BRPI0510177B1 - composto, composição farmacêutica e uso do mesmo - Google Patents
composto, composição farmacêutica e uso do mesmo Download PDFInfo
- Publication number
- BRPI0510177B1 BRPI0510177B1 BRPI0510177A BRPI0510177A BRPI0510177B1 BR PI0510177 B1 BRPI0510177 B1 BR PI0510177B1 BR PI0510177 A BRPI0510177 A BR PI0510177A BR PI0510177 A BRPI0510177 A BR PI0510177A BR PI0510177 B1 BRPI0510177 B1 BR PI0510177B1
- Authority
- BR
- Brazil
- Prior art keywords
- mmol
- fluorophenyl
- concentrated
- solution
- nmr
- Prior art date
Links
- 0 C*Cc(cc(cc1)[N+]([O-])=O)c1OC(C=C(N)N)=NC=C Chemical compound C*Cc(cc(cc1)[N+]([O-])=O)c1OC(C=C(N)N)=NC=C 0.000 description 15
- MXJQJURZHQZLNN-UHFFFAOYSA-N Nc(cc1)cc(F)c1O Chemical compound Nc(cc1)cc(F)c1O MXJQJURZHQZLNN-UHFFFAOYSA-N 0.000 description 3
- ORLGLBZRQYOWNA-UHFFFAOYSA-N Cc1cc(N)ncc1 Chemical compound Cc1cc(N)ncc1 ORLGLBZRQYOWNA-UHFFFAOYSA-N 0.000 description 2
- DKHASFYGUUDZOS-UHFFFAOYSA-N CC(C)(C)C(Nc(ccc(O)c1)c1F)=O Chemical compound CC(C)(C)C(Nc(ccc(O)c1)c1F)=O DKHASFYGUUDZOS-UHFFFAOYSA-N 0.000 description 1
- RTYDQRGWCSTEIQ-UHFFFAOYSA-N CC(N1)=CC=C(C(Nc(cc2)cc(F)c2O)O)C1=O Chemical compound CC(N1)=CC=C(C(Nc(cc2)cc(F)c2O)O)C1=O RTYDQRGWCSTEIQ-UHFFFAOYSA-N 0.000 description 1
- NNMNRLMVVILNGK-UHFFFAOYSA-N CC(Nc(cc1)cc(F)c1Oc1cc(Nc(cc2)ccc2F)ncc1)=O Chemical compound CC(Nc(cc1)cc(F)c1Oc1cc(Nc(cc2)ccc2F)ncc1)=O NNMNRLMVVILNGK-UHFFFAOYSA-N 0.000 description 1
- ISIBRYHFSVXOHS-CLTKARDFSA-N CC/C=N\CC(O)Cl Chemical compound CC/C=N\CC(O)Cl ISIBRYHFSVXOHS-CLTKARDFSA-N 0.000 description 1
- MLYZRAAPMRNQMB-UHFFFAOYSA-N CC1NC(N)=CC(C)=C1 Chemical compound CC1NC(N)=CC(C)=C1 MLYZRAAPMRNQMB-UHFFFAOYSA-N 0.000 description 1
- MCYCJGGYLUCZSO-UHFFFAOYSA-N CCc(cc(C)cc1)c1Oc(cccc1)c1-c(cc1)ccc1C(NI)=O Chemical compound CCc(cc(C)cc1)c1Oc(cccc1)c1-c(cc1)ccc1C(NI)=O MCYCJGGYLUCZSO-UHFFFAOYSA-N 0.000 description 1
- UZJIRAOXRCPKHP-UHFFFAOYSA-N CCc1c(N(C(C)(C)O2)C2=O)nccc1C Chemical compound CCc1c(N(C(C)(C)O2)C2=O)nccc1C UZJIRAOXRCPKHP-UHFFFAOYSA-N 0.000 description 1
- YVXDLRYGISTVMO-UHFFFAOYSA-N COC(C(C1O)=CC=CN1c1ccccc1)=O Chemical compound COC(C(C1O)=CC=CN1c1ccccc1)=O YVXDLRYGISTVMO-UHFFFAOYSA-N 0.000 description 1
- XMYPFXMZSPVINW-UHFFFAOYSA-N COC(C1=CC=CN(c2ccccc2)C1=O)=O Chemical compound COC(C1=CC=CN(c2ccccc2)C1=O)=O XMYPFXMZSPVINW-UHFFFAOYSA-N 0.000 description 1
- JLDQDOJXDWJSQR-UHFFFAOYSA-N COC(c(cc1)ccc1-c1cnccc1Oc(ccc(N=O)c1)c1F)=O Chemical compound COC(c(cc1)ccc1-c1cnccc1Oc(ccc(N=O)c1)c1F)=O JLDQDOJXDWJSQR-UHFFFAOYSA-N 0.000 description 1
- YYUSFELHBJJWPU-UHFFFAOYSA-N Cc(c(C(F)(F)F)c1)ccc1O Chemical compound Cc(c(C(F)(F)F)c1)ccc1O YYUSFELHBJJWPU-UHFFFAOYSA-N 0.000 description 1
- FDQUEAPRNNBTRX-UHFFFAOYSA-N Cc(c(F)c(cc1)O)c1N Chemical compound Cc(c(F)c(cc1)O)c1N FDQUEAPRNNBTRX-UHFFFAOYSA-N 0.000 description 1
- LSBIUXKNVUBKRI-UHFFFAOYSA-N Cc1cc(C)ncn1 Chemical compound Cc1cc(C)ncn1 LSBIUXKNVUBKRI-UHFFFAOYSA-N 0.000 description 1
- OMMAWIUZUACEOB-UHFFFAOYSA-N Cc1ncnc(C2=CC2)c1 Chemical compound Cc1ncnc(C2=CC2)c1 OMMAWIUZUACEOB-UHFFFAOYSA-N 0.000 description 1
- IHRZTUDVYFWMJX-UHFFFAOYSA-N Cc1ncnc(NCc(cc2)ccc2OC)c1 Chemical compound Cc1ncnc(NCc(cc2)ccc2OC)c1 IHRZTUDVYFWMJX-UHFFFAOYSA-N 0.000 description 1
- DVMBIRDILLVLIL-UHFFFAOYSA-N NC(Cc(cc1)ccc1-c1c(N)nccc1Oc(ccc(N)c1)c1F)=O Chemical compound NC(Cc(cc1)ccc1-c1c(N)nccc1Oc(ccc(N)c1)c1F)=O DVMBIRDILLVLIL-UHFFFAOYSA-N 0.000 description 1
- NGBYYNAXOPCQCG-UHFFFAOYSA-N Nc(cc1)cc(F)c1Oc(ccnc1N)c1C#CCO Chemical compound Nc(cc1)cc(F)c1Oc(ccnc1N)c1C#CCO NGBYYNAXOPCQCG-UHFFFAOYSA-N 0.000 description 1
- RXGKXMHZCFAIEX-UHFFFAOYSA-N Nc(cc1)cc(F)c1Oc1ccncc1C#CCNC(CN1CCCC1)=O Chemical compound Nc(cc1)cc(F)c1Oc1ccncc1C#CCNC(CN1CCCC1)=O RXGKXMHZCFAIEX-UHFFFAOYSA-N 0.000 description 1
- PLIKAWJENQZMHA-UHFFFAOYSA-N Nc(cc1)ccc1O Chemical compound Nc(cc1)ccc1O PLIKAWJENQZMHA-UHFFFAOYSA-N 0.000 description 1
- AZANUDOYLBHLBZ-UHFFFAOYSA-N Nc1cc(Oc(cc2)cc(C(F)(F)F)c2NC(NC(Cc(cc2)ccc2F)=O)=O)ccn1 Chemical compound Nc1cc(Oc(cc2)cc(C(F)(F)F)c2NC(NC(Cc(cc2)ccc2F)=O)=O)ccn1 AZANUDOYLBHLBZ-UHFFFAOYSA-N 0.000 description 1
- VYHHGEHFXOLMCB-UHFFFAOYSA-N Nc1cc(Oc(ccc(NC(NC(Cc2cc(O)ccc2)=O)=O)c2)c2F)ccn1 Chemical compound Nc1cc(Oc(ccc(NC(NC(Cc2cc(O)ccc2)=O)=O)c2)c2F)ccn1 VYHHGEHFXOLMCB-UHFFFAOYSA-N 0.000 description 1
- BLKOFLKFWIYXMG-UHFFFAOYSA-N O=C(C1=CC=CN(Cc2ccccc2)C1=O)Nc(cc1)cc(F)c1Oc1ccnc(NI)c1 Chemical compound O=C(C1=CC=CN(Cc2ccccc2)C1=O)Nc(cc1)cc(F)c1Oc1ccnc(NI)c1 BLKOFLKFWIYXMG-UHFFFAOYSA-N 0.000 description 1
- PNJKMGLQFQUVKJ-UHFFFAOYSA-N O=C(CNCC#Cc1cnccc1Oc(ccc(NC(NC(Cc(cc1)ccc1F)=O)=O)c1)c1F)CN1CCCC1 Chemical compound O=C(CNCC#Cc1cnccc1Oc(ccc(NC(NC(Cc(cc1)ccc1F)=O)=O)c1)c1F)CN1CCCC1 PNJKMGLQFQUVKJ-UHFFFAOYSA-N 0.000 description 1
- UFPOSTQMFOYHJI-UHFFFAOYSA-N O=Cc1ccnc(Cl)c1 Chemical compound O=Cc1ccnc(Cl)c1 UFPOSTQMFOYHJI-UHFFFAOYSA-N 0.000 description 1
- WVROKKAXTJAKJG-UHFFFAOYSA-O OC(c1cccc(-c(cc2)ccc2F)[n+]1O)=O Chemical compound OC(c1cccc(-c(cc2)ccc2F)[n+]1O)=O WVROKKAXTJAKJG-UHFFFAOYSA-O 0.000 description 1
- NIZVDIMAAVITCT-UHFFFAOYSA-N Oc(ccc(NC(C1=CC=CN(C(CC2)=CC=C2F)C1=O)=O)c1)c1F Chemical compound Oc(ccc(NC(C1=CC=CN(C(CC2)=CC=C2F)C1=O)=O)c1)c1F NIZVDIMAAVITCT-UHFFFAOYSA-N 0.000 description 1
- ORPHLVJBJOCHBR-UHFFFAOYSA-N [O-][N+](c(cc1)cc(F)c1O)=O Chemical compound [O-][N+](c(cc1)cc(F)c1O)=O ORPHLVJBJOCHBR-UHFFFAOYSA-N 0.000 description 1
- JNEVSBRZTCYCLT-UHFFFAOYSA-N [O-][N+](c(cc1)cc(F)c1Oc(ccnc1)c1-c1cnccc1)=O Chemical compound [O-][N+](c(cc1)cc(F)c1Oc(ccnc1)c1-c1cnccc1)=O JNEVSBRZTCYCLT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
- C07D213/82—Amides; Imides in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/4155—1,2-Diazoles non condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/63—One oxygen atom
- C07D213/68—One oxygen atom attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/73—Unsubstituted amino or imino radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/74—Amino or imino radicals substituted by hydrocarbon or substituted hydrocarbon radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/75—Amino or imino radicals, acylated by carboxylic or carbonic acids, or by sulfur or nitrogen analogues thereof, e.g. carbamates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/89—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members with hetero atoms directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/34—One oxygen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/42—One nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/47—One nitrogen atom and one oxygen or sulfur atom, e.g. cytosine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oncology (AREA)
- Virology (AREA)
- Neurology (AREA)
- Communicable Diseases (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Diabetes (AREA)
- Epidemiology (AREA)
- Physical Education & Sports Medicine (AREA)
- Immunology (AREA)
- Hematology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Rheumatology (AREA)
- Heart & Thoracic Surgery (AREA)
- Urology & Nephrology (AREA)
- Cardiology (AREA)
- Molecular Biology (AREA)
- Pulmonology (AREA)
- Dermatology (AREA)
- AIDS & HIV (AREA)
- Hospice & Palliative Care (AREA)
- Biotechnology (AREA)
- Psychiatry (AREA)
- Psychology (AREA)
- Ophthalmology & Optometry (AREA)
Abstract
heterociclos monocíclicos como inibidores de cinase. a presente invenção é direcionada a compostos tendo a fórmula e métodos para empregá-los para o tratamento de câncer.
Description
CAMPO DA INVENÇÃO
Esta invenção refere-se a compostos que inibem a atividade de 10 proteína tirosina cinase de receptores de fator de crescimento, tal como cMet, desse modo tornando-os úteis como agentes anticâncer. As composições farmacêuticas que compreendem estes compostos são também úteis no tratamento de doenças, exceto câncer, que são associados com trilhas de transdução de sinal operando através do fator de crescimento e recepto15 res antiangiogênese tal como c-Met.
RESUMO DA INVENÇÃO
A presente invenção é direcionada a compostos tendo fórmulas I e II como descrito abaixo, que são úteis no tratamento de câncer.

ou um enantiômero, diastereômero, hidrato, solvato ou sal farmacêutica20 mente aceitável deste, em que:
R1 é H, alquila, alquila substituída, cicloalquila, cicloalquila substituída, arilalquila, arilalquila substituída, aríla, arila substituída, alquenila, alquenila substituída, alquinila, alquinila substituída, heteroarila, heteroarila substituída, heterociclo, heterociclo substituído, heteroarilalquila, heteroari25 lalquila substituída, heterocicloalquila, ou heterocicloalquíla substituída;
cada R2 é independentemente H, halogênio, ciano, NO2:, OR5,
NR6R7, alquila, alquila substituída,cicloalquila, cicloalquila substituída, arila, aríla substituída, heteroarila, heteroarila substituída, heterociclo, heterociclo substituído, arilquila, arilalquila substituída, heterocicloalquila, ou heterocicloalquila substituída;
B é O, NRS, NR8CH2, S, SO, SO2, ou CR9R10;
V for NR11 ou -(ÇR37R38)p- contate que quando VR11 é N, R1 é uma alquila ou cicloalquila;
W e X são cada independentemente C ou N;
Y é selecionado de O, S, e NR12;
Z é -CR13R14-, ou -(CR13R14),NR1S-;
é 0 a 2;
n é 0 a 4 se W e X ambos forem C, 0 a 3 se um de X ou W for
N, eOa 2 se Xe W ambos forem N;
p é 1 a 4;
R3, R5, R·, R7, R8, R11 e R15 são independentemente selecionado de H, alquila, alquila substituída, alquenila, alqueniia substituída, alquinila, alquinila substituída, cicloalquila, cicloalquila substituída, aríla, aríla substituída, heteroarila, heteroarila substituída, heterociclo, heterociclo substituído;
R4 é selecionado de aríla, aríla substituída, heteroarila, heteroarila substituída, heterocicloalquila, e heterocicloalquila substituída, contanto que (a) se R4 for fenila (i) R* não é substituído por ambos hidróxi e amido; e (ii) R4 não é substituído por -NRSO2R- em que R é alquila ou ante;
(b) se R4 é piridila, R4 não é substituído por ambos hidróxi e metóxi; e (c) se R4 é pirimidinila, ela não é substituída por =O;
R® e Rw são independentemente selecionados de H, halogênio, alquila, alquila substituída, cicloalquila, cicloalquila substituída, aríla, aríla substituída, heteroarila, heteroarila substituída, heterocicloalquila, ou heterocicloalquila substituída;
R12 é selecionado de H, alquila, alquila substituída, alquenila, alquenila substituída, alquimia, alquinila substituída, GN, NOaou SOjNHa;
R13 e R14 são independentemente selecionado de H, halogênio, alquila, alquila substituída,alquenila, alquenila substituída, alquinila, alquinila substituída, cicloalquila, cicloalquila substituída, arila, arila substituída, hete5 roarila, heteroarila substituída, heterocicloalquila, heterocicloalquila substituída ou tomados juntos para formarem um anel carbocíclico ou heterocíclico de 3 a 8 átomos;
A é selecionado de um dos seguintes:




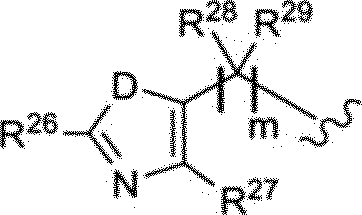
em que
DéSouO;
' m é 0 to 6;
_ Rw, R17, R18, R’®, R20, R21, R22, R23, R24, R25, R26 e R27 são inde' pendentemente selecionados de H, halogênio, NR^R31, OR32, COaR®, CONR34R35, SO2R36, alquila, alquila substituída, cicloalquila, cicloalquila substituída, alquenila, alquenila substituída, alquinila, alquinila substituída, -CN, arila, arila substituída, heteroarila, heteroarila substituída, heterocicloalquila, ou heterocicloalquila substituída;
p2s e |^29 são independentemente selecionados de H, alquila, alquila substituída,cicloalquila, cicloalquila substituída, arila, arila substituída, ou tomados juntos para formarem um anel carbocíclico ou heterocíclico de 3 a 8 átomos;
R30, R31, R32, R33, R34, R35, e R36 são independentemente selecionados de H, alquila, alquila substituída,alquenila, alquenila substituída, alquinila, alquinila substituída, cicloalquila, cicloalquila substituída, alcoxicar25 bonila, arila, arila substituída, heteroarila, heteroarila substituída, heterociclo, heterociclo substituído, heterocicloalquila, ou heterocicloalquila substituída;
e
R37 e R38 são cada independentemente H, halogênio, ou alquila.
DESCRIÇÃO DA INVENÇÃO
A presente invenção fornece para compostos de Fórmulas I e II definidos acima, composições farmacêuticas empregando-se tais compostos, e métodos de uso de tais compostos no tratamento de câncer.
O termo alquila aqui sozinho ou como parte de outro grupo refere-se a um alcano monovalente (hidrocarboneto) radical derivado contendo de 1 a 12 átomos de carbono exceto se de outro modo definido. Grupos preferíveis de alquila são grupos de alquila inferior tendo de 1 a 6 átomos de carbono. Um grupo de alquila é opcionalmente um grupo de hidrocarboneto saturado linear, ramificado ou cíclico substituído. Grupos de alqui10 Ia podem ser substituídos em qualquer ponto de ligação disponível. Um grupo de alquila substituída por outro grupo de alquila é também referido grupo de alquila ramificada. Os grupos de alquila exemplares incluem metila, etila, propila, isopropila, n-butila, t-butila, isobutila, pentila, hexila, isoexila, heptila, 4,4-dimetilpentila, Ctila, 2,2,4-trimetilpentila, nonila, decila, undecíla, dodeci15 Ia, e similares. Substituintes exemplares incluem, porém não são limitados a um ou mais dos seguintes grupos: alquila, cicloalquila, heterocicloalquila, CN, arila, heteroarila, halo (tais como F, Cl, Br, I), haloalquila (tais cornos
CCI3 ou CF3), hidroxila, alcóxi, alquiltio, alquilamino, -COOH, -C0GR, C(O)R, -OCOR, amino, carbamoíla (-NHCOOR- ou -OCONHR-), uréia (20 NHCONHR-) ou tiol (-SH).
O termo alquenila aqui sozinho ou como parte de outro grupo refere-se a um radical de hidrocarboneto linear, ramificado ou cíclico contendo de 2 a 12 átomos de carbono e pelo menos uma ligação dupla de carbono a carbono. Grupos de alquenila podem também ser substituído em 25 qualquer ponto de ligação disponível. Substituintes exemplares de alquenila incluem aqueles listados acima para grupos de alquila.
O termo alquinila aqui sozinho ou como parte de outro grupo refere-se a um radical de hidrocarboneto linear, ramificado ou cíclico contendo de 2 a 12 átomos de carbono e pelo menos uma ligação tripla de car30 bono a carbono. Grupos de alquinila podem também ser substituídos em qualquer ponto disponível de ligação. Substituintes exemplares para grupos de alquinila incluem aqueles descritos acima para grupos de alquila.
Os números no subscrito depois do símbolo ”C” definem o número de átomos de carbono que um grupo particular pode conter. Por exemplo ttCt.g alquila significa uma cadeia de carbono saturada, linear ou ramificada tendo de um a seis átomos de carbono; exemplos incluem metila, etila, n-propila, isopropila, n-butila, sec-butila, isobutila, t-butila, n-pentila, sec-pentita, isopentila, e n-hexila. Dependendo do contexto, CM alquila*’ pode também referir-se a Cv6 alquileno que liga-se a dois grupos; exemplos incluem propano-1,3-diila, butano-1,4-cW/a, 2-metil-butano-1,4-diila, etc. C2^ alquenila significa uma cadeia de carbono linear ou ramificada tendo peto menos uma ligação dupla de carbono a carbono, e tendo de dois a seis átomos de carbono; exemplos incluem, propenila, isopropenila, butenila, isobutenila, pentenila, e hexenila. Dependendo do contexto, C^ alquenila pode também referir-se a CM alquenodiila que liga-se a dois grupos; Exemplos incluem etileno-1,2-diila(vinileno), 2-metil-2-buteno-1,4-diila, 2-hexeno-1,6diila, etc. C^ alquinila significa uma cadeia de carbono linear ou ramificada contendo pel© menos uma ligação tripla de carbono a carbono, e de dois a seis átomos de carbono; exemplos incluem etinila, propinila, butinila, e hexinila.
O termo cicloalquila aqui sozinho ou como parte de outro grupo é uma espécie de alquila contendo de 3 a 15 átomos de carbono, sem ligações duplas alternantes ou ressonantes entre os átomos de carbono. Ela pode conter de 1 a 4 anéis. Grupos exemplares incluem ciclopropila, ciclobutila, ciclopentila, cicloexila, adamantila, etc. Grupos de cicloalquila podem ser substituídos em qualquer ponto disponível de ligação. Substitutes exemplares incluem um ou mais dos seguintes grupos: halogênio, tais como F, Br, ou Cl, hidroxila, alquila, alcóxi, amino, nitro, ciano, tiol, alquiltio, e qualquer dos substituintes descritos acima para grupos de alquila.
Os termos alcóxi ou alquiltio” aqui sozinhos ou como parte de outro grupo denotam um grupo de alquila como descrito acima ligado através de uma ligação de oxigênio (-O-) ou uma ligação de enxofre (-S-), respectivamente.
Θ termo alquiloxicarbonila aqui sozinho ou como parte de outro grupo denota um grupo de alcóxi ligado através de um grupo de carbonila. Um radical de alcoxicarbonila é representado pela Fórmula: -C(O)OR, onde o grupo R é um grupo de alquila ramificada ou linear, cicloalquila, arila ou heteroarila.
O termo alquilcarbonila aqui sozinho ou como parte de outro grupo refere-se a um grupo de alquila ligado através de um grupo de carbonila.
O termo alquilcarbonilóxi aqui sozinho ou como parte de outro grupo denota um grupo de alquicarboníla ligado através de uma ligação de 10 oxigênio.
O termo arila aqui sozinho ou como parte de outro grupo refere-se a anéis aromáticos monocíclicos ou bicíclicos, por exemplo, fenila, fenila substituída e similar, bem como grupos que são fundidos, por exemplo, naftila, fenantrenila e similares. Um grupo de arila, desse modo contém pelo 15 menos um anel tendo pelo menos 6 átomos, com até cinco dos tais anéis estando presentes, contendo até 22 átomos neles, com ligações duplas alternantes (ressonantes) entre átomos de carbono adjacentes ou heteroátomos adequados. Grupos de arila podem opcionalmente ser substituído por um ou mais grupos incluindo, porém não limitado a halogênio, alquila, alcóxi, hidróxi, carbóxi, carbamoíla, alquiloxicarbonila, nitro, alquenilóxi, trifluorometila, amino, cicloalquila, arila, heteroarila, ciano, alquila S(O)m (m = 0, 1, 2), ou tiol.
Os termos arilalquila ou aralquila aqui sozinhos ou como partes de outro grupo denotam um grupo de arila como descrito acima ligado 25 através um grupo de alquila, como descrito acima. exemplo de um grupo de aralquila é um grupo de benzila.
O termo “amino aqui sozinho ou como parte de outro grupo refere-se a -NH2, Um amíno pode opcionalmente ser substituído por um ou dois substituintes, que podem ser iguais ou diferentes, tais como alquila, arila, arilalquila, alquenila, alquinila, heteroarila, heteroarilalquila, cicloeteroalquila, cicloeteroalquilalquila, cicloalquila, ctcloalquilalquila, haloalquila, hidroxialquila, alcoxialquila, tioalquila, carbonila ou carboxila. Estes substituin tes podem ser também substituídos com um ácido carboxílico, qualquer dos substituintes alquila ou ante mencionados aqui. Em algumas modalidades, os grupos de amino são substituídos com carboxila ou carbonila para formarem derivados de /V-acila ou A/-carbamoíla.
O termo heteroarila aqui sozinho ou como parte de outro grupo refere-se um grupo monocíclico de 5 a 6 membros aromáticos substituído ou não-substituído, grupos bicíclicos de 9 a 10 membros, e grupos tricíclicos de 11 a 14 membros que têm peto menos um heteroátomo (O, S ou N) em pelo menos um dos anéis. Cada anel do grupo de heteroarila contendo um heteroátomo pode conter um ou dois átomos de oxigênio ou enxofre e/ou de um a quatro átomos de nitrogênio, contato que o número total de heteroátomos em cada anel seja quatro ou menos e cada anel tenha pelo menos um átomo de carbono. Os anéis fundidos completando os grupos bicíclicos e tricíclicos podem conter apenas um átomo de carbono e podem ser saturados, parcialmente saturados, ou não saturados. Os átomos de nitrogênio e enxofre podem opcionalmente ser oxidados e os átomos de nitrogênio podem opcionalmente ser quarternizados. Grupos de heteroarila que são bicíclicos ou tricíclicos devem incluir pelo menos um anel totalmente aromático porém o outro anel ou anéis fundidos podem ser aromático ou não aromático. O grupo de heteroarila pode ser ligado em qualquer átomo de carbono ou nitrogênio disponível de qualquer anel. O sistema de anel de pode conter zero, um, dois ou três substituintes selecionados do grupo consistindo em halo, alquila, alquila substituída,alquenila, alquinila, arila, nitro, ciano, hidróxi, alcóxi, tioalquila, =O, -CO2H, -C(=O)H, -CO2-alquila, -C(=O)alquila, fenila, benzila, feniletila, fenilóxi, feniltio, cicloalquifa, cicloalquila substituída, heterociclo, heteroarila, -NR’R* -G(=Q)NRW, -CO2NR’R· -C^O)NR’R, -NRGO2R”, -NR’C(=O)R, -SO2NR’R, e -NR’SO2R”, onde cada de R’ e R é selecionado de hidrogênio, alquila, alquila substituída, e cicloalquila, ou R’ e R juntamente formando um anel de heterociclo ou heteroarila.
Grupos de heteroarila monocíclicos exemplares incluem pirroíila, pirazolila, pirazolinila, imidazolila, oxazolila, diazolila, isoxazolila, tiazolila, tiadiazolila, isotiazolila, furanila, tienila, oxadiazolila, piridíla, pirazinila, pirimi dinila, piridazinila, triazinila e similares.
Grupos de heteroarila bicíclicos exemplares incluem indolila, benzotiazolila, benzodioxolila, benzoxaxolila, benzotienila, quinolinila, tetraidroisoquinolinila, isoquinolinila, benzimidazolila, benzopiranila, indolizinila, benzofuranila, cromonila, cumarinila, benzopiranila, cinolinila, quinoxalinila, indazolila, pirrolopiridila, furopiridinila, diidroisoindolila, tetraidroquinolinila e similares.
Grupos de heteroarila tricíclicos exemplares incluem carbazolila, benzidolila, fenantrolinila, acridinila, fenantridinila, xantenila e similares.
O termo anel heterociclico aqui sozinho ou como parte de outro grupo refere-se a um sistema de anel monocíclico saturado ou parcialmente não saturado estável contendo 5 a 7 membros de anel de átomo de carbono e outros átomos selecionados de nitrogênio, enxofre e/ou oxigênio. Preferivelmente, um anel heterociclico é um anel monocíclico de 5 a 6 membros e contém um, dois, ou três heteroátomos selecionados de nitrogênio, oxigênio e/ou enxofre. O anel heterociclico pode ser opcionalmente substituído que significa que o anel heterociclico pode ser substituído em uma ou mais posições de anéis substituíveis por um mais grupos independentemente selecionados de alquila (preferivelmente alquila inferior), alcóxi (preferivelmente alcóxi inferior), nitro, monoalquilamino (preferivelmente um alquinoamino inferior), dialquilamino (preferivelmente a di[inferior]alquilamino), cíano, halo, haloalquila (preferivelmente trífluorometila), alcanoíla, aminocarbonila, monoalquilamínocarbonila, dialquilaminocarbonila, alquilamido (preferivelmente alquila inferior amido), alcoxialquila (preferivelmente um alcóxi inferior[inferior]alquila), alcoxicarbonila (preferivelmente uma alcoxicarbonila inferior), alquilcarbonilóxi (preferivelmente uma alquilcarbonilóxi inferior) e arila (preferivelmente fenila), a referida arila sendo opcionalmente substituído por grupos de halo, alquila inferior e alcóxi inferior. Exemplos de tais anéis heterocíclicos são ísoxazolila, imidazolinila, tiazolinila, imidazolidinila, pirrolila, pirrolinila, piranila, pirazinila, piperidila, morfolinila e triazolila. O anel heterociclico pode ser ligado à estrutura origem através de um átomo de carbono ou através qualquer heteroátomo de heterociclico que resulta na estrutura estável.
O termo heteroátomo” significa O, S ou N, selecionado em uma base independente. Deve-se observar que qualquer heteroátomo com valências não satisfeitas é assumido ter o átomo de hidrogênio para satisfazer as valências.
O termo halogênio ou halo refere-se a cloro, bromo, flúor ou iodo selecionado em urna base independente.
Quando um grupo funcional é denominado protegido”, isto significa que o grupo está em forma modificada para impedir reações colaterais indesejadas no sítio protegido. Grupos de proteção adequados para os compostos da presente invenção serão reconhecidos a partir do presente pedido, considerando o nível de experiência na técnica, e com referência a livros didáticos padrão, tal como Greene, T.W. e outro, Protective Groups in Organic Synthesis, Wiley, N.Y. (1091).
Como empregado aqui, o termo paciente abrange todas as espécies de mamíferos.
Exemplos adequados de sais dos compostos de acordo com a invenção com ácidos inorgânicos ou orgânicos são cloridrato, bromidrato, sulfato, metanossulfonato, maleato, fumarato, e fosfato. Sais que são inadequados para usos farmacêuticos, porém que podem ser empregados, por exemplo, para a isolação ou purificação de compostos livres I ou II, seus sais farmaceuticamente aceitáveis, são também inclusos.
Em geral, a presente invenção compreende compostos tendo
Fórmula I ou II:

ou um enantiômero, diastereômero, hidrato, solvato ou sal farmaceuticamente aceitável deste em que:
R1 é H, alquila, alquila substituída, cicloalquila, cicloalquila substituída, arilalquila, arilalquila substituída, arila, arila substituída, alquenila, alquenila substituída, alquinila, alquinila substituída, heteroarila, heteroarila substituída, heterociclo, heterociclo substituído, heteroarilalquila, heteroarilalquila substituída, heterocicloalquila, ou heterocicloalquila substituída;
cada R2 é independentemente H, halogênio, ciano, NO2, OR5,
NR6R7, alquila, alquila substituída,cicloalquila, cicloalquila substituída, arila, arila substituída, heteroarila, heteroarila substituída, heterociclo, heterociclo substituído, arilquila, arilalquila substituída, heterocicloalquila, ou heterocicloalquila substituída;
B é O, NR8, NR8GH2, S, SO, SO2, ouCRW0;
V for NR11 ou -(CR^R38)^ contato que quando V for N, R1 seja uma alquila ou cicloalquila;
W e X são cada independentemente C ou N;
Y é selecionado de O, S, e NR12;
Z é -GR13R14-, ou -(GR13R14),NR1S15 1 é/OaZ;
n é 0 a 4 se W e X ambos forem G, 0 a 3 se um de XouW for N, e0a2seXeW ambos forem N;
pé 1 a 4;
R®, R5, R®, R7, R8, R11 e R15 sâo independentemente seleciona20 dos de H, alquila, alquila substituída,alquenila, alquenila substituída, alquinila, alquinila substituída, cicloalquila, cicloalquila substituída, arila, arila substituída, heteroarila, heteroarila substituída, heterociclo, heterociclo substituído;
R4é selecionado de arila, arila substituída, heteroarila, heteroari25 Ia substituída, heterocicloalquila, e heterocicloalquila substituída, contanto que (a) se R4 for fenila (i) R4 não é substituído por ambos hidréxi e amido; e (ii) R4 não é substituído por -NRSO2R- em que R é alqui-
Ia ou arila;
(b) se R4 for piridlla ,R4 não é substituída por ambos hidróxi e metóxi; e (c) se R4forpirimidinila, ela não é substituído por =O;
R9 e R10 são independentemente selecionado de H, halogênio, alquila, alquila substituída,cicloalquila, cicloalquila substituída, arila, arila substituída, heteroarila, heteroarila substituída, heterocicloalquila, ou heterocicloalquila substituída;
R12 é selecionado de H, alquila, alquila substituída,alquenila, alquenila substituída, alquinila, alquinila substituída, CN, NO2ouSO2NH2;
R13 e R14 são independentemente selecionado de H, halogênio, alquila, alquila substituída,alquenila, alquenila substituída, alquinila, aíquinila substituída, cicloalquila, cicloalquila substituída, arila, arila substituída, heteroarila, heteroarila substituída, heterocicloalquila, heterocicloalquila substituída ou tomados juntos para formarem um anel carbocíclico ou heterociclico de 1 a 8 átomos;
A é selecionado de um dos seguintes:



em que,
D é S ou O;
m é0to6;
pendentemente selecionado de H, halogênio, NR3OR31, OR32, CO2R33, CONR34R35, SO2R36, alquila, alquila substituída, cicloalquila, cicloalquila substituída, alquenila, alquenila substituída, alquinila, alquinila substituí da, -CN, arila, arila substituída, heteroarila, heteroarila substituída, heteroci cloalquila, ou heterocicloalquila substituída;
R28 e R® são independentemente selecionado de H, alquila, alquila substituída,cicloalquila, cicloalquila substituída, arila, arila substituída, ou tomados juntos para formarem um anel carbocíclico ou heterociclico de 3 a 8 átomos;
R30, R31, R32, R33, R34, R35, e R36 são independentemente selecionado de H, alquila, alquila substituída,alquenila, alquenila substituída, alqui nila, alquinila substituída, cicloalquila, cicloalquila substituída, alcoxicarbonila, arila, arila substituída, heteroarila, heteroarila substituída, heterociclo, heterociclo substituído, heterocicloalquila, ou heterocicloalquila substituída; e
R37 e R38 são cada independentemente H, halogênio, ou alquila.
Em algumas modalidades da presente invenção, R1 é uma fenila substituída ou nâo-substituída, tais como fluorofenila, uma C-, a C4 alquila substituída ou nâo-substituída, tal como metila, ou uma C3 a C8 cicloalquila substituída ou nâo-substituída, tal como cicloexila ou ciclopentila.
Em algumas modalidades da presente invenção, R2 é C, a C4 alquila, halogênio, ou haloalquila.
Em algumas modalidades da presente invenção, R4 é opcionalmente fenila substituída, ou um nitrogênio de 5 a 6 membros contendo um grupo de heteroarila, tal como piridila, piridinona, pirazolila, ou pirrolidila.
De acordo com uma modalidade da presente invenção, B é O, NHCH2, CH2 ou CH(OH); Y é O ou S e Z é -CR13R14 ou -NR15 em que R13, R14, e R1S são cada H.
Em algumas modalidades da presente invenção, A é uma piridina ou pirimidina opcionalmente substituída, em que o substituinte é alquila, alqueníla, alquinila, halogênio, cicloalquila, heterocicloalquila, -NR38COR*, NR^CÍO^R40, -NR41R42, ou -C(O)NR43R44, em que R39 R40, R41, R42, R43, e R44 sâo independentemente H, alquila inferior, alquila substituída inferior, hidroxialquila, aminoalquila, cicloalquila, cicloalquila substituída, heterocicloalquila, heterocicloalquila substituída, cicloalquenila, cicloalquenila substituída, arila, arila substituída, heteroarila, heteroarila substituída, ou NR^R^formam uma heterocicloalquila.
De acordo com algumas modalidades da presente invenção, A é uma piridina substituída por -NR41R42, -NR^COR40, -CXOJNR^R44, halogênio, O, a C4 alquila, opcionalmente substituída por hidróxi, hidroxialquilamino, alquilamino, aminoalquilamino, ou heteroarilalquila; ou -C=G-R45, -CsC-R*, em que R45 e R* são alquila, hidroxialquila, aminoalquila, cicloalquila, heterocicloalquila, -C(O)R47, -NR^COR40, arila, ou heteroarila; ou a piridina é substituída por arila, tal como fenila, que pode ser também substituída por CONH2, metila, aminoetila, hidroxietila, -CONHCH .CH.NHCH,, ou CH2CONH2. a piridina pode também ser substituída por grupos de piridila ou piperidila.
De acordo com algumas modalidades da presente invenção, A é uma pirimidina opcionalmente substituída. Substituintes preferíveis incluem -NR41R42, ou -NR39CO2R40, em que R41 e Rzsâo preferivelmente H ou metila e r39 e R4o são preferivelmente, H ou alquila.
Em uma modalidade da presente invenção, compostos tendo a seguinte fórmula III:

em que,
R1 é opcionalmente fenila substituída ou alquila; Z é NH ou NCHc R‘ é F, Cl. CH, ou CF, R' é H: e Y é O ou S. Em algumas modalidades, R1 é C3 a C7 cicloalquila, fenila substituída ou não-substituída, ou (CHA-R50 em que n é 1 a 3, R50 é H, fenila substituída ou não-substituída, amino, amido, CN, -C(O)2H, ou -Ο(Ο)2ΟΗλ
Em algumas modalidades da presente invenção, compostos tendo a seguinte Fórmula IV:

em que R2 é halo ou H; R3 é H; R4 é opcionalmente fenila substituída, opcionalmente pirazol substituído, ou opcionalmente piridila, piridinona ou piridina-N-óxido substituído.
Em uma modalidade da presente invenção, compostos são da seguinte Fórmula V:

A
V em que R1 é opcionalmente alquila ou cicloalquila; A é opcionalmente piridina pirimidina substituída; e R2 é halo ou H; e R’3 e RM são ou H ou juntamente com o carbono ao qual eles são ligados formam uma cicloalquila, tal como ciclopropila.
A invenção também fornece métodos para tratar uma doença proliferativa, tal como câncer administrando-se a uma espécie de mamífero em necessidade de tratamento uma quantidade efetiva de um composto de fórmulas I ou II, como definido acima. Em outra modalidade, a invenção fornece um método para tratar uma doença proliferativa por meio de modulação de Met cinase administrando-se a uma espécie de mamífero em necessidade de tal tratamento uma quantidade efetiva de um composto de fórmulas 1 ou II, como definido acima, em combinação (simultaneamente ou sequencialmente) com peto menos um outro agente anticâncer. Em uma modalidade preferida, a doença proliferativa é câncer.
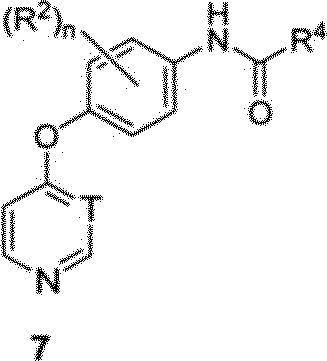
Certas compostos de Fórmulas I e II podem geralmente ser preparados de acordo com os seguintes esquemas 1-14, Os compostos são sintetizados facilmente empregando-se métodos sintéticos conhecidos por alguém versado na técnica. Solvatos (por exemplo, hidratas) dos compostos de Fórmulas I e II incluem-se também no escopo da presente invenção. Métodos de solvação são geralmente conhecidos na técnica. Consequentemente, os compostos da presente invenção podem ser na forma livre ou forma de hidrato, e podem ser obtidos por métodos exemplificados pelos seguintes esquemas abaixo.
Vias gerais para análogo de piridina e pirimidina descritos na invenção são ilustrados no Esquema 1, Uma pirimidina ou piridina apropriadamente substituída 1 pode ser tratada com fenóis funcionalizados 2, 4, e 8 na presença de uma base, tal como hidreto de sódio, hidróxido de sódio, ou carbonato de potássio, para fornecer os éteres desejados 3, 5, e 9, respectivamente. A remoção do grupo de proteção de acetamida do composto 3 com HC1 aquoso em metanol fornecendo a chave intermediária 5. Alternativamente, anilina 5 pode ser obtida do composto 9 por meio de redução do grupo de nitro com ou pó de zinco e cloreto de amônio ou catalisador Adams (óxido de platina (IV)) sob condições de hidrogenação. Análogos 6 e 7 podem então ser preparados por acilação de anilina 5 com, por exemplo, isocianatos, cloreto de ácidos ou por tratamento com um ácido carboxílico e um reagente de acoplamento, tal como: hexafluorofosfato de benzotriazol-110 iloxitris(trimetilamino)fosfônio (Reagente BOP), hexafluorofosfato de bromotripirrolidinofosfônio (PiBroP), tetrafluoroborato de O-(1H-benzotriazol-1-il)Λ/,Λ/,Λ/',/V-tetrametilurônio (TBTU). Formação da aciltiuréia de 6 (Y = S, Z =
NH) pode ser realizado tratando-se anilina 5 com um isotiocianato apropriadamente substituído
ESQUEMA 1


1N aq. HCI/MeOH
Rcfluxo

nh2 adlaçiooii formação de tiourcia
Zn, NHtCI
MeOHTHf oe
HjPtOa.MeOH


T = CR19ouN t - grupo de partida, tal como um halogênio ou KOj.
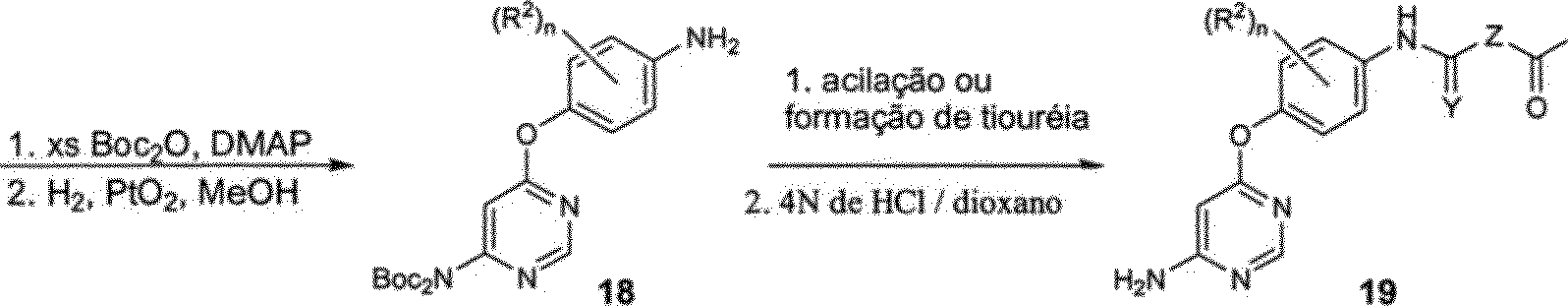
Os dois diferentes análogos de aminopirimidina regtoisoméricos e 19 podem ser preparados empregando-se as vias delineadas nos ESQUEMAS 2 e 3, Aminopirimidina protegida por PMB 11, deriva de 2,45 dicloropirimidina comercialmente disponível (10, Aldrich), pode ser convertido em éter 13 por meio de anilina 12 empregando-se a mesma química delineadas no ESQUEMA 1, A remoção do grupo de PMB de 13 pode ser realizada com ácido trifluoroacético e anisol para gerar o composto 14,
ESQUEMA 2

Cl

«
AMfiA pCH3OCeH4CHaNH2


PMB =p-CH9OCeH4CH2

Similarmente, aminopirimidina protegida por PMB 16, derivado de 4,6-dteloropirimidina comercialmente disponível (15, TCI America), pode ser convertido em éter 17 seguindo a etapa de desproteçâo de PMB (ES5 QUEMA 3). Bis-Boc (Pbutiloxicarbonila) proteção da amina de 17 com excesso de dicarbonato de di-terc-butila seguido por hidrogenização com catalisador Adams fornece anilina 18, Amina 19 pode ser obtida de composto 18 seguindo uma etapa de acilação ou formação de tiouréia e a remoção dos grupos de proteção Boc sob condições acídicas.
ESQUEMA3

ρ-ΟΗ3ΟΟβΗ4ΟΗ2ΝΗ2


NaH. DMF
IFAanwl

TCIAmerica PMB ^p-CHgOCeHtoHa
Derivados de aminopiridazina 26 podem ser preparados empregando-se a via sintética definida no ESQUEMA 4 que é baseado na química similar citada nas seguintes referências: Chung, H.-A. e outro, J. Heterocy18 die Ghem. 1999, 36, 905-910 e Bryant R. D. e outro, J. Heterocydic Ghem.
1995, 32, 1473-1476, a descrição dos quais estão aqui incorporados por referência. 4,5-dicloropiridazin-3(2H)-ona (20, Aldrich) pode ser protegido com, por exemplo um grupo de tetraidropirano (THP) para fornecer o inter5 mediário 21, Tratamento de composto 21 com um fenol apropriadamente substituído e uma base (isto é, hidreto de sódio) seguido por redução do nitro contendo intermediário sob condições de hidrogenação catalística pode fornecer anilina 22. Proteção do grupo de anilina 22 como carbamato de bisbenzila (Cbz) seguido por remoção do grupo THP sob condições acídicas podem fornecer o composto 23. Tratamento de composto 23 com ou anidrido trifluoroacético (TFAA), oxicloreto fosforoso ou brometo fosforoso na presença de uma base, tal como trietilamina ou diisopropiletilamina pode introduzir o grupo de partida necessário na posição 3 de composto 24, Deslocamento do grupo de partida X de composto 24 com uma amina apropriada15 mente substituída, seguido por remoção de grupos Cbz podem gerar intermediário 25, Anilina 25 pode ser convertida em análogo de 3-aminopiridazina desejada 26 empregando-se química previamente descrita nos ESQUEMAS 1-3,
ESQUEMA 4

AUrich THP s tetrahidropitano
1. xsPhCH2OC{O)CI b3n. dmap. ch2ci2
2. HCI Dioxano

X = OTf,CI, Br


Derivados de 2-aminopiridina podem ser preparados empregando-se as vias delineadas nos ESQUEMAS 5 e 6, Anilina 27, derivada química descrita no ESQUEMA 1 pode ser convertida em intermediário 28 em aquecimento com pó de Cobre e carbonato de potássio em benzilamina (ESQUEMA 5). A remoção de grupo de proteção de benzila de composto 28 sob condições de hidrogenaçâo catalística com paládio sobre carbono fornece aminopiridina 29, Os intermediários 28 ou 29 podem ser tratados com isotiocianatos 30, isocianatos 32, e ácidos carboxílicos 34 na presença de 10 um reagente de acoplamento para fornecer aciltiouréia 31, aciluréia 33, e amída 35, respectivamente.
ESQUEMA 5


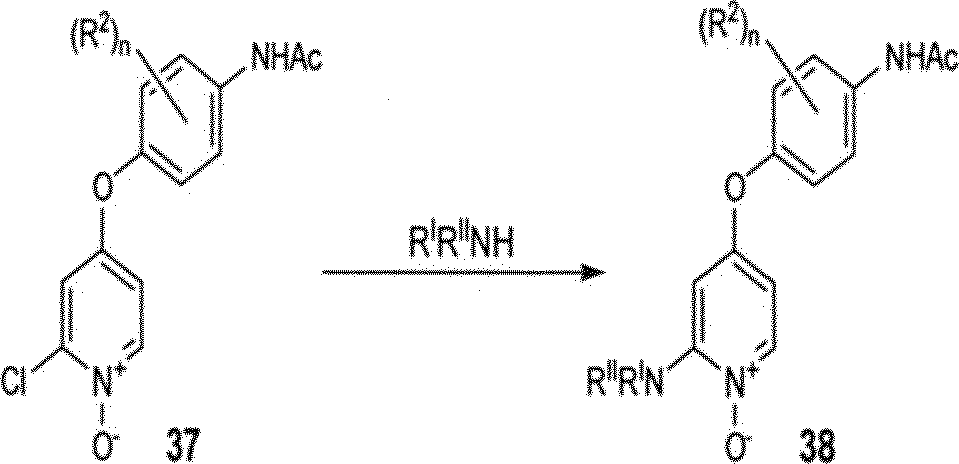
Em um método relacionado, o intermediário 2-ctoropiridina 36, obtido empregando-se a química descrita no ESQUEMA 1 pode ser convertido no /V-óxido 37 empregando-se o ácido 3-cloroperoxibenzóico (m-CPBA) em clorofórmio (veja, W02004/002410) (ESQUEMA 6). Tratamento do composto 37 com uma amína propriamente substituída pode fornecer intermediário 38. A redução de /V-óxido do composto 38 com, por exemplo trifenilfosfina, seguido por remoção do grupo de proteção de acetamida sob condições acidicas pode fornecer anilina 39. A conversão de anilína 39 em análogo desejado 40 pode ser realizada empregando-se química previamente descrita nos ESQUEMAS 1-5,
ESQUEMA 6
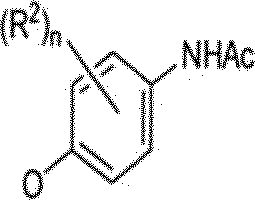

w-CPBA
CHCI3



Em ura método alternativo para os compostos relacionados com
40, os derivados de 2-aminopiridina 47 e 48 pode ser preparado de acordo com a sequência sintética ilustrada no ESQUEMA 7. Para esta finalidade, o ácido 4-cloropicolínico (41, TCI América) pode ser convertido em 4cloropicolinamida (42) empregando-se um procedimento de duas etapas envolvendo tionilcloreto seguido por amônia em metanol. O acoplamento de intermediário 42 com derivado de 4-aminofenol 43, na presença de uma base, tal como f-butóxido de potássio pode fornecer o derivado de picolinamida
44. Uma acilaçâo ou formação de aciluréia do intermediário 44 pode fornecer intermediários tais como 45 e 46. O tratamento dos derivados picolinamida 45 e 46 com ou bis-(trifluoroacetóxi)-iodobenzeno, piridina e água em DMF ou bromo, hidróxido de potássio em água promove um redisposição Hofmann para gerar derivados desejáveis de 2-aminopiridina 47 e 48,
ESQUEMA7

TCIAmerica


PM(OCF,), pirídina
Ji-O. DMFou &Γκο0^Γ* dioxanoem seguida AcOH

acilaçeoou —Qj.
formação de tourtia

Br2, KOH. H20, dioxanoem seguida AcOH
Phl(OCBj), piridina
HA DMF ou

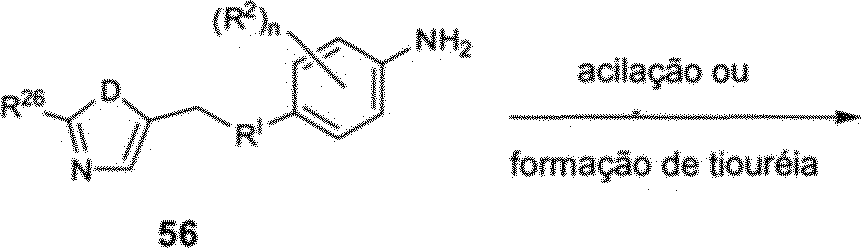
O tiazol contendo compostos 53, 57 e 62 pode ser preparado empregando-se as vias sintéticas descritas nos ESQUEMAS 8-10, O deslocamento dos grupos de saída de 49 (ESQUEMA 8) ou 54 (ESQUEMA 9) 5 com uma anilina/fenol 50 pode fornecer intermediários 51 e 55, respectivamente. A redução dos substituintes de nitro de 51 e 55 com pó de zinco e cloreto de amônio em uma mistura de THF-MeOH deve gerar anilhas 52 e 56, respectiva mente. A conversão de anilinas 52 e 56 nos compostos desejados 53 e 57 pode ser realizada empregando-se química previamente des10 crita (vide supra).
ESQUEMA 8
Zn. NH4CI.
THF-MeOH

1» Grupo de partida tal como Cl ou BrR‘=O,SouNH

ESQUEMA 9

L=Grupo de saída tal c»mo Cl ou Br 55 R'*OouNH
Zn, NH4CI
THF-MeOH

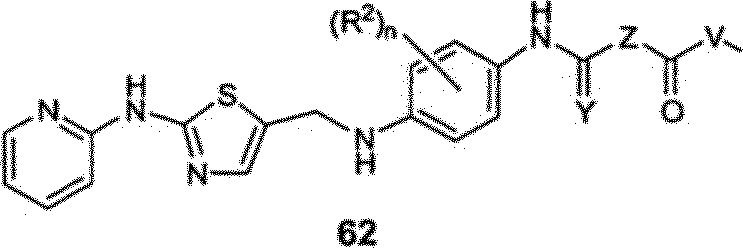
A aminação redutiva de aldeído 58 pode ser obtida empregando-se métodos descritos em WO 2004/001059, aqui incorporado por refe5 rências em sua totalidade, empregando-se uma anilina apropriadamente substituída 59 pode fornecer o intermediário de nitro 60 (ESQUEMA 10). O intermediário de aminotiazol desejado 62 pode então ser obtido empregando-se a química similar aquela que foi descrita nos ESQUEMAS 8 e 9,
ESQUEMA 10

(WO 2004/001059)

ei3síh. tfa
Ζη,ΝΗ^,
THF-MeOH eo

A incorporação de vários substituintes na posição 3 do núcleo de piridina pode ser realizada empregando-se a química previamente delineada no ESQUEMA 11, Para esta finalidade, 4-cloro-3-íodopiridina (63, Tabanella, S. e outro, Org. Biomol. Chem. 2003, 1, 4254-4261), pode ser 5 acoplado com o derivado de 4-nitrofenol 8 na presença de uma base, tal como diisopropiletilamina (base de Hunig) para fornecer o intermediário de iodeto desejado 64, Uma variedade de reações de acoplamento mediado organometálico pode então ser realizada com o derivado de iodeto 64, exemplos dos quais são ilustrados no ESQUEMA 11,0 iodeto 64 pode ser 10 tratado com as aminas (RR’NH), alquinas substituídas 66, arilboronatos 67, vinilestananos, e ésteres α,β-insaturados na presença de um catalisador de paládio ou cobre para fornecer os intermediários 65, 68-71, respectivamente. A porção de nitro dos compostos 65 e 68-71 pode ser reduzida com, por exemplo, pó de zinco e cloreto de amônio em uma mistura de THF-MeOH, e 15 os intermediários de anilina resultantes podem ser adiados empregando-se a química previamente empregando-se descrita nos ESQUEMAS 1-5,
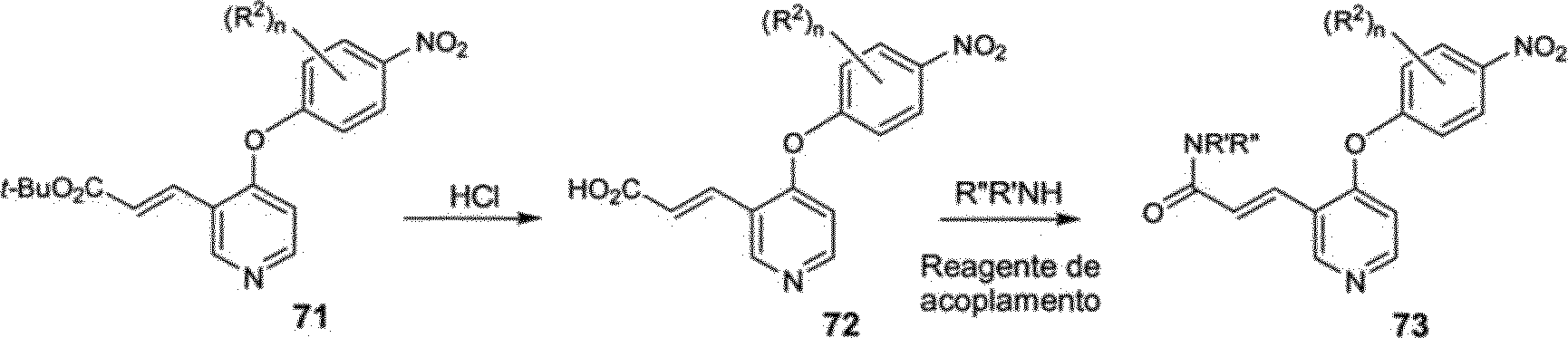
O intermediário 71 pode então ser convertido em amidas α,βinsaturadas 73 (ESQUEMA 12). O composto 72, derivado de ácido que promoveu a hidrólise de éster 71, pode ser acoplado com várias aminas 20 (RR’NH) na presença de um reagente de acoplamento tal como, porém não limitado a EDCI, TBTU, DCC, para fornecer a amida intermediária desejada 73, A redução da porção de nitro de 73 e acilação subsequente do intermediário de anilina requisitado pode ser realizado empregando-se a química previamente descrita nos ESQUEMAS 1-5,
ESQUEMA 11


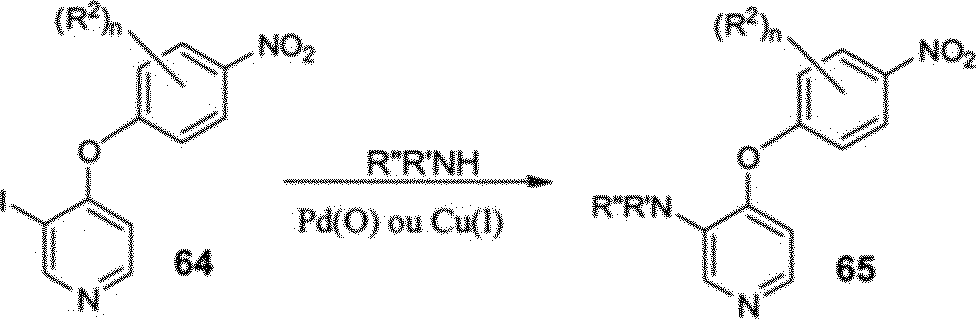
| R* — | Ar.B(OR)2 67 | C' ’SrsBu-, | «yO^C^· |
| 66 | Pd(PPh3)4. | Pd(PPh3)4, | ROAch |
| Cul. Et3N | Na2CO3, | CsF. Cul, | NBu, |
| Dioxano/HjO * | Et3N. DMF | DMF. 100 °C | |
| THF, ’ rvfluKM ' | X0C ( | 100 «c |
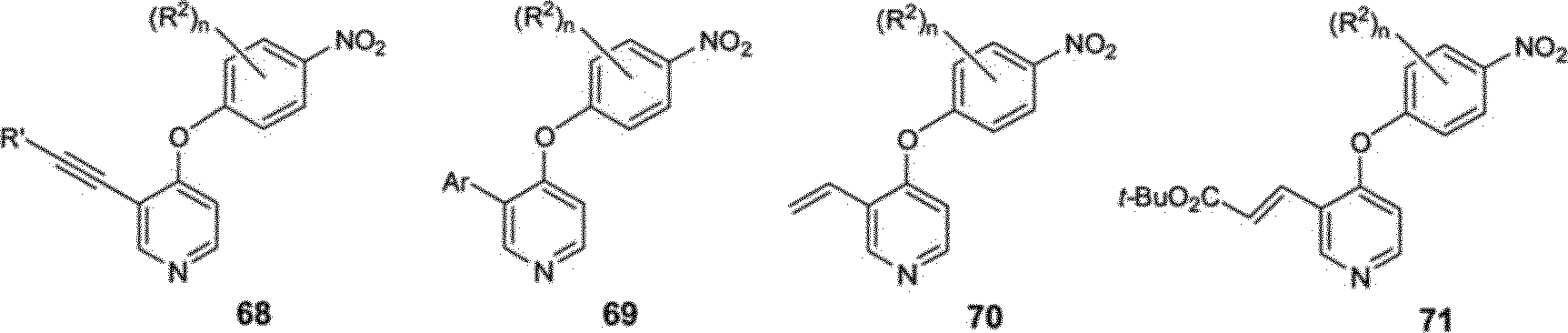
ESQUEMA 12
O intermediário 74 pode também ser ainda modificado para pre5 parar aminas propargílicas 76 (ESQUEMA 13). A mesilação do álcool propargílico 74, pode ser realizada com cloreto de metanossulfonila na presença de uma base, tal como diisopropiletilamina (base de Hunig) para fornecer o mesilato 75. O deslocamento do grupo de mesilato do composto 75 com várias aminas (RR’NH) pode fornecer as aminas propargílicas 76. A redu10 ção da porção de nitro de 76 e acilação subsequente do intermediário de anilina requisitado pode ser realizada empregando-se a química previamente descrita nos ESQUEMAS 1-5,
ESQUEMA 13

Os derivados de 3-aminopiridina 79 e 80 podem ser preparados de acordo com a via sintética descrita no ESQUEMA 14. Para esta finalidade, 4-cloro-3-nitropiridina (77, Lancaster Synthesis Ltd.) pode ser acoplado com 4-aminofenol na presença de uma base, tal como hidreto de sódio em
DMF para fornecer o intermediário de nitro 78, A química previamente descrita acima, pode ser empregada para converter o intermediário 78 para os compostos desejados 79 e 80, O substituinte de amino de 79 e 80 pode também ser ainda modificado, por exemplo, por meio de alquilaçâo, acilação, arilação ou sulfonilação.

A incorporação de substituintes na posição 5 ou 3 do anel de 215 aminopiridina pode ser realizada empregando-se os intermediários de iodeto e 86, respectivamente (ESQUEMAS 15 e 16). O derivado de 2carboxamida 81 pode ser convertido no derivado de 2-aminopiridina 82 empregando-se o protocolo de redisposíção Hofmann previamente descrito no
ESQUEMA 7, A iodinação da posição 5 do composto 82 pode ser obtido com M-iodossucdnimida em uma mistura de acetonitril-isopropanoi para fornecer o intermediário de iodeto 83, Alternativamente, 4-cloropiridin-2Ifcarbamato de f-butila (84, CB Research e Development Inc.) pode ser convertido em 4-cloro-3-iodopiridin-2-ilcarbamato de f-butila (85) por meio de processo de duas etapas envolvendo n-butillítio em THF em baixa temperatura seguido peta adição de iodo. A remoção do grupo de proteção de M-Boc (Fbutilcarbamato) de 85 com brometo de hidrogênio aquoso com refluxo seguido pelo acoplamento do intermediário de cloreto com o derivado de 4nitrofenol 8 na presença de ditsopropilamina (base de Hunig) em /V10 metilpirrolidinona (NMP) em elevada temperatura pode fornecer o intermediário de iodeto 86, Os intermediários de iodeto 83 e 86 pode ainda ser processados empregando-se a química similar aquela previamente descrita no ESQUEMA 11,
ESQUEMA 15

ESQUEMA 16

D^vel^msntlns.
Os análogos 93 e 94 ligados a metileno (B = CH2) podem ser preparados de acordo com a seqüência sintética delineada no ESQUEMA
17, O composto 88, derivado de proteção de W-Boc dos derivados de 4bromoanilina 87, pode ser tratado com brometo de metilmagnésio seguido >
pelos terc-butillítio e 2-cloroisonicotinaldeído (Frey, L. F. e outro, Tetrahedron Lett. 2001, 42, 6815-6818) em baixa temperatura para fornecer o intermediário 90. A oxidação do anel de piridina de 90 com ácido 3-cloroperoxibenzóico (m-CPBA), seguida pelo deslocamento do substituinte de cloro com uma amina (R’NH2) e a redução subsequente do intermediário de ΛΖ-óxido intermediário com zinco e formiato de amônio em metanol pode fornecer o intermediário 91. Quando a alilamina for empregada como a amina nucleofílica (R’NHa), o grupo de alila pode ser removido da amina de 91 empregando-se o catalisador de ródio em uma mistura de etanol-água. A re10 moção do grupo de hidroxila de 91 pode ser realizada por dois métodos diferentes. Por exemplo, a hidrogenólise de composto 91 na presença de um catalisador de paládio, seguida pela desproteção do grupo W-Boc na anilina sob condições acídicas (HCI em metanol) pode fornecer o composto 92, Altemativamente, o composto 92 pode ser obtido pela acilação do álcool de 91 e a hidrólise subsequente do intermediário na presença de um catalisador de paládio e a remoção do grupo de proteção de /V-Boe sob condições acídicas (ácido trifluoroacético em cloreto de metileno). O intermediário 92 pode ser acilado para fornecer os compostos desejados 93 e 94 empregando-se a química previamente descrita nos ESQUEMAS 1-5,
ESQUEMA 17



R1 = H,3-dimettlamír»opropila
RhíPPhahCI Γ~ R’^ alila EtOH.HzO1— R' = H

Os derivados de amida heterocíclica 100 e 105 podem ser preparados de acordo com as vias sintéticas descritas nos ESQUEMAS 18 e 10, Para esta finalidade, metil 2-oxo-1-fenil-1,2-diidropiridina-3-carboxilato (97) pode ser obtido em um processo de duas etapas começando com 2-(3metoxialilideno)malonato de (E)-dimetila comercialmente disponível (95) (ESQUEMA 18). Desse modo, o tratamento do composto 95 com anilina em temperatura ambiente pode fornecer o intermediário 96, que pode então ser ciclizado na presença de uma base, tal como hidreto de sódio em dimetilsul10 fóxido para gerar o 97, A hidrólise do intermediário 97 sob condições básicas pode fornecer o ácido 2-oxo-1-fenil-1,2-diidropiridina-3-carboxílico (98).
O ácido carboxílico 98 pode então ser acoplado com o derivado de anilina na presença de um reagente de acoplamento, tal como cloridrato de 1(dimetilaminopropil)-3-etilcart)odiimida (EDGI) e hidroxibenzotriazol (HOBt) em DMF para fornecer o composto desejado 100,
ESQUEMA 18

anilina
Acros Organics
THF temperatura


......UOH ,
MeOH-HjO temperatura ambiente
NaH.....x
0MSO temperatura ambiente

O intermediário de N-óxido de piridila 104 (ESQUEMA 19) pode ser obtido pelo processo de duas etapas em que o ácido 6-bromopicolínico comercialmente disponível (101) é acoplado com o fenil-1,3,2-dioxaborinano
102 (Aldrich) na presença de um catalisador de paládio (Q) e carbonato de sódio, seguido pela oxidação dos intermediários requisitados 103 em elevada temperatura. O acoplamento do intermediário 104 com o derivado de anilina 99 pode fornecer o composto desejado 105,
4#
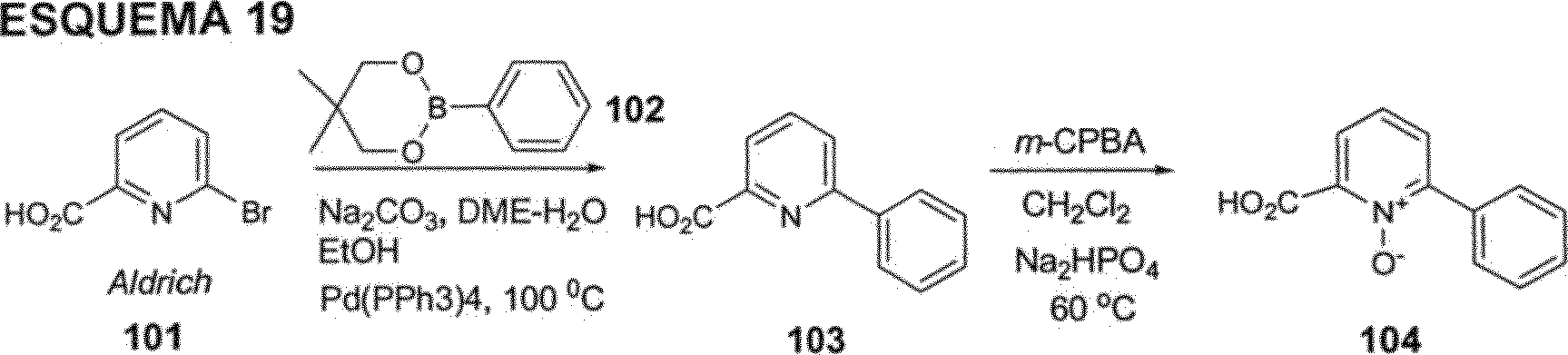

Os compostos das Fórmulas I e II são úteis no tratamento de uma variedade de cânceres, incluindo, porém não limitados aos seguintes:
(a) carcinoma, incluindo aqueles da bexiga, mama, colo, rim, fígado, pulmão, incluindo câncer de pulmão de célula pequena, esôfago, vesícula biliar, ovário, pâncreas, estômago, cérvix, tireóide, próstata, e pele, incluindo carcinoma de célula escamosa;
(b) Tumores hematopoiéticos de linhagem linfóide, incluindo leucemia, leucemia linfocítica aguda, leucemia línfoblástica aguda, linfoma de célula B, linfoma de célula T, linfoma de Hodgkin, linfoma não-Hodgkins, linfoma de célula pilosa e linfoma de Burkett;
(c) tumores hematopoiéticos de linhagem mielóide, incluindo leucemias mielogenosas agudas e crônicas, síndrome mielodisplásica e leucemia promielocítica;
(d) tumores de origem mesenquimal, incluindo fibrossarcoma e rabdomiossarcoma;
(e) tumores do sistema nervoso central e periférico, incluindo astrocitoma, neuroblastoma, glioma e schwanomas; e (f) outros tumores, incluindo melanoma, seminoma, teratocar hui cínoma, osteossarcoma, xenoderoma pigmentosa, ceratocantoma, câncer folicular de tireóide e sarcoma de Kaposi.
Devido ao papel chave das proteínas cinases na regulação de proliferação celular em geral, inibidores podem agir como agentes citostáti5 cos reversíveis que podem ser úteis no tratamento de qualquer processo de doença que caracterize a proliferação celular anormal, por exemplo, hiperplasia prostática benigna, polipose de adenomatose familiar, neuro-fibromatose, aterosclerose, fibrose pulmonar, artrite, psoríase, glomerulo-nefrite, restenose seguindo angioplastia ou cirurgia vascular, formação de cicatriz hipertrofica, doença do intestino inflamatório, rejeição de transplante, choque endotóxico, e infecções fúngicas.
Compostos de Fórmulas I e II como moduladores de apoptose, serão úteis no tratamento de câncer (incluindo, porém não limitado àqueles tipos aqui mencionados acima), infecções virais (incluindo, porém não limi15 tado um herpesvirus, poxvirus, vírus Epstein-Barr, adenovirus e vírus Sindbis), prevenção de desenvolvimento de AIDS em indivíduos infectados por HIV, doenças autoimunes (incluindo, porém não limitadas a lúpus sistêmico, eritematoso, glomerulonefrite autoimuno-mediada, artrite reumatóide, psoríase, doença do intestino inflamatório, e diabetes autoimune), distúrbios neu20 rodegenerativos (incluindo, porém não limitado a doença de Alzheimer, demência relacionada com a AIDS, doença de Parkinson, esclerose lateral amiotrófica, retinite pigmentosa, atrofia muscular espinhal e degeneração cerebelar), síndromes mielodisplásica, anemia aplásica, tesão isquêmica associada com infarto miocárdico, acidente vascular cerebral e lesão de reperfu25 são, arritmia, aterosclerose, doenças do fígado relacionada com álcool ou induzida por toxina, doenças hematológicas (incluindo, porém não limitado a anemia crônica e anemia aplásica), doenças degenerativas do sistema musculoesqueletal (incluindo, porém não limitado a osteoporose e artrite), rinossinusite sensível à aspirina, fibrose cistica, esclerose múltipla, doença dos 30 rins e doença do câncer.
Compostos de Fórmulas I e II podem modular no nível de síntese de RNA e DNA celular. Estes agentes portanto seriam úteis no tratamen to de infecções virais (incluindo, porém não limitado a HIV, vírus d© papiloma humano, herpesvirus, poxvirus, vírus Epstein-Barr, vírus Sindbis e adenovirus).
Os compostos de Fórmulas I e II podem ser úteis na quimioprevenção de câncer. A quimioprevenção é definida como inibindo o desenvolvimento de câncer invasivo ou bloqueando o evento mutagênico inicial ou bloqueando a progressão de células pré-malígnas que já sofreram um insulto ou inibição de reincidência de tumor.
Compostos de Fórmulas I e II podem também ser úteis na inibição de angiogênese de tumor e podem também ser úteis na inibição de angiogênese e metástese de tumor.
O termo agente anticâncer'* inclui qualquer agente conhecido que é útil para o tratamento de câncer incluindo 17a-Etínilestradiol, Dietilstilbestrol, Testosterona, Prednisona, Fluoximesterona, Propionato de dromostanolona, Testolactona, Megestrolacetato, Metilprednisolona, Metil-testosterona, Prednisolona, Triamcinolona, Clorotrianisene, Hidroxiprogesterona, Aminoglutetimida, Estramustina, Medroxiprogesteroneacetato, Leuprolida, Flutamida, Toremifeno, Zoladex, inibidores de metaloproteinase matriz, inibidores de VEGF, incluindo como anticorpos anti-VEGF tal como Avastina, e pequenas moléculas tal como ZD6474 e SU6668, vatalanib, BAY-43-9006, SU11248, CP-547632, e GEP-7G55 são também incluídos. Anticorpos antiHer2 de Genentech (tal como Herceptina) podem também ser utilizados. Inibidores de EGFR adequados incluem gefitinib, erlotinib, e cetuximab. Os inibidores Pan Her incluem canertinib. EKB-569, e GW-572016, São também incluídos os inibidores de Src bem como Casodex (bicalutamida, Astra Zeneca), Tamoxifen, inibidores de MEK-1 cínase, inibidores de MAPK cinase, inibidores de PI3, e inibidores de PDGF, tal como imatiníb. São também incluídos agentes anti-angiogênicos e antivasculares que, interrompendo o fluxo sanguíneo para tumores sólidos, tornam as células de câncer quiescentes privando-as de nutrição. A castração, que também torna os carcinomas dependentes de androgênio não proliferativos, pode também ser utilizada. São também incluídos os inibidores de IGF1R, inibidores de tirosina cinases não receptoras e receptoras, e inibidores de sinalização de integrina. Agentes anticâncer adicionais incluem agentes de estabilização de microtúbulo tais como paclitaxel (também conhecido como Taxol®), docetaxel (também conhecido como-Taxotere®), 7-O-metiltiometilpaclitaxel (descrito na U.S. 5,646,176), 4-desacetil-4-metilcarbonatopaclitaxel, 3’-terc-butil-3’-Nterc-butiloxicarbonila-4-deacetil-3’-defenil-3’-N-debenzoil-4-0-metoxicart)onila-paclitaxel (descrito na USSN 09/712,352 depositada em 14 de novembro de 2000), paclitaxel de carbonato de C-4 metila, epotilona A, epotilona B, epotilona C, epotilona D, desoxiepotilona A, desoxiepotilona B, (1S-[1R*,3R*(E), 7R*, 1OS*, 11R*, 12R*, 16S*]]-7-11 -diid róxi-8,8,10,12,16-pentametil-3-[1 -metil2-(2-metil-4-tiazolil)etenil]-4-aza-17-oxabiciclo [14,1,0]heptadecano-5,9-diona (descrito em WO 99/02514), [1S-[1R*,3R*(E),7R*,10S*, 11R*,12R*, 16S*]]-3[2-p-(aminometil)-4-tiazolil]-1-metiletenil]-7,11-diídróxi-8,8,10,12,16pentametil-4-17-díoxabiciclo[14,1,0]-heptadecano-5,9-diona (descrito em USP 6,262,094) e derivados deste; e agentes rompedores de microtúbulo. São também adequados os inibidores de CDK, um inibidor de ciclo de célula antiproliferativa, epidofilotoxina; uma enzima antineoplásica; um inibidor de topoisomerase; procarbazine; mitoxantrona; complexos de coordenação de platina tais como cis-platina e carboplatina; modificadores de resposta biológica; inibidores de crescimento; agentes terapêuticos anti-hormonais; leucovorina; tegafur; e fatores de crescimento hematopoiéticos.
Agentes citotóxicos adicionais incluem, derivados de melfalan, melamina hexametila, tiotepa, citarabina, idatrexato, trimetrexato, dacarbazina, L-asparaginase, camptotecina, topotecan, bicalutamida, flutamida, leuprolida, piridobenzoindol, interferons, e interleucinas.
As composições farmacêuticas contendo o ingrediente ativo podem ser em uma forma adequada para uso oral, por exemplo, como comprimidos, trociscos, lozangos, suspensões aquosas ou oleosas, grânulos ou pós dispersíveis, emulsões, cápsulas duras ou macias, ou xaropes ou elixires. As composições destinadas ao uso oral podem ser preparadas de acordo com qualquer método conhecido na técnica para a preparação de composições farmacêuticas e tais composições podem conter um ou mais agen tes selecionados do grupo que consiste em agentes adoçantes, agentes aromatizantes, agentes colorantes e agentes convervantes a fim de fornecer preparações farmaceuticamente elagantes e palatáveis. Comprimidos contêm o ingrediente ativo em mistura com excipientes farmaceuticamente aceitáveis não-tóxicos que são adequados para a fabricação de comprimidos. Estes excipientes podem ser, por exemplo, diluentes inertes, tais como carbonato de cálcio, carbonato de sódio, lactose, fosfato de cálcio ou fosfato de sódio; agentes de granulação e desintegração, por exemplo, celulose microcristalina, croscarmelose sódica, amido de milho, ou ácido algínico; agentes de ligação, por exemplo, amido, gelatina, polivinil-pirrolidona ou acácia, e agentes lubrificantes, por exemplo, estearato de magnésio, ácido esteárico ou talco. Os comprimidos podem ser não-revestidos ou eles podem ser revestidos com técnicas conhecidas para mascarar o sabor desagradável do fármaco ou retardar a desintegração e absorção no trato gastrointestinal e desse modo fornecer uma ação prolongada durante um período maior. Por exemplo, um material de mascaramento de sabor solúvel em água tal como um material de mascaramento de sabor solúvel em água tal como tal como hidroxipropil-metilcelulose ou hidroxipropil-celulose, ou um material de retardamento do tempo tal como etil celulose, butirato de acetato de celulose pode ser empregado.
Formulações de uso oral podem também ser apresentadas como cápsulas de gelatina dura em que o ingrediente ativo é misturado com um diluente sólido inerte, por exemplo, carbonato de cálcio, fosfato cálcio ou caulim, ou como cápsulas de gelatina macia em que o ingrediente ativo é misturado com portador solúvel em água tal como polietileno glicol ou um meio oleoso, por exemplo óleo de amendoim, parafina líquida, ou azeite.
As suspensões aquosas contêm o material ativo em mistura com excipientes adequados para a fabricação de suspensões aquosas. Tais excipientes são agentes de suspensão, por exemplo carboximetilcelulose de sódio, metílcelulose, hidroxípropilmetil-celulose, alginato de sódio, polivinilpirrolidona, goma tragacanto e goma acácia; agentes díspersantes ou umectantes podem ser fosfatídeo de°Corrência natural, por exemplo lecitina, ou
Ag produtos de condensação de um óxido de alquileno com ácidos graxos, por exemplo, estearato de polioxietileno, ou produtos de condensação de óxido de etiteno com álcoois alifáticos de cadeia longa, por exemplo, heptadecaetileno-oxicetanol, ou produtos de condensação de óxido de etiteno com éste5 res parciais derivados de ácidos graxos e um hexitol tal como monooleato de sorbitol de polioxietileno, ou produtos de condensação de óxido de etileno com ésteres parciais derivados de ácidos graxos e anidridos de hexitol, por exemplo, monooleato de sorbitan de polietileno. As suspensões aquosas podem também conter um ou mais conservantes, por exemplo p-hidro10 xibenzoato de n-propila ou etila, um ou mais agentes de coloração, um ou mais agentes aromatizantes, e um ou mais agentes adoçantes, tais como sacarose, sacarina ou aspartame.
Suspensões oleosas podem ser formuladas por suspender o ingrediente ativo em um óleo vegetal, por exemplo, óleo de amendoim, azei15 te, óleo de gergelim ou óleo de coco, ou em óleo -mineral tal como, parafina líquida. As suspensões oleosas podem conter um agente espessante, por exemplo cera de abelha, parafina dura ou álcool de cetila. Agentes adoçantes tal como aqueles mencionados acima, e agentes aromatizantes podem ser adicionados para fornecer uma preparação oral palatável. Estas compo20 sições podem ser conservadas pela adição de um antioxidante tal como, hidroxianisol butilado ou alfa-tocoferol.
Pós e grânulos díspersiveis adequados para a preparação de uma suspensão aquosa pela adição de água fornece o ingrediente ativo na mistura com um agente dispersante ou umectante, agente de suspensão e 25 um ou mais conservantes. Agentes dispersantes ou umeçtantes adequadas e agentes de suspensão são exemplificados por aqueles já mencionados acima. Excipientes adicionais, por exemplo agentes adoçantes, aromatizantes e colorantes, podem também estar presentes. Estas composições podem ser conservadas pela adição de um antioxidante tal como, ácido ascór30 bico.
As composições farmacêuticas da invenção podem também ser na forma de uma emulsão de óleo-em-água. A fase oleosa pode ser um óleo dequados podem ser fosfatídeos de°Corrência natural, por exemplo lecitina de soja, e ésteres ou ésteres parciais derivados de ácidos graxos e anidridos de hexitol, por exemplo, monooleato de sorbitan, e produtos de condensação dos referidos ésteres parciais com oxido de etileno, por exemplo monooleato de sorbitan de polietileno. As emulsões podem também conter agentes aromatizantes, adoçantes, conservantes e antioxidantes.
Xaropes e elixires podem ser formulados com agentes adoçantes, por exemplo, glicerol, propileno glicol, sorbitol ou sacarose. Tais formulações podem também conter um agente demulcente, conservantes, aromatizante e colorante e antioxidante.
As composições farmacêuticas podem ser na forma de soluções aquosas injetáveis estéreis. Entre os veículos aceitáveis e solventes que podem ser empregados estão a água, solução de Ringer e solução de cloreto de sódio isotônica.
A preparação injetável estéril pode também ser uma microemulsão de óleo-em-água injetável estéril na qual o ingrediente ativo é dissolvido na fase oleosa. Por exemplo, o ingrediente ativo pode ser primeiro dissolvido em uma mistura de óleo de soja e lecitina. A solução oleosa em seguida introduzida em uma mistura de água e glicerol e processada para formar uma microemulsao
As soluções injetáveis ou microemulsões podem ser introduzidas na corrente sanguínea do paciente pela injeção de bolo local. Alternativamente, elas podem ser vantajosas para administrar a solução ou a microemulsao de um tal modo para manter uma concentração circulante constante do presente composto. Afim de manter uma tal concentração constante, um dispositivo de liberação contínua pode ser utilizado. Um exemplo de tal dispositivo é a bomba íntravenosa Deltec CADD-PLUS.TM. modelo 5400,
As composições farmacêuticas podem ser na forma de uma suspensão oleaginosa ou aquosa injetável estéril para administração intramuscular e subcutânea. Esta suspensão pode ser formulada de acordo com a técnica conhecida empregando-se agentes dispersantes ou umectantes adequados e agentes de suspensão que foram mencionados acima. A preparação injetável estéril pode também ser uma solução injetável estéril ou suspensão em um solvente ou diluente parenteralmente aceitável nãotóxico, por exemplo como uma solução em diol de 1,3-butano. Além disso, óleos fixos estéreis são convencionalmente empregados como um meio solvente ou de suspensão. Para este propósito qualquer óleo fixo suave pode ser empregado incluindo mono- ou diglicerídeos sintéticos. Além disso, ácidos graxos, tais como ácido oléico encontram uso na preparação de injetáveis.
Os compostos de Fórmulas I e II podem também ser administrados na forma de supositórios para administração retal do fármaco. Estas composições podem ser preparadas misturando-se o fármaco com um excipiente não irritante adequado que é sólido em temperaturas usuais, porém líquido na temperatura retal e portanto derreterá no reto para liberar o ®rmaco. Tais materiais incluem manteiga de cacau, gelatina glicerinada, óleos vegetais hidrogenados, misturas de polietilenos glicóis de vários pesos moleculares e ésteres ácidos graxos de polietileno glicol.
Para uso tópico, cremes, ungüento, geléias, soluções ou suspensões, etc., contendo os compostos de Fórmula I são empregados. (Para os propósitos desta aplicação, a aplicação tópica deve incluir anti-sépticos bucais e gargarejes.)
Os compostos da presente invenção podem ser administrados em forma intranasal por meio de uso tópico de veículos intranasais adequados e dispositivos de liberação, ou por vias transdérmicas, empregando-se aquelas formas de emplastros de pele transdérmicos bem conhecidas por aqueles versados na técnica. Para ser administrada na forma de um sistema de liberação transdérmica, a administração de dosagem, de fato, será contínua em vez de intermitente em todo o regime de dosagem. Os compostos da presente invenção podem também ser liberados como um -supositórios empregando-se bases, tais como, manteiga de cacau, gelatina glicerinada, óleos vegetais hidrogenados, misturas de polietileno glicóis de vários pesos moleculares e ésteres ácidos graxos de poiietileno glicol.
Quando um composto de acordo com está invenção for administrado em um indivíduo humano, a dosagem diária será normalmente determinada peto médico prescrevente com a dosagem geralmente variando de acordo com a idade, peso, sexo e resposta do paciente individual, bem como a severidade dos sintomas do paciente.
Se formulados como uma dose fixa, tais produtos de combinação empregam os compostos desta invenção dentro da faixa de dosagem acima descrita e o outro agente farmaceuticamente ativo ou tratamento dentro de sua faixa de dosagem aprovada. Os compostos de Fórmulas I e II podem também ser administrados sequencialmente com agentes anticâncer ou citotóxicos conhecidos quando a formulação de combinação é imprópria. A invenção não está limitada na sequência de administração. Os compostos de fórmulas I e II podem ser administrados antes ou após a administração de agente(s) anticâncer ou citotóxicos conhecidos.
ENSAIOS
As propriedades farmacológicas dos compostos desta invenção podem ser confirmadas por diversos ensaios farmacológicos. Os ensaios farmacológicos exemplificados que seguem funcionaram com os compostos de acordo com a invenção e os sais destes.
Ensaio de Met Cinase
| Reagentes | Concentração Final de Mistura de Substrato. |
| Solução de Estoque | |
| Tris-HCI, (1M, pH 7,4) | 20 mM |
| MnCI2(1M) | 1 mM |
| DTT(1M) | 1 mM |
| BSA(100 mg/ml) | 0,1 mg/ml |
| polyGluVtyrfWmg/m!) | 0,1 mg/mL |
| ATP(1mM) | 1 μΜ |
| y-ATP (10μΟί/μΙ) | 0,2 pCi/ml |
| Tampão | Mistura de Enzima |
| 20ul 1MDTT | 4ul GST/Met enzima (3,2mg/ml) = 10 ng/rxn |
| 200 ul 1M Tris-HCL, pH 7,4 | qs 12 ml de Tampão |
| 20 ul 100 mg/ml BSA | |
| qs 20ml H20 |
Misturas de incubação empregadas para o ensaio de Met cinase contêm substrato sintético polyGlu:Tyr, (4:1), ΑΤΡ, ΑΤΡ-γ-33Ρ e tampão contendo Mn** *” Mg**, DTT, BSA, e tampão de Tris. As reações foram incubadas durante 60 minutos a 27°C e interrompidas pela adição de ácido tricloroacético frio (TCA) em uma concentração final de 4%. Os precipitados de TCA foram coletados sobre placas Unifilter GF/C (Packard Instrument Co., Meriden, CT) empregando-se uma coletora universal Filtermate (Packard Instrument Co., Meriden, CT) e os filtros são quantificados empregando-se uma registradora de cintilação liquida de 96 cavidades TopCount 96 (Packard Instrument Co., Meriden, CT). As curvas de dosagem foram geradas para determinar a concentração requerida para inibir 50% de atividade de cinase (ICM). Os compostos foram dissolvidos a 10 mM em sulfóxido de dimetila (DMSO) e avaliados em seis concentrações, cada qual em quadruplicate. A concentração final de DMSO no ensaio é de 1%. Os valores IC50 foram derivados por análise de regressão não-linear e tiveram um coeficiente de variação (Desvio Padrão/média, n=6) = 16%.
Os compostos preferidos da invenção inibem Met cinase com valores IC^ entre 0,01 a 100 μΜ. Os compostos mais preferidos têm valores ICS menores do que 0,5 μΜ.
Outra matéria objeto da invenção também inclui produtos farmacêuticos para uso como descrito acima incluindo controle de câncer, inflamação e artrite, que contêm peto menos um dos compostos de Fórmulas I e II como definido acima ou peto menos um de seus saís de adição de ácido farmaceuticamente aceitáveis, e o uso de um composto das Fórmulas I e II como definido acima para a preparação de um produto farmacêutico tendo atividade contra doenças proliferativas como descrito previamente incluindo contra o câncer, inflamação e/ou artrite.
Os seguintes exemplos e preparações descrevem a maneira e processo de preparar e empregar a invenção e são ilustrativos em vez de limitantes. Deve ser entendido que aí pode existir outras modalidades que incluem-se no espírito e escopo da invenção, como definido pelas reivindicações anexas a ela.
EXEMPLOS
A invenção será agora também descrita petos seguintes exemplos de preparação, que são modalidades preferidas da invenção. Todas as reações foram realizadas com agitação magnética contínua sob uma atmosfera de argônio ou nitrogênio seco. Todas as evaporações e concentrações foram realizadas em um evaporador rotativo sob pressão reduzida. Os reagentes comerciais -foram empregados como recebidos sem purificação adicional. Solventes foram de graus anidros comerciais e foram empregados sem outra secagem ou purificação. Cromatografia instantânea foi realizada empregando-se síltca-gel (EMerck Kieselgel 600,040 - 0,060 mm).
HPLC de Fase Reversa Analítica (RP) HPLC foi realizada empregando-se uma coluna Phenomonex Luna C18 -S5 4,6 mm χ 50 mm ou uma coluna YMC S5 ODS 4,6 x 50 mm. Em cada caso um gradiente linear de 4 minutos (de 100% A:%0 Ba 0% A: 100% B) foi empregado com o seguinte sistema de fase móvel: A = 90% H2O/MeOM + 0,2% B = 90% MeOH/H2O + 0,2% H3PO4 em taxa de fluxo = 4 mL/minuto e detecção a 220 nm.
HPLC de Fase Reversa Preparativa (RP) HPLC foi realizada com uma eluição gradiente linear empregando-se metanol a 10%, -água a 90%, TFA a 0,1% (solvente A) e metanol a 90%, água a 10%, TFA a 0,1% (solvente B) e detecção a 220 nm em uma das seguintes colunas: A - coluna Shimadzu S5- ODS-VP 20 χ 100 mm com uma taxa de fluxo de 20 mL/minuto; B - coluna YMC S5 ODS 30 χ 100 mm com uma taxa de fluxo de 20 mL/minuto; C - coluna Phenomonex 30 χ 250 mm com uma taxa de fluxo de 10 mL/min; D - coluna YMC S5 ODS 20 χ 250 mm com uma taxa de fluxo de 10 mL/minuto; E - coluna YMC S10 ODS 50 χ 500 mm com uma taxa de fluxo de 50 mL/minuto; ou F - coluna YMC S10 ODS 30 χ 500 mm com uma taxa de fluxo de 20 mL/minuto.
Todos os produtos finais foram caracterizados por 1H RMN, RP HPLC, ionização por eletrovaporização (ESI MS) ou espectrometria de massa por ionização de pressão atmosférica (API MS). Os espectros 1H RMN foram obtidos em um instrumento 500 MHz JEOL ou 400 MHz Broker. Os espectros de 13C RMN foram registrados a 100 ou 125 MHz. Resistência de
Campo são expressas em unidades de δ (partes por milhão, ppm) com relação aos picos de solvente, e multiplicidades de pico são designados como segue: s, singleto; d, dubleto; dd, dubleto de dubleto; dm, dubleto de multipleto; t, tripleto; q, quarteto;· br s, singleto amplo; m, multipleto.
As seguintes abreviações são empregadas para reagentes comumente empregados: Boc ou BOC: carbamate de t-butila; Fmoc: carbamate de 9H-fluorenÍlmetila; TEA: trietilamina; NMM: /V-metilmorfolina; Ms: metanossulfonila; DIEA ou DIPEA: diisopropiletilamina ou base de Hunig; NMP: /V-metilpirrolidinona; Reagents BOP: hexafluorofosfato de benzotriazol-1iloxitris(trimetílamino)fosfônio; DGG: 1,3-dÍcicloexilcart)odiimída; EDGI: cloridrato de 1-(dimetilaminopropil)-3-etilcarbodiimida; TA ou ta: temperatura ambiente; tR: tempo de retenção; h: hora(s); min: minuto(s); PyBroP: hexafluorofosfato de bromotripirrolidinofosfônio; TBTU: tetrafluoroborato de O(1 H-benzotriazol-1 A/’,A/-tetrametilurônio; DMAP: 4-M,A/-dimetilaminopiridina; HOBt ou HOBT: hidroxibenzotriazol; Na(OAc)3BH: triacetoxiboroidreto de sódio; HOAc: ácido acético; TFA: ácido trifluoroacético; LiHMDS: bis(trimetilsilil)amida de lítio; DMSO: sulfóxido de dimetila; MeGN: acetonitrila; MeOH: metanol; EtOAc: acetato de etila; DMF: dimetil formamida; THF: tetraidrofurano; DCE: 1,2-dictoroetano; Et2O: éter de dietila; DCM: diclorometano ou cloreto de metileno; m-GPBA: ácido de 4-cloroperoxibenzóico.
Exemplo 1


âA(4-Fluorofenil)-M-(4-(piridin-4-ilóxi)feníl)malonamida.
NH2


A) 4-(4-Aminofenóxi)piridina
Uma solução de cloridrato de 4-cloropiridina (Aldrich, 3,0 g, 20,0 mmoles) em sulfoxide de dimetila (40 mL) foi tratada com 4-aminofenol (Aldrich, 2,1 g, 20,0 mmoles) e péletes de hidróxido de sódio (2,0 g, 50,0 mmoles) e a mistura foi aquecida a 100°C durante 18 horas. A mistura foi resfriada para a temperatura ambiente, despejada sobre uma mistura de águaágua (300 g) e extraída com Et2O (3 x 150 mL). Os extratos combinados foram lavados com salmoura, secados (MgSQ4) e concentrados para fornecer o 4-(4-aminofenóxi)aniltna como um sólido amarelo pálido (3,5 g, 94%). 1H RMN (DMSO-d6) δ 8,38 (dd, 2H, J = 5,5,1,5 Hz), 6,83 - 6,79 (m, 4H), 6,63 - 6,59 (m, 2H), 5,13 (br s, 2H); MS (ESI*) m/z 187,2 (M + H)*.
G O

F
B) Ácido 3-(4-fluorofenilamino)-3-oxopropanóico.
A uma solução de 3-cloro-3-oxopropanoato de etila (Aldrich, 5,0 mL, 40 mmoles) em cloreto de metileno (100 mL) a 0°C foi adicionado diisopropiletilamina (8,4 mL, 48 mmoles) seguido por 4-fluoroanilina (Aldrich, 3,6 mL, 38 mmoles). A mistura de reação foi agitada em temperatura ambiente durante a noite e foi então saciada com 100 mL de solução de NaHCO3 saturada. A camada aquosa foi extraída com clorofórmio (3 x 100 mL). Os extratos orgânicos combinados foram secados em Ma2SO4 anidro e concentrados em vácuo para produzir o produto bruto como um óleo amarelo que solidificou-se em repouso (10 g). 1H RMN (CDCI3) δ 9,30 (br s, 1H), 7,55 (m, 2H), 7,05 (t, 2H, J = 8,8 Hz), 4,28 (q, 2H, J = 7,2 Hz), 3,49 (s, 2H), 1, 35 (t, 3H, J - 7,1 Hz); MS (ESI*) m/z 226,11 (M + Η)*. O éster acima foi dissolvido em 100 mL de etanol e resfriado a 0 °C. 1 N de solução de NaOH aquosa (100 mL) foi adicionada e a reação foi agitada a 0 °C durante 1 hora. A reação foi concentrada em vácuo para remover o etanol. A solução aquosa foi extraída com EtOAc (50 mL) e foi em seguida tornada acídica com 1N de solução de HCl aquosa. A solução aquosa foi extraída com EtOAc (5 x 100 mL). Os extratos orgânicos combinados foram secados em Na2SO4 anidro e concentrados em vácuo para produzir o produto bruto como um sólido amarelo (6,31 g, 84%) que foi empregado sem outra purificação. Ή RMN (DMSO-dâ) δ 12,9 (br s, 1H), 10,3 (br s. 1H), 7,59 (m, 2H), 7,16 (t, 2H, J = 8,9 Hz), 3,34 (s, 2H); MS(ESf) m/z 198,43 (M+ H)\
C) N-(4-Fluorofenil)-/V-(4-(piridin-4-ilóxi)fenil)malonamida
Uma solução de 4-(4-aminofenóxi)piridína (93 mg, 0,50 mmol) em DMF foi tratada com ácido 3-(4-fluorofenilamino)-3-oxopropanóico (99 mg , 0,50 mmol), DIPEA (113 μΙ_, 0,65 mmol) e TBTU (209 mg, 0,65 mmol) e a mistura foi agitada em temperatura ambiente durante 2 horas. A mistura foi concentrada para remover o DMF e o resíduo dividido entre EtOAc e so lução de bicarbonate de sódio saturada. A fase EtOAc foi lavada com solução de bicarbonate de sódio saturada, salmoura, secada (MgSO4) e concentrados para fornecer o composto do título como uma espuma não totalmente branca (140 mg, 76%). Ή RMN (DMSO-d6) Õ 10,30 (s, 1H), 10,24 (s, 1H),
8,43 (dd, 2H , J = 5,5, 1,5 Hz), 7,70 (d, 2H, J = 9,1 Hz), 7,63-7,60 (m, 2H), 7,17-7,14 (m, 4H), 6,89 (dd, 2H, J = 5,5, 1,5 Hz), 3,46 (s, 2H); MS(ESF) m/z 365,0 (M + H)+.
Exemplo 2



ia


A) 4-Cloro-6-(2-flúor-4-nitrofenóxi)pirimidina
Uma mistura de 4,6-dicloropÍrimidina (Aldrich, 0,74 g, 5,0 mmoles), 2-flúor-4-nitrofenol (Aldrich, 0,79 g, 5,0 mmoles) e DMF (10 ml) foi tratada com carbonato de potássio (0,72 g, 5,2 mmoles) e aquecida a 80°C durante 3 horas. A mistura foi resfriada, diluída com água e extraída com acetato de etila. O acetato de etila extraído foi lavado com salmoura, secado (MgSO4), e concentrado para fornecer o produto bruto como um sólido amarelo. O produto bruto foi triturado com éter de isopropila para fornecer 4cloro-6-(2-flúor-4-nitrofenòxi)pirimidina (1,3 g, 94%). 1H RMN (DMSO-d6) 8 8,80 (s, 1H), 8,51 (dd, 1H, J = 8,6, 2,5 Hz), 8,31 (d, 1H, J = 9,1 Hz), 7,87 (d, 1H, J = 9,1 Hz), 7,84 (s, 1H).


α
B) 4-Cloro-6-(2-aminO’2-fluorofenóxi)plrimldina
Uma solução de 4-cloro-6-(2-flúor-4-nitrofenóxi)pirimidina (1,3 g, 4,8 mmoles) em metanol (120 mL) foi tratada com níquel Raney (1,5 g, suspensão aquosa) e a mistura de reação foi agitada sob uma manta de hidrogênio (de balão de látex) em temperatura ambiente durante 3 horas. O catalisador foi filtrado, o filtrado concentrado, e o resíduo dividido entre CH2CI2 e água. A fase de cloreto de metileno foi separada, secada (MgSO4) e concentrada. O produto bruto foi purificado por cromatografía instantânea em síiica-gel empregando-se 1- 2% MeOH em CH2CI2 como o eluente para fornecer 4cloro-6-(2-amino-2-fíuorofenóxi)pirirnidina como um sólido branco (600 mg, 52%). Ή RMN (DMSO-dg) 5 8,64 (s, 1H), 7,39 (s, 1H), 6,97 (dd, 1H, J = 8,8, 8,8 Hz), 6,46 (dd, 1H, J = 13,1, 2,5 Hz), 6,38 (dd, 1H, 8,6, 2,5 Hz), 5,44 (br s, 2H); MS(ESI*)m/z 240,04 (M + H)\
¢) 1-(4-(6-CloroplrlmMlr»-4-ilóxi)-3-fluorofenil)-3’(2-(4fluorofenil)acetil)tiouréia
Cloreto de 4-fluorofenilacetila (Lancaster, 0,52 g, 3,0 mmoles) foi adicionado a uma mistura de NaSCN (0,27 g, 3,3 mmoles) e EtOAc (12 mL) e a mistura resultante agitada em temperatura ambiente durante 30 minutos. Esta mistura foi adicionada a uma solução de 4-cloro-6-(2-amino-2-fIuorofenóxi) pirimidina em 1:1 EtOAc/CH2CI2 (5 ml) e a mistura resultante agitada em temperatura ambiente durante a noite. A mistura foi concentrada e o re síduo dividido entre EtOAc/ H2Q. A fase EtOAc foi separada, lavada com salmoura, secada (MgSOJ e concentrada. O produto bruto foi purificado por cromatografia instantânea empregando-se 10 - 35% de EtOAc / hexanos como o eluente para fornecer o composto do título como um sólido cristalino amarelo (0,85 g, 65%). Ή RMN (DMSO-c^ 8 12,40 (s, 1H), 11,81 (s, 1H), 8,67 (s, 1H), 7,90 (dd, 1H, J = 12,1, 2,0 Hz), 7,62 (s, 1«), 7,47-7,41 (m, 2H), 7,38-7,35 (m, 2H), 7,17 (t, 2H, J 8,8 Hz), 3,81 (s, 2H); MS(ESF) m/z 434,98 (M + H)*.
Exemplo3

1-(2-(4-Fluorofenil)acetil)-3-(4-(piridin-4-ilóxi)fenil)tiouréla.
O composto do título foi preparado empregando-se 4-(4aminofenóxQpiridina (Composto A de Exemplo 1) e um procedimento similar delineado para a preparação de Composto C de Exemplo 2, Produção: 10%. Ή RMN (CDCI3) 8 12,3 (s, 1H), 8,63 (s, 1H), 8,49, (d, 2H, J = 6,2 Hz), 7,71 (d, 2H, J = 8,9 Hz), 7,31-7,27 (m, 2H), 7,14-7,09 (m, 4H), 6,90 (dd, 2H, J = 4,8,1,4 Hz), 3,73 (s, 2H); MS(ESI*) m/z 382,2 (M + H)*.
Exemplo 4


^í-(4-(6-Cloropirimidin-4-iléxl)-3-fluorofenil)-/V’-(4-fluorofenil) malonamida
Uma solução de 4-cloro-6-(2-amino-2-fluorofenóxi)pirimidina (29 mg, 0,12 mmol, Composto B de Exemplo 2), ácido 3-(4-fluorofenilamino>3-oxopropanóico (26 mg, 0,13 mmol, Composto B de Exemplo 1) em DMF (1,5 mL) foi tratada com DIPEA (24 μΙ, 0,14 mmol) e TBTU (46 mg,
0,14 mmol). A mistura de reação foi agitada em temperatura ambiente durante a noite, diluída com EtOAc (25 mL), e a fase orgânica foi lavada com salmoura (3 x 20 mL), secada (MgSO4) e concentrada. O produto bruto foi purificado por cromatografia instantânea empregando-se 1-3% MeOH em CH2CI2 como o eluente para fornecer o composto do título como um sólido branco (35 mg, 78%). 1H RMN (DMSO-<) δ 10,49 (s, 1H), 10,25 (s, 1H), 8,65 (s, 1H), 7,78 (d, 1H, J = 12,1 Hz), 7,63 - 7,57 (m, 3H), 7,40 - 7,34 (m, 2H), 7,16 (t, 2H, J-8,8 Hz), 3,48 (s, 2H); MS (ESI*) m/z 419,21 (M + H)*.
Exemplo 5


â/L(3-flóor-4-(6-(metitamino)pirimidln-4-ilóxi)fenll>WL(4-flu©rofcnil) ma· lonamida
Uma solução de /Vf-(4-(6-cloropirimidin-4-ilóxi)-3-fluorofeníl)-/V3(4-fluorofenil)malonamida (100 mg, 0,42 mmol, Exemplo 4) em n-BuOH (3 mL) foi tratada com 2 M metílamina/THF (0,2 mL) e aquecida em um frasconete de tampa de rosca 80°C durante 12 horas. A mistura foi concentrada e o resíduo purificado por HPLC preparativa empregando-se um gradiente de MeOH-H2O contendo 0,1% TFA. A fração contendo o produto foi liofilizado para fornecer o composto do título como um sólido amarelo pálido (60 mg, 34%). 1H RMN (DMSO-cg δ 10,58 (s, 1H), 10,36 (br s, 2H), 8,19 (br s, 1H), 7,76 (d, 1H, J - 12,1 Hz), 7,64-7,62 (m, 2H), 7,36-7,27 (m, 2H), 7,15 (dd, 2H, J = 8,8, 8,8 Hz), 5,95 (br s, 1H), 3,49 (s, 2H), 2,80 (s, 3H); MS(ESI*) m/z 414,16 (M + H)*.
Exemplo 6

F
H
6-(2-flúor-4-(3-(4-fluorofenilamino)-3-oxopropanamic!o)fenóxi) pirimidin4-ilcarbamato de terc-butila.

A) W-(2,4,e<rtmetexlbenzil)-e-cteroplrimidin-4-amina
A mistura de 4,6-dicloropirimidina (Aldrich, 1,48 g, 10,0 mmoles), cloridrato de 2,4,6-trimetoxibenzilamina (2,33 g, 10,0 mmoles), DIPEA (4,8 mL, 27,7 mmoles), e n-BuOH (50 mL) foi aquecida a 100°C durante 2 horas.
A mistura foi resfriada, diluída com água (200 mL) e o produto precipitado foi coletado por filtração a vácuo em um funil Büchner. O produto foi lavado com água fria, éter, e secada a vácuo para fornecer um sólido esbranquiça10 do (2,8 g, 90%). 1H RMN (DMSO-ds) S 8,28 (s, 1H), 7,38 (s, 1H), 6,49 (s,
1H), 6,25 (s, 2H), 4,34 (s, 2H), 3,77 (s, 9H).
oAz rí^N h2n^n^
B) 6-(2-flúor-4-mtrofenóxi)pirimidin-4-amina
A mistura de /V-(2,4,6-trimetoxibenzil)-6-ctoropirimidin-4-amina (2,2 g, 7,11 mmoles), 2-flúor-4-nitrofenol (1,1 g, 7,0 mmoles), e 2-metoxietil 15 éter (50 mL) foi aquecida a 160°C durante 60 horas. A mistura de reação foi resfriada em temperatura ambiente e despejada em H2O (200 mL). O sólido foi coletado, lavado com 2: M de Na2CO3 e H2O, aquoso e em seguida, secado a vácuo em um funil Büchner. O produto bruto foi tratado com TFA (20 mL) em dioxano (40 mL) e agitado em temperatura ambiente durante 4 ho20 ras. A mistura de reação foi concentrada, o resíduo dividido entre EtOAc e solução de NaHCO3 saturada. A fase EtOAc foi separada, secada (MgSO4), concentrada e o produto bruto purificado por cromatografia instantânea empregando-se 1-2% de MeOH em CH2GIZ como o eluente para fornecer 6-(2flúor-4-nitrofenóxi)pirimidin-4-amina (440 mg, 31%). 1H RMN (DMSO-de) δ
8,31 (dd, 1H, J = 10,4, 2,5 Hz), 8,14 (dd, 1H, J = 9,8, 2,0 Hz), 8,04 (s, 1H),
7,61 (dd, 1H, J = 8,3, 8,3 Hz), 7,07 (s, 2H), 6,02 (s, 1H); MS(ESi*) m/z 251, 15 (M + Hf.
C) 6-(2-flúor-4-nitrofenóxi)pirimidin-4-ilcart>amato de tercbutlla.
Uma mistura de 6-(2-flúor-4-nitrofenóxi)pirimidin-4-amina (439 mg, 1,2 mmol), BOC2O (261 mg, 1,2 mmol), DMAP (10 mg), e THF (10 mL) foi agitada em temperatura ambiente durante 1 hora em seguida concentrada em vácuo para fornecer o produto bruto. O produto bruto foi purificado por cromatografia instantânea empregando-se 1-2% MeOH em CH2CI2 como o eluente para fornecer 6-(2-flúor-4-nitrofenóxi)pirimidin-4-ilcart)amato de terc-butila como um sólido branco (110 mg, 26%). 1H RMN (DMSO-de) δ 10,59 (s, 1H), 8,39 (dd, 1H, J = 8,8, 1,1 Hz), 8,32 (dd, 1H, J = 10,3,2,4 Hz), 8,15 (ddd, 1H, J = 9,1, 2,5, 1,0 Hz), 7,67 (dd, 1H, J = 8,8, 8,8 Hz), 7,45 (s, 1H), 1,44 (s, 9H); MS(ESI) mlz 349,08 (Μ - H).

D) 6-(4-amino-2-fluorofenóxi)pirimidin-4-ilcarbamato de terc-butiia.
Uma solução de 6-(2-flúor-4-nitrofenóxi)pirimidin-4-ilcarbamato de terc-butila (11 mg, 0,031 mmol) em MeOH (2 mL) foi tratada com PtO2 e a mistura de reação foi agitada sob uma manta de hidrogênio (de balão de látex) durante 2 horas. O catalisador foi filtrado e o filtrado concentrado para fornecer 6-(4-amino-2-fluorofenóxi)pirimidin-4-ilcarbamato de terc-butila (8 mg, 81%). ’H RMN (DMSO-cy δ 10,62 (s, 1H), 8,43 (d, 1H, J - 2,5 Hz), 8,36 (dd, 1H, J = 9,8, 2,5 Hz), 8,36 (dd, 1H, J = 9,8, 2,5 Hz), 8,20-8,17 (m, 1H), 7,71 (dd, 1H, J = 8,8, 8,8 Hz), 7,49 (s, 1H), 1,48 (s, 9H).
E) 6-(2-flúor-4-(3-(4-fluorofenilamino)-3-oxopropanamido) fenóxi)pirimidin-4-ilcarbamato de terc-butila.
O composto do título foi preparado de 6-(4-amino-2-fluorofenóxi) pirimidin-4-ilcarbamato de terc-butila (8 mg, 0,025 mmol) e ácido (4-fluorofenilamino)-3-oxopropanóico (6 mg, 0,031 mmol, Composto B de Exemplo 1), TBTU (11 mg, 0,034 mmol), e DIPEA (6 pL, 0,030 mmol) empregando-se um procedimento similar descrito para a preparação de Composto C, Exemplo 1, Cromatografia instantânea empregando-se 1-1, 5% de MeOH em CH2CI2 como o eiuente fornece o composto do título como um sólido branco (10 mg, 80%). 1H RMN (DMSO-cg δ 10,48 (s, 1H), 10,46 (s, 1H), 10,25 (s, 1H), 8,39 (s, 1H), 7,74 - 7,17 (m, 1H), 7,62 (d, 1H, J = 5,5 Hz), 7,61 (d, 1H, J = 4,9 Hz), 7,36 - 7,30 (m, 2H), 7,17 - 7,13 (m, 2H), 3,48 (s, 2H), 1,46 (s, 9H); MS(ESE) m/z 500,12 (M + H)*.
Exemplo 7

ch3
6-(2-flúor-4-(3-(4-fluorofenilamino)-3-oxopropanamWo)fenóxi) pirimtdin-4-il(metil)carbamato de terc-butila.
no2


ch3
A) 6-(2-flúor-4-nitrofen6xi)pirimidin-4-ll(metil)carbamato de terc-butila.
Uma solução de 6-(2-flúor-4-nitrofenóxi)pirimidin-4-ilcarbamato de terc-butila (Composto C de Exemplo 6, 44 mg, 0,13 mmol) em DMF ani dro (1 mL) foi resfriada em um banho de gelo e tratada com 60% de NaH (44 mg, 0,16 mmol) e agitada na mesma temperatura durante 30 minutos. A mistura de reação foi tratada com iodometano (10 pL, 0,15 mmol) e agitada a 0-5°C durante 10 minutos. A mistura de reação foi deixada aquecer para temperatura ambiente e agitada durante 30 minutos. A mistura foi diluída com H2O (10 mL) e extraída com EtOAc (2x10 mL). Os extratos combinados foram secados (MgSO4) e concentrados em vácuo para fornecer o produto (35 mg, 74%) como um sólido amarelo pálido. Ή RMN (DMSO-d6) δ 8,57 (d, 1H, J = 1,1 Hz), 8,37 (dd, 1H, J = 9,8, 3,0 Hz), 8,19 (ddd, 1H, J = 9,1, 2,5, 1,0 Hz), 7,71 (dd, 1H, J = 8,8, 8,8 Hz), 7,66 (s, 1H), 3,38 (s, 3H),
1,51 (s, 9H)

de íerc-butila
Uma mistura de 6-(2-flúor-4-nitrofenóxi)pirimidin-4-il(metil)carbamato de terc-butila em 1:1 EtOH/MeOH (2 mL) foi tratada com PtO2 (10 mg) e a mistura de reação foi agitada sob uma manta de H2 (de balão de látex) durante 2 horas. A mistura de reação foi filtrada e concentrada para fornecer o produto desejado (30 mg, 75%) como um sólido marrom claro. MS(ESf) m/z 365,13 (Μ + ΗΓ.
C) 6-(2-flúor-4-(3-(4-fluorofenilamino)-3-oxopropanamido) fenóxi)pirimidín-4-il(metil)carbamato de terobutila.
O composto do título foi preparado de 6-(4-amino-2-fluorofenóxi) pirimidin-4-il(metil)carbamato de terc-butila (30 mg, 0,068 mmol), cloreto de 4-fluorofenilacetila (Lancaster, 15 mg, 0,088 mmol) e NaSCN (9 mg, 0,11 mmol) em EtOAc/ CH2CI2 empregando-se um procedimento similar descrito para a preparação de Composto C de Exemplo 2, Cromatografia instantânea em SiO2 empregando-se 1 - 40% de EtOAc em hexanos como o eluente fornece o composto do título (30 mg, 83%) como um sólido branco. 1H RMN (DMSO-cQ δ 12,40 (s, 1H), 11,79 (s, 1H), 8,55 (s, 1H), 7,86 (dd, 1H, J = 12,1, 2,5 Hz), 7,54 (d, 1H, J = 1, 1 Hz), 7,45 - 7,35 (m, 4H), 7,20 - 7,14 (ddd, 2H, J = 8,8, 8,8, 2,1 Hz). 3,81 (s, 2H), 3,36 (s, 3H), 1,49 (s, 9H); MS(ESI+) m/z 530,09 (M + H)+.
Exemplo 8

1-(3-flúor-4-(6-(metilamino)pirimidin-4-ilóxi)fenil)-3-(2-(4fluorofenil)acetil)tiouréia.
6-(2-flúor-4-(3-(4-fluorofenilamino)-3-oxopropanamido)fenóxi) pirimidin-4-il(metil)carbamato de terc-butila (Exemplo 7, 25 mg, 0,047 mmol) foi tratado com 4 M de HCI em 1,4-dioxano (3 mL), agitado em temperatura ambiente durante 4 horas e concentrado em vácuo. O resíduo foi dividido entre solução de NaHCO3 aquosa saturada e EtOAc. A fase EtOAc foi separada, secada (MgSOJ e concentrada em vácuo. Cromatografia instantânea em SiO2 empregando-se 1 % de MeOH em CH2CI2 como o eluente fornece o composto do título (10 mg, 50%) como um sólido branco. 1H RMN (DMSOd6) S 12,37 (s, 1H), 11,77 (s, 1H), 8,09 (s, 1H), 7,82 (d, 1H, J = 11,6 Hz), 7,41-7,35 (m, 5H), 7,32-7,28 (m, 1H), 7,19-7,14 (m, 2H), 3,81 (s, 2H), 2,78 (s, 3H); MS(ESE) m/z 430,07 (M + H)\
Exemplo 9
Η H


1-(4-(6-Aminopirlmldin-4-llóxi)-3-fluorofenil)-3-(2-(4-fluorofe- nil) acetii) tiouréia

A) 6-(W,^-di-terc-butiloxicarbonil)amino-4-(2-flúor-4-nitro- fenóxi)pirimidina.
Uma mistura de 6-(2-flúor-4-nitrofenóxi)pirimidÍn-4-amina (Composto B de Exemplo 6, 150 mg, 0,60 mmol), BOC2O (275 mg, 1,26 mmol), 5 DMAP (5 mg), e THF (20 mL) foi agitada em temperatura ambiente durante
2,5 horas. A mistura de reação foi concentrada em vácuo para fornecer o produto bruto. A cromatografia instantânea em SiO2 empregando-se 5 -15% de EtOAc em hexanos como o eluente fornece o composto do título (180 mg, 67%) como um sólido branco. 1H RMN (DMSO-<) δ 8,62 (d, 1H, J = 1,0
Hz), 8,42-8,39 (m, 1H), 8,21 (ddd, 1H, J = 9,1, 2,5, 1,0 Hz), 7,77 (dd, 1H, J =
8,8, 8,8 Hz), 7,49 (d, 1H, J = 1,1 Hz), 1,49 (s, 18H); MS(ESI*) m/z 451,12 (M + ΗΓ

B) 6-(/V,/V-di-terc-Butiloxlcarbonil)amino-4-(4-amino-2fluorofenóxijpirimidina.
A mistura do 6-(MWdi-terG-butiloxicatoonila)amino-4-(2-flúor-4nitrofenóxi)pirimidína (175 mg, 0,38 mmol) em tolueno (5 mL) e MeOH (3 mL) foi tratada com PtO2 (35 mg) e a mistura de reação foi agitada sob uma manta de H2 (de balão de látex) durante 15 horas. O catalisador foi filtrado, © filtrado concentrado em vácuo e o resíduo foi purificado por cromatografia instantânea em SiO2 empregando-se 1-10% de MeOH em CH2CI2 como o eluente para fornecer o produto (110 mg, 68%) como um sólido branco. 1H
RMN (DMSO-O δ 8,56 (s, 1H), 7,17 (s, 1H), 6,97 (dd, 1H, /= 8,8, 8,8 Hz), 6,46 (dd, 1H, J = 12,6, 2,5 Hz), 6,38 (dd, 1H, J = 8,8, 2,5 Hz), 5,40 (s, 2H),
1,89 (s, 18H).
C) 1 -(4-« AFdi-tenc-butiloxicarbonila 6-aminopirimidin-4-ilóxi)3-fluorofenil>3-(2-(4*fluorofenil)acetil)tiouréia.
O composto do título foi preparado de 6-(MA/-di-ten>butiloxicarbonil)amíno-4-(4-amino-2-fluorofenóxi)pirimidina (20 mg, 0,048 mmol), cloreto de 4-fluorofenilacetila (Lancaster, 10 mg, 0,062 mmol), e NaSCN (95 mg, 0,062 mmol) em EtOAc / CH2CI2 empregando-se um procedimento similar descrito para a preparação de Composto G de Exemplo 2, cromatografia instantânea em SiO2 empregando-se 10-20% EtOAc em hexanos como o eluente fornece o composto do título (23 mg, 77%) como um sólido branco. 1H RMN (DMSO-dg) δ 12,40 (s, 1H), 11,79 (s, 1H), 8,59 (s, 1H), 7,88 (dd, 1H, J = 12,5, 1, 8 Hz), 7,46 - 7,41 (m, 2H), 7,38 - 7,35 (m, 3H), 7,17 (dd, 2H, J = 8,8, 8,8 Hz), 3,81 (s, 2H), 1,47 (s, 18H); MS(ESF) m/z 616,12 (M + H)*.
D) 1-(4-(6-Aminopirimidin-4-ilóxO-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)tiouréia.
A mistura de 1-(4-( M/V-di-terc-butilcarboníla 6-aminopirimidin-4ilóxi)-3-fluorofeni!)-3-(2-(4-fluorofenil)acetil)tiouréia (18 mg, 0,029 mmol) e 4 M de HCI em dioxano (1,5 mL) foi agitada em temperatura ambiente durante 18 horas e em seguida concentrados para fornecer o produto bruto. O produto bruto foi dividido entre EtOAc e solução de NaHCO3 aquosa -saturada. A fase de EtOAc separada, secada (MgSO„), concentrada em vácuo e o resíduo foi purificado por cromatografia instantânea em SÍO2 empregando-se 1- 2% de MeOH em CH2CI2 para fornecer o composto do título (12 mg, 99%) como um sólido branco. Ή RMN (DMSO-d6) δ 12,38 (s, 1H), 11,77 (s, 1H), 8,05 (s, 1H), 7,83 (dd, 1H, J = 12,3, 1,8 Hz), 7,41 - 7,35 (m, 3H), 7,31 (dd, 2H, J = 8,8, 8,8 Hz), 7,17 (t, 1H, J = 8,8 Hz), 7,02 (s, 2H), 5,89 (s, 1H), 3,81 (s, 2H); MS(ESI*) m/z 416,06 (M + H)*.
Exemplo 10

^-1^4-(6-Aminoptrimidin-4-ilóxi)-3-fluorofenil)-^-(4-fluorofenil)malonamida
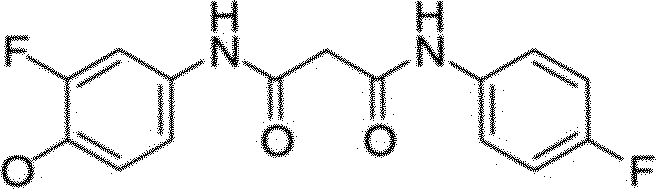

I
Bog
A) /^-(A-CM^-di-terc-Butilcarbonil 6-aminopirimiclir»-4-ilóxi)3-fluorofenil)-NJ-(4-fluorofenil)malonamida.
O composto do título foi preparado de uma mistura de 6-(A/,N-diterc-butiloxicarbonil)amino-4-(4-amino-2-fluorofenóxi)pirÍmidina (Composto B de Exemplo 9, 20 mg, 0,048 mmol), 3-(4-fluorofenilamino)-3-oxopropanóíco (Composto B de Exemplo 1, 14 mg, 0,072 mmol), DIPEA (12 pL, 0,069 mmol) em DMF empregando-se um procedimento similar descrito para a preparação de Composto C de Exemplo 1, Cromatografia instantânea em SiO2 empregando-se 15-50% EtOAc em hexanos como o eluente fornece o composto do título (23 mg, 80%) como um sólido branco. Ή RMN (DMSOδ 10,47 (s, 1H), 10,24 (s, 1H), 8,58 (d, 1H, J = 1 Hz), 7,77 (dd, 1H, J = 12,9, 1,8 Hz), 7,63-7,60 (m, 2H), 7,37-7,36 (m, 2H), 7,32 (s, 1H), 7,15 (t, 2H, J = 8,8 Hz), 1,47 (s, 18H); MS(ESI+) m/z 600,17 (M + Hf.
B) N,-(4-(6-Amínopirimidin-4-ilóxi)-3-fluorofenil)-/V3-(4-fluorofenil)malonamida
O composto do título foi preparado de /V-l^-CM/V-di-terc-butilcarbonil 6-aminopÍrimidin-4-ilóxi)-3-fluorofenil)-/V-3-(4-fluorofenil)malonamida (20 mg, 0,032 mmol) empregando-se um procedimento similar descrito para a preparação de Composto D de Exemplo 9 para fornecer o composto do título (13 mg, 98%) como um sólido branco. 1H RMN (DMSO-cy δ 10,42 (s, 1H), 10,24 (s, 1H), 8,03 (s, 1H), 7,73 (d, 1H, d = 11,0 Hz), 7,61 (dd, 2H, J = 0,2, 4,9 Hz), 7,33 - 7,30 (m, 1H), 7,28 - 7,23 (m, 1H), 7,18 - 7,13 (m, 2H),
Exemploll

1-(4-(6-(4-Metoxibenzilamino)pirimidin-4-ilóxí)’3-fluorofenil)-
3-(2(4-fluorofenil)acetil)uréia.

A) /V-(4-Metoxibenzil)-6-cloropirimidin-4-amina
A mistura de 4,6-dicloropirimidina (Aldrich, 3,6 g, 24,2 mmoles), 4-metoxibenzilamina (2,7 g, 19,7 mmoles), DIPEA (5 ml, 28,8 mmoles), e nBuOH foi aquecida até o refluxo durante 3 horas. A mistura foi concentrada separada, lavada com solução de NaHCO3 aquosa saturada e salmoura, secada (MgSO4), e concentrados para fornecer o composto do título (4,5 g) que foi empregado sem outra purificação. Ή RMN (DMSO-d§) δ 8,29 (s, 1H), ^3,1 3 (txr , 1II), ,12I5 (, , 8 , IΊ :£), ,^^^3 (d, 12H, ,12 111^»), f3, füfS (,

B)A/-(4-Metoxibenzil)-6-(2-flúor’4-nitrofenóxi)pirimidin-4-amina
Uma mistura de N-(4-metoxibenzil)-6-cloropirimidin-4-amina (2,3 g
9,2 mmoles), 2-fiúor-4-nitrofenol (1,45 g, 9,2 mmoles), DIPEA (15 mL), e éter de 2-metoxietila (75 mL) foi aquecida a 160°C em um fiasco de pressão selado durante 50 horas. A mistura foi resfriada, despejada em gelo esmagado (200 g), tratada com EtOAc (200 mL). Após agitar vigorosamente durante 10 minutos, o material insolúvel foi filtrado. A fase EtOAc foi lavada com de solução de Na2CO3 aquosa saturada (100 mL), salmoura (3 x 100 mL), secada (MgSOJ e concentrada em vácuo. O sólido gomoso obtido foi triturado com éter de isopropila para fornecer o composto do título (1,75 g, 76%) como um sólido marrom. 1H RMN (DMSO-<)f 8,32 (dd, 1H, J = 10,2,2,0 Hz), 8,15 (d, 2H, J = 9,2 Hz), 8,05 (br s, 1H), 7,61 (dd, 1H, J = 8,4, 8,4 Hz), 7,26 (d, 2H, J = 8,5 Hz), 6,91 (d, 2H, J = 8,5 Hz), 6,14 (br s, 1H), 4,48 (br s, 2H), 3,74 (s, 3H).

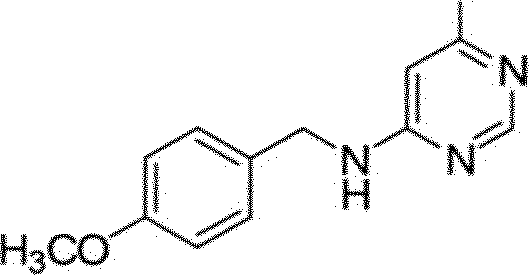
C) M(4-Metoxibenzil)-6-(4-amino-2-fluorofenóxi)pirimiciin-4amina.
Uma solução de AI-(4-metoxibenzil)-6-(2-flúor-4-nitrofenóxi) pirimidin-4-amina (150 mg, 0,41 mmol) em 1:1 MeOH /THF (20 mL) foi tratada com cloreto de amônio (0,22 g, 4,1 mmoles), e pó de zinco (< 20 microns, 0,27 g, 4,2 mmoles). A mistura de reação foi agitada em temperatura ambiente durante 1 hora. Uma porção adicional de pó de zinco (150 mg) foi adicionada a uma mistura e a mistura de reação foi agitada em temperatura ambiente durante 1 hora e aquecida a 70°C durante 20 minutos. A mistura foi filtrada para remover os sólidos inorgânicos, concentrada em vácuo, e o resíduo dividido entre EtOAc e salmoura. A fase EtOAc foi separada, lavada com salmoura, secada (MgSOJ e concentrada em vácuo para fornecer o composto do «tufo (145 mg, 99%). Ή RMN (DMSO-<^) δ 8,10 (br s, 1H), 7,76 (br s, 1H), 7,21 (d, 2H, J = 8,6 Hz), 6,88 (d, 3H. J = 8,6 Hz), 6,44 (dd, 1H, J= 12,7, 2,0 Hz), 6,36 (dd, 1H, J = 8.3, 2,3 Hz), 5,79 (s, 1H), 4,41 (br s,
F
2H), 3,73 (s, 3H); MS(ESP) m/z 341,18 (M + H)*.
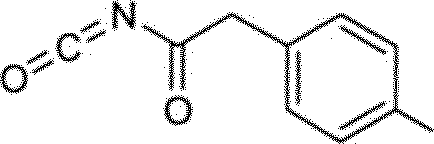
D) isocianato de 2-(4-Fluorofenil)acetila.
Cianato de prata (0,912 g, 6,08 mmoles, 1,05 equivalente) foi adicionado a uma solução cloreto de 4-fluorofenilacetila (Lancaster, 0,794 ml, 5,79 mmoles, 1,0 eq) em tolueno (16 ml) em temperatura ambiente. A mistura de reação foi protegida da luz e aquecida até o refluxo. Apôs 60 minutos, a mistura foi resfriada para a temperatura ambiente e filtrada (Acrodisc, PTFE 0,2 μΜ) para fornecer uma solução de 0,36 M de isocianato de
2-(4-fluorofenil)acetila em tolueno, que foi empregado sem outra purificação.
E) 1-(4-(6-(4-Metoxibenzilamino)pirimidin-4-ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia.
Uma solução de A/-(4-metoxíbenzil)-6-(4-amino-2-fluorofenóxi) pirimidin-4-amina (88 mg, 0,26 mmol) em THF (2 mL) foi tratada com 0,36 M de isocianato de 2-(4-fluorofenil)acetila em tolueno (0,72 mL, 0,26 mmol) e a mistura de reação foi agitada em temperatura ambiente durante 1 hora. A mistura foi concentrada em vácuo e o sólido obtido foi triturado com éter de isopropila para fornecer o composto do título (125 mg, 93%) como um sólido esbranquiçado. Ή RMN (DMSO-O δ 11,00 (s, 1H), 10,50 (s, 1H), 8,09 (s, 1H), 7,85 (br s, 0,5 H), 7,66 (d, 1H, J = 12,6 Hz), 7,36-7,15 (m, 9,5 H), 6,88 (d, 1H, J = 8,1 Hz), 5,91 (s, 1H), 4,42 (br s, 2H), 3,72 (s, 2H), 3,71 (s, 3H); MS(ESI*) m/z 520,14 (M + H)*.
ExemploH
N1-(4-(6-(4-Metoxibenzilamino)pirimldin-4-ilóxi)-3-fluorofenil>· /Ví-(4-fluorofenll)malonamída.
O composto do título foi preparado de A/-(4-metoxibenzil)-6-(4amino-2-fluorofenóxi)pirimiciin-4-amina (Composto C de Exemplo 11, 200 mg, 0,59 mmol), ácido 3-(4-fluorofenilamino)-3-oxopropanóico (Composto B de Exemplo 1, 128 mg, 0,65 mmol), TBTU (228 mg, 0,71 mmol), e DIPEA (123 pL, 0,71 mmol) em DMF empregando-se um procedimento similar descrito para a preparação de Composto C de Exemplo 1. O produto bruto foi purificado por trituração com éter de isopropila para fornecer o composto do titulo (250 mg, 82%) como um sólido esbranquiçado. Ή RMN (DMSO-cQ δ 10,43 (s, 1H), 10,25 (s, 1H), 8,09 (s, 1H), 7,85 (s, 1H), 7,72 (dd, 1H, /= 9,1, 5,0 Hz), 7,62 (dd, 2H, J = 5,0 Hz), 7,31 - 7,21 (m, 4H), 7,15 (dd, 2H, J = 8,8, 8,8
Hz), 6,88 (m, 2H), 5,90 (s, 1H), 4,42 (br s, 2H), 3,71 (s, 3H), 3,46 (s, 2H); MS(ESF) m/z 520,14 (M + H)*.
Exemplo 13
Η2Ν^Ι<
1-(4-(6-Amlnopirimidin-4«ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenll) acetil)uréia.
A) /V-(3-flúor-4-hidroxifenil)acetamlda
O composto do titulo foi preparado do 2-fiüor-4-nitrofenol comercialmente disponível de acordo com o procedimento de Burckhalter, J. H. e outro, (/ Am. Chem. Soe. 1948, 70,1363). Nitrofenol (5,73 g, 36,5 mmoles) e anidrido acético (3,72 g, 36,5 mmoles) foram dissolvidos em ácido acético (20 mL) e PtO2 (150 mg) foi em seguida adicionado. A mistura de reação foi agitada sob atmosfera de H2 (3,51 Kg/cm2) em temperatura ambiente durante 24 horas. O precipitado que se formou foi coletado por filtração a vácuo e o papel filtro foi lavado com ácido acético (25 mL). O filtrado combinado e lavado foi concentrado em vácuo para fornecer o composto do título (2,0 g). O sólido que permaneceu sobre papel filtro foi tratado com MeOH para dis solver o produto e o Pt2O filrado. O filtrado foi concentrado em vácuo e o sólido obtido foi triturado com 1:1 EtOAc/hexanos (200 mL) para fornecer uma segunda coleta do composto do titulo (1,8 g, 62% total). 1H RMN (DMSO-<) δ 9,83 (s, 1H), 9,51 (s, 1H), 7,50 (dd, 1H, J- 13,6, 2,5 Hz), 7,03 (d, 1H, J - 8,5 Hz), 6,84 (dd, 1H, J = 9,3, 9,3 Hz), 1,98 (s, 3H); MS(ESF) m/z 170,23 (M + H)T

B) #-(4-(6-Gloroplrlmiclin-4-ilóxi)-3-fluorofenil)acetamída
A mistura de 4,6-dicloropirimidina (1,50 g, 10,0 mmoles), #-(3flúor-4-hidroxifenil)acetamida (1,70 g, 10,0 mmoles), I^CO3 (1,8 g, 13,0 mmoles), e DMF (15 mL) foi aquecida a 70°C durante 1,5 hora. A mistura foi concentrada para a metade de seu volume original e resfriada em um banho de gelo. A mistura foi tratada com H2O (100 mL) para precipitar o produto que foi coletado por filtração a vácuo. O produto foi lavado com H2O e secado a vácuo no funil durante a note para fornecer o composto do título (2,0 g, 71%) como um sólido cinza. Ή RMN (DMSO-de) 5 10, 26 (s, 1H), 8,67 (s, 1H), 7,78 (dd, 1H, J = 12,6, 2,0 Hz), 7,56 (s, 1H), 7,37-7,30 (m, 2H), 2,08 (s, 3H); MS(ESI*) m/z 282,10 (M + H)*.

G) #-(4-(6-Aminopirimiciin-4-ilóxi)-3-fluorofenil)acetami<ia.
A mistura de A/-(4-(6-aminopÍrimidin-4-ilóxi)-3-fluorofenil)acetamida (1,0 g, 3,5 mmoles) e aproximadamente. 7 M NH3 em MeOH (5 mL) foi aquecida a 100°C em um frasco de pressão selado durante 2 horas. A mistura foi concentrada em vácuo e o resíduo dividido entre EtOAc e solução de NaHCO3 aquosa saturada. A fase EtOAc foi separada, lavada com salmou ra, secada (MgSOJ e concentrada. Cromatografia instantânea em SiO2 empregando-se primeiro 50% de EtOAc em hexanos em seguida 5% de MeOH em engole como eluentes fornece o composto do título (175 mg, 20%) como um sólido marrom. 1H RMN (DMSO-dg) 5 10,17 (s, 1H), 8,02 (s, 1H), 7,70 (dd, 1H, J = 13,2, 2,2 Hz), 7,26 (dd, 1H, J = 8,8, 2,2 Hz), 7,22 (dd, 1H, J =
8,8, 8,8 Hz), 6,89 (brs, 2H), 2,05 (s,3H); MS(ESQm/z 263,15 (M + H)L

D) 6-(4-Amino-2-fluorofenóxi)pirímiciin-4-amina
A mistura de N-(4-(6-aminopirimidin-4-ilóxi)-3-fluorofenil) acetamida (175 mg, 0,67 mmol), 1 M de HOI (6 mL), e MeOH (2 mL) foi aquecida até o refluxo durante 3 horas. A mistura de reação foi resfriada, tomada básica (pH 8) com solução de Na2CO3 aquosa e extraída com EtOAc (2 x 25 mL). Os extratos combinados foram secados (MgSO4) e concentrados em vácuo para fornecer o composto do título (140 mg, 96%) como um sólido marram. 1H RMN (DMSO<) δ 8,02 (s, 1H), 6,89 (dd, 1H, J = 9,0, 9,0 Hz), 6,80 (br s, 2H), 6,43 (dd, 1H, J = 12,7,2,8 Hz), 6,35 (dd ,1H, 8,8, 2,2 Hz),
5,67 (s, 1H), 5,34 (brs, 2H).
E) 1-(4-(6-Aminopirimidin-4-ilóxi)-3-fluorofenll)-3-(2-(4-fluorofenil)aceW)uréia.
O composto do titulo foi preparado de 6-(4-amino-2-fluorofenóxi) pirimidin-4-amina (92 mg, 0,42 mmol) e 0,36 M de isocianato de 2-(4-fluorofenil)acetila em tolueno (Composto D de Exemplo 11, 1,3 mL, 0,45 mmol) em THF como descrito acima por exemplo 11.0 produto bruto foi purificação por trituração empregando-se 1:1 EtOH / H2O seguido por EtOH absoluto. O produto foi secado a vácuo para fornecer o composto do título (100 mg, 60%). Uma segunda coleta levemente mais pura do produto (45 mg, 27%) foi obtido por extração dos filtrados combinados e lavagem com EtOAc. 'H RMN (DMSO-cy δ 10,99 (s, 1H), 10,50 (s, 1H), 8,01 (s, 1H), 7,65 (dd, 1H, J = 12,6, 2,1 Hz), 7,33 (dd, 2H, J = 8,1, 6,0 Hz), 7,28 (dd, 1H, J = 8,6,
2,0 Hz), 7,22 (dd, 1H, J = 8,8, 8,8 Hz), 7,17 - 7,12 (m, 2H), 6,89 (br s, 2H),
5,8O(s, 1H),3,71 (s, 2H); MS(ESr) m/z 400,09 (M + H)*.
Exemplo 14

/^-(4-(2-(4-Metoxibenzllamlno)pirimidin-4-ilóxi)-3-fluorofeníl)5 ^-(A-fluorofenllJmalonamicla

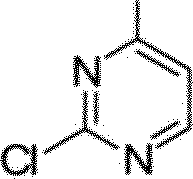
A) 2-Cloro-4-(2-11úor*4-nitrofenóxi)pirimidina
A mistura de 2,4-dicloropirimidina (Aldrich, 0,74 g, 5,0 mmoles),
2-flúor-4-nitrofenol (Avacado, 0,79 g, 5,0 mmoles), K2CO3 (0,76 g, 5,5 mmoles), e DMF (50 mL) foi aquecida a 100°C durante 2 horas. A mistura foi res10 friada e diluída com solução de NaHCO3 saturada (100mL) e extraída com
EtOAc. O extrato EtOAc foi lavado com salmoura, secado (MgSO4) e concentrado em vácuo para fornecer a mistura dos regioisômeros de 2-fenóxi- e
4-fenoxipirimidina como um sólido amarelo. Os regioisômeros foram separados por cromatografia instantânea empregando-se 10-40% EtOAc em hexa15 nos como o eluente para fornecer o composto do título (0,71 g, 53%) como um sólido branco. Ή RMN (DMSO-de) δ 8,76 (dd, 1H, J = 6,0,1,6 Hz), 8,43 (dt, 1H, */=9,8, 2,2 Hz), 8,23 (dd, 1H, */ = 8,8,1,6 Hz), 7,80 (dt, 1H, J = 9,8,

B) âF(4-Metoxibenzil)-4-(2-flúor-4-nitrofenóxi)pirimidin-2- amina
A mistura de 2-cloro-4-(2-flúor-4-nitrofenóxi)pirimidina (0,66 g, 2,44 mmoles), 4-metoxibenzilamina (0,34 g, 3,45 mmoles), K2CO3 (0,37 g, 2,66 mmoles), e DMF (15 mL) foi aquecida a 100°C durante 1 hora. A mistura foi resfriada, diluída com H2O (100 mL) e extraída com EtOAc (100 mL). A fase orgânica foi lavada duas vezes cada com solução de NaHCO3 saturada e salmoura. Os orgânicos foram secados (MgSO4) e concentrados para fornecer o produto bruto. Cromatografia instantânea em SiO2 empregando-se 1- 3% de MeOH em CH2CI2 como o eluente fornece o composto do título (275 mg, 29%) como um sólido amarelo pálido. Ή RMN (DMSO-d6) δ 8,408,21 (m, 2H), 8,16 (dd, 1H, J = 8,8, 1,7 Hz), 7,98 (br s, 0,5H), 7,73-7,55 (m, 1,5 H), 7,16 (br s, 1H), 6,85-6,71 (m, 3H), 6,37 (s, 1H), 4,43 (br s, 1H), 3,95 (br s, 1H), 3,69 (s, 3H).
NH2
F.
O'
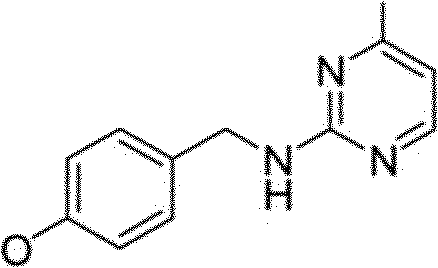
h3c
C) JV-(4-Metoxibenzil)-4-(4-amino-2-fluorofenóxi)pirimidin-2- amina.
O composto do título foi obtido pela redução de A/-(4-metoxibenzil)-4-(2-flúor-4-nitrofenóxí)pirimidin-2-amina (270 mg, 0,73 mmol) com pó de zinco (475 mg, 7,3 mmoles) e NH4CI (387 mg, 7,3 mmoles) em 1:1
THF/MeOH (20 mL) empregando-se um procedimento similar descrito para o Composto C de Exemplo 11, Cromatografia instantânea em SíO2 empregando-se 1-3% MeOH em CH2CI2 como o eluente fornece © composto do título (235 mg, 95%) como uma película marrom. 1H RMN (DMSO-d6) δ 8,01 25 (s, 1H), 7,65 (br s, 0,5 H), 7,40 (br s, 0,5H), 7,08 (br s, 1H), 6,83 (br s, 2H),
6,81 (m, 1H), 6,69 (br s, 2H), 6,39 (br s, 0,5H), 6,31 (br s, 0,5H), 6,01 (m,
1H), 5,26 (br s, 2H), 4,24 (br s, 1H), 3,95 (br s, 1H), 3,61 (s, 3H); MS(ESF) m/z341,16(M + H)*.
D) A/’-(4-(2-(4-Metoxibenzilamino)pirimidin-4-ilóxi)-3-fluorofenil)-A/’-(4-fluorofenil)malonamida
O composto do título foi obtido de A/-(4-metoxibenzilH-(4amÍno-2-fluorofenóxi)pirímidin-2-amina (34 mg, 0,10 mmol), ácido 3-(4-fluorofenilamino)-3-oxopropanóico (Composto B de Exemplo 1, 22 mg, 0,11 mmol), TBTU (39 mg, 0,12 mmol) e DIPEA (23 mL, 0,17 mmol) empregando-se um procedimento similar descrito para a preparação de Composto C de Exemplo 1, o produto bruto foi triturado com 3:1 de éter de isopropila / EtOAc para fornecer o composto do título (35 mg, 61%) como um sólido esbranquiçado. fH RMN (DMSO-de) δ 10,47 (br s, 1H), 10,26 (s, 1H), 8,15 (s, 1H), 7,77 (m, 3H), 7,61 (m, 2H), 7,35-7,25 (m, 2H), 7,15 (dd, 2H, J = 8,8, 8,8 Hz), 6,81-6,73 (m, 2H), 6,24 (s, 1H), 4,32 (br s, 1H), 3,96 (br s, 1H), 3,68 (s, 1H), 3,48 (s, 2H);MS(ESI*) m/z 520,14 (M + H)*.
Exemplo 15 h3cct
1-(4-(2-(4-Metoxibenzilamlno)pirlmiciln-4-ilóxi)-3-0uorofenil)-
3-(2-(4-fluorofenit)acetil)uréia.
Uma solução de A/-(4-metoxibenzil)-4-(4-amino-2-fluorofenóxi) pirimidin-2-amina (Composto C de Exemplo 14, 34 mg, 0,10 mmol) em THF (1 ml) foi tratada com 0,36 M de isocianato de 2-(4-fluorofenil)acetila em tolueno (Composto D de Exemplo 11, 0,31 mL, 0,11 mmol) e a mistura agitada em temperatura ambiente durante 1 hora. A mistura foi concentrada em vácuo e o sólido obtido foi triturado primeiro com 3:1 de éter de isopropila / EtOAc e em seguida CH2CI2 para fornecer o composto do título (32 mg, 62%) como um sólido esbranquiçado. 1H RMN (DMSO-de) δ 10,47 (s, 1H), 10,26 (s,1H), 8,15 (s, 1H), 7,76 (d, 1H, 12,6 Hz), 7,61 (dd, 3H, J = 9,1,
5,0 Hz), 7,29 (s, 2H), 7,31 - 7,21 (m, 3H), 6,81 (s, 3H), 6,24 (s, 1H), 4,32 (s, 1H), 3,96 (s, 1H), 3,68 (s, 3H), 3,48 (s, 2H); MS(ESF) m/z 520,14 (M ♦ H)*
Exemplo 16

/V,-(4-(2-Aminoplrimiclin-4-llóxi)-3-fluorofenil)-W3-(4-fluorofenil)malonamida
A mistura de N’-(4-(2-(4-metoxibenzilamino)ptrimidin-4-ilóxi)-3f!uorofenil)-/V3-(4-fluorofenil)malonamida (Exemplo 14, 25 mg, 0,048 mmol), anisol (52 mg, 0,48 mmol) em TFA (1 mL) foi aquecida a 85°C durante 6 horas. O TFA foi removido sob vácuo e o resíduo dividido entre EtOAc e solução de NaHCO3 aquosa saturada. A fase EtOAc foi lavada com salmoura, secada (MgSOJ, e concentrada em vácuo. Cromatografia instantânea em SiO2 empregando-se EtOAc em seguida 1-2% MeOH in CH2CI2 como eluentes fornece o composto do título (12 mg, 63%) como um sólido esbranquiçado. ’H RMN (OMSO-^)l 11,00 (s, 1H), 10,51 (s, 1H), 8,02 (s, 1H), 7,66 (dd, 1H, 12,6, 2,0 Hz), 7,42-7,31 (m, 2H), 7,32-7,27 (m, 1H), 7,23 (t, 2H, J =
8,6 Hz), 7,16 (t, 2H, J = 9,1 Hz), 6,91 (s, 2H), 3,73 (s, 2H); MS(ESF) m/z 400,11 (M + H)*.
Exemplo 17


h2n
1-(4-(2-Aminopirimidin-4-llóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia
O composto do título foi preparado de 1-(4-(2-(4-metoxibenzilamino) pirimidin-4-ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia (Exemplo 15, 20 mg, 0,039 mmol) empregando-se um procedimento similar descrito por exemplo 16, Cromatografia instantânea em SiO2 empregando-se EtOAc em seguida 1-2% de MeOH em CH2CI2 como eluentes fornece o composto do título (10 mg, 62%) como um sólido esbranquiçado. 1H RMN (DMSO-d6) δ
11,01 (s, 1H), 10,52 (s, 1H), 8,20 (d, 1H, 6,0 Hz), 7,70 (dd, 1H, J = 12,1,
2,0 Hz), 7,36 - 7,30 (m, 6H), 7,16 (dd, 2H, J = 8,8, 8,8 Hz), 6,45 (d, 1H, J-
6,1 Hz), 3,73 (s, 2H); MS(ESI*) m/z 400,00 (M + H)*.
Exemplo 18

/V,-(3-flúor-4-(2-(2-morfolinoetllamino)pirldin-4-ilóxi)fenll)-^(4-fluorofenil)malonamida, sal de cloridrato.
crw
A) /V»(4-(2-Cloropiridin-4-il0xi)-3-fluorofenil)acetamida,
Uma mistura de A/-(3-flúor-4-hidroxifenil)acetamida (Composto A 10 de Exemplo 13, 1,33 g, 7,87 mmoles), 2-cloro-4-nitropiridina (Aldrich, 1,24 g,
7,87 mmoles), K2CO3 (1,6 g, 11,8 mmoles), e DMF (25 mL) foi aquecida a
100°C durante 9 horas. A mistura de reação foi concentrada em vácuo e ο resíduo dividido entre EtOAc e solução de NaHCO3 saturada. A fase EtOAc foi lavada com salmoura, secada (MgSO4), e concentrada. Cromatografia 15 instantânea empregando-se 30 -80% EtOAc em hexanos como o eluente fornece o composto do título (1,6 g, 73%) como um sólido amarelo pálido. 1H
RMN (DMSO-cy δ 10,24 (s, 1HJ, 8,65 (s, 1H), 7,89-7,64 (m, 1H), 7,56 (s, 1H), 7,46-7,19 (m, 2H). 2,06 (s, 3H); MS(ESf) m/z 281, 16 (M + H)\

B) N-(4-(2-Cloropiridin-4-ilóxi-1 -óxido)-3-fluorofenil)acetamida.
A mistura de N-(4-(2-cloropiridin-4-ilóxi)-3-fluorofenil)acetamida (0,98 g, 3,5 mmoles), ácido m-cloroperoxibenzóico {> 90%, 1,3 g, 7,6 mmoles), e CHCI3 (50 mL) foi agitada em temperatura ambiente durante 60 horas. A mistura foi concentrada e o resíduo triturado com E^O (2 x 100 mL) para fornecer o composto do título (0,89 g, 87%) como um sólido amarelo pálido. Ή RMN (DMSO-<) δ 10,25 (s, 1H), 8,34 (d, 1H, /= 7,1 Hz), 7,80 (d, 1H, J = 13,2 Hz), 7,40 (d, 1H, J = 3,3 Hz), 7,33 (d, 2H, J = 4,9 Hz), 7,02 (dd, 1H, J = 7,1, 3,3 Hz), 2,06 (s, 3H); MS(ESI) m/z 295,04 (Μ - H).


C) A/-(3-flúor-4-(2-(2-morfolinoetilamino)piridin-4-llóxi-1óxido)fenil)acetamida.
Uma mistura de /V-(4-(2-cloropiridin-4-ilóxi-1-óxido)-3-fluorofenil) acetamida (205 mg, 0,62 mmol), 4-(2-aminoetil)morfolina (Aldrich, 169 mg, 1,30 mmol), e o EtOH absoluto foi aquecido até o refluxo durante 16 horas. A mistura de reação foi concentrada em vácuo, o resíduo tratado com H2O (3 mL) e aplicado a um cartucho Varian C-18 de 10g. O cartucho foi eluído primeiro com H2O em seguida com 30% de MeOH em H2Q. As frações que continham o produto desejado foram agrupadas, concentradas para um volume de 5 mL, e extraídas 3 vezes com EtOAc. Os extratos combinados foram lavados com salmoura, secados (MgSO4) e concentrados para fornecer o composto do título (100 mg, 40%). Ή RMN (DMSO-cQ δ 10,22 (s, 1H), 7,84 (d, 1H, J = 6,1 Hz), 7,77 (dd, 1H, J = 13,2, 2,2 Hz), 7,31 (dd, 1H, J = 8,8, 2,2 Hz), 7,24 (t, 1H, J = 8,8 Hz), 6,41 (m, 1H), 6,13 (dd, 1H, U = 5,5, 2,2 Hz), 5,81 (d, 1H, J = 2,2 Hz), 3,60-3,52 (m, 4H), 3,31 - 3,28 (m, 2H), 2,38 (t, 2H, J = 7,1 Hz), 2,34 (m, 4H), 2,06 (s, 3H); MS(ESr) m/z 405,22 (M + H)+.

D) /A(3-flúor-4-(2-(2-morfolinoetilamino)piridin-4-ilóxi) fenil) acetamida, sal de ácido trifluoroacético.
A mistura de N-(3-flúor-4-(2-(2-morfolinoetilamino)piridin-4-ilóxi1-óxido)fenil)acetamida (100 mg, 0,26 mmol), e polímero de trifenilfosfina sustentada (1,4 -2,0 mmoles/g) em polistireno (500 mg) e DMF (2 mL) foi agitada a 135°C durante 15 horas. A mistura foi filtrada para remover a resina, e a resina lavada com DMF e EtOAc. Os filtrados e lavados foram combinados e concentrados. O produto bruto foi purificado por HPLC preparativa (Shimadzu S5 VP-ODS 20 x 100 mm) para fornecer o composto do titulo (45 10 mg, 46%) como um sólido branco. Ή RMN (DMSO-d6) δ 10,33 (s, 1H), 8,02 (d, 1H, 6,6 Hz) 7,84 (dd, 1H, J = 13,2, 2,0 Hz), 7,39 - 7,31 (m, 2H), 6,52 (s, 1H), 6,10 (s, 1H), 3,83 (br s, 4H), 3,64 (m, 2H), 3,28 (m, 6H), 2,08 (s, 3H); MS(ESF) m/z 375,12 (M + H)*.


E) 4-(4-AminO’2-fluorofenóxi)-^(2-morfolirioetil)pirídin-2- amina, sal de cioridrato.
A mistura de trífluoroacetato de A/-(3-flúor-4-(2-(2-morfolinoetilamÍno)piridÍn-4-ilóxi)fenil)acetamida (40 mg), MeOH (1 mL), e 6 M HCl (0,2 mL) foi aquecida até o refluxo durante 3 horas. A mistura de reação foi concentrada em um evaporador .giratório e o resíduo liofllizado para fornecer o composto do título (30 mg, 76%) como um sólido branco. 1H RMN (DMSOcQ δ 11,12 (br s, 1H), 8,85 (br s, 1H), 7,95 (d, 1H, 7,2 Hz), 7,08 (dd, 1H,
J = 8,8, 8,8 Hz), 6,65-6,63 (m, 2H), 6,54 (d, 1H, J = 8,3 Hz), 6,31 (br s, 1H), 3,85 (m, 6H), 3,33 (m, 6H); MS(ESL) m/z 373,14 (Μ - H).
F) N?-(3-flúor-4-(2-(2-morfolinoetilamino)piridin-4-ilóxi)fenil)A/’-(4-fluorofenil)malonamida.
O composto do título foi preparado de uma mistura de 4-(4amino-2-fluorofenóxi)-/V-(2-morfolinoetil)piridin-2-amina, sal de cloridrato (15 mg, 0,043 mmol), ácido 3-(4-fluorofenilamino)-3-oxopropanóico (Composto B de Exemplo 1,10 mg, 0,052 mmol), TBTU (17 mg, 0,052 mmol), DIPEA (30 pL), e DMF (1 mL) empregando-se um procedimento similar descrito para a preparação de Composto C de Exemplo 1, o produto bruto foi purificado por HPLC preparativa (Shimadzu S5 VP-ODS 20 x 100 mm). O produto obtido de purificação por HPLC foi tratado com 1 M de HCI e liofilizado para fornecer o composto do título (10 mg, 40%) como um sólido branco. Ή RMN (DMSO-cÇ)ô 10,37 (s, 1H), 10,05 (s, 1H), 9,90 (brs, 1H), 7,90 (d, 1H, J = 6,1 Hz), 7,75 (d, 1H, /= 13,2 Hz), 7,58 - 7,55 (m, 2H), 7,53 - 7,50 (m, 1H), 7,40 (d, 1H, J = 8,8 Hz), 7,24 (t, 1H, J = 8,8 Hz), 7,08 - 7,03 (m, 3H), 6,39 (d, 1H, J = 6,1 Hz), 6,11 (s, 1H), 3,80 -3,81 (m, 4H), 3,67- 3,65 (m, 2H), 3,47 (br s, 2H), 3,20 (br s, 4H); MS(ESI+) m/z 512,12 (M + Hf.
Exemplo 19

A/1-(3-flúor-4-(piridin-4-ilóxi)fenil)-A/3-(4-fluorofenil)malonamida.

A) N’-(3-flúor-4-hidroxifenil)-/V3-(4-fluorofenil)malonamida.
A uma solução de 2-flúor-4-nitrofenol (Avacado, 1,00 g, 6,37 mmoles) em 4 mL de tetraidrofurano e 6 mL de metanol a 0°C foi adicionado pó de zinco (2,08 g, 31,8 mmoles, <10 micron) seguido por cloreto de amônio (1,70 g, 31,8 mmoles). A mistura foi agitada em temperatura ambiente durante a noite. A mistura heterogênea foi filtrada através de uma almofada fina de Celite® com metanol e o filtrado foi concentrado em vácuo para fornecer 4-amino-2-fluorofenol como um sólido marrom que foi empregado sem outra purificação (656 mg, 81 %).
Ácido 3-(4-Fluorofenilamino)-3-oxopropanóico (Composto B de Exemplo 1, 197 mg, 1,00 mmol) foi dissolvido em dimetilformamida (4 mL). Trietilamina (140 pL, 1,00 mmol) foi adicionado e a solução foi resfriada a 0°C, 4-Amino-2-fluorofenol (Etapa A de Exemplo 19, 127 mg, 1,00 mmol) foi adicionado seguido por hexafluorofosfato de benzotriazol-1-iloxitris(dimetilamino) fosfônio (Reagente BOP, 442 mg, 1,00 mmol). A reação foi deixada aquecer para temperatura ambiente e foi em seguida agitada em temperatura ambiente durante 3 horas. A mistura de reação foi concentrada para remover o cloreto de metileno e água foi adicionada para precipitar o produto. A filtração e a trituração com água fornecem o composto do título (211 mg, 69%) como um sólido branco. 1H RMN (CD3OD) δ 7,61-7,57 (m, 2H), 7,51 (dd, 1H, 13, 2,5 Hz), 7,08-6,99 (m, 3H), 6,88 (t, 1H, J= 9,4 Hz), 3,51 (s,
2H); MS(ESI+) m/z 307,44 (M+ H)+.
B) A/1-(3-’flúor“4“(piridin-’4-ilóxi)fenil)-/V3-(4-fluorofenil)malonamida.
/VL(3-flúor-4-hidroxifenil)-/\^(4-fluorofenil)malonamida (31 mg, 0,10 mmol), acetato de cobre (II) (27 mg, 0,15 mmol), ácido piridin-4ilborônico (25 mg, 0,20 mmol), e piridina (16 pL, 0,20 mmol) foram colocados em um tubo de pressão naquela ordem. O tubo foi camegado com cloreto de metileno (0,5 mL) e selado. A reação foi agitada a 120°C durante 5 horas. A mistura de reação foi filtrada através de sílica-gel empregando-se 5% de metanol / acetato de etila. Após a concentração, o produto bruto foi purificado por HPLC preparativa. A fração apropriada foi concentrada para remover metanol e a solução aquosa resultante foi tornada básica com a solução NaHCO3 de saturada (5 mL). A solução aquosa foi extraída com EtOAc (3 x 10mL) e os extratos orgânicos combinados foram secados em Na2SO4 anidro concentrada em vácuo, O produto foi tratado com 4N HCI em dioxano e concentrada. A liofilização com água fornece o composto do título (8 mg, 21%) como um sólido amarelo. 1H RMN (CD3OD) δ 8,31 (d, 2H, J= 6,1 Hz),
7,72 (dd, 1H, J= 12,7, 2,4 Hz), 7,49-7,46 (m, 2H), 7,27-7,25 (m, 1H), 7,15 (t, 1H, J = 6,8 Hz), 6,97 (t, 2H, J = 8,7 Hz), 6,85 (dd, 2H, J = 5,1, 1,2 Hz), 3,46 (s, 2H); MS(ESr) m/z 384,21 (M + Hy.
Exemplo 20

ci /Ví-(4-(2-Cloropiridiri-4-ilóxi)-3-fluorofenil)-N3-(4-fluorofenil) malonamida.

A) 2-flúor-4-aminofenol
A mistura de óxido de platina (0,010 g) e 2-flúor-4-nitrofenol (Aldrich, 1,24 g, 7,78 mmoles, 1,0 eq.) em MeOH (100 ml) foram agitadas sob uma atmosfera de H2 a 3,51 Kg/cm2 em temperatura ambiente. A mistura de reação foi-filtrada através de celita e o filtrado concentrado em vácuo para fornecer o composto do título (1,00 g, 100%), como um sólido que foi empregado sem outra purificação. Ή RMN (DMSO-d6) δ 8,57 (s, 1H), 6,46-6,47 (m, 1H), 6,33-6,46 (m, 1H), 6,19-6,21 (m, 1H), 4,79 (s, 2H); MS(ESI*) m/z 128 (M+ H)\


B) 4-(2-Cloropiridin-4-ilóxi)-3-fluorobenzenamina.
Hidrato de sódio (60%, 0,104 g, 2,60 mmoles, 1,1 eq.) foi adicionado a uma solução de 2-flúor-4-aminofenol (0,30 g, 2,36 mmoles, 1,0 eq.) em DMF (6,5 mL) em temperatura ambiente e a mistura de reação foi agitada durante 30 minutos. 2-Cloro-4-nitropiridina (Aldrich, 0,374 g, 2,36 mmol, 1,0 eq) foi adicionado e a mistura de reação foi aquecida para 90°C durante 12 horas. A mistura de reação foi resfriada a temperatura ambiente, extin güida com solução de NaC! aquosa saturada e extraída com acetato de etila (3 x 70 mL). Os extratos orgânicos combinados foram lavados com solução de LiCI aquosa a 10% (3 x 70 mL), secados sobre Na2SO4, filtrados e o filtrado concentrado em vácuo para fornecer o composto do título (0,430 g, 76%) que foi empregado sem outra purificação. Ή RMN (DMSO-dfi) δ 8,27 (d, 1H, J = 5,7 Hz), 6,90-7,04 (m, 3H), 6,42-6,54 (m, 2H), 5,54 (s, 2H); MS(ESO) m/z 239 (M + H)+; HRMS (ESI+) calculada.: 239,0387, encontrado: 239,0301,
C) /V,-(4-(2-Cloropiri<iin-4-ilóxi)-3-fluorofenil)-AP-(4-fluorofenilmalonamida.
Diisopropiletilamina (0,091 mL, 0,525 mmol, 2,5 eq) foi adicionado a uma solução de 4-(2-cloropiridin-4-ilóxi)-3-fluorobenzenamina (0,050 g, 0,21 mmol, 1,0 eq), ácido 3-(4-fluorofenilamino)-3-oxopropanóico (Composto B de Exemplo 1,0, 041 g, 0,21 mmol, 1,0 eq.), e PyBroP (0,117 g, 0,252 mmol, 1,2 eq) in CH2CI2 (1,0 mL) a 0’C. A mistura de reação foi aquecer para temperatura ambiente e agitada durante 12 horas. A mistura de reação foi extingüida com solução de NaCI aquosa saturada, e a mistura foi extraída com CH2CI2 (3 x 20 mL). Os extratos orgânicos combinados foram secados em Na2SO4, filtrados e os filtrados concentrados em vácuo. O resíduo foi purificado por cromatografia instantânea de sílica-gel (Merck, 40-63 μΜ, 230-240 mesh, eluindo 3/1 acetato de etila / hexano) para fornecer o composto do título (0,056 g, 64%) como um sólido. 1H RMN (DMSO-cg δ 10,61 (s, 1H), 10,34 (s, 1H), 8,36-8,38 (m, 1H), 7,91-7,93 (m, 1H), 7,67-7,71 (m, 2H), 7,46-7,48 (m, 2H), 7,04-7,26 (m, 4H), 3,56 (s, 2H); MS(ESP) m/z 418 (M + H)+; HRMS (ESI+) calculada.; 418,0770, encontrado: 418,0767,
Exemplo 21
1-(4-(2-Cloropiridin-4-ilóxi)-3-fluorofenil)-3-(2-(4-fluoiOfenil) acetil) tiouréia
4-Fluorofenilacetilcloreto (Aldrich, 0,072 mL, 0,525 mmol, 2,5 eq.) foi adicionado a uma solução de tiocianato de sódio (0,056 g, 0,605 mmol, 3,3 eq.) em acetato de etila (2,0 mL) em temperatura ambiente e a mistura de reação foi agitada durante 1,5 hora para fornecer uma solução de isotio5 cianato de 2-(4-fluorofenil)etanoíla (0,263 M). Uma solução de 4-(2-cloropiridin-4-ilóxi)-3-fluorobenzenamina (0,050 g, 0,21 mmol, 1,0 eq.) em CH2CI2 (1,0 mL) foi adicionado em gotas à solução de isotiocianato de 2-(4fluorofenil)etanoíla e a mistura de reação foi agitada em temperatura ambiente durante 12 horas. A mistura de reação foi concentrada em vácuo e o 10 resíduo resultante foi purificado por cromatografia instantânea de sílica-gel (Merck, 40-63 μΜ, malha 230-240, eluindo 3/1 hexano/ acetato de etila) para fornecer o composto do título (0,058 g, 64%) como um sólido. 1H RMN (DMSO-O- 3 12,46 (s, 1H), 11,84 (s, 1H), 8,35 - 8,33 (m, 1H), 8,02-8,33 (m, 1H),
6,99 - 7,52 (m, 8H), 3,84 (s, 2H); MS(ESF) m/z 434 (Μ + H)*; HRMS (ESF) 15 calculada: 434,0542, encontrado: 434,0547,
Exemplo 22


/^’-(4-(2-(Benzilamino)plridm-4-ilóxi)-3-fluorofenil)-^í-(4-fluorofenil) malonamída.
A) 4-(2-cloropirldin-4-ilóxl)-3-fluorofer»llcarbamato de terc20 butíla.
Dicarbonato de di-terc-butila (0,920 g, 4,22 mmoles, 4,5 eq.) foi adicionado a uma solução de 4-(2-cloropiridin-4-ilóxi)-3-fluorobenzenamina (Composto B de Exemplo 20, 0,224 g, 0,939 mmol, 1,0 eq.) e trietilamina (0,391 mL, 3,00 mmoles, 3,0 eq) em THF (10 mL) e a mistura de reação foi aquecida a 55*C durante 14 horas. A mistura de reação foi resfriada a temperatura ambiente e finalizada com 1N de HGI. A solução foi extraída com CH2CI2 (3 x 70 mL), os extratos orgânicos combinados lavados com 1N NaOH (100 mL), secados (Na2SO4), filtrados e concentrados em vácuo. O resíduo foi purificado por cromatografia instantânea de sílica-gel (Merck, 40-63 μΜ, 230-240 mesh, eluindo 4:1 hexano/ acetato de etila) para fornecer o composto do título (0,270 g, 85%). Ή RMN (DMSO-d6) δ 8,35-8,36 (m, 1H), 7,55-7,57 (m, 1H), 7,45-7,46 (m, 1H), 7,21-7,24 (m, 1H), 6,96-6,97 (m, 2H), 1,40 (s, 9H); MS(ESF) m/z 339 (M + H)*; HRMS (ESF) calculada.: 339,0912, encontrado: 339,0915,

B) -4-(2-(benzilamino)piridin-4-ilóxi)-3-fluorofenilcarbamato de terc-butila.
4-(2-cloropiridin-4-ílóxi)-3-fluorofenilcarbamato de terc-butila (0,100 g, 0,295 mmol, 1,0 eq) foi adicionado a uma solução desgaseificada de dppf.PdGI2 (Matrix Scientific, 0,011 g, 0,0148 mmol, 0,05 eq), dppf (0,012 g, 0,022 mmol, 0,075 eq), e NaOf-Bu (0,040 g, 0,414 mmol, 1,4 eq) em tolueno em temperatura ambiente. Benzilamina (0,045 mL, 0,414 mmol, 1,4 eq) foi adicionada a uma mistura de reação e a solução resultante foi -agitada a 80eG durante 4 horas. A mistura de reação foi resfriada a temperatura ambiente, finalizada com 1N de HCI e a solução extraída com CHCI3 (3 x 50 mL). Os extratos orgânicos combinados foram lavados com 1 N de NaOH (70 mL), secados (Na2SO4), filtrados e concentrados em vácuo. O resíduo foi purificado por cromatografia instantânea de sílica-gel (Merck, 40-63 μΜ, malha 230-240, eluindo 2:1 hexano / acetato de etila) para fornecer o composto do título (0,020 g, 17%). 'H RMN (CDCI3) δ 7,80 - 7,90 (m, 1H), 7,35-7,45 (m, 1H), 7,19-7,24 (m, 3H), 6,91-6,93 (m, 2H), 6,59 (br m, 1H), 6,10-6,20 (m, 1H), 5,75 (br m, 1H), 5,30-5,40 (m, 1H), 4,34 (s, 2H), 1,46 (s, 9H); MS(ESP) m/z 410 (M + Η)*; HRMS (EST) calculada.: 410,1880, encontrado: 410,1884,


G) 4-(4-Amino-2-fluorofenóxi)-/V-benzílpiridin-2-amina, sal de cloridrato.
HCI anidro em dioxano (4N, 2,00 mL, 8,00 mmoles, 165 eq) fol adicionado a 4-(2-(benzílamino)piridin-4-ilóxi)-3-fluorofenilcarbamato de tercbutila (0,020 g, 0,0489 mmol, 1,0 eq) e a mistura de reação foi agitada em temperatura ambiente durante 12 horas. A mistura de reação foi concentrada em vácuo para fornecer o composto do titulo (0,017 g, 100%) um sólido que foi empregado sem outra purificação. MS(ESL) m/z 310 (M+ H)+; HRMS (ESI*) calculada.: 310,1356, encontrado: 310,1364,
D) #-(4»(2-(Benzilamino)piridin-4-ilóxl)-3-fluorofenil)-^-(4fluorofeníl) malonamida
Diisopropiletilamina (0,014 mL, 0,081 mmol, 3,5 eq)foi adicionada a uma solução de 4-(4-amino-2-fluorofenóxi)-A/-benzÍlpiridin-2-amina, sal de cloridrato (0,008 g, 0,023 mmol, 1,0 eq), ácido 3-(4-fluorofenilamino)-3oxopropanóico (Composto B de Exemplo 1,0,005 g, 0,023 mmol, 1,0 eq), e PyBroP (0,013 g, 0,028 mmol, 1,2 eq) em CH2CI2 (1,0 mL) a 0°C. A mistura de reação foi aquecida para temperatura ambiente e agitada durante 16 horas. A mistura de reação foi concentrada em vácuo e o resíduo foi purificado por cromatografia de HPLC de fase reversa (YMC-ODS-A, C-18. S10. 30x500 mm, eluindo 20-90% MeOH aquoso com 0,1% de TFA, gradiente de 30 minutos). As frações apropriadas foram concentradas em vácuo, neutralizadas com solução de NaHCO3 aquosa saturada e a mistura e extraída com CHCI3 (3 x 10 mL). Os extratos orgânicos combinados foram secados (Na2SO4), filtrados e concentrados em vácuo para fornecer o composto do título (0,0025 g, 45%) como um sólido. MS(ESI+) m/z 489 (M + Hf; HRMS(ESI+) calculada.: 489,1738, encontrado: 489,1743,
Exemplo 23


1-(3-flúor-4-(plridin-4-ilóxi)fenil)-3-(2-(4-fluorofenil)acetil)uréia.
o'

A) 3-flúor-4-(piridin-4-ilóxi)benzenamina.
Hidrato de potássio (30%, 0,520 g, 3*90 mmoles, 3,0 eq) foi adicionado a uma solução de 2-flúor-4-aminofenol (Composto A de Exemplo 20, 0,254 g, 2,00 mmoles, 1,5 eq) em DMF (5,0 mL) em temperatura ambiente e a mistura de reação foi agitada durante 15 minutos. 4-Cloro-piridina (Aldrich, 0,200 g, 1,30 mmol, 1,0 eq) foi adicionado e a mistura de reação foi aquecida para 150°C durante 2 horas. A mistura de reação foi resfriada a temperatura ambiente, extinguida com 1N NaOH e a solução extraída com acetato de etila (3 x 50 mL). Os extratos orgânicos combinados foram lavados com 1N de NaOH aquoso (2 x 30 mL) seguido por LiCI aquoso a 10% (3 x 50 mL). Os extratos orgânicos combinados foram secados (Na2SO4), filtrados e concentrados em vácuo para fornecer o composto do título como um sólido. ’H RMN (DMSO-cy δ 8,44 - 8,46 (m, 2H), 6,89 - 7,03 (m, 1H), 6,876,88 (m, 2H), 6,44 - 6,56 (m, 2H), 5,51 (s, 2H); MS(ESI+) m/z 205 (M + H)*; HRMS(ESI+) calculada.: 205,0777, encontrado: 205,0775,
B) 1-(3-flúoM-(piridin-4-ilóxi)fem!)-3-(2-(4-flyorofenil) acetil) uréia.
Cianato de prata (0,912 g, 6,08 mmoles, 1,05 eq) foi adicionado a uma solução de cloreto de 4-fluorofenilacetila (Aldrich, 0,794 mL, 5,79 mmoles, 1,0 eq) em tolueno (16 mL) em temperatura ambiente protegida da luz. A mistura de reação foi aquecida para refluxo durante 60 minutos e em seguida resfriado a temperatura ambiente. A mistura de reação foi filtrada (Acrodisc, PTFE 0,2 μΜ) e a solução resultante de isocianato 2-(4-fluorofenil) acetila (0,36 M, 0,75 mL, 0,27 mmol, 1,1 eq) fol adicionado a uma solução de 3-flúor-4-(pirídin-4-ilóxi)benzenamina (0,050 g, 0,245 mmol, 1,0 eq) em CH2CI2 (2,0 mL) em temperatura ambiente. A mistura de reação foi agitada em temperatura ambiente durante 1 hora, finalizada com solução de NaCl aquosa saturada e a mistura foi extraída com CH2CI2 (3 x 30 mL). Os extratos orgânicos combinados foram secados (Na2SO4), filtrados e concentrados em vácuo. O resíduo foi purificado por cromatografia instantânea de sílica-gel (Merck, 40-63 μΜ, 230-240 mesh, eluindo 0-5% de MeOH em CHCl3) para fornecer o composto do título (0,043 g, 46%) como um sólido. Ή RMN (DMSO-dg) δ 11,06 (s, 1H), 10,60 (s, 1H), 8,47 (s, 2H), 7,77-7,80 (m, 1H), 6,92-7,48 (m, 8H), 3,75 (s, 2H); MS(ESI*) m/z 384 (M + H)*; HRMS(ESF) calculada.: 384,1160, encontrado: 384,1147,
Exemplo 24


1-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofeníl)-3-(2-(4-fluorofenil) acetil)uréia.


A) 4-(4-Amino-2-fluorofenóxi)-Af-benzilplriciin-2-amina.
Benzilamina (9,1 mL, 83,8 mmoles, 20 eq) foi adicionada a 4-(2cloropiridin-4-ilóxi)-3-fluorobenzenamina (Composto B de Exemplo 20,1,0 g, 4,19 mmoles, 1,0 eq), pó de cobre (0,266 g, 4,19 mmoles, 1,0 eq) e K2CO3 (0,578 g, 4,19 mmoles, 1,0 eq) em um tubo selado e a mistura de reação foi aquecida para 160°C durante 12 horas. A mistura de reação foi resfriada a temperatura ambiente e extinguida com solução de NaCl aquosa saturada.
A solução foi extraída com acetato de etila (3 x 100 mL), Os extratos orgânicos combinados secados (Na2SO4), filtrados e concentrados em vácuo. O resíduo foi purificado por HPLC preparativa de fase reversa (YMC C-18 ODS-A S10 50 x 500 mm, eluíndo 10-90% de MeOH aquoso com 0,1% de
TFA, gradiente de 30 minutos) e as frações apropriadas foram concentradas em vácuo. O concentrado foi neutralizado com solução de NaHCO3 aquosa saturada e extraído com CH2CÍ2 (3 x 100 mL). Os extratos orgânicos combinados foram secados (Na2SO4), filtrada e concentrada em vácuo para fornecer o composto do título (0,675 g, 52%) como um sólido. Ή RMN (CD3OD) δ
7,78 - 7,80 (m, 1H), 7,28 - 7,30 (m, 5H), 6,80 - 6,90 (m, 1H), 6,52 - 6,55 (m,
2H), 6,18 - 6,20 (m, 1H), 5,87 - 5,88 (m, 1H), 4,40 (s,2H); MS(ESÇ) m/z 310 (M + H)*; HRMS(ESr) calculada.: 310,1356, encontrado: 310,1360,
ΡχζΤχ. NH2
B) 4-(4-Amino-2-fluorofenóxi)piridin-2-amina.
Hidróxido de paládio sobre carbono (10%, 0,050 g) foi adiciona15 do a uma solução de 4-(4-amino-2-fluorofenóxi)-/V-benzilpiridin-2-amina (0,245 g, 0,790 mmol, 1,0 eq) em 5% de HCO2H-MeOH (10 mL) sob uma manta de hidrogênio (de um balão) em temperatura ambiente. A mistura de reação foi agitada em temperatura ambiente durante 12 horas, filtrada através de Celite* e o filtrado concentrado em vácuo. O resíduo foi purificado 20 por HPLC preparativa de fase reversa (YMC ODS-A S10 30 x 500 mm. 1090% MeOH aquoso com 0,1% de TFA, gradiente de 30 minutos) e as frações apropriadas foram concentradas em vácuo. O concentrado foi neutralizado com solução de NaHCO3 aquosa saturada e a mistura foi extraída com CHÇI3 (3 x 35 mL). Os extratos orgânicos combinados foram secados (Na2SO4), filtrados e concentrados em vácuo para fornecer o composto do título (0,045 g, 26%) como um sólido. Ή RMN (CD3OD) δ 7,62 - 7,63 (m,
1H), 6,77-6,82 (m, 1H), 6,38 - 6,47 (m, 2H), 6,09 - 6,11 (m, 1H), 5,83-5,84 (m, 1H); MS(ESI+) m/z 220 (M + H)+; HRMS(ESP) calculada.: 220,0886, en79 contrado: 220,0877,
C) 1-(4-(2-Aminopiridin-4-ilóxi)-3-fiuorofenil)-3-(2-(4-fluorofenil) acetiljuréía.
Isocianato de 2-(4-Fluorofenil)acetila (Composto D de Exemplo 11,0,362 M, 0,351 mL, 0,127 mmol, 1,3 eq) foi adicionado a uma solução de 4-(4-amino-2-fluorofenóxi)piridin-2-amina (0,022 g, 0,100 mmol, 1,0 eq) em CH2CI2 (2,0 ml) em temperatura ambiente. A mistura de reação foi agitada durante 13 horas em temperatura ambiente e em seguida concentrada em vácuo. O resíduo foi purificado por cromatografia instantânea de sílica-gel (Merck gel 40-63 pM, 230-240 mesh, 1:1 acetato de etila/hexano) para fornecer o composto do título (0,025 g, 64%) como um sólido. 1H RMN (CD3OD) δ 7,62 - 7,67 (m, 2H), 7,23 - 7,29 (m, 2H), 7,07-7,12 (m, 2H), 6,95 6,99 (m,2H), 6,12 - 6,14 (m, 1H), 5,86 - 5,87 (m, 1H), 3,61 (s, 2H); MS(ESF) m/z 399 (M+ H)*; HRMS(ESC) calculada: 399,1269, encontrado: 399,1269,
Alternativamente, Exemplo 24 foi preparado da seguinte maneira:
h2noc ci

A*) 4-Cloroplcolinamida
Uma mistura heterogênea de ácido 4-cloropícolínico (TCI America, 5,4 g, 34,2 mmoles, 1,0 eq) e cloreto de tionila (30 mL) foram aquecidas a 80°C durante 2 horas. A mistura de reação foi resfriada a temperatura ambiente e concentrada em vácuo. O resíduo foi tratada com uma solução de MeOH em amônia (7N, 45 mL) em um banho de gelo e a mistura de reação foi agitada durante 15 minutos. O banho de gelo foi em seguida removido e a reação foi aquecer para temperatura ambiente e em seguida agitada durante 3 horas. A mistura de reação foi concentrada em vácuo e o resíduo purificado por recristalização de EtOAc para fornecer o produto (5,14 g, 96%) como um sólido. Ή RMN (DMSO-cy δ 8,61-8,63 (m, 1H), 8,21 (m, 1H), 8,03-8,04 (m, 1H), 7,76-7,83 (m, 2H); MS(ESI*) mfe 157 (M + H)*.

B’) 4-(4-Amino-2-fluorofenóxi)picolinamida.
Uma solução de 4-amino-2-fluorofenol (Composto A de Exemplo
20, 0,81 g, 6,4 mmoles, 1,0 eq) em DMF (6,5 mL) foi tratada com tem* butóxido de potássio (0,79 g, 7,1 mmoles, 1,1 eq) em temperatura ambiente e a mistura de reação foi agitada durante 1 hora. 4-Cloropicolinamida (1,0 g,
6,4 mmoles, 1,0 eq) foi adicionada e a mistura de reação foi aquecida para 110°C durante 8 horas. A reação foi resfriado a temperatura ambiente e a mistura de reação finalizada com água. A solução heterogênea resultante foi filtrada e o material sólido foi lavada com água. O sólido foi triturado com uma pequena quantidade de MeOH seguido por Et2O. O sólido foi filtrado e secado em vácuo para fornecer o produto (1,3 g, 82%) como um sólido. Ή
RMN (DMSO-cg 5 8,49-8,50 (m, 1H), 8,12 (br s, 1H), 7,71 (br s, 1H), 7,35
7,36 (m, 1H), 7,14-7,16 (m, 1H), 7,01-7,06 (m, 1H), 6,44-6,47 (m, 2H), 5,53
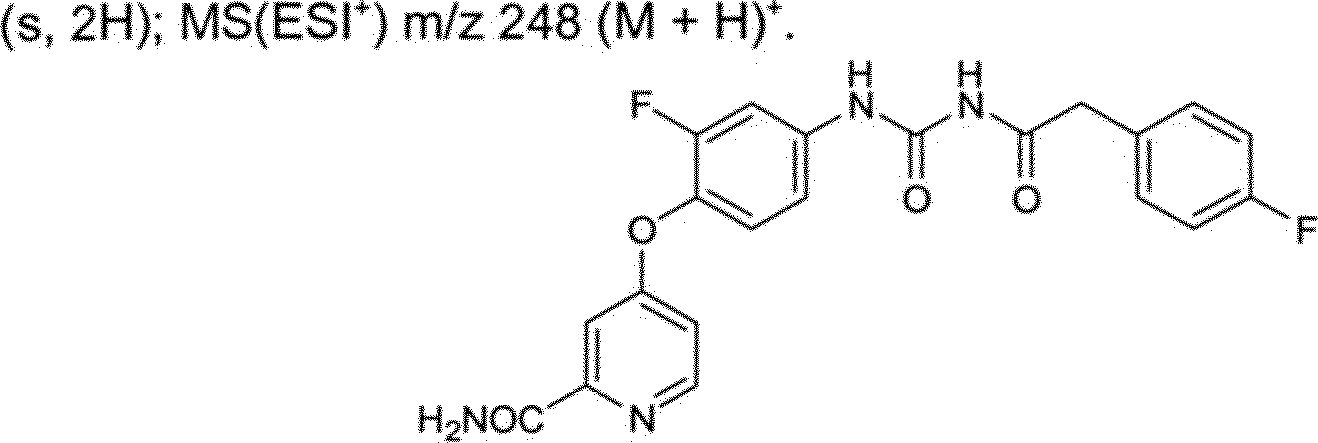
C’) 1 -(4-(2-Garbamoilpiridin-4-ilóxi)-3-fluoroferiil)-3-(2-(4-fluorofeml)acetil)uréia.
Uma solução de isocianato de 2-(4-fluorofenil)acetila (Composto
D de Exemplo 11,0,29 M em tolueno, 54,9 mL, 15,9 mmoles, 2,1 eq) foi adicionado à 4-(4-amino-2-fluorofenóxi)picolinamida (1,86 g, 7,53 mmoles, 1,0 eq) em 10/3 DCM/DMF (65 mL) em temperatura ambiente e a mistura de reação foi agitada durante 17 horas. A mistura de reação foi concentrada em vácuo e o resíduo redissolvido em GHGI3i A camada orgânica foi lavada com
NaGI aquosa saturada, a fração orgânica separada, secada (Na2SO4), filtra da e concentrada em vácuo. O resíduo foi purificado por cromatografia de sílica-gel (eluindo 1/3 hexano/EtOAc, em seguida para eluir o produto de MeOH a 5% em CHCI3), e as frações apropriadas concentradas em vácuo para fornecer o produto (2,2 g, 69%) como um sólido. Ή RMN (DMSO-<) δ 11,07 (s, 1H), 10,62 (s, 1H), 8,54 (d, 1H, J = 5,60 Hz), 8,16-8,19 (m, 1H), 7,76-7,84 (m, 2H), 7,35-7,49 (m, 5H), 7,16-7,23 (m, 3H), 3,76 (s, 2H); MS(ESf) m/z 427 (M + Hf. HRMS (ESF) calculada.: 427,1218, encontrado: 427,1214,
D*) 1 -(4-(2-Aminoplriciin-4-ilóxi)-3-fluorofeiiil)-3-(2-(4-fluorofenii)acetil)uréia, sal de cloridrato.
Bis-(trífluoroacetóxi)-iodobenzeno (Aldrich, 3,09 g, 7,20 mmoles,
1,4 eq) foi adicionado a uma solução de 1-(4-(2-carbamoilptridin-4-ilóxÍ)-3fluorofenil)-3-(2-(4-fluorofenil)acetíl)uréia (2,19 g, 5,14 mmoles, 1,0 eq), água (0,241 mL, 13,4 mmoles, 2,6 eq) e piridina (1,62 mL, 20 mmoles, 3,9 eq) em DMF (20 mL) em temperatura ambiente e a mistura de reação foi agitada durante 5 horas. A mistura de reação foi finalizada com 1 N de HCI e a solução aquosa extraída com E^O, descartando a camada orgânica. A camada aquosa foi neutralizada com 1 N de NaOH e extraída com EtOAc. As camadas orgânicas combinadas foram lavadas com LiCI aquosa a 10%, secadas (Na2SO4), filtradas e concentradas em vácuo. O resíduo foi purificado por cromatografia de sílica-gel (eluindo com 0-5% MeOH em CHCI3) e as frações apropriadas foram concentradas em vácuo. O resíduo foi dissolvido em THF (50 mL) resfriado a 0 °C e tratado com HCI anidro (4N, 10 mL, 40 mmol, 7,8 eq). A mistura de reação foi deixada aquecer para temperatura ambiente e agitada durante 2 horas resultando em uma solução heterogênea. A solução foi filtrada e o sólido lavado com EtzO e secado em vácuo para fornecer o composto do título (1,38 g, 63%) como um sólido. 1H RMN (DMSOO S 11,09 (s, 1H), 10,65 (s, 1H), 7,97-8,00 (m, 1H), 7,83-7,90 (m, 3H), 7,35-7,48 (m, 4H), 7,15-7,21 (m, 2H), 6,70-6,72 (m, 1H), 6,16-6,17 (m, 1H), 3,77 (s, 2H); MS(ESF) m/z 399 (M + H)*. HRMS (ESF) calculada.: 399,1269; encontrado: 399,1258, análise elementar para C20H16N4O3F2 1,0 HCI. 0,22 H2O Calculada.: C; 54,75, H; 4,01, N; 12,77, Cl; 8,08, encontrado:
C; 54,75, Η; 4,35, N; 4,35, Cl; 8,06,
Exemplo 25


h2n
IV-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenil)-2-flúor-5-metilbenzamida.
Diisopropiletilamina (0,035 mL, 0,200 mmol, 2,0 eq) foi adicionado a uma solução de 4-(4-amino-2-fluorofenóxi)piridin-2-amina (Composto B de Exemplo 24, 0,022 g, 0,100 mmol, 1,0 eq), ácido 2-flúor-5-metil-benzóico (Aldrich, 0,015 g, 0,100 mmol, 1,0 eq), EDO (0,021 g, 0,11 mmol, 1,1 eq) e HOBT (0,014 g, 0,100 mmol, 1,0 eq) em DMF (0,700 mL) em temperatura ambiente. A mistura de reação foi agitada em temperatura ambiente durante horas, extinguida com solução de NaHCO3 aquosa saturada e extraída com CHCI3 (3 x 10 mL), Os extratos orgânicos combinados foram secados (Na2SO4), filtrados e concentrados em vácuo. O resíduo foi purificado por HPLC preparativa de fase reversa (YMC ODS-A S10 30 x 500 mm, 30-90% 15 de MeOH aquoso com 0,1% TFA, gradiente de 30 minutos) e as frações apropriadas foram concentradas em vácuo. O concentrado foi neutralizado com solução de NaHCO3 aquosa saturada e a mistura extraída com CHCI3 (3 x 30 mL). Os orgânicos combinados foram secados (NazSO4), filtrados e concentrados em vácuo para fornecer o composto do título (0,014 g, 40%) 20 como um sólido. 1H RMN (CD3OD) δ 7,67 - 7,80 (m, 2H), 7,36 - 7,45 (m, 3H),
7,03 - 7,14 (m, 2H), 6,14 - 6,16 (m, 1H), 5,89 - 5,90 (m, 1H), 2,29 (s, 3H);
MS(ESI+) m/z 356 (M + Hf; HRMS(ESF) calculada: 356,1211, encontrado: 356,1203,
Exemplo 26


h2n ^*-(4-(2-Aminoplridin-4-ilóxi)-3-fluorofenil)-^3-(4-fluorofenilmalonamida
Diisopropiletilamina (0,105 mL, 0,604 mmol, 3,3 eq) foi adicionada a uma solução de 4-(4-amino-2-fluorofenóxi)piridin-2-amina (Composto B de Exemplo 24, 0,040 g, 0,183 mmol, 1,0 eq), ácido 3-(4-fluorofenilamino)-3oxopropanóico (Composto B de Exemplo 1,0,054 g, 0,274 mmol, 1,5 eq), e PyBroP (0,139 g, 0,298 mmol, 1,6 eq) em CH2CI2 (2,0 mL) a 0°C. A mistura de reação foi aquecer para temperatura ambiente e agitada durante 18 horas, A mistura de reação foi extingüida com uma solução de NaHCO3 aquosa 10 saturada e a solução extraída com CHCI3 (3x10 mL). Os extratos orgânicos combinados foram secados (Na2SOJ, filtrada e concentrada em vácuo. O resíduo foi purificado por cromatografia instantânea de sílica-gel (Merck 4063 μΜ, malha 230-240, eluindo 0-6% MeOH in CHCI3 para fornecer o composto do título (0,056 g, 77%) como um sólido. 1H RMN (CD3OD) δ 7,67 15 7,68 (m, 2H), 7,48 - 7,52 (m, 2H), 7,13 - 7,25 (m, 1H), 7,10 - 7,12 (m, 1H), 6,94 - 6,99 (m, 2H), 6,16 - 6,17 (m, 1H), 5,88 - 5,89 (m, 1H), 3,30 (s, 2H);
MS(ESI) mfe 399 (M - H+); HRMS (ESI*) calculada: 390,1260, encontrado: 399,1261,
Exemplo 27


1-(4-(2-Aminopiridin-4-ilóxl)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)tiouréia.
4-Fluorofenilacetílcloreto (Aldrich, 0,017 mL, 0,126 mmol, 2,5 eq) foi adicionado a uma solução de tiocianato de sódio (0,014 g, 0,176 mmol,
3,5 eq) em acetate de etila (1,0 mL) em temperatura ambiente e a mistura de reação foi agitada durante 1,5 hora para fornecer uma solução de isotiocianato de 2-(4-fluorofenil)etanoíla (0,126 M). 4-(4-Amino-2-fluorofenóxi) piridin-2-amina (Composto B de Exemplo 24, 0,011 g, 0,050 mmol, 1,0 eq) foi dissolvido em CH2CI2 (1,0 mL) e isotiocianato de 2-(4-fluorofenil)etanoíla (0,126 M, 0,50 mL, 0,063 mmol, 1,3 eq) foi adicionado e a mistura de reação foi agitada em temperatura ambiente durante 20 horas. A mistura de reação foi concentrada em vácuo e o resíduo resultante foi purificado por cromatografia instantânea de sílica-gel (Merck, 40-63 μΜ, malha 230-240, eluindo 06% MeOH em CHCI3) para fornecer o composto do título (0,008 g, 38%) como um sólido. Ή RMN (CD3OD) δ 7,85-7,95 (m, 1H), 7,67-7,69 (m, 1H), 7,13-7,28 (m, 4H), 6,95-7,00 (m, 2H), 6,05-6,15 (m, 1H), 5,90-5,91 (m, 1H), 3,65 (s, 2H); MS(ESI*) m/z 415 (M + H)*; HRMS(ESF) calculada: 415,1040, encontrado: 415,1041,
Exemplo 28
1-(3-flúor-4-(2-(4-fluorofenilamino)piridin-4-ilóxi)fenil)-3-(2-(4-flyorofenil)acetil)uréia.
A) A/-(3-flúor-4-(2-(4-fluorofenilamino)piridin-4-ilóxl-1-óxido) fenil)acetamida.
Uma mistura /M-(4-(2-cloropiridin-4-ilóxi-1-óxido)-3-fluorofenil) acetamida (Composto B de Exemplo 18, 62 mg, 0,21 mmol), 4-fluoroanilina (47 mg, 0,42 mmol), e éter de 2-metoxietila (91 mL) foi aquecida a 140°C durante 15 minutes. A mistura foi resfriada em temperatura ambiente, diluída com EtOAc (20 mL), lavada com solução NaHCO3 saturada e salmoura (diversas vezes), secada (MgSO4), e concentrada em vácuo para fornecer 4:1 de uma mistura de o composto do título e a piridina de origem como um óleo marrom claro (45 mg, 58%). O produto foi empregado na etapa subseqüente sem qualquer outra purificação. MS(ESF) m/z 372,1 (M + H)+.
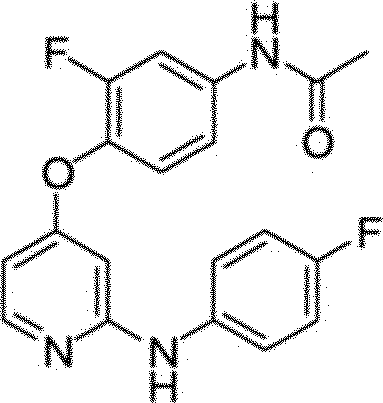
B)M(3-flúor-4-(2-(4-fluorofenilamino)piridm-4-ilóxi)fenil) acetamide.
Uma mistura de N-(3-flúor-4-(2-(4-fluorofenilamino)piridin-4-ilóxi1-óxido)fenil)acetamida (45 mg), polímero de trifenilfosfina suportada (-3 mmol/g) em poliestireno (200 mg, Fluka) e DMF (3 mL) foi aquecida a 135°C durante 48 horas. A resina foi filtrada, lavada com DMF e EtOAc. Os filtrados e lavados foram combinados e concentrados em vácuo. O produto bruto foi purificado por cromatografia instantânea empregando-se 30-80% EtOAc em hexanos como o eluent© para fornecer o composto do título (22 mg, 51%) como um sólido rosa. Ή RMN (DMSO-<) δ 10,24 (s, 1H), 8,99 (s, 1H), 8,03 (d, 1H. J = 6,3 Hz), 7,80 (dd, 1H, J = 13.0, 2.1 Hz), 7.63-7.60 (m, 2H), 7,367,29 (m, 2H), 7,05 (dd, 1H, J =:9,1, 8,6 Hz), 6,44 (dd, 1H, J = 5,5, 2,2 Hz), 6,09 (d, 1H, J = 2 Hz), 2,07 (s, 3H); MS(ESF) m/z 356,7 (M + H)+.
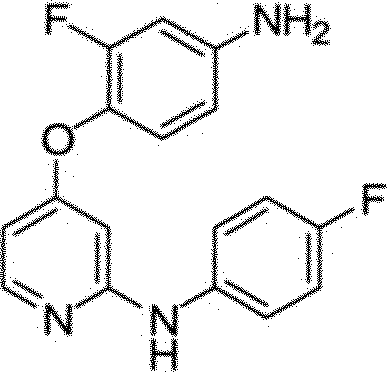
C) 4-(4-Amino-2-fluorofenóxi)-N-(4-fluorofenil)pirÍdin-2amina.
A mistura de N-(3-flúor-4-(2-(4-fluorofenilamino)piridín-4-ilóxi) fenil) açetamida (18 mg, 0,051 mmol), 6 M HGI (0,1 mL, 0,60 mmol) e MeOH (1,5 mL) foi aquecida até o refluxo durante 2 horas. A mistura foi concentrada em vácuo e o resíduo tornado básico com solução NaHCO3 aquosa saturada em seguida extraída com EtOAc. O extrato foi secado (MgSO4) e concentrado em vácuo para fornecer o composto do título (14 mg, 88%) como uma goma vermelha. ’H RMN (DMSO-dg) δ 8,97 (s, 1H), 7,98 (d, 1H, J = 5,8 Hz), 7,64 -7,60 (m, 2H), 7,05 (dd, 2H, J = 9,1, 8,8 Hz), 6,97 (dd, 1H, J = 9,4, 8,8 Hz), 6,51 (dd, 1H, J = 13,3, 2,6 Hz), 6,40 (ddd, 2H, J = 9,0, 6,2, 2,1 Hz), 6,08 (d, 1H, J = 2,0 Hz), 5,44 (br s, 2H); MS(ESL) m/z 314,17 (M + H)\
D) 1-(3-flúor-4-(2-(4-fluorofenilamino)piridin-4-ilóxi)fenil)-3(2-(4-fluorofenll)acetil)uréia.
Uma solução de 4-(4-amino-2-fluorofenóxi)-N-(4-fluorofenil) piridin-2-amina (11 mg, 0,035 mmol) em THF (1 mL) foi resfriada em um banho de gelo e tratada com uma solução de isocianato de 2-(4-fluorofenil)acetíla em tolueno (Composto D de Exemplo 11, 250 pL, 0,070 mmol) e agitada em temperatura ambiente durante 2 horas. A mistura foi concentrada em vácuo e o resíduo triturado com éter de isopropila para fornecer o composto do título (11 mg, 65%) como um sólido branco. ’H RMN (DMSO-dg) δ 11,04 (s, 1H), 10,56 (s, 1H), 9,01 (s, 1H), 8,03 (d, 1H, J = 5,6 Hz), 7,77 (dd, 1H, d = 13,3, 2,0 Hz), 7,63-7,60 (m, 2H), 7,41-7,31 (m, 5H), 7,19-7,14 (m, 2H), 7,05 (dd, 1H, J = 9,1, 8,5 Hz), 6,43 (dd, 1H, d = 6,2, 2,1 Hz), 6,10 (d, 1H, J = 2,1 Hz), 3,74 (s, 2H); MS(ESI*) m/z 493,2 (M + H)*.
Exemplo 29

M,-(4-(6-(4-(Beraii6xi)fenilamino)pirlmMin-4-ilóxl)-3-fluorofenil)-A^-(4-fluorofeníl)malonamida.

A) Af/4-(6-(4-(Benzilóxi)fenilammo)pirimidm-4-ilóxi)«3-flu<>· rofeni!)acetamida.
A mistura de A/-(4-(6-cloropirimidin-4-ilóxi)-3-fluorofenil) acetamída (Composto B de Exemplo 13, 281 mg, 1,00 mmol), 4-benziloxiani!ina (Aldrich, 398 mg, 2,00 mmol), e éter de 2-metoxietila (2 mL) foi aquecida a 160°C durante 45 minutos. A mistura resfriada foi tratada com H2O (50 mL) e extraída com EtOAc (100 mL). O extrato de EtOAc foi lavado com salmoura (3 x 25 mL), secado (MgSOJ e concentrado em vácuo para fornecer o composto do título (200 mg, 22%) como um sólido púrpuro. Ή RMN (400 MHz, DMSO<) δ 10,19 (s, 1H), 9,43 (s, 1H), 8,23 (s, 1H), 7,72 (dd, 1H, /= 12,5, 2,0 Hz), 7,44-7,42 (m, 4H), 7,38 (dd, 2H, /= 8,0, 6,9 Hz), 7,33-7,23 (m, 3H), 6,98 (d, 2H, J- 9,0 Hz), 6,07 (s, 1H), 5,07 (s, 2H), 2,05 (s, 3H); MS(ESI*) m/z 445,13 (M + H)*.

B) 6-(4-Amino-2-fluorofenóxi)-AF(4-(benzilóxi)fenil)plrimidin4-amina.
A mistura de A/»(4-(6-(4-(benzilóxi)fenilamino)pirimidín-4-ilóxí)-3fluorofenil)acetamida (150 mg, 0,34 mmol), 6 M HCI (0,5 mL) e MeOH (3 mL) foi aquecida até refluxo durante 2 horas. A mistura foi concentrada para remover o MeOH e o resíduo tratado com solução de NaHCO3 saturada e extraída com EtOAc. A fase orgânica foi secada (MgSO4) e concentrada em vácuo para fornecer o composto do título (123 mg, 90%) como um sólido rosa. Ή RMN (DMSO-cy δ 9,37 (s, 1H), 8,24 (s, 1H), 7,46 - 7,31 (m, 7H), 6,99 - 6,92 (m, 3H), 6,48 (dd, 1H, J = 12,5, 2,7 Hz), 6,39 (dd, 1H, /= 8,6, 2,7
Hz), 5,07 (s, 1H), 5,39 (br s, 2H), 5,08 (s, 2H); MS(ESI*) m/z 403,00 (M +
H)+.
C) ^^(e^-ÍBenzilóxiJfenilaminoÍpirimidin^-ilóxiJ-a-fluorofenil)-N3-(4-fluorofeml)malonamida.
O composto do título foi preparado de uma mistura de 6-(4amino-2-fluorofenóxi)-N-(4-(benzilóxi)fenil)pirimidin-4-amina (45 mg, 0,11 mmol), ácido 3-(4-fluorofenilamino)-3-oxopropanóico (Composto B de Exemplo 1, 24 mg, 0,12 mmol), TBTU (48 mg, 0,15 mmol), DIPEA (0,26 mL,
0,15 mmol), e DMF (1 mL) empregando-se um procedimento similar descrito para a preparação de Composto C de Exemplo 1, o produto bruto foi triturado com éter de isopropila para fornecer o composto do título (56 mg, 88%) como um sólido rosa. 1H RMN (DMSO-c® 6 10,47 (s, 1H), 10,27 (s, 1H), 9,45 (s, 1H), 8,25 (s, 1H), 7,77 (dd, 1H, J = 12,7, 2,0 Hz), 7,65 - 7,62 (m, 2H), 7,46 (d, 4H, 7,3 Hz), 7,40 (dd, 2H, J = 7,6, 7,3 Hz), 7,37 - 7,29 (m,
3H), 7,17 (dd, 2H, J = 9,0, 8,3 Hz), 7,00 (d, 2H, J- 9,0 Hz) 6,09 (s, 1H), 5,09 (s, 2H) 3,49 (s, 2H); MS(ESI+) m/z 582,3 (M + H)*.
Exemplo 30

1-(4-(6-(4-(Benzilóxi)femlamino)pirlmidin-4-ilóxi)-3-fluorofe- nil)-3-(2-(4-fluorofenil)acetil)uréia.
O composto do título foi preparado de 6-(4-amino-2-fluorO5 fenóxi)-A/-(4-(benzilóxi)fenil)pirimidin-4-amina (Composto B de Exemplo 29, mg, 0,11 mmol) e uma solução de isocianato de 2-(4-fluorofenil)acetila em tolueno (Composto D de Exemplo 11, 0,13 mmol) em THF empregandose um procedimento similar descrito para a preparação de Composto E de
Exemplo 11.0 produto broto foi triturado com éter de isopropila para fome10 cer o composto do título (58 mg, 90%) como um sólido rosa. 1H RMN (DMSO-<)3 11,02 (s, 1H), 10,54 (s, 1H), 9,46 (s, 1H), 8,24 (s,1H), 7,70 (dd, 1H, J = 12,7, 2,4 Hz), 7,46-7,26 (m, 9H), 7,18 (dd, 2H, J = 9,6, 8,3 Hz), 7,00 (d, 2H, J = 9,6 Hz), 6,11 (s, 1H), 5,09 (s, 2H), 3,75 (s, 2H); MS(ESF) m/z 582,3 (M + H)*.
Exemplo 31
1-(3-fiúor-4-(2-(4-fluorofenilamino)pirimidin-4-ilóxi)fenil)-3-(2- (4-fluorofenil)acetil)ureÍa.
n a
A) #-(4-(2-Cloropirimidin-4-ilóxi)-3-fluorofenil)acetamicia.
Uma mistura de 2,4-dicloropirimidina (Aldrich, 1,5 g, 10,0 mmo les), A/-(3-flúor-4-hidroxifenil)acetamida (0,85 g, 5,0 mmoles), K2CO3 (0,76 g, 5,5 mmoles), e CH3CN (100 ml) foi aquecida até o refluxo durante 2 horas.
A mistura foi concentrada e o resíduo dividido entre EtOAc e solução de NaHCO3 saturada. A de fase EtOAc foi lavada com solução de NaHCO3 sa5 furada, salmoura, secada (MgSO4) e concentrada em vácuo. O produto bruto foi purificado por cromatografia instantânea empregando-se uma forma gradiente de EtOAc a 30% em hexanos para EtOAc a 100% para fornecer o composto do título (1,1 g, 78%) como um sólido branco. Ή RMN (DMSO-ds) δ 10,22 (s, 1H), 8,63 (d, 1H, J 5,6 Hz), 7,74 (dd, 1H, J = 12,6, 2,4 Hz), 10 7,34-7,26 (m, 3H), 2,01 (s, 3H).


B) /V-(3-flúor-4-(2-(4-fluorofenilamino)pinmidin-4-ilóxi)fenil) acetamida.
Uma mistura de M-(4-(2-ciorOpirimidin-4-ilóxi)-3-fluorofenil) acetamida (100 mg, 0,36 mmol), 4-fluoroanilina (Aldrich, 40 mg, 0,36 mmol), e
1,4-dioxano (3 mL) foi aquecida até o refluxo durante 2 horas. A mistura foi concentrada em vácuo e o resíduo triturado com éter para fornecer um sólido cinza. O produto foi dissolvido em MeOH, tratada com sílica-gel (150 mg) e a mistura concentrada para secagem. O composto foi concentrado em sílica-gel e aplicado a uma coluna de sílica-gel e eluindo primeiro com EtOAc em seguida com 100:1 MeOH / NH4OH em CH2CI2 para fornecer o composto do título (40 mg, 31%) como um sólido branco. 1H RMN (DMSO-de) δ 10,19 (s, 1H), 9,61 (s, 1H), 8,33 (d, 1H, 5,6 Hz), 7,71 (d, 1H, U = 12,7 Hz), 7,40 (s, 2H), 7,30-7,26 (m, 2H), 6,86 (dd, 2H, 8,3, 8,3 Hz), 6,50 (d, 1H, J = 5,4
Hz), 2,05 (s, 3H); MS(ESr) m/z 357,13 (M + H)*.


C) 4-(4-Amino-2-fluorofenóxi)-/V-(4-fluorofeníl)pirimidín-2· arnina.
A mistura de A/-(3-flúor-4-(2-(4-fluorofenilamino)pinmídin-4-ilóxi) feníl)acetamida (32 mg, 0,09 mmol), 6 M de HOI (0,2 mL) e MeOH (2 mL)foi aquecida até o refluxo durante 2 horas. A mistura foi resfriada, diluída com EtOAc (20 mL), lavada com solução de NaHGO3 saturada e salmoura, secada (MgSO4), e concentrada em vácuo. Cromatografia instantânea em SiO2 empregando-se 30-40% de EtOAc em hexanos contendo 1 % de Et3N fornece o composto do título (15 mg, 46%) como um sólido branco. 1H RMN (DMSO-O δ 9,55 (s, 1H), 8,25 (d, 1H, J = 5,5 Hz), 7,43 (br s, 2H), 6,02-6,85 (m, 3H), 6,45 (dd, 1H, J = 13,5, 2,1 Hz), 6,38-6,35 (m, 2H), 5,35 (br s, 2H). MS(ESI*) m/z 315,17 (M + H)+.
D) 1-(3-flúor-4-(2-(4-fluorofenilamino)pírimiclin-4-ilóxi)fenil)-
3-(2-(4-fluorofenil)acetil)uréia.
Uma solução de 4-(4-amino-2-fluorofenóxi)-A/-(4-fluorofenil) pirimidin-2-amina (10 mg, 0,032 mmol) em THF (1 mL) foi resfriada em um banho de gelo e tratada com uma solução de isocianato de 2-(4-fluorofenil)acetila em tolueno (Composto 0 de Exemplo de 11, 228 >L, 0,064 mmol) e agitada em temperatura ambiente durante 2 horas. A mistura de reação foi concentrada em vácuo e o resíduo triturado com éter de isopropila para fornecer o composto do título (15 mg, 93%) como um sólido branco. Ή RMN (DMSO-O δ 11,06 (s, 1H), 10,56 (s, 1H), 9,68 (s, 1H), 8,39 (d, 1H, 5,7
Hz), 7,76 (dd, 1H, J = 13,5, 2,1 Hz), 7,43 (br s, 2H), 7,46-7,35 (m, 6H), 7,18 (dd, 2H, d = 8,8, 8,8 Hz), 6,57 (d, 1H, J = 5,4 Hz), 3,76 (s, 2H); MS(ESI*) m/z 492,0 (M + H)*.
Exemplo 32

1-(2-(4-Fluorofenil)acetil)-3-(4-((2-(piriclin-2-ilamino)tiazol-5il)metilammo)fenil)tiouréia.

A) /V1-((2-(Piridin-2-ilamino)tiazol-5-il)metíl)benzeno-1,4- diamina.
Uma solução de 2-(piridina-2-ilamino)-tias>b5-carbaldeído (0,10 g,
0,49 mmol, W02004/001059), benzeno-1,4-diamina (0,105 g, 0,97 mmol) e trietilsilano (0,19 mL, 1,2 mmoles) em CH2CI2-TFA (3:1, 4 mL) foi agitada em temperatura ambiente durante 4 horas. A mistura de reação foi concentrada 10 em vácuo e o resíduo dividido entre CH2CI2 e solução de NaHCO3 aquosa saturada. A fase orgânica foi lavada com solução de NaHCO3 aquosa saturada, salmoura, secada (MgSO4) e concentrada em vácuo. O produto bruto que continha o composto do título juntamente com benzeno-1,4-diamina e aldeído de partida e foi levado diretamente para a etapa seguinte.
B) 1-(2-(4-Fluorofeníl)acetiiy3-(4-((2-(piridlin-2-llamino)tiazol-
5-il)metilamino)fenil)tiouréia.
Cloreto de 4-fluorofenilacetila (7,4 μί, 0,053 mmol) foi adicionado a uma suspensão de NaSCN (4,5 mg, 0,055 mmol) em EtOAc (0,5 mL) e a mistura resultante foi agitada em temperatura ambiente durante 30 minu20 tos. Esta mistura foi em seguida adicionada à uma solução de mistura acima obtida em A (14,5 mg) in CH2CI2 (0,5 ml) e a mistura resultante foi agitada em temperatura ambiente durante 2 horas. A mistura de reação foi concentrada em vácuo e o resíduo foi purificado por cromatografia instantânea em SiO2 empregando-se uma eluição gradiente 2-5% MeOH-CHCI3 para forne25 cer o composto do título (2 mg) como uma película laranja. MS(ESI+) m/z 493,2 (M + H)*.
Exemplo 33


h3c
1-(4-(3-Etilpiridin-4-llóxi)-3-fluorofenil)-3-(2-(4-fluorofenil) acetil) uréia, sal de cloridrato.
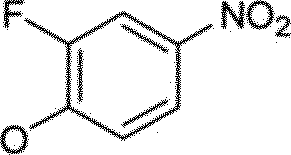

A) 4-(2-flúor-4-nitrofenóxi)-3«iociopiridina.
A mistura de 4-cloro-3-iodopiridina (1,50 g, 6,30 mmoles, preparada de acordo com Tabanella, S. e outro, Org. Biomol. Chem. 2003, 1, 4254-4261), 2-flúor-nitrofenol (Lancaster, 2,0 g, 12,7 mmoles), DIPEA (5 mL), e
NMP (10 mL) foi aquecida a 150°C. Apôs 12 horas, mais 2-flúor-nitrofenol (0,50 g, 3,18 mmoles) foi adicionado para a mistura de reação e o aqueci10 mento foi continuado durante 4 horas. A maioria dos componentes voláteis foi removida sob vácuo a 75eC, o resíduo tratado com solução de NaHCO3 aquosa saturada (150 mL) e extraído com EtOAc (2 x 100 mL). Os extratos combinados foram lavados com salmoura, secados (MgSQ4) e concentrados em vácuo para fornecer o produto· bruto. A purificação por cromatografia ins15 tantânea em sílica-gel, empregando-se 0 -100% CH2Cl2/ hexanos em seguida 2% de MeOH / CH2CI2 fornece o composto do título (1,0 g, 43%) como um sólido amarelo. Ή RMN (DMSO-de) δ 8,96 (s, 1H), 8,47 (d, 2H, J 5,5
Hz), 8,44 (dd, 1H, J = 2,7, 9,2 Hz), 7,49 (dd, 1H, J = 8,8, 8,2 Hz), 7,07 (d,
1H, J = 5,5 Hz); MS(ESI*): m^ 361,05 (M + H)*.


B) 4-(2-flúor-4-nitrofenóxi)-3-vinllpiri(iina.
Uma solução de 4-(2-flúor-4-nitrofenóxi)-3-iodopiridina (200 mg,
0,56 mmol), tributilvinilestanho (212 mg, 0,67 mmol) em DMF (1 mL) foi tratada com CsF (169 mg, 1,12 mmol) seguida por (Ph3P)4Pd (36 mg, 0,031 mmol) e Cul (10 mg, 0,056 mmol) e a mistura foi aquecida a 45°C durante 1 hora. A mistura foi resfriada, diluída com CH2CI2 (15 mL) e H2O (10 mL), agitada vigorosamente e em seguida filtrada através de Celite*. A massa filtrante foi lavada com 1:1 CH2CI2 / EtOAc e as lavagens foram combinadas com o filtrado. A solução foi lavada com salmoura, secada (MgSO4) e concentra10 da em vácuo para fornecer um óleo marrom. O produto bruto foi purificado por cromatografia instantânea em SiO2 empregando-se 0-2% MeOH / CH2CI2 para fornecer um produto semi-puro. O produto foi tratado com 2 M HCl / Et2O (10 mL) e o derivado de cloridrato precipitado coletado por filtragem e lavagem com Et2O e EtOAc para um sólido amarelo (145 mg, 87%). 1H RMN (DMSO-O δ 9,11 (s, 1H), 8,64 (s, 1H), 8,51-8,48 (m, 1H), 8,24 (d, 1H, J = 7,7 Hz), 7,83-7,79 (m, 1H), 7,28 (d, 1H, J ~ 6,0 Hz), 7,02-6,95 (m, 1H), 6,24 (d, 1H, J = 17,6 Hz), 5,68 (d, 1H, 11,5 Hz); MS(ESF): m&261,18 (M + Hf.
O sal de cloridrato acima foi convertido na base livre como segue: O cloridrato de piridina (230 mg) foi agitado com NaHCO3 (25 mL) e 20 EtOAc (20 mL) até tomar-se homogêneo e a fase EtOAc separada, lavada com salmoura, secada (MgSO4) e concentrada, o composto do título (190· mg) foi obtido como um óleo amarelo.
FV%zNH2
C) 4-(3-Etilpiridin-4-iIóxi)-3-fluorobenzenamina.
Uma solução de 4-(2-flúor-4-nitrofenóxi)-3-vinilpiridina (80 mg,
0,30 mmol) em 1:1 EtOAc / MeOH (2 mL) foi hidrogenado sobre paládiocarbono a 10% (30 mg) durante 1 hora empregando-se balão de látex de H2.
Pt2O (10 mg) foi adicionado a uma mistura e a reação continuada durante 1 hora. A mistura foi filtrada através de Celite® e concentrada em vácuo para fornecer o composto do título (50 mg, 63%) como um óleo amarelo. 1H RMN (DMSO-cy δ 8,33 (s, 1H), 8,22 (d. 1H, J = 5,6 Hz), 6,96 (dd, 1H, J = 8,7, 9,1 Hz), 6,50 (dd, 1H, J = 2,0, 13,7 Hz), 6,56 (d, 1H, J = 5,6 Hz), 6,41 (dd, 1H, J = 2,5. 6,1 Hz), 2,69 (q, 2H, J = 7fi Hz), 1,21 (t, 3H, J = 7,6 Hz).
D) 1-(4-(3-Etilpiridln-4-ilóxi)-3-fluorofenll)-3-(2-(4-fluorofenil)acetii)ureia, sal de cloridrato.
Uma solução de 4-(3-etilpiridin-4-ilóxi)-3-fluorobenzenamina (23 mg, 0,10 mmol) em CH2CI2 (1 mL) foi tratada com uma solução de solução de 0,3 de isocianato de 2-(4-fluorofenil)acetila em tolueno (Composto D de Exemplo 11,0,33 mL, 0,11 mmol) e a mistura foi agitada em temperatura ambiente durante 2,5 horas. A mistura foi concentrada sob vácuo e o resíduo triturado com 1:1 éter de isopropila / EtOAc para fornecer um sólido amarelo. Θ produto foi tratado com MeOH absoluto (1 mL) e 2 M HCl / Et2O (1 mL), agitado em temperatura ambiente durante 5 minutos e concentrado sob vácuo para fornecer o composto do título (15 mg, 36%) como um sólido amarelo pálido. ’H RMN (DMSO-c^) δ 11,04 (s, 1H), 10,57 (s, 1H), 8,41 (s, 1H), 8,26 (d, 1H, J = 5,6 Hz), 7,76 (dd, 1H, J - 2,0, 12,7 Hz), 7,40 - 7,28 (m, 4H), 7,19-7,14 (m, 3H), 6,54 (d, 1H, J = 5,6 Hz), 3,73 (s, 2H), 2,72 (q, 2H, J = 7,6 Hz), 1,23 (t, 3H, 7,6 Hz); MS(ESI*): m/z 412,20 (Μ + H)*.
Exemplo 34 h2n n
1-(4-(2-Amino-3-eti!piridln-4-ilóxi)-3-fluorofenil)«3-(2-(4- fluorofenil)acetil)uréia, sal de ácido trifluoroacético.
Cl
A) Éster de terc-butila de ácido (4-Cloro-3-iodopiridin-2-H)- carbâmico.
Uma solução de éster de terc-butila de ácido (4-cloro-piridin-2-il)4W carbâmíco (CB Research e Development Inc., 5,0 g, 22,0 mmoles), TMEDA (8 mL) em THF anidro (100 mL) foi colocada sob uma atmosfera de nitrogênio e resfriada a -70°C e tratada em gotas com 2,5 M n-BuLi em hexanos (22,0 mL, 54,8 mmoles) durante urn periodo de 30 minutos. A mistura foi agitada a -70°C durante 1 hora em seguida tratada em gotas com uma solução de l2 (14 g, 110 mmoles) em THF anidro (16 mL) a -70°C. Após a adição ser completada, a reação foi agitada a -70°C durante 30 minutos em seguida permitida aquecer para temperatura ambiente. A mistura foi tratada com uma solução de hidrogeniossulfito de sódio (16 g) em H2O (100 mL) e agitada durante 30 minutos em seguida extraída com EtOAc. O extrato foi lavado com salmoura, secado (MgSOJ e concentrado em vácuo. O produto bruto foi purificado por cromatografia instantânea em SiO2 eluindo com 0 - 5% MeOH / CH2CI2 para fornecer o composto do título (5,8 g, 78%) como um sólido branco. 1H RMN (DMSO-cy 5 9,46 (s, 1H), 8,29 (d, 1H, J = 5,6 Hz), 7,46 (d, 1H, J = 5,0 Hz), 1,44 (s, 0H); MS(ESI ): rrtó 352,99 ( M-H)‘.
Cl

B) 4-Cloro-3-iodopiriidin-2*-amina
A suspensão de éster de terc-butila de ácido (4-cloro-3-iodopiridin-2-il)-cart)âmico (5,6 g, 15,8 mmoles) em 48% de ácido hidrobrômico foi aquecido a 100°C durante 10 minutos para fornecer uma solução clara. A mistura foi resfriada, tratada com gelo amassado e tornada básica com 6 M de NaOH. O produto precipitado foi coletado por filtração a vácuo, lavado com HjO e parcialmente sorvido no funil para fornecer um sólido branco. O produto foi dissolvido em THF e a solução secada sobre MgSO4 e concentrada em vácuo para fornecer o composto do título (3,7 g, 93%) como um sólido branco. Ή RMN (DMSO-d6) δ 7,84 (d, 1H, J = 5,1 Hz), 6,73 (d, 1H, J = 5,6 Hz), 6,51 (br s, 2H); MS(ESF): m/z 254,97 (M + Hf.
HgN'ir
C) 4-(2-flúor-4-m'trofenóxí)-3-iociopiridir>-2-amina.
A mistura de 4-cioro-3-iodopiridin-2-amina (3,6 g, 14,2 mmoles) e 2-flúor-4-nitrofenol (Lancaster, 4,5 g, 28,4 mmoles), DIPEA (3,6 mL, 20,7 mmoles) e NMP (8 mL) foi colocada em um vaso de pressão de vidro e aquecida rapidamente para 170°C e o aquecimento continuado durante 18 horas. Os componentes voláteis foram destilados sob pressão reduzida e o resíduo viscoso despejado em água-gelada (150 mL). A mistura foi sonicada durante 15 minutos afim de romper o sólido gomoso e o pH da mistura foi ajustado para 7,5 solução de NaHCO3 aquosa saturada. O sólido foi coletado por filtração a vácuo, lavado com H2O, sorvido parcialmente seco no funil. O sólido parcialmente secado foi suspenso em tolueno (150 mL) e a mistura concentrada em vácuo e o processo repetido 3 vezes para fornecer um sólido marrom. O produto foi dissolvido em MeOH (150 mL), tratado com 4 M HCI /1,4-dioxano (8 mL) e agitado em temperatura ambiente durante 5 minutos e em seguida a mistura foi concentrada em vácuo. O cloridrato desse modo obtido, foi lavado e triturado com EtOAc e dividido entre EtOAc e solução de NaHCO3 aquosa saturada. A fase de EtOAc foi separada, lavada com salmoura, e em seguida secada (MgSO4). A solução de EtOAc foi tratada com carvão vegetal ativado, agitada em temperatura ambiente durante 10 minutos e o carvão vegetal filtrado. A solução foi concentrada em vácuo para fornecer o composto do título (3,9 g, 74%) como um sólido amarelo. Ή RMN (DMSOO δ 8,39 (dd, 1H, 2,5, 10,7 Hz), 8,12 (dd, 1H, J = 1,5, 0,2
Hz), 7,86 (d, 1H, J = 5,6 Hz), 7,32 (dd, 1H, J = 8,6, 8,6 Hz), 6,40 (br s, 2H), 6,18 (d, 1H, J - 5,6 Hz).

D) 4-(2-flúor-4-nitrofenóxi)-3-vinilplridin-2-amina.
O composto do título foi preparado de 4-(2-flúor-4-nitrofenóxi)-3iodopirídin-2-amina e tributílvinilestanho por meio de uma reação de acoplamento Stille da mesma maneira com descrito na Etapa B de Exemplo 33, Ή RMN (DMSO<) δ 8,35 (dd, 1H, J = 10,7, 3,1 Hz), 8,09 (d, 1H, J = 9,2 Hz), 7,85 (d, 1H, J = 5,6 Hz), 7,31-7,15 (m, 1H), 6,54 (dd, 1H, J - 17,8,11,7 Hz), 6,24 (br s, 2H), 6,20 (d, 1H, J = 5,6 Hz), 5,71 (d, 1H, J = 17,8 Hz), 5,46 (d, 1H, J = 11,7 Hz); MS(ESf): m/z 276,17 (M + H)\ o o
E) 4-(2-flúor-4-nitrofer»óxi)-3-vlnilpiriciin-2-ilcarbamato de terc-butila.
Uma solução de 4-(2-flúor-4-nitrofenóxi>3-vinilpiridin-2-amina (60 mg, 0,22 mmol) em 1,4-dioxano (0,5 mL) e álcool de terc-butíla (1,5 mL) foi tratada com Boc2O (140 mg, 0,64 mmol) e aquecida a 65*C durante 5 horas. A mistura foi resfriada, dividida entre EtOAc e solução de NaHCO, aquosa saturada. A fase EtOAc foi separada, lavada com salmoura, secada (MgSO4) e concentrada em vácuo. O produto bruto foi purificado por cromatografia instantânea em SiO2 para fornecer o composto do título (50 mg, 60%) como um sólido amarelo. Ή RMN (DMSQ-de) δ 9,37 (s, 1H), 8,41 (dd, 1H, J = 10,7, 2,5 Hz), 8,22 (d, 1H, J = 5,6 Hz), 8,15 (d, 1H, J = 8,6 Hz), 7,42 (t, 1H, U = 8,6 Hz), 6,86 (d, 1H, J = 5,6 Hz), 6,58 (dd, 1H, J = 17,8,11,7 Hz), 5,82 (d, 1H, J = 16,3 Hz), 5,52 (d, 1H, J = 11,7 Hz), 1,42 (s, 9H); MS(ESF): m/z 376,18 (M + H)*.
F) 4-(4-amÍno-2-fluorofenóxi)-3-etilplridin-2-ilcarbamato de terc-butila.
Uma solução de 4-(2-flúor-4-nitrofenóxi)-3-vinilpiridin-2-iteart>amato de terc-butila (48 mg, 0,13 mmol) foi hidrogenada sobre paládiocarbono a 10% (10 mg) e Pt2O (5 mg) durante 1,5 hora empregando-se H2 de um balão de borracha. A mistura foi filtrada através de Celite® e o filtrado concentrado em vácuo para fornecer o composto do título (40 mg, 89%) como um sólido amarelo pálido. Ή RMN (DMSO-ds) δ 9,04 (s, 1H), 8,03 (d, 1H,
J = 5,6 Hz), 6,95 (dd, 1H, J = 8,6, 8,6 Hz), 6,50 (dd, 1H, J = 2,5, 13,2 Hz), 6,41 (dd, 1H, 2,5, 9,4 Hz), 6,36 (d, TH, J = 5,6 Hz), 5,44 (s, 2H), 2,672,62 (m, 2H), 1,43 (s, 0H), 1,11 (t, 3H, J - 7,1 Hz); MS(ESr): rtó 348,22 (M + H)T


G) 3-etil-4-(2-flúor-4-(3-(2-(4-fluorofenil)acetil)ureido) fenóxi) plridin-2-ilcarbamato de terc-butila.
O composto do título foi preparado de 4-(4-amino-2fluorofenóxi)-3-etilpiridin-2-ilcarbamato de terc-butila (20 mg, 0,058 mmol) e solução de 0,3 M de isocianato de 2-(4-fluorofenil)acetila em tolueno (232 μί, 0,070 mmol) em THF da mesma maneira como a Etapa D de Exemplo 33, MS(ESr): ot 527,31 (M + H)*.
H) 1-(4-(2-Amino-3-etilpiridin-4-ilóxl)-3-fluorofenil)-3-(2-(4fluorofenil)acetil)uréiat sal de ácido trifluoroacético.
Uma solução de 3-etil-4-(2-flúor-4-(3-(2-(4-fluorofenil)acetil) ureído)fenóxi)piridin-2-ilcarbamato de terc-butila (16 mg, 0,03 mmol) foi dissolvida em THF anidro (0,5 mL) e tratada com 4 M de HCI / 1,4-díoxano (1,5 mL) e agitada em temperatura ambiente durante 3 horas. A mistura foi concentrada em vácuo e o produto purificado por método de HPLC preparativa A para fornecer o composto do título (5 mg, 36%) como um sólido branco. 1H
100
RMN (DMSO-O δ 11,06 (s, 1H), 10,6 (s, 1H), 7,80 - 7,79 (m, 4H), 7,43 7,33 (m, 4H), 7,16 (dd, 2H, J = 8,9, 8,9 Hz), 6,19 ( d, 1H, J = 7,1 Hz), 3,73 (s, 2H), 2,71 -2,66 (m, 2H), 1,10 (t, 3H, J = 7,1 Hz); MS(ESF): m/z 427,18 (M + H)*.
Exemplo 35

1-(4-(3-(2«(4-Aminocicloex-1-enil)etinil)piridln-4-ll0xl)-3fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia.
BocHN

OTf
A) 4-(ferc-Butox»carbonil)cicloex-1-enil trifluorometanossulfonato.
Uma solução de MBoc-4-aminocicloexanona (Astatech Inc., 213 mg, 1,0 mmol) em THF (7 mL) foi resfriado a -70°C e tratada com 0,5 M de uma solução de KHMDS em tolueno (2,4 ml, 1,2 mmol). A mistura foi agitada a -70°C durante 20 minutos, tratada em gotas com uma solução de feniltrifluorometanossulfonimida (392 mg, 1,1 mmol) em THF (4 mL) e agitada a 15 70°C durante 25 minutos. A mistura foi finalizada com solução de NH4Ci aquosa saturada, diluída com EtOAc, lavada com Na2CO3 a 10% e salmoura, secada (MgSOJ e concentrada em vácuo. O produto bruto foi purificado por cromatografia instantânea em SiO2 eluindo com 10 -25% EtOAc / hexanos para fornecer o composto do título (180 mg, 52%) como um sólido branco.
1H RMN (DMSO-cy δ 5,68 (s, 1H), 4,50 (s, 1H), 3,82 (s, 1H), 2,68 - 2,25 (m,
3H), 2,22 -1,89 (m, 2H), 1,87 - 1,63 (m, 1H), 1,43 (s, 9H).
BocHH
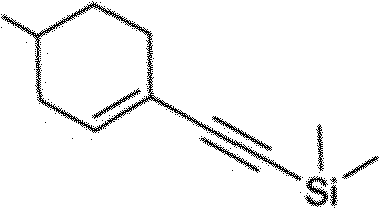
B) 4-(2-(trimetilsilil)etinil)cicloex-3-enilcarbamato de ten>
101 botila.
Uma mistura de trifluorometanossulfonato de 4-(terc-butoxicarbonila)cicloex-1-enila (170 mg, 0,49 mmol), trimetilsililacetileno (138 pL, 0,98 mmol), Et3N (0,68 mL) e THF (8 mL) em um frasco de reação foi purgada com argônio e tratada sucessivamente com Gul (14 mg, 0,072 mmol) e (Ph3P)4Pd (27 mg, 0,024 mmol). A mistura de reação foi agitada em temperatura ambiente durante 25 minutos, e em seguida diluída com EtOAc (50 mL), lavada com solução de NaHGO3 aquosa saturada e salmoura, secada (MgSO4) e concentrada em vácuo. O produto bruto foi purificado por cromatografia instantânea em SiO2 eluindo com 0 -25% de EtOAc / hexanos para fornecer o composto do título (116 mg, 81%) como um sólido amarelo. Ή RMN (DMSO-cy δ 6,06 (s, 1H), 4,50 (s, 1H), 3,76 (s, 1H), 2,46 (d, 1H, J 18,8 Hz), 2,36-2,14 (m, 2H), 2,00-1,78 (m, 2H), 1,66-1,50 (m, 1H), 1,43 (s, 9H), 0,27-0,05 (m, 9H).

C) 4-etinilcicloex-3-enilcarbamato de terc-butila.
Uma solução de 4-(2-(trimetilsi!H)etinil)cícloex-3-enilcarbamato de terc-butila (112 mg, 0,38 mmol) em THF foi resfriada a -15°G, tratada com 1,0 M fluoreto de tetrabutilamônio em THF (Aldrich, 440 pL, 0,44 mmol) e a mistura agitada a -15°C durante 40 minutos. A mistura foi tratada com Na2CO3 a 5% (25 mL) e extraída com éter. O éter extraído foi lavado com Na2GO3 a 5% salmoura, secada (MgSQ4) e concentrada em vácuo para fornecer o composto do título (83 mg, 99%) como um óleo marrom. *H RMN (DMSO-O S 6,09 (S, 1H), 4,51 (s, 1H), 3,77 (s, 1H), 2,82 (s, 1H), 2,47 (d, 1H, J = 18,3 Hz), 2,35-2,16 (m, 2H), 2,04-1,79 (m, 2H), 1,72 - 1,51 (m, 1H), 1,43 (s. 0H).

D) 4-(2-(4-(2-flúor-4-nitrofenóxi)piridin-3-il)etinil)cicloex-3
102 enilcarbamato de terc-butila.
Uma solução de 4-(2-flúor-4-nitrofenóxi)-3-iodopiridina (Composto A de Exemplo 33, 130 mg, 0,36 mmol) e /V-Boc-4-etinilcicloex-3-enamina (80 mg, 0,36 mmol) em THF anidro (2 mL) foi tratada com Et3N (2 mL) e o desgaseificado por vácuo/purga de argônio. A solução foi tratada com paládio de tetracistrifenilfosfina (20 mg, 0,0018 mmol) e Cui (10 mg, 0,054 mmol) e em seguida aquecida até o refluxo durante 2 horas. A mistura foi resfriada e dividida entre solução de NaHCO3 aquosa saturada e EtOAc. A fase EtOAc foi separada, lavada com salmoura, secada (MgSO4) e concentrada em vácuo, O produto bruto foi purificado por cromatografia instantânea em sílica-gel eluindo com 0-40% EtOAc / hexanos para fornecer o composto do título (124 mg, 76%) como um sólido amarelo. Ή RMN (DMSO-d6) δ 8,68 (s, TH), 8,51 (d, 1H, J = 5,6 Hz), 8,43 (dd, 1H, J = 2,5, 10, 7 Hz), 8,15 (d, 1H, J = 9,2 Hz), 7,49 (dd, 1H, J = 8,6, 8,6 Hz), 7,14 (d, 1H, J = 5,6 Hz), 6,85 (d, 1H, J = 7,1 Hz), 6,04- 6,00 (m, 1H), 3,48 - 3,35 (m, 1H), 2,36 - 2,25 ( m, 1H), 2,17 - 2,04 (m, 2H), 2,03 - 1,89 (m, 1H), 1,78 -1,69 (m, 1H), 1,46 - 1,35 (m, 1H), 1,36 (s, 9H); MS(ESI*| m/z 454,27 (M + H)*.
E) 4-(2-(4-(4-amino-2-fIuorofenóxi)piridin-3-il)etinil)cicloex3-enilcarbamato de terc-butila.
A mistura 4-(2-(4-(2-flúor-4-nitrofenóxi)piridin-3-il)etinil)cicloex-3enilcarbamato de terc-butila (110 mg, 0,24 mmol), pó de ferro, -325 mesh(150 mg, 2,7 mmoles), NH4CI (280 mg, 5,3 mmoles), DMF(1 mL), H2O (1 mL) e EtOH (1 mL) foi aquecida a 100°C durante 30 minutos. A mistura foi filtrada através de uma almofada de Celite® empregando-se DMF para amassar a massa filtrante e o filtrado tomado básico para o pH 8 com solução de NaHCO3 aquosa saturada. A mistura foi extraída duas vezes com EtOAc e secada (MgSO4) e concentrada em vácuo para fornecer o composto do título (105 mg) que foi empregado sem qualquer outra purificação. MS(ESF):
103


F) 4-(2-(4-(2-flúor-4-(3-(2-(4-fluorofenil) acetil) ureído)fenóxi) pirldin-3-il)etinil)eicteex-3-enileartamato de terc-butila.
Uma solução de 4-(2-(4-(4-amino-2-fluorofenóxí)píridin-3-il)etinil) cicloex-3-enílcarbamato de terc-butila (50 mg, 0,12 mmol) em CH2CI2 seco (2 mL) foi tratada com uma solução de 0,3 M de isocianato 2-(4-fluorofenil) acetila em tolueno (Composto D de Exemplo 11,0,8 mL, 0,24 mmol) e a mistura agitada em temperatura ambiente durante 1 hora. Os solventes foram evaporados sob vácuo e o resíduo purificado por cromatografia instantânea em sílica-gel eluindo com 10-60% EtOAc / hexanos para fornecer o composto do título (50 mg, 69%) como um sólido branco. Ή RMN (DMSO-d6) δ 11,03 (s, 1H), 10,57 (s, 1H), 8,57 (s, 1H), 8,36 (d, 1H, J = 5,7 Hz), 7,78 (dd, 1H, J = 1,8, 13,1 Hz), 7,41 - 7,29 (m, 3H), 7,16 (dd, 3H, J = 8,6, 8,6 Hz), 6,85 (d, 1H, J = 8,3 Hz), 6,70 (d, 1H, J = 5,7 Hz), 6,13 - 6,08 (m, 1H), 3,73 (s, 2H), 3,51 - 3,41 (m, 1H), 2,38 - 2,27 (m, 1H), 2,27 - 2,20 (m, 2H), 1,82 -1,72 (m, 1H), 1,54 -1,28 (m, 2H), 1,37 (s, 9H); ESI MS): m/z 603,24 (Μ + H)*.
G) 1-(4-(3-(2-(4-Aminocicloex-1-enil)etinil)piridin-4-ilóxi)-3fluorofenil)-3-(2-(4-fiuorofenil)acetil)uréía, sal de dicloridrato.
Uma solução de 4-(2-(4-(2-flúor-4-(3-(2-(4-fluorofenil) acetil) ureído)fenóxi)píridin-3-il)etinil)cícloex-3-enilcarbamato de terc-butila (40 mg, 0,066 mol) em 1,4-dioxano anidro (2 mL) foi resfriada a -10°C e tratada com 4 M HCI/1,4-dioxano (4 mL). A mistura foi agitada a -5°C durante 2,5 horas em seguida em temperatura ambiente durante 1 hora. A mistura foi concentrada sob vácuo sem qualquer aquecimento para fornecer o composto do título (32 mg, 84%) como um sólido amarelo. 1H RMN (DMSO-d6) δ 11,05 (s, 1H), 10,61 (s, 1H), 8,69 (s, 1H), 8,44 (d, 1H, J = 6,1 Hz), 8,06 (d, 1H, 2,0
Hz), 7,80 (dd, 1H, J = 12,7, 2,0 Hz), 7,46 - 7,38 (m, 1H), 7,35 (dd, 1H, J 8,6, 5,6 Hz), 7,19 - 7,13 (m, 1H), 6,82 (d, 1H, J = 5,6 Hz), 6,17 (s, 1H), 3,74
104 (s, 2H), 3,73 - 3,62 (m, 2H), 3,62 - 3,54 (m, 1H), 3,34 - 3,22 (m, 1H), 2,31 (s,
1H), 1,99 - 1,96 (m, 1H), 1,71 - 1,66 (m, 1H); MS(ESf): m/z 503,12 (M + H)\ Exemplo 36

1-(4-(3-(3-(3-(Aminometil)azetidin-1-il)prop-1-inil)piridin-4ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de tricloridrato.
FV\rzNG2 pH JL T c/W
A) 3-(4-(2-flúor-4*nitrofenóxl)piriclin-3-il)prop-2-in-1 -ol.
Uma solução de 4-(2-flúor-4-nitrofenóxi)-3-iodopiridina (Composto A de Exemplo 33, 300 mg, 0,83 mmol), álcool propargilico (Aldrich, 145 pL, 2,50 mmol), Et3N (2 mL) e THF anidro (2 mL) foi desgaseificado por vácuo/purga de argônio e tratada com Pd(Ph3P)4 (31 mg, 0,027 mmol) e Cui (10 mg, 0,054 mmol). A mistura foi aquecida até o refluxo sob atmosfera de argônio durante 10 minutos, resfriada em temperatura ambiente e diluída com EtOAc (25 mL) e H2O (20 mL). A fase EtOAc foi lavada com água e salmoura, secada (MgSO4) e concentrada em vácuo. O resíduo bruto foi purificado por cromatografia instantânea em sílica-gel empregando-se 0-3% MeOH / CH2CI2 para fornecer o produto desejado (185 mg, 77%) como um sólido amarelo pálido. Ή RMN (DMSO-d6) δ 8,69 (s, 1H), 8,49 (d, 1H, J = 5,6
Hz), 8,43 (dd, 1H, J ~ 10,7, 2,5 Hz), 8,17 (d, 1H, J = 9,2 Hz), 7,57 (t, 1H, J =8,6 Hz), 7,04 (d, 1H, J = 5,6 Hz), 5,40 (t, 1H, J = 6,1), 4,28 (d, 2H, J = 6,1 Hz); MS(ESF): 289,13 (M + H)*.
105

B) (1-(3-(4-(2-flúor-4-nitrofenóxi)piridin-3-il)prop-2-mil)azetidin-3-il)metilcarbamato de ferobutila.
Uma solução de 3-(4-(2-flúor-4-nitrofenóxi)piridin-3-il)prop-2-in-1ol (43 mg, 0,15 mmol) e DIPEA (45 pL, 0,26 mmol) em THF anidro (1,5 mL) foi resfriada a 0°C e tratada com cloreto de metanossulfonila (15 mg, 0,11 mmol) em porções. Após agitar a 0°C durante 1 hora, a mistura foi concentrada sob pressão reduzida. O resíduo foi tratado com DMF (1,0 mL), DIPEA (45 pL, 0,26 mmol) e éster de terc-butila de ácido azetidin-3-ilmetilcarbâmico (Beta Pharma Inc., 145 mg, 0,78 mmol) e agitada em temperatura ambiente durante 2 horas. A mistura de reação foi dividido entre EtOAc e solução de NaHCO3 aquosa saturada e a fase EtOAc separada, lavada com salmoura secada (MgSOJ e concentrada em vácuo. O resíduo foi purificado por cromatografia instantânea em SiO2 eluindo com 1-5% MeOH / CH2CI2 para fornecer o composto do título (33 mg, 48%) como um óleo incolor. Ή RMN (DMSCMg) 8 8,74 (s, 1H), 8,51 (d, 1H, J - 5,6 Hz), 8,41 (dd, 1H, J 10,7,2,5 Hz), 8,15 (d, 1H, J = 9,2 Hz), 7,53 (t, 1H, J = 8,6 Hz), 7,09 (d, 1H, J = 6,1 Hz), 6,86 (t, 1H, J = 5,6 Hz), 3,39 (s, 2H), 3,24-3,14 (m, 2H), 3,07 2,98 (m, 2H), 2,94 - 2,87 (m, 2H), 2,37 - 2,26 (m, 1H), 1,33 (s, 9H); MS(ESF): m/z 401,20 (100), [(M-C4H9)]+; m/z 457.20 (25), (M + H)+.

C) (1 -(3-(4-(4-amíno-2-fluorofenóxl)piridin-3-il)prop-2-inil) azetidin-3-il)metilcarbamato de terc-butila.
106
O composto do título foi preparado peta redução de (1-(3-(4-(2flúor-4-nitrofenóxi)piridin-3-il)prop-2-inÍI)azetidin-3-il)metilcarbamato de tercbutila (30 mg, 0,66 mmol) da mesma maneira como na Etapa E de Exemplo 35 empregando-se pó de Fe (50 mg, 0,091 mmol) e NH4CI (96 mg, 1,82 mmoles). O produto foi empregado em reações subsequentes sem qualquer purificação. MS(ESF): m^ 371,24 (100), KM-C4H8)J+; m/z 427,27 (25), (M +
H)*.

D) (1 -(3-(4-(2-flúor-4-(3-(2-(4-fluorofeni!)acetiI)ureído)ferióxi) piridin-3-il)prop-2-inil)azetidÍn-3-il)metilcarbamato de terc-butila.
O composto do título foi preparado de (1-(3-(4-(4-amino-2fluorofenóxi)piridin-3-il)prop-2-inil)azetidin-3-il)metilcarbamato de terc-butila (25 mg, 0,059 mmol) e uma solução de 0,3 M de isocianato de 2-(4-fluorofenil)acetila em tolueno (Composto D de Exemplo 11,0,37 mL, 0,11 mmol) da mesma maneira como a Etapa D de Exemplo 33 para fornecer o composto do título como um sólido branco (20 mg, 57%). 1H RMN (DMSO-cQ δ 11,04 (s, 1H), 10,58 (s, 1H), 8,63 (s, 1H), 8,37 (d, 1H, J = 5,5 Hz), 7,78 (d, 1H, 12,6 Hz), 7,40 - 7,33 (m, 4H), 7,16 (dd, 2H, J = 8,8, 8,9 Hz), 6,89 6,87 (m, 1H), 6,68 (d, 1H, J = 5,5 Hz), 3,73 (s, 2H), 3,45 (s, 2H), 3,26-3,24 (m, 2H), 3,07-3,04(m, 2H), 2,98-2,96 (m, 2H), 2,38 - 2,35(m, 2H), 1,32 (s, 9H); MS(ESr): m/z 606,26 (M + H)*.
E) l-(4-(3-(3-(3-(Aminometil)azetidin-1-il)prop-1-inil)piriciin-
4-Ílóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de tricloridrato.
(l-(3-(4-(2-flúor-4-(3-(2-(4-fluorofenil)acetil)ureído)fenóxi)piridin3-ÍI)prop-2-inil)azetidin-3-il)metilcarbarnato de terc-butila (20 mg, 0,033 mmol) foi dissolvido em CH2CI2 (2 mL) e tratado com TFA (0,5 mL) e a mistura agitada em temperatura ambiente durante 1,5 bora. A mistura foi concentrada em vácuo e purificada por HPLC preparativa (Coluna A) para fornecer o sal de
107
TFA. O sal de TFA foi dissolvido em MeOH absoluto e tratado com 1,0 M de HCI / éter agitado durante 5 minutes e concentrado em vácuo para fornecer o composto do título (9 mg, 45%) como um sólido branco. Ή RMN (DMSOde) 5 11,07 (s, 1H), 10,06 (s, 1H), 8,96 (m, 1H), 8,61 - 8,52 (m, 1H), 8,36 8,25 (s, 2H), 7,82 (d, 1H, J = 12,2 Hz), 7,45 - 7,42 (m, 2H), 7,37 - 7,33 (m, 2H), 7,18 - 7,14 (m, 2H), 6,92 (d, 1H, J = 6,1 Hz), 4,48 (s, 2H), 4,27 - 3,98 (m, 2H), 3,76 (s, 2H), 3,30 - 3,20 (m, 1H), 3,16 - 3,00 (m, 2H); MS(ESI+): mA 506,18 (M + Hf.
Os exemplos 37-40 foram preparados de uma maneira similar ao qual foi descrita no Exemplo 36,
Exemplo 37
-(4-(3-(3-(3-Amlnoazetidin-1 -il)prop-1 -inil)plriciin-4-ilóxi)-3fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de tricloridrato.
MS(ESI*): mà 492,17 (Μ * H)*
Exemplo 38
-(3-flúor-4-(3-(3-(piperazin-1 -il)prop-1 -iniljpirid in-4ilóxi)fenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de tricloridrato.
Ή RMN (DMSOO δ 11,07 (s, 1H), 10,63 (s, 1H), 9,44 (s, 1H),
8,91 (s, 1H), 8,50 (d, 1H, J-6,2 Hz), 7,84-7,78 (m, 1H), 7,45-7,39 (m, 2H), 7,38-7,32 (m, 2H), 7,16 (t, 2H, J = 8,8 Hz), 6,85 (d, 1H, J = 6,2 Hz), 4,26 (s, 2H), 3,75 (s, 2H), 3,34 (br s, 4H), 2,49 (br s, 4H); MS(ESF): mA 506,23 (M + H)*.
108
Exemplo 39
NH?
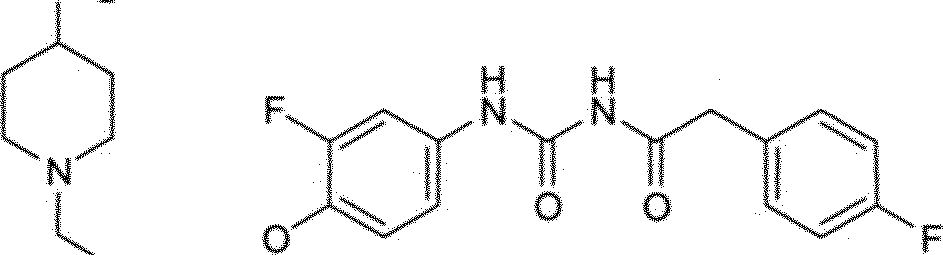
ir
1-(4-(3-(3-(4-Aminopiperidin-1-il)prop-1-inil)piridin-4-ilóxi)-3fluorofenil)-3-(2-(4-fluorofenii)acetil)uréia. sal de tricloridrato.
1H RMN (DMSO-cy 5 11,07 (s, 1H), 10,62 (s, 1H), 8,81 (s, 1H),
8,48 (d, 1H, J = 6,1 Hz), 8,31 (s, 2H), 7,80 (dd, 1H, J = 2,2, 12,7 Hz), 7,44 7,33 (m, 4H), 7,19 - 7,13 (m, 2H), 6,78 (d, 1H, J = 5,7 Hz), 4,40 (s, 2H), 3,74 (s, 2H), 3,64 - 3,60 (m, 2H), 3,34-3,22 (m, 1H), 3,19 - 3,13 (m, 2H), 2,16 2,13 (m, 2H), 1,99 -1,88 (m, 2H); MS(ESf): m/z 506,23 (M + H)*.
Exemplo 40

(±)-1 -(4-(3-(3-(3-Ammopirrolidin-1 -il)prop-1 -i n i l)pi ridi n-4ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sai de tricloridrato.
Ή RMN (DMSO-dg) δ 11,08 (s, 1H), 10,65 (s, 1H), 8,96 (s, 1H),
8,54 (d, 1H, J = 6,1 Hz), 7,83 (d, 1H, J = 12,7 Hz), 7,43 (s, 2H), 7,37 - 7,33 (m, 2H), 7,16 - 7,14 (m, 2H), 6,90 (d, 1H, J = 5,6 Hz), 4,62 (s, 2H), 4,06 15 3,87 (m, 1H), 3,75 (s, 2H), 3,70 - 3,55 (m, 3H), 3,49 - 3,44 (m, 2H), 2,25 2,08 (m, 1H); MS(ESf): m/z 506,22 (M + H)*.
109
Exemplo 41

1-(3-flúor-4-(3-(3-((3R,4R)-3-hidróxi-4-(plrrolidin-1-il)plrroli- din-1-il)prop-1-inil)piridin-4-ilóxí)fenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de tricloridrato.

A) (3R,4R)-1-(3-(4-(2-flúor-4-nitrofenóxi)pirldin-3-ll)prop-2inil)-4-(pirrolidin-1*il)pirrolidin-3-ol.
Uma solução de 3-(4-(2-flúor-4-nitrofenóxi)piridin-3-il)prop-2-in-1ol (Composto A de Exemplo 36, 43 mg, 0,15 mmol) e DIPEA (45 pL, 0,26 mmol) em THF anidro (1,5 mL) foi resfriada a 0°C e tratada com cloreto de metanossulfonila (15 mg, 0,11 mmol) em porções. Após agitar a 0°C durante hora, a mistura foi concentrada sob pressão reduzida. O resíduo foi tratado com DMF (1,0 mL), DIPEA (45 pL, 0,26 mmol) e (3R,4R)-4-(pirrolidin-1il)pirroltdin-3-ol (Lexicon Pharmaceutical Corp., 94 mg, 0,6 mmol) e agitada em temperatura ambiente durante 2 horas. A mistura de reação foi dividido entre EtOAc e solução de NaHCO3 aquosa saturada e a fase EtOAc separada, lavada com salmoura, secada (MgSOJ e concentrada em vácuo. O produto bruto foi purificado por cromatografia instantânea em SiO2 eluindo com 0-1, 5% MeOH / CH2CI2 para fornecer o composto do título (38 mg, 59%) como um óleo marrom. Ή RMN (DMSO<) δ 8,72 (s, 1H), 8,54 (d, 1H,
J = 5,6 Hz), 8,40 (dd, 1H, J = 10,7, 2,5 Hz), 8,14 (d, 1H, J - 9,2 Hz), 7,43 (t,
1H, J = 8,6 Hz), 7,15 (d, 1H, 5,6 Hz), 4,99-4,81 (m, 1H), 4,11-4,10 (m,
0,5H), 3,92-3,84 (m, 1H), 3,54 (s, 2H), 3,59-3,50 (m, 0,5H), 3,16-3,15 (m.
110
1H), 2,79-2,75 (m, 1H), 2,60-2,32 (m, 4H), 1,70-1,59 (m, 4H); MS(ESF): m/z
427,24 (M + H)*.
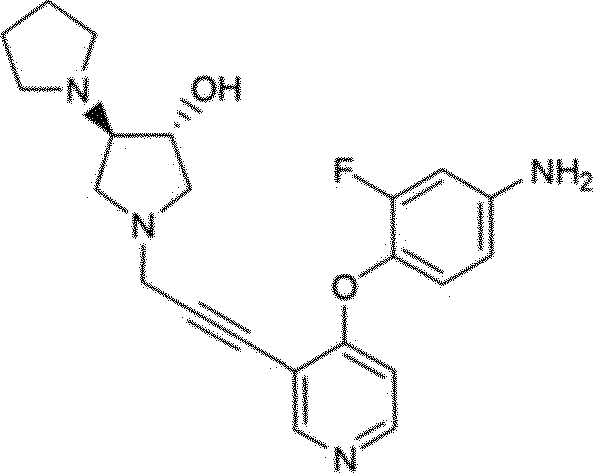
B) (3/?,4/^-1«(3-(4-(4-Amino-2-fluorofenóxi)piridin-3-il)prop-
2-inil)-4-(pirrolidin-1-il)pirrolidin-3-ol
A mistura de (3R,4R)-1-(3-(4-(2-flúor-4-nitrofenóxÍ)piridin-3-il) prop-2-inil)-4-(pirrolidin-1-il)pirrolidin-3-ol (35 mg, 0,082 mmol), DMF (1 mL),
EtOH (1 mL) e H2O (1 mL) foi tratada com pó de Fe (67 mg, 1,2 mmol, 2,4 mmoles) e aquecida a 100°C durante 45 minutos. A mistura foi filtrada através de Celite* tomada básica com NaHCO3 e concentrada em vácuo. O 10 resíduo foi dividido entre EtOAc e solução de NaHCO3 aquosa saturada. A fase EtOAc foi secada (MgSO4) e concentrada em vácuo para fornecer a anilina brutoa (16 mg, 50%) que foi empregada diretamente na etapa seguinte sem outra purificação. MS(ESr): m/z 397,28 (M + H)*.
C) 1-(3-flúor-4-(3-(3-((3R,4R)-3-hidróxi-4-(pirrolidin-1-il) pir15 rolidin-1-il)prop-1-inii)piridin-4-ilóxi)fenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de tricloridrato,
Q composto do título foi preparado de (3R,4R)-l-(3-(4-(4-amino-
2-fluorofenóxi)piridin-3-il)prop-2-ÍnÍI)-4-(pirrolidin-1-il)pirrolidin-3-ol (16 mg,
0,04 mmol) e uma solução de 0,3 M de isocianato2-(4-fluorofenil)acetila em tolueno (Composto D de Exemplo 11,0,13 mL, 0,04 mmol) de uma maneira similar àquela de Etapa D de Exemplo 33, o produto bruto foi purificado por
HPLC preparativa (Coluna A) e convertido no sal de cloridrato da mesma maneira como na Etapa E de Exemplo 36 para fornecer o composto do título (9 mg, 33%) como um sólido branco. Ή RMN (DMSO-d6) δ 11,06 (s, 1H), 25 10,62 (s, 1H), 8,86 (s, 1H), 8,45 (d, 1H, J = 6,1 Hz), 7,81 (d, 1H, J = 12,2
Hz), 7,45 - 7,40 (m, 4H), 7,33 - 7,28 (m, 4H), 7,16 (dd, 2H, d = 9,2, 8,6 Hz),
6,80 (d, 1H, J = 6,1 Hz), 4,64 (s, 1H), 4,34 (s, 2H), 3,84 - 3,70 (m, 4H), 2,70 111
3,55 (m, 2H), 3,55 - 2,98 (m, 3H), 2,08 - 1,92 (m, 2H), 1,92 - 1,75 (m, 2H);
MS(ESF): m/z 576,25 (M + H)*.
Exemplo 42

1-(3-flúor-4-(3-(3-(2-(pirrolidin-1-il)acetamido)prop-1-inil) pl- ridin-4-ilóxi)fenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de dicloridrato.
BocHN

A) /V-Boc-propargilamina.
Di-terc-butil-dicarbonato (21, 8 mg, 100,0 mmoles) foi dissolvido em THF (25 mL) e a solução resfriada a 0°C e tratada em gotas com uma solução de propargilamina (Aldrich, 5,0 g, 90,0 mmoles) mantendo a temperatura abaixo de 15°C. A mistura foi agitada em temperatura ambiente durante 1,5 hora em seguida concentrada sob vácuo. O resíduo foi dissolvido em hexanos e filtrada através de uma coluna de sílica-gel empregando-se ΟΙ 00% de CH2CI2 / hexanos para eluir o produto. O eluente contendo o produto foi concentrado em vácuo para fornecer um óleo incolor que foi dissolvido em hexanos (150 mL) e resfriado a 0°C para fornecer cristais brancos. Os cristais foram coletados por filtração e secados sob vácuo para fornecer o composto do título (10,5 g, 75%). 1H RMN (CDCI3) δ 4,75 (s, 1H), 3,95 (s, 2H), 2,25-2,24 (m, 1H), 1,48 (s, 9H).
FXrííí;X^-NO2
BocHN
B) 3-(4-(2-flúor-4-nitrofenóxl)piridin-3-il)prop-2-inilcarbamato de terc-butila.
O composto do título foi preparado de AFBoc-propargilamina (98 mg, 0,63 mmol) e 4-(2-flúor-4-nitrofenóxi)-3-iodopiridina (Composto A de Exemplo 33, 150 mg, 0,42 mmol) por meio de reação de acoplamento bruto-

112 zado Sonagashira empregando-se Pd(Ph3P)4 (9 mg, 0,008 mmol) e Cui (1,5 mg, 0,008 mmol) em 1:1 Et3N / THF (3 mL) de acordo com a etapa C de Exemplo 35, o composto do título (124 mg, 76%) foi obtido como um óleo vermelho. 1H RMN (DMSO-cy & 8,66 (s, 1H), 8,50 (d, IM, J = 5,6 Hz), 8,40 (dd, 1H, J = 2,5,10,7 Hz), 8,15 (d, 1H, J = 9,2 Hz), 7,52 (dd, 1H, J = 8,1, 8,6
Hz), 7,33-7,30 (m, 1H), 7,07 (d, 1H, J = 5,6 Hz), 3,95 (d, 2H, J = 5,6 Hz), 1,35 (s, 9H); MS(ESI+): m/z 388,21 (M + H)‘.

C) 3-(4-(2-flúor-4-nitrofenóxi)piridin-3-il)prop-2-in-1-amina.
Uma solução de 3-(4-(2-flúor-4-nitrofenóxi)píridin-3-il)prop-210 inilcarbamato de terc-butila (300 mg, 0,78 mmol) em CH2CI2 (10 mL) foi tratada com TFA (2 mL) e agitada em temperatura ambiente durante 45 minutos. A mistura foi concentrada sob vácuo e o resíduo foi dividido entre EtOAc e solução de NaHCO3 aquosa saturada. A fase EtOAc foi separada e lavada com salmoura, secada (MgSO4) e concentrada em vácuo para fornecer o composto do título (180 mg, 80%) como um óleo vermelho. 1H RMN (DMSOφ) δ 8,65 (s, 1H), 8,47 (d, 1H, J = 5,6 Hz), 8,43 (dd, 1H, J = 10,7, 2,5 Hz), 8,17 (d, 1H, J — 8,6 Hz), 7,54 (t, 1H, J = 8,6 Hz), 7,04 (d, 1H, J = 5,6 Hz), 3,49 (s, 2H); MS(ESF): m/z 288,17 (M + H)+.

D) M-(3-(4-(2-0úor-4-nitrofenóxi)piridin-3-il)prop-2-inil)-2- (pirrolidin-l-il)acetamida
Uma solução de 3-(4-(2-flúor-4-nitrofenóxi)píridín-3-il)prop-2-in-1amina (80 mg, 0,26 mmol) em CH2CI2 anidro (2,5 mL) foi resfriado a 0°C e tratada com cloreto de cloroacetila (40 mg, 0,37 mmol) a mistura foi agitada em temperatura ambiente durante 1 hora. A mistura foi concentrada sob vá
113 cuo para remover o solvente e o excesso de reagent© e o resíduo redissolvido em CH3CN (1,5 mL), tratada com pirrolidona (55 mg, 0,78 mmol) e agitada em temperatura ambiente durante 4 horas. A mistura foi dividida entre EtOAc e solução de NaHCO3 aquosa saturada e a fase orgânica separada, lavada com salmoura, secada (MgSO4) e concentrada para fornecer produto bruto. O resíduo foi purificado por cromatografia instantânea em sílica-gel eluindo com 0-10% MeOH/ CH2CI2 para fornecer o composto do título (40 mg, 39%) como um óleo marrom. Ή RMN (DMSO-d6) δ 8,66 (s, 1H), 8,50 (d, 1H, J = 6,1 Hz), 8,41 (dd, 1H, J = 2,8, 10,4 Hz), 8,18 - 8,15 (m, 2H), 7,51 (dd, 1H, J = 8,3, 8,8 Hz), 7,06 (d, 1H, J = 5,5 Hz) 4,10 (d, 2H, J = 5,5 Hz), 3,01 (s, 2H), 2,47 - 2,43 (m, 4H), 1,67 - 1,63 (m, 4H); MS(ESL): m/z 399,27 (Μ + H)*.

E) A/-(3-(4-(4-Amino-2-fluorofénóxi)piridin-3-il)prop-2-inil)-2(pirrolidin-l-il)acetamida.
O composto do título foi preparado pela redução de N-(3-(4-(2flúor-4-nitrofenóxi)piridin-3-il)prop-2-inil)-2-(pirrolidin-1 -il)acetamida (35 mg, 0,088 mmol) de maneira similar da Etapa E de Exemplo 35 empregando-se pó de Fe (67 mg, 1,21 mmol) e NH4CI (128 mg, 2,42 mmoles). O produto foi empregado em reações subseqüentes sem qualquer purificação. Óleo amarelo (30 mg, 93%). MS(ESr): 319,24 (M + H)*.
F) 1-(3-flúor-4-(3-(3-(2-(pirrolidin-1-il)acetamido)prop-1-inil) piridin-4-ilóxi)fenll)-3-(2-(4-fluorofenil)acetB)uriia, sal de dicloridrato.
O composto do título foi preparado de A/-(3(4-(4-amino-2-fluorofenóxi)piridin-3-il)prop-2-inil)-2-(pirrolidin-1-il)acetamida (32 mg, 0,088 mmol) e solução de 0,3 de isocianato de 2-(4-fluorofenil)acetila em tolueno (Composto D de Exemplo 11, 0,40 ml, 0,12 mmol) empregando-se THF (0,5 ml) de uma maneira similar àquela de Etapa D de Exemplo 33. O produto bruto foi purificado por HPLC preparativa (Coluna B). A fração contendo o produto foi
114 tratado com excesso de 1 M ácido clorídrico, concentrado em vácuo e íiofilizado para fornecer o composto do título (30 mg, 63%) como um sólido amarelo pálido. Ή RMN (DMSO-ds) δ 11,07 (s, 1H), 10,62 (s, 1H), 10,09 (s, 1H), 9,17 - 9,14 (m, 1H), 8,65 (s, 1H), 8,43 (d, 1H, J = 5,6 Hz), 7,81 (dd, 1H, J = 2,5, 12,7 Hz), 7,44 - 7,33 (m, 4H), 7,19 - 7,12 (m, 2H), 4,31 (d, 2H, J = 5,6 Hz), 4,05 (d, 2H, J = 5,6 Hz), 3,74 (s, 2H), 3,56 - 3,51 (m, 2H), 3,05 - 2,99 (m, 2H), 1,96 - 1,90 (m, 2H), 1,88 - 1,79 (m, 2H); MS(ESF): m/z 548,26 (M + H)+.
Exemplo 43

1-(3-flúor-4-(3-(3-(2-(4-hidroxipiperidin-1-il)acetamlclo)prop-1- inil)píridin-4-ilóxi)fenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de dicloridrato.

A) AF(3-(4-(2-flúor-4-nitrofenóxi)piridin-3-il)prop-2-inil)-2-(4hidroxipiperidin-1 -il)acetamlda.
O composto do título foi preparado de 3-(4-(2-flúoM-nitrofenóxi) piridin-3-il)prop-2-in-1-amina (Composto A de Exemplo 36, 80 mg, 0,26 mmol), 4-hidroxipiperidina (79 mmoles, 0,78 mmol) e cloreto de cloroacetila (40 mg, 0,36 mmol) da mesma maneira como para a Etapa D de Exemplo 42. O resíduo foi purificado por cromatografia instantânea em sílica-gel eluindo com 1-3% de MeOH / CH2CI2 para fornecer uma espuma branca (40 mg, 36%). 1H RMN (DMSO-cg δ 8,65 (s, 1H), 8,49 (d, 1H, J = 5,5 Hz), 8,41 (dd, 1H, J = 10,4, 2,7 Hz), 8,18 - 8,12 (m, 2H), 7,55-7,50 (m, 1H), 7,05 (d,
1H. J = 6,0 Hz), 4,54 (d, 1H, J = 3,8 Hz), 4,11 (d, 2H, /= 6,0 Hz), 3,43-3,36
115 (m, 1H), 2,86 (s, 2H), 2,64-2,58 (m, 2H), 2,12-2,05 (m, 2H), 1,67-1,61 (m, 2H), 1,44-1,36 (m, 2H); MS(ESI+): m/z 429,18 (M + H)+.

B) ^-(3-(4-(4-Amino-2-fluoiOfenóxi)pirldin-3-il)prop-2-irjil)-2(4-hidroxipiperidin«1-il)acetamida.
Θ composto do título foi preparado pela redução de AP(3-(4-(2flúor-4-nitrofenóxi)piridin-3-il)prop-2-inil)-2-(4-hidroxipiperidin-1-il)acetamida (33 mg, 0,077 mmol) de uma maneira similar àquela de Etapa E de Exemplo empregando-se Fe (pó, 67 mg, 1,21 mmol), NH4CI (128 mg, 2,42 mmoles). O produto (30 mg, 100%) foi obtido como um óleo amarelo que foi em10 pregado diretamente na etapa subseqüente. MS(ESr): m/z 399,27 (M + H)*.
C) 1-(3-flúor-4-(3-(3-(2-(4-hidroxipiperidin-1-íl)acetamido) prop-1-inil)pirídin-4-ilóxi)fenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de dicloridrato.
O composto do título foi preparado de N-(3-(4-(4-amino-215 fluorofenóxi) piridin-3-il)prop-2-inil)-2-(4-hidroxipiperidin-1-il)acetamida (25 mg,
0,063 mmol) e solução de isocianato de 0,3 M de 2-(4-fluorofenil)acetila em tolueno (Composto D de Exemplo 11, 0,40 mL, 0,12 mmol) empregando-se
THF (0,5 ml) de uma maneira similar àquela de Etapa D de Exemplo 33. O produto bruto foi purificado por HPLC preparativa (Coluna B). A fração con20 tendo o produto foi tratado com excesso de 1 N de ácido clorídrico, concentrado e liofilizado para fornecer o composto do título (10 mg, 30%) como um sólido amarelo pálido. Ή RMN (DMSO-cy δ 11,08 (s, 1H), 10,64 (s, 1H),
9.83 - 9,72 (m, 1H), 9,25 - 9,20 (m, 1H), 8,66 (s, 1H), 8,45 (d, 1H, J =6,1 Hz),
7.83 (dd, 1H, J = 2,0,13,2 Hz), 7,46 - 7,35 (m, 3H), 7,21 - 7,11 (m, 2H), 6,77 (d, 1H, J = 6,1 Hz), 4,33 -4,31 (m, 2H), 3,99 - 3,04 (m, 2H), 3,92 - 3,89 (m,
1H), 3,76 (s, 2H), 3,46 - 3,40 (m, 2H), 3,29 - 3,21 (m, 2H), 3,10 - 3,00 (m,
1H), 1,98 -1,87 (m, 2H), 1,72 -1,62 (m, 2H); MS(ESr): m/z 528,25 (M + Hf.
116
Exemplo 44

(S)-1 -(3-flúor-4-(3-(3-(2-(pirroliclin-1 -ilmetíl)pirrolidina-1 carboxamido)prop-1-inil)piridin-4-ilóxi)fenil)-3-(2-(4-fluorofenil)acetil) uréia, sal de dicloridrato.

A) (S)-/V-(3-(4-(2-flúor-4-nitrofenóxi)piridin-3-il)prop-2-inil)-2(pirrolidln-1«ilmetll)pirrolidina-1-carboxiamlda.
3-(4-(2-flúoM-nítrofenóxi)piridin-3-il)prop-2-in-1 -amina (Compos- to A de Exemplo 36, 55 mg, 0,19 mmol) foi dissolvido em CH2CI2 (5 mL), tratada com cloroformiato de 4-nitrofenila (0,38 mg, 0,19 mmol) e piridina (15 pL, 0,19 mmol). A mistura de reação foi agitada em temperatura ambiente.
Após 1 hora, a mistura foi tratada com Et3N (30 mL, 0,20 mmol) e (S)-(+)-l(2-pírrolidinilmetil)pirrolidona (Aldrich, 32 mg, 0,21 mmol) e agitada em temperatura ambiente durante 15 horas. A mistura foi em seguida diluída com CH2CI2 (50 mL), lavada com 1 M NaOH e salmoura, secada (MgSO4) e con15 centrada em vácuo para fornecer o produto bruto. O resíduo foi purificado por cromatografia instantânea em sílica-gel eluindo com 0-10% MeOH / CH2CI2 para fornecer o composto do título (53 mg, 60%) como um óleo amarelo. Ή RMN (DMSO-d6) δ 8,65 (s, 1H), 8,49 (d, 1H, J = 6,1 Hz), 8,41 (dd,
1H, J = 2,8 Hz, 10,5 Hz), 8,16 (d, 1H, J = 7,7 Hz), 7,52 (dd, 1H, J = 8,3, 8,8 20 Hz), 7,05 (d, 1H, J = 6,1 Hz), 4,11 - 4,06 (m, 2H), 3,99 - 3,95 (m, 2H), 3,76 (s, 2H), 3,16 - 3,11 (m, 2H), 2,57 - 2,48 (m, 2H), 2,43 - 2,41 (m, 2H), 3,36 2,34 (m, 2H), 1,90 -1,86 (m, 2H), 1,76 - 1,62 (m, 3H); MS(ESI*): m/z 468,27
117 (M + H)+.

Β) (S)-/V-(3-(4-(4-Amino-2-fluorofenóxi)pindin-3-íl)prop-2inil)-2-(pírrolidin-1-ilmetil)pirrolidina-1-carboxamida
Ο composto do título foi preparado pela redução de (S)-N-(3-(4(2-flúor-4-nitrofenóxi)piridin-3-il)prop-2-inil)-2-(pirrolidin-1-ilmetil)pirrolidina-1carboxamida (50 mg, 0,11 mmol) de uma maneira similar àquela de Etapa E de Exemplo 35 empregando-se Fe (pó, 67 mg, 1,21 mmol), NH4CI (128 mg, 2,42 mmoles). O produto (36 mg, 7596) foi obtido como um óleo amarelo que foi empregado diretamente na etapa subseqüente. MS(ESr): m/z 438,30 (M + Hy.
C) (S)-1-(3-flúor-4-(3-(3-(2-(plrrolidin-1-ilmetil)plriOliclina-1carboxamido)prop-1-inil)piridin-4-iIóxi)fenil)-3-(2-(4-fluorofenil)acetil) uréia, sal dedicloridrato.
O composto do título foi preparado de (S)-N-(3-(4-(4-amíno-2fluorofenóxi)piridin-3-il)prop-2-Ínil)-2-(pirrolidin-1-ilmetií)pirrolídina-1-carboxamida (36 mg, 0,057 mmol) e solução de isocianato 0,3 M de 2-(4-fluorofenil) acetila em tolueno (Composto D de Exemplo 11,0,37 mL, 0,11 mmol) da mesma maneira como na Etapa D de Exemplo 33. O produto bruto foi purificado por HPLC preparativa (Coluna B) e convertido para seu cloridrato de acordo com a Etapa C de Exemplo 43 para fornecer o composto do título (10 mg, 25%) com um óleo de cor âmbar. Ή RMN (DMSO-d6) δ 11,06 (s, 1H), 10,60 (s, 1H), 9,56 (s, 1H), 8,58 (s, 1H), 8,38 (d, 1H, J =5,6 Hz). 7,80 (dd, 1H, J = 2,6, 12,7 Hz), 7,42 (dm, 1H, J = 10,6 Hz), 7,38 - 7,31 (m, 2H), 7,30 - 7,25 (m, 1H), 7,20 - 7,10 (m, 2H), 6,69 (d, 1H, J = 6,1 Hz). 4,26 -4,12 (m, 2H), 3,79 - 3,69 (m, 3H), 3,56 (s, 2H), 3,31 - 3,18 (m, 2H), 3,17- 2,93 (m, 2H), 2,14 - 2,02 (m, 4H), 2,03 -1,76 (m, 5H), 1,71 -1,61 (m, 1H); MS(ESI*): 617,20 (M + H)*.
118
Exemplo 45


(^-1-(4-(3-(3-(4-Aminopiperidin-1-i)-3-oxoprop-1-enil)piriclin-
4-ilóxí)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de ácido ditriflu- oroacétíco.

A) 3-(4-(2-flúor-4-nltrofenóxi)piri<lin-3-il)acrilato (E)-terc-Butila.
Uma mistura de 4-(2flúor-4-nitrofenóxi)-3-iodopiridina (Composto A de Exemplo 33,150 mg, 0,42 mmol), terc-butilacrilato (Aldrich, 107 mg,
0,84 mmol), tri-n-butilamina (0,21 mL, 0,92 mmol) e DMF (2 mL) foi desga10 seificada por vácuo/purga de argônio e em seguida tratada com Pd(OAc)2 (17 mg, 0,078 mmol). A mistura foi aquecida a 100-130°C sob argônio durante 45 minutos, em seguida a mistura foi resfriada em temperatura ambiente, dividido entre EtOAc e solução de NaHCO3 aquosa saturada. As fases foram separadas e os extratos de EtOAcs foram lavados com solução de 15 NaHCO3 aquosa saturada e salmoura, secados (MgSO4) e concentrados em vácuo. O produto bruto foi purificado por cromatografia instantânea em SiO2 eluindo com 0-20% EtOAc / CH2CI2 para fornecer o composto do título (118 mg, 78%) como um óleo amarelo pálido que solidificou-se em temperatura ambiente. Ή RMN (DMSO-φ) δ 9,02 (s, 1H), 8,49 (d, 1H, J = 5,5 Hz), 8,46 20 (dd, 1H, J = 2,5, 11,7 Hz), 8,21 (dm, 1H, J = 9,2 Hz), 7,74 (d, 1H, J - 16,3
Hz), 7,65 (dd, 1H, J = 8,1,8,6 Hz), 6,95 (d, 1H, J =6,1 Hz), 6,77 (d, 1H, J =
16,3 Hz), 1,47 (s, OH); MS(ESF): mA 361,15 (M + H)*.
110

B) Ácido (E)-3-(4-(2-flúor-4-nitrofenóxi)piridln-3-ii)acrílico.
3-(4-(2-flúor-4-nitrofenóxi)piridin-3-il)acrilato (E)-terc-butila (115 mg, 0,32 mmol) foi tratado com 1:1 TFA / CH2CI2 (6 mL) e agitado em temperatura ambiente durante 1,5 hora. A mistura foi concentrada sob vácuo e o resíduo tratado com MeOH (5 mL) e 2 M HCI / Et2O (15 mL) e concentrada sob vácuo. Um segundo tratamento com MeOH (5 mL) e 2 M de HCI / Et2O (15 mL) e a reconcentração fornece o composto do título (120 mg). 1H RMN (DMSOδ 9,17 (s, 1H), 8,62 (d, 1H, J = 6,6 Hz), 8,50 (dd, 1H, J = 2,6, 10,1 Hz),
8,25 (d, 1H, J = 9,1 Hz), 7,78 (d, 1H, J = 16,3 Hz), 7,74 (dd, 1H, J =8,1, 8,6
Hz), 7,17 (d, 1H, J = 6,1 Hz), 6,87 (d, 1H, J = 16,3 Hz); MS(ESI+): m/z 305,11 (M + H)*·
NHBoc
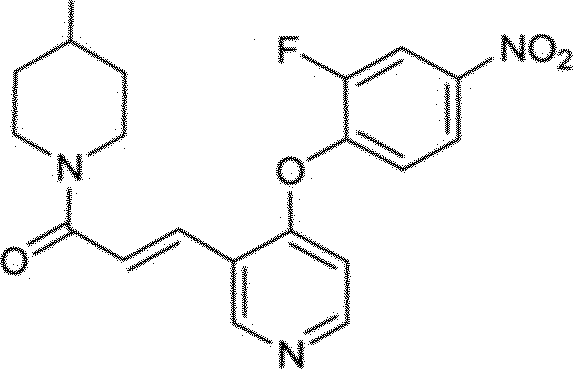
C) 1-(3-(4-(2-flúor-4-nitrofenóxi)piriclln-3-il)acriloil) piperidin-
4-ilcarbamato de (E)-terc-Butila
Uma solução de ácido (E)-3-(4-(2-flúor-4-nitrofenóxi)piridin-315 il)acrílico (143 mg, 0,42 mmol), 4-N-Boc-aminopiperidina (Aldrich, 84 mg,
0,42 mmol) em DMF (1,5 mL) foi tratada com DIPEA (160 pL, 0,92 mmol), e
TBTU (160 mg, 0,50 mmol) e a mistura agitada em temperatura ambiente durante 2 horas. A mistura foi diluída com EtOAc, lavada com solução de
NaHCO3 aquosa saturada e salmoura, secada (MgSOJ e concentrada em vácuo. Q produto bruto foi purificado por cromatografia instantânea em sílíca-gel eluindo primeiro com 30-100% EtOAc / hexanos em seguida 5% MeOH / CH2CI2 para fornecer o composto do título (110 mg, 54%) como um sólido marrom claro. 'H RMN (DMSO-de) δ 9,15 (s, 1H), 8,48 (d, 1H, J = 5,6
1H), 3,20 - 3,14 (m, 1H), 2,83 - 2,81 (m, 1H), 1,82 -1,71 (m, 2H), 1,34 -1,18 (m, 2H), 1,37 (s, 9H); MS(ESf): m/z 431,04 (100) [(M-C4H9) +Hf; m/z 487,10 (90) (M + H)\
NHBoc


D) 1-(3-(4-(4-amino-2-flyorofenóxi)piridin-3-il)acriloil)piperidin-4-ilcarbamato (£)-ierc-butila.
O composto do título foi preparado pela redução de 1-(3-(4-(210 fiúor-4-nitrofenóxi)píridin-3-il)acriioil)piperidin-4-ilcarbamato de (E)-terc-butila (100 mg, 0,21 mmol) de uma maneira similar àquela de Etapa E de Exemplo 35 empregando-se pó de Fe (55 mg, 2,7 mmoles), NH4CI (280 mg, 5,3 mmoles). O produto (90 mg, 95%) foi obtido como um sólido marrom claro que foi empregado diretamente na etapa subseqüente. MS(ESr): m/z


E) 1-(3-(4-(2-flúor-4-(3-(2-(4-fluorofenil)acetil) ureído)fenóxi) pirtdin-3-il)acriloil)piperidin-4-iicarbamato (E)-terc-butila.
O composto do título foi preparado de 1-(3-(4-(4-amíno-2fluorofenóxi)piridin-3-i!)acriloil)piperidin-4-ilcarbamato de (E)-terc-butila (42 mg,
0,092 mmol) e solução de 0,3 M de isocianato de 2-(4-fluorofenÍI)acetila em tolueno (Composto D de Exemplo 11,0,50 mL, 0,15 mmol) de uma maneira similar àquela de Etapa D de Exemplo 33, o produto bruto foi absorvido em sílica-gel e purificado por cromatografia instantânea eluíndo com 0-5% de Me
121
OH / EtOAc para fornecer O produto (20 mg, 33%) como um sólido branco. 1H RMN (DMSO-de) 5 11,05 (s, 1H), 10,59 (s, 1H), 9,03 (s, 1H), 8,37 (d, 1H, J = 5,6 Hz), 7,82-7,74 (m, 2H), 7,47 (d, 1H, J -15,8 Hz), 7,42-7,33 (m, 4H), 7,20-7,13 (m, 2H), 6,89 (d, 1H, J = 8,1 Hz), 6,63 (d, 1H, J = 5,6 Hz), 4,31 (d, 1H, J = 13,7 Hz), 4,19 (d, 1H, J = 12,7 Hz), 3,74 (s, 2H), 3,56-3,47 <m, 1H), 3,22-3,13 (m, 1H), 2,84-2,76 (m, 1H), 1,76 (s, 2H), 1,37 (s, 9H), 1,32-1,20 (m, 2H); MS(ESr): 636,23 (M + H)+.
F) (E)-1-(4-(3-(3-(4-Aminopiperidin-1-il)-3-oxoprop-1-enil) plridin-4-ilóxi)-3-fluorofenil)-3-(2-(4-fIuorofenil)acetil)uréia, sal de ácido ditrifluoroacético.
1-(3-(4-(2-flúor-4-(3-(2-(4-fluorofeníl)acetil)ureído)fenóxi)piridin-3il)acriloil)piperidin-4-ilcarbamato de (E)-terc-butila (15 mg, 0,024 mmol) foi dissolvido em MeOH anidro (0,5 mL), tratado com 4 M de HCI / 1,4-dioxano (1,5 mL) e agitado em temperatura ambiente durante 1 hora. A mistura foi concentrada sob vácuo para fornecer o produto bruto que foi purificado por HPLC preparativa (Coluna A). A fração contendo o produto foi tratada com excesso de 1 M de ácido clorídrico, concentrada e liofilizado para fornecer o composto do título (8 mg, 44%) como um sólido amarelo pálido. ’H RMN (DMSO-dg) § 11,07 (s, 1H), 10,61 (s, 1H), 9,10 (s, 1H), 8,44 (s, 1H), 7,91 (m, 3H), 7,86 - 7,69 (m, 2H), 7,55 - 7,28 (m, 5H), 7,23 - 7,08 (m, 2H), 6,80 - 6,72 (m, 1H), 5,61 - 5,33 (m, 1H), 4,45 - 4,20 (m, 2H), 3,74 (s, 2H), 3,39 - 3,07 (m, 2H), 2,82 - 2,68 (m, 1H), 1,91 -1,51 (m, 2H), 1,50 - 1,15 (m, 1H); MS(ESI*): flwfc 536,16 (M + ΗΓ.
Exemplo 46
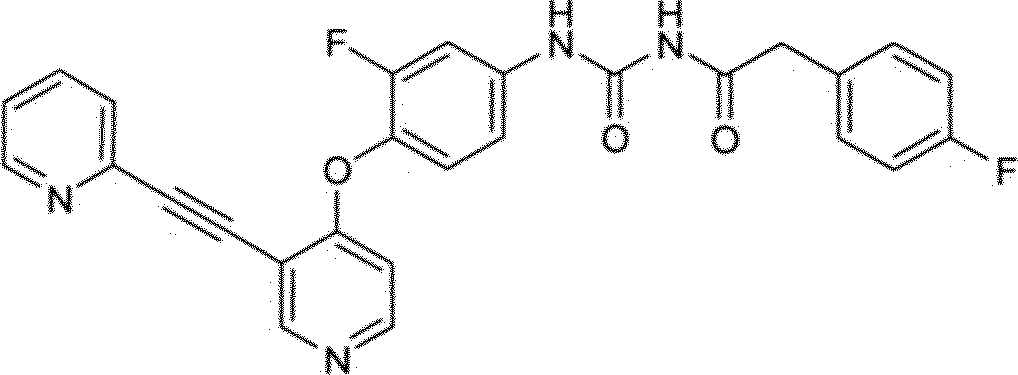
1-(3-flúor-4-(3-(2-(piridin-2-il)etinil)piridin-4-ilóxi)fenil)-3-(2-(4- fluorofenil)aceti!)ureia, sal de dicloridrato.
122

no2
A) 2-(2-(4-(2-flúor-4-nitrofenóxi)piridin-3-il)etinil)piri<iina.
Uma mistura de 4-(2-flúor-4-nitrofenóxi)-3-iodopiridina (Composto A de Exemplo 33, 50 mg, 0,14 mmol) e 2-etWlpiridina (Aldrich, 57 mg, 0,54 mmol), THF (1 mL) e Et3N (1 mL) foi desgaseificada por vácuo/purga de argônio e tratada sucessivamente com Cui (3 mg, 0,016 mmol) e (Ph3P)4Pd (10 mg, 0,009 mmol). A mistura foi aquecida a 60“C durante 45 minutos, resfriada, dividida entre EtOAc e bicarbonate de sódio saturado e a fase EtOAc secada (MgSOJ e concentrada em vácuo para fornecer o produto bruto. A purificação do resíduo por cromatografia de coluna instantânea em SiO2 eluindo com 0-1,5% de MeOH / CH2CI2 fornece o composto do título (42 mg, 89%) como um sólido marrom. 1H RMN (DMSO-d6) δ 8,91 (s, 1H), 8,60 (d, 1H, J = 4,5 Hz), 8,45 (dd, 1H, J = 2,6,10,7 Hz), 8,19 (d, 1H, J = 9,1 Hz), 7,86 - 7,83 (m, 1H), 7,66 (dd, 1H, J = 8,7, 8,7 Hz), 7,57 (d, 1H, J = 7,6 Hz), 7,45 -7,42 (m, 2H), 7,14 (d, 1H, J = 4,5 Hz); MS(ESF): m/z 336,20 (M + H)*.
B) 3-flúor-4-(3-(2-(pirídin-2-il)etinil)piriciin-4-ilóxi)benzena- mina
O composto do título foi preparado de 2-(2-(4-(2-flúor-4-nitrOfenóxi) piridÍn-3-il)etinil)piridina (30 mg, 0,090 mmol) de uma maneira similar àquela de Etapa E de Exemplo 35 para fornecer o composto do título (20 mg) como um sólido marrom. MS(ESL): m/z 306,20 (M + H)+.
C) 1-(3-flúor-4-(3-(2-(piridin-2-il)etinil)piridin-4-ilóxi)fenil)-3(2-(4-fluorofenil)acetil)uréia, sal de dicloridrato.
O composto do título foi preparado de 3-flúor-4-(3-(2-(piridin-2il)etinil)piridin-4-ilóxí)benzenamina (19 mg, 0,062 mmol) e solução de 0,3 M
123 de isocianato de 2-(4-fluorofenil)acetila em toluene (Composto D de Exemplo 11,0,50 mL, 0,15 mmol) de uma maneira similar àquela de Etapa D de
Exemplo 33, a purificação de uma mistura de reação por cromatografia instantânea em SiO2 eluindo com 0 - 100% de EtOAc / CH2CI2 fornece um sóli5 do branco que foi convertido em cloridrato de uma maneira similar à Etapa D de Exemplo 33 para fornecer o composto do título (19 mg, 60%) como um sólido branco. 1H RMN (DMSO-d6) δ 11,07 (s, 1H), 10,61 (s, 1H), 8,79 (s,
1H), 8,63 (d, 1H, J = 5,6 Hz), 8,47 (d, 1H, J = 5,6 Hz), 7,89 - 7,86 (m, 1H),
7,83 (dd, TH, 1,5, 12,7 Hz), 7,67 (d, 1H, J = 7,6 Hz), 7,47 - 7,43 (m, 2H), 10 7,39 - 7,35 (m, 1H), 7,30 - 7,27 (m, 1H), 7,20 - 7,16 (m, 2H), 7,14 - 7,10 (m,
1H), 6,77 (d, TH, J - 5,6 Hz), 3,75 (s, 2H); MS(ESI+): rwfc 485,17 (M + H)*.
Exemplo 47
1-(3-flúor-4-(3-(2-(piridin-3-il)etinil)plridln-4-ilóxi)fenll)-3-(2-(4- fluorofenil)acetil)uréia, sal de dicloridrato.
A) 3-(2-(4-(2-flúor-4-nitrofenóxi)piri<iin-3-li)etinil)piri(iina.
A mistura de 4-(2-flúor-4-nitrofenóxi)-3-iodopiridina (Composto A de Exemplo 33, 50 mg, 0,14 mmol) e 3-etinilpiridina (57 mg, 0,54 mmol),
THF (1 mL) e EtaN (1 mL) foi desgaseificada por vácuo/purga de argônio e tratada sucessivamente com Cul (3 mg, 0,016 mmol), e (Ph3P)4Pd (10 mg,
0,009 mmol). A mistura foi aquecida a 60°C durante 45 minutos, resfriada, dividida entre EtOAc e bicarbonate de sódio saturado e a fase EtOAc secada (MgSO4) e concentrada em vácuo. A purificação do resíduo por cromatografia instantânea em SiO2 eluindo com 0-1, 5% MeOH / CH2CI2 fornece o composto do título (33 mg, 77%) como um sólido marrom. Ή RMN (DMSO8,7, 8,7 Hz), 7,46 (dd, 1H, J = 4,6 , 8,1 Hz), 7,17 (d, 1H, J = 5,6 Hz);
MS(ESI+): m/z 336,19 (M + H)*.
/Ύ

B) 3-flúor-4-(3-(2-(piridin-3-il)etinil)piríciin-4-ilóxi)benzenamina.
O composto do título foi preparado de 3-(2-(4-(2-flúor-4-nitrofenóxi) piridin-3-il)etinil)piridina (30 mg, 0,090 mmol) de uma maneira similar à Etapa E de Exemplo 35 para fornecer o composto do título como um sólido marrom (25 mg, 93%). MS(ESF): m/z 306,20 (M + H)+.
C) 1-(3-flúor-4-(3-(2-(piridin-3-il)etinil)piridin-4-ilóxi)fenil)-3(2-(4-fluorofenil)acetil)uréia, sal de dicloridrato.
O composto do título foi preparado de 3-flúor-4-(3-(2-(piridin-3il)etinil)piridin-4-ilóxi)benzenamina (22 mg, 0,072 mmol) e solução de 0,3 M de isocianato de 2-(4-fluorofenil)acetila em tolueno (Composto D de Exemplo 11, 0,50 mL, 0,15 mmol) de uma maneira similar à Etapa D de Exemplo 33, a purificação de uma mistura de reação por cromatografia de coluna instantânea em SÍO2 eluindo com 0-100% EtOAc / CH2CI2 fornece um sólido branco que foi convertido em cloridrato de uma maneira similar à Etapa D de
125
Exemplo 48
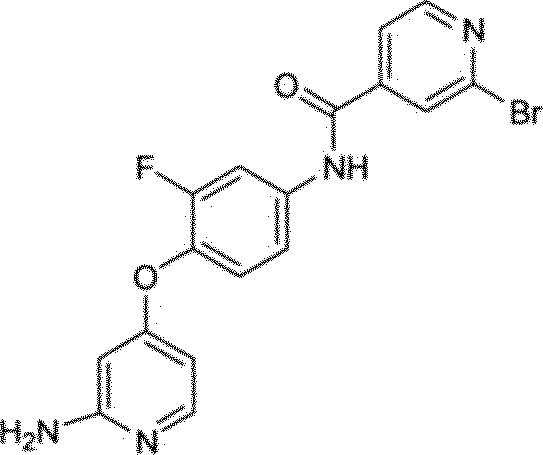
/V-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenil)-2-bromoisomcotínamida, sal de ácido trifluoroacético.
coa

A) Cloreto de 2-Bromo-isonicotinicacila.
Uma solução de ácido 2-bromo-isonicotínico (Lancaster, 70 mg, 0,34 mmol) em cloreto de tionila (1,2 mL) foi aquecida até a temperatura de refluxo durante 1,5 hora. A mistura foi concentrada e o produto bruto de cloreto de 2-bromoisonicotinoila foi empregado diretamente na etapa seguinte sem outra purificação.
B) JV-(4-(2-Aminopiridin-4-ll0xi)“3-fluorofenil)-2-bromoisonicotinamida.
Ao resíduo acima foi adicionado uma solução de 4-(4-amino-2fluorofenóxi)piridina-2-amina (Composto B de Exemplo 24, 70 mg, 0,32 mmol) em CH2CI2 (3 mL) em temperatura ambiente, e a reação foi agitada durante 1 hora. A mistura de reação foi concentrada e o resíduo foi purificado por HPLC preparativa para obter o composto do título (75 mg, 45%) como um sólido amarelo (sal de TFA ). 1H RMN (DMSO-d6) δ 11,03 (s, 1H), 8,62 (d, 1H, J = 5,0 Hz), 8,17 (s, 1 H), 7,89-8,04 (m, 4H), 7,71 (d, 1H, J = 8,8 Hz), 7,51 (t, 1H, J = 9,3 Hz), 6,72 (dd, 1H, J = 7,1, 2,2 Hz), 6,18 (d, 1H, J =
2,2 Hz); MS(ESr) m/z 403, 405 (M + H)*.
126

Exemplo 49


M(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenil)-2-(4-fluorofenilamino) isonicotinamida, sal de ácido bis-trifluoroacético.
F

ho2c

A) Ácido 2-(4-Fluorofenilamino)isonicotínico.
A uma mistura de ácido 2-flúor-isonicotínico (Aldrich, 423 mg,
3,0 mmoles) e 4-fluoroanÍlína (555 mg, 5,0 mmoles) em DMF (18 mL) em temperatura ambiente foi adicionado NaH (500 mg de 60% em óleo), e a mistura foi aquecida a 85°C durante 75 minutes. Q ácido acético (0,7 mL) foi adicionado a uma mistura de reação e foi concentrado em vácuo. Aos resíduos foram adicionados EtOAc (100 mL) e água (20 mL), agitados durante 20 minutos e o sólido foi filtrado, lavado com EtOAc e secado para obter o produto desejado (600 mg, 50%) como um sólido castanho. 1H RMN (DMSO-de) δ 8,98 (s, 1H), 8,43 (s, 1H), 8,04 (d, 1H, J = 4,9 Hz), 7,69 (dd, 2H, J = 8,8, 4,9 Hz), 7,24 (s, 1H), 7,05 (dd, 2H, J = 8,2, 6,0 Hz); MS(ESf) m/z 233,3 (Μ + ΗΓ·

B) Cloreto 2-(4-Fluorofenilamino)ispnicotinicacila.
A mistura de ácido 2-(4-fluorofenilamino)isonicotínico (464 mg, 2,0 mmoles) e cloreto de tionila (10 mL) foi aquecida até a temperatura de refluxo durante 2 horas. A reação foi concentrada em vácuo, e o produto bruto foi empregado diretamente na etapa seguinte.
C) /V-(4-(2-Aminopiridin-4-ilóxi)-3-fiuorofenil)-2-(4-fluorofenilamino)isonicotinamida
Uma solução de 4-(4-amino-2-fluorofenóxi)piridina-2-amina (Com
127 posto B de Exemplo 24, 450 mg, 2,1 mmol) em 1,2-dicloroetano (10 mL) foi adicionado lentamente a uma solução de acilcloreto obtido acima em 1,2dicloroetano(10 mL) em temperatura de banho gelado com agitação. A mistura de reação foi deixada descansar temperatura ambiente durante a noite, a uma mistura de reação foram adicionados EtOAc (150 mL) e solução de NaHCO3 aquosa saturada(50 mL). A camada de EtOAc foi separada, secada sobre MgSO4 e concentrada em vácuo. O resíduo foi purificado por HPLC preparativa para obter o composto do título (69 mg, 6,3%) como um sólido amarelo (sal de bis-TFA). 1H RMN (DMSO-<) δ 10,90 (s, 1H), 9,32 (s, 1H), 8,27 (d, 1H, J = 5,5 Hz), 7,92 -7,07 (m, 11H), 6,68 (dd, 1H, J = 7,1, 2,2 Hz), 6,10 (d, 1H, J = 2,2 Hz); MS(ESF) m/z 217,9 (M + H)*.
Exemplo 50 /V-(4-(2-Aminopirldin-4-llóxi)-3-fluorofenil)-6-oxo-1,6-diidropiridina-2-carboxamida, sal de ácido trifluoroacético.
A uma solução de ácido 6-hidroxipicolínico (Aldrich, 28 mg, 0,20 mmol) e HOBt (28 mg, 0,21 mmol) em DMF (2 mL) em temperatura ambiente foi adicionado EDCI.HCI (50 mg, 0,26 mmol) seguido por 4-(4-amino-2fluorofenóxi)piridina-2-amÍna (Composto B de Exemplo 24, 42 mg, 0,19 mmol), e a mistura de reação foi agitada durante a noite em temperatura ambiente. A purificação de uma mistura de reação por HPLC preparativa fornecendo o produto desejado (24 mg, 25%) como um sólido marrom claro (sal de TFA). Ή RMN (CD3OÇ) δ 8,03 (dd, 1H, J = 12,6, 2,2 Hz), 7,79 - 7,36 (m, 5H), 6,85 (d, 1H, J = 8,8 Hz), 6,67 (dd, 1H, J = 7,2, 4,4 Hz), 6,22 (d, 1H, J = 2,2 Hz); MS(ESF) m/z 341, 3 (M + H)*.
128
Exemplo 51

/V-(4-(2-amínopiridin-4-ilóxi)-3-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxamida, sal de ácido trifluoroacético.
A uma solução de ácido 2-hidroxinicotinico (Aldrich, 42 mg, 0,30 mmol) e HOBt (18 mg) em DMF (2 mL) em temperatura ambiente foi adicionado EDGI.HGI (80 mg, 0,42 mmol) seguido por 4-(4-amino-2-fluorofenóxi) piridina-2-amina (Composto B de Exemplo 24, 65 mg, 0,30 mmol), e a mistura de reação foi agitada durante 20 horas em temperatura ambiente. A purificação de uma mistura de reação por HPLC preparativa fornecendo o produto desejado (70 mg, 49%) como um sólido de cor bege (sal de TFA ). 1H RMN (GD3OD) δ 8,59 (dd, 1H, J = 7,1, 2,2 Hz), 8,03 (dd, 1H, J = 12,6, 2,2 Hz), 7,83 (d, 1H, J = 7,7 Hz), 7,75 (dd, 1H, J = 6,6, 2,2 Hz), 7,44 (d, 1H, J = 8,8 Hz), 7,32 (t, 1H, J * 8,8 Hz), 6,67 (m, 2H), 6,21 (d, 1H, J = 2,2 Hz); MS(ESF) m/z 341,2 (M + H)+.
Exemplo 52

h2n /V-(4-(2-Ammopiridin-4-ilóxl)-3-fluorofenll)-6-metil-2-oxo-1,2diidroplridina-3-carboxamida, sal de ácido trifluroacético.
A uma solução de ácido 2-hidróxi-6-metilnicotínico (Lancaster, 72 mg, 0,47 mmol) e HOBt (50 mg) em DMF (5 mL) em temperatura ambiente foi adicionado EDGI.HGI (130 mg, 0,68 mmol) seguido por 4-(4-amino-2fluorofenóxi)piridina-2-amina (Composto B de Exemplo 24, 110 mg, 0,50 mmol), e a mistura de reação foi agitada em temperatura ambiente durante
129 horas. A purificação de uma mistura de reação por HPLC preparativa fornece o produto desejado (125 mg, 55%) como um sólido de cor bege (sal de
TFA). Ή RMN (CD3OD) δ 8,46 (d, 1H, J = 7,9 Hz), 8,03 (dd, 1H. J = 12,7, 2,8
Hz), 7,83 (d, 1H, J= 7,1Hz), 7,44 (dd, 1H, J = 8,8, 2,2 Hz), 7,33 (t, 1H, J = 5 8,8 Hz), 6,67 (dd, 1H, J = 7,7, 2,7 Hz), 6,45 (d, 1H, J = 7,7 Hz), 6,21 (d, 1H,
J = 2,8 Hz), 2,40 (s, 3H); MS(ESF) m/z 355,2 (M + Hf.
Exemplo 53

A/-(4-(2-Aminopiriclin-4-ilóxi)-3-fluorofenil)-5-cloro-2-oxo-1,2diidropiridina-3-carboxamida, sal de ácido trlfluroacético.
A uma solução de ácido 2-hidróxi-5-cloronicotínico (Avocado, 87 mg, 0,50 mmol) e HOBt (40 mg) em DMF (4 mL) em temperatura ambiente foi adicionado EDCI.HGI (130 mg, 0,68 mmol) seguido por 4-(4-amino-2fluorofenóxi)piridina-2-amina (Composto B de Exemplo 24, 110 mg, 0,50 mmol), e a mistura de reação foi agitada durante 72 horas. A purificação de uma mistura de reação por HPLC preparativa fornece o produto desejado (115 mg, 45%) como um sólido de cor bege (Sal de TFA). 1H RMM (CD3OD)
8,50 (d, 1H, J=3,3 Hz), 8,03 (dd, 1H, J = 12,6, 2,1 Hz), 7,84 (m, 2H), 7,46 (d, 1H, J = 8,8 Hz), 7,34 (t, 1H, J = 8,8 Hz), 6,67 (dd, 1H, J - 7,2, 2,2 Hz), 6,21 (d, 1H, /= 2,2 Hz); MS(ESF) m/z 375,1, 377,1 (M+H)*.
Exemplo 54

W-(4-(2-AminopÍridin-4-ilóxl)-3-fluorofenil)-5-bromo-2-oxo130
1,2-diidropiriciina-3-carboxarnida, saí de ácido trifluoroacético
A uma solução de ácido 2-hidróxi-5-bromo-nicotínico (147 mg, 0,67 mmol, Syn. Comm., 1989, 19, 553-559) e HOBt (30 mg) em DMF (4 mL) em temperatura ambiente foi adicionado EDCI.HCI (160 mg, 0,83 mmol) seguido por 4-(4-amino-2-fluorofenóxi)pÍridina-2-amina (Composto B de Exemplo 24, 147 mg, 0,67 mmol), e a mistura de reação foi agitada em temperatura ambiente durante a noite. A purificação de uma mistura de reação por HPLC preparativa fornece o composto do título (120 mg, 33%) como um sólido de cor bege (Sal de TFA). 'H RMN (DMSO-cy S 13,22 (s, 1H), 12,23 (s, 1H), 8,40 (d, 1H, J = 2,8 Hz), 8,14 (d, 1H, J = 2,8 Hz), 8,12-7,46 (m, 5H), 6,68 (dd, 1H, J = 7,2, 2,2 Hz), 6,14 (d, 1H, J = 2,2 Hz); MS(ESF) m/z 410 / 421 (M + H)*.
Exemplo 55


h2n n
A/-(4-(2-Aminopiriclin-4-ilóxi)-3-fluorofenil)-6-metil-4-oxo-1,4diidropiridina-3-carboxamida, sal de ácido trifluoroacético.
A uma solução de ácido 4-hidróxi-6-metil-nicotínico (Wako, 77 mg, 0,50 mmol) e HOBt (50 mg) em DMF (5 mL) em temperatura ambiente foi adicionado EDCI.HCI (130 mg, 0,68 mmol) seguido por 4-(4-amino-2fluorofenóxi)piridina-2-amina (Composto B de Exemplo 24, 110 mg, 0,50 mmol), e a mistura de reação foi agitada em temperatura ambiente durante a noite, e em seguida aquecida a 75 °C durante 1,5 hora. Após resfriamento a mistura de reação para purificação em temperatura ambiente por HPLC preparativa fornece o composto do título (70 mg, 29%) como um sólido branco (Sal de TFA). Ή RMN (DMSO-<) δ 13,22 (br s, 1H), 12,53 (s, 1H), 8,47 (d, 1H, J = 5,5 Hz), 8,04 (d, 1H, J = 2,2 Hz), 7,95 (d, 1H, J = 8,2 Hz), 7,82 (s, 2H), 7,46-7,42 (m, 2H), 6,70 (dd, 1H, J = 7,7, 2,7 Hz), 6,39 (s, 1H),
131
6,14 (d, 1H, J = 2,2 Hz); MS(ESF) m/z 355,3 (M + H)*.
Exemplo 56


/V-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenii)-5-benzii-4-oxo-1,4diidropiridina-3-carboxamida, sal de ácido trifluoroacético.
OH OH

A) 3-Bromo-5-(hidróxi(feni!)metil)piridln-4-ol
A uma mistura heterogênea de 3,5-dtbromo-4-hidroxipiridina (2,53 g, 10 mmoles, preparada seguindo o procedimento em Synthesis, 2001, 14, 2175-2170) em THF anidro (20 mL) a -78°G sob Ar-atm. foi adicionada solução de brometo de fenilmagnésio (11 mL de 1 M de solução em THF, 11 mmoles). Após agitar durante 15 minutos, foi adicionada a solução de n-BuLi (5,5 mL de 2 M de solução em cicloexano), e a mistura de reação foi agitada durante 15 minutos a -78 °C sob Ar-atm. A esta mistura de benzaldeído (2,15 mL) foi adicionada e a mistura de reação foi agitada durante 2 horas -78°C sob Ar-atm. A mistura de reação foi extinguida por adição de HOAc (3 mL) e TFA (3 mL), concentrada em vácuo e o resíduo foi purificado por cromatografia de coluna instantânea em sílica-gel eluindo com hexano/EtOAc/MeOH // 750:250:50 seguido por hexano/EtOAc/MeOH/Et3N // 460:460:50:10 para fornecer o produto desejado (2,85 g, 91%) como um sólido branco. 1H RMN (GD3OD) δ 8,13 (s, 1H), 8,04 (s, 1H), 7,41-7,20 (m, 5H), 5,94 (s, 1H); MS(ESI*) m/z 280, 282 (M + Hf.
OH
Biv A.
132
B) 3-Benzil-5-bromopiridin-4-ol.
Uma mistura de 3-bromo-5-(hidróxi(fenil)metil)piridÍn-4-ol (2,55g, 91 mmoles), TEA (16 mL) e Et3SiH em CH2C12 (30 mL) foi agitada em temperatura ambiente durante 10 horas. A mistura de reação foi concentrada em vácuo e o resíduo foi purificado por cromatografia de coluna instantânea em sílica-gel eluindo com hexano/EtOAc/MeOH // 600:300:50 seguido por hexano/EtOAc/MeOH // 400:400:50:10 para fornecer um produto impuro que foi triturado com uma pequena quantidade de MeOH e EtaO para obter o produto desejado (255 mg, 10%) como um sólido branco. 1H RMN (DMSO<) 8 11,75 (br s, IH), 8,13 (s, 1H), 7,54 (s, 1H), 7,26-7,14 (m, 5H), 2,49 (s, 2H).
OH
C) Ácido 5-Benztl-4-hídroxinicotínico.
A uma solução de 3-benzil-5-bromopiridin-4-ol (220 mg, 0,83 mmol) em THF anidro (8 mL) a -78°C sob Ar-atm foi adicionado solução de MeLi (0,61 mL de 1,5 M de solução em THF, 0,92 mmol). Após agitar durante 5 minutos, a solução de n-BuLi (0,5 mL de 2 M de solução em cicloexano, 1,0 mmol) foi adicionado e mistura foi agitada durante 15 minutos a -78°C sob Ar-atm. Dióxido de carbono foi borbulhado através de uma mistura de reação durante 20 minutos a -78 °C. A mistura de reação foi então saciada por adição HOAc (2 mL), concentrada em vácuo e o resíduo foi purificado por HPLC preparativa para fornecer o produto desejado (100 mg, 35%) como um sólido branco (Sal de TFA). Ή RMN (DMF-<) δ 12,99 (br s, IH), 8,69 (s, 1H), 8,28 (s, IH), 7,35-7,19 (m, 5H), 3,90 (s, 2H); MS(ESF) m/z 230,1 (Μ + ΗΓ.
D) N-(4-(2-Aminoplridm-4-ilóxi)-3-fluorofenil)-5-benzil-4-oxo-
1,4-diidropiridina-3-carboxamida, sal de ácido trifluoroacétlco
A uma solução de ácido 4-hidróxi-5-benzilnicotínico (35 mg, 0,15 mmol) e HOBt (30 mg) em DMF (2,5 mL) em temperatura ambiente foi adicionado EDCLHCi (80 mg, 0,42 mmol) seguido por 4-(4-amino-2-fluorofe133 nóxi)piridina-2-amtna (Composto B de Exemplo 24, 35 mg, 0,16 mmol), e a mistura de reação foi agitada durante 40 horas em temperatura ambiente. A purificação de uma mistura de reação por HPLC preparativa fornece o produto desejado (35 mg» 43%) como um sal sólido de TFA branco. 1H RMN 5 (DMSO-O 8 13,25 (br s, 1H), 12,44 (s, 1H), 8,59 (d, 1H, J = 4,9 Hz), 8,08 (dd, 1H, J- 13,2, 2,2 Hz), 7,97 (d, 1H, J = 7,1 Hz), 7,81 (d, 1H, J = 9,4 Hz), 7,52-7,19 (m, 7H), 6,72 (dd, 1H, J = 7,2, 2,2 Hz), 6,15 (d, 1H, J = 2,5 Hz), 3,81 (s, 2H), 3,51 (br s, 2H); MS(ESI*) m/z 431,2 (M + H)*.
Exemplo 57
M-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenil)-2-oxo-1-fenil-1,2diidropiridina-3-carboxamida, sal de ácido trifluoroacético
HgCOjC CQ2CH3
A) 2-(3-(feniiamino)alilideno)malonato de (E)-Dimetila
A uma solução de éster de dimetila de ácido 2-(3-metoxialilideno)malônico (Acros Organics, 200 mg, 1,0 mmol) em THF (2 mL) em temperatura ambiente foi adicionada anilina (300 mg, 3,2 mmoles) e a mistura de reação foi aquecida a 60°C durante 8,5 horas. A purificação de uma mistura de reação por HPLC preparativa forneceu o produto desejado (150 mg, 57%) como um sólido amarelo. Ή RMN (DMSO-d6) δ 10,16 (d, 1H, J =
12,7 Hz), 8,06 (t, 1H, J = 12,7 Hz), 7,74 (d, 1H, J = 12,7 Hz), 7,30 (t, 2H, J = 20 8,7 Hz), 7,16 (d, 2H, J - 7,7 Hz), 6,98 (t, 1H, J = 7,7 Hz), 6,35 (t, 1H, J =
12,1 Hz), 3,69 (s, 3H), 3,65 (s, 3H).
134

B) 2-oxo-1-fenil-1,2-dlMropirMina-3-carboxllato de metila.
A uma solução do aduzido de anilina obtido acima (130 mg, 0,50 mmol) em metanol (8 mL) em temperatura ambiente foi adicionado NaH (50 mg do NaH a 60% em óleo, 1,2 mmol) e a mistura foi agitada. O ácido acético (0,3 mL) foi adicionado a uma mistura, concentrada a um volume de - 4 mL, e a purificação de uma mistura de reação por HPLC preparativa fornece o produto desejado (105 mg, 92%) como um sólido amarelo. Ή RMN (CD3OB) 5 8,30(dd, 1H, J = 7,2,2,2 Hz), 7,87 (dd, 1H, J = 6,6,1,7 Hz), 7,577,38 (m, 5H), 6,53 (t, 1H, J = 7,0 Hz), 3,84 (s, 3H); MS(ESP) m/z 230,3 (M + H)L ho2c

C) Ácido 2-oxo-1-fenil-1,2-diidropiridlna-3-carboxílico.
A mistura de 2-oxo-1-fenil-1,2-diidropiridina-3-carboxilato de metila (70 mg, 0,31 mmol) e LiOH (40 mg) em metanol (6 mL) e água (1 mL) foi agitada em temperatura ambiente durante a noite, a uma mistura de reação foram adicionados EtOAc (50 mL) e 1 N de HCI aquosa (15 mL). A camada de EtOAc foi separada, secada sobre MgSO4, e concentrada em vácuo para fornecer o produto (55 mg, 83%) como um sólido amarelo claro. 1H RMN (DMF-cÇ) Ô 11,77 (br s, 1H), 8,57 (dd, 1H, J = 7,4, 2,0 Hz), 8,26 (dd, 1H, J = 6,6, 1,6 Hz), 7,64-7,55 (m, 5H), 6,88 (t, 1H, J = 7,0 Hz); MS(ESF) m/z 216,2 (M + H)C
D) W-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenil)-2-oxo-1 -fenil-
1,2-diidropiridina-3-carboxamida.
A uma solução de ácido 2-oxo-1-fenil-1,2-diidropiridina-3-carboxílico (36 mg, 0,17 mmol) e HOBt (18 mg) em DMF (3 mL) em temperatura ambiente foi adicionado EDCI.HCI (45 mg, 0,23 mmol) seguido por 4-(4amino-2-fluorofenóxÍ)piridina-2-amina (Composto B de Exemplo 24, 36 mg, 0,17 mmol), e a mistura de reação foi agitada em temperatura ambiente du135 rante a noite. A purificação de uma mistura de reação por HPLC preparativa fornece o composto do título (32 mg, 36%) como um sólido de cor bege (Sal de TFA). 1H RMN (DMSO-d6) δ 13,35 (br s, 1H), 12,11 (s, 1H), 8,52 (dd, 1H, J=7,3, 2,1 Hz), 8,08 (dd, 1H, J=6,6, 2,1 Hz), 8,03 (d, 1H, J = 2,3 Hz), 7,89 (d, 1H, J= 7,2 Hz), 7,81 (s, 1H), 7,54-7,36 (m, 6H), 6,69-6,63 (m, 2H), 6,08 (d, 1H, J = 2,4 Hz), 3,55 (br s, 1H); MS(ESl+) m/z 417,2 (M + H)+.
Exemplo 58


h2n
N-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenil)-6-(4-fluorofenil) piridil-ZV-óxido-amída, sal de ácido trifluoroacético.
HO2C/^
A) Ácido 6-(4-Fluorofenil)picolínico.
Uma solução de ácido 2-bromo-picolínico (Aldrich, 2,02 g, 10 mmoles) em DME contendo 4 mL de Na2CO3aquoso a 10% foi purgado com gás Ar. A esta mistura foi adicionado Pd(PPh3)4 seguido por 2-(4-fluorofenil)-
5,5-dÍmetíl-1,3,2-dioxaborinano (Aldrich, 2,40 g, 11,5 mmoles) e EtOH (20 mL), e a mistura foi purgada com gás Ar. A mistura de reação foi aquecida a 100°C durante 2,5 horas em um tubo selado. Ácido 2-bromo-picolínico adicional (900 mg) e Pd(PPh3)4 foi adicionado, e depois purgado com gás Ar, foi aquecido a 100 °C durante 4,5 horas. Ácido trifluoroacético (20 mL) foi adicionado a uma reação e a mistura foi concentrada em vácuo. MeOH (150 mL) foi adicionado ao resíduo e o material insolúvel foi filtrado, e a solução filtrada foi concentrada em vácuo. A purificação do resíduo resultante por cromatografia de coluna instantânea em sílica-gel eluindo com EtOAc/MeOH // 900:100 seguido por EtOAc/MeOH/HOAc // 700:1500:50 fornece o produto
136 desejado (1,0 g, 40% baseado no material de partida de borinano) como um sólido branco. Ή RMN (CD3OD) δ 8,01 (d, 1H, J = 7,7 Hz), 7,94 - 7,87 (m, 3H), 7,73 (d, 1H, J = 7,7 Hz), 7,13 (t, 2H, J = 8,8 Hz); MS(ESr) m/z 234 (M + H)*.

B) W-oxido de ácido 6-(4-Fluorofenil)picolinico.
A mistura de derivado de ácido picolinic© (1-,0 g, 4,6 mmoles), Na2HPO4 (1,2 g) e m-GPBA (1,1 g, -70% de Aldrich) em CH2GICH2GI (30 mL) foi agitada em temperatura ambiente durante 2 horas. Na2HPO4 adicional (0,8 g) e m-GPBA <1,0 g) foi adicionado a uma mistura de reação e fbi agitada durante 3 horas em temperatura ambiente. Outros Na2HPO4 (0,5 g) e m-CPBA (0,5 g) foram adicionados a uma mistura de reação e foi agitada em temperatura ambiente durante a noite. CHCI3(160 mL) e 2 N de solução HGI aquosa (50 mL) foram adicionados à mistura de reação e a camada orgânica foi separada, secada sobre MgSO4 e concentrada em vácuo. O resíduo foi purificado por cromatografia de coluna instantânea em sílica-gel eluindo com EtOAc/MeOH /HOAc f 700:240:60 para obter o produto desejado que foi contaminado com m-GPBA. O material impuro foi purificado por HPLC preparativa para obter o produto desejado (175 mg, 16%) como um sólido branco. 1H RMN (DMF-dz) 8,45 (dd, 1H, J = 8,3, 2,2 Hz), 8,15 (d, 1H, J = 2,2 Hz), 8,13 - 8,00 (m, 4H), 7,45 (t, 2H, J = 8,7 Hz).
C) /V-(4-(2-Aminopiridin-4-iíóxi)-3-fluorofenil)-6-(4-fluorofenil) piridil-AFóxido-amicla, sal de ácido trifluoroacético.
A uma solução de /V-óxido de ácido 6-(4-fluorofenil)picolínico(23 mg, 0,1 mmol) e HOBt (10 mg) em DMF (2 mL) em temperatura ambiente foi adicionado EDCI.HGI (30 mg, 0,16 mmol) seguido por 4-(4-amino-2fluorofenóxi)piridina-2-amina (Composto B de Exemplo 24, 22 mg, 0,1 mmol), e a mistura de reação foi agitada em temperatura ambiente durante a noite. A purificação de uma mistura de reação por HPLG preparativa fornece o composto do título (25 mg, 46%) como um sólido branco (Sal de TFA). 1H RMN (DMF-cM δ 14,00 (s, 1H), 8,43 (dd, 1H, J = 8,0, 2,2 Hz), 8,15 (dd, 1H, J = 12,8, 2,4 Hz), 8,08 (d, 1H, J = 7,1 Hz), 7,99-7,37 (m, 9H), 6,72 (dd, 1H, J = 7,0, 2,4 Hz), 6,32 (d, 1H, J = 2,3 Hz), 3,7 (br s, 2H); MS(ESF) m/z 417,2 (M + H)*.
Exemplo 5®

-(4-(3-(4-(2-Amino-2-ox oe t i I )feni I)pi ridi n-4-i I óxi)-3-f!uorofenil)-3-(2-(4fluorofenil)acetíl)uréia, sal de cloridrato.

A) Ácido 2-(4-(4-(2-flúor-4-nitrofenóxi)piridin-3-ii)ferjil) acé- tico.
Um frasco de base redonda de o eter impuro de coluna de borinano 25 mL foi carregado com 4-(2-f!úor-4-nitrofenoxi)-3-iodopiridina (Composto A de Exemplo 33, 120 mg, 0,33 mmol), ácido 2-(4-(4,4,5,5-tetrametil-
1,3,2-dioxaborolan-2-il)fenil)acético (Frontier Scientific, 131 mg, 0,50 mmol), tetracis(trifenilfosfina)paládio(0) (Strem Chemicals, 38 mg, 0,033 mmol), e carbonato de sódio (245 mg, 2,3 mmoles). O fiasco foi inundado com nitro gênio e em seguida carregado com dioxano e água (1 mL cada). Após agitar a 80°C durante 10 horas, a mistura foi resfriada para a temperatura ambien te e em seguida concentrada em vácuo. O produto bruto foi purificado por cromatografia de coluna instantânea em sílica-gel (30% MeOH / EtOAc) para fornecer o composto do título (120 mg, 99%) como um sólido branco. 1H
138 h2n
N02
E
0'

N
B) 2-(4-(4-(2-flúor-4-nitrofenóxi)piridin-3-il)fenil)acetamida.
Um frasco de base redonda de 25 mL foi carregado com ácido
2-(4-(4-(2-flúor-4-nitrofenóxi)piridin-3-il)fenil)acético (50 mg, 0,136 mmol), HOST (46 mg, 0,34 mmol), e EDO (65 mg, 0,34 mmol). O frasco foi inundado com nitrogênio e em seguida DMF foi adicionado (1 mL). Após agitar em temperatura ambiente durante 1 hora, a solução foi resfriada a 0°C e em seguida carregada com hidróxido de amônio (0,5 mL). A reação foi agitada a 0°C durante 1 hora e foi em seguida diluída com salmoura (5 mL) e extraída com acetato de etila (3x5 mL). Os extratos orgânicos combinados foram secados em Na2SO4 anidro e concentrados em vácuo. O produto bruto foi purificado por cromatografia de coluna instantânea em sílica-gel (20% MeOH / EtOAc) para fornecer o composto do título (33 mg, 66%) como um óleo incolor. Ή RMN (CD3OD) δ 8,49 (s, 1H), 8,38 (d, 1H, J = 5,6 Hz), 8,10 (dd, 1H, J ~ 10,4, 2,4 Hz), 7,99 (m, 1H), 7,45 (d, 2H, J = 8,2 Hz), 7,30 (d, 2H, J =
8,2 Hz), 7,28 (m, 1H), 6,93 (d, 1H, J = 5,6 Hz), 3,44 (s, 2H); MS(ESF) m/z 368,18 (M+ H)*.
C) 1-(4-(3-(4-(2-Amino-2-oxoetil)fenil)piridin-4-ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetii)ureia.
Uma solução de 2-(4-(4-(2-flúor-4-nitrofenóxi)piridin-3-il)fenil) acetamida (33 mg, 0,09 mmol) em THF (0,8 mL) e metanol (1,2 mL) foi tratada com pó de Zn (59 mg, 0,9 mmol) seguido por cloreto de amônio (48 mg, 0,9 mmol). A mistura foi agitada em temperatura ambiente durante 4 horas e foi em seguida filtrada através de uma almofada fina de Celíte® com metanol. O filtrado fot concentrado e o resíduo dividido entre EtOAc e solução de bicarbonate de sódio aquosa saturada. A fase EtOAc foi secada sobre NazSO4 anidro e concentrada em vácuo para produzir o produto bruto (25 mg, 82%) como um óleo amarelo que foi suficientemente puro para o emprego na etapa seguinte sem outra purificação. 1H RMN (CD3OD) δ 8,33
139
Β· lv

i

Β (s, 1 Η), 8,19 (d, 1H, J = 5,6 Hz), 7,49 (d, 2H, J = 8,1 Hz), 7,32 « 2H, J = 8,2 Hz), 6,82 (t, 1H, J = 8,8 Hz), 6,61 (d, 1H, J = 5,6 Hz), 6,44 (qd, 1H, J = 12,8, 2,8 Hz), 3,48 (s, 2H); MS(ESF) m/z 338,25 (M + H)B
A amina acima foi dissolvida em THF (1 mL) e em seguida carregada com isocianato de 2-(4-fluorofenil)acetila (Composto D de Exemplo 11, 250 pL, 0,074 mmol, 0,3 M em tolueno). Após agitar em temperatura ambiente durante 1 hora, A reação foi purificada diretamente por cromatografia instantânea em sílica-gel (10% de MeOH / EtOAc) para fornecer o composto do título como um sólido branco. O sólido foi dissolvido em dioxano (2 mL) e resfriado a 0°C. O HCI anidro (2 mL, 1 N ín éter) foi adicionado. Após agitar a 0°C durante 15 minutos, a solução foi concentrada em vácuo. O sal' de HCI resultante foi lioffeado de acetonitrila/água para fornecer o composto do título (23 mg, 57%) como um sólido branco. 1H RMN (DMSOd6) 8 11,02 (s, 1H), 10,59 (s, 1H), 8,83 (s, 1H), 8,58 (d, 1H, J = 6,4 Hz), 7,79 (dd, 1H, J = 12,8, 2,4 Hz), 7,60 (d, 2H. J = 8,4 Hz), 7,46-7,34 (m, 4H), 7,31 7,28 (m, 2H), 7,11 (t, 2H, J = 8,7 Hz), 6,88 (d, 1H, J = 5,6 Hz), 3,70 (s, 2H), 3,40 (s. 2H); MS(ESF) m/z 517,19 (M + H)B
Exemplo 60
1-(4-P-(4-(Aminometil)fenil)pirlciin-4-ilóxi)-3-flu©rofenll)-3-(2- (4-fluorofenil)acetil)uréia, sal de ácido trifluoroacético.
A) 4-(4-(2-Fluoro4-nitrofenóxi)piridin-3-il)benzaldeído.
Preparado de uma maneira similar à Etapa A de Exemplo 59 para fornecer o composto do título (86%) como um óleo incolor. Ή RMN (CD3OD) 8 9,92 (s, 1H), 8,56 (s, 1H), 8,41 (d, 1H, J = 6 Hz), 8,12 (dd, 1H, J =
B) 4-(4-(2-flúor-4-nitrofenóxi)piridin-3-il)benzilcarbamato de terc-butila.
| A 4-(4-(2-f!úor-4-nitrofenóxi)piridin-3-il)benzaldeído (81 mg, 0,24 | |
| mmol) err | i metanol (2 mL) foi adicionado acetato de amônio (185 mg, 2,4 |
mmoles) seguido por cianoboroidreto de sódio (16 mg, 0,24 mmol). A reação foi agitada em temperatura ambiente durante 4 horas e foi em seguida con-
| centrada < | am vácuo. O resíduo foi dissolvido em água (5 mL) e extraído com |
EtOAc (2x5 mL). Os extratos orgânicos combinados foram lavados com solução de bicarbonato de sódio aquosa saturada, secados sobre Na2SO4, anidro e concentrados em vácuo. O produto bruto foi dissolvido em dicloro metano (2 mL) e em seguida trietííamina (50 uL, 0,36 mmol), DMAP (ponta de espátula), e dicarbonato de dí-terc-butila (Aldrich, 57 mg, 0,26 mmol) foram sequencialmente adicionados. A reação foi agitada em temperatura ambiente durante 2 horas e foi em seguida purificada diretamente por cromatografia de coluna instantânea em sílica-gel (EtOAc) para fornecer o composto do título (13 mg, 12%) como um óleo incolor. 1H RMN (CD3OD) δ 8,50 (s, 1H), 8,37 (d, 1H, J = 5,6 Hz), 8,09 (dd, 1H, J = 6,1, 2,6 Hz), 8,017,99 (m, 1H), 7,45 (d, 2H, J = 8 Hz), 7,26 (d, 2H, J = 8,2 Hz), 7,25 (m, 1H), 6,95 (d, 1H, J = 5,6 Hz), 4,16 (s, 2H), 1,35 (s, 9H); MS(ESf) m/z 440,19 (M +
C) 1-(4-(3-(4-(Aminometil)fenil)piridin-4-ilóxi)-3-fluorofenil)-
3-(2-(4-fluorofenil)acetil)uréia.
Preparado de uma maneira similar à Etapa C de Exemplo 59, após a formação de aciluréia, 4 N de HCI em dioxano (5 mL) foi adicionado. Após agitar em temperatura ambiente durante 5 minutos, a reação foi con
141 centrada em vácuo. O resíduo foi suspenso em EtOAc, lavado com solução de bicarbonate de sódio aquosa saturada, secado sobre NazSO4 anidro, e concentrado em vácuo. O material bruto foi purificado por HPLC preparativa. As frações apropriadas foram concentradas em vácuo para remover metanol. O tolueno foi adicionado e em seguida concentrada (2x5 mL). O sólido resultante foi liofilizado de acetonitrilo / água para fornecer o sal de TFA do composto do título (6 mg, 25%) como um sólido branco. Ή RMN (DMSO-d6) δ 11,00 (s, 1H), 10,53 (s, 1H), 8,55 (s, 1H), 8,40 (d, 1H, J = 4 Hz), 7,73 (dd, 1H, J = 12, 4 Hz), 7,67 (d, 2H, J = 8 Hz), 7,51 (d, 2H, J = 8 Hz), 7,34-7,27 (m, 4H), 7,13-7,09 (m, 2H), 6,73 (d, 1H, J = 4 Hz), 4,03 (s, 2H), 3,68 (s, 2H); MS(ESF) m/z 489,18 (M + Hf.
Exemplo 61

1-(3-fiúor-4-(3-(6-(piperazin-1-il)piridin-3-il)piriclin-4-ilóxi)fenil)-3-(2-(4fluorofeml)acetil)uréia, sal de cloridrato.

c
A) 4-(5-(4-(2-flúor-4-nltrofenóxi)piridin-3-il)piridm-2-il)piperazina-1-carboxilato de íerc-butila.
Preparado de uma maneira similar à Etapa A de Exemplo 59 para fornecer o composto do título (87%) como um óleo incolor. 1H RMN (CBjOD) δ 8,62 (s, 1H), 8,46 (d, 1H, J = 6 Hz), 8,36 (d, 1H, J = 2,4 Hz), 8,24 (dd, 1H, J = 10,4, 2,8 Hz), 8,15-8,11 (m, 1H), 7,84 (dd, 1H, J = 8,8, 2,4 Hz), 7,41 (t, 1H, J = 8,4 Hz), 7,04 (d, 1H, J = 5,6 Hz), 6,92 (d, 1H, J = 8,8 Hz), 3,61-3,59 (m, 4H), 3,56-3,54 (m, 4H), 1,50 (s, 9H); MS(ESP) m/z 496,23 (M + HF.
142

N
Β) 4-(5-(4-(4-amino-2-fluorofenóxi)piriclin-3-il)piridin-2-ll) piperazina-1-carbOxilato de terc-butila.
Preparado de uma maneira similar à Etapa C de Exemplo 59 para fornecer o composto do título (96%) como um óleo incolor. 1H RMN (GD3OD) δ 8,34 (S, 1H), 8,29 (d, 1H, J = 2 Hz), 8,18 (d, 1H, J- 6 Hz), 7,78 (dd, 1H, J = 9,2, 2,8 Hz), 6,87-6,83 (m, 2H), 6,61 (d, 1H, J = 5,6 Hz), 6,48 (dd, 1H, J = 12,8, 2,8 Hz), 6,44-6,41 (m, 1H), 3,50-3,46 (m, 8H), 1,38 (s, 9H); MS(ESf) m/z 466,25 (M+ H)“.
C) 1-(3-flúor-4-(3-(6-(plperazin-1-il)piridin-3-il)plridin-4ilóxi)fenil)-3-(2-(4-fluorofenil)acetil)ureia.
Preparado de uma maneira similar à Etapa G de Exemplo 59 para fornecer o composto do título (28%) como o sal de HCI. Ή RMN (DMSO-< 8 11,33 (s, 1H), 10,76 (s, 1H), 9,09 (s, 1H), 8,74 (d, 1H, J = 6,8 Hz), 8,57 (d, 1H, J = 2,4 Hz), 8,16 (dd, 1H, J~ 8,8, 2 Hz), 7,91 (dd, 1H, J-12,8, 2: Hz), 7,61 (t, 1H, J = 8,8 Hz), 7,55-7,50 (m, 1H), 7,42-7,38 (m, 2H), 7,28 (d, 1H, J = 6,5 Hz), 7,27-7,18 (m, 3H), 3,98 (m, 4H), 3,82 (s, 2H), 3,23 (m, 4H); MS(ESI+) m/z 545,19 (M + H)+.
Exemplo 62 /V-(3-flúor-4-(3-(6-(piperazin-1-il)piridin-3-il)piridin-4-i1óxl) fenil)-2-oxo-1-fenil-1,2-diidropiridina-3-carboxamida, sal de cloridrato.
A 4-(5-(4-(4-amino-2-fluorofenóxi)piridín-3-il)piridin-2-íl)piperazina-1-carboxilato de terc-butila (Composto B de Exemplo 61, 32 mg, 0,069 mmol) em THF / DMF (1 mL cada) foi adicionado ácido 2-oxo-1-fenil-1,2diidropiridina-3-carboxilico (Composto C de Exemplo 57, 15 mg, 0,069
143 mmol), DIPEA (60 uL, 0,35 mmol), em seguida TBTU (Fluka, 33 mg, 0,10 mmol). Após agitar em temperatura ambiente durante 18 horas, a reação foi diluída com EtOAc (5 mL), lavada com solução de cloreto de lítio aquosa a 10% (2x5 mL) seguido por solução de bicarbonate de sódio saturada (1x5 mL), secada sobre Na2SO4 anidro, e concentrada em vácuo. O resíduo foi suspenso em éter, resfriado a 0°C, e tratado com 4 N de HGI em dioxano (5 mL). A solução foi deixada aquecer para temperatura ambiente e em seguida agitada em temperatura ambiente durante 2 horas. A solução foi concentrada em vácuo e o produto bruto resultante, foi purificado por HPLC prepaid ratíva. As frações apropriadas foram concentradas para remover o metanol e em seguida tornadas básicas com solução de bicarbonate de sódio aquosa saturada. A solução aquosa foi extraída com EtOAc (2 χ 10 mL) e os extratos orgânicos agrupados foram secados sobre Na2SO4 anidro e em seguida concentrados em vácuo. O resíduo foi dissolvido em THF (2 mL), resfriado a 15 0°C, e tratado com 1N de HCI em éter (0,5 mL). Após agitar a 0°C durante 5 minutos, a mistura foi concentrada. O sólido branco resultante foi liofilizado de acetronitrila / água para fornecer o sal de HCI do composto do título (26 mg, 56%) como um sólido amarelo pálido. Ή RMN (DMSO-d6) § 12,16 (s, 1H), 8,92 (s, 1H), 8,59 (d, 1H, J = 6,4 Hz), 8,53 (dd, 1H, J = 7,2, 2 Hz), 8,47 20 (d, 1H, J = 2,4 Hz), 8,10 (dd, 1H, J = 6,4, 2 Hz), 8,04 (d, 1H, J = 12 Hz), 7,99 (dd, 1H, J = 8,8, 2,4 Hz), 7,54-7,46 (m,7H), 7,20 (d, 1H, 6,4 Hz), 7,97 (d,
1H, J = 9,2 Hz), 6,68 (t, 1H, J = 6,8 Hz), 3,82-3,80 (m, 4H), 3,12 (m, 4H); MS(ESF) m/z 545,19 (M + H)+.
Exemplo 63

W*-((R)-2-Amino-2-ox©-1 -feniletil)-/^-(3-flúor-4-(3-(6-(piperazin-1-i!)piridin-3-il)piridin-4-ilóxi)fenil)malonamida, sal de cioridrato.
A 4-(5-(4-( 4-amino-2-fluorofenóxi)piridin-3-il)piridin-2-il)pÍperazina-1-carboxilato de terc-butila (Composto B de Exemplo 61, 32 mg, 0,069 mmol) em THF (1 ml) foi adicionado DIPEA (60 uL, 0,35 mmol) em seguida 3-cloro-3-oxopropanoato de etila (Aldrich, 10 uL, 0,076 mmol). Após agitar em temperatura ambiente durante 2 horas, A reação foi diluída com EtOAc (5 mL), lavada com solução de bicarbonate de sódio aquosa saturada (1x5 mL), secada sobre Na2SO4 anidro, e concentrada em vácuo. O óleo amarelo resultante (65 mg) foi dissolvido em THF (2 mL) e em seguida carregado com 1 N de solução de hidróxido de sódio aquosa (2 mL). A solução foi agitada em temperatura ambiente durante 6 horas. A solução foi concentrada para remover THF e em seguida acidificada para o pH 4-5 com 1 N de solução de HC1 aquosa. O sólido foi coletado por filtração a vácuo e lavado com água para fornecer o ácido correspondente como um sólido branco (30 mg, 78% 2 Etapas). MS(ESF) m/z 552,21 (M + H)\
O ácido acima foi acoplado com D(-)-fenilglicinamida (Bachem) empregando-se TBTU como descrito acima para fornecer o sal de HCI do composto do título (32%) como um sólido branco. 1H RMN (DMSO-d6) 8 10,64 (s, 1H), 8,89 (s, 1H), 8,74 (d, 1H, J = 8 Hz), 8,56 (d, 1H, J = 5,6 Hz), 8,46 (d, 1H, J = 2,4 Hz), 7,96 (d, 1H, J = 8,8 Hz), 7,84 (d, 1H, J = 12 Hz), 7,74 (s, 1H), 7,46-7,37 (m, 4H), 7,31-7,22 (m, 5H), 7,06 (d, 1H, J = 9,2 Hz), 5,34 (d, 1H, J = 8 Hz), 3,81-3,78 (m, 4H), 3,40 (s, 2H), 3,12 (m, 4H); MS(ESF) m/z 584,25 (M + H)3
Exempio 64
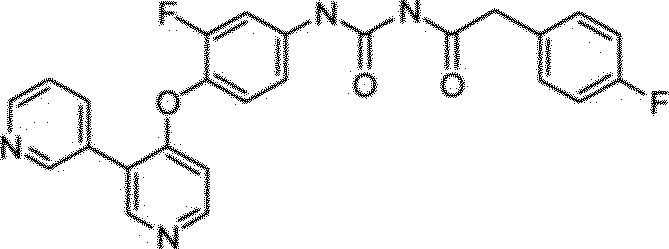
1-(3-flúor-4-(3-(piridin-3-il)piridin-4-ilóxi)fenil)-3-(2-(4- f!uorofenil)acetil)uréia, sal de cloridrato.

A) 3-(4-(2-flúor-4nitrofenóxi)piriciin-3-il)piriclina
Preparado de uma maneira similar à Etapa A de Exemplo 59
B) 3-flúor-4-(3-(piridin-3-il)piridin-4-ilóxi)benzenamina
Preparado de uma maneira similar à Etapa C de Exemplo 59 fornece o composto do título como um óleo incolor. MS(ESF) m/z 282,12 (M + H)\
G) 1 -(3-flúor-4-(3-(píridin-3-íl)piridin-4-ilóxi)fenil)-3-(2-(4fluorofenil)acetil)uréia.
Preparado de uma maneira similar à Etapa C de Exemplo 59 para fornecer o sal de HCl do composto do título (47% de Produção combinada para as Etapas BeC) como um sólido amarelo. 1H RMN (DMSO-d6) δ 11,14 (s, 1H), 10,73 (s, 1H), 9,16 (d, 1H, J = 1,6 Hz), 9,08 (s, 1H), 8,90 (dd, 1H, J = 5,2, 1,2 Hz), 8,78 (d, 1H, J = 6,4 Hz), 8,56 (d, 1H, J = 8 Hz), 7,957,90 (m, 2H), 7,60 (t, 1H, J = 8,8 Hz), 7,55-7,52 (m, 1H), 7,44 - 7,41 (m, 2H), 7,26-7,21 (m, 3H), 3,82 (s, 2H); MS (ESP) m/z 461, 16 (M + H)+.
Exemplo 65

1-(3-flúor-4-(3-(4-(piperazin-1-i!)fenil)piridln-4-ilóxi)fenil)-3-(2(4-fluorofenil)acetil)uréia!( sal de tricloridrato.
_ .Fv .NO,
146
A) 4-(4-(4-(2-flúor-4-nitrofenóxi)piridin-3-il)fenil)piperazína-
1-carboxilato de íerc-butíla.
Preparado de uma maneira similar à Etapa A de Exemplo 50 fornece o composto do título como um óleo incolor. 1H RMN (CD3OD) δ 8,48 (s, IH), 8,32 (d, 1H, J = 5,6 Hz), 8,08 (dd, 1H, J = 12, 4 Hz), 7,97 (d, IH, J = 8 Hz), 7,38 (d, 2H, J = 8,8 Hz), 7,21-7,18 (m, 1H), 6,95-6,92 (m, 3H), 3,46 (m, 4H), 3,09-3,06 (m, 4H), 1,10 (s, 9H); MS(ESP) m/z 495,23 (M + H)*.

B) 4-(4-(4-(4-amino-2-fluorofenóxi)plridin-3-il)fenil) plperazina-1 -carboxilato de terc-butila.
Preparado de -uma maneira similar à Etapa C de Exemplo 59 para fornecer o composto do título (94% de Produção combinada para as Etapas A e B) como um óleo incolor. 1H RMN (CD3OD) δ 8,30 (s, 1H), 8,14 (d, 1H, J = 5,6 Hz), 7,45 (d, 2H, J = 8,8 Hz), 6,98 (d, 2H, J = 8,8 Hz), 6,83 (t, 1H, J = 8,8 Hz), 6,58 (d, IH, J = 5,6 Hz), 6,48 (dd, IH, J = 12,8, 2,4 Hz), 6,43-6,41 (m, IH), 3,49 (m, 4H), 3,11-3,09 (m, 4H), 1,16 (s, 9H); MS(ESF) m/z 465,24 (M + H)+.
C) 1-(3-flúor-4-(3-(4-(Piperazin-1-il)fenll)piridin-4-ilóxi) fenil)3-(2-(4-fluoroferiil)acetil)uréia.
Preparado de uma maneira similar à Etapa C de Exemplo 59 para fornecer o sal de HCI do composto do título (37%) como um sólido amarelo pálido. 1H RMN (DMSOO δ 11,01 (s, IH), 10,60 (s, 1H), 8,77 (s, IH), 8,51 (d, IH, J = 6,4 Hz), 7,78 (d, IH, J = 12 Hz), 7,59 (d, 2H, J = 8,4 Hz), 7,44-7,40 (m, 2H), 7,31-7,28 (m, 2H), 7,13-7,08 (m, 4H), 7,03 (d, 1H, J = 6,4 Hz), 3,70 (s, 2H), 3,42 (m, 4H), 3,15 (m, 4H); MS(ESF) mfe 544,26 (M
Exemplo 66
147

N-(3-flúor-4-(3-(4-(piperazin-1 -il)fenil)pirídin-4-ilóxi)fenil)-2oxo-1-fenil-1t2-diidropiridina-3-carboxamida. sal de tricloridrato.
Preparado de uma maneira- similar àquela de Exemplo 62 para fornecer o sal de HCI do composto do título (43%) como um sólido amarelo pálido. 1H RMN (DMSO-d6) δ 12,15 (s, 1H), 8,83 (s, 1H), 8,56-8,52 (m, 2H),
8,10 (dd, 1H, J = 6,8, 2,4 Hz), 8,04 (dd, 1H, J = 11,6, 2 Hz), 7,61 (d, 2H, J =
8,8 Hz), 7,55 - 7,45 (m, 7H), 7,15 (d, 1H, J = 6,8 Hz), 7,09 (d, 2H, J = 8,8
Hz), 6,61 (t, 1H, J = 6,8 Hz), 3,44 (m, 4H), 3,15 (m, 4H); MS(ESF) m/z 562,36 (Μ + H)*.
Exemplo 67


/V-(3-flúor-4-(3-(4-(2-hidroxietll)fenil)piriciin-4-ilóxí)fenil)-2oxo-1-fenil-1,2-dlidropiridina-3-carboxamlda.
tr
A) 3-(4-(2-(terc-butildimetilsilllóxi)etil)fenil)-4-(2-flúor-4nitrofenóxi)piridina.
Preparado de uma maneira similar à Etapa A de Exemplo 59 para fornecer o composto do título (77%) como um óleo incolor. 1H RMN (CD3OD) 5 8,67 (s, 1H), 8,56 (d, 1H, J- 8 Hz), 8,27 (dd, 1H, J = 12, 4 Hz), 8,16 (d, 1H, J = 8 Hz), 7,58 (d, 2H, J = 8 Hz), 7,39 (d, 2H, J = 8 Hz), 7,36 (m,
1H), 7,15 (d, 1H, 4= 4 Hz), 3,91 (t, 2H, 4 = 8 Hz), 2,90 (t, 2H, 4 = 8 Hz), 0,90 (s, 9H), 0,00 (s, 6H); MS(ESI*) mfe 469,25 (M + H)*.
148
FTBSO.

nh2
B) 3-(4-(2-(terc-butildimetilsllilóxi)etil)fenil)-4-(2-flúor-4nitrofenóxi)piridina.
Preparado de uma maneira similar à Etapa C de Exemplo 59 para fornecer o composto do título (76%) como um óleo amarelo pálido. ΤΗ RMN (CD3OD) δ 8,44 (s, 1H), 8,32 (d, 1H, J = 4 Hz), 7,57 (d, 2H, J = 8 Hz), 7,36 (d, 2H, J = 8,4 Hz), 6,95 (t, 1H, J = 8 Hz), 6,75 (d, 1H, J = 4 Hz), 6,61 (d, 1H, J = 8 Hz), 6,55 (d, 1H, J = 4 Hz), 3,90 (t, 2H, J = 6,8 Hz), 2,89 (t, 2H, J = 6,4 Hz), 0,87 (s, 9H), 0,00 (s, 6H); MS(ESf) m/z 439,26 (M+ H)\
C) IV-(3-flúor-4-(3-(4-(2-hidroxietil)fenil)pirldln-4-ilóxi) fenil)-
2-OXO-1 -fenil-1,2-diidropiridina-3-carboxamida.
Preparado de uma maneira similar à Etapa C de Exemplo 62, após a formação da amida, o óleo amarelo resultante foi dissolvido em THF (2 mL) e em seguida tratado com TBAF (Aldrich, 180 uL, 1 M em THF) em temperatura ambiente durante 1 hora. A reação foi diluída com EtOAc (10 mL), lavado sucessivamente com água e salmoura (5 mL each), secada sobre Na2SO4 anidro, e concentrada em vácuo. O produto bruto foi purificado por cromatografia instantânea em sílica-gel (metanol/EtOAc a 10%) para fornecer o composto do título (72%) como um sólido amarelo pálido. Ή RMN (CD3OD) δ 8,59 (dd, 1H, J = 7,6, 2,0 Hz), 8,41 (s, 1H), 8,26 (d, 1H, J = 5,6 Hz), 7,91-7,85 (m, 2H), 7,55-7,46 (m, 5H), 7,41 (d, 2H, J = 6,7 Hz), 7,29 (d, 2H, J = 8,1 Hz), 7,26 (m, 1H), 7,13 (t, 1H, J = 8,7 Hz), 6,69 (d, 1H, J - B Hz), 6,64 (t, 1H, J = 7,2 Hz), 3,73 (t, 2H, J = 7,2 Hz), 2,82 (t, 2H, J = 6,8 Hz); MS(ESF) m/z 522,27 (M + H)+.
Exemplo 68
JW
149
A^(4-(3-(4-(2-Aminoetíl)fenil)piridin-4-ilóxi)-3-fluorQfenil)-2oxo-1-fenil-1.2-diidropiridina-3-carboxamida, sal de dicloridrato.
A /V-(3-flúor-4-(3-(4-(2-hidroxietil)fenil)piridín-4-ilóxi)fenÍl)-2-oxo-1fenil-1,2-diidropiridina-3-carboxamida (Composto C de Exemplo 67, 40 mg, 0,077 mmol) em THF (1 mL) foi adicionado DIPEA (27 uL, 0,154 mmol) seguido por cloreto de metanossulfonila (Aldrich, 7 uL, 0,092 mmol). Após agitar em temperatura ambiente durante 30 minutos, a reação foi concentrada em vácuo. O resíduo foi dissolvido em 3 mL de etanol e transferido a um tubo de pressão. O hidróxido de amônio (7 mL) foi adicionado e o tubo foi selado e aquecido a 50*C durante 8 horas. Após o resfriamento para temperatura ambiente, a reação foi diluída com EtOAc (10 mL), lavada com água (2x10 mL) em seguida salmoura (1x10 mL), secada sobre Na2SO4 anidro, e concentrada em vácuo. O produto broto foi purificado por HPLC preparativa. As frações apropriadas foram concentradas para remover metanol e basiflcadas com solução de bicarbonate de sódio saturada. A camada aquosa foi extraída com EtOAc (2 χ 20 mL) e os extratos orgânicos agrupados foram lavados com salmoura (1 χ 10 mL), secada sobre Na2SO4 anidro, e concentrada. O resíduo foi dissolvido em dioxano (2 mL) e carregado com IN de HCl em éter (1 mL). A solução foi concentrada e o sólido resultante foi liofilizado de acetronitrila/água para fornecer o sal de HCl do composto do título (24 mg, 53%) como um sólido branco. 1H RMN (DMSO-d6) δ 12,13 (s, 1H), 8,74 (s, 1H), 8,55-8,51 (m, 2H), 8,09 (dd, 1H, J = 6,4, 2 Hz), 8,03 (m, 1H), 7,64 (d, 2H, J = 6 Hz), 7,63 (m, 1H), 7,54-7,41 (m, 6H), 7,38 (d, 2H, J = 8,4 Hz), 7,04 (d, 1H, J = 6 Hz), 6,68 (t, 1H, J = 6,8 Hz), 3,02 (m, 2H), 2,89 (m, 2H); MS(ESF) m/z 521,27 (M + H)*.
Exemplo 69

N-(4-(3-(4-((2-(Metllamino)etil)carbamoil)fenil)piridin-4-llóxi)-
3-fluorofentl)-1-(4-fluorofenil)-2-oxo-1,2-diidroplridlna-3-carboxamida,
150 sal de dicloridrato.

A) 4-(4-(2-flúor-4-nitrofenóxi)piridin-3-il)benzoato metila.
Preparado de uma maneira similar à Etapa A de Exemplo 59 para fornecer o composto do título (77%) como um óleo incolor. 1H RMN (CD3OD) δ 8,66 (s, 1H), 8,52 (d, 1H, J = 6 Hz), 8,22 (dd, 1H, J = 10,4, 2,8 Hz), 8,16-8,13 (m, 1H), 8,10 (d, 2H, J = 8,4 Hz), 7,97 (d, 2H, J = 8 Hz), 7,44 (t, 1H, J = 8,5 Hz), 7,06 (d, 1H, J = 6 Hz), 3,93 (s,3H); MS(ESP) m/z 369,22 (M + Η)*.

B) 4-(4-(4-amino-2-fluorofenóxi)piridln-3-il)benzoato de me10 tila.
Preparado de uma maneira similar à Etapa C de Exemplo 59 para fornecer o composto do título (99%) como um óleo amarelo. 1H RMN (CD8QD) i 8,38 (s, 1H), 8,24 (d, 1H, J = 6 Hz), 8,01 (d, 2H, J = 8,4 Hz), 7,66 (d, 2H, J = 8,4 Hz), 6,85 (t, 1H, J = 9,2 Hz), 6,65 (d, 1H, J = 6 Hz), 6,48 (dd, 15 1H, J = 12,8, 2,4 Hz), 6,43 (d, 1H, J = 2,8 Hz), 3,82 (s, 3H); MS(ESI*) m/z
339,28 (M+Hy.

C) 4-(4-(2-f!úor-4-(1 -(4-fluorofenil)-2-oxo-1,2-diidropiridlna-3carboxamido)fenóxi)piridin-3-il)benzoato de metila.
Preparado de uma maneira similar à Etapa G de Exemplo 62
151 para fornecer o composto do título (81 %) como um óleo amarelo. Ή RMN (CD3OD) δ 8,66 (dd, 1H, J = 7,2, 2 Hz), 8,56 (s. 1H), 8,40 (d. 1H, J = 6 Hz), 8,13 (d, 2H, J = 8,4 Hz), 7,97-7,95 (m. 2H), 7,78 (d, 2H. J = 8,4 Hz), 7,557,52 (m, 2H). 7,38-7,31 (m, 3H), 7,26 (t, 1H, J = 7.2 Hz), 6,82 (d, 1H, J = 5,6 Hz), 6,72 (t, 1H, J = 6,7 Hz), 3,94 (s, 3H); MS(ESI*) m/z 554,21 (M + Hf.
D) #-(4-(3-(4-((2-(Metilamino)etil)carbamoil)feníl)piridin-4ilóxi)-3-fluorofenll)-1“(4-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxamida.
Ao éster acima (159 mg, 0,29 mmol) em THF (5 mL) foi adicionado 1 N de NaOH aquoso (5 mL). Após agitar em temperatura ambiente durante 20 horas, a reação foi concentrada para remover THF. A solução aquosa foi acidificada para o pH 4 com 1 N de HCI aquoso. O ácido foi coletado por filtração e lavagem com água para fornecer o produto desejado (144 mg, 92%) como um sólido castanho. MS(ESF) m/z 540,21 (M + H)*.
A amida foi preparada como descrito acima empregando-se TBTU para fornecer o sal de HCI do composto do título (62%) como um sólido branco. 1H RMN (DMSO-<) δ 12,21 (s, 1H), 8,93-8,90 (m, 2H), 8,68-8,64 (m, 2H), 8,21 (dd, 1H. J = 6,4, 2 Hz), 8,16 (m, 1H), 8,11 (d, 2H, J = 8 Hz), 7,90 (d, 2H, J = 8,4 Hz), 7,69-7,65 (m, 2H), 7,60-7,47 (m, 4H), 7,16 (d, 1H. J = 5,6 Hz), 6,80 (t, 1H, J = 7 Hz), 3,64 (t, 2H, J = 5,2 Hz), 3,17 (t, 2H, J = 5,2 Hz), 2,65 (s, 3H); MS(ESF) m/z 596,37 (M + H)*.
Exemplo 70


^(4-(3-(4-((2-aminoetil)carbam©ll)fenil)piridin-4-ilóxl)-3fluorofenilH -(4-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxamida, sal de dicforidrato.
Preparado de uma maneira similar ao Exemplo 69 para fornecer o sal de HCI do composto do título como um sólido esbranquiçado. Ή RMN (DMSO-de) δ 12,17 (s, 1H). 8,87 - 8,85 (m, 2H), 8,64 (d, 1H, J = 6,4 Hz), 8,59 (dd, 1H, J = 7,6, 2,4 Hz), 8,16 (dd, 1H, J = 6,8, 2,4 Hz), 8,10 (m, 1H). 8,06 (d, 2H, J = 8,4 Hz), 8,00 (br s, 2H), 7,85 (d, 2H, J = 8,4 Hz), 7,64 - 7,60 (m, 2H), 7,55- 7,42 (m, 4H), 7,13 (d, 1H, J = 6 Hz), 6,75 (t, 1H, J = 7,2 Hz), 3,563,53 (m, 2H), 3,04 - 2.99 (m, 2H); MS(ESF) m/z 582,32 (M + H)+.
Exemplo 71

/V-(4-(3-(4-((2-(dimetilamino)etil)carbamoil)fenil)piridin-4ilóxi)-3-fluorofenil)-1-(4-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxamida, sal de dicloridrato.
Preparado de uma maneira similar ao Exemplo 69 para fornecer o sal de HCI do composto do título (56%) como um sólido esbranquiçado. 1H RMN (DMSO-d6) Ô 12,08 (s, 1H), 8,87 (br s, 1H), 8,75 (s, 1H), 8,54 - 8,51 (m, 2H), 8,08 (dd, 1H, J = 6,8, 2,4 Hz), 8,02 (m, 1H), 7,99 (d, 2H, J = 8,4 Hz),
7.77 (d, 2H, J = 8,4 Hz), 7,56-7,53 (m, 2H), 7,47 - 7,34 (m, 4H), 6,99 (d, 1H, J = 5,6 Hz), 6,67 (t, 1H, J = 6,8 Hz), 3,61-3,57 (m, 2H), 3,23 - 3,21 (m, 2H),
2.77 (s, 3H), 2,76 (s, 3H); MS(ESF) m/z 610,30 (M + H)+.
Exemplo 72


1-(4-(3-Aminopiridin-4-ilóxi)-3-fluorofenii)-3-(2-(4-fluorofenil)acetil)uréia.
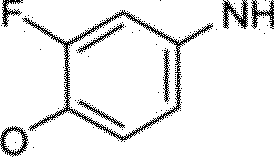

A) 3-flúor-4-(3-nitropiridín-4-ilóxi)benzenamina.
153
A 4-amino-2-fluorofenol (veja Etapa A de Exemplo 19, 127 mg, 1,0 mmol) em DMF (5 mL) em temperatura ambiente sob nitrogênio foi adicionado hidreto de sódio (80 mg, 2 mmoles, 60%). Após agitar em temperatura ambiente durante 10 minutos, cloridrato 4-cloro-3-nitropiridina (Lancaster, 195 mg, 1,0 mmol) foi adicionado. A mistura foi agitada em temperatura ambiente durante 1 hora e foi em seguida diluída com EtOAc (50 mL) e lavada com água, solução de cloreto de sódio aquosa a 10%, e em seguida salmoura (1 χ 30 mL cada). A camada orgânica fbi secada sobre Na2SO4 anidro e concentrada em vácuo. O produto bruto fbi purificado por cromatografia de coluna instantânea em sílica-gel eluindo com EtOAc para fornecer o composto do título (150 mg, 60%) como um sólido laranja-amarelado. 1H RMN (DMSO-O δ 9,13 (s, 1H), 8,63 (d, 1H, J = 6 Hz), 7,09 (t, 1H, J = 9,2 Hz), 6,92 (d, 1H, J = 6 Hz), 6,55 (dd, 1H, J = 13,2, 2,4 Hz), 6,45 (dd, 1H, J = 9,2, 2,4 Hz), 5,61 (s, 2H); MS(ESI+) m/z 250,18 (Μ * H)*.

θ2Νγ\
B) 1-(3-flÚGr-4-(3-nitropiridin-4-ilóxi)fenil)-3-(2-(4-fluorofenil)acetil)uréia.
Uma solução de 3-flúor-4-(3-nitropiridin-4-ilóxi)benzenamina (158 mg, 0,63 mmol) em THF (3 mL) foi tratada com uma solução de isocianato de 2-(4-fluorofenil)acetila em tolueno (Composto D de Exemplo 11,1,3 mmol) e agitada em temperatura ambiente durante 2 horas em seguida a 50°C durante 5 minutos. A mistura foi concentrada e o resíduo tratado com DMF (15 mL) e SiO2 (150 mg) e a mistura concentrada para secagem sob vácuo e aplicada a uma coluna de SiO2. A coluna foi eluída com 20 a 60% EtOAc / hexanos para fornecer o produto, que foi novamente purificada por trituração com éter de isopropila para fornecer um sólido amarelo pálido (120 mg, 25%). 1H RMN (DMSO-de) δ 11,07 (s, 1H), 10,63 (s, 1H), 9,19 (s, 1H), 8,67 (d, 1H, J = 5,6 Hz), 7,85 (d, 1H, J = 11,7 Hz), 7,46 - 7,45 (m, 2H), 7,39 - 7,35 (m, 2H), 7,18 (dd, 2H, J = 8,6, 8,6 Hz), 7,01 (d, 1H, J = 6,1 Hz),
SdG
154
3,76 (s, 2H).
C) 1-(4-(3-Aminopiridin-4-ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia,
A suspensão de 1 -(3-flúor-4-(3-nÍtropiridin-4-ilóxi)fenil)-3-(2-(45 fluorofeni!)acetil)uréia (125 mg, 0,29 mmol) em 3:1 MeOH / THF (20 mL) foi hidrogenada sob Pt2O (50 mg) empregando-se balão de látex de H2 durante horas. O catalisador foi filtrado com o auxílio de Celite* e o filtrado concentrado para fornecer o composto do título (85 mg, 74%) como um sólido branco. Ή RMN (DMSO-d6)ô 11,03 (s, 1H), 10,55 (s, 1H), 8,03 (s, 1H), 7,75 (dd, 1H, J = 2,5, 13,2 Hz), 7,65 (d, 1H, J = 5,1 Hz), 7,38 - 7,35 (m, 3H), 7,23 - 7,16 (m, 3H), 6,42 (d, 1H, 5,1 Hz), 5,26 (s, 2H), 3,75 (s, 2H); MS(ESI*):
m/z 399,35 (M + H)*. '
Exemplo 73

1-(4-(3-((1 S,4S)-4-aminocicloexanocarboxamido)piridin-4- ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de ácido trifluoroacético.

A) (1 S,4S)-4-((4-(2-flúor-4-(3-(2-(4-fluorofenil)acetil)ureído) fenóxi)piridin-3-il)carbamoil)cicloexilcarbamato de terç-butila.
Uma solução de ácido carboxílico N-Boc-c/s-1,4-diaminociclo20 exano (Chem-lmprex International, 24 mg, 0,10 mmol) em THF (1 mL) foi resttada a 0®C, e tratada com Et3N e em seguida isobutilcloroformiato. Após 5 minutos, a mistura foi tratada com uma solução de 1-(4-(3-aminopiridin-4ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia (Composto C de Exemplo
155
72, 27 mg, 0,068 mmol) em THF (0,5 mL) e a agitação continuada a 0°C durart® 10 minutos e em seguida em temperatura ambiente durante 2 horas, A mistura foi dividida entre EtOAc e solução de NaHGO3 aquosa saturada e a fase EtOAc separada, secada (MgSO4) e concentrada em vácuo 5 para fornecer o produto bruto. A purificação do resíduo por cromatografia de coluna instantânea em SiO2 eluindo com 50 -100% de EtOAc / hexanos fornece o composto do título (13 mg, 21%) como um sólido branco. MS(ESr): m/z 624,25 (M + H)*.
B) 1-(4-(3-((1 S,4S)-4-Aminocic!oexanocarboxamido)piridin-
4-ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de ácido trifluoroacético.
Uma solução de (1S,4S)-4-((4-(2-flúor-4-(3-(2-(4-fluorofenil) acetil)ureído)fenóxi)piridin-3-il)carbamoil)cicloexilcarbamato de terc-butila. (10 mg, 0,016 mmol) em MeOH anidro (0,5 mL) foi resfriada a 0°G e tratada com
4 M HCI /1,4-dioxano (2 mL). A mistura foi agitada a 0°G durante 1,5 hora e em seguida em temperatura ambiente durante 20 minutos e finalmente concentrada em vácuo para fornecer o produto bruto. A purificação do resíduo por HPLC preparativa (Coluna A) fornece o composto do título (4 mg, 33%) como um sólido amarelo. 1H RMN (DMSO-dg) δ 11,07 (s, 1H), 10,64 (s, 1H),
9,28 (s, 1H), 8,40 - 8,37 (m, 1H), 7,94 (s, 1H), 7,83 (dd, 1H, J = 2,1, 12,7 Hz), 7,47 - 7,33 (m, 5H), 7,19 - 7,14 (m, 3H), 7,07 - 7,02 (m, 1H), 3,75 (s, 2H), 3,25 - 3,15 (m, 1H), 2,85 - 2,76 (m, 1H), 1,97 - 1,84 (m, 2H), 1,85 - 1,61 (m, 5H); MS(ESF): m/z 524,26 (M + H)*.
Exemplo 74

1-(4-(3-((1 R,4^4-Aminocicloexanocarboxamido)pirídin-4ilóxi)-3-fIuorofenil)-3-{2-(4-fluorofenil)acetil)uréia, sal de ácido bis-trifluoroacético.
156

'Ato
A) (1R,4R)-4-((4-(2-flúor-4-(3-(2-(4-fluorofenil)acetil)ureído) fenóxi) piridin-3-il)carbarnoil)cicloexilcarbamato de terc-butila.
O composto do título foi preparado de 1-(4-(3-aminopiridin-4ilóxi)-3-fluorofenit)-3-(2-(4-fluorofeni!)acetil)uréia (Composto C de Exemplo 72, 57 mg, 0,14 mmoi)e ácido 5/-Boc-teans-4-amÍnocicloexano-1-carboxílico. (Anaspec Inc., 51 mg, 0,21 mmol) de uma maneira similar como descrito à Etapa A de Exemplo 73 para fornecer o composto do título (32 mg, 66%) como um sólido branco. MS(ESF): m/z 624,41 (M + H)*.
B) 1-(4-(3-((1R,4R)-4-Aminocicloexanocarboxamicio)piridin-
4-ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de ácido bistrifluoroacético.
O composto do título foi preparado de (1S,4S)-4-((4-(2-flúor-4-(3(2-(4-fluorofenil)acetil)ureído)fenóxi)piridin-3-il)carbamoil)cÍcloexilcarbamato de terc-butila (25 mg) de uma maneira similar como descrito por exemplo 73, a purificação de uma mistura de reação por HPLC preparativa (Coluna A) fornece o composto do título (7 mg, 23%) como um sólido branco. Ή RMN (DMSO-de) δ 11,06 (s, 1H), 10,61 (s, 1H), 9,94 (s, 1H), 9,19 (s, 1H), 8,28 (d, 1H, J = 5,6 Hz), 7,82 (dd, 1H, J * 2,0, 13,2 Hz), 7,79 - 7,76 (m, 3H), 7,43 (dd, 1H, J = 2,0, 8,6 Hz), 7,38 - 7,33 (m, 2H), 7,17 (dd, 2H, J = 9,2, 6,6 Hz), 6,86 (d, 1H, J = 5,6 Hz), 3,74 (s, 2H), 3,08 - 2,95 (m, TH), 2,67 - 2,43 (m, 1H), 2,01 - 1,88 (m, 4H), 1,53 -1,43 (m, 2H), 1,37 - 1,27 (m, 2H); MS(ESI*): m/z 524,35 (M + Hf.
Exemplo75

1-(4-(3-((1 R,4R)-4-(AminometiÍ)cicloexanocarboxamido) piri- din-4-ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetll)uréia.
157

A) ((1R,4R)-4-((4-(2-flúor-4-(3-(2-(4-fluorofenll)acetíl) ureído) fenóxi)piridin-3-il) carbamoil)cicloexil)metilcarbamato de benzila.
O composto do título foi preparado de 1-(4-( 3-aminopiridin-45 ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia (Composto C de Exemplo 72, 50 mg, 0,13 mmol) e ácido· frans-4-(benziloxicarbonil)metil) cicloexanocarboxilico (40 mg, 0,14 mmol, preparado de acordo com a via sintética descrita em Schaus, J. M. e outro, J. Med. Chem. 1998, 41, 1943-1955) de acordo com a Etapa A de Exemplo 73 para fornecer o composto do título (30 mg, 34%) como um sólido branco. MS(ESr): m/z 672,34 (M + Hf.
B) 1 -(4-(3-(( 1 R4R)-4-(Aminometil)cicloexanocarboxamido) piridín-4-»lóxO-3-fluorofenil)-3-(2-(4-fluorofenil)acetH)uréia.
Uma solução de ((1R,4/?)-4-((4-(2-flúor-4-(3-(2-(4-fluorofenil) acetil)ureído)fenóxi)piridin-3-il)carbamoil)cicloexÍI)metilcarbamato de benzila 15 (25 mg 0,037 mmol) em MeOH (1,5 mL) foi hidrogenada sobre paládiocarbono a 10% (15 mg) durante 4 horas empregando-se H2 de um balão de borracha. O catalisador foi filtrado e o filtrado concentrado em vácuo para fornecer o composto do título (18 mg, 90%) como um sólido amarelo. Ή RMN (DMSO-cg δ 10,62 (s, 1H), 9,64 (s, 1H), 9,01 (s, 1H), 8,17 (d, 1H, J = 20 5,6 Hz), 7,79 (dd, 1H, J = 2,5, 12,7 Hz), 7,42 - 7,35 (m, 4H), 7,30 (dd, 1H, J = 8,6, 9,2 Hz), 7,18 (m, 2H), 6,66 (d, 1H, J = 5,6 Hz), 3,75 (s, 2H), 2,39 (d, 2H, J = 6,6 Hz), 1,87 - 1,81 (m, 4H), 1,46 -1,35 (m, 1H), 1,33 - 1,10 (m, 1H), 0,95 - 0,77 (m, 4H); MS(ESI+): m/z 533,28(M + H>*.
Exemplo 76

1-(4-(3-(Cicloexanocarboxamiclo)piriciiri-4-ilóxi)-3-fluorofeml)-3-(2-(4-fluorofenll)acetil)uréia.
158
Uma solução de 1-(4-(3-aminopiridin-4-ilóxi)-3-fluorofenil)-3-(2(4-fluorofenil)acetil)uréia (Composto C de Exemplo 72, 25 mg, 0,062 mmol) em CH2CI2 (2 mL) foi tratada com Et3N (10 pL, 0,074 mmol) e cloreto de cícloexanocarbonila (Aldrich, 11 mg, 0,074 mmol) e agitada em temperatura ambiente durante 2 horas. Uma porção adicional de cloreto de cicloexanocarbonila (11 mg, 0,074 mmol) foi adicionado a uma mistura e a reação continuada durante 18 horas. A mistura foi diluída com CH2CI2, lavada com solução de NaHCO3 aquosa saturada, secada (MgSO4) e concentrada em vácuo. O resíduo foi purificado por cromatografia de coluna instantânea em 10 SiO2 eluindo com 50 - 100% de EtOAc / hexanos para fornecer o composto do título (19 mg, 61%) como um sólido branco. 1H RMN (DMSO-cy δ 11,03 (s, 1H), 10,57 (m, 1H), 9,60 (m, 1H), 8,99 (s, 1H), 8,15 (d, 1H, J = 5,6 Hz), 7,76 (dd, 1H, J = 2,0,13,2 Hz), 7,39 - 7,33 (m, 3H), 7,28 (dd, 1H, J = 8,6, 9,2
Hz), 7,18 - 7,14 (m, 2H), 6,65 (d, 1H, J = 5,1 Hz), 3,73 (s, 2H), 1,81 - 1,71 15 (m, 5H), 1,64 -1,61 (m, 1H), 1,43 - 1,34 (m, 2H), 1,29 - 1,14 (m, 3H);
MS(ESI+): m/z 509,27 (M + H)*.
Exemplo-77

- 1-(4-(3-(4-Aminopiperidina-1-carboxamido)piriciin-4-ilóxi)-3fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de ácido bis-trifluoroacé20 tico.

A) 1 -(3-flúor-4-(3-(4-(2-fenoxiacetamido)pipericlina-1 -carboxamído)piriclin-4-ilóxi)fem*l)-3-(2-(4-fluorofeni!)acetil)uréia
Uma solução de trifosgênio (50 mg, 0,17 mmol), em CH2CI2 (0,4 mL) foi resfriado a -10°C e tratada com uma solução de 1-(4-(3-aminopiridin25 4-ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetíl)uréia (Composto C de Exemplo
159
72, 67 mg, 0,17 mmol) e DIPEA (65 pL, 0,37 mmol) em CH2CI2 (0,4 mL). A mistura foi agitada a -10°C durante 10 minutos e em seguida tratada com uma solução de 4-((carbobenzilóxí)amido)piperidina (40 mg, 0,17 mmol, preparada empregando-se o procedimento descrito em Schaus, J. M. e ou5 tro, J. Med. Chem. 1998, 41, 1943-1955) e DIPEA (65 μΙ, 0,37 mmol) em CH2CI2 (0,4 mL). Após agitar durante 2 minutos, a mistura foi aquecer para temperatura ambiente em seguida aquecida para 40°C durante 10 minutos.
A mistura foi diluída com EtOAc, lavada com NaHCO3 aquosa saturada e salmoura, secada (MgSO4) e concentrada em vácuo. O produto bruto foi 10 purificado por cromatografia de coluna instantânea em SiO2 eluindo com 0 5% de MeOH / CH2CI2 para fornecer o composto do título (50 mg, 45%) como sólido amarelo. MS(ESI+) m/z 659,29 (M + H)+.
B) 1-(4-(3-(4-Aminopiperidina-1-carboxamido)piridin-4-iió- xi)-3-fluorofenil)-3-(2-(4-fluorofenÍI)acetil)uréia, sal de ácido bis-trifluo15 roacético.
Uma solução de 1-(3-flúor-4-(3-(4-(2-fenoxiacetamido) piperidina-1-carboxamido)pÍridin-4-ilóxi)feni!)-3-(2-(4-fluorofenil)acetil)uréia (45 mg, 0,068 mmol) em MeOH absoluto (2,5 mL) foi hidrogenado sobre paládiocarbono a 10% (15 mg) empregando-se um balão de borracha de H2 duran20 te 2,5 horas. O catalisador foi filtrado e o filtrado concentrado em vácuo e o resíduo foi purificado por HPLC preparativa (Coluna A) para fornecer o composto do título. 1H RMN (DMSO-c© 1 10,57 (s, 1H), 8,55 (s, 1H), 8,27 (m, 1H), 8,14 (d, 1H, J = 5,6 Hz), 7,75 (dd, 1H, J = 2,0, 12,7 Hz), 7,37 - 7,33 (m, 3H), 7,23 - 7,14 (m, 3H), 6,65 (d, 1H, J = 5,1 Hz), 4,05 - 3,98 (m, 2H), 3,73 25 (s, 2H), 3,05 - 2,91 (m, 1H), 2,88 - 2,83 (m, 2H), 1,83 - 1,74 (m, 2H), 1,34 1,20 (m, 2H); MS(ESI+) 525,35 (M + H)*.
Exemplo 78

1-(4-(2-Amino-3-(4-(2-amino-2-oxoetil)fenil)pirídin-4-ilóxi)-3-
160 fluorofenil)-3-(2-(4-fiuorofenil)acetil)uréia. sai de ácido trifluoroacético.

A) Ácido 2-(4-(2-Amino-4-(2-flúor-4-nitrofenóxi)piridin-3-il) feniljacético.
A mistura de 4-(2-flúor-4-nttrofenóxi)-3-todopiridÍn-2-amina (Composto C de Exemplo 34, 88 mg, 0,23 mmol),éster de picanol de ácido
4-(diidroxiborano)fenilacético (Frontier Scientific Inc., 92 mg, 0,35 mmol), Na2CO3 (170 mg, 1,61 mmol), 1,4-dioxano (2 mL) e H2O (2 mL) foi desgaseificada por vácuo/purga de argônio e tratada com tetracís(trifenilfosfina) paládio (27 mg, 0,023 mmol). Após aquecer a 100°C durante 3 horas, o pH da mistura foi ajustado para o pH 6 empregando-se 1 N de ácido clorídrico. A mistura foi concentrada em vácuo e o resíduo dividido entre EtOAc e tampão de fosfato de pH 7. A fase aquosa foi extraída com EtOAc e os extratos combinados foram secados (MgSOJ e concentrada em vácuo para fornecer o produto bruto. O produto foi triturado com 2:1 EtOAc/ MeOH para fornecer o produto desejado (70 mg, 80%) como um sólido marrom-alaranjado. 1H RMN (DMSO-de) δ 12,34 (s, 1H), 8,23 (dd, 1H, J = 3,1,10,5 Hz), 8,05 (d, 1H, d = 10,2 Hz), 7,94 (d, 1H, J = 6,1 Hz), 7,32 - 7,25 (m, 5H), 6,26 (d, 1H, J = 6,1 Hz), 5,62 (s, 2H), 3,57 (s, 2H); MS(ESF): m/z 384,16 (M + H)*.

B) 2-(4-(2-Amino-4-(2-flúor-4-nitrofenóxi)piridin-3-il) fenil) acetamide.
Uma solução de ácido 2-(4-(2-amíno-4-(2-flúor-4-nitrofenóxi) piridin-3-ii)fenil)acético (65 mg, 0,17 mmol) em DMF anidro (1,2 mL) foi tratada com PyBOP (125 mg, 0,24 mmol) e HOBt (32 mg, 0,24 mmol) seguido por DIPEA (60 pL, 0,35 mmol) e NH4CI (19 mg, 0,35 mmol). Após agitar em
161 temperatura ambiente durante 20 minutos, a mistura foi concentrada sob vácuo e o resíduo dividido entre EtOAc e solução de NaHCO3 aquosa saturada. A fase de EtOAc foi lavada com salmoura, secada (MgSO4) e concentrada em vácuo. O produto bruto foi purificado por cromatografia de coluna instantânea em SiO2 eluindo com 0-8% de MeOH / CH2CI2 para fornecer o composto do título (40 mg, 62%) com um óleo de cor âmbar. 1H RMN (DMSO-<) δ 8,23 (dd, 1H, J = 10,7, 2,5 Hz), 8,05 (d, 1H, J = 9,2 Hz), 7,03 (d, 1H, J = 6,1 Hz), 7,42-7,32 (m, 2H), 7,33-7,25 (m, 4H), 6,92 (s, 1H), 6,25 (d, 1H, J = 5,6 Hz), 5,64 (s, 2H), 3,36 (s, 2H); MS(ESF): 383,17 (M + Hf.

C) 2-(4-(2-Amino-4-(4-amino-2-fluorofenóxi)piridÍn-3-tl)fe- nil)acetamida. .
A mistura de 2-(4-(2-amino-4-(2-flúor-4-nitrofenóxi)piridin-3-il) fenil)acetamida (32 mg, 0,086 mmol), DMF (1 mL), EtOH (1 mL) e H2O (1 mL) foi tratada com pó de Fe (67 mg, 1,2 mmol), e NH4GI (128 mg, 2,4 mmoles) e a mistura aquecida a 100°C durante 20 minutos. A mistura foi filtrada através de Celite®, o pH do filtrado ajustado para o pH 7 empregando-se tampão de fosfato e em seguida a mistura foi extraída asm EtOAc. O extrato orgânico foi secado (MgSO4) e concentrado para fornecer o produto desejado (20 mg, 67%) como um sólido marrom-amarelado. MS(ESI+): m/z 353,32 (M + H)‘.
D) 1-(4-(2-Amíno-3-(4-(2-amino-2-oxoetil)fenil)plridir»-4-ióxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia sai de ácido trifluoroacético.
O composto do título foi preparado de 2-(4-(2-amino-4-(4-amino-
2-fluorofenóxi)piridín-3-il)fenil)acetamida (19 mg, 0,054 mmol) e solução de 0,3 M de isocianato de 2-(4-fluorofenil)acetila em tolueno (Composto D de Exemplo 11,0,27 mL, 0,081 mmol) de uma maneira similar àquela descrita para a Etapa D de Exemplo 33, a purificação de uma mistura de reação por HPLC preparativa (Coluna A) fornece o composto do título (9 mg, 26%) co-
7,37 - 7,29 (m, 6H), 7,16 (dd, 2H, J = 8,6, 8,8 Hz), 6,94 (s, 1H), 6,31 (d, 1H, J = 7,1 Hz), 3,72 (s, 2H), 3,43 (s, 2H); MS(ESf): m/z 532,24 (M + Hf. Exemplo 79


1-(4-(2-Amino-3-(2-(piridin-2-il)etinil)piridin-4-ilóxi)-3-fluoro- fenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de dicloridrato.
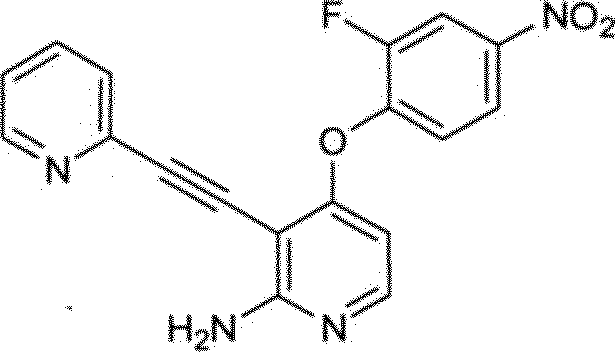
A) 4-(2-flúor-4-nitrofenóxi)-3-(2-(piriclln-2-il)etinil)piridin-2amina.
A mistura de 4-(2-flúor-4-nitrofenóxi)-3-iodopiridin-2-amina (Composto C de Exemplo 34, 100 mg, 0,27 mmol) e 2-etinilpiridina (Aldrich, 57 mg, 0,54 mmol), THF (2 mL) e Et3N (2 mL) foi desgaseificada por vácuo/purga de argônio e tratada sucessivamente com Cui (6 mg, 0,032 mmol) e (Ph3P)4Pd ( 20 mg, 0,017 mmol). A mistura foi aquecida a 60°C durante 45 minutos, resfriada, dividido entre EtOAc e solução de bicarbonato de sódio aquosa saturada. A fase orgânica foi secada (MgSOJ e concentrada em vácuo para fornecer o produto bruto. A purificação do resíduo por cromatografia de coluna instantânea em SiO2 eluindo com 0-1, 5% de MeOH / CH2CI2 fornece o composto do título (55 mg, 58%) como um sólido marrom. Ή RMN (DMSO-dtí) δ 8,53 (d, 1H, J = 5,1 Hz), 8,39 (dd, 1H, J = 2,5, 10,7 Hz), 8,15 (dm, 1H, J = 8.1 Hz.), 7,97 (d, 1H, J = 5,6 Hz), 7,81 (d, IH, J = 8,1
163

B) 4-(4-Amino-2-fluorofenóxi)-3-(2<*(piriciin-24l)etinil)piridin-
2-amina.
A mistura de 4-(2-flúor-4-nitrofenóxi)-3-(2“(piridin-2-il)etinil) piridin-2-amina (35 mg, 0,1 mmol), THF (1,5 mL) e MeOH (1,5 ml) foi tratada com pó de zinco (65 mg, 1,0 mmol) e NH4CI (53 mg, 1,0 mmol) e aquecida a 60°C durante 45 minutos. A mistura de reação foi resfriada, filtrada e concentrada sob vácuo. O resíduo foi dividido entre EtOAc e solução de NaHCO3 aquosa saturada. A fase orgânica foi separada, lavada com salmoura, secada (MgSOJ e concentrada para fornecer o composto do título (25 mg, 78%) como um sólido marrom. MS(ESr): m/z 321,2 (M + H)+.
C) 1-(4-(2-Amino-3-(2-(piridin-2-il)etiníl)piridin-4-ilóxi)-3fluorofenil)-3-(2-(4-fluorofeml)acetil)uréia, sal de dicloridrato.
Uma solução de 4-(4-amino-2-fluorofenóxi)^3-(2-(piridin-2-il) etínil)piridin-2-amina (25 mg, 0,078 mmol) em THF (2 mL) foi resfriada a 0°C e tratada com solução de 0,3 M de isocianato de 2-(4»fluorofenil)acetila em tolueno (Composto D de Exemplo 11, 0,26 mL, 0,078 mmol). Depois de 1 hora, a mistura foi aquecida para temperatura ambiente e agitada durante 15 minutos. A mistura foi concentrada sob vácuo e o resíduo purificado por HPLC preparativa (Coluna A) para fornecer o composto do título como um sal de TFA. O sal de TFA foi dissolvido em MeOH anidro e tratado com 1 M HCI / Et2O a 0°C e agitado durante 5 minutos. A mistura foi em seguida concentrada em vácuo para fornecer o composto do título (18 mg, 41%) como um sólido marrem. ’H RMN (DMSO-c^) δ 11,06 (s, 1H), 10,63 (s, 1H), 8,62 (d, 1H, J = 4,5 Hz), 8,22 (s, 2H), 7,99 (d, 1H, J= 7,1 Hz), 7,94 - 7,81 (m, 3H),
7,48 - 7,44 (m, 3H), 7,37 - 7,33 (m, 2H), 7,16 (dd, 2H, J = 6,2, 9,2 Hz), 6,30 (d, 1H, J = 7,1 Hz), 3,74 (s, 2H); MS(ESL): m/z 500,21 (M + Hf.
Exemplo 80
164


1-(4-(2-Acetamidopfridin-4-ilóxi)-3-fluorofenil)-3-(2-(4-fluoro- feni)acetil)uréía, sal de ciorídrato.


o
A) 4-(2-carbamoilpiridin-4-ilóxi)-3-fIuorofenilcarbamato de ferc-butila.
A mistura de 4-(4-amino-2-fluorofenóxi)picolinamida (Composto
B’ de Exemplo 24, 190 mg, 0,76 mmol), álcool de ferc-butila (2 mL), 1,4dioxano (1 mL), DMF (1 mL) e Boc2O (167 mg, 0,76 mmol) foi aquecida a 65°C durante 16 horas. Porções adicionais de Boc2O (85 mg e 60 mg ) foram adicionados após 16 horas e 32 horas, respectivamente e a mistura a10 quecida durante um total de 40 horas. A mistura foi concentrada sob vácuo e o resíduo dividido entre EtOAc e solução de NaHCO3 aquosa saturada. A fase EtOAc foi secada (MgSO4) e concentrada em vácuo para fornecer o produto bruto. A purificação do resíduo por cromatografia de coluna instantânea em SiO2 eluindo com 30 - 60% de EtOAc / hexanos fornece o compos15 to do título (180 mg, 68%) como um sólido marrom. Ή RMN (DMSO-cy δ 9,74 (s, 1H), 8,52 (d, 1H, J = 5,6 Hz), 8,13 (s, 1H), 7,72 (s, 1H), 7,62 (d, 1H, J = 13,7 Hz), 7,35-7,31 (m, 3H), 7,18 (dd, 1H, J = 5,6, 2,5 Hz), 1,39 (s, 9H); MS(ESF): m/z 348,22 (M + H)*.
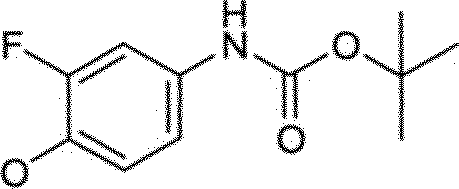
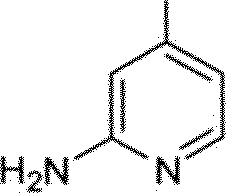
165
B) 4-(2-amínopiridin-4-ilóxi)-3-fluorofenilcarbamato de terc- butila.
Uma solução de KOH (280 mg, 5,0 mmol) in H2O (2 mL) foi resfriada a 0 a 5°C e tratada em gotas com brometo (162 mg, 1,0 mmol) e a mistura agitada durante 5 minutos. 4-(2-carbamoilpiridin-4-ilóxi)-3-fluorofenilcarbamato de terc-butila (347 mg, 1,0 mmol) foi adicionado a uma mistura em uma porção como um sólido e em seguida 1,4-dioxano (3 mL) foi adicionado para dissolver os sólidos. A mistura de reação foi agitada em temperatura ambiente durante 30 minutos em seguida a 55°C durante 45 minu10 tos. A mistura foi em seguida resfriada a temperatura ambiente, tratada com
HOAc (0,5 mL) e agitada até a espumação baixar. A mistura foi reaquecida para 55°C durante 20 minutos, resfriada em temperatura ambiente, tratada com KOH (350 mg) e extraída com CH2CI2 o extrato orgânico foi secado (MgSO4) e concentrado sob vácuo. O resíduo foi purificado por cromatogra15 fia de coluna instantânea em SiO2 eluindo com 30 a 70% EtOAc / hexanos para fornecer o composto do título (265 mg, 83%). M RMN (DMSO-d6) δ
9,67 (s, 1H), 7,77 (d, 1H, J = 6,1 Hz), 7,56 (d, 1H, J = 11,7 Hz), 7,26 - 7,18 (m, 2H), 6,12 (dd, 1H, 4 = 2,0,6,1 Hz), 5,93 (s, 2H), 5,74 (d, 1H, 4 = 2,5 Hz), 1,47 (s, 9H); MS(ESI*): m/z320t23 (M + H)*.
C) 4-(2-acetamidopiridin-4-Hóxi)-3-fluorofenilcarbamato de terc-butila.
4-(2-aminopirídin-4-ilóxi)-3-flyorofenilcarbamato de terc-butila (150 mg, 0,47 mmol)· em piridina anidra (0,5 mL) foi resfriado a 10qC e tratado com cloreto de acetila (33 pL, 0,47 mmol) e a mistura agitada durante 45 25 minutos. Uma porção adicional de cloreto de acetila (16 pL, 0,24 mmol) foi adicionada â reação e agitada continuamente durante 25 minutos. A mistura foi diluída com EtOAc (20 mL), lavada com salmoura, secada (MgSO4) e
166 concentrada sob vácuo para fornecer o composto do título (115 mg, 68%). Ή RMN (DMSO-d6) δ 10,55 (s, 1H), 9,71 (s, 1H), 8,16 (d, 1H, J = 5,5 Hz), 7,63 - 7,55 (m, 2H), 7,29 - 7,23 (m, 2H), 6,68 - 6,63 (m, 1H), 2,02 (s, 3H),
1,48 (s, 9H); MS(ESF): m/z 362,22 (M + H)+.
qAz

D) N-(4-(4-Amino-2-fluorofenóxi)piridin-2-il)acetamida.
Uma solução de 4-(2-acetamidopiridin-4-ilóxi)-3-fluorofenilcarbamato de terc-butila (110 mg, 0,30 mmol) em 4 M de HCI / 1,4-dioxano (1,5 mL) foi agitada a 0°C durante 20 minutos em seguida em temperatura ambiente durante 25 minutos. A mistura foi diluída com EtOAc (25 mL) e solução de NaHCO3 aquosa saturada(20 mL), e agitada vigorosamente durante 5 minutos. A fese EtOAc foi lavada com salmoura, secada (MgSO4) e concentrada em vácuo para fornecer o composto do título (69 mg, 87%) como um sólido amarelo. 1H RMN (DMSO-cQ δ 10,49 (s, 1H), 8,13 (d, 1H, J= 5,6 Hz), 7,60 (m, 1H), 6,95 (dd, 1H, J = 8,6, 9,2 Hz), 6,60 (dd, 1H, J = 2,5, 5,6 Hz),
6,48 (dd, 1H, J = 2,5, 13,2 Hz), 6,40 (dd, 1H, J = 2,0, 8,6 Hz), 5,44 (s, 2H), 2,02 (s, 3H).
E) 1-(4-(2-Acetamidoplrldln-4-ilóxi)-3-fluorofenil)-3-(2-(4fluorofenil)acetil)uréia, sal de cloridrato.
Uma solução de A/-(4-(4-amino-2-fluorofenóxi)piridin-2-il)acetamida (20 mg, 0,077 mmol) em THF (1 mL) foi tratada com solução de 0,3 M de isocianato de 2-(4-fluorofenil)acetila em tolueno (Composto D de Exemplo 11,0,26 mL, 0,77 mmol) e a mistura foi-agitada em temperatura ambiente durante 1 hora. A mistura foi concentrada sob vácuo eo resíduo purificado por HPLC preparativa (Coluna A) para fornecer o composto do título como um sal de TFA. O sal de TFA foi dissolvido em MeOH anidro e tratado com 1 M HCI/Et2O a 0°C e agitado durante 5 minutos. A mistura foi em seguida concentrada em vácuo para fornecer o composto do título (12 mg, 33%) como um sólido branco. ’H RMN (DMSO-d6): 1 11,04 (s, 1H), 10,65 (s,
Η), 10,58 (s, 1Η), 8,18 (d, 1H, J = 6,1 Hz), 7,77 (dd, 1H, J = 2,0, 12,7 Hz), 7,56 (m, 1H), 7,40 - 7,30 (m, 5H), 7,18 - 7,14 (m, 2H), 6,71 (dd, 1H, J = 2,5,
6,1 Hz), 3,74 (s, 2H), 2,03 (s, 3H); MS(ESF): m/z 441,18 (M + H)*.
Exemplo 81


/V-(4-(2-Acetamidopiridin-4-ilóxi)-3-fluorofenil)-2,6-difluoro· benzamida, sal de cloridrato.
Uma solução de A/-(4-(4-amino-2-fluorofenóxi)piridin-2-ÍI)acetamida (Composto B' de Exemplo 24,15 mg, 0,057 mmol) em THF (0,5 mL) foi tratada com DIPEA (15 μί, 0,086 mmol) e cloreto de 2-6-difluorobenzoila (10 mg, 0,057 mmol) e a mistura agitada em temperatura ambiente durante 1,5 hora. A mistura foi concentrada sob vácuo e o resíduo purificado por HPLC preparativa (Coluna A) para fornecer o composto do título como um sal de TFA. O sal de TFA foi dissolvido em MeOH anidro e tratado com 1 M de HCI / Et2O a 0°C e agitado durante 5 minutos. A mistura foi em seguida concentrada em vácuo para fornecer o composto do título (15 mg, 60%) como um sólido esbranquiçado. Ή RMN (DMSO-d6) δ 11,17 (s, 1H), 10,79 (s, 1H), 8,21 (d. 1H, J = 6.1 Hz), 7,89 (dd. 1H. J = 2.0. 12,7 Hz), 7,66 - 7.59 (m, 1H). 7,53 - 7,50 (m, 2H), 7,42 (dd, 1H. J = 8.6, 9,2 Hz), 7.28 (dd, 2H, J = 8.1,
8,1 Hz), 6,80 (dd, 1H, J = 2,0, 6,1 Hz), 2,06 (s, 3H); MS(ESF): m/z 402,13 (M + HJ*.
Exemplo 82



1-(3-flúor-4-(2-(2-morfolinoetilamino)plridin-4-iloxi)fenll)-3-(2-
168


A) N-(4-(2-Cloropiridin-4-ilóxi)-3-fluorofenil)acetamida.
A mistura de /V-(3-flúor-4-hídroxifenil)acetamÍda (Composto A de
Exemplo 13, 1,33 g, 7,87 mmoles), 2-cloro-4-nitropiridina (Aldrich, 1,24 g,
7,87 mmoles), K2CO3 (1,6 g, 11,8 mmoles), e DMF (25 mL) foi aquecida a
100°C durante 9 horas. A mistura foi concentrada em vácuo e o resíduo foi dividido entre EtOAc e solução de NaHCO3 aquosa saturada. A fase EtOAc foi lavada com salmoura, secada (MgSO4), e concentrada em vácuo. O resíduo foi purificado por cromatografia de coluna instantânea em SiO2 eluindo com 30 -80% de EtOAc em hexanos para fornecer o composto do título (1,6 10 g, 73%) como um sólido amarelo pálido. 1H RMN (DMSO-d6) δ 10,25 (s, 1H),
8,35 (d, 1H, J = 7 Hz), 7,80 (d, 1H, J = 14 Hz), 7,50 (d, 1H, J = 3 Hz), 7,33 (m, 2H), 7,02 (m, 1H), 2,06 (s, 3H); MS(ESF): m/z 281,16 (M + H)L


t
O
B) AF(4-(2-Cloropirldin-4-í lóxi-1 -óxido)-3-fluorofenil)acetamida.
A mistura de ácido N-(4-(2-cloropíridin-4-ilóxi)-3-fluorofenil) acetamida (0,98 g, 3,5 mmoles), >90%, m-cloroperoxibenzóico (1,3 g, 7,6 mmoles), e CHCI3 (50 mL) foi agitada em temperatura ambiente durante 60 horas. A mistura foi concentrada em vácuo e o resíduo triturado com EtzO (2 x
100 mL) para fornecer o composto do título (0,89 g, 86%) como um sólido amarelo pálido. 1H RMN (DMSO<) δ 10,25 (s, 1H), 8,35 (d, 1H, J = 7,3 Hz),
7,80 (d, 1H, J = 13 Hz), 7,33-7,32 (m, 3H), 7,02 (dd, 1H, J = 3,5, 7,5 Hz), 2,06 (s, 3H); MS(ESI ): m/z 295,04 (M + H)*.
169

C) AF(3-flúor-4-(2-(2-morfolmoetilamino)piridm-4-ilóxi-1 - óxido)fenil)acetamida.
A mistura de cloridrato de M(4-(2-cloropiridin-4-ilóxi-1-óxido)-3fluorofenil)acetamida (205 mg, 0,62 mmol), 4-(2-arninoetil)morfolina (Aldrich,
169 mg, 1,30 mmol), e EtOH absoluto foi aquecido até o refluxo 16 horas. A mistura de reação foi concentrada em vácuo, e o resíduo foi tratado com
H2O (3 mL) e aplicado para um cartucho Varian 018 de 10 g. O cartucho foi eluído primeiro com H2O em seguida com 30% de MeOH em H2O. O eluente que continha o produto· desejado foi agrupado, concentrado para um volume de 5 mL, e extraído 3 vezes com EtOAc. Os extratos combinados foram lavados com salmoura, secados (MgSO4) e concentrados em vácuo para fornecer o composto do título (100 mg, 40%). 1H RMN (DMSO-cy δ 10,22 (s,
1H), 7,84 (d, 1H, J = 6 Hz), 7,77 (dd, 1H, J = 2, 12 Hz), 7,31 (dd, 1H, J = 2, 9 Hz), 7,24 (dd, 1H, J = 9, 9 Hz), 6,41 (m, 1H), 6,13 (dd, TH, J = 2, 6 Hz), 5,81 (d, 1H, J = 2,5 Hz), 3,56-3,48 (m, 2H), 3,31-3,19 (m, 4H), 2,38 (t, 2H, J = 7
Hz), 2,40-2,28 (m, 4H), 2,06 (s, 3H); MS(ESI*): 405,22 (M + H)t


D) N-(3-flúor-4-(2-(2-morfolinoetilamino)piridin-4-ilóxi)fenil) acetamida, sal de ácido trifluoroacético.
A mistura de Af-(3-flúor-4-(2-(2-morfolinoetilamino)piridin-4-ilóxi20 1-óxido)fenil)acetamida (100 mg, 0,26 mmol), e polímero de trifenilfosfina suportado (1,4 -2,0 mmol/g) em poliestireno (500 mg) e DMF (2 mL) foi agitada a 135°C durante 15 horas. A mistura foi filtrada para remover a resina e a resina lavada com DMF e EtOAc. O filtrado e as lavagens foram combina
170 dos e concentrados. O produto bruto foi purificado por HPLC preparativa (Coluna A) para fornecer o composto do título (45 mg, 24%) como um sólido branco. Ή RMN (DMSO-cg δ 10,33 (s, 1H), 8,02 (d, 1H, J = 7 Hz) 7,84 (dd, 1H, J = 2, 13 Hz), 7,39-7,31 (m, 2H), 6,52 (s, I H), 6,10 (s, 1H), 3,83 (s, 4H), 3,60-3,48 (m, 2H), 3,32-3,18 (m, 6H), 2,08 (s, 3H); MS(ESI*): m/z 375,12 (M + H)+.
FXíí^V'NH2

E) 4-(4-Amino-2-fluorofenóxi)-N-(2-morfolinoetil)piridin-2amina, sal de cloridrato.
Uma mistura de trifluoroacetato de AF(3-flúoM-(2-(2-morfolinoetilamino)pirídin-4-ílóxi)fenil)acetamida (40 mg), MeOH (1 mL), e 6 M HCI (0,2 mL) foi aquecida até o refluxo durante 3 horas. A mistura foi concentrada em um evaporador giratório e o resíduo foi liofilizado para fornecer o composto do título (30 mg) como um sólido branco. -H RMN (DMSO-de) δ 11,12 (s, 1H), 8,85 (s, 1H), 7,95 (d, 1H,U=7Hz), 7,08 (dd, I H, J = 9,9 Hz), 6,65-6,63 (m, 2H), 6,54 (d, 1H, J = 8 Hz), 6,31 (s, 1H), 3,90-3,75 (m 6H), 3,37-3,21 (m, 6H); MS(ESI): m/z 373,14 (M + H)*.
F) 1-(341úor-4-(2-(2-morfolinoetilamino)piridin-4-ilóxi) fenll)-
3-(2-(4-fluorofer»il)acetil)uréia, sal de dicloridrato.
Uma solução de cloridrato 4-(4-amino-2-fluorofenóxi)-/V-(2-morfolinoetil) piridin-2-amina (15 mg, 0,045 mmol) em MeOH (5 mL) foi tratada com Et3N (2 mL) e a mistura agitada em temperatura ambiente durante 5 minutos. A mistura foi concentrada em vácuo para remover o MeOH, o resíduo suspenso em THF (1 mL) e tratado com uma solução de solução de 0,3 M de isocianato de 2-(4-fluorofenil)acetila em tolueno (Composto D de Exemplo 11, 180 mL, 0,054 mmol). Após agitação, a mistura foi concentrada em vácuo e o resíduo dividido entre EtOAc e NaHCO3 saturado. A fase EtOAc foi separada, lavada com salmoura, secada (MgSOJ e concentrada. A mistura foi concentrada sob vácuo e o resíduo purificado por HPLC prepara
171 tíva (Coluna A) para fornecer o composto do título como um sal de TFA. O sal de TFA foi dissolvido em MeOH anidro e tratado com 1 N de HCI / Et2O a 0°C e agitada durante 5 minutos. A mistura foi em seguida concentrada em vácuo para fornecer o composto do título (10 mg, 43%) como um sólido branco. Ή RMN (DMSO-de) δ 11,05 (s, 1H), 10,61 (s, 1H), 7,98 (d, 1H, J =
7,1 Hz), 7,86-7,73 (m, 1H), 7,48-7,38 (m, 1H), 7,37-7,30 (m, 3H), 7,24-7,04 (m, 2H), 6,60 (s, 1H), 6,26 (s, 1H), 3,98-3,60 (m, 8H), 3,74 (s, 2H), 3,39-3,19 (m, 4H); MS(ESI*): rrtó 512,2 (M + H)*.
Os Exemplos 83-85 foram preparados de uma maneira similar como descrito para o exemplo 82.
Exemplo 83
1-(3-flúor-4-(2-(3-morfolinopropilamino)piriclin-4-ilóxi) fenil)-
3-(2-(4-fluorofenil)acetil)uréia, sai de cloridrato.
Ή RMN (DMSO-cy δ 11,06 (s, 1H), 10,62 (s, 1H), 7,93 (d, 1H, J = 7,1 Hz), 7,83 (d, 1H, J = 12,7 Hz), 7,45 - 7,33 (m, 4H), 7,16 (dd, 2H, J ~
8,6, 9,2 Hz), 6,64 (s, 1H), 6,23 (s, 1H), 3,95 - 3,76 (m, 4H), 3,74 (s, 2H), 3,70 -3,48 (m, 4H), 3,48 - 3,35 (m, 2H), 3,20 - 3,-04 (m, 2H), 2,02 - 1,93 (m, 2H).
Exemplo 84
1-(4-(2-(3-(Dimetilamino)propilamino)plridln-4-ilóxO-3-fluorofenil)-3-(2-(4-fluorofenil)aceti0uréia, sal de cloridrato.
1H RMN (DMSO-de) δ 11,06 (s, 1H). 10,62 (s, 1H), 10,37 (s, 1H), 7,93 (d, 1H, J - 7,1 Hz), 7,82 (dd, 1H, J = 2,0, 12,7 Hz), 7,45 (dd, 1H, J =
2,6, 8,6 Hz), 7,40 (d, 1H, J = 8,6 Hz), 7,37 - 7,33 (m, 2H), 7,16 (dd, 2H, J =
172
8,7, 9,1 Hz), 6,65 (s, 1H), 6,24 (s, 1H), 3,75 (s, 2H), 3,45 - 3,36 (m, 2H), 3,13 -3,03 (m, 2H), 2,73 (s, 3H), 2,72 (s, 3H), 1,94 -1,90 (m, 2H).
Exemplo 85


1-(4-(2-(4-(Dlmetilamino)butllamino)piriciin-4-ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de cloridrato.
MS(ESF): m/z 498,2 (M + Hf.
Exemplo 86

1-(4-(2,6-Diaminoplridin-4-ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uráia, sal de cloridrato.
g
Ó 0
A) 4-Cloroplridina-2,6-dicarboxamida
A mistura de ácido quelidâmico (3,19 g, 17,0 mmoles), PCI5 (2,1 g) e CCI4 (30 mL) fol refluxada durante 6 horas e em seguida resfriada a 65eG e tratada com MeOH (5 mL) sob reflux© suave. A mistura foi refluxada durante 5 horas e em seguida concentrada em vácuo. O resíduo foi tratado com água-água (50 mL) e o sólido precipitado foi coletado por filtraçâo e sorvido seco sobre o funil para fornecer agulhas brancas de 2,6-biscarbometóxi-4cloropiridina (2,4 g). O produto foi tratado com - 7 M NH3 / MeOH e agitado em temperatura ambiente durante 1 hora. A mistura foi filtrada para coletar o composto do título como um sólido branco (1,8 g, 53%). 1H RMN (DMSO-dô) δ 8,91 (s, 2H), 8,15 (s, 2H), 7,87 (s, 2H).
173


B) 4-(2,6-dicarbamoilpiridin-4-ilõxi)-3-fluorofenilcarbamato de íerc-butila.
Uma solução de N-Boc-4-amino-2-fluorofenol (228 mg, 1,0 mmol) em DMF (2 mL) foi tratada com FBuOK (124 mg, 1,1 mmol) e a mistura agi5 tada em temperatura ambiente durante 2 horas. A mistura foi tratada com 4cloropÍridina-2,6-dtcarboxamida (200 mg, 1,0 mmol) e K2CO3 (35 mg, 0,5 mmol) e aquecida a 80°C durante 1,5 hora. A mistura foi concentrada em vácuo, tratada com EtOAc (10 mL) e H2O (10 mL) e filtrada para remover o material insolúvel. A fase EtOAc foi lavada com salmoura, secada (MgSO4) e concen10 trada em vácuo. A purificação do resíduo por cromatografia de coluna instantânea em SiO2 eluindo com 30 -100% de EtOAc / hexanos fornece o composto do título (170 mg, 44%) como um sólido branco contendo da cloropiridina de partida a 10%. Ή RMN (DMSO<) δ 9,77 (s, 1H), 8,86 (s, 2H).
7,87 (s, 1H), 7,63 (d, 1H, J = 12,1 Hz), 7,55 (s, 2H), 7,38 - 7,31 (m, 2H), 1,48 (s, 9H).
h2n

NHz
NHBoc
C) 4-(2,6-diaminopíridin-4-ilóxi)-3-fluorofem’lcarbamato de terc-butila.
O composto do título foi preparado de 4-(2,6-dfcarbamoilpiridin-
4-i!óxi)-3-fluorofeni!carbamato de terc-butila (110 mg, 0,28 mmol) empregan- do-se um procedimento similar como descrito para a Etapa B de Exemplo
80, Cromatografia instantânea em SiO2 eluindo com 0 - 2% de MeOH / EtOAc fornece o composto do título (60 mg, 63%) como um sólido branco. 1H
RMN (DMSO-de) δ 9,60 (s, 1H), 7,50 (dd, 1H, /= 1,8, 13,6 Hz), 7,21 (dd, 1H,
174
J = 2,2, 8,7 Hz), 7,13 (dd, 1H, J = 8,7, 9,2 Hz), 5,40 (s, 4H), 5,13 (s, 2H),
1,47 (s, 9H); MS(ESC): mA 335,23 (M + H)L

H2N/<^NH2
D) 4-(4-Amino-2-fluorofenóxi)piriciina-2,6-diamlna.
O composto do título foi preparado de 4-(2,6-diaminopiridin-4ilóxi)-3-fluorofeni1carbamato de terc-butila (30 mg, 0,089 mmol) de uma maneira similar àquela descrita na Etapa D de Exemplo 80 para fornecer um óleo claro (20 mg, 100%). MS(ESF): mA 235,22 (M + H)*.
E) 1-(4-(2,6-Diaminopiridin-4-ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de cloridrato.
O composto do título foi preparado de 4-(4-amino-2-fluorofenóxi) piridina-2,6-diamina (19 mg, 0,081 mmol) e uma solução de isocianato de 2(4-fluorofenil)acetila em tolueno (0,3 M, Composto D de Exemplo 11, 0,27 mL, 0,081 mmol) de uma maneira similar com a Etapa D de Exemplo 33, a mistura de reação foi purificado por HPLC preparativa (Coluna A) para fornecer o composto do título como um sal de TFA. O sal de TFA foi dissolvido em MeOH anidro e tratado com 1 M de HCI / Et2O a 0°C e agitada durante 5 minutos. A mistura foi em seguida concentrada em vácuo para fornecer o composto do título (8 mg, 24%) como um sólido amarelo pálido. 1H RMN (DMSO-O 5 11,01 (s, 1H), 10,51 (s, 1H), 7,68 (dd, 1H, J - 2,6, 12,7 Hz), 7,36 - 7,30 (m, 3H), 7,22 - 7,14 (m, 3H), 5,52 (s, 4H), 5,15 (s, 2H), 3,73 (s, 2H); MS(ESI*): m£414,09 (M + H)+.
Exemplo 87

1-(4-((2-(3-(Dimetilamino)propilamino)piridin-4-il)metil)-3fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de dicloridrato.
175
Br'

NHBoc
A) 4-bromo-3-fluorofenilcarbamato de terc-butila.
A uma solução de 4-bromo-3-fluorobenzenamina (Lancaster,
7,05 g, 37,1 mmoles) em tetraidrofurano anidro (40 mL) em temperatura ambiente foi adicionado (Boc)2O (8,10 g, 37,1 mmoles) e trietilamina (5,17 mL, 37,1 mmoles). A mistura de reação foi aquecida até o refluxo durante a noite. Após o baixo resfriamento, a mistura de reação foi concentrada sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna instantânea em SiO2 eluindo com diclorometano a 20% em hexano, em seguida acetato de etila a 20% em hexano para fornecer 4-bromo-3-fluorofenilcarbamato de terc-butila (5,30 g, 49% Produção). MS(ESr): rwk 290,2 (M + H)*.
NHBoc
CI^N^
B) 4-((2-cloropiridin-4-ilXhídróxi)metil)-3-fluorofenilcarbamato de terc-butila.
A uma solução de 4-bromo-3-fluorofenitearbamato de terc-butila (2,60 g, 9,0 mmoles) em THF anidro (30 mL) a -78°C foi adicionado MeMgBr (3,0 M em Et2O, 3,1 mL, 9,3 mmoles) por meio de seringa. A solução foi agitada durante 10 minutos naquela temperatura e em seguida aquecida para 0°C durante 0,5 hora. Após a solução ser resfriada novamente para -78°C, uma solução de t-BuLi (1,7 M em hexano, 10,6 mL, 18,1 mmoles) foi adicionado durante 4 minutos. A solução resultante foi deixada agitar durante 5 minutos antes de uma solução de 2-cloroisonicotinaldeído (1,41 g, 10 mmoles) (para preparação, veja Frey, L. F. e outro. Tetrahedron Lett. 2001, 42, 6815) em THF anidro (25 mL) ser adicionada em 3 minutos. A mistura de reação foi agitada a -78°G durante 20 minutos e 2,0 mL de MeGH foi adicionado. A solução foi em seguida concentrada sob pressão reduzida e o resíduo foi dissolvido em 200 mL de EtOAc. Foi subsequentemente lavada com H2O (2 x 50 mL), salmoura (2 χ 50 mL) e secada sobre MgSG4i apôs a filtra
176 ção e concentração, O resíduo foi purificado por cromatografia instantânea em SiO2 eluindo com 0%-50% EtOAc em hexano para fornecer 4-((2cloropiridÍn-4-il)(hidróxi)metil)-3-fluorofenilcarbamato de ferc-butila (1,30 g,
41% Produção). MS(ESI+): m/z 353,28 / 355,24 (M + H)L

NHBoc
0“
C) 4-((2-cloropiriciin-#óxido-4-ll)(hidróxi)metil)-3-fluorofenilcarbamato de terc-butila.
A uma solução de 4-((2-cloropiridin-4-il)(hidróxi)metil)-3-fluorofenilcarbamato de terc-butila (1,20 g, 3,40 mmoles) em uma mistura de diclorometano (100 mL) e acetato de etila (10 mL) foi adicionada m-CPBA 10 (70%, 2,34 g, 9,48 mmoles). A mistura de reação foi agitada em temperatura ambiente durante 2 horas, e em seguida aquecida até o refluxo durante 5 horas. O solvente foi removido sob pressão reduzida e o resíduo foi purificado por cromatografia instantânea em SiO2 eluindo com 50% EtOAc em hexano, 100% de EtOAc e em seguida 10% de MeOH em EtOAc, para forne15 cer 4-((2-cloropíridin-A/-óxido-4-il)(hidróxi)metil)-3-fluorofenilcarbamato de terc-butila (840 mg, 67% Produção). MS(ESL): m/z 369,13 / 371, 13 (M +
Hy.

D) 4-((2-(3-(dimetilamino)propilamino)piridina -4-il)(hidróxi) metil)-3-fIuorofenilcarbamato de terc-butila.
A uma solução de 4-((2-cloropiridin-N-óxido-4-il)(hidróxi)metil)-3fluorofenilcarbamato de terc-butila (80 mg, 0,22 mmol) em EtOH (2,0 mL) foi adicionado #,/VMímetilpropano-1,3-diamina (225 mg, 2,2 mmoles). A mistura de reação foi aquecida a 80°C durante 12 horas e o solvente foi removido
177 para fornece 4-((2-(3-(dimetilamino)propilamino)piridina-AFóxido-4-ii) (hidróxi)metil)-3-fluorofenilcarbamato de terc-butila bruto, que foi diretamente empregado na etapa seguinte. MS(ESr): m/z 435,37 (M + H)+.
A uma solução de 4-((2-(3-(dimetÍlamino)propilamino)piridina-/Vóxido-4-il)(hidróxi)metil)-3-fluorofenilcarbamato de terc-butila (-0,22 mmol) em MeOH (2,0 mL) foi adicionado zinco (114 mg, 1,75 mmol) e NH4CO2H (139 mg, 2,20 mmoles). A suspensão foi refluxada durante a noite. Mais zinco (114 mg) e NH4CO2H (139 mg) foram adicionados e a suspensão foi refluxada durante 2 horas. Após o baixo resfriamento, a solução foi filtrada e o filtrado foi concentrado sob pressão reduzida. O resíduo foi em seguida purificado por cromatografia instantânea em SiO2 eluindo com 10-30% MeOH em DCM para fornecer 4-((2-(3-(dímetilamino)propilamino)piridina-4-il) (hidróxi) meti)-3-fluorofenílcarbamato de terc-butila (80 mg, 87% Produção). MS(ESI+): m/z 419,34 (M + H)+.

H
E) 4-(4-Amino-2-fluorobenzil)-W-(3-(dimetilamino)propil) plridin-2-amina
A uma solução de 4-((2-(3-(dimetilamino)propilamino)piridina- 4il)(hidróxi)metíl)-3-fluorofenilcarbamato de terc-butila (80 mg, 0,19 mmol) em MeOH (5,0 mL) foi adicionado 2 mL de HCI concentrado e paládio em carvão vegetal (10%, 200 mg). A suspensão foi aquecida a 75°C sob atmosfera de Hz durante 24 horas. A mistura foi resfriada, filtrada e concentrada em vácuo. O resíduo foi dissolvido em 1 mL de NH4OH concentrado e foi extraída com DCM (5x5 mL). A camada orgânica combinada foi secada sobre Na2SO4i após filtração, foi concentrada em vácuo para fornecer 4-(4-amino-
2-fluorobenzil)-N-(3-(dimetilamino)propil)piridin-2-amina (31 mg, 40% Produção). MS(ESP): 303,31 (M + H)*.
F) 1-(4-((2-(3-(Dimetilamino)propilamino)plridin-4-il)metil)-3-
178 fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de dicloridrato.
A uma solução de 4-(4-amino-2-fluorobenzil)-/V-(3-(dimetilamino) propil)piridin-2-amina (30 mg, 0,1 mmol) em DCM (2 mL) foi adicionado uma solução de isocianato de 2-(4-fiuorofenil)acetila (Composto D de Exemplo 11, 0,347 M em tolueno, 0,25 mL). A mistura foi agitada em temperatura ambiente durante 0,5 hora, antes foi extinguida com MeOH. A solução foi concentrada em vácuo e o resíduo foi purificada por HPLC preparativa. As frações desejadas foram coletadas e concentradas em vácuo. O resíduo foi dissolvido em MeOH e triamida de dietileno ligada a polímero (50 mg) foi adicionado para remover o ácido trifluoroacético. Após a fíltração e concentração, o resíduo foi convertido sal de cloridrato por adição de 1 N de HCI (0,5 mL) e liofilizado para fornecer cloridrato de 1-(4-((2-(3-(dimetilamino) propilamino)piridin-4-il)metil)-3-fluorofeni!)-3-(2-(4-fluorofenil)acetil)uréia (8,0 mg, 14% de Produção), MS(ESC): m/z 482,24 (M + H)*.
Exemplo 88

1-(4-((2-(3-(Dimetilamino)propilamino)piridin-4-il)(hidróxi) metil) fenil)-3-(2-(4-fluorofenil)acetil)uréia.
gi- r
A) 4-((2-cloroplridin-4-il)(hidróxi)metil)fenilcarbamato de terc-butila.
Preparado de maneira similar àquela que é descrita na Etapa B de Exemplo 87, 2-Cloroisonicotinaldeído (141 mg, 1,0 mmol) foi convertido em 4-((2-cloropiridin-4-il)(hidróxi)metil)fenilcarbamato de terc-butila (190 mg, 57% de Produção). MS(ESr): 335,27/337,27 (M + H)*.
179

B) 4-((2-cloropiriclin-W-óxido-4-il)(hiciróxi)metil) fenilcarba- mato de terc-butila.
Preparado de maneira similar àquela que é descrita na Etapa C de Exemplo 87, de 4-((2-cloropiridin-4-il)(hidróxi)metil)fenilcarbamato terc5 butila (78 mg, 0,23 mmol) foi convertido a 4-((2-cloropiridin-A/-óxido-4-ÍI) (hidróxi)metil) fenilcarbamato de terc-butila (36 mg, 44% Produção). MS(ESF):
m/z 351,28/ 353,27 (M + H)+.
HO

NHBoc
C) 4-((2-(3-(dimetilamino)propilamino)piridin-4-il)(hidróxi) metil)fenilcarbamato de terc-butila.
Preparado de maneira similar àquela que é descrita na Etapa D de Exemplo 87, 4-((2-cloropiridin-/V-óxido-4-il)(hidróxi)metÍI) fenilcarbamato de terc-butila (36 mg, 0,1 mmol) foi convertido em 4-((2-(3-(dimetilamino) propilamino)piridin-4-il)(hidróxi)metil)fenilcarbamato de terc-butila (16 mg.
40% Produção). MS(ESF)· mâr 401, 38 (M + H)*.
nh2
HO

D) (4-Aminofenil)(2-(3-(dimetilamino)propilamino)piridin-4-il) metanol.
A uma solução de 4-((2-(3-(dimetilamino)propilamino)piridin -4il)(hidróxi)metil)fenilcarbamato de terc-butila (16 mg, 0,04 mmol) em 1 mL de
180
DCM foram adicionados Et3SiH (0,1 mL)/TFA em DGM (10%, 0,2 mL). A mistura foi agitada durante % hora e nenhuma reação foi detectada por LCMS. Mais 0,1 mL de Et3SiH e 0,8 mL de TFA em DCM (10%) foram adicionados e a mistura foi agitada durante 2 horas. O solvente foi removido e foi purificado pela extração de sólido (cartucho de extração Waters Oasis®MCX ) para fornecer (4-aminofenil)(2-(3-(dimetilamino)propilamino)piridin-4il)metanol (6,0 mg, 50% de Produção). MS(ESr): m/z 301, 40 (M + H)+.
E) 1-(4-((2-(3-(Dimetilamino)propilamino)piridin-4-il)(hidiOxi) metil)fenil)-3-(2-(4-fluorofenil)acetil)uréia.
Preparado de maneira similar àquela que é descrita na Etapa F de Exemplo 87, 4-AminofenÍI-(2-(3-(dimetilamÍno)propilamino)piridin-4-il) metanol (6,0 mg, 0,02 mmol) foi convertido a 1-(4-((2-(3-(dimetilamino) propilamíno)piridin-4-il)(htdróxi)metil)fenil)-3-(2-(4-fluorofenil)acetil)uréia, ácido bistrifluoroacético (6,1 mg, 43% de Produção). Ή RMN (CD3OD) ô 7,82 (d, 1H, J = 6,4 Hz), 7,50 (m, 2H), 7,36 (m, 4H), 7,07 (m, 3H), 6,75 (m, 1H), 5,68 (s, 1H), 3,71 (s, 2H), 3,21-3,49 (m, 4H), 2,90 (s, 6H), 2,08 (m, 2H); MS(ESF): m/z 480,31 (M + H)*.
Exemplo 89
H2NAIÍ
1-(4-((2-Amlnopiridln-4-íl)metil)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de ácido trifluoroacético.
Fx^x^NHBoc
A) 4-((2-(alilamino)piridin-/^-óxido-4-il)(hidróxÍ)metil)-3fluorofenllcarbamato de terc-butila.
A uma solução de 4-((2-cloropiridin-N-óxído-4-ilXhidróxi)metil)-3
181 fluorofenilcarbamato de terc-butila (Etapa G de Exemplo 87, 500 mg, 1,36 mmol) em EtOH (14 mL) foi adicionado alilamina (1,0 mL, 13,6 mmoles). A mistura foi aquecida a 80°C durante a noite. Após baixo resfriamento, o solvente foi removido e o resíduo foi purificado por cromatografia instantânea em SiO2 eluindo com 0%-15% de MeOH em DGM para fornecer 4-((2(alilamino) piridin-/V-óxido-4-il)(hidróxi)metil)-3-fluorofenilcarbamato de tercbutila (440 mg, 83% de Produção). MS(ESr): m/z 390,19 (M + H)L

B) 4-((2-(alilamino)piridin-4-il)(hidróxi)metil)-3-fluorofenilcarbamato de terc-butila.
Preparado de maneira similar àquela que é descrita na Etapa D de Exemplo 87, 4-((2-(alilamino)piridin-A/-óxido-4-ilXhidróxi)metil)-3-fluorofenilcarbamato de terc-butila (440 mg, 1,13 mmol) foi convertido a 4-((2(alilamino)piridin-4-il)(hidróxi)metil)-3-fluorofenilcarbamato de ferc-butila (400 mg, 95% Produção). MS(EST): m/z 374,33 (M + H)+.

C) Acetato de (2-(Alilamino)piridin-4-ilX4-(terc-butoxlcarbonil)-2-fluorofenil)metila.
A uma solução de 4-((2-(alilamino)pÍridin-4-il)(hidróxi)metii)-3fluorofenilcarbamato de terc-butila (400 mg, 1,1 mmol) em THF (10 mL) foram adicionados diisopropiletilamina (DIEA) (0,2 mL, 1,1 mmol), 4-dimet‘ilaminopiridina (DMAP) (360 mg, 3,0 mmoles) e Ac2O (0,29 mL, 3,0 mmoles). A mistura foi agitada durante a noite e em seguida aquecida até o refluxo durante 1 hora. Após baixo resfriamento, o solvente foi removido sob pressão reduzida e o resíduo foi purificado por cromatografia instantânea em SiO2
182 eluindo com 0% -100% de EtOAc em hexano para fornecer acetate de (2(alilamino)piridin-4-il)(4-(terc-butoxicarbonila)-2-fluorofenil)metila (300 mg,
85% de Produção). MS(ESF): m/z 416,33 (M + H)+.
NHBoc

D) Acetato de (2-Aminopiridin-4-il)(4-(terc-butoxicaAonil)-2· fluorofenil)metila.
Uma solução de acetato de (2-(alilamino)piridin-4-il)(4-(tefcbutoxicarbonil)-2-fluorofenil)metila (380 mg, 0,91 mmol) em uma mistura de EtOH/H2O (10: 1,40 mL) foi desgaseificada por meio de borbulhamento em uma solução de N2 durante 1 hora. A uma mistura foi adicionado Rh(PPh3)3GI (80 mg, 0,09 mmol). A solução foi refluxada para remover o solvente e o resíduo foi purificado por cromatografia instantânea em SiO2, seguido por purificação por HPLC preparativa, para fornecer acetato de (2aminopiridin-4-il)(4-(terc-butoxicarbonil)-2-fluorofenil)metila, sal de ácido trifluoroacético (185 mg, 42% Produção). MS(ESL): m/z 376,26 (M + H)\
NHBoc
H2N

E) 4-((2-aminopiridin-4-il)metil)-3-fluorofenilcarbamato de terc-butila.
A uma solução de acetato de (2-aminopiridin-4-H)(4-(terc-butoxicarbonil)-2-fluorofenil)metila como um sal de TFA (180 mg, 0,37 mmol) em MeOH (10 mL) foi adicionado 10% de Pd/C (90 mg). A suspensão foi agitada sob atmosfera de H2 durante 1 hora. O catalisador foi removido e o filtrado foi concentrado em vácuo. O resíduo foi em seguida purificado por cromatografia instantânea em SiO2 eluindo com 3% de MeOH em DGM, para fornecer 4-((2-aminopiridin-4-il)rnetil)-3-fluorofenilcarbamato de terc-butila como um sal de TFA (73 mg, 46% de Produção). MS(ESI+): m/z 318,24 (M +
183
ΗΓ

F) 4-(4-Amino-2-fluorobenzil)piridin-2-amina.
A uma solução de 4-((2-aminopiridin-4-il)metil)-3-fluorofenilcarbamato de terc-butila como um sal de TFA (73 mg, 0,17 mmol) em DCM (4,0 mL) foi adicionado TFA (1,0 mL). A solução foi agitada em temperatura ambiente durante 2 horas e o solvente foi removido em vácuo para fornecer
4-(4-amino-2-Wuorobenzil)piridin-2-amina, -ácido bis-trifluoroacético (70 mg, 93% Produção). MS(ESI+): m/z 218,12 (M + H)L
G) 1-(4-((>Aminopiridin-4-il)metil)-3-fluorofenil)-3-(2-(4fluorofenil)acetH)uréia, sal de ácido trifluoroacético.
Preparado de maneira similar àquela que é descrita na Etapa F de Exemplo 87, 4-(4-AmÍno-2-fluorobenzil)piridin-2-amina como 2 Sal de
TFA (19 mg, 0,042 mmol) foi convertido em 1-(4-((2-aminopiridin-4-il)metil>-
3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de ácido trifluoroacético (19 mg, 88% de Produção). Ή RMN (DMSO-dg) § 10,94 (s, 1HJ, 10,47 (s, 1H),
7,76 (m, 3H), 7,50 (d, 1H, J = 11,5 Hz), 7,10-7,26 (m, 4H), 7,10 (m, 2H), 6,65 (d, 1H, /= 6,5 Hz), 6,55 (s, 1H), 3,89 (s, 2H), 3,65 (s, 2H); MS(ESF):
397,26 (M + H)+.
Exemplo 00

1-(4-(2-Oarbamollpiridln-4-ilâxl)-3-ctorofenil)-3-(2-(4-fluor©fenil)acetil)uréia.
184

Ο
A) 4-(4-Amino-2-clorofenóxi)picolinamida
A uma solução de 4-amino-2“Clorofenol (Aldrich, 430 mg, 3,0 mmoles) em DMF (2,0 mL) em temperatura ambiente foi adicionado KOt-Bu (352 mg, 3,2 mmoles). A mistura foi deixada agitar em temperatura ambiente durante 1 hora. A uma solução foram em seguida adicionados 4-cloropicolinamida (468 mg, 3,0 mmoles) e KgCCXj (221 mg, 1,6 mmol). A suspensão resultante foi aquecida a 90°C durante a noite. Após baixo resfriamento, a suspensão foi diluída com 100 mL de EtOAc e 50 mL of H2O. A camada orgânica foi separada e lavada com salmoura (2 χ 25 mL) e secada sobre MgSO4 após a filtração e concentração, o sólido foi triturado com 50 mL de DCM. O sólido foi em seguida coletado e lavado com DCM (2 χ 20 mL), EtOAc (5,0 mL) e secada para fornecer 4-(4-amino-2-clorofenóxi) picolinamida (320 mg, 40% de Produção). MS(ESI+): mZz 264,12 / 266,07 (M + H)L


O
B) 1-(4-(2-Carbamoilpiridin-4-ilóxi)-3-clorofenil)-3-(2-(4-fluorofeml)acetil)uréia.
Preparado de maneira similar àquela que é descrita na Etapa F de Exemplo 87, 4-(4-Amino-2“Clorofenóxi)picolinamida (79 mg, 0,30 mmol) em DMF (1,0 mL) foi convertido em 1-(4-(2-carbamoilpiridin-4-ilóxi)-3clorofenil>3-(2-(4-fluorofenil)acetil)uréia (65 mg, 49% Produção). 1H RMN (DMSO-d6) δ 11,05 (s, 1H), 10,58 (s, 1H), 8,52 (d, 1H, J = 4,5 Hz), 8,15 (s, 1H), 7,98 (s, 1H), 7,70 (s, 1H), 7,55 (m, 1H), 7,39 (m, 3H), 7,27 (m, 1H), 7,16 (m, 3H), 3,73 (s, 2H); MS(ESI+): m/z 443,17 (M + H)\
185
Exemplo 91


h2n
1-(4-(2-AmÍnopiridin-4-ilóxi)-3-clorofenil)-3-(2-(4-fluorofenil) acetil) uréia, sal de cloridrato.
A uma solução de 1-(4-(2-carbamoilpiridin-4-ilóxÍ)-3-clorofenil)-35 (2-(4-fluorofenil)acetil)uréia (Exemplo 90, 27 mg, 0,06 mmol) em DMF (1,0 mL) foram adicionados H2O (2,2 mg, 0,12 mmol), piridina (0,04 mL) e bis(trifluoroacetóxi)iodobenzeno (Aldrich, 39 mg, 0,09 mmol) em temperatura ambiente. A solução foi deixada agitar durante a noite e em seguida foi purificada em HPLC preparativa para fornecer o produto desejado, que foi no10 vamente convertido em cloridrato de 1-(4-(2-aminopiridin-4-ilóxi)-3-clorofenil)-3-(2-(4-fluorofenil)acetil)uréia (19 mg, 70% Produção) pela adição de solução de 1N de HCI (0,5 mL). Ή RMN (DMSO-d6) δ 13,50 (s, 1H), 11,02 (s, 1H), 10,57 (s, 1H), 7,80-7,95 (m, 4H), 7,55 (m, 1H), 7,37 (m, 1H), 7,30 (m, 2H), 7,11 (m, 2H), 6,60 (m, 1H), 6,00 (s, 1H), 3,70 (s, 2H); MS(ESr): m/z
415,16 (M + Hy.
Exemplo 92


H2N
1-(4-(2-Aminopiridin-4-ilóxi)-2-clorofenil)-3-(2-(4-fluorofenil) acetilforéia.
186


Ο
A) 1-(4-(2-Garbamoilpiridin-4-ilóxi)-2-clorofenil)-3-(2-(4>fluoroffenil)acetil)uréia.
Preparado de maneira similar àquela que é deserte na Etapa A de Exemplo 00, 4-(4-Amino-3-clorofenóxi)pícolinamida (39 mg, 0,19 -mmol) em DMF (1,0 mL) foi convertido em 1-(4-(2-carbamoilpiridin-4-Ílóxi)-2clorofenil)-3-(2-(4-fluorofenil)acetil)uréia (18 mg, 41% Produção) após purificação por HPLC preparativa. MS(ESr): 443,13 / 445,14 (M + H)*.
B) 1-(4-(2-Aminopiridin-4-ilóxi)-2-clorofenil)-3-(2-(4-fluorofenil) acetiljuréia.
Preparado de maneira similar àquela descrita para Exemplo 91, 1-(4-(2-Carbamoilpiridin-4-ilóxi)-2-clorofenil)-3-(2-(4-fluorofenil)acetil)uréia (18 mg, 0,04 mmol) em DMF (1,0 mL) foi convertido em 1-(4-(2-aminopiridin-
4-ilóxi)-2-clorofenil)-3-(2-(4-fluorofenil)acetil)uréia (10 mg, 55% de Produção). Ή RMN (DMSO-O 113,47 (s, IH), 11,26 (s, IH), 11,08 (s, IH), 8,37 (d, 1H, J = 8,5 Hz), 7,95 (d, 1H, J = 7,5 Hz), 7,88 (s, 2H), 7,62 (s, 1H), 7,35 (m, 3H), 7,17 (m, 2H), 6.64 (d, IH, J = 7,5 Hz), 6,13 (s, IH), 3,76 (s, 2H)· MS(ESI+): m/z 415,18 /417,17 (M + H)*.
Exemplo 93


O
1-(4-(2-Carbarnoilpiridin-4-ilóxl)-3-metWfenil)-3-(2-(4-fluorofenil) acetil)uréia.
187 h2n

O
NH2
A) 4-(4-Amino<-2-metilfenóxi)picolinamida.
Preparado de maneira similar àquela que é descrita na Etapa A de Exemplo 90, 4-Amino-2-metilfenol (246 mg, 2,0 mmoles) foi convertido em 4-(4-amino-2-metilfenóxi)picolinamída (230 mg, 47% Produção). MS (ESF): m/z 244,15 (M + H)*.
B) 1-(4-(2-Oarbamoilpiridin-4-ilóxi)-3-metilfenil)-3-(2-(4-fluorofenil)acetil)uréia.
Preparado de maneira similar àquela que é descrita na Etapa F de Exemplo 87.4-(4-Amino-2-metilfenóxi)picolinamida (48 mg, 0,2 mmol) em DMF (1,0 mL) foi convertido em 1-(4-(2-carbamoilpiridin-4-ilóxi)-3-metilfenil)3-(2-(4-fluorofenil)acetil)uréia (35 mg, 41% Produção. 1H RMN (DMSO-c^) i 10,92 (s, 1H), 10,44 (s, 1H), 8,44 (d, 1H, J = 5,5 Hz), 8,06 (s, 1H), 7,63 (s, 1H), 7,50 (s, 1H), 7,42 (m, 1H), 7,31 (m, 2H), 7,24 (d, 1H, /= 2,0 Hz), 7,067,12 (m, 4H), 3,69 (s,2H), 2,02 (s, 3H);). MS(ESF): m/z 423,17 (M + H)*.
Exemplo94
1-(4-(2-Aminopiridin-4-ilóxl)-3-metilfenil)-3-(2-(4-fluorofenil) acetil)uréia, sal de cloridrato.
Preparado de maneira similar àquela que é descrita na Etapa A de Exemplo 91, 1-(4-(2-Carbamoilpiridin-4-ilóxi)-3-metilfenil)-3-(2-(4-fluorofenil) acetil)uréia (27 mg, 0,06 mmol) em DMF (1,0 mL) foi convertido em 1(4-(2-aminopiridin-4-ilóxi)-3-metilfenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de cloridrato (24 mg, 88% Produção) após purificação por HPLC. 1H RMN (DMSO-<)> 13,18 (s, 1H), 10,93 (s, 1H), 10,45 (s, 1H), 7,88 (d, 1H, J = 7,0 Hz),
188
7,73 (s, 2H), 7,50 (m, 2H), 7,29 (m, 2H), 7,10 (m, 3H), 6,56 (d, 1H, J = 7,0
Hz), 5,91 (d, 1H, J = 2,5 Hz), 3,68 (s, 2H), 2,02 (s, 3H); MS(ESF): m/z
395,20 (M + H)+.
Exemplo 95

1-(4-(2-Aminopiridin-4-ilóxi)-2-(trlfluorometil)fenil)-3-(2-(4fluorofen»l)acetil)uréia, sal de cloridrato.
HO

NH2
A) 4-Amino-3-(trifluorometil)fenol
Uma solução de 4-nitro-3-(trifluorometil)fenol (Aldrich, 414 mg,
2,0 mmoles) em 10 mL de MeOH foi adicionado 10% de Pd/C (100 mg). A suspensão foi agitada sob atmosfera H2 durante 12 horas e foi em seguida filtrada e concentrada em vácuo para fornecer 4-amino-3-(trifluorometil)fenol (350 mg, 95% Produção), que foi suficientemente pura para emprego na
Etapa seguinte. MS(ESI*): m/z 178,02 (M + H)*.


B) 4-(4-Amino-3-(trifluorometÍI)fenóxi)picolinamida
Preparado de maneira similar àquela que é descrita na Etapa A de Exemplo 90, 4-Amino-3-(trifluorometil)fenol (177 mg, 1,0 mmol) em DMF (2,0 mL) foi convertido em 4-(4-amino-3-(trifluorometil)fenóxi)picolinamida 1
189 (180 mg, 61% Produção). MS(ESL): m/z 298,20 (M + H)+.

F
H2N 0
C) 1 -(4-(2-Carbamoílpiridin-4-ilóxi)-2-(trifluorometll)feriil)-3(2-(4“fluorofenil)acetil)urtla.
Preparado de maneira similar àquela que é descrita na Etapa F de Exemplo 87, 4-(4-Amino-3-(trifluorometil)fenóxi)picolinamida (30 mg, 0,1 mmol) em DMF (1,0 mL) foi convertido em 1-(4-(2-carbamoilpiridin-4Ílóxi)-2-(trifluorometil)fenil)«3-(2-(4-fluorofenfl)acetíl)uriia (30 mg, 63% Produção). MS(ESI*): mór 477,12 (M + H)*.
D) 1-(4-(2-Aminopíridin-4-ilóxi)-2-(trifluorometil)fenil)-3-(2-(4fluorofenil)acetil)uréia.
Preparado de maneira similar àquela que é descrita na Etapa A de Exemplo 91, 1-(4-(2-Carbamoilpiridin-4-ilóxi)-2-(trifiuorometil)fenil)-3-(2-(4 fluorofenil)acetil)uréia (26 mg, 0,055 mmol) em DMF (1,0 mL) foi convertido em 1 -(4-(2-aminopiridin-4-ilóxi)-2-(trifluorometil)fenil)-3-(2-(4-fluorofenil) acetil)uréia ctoridrato (15 mg, 56% Produção) após purificação por HPLC preparativa. Ή RMN (DMSO-cy § 13,40 (s, 1H), 11,28 (s, 1H), 10,95 (s, 1H), 8,25 (d, 1H, 4 = 8,5 Hz), 7,97 (d, 1H, 4 = 7,0 Hz), 7,88 (s, 2H), 7,72 (d, 1H, 4 = 2,5 Hz), 7,65 (m, 1H), 7,35 (m, 2H), 7,19 (m, 2H), 6,66 (d, 1H, 4 = 2,5 Hz), 3,75 (s, 2H); MS(ESI*): m/z 449,14 (M + H)*.
Exemplo 96

h2n
1-(4-(2-Aminopiridin-4-ilóxi)-2-fluorofenil)-3-(2-(4-fluorofenil)
190 acetil) uréia, sai de ácido trifluoroacético.


O
A) 4-(3-flúor-4-pivalamidofenóxi)picolinamicla.
A uma solução de 4-amino-3-fluorofenol (Oakwood Products
Inc., 252 mg, 2,0 mmoles) em NMP (4,0 mL) foram adicionados 4-cloropi5 colinamida (312 mg, 2,0 mmoles) e DIEA (0,3 mL). A solução foi aquecida a 250°C em um forno de microondas. Após baixo resfriamento, a solução foi diluída com H2O e a solução foi extraída com EtOAc (3 χ 40 mL). As camadas orgânicas combinadas foram lavadas com salmoura, secada sobre
MgSO4< após a filtração e concentração, o resíduo foi purificado por croma10 tografla instantânea em SiO2 eluindo com 0-30% MeOH em DOM para fornecer uma fração contendo 4-(4-amino-3-fluorofenóxi)picolinamida (50% puro, detecção por HPLC-UV). MS(ESP): 248,12 (M + H)*.
A uma solução de 4-(4-amino-3-fluorofenóxi)picolÍnamida, obtido previamente da Etapa em THF (3,0 mL) e DCM (10,0 mL) foram adicionados
1 N de NaOH (5,0 mL) e cloreto de trimetilacetila (0,25 mL, 2 mmoles) em temperatura ambiente. A solução foi agitada durante 2 horas e foi em seguida extraída com EtOAc. A camada orgânica foi lavada com salmoura e secada sobre MgSO4 Após a filtração e concentração, o resíduo foi purificado por cromatografia instantânea em SiO2 eluindo com 0%-100% de EtOAc em hexano para fornecer 4-(3-flúor-4-pivalamidofenóxi)picolinam»da (110 mg, 17% de Produção para duas Etapas). MS(ESF): m/z 332,18 (M + H)*.
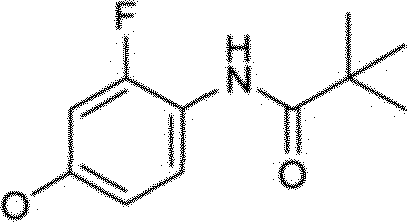

h2n
B) /V-(4-(2-Aminopiridin-4-ilóxi)-2-fluorofeml)pivalamida.
191
Preparado de maneira similar àquela que é descrita na Etapa A de Exemplo 91, 4-(3-flúor-4-pivalamÍdofenóxi)picolinamida (110 mg, 0,33 mmol) em acetonitrilo (4 mL) foi convertido em N-(4-(2-aminopÍrtdin-4-ilóxi)-2fluorofenil)piva!amida (70 mg, 70% de Produção). MS(ESF): m/z 304,21 (M + H)*.


H2N
C) 4-(4-Amino-3-fluorofenóxi)piridin-2-amina.
Uma solução de N-(4-(2-aminopiridin-4-ilóxi)-2-fluorofenil) pivalamida (70 mg, 0,23 mmol) em 3 mL de MeOH foi adicionado 2 mL de 6 N de HOI. A mistura foi em seguida aquecida até o refluxo durante 48 horas.
Após baixo resfriamento, o solvente foi removido sob pressão reduzida e o resíduo foi purificado por extração de sólido (cartucho de extração Waters Oasis®MCX ) para fornecer 4-(4-amino-3-fluorofenóxi)piridin-2-amina (27 mg, 54% Produção). MS(ESr): m/z 220,21 (M + H)+.
D) 1-(4«(2‘Aminopiridin-4-ilóxi)-2-fluorofenil)-3-(2-(4-fluoro- feniljacetiljuréia, sal de ácido trifluoroacético.
Preparado de maneira similar àquela que é descrita na Etapa F de Exemplo 87, 4-(4-Amino-3-fluorofenóxi)piridin-2-amina (28 mg, 0,095 mmol) em THF (2,0 mL) foi convertido em ácido trifluoroacético de 1-(4-(2amtnopiridin-4-ilóxi)-2-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia (23 mg, 47% 20 Produção) após a purificação por HPLC preparativa. 1H RMN (DMSO-cy δ 11,20 (s, 1H), 10,77 (s, 1H), 8,23 (m, 1H), 7,94 (d, 1H, J = 6,5 Hz), 7,70 (s, 2H), 7,45 (m, 1H), 7,35 (m, 2H), 7,16 (m, 3H), 6,64 (d, 1H, d = 2,5 Hz), 6,11 (s, 1H), 3,75 (s, 2H); MS(ESI*): 399,12 (M + H)+.
Exemplo 97
192

1-(4-(2-Aminopiridín-4-ilóxl)-2,3-difluorofenil>3-(2-(4-fluor<> feml)acetil)uréia, sal de ácido trifluoroacétíco.

A) 4-Amino-2,3-difluorofenol.
A uma solução de 1,2,3-trifluoro-4-nitrobenzeno (Aldrich, 15,0 g,
84,7 mmoles) em DMF (25,0 mL) foram adicionados K2CO3 (17,6 g, 127,8 mmoles) e álcool benzílico (8,8 mL, 85,0 mmoles). A suspensão foi agitada durante a noite. A uma mistura de reação foi em seguida adicionado H2O (100 mL) e a solução foi mantida a 4°C durante a noite. O precipitado foi em seguida coletado e lavado com H2O para fornecer a mistura de dois isômeros (22,4 g) [1-(benzilóxi)-2,3-difluoro-4-nitrobenzeno e 2-(benzilóxi)-3,4difluoro-1-nitrobenzene em uma relação de 1:1].
A uma solução de [1-(benzilóxi)-2,3-dífluoro-4-nitrobenzeno e 2(benzilóxi)-3,4-difluoro-1-nitrobenzene] (22,4 g, 84,5 mmoles) em EtOAc (20,0 mL) e MeOH (100,0 mL) foi adicionado 10% Pd/C (1,0 g). A suspensão foi agitada sob atmosfera de H2 durante 12 horas. A suspensão foi em seguida filtrada e concentrada em vácuo para fornecer a mistura de dois isômeros (12,6 g) [4-amino-2,3-difluorofenol e 6-amino-2,3-difluorofenol em uma relação de 1:1). MS(ESL): m/z 146,00 (M + H)+.

h2n

B) N-(4-(2-Amlnopiridir»-4-ilóxi)-2,3-difluorofenil)pivalamicla.
193
Preparado de maneira similar àquela que é descrita na Etapa A de Exemplo 90. A mistura de 4-amino-2,3-difluorofenol e 6-arnino-2,3-difluorofenol (580 mg, 4,0 mmoles) em DMF (3,0 mL) foi convertida a uma mistura de 4-(4-amino-2,3-difluorofenóxi)picolinamida e 4-(6-amino-2,3-difluorofenóxi) picolinamida (300 mg). MS(ESF): m/z 266,13 (M + H)+.
Preparado de maneira similar àquela que é descrita na Etapa A de Exemplo 96. A mistura de 4-(4-amino-2,3-difluorofenóxi)picolinamtda e 4(6-amino-2,3-difluorofenóxi)picolinamida (300 mg, 1,13 mmol) foi convertida em uma mistura de 4-(2,3-difluoro-4-pivalamidofenóxi)picolinamida e 4-(2,3difluoro-6-pivalamidofenóxi)picolinamida (406 mg). MS(ESF): m/z 350,20 (M + H)*.
Preparado de maneira similar àquela que é descrita na Etapa A de Exemplo 91, a mistura de 4-(2,3-difluoro-4-pivalamidofenóxi)picolinamida e 4-(2,3-difluoro-6-pivalamidofenóxi)picolinamida (400 mg) foi reagida com bis(trifluoroacetóxi)iodobenzeno para fornecer A/-(4-(2-aminopiridin-4-ilóxí)-
2,3-difluorofenil)pivalamida, sal de ácido trifluoroacético (120 mg, 24% Produção) após a purificação por HPLC preparativa. MS(ESI*): m/z 322,23 (M +
H)+.


H2N
C) 4-(4-Amino-2,3-difluorofenóxi)piridin-2-amina.
Preparado de maneira similar àquela que é descrita na Etapa C de Exemplo 96, /V-(4-(2-Aminopiridin-4-ilóxi)-2,3-difluorofenil)pivalamida, sal de ácido trifluoroacético (120 mg, 0,27 mmol) foi convertido em 4-(4-amino-
2,3-difluorofenóxi)píridin-2-amina (52 mg, 81% de Produção). MS(ESF): m/z 238,11 (M + H)*.
D) 1 -(4-(2-Amlnoplridin-4-ilóxl)-2,3-difluorofenil)-3-(2-(4fluorofenil)acetil)uréia, sal de ácido-trifluoroacético.
Preparado de maneira similar àquela que é descrita na Etapa F
194 de Exemplo 87, 4-(4-Amino-2,3-difluoroferióxí)piridin-2-amina (24 mg, 0,10 mmol) em THF (3,0 mL) foi convertido em ácido trifluoroacético 1-(4-(2aminopiridin-4-ilóxi)-2,3-difluorofenil)-3-(2-(4-fluorofenil)aceti!)uréia, (21 mg, 40% de Produção). 1H RMN (DMSO-d6) §11,27 (s, 1H), 10,84 (s, 1H), 8,02 (m, 1H), 7,96 (d, 1H, J = 8,5 Hz), 7,73 (s, 2H), 7,34 (m, 3H), 7,17 (m, 2H),
6,70 (m, 1H), 6,20 (d,1H, J = 2,0 Hz), 3,75 (s, 2H); MS(ESr): m/z 417,10 (M + H)*.
Exemplo 98
H2N' w
W-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenil)-1-benzil-5-metil10 1H-pirazol-3-carboxamlda, sal de cloridrato.
A) Ácido 1-Benzil-5-metil-1 H-pirazol-3-carboxílico.
A uma solução de dicloridrato de 1-benzilidrazina (Aldrich,
0,98 g, 5,0 mmoles) em EtOH (30 mL) foram adicionados DlEA (2,0 mL) e
2,4-dioxopentanoato de etila (0,70 mL, 5,0 mmoles). A mistura foi agitada em temperatura ambiente durante 12 horas e foi concentrada em vácuo. O resíduo foi dissolvido em 1 N de NaOH (10 mL). A solução fei aquecida a 60°C durante 1 hora. Após baixo resfriamento, a solução foi extraída com
DCM (3 x 50 mL). A camada aquosa foi neutralizada o pH 2,0 e em seguida foi extraída com EtOAc. A camada orgânica foi lavada com salmoura e se20 cada sobre MgSO4 foi filtrada e concentrada para fornecer ácido 1-benzii-5metil-1H-pirazol-3-carboxí!ico (1,0 g, 92% de Produção). MS(ESP): m/z 217,12 (M + H)*.
B) /V-(4-(2--Amiriopirl<iln-4-ilóxi)-3-fluorofenll)-1-benzil-5-me
195 til-1 W-plrazol-3-carboxamida, sal de cloridrato.
4-(4-Amino-2-fluorofenóxi)piridin-2-amina (Composto B de Exemplo
24, 25 mg, 0,11 mmol) foi acoplado com ácido 1-benzil-5-metil-1H-pirazole3-carboxilico (25 mg, 0,11 mmol) de uma maneira similar àquela que é des5 crita na Etapa C de Exemplo 1 para fornecer cloridrato de AH4-(2aminopiridin-4-ilóxi)-3-fluorofenil)-1-benzil-5-metil-1H-pirazol-3-carboxamida (10 mg, 20% Produção) após apurificação por HPLC preparativa. 1H RMN (DMSO-dg) 6 10,64 (s, 1H), 7,8-7,98 (m, 3H), 7,00-7,65 (m, 9H), 6,71 (m, 1H), 6,15 (s,1H), 5,65 (s, 2H), 2,24 (s, 3H); MS(ESF): mZz 418,21 (M + H)*.
Exemplo 99
H2N


2-(4-Fluorobenzilsulfínil)-Af-(4-(2-aminopiridin-4-ilóxi)-3- fluorofeniljacetamida, sal de cloridrato.

A) 2-(4-fluorobenziltio)acetato de etila.
A uma solução de 2-mercaptoacetato de etila (Aldrich, 1,0 mL,
9,1 mmoles) em acetonitrila (10,0 mL) foram adicionados K2CO3 (2,76 g, 20,0 mmoles) e 1-(bromometil)-4-fluorobenzeno (2,27 g, 12,0 mmoles). A mistura foi agitada em temperatura ambiente durante 12 horas. Após a filtração e concentração, o resíduo foi purificado por cromatografia de coluna instantânea em SiCt, para fornecer 2-(4-fluorobenzlio)acetato de etila (1,89 g,
91 % de Produção). MS(ESF): m/z 251,08 (M + H)*.

B) 2-(4-fluorobenzilsulfinil)acetato de etila.
A uma solução de 2-(4-fluorobenziltfo)acetato de etila (1,89 g,
8,29 mmoles) em DOM (20,0 mL) a -40°C foi adicionado Uma solução de m196

CPBA (77%, 1,86 g, 8,29 mmoles) em DCM (20,0 mL) em gotas. A solução foi agitada de -40°C para temperatura ambiente durante a noite. A solução foi então saciada com triamina de dietileno ligada a polímero. Após a filtração e concentração, o resíduo foi purificado por cromatografia instantânea em SiO2 para fornecer 2-(4-fluorobenzilsulfínil)acetato de etila (2,0 g, 98% Produção). MS(EST): m/z 267,09 (M + H|.

C) Ácido 2-(4-Fluorobenzilsulfinil)acético.
A uma solução de 2-(4-fluorobenzilsuifiníl)acetato de etila (1,60 g,
6,55 mmoles) em THF (10,0 mL) e MeOH (20,0 mL) foi adicionado 1 N NaOH (20,0 mmoles). A mistura foi agitada em temperatura ambiente durante 2 horas. Após a remoção do solvente orgânico sob pressão reduzida, a solução aquosa restante foi neutralizada com 1 N de HCI (25,0 mL). Ela foi extraída com EtOAc (3 χ 100 mL) e a camada orgânica combinada foi secada sobre MgSO4 a solução foi em seguida filtrada e concentrada em vácuo para fornecer ácido 2-(4-fluorobenzilsulfinil)acétíco (1,25 g, 88% de Produção). MS(ESF): m/z217,05 (M + H)+,
D) 2-(4-Fluorobenzilsulfiriil)-M‘(4-(2-amiriopiridin-4-ilóxi)-3- . fluorofenil)acetamidar sal de cloridrato.
4-(4-Amino-2-fluorofenóxi)piridin-2-amina dicloridrato (Composto
B de Exemplo 24, 29 mg, 0,10 mmol) foi acoplado com ácido 2-(4-fluorobenzi!sulfinil)acético (22 mg, 0,1 mmol) de uma maneira similar àquela que é descrita na Etapa C de Exemplo 1 para fornecer 2-(4-fluorobenzilsulfinil)/V-(4-(2-aminopiridin-4-ilóxi)-3-fluorofenil)acetamida, sal de cloridrato (17 mg, 37% de Produção). Ή RMN (DMSO-dJ δ 10,94 (s, 1H), 7,97 (d, 1H, J = 7,5 Hz), 7,85 (m, 3H), 7,39 - 7,45 (m, 4H), 7,23 (t, 2H, J- 7,5 Hz), 6,70 (m, 1H),
6,13 (d, 1H, d = 2,5 Hz), 4,32 (d, 1H, J = 11,0 Hz), 4,11 (d, 1H, J = 11,0 Hz),
3,98 (d, 1H, J = 13,0 Hz), 3,65 (d, 1H, J = 13,0 Hz); MS(ESP): m/z 418,26 (M + H)*.
Exemplo 100
107
H2N


2-(4-Fluorobenzllsulfonil)-àF(4-(2-amlnopiridin-4-ilóxl)-3fluorofenii)acetamida, sal de cloridrato.

A) 2-(4-fluorobenzilsulfonil)acetato de etila.
A uma solução de 2-(4-fluorobenzilsulfinil)acetato de etila (370 mg, 1,52 mmol) em DCM (5,0 mL) foi adicionado m-CPBA (77%, 450 mg, 2,0 mmol). A mistura foi agitada em temperatura ambiente durante 2 horas e foi então saciada com triamida de dietileno ligada a polímero (1,5 g). A mistura de reação foi filtrada e concentrada em vácuo para fornecer 2-(4-fluorobenzílsulfonil)acetato de etila (360 mg, 91% de Produção). MS(ESL): m/z 283,10 (M + H)*.

B) Ácido 2-(4-Fluorobenzilsulfonll)acético.
Preparado de maneira similar àquela que é descrita na Etapa C de Exemplo 99,2-(4-fluorobenziteulfonil)acetato etila (340 mg, 1,31 mmol) foi convertido em ácido 2-(4-fluorobenzilsulfonil)acético (270 mg, 81% Produção). MS(ESL): m/z 255,05 (M + Hf.
C) 2-(4-Fluorobenzilsulfonil)-N-(4-(2-aminopiridin-4-ilóxi)-3fluorofenil)acetamida, sal de cloridrato.
4-(4-Amino-2-fluorofenóxí)piridin-2-amina dicloridrato (50 mg,
0,17 mmol) foi acoplado com ácido 2-(4-fluorobenzilsulfonil)acético (33 mg, 0,14 mmol) de uma maneira similar àquela que é descrita na Etapa C de Exemplo 1 para fornecer 2-(4-fluorobenzilsulfonil)-/V-(4-(2-aminopiridin-4ilóxi)-3-fluorofenil)acetamida, sal de cloridrato (30 mg, 45% Produção). Ή RMN (DMSO-O | 13,40 (s, 1H), 11,15 (s, 1H), 7,97 (d, 1H, J - 7,0 Hz),
7.80-7,90 (m, 3H), 7,47 (m, 4H), 7,26 (t, 2H, J = 8,5 Hz), 6,72 (d, 1H, J = 7.0
Hz), 6,14 (d, 1H, J = 2,0 Hz), 4,69 (s, 2H), 4,27 (s, 2H); MS(ESf): m/z
434,15 (M + H)*
Exemplo 101


A/-(4-(2-Aminopiridin-4-iióxi)-3-fluorofenil)-1-(4-fluorofenil)-2oxo-1,2-diidropiridina-3-carboxamida, sal de cloridrato.
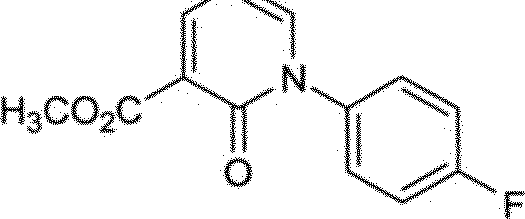
A) 1-(4-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxilato de metila.
A uma solução de 2-oxo-2H-piran-3-carboxilato de metila (Aldrich, 2,31 g, 15 mmoles) em THF (40 mL) e DMF (10 mL) em temperatura ambiente foi adicionado 4-fluoroanilina (1,67 g, 15 mmoles), e a mistura de reação foi agitada durante 2,5 horas. A precipitação sólida foi observada. Ao intermediário de aduzido de 4-fluoroanilina por meio de adição de Michael obtida in situ foi adicionado EDCLHCI (3,85 g, 20 mmoles) e DMAP (120 mg) em temperatura ambiente. A mistura de reação foi agitada em temperatura ambiente durante a noite. A uma mistura de reação foram adicionados 1N HCI aquosa (50 mL) e EtOAc (150 mL), a camada de EtOAc foi separada, e a camada aquosa foi lavada com EtOAc (150 mL), a camada de EtOAc combinada foi secada sobre MgSO4 e concentrada em vácuo para obter o material semi-sólido (-4,4 g). A este produto bruto foram adicionados éter (100 mL) e metanol (15 mL), agitado, e o sólido foi filtrada para obter o pro duto sólido indesejado (870 mg). A solução filtrada foi concentrada para obter um produto bruto semi-sólido desejado (2,95 g, bruto 80%) que foi puro o
190 (DMSO-cg δ 8,23 (dd, 1H, J = 7,2, 2,2 Hz), 7,57 (dd, 1H, J = 6,6, 1,7 Hz ),
7,32-7,34 (m, 2H), 7,17 (t, 2H, J = 8,8 Hz), 6,32 (t, 1H, J = 7,1 Hz), 3,89 (s, 3H); MS(ESr) m/z 248,2 (M + H)2
B) Ácido 1-(4-Fluorofenil)-2-oxo-1,2-diidropiridlna-3-carbo- xilico.
Uma mistura de 1-(4-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxilato de metila (cru 2,45 g, 12 mmoles) e 6 N de NaOH aquosa (2,5 mL) em metanol (60 mL) foi agitada em temperatura ambiente durante 4 horas. A uma mistura de reação foi adicionado HCI concentrado (1 mL) lentamente com agitação em temperatura ambiente, e o sólido precipitado foi filtrado, lavado com pequena quantidade água e secado para obter o produto ácido desejado (2,1 g) como um sólido amarelo. A solução filtrada foi concentrada em vácuo. O resíduo foi misturado com água (50 mL) e lavado com EtOAc (2 χ 130 mL). As camadas de EtOAc foram secadas sobre MgSO4 e concentradas em vácuo. O resíduo foi triturado com uma pequena quantidade de éter para obter a 2* coleta do produto (195mg, total 2,30 g, 82%). 1H RMN (DMSO-cg s 8,47 (dd, 1H, J = 7,2, 2,2 Hz), 8,19 (dd, 1H, J = 6,6, 1,7 Hz), 7,62-7,60 (m, 2H), 7,42 (t, 2H, J = 8,8 Hz), 6,78 (t, 1H, J = 7,1 Hz); MS(ESF) m/z 234,2 (Μ + Hf.
C) ^-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenil)-1-(4-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxamida, sal de cloridrato.
4-(4-Amino-2-fluorofenóxi)piridín-2-amina (Composto B de Exemplo 24, 58 mg, 0,20 mmol) foi acoplado com ácido 1-(4-fluorofenil)-2-oxo-1,2diidropiridina-3-carboxílteo (47 mg, 0,20 mmol) de uma maneira similar àquela que é descrita na Etapa C de Exemplo 1 para fornecer /V-(4-(2aminopiridin-4-ilóxi)-3-fluorofenil)-1-(4-fluorofenil)-2-oxo-1,2-diidropiridina-3carboxamida, sal de cloridrato (22 mg, 23% Produção). Ή RMN (DMSO-cy 5 13,40 (s, 1H), 12,13 (s, 1H), 8,58 (d, 1H, d = 5,0 Hz), 8,13 (d, 1H, J = 5,0 Hz), 8,07 (d, 1H, J = 10,0 Hz), 7,98 (d, 1H, J = 7,5 Hz), 7,89 (s, 2H), 7,40
200
7,60 (m, 6H), 6,72 (m, 2H), 6,17 (d, 1H, J = 2,5 Hz); MS(ESI+) m/z 435,18 (M + H)*.
Exemplo 102

H2hr N (SyAf-fS-Amlno-Z-oxo-l-feniletill-AF-íé-íZ-aminopiridin-^· ilóxi)-3-fluorofeníl)malonamida, sal de cloridrato.
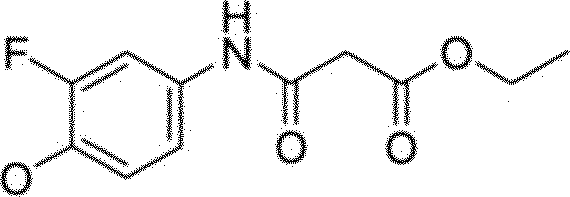
A) 3-(4-(2-carbamoilpiridin-4-ilóxi)-3-fluorofenilamino)-3oxopropanoato de etila.
A uma solução de 4-(4-amino-2-fluorofenóxi)picolinamida (Composto B’ de Exemplo 24, 1,0 g, 4,0 mmoles) em DMF (10,0 mL) foram adicionados DIEA (2,0 mL) e 3-cioro-3-oxopropanoato de etila (Aldrich, 0,75 mL, 6,0 mmoles). A mistura foi agitada em temperatura ambiente durante 12 ' horas e mais 3-cloro-3-oxopropanoato de etila (0,20 mL, 1,6 mmol) foi adicionado. A mistura foi agitada durante 2 horas e foi em seguida diluída com EtOAc (200 mL). Ela foi lavada com H2O e salmoura e em seguida secada sobre MgSO4 Após a filtração e concentração, O resíduo foi triturado com DCM e filtrado para fornecer 3-(4-(2-carbamoilpiridin-4-ilóxi)-3-fluorofenilamino)-3-oxopropanoato de etila (900 mg, 62% Produção). MS(ESI+) m/z 362,28 (M + Hf.
201


h2n
B) 3-(4-(2-aminopiridin-4-ilóxi)-3-fluorofeni!ammo)-3-oxopropanoato de etila.
Preparado de maneira similar àquela que é descrita na Etapa A de Exemplo 91, 3-(4-(2-carbamoilpiridin-4-ilóxi)-3-fluorofenilamino)-3-oxopropanoato de etila (900 mg, 2,5 mmoles) em DMF (10,0 mL) foi convertido em 3-(4-(2-aminopiridin-4-ilóxi)-3-fluorofenilamino)-3-oxopropanoato de etila (710 mg, 86% de Produção). MS(ESI*) m/z 334,26 (M + H)*.


H2n
C) Ácido 3-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenilamino)-3oxopropanóico.
Preparado de maneira similar àquela que é descrita na Etapa C de Exemplo 99, 3-(4-(2-aminopirÍdin-4-ilóxi)“3-fluorofenilamino>3-oxopropanoato de etila (700 mg, 2,10 mmoles) foi convertido em ácido 3-(4-(2aminopÍridin-4-ilóxi)-3-fluorofenilamino)-3-oxopropanóÍco (630 mg, 98% Produção). MS(ESF) m/z 306,20 (M + H)*.
D) (S)-WH2-Amino-2-oxo-1 -feniletil)-/^-(4-(2-aminopiridín-4ilóxi)-3-fluorofenil)malonamida, sal de cloridrato.
Ácido 3-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenilamino)-3-oxopropanóico (30 mg, 0,10 mmol) foi acoplado com cloridrato de (S>2-amino-2fenílacetamida (Acros, 28 mg, 0,15 mmol) de uma maneira similar àquela que é descrita na Etapa C de Exemplo 1 para fornecer (S>/^-(2-amino-2oxo-1 -feniletil)-N3-(4-(2-aminopiridin-4-ilóxi)-3-fluorofenil)malonamida, sal de cloridrato (25 mg , 53% Produção). 1H RMN (DMSO-c^) 8 10,68 (s, 1H), 8,80 (d, 1H, J = 8,0 Hz), 7,96 (d, 1H, J = 7,5 Hz), 7,77 - 7,90 (m, 4H), 7,20 - 7,45
202 (m, 8H), 6,70 (m, 1H), 6,12 (s, 1H), 5,39 (d, 1H, J = 7,5 Hz), 3,48 (d, 1H, J = 15,0 Hz), 3,41 (d, 1H, J= 15,0 Hz); MS(ESI+) m/z 438,26 (M + H)*.
Exemplo 103
IT (R).A^-(2-Amino-2-oxo-14eniletil)-AF-(4-(2-aminopiridin-4ilóxi)-3-fluorofenil)malonamida, sal de cloridrato.
Ácido 3-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenilamino)-3-oxopropanóico (Composto C de Exemplo 102, 30 mg, 0,10 mmol) foi acoplado com cloridrato de (R)-2-amino-2-fenilacetamida (Bachem, 28 mg, 0,15 mmol) de uma maneira similar àquela que é descrita na Etapa G de Exemplo 1 para fornecer (R)-A/1-(2-amino-2-oxo-1-feniletil)-AP-(4-(2-aminopiridin-4-ílóxi)-3fluorofenil)malonamida, sal de cloridrato (14 mg, 30% Produção). 1H RMN (DMSO-de) § 10,65 (s, 1H), 8,76 (d, 1H, 4= 8,0 Hz), 7,92 (d, 1H, 4=7,0 Hz), 7,75 - 7,88 (m, 4H), 7,20 - 7,43 (m, 8H), 6,66 (m, 1H), 6,09 (s, 1H), 5,35 (d, 1H, J = 8,0 Hz), 3,45 (d, 1H, J = 15,0 Hz), 3,37 (d, 1H, J 15,0 Hz); MS(ESF) m/z 438,23 (M + H)+.
Exemplo 104
H2N^r
2-(3-(4-(2-aminopirldin-4-il6xi)-3-fluorofenilamino)-3-oxopropanamido)-2-fenilacetato de (S)-Metila, sal de cloridrato.
Ácido 3-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenilamino)-3-oxopropanóico (Composto C de Exemplo 102, 30 mg, 0,10 mmol) foi acoplado com cloridrato de (S)-metil 2-amino-2-fenilacetato (Aldrich, 30 mg, 0,10 mmol) de uma maneira similar àquela que é descrita na Etapa C de Exemplo 1 para
203 fornecer 2-(3-(4-(2-aminopiridin-4-ilóxi)-3-fluorofenilamino)-3-oxopropanamido)-2-fenilacetato de (S>metila, sal de cloridrato (21 mg, 43% Produção). Ή RMN (DMSO-cy 8 10,58 (s, IH), 9,04 (d, IH, J = 7,0 Hz), 7,97 (d, 1H, J = 7,0 Hz), 7,75-7,88 (m, 3H), 7,42 (m, 7H), 6,72 (d, IH, J = 7,0 Hz), 6,12 (s, 5 IH), 5,45 (d, IH, J = 7,0 Hz), 3,63 (s, 3H), 3,43-3,38 (m, 2H); MS(ESF) m/z
453,26 (Μ + ΗΓExemplo 105
H2Im hT
2-(3-(4-(2-aminopiridin-4-ilóxi)-3-fluorofenilamino)-3-oxo- propanamido)-2-fenilacetato de (R)-Metila, sal de cloridrato
Ácido 3-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenilamino)-3-oxopropanóico (Composto G de Exemplo 102, 30 mg, 0,10 mmol) foi acoplado com
2-amino-2-fenilacetato, sal de cloridrato de (R)-metila (Aldrich, 30 mg, 0,10 mmol) de uma maneira similar àquela que é descrita na Etapa C de Exemplo para fornecer 2-(3-(4-(2-aminopiridín-4-ilóxÍ)-3-fluorofenilamino)-3-oxopro15 panamido)-2-fenilacetato de (R)-metila, sal de cloridrato (25 mg, 51% Produção). M RMN (DMSO-cy δ 10,58 (s, IH), 9,03 (d, IH, J - 7,0 Hz), 7,96 (d,
1H, J ~ 7,0 Hz), 7,77 - 7,88 (m, 3H), 7,42 (m, 7H), 6,71 (d, 1H, J = 7,5 Hz), 6,12 (s, IH), 5,44 (d, IH, J = 7,0 Hz), 3,63 (s, 3H), 3,44 - 3,38 (m, 2H); MS (ESF) m/z 453,29 (M + H)*.
Exemplo 106 ^’-(4-(2-Aminopíridin-4-ilóxi)-3-fluorofenil)-^-cictepentilmalonamida, sal de cloridrato.
204
Ácido 3-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenilamino)-3-oxopropanóico (Composto C de Exemplo 102, 30 mg, 0,10 mmol) foi acoplado com ciclopentanamina (Aldrich, 17 mg, 0,2 mmol) de uma maneira similar àquela que é descrita na Etapa C de Exemplo 1 para fornecer A/1-(4-(2-aminopiridin4-ilóxi)-3-fluorofenil)-A/3-ciclopentilmalonamida, sal de cloridrato (18 mg, 44% Produção). 1H RMN (DMSO-de) .8 13,34 (s, 1H), 10,66 (s, 1H), 8,15 (d, 1H, J = 7,0 Hz), 7,96 (d, 1H, J = 7,0 Hz), 7,77-7,88 (m, 3H), 7,42 (m, 2H), 6,70 (d, 1H, J = 7,5 Hz), 6,12 (s, 1H), 3,98 (m, 1H), 3,69 (s, 2H), 1,78 (m, 2H), 1,63 (m, 2H), 1,49 (m, 2H), 1,37 (m, 2H); MS(ESf) m/z 373,30 (M + H)\ Exemplo 107

h2n
W’-(4-(2-Aminopiridiri-4ilóxi)-3-fluorofenll)-/V3-cicloexilmaio· namida, sal de cloridrato.
Ácido 3-(4-(2-Aminopiridtn-4-ilóxi)-3-fluorofenilamino)-3-oxopropanóico (Composto C de Exemplo 102, 30 mg, 0,10 mmol) foi acoplado com cicloexanamina (Aldrich, 20 mg, 0,2 mmol) de uma maneira similar àquela que é descrita na Etapa C de Exemplo 1 para fornecer /\f-(4-(2-aminopiridin4-ilóxi)-3-fluorofenil)-A/J-cicloexilmalonamida, sal de cloridrato (22 mg, 52% de Produção). Ή RMN (DMSO-ds) δ 14,00 (s, 1H), 10,72 (s, 1H), 8,10 (d, 1H, J = 7,0 Hz), 7,97 (d, 1H, J = 7,0 Hz), 7,88 (m, 3H), 7,44 (m, 2H), 6,70 (m, 1H), 6,14 (d, 1H, J = 2,0 Hz), 3,54 (m, 1H), 3,27 (s, 2H), 1,66-1,75 (m, 4H), 1,52 (m, 1H), 1,15 -1,25 (m, 5H); MS(ESI*) m/z 387,32 (M + H)*.
Exemplo 108


h2n
M-(4-(2-Aminoplridin-4-ilóxi)-3-fluorofenil)-^!-neopentilma
205 lonamida, sal de cloridrato.
Ácido 3-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenilamino)-3-oxopropanóico (Composto C de Exempto 102, 30 mg, 0,10 mmol) foi acoplado com
2,2-dimetilpropan-1-amina (Aldrich, 12 mg, 0,2 mmol) de uma maneira simi- lar àquela que é descrita na Etapa C de Exempto 1 para fornecer W*-(4-(2aminopiridin^-ilóxij-S-fluorofeniO-A^-neopentilmalonamida, sal de cloridrato (13 mg, 32% Produção). 1H RMN (DMSO-d6) δ 13,34 (s, 1H), 10,69 (s, 1H), 8,08 (m, 1H), 7,96 (d, 1H, J = 7,0 Hz), 7,87 (m, 3H), 7,44 (m, 2H), 6,70 (m,
1H), 6,13 (d, 1H, J = 2,0 Hz), 3,34 (s, 2H), 2,91 (d, 2H, J = 6,5 Hz), 0,84 (s, 10 9H); MS(ESF) m/z 375,32 (M + H)*.
Exemplo 109
OH
H2N N
Ácido (S)-2-(3-(4-(2-Aminopiridín“4-ilóxi)-3-fluorofenilamirio)-
3-oxopropanarmdo)-2-fer»ílacético, sal de cloridrato
Segundo um procedimento similar aquele que é descrito na E15 tapa C de Exemplo 99, cloridrato de 2-(3-(4-(2-aminopiridin-4-ilóxi)-3fluorofenilamino)-3-Gxopropanamido)-2-fenilacetato de (S)-metila (Composto
D de Exempto 102, 14 mg, 0,028 mmol) foi hidrolisado para fornecer ácido (S)-2-(3-(4-(2-aminopiridin-4-ilóxi)-3-fluorofenilamino)-3-oxopropanamido)-2fenilacético, sal de cloridrato (13 mg, 97% de Produção). 1H RMN (DMSO-ds)
113,20 (s, 1H), 10,57 (s, 1H), 8,92 (d, 1H, J = 7,0 Hz), 7,95 (d, 1H, J = 7,0
Hz), 7,87 (d, 1H, J = 11,0 Hz), 7,70 (s, 2H), 7,41 (m, 8H), 6,69 (d, 1H, J = 7,5
Hz), 6,12 (d, 1H, 2,0 Hz), 5,35 (d, 1H, J - 7,5 Hz), 3,42 (s, 2H); MS(ESP) m/z 439,27 (M + H)\
Exemplo 110
206

N-(4-(2“(3-(Dimetilamino)propilamino)piridin-4-ilóxi)-3-fluorofenil)-2,6-difluorobenzamida, sal de cloridrato.



A) M(4-(2-Cloropiridin-4-ilóxi)-3-fluorofenil)-2,6-difluorobenzamida.
Uma solução de 4-(2-cloropiridin-4-ilóxi)-3-fluorobenzenamina (Composto B de Exemplo 20, 64 mg, 0,27 mmol), THF (1 ml), Et3N (100 pL) foi tratada em gotas com cloreto de 2,6-difluorobenzoíla (Aldrich, 33 μί, 0,27 mmol) e a mistura foi agitada em temperatura ambiente durante 30 minutos. A mistura foi dividida entre EtOAc e NaHCO3 aquosa saturada e a fase de EtOAc foi separada, secada (MgSOJ e concentrada em vácuo para fornecer o composto do título (102 mg, 100%) como um sólido branco. MS(ESI+): m/z 418,18
B) N-(4-(2-(3-(Dimetilamino)propilamino)piridin-4-ilóxi)-3fluorofenil)-2,6-difluorobenzamida, sal de cloridrato.
Uma mistura de N-(4-(2-cloropiridin-4-ilóxi)-3-fluorofenil)-2,6difluorobenzamida (70 mg, 0,19 mmol), 3-(dimetilamino)propilamÍna (44 mL, 0,35 mmol), Cs2CO3 (85 mg, 0,26 mmol) e CuCI (17 mg, 0,17 mmol) em um frasconete de tampa de rosca foi purgado com N2, NMP e 2,2,6,6,-tetrametil-
3,5-heptanodiona (31 mg, 0,17 mmol) foram adicionados a uma mistura que foi em seguida aquecida a 120°C durante 4 horas. A mistura foi resfriada,
207 dividida entre EtOAc e solução de NaHCO3 aquosa saturada e a fase EtOAc foi separada, secada (MgSO4) e concentrada em vácuo para fornecer o produto bruto. A purificação do resíduo por HPLC preparativa (Coluna C) e conversão no sal de cloridrato foram realizadas de maneira similar à Etapa D de 5 Exemplo 33 para fornecer o composto do título (7 mg, 7%) como um sólido esbranquiçado. MS(ESr): m/z 517,37 (M + H)+.
Exemplo 111

B-(4-(2-Amtoo-3-(4-(2-amin©-2-oxoetW)fenil)piridin-4-ilóxi)-3fluorofenil)-1-(4-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxamida, sal 10 de cloridrato.
Uma solução d© 2-(4-(2-amino-4-(4-amino-2-fluorofenóxi)pÍridin-
3-il)fenil)acetamida (Composto C de Exemplo 78, 18 mg, 0,05 mmol) em DMF (1,5 mL) foi tratada com ácido 1-(4-fluorofeníl)-2-oxo-1,2-diidropiridina-
3-carboxílico (Composto B de Exemplo 101, 11 mg, 0,05 mmol), DIPEA (10 pL, 0,06 mmol) e TBTU (19 mg, 0,06 mmol). A mistura foi agitada em temperatura ambiente durante 40 horas. A mistura foi concentrada sob vácuo e o resíduo foi purificado por HPLC preparativa (Coluna A) para fornecer o composto do título como um sal de TFA. O sal de TFA foi dissolvido em MeOH anidro e tratado com 1 M HCl / Β8Θ a 0°C e agitado durante 5 minutos. A mistura foi em seguida concentrada em vácuo para fornecer o composto do título (15 mg, 47%) como um sólido amarelo. 1H RMN (DMSO-d6) δ 12,11 (s,
1H), 8,56 (dd, 1H, J = 2,2, 7,1 Hz), 8,13 (dd, 1H, J = 2,2, 6,7 Hz), 8,00 (dd, 1H, J = 2,2, 12,6 Hz), 7,96 « 1H, J = 7,7 Hz), 7,60 - 7,57 (m, 2H), 7,48 7,33 (m, 10H), 6,94 (s, 1H), 6,72 (dd, 1H, J = 7,2, 7,2 Hz), 6,38 (d, 1H, J =
7,2 Hz), 3,95 (s, 2H); MS(ESI*): m/z 568,23 (M + H)*.
Exemplo 112
208

1-(4-(2-Aminopiriclin-4-ilóxi)-2,5-clifluorofenil)-3-(2-(4-fluorofeml)-acetil)uréia, sal de ácido clorídrico.

A) 1 -((2,5-Difluoro-4-mtrofenóxi)metil)benzeno
Uma mistura de 2,4,5-trifluoronitrobenzeno (5,4 g, 30,8 mmoles), álcool benzílico (3,2 mL, 30,8 mmoles) e carbonato de potássio (6,4 g, 46,1 mmoles), em DMF (20 mL), foi agitada em temperatura ambiente durante 72 horas. Água (60 mL) foi adicionada e a mistura foi resfriada a 4°C durante horas. O precipitado resultante foi filtrado, enxagüado com água e seca* do em vácuo para fornecer o produto (7,5 g, 92%) como um sólido amarelo pálido. 1H RMN (CDCI3) δ 7,90 - 7,94 (m, 1H), 7,38 - 7,44 (m, 5H), 6,85 6,90 (m, 1H), 5,22 (s, 2H).

B) 4-Amino-2,5-difluorofenol
A um frasco carregado com 1-((2,5-difluoro-4-nitrofenóxi) metil)benzeno (4,1 g, 15,6 mmoles), que foi seqüencialmente evacuado e em seguida purgado com nitrogênio três vezes, foi adicionado paládio sob carbono a 10% (0,40 g). Para os sólidos, sob uma atmosfera de nitrogênio, foi adicionado metanol anidro (100 mL). A mistura foi em seguida agitada sob uma atmosfera de hidrogênio durante 16 horas. O nitrogênio foi borbulhado através da mistura de reação durante trinta minutos, antes a mistura foi filtrada através de uma almofada de Celité®, que foi em seguida enxaguada com metanol. O filtrado foi concentrado em vácuo, em seguida azeotropado com tolueno para fornecer o composto do título como um sólido marrom escuro (2,2 g, 99%). 1H RMN (DMSO-<) δ 9,05 (br s, 1H), 6,53-6,65 (m, 2H),
209
4,68 (s, 2H); MS(ESF) m/z 146 (M+H)*.
ir^CONHa
G) 4-(4-Amino-2,5-difluorofenóxi)picolinamida.
Para a mistura de Hidreto de potássio (30-35% dispersão em óleo mineral, 1,9 g, 13,9 mmoles) em DMF (30 mL) foi adicionado 4-amino-
2,5-difluorofenol (1,7 g, 11,6 mmoles) como uma solução em DMF (5 mL). Após uma hora de agitação em temperatura ambiente, 4-cloropicolinamida (1,8 g, 11,6 mmoles) foi adicionado e a mistura de reação foi aquecida para 100°C durante 135 horas. A mistura foi resfriada para a temperatura ambiente , extinguida com cloreto de lítio aquoso a 10% e em seguida extraída três vezes com EtOAc. As camadas orgânicas combinadas foram secadas (MgSOJ, filtradas e concentradas em vácuo. O sólido resultante foj dividido entre clorofórmio e água. A camada orgânica foi lavada com salmoura, secada (MgSCL), filtrada e concentrada em vácuo para um sólido (3,0 g, 98%). rH RMN (DMSO-de) δ 8,51 - 8,57 (m, IH), 8,14 (br s, IH), 7,74 (br s, IH), 7,37 - 7,38 (m, IH), 7,17 - 7,30 (m, 2H), 6,74-6,80 (m, 1H), 5,62 (s, 2H); MS(ESI*) 266 (M+H)*· ^n^conh2
D) 1-(4-(2-Garbamoílpiridin-4-ilóxi)-2,5-difluorofenil)-3-(2-(4fluorofenil)acetil)uréia,
Para uma mistura homogênea mistura de 4-(4-amino-2,5-difluorofenóxi)picoltnamida (0,15 g, 0,57 mmol) em THF (5 mL) foi adicionado diisopropiletilamina (0,10 mL, 0,57 mmol). A mistura foi agitada durante dois minutos em temperatura ambiente antes o isocianato de 2-(4-fluorofenil) acetila (Composto D de Exemplo 11, 0,36 M em tolueno, 2,0 mL, 0,72 mmol)
210 foi adicionado. Após 3,5 horas, o isocianato de 2-(4-fluorofenil)acetila (0,36 M em tolueno, 2,0 mL, 0,72 mmol) foi adicionado a uma mistura de reação.
Após mais duas horas, o isiocianato de 2-(4-fluorofenil)acetila (0,36 M em tolueno, 2,0 mL, 0,72 mmol) foi adicionado a uma mistura de reação. A mis5 tura foi em seguida agitada durante 16 horas antes de ser concentrada em vácuo. O resíduo foi tratado com Et2O e sonicação e o sólido branco resultante foi removido por filtração. O sólido foi tratado com Et2O e sonicação mais duas vezes antes da filtragem a vácuo fornece um sólido branco (0,23 g, 91%). 1H RMN (DMSO-ds) δ 11,29 (s, 1H), 10,92 (s, 1H), 8,56 (d, 1H, J = 10 5,6 Hz), 8,21 - 8,26 (m, 1H), 8,16 (br s, 1H), 7,76 (br s, 1H), 7,66-7,71 (m,
1H), 7,12 - 7,44 (m, 6H), 3,77 (s, 2H); MS(ESF) m/z 445 (M+H)*.
E) 1-(4-(2-Aminopiridin-4-ilóxi)-2.5-difluorofenil)-3-(2-(4fluorofenil)-acetil)uréia, sal de ácido clorídrico.
Bis(trifluoroacetóxi)iodobenzeno (Aldrich, 0,18 g, 0,42 mmol) foi adicionado a uma solução de 1-(4-(2-carbamoilpiridÍn-4-ilóxi)-2,5-difluorofenil)-3-(2-(4-fluorofeni!)-acetil)uréia (0,13 g, 0,30 mmol), água (0,01 mL,
0,60 mmol) e piridina (0,05 mL, 0,66 mmol) em DMF (2 mL) em temperatura ambiente. Após dez minutos, DMF adicional (2 mL) foi adicionado. A mistura de reação foi em seguida agitada durante 16 horas antes de ser concentra20 da em vácuo para aproximadamente metade de seu volume original. A mistura resultante foi dividida entre 6 N HCI e Et2O, a solução aquosa extraída com Et2O e as camadas orgânicas combinadas descartadas. A camada aquosa foi neutralizada com NaHCO3 (aq) e extraída com EtOAc. As camadas orgânicas combinadas foram secadas (Na2SO4), filtradas e concentradas em vácuo. O resíduo foi purificado por cromatografia de sílica-gel (eluindo com 0-5% MeOH em CHCI3) e as frações apropriadas foram concentradas em vácuo. O resíduo foi dissolvido em THF (1 mL), resfriado a 0 °C e tratado com HCI (4 N em dioxano, 0,5 mL, 2,0 mmoles). A mistura de reação foi deixada aquecer para temperatura ambiente e agitada durante uma hora, antes de ser liofilizada para fornecer o composto do título (73 mg, 53%) como um sólido branco. Ή RMN (DMSO-de)8 13,54 (brs, 1H), 11,35 (s, 1H), 10,95 (s, 1H), 8,24-8,28 (m, 1H), 7,95-8,01 (m, 3H), 7,73-7,77 (m, 1H), 7,35-7,39 (m,
211
2H), 7,16-7,20 (m, 2H), 6,72-6,74 (m, 1H), 6,25 (s, 1H), 3,78 (s, 2H);
HRMS(ESr): 417,1175 (M+H)* calculada., 417,1187 (M+H)* encontrado.
Exemplo 113
1-(4-(2-Aminopiridín-4-ilóxi)-3,5-difluorofenil)-3-(2-(45 fluorofenil)-acetil)uréia, sal de ácido trifluoroacético.
ΗΟ'^'γ f
A) 2,6-Difluoro-4-nitrofenol
2,6-Difluorofenol (10,0 g, 76,9 mmol) foi convertido no composto do título (12,7 g, 94%) de uma maneira similar às condições descritas por
Kirk e outro, J. Heterocyclic Chem. 1976, 13,1253,1H RMN (CDCIS) 5 12,15 10 (br s, 1H), 8,01 - 8,10 (m, 2H).

F
B) 4-Amino-2,6-difluorofenol.
2,6-Difluoro-4-nitrofenol (2,1 g, 12,1 mmoles) foi convertido no composto do título (1,7 g, 99%) de uma maneira similar àquela descrita por Demopoulos e outro, d. Med. Chem. 2004, 47, 2706, MS(ESF) m/z 146 15 (M+Hy.

C) 4-(4-Amino-2,6-difluorofenóx0picolinamida
4-Cloropicolinamida (0,47 g, 3,0 mmoles) foi convertido no com- posto do título (0,23 g, 29%) de uma maneira similar a uma preparação de
212
Composto C de Exemplo 112, exceto que 4-amino-2,6-djfluorofenol (0,44 g, 3,0 mmoles) foi empregado em lugar de 4-amíno-2,5-difluorofenol. 1H RMN (DMSO-cg δ 8,60 (d, 1H, J = 5,6 Hz), 8,22 (br s, 1H), 7,83 (br s, 1H), 7,457,46 (m, 1H), 7,30-7,32 (m, 1H), 6,43-6,49 (m, 2H), 5,94 (s, 2H); MS(ESf ) m^266 (M+H)Ç 'CONH2
D) 1-(4-(2-Carbamoilpiridin-4-Hóxi)-3,5-difluorofenil>3-(2-(4fluorofenil)acetíl)uréia.
4-(4-Amino-2,6-difluorofenóxi)pícolinamida (104 mg, 0,39 mmol) foi convertido no composto do título (91 mg, 52%) de uma maneira similar a uma preparação de Composto D de -Exemplo 112, Ή RMN (DMSO-c^) δ 11,07 (s, 1H), 10,62 (s, 1H), 8,50 (d, 1H, J = 5,6 Hz), 8,11 (br s, 1H), 7,72 (br s, 1H), 7,61 (m, 2H), 7,36-7,37 (d, 1H, d = 2,3 Hz), 7,23-7,31 (m, 3H), 7,11 (m, 2H), 3,69 (s, 2H); HRMS(ESF), 445,1124 (M+H)* calculada., 445,1117 (M+Hf encontrado.
E) 1 -(4-(2-Aminopiriciin-4-ilóxi)-3,5-clifluorofenil)-3-(2-(4fluorofenil)-acetíl)uréia, sal de ácido trifluoroacético.
1-(4-(2-Carbamoilpiridin-4-ilóxi)-3,5-difluorofenÍl)-3-(2-(4-fluorofenil)acetil)uréia (87 mg, 0,20 mmol) foi convertido no composto do título de uma maneira similar a uma preparação de Composto E de Exemplo 112, exceto que o produto bruto foi purificado por HPLC preparativa (YMC S10 ODS, 30 x 500mm, gradiente de 30 minutos de 58% a 90% metanol aquoso com 0,1%TFA). As frações apropriadas foram combinadas e Itofilizadas para fomecer o composto do título (23 mg, 22%) como um sólido branco. 1H RMN (DMSO-c^) δ 11,09 (s, 1H), 10,63 (s, 1H), 7,92 (d, 1H, J = 7,2 Hz), 7,62 7,72 (m, 4H), 7,27 - 7,31 (m, 2H), 7,09 - 7,14 (m, 2H), 6,68 - 6,70 (m, 1H), 6,17 - 6,18 (m, 1H), 3,69 (s, 2H); MS(ESF) m/z 417 (M+H)*
Exemplo 114
213

Af-(4-(2-Aminoplridin-4-ilóxi)-2,5-difluorofenil)-2-oxo-1-fenil-
1,2-diidropiridina-3-carboxamida, sai de ácido clorídrico.

Krf^CONH:2
A) 4-(2,5-Difluoro-4-(2-oxo-1-feml-1,2-diidropiridina-3-carboxamido)-fenóxi)picolinamida
A uma mistura homogênea de ácido 2-oxo-1-fenil-1,2-diidropiridina-3-carboxílico (Composto C de Exemplo 57, 43 mg, 0,20 mmol) em DMF (4 mL) foi adicionado hidrate de 1-hidróxi-benzotriazol (22 mg, 0,16 mmol). A mistura foi agitada até tornar-se homogênea antes do hidreto de 1(3-dtaetitamínopropí0-3-etil-carbociiímida (102 mg, 0,53 mmol) ser adicionado. Após dois minutos, 4-(4-amino-2,5-difluorofenóxi)picolinamida (Composto C de Exemplo 112, 53 g, 0,20 mmol) foi adicionado e a mistura de reação agitada, em temperatura ambiente durante 17 horas. A mistura de reação foi em seguida aquecida para 40°C e agitada durante 143 horas adicionais. Após resfriamento para temperatura ambiente, a mistura foi dividida entre EtOAc e 10% LiCI (aq). A camada orgânica foi lavada duas vezes com LiCI a 10% (aq), em seguida concentrada em vácuo. O resíduo foi purificado por cromatografia de sílica-gel (eluindo com 1:3 hexano / EtOAc) e as frações apropriadas foram concentradas em vácuo para fornecer o composto do título (45 mg, 49%). MS(ESL) m/z 463 (M+H)*.
B) ^H4-(2-Aminopiridm-4-ilóxi)-2,5-difluorofeml)-2-oxo-1 fenil-1,2-diidropiridina-3-carboxamida, sal de ácido clorídrico.
4-(2,5-Difluoro-4-(2-oxo-1 -fenil-1,2-diidropiridina-3-carboxamido)
214 fenóxi)picolinamida (45 mg, 0,10 mmol) foi convertido no composto do título (19 mg, 40%) de uma maneira similar a uma preparação de Composto E de Exemplo 112, 1H RMN (DMSO-d6) δ 13,40 (br s, 1H), 12,47 (s, 1H), 8,538,57 (m, 2H), 8,12-8,13 (m, 1H), 7,92-7,93 (m, 1H), 7,83 (s, 2H), 7,66-7,71 (m, 1H), 7,46-7,53 (m, 5H), 6,66-6,72 (m, 2H), 6,19 (s, 1H); HRMS(ESr), 435,1269 (M+H)* calculada., 435,1258 (M+H)* encontrado.
Exemplo 115

W-(4-(2-AminoplrWin-4-ilóxi)-2,5-difluorofenil)-1-(4-fluorofenil)-2-oxo-1.2-diidropiridina-3-carboxamida, sal de ácido clorídrico.

A) 4-(2,5-Dlfluoro-4-(1 -(4-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxamido)fenóxi)picolinamida
A uma mistura homogênea de ácido 1-(4-fluorofenil)-2-oxo-1,2diidropiridina-3-carboxílico (Composto B de Exemplo 101, 50 mg, 0,21 mmol) e 4-(4-amino-2,5-difluorofenóxi)picolinamida (Composto C de Exemplo 112, 69 mg, 0.26 mmol) em DMF (3 mL) foi adicionado DIPEA (0,05 mL, 0,26 mmol) e hexafluorofosfato de O-benzotriazol-l-il-MMWÇM-bisítetrametileno)-urânio (TBTU) (83 mg, 0,26 mmol). A solução resultante foi agitada durante 18 horas antes de ser extinguida com LiCI a 10% (aq). A mistura foi dividida entre EtOAc e LiCI a 10% (aq.), as camadas separadas, e a camada aquosa extraída com EtOAc. As camadas orgânicas combinadas foram lavadas duas vezes com LiCI a 10% (aq.), em seguida concentrada em vácuo. O resíduo foi purificado por cromatografia de sílica-gel (eluindo com 1:3 he
215 xano / EtOAc) e as frações apropriadas foram concentradas em vácuo para fornecer o composto do título (22 mg, 22%). MS(EST) m/z 481 (M+H)*.
B) ^(4-(2-Amínopiridin-4-ilóxi)-2,5-difluorofenll)«1-(4-fluorofenil)-2-oxo-1.2-diidropiridina-3-carboxamida, sal de ácido clorídrico.
4-(2,5-Difluoro-4-(1-(4-fluorofenil)-2-oxo-1,2-diidropíridina-3-carboxamido)fenóxi)picolinamida (22 mg, 0,04 mmol) foi convertido no composto do título (21 mg, 95%) de uma maneira similar a uma preparação de Composto E de Exemplo 112, 1H RMN (DMSO-cg δ 13,71 (br s, 1H), 12,43 (s, 1H), 8,48-8,57 (m, 2H), 8,10-8,13 (m, 1H), 7,93-7,95 (m, 3H), 7,61-7,70 (m, 1H), 7,53-7,56 (m, 2H), 7,29-7,39 (m, 2H), 6,64-6,72 (m, 2H), 6,10 (s, 1H); HRMS(ESI*), 453,1175 (M+H)* calculada., 453,1168 (M+H)* encontrado.
Exemplo 1-16 (±)-â/*-(4-(2-Aminopiridin-4-ílóxl)-2,5-difluorofenil)-^-(1-feniletiljmalonamida, sal de ácido clorídrico.
A) 3-(4-(2-carbamoilpiridin-4-ilóxi)-2,5-difluorofenilamino)-3oxopropanoato de elite.
4-(4-Amino-2,5-difluorofenóxi)picolinamida (Composto C de Exemplo 112, 1,0 g, 3,9 mmoles) foi convertido no composto do título (320 mg, 22%) de uma maneira similar a uma preparação de Composto A de Exemplo 102, Ή RMN (DMSO-d6) δ 10,32 (s, 1H), 8,54 (d, 1H, J = 5,5 Hz), 8,14-8,17 (m, 2H), 7,75 (br s, 1H), 7,61-7,65 (m, 1H), 7,42-7,43 (m, 1H), 7,24-7,25 (m, 1H), 4,12 (q, 2H, J = 7,2 Hz), 3,60 (s, 2H), 1,20 (t, 3H, J = 7,2 Hz); MS(ESI*) m/z 380 (M+H)+.
216
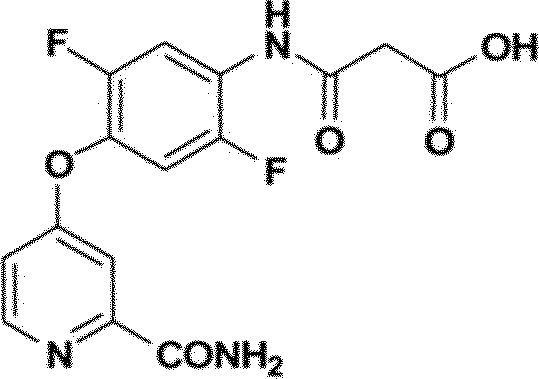
B) Ácido 3-(4-(2-Carbamoilpiridin-4-ilóxi)-2,5-difluorofenilamino)-3-oxopropanóico.
A uma mistura heterogênea de 3-(4-(2-carbamoilpiridin-4-ílóxi)2,5-difluorofenilamino)-3-oxopropanoato de etila (305 mg, 0,80 mmol) em MeOH (8 mL) foi adicionado 1 M de NaOH aquoso (1,70 mL, 1,70 mmol). Após uma hora de agitação, a mistura foi acidificada com 1N de HCl aquoso (5 mL). A reação foi extraída com EtOAc antes as camadas orgânicas combinadas foram secadas (MgSO4), filtradas e concentradas em vácuo para fornecer o composto do título (317 mg) que foi empregado sem outra purificação. HRMS (ESF), 352,0745 (M+H)* calculada., 352,0752 (M+H)* encontrado.

C) (±)-4-(2,5-Difluoro-4-(3-oxo-3-(1 -feniletilamino)propanamidoHenóxi)picolinamida.
A uma mistura homogênea de ácido 3-(4-(2-Garbamoilpiridin-4ilóxi)-2,5-difluorofenilamino)-3-oxopropanóico (89 mg, 0,25 mmol) e (±)-1fenil-etanamina (Aldrich, 0,05 mL, 0,38 mmol) em DMF (3 mL) foi adicionado DIPEA (0,07mL, 0,38 mmol) e hexafluorofosfato de O-benzotriazol-1-iiMMN(N-bis(tetrametilene)urânio (TBTU) (121 mg, 0,38 mmol). A solução resultante foi agitada durante 15 horas antes de ser extinguida com LiCI a 10% (aq). A mistura foi dividida entre EtOAc e 10% LiCI (aq), as camadas separadas, e a camada aquosa extraída com EtOAc. As camadas orgânicas combinadas foram lavadas duas vezes com LiCI a 10% (aq), em seguida concentrada em vácuo. O resíduo foi purificado por cromatografia de silica
217 gel (eluindo com 1:3 hexano / EtOAc) e as frações apropriadas foram concentradas em vácuo para fornecer o composto do título (42 mg, 37%). HRMS(ESr), 455,1532 (M+H)* calculada., 455,1528 (M+Hf encontrado.
D) (±)-A/,-(4-(2«Aminopiridin-4-ilóxi)-2,5-difluorofenll)-^-(1fenil-etil)malonamida, sal de ácido clorídrico.
(±H“(2»5-Difluoro-4-(3-oxo-3-(1 -feníletilaminojpropanamido) fenóxi)-picolinamida (41 mg, 0,09 mmol) foi convertido no composto do título (26 mg, 62%) de uma maneira similar a uma preparação de Composto E de Exemplo 112, 1H RMN (DMSO-d6) δ 10,46 (s, 1H), 8,70 (d, 1H, J = 7,8 Hz), 8,21 - 8,26 (m, 1H), 8,00 (d, 1H, J = 7,2 Hz), 7,89 (s, 2H), 7,67 - 7,72 (m, 1H), 7,22 - 7,36 (m, 5H), 6,73 - 6,76 (m, 1H), 6,21 - 6,22 (m, 1H), 4,93 - 4,96 (m, 1H), 3,57 (s, 2H), 1,35 (d, 3H, J = 7,0 Hz); HRMS(ESI*), 427,1582 (M+H)* calculada, 427,1574 (M+H)* encontrado.
Exemplo 117

(±)-W*-<4-p-Ammopiridin-4-ilóxiy2,5-difluorofonil)-^-(ciano (fenii)-meti!)ma!onamida, sal de ácido trifluoroacético.

A) (±)-4-(4-(3“(Ciano(fenil)metilamino)-3-oxopiOpanamidoy· 2,5-difluorofenóxi)picolinamida.
Ácido 3-(4-(2-Carbamoilpiridin-4-ilóxi)-2,5-difluorofenilamino)-3oxopropanóico (Composto B de Exemplo 116, 70 mg, 0,20 mmol) foi convertido no composto do título (67 mg, 72%) de uma maneira similar a uma preparação de Composto C de Exemplo 116, exceto que cloridrato de (±)-2amíno-2-fenilacetonitrila (Aldrich, 47 mg, 0,28 mmol) foi empregado em vez
218 de (±)-1-feniletanamina. MS(ESF) m/z 466 (M+H)+.
B) (±)-AH-(4-(2-Aminopiridin-4-ilóxi)-2,5-difluorofenil)-W3(ciano-(fenil)metil)malonamida, sal de ácido trifluoroacético.
(±)-4-(4-(3-(Ciano(fenil)metiiamino)-3-oxopropanamido)-2,5diflúor-fenóxi)picolinamida (65 g, 0,14 mmol) foi convertido no composto do título de uma maneira similar a uma preparação de Composto E de Exemplo 112, exceto que o produto bruto foi purificado por HPLC preparativa (YMC S10 ODS, 30 X 500mm, gradiente de 30 minutos de 34% a 90% de metanol aquoso com 0,1% de TFA). As frações apropriadas foram combinadas e liofilizadas para fornecer o composto do título (38 mg, 49%) como um sólido branco. Ή RMN (DMSO-cg 5 10,40 (s, 1H), 9,47 (d, 1H, J - 7,63 Hz), 8,218,26 (m, 1H), 7,99 (d, 1H, J = 7,2 Hz), 7,86 (br s, 2H), 7,68-7,73 (m, 1H), 7,43-7,54 (m, 5H), 6,74-6,77 (m, 1H), 6,20-6,22 (m, 2H), 3,55 (m, 2H); HRMS(ESF), 438,1378 (M+Hf calculada., 438,1374 (M+H)* encontrado.
Exemplo 118 ^n^nh2 (±)-^-(2-Amino-1 -feniletil)-^5-(4-(2-aminopiridin-4-llóxi)-2í5difluorofenil)malonamida, sal de ácido bistrifluoroacético.
(±)-/V,-(4-(2-Aminopiridin-4-ilóxi)-2,5-di11uorofenil)-/\^-(ciano (fenil)-metil)malonamida, sal de ácido trifluoroacético (Composto B de Exemplo 117, 21 mg, 0,04 mmol) foi convertido no composto do título de uma maneira similar às condições descritas por Campiani, e outro, (Tetrahedron 2002, 58, 3689). O boreto de cobalto foi comercializado de Alfa Aesar. O produto bruto foi purificado por HPLC preparativa (YMC S5 ODS, 10 x 250mm, gradiente de 30 minutos de 10% a 90% de metanol aquoso com 0,1% de TFA). Frações apropriadas foram combinadas e liofilizadas para fornecer o composto do título (5 mg, 21%) como um sólido branco. ’H RMN (DMSO-de) δ 10,39 (s, 1H), 8,72 (d, 1H, J = 8,5 Hz), 8,12 - 8,17 (m, 1H), 7,90 - 7,94 (m,
3H), 7,59 - 7,63 (m, 2H), 7,27-7,34 (m, 4H), 6,60 - 6,62 (m, 1H), 6,09-6,10 (m, 1H), 5,09 - 5,10 (m, 1H), 3,10-3-50 (m, 6H); HRMS(ESf), 442,1691 (M+H)‘ calculada., 442,1678 (M+H)+ encontrado.
Exemplo 119
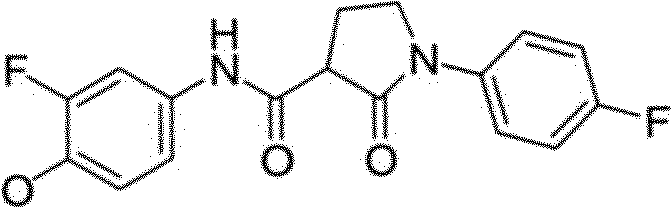

h2n
N-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenil)-1-(4-fluorofeníl)-2oxopirrolidina-3-carboxamida
Ο

A) ácido 1-(4-Fluorofenil)-2-oxopirrolidina-3-carboxílico.
A uma solução de 6,6-dimetil5,7-dioxaespiro[2,5]-octano-4,8 diona (Aldrich, 51 mg, 0,3 mmol) em DMF (0,5 mL) em temperatura ambiente foi adicionado 4-fluoroanilina (Aldrich, 33 mg, 0,3 mmol). A mistura de reação foi aquecida a 90°C durante 2 horas, resfriada a temperatura ambiente, e empregada diretamente na etapa seguinte.
B) /V-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenil)-1-(4-fluorofenil)-2-oxopirrolidina-3-carboxamida.
A uma mistura de ácido 1-(4-fluorofenil)-2-oxopirrolidina-3carboxílico (0,3 mol), 4-(4-amino-2-fluorofenóxi)piridÍn-2-amina (Composto B de Exemplo 24, 21,9 mg, 0,1 mmol) em DMF (0,5 ml), foi adicionado HATU (76 mg, 0,2 mmol) e seguido por diisopropiletilamina (0,1 mL, 0,57 mmol). A mistura de reação foi agitada em temperatura ambiente durante a noite e foi então saciada com 2 mL de metanol. A mistura de reação foi purificado por HPLC preparativa. As frações desejadas foram combinadas, neutralizadas com solução de NaHCO3 aquosa saturada, e concentrada em vácuo para fornecer o composto do título (18 mg, 43%) como um sólido branco. 1H RMN (DMSO-cy δ 10,76 (br s, 1H), 7,95 (d, 1H, J = 7,2 Hz), 7,91 (m, 1H), 7,68 (m, 2H), 7,50(m, 1H), 7,44 (t, 1H, J = 10,0 Hz), 7,24 (m, 2H), 6,70 (m, 1H), 6,11 (d, 1H, J = 2,8 Hz), 3,91 (m, 2H), 3,78 (t, 1H, J = 5,0 Hz), 2,41 (m, 2H); MS(ESr) m/z 425,15 (M+ H)*.
Exemplo 120


A/-(4-(3-Aminopiridin-4-ilóxl)-3-fluorofenil)-1-(4-fluorofenil)-2oxo-1,2-diidropiridina-3-carboxamicia.
_ H if J.


A) N-(3-flúor-4-(3-nitropiridin-4-ilóxi)fenil)-1-(4-fluorofenil)-2oxo-1,2-diidropiridina-3-carboxamida.
Preparados de 3-flúor-4-(3-nitropiridin-4-ilóxi)benzenamina (Composto A de Exemplo 72) de uma maneira similar àquela de Exemplo 62 para fornecer o composto do título (89%) como um sólido castanho. 1H RMN (CD3OD) δ 9,13 (s, 1H), 8,72 (dd, 1H, J = 8, 4 Hz), 8,60 (d, 1H, J = 6 Hz), 8,07 (d, 1H, J = 12 Hz), 8,01 - 7,99 (m, 1H), 7,58 - 7,55 (m, 2H), 7,45 (t, 1H, J = 8 Hz), 7.40 - 7,32 (m. 3H). 6,99 (d, 1H, J = 4 Hz), 6,76 (t. 1H, J = 8 Hz): MS(ESI+) m/z 465,18 (M + H)+.
B) /V-(4-(3-Aminopiridin-4-ilóxi)-3-fluorofenil)-1-(4-fluorofeni!)-2-oxo-1,2-diidropiridína-3-carboxamida.
Preparado de uma maneira similar à Etapa C de Exemplo 59, O produto bruto foi purificado por cromatografia instantânea em sílica-gel (10% de MeOH / EtOAc) para fornecer 0 sal de HCI do composto do título (58%) como um sólido esbranquiçado. 1H RMN (DMSO-de) δ 12,07 (s, 1H), 8,59
Exemplo 121


Af-(3-flúor-4-(3-(pirroliclin-3-ilmetllamino)pirlclln-4-ilóxl) fenil)1-(4-fluorofenil)-2-oxo-1.2-diidropiridina-3-carboxamída, sal de cloridrato.
A /V-(4-(3-aminopiridin-4-ilóxi)-3-fluorofenil)-1-(4-fluorofenil)-2oxo-1,2-diidropiridina-3-carboxamida (Exemplo 120, 30 mg, 0,07 mmol) em DCE (1 mL) foi adicionado éster de terc-butila de ácido 3-formil-pírrolidina-1 carboxílico (CB Research e Development Inc., 28 mg, 0,14 mmol), ácido acético (5 pL, 0,084 mmol), em seguida triacetoxiboroidreto de sódio (23 mg, 0,104 mmol). Após agitar em temperatura ambiente durante 6 horas, uma segunda porção (23 mg) de triacetoxiboroidreto de sódio foi adicionado. Após agitar em temperatura ambiente durante 2 horas, a mistura de reação foi carregada com 4N de HCI em dioxano (5 mL) e agitada por mais 1 hora em temperatura ambiente. A reação foi diluída com 10% de MeOH / EtOAc (10 mL) e lavada com solução de bicarbonato de sódio saturada (10 mL). A fase aquosa foi novamente extraída com 10 mL de 10% de MeOH / EtOAc e as camadas orgânicas combinadas foram secadas sobre Na2SO4 anidro e em seguida concentrada em vácuo. O produto bruto resultante foi purificado por HPLC preparativa. As frações apropriadas foram concentradas para remover o metanol e em seguida tornadas básicas com solução de bicarbonato de sódio aquosa saturada. A solução aquosa foi extraída com 10% de MeOH / EtOAc (3 χ 20 mL) e os extratos orgânicos agrupados foram secados sobre Na2SO4 anidro e em seguida concentrada em vácuo. O resíduo foi
222 liofllizado de acetronitriia (1 mL) / água (3 mL) / 1N HCI aquoso (0,2 mL) para fornecer o sal de HCI do composto do título (25 mg, 60%) como um sólido branco. 1H RMN (DMSO-ds) δ 12,17 (s, 1H), 8,59 (dd, 1H, J « 7,6, 2 Hz), 8,30 (s, 1H), 8,16 (dd, 1H, J~ 6,4, 2 Hz), 8,10 (dd, 1H, J = 12,8, 2 Hz), 8,00 (d, 1H, J* 6,4 Hz), 7,64 - 7,58 (m, 3H), 7,52 (t, 1H, J = 9,2 Hz), 7,46 - 7,42 (m, 2H), 7,00 (d, 1H, J = 6 Hz), 6,75 (t, 1H, J = 7,2 Hz), 3,72-3,64 (m, 3H),
3,37 - 3,26 (m, 1H), 3,17 - 3,10 (m, 1H), 2,97 - 2,92 (m, 1H), 2,74-2,66 (m,
1H), 2,10 - 2,03 (m, 1H), 1,75 - 1,68 (m, 1H); MS(ESE) m/z 518,29 (M + H)*.
Exemplo 122

W-(4-(3-(2-Aminoetilamlno)piridin-4-ilóxl)-3-fluorofenll)-1-(4fluorofeml)-2-oxo-1,2-diidroplridina-3-carboxamida, sal de clorldrato.
Preparado de uma maneira similar ao Exemplo 121 para fornecer o sal de HCI do composto do título (52%) como um sólido branco. 1H
RMN (DMSO-de) δ 12,10 (s, TH), 8,52 (dd, 1H, J = 7,6, 2,4 Hz), 8,25 (s, 1H),
8,09 (dd, 1H, J = 6,8, 2,4 Hz), 8,06 - 8,02 (m, 1H), 7,96 (d, 1H, J = 6,4 Hz), 7,56 - 7,52 (m, 3H), 7,42-7,34 (m, 3H), 6,92 (d, 1H, J ~ B Hz), 6,68 (t, 1H, J 6,8 Hz), 3,50 - 3,48 (m, 2H), 3,00 - 2,99 (m, 2H); MS(ESI+) m/z 478,29 (M + HF.

M*(3-flúor-4-(3’(piperldin-2-ilmetitomlno)piridin-4-ilóxi) fenil)1-(4-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxamida, sai de cloridrato.
223
Preparado de uma maneira similar ao Exemplo 121 para fornecer o sal de HCI do composto do título (71%) como um sólido amarelo. Ή RMN (DMSO-cQ δ 12,17 (s, 1H), 8,59 (dd, 1H, J = 7,2, 2 Hz), 8,51 (s, 1H), 8,16 (dd, 1H, J = 6,8, 2 Hz), 8,11 (dd, 1H, J = 13,2, 2,4 Hz), 8,02 (d, 1H, J =
6,4 Hz), 7,63 - 7,58 (m, 3H), 7,51 (t, 1H, J = 8,8 Hz), 7,46 - 7,41 (m, 2H), 7,01 (d, 1H, J = 6 Hz), 6,75 (t, 1H, J = 6,8 Hz), 3,70 - 3,63 (m, 1H), 3,52 3,45 (m, 1H), 3,30-3,27 (m, 2H), 2,86 - 2,84 (m, 1H), 1,95 - 1,93 (m, 1H), 1,80 - 1,62 (m, 3H), 1,58- 1,44 (m, 2H); MS(ESP) m/z 532,31 (M + H)5 Exemplo 124

HN
AH4-(3-(3-(Dimetilamino)-2,2-dimetilpropilamirio)piridin-4> ilóxi)-3-fluorofenil)-1-(4-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxamida, sal de cloridrato.
Preparado de uma maneira similar ao Exemplo 121 para fornecer o sal de HCI do composto do título (58%) como um sólido branco. 1H RMN (DMSO-dg) δ 12,10 (s, 1H), 8,57 (s, 1H), 8,52 (dd, 1H, J = 7,6, 2 Hz), 8,09 (dd, 1H, d = 6,8, 2,4 Hz), 8,03 (dd, 1H, J = 13,2,2,4 Hz), 7,90 (d, 1H, J = 6,4 Hz), 7,56-7,51 (m, 3H), 7,44 (t, 1H, J = 8,8 Hz), 7,39-7,35 (m, 2H), 6,92 (d, 1H, J = 6,4 Hz), 6,68 (t, 1H, J= 7,2 Hz), 3,33 (s, 2H), 3,10 (s, 2H), 2,77 (s, 3H), 2,76 (s, 3H), 1,07 (s, 6H); MS(ESI+) m/z 548,34 (M + H)\
Exemplo 125

M-(4-(3-(2-Amino-3-hidroxipropitamino)piridin-4-ilóxl}-3fluorofenilM(^-fluorofenil)-2-oxo-1,2-díidroplridma-3-cart>oxamida, sal
224 de cloridrato.
Preparado de uma maneira similar ao Exemplo 121 para fornecer o sal de HCI do composto do título (65%) como um sólido branco. 1H RMN (DMSO-cO δ 12,10 (s, 1H), 8,52 (dd, 1H, J = 7,2, 2 Hz), 8,30 (s, 1H), 8,09 (dd, 1H, J = 6,8, 2,4 Hz), 8,03 (dd, 1H, J = 12,8, 2 Hz), 7,96 (d, 1H, J =
6,4 Hz), 7,56 - 7,52 (m, 3H), 7,43 - 7,35 (m, 3H), 6,93 (d, IH, B Hz), 6,68 (t, IH, J = 7,2 Hz), 3,65 - 3,56 (m, 2H), 3,48 - 3,45 (m, 3H); MS(ESI+) m/z 508,27 (M + H)*.
Exemplo 126

HO I
HaN^lV
1-(4-(2-Amino-3-(hidroximetil)piridin-4-ilóxi)-3-fluorofenil)-3(2-(4-fluorofenil)acetll)uréia, sal de cloridrato.
Cl
BocHN^fT
A) 4-cloro-3-formilpiridin-2*ilcarbamato de terc-butila.
A éster de terc-butila de ácido (4-cloro-piridin-2-il)-carbâmico (CB Research e Development Inc., 2,0 g, 8,75 mmoles) em THF (18 mL) sob nitrogênio a -78°C, foi adicionado n-BuLi (13,7 mL, 21,9 mmoles, 1,6 M em hexanos) em gotas. Após agitar a -78®C durante 45 minutos, uma solução de DMF (1,93 mL) em THF (2 mL) foi adicionado em gotas. A reação foi agitada a -78°C durante 30 minutos e foi em seguida deixada aquecer lentamente para temperatura ambiente. A reação foi extinguida com 1N solução de HCI aquosa e em seguida basificada com solução de bicarbonate de sódio aquosa saturada (50 mL) e extraída com acetato de etila (3 x 50 mL). Os extratos orgânicos combinados foram secados era Na2SO4 anidro e concentrado em vácuo. O produto bruto foi purificado por cromatografía de coluna instantânea em sílica-gel (EtOAc) para fornecer o composto do título (0,95 g, 42%) como um sólido branco. Ή RMN (DMSO-de) δ 10,44 (s, IH), 10,13 (s,
225
1H), 8,47 (d, 1H, J= 5,2 Hz), 7,40 (d, 1H, J = 5,6 Hz), 1,48 (s, OH).

1Γ J
BocHN^IV
B) 4-(2-flúor-4-nitrofenóxi)-3-formilpirídin-2-ilcarbamato de terc-Butila.
A 2-flúor-nitrofenol (Aldrich, 700 mg, 4,44 mmoles) em DMF (5 mL) foi adicionado hidreto de sódio (60%, 180 mg, 4,44 mmoles). Após agitar em temperatura ambiente durante 5 minutos, uma solução de 4-cloro-3formilpiridin-2-ilcarbamato de terc-butila (0,95 g, 3,7 mmoles) em 5 mL de
DMF foi adicionado a uma mistura. A mistura de reação foi agitada a 60°C durante 20 horas. Após resfriar para a temperatura ambiente, a reação foi diluída com EtOAc (50 mL), lavada com solução de cloreto de lítio aquosa a 10% (2 x 40 mL) seguido por solução de bicarbonato de sódio aquosa saturada (40 mL), secada sobre Na2SO4 anidro, e concentrada em vácuo. O produto bruto foi purificado por cromatografia instantânea em sílica-gel (50% de
EtOAc / hexanos) para fornecer o composto do título (1,0 g, 72%) como um 15 óleo amarelo. TH RMN (CD3OD) δ 10,59 (s, 1H), 8,37 - 8,25 (m, 2H), 7,67 (t, 1H, 7,6 Hz), 6,60 (d, 1H, J = 6 Hz), 1,59 (s, 0H).

C)4-(2-flúor-4-nitiOfenóxi)-3-(hidroximetil)piridin-2-ilcarbamato de terc-butila.
A 4-(2-flúor-4-nitrofenóxi)-3-formilpiridin-2-itcarbamato de terc20 butila (75 mg, 0,2 mmol) em metanol (1 mL) a 0°Ç foi adicionado boroidreto de sódio (7,6 mg, 0,20 mmol). Após agitar a 0°C durante 30 minutos, a reação foi extinguida com solução de cloreto de amônio aquosa saturada (1 mL). A reação foi diluída com EtOAc (5 mL) e as camadas foram separadas.
A camada orgânica foi secada sobre anidro Na2SO4 e concentrada em vá
226 cuo para fornecer o composto do título (65 mg, 86%) como um sólido branco que foi empregado sem outra purificação. Ή RMN (CD3OD) δ 8,28 (dd, 1H, J = 10,4, 2,8 Hz), 8,22- 8,18 (m, 2H), 7,45 (t, 1H, J-8,4 Hz), 6,68 (d, 1H,
Hz), 4,81 (s, 2H), 1,57 (s, 9H); MS(ESF) m/z 380,27 (M + H)*.
BocHN^N'
D) 4-(4-aminO“2-fluGrofenóxi)-3-(hidroxlmetil)piridin-2-ilcarbamato de terc-butíla.
Preparado de uma maneira similar à Etapa C de Exemplo 59 para fornecer o composto do título como um sólido branco. Ή RMN (CD3OD) δ 8,14 (d, 1H, J = 6 Hz), 6,97 (t, 1H, J = 8,8 Hz), 6,62 - 6,54 (m, 2H), 6,46 (d, 1H, J= 6,4 Hz), 4,64 (s, 2H), 1,55 (s, 9H); MS(ESI*) m/z 350,11 (M + H)*.
E) 1-(4-(2-Amino-3-(hidrGximetil)piridin-4-ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de cloridrato.
Preparado de uma maneira similar à Etapa C de Exemplo 59 para fornecer o sal de HCI do composto do título como um sólido branco. Ή RMN (DMSO-d6)Ô 10,99 (s, 1H), 10,56 (s, 1H), 7,82 (d, 1H, J= 7,2 Hz), 7,74 (dd, 1H, J = 13,6, 2,8 Hz), 7,31 - 26 (m, 3H), 7,19 - 7,02 (m, 3H), 6,16 (d, 1H, J = 7,2 Hz), 4,57 (s, 2H), 3,69 (s, 2H); MS (ESI*) m/z 429,16 (M + H)*.
Exemplo 127


1-(4-(2-Amino-3-((metilamiiio)metil)piridin-4-ilGxi)-3-fluGrofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de cloridrato.
227
I Q>
A) (2-bis-BOG-amino-4-(2-flúor«4-nitrofenóxi)piriciiri-3il)metil(metil)carbamato de ierc-butila.
A 4-(2-flúor-4-nitrofenóxi)-3-formilpiridin-2-Ílcarbamato de tercbutila (Composto B de Exemplo 126, 75 mg, 0,2 mmol) em dicloroetano (1 mL) a 0°C foi adicionado metilamina (240 pL, 0,24 mmol, 2M em THF), ácido acético (14 pL, 0,24 mmol), seguido por triacetoxiboroidreto sódio (400 mg, 1,89 mmol). Após agitar em temperatura ambiente durante 16 horas, a mistura de reação foi diluída com EtOAc (5 mL), lavada com solução de bicarbonate de sódio aquosa saturada (5 mL), secada sobre NagSO^ anidro, e 10 concentrada em vácuo. O produto bruto foi purificado por cromatografia de coluna instantânea em sílica-gel (20% de MeOH / EtOAc) para fornecer 4-(2flúor-4-nitrofenóxi)-3-((metilamino)rnetil)piridin-2-ilcarbamato de terc-butila (37 mg, 47%) como um óleo amarelo.
A 4-(2-flúor-4-nitrofenóxi)-3-((metilamino)metil)piridin-2-ilcarba15 mato de íero-butila (48 mg, 0,122 mmol) e DMAP (16 mg, 0,134 mmol) em diclorometano (1 mL) foi adicionado dicarbonato de di-terc-butila (Aldrich, . 32 mg, 0,15 mmol). Após agitar em temperatura ambiente 30 minutos, a mistura de material bis- e tris- BOC (2:1) foi observada. Uma porção adicional de DMAP e dicarbonato de di-terc-butila foi adicionado. Após agitar em tem20 peratura ambiente durante 30 minutos, a reação foi purificada diretamente por cromatografia de coluna instantânea em sílica-gel (EtOAc) para fornecer o composto do título (44 mg, 61%) como um óleo amarelo. Ή RMN (GD3OD) 5 8,50 (d, 1H, J ~ 5,2 Hz), 8,15 - 7,97 (m, 2H), 7,43 (d, IH, J = 5,6 Hz), 7,07 (t, IH, 8,4 Hz), 4,64 (s, 2H), 2,83 (s, 3H), 1,44 (s, 27H); MS (ESF) m/z 25 593,34 (M + HB
B) 1 -(4-(2-Amino-3-((metilamino)metil)piriclin-4-ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de cloridrato.
Preparado de uma maneira similar à Etapa G de Exemplo 59
228 para fornecer o sal de HCI do compost© do título (60%) como urn sólido branco. Ή RMN (DMSO-<) 8 10,88 (s, 1H), 10,35 (s, 1H), 7,45 - 7,42 (m,
1H), 7,30 (m, 3H), 7,11 - 7,07 (m, 4H), 6,52 (d, 1H, J = 7,2 Hz), 4,02 (s, 2H), 3,66 (s, 2H), 2,54 (s, 3H); MS(ESF) m/z 442,30 (M + H)+.
Exemplo 128

1-(4-(2-Amino-3-((2-hidroxietilamino)metil)piridin-4-ilóxi)-3fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de ácido trifluoroacético.
O'

- A) (2-BOC-amino-4-(2-flúor-4-nitrofenóxi)piridin-3-íl)metil (2hidroxietil)carbamato de terc-butila.
Preparado de uma maneira similar à Etapa A de Exemplo 127 para fornecer o composto do título (14%) como um óleo amarelo. Ή RMN (CD3OD) 88,30 (dd, 1H, J = 10,4, 2,8 Hz), 8,23-8,20 (m, 2H), 7,52 (t, 1H, J =
8,4 Hz), 6,64 (d, 1H, J = 7,2 Hz), 4,71 (s, 2H), 3,65 (t, 2H, J = 6 Hz), 3,40 (m, 2H), 1,57 (s, 18H); MS(ESF) m/z 523,32 (M + H)+.
B) 1-(4-(2’Amino-3-((2-hidroxietilamino)metil)piridin-4-ilóxi)-
3-fluorofenil)-3-(2-(4-fluorofenil)acetll)uréia, sal de ácido trífluoroacéti- co.
Preparado de uma maneira similar à Etapa C de Exemplo 59 para fornecer o sal de TFA do composto do título (45%) como um sólido 20 branco. 1H RMN (DMSO-d6) δ 11,12 (s, 1H), 10,69 (s, 1H), 8,01 (d, 1H, J =
6,8 Hz), 7,87 (dd, 1H, 12,8, 2,4 Hz), 7,53 - 7,40 (m, 4H), 7,26 - 7,20 (m,
2H), 6,68 (d, 1H, J = 7,2 Hz), 4,34 (s, 2H), 3,81 (s, 2H), 3,75 (t, 2H, J = 7,2 Hz), 3,18 (m, 2H); MS(ESE) 472,24 (M + H)*.
229
Exemplo 128


AF(4-(2-Amino-3-((2«amlnoetilamino)metil)piridin-4-ilóxi)-3fluorofenil>1-(4-fluoiOfenil)-2-oxo-1,2-diidropiridina-3-carboxamida, sal de ácido trifluoroacético.


A) (2-BOÇ-amino-4-(2-flúor-4-nitrofenóxi)piridin-3-il)metil (2BOC-aminoetil)carbamato de terc-butila.
Preparado de uma maneira similar à Etapa A de Exemplo 127 para fornecer o composto do título (19%) como um óleo amarelo. Ή RMN (CD300) δ 8,30 (dd, 1H, d = 10, 2,4 Hz), 8,23 - 8,20 (m,2H), 7,52 (t, 1H, J = 10 8,4 Hz), 6,61 (d, 1H, J = 7,2 Hz), 4,65 (s, 2H), 3,39 (m, 2H), 3,25 (m, 2H),
1,61 (s, 27H); MS (ESF) m/z 622,47 (M + H)*.
B) /V-(4-(2-Amino-3-((2-aminoetilamino)metil)piridin-4-ilóxi)-
3-fluorofenil)-1-(4-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxamida, sal de ácido trifluoroacético.
O grupo de nitro foi reduzido de uma maneira similar com a Etapa C de Exemplo 59 e em seguida a amida foi formada de uma maneira similar ao Exemplo 62 para fornecer o sal de TFA do composto do título (28%) como um sólido branco. Ή RMN (DMSO-d6) δ 12,09 (s, 1H), 8,52 (dd, 1H. J = 7,2, 2 Hz), 8,09 (dd, 1H, J = 6,4, 2 Hz), 7,99 (dd, 1H, J = 12,8, 2,4 Hz), 20 7,89 (d, 1H, J = 6,4 Hz), 7,56-7,52 (m, 2H), 7,49-7,47 (m, 1H), 7,39-7,33 (m,
3H), 6,68 (t, 1H, /= 7,2 Hz), 6,07 (d, 1H, J = 7,2 Hz), 4,25 (s, 2H), 3,20 (m, 2H), 3,08 (m, 2H); MS(ESI*) m^ 507,23 (M + H)*.
Exemplo 130
230


N-(4-(2-Amino-3-(hidroximetil)piridin-4-ilóxi)-3-fluorofenil)-1(4-fluorofeníl)-2-oxo-1,2-diidropiridina-3’Carboxamida, sal de ácido trifluoroacético.

A) 3-((terc-butildimetilsililóxi)metil)-4-(2-flúor-4-nitrofenóxi) piridin-2-ilcarbamato de terc-butila.
A 4-(2-flúor-4-nitrofenóxi)-3-(hidroximetil)piridin-2-ilcarbamato de terc-butila (Composto C de Exemplo 126,100 mg, 0,26 mmol) em dlclorometano (3 mL) foi adicionado imídazol (21 mg, 0,31 mmol) seguido por cloreto de terc-butildimetilsilila (40 mg, 0,26 mmol). Após agitar em temperatura ambiente durante 1 hora, um segundo equivalente de cloreto de terc-butildimetiteiliía (40 mg, 0,26 mmol) foi adicionado. A reação foi agitada em temperatura ambiente durante 3 horas e foi em seguida diluída com diclorometano (10 mL), lavada com água (10 mL), secada sobre anidro Na2SO4, e concentrada em vácuo. O produto bruto foi purificado por cromatografia de coluna instantânea em sílica-gel (50% EtOAc / hexanos) para fornecer o composto do título (102 mg, 79%) como um sólido branco. 1H RMN (CD3OD) δ 8,16 (dd, 1H, J = 10,4, 2,8 Hz), 8,09 - 8,05 (m, 2H), 7,29 (t, 1H, J = 8,4 Hz), 6,53 (d, 1H, 5,6 Hz), 4,84 (s, 2H), 1,43 (s, 9H), 0,84 (s, 9H), 0,00 (s, 6H);
MS(ESr) m/z 494,29 (M + H)+.


231
B) 4-(4-amino-2-fluorofenóxi)-3-((terc-butildimetilsililóxi)metil)pirídin-2-ilcarbamato de terc-butila.
Preparado de uma maneira similar à Etapa C de Exemplo 59 fornece o composto do título (95 mg, 98%) como um óleo amarelo. 1H RMN (CD3OD) δ 7,90 (d, 1H, J = 6 Hz), 6,76 (t, 1H, J = 8,8 Hz), 6,43 (dd, 1H, J = 12,8, 2,8 Hz), 6,39-6,37 (m, 1H), 6,21 (d, 1H, J = 5,6 Hz), 4,85 (s, 2H), 1,39 (s, 9H), 0,81 (s, 9H), 0,01 (s, 6H); MS(ESF) m/z 464,34 (M + H)*.


C) 3-((terc-butildimetilsililóxi)metil)-4-(2-flúor-4-(1-(4-fluorofenil)-2-oxo-1,2-diidropiridÍna-3-carboxamido)fenóxi)piridin-2-ilcarbamato de terc-butila.
Preparado de uma maneira similar àquela de Exemplo 62 para fornecer o composto do título (86%) como um óleo incolor. 1H RMN (CD3OD) δ 8,51 (dd, 1H, J = 7,6, 2,4 Hz), 7,93 (d, 1H, J = 6 Hz), 7,84 - 7,80 (m, 2H), 7,40 - 7,37 (m, 2H), 7,21 - 7,15 (m, 3H), 7,06 (t, 1H, J = 8,8 Hz), 6,57 (t, 1H,
J = 6,8 Hz), 6,26 (d, 1H, J = 5,6 Hz), 4,86 (s, 2H), 1,40 (s, 0H), 0,82 (s, 9H),
0,00 (s, 6H); MS(ESr) m/z 679,34 (M + H)*.
BocHN
HO
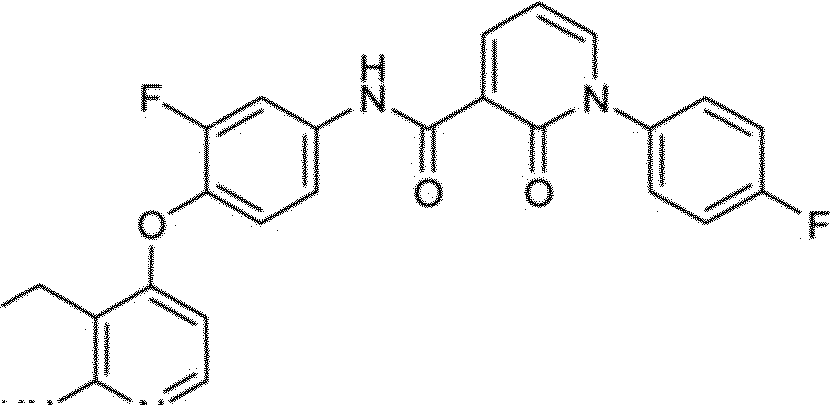
D) 4-(2-flúor-4-(1 -(4-fluorofenil)-2-oxo-1,2-diidropiridina-3carboxamido)fenóxi)-3-(hidroximetil)piriclin-2-ilcarbamato de terc-butila.
A 3-((terc-butildimetilsililóxi)metil)-4-(2-flúor-4-(1 -(4-fluorofenil)-2oxo-1,2-díidropiridina-3-cart>oxamido)fenóxi)piridin-2-ilcart>amato de tercbutila (119 mg, 0,176 mmol) em THF (2 mL) em temperatura ambiente foi adicionado fluoreto de tetrabutilamônio (260 μί, 0,264 mmol, 1 M em THF).
232
Após agitar em temperatura ambiente durante 30 minutos, a reação foi diluída com acetato de etila (20 mL), lavada com água seguido por salmoura (10 mL cada), secada sobre MgSO4 anidro, e concentrada em vácuo. O produto bruto foi purificado por cromatografia de coluna instantânea em sílica-gel (5% de MeOH / EtOAc) para fornecer o composto do título (66 mg, 66%) como um óleo amarelo. Ή RMN (CD3OD)5 8,62 (dd, 1H, J -7,2,2 Hz), 8,04 (d, 1H, J = 6 Hz), 7,94 - 7,90 (m, 2H), 7,50 - 7,46 (m, 2H), 7,33 - 7,25 (m, 3H), 7,20 (t, 1H, J = 8,8 Hz), 6,67 (t, 1H, J = 6,8 Hz), 6,40 (d, 1H, J = 5,2 Hz), 4,78 (s, 2H), 1,49 (s, 9H); MS(ESI*)?rwfe 565,17 (M + H)+.
E) /V-(4-(2-Amino-3-(hidroximetil)piridin-4-ilóxi)-3-fluorofe- nil)-1-(4-fluorofenil)-2-oxo-1#2-díidropiridina-3-carboxamida, sal de ácido trifluoroacético.
A 4-(2-flúor-4-(1 -(4-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxamido)fenóxi)-3-(hidroxímetil)píridin-2-ílcarbamato de terc-butila (33 mg
0,058 mmol) em THF (2 mL) em temperatura ambiente foi adicionado 4N HGI em dioxano (10 mL). Após agitar em temperatura ambiente durante 8 horas, a reação foi concentrada em vácuo. O produto resultante bruto foi purificado por HPLC preparativa, as frações apropriadas foram concentradas e o tolueno foi adicionado (2 x 3 mL) e a mistura resultante foi concentrada novamente. O resíduo foi liofilizado de acetronitrila (1 mL) / água (3 mL) para fornecer o sal de TFA do composto do título (14 mg, 42%) como um sólido branco. 1H RMN (DMSO-de) δ 12,08 (s, 1H), 8,52 (dd, 1H, J = 7,2, 2 Hz), 8,09 (dd, 1H, J = 6,8, 2 Hz), 7,99 (dd, 1H, J = 12,8, 2,4 Hz), 7,81 (d, 1H, J -
7,2 Hz), 7,56-7,52 (m, 2H), 7,48-7,46 (m, 1H), 7,39 - 7,34 (m, 2H), 7,30 (t, 1H, J = 9,2 Hz), 6,67 (t, 1H, J = 7,2 Hz), 6,21 (d, 1H, J = 7,2 Hz), 4,58 (s, 2H); MS(ESF) m/z 465,17 (M + H)*,
Exemplo 131 h2n n
1-(4-(2-Amino-3-(2-(piridin-2-il)etinll)piridin-4-ilóxi)-3233 fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sai de dicloridrato.
»2

HgM N
A) 4-(2-flúor-4-nitrofenóxi)-3-(2-(piridin-2-il)etinil)piridin-2amina.
Uma solução de 4-(2-flúor-4-nitrofenóxi)-3-iodopiridin-2-amina (Composto C de Exemplo 34, 100 mg, 0,27 mmol), 2-etinilpiridina (Aldrich, 56 μί, 0,54 mmol), Et3N (2 mL), e THF (2 mL) foi desgaseificada por vácuo/purga de argônio e em seguida tratada com Cul (6 mg, 0,032 mmol) e (Ph3P)4Pd (20 mg, 0,017 mmol). A mistura de reação foi aquecida a 60°C durante 45 minutos, resfriado em temperatura ambiente e diluída com EtO10 Ac. A mistura foi lavada com solução de NaHCO3 aquosa saturada, salmoura, secada (MgSO4) e concentrada em vácuo. O produto bruto foi purificado por cromatografia instantânea em SiO2 empregando-se 0-1, 5% de MeOH /
CH2CI2 para fornecer o composto do título (55 mg, 57%) como um sólido verde oliva. Ή RMN (DMSO-d6) δ 8,53 (d, 1H, J = 5,1 Hz), 8,42 - 8,37 (m, 15 1H), 8,19 - 8,12 (m, 1H), 7,96 (d, 1H, J = 5,6 Hz), 7,85-7,78 (m, 1H), 7,71 (d,
1H, J = 7,6 Hz), 7,52 (t, 1H, J = 8,6 Hz), 7,36 (ddd, 1H, J = 7,6, 5,1, 1,0 Hz), 6,71 (s, 2H), 6,21 (d, 1H, J = 5,6 Hz); MS (ESF): m/z 351,25 (M+H)*.
h2k n
B) 4-(4-Amino-2-fluorofenóxi)-3-(2-(piridin-2-il)etiriil)piridin-
2-amina.
nitrofenóxi)-3-(2-(piridin-2-il)etinil)pÍridin-2-amina (35 mg, 0,10 mmol) empregando-se pó de zinco (65 mg, 1,0 mmol) e NH4CI (53 mg, 1,0 mmol) da mesma maneira como a Etapa C de Exemplo 11, este fornece o composto do título (25 mg, 78%) como óleo marrom. MS (ESI*): m/z 321, 25 (M+H)*.
234
C) 1-(4-(2-Ammo-3-(2-(piridin-2-ll)etinil)pirldin-4-Hóxi)-3fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de dicloridrato.
O composto do título foi preparado de 4-(4-amino-2-fluorofenoxi)-3-(2-(piridin-2-il)etinil)pfridin-2-amina (25 mg, 0,078 mmol) e uma solução de 0,3 M de isocianato de 2-(4-fluorofenil)acetila em tolueno (Composto D de Exemplo 11,0,26 mL, 0,078 mmol) da mesma maneira como a Etapa E de Exemplo 11,0 produto bruto foi preparado por HPLC preparativa (Coluna B). O sal de TFA foi convertido ao cloridrato da mesma maneira como na Etapa E de Exemplo 36 para fornecer o composto do título (18 mg, 40%) como um sólido marrom. 1H RMN(DMSO-d6) δ 11,06 (s, 1H), 10,63 (s, 1H), 8,62 (d, 1H, J = 4,6 Hz), 8,23 (s, 1H), 7,99 (d, 1H, J = 7,1 Hz), 7,94 - 7,84 (m, 3H), 7,87 - 7,76 (m, 1H), 7,51 - 7,42 (m, 3H), 7,39 - 7,32 (m, 2H), 7,21 - 7,13 (m, 2H), 6,30 (d, 1H, J = 7,1 Hz), 3,74 (s, 2H) MS(ESI+)i m/z 498,11 (M+H)+235
Exemplo 132

W“(4»(2-Amino-3-(4-(2-amino-2-oxoetil)feníl)piridin-4-ilóxí)-3fluorofenil)-1-(4-fluorofenil)-2-oxo-1,2-diidropiridma-3»carboxamida, sal de cloridrato.
O composto do título foi preparado de 2-(4-(2-amino-4-(4-amino-
2-fluorofenóxi)piridin-3-ilXenil)acetamida (Composto C de Exemplo 78, 18 mg, 0,05 mmol) e ácido 1-(4-fluorofenilX2-oxo-1,2-d«idropiridina-3-carboxílico (Composto B de Exemplo 101,11 mg, 0,05 mmol) da mesma maneira que o Exemplo 62, o produto bruto foi preparado por HPLC preparativa (Coluna B) seguido por conversão do sal de TFA para o sal de cloridrato (15 mg, 50%).
1H RMN (DMSO-d6) δ 12,11 (s, 1H), 8,58 - 8,54 (m, 1H), 8,13 (dd, 1H, J =
6,6, 2,2 Hz), 8,00 (dd, 1H, J = 12,6, 2,2 Hz), 7,96 (d, 1H, J = 7,7 Hz), 7,59 (dd, 2H, J = 8,8. 4,9 Hz), 7,47 - 7,41 (m, 6H), 7,40 - 7,35 (m, 3H), 6,94 (s,
1H), 6,72 (t, 1H, J= 7,1 Hz), 6,38 (d, 1H, 7,1 Hz), 3,44 (s, 2H); MS(ESI*):
m/z 568,23 (M+H)*.
Exemplo 133

1-(4-(2-Amino-3-(2-(5-aminopiridin-2-il)etinil)piridin-4-ilóxi)-3fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de dicioridrato.

SiíCHsh
A) 6-(2-(Trimetilsilil)etinil)piridin-3-amlna.
Uma mistura de 3-amino-6-bromopiridina (Alfa Aesar, 1,0 g, 5,8
236 mmoles), trimetilsilano de etinila (1,7 mL, 17,3 mmoles), CH3CN (3 mL), DMF (2 mL) e Et3N (2 mL) fei tratada com Cui (60 mg, 0,32 mmol) e (Ph3P)4Pd (114 mg, 0,10 mmol) e a mistura agitada a 45°C durante 1,5 hora. Mais etiniltrimetilsilano (1,7 mL, 17,3 mmoles) fbi adicionado à reação e a mistura foi agitada durante 2 horas. A mistura foi concentrada em vacuo e o resíduo dividido entre EtOAc e solução de NaHCO3 aquosa saturada. A fase EtOAc foi lavada com salmoura, secada (MgSO4) e concentrada em vácuo. O produto bruto fei purificado por cromatografia instantânea em SiO2 empregando-se 0-6% de EtOAc/hexanos para fornecer o composto do título (0,75 g, 68%) como um sólido marrom. 1H RMN (DMSO-de) δ 7,85 (s, TH), 7,15 (d, 1H, 4 = 8,1 Hz), 6,81 (d, 1H, J = 8,1 Hz), 5,76 (s, 2H), 0,19(s, 9H); MS(ESI*): m/z 191, 20 (M+H)L

SI(CH3)3
B) 6-(2-(trimetílsilil)etinil)piridin-3-ilcarbamato de terc-butila.
Uma solução de 6-(2-(Wme®s®)etinil)piridin-3-amina (0,5 g, 2,6 mmoles) em THF foi resfriada a -50°C e tratada em gotas com 1,0 M NaHMDS em THF (5,3 mL, 5,5 mmoles). A mistura foi aquecida para -20°G, tratada com anidrido de BOG em uma porção e permitida aquecer para temperatura ambiente durante 25 minutos. A mistura foi dividida entre EtOAc e solução de NaHGO3 aquosa saturada e a fase EtOAc foi separada e lavada com salmoura. A solução de EtOAc foi secada (MgSO4) e concentrada em vácuo. O produto bruto foi purificado por cromatografia instantânea em SiO2 empregando-se 0-25% de EtOAc / hexanos para fornecer o produto (0,5 g, 67%) como um sólido branco. 1H RMN (DMSO-de) δ 9,80 (s, 1H), 8,58 (d, 1H, 4= 2,0 Hz), 7,86 (dd, J~ 8,6, 2,5 Hz, 1 H), 7,44 (d, 1H, J = 8,6 Hz), 1,47 (s, 9H), 0,22 (s, 9H); MS(ESF): m/z 291, 34 (M+H)*·
237
C) 6-etinilpiridin-3-ilcarbamato de terc-butila.
Uma solução de 6-(2-(tr/met//s////)etinil)piridin-3-ilcarbamato de terc-butila (62 mg, 0,21 mmol) em THF (5 mL) foi resfriado a -15°C e tratada com 1,0 M fluoreto de tetrabutilamônio (Aldrich, 0,25 mL, 0,25 mmol) e agi5 tada durante 40 minutos. A mistura foi concentrada em vácuo e dividida entre EtOAc e solução de NaHCO3 aquosa saturada. A fase de EtOAc foi lavada com salmoura, secada (MgSO4) e concentrada em vácuo para fornecer o composto do título (45 mg, 100%) como um sólido esbranquiçado. Ή RMN (DMSO-4) δ 9,78 (s, 1H), 8,58 (d, 1H, J = 2,5 Hz), 7,92 - 7,84 (m, 1H), 7,46 (d, 1H, J= 8,6 Hz), 4,17 (s, 1H), 1,47 (s, 9H); MS (ESF): m/z 219,20 (M+H)+.
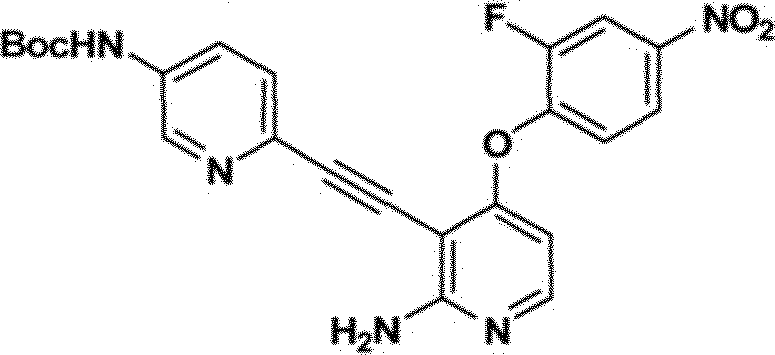
D) 6-(2-(2-amino-4-(2-flúor-4-nitrofenóxi)piridin-3-il)etinil) piridin-3-ilcarbamato de terc-butila.
O composto do título foi preparado de 6-etinilpiridin-3-ilcarbamato de terc-butila e 4-(2-flúor-4-nitrofenóxi)-3-iodopiridin-2-amina (Com15 posto C de Exemplo 34) da mesma maneira como a Etapa A de Exemplo 46 em 44% de Produção. Ή RMN (DMSO-cg δ 9,77 (s, 1H), 8,56 (s, 1H), 8,408,36 (m, 1H), 8,14 (d, 1H, J = 8,8 Hz), 7,94 (d, 1H, J = 5,5 Hz), 7,89 (d, 1H, J = 8,2 Hz), 7,59 (d, 1H, J * 8,8 Hz), 7,53-7,47 (m, 1H), 6,63 (br s, 2H), 6,21 (d, 1H, 6,0 Hz), 1,47 (s, 9H); MS(ESF): mór 466,22 (M+H)*.

E) 6-(2-(2-amino-4-(4awino-2-fluorofenóxi)piridin-3-il)etinil) piridin-3-ilcarbamato de terc-butila.
O composto do título foi preparado de 6-(2-(2-amino-4-(2-flúor-4nitrofenóxi)piridin-3-il)etinil)piridin-3-ilcarbamato de terc-butila da mesma
238 maneira como a Etapa C de
Exemplo 11 em Produção quantitativa.
MS(ESF): m/z 436,29 (M+H)+.

F)6-(2-(2-amino-4-(2-flúor-4-(3-(2-(4-fluorofenil)acetil)ureído) fenóxi)piridin-3-il)etinil)piridin-3-ilcarbamato de terc-butila.
O composto do título foi preparado de 6-(2-(2-amino-4-(2-flúor-4nitrofenóxi)piridin-3-il)etinil)piridin-3-ilcarbamato de terc-butila da mesma maneira como a Etapa E de Exemplo 11 em 80% de Produção. ’H RMN (DMSO-de) δ 11,02 (s, 1H), 10,56 (s, 1H), 9,78 (s, 1H), 8,59 (d, 1H, J = 2,5
Hz), 7,91 (dd, 1H, J = 8,6, 2,5 Hz), 7,81 (d, 1H, J = 5,6 Hz), 7,75 (dd, 1H, J = 10 12,7, 2,5 Hz), 7,68 (d, 1H, J - 8,6 Hz,), 7,44-7,27 (m, 5H), 7,19-7,13 (m, 2H),
6,47 (s, 2H), 5,86-5,81 (m, 1H), 1,48 (s, 9H); MS(ESI*): m£615,34(M+H)+.
G) 1 -(4-(2-Amino-3-(2-(5-aminopiridin-2-il)etinil)piridin-4ilóxi)-3-fluorofenll)-3-(2-(4-fluorofenil)acetil)uréia, sal de dicloridrato.
Uma solução de 6-(2-(2-amino-4-(2-flúor-4-(3-(2-(4-fluorofenil) 15 acetil)ureído)fenóxí)piridin-3-il)etinil)piridin-3-ilcarbamato de terc-butila (23 mg, 0,037 mmol) em CH2CI2(2 mL) e tratada com TFA (0,5 mL) e agitada em temperatura ambiente durante 1 hora. A mistura foi concentrada em vácuo e o produto bruto purificado por HPLC preparativa (Coluna B) e convertido em seu sal de cloridrato (11 mg, 52%) da mesma maneira como a Etapa E de 20 Exemplo 36, Ή RMN (DMSO-oy δ 11,05 (s, 1H), 10,62 (s, 1H), 8,05 (s, 1H),
7,97 (d, 1H, J = 2,7 Hz), 7,94 (d, 1H, J = 7,1 Hz), 7,82 (d, 1H, 12,1 Hz),
7,66 (d, 1H, J = 8,8 Hz), 7,45-7,45 (m, 2H), 7,35 (dd, 2H, J = 8,2, 5,5 Hz), 7,20 (d, 1H, J 6,6 Hz), 7,16 (t, 2H, J = 8,8 Hz), 6,24 (d, 1H, J = 6,6 Hz),
3,74 (s, 2H); MS(ESr): m/z 515,19 (M+H)*.
230
Exemplo 134


/V-(4-(2-Aniino-3-(3-((3R,4R)-3-hidróxi-4-(pirrolidin-1-il) pirrolidin-1-il)-3-metilbut-1-inil)piridin-4-ilóxi)-3-fluorofenil)-1-(4-fluorofenil)-2oxo-1,2-diidropiridina-3-carboxamida, sal de tricloridrato.
OH

A) (3/?,4R)-1-(2-Metilbub3-in-2-ilH(PinOlWin-1-il)pirroliciin-
3-ol
Uma mistura de (3R,4R)-4-{pirTOlidín-1-il)pirrolidin-3-ol (Lexicon
Pharma, 1,56 g, 10,0 mmoles) e 3-cloro-3-metil-1-butina (GFS Chemical, Inc., 1,36 g, 13,2 mmoles), THF (15 mL) e Et3N (13,3 mmoles) foi tratada com Cul (77 mg, 0,78 mmol). Uma reação exotérmica segui-se com concomitante formação de um precipitado. Após agitar em temperatura ambiente durante 15 horas, a mistura foi concentrada em vácuo e dividido entre EtOAc e solução de NaHCO. aquosa saturada. A fase aquosa foi extraída duas vezes com EtOAc e as fases de EtOAc combinadas foram secadas (MgSOJ e concentrada para fornecer um sólido marrom claro (1,0 g, 45%). 1H RMN (CDCI3) δ 4,20 (br s, 1H), 3,13-3,04 (m, 1H), 2,95 (dd, 1H, J=10,1,
6,6 Hz), 2,77 (dd, IH, J = 10,1, 2,6 Hz), 2,64 (m, 3H), 2,61 (m, 2H), 2,55 2,46 (m, IH), 2,26 (s, 1H), 1,85 - 1,74 (m, 4H), 1,38 (s, 3H), 1,36 (s, 3H); MS(ESI*): m/z 223,29 (M+H)*.

240
B) (3/?,4^-1-(4-(2-Amino-4-(2-flúor-4-nitrofenóxi)piridin-3-il)-
2-metilbut-3-in-2-il)-4-(pirrolidin-1-il)pirrolidin-3-ol.
O composto do título foi preparado de (3R,4R)-1-(2-rnetílbut-3-in-
2-il)-4-(pirrolidin-1-il)pirrolidin-3-ol e 4-(2-flúor-4-nitrofenóxi)-3-iodopÍridin-2- amina (Composto C de Exemplo 34) da mesma maneira como a Etapa D de
Exemplo 35 em 57% de Produção. Ή RMN (CDCI3) δ 8,10 (dd, 1H, J = 10,2,
2,5 Hz), 8,03 (d, 1H, J = 9,2 Hz), 7,99 (d, 1H, J = 5,6 Hz), 7,13-7,06 (m, 1H),
6,26 (d, 1H, J = 6,1 Hz), 5,18 (br s, 2H), 4,21 (m, 1H), 3,10-2,99 (m, 1H),
2,96 (s, 1H), 2,70 (dd, 2H, J= 9,7, 3,1 Hz), 2,66-1,15 (m, 2H), 1,87-1,76 (m,
5H), 133-1,32 (m, 2H), 1,32 (m, 3H); 1,34 (m, 3H); MS(ES1*): m/z 470,20 (M+H)*.

C) (3R,4^-1-(4-(2-Ammo-4-(4-amino-2-fluorofenóxl)piridiii-
3-il)-2-metilbut-3-in-2-il)-4-(pirrolidin-1-il)pirrolidin-3-ol.
O composto do título foi preparado de (3R,4R>-1-(4-(2-amino-415 (2-flúor-4-nitrofenóxi)pirÍdin-3-il)-2-metilbut-3-in-2-il)-4-(pirrolidin-1 -il )pirrolidin-
3-ol da mesma maneira como a Etapa C de Exemplo 11 em 95% de Produção. MS(ESF): m/Z 440,37 (M+H)*.
B) A/-(4-(2-Amino-3-(3-((3/^4/?)-3-hidróxi-4-(pirroiidin-1-il) pirrolidin-1-i)-3-metilbut-1-inil)plridln-4-ilóxi>3-fluorofenil)-1-(4*flu©ro* fenil)-2-oxo-1,2-diidropiridina-3-carboxamida, sal de tricloridrato.
O composto do título foi preparado de (3R,4R)-1 -(4-(2-amino-4(4-amíno-2-fluorofenóxi)piridin-3-il)-2-metilbut-3-in-2-il)-4-(pirrolidin-1-íl) pirrolidin-3-ol e ácido 1-(4-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxílico (Composto B de Exemplo 101) da mesma maneira como o Exemplo 62 em 50% 25 de Produção. 'H RMN (DMSO-cü δ 12,14 (s, 1H), 8,62 - 8,54 (m, 1H), 8,14 (dd, 3H, J -6,6, 2,0 Hz), 8,05 (d, 1H, J = 13,2 Hz), 7,99 (d, 1H, J = 7,1 Hz),
7,62 - 7,51 (m, 5H), 7,42 (t, 2H, J = 8,6 Hz), 6,78 - 6,70 (m, 1H), 6,32 (d, 1H, J = 7,1 Hz), 4,65 (br s, 1H), 3,94-3,84 (m, 1H), 3,71 (d, 2H, J = 16,8 Hz),
3,60 (m, 2 H), 3,52 (m, 1H), 3,22 - 3,08 (m, 2H), 2,00 - 1,91 (m, 2H), 1,88 1,78 (m, 2H), 1,70 (s, 6H); MS(ESF): m/z 655,40 (M+Hf.
Exemplo 135
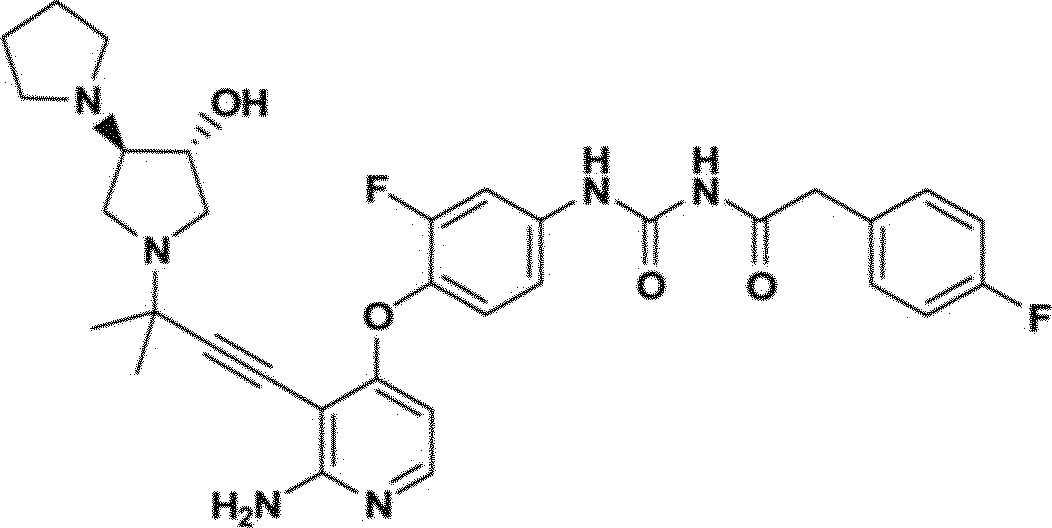
1-(4-(2-Amino-3-(3-((3R,4R)-3-hidróxi-4-(pirroIidin-1-il) pirrolidin-1-ll)-3-metilbut-1-inil)piridin-4-ilóxi)-3-fluorofenil)-3-(2-(4-fiuorofenil) acetil)uréia, sal de tricloridrato.
O composto do título foi preparado de (3R,4R)-1-(4-(2-amino-410 (4-amino-2-fluorofenóxi)piridin-3-il)-2-metilbut-3-in-2-il)-4-(pirroiidin-1-il)pirrolidin-3-ol em 40% de Produção da mesma maneira como a Etapa E de
Exemplo 11, Ή RMN (DMSO-d6) δ 11,06 (s, 1H), 10,62 (s, 1H), 7,97 (d, 2H,
J = 6,6 Hz), 7,80 (d, 1H, J = 11,7 Hz), 7,51-7,40 (m, 2H), 7,38-7,32 (m, 2H), 7.16 (t, 2H, J = 8.6 Hz), 6,25 (d, 1H, J = 6,6 Hz). 4,63-4,53 (m, 1H). 3,793,68 (m, 1H), 3,74 (s, 2H), 3,68-3,56 (m, 2 H), 3,51-3,43 (m, 2H), 3,08-2,99 (m, 4H), 1,98-1,92 (m, 2H), 1,89-1,77 (m, 2H), 1,64 (s, 6H); MS(ESf): m/z 619,41 (M+H)+.
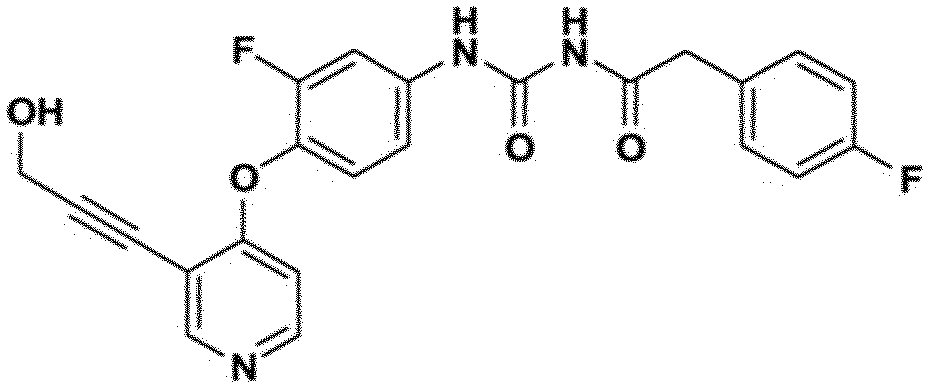
1-(3-flúor-4-(3-(3-hidroxiprop-1-inil)plridin-4-ilóxí)fenil)-3-(2242

A) 3-(4-(2-flúor-4-nitrofenóxi)piridin-3-il)prop-2-in-1-ol
O composto do título foi preparado de 4-(2-flúor-4-nitrofenóxi)-3iodopiridina (Composto A de Exemplo 33) e álcool de propargila (Aldrich) em 77% de Produção da mesma maneira como a Etapa A de Exemplo 36, RMN (DMSO-cg 1 8,69 (s, 1H), 8,49 (d, 1H, J = 5,6 Hz), 8,43 (dd, 1H, J ~ 10,7, 2,5 Hz), 8,17 (d, 1H, U = 9,2 Hz), 7,57 (t, 1H, d = 8,6 Hz), 7,04 (d, 1H, J = 5,6 Hz), 5,40 (t, 1H, J = 6,1 Hz), 4,28 (d, 2H, J = 6,1 Hz); MS(ESF): m/z
289,15 (M+H)*.

B) 3-(4-(4-Amino-2-fluorofenóxi)piricHn-3-ll)prop-2-in-1 -ol
O composto do título foi preparado de 3-(4-(2-flúor-4-nitrofenóxi) piridin-3-il)prop-2-in-1 -ol em 95% de Produção da mesma maneira como a Etapa C de Exemplo 11, MS(ESf): m/z 259,21 (M+Hf.
C) 1-(3-flúor-4-(3-(3-hidroxiprop-1-inil)piridin-4-ilóxi)fenil)-3(2-(4-fluorofenil)acetil)uréia, sa! de cloridrato.
O composto do título foi preparado de 3-(4-(4-amino-2-fluorofenóxi)piridÍn-3-il)prop-2-in-1-ol em 36% de Produção da mesma maneira como a Etapa E de Exemplo 11,1H RMN (DMSO-<) δ 11,06 (s, 1H), 10,63 (s, 1H), 8,79 (s, 1H), 8,49 (d, 1H, J = 6,1 Hz), 7,82 (d, 1H, J = 12,2 Hz), 7,457,40 (m, 2H), 7,38-7,33 (m, 2H), 7,23-7,13 (m, 2H), 6,89 (d, 1H, J = 6,1 Hz), 4,37 (s, 2H), 3,74 (s, 2H); MS(ESI*): m/z 438,23 (M+H)*.
243
Exemplo 137

M-(4-(2-Amlno-3-(3-aminoprop-1-inil)pirtdin-4-ilóxi)-3-fluorofenil)-1-(4-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxamida, sal de dicloridrato.
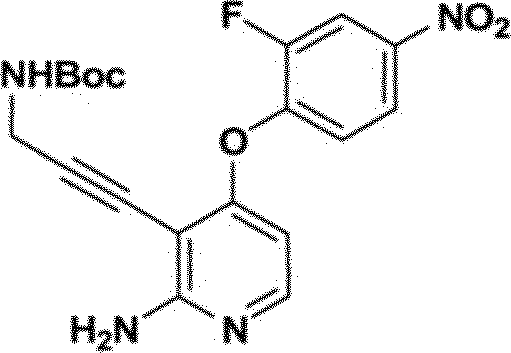
A) 3-(2-amlno-4-(2-flúor-4-nitrofenóxi)piriciin-3-il)prop-2inilcarbamato de terobutila.
O composto do título foi preparado de propargilamina N-Boc (Composto A de Exemplo 42) e 4-(2-flúor-4-nitrofenóxi)-3-iodopiridin-2amina (Composto C de Exemplo 34) da mesma maneira como a Etapa A de Exemplo 42 em 59% Produção. 1H RMN (DMSO-d6) δ 8,35 (dd, 1H, J = 10,7,
2,5 Hz), 8,12 (d, 1H, J= 9,2 Hz), 7,88 (d, 1H, 5,6 Hz), 7,39 (t, 1H, J=8,6
Hz), 7,31 (t, 1H, J = 5,1 Hz), 6,53 (s, 2H), 6,15 (d, 1H, J = 5,6 Hz), 3,92 (d, 2H, J = 5,6 Hz), 1,36 (s, 9H); MS(ESE): m/z 403,34 (M+H)*.
NHBoc 1 T

B) 3-(2-amino-4-(4-amino-2-fluorofenóxi)piridin-3-il)prop-2inilcarbamato de terc-butila.
O composto do título foi preparado de 3-(2-amíno-4-(2flúor-4nitrofenóxi)piridin-3-il)prop-2-inilcarbamato de terc-butila em Produção quantitativa da mesma maneira como a Etapa C de Exemplo 11, MS(ESI*): m/z 373,35 (M+H)*.
244


C) 3-(2-amino-4-(2-flyor-4-(1-(4-fluorofenil)-2-oxo-1,2-diiciropiridina-3-carboxamido)fenóxi)piridin-3-il)prop-2-inilcarbamato de tercbutila.
O composto do título foi preparado de 3-(2-amino-4-(4-amino-2fluorofenóxi)piridin-3-il)prop-2-ínilcarbamato de terc-butila e ácido 1-(4-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxílico (Composto B de Exemplo 101) da mesma· maneira como o Exemplo 62 em 48% Produção. Ή RMN (DMSO-d6) δ 12,07 (s, 1H), 8,57 (dd, 1H, J = 7,1, 2,0 Hz), 8,12 (dd, 1H, J = 6,6, 2,0 Hz), 8,00 - 7,93 (m, 1H), 7,75 (d, 1H, J = 6,1 Hz), 7,61 (d, 2H, J = 5,1 Hz), 7,59 (d, 1H, J = 4,6 Hz), 7,48-7,37 (m, 4 H), 7,30 - 7,25 (m, 1 H), 6,72 (t, 1H, J = 7,1 Hz), 6,37 (br s, 1H), 5,80 (d, 1H, J = 6,1 Hz), 4,00 (d, 2H, J = 5,1 Hz), 1,38 (s, 9H); MS(ESI*): m/z 588,26 (M+H)*.
D) M»(4-(2-Amino-3’(3-aminoprop-1-inil)piridin-4-il0xi)-3fluorofenil)-1-(4-fluorofenil)-2-oxo-1 ^-diidropiridina-S-carboxamida, sal de dicloridrato.
O composto do título foi-preparado de 3-(2-amino-4-(2-flúor-4-(1(4-fiuorofenil)-2-oxo-1,2-diidropiridina-3-carboxamido)fenóxi)piridin-3-il) prop-
2-inilcarbamato de terc-butila em 80% de Produção da mesma maneira como a Etapa E de Exemplo 36,1H RMN (DMSO-cO δ 12,13 (s, 1H), 8,57 (dd, 1H, J = 71, 2,0 Hz), 8,51 (s, 2 H), 8,14 (dd, 1H, J ~ 6,6, 2,0 Hz), 8,11 - 8,02 (m, 1H), 7,98 - 7,90 (m, 1H), 7,60 (dd, 2H, J = 9,2, 5,1 Hz), 7,53 (d, 1H, J =
9,2 Hz), 7,46 - 7,37 (m, 3H), 6,76 - 6,71 (m, 1H), 6,20 (d, 1H, J = 6,6 Hz), 4,11-4,04 (m, 2H); MS(ESf): m/z 488,16 (M+H)*.
245
Exemplo 138

1-(4-(2-Amino-3-(3-aminoprop-1-iníl)pirldin-4-ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de dicloridrato.

A) 3-(2-amino-4-(2-flúor-4-(3-(2-(4-fluorofenil)acetil)ureído) fenóxi)piridin-3-il)prop-2-inilcarbamato de terc-butila.
O composto do título foi preparado de 3 -(2-amino-4-(4-amino-2fluorofenóxi)piridin-3-il)prop-2-inilcarbamato de terc-butila em 38% de Produção da mesma maneira como a Etapa E de Exemplo 11,1H RMN (DMSO§ 11,02 (s, 1H), 10,55 (s, 1H), 7,80-7,73-7,69 (m, 2H), 7,38-7,29 (m, 3H), 7,33 (d, 1H, J = 5,1 Hz), 7,30-7,21 (m, 1H), 7,21-7,13 (m, 2H), 6,37 (br s, 1H), 5,76 (d, 1H, J = 6,1 Hz), 4,00 (s, 2H), 3,73 (s, 2H), 1,38 (s, 0H); MS(ESF): m/z 552,24 (M+H)*.
B) 1 -(4-(2-Ammo-3-(3-amlfioprop-1-lnll)pirldin-4-ilóxi)-3fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de dicloridrato
O composto do título foi preparado de 3-(2-amíno-4-(2-flúor-4-(3(2-(4-fluorofenil)acetil)urefdo)fenóxi)piridin-3-il)prop-2-inilcarbamato de torebutila em 65% de Produção da mesma maneira como a Etapa E de Exemplo 11JH RMN (DMSO-dg) δ 11,05 (s, 1H), 10,61 (s, 1H), 8,48 (br s, 3H), 7,90 (d, 1H, J = 6,6 Hz), 7,84-7,76 (m, 2H), 7,45-7,39 (m, 1H), 7,39-7,32 (m, 3H), 7,20-7,13 (m, 2H), 6,11 (d, 1H, J -6,1 Hz), 4,09-4,03 (ra, 2H), 3,74 (s, 2H); MS(ESF):m^ 452,12 (M+H)*.
246
Exemplo 139
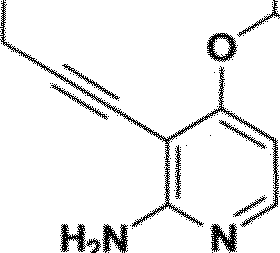

1-(4-(2-Amlno-3-(3-hidroxiprop-1-mil)piridin-4-ilóxi)-3-fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de cloridrato.
OH

A) 3-(2-Amino-4-(2-flúor-4-nitrofenóxi)piridin-3-il)prop-2-in5 l-ol
O composto do título foi preparado de álcool de propargila (Aldrich) e 4-(2-flúor-4-nitrofenóxí)-3-iodopiridin-2-amina (Composto C de Exemplo 34) em 46% da mesma maneira como a Etapa A de Exemplo 36,1H
RMN (DMSO-O δ 8,40-8,34 (m, 1H), 8,13 (d, 1H, J = 8,6 Hz), 7,88 (d, 1H, J 10 = 5,6 Hz), 7,42 (t, 1H, J = 8,6 Hz), 6,49 (s, 2H), 6,14 (d, 1H, J = 6,1 Hz),
5,26-5,20 (m, 1H), 4,26 (d, 2H, J = 6,1 Hz); MS(ESF): m/z 304,23 (M+H)\

B) 3-(2-Amino-4«(4-amlno-2-fluorofenóxi)plridin-3-il)prop-2in-1-ol.
O composto do título foi preparado de 3-(2-amino-4-(2-flúor-415 nitrofenóxi)piridin-3-il)prop-2-in-1-ol em 65% de Produção. MS(ESr): m/z 274,21 (M+H)*.
C) 1-(4-(2-Amino-3-(3-hidroxlprop-1-inil)piridin-4-ilóxi)-3fluorofenil)-3-(2-(4-fluorofenil)aceti!)uréia, sal de cloridrato.
O composto do título foi preparado de 3-(2-amino-4-(4-amino-2 fluorofenóxi)piridin-3-il)prop-2-in-1 -ol em 48% de Produção. 1H RMN (DMSOd6) δ 11,05 (s, 1H), 10,60 (s, 1H), 7,86 (d, 1H, J = 7,1 Hz), 7,83-7,77 (m, 1H), 7,44 - 7,37 (m, 2H), 7,37 - 7,32 (m, 3 H), 7,20 - 7.13 (m, 2H), 6,14 (d, J =
6,6 Hz, 1H), 4,37 (s, 2H), 3,74 (s, 2H); MS(ESI+): m/z 453,28 (M+H)+. Exemplo 140

A/-(4-(2-Amino-3-(3-(dimetilamino)prop-1-inil)piridin-4-ilóxi)-
3-fluorofenil)-1-(4-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxamida, sal de dicloridrato.

piridin-2-amina.
O composto do título foi preparado de 1 -dimetilamin-2-propina (Aldrich) e 4-(2-flúor-4-nitrofenóxi)-3-íodopiridÍn-2-amina (Composto C de Exemplo 34) em 64% de Produção da mesma maneira como a Etapa A de Exemplo 42, Ή RMN (DMSO-d6) δ 8,36 (dd, 1H, J = 10,7, 2,5 Hz), 8,11 (d, 1H, J = 8,6 Hz), 7,93 (d, 1H, J = 5,6 Hz), 7,30 (t, 1H, J = 8,6 Hz), 6,43 (br s, 2H), 6.28 (d, 1H, J = 6,1 Hz), 3,41 (s, 2H), 2,05 (s, 6H); MS(ESI‘): m/z 331, 28 (M+H)7

B) 4-(4-Amino-2-fluorofenóxi)-3-(3-(dimetilamino)prop-1inil)piridin-2-amina
248
O composto do título foi preparado de 3-(3-(dimetilamino)prop-1inil)-4-(2-flúor-4-nitrofenóxi)piridin-2-amina em 77% de Produção da mesma maneira como a Etapa C de Exemplo 11, MS(ESF): m/z 301, 30 (M+H)+.
C) N-(4-(2-Amino-3-(3-(dimetilamino)prop-1-inil)piridin-4ílóxi)-3-fluorofenil)-1-(4-fluorofenil)-2-oxo-1,2-diidropiridina-3-carboxamida, sal de dícloridrato.
O composto do título foi preparado de 4-(4-amino-2-fluorofenóxi)-3-(3-(dimetilamino)prop-1-inil)piridin-2-amina e ácido 1-(4-fluorofeníl)-
2-oxo-1,2-diidropiridina-3-carboxílico (Composto B de Exemplo 101) da mesma maneira como o Exemplo 62 em 71% de Produção. Ή RMN (DMSO-dg) δ 12,14 (s, 1H), 11,39 (s, 1 H), 8,60-8,53 (m, 1H), 8,33 (s, 1H), 8,14 (dd, 1H, J = 6,6, 2,2 Hz), 8,05 (dd, 1H, J=12,7, 2,2 Hz), 7,98 (d, 1H, J=7,0 Hz), 7,61 (d, 1H, J == 5,3 Hz), 7,58 (d, 1H, J = 4,8 Hz), 7,56-7,50 (m, 1H), 7,37-7,47 (m, 3H), 6,76-6,69 (m, 1H), 6,30 (d, 1H, J = 7,0 Hz), 4,39 (s, 2H), 2,84 (s, 6H); MS(ESF): m/z 480,28 (M+H)*.
Exemplo 141 h2n tr
-(4-(2-Amino-3-(3-(dimetilamino)prop-1 -inil)piridin-4-ilóxi)-3fluorofenil)-3-(2-(4-fluorofenil)acetil)uréia, sal de dicloridrato.
O composto do título foi preparado de 4-(4-amino-2-fluorofenóxi)-3-(3-(dimetilamÍno)prop-1-inil)piridin~2-arnina de uma maneira similar com a Etapa E de Exemplo 11 em 70% de Produção. Ή RMN (DMSO-d6) δ 11,46 (br s, 1H), 11,07 (s, 1H), 10,64 (s, 1H), 8,37 br (s, 1H), 7,98 (t, 1H, J = 7,5 Hz), 7,81 (d, 1H, J = 13,6 Hz), 7,43-7,32 (m, 4H), 7,20-7,09 (m, 2H), 6,27 (d, 1H, J = 7,0 Hz). 4,38 (s, 2H), 3,75 (s, 2H), 2,84 (s, 6H); MS(ESF): m/z 516,31 (M+H)*.
249
Exemplo 142

1-(4-(2-Aminopiridin-4-ilóxi)feml)-3-(2-(4-fluorofenil)acetil) uréia, sal de cloridrato.
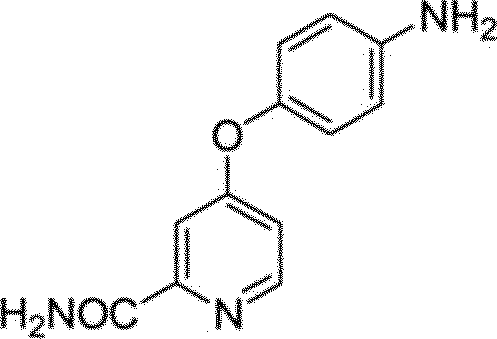
A) 4-(4-Amlnofenóxl)picolinamida
O composto do título foi preparado de uma maneira similar descrita na Etapa B’ de Exemplo 24, partindo de 4-aminofenol. Produção: 85%.
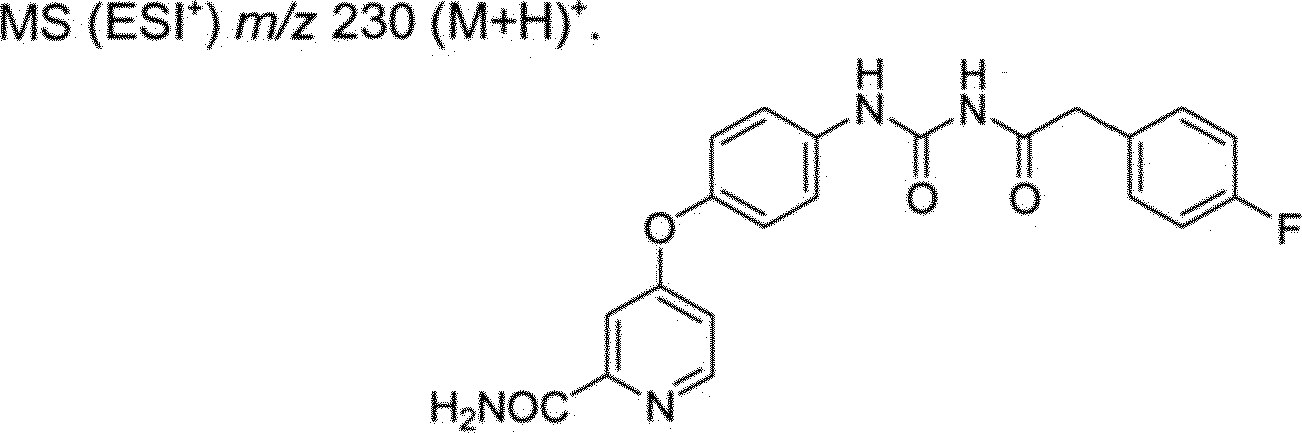
B) 1-(4-(2-Carbamoilpiridin-4-ilóxi)fenil)-3-(2-(4-fluorofenil) acetil)uréia.
© composto do título foi preparado de uma maneira descrita na
Etapa C’ de Exemplo 24, Produção: 95%. MS (ESF) m/z 409 (M+H)*.
C) 1>(4«(2-Aminopiridin-4-*ilóxi)fenil)-3-(2«(4-fluoro1enil) acetil) uréia, sal de cloridrato.
O composto do título foi preparado de uma maneira similar des15 crita na Etapa D’ de Exemplo 24, Produção: 58%. 1H RMN (DMSO-d6) δ
12,86 (s, 1H), 10,97 (s, 1H), 10,51 (s, 1H), 7,91 (d, 1H, 6,0 Hz), 7,67 (d,
3H, J=9,0 Hz), 7,34 (dd, 2H, J = 9,0, 5,0 Hz), 7,21 (d, 2H, J = 9,0 Hz), 7,16 (t, 2H, J = 9,0 Hz), 6,63 (dd, 1H, J 9,5, 2,0 Hz), 6,05 (d, 1H, J = 2,0 Hz),
3,73 (s, 2H); MS (ESF) m/z 381 (M+H)*.
250
Exemplo 143

1-(4-(2-Aminopiridin-4-ilóxi)-3-fIuorofenil)-3-(2-(3-hidroxifenil) aeetiljuréia.

OBn
A) Ácido 2-(3-(Benzilóxi)fenM)acético.
A uma solução de ácido 3-hidroxifeniíacético (Acros, 3,04 g, 20 mmoles) em 20 mL de DMF foram adicionados K^GOg (6,90 g, 50 mmoles) e brometo de benzia (4,75 mL, 40 mmoles) em temperatura ambiente. A mistura de reação foi agitada durante 24 horas. A suspensão foi filtrada e lavada com éter de dietila. O filtrado foi em seguida lavado com salmoura e se cado sobre filtração de MgSO4, seguido por concentração, fornece 2-(3(benzilóxí)fenil)acetato de benzila bruto que foi empregado na Etapa seguinte. MS (ESF)/7^355 (M+Na)*.
O 2-(3-(benzilóxi)fenil)acetato de benzila bruto foi dissolvido em uma mistura de MeOH (20 mL) e THF (50 mL). A esta solução foram adicio nados 40 mL de 1N de NaOH (40 mmoles). A mistura de reação foi agitada em temperatura ambiente durante 2 horas. O solvente orgânico foi removido em vácuo. A solução aquosa restante foi extraída com éter de dietila (2 x 50 mL). A solução aquosa foi em seguida acidificada com 1N de HCI (50 mL) e o composto do título foi precipitado O sólido foi coletado por ffltraçâo (4,35 g, 90% de duas etapas). MS(ESF) m/z 265 (M+Naf.

OBn
B) Isocianato de 2-(3-(Benzilóxi)fenil)acetila.
A uma solução de ácido 2-(3-(benzílóxi)fenil)acético (484 mg,
251
2,0 mmoles) em DOM (10 mL) em temperatura ambiente foi adicionado 1 gota de DMF e cloreto de tionila (0,30 mL, 4 mmoles). A mistura de reação foi agitada em temperatura ambiente durante 1 hora e em seguida a 50°C durante 0,5 hora. A mistura foi resfriada e o solvente foi removido em vácuo.
O resíduo fbi dissolvido em 5 mL de tolueno e AgOGN (600 mg, 4,0 mmoles) foi adicionado. A suspensão foi agitada durante 0,5 hora e ela foi filtrada para fornecer uma solução de isocianato de 2-(3-(benzilóxi)fenil)acetila em tolueno (0,40 M).

C) 1 -(2-(3-(Benzilóxi)fenil)acetil)-3-(4“(2-carbamoilpiridin-4iíóxi)“3-fluorofenil)uréia.
O composto do título foi preparado de uma maneira similar descrita na Etapa C’ de Exemplo 24, empregando-se a solução de Etapa B deste Exemplo. Produção: 63%. MS(ESI*) m/z 515 (M+H)*.

D) 1-(4-(2-Aminopiridin-4-ílóxi)-3-fluorofenil)-3-(2-(3-(benzilóxi)feml)acetil)uréia.
O composto do título foi preparado de uma maneira similar descrita na Etapa D’ de Exemplo 24, Produção: 61%. MS(ESI*) m/z 487 (M+H)*.
E) 1-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenil)-3-(2-(3-hidroxifenil)acetÍI)uréia.
A uma solução de 1-(4-(2-aminopindin-4-ilóxi)-3-fluorofenil)-3-(2(3-(benzitóxi)fenil)acetíl)uréia (150 mg, 0,31 mmol) em uma mistura de 5 mL de EtOAc e 3 mL de MeOH foi adicionado 10% de Pd/G (200 mg). A suspensão foi agitada sob atmosfera de H2 durante 1 hora. A filtração, seguida por concentração, fornece o composto do título (77 mg, 63%). 1H RMN (DMSOO δ 10,95 (s, 1H), 10,54 (s, 1H), 9,35 (s, 1H), 7,74 (d, 1H, J = 6,0 Hz), 7,68 (dd, 1H, J = 13,0, 2,0 Hz), 7,30 (dd, 1H, J = 9,0, 1,1 Hz), 7,23 (t, 1H, J = 9,0 Hz), 7,07 (t, 1H, J = 8,0 Hz), 6,69 (s, 1H), 6,68 (d, 1H, J = 7,0 Hz), 6,62 (dd, 1H, J = 7,0, 2,0 Hz), 6,10 (dd, 1H, J = 6,0, 2,0 Hz), 5,90 (s, 2H), 5,73 (d, 1H, J = 2,0 Hz), 3,27 (s, 2H); MS(ESF) m/z 397 (M+H)+.
Exemplo 144

3-(4-(2-Aminopiridin-4-ilõxi)-3«fluorofenil)-1-(2-(4-flyorofenil) acetil)-1-metiluréia, sal de cloridrato.
o TB
XX- p
A) 2-(4-Fluorofenil)-/V-metilacetamida.
A uma solução de metilamina em THF (2,0 M, 5 mL, 10 mmoles) foi adicionado cloreto de 4-fluorofenilacetila (518 mg, 3,0 mmoles) a -78°C. A mistura de reação foi agitada de -78°C para temperatura ambiente durante 1 hora. A solução foi diluída com H2O e extraída com EtOAc. A camada orgânica foi lavada com salmoura e secada sobre MgSO4 Filtração, seguida por concentração, fornece o composto do título (490 mg, 98%). MS(ESF) m/z 168 (M+HB
H


B) 3-(4-(2-Carbamoilpiridín-4-ilóxi)-3-fluorofenil)-1-(2-(4fluorofenil)acetil)-1-metiluréia.
A uma solução de 2-(4-fluorofenil)-A/-metilacetamida (89 mg,
253
0,34 mL, 0,55 mmol). A solução foi agitada a -78°C durante 5 minutos e em seguida 20% de fosgênio em toluene (1,9 M, 0,29 mL, 0,55 mmol) foi introduzido rapidamente. Após 2 minutos, 4-(4-amino-2-fluorofenóxi)picolinamida (Composto B’ de Exemplo 24, 100 mg, 0,40 mmol) foi adicionado, seguido pela adição de DMF (2 mL) e DIEA (0,4 mL). A mistura de reação foi agitada em temperatura ambiente durante 1 hora e extinguida com H2O. A solução foi em seguida extraída com EtOAc e a camada orgânica foi lavada com salmoura, secada sobre MgSO4. Após a filtração e concentração, o resíduo foi purificado por cromatografia de sílica-gel para fornecer o composto do título (77 mg, 33%). MS(ESF) m^441 (M+Hf.
G) 3-(4-(2«Aminopiridin-4-ilóxi)-3-fIuorofeníl)-1-(2-(4-fluorofenil)acetllH1-metiuréia, sal de cloridrato.
O composto do título foi preparado de uma maneira similar descrita na Etapa D’ de Exemplo 24, Produção: 24%. 1H RMN (DMSO-de) δ . 13,20 (s, 1H), 11,17 (s, 1H), 7,90 (d, 1H, J = 7,0 Hz), 7,78 - 7,74 (m, 3H),
7,39 - 7,34 (m, 2H), 7,23-7,21 (m, 2H), 7,10 - 7,05 (m,2H), 6,63 (dd, 1H, J = 7,0, 2,0 Hz), 6,08 (d, 1H, J = 2,0 Hz), 4,00 (s, 2H), 3,24 (s, 3H); MS (ESf) mZz413(M+H)L
Exemplo 145 (Ri-Af’-CZ-Amino-Z-oxo-l •toniletil)-A^-(4-(2-aminopiridin-4ilóxi)-2,5-difluorofenil)malonamida, sal de ácido trifluoroacético.
A) Etil 3-(4-(2-aminopiridin-4-ilóxi)-2,5-dlfluorofenilamino)-3oxopropanoato
254
3-(4-(2-carbamoilpiridin-4-ilóxi)-2,5-difluorofeniiam!no)-3-oxopro- panoato de etila (Composto A de Exemplo 116, 0,73 g, 1,9 mmol) foi convertido no composto do título (0,24 g, 35%) de uma maneira similar a uma preparação de Composto E de Exemplo 112, MS (ESI*) m/z 352 (M+H)*.

B) ácido 3-(4-(2-Aminopiridin-4-ilóxi)-2,5-difluorofenilarnino)-3-oxopropanóico.
3-(4-(2-aminopiridin-4-ilóxi)-2,5-difluorofenilamino)-3-oxopropanoato de etila (0,24 g, 0,68 mmol) foi convertido no composto do título (0,039 mg, 18%) de uma maneira similar a uma preparação de Composto B 10 de Exemplo 116, HRMS (ESI*), calculada: 324,0796 (M+H)*, encontrado:
324,0795 (M+H)*.

C) sal de ácido trifluoroacético (/^-^-(Z-Amino-Z-oxo-lfeniletil)-N3-(4-(2-aminopindin-4-iióxi)-2.5-difiuorofenil)malonamida.
Ácido 3-(4-(2-AminopÍridin-4-ilóxi)-2,5-difluorofenilamino)-3-oxo15 propanóico (0,039 g, 0,12 mmol) foi acoplado com cloridrato de (R)-2-amino-
2-fenilacetamida (Bachem, 0,023 g, 0,12 mmol), de uma maneira similar ô preparação de Exemplo 103, para fornecer o composto do título (0,0048 g, 7%). 1H RMN (DMSO-d§) δ 10,33 (s, 1H), 8,77 (d, 1H, J = 7,8 Hz), 8,10 -
8,19 (m, 1H), 7,90 (d, 1H, J = 7,2 Hz), 7,60 - 7,75 (m, 4H), 7,17 - 7,39 (m, 20 6H), 6,66 - 6,69 (m, 1H), 6,11 (s, 1H), 5,35 (d, 1H, J = 7,8 Hz); HRMS(ESI*), calculada: 456,1483 (M+H)*, encontrado: 456,1487 (M+HJ*.
V
255
Exemplo 146


NHf
N-(4-(2-Aminopiridin-4-ilóxi)fenil)-2-oxo-1-fenil-1,2-diidropiridina-3-carboxamida, sal de cloridrato.


CONH2
A) 4-(4-(2-Oxo-1-fenil-1,2-diidropiridina-3-carboxamido)fe5 nóxi) picolinamida.
4-(4-Aminofenóxi)picolinamida (Composto A de Exemplo 142,
0,030 g, 0,13 mmol) foi acoplado com Composto C de Exemplo 57 (0,028 g,
0,13 mmol), de uma maneira similar a uma preparação de Composto A de Exemplo 115, para fornecer o composto do título (0,057 g, 100%) que foi empregado sem outra purificação. MS(ESI+) m/z 427 (M+H)\


nh2
B) /V-(4-(2-Aminoplridin-4-ilóxi)fenil)-2-oxo-1 -fenil-1,2-diidropiridina-3-carboxamida, sal de cloridrato.
4-(4-(2-Oxo-1-fenil-1,2-diidropiridina-3-carboxamido)fenóxi)picolinamida (0,055, 0,13 mmol) foi convertido no composto do título (0,0093 g,
16%) de uma maneira similar a uma preparação de Composto E de Exemplo
112, 1H RMN (CD3OD) δ 12,10 (s, 1H), 8,59 - 8,61 (m, 1H), 7,88 - 7,90 (m,
1H), 7,69 - 7,76 (m, 3H), 7,39 - 7,53 (m, 5H), 7,10-7,13 (m, 2H), 6,66 (t, 1H,
256
J 6,9 Hz), 6,53 - 6,55 (m, 1H), 6,07-6,08 (m, 1H), 4,75 (br s, 2H);
HRMS(ESF), calculada.· 399,1457 (M+H)*, encontrado: 399,1453 (M+H)*.
Exemplo 147

Af-(4-(2-Aminopiridin-4-ilóxi)-3-fluorofenil)-1 -benzil-2-oxo-1,2diidropiridina-3-carboxamidaf sal de cloridrato.

A) 1-benzil-2-oxo-1,2-diidropiridina-3-carboxilato de metila
Uma mistura heterogênea de 2-oxo-2H-piran-3-carboxilato de metila (Aldrich, 2,0 g, 13 mmoles, 1,0 eq.) e 4-fIuorobenzilamina (1,5 mL, 13 mmoles, 1,0 eq.) em DMF (10 ml) foram agitadas em temperatura ambiente durante 3 horas. A mistura de reação foi tratada com EDCI (3,4 g, 18 mmoles, 1,4 eq) e DMAP (0,11 g, 9,91 mmoles, 0,07 eq.) em temperatura ambiente e a solução resultante foi agitada durante 12 horas. A mistura de reação foi extinguida com 1N de HCl aquoso e a solução foi extraída com acetato de etila (4 x 50 mL). Os extratos orgânicos combinados foram lavados com LiCI aquoso a 10% (3 x 70 mL), secados (Na2SO4), filtrados e o filtrado concentrado em vácuo para fornecer o produto (2,5 g, 73%) como um sólido, que foi empregado sem outra purificação. 1H RMN (DMSO-d6) δ 8,17 8,20 (m, 1H), 8,03 - 8,05 (m, 1H), 7,38 - 7,46 (m, 2H), 7,16 - 7,22 (m, 2H), 6,37 (dd, 1H, d = 6,94 Hz), 5,13 (s, 2H), 3,73 (s, 3H); HRMS (ESF), calculada: 262,0879, encontrado: 262,0885,
257
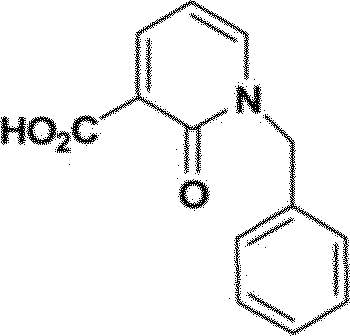
B) Ácido 1-Benzll-2-oxo-1,2-diidropiridlna-3-carboxílico.
Uma solução de 1-benzil-2-oxo-1,2-düdropiridina-3-carboxilato de metila (2,4 g, 9,2 mmoles, 1,0 eq.) em MeOH (25 mL) foi tratada com 5N de hidróxido de sódio aquoso (4,6 mL, 24 mmoles, 2,6 eq.) em temperatura ambiente e a mistura de reação foi agitada durante 15 horas. A mistura de reação foi em seguida concentrada em vácuo, diluída com água e a solução foi extraída com acetato de etila, descartando a fração orgânica. A fração aquosa foi resfriada a 0°C e foi acidificada com HCI concentrado. O sólido resultante foi filtrado, lavado com água e o sólido secado em vácuo para fornecer o produto (1,6 g, 70%), que foi empregado sem outra purificação.. 1H RMN (DMSO-dg) 8 8,39-8,44 (m, 2H), 7,42-7,46 (m, 2H), 7,18-7,24 (m, 2H), 6,78 (dd, 1H J = 6,98 Hz), 5,31 (s, 2H); HRMS(ESL), calculado: 248,0723, encontrado: 248,0718, dd, 1H, J = 6,98 Hz), 5,31 (s, 2H); HRMS(ESF), Calculada: 248,0723, encontrado: 248,0718.

C) 4-(4-(1-Benzil-2-oxo-1,2-diidropiridina-3-carboxamido)-2fluorofenóxi)picolinamida.
Uma solução homogênea de ácido 1-benzil-2-oxo-1,2-diidropiridina-3-carboxílico (0,10 g, 0,41 mmol, 1,0 eq.), 4-(4-amino-2-fluorofenóxi)pícolinamida (0,10 g, 0,41 mmol, 1,0 eq.) e TBTU (0,17 g, 0,45 mmol, 1,1 eq.) em DMF (2 mL) foi tratada com DIPEA (0,18 mL, 1,0 mmol, 2,5 eq.) em temperatura ambiente e a mistura de reação foi agitada durante 12 horas. A mistura de reação foi extinguida com LiCI aquoso a 10% (15 mL) e a solução resultante foi extraída com acetato de etila (4 x 40 mL). Os extratos
258 orgânicos combinados foram lavados com LiCI aquoso a 10% (4 x 50 mL), secados (Na2SO4), filtrados e o filtrado concentrado em vácuo. O resíduo foi purificado por cromatografia de coluna instantânea (SiO2, eluindo com acetato de etila) para fornecer o produto (0,13 g, 67%) como um sólido. 1H RMN (DMSO-cg § 12,26 (s, 1H), 8,49-8,56 (m, 2H), 8,33-8,36 (m, 1H), 8,15 (br m, 1H), 8,03-8,07 (m, 1H), 7,74-7,75 (m, 1H), 7,51-7,54 (m, 1H), 7,41-7,46 (m, 4H), 7,20-7,24 (m, 3H), 6,71 (dd, 1H, J = 6,89 Hz), 5,32 (s, 2H) HRMS(ESF), calculada: 477,1374, encontrado: 477,1378,
D) /V-(4-(2-aminopíriciín-4-ilóxi)-3-fluorofenil)-1-benzil-2-oxo-
1,2-diidropiridina-3-carboxamida, sal de cloridrato.
Bis (trifluoroacetóxi) iodobenzeno (0,12 g, 0,28 mmol, 1,1 eq) foi adicionado a uma solução de 4-(4-(1-benzil-2-oxo-1,2-diidropiridina-3-carboxamido)-2-fluorofenóxi)picolinamida (0,12 g, 0,26 mmol, 1,0 eq), e água (0,01 mL, 0,51 mmol, 2,0 eq.) em DMF (1 mL) em temperatura ambiente. A piridína (0,065 mL, 0,77 mmol, 3,0 eq) foi adicionada à mistura homogênea e a mistura de reação foi agitada em temperatura ambiente durante 12 horas. A mistura de reação foi extinguida com IN de HCI aquoso (1 mL) e a solução resultante foi extraída com éter de dietila (3 x 5 mL), descartando a camada orgânica. A fração aquosa foi neutralizada com 1N de NaOH aquoso e a solução resultante foi extraída com 9/1 CHCL/MeOH (4x10 mL). Os extratos orgânicos combinados foram secados (Na2SO4), filtrada e o filtrado foi concentrada em vácuo. O resíduo foi purificado por cromatografia instantânea (SiO2, eluindo 0-3% MeOH em CHGI3) e as frações apropriadas foram concentradas em vácuo. A base livre foi dissolvida em THF, resfriada a 0°G e a solução homogênea tratada com 4N HCI anidro em dioxano. A mistura de reação foi aquecer para temperatura ambiente, concentrada em vácuo e o resíduo triturado com éter de dietila, descartando o filtrado. O sólido foi secado em vácuo para fornecer o composto do título (0,082 g, 66%) como um sal de HCI. 1H RMN (DMSO-<) § 13,66 (br s, 1H), 8,49 - 8,51 (m, 1H), 8,39 - 8,49 (m, 1H), 8,37 - 8,39 (m, 1H), 8,00 - 8,09 (m, 3H), 7,54-7,56 (m, 1H), 7,43 - 7,48 (m, 3H), 7,19-7,24 (m, 2H), 6,69 - 6,72 (m, 2H), 6,21 - 6,22 (m, 1H), 5,32 (s, 2H); HRMS(ESr), calculada.: 449,1425, encontrado:
259
440,1406,
Exemplo 148
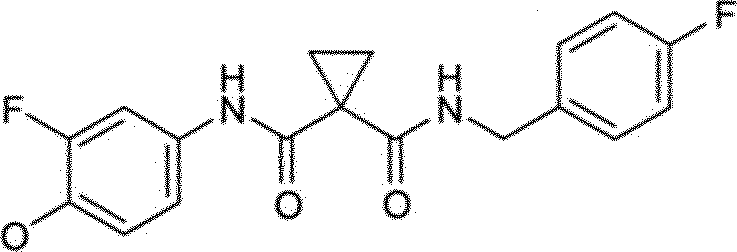

M-(4-Fluorobenzil)-1V-(4-(2-aminoplridin-4-ilóxi)-3-fluorofenil)cíclopropano1,1-dicarboxamida.

A) Ácido 1 -((4-Fluorobenzil)carbamoil)ciclopropanocarboxílico.
A uma solução de ácido 1,1-ciclopropanocarboxílico (Aldrich,
390 mg, 3,0 mmoles) em THF (5 mL) a 0°C foi adicionado trietilamina (0,42 mL, 3,0 mmoles). Após agitar durante 30 minutos a 0°C, o cloreto de tíonila (0,219 mL, 3,0 mmoles) foi adicionado a uma mistura de reação. A mistura foi agitada a 0®C durante 30 minutos adicionais e uma solução de 4fluorobenzilamina (Aldrich, 375 mg, 3,0 mmol) em THF (2 mL) foi adicionado. A mistura de reação foi agitada a 0°C durante 2 horas, diluída com acetato de etila (100 mL) e extraída com 1 N NaOH (10 mL). A fase aquosa foi acidificada com 1 N de HCI para pH 1-2, o sólido que formou-se foi coletado por filtração (343 mg, 48%). MS(ESF) m/z 238,24 (M + Hf.


B) W-(4-Fluoroberizll)-/V-(4-(2-carbamoi!piridin-4-ilóxi)-3fluorofenil)clclopropano-1,1 -dicarboxamida.
A uma solução de 4-(4-amino-2-fluorofenóxi)picolinamída (Com
260 posto B’ de Exemplo 24, 49 mg, 0,2 mmol) em DMF (2 mL) em temperatura ambiente foi adicionado ácido 1-((4-fluorobenzil)carbamoil) ciclopropanocarboxílico (47 mg, 0,2 mmol), HATU (Perseptive Biosystem, 114 mg, 0,3 mmol), e DIEA (0,2 mL, 1,1 mmol). A mistura de reação foi agitada em tem5 peratura ambiente durante 2 horas, e em seguida extinguida por adição de 4 mL de metanol A mistura de reação foi purificada por HPLC preparativa. As frações desejadas foram combinadas, neutralizadas com K2HPO4 aquosa, e concentrada em vácuo. O sólido que se formou foi coletado por filtração (29 mg, 31%). Ή RMN (DMSO-cg δ 10,78 (br s, 1H), 8,53 (d, 1H, J = 5,5 Hz),
8,47 (t, 1H, J= 5,5 Hz), 8,11 (s, 1H), 7,88 (dd, 1H, J = 13,2, 2,3 Hz), 7,70 (s,
1H), 7,47 (d, 1H, J = 9,2 Hz), 7,38 (t, 1H, J = 9,2 Hz), 7,34 - 7,29 (m, 3H),
7,22 (dd, 1H, J = 5,5, 2,8 Hz), 7,13 (t, 2H, J = 8,8 Hz), 4,30 (d, 2H, J = 5,5
Hz), 1,37 (d, 4H, J- 10,6 Hz); MS (ESF) m/z 467,12 (M + Hf.
C) /V-(4-Fluorobenzil)-/V-(4-(2-aminopiridin-4-ilóxi)-3-fluoro- fenil)ciclopropano-1,1-clicarboxamida.
A uma solução de /V-(4-fluorobenzil)-AH4-(2-carbarnoilpiridin-4ilóxi)-3-fluorofenil)ciclopropano-1,1-dicarboxamida (25 mg, 0,05 mmol) em
DMF (1 mL) em temperatura ambiente foi adicionado piridina (0,2 mL), água (0,1 mL), e (bis(trifluoroacetóxi)-iodo]benzeno (Aldrich, 34 mg, 0,08 mmol). A mistura de reação foi agitada em temperatura ambiente durante 2 horas, e em seguida extinguida por adição de 2 mL de metanol. A mistura de reação foi purificada por HPLC preparativa. As frações desejadas foram combinadas, neutralizadas com K2HPO4 aquosa, concentradas e extraídas com acetato de etila. Os extratos orgânicos foram secados sobre MgSO4 e concen25 trados em vácuo, O resíduo foi dissolvido em uma pequena quantidade de CH3CN/H2O e liofilizado até a secura para fornecer o composto do título (21 mg, 90%) como um sólido branco. Ή RMN (DMSO-oy δ 10,87 (br s, 1H), 8,44 (t, 1H, J = 6,0 Hz), 7,91 (d, 1H, U = 6,6 Hz), 7,88 (d,1 H, d = 13,2 Hz), 7,37 - 7,29 (ffi, 6H), 7,13 (t, 2H, J = 8,8 Hz), 4,29 (d, 2H, J = 6,1 Hz), 1,38 (d,
4H, J = 2,2 Hz); MS(ESF) mór 439,14 (M + H)*.
Claims (10)
- REIVINDICAÇÕES1. Composto, caracterizado pelo fato de que apresenta a fórmula (II):
 AII ou um enantiômero, ou sal farmaceuticamente aceitável do mesmo, em que:cada R2 é independentemente H ou halogênio;B é O;W e X são cada um independentemente CH;n é 0 a 4;R3 é H;R4 é piridinona, piridila ou piridil-N-óxido não substituído ou substituído com oxigênio, fenil, fluorofenil, Ci a C4 alquil, Ci a C6 alcoxi, C1 a C4 alquil substituído com fenil ou halogênio;A é:
AII ou um enantiômero, ou sal farmaceuticamente aceitável do mesmo, em que:cada R2 é independentemente H ou halogênio;B é O;W e X são cada um independentemente CH;n é 0 a 4;R3 é H;R4 é piridinona, piridila ou piridil-N-óxido não substituído ou substituído com oxigênio, fenil, fluorofenil, Ci a C4 alquil, Ci a C6 alcoxi, C1 a C4 alquil substituído com fenil ou halogênio;A é: em queR16, R17, e R18 são independentemente H, flúor, cloro, -NH2, N(CH3)2, C1 a C6 alquila ou fenila substituída com -CONH2, metila, aminoetila, hidroxietila, -CONHCH2CH2NHCH3 ou -CH2CONH2;R19 é H; eR32 é H ou C1 a C4 alquila.
em queR16, R17, e R18 são independentemente H, flúor, cloro, -NH2, N(CH3)2, C1 a C6 alquila ou fenila substituída com -CONH2, metila, aminoetila, hidroxietila, -CONHCH2CH2NHCH3 ou -CH2CONH2;R19 é H; eR32 é H ou C1 a C4 alquila. - 2. Composto de acordo com a reivindicação 1, caracterizado pelo fato de que R4 é piridinona não substituída ou substituída com oxigênio, fenila, fluorofenila, Ci a C4 alquila, Ci a C2 alcóxi, Ci a C4 alquila substituída com fenila ou halogênio.Petição 870200007188, de 16/01/2020, pág. 4/10
- 3. Composto de acordo com a reivindicação 1, caracterizado pelo fato de que R4 é piridil-N-óxido não substituído ou substituído com oxigênio, fenila, fluorofenila, Ci a C4 alquila, Ci a C2 alcóxi, Ci a C4 alquila substituída com fenila ou halogênio.
- 4. Composto, de acordo com a reivindicação 1, caracterizado pelo fato de que R4 é substituído por oxigênio, fenila, fluorofenila, Ci a C4 alquila, Ci a C2 alcóxi ou halogênio.
- 5. Composto, de acordo com a reivindicação 1, caracterizado pelo fato de que R16, R17, e R18 são independentemente H, F, Cl, -NH2, C1 a C6 alquila ou fenila substituída com -CONH2, metila, aminoetila, hidroxietila, CONHCH2CH2NHCH3 ou -CH2CONH2.
- 6. Composto, de acordo com a reivindicação 5, caracterizado pelo fato de que R16 ou R17 são independentemente -NH2, F, Cl ou Ci a C6 alquila.
- 7. Composto, de acordo com a reivindicação 1, caracterizado pelo fato de que um de R16, R17, e R18 é um grupo fenila substituído com CONH2, metila, aminoetila, hidroxietila, -CONHCH2CH2NHCH3 ou CH2CONH2.
- 8 Composto, de acordo com a reivindicação 5, caracterizado pelo fato de que um de R16 ou R17 é -NH2 ou -N(CH3)2.
- 9. Composição farmacêutica, caracterizada pelo fato de que contém um composto como definido em qualquer uma das reivindicações 1 a 8, ou enantiômero, ou sal farmaceuticamente aceitável do mesmo.
- 10. Uso do composto como definido em qualquer uma das reivindicações 1 a 8, ou enantiômero, ou sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de ser para preparação de uma composição farmacêutica para tratar e/ou prevenir êmese, vômito e/ou constipação.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US56484204P | 2004-04-23 | 2004-04-23 | |
| US63917804P | 2004-12-23 | 2004-12-23 | |
| US11/111,144 US7459562B2 (en) | 2004-04-23 | 2005-04-21 | Monocyclic heterocycles as kinase inhibitors |
| PCT/US2005/014120 WO2005117867A2 (en) | 2004-04-23 | 2005-04-22 | Monocyclic heterocycles as kinase inhibitors |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| BRPI0510177A BRPI0510177A (pt) | 2007-10-02 |
| BRPI0510177B1 true BRPI0510177B1 (pt) | 2020-04-07 |
| BRPI0510177B8 BRPI0510177B8 (pt) | 2021-05-25 |
Family
ID=35187908
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0510177A BRPI0510177B8 (pt) | 2004-04-23 | 2005-04-22 | composto, composição farmacêutica e uso do mesmo |
Country Status (21)
| Country | Link |
|---|---|
| US (3) | US7459562B2 (pt) |
| EP (1) | EP1737451B1 (pt) |
| JP (1) | JP4778959B2 (pt) |
| KR (1) | KR100918727B1 (pt) |
| AT (1) | ATE499097T1 (pt) |
| AU (1) | AU2005249382B2 (pt) |
| BR (1) | BRPI0510177B8 (pt) |
| CA (1) | CA2563831C (pt) |
| CY (1) | CY1111640T1 (pt) |
| DE (1) | DE602005026508D1 (pt) |
| GE (1) | GEP20104884B (pt) |
| HK (1) | HK1093159A1 (pt) |
| HR (1) | HRP20110156T1 (pt) |
| IL (1) | IL178675A (pt) |
| MX (1) | MXPA06012185A (pt) |
| NO (1) | NO339960B1 (pt) |
| NZ (1) | NZ550614A (pt) |
| PL (1) | PL1737451T3 (pt) |
| PT (1) | PT1737451E (pt) |
| SI (1) | SI1737451T1 (pt) |
| WO (1) | WO2005117867A2 (pt) |
Families Citing this family (121)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005082854A1 (ja) * | 2004-02-27 | 2005-09-09 | Eisai Co., Ltd. | 新規ピリジン誘導体およびピリミジン誘導体(1) |
| TW200538453A (en) * | 2004-04-26 | 2005-12-01 | Bristol Myers Squibb Co | Bicyclic heterocycles as kinase inhibitors |
| US7439246B2 (en) * | 2004-06-28 | 2008-10-21 | Bristol-Myers Squibb Company | Fused heterocyclic kinase inhibitors |
| US7432373B2 (en) * | 2004-06-28 | 2008-10-07 | Bristol-Meyers Squibb Company | Processes and intermediates useful for preparing fused heterocyclic kinase inhibitors |
| US7173031B2 (en) | 2004-06-28 | 2007-02-06 | Bristol-Myers Squibb Company | Pyrrolotriazine kinase inhibitors |
| TW200600513A (en) * | 2004-06-30 | 2006-01-01 | Bristol Myers Squibb Co | A method for preparing pyrrolotriazine compounds |
| WO2006010264A1 (en) | 2004-07-30 | 2006-02-02 | Methylgene, Inc. | Inhibitors of vegf receptor and hgf receptor signaling |
| US7504521B2 (en) * | 2004-08-05 | 2009-03-17 | Bristol-Myers Squibb Co. | Methods for the preparation of pyrrolotriazine compounds |
| US7148348B2 (en) | 2004-08-12 | 2006-12-12 | Bristol-Myers Squibb Company | Process for preparing pyrrolotriazine aniline compounds useful as kinase inhibitors |
| US7776869B2 (en) * | 2004-10-18 | 2010-08-17 | Amgen Inc. | Heteroaryl-substituted alkyne compounds and method of use |
| US7470693B2 (en) * | 2005-04-21 | 2008-12-30 | Bristol-Myers Squibb Company | Oxalamide derivatives as kinase inhibitors |
| JO2787B1 (en) * | 2005-04-27 | 2014-03-15 | امجين إنك, | Alternative amide derivatives and methods of use |
| ES2473341T3 (es) | 2005-05-20 | 2014-07-04 | Methylgene Inc | Inhibidores de la se�alizaci�n del receptor del VEGF y del receptor del HGF |
| MX2007014616A (es) | 2005-05-20 | 2009-08-12 | Methylgene Inc | Inhibidores de señalizacion del receptor del factor a de crecimiento endotelial vascular y del receptor del factor de crecimiento de hepatocitos. |
| PT1889836E (pt) * | 2005-08-24 | 2013-08-28 | Eisai R&D Man Co Ltd | Novo derivado de piridina e derivado de pirimidina (3) |
| CN101522687A (zh) * | 2006-01-30 | 2009-09-02 | 阿雷生物药品公司 | 用于癌症治疗的杂二环噻吩化合物 |
| EP2001880A2 (en) | 2006-03-07 | 2008-12-17 | Array Biopharma, Inc. | Heterobicyclic pyrazole compounds and methods of use |
| AR059898A1 (es) * | 2006-03-15 | 2008-05-07 | Janssen Pharmaceutica Nv | Derivados de 3-ciano-piridona 1,4-disustituida y su uso como moduladores alostericos de los receptores mglur2 |
| EP2023926B1 (en) * | 2006-05-19 | 2011-10-12 | Bayer Pharma Aktiengesellschaft | Pyridonecarboxamide derivatives useful in treating hyper-proliferative and angiogenesis disorders |
| US20080004273A1 (en) * | 2006-05-30 | 2008-01-03 | Stephane Raeppel | Inhibitors of protein tyrosine kinase activity |
| CN101528702A (zh) * | 2006-06-08 | 2009-09-09 | 阿雷生物药品公司 | 喹啉化合物和使用方法 |
| US8778977B2 (en) | 2006-06-30 | 2014-07-15 | Sunesis Pharmaceuticals, Inc. | Pyridinonyl PDK1 inhibitors |
| MX2009000661A (es) * | 2006-07-19 | 2009-03-27 | Univ Georgia Res Found | Piridinon-diceto-acidos: inhibidores de replicacion de vih en terapia de combinacion. |
| ES2375284T3 (es) * | 2006-08-23 | 2012-02-28 | Eisai R&D Management Co., Ltd. | Sal de un derivado de fenoxipiridina, o cristal de la misma, y procedimiento de producción de la misma. |
| US7790885B2 (en) * | 2006-08-31 | 2010-09-07 | Eisai R&D Management Co., Ltd. | Process for preparing phenoxypyridine derivatives |
| US8148391B2 (en) | 2006-10-23 | 2012-04-03 | Cephalon, Inc. | Fused bicyclic derivatives of 2,4-diaminopyrimidine as ALK and c-Met inhibitors |
| PT2089364E (pt) * | 2006-11-08 | 2013-08-26 | Bristol Myers Squibb Co | Compostos de piridinona |
| ES2392156T3 (es) * | 2006-12-20 | 2012-12-05 | Amgen Inc. | Heterociclos sustituidos y métodos de uso |
| WO2008102870A1 (ja) * | 2007-02-23 | 2008-08-28 | Eisai R & D Management Co., Ltd. | Hgfr遺伝子増幅細胞株に優れた細胞増殖阻害効果および抗腫瘍効果を示すピリジン誘導体またはピリミジン誘導体 |
| TW200845978A (en) * | 2007-03-07 | 2008-12-01 | Janssen Pharmaceutica Nv | 3-cyano-4-(4-tetrahydropyran-phenyl)-pyridin-2-one derivatives |
| TW200900065A (en) | 2007-03-07 | 2009-01-01 | Janssen Pharmaceutica Nv | 3-cyano-4-(4-pyridinyloxy-phenyl)-pyridin-2-one derivatives |
| KR20100065191A (ko) | 2007-09-14 | 2010-06-15 | 오르토-맥닐-얀센 파마슈티칼스 인코포레이티드 | 1,3-이치환된 4-(아릴-x-페닐)-1h-피리딘-2-온 |
| CN101801951B (zh) * | 2007-09-14 | 2013-11-13 | 杨森制药有限公司 | 1’,3’-二取代的-4-苯基-3,4,5,6-四氢-2h,1’h-[1,4’]二吡啶-2’-酮 |
| TW200927731A (en) | 2007-09-14 | 2009-07-01 | Ortho Mcneil Janssen Pharm | 1,3-disubstituted-4-phenyl-1H-pyridin-2-ones |
| ES2637794T3 (es) * | 2007-11-14 | 2017-10-17 | Janssen Pharmaceuticals, Inc. | Derivados de imidazo[1,2-A]piridina y su uso como moduladores alostéricos positivos de receptores MGLUR2 |
| JP2009132660A (ja) * | 2007-11-30 | 2009-06-18 | Eisai R & D Management Co Ltd | 食道癌治療用組成物 |
| ES2531396T3 (es) * | 2008-01-23 | 2015-03-13 | Bristol Myers Squibb Co | Proceso para preparar compuestos de piridinona |
| DK2235002T3 (da) * | 2008-01-23 | 2013-03-11 | Bristol Myers Squibb Co | 4-pyridinonforbindelser og anvendelse heraf til behandling af cancer |
| JP2009203226A (ja) * | 2008-01-31 | 2009-09-10 | Eisai R & D Management Co Ltd | ピリジン誘導体およびピリミジン誘導体を含有するレセプターチロシンキナーゼ阻害剤 |
| WO2009104520A1 (ja) * | 2008-02-18 | 2009-08-27 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | フェノキシピリジン誘導体の製造方法(2) |
| EP2262815A4 (en) | 2008-03-05 | 2012-04-11 | Methylgene Inc | Inhibitors of protein intolerance activity |
| CN103333157A (zh) | 2008-09-02 | 2013-10-02 | 诺瓦提斯公司 | 作为激酶抑制剂的吡啶甲酰胺衍生物 |
| RU2510396C2 (ru) | 2008-09-02 | 2014-03-27 | Янссен Фармасьютикалз, Инк. | 3-азабицикло[3.1.0]гексильные производные в качестве модуляторов метаботропных глутаматных рецепторов |
| AU2009303602B2 (en) | 2008-10-14 | 2012-06-14 | Sunshine Lake Pharma Co., Ltd. | Compounds and methods of use |
| BRPI0920354A2 (pt) | 2008-10-16 | 2017-06-27 | Addex Pharmaceuticals Sa | derivados de indol e benzomorfolina como moduladores de receptores de glutamato metabotrópicos |
| BRPI0920765A2 (pt) * | 2008-10-29 | 2015-08-18 | Deciphera Pharmaceuticals Llc | Amidas de cilopropano e análogos que exibem atividades anticâncer e antiproliferativas |
| AU2009319387B2 (en) | 2008-11-28 | 2012-05-10 | Addex Pharma S.A. | Indole and benzoxazine derivatives as modulators of metabotropic glutamate receptors |
| EP2408300B1 (en) * | 2009-03-21 | 2016-05-11 | Sunshine Lake Pharma Co., Ltd. | Amino ester derivatives, salts thereof and methods of use |
| AR076332A1 (es) * | 2009-04-21 | 2011-06-01 | Boehringer Ingelheim Int | Derivados heterociclicos de 5-alquinil-piridinas, composiciones farmaceuticas que los comprenden y uso de los mismos para el tratamiento y/o prevencion del cancer, procesos inflamatorios, autoinmunes, y/o infecciones. |
| MY153913A (en) | 2009-05-12 | 2015-04-15 | Janssen Pharmaceuticals Inc | 7-aryl-1,2,4-triazolo[4,3-a]pyridine derivatives and their use as positive allosteric modulators of mglur2 receptors |
| RS53075B (en) | 2009-05-12 | 2014-04-30 | Janssen Pharmaceuticals Inc. | 1,2,4-TRIAZOLO [4,3-A] PYRIDINE DERIVATIVES AND THEIR USE IN THE TREATMENT OR PREVENTION OF NEUROLOGICAL AND PSYCHIATRIC DISORDERS |
| JP5707390B2 (ja) | 2009-05-12 | 2015-04-30 | ジャンセン ファーマシューティカルズ, インコーポレイテッド | 1,2,4−トリアゾロ[4,3−a]ピリジン誘導体およびmGluR2受容体の正のアロステリック調節因子としてのその使用 |
| EP2261211A1 (en) * | 2009-05-20 | 2010-12-15 | Université de Lille 2 Droit et Santé | 1,4-dihydropyridine derivatives and their uses |
| JP2013503170A (ja) * | 2009-08-28 | 2013-01-31 | グラクソスミスクライン・リミテッド・ライアビリティ・カンパニー | 化合物および方法 |
| CA2800569A1 (en) | 2010-04-29 | 2011-11-03 | Deciphera Pharmaceuticals, Llc | Cyclopropyl dicarboxamides and analogs exhibiting anti-cancer and anti-proliferative activities |
| EP2563773A1 (en) | 2010-04-29 | 2013-03-06 | Deciphera Pharmaceuticals, LLC | Pyridone amides and analogs exhibiting anti-cancer and anti-proliferative activites |
| US8911785B2 (en) | 2010-04-30 | 2014-12-16 | Bristol-Myers Squibb Company | Pharmaceutical compositions comprising N-(4-(2-amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl)-2-OXO-1,2-dihydropyridine-3-carboxamide |
| JP5960688B2 (ja) | 2010-05-17 | 2016-08-02 | インコゼン セラピューティクス プライベート リミテッド | プロテインキナーゼ調節物質としての新規3,5−二置換−3h−[1,2,3]トリアゾロ[4,5−b]ピリジン化合物 |
| CN102958523B (zh) | 2010-06-25 | 2014-11-19 | 卫材R&D管理有限公司 | 使用具有激酶抑制作用的组合的抗肿瘤剂 |
| US8664244B2 (en) | 2010-09-12 | 2014-03-04 | Advenchen Pharmaceuticals, LLC | Compounds as c-Met kinase inhibitors |
| EP2661435B1 (en) | 2010-11-08 | 2015-08-19 | Janssen Pharmaceuticals, Inc. | 1,2,4-TRIAZOLO[4,3-a]PYRIDINE DERIVATIVES AND THEIR USE AS POSITIVE ALLOSTERIC MODULATORS OF MGLUR2 RECEPTORS |
| EP2643320B1 (en) | 2010-11-08 | 2015-03-04 | Janssen Pharmaceuticals, Inc. | 1,2,4-TRIAZOLO[4,3-a]PYRIDINE DERIVATIVES AND THEIR USE AS POSITIVE ALLOSTERIC MODULATORS OF MGLUR2 RECEPTORS |
| EP2649069B1 (en) | 2010-11-08 | 2015-08-26 | Janssen Pharmaceuticals, Inc. | 1,2,4-TRIAZOLO[4,3-a]PYRIDINE DERIVATIVES AND THEIR USE AS POSITIVE ALLOSTERIC MODULATORS OF MGLUR2 RECEPTORS |
| US8466162B2 (en) * | 2011-01-26 | 2013-06-18 | Boehringer Ingelheim International Gmbh | 5-alkynyl-pyridines |
| TW201245176A (en) | 2011-01-26 | 2012-11-16 | Boehringer Ingelheim Int | New 5-alkynyl-pyridines |
| US8759530B2 (en) | 2011-03-29 | 2014-06-24 | Eisai R&D Management Co., Ltd. | Method for producing phenoxypyridine derivative |
| US8841301B2 (en) | 2011-09-26 | 2014-09-23 | Bristol-Myers Squibb Company | Selective NR2B antagonists |
| WO2013049119A1 (en) * | 2011-09-30 | 2013-04-04 | Bristol-Myers Squibb Company | Selective nr2b antagonists |
| KR20140072177A (ko) | 2011-10-06 | 2014-06-12 | 바이엘 인텔렉쳐 프로퍼티 게엠베하 | 헤테로사이클릴피리(미)디닐피라졸 |
| CA2851142A1 (en) | 2011-10-06 | 2013-04-11 | Bayer Intellectual Property Gmbh | Heterocyclylpyri (mi) dinylpyrazole as fungicidals |
| KR20140097402A (ko) * | 2011-11-22 | 2014-08-06 | 데시페라 파마슈티칼스, 엘엘씨. | 항암 및 항증식 활성을 나타내는 피리돈 아미드 및 유사체 |
| WO2013113781A1 (en) | 2012-02-03 | 2013-08-08 | Basf Se | Fungicidal pyrimidine compounds i |
| WO2013113716A1 (en) | 2012-02-03 | 2013-08-08 | Basf Se | Fungicidal pyrimidine compounds |
| WO2013113788A1 (en) | 2012-02-03 | 2013-08-08 | Basf Se | Fungicidal pyrimidine compounds |
| JP2015511940A (ja) | 2012-02-03 | 2015-04-23 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | 殺菌性ピリミジン化合物 |
| WO2013113719A1 (en) | 2012-02-03 | 2013-08-08 | Basf Se | Fungicidal pyrimidine compounds ii |
| EP2809659A1 (en) | 2012-02-03 | 2014-12-10 | Basf Se | Fungicidal pyrimidine compounds |
| WO2013113782A1 (en) | 2012-02-03 | 2013-08-08 | Basf Se | Fungicidal pyrimidine compounds |
| WO2013113776A1 (en) | 2012-02-03 | 2013-08-08 | Basf Se | Fungicidal pyrimidine compounds |
| WO2013113773A1 (en) | 2012-02-03 | 2013-08-08 | Basf Se | Fungicidal pyrimidine compounds |
| WO2013135672A1 (en) | 2012-03-13 | 2013-09-19 | Basf Se | Fungicidal pyrimidine compounds |
| US9462809B2 (en) | 2012-03-13 | 2016-10-11 | Basf Se | Fungicidal pyrimidine compounds |
| WO2013144737A2 (en) | 2012-03-30 | 2013-10-03 | Rhizen Pharmaceuticals Sa | Novel 3,5-disubstitued-3h-imidazo[4,5-b]pyridine and 3,5- disubstitued -3h-[1,2,3]triazolo[4,5-b] pyridine compounds as modulators of c-met protein kinases |
| US9173883B2 (en) | 2012-05-21 | 2015-11-03 | Novartis Ag | Ring-substituted N-pyridinyl amides as kinase inhibitors |
| EP2879677B1 (en) | 2012-07-28 | 2017-06-14 | Calitor Sciences, LLC | Substituted pyrazolone compounds and methods of use |
| US8975282B2 (en) | 2012-07-28 | 2015-03-10 | Sunshine Lake Pharma Co., Ltd. | Substituted pyrazolone compounds and methods of use |
| BR112015009004A8 (pt) | 2012-12-21 | 2021-07-20 | Eisai R&D Man Co Ltd | forma amorfa de derivado de quinolina e método de produção da mesma |
| MX368099B (es) | 2013-05-14 | 2019-09-19 | Eisai R&D Man Co Ltd | Biomarcadores para predecir y evaluar el grado de respuesta de sujetos con cancer de endometrio a compuestos de tipo lenvatinib. |
| JO3368B1 (ar) | 2013-06-04 | 2019-03-13 | Janssen Pharmaceutica Nv | مركبات 6، 7- ثاني هيدرو بيرازولو [5،1-a] بيرازين- 4 (5 يد)- اون واستخدامها بصفة منظمات تفارغية سلبية لمستقبلات ميجلور 2 |
| JO3367B1 (ar) | 2013-09-06 | 2019-03-13 | Janssen Pharmaceutica Nv | مركبات 2،1، 4- ثلاثي زولو [3،4-a] بيريدين واستخدامها بصفة منظمات تفارغية موجبة لمستقبلات ميجلور 2 |
| DK3431106T3 (da) | 2014-01-21 | 2021-03-15 | Janssen Pharmaceutica Nv | Kombinationer omfattende positive allosteriske modulatorer eller orthosteriske agonister af metabotrop glutamaterg subtype 2-receptor og anvendelse af disse |
| ES2860298T3 (es) | 2014-01-21 | 2021-10-04 | Janssen Pharmaceutica Nv | Combinaciones que comprenden moduladores alostéricos positivos del receptor glutamatérgico metabotrópico de subtipo 2 y su uso |
| HRP20221047T1 (hr) | 2014-08-28 | 2022-11-11 | Eisai R&D Management Co., Ltd. | Derivat kinolina visoke čistoće i postupak za njegovu proizvodnju |
| CN107427505A (zh) | 2015-02-25 | 2017-12-01 | 卫材R&D管理有限公司 | 用于抑制喹啉衍生物的苦味的方法 |
| WO2016140717A1 (en) | 2015-03-04 | 2016-09-09 | Merck Sharp & Dohme Corp. | Combination of a pd-1 antagonist and a vegfr/fgfr/ret tyrosine kinase inhibitor for treating cancer |
| WO2016159049A1 (ja) * | 2015-03-31 | 2016-10-06 | 武田薬品工業株式会社 | 単環式化合物 |
| US11369623B2 (en) | 2015-06-16 | 2022-06-28 | Prism Pharma Co., Ltd. | Anticancer combination of a CBP/catenin inhibitor and an immune checkpoint inhibitor |
| EP3319968A1 (en) | 2015-07-06 | 2018-05-16 | Rodin Therapeutics, Inc. | Heterobicyclic n-aminophenyl-amides as inhibitors of histone deacetylase |
| PL3319959T3 (pl) | 2015-07-06 | 2022-02-14 | Alkermes, Inc. | Hetero-haloinhibitory deacetylazy histonowej |
| JP6966996B2 (ja) * | 2015-09-17 | 2021-11-17 | シティ・オブ・ホープCity of Hope | Pcna阻害剤 |
| ES2916582T3 (es) | 2015-11-26 | 2022-07-01 | Novartis Ag | Derivados de diaminopiridina |
| US11970486B2 (en) | 2016-10-24 | 2024-04-30 | Janssen Pharmaceutica Nv | Compounds and uses thereof |
| WO2018132533A1 (en) | 2017-01-11 | 2018-07-19 | Rodin Therapeutics, Inc. | Bicyclic inhibitors of histone deacetylase |
| WO2018166855A1 (en) | 2017-03-16 | 2018-09-20 | Basf Se | Heterobicyclic substituted dihydroisoxazoles |
| SI3664802T1 (sl) | 2017-08-07 | 2022-10-28 | Alkermes, Inc. | Biciklični zaviralci histon deacetilaze |
| WO2019084157A1 (en) | 2017-10-24 | 2019-05-02 | Yumanity Therapeutics, Inc. | COMPOUNDS AND USES THEREOF |
| CN114174273A (zh) * | 2019-03-22 | 2022-03-11 | 优曼尼蒂治疗公司 | 化合物和其用途 |
| CN114072148A (zh) | 2019-05-03 | 2022-02-18 | 金耐特生物制药公司 | Raf激酶的抑制剂 |
| WO2021224186A1 (en) * | 2020-05-04 | 2021-11-11 | Institut Curie | New pyridine derivatives as radiosensitizers |
| JP2023532010A (ja) * | 2020-07-10 | 2023-07-26 | オーセントラ セラピュティクス ピーティーワイ エルティーディー | 治療用のタンパク質キナーゼ阻害剤として使用するための、2-オキソ-n-(4-(ピリミジン-4-イルオキシ/チオ)フェニル)-1,2-ジヒドロピリジン-3-カルボキサミドの誘導体 |
| US11407737B2 (en) | 2020-09-18 | 2022-08-09 | Kinnate Biopharma Inc. | Inhibitors of RAF kinases |
| WO2022081469A1 (en) * | 2020-10-12 | 2022-04-21 | Kinnate Biopharma Inc. | Inhibitors of raf kinases |
| WO2022189618A1 (en) * | 2021-03-12 | 2022-09-15 | Institut Curie | Nitrogen-containing heterocycles as radiosensitizers |
| CA3216104A1 (en) | 2021-04-23 | 2022-10-27 | Aleksandra Franovic | Treatment of cancer with a raf inhibitor |
| KR20220170766A (ko) * | 2021-06-22 | 2022-12-30 | 주식회사 엘지화학 | 단백질 키나아제 억제제로서의 신규한 화합물 |
| TW202315628A (zh) * | 2021-06-24 | 2023-04-16 | 南韓商Lg化學股份有限公司 | 作為ron抑制劑之新穎吡啶衍生物化合物 |
| WO2023114809A1 (en) | 2021-12-16 | 2023-06-22 | Kinnate Biopharma Inc. | Inhibitors of met kinase |
| CN114031619A (zh) * | 2021-12-17 | 2022-02-11 | 山东汇海医药化工有限公司 | 一种图卡替尼中间体的制备方法 |
| US20230257364A1 (en) * | 2022-02-16 | 2023-08-17 | Cmg Pharmaceutical Co., Ltd. | Pyridazinone-based compounds as axl, c-met, and mer inhibitors and methods of use thereof |
| EP4289427A1 (en) | 2022-06-10 | 2023-12-13 | Anagenesis Biotechnologies | Dihydro[1,8]naphthyridin-7-one and pyrido[3,2-b][1,4]oxazin-3-one for use in treating cancer, and metastases in particular. |
| WO2024077057A1 (en) * | 2022-10-05 | 2024-04-11 | The Translational Genomics Research Institute | Phenyl oxy amide kinase inhibitors |
Family Cites Families (97)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3646202A (en) * | 1967-11-22 | 1972-02-29 | Merck & Co Inc | Liver fluke compositions containing salicylic acid derivatives |
| JPS54115384A (en) | 1978-02-28 | 1979-09-07 | Hokko Chem Ind Co Ltd | Ryrazolyl pyrimidine derivative, and agricultural and horticultural fungicides |
| CH648611A5 (de) | 1980-08-18 | 1985-03-29 | Staeubli Ag | Verfahren und schaftmaschine zur gleichstellung aller schaefte einer webmaschine. |
| GB2106500B (en) | 1981-09-18 | 1986-01-02 | Dow Chemical Co | Method for making benzoylphenylureas |
| DE3139457A1 (de) | 1981-10-03 | 1983-04-21 | Bayer Ag, 5090 Leverkusen | Neue substituierte benzanilidether, verfahren zu ihrer herstellung, ihre verwendung als fungizide und zwischenprodukte hierfuer |
| DE3464368D1 (de) | 1983-03-09 | 1987-07-30 | Beecham Group Plc | Anti-inflammatory pyrazolo pyridines |
| EP0141104A1 (en) * | 1983-09-28 | 1985-05-15 | Becton Dickinson and Company | Culture Flask |
| EP0151962A3 (en) | 1984-01-25 | 1985-10-02 | Beecham Group Plc | Pyrazolopyridine derivatives |
| US4663341A (en) * | 1984-02-16 | 1987-05-05 | Rohm And Haas Company | Insecticidal n-aryl-3-aryl-4,5-dihydro-1h-pyrazole-1-carboxamides |
| GB8404584D0 (en) | 1984-02-22 | 1984-03-28 | Beecham Group Plc | Compounds |
| CH657015A5 (de) * | 1984-06-27 | 1986-08-15 | Ciba Geigy Ag | Motten- und kaeferschutzmittel. |
| JPS6133176A (ja) * | 1984-07-24 | 1986-02-17 | Ishihara Sangyo Kaisha Ltd | N−ニトロベンゾイル−n’−ピリダジニルオキシフエニルウレア系化合物、それらの製造方法及びそれらを含有する抗癌剤 |
| US4753940A (en) * | 1985-02-15 | 1988-06-28 | Ciba-Geigy Corporation | Barbituric acid derivatives |
| JPS62135463A (ja) | 1985-12-09 | 1987-06-18 | Ishihara Sangyo Kaisha Ltd | N−ベンゾイルウレア系化合物、それらを含有する抗癌剤並びにそれらの製造方法 |
| JPS625960A (ja) | 1986-03-05 | 1987-01-12 | Ishihara Sangyo Kaisha Ltd | ベンゾイルウレア系化合物の製造方法 |
| JPS6262A (ja) | 1986-03-05 | 1987-01-06 | Ishihara Sangyo Kaisha Ltd | ベンゾイルウレア系化合物の製造方法 |
| JPS625959A (ja) | 1986-03-05 | 1987-01-12 | Ishihara Sangyo Kaisha Ltd | ベンゾイルウレア系化合物 |
| US4908056A (en) * | 1986-04-25 | 1990-03-13 | E. I. Du Pont De Nemours And Company | Heterocyclic acyl sulfonamides |
| DE3830054A1 (de) * | 1988-09-03 | 1990-03-15 | Boehringer Mannheim Gmbh | Phenylamide - verfahren zu ihrer herstellung sowie diese verbindungen enthaltende arzneimittel |
| EP0420804A3 (en) * | 1989-09-26 | 1991-11-27 | Ciba-Geigy Ag | Anthelmintic compounds |
| EP0430885A3 (en) | 1989-12-01 | 1991-11-06 | Ciba-Geigy Ag | Anthelmintical compounds |
| DE4107829A1 (de) * | 1990-03-13 | 1991-09-19 | Ciba Geigy Ag | Diazaphosphorinderivate |
| SU1761753A1 (ru) | 1990-07-16 | 1992-09-15 | Ф. С. Михайлицын и Н. С. Уварова | 5-(2-Хлор-4-нитро(амино)фенокси) бензо-2,1,3-тиадиазол в качестве промежуточного соединени в синтезе 5- @ 2-хлор-4-[(2-окси-3,5-дибромбензоил)амино]фенокси @ бензо-2,1,3-тиадиазола, про вл ющего активность при гименолепидозе белых мышей |
| JP3020277B2 (ja) * | 1992-09-22 | 2000-03-15 | アメリカ合衆国 | リンパ腫および乳癌を治療するためのタキソールの使用 |
| US5646176A (en) * | 1992-12-24 | 1997-07-08 | Bristol-Myers Squibb Company | Phosphonooxymethyl ethers of taxane derivatives |
| EA000304B1 (ru) | 1995-02-02 | 1999-04-29 | Смитклайн Бичам Плс | Производные индола как антагонисты рецептора 5-ht |
| US6214344B1 (en) * | 1995-06-02 | 2001-04-10 | Genetech, Inc. | Hepatocyte growth factor receptor antagonists and uses thereof |
| US6143764A (en) * | 1995-11-07 | 2000-11-07 | Kirin Beer Kabushiki Kaisha | Quinoline and quinazoline derivatives inhibiting platelet-derived growth factor receptor autophosphorylation and pharmaceutical compositions containing the same |
| DE19710609A1 (de) | 1997-03-14 | 1998-09-17 | Bayer Ag | Substituierte Aminosalicylsäureamide |
| CN1268137A (zh) | 1997-07-03 | 2000-09-27 | 杜邦药品公司 | 治疗神经失调的咪唑并嘧啶和咪唑并吡啶 |
| US6605599B1 (en) | 1997-07-08 | 2003-08-12 | Bristol-Myers Squibb Company | Epothilone derivatives |
| US6022884A (en) | 1997-11-07 | 2000-02-08 | Amgen Inc. | Substituted pyridine compounds and methods of use |
| TW515786B (en) | 1997-11-25 | 2003-01-01 | Nihon Nohyaku Co Ltd | Phthalic acid diamide derivatives, agricultural and horticultural insecticides, and a method for application of the insecticides |
| US6362369B2 (en) * | 1997-11-25 | 2002-03-26 | Nihon Nohyaku Co., Ltd. | Phthalic acid diamide derivatives fluorine-containing aniline compounds as starting material, agricultural and horticultural insecticides, and a method for application of the insecticides |
| US6200995B1 (en) * | 1998-01-29 | 2001-03-13 | Tularik Inc. | PPAR-γ modulators |
| US6232320B1 (en) * | 1998-06-04 | 2001-05-15 | Abbott Laboratories | Cell adhesion-inhibiting antiinflammatory compounds |
| EP1087937A1 (en) * | 1998-06-17 | 2001-04-04 | Du Pont Pharmaceuticals Company | Cyclic hydroxamic acids as metalloproteinase inhibitors |
| CZ299375B6 (cs) * | 1998-11-30 | 2008-07-09 | Nihon Nohyaku Co., Ltd. | Ftalamidové deriváty nebo jejich soli, zemedelsko-zahradnický insekticid je obsahující a jeho použití |
| US20020065296A1 (en) | 1999-01-13 | 2002-05-30 | Bayer Corporation | Heteroaryl ureas containing nitrogen hetero-atoms as p38 kinase inhibitors |
| NZ513006A (en) | 1999-01-22 | 2003-10-31 | Kirin Brewery | Quinoline derivatives and quinazoline derivatives |
| AU771089B2 (en) * | 1999-02-22 | 2004-03-11 | Bristol-Myers Squibb Company | C-21 modified epothilones |
| GB9904103D0 (en) | 1999-02-24 | 1999-04-14 | Zeneca Ltd | Quinoline derivatives |
| GB9906566D0 (en) | 1999-03-23 | 1999-05-19 | Zeneca Ltd | Chemical compounds |
| US6982265B1 (en) | 1999-05-21 | 2006-01-03 | Bristol Myers Squibb Company | Pyrrolotriazine inhibitors of kinases |
| ES2328269T3 (es) | 1999-05-21 | 2009-11-11 | Bristol-Myers Squibb Company | Pirrolotriazinas como inhibires de quinasas. |
| EP1181296A1 (en) | 1999-06-03 | 2002-02-27 | Abbott Laboratories | Cell adhesion-inhibiting antiinflammatory compounds |
| WO2001005769A2 (en) * | 1999-07-20 | 2001-01-25 | Dow Agrosciences Llc | Fungicidal heterocyclic aromatic amides and their compositions, methods of use and preparation |
| US6355660B1 (en) * | 1999-07-20 | 2002-03-12 | Dow Agrosciences Llc | Fungicidal heterocyclic aromatic amides and their compositions, methods of use and preparation |
| WO2001021596A1 (en) | 1999-09-21 | 2001-03-29 | Astrazeneca Ab | Quinazoline derivatives and their use as pharmaceuticals |
| BR0014139B1 (pt) | 1999-09-24 | 2011-09-20 | derivado de diamida aromática ou um seu sal, composição agrìcola e hortìcola, e, método para utilizar uma composição agrìcola e hortìcola. | |
| GB9924092D0 (en) | 1999-10-13 | 1999-12-15 | Zeneca Ltd | Pyrimidine derivatives |
| AU2576501A (en) | 1999-12-08 | 2001-06-18 | Advanced Medicine, Inc. | Protein kinase inhibitors |
| EP1243582A4 (en) | 1999-12-24 | 2003-06-04 | Kirin Brewery | CHINOLINE AND CHINAZOLINE DERIVATIVES AND MEDICATIONS CONTAINING THEM |
| US20020065270A1 (en) * | 1999-12-28 | 2002-05-30 | Moriarty Kevin Joseph | N-heterocyclic inhibitors of TNF-alpha expression |
| US6906067B2 (en) * | 1999-12-28 | 2005-06-14 | Bristol-Myers Squibb Company | N-heterocyclic inhibitors of TNF-α expression |
| WO2001047921A1 (en) | 1999-12-28 | 2001-07-05 | Pharmacopeia, Inc. | Pyrimidine and triazine kinase inhibitors |
| US6750246B1 (en) * | 2000-02-03 | 2004-06-15 | Bristol-Myers Squibb Company | C-4 carbonate taxanes |
| EP1149583A3 (en) * | 2000-04-13 | 2001-11-14 | Pfizer Products Inc. | Combinations of corticotropin releasing factor antagonists and growth hormone secretagogues |
| EP1287001B1 (en) | 2000-06-06 | 2004-09-29 | Pfizer Products Inc. | Thiophene derivatives useful as anticancer agents |
| FR2812633A1 (fr) * | 2000-08-04 | 2002-02-08 | Aventis Cropscience Sa | Derives de phenyl(thio)urees et phenyl(thio)carbamates fongicides |
| EP1326835A1 (en) * | 2000-10-05 | 2003-07-16 | Fujisawa Pharmaceutical Co., Ltd. | Benzamide compounds as apo b secretion inhibitors |
| EP1415987B1 (en) | 2000-10-20 | 2007-02-28 | Eisai R&D Management Co., Ltd. | Nitrogenous aromatic ring compounds as anti cancer agents |
| CA2429628A1 (en) | 2000-11-17 | 2002-05-23 | Bristol-Myers Squibb Company | Methods of treating p38 kinase-associated conditions and pyrrolotriazine compounds useful as kinase inhibitors |
| WO2002044156A2 (en) | 2000-11-29 | 2002-06-06 | Glaxo Group Limited | Benzimidazole derivatives useful as tie-2 and/or vegfr-2 inhibitors |
| CA2432713C (en) * | 2000-12-22 | 2009-10-27 | Ishihara Sangyo Kaisha, Ltd. | Aniline derivatives or salts thereof and cytokine production inhibitors containing the same |
| US7081454B2 (en) * | 2001-03-28 | 2006-07-25 | Bristol-Myers Squibb Co. | Tyrosine kinase inhibitors |
| US6995171B2 (en) | 2001-06-21 | 2006-02-07 | Agouron Pharmaceuticals, Inc. | Bicyclic pyrimidine and pyrimidine derivatives useful as anticancer agents |
| CN100415720C (zh) | 2001-06-22 | 2008-09-03 | 麒麟医药株式会社 | 喹啉衍生物和喹唑啉衍生物以及含有这些化合物的药物组合物 |
| CA2451128A1 (en) * | 2001-06-26 | 2003-01-09 | Bristol-Myers Squibb Company | N-heterocyclic inhibitors of tnf-alpha expression |
| SE0102384D0 (sv) * | 2001-07-03 | 2001-07-03 | Pharmacia Ab | New compounds |
| WO2003011028A1 (en) | 2001-08-01 | 2003-02-13 | Nissan Chemical Industries, Ltd. | Substituted amides and pest controllers |
| WO2003033472A1 (fr) | 2001-10-17 | 2003-04-24 | Kirin Beer Kabushiki Kaisha | Derives de quinoline ou de quinazoline inhibant l'autophosphorylation de recepteurs du facteur de croissance des fibroblastes |
| TW200300350A (en) | 2001-11-14 | 2003-06-01 | Bristol Myers Squibb Co | C-5 modified indazolylpyrrolotriazines |
| JP2005510564A (ja) * | 2001-11-28 | 2005-04-21 | 藤沢薬品工業株式会社 | アポリポタンパク質b阻害剤としての複素環式アミド化合物 |
| ES2393900T3 (es) * | 2001-12-03 | 2012-12-28 | Bayer Healthcare Llc | Compuestos de aril-urea en combinación con otros agentes citostáticos o citotóxicos para tratamiento de cánceres humanos |
| JP2003321472A (ja) | 2002-02-26 | 2003-11-11 | Takeda Chem Ind Ltd | Grk阻害剤 |
| US6900208B2 (en) | 2002-03-28 | 2005-05-31 | Bristol Myers Squibb Company | Pyrrolopyridazine compounds and methods of use thereof for the treatment of proliferative disorders |
| EP2289894A3 (en) * | 2002-04-23 | 2011-07-20 | Bristol-Myers Squibb Company | Pyrrolo-triazine compounds useful as kinase inhibitors |
| WO2003091229A1 (en) * | 2002-04-23 | 2003-11-06 | Bristol-Myers Squibb Company | Aryl ketone pyrrolo-triazine compounds useful as kinase inhibitors |
| US7388009B2 (en) * | 2002-04-23 | 2008-06-17 | Bristol-Myers Squibb Company | Heteroaryl-substituted pyrrolo-triazine compounds useful as kinase inhibitors |
| TW200407143A (en) | 2002-05-21 | 2004-05-16 | Bristol Myers Squibb Co | Pyrrolotriazinone compounds and their use to treat diseases |
| TW200401638A (en) | 2002-06-20 | 2004-02-01 | Bristol Myers Squibb Co | Heterocyclic inhibitors of kinases |
| TWI329112B (en) * | 2002-07-19 | 2010-08-21 | Bristol Myers Squibb Co | Novel inhibitors of kinases |
| AR043059A1 (es) * | 2002-11-12 | 2005-07-13 | Bayer Pharmaceuticals Corp | Derivados de indolil pirazinona utiles para el tratamiento de trastornos hiper-proliferativos |
| US7030112B2 (en) * | 2003-03-25 | 2006-04-18 | Bristol-Myers Squibb Company | Pyrrolopyridazine compounds and methods of use thereof for the treatment of proliferative disorders |
| CA2531485C (en) * | 2003-07-07 | 2013-03-26 | Merck Patent Gmbh | Malonamide derivatives |
| ES2465624T3 (es) * | 2003-07-11 | 2014-06-06 | Merck Patent Gmbh | Benzimidazol carboxamidas como inhibidores de RAF quinasa |
| US7419978B2 (en) * | 2003-10-22 | 2008-09-02 | Bristol-Myers Squibb Company | Phenyl-aniline substituted bicyclic compounds useful as kinase inhibitors |
| DE10354060A1 (de) * | 2003-11-19 | 2005-06-02 | Merck Patent Gmbh | Pyrrolderivate |
| TW200538453A (en) * | 2004-04-26 | 2005-12-01 | Bristol Myers Squibb Co | Bicyclic heterocycles as kinase inhibitors |
| US7439246B2 (en) * | 2004-06-28 | 2008-10-21 | Bristol-Myers Squibb Company | Fused heterocyclic kinase inhibitors |
| US7432373B2 (en) * | 2004-06-28 | 2008-10-07 | Bristol-Meyers Squibb Company | Processes and intermediates useful for preparing fused heterocyclic kinase inhibitors |
| US7173031B2 (en) * | 2004-06-28 | 2007-02-06 | Bristol-Myers Squibb Company | Pyrrolotriazine kinase inhibitors |
| WO2006044687A2 (en) | 2004-10-15 | 2006-04-27 | Takeda San Diego, Inc. | Kinase inhibitors |
| US7470693B2 (en) * | 2005-04-21 | 2008-12-30 | Bristol-Myers Squibb Company | Oxalamide derivatives as kinase inhibitors |
| US7444690B2 (en) * | 2005-07-15 | 2008-11-04 | Marilyn Holland | Sitz bath for treating hemorrhoids |
| WO2007035428A1 (en) * | 2005-09-15 | 2007-03-29 | Bristol-Myers Squibb Company | Met kinase inhibitors |
-
2005
- 2005-04-21 US US11/111,144 patent/US7459562B2/en active Active
- 2005-04-22 MX MXPA06012185A patent/MXPA06012185A/es active IP Right Grant
- 2005-04-22 SI SI200531288T patent/SI1737451T1/sl unknown
- 2005-04-22 WO PCT/US2005/014120 patent/WO2005117867A2/en active Application Filing
- 2005-04-22 AT AT05779444T patent/ATE499097T1/de active
- 2005-04-22 AU AU2005249382A patent/AU2005249382B2/en active Active
- 2005-04-22 JP JP2007509726A patent/JP4778959B2/ja active Active
- 2005-04-22 EP EP05779444A patent/EP1737451B1/en active Active
- 2005-04-22 PT PT05779444T patent/PT1737451E/pt unknown
- 2005-04-22 NZ NZ550614A patent/NZ550614A/en unknown
- 2005-04-22 PL PL05779444T patent/PL1737451T3/pl unknown
- 2005-04-22 BR BRPI0510177A patent/BRPI0510177B8/pt active IP Right Grant
- 2005-04-22 GE GEAP20059707A patent/GEP20104884B/en unknown
- 2005-04-22 KR KR1020067024515A patent/KR100918727B1/ko active IP Right Grant
- 2005-04-22 DE DE602005026508T patent/DE602005026508D1/de active Active
- 2005-04-22 CA CA2563831A patent/CA2563831C/en active Active
-
2006
- 2006-10-17 IL IL178675A patent/IL178675A/en active IP Right Grant
- 2006-11-08 NO NO20065148A patent/NO339960B1/no unknown
-
2007
- 2007-01-04 HK HK07100116.2A patent/HK1093159A1/xx unknown
-
2008
- 2008-10-17 US US12/253,491 patent/US7714138B2/en active Active
-
2010
- 2010-03-19 US US12/727,534 patent/US7989477B2/en active Active
-
2011
- 2011-03-02 HR HR20110156T patent/HRP20110156T1/hr unknown
- 2011-05-20 CY CY20111100491T patent/CY1111640T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| BRPI0510177B1 (pt) | composto, composição farmacêutica e uso do mesmo | |
| ES2359836T3 (es) | Heterociclos monociclicos como inhibidores de quinasa. | |
| AU2005259894B2 (en) | Pyrrolotriazine kinase inhibitors | |
| US7439246B2 (en) | Fused heterocyclic kinase inhibitors | |
| EP1937682B1 (en) | Met kinase inhibitors | |
| TWI310378B (en) | Pyrazine-2-carboxamide derivatives as mglur5 antagonists | |
| ES2928084T3 (es) | Inhibidores de GDF-8 | |
| CA2543861A1 (en) | Novel pyridine derivative and pyrimidine derivative (2) | |
| CA2700559A1 (fr) | Derives de nicotinamide, leur preparation et leur application en therapeutique | |
| PT2178840E (pt) | Derivados do ácido azabifenilaminobenzóico como inibidores da dhodh | |
| CA2774515A1 (en) | Substituted phenylamine carboxamide analogs as mglur5 negative allosteric modulators and methods of making and using the same | |
| US11370772B2 (en) | 6-Substituted 3-Fluoro-2-Pyridinaldoxime, 3-Fluoro-2-pyridine hydroxamic acid, and 3-Fluoro-2-Pyridinamidoxime scaffolds | |
| WO2020002969A1 (en) | Triazolotriazine derivatives as a2a receptor antagonists | |
| TWI324926B (en) | Monocyclic heterocycles as kinase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] | ||
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B07E | Notification of approval relating to section 229 industrial property law [chapter 7.5 patent gazette] |
Free format text: NOTIFICACAO DE ANUENCIA RELACIONADA COM O ART 229 DA LPI |
|
| B07A | Application suspended after technical examination (opinion) [chapter 7.1 patent gazette] | ||
| B06A | Patent application procedure suspended [chapter 6.1 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 10 (DEZ) ANOS CONTADOS A PARTIR DE 07/04/2020, OBSERVADAS AS CONDICOES LEGAIS. |
|
| B16C | Correction of notification of the grant [chapter 16.3 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 22/04/2005 OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF |