同强度的片剂。
依据本发明的片剂优选地具有在0.01-1.5 g范围内,更优选地在0.05-1.2
g范围内,仍然更优选地在0.1 g-1.0 g范围内,还更优选地在0.2 g-0.9 g范围内,和最优选地在0.3
g-0.8 g范围内的总重量。在一个优选的实施方案中,片剂总重量在500±450 mg,更优选地在500±300 mg,仍然更优选地在500±200 mg,还更优选地在500±150 mg,最优选地在500±100 mg,并且特别是在500±50 mg范围内。
已经令人惊讶地发现,为片剂总大小的函数的片剂总重量可被优化,以在抗破碎性、崩解时间和药物释放、载药量、加工性能(尤其是成片性(tablettability))和患者依从性之间提供最好的折中。
在一个优选的实施方案中,依据本发明的片剂为圆形片剂。该实施方案的片剂优选地具有在约1 mm-约30 mm,特别是在约2 mm-约25 mm范围内,更具体地讲在约5 mm-约23 mm,甚至更具体地讲在约7 mm-约13 mm范围内的直径;和在约1.0 mm-约12 mm,特别是在约2.0 mm-约10 mm,甚至更具体地讲是在3.0 mm-约9.0 mm,甚至更具体地讲为在约4.0 mm-约8.0 mm范围内的厚度。
在另一个优选的实施方案中,依据本发明的片剂为椭圆形片剂。该实施方案的片剂优选地具有约1 mm-约30 mm,特别是在约2 mm-约25 mm,更具体地讲在约5 mm-约23 mm,甚至更具体地讲在约7 mm-约20 mm范围内的纵向延伸(纵向延伸);在约1 mm-约30 mm范围内,特别是在约2 mm-约25 mm,更具体地讲在约5 mm-约23 mm,甚至更具体地讲在约7 mm-约13 mm范围内的宽度;和在约1.0 mm-约12 mm范围内,特别是在约2.0 mm-约10 mm,甚至更具体地讲在3.0 mm-约9.0 mm,甚至进一步具体地讲在约4.0 mm-约8.0 mm范围内的厚度。
依据本发明的片剂可任选地部分或完全地提供常规包衣。依据本发明的片剂优选地用常规薄膜包衣组合物进行薄膜包衣。合适的包衣材料可例如以商标Opadry®
和Eudragit®经市售得到。
合适材料的实例包括纤维素酯和纤维素醚,例如甲基纤维素(MC)、羟丙基甲基纤维素(HPMC)、羟丙基纤维素(HPC)、羟乙基纤维素(HEC)、羧甲基纤维素钠(Na-CMC);聚(甲基)丙烯酸酯,例如甲基丙烯酸氨基烷基酯共聚物、甲基丙烯酸甲基丙烯酸甲酯共聚物、甲基丙烯酸甲基丙烯酸甲酯共聚物;乙烯基聚合物,例如聚乙烯吡咯烷酮、聚乙烯醇、聚醋酸乙烯酯;以及天然的成膜剂。
在一个特别优选的实施方案中,包衣为水溶性的。在一个优选的实施方案中,包衣为基于聚乙烯醇,例如聚乙烯醇-部分水解的,并可另外含有聚乙二醇,例如聚乙二醇3350和/或颜料。在另一个优选的实施方案中,包衣为基于羟丙基甲基纤维素的,优选地为具有3-15 mPas粘度的羟丙甲纤维素(hypromellose)型2910。
包衣可耐胃液,并按释放环境的pH值的函数溶解。通过这种包衣,可以确保依据本发明的片剂通过胃而不溶解,并且活性化合物仅在肠中释放。耐胃液的包衣优选地在5-7.5之间的pH值下溶解。
包衣也可用于例如改善片剂的美学印象和/或味道和它们可吞咽的容易程度。将依据本发明的片剂包衣也可用于其它目的,例如改善稳定性和贮藏寿命。合适的包衣制剂包含成膜聚合物,例如聚乙烯醇或羟丙基甲基纤维素,例如羟丙甲纤维素;塑化剂例如二醇,例如丙二醇或聚乙二醇;遮光剂,例如二氧化钛;和薄膜增滑剂,例如滑石。合适的包衣溶剂为水以及有机溶剂。有机溶剂的实例为醇,例如乙醇或异丙醇;酮,例如丙酮或者卤代烃,例如二氯甲烷。依据发明的包衣片剂优选地通过首先制备片芯,并随后使用常规技术,例如在包衣锅中包衣来把所述片芯包衣而制备。
本文使用的术语“抗破碎剂(tamper-resistant)”指可抵抗通过常规手段例如以研钵研磨或通过锤子破碎转化为适合于误用或滥用,特别是适合于鼻腔和/或静脉内给予的形式的片剂。在这方面,片剂本身可通过常规手段破碎。然而,在依据本发明的片剂中含有的包衣颗粒呈现机械性能,使得它们不能通过常规手段进行任何进一步的粉碎。因为包衣颗粒具有宏观尺寸和含有药理学活性化合物,它们不能经鼻腔给予,从而使得片剂能抗破碎。优选地,当试图篡用(tamper)剂型以制备适合于通过静脉内给予滥用的制剂时,可通过注射器从剩余部分分离的制剂的液体部分尽可能地少,优选地其含有不多于20
wt.-%,更优选地不多于15 wt.-%,还更优选地不多于10
wt.-%,和最优选地不多于5 wt.-%的最初含有的药理学活性化合物。优选地,这种性质通过以下步骤测试:(i) 把或者完整的或者已通过两个勺子手动粉碎的片剂分配到5 ml的纯净水中,(ii) 把液体加热至高达其沸点,(iii) 使液体在覆盖的容器中沸腾5分钟而不加入其它的纯净水,(iv) 把热的液体吸到注射器中(配备有卷烟过滤嘴的针21G),(v) 测定在注射器内的液体中含有的药理学活性化合物的量。
进一步地,当试图通过锤子或研钵破裂片剂时,包衣颗粒也可能依环境而定趋于相互粘附,从而分别形成聚集物和附聚物,其大小大于未处理的包衣颗粒。
依据本发明的片剂可被给予的受试者不受具体限制。优选地,受试者为动物,更优选地为人类。
在依据本发明的片剂中,包衣颗粒被掺入到基质材料中。从宏观的角度来看,基质材料优选地形成其中包衣颗粒作为非连续相嵌入的连续相。
优选地,基质材料为均匀一致的物质,优选地为固体组分的均匀混合物,其中嵌入了包衣颗粒,从而使包衣颗粒彼此在空间上分开。尽管可能包衣颗粒的表面相互接触或至少非常密切地接近,多个包衣颗粒优选地不能视为片剂内的单一的连续一致的物质。
换句话说,依据本发明的片剂包含作为第一类型的体积要素的包衣颗粒,其中优选均匀地含有药理学活性化合物和生理学上可接受的聚合物,优选聚环氧烷;和作为不同于形成包衣颗粒的材料的第二类型的体积要素的基质材料,优选地既不含有药理学活性化合物也不含有生理学上可接受的聚合物,优选地没有聚环氧烷,但是任选地为其分子量不同于聚环氧乙烷的聚乙二醇。
依据本发明的片剂中的基质材料的目的是确保快速崩解和药理学活性化合物自崩解的片剂,即自包衣颗粒的随后的释放。因此,基质材料优选地不含有可能分别对崩解和药物释放具有阻滞效应的任何赋形剂。因此,基质材料优选地不含有一般用作延长释放制剂中的基质材料的任何聚合物。
图1图解说明依据本发明的片剂的优选实施方案。片剂(1)含有具有片芯(2a)和包衣(2b),形成基质材料(3)中的非连续相的多个包衣颗粒(2),基质材料依次形成连续相。
依据本发明的抗破碎片剂包含超过片剂总重量的三分之一的量的基质材料。
已经令人惊讶地发现,片剂中基质材料的含量可被优化,以在抗破碎性、崩解时间和药物释放、载药量、加工性能(尤其是成片性)和患者依从性之间提供最好的折中。
优选地,基质材料的含量为基于片剂总重量的至少35 wt.-%、至少37.5
wt.-%或至少40 wt.-%,更优选地为至少42.5
wt.-%、至少45 wt.-%、至少47.5
wt.-%或至少50 wt.-%,仍然更优选地为至少52.5
wt.-%、至少55 wt.-%、至少57.5
wt.-%或至少60 wt.-%,还更优选地为至少62.5
wt.-%、至少65 wt.-%、至少67.5
wt.-%或至少60 wt.-%,最优选地为至少72.5
wt.-%、至少75 wt.-%、至少77.5
wt.-%或至少70 wt.-%,并且特别是至少82.5
wt.-%、至少85 wt.-%、至少87.5
wt.-%或至少90 wt.-%。
优选地,基质材料的含量为基于片剂总重量的至多90 wt.-%、至多87.5
wt.-%、至多85 wt.-%或至多82.5
wt.-%,更优选地为至多80 wt.-%、至多77.5
wt.-%、至多75 wt.-%或至多72.5
wt.-%,仍然更优选地为至多70 wt.-%、至多67.5
wt.-%、至多65 wt.-%或至多62.5
wt.-%,还更优选地为至多60 wt.-%、至多57.5
wt.-%、至多55 wt.-%或至多52.5
wt.-%,最优选地为至多50 wt.-%、至多47.5
wt.-%、至多45 wt.-%或至多42.5
wt.-%,并且特别是至多40 wt.-%、至多37.5
wt.-%或至多35 wt.-%。
在一个优选的实施方案中,基质材料的含量基于片剂的总重量计在40±5
wt.-%,更优选地在40±2.5
wt.-%的范围内。在另一个优选的实施方案中,基质材料的含量基于片剂的总重量计在45±10 wt.-%,更优选地在45±7.5 wt.-%,仍然更优选地在45±5 wt.-%,和最优选地在45±2.5 wt.-%的范围内。在另一个更优选的实施方案中,基质材料的含量基于片剂的总重量计在50±10 wt.-%,更优选地在50±7.5 wt.-%,仍然更优选地在50±5 wt.-%,和最优选地在50±2.5 wt.-%的范围内。在还有的另一个优选的实施方案中,基质材料的含量基于片剂的总重量计在55±10 wt.-%,更优选地在55±7.5 wt.-%,还更优选地在55±5 wt.-%,和最优选地在55±2.5 wt.-%的范围内。
优选地,基质材料为混合物,优选地为至少两种不同组分,更优选地为至少三种不同组分的混合物。在一个优选的实施方案中,基质材料的所有组分均匀分布于由基质材料形成的连续相中。
在一个优选的实施方案中,基质材料的所有组分的混合物作为粉末即非预先压实的形式掺和和使用,随后与含有药理学活性化合物和聚环氧烷的颗粒混合,并然后压制成片剂。当适当调整压片机时,可得到按照欧洲药典
2.9.40“剂量单位的均匀性(Uniformity of Dosage Units)”(UDU)具有约5-6之间的接受值的片剂。应在最大程度上避免振动(例如通过使料斗和压片机分开),并且设备部件的空隙应尽可能地小。例如,在加上26个工位的旋转式压片机IMA
S250上,以下参数是合适的:圆形凸模直径10 mm;曲率半径8 mm,没有凹陷;填充曲线(fill curve) 13
mm;片剂重量500 mg,速率:13700-13800片每小时;预压制力4.7 kN;主压制力6.7 kN和8.7 kN;填充深度14.5 mm和15 mm; 片块高度(预压制):3.5 mm;片块高度(主压制):3.3 mm和3.1 mm;进料器的转速(Filomat):40 rmp。
在另一个优选的实施方案中,基质材料也以颗粒形式提供,即在制备依据本发明的片剂的过程中,基质材料的组分优选地被加工成颗粒,随后与含有药理学活性化合物和生理学上可接受的聚合物(优选聚环氧烷)的包衣颗粒混合,然后压制成片剂。
优选地,基质材料的颗粒的平均大小在包衣颗粒的平均大小的±60%,更优选地在±50%,仍然更优选地在±40%,还更优选地在±30%,最优选地在±20%,并且特别是在±10%的范围内,所述包衣颗粒含有药理学活性化合物和生理学上可接受的聚合物(优选聚环氧烷)。
已经令人惊讶地发现,当这种方式进行时,在掺和包衣颗粒与以颗粒形式存在的基质材料时的分离现象可被减小或者甚至完全抑制,从而基本上改善依据本发明的片剂的含量均匀性。
特别令人惊讶的是,当打算混合和压制成片剂的颗粒越大,一般地越难以满足含量均匀性要求。与常规片剂相比较,依据本发明的片剂自基质材料的比较大的包衣颗粒和任选地也自比较大的预压实颗粒制备。优选地,就依据本发明的片剂的含量均匀性而言,AV (合格值)为至多15,更优选地为至多14,仍然更优选地为至多13,还更优选地为至多12,甚至更优选地为至多11,最优选地为至多10和特别是为至多9。测定AV的方法为技术人员已知的。优选地,AV按照欧洲药典进行测定。
依据本发明的片剂的这种优选实施方案在图2中图解说明。片剂(1)含有具有片芯(2a)和包衣(2b)的多个包衣颗粒(2),其形成基质材料(3)中的非连续相,基质材料依次形成连续相并且也以颗粒形式提供,各个颗粒在边界(4)相互紧密接触。因为基质材料的颗粒一般具有低于包衣颗粒(2)的机械强度,基质材料的颗粒在通过压制制备片剂的过程中为变形的。
基质材料的颗粒大部分也可任选地用依据本发明的包衣颗粒的相同包衣材料或用另一种包衣材料进行包衣。然而,优选地,基质材料的颗粒为未包衣的。
基质材料的颗粒可通过用于自粉末混合物制备聚集物和附聚物的常规方法例如制粒和压实进行制备。
在一个优选的实施方案中,基质材料的所有组分的混合物被掺混和压实,从而产生预先压实的基质材料。
用于制备这种预先压实的基质材料的合适方法为技术人员已知的。优选地,预先压实通过干法制粒,优选地通过击压(slugging)或碾压进行。当这种方式进行时,一般将调整工艺参数以获得期望的性能(参见以下)。典型的工艺参数为压实力(优选地调整在2-12 kN的范围内)、辊位移(优选地调整在2-5 mm的范围内)和颗粒筛(优选地调整在1.0-2.0 mm的范围内)。预先压实的材料的期望性能主要包括粒度和细粒含量。密度也可能起作用。粒度优选地在颗粒大小的范围内(对于具有0.8 x 0.8 mm尺寸的颗粒优选地为至少60%
> 700 µm)。细粒(即具有小于600
µm大小的颗粒)的含量优选地为至多40%,更优选地为至多30%,最优选地为至多20%。所述工艺参数对所述期望的性能的影响可易于由技术人员通过常规试验来测定。
在另一个优选的实施方案中,基质材料的所有组分的混合物进行干法制粒,从而产生粒状基质材料。在还有的另一个优选的实施方案中,基质材料的所有组分的混合物通过非水性溶剂例如乙醇进行湿法制粒,从而产生另一种粒状基质材料。然而,优选地避免水性制粒,因为这通常对片剂的崩解具有有害的影响。在还有的另一个优选的实施方案中,基质材料的所有组分的混合物例如通过挤出机、可加热的高剪切混合器或制粒机进行熔体制粒(melt
granulated)。
如以上已经提及的那样,依据本发明的片剂中的基质材料应确保快速崩解和药理学活性化合物自崩解的片剂即自包衣颗粒的随后释放。因此,基质材料优选地不含有可能分别对崩解和药物释放具有阻滞效应的任何赋形剂。进一步地,基质材料优选地不含有任何药理学活性化合物。
优选地,基质材料包含崩解剂。合适的崩解剂为技术人员已知的,并且优选地选自交联羧甲基纤维素钠(Na-CMC) (例如交联羧甲基纤维素(Crosscarmellose),Ac-Di-Sol®)、交联酪蛋白(例如Esma-Spreng®)、得自大豆的多糖混合物(例如Emcosoy®)、预处理的玉米淀粉(例如Amijel®)、海藻酸钠、聚乙烯吡咯烷酮(PVP) (例如
Kollidone®、Polyplasdone®、Polydone®)、交联聚乙烯吡咯烷酮(PVP
CI) (例如Polyplasdone® XL)、淀粉和预处理的淀粉例如羧甲基淀粉钠(例如Explotab®、Prejel®、Primotab®
ET、Starch® 1500、Ulmatryl®)。交联的聚合物为特别优选的崩解剂,尤其是交联羧甲基纤维素钠(Na-CMC)或交联聚乙烯吡咯烷酮(PVP CI)。
优选地,崩解剂包含在依据本发明的片剂的基质材料中而不是在包衣颗粒中。
在一个优选的实施方案中,基质材料中的崩解剂的含量在基于基质材料总重量的5±4 wt.-%,更优选地在5±3 wt.-%,仍然更优选地在5±2.5 wt.-%,还更优选地在5±2 wt.-%,最优选地在5±1.5 wt.-%,并且特别是在5±1 wt.-%的范围内。在另一个优选的实施方案中,基质材料中的崩解剂的含量在基于基质材料总重量的7.5±4 wt.-%,更优选地在7.5±3 wt.-%,仍然更优选地在7.5±2.5 wt.-%,还更优选地在7.5±2 wt.-%,最优选地在7.5±1.5 wt.-%,并且特别是在7.5±1 wt.-%的范围内。在还有的另一个优选的实施方案中,基质材料中的崩解剂的含量在基于基质材料总重量的10±4 wt.-%,更优选地在10±3 wt.-%,仍然更优选地在10±2.5 wt.-%,还更优选地在10±2 wt.-%,最优选地在10±1.5 wt.-%,并且特别是在10±1 wt.-%的范围内。在另一个优选的实施方案中,基质材料中的崩解剂的含量在基于基质材料总重量的12.5±4 wt.-%,更优选地在12.5±3 wt.-%,仍然更优选地在12.5±2.5 wt.-%,还更优选地在12.5±2 wt.-%,最优选地在12.5±1.5 wt.-%,并且特别是在12.5±1 wt.-%的范围内。
在一个优选的实施方案中,片剂中的崩解剂的含量在基于片剂总重量的2±1.8
wt.-%,更优选地在2±1.5
wt.-%,仍然更优选地在2±1.3
wt.-%,还更优选地在2±1.0
wt.-%,最优选地在2±0.8
wt.-%,并且特别是在2±0.5
wt.-%的范围内。在另一个优选的实施方案中,片剂中的崩解剂的含量在基于片剂总重量的4±1.8 wt.-%,更优选地在4±1.5 wt.-%,仍然更优选地在4±1.3 wt.-%,还更优选地在4±1.0 wt.-%,最优选地在4±0.8 wt.-%,并且特别是在4±0.5 wt.-%的范围内。在还有的另一个优选的实施方案中,片剂中的崩解剂的含量在基于片剂总重量的6±1.8 wt.-%,更优选地在6±1.5 wt.-%,仍然更优选地在6±1.3 wt.-%,还更优选地在6±1.0 wt.-%,最优选地在6±0.8 wt.-%,并且特别是在6±0.5 wt.-%的范围内。在另一个优选的实施方案中,片剂中的崩解剂的含量基于片剂总重量的8±1.8 wt.-%,更优选地在8±1.5 wt.-%,仍然更优选地在8±1.3 wt.-%,还更优选地在8±1.0 wt.-%,最优选地在8±0.8 wt.-%,并且特别是在8±0.5 wt.-%的范围内。
优选地,基质材料包含与一种或多种水不溶性药用赋形剂,优选地为填充剂/粘合剂和/或润滑剂组合的崩解剂。
优选地,基质材料包含填充剂或粘合剂。因为许多填充剂可视为粘合剂并且反之亦然,为了说明书的目的,“填充剂/粘合剂”指适合作为填充剂、粘合剂或或两者兼而有之的任何赋形剂。因此,基质材料优选地包含填充剂/粘合剂。
优选的填充剂(=填充剂/粘合剂)选自二氧化硅(e.g. Aerosil®)、微晶纤维素(例如Avicel®、Elcema®、Emocel®、ExCel®、Vitacell®)、纤维素醚(例如Natrosol®、Klucel®、Methocel®、Blanose®、Pharmacoat®、Viscontran®)、甘露醇、糊精、右旋糖、磷酸氢钙(例如Emcompress®)、麦芽糊精(例如Emdex®)、乳糖(例如Fast-Flow Lactose®、Ludipress®、Tablettose®、Zeparox®)、聚乙烯吡咯烷酮(PVP)
(例如Kollidone®、Polyplasdone®、Polydone®)、蔗糖(例如Nu-Tab®、Sugar Tab®)、镁盐(例如MgCO3、MgO、MgSiO3)、淀粉和预处理的淀粉(例如Prejel®、Primotab®
ET、Starch® 1500)。优选的粘合剂选自海藻酸盐、壳聚糖;及以上提及的任何一种填充剂(=填充剂/粘合剂)。
一些填充剂/粘合剂也可用于其它目的。已知例如二氧化硅作为助流剂呈现优良的功能。因此,优选地,基质材料包含助流剂例如二氧化硅。
在一个优选的实施方案中,基质材料中的填充剂/粘合剂或填充剂/粘合剂的混合物的含量基于基质材料的总重量计在50±25
wt.-%,更优选地在50±20
wt.-%,仍然更优选地在50±15
wt.-%,还更优选地在50±10
wt.-%,最优选地在50±7.5
wt.-%,并且特别是在50±5
wt.-%的范围内。在另一个优选的实施方案中,基质材料中的填充剂/粘合剂或填充剂/粘合剂的混合物的含量基于基质材料的总重量计在65±25 wt.-%,更优选地在65±20 wt.-%,仍然更优选地在65±15 wt.-%,还更优选地在65±10 wt.-%,最优选地在65±7.5 wt.-%,并且特别是在65±5 wt.-%的范围内。在还有的另一个优选的实施方案中,基质材料中的填充剂/粘合剂或填充剂/粘合剂的混合物的含量基于基质材料的总重量计在80±19 wt.-%,更优选地在80±17.5 wt.-%,仍然更优选地在80±15 wt.-%,还更优选地在80±10 wt.-%,最优选地在80±7.5 wt.-%,并且特别是在80±5 wt.-%的范围内。在另一个优选的实施方案中,基质材料中的填充剂/粘合剂或填充剂/粘合剂的混合物的含量基于基质材料的总重量计在90±9 wt.-%,更优选地在90±8 wt.-%,仍然更优选地在90±7 wt.-%,还更优选地在90±6 wt.-%,最优选地在90±5 wt.-%,并且特别是在90±4 wt.-%的范围内。
在一个优选的实施方案中,片剂中的填充剂/粘合剂或填充剂/粘合剂的混合物的含量基于片剂的总重量计在25±24
wt.-%,更优选地在25±20
wt.-%,仍然更优选地在25±16
wt.-%,还更优选地在25±12
wt.-%,最优选地在25±8
wt.-%,并且特别是在25±4
wt.-%的范围内。在另一个优选的实施方案中,片剂中的填充剂/粘合剂或填充剂/粘合剂的混合物的含量基于片剂的总重量计在30±29 wt.-%,更优选地在30±25 wt.-%,仍然更优选地在30±20 wt.-%,还更优选地在30±15 wt.-%,最优选地在30±10 wt.-%,并且特别是在30±5 wt.-%的范围内。在还有的另一个优选的实施方案中,片剂中的填充剂/粘合剂或填充剂/粘合剂的混合物的含量基于片剂的总重量计在35±34 wt.-%,更优选地在35±28 wt.-%,仍然更优选地在35±22 wt.-%,还更优选地在35±16 wt.-%,最优选地在35±10 wt.-%,并且特别是在35±4 wt.-%的范围内。在另一个优选的实施方案中,片剂中的填充剂/粘合剂或填充剂/粘合剂的混合物的含量基于片剂的总重量计在40±39 wt.-%,更优选地在40±32 wt.-%,仍然更优选地在40±25 wt.-%,还更优选地在40±18 wt.-%,最优选地在40±11 wt.-%,并且特别是在40±4 wt.-%的范围内。
优选地,填充剂/粘合剂包含在依据本发明的片剂的基质材料中而不是在包衣颗粒中。
在一个优选的实施方案中,一部分(例如总片剂质量的10%)基质在包衣颗粒上形成颗粒(优选地通过非水性湿法制粒,例如用异丙醇(isopropylic
alcohol)),并把剩余的基质材料在压制/加工成片剂之前加入到如此形成颗粒的包衣颗粒中并进行掺混。因此,按照该实施方案,颗粒用依次给一部分基质材料加外涂层的包衣料进行包衣,而基质材料的剩余部分优选地以非粒状形式使用。
优选地,基质材料包含优选地选自以下的稀释剂或润滑剂:硬脂酸钙、硬脂酸镁、甘油单山嵛酸酯(例如Compritol®)、Myvatex®、Precirol®、Precirol®
Ato5、硬脂酰醇富马酸钠(例如Pruv®)和滑石。硬脂酸镁为特别优选的。优选地,基质材料中的润滑剂的含量基于基质材料的总重量和基于片剂的总重量为至多10.0
wt.-%,更优选地为至多7.5 wt.-%,仍然更优选地为至多5.0
wt.-%,还更优选地为至多2.0 wt.-%,甚至更优选地为至多1.0
wt.-%,并且最优选地为至多0.5 wt.-%。
在特别优选的实施方案中,基质材料包含崩解剂、填充剂/粘合剂和润滑剂的组合。
基质材料的崩解剂、填充剂/粘合剂和润滑剂相对于基质材料的总重量的特别优选的含量,作为实施方案A1-A6在此概述在下表中:
| wt.-% | A1 | A2 | A3 | A4 | A5 | A6 |
| 崩解剂 | 11±10 | 11±7.5 | 11±5.0 | 11±3.5 | 11±2.5 | 11±1.5 |
| 填充剂/粘合剂 | 88±12 | 88±10 | 88±8 | 88±6 | 88±4 | 88±2.5 |
| 润滑剂 | 0.30±0.28 | 0.30±0.26 | 0.30±0.24 | 0.30±0.22 | 0.30±0.20 | 0.30±0.15 |
其中崩解剂优选地为交联羧甲基纤维素钠(Na-CMC)或交联聚乙烯吡咯烷酮(PVP CI);填充剂/粘合剂优选地为微晶纤维素或微晶纤维素与胶态二氧化硅的组合;和润滑剂优选地为硬脂酸镁。
依据本发明的片剂的基质材料可另外含有本领域常规的其它赋形剂,例如稀释剂、粘合剂、制粒助剂、着色剂、矫味剂、造孔剂、表面活性剂、助流剂、湿法制粒剂和崩解剂。技术人员将易于能够确定这些赋形剂中每一种的合适量。
优选的造孔剂包括,但不限于葡萄糖、果糖、甘露醇、甘露糖、半乳糖、山梨醇、支链淀粉、右旋糖酐、水溶性的亲水性聚合物、羟基烷基纤维素、羧基烷基纤维素、羟丙基甲基纤维素、纤维素醚、丙烯酸树脂、聚乙烯吡咯烷酮、交联聚乙烯吡咯烷酮、聚环氧乙烷、聚乙二醇、卡波普、二醇、多元醇(polyols)、多元醇(polyhydric alcohols)、聚亚烷基二醇、聚乙二醇、聚丙二醇或其嵌段共聚物、聚二醇、聚(α-ω)亚烷基二醇、无机化合物、碱金属盐、碱土金属盐,或其组合。
优选的表面活性剂为非离子型、阴离子型、阳离子型或两性表面活性剂。
在一个优选的实施方案中,基质材料含有离子型表面活性剂,特别是阴离子型表面活性剂。
合适的阴离子型表面活性剂包括,但不限于硫酸酯例如十二烷基硫酸钠(十二烷基硫酸钠,例如Texapon®
K12)、十六烷基硫酸钠(例如Lanette
E®)、鲸蜡基硬脂基硫酸钠、十八烷基硫酸钠、二辛基磺基琥珀酸钠(多库酸酯钠(docusate sodium)),及其相应的钾或钙盐。
优选地,阴离子型表面活性剂具有通式(II-a)

其中n为8-30,优选地为10-24,更优选地为12-18的整数;和M选自Li+、Na+、K+、NH4 + 1/2 Mg2+和1/2 Ca2+。
进一步合适的阴离子型表面活性剂包括胆酸的盐,包括甘氨胆酸钠(例如Konakion®
MM, Cernevit®);牛磺胆酸钠及相应的钾或铵盐。
在另一个优选的实施方案中,基质材料含有非离子型表面活性剂。合适的非离子型表面活性剂包括,但不限于
- 可为线形或分支的脂肪醇,例如鲸蜡醇、硬脂醇、鲸蜡硬脂醇(cetylstearyl
alcohol)、2-辛基十二烷-1-醇和2-己基癸烷-1-醇;
- 甾醇,例如胆固醇;
- 脱水山梨糖醇的脂肪酸偏酯,例如脱水山梨糖醇单月桂酸酯、脱水山梨糖醇单棕榈酸酯、脱水山梨糖醇单硬脂酸酯、脱水山梨糖醇三硬脂酸酯、脱水山梨糖醇单油酸酯、脱水山梨糖醇倍半油酸酯和脱水山梨糖醇三油酸酯;
- 聚氧乙烯脱水山梨糖醇的脂肪酸偏酯(聚氧乙烯-脱水山梨糖醇-脂肪酸酯),优选地为聚氧乙烯脱水山梨糖醇的脂肪酸单酯、聚氧乙烯脱水山梨糖醇的脂肪酸二酯或聚氧乙烯脱水山梨糖醇的脂肪酸三酯,例如单-和三-十二烷基、棕榈基、硬脂基和油基酯,例如以名称“polysorbat”已知的和以商品名“Tween”可市售得到的类型,包括Tween®
20 [聚氧乙烯(20)脱水山梨糖醇单月桂酸酯]、Tween® 21 [聚氧乙烯(4)脱水山梨糖醇单月桂酸酯]、Tween®
40 [聚氧乙烯(20)脱水山梨糖醇单棕榈酸酯]、Tween® 60 [聚氧乙烯(20)脱水山梨糖醇单硬脂酸酯]、Tween®
65 [聚氧乙烯(20)脱水山梨糖醇三硬脂酸酯]、Tween® 80 [聚氧乙烯(20)脱水山梨糖醇单油酸酯]、Tween 81 [聚氧乙烯(5)脱水山梨糖醇单油酸酯]和Tween® 85 [聚氧乙烯(20)脱水山梨糖醇三油酸酯];优选地为依据通式(II-b)的聚氧乙烯脱水山梨醇的脂肪酸单酯

其中(w+x+y+z)在15-100,优选地在16-80,更优选地在17-60,还更优选地在18-40,和最优选地在19-21的范围内;和亚烷基任选地为包含6-30个碳原子,更优选地为8-24个碳原子和最优选地为10-16个碳原子的不饱和亚烷基;
- 聚氧乙烯甘油脂肪酸酯,例如甘油的单-、二-和三酯与具有在200-4000 g/mol范围内的分子量的聚乙二醇的二-和单酯的混合物,例如聚乙二醇甘油辛酰基癸酸酯、聚乙二醇甘油月桂酸酯、聚乙二醇甘油椰油酸酯、聚乙二醇甘油亚油酸酯、聚乙二醇-20-甘油单硬脂酸酯、聚乙二醇-6-甘油辛酰基癸酸酯、聚乙二醇甘油油酸酯、聚乙二醇甘油硬脂酸酯、聚乙二醇甘油羟基硬脂酸酯(例如Cremophor® RH 40)和聚乙二醇甘油蓖麻油酸酯(例如Cremophor®
EL);
- 聚氧乙烯脂肪酸酯,脂肪酸优选地具有约8-约18个碳原子,例如油酸聚乙二醇酯、硬脂酸聚乙二醇酯、聚乙二醇-15-羟基硬脂酸酯、12-羟基硬脂酸的聚氧乙烯酯,例如以商品名“Solutol
HS 15”已知和可市售得到的类型,优选地为依据通式(II-c)的类型:

其中n为6-500,优选地为7-250,更优选地为8-100,仍然更优选地为9-75,还更优选地为10-50,甚至更优选地为11-30,最优选地为12-25,并且特别是13-20的整数;和
其中m为6-28,更优选地为6-26,仍然更优选地为8-24,还更优选地为10-22,甚至更优选地为12-20,最优选地为14-18的整数,并且特别是16;
- 聚氧乙烯脂肪醇醚,例如聚乙二醇鲸蜡基硬脂基醚、聚乙二醇月桂基醚、聚乙二醇油基醚、聚乙二醇硬脂基醚;
- 聚氧丙烯-聚氧乙烯嵌段共聚物(泊咯沙姆);
- 蔗糖的脂肪酸酯,例如蔗糖二硬脂酸酯、蔗糖二油酸酯、蔗糖二棕榈酸酯、蔗糖单硬脂酸酯、蔗糖单油酸酯、蔗糖单棕榈酸酯、蔗糖单肉豆蔻酸酯和蔗糖单月桂酸酯;
- 聚甘油的脂肪酸酯,例如聚甘油油酸酯;
- α-生育酚琥珀酸酯的聚氧乙烯酯,例如D-α-生育酚-PEG-1000-琥珀酸酯(TPGS);
- 多糖酵解的甘油酯,例如以商品名“月桂酸聚乙二醇甘油酯(Gelucire) 44/14”、“月桂酸聚乙二醇甘油酯(Gelucire) 50/13”和“辛酸癸酸聚乙二醇甘油酯(Labrasol)”已知和可市售得到的类型;
- 天然或氢化蓖麻油与环氧乙烷的反应产物,例如以商品名“聚氧乙烯蓖麻油(Cremophor)”已知和可市售得到的各种液体表面活性剂;和
- 多官能醇的脂肪酸偏酯,例如甘油脂肪酸酯,例如单-和三-十二烷基、棕榈基、硬脂基和油基酯,例如单硬脂酸甘油酯、单油酸甘油酯,例如以商品名“单油酸甘油酯(Peceol)”已知和可市售得到的单油酸甘油酯40;二山嵛酸甘油酯、二硬脂酸甘油酯、单亚油酸甘油酯、单硬酯酸乙二醇酯、单棕榈硬脂酸乙二醇酯、单硬脂酸季戊四醇酯。
在一个优选的实施方案中,依据本发明的基质材料包含可通过以下得到的表面活性剂或不同表面活性剂的混合物
(i) 用聚乙二醇和任选地用甘油酯化任选地带有羟基的饱和或不饱和的C12-C18-脂肪酸,其中聚乙二醇任选地包含10-40个氧化乙烯单位(-CH2CH2O-);和/或
(ii) 用环氧乙烷酯化带有羟基的饱和或不饱和的C12-C18-脂肪酸的甘油三酯,以致聚乙二醇部分经醚键连接于C12-C18-脂肪酸的羟基,其中聚乙二醇部分优选地包含30-50个氧化乙烯单位(-CH2CH2O-)。
在一个优选的实施方案中,表面活性剂的含量为基于片剂总重量的至少0.001 wt.-%或至少0.005 wt.-%,更优选地为至少0.01
wt.-%或至少0.05 wt.-%,仍然更优选地为至少0.1
wt.-%、至少0.2 wt.-%或至少0.3
wt.-%,还更优选地为至少0.4 wt.-%、至少0.5
wt.-%或至少0.6 wt.-%,并且特别是至少0.7
wt.-%、至少0.8 wt.-%、至少0.9
wt.-%或至少1.0 wt.-%。
然而,在一个优选的实施方案中,依据本发明的片剂的基质材料由一种或多种崩解剂、一种或多种填充剂/粘合剂和一种或多种润滑剂组成,但是不含任何其它组分。
在一个特别优选的实施方案中,依据本发明的片剂的基质材料不含一种或多种胶凝剂和/或硅酮。
本文使用的术语“胶凝剂”用于指一种化合物,其在与溶剂(例如水)接触时吸收该溶剂并膨胀,从而形成粘性或半粘性物质。优选的胶凝剂不是交联的。这种物质可减慢药理学活性化合物自嵌入的颗粒在水性和含水醇介质两者中的释放。在完全水化时,一般产生稠的粘性溶液或分散液,这显著地减少游离溶剂的量和/或使游离溶剂的量最小化,这种游离溶剂可含有一定量的溶解的药理学活性化合物并可抽吸到注射器中。所形成的凝胶也可通过捕获凝胶结构内的药理学活性化合物减少可用溶剂提取的药理学活性化合物的总量。因此胶凝剂可在赋予依据本发明的片剂的抗破碎特性上起重要作用。
优选地在基质材料中不含有的胶凝剂包括药学上可接受的聚合物,一般地为亲水性聚合物,例如水凝胶。胶凝剂的代表性实例包括聚环氧乙烷、聚乙烯醇、羟丙基甲基纤维素、卡波姆、聚糖醛酸(poly(uronic)
acids)及其混合物。
因此,包含在依据本发明的片剂的包衣颗粒中的生理学上可接受的聚合物(优选地为聚环氧烷)优选地也不包含在基质材料中。
优选地,包含在依据本发明的片剂的包衣颗粒中的药理学活性化合物优选地也不包含在基质材料中。
因此,在一个优选的实施方案中,在依据本发明的片剂中含有的药理学活性化合物的总量存在于包衣颗粒中,包衣颗粒形成基质材料中的非连续相,并且形成连续相的基质材料不含任何药理学活性化合物。
依据本发明的片剂含有多个包衣颗粒。包衣颗粒包含药理学活性化合物和生理学上可接受的聚合物(优选聚环氧烷)。优选地,药理学活性化合物分散于生理学上可接受的聚合物(优选聚环氧烷)中。
为了说明书的目的,术语“颗粒”指例如在20℃下或者在室温或环境温度下为固体的离散质量的物质。优选地颗粒在20℃下为固体。优选地,包衣颗粒为单片的(monoliths)。优选地,药理学活性化合物和生理学上可接受的聚合物(优选地为聚环氧烷),紧密地均匀分布于包衣颗粒中,以致包衣颗粒不含任何这样的片段,即在这样的片段中,或者在生理学上可接受的聚合物(优选聚环氧烷)不存在下,存在药理学活性化合物,或者在片段中不存在药理学活性化合物,而存在生理学上可接受的聚合物(优选聚环氧烷)。
颗粒为薄膜包衣的,并且生理学上可接受的聚合物(优选聚环氧烷)优选地均匀分布于药用剂型(片剂)的芯中,即薄膜包衣优选地不含生理学上可接受的聚合物(优选聚环氧烷),但是任选地含有以其低分子量存在的不同于聚环氧烷的聚亚烷基二醇。尽管如此,薄膜包衣本身当然可含有一种或多种聚合物,然而其优选地不同于芯中含有的聚环氧烷。
包衣颗粒具有宏观尺寸,一般地平均直径在100 µm-1500 µm,优选地在200 µm-1500 µm,更优选地在300
µm-1500 µm,仍然更优选地在400 µm-1500 µm,最优选地在500 µm-1500 µm,并且特别是在600
µm-1500 µm的范围内。依据本发明的片剂包含作为非连续相的颗粒,即包衣颗粒形成基质材料中的非连续相,这种基质材料依次优选地形成连续相。在这方面,非连续的意指不是各个和每个颗粒与另一个颗粒密切接触,而是包衣颗粒至少部分地通过其中嵌入包衣颗粒的基质材料相互分开。换句话说,包衣颗粒优选地不形成依据本发明的片剂中的单一一致的物质。
依据本发明的片剂包含少于片剂总重量的三分之二的量的颗粒。
已经令人惊讶地发现,片剂中的颗粒含量可被优化,以在抗破碎性、崩解时间和药物释放、载药量、加工性能(尤其是成片性)和患者依从性之间提供最好的折中。
优选地,依据本发明的片剂中的包衣颗粒的含量为基于片剂的总重量的至多65 wt.-%,更优选地为至多62.5 wt.-%,仍然更优选地为至多60
wt.-%,还更优选地为至多57.5 wt.-%,最优选地为至多55
wt.-%,并且特别是为至多52.5 wt.-%。
优选地,依据本发明的片剂中的包衣颗粒的含量为基于片剂的总重量的至少10 wt.-%、至少12.5 wt.-%、至少15
wt.-%或至少17.5 wt.-%;更优选地为至少20
wt.-%、至少22.5 wt.-%、至少25
wt.-%或至少27.5 wt.-%;最优选地为至少30
wt.-%、至少32.5 wt.-%、至少35 wt.-%或至少37.5 wt.-%;并且特别是为至少40
wt.-%、至少42.5 wt.-%、至少45
wt.-%或至少47.5 wt.-%。
在一个优选的实施方案中,依据本发明的片剂中的包衣颗粒的含量在基于片剂总重量的35±30 wt.-%,更优选地在35±25 wt.-%,仍然更优选地在35±20 wt.-%,还更优选地在35±15 wt.-%,最优选地在35±10 wt.-%,并且特别是在35±5 wt.-%的范围内。在另一个优选的实施方案中,依据本发明的片剂中的包衣颗粒的含量在基于片剂总重量的40±30 wt.-%,更优选地在40±25 wt.-%,仍然更优选地在40±20 wt.-%,还更优选地在40±15 wt.-%,最优选地在40±10 wt.-%,并且特别是在40±5 wt.-%的范围内。在还有的另一个优选的实施方案中,依据本发明的片剂中的包衣颗粒的含量在基于片剂总重量的45±30 wt.-%,更优选地在45±25 wt.-%,仍然更优选地在45±20 wt.-%,还更优选地在45±15 wt.-%,最优选地在45±10 wt.-%,并且特别是在45±5 wt.-%的范围内。在还有的另一个优选的实施方案中,依据本发明的片剂中的包衣颗粒的含量在基于片剂总重量的50±30 wt.-%,更优选地在50±25 wt.-%,仍然更优选地在50±20 wt.-%,还更优选地在50±15 wt.-%,最优选地在50±10 wt.-%,并且特别是在50±5 wt.-%的范围内。在另一个优选的实施方案中,依据本发明的片剂中的包衣颗粒的含量在基于片剂总重量的55±30 wt.-%,更优选地在55±25 wt.-%,仍然更优选地在55±20 wt.-%,还更优选地在55±15 wt.-%,最优选地在55±10 wt.-%,并且特别是在55±5 wt.-%的范围内。在还有的另一个优选的实施方案中,依据本发明的片剂中的包衣颗粒的含量在基于片剂总重量的60±30 wt.-%,更优选地在60±25 wt.-%,仍然更优选地在60±20 wt.-%,还更优选地在60±15 wt.-%,最优选地在60±10 wt.-%,并且特别是在60±5 wt.-%的范围内。
包衣颗粒的形状不受特别限制。因为包衣颗粒优选地通过热熔挤出法制备,存在于依据本发明的片剂中的优选的包衣颗粒形状通常为圆柱形的。这样的包衣颗粒的直径因此为其圆形截面的直径。圆柱形状通过挤出过程引起,据此圆形截面的直径为挤压模的函数和圆柱状的长度为切割长度的函数,据此材料的挤出束被切割成优选地或多于或少于预定长度的条块。
用于制备依据本发明的片剂的圆柱形即球形颗粒的适用性是意想不到的。一般地,长宽比被视为球形的重要量度。长宽比被定义为最大直径(d最大)与其正交弗雷特直径(orthogonal
Feret-diameter)的比率。对于球形颗粒,长宽比具有高于1的数值。数值越小,颗粒越为球形。长宽比低于1.1一般认为是令人满意的,然而长宽比高于1.2一般认为不适合于制备常规片剂。发明人已经令人惊讶地发现,当制备依据本发明的片剂时,甚至可加工具有高于1.2的长宽比的颗粒而没有困难,并且没有必要提供球形颗粒。在一个优选的实施方案中,包衣颗粒的长宽比为至多1.40,更优选地为至多1.35,仍然更优选地为至多1.30,还更优选地为至多1.25,甚至更优选地为至多1.20,最优选地为至多1.15,并且特别是为至多1.10。在另一个优选的实施方案中,包衣颗粒的长宽比为至少1.10,更优选地为至少1.15,仍然更优选地为至少1.20,还更优选地为至少1.25,甚至更优选地为至少1.30,最更优选地为至少1.35,并且特别是为至少1.40。
依据本发明的片剂中的包衣颗粒具有宏观尺寸,即一般具有至少50 µm,更优选地为至少100
µm,仍然更优选地为至少150 µm或至少200
µm,还更优选地为至少250 µm或至少300
µm,最优选地为至少400 µm或至少500
µm,并且特别是为至少550 µm或至少600
µm的平均粒度。
优选的包衣颗粒具有约1000 µm或更少的平均长度和平均直径。当通过挤出技术制备包衣颗粒时,包衣颗粒的“长度”为与挤出方向平行的包衣颗粒的尺寸。包衣颗粒的“直径”为与挤出方向垂直的最大尺寸。
特别优选的包衣颗粒具有小于约1000 μm,更优选地小于约800 μm,仍然更优选地小于约650 μm的平均直径。尤其优选的包衣颗粒具有小于700 μm,特别是小于600 μm,还更特别是小于500 μm,例如小于400 μm的平均直径。特别优选的颗粒具有在200-1000 μm,更优选地在400-800 μm,仍然更优选地在450-700 μm,还更优选地在500-650 μm,例如在约500-600 μm范围内的平均直径。进一步优选的颗粒具有在约300 μm-约400 μm之间,在约400 μm-500 μm之间,或在约500 μm-600 μm之间,或在600 µm-700 µm之间或者在700
µm-800 µm之间的平均直径。
存在于依据本发明的片剂中的优选颗粒具有少于约1000 μm的平均长度,优选地少于约800 μm的平均长度,仍然更优选地少于约650 μm的平均长度,例如约800 µm、约700 µm、约600 μm、约500 μm、约400 μm或约300 μm的长度。尤其优选的颗粒具有少于700 μm,特别是少于650 μm,还更特别是少于550 μm,例如少于450 μm的平均长度。特别优选的颗粒因此具有在200-1000 μm,更优选地在400-800 μm,仍然更优选地在450-700 μm,还更优选地在500-650 μm,例如在约500-600 μm范围内的平均长度。微粒的最小平均长度通过切割步骤测定,并可为例如500 μm、400 μm、300 μm或200 μm。
在一个优选的实施方案中,包衣颗粒具有(i) 约1000±300 μm,更优选地为1000±250 μm,仍然更优选地为1000±200 μm,还更优选地为100±150 μm,最优选地为1000±100 μm,并且特别是为1000±50 μm的平均直径;和/或(ii) 约750±300 μm,更优选地为750±250 μm,仍然更优选地为750±200 μm,还更优选地为750±150 μm,最优选地为750±100 μm,并且特别是为750±50 μm的平均长度。
在另一个优选的实施方案中,包衣颗粒具有(i) 约750±300 μm,更优选地为750±250 μm,仍然更优选地为750±200 μm,还更优选地为750±150 μm,最优选地为750±100 μm,并且特别是为750±50 μm的平均直径;和/或(ii) 约 750±300 μm,更优选地为750±250 μm,仍然更优选地为750±200 μm,仍然更优选地为750±150 μm,最优选地为750±100 μm,并且特别是为750±50 μm的平均长度。
已经令人惊讶地发现,片剂中的包衣颗粒的大小可被优化,以在抗破碎性、崩解时间和药物释放、载药量、加工性能(尤其是成片性)和患者依从性之间提供最好的折中。
颗粒的大小可通过本领域已知的任何常规程序例如激光散射、筛分分析、光学显微镜或图像分析测定。
优选地,在依据本发明的片剂中含有的多个包衣颗粒具有算术平均重量,在以下称为“aaw”,其中在所述多个包衣颗粒中含有的各个颗粒的至少70%,更优选地为至少75%,仍然更优选地为至少80%,还更优选地为至少85%,最优选地为至少90%,并且特别是至少95%,具有在aaw±30%,更优选地在aaw±25%,仍然更优选地在aaw±20%,还更优选地在aaw±15%,最优选地在aaw±10%,并且特别是在aaw±5%范围内的单个重量。例如,如果依据本发明的片剂含有100个颗粒中的大多数,并且所述多个包衣颗粒的aaw为1.00 mg,那么至少75个单个颗粒(即75%)具有在0.70-1.30 mg (1.00 mg ±30%)范围内的单个重量。
颗粒被包衣,优选地被薄膜包衣。已经令人惊讶地发现,当颗粒被薄膜包衣时,崩解时间和/或药物自片剂释放可进一步加速,这对于药物立即释放的片剂特别显著。
优选地,在依据本发明的剂型中含有的多个包衣颗粒包括含有药理学活性化合物的全部量的颗粒,即优选地所有含有活性化合物的颗粒被包衣。
优选地,包衣材料不含崩解剂。
依据本发明的颗粒部分或优选地全部被提供包衣。依据本发明的颗粒优选地用薄膜包衣组合物进行薄膜包衣。合适的包衣材料可例如以商标Opadry®和Eudragit®经市售得到。
优选地,包衣材料包含水溶性聚合物。为了说明书的目的,水溶性聚合物优选地为非肠溶的聚合物,其在暴露于酸性介质例如胃液时快速溶解。优选地,聚合物在pH
1.2和21℃下的100 g人工胃液(HCl水溶液)中的水溶性为至少1.0 g,更优选地为至少2.0 g,仍然更优选地为至少3.0 g,还更优选地为至少4.0 g,最优选地为至少5.0 g,并且特别是为至少6.0 g。
合适的包衣材料的实例包括纤维素酯和纤维素醚,例如甲基纤维素(MC)、羟丙基甲基纤维素(HPMC)、羟丙基纤维素(HPC)、羟乙基纤维素(HEC)、羧甲基纤维素钠(Na-CMC)、乙基纤维素(EC)、邻苯二甲酸醋酸纤维素(CAP)、羟丙基甲基纤维素邻苯二甲酸酯(HPMCP);聚(甲基)丙烯酸酯,例如甲基丙烯酸氨烷基酯共聚物、丙烯酸乙酯甲基丙烯酸甲酯共聚物、甲基丙烯酸甲基丙烯酸甲酯共聚物、甲基丙烯酸甲基丙烯酸甲酯共聚物;乙烯基聚合物,例如聚乙烯吡咯烷酮、聚乙烯醋酸邻苯二甲酸酯、聚乙烯醇、聚乙烯醇-聚乙二醇接枝共聚物、聚醋酸乙烯酯;及天然成膜剂。
包衣材料可含有赋形剂例如稳定剂(例如表面活性剂,例如聚乙二醇鲸蜡硬脂基醚、十二烷基硫酸钠等)。薄膜包衣材料的合适赋形剂为技术人员已知的。
在一个特别优选的实施方案中,包衣为水溶性的。在一个优选的实施方案中,包衣为基于聚乙烯醇例如聚乙烯醇-部分水解的,并可另外含有聚乙二醇,例如聚乙二醇3350和/或颜料。在另一个优选的实施方案中,包衣为基于羟丙基甲基纤维素,优选地为具有3-15 mPas的粘度的羟丙甲纤维素型2910。
一种特别优选的包衣含有聚乙烯醇,并且任选地含有其它的赋形剂例如黄原胶和/或滑石。
颗粒被薄膜包衣,并且干的薄膜包衣的含量优选地为基于包衣颗粒的总重量的至多5 wt.-%,更优选地为至多4 wt.-%,仍然更优选地为至多3.5 wt.-%,还更优选地为至多3 wt.-%,最优选地为至多2.5 wt.-%,并且特别是为至多2 wt.-%。在一个特别优选的实施方案中,相对于颗粒总重量(未包衣的起始材料)的重量增加在3.0-4.7 wt.-%,更优选地在3.1-4.6 wt.-%,仍然更优选地在3.2-4.5
wt.-%,还更优选地在3.3-4.4 wt.-%,最优选地在3.4 -4.3 wt.-%,并且特别是在3.5-4.2
wt.-%的范围内。
已经令人惊讶地发现,片剂中的基质材料:颗粒的相对重量比可被优化,以在抗破碎性、崩解时间和药物释放、载药量、加工性能(尤其是成片性)和患者依从性之间提供最好的折中。
优选地,所述相对重量比在1:1.00±0.75,更优选地在1:1.00±0.50,仍然更优选地在1:1.00±0.40,还更优选地在1:1.00±0.30,最优选地在1:1.00±0.20,并且特别是在1:1.00±0.10的范围内。
包衣颗粒至少含有药理学活性化合物和生理学上可接受的聚合物(优选聚环氧烷)。然而,优选地,包衣颗粒含有另外的药用赋形剂例如抗氧化剂和塑化剂。
药理学活性化合物不受特别限制。优选地,药理学活性化合物为阿片样物质。
在一个优选的实施方案中,包衣颗粒和片剂分别仅含有单一的药理学活性化合物。在另一个优选的实施方案中,包衣颗粒和片剂分别含有两种或更多种药理学活性化合物的组合。
优选地,药理学活性化合物为具有滥用可能性的活性成分。具有滥用可能性的活性成分为本领域技术人员已知的,并且包含例如镇静剂、兴奋剂、巴比妥类、麻醉药、阿片样物质或阿片样物质衍生物。
优选地,药理学活性化合物呈现精神作用。
优选地,药理学活性化合物选自鸦片(opiates)、阿片样物质、兴奋剂、镇静剂及其它麻醉药。
特别优选地,药理学活性化合物为阿片样物质。根据ATC指数,阿片样物质被分为天然阿片生物碱、苯基哌啶衍生物、二苯基丙胺衍生物、苯并吗吩酚衍生物、东罂粟碱衍生物、吗啡喃衍生物等。
以下鸦片、阿片样物质、镇静剂或其它麻醉药为具有精神作用,即具有滥用可能性的物质,并因此优选地分别包含在片剂和包衣颗粒中:阿芬太尼、阿洛巴比妥、烯丙罗定、阿法罗定、阿普唑仑、安非拉酮、安非他明、安非他尼(amphetaminil)、异戊巴比妥、阿尼利定、阿朴可特因、阿索马多(axomadol)、巴比妥、羟基派替啶、苄吗啡(benzylmorphine)、苯腈米特、溴西泮、溴替唑仑、丁丙诺啡、丁巴比妥、布托啡诺、卡马西泮、卡芬太尼、去甲伪麻黄碱/D-去甲伪麻黄碱、氯氮卓、氯巴占、氯苯达诺、氯硝西泮、氯尼他秦、氯氮、氯噻平、氯噁唑仑、可卡因、可待因、环己巴比妥、环啡烷(cyclorphan)、环丙诺啡、地洛西泮、二氢脱氧吗啡、右吗拉胺、右丙氧酚、地佐辛、地恩丙胺、二醋吗啡酮(diamorphone)、地西泮、二氢可待因、二氢吗啡、二氢吗啡酮、地美沙朵、地美庚醇(dimephetamol)、二甲噻丁、吗苯丁酯、地匹哌酮、屈大麻酚、依他佐辛、艾司唑仑、依索庚嗪、乙甲噻丁、氯氟卓乙酯、乙基吗啡、依托尼秦、埃托啡、对乙酰氨基酷盐酸曲马多(faxeladol)、芬莰法明、苯甲锡林、芬哌酰胺、芬普雷司、芬太尼、氟地西泮、氟硝西泮、氟西泮、哈拉西泮、卤沙唑仑、海洛因、氢可酮、氢吗啡酮、羟哌替啶、异美沙酮、羟甲基吗啡喃、凯他唑仑、凯托米酮、左醋美沙朵(LAAM)、左美沙酮、左啡诺、左芬啡烷、立福沙辛(levoxemacin)、二甲磺酸赖右苯丙胺(lisdexamfetamine dimesylate)、洛芬太尼、氯普唑仑、氯羟去甲安定、氯甲西泮、马吲哚、美达西泮、美芬雷司、哌替啶、眠尔通、甲基二氢吗啡酮(metapon)、美普他酚、美他佐辛、甲基吗啡、甲基苯丙胺、美沙酮、安眠酮、3-甲基芬太尼、4-甲基芬太尼、哌醋甲酯、甲苯巴比妥、甲乙哌酮、甲基二氢吗啡酮、咪达唑仑、莫达非尼、吗啡、麦罗啡、大麻隆、纳布啡(nalbuphene)、烯丙吗啡、罂粟碱、尼可吗啡、尼美西泮、硝基安定、去甲西泮、去甲左啡诺、去甲美沙酮、去甲吗啡、诺匹哌酮、鸦片、去甲羟基安定、噁唑仑、羟考酮、羟吗啡酮、罂粟、阿片全碱、匹莫林(pernoline)、喷他佐辛、戊巴比妥、哌替啶、苯吗庚酮、非诺啡烷、非那佐辛、苯哌利定、匹米诺定、福尔可定、苯甲吗啉、苯巴比妥、芬特明、匹那西泮、哌苯甲醇、哌腈米特、普拉西泮、普罗法朵、普罗庚嗪、二甲哌替啶、丙哌利定、丙氧芬、瑞芬太尼、异丁比妥、司可巴比妥、舒芬太尼、他喷他多、替马西泮、四氢西泮、替利定(顺式和反式)、曲马多、三唑仑、乙烯比妥、N-(1-甲基-2-哌啶子基乙基)-N-(2-吡啶基)丙酰胺、(1R,2R)-3-(3-二甲基氨基-1-乙基-2-甲基-丙基)苯酚、(1R,2R,4S)-2-(二甲基氨基)甲基-4-(对-氟苄氧基)-1-(间-甲氧基苯基)环己醇、(1R,2R)-3-(2-二甲基氨基甲基-环己基)苯酚、(1S,2S)-3-(3-二甲基氨基-1-乙基-2-甲基-丙基)苯酚、(2R,3R)-1-二甲基氨基-3(3-甲氧基苯基)-2-甲基-戊-3-醇、(1RS,3RS,6RS)-6-二甲基氨基甲基-1-(3-甲氧基苯基)-环己烷-1,3-二醇(优选地作为外消旋体)、2-(4-异丁基-苯基)丙酸3-(2-二甲基氨基甲基-1-羟基-环己基)苯基酯、2-(6-甲氧基-萘-2-基)丙酸3-(2-二甲基氨基甲基-1-羟基-环己基)苯基酯、2-(4-异丁基-苯基)丙酸3-(2-二甲基氨基甲基-环己-1-烯基)-苯基酯、2-(6-甲氧基-萘-2-基)丙酸3-(2-二甲基氨基甲基-环己-1-烯基)-苯基酯、(RR-SS)-2-乙酰氧基-4-三氟甲基-苯甲酸3-(2-二甲基氨基甲基-1-羟基-环己基)-苯基酯、(RR-SS)-2-羟基-4-三氟甲基-苯甲酸3-(2-二甲基氨基甲基-1-羟基-环己基)-苯基酯、(RR-SS)-4-氯-2-羟基-苯甲酸3-(2-二甲基氨基甲基-1-羟基-环己基)-苯基酯、(RR-SS)-2-羟基-4-甲基-苯甲酸3-(2-二甲基氨基甲基-1-羟基-环己基)-苯基酯、(RR-SS)-2-羟基-4-甲氧基-苯甲酸3-(2-二甲基氨基甲基-1-羟基-环己基)-苯基酯、(RR-SS)-2-羟基-5-硝基-苯甲酸3-(2-二甲基氨基甲基-1-羟基-环己基)-苯基酯、(RR-SS)-2’,4’-二氟-3-羟基-联苯-4-羧酸3-(2-二甲基氨基甲基-1-羟基-环己基)-苯基酯,及相应的立体异构化合物,在每一种情况下其相应的衍生物、其生理学上可接受的对映体、立体异构体、非对映体和外消旋体及生理学上可接受的衍生物,例如醚、酯或酰胺,和在每一种情况下其生理学上可接受的化合物,特别是其酸或碱加成盐和溶剂合物,例如盐酸盐。
在一个优选的实施方案中,药理学活性化合物选自DPI-125、M6G
(CE-04-410)、ADL-5859、CR-665、NRP290和癸二酰基双纳布啡酯。
在一个优选的实施方案中,药理学活性化合物选自羟吗啡酮、氢吗啡酮和吗啡。
在另一个优选的实施方案中,药理学活性化合物选自他喷他多、对乙酰氨基酷盐酸曲马多(faxeladol)和阿索马多(axomadol)。
在还一个优选的实施方案中,药理学活性化合物选自1,1-(3-二甲基氨基-3-苯基五亚甲基)-6-氟-1,3,4,9-四氢吡喃并[3,4-b]吲哚,特别是其半枸橼酸盐;1,1-[3-二甲基氨基-3-(2-噻吩基)五亚甲基]-1,3,4,9-四氢吡喃并[3,4-b]吲哚,特别是其枸橼酸盐;以及1,1- [3-二甲基氨基-3-(2-噻吩基)五亚甲基]-1,3,4,9-四氢吡喃并[3,4-b]-6-氟吲哚,特别是其半枸橼酸盐。这些化合物自例如WO
2004/043967、WO 2005/066183已知。
药理学活性化合物可以生理学上可接受的盐,例如以生理上可接受的酸加成盐的形式存在。
生理上可接受的酸加成盐包含可通过用合适的有机和无机酸处理活性成分的碱形式便利地得到的酸加成盐形式。通过用合适的有机和无机碱处理可将含有酸性质子的活性成分转化为其非毒性的金属或胺加成盐形式。术语加成盐也包含活性成分能够形成的水合物和溶剂加成形式。这样的形式的实例为例如水合物、醇化物等。
已经令人惊讶地发现,片剂和包衣颗粒中的药理学活性化合物的含量分别可被优化,以在抗破碎性、崩解时间和药物释放、载药量、加工性能(尤其是成片性)和患者依从性之间提供最好的折中。
药理学活性化合物以治疗有效量存在于片剂中。构成治疗有效量的量依据所使用的活性成分、受治疗的病症、所述病症的严重性、受治疗的患者以及给药频率而变化。
片剂中的药理学活性化合物的含量不受限制。适合用于给药的药理学活性化合物的剂量优选地在0.1 mg-500 mg的范围内,更优选地在1.0 mg-400 mg的范围内,甚至更优选地在5.0
mg-300 mg的范围内,和最优选地在10 mg-250 mg的范围内。在一个优选的实施方案中,在片剂中含有的药理学活性化合物的总量在0.01-200 mg,更优选地在0.1-190 mg,仍更优选地在1.0-180
mg,还更优选地在1.5-160 mg,最优选地在2.0-100
mg,并且特别是在2.5-80 mg的范围内。
优选地,药理学活性化合物的含量在基于片剂的总重量的0.01-80 wt.-%,更优选地在0.1-50 wt.-%,仍更优选地在1-25
wt.-%的范围内。
在一个优选的实施方案中,药理学活性化合物的含量在5.0±4.5
wt.-%、或7.5±7.0
wt.-%、或10±9.0
wt.-%、或12.5±12.0
wt.-%、或15±14
wt.-%、或17.5±17.0
wt.-%、或20±19
wt.-%、或22.5±22.0
wt.-%、或25±24
wt.-%,更优选地在5.0±4.0
wt.-%、或7.5±6.0
wt.-%、或10±8.0
wt.-%、或12.5±12.0
wt.-%、或15±12
wt.-%、或17.5±15.0
wt.-%、或20±19
wt.-%、或22.5±22.0
wt.-%、或25±24
wt.-%,仍然更优选地在5.0±3.5 wt.-%、或7.5±5.0 wt.-%、或10±7.0 wt.-%、或12.5±10.0 wt.-%、或15±10 wt.-%、或17.5±13.0 wt.-%、或20±17 wt.-%、或22.5±19.0 wt.-%、或25±21 wt.-%,还更优选地在5.0±3.0 wt.-%、或7.5±4.0 wt.-%、或10±6.0 wt.-%、或12.5±8.0 wt.-%、或15±8.0 wt.-%、或17.5±11.0 wt.-%、或20±15 wt.-%、或22.5±16.0 wt.-%、或25±18 wt.-%,甚至更优选地在5.0±2.5 wt.-%、或7.5±3.0 wt.-%、或10±5.0 wt.-%、或12.5±6.0 wt.-%、或15±6.0 wt.-%、或17.5±9.0 wt.-%、或20±13 wt.-%、或22.5±13.0 wt.-%、或25±15 wt.-%,最优选地在5.0±2.0 wt.-%、或7.5±2.0 wt.-%、或10±4.0 wt.-%、或12.5±4.0 wt.-%、或15±4.0 wt.-%、或17.5±7.0 wt.-%、或20±11 wt.-%、或22.5±10.0 wt.-%、或25±12 wt.-%,并且特别是在5.0±1.5 wt.-%、或7.5±1.0 wt.-%、或10±3.0 wt.-%、或12.5±2.0 wt.-%、或15±2.0 wt.-%、或17.5±5.0 wt.-%、或20±9 wt.-%、或22.5±7.0 wt.-%、或25±9 wt.-%的范围内,在每一种情况下均基于片剂的总重量计。
在进一步优选的实施方案中,药理学活性化合物的含量在基于片剂的总重量的20±6 wt.-%,更优选地在20±5 wt.-%,仍然更优选地在20±4 wt.-%,最优选地在20±3 wt.-%,并且特别是在20±2 wt.-%的范围内。在另一个优选的实施方案中,药理学活性化合物的含量在基于片剂的总重量的25±6 wt.-%,更优选地在25±5 wt.-%,仍然更优选地在25±4 wt.-%,最优选地在25±3 wt.-%,并且特别是在25±2 wt.-%的范围内。
技术人员可易于确定要在片剂中包含的药理学活性化合物的合适量。例如,在镇痛剂的情况下,存在于片剂中的药理学活性化合物的总量为足以提供镇痛的量。要以一个剂量给予患者的药理学活性化合物的总量将依众多因素而变化,包括药理学活性化合物的性质、患者的重量、疼痛的严重性、正给予的其它治疗剂的性质等。
在一个优选的实施方案中,药理学活性化合物以7.5±5 mg、10±5 mg、20±5 mg、30±5 mg、40±5 mg、50±5 mg、60±5 mg、70±5 mg、80±5 mg、90±5 mg、100±5 mg、110±5 mg、120±5 mg、130±5、140±5 mg、150±5 mg、160±5 mg、170±5 mg、180±5 mg、190±5 mg、200±5 mg、210±5 mg、220±5 mg、230±5 mg、240±5 mg、250±5 mg、260±5 mg、270±5 mg、280±5 mg、290±5 mg或300±5 mg的量包含在片剂中。在另一个优选的实施方案中,药理学活性化合物以5±2.5
mg、7.5±2.5
mg、10±2.5
mg、15±2.5
mg、20±2.5
mg、25±2.5
mg、30±2.5
mg、35±2.5
mg、40±2.5
mg、45±2.5
mg、50±2.5
mg、55±2.5
mg、60±2.5
mg、65±2.5
mg、70±2.5
mg、75±2.5
mg、80±2.5
mg、85±2.5
mg、90±2.5
mg、95±2.5
mg、100±2.5
mg、105±2.5
mg、110±2.5
mg、115±2.5
mg、120±2.5
mg、125±2.5
mg、130±2.5
mg、135±2.5
mg、140±2.5
mg、145±2.5
mg、150±2.5
mg、155±2.5
mg、160±2.5
mg、165±2.5
mg、170±2.5
mg、175±2.5
mg、180±2.5
mg、185±2.5
mg、190±2.5
mg、195±2.5
mg、200±2.5
mg、205±2.5
mg、210±2.5
mg、215±2.5
mg、220±2.5
mg、225±2.5
mg、230±2.5
mg、235±2.5
mg、240±2.5
mg、245±2.5
mg、250±2.5
mg、255±2.5
mg、260±2.5
mg或265±2.5
mg的量包含在片剂中。
在一个特别优选的实施方案中,药理学活性化合物为他喷他多,优选地为其HCl盐,并且这种片剂适合于每天一次、每天两次、每天三次或更加频繁地给予。在该实施方案中,药理学活性化合物优选地以25-100
mg的量包含在片剂中。
在一个特别优选的实施方案中,药理学活性化合物为羟吗啡酮,优选地为其HCl盐,并且这种片剂适合于每天一次、每天两次、每天三次或更加频繁地给予。在该实施方案中,药理学活性化合物优选地以5-40
mg的量包含在片剂中。在另一个特别优选的实施方案中,药理学活性化合物为羟吗啡酮,优选地为其HCl盐,并且这种片剂适合于给予每天一次。在该实施方案中,药理学活性化合物优选地以10-80 mg的量包含在片剂中。
在另一个特别优选的实施方案中,药理学活性化合物为羟考酮,优选地为其HCl盐,并且这种片剂适合于每天一次、每天两次、每天三次或更加频繁地给予。在该实施方案中,药理学活性化合物优选地以5-80
mg的量包含在片剂中。
在还一个特别优选的实施方案中,药理学活性化合物为氢吗啡酮,优选地为其HC1,并且这种片剂适合于每天一次、每天两次、每天三次或更加频繁地给予。在该实施方案中,药理学活性化合物优选地以2-52
mg的量包含在片剂中。在另一个特别优选的实施方案中,药理学活性化合物为氢吗啡酮,优选地为其HCl,并且这种片剂适合于每天一次、每天两次、每天三次或更加频繁地给予。在该实施方案中,药理学活性化合物优选地以4-104
mg的量包含片剂中。
存在于依据本发明的片剂中的包衣颗粒优选地包含基于颗粒的总重量的3-75 wt.-%的药理学活性化合物,更优选地为5-70 wt.-%的药理学活性化合物,仍然更优选地为7.5-65
wt.-% 的药理学活性化合物。
优选地,药理学活性化合物的含量为基于颗粒的总重量的至少25 wt.-%,更优选地为至少30
wt.-%,仍然更优选地为至少35 wt.-%,还更优选地为至少40
wt.-%,最优选地为至少45 wt.-%。
优选地,药理学活性化合物的含量为基于颗粒的总重量的至多70 wt.-%,更优选地为至多65
wt.-%,仍然更优选地为至多60 wt.-%,还更优选地为至多55
wt.-%,最优选地为至多50 wt.-%。
在一个优选的实施方案中,药理学活性化合物的含量在基于颗粒的总重量的35±30
wt.-%,更优选地在35±25
wt.-%,仍然更优选地在35±20 wt.-%,还更优选地在35±15 wt.-%,最优选地在35±10 wt.-%,并且特别是在35±5 wt.-%的范围内。在另一个优选的实施方案中,药理学活性化合物的含量在基于颗粒的总重量的45±30 wt.-%,更优选地在45±25 wt.-%,仍然更优选地在45±20 wt.-%,还更优选地在45±15 wt.-%,最优选地在45±10 wt.-%,并且特别是在45±5 wt.-%的范围内。在还一个优选的实施方案中,药理学活性化合物的含量在基于颗粒的总重量的55±30 wt.-%,更优选地在55±25 wt.-%,仍然更优选地在55±20 wt.-%,还更优选地在55±15 wt.-%,最优选地在55±10 wt.-%,并且特别是在55±5 wt.-%的范围内。
在依据本发明的片剂制剂中包含的药理学活性化合物具有小于500微米,仍然更优选地小于300微米,还更优选地小于200或100微米的平均粒度。对于平均粒度没有下限,并且其可为例如50微米。药理学活性化合物的粒度可通过本领域常规的任何技术例如激光散射、筛分分析、光学显微镜或图像分析测定。一般而言优选的是药理学活性化合物颗粒的最大尺寸小于包衣颗粒的尺寸(例如小于包衣颗粒的最小尺寸)。
技术人员知道如何测定药代动力学参数例如t1/2、T最大、C最大、AUC和生物利用度。对于本说明书的目的,如下定义可自3-(2-二甲基氨基甲基环己基)苯酚的血浆浓度测定的药代动力学参数:
| C最大 | 在单次给予后测量的活性成分的血浆浓度的最大值(≡平均峰值血浆水平) |
| T最大 | 自给予活性成分直到达到C最大的时间间隔 |
| AUC | 包括来自外推至无穷大的最终测量值的分区的血浆浓度/时间曲线的总面积 |
| t1/2 | 半衰期 |
以上参数在每一种情况下表示为对于所有研究的患者/受试对象的各个值的平均值。
本领域的技术人员知道可如何自所测量的血浆中活性成分的浓度计算活性成分的药代动力学参数。关于这一点,可参照例如Willi
Cawello (ed.) Parameters for Compartment-free Pharmacokinetics, Shaker
Verlag Aachen (1999)。
在一个优选的实施方案中,药理学活性化合物为他喷他多或其生理学上可接受的盐,例如盐酸盐。优选地,依据本发明的片剂提供至少22%,更优选地为至少24%,仍然更优选地为至少26%,仍然更优选地为至少28%,最优选地为至少30%,并且特别是至少32%的他喷他多的平均绝对生物利用度。他喷他多的T最大优选地在1.25±1.20
h,更优选地在1.25±1.00
h,仍然更优选地在1.25±0.80
h,还更优选地在1.25±0.60
h,最优选地在1.25±0.40
h,并且特别是在1.25±0.20
h的范围内。他喷他多的t1/2优选地在4.0±2.8 h,更优选地在4.0±2.4 h,仍然更优选地在4.0±2.0 h,还更优选地在4.0±1.6 h,最优选地在4.0±1.2 h,并且特别是在4.0±0.8 h的范围内。优选地,当标准化为100 mg他喷他多的剂量时,他喷他多的C最大优选地在90±85
ng/mL,更优选地在90±75
ng/mL,仍然更优选地在90±65
ng/mL,还更优选地在90±55
ng/mL,最优选地在90±45
ng/mL,并且特别是在90±35
ng/mL的范围内;和/或他喷他多的AUC优选地在420±400 ng/mL•h,更优选地在420±350 ng/mL•h,仍然更优选地在420±300 ng/mL•h,还更优选地在420±250 ng/mL•h,最优选地在420±200 ng/mL•h,并且特别是在420±150 ng/mL•h的范围内。
在另一个优选的实施方案中,药理学活性化合物为羟吗啡酮或其生理学上可接受的盐,例如盐酸盐。优选地,依据本发明的片剂提供至少1%,更优选地为至少2%,仍然更优选地为至少4%,还更优选地为至少6%,最优选地为至少8%,并且特别是至少19%的羟吗啡酮的平均绝对生物利用度。羟吗啡酮的T最大优选地在0.5±0.45
h,更优选地在0.5±0.40
h,仍然更优选地在0.5±0.35 h,还更优选地在0.5±0.30 h,最优选地在0.5±0.25 h,并且特别是在0.5±0.20 h的范围内。羟吗啡酮的t1/2优选地在9.5±8.0 h,更优选地在9.5±7.0 h,仍然更优选地在9.5±6.0 h,还更优选地在9.5±5.0 h,最优选地在9.5±4.0 h,并且特别是在9.5±3.0 h的范围内。优选地,当标准化为20 mg羟吗啡酮的剂量时,羟吗啡酮的C最大优选地在4.4±3.5 ng/mL,更优选地在4.4±3.0 ng/mL,仍然更优选地在4.4±2.5 ng/mL,还更优选地在4.4±2.0 ng/mL,最优选地在4.4±1.5 ng/mL,并且特别是在4.4±1.0 ng/mL的范围内;和/或羟吗啡酮的AUC优选地在20.0±15.0 ng/mL•h,更优选地在20.0±12.5 ng/mL•h,仍然更优选地在20.0±10.0 ng/mL•h,还更优选地在20.0±7.5 ng/mL•h,最优选地在20.0±6.0 ng/mL•h,并且特别是在20.0±5.0 ng/mL•h的范围内。
在另一个优选的实施方案中,药理学活性化合物为羟考酮或其生理学上可接受的盐,例如盐酸盐。优选地,依据本发明的片剂提供至少40%,更优选地为至少45%,仍然更优选地为至少50%,还更优选地为至少55%,最优选地为至少60%,并且特别是至少70%的羟考酮的平均绝对生物利用度。羟考酮的T最大优选地在2.6±2.5 h,更优选地在2.6±2.0 h,仍然更优选地在2.6±1.8 h,还更优选地在2.6±1.6 h,最优选地在2.6±1.4 h,并且特别是在2.6±1.20 h的范围内。羟考酮的t1/2优选地在3.8±3.5 h,更优选地在3.8±3.0 h,仍然更优选地在3.8±2.5 h,还更优选地在3.8±2.0 h,最优选地在3.8±1.5 h,并且特别是在3.8±1.0 h的范围内。优选地,当标准化为30 mg羟考酮的剂量时,羟考酮的C最大优选地在40±35 ng/mL,更优选地在40±30 ng/mL,仍然更优选地在40±25 ng/mL,还更优选地在40±20 ng/mL,最优选地在40±15 ng/mL,并且特别是在40±10 ng/mL的范围内;和/或羟考酮的AUC优选地在270±250 ng/mL•h,更优选地在270±200 ng/mL•h,仍然更优选地在270±150 ng/mL•h,还更优选地在270±100 ng/mL•h,最优选地在270±75 ng/mL•h,并且特别是在270±50 ng/mL•h的范围内。
在还有的另一个优选的实施方案中,药理学活性化合物为吗啡或其生理学上可接受的盐,例如硫酸盐。优选地,依据本发明的片剂提供至少15%,更优选地为至少20%,仍然更优选地为至少25%,还更优选地为至少30%,最优选地为至少35%,并且特别是至少40%的吗啡的平均绝对生物利用度。吗啡的T最大优选地在0.625±0.60
h,更优选地在0.625±0.50
h,仍然更优选地在0.625±0.40
h,还更优选地在0.625±0.30
h,最优选地在0.625±0.20
h,并且特别是在0.625±0.15
h的范围内。优选地,当标准化为30 mg硫酸吗啡的剂量时,吗啡的C最大优选地在25±20 ng/mL,更优选地在25±15 ng/mL,仍然更优选地在25±10 ng/mL,还更优选地在25±5 ng/mL的范围内;和/或吗啡的AUC优选地在50±45 ng/mL•h,更优选地在50±40 ng/mL•h,仍然更优选地在50±35 ng/mL•h,还更优选地在50±30 ng/mL•h,最优选地在50±25 ng/mL•h,并且特别是在50±20 ng/mL•h的范围内。
依据本发明的片剂也可包含一种或多种另外的药理学活性化合物。所述另外的药理学活性化合物可能易被滥用或为另一种药物。另外的药理学活性化合物可存在于包衣颗粒内(“颗粒内的”)或基质内(例如“颗粒外的”,或者当基质材料也以颗粒形式提供时也为“颗粒内的”)。当颗粒内存在另外的药理学活性化合物时,其可与一种或多种药理学活性化合物组合存在于相同的颗粒内或单独存在于离散的颗粒群中,并与存在于片剂中的任何其它药理学活性化合物分开。
在一个优选的实施方案中,依据本发明的片剂,优选地为包衣颗粒包含阿片样物质(激动剂)以及阿片样物质拮抗剂。
可存在任何常规的阿片样物质拮抗剂,例如纳曲酮或纳洛酮或其药学上可接受的盐。纳洛酮,包括其盐,为特别优选的。阿片样物质拮抗剂可存在于包衣颗粒内或基质内。或者,阿片样物质拮抗剂可以分开的颗粒提供给药理学活性化合物。这样的颗粒的优选组成与对于含有药理学活性化合物的颗粒描述的相同。
依据本发明的片剂中的阿片样物质激动剂与阿片样物质拮抗剂的比率优选地按重量计为1:1-3:1,例如按重量计为约2:1。
在另一个优选的实施方案中,包衣颗粒和片剂均不包含任何阿片样物质拮抗剂。
依据本发明的包衣颗粒含有生理学上可接受的聚合物(优选聚环氧烷)。
优选地,生理学上可接受的聚合物选自聚环氧烷,优选地为聚甲醛(polymethylene oxide)、聚环氧乙烷、聚环氧丙烷;聚乙烯、聚丙烯、聚氯乙烯、聚碳酸酯、聚苯乙烯、聚乙烯吡咯烷酮、聚(烷基)丙烯酸酯;聚(羟基脂肪酸),例如聚(3-羟基丁酸酯共聚-3-羟基戊酸酯) (Biopol®)、聚(羟基戊酸);聚已内酯、聚乙烯醇、聚酰胺酯、聚丁二酸亚乙酯、聚内酯(polylactone)、聚乙交酯、聚氨酯、聚酰胺、聚丙交酯、聚缩醛(例如任选地具有改性侧链的多糖)、聚丙交酯/乙交酯、聚内酯、聚乙交酯、聚原酸酯、聚酐、聚乙二醇与聚对苯二甲酸丁二醇酯的嵌段共聚物(Polyactive®)、聚酐(Polifeprosan)、其共聚物、其嵌段共聚物(例如Poloxamer® (泊洛沙姆))和至少两种所述聚合物的混合物或具有以上特性的其它聚合物。聚环氧烷为特别优选的。
优选地,生理学上可接受的聚合物为聚环氧烷,更优选地选自聚甲醛、聚环氧乙烷和聚环氧丙烷,或其共聚物。聚环氧乙烷为优选的。
在一个优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)具有至少200,000或至少500,000 g/mol,优选地为至少1,000,000 g/mol或至少2,500,000
g/mol,更优选地在约1,000,000 g/mol-约15,000,000 g/mol范围内,和最优选地在约5,000,000
g/mol-约10,000,000 g/mol范围内的重均分子量(MW)或粘均分子量(Mη)。测定MW和Mη的合适方法为本领域技术人员已知的。Mη优选地通过流变学测试测定,而MW可通过凝胶渗透色谱(GPC)测定。
生理学上可接受的聚合物(优选聚环氧烷)可包含具有特定平均分子量的单一聚合物,或不同聚合物例如2、3、4或5种聚合物的混合物(掺混物),所述聚合物为例如具有相同化学性质但是不同平均分子量的聚合物、不同化学性质但是相同平均分子量的聚合物、或不同化学性质以及不同分子量的聚合物。
为了说明书的目的,聚亚烷基二醇具有最多20,000 g/mol的分子量,而聚环氧烷具有大于20,000 g/mol的分子量。在一个优选的实施方案中,在片剂中含有的所有聚环氧烷的所有分子量的重量平均为至少200,000
g/mol。因此,当测定聚环氧烷的重均分子量时,如果有的话,优选地不考虑聚亚烷基二醇。
在一个优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)均匀分布于依据本发明的包衣颗粒中。优选地,药理学活性化合物和生理学上可接受的聚合物(优选聚环氧烷)紧密地均匀分布于包衣颗粒中,以致包衣颗粒不含任何这样的片段,即在这样的片段中,或者在生理学上可接受的聚合物(优选聚环氧烷)不存在下,存在药理学活性化合物,或者在片段中不存在药理学活性化合物,而存在生理学上可接受的聚合物(优选聚环氧烷)。
颗粒为薄膜包衣的,并且生理学上可接受的聚合物(优选聚环氧烷)优选地均匀分布于包衣颗粒的芯中,即薄膜包衣优选地不含生理学上可接受的聚合物(优选聚环氧烷)。尽管如此,薄膜包衣本身当然可含有一种或多种聚合物,然而其优选地不同于芯中含有的生理学上可接受的聚合物,优选聚环氧烷。
生理学上可接受的聚合物(优选聚环氧烷)可结合一种或多种选自以下的不同聚合物:聚环氧烷,优选地为聚甲醛、聚环氧乙烷、聚环氧丙烷;聚乙烯、聚丙烯、聚氯乙烯、聚碳酸酯、聚苯乙烯、聚乙烯吡咯烷酮、聚(烷基)丙烯酸酯;聚(羟基脂肪酸),例如聚(3-羟基丁酸酯共聚-3-羟基戊酸酯) (Biopol®)、聚(羟基戊酸);聚已内酯、聚乙烯醇、聚酰胺酯、聚丁二酸亚乙酯、聚内酯、聚乙交酯、聚氨酯、聚酰胺、聚丙交酯、聚缩醛(例如任选地具有改性侧链的多糖)、聚丙交酯/乙交酯、聚内酯、聚乙交酯、聚原酸酯、聚酐、聚乙二醇与聚对苯二甲酸丁二醇酯的嵌段共聚物(Polyactive®)、聚酐(Polifeprosan)、其共聚物、其嵌段共聚物(例如Poloxamer® (泊洛沙姆))和至少两种所述聚合物的混合物,或具有以上特性的其它聚合物。
优选地,生理学上可接受的聚合物(优选聚环氧烷)的分子量分散性Mw/Mn在2.5±2.0,更优选地在2.5±1.5,仍然更优选地在2.5±1.0,还更优选地在2.5±0.8,最优选地在2.5±0.6,并且特别是在2.5±0.4的范围内。
生理学上可接受的聚合物(优选聚环氧烷)优选地在25℃下具有以下粘度:30-17600 cP,更优选地为55-17,600 cP,仍然更优选地为600-17600
cP和最优选地为4500 -17600 cP (以5 wt.-%水溶液,使用型号RVF Brookfield粘度计(主轴编号2 /转速2 rpm)测量);400-4000 cP,更优选地为400-800
cP或2000-4000 cP (以2 wt.-%水溶液,使用所述粘度计(主轴编号1或3 /转速10 rpm)测量);或1650-10000 cP,更优选地为1650-5500
cP、5500-7500 cP或7500-10000 cP (以1 wt.-%水溶液,使用所述粘度计(主轴编号2 /转速2 rpm)测量)。
适合用于依据本发明的片剂的聚环氧乙烷可自Dow市售得到。例如,Polyox
WSR N-12K、Polyox N-60K、Polyox WSR 301 NF或Polyox
WSR 303NF可用于依据本发明的片剂。关于这些产品性能的详情,可参照例如产品说明书。
优选地,生理学上可接受的聚合物(优选聚环氧烷)的含量,在基于片剂的总重量的1-60 wt.-%,更优选地在3-55
wt.-%,仍然更优选地在5-50 wt.-%,还更优选地在7-45
wt.-%,最优选地在10-40 wt.-%,并且特别是在15-35 wt.-%的范围内。在一个优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)的含量在基于片剂的总重量的至少2 wt.-%,更优选地为至少5 wt.-%,仍然更优选地为至少10 wt.-%,还更优选地为至少15 wt.-%,并且特别是为至少20 wt.-%。
在一个优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)的总含量,在基于片剂的总重量的10±8 wt.-%,更优选地在10±6 wt.-%,最优选地在10±4 wt.-%,并且特别是在10±2 wt.-%的范围内。在另一个优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)的总含量,在基于片剂的总重量的15±12 wt.-%,更优选地在15±10 wt.-%,最优选地在15±7 wt.-%,并且特别是在15±3 wt.-%的范围内。在还一个优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)的总含量,在基于片剂的总重量的20±16 wt.-%,更优选地在20±12 wt.-%,最优选地在20±8 wt.-%,并且特别是在20±4 wt.-%的范围内。在又一个优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)的总含量,在基于片剂的总重量的25±20 wt.-%,更优选地在25±15 wt.-%,最优选地在25±10 wt.-%,并且特别是在25±5 wt.-%的范围内。在进一步优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)的总含量,在基于片剂的总重量的30±20 wt.-%,更优选地在30±15 wt.-%,最优选地在30±10 wt.-%,并且特别是在30±5 wt.-%的范围内。在更进一步优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)的总含量在35±20 wt.-%,更优选地在35±15 wt.-%,最优选地在35±10 wt.-%,并且特别是在35±5 wt.-%的范围内。在还更进一步优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)的总含量,在基于片剂的总重量的40±20 wt.-%,更优选地在40±15 wt.-%,和最优选地在40±10 wt.-%,并且特别是在40±5 wt.-%的范围内。
优选地,生理学上可接受的聚合物(优选聚环氧烷)的含量,在基于包衣颗粒的总重量的1-99 wt.-%,更优选地在5 to
95 wt.-%,仍然更优选地在10-90 wt.-%,还更优选地在15-85 wt.-%,最优选地在20-80
wt.-%,并且特别是在25-75 wt.-%的范围内。在一个优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)的含量,在基于包衣颗粒的总重量的至少10 wt.-%,更优选地为至少15
wt.-%,仍然更优选地为至少20 wt.-%,还更优选地为至少25
wt.-%,并且特别是为至少30 wt.-%。
在一个优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)的总含量,在基于包衣颗粒的总重量的30±20
wt.-%,更优选地在30±15
wt.-%,最优选地在30±10
wt.-%,并且特别是在30±5
wt.-%的范围内。在另一个优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)的总含量,在基于包衣颗粒的总重量的35±20 wt.-%,更优选地在35±15 wt.-%,最优选地在35±10 wt.-%,并且特别是在35±5 wt.-%的范围内。在还一个优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)的总含量,在基于包衣颗粒的总重量的40±20 wt.-%,更优选地在40±15 wt.-%,最优选地在40±10 wt.-%,并且特别是在40±5 wt.-%的范围内。在又一个优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)的总含量,在基于包衣颗粒的总重量的45±20 wt.-%,更优选地在45±15 wt.-%,最优选地在45±10 wt.-%,并且特别是在45±5 wt.-%的范围内。在进一步优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)的总含量,在基于包衣颗粒的总重量的50±20 wt.-%,更优选地在50±15 wt.-%,最优选地在50±10 wt.-%,并且特别是在50±5 wt.-%的范围内。在更进一步优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)的总含量在55±20 wt.-%,更优选地在55±15 wt.-%,最优选地在55±10 wt.-%,并且特别是在55±5 wt.-%的范围内。在还更进一步优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)的总含量,在基于包衣颗粒的总重量的60±15 wt.-%,更优选地在60±10 wt.-%,最优选地在60±5 wt.-%,并且特别是在60±5 wt.-%的范围内。
优选地,生理学上可接受的聚合物(优选聚环氧烷)与药理学活性化合物的相对重量比在1:1.00±0.75,更优选地在1:1.00±0.50,仍然更优选地在1:1.00±0.40,还更优选地在1:1.00±0.30,最优选地在1:1.00±0.20,并且特别是在1:1.00±0.10的范围内。
依据本发明的包衣颗粒可含有以常规量在片剂中常规含有的另外的药用赋形剂,例如抗氧化剂、防腐剂、润滑剂、塑化剂、填充剂、粘合剂等。
技术人员将可易于确定合适的其它赋形剂以及这些赋形剂中每一种的量。可用于配制依据本发明的片剂的药学上可接受的载体和赋形剂的具体实例描述于药用赋形剂手册(Handbook
of Pharmaceutical Excipients), 美国制药协会(American
Pharmaceutical Association) (1986)中。
在一个优选的实施方案中,包衣颗粒不含崩解剂。
优选地,包衣颗粒进一步包含抗氧化剂。合适的抗氧化剂包括抗坏血酸、丁羟茴醚(BHA)、丁羟甲苯(BHT)、抗坏血酸的盐、单硫代甘油(monothioglycerol)、亚磷酸、维生素C、维生素E及其衍生物、苯甲酸松柏醇酯、去甲二氢愈创木酸、没食子酸酯(gallus acid esters)、亚硫酸氢钠,特别优选地为丁羟甲苯或丁羟茴醚和α-生育酚。抗氧化剂优选地以基于包衣颗粒的总重量的0.01
wt.-%至10 wt.-%,更优选地0.03
wt.-%至5 wt.-%,最优选地0.05
wt.-%至2.5 wt.-%的量存在。
在一个优选的实施方案中,包衣颗粒进一步包含酸,优选地为枸橼酸。酸的量优选地在基于包衣颗粒总重量的0.01 wt.-%至约20 wt.-%的范围内,更优选地在0.02 wt.-%至约10 wt.-%的范围内,和仍然更优选地在0.05 wt.-%至约5 wt.-%的范围内,并且最优选地在0.1 wt.-%至约1.0 wt.-%的范围内。
在一个优选的实施方案中,包衣颗粒进一步包含优选地选自以下的另一种聚合物:纤维素酯和纤维素醚,特别是羟丙基甲基纤维素(HPMC)。
其它优选的聚合物为聚乙烯基己内酰胺-聚醋酸乙烯酯-聚乙二醇接枝共聚物,例如可以商品名Soluplus®市售得到的聚合物。
其它聚合物(优选地羟丙基甲基纤维素)的量优选地在基于包衣颗粒的总重量的0.1 wt.-%至约30
wt.-%的范围内,更优选地在1.0 wt.-%至约20
wt.-%的范围内,最优选地在2.0 wt.-%至约15
wt.-%的范围内,并且特别是在3.5 wt.-%至约10.5
wt.-%的范围内。
在一个优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)与其它聚合物的相对重量比在4.5±2:1,更优选地在4.5±1.5:1,仍然更优选地在4.5±1:1,还更优选地在4.5±0.5:1,最优选地在4.5±0.2:1,并且特别是在4.5±0.1:1的范围内。在另一个优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)与其它聚合物的相对重量比在8±7:1,更优选地在8±6:1,仍然更优选地在8±5:1,还更优选地在8±4:1,最优选地在8±3:1,并且特别是在8±2:1的范围内。在还一个优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)与其它聚合物的相对重量比在11±8:1,更优选地在11±7:1,仍然更优选地在11±6:1,还更优选地在11±5:1,最优选地在11±4:1,并且特别是11±3:1的范围内。
在另一个优选的实施方案中,依据本发明的包衣颗粒除了生理学上可接受的聚合物(优选聚环氧烷)和任选的聚乙二醇外,不含任何其它的聚合物。
在一个优选的实施方案中,包衣颗粒含有至少一种润滑剂。在另一个优选的实施方案中,包衣颗粒不含润滑剂。尤其优选的润滑剂选自:
- 硬脂酸镁和硬脂酸;
- 脂肪酸的甘油酯,包括脂肪酸的甘油一酯、甘油二酯、甘油三酯及其混合物;优选地为C6-C22脂肪酸的甘油一酯、甘油二酯、甘油三酯及其混合物;尤其优选的是C16-C22脂肪酸的部分甘油酯,例如甘油山嵛酸酯(glycerol behenat)、棕榈硬脂酸甘油酯(glycerol palmitostearate)和单硬脂酸甘油酯;
- 聚氧乙烯甘油脂肪酸酯,例如甘油的单-、二-和三酯的混合物及具有在200-4000 g/mol范围内的分子量的聚乙二醇的二-和单酯,例如聚乙二醇甘油辛酸癸酸酯(macrogolglycerolcaprylocaprate)、聚乙二醇甘油月桂酸酯、聚乙二醇甘油椰油酸酯(macrogolglycerolococoate)、聚乙二醇甘油亚油酸酯、聚乙二醇-20-甘油单硬脂酸酯、聚乙二醇-6-甘油辛酸癸酸酯、聚乙二醇甘油油酸酯、聚乙二醇甘油硬脂酸酯、聚乙二醇甘油羟基硬脂酸酯和聚乙二醇甘油蓖麻油酸酯;
- 多糖酵解的(polyglycolyzed)甘油酯,例如以商品名“Labrasol”已知并可市售得到的多糖酵解的甘油酯;
- 可为线形或分支的脂肪醇,例如鲸蜡醇、硬脂醇、鲸蜡基硬脂醇、2-辛基十二烷-1-醇和2-己基癸烷-1-醇;
- 具有在10.000-60.000 g/mol之间的分子量的聚乙二醇;和
- 天然的半合成或合成蜡,优选地为具有至少50℃,更优选地为60℃的软化点的蜡,并且特别是为巴西棕榈蜡和蜂蜡。
优选地,润滑剂的量在基于包衣颗粒的总重量的0.01 wt.-%至约10
wt.-%的范围内,更优选地在0.05 wt.-%至约7.5
wt.-%的范围内,最优选地在0.1 wt.-%至约5
wt.-%的范围内,并且特别是在0.1 wt.-%至约1
wt.-%的范围内。
优选地,包衣颗粒进一步包含塑化剂。塑化剂改善生理学上可接受的聚合物(优选聚环氧烷)的加工性能。优选的塑化剂为聚亚烷基二醇,像聚乙二醇、三醋精、脂肪酸、脂肪酸酯、蜡和/或微晶蜡。特别优选的塑化剂为聚乙二醇,例如PEG 6000。
优选地,塑化剂的含量在基于包衣颗粒的总重量的0.5-30 wt.-%,更优选地在1.0-25 wt.-%,仍然更优选地在2.5
wt.-%至22.5 wt.-%,仍然更优选地在5.0
wt.-%至20 wt.-%,最优选地在6-20
wt.-%,并且特别是在7 wt.-%至17.5
wt.-%的范围内。
在一个优选的实施方案中,塑化剂为聚亚烷基二醇,所述聚亚烷基二醇具有在基于包衣颗粒总重量的7±6 wt.-%,更优选地在7±5 wt.-%,仍然更优选地在7±4 wt.-%,还更优选地在7±3 wt.-%,最优选地在7±2 wt.-%,并且特别是在7±1 wt.-%的范围内的含量。
在另一个优选的实施方案中,塑化剂为为聚亚烷基二醇,所述聚亚烷基二醇具有在基于包衣颗粒总重量的10±8 wt.-%,更优选地在10±6 wt.-%,仍然更优选地在10±5 wt.-%,还更优选地在10±4 wt.-%,最优选地在10±3 wt.-%,并且特别是在10±2 wt.-%的范围内的含量。
在一个优选的实施方案中,生理学上可接受的聚合物(优选聚环氧烷)与聚亚烷基二醇的相对重量比在5.4±2:1,更优选地在5.4±1.5:1,仍然更优选地在5.4±1:1,还更优选地在5.4±0.5:1,最优选地在5.4±0.2:1,并且特别是在5.4±0.1:1的范围内。该比率满足相对高的聚合物含量和良好可挤压性的要求。
塑化剂有时可起润滑剂的作用,并且润滑剂有时可起塑化剂的作用。
依据本发明的片剂的包衣颗粒和基质材料优选地不含选自以下的任何聚合物:
• 丙烯酸酯(例如丙烯酸和甲基丙烯酸聚合物,包括丙烯酸和甲基丙烯酸共聚物、甲基丙烯酸甲酯共聚物、甲基丙烯酸乙氧基乙酯、甲基丙烯酸氰乙基酯、聚(丙烯酸)、聚(甲基丙烯酸)、甲基丙烯酸烷基酰胺共聚物、聚(甲基丙烯酸甲酯)、聚甲基丙烯酸酯、聚(甲基丙烯酸甲酯)共聚物、聚丙烯酰胺、甲基丙烯酸氨烷基酯共聚物、聚(甲基丙烯酸酐)和甲基丙烯酸缩水甘油酯共聚物,例如Eudragit®
NE、NM、RS或RL)。
• 烷基纤维素和羟基烷基纤维素(例如甲基纤维素、乙基纤维素、羟丙基纤维素和羟丙基甲基纤维素);和
• 水合形成凝胶以控制水移动的胶凝剂,例如高分子量等级(高粘度)的羟丙基甲基纤维素(HPMC)、果胶、刺槐豆胶和黄原胶。
在一个优选的实施方案中,依据本发明的片剂不含刺激鼻道和/或咽的物质,即当经鼻道和/或经咽给予时造成对患者如此不愉快以致他/她不希望或不能继续给予的身体反应(例如烧灼)或在生理上阻碍相应的活性化合物的摄取(例如由于鼻分泌物或打喷嚏增多)的物质。刺激鼻道和/或咽的物质的其它实例为引起烧灼感、瘙痒、打喷嚏的冲动、增加分泌物的形成或这些刺激中至少两种的组合的那些物质。常规使用的相应物质及其量为本领域技术人员已知的。刺激鼻道和/或咽的一些物质因此基于辛辣物质药物(hot substance drug)的一种或多种组分或者一种或多种植物部分。相应的辛辣物质药物本身为本领域技术人员已知的,并描述于例如Prof. Dr.
Hildebert Wagner的“Pharmazeutische
Biologie-Drogen und ihre Inhaltsstoffe”, 第2次修订版, Gustav Fischer
Verlag, Stuttgart-New York, 1982, 第82页等中。相应的描述作为参考文献引入本文,并且视为本公开的部分。
此外,依据本发明的片剂优选地不含药理学活性化合物的拮抗剂,优选地不含针对治疗精神病物质的拮抗剂,特别是不含抑制阿片样物质的拮抗剂。适合于给定的药理学活性化合物的拮抗剂为本领域技术人员已知的,并可以本身或以相应的衍生物,特别是酯或醚的形式存在,或在每一种情况下以相应的生理学上可接受的化合物的形式,特别是以其盐或溶剂合物的形式存在。依据本发明的片剂优选地不含选自以下的拮抗剂:纳洛酮、纳曲酮、纳美芬、纳立德(nalide)、纳美酮、烯丙吗啡或纳布芬(naluphine),在每种情况下任选地以相应的生理学上可接受的化合物的形式,特别是以碱、盐或溶剂合物的形式存在;并且不含精神抑制药,例如选自以下的化合物:氟哌啶醇、异丙嗪(promethacine)、氟奋乃静、奋乃静、左美丙嗪、硫利达嗪、培拉嗪、氯丙嗪、氯普噻吨(chlorprothixine)、珠氯噻醇(zuclopenthixol)、氟哌噻吨、丙硫喷地、佐替平、苯哌利多、匹泮哌隆、美哌隆和溴哌醇。
此外,依据本发明的片剂优选地不含催吐剂。催吐剂为本领域技术人员已知的,并可以本身或以相应的衍生物,特别是酯或醚的形式存在,或在每一种情况下以相应的生理学上可接受的化合物的形式,特别是以其盐或溶剂合物的形式存在。依据本发明的片剂优选地不含基于吐根树(ipecacuanha
(吐根植物))的根的一种或多种组分的催吐剂,例如基于如例如Prof.Dr.Hildebert
Wagner 的“Pharmazeutische Biologie-Drogen und
ihreInhaltsstoffe”, 第2次修订版,Gustav Fischer
Verlag, Stuttgart-New York, 1982中描述的成分吐根碱的催吐剂。相应的文献描述作为参考文献引入本文,并且视为本公开的部分。依据本发明的片剂优选地也不含阿扑吗啡作为催吐剂。
最后,依据本发明的片剂优选地也不含苦味物质。苦味物质和有效用量可见于US-2003/0064099
Al中,其相应的公开应视为本申请的公开内容,并作为参考文献引入本文。苦味物质的实例为芳香油,例如薄荷油、桉树油、苦杏仁油、薄荷醇、果实芳香物质;来自柠檬、橙、酸柚、葡萄柚的芳香物质或其混合物,和/或地那铵苯甲酸盐。
依据本发明的片剂因此优选地既不含刺激鼻道和/或咽的物质,也不含药理学活性化合物的拮抗剂,也不含催吐剂,也不含苦味物质。
包衣颗粒的药理学活性化合物、生理学上可接受的聚合物(优选聚环氧烷)、塑化剂和抗氧化剂相对于包衣颗粒总重量的特别优选的含量,作为实施方案B1-B6在此概述于下表中:

其中药理学活性化合物优选地为阿片样物质,特别优选地为他喷他多或其生理学上可接受的盐;聚合物优选地为聚环氧烷,更优选地为具有至少500,000
g/mol的重均分子量的聚环氧乙烷;塑化剂优选地为聚乙二醇;和抗氧化剂优选地为α-生育酚。
除了包衣颗粒和优选地预先压实或粒状的基质材料外,依据本发明的片剂还可包含一种或多种药用赋形剂,例如粘合剂、填充剂、润滑剂等。
在一个优选的实施方案中,片剂另外包含润滑剂。硬脂酸镁为优选的。以上描述了进一步优选的润滑剂,因此下文不再重复。
如果片剂在优选地预先压实或预先制粒的基质材料外部含有另外的润滑剂,其含量基于片剂的总重量计优选地不多于1
wt.-%,更优选地不多于0.5 wt.-%。
尽管在依据本发明的片剂中含有的包衣颗粒优选地呈现增加的机械强度,片剂本身优选地具有常规的机械性能。一般地,依据本发明的片剂可例如通过锤子进行粉碎,从而得到含有基质材料、包衣颗粒和在片剂中含有的任何其它成分的破裂组合物。然而,由此以或多或少的分离形式得到的包衣颗粒优选地不能通过锤子进一步粉碎和破裂。
优选地,包衣颗粒被热熔挤出和/或具有至少300 N的断裂强度。
依据本发明的片剂为抗破碎的。优选地,基于包衣颗粒的机械性能实现抗破碎性,以致避免或至少基本上阻碍粉碎。根据本发明,术语粉碎意指采用滥用者通常可利用的常规手段,例如杵和研钵、锤子、槌或用于在力作用下粉碎的其它常规手段使包衣颗粒粉末化。因此,抗破碎性优选地意指避免或至少基本上阻碍使用常规手段使包衣颗粒粉末化。
优选地,依据本发明的包衣颗粒的机械性能,特别是其断裂强度和可变形性,基本上依赖于生理学上可接受的聚合物(优选聚环氧烷)的存在和空间分布,尽管其纯粹的存在一般地不足实现所述性能。通过借助于用于制备片剂的常规方法简单加工药理学活性化合物、生理学上可接受的聚合物(优选聚环氧烷)和任选的其它赋形剂,可能无法自动地获得依据本发明的包衣颗粒的有利机械性能。事实上,通常必须选择合适的装置用于制备,并且必须调节关键的工艺参数,特别是压力/力、温度和时间。因此,即使使用常规装置,通常还必须调整加工方案以满足所要求的标准。
通常,只有在颗粒制备期间才可得到呈现期望的性能的包衣颗粒:
- 合适的组分
- 以合适的量
暴露于
- 足够的压力
- 在足够的温度下
- 持续足够的时间段。
因此,不管所使用的装置,必需调整加工方案以满足所要求的标准。因此,颗粒的断裂强度和可变形性与组成是可区分的。
在依据本发明的片剂中含有的包衣颗粒优选地具有至少300 N、至少400 N或至少500 N,优选地为至少600 N,更优选地为至少700 N,仍然更优选地为至少800 N,还更优选地为至少1000 N,最优选地为至少1250 N,并且特别是至少1500 N的断裂强度。
为了验证颗粒是否呈现例如300 N或500 N的特定断裂强度,通常没有必要使所述颗粒分别受到远高于300 N和500 N的力。因此,一旦略微超过对应于期望的断裂强度的力,例如分别在例如330 N和550 N的力下,通常可以终止断裂强度试验。
片剂和颗粒的“断裂强度”(抗压碎性)为技术人员已知的。在这方面可参照例如W.A. Ritschel, Die Tablette, 2. Auflage,
Editio Cantor Verlag Aulendorf, 2002; H Liebermann et al., Tablets: Tablets,第2卷,Informa
Healthcare;第2版,1990;和制药技术百科全书(Encyclopedia of Pharmaceutical Technology), Informa
Healthcare;第1版。
为了说明书的目的,断裂强度优选地定义为使颗粒断裂所必需的力(=破坏力)的量。因此,对于说明书的的目,颗粒优选地在其破裂,即断裂成至少两个相互分离的独立部分时不呈现期望的断裂强度。然而,在另一个优选的实施方案中,如果力降低了在测量(见下文)期间测量的最高力的2550%(阈值),则颗粒视为破裂。
为了说明书的目的,依据本发明的包衣颗粒的机械性能本质上指所述包衣颗粒的芯而不是其包衣的机械性能。因此,如果依据本发明的包衣颗粒被施加外力,引起材料部分或全部与芯分离,然而其反过来不破裂,那么包衣颗粒仍然呈现期望的性能。
依据本发明的包衣颗粒与可在片剂中含有的常规颗粒的区别在于,由于其断裂强度,它们不能通过用常规手段,例如杵和研钵、锤子、槌或用于粉碎的其它常规手段,特别是为此目的开发的装置(片剂粉碎机)施加力来粉末化。在这方面,“粉碎”意指粉碎成小颗粒。避免粉末化事实上防止口服或非肠道,特别是静脉内或经鼻滥用。
常规颗粒一般地具有远低于200 N的断裂强度。
可按照以下经验公式估测常规圆形片剂/颗粒的断裂强度:
断裂强度[以N计] = 10 x 片剂/颗粒直径 [以mm计]。
因此,按照所述经验公式,具有至少300 N的断裂强度的圆形片剂/颗粒将需要至少30 mm的直径。然而,这种片剂无法吞咽,更不必说含有多个这样的颗粒的片剂。以上经验公式优选地不适用于依据本发明的包衣颗粒,依据本发明的包衣颗粒不是常规的而是特殊的。
进一步地,实际平均咀嚼力为约220 N (参见例如P.A.
Proeschel et al.,J Dent Res, 2002,81(7),464-468)。这意味着具有远低于200 N的断裂强度的常规颗粒可以在自发咀嚼时被粉碎,而依据本发明的包衣颗粒则优选地不能被嚼碎。
更进一步地,当施加约9.81 m/s2的重力加速度时,300 N相当于大于30 kg的重力,即依据本发明的包衣颗粒可优选地承受大于30 kg的重量而不粉碎。
用于测量片剂的断裂强度的方法为技术人员已知的。合适的装置为市售可得到的。
例如,可按照欧洲药典5.0、2.9.8或6.0、2.09.08“抗片剂粉碎性(Resistance to Crushing of Tablets)”测量断裂强度(抗压碎性)。该试验打算在所限定的条件下分别测定片剂和颗粒的抗压碎性,这通过经粉碎破坏它们所需的力来测量。该装置由相互面对的2个钳口(jaws)组成,其中一个朝另一个运动。钳口的平坦表面垂直于运动方向。钳口的压碎表面为平坦的并分别大于与片剂和颗粒的接触区域。使用精密度为1牛顿的系统校准装置。将片剂和颗粒分别置于钳口之间,如果适用,将形状、疵点(break-mark)和刻痕(inscription)考虑进去;对每一个测量而言,分别使片剂和颗粒以与施力方向(和其中要测量断裂强度的延伸方向)相同的方式定向。分别对10个片剂和颗粒进行测量,注意在每次测定之前去除所有碎片。结果表示为测量的力的平均值、最小值和最大值,均以牛顿表示。
断裂强度(破裂力)的类似描述可见于USP。或者可按照其中描述的方法(其中指出,断裂强度为分别使片剂和颗粒在特定平面破坏(即破裂)需要的力)测量断裂强度。通常分别将片剂和颗粒置于两个压板之间,其中一个移动以分别向片剂和颗粒施加足以造成破裂的力。分别对于常规圆形(圆形截面)片剂和颗粒而言,负载(loading)横跨(across)其直径存在(有时称为径向负载(diametral loading)),并在该平面发生断裂。片剂和颗粒的破裂力在制药文献中通常分别称为硬度;然而,该术语的使用为误导性的。在材料科学中,术语硬度指表面对小探针穿入或压入的抗性。术语压碎强度也频繁地用于描述片剂和颗粒分别对施加压缩负载的抗性。尽管该术语比硬度更准确地描述该试验的真实性质,但其意味着片剂和颗粒分别在试验期间被实际粉碎,但其常常不是这样的情况。
或者,可按照WO 2008/107149测量断裂强度(抗压碎性),其可视为对在欧洲药典中描述的方法的改进。用于测量的装置优选地为“Zwick
Z 2.5”材料测试器,在1150 mm的最大拉伸下F最大= 2.5 kN,其应用以下来设置:一个柱和一个主轴,间隙低于100
mm,和测试速度可调整在0.1-800 mm/分钟之间,附有testControl软件。技术人员知道如何把测试速度适当调整至例如10
mm/分钟、20 mm/分钟或40
mm/分钟。使用以下进行测量:带有旋入式插入物和气缸(直径10 mm)的压力活塞、测力传感器、F最大.1 kN,直径=8 mm,0.5级从10 N开始,1级从2 N开始至ISO 7500-1,具有按照DIN
55350-18 (Zwick总力F最大=1.45 kN)的制备商试验证书M (所有装置得自Zwick GmbH & Co. KG, Ulm, 德国) (测试器的订单号BTC-FR 2.5 TH.
D09,测力传感器的订单号BTC-LC 0050N. P01,定心装置的订单号BO 70000 S06)。
当使用testControl软件(testXpert
V10.11)时,以下示例性的设置和参数显示是有用的:LE-位置:夹具距离150 mm。LE-速度:500 mm/分钟,预行程后的夹具距离:195 mm,预行程速度(pre-travel speed):500
mm/分钟,没有预先加力(pre-force)控制-预先加力: 预先加力1 N,预先加力速度10 mm/分钟-样品数据:无样品形式,测量长度导线距离(traverse
distance) 10 mm,在测试-测试/测试结束之前不需要输入;测试速度:位置控制的10
mm/分钟,延迟速度变化:1,力关闭阈值50% F最大,对于断裂试验没有力阈值,没有最大长度变化,力上限:600 N-膨胀补偿:没有校正测量长度-测试后操作:LE在测试后设置,没有卸载样品-TRS:数据存储:TRS距离间隔直到打破1 µm,TRS时间间隔0.1s,TRS力间隔1 N-机器;导线距离控制器:软上端358 mm,软下端192 mm-较低的测试空间。应确保上板和砧骨(ambos)平行排列-这些部件在测试期间或测试后不能触摸。测试后,在与所测试的颗粒密切接触的两个支架之间仍应存在小间隙(例如0.1或0.2 mm),代表变形颗粒的剩余厚度。
在一个优选的实施方案中,颗粒如果断裂成类似形态学的至少两个分离的碎片则被视为破裂。具有不同于变形颗粒的形态学的分开的物质(例如粉尘)不认为是符合破裂的定义的碎片。
依据本发明的包衣颗粒优选地在宽的温度范围内呈现机械强度,除了断裂强度(抗压碎性)外,任选地还有足够的硬度、屈服强度、疲劳强度、抗冲击性、冲击弹性、抗拉强度、压缩强度和/或弹性模量,任选地还在低温下(例如低于-24℃、低于-40℃或者甚至可能在液氮中)具有以上性质,以使其几乎不能通过自发的咀嚼、在研钵中研磨、捣碎等进行粉碎。因此,优选地,甚至在低或非常低的温度下,例如当将该片剂初始冷冻以提高其脆性,例如冷冻至低于-25℃、低于-40℃的温度或者甚至在液氮中时保持依据本发明的颗粒的比较高的断裂强度。
依据本发明的颗粒的特征在于一定程度的断裂强度。这并不意味着颗粒也必须呈现一定程度的硬度。硬度和断裂强度为不同的物理性质。因此,片剂的抗破碎性不一定依赖于包衣颗粒的硬度。例如,分别由于其断裂强度、冲击强度、弹性模量和抗拉强度,包衣颗粒在例如使用锤子施加外力时优选地可变形,例如塑性变形,但不能粉末化,即粉碎成大量碎片。换句话说,依据本发明的包衣颗粒的特征在于一定程度的断裂强度,但不一定特征也在于一定程度的形状稳定性。
因此,在说明书的含义中,当暴露于特定延伸方向上的力时变形但不破裂(塑性变形或塑性流动)的颗粒,优选地被视为在所述延伸方向上具有期望的断裂强度。
存在于依据本发明的片剂中的优选颗粒为具有如经本领域目前接受的测试方法测定的合适的抗拉强度的那些颗粒。进一步优选的颗粒为具有如经本领域的测试方法测定的杨氏模量(Youngs
Modulus)的那些颗粒。仍然进一步优选的颗粒为在破裂时具有可接受的伸长的那些颗粒。
不论依据本发明的颗粒是否具有增加的断裂强度与否,依据本发明的颗粒优选地呈现一定程度的可变形性。在依据本发明的片剂中含有的颗粒优选地具有可变形性,以致它们在经受如以上描述的断裂强度试验时,在力-位移-图(力-距离图)中,在位移相应减小下显示力的增加,优选地为力的稳定增加。
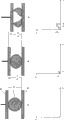
各个颗粒的这种机械性能即可变形性在图5和6中图解说明。
图5图解说明测量和相应的力-位移-图(力-距离图)。具体地讲,图5A显示测量开始时的初始状况。把样品颗粒(9)置于上部钳口(8a)和下部钳口(8b)之间,每一个钳口各自与颗粒(9)的表面密切接触。上部钳口(8a)与下部钳口(8b)之间的初始位移d0对应于与上部钳口(8a)和下部钳口(8b)的表面正交的颗粒延伸。此时,根本没有施加力,并且因此在下面的力-位移-图(力-距离图)中没有图表显示。当测量开始时,上部钳口优选地按恒速在下部钳口(8b)的方向移动。图5B显示一种状况,其中由于上部钳口(8a)向下部钳口(8b)运动,对颗粒(9)施加力。由于其可变形性,颗粒(9)变平而不断裂。力-位移-图(力-距离图)表明,在上部钳口(8a)与下部钳口(8b)的位移d0减小距离x1后,即在位移d1=d0- x1时,测量力F1。图5C显示一种状况,其中由于上部钳口(8a)向下部钳口(8b)连续运动,对颗粒(9)施加的力引起进一步变形,尽管颗粒(9)不断裂。力-位移-图(力-距离图)表明,在上部钳口(8a)与下部钳口(8b)的位移d0减小距离x2后,即在位移d2=d0-
x2时,测量力F2。在这些情况下,颗粒(9)未破裂(断裂),并且在力-位移-图(力-距离图)中测量到力的明显稳定增加。
与此相反,图6图解说明没有如同依据本发明的颗粒的可变形程度的常规比较性颗粒的测量和相应的力-位移-图(力-距离图)。图6A显示测量开始时的初始状况。把比较性样品颗粒(9)置于上部钳口(8a)和下部钳口(8b)之间,每一个钳口各自与比较性颗粒(9)的表面密切接触。上部钳口(8a)与下部钳口(8b)之间的初始位移d0对应于与上部钳口(8a)和下部钳口(8b)的表面正交的比较性颗粒的延伸。此时,根本没有施加力,并且因此在下面的力-位移-图(力-距离图)中没有图表显示。当测量开始时,上部钳口优选地按恒速在下部钳口(8b)的方向移动。图6B显示一种状况,其中由于上部钳口(8a)向下部钳口(8b)运动,对比较性颗粒(9)施加力。由于一些可变形性,比较性颗粒(9)稍微变平而不断裂。力-位移-图(力-距离图)表明在上部钳口(8a)与下部钳口(8b)的位移d0减小距离x1后,即在位移d1=d0- x1时,测量力F1。图6C显示一种状况,其中由于上部钳口(8a)向下部钳口(8b)连续运动,对颗粒(9)施加的力引起比较性颗粒(9)突然断裂。力-位移-图(力-距离图)表明,在上部钳口(8a)与下部钳口(8b)的位移d0减小距离x2后,即在位移d2=d0-
x2时,测量当颗粒断裂时突然下降的力F2。在这些情况下,颗粒(9)已经破裂(断裂),并且在力-位移-图(力-距离图)中没有测量到力的稳定增加。力的突然下降(减小)可易于觉察到,并不需要对测量量化。力-位移-图(力-距离图)中的稳定增加在颗粒破裂时的位移d2=d0-
x2结束。
在一个优选的实施方案中,在依据本发明的片剂中含有的颗粒具有可变形性,以致它们在经受如以上描述的断裂强度试验(“Zwick Z 2.5”材料测试器,恒速)时,在力-位移-图(力-距离图)中的位移相应减小时显示力的增加,优选地为力的明显稳定的增加,优选地至少直到上部钳口(8a)与下部钳口(8b)的位移d减小至初始位移d0的90%的数值(即d=0.9 • d0),优选地至初始位移d0的80%的位移d,更优选地至初始位移d0的70%的位移d,仍然更优选地至初始位移d0的60%的位移d,还更优选地至初始位移d0的50%的位移d,甚至更优选地至初始位移d0的40%的位移d,最优选地至初始位移d0的30%的位移d,并且特别是至初始位移d0的20%的位移d,或者至初始位移d0的15%的位移d、至初始位移d0的10%的位移d或至初始位移d0的5%的位移d。
在另一个优选的实施方案中,在依据本发明的片剂中含有的颗粒具有可变形性,以致它们在经受如以上描述的断裂强度试验(“Zwick Z 2.5”材料测试器,恒速)时,在力-位移-图(力-距离图)中的位移相应减小时显示力的增加,优选地为力的明显稳定的增加,优选地至少直到上部钳口(8a)与下部钳口(8b)的位移d减小至0.80 mm或0.75 mm,优选地为0.70 mm或0.65 mm,更优选地为0.60 mm或0.55 mm,仍然更优选地为0.50 mm或0.45 mm,还更优选地为0.40 mm或0.35 mm,甚至更优选地为0.30 mm或0.25 mm,最优选地为0.20 mm或0.15 mm,并且特别是为0.10或0.05 mm。
在还一个优选的实施方案中,在依据本发明的片剂中含有的颗粒具有可变形性,以致它们在经受如以上描述的断裂强度试验(“Zwick Z 2.5”材料测试器,恒速)时,在力-位移-图(力-距离图)中的位移相应减小时显示力的增加,优选地为力的明显稳定增加,至少直到上部钳口(8a)与下部钳口(8b)的位移d减小至初始位移d0的50% (即d=d0/2),而在所述位移(d=d0/2)下测得的力为至少25 N或至少50 N,优选地为至少75 N或至少100 N,仍然更优选地为至少150 N或至少200 N,还更优选地为至少250 N或至少300 N,甚至更优选地为至少350 N或至少400 N,最优选地为至少450 N或至少500 N,并且特别是为至少625 N、或至少750 N、或至少875 N、或至少1000 N、或至少1250 N、或至少1500 N。
在另一个优选的实施方案中,在依据本发明的片剂中含有的颗粒具有可变形性,以致它们在经受如以上描述的断裂强度试验(“Zwick Z 2.5”材料测试器,恒速)时,在力-位移-图中的位移相应减小时显示力的增加,优选地为力的明显稳定的增加,至少直到上部钳口(8a)与下部钳口(8b)的位移d减小至至少0.1 mm,更优选地为至少0.2 mm,仍然更优选地为至少0.3 mm,还更优选地为至少0.4
mm,甚至更优选地为至少0.5 mm,最优选地为至少0.6
mm,并且特别是为至少0.7 mm,而在所述位移下测得的力在5.0
N-250 N,更优选地在7.5 N-225 N,仍然更优选地在10 N-200 N,还更优选地在15
N-175 N,甚至更优选地在20 N-150 N,最优选地在25
N-125 N,并且特别是在30 N-100 N的范围内。
在仍然另一个实施方案中,在依据本发明的片剂中含有的颗粒具有可变形性,以致它们在如以上描述的断裂强度试验(“Zwick Z 2.5”材料测试器,恒速)中经受例如50 N、100 N、200 N、300 N、400 N、500 N或600 N的恒力时变形而不断裂,直到上部钳口(8a)与下部钳口(8b)的位移d减小,以致在所述恒力下不发生进一步的变形,而在这种平衡状态下,上部钳口(8a)与下部钳口(8b)的位移d为初始位移d0的至多90% (即d≤ 0.9 • d0),优选地为初始位移d0的至多80% (即d≤ 0.8 • d0),更优选地为初始位移d0的至多70% (即d≤ 0.7 • d0),仍然更优选地为初始位移d0的至多60% (即d≤ 0.6 • d0),还更优选地为初始位移d0的至多50% (即d≤ 0.5 • d0),甚至更优选地为初始位移d0的至多40% (即d≤ 0.4 • d0),最优选地为初始位移d0的至多30% (即d≤ 0.3 • d0),并且特别是初始位移d0的至多20% (即d≤ 0.2 • d0),或者初始位移d0的至多15% (即d≤ 0.15 • d0)、初始位移d0的至多10% (即d≤ 0.1 • d0)、或初始位移d0的至多5% (即d≤ 0.05 • d0).
优选地,在依据本发明的片剂中含有的颗粒具有可变形性,以致它们在如以上描述的断裂强度试验(“Zwick Z 2.5”材料测试器,恒速)中经受例如50 N、100 N、200 N、300 N、400 N、500 N或600 N的恒力时变形而不断裂,直到上部钳口(8a)与下部钳口(8b)的位移d减小,以致在所述恒力下不发生进一步的变形,而在这种平衡状态下,上部钳口(8a)与下部钳口(8b)的位移d为至多0.80 mm或至多0.75 mm,优选地为至多0.70 mm或至多0.65 mm,更优选地为至多0.60 mm或至多0.55 mm,仍然更优选地为至多0.50 mm或至多0.45
mm,还更优选地为至多0.40 mm或至多0.35
mm,甚至更优选地为至多0.30 mm或至多0.25
mm,最优选地为至多0.20 mm或至多0.15
mm,并且特别是为至多0.10或至多0.05
mm。
在另一个实施方案中,在依据本发明的片剂中含有的颗粒具有可变形性,以致它们在如以上描述的断裂强度试验(“Zwick Z 2.5”材料测试器,恒速)中经受例如50 N、100 N、200 N、300 N、400 N、500 N或600 N的恒力时变形而不断裂,直到上部钳口(8a)与下部钳口(8b)的位移d减小,以致在所述恒力下不发生进一步的变形,而在这种平衡状态下,上部钳口(8a)与下部钳口(8b)的位移d为初始位移d0的至少5% (即d ≥0.05 • d0),优选地为初始位移d0的至少10% (即d ≥0.1 • d0),更优选地为初始位移d0的至少15% (即d ≥0.15 • d0),仍然更优选地为初始位移d0的至少20% (即d ≥0.2 • d0),还更优选地为初始位移d0的至少30% (即d ≥0.3 • d0),甚至更优选地为初始位移d0的至少40% (即d ≥0.4 • d0),最优选地为初始位移d0的至少50% (即d ≥0.5 • d0),并且特别是为初始位移d0的至少60% (即d ≥0.6 • d0),或者初始位移d0的至少70% (即d ≥0.7 • d0)、初始位移d0的至少80% (即d ≥0.8 • d0)或初始位移d0的至少90% (即d ≥0.9 • d0)。
优选地,在依据本发明的片剂中含有的颗粒具有可变形性,以致它们在如以上描述的断裂强度试验(“Zwick Z 2.5”材料测试器,恒速)中经受例如50 N、100 N、200 N、300 N、400 N、500 N或600 N的恒力时变形而不断裂,直到上部钳口(8a)与下部钳口(8b)的位移d减小,以致在所述恒力下不发生进一步的变形,而在这种平衡状态下,上部钳口(8a)与下部钳口(8b)的位移d为至少0.05 mm或至少0.10 mm,优选地为至少0.15 mm或至少0.20 mm,更优选地为至少0.25 mm或至少0.30 mm,仍然更优选地为至少0.35 mm或至少0.40
mm,还更优选地为至少0.45 mm或至少0.50
mm,甚至更优选地为至少0.55 mm或至少0.60
mm,最优选地为至少0.65 mm或至少0.70
mm,并且特别是为至少0.75或至少0.80
mm。
优选地,按照欧洲药典,依据本发明的片剂在体外条件下提供药理学活性化合物的立即释放。
用于片剂的术语“立即释放”可被本领域的技术人员理解为其对各片剂具有结构含义。该术语例如在最新一期的美国药典(USP),General Chapter 1092,“溶解过程:开发和验证(THE
DISSOLUTION PROCEDURE: DEVELOPMENT AND VALIDATION)”,标题“研究设计(STUDY DESIGN)”、“时间点(Time
Points)”中定义。对于即释剂型,过程的持续时间一般为30-60分钟;在大多数情况下,单一时间点规范对于药典目的是足够的。产品可比性和性能的工业和监管理念可能需要另外的时间点,这对于产品注册或批准也可能是需要的。应选择足够数目的时间点,以充分表征溶出曲线的上升和平台期。根据在几个FDA指南中涉及的生物药剂学分类系统,用快速溶解产品配制的高可溶性、高渗透性药物,如果它们可在15分钟内显示释放85%或更多的活性药物物质,则不需经受曲线比较。对于这些类型的产品,一点测试就足够了。然而,大多数产品不属于这一类。即释产品的溶出曲线一般显示逐渐增加,在约30-45分钟时达到85%-100%。因此,在15、20、30、45和60分钟范围内的溶出时间点通常用于大多数即释产品。
优选地,在生理条件下,依据本发明的片剂在30分钟后已经释放至少70%,更优选地至少75%,仍然更优选地至少80%,还更优选地至少82%,最优选地至少84%,并且特别是至少86%的在片剂中最初含有的药理学活性化合物。
优选地,在生理条件下,依据本发明的片剂在10分钟后已经释放至少70%,更优选地至少73%,仍然更优选地至少76%,还更优选地至少78%,最优选地至少80%,并且特别是至少82%的在片剂中最初含有的药理学活性化合物。
进一步优选的释放曲线C1-C10在此概述于下表中[所有数据以所释放的药理学活性化合物的wt.-%表示]:

优选地,依据本发明的片剂的释放曲线、药物和药用赋形剂在储存时,优选地于密闭容器中,在升高的温度例如40℃下储存3个月时是稳定的。
关于释放曲线“稳定的”意指当比较初始释放曲线与储存后的释放曲线时,在任何给定的时间点,释放曲线相互偏离不多于20%,更优选地不多于15%,仍然更优选地不多于10%,还更优选地不多于7.5%,最优选地不多于5.0%,并且特别是不多于2.5%。
关于药物和药用赋形剂“稳定的”意指就药用产品的保质期而言,片剂满足EMEA的要求。
合适的体外条件为技术人员已知的。在这方面可参照例如欧洲药典。优选地,在以下条件下测量释放曲线:不配备铅锤(sinker)的浆式装置,50 rpm,37±5℃,900 mL人工肠液pH 6.8 (磷酸盐缓冲液)或pH 4.5。在一个优选的实施方案中,桨的转速增加至75
rpm。
在一个优选的实施方案中,依据本发明的片剂适合于每天给予一次。在另一个优选的实施方案中,依据本发明的片剂适合于每天给予两次。在还一个优选的实施方案中,依据本发明的片剂适合于每天给予三次。在又一个优选的实施方案中,依据本发明的片剂适合于比每天三次更加频繁地给予,例如每天4次、每天5次、每天6次、每天7次或每天8次。
为了说明书的目的,“每天两次”意指在各次给药之间相等或几乎相等的时间间隔,即约每12小时,或不同的时间间隔,例如8和16小时或10和14小时。
为了说明书的目的,“每天三次”意指在各次给药之间相等或几乎相等的时间间隔,即约每8小时,或不同的时间间隔,例如6、6和12小时或7、7和10小时。
优选地,依据本发明的片剂在体外条件下具有至多5分钟,更优选地为至多4分钟,仍然更优选地为至多3分钟,还更优选地为至多2.5分钟,最优选地为至多2分钟,并且特别是至多1.5分钟的按照欧洲药典测量的崩解时间。
已经惊讶地发现,口服剂型可被设计在抗破碎性、崩解时间和药物释放、载药量、加工性能(尤其是成片性)和患者依从性之间提供最好的折中。
已经发现依据本发明的片剂的崩解时间可能受到基质材料:颗粒的相对重量比的影响。通常,据观察该比率越高,则崩解越快。然而,该比率不能无限地(ad
ultimo)增加,因为需要考虑其它的片剂性能,特别是载药量及总的片剂大小和重量。因为需要给予一定剂量的药理学活性化合物,颗粒的含量仍应足够高,并且总的片剂重量不应超过一定限度,因为这将恶化患者的依从性,例如可吞咽性。
情况由于相反方向的趋势而更加复杂。具体地讲,已经发现依据本发明的片剂的成片性也可能受到基质材料:颗粒的相对重量比的影响。通常,据观察该比率越低,则成片性越好。该趋势平行于载药量的趋势。
因此,一方面崩解时间和另一方面成片性/载药量可通过发现最佳的折中进行优化。
类似地,抗破碎性和药物释放也相互对抗。尽管较小的颗粒一般应显示药理学活性化合物的更快释放,抗破碎性需要一些最小体积的包衣颗粒,以有效地防止滥用,例如静脉内给予。包衣颗粒越大,它们越不适合于鼻腔滥用。包衣颗粒越小,发生凝胶形成越快。
因此,一方面药物释放和另一方面抗破碎性可通过发现最佳的折中进行优化。
依据本发明的片剂的优选实施方案D1-D4在此概述于下表中:

依据本发明的包衣颗粒优选地通过熔融挤出进行制备,尽管为了制备依据本发明的包衣颗粒也可使用其它热成形方法,例如在升高的温度下压模或加热颗粒,所述颗粒在第一步骤中通过常规压制制备,然后在第二步骤中,在高于包衣颗粒中的生理学上可接受的聚合物(优选聚环氧烷)的软化温度下加热,以形成硬的片剂。在这方面,热成形意指丸块在施加热之后的成型或模制。在一个优选的实施方案中,包衣颗粒通过热熔挤出而被热成形。
在一个优选的实施方案中,包衣颗粒通过热熔挤出,优选地通过双螺杆挤出机制备。熔融挤出优选地提供熔融挤出的束,后者优选地被切割成单片,其然后任选地压制并形成为颗粒。优选地,通过阴模和阳模,优选地自通过熔融挤出得到的单片块实现压制。如果经熔融挤出得到,压制步骤优选地用显示环境温度,即在20-25℃范围内的温度的单片块实施。通过挤出得到的束可或者本身经受压制步骤,或者可在压制步骤之前进行切割。可通过常用技术,例如使用转刀或压缩空气,在升高的温度下,例如当挤出的束由于热熔挤出仍然温热,或者在环境温度下,即在已经使挤出的束冷却下来后实施这种切割。当挤出的束仍然温热时,优选地通过在挤出的束退出挤压模后立即切割挤出的束,实施把挤出的束单颗化(singulation)为挤出的颗粒。然而,当挤出的束以冷却状态切割时,优选地通过任选地把仍热的挤出的束经传送带运送,使之冷却下来并凝固,随后将其切割成为挤出颗粒,实施把挤出的束随后单颗化为挤出颗粒。或者,可如在EP-A
240906中描述的那样,通过在两个反向旋转压辊之间传送挤出物,并直接成形为颗粒发生成形。当然也可使挤出的束在仍然温热时经受压制步骤或经受切割步骤,其或多或少地在挤出步骤之后立即进行。挤出优选地通过双螺杆挤出机进行。
依据本发明的包衣颗粒可通过不同方法制备,以下更详细地解释其中特别优选的方法。先前技术已经描述了几种合适的方法。在这方面可参考例如WO
2005/016313、WO2005/016314、WO 2005/063214、WO
2005/102286、WO 2006/002883、WO 2006/002884、WO2006/002886、WO 2006/082097 和 WO
2006/082099。
通常,用于产生依据本发明的包衣颗粒的方法优选地包括以下步骤:
(a) 混合所有成分;
(b) 任选地预成形得自步骤(a)的混合物,优选地通过对得自步骤(a)的混合物施加热和/或力,所供给的热量优选地不足以将生理学上可接受的聚合物(优选聚环氧烷)加热至高达其软化点;
(c) 通过施加热和力使混合物硬化,可于施力期间和/或之前供热,并且所供给的热量足以将生理学上可接受的聚合物(优选聚环氧烷)加热至至少高达其软化点;并且之后使材料冷却并去除力;
(d) 任选地将所硬化的混合物单颗化(singulating);
(e) 任选地将颗粒成形;和
(f) 提供包衣,优选地为薄膜包衣。
可例如通过接触或借助于热气体例如热空气,或借助于超声直接供热;或者通过摩擦和/或剪切间接供热。可施加力和/或可例如通过直接压片或借助于合适的挤出机,特别是借助于配备有一或两个螺杆的螺杆挤出机(分别为单螺杆挤出机和双螺杆挤出机)或借助于行星齿轮式挤出机来将颗粒成形。
可在通过施加热和力的混合物硬化过程中(步骤(c))或在后续步骤(步骤(e))中提供颗粒的最终形状。在这两者情况下,所有组分的混合物优选地以塑化状态存在,即优选地在至少高于生理学上可接受的聚合物(优选聚环氧烷)的软化点的温度下进行成形。然而,在较低温度例如环境温度下挤出也是可能的并且可为优选的。
可例如借助于包含适当形状的阴模和阳模的压片机进行成形。
用于提供具有包衣,优选地具有薄膜包衣的颗粒的合适方法为技术人员已知的,例如流化床包衣、锅包衣法、聚析、干粉包衣、挤压包衣和phoqus技术(复制包衣)。优选地,颗粒例如在流化床喷雾干燥制粒机中通过喷雾(顶部喷雾或底部喷雾)进行包衣。
用于制备依据本发明的包衣颗粒的特别优选的方法包括热熔挤出。在这种方法中,通过借助于挤出机热成形来产生依据本发明的包衣颗粒,优选地没有任何可观察到的挤出物的随后变色。
该方法的特征在于
a) 混合所有组分;
b) 在挤出机中将生成的混合物至少加热至达到生理学上可接受的聚合物(优选聚环氧烷)的软化点,并通过施加力经挤出机的出口孔挤出,
c) 将仍塑性的挤出物单颗化并成形为颗粒或
d) 将冷却和任选地重新加热的单颗化的挤出物成形为颗粒。
根据工艺步骤a)的组分的混合也可在挤出机中进行。
组分也可在本领域技术人员已知的混合机中混合。混合机可例如为辊式混合机、摇动混合机、剪切混合机或强制式混合机。
已经在挤出机中至少加热至达到生理学上可接受的聚合物(优选聚环氧烷)的软化点的优选地熔融的混合物,经由具有至少一个孔的模具自挤出机挤出。
依据本发明的方法要求使用合适的挤出机,优选地为螺杆挤出机。配有两个螺杆的螺杆挤出机(双螺杆挤出机)为特别优选的。
优选地,在不存在水的情况下进行挤出,即不加入水。然而,可存在痕量的水(例如由大气湿度造成)。
挤出机优选地包含至少两个温度区,在第一个区中进行将混合物至少加热至达到生理学上可接受的聚合物(优选聚环氧烷)的软化点,所述第一个区在进料区和任选的混合区的下游。混合物的生产量优选地为1.0
kg-15 kg/小时。在一个优选的实施方案中,生产量为0.5/小时-3.5 kg/小时。在另一个优选的实施方案中,生产量为4-15
kg/小时。
在一个优选的实施方案中,模头压力在25-200巴的范围内。尤其可通过挤出机中的模具几何形状、温度分布曲线、挤出速度、模具中的孔数、螺杆构型、第一进料步骤等调节模头压力。
模具几何形状或孔的几何形状可自由选择。模具或孔可因此呈现圆形、长椭圆形或椭圆形横截面,其中圆形横截面优选地具有0.1
mm-2 mm的直径。优选地,模具或孔具有圆形横截面。根据本发明使用的挤出机的套管(casing)可加热或冷却。设置相应的温度控制,即加热或冷却,以使要挤出的混合物呈现至少与生理学上可接受的聚合物(优选聚环氧烷)的软化温度相当的平均温度(产品温度),并且不升至高于可能破坏要加工的药理学活性化合物的温度。优选地,将要挤出的混合物的温度调节至低于180℃,优选地低于150℃,但是至少调节至生理学上可接受的聚合物(优选聚环氧烷)的软化温度。典型的挤出温度为120℃和150℃。
在一个优选的实施方案中,挤出机转矩在30-95%的范围内。尤其可通过挤出机中的模具几何形状、温度分布曲线、挤出速度、模具中的孔数、螺杆构型、第一进料步骤等调节挤出机转矩。
在挤出熔化的混合物并任选冷却挤出的束或挤出的多条束后,优选地将挤出物单颗化。可优选地通过借助于回转或旋转切刀、金属线、刀片或借助于激光切割机切割挤出物来进行这种单颗化。
优选地,任选地单颗化的挤出物或最终形状的依据本发明的颗粒的中间或最终储存在无氧气氛下进行,所述无氧气氛可例如通过借助于除氧剂来实现。
可将单颗化的挤出物压制成形为颗粒,以赋予颗粒的最终形状。
通过控制挤出机中的传送装置的转速及其几何学,和通过以在挤出机中积聚用于挤出塑化的混合物必需的压力(优选在临挤出之前)的这种方式设置出口孔的尺寸,来调节在挤出机中施加到所述至少塑化的混合物上的力。可通过简单的初步测试确定对每一种特定组合物而言产生具有期望的机械性能的片剂必需的挤出参数。
例如,但不受限制,可借助于ZSE 18或ZSE
27型双螺杆挤出机(Leistritz, Niirnberg, 德国,螺杆直径为18或27 mm)进行挤出。可使用具有偏心或钝端的螺杆。可使用具有圆孔或具有许多孔的可加热模具,每一个孔具有0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9或1.0 mm的直径。可将挤出参数调节至例如以下值:螺杆转速:120 Upm;传送速度:对于ZSE
18为2 kg/小时,或者对于ZSE
27为3 kg/h、8
kg/h或者甚至10 kg/h和更多;产品温度:模具前125℃和模具后135℃;和夹套温度:110℃。通常可通过增加挤出机出口的模具数目来增加生产量。
优选地,借助于双螺杆挤出机或行星齿轮挤出机进行挤出,双螺杆挤出机(正转或反转)为特别优选的。
优选地通过借助于挤出机热成形来制备依据本发明的颗粒,没有任何可观察到的挤出物的随后变色。
用于制备依据本发明的包衣颗粒的方法优选地连续进行。优选地,该方法包括挤出所有组分的均匀混合物。如果如此得到的中间体(例如通过挤出得到的束)呈现均匀特性,则是特别有利的。特别期望的是均匀的密度、活性化合物的均匀分布、均匀的机械性质、均匀的孔隙度、表面的均匀外观等。仅在这些情况下才可确保药理学性质的一致性,例如释放曲线的稳定性,并可保持低的次品量。
优选地,依据本发明的包衣颗粒可视为“挤出丸粒”。术语“挤出丸粒”具有本领域技术人员理解的结构含义。本领域的技术人员知道丸状剂型可通过许多技术制备,包括:
• 药物在极品糖(nonpareil
sugar)或微晶纤维素珠上分层,
• 喷雾干燥,
• 喷雾冷凝,
• 转筒式造粒(rotogranulation),
• 热熔挤出,
• 低熔点材料的滚圆(spheronization),或
• 挤出-滚圆湿块。
因此,“挤出丸粒”可通过热熔挤出或通过挤出-滚圆得到。
“挤出丸粒”可区别于其它类型的丸粒,因为挤出丸粒一般地具有不同形状。挤出丸粒的形状一般地比完全球状的圆形更加切杆样(cut-rod-like)。
“挤出丸粒”可区别于其它类型的丸粒,因为它们结构上不同。例如,药物在极品上分层得到具有芯的多层丸粒,而挤出一般地产生包含所有成分的均匀混合物的单片块(monolithic
mass)。类似地,喷雾干燥和喷雾冷凝一般地产生球体,而挤出一般地产生可随后滚圆的圆柱形挤出物。
“挤出丸粒”与“凝聚丸粒”之间的结构差异是显著的,因为它们可影响活性物质自丸粒释放,并因此导致不同的药理学特征。因此,药学配制领域的技术人员不应认为“挤出丸粒”等同于“凝聚丸粒”。
依据本发明的片剂可通过任何常规方法制备。然而,优选地,片剂通过压制制备。因此,上文定义的包衣颗粒优选地与基质材料混合,例如掺和和/或制粒(例如湿法制粒),然后优选地以模具压制生成的混合物(例如掺合或制粒)以形成片剂。还设想本文描述的包衣颗粒可使用其它方法,例如通过熔体制粒(例如使用脂肪醇和/或水溶性蜡和/或水不溶性蜡)或高剪切制粒,随后通过压制结合到基质中。
当通过偏心式压片机制备依据本发明的片剂时,压制力优选地在5-15 kN的范围内。当通过旋转式压片机制备依据本发明的片剂时,压制力优选地在5-40
kN的范围内,在某些实施方案中>25 kN,在其它的实施方案中为约13 kN。
依据本发明的片剂可任选地包含包衣,例如装饰性包衣。包衣优选地在形成片剂之后应用。包衣可在固化过程之前或之后应用。优选的包衣为可自Colorcon得到的Opadry®包衣。其它优选的包衣为也可自Colorcon经市售得到的Opaglos®包衣。
依据本发明的片剂的特征在于良好的储存稳定性。优选地,在于40℃和75%相对湿度下储存4周后,药理学活性化合物的含量总计为其储存之前初始含量的至少98.0%,更优选地为至少98.5%,仍然更优选地为至少99.0%,还更优选地为至少99.2%,最优选地为至少99.4%,并且特别是为至少99.6%。用于测量片剂中的药理学活性化合物的含量的合适方法为技术人员已知的。在这方面指欧洲药典或美国药典,尤其是指反相HPLC分析。优选地,片剂储存于密闭的,优选地密封的容器中。
依据本发明的其它方面-另外权利要求类别的基础。
依据本发明的包衣颗粒和片剂可用于医学,例如作为镇痛剂。包衣颗粒和片剂因此特别适合于治疗或控制疼痛。在这样的片剂中,药理学活性化合物优选地为镇痛剂。
依据本发明的其它方面涉及如以上描述的用于治疗疼痛的片剂。
依据本发明的其它方面涉及如以上描述的片剂用于避免或阻碍其中含有的药理学活性化合物的滥用的用途。
依据本发明的其它方面涉及如以上描述的片剂用于避免或阻碍其中含有的药理学活性化合物的非故意超剂量的用途。
在这方面,本发明也涉及如以上描述的药理学活性化合物和/或如以上描述的生理学上可接受的聚合物(优选聚环氧烷)在制备依据本发明的片剂中的用途,所述片剂用于预防和/或治疗疾病,从而防止特别是由于通过机械作用粉碎片剂来超剂量使用药理学活性化合物。
实施例
以下实施例进一步说明本发明,但不视为对其范围的限制。
实施例1
研究颗粒大小对抗破碎性的相关性。
发现比较小的颗粒,例如具有0.5 mm x 0.5 mm的直径和长度的颗粒,已经提供一定程度的抗破碎性:当鼻腔给予时它们引起不愉快的感觉,并且此外由于粘膜上缺水,不能像口服给予时那么快地释放药理学活性化合物。因此,通过鼻腔冲击或急速(kick
or rush)给予这样的颗粒不太可能实现。因此,甚至当鼻腔给予时,这样的比较小的颗粒已经提供抗破碎性,即避免药物滥用或至少使药物滥用明显更加困难。此外,这样的比较小的颗粒具有优良的膨胀性,从而有效地防止转化为用于静脉内给予的液体剂型。
发现抗破碎性甚至可通过把颗粒大小增加至例如1.0 mm x 1.0 mm的直径和长度而得到进一步的改善。当鼻腔给予和缺乏足够的水时,这样的颗粒甚至提供更加不愉快的感觉,相当缓慢地释放药理学活性化合物。进一步地,它们也不能容易地转化为用于静脉内给予的液体剂型。
然而,这样在开具口服给予所述片剂的处方时更明显的阻滞效应对于期望的立即释放是不利的,必须在一方面抗破碎性和另一方面开具口服给予的处方时的药物立即释放之间,特别是关于崩解时间和药物释放动力学发现折中。此外,载药量、加工性能(尤其是成片性)和患者依从性也是要满足的重要要求。
认为800 µm x 800 µm的预定颗粒大小是最合适的,即考虑到在挤出过程期间,特别是当所述束退出模具时可能发生离模膨胀,以致挤出的束的直径实际上扩大至约1000
µm的直径(这取决于组成和挤出参数),认为最合适的是把挤压模的直径以及挤出的束的切割长度调整至800 µm。因此,当这种方式进行时,认为最合适的是制备具有约1000 µm直径(在离模膨胀后,挤压模的直径为800 µm)和约800
µm长度的挤出颗粒。
实施例2:
研究不同的颗粒组成并由此制备不同大小的颗粒。
颗粒组成在此概述于下表中:
把所有材料称重,筛分(手动筛分,1 mm),以14 rpm掺混(Bohle LM40具有MC5或MC10,依槽的大小而定) 15分钟,并热熔挤出(Leistritz挤出机ZSE18型,具有不同构型的螺杆)。
在以下挤出条件下挤出组合物1-9:

对于较大规模,可采用螺杆构型和可升高温度(例如HZ8和10:130℃,HZ11:145℃;或HZ11:150℃和极端剪切构型,生产量25 g/分钟)。
使用桨式装置50 rpm,在37℃下,以900 mL 0.1N HCl监测体外释放特性。结果描绘于图3中。
实施例3:
研究片剂中颗粒含量的影响。
测试以下组成:
具有总重量600 mg的片剂中的300
mg颗粒
具有总重量600 mg的片剂中的250
mg颗粒
具有总重量600 mg的片剂中的200
mg颗粒
在成片性与大小之间最有前途的折中显示具有总重量500 mg的片剂中为250 mg颗粒。认为对于患者依从性而言具有总重量600
mg的片剂太大,尽管颗粒与基质材料的相对重量比约1:1对于崩解时间和溶出时间而言似乎是有利的。
实施例4-1:
研究基质材料的影响-湿法制粒。
制备具有以下组成的颗粒用于制备丸粒-片剂。相外的颗粒即基质材料通过湿法制粒来制备。掺混颗粒和丸粒。评价片剂在压制后的分离(任选地)和崩解。使用单工位压片机(Korsch EK0)“手工”制备片剂(对于每种片剂组分单独称重,并在成片之前直接混合)。
| a | Galen IQ,在Diosna羧甲基淀粉钠(5%)水性制粒 | 可检测到混合物没有分离 | 崩解试验:3分钟后不可检测到崩解。 |
| b | Galen IQ,在Diosna聚乙烯吡咯烷酮(Diosna Kollidon) CL (5%)水性制粒 | 可检测到混合物没有分离在已经压制3片时混合物显示明显的阳模沉积 | 崩解试验:3分钟后表面略溶。 |
| c | 微晶纤维素(Avicel),具有PVP-制粒溶液 | 可检测到混合物明显分离 | 崩解试验:3分钟后部分崩解。 |
| d | MCC+乳糖(20:80),具有PVP-制粒溶液 | 可检测到混合物没有分离 | 崩解试验:3分钟后不可检测到崩解。 |
| d | MCC+乳糖(50:50),具有PVP-制粒溶液 | 可检测到混合物轻微分离 | 崩解试验:3分钟后部分崩解。 |
| e | Gelcarin +乳糖(20%+80%) +水(57% + 43%) | 可检测到混合物没有分离 | 崩解试验:3分钟后不可检测到崩解。 |
| f | 糖酯S-1570 +磷酸三钙+微晶纤维素(Acivel) + Gelcarin | 可检测到混合物明显分离 | 崩解试验:3分钟后不可检测到崩解。 |
| g | 来自蔗糖的结壳颗粒 | 颗粒不能制备或仅有困难与颗粒掺混不可能->因此,没有片剂制备 | 没有片剂制备 |
不能自以上组成制备快速崩解片剂,可能是因为崩解剂在湿法制粒过程期间失去崩解能力。
实施例4-2:
研究基质材料的影响-干法制粒-碾压。
通过重击处理以下组成,包括以下步骤:
• 称重/调剂组分
• 筛分/掺混
• 使用单工位压片机(Korsch EK0),25 kN压制力制备20 mm直径的双平面片剂
• 把片剂破碎成许多部分(手动)并使用Frewitt筛分机(Frewitt Sieving machine)筛分(1.5
mm筛孔尺寸)
• 使用颗粒作为丸粒-片剂的外相/基质材料
试验结果概述于下表中:


研究含有如此压实的基质材料的片剂释放特性。结果描绘于图4中(900 mL HCl, 50 rpm, 没有铅锤的浆式装置)。
实施例4-3:
因为压制药片(slugging)方法不是干法制粒的技术状态,借助于碾压机进行关于干法制粒的相应试验。这具有所有的相关参数(辊位移、压制力、制粒机尺寸)可进行调整,以致得到具有期望的性能(粒度、硬度、压缩性、密度)的颗粒的优点。
参数(Gerteis MiniPactor):
辊位移:2-3 mm
转速:2-5 rpm
压实力:3-15 kN/cm
筛号:1.0-1.25-1.5-2.0 mm
如此制备的坯块(compacts) (干颗粒)与颗粒掺混并压制成片剂。在掺混时,作为既不在坯块中含有也不在颗粒中含有的外部赋形剂加入润滑剂(分别为硬脂酸镁和硬脂基富马酸钠)。

试验显示自坯块制备的和自压制颗粒制备的片剂显示类似的快速释放。
确认实验:

实施例4-4:
自依据实施例2-5的颗粒(250
mg)和依据实施例4-3 #12的基质材料(250
mg)制备片剂(500 mg)。
按照欧洲药典测定体外释放。

把片剂的体外释放与含有盐酸他喷他多的非抗破碎性市售的产品(薄膜包衣片剂)相比较。30分钟后(依据欧洲药典2.9.3),两种剂型释放整个量的药理学活性化合物(100%)。
实施例5:
在以下条件下研究常规的市售中性丸粒的机械性能:

测量板与砧骨之间的位移减少x (以mm计) (=“压缩[c]”)和相应的力f (以N计)。在测量期间测量的最大力f最大和位移的相应减小x最大在此概述于下表中:
自以上数据很明显,实施例5-1的比较性颗粒在仅为约5 N的很低的力下破裂,并可变形达少于0.1 mm。与此相反,实施例5-2和5-3的本发明颗粒根本不破裂,并可变形(变平)达多于0.8 mm。
相应的力位移图分别显示于图7、8和9中。
Claims (14)
1. 一种抗破碎片剂,其包含
(i) 超过片剂总重量的三分之一的量的基质材料;和
(ii) 少于片剂总重量的三分之二的量的多个包衣颗粒;其中所述颗粒包含药理学活性化合物和生理学上可接受的聚合物;并形成基质材料中的非连续相;
其按照欧洲药典,在体外条件下提供药理学活性化合物的立即释放。
2. 依据权利要求1的片剂,其在体外条件下具有按照欧洲药典测量的至多3分钟的崩解时间。
3. 依据前述权利要求中任何一项的片剂,其中所述基质材料的含量为基于片剂的总重量的至少40
wt.-%。
4. 依据前述权利要求中任何一项的片剂,其中所述药理学活性化合物为阿片样物质。
5. 依据前述权利要求中任何一项的片剂,其中所述颗粒具有约1000±250 μm的平均直径和/或约750±250
μm的平均长度。
6. 依据前述权利要求中任何一项的片剂,其中所述聚合物的含量为基于颗粒的总重量的至少25
wt.-%。
7. 依据前述权利要求中任何一项的片剂,其中所述药理学活性化合物含量为基于颗粒的总重量的至少25
wt.-%。
8. 依据前述权利要求中任何一项的片剂,其具有500±300 mg的总重量和含有
(i) 50±15 wt.-%的具有平均粒度800±400 µm的包衣颗粒,其中所述包衣颗粒包含
• 23±20 wt.-%药理学活性化合物,
• 22±12 wt.-%生理学上可接受的聚合物,
• 任选地,4.0±3.5 wt.-%塑化剂,和
• 任选地,0.05±0.05 wt.-%其它赋形剂;和
(ii) 49±15 wt.-%基质材料,其中所述基质材料包含
• 43±10 wt.-%填充剂和/或粘合剂,
• 任选地,5±4 wt.-%崩解剂和
• 任选地,0.15±0.15 wt.-%润滑剂;
所有的wt.-%均相对于片剂的总重量表示。
9. 依据前述权利要求中任何一项的片剂,其中所述颗粒被热熔挤出。
10. 依据前述权利要求中任何一项的片剂,其中所述聚合物为聚环氧烷。
11. 依据前述权利要求中任何一项的片剂,其中所述基质材料也以颗粒形式存在。
12. 依据前述权利要求中任何一项的片剂,其中所述颗粒用包含水溶性聚合物的包衣材料进行包衣。
13. 依据权利要求13的片剂,其中所述水溶性聚合物选自纤维素酯、纤维素醚、聚(甲基)丙烯酸酯、乙烯基聚合物和天然成膜剂。
14. 依据前述权利要求中任何一项的片剂,其中所述基质材料包含粘合剂、填充剂、崩解剂和/或润滑剂。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11006254.4 | 2011-07-29 | ||
| EP11006254 | 2011-07-29 | ||
| PCT/EP2012/003188 WO2013017234A1 (en) | 2011-07-29 | 2012-07-27 | Tamper-resistant tablet providing immediate drug release |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN103841964A true CN103841964A (zh) | 2014-06-04 |
Family
ID=45034267
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201280038092.1A Pending CN103841964A (zh) | 2011-07-29 | 2012-07-27 | 提供药物立即释放的抗破碎片剂 |
Country Status (30)
| Country | Link |
|---|---|
| US (2) | US10201502B2 (zh) |
| EP (1) | EP2736495B1 (zh) |
| JP (1) | JP2014524925A (zh) |
| KR (1) | KR20140053159A (zh) |
| CN (1) | CN103841964A (zh) |
| AR (1) | AR087360A1 (zh) |
| AU (1) | AU2012289764B2 (zh) |
| BR (1) | BR112014002022A2 (zh) |
| CA (1) | CA2839123A1 (zh) |
| CL (1) | CL2013003363A1 (zh) |
| CO (1) | CO6811869A2 (zh) |
| CY (1) | CY1119500T1 (zh) |
| DK (1) | DK2736495T3 (zh) |
| EA (1) | EA201400172A1 (zh) |
| EC (1) | ECSP14013168A (zh) |
| ES (1) | ES2655900T3 (zh) |
| HK (1) | HK1198132A1 (zh) |
| HR (1) | HRP20171506T1 (zh) |
| HU (1) | HUE034711T2 (zh) |
| IL (1) | IL229517A0 (zh) |
| LT (1) | LT2736495T (zh) |
| MX (1) | MX348491B (zh) |
| NO (1) | NO2736495T3 (zh) |
| PE (1) | PE20141638A1 (zh) |
| PL (1) | PL2736495T3 (zh) |
| PT (1) | PT2736495T (zh) |
| RS (1) | RS56528B1 (zh) |
| SI (1) | SI2736495T1 (zh) |
| WO (1) | WO2013017234A1 (zh) |
| ZA (1) | ZA201309297B (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107889459A (zh) * | 2015-04-24 | 2018-04-06 | 格吕伦塔尔有限公司 | 具有立即释放和对溶剂萃取的抗性的抗篡改剂型 |
Families Citing this family (57)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1429744A1 (en) | 2001-09-21 | 2004-06-23 | Egalet A/S | Morphine polymer release system |
| WO2003024429A1 (en) | 2001-09-21 | 2003-03-27 | Egalet A/S | Polymer release system |
| US7776314B2 (en) | 2002-06-17 | 2010-08-17 | Grunenthal Gmbh | Abuse-proofed dosage system |
| ATE495732T1 (de) | 2003-03-26 | 2011-02-15 | Egalet As | Morphin-system mit kontrollierter freisetzung |
| DE10361596A1 (de) * | 2003-12-24 | 2005-09-29 | Grünenthal GmbH | Verfahren zur Herstellung einer gegen Missbrauch gesicherten Darreichungsform |
| DE102005005446A1 (de) | 2005-02-04 | 2006-08-10 | Grünenthal GmbH | Bruchfeste Darreichungsformen mit retardierter Freisetzung |
| DE10336400A1 (de) | 2003-08-06 | 2005-03-24 | Grünenthal GmbH | Gegen Missbrauch gesicherte Darreichungsform |
| US20070048228A1 (en) | 2003-08-06 | 2007-03-01 | Elisabeth Arkenau-Maric | Abuse-proofed dosage form |
| DE102004032049A1 (de) | 2004-07-01 | 2006-01-19 | Grünenthal GmbH | Gegen Missbrauch gesicherte, orale Darreichungsform |
| DE102005005449A1 (de) | 2005-02-04 | 2006-08-10 | Grünenthal GmbH | Verfahren zur Herstellung einer gegen Missbrauch gesicherten Darreichungsform |
| US8445018B2 (en) | 2006-09-15 | 2013-05-21 | Cima Labs Inc. | Abuse resistant drug formulation |
| AU2014200915A1 (en) * | 2006-10-03 | 2014-03-13 | Tekmira Pharmaceuticals Corporation | Lipid containing formulations |
| DE102007011485A1 (de) * | 2007-03-07 | 2008-09-11 | Grünenthal GmbH | Darreichungsform mit erschwertem Missbrauch |
| WO2008148798A2 (en) | 2007-06-04 | 2008-12-11 | Egalet A/S | Controlled release pharmaceutical compositions for prolonged effect |
| KR101616246B1 (ko) | 2008-01-25 | 2016-05-02 | 그뤼넨탈 게엠베하 | 약제학적 투여형 |
| MX2010012039A (es) | 2008-05-09 | 2010-11-30 | Gruenenthal Gmbh | Proceso para la preparacion de una formulacion de polvo intermedia y una forma de dosificacion solida final bajo el uso de un paso de congelacion por rocio. |
| NZ594207A (en) | 2009-02-06 | 2013-03-28 | Egalet Ltd | Immediate release composition resistant to abuse by intake of alcohol |
| NZ597283A (en) | 2009-06-24 | 2013-07-26 | Egalet Ltd | Controlled release formulations |
| MX2012000317A (es) | 2009-07-22 | 2012-02-08 | Gruenenthal Gmbh | Forma de dosificacion de liberacion controlada extruida por fusion en caliente. |
| RU2015138422A (ru) | 2009-07-22 | 2018-12-25 | Грюненталь Гмбх | Стабильная при окислении, прочная на излом лекарственная форма |
| WO2011095314A2 (en) * | 2010-02-03 | 2011-08-11 | Grünenthal GmbH | Preparation of a powdery pharmaceutical composition by means of an extruder |
| CA2808219C (en) | 2010-09-02 | 2019-05-14 | Gruenenthal Gmbh | Tamper resistant dosage form comprising inorganic salt |
| ES2487244T3 (es) | 2010-09-02 | 2014-08-20 | Grünenthal GmbH | Forma de dosificación resistente a la manipulación que comprende un polímero aniónico |
| AU2012292418B2 (en) | 2011-07-29 | 2017-02-16 | Grunenthal Gmbh | Tamper-resistant tablet providing immediate drug release |
| CA2839123A1 (en) | 2011-07-29 | 2013-02-07 | Grunenthal Gmbh | Tamper-resistant tablet providing immediate drug release |
| ES2703688T3 (es) * | 2011-09-16 | 2019-03-12 | Purdue Pharma Lp | Formulaciones de liberación inmediata resistentes a alteración |
| MX356421B (es) | 2012-02-28 | 2018-05-29 | Gruenenthal Gmbh | Forma de dosificacion resistente a la manipulacion indebida que comprende un compuesto farmacologicamente activo y un polimero anionico. |
| CN104394851B (zh) | 2012-04-18 | 2017-12-01 | 格吕伦塔尔有限公司 | 抗篡改和抗剂量‑倾泻药物剂型 |
| US10064945B2 (en) | 2012-05-11 | 2018-09-04 | Gruenenthal Gmbh | Thermoformed, tamper-resistant pharmaceutical dosage form containing zinc |
| KR20150059167A (ko) | 2012-07-06 | 2015-05-29 | 에갈렛 리미티드 | 제어된 방출을 위한 남용 제지 약학적 조성물 |
| WO2014011830A1 (en) | 2012-07-12 | 2014-01-16 | Mallinckrodt Llc | Extended release, abuse deterrent pharmaceutical compositions |
| AR096438A1 (es) | 2013-05-29 | 2015-12-30 | Gruenenthal Gmbh | Forma de dosificación resistente al uso indebido con perfil de liberación bimodal, proceso |
| CA2907950A1 (en) * | 2013-05-29 | 2014-12-04 | Grunenthal Gmbh | Tamper-resistant dosage form containing one or more particles |
| BR112016000194A8 (pt) | 2013-07-12 | 2019-12-31 | Gruenenthal Gmbh | forma de dosagem resistente à violação contendo o polímero de acetato de etileno-vinila |
| US10195153B2 (en) | 2013-08-12 | 2019-02-05 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded immediate release abuse deterrent pill |
| ES2774304T3 (es) * | 2013-08-12 | 2020-07-20 | Pharmaceutical Manufacturing Res Services Inc | Píldora extruida disuasoria de abuso, de liberación inmediata |
| US20160199369A1 (en) * | 2013-08-12 | 2016-07-14 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded Immediate Release Abuse Deterrent Pill |
| CN105934241B (zh) | 2013-11-26 | 2020-06-05 | 格吕伦塔尔有限公司 | 通过低温研磨制备粉末状药物组合物 |
| CA2934078C (en) * | 2013-12-17 | 2019-08-06 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US10172797B2 (en) | 2013-12-17 | 2019-01-08 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US9492444B2 (en) | 2013-12-17 | 2016-11-15 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| WO2015173195A1 (en) | 2014-05-12 | 2015-11-19 | Grünenthal GmbH | Tamper resistant immediate release capsule formulation comprising tapentadol |
| JP2017516789A (ja) | 2014-05-26 | 2017-06-22 | グリュネンタール・ゲゼルシャフト・ミト・ベシュレンクテル・ハフツング | エタノール過量放出に対して防護されている多粒子 |
| ES2809458T3 (es) | 2014-07-17 | 2021-03-04 | Pharmaceutical Manufacturing Res Services Inc | Forma de dosificación llena de líquido, disuasoria del abuso y de liberación inmediata |
| US9132096B1 (en) | 2014-09-12 | 2015-09-15 | Alkermes Pharma Ireland Limited | Abuse resistant pharmaceutical compositions |
| JP2017531026A (ja) | 2014-10-20 | 2017-10-19 | ファーマシューティカル マニュファクチュアリング リサーチ サービシズ,インコーポレーテッド | 徐放性乱用抑止性液体充填剤形 |
| EP3285748A1 (en) * | 2015-04-24 | 2018-02-28 | Grünenthal GmbH | Tamper-resistant fixed dose combination providing fast release of two drugs from different particles |
| US20160310486A1 (en) * | 2015-04-24 | 2016-10-27 | Grünenthal GmbH | Tamper-resistant fixed dose combination providing fast release of two drugs from particles and a matrix |
| WO2016170094A1 (en) * | 2015-04-24 | 2016-10-27 | Grünenthal GmbH | Tamper-resistant fixed dose combination providing fast release of two drugs from particles |
| US10383825B2 (en) | 2015-08-13 | 2019-08-20 | Temple University—Of the Commonwealth System of Higher Education | Calcium alginate dosage formulations, and methods of making and using thereof |
| CA2998259A1 (en) * | 2015-09-10 | 2017-03-16 | Grunenthal Gmbh | Protecting oral overdose with abuse deterrent immediate release formulations |
| WO2018029327A1 (en) | 2016-08-12 | 2018-02-15 | Grünenthal GmbH | Tamper resistant formulation of ephedrine and its derivatives |
| TW202002957A (zh) | 2018-02-09 | 2020-01-16 | 德商歌林達有限公司 | 包含轉化抑制劑之麻黃素及其衍生物之抗損壞調配物 |
| EP3698776A1 (en) | 2019-02-19 | 2020-08-26 | Grünenthal GmbH | Tamper-resistant dosage form with immediate release and resistance against solvent extraction |
| GR1009751B (el) * | 2019-03-22 | 2020-05-29 | "Φαρματεν Α.Β.Ε.Ε." | Σκευασμα παρατεταμενης αποδεσμευσης που περιλαμβανει οξαλικη ταπενταδολη και μεθοδος παρασκευης αυτου |
| WO2023145868A1 (ja) * | 2022-01-31 | 2023-08-03 | 住友精化株式会社 | 製剤用組成物及び製剤並びに製剤用組成物の製造方法 |
| WO2023145870A1 (ja) * | 2022-01-31 | 2023-08-03 | 住友精化株式会社 | 製剤用組成物及び製剤並びに製剤用組成物の製造方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090004267A1 (en) * | 2007-03-07 | 2009-01-01 | Gruenenthal Gmbh | Dosage Form with Impeded Abuse |
Family Cites Families (583)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2524855A (en) | 1950-10-10 | Process for the manufacture of | ||
| CA722109A (en) | 1965-11-23 | W. Mock Henry | Extrusion of ethylene oxide polymers | |
| US2806033A (en) | 1955-08-03 | 1957-09-10 | Lewenstein | Morphine derivative |
| US2987445A (en) | 1958-10-10 | 1961-06-06 | Rohm & Haas | Drug composition |
| US3370035A (en) | 1961-06-23 | 1968-02-20 | Takeda Chemical Industries Ltd | Stabilization of polyalkylene oxide |
| US3332950A (en) | 1963-03-23 | 1967-07-25 | Endo Lab | 14-hydroxydihydronormorphinone derivatives |
| GB1147210A (en) | 1965-06-30 | 1969-04-02 | Eastman Kodak Co | Improvements in or relating to vitamins |
| US3652589A (en) | 1967-07-27 | 1972-03-28 | Gruenenthal Chemie | 1-(m-substituted phenyl)-2-aminomethyl cyclohexanols |
| US3806603A (en) | 1969-10-13 | 1974-04-23 | W Gaunt | Pharmaceutical carriers of plasticized dried milled particles of hydrated cooked rice endosperm |
| CH503520A (de) | 1969-12-15 | 1971-02-28 | Inventa Ag | Verfahren zum Vermahlen von körnigen Materialien, insbesondere von Kunststoffgranulaten, bei tiefen Temperaturen |
| DE2210071A1 (de) | 1971-03-09 | 1972-09-14 | PPG Industries Inc., Pittsburgh, Pa. (V.StA.) | Verfahren zum Auftragen und Härten einer Vielzahl von Überzügen |
| US3865108A (en) | 1971-05-17 | 1975-02-11 | Ortho Pharma Corp | Expandable drug delivery device |
| US3966747A (en) | 1972-10-26 | 1976-06-29 | Bristol-Myers Company | 9-Hydroxy-6,7-benzomorphans |
| US4014965A (en) | 1972-11-24 | 1977-03-29 | The Dow Chemical Company | Process for scrapless forming of plastic articles |
| US3980766A (en) | 1973-08-13 | 1976-09-14 | West Laboratories, Inc. | Orally administered drug composition for therapy in the treatment of narcotic drug addiction |
| US3941865A (en) | 1973-12-10 | 1976-03-02 | Union Carbide Corporation | Extrusion of ethylene oxide resins |
| US4002173A (en) | 1974-07-23 | 1977-01-11 | International Paper Company | Diester crosslinked polyglucan hydrogels and reticulated sponges thereof |
| DE2530563C2 (de) | 1975-07-09 | 1986-07-24 | Bayer Ag, 5090 Leverkusen | Analgetische Arzneimittel mit vermindertem Mißbrauchspotential |
| JPS603286B2 (ja) | 1977-03-03 | 1985-01-26 | 日本化薬株式会社 | 定速溶出性製剤 |
| US4207893A (en) | 1977-08-29 | 1980-06-17 | Alza Corporation | Device using hydrophilic polymer for delivering drug to biological environment |
| US4175119A (en) | 1978-01-11 | 1979-11-20 | Porter Garry L | Composition and method to prevent accidental and intentional overdosage with psychoactive drugs |
| DE2822324C3 (de) | 1978-05-22 | 1981-02-26 | Basf Ag, 6700 Ludwigshafen | Herstellung von Vitamin-E-Trockenpulver |
| US4211681A (en) | 1978-08-16 | 1980-07-08 | Union Carbide Corporation | Poly(ethylene oxide) compositions |
| US4200704A (en) | 1978-09-28 | 1980-04-29 | Union Carbide Corporation | Controlled degradation of poly(ethylene oxide) |
| NO793297L (no) | 1978-10-19 | 1980-04-22 | Mallinckrodt Inc | Fremgangsmaate til fremstilling av oksymorfon |
| US4215104A (en) | 1979-03-26 | 1980-07-29 | Mead Johnson & Company | Multi-fractionable tablet structure |
| US4258027A (en) | 1979-03-26 | 1981-03-24 | Mead Johnson & Company | Multi-fractionable tablet structure |
| CA1146866A (en) | 1979-07-05 | 1983-05-24 | Yamanouchi Pharmaceutical Co. Ltd. | Process for the production of sustained release pharmaceutical composition of solid medical material |
| US4353887A (en) | 1979-08-16 | 1982-10-12 | Ciba-Geigy Corporation | Divisible tablet having controlled and delayed release of the active substance |
| CH648754A5 (en) | 1979-08-16 | 1985-04-15 | Ciba Geigy Ag | Pharmaceutical slow release tablet |
| US4457933A (en) | 1980-01-24 | 1984-07-03 | Bristol-Myers Company | Prevention of analgesic abuse |
| JPS56169622A (en) | 1980-06-03 | 1981-12-26 | Kissei Pharmaceut Co Ltd | Method of making solid preparation from oily substance |
| DE3024416C2 (de) | 1980-06-28 | 1982-04-15 | Gödecke AG, 1000 Berlin | Verfahren zur Herstellung von Arzneimitteln mit retardierter Wirkstoff-Freisetzung |
| US4473640A (en) | 1982-06-03 | 1984-09-25 | Combie Joan D | Detection of morphine and its analogues using enzymatic hydrolysis |
| US4462941A (en) | 1982-06-10 | 1984-07-31 | The Regents Of The University Of California | Dynorphin amide analogs |
| US4427778A (en) | 1982-06-29 | 1984-01-24 | Biochem Technology, Inc. | Enzymatic preparation of particulate cellulose for tablet making |
| US4485211A (en) | 1982-09-15 | 1984-11-27 | The B. F. Goodrich Company | Poly(glycidyl ether)block copolymers and process for their preparation |
| US4427681A (en) | 1982-09-16 | 1984-01-24 | Richardson-Vicks, Inc. | Thixotropic compositions easily convertible to pourable liquids |
| US4529583A (en) | 1983-03-07 | 1985-07-16 | Clear Lake Development Group | Composition and method of immobilizing emetics and method of treating human beings with emetics |
| US4603143A (en) | 1983-05-02 | 1986-07-29 | Basf Corporation | Free-flowing, high density, fat soluble vitamin powders with improved stability |
| US4783337A (en) | 1983-05-11 | 1988-11-08 | Alza Corporation | Osmotic system comprising plurality of members for dispensing drug |
| US4765989A (en) | 1983-05-11 | 1988-08-23 | Alza Corporation | Osmotic device for administering certain drugs |
| US4612008A (en) | 1983-05-11 | 1986-09-16 | Alza Corporation | Osmotic device with dual thermodynamic activity |
| US5082668A (en) | 1983-05-11 | 1992-01-21 | Alza Corporation | Controlled-release system with constant pushing source |
| US4599342A (en) | 1984-01-16 | 1986-07-08 | The Procter & Gamble Company | Pharmaceutical products providing enhanced analgesia |
| US4629621A (en) | 1984-07-23 | 1986-12-16 | Zetachron, Inc. | Erodible matrix for sustained release bioactive composition |
| AU592065B2 (en) | 1984-10-09 | 1990-01-04 | Dow Chemical Company, The | Sustained release dosage form based on highly plasticized cellulose ether gels |
| GB8507779D0 (en) | 1985-03-26 | 1985-05-01 | Fujisawa Pharmaceutical Co | Drug carrier |
| WO1987000045A1 (en) | 1985-06-24 | 1987-01-15 | Ici Australia Limited | Ingestible capsules |
| EP0227806B1 (en) | 1985-06-28 | 1989-08-30 | Carrington Laboratories, Inc. | Processes for preparation of aloe products, products produced thereby and compositions thereof |
| US4992279A (en) | 1985-07-03 | 1991-02-12 | Kraft General Foods, Inc. | Sweetness inhibitor |
| US4851521A (en) | 1985-07-08 | 1989-07-25 | Fidia, S.P.A. | Esters of hyaluronic acid |
| DE3689650T2 (de) | 1985-12-17 | 1994-05-26 | United States Surgical Corp | Bioresorbierbare Polymere von hohem Molekulargewicht und Implantate davon. |
| US5229164A (en) | 1985-12-19 | 1993-07-20 | Capsoid Pharma Gmbh | Process for producing individually dosed administration forms |
| US4711894A (en) | 1986-01-16 | 1987-12-08 | Henkel Corporation | Stabilized tocopherol in dry, particulate, free-flowing form |
| US5198226A (en) | 1986-01-30 | 1993-03-30 | Syntex (U.S.A.) Inc. | Long acting nicardipine hydrochloride formulation |
| US4940556A (en) | 1986-01-30 | 1990-07-10 | Syntex (U.S.A.) Inc. | Method of preparing long acting formulation |
| US4764378A (en) | 1986-02-10 | 1988-08-16 | Zetachron, Inc. | Buccal drug dosage form |
| AU7077287A (en) | 1986-03-31 | 1987-10-08 | Union Carbide Corporation | Alkylene oxide polymerisation catalyst |
| DE3612211A1 (de) | 1986-04-11 | 1987-10-15 | Basf Ag | Kontinuierliches verfahren zum tablettieren |
| US4667013A (en) | 1986-05-02 | 1987-05-19 | Union Carbide Corporation | Process for alkylene oxide polymerization |
| USRE33093E (en) | 1986-06-16 | 1989-10-17 | Johnson & Johnson Consumer Products, Inc. | Bioadhesive extruded film for intra-oral drug delivery and process |
| US4713243A (en) | 1986-06-16 | 1987-12-15 | Johnson & Johnson Products, Inc. | Bioadhesive extruded film for intra-oral drug delivery and process |
| USRE34990E (en) | 1986-08-07 | 1995-07-04 | Ciba-Geigy Corporation | Oral therapeutic system having systemic action |
| CA1335748C (en) | 1986-09-25 | 1995-05-30 | Jeffrey Lawrence Finnan | Crosslinked gelatins |
| US5227157A (en) | 1986-10-14 | 1993-07-13 | Board Of Regents, The University Of Texas System | Delivery of therapeutic agents |
| JP2962731B2 (ja) | 1986-11-10 | 1999-10-12 | バイオピュアー、コーポレーション | 超純枠半合成代用血液 |
| US4892889A (en) | 1986-11-18 | 1990-01-09 | Basf Corporation | Process for making a spray-dried, directly-compressible vitamin powder comprising unhydrolyzed gelatin |
| JPH0831303B2 (ja) | 1986-12-01 | 1996-03-27 | オムロン株式会社 | チツプ型ヒユ−ズ |
| ATE72111T1 (de) | 1987-01-14 | 1992-02-15 | Ciba Geigy Ag | Therapeutisches system fuer schwerloesliche wirkstoffe. |
| US4892778A (en) | 1987-05-27 | 1990-01-09 | Alza Corporation | Juxtaposed laminated arrangement |
| US5051261A (en) | 1987-11-24 | 1991-09-24 | Fmc Corporation | Method for preparing a solid sustained release form of a functionally active composition |
| JP2719020B2 (ja) | 1987-12-17 | 1998-02-25 | ジ・アップジョン・カンパニー | 三本の溝付き錠剤 |
| DE3812567A1 (de) | 1988-04-15 | 1989-10-26 | Basf Ag | Verfahren zur herstellung pharmazeutischer mischungen |
| US4954346A (en) | 1988-06-08 | 1990-09-04 | Ciba-Geigy Corporation | Orally administrable nifedipine solution in a solid light resistant dosage form |
| US4960814A (en) | 1988-06-13 | 1990-10-02 | Eastman Kodak Company | Water-dispersible polymeric compositions |
| US5350741A (en) | 1988-07-30 | 1994-09-27 | Kanji Takada | Enteric formulations of physiologically active peptides and proteins |
| JPH0249719A (ja) | 1988-08-11 | 1990-02-20 | Dai Ichi Kogyo Seiyaku Co Ltd | 易水分散・可溶性能を有する油溶性ビタミン粉末 |
| GB8820327D0 (en) | 1988-08-26 | 1988-09-28 | May & Baker Ltd | New compositions of matter |
| DE3830353A1 (de) | 1988-09-07 | 1990-03-15 | Basf Ag | Verfahren zur kontinuierlichen herstellung von festen pharmazeutischen formen |
| US5139790A (en) | 1988-10-14 | 1992-08-18 | Zetachron, Inc. | Low-melting moldable pharmaceutical excipient and dosage forms prepared therewith |
| US5004601A (en) | 1988-10-14 | 1991-04-02 | Zetachron, Inc. | Low-melting moldable pharmaceutical excipient and dosage forms prepared therewith |
| US4957668A (en) | 1988-12-07 | 1990-09-18 | General Motors Corporation | Ultrasonic compacting and bonding particles |
| US5190760A (en) | 1989-07-08 | 1993-03-02 | Coopers Animal Health Limited | Solid pharmaceutical composition |
| US5169645A (en) | 1989-10-31 | 1992-12-08 | Duquesne University Of The Holy Ghost | Directly compressible granules having improved flow properties |
| US5200197A (en) | 1989-11-16 | 1993-04-06 | Alza Corporation | Contraceptive pill |
| GB8926612D0 (en) | 1989-11-24 | 1990-01-17 | Erba Farmitalia | Pharmaceutical compositions |
| EP0449775A3 (en) | 1990-03-29 | 1992-09-02 | Ciba-Geigy Ag | Polyether-polyester block copolymers and their use as dispersing agents |
| SU1759445A1 (ru) | 1990-06-15 | 1992-09-07 | Ленинградский Технологический Институт Им.Ленсовета | Способ получени капсулированных гидрофобных веществ |
| FR2664851B1 (fr) | 1990-07-20 | 1992-10-16 | Oreal | Procede de compactage d'un melange pulverulent permettant d'obtenir un produit compact absorbant ou partiellement delitable et produit obtenu par ce procede. |
| EP0477135A1 (en) | 1990-09-07 | 1992-03-25 | Warner-Lambert Company | Chewable spheroidal coated microcapsules and methods for preparing same |
| US5126151A (en) | 1991-01-24 | 1992-06-30 | Warner-Lambert Company | Encapsulation matrix |
| US5273758A (en) | 1991-03-18 | 1993-12-28 | Sandoz Ltd. | Directly compressible polyethylene oxide vehicle for preparing therapeutic dosage forms |
| US5149538A (en) | 1991-06-14 | 1992-09-22 | Warner-Lambert Company | Misuse-resistive transdermal opioid dosage form |
| JP3073054B2 (ja) | 1991-07-11 | 2000-08-07 | 住友精化株式会社 | アルキレンオキシド重合体の製造方法 |
| CA2116563C (en) | 1991-08-30 | 2001-07-03 | Yoji Ito | Dry gel pharmaceutical composition |
| AU2670292A (en) | 1991-10-04 | 1993-05-03 | Olin Corporation | Fungicide tablet |
| ATE183642T1 (de) | 1991-10-04 | 1999-09-15 | Yoshitomi Pharmaceutical | Tablette mit verzögerter freisetzung |
| DE4138513A1 (de) | 1991-11-23 | 1993-05-27 | Basf Ag | Feste pharmazeutische retardform |
| US5266331A (en) | 1991-11-27 | 1993-11-30 | Euroceltique, S.A. | Controlled release oxycodone compositions |
| DK0615438T3 (da) | 1991-12-05 | 1996-11-11 | Mallinckrodt Veterinary Inc | En carbohydratglasmatrix til langvarig frigivelse af et terapeutisk middel |
| US5200194A (en) | 1991-12-18 | 1993-04-06 | Alza Corporation | Oral osmotic device |
| ATE157864T1 (de) | 1991-12-18 | 1997-09-15 | Warner Lambert Co | Verfahren für die herstellung einer festen dispersion |
| US5225417A (en) | 1992-01-21 | 1993-07-06 | G. D. Searle & Co. | Opioid agonist compounds |
| IL105553A (en) | 1992-05-06 | 1998-01-04 | Janssen Pharmaceutica Inc | Solid dosage forms consisting of a porous network of matrix that releases a substance that dissipates rapidly in water |
| US5792474A (en) | 1992-05-22 | 1998-08-11 | Goedecke Aktiengesellschaft | Process for the production of retarded pharmaceutical compositions |
| GB9217295D0 (en) | 1992-08-14 | 1992-09-30 | Wellcome Found | Controlled released tablets |
| DE4227385A1 (de) | 1992-08-19 | 1994-02-24 | Kali Chemie Pharma Gmbh | Pankreatinmikropellets |
| DE4229085C2 (de) | 1992-09-01 | 1996-07-11 | Boehringer Mannheim Gmbh | Längliche, teilbare Tablette |
| RU2121830C1 (ru) | 1992-09-18 | 1998-11-20 | Яманоути Фармасьютикал Ко., ЛТД | Гидрогелевый препарат с длительным высвобождением лекарства |
| US5472943A (en) | 1992-09-21 | 1995-12-05 | Albert Einstein College Of Medicine Of Yeshiva University, | Method of simultaneously enhancing analgesic potency and attenuating dependence liability caused by morphine and other opioid agonists |
| FI101039B (fi) | 1992-10-09 | 1998-04-15 | Eeva Kristoffersson | Menetelmä lääkepellettien valmistamiseksi |
| AU679937B2 (en) | 1992-11-18 | 1997-07-17 | Johnson & Johnson Consumer Products, Inc. | Extrudable compositions for topical or transdermal drug delivery |
| EP0675710B1 (en) | 1992-12-23 | 1999-08-25 | Saitec S.R.L. | Process for preparing controlled release pharmaceutical forms and the forms thus obtained |
| GB2273874A (en) | 1992-12-31 | 1994-07-06 | Pertti Olavi Toermaelae | Preparation of pharmaceuticals in a polymer matrix |
| US6071970A (en) | 1993-02-08 | 2000-06-06 | Nps Pharmaceuticals, Inc. | Compounds active at a novel site on receptor-operated calcium channels useful for treatment of neurological disorders and diseases |
| US5914132A (en) | 1993-02-26 | 1999-06-22 | The Procter & Gamble Company | Pharmaceutical dosage form with multiple enteric polymer coatings for colonic delivery |
| DE4309528C2 (de) | 1993-03-24 | 1998-05-20 | Doxa Gmbh | Folie oder Folienschlauch aus Casein, Verfahren zu deren Herstellung und deren Verwendung |
| NZ260408A (en) | 1993-05-10 | 1996-05-28 | Euro Celtique Sa | Controlled release preparation comprising tramadol |
| IL109944A (en) | 1993-07-01 | 1998-12-06 | Euro Celtique Sa | Continuous release dosage form containing morphine and a method of preparing such sustained release unit dosage forms |
| DE4329794C2 (de) | 1993-09-03 | 1997-09-18 | Gruenenthal Gmbh | Tramadolsalz enthaltende Arzneimittel mit verzögerter Wirkstofffreisetzung |
| HU218673B (hu) | 1993-10-07 | 2000-10-28 | Euroceltique S.A. | Opioid analgetikumot tartalmazó elnyújtott hatóanyag-felszabadítású orális gyógyszerkészítmény és eljárás előállítására |
| KR100354702B1 (ko) | 1993-11-23 | 2002-12-28 | 유로-셀티크 소시에떼 아노뉨 | 약학조성물의제조방법및서방형조성물 |
| ES2168290T3 (es) | 1993-11-23 | 2002-06-16 | Euro Celtique Sa | Metodo para preparar una composicion de liberacion sostenida. |
| WO1995017174A1 (en) | 1993-12-20 | 1995-06-29 | The Procter & Gamble Company | Process for making laxatives containing dioctyl sulfosuccinate |
| IL112106A0 (en) | 1993-12-22 | 1995-03-15 | Ergo Science Inc | Accelerated release composition containing bromocriptine |
| GB9401894D0 (en) | 1994-02-01 | 1994-03-30 | Rhone Poulenc Rorer Ltd | New compositions of matter |
| MX9603480A (es) | 1994-02-16 | 1997-12-31 | Abbott Lab | Procedimiento para preparar formulaciones farmaceuticas en particulas finas. |
| SE9503924D0 (sv) | 1995-08-18 | 1995-11-07 | Astra Ab | Novel opioid peptides |
| US5458887A (en) | 1994-03-02 | 1995-10-17 | Andrx Pharmaceuticals, Inc. | Controlled release tablet formulation |
| DE4413350A1 (de) | 1994-04-18 | 1995-10-19 | Basf Ag | Retard-Matrixpellets und Verfahren zu ihrer Herstellung |
| MX9605419A (es) | 1994-05-06 | 1997-12-31 | Pfizer | Formas de dosificacion de liberacion controlada de azitromicina. |
| DE19509807A1 (de) | 1995-03-21 | 1996-09-26 | Basf Ag | Verfahren zur Herstellung von Wirkstoffzubereitungen in Form einer festen Lösung des Wirkstoffs in einer Polymermatrix sowie mit diesem Verfahren hergestellte Wirkstoffzubereitungen |
| AT403988B (de) | 1994-05-18 | 1998-07-27 | Lannacher Heilmittel | Festes orales retardpräparat |
| US5460826A (en) | 1994-06-27 | 1995-10-24 | Alza Corporation | Morphine therapy |
| DE4426245A1 (de) | 1994-07-23 | 1996-02-22 | Gruenenthal Gmbh | 1-Phenyl-3-dimethylamino-propanverbindungen mit pharmakologischer Wirkung |
| IT1274879B (it) | 1994-08-03 | 1997-07-25 | Saitec Srl | Apparecchio e metodo per preparare forme farmaceutiche solide a rilascio controllato del principio attivo. |
| JP3285452B2 (ja) | 1994-08-11 | 2002-05-27 | サンスター株式会社 | 歯磨組成物 |
| US5837790A (en) | 1994-10-24 | 1998-11-17 | Amcol International Corporation | Precipitation polymerization process for producing an oil adsorbent polymer capable of entrapping solid particles and liquids and the product thereof |
| AUPM897594A0 (en) | 1994-10-25 | 1994-11-17 | Daratech Pty Ltd | Controlled release container |
| US5965161A (en) | 1994-11-04 | 1999-10-12 | Euro-Celtique, S.A. | Extruded multi-particulates |
| DE4446470A1 (de) | 1994-12-23 | 1996-06-27 | Basf Ag | Verfahren zur Herstellung von teilbaren Tabletten |
| DE19504832A1 (de) | 1995-02-14 | 1996-08-22 | Basf Ag | Feste Wirkstoff-Zubereitungen |
| US5945125A (en) | 1995-02-28 | 1999-08-31 | Temple University | Controlled release tablet |
| US6117453A (en) | 1995-04-14 | 2000-09-12 | Pharma Pass | Solid compositions containing polyethylene oxide and an active ingredient |
| US6348469B1 (en) | 1995-04-14 | 2002-02-19 | Pharma Pass Llc | Solid compositions containing glipizide and polyethylene oxide |
| US5900425A (en) | 1995-05-02 | 1999-05-04 | Bayer Aktiengesellschaft | Pharmaceutical preparations having controlled release of active compound and processes for their preparation |
| DE19522899C1 (de) | 1995-06-23 | 1996-12-19 | Hexal Pharmaforschung Gmbh | Verfahren zum kontinuierlichen Ersintern eines Granulats |
| US5759583A (en) | 1995-08-30 | 1998-06-02 | Syntex (U.S.A.) Inc. | Sustained release poly (lactic/glycolic) matrices |
| US6063405A (en) | 1995-09-29 | 2000-05-16 | L.A.M. Pharmaceuticals, Llc | Sustained release delivery system |
| US5811126A (en) | 1995-10-02 | 1998-09-22 | Euro-Celtique, S.A. | Controlled release matrix for pharmaceuticals |
| DE19539361A1 (de) | 1995-10-23 | 1997-04-24 | Basf Ag | Verfahren zur Herstellung von mehrschichtigen, festen Arzneiformen zur oralen oder rektalen Verabreichung |
| US5908850A (en) | 1995-12-04 | 1999-06-01 | Celgene Corporation | Method of treating attention deficit disorders with d-threo methylphenidate |
| US6355656B1 (en) | 1995-12-04 | 2002-03-12 | Celgene Corporation | Phenidate drug formulations having diminished abuse potential |
| DE19547766A1 (de) | 1995-12-20 | 1997-06-26 | Gruenenthal Gmbh | 1-Phenyl-2-dimethylaminomethyl-cyclohexan-1-ol-verbindungen als pharmazeutische Wirkstoffe |
| ES2168610T3 (es) | 1996-03-12 | 2002-06-16 | Alza Corp | Composicion y forma galenica que contiene un antagonista opioide. |
| US6461644B1 (en) | 1996-03-25 | 2002-10-08 | Richard R. Jackson | Anesthetizing plastics, drug delivery plastics, and related medical products, systems and methods |
| ES2175385T5 (es) | 1996-04-05 | 2006-06-01 | Takeda Chemical Industries, Ltd. | Combinacion farmaceutica que contiene un compuesto que tiene actividad antagonista de angiotensina ii y un compuesto que aumenta la sensibilidad a la insulina. |
| US6096339A (en) | 1997-04-04 | 2000-08-01 | Alza Corporation | Dosage form, process of making and using same |
| US20020114838A1 (en) | 1996-04-05 | 2002-08-22 | Ayer Atul D. | Uniform drug delivery therapy |
| US5817343A (en) | 1996-05-14 | 1998-10-06 | Alkermes, Inc. | Method for fabricating polymer-based controlled-release devices |
| IL127378A (en) | 1996-06-06 | 2003-07-31 | Bifodan As | Enteric coating for an oral preparation |
| EP1014941B2 (en) | 1996-06-26 | 2017-05-17 | The Board Of Regents, The University Of Texas System | Hot-melt extrudable pharmaceutical formulation |
| ES2322405T3 (es) | 1996-07-08 | 2009-06-19 | Penwest Pharmaceuticals Co. | Matriz de liberacion controlada para farmacos insolubles en dosis elevadas. |
| DE19629753A1 (de) | 1996-07-23 | 1998-01-29 | Basf Ag | Verfahren zur Herstellung von festen Arzneiformen |
| NL1003684C2 (nl) | 1996-07-25 | 1998-01-28 | Weterings B V H | Inrichting voor het afgeven van een vloeistof. |
| DE19630236A1 (de) | 1996-07-26 | 1998-01-29 | Wolff Walsrode Ag | Biaxial gereckte, biologisch abbaubare und kompostierbare Wursthülle |
| BE1010353A5 (fr) | 1996-08-14 | 1998-06-02 | Boss Pharmaceuticals Ag | Procede pour la fabrication de produits pharmaceutiques, dispositif pour un tel procede et produits pharmaceutiques ainsi obtenus. |
| DE69730852T2 (de) | 1996-11-05 | 2005-09-22 | Novamont S.P.A. | Biologisch abbaubare polymerzusammensetzungen, die stärke und ein thermoplastisches polymer enthalten |
| US5991799A (en) | 1996-12-20 | 1999-11-23 | Liberate Technologies | Information retrieval system using an internet multiplexer to focus user selection |
| DE19705538C1 (de) | 1997-02-14 | 1998-08-27 | Goedecke Ag | Verfahren zur Trennung von Wirkstoffen in festen pharmazeutischen Zubereitungen |
| US5948787A (en) | 1997-02-28 | 1999-09-07 | Alza Corporation | Compositions containing opiate analgesics |
| DE19710213A1 (de) | 1997-03-12 | 1998-09-17 | Basf Ag | Verfahren zur Herstellung von festen Kombinationsarzneiformen |
| DE19710009A1 (de) | 1997-03-12 | 1998-09-24 | Knoll Ag | Mehrphasige wirkstoffhaltige Zubereitungsformen |
| DE19710008A1 (de) | 1997-03-12 | 1998-09-17 | Basf Ag | Feste, mindestens zweiphasige Zubereitungsformen eines Opioid-Analgeticums mit verzögerter Freisetzung |
| US6139770A (en) | 1997-05-16 | 2000-10-31 | Chevron Chemical Company Llc | Photoinitiators and oxygen scavenging compositions |
| DE19721467A1 (de) | 1997-05-22 | 1998-11-26 | Basf Ag | Verfahren zur Herstellung kleinteiliger Zubereitungen biologisch aktiver Stoffe |
| US6635280B2 (en) | 1997-06-06 | 2003-10-21 | Depomed, Inc. | Extending the duration of drug release within the stomach during the fed mode |
| ES2248908T7 (es) | 1997-06-06 | 2014-11-24 | Depomed, Inc. | Formas de dosificación de fármacos por vía oral y retención gástrica para liberación continuada de fármacos altamente solubles |
| WO1999001111A1 (en) | 1997-07-02 | 1999-01-14 | Euro-Celtique, S.A. | Stabilized sustained release tramadol formulations |
| IE970588A1 (en) | 1997-08-01 | 2000-08-23 | Elan Corp Plc | Controlled release pharmaceutical compositions containing tiagabine |
| CA2303036A1 (en) | 1997-09-10 | 1999-03-18 | Alliedsignal Inc. | Injection molding of structural zirconia-based materials by an aqueous process |
| US6009390A (en) | 1997-09-11 | 1999-12-28 | Lucent Technologies Inc. | Technique for selective use of Gaussian kernels and mixture component weights of tied-mixture hidden Markov models for speech recognition |
| DK1033975T3 (da) | 1997-11-28 | 2002-05-27 | Knoll Ag | Fremgangsmåde til fremstilling af opløsningsmiddelfrie ikke-krystallinske aktive stoffer |
| DE19753534A1 (de) | 1997-12-03 | 1999-06-10 | Bayer Ag | Schnell kristallisierende, biologisch abbaubare Polyesteramide |
| JP2001525433A (ja) | 1997-12-03 | 2001-12-11 | バイエル・アクチエンゲゼルシヤフト | ポリエーテルエステルアミド類 |
| BR9813826A (pt) | 1997-12-22 | 2000-10-10 | Euro Celtique Sa | Potencial de uso abusivo de administração oral de opióide analgésico |
| US6375957B1 (en) | 1997-12-22 | 2002-04-23 | Euro-Celtique, S.A. | Opioid agonist/opioid antagonist/acetaminophen combinations |
| DE19800689C1 (de) | 1998-01-10 | 1999-07-15 | Deloro Stellite Gmbh | Formkörper aus einem verschleißfesten Werkstoff |
| DE19800698A1 (de) | 1998-01-10 | 1999-07-15 | Bayer Ag | Biologisch abbaubare Polyesteramide mit blockartig aufgebauten Polyester- und Polyamid-Segmenten |
| US6251430B1 (en) | 1998-02-04 | 2001-06-26 | Guohua Zhang | Water insoluble polymer based sustained release formulation |
| CN1296516A (zh) | 1998-02-06 | 2001-05-23 | 联合碳化化学品及塑料技术公司 | 烯化氧聚合物组合物 |
| US6235825B1 (en) | 1998-03-05 | 2001-05-22 | Mitsui Chemicals, Inc. | Polylactic acid resin composition and film therefrom |
| US6245357B1 (en) | 1998-03-06 | 2001-06-12 | Alza Corporation | Extended release dosage form |
| US6090411A (en) | 1998-03-09 | 2000-07-18 | Temple University | Monolithic tablet for controlled drug release |
| US6110500A (en) | 1998-03-25 | 2000-08-29 | Temple University | Coated tablet with long term parabolic and zero-order release kinetics |
| JP2002510878A (ja) | 1998-04-02 | 2002-04-09 | アプライド マテリアルズ インコーポレイテッド | 低k誘電体をエッチングする方法 |
| AU3024399A (en) | 1998-04-03 | 1999-10-25 | Bm Research A/S | Controlled release composition |
| US5962488A (en) | 1998-04-08 | 1999-10-05 | Roberts Laboratories, Inc. | Stable pharmaceutical formulations for treating internal bowel syndrome containing isoxazole derivatives |
| DE19822979A1 (de) | 1998-05-25 | 1999-12-02 | Kalle Nalo Gmbh & Co Kg | Folie mit Stärke oder Stärkederivaten und Polyesterurethanen sowie Verfahren zu ihrer Herstellung |
| US6333087B1 (en) | 1998-08-27 | 2001-12-25 | Chevron Chemical Company Llc | Oxygen scavenging packaging |
| DE19841244A1 (de) | 1998-09-09 | 2000-03-16 | Knoll Ag | Verfahren und Vorrichtung zum Herstellen von Tabletten |
| GT199900148A (es) | 1998-09-10 | 2001-02-28 | Desnaturalizantes para las sales aminas simpaticomimeticas. | |
| US6268177B1 (en) | 1998-09-22 | 2001-07-31 | Smithkline Beecham Corporation | Isolated nucleic acid encoding nucleotide pyrophosphorylase |
| CN1327384A (zh) | 1998-10-20 | 2001-12-19 | 韩国科学技术研究院 | 作为血浆高密度脂蛋白浓度增高剂的生物类黄酮 |
| US6322819B1 (en) | 1998-10-21 | 2001-11-27 | Shire Laboratories, Inc. | Oral pulsed dose drug delivery system |
| US20060240105A1 (en) | 1998-11-02 | 2006-10-26 | Elan Corporation, Plc | Multiparticulate modified release composition |
| ES2141688B1 (es) | 1998-11-06 | 2001-02-01 | Vita Invest Sa | Nuevos esteres derivados de compuestos fenil-ciclohexil sustituidos. |
| DE19855440A1 (de) | 1998-12-01 | 2000-06-08 | Basf Ag | Verfahren zum Herstellen fester Darreichungsformen mittels Schmelzextrusion |
| DE19856147A1 (de) | 1998-12-04 | 2000-06-08 | Knoll Ag | Teilbare feste Dosierungsformen und Verfahren zu ihrer Herstellung |
| EP1005863A1 (en) | 1998-12-04 | 2000-06-07 | Synthelabo | Controlled-release dosage forms comprising a short acting hypnotic or a salt thereof |
| US6419960B1 (en) | 1998-12-17 | 2002-07-16 | Euro-Celtique S.A. | Controlled release formulations having rapid onset and rapid decline of effective plasma drug concentrations |
| US6238697B1 (en) | 1998-12-21 | 2001-05-29 | Pharmalogix, Inc. | Methods and formulations for making bupropion hydrochloride tablets using direct compression |
| WO2000040205A2 (en) | 1999-01-05 | 2000-07-13 | Copley Pharmaceutical Inc. | Sustained release formulation with reduced moisture sensitivity |
| WO2000045830A1 (fr) | 1999-02-04 | 2000-08-10 | Nichimo Co., Ltd. | Substances permettant d'eviter la survenue de l'arteriosclerose, substances immunostimulantes, vertebres nourris a l'aide ces substances et oeufs de ces vertebres |
| US7374779B2 (en) | 1999-02-26 | 2008-05-20 | Lipocine, Inc. | Pharmaceutical formulations and systems for improved absorption and multistage release of active agents |
| US6375963B1 (en) | 1999-06-16 | 2002-04-23 | Michael A. Repka | Bioadhesive hot-melt extruded film for topical and mucosal adhesion applications and drug delivery and process for preparation thereof |
| US6384020B1 (en) | 1999-07-14 | 2002-05-07 | Shire Laboratories, Inc. | Rapid immediate release oral dosage form |
| US20030118641A1 (en) | 2000-07-27 | 2003-06-26 | Roxane Laboratories, Inc. | Abuse-resistant sustained-release opioid formulation |
| WO2001008661A2 (en) | 1999-07-29 | 2001-02-08 | Roxane Laboratories, Inc. | Opioid sustained-released formulation |
| US6562375B1 (en) | 1999-08-04 | 2003-05-13 | Yamanouchi Pharmaceuticals, Co., Ltd. | Stable pharmaceutical composition for oral use |
| RU2220715C2 (ru) | 1999-08-04 | 2004-01-10 | Яманоути Фармасьютикал Ко., Лтд. | Стабильная пероральная, образующая гидрогель, фармацевтическая композиция, способ ее получения, способ предотвращения изменений высвобождения лекарственного средства, способ увеличения физической стабильности пероральной композиции |
| KR100345214B1 (ko) | 1999-08-17 | 2002-07-25 | 이강춘 | 생체적합성 고분자가 수식된 펩타이드의 비점막 전달 |
| NZ517559A (en) | 1999-08-31 | 2004-08-27 | Gruenenthal Chemie | Sustained release pharmaceutical composition containing tramadol saccharinate |
| DE19940944B4 (de) | 1999-08-31 | 2006-10-12 | Grünenthal GmbH | Retardierte, orale, pharmazeutische Darreichungsformen |
| DE19940740A1 (de) | 1999-08-31 | 2001-03-01 | Gruenenthal Gmbh | Pharmazeutische Salze |
| DE19960494A1 (de) | 1999-12-15 | 2001-06-21 | Knoll Ag | Vorrichtung und Verfahren zum Herstellen von festen wirkstoffhaltigen Formen |
| ES2160534B1 (es) | 1999-12-30 | 2002-04-16 | Vita Invest Sa | Nuevos esteres derivados de (rr,ss)-2-hidroxibenzoato de 3-(2-dimetilaminometil-1-hidroxiciclohexil) fenilo. |
| US6680070B1 (en) | 2000-01-18 | 2004-01-20 | Albemarle Corporation | Particulate blends and compacted products formed therefrom, and the preparation thereof |
| SI1299104T1 (sl) | 2000-02-08 | 2009-10-31 | Euro Celtique Sa | Oralne formulacije opioidnih agonistov, varne pred zlorabo |
| US20020015730A1 (en) | 2000-03-09 | 2002-02-07 | Torsten Hoffmann | Pharmaceutical formulations and method for making |
| DE10015479A1 (de) | 2000-03-29 | 2001-10-11 | Basf Ag | Feste orale Darreichungsformen mit retardierter Wirkstofffreisetzung und hoher mechanischer Stabilität |
| US8012504B2 (en) | 2000-04-28 | 2011-09-06 | Reckitt Benckiser Inc. | Sustained release of guaifenesin combination drugs |
| US6572887B2 (en) | 2000-05-01 | 2003-06-03 | National Starch And Chemical Investment Holding Corporation | Polysaccharide material for direct compression |
| US6419954B1 (en) | 2000-05-19 | 2002-07-16 | Yamanouchi Pharmaceutical Co., Ltd. | Tablets and methods for modified release of hydrophilic and other active agents |
| AU7494701A (en) | 2000-05-23 | 2001-12-03 | Cenes Pharmaceuticals Inc | Nrg-2 nucleic acid molecules, polypeptides, and diagnostic and therapeutic methods |
| DE10029201A1 (de) | 2000-06-19 | 2001-12-20 | Basf Ag | Verfahren zur Herstellung fester oraler Darreichungsformen mit retardierender Wirkstoffreisetzung |
| US6488962B1 (en) | 2000-06-20 | 2002-12-03 | Depomed, Inc. | Tablet shapes to enhance gastric retention of swellable controlled-release oral dosage forms |
| US6607748B1 (en) | 2000-06-29 | 2003-08-19 | Vincent Lenaerts | Cross-linked high amylose starch for use in controlled-release pharmaceutical formulations and processes for its manufacture |
| DE10036400A1 (de) | 2000-07-26 | 2002-06-06 | Mitsubishi Polyester Film Gmbh | Weiße, biaxial orientierte Polyesterfolie |
| AU2001292993A1 (en) | 2000-09-25 | 2002-04-08 | Pro-Pharmaceuticals, Inc. | Compositions for reducing side effects in chemotherapeutic treatments |
| WO2002026061A1 (en) | 2000-09-27 | 2002-04-04 | Danisco A/S | Antimicrobial agent |
| WO2002026928A1 (en) | 2000-09-28 | 2002-04-04 | The Dow Chemical Company | Polymer composite structures useful for controlled release systems |
| GB0026137D0 (en) | 2000-10-25 | 2000-12-13 | Euro Celtique Sa | Transdermal dosage form |
| US6344215B1 (en) | 2000-10-27 | 2002-02-05 | Eurand America, Inc. | Methylphenidate modified release formulations |
| EP2283829A1 (en) | 2000-10-30 | 2011-02-16 | Euro-Celtique S.A. | Controlled release hydrocodone formulations |
| AU2002226098A1 (en) | 2000-10-30 | 2002-05-15 | The Board Of Regents, The University Of Texas System | Spherical particles produced by a hot-melt extrusion/spheronization process |
| DE10109763A1 (de) | 2001-02-28 | 2002-09-05 | Gruenenthal Gmbh | Pharmazeutische Salze |
| JP2002265592A (ja) | 2001-03-07 | 2002-09-18 | Sumitomo Seika Chem Co Ltd | アルキレンオキシド重合体の製造方法 |
| WO2002071860A1 (en) | 2001-03-13 | 2002-09-19 | L.A. Dreyfus Co. | Gum base and gum manufacturing using particulated gum base ingredients |
| JP3967554B2 (ja) | 2001-03-15 | 2007-08-29 | 株式会社ポッカコーポレーション | フラボノイド化合物及びその製造方法 |
| US20020132395A1 (en) | 2001-03-16 | 2002-09-19 | International Business Machines Corporation | Body contact in SOI devices by electrically weakening the oxide under the body |
| EP1241110A1 (en) | 2001-03-16 | 2002-09-18 | Pfizer Products Inc. | Dispensing unit for oxygen-sensitive drugs |
| WO2002085335A1 (en) | 2001-04-18 | 2002-10-31 | Nostrum Pharmaceuticals Inc. | A novel coating for a sustained release pharmaceutical composition |
| US20020187192A1 (en) | 2001-04-30 | 2002-12-12 | Yatindra Joshi | Pharmaceutical composition which reduces or eliminates drug abuse potential |
| DK1385898T3 (da) | 2001-05-01 | 2006-10-02 | Union Carbide Chem Plastic | Farmaceutisk sammensætning omfattende poly(alkylenoxider) med reducerede mængder af myresyreforbindelser |
| UA81224C2 (uk) | 2001-05-02 | 2007-12-25 | Euro Celtic S A | Дозована форма оксикодону та її застосування |
| WO2002090316A1 (en) | 2001-05-08 | 2002-11-14 | The Johns Hopkins University | Method of inhibiting methamphetamine synthesis |
| CA2778114A1 (en) | 2001-05-11 | 2002-11-21 | Endo Pharmaceuticals, Inc. | Abuse-resistant opioid dosage form |
| EP1387673B1 (en) | 2001-05-11 | 2010-12-29 | Endo Pharmaceuticals Inc. | Abuse-resistant controlled-release opioid dosage form |
| US6623754B2 (en) | 2001-05-21 | 2003-09-23 | Noveon Ip Holdings Corp. | Dosage form of N-acetyl cysteine |
| US7125561B2 (en) | 2001-05-22 | 2006-10-24 | Euro-Celtique S.A. | Compartmentalized dosage form |
| US20030064122A1 (en) | 2001-05-23 | 2003-04-03 | Endo Pharmaceuticals, Inc. | Abuse resistant pharmaceutical composition containing capsaicin |
| US7968119B2 (en) | 2001-06-26 | 2011-06-28 | Farrell John J | Tamper-proof narcotic delivery system |
| US20030008409A1 (en) | 2001-07-03 | 2003-01-09 | Spearman Steven R. | Method and apparatus for determining sunlight exposure |
| US8329216B2 (en) | 2001-07-06 | 2012-12-11 | Endo Pharmaceuticals Inc. | Oxymorphone controlled release formulations |
| DK1404331T3 (da) | 2001-07-06 | 2008-01-28 | Penwest Pharmaceuticals Co | Sustained release-formuleringer af oxymorphon |
| CA2452874A1 (en) | 2001-07-06 | 2003-01-16 | Endo Pharmaceuticals, Inc. | Oral administration of 6-hydroxy-oxymorphone for use as an analgesic |
| JP2003020517A (ja) | 2001-07-10 | 2003-01-24 | Calp Corp | 複合繊維用樹脂組成物 |
| SI1416842T1 (sl) | 2001-07-18 | 2009-06-30 | Euro Celtique Sa | Farmacevtske kombinacije oksikodona in naloksona |
| US6883976B2 (en) | 2001-07-30 | 2005-04-26 | Seikoh Giken Co., Ltd. | Optical fiber ferrule assembly and optical module and optical connector using the same |
| AU2002321879A1 (en) | 2001-08-06 | 2003-03-03 | Thomas Gruber | Pharmaceutical formulation containing dye |
| KR20040060917A (ko) | 2001-08-06 | 2004-07-06 | 유로-셀티크 소시에떼 아노뉨 | 오피오이드 남용을 방지하기 위한 조성물 및 방법 |
| US7144587B2 (en) | 2001-08-06 | 2006-12-05 | Euro-Celtique S.A. | Pharmaceutical formulation containing opioid agonist, opioid antagonist and bittering agent |
| IL160217A0 (en) | 2001-08-06 | 2004-07-25 | Euro Celtique Sa | Compositions and methods to prevent abuse of opioids |
| US20030068375A1 (en) | 2001-08-06 | 2003-04-10 | Curtis Wright | Pharmaceutical formulation containing gelling agent |
| US7842307B2 (en) | 2001-08-06 | 2010-11-30 | Purdue Pharma L.P. | Pharmaceutical formulation containing opioid agonist, opioid antagonist and gelling agent |
| US7157103B2 (en) | 2001-08-06 | 2007-01-02 | Euro-Celtique S.A. | Pharmaceutical formulation containing irritant |
| WO2003013433A2 (en) | 2001-08-06 | 2003-02-20 | Euro-Celtique S.A. | Sequestered antagonist formulations |
| US20030044458A1 (en) | 2001-08-06 | 2003-03-06 | Curtis Wright | Oral dosage form comprising a therapeutic agent and an adverse-effect agent |
| US7332182B2 (en) | 2001-08-06 | 2008-02-19 | Purdue Pharma L.P. | Pharmaceutical formulation containing opioid agonist, opioid antagonist and irritant |
| US7141250B2 (en) | 2001-08-06 | 2006-11-28 | Euro-Celtique S.A. | Pharmaceutical formulation containing bittering agent |
| JP3474870B2 (ja) | 2001-08-08 | 2003-12-08 | 菱計装株式会社 | 昇降機 |
| US20030049272A1 (en) | 2001-08-30 | 2003-03-13 | Yatindra Joshi | Pharmaceutical composition which produces irritation |
| US6691698B2 (en) | 2001-09-14 | 2004-02-17 | Fmc Technologies Inc. | Cooking oven having curved heat exchanger |
| US20030059467A1 (en) | 2001-09-14 | 2003-03-27 | Pawan Seth | Pharmaceutical composition comprising doxasozin |
| US20030068276A1 (en) | 2001-09-17 | 2003-04-10 | Lyn Hughes | Dosage forms |
| US20030059397A1 (en) | 2001-09-17 | 2003-03-27 | Lyn Hughes | Dosage forms |
| US20030092724A1 (en) | 2001-09-18 | 2003-05-15 | Huaihung Kao | Combination sustained release-immediate release oral dosage forms with an opioid analgesic and a non-opioid analgesic |
| ATE381924T1 (de) | 2001-09-21 | 2008-01-15 | Egalet As | Feste dispersionen mit kontrollierter freisetzung von carvedilol |
| EP1429744A1 (en) | 2001-09-21 | 2004-06-23 | Egalet A/S | Morphine polymer release system |
| US20030091635A1 (en) | 2001-09-26 | 2003-05-15 | Baichwal Anand R. | Opioid formulations having reduced potential for abuse |
| EP1429735A2 (de) | 2001-09-26 | 2004-06-23 | Klaus-Jürgen Steffens | Verfahren und vorrichtung zur herstellung von granulaten umfassend mindestens einen pharmazeutischen wirkstoff |
| US6837696B2 (en) | 2001-09-28 | 2005-01-04 | Mcneil-Ppc, Inc. | Apparatus for manufacturing dosage forms |
| JP2005529059A (ja) | 2001-09-28 | 2005-09-29 | マクニール−ピーピーシー・インコーポレイテッド | 改質した放出用の投薬形態 |
| JP2005505657A (ja) | 2001-10-09 | 2005-02-24 | ザ プロクター アンド ギャンブル カンパニー | 表面処理用水性組成物 |
| US6592901B2 (en) | 2001-10-15 | 2003-07-15 | Hercules Incorporated | Highly compressible ethylcellulose for tableting |
| JP2003125706A (ja) | 2001-10-23 | 2003-05-07 | Lion Corp | 口中清涼製剤 |
| PE20030527A1 (es) | 2001-10-24 | 2003-07-26 | Gruenenthal Chemie | Formulacion farmaceutica con liberacion retardada que contiene 3-(3-dimetilamino-1-etil-2-metil-propil) fenol o una sal farmaceuticamente aceptable del mismo y tabletas para administracion oral que la contienen |
| US20030104052A1 (en) | 2001-10-25 | 2003-06-05 | Bret Berner | Gastric retentive oral dosage form with restricted drug release in the lower gastrointestinal tract |
| US20030091630A1 (en) | 2001-10-25 | 2003-05-15 | Jenny Louie-Helm | Formulation of an erodible, gastric retentive oral dosage form using in vitro disintegration test data |
| TWI312285B (en) | 2001-10-25 | 2009-07-21 | Depomed Inc | Methods of treatment using a gastric retained gabapentin dosage |
| US6723340B2 (en) | 2001-10-25 | 2004-04-20 | Depomed, Inc. | Optimal polymer mixtures for gastric retentive tablets |
| CA2409552A1 (en) | 2001-10-25 | 2003-04-25 | Depomed, Inc. | Gastric retentive oral dosage form with restricted drug release in the lower gastrointestinal tract |
| US20030152622A1 (en) | 2001-10-25 | 2003-08-14 | Jenny Louie-Helm | Formulation of an erodible, gastric retentive oral diuretic |
| EP1441703B1 (en) | 2001-10-29 | 2018-01-03 | Massachusetts Institute of Technology | Zero-order release profile dosage form manufactured by three-dimensional printing |
| EP1450824A4 (en) | 2001-11-02 | 2005-09-28 | Elan Corp Plc | PHARMACEUTICAL COMPOSITION |
| US20040126428A1 (en) | 2001-11-02 | 2004-07-01 | Lyn Hughes | Pharmaceutical formulation including a resinate and an aversive agent |
| EP1463515A4 (en) | 2001-12-06 | 2005-01-12 | Scolr Pharma Inc | ISOFLAVONAL COMPOSITION FOR ORAL ADMINISTRATION |
| FR2833838B1 (fr) | 2001-12-21 | 2005-09-16 | Ellipse Pharmaceuticals | Procede de fabrication d'un comprime incluant un analgesique de type morphinique et comprime obtenu |
| AUPS044502A0 (en) | 2002-02-11 | 2002-03-07 | Commonwealth Scientific And Industrial Research Organisation | Novel catalysts and processes for their preparation |
| US20040033253A1 (en) | 2002-02-19 | 2004-02-19 | Ihor Shevchuk | Acyl opioid antagonists |
| US20030158265A1 (en) | 2002-02-20 | 2003-08-21 | Ramachandran Radhakrishnan | Orally administrable pharmaceutical formulation comprising pseudoephedrine hydrochloride and process for preparing the same |
| US20030190343A1 (en) | 2002-03-05 | 2003-10-09 | Pfizer Inc. | Palatable pharmaceutical compositions for companion animals |
| US6572889B1 (en) | 2002-03-07 | 2003-06-03 | Noveon Ip Holdings Corp. | Controlled release solid dosage carbamazepine formulations |
| US6753009B2 (en) | 2002-03-13 | 2004-06-22 | Mcneil-Ppc, Inc. | Soft tablet containing high molecular weight polyethylene oxide |
| DE20308437U1 (de) | 2002-04-05 | 2003-11-13 | Euro Celtique Sa | Matrix zur verzögerten, gleichbleibenden und unabhängigen Freisetzung von Wirkstoffen |
| DE10217232B4 (de) | 2002-04-18 | 2004-08-19 | Ticona Gmbh | Verfahren zur Herstellung gefüllter Granulate aus Polyethylenen hohen bzw. ultrahohen Molekulargewichts |
| WO2003089506A1 (en) | 2002-04-22 | 2003-10-30 | Purdue Research Foundation | Hydrogels having enhanced elasticity and mechanical strength properties |
| US20050106249A1 (en) | 2002-04-29 | 2005-05-19 | Stephen Hwang | Once-a-day, oral, controlled-release, oxycodone dosage forms |
| CA2483655A1 (en) | 2002-04-29 | 2003-11-13 | Alza Corporation | Methods and dosage forms for controlled delivery of oxycodone |
| WO2003094812A1 (en) | 2002-05-13 | 2003-11-20 | Endo Pharmaceuticals Inc. | Abuse-resistant opioid solid dosage form |
| AU2003245345A1 (en) | 2002-05-31 | 2003-12-19 | Alza Corporation | Dosage forms and compositions for osmotic delivery of variable dosages of oxycodone |
| US7776314B2 (en) | 2002-06-17 | 2010-08-17 | Grunenthal Gmbh | Abuse-proofed dosage system |
| DE10250083A1 (de) | 2002-06-17 | 2003-12-24 | Gruenenthal Gmbh | Gegen Missbrauch gesicherte Darreichungsform |
| WO2004004693A1 (en) | 2002-07-05 | 2004-01-15 | Collgegium Pharmaceutical | Abuse-deterrent pharmaceutical compositions of opiods and other drugs |
| US20040011806A1 (en) | 2002-07-17 | 2004-01-22 | Luciano Packaging Technologies, Inc. | Tablet filler device with star wheel |
| US20070196481A1 (en) | 2002-07-25 | 2007-08-23 | Amidon Gregory E | Sustained-release tablet composition |
| US7388068B2 (en) | 2002-08-21 | 2008-06-17 | Clariant Produkte (Deutschland) Gmbh | Copolymers made of alkylene oxides and glycidyl ethers and use thereof as polymerizable emulsifiers |
| JP2006501234A (ja) | 2002-08-21 | 2006-01-12 | フォークス ファーマシューティカルズ リミテッド | 錠剤製造における、クエン酸およびラクチトール等の水溶性糖水溶液の、造粒液としての使用 |
| US20040052844A1 (en) | 2002-09-16 | 2004-03-18 | Fang-Hsiung Hsiao | Time-controlled, sustained release, pharmaceutical composition containing water-soluble resins |
| US20040077677A1 (en) | 2002-09-17 | 2004-04-22 | Wyeth | Oral formulations |
| JP4195447B2 (ja) | 2002-09-20 | 2008-12-10 | エフ エム シー コーポレーション | ミクロ結晶性セルロースを含有する化粧品用組成物 |
| DE60319252T2 (de) | 2002-09-21 | 2009-03-05 | Zhang, Shuyi | Formulierung von acetaminophen und tramadol mit verzögerter freisetzung |
| JP5189242B2 (ja) * | 2002-09-23 | 2013-04-24 | アルケルメス ファーマ アイルランド リミテッド | 乱用抵抗性の医薬組成物 |
| JP2004143071A (ja) | 2002-10-23 | 2004-05-20 | Hosokawa Funtai Gijutsu Kenkyusho:Kk | 薬物含有複合粒子の製造方法および薬物含有複合粒子 |
| US20050191244A1 (en) | 2002-10-25 | 2005-09-01 | Gruenenthal Gmbh | Abuse-resistant pharmaceutical dosage form |
| AU2003275855B2 (en) | 2002-10-25 | 2009-04-23 | Paladin Labs (Barbados) Inc. | Sustained-release tramadol formulations with 24-hour efficacy |
| DE10250087A1 (de) | 2002-10-25 | 2004-05-06 | Grünenthal GmbH | Gegen Missbrauch gesicherte Darreichungsform |
| DE10250088A1 (de) | 2002-10-25 | 2004-05-06 | Grünenthal GmbH | Gegen Missbrauch gesicherte Darreichungsform |
| US20050186139A1 (en) | 2002-10-25 | 2005-08-25 | Gruenenthal Gmbh | Abuse-proofed dosage form |
| DE10250084A1 (de) | 2002-10-25 | 2004-05-06 | Grünenthal GmbH | Gegen Missbrauch gesicherte Darreichungsform |
| DE10252667A1 (de) | 2002-11-11 | 2004-05-27 | Grünenthal GmbH | Spirocyclische Cyclohexan-Derivate |
| US20040091528A1 (en) | 2002-11-12 | 2004-05-13 | Yamanouchi Pharma Technologies, Inc. | Soluble drug extended release system |
| US7018658B2 (en) | 2002-11-14 | 2006-03-28 | Synthon Bv | Pharmaceutical pellets comprising tamsulosin |
| US20040121003A1 (en) | 2002-12-19 | 2004-06-24 | Acusphere, Inc. | Methods for making pharmaceutical formulations comprising deagglomerated microparticles |
| US20040185097A1 (en) | 2003-01-31 | 2004-09-23 | Glenmark Pharmaceuticals Ltd. | Controlled release modifying complex and pharmaceutical compositions thereof |
| US7442387B2 (en) | 2003-03-06 | 2008-10-28 | Astellas Pharma Inc. | Pharmaceutical composition for controlled release of active substances and manufacturing method thereof |
| ATE454169T1 (de) | 2003-03-13 | 2010-01-15 | Controlled Chemicals Inc | Oxycodon- konjugate mit niedrigerem missbrauch- potential und ausgedehnter tätigkeitsdauer |
| ATE495732T1 (de) | 2003-03-26 | 2011-02-15 | Egalet As | Morphin-system mit kontrollierter freisetzung |
| EP1974726B1 (en) | 2003-03-26 | 2010-01-13 | Egalet A/S | Matrix compositions for controlled delivery of drug substances |
| MXPA05011071A (es) | 2003-04-21 | 2005-12-12 | Euro Celtique Sa | Forma de dosificacion resistente a la alteracion que comprende particulas co-extrusionadas de agente adverso y proceso de fabricacion de las misma. |
| MY135852A (en) | 2003-04-21 | 2008-07-31 | Euro Celtique Sa | Pharmaceutical products |
| AR047936A1 (es) | 2003-04-30 | 2006-03-15 | Purdue Pharma Ltd | Forma de dosificacion transdermica resistente a la manipulacion |
| US8906413B2 (en) | 2003-05-12 | 2014-12-09 | Supernus Pharmaceuticals, Inc. | Drug formulations having reduced abuse potential |
| CN1473562A (zh) | 2003-06-27 | 2004-02-11 | 辉 刘 | 儿用口腔速溶、速崩冻干片及其制备方法 |
| HU227142B1 (en) | 2003-07-02 | 2010-08-30 | Egis Gyogyszergyar Nyilvanosan | Capsule of improved release containing fluconazole |
| US20050015730A1 (en) | 2003-07-14 | 2005-01-20 | Srimanth Gunturi | Systems, methods and computer program products for identifying tab order sequence of graphically represented elements |
| NZ545202A (en) | 2003-08-06 | 2010-03-26 | Gruenenthal Chemie | Abuse-proofed dosage form comprising opiods and a high molecular weight polyethylene oxide |
| US8075872B2 (en) | 2003-08-06 | 2011-12-13 | Gruenenthal Gmbh | Abuse-proofed dosage form |
| DE102004020220A1 (de) | 2004-04-22 | 2005-11-10 | Grünenthal GmbH | Verfahren zur Herstellung einer gegen Missbrauch gesicherten, festen Darreichungsform |
| DE102005005446A1 (de) | 2005-02-04 | 2006-08-10 | Grünenthal GmbH | Bruchfeste Darreichungsformen mit retardierter Freisetzung |
| DE10336400A1 (de) | 2003-08-06 | 2005-03-24 | Grünenthal GmbH | Gegen Missbrauch gesicherte Darreichungsform |
| US20070048228A1 (en) | 2003-08-06 | 2007-03-01 | Elisabeth Arkenau-Maric | Abuse-proofed dosage form |
| RU2339365C2 (ru) | 2003-08-06 | 2008-11-27 | Грюненталь Гмбх | Защищенная от применения не по назначению лекарственная форма |
| CL2004002016A1 (es) | 2003-08-06 | 2005-05-20 | Gruenenthal Chemie | Forma de dosificacion termoformada a prueba de abuso que contiene (a) uno o mas principios activos susceptibles de abuso, (b) opcionalmente sustancias auxiliares, (c) al menos un polimero sintetico o natural definido y (d) opcionalmente al menos una |
| DE10361596A1 (de) | 2003-12-24 | 2005-09-29 | Grünenthal GmbH | Verfahren zur Herstellung einer gegen Missbrauch gesicherten Darreichungsform |
| DE102004032051A1 (de) | 2004-07-01 | 2006-01-19 | Grünenthal GmbH | Verfahren zur Herstellung einer gegen Missbrauch gesicherten, festen Darreichungsform |
| US20050063214A1 (en) | 2003-09-22 | 2005-03-24 | Daisaburo Takashima | Semiconductor integrated circuit device |
| AU2004277898B2 (en) | 2003-09-25 | 2009-04-02 | Euro-Celtique S.A. | Pharmaceutical combinations of hydrocodone and naltrexone |
| EP1682099A2 (en) | 2003-09-30 | 2006-07-26 | ALZA Corporation | Osmotically driven active agent delivery device providing an ascending release profile |
| US20060172006A1 (en) | 2003-10-10 | 2006-08-03 | Vincent Lenaerts | Sustained-release tramadol formulations with 24-hour clinical efficacy |
| US20060009478A1 (en) | 2003-10-15 | 2006-01-12 | Nadav Friedmann | Methods for the treatment of back pain |
| EP1677798A2 (en) | 2003-10-29 | 2006-07-12 | Alza Corporation | Once-a-day, oral, controlled-release, oxycodone dosage forms |
| US7201920B2 (en) | 2003-11-26 | 2007-04-10 | Acura Pharmaceuticals, Inc. | Methods and compositions for deterring abuse of opioid containing dosage forms |
| JP2007513147A (ja) | 2003-12-04 | 2007-05-24 | ファイザー・プロダクツ・インク | 押し出し機を使用して、好ましくはポロキサマーとグリセリドを含有する多粒子結晶性医薬組成物を製造するための噴霧凝結方法 |
| CA2548834C (en) | 2003-12-09 | 2009-08-11 | Euro-Celtique S.A. | Tamper resistant co-extruded dosage form containing an active agent and an adverse agent and process of making same |
| WO2005060942A1 (en) | 2003-12-19 | 2005-07-07 | Aurobindo Pharma Ltd | Extended release pharmaceutical composition of metformin |
| DE10360792A1 (de) | 2003-12-23 | 2005-07-28 | Grünenthal GmbH | Spirocyclische Cyclohexan-Derivate |
| EP1701706A2 (en) | 2003-12-29 | 2006-09-20 | Alza Corporation | Novel drug compositions and dosage forms |
| WO2005079752A2 (en) | 2004-02-11 | 2005-09-01 | Rubicon Research Private Limited | Controlled release pharmaceutical compositions with improved bioavailability |
| TWI350762B (en) | 2004-02-12 | 2011-10-21 | Euro Celtique Sa | Particulates |
| GB0403100D0 (en) | 2004-02-12 | 2004-03-17 | Euro Celtique Sa | Particulates |
| GB0403098D0 (en) | 2004-02-12 | 2004-03-17 | Euro Celtique Sa | Extrusion |
| SI1718258T1 (sl) | 2004-02-23 | 2009-08-31 | Euro Celtique Sa | Naprava za transdermalno dajanje opioida, varna pred zlorabo |
| TWI483944B (zh) | 2004-03-30 | 2015-05-11 | Euro Celtique Sa | 含有小於25ppm14-羥可待因酮之羥可酮鹽酸鹽組成物、醫藥劑型、延遲釋出口服劑型及醫藥上可以接受的包裝 |
| US20050220877A1 (en) | 2004-03-31 | 2005-10-06 | Patel Ashish A | Bilayer tablet comprising an antihistamine and a decongestant |
| JP2005314407A (ja) | 2004-03-31 | 2005-11-10 | Nippon Nohyaku Co Ltd | 新規なハロアルキルスルホンアニリド誘導体、除草剤及びその使用方法並びにその中間体 |
| DE102004019916A1 (de) | 2004-04-21 | 2005-11-17 | Grünenthal GmbH | Gegen Missbrauch gesichertes wirkstoffhaltiges Pflaster |
| JP5064209B2 (ja) | 2004-04-22 | 2012-10-31 | グリューネンタール・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング | 乱用防止固体剤形を製造する方法 |
| WO2005105036A1 (en) | 2004-04-28 | 2005-11-10 | Natco Pharma Limited | Controlled release mucoadhesive matrix formulation containing tolterodine and a process for its preparation |
| US20050271594A1 (en) | 2004-06-04 | 2005-12-08 | Groenewoud Pieter J | Abuse resistent pharmaceutical composition |
| TWI356036B (en) | 2004-06-09 | 2012-01-11 | Smithkline Beecham Corp | Apparatus and method for pharmaceutical production |
| SI1612203T1 (sl) | 2004-06-28 | 2007-12-31 | Gruenenthal Chemie | Kristalne oblike (-)-(1R,2R)-3-(3-dimetilamino-1-etil-2-metilpropil)-fenol hidroklorida |
| ITMI20041317A1 (it) | 2004-06-30 | 2004-09-30 | Ibsa Inst Biochimique Sa | Formulazioni farmaceutiche per la somministrazione sicura di farmaci utilizzati nel trattamento della tossicodipendenza e procedimento per il loro ottenimento |
| PL1765303T5 (pl) | 2004-07-01 | 2023-05-22 | Grünenthal GmbH | Tabletka doustna zabezpieczona przed nadużywaniem |
| EP1765298B1 (de) | 2004-07-01 | 2012-10-24 | Gruenenthal Gmbh | Verfahren zur herstellung einer gegen missbrauch gesicherten, festen darreichungsform unter verwendung eines planetwalzenextruders |
| DE102004032049A1 (de) | 2004-07-01 | 2006-01-19 | Grünenthal GmbH | Gegen Missbrauch gesicherte, orale Darreichungsform |
| DE102004032103A1 (de) | 2004-07-01 | 2006-01-19 | Grünenthal GmbH | Gegen Missbrauch gesicherte, orale Darreichungsform |
| AU2005259478B2 (en) | 2004-07-01 | 2010-07-15 | Gruenenthal Gmbh | Oral dosage form safeguarded against abuse containing (1R, 2R)-3-(3-dimethylamino-1-ethyl-2-methyl-propyl)-phenol |
| KR20060007225A (ko) | 2004-07-19 | 2006-01-24 | 삼성전자주식회사 | 촬상소자 구동제어와 메모리 읽기제어를 이용한 손떨림보정방법 및 이를 적용한 촬영장치 |
| WO2006010440A1 (en) | 2004-07-27 | 2006-02-02 | Unilever Plc | Hair care compositions |
| GB2418854B (en) | 2004-08-31 | 2009-12-23 | Euro Celtique Sa | Multiparticulates |
| US20060068009A1 (en) | 2004-09-30 | 2006-03-30 | Scolr Pharma, Inc. | Modified release ibuprofen dosage form |
| US20070077297A1 (en) | 2004-09-30 | 2007-04-05 | Scolr Pharma, Inc. | Modified release ibuprofen dosage form |
| US7426948B2 (en) | 2004-10-08 | 2008-09-23 | Phibrowood, Llc | Milled submicron organic biocides with narrow particle size distribution, and uses thereof |
| US20060177380A1 (en) | 2004-11-24 | 2006-08-10 | Acura Pharmaceuticals, Inc. | Methods and compositions for deterring abuse of orally administered pharmaceutical products |
| US20080152595A1 (en) | 2004-11-24 | 2008-06-26 | Acura Pharmaceuticals, Inc. | Methods and compositions for deterring abuse of orally administered pharmaceutical products |
| US20070231268A1 (en) | 2004-11-24 | 2007-10-04 | Acura Pharmaceuticals, Inc. | Methods and compositions for deterring abuse of orally administered pharmaceutical products |
| ES2630002T3 (es) | 2005-01-26 | 2017-08-17 | Taiho Pharmaceutical Co., Ltd. | Fármaco anticanceroso que contiene alfa,alfa,alfa-trifluorotimidina e inhibidor de timidina fosforilasa |
| AP2274A (en) | 2005-01-28 | 2011-08-19 | Euro Celtiques Sa | Alcohol resistant dosage forms. |
| DE102005005449A1 (de) | 2005-02-04 | 2006-08-10 | Grünenthal GmbH | Verfahren zur Herstellung einer gegen Missbrauch gesicherten Darreichungsform |
| FR2889810A1 (fr) | 2005-05-24 | 2007-02-23 | Flamel Technologies Sa | Forme medicamenteuse orale, microparticulaire, anti-mesurage |
| US7292616B2 (en) | 2005-02-09 | 2007-11-06 | Ultratech, Inc. | CO2 laser stabilization systems and methods |
| CA2597492A1 (en) | 2005-02-10 | 2006-08-17 | Lifecycle Pharma A/S | A stable pharmaceutical composition comprising a fixed dose combination of fenofibrate and an hmg-coa reductase inhibitor |
| US20060194759A1 (en) | 2005-02-25 | 2006-08-31 | Eidelson Stewart G | Topical compositions and methods for treating pain and inflammation |
| EP1695700A1 (en) | 2005-02-28 | 2006-08-30 | Euro-Celtique S.A. | Dosage form containing oxycodone and naloxone |
| WO2006094672A1 (en) | 2005-03-04 | 2006-09-14 | Euro-Celtique S.A. | Method of reducing alpha, beta unsaturated ketones in opioid compositions |
| US20060204575A1 (en) | 2005-03-11 | 2006-09-14 | Hengsheng Feng | Amphetamine formulations |
| US7732427B2 (en) | 2005-03-31 | 2010-06-08 | University Of Delaware | Multifunctional and biologically active matrices from multicomponent polymeric solutions |
| JP5256425B2 (ja) | 2005-04-08 | 2013-08-07 | リングアル コンセグナ ピーティーワイ エルティーディー | 口腔送達システム |
| NZ585116A (en) | 2005-05-10 | 2011-12-22 | Novartis Ag | Extrusion process for making compositions with poorly compressible therapeutic compounds e.g. metformin hydrochloride |
| JP5161075B2 (ja) | 2005-06-03 | 2013-03-13 | エガレット エイ/エス | 分散媒体の第1フラクションとマトリックスの第2フラクションを有し、第2フラクションが胃腸の液体に少なくとも部分的に最初に曝される固形の医薬組成物 |
| WO2007005716A2 (en) | 2005-06-30 | 2007-01-11 | Cinergen, Llc | Methods of treatment and compositions for use thereof |
| NZ565108A (en) | 2005-07-07 | 2011-10-28 | Farnam Co Inc | Sustained release pharmaceutical compositions for highly water soluble drugs |
| DE102005032806A1 (de) | 2005-07-12 | 2007-01-18 | Röhm Gmbh | Verwendung eines teilneutralisierten, anionischen (Meth)acrylat-Copolymers als Überzug für die Herstellung einer Arzneiform mit einer Wirkstofffreisetzung bei erniedrigten pH-Werten |
| US8858993B2 (en) | 2005-07-25 | 2014-10-14 | Metrics, Inc. | Coated tablet with zero-order or near zero-order release kinetics |
| JP4809952B2 (ja) | 2005-07-29 | 2011-11-09 | キョーラク株式会社 | コンテナおよびその製造方法 |
| AU2006275476A1 (en) | 2005-08-01 | 2007-02-08 | Alpharma Inc. | Alcohol resistant pharmaceutical formulations |
| CN101232871A (zh) | 2005-08-03 | 2008-07-30 | 伊士曼化工公司 | 生育酚聚乙二醇琥珀酸酯粉末及其制备方法 |
| US20070048373A1 (en) | 2005-08-30 | 2007-03-01 | Cima Labs Inc. | Dried milled granulate and methods |
| EA015737B1 (ru) | 2005-10-14 | 2011-10-31 | Дзе Китасато Инститьют | Новые производные дигидропсевдоэритромицина |
| US8710233B2 (en) | 2005-10-19 | 2014-04-29 | Gruenenthal Gmbh | Vanilloid receptor ligands and use thereof for the production of pharmaceutical preparations |
| US20070092573A1 (en) | 2005-10-24 | 2007-04-26 | Laxminarayan Joshi | Stabilized extended release pharmaceutical compositions comprising a beta-adrenoreceptor antagonist |
| PL116330U1 (en) | 2005-10-31 | 2007-04-02 | Alza Corp | Method for the reduction of alcohol provoked rapid increase in the released dose of the orally administered opioide with prolonged liberation |
| US8329744B2 (en) | 2005-11-02 | 2012-12-11 | Relmada Therapeutics, Inc. | Methods of preventing the serotonin syndrome and compositions for use thereof |
| WO2008134071A1 (en) | 2007-04-26 | 2008-11-06 | Theraquest Biosciences, Inc. | Multimodal abuse resistant extended release formulations |
| US8652529B2 (en) | 2005-11-10 | 2014-02-18 | Flamel Technologies | Anti-misuse microparticulate oral pharmaceutical form |
| FR2892937B1 (fr) | 2005-11-10 | 2013-04-05 | Flamel Tech Sa | Forme pharmaceutique orale microparticulaire anti-mesusage |
| DE102005058569B4 (de) | 2005-12-08 | 2010-07-15 | Lts Lohmann Therapie-Systeme Ag | Schaumwafer mit Polyvinylalkohol-Polyethylenglycol-Pfropfcopolymer |
| SG169334A1 (en) | 2006-01-21 | 2011-03-30 | Abbott Gmbh & Co Kg | Dosage form and method for the delivery of drugs of abuse |
| US20090022798A1 (en) | 2007-07-20 | 2009-01-22 | Abbott Gmbh & Co. Kg | Formulations of nonopioid and confined opioid analgesics |
| US20090317355A1 (en) | 2006-01-21 | 2009-12-24 | Abbott Gmbh & Co. Kg, | Abuse resistant melt extruded formulation having reduced alcohol interaction |
| US20100172989A1 (en) | 2006-01-21 | 2010-07-08 | Abbott Laboratories | Abuse resistant melt extruded formulation having reduced alcohol interaction |
| EP1813276A1 (en) | 2006-01-27 | 2007-08-01 | Euro-Celtique S.A. | Tamper resistant dosage forms |
| FR2897267A1 (fr) | 2006-02-16 | 2007-08-17 | Flamel Technologies Sa | Formes pharmaceutiques multimicroparticulaires pour administration per os |
| ZA200807571B (en) | 2006-03-01 | 2009-08-26 | Ethypharm Sa | Crush-resistant tablets intended to prevent accidental misuse and unlawful diversion |
| EP2086515A2 (en) | 2006-03-02 | 2009-08-12 | Vaunnex, Inc. | Rate-controlled bioadhesive oral dosage formulations |
| CN101395159A (zh) | 2006-03-02 | 2009-03-25 | 马林克罗特公司 | 制备含有低含量α,β-不饱和酮化合物的吗啡烷-6-酮产物的方法 |
| US20070224637A1 (en) | 2006-03-24 | 2007-09-27 | Mcauliffe Joseph C | Oxidative protection of lipid layer biosensors |
| WO2007112285A2 (en) | 2006-03-24 | 2007-10-04 | Auxilium Pharmaceuticals, Inc. | Process for the preparation of a hot-melt extruded laminate |
| NZ572207A (en) | 2006-03-24 | 2012-02-24 | Auxilium Int Holdings Inc | Stabilized compositions containing alkaline labile drugs |
| US10960077B2 (en) | 2006-05-12 | 2021-03-30 | Intellipharmaceutics Corp. | Abuse and alcohol resistant drug composition |
| US9023400B2 (en) | 2006-05-24 | 2015-05-05 | Flamel Technologies | Prolonged-release multimicroparticulate oral pharmaceutical form |
| WO2007138466A2 (en) | 2006-06-01 | 2007-12-06 | Wockhardt Ltd | Pharmaceutical compositions comprising meloxicam and tramadol combination |
| US20070292508A1 (en) | 2006-06-05 | 2007-12-20 | Balchem Corporation | Orally disintegrating dosage forms |
| US20080069891A1 (en) | 2006-09-15 | 2008-03-20 | Cima Labs, Inc. | Abuse resistant drug formulation |
| HUE032156T2 (en) | 2006-06-19 | 2017-09-28 | Alpharma Pharmaceuticals Llc | Pharmaceutical preparations |
| CN101091721A (zh) | 2006-06-22 | 2007-12-26 | 孙明 | 阿胶新剂型的制备方法 |
| WO2008008120A1 (en) | 2006-07-14 | 2008-01-17 | Fmc Corporation | Solid form |
| JP4029109B1 (ja) | 2006-07-18 | 2008-01-09 | タマ生化学株式会社 | ビタミンeとプロリンの複合体粉末及びその製造方法 |
| WO2008011596A2 (en) | 2006-07-21 | 2008-01-24 | Lab International Srl | Hydrophilic abuse deterrent delivery system |
| SA07280459B1 (ar) | 2006-08-25 | 2011-07-20 | بيورديو فارما إل. بي. | أشكال جرعة صيدلانية للتناول عن طريق الفم مقاومة للعبث تشتمل على مسكن شبه أفيوني |
| US8445018B2 (en) | 2006-09-15 | 2013-05-21 | Cima Labs Inc. | Abuse resistant drug formulation |
| US8187636B2 (en) | 2006-09-25 | 2012-05-29 | Atlantic Pharmaceuticals, Inc. | Dosage forms for tamper prone therapeutic agents |
| KR101400824B1 (ko) | 2006-09-25 | 2014-05-29 | 후지필름 가부시키가이샤 | 레지스트 조성물, 이 레지스트 조성물에 사용되는 수지, 이수지의 합성에 사용되는 화합물, 및 상기 레지스트조성물을 사용한 패턴형성방법 |
| US20080085304A1 (en) | 2006-10-10 | 2008-04-10 | Penwest Pharmaceuticals Co. | Robust sustained release formulations |
| EP2079453A1 (en) | 2006-10-10 | 2009-07-22 | Penwest Pharmaceuticals Co. | Robust sustained release formulations |
| GB0624880D0 (en) | 2006-12-14 | 2007-01-24 | Johnson Matthey Plc | Improved method for making analgesics |
| DE102006062120A1 (de) | 2006-12-22 | 2008-06-26 | Grünenthal GmbH | Pharmazeutische Zusammensetzung zur Aknebehandlung |
| CA2673511A1 (en) | 2006-12-22 | 2008-07-03 | Combinatorx, Incorporated | Pharmaceutical compositions for treatment of parkinson's disease and related disorders |
| AU2008207200B2 (en) | 2007-01-16 | 2011-02-17 | Egalet Ltd | Use of i) a polyglycol and ii) an active drug substance for the preparation of a pharmaceutical composition for i) mitigating the risk of alcohol induced dose dumping and/or ii) reducing the risk of drug abuse |
| WO2008094877A2 (en) | 2007-01-30 | 2008-08-07 | Drugtech Corporation | Compositions for oral delivery of pharmaceuticals |
| CN100579525C (zh) | 2007-02-02 | 2010-01-13 | 东南大学 | 盐酸尼卡地平缓释制剂及其制备方法 |
| ATE552861T1 (de) | 2007-02-08 | 2012-04-15 | Kempharm Inc | Polare hydrophile prodrugs von amphetamin und anderen stimulantien sowie herstellungs- und verwendungsverfahren |
| CN101057849A (zh) | 2007-02-27 | 2007-10-24 | 齐齐哈尔医学院 | 含有盐酸二甲双胍和格列吡嗪的缓释制剂及其制备方法 |
| EP2144599B1 (en) | 2007-03-02 | 2010-08-04 | Farnam Companies, Inc. | Sustained release pellets comprising wax-like material |
| EP1980245A1 (en) | 2007-04-11 | 2008-10-15 | Cephalon France | Bilayer lyophilized pharmaceutical compositions and methods of making and using same |
| US20080260836A1 (en) | 2007-04-18 | 2008-10-23 | Thomas James Boyd | Films Comprising a Plurality of Polymers |
| EP2061587A1 (en) | 2007-04-26 | 2009-05-27 | Sigmoid Pharma Limited | Manufacture of multiple minicapsules |
| US20110020408A1 (en) | 2007-05-17 | 2011-01-27 | Ranbaxy Laboratories Limited | multilayered modified release formulation comprising amoxicillin and clavulanate |
| US8202542B1 (en) | 2007-05-31 | 2012-06-19 | Tris Pharma | Abuse resistant opioid drug-ion exchange resin complexes having hybrid coatings |
| WO2008148798A2 (en) | 2007-06-04 | 2008-12-11 | Egalet A/S | Controlled release pharmaceutical compositions for prolonged effect |
| US20100035886A1 (en) | 2007-06-21 | 2010-02-11 | Veroscience, Llc | Parenteral formulations of dopamine agonists |
| DE102007030308A1 (de) | 2007-06-29 | 2009-01-02 | Printed Systems Gmbh | Verfahren zum Herstellen einer Speicherstruktur |
| JP2010532358A (ja) | 2007-07-01 | 2010-10-07 | ピーター ハバウシ,ジョセフ | 咀嚼可能外層を有する配合剤 |
| MX2010000803A (es) | 2007-07-20 | 2010-06-23 | Abbott Gmbh & Co Kg | Formulaciones de analgesicos no opioides y opioides confinados. |
| EP2200591A2 (en) | 2007-09-11 | 2010-06-30 | Ranbaxy Laboratories Limited | Controlled release pharmaceutical dosage forms of trimetazidine |
| WO2009035474A1 (en) | 2007-09-13 | 2009-03-19 | Cima Labs Inc. | Abuse resistant drug formulation |
| US20100303883A1 (en) | 2007-10-17 | 2010-12-02 | Axxia Pharmaceuticals, Llc | Polymeric drug delivery systems and thermoplastic extrusion processes for producing such systems |
| CN104958282B (zh) | 2007-11-23 | 2018-05-29 | 格吕伦塔尔有限公司 | 他喷他多组合物 |
| JP2011506319A (ja) | 2007-12-06 | 2011-03-03 | デュレクト コーポレーション | 疼痛、関節炎症状、または慢性疾患に伴う炎症の治療に有用な方法 |
| MX2010006005A (es) | 2007-12-12 | 2010-06-15 | Basf Se | Sales de ingredientes activos con contraiones polimericos. |
| CA2707980C (en) | 2007-12-17 | 2015-05-12 | Labopharm Inc. | Misuse preventative, controlled release formulation |
| KR101616246B1 (ko) | 2008-01-25 | 2016-05-02 | 그뤼넨탈 게엠베하 | 약제학적 투여형 |
| KR100970665B1 (ko) | 2008-02-04 | 2010-07-15 | 삼일제약주식회사 | 알푸조신 또는 그의 염을 함유하는 서방성 정제 |
| BRPI0909030A2 (pt) | 2008-03-05 | 2018-03-13 | Panacea Biotec Ltd | composições farmacêuticas de liberação modificada compreendendo micofenolato e processos para as mesmas. |
| US8372432B2 (en) | 2008-03-11 | 2013-02-12 | Depomed, Inc. | Gastric retentive extended-release dosage forms comprising combinations of a non-opioid analgesic and an opioid analgesic |
| EP2100598A1 (en) | 2008-03-13 | 2009-09-16 | Laboratorios Almirall, S.A. | Inhalation composition containing aclidinium for treatment of asthma and chronic obstructive pulmonary disease |
| TWI519322B (zh) | 2008-04-15 | 2016-02-01 | 愛戴爾製藥股份有限公司 | 包含弱鹼性藥物及控制釋放劑型之組合物 |
| MX2010012039A (es) | 2008-05-09 | 2010-11-30 | Gruenenthal Gmbh | Proceso para la preparacion de una formulacion de polvo intermedia y una forma de dosificacion solida final bajo el uso de un paso de congelacion por rocio. |
| PT2309987E (pt) | 2008-07-03 | 2012-12-13 | Novartis Ag | Processo para granulação por fusão |
| CA2734646C (en) | 2008-08-20 | 2016-06-28 | James W. Mcginity | Hot-melt extrusion of modified release multi-particulates |
| FR2936709B1 (fr) | 2008-10-02 | 2012-05-11 | Ethypharm Sa | Comprimes alcoolo-resistants. |
| WO2010044842A1 (en) | 2008-10-16 | 2010-04-22 | University Of Tennessee Research Foundation | Tamper resistant oral dosage forms containing an embolizing agent |
| CN102197067B (zh) | 2008-10-24 | 2013-08-14 | 花王株式会社 | 树脂组合物的制造方法 |
| UA102706C2 (ru) | 2008-10-27 | 2013-08-12 | Алза Корпорейшн | Лекарственная форма ацетаминофена и комплекса трамадола с замедленным действием для перорального приема |
| US20100260844A1 (en) | 2008-11-03 | 2010-10-14 | Scicinski Jan J | Oral pharmaceutical dosage forms |
| TW201022253A (en) | 2008-11-14 | 2010-06-16 | Portola Pharm Inc | Solid composition for controlled release of ionizable active agents with poor aqueous solubility at low pH and methods of use thereof |
| ES2414856T3 (es) | 2008-12-12 | 2013-07-23 | Paladin Labs Inc. | Formulaciones de fármaco narcótico con potencial de adicción disminuido |
| JP5667575B2 (ja) | 2008-12-16 | 2015-02-12 | パラディン ラブス インコーポレーテッド | 誤用を防止する放出制御製剤 |
| WO2010083843A1 (en) | 2009-01-26 | 2010-07-29 | Egalet A/S | Controlled release formulations with continuous efficacy |
| NZ594208A (en) | 2009-02-06 | 2012-10-26 | Egalet Ltd | Pharmaceutical compositions resistant to abuse |
| EP2408436B1 (en) | 2009-03-18 | 2017-02-22 | Evonik Röhm GmbH | Controlled release pharmaceutical composition with resistance against the influence of ethanol employing a coating comprising neutral vinyl polymers and excipients |
| EP2246063A1 (en) | 2009-04-29 | 2010-11-03 | Ipsen Pharma S.A.S. | Sustained release formulations comprising GnRH analogues |
| GB0909680D0 (en) | 2009-06-05 | 2009-07-22 | Euro Celtique Sa | Dosage form |
| NZ597283A (en) | 2009-06-24 | 2013-07-26 | Egalet Ltd | Controlled release formulations |
| WO2011008298A2 (en) | 2009-07-16 | 2011-01-20 | Nectid, Inc. | Novel axomadol dosage forms |
| RU2015138422A (ru) | 2009-07-22 | 2018-12-25 | Грюненталь Гмбх | Стабильная при окислении, прочная на излом лекарственная форма |
| MX2012000317A (es) | 2009-07-22 | 2012-02-08 | Gruenenthal Gmbh | Forma de dosificacion de liberacion controlada extruida por fusion en caliente. |
| AU2010300641B2 (en) | 2009-09-30 | 2016-03-17 | Acura Pharmaceuticals, Inc. | Methods and compositions for deterring abuse |
| EP2535114A4 (en) | 2009-11-13 | 2015-11-18 | Moriroku Chemicals Company Ltd | PROCESS FOR PRODUCING FINE POWDER AND FINE POWDER PRODUCED ACCORDING TO THIS METHOD |
| EP2506838A1 (en) | 2009-12-01 | 2012-10-10 | Noven Pharmaceuticals, INC. | Transdermal testosterone device and delivery |
| WO2011095314A2 (en) | 2010-02-03 | 2011-08-11 | Grünenthal GmbH | Preparation of a powdery pharmaceutical composition by means of an extruder |
| AU2011223790A1 (en) | 2010-03-01 | 2012-08-30 | Myrexis, Inc. | Compounds and therapeutic uses thereof |
| GB201003731D0 (en) | 2010-03-05 | 2010-04-21 | Univ Strathclyde | Immediate/delayed drug delivery |
| CA2792523C (en) | 2010-03-09 | 2018-01-09 | Alkermes Pharma Ireland Limited | Alcohol resistant enteric pharmaceutical compositions |
| ES2592277T3 (es) | 2010-04-02 | 2016-11-29 | Buzzz Pharmaceuticals Limited | Formulaciones transdérmicas disuasorias del abuso de agonistas y agonistas-antagonistas opiáceos |
| AU2011236548A1 (en) | 2010-04-07 | 2012-11-01 | Lupin Limited | Controlled release pharmaceutical compositions of tapentadol |
| GB201006200D0 (en) | 2010-04-14 | 2010-06-02 | Ayanda As | Composition |
| EP2560624B1 (en) | 2010-04-23 | 2018-07-04 | KemPharm, Inc. | Therapeutic formulation for reduced drug side effects |
| US20130059010A1 (en) | 2010-05-14 | 2013-03-07 | Ethypharm | Alcohol-resistant oral pharmaceutical form |
| FR2960775A1 (fr) | 2010-06-07 | 2011-12-09 | Ethypharm Sa | Microgranules resistants au detournement |
| ES2487244T3 (es) | 2010-09-02 | 2014-08-20 | Grünenthal GmbH | Forma de dosificación resistente a la manipulación que comprende un polímero aniónico |
| CA2808219C (en) | 2010-09-02 | 2019-05-14 | Gruenenthal Gmbh | Tamper resistant dosage form comprising inorganic salt |
| AR082861A1 (es) | 2010-09-02 | 2013-01-16 | Gruenenthal Gmbh | Forma de dosificacion resistente a alteracion que comprende un polimero anionico |
| US20120202838A1 (en) | 2010-11-04 | 2012-08-09 | Abbott Laboratories | Drug formulations |
| US20120231083A1 (en) | 2010-11-18 | 2012-09-13 | The Board Of Trustees Of The University Of Illinois | Sustained release cannabinoid medicaments |
| GB201020895D0 (en) | 2010-12-09 | 2011-01-26 | Euro Celtique Sa | Dosage form |
| AU2011346758C1 (en) | 2010-12-23 | 2015-09-03 | Purdue Pharma L.P. | Tamper resistant solid oral dosage forms |
| WO2012112952A1 (en) | 2011-02-17 | 2012-08-23 | QRxPharma Ltd. | Technology for preventing abuse of solid dosage forms |
| PL3287123T3 (pl) | 2011-03-04 | 2020-11-02 | Grünenthal GmbH | Wodny preparat farmaceutyczny tapentadolu do podawania doustnego |
| RS56523B1 (sr) | 2011-04-29 | 2018-02-28 | Gruenenthal Gmbh | Tapentadol za sprečavanje i lečenje depresije i anksioznosti |
| US8858963B1 (en) | 2011-05-17 | 2014-10-14 | Mallinckrodt Llc | Tamper resistant composition comprising hydrocodone and acetaminophen for rapid onset and extended duration of analgesia |
| CA2837077A1 (en) | 2011-06-01 | 2012-12-06 | Fmc Corporation | Controlled release solid dose forms |
| WO2013003845A1 (en) | 2011-06-30 | 2013-01-03 | Neos Therapeutics, Lp | Abuse resistant drug forms |
| CA2839123A1 (en) | 2011-07-29 | 2013-02-07 | Grunenthal Gmbh | Tamper-resistant tablet providing immediate drug release |
| AU2012292418B2 (en) | 2011-07-29 | 2017-02-16 | Grunenthal Gmbh | Tamper-resistant tablet providing immediate drug release |
| IN2014CN00827A (zh) | 2011-08-16 | 2015-04-03 | Merck Sharp & Dohme | |
| FR2979242A1 (fr) | 2011-08-29 | 2013-03-01 | Sanofi Sa | Comprime contre l'usage abusif, a base de paracetamol et d'oxycodone |
| CN103998025A (zh) | 2011-10-06 | 2014-08-20 | 格吕伦塔尔有限公司 | 包含阿片样物质激动剂和阿片样物质拮抗剂的防篡改口服药物剂型 |
| JP6085307B2 (ja) | 2011-11-17 | 2017-02-22 | グリュネンタール・ゲゼルシャフト・ミト・ベシュレンクテル・ハフツング | 薬理学的に活性な成分、オピオイドアンタゴニストおよび/または嫌忌剤(aversiveagent)、ポリアルキレンオキシドおよび陰イオン性ポリマーを含むタンパーレジスタント経口医薬剤形 |
| TW201336529A (zh) | 2011-12-09 | 2013-09-16 | Purdue Pharma Lp | 包含聚(ε-己內酯)和聚氧化乙烯之藥物劑量型 |
| JP2013155124A (ja) | 2012-01-30 | 2013-08-15 | Moriroku Chemicals Co Ltd | 医薬品の原末及びその製造方法 |
| WO2013127830A1 (en) | 2012-02-28 | 2013-09-06 | Grünenthal GmbH | Tamper-resistant pharmaceutical dosage form comprising nonionic surfactant |
| MX356421B (es) | 2012-02-28 | 2018-05-29 | Gruenenthal Gmbh | Forma de dosificacion resistente a la manipulacion indebida que comprende un compuesto farmacologicamente activo y un polimero anionico. |
| SG11201405011PA (en) | 2012-03-02 | 2014-09-26 | Rhodes Pharmaceuticals Lp | Tamper resistant immediate release formulations |
| CN104394851B (zh) | 2012-04-18 | 2017-12-01 | 格吕伦塔尔有限公司 | 抗篡改和抗剂量‑倾泻药物剂型 |
| MX2014011815A (es) | 2012-04-18 | 2014-12-05 | Mallinckrodt Llc | Composiciones farmaceuticas de liberacion inmediata con propiedades disuasivas de abuso. |
| WO2013167735A1 (en) | 2012-05-11 | 2013-11-14 | Grünenthal GmbH | Thermoformed, tamper-resistant pharmaceutical dosage form containing zinc |
| US10064945B2 (en) | 2012-05-11 | 2018-09-04 | Gruenenthal Gmbh | Thermoformed, tamper-resistant pharmaceutical dosage form containing zinc |
| CA2880163A1 (en) | 2012-08-01 | 2014-02-06 | Acura Pharmaceuticals, Inc. | Stabilization of one-pot methamphetamine synthesis systems |
| JP6150896B2 (ja) | 2012-08-27 | 2017-06-21 | エボニック レーム ゲゼルシャフト ミット ベシュレンクテル ハフツングEvonik Roehm GmbH | エタノールの影響に対して耐性を有する胃液抵抗性の医薬組成物又は栄養補助組成物 |
| JP6161701B2 (ja) | 2012-08-27 | 2017-07-12 | エボニック レーム ゲゼルシャフト ミット ベシュレンクテル ハフツングEvonik Roehm GmbH | 徐放特性及びエタノールの影響に対する耐性を有する医薬又は栄養補助組成物 |
| US9463165B2 (en) | 2012-09-05 | 2016-10-11 | Teika Pharmaceutical Co., Ltd. | Granular material for orally fast disintegrating tablets |
| EP2906202A4 (en) | 2012-10-15 | 2016-04-27 | Isa Odidi | DRUG FORMULATIONS FOR ORAL ADMINISTRATION |
| US10420729B2 (en) | 2013-03-15 | 2019-09-24 | R.P. Scherer Technologies, Llc | Abuse resistant capsule |
| EP2968169A1 (en) | 2013-03-15 | 2016-01-20 | Mallinckrodt LLC | Compositions comprising an opioid and an additional active pharmaceutical ingredient for rapid onset and extended duration of analgesia that may be administered without regard to food |
| US9517208B2 (en) | 2013-03-15 | 2016-12-13 | Purdue Pharma L.P. | Abuse-deterrent dosage forms |
| CA2907950A1 (en) | 2013-05-29 | 2014-12-04 | Grunenthal Gmbh | Tamper-resistant dosage form containing one or more particles |
| AR096438A1 (es) | 2013-05-29 | 2015-12-30 | Gruenenthal Gmbh | Forma de dosificación resistente al uso indebido con perfil de liberación bimodal, proceso |
| CA2817728A1 (en) | 2013-05-31 | 2014-11-30 | Pharmascience Inc. | Abuse deterrent immediate release formulation |
| BR112016000194A8 (pt) | 2013-07-12 | 2019-12-31 | Gruenenthal Gmbh | forma de dosagem resistente à violação contendo o polímero de acetato de etileno-vinila |
| US10195153B2 (en) | 2013-08-12 | 2019-02-05 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded immediate release abuse deterrent pill |
| US9770514B2 (en) | 2013-09-03 | 2017-09-26 | ExxPharma Therapeutics LLC | Tamper-resistant pharmaceutical dosage forms |
| WO2015048597A1 (en) | 2013-09-30 | 2015-04-02 | Daya Drug Discoveries, Inc. | Prevention of illicit methamphetamine manufacture from pseudoephedrine using food flavor excipients |
| US20150118300A1 (en) | 2013-10-31 | 2015-04-30 | Cima Labs Inc. | Immediate Release Abuse-Deterrent Granulated Dosage Forms |
| WO2015103379A1 (en) | 2013-12-31 | 2015-07-09 | Kashiv Pharma, Llc | Abuse-resistant drug formulations |
| WO2015120201A1 (en) | 2014-02-05 | 2015-08-13 | Kashiv Pharma, Llc | Abuse-resistant drug formulations with built-in overdose protection |
| US20160089439A1 (en) | 2014-09-28 | 2016-03-31 | Satara Pharmaceuticals, LLC | Prevention of Illicit Manufacutre of Methamphetamine from Pseudoephedrine Using Food Flavor Excipients |
| US20170112766A1 (en) | 2015-04-24 | 2017-04-27 | Grünenthal GmbH | Tamper-resistant dosage form with immediate release and resistance against solvent extraction |
| EP3285745A1 (en) | 2015-04-24 | 2018-02-28 | Grünenthal GmbH | Tamper-resistant dosage form with immediate release and resistance against solvent extraction |
| US20170296476A1 (en) | 2016-04-15 | 2017-10-19 | Grünenthal GmbH | Modified release abuse deterrent dosage forms |
-
2012
- 2012-07-27 CA CA2839123A patent/CA2839123A1/en not_active Abandoned
- 2012-07-27 PE PE2013002883A patent/PE20141638A1/es not_active Application Discontinuation
- 2012-07-27 WO PCT/EP2012/003188 patent/WO2013017234A1/en active Application Filing
- 2012-07-27 EA EA201400172A patent/EA201400172A1/ru unknown
- 2012-07-27 AR ARP120102742A patent/AR087360A1/es unknown
- 2012-07-27 LT LTEP12751251.5T patent/LT2736495T/lt unknown
- 2012-07-27 ES ES12751251.5T patent/ES2655900T3/es active Active
- 2012-07-27 KR KR1020147002756A patent/KR20140053159A/ko not_active Application Discontinuation
- 2012-07-27 DK DK12751251.5T patent/DK2736495T3/da active
- 2012-07-27 HU HUE12751251A patent/HUE034711T2/en unknown
- 2012-07-27 BR BR112014002022A patent/BR112014002022A2/pt active Search and Examination
- 2012-07-27 CN CN201280038092.1A patent/CN103841964A/zh active Pending
- 2012-07-27 JP JP2014521993A patent/JP2014524925A/ja active Pending
- 2012-07-27 AU AU2012289764A patent/AU2012289764B2/en not_active Ceased
- 2012-07-27 PT PT127512515T patent/PT2736495T/pt unknown
- 2012-07-27 SI SI201231108T patent/SI2736495T1/sl unknown
- 2012-07-27 MX MX2014001104A patent/MX348491B/es active IP Right Grant
- 2012-07-27 PL PL12751251T patent/PL2736495T3/pl unknown
- 2012-07-27 EP EP12751251.5A patent/EP2736495B1/en active Active
- 2012-07-27 RS RS20171149A patent/RS56528B1/sr unknown
- 2012-07-27 NO NO12751251A patent/NO2736495T3/no unknown
- 2012-07-27 US US13/559,644 patent/US10201502B2/en not_active Expired - Fee Related
-
2013
- 2013-11-20 IL IL229517A patent/IL229517A0/en unknown
- 2013-11-22 CL CL2013003363A patent/CL2013003363A1/es unknown
- 2013-12-04 CO CO13284675A patent/CO6811869A2/es not_active Application Discontinuation
- 2013-12-10 ZA ZA2013/09297A patent/ZA201309297B/en unknown
-
2014
- 2014-01-23 EC ECSP14013168 patent/ECSP14013168A/es unknown
- 2014-11-19 HK HK14111670.8A patent/HK1198132A1/zh not_active IP Right Cessation
-
2017
- 2017-10-06 HR HRP20171506TT patent/HRP20171506T1/hr unknown
- 2017-10-26 CY CY20171101115T patent/CY1119500T1/el unknown
-
2018
- 2018-12-19 US US16/225,418 patent/US10864164B2/en active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090004267A1 (en) * | 2007-03-07 | 2009-01-01 | Gruenenthal Gmbh | Dosage Form with Impeded Abuse |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107889459A (zh) * | 2015-04-24 | 2018-04-06 | 格吕伦塔尔有限公司 | 具有立即释放和对溶剂萃取的抗性的抗篡改剂型 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10864164B2 (en) | Tamper-resistant tablet providing immediate drug release | |
| US10695297B2 (en) | Tamper-resistant tablet providing immediate drug release | |
| US20200276188A1 (en) | Tamper-resistant dosage form with immediate release and resistance against solvent extraction | |
| AU2012289764A1 (en) | Tamper-resistant tablet providing immediate drug release | |
| US20200009055A1 (en) | Tamper-resistant dosage form with immediate release and resistance against solvent extraction | |
| CN104394851A (zh) | 抗篡改和抗剂量-倾泻药物剂型 | |
| CN103179954A (zh) | 包含阴离子聚合物的抗破碎剂型 | |
| US20200268748A1 (en) | Tamper-resistant dosage form with immediate release and resistance against solvent extraction | |
| US20200338069A1 (en) | Tamper-resistant dosage form with immediate release and resistance against solvent extraction | |
| EP3698776A1 (en) | Tamper-resistant dosage form with immediate release and resistance against solvent extraction | |
| NZ618168B2 (en) | Tamper-resistant tablet providing immediate drug release | |
| NZ619016B2 (en) | Tamper-resistant tablet providing immediate drug release |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20140604 |
|
| RJ01 | Rejection of invention patent application after publication |