WO2019021890A1 - ガスバリア性樹脂組成物及びその用途 - Google Patents
ガスバリア性樹脂組成物及びその用途 Download PDFInfo
- Publication number
- WO2019021890A1 WO2019021890A1 PCT/JP2018/026766 JP2018026766W WO2019021890A1 WO 2019021890 A1 WO2019021890 A1 WO 2019021890A1 JP 2018026766 W JP2018026766 W JP 2018026766W WO 2019021890 A1 WO2019021890 A1 WO 2019021890A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- gas barrier
- resin composition
- copolymer
- barrier resin
- group
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/02—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids
- C08G69/26—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from polyamines and polycarboxylic acids
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/02—Ethene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
- C08F8/12—Hydrolysis
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L101/00—Compositions of unspecified macromolecular compounds
- C08L101/12—Compositions of unspecified macromolecular compounds characterised by physical features, e.g. anisotropy, viscosity or electrical conductivity
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L23/04—Homopolymers or copolymers of ethene
- C08L23/08—Copolymers of ethene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L29/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an alcohol, ether, aldehydo, ketonic, acetal or ketal radical; Compositions of hydrolysed polymers of esters of unsaturated alcohols with saturated carboxylic acids; Compositions of derivatives of such polymers
- C08L29/02—Homopolymers or copolymers of unsaturated alcohols
- C08L29/04—Polyvinyl alcohol; Partially hydrolysed homopolymers or copolymers of esters of unsaturated alcohols with saturated carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L67/00—Compositions of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Compositions of derivatives of such polymers
- C08L67/02—Polyesters derived from dicarboxylic acids and dihydroxy compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L77/00—Compositions of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Compositions of derivatives of such polymers
- C08L77/06—Polyamides derived from polyamines and polycarboxylic acids
Definitions
- the present invention relates to a gas barrier resin composition in which the physical properties of a gas barrier resin represented by a vinyl alcohol resin, a polyamide resin, and a polyester resin are improved, a sheet, a film, or a bag, a bottle, a tank, etc. And a method of modifying the gas barrier resin composition.
- a gas barrier resin composition in which the physical properties of a gas barrier resin represented by a vinyl alcohol resin, a polyamide resin, and a polyester resin are improved, a sheet, a film, or a bag, a bottle, a tank, etc. And a method of modifying the gas barrier resin composition.
- Vinyl represented by polyvinyl alcohol hereinafter sometimes abbreviated as “PVOH”), ethylene / vinyl alcohol copolymer (hereinafter sometimes abbreviated as “EVOH”), etc.
- Alcohol-based resins have very strong intermolecular force due to the formation of hydrogen bonds between hydroxyl groups present in the polymer chain. Therefore, since the crystallinity is high and the intermolecular force is high even in the amorphous part, vinyl alcohol resins exhibit high gas barrier properties, and gas molecules such as oxygen and nitrogen, and vapors of organic solvents, etc. It is known that it can not easily permeate a molded article (barrier layer) molded using an alcohol-based resin.
- EVOH can be melt-molded and has high gas barrier properties, oil resistance, organic solvent resistance, transparency and the like, so its molded articles (films, sheets, bottle containers, etc.) It is used in a wide range of fields such as food packaging materials, pharmaceutical packaging materials, industrial chemical packaging materials, agrochemical packaging materials, fuel containers and the like.
- EVOH has a disadvantage that it is a hard and brittle resin and has poor flexibility because it has high crystallinity and crystallization rate. Therefore, the processability at the time of molding into packaging materials such as food, especially heat stretchability is low, cracks may occur in the barrier layer at the time of molding, and the product yield may decrease, or mechanical strength due to thickness unevenness.
- Patent Document 1 describes a method of adding water or a plasticizer to a resin composition for forming a sheet containing EVOH as a main component.
- Patent Document 2 JP-A-52-141785 (Patent Document 2) and JP-A-59-20345 (Patent Document 3) disclose polyamides as resin compositions for forming a sheet or the like mainly containing EVOH.
- Patent Document 3 disclose polyamides as resin compositions for forming a sheet or the like mainly containing EVOH.
- methods of blending aromatic polyesters are described. Although the improvement of the flexibility was seen with any of the methods, the gas barrier property was greatly reduced, and the original gas barrier properties of EVOH could not be maintained.
- JP-A-8-239528 discloses a saponified product of two ethylene / vinyl acetate copolymers having a saponification degree of 95 mol% or more and 70 mol% or more and a polyamide having a terminal carboxyl group adjusted.
- a resin composition is disclosed, and JP-A-2000-212369 (Patent Document 5) contains EVOH having a saponification degree of 98 mol% or more and EVOH having a reduced saponification degree by reacetylation.
- Resin compositions are disclosed.
- Polyamide (alias: nylon) resin is excellent in various physical properties such as strength, heat resistance, gas barrier property, optical characteristics, or oil resistance, and parts for automobiles and vehicles, parts for electricity and electronics, films for packaging, etc. , Is used in various fields. Furthermore, in recent years, particularly in the automobile industry, for the purpose of improving fuel efficiency by reducing the weight of the vehicle, it has been adopted in each part as a substitute for metal parts.
- a polyamide resin is a polymer produced by condensation polymerization of a dicarboxylic acid and a diamine, and having a repeating unit of an amide bond. By changing the chemical structure of the dicarboxylic acid and diamine used as the raw materials, it is possible to control basic physical properties such as heat resistance and strength of the resulting polyamide resin, and the structure is designed according to the required physical properties of the application. ing.
- Polyamide MXD6 (alias: nylon MXD6, MXD6-nylon) is an aromatic polyamide-based resin obtained by condensation polymerization of adipic acid and metaxylylene diamine, and is superior in gas barrier properties to other polyamide-based resins. Therefore, polyamide MXD6 is used as a gas barrier layer in food packaging, PET bottles and the like. However, polyamide MXD6 is a hard and brittle resin, and has a disadvantage that it has poor flexibility. Therefore, the processability at the time of molding into packaging materials such as food, especially heat stretchability is low, cracks may occur in the barrier layer at the time of molding, and the product yield may decrease, or mechanical strength due to thickness unevenness.
- Patent Document 6 Japanese Patent Publication No. 7-15059 (Patent Document 6) and JP-A-9-183900 (US Patent No. 5780577; Patent Document 7), acid modification of an ethylene-based copolymer is carried out to improve pinhole resistance.
- Products and acid-modified products of partially saponified ethylene / vinyl acetate copolymers are added.
- these copolymers have a carboxy group in the molecule, and when mixed with a polyamide resin, the filter installed in the passage of the molten resin in the extruder tends to be clogged during film forming, and filter replacement As a result, there is a problem that the impact on film productivity is enormous.
- Polyester-based resins represented by polyethylene terephthalate (abbreviation: PET), polybutylene terephthalate (abbreviation: PBT), polyethylene naphthalate (abbreviation: PEN), etc. are excellent in mechanical properties and chemical properties, and their respective properties Therefore, it is used in various fields such as automobile and vehicle parts, electric and electronic parts, and packaging films. Further, a bottle obtained by hollow molding from saturated polyester such as polyethylene terephthalate in particular is excellent in mechanical strength, transparency and gas barrier properties, and thus containers for juice, carbonated beverages, soft drinks, cosmetics, eye drops It is also used as a container for liquids.
- a polyester resin is a polymer produced by condensation polymerization of a dicarboxylic acid and a diol, and having a repeating unit of an ester bond.
- Polybutylene terephthalate is a saturated polyester produced by condensation polymerization of terephthalic acid and 1,4-butanediol. It is a hair dryer, a telephone, an electric / electronic component such as a connector, a switch, a door handle, an ignition coil, a side mirror , Valves, switches and other automotive parts. Further, in film applications, unstretched polybutylene terephthalate films by a cast molding method mainly for food packaging, uniaxially stretched polybutylene terephthalate films for shrink labels of beverage bottles, and the like are manufactured. However, polybutylene terephthalate is a hard and brittle resin because of its high crystallinity, and has the disadvantage of being less flexible.
- the processability at the time of molding into packaging materials such as food, especially heat stretchability is low, cracks may occur in the barrier layer at the time of molding, and the product yield may decrease, or mechanical strength due to thickness unevenness. This may cause a decrease in the gas barrier property and the like, resulting in a lack of quality stability. Furthermore, repeated use of bending as a packaging material or a molding material has the problem that cracks and pinholes easily occur due to bending fatigue and the like, and the excellent performance can not be maintained.
- biaxial stretch molding is used for general purpose plastics such as polypropylene and polyamide 6, in the case of polybutylene terephthalate, the low heat stretchability resulting from the high crystallization rate leads to the commercialization of the biaxial stretched film Not in.
- Patent Document 8 Japanese Patent Application Laid-Open No. 2006-241398 (Patent Document 8) and Japanese Patent Application Laid-Open No. 2016-191009 (Patent Document 9) describe a method of blending polyester elastomers and polycarbonates in order to improve flexibility and heat drawability.
- Patent Document 8 Japanese Patent Application Laid-Open No. 2006-241398
- Patent Document 9 Japanese Patent Application Laid-Open No. 2016-191009
- Japanese Patent Application Laid-Open No. 53-088067 JP-A-52-141785 Japanese Patent Application Laid-Open No. 59-20345 JP-A-8-239528 JP 2000-212369 A Japanese Examined Patent Publication 7-15059 Unexamined-Japanese-Patent No. 9-183900 (US Patent No. 5780577) JP, 2006-241398, A JP, 2016-191009, A
- An object of the present invention is to provide a resin composition having improved low flexibility and low impact resistance, which is a defect of gas barrier resin, without impairing the excellent gas barrier property of gas barrier resin, and a resin composition molded using the resin composition. It is to provide a film, a sheet, or a container etc.
- the inventors of the present invention conducted intensive studies to solve the above problems, and as a result, by blending an ethylene / hydroxyl group-containing allyl monomer copolymer having high flexibility with the gas barrier resin, the superiority derived from the gas barrier resin It has been found that a resin composition to which excellent flexibility and impact resistance have been imparted without impairing the gas barrier properties and molded articles thereof can be obtained, and the present invention has been completed.
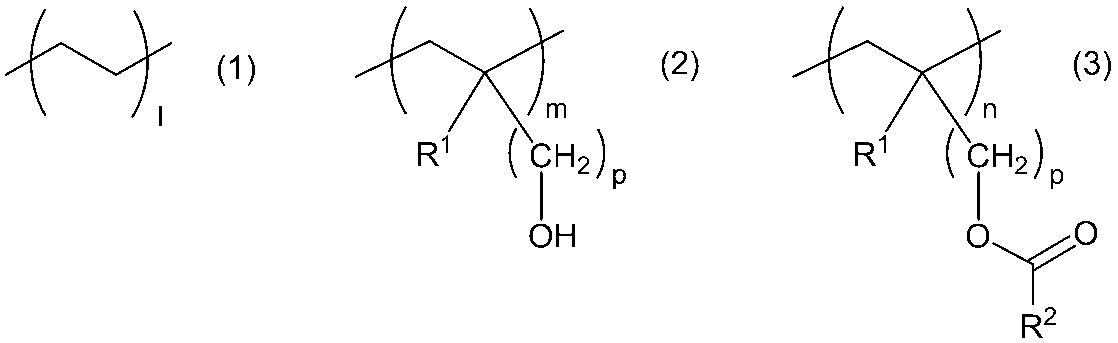
- Gas-barrier resin (A) having an oxygen permeability coefficient of 1.0 ⁇ 10 -14 (cm 3 ⁇ cm / cm 2 ⁇ s ⁇ Pa) or less and the general formula (1), the general formula (2), and the general formula Formula (3) (Wherein, R 1 represents a hydrogen atom or a methyl group, and R 2 represents a halogen atom, a hydroxyl group, an alkoxy group, or a hydrocarbon group having 1 to 20 carbon atoms which may be substituted with an amino group. , M and n are numerical values representing the molar ratio of the respective monomer structural units, n may be 0.
- a resin composition comprising a copolymer (B) containing a monomer structural unit represented by: a copolymer based on the total mass of the gas barrier resin (A) and the copolymer (B) A gas barrier resin composition characterized in that the proportion of the mass of B) is 1 to 40 mass%.
- R 2 The gas barrier resin composition according to the above item 1, wherein the hydrocarbon group having 1 to 20 carbon atoms represented by R 2 is an alkyl group having 1 to 20 carbon atoms or an aryl group having 6 to 20 carbon atoms.
- the molar ratio m of the monomer structural unit represented by the general formula (2) of the copolymer (B) and the molar ratio n of the monomer structural unit represented by the general formula (3) are represented by the following formulas:
- [5] The gas barrier resin composition according to any one of the above items 1 to 3, wherein n 0 in the monomer structural unit represented by the general formula (3) of the copolymer (B).
- the number average molecular weight (Mn) of the copolymer (B) is 1,000 to 1,000,000, and the ratio Mw / Mn of the weight average molecular weight (Mw) to the number average molecular weight (Mn) is 1.5 to 4.0 6.
- the gas barrier resin (A) is a polyamide resin (A2).
- the polyamide resin (A2) is at least one selected from polyamide 6, polyamide 66, and polyamide MXD6.
- a container comprising the gas barrier resin composition described in any one of the above items 1 to 13 as a barrier layer.
- R 1 represents a hydrogen atom or a methyl group
- R 2 represents a halogen atom, a hydroxyl group, an alkoxy group, or a hydrocarbon group having 1 to 20 carbon atoms which may be substituted with an amino group.
- M and n are numerical values representing the molar ratio of the respective monomer structural units, n may be 0.
- p represents an integer of 1 to 4.
- the gas barrier resin composition of the present invention in which an ethylene / hydroxyl group-containing allyl monomer copolymer having high flexibility is blended with the gas barrier resin of the present invention is flexible and shock resistant without impairing the excellent gas barrier property derived from the gas barrier resin.
- the properties are improved, and it is useful as a sheet, film, and a barrier layer of containers such as bags, bottles, and tanks.
- the resin composition of the present invention has a gas barrier resin (A) having an oxygen permeability coefficient of 1.0 ⁇ 10 -14 (cm 3 ⁇ cm / cm 2 ⁇ s ⁇ Pa) or less, a general formula (1), a general formula 2) and general formula (3) Wherein the ratio of the mass of the copolymer (B) to the total mass of the gas barrier resin (A) and the copolymer (B) is 1 to 40 mass, including the copolymer (B) containing the monomer structural unit represented by %.
- R 1 represents a hydrogen atom or a methyl group
- R 2 represents a halogen atom, a hydroxyl group, an alkoxy group or an amino group and may have 1 to 20 carbon atoms
- l, m and n are numerical values representing the molar ratio of the respective monomer structural units, and n may be 0.
- p represents an integer of 1 to 4;
- the gas barrier resin (A) constituting the gas barrier resin composition of the present invention is a resin having an oxygen permeation coefficient of 1.0 ⁇ 10 -14 (cm 3 ⁇ cm / cm 2 ⁇ s ⁇ Pa) or less.
- the oxygen permeation coefficient is a value measured by a differential pressure method in accordance with JIS K7126.
- the oxygen gas permeation amount (cm 3 ) is a value at STP (standard temperature and pressure; 0 ° C., 1 atm). The specific measurement method is shown in the Example section.
- Such gas barrier resins (A) include non-crystalline polyethylene terephthalate, polyvinyl chloride, nylon-6, polyvinyl fluoride, polyvinylidene chloride, polyacrylonitrile, ethylene-vinyl alcohol copolymer, polyvinyl alcohol and the like.
- Table 1 shows the oxygen permeability coefficients of the main resins (Source: Polymer handbook 4th Edition, John Wiley & Sons, Inc. (1999) et al.).
- the gas barrier resin (A) may be a combination of a plurality of resins having an oxygen permeability coefficient of 1.0 ⁇ 10 -14 (cm 3 ⁇ cm / cm 2 ⁇ s ⁇ Pa) or less.
- other gas barrier resins may be contained as long as the effects of the present invention are not impaired.
- vinyl alcohol resins (A1) such as ethylene-vinyl alcohol copolymer, polyamide resins (A2) such as nylon-6, poly Preferred are polyester resins (A3) such as butylene terephthalate.
- the vinyl alcohol resin (A1) constituting the gas barrier resin composition of the present invention is a polymer containing a structural unit derived from a vinyl alcohol monomer.
- the vinyl alcohol monomer is a monomer containing a carbon-carbon double bond and a hydroxyl group (but excluding the monomer giving the structure of the general formula (2)), and examples thereof include vinyl alcohol and 1-butene-3-ol. And 2-methyl-2-propen-1-ol, 1-butene-3,4-diol and the like.
- the vinyl alcohol resin (A1) is preferably a polymer containing a vinyl alcohol structural unit, and may be a vinyl alcohol copolymerized with ethylene, 1-butene-3,4-diol or the like.
- the vinyl alcohol resin (A1) is an ethylene-vinyl alcohol copolymer
- the ethylene content is preferably 10 to 60 mol%, and from the viewpoint of gas barrier properties and polymer physical properties under high humidity, 20 to 50 mol% Is more preferred. If the ethylene content is less than 10 mol%, the heat resistance and the extrusion moldability are reduced. When the ethylene content exceeds 60 mol%, the gas barrier properties are significantly reduced.
- the vinyl alcohol resin (A1) is produced by a hydrolysis reaction under acidic conditions or a saponification reaction under basic conditions with respect to the vinyl ester (co) polymer.
- EVOH is obtained by hydrolysis or saponification reaction using an ethylene-vinyl ester copolymer represented by ethylene-vinyl acetate copolymer as a raw material.
- the ratio of the number of vinyl alcohol structural units to the total number of vinyl alcohol structural units and the number of vinyl ester structural units in the vinyl alcohol resin (A1) (hydrolysis ratio or saponification ratio) is from the viewpoint of thermal stability and gas barrier properties. 85 mol% or more is preferable, 90 mol% or more is more preferable, and 98 mol% or more is more preferable.
- the (co) polymerization method of the vinyl ester compound for obtaining the vinyl ester (co) polymer is not particularly limited, and known methods such as solution polymerization method, suspension polymerization method, emulsion polymerization method, bulk polymerization method, etc. It can be done in a way. Also, the polymerization mode may be batch mode or continuous mode, and single stage polymerization or multistage polymerization may also be performed.
- the (co) polymerization type of the vinyl ester compound is not particularly limited, and, for example, radical polymerization method using an organic / inorganic peroxide or an azo compound as a catalyst, Lewis acid or Bronsted acid etc.
- a cationic polymerization method used as a catalyst, an anionic polymerization method using a Lewis base or the like as a catalyst, and a coordination anionic polymerization method using a metal complex catalyst or the like can be used.
- the radical polymerization method is particularly preferred.
- the vinyl ester compound used in the polymerization include vinyl acetate, vinyl propionate, vinyl trifluoroacetate, vinyl benzoate and the like, and vinyl acetate is particularly preferable from the viewpoint of industrial availability.
- a vinyl alcohol resin (A1) a polymer obtained by formalizing a part of vinyl alcohol structural unit of PVOH or EVOH with formaldehyde, butyralizing with butyraldehyde, or grafting other monomers to PVOH or EVOH It may be a polymer obtained by polymerization.
- the vinyl alcohol resin (A1) may contain other structural units derived from monomers other than ethylene and vinyl ester.
- a monomer which gives such another structural unit a (meth) acrylic acid ester type compound, a (meth) acrylic acid compound, a vinyl ether type compound, a vinylsilane type compound, a vinyl siloxane type compound etc. are mentioned.
- the polyamide resin (A2) constituting the gas barrier resin composition of the present invention is a polymer having a repeating unit of an amide bond formed by condensation of a carboxy group and an amino group, and is also called nylon.
- polyamide resin (A2) polyamide 6 (alias: 6-nylon, nylon 6) obtained by ring-opening polymerization of ⁇ -caprolactam, polyamide 66 obtained by condensation polymerization of adipic acid and hexamethylene diamine (alias 6,6-nylon, nylon 6,6), polyamide MXD 6 (alias: MXD 6-nylon, nylon MXD 6) obtained by condensation polymerizing adipic acid and metaxylylenediamine, polycondensation of sebacic acid and metaxylylene diamine
- Polyamide MXD10 alias: MXD10-nylon, nylon MXD10
- polyamide 610 obtained by condensation polymerization of sebacic acid and hexamethylenediamine
- the production method of the polyamide resin (A2) is roughly classified into a method of condensation polymerization of dicarboxylic acid and diamine and a method of ring opening polymerization of cyclic lactam, and the polymerization is a melt polymerization method, a solid phase polymerization method, It can be carried out by a known method such as a solution polymerization method or a bulk polymerization method. Also, the polymerization mode may be batch mode or continuous mode, and single stage polymerization or multistage polymerization may also be performed.
- polyester-based resin (A3) the method of condensation-polymerizing dicarboxylic acid or diester, and diol on acidic conditions or basic conditions is common.
- the polymerization can be carried out by a known method such as a melt polymerization method, a solid phase polymerization method, a solution polymerization method or a bulk polymerization method.
- the polymerization mode may be batch mode or continuous mode, and single stage polymerization or multistage polymerization may also be performed.
- dicarboxylic acid used in the production of the polyester resin (A3) include phthalic acid, isophthalic acid, terephthalic acid, 1,2-naphthalenedicarboxylic acid, 1,3-naphthalenedicarboxylic acid and 1,4-naphthalene Dicarboxylic acid, 1,5-naphthalenedicarboxylic acid, 1,6-naphthalenedicarboxylic acid, 1,7-naphthalenedicarboxylic acid, 1,8-naphthalenedicarboxylic acid, 2,3-naphthalenedicarboxylic acid, 2,4-naphthalenedicarboxylic acid 2,6-naphthalenedicarboxylic acid, 2,7-naphthalenedicarboxylic acid, 4,4'-biphenyldicarboxylic acid, cis-1,2-cyclopropanedicarboxylic acid, trans-1,2-cyclopropanedicarboxylic acid, cis- 1,2-cyclobutanedicar
- the diester used by manufacture of polyester-based resin (A3) is a compound obtained by condensation reaction of said dicarboxylic acid and alcohol.
- the alcohol include methanol, ethanol, n-propanol, isopropanol, n-butanol, isobutanol, sec-butanol, t-butanol, phenol and the like.
- the dicarboxylic acids or diesters may be used in combination of two or more.
- diol used in the production of the polyester resin (A3) include ethylene glycol, 1,2-propanediol, 1,3-propanediol, 1,2-butanediol, 1,3-butanediol, 1,4-butanediol, 2,3-butanediol, 1,2-pentanediol, 1,3-pentanediol, 1,4-pentanediol, 1,5-pentanediol, 2,3-pentanediol, 2 , 4-Pentanediol, cis-1,2-cyclohexanedimethanol, trans-1,2-cyclohexanedimethanol, cis-1,3-cyclohexanedimethanol, trans-1,3-cyclohexanedimethanol, cis-1, 4-cyclohexanedimethanol, trans-1,4-cyclohexanedimethano Le, and
- the polyester resin (A3) constituting the gas barrier resin composition of the present invention may be used in combination of two or more.
- the polyester resin (A3) constituting the gas barrier resin composition of the present invention is polyethylene terephthalate produced by condensation polymerization of terephthalic acid or its ester with ethylene glycol from the viewpoint of versatility and strength, terephthalic acid or Polytrimethylene terephthalate (abbreviation: PTT) produced by condensation polymerization of the ester and 1,3-propanediol, polybutylene terephthalate produced by condensation polymerization of terephthalic acid or its ester and 1,4-butanediol Polyethylene naphthalate produced by condensation polymerization of 2,6-naphthalene dicarboxylic acid or its ester and ethylene glycol, and condensation polymerization of 2,6-naphthalene dicarboxylic acid or its ester and 1,4-butanediol Polybutylene naphthalate Abbreviation: PBN) is preferable.
- PTT Polytrimethylene terephthalate
- PTT Polytrimethylene
- the copolymer (B) contained in the gas barrier resin composition of the present invention is blended to improve the flexibility and impact resistance of the gas barrier resin composition, and is represented by the following general formula (1) , General formula (2) and, if necessary, general formula (3) It is a copolymer containing the monomer structural unit shown by these. The details of the copolymer (B) will be described below.
- R 1 in the general formula (2) and the general formula (3) represents a hydrogen atom or a methyl group, and p represents an integer of 1 to 4.
- R 1 is preferably a hydrogen atom, and p is preferably 1.
- R 2 in the general formula (3) represents a halogen atom, a hydroxyl group, an alkoxy group, or a hydrocarbon group having 1 to 20 carbon atoms which may be substituted with an amino group.
- the hydrocarbon group having 1 to 20 carbon atoms is preferably an alkyl group having 1 to 20 carbon atoms or an aryl group having 6 to 20 carbon atoms.
- the aryl group also includes an aromatic ring to which an alkyl group is added.
- the halogen atom as a substituent is preferably a fluorine, chlorine or bromine atom, more preferably a fluorine atom.
- the alkoxy group as a substituent is preferably an alkoxy group having 1 to 3 carbon atoms.
- methyl, ethyl, phenyl, trifluoromethyl and trichloromethyl are preferable, and methyl and trifluoromethyl are more preferable, from the viewpoint of cost and industrial availability of monomers as raw materials. .
- l, m and n are molar ratios of the monomer structural unit represented by the general formula (1), the monomer structural unit represented by the general formula (2), and the monomer structural unit represented by the general formula (3), respectively n may be 0.
- Molar ratio of the sum of the number of monomer structural units represented by the general formula (2) and the number of monomer structural units represented by the general formula (3) relative to the total number of monomer structural units ⁇ (m + n) / (l + m + n) ⁇ ⁇ 100 Is preferably 0.1 to 80 mol%, more preferably 5 to 50 mol%, and 10 to 40 mol% from the viewpoint of easiness of mixing with the gas barrier resin (A) and physical properties of the gas barrier resin composition More preferable.
- the copolymer (B) imparts flexibility and impact resistance to the gas barrier resin (A), and the copolymer (B) itself preferably has flexibility and impact resistance.
- the molecular weight and molecular weight distribution of the copolymer (B) There are no particular limitations on the molecular weight and molecular weight distribution of the copolymer (B).
- the number average molecular weight (Mn) is preferably 1,000 to 1,000,000, more preferably 2,000 to 300,000, and still more preferably 3,000 to 100,000.
- the ratio (Mw / Mn) of number average molecular weight (Mn) to weight average molecular weight (Mw), which is an index of molecular weight distribution is preferably 1.5 to 4.0, and more preferably 1.5 to 3.0. preferable.
- the copolymer (B) may contain structural units other than the monomer structural units represented by the general formula (1), the general formula (2) and the general formula (3).
- a monomer which gives such another structural unit a (meth) acrylic acid ester type compound, a (meth) acrylic acid compound, a vinyl ether type compound, a vinylsilane type compound, a vinyl siloxane type compound etc. are mentioned.
- These other structural units are preferably 5 mol% or less in the copolymer (B) from the viewpoint of gas barrier properties.
- the method for producing the copolymer (B) is not particularly limited.
- Copolymer (B) by a method of copolymerizing a monomer having an ester group represented by or a method of performing a hydrolysis or saponification reaction after copolymerizing a monomer having an ester group represented by ethylene or the general formula (5) ) Can be obtained.
- R 1 , R 2 and p in the general formula (4) and the general formula (5) have the same meanings as described above.
- Examples of the hydroxyl group-containing monomer represented by the general formula (4) include allyl alcohol, 3-buten-1-ol, 4-penten-1-ol, 5-hexen-1-ol, methallyl alcohol, 3-methyl -3-buten-1-ol, 4-methyl 4-penten-1-ol, 5-methyl-5-hexen-1-ol.
- Preferred is allyl alcohol or methallyl alcohol, and more preferred is allyl alcohol.
- the monomer having an ester group represented by the general formula (5) include allyl acetate, 3-butenyl acetate, 4-pentenyl acetate, 5-hexenyl acetate, methallyl acetate, acetic acid (3-methyl 3-butenyl), Acetic acid (4-methyl-4-pentenyl), acetic acid (5-methyl 5-hexenyl), allyl propionate, 3-butenyl propionate, 4-pentenyl propionate, 5-hexenyl propionate, methallyl propionate, propionic acid ( 3-Methyl 3-butenyl), propionic acid (4-methyl-4-pentenyl), propionic acid (5-methyl 5-hexenyl), allyl butyrate, 3-butenyl butyrate, 4-pentenyl butyrate, 5-hexenyl butyrate, butyric acid Methallyl, butyric acid (3-methyl 3-butenyl), butyric acid (4-methyl-4--
- the polymerization method of ethylene and the monomer represented by the general formula (4) and the general formula (5) is not particularly limited, and, for example, a solution polymerization method, a suspension polymerization method, an emulsion polymerization method, a bulk polymerization method, a gas phase It can be carried out by a known method such as a polymerization method. Also, the polymerization mode may be batch mode or continuous mode, and single stage polymerization or multistage polymerization may also be performed.
- the polymerization form of ethylene and the monomers represented by the general formula (4) and the general formula (5) is not particularly limited.
- a radical using an organic / inorganic peroxide or an azo compound as a catalyst Polymerization method, cationic polymerization method using Lewis acid or Bronsted acid as catalyst, anionic polymerization method using Lewis base as catalyst, coordination anion polymerization method using metal complex catalyst, etc. it can.
- a cationic polymerization method, an anionic polymerization method or a coordinated anionic polymerization method is preferable, and a coordinated anionic polymerization method is particularly preferable.
- the polymerization catalyst used is not particularly limited, but the polymerization activity and From the viewpoint of the properties of the polymer to be obtained, metal complex catalysts described in JP-A-2014-159540, Retable 2013/168626, etc. are particularly preferable.
- the main features of the copolymer (B) obtained by polymerization using the above-mentioned metal complex catalyst, as compared with a polymer obtained by a radical polymerization method used for general polymer production, are shown below. 1) A high molecular weight product can be obtained, and the molecular weight distribution (Mw / Mn) is narrow. 2) The main chain structure is substantially linear.
- the monomer represented by the general formula (4) or (5) has a methylene group at the ⁇ -position of a vinyl group or a vinylidene group, and in radical polymerization, a degenerative chain by hydrogen radical abstraction of the methylene group of the propagating radical Because of the migration, the polymerization reaction stops and the molecular weight is also low. If the molecular weight of the copolymer (B) is low, the melt viscosity of the copolymer (B) will be low, so uniform kneading is difficult at the time of heat melting and kneading with the gas barrier resin (A). Moreover, generally, it is known that the polymer obtained by the radical polymerization method has a wide molecular weight distribution, and Mw / Mn is 4.0 or more.
- radical polymerization In radical polymerization, a backbiting reaction generates radicals in the middle part of the polymer main chain, and a growth reaction proceeds from there as a starting point, so the produced polymer has many long chain branch structures and short chain branches. It is known. When the number of branched structures is large, relatively high reactivity of methine carbon is increased, so that resin deterioration such as oxidation deterioration is likely to occur. Therefore, it can be said that the copolymer (B) obtained by polymerization using the above metal complex catalyst is superior to radical polymerization from the viewpoint of high temperature stability and weather resistance.
- the ratio of the mass of the copolymer (B) to the total mass of the gas barrier resin (A) and the copolymer (B) is It is 1 to 40% by mass, preferably 10 to 35% by mass, and more preferably 15 to 30% by mass.
- the blend ratio of the copolymer (B) is less than 1% by mass, the resin composition obtained is inferior in flexibility and impact resistance.
- the compounding ratio of the copolymer (B) exceeds 40% by mass, the obtained resin composition has inferior mechanical strength.
- gas-barrier resin composition of this invention As a component which comprises the gas-barrier resin composition of this invention, several compounds other than gas-barrier resin (A) and copolymer (B) may exist.
- other polymers antioxidants, light stabilizers, metal deactivators, plasticizers, flame retardants, preservatives, antistatic agents, lubricants, mold release agents, inorganic fillers, glass fibers, foams Agents, coloring agents and the like.
- the gas barrier resin composition of the present invention can be produced by kneading the gas barrier resin (A) and the copolymer (B) by a conventional heat melting and kneading method.
- the heat melting and kneading method include a heat melting and kneading method using a single-screw or twin-screw extruder, a kneader, a mill, Brabender and the like, and a heat-melt kneading method using a twin-screw extruder excellent in kneading ability preferable.
- mixing temperature in this case is not specifically limited, According to the melting temperature and melt viscosity of gas barrier resin (A) and copolymer (B) to knead
- the kneading temperature in the case where the gas barrier resin (A) is a vinyl alcohol resin (A1) can be arbitrarily selected from the range of 150 to 350 ° C., preferably 160 to 300 ° C., and more preferably 180 to 280 ° C.
- the kneading temperature when the gas barrier resin (A) is a polyamide resin (A2) can be arbitrarily selected from the range of 150 to 350 ° C., preferably 160 to 320 ° C., and more preferably 180 to 300 ° C.
- the kneading temperature in the case where the gas barrier resin (A) is a polyester resin (A3) can be arbitrarily selected from the range of 150 to 350 ° C., preferably 160 to 300 ° C., and more preferably 180 to 280 ° C.
- the gas barrier resin composition of the present invention contains a polyamide resin (A2) or a polyester resin (A3)
- the polyamide resin (A2) or the polyester resin (A3) and the copolymer (B) are mixed by kneading. Part or all of may be reacted.
- the gas barrier resin composition of the present invention is formed into various resin molded articles such as films, sheets, containers, pipes, fibers, etc. by melt molding or the like.
- a film means what has a thickness of 300 micrometers or less normally
- seat means what has a thickness which exceeds 300 micrometers normally.
- a container a bag, a tank, a bottle etc. are mentioned.
- melt molding is not particularly limited, for example, extrusion molding, cast molding, inflation extrusion molding, compression molding, blow molding, melt spinning, injection molding, injection blow molding, stretch molding (stretch blow molding, stretched film molding, etc.) Etc.
- melt molding temperature it changes with melt temperature etc. of gas barrier resin (A) and copolymer (B).
- vinyl alcohol resin (A1) 160 to 300 ° C. is preferable, and 180 to 280 ° C. is more preferable.
- polyamide resin (A2) 160 to 320 ° C. is preferable, and 180 to 300 ° C. is more preferable.
- polyester resin (A3) 160 to 300 ° C. is preferable, and 180 to 280 ° C. is more preferable.
- the resin molded product obtained by the above-mentioned melt molding is subjected to secondary processing such as bending, thermoforming (vacuum forming, hot plate pressure forming, vacuum pressure forming), if necessary, to obtain the target resin forming It may be an article.
- the resin molded article may be a resin molded article having a single-layer structure consisting of only a barrier layer (hereinafter, described only as a "barrier layer”) formed of the gas barrier resin composition, but the functional improvement is possible. From the viewpoint of the above, it is also possible to form a molded body of a laminated structure (hereinafter, simply referred to as “laminated body”) having a layer composed of another component on at least one surface of the barrier layer. Examples of the laminate include multilayer films, multilayer sheets, multilayer containers, multilayer pipes, multilayer hoses, multilayer fibers and the like.
- the thermoplastic resin layer formed from a thermoplastic resin is preferable as a layer which consists of another component which comprises the said laminated body.
- the laminate is excellent in appearance, retort resistance, and processing characteristics by combining the barrier layer and the thermoplastic resin layer.
- the layer structure of the laminate is not particularly limited, but the layer comprising the barrier layer is E, the layer obtained from the adhesive resin is Ad, and the layer obtained from the thermoplastic resin is T. Layer structures such as T / E / T, E / Ad / T, T / Ad / E / Ad / T, etc. may be mentioned.
- the method for producing the laminate is not particularly limited, but a method of melt-extruding a thermoplastic resin to a resin molded product (film, sheet, etc.) obtained from the gas barrier resin composition, the resin composition and the other methods
- Method of co-extrusion with a thermoplastic resin method of co-injection of the resin composition with a thermoplastic resin, method of co-blow molding of the resin composition with a thermoplastic resin, the resin composition with a thermoplastic resin
- a method of co-interior molding, a known adhesive such as an isocyanate compound, an organic titanium compound, or a polyester compound with the barrier layer or laminate obtained from the resin composition and a film, sheet or the like of another substrate The method of using and laminating etc. are mentioned.
- thermoplastic resin used for the layer which consists of another component in a laminated body linear low density polyethylene, low density polyethylene, high density polyethylene, ethylene vinyl acetate copolymer, ethylene propylene copolymer, polypropylene, Homopolymers of olefins such as polybutene and polypentene or copolymers thereof, polyesters such as polyethylene terephthalate and polybutylene terephthalate, polyester elastomers, polyamides such as polyamide 6 and polyamide 66, polystyrene, polyvinyl chloride, polyvinylidene chloride, acrylic resin, Polyurethane elastomer, polycarbonate, chlorinated polyethylene, chlorinated polypropylene and the like can be mentioned.
- polypropylene, polyethylene, ethylene / propylene copolymer, ethylene / vinyl acetate copolymer, polyamide, polystyrene and polyester are preferably used.
- the polyamide and polyester here may be the same type as the polyamide resin (A2) and polyester resin (A2) of the present invention.
- Adhesive resin containing a carboxylic acid modified polyolefin is preferable.
- a carboxylic acid-modified polyolefin a modified polymer containing a carboxyl group obtained by chemically bonding (for example, addition reaction, grafting reaction, etc.) an ethylenically unsaturated carboxylic acid, an ester thereof or an anhydride thereof to an olefin polymer
- An olefin polymer can be used suitably.
- the olefin polymer means polyethylene (low pressure, high pressure), linear low density polyethylene, polypropylene, polyolefin such as polypropylene, polybutene, olefin and other monomer (vinyl ester, unsaturated carboxylic acid ester, etc.)
- a polymer eg, ethylene / vinyl acetate copolymer, ethylene / acrylic ester copolymer, etc.
- linear low density polyethylene, ethylene / vinyl acetate copolymer, ethylene / acrylic ester copolymer are preferable, and linear low density polyethylene and ethylene / vinyl acetate copolymer are particularly preferable.
- Ethylenically unsaturated carboxylic acids, esters thereof or anhydrides thereof include ethylenically unsaturated monocarboxylic acids, or esters thereof, ethylenically unsaturated dicarboxylic acids, or mono or diesters thereof, or anhydrides thereof.
- ethylenically unsaturated dicarboxylic acid anhydride is preferable. Specific examples thereof include maleic acid, fumaric acid, itaconic acid, maleic anhydride, itaconic acid anhydride, monomethyl ester of maleic acid, monoethyl ester of maleic acid, diethyl ester of maleic acid, monomethyl ester of fumaric acid, etc.
- maleic anhydride Acids are preferred.
- the gas barrier resin composition and the thermoplastic resin have adhesiveness, the adhesive resin may not be present.
- polyamide layer a layer formed of the polyamide-based resin composition (hereinafter referred to as “polyamide layer”) as the resin molded article
- the resin molded article may have a single layer structure consisting of only a single layer structure, but from the viewpoint of gas barrier property improvement, it may be a resin molded article of a laminated structure having a barrier layer on at least one surface of the polyamide layer is there.
- a resin molded article of a laminated structure having a barrier layer many of the resins forming the barrier layer are inferior in flexibility and moisture resistance, so in the opposite surface of the barrier layer having a polyamide layer, flexibility and moisture resistance It is preferable to have a layer made of an excellent thermoplastic resin.
- a resin which forms a barrier layer polyvinyl alcohol, polyamide MXD6, polyacrylonitrile, polyvinylidene chloride etc. are mentioned.
- thermoplastic resin which forms a layer in the opposite surface of the barrier layer which has a polyamide layer
- the layer obtained from the adhesive resin mentioned above is mentioned.
- polyester layer a layer formed of the polyester-based resin composition (hereinafter referred to as "polyester layer”) as the resin molded article
- A3 polyester-based resin
- polybutylene terephthalate a layer formed of the polyester-based resin composition
- It may be a resin molded article having a single layer structure consisting of only a single layer structure, but it is also possible to obtain a resin molded article of a laminated structure having a barrier layer on at least one surface of the polyester layer from the viewpoint of gas barrier property improvement. .
- a layer made of an excellent thermoplastic resin As a resin which forms a barrier layer, polyvinyl alcohol, polyamide MXD6, polyacrylonitrile, polyvinylidene chloride etc. are mentioned.
- thermoplastic resin forming a layer on the opposite surface of the barrier layer having a polyester layer examples include those described above. Moreover, you may add the layer obtained from the adhesive resin mentioned above as needed.
- the method of coextrusion with the gas barrier resin composition resin composition of the present invention, another gas barrier resin, adhesive resin, thermoplastic resin, etc. is not particularly limited, and a multi-manifold merging method T-die method Feed block merging method T-die method, inflation method and the like.
- the film and the sheet are formed from the gas barrier resin composition. Films and sheets formed from the resin composition are excellent in appearance characteristics, various gas barrier properties, impact resistance, repeated bending resistance, and film / sheet breakage.
- the film sheet includes single layers and multiple layers.
- Films and sheets can be produced by the same method as that described above for producing the resin molded article. From the viewpoint of film- and sheet-breakability improvement of the obtained film and sheet, the unstretched film or sheet obtained through a cast-forming step of melt-extruding the resin composition onto a casting roll is stretched (uniaxial stretching step, sequentially)
- the biaxial process, simultaneous biaxial stretching process, inflation molding process) method is particularly preferred.
- the single layer or multilayer film or sheet obtained from the gas barrier resin composition of the present invention can be used in various applications.
- a film, a sheet, a tube, a bottle container or the like may be obtained by secondary processing of the film or sheet.
- resin molded products obtained by this secondary processing include: (1) multilayer co-oriented sheets or films obtained by uniaxially or biaxially stretching and heat treating a single layer or multilayer film or sheet; (2) Multilayer rolled sheet or film obtained by rolling single layer or multilayer film or sheet, (3) Thermoforming processing such as vacuum forming, pressure forming, vacuum pressure forming of single layer or multilayer film or sheet (4)
- a bottle obtained by performing stretch blow molding etc. to a laminated body, a cup-shaped container, etc. are mentioned.
- resin molded articles obtained from the gas barrier resin composition of the present invention are packaging materials for food and drink, packaging materials for pharmaceuticals, packaging materials for cosmetics, packaging materials for industrial chemicals, packaging materials for agricultural chemicals, organic liquids It can be suitably used for transport pipes, medical infusion bags, fuel containers and the like.
- the content ratio of the monomer structural units represented by the general formula (2) and the general formula (3) of the copolymer (B) is a nuclear magnetic resonance apparatus JNM-ECS400 manufactured by JEOL. It was determined by 1 H and 13 C NMR analysis at 120 ° C. using 1,1,2,2-tetrachloroethane-d4 as solvent.
- Thermophysical properties of polymer were measured by a method according to JIS K 7121 using an X-DSC 7000 differential scanning calorimeter manufactured by SII Nano Technology Inc. About 3 mg of a powdery sample is packed in an aluminum pan, heated from 30 ° C to 200 ° C at a heating rate of 10 ° C / min, held for 5 minutes, and cooled to -150 ° C at 10 ° C / min. The melting curve was obtained by raising the temperature to 200 ° C. at 10 ° C./min.
- a sheet of 1 mm in thickness was prepared by the method described in JIS K 7151 (1995) (cooling method A), and the resin of each example and each comparative example was punched out to prepare JIS K 7162 (1994).
- the tensile test was performed according to JIS K7161 (1994) using the described 5B type small test piece, and the tensile elastic modulus and the breaking elongation were measured. In addition, it carried out on test conditions of temperature 23 ° C, relative humidity 50% RH, distance between chucks 21 mm, measurement speed 10 mm / min, using tensile tester Tensilon RTG-1250 made from A & D as a measuring device. .
- the resulting molded plate was conditioned at a temperature of 23 ⁇ 2 ° C. and a humidity of 50 ⁇ 5 ° C. for 48 hours or more.
- a test piece of the shape of ASTM D1822 Type-S was punched from the conditioned press plate and used as a test sample.
- test conditions Using the above test pieces, the tensile impact strength was measured with reference to method B of JIS K 7160-1996. The only difference from JIS K 7160-1996 is the shape of the test piece. With respect to other measurement conditions, etc., tests were conducted according to JIS K 7160-1996.
- the resulting molded plate was conditioned at a temperature of 23 ⁇ 2 ° C. and a humidity of 50 ⁇ 5 ° C. for 48 hours or more.
- a 5.5 cm-diameter circular test piece was produced from the conditioned plate and used as a test sample.
- Oxygen permeability coefficient (cm 3 ⁇ cm / cm 2 ⁇ s ⁇ Pa) ⁇ oxygen permeation amount (cm 3 ) ⁇ sample thickness (cm) ⁇ / ⁇ transmission area (cm 2 ) ⁇ time (s) ⁇ oxygen partial pressure difference ( Pa) ⁇
- Ethylene / vinyl alcohol copolymer (A1-1) (hereinafter abbreviated as EVOH (A1-1)): manufactured by Kuraray Co., Ltd., trade name Eval (registered trademark) F101B, ethylene content 32 mol%, vinyl Alcohol content 68 mol%.
- Ethylene / vinyl alcohol copolymer (A1-2) (hereinafter abbreviated as EVOH (A1-2)): manufactured by Nippon Synthetic Chemical Industry Co., Ltd., trade name Soarnol (registered trademark) A4412, ethylene content 44 mol %, Vinyl alcohol content 56 mol%.
- Polyamide-based resin (A2-1) (hereinafter abbreviated as polyamide (A2-1)): Mitsubishi Gas Chemical Co., Ltd., S6007, polyamide MXD6.
- Polyester-based resin (A3-1) (hereinafter abbreviated as polyester (A3-1)): Mitsubishi Engineering Plastics Co., Ltd., trade name Novadurane (registered trademark) 5008, polybutylene terephthalate.
- Ethylene / vinyl alcohol copolymer (C-1) (hereinafter abbreviated as polymer (C-1)): Tosoh Corp. product name: Mercen (registered trademark) H-6051, vinyl alcohol content 9 .3 mol%.
- Ethylene / propylene / 1-hexene copolymer (C-2) (hereinafter abbreviated as polymer (C-2)): Kernel KEL 640T (trade name) manufactured by Japan Polyethylene Corporation.
- Ethylene / methyl acrylate copolymer (C-3) (hereinafter abbreviated as polymer (C-3)): manufactured by Nippon Polyethylene Co., Ltd., trade name Lexpearl (registered trademark) EB440H.
- polymer (C-3) manufactured by Nippon Polyethylene Co., Ltd., trade name Lexpearl (registered trademark) EB440H.
- Synthesis Example 1 Synthesis of copolymer (B-1) The following metal complex catalyst was added to allyl acetate (1 L) charged with ethylene gas (0.5 MPa) at 65 ° C. in a 2 L autoclave under a nitrogen gas atmosphere. (1.0 g, 1.4 mmol, published in Japanese Patent Application Publication No. 2014-159540) The toluene solution of (40 mL) was added and stirred at 65 ° C. for 30 hours. Ethylene gas was purged with nitrogen gas, and after cooling to room temperature, the reaction solution in the autoclave was concentrated under reduced pressure to about 100 mL. The concentrate was added to methanol (1 L) to precipitate a polymer.
- the resulting polymer was recovered by filtration, washed with methanol and then dried under reduced pressure to obtain an ethylene / allyl acetate copolymer.
- the yield was 13.3 g.
- the number average molecular weight was 49,000 and the weight average molecular weight was 90000, and Mw / Mn was 1.84 according to Molecular Weight Measurement Method-1. According to differential scanning calorimetry, the melting point was 50.1 ° C., the crystallization temperature was 33.5 ° C., and the glass transition temperature was -41.8 ° C.
- the resulting polymer was recovered by filtration, washed with methanol and then dried under reduced pressure to obtain a copolymer (B-1) which is an ethylene / allyl alcohol copolymer.
- the yield was 8.1 g.
- the number average molecular weight was 46000 and the weight average molecular weight was 70000, and Mw / Mn was 1.53.
- the melting point was 67.4 ° C.
- the crystallization temperature was 47.8 ° C.
- the glass transition point was 4.3 ° C.
- Synthesis Example 2 Synthesis of Copolymer (B-2) Synthesis Example 1 in allyl acetate (1.0 L) filled with ethylene gas (0.12 MPa) at 40 ° C. in a 2 L autoclave under a nitrogen gas atmosphere. An allyl acetate solution (90 mL) of the same metal complex catalyst (0.69 g, 1.00 mmol) as that used in 4. was added, and the mixture was stirred at 40 ° C. for 90 hours. Ethylene gas was purged with nitrogen gas, and after cooling to room temperature, the reaction solution in the autoclave was concentrated under reduced pressure to obtain an ethylene / allyl acetate copolymer. The yield was 9.0 g.
- the number average molecular weight was 7100 and the weight average molecular weight was 11000, and Mw / Mn was 1.55 according to Molecular Weight Measurement Method-1.
- the glass transition temperature was -37.0 ° C by differential scanning calorimetry, and no melting point or crystallization temperature was observed.
- the resulting polymer was recovered by filtration, washed with methanol and then dried under reduced pressure to obtain a copolymer (B-2) which is an ethylene / allyl alcohol copolymer.
- the yield was 3.0 g. Mw / Mn was 1.57, which was calculated as a number average molecular weight of 4800 and a weight average molecular weight of 7600 according to Molecular Weight Measurement Method-3.
- the glass transition temperature was 15.0 ° C. by differential scanning calorimetry, and no melting point or crystallization temperature was observed.
- Example 1-1 In the hopper of a twin-screw kneader-extruder (small kneader manufactured by Xplore Instruments) heated at a cylinder temperature of 220 ° C. by blending 80% by mass of EVOH (A1-1) and 20% by mass of the copolymer (B-1) It was thrown in. After melt-kneading for 3 minutes at a screw rotational speed of 100 rpm, the molten composition flowing out from the die was cooled and cut to prepare a pellet-like vinyl alcohol resin composition 1-1. The obtained resin composition 1-1 was subjected to a tensile test, a tensile impact test, and a gas barrier property (oxygen permeation coefficient) evaluation by the above-mentioned method. The measurement results are shown in Table 2.
- Example 1-2 A vinyl alcohol-based resin composition 1-2 was prepared by the same method as in Example 1-1 except that the compounding ratio of EVOH (A1-1) and copolymer (B-1) was changed, and a tensile test was conducted. , Tensile impact test, and gas barrier property evaluation were performed. The measurement results are shown in Table 2.
- Example 1-3 A vinyl alcohol-based resin composition 1-3 was produced in the same manner as in Example 1-1 except that the copolymer (B-2) was used instead of the copolymer (B-1), and tension was applied. Tests, tensile impact tests, and gas barrier properties were evaluated. The measurement results are shown in Table 2.
- Comparative Example 1-1 A vinyl alcohol resin composition 1-4 was prepared by the same method as Example 1-1 using only EVOH (A1-1) without adding the copolymer (B-1), and a tensile test was conducted. , Tensile impact test, and gas barrier property evaluation were performed. However, the resin composition was brittle, and a sample for tensile impact test could not be prepared. The measurement results are shown in Table 2.
- Comparative Examples 1-2 to 1-4 In the same manner as in Example 1-1 except that the polymer (C-1) was used instead of the copolymer (B-1), a vinyl alcohol resin composition 1- was obtained in the same manner as in Example 1-1. 5 to 1-7 were prepared, and a tensile test, a tensile impact test, and a gas barrier property evaluation were performed. The measurement results are shown in Table 2.
- Comparative Example 1-5 A vinyl alcohol resin composition 1-8 is prepared in the same manner as in Example 1 except that the polymer (C-2) is used instead of the copolymer (B-1), and a tensile test and a tensile test are carried out. Impact test and gas barrier property evaluation were performed. The measurement results are shown in Table 2.
- Comparative Example 1-6 A vinyl alcohol-based resin composition 1-9 is produced in the same manner as in Example 1 except that the polymer (C-3) is used instead of the copolymer (B-1), and a tensile test and a tensile test are carried out. Impact test and gas barrier property evaluation were performed. The measurement results are shown in Table 2.
- the resin composition of the present invention containing the copolymer (B) is compared to the resin composition not containing the copolymer (B) It was found that the flexibility and impact resistance were high. Moreover, it also became clear that the resin composition of this invention does not impair the outstanding oxygen barrier property which vinyl alcohol-type resin (A1) has. From the results of Comparative Examples 1-2 to 1-6, even in the resin composition in which the other polymer (C) is mixed, improvement in flexibility and impact resistance is seen compared to the resin composition of Comparative Example 1-1. However, the improvement effect was small compared to the resin composition of the present invention. Furthermore, any resin composition also impairs the excellent oxygen barrier property of the vinyl alcohol resin (A1), and it can be said that the resin composition of the present invention is superior.
- an ethylene / vinyl alcohol copolymer EVOH (A1-2) having an ethylene content of 44 mol% and a vinyl alcohol content of 56 mol% is used as a vinyl alcohol resin (A1), and a vinyl alcohol resin composition is prepared. Production and evaluation.
- Examples 1-4, 1-5 and Comparative Examples 1-7, 1-8 A vinyl alcohol-based resin composition was prepared in the same manner as in Example 1-1 except that EVOH (A1-2) was used instead of EVOH (A1-1) and the formulation was otherwise described in Table 3. 1-10 to 1-13 were prepared, and a tensile test, a tensile impact test, and a gas barrier property evaluation were performed. The measurement results are shown in Table 3.
- Example 2-1 In the hopper of a twin-screw kneader-extruder (small kneader manufactured by Xplore Instruments) heated at a cylinder temperature of 280 ° C. by blending 80 mass% of polyamide (A2-1) and 20 mass% of copolymer (B-1) It was thrown in. After melt-kneading for 2 minutes at a screw rotational speed of 100 rpm, the molten composition flowing out from the die was cooled and cut to prepare a polyamide resin composition 2-1 in the form of pellets. The obtained resin composition 2-1 was subjected to a tensile test, a tensile impact test, and a gas barrier property (oxygen permeation coefficient) evaluation by the above-mentioned method. The measurement results are shown in Table 4.
- Example 2-2 A pellet-like polyamide resin composition 2-2 is prepared in the same manner as in Example 2-1 except that the copolymer (B-2) is used instead of the copolymer (B-1), and a tensile test is performed. , Tensile impact test, and gas barrier property evaluation were performed. The measurement results are shown in Table 4.
- Comparative Example 2-1 A polyamide resin composition 2-2 was produced by the same method as in Example 2-1 using only the polyamide (A2-1) without adding the copolymer (B-1), and a tensile test, Tensile impact test and gas barrier property evaluation were performed. The measurement results are shown in Table 4.
- Comparative Example 2-2 Using the polymer (C-1) instead of the copolymer (B-1), a polyamide resin composition 2-3 was produced in the same manner as in Example 2-1, and subjected to a tensile test and a tensile impact. Tests and gas barrier properties were evaluated. The measurement results are shown in Table 4.
- the polyamide resin composition of the present invention containing the copolymer (B) is a polyamide resin not containing the copolymer (B). It was found that the resin composition was superior in flexibility, ease of elongation and impact resistance as compared with the resin composition. That is, by kneading the copolymer (B), the polyamide resin (A2) could be modified. In addition, it has also been revealed that the excellent gas barrier properties possessed by the polyamide resin (A2) are not impaired even if the copolymer (B) is kneaded.
- Example 2-1 and Comparative Example 2-2 when the results of Example 2-1 and Comparative Example 2-2 are compared, the present invention in which the copolymer (B) is kneaded compared to the polyamide resin composition in which the other polymer (C) is kneaded It was clarified that the polyamide resin composition of the present invention is superior in flexibility, elongation, impact resistance and gas barrier properties.
- Example 3-1 In the hopper of a twin-screw kneader-extruder (small kneader manufactured by Xplore Instruments) heated at a cylinder temperature of 250 ° C. by blending 80% by mass of polyester (A3-1) and 20% by mass of copolymer (B-1) It was thrown in. After melt-kneading for 2 minutes at a screw rotational speed of 100 rpm, the molten composition flowing out from the die was cooled and cut to prepare a polyester resin composition 3-1 in pellet form. The obtained resin composition 3-1 was subjected to a tensile test, a tensile impact test, and a gas barrier property (oxygen permeability coefficient) evaluation by the above-mentioned method. The measurement results are shown in Table 5.
- Example 3-2 A pellet-like polyester resin composition 3-2 is prepared in the same manner as in Example 3-1 except that the copolymer (B-2) is used instead of the copolymer (B-1), and a tensile test is performed. , Tensile impact test, and gas barrier property evaluation were performed. The measurement results are shown in Table 5.
- Comparative Example 3-1 A polyester-based resin composition 3-3 was produced in the same manner as in Example 3-1 using only the polyester (A3-1) without the addition of the copolymer (B-1), and a tensile test, Tensile impact test and gas barrier property evaluation were performed. The measurement results are shown in Table 5.
- Comparative Example 3-2 A polyester-based resin composition 3-4 was produced in the same manner as in Example 3-1, using the polymer (C-1) instead of the copolymer (B-1), and subjected to a tensile test and a tensile impact test. Tests and gas barrier properties were evaluated. The measurement results are shown in Table 5.
- the polyester resin composition of the present invention containing the copolymer (B) is a polyester type not containing the copolymer (B) It was found that the resin composition was superior in flexibility, ease of elongation and impact resistance as compared with the resin composition. That is, by kneading the copolymer (B), it was possible to modify the polyester resin (A3) without substantially deteriorating the gas barrier properties.
- Example 3-1 and Comparative Example 3-2 when the results of Example 3-1 and Comparative Example 3-2 are compared, the present invention in which the copolymer (B) is kneaded compared to the polyester resin composition in which the other polymer (C) is kneaded It has been revealed that the polyester resin composition of the present invention is more excellent in flexibility, elongation, impact resistance, and gas barrier properties.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Containers Having Bodies Formed In One Piece (AREA)
- Laminated Bodies (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Manufacture Of Macromolecular Shaped Articles (AREA)
Abstract
本発明は、酸素透過係数が1.0×10-14(cm3・cm/cm2・s・Pa)以下のガスバリア性樹脂(A)並びに一般式(1)、一般式(2)、及び一般式(3)(式中、R1は水素原子またはメチル基を表し、R2はハロゲン原子、水酸基、アルコキシ基、またはアミノ基で置換されていてもよい炭素原子数1~20の炭化水素基を表す。l、m、及びnはそれぞれのモノマー構造単位のモル比を表す数値であり、nは0であってもよい。pは1~4の整数を表す。)で示されるモノマー構造単位を含む共重合体(B)を含む樹脂組成物であって、ガスバリア性樹脂(A)と共重合体(B)の合計質量に対する共重合体(B)の質量の割合が1~40質量%であるガスバリア性樹脂組成物に関する。ガスバリア性樹脂(A)としては、ビニルアルコール系樹脂(A1)、ポリアミド系系樹脂(A2)、ポリエステル系樹脂(A3)が好適である。 本発明によれば、前記ガスバリア性樹脂の優れたガスバリア性を損なわずに、ガスバリア性樹脂の欠点である低柔軟性及び低耐衝撃性を改善した樹脂組成物を提供することができる。
Description
本発明は、ビニルアルコール系樹脂、ポリアミド系樹脂、ポリエステル系樹脂に代表されるガスバリア性樹脂の物性が改善されたガスバリア性樹脂組成物、それを含有するシート、フィルム、または袋、ボトル、タンク等の容器、及びガスバリア性樹脂組成物の改質方法に関する。
ポリビニルアルコール(以下、「PVOH」と省略して記載することがある。)やエチレン・ビニルアルコール共重合体(以下、「EVOH」と省略して記載することがある。)等に代表されるビニルアルコール系樹脂は、高分子鎖に存在する水酸基同士の水素結合形成のため、非常に強い分子間力を有する。それゆえに、結晶性が高く、かつ非晶部分においても分子間力が高いため、ビニルアルコール系樹脂は高いガスバリア性を示し、酸素や窒素等の気体分子や有機溶媒の蒸気等は、上記のビニルアルコール系樹脂を使用して成形された成形品(バリア層)を容易には透過できないことが知られている。
特に、EVOHは、溶融成形が可能であり、かつ高いガスバリア性、耐油性、耐有機溶剤性、透明性等を有していることから、その成形品(フィルム、シート、ボトル容器等)は、食品包装材料、医薬品包装材料、工業薬品包装材料、農薬包装材料、燃料容器等の幅広い分野で使用されている。
しかしながら、EVOHは、高い結晶性及び結晶化速度を有するため、硬くて脆い樹脂であり、柔軟性に乏しいという欠点がある。そのため、食品等の包装材料等に成形する際の加工性、特に加熱延伸性が低く、成形時にバリア層にクラックが発生し、製品の歩留まりが低下するおそれや、厚み斑に起因する機械強度の低下、ガスバリア性の低下等が起こり、品質安定性に欠けるおそれがある。さらに、包装材料や成形材料として使用したときに、折り曲げを繰り返して使用すると、屈曲疲労等により容易にクラックやピンホールを生じ、その優れた性能を保持することができなくなる等の問題もある。
しかしながら、EVOHは、高い結晶性及び結晶化速度を有するため、硬くて脆い樹脂であり、柔軟性に乏しいという欠点がある。そのため、食品等の包装材料等に成形する際の加工性、特に加熱延伸性が低く、成形時にバリア層にクラックが発生し、製品の歩留まりが低下するおそれや、厚み斑に起因する機械強度の低下、ガスバリア性の低下等が起こり、品質安定性に欠けるおそれがある。さらに、包装材料や成形材料として使用したときに、折り曲げを繰り返して使用すると、屈曲疲労等により容易にクラックやピンホールを生じ、その優れた性能を保持することができなくなる等の問題もある。
特開昭53-088067号公報(特許文献1)には、EVOHを主成分とするシート等を形成するための樹脂組成物に水または可塑剤を添加させる方法が記載されている。また、特開昭52-141785号公報(特許文献2)及び特開昭59-20345号公報(特許文献3)には、EVOHを主成分とするシート等を形成するための樹脂組成物にポリアミドまたは芳香族ポリエステルをブレンドさせる方法が記載されている。いずれの方法も柔軟性の改善は見られるが、ガスバリア性の低下が大きく、本来のEVOHの優れたガスバリア性を維持できなかった。
上記の加工性を改善するために、EVOHにエチレン・酢酸ビニル共重合体等の各種エラストマーをブレンドすることが試みられている。しかしながら、これらのエラストマーはEVOHとの相溶性が低く、得られる組成物は透明性が低くなり、かつEVOHそのもののガスバリア性を低下させてしまうという不都合があった。
上記の加工性を改善するために、EVOHにエチレン・酢酸ビニル共重合体等の各種エラストマーをブレンドすることが試みられている。しかしながら、これらのエラストマーはEVOHとの相溶性が低く、得られる組成物は透明性が低くなり、かつEVOHそのもののガスバリア性を低下させてしまうという不都合があった。
かかる点に鑑み、上記加工適性を確保しつつ透明性を改善する方法として、エチレン含有量の異なるEVOHをブレンドする方法が検討されている。特開平8-239528号公報(特許文献4)には、ケン化度が95モル%以上及び70モル%以上の2種のエチレン・酢酸ビニル共重合体ケン化物及び末端カルボキシ基を調整したポリアミドを含有する樹脂組成物が開示され、特開2000-212369号公報(特許文献5)には、ケン化度98モル%以上のEVOHと再酢化することでケン化度を下げたEVOHとを含有する樹脂組成物が開示されている。これらの組成物では透明性及び加熱延伸性は改善されるが、ポリアミドや再酢化したEVOHを使用しているため、ロングラン性が低く、ロングラン時のゲル状ブツの発生が多くなり、環境面の観点から、成形時の酢酸等の分解生成物の臭気についての配慮が必要となる等の不都合があった。また、ブレンドさせる2種類以上のEVOH自体が硬く脆い樹脂であり、ブレンドさせてもその性質は変わらなかった。
また、食品容器中のバリア層にクラック等が発生すると、賞味期限よりも短い期間でその部分の食品が腐敗・変色するなど、食品容器としては致命的欠陥となるため、エチレン含有量が比較的高いEVOHを使用することがある。この場合、ガスバリア性が低下するため、その分厚みを厚くする必要があり、コストアップに繋がる。
以上のように、ビニルアルコール系樹脂の柔軟性に乏しく、硬くて脆い欠点に対して、高いガスバリア性等のビニルアルコール系樹脂が有する優れた性質を損なわずに改善させるための満足のいく方法はこれまでなかった。
以上のように、ビニルアルコール系樹脂の柔軟性に乏しく、硬くて脆い欠点に対して、高いガスバリア性等のビニルアルコール系樹脂が有する優れた性質を損なわずに改善させるための満足のいく方法はこれまでなかった。
ポリアミド(別名:ナイロン)系樹脂は、強度、耐熱性、ガスバリア性、光学的特性、または耐油性等の諸物性が優れており、自動車・車両用部品、電気・電子用部品、包装用フィルム等、様々な分野で使用されている。さらに、近年では、特に自動車業界において、車両の軽量化による燃費向上を目的に、金属部品の代替として各部品に採用されている。
一般的に、ポリアミド系樹脂は、ジカルボン酸とジアミンを縮重合させて製造され、アミド結合の繰り返し単位を有する重合体である。原料とするジカルボン酸及びジアミンの化学構造を変えることで、得られるポリアミド系樹脂の耐熱性や強度等の基礎物性を制御することが可能であり、用途の要求物性に合わせて、構造が設計されている。
一般的に、ポリアミド系樹脂は、ジカルボン酸とジアミンを縮重合させて製造され、アミド結合の繰り返し単位を有する重合体である。原料とするジカルボン酸及びジアミンの化学構造を変えることで、得られるポリアミド系樹脂の耐熱性や強度等の基礎物性を制御することが可能であり、用途の要求物性に合わせて、構造が設計されている。
ポリアミドMXD6(別名:ナイロンMXD6、MXD6-ナイロン)は、アジピン酸とメタキシリレンジアミンを縮重合させた芳香族ポリアミド系樹脂であり、他のポリアミド系樹脂に比べて、ガスバリア性に優れている。そのため、ポリアミドMXD6をガスバリア層として、食品包装やPETボトル等に使用されている。
しかしながら、ポリアミドMXD6は、硬くて脆い樹脂であり、柔軟性に乏しいという欠点がある。そのため、食品等の包装材料等に成形する際の加工性、特に加熱延伸性が低く、成形時にバリア層にクラックが発生し、製品の歩留まりが低下するおそれや、厚み斑に起因する機械強度の低下、ガスバリア性の低下等が起こり、品質安定性に欠けるおそれがある。さらに、包装材料や成形材料として使用したときに、折り曲げを繰り返して使用すると、屈曲疲労等により容易にクラックやピンホールを生じ、その優れた性能を保持することができなくなる等の問題もある。
しかしながら、ポリアミドMXD6は、硬くて脆い樹脂であり、柔軟性に乏しいという欠点がある。そのため、食品等の包装材料等に成形する際の加工性、特に加熱延伸性が低く、成形時にバリア層にクラックが発生し、製品の歩留まりが低下するおそれや、厚み斑に起因する機械強度の低下、ガスバリア性の低下等が起こり、品質安定性に欠けるおそれがある。さらに、包装材料や成形材料として使用したときに、折り曲げを繰り返して使用すると、屈曲疲労等により容易にクラックやピンホールを生じ、その優れた性能を保持することができなくなる等の問題もある。
特公平7-15059号公報(特許文献6)や特開平9-183900号公報(米国特許第5780577号;特許文献7)では、耐ピンホール性を向上させるため、エチレン系共重合体の酸変性品やエチレン・酢酸ビニル共重合体の部分けん化体の酸変性品を添加させている。しかしながら、これら共重合体は分子内にカルボキシ基を有しており、ポリアミド系樹脂に混合すると、フィルム成形時に、押出機内の溶融樹脂の通路に設置されているフィルターが閉塞しやすく、またフィルター交換のために、フィルム生産性に与える影響が甚大であるという問題が生じた。
以上のように、ポリアミド系樹脂の柔軟性に乏しく、硬くて脆いという欠点を、高いガスバリア性等のポリアミド系樹脂が有する優れた性質を損なわずに改善させるための満足のいく方法はこれまでなかった。
以上のように、ポリアミド系樹脂の柔軟性に乏しく、硬くて脆いという欠点を、高いガスバリア性等のポリアミド系樹脂が有する優れた性質を損なわずに改善させるための満足のいく方法はこれまでなかった。
ポリエチレンテレフタレート(略称:PET)、ポリブチレンテレフタレート(略称:PBT)、ポリエチレンナフタレート(略称:PEN)等に代表されるポリエステル系樹脂は、機械的特性や化学的特性に優れており、それぞれの特性に応じて、自動車・車両用部品、電気・電子用部品、包装用フィルム等、様々な分野で使用されている。また、特にポリエチレンテレフタレート等の飽和ポリエステルから中空成形されて得られるボトルは、機械的強度、透明性、及びガスバリア性に優れているため、ジュース、炭酸飲料、清涼飲料水等の容器や化粧品、点眼液等の容器としても使用されている。
一般的に、ポリエステル系樹脂は、ジカルボン酸とジオールを縮重合させて製造され、エステル結合の繰り返し単位を有する重合体である。原料とするジカルボン酸及びジオールの化学構造を変えることで、得られるポリエステル系樹脂の耐熱性や強度等の基礎物性を制御することが可能であり、用途の要求物性に合わせて構造が設計されている。
一般的に、ポリエステル系樹脂は、ジカルボン酸とジオールを縮重合させて製造され、エステル結合の繰り返し単位を有する重合体である。原料とするジカルボン酸及びジオールの化学構造を変えることで、得られるポリエステル系樹脂の耐熱性や強度等の基礎物性を制御することが可能であり、用途の要求物性に合わせて構造が設計されている。
ポリブチレンテレフタレートは、テレフタル酸と1,4-ブタンジオールを縮重合させて製造される飽和ポリエステルであり、ヘアドライヤー、電話機、コネクター、スイッチ等の電気・電子部品、ドアハンドル、イグニッションコイル、サイドミラー、バルブ、スイッチ等の自動車部品等に使用されている。
また、フィルム用途では、主に食品包装向けにキャスト成形法による未延伸ポリブチレンテレフタレートフィルムや飲料ボトルのシュリンクラベル向けに一軸延伸ポリブチレンテレフタレートフィルム等が製造されている。しかしながら、ポリブチレンテレフタレートは、その高い結晶性のため硬くて脆い樹脂であり、柔軟性に乏しいという欠点がある。そのため、食品等の包装材料等に成形する際の加工性、特に加熱延伸性が低く、成形時にバリア層にクラックが発生し、製品の歩留まりが低下するおそれや、厚み斑に起因する機械強度の低下、ガスバリア性の低下等が起こり、品質安定性に欠けるおそれがある。さらに、包装材料や成形材料として折り曲げを繰り返して使用すると、屈曲疲労等により容易にクラックやピンホールを生じ、その優れた性能を保持することができなくなる等の問題もある。また、ポリプロピレンやポリアミド6等の汎用プラスチックでは二軸延伸成形が用いられているが、ポリブチレンテレフタレートの場合、高い結晶化速度に起因する低加熱延伸性により、二軸延伸フィルムの実用化に至っていない。
また、フィルム用途では、主に食品包装向けにキャスト成形法による未延伸ポリブチレンテレフタレートフィルムや飲料ボトルのシュリンクラベル向けに一軸延伸ポリブチレンテレフタレートフィルム等が製造されている。しかしながら、ポリブチレンテレフタレートは、その高い結晶性のため硬くて脆い樹脂であり、柔軟性に乏しいという欠点がある。そのため、食品等の包装材料等に成形する際の加工性、特に加熱延伸性が低く、成形時にバリア層にクラックが発生し、製品の歩留まりが低下するおそれや、厚み斑に起因する機械強度の低下、ガスバリア性の低下等が起こり、品質安定性に欠けるおそれがある。さらに、包装材料や成形材料として折り曲げを繰り返して使用すると、屈曲疲労等により容易にクラックやピンホールを生じ、その優れた性能を保持することができなくなる等の問題もある。また、ポリプロピレンやポリアミド6等の汎用プラスチックでは二軸延伸成形が用いられているが、ポリブチレンテレフタレートの場合、高い結晶化速度に起因する低加熱延伸性により、二軸延伸フィルムの実用化に至っていない。
特開2006-241398号公報(特許文献8)や特開2016-191009号公報(特許文献9)には、柔軟性や加熱延伸性を向上させるために、ポリエステルエラストマーやポリカーボネートをブレンドさせる方法が記載されているが、透明性やガスバリア性が大きく低下する等、本来のポリブチレンテレフタレートの特性を損なってしまうという問題があった。
以上のように、ポリエステル系樹脂の柔軟性に乏しく硬くて脆いという欠点を、高いガスバリア性等のポリエステル系樹脂が有する優れた性質を損なわずに改善する満足のいく方法はこれまでなかった。
以上のように、ポリエステル系樹脂の柔軟性に乏しく硬くて脆いという欠点を、高いガスバリア性等のポリエステル系樹脂が有する優れた性質を損なわずに改善する満足のいく方法はこれまでなかった。
本発明の課題は、ガスバリア性樹脂の優れたガスバリア性を損なわずに、ガスバリア性樹脂の欠点である低柔軟性及び低耐衝撃性が改善された樹脂組成物及びそれを使用して成形されたフィルム、シート、または容器等を提供することにある。
本発明者らは、上記の課題を解決すべく鋭意検討を重ねた結果、ガスバリア性樹脂に、柔軟性が高いエチレン・水酸基含有アリルモノマー共重合体を配合させることで、ガスバリア性樹脂由来の優れたガスバリア性を損なわずに優れた柔軟性及び耐衝撃性が付与された樹脂組成物及びその成形品が得られることを見出し、本発明を完成するに至った。
すなわち、本発明は以下の[1]~[19]に関する。
[1] 酸素透過係数が1.0×10-14(cm3・cm/cm2・s・Pa)以下のガスバリア性樹脂(A)並びに一般式(1)、一般式(2)、及び一般式(3)
(式中、R1は水素原子またはメチル基を表し、R2はハロゲン原子、水酸基、アルコキシ基、またはアミノ基で置換されていてもよい炭素原子数1~20の炭化水素基を表す。l、m、及びnはそれぞれのモノマー構造単位のモル比を表す数値であり、nは0であってもよい。pは1~4の整数を表す。)
で示されるモノマー構造単位を含む共重合体(B)を含むことを特徴とする樹脂組成物であって、前記ガスバリア性樹脂(A)と共重合体(B)の合計質量に対する共重合体(B)の質量の割合が1~40質量%であることを特徴とするガスバリア性樹脂組成物。
[2] R2が示す炭素原子数1~20の炭化水素基が、炭素原子数1~20のアルキル基または炭素数6~20のアリール基である前項1に記載のガスバリア性樹脂組成物。
[3] 共重合体(B)の一般式(1)で示されるモノマー構造単位のモル比l、一般式(2)で示されるモノマー構造単位のモル比m、及び一般式(3)で示されるモノマー構造単位のモル比nが、下記式:
80≧{(m+n)/(l+m+n)}×100≧0.1 を満たす前項1または2に記載のガスバリア性樹脂組成物。
[4] 共重合体(B)の一般式(2)で示されるモノマー構造単位のモル比mと一般式(3)で示されるモノマー構造単位のモル比nが、下記式:
100≧{m/(m+n)}×100≧50 を満たす前項1~3のいずれかに記載のガスバリア性樹脂組成物。
[5] 共重合体(B)の一般式(3)で示されるモノマー構造単位において、n=0である前項1~3のいずれかに記載のガスバリア性樹脂組成物。
[6] 共重合体(B)の数平均分子量(Mn)が1000~1000000であり、かつ重量平均分子量(Mw)と数平均分子量(Mn)の比Mw/Mnが1.5~4.0である前項1~5のいずれかに記載のガスバリア性樹脂組成物。
[7] 一般式(2)及び一般式(3)中のR1が水素原子であり、pが1である前項1~6のいずれかに記載のガスバリア性樹脂組成物。
[8] ガスバリア性樹脂(A)が、ビニルアルコール系樹脂(A1)である前項1~7のいずれかに記載のガスバリア性樹脂組成物。
[9] ビニルアルコール系樹脂(A1)が、エチレン構造単位を10~60モル%含むエチレン・ビニルアルコール系共重合体である前項8に記載のガスバリア性樹脂組成物。
[10] ガスバリア性樹脂(A)が、ポリアミド系樹脂(A2)である前項1~7のいずれかに記載のガスバリア性樹脂組成物。
[11] ポリアミド系樹脂(A2)が、ポリアミド6、ポリアミド66、及びポリアミドMXD6の中から選ばれる少なくとも一種である前項10に記載のガスバリア性樹脂組成物。
[12] ガスバリア性樹脂(A)が、ポリエステル系樹脂(A3)である前項1~7のいずれかに記載のガスバリア性樹脂組成物。
[13] ポリエステル系樹脂(A3)が、ポリエチレンテレフタレート及びポリブチレンテレフタレートから選ばれる少なくとも一種である前項12に記載のガスバリア性樹脂組成物。
[14] 前項1~13のいずれかに記載されたガスバリア性樹脂組成物をバリア層として有する容器。
[15] 前項1~13のいずれかに記載されたガスバリア性樹脂組成物を成形してなる樹脂成形品。
[16] 前記成形が、射出成形法または押出成形法である前項15に記載の樹脂成形品。
[17] 前記樹脂成形品が、シート、フィルム、チューブ、パイプ、ボトル、またはタンクのいずれかである前項15または16に記載の樹脂成形品。
[18] 一般式(1)、一般式(2)、及び一般式(3)

(式中、R1は水素原子またはメチル基を表し、R2はハロゲン原子、水酸基、アルコキシ基、またはアミノ基で置換されていてもよい炭素原子数1~20の炭化水素基を表す。l、m、及びnはそれぞれのモノマー構造単位のモル比を表す数値であり、nは0であってもよい。pは1~4の整数を表す。)
で示されるモノマー構造単位を含む共重合体を成分として含むことを特徴とする、酸素透過係数が1.0×10-14(cm3・cm/cm2・s・Pa)以下のガスバリア性樹脂の改質材。
[19] 一般式(1)、一般式(2)、及び一般式(3)

(式中、R1は水素原子またはメチル基を表し、R2はハロゲン原子、水酸基、アルコキシ基、またはアミノ基で置換されていてもよい炭素原子数1~20の炭化水素基を表す。l、m、及びnはそれぞれのモノマー構造単位のモル比を表す数値であり、nは0であってもよい。pは1~4の整数を表す。)
で示されるモノマー構造単位を含む共重合体を酸素透過係数が1.0×10-14(cm3・cm/cm2・s・Pa)以下のガスバリア性樹脂(A)に混合することを特徴とするガスバリア性樹脂の改質方法。
[1] 酸素透過係数が1.0×10-14(cm3・cm/cm2・s・Pa)以下のガスバリア性樹脂(A)並びに一般式(1)、一般式(2)、及び一般式(3)
で示されるモノマー構造単位を含む共重合体(B)を含むことを特徴とする樹脂組成物であって、前記ガスバリア性樹脂(A)と共重合体(B)の合計質量に対する共重合体(B)の質量の割合が1~40質量%であることを特徴とするガスバリア性樹脂組成物。
[2] R2が示す炭素原子数1~20の炭化水素基が、炭素原子数1~20のアルキル基または炭素数6~20のアリール基である前項1に記載のガスバリア性樹脂組成物。
[3] 共重合体(B)の一般式(1)で示されるモノマー構造単位のモル比l、一般式(2)で示されるモノマー構造単位のモル比m、及び一般式(3)で示されるモノマー構造単位のモル比nが、下記式:
80≧{(m+n)/(l+m+n)}×100≧0.1 を満たす前項1または2に記載のガスバリア性樹脂組成物。
[4] 共重合体(B)の一般式(2)で示されるモノマー構造単位のモル比mと一般式(3)で示されるモノマー構造単位のモル比nが、下記式:
100≧{m/(m+n)}×100≧50 を満たす前項1~3のいずれかに記載のガスバリア性樹脂組成物。
[5] 共重合体(B)の一般式(3)で示されるモノマー構造単位において、n=0である前項1~3のいずれかに記載のガスバリア性樹脂組成物。
[6] 共重合体(B)の数平均分子量(Mn)が1000~1000000であり、かつ重量平均分子量(Mw)と数平均分子量(Mn)の比Mw/Mnが1.5~4.0である前項1~5のいずれかに記載のガスバリア性樹脂組成物。
[7] 一般式(2)及び一般式(3)中のR1が水素原子であり、pが1である前項1~6のいずれかに記載のガスバリア性樹脂組成物。
[8] ガスバリア性樹脂(A)が、ビニルアルコール系樹脂(A1)である前項1~7のいずれかに記載のガスバリア性樹脂組成物。
[9] ビニルアルコール系樹脂(A1)が、エチレン構造単位を10~60モル%含むエチレン・ビニルアルコール系共重合体である前項8に記載のガスバリア性樹脂組成物。
[10] ガスバリア性樹脂(A)が、ポリアミド系樹脂(A2)である前項1~7のいずれかに記載のガスバリア性樹脂組成物。
[11] ポリアミド系樹脂(A2)が、ポリアミド6、ポリアミド66、及びポリアミドMXD6の中から選ばれる少なくとも一種である前項10に記載のガスバリア性樹脂組成物。
[12] ガスバリア性樹脂(A)が、ポリエステル系樹脂(A3)である前項1~7のいずれかに記載のガスバリア性樹脂組成物。
[13] ポリエステル系樹脂(A3)が、ポリエチレンテレフタレート及びポリブチレンテレフタレートから選ばれる少なくとも一種である前項12に記載のガスバリア性樹脂組成物。
[14] 前項1~13のいずれかに記載されたガスバリア性樹脂組成物をバリア層として有する容器。
[15] 前項1~13のいずれかに記載されたガスバリア性樹脂組成物を成形してなる樹脂成形品。
[16] 前記成形が、射出成形法または押出成形法である前項15に記載の樹脂成形品。
[17] 前記樹脂成形品が、シート、フィルム、チューブ、パイプ、ボトル、またはタンクのいずれかである前項15または16に記載の樹脂成形品。
[18] 一般式(1)、一般式(2)、及び一般式(3)
で示されるモノマー構造単位を含む共重合体を成分として含むことを特徴とする、酸素透過係数が1.0×10-14(cm3・cm/cm2・s・Pa)以下のガスバリア性樹脂の改質材。
[19] 一般式(1)、一般式(2)、及び一般式(3)
で示されるモノマー構造単位を含む共重合体を酸素透過係数が1.0×10-14(cm3・cm/cm2・s・Pa)以下のガスバリア性樹脂(A)に混合することを特徴とするガスバリア性樹脂の改質方法。
ガスバリア性樹脂に、柔軟性が高いエチレン・水酸基含有アリルモノマー共重合体を配合させた本発明のガスバリア性樹脂組成物は、ガスバリア性樹脂由来の優れたガスバリア性を損なわずに柔軟性及び耐衝撃性が改善され、シート、フィルム、及び袋、ボトル、タンク等の容器のバリア層等として有用である。
本発明の樹脂組成物は、酸素透過係数が1.0×10-14(cm3・cm/cm2・s・Pa)以下のガスバリア性樹脂(A)並びに一般式(1)、一般式(2)、及び一般式(3)

で示されるモノマー構造単位を含む共重合体(B)を含み、前記ガスバリア性樹脂(A)と共重合体(B)の合計質量に対する共重合体(B)の質量の割合が1~40質量%である。
上記一般式(1)~(3)中、R1は水素原子またはメチル基を表し、R2はハロゲン原子、水酸基、アルコキシ基、アミノ基で置換されていてもよい炭素原子数1~20の炭化水素基を表す。l、m、及びnはそれぞれのモノマー構造単位のモル比を表す数値であり、nは0であってもよい。pは1~4の整数を表す。
上記一般式(1)~(3)中、R1は水素原子またはメチル基を表し、R2はハロゲン原子、水酸基、アルコキシ基、アミノ基で置換されていてもよい炭素原子数1~20の炭化水素基を表す。l、m、及びnはそれぞれのモノマー構造単位のモル比を表す数値であり、nは0であってもよい。pは1~4の整数を表す。
[ガスバリア性樹脂(A)]
本発明のガスバリア性樹脂組成物を構成するガスバリア性樹脂(A)は、その酸素透過係数が1.0×10-14(cm3・cm/cm2・s・Pa)以下の樹脂である。酸素透過係数は、JIS K7126に準拠した差圧法により測定された値である。酸素ガス透過量(cm3)は、STP(standard temperature and pressure;0℃、1気圧)での値である。具体的測定方法は実施例の項に示す。
本発明のガスバリア性樹脂組成物を構成するガスバリア性樹脂(A)は、その酸素透過係数が1.0×10-14(cm3・cm/cm2・s・Pa)以下の樹脂である。酸素透過係数は、JIS K7126に準拠した差圧法により測定された値である。酸素ガス透過量(cm3)は、STP(standard temperature and pressure;0℃、1気圧)での値である。具体的測定方法は実施例の項に示す。
このようなガスバリア性樹脂(A)としては非晶性ポリエチレンテレフタレート、ポリ塩化ビニル、ナイロン-6、ポリフッ化ビニル、ポリ塩化ビニリデン、ポリアクリロニトリル、エチレン-ビニルアルコール共重合体、ポリビニルアルコール等が挙げられる。表1に主な樹脂の酸素透過係数を示す(出典:Polymer handbook 4th Edition,John Wiley & Sons,Inc. (1999)他)。

本発明では、ガスバリア性樹脂(A)は、酸素透過係数が1.0×10-14(cm3・cm/cm2・s・Pa)以下の樹脂を複数種組み合わせたものであってもよい。また、本発明の効果を損なわない範囲で、他のガスバリア性樹脂を含んでいてもよい。
本発明の共重合体(B)との相溶性がよいという観点から、特にエチレン-ビニルアルコール共重合体などのビニルアルコール系樹脂(A1)、ナイロン-6などのポリアミド系樹脂(A2)、ポリブチレンテレフタレートなどのポリエステル系樹脂(A3)が好ましい。
[ビニルアルコール系樹脂(A1)]
本発明のガスバリア性樹脂組成物を構成するビニルアルコール系樹脂(A1)は、ビニルアルコール系モノマー由来の構造単位を含む重合体である。ビニルアルコール系モノマーとは炭素-炭素二重結合と水酸基とを含むモノマー(ただし、一般式(2)の構造を与えるモノマーは除く。)であり、例えば、ビニルアルコールや1-ブテン-3-オール、2-メチル-2-プロペン-1-オール、1-ブテン-3,4-ジオールなどが挙げられる。ビニルアルコール系樹脂(A1)は、好ましくはビニルアルコール構造単位を含む重合体であり、ビニルアルコールに、エチレンや1-ブテン-3,4-ジオールなどが共重合されたものであってもよい。具体的には、ポリビニルアルコール(PVOH)やエチレンを共重合させたエチレン・ビニルアルコール共重合体(EVOH)等が挙げられる。ビニルアルコール系樹脂(A1)がエチレン・ビニルアルコール共重合体である場合、エチレン含有量は10~60モル%が好ましく、ガスバリア性及び高湿下でのポリマー物性の観点から、20~50モル%がより好ましい。エチレン含有量が10モル%未満の場合、耐熱性及び押出成形性が低下する。エチレン含有量が60モル%を超える場合、ガスバリア性が著しく低下する。
本発明のガスバリア性樹脂組成物を構成するビニルアルコール系樹脂(A1)は、ビニルアルコール系モノマー由来の構造単位を含む重合体である。ビニルアルコール系モノマーとは炭素-炭素二重結合と水酸基とを含むモノマー(ただし、一般式(2)の構造を与えるモノマーは除く。)であり、例えば、ビニルアルコールや1-ブテン-3-オール、2-メチル-2-プロペン-1-オール、1-ブテン-3,4-ジオールなどが挙げられる。ビニルアルコール系樹脂(A1)は、好ましくはビニルアルコール構造単位を含む重合体であり、ビニルアルコールに、エチレンや1-ブテン-3,4-ジオールなどが共重合されたものであってもよい。具体的には、ポリビニルアルコール(PVOH)やエチレンを共重合させたエチレン・ビニルアルコール共重合体(EVOH)等が挙げられる。ビニルアルコール系樹脂(A1)がエチレン・ビニルアルコール共重合体である場合、エチレン含有量は10~60モル%が好ましく、ガスバリア性及び高湿下でのポリマー物性の観点から、20~50モル%がより好ましい。エチレン含有量が10モル%未満の場合、耐熱性及び押出成形性が低下する。エチレン含有量が60モル%を超える場合、ガスバリア性が著しく低下する。
ビニルアルコール系樹脂(A1)は、ビニルエステル(共)重合体に対して、酸性条件での加水分解反応あるいは塩基性条件でのけん化反応によって製造される。例えば、EVOHはエチレン・酢酸ビニル共重合体に代表されるエチレン・ビニルエステル共重合体を原料として使用し、加水分解あるいはけん化反応によって得られる。ビニルアルコール系樹脂(A1)中のビニルアルコール構造単位数とビニルエステル構造単位数の総和に対するビニルアルコール構造単位数の割合(加水分解率あるいはけん化率)は、熱安定性及びガスバリア性の観点から、85モル%以上が好ましく、90モル%以上がより好ましく、98モル%以上がさらに好ましい。
ビニルエステル(共)重合体を得るためのビニルエステル化合物の(共)重合方法としては特に制限されるものではなく、溶液重合法、懸濁重合法、乳化重合法、バルク重合法等の公知の方法で行うことができる。また、重合様式は、バッチ様式でも連続様式でも可能であり、一段重合でも、多段重合でも行うこともできる。
また、ビニルエステル化合物の(共)重合形式は特に制限されるものではなく、例えば、有機・無機過酸化物やアゾ系化合物等を触媒として使用するラジカル重合法、ルイス酸やブレンステッド酸等を触媒として使用するカチオン重合法、ルイス塩基等を触媒として使用するアニオン重合法、金属錯体触媒等を使用する配位アニオン重合法等の方法で行うことができる。ビニルエステル化合物の重合反応性の観点から、ラジカル重合法が特に好ましい。
重合で使用するビニルエステル化合物としては、酢酸ビニル、プロピオン酸ビニル、トリフルオロ酢酸ビニル、安息香酸ビニル等が挙げられるが、工業的入手の容易さの観点から、酢酸ビニルが特に好ましい。
重合で使用するビニルエステル化合物としては、酢酸ビニル、プロピオン酸ビニル、トリフルオロ酢酸ビニル、安息香酸ビニル等が挙げられるが、工業的入手の容易さの観点から、酢酸ビニルが特に好ましい。
また、ビニルアルコール系樹脂(A1)として、PVOHやEVOHの一部のビニルアルコール構造単位をホルムアルデヒドによるホルマール化やブチルアルデヒドによるブチラール化させた重合体、またはPVOHやEVOHに対して他のモノマーをグラフト重合させて得られた重合体であってもよい。
上記ビニルアルコール系樹脂(A1)には、エチレン及びビニルエステル以外のモノマーに由来する他の構造単位を含んでいてもよい。このような他の構造単位を与えるモノマーとしては、(メタ)アクリル酸エステル系化合物、(メタ)アクリル酸化合物、ビニルエーテル系化合物、ビニルシラン系化合物、ビニルシロキサン系化合物等が挙げられる。
上記ビニルアルコール系樹脂(A1)には、エチレン及びビニルエステル以外のモノマーに由来する他の構造単位を含んでいてもよい。このような他の構造単位を与えるモノマーとしては、(メタ)アクリル酸エステル系化合物、(メタ)アクリル酸化合物、ビニルエーテル系化合物、ビニルシラン系化合物、ビニルシロキサン系化合物等が挙げられる。
[ポリアミド系樹脂(A2)]
本発明のガスバリア性樹脂組成物を構成するポリアミド系樹脂(A2)は、カルボキシ基とアミノ基が縮合して形成されるアミド結合の繰り返し単位を有する重合体であり、ナイロンとも呼ばれる。ポリアミド系樹脂(A2)としては、ε-カプロラクタムを開環重合させて得られるポリアミド6(別名:6-ナイロン、ナイロン6)、アジピン酸とヘキサメチレンジアミンを縮重合させて得られるポリアミド66(別名:6,6-ナイロン、ナイロン6,6)、アジピン酸とメタキシリレンジアミンを縮重合させて得られるポリアミドMXD6(別名:MXD6-ナイロン、ナイロンMXD6)、セバシン酸とメタキシリレンジアミンを縮重合させて得られるポリアミドMXD10(別名:MXD10-ナイロン、ナイロンMXD10)、セバシン酸とヘキサメチレンジアミンを縮重合させて得られるポリアミド610(別名:6,10-ナイロン、ナイロン6,10)、ドデカン二酸とヘキサメチレンジアミンを縮重合させて得られるポリアミド612(別名:6,12-ナイロン、ナイロン6,12)、ω-アミノウンデカン酸を縮重合させて得られるポリアミド11(別名:11-ナイロン、ナイロン11)、ラウロラクタムを開環重合させて得られるポリアミド12(別名:12-ナイロン、ナイロン12)、アジピン酸と1,4-ジアミノブタンを縮重合させて得られるポリアミド46(別名:4,6-ナイロン、ナイロン4,6)、等が挙げられる。ポリアミド系樹脂(A)にはこれら重合体の複数種を含んでいてもよい。ポリアミド系樹脂としては、汎用性、ガスバリア性の観点から、ポリアミド6、ポリアミド66、及びポリアミドMXD6が好ましい。
本発明のガスバリア性樹脂組成物を構成するポリアミド系樹脂(A2)は、カルボキシ基とアミノ基が縮合して形成されるアミド結合の繰り返し単位を有する重合体であり、ナイロンとも呼ばれる。ポリアミド系樹脂(A2)としては、ε-カプロラクタムを開環重合させて得られるポリアミド6(別名:6-ナイロン、ナイロン6)、アジピン酸とヘキサメチレンジアミンを縮重合させて得られるポリアミド66(別名:6,6-ナイロン、ナイロン6,6)、アジピン酸とメタキシリレンジアミンを縮重合させて得られるポリアミドMXD6(別名:MXD6-ナイロン、ナイロンMXD6)、セバシン酸とメタキシリレンジアミンを縮重合させて得られるポリアミドMXD10(別名:MXD10-ナイロン、ナイロンMXD10)、セバシン酸とヘキサメチレンジアミンを縮重合させて得られるポリアミド610(別名:6,10-ナイロン、ナイロン6,10)、ドデカン二酸とヘキサメチレンジアミンを縮重合させて得られるポリアミド612(別名:6,12-ナイロン、ナイロン6,12)、ω-アミノウンデカン酸を縮重合させて得られるポリアミド11(別名:11-ナイロン、ナイロン11)、ラウロラクタムを開環重合させて得られるポリアミド12(別名:12-ナイロン、ナイロン12)、アジピン酸と1,4-ジアミノブタンを縮重合させて得られるポリアミド46(別名:4,6-ナイロン、ナイロン4,6)、等が挙げられる。ポリアミド系樹脂(A)にはこれら重合体の複数種を含んでいてもよい。ポリアミド系樹脂としては、汎用性、ガスバリア性の観点から、ポリアミド6、ポリアミド66、及びポリアミドMXD6が好ましい。
ポリアミド系樹脂(A2)の製造方法としては、ジカルボン酸とジアミンを縮重合させる方法と環状のラクタムを開環重合させる方法の2つに大別され、重合は溶融重合法、固相重合法、溶液重合法、バルク重合法等の公知の方法で行うことができる。また、重合様式は、バッチ様式でも連続様式でも可能であり、一段重合でも、多段重合でも行うこともできる。
[ポリエステル系樹脂(A3)]
本発明のガスバリア性樹脂組成物を構成するポリエステル系樹脂(A3)は、カルボキシ基と水酸基が縮合して形成されるエステル結合(-C(=O)-O-)の繰り返し単位を有する重合体である。
本発明のガスバリア性樹脂組成物を構成するポリエステル系樹脂(A3)は、カルボキシ基と水酸基が縮合して形成されるエステル結合(-C(=O)-O-)の繰り返し単位を有する重合体である。
ポリエステル系樹脂(A3)の製造方法としては、ジカルボン酸またはジエステルとジオールを、酸性条件または塩基性条件下で縮重合させる方法が一般的である。重合は溶融重合法、固相重合法、溶液重合法、バルク重合法等の公知の方法で行うことができる。また、重合様式は、バッチ様式でも連続様式でも可能であり、一段重合でも、多段重合でも行うこともできる。
ポリエステル系樹脂(A3)の製造で使用されるジカルボン酸の具体例としては、フタル酸、イソフタル酸、テレフタル酸、1,2-ナフタレンジカルボン酸、1,3-ナフタレンジカルボン酸、1,4-ナフタレンジカルボン酸、1,5-ナフタレンジカルボン酸、1,6-ナフタレンジカルボン酸、1,7-ナフタレンジカルボン酸、1,8-ナフタレンジカルボン酸、2,3-ナフタレンジカルボン酸、2,4-ナフタレンジカルボン酸、2,6-ナフタレンジカルボン酸、2,7-ナフタレンジカルボン酸、4,4’-ビフェニルジカルボン酸、cis-1,2-シクロプロパンジカルボン酸、trans-1,2-シクロプロパンジカルボン酸、cis-1,2-シクロブタンジカルボン酸、trans-1,2-シクロブタンジカルボン酸、cis-1,3-シクロブタンジカルボン酸、trans-1,3-シクロブタンジカルボン酸、cis-1,2-シクロペンタンジカルボン酸、trans-1,2-シクロペンタンジカルボン酸、cis-1,3-シクロペンタンジカルボン酸、trans-1,3-シクロペンタンジカルボン酸、cis-1,2-シクロヘキサンジカルボン酸、trans-1,2-シクロヘキサンジカルボン酸、cis-1,3-シクロヘキサンジカルボン酸、trans-1,3-シクロヘキサンジカルボン酸、cis-1,4-シクロヘキサンジカルボン酸、trans-1,4-シクロヘキサンジカルボン酸、シュウ酸、マロン酸、コハク酸、グルタル酸、アジピン酸、セバシン酸、ドデカン二酸等が挙げられる。
ポリエステル系樹脂(A3)の製造で使用されるジエステルは、上記のジカルボン酸とアルコールの縮合反応によって得られる化合物である。アルコールの具体例としては、メタノール、エタノール、n-プロパノール、イソプロパノール、n-ブタノール、イソブタノール、s-ブタノール、t-ブタノール、フェノール等が挙げられる。なお、ジカルボン酸またはジエステルは、2つ以上組み合わせて使用してもよい。
ポリエステル系樹脂(A3)の製造で使用されるジオールの具体例としては、エチレングリコール、1,2-プロパンジオール、1,3-プロパンジオール、1,2-ブタンジオール、1,3-ブタンジオール、1,4-ブタンジオール、2,3-ブタンジオール、1,2-ペンタンジオール、1,3-ペンタンジオール、1,4-ペンタンジオール、1,5-ペンタンジオール、2,3-ペンタンジオール、2,4-ペンタンジオール、cis-1,2-シクロヘキサンジメタノール、trans-1,2-シクロヘキサンジメタノール、cis-1,3-シクロヘキサンジメタノール、trans-1,3-シクロヘキサンジメタノール、cis-1,4-シクロヘキサンジメタノール、trans-1,4-シクロヘキサンジメタノール等が挙げられる。なお、ジオールは、2つ以上組み合わせて使用してもよい。
本発明のガスバリア性樹脂組成物を構成するポリエステル系樹脂(A3)は、2種以上のものを組み合わせて使用してもよい。
本発明のガスバリア性樹脂組成物を構成するポリエステル系樹脂(A3)としては、汎用性や強度の観点から、テレフタル酸またはそのエステルとエチレングリコールとの縮重合で製造されるポリエチレンテレフタレート、テレフタル酸またはそのエステルと1,3-プロパンジオールとの縮重合で製造されるポリトリメチレンテレフタレート(略称:PTT)、テレフタル酸またはそのエステルと1,4-ブタンジオールとの縮重合で製造されるポリブチレンテレフタレート、2,6-ナフタレンジカルボン酸またはそのエステルとエチレングリコールとの縮重合で製造されるポリエチレンナフタレート、2,6-ナフタレンジカルボン酸またはそのエステルと1,4-ブタンジオールとの縮重合で製造されるポリブチレンナフタレート(略称:PBN)が好ましい。中でも、汎用性、ガスバリア性の観点からポリエチレンテレフタレート及びポリブチレンテレフタレートが特に好ましい。
[共重合体(B)]
本発明のガスバリア性樹脂組成物に含まれる共重合体(B)は、ガスバリア性樹脂組成物の柔軟性及び耐衝撃性を向上させるために配合されるものであり、以下の一般式(1)、一般式(2)及び必要に応じて一般式(3)

で示されるモノマー構造単位を含む共重合体である。
共重合体(B)について、以下に詳細を述べる。
本発明のガスバリア性樹脂組成物に含まれる共重合体(B)は、ガスバリア性樹脂組成物の柔軟性及び耐衝撃性を向上させるために配合されるものであり、以下の一般式(1)、一般式(2)及び必要に応じて一般式(3)
共重合体(B)について、以下に詳細を述べる。
一般式(2)及び一般式(3)中のR1は水素原子またはメチル基を表し、pは1~4の整数を表す。R1は水素原子が好ましく、pは1が好ましい。
一般式(3)中のR2は、ハロゲン原子、水酸基、アルコキシ基、またはアミノ基で置換されていてもよい炭素原子数1~20の炭化水素基を表す。炭素原子数1~20の炭化水素基としては、炭素原子数1~20のアルキル基または炭素数6~20のアリール基が好ましい。なお、アリール基にはアルキル基が付加した芳香環も含むものとする。置換基としてのハロゲン原子はフッ素、塩素、臭素原子が好ましく、フッ素原子がより好ましい。置換基としてのアルコキシ基としては炭素数1~3のアルコキシ基が好ましい。
一般式(3)中のR2は、ハロゲン原子、水酸基、アルコキシ基、またはアミノ基で置換されていてもよい炭素原子数1~20の炭化水素基を表す。炭素原子数1~20の炭化水素基としては、炭素原子数1~20のアルキル基または炭素数6~20のアリール基が好ましい。なお、アリール基にはアルキル基が付加した芳香環も含むものとする。置換基としてのハロゲン原子はフッ素、塩素、臭素原子が好ましく、フッ素原子がより好ましい。置換基としてのアルコキシ基としては炭素数1~3のアルコキシ基が好ましい。
R2が表すハロゲン原子、水酸基、アルコキシ基、またはアミノ基で置換されていてもよい炭素原子数1~20の炭化水素基の具体例としては、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、n-ペンチル基、ネオペンチル基、n-ヘキシル基、トリフルオロメチル基、トリクロロメチル基、トリブロモメチル基、ペンタフルオロエチル基、ペンタクロロエチル基、ペンタブロモエチル基、フェニル基、2-メチルフェニル基、3-メチルフェニル基、4-メチルフェニル基、2,6-ジメチルフェニル基、2,4,6-トリメチルフェニル基、2-メトキシフェニル基、3-メトキシフェニル基、4-メトキシフェニル基、2,3-ジメトキシフェニル基、2,4-ジメトキシフェニル基、2,5-ジメトキシフェニル基、2,6-ジメトキシフェニル基、2,4,6-トリメトキシフェニル基、2-エトキシフェニル基、3-エトキシフェニル基、4-エトキシフェニル基、2,3-ジエトキシフェニル基、2,4-ジエトキシフェニル基、2,5-ジエトキシフェニル基、2,6-ジエトキシフェニル基、2,4,6-トリエトキシフェニル基、2-プロポキシフェニル基、3-プロポキシフェニル基、4-プロポキシフェニル基、2,3-ジプロポキシフェニル基、2,4-ジプロポキシフェニル基、2,5-ジプロポキシフェニル基、2,6-ジプロポキシフェニル基、2,4,6-トリプロポキシフェニル基、2-イソプロポキシフェニル基、3-イソプロポキシフェニル基、4-イソプロポキシフェニル基、2,3-ジイソプロポキシフェニル基、2,4-ジイソプロポキシフェニル基、2,5-ジイソプロポキシフェニル基、2,6-ジイソプロポキシフェニル基、2,4,6-トリイソプロポキシフェニル基、2-ヒドロキシフェニル基、3-ヒドロキシフェニル基、4-ヒドロキシフェニル基、2,3-ジヒドロキシフェニル基、2,4-ジヒドロキシフェニル基、2,5-ジヒドロキシフェニル基、2,6-ジヒドロキシフェニル基、2,4,6-トリヒドロキシフェニル基、ヒドロキシメチル基、2-ヒドロキシエチル基、2-ヒドロキシ-n-プロピル基、3-ヒドロキシ-n-プロピル基、ペンタフルオロフェニル基、2-トリフルオロメチルフェニル基、3-トリフルオロメチルフェニル基、4-トリフルオロメチルフェニル基、3,5-ジ(トリフルオロメチル)フェニル基、2,3-ジ(トリフルオロメチル)フェニル基、2,4-ジ(トリフルオロメチル)フェニル基、3,4-ジ(トリフルオロメチル)フェニル基、2,4,6-トリフルオロメチルフェニル等が挙げられる。これらの中でも原料となるモノマーのコスト及び工業的な入手容易性の観点から、メチル基、エチル基、フェニル基、トリフルオロメチル基、トリクロロメチル基が好ましく、メチル基、トリフルオロメチル基がより好ましい。
l、m及びnは、それぞれ一般式(1)で示されるモノマー構造単位、一般式(2)で示されるモノマー構造単位、及び一般式(3)で示されるモノマー構造単位のモル比であり、nは0であってもよい。
全モノマー構造単位数に対する一般式(2)で示されるモノマー構造単位数と一般式(3)で示されるモノマー構造単位数の和のモル比率
{(m+n)/(l+m+n)}×100
は、ガスバリア性樹脂(A)との混ざりやすさ及びガスバリア性樹脂組成物の物性の観点から、0.1~80モル%が好ましく、5~50モル%がより好ましく、10~40モル%がさらに好ましい。
全モノマー構造単位数に対する一般式(2)で示されるモノマー構造単位数と一般式(3)で示されるモノマー構造単位数の和のモル比率
{(m+n)/(l+m+n)}×100
は、ガスバリア性樹脂(A)との混ざりやすさ及びガスバリア性樹脂組成物の物性の観点から、0.1~80モル%が好ましく、5~50モル%がより好ましく、10~40モル%がさらに好ましい。
一般式(2)で示されるモノマー構造単位数と一般式(3)で示されるモノマー構造単位数の和に対する一般式(2)で示されるモノマー構造単位数のモル比率
{m/(m+n)}×100
は、ガスバリア性樹脂(A)との混ざりやすさ及びガスバリア性樹脂組成物の物性の観点から、50モル%以上が好ましく、70モル%以上がより好ましく、90モル%以上がさらに好ましい。
{m/(m+n)}×100
は、ガスバリア性樹脂(A)との混ざりやすさ及びガスバリア性樹脂組成物の物性の観点から、50モル%以上が好ましく、70モル%以上がより好ましく、90モル%以上がさらに好ましい。
共重合体(B)は、ガスバリア性樹脂(A)に柔軟性や耐衝撃性を付与するものであり、共重合体(B)自体が柔軟性や耐衝撃性を有するものが好ましい。
共重合体(B)の分子量及び分子量分布に特に制限はない。ガスバリア性樹脂(A)との混ざりやすさの観点から、数平均分子量(Mn)は、1000~1000000が好ましく、2000~300000がより好ましく、3000~100000がさらに好ましい。同様に、分子量分布の指標である数平均分子量(Mn)と重量平均分子量(Mw)の比(Mw/Mn)は、1.5~4.0が好ましく、1.5~3.0がより好ましい。
共重合体(B)の分子量及び分子量分布に特に制限はない。ガスバリア性樹脂(A)との混ざりやすさの観点から、数平均分子量(Mn)は、1000~1000000が好ましく、2000~300000がより好ましく、3000~100000がさらに好ましい。同様に、分子量分布の指標である数平均分子量(Mn)と重量平均分子量(Mw)の比(Mw/Mn)は、1.5~4.0が好ましく、1.5~3.0がより好ましい。
共重合体(B)には、一般式(1)、一般式(2)、一般式(3)で示されるモノマー構造単位以外の構造単位を含んでいてもよい。このような他の構造単位を与えるモノマーとしては、(メタ)アクリル酸エステル系化合物、(メタ)アクリル酸化合物、ビニルエーテル系化合物、ビニルシラン系化合物、ビニルシロキサン系化合物等が挙げられる。これら他の構造単位は、ガスバリア性の観点から共重合体(B)中、5モル%以下とするのが好ましい。
共重合体(B)の製造方法は特に限定されない。例えば、エチレン、一般式(4)

で示される水酸基含有モノマー、及び一般式(5)
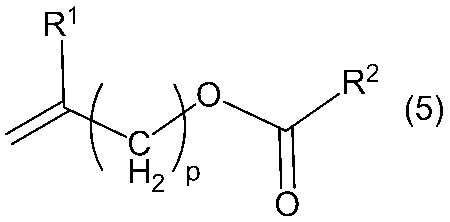
で示されるエステル基を有するモノマーを共重合させる方法、あるいはエチレン、一般式(5)で示されるエステル基を有するモノマーを共重合させた後に加水分解あるいはけん化反応を行う方法で共重合体(B)を得ることができる。
一般式(4)及び一般式(5)中のR1、R2及びpは、上述と同様の意味を表す。
一般式(4)及び一般式(5)中のR1、R2及びpは、上述と同様の意味を表す。
一般式(4)で示される水酸基含有モノマーの例としては、アリルアルコール、3-ブテン-1-オール、4-ペンテン-1-オール、5-ヘキセン-1-オール、メタリルアルコール、3-メチル-3-ブテン-1-オール、4-メチル4-ペンテン-1-オール、5-メチル-5-ヘキセン-1-オールが挙げられる。好ましくはアリルアルコールあるいはメタリルアルコールであり、より好ましくはアリルアルコールである。
一般式(5)で示されるエステル基を有するモノマーの具体例としては、酢酸アリル、酢酸3-ブテニル、酢酸4-ペンテニル、酢酸5-ヘキセニル、酢酸メタリル、酢酸(3-メチル3-ブテニル)、酢酸(4-メチル-4-ペンテニル)、酢酸(5-メチル5-ヘキセニル)、プロピオン酸アリル、プロピオン酸3-ブテニル、プロピオン酸4-ペンテニル、プロピオン酸5-ヘキセニル、プロピオン酸メタリル、プロピオン酸(3-メチル3-ブテニル)、プロピオン酸(4-メチル-4-ペンテニル)、プロピオン酸(5-メチル5-ヘキセニル)、酪酸アリル、酪酸3-ブテニル、酪酸4-ペンテニル、酪酸5-ヘキセニル、酪酸メタリル、酪酸(3-メチル3-ブテニル)、酪酸(4-メチル-4-ペンテニル)、酪酸(5-メチル5-ヘキセニル)、吉草酸アリル、吉草酸3-ブテニル、吉草酸4-ペンテニル、吉草酸5-ヘキセニル、吉草酸メタリル、吉草酸(3-メチル3-ブテニル)、吉草酸(4-メチル-4-ペンテニル)、吉草酸(5-メチル5-ヘキセニル)、安息香酸アリル、安息香酸3-ブテニル、安息香酸4-ペンテニル、安息香酸5-ヘキセニル、安息香酸メタリル、安息香酸(3-メチル3-ブテニル)、安息香酸(4-メチル-4-ペンテニル)、安息香酸(5-メチル5-ヘキセニル)、トリフルオロ酢酸アリル、トリフルオロ酢酸3-ブテニル、トリフルオロ酢酸4-ペンテニル、トリフルオロ酢酸5-ヘキセニル、トリフルオロ酢酸メタリル、トリフルオロ酢酸(3-メチル3-ブテニル)、トリフルオロ酢酸(4-メチル-4-ペンテニル)、トリフルオロ酢酸(5-メチル5-ヘキセニル)、トリクロロ酢酸アリル、トリクロロ酢酸3-ブテニル、トリクロロ酢酸4-ペンテニル、トリクロロ酢酸5-ヘキセニル、トリクロロ酢酸メタリル、トリクロロ酢酸(3-メチル3-ブテニル)、トリクロロ酢酸(4-メチル-4-ペンテニル)、トリクロロ酢酸(5-メチル5-ヘキセニル)、トリブロモ酢酸アリル、トリブロモ酢酸3-ブテニル、トリブロモ酢酸4-ペンテニル、トリブロモ酢酸5-ヘキセニル、トリブロモ酢酸メタリル、トリブロモ酢酸(3-メチル3-ブテニル)、トリブロモ酢酸(4-メチル-4-ペンテニル)、トリブロモ酢酸(5-メチル5-ヘキセニル)等が挙げられる。工業的な入手容易性及びポリマー生産性の観点から、酢酸アリル、酢酸メタリル、トリフルオロ酢酸アリル、トリフルオロ酢酸メタリル、安息香酸アリル、安息香酸メタリルが好ましく、酢酸アリル及び酢酸メタリルがより好ましい。
エチレン、一般式(4)及び一般式(5)で示されるモノマーの重合方法は特に制限されるものではなく、例えば、溶液重合法、懸濁重合法、乳化重合法、バルク重合法、気相重合法等の公知の方法で行うことができる。また、重合様式は、バッチ様式でも連続様式でも可能であり、一段重合でも、多段重合でも行うこともできる。
エチレン、一般式(4)及び一般式(5)で示されるモノマーの重合方法は特に制限されるものではなく、例えば、溶液重合法、懸濁重合法、乳化重合法、バルク重合法、気相重合法等の公知の方法で行うことができる。また、重合様式は、バッチ様式でも連続様式でも可能であり、一段重合でも、多段重合でも行うこともできる。
また、エチレン、一般式(4)及び一般式(5)で示されるモノマーの重合形式は特に制限されるものではなく、例えば、有機・無機過酸化物やアゾ系化合物等を触媒として使用するラジカル重合法、ルイス酸やブレンステッド酸等を触媒として使用するカチオン重合法、ルイス塩基等を触媒として使用するアニオン重合法、金属錯体触媒等を使用する配位アニオン重合法等の方法で行うことができる。一般式(4)及び一般式(5)で示されるモノマーの重合反応性の観点から、カチオン重合法、アニオン重合法、または配位アニオン重合法が好ましく、配位アニオン重合法が特に好ましい。
エチレン、一般式(4)及び一般式(5)で示されるモノマーの重合形式が配位アニオン重合法により製造される場合、使用される重合触媒は特に限定されるものではないが、重合活性及び得られる重合体の特性の点から、特開2014-159540号、再表2013/168626号等に記載されている金属錯体触媒が特に好ましい。一般的な重合体製造に利用されているラジカル重合法で得られる重合体と比較した、上記金属錯体触媒を使用した重合により得られる共重合体(B)の主な特徴を以下に示す。
1)高分子量体を得ることができ、分子量分布(Mw/Mn)が狭い。
2)主鎖構造が実質的に直鎖状である。
1)高分子量体を得ることができ、分子量分布(Mw/Mn)が狭い。
2)主鎖構造が実質的に直鎖状である。
一般式(4)または(5)で示されるモノマーは、ビニル基またはビニリデン基のα位にメチレン基を有しており、ラジカル重合法では、生長ラジカルのメチレン基の水素ラジカル引き抜きによる退化的連鎖移動のため、重合反応が停止し、分子量も低い。共重合体(B)の分子量が低いと、共重合体(B)の溶融粘度が低くなるため、ガスバリア性樹脂(A)との加熱溶融混練時に均一混練が困難である。また、一般的に、ラジカル重合法で得られる重合体は分子量分布が広いことが知られており、Mw/Mnが4.0以上である。
ラジカル重合では、バックバイティング反応により、重合体主鎖の中間部にラジカルが発生し、そこを起点に生長反応が進行するため、生成する重合体は長鎖分岐構造及び短鎖分岐を多く有することが知られている。分岐構造が多いと、比較的反応性が高いメチン炭素が多くなるため、酸化劣化等の樹脂劣化が起きやすくなる。
従って、高温安定性や耐候性の点から、ラジカル重合よりも上記金属錯体触媒を使用した重合により得られる共重合体(B)の方が優れているといえる。
従って、高温安定性や耐候性の点から、ラジカル重合よりも上記金属錯体触媒を使用した重合により得られる共重合体(B)の方が優れているといえる。
本発明のガスバリア性樹脂組成物を構成する共重合体(B)の配合比について、ガスバリア性樹脂(A)と共重合体(B)の合計質量に対する共重合体(B)の質量の割合は1~40質量%であり、10~35質量%が好ましく、15~30質量%がさらに好ましい。共重合体(B)の配合比が1質量%未満になると得られる樹脂組成物は、柔軟性及び耐衝撃性に劣るものとなる。一方で、該共重合体(B)の配合比が40質量%を超える場合、得られる樹脂組成物は、機械強度に劣るものとなる。
本発明のガスバリア性樹脂組成物を構成する成分として、ガスバリア性樹脂(A)及び共重合体(B)以外の複数の化合物が存在していてもよい。具体例として、他の重合体、酸化防止剤、光安定化剤、金属不活性化剤、可塑剤、難燃剤、防腐剤、帯電防止剤、滑剤、離型剤、無機フィラー、ガラスファイバー、発泡剤、着色剤等が挙げられる。
本発明のガスバリア性樹脂組成物は、ガスバリア性樹脂(A)及び共重合体(B)を従来の加熱溶融混練方法で混練することにより製造することができる。加熱溶融混練方法の具体例として、単軸または二軸押出機、ニーダー、ミル、ブラベンダー等による加熱溶融混練方法が挙げられ、混練能力に優れた二軸押出機を使用した加熱溶融混練方法が好ましい。また、この際の混練温度は特に限定されるものではなく、混練させるガスバリア性樹脂(A)及び共重合体(B)の溶融温度及び溶融粘度に合わせて選定される。
ガスバリア性樹脂(A)がビニルアルコール系樹脂(A1)の場合の混練温度は、150~350℃の範囲から任意に選ぶことができ、160~300℃が好ましく、180~280℃がより好ましい。
ガスバリア性樹脂(A)がポリアミド系樹脂(A2)の場合の混練温度は、150~350℃の範囲から任意に選ぶことができ、160~320℃が好ましく、180~300℃がより好ましい。
ガスバリア性樹脂(A)がポリエステル系樹脂(A3)の場合の混練温度は、150~350℃の範囲から任意に選ぶことができ、160~300℃が好ましく、180~280℃がより好ましい。
ガスバリア性樹脂(A)がポリアミド系樹脂(A2)の場合の混練温度は、150~350℃の範囲から任意に選ぶことができ、160~320℃が好ましく、180~300℃がより好ましい。
ガスバリア性樹脂(A)がポリエステル系樹脂(A3)の場合の混練温度は、150~350℃の範囲から任意に選ぶことができ、160~300℃が好ましく、180~280℃がより好ましい。
本発明のガスバリア性樹脂組成物がポリアミド系樹脂(A2)、ポリエステル系樹脂(A3)を含む場合は、混錬によりポリアミド系樹脂(A2)あるいはポリエステル系樹脂(A3)と共重合体(B)の一部またはすべてが反応していてもよい。
本発明のガスバリア性樹脂組成物は、溶融成形等により、フィルム、シート、容器、パイプ、繊維等、各種樹脂成形品に形成される。ここで、フィルムとは、通常300μm以下の厚みを有するものをいい、シートとは、通常300μmを超える厚みを有するものをいう。容器としては、袋、タンク、ボトル等が挙げられる。
溶融成形の方法は特に限定されないが、例えば、押出成形、キャスト成形、インフレーション押出成形、圧縮成形、ブロー成形、溶融紡糸、射出成形、射出ブロー成形、延伸成形(延伸ブロー成形、延伸フィルム成形等)等が挙げられる。溶融成形温度としては、ガスバリア性樹脂(A)及び共重合体(B)の溶融温度等により異なる。ビニルアルコール系樹脂(A1)の場合は、160~300℃が好ましく、180~280℃がより好ましい。ポリアミド系樹脂(A2)の場合は、160~320℃が好ましく、180~300℃がより好ましい。ポリエステル系樹脂(A3)の場合は、160~300℃が好ましく、180~280℃がより好ましい。
樹脂成形品は、再利用の目的で、粉砕し、再度成形することも可能である。上記溶融成形によって得られた樹脂成形品は、必要に応じて、曲げ加工、熱成形(真空成形、熱板圧空成形、真空圧空成形)等の二次加工成形を行って、目的とする樹脂成形品としてもよい。
上記樹脂成形品としては、当該ガスバリア性樹脂組成物から形成されるバリア層(以下、「バリア層」とのみ記載する。)のみからなる単層構造の樹脂成形品としてもよいが、機能性向上の観点から、バリア層の少なくとも一方の面に他の成分からなる層を有する積層構造の成形体(以下、単に「積層体」と記載する。)とすることもできる。積層体としては、多層フィルム、多層シート、多層容器、多層パイプ、多層ホース、多層繊維等が挙げられる。
上記積層体を構成する他の成分からなる層としては、熱可塑性樹脂から形成される熱可塑性樹脂層が好ましい。積層体は、バリア層と熱可塑性樹脂層とを兼ね備えることで、外観性、耐レトルト性、及び加工特性に優れる。
上記積層体の層構造としては、特に制限されるものではないが、上記バリア層からなる層をE、接着性樹脂から得られる層をAd、熱可塑性樹脂から得られる層をTで表わすと、T/E/T、E/Ad/T、T/Ad/E/Ad/T等の層構造が挙げられる。
上記積層体の層構造としては、特に制限されるものではないが、上記バリア層からなる層をE、接着性樹脂から得られる層をAd、熱可塑性樹脂から得られる層をTで表わすと、T/E/T、E/Ad/T、T/Ad/E/Ad/T等の層構造が挙げられる。
積層体を製造する方法は特に制限されるものではないが、ガスバリア性樹脂組成物から得られる樹脂成形品(フィルム、シート等)に熱可塑性樹脂を溶融押出する方法、当該樹脂組成物と他の熱可塑性樹脂とを共押出する方法、当該樹脂組成物と熱可塑性樹脂とを共射出する方法、当該樹脂組成物と熱可塑性樹脂とを共ブロー成形する方法、当該樹脂組成物と熱可塑性樹脂とを共インレーション成形する方法、当該樹脂組成物から得られる上記バリア層もしくは積層体と他の基材のフィルム、シート等とをイソシアネート化合物、有機チタン化合物、ポリエステル系化合物等の公知の接着剤を用いてラミネートする方法等が挙げられる。
積層体における他の成分からなる層に用いられる熱可塑性樹脂としては、直鎖状低密度ポリエチレン、低密度ポリエチレン、高密度ポリエチレン、エチレン・酢酸ビニル共重合体、エチレン・プロピレン共重合体、ポリプロピレン、ポリブテン、ポリペンテン等のオレフィンの単独またはその共重合体、ポリエチレンテレフタレート、ポリブチレンテレフタレート等のポリエステル、ポリエステルエラストマー、ポリアミド6、ポリアミド66等のポリアミド、ポリスチレン、ポリ塩化ビニル、ポリ塩化ビニリデン、アクリル系樹脂、ポリウレタンエラストマー、ポリカーボネート、塩素化ポリエチレン、塩素化ポリプロピレン等が挙げられる。これらの中でも、ポリプロピレン、ポリエチレン、エチレン・プロピレン共重合体、エチレン・酢酸ビニル共重合体、ポリアミド、ポリスチレン、ポリエステルが好ましく用いられる。なお、ここでのポリアミド、ポリエステルは、本発明のポリアミド系樹脂(A2)、ポリエステル系樹脂(A2)と同一種であってもよい。
上記Ad層を構成する接着性樹脂としては、特に限定されないが、カルボン酸変性ポリオレフィンを含有する接着性樹脂が好ましい。カルボン酸変性ポリオレフィンとしては、オレフィン系重合体にエチレン性不飽和カルボン酸、そのエステルまたはその無水物を化学的(例えば、付加反応、グラフト反応等)に結合させて得られるカルボキシル基を含有する変性オレフィン系重合体を好適に用いることができる。ここで、オレフィン系重合体とは、ポリエチレン(低圧、高圧)、直鎖状低密度ポリエチレン、ポリプロピレン、ボリブテン等のポリオレフィン、オレフィンと他のモノマー(ビニルエステル、不飽和カルボン酸エステルなど)との共重合体(例えば、エチレン・酢酸ビニル共重合体、エチレン・アクリル酸エステル共重合体等)を意味する。これらの中でも、直鎖状低密度ポリエチレン、エチレン・酢酸ビニル共重合体、エチレン・アクリル酸エステル共重合体が好ましく、直鎖状低密度ポリエチレン及びエチレン・酢酸ビニル共重合体が特に好ましい。エチレン性不飽和カルボン酸、そのエステルまたはその無水物としては、エチレン性不飽和モノカルボン酸、またはそのエステル、エチレン性不飽和ジカルボン酸、またはそのモノもしくはジエステル、もしくはその無水物が挙げられ、これらの中でもエチレン性不飽和ジカルボン酸無水物が好ましい。具体的にはマレイン酸、フマル酸、イタコン酸、無水マレイン酸、無水イタコン酸、マレイン酸モノメチルエステル、マレイン酸モノエチルエステル、マレイン酸ジエチルエステル、フマル酸モノメチルエステルなどが挙げられ、特に、無水マレイン酸が好適である。また、当該ガスバリア性樹脂組成物及び上記熱可塑性樹脂が接着性を有していれば、接着性樹脂はなくともよい。
また、ポリアミド6等のポリアミド系樹脂(A2)を含むポリアミド系樹脂組成物を使用した場合、上記樹脂成形品としては、ポリアミド系樹脂組成物から形成される層(以下、「ポリアミド層」と記載する。)のみからなる単層構造の樹脂成形品としてもよいが、ガスバリア性向上の観点から、ポリアミド層の少なくとも一方の面にバリア層を有する積層構造の樹脂成形品体とすることも可能である。バリア層を有する積層構造の樹脂成形品とする場合、バリア層を形成する樹脂は柔軟性や耐湿性に劣るものが多いため、ポリアミド層を有するバリア層の反対面に、柔軟性及び耐湿性に優れた熱可塑性樹脂からなる層を持つことが好ましい。
バリア層を形成する樹脂として、ポリビニルアルコール、ポリアミドMXD6、ポリアクリロニトリル、ポリ塩化ビニリデン等が挙げられる。
バリア層を形成する樹脂として、ポリビニルアルコール、ポリアミドMXD6、ポリアクリロニトリル、ポリ塩化ビニリデン等が挙げられる。
ポリアミド層を有するバリア層の反対面に層を形成する熱可塑性樹脂の例としては、上述したものが挙げられる。
また、必要に応じて、上述した接着性樹脂から得られる層を加えてもよい。
また、必要に応じて、上述した接着性樹脂から得られる層を加えてもよい。
ポリブチレンテレフタレート等のポリエステル系樹脂(A3)を含むポリエステル系樹脂組成物を使用した場合、上記樹脂成形品としては、ポリエステル系樹脂組成物から形成される層(以下、「ポリエステル層」と記載する。)のみからなる単層構造の樹脂成形品としてもよいが、ガスバリア性向上の観点から、ポリエステル層の少なくとも一方の面にバリア層を有する積層構造の樹脂成形品体とすることも可能である。バリア層を有する積層構造の樹脂成形品とする場合、バリア層を形成する樹脂は柔軟性や耐湿性に劣るものが多いため、ポリエステル層を有するバリア層の反対面に、柔軟性及び耐湿性に優れた熱可塑性樹脂からなる層を持つことが好ましい。
バリア層を形成する樹脂として、ポリビニルアルコール、ポリアミドMXD6、ポリアクリロニトリル、ポリ塩化ビニリデン等が挙げられる。
バリア層を形成する樹脂として、ポリビニルアルコール、ポリアミドMXD6、ポリアクリロニトリル、ポリ塩化ビニリデン等が挙げられる。
ポリエステル層を有するバリア層の反対面に層を形成する熱可塑性樹脂の例としては、上述したものが挙げられる。
また、必要に応じて、上述した接着性樹脂から得られる層を加えてもよい。
また、必要に応じて、上述した接着性樹脂から得られる層を加えてもよい。
本発明のガスバリア性樹脂組成物樹脂組成物、他のガスバリア性樹脂、接着性樹脂、熱可塑性樹脂等との共押出の方法としては、特に制限されるものではなく、マルチマニホールド合流方式Tダイ法、フィードブロック合流方式Tダイ法、インフレーション法等を挙げることができる。
フィルム及びシートは、当該ガスバリア性樹脂組成物から形成される。当該樹脂組成物から形成されるフィルム及びシートは、外観特性、各種ガスバリア性、耐衝撃性、耐繰り返し屈曲性、及び耐フィルム・シート破断性に優れる。当該フィルム・シートには、単層及び多層のものが含まれる。
フィルム及びシートは、当該ガスバリア性樹脂組成物から形成される。当該樹脂組成物から形成されるフィルム及びシートは、外観特性、各種ガスバリア性、耐衝撃性、耐繰り返し屈曲性、及び耐フィルム・シート破断性に優れる。当該フィルム・シートには、単層及び多層のものが含まれる。
フィルム及びシートは、上述の樹脂成形品を製造する方法として示したものと同様の方法で製造することができる。得られるフィルム及びシートの耐フィルム・シート破断性向上の観点から、当該樹脂組成物をキャスティングロール上に溶融押出するキャスト成形工程を経て得られる無延伸フィルム・シートを延伸する(一軸延伸工程、逐次二軸工程、同時二軸延伸工程、インフレーション成形工程)方法が特に好ましい。
本発明のガスバリア性樹脂組成物から得られた単層あるいは多層のフィルムまたはシートは、各種用途に使用することができる。さらに、上記フィルムまたはシートを二次加工することにより、フィルム、シート、チューブ、ボトル容器等を得てもよい。この二次加工することで得られる樹脂成形品としては、例えば、(1)単層あるいは多層のフィルムまたはシートを一軸または二軸方向に延伸及び熱処理することにより得られる多層共延伸シートまたはフィルム、(2)単層または多層のフィルムまたはシートを圧延することにより得られる多層圧延シートまたはフィルム、(3)単層あるいは多層のフィルムまたはシートを真空成形、圧空成形、真空圧空成形等の熱成形加工することにより得られる多層トレーカップ状容器、(4)積層体にストレッチブロー成形等を行って得られるボトル、カップ状容器等が挙げられる。
本発明のガスバリア性樹脂組成物から得られる樹脂成形品は、具体的には、飲食品用包装材料、医薬品用包装材料、化粧品用包装材料、工業薬品用包装材料、農薬用包装材料、有機液体輸送用パイプ、医療用輸液バッグ、または燃料容器等に好適に使用することができる。
以下、実施例及び比較例を挙げて本発明をより詳細に説明するが、本発明は下記の例に限定されるものではない。
[共重合体(B)の構造解析]
[1]分子量測定
共重合体(B)の数平均分子量(Mn)及び重量平均分子量(Mw)は、以下に記載した3つの測定方法のうち、重合体の溶解性に適した方法を用いて算出した。
1)分子量測定方法-1
昭和電工(株)製AT-806MSカラム(GPC用、2本直列)を備えた東ソー(株)製高温GPC装置HLC-8121GPC/HTを用い、ポリスチレンを分子量の標準物質とするサイズ排除クロマトグラフィー(溶媒:1,2-ジクロロベンゼン、温度:145℃)により測定した。
2)分子量測定方法-2
昭和電工(株)製KF-806Mカラム(GPC用、2本直列)及び昭和電工(株)製SE-61RI検出器を備えた液体クロマトグラフィー装置を用い、ポリスチレンを分子量の標準物質とするサイズ排除クロマトグラフィー(溶媒:テトラヒドロフラン、温度:40℃)により算出した。
3)分子量測定方法-3
昭和電工(株)製Asahipak GF-310HQカラム(2本直列)を備えたウォーターズ(株)製GPC装置Alliance e2695を用い、プルランを分子量の標準物質とするサイズ排除クロマトグラフィー(溶媒:メタノール/水=1:1、温度:40℃)により算出した。
[1]分子量測定
共重合体(B)の数平均分子量(Mn)及び重量平均分子量(Mw)は、以下に記載した3つの測定方法のうち、重合体の溶解性に適した方法を用いて算出した。
1)分子量測定方法-1
昭和電工(株)製AT-806MSカラム(GPC用、2本直列)を備えた東ソー(株)製高温GPC装置HLC-8121GPC/HTを用い、ポリスチレンを分子量の標準物質とするサイズ排除クロマトグラフィー(溶媒:1,2-ジクロロベンゼン、温度:145℃)により測定した。
2)分子量測定方法-2
昭和電工(株)製KF-806Mカラム(GPC用、2本直列)及び昭和電工(株)製SE-61RI検出器を備えた液体クロマトグラフィー装置を用い、ポリスチレンを分子量の標準物質とするサイズ排除クロマトグラフィー(溶媒:テトラヒドロフラン、温度:40℃)により算出した。
3)分子量測定方法-3
昭和電工(株)製Asahipak GF-310HQカラム(2本直列)を備えたウォーターズ(株)製GPC装置Alliance e2695を用い、プルランを分子量の標準物質とするサイズ排除クロマトグラフィー(溶媒:メタノール/水=1:1、温度:40℃)により算出した。
[2]モノマー構造単位含有率
共重合体(B)の一般式(2)及び一般式(3)で示されるモノマー構造単位の含有率は、日本電子(株)製核磁気共鳴装置JNM-ECS400を使用して、溶媒として1,1,2,2-テトラクロロエタン-d4を使用した120℃における1H及び13C-NMR解析によって決定した。
共重合体(B)の一般式(2)及び一般式(3)で示されるモノマー構造単位の含有率は、日本電子(株)製核磁気共鳴装置JNM-ECS400を使用して、溶媒として1,1,2,2-テトラクロロエタン-d4を使用した120℃における1H及び13C-NMR解析によって決定した。
[重合体の熱物性]
重合体の融点、結晶化温度、及びガラス転移点は、エスアイアイ・ナノテクノロジー(株)製X-DSC7000示差走査熱量測定装置を使用して、JIS K7121に準拠した方法で測定した。粉末状のサンプル3mg程度をアルミパンに詰め、30℃から一旦200℃まで昇温速度10℃/分で昇温し、5分間保持した後に、10℃/分で-150℃まで冷却させた後に、10℃/分で200℃まで昇温することにより融解曲線を得た。
重合体の融点、結晶化温度、及びガラス転移点は、エスアイアイ・ナノテクノロジー(株)製X-DSC7000示差走査熱量測定装置を使用して、JIS K7121に準拠した方法で測定した。粉末状のサンプル3mg程度をアルミパンに詰め、30℃から一旦200℃まで昇温速度10℃/分で昇温し、5分間保持した後に、10℃/分で-150℃まで冷却させた後に、10℃/分で200℃まで昇温することにより融解曲線を得た。
[引張弾性率、破断伸びの測定]
各実施例及び各比較例の樹脂を、JIS K7151(1995年)に記載の方法(冷却方法A)で厚さ1mmのシートを作製し、これを打抜いて作製したJIS K7162(1994年)に記載の5B形小型試験片を用いて、JIS K7161(1994年)に従って引張試験を行い、引張弾性率及び破断伸びを測定した。なお、測定装置として(株)エー・アンド・デイ製 引張試験機テンシロンRTG-1250を用い、温度23℃、相対湿度50%RH、チャック間距離21mm、測定速度10mm/分の試験条件で行った。
各実施例及び各比較例の樹脂を、JIS K7151(1995年)に記載の方法(冷却方法A)で厚さ1mmのシートを作製し、これを打抜いて作製したJIS K7162(1994年)に記載の5B形小型試験片を用いて、JIS K7161(1994年)に従って引張試験を行い、引張弾性率及び破断伸びを測定した。なお、測定装置として(株)エー・アンド・デイ製 引張試験機テンシロンRTG-1250を用い、温度23℃、相対湿度50%RH、チャック間距離21mm、測定速度10mm/分の試験条件で行った。
[引張衝撃強度の測定]
1)試験サンプルの作製方法
各実施例及び各比較例の樹脂を、厚さ1mmの加熱プレス用モールドに入れ、表面温度230℃の熱プレス機中で5分間予熱後、加圧と減圧を繰り返すことにより樹脂を溶融すると共に溶融樹脂中の残留気体を脱気し、さらに4.9MPaで加圧し、5分間保持した。その後、4.9MPaの圧力をかけた状態で、10℃/分の速度で徐々に冷却し、温度が室温付近まで低下したところでモールドから成形板を取り出した。得られた成形板を温度23±2℃、湿度50±5℃の環境下で48時間以上状態調節した。状態調節後のプレス板からASTM D1822 Type-Sの形状の試験片を打ち抜き、試験サンプルとした。
1)試験サンプルの作製方法
各実施例及び各比較例の樹脂を、厚さ1mmの加熱プレス用モールドに入れ、表面温度230℃の熱プレス機中で5分間予熱後、加圧と減圧を繰り返すことにより樹脂を溶融すると共に溶融樹脂中の残留気体を脱気し、さらに4.9MPaで加圧し、5分間保持した。その後、4.9MPaの圧力をかけた状態で、10℃/分の速度で徐々に冷却し、温度が室温付近まで低下したところでモールドから成形板を取り出した。得られた成形板を温度23±2℃、湿度50±5℃の環境下で48時間以上状態調節した。状態調節後のプレス板からASTM D1822 Type-Sの形状の試験片を打ち抜き、試験サンプルとした。
2)試験条件
上記試験片を用い、JIS K7160-1996のB法を参考として引張衝撃強度を測定した。なお、JIS K7160-1996と異なるのは、試験片の形状のみである。その他測定条件等に関しては、JIS K7160-1996に準じた方法で試験を実施した。
上記試験片を用い、JIS K7160-1996のB法を参考として引張衝撃強度を測定した。なお、JIS K7160-1996と異なるのは、試験片の形状のみである。その他測定条件等に関しては、JIS K7160-1996に準じた方法で試験を実施した。
[酸素透過係数]
1)試験サンプルの作製方法
各実施例及び各比較例の樹脂を、厚さ0.3mmの加熱プレス用モールドに入れ、表面温度230℃の熱プレス機中で5分間予熱後、加圧と減圧を繰り返すことにより樹脂を溶融すると共に溶融樹脂中の残留気体を脱気し、さらに4.9MPaで加圧し、3分間保持した。その後、4.9MPaの圧力をかけた状態で、10℃/分の速度で徐々に冷却し、温度が室温付近まで低下したところでモールドから成形板を取り出した。得られた成形板を温度23±2℃、湿度50±5℃の環境下で48時間以上状態調節した。状態調節後のプレス板から直径5.5cmの円状試験片を作製し、試験サンプルとした。
1)試験サンプルの作製方法
各実施例及び各比較例の樹脂を、厚さ0.3mmの加熱プレス用モールドに入れ、表面温度230℃の熱プレス機中で5分間予熱後、加圧と減圧を繰り返すことにより樹脂を溶融すると共に溶融樹脂中の残留気体を脱気し、さらに4.9MPaで加圧し、3分間保持した。その後、4.9MPaの圧力をかけた状態で、10℃/分の速度で徐々に冷却し、温度が室温付近まで低下したところでモールドから成形板を取り出した。得られた成形板を温度23±2℃、湿度50±5℃の環境下で48時間以上状態調節した。状態調節後のプレス板から直径5.5cmの円状試験片を作製し、試験サンプルとした。
2)試験条件
JIS K7126(2006年)に準拠した差圧法により、差圧式ガス・水蒸気透過率測定装置(差圧式ガス透過装置:GTRテック(株)製「GTR-30XAD2」、ガスクロマトグラフィー検出器:ヤナコテクニカルサイエンス(株)製「G2700T・F」)を使用して、温度:26℃あるいは40℃、相対湿度:0%RHあるいは90%RH、透過面積:15.2cm2の条件で、試験サンプルの酸素透過量を測定した。酸素透過係数は、以下の式を使用して算出した。
酸素透過係数(cm3・cm/cm2・s・Pa)={酸素透過量(cm3)×サンプル厚み(cm)}/{透過面積(cm2)×時間(s)×酸素分圧差(Pa)}
JIS K7126(2006年)に準拠した差圧法により、差圧式ガス・水蒸気透過率測定装置(差圧式ガス透過装置:GTRテック(株)製「GTR-30XAD2」、ガスクロマトグラフィー検出器:ヤナコテクニカルサイエンス(株)製「G2700T・F」)を使用して、温度:26℃あるいは40℃、相対湿度:0%RHあるいは90%RH、透過面積:15.2cm2の条件で、試験サンプルの酸素透過量を測定した。酸素透過係数は、以下の式を使用して算出した。
酸素透過係数(cm3・cm/cm2・s・Pa)={酸素透過量(cm3)×サンプル厚み(cm)}/{透過面積(cm2)×時間(s)×酸素分圧差(Pa)}
実施例及び比較例で使用したビニルアルコール系樹脂(A1)、ポリアミド系樹脂(A2)、ポリエステル系樹脂(A3)、共重合体(B)、及びその他重合体(C)の詳細を以下に示す。
<ビニルアルコール系樹脂(A1)>
エチレン・ビニルアルコール共重合体(A1-1)(以下、EVOH(A1-1)と略記する。):(株)クラレ製、商品名エバール(登録商標)F101B、エチレン含有率32モル%、ビニルアルコール含有率68モル%。
エチレン・ビニルアルコール共重合体(A1-2)(以下、EVOH(A1-2)と略記する。):日本合成化学工業(株)製、商品名ソアノール(登録商標)A4412、エチレン含有率44モル%、ビニルアルコール含有率56モル%。
エチレン・ビニルアルコール共重合体(A1-1)(以下、EVOH(A1-1)と略記する。):(株)クラレ製、商品名エバール(登録商標)F101B、エチレン含有率32モル%、ビニルアルコール含有率68モル%。
エチレン・ビニルアルコール共重合体(A1-2)(以下、EVOH(A1-2)と略記する。):日本合成化学工業(株)製、商品名ソアノール(登録商標)A4412、エチレン含有率44モル%、ビニルアルコール含有率56モル%。
<ポリアミド系樹脂(A2)>
ポリアミド系樹脂(A2-1)(以下、ポリアミド(A2-1)と略記する。):三菱ガス化学(株)製、S6007、ポリアミドMXD6。
ポリアミド系樹脂(A2-1)(以下、ポリアミド(A2-1)と略記する。):三菱ガス化学(株)製、S6007、ポリアミドMXD6。
<ポリエステル系樹脂(A3)>
ポリエステル系樹脂(A3-1)(以下、ポリエステル(A3-1)と略記する。):三菱エンジニアリングプラスチックス(株)製、商品名ノバデュラン(登録商標)5008、ポリブチレンテレフタレート。
ポリエステル系樹脂(A3-1)(以下、ポリエステル(A3-1)と略記する。):三菱エンジニアリングプラスチックス(株)製、商品名ノバデュラン(登録商標)5008、ポリブチレンテレフタレート。
<共重合体(B)>
エチレン・アリルアルコール共重合体(B-1)(以下、共重合体(B-1)と略記する。):数平均分子量Mn=46000、重量平均分子量Mw=70000、Mw/Mn=1.53、アリルアルコール含有率18モル%(R1=H、n=0、{(m+n)/(l+m+n)}×100=18、{m/(m+n)}×100=100)。
エチレン・アリルアルコール共重合体(B-2)(以下、共重合体(B-2)と略記する。):数平均分子量Mn=4700、重量平均分子量Mw=7600、Mw/Mn=1.57、アリルアルコール含有率41モル%(R1=H、n=0、{(m+n)/(l+m+n)}×100=41、{m/(m+n)}×100=100)。
エチレン・アリルアルコール共重合体(B-1)(以下、共重合体(B-1)と略記する。):数平均分子量Mn=46000、重量平均分子量Mw=70000、Mw/Mn=1.53、アリルアルコール含有率18モル%(R1=H、n=0、{(m+n)/(l+m+n)}×100=18、{m/(m+n)}×100=100)。
エチレン・アリルアルコール共重合体(B-2)(以下、共重合体(B-2)と略記する。):数平均分子量Mn=4700、重量平均分子量Mw=7600、Mw/Mn=1.57、アリルアルコール含有率41モル%(R1=H、n=0、{(m+n)/(l+m+n)}×100=41、{m/(m+n)}×100=100)。
<その他重合体(C)>
エチレン・ビニルアルコール共重合体(C-1)(以下、重合体(C-1)と略記する。):東ソー(株)製、商品名メルセン(登録商標)H-6051、ビニルアルコール含有率9.3モル%。
エチレン・プロピレン・1-ヘキセン共重合体(C-2)(以下、重合体(C-2)と略記する。):日本ポリエチレン(株)製、商品名カーネル(登録商標)KJ640T。
エチレン・アクリル酸メチル共重合体(C-3)(以下、重合体(C-3)と略記する。):日本ポリエチレン(株)製、商品名レクスパール(登録商標)EB440H。
以下に、共重合体(B-1)及び(B-2)の合成例を示す。
エチレン・ビニルアルコール共重合体(C-1)(以下、重合体(C-1)と略記する。):東ソー(株)製、商品名メルセン(登録商標)H-6051、ビニルアルコール含有率9.3モル%。
エチレン・プロピレン・1-ヘキセン共重合体(C-2)(以下、重合体(C-2)と略記する。):日本ポリエチレン(株)製、商品名カーネル(登録商標)KJ640T。
エチレン・アクリル酸メチル共重合体(C-3)(以下、重合体(C-3)と略記する。):日本ポリエチレン(株)製、商品名レクスパール(登録商標)EB440H。
以下に、共重合体(B-1)及び(B-2)の合成例を示す。
合成例1:共重合体(B-1)の合成
窒素ガス雰囲気下、2Lオートクレーブ中で、65℃でエチレンガス(0.5MPa)が充填された酢酸アリル(1L)に、下記の金属錯体触媒(1.0g,1.4mmol、公開公報:特開2014-159540号に記載)

のトルエン溶液(40mL)を加え、65℃で30時間撹拌した。エチレンガスを窒素ガスでパージさせて、室温まで冷却後、オートクレーブ内の反応液を100mL程度になるまで減圧濃縮させた。濃縮液をメタノール(1L)に加え重合体を析出させた。生じた重合体をろ過によって回収し、メタノールで洗浄した後に減圧下乾燥して、エチレン・酢酸アリル共重合体を得た。収量は13.3gであった。分子量測定方法-1により、数平均分子量49000、重量平均分子量90000と算出し、Mw/Mnは1.84であった。示差走査熱量測定により融点は50.1℃、結晶化温度33.5℃、ガラス転移点-41.8℃であった。共重合体中の酢酸アリル含有率は、1H-NMR及び13C-NMR測定により、エチレン:酢酸アリルのモル比は82.0:18.0(酢酸アリルモル分率=18.0%)と決定した。
窒素ガス雰囲気下、2Lオートクレーブ中で、65℃でエチレンガス(0.5MPa)が充填された酢酸アリル(1L)に、下記の金属錯体触媒(1.0g,1.4mmol、公開公報:特開2014-159540号に記載)
続いて、得られたエチレン・酢酸アリル共重合体のけん化反応を行った。窒素ガス雰囲気下、エチレン・酢酸アリル共重合体(10.3g)、トルエン(140mL)及びメタノール(75mL)を含む1Lセパラブルナスフラスコに、水酸化ナトリウム(和光純薬工業製、0.088g、2.2mmol)のメタノール溶液(10mL)を加え、加熱還流下、2.5時間撹拌させた。室温まで冷却後、反応液をメタノール(1L)に加え、重合体を析出させた。生じた重合体をろ過によって回収し、メタノールで洗浄した後に減圧下乾燥して、エチレン・アリルアルコール共重合体である共重合体(B-1)を得た。収量は8.1gであった。分子量測定方法-2により、数平均分子量46000、重量平均分子量70000と算出し、Mw/Mnは1.53であった。示差走査熱量測定により融点は67.4℃、結晶化温度47.8℃、ガラス転移点4.3℃であった。共重合体中のアリルアルコール含有率は、1H-NMR及び13C-NMR測定により、エチレン:アリルアルコールのモル比は82.0:18.0(アリルアルコールモル分率=18.0%)と決定した。
合成例2:共重合体(B-2)の合成
窒素ガス雰囲気下、2Lオートクレーブ中で、40℃でエチレンガス(0.12MPa)が充填された酢酸アリル(1.0L)に、合成例1で使用したものと同じ金属錯体触媒(0.69g,1.00mmol)の酢酸アリル溶液(90mL)を加え、40℃で90時間撹拌した。エチレンガスを窒素ガスでパージさせて、室温まで冷却後、オートクレーブ内の反応液を減圧濃縮させ、エチレン・酢酸アリル共重合体を得た。収量は9.0gであった。分子量測定方法-1により、数平均分子量7100、重量平均分子量11000と算出し、Mw/Mnは1.55であった。示差走査熱量測定によりガラス転移点-37.0℃であり、融点及び結晶化温度は観測されなかった。共重合体中の酢酸アリル含有率は、1H-NMR及び13C-NMR測定により、エチレン:酢酸アリルのモル比は60.0:40.0(酢酸アリルモル分率=40.0%)と決定した。
続いて、得られたエチレン・酢酸アリル共重合体のけん化反応を行った。窒素ガス雰囲気下、エチレン・酢酸アリル共重合体(5.3g)、トルエン(120mL)及びメタノール(65mL)を含む1Lセパラブルナスフラスコに、水酸化ナトリウム(和光純薬工業製、0.075g、1.9mmol)のメタノール溶液(10mL)を加え、加熱還流下、3時間撹拌させた。室温まで冷却後、反応液を水・アセトン混合溶媒(1:1vol/vol、1L)に加え、重合体を析出させた。生じた重合体をろ過によって回収し、メタノールで洗浄した後に減圧下乾燥して、エチレン・アリルアルコール共重合体である共重合体(B-2)を得た。収量は3.0gであった。分子量測定方法-3により、数平均分子量4800、重量平均分子量7600と算出し、Mw/Mnは1.57であった。示差走査熱量測定によりガラス転移点は15.0℃であり、融点及び結晶化温度は観測されなかった。共重合体中のアリルアルコール含有率は、1H-NMR及び13C-NMR測定により、エチレン:アリルアルコールのモル比は60.0:40.0(アリルアルコールモル分率=40.0%)と決定した。
窒素ガス雰囲気下、2Lオートクレーブ中で、40℃でエチレンガス(0.12MPa)が充填された酢酸アリル(1.0L)に、合成例1で使用したものと同じ金属錯体触媒(0.69g,1.00mmol)の酢酸アリル溶液(90mL)を加え、40℃で90時間撹拌した。エチレンガスを窒素ガスでパージさせて、室温まで冷却後、オートクレーブ内の反応液を減圧濃縮させ、エチレン・酢酸アリル共重合体を得た。収量は9.0gであった。分子量測定方法-1により、数平均分子量7100、重量平均分子量11000と算出し、Mw/Mnは1.55であった。示差走査熱量測定によりガラス転移点-37.0℃であり、融点及び結晶化温度は観測されなかった。共重合体中の酢酸アリル含有率は、1H-NMR及び13C-NMR測定により、エチレン:酢酸アリルのモル比は60.0:40.0(酢酸アリルモル分率=40.0%)と決定した。
続いて、得られたエチレン・酢酸アリル共重合体のけん化反応を行った。窒素ガス雰囲気下、エチレン・酢酸アリル共重合体(5.3g)、トルエン(120mL)及びメタノール(65mL)を含む1Lセパラブルナスフラスコに、水酸化ナトリウム(和光純薬工業製、0.075g、1.9mmol)のメタノール溶液(10mL)を加え、加熱還流下、3時間撹拌させた。室温まで冷却後、反応液を水・アセトン混合溶媒(1:1vol/vol、1L)に加え、重合体を析出させた。生じた重合体をろ過によって回収し、メタノールで洗浄した後に減圧下乾燥して、エチレン・アリルアルコール共重合体である共重合体(B-2)を得た。収量は3.0gであった。分子量測定方法-3により、数平均分子量4800、重量平均分子量7600と算出し、Mw/Mnは1.57であった。示差走査熱量測定によりガラス転移点は15.0℃であり、融点及び結晶化温度は観測されなかった。共重合体中のアリルアルコール含有率は、1H-NMR及び13C-NMR測定により、エチレン:アリルアルコールのモル比は60.0:40.0(アリルアルコールモル分率=40.0%)と決定した。
実施例1-1:
EVOH(A1-1)80質量%及び共重合体(B-1)20質量%を配合して、シリンダー温度220℃に加熱した二軸混練押出機(Xplore Instruments社製 小型混練機)のホッパーに投入した。スクリュー回転数100rpmにて3分間溶融混練を行った後、ダイより流出する溶融組成物を冷却後裁断し、ペレット状のビニルアルコール系樹脂組成物1-1を作製した。
得られた樹脂組成物1-1は、上述の方法で引張試験、引張衝撃試験、ガスバリア性(酸素透過係数)評価を行った。測定結果を表2に示した。
EVOH(A1-1)80質量%及び共重合体(B-1)20質量%を配合して、シリンダー温度220℃に加熱した二軸混練押出機(Xplore Instruments社製 小型混練機)のホッパーに投入した。スクリュー回転数100rpmにて3分間溶融混練を行った後、ダイより流出する溶融組成物を冷却後裁断し、ペレット状のビニルアルコール系樹脂組成物1-1を作製した。
得られた樹脂組成物1-1は、上述の方法で引張試験、引張衝撃試験、ガスバリア性(酸素透過係数)評価を行った。測定結果を表2に示した。
実施例1-2:
EVOH(A1-1)及び共重合体(B-1)の配合比を変えたこと以外は実施例1-1と同様の方法で、ビニルアルコール系樹脂組成物1-2を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表2に示した。
EVOH(A1-1)及び共重合体(B-1)の配合比を変えたこと以外は実施例1-1と同様の方法で、ビニルアルコール系樹脂組成物1-2を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表2に示した。
実施例1-3:
共重合体(B-1)の代わりに共重合体(B-2)を使用したこと以外は実施例1-1と同様の方法で、ビニルアルコール系樹脂組成物1-3を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表2に示した。
共重合体(B-1)の代わりに共重合体(B-2)を使用したこと以外は実施例1-1と同様の方法で、ビニルアルコール系樹脂組成物1-3を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表2に示した。
比較例1-1:
共重合体(B-1)を加えずに、EVOH(A1-1)のみを使用して、実施例1-1と同様の方法でビニルアルコール系樹脂組成物1-4を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。ただし、樹脂組成物が脆く、引張衝撃試験用のサンプルは作製できなかった。測定結果を表2に示した。
共重合体(B-1)を加えずに、EVOH(A1-1)のみを使用して、実施例1-1と同様の方法でビニルアルコール系樹脂組成物1-4を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。ただし、樹脂組成物が脆く、引張衝撃試験用のサンプルは作製できなかった。測定結果を表2に示した。
比較例1-2~1-4:
共重合体(B-1)の代わりに重合体(C-1)を使用して、表2に記載の配合比で、実施例1-1と同様の方法でビニルアルコール系樹脂組成物1-5~1-7を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表2に示した。
共重合体(B-1)の代わりに重合体(C-1)を使用して、表2に記載の配合比で、実施例1-1と同様の方法でビニルアルコール系樹脂組成物1-5~1-7を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表2に示した。
比較例1-5:
共重合体(B-1)の代わりに重合体(C-2)を使用したこと以外は実施例1と同様の方法で、ビニルアルコール系樹脂組成物1-8を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表2に示した。
共重合体(B-1)の代わりに重合体(C-2)を使用したこと以外は実施例1と同様の方法で、ビニルアルコール系樹脂組成物1-8を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表2に示した。
比較例1-6:
共重合体(B-1)の代わりに重合体(C-3)を使用したこと以外は実施例1と同様の方法で、ビニルアルコール系樹脂組成物1-9を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表2に示した。
共重合体(B-1)の代わりに重合体(C-3)を使用したこと以外は実施例1と同様の方法で、ビニルアルコール系樹脂組成物1-9を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表2に示した。

実施例1-1~1-3及び比較例1-1の結果より、共重合体(B)を含む本発明の樹脂組成物が、共重合体(B)を含まない樹脂組成物に比べて、柔軟性及び耐衝撃性が高いことが判った。また、本発明の樹脂組成物は、ビニルアルコール系樹脂(A1)が持つ優れた酸素バリア性を損なわないことも明らかとなった。
比較例1-2~1-6の結果より、その他重合体(C)を混合させた樹脂組成物でも比較例1-1の樹脂組成物に比べて、柔軟性や耐衝撃性の改善が見られるが、本発明の樹脂組成物に比べると改善効果は小さかった。さらに、いずれの樹脂組成物も、ビニルアルコール系樹脂(A1)が持つ優れた酸素バリア性を損なうものであり、本発明の樹脂組成物の方が優れているといえる。
比較例1-2~1-6の結果より、その他重合体(C)を混合させた樹脂組成物でも比較例1-1の樹脂組成物に比べて、柔軟性や耐衝撃性の改善が見られるが、本発明の樹脂組成物に比べると改善効果は小さかった。さらに、いずれの樹脂組成物も、ビニルアルコール系樹脂(A1)が持つ優れた酸素バリア性を損なうものであり、本発明の樹脂組成物の方が優れているといえる。
続いて、エチレン含有率44モル%、ビニルアルコール含有率56モル%であるエチレン・ビニルアルコール共重合体EVOH(A1-2)をビニルアルコール系樹脂(A1)として使用し、ビニルアルコール系樹脂組成物の作製及び評価を行った。
実施例1-4、1-5及び比較例1-7、1-8:
EVOH(A1-1)の代わりにEVOH(A1-2)を使用して、それ以外は表3に記載の配合とした以外は実施例1-1と同様の方法で、ビニルアルコール系樹脂組成物1-10~1-13を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表3に示した。
EVOH(A1-1)の代わりにEVOH(A1-2)を使用して、それ以外は表3に記載の配合とした以外は実施例1-1と同様の方法で、ビニルアルコール系樹脂組成物1-10~1-13を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表3に示した。

実施例1-4~1-5及び比較例1-1の結果より、ビニルアルコール系樹脂(A1)がエチレン及びビニルアルコール含有率が異なるEVOH(A1-2)であっても、本発明の樹脂組成物が、共重合体(B)を含まない樹脂組成物に比べて、靱性及び耐衝撃性が高く、かつビニルアルコール系樹脂(A1)が持つ優れた酸素バリア性を損なわないことが判った。
比較例1-8の結果より、その他重合体(C-1)を混合させた樹脂組成物では、比較例1-1の樹脂組成物に比べて、耐衝撃性の改善は見られるが、柔軟性の改善は全く見られなかった。また、酸素バリア性の低下が見られた。
比較例1-8の結果より、その他重合体(C-1)を混合させた樹脂組成物では、比較例1-1の樹脂組成物に比べて、耐衝撃性の改善は見られるが、柔軟性の改善は全く見られなかった。また、酸素バリア性の低下が見られた。
実施例2-1:
ポリアミド(A2-1)80質量%及び共重合体(B-1)20質量%を配合して、シリンダー温度280℃に加熱した二軸混練押出機(Xplore Instruments社製 小型混練機)のホッパーに投入した。スクリュー回転数100rpmにて2分間溶融混練を行った後、ダイより流出する溶融組成物を冷却後裁断し、ペレット状のポリアミド系樹脂組成物2-1を作製した。
得られた樹脂組成物2-1は、上述の方法で引張試験、引張衝撃試験、ガスバリア性(酸素透過係数)評価を行った。測定結果を表4に示す。
ポリアミド(A2-1)80質量%及び共重合体(B-1)20質量%を配合して、シリンダー温度280℃に加熱した二軸混練押出機(Xplore Instruments社製 小型混練機)のホッパーに投入した。スクリュー回転数100rpmにて2分間溶融混練を行った後、ダイより流出する溶融組成物を冷却後裁断し、ペレット状のポリアミド系樹脂組成物2-1を作製した。
得られた樹脂組成物2-1は、上述の方法で引張試験、引張衝撃試験、ガスバリア性(酸素透過係数)評価を行った。測定結果を表4に示す。
実施例2-2:
共重合体(B-1)の代わりに共重合体(B-2)を用いた以外は実施例2-1と同様にしてペレット状のポリアミド系樹脂組成物2-2を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表4に示す。
共重合体(B-1)の代わりに共重合体(B-2)を用いた以外は実施例2-1と同様にしてペレット状のポリアミド系樹脂組成物2-2を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表4に示す。
比較例2-1:
共重合体(B-1)を加えずに、ポリアミド(A2-1)のみを使用して、実施例2-1と同様の方法でポリアミド系樹脂組成物2-2を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表4に示す。
共重合体(B-1)を加えずに、ポリアミド(A2-1)のみを使用して、実施例2-1と同様の方法でポリアミド系樹脂組成物2-2を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表4に示す。
比較例2-2:
共重合体(B-1)の代わりに重合体(C-1)を使用して、実施例2-1と同様の方法でポリアミド系樹脂組成物2-3を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表4に示す。
共重合体(B-1)の代わりに重合体(C-1)を使用して、実施例2-1と同様の方法でポリアミド系樹脂組成物2-3を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表4に示す。
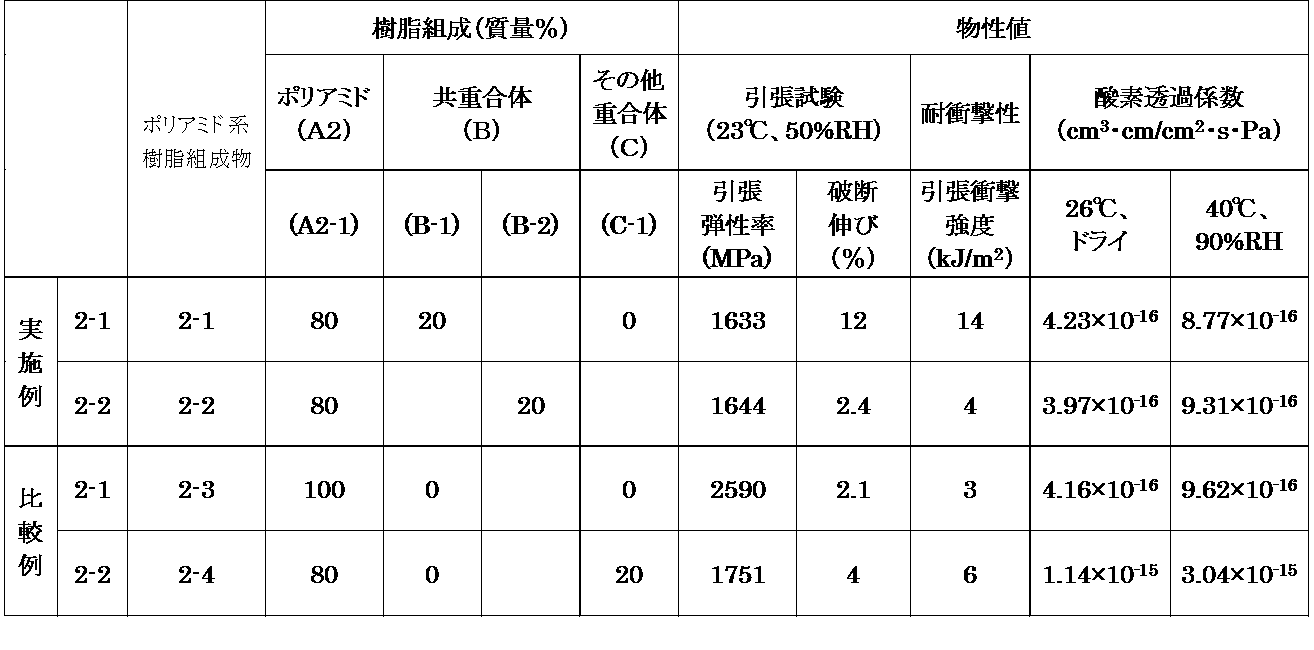
実施例2-1、実施例2-2及び比較例2-1の結果から、共重合体(B)を含む本発明のポリアミド系樹脂組成物は、共重合体(B)を含まないポリアミド系樹脂組成物に比べて、柔軟性、伸びやすさ及び耐衝撃性に優れることが判った。すなわち、共重合体(B)を混練させることで、ポリアミド系樹脂(A2)を改質することができた。また、共重合体(B)を混練させても、ポリアミド系樹脂(A2)が持つ優れたガスバリア性を損なわないことも明らかとなった。
また、実施例2-1及び比較例2-2の結果を比較すると、その他重合体(C)を混練させたポリアミド系樹脂組成物に比べて、共重合体(B)を混練させた本発明のポリアミド系樹脂組成物の方が、柔軟性、伸びやすさ、耐衝撃性、及びガスバリア性に優れていることが明確となった。
また、実施例2-1及び比較例2-2の結果を比較すると、その他重合体(C)を混練させたポリアミド系樹脂組成物に比べて、共重合体(B)を混練させた本発明のポリアミド系樹脂組成物の方が、柔軟性、伸びやすさ、耐衝撃性、及びガスバリア性に優れていることが明確となった。
実施例3-1:
ポリエステル(A3-1)80質量%及び共重合体(B-1)20質量%を配合して、シリンダー温度250℃に加熱した二軸混練押出機(Xplore Instruments社製 小型混練機)のホッパーに投入した。スクリュー回転数100rpmにて2分間溶融混練を行った後、ダイより流出する溶融組成物を冷却後裁断し、ペレット状のポリエステル系樹脂組成物3-1を作製した。
得られた樹脂組成物3-1は、上述の方法で引張試験、引張衝撃試験、ガスバリア性(酸素透過係数)評価を行った。測定結果を表5に示す。
ポリエステル(A3-1)80質量%及び共重合体(B-1)20質量%を配合して、シリンダー温度250℃に加熱した二軸混練押出機(Xplore Instruments社製 小型混練機)のホッパーに投入した。スクリュー回転数100rpmにて2分間溶融混練を行った後、ダイより流出する溶融組成物を冷却後裁断し、ペレット状のポリエステル系樹脂組成物3-1を作製した。
得られた樹脂組成物3-1は、上述の方法で引張試験、引張衝撃試験、ガスバリア性(酸素透過係数)評価を行った。測定結果を表5に示す。
実施例3-2:
共重合体(B-1)の代わりに共重合体(B-2)を用いた以外は実施例3-1と同様にしてペレット状のポリエステル系樹脂組成物3-2を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表5に示す。
共重合体(B-1)の代わりに共重合体(B-2)を用いた以外は実施例3-1と同様にしてペレット状のポリエステル系樹脂組成物3-2を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表5に示す。
比較例3-1:
共重合体(B-1)を加えずに、ポリエステル(A3-1)のみを使用して、実施例3-1と同様の方法でポリエステル系樹脂組成物3-3を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表5に示す。
共重合体(B-1)を加えずに、ポリエステル(A3-1)のみを使用して、実施例3-1と同様の方法でポリエステル系樹脂組成物3-3を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表5に示す。
比較例3-2:
共重合体(B-1)の代わりに重合体(C-1)を使用して、実施例3-1と同様の方法でポリエステル系樹脂組成物3-4を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表5に示す。
共重合体(B-1)の代わりに重合体(C-1)を使用して、実施例3-1と同様の方法でポリエステル系樹脂組成物3-4を作製し、引張試験、引張衝撃試験、ガスバリア性評価を行った。測定結果を表5に示す。

実施例3-1、実施例3-2及び比較例3-1の結果から、共重合体(B)を含む本発明のポリエステル系樹脂組成物は、共重合体(B)を含まないポリエステル系樹脂組成物に比べて、柔軟性、伸びやすさ及び耐衝撃性に優れることが判った。すなわち、共重合体(B)を混練させることで、ガスバリア性をほとんど悪化させずにポリエステル系樹脂(A3)を改質することができた。
また、実施例3-1及び比較例3-2の結果を比較すると、その他重合体(C)を混練させたポリエステル系樹脂組成物に比べて、共重合体(B)を混練させた本発明のポリエステル系樹脂組成物の方が、柔軟性、伸びやすさ、耐衝撃性、及びガスバリア性に優れていることが明らかとなった。
また、実施例3-1及び比較例3-2の結果を比較すると、その他重合体(C)を混練させたポリエステル系樹脂組成物に比べて、共重合体(B)を混練させた本発明のポリエステル系樹脂組成物の方が、柔軟性、伸びやすさ、耐衝撃性、及びガスバリア性に優れていることが明らかとなった。
Claims (19)
- 酸素透過係数が1.0×10-14(cm3・cm/cm2・s・Pa)以下のガスバリア性樹脂(A)並びに一般式(1)、一般式(2)、及び一般式(3)

で示されるモノマー構造単位を含む共重合体(B)を含むことを特徴とする樹脂組成物であって、前記ガスバリア性樹脂(A)と共重合体(B)の合計質量に対する共重合体(B)の質量の割合が1~40質量%であることを特徴とするガスバリア性樹脂組成物。 - R2が示す炭素原子数1~20の炭化水素基が、炭素原子数1~20のアルキル基または炭素数6~20のアリール基である請求項1に記載のガスバリア性樹脂組成物。
- 共重合体(B)の一般式(1)で示されるモノマー構造単位のモル比l、一般式(2)で示されるモノマー構造単位のモル比m、及び一般式(3)で示されるモノマー構造単位のモル比nが、下記式:
80≧{(m+n)/(l+m+n)}×100≧0.1 を満たす請求項1または2に記載のガスバリア性樹脂組成物。 - 共重合体(B)の一般式(2)で示されるモノマー構造単位のモル比mと一般式(3)で示されるモノマー構造単位のモル比nが、下記式:
100≧{m/(m+n)}×100≧50 を満たす請求項1~3のいずれかに記載のガスバリア性樹脂組成物。 - 共重合体(B)の一般式(3)で示されるモノマー構造単位において、n=0である請求項1~3のいずれかに記載のガスバリア性樹脂組成物。
- 共重合体(B)の数平均分子量(Mn)が1000~1000000であり、かつ重量平均分子量(Mw)と数平均分子量(Mn)の比Mw/Mnが1.5~4.0である請求項1~5のいずれかに記載のガスバリア性樹脂組成物。
- 一般式(2)及び一般式(3)中のR1が水素原子であり、pが1である請求項1~6のいずれかに記載のガスバリア性樹脂組成物。
- ガスバリア性樹脂(A)が、ビニルアルコール系樹脂(A1)である請求項1~7のいずれかに記載のガスバリア性樹脂組成物。
- ビニルアルコール系樹脂(A1)が、エチレン構造単位を10~60モル%含むエチレン・ビニルアルコール系共重合体である請求項8に記載のガスバリア性樹脂組成物。
- ガスバリア性樹脂(A)が、ポリアミド系樹脂(A2)である請求項1~7のいずれかに記載のガスバリア性樹脂組成物。
- ポリアミド系樹脂(A2)が、ポリアミド6、ポリアミド66、及びポリアミドMXD6の中から選ばれる少なくとも一種である請求項10に記載のガスバリア性樹脂組成物。
- ガスバリア性樹脂(A)が、ポリエステル系樹脂(A3)である請求項1~7のいずれかに記載のガスバリア性樹脂組成物。
- ポリエステル系樹脂(A3)が、ポリエチレンテレフタレート及びポリブチレンテレフタレートから選ばれる少なくとも一種である請求項12に記載のガスバリア性樹脂組成物。
- 請求項1~13のいずれかに記載されたガスバリア性樹脂組成物をバリア層として有する容器。
- 請求項1~13のいずれかに記載されたガスバリア性樹脂組成物を成形してなる樹脂成形品。
- 前記成形が、射出成形法または押出成形法である請求項15に記載の樹脂成形品。
- 前記樹脂成形品が、シート、フィルム、チューブ、パイプ、ボトル、またはタンクのいずれかである請求項15または16に記載の樹脂成形品。
- 一般式(1)、一般式(2)、及び一般式(3)
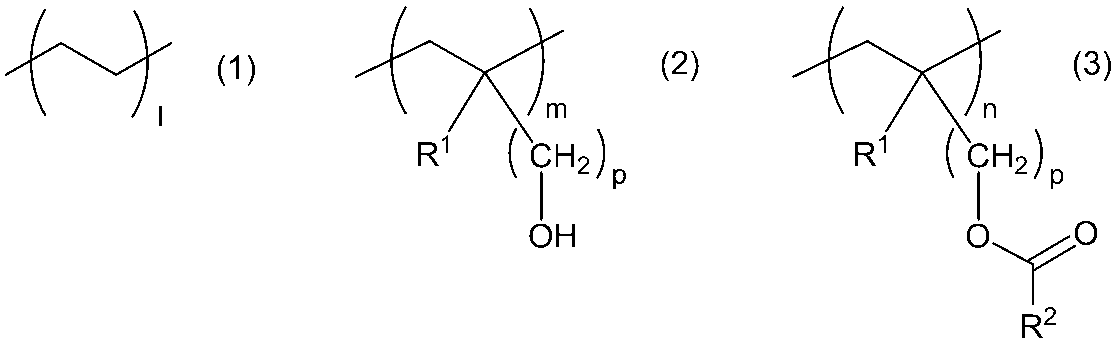
で示されるモノマー構造単位を含む共重合体を成分として含むことを特徴とする、酸素透過係数が1.0×10-14(cm3・cm/cm2・s・Pa)以下のガスバリア性樹脂の改質材。 - 一般式(1)、一般式(2)、及び一般式(3)

で示されるモノマー構造単位を含む共重合体を酸素透過係数が1.0×10-14(cm3・cm/cm2・s・Pa)以下のガスバリア性樹脂(A)に混合することを特徴とするガスバリア性樹脂の改質方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201880042296.XA CN110799600B (zh) | 2017-07-25 | 2018-07-17 | 阻气性树脂组合物及其用途 |
| EP18839085.0A EP3660105B1 (en) | 2017-07-25 | 2018-07-17 | Gas-barrier resin composition and use thereof |
| JP2019532525A JP7289025B2 (ja) | 2017-07-25 | 2018-07-17 | ガスバリア性樹脂組成物及びその用途 |
| US16/624,055 US11655360B2 (en) | 2017-07-25 | 2018-07-17 | Gas-barrier resin composition and use thereof |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017143454 | 2017-07-25 | ||
| JP2017-143454 | 2017-07-25 | ||
| JP2017243014 | 2017-12-19 | ||
| JP2017-243014 | 2017-12-19 | ||
| JP2017-243015 | 2017-12-19 | ||
| JP2017243015 | 2017-12-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019021890A1 true WO2019021890A1 (ja) | 2019-01-31 |
Family
ID=65040797
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2018/026766 WO2019021890A1 (ja) | 2017-07-25 | 2018-07-17 | ガスバリア性樹脂組成物及びその用途 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US11655360B2 (ja) |
| EP (1) | EP3660105B1 (ja) |
| JP (1) | JP7289025B2 (ja) |
| CN (1) | CN110799600B (ja) |
| WO (1) | WO2019021890A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019069597A (ja) * | 2017-10-06 | 2019-05-09 | 昭和電工株式会社 | ガスバリア性樹脂積層体及びその製造方法 |
| WO2021192870A1 (ja) * | 2020-03-25 | 2021-09-30 | パナソニックIpマネジメント株式会社 | ガス吸着体、及びガスセンサ |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS52141785A (en) | 1976-05-20 | 1977-11-26 | Toyo Seikan Kaisha Ltd | Container |
| JPS5388067A (en) | 1977-01-13 | 1978-08-03 | Kuraray Co | Production of ethylenee vinylalcohol copolymer film with improved property |
| JPS5920345A (ja) | 1982-07-23 | 1984-02-02 | アメリカン・カン・カンパニ− | 可塑化されたエチレン「あ」ビニルアルコ−ル共重合体の配合物ならびにその利用方法および製品 |
| JPH0715059B2 (ja) | 1987-12-26 | 1995-02-22 | ユニチカ株式会社 | 耐ピンホール性に優れたポリアミドフィルム |
| JPH08239528A (ja) | 1994-12-07 | 1996-09-17 | Nippon Synthetic Chem Ind Co Ltd:The | エチレン−酢酸ビニル共重合体ケン化物系樹脂組成物及びその用途 |
| JPH09183900A (ja) | 1995-11-02 | 1997-07-15 | Ube Ind Ltd | ポリアミド樹脂組成物 |
| JP2000212369A (ja) | 1999-01-27 | 2000-08-02 | Nippon Synthetic Chem Ind Co Ltd:The | 樹脂組成物およびその用途 |
| JP2002145947A (ja) * | 2000-11-13 | 2002-05-22 | Mitsui Chemicals Inc | 極性基含有オレフィン共重合体、該共重合体を含む熱可塑性樹脂組成物およびこれらの用途 |
| JP2006241398A (ja) | 2005-03-07 | 2006-09-14 | Teijin Dupont Films Japan Ltd | 二軸延伸フィルム |
| WO2013168626A1 (ja) | 2012-05-11 | 2013-11-14 | 国立大学法人東京大学 | ポリオレフィン類合成用触媒 |
| JP2014159540A (ja) | 2013-01-23 | 2014-09-04 | Univ Of Tokyo | 極性基含有オレフィン系共重合体の製造方法 |
| JP2016191009A (ja) | 2015-03-31 | 2016-11-10 | 興人フィルム&ケミカルズ株式会社 | 易引裂性二軸延伸ポリブチレンテレフタレートフィルム |
| JP2017031300A (ja) * | 2015-07-31 | 2017-02-09 | 国立大学法人 東京大学 | エチレン・α−オレフィン・極性基含有アリルモノマー三元共重合体及びその製造方法 |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6246643A (ja) * | 1985-08-23 | 1987-02-28 | 株式会社クラレ | 耐気体透過性積層体 |
| FR2731005B1 (fr) * | 1994-12-29 | 1997-04-04 | Atochem Elf Sa | Composition en poudre a base de polyamide pour le revetement de substrats metalliques |
| US5646225A (en) * | 1996-05-13 | 1997-07-08 | Arco Chemical Technology, L.P. | Water-reducible resins for coatings and inks |
| JPH10264318A (ja) * | 1997-03-28 | 1998-10-06 | Kuraray Co Ltd | 積層体 |
| JPH11293080A (ja) * | 1998-04-09 | 1999-10-26 | Kuraray Co Ltd | アリルアルコール系重合体中空成形容器 |
| JPH11291405A (ja) * | 1998-04-14 | 1999-10-26 | Kuraray Co Ltd | アリルアルコール系重合体積層シュリンクフィルム |
| JP5073137B2 (ja) * | 2000-09-01 | 2012-11-14 | 株式会社クラレ | エチレン−ビニルアルコール共重合体組成物およびそれを用いた多層容器 |
| EP1186619B1 (en) | 2000-09-07 | 2006-11-29 | Mitsui Chemicals, Inc. | Polar group-containing olefin copolymer, process for preparing the same, thermoplastic resin composition contaning the copolymer, and uses thereof |
| US7585516B2 (en) | 2001-11-12 | 2009-09-08 | Advanced Cardiovascular Systems, Inc. | Coatings for drug delivery devices |
| JP2004091775A (ja) * | 2002-08-09 | 2004-03-25 | Kuraray Co Ltd | オレフィン系重合体組成物 |
| WO2006053297A1 (en) * | 2004-11-08 | 2006-05-18 | E.I. Dupont De Nemours And Company | Toughened polyamide for food packaging and health care applications |
| EP2014712B1 (en) * | 2006-04-25 | 2010-06-02 | The Nippon Synthetic Chemical Industry Co., Ltd. | Resin composition and multilayer structure making use of the same |
| CN102731877A (zh) * | 2006-10-19 | 2012-10-17 | 三菱瓦斯化学株式会社 | 阻隔性良好的注塑成型体 |
| JP5909846B2 (ja) * | 2010-08-26 | 2016-04-27 | 横浜ゴム株式会社 | 熱可塑性樹脂組成物およびそれを用いたタイヤ |
| JP6298741B2 (ja) * | 2014-08-27 | 2018-03-20 | 株式会社クラレ | (メタ)アリルアルコール共重合体およびその製造方法 |
-
2018
- 2018-07-17 WO PCT/JP2018/026766 patent/WO2019021890A1/ja unknown
- 2018-07-17 CN CN201880042296.XA patent/CN110799600B/zh active Active
- 2018-07-17 US US16/624,055 patent/US11655360B2/en active Active
- 2018-07-17 JP JP2019532525A patent/JP7289025B2/ja active Active
- 2018-07-17 EP EP18839085.0A patent/EP3660105B1/en active Active
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS52141785A (en) | 1976-05-20 | 1977-11-26 | Toyo Seikan Kaisha Ltd | Container |
| JPS5388067A (en) | 1977-01-13 | 1978-08-03 | Kuraray Co | Production of ethylenee vinylalcohol copolymer film with improved property |
| JPS5920345A (ja) | 1982-07-23 | 1984-02-02 | アメリカン・カン・カンパニ− | 可塑化されたエチレン「あ」ビニルアルコ−ル共重合体の配合物ならびにその利用方法および製品 |
| JPH0715059B2 (ja) | 1987-12-26 | 1995-02-22 | ユニチカ株式会社 | 耐ピンホール性に優れたポリアミドフィルム |
| JPH08239528A (ja) | 1994-12-07 | 1996-09-17 | Nippon Synthetic Chem Ind Co Ltd:The | エチレン−酢酸ビニル共重合体ケン化物系樹脂組成物及びその用途 |
| JPH09183900A (ja) | 1995-11-02 | 1997-07-15 | Ube Ind Ltd | ポリアミド樹脂組成物 |
| US5780577A (en) | 1995-11-02 | 1998-07-14 | Ube Industries, Ltd. | Polyamide resin composition |
| JP2000212369A (ja) | 1999-01-27 | 2000-08-02 | Nippon Synthetic Chem Ind Co Ltd:The | 樹脂組成物およびその用途 |
| JP2002145947A (ja) * | 2000-11-13 | 2002-05-22 | Mitsui Chemicals Inc | 極性基含有オレフィン共重合体、該共重合体を含む熱可塑性樹脂組成物およびこれらの用途 |
| JP2006241398A (ja) | 2005-03-07 | 2006-09-14 | Teijin Dupont Films Japan Ltd | 二軸延伸フィルム |
| WO2013168626A1 (ja) | 2012-05-11 | 2013-11-14 | 国立大学法人東京大学 | ポリオレフィン類合成用触媒 |
| JP2014159540A (ja) | 2013-01-23 | 2014-09-04 | Univ Of Tokyo | 極性基含有オレフィン系共重合体の製造方法 |
| JP2016191009A (ja) | 2015-03-31 | 2016-11-10 | 興人フィルム&ケミカルズ株式会社 | 易引裂性二軸延伸ポリブチレンテレフタレートフィルム |
| JP2017031300A (ja) * | 2015-07-31 | 2017-02-09 | 国立大学法人 東京大学 | エチレン・α−オレフィン・極性基含有アリルモノマー三元共重合体及びその製造方法 |
Non-Patent Citations (2)
| Title |
|---|
| "Polymer handbook", 1999, JOHN WILEY & SONS, INC. |
| See also references of EP3660105A4 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019069597A (ja) * | 2017-10-06 | 2019-05-09 | 昭和電工株式会社 | ガスバリア性樹脂積層体及びその製造方法 |
| JP7080151B2 (ja) | 2017-10-06 | 2022-06-03 | 昭和電工株式会社 | ガスバリア性樹脂積層体及びその製造方法 |
| WO2021192870A1 (ja) * | 2020-03-25 | 2021-09-30 | パナソニックIpマネジメント株式会社 | ガス吸着体、及びガスセンサ |
Also Published As
| Publication number | Publication date |
|---|---|
| JP7289025B2 (ja) | 2023-06-09 |
| CN110799600A (zh) | 2020-02-14 |
| EP3660105A1 (en) | 2020-06-03 |
| US11655360B2 (en) | 2023-05-23 |
| US20210147669A1 (en) | 2021-05-20 |
| EP3660105A4 (en) | 2021-04-21 |
| JPWO2019021890A1 (ja) | 2020-05-28 |
| EP3660105B1 (en) | 2022-03-16 |
| CN110799600B (zh) | 2022-06-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100852313B1 (ko) | 폴리에스테르계 수지 조성물 및 이에 의한 성형체 | |
| KR101257521B1 (ko) | 방향족 폴리아미드 연신 필름 | |
| JP5615343B2 (ja) | 樹脂組成物およびその成形品 | |
| KR20160102227A (ko) | 푸란계 중합체성 탄화수소 연료 배리어 구조체 | |
| JP7160029B2 (ja) | 樹脂組成物、成形品、フィルムおよび多層フィルム | |
| JP5471009B2 (ja) | 多層容器 | |
| CN107848657B (zh) | 杯型多层容器 | |
| JPH10204174A (ja) | コポリアミドpa−6,i/6,t/6,6および/またはpa−6,i/6,tをベースとした耐延伸および/または耐熱成形性のある耐湿バリヤー材料 | |
| JP7289025B2 (ja) | ガスバリア性樹脂組成物及びその用途 | |
| CN102307936A (zh) | 热收缩性薄膜 | |
| JPH04226556A (ja) | エチレンビニルアルコール共重合体および選択された非晶質ポリアミド類を基とするブレンド物および構造物 | |
| JP2011089007A (ja) | 柔軟性とバリア性に優れるポリアミド樹脂及び成形体 | |
| JP5407521B2 (ja) | 樹脂組成物および包装材料 | |
| JP2005111926A (ja) | 多層積層体およびそれから成る成形体 | |
| JP2022077210A (ja) | ポリアミド系二軸延伸フィルムおよび包装体 | |
| JPH04178447A (ja) | 樹脂組成物 | |
| JP2018053033A (ja) | ポリアミド樹脂組成物および多層成形体 | |
| JPS63213529A (ja) | ポリエステル中空成形体 | |
| JPH05147177A (ja) | 積層体 | |
| WO2023171792A1 (ja) | 変性ポリエステル系エラストマー及びその製造方法 | |
| JP2010241910A (ja) | 樹脂組成物および多層構造物 | |
| CN118749005A (zh) | 改性聚酯系弹性体和其制造方法 | |
| JP2007084642A (ja) | ポリエステル樹脂組成物 | |
| JP6661914B2 (ja) | ポリアミド樹脂組成物、成形品および成形品の製造方法 | |
| JPS61276852A (ja) | ポリエステル組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18839085 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2019532525 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2018839085 Country of ref document: EP Effective date: 20200225 |