WO2017010089A1 - 焼成アパタイトを含む歯表面膜形成用粉体 - Google Patents
焼成アパタイトを含む歯表面膜形成用粉体 Download PDFInfo
- Publication number
- WO2017010089A1 WO2017010089A1 PCT/JP2016/003297 JP2016003297W WO2017010089A1 WO 2017010089 A1 WO2017010089 A1 WO 2017010089A1 JP 2016003297 W JP2016003297 W JP 2016003297W WO 2017010089 A1 WO2017010089 A1 WO 2017010089A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- film
- powder
- forming
- hydroxyapatite
- forming powder
- Prior art date
Links
Images









Classifications
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B25/00—Phosphorus; Compounds thereof
- C01B25/16—Oxyacids of phosphorus; Salts thereof
- C01B25/26—Phosphates
- C01B25/32—Phosphates of magnesium, calcium, strontium, or barium
- C01B25/327—After-treatment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K6/00—Preparations for dentistry
- A61K6/15—Compositions characterised by their physical properties
- A61K6/17—Particle size
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K6/00—Preparations for dentistry
- A61K6/20—Protective coatings for natural or artificial teeth, e.g. sealings, dye coatings or varnish
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K6/00—Preparations for dentistry
- A61K6/70—Preparations for dentistry comprising inorganic additives
- A61K6/78—Pigments
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K6/00—Preparations for dentistry
- A61K6/80—Preparations for artificial teeth, for filling teeth or for capping teeth
- A61K6/802—Preparations for artificial teeth, for filling teeth or for capping teeth comprising ceramics
- A61K6/816—Preparations for artificial teeth, for filling teeth or for capping teeth comprising ceramics comprising titanium oxide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K6/00—Preparations for dentistry
- A61K6/80—Preparations for artificial teeth, for filling teeth or for capping teeth
- A61K6/831—Preparations for artificial teeth, for filling teeth or for capping teeth comprising non-metallic elements or compounds thereof, e.g. carbon
- A61K6/838—Phosphorus compounds, e.g. apatite
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B05—SPRAYING OR ATOMISING IN GENERAL; APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05B—SPRAYING APPARATUS; ATOMISING APPARATUS; NOZZLES
- B05B1/00—Nozzles, spray heads or other outlets, with or without auxiliary devices such as valves, heating means
- B05B1/30—Nozzles, spray heads or other outlets, with or without auxiliary devices such as valves, heating means designed to control volume of flow, e.g. with adjustable passages
- B05B1/3006—Nozzles, spray heads or other outlets, with or without auxiliary devices such as valves, heating means designed to control volume of flow, e.g. with adjustable passages the controlling element being actuated by the pressure of the fluid to be sprayed
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B05—SPRAYING OR ATOMISING IN GENERAL; APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D—PROCESSES FOR APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D1/00—Processes for applying liquids or other fluent materials
- B05D1/02—Processes for applying liquids or other fluent materials performed by spraying
- B05D1/12—Applying particulate materials
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B35/00—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/622—Forming processes; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/626—Preparing or treating the powders individually or as batches ; preparing or treating macroscopic reinforcing agents for ceramic products, e.g. fibres; mechanical aspects section B
- C04B35/62605—Treating the starting powders individually or as mixtures
- C04B35/6261—Milling
- C04B35/6262—Milling of calcined, sintered clinker or ceramics
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B35/00—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/622—Forming processes; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/626—Preparing or treating the powders individually or as batches ; preparing or treating macroscopic reinforcing agents for ceramic products, e.g. fibres; mechanical aspects section B
- C04B35/62605—Treating the starting powders individually or as mixtures
- C04B35/62645—Thermal treatment of powders or mixtures thereof other than sintering
- C04B35/62665—Flame, plasma or melting treatment
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B35/00—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/622—Forming processes; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/626—Preparing or treating the powders individually or as batches ; preparing or treating macroscopic reinforcing agents for ceramic products, e.g. fibres; mechanical aspects section B
- C04B35/62605—Treating the starting powders individually or as mixtures
- C04B35/62645—Thermal treatment of powders or mixtures thereof other than sintering
- C04B35/62675—Thermal treatment of powders or mixtures thereof other than sintering characterised by the treatment temperature
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B35/00—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/622—Forming processes; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/626—Preparing or treating the powders individually or as batches ; preparing or treating macroscopic reinforcing agents for ceramic products, e.g. fibres; mechanical aspects section B
- C04B35/62605—Treating the starting powders individually or as mixtures
- C04B35/62645—Thermal treatment of powders or mixtures thereof other than sintering
- C04B35/6268—Thermal treatment of powders or mixtures thereof other than sintering characterised by the applied pressure or type of atmosphere, e.g. in vacuum, hydrogen or a specific oxygen pressure
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B05—SPRAYING OR ATOMISING IN GENERAL; APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D—PROCESSES FOR APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D2401/00—Form of the coating product, e.g. solution, water dispersion, powders or the like
- B05D2401/30—Form of the coating product, e.g. solution, water dispersion, powders or the like the coating being applied in other forms than involving eliminable solvent, diluent or dispersant
- B05D2401/32—Form of the coating product, e.g. solution, water dispersion, powders or the like the coating being applied in other forms than involving eliminable solvent, diluent or dispersant applied as powders
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/61—Micrometer sized, i.e. from 1-100 micrometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/62—Submicrometer sized, i.e. from 0.1-1 micrometer
Definitions
- the present invention is used in an apparatus for forming a film on a tooth surface by injecting powder onto the tooth to form a film with high hardness and extremely low acid solubility on the tooth surface in a short time.
- suitable for forming a film suitable for the color tone of the tooth, and the film forming powder containing the hydroxyapatite powder which is the main component of the tooth in a short time The present invention relates to a film-forming powder using a hydroxyapatite powder blended with a color tone adjusting agent.
- apatite is a main component constituting teeth and bones, has excellent biocompatibility, and is a suitable material for replacement or repair of damaged hard tissue.
- Development of dentistry and medical materials containing In dentistry, toothpaste containing hydroxyapatite (Patent Documents 1 and 2) and glass powder for glass ionomer cement containing hydroxyapatite (Patent Document 3) for the purpose of caries prevention, caries treatment and tooth bleaching. ), And a tooth bleaching agent (Patent Document 4) in which a hydroxyapatite powder and a strong acid aqueous solution are mixed and applied as a dental paste has been developed.
- a plasma spraying method Patent Document 5
- a sputtering method Patent Document 6
- a thermal decomposition method Patent Documents 7 and 8
- hydroxyapatite powder which is the main component of enamel and dentin, is sprayed onto the tooth surface at high speed to form a hydroxyapatite film on the surface of enamel and dentin.
- Devices Patent Documents 9 to 12 have been proposed.
- a technique for injecting a powder onto a target as in the present invention can form a hydroxyapatite powder layer on a metal surface, for example, an implant.
- Uniform coating of hydroxyapatite on the body surface makes it possible to further improve biocompatibility, and can sufficiently contribute to prevention of peri-implantitis, long-term stabilization of treatment prognosis, and improvement of maintenance. is there.
- hydroxyapatite powder which is the main component of enamel and dentin
- hydroxyapatite is applied to the surface of enamel and dentin.
- a method of forming a film has been studied, in order to form and integrate a hydroxyapatite film on the surface of enamel or dentin, the film formation efficiency is poor with respect to the amount of powder injection, and it takes a long time for film formation.
- the formed film has not been practically used because it has high acid solubility.
- the background of the present invention is that the patient's demand for aesthetic dental treatment has increased in recent years, and there are treatments by the bleaching method and porcelain laminate veneer method as the treatment methods. Since the quality is invasive, the burden on patients is regarded as a problem. On the other hand, if the film is formed on the tooth by injection of hydroxyapatite powder, it is possible to adjust the color tone of the crown with the same components as the tooth, so there is no invasion to the healthy tooth, Therefore, treatment that greatly reduces the burden on the patient is possible.
- the method for producing a color tone adjusting material it has been performed by mixing powders with a powder mixer or the like.
- powder mixing is insufficient.
- problems such as unevenness in the color tone of the formed film or fading may occur.
- Non-patent Document 1 Regarding the Vickers hardness of dental enamel, 270-366Hv was reported by the Japan Dental Science and Technology Society Dental Equipment Research Committee, and the properties of crown restorations are equivalent to the physical properties of the teeth. It has been reported that a property higher than that is required (Non-patent Document 1). In addition, there are reports on plasma irradiation (Non-Patent Documents 2 to 4).
- the object of the present invention is to solve the above-mentioned problems, and to quickly and evenly adjust the color tone of a film having a high hardness and a very low solubility in acid on a base material such as a tooth surface.
- An object of the present invention is to provide a film forming powder.
- the present inventors have added and mixed a film-forming powder obtained by baking hydroxyapatite powder using an inert gas, and a color tone adjusting agent for adjusting the color tone of the crown to the film-forming powder.
- a film-forming powder obtained by baking hydroxyapatite powder using an inert gas, and a color tone adjusting agent for adjusting the color tone of the crown to the film-forming powder.
- the film-forming powders and the film-forming powders are subjected to plasma irradiation with a low-temperature plasma processing apparatus, and mechanical energy such as compression and shearing is applied.
- a hydroxyapatite film with a high strength and low acid solubility on a base material such as a tooth surface and a hydroxyapatite film that can adjust the color of the crown can be formed.
- a powder was developed.
- film-forming powder of the present invention film formation is good with respect to the amount of powder sprayed, so that a film can be formed in a short time
- the present invention is as follows.
- a powder for forming a film for use in a device for spraying on teeth to form a film on the tooth surface and Ca 10-X ⁇ M X (ZO 4 ) 6 Y 2 (where X Is 0 to 10, M is a metal or hydrogen, ZO 4 is PO 4 , CO 3 , CrO 4 , AsO 4 , VO 4 , SiO 4 , SO 4 or GeO 4 , Y is a hydroxyl group, a halogen element or a carbonate group)
- an average particle diameter produced by firing apatite represented by Ca 10-X ⁇ M X (ZO 4 ) 6 Y 2 at 600 to 1350 ° C. in an inert gas atmosphere is used for a method for forming a film on a tooth surface using a device for spraying teeth, and [2] used for a device for spraying teeth.
- the apatite represented by Ca 10-X ⁇ M X (ZO 4 ) 6 Y 2 for use as a film forming powder for forming a film on the tooth surface is subjected to 600 to 1350 ° C. in an inert gas atmosphere.
- inert gas atmosphere is 0.5 [mu] m ⁇ 30 [mu] m, with respect to [4] teeth
- an apatite film By spraying the powder for forming a film of the present invention onto the tooth surface at a high speed, an apatite film can be rapidly formed without giving a burden to the patient. Bleaching and esthetic treatment with a film close to the color of the tooth surface can be easily performed.
- the hydroxyapatite-only film-forming layer is translucent, so it is difficult to identify the treatment site when the film is formed on caries, hypersensitive sites, or exposed root surfaces. Therefore, in order to clarify the treatment field, it is also important to give color tone to the film formation layer.
- the film since it was confirmed that the solubility of the film formation layer was suppressed and the hardness was improved, the film was stably maintained for a long period of time, that is, the color unevenness of the film formation layer was suppressed, and the color tone was stabilized.
- a film-forming powder blended with a color tone adjusting agent suitable for obtaining the obtained film it becomes possible to form a film-forming layer having various hues desired by the individual patient for a patient who is troubled by the hue of teeth, which greatly contributes to aesthetic dental treatment.
- FIG. 3 is a graph showing the average particle size and particle size distribution of the film-forming hydroxyapatite powder produced in Example 2-1. It is a figure which shows the diffraction pattern by the powder X-ray-diffraction apparatus of Example 9-1.
- FIG. 9 shows the change in spectrum of the film forming powder of Example 9-2 as measured by a laser Raman spectroscopic analyzer.
- FIG. It is the film-forming layer (photograph) using the powder for film formation which mix
- Multi-layered film using the film-forming powder produced in Example 2 and the film-forming powder blended with the color tone adjusting agent (containing 1% titanium oxide in the first layer and zinc oxide 5 in the second layer) It is a figure (photograph) of% mixing
- the film-forming powder of the present invention is a powder having an average particle diameter of 0.5 to 30 ⁇ m for use in an apparatus for spraying teeth, which is used for forming a film on the tooth surface.
- Ca 10-X ⁇ M X (ZO 4 ) 6 Y 2 (where X is 0 to 10, M is metal or hydrogen, ZO 4 is PO 4 , CO 3 , CrO 4 , AsO 4 , VO 4 , It is characterized by being produced by firing apatite represented by SiO 4 , SO 4 or GeO 4 , Y being a hydroxyl group, a halogen element or a carbonate group) at 600 to 1350 ° C. in an inert gas atmosphere.
- the apatite represented by Ca 10-X ⁇ M X (ZO 4 ) 6 Y 2 is fired at 600 to 1350 ° C. in an inert gas atmosphere, and then pulverized and classified. And spray the powder onto the teeth
- the apatite is not particularly limited as long as it is a method for producing a film-forming powder having an average particle size of 0.5 to 30 ⁇ m for use in an apparatus for forming a film on the tooth surface.
- hydroxyapatite which is basic calcium phosphate represented by the chemical formula Ca 10 (PO 4 ) 6 (OH) 2 , is particularly preferable.
- the hydroxyapatite is a kind of calcium phosphate, has good biocompatibility, and is contained in a large amount in bones, teeth and the like.
- natural hard tissues can be obtained from fish bones, pork bones, beef bones, etc. of edible fish such as salmon.
- the method for synthesizing hydroxyapatite used in the present invention is not particularly limited and can be appropriately selected.
- it can be obtained by reacting calcium salt and phosphate in an aqueous solution and drying at a predetermined temperature.
- calcium salts include general calcium salts such as calcium hydroxide, calcium acetate, calcium carbonate, calcium chloride, calcium citrate, and calcium lactate.
- phosphate include phosphoric acid, ammonium phosphate, Common phosphates such as sodium phosphate, potassium phosphate, pyrophosphoric acid and sodium hexametaphosphate can be mentioned.
- calcium nitrate tetrahydrate is dissolved in pure water, and then an aqueous solution of ammonium dihydrogen phosphate is slowly added to a solution in which the pH of the solution is adjusted to pH 10 with aqueous ammonia. Then, a small amount of aqueous ammonia was added so that the pH in the solution was 10, and after adding all the aqueous solution of diammonium hydrogen phosphate, the solution was aged at 90 ° C. while stirring, and the precipitate was filtered, A method of washing in pure water by ultrasonic treatment and drying the obtained solid at 80 ° C. can be mentioned.
- a phosphoric acid aqueous solution is dropped into a 0.5 M calcium hydroxide aqueous suspension at room temperature to prepare an apatite suspension, and the pH of the reaction solution is adjusted to 10.5 using an aqueous ammonia solution. Then, after confirming that the solution is completely mixed, the suspension is aged overnight, the resulting precipitate is filtered, and the solid is dried at 80 ° C.
- Hydroxyapatite can also be synthesized.
- the hydroxyapatite hydroxyl group is halogenated by coexisting a halogen element source such as calcium fluoride, sodium fluoride or calcium chloride during the production of hydroxyapatite.
- a halogen element source such as calcium fluoride, sodium fluoride or calcium chloride
- Fluoroapatite Ca 10 (PO 4 ) 6 F 2 and chloroapatite Ca 10 (PO 4 ) 6 Cl 2 in which Y is a halogen element can be produced.
- the hydroxyapatite after the hydroxyapatite is formed, it can be replaced by mixing with a solvent containing a halogen element source.
- Halogen-substituted apatite can also be synthesized by dry synthesis of hydroxyapatite with a halogen compound such as calcium fluoride and a phosphate compound. Fluorine apatite substituted with fluorine can be used as a tooth surface reinforcing agent.
- hydroxyapatite for example, sodium, lithium, magnesium, barium, strontium, zinc, cadmium, lead, vanadium, silicon
- water-soluble salts such as germanium, iron, arsenic, manganese, aluminum, rare earth elements, cobalt, silver, chromium, antimony, tungsten, molybdenum, etc.
- apatite in which at least a part of Ca is replaced with a metal element is synthesized. can do.
- the film-forming powder of the present invention is obtained by, for example, calcining apatite such as hydroxyapatite produced by the general method as described above in an inert gas atmosphere at 600 to 1350 ° C., preferably 800 to 1350 ° C. It can obtain by baking with. After firing at 600 to 1350 ° C. in an inert gas atmosphere, pulverization and classification, preferably pulverization, classification and mixing, to obtain a film forming powder having an average particle size of 0.5 ⁇ m to 30 ⁇ m, preferably 1 ⁇ m to 10 ⁇ m. Can do. As long as the average particle size is 0.5 ⁇ m to 30 ⁇ m, the shape, structure, etc. are not particularly limited, and can be appropriately selected according to the purpose.
- the film-forming powder obtained by this baking is subjected to a plasma irradiation process or a process of imparting mechanical energy, whereby a powder more suitable for film formation can be obtained.
- the film forming powder of the present invention when the performance of the film formed on the substrate such as the tooth surface was investigated, it was possible to form the film in a short time, and further, the hiding power against discoloration of the crown was examined. It has become clear that it is preferable to form a film thickness of 30 ⁇ m or more, and that the Vickers hardness is preferably 340 Hv or more. Therefore, it is necessary to bake apatite in an inert gas atmosphere at 600 to 1350 ° C. I understood. Moreover, the film forming powder of the present invention can be blended with a color tone adjusting agent for adjusting the color tone of the crown. Compounding hydroxyapatite powder and various color tone modifiers to obtain a film-forming powder containing a color tone modifier for forming a film-forming layer that imparts various color tones to teeth Can do.
- the particle size of the color tone adjusting material is hydroxyapatite. From the viewpoint of imparting better mixing properties, it is preferably from 0.05 ⁇ m to 10 ⁇ m. preferable.
- inorganic and organic pigments known for dentistry can be used without any limitation.
- Inorganic pigments include oxides, hydroxides, sulfides, chromates, silicates, sulfates, carbonates, ferrocyan compounds, phosphates, carbons, etc. Among them, oxides are preferably used. It is done.
- the organic pigment include tar dyes, azo pigments, phthalocyanine pigments, condensed polycyclic pigments, nitro pigments, nitroso pigments, and fluorescent pigments. Among them, azo pigments and phthalocyanine pigments are preferably used. These inorganic pigments and organic pigments can be mixed and used.
- red pigments and / or dyes red rose, molybdenum red, chromophone, etc. Tarred, Red No. 2 (Amaranth), Red No. 104 (Phloxine), Red No. 105 (Rose Bengal), Red No. 106 (Acid Red), Red No. 201 (Risor Rubin BCA), Red No. 202 (Risor Rubin BCA), Red 203 No. (Rake Red C), Red No. 204 (Rake Red CBA), Red No. 205 (Risor Red), Red No.
- Red CA Red No. 207
- Red No. 208 Red SR
- Red 213 Red No. 207
- red 215 rhodamine B stearate
- red 218 tetrachlorotetrabromofluorescein
- red 219 brilliant lake red R
- red 220 deep maroon
- red 221 toluidine red
- Red 223 Tetrabromofluorescein
- Red 225 Sud III
- Red 226 Herringdon Pink CN
- Red 227 Flust Acid Magenta
- Red 228 Permaton Red
- Naphthol AS Naphthol Rubin, Naphthol Red FGR, Naphthol Carmine FBB, Naphthol Carmine F3B, Naphthol Red F5RK, Naphthol Red HF4B
- BONA rake BONA barium rake, BONA calcium rake, BONA strontium rake, BONA manganese rake, BONA magnesium rake
- risol rubin brilliant carmine 6B
- Blue No. 201 Indigo
- Blue No. 202 Patent Blue NA
- Blue No. 203 Patent Blue CA
- Blue No. 204 Carbanthrene Blue
- Blue 205 No. Alphahazulin FG
- black pigments such as black iron oxide and carbon black.
- silicon dioxide and resin fine particles can be used as a color tone adjusting agent for imparting gloss. .
- the film-forming powder of the present invention contains, in addition to the above components, other components usually used in dental materials, for example, silica, magnesium phosphate, calcium carbonate, zirconia and the like, if necessary. It can mix
- the film-forming powder of the present invention further includes a powder having the same average particle diameter or a mixture of powders having different particle diameters, a powder having the same firing inert gas atmosphere, or an inert powder having different firing atmospheres.
- Plasma irradiation using an apparatus for performing plasma irradiation such as low-temperature plasma in the production of the film-forming powder according to the present invention is not limited to treatment of apatite powder alone, mixing treatment of apatite powders, or color adjustment with apatite powder. It is also preferable to carry out the mixing treatment of powders other than apatite such as an agent. For example, for a method for producing a hydroxyapatite powder blended with a color tone adjusting agent for adjusting the color tone of a crown, usually the hydroxyapatite powder and the color tone adjusting agent are mixed and processed. It is preferable to use an apparatus that performs plasma irradiation such as low-temperature plasma during mixing. .
- the treatment using an apparatus that imparts mechanical energy such as compression or shearing in addition to plasma irradiation.
- plasma irradiation By performing plasma irradiation, the surface of the particles can be cleaned and activated, and by applying mechanical energy, the particles can be combined firmly and densely, and the functionality of the powder can be improved. Enhanced particle design is possible.
- no change was observed in the properties of the formed film even when the single treatment was performed using an apparatus for applying mechanical energy.
- the crystallization of the particle surface is further increased. It was confirmed that the physicochemical properties changed. Good results have been found in the properties of the film formed using the powder for film formation of the present invention that has been subjected to treatment by applying mechanical energy and treatment by plasma irradiation, and is suitable for use in the oral environment It was confirmed that can be formed.
- a plasma surface treatment apparatus As the plasma irradiation apparatus, a plasma surface treatment apparatus (Asakusa Seisakusho), a multi-gas plasma jet (Plasma Factory), a plasma mixer PMR (Alpha Corporation), or the like can be used. Further, by applying mechanical energy such as grinding, friction, stretching, compression, and shearing to the particles, changes in structure, phase transition, reactivity, adsorptivity, catalytic activity, etc. can be caused. As a device for applying such mechanical energy, a hybridization system (Nara Machinery Co., Ltd.), mechano-fusion, nobilta (Hosokawa Micron) and the like can be used.
- mechanofusion has the effect of crushing the convex portions of the particles due to shearing force and the like, and particles in a state of being chamfered by grinding and reattachment are obtained.
- the applied voltage When applying low-temperature plasma that promotes cleaning and activation of the particle surface, it is preferable to set the applied voltage to 5 to 20 kV and the rotational speed of the rotor head to 1500 to 6000 rpm, and the processing time depends on the processing powder etc. Although different, 5 to 20 minutes can be exemplified.
- the plasma gas species helium gas, argon gas, oxygen gas, nitrogen gas, neon gas, carbon dioxide gas, air and the like can be used, and helium gas can be preferably exemplified.
- the rotational speed of the rotor head that applies compression and shearing force is preferably 100 to 6000 rpm, and the processing time varies depending on the processing powder and the like.
- the form of the powder for film formation is not limited to powder, but may be in the form of pellets obtained by pressing and compacting the powder, and further baked pellets obtained by firing the powder. It is also possible to use it.
- the form of the pellets one kind of film forming powder may be pelletized or two or more kinds of film forming powders may be laminated. Included in powder for use.
- the film-forming powder of the present invention is used for an application in which a film is formed on a tooth surface using an apparatus for spraying on teeth.
- a film forming apparatus by powder injection the powder injection apparatuses described in Patent Documents 9 to 12 can be used.
- the film forming conditions when using a powder injection device manufactured in-house are: handpiece tip nozzle inner diameter: 0.5 to 5.0 mm, injection pressure: 0.2 to 0.8 MPa, injection nozzle tip-tooth surface
- the distance between the nozzles is 0.1 to 30 mm (the nozzle tip is kept perpendicular to the tooth surface), and the spray nozzle moving speed is 0 to 10 mm / s.
- the powder injection devices described in Patent Documents 9 to 12 can also be used under similar conditions.
- the obtained film-forming layer is preferably subjected to surface polishing with a diamond polisher paste.
- the upper layer may be formed using a film forming powder blended with another color tone adjusting agent, or a color tone adjusting agent may be used.
- a multilayer film-forming layer can be formed on the surface of the crown, for example, by using a film-forming powder that is not blended.
- a firing atmosphere furnace As a firing atmosphere furnace, a vacuum substitution atmosphere furnace 2024-V type (Marusho Denki) was used. Further, a fluidized bed type opposed jet mill, counter jet mill 100AFG type (Hosokawa Micron) was used as a pulverization and classification device.
- Apatite powder for film formation Hydroxyapatite, fluorapatite, carbonate apatite and magnesium solid solution apatite synthesized above are pulverized in a mortar and 200 to 1350 ° C. in the atmosphere, argon gas and nitrogen gas atmosphere, or Baking was performed at 600 to 1350 ° C.
- the fired samples were pulverized and classified with an opposed airflow pulverizer to obtain hydroxyapatite powder having an average particle size of 0.5 to 30 ⁇ m for each sample.
- Titanium oxide is "Kishida Chemical Co., Ltd .: Special”
- Zinc oxide is "Haksuitec Co., Ltd .: Pharmacopoeia Zinc Oxide”
- Ultramarine is “Pinoa, Ultra Marine”
- Iron Oxide is "Kanto Chemical ( Co., Ltd .: Deer Grade 1 ”, and Red No. 204 used“ Tokyo Chemical Industry Co., Ltd., Lake Red CBA ”.
- 1 is an AC / DC converter (AC100V ⁇ DC24V)
- 2 is a cold cathode tube inverter (DC24V ⁇ AC1000V)
- 3 is a booster circuit (cockcroft-Walton circuit; AC1000V ⁇ AC10KV)
- 4 is a plasma nozzle
- 5 is a gas
- Each flow meter is shown.
- Mechano-Fusion AMS-MINI Hosokawa Micron
- Nanocura NC-ALB Hosokawa Micron
- hydroxyapatite powder for plasma irradiation treatment and mechanical energy addition treatment film formation (individual treatment) Film-forming hydroxyapatite powder produced in Example 2-1, film-forming silica-containing hydroxyapatite powder produced in Example 2-2, and film-forming color tone adjusting agent produced in Example 2-3
- the hydroxyapatite powder was treated with an apparatus for applying mechanical energy (Mechanofusion AMS-MINI, Hosokawa Micron) and then subjected to plasma irradiation to obtain a film forming powder.
- the film-forming hydroxyapatite powder produced in Example 2-1 was subjected to a plasma irradiation treatment, and then treated with an apparatus that applies mechanical energy to obtain a film-forming powder. .
- Example 3-4 Production of Hydroxyapatite Powder for Mechanically Energy-added Treatment Film Formation Hydroxyapatite Powder for Film Formation Produced in Example 2-1 and Silica-Containing Hydroxyapatite Powder for Film Formation Produced in Example 2-2
- the film-forming color adjusting agent-containing hydroxyapatite powder produced in Example 2-3 was subjected to a process of applying mechanical energy to obtain a film-forming powder.
- FIG. 6 shows a film-forming layer (photograph) using the film-forming hydroxyapatite powder blended with the above color tone adjusting agent.
- Example 4-1 Particle Size of Film Forming Powder The average particle size and particle size distribution of the film forming powder produced in Example 2-1 are shown in FIG.
- a particle size distribution measuring device LA-950, manufactured by Horiba, Ltd. was used for measuring the particle size distribution of the film forming powder.
- a dry unit was used for the measurement.
- particle size 0.5 ⁇ m indicates a powder having an average particle size of 0.4 to 0.6 ⁇ m
- particle size 1 ⁇ m indicates an average particle size of 0.9 to 1.1 ⁇ m powder
- particle diameter 5 ⁇ m is an average particle diameter of 4.0 to 6.0 ⁇ m
- particle diameter 10 ⁇ m is an average particle diameter of 9.0 to 11.0 ⁇ m
- Particle diameter 20 ⁇ m means a powder having an average particle diameter of 19.0 to 21.0 ⁇ m
- particle diameter 30 ⁇ m means a powder having an average particle diameter of 29.0 to 31.0 ⁇ m.
- the enamel block for measuring the Ca ion elution amount prepared above is immersed in a lactic acid buffer solution for 30 minutes under a test condition of 37 ° C. Subsequently, a total of 3 cycles of 90-minute immersion in the HEPES buffer solution was performed. After completion of the test, the Ca ion concentration eluted in the solution was measured by ion chromatography (cation chromatography method). Moreover, the measuring method was performed on the following measuring conditions.
- the film produced by the film formation process was measured using a microhardness meter FM-700 (Future Tech Co., Ltd.) with an indentation load of 100 g and a load holding time of 30 seconds.
- the in-house manufactured powder injection device includes an AC / DC converter (AC 100 V ⁇ DC 24 V), a solenoid valve, a mist separator, an air regulator, a speed controller, and the like.
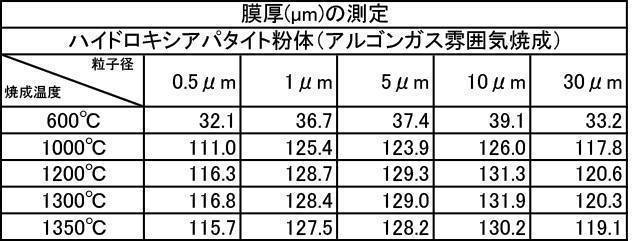
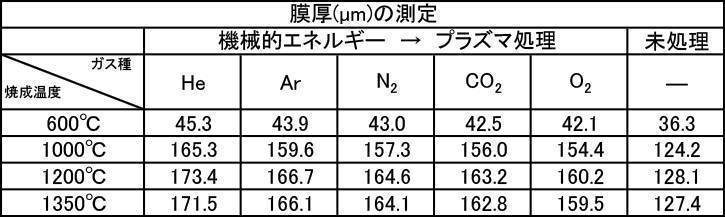
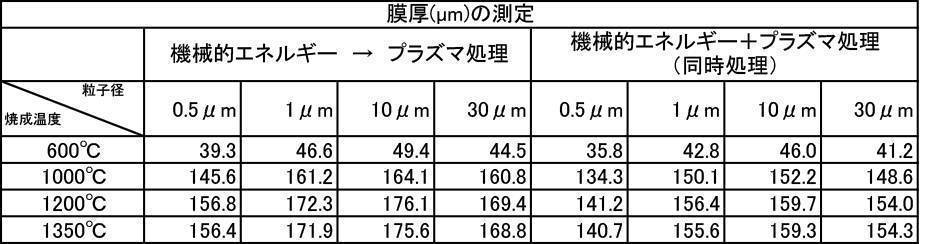
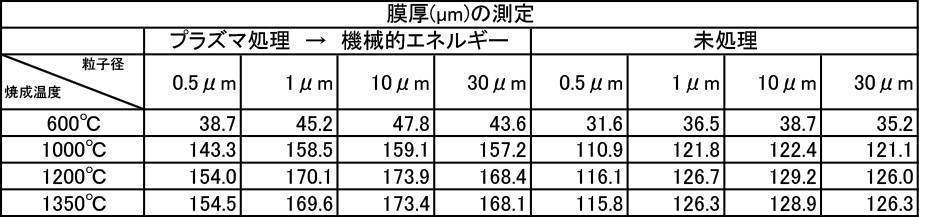
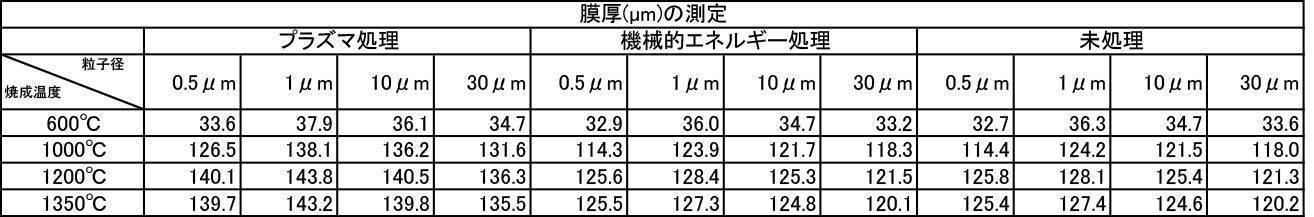
- Table 1 shows the measurement results of film thickness at various particle diameters of hydroxyapatite powder fired at 200 to 1350 ° C. in an air atmosphere, and various hydroxyapatite powders fired at 600 to 1350 ° C. in an argon gas atmosphere.
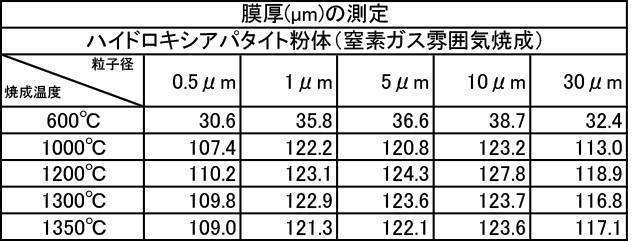
- Table 2 shows the measurement results of the film thickness at the particle diameter, and Table 3 shows the measurement results of the film thickness at various particle diameters of the hydroxyapatite powder fired at 600 to 1350 ° C. in a nitrogen gas atmosphere.
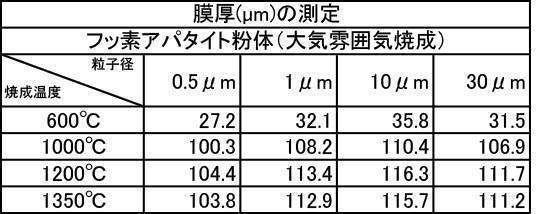
- Table 4 shows the measurement results of film thicknesses at various particle sizes of the fluorapatite powder fired at 600 to 1350 ° C.
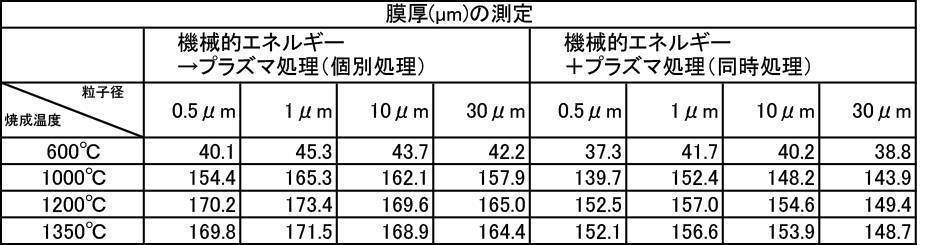
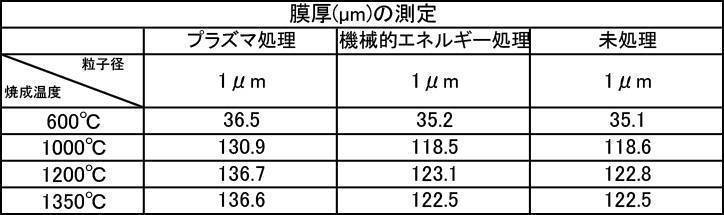
- the film thickness was less than 30 ⁇ m when the film was formed with a hydroxyapatite powder or a fluorapatite powder having an average particle size of 0.5 ⁇ m that was fired at 600 ° C. in an air atmosphere.
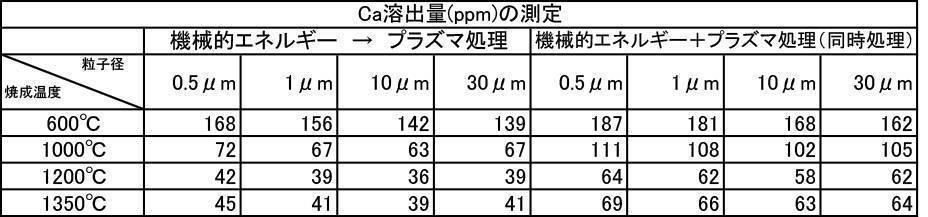
- All hydroxyapatite powders having an average particle size of 0.5 to 30 ⁇ m that were calcined at 600 to 1350 ° C. in a nitrogen gas atmosphere had a film thickness of 30 ⁇ m or more. From this, it is understood that when a hydroxyapatite powder produced by firing in an inert gas atmosphere, particularly an argon gas atmosphere, is used as a film forming powder, an excellent film thickness can be obtained.
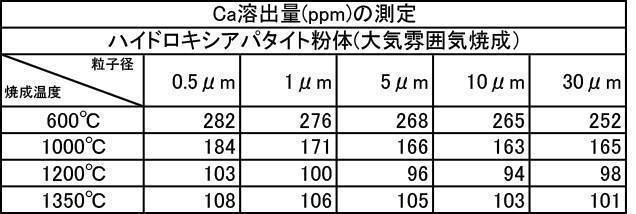
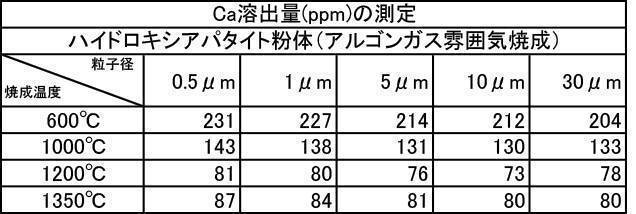
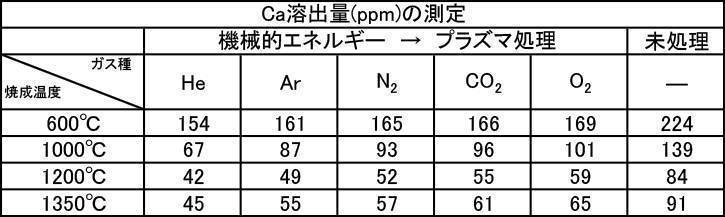
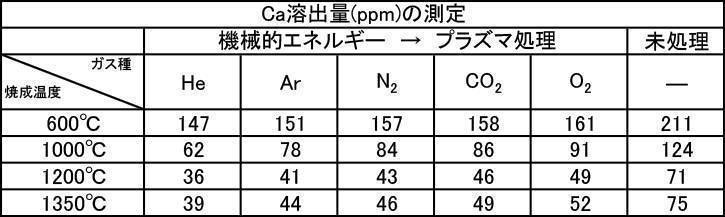
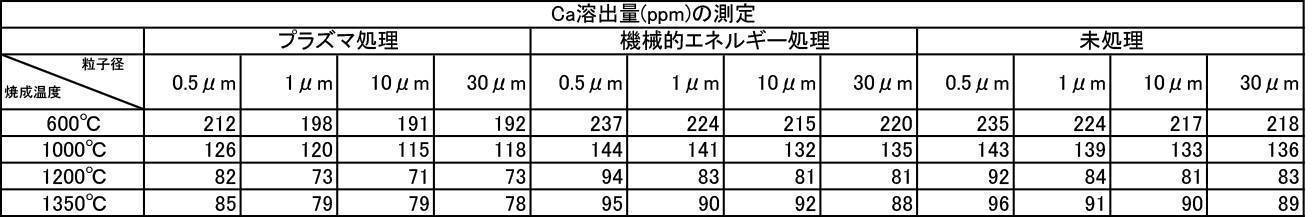
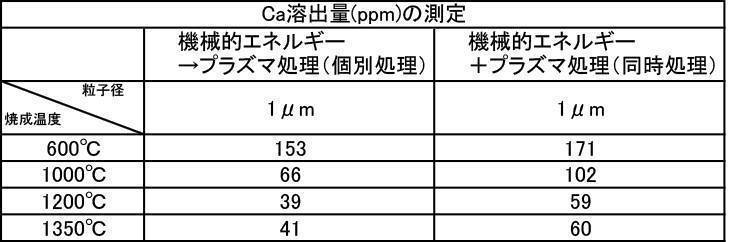
- the argon gas atmosphere and the nitrogen gas atmosphere are compared with the Ca elution amount when the film is formed with the hydroxyapatite powder having an average particle diameter of 0.5 to 30 ⁇ m, which is fired at 600 to 1350 ° C. in the air atmosphere.
- the amount of Ca elution was reduced.
- the Ca elution amount is suppressed by about 20%. I understand.
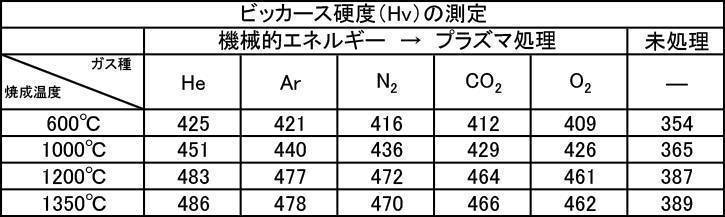
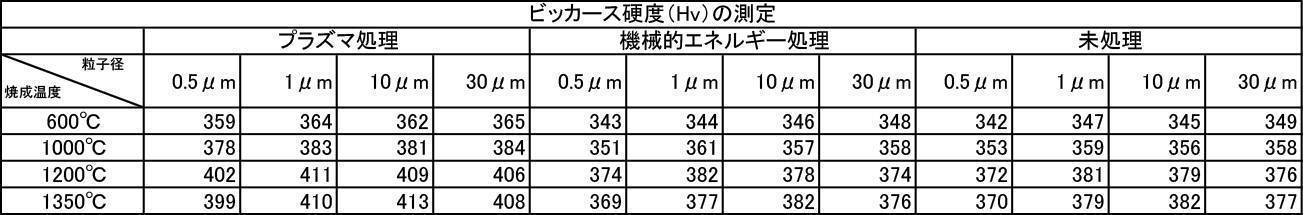
- Example 5-3 Vickers Hardness The hydroxyapatite powder for film formation produced in Example 2-1 was subjected to film formation treatment, and Vickers hardness was measured.
- [Table 8] shows the measurement results of Vickers hardness at various particle sizes of hydroxyapatite powder fired at 600 to 1350 ° C. in the air atmosphere, and various hydroxyapatite powders fired at 600 to 1350 ° C. in an argon gas atmosphere. The measurement results of Vickers hardness at the particle size are shown in [Table 9], and the measurement results of Vickers hardness at various particle sizes of the hydroxyapatite powder fired at 600 to 1350 ° C. in a nitrogen gas atmosphere are shown in [Table 10].
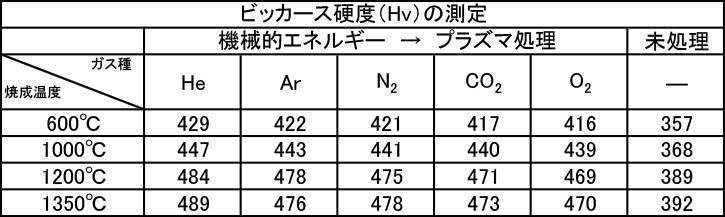
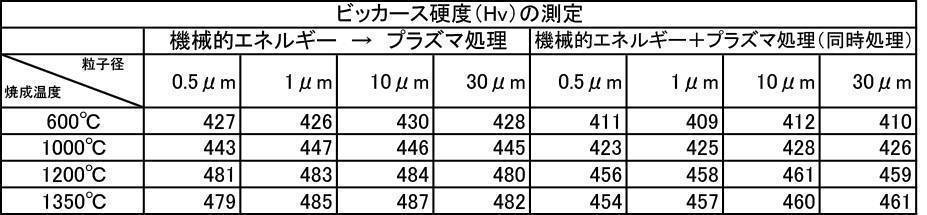
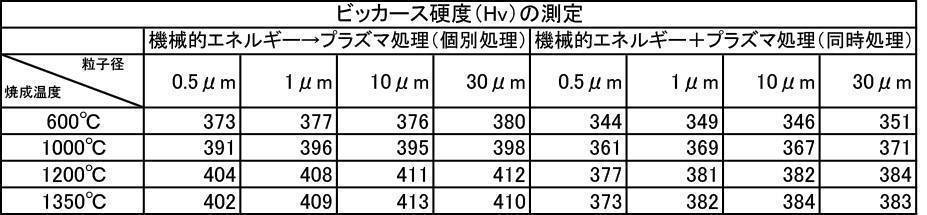
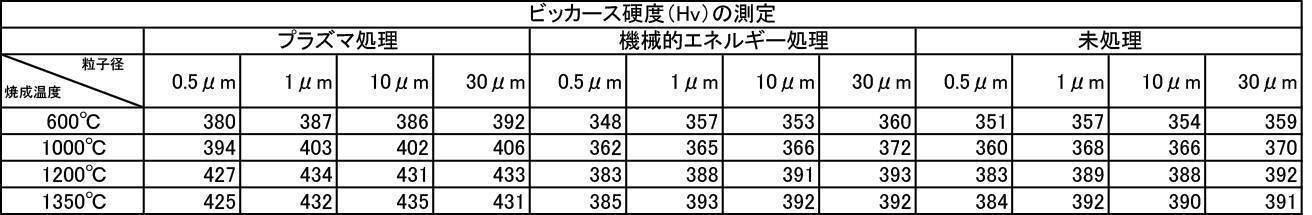
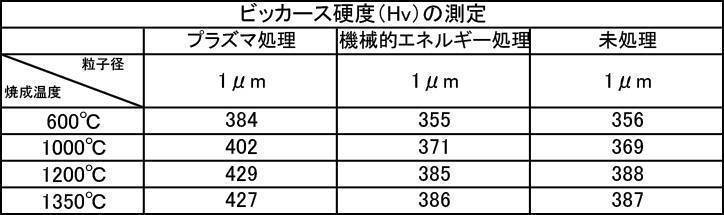
- the Vickers hardness in the case of forming a film with a hydroxyapatite powder having an average particle size of 0.5 to 30 ⁇ m, which was fired at 600 to 1350 ° C. in the air atmosphere was 302 to 330 Hv, and all were 330 Hv or less. there were. This value was equivalent to the low level of Vickers hardness of enamel reported in the literature.
- the Vickers hardness is 351 to 391 Hv and 351 to 391 Hv, respectively, when the hydroxyapatite powder having an average particle size of 0.5 to 30 ⁇ m is fired at 600 to 1350 ° C. in an argon gas atmosphere or a nitrogen gas atmosphere.
- the Vickers hardness of the film formed with the hydroxyapatite powder fired at 600 to 1350 ° C. in the air atmosphere was 330 Hv or less, but the hydroxyapatite powder fired at 600 to 1350 ° C. in the inert gas atmosphere
- a strength of 340 Hv or more was recognized at any particle size, and the film was effective as a film forming powder.
- the film thickness, Ca elution amount, and Vickers hardness of the film formed with a powder having an average particle diameter of 0.5 to 30 ⁇ m are all measured. The best result was obtained in the measurement results.
- a powder with an average particle size of 0.5 to 30 ⁇ m and fired in an argon gas atmosphere at 600 to 1350 ° C. and having a high shielding property (film thickness: 30 ⁇ m or more) can be formed in a short time.
- film thickness 30 ⁇ m or more
- the film thickness and the film thickness of the hydroxyapatite powder blended with 5% by mass of zinc oxide are blended.
- the film thickness in the case of not being added is approximately the same, and the film thickness in the case of the hydroxyapatite powder containing 1% by mass of titanium oxide is superior to the film thickness in the case of not being added. all right.
- Example 2 The film-forming apatite powder produced in Example 2 is subjected to mechanical energy addition treatment and then subjected to plasma irradiation treatment, or untreated powder that has not been subjected to mechanical energy addition treatment or plasma irradiation treatment. Each film was subjected to film formation treatment, and the film thickness, Ca elution amount, and Vickers hardness were measured for each sample. In the same manner as in Example 4-2, the film was formed by cutting a smooth enamel surface from the extracted human tooth and polishing the surface. Using a film forming apparatus by powder injection, film forming treatment was performed on the polished surface using various hydroxyapatite powders.
- the film forming conditions are as follows: handpiece tip nozzle inner diameter: 3.0 mm, spray pressure: 0.4 MPa, spray nozzle tip-substrate distance is 10 mm (nozzle tip is held perpendicular to the substrate), spray nozzle moving speed is It was 2 mm / s.
- the obtained film formation layer was surface-polished with a diamond polisher paste.
- Plasma irradiation treatment after mechanical energy addition treatment was performed for 30 minutes at a rotor rotation speed of 500 rpm using a mechanical energy application device (Mechanofusion AMS-MINI, Hosokawa Micron).
- the plasma treatment is performed by irradiating plasma with plasma generation conditions (applied voltage of 20 kV) from the tip of the plasma nozzle in a state where the container charged with the powder after mechanical energy treatment is rotated at a rotor rotational speed of 150 rpm. Processing was performed for a minute.
- As the plasma gas helium, argon, nitrogen, carbonic acid, and oxygen were used.
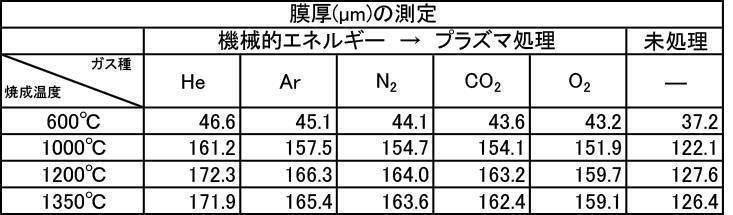
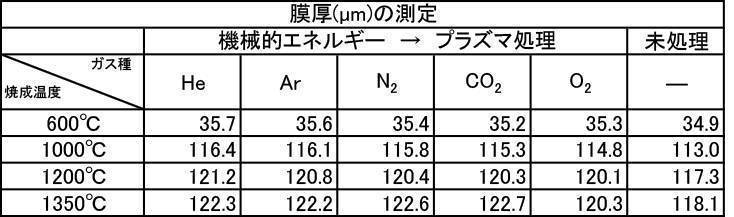
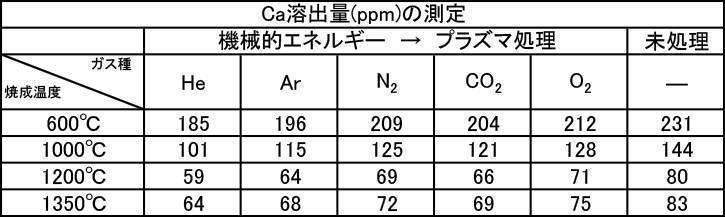
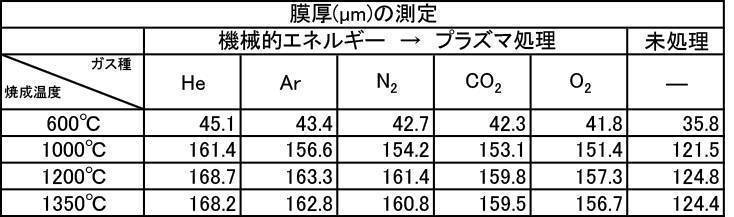
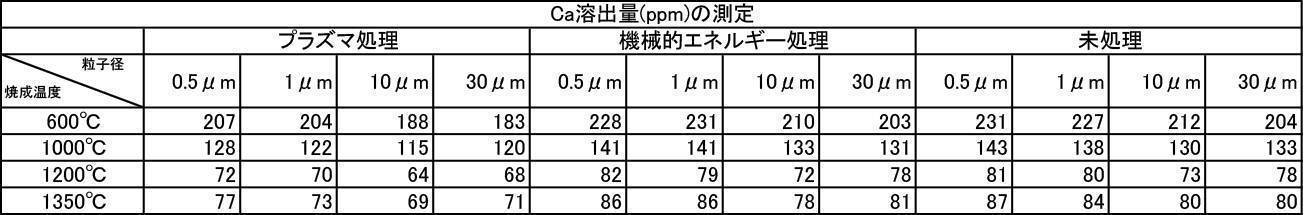
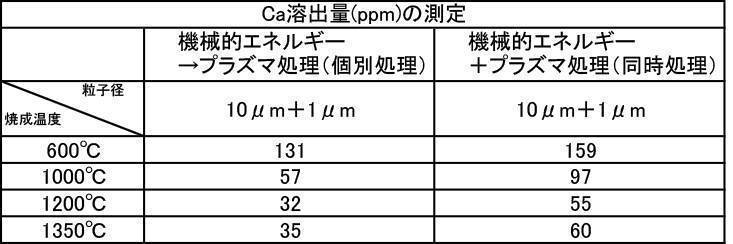
- the film thickness produced by the powder subjected to the plasma irradiation treatment after the mechanical energy addition treatment the film thickness, Ca elution amount, and Excellent in all items of Vickers hardness.
- the influence of the plasma gas species was excellent in the order of He>Ar> N 2 > CO 2 > O 2 .
- the film thickness produced by the mixed powder that was subjected to the plasma irradiation treatment after the mechanical energy addition treatment was compared with the film thickness produced by the untreated powder. And excellent in all items of Vickers hardness.
- the influence of the plasma gas type is not different in the film thickness, the Ca elution amount is excellent in the order of He>Ar> CO 2 > N 2 > O 2 , and the Vickers hardness is He>Ar> N 2 > CO. 2> it was excellent in the order of O 2.
- the film thickness produced by the mixed powder that was subjected to the plasma irradiation treatment after the mechanical energy addition treatment was compared with the film thickness produced by the untreated powder. And excellent in all items of Vickers hardness.
- the influence of the plasma gas species is that He is excellent in film thickness, Ca elution is excellent in the order of He>Ar> CO 2 > N 2 > O 2 , and He>Ar> N 2 > CO in Vickers hardness. 2> it was excellent in the order of O 2.
- the one-to-one mixed powder of argon gas atmosphere baking and nitrogen gas atmosphere baking is more than the one-to-one mixed powder of argon gas atmosphere baking and air baking, and the film thickness, Ca elution amount, and Vickers. Excellent in all items of hardness.
- Example 6-2 Plasma irradiation treatment after mechanical energy addition treatment (with color tone modifier)
- the film-forming color adjusting agent-containing hydroxyapatite powder produced in Example 2-3 was subjected to mechanical energy addition treatment, and then formed into a film using the powder subjected to plasma irradiation treatment and the untreated powder.
- the film thickness, Ca ion elution amount from the film, and the Vickers hardness of the film were measured.
- the mechanical energy treatment was carried out for 5 minutes at a rotor rotational speed of 5,000 rpm using an apparatus for applying mechanical energy (Mechanofusion AMS-MINI, Hosokawa Micron).
- the plasma treatment is performed for 20 minutes by irradiating the plasma under the plasma generation condition (applied voltage 5 kV) from the tip of the plasma nozzle with the container charged with the mechanical energy treated powder being rotated at a rotor rotation speed of 150 rpm.
- the plasma gas helium, argon, nitrogen, carbonic acid, and oxygen were used.
- the enamel smooth surface was cut out from the extracted human tooth and subjected to surface polishing in the same manner as in Example 4-2.
- film forming treatment was performed on the polished surface using various hydroxyapatite powders.
- the film forming conditions are as follows: the inner diameter of the nozzle of the hand piece tip: 0.5 mm, the injection pressure: 0.2 MPa, the distance between the tip of the injection nozzle and the substrate is 30 mm (the nozzle tip is held perpendicular to the substrate), The spray nozzle moving speed was 5 mm / s.
- the obtained film formation layer was surface-polished with a diamond polisher paste.
- the measurement results of the film thickness of the film formed with the powder are shown in [Table 22]
- the measurement results of the Ca elution amount are shown in [Table 23]
- the measurement results of the Vickers hardness are shown in [Table 24] for each plasma gas type.
- a film-forming powder in which 5% by mass of zinc oxide as a color tone adjusting agent is blended with a film-forming powder having an average particle diameter of 1 ⁇ m fired in an argon gas atmosphere is subjected to mechanical energy addition treatment, and then plasma irradiation treatment.
- Table 25 shows the measurement results of the film thickness of the film formed from the powder subjected to the above
- Table 26 shows the measurement results of the Ca elution amount
- Table 27 shows the measurement results of the Vickers hardness. Shown for each gas type.
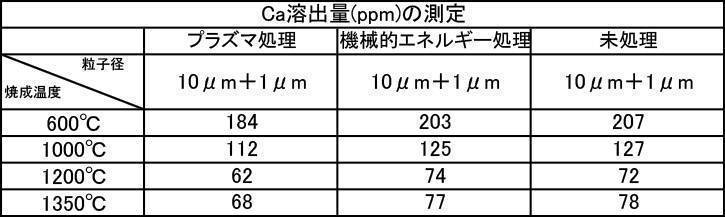
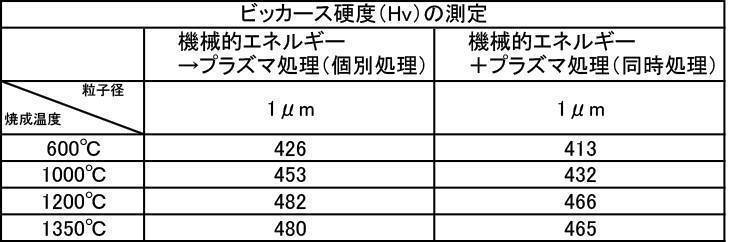
- the film formed with the powder that has been subjected to the plasma energy treatment after the mechanical energy addition treatment is more suitable for any plasma gas type than the film produced with the untreated powder. It was excellent in all items of thickness, Ca elution amount, and Vickers hardness.
- the film formation rate is high, the Ca elution amount is suppressed, and high Vickers hardness is achieved. From the results, it can be seen that a more stable and stable film can be formed.
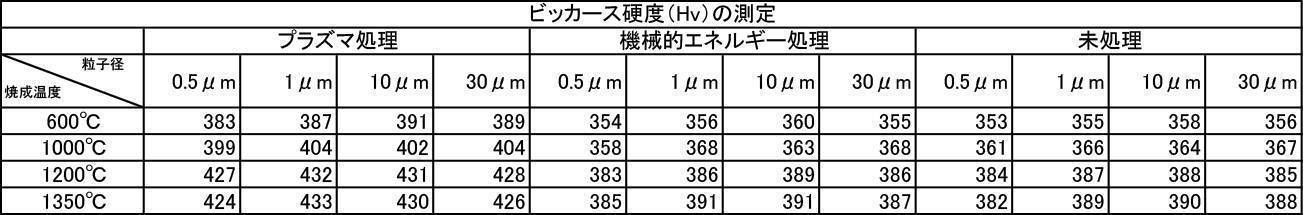
- the film-forming powder subjected to mechanical energy treatment and plasma treatment, and the film formed from the film-forming powder blended with the color tone adjusting agent have a Vickers hardness of 380 Hv or more, particularly mechanical.
- the film-forming powders that were plasma-treated after energy treatment all showed a value of 400 Hv or higher, and it was recognized that natural teeth were strengthened, which prolonged the life of the teeth. It is a great advantage in the sense of doing.
- the film thickness formed with the film forming powder blended with the color adjusting agent subjected to mechanical energy treatment and plasma treatment is almost the same as the film thickness formed with the film forming powder. This indicates that the color tone adjusting agent does not affect the film formation.
- the Ca elution amount of the film-forming powder that has been subjected to mechanical energy treatment and plasma treatment, and the film-forming powder that is blended with a color tone adjusting agent is untreated (mixing only) or mechanical energy. It shows a low value compared to the Ca elution amount when formed by the treatment, and the stability of the formed film is improved, and a dense film having high acid resistance can be obtained. It is considered that a film that stably exists in the oral environment can be provided.
- the mechanical energy addition treatment was performed for 10 minutes at a rotor rotational speed of 2500 rpm using a mechanical energy application device (Mechanofusion AMS-MINI, Hosokawa Micron).
- a mechanical energy application device Mechanism for generating plasma (applied voltage: 10 kV, plasma gas: helium) in a state where the container charged with the powder before and after the mechanical energy application treatment is rotated at a rotor rotation speed of 150 rpm, Irradiation from the tip of the nozzle was performed for 10 minutes.
- the enamel smooth surface was cut out from the extracted human tooth and subjected to surface polishing in the same manner as in Example 4-2.
- film formation treatment was performed on the polished surface using various hydroxyapatite powders and color adjuster-containing hydroxyapatite powders.
- the film formation conditions are as follows: handpiece tip nozzle inner diameter: 1.8 mm, spray pressure: 0.5 MPa, spray nozzle tip-substrate distance is 5 mm (the nozzle tip is held perpendicular to the substrate), and the spray nozzle moving speed is 1 mm / s.
- the obtained film formation layer was surface-polished with a diamond polisher paste.
- film formation with a film thickness of 30 ⁇ m or more is recognized, and the film can be effectively used as a film-forming powder. all right.
- the film thickness produced by the one-to-one mixed hydroxyapatite powder of argon gas atmosphere firing and air atmosphere firing is larger than the film thickness produced by argon gas atmosphere firing hydroxyapatite powder, and the film thickness and Ca elution amount. , And inferior in all items of Vickers hardness.
- the film thickness, Ca elution amount, and Vickers hardness were superior in all items, but the film thickness, Ca elution amount, and Vickers were better than those formed by argon gas atmosphere calcined hydroxyapatite powder. It was inferior in all items of hardness.
- Example 7-6 Silica-Containing Hydroxyapatite Powder for Film Formation
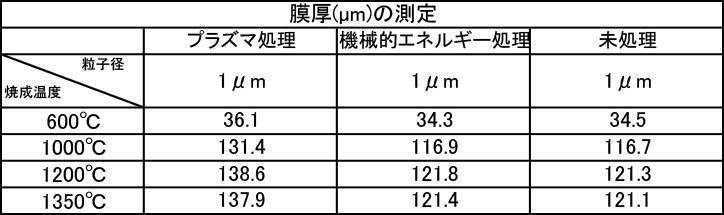
- a powder obtained by adding 1% of silica powder to hydroxyapatite powder having an average particle diameter of 1 ⁇ m was prepared in an argon atmosphere.
- the powder for film formation having a particle diameter of 1 ⁇ m to which silica that had been baked, pulverized and classified with an opposed airflow pulverizer was added, the same test as in the above 7-2 was performed.
- the results for the film thickness are shown in [Table 49] and [Table 50]
- the results for the Ca elution amount are shown in [Table 51] and [Table 52]
- the results for the Vickers hardness are shown in [Table 53] and [Table 50]. 54].
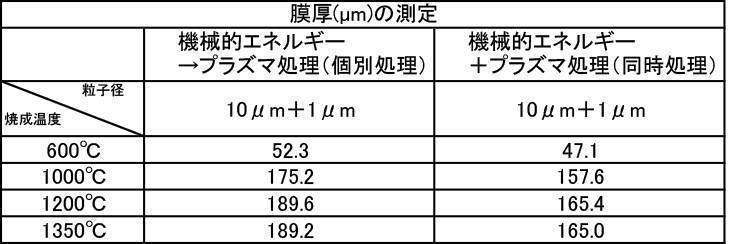
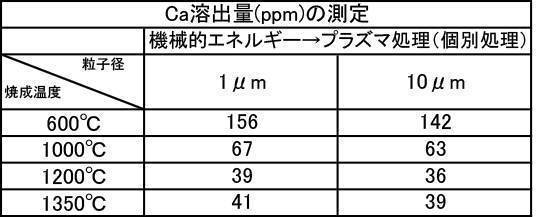
- a hydroxyapatite powder obtained by mixing 1: 1 a film-forming hydroxyapatite powder having an average particle diameter of 1 ⁇ m and 10 ⁇ m was used, and the powder was prepared by performing a plasma irradiation process after a mechanical energy addition process.
- the film formation is compared with a film formed by using a hydroxyapatite powder for forming each film having an average particle diameter of 1 ⁇ m and an average particle diameter of 10 ⁇ m, and applying a plasma irradiation process after a mechanical energy addition process.
- the film thickness, Ca elution amount, and Vickers hardness were all excellent.
- plasma irradiation process by performing the plasma irradiation process (plasma process), it is suitable for forming a film having high hardness and low acid solubility (a film with a small amount of Ca elution) in a short time.
- a film-forming powder was obtained.
- plasma irradiation mechanical energy ⁇ plasma processing; individual processing
- plasma irradiation mechanical energy ⁇ plasma processing; individual processing
- each of the samples was subjected to a film formation process (non-processed) without applying energy, and each sample was subjected to film formation thickness, Ca elution amount, Vickers hardness as in Example 7. Constant was carried out. The measurement methods of the film thickness, Ca elution amount, and Vickers hardness were performed in the same manner as in Example 5.
- Powder containing 1% by mass of titanium oxide as a color tone adjusting agent 1% by mass of titanium oxide as a color tone adjusting agent is formed into a powder for film formation which is baked in an argon atmosphere and pulverized and classified by an opposed airflow crusher.
- a film was formed using the powder obtained by performing the above treatments 1) and 3) to 6) on the powder blended in%.
- the results for the film thickness are shown in [Table 64] and [Table 65]
- the results for the Ca elution amount are shown in [Table 66] and [Table 67]
- the results for the Vickers hardness are shown in [Table 68] and [Table 65]. 69].
- Powder containing 5% by mass of zinc oxide as a color tone adjusting agent 5% by mass of zinc oxide as a color tone adjusting agent is formed into a powder for film formation which is baked in an argon atmosphere and pulverized and classified by an opposed airflow pulverizer.
- a film was formed using the powder obtained by performing the above treatments 1) and 3) to 6) on the powder blended in%.
- the results for the film thickness are shown in [Table 70] and [Table 71]
- the results for the Ca elution amount are shown in [Table 72] and [Table 73]
- the results for the Vickers hardness are shown in [Table 74] and [Table 71]. 75].
- Example 7-2 Laser Raman spectroscopic test In the powder X-ray diffraction test, the change in crystallinity on the near surface of the powder particles could not be confirmed.
- a film of a tooth crown color adjusting agent (white; containing 1% titanium oxide) is formed on a glass plate, and a film of a tooth crown color adjusting agent (as a tooth color) is formed as a second layer on the first layer. Close color; zinc oxide 5% blended) was formed, and on this second layer, a crown color tone modifier film (transparent color (as top coat); only hydroxyapatite) was formed as the third layer. .
- the film formation conditions were the same for the first layer, the second layer, and the third layer, and the handpiece tip nozzle inner diameter was 1.8 mm and the injection pressure was 0.5 MPa.
- FIG. 8 shows a cross-sectional image of the multilayer film formation layer shown in FIG. 7 by a laser microscope.
- the film-forming powder of the present invention is useful in the field of dental treatment.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Oral & Maxillofacial Surgery (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Ceramic Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Plastic & Reconstructive Surgery (AREA)
- Materials Engineering (AREA)
- Structural Engineering (AREA)
- Physics & Mathematics (AREA)
- Thermal Sciences (AREA)
- Geology (AREA)
- General Life Sciences & Earth Sciences (AREA)
- Environmental & Geological Engineering (AREA)
- Plasma & Fusion (AREA)
- Dental Preparations (AREA)
- Cosmetics (AREA)
- Materials For Medical Uses (AREA)
- Dental Prosthetics (AREA)
Abstract
Description
他方、エナメル質や象牙質と一体化できる方法として、エナメル質や象牙質の主成分であるハイドロキシアパタイト粉体を高速で歯表面に噴射してエナメル質や象牙質の表面にハイドロキシアパタイト膜を形成する装置(特許文献9~12)が提案されている。
また、本発明の背景には、近年、審美歯科治療に対する患者の要望が高まっており、その治療法として、ブリーチング法やポーセレンラミネートベニヤ法による処置があるが、これらの治療法は、健全歯質を侵襲することから患者の負担が大きいことが問題視されている。これに対し、ハイドロキシアパタイト粉体の噴射により、歯に膜成形を行えば、歯と同様の成分で歯冠の色調調整をすることが可能となるため、健全な歯質に対する侵襲がなく、逆に歯質強化ができるなど、患者の負担を大きく低減した治療が可能となる。
(1)歯に対して噴射する装置に使用して歯表面に膜を形成するための膜形成用粉体であって、Ca10-X・MX(ZO4)6Y2(ただし、Xは0~10、Mは金属、又は水素、ZO4はPO4、CO3、CrO4、AsO4、VO4、SiO4、SO4又はGeO4、Yは水酸基、ハロゲン元素又は炭酸基)で示されるアパタイトを、不活性ガス雰囲気下600~1350℃で焼成することにより製造したことを特徴とする平均粒子径が0.5~30μmである膜形成用粉体。
(2)アパタイトが、ハイドロキシアパタイトであることを特徴とする上記(1)記載の膜形成用粉体。
(3)不活性ガスがアルゴンガス又は窒素ガスであることを特徴とする上記(1)又は(2)記載の膜形成用粉体。
(4)さらに、歯冠の色調を調整するための色調調整剤を配合したことを特徴とする上記(1)~(3)のいずれか記載の膜形成用粉体。
(5)歯冠の色調調整剤が、酸化チタン、酸化亜鉛、群青及び赤色顔料から選ばれる少なくとも1種であることを特徴とする上記(4)記載の膜形成用粉体。
(6)プラズマ照射を行うことにより製造したことを特徴とする上記(1)~(5)のいずれか記載の膜形成用粉体。
(7)さらに、機械的なエネルギーを加えることにより製造したことを特徴とする上記(6)記載の膜形成用粉体。
(8)機械的なエネルギーを加えた後で、プラズマ照射を行うことにより製造したことを特徴とする上記(7)記載の膜形成用粉体。
(9)ヘリウムガスを照射ガスとしたプラズマ照射であることを特徴とする上記(6)~(8)のいずれか記載の膜形成用粉体。
(10)ハンドピース先端ノズル内径:0.5~5.0mm、噴射圧:0.2~0.8MPa、噴射ノズル先端-基板間距離0.1~3.0cm、噴射ノズル移動速度0~10mm/sの条件で、粉体を基板に噴射した場合に、形成された膜の膜厚が30μm以上で、ビッカース硬度が340Hv以上であることを特徴とする上記(1)~(9)のいずれか記載の膜形成用粉体。
(11)Ca10-X・MX(ZO4)6Y2(ただし、Xは0~10、Mは金属、又は水素、ZO4はPO4、CO3、CrO4、AsO4、VO4、SiO4、SO4又はGeO4、Yは水酸基、ハロゲン元素又は炭酸基)で示されるアパタイトを不活性ガス雰囲気下600~1350℃で焼成した後、粉砕及び分級することを特徴とする、歯に対して噴射する装置に使用して歯表面に膜を形成するための、平均粒子径が0.5~30μmである膜形成用粉体の製造方法。
(12)アパタイトが、ハイドロキシアパタイトであることを特徴とする上記(11)記載の膜形成用粉体の製造方法。
(13)不活性ガスがアルゴンガス又は窒素ガスであることを特徴とする上記(11)又は(12)記載の膜形成用粉体の製造方法。
(14)さらに、歯冠の色調を調整するための色調調整剤を配合したことを特徴とする上記(11)~(13)のいずれか記載の膜形成用粉体の製造方法。
(15)歯冠の色調調整剤が、酸化チタン、酸化亜鉛、群青及び赤色顔料から選ばれる少なくとも1種であることを特徴とする上記(14)記載の膜形成用粉体の製造方法。
(16)粉砕及び分級後に、プラズマ照射を行うことを特徴とする上記(11)~(15)のいずれか記載の膜形成用粉体の製造方法。
(17)さらに、機械的なエネルギーを加えることを特徴とする上記(16)記載の膜形成用粉体の製造方法。
(18)機械的なエネルギーを加えた後で、プラズマ照射を行うことを特徴とする上記(17)記載の膜形成用粉体の製造方法。
(19)ヘリウムガスを照射ガスとしたプラズマ照射であることを特徴とする上記(16)~(18)のいずれか記載の膜形成用粉体の製造方法。
(20)ハンドピース先端ノズル内径:0.5~5.0mm、噴射圧:0.2~0.8MPa、噴射ノズル先端-基板間距離0.1~3.0cm、噴射ノズル移動速度0~10mm/sの条件で、粉体を基板に噴射した場合に、形成された膜の膜厚が30μm以上で、ビッカース硬度が340Hv以上であることを特徴とする上記(11)~(19)のいずれか記載の膜形成用粉体の製造方法。
(21)上記(1)~(10)のいずれか記載の膜形成用粉体を含むペレット。
1-1 ハイドロキシアパタイトの合成
室温下、0.5Mの水酸化カルシウム水懸濁液(2L)中に、0.3Mのリン酸水溶液(2L)を滴下してアパタイトの懸濁液を作製した。反応溶液のpHはアンモニア水溶液を用いて10.5に調整した。溶液が完全に混合したのを確認した後、この懸濁液を一晩熟成させた。得られた沈殿物をろ過、固形物を80℃で乾燥させた。
0.25Mの水酸化カルシウム懸濁液(2L)と、0.3molのリン酸と0.1molのフッ化水素を混合した水溶液(2L)を調製した。室温下、この水酸化カルシウム懸濁液中に、リン酸とフッ化水素の混合水溶液を2時間かけて滴下した。滴下終了後、懸濁液を攪拌しながら80℃で5時間熟成させた。得られた沈殿物をろ過して、固形分を80℃で乾燥させた。
0.75Lの純水中に炭酸ガスを30分間バブリングした。この溶液のpHは7から4に低下した。得られた溶液に0.3molのリン酸を加え、全量を純水で1Lにメスアップした。この溶液を0.5Mの水酸化カルシウム水溶液(1L)中に、1L/3hの速度で滴下した。懸濁液を2時間攪拌後、一晩熟成を行い、ろ過、得られた固形物を80℃で乾燥させた。
0.19molの硝酸カルシウム四水和物と0.01molのMg(OH)2を500mLの純水で溶解させた。その後、本溶液のpHをアンモニア水でpH10に調整した。この溶液に、0.12Mのリン酸二水素アンモニウム水溶液(500mL)をゆっくりと加えた。この時、溶液中のpHが10になるように少量のアンモニア水を加えた。リン酸水素二アンモニウムの水溶液を全て加えた後、溶液を攪拌しながら90℃で5時間熟成を行った。沈殿物をろ過して、超音波処理により純水中で3回洗浄を行った。得られた固形物は80℃で乾燥させた。
焼成用雰囲気炉として、真空置換式雰囲気炉2024-V型(丸祥電器)を用いた。また、粉砕、分級装置として、流動層式対向型ジェットミル カウンタージェットミル 100AFG型(ホソカワミクロン)を用いた。
上記で合成したハイドロキシアパタイト、フッ素アパタイト、炭酸アパタイト及びマグネシウム固溶アパタイトを、乳鉢で粉砕し、大気中、アルゴンガス及び窒素ガス雰囲気中、200~1350℃、又は600~1350℃で焼成した。焼成した試料を、対向式気流粉砕機で粉砕、分級して、各々の試料について平均粒子径が0.5~30μmのハイドロキシアパタイト粉体を得た。
また、上記2-1で合成した膜形成用アパタイト粉体に、アパタイト以外の成分としてシリカを1重量%添加して同様の処理を行い、シリカを配合したハイドロキシアパタイト粉体を得た。シリカは、「堺化学工業(株)のSciqasシリーズ 粒子径:1.0μm」を用いた。
上記2-1で作製した膜形成用ハイドロキシアパタイト粉体に、各種の色調調整剤を配合して、色調調整剤を配合した膜形成用粉体を得た。酸化チタンは「キシダ化学(株):特製」を、酸化亜鉛は「ハクスイテック(株):局方酸化亜鉛」を、群青は「(株)ピノア、ウルトラマリーン」を、酸化鉄は「関東化学(株):鹿1級」を、赤色204号は「東京化成工業(株)、レーキレッドCBA」をそれぞれ使用した。
プラズマ照射装置として、自社で作製したプラズマ発生装置を用いた。プラズマ照射時に使用する粉体の混合機は、300ccのビーカーを、傾斜をつけた状態のターンテーブル電動式T-AU上に固定し、回転させて用いた。
プラズマ発生装置を図1及び図2に示す。図中、1はAC/DCコンバータ(AC100V→DC24V)、2は冷陰極管インバータ(DC24V→AC1000V)、3は昇圧回路(コッククロフト・ウォルトン回路;AC1000V→AC10KV)、4はプラズマノズル、5はガス流量計をそれぞれ示す。また、機械的エネルギーを加える装置として、メカノフュージョンAMS-MINI(ホソカワミクロン)を用い、機械的エネルギーとプラズマ照射を同時に行える装置として、ナノキュラNC-ALB(ホソカワミクロン)を用いた。
実施例2-1で製造した膜形成用ハイドロキシアパタイト粉体、実施例2-2で製造した膜形成用シリカ配合ハイドロキシアパタイト粉体、及び実施例2-3で製造した膜形成用色調調整剤配合ハイドロキシアパタイト粉体について、自社製プラズマ発生装置を用いて、プラズマ照射処理を行った。混合機(300ccのビーカーを、ターンテーブル電動式 T-AUで回転)で膜形成用粉体を混合しながら、プラズマ照射処理を行い、プラズマ照射処理膜形成用粉体を得た。
実施例2-1で製造した膜形成用ハイドロキシアパタイト粉体、実施例2-2で製造した膜形成用シリカ配合ハイドロキシアパタイト粉体、及び実施例2-3で製造した膜形成用色調調整剤配合ハイドロキシアパタイト粉体について、機械的エネルギーを加える装置(メカノフュージョン AMS-MINI、ホソカワミクロン)で処理を行なった後、プラズマ照射を行う処理を行なって、膜形成用粉体を得た。同様に、実施例2-1で製造した膜形成用ハイドロキシアパタイト粉体について、プラズマ照射を行う処理を行なった後、機械的エネルギーを加える装置で処理を行なって、膜形成用粉体を得た。
実施例2-1で製造した膜形成用ハイドロキシアパタイト粉体、実施例2-2で製造した膜形成用シリカ配合ハイドロキシアパタイト粉体、及び実施例2-3で製造した膜形成用色調調整剤配合ハイドロキシアパタイト粉体について、機械的エネルギー付加処理とプラズマ照射処理を同時に行える装置(ナノキュラ NC-ALB、ホソカワミクロン)で処理を行い、膜形成用粉体を得た。
実施例2-1で製造した膜形成用ハイドロキシアパタイト粉体、実施例2-2で製造した膜形成用シリカ配合ハイドロキシアパタイト粉体、及び実施例2-3で製造した膜形成用色調調整剤配合ハイドロキシアパタイト粉体について、機械的エネルギーを加える処理を行ない、膜形成用粉体を得た。
実施例2-1で製造した膜形成用ハイドロキシアパタイト粉体、実施例2-2で製造した膜形成用シリカ配合ハイドロキシアパタイト粉体、及び実施例2-3で製造した膜形成用色調調整剤配合ハイドロキシアパタイト粉体により膜を形成し、それぞれ膜厚、Ca溶出量、及びビッカース硬度の測定を行った。また、上記色調調整剤を配合した膜形成用ハイドロキシアパタイト粉体を用いた成膜層(写真)を図6に示す。
実施例2-1で製造した膜形成用粉体の平均粒子径及び粒度分布を図3に示す。膜形成用粉体の粒度分布の測定には、粒度分布測定装置(LA-950, 堀場製作所社製)を使用した。また測定には乾式ユニットを使用した。なお、これ以降に記載する[表]等における「粒子径0.5μm」は平均粒子径が0.4~0.6μmの粉体を、「粒子径1μm」は平均粒子径が0.9~1.1μmの粉体を、「粒子径5μm」は平均粒子径が4.0~6.0μmの粉体を、「粒子径10μm」は平均粒子径が9.0~11.0μmの粉体を、「粒子径20μm」は平均粒子径が19.0~21.0μmの粉体を、「粒子径30μm」は平均粒子径が29.0~31.0μmの粉体をそれぞれ意味する。
ヒト抜去歯からエナメル質平滑面を切り出して、表面研磨を行った。粉体噴射による膜形成装置により、上記の研磨表面に、各種の上記膜形成用ハイドロキシアパタイト粉体を用いて成膜処置を行った。成膜条件は、ハンドピース先端ノズル内径:5.0mm、噴射圧:0.6MPaとした。噴射ノズル先端-基板間距離は0.5cm(ノズル先端は基板に垂直に保持)、噴射ノズル移動速度は10mm/sとした。得られた成膜層はダイヤポリッシャーペーストで表面研磨を行った。なお、研磨処理による成膜層の厚さは変化しないことを、デジタルマイクロスコープVHX-1000(株式会社キーエンス)を用いて確認した。
上記4-2の成膜処理により形成した膜厚の測定は、デジタルマイクロスコープVHX-1000(株式会社キーエンス)を用いて3D計測から膜厚を求めた。
上記4-2の成膜処置を行った成膜面(約2mm×2mmのウインドウ)以外の試料面を、全てネイルエナメルでマスキングして、Ca溶出量の測定用のエナメル質ブロックを作製した。膜のCa溶出量の評価は、口腔内のpH変動を模擬したpHサイクル試験によって、成膜層から溶出するCaイオン濃度を測定した。試験溶液には0.2mol/Lの乳酸緩衝溶液(pH4.5)及び0.02mol/LのHEPES緩衝溶液(pH7.0)を使用した。この溶液に、上記で作製したCaイオン溶出量の測定用のエナメル質ブロックを、37℃の試験条件下で、乳酸緩衝溶液に30分浸漬する。続いてHEPES緩衝溶液に90分浸漬というサイクルを計3サイクル行った。試験終了後、溶液中に溶出したCaイオン濃度をイオンクロマトグラフィーで測定した(陽イオンクロマトグラフィー法)。また、測定方法は、以下の測定条件で行なった。
装置名:Intelligent HPLC LC-2000 Plus(日本分光社)
測定用カラム:陽イオン測定用カラム IC YK-421(Shodex社)
溶離液:5mM酒石酸+1mMジピコリン酸+1.5g/Lホウ酸
流量:1.0ml/min
試料注入量:20μl
カラム温度:40℃
検出器:電気伝導度検出器
成膜処理により作製した膜について、微小硬度計FM-700(株式会社フューチュアテック)を用いて、押し込み荷重:100g、荷重保持時間:30秒で測定した。
実施例2で製造した膜形成用アパタイト粉体について成膜処理を行い、各試料について、成膜厚さ、Ca溶出量、ビッカース硬度の測定を行った。成膜には、自社製の粉体噴射装置を用いた。自社製の粉体噴射装置は、AC/DCコンバータ(AC100V→DC24V)、ソレノイドバルブ、ミストセパレータ、エアーレギュレータ、スピードコントローラなどを備えている。
実施例2-1で製造した膜形成用アパタイト粉体について成膜処理を行い、成膜厚さを測定した。大気雰囲気中で200~1350℃で焼成したハイドロキシアパタイト粉体の各種粒径における膜厚の測定結果を[表1]に、アルゴンガス雰囲気中で600~1350℃で焼成したハイドロキシアパタイト粉体の各種粒径における膜厚の測定結果を[表2]に、窒素ガス雰囲気中で600~1350℃で焼成したハイドロキシアパタイト粉体の各種粒径における膜厚の測定結果を[表3]に、大気雰囲気中で600~1350℃で焼成したフッ素アパタイト粉体の各種粒径における膜厚の測定結果を[表4]にそれぞれ示す。

実施例2-1で製造した膜形成用ハイドロキシアパタイト粉体について成膜処理を行い、Ca溶出量を測定した。大気雰囲気中で600~1350℃で焼成したハイドロキシアパタイト粉体の各種粒径におけるCa溶出量の測定結果を[表5]に、アルゴンガス雰囲気中で600~1350℃で焼成したハイドロキシアパタイト粉体の各種粒径におけるCa溶出量の測定結果を[表6]に、窒素ガス雰囲気中で600~1350℃で焼成したハイドロキシアパタイト粉体の各種粒径におけるCa溶出量の測定結果を[表7]にそれぞれ示す。

実施例2-1で製造した膜形成用ハイドロキシアパタイト粉体について成膜処理を行い、ビッカース硬度を測定した。大気雰囲気中で600~1350℃で焼成したハイドロキシアパタイト粉体の各種粒径におけるビッカース硬度の測定結果を[表8]に、アルゴンガス雰囲気中で600~1350℃で焼成したハイドロキシアパタイト粉体の各種粒径におけるビッカース硬度の測定結果を[表9]に、窒素ガス雰囲気中で600~1350℃で焼成したハイドロキシアパタイト粉体の各種粒径におけるビッカース硬度の測定結果を[表10]にそれぞれ示す。



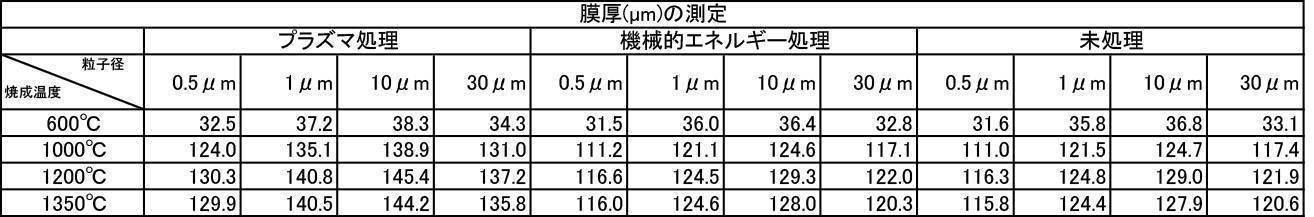
特に、アルゴンガス雰囲気中600~1350℃で焼成したハイドロキシアパタイト粉体においては、平均粒子径が0.5~30μmの粉体で成膜した膜の膜厚、Ca溶出量、ビッカース硬度の全ての測定結果において最も良好な結果が得られた。
実施例2-3で製造した膜形成用色調調整剤配合ハイドロキシアパタイト粉体について成膜処理を行い、成膜厚さを測定した。アルゴンガス雰囲気中で200~1350℃で焼成した酸化チタン1質量%配合ハイドロキシアパタイト粉体の各種粒径における膜厚の測定結果を[表11]に、アルゴンガス雰囲気中で200~1350℃で焼成した酸化亜鉛5質量%配合ハイドロキシアパタイト粉体の各種粒径における膜厚の測定結果を[表12]にそれぞれ示す。


実施例2で製造した膜形成用アパタイト粉体に、機械的エネルギー付加処理をした後にプラズマ照射処理を施した粉体、又は機械的エネルギー付加処理やプラズマ照射処理を施していない未処理の粉体でそれぞれ成膜処理を行い、各試料について、成膜厚さ、Ca溶出量、ビッカース硬度の測定を行った。膜の形成方法は、前記実施例4-2と同様に、ヒト抜去歯からエナメル質平滑面を切り出して、表面研磨を行った。粉体噴射による膜形成装置により、上記の研磨表面に、各種のハイドロキシアパタイト粉体を用いて成膜処置を行った。また、成膜条件は、ハンドピース先端ノズル内径:3.0mm、噴射圧:0.4MPaとして、噴射ノズル先端-基板間距離は10mm(ノズル先端は基板に垂直に保持)、噴射ノズル移動速度は2mm/sとした。得られた成膜層はダイヤポリッシャーペーストで表面研磨を行った。
機械的エネルギー付加処理は、機械的エネルギーを加える装置(メカノフュージョン AMS-MINI、ホソカワミクロン)を用いて、ローター回転数500rpmとして30分間処理した。また、プラズマ処理は、機械的エネルギー処理後の粉体を投入した容器をローター回転数150rpmで回転させた状態で、プラズマ発生条件(印加電圧20kV)のプラズマを、プラズマノズル先端から照射させて5分間処理を行なった。プラズマガスは、ヘリウム、アルゴン、窒素、炭酸、酸素を用いた。
実施例2-1で製造したアルゴンガス雰囲気中で600~1350℃で焼成した粒子径1μmのハイドロキシアパタイト粉体に機械的エネルギー付加処理をし、その後にプラズマ照射処理を施した粉体で作製した成膜の膜厚の測定結果を[表13]に、Ca溶出量の測定結果を[表14]に、ビッカース硬度の測定結果を[表15]に、それぞれプラズマガス種ごとに示す。


アルゴンガス雰囲気で焼成した粒子径1μmのハイドロキシアパタイト粉体と、大気雰囲気で焼成した粒子径1μmのハイドロキシアパタイト粉体との1対1混合粉体に機械的エネルギー付加処理をし、その後にプラズマ照射処理を施した混合粉体で作製した成膜の膜厚の測定結果を[表16]に、Ca溶出量の測定結果を[表17]に、ビッカース硬度の測定結果を[表18]に、それぞれプラズマガス種ごとに示す。


アルゴンガス雰囲気で焼成した粒子径1μmの膜形成用粉体と、窒素ガス雰囲気で焼成した粒子径1μmの膜形成用粉体との1対1混合粉体に、機械的エネルギー付加処理をし、その後にプラズマ照射処理を施した混合粉体で作製した成膜の膜厚の測定結果を[表19]に、Ca溶出量の測定結果を[表20]に、ビッカース硬度の測定結果を[表21]に、それぞれプラズマガス種ごとに示す。


実施例2-3で製造した膜形成用色調調整剤配合ハイドロキシアパタイト粉体に機械的エネルギー付加処理をし、その後にプラズマ照射処理を施した粉体、及び未処理粉体を用いて成膜して、膜厚、膜からのCaイオン溶出量、及び膜のビッカース硬度の測定を行った。機械的エネルギー処理は、機械的エネルギーを加える装置(メカノフュージョン AMS-MINI、ホソカワミクロン)を用いて、ローター回転数5,000rpmとして5分間処理した。プラズマ処理は、機械的エネルギー処理後の粉体を投入した容器をローター回転数150rpmで回転させた状態で、プラズマ発生条件(印加電圧5kV)のプラズマを、プラズマノズル先端から照射させて20分間処理を行なった。プラズマガスは、ヘリウム、アルゴン、窒素、炭酸、酸素を用いた。
[焼成雰囲気が異なる粉体の処理方法による効果の違い]
7-1 実験条件
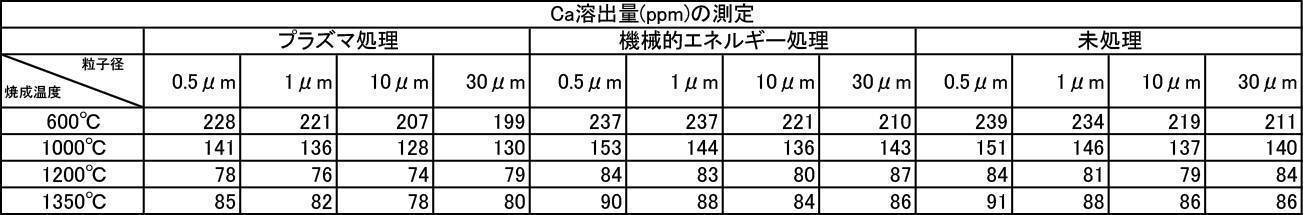
実施例2で製造した膜形成用アパタイト粉体に、1)機械的エネルギーを加えた後にプラズマ照射を行う処理(機械的エネルギー→プラズマ処理;個別処理)、2)プラズマ照射を行った後に機械的エネルギーを加える処理(プラズマ処理→機械的エネルギー;個別処理)、3)機械的エネルギーとプラズマ照射を同時に加える処理(機械的エネルギー=プラズマ処理;同時処理)、4)プラズマ照射を行う処理(プラズマ処理)、5)機械的エネルギーを加える処理(機械的エネルギー処理)を各々行った粉体、及び6)焼成、粉砕、分級、混合するだけで、プラズマ照射及び/又は機械的エネルギーを加える処理を行わない粉体(未処理)でそれぞれ成膜処理を行い、各試料について、成膜厚さ、Ca溶出量、ビッカース硬度の測定を行った。成膜厚さ、Ca溶出量、ビッカース硬度の測定方法は、実施例5と同様の方法により行った。
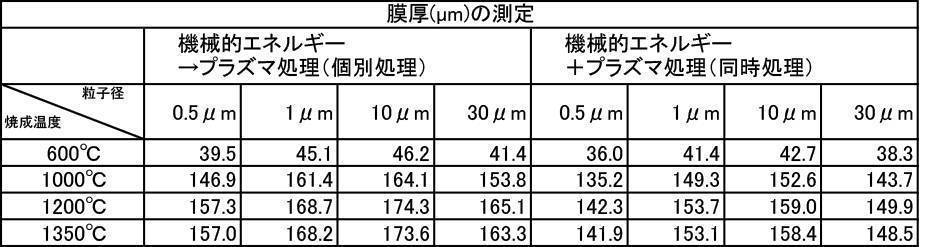
アルゴン雰囲気で焼成して、対向式気流粉砕機で粉砕、分級した膜形成用粉体に上記1)~6)の処理を施した粉体を用いて成膜した。成膜厚さについての結果を[表28]~[表30]に、Ca溶出量についての結果を[表31]~[表33]に、ビッカース硬度についての結果を[表34]~[表36]に示す。



実施例2で合成したフッ素アパタイト、炭酸アパタイト、マグネシウム固溶アパタイトについても、上記7-2と同様の試験を行なった。その結果、プラズマ照射を行う処理(プラズマ処理)、機械的エネルギーを加える処理とプラズマ照射の両方を行う処理、特に、機械的なエネルギーを加える処理の後で、プラズマ照射を行う処理(機械的エネルギー→プラズマ処理(個別処理))を行った場合には、焼成、粉砕、分級、混合するだけで、プラズマ照射、及び機械的エネルギーを加える処理を行わない(未処理)の膜形成用粉体に比べて、膜厚、Ca溶出量、ビッカース硬度において、7-2の場合と同様の結果が得られた。
アルゴン雰囲気で焼成して、対向式気流粉砕機で粉砕、分級した膜形成用ハイドロキシアパタイト粉体と、大気雰囲気で焼成して、対向式気流粉砕機で粉砕、分級したハイドロキシアパタイト粉体を1:1で混合した粉体を用いて、上記7-2と同様の試験を行なった。成膜厚さについての結果を[表37]と[表38]に、Ca溶出量についての結果を[表39]と[表40]に、ビッカース硬度についての結果を[表41]と[表42]に示す。


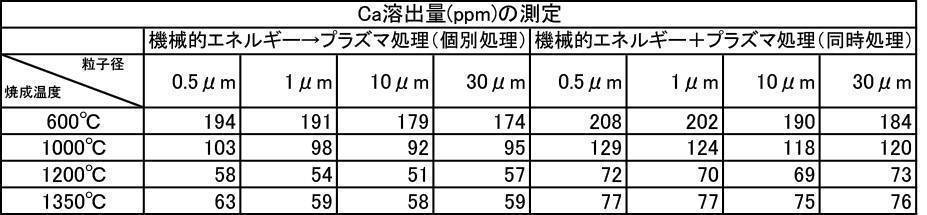

アルゴン雰囲気で焼成して、対向式気流粉砕機で粉砕、分級した膜形成用ハイドロキシアパタイト粉体と、窒素雰囲気で焼成して、対向式気流粉砕機で粉砕、分級したハイドロキシアパタイト粉体を1:1で混合した粉体を用いて、上記7-2と同様の試験を行なった。成膜厚さについての結果を[表43]と[表44]に、Ca溶出量についての結果を[表45]と[表46]に、ビッカース硬度についての結果を[表47]と[表48]に示す。



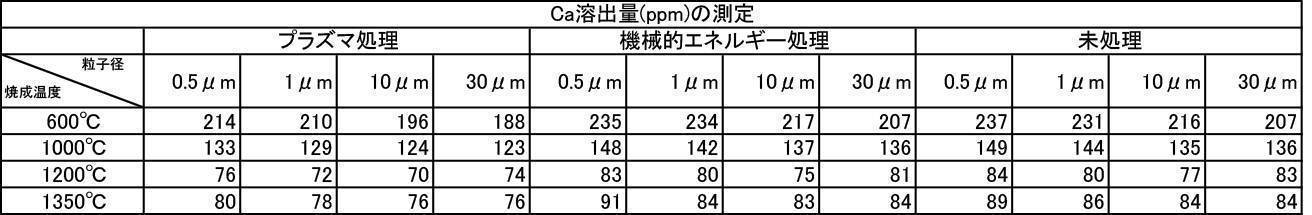

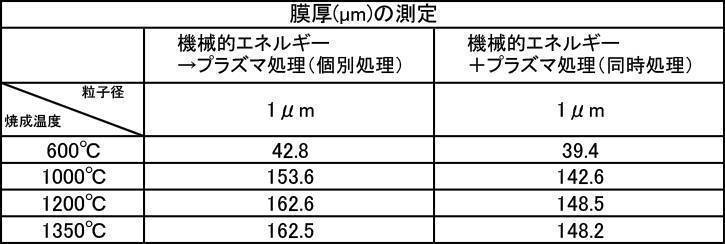
上記実施例2-2に示されるように、平均粒子径1μmのハイドロキシアパタイト粉体に、シリカ粉体を1%添加した粉体を、アルゴン雰囲気で焼成して、対向式気流粉砕機で粉砕、分級したシリカを添加した粒子径1μmの膜形成用粉体を用いて、上記7-2と同様の試験を行なった。成膜厚さについての結果を[表49]と[表50]に、Ca溶出量についての結果を[表51]と[表52]に、ビッカース硬度についての結果を[表53]と[表54]に示す。




アルゴン雰囲気で焼成して、対向式気流粉砕機で粉砕、分級した平均粒子径が10μmの膜形成用ハイドロキシアパタイト粉体と、アルゴン雰囲気で焼成して、対向式気流粉砕機で粉砕、分級した平均粒子径が1μmの膜形成用ハイドロキシアパタイト粉体を1:1で混合した粉体を用いて、上記7-2と同様の試験を行なった。成膜厚さについての結果を[表55]と[表56]、及びその比較を[表57]に、Ca溶出量についての結果を[表58]と[表59]、及びその比較を[表60]に、ビッカース硬度についての結果を[表61]と[表62]、及びその比較を[表63]にそれぞれ示す。





前記実施例2-3に示されるように、アルゴン雰囲気で焼成して、対向式気流粉砕機で粉砕、分級した膜形成用粉体に、色調調整剤を配合した粉体に、1)機械的エネルギーを加えた後にプラズマ照射を行う処理(機械的エネルギー→プラズマ処理;個別処理)、3)機械的エネルギーとプラズマ照射を同時に加える処理(機械的エネルギー=プラズマ処理;同時処理)、4)プラズマ照射を行う処理(プラズマ処理)、5)機械的エネルギーを加える処理(機械的エネルギー処理)を各々行った粉体、及び6)焼成、粉砕、分級、混合するだけで、プラズマ照射及び/又は機械的エネルギーを加える処理を行わない粉体(未処理)でそれぞれ成膜処理を行い、各試料について、実施例7と同様に、成膜厚さ、Ca溶出量、ビッカース硬度の測定を行った。成膜厚さ、Ca溶出量、ビッカース硬度の測定方法は、実施例5と同様の方法により行った。
アルゴン雰囲気で焼成して、対向式気流粉砕機で粉砕、分級した膜形成用粉体に、色調調整剤として酸化チタンを1質量%配合した粉体に上記1)及び3)~6)の処理を施した粉体用いて成膜した。成膜厚さについての結果を[表64]と[表65]に、Ca溶出量についての結果を[表66]と[表67]に、ビッカース硬度についての結果を[表68]と[表69]に示す。



アルゴン雰囲気で焼成して、対向式気流粉砕機で粉砕、分級した膜形成用粉体に、色調調整剤として酸化亜鉛を5質量%配合した粉体に上記1)及び3)~6)の処理を施した粉体用いて成膜した。成膜厚さについての結果を[表70]と[表71]に、Ca溶出量についての結果を[表72]と[表73]に、ビッカース硬度についての結果を[表74]と[表75]に示す。
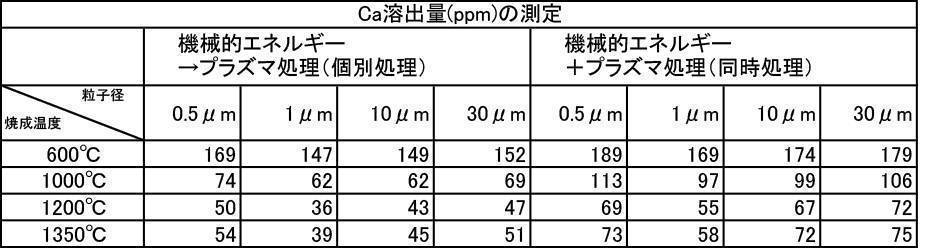


アルゴン雰囲気で焼成して、対向式気流粉砕機で粉砕、分級した膜形成用粉体に、色調調整剤として赤色204号を0.1質量%配合した粉体に上記1)及び3)~6)の処理を施した粉体用いて成膜した。成膜厚さについての結果を[表76]と[表77]に、Ca溶出量についての結果を[表78]と[表79]に、ビッカース硬度についての結果を[表80]と[表81]に示す。


各処理における膜形成用ハイドロキシアパタイト粉体の粉体性状の違いを検討するために、粉末X線回折試験、レーザーラマン分光分析試験により、膜形成用粉体の特性を調査した。
実施例2-1で製造した(焼成、粉砕、分級、混合するだけで、プラズマ照射及び/又は機械的エネルギーを加える処理を行わない)平均粒径1μmのハイドロキシアパタイト粉体(HAP);実施例7-2で製造した、このハイドロキシアパタイト粉体に機械的エネルギーを加えた後にプラズマ照射を行う処理を行った平均粒径1μmの膜形成用粉体、及び実施例実施例8-1で製造した、酸化チタンを1質量%配合した膜形成用ハイドロキシアパタイト粉体に機械的エネルギーを加えた後にプラズマ照射を行う処理を行った平均粒径1μmの膜形成用粉体;の3つの試料について、粉末X線回折装置(Empyrean、PANalytical製)を用いて、ターゲット:Cu、管電圧:45kV、管電流:40mA、走査範囲:2θ=5~60°の条件で粉末X線回折試験を行なった。結果を図4に示す。その結果、全ての回折パターンは同様であり、これら試料における粉体の違いを確認することはできなかった。
粉末X線回折試験では、粉体粒子の近表面における結晶性の変化を確認することができないことから、ラマン分光分析法による検討を行なった。実施例7-2で製造した、平均粒径1μmの1)機械的エネルギーを加えた後にプラズマ照射を行う処理(機械的エネルギー→プラズマ処理;個別処理)、3)機械的エネルギーとプラズマ照射を同時に加える処理(機械的エネルギー=プラズマ処理;同時処理)、4)プラズマ照射を行う処理(プラズマ処理)、5)機械的エネルギーを加える処理(機械的エネルギー処理)を各々行った粉体、及び6)焼成、粉砕、分級、混合するだけで、プラズマ照射及び/又は機械的エネルギーを加える処理を行わない粉体(未処理)の5つの試料について、レーザーラマン分光分析装置(Invia Reflex、RENISHAW製)を用いてその特性を調査した。

歯冠色調調整剤料の膜(白色;酸化チタン1%配合)をガラス板上に成膜し、この第1層上に、第2層として歯冠色調調整剤料の膜(歯の色に近い色;酸化亜鉛5%配合)を成膜し、この第2層上に、第3層として歯冠色調調整剤料の膜(透明色(トップコートとして);ハイドロキシアパタイトのみ)を成膜した。成膜条件は、第1層、第2層、第3層は全て同じであり、ハンドピース先端ノズル内径:1.8mm、噴射圧:0.5MPaとした。噴射ノズル先端-基板間距離は1.0mm(ノズル先端は基板に垂直に保持)、噴射ノズル移動速度は5mm/sで行なった。結果(写真)を図7に示す。また、図7に示す多層の成膜層のレーザー顕微鏡による断面像を図8に示す。
2 プラズマ発生装置の冷陰極管インバータ
3 プラズマ発生装置の昇圧回路(コッククロフト・ウォルトン回路)
4 プラズマ発生装置のプラズマノズル
5 プラズマ発生装置のガス流量計
Claims (21)
- 歯に対して噴射する装置に使用して歯表面に膜を形成するための膜形成用粉体であって、Ca10-X・MX(ZO4)6Y2(ただし、Xは0~10、Mは金属又は水素、ZO4はPO4、CO3、CrO4、AsO4、VO4、SiO4、SO4又はGeO4、Yは水酸基、ハロゲン元素又は炭酸基)で示されるアパタイトを、不活性ガス雰囲気下600~1350℃で焼成することにより製造したことを特徴とする平均粒子径が0.5~30μmである膜形成用粉体。
- アパタイトが、ハイドロキシアパタイトであることを特徴とする請求項1記載の膜形成用粉体。
- 不活性ガスがアルゴンガス又は窒素ガスであることを特徴とする請求項1又は2記載の膜形成用粉体。
- さらに、歯冠の色調を調整するための色調調整剤を配合したことを特徴とする請求項1~3のいずれか記載の膜形成用粉体。
- 歯冠の色調調整剤が、酸化チタン、酸化亜鉛、群青、及び赤色顔料から選ばれる少なくとも1種であることを特徴とする請求項4記載の膜形成用粉体。
- プラズマ照射を行うことにより製造したことを特徴とする請求項1~5のいずれか記載の膜形成用粉体。
- さらに、機械的なエネルギーを加えることにより製造したことを特徴とする請求項6記載の膜形成用粉体。
- 機械的なエネルギーを加えた後で、プラズマ照射を行うことにより製造したことを特徴とする請求項7記載の膜形成用粉体。
- ヘリウムガスを照射ガスとしたプラズマ照射であることを特徴とする請求項6~8のいずれか記載の膜形成用粉体。
- ハンドピース先端ノズル内径:0.5~5.0mm、噴射圧:0.2~0.8MPa、噴射ノズル先端-基板間距離0.1~3.0cm、噴射ノズル移動速度0~10mm/sの条件で、粉体を基板に噴射した場合に、形成された膜の膜厚が30μm以上で、ビッカース硬度が340Hv以上であることを特徴とする請求項1~9のいずれか記載の膜形成用粉体。
- Ca10-X・MX(ZO4)6Y2(ただし、Xは0~10、Mは金属又は水素、ZO4はPO4、CO3、CrO4、AsO4、VO4、SiO4、SO4又はGeO4、Yは水酸基、ハロゲン元素又は炭酸基)で示されるアパタイトを不活性ガス雰囲気下600~1350℃で焼成した後、粉砕及び分級することを特徴とする、歯に対して噴射する装置に使用して歯表面に膜を形成するための、平均粒子径が0.5~30μmである膜形成用粉体の製造方法。
- アパタイトが、ハイドロキシアパタイトであることを特徴とする請求項11記載の膜形成用粉体の製造方法。
- 不活性ガスがアルゴンガス又は窒素ガスであることを特徴とする請求項11又は12記載の膜形成用粉体の製造方法。
- さらに、歯冠の色調を調整するための色調調整剤を配合したことを特徴とする請求項11~13のいずれか記載の膜形成用粉体の製造方法。
- 歯冠の色調調整剤が、酸化チタン、酸化亜鉛、群青、及び赤色顔料から選ばれる少なくとも1種であることを特徴とする請求項14記載の膜形成用粉体の製造方法。
- 粉砕及び分級後に、プラズマ照射を行うことを特徴とする請求項11~15のいずれか記載の膜形成用粉体の製造方法。
- さらに、機械的なエネルギーを加えることを特徴とする請求項16記載の膜形成用粉体の製造方法。
- 機械的なエネルギーを加えた後で、プラズマ照射を行うことを特徴とする請求項17記載の膜形成用粉体の製造方法。
- ヘリウムガスを照射ガスとしたプラズマ照射であることを特徴とする請求項16~18のいずれか記載の膜形成用粉体の製造方法。
- ハンドピース先端ノズル内径:0.5~5.0mm、噴射圧:0.2~0.8MPa、噴射ノズル先端-基板間距離0.1~3.0cm、噴射ノズル移動速度0~10mm/sの条件で、粉体を基板に噴射した場合に、形成された膜の膜厚が30μm以上で、ビッカース硬度が340Hv以上であることを特徴とする請求項11~19のいずれか記載の膜形成用粉体の製造方法。
- 請求項1~10のいずれか記載の膜形成用粉体を含むペレット。
Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016568065A JP6163641B2 (ja) | 2015-07-13 | 2016-07-12 | 焼成アパタイトを含む歯表面膜形成用粉体 |
| KR1020177036394A KR102262274B1 (ko) | 2015-07-13 | 2016-07-12 | 소성 아파타이트를 포함하는 치아 표면막 형성용 분체 |
| CN201680036565.2A CN107735070B (zh) | 2015-07-13 | 2016-07-12 | 包含经烧成的磷灰石的牙齿表面膜形成用粉体 |
| EP16824074.5A EP3323406B1 (en) | 2015-07-13 | 2016-07-12 | Tooth-surface-membrane-forming powder containing sintered apatite |
| CA2994374A CA2994374C (en) | 2015-07-13 | 2016-07-12 | Powder containing apatite for forming a film on tooth surface |
| RU2018104562A RU2688152C1 (ru) | 2015-07-13 | 2016-07-12 | Порошок для формирования мембраны на поверхности зуба, содержащий спеченный апатит |
| ES16824074T ES2912114T3 (es) | 2015-07-13 | 2016-07-12 | Polvo formador de membranas dentales que contiene apatita sinterizada |
| SG11201710905WA SG11201710905WA (en) | 2015-07-13 | 2016-07-12 | Tooth-surface-membrane-forming powder containing sintered apatite |
| KR1020207014673A KR20200060535A (ko) | 2015-07-13 | 2016-07-12 | 소성 아파타이트를 포함하는 치아 표면막 형성용 분체 |
| US15/742,747 US11007125B2 (en) | 2015-07-13 | 2016-07-12 | Tooth-surface-membrane-forming powder containing sintered apatite |
| AU2016293655A AU2016293655B2 (en) | 2015-07-13 | 2016-07-12 | Tooth-surface-membrane-forming powder containing sintered apatite |
| HK18105833.0A HK1246198A1 (zh) | 2015-07-13 | 2018-05-07 | 包含經燒成的磷灰石的牙齒表面膜形成用粉體 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015139692 | 2015-07-13 | ||
| JP2015-139692 | 2015-07-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017010089A1 true WO2017010089A1 (ja) | 2017-01-19 |
Family
ID=57757244
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/003298 WO2017010090A1 (ja) | 2015-07-13 | 2016-07-12 | 焼成アパタイトを含む歯表面膜形成用粉体 |
| PCT/JP2016/003297 WO2017010089A1 (ja) | 2015-07-13 | 2016-07-12 | 焼成アパタイトを含む歯表面膜形成用粉体 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/003298 WO2017010090A1 (ja) | 2015-07-13 | 2016-07-12 | 焼成アパタイトを含む歯表面膜形成用粉体 |
Country Status (13)
| Country | Link |
|---|---|
| US (3) | US20180200154A1 (ja) |
| EP (2) | EP3323406B1 (ja) |
| JP (2) | JP6770517B2 (ja) |
| KR (4) | KR102264866B1 (ja) |
| CN (2) | CN107735070B (ja) |
| AU (2) | AU2016293655B2 (ja) |
| CA (2) | CA2994374C (ja) |
| ES (1) | ES2912114T3 (ja) |
| HK (2) | HK1246198A1 (ja) |
| RU (2) | RU2688152C1 (ja) |
| SG (2) | SG11201710905WA (ja) |
| TW (2) | TWI698408B (ja) |
| WO (2) | WO2017010090A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2022015639A (ja) * | 2020-07-09 | 2022-01-21 | 日東紡績株式会社 | ハイドロキシアパタイト粒子分散液及びハイドロキシアパタイト付着基材の製造方法 |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102264866B1 (ko) | 2015-07-13 | 2021-06-14 | 가부시키가이샤 상기 | 소성 아파타이트를 포함하는 치아 표면막 형성용 분체 |
| JP6778295B2 (ja) * | 2019-04-09 | 2020-10-28 | 学校法人近畿大学 | ヒドロキシアパタイトをチタン系金属基材に固定化する方法及びヒドロキシアパタイト被覆金属材 |
| CN110200709B (zh) * | 2019-07-15 | 2021-12-14 | 杨桐 | 一种免于备牙的骨架瓷牙贴面及其制备方法 |
| CN110694093A (zh) * | 2019-11-18 | 2020-01-17 | 袭晓冰 | 一种感染科病房消毒液喷洒装置 |
| CN112592171A (zh) * | 2020-12-16 | 2021-04-02 | 昆明理工大学 | 一种氧化镁/羟基磷灰石多孔复合材料的制备方法 |
| WO2023194316A1 (en) * | 2022-04-05 | 2023-10-12 | Ferton Holding S.A. | Powder for use in treating tooth surfaces |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63282171A (ja) * | 1987-05-12 | 1988-11-18 | Asahi Optical Co Ltd | リン酸カルシウム系材料の製造方法 |
| JPH021285A (ja) * | 1988-01-11 | 1990-01-05 | Asahi Optical Co Ltd | 固着可能な歯科用及び医科用顆粒状骨補填材、その固着方法及び骨補填物 |
Family Cites Families (55)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3340265A (en) | 1964-04-23 | 1967-09-05 | Millmaster Onyx Corp | Quaternary ammonium salts of acetylenenic carboxylic acids |
| JPS5031398B2 (ja) | 1972-06-13 | 1975-10-09 | ||
| JPS5611759B2 (ja) | 1973-11-20 | 1981-03-17 | ||
| US3971877A (en) | 1975-09-29 | 1976-07-27 | Lawrence Y. Lee | Removable filters on electronic chassis and method of assembling same |
| WO1981002670A1 (en) | 1980-03-17 | 1981-10-01 | Pennwalt Corp | Dental prophylaxis compositions and their use |
| JPS6399867A (ja) | 1986-10-17 | 1988-05-02 | ペルメレツク電極株式会社 | リン酸カルシウム化合物被覆複合材及びその製造方法 |
| JPS6486975A (en) | 1987-09-29 | 1989-03-31 | Permelec Electrode Ltd | Preparation of calcium phosphate compound coated composite material |
| JPH0339165A (ja) | 1989-07-05 | 1991-02-20 | Nippon Seisen Co Ltd | 人体内埋込材の焼結方法 |
| WO1992009715A1 (en) * | 1990-11-21 | 1992-06-11 | Leningradskaya Assotsiatsia 'poliplazma' | Method for plasma jet spraying of biologically active coatings on an implant |
| EP0639366A1 (en) * | 1993-08-19 | 1995-02-22 | Kingstar Technology Limited (Uk) | Hydroxyapatite cement as bone or tooth replacement |
| JP3340265B2 (ja) | 1994-11-21 | 2002-11-05 | 一枝 山岸 | 歯の漂白剤 |
| JP4040705B2 (ja) | 1996-01-24 | 2008-01-30 | 株式会社サンギ | 口腔組成物 |
| US5702677A (en) * | 1996-07-10 | 1997-12-30 | Osteotech, Inc. | Spherical hydroxyapatite particles and process for the production thereof |
| JP3971877B2 (ja) | 1999-10-25 | 2007-09-05 | 株式会社サンギ | 口腔用組成物 |
| JP2001178813A (ja) | 1999-12-22 | 2001-07-03 | Miimu:Kk | 生体親和性薄膜を持った医療材料 |
| JP4672112B2 (ja) | 2000-06-13 | 2011-04-20 | 株式会社ジーシー | グラスアイオノマーセメント用ガラス粉末 |
| JP4279077B2 (ja) | 2003-07-04 | 2009-06-17 | Hoya株式会社 | 焼結体の製造方法および焼結体 |
| GB2422843B (en) | 2002-08-12 | 2007-03-14 | Pentax Corp | Cell culture base |
| JP4334303B2 (ja) | 2003-09-03 | 2009-09-30 | 山八歯材工業株式会社 | スパッタリング装置用被覆体回転装置 |
| KR100591762B1 (ko) | 2004-01-19 | 2006-06-22 | 삼성전자주식회사 | 증착 장치 및 증착 방법 |
| KR100583849B1 (ko) * | 2004-01-20 | 2006-05-26 | 재단법인서울대학교산학협력재단 | 인산칼슘 화합물의 폴리메릭 졸 제조방법 |
| JP3962061B2 (ja) | 2005-01-24 | 2007-08-22 | 常元 厨川 | 目標物に対する膜の形成方法及び装置 |
| CN1282629C (zh) * | 2005-02-22 | 2006-11-01 | 清华大学 | 可加工羟基磷灰石/钛硅碳生物陶瓷复合材料的制备方法 |
| US9072672B2 (en) * | 2005-06-28 | 2015-07-07 | Colgate-Palmolive Company | Compositions and methods for altering the color of teeth |
| KR100672112B1 (ko) | 2005-10-11 | 2007-01-19 | 주식회사 히타치엘지 데이터 스토리지 코리아 | 광디스크장치의 오피씨 수행방법 |
| GB2436067A (en) * | 2006-03-17 | 2007-09-19 | Apatech Ltd | A flowable biomedical filler resisiting flow at higher shear stress or compressive force |
| JP5031398B2 (ja) | 2007-02-21 | 2012-09-19 | 独立行政法人科学技術振興機構 | 粉体噴射装置及び同粉体噴射装置を用いた加工方法 |
| CN101254314A (zh) * | 2007-03-02 | 2008-09-03 | 北京奥精医药科技有限公司 | 羟基磷灰石涂层镁合金医用内植入材料及其制备方法 |
| US20080317807A1 (en) * | 2007-06-22 | 2008-12-25 | The University Of Hong Kong | Strontium fortified calcium nano-and microparticle compositions and methods of making and using thereof |
| JP5248848B2 (ja) * | 2007-12-11 | 2013-07-31 | 山八歯材工業株式会社 | インプラントの製造方法及び人工歯根の製造方法 |
| GB0724896D0 (en) * | 2007-12-20 | 2008-01-30 | Imp Innovations Ltd | Bioactive Glass coatings |
| CN101658693B (zh) * | 2008-08-25 | 2013-03-13 | 中国科学院上海硅酸盐研究所 | 一种硅酸镁涂层-钛或钛合金硬组织替代材料及制备方法 |
| CN101428844B (zh) * | 2008-12-04 | 2010-11-17 | 东华大学 | 纳米氧化锌表面大气压、常温等离子体改性处理方法 |
| US8481678B2 (en) * | 2009-03-30 | 2013-07-09 | E I Du Pont De Nemours And Company | Peptide-based tooth whitening reagents |
| US10299887B2 (en) | 2009-04-23 | 2019-05-28 | Nanova, Inc. | Atmospheric non-thermal gas plasma method for dental surface treatment |
| CN102181842A (zh) * | 2011-04-14 | 2011-09-14 | 中国科学院上海硅酸盐研究所 | 一种对钛金属表面进行改性的方法 |
| JP6002942B2 (ja) | 2011-05-11 | 2016-10-05 | 株式会社ブイ・テクノロジー | レンズおよびそのレンズを搭載したレーザ加工装置 |
| KR20120137465A (ko) | 2011-06-12 | 2012-12-21 | 김영균 | 치과용 수복재 및 그 제조방법 |
| CN102240234B (zh) * | 2011-06-29 | 2013-07-10 | 西安交通大学 | 一种可调式人工颈椎及椎间连接复合体 |
| KR101654381B1 (ko) * | 2011-07-15 | 2016-09-05 | 단국대학교 산학협력단 | 생체 세라믹을 내포하는 의료용 고분자 및 이의 제조방법 |
| KR101305382B1 (ko) | 2011-10-12 | 2013-09-06 | 한국과학기술연구원 | 생체용 재료의 표면개질 장치 및 표면개질 방법 |
| JP2013215240A (ja) * | 2012-04-04 | 2013-10-24 | Osaka Univ | バイオセラミックスを含む人工骨の改質方法と、その方法で改質された人工骨 |
| JP5891150B2 (ja) | 2012-09-07 | 2016-03-22 | 株式会社アドバンス | 歯科用インプラントの製造方法 |
| CN102921042A (zh) * | 2012-11-26 | 2013-02-13 | 中国科学院上海硅酸盐研究所 | 一种硬组织替代材料及其制备方法 |
| CN103110979B (zh) * | 2013-02-09 | 2015-06-17 | 复旦大学 | 表面沉积类骨磷灰石的高分子多孔材料及其制备方法和应用 |
| KR101405859B1 (ko) | 2013-04-24 | 2014-06-12 | 오스템임플란트 주식회사 | pH 완충 물질과 술폰기를 갖는 유기 양친성 물질의 혼합 용액으로 코팅된 치과용 임플란트 및 그 제조방법 |
| JP6417596B2 (ja) | 2013-06-05 | 2018-11-07 | 株式会社サンギ | 粉体噴射用ハンドピース |
| DE102013109758B4 (de) | 2013-09-06 | 2017-07-13 | Ferton Holding S.A. | Pulvergemisch, Verwendung des Pulvergemischs, Pulverstrahlgerät und Verfahren zur Remineralisierung von Zähnen |
| JP6284222B2 (ja) | 2013-11-28 | 2018-02-28 | 株式会社サンギ | 粉体流通装置 |
| CN103668940B (zh) * | 2013-12-18 | 2016-08-17 | 华东理工大学 | 一种表面改性纤维增强复合骨水泥及其制备方法和应用 |
| JP5827357B2 (ja) * | 2014-03-04 | 2015-12-02 | 株式会社ビー・アイ・テック | リン酸カルシウム含有複合層をもつ樹脂複合体及びその製造方法 |
| KR20150104429A (ko) | 2014-03-05 | 2015-09-15 | 엘지전자 주식회사 | 태양 전지 및 이의 제조 방법 |
| KR20150013095A (ko) | 2014-09-29 | 2015-02-04 | 주식회사 제이투엔 | 코드 기반의 장치 액세스 방법 및 코드 기반의 장치 액세스를 위한 사용자 장치 |
| CN104587527B (zh) * | 2015-01-16 | 2017-03-08 | 中国石油大学(华东) | 一种生物功能化的碳/碳复合材料及其制备方法 |
| KR102264866B1 (ko) * | 2015-07-13 | 2021-06-14 | 가부시키가이샤 상기 | 소성 아파타이트를 포함하는 치아 표면막 형성용 분체 |
-
2016
- 2016-07-12 KR KR1020177036388A patent/KR102264866B1/ko active IP Right Grant
- 2016-07-12 ES ES16824074T patent/ES2912114T3/es active Active
- 2016-07-12 KR KR1020177036394A patent/KR102262274B1/ko active IP Right Grant
- 2016-07-12 WO PCT/JP2016/003298 patent/WO2017010090A1/ja active Application Filing
- 2016-07-12 JP JP2017528290A patent/JP6770517B2/ja active Active
- 2016-07-12 CA CA2994374A patent/CA2994374C/en active Active
- 2016-07-12 CA CA2994385A patent/CA2994385C/en active Active
- 2016-07-12 US US15/742,763 patent/US20180200154A1/en not_active Abandoned
- 2016-07-12 CN CN201680036565.2A patent/CN107735070B/zh active Active
- 2016-07-12 EP EP16824074.5A patent/EP3323406B1/en active Active
- 2016-07-12 KR KR1020207014673A patent/KR20200060535A/ko active Application Filing
- 2016-07-12 SG SG11201710905WA patent/SG11201710905WA/en unknown
- 2016-07-12 KR KR1020207014672A patent/KR20200062357A/ko active Application Filing
- 2016-07-12 SG SG11201710906QA patent/SG11201710906QA/en unknown
- 2016-07-12 AU AU2016293655A patent/AU2016293655B2/en active Active
- 2016-07-12 RU RU2018104562A patent/RU2688152C1/ru active
- 2016-07-12 WO PCT/JP2016/003297 patent/WO2017010089A1/ja active Application Filing
- 2016-07-12 EP EP16824075.2A patent/EP3323407A4/en not_active Withdrawn
- 2016-07-12 US US15/742,747 patent/US11007125B2/en active Active
- 2016-07-12 RU RU2018104564A patent/RU2689789C1/ru active
- 2016-07-12 JP JP2016568065A patent/JP6163641B2/ja active Active
- 2016-07-12 CN CN201680036638.8A patent/CN107708650A/zh active Pending
- 2016-07-12 AU AU2016293656A patent/AU2016293656B2/en active Active
- 2016-07-13 TW TW105122048A patent/TWI698408B/zh active
- 2016-07-13 TW TW105122046A patent/TWI698256B/zh active
-
2018
- 2018-05-07 HK HK18105833.0A patent/HK1246198A1/zh unknown
- 2018-05-07 HK HK18105832.1A patent/HK1246197A1/zh unknown
-
2019
- 2019-11-25 US US16/693,844 patent/US20200179238A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63282171A (ja) * | 1987-05-12 | 1988-11-18 | Asahi Optical Co Ltd | リン酸カルシウム系材料の製造方法 |
| JPH021285A (ja) * | 1988-01-11 | 1990-01-05 | Asahi Optical Co Ltd | 固着可能な歯科用及び医科用顆粒状骨補填材、その固着方法及び骨補填物 |
Non-Patent Citations (5)
| Title |
|---|
| AKATSUKA, RYO ET AL.: "Characteristics of hydroxyapatite film formed on human enamel with the powder jet deposition technique", J BIOMED MATER RES B APPL BIOMATER, vol. 98 B, no. 2, 2011, pages 210 - 216, XP055344827, ISSN: 1552-4981 * |
| AKATSUKA, RYO ET AL.: "Effect of hydroxyapatite film formed by powder jet deposition on dentin permeability", EUR J ORAL SCI, vol. 120, no. 6, 2012, pages 558 - 562, XP055344826, ISSN: 1600-0722 * |
| KEI SATO ET AL.: "Creation of Hydroxyapatite Film on Human Enamel Utilized Powder Jet Deposition", TRANSACTIONS OF THE JAPAN SOCIETY OF MECHANICAL ENGINEERS (SERIES C, vol. 79, no. 808, 25 December 2013 (2013-12-25), pages 4634 - 4642, XP055344989, ISSN: 1884-8354, DOI: doi:10.1299/kikaic.79.4634 * |
| NOJI, MIYOKO ET AL.: "Characteristics of the hydroxyapatite film deposited on human enamel: deposition of a ceramic film by powder jet deposition technique", INT J ABRASIVE TECHNOLOGY, vol. 2, no. 1, 2009, pages 83 - 96, XP009508338, ISSN: 1752-265X * |
| See also references of EP3323406A4 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2022015639A (ja) * | 2020-07-09 | 2022-01-21 | 日東紡績株式会社 | ハイドロキシアパタイト粒子分散液及びハイドロキシアパタイト付着基材の製造方法 |
| JP7294260B2 (ja) | 2020-07-09 | 2023-06-20 | 日東紡績株式会社 | ハイドロキシアパタイト粒子分散液及びハイドロキシアパタイト付着基材の製造方法 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6163641B2 (ja) | 焼成アパタイトを含む歯表面膜形成用粉体 | |
| AU2008248087B2 (en) | Hybrid organic/inorganic chemical hybrid systems, including functionalized calcium phosphate hybrid systems, and a solid-state method of producing the same | |
| JP6564457B2 (ja) | フッ化物でドーピングされておりクエン酸塩で被覆されている非晶質リン酸カルシウムナノ粒子を得るための製法 | |
| Ren et al. | Synergistic remineralization of enamel white spot lesions using mesoporous bioactive glasses loaded with amorphous calcium phosphate | |
| KR20130049941A (ko) | 과민감성 치아의 완화를 위한 치약 조성물 | |
| WO2020138422A1 (ja) | ヒドロキシアパタイト微粒子 | |
| Sun et al. | A new approach to prepare well‐dispersed CaF2 nanoparticles by spray drying technique | |
| Dobrota et al. | Dynamics of Dental Enamel Surface Remineralization under the Action of Toothpastes with Substituted Hydroxyapatite and Birch Extract | |
| Bashir et al. | Stabilization of Zirconia Ceramics for Dental Coatings-Effect of Aging Conditions |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2016568065 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16824074 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20177036394 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15742747 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2994374 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2016293655 Country of ref document: AU Date of ref document: 20160712 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2018104562 Country of ref document: RU Ref document number: 2016824074 Country of ref document: EP |