WO2015098099A1 - 抗trop2抗体-薬物コンジュゲート - Google Patents
抗trop2抗体-薬物コンジュゲート Download PDFInfo
- Publication number
- WO2015098099A1 WO2015098099A1 PCT/JP2014/006421 JP2014006421W WO2015098099A1 WO 2015098099 A1 WO2015098099 A1 WO 2015098099A1 JP 2014006421 W JP2014006421 W JP 2014006421W WO 2015098099 A1 WO2015098099 A1 WO 2015098099A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- antibody
- amino acid
- ggfg
- drug
- cancer
- Prior art date
Links
- 0 CC(C1)C(C)C11*CCCC1 Chemical compound CC(C1)C(C)C11*CCCC1 0.000 description 2
- YLGVBUAQTQSHCD-DEOSSOPVSA-N CC[C@](C(C=C1N2Cc3c(CCCc4c(C)c(F)c5)c4c5nc13)=C(CO1)C2=O)(C1=O)O Chemical compound CC[C@](C(C=C1N2Cc3c(CCCc4c(C)c(F)c5)c4c5nc13)=C(CO1)C2=O)(C1=O)O YLGVBUAQTQSHCD-DEOSSOPVSA-N 0.000 description 2
- SEEYREPSKCQBBF-UHFFFAOYSA-N CN(C(C=C1)=O)C1=O Chemical compound CN(C(C=C1)=O)C1=O SEEYREPSKCQBBF-UHFFFAOYSA-N 0.000 description 2
- UYOSDILTAHHRLF-IYPRWPFWSA-O C=NC(C[U]CNC(CNC([C@H](Cc1ccccc1)NC(CNC(CN)[OH2+])=O)=O)=O)=O Chemical compound C=NC(C[U]CNC(CNC([C@H](Cc1ccccc1)NC(CNC(CN)[OH2+])=O)=O)=O)=O UYOSDILTAHHRLF-IYPRWPFWSA-O 0.000 description 1
- NIXKACOKLPRDMY-UHFFFAOYSA-N CC(CC(N1C)=O)C1=O Chemical compound CC(CC(N1C)=O)C1=O NIXKACOKLPRDMY-UHFFFAOYSA-N 0.000 description 1
- MRMAUBGOAHLOSR-UHFFFAOYSA-N CC(CNC(OCC1c2ccccc2-c2c1cccc2)=[U])NCOCC(O)=O Chemical compound CC(CNC(OCC1c2ccccc2-c2c1cccc2)=[U])NCOCC(O)=O MRMAUBGOAHLOSR-UHFFFAOYSA-N 0.000 description 1
- NKDSPEFADHLMHN-UHFFFAOYSA-N CC(OCNC(CNC(OCC1c2ccccc2-c2c1cccc2)=O)=O)=O Chemical compound CC(OCNC(CNC(OCC1c2ccccc2-c2c1cccc2)=O)=O)=O NKDSPEFADHLMHN-UHFFFAOYSA-N 0.000 description 1
- APLGBTWKEBDOKC-UHFFFAOYSA-N CCC(C)(CC)C(CC(N1OC)=O)C1=O Chemical compound CCC(C)(CC)C(CC(N1OC)=O)C1=O APLGBTWKEBDOKC-UHFFFAOYSA-N 0.000 description 1
- IQOYLCABBAEGEO-XEDFXBRGSA-N CC[C@](C(C=C1N2Cc3c([C@H](CCc4c(C)c(F)c5)NC(COCNC(CNC(OCC6c7ccccc7-c7c6cccc7)=O)=O)=O)c4c5nc13)=C(CO1)C2=O)(C1=O)O Chemical compound CC[C@](C(C=C1N2Cc3c([C@H](CCc4c(C)c(F)c5)NC(COCNC(CNC(OCC6c7ccccc7-c7c6cccc7)=O)=O)=O)c4c5nc13)=C(CO1)C2=O)(C1=O)O IQOYLCABBAEGEO-XEDFXBRGSA-N 0.000 description 1
- WXNSCLIZKHLNSG-MCZRLCSDSA-N CC[C@](C(C=C1N2Cc3c([C@H](CCc4c(C)c(F)c5)NC(COCNC(CNC([C@H](Cc6ccccc6)NC(CNC(CNC(CCCCCN(C(C=C6)=O)C6=O)=O)=O)=O)=O)=O)=O)c4c5nc13)=C(CO1)C2=O)(C1=O)O Chemical compound CC[C@](C(C=C1N2Cc3c([C@H](CCc4c(C)c(F)c5)NC(COCNC(CNC([C@H](Cc6ccccc6)NC(CNC(CNC(CCCCCN(C(C=C6)=O)C6=O)=O)=O)=O)=O)=O)=O)c4c5nc13)=C(CO1)C2=O)(C1=O)O WXNSCLIZKHLNSG-MCZRLCSDSA-N 0.000 description 1
- ODCYLJDZBUQXIS-GKVSMKOHSA-N CC[C@](C(C=C1N2Cc3c([C@H](CCc4c(C)c(F)c5)NC)c4c5nc13)=C(CO1)C2=O)(C1=O)O Chemical compound CC[C@](C(C=C1N2Cc3c([C@H](CCc4c(C)c(F)c5)NC)c4c5nc13)=C(CO1)C2=O)(C1=O)O ODCYLJDZBUQXIS-GKVSMKOHSA-N 0.000 description 1
- KZNICNPSHKQLFF-UHFFFAOYSA-N O=C(CC1)NC1=O Chemical compound O=C(CC1)NC1=O KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 1
- MZOSSDIBSRKPCO-UHFFFAOYSA-N O=C(CNC(OCC1c2ccccc2-c2ccccc12)=O)NCOCC(OCc1ccccc1)=O Chemical compound O=C(CNC(OCC1c2ccccc2-c2ccccc12)=O)NCOCC(OCc1ccccc1)=O MZOSSDIBSRKPCO-UHFFFAOYSA-N 0.000 description 1
- FBKUOPULLUJMOC-UHFFFAOYSA-N OC(CNC(CNC(OCC1c2ccccc2-c2ccccc12)=O)=O)=O Chemical compound OC(CNC(CNC(OCC1c2ccccc2-c2ccccc12)=O)=O)=O FBKUOPULLUJMOC-UHFFFAOYSA-N 0.000 description 1
- VPYJBEIOKFRWQZ-UHFFFAOYSA-N OCC(OCc1ccccc1)=O Chemical compound OCC(OCc1ccccc1)=O VPYJBEIOKFRWQZ-UHFFFAOYSA-N 0.000 description 1
- WOJKKJKETHYEAC-UHFFFAOYSA-O [OH2+]C(CCCCCN(C(C=C1)=O)C1=O)=O Chemical compound [OH2+]C(CCCCCN(C(C=C1)=O)C1=O)=O WOJKKJKETHYEAC-UHFFFAOYSA-O 0.000 description 1
Images





















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6857—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from lung cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6859—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from liver or pancreas cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6863—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from stomach or intestines cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6865—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from skin, nerves or brain cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6869—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from a cell of the reproductive system: ovaria, uterus, testes, prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/22—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains four or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
- C07K16/3023—Lung
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
- C07K16/303—Liver or Pancreas
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
- C07K16/3046—Stomach, Intestines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
- C07K16/3053—Skin, nerves, brain
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
- C07K16/3069—Reproductive system, e.g. ovaria, uterus, testes, prostate
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/77—Internalization into the cell
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
Definitions
- the present invention relates to an antibody-drug conjugate useful as an antitumor drug, in which an anti-TROP2 antibody and an antitumor drug are bound via a linker structure moiety.
- An antibody-drug conjugate in which a cytotoxic drug is bound to an antibody that binds to an antigen that is expressed on the surface of a cancer cell and can be internalized in the cell is selectively applied to a cancer cell. By being able to deliver a drug, it can be expected to accumulate the drug in cancer cells and kill the cancer cells (see Non-Patent Documents 1 to 3).
- ADC for example, Mylotarg (registered trademark; gemtuzumab ozogamicin) in which calicheamicin is bound to an anti-CD33 antibody is approved as a therapeutic agent for acute myeloid leukemia.
- Adcetris registered trademark; brentuximab bedotin
- auristatin E is bound to an anti-CD30 antibody
- Drugs contained in previously approved ADCs target DNA or tubulin.
- a camptothecin derivative that is a compound that inhibits topoisomerase I and exhibits an antitumor action is known as an antitumor low molecular weight compound.
- SN-38 which is a medicinal substance of irinotecan, and topoisomerase I inhibitory activity are stronger than topotecan also used in clinical practice, and it has stronger cytotoxic activity against various cancer cells in vitro. Yes. In particular, it was effective against cancer cells that were resistant to SN-38 and the like due to the expression of P-glycoprotein.
- a mouse human tumor subcutaneous transplantation model shows a strong antitumor effect, and clinical trials have not been performed yet (see Non-Patent Documents 5 to 10). It was not clear whether exatecan effectively acts as an ADC.
- DE-310 is a complex in which exatecan is bound to a biodegradable carboxymethyldextran polyalcohol polymer via a GGFG peptide spacer (Patent Document 3).
- exatecan By making exatecan into a polymeric prodrug, it retains high blood retention and passively increases the directivity to the tumor site by using increased permeability of tumor neovascularization and tumor tissue retention It is a thing.
- exatecan which is the active body, and exatecan in which glycine is bonded to the amino group are continuously released, and as a result, pharmacokinetics are improved.
- DE-310 is more effective than the single agent administration, despite the fact that the total amount of exatecan contained therein is reduced compared to the administration of exatecan alone. It showed higher effectiveness.
- clinical trials have been conducted and effective cases have been confirmed, and it has been confirmed that the active substance accumulates in the tumor rather than the normal tissue, while DE-310 and active substance in tumors in humans have been reported There is a report that the accumulation of is not much different from the accumulation in normal tissues, and passive targeting was not seen in humans (see Non-Patent Documents 11 to 14). As a result, DE-310 also did not enter the market, and it was not clear whether Exatecan effectively functions as a drug aimed at such targeting.
- TROP2 tumor-associated calcium signal transducer 2, GA733-1, EGP-1, M1S1; hereinafter referred to as hTROP2
- hTROP2 tumor-associated calcium signal transducer 2, GA733-1, EGP-1, M1S1; hereinafter referred to as hTROP2
- TACSTD2 tumor-associated calcium signal transducer 2, GA733-1, EGP-1, M1S1; hereinafter referred to as hTROP2
- hTROP2 tumor-associated calcium signal transducer 2
- Non-patent Document 17 tumor antigen GA733-1
- Non-patent Document 18 epithelial glycoprotein
- RS7-3G11 mouse monoclonal antibody
- the hTROP2 gene constitutes the TACSTD gene family together with human Trop-1 (EpCAM, EGP-2, TACSTD1) having about 50% homology (Non-patent Document 21).
- the hTROP2 protein is composed of a signal sequence composed of N-terminal 26 amino acid residues, an extracellular domain composed of 248 amino acid residues, a transmembrane domain composed of 23 amino acid residues, and an intracellular domain composed of 26 amino acid residues. There are four N-linked glycosylation sites in the extracellular domain, and the apparent molecular weight is known to increase around 10 kilodaltons from the theoretically calculated value of 35 kilodaltons (Non-patent Document 19).
- Non-patent Document 20 Protein kinase C phosphorylates intracellular serine 303 residue (Non-patent Document 18) and has a PIP 2 binding sequence in the intracellular domain, suggesting a signal transduction function in tumor cells. (Non-patent document 22).
- hTROP2 is overexpressed in various epithelial cell carcinoma types, and in normal tissues, it is limited to expression in epithelial cells of some tissues, and its expression level Has also been shown to be low compared to tumor tissue (Non-Patent Documents 23 to 27). Moreover, hTrop2 expression is poor in prognosis in colorectal cancer (Non-patent document 23), stomach cancer (Non-patent document 24), pancreatic cancer (Non-patent document 25), oral cancer (Non-patent document 26), and glioma (Non-patent document 27). It has also been reported to correlate with Furthermore, from a model using colon cancer cells, it has been reported that the expression of hTROP2 is involved in anchorage-independent cell proliferation of tumor cells and tumor formation in immunodeficient mice (Non-patent Document 28).
- an object of the present invention is to obtain and provide an antitumor drug having an excellent therapeutic effect and excellent antitumor effect and safety.
- anti-TROP2 antibody is an antibody capable of targeting tumor cells, that is, a property capable of recognizing tumor cells, a property capable of binding to tumor cells, a property capable of being internalized in tumor cells, and the like. Since it is an antibody, cytotoxicity using the antibody can be obtained by converting exatecan, which is an antitumor compound, to an antibody-drug conjugate conjugated to the antibody via a linker structure moiety, Furthermore, the antitumor compound can be surely moved by the tumor cells, and the antitumor effect of the compound can be exhibited specifically in the tumor cells. Therefore, the dose of the antitumor compound can be increased while the antitumor effect is reliably exhibited.
- the present inventors have created a linker having a specific structure and succeeded in obtaining an antibody-drug conjugate in which an anti-TROP2 antibody and exatecan are bound via this linker.
- the present invention was completed by finding that the antitumor effect was exhibited.
- the present invention relates to an antibody-drug conjugate characterized in that it is bound by a thioether bond formed in a disulfide bond part present in the hinge part of an anti-TROP2 antibody via a linker having a structure represented by
- the anti-TROP2 antibody binds at the end of L 1 , and the antitumor compound has a — (CH 2 ) n 2 —C ( ⁇ O) — moiety with the nitrogen atom of the amino group at position 1 as the binding site. Binds to the carbonyl group.
- n 1 represents an integer of 0 to 6
- n 2 represents an integer of 0 to 5
- n 3 represents an integer of 2 to 8
- L 2 represents —NH— (CH 2 CH 2 —O) n 4 —CH 2 CH 2 —C ( ⁇ O) — or a single bond
- n 4 represents an integer of 1 to 6
- L P represents a peptide residue composed of 2 to 7 amino acids
- L a represents -O- or a single bond
- -(Succinimid-3-yl-N)- is the following formula: It binds to the anti-TROP2 antibody at the 3-position of this, and binds to the methylene group in the linker structure containing it on the nitrogen atom at the 1-position.
- peptide residue between L P is, phenylalanine, glycine, valine, lysine, citrulline, serine, glutamic acid, the antibody according to a peptide residue consisting of amino acid selected from aspartic acid [1] - drug conjugates.
- [3] The antibody-drug conjugate according to [1] or [2], wherein L P is a peptide residue selected from the following group: -GGF-, -DGGF-, -(D-) D-GGF-, -EGGF-, -GGFG-, -SGGF-, -KGGF-, -DGGFG-, -GGFGG-, -DDGGFG-, -KDGGFG-, and -GGFGGGF-;
- “(D-) D” represents D-aspartic acid.
- L P is a peptide residue composed of 4 amino acids.
- [5] The antibody-drug conjugate according to any one of [1] to [4], wherein L P is a tetrapeptide residue -GGFG-.
- n 3 is an integer of 2 to 5
- L 2 is a single bond.
- n 3 is an integer of 2 to 5
- L 2 is —NH— (CH 2 CH 2 —O) n 4 —CH 2 CH 2 —C ( ⁇ O) —
- n 4 is 2
- the antibody-drug conjugate according to any one of [1] to [9], which is —NH—CH 2 CH 2 —O—CH 2 —C ( ⁇ O) —.
- the antibody-drug conjugate according to any one of [1] to [9].
- a drug-linker structure portion in which a drug is bound to -L 1 -L 2 -L P -NH- (CH 2 ) n 1 -L a- (CH 2 ) n 2 -C ( O)-
- a drug-linker structure portion in which a drug is bound to -L 1 -L 2 -L P -NH- (CH 2 ) n 1 -L a- (CH 2 ) n 2 -C ( O)-
- An antibody-drug conjugate which is bonded via a thioether bond formed in a disulfide bond moiety present in the hinge part of an anti-TROP2 antibody via a linker having a structure represented by
- the anti-TROP2 antibody binds at the end of L 1 and the antitumor compound binds to the carbonyl group of the — (CH 2 ) n 2 —C ( ⁇ O) — moiety.
- n 1 represents an integer of 0 to 6
- n 2 represents an integer of 0 to 5
- n 3 represents an integer of 2 to 8
- L 2 represents —NH— (CH 2 CH 2 —O) n 4 —CH 2 CH 2 —C ( ⁇ O) — or a single bond
- n 4 represents an integer of 1 to 6
- L P represents a tetrapeptide residue of -GGFG- L a represents -O- or a single bond
- -(Succinimid-3-yl-N)- is the following formula: It binds to the anti-TROP2 antibody at the 3-position of this, and binds to the methylene group in the linker structure containing it on the nitrogen atom at the 1-position.
- n 1 is 3, n 2 is 0, n 3 is 2, and L 2 is —NH— (CH 2 CH 2 —O) n 4 —CH 2 CH 2 —C ( ⁇ O )-, Where n 4 is 2 and La is a single bond, n 1 is 1, n 2 is is 1, n 3 is 5, L 2 is a single bond, or L a is -O-, and or n 1 is 2, n 2 is is 1, n 3 is 5, L 2 is a single bond, an antibody according to L a is -O- [14] - drug conjugates. [16] The antibody-drug conjugate according to [14] or [15], wherein n 3 is 2 or 5, and L 2 is a single bond.
- n 3 is 2 or 5
- L 2 is —NH— (CH 2 CH 2 —O) n 4 —CH 2 CH 2 —C ( ⁇ O) —
- n 4 is 2 or 4
- a drug-linker structure portion in which a drug is bound to -L 1 -L 2 -L P -NH- (CH 2 ) n 1 -L a- (CH 2 ) n 2 -C ( O)-
- a drug-linker structure portion in which a drug is bonded to -L 1 -L 2 -L P -NH- (CH 2 ) n 1 -L a- (CH 2 ) n 2 -C ( O)-
- a medicament comprising the antibody-drug conjugate according to any one of [1] to [23], a salt thereof, or a hydrate thereof.
- An antitumor drug and / or an anticancer drug comprising the antibody-drug conjugate according to any one of [1] to [23], a salt thereof, or a hydrate thereof.
- [27] The antibody-drug conjugate according to any one of [1] to [23], a salt thereof, or a hydrate thereof as an active ingredient, and a pharmaceutically acceptable formulation component Pharmaceutical composition.
- [28] Lung cancer, renal cancer, urothelial cancer, colon cancer, prostate cancer, glioblastoma multiforme, ovarian cancer, pancreatic cancer, breast cancer, melanoma, liver cancer, bladder cancer, stomach cancer, cervical cancer, head and neck cancer, Or the pharmaceutical composition as described in [27] for applying to esophageal cancer.
- a method for treating a tumor and / or cancer comprising administering the antibody-drug conjugate according to any one of [1] to [23], a salt thereof, or a hydrate thereof.
- An antibody-drug wherein a drug-linker moiety is bound to the antibody by a method of reacting with an anti-TROP2 antibody or a reactive derivative thereof to form a thioether bond at a disulfide bond moiety present in the hinge part of the antibody A method for producing a conjugate.
- n 3 represents an integer of 2 to 8
- L 2 represents —NH— (CH 2 CH 2 —O) n 4 —CH 2 CH 2 —C ( ⁇ O) — or a single bond
- n 4 represents an integer of 1 to 6
- L P represents a peptide residue composed of 2 to 7 amino acids selected from phenylalanine, glycine, valine, lysine, citrulline, serine, glutamic acid, aspartic acid
- n 1 represents an integer of 0 to 6
- n 2 represents an integer of 0 to 5
- L a represents -O- or a single bond
- (maleimid-N-yl)- is the following formula And a group having a nitrogen atom as a binding site.
- -(NH-DX) is And a group in which the nitrogen atom of the amino group at the 1-position is a binding site.
- an excellent antitumor effect and safety can be achieved by the anti-TROP2 antibody-drug conjugate in which the antitumor compound exatecan is bound via a linker having a specific structure.
- the nucleotide sequence (SEQ ID NO: 7) and amino acid sequence (SEQ ID NO: 8) of the cTINA1 antibody heavy chain are shown.
- the nucleotide sequence (SEQ ID NO: 9) and amino acid sequence (SEQ ID NO: 10) of the cTINA1 antibody light chain are shown.
- the nucleotide sequence (SEQ ID NO: 11) and amino acid sequence (SEQ ID NO: 12) of the hTINA1-H1 heavy chain are shown.
- the nucleotide sequence (SEQ ID NO: 13) and amino acid sequence (SEQ ID NO: 14) of the hTINA1-H2 heavy chain are shown.
- the nucleotide sequence (SEQ ID NO: 15) and amino acid sequence (SEQ ID NO: 16) of the hTINA1-H3 heavy chain are shown.
- the nucleotide sequence (SEQ ID NO: 17) and amino acid sequence (SEQ ID NO: 18) of the hTINA1-L1 light chain are shown.
- the nucleotide sequence (SEQ ID NO: 19) and amino acid sequence (SEQ ID NO: 20) of the hTINA1-L2 light chain are shown.
- the nucleotide sequence (SEQ ID NO: 21) and amino acid sequence (SEQ ID NO: 22) of the hTINA1-L3 light chain are shown.
- CDRA1 amino acid sequence (SEQ ID NO: 23), CDRH2 amino acid sequence (SEQ ID NO: 24), CDRH3 amino acid sequence (SEQ ID NO: 25), CDRL1 amino acid sequence (SEQ ID NO: 26), CDRL2 amino acid sequence (SEQ ID NO: 23) 27) and the amino acid sequence of CDRL3 (SEQ ID NO: 28).
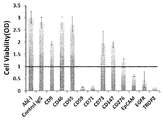
- the antibody-drug conjugate (1), (6), or (12) shows the antitumor effect shown against the human colon cancer cell line COLO205 subcutaneously transplanted BALB / c-nu / nu mice.
- the antibody-drug conjugate (1), (6), or (12) shows the antitumor effect shown against the human pancreatic adenocarcinoma cell line BxPC-3 subcutaneously transplanted BALB / c-nu / nu mice.
- the antibody-drug conjugate (1), (6), or (12) shows the antitumor effect shown against the human pancreatic adenocarcinoma cell line Capan-1 subcutaneously transplanted BALB / c-nu / nu mice.
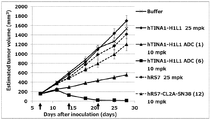
- the antibody-drug conjugate (2), (5), (7), or (10) shows the antitumor effect shown against the human colon cancer cell line COLO205 subcutaneously transplanted BALB / c-nu / nu mice.
- Antibody-drug conjugate (2), (5), (7), or (10) shows the antitumor effect shown against human pancreatic cancer cell line BxPC-3 subcutaneously transplanted BALB / c-nu / nu mice .
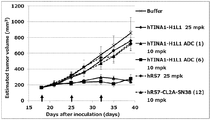
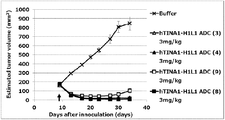
- the antibody-drug conjugate (3), (4), (8), or (9) shows the antitumor effect exhibited against the human colon cancer cell line COLO205 subcutaneously transplanted BALB / c-nu / nu mice.
- Antibody-drug conjugate (3), (4), (8), or (9) shows the antitumor effect shown against human pancreatic cancer cell line BxPC-3 subcutaneously transplanted BALB / c-nu / nu mice .
- Antibody-drug conjugate (3), (4), (8), or (9) shows the antitumor effect shown against human gastric cancer cell line NCI-N87 subcutaneously transplanted BALB / c-nu / nu mice.
- Antibody-drug conjugate (3), (4), (8), or (9) shows the anti-tumor effect shown against human lung cancer cell line NCI-H292 subcutaneously transplanted BALB / c-nu / nu mice.
- Antibody-drug conjugate (3), (4), (8), or (9) shows the anti-tumor effect shown against human pharyngeal cancer cell line FaDu subcutaneously transplanted BALB / c-nu / nu mice.
- the antibody-drug conjugate (3), (4), (8), or (9) exhibits an antitumor effect on human pancreatic adenocarcinoma cell line CFPAC-1 subcutaneously transplanted BALB / c-nu / nu mice Show.
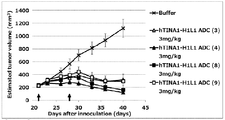
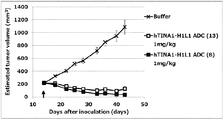
- Antibody-drug conjugate (8) or (13) shows the antitumor effect shown against human pancreatic adenocarcinoma cell line CFPAC-1 subcutaneously transplanted BALB / c-nu / nu mice.
- the antibody-drug conjugate (8) or (13) shows the antitumor effect shown against human pancreatic adenocarcinoma cell line HPAC subcutaneously transplanted BALB / c-nu / nu mice.
- the antibody-drug conjugate (8) or (13) shows the antitumor effect shown against NOD-scid mice transplanted subcutaneously into human esophageal cancer tissue.
- the anti-TROP2 antibody-drug conjugate of the present invention is an antitumor drug obtained by binding an antitumor compound to an anti-TROP2 antibody via a linker structure moiety, and will be described in detail below.
- the anti-TROP2 antibody used in the anti-TROP2 antibody-drug conjugate of the present invention may be derived from any species, but preferably humans, rats, mice, and rabbits can be exemplified. When derived from species other than human, it is preferable to chimerize or humanize using well-known techniques.
- the antibody of the present invention may be a polyclonal antibody or a monoclonal antibody, but a monoclonal antibody is preferred.
- the anti-TROP2 antibody is an antibody that can target tumor cells, that is, has the property of recognizing tumor cells, the property of binding to tumor cells, the property of being taken up and internalized in tumor cells, and the like. Can be linked via a linker to form an antibody-drug conjugate.
- the binding property of the antibody to tumor cells can be confirmed using flow cytometry.
- Methods for confirming the uptake of antibodies into tumor cells include (1) an assay that uses a secondary antibody (fluorescent label) that binds to a therapeutic antibody to visualize the antibodies taken into the cells with a fluorescence microscope (Cell Death and Differentiation (2008) 15, 751-761), (2) Assays that measure the amount of fluorescence taken into cells using secondary antibodies (fluorescent labels) that bind to therapeutic antibodies (Molecular Biology of the Cell Vol. 15 , 5268-5282, December 2004), or (3) Mab-ZAP assay (Bio Techniques 28), which uses an immunotoxin that binds to a therapeutic antibody to release the toxin and inhibit cell proliferation when taken up into the cell.
- the immunotoxin a recombinant complex protein of the catalytic region of diphtheria toxin and protein G can also be used. Since the antibody-drug conjugate is conjugated with a compound that exhibits an antitumor effect, it is preferable but not essential that the antibody itself has an antitumor effect. For the purpose of specifically and selectively exerting the cytotoxicity of the antitumor compound in tumor cells, it is important and preferable that the antibody has a property of internalizing and transferring into the tumor cells.
- the anti-TROP2 antibody can be obtained by immunizing an animal with a polypeptide serving as an antigen, and collecting and purifying the antibody produced in the living body, using a method commonly practiced in this field.
- the origin of the antigen is not limited to humans, and animals can be immunized with antigens derived from animals other than humans such as mice and rats.
- an antibody applicable to a human disease can be selected by examining the cross-reactivity between an antibody that binds to the obtained heterologous antigen and a human antigen.
- a hybridoma can be established by fusing an antibody-producing cell that produces an antibody against an antigen and a myeloma cell to obtain a monoclonal antibody.
- the antigen can be obtained by causing a host cell to produce a gene encoding an antigen protein by genetic manipulation.
- a vector capable of expressing an antigen gene may be prepared, introduced into a host cell to express the gene, and the expressed antigen may be purified.
- An antibody can also be obtained by using a method for immunizing an animal with an antigen-expressing cell or a cell line expressing the antigen by genetic manipulation as described above.
- Anti-TROP2 antibody can be obtained by known means.
- the anti-TROP2 antibody that can be used in the present invention is not particularly limited, but, for example, those specified by the amino acid sequences shown in the sequence listing of the present application can be preferably used.
- the TROP2 antibody used in the present invention preferably has the following characteristics. (1) an antibody having the following characteristics; (A) specifically binds to TROP2 (b) has the activity of internalizing in TROP2 expressing cells by binding to TROP2 (2) The antibody according to (1) above, wherein TROP2 is human TROP2.
- a heavy chain variable region comprising the amino acid sequence set forth in amino acid numbers 20 to 140 in SEQ ID NO: 12 and a light chain variable region comprising the amino acid sequence set forth in amino acid numbers 21 to 129 in SEQ ID NO: 18, SEQ ID NO: 12
- variable region of the heavy chain consisting of the amino acid sequence set forth in amino acid numbers 20 to 140 in SEQ ID NO: 12 and the variable region of the light chain consisting of the amino acid sequence set forth in amino acid numbers 21 to 129 in SEQ ID NO: 18, SEQ ID NO: 14 A variable region of a heavy chain consisting of the amino acid sequence set forth in amino acid numbers 20 to 140 in FIG.
- variable region 8 and a light chain variable region consisting of the amino acid sequence set forth in amino acid numbers 21 to 129 of SEQ ID NO: 18;
- the variable region of the heavy chain consisting of the amino acid sequence described in the above, the variable region of the light chain consisting of the amino acid sequence described in amino acid numbers 21 to 129 in SEQ ID NO: 20, and the amino acid sequence described in amino acid numbers 20 to 140 of SEQ ID NO: 16 In the variable region of the heavy chain consisting of Antibody according to (7) having a heavy chain variable region and a variable region of the light chain of light chain consisting of the amino acid sequence selected from the group consisting of the variable regions as set forth in amino acid number 21 to 129 Te.
- a heavy chain consisting of the amino acid sequence described in amino acid numbers 20 to 470 in SEQ ID NO: 12 and SEQ ID NO: 22 is a light chain comprising the amino acid sequence of amino acid numbers 21 to 234,
- SEQ ID NO: 14 is the heavy chain comprising the amino acid sequence of amino acid numbers 20 to 470, and
- SEQ ID NO: 18 is the amino acid sequence of amino acid numbers 21 to 234.
- a light chain consisting of: SEQ ID NO: 14 A heavy chain consisting of the amino acid sequence described in amino acid numbers 20 to 470, a light chain consisting of the amino acid sequence described in amino acid numbers 21 to 234 in SEQ ID NO: 20, and an amino acid sequence described in amino acid numbers 20 to 470 in SEQ ID NO: 14
- a heavy chain consisting of the amino acid sequence set forth in amino acid numbers 21 to 234 in SEQ ID NO: 22 a heavy chain consisting of the amino acid sequence set forth in amino acid numbers 20 to 470 in SEQ ID NO: 16 and amino acid number 21 in SEQ ID NO: 18
- the above heavy chain and light chain selected from the group consisting of the heavy chain consisting of the amino acid sequence set forth in acid numbers 20 to 470 and the light chain consisting of the amino acid sequence set forth in amino acid numbers 21 to 234 in SEQ ID NO: 22 (6 Or the antibody according to (7).
- a heavy chain consisting of the amino acid sequence set forth in SEQ ID NO: 12 and a light chain consisting of the amino acid sequence set forth in SEQ ID NO: 18, a heavy chain consisting of the amino acid sequence set forth in SEQ ID NO: 12 and an amino acid sequence set forth in SEQ ID NO: 20 A light chain consisting of the amino acid sequence set forth in SEQ ID NO: 12, a light chain consisting of the amino acid sequence set forth in SEQ ID NO: 22, a heavy chain consisting of the amino acid sequence set forth in SEQ ID NO: 14, and the sequence described in SEQ ID NO: 18
- a light chain consisting of an amino acid sequence, a heavy chain consisting of an amino acid sequence set forth in SEQ ID NO: 14 and a light chain consisting of an amino acid sequence set forth in SEQ ID NO: 20 a heavy chain consisting of an amino acid sequence set forth in SEQ ID NO: 14 and A light chain consisting of the described amino acid sequence, a heavy chain consisting of the amino acid sequence set forth in SEQ ID NO: 16 and a
- (11) A heavy chain consisting of the amino acid sequence described in amino acid numbers 20 to 470 in SEQ ID NO: 12 and a light chain consisting of the amino acid sequence described in amino acid numbers 21 to 234 in SEQ ID NO: 18, and amino acid numbers 20 to 470 in SEQ ID NO: 14
- the anti-TROP2 antibody used in the present invention will be described below.
- cancer and “tumor” are used interchangeably.
- the term “gene” includes not only DNA but also mRNA, cDNA, and cRNA thereof.
- polynucleotide is used interchangeably with nucleic acid, and includes DNA, RNA, probes, oligonucleotides, and primers.
- polypeptide and “protein” are used without distinction.
- the “cell” includes a cell in an animal individual and a cultured cell.
- TROP2 is used in the same meaning as TROP2 protein.
- CDR in this specification means a complementarity determining region (CDR). It is known that there are three CDRs in each of the heavy and light chains of an antibody molecule. CDRs, also called hypervariable domains, are sites in the variable regions of antibody heavy and light chains that have particularly high primary structure variability, and are heavy and light chain polypeptide chains. In the primary structure of each, it is separated into three locations.
- CDRH1, CDRH2, CDRH3 from the amino terminal side of the heavy chain amino acid sequence
- CDRL1 from the amino terminal side of the light chain amino acid sequence.
- hybridize under stringent conditions means to hybridize at 68 ° C. in a commercially available hybridization solution ExpressHyb Hybridization Solution (manufactured by Clontech) or using a filter on which DNA is fixed. After hybridization at 68 ° C. in the presence of 0.7-1.0 M NaCl, 0.1-2 fold SSC solution (1 fold SSC consists of 150 mM NaCl, 15 mM sodium citrate) Used, hybridization under conditions that can be identified by washing at 68 ° C. or equivalent conditions.
- TROP2 TROP2 is one of the TACSTD family expressed in human trophoblasts and is a single-transmembrane type 1 cell membrane protein involved in immune resistance common to human trophoblast cells and cancer cells.
- the TROP2 protein used in the present invention can be directly purified from TROP2-expressing cells of humans and non-human mammals (rats, mice, etc.) or can be used by preparing a cell membrane fraction of the cells. Alternatively, it can be obtained by synthesizing TROP2 in vitro or producing it in a host cell by genetic manipulation.
- TROP2 cDNA is incorporated into a vector that can be expressed and then synthesized in a solution containing enzymes, substrates, and energy substances necessary for transcription and translation, or other prokaryotic or true organisms.
- the protein can be obtained by expressing TROP2 by transforming a nuclear host cell. It is also possible to use a TROP2 expressing cell or a cell line expressing TROP2 by the above-described genetic manipulation as a TROP2 protein.
- the DNA sequence and amino acid sequence of TROP2 are published on public databases and can be referred to by accession numbers such as NM_002353, NP_002344 (NCBI), for example.
- TROP2 protein having a biological activity equivalent to the protein consisting of an amino acid sequence in which one or several amino acids are substituted, deleted, and / or added in the amino acid sequence of TROP2 is also included in TROP2.
- Human TROP2 protein is composed of a signal sequence consisting of N-terminal 26 amino acid residues, an extracellular domain consisting of 248 amino acid residues, a transmembrane domain consisting of 23 amino acid residues, and an intracellular domain consisting of 26 amino acid residues .
- the antibody against TROP2 of the present invention is produced in vivo by immunizing an animal with any polypeptide selected from TROP2 or the amino acid sequence of TROP2 using a method commonly practiced in this field. Can be obtained by collecting and purifying the antibody.
- the biological species of TROP2 serving as an antigen is not limited to humans, and it is also possible to immunize animals with TROP2 derived from animals other than humans such as mice and rats.
- antibodies applicable to human diseases can be selected by examining the cross-reactivity between the obtained antibody binding to heterologous TROP2 and human TROP2.
- known methods for example, Kohler and Milstein, Nature (1975) 256, p. 495-497; Kennet, R.
- Hybridomas can be established by fusing antibody-producing cells that produce antibodies against TROP2 and myeloma cells to obtain monoclonal antibodies.
- the TROP2 serving as an antigen can be obtained by expressing the TROP2 gene in a host cell by genetic manipulation. Specifically, a vector capable of expressing the TROP2 gene may be prepared, introduced into a host cell to express the gene, and the expressed TROP2 may be purified. It is also possible to use a TROP2 expressing cell by the above-described genetic manipulation or a cell line expressing TROP2 as a TROP2 protein.
- a method for obtaining an antibody against TROP2 will be specifically described.
- Anti-TROP2 antibody is prepared by using TROP2 or a polypeptide comprising at least six consecutive partial amino acid sequences, or a derivative in which any amino acid sequence or carrier is added thereto.
- TROP2 can be used by directly purifying from human tumor tissue or tumor cells, and can be obtained by synthesizing TROP2 in vitro or producing it in a host cell by genetic engineering. Specifically, in genetic manipulation, TROP2 cDNA is incorporated into a vector capable of expression and then synthesized in a solution containing enzymes, substrates and energy substances necessary for transcription and translation, or other prokaryotic or true organisms. An antigen can be obtained by transforming a nuclear host cell to express TROP2.
- TROP2 cDNA is, for example, a polymerase chain reaction (hereinafter referred to as “PCR”) using a cDNA library expressing the TROP2 cDNA as a template and a primer that specifically amplifies the TROP2 cDNA (Saiki, R. K., et al., Science (1988) 239, pp. 487-489).
- PCR polymerase chain reaction
- examples of in vitro synthesis of polypeptides include, but are not limited to, a rapid translation system (RTS) manufactured by Roche Diagnostics.
- prokaryotic host examples include Escherichia coli and Bacillus subtilis.
- the host cell is transformed with a plasmid vector containing a replicon or origin of replication from a species compatible with the host and regulatory sequences.
- the vector preferably has a sequence capable of imparting phenotypic (phenotypic) selectivity to transformed cells.
- eukaryotic host cells include vertebrates, insects, yeast, and the like. Examples of vertebrate cells include COS cells (Gluzman, Y. Cell (1981) 23, p. 175-182, ATCC CRL-1650; ATCC: American Type Culture Collection, mouse fibroblast NIH3T3 (ATCC No.
- CHO cells ATCC CCL-61
- dihydrofolate reductase Deficient strains Urlauub, G. and Chasin, LA Proc. Natl. Acad. Sci. USA (1980) 77, p. 4126-4220
- the transformant obtained as described above can be cultured according to a method commonly practiced in this field, and the desired polypeptide is produced intracellularly or extracellularly by the culture.
- the medium used for the culture can be appropriately selected from those commonly used according to the host cell employed by those skilled in the art. In the case of Escherichia coli, for example, ampicillin can be added to the LB medium as necessary.
- Antibiotics such as these and IPMG can be added and used.
- the recombinant protein produced inside or outside the transformant by the above culture can be separated and purified by various known separation procedures utilizing the physical and chemical properties of the protein. it can.
- Specific examples of the method include various liquid chromatography such as treatment with an ordinary protein precipitating agent, ultrafiltration, molecular sieve chromatography (gel filtration), adsorption chromatography, ion exchange chromatography, affinity chromatography and the like. , Dialysis methods, combinations thereof and the like.
- a histidine tag consisting of 6 residues to a recombinant protein to be expressed, it can be efficiently purified with a nickel affinity column.
- the desired polypeptide can be easily produced in large quantities with high yield and high purity. It is also possible to use the transformant itself described above as an antigen. It is also possible to use a cell line expressing TROP2 as an antigen.
- Such cell lines include humanized lung cancer lines NCI-H322, PC14, NCIH-H2122, or LCAM1, human prostate cancer line PC3, human pancreatic cancer line BxPC-3, Capan-1, or PK-1, human ovary
- Examples include the cancer line SKOV3 and the human colon cancer line COLO205, but are not limited to these cell lines as long as TROP2 is expressed.
- TROP2 Purification of antigen TROP2 prepared by the method as described above or a part thereof can be used as the antigen.
- a membrane fraction prepared by a TROP2-expressing recombinant cell, or a TROP2-expressing recombinant cell itself, and a partial peptide of the protein of the present invention chemically synthesized using methods well known to those skilled in the art are used as antigens. You can also.
- a TROP2-expressing cell line can also be used as an antigen.
- step (B) Preparation of antibody-producing cells
- the antigen obtained in step (a) is mixed with Freund's complete or incomplete adjuvant, or an auxiliary agent such as potassium alum, and an experimental animal is immunized as an immunogen.
- an experimental animal there is a method of immunizing experimental animals using antigen-expressing cells as an immunogen.
- an animal used in a known hybridoma production method can be used without any problem. Specifically, for example, mouse, rat, goat, sheep, cow, horse and the like can be used. However, from the viewpoint of easy availability of myeloma cells to be fused with the extracted antibody-producing cells, it is preferable to use mice or rats as immunized animals.
- mice there are no particular limitations on the mouse and rat strains actually used.
- rats for example, Wistar, Low, Lewis, Sprague, Dawley, ACI, BN Fischer or the like can be used.
- mice and rats can be obtained, for example, from laboratory animal breeding and sales companies such as Nippon Clare Co., Ltd. and Japan Charles River Co., Ltd.
- the BALB / c strain is particularly preferable for mice and the Wistar and Low strains are particularly preferable for rats, considering fusion compatibility with myeloma cells described later.
- mice with reduced biological mechanisms for removing autoantibodies that is, autoimmune disease mice.
- the age at the time of immunization of these mice or rats is preferably 5 to 12 weeks old, more preferably 6 to 8 weeks old.
- a preferable method in the present invention is specifically shown as follows. That is, first, a membrane protein fraction that is an antigen or a cell in which the antigen is expressed is administered intradermally or intraperitoneally in an animal. However, in order to increase the immune efficiency, the combination of both is preferable. When the first half is intradermally administered and the second half or the last is intraperitoneally administered, the immune efficiency can be particularly enhanced.
- the antigen administration schedule varies depending on the type of animal to be immunized, individual differences, and the like, but in general, the number of antigen administrations is preferably 3 to 6 times, and the administration interval is 2 to 6 weeks. More preferably 4 weeks.
- the dose of the antigen varies depending on the kind of animal, individual differences, etc., but is generally 0.05 to 5 mg, preferably about 0.1 to 0.5 mg.
- the booster immunization is performed 1 to 6 weeks after the antigen administration as described above, preferably 1 to 4 weeks, and more preferably 1 to 3 weeks. When the immunogen is a cell, 1 ⁇ 10 6 to 1 ⁇ 10 7 cells are used.
- the dose of antigen for booster immunization varies depending on the type and size of the animal, but generally 0.05 to 5 mg, preferably 0.1 to 0.5 mg, more preferably, for example, in the case of mice. Is about 0.1 to 0.2 mg.
- the immunogen is a cell
- 1 ⁇ 10 6 to 1 ⁇ 10 7 cells are used.
- Spleen cells or lymphocytes containing antibody-producing cells are aseptically removed from the immunized animal 1 to 10 days after the boost, preferably 2 to 5 days, and more preferably 2 to 3 days later. In this case, if the antibody titer is measured and an animal having a sufficiently high antibody titer is used as a source of antibody-producing cells, the efficiency of subsequent operations can be increased.
- Examples of the antibody titer measurement method used here include, but are not limited to, the RIA method and the ELISA method.
- the antibody titer in the present invention can be measured according to the procedure described below, for example, according to the ELISA method.
- a purified or partially purified antigen is adsorbed on a solid phase surface such as a 96-well plate for ELISA, and the solid phase surface on which no antigen is adsorbed is covered with a protein unrelated to the antigen, such as bovine serum albumin (BSA), After washing the surface, it is contacted with a serially diluted sample (eg, mouse serum) as the first antibody, and the antibody in the sample is bound to the antigen.
- a serially diluted sample eg, mouse serum
- an antibody against a mouse antibody labeled with an enzyme as a second antibody is added and bound to the mouse antibody.
- the substrate of the enzyme is added, and the change in absorbance due to color development based on the decomposition of the substrate is measured. calculate.
- Separation of antibody-producing cells from the spleen cells or lymphocytes of the immunized animal can be performed by known methods (for example, Kohler et al., Nature (1975) 256, p. 495; Kohler et al., Eur. J. Immunol. (1977) 6, p. 511; Milstein et al., Nature (1977), 266, p. 550; Walsh, Nature, (1977) 266, p. 495).
- spleen cells a general method of separating antibody-producing cells by chopping the spleen and filtering the cells through a stainless mesh and then suspending them in the Eagle's minimum essential medium (MEM) can be employed.
- MEM Eagle's minimum essential medium
- myeloma Preparation of myeloma cells (hereinafter referred to as “myeloma”)
- Myeloma cells used for cell fusion are not particularly limited and can be appropriately selected from known cell lines.
- HGPRT Hydropoxanthine-guanine phosphoryltransferase
- 8-azaguanine medium RPMI-1640 medium supplemented with glutamine, 2-mercaptoethanol, gentamicin, and fetal bovine serum (hereinafter referred to as “FBS”).
- FBS fetal bovine serum
- IMDM Iscove's Modified Dulbecco's Medium
- DMEM Dulbecco's Modified Eagle Medium
- the antibody-producing cells are used in a polyethylene glycol solution having a molecular weight of 1500 to 6000, preferably 2000 to 4000, at a temperature of 30 to 40 ° C., preferably 35 to 38 ° C. Mix with myeloma cells for 1-10 minutes, preferably 5-8 minutes.
- hybridoma group The selection method of the hybridoma obtained by the above-mentioned cell fusion is not particularly limited. 495; Milstein et al., Nature (1977) 266, p. 550). This method is effective when hybridomas are obtained using myeloma cells of HGPRT-deficient strains that cannot survive with aminopterin. That is, by culturing unfused cells and hybridomas in a HAT medium, only hybridomas having resistance to aminopterin can be selectively left and grown.
- a method for cloning a hybridoma As a method for cloning a hybridoma, a known method such as a methyl cellulose method, a soft agarose method, a limiting dilution method, or the like can be used (for example, Barbara, B. M. and Stanley, MS: Selected Methods in Cellular Immunology, WH Freeman and Company, San Francisco (1980)). Among these methods, a three-dimensional culture method such as a methyl cellulose method is particularly preferable.
- a hybridoma group formed by cell fusion is suspended and cultured in a methylcellulose medium such as ClonCell-HY Selection Medium D (StemCell Technologies # 03804), and the formed hybridoma colonies are recovered to recover the monoclonal hybridoma. Acquisition is possible.
- a methylcellulose medium such as ClonCell-HY Selection Medium D (StemCell Technologies # 03804)
- the formed hybridoma colonies are recovered to recover the monoclonal hybridoma. Acquisition is possible.
- Each of the collected hybridoma colonies is cultured, and those in which the antibody titer is stably recognized in the obtained hybridoma culture supernatant are selected as TROP2 monoclonal antibody-producing hybridoma strains.
- TROP2 hybridoma TINA1 can be mentioned.
- an antibody produced by TROP2 hybridoma TINA1 is referred to as “TINA1 antibody” or simply “TINA1”.
- the heavy chain variable region of the TINA1 antibody has the amino acid sequence shown in SEQ ID NO: 2 in the sequence listing.
- the light chain variable region of the TINA1 antibody has the amino acid sequence shown in SEQ ID NO: 4 in the sequence listing.
- (G) Preparation of monoclonal antibody by culturing hybridoma
- the hybridoma selected in this manner can be efficiently obtained by culturing the hybridoma, but the target monoclonal antibody is produced prior to culturing. It is desirable to screen for hybridomas. For this screening, a method known per se can be employed.
- the antibody titer in the present invention can be measured by, for example, the ELISA method described in the item (b) above.
- the hybridoma obtained by the above method can be stored in a frozen state in liquid nitrogen or in a freezer at ⁇ 80 ° C. or lower.
- the hybridoma that has been cloned is cultured by changing the medium from the HT medium to the normal medium.
- Mass culture is performed by rotary culture using a large culture bottle or spinner culture.
- a monoclonal antibody that specifically binds to the protein of the present invention can be obtained by purifying the supernatant in this mass culture using a method well known to those skilled in the art, such as gel filtration.
- a large amount of the monoclonal antibody of the present invention is contained by injecting a hybridoma into the abdominal cavity of a mouse of the same strain (for example, the above-mentioned BALB / c) or Nu / Nu mouse, and proliferating the hybridoma. Ascites can be obtained.
- a monoclonal antibody having a concentration of about 100 times or more compared with that in the culture solution can be obtained.
- Monoclonal antibodies obtained by the above method are described in, for example, Weir, D. et al. M.M. : Handbook of Experimental Immunology, Vol. I, II, III, Blackwell Scientific Publications, Oxford (1978). The monoclonal antibody thus obtained has a high antigen specificity for TROP2.
- the isotype and subclass of the monoclonal antibody thus obtained can be determined as follows.
- the identification method include an octerlony method, an ELISA method, and an RIA method.
- the octerulony method is simple, but concentration is necessary when the concentration of the monoclonal antibody is low.
- the ELISA method or the RIA method is used, the culture supernatant is reacted with the antigen-adsorbing solid phase as it is, and further, antibodies corresponding to various immunoglobulin isotypes and subclasses are used as secondary antibodies. Isotypes and subclasses can be identified.
- a commercially available identification kit for example, mouse typer kit; manufactured by Bio-Rad
- an antibody having cytotoxic activity equivalent to that of the TINA1 antibody can be obtained.
- An example of such an antibody is an antibody that binds to the same epitope as the TINA1 antibody.
- the monoclonal antibody binds to the partial peptide or partial conformation to which the TINA1 antibody binds, it can be determined that the monoclonal antibody binds to the same epitope as the TINA1 antibody. Further, by confirming that the monoclonal antibody competes for binding of TINA1 antibody to TROP2 (that is, the monoclonal antibody prevents binding of TINA1 antibody to TROP2), the sequence or structure of a specific epitope is determined. Even if not, it can be determined that the monoclonal antibody binds to the same epitope as the anti-TROP2 antibody. When it is confirmed that the epitope is the same, it is strongly expected that the monoclonal antibody has the same antigen binding ability or biological activity as the TINA1 antibody.
- the antibodies of the present invention include genetically engineered antibodies that have been artificially modified for the purpose of reducing the heterologous antigenicity against humans, such as chimera (Chimeric ) Antibodies, humanized antibodies, human antibodies and the like. These antibodies can be produced using known methods.
- Examples of the chimeric antibody include antibodies in which the variable region and the constant region of the antibody are different from each other, for example, a chimeric antibody in which the variable region of a mouse or rat-derived antibody is joined to a human-derived constant region (Proc. Natl. Acad). Sci.U.S.A., 81, 6851-6855, (1984)).
- humanized antibody an antibody in which only the complementarity determining region (CDR) is incorporated into a human-derived antibody (see Nature (1986) 321; p.522-525), in addition to the CDR sequence by CDR grafting, Examples of the amino acid residues of some frameworks include antibodies grafted to human antibodies (WO 90/07861).
- the humanized antibody derived from the TINA1 antibody is not limited to a specific humanized antibody as long as it retains all six CDR sequences of the TINA1 antibody.
- the heavy chain variable region of the TINA1 antibody is shown in CDRH1 (TAGMQ) consisting of the amino acid sequence shown in SEQ ID NO: 23 in the sequence listing, CDRH2 (WINTHSGVPKYAEDFKG) consisting of the amino acid sequence shown in SEQ ID NO: 24, and SEQ ID NO: 25.
- CDRH3 SGFGSSYWYFDV having the amino acid sequence described above is possessed.
- the light chain variable region of the TINA1 antibody is represented by CDRL1 (KASQDVSTAVA) consisting of the amino acid sequence shown in SEQ ID NO: 26 of the sequence listing, CDRL2 (SASYRYT) consisting of the amino acid sequence shown by SEQ ID NO: 27, and SEQ ID NO: 28.
- CDRL1 KASQDVSTAVA
- CDRL2 SASYRYT
- Examples of the humanized antibody of mouse antibody TINA1 include (1) an amino acid sequence consisting of amino acid residues 20 to 140 of SEQ ID NO: 12, 14, or 16 in the sequence listing, and (2) the amino acid sequence of (1) above.
- “several” means 1 to 10, 1 to 9, 1 to 8, 1 to 7, 1 to 6, 1 to 5, 1
- amino acid substitution is preferable as the amino acid substitution in this specification.
- Such amino acid substitution is preferably performed within a range that does not deteriorate the properties of the substance having the original amino acid sequence.
- the heavy chain having a variable region consisting of the amino acid sequence described in amino acid numbers 20 to 140 in SEQ ID NO: 12 and the amino acid numbers 21 to 129 in SEQ ID NO: 18 An antibody consisting of a light chain having a variable region consisting of the amino acid sequence of SEQ ID NO: 12, a heavy chain having a variable region consisting of the amino acid sequence described in amino acid numbers 20 to 140 in SEQ ID NO: 12 and amino acid numbers 21 to 129 in SEQ ID NO: 20
- An antibody consisting of a light chain having a variable region consisting of an amino acid sequence, a heavy chain having a variable region consisting of an amino acid sequence described in amino acid numbers 20 to 140 in SEQ ID NO: 12 and an amino acid described in amino acid numbers 21 to 129 in SEQ ID NO: 22 An antibody comprising a light chain having a variable region comprising a sequence
- Amino acids in the chain and SEQ ID NO: 18 An antibody consisting of a light chain consisting of the amino acid sequence set forth in acid numbers 21 to 234; a heavy chain consisting of the amino acid sequence set forth in amino acid numbers 20 to 470 in SEQ ID NO: 14; and an amino acid sequence set forth in amino acid numbers 21 to 234 in SEQ ID NO: 20
- Antibody consisting of light chain consisting of amino acid sequence antibody consisting of heavy chain consisting of amino acid sequence described in amino acid numbers 20 to 470 in SEQ ID NO: 14 and light chain consisting of amino acid sequence described in amino acid numbers 21 to 234 in SEQ ID NO: 22
- a heavy chain having a variable region consisting of the amino acid sequence described in amino acid numbers 20 to 140 in SEQ ID NO: 12 and a variable region consisting of the amino acid sequence described in amino acid numbers 21 to 129 in SEQ ID NO: 18 are used.
- Another preferred combination includes an antibody consisting of a heavy chain consisting of the amino acid sequence set forth in SEQ ID NO: 12 and a light chain consisting of the amino acid sequence set forth in SEQ ID NO: 18, and a heavy chain consisting of the amino acid sequence set forth in SEQ ID NO: 12.
- An antibody comprising a chain and a light chain comprising the amino acid sequence set forth in SEQ ID NO: 20, a heavy chain comprising the amino acid sequence set forth in SEQ ID NO: 12 and a light chain comprising the amino acid sequence set forth in SEQ ID NO: 22, SEQ ID NO: 14
- An antibody comprising a chain, a heavy chain comprising the amino acid sequence set forth in SEQ ID NO: 14 and an antibody comprising the light chain comprising the amino acid sequence set forth in SEQ ID NO: 22 An antibody comprising a heavy chain comprising the amino acid sequence set forth in SEQ ID NO: 16 and a light chain comprising the amino acid sequence set forth in SEQ ID NO: 18, a heavy chain comprising the amino acid sequence set forth in SEQ ID NO: 16 and an amino acid sequence set forth in SEQ ID NO: 20. And an antibody consisting of a heavy chain consisting of the amino acid sequence set forth in SEQ ID NO: 16 and an antibody consisting of the light chain consisting of the amino acid sequence set forth in SEQ ID NO: 22.
- An excellent and preferred combination is an antibody comprising a heavy chain consisting of the amino acid sequence described in amino acid numbers 20 to 470 in SEQ ID NO: 12 and a light chain consisting of the amino acid sequence described in amino acid numbers 21 to 234 in SEQ ID NO: 18; An antibody consisting of a heavy chain consisting of the amino acid sequence of amino acid numbers 20 to 470 in No. 14 and a light chain consisting of the amino acid sequence of amino acid numbers 21 to 234 in SEQ ID No. 18, and an amino acid number of 20 to 470 in SEQ ID No.
- an antibody comprising a heavy chain consisting of the amino acid sequence described in amino acid numbers 20 to 469 in SEQ ID NO: 12 and a light chain consisting of the amino acid sequence described in amino acid numbers 21 to 234 in SEQ ID NO: 18,
- an antibody having a biological activity equivalent to that of each of the above antibodies by combining a sequence having high homology with the above heavy chain amino acid sequence and light chain amino acid sequence. Such homology is generally 80% or more, preferably 90% or more, more preferably 95% or more, most preferably 99% or more. Homology.
- An antibody having biological activity equivalent to that of each of the above antibodies can also be selected by combining an amino acid sequence in which one to several amino acid residues are substituted, deleted, or added to the heavy chain or light chain amino acid sequence. Is possible.
- Blast algorithm version 2.2.2 (Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaeffer, Jinghui Zhang, Zhangband, Zhangbhang. (1997), “Gapped BLAST and PSI-BLAST: a new generation of protein database search programs”, Nucleic Acids Res. 25: 3389-3402).
- Blast algorithm can also be used by accessing www.ncbi.nlm.nih.gov/blast on the Internet.
- the amino acid sequence consisting of the 1st to 19th amino acid residues is a signal sequence, and from the 20th to 140th amino acid residues.
- the amino acid sequence consisting of the amino acid sequence consisting of the 141st to 470th amino acid residues is the constant region.
- the sequence of SEQ ID NO: 12 is described in FIG. 3, the sequence of SEQ ID NO: 14 is described in FIG. 4, and the sequence of SEQ ID NO: 16 is described in FIG.
- the amino acid sequence consisting of the 1st to 20th amino acid residues is a signal sequence, and from the 21st to 129th amino acid residues.
- the amino acid sequence consisting of the amino acid sequence consisting of the 130th to 234th amino acid residues is the constant region.
- the sequence of SEQ ID NO: 18 is described in FIG. 6, the sequence of SEQ ID NO: 20 is described in FIG. 7, and the sequence of SEQ ID NO: 22 is described in FIG.
- Examples of the antibody of the present invention further include a human antibody that binds to TROP2.
- An anti-TROP2 human antibody means a human antibody having only the gene sequence of an antibody derived from a human chromosome.
- the anti-TROP2 human antibody is a method using a human antibody-producing mouse having a human chromosome fragment containing the heavy and light chain genes of a human antibody (Tomizuka, K. et al., Nature Genetics (1997) 16, p. 133. -143; Kuroiwa, Y. et.al., Nucl.Acids Res. (1998) 26, p.3447-3448; Yoshida, H.
- the endogenous immunoglobulin heavy chain and light chain loci are disrupted, and instead, the human immunoglobulin is transferred via a yeast artificial chromosome (YAC) vector or the like.
- YAC yeast artificial chromosome
- genetically modified animals into which heavy and light chain loci have been introduced they can be produced by creating knockout animals and transgenic animals and crossing these animals together.
- eukaryotic cells are transformed with cDNA encoding each of the heavy chain and light chain of such a human antibody, preferably a vector containing the cDNA, to produce a gene recombinant human monoclonal antibody.
- This antibody can also be obtained from the culture supernatant by culturing the transformed cells.
- eukaryotic cells preferably CHO cells
- mammalian cells such as lymphocytes and myeloma can be used as the host.
- a method for obtaining a human antibody derived from phage display selected from a human antibody library (Wormstone, IM et al, Investigative Ophthalmology & Visual Science. (2002) 43 (7), p. 2301-2308; Mé, S. et.al., Briefings in Functional Genomics and Proteomics (2002), 1 (2), p.189-203; Siriwardena, D. et al., Ophthalmology (2002) 109 (3), p. 427-431 etc.) are also known.
- a phage display method (Nature Biotechnology (2005), 23, (9), p.
- a variable region of a human antibody in which a variable region of a human antibody is expressed on a phage surface as a single chain antibody (scFv) and a phage that binds to an antigen is selected.
- scFv single chain antibody
- a phage that binds to an antigen ⁇ 1116
- the DNA sequence encoding the variable region of the human antibody that binds to the antigen can be determined. If the DNA sequence of scFv binding to the antigen is clarified, a human antibody can be obtained by preparing an expression vector having the sequence, introducing it into a suitable host and expressing it (WO 92/01047). No.
- a newly produced human antibody binds to the partial peptide or partial conformation to which the TINA1 antibody binds, it can be determined that the human antibody binds to the same epitope as the TINA1 antibody.
- the human antibody competes for binding of TINA1 antibody to TROP2 (that is, the human antibody prevents binding of TINA1 antibody to TROP2), the sequence or structure of a specific epitope can be determined.
- the human antibody binds to the same epitope as the TINA1 antibody.
- the epitope is the same, it is strongly expected that the human antibody has a biological activity equivalent to that of the TINA1 antibody.
- the chimeric antibody, humanized antibody, or human antibody obtained by the above method can be evaluated for binding to an antigen by a known method or the like, and a suitable antibody can be selected.
- An example of another index for comparing the properties of antibodies is antibody stability.
- DSC Differential scanning calorimetry
- Tm thermal denaturation midpoint
- the difference in thermal stability can be compared by measuring the Tm value using DSC and comparing the values.
- Tm thermal denaturation midpoint
- a suitable antibody can be selected using stability as an index.
- Other indicators for selecting antibodies include high yields in suitable host cells and low aggregation in aqueous solutions. For example, since the antibody with the highest yield does not necessarily exhibit the highest thermal stability, it is necessary to select the most suitable antibody for human administration based on a comprehensive judgment based on the above-mentioned indicators. .
- the antibody of the present invention includes a modified antibody.
- the modified product means a product obtained by chemically or biologically modifying the antibody of the present invention.
- Chemical modifications include chemical modifications to the amino acid backbone, chemical modifications of N-linked or O-linked carbohydrate chains, and the like.
- Biological modifications include post-translational modifications (eg, glycosylation to N- or O-links, N- or C-terminal processing, deamidation, aspartic acid isomerization, methionine oxidation) And those obtained by adding a methionine residue to the N-terminal by expression using a prokaryotic host cell.
- modified products of the antibody of the present invention for example, an enzyme label, a fluorescent label, and an affinity label are also included in the meaning of such a modified product.
- Such a modified product of the antibody of the present invention is useful for improvement of antibody stability and blood retention, reduction of antigenicity, detection or isolation of antibody or antigen, and the like.
- the antibody of the present invention includes an antibody whose sugar chain modification is regulated.
- an antibody gene is once isolated and then introduced into an appropriate host to produce an antibody, a combination of an appropriate host and an expression vector can be used.
- Specific examples of the antibody gene include a combination of a gene encoding the heavy chain sequence of the antibody described herein and a gene encoding the light chain sequence.
- the heavy chain sequence gene and the light chain sequence gene can be inserted into the same expression vector, or can be inserted into separate expression vectors. is there.
- eukaryotic cells animal cells, plant cells, and eukaryotic microorganisms can be used.
- animal cells mammalian cells such as COS cells (Gluzman, Y. Cell (1981) 23, p.175-182, ATCC CRL-1650) which are monkey cells, mouse fibroblasts NIH3T3 (ATCC No. CRL-1658) and Chinese hamster ovary cells (CHO cells, ATCC CCL-61) dihydrofolate reductase-deficient strain (Urlauub, G.
- the antibody of the present invention includes a step of culturing the transformed host cell and a step of collecting the target antibody or a functional fragment of the antibody from the culture obtained in the step. An antibody obtained by the method for producing the antibody is also included.
- the antibody according to the present invention also includes the antibody subjected to the modification and a functional fragment of the antibody, a deletion in which 1 or 2 amino acids are deleted at the heavy chain carboxyl terminus, and amidated Such deletion forms (for example, heavy chain in which the proline residue at the carboxyl terminal site is amidated) and the like are also included.
- the carboxyl-terminal deletion of the heavy chain of the antibody according to the present invention is not limited to the above type.
- the two heavy chains constituting the antibody according to the present invention may be either one of the heavy chains selected from the group consisting of the full length and the above-mentioned deletion, or a combination of any two of them. It may be a thing.
- the amount ratio of each deletion can be influenced by the type and culture conditions of cultured mammalian cells producing the antibody according to the present invention, but the main component of the antibody according to the present invention is the carboxyl in both of the two heavy chains. A case where one terminal amino acid residue is deleted can be mentioned.
- Examples of the isotype of the antibody of the present invention include IgG (IgG1, IgG2, IgG3, IgG4) and the like, and preferably IgG1 or IgG2.
- the biological activity of an antibody generally includes an antigen-binding activity, an activity that is internalized in a cell that expresses the antigen by binding to the antigen, an activity that neutralizes the activity of the antigen, an activity that enhances the activity of the antigen, Examples include antibody-dependent cytotoxicity (ADCC) activity, complement-dependent cytotoxicity (CDC) activity, and antibody-dependent cell-mediated phagocytosis (ADCP).
- the function of the antibody according to the present invention is directed to TROP2. It is a binding activity, preferably an activity that is internalized in a TROP2-expressing cell by binding to TROP2.
- the antibody of the present invention may have ADCC activity, CDC activity and / or ADCP activity in addition to cell internalization activity.
- the obtained antibody can be purified to homogeneity. Separation and purification of antibodies may be carried out using separation and purification methods used for ordinary proteins. For example, antibodies can be separated and purified by appropriately selecting and combining column chromatography, filter filtration, ultrafiltration, salting out, dialysis, preparative polyacrylamide gel electrophoresis, isoelectric focusing, etc. (Stratesies) for Protein Purification and Characterization: A Laboratory Course Manual, Daniel R.Marshak et al.eds, Cold Spring Harbor Laboratory Press (1996); Antibodies:. A Laboratory Manual.Ed Harlow and David Lane, Cold Spring Harbor Laboratory (1988)) Is limited to these Not.
- Examples of the chromatography include affinity chromatography, ion exchange chromatography, hydrophobic chromatography, gel filtration chromatography, reverse phase chromatography, adsorption chromatography and the like. These chromatography can be performed using liquid chromatography, such as HPLC and FPLC.
- Examples of the column used for affinity chromatography include a protein A column and a protein G column. For example, as a column using a protein A column, Hyper D, POROS, Sepharose F. F. (Pharmacia) and the like. It is also possible to purify an antibody using a carrier on which an antigen is immobilized, utilizing the binding property to the antigen.
- the antitumor compound bound to the anti-TROP2 antibody-drug conjugate of the present invention is described.
- the antitumor compound used in the present invention is not particularly limited as long as it is a compound having an antitumor effect and has a substituent and a partial structure that can be bonded to a linker structure.
- a part or all of the linker is cleaved in the tumor cell, and the antitumor compound portion is released to exhibit an antitumor effect.
- the linker is cleaved at the binding site with the drug, the antitumor compound is released in an unmodified structure, and its original antitumor effect is exhibited.
- exetecan ((1S, 9S) -1-amino-9-ethyl-5-fluoro-2,3-dihydro-9-hydroxy-4-methyl-, which is a camptothecin derivative, is used.
- Exatecan may be preferably used. Although this exatecan has excellent antitumor activity, it has not been marketed as an antitumor drug.
- the compound can be easily obtained by a known method, and the amino group at the 1-position can be suitably used as a binding site to the linker structure. Exatecan may be released in tumor cells in a state in which a part of the linker is bound, and even if it has such a structure, it is an excellent compound that exhibits an excellent antitumor effect.
- Exatecan has a camptothecin structure, so in an acidic aqueous medium (for example, about pH 3), the equilibrium is biased to a structure in which a lactone ring is formed (ring-closed), whereas in a basic aqueous medium (for example, about pH 10), the lactone ring is It is known that the equilibrium is biased to the ring-opened structure (ring-opened body). Even a drug conjugate into which an exatecan residue corresponding to such a ring-closed structure and a ring-opened structure is introduced is expected to have an equivalent antitumor effect, and any state is included in the scope of the present invention. Needless to say.
- antitumor compounds examples include doxorubicin, daunorubicin, mitomycin C, bleomycin, cyclocytidine, vincristine, vinblastine, methotrexate, platinum antitumor agents (cisplatin or derivatives thereof), taxol or derivatives thereof, other camptothecins or derivatives thereof (Anti-tumor agent described in JP-A-6-87746) and the like.
- antibody-drug conjugates the number of drugs bound to one antibody molecule is an important factor affecting the effectiveness and safety of the antibody-drug conjugate.
- Antibody-drug conjugates are manufactured by specifying reaction conditions such as the amount of raw materials and reagents to be reacted so that the number of drug bonds is constant. What is a chemical reaction of low molecular weight compounds? In contrast, antibody-drug conjugates are usually obtained as a mixture of different numbers of drugs attached. The number of drugs bound to one antibody molecule is specified and expressed as an average value, that is, the average number of drug bonds.
- the number of drug bindings is shown unless an antibody-drug conjugate having a specific drug binding number contained in an antibody-drug conjugate mixture having a different drug binding number is indicated. Mean value.
- the number of Exatecan binding to the antibody molecule is controllable, and about 1 to 10 Exatecan can be bound as the average number of drugs bound per antibody, preferably 2 to 8, more preferably Is from 3 to 8.
- a person skilled in the art can design a reaction for binding the required number of drugs to the antibody from the description of the examples of the present application, and can obtain an antibody-drug conjugate in which the number of bindings of exatecan is controlled. .
- Linker structure A linker structure for binding an antitumor compound to an anti-TROP2 antibody in the anti-TROP2 antibody-drug conjugate of the present invention will be described.
- n 1 represents an integer of 0 to 6, preferably an integer of 1 to 5, and more preferably 1 to 3.
- the position 3 in this partial structure is a binding site to the anti-TROP2 antibody.
- This binding to the antibody at the 3-position is characterized by binding by forming a thioether.
- n 3 is an integer of 2 to 8, preferably 2 to 5.
- N 4 is an integer of 1 to 6, preferably 2 to 4.
- L 2 binds to L 1 at the terminal amino group and binds to L P at the opposite terminal carbonyl group.
- L P L P is a peptide residue composed of 2 to 7 amino acids. That is, it is composed of oligopeptide residues in which 2 to 7 amino acids are peptide-bonded. L P binds to L 2 at the N-terminus and to the amino group of the linker —NH— (CH 2 ) n 1 -L a — (CH 2 ) n 2 —C ( ⁇ O) — moiety at the C-terminus To do.
- the amino acid constituting L P is not particularly limited, and is, for example, an L- or D-amino acid, preferably an L-amino acid.
- amino acids having structures such as ⁇ -alanine, ⁇ -aminocaproic acid, and ⁇ -aminobutyric acid may also be used. There may be.
- the amino acid sequence of L P is not particularly limited, but as constituent amino acids, phenylalanine (Phe; F), tyrosine (Tyr; Y), leucine (Leu; L), glycine (Gly; G), alanine (Ala; A ), Valine (Val; V), lysine (Lys; K), citrulline (Cit), serine (Ser; S), glutamic acid (Glu; E), aspartic acid (Asp; D) and the like.
- phenylalanine, glycine, valine, lysine, citrulline, serine, glutamic acid, and aspartic acid can be preferably used.
- the number of amino acids may be 2 to 7.
- L P As a specific example of L P , -GGF-, -DGGF-, -(D-) D-GGF-, -EGGF-, -GGFG-, -SGGF-, -KGGF-, -DGGFG-, -GGFGG-, -DDGGFG-, -KDGGFG-, -GGFGGGF- Can be mentioned.
- (D-) D means D-aspartic acid.
- Antibodies of the present invention - as particularly preferred L P drug conjugates may include the tetrapeptide residues -GGFG-.
- n 2 is an integer of 0 to 5, preferably 0 to 3, and more preferably 0 or 1.
- L a — (CH 2 ) n 2 —C ( ⁇ O) — includes the following structures.
- the linker —NH— (CH 2 ) n 1 -L a — (CH 2 ) n 2 —C ( ⁇ O) — preferably has a chain length of 4 to 7 atoms, more preferably It has a chain length of 5 or 6 atoms.
- Antitumor derivatives that are released from the antibody-drug conjugate of the present invention and exhibit an antitumor effect include -NH- (CH 2 ) n 1 -L a- (CH 2 ) n 2 of the above-mentioned linkers.
- NH 2 -CH 2 CH 2 -C ( O)-(NH-DX)
- NH 2 -CH 2 CH 2 CH 2 -C ( O)-(NH-DX)
- the drug-linker structure moiety [-L 1 -L 2 -L P -NH- (CH 2 ) n 1 -L a- (CH 2 ) n 2 —C ( ⁇ O) — (NH—DX)] is preferably bound to an antibody.
- These drug-linker structure moieties may be bound to 1 to 10 as an average number of bonds per antibody, but preferably 2 to 8, more preferably 3 to 8.
- the linker structure that binds the anti-TROP2 antibody and the drug can be constructed by linking the preferred structures shown in the above-mentioned linker parts.
- the following structure can be preferably used. The left end of the structure is the binding site with the antibody, and the right end is the binding site with the drug.
- An antibody-drug conjugate represented by formula (1) in which an antibody and a drug-linker structure are bonded via a thioether can be produced, for example, by the following method.
- AB represents an antibody having a sulfhydryl group, L 1 ', in the linker structure represented by L 1, the linker terminal maleimidyl group (following formula)
- the compound of the formula (1) can be interpreted as a structure in which one structural portion from the drug to the linker end is bound to one antibody, but this is for convenience of explanation. In practice, there are many cases in which a plurality of the structural portions are actually bound to one antibody molecule. This situation is the same in the following description of the manufacturing method.
- the antibody-drug conjugate (1) can be produced by reacting the compound (2), which can be obtained by the method described later, with the antibody (3a) having a sulfhydryl group.
- the antibody (3a) having a sulfhydryl group can be obtained by a method well known to those skilled in the art (Hermanson, GT, Bioconjugate Techniques, pp. 56-136, pp. 456-493, Academic Press (1996)).
- Traut's reagent is allowed to act on the amino group of an antibody; N-succinimidyl S-acetylthioalkanoates are allowed to act on the antibody's amino group, and then hydroxylamine is allowed to act; N-succinimidyl 3- (pyridyldithio) ) Propionate is allowed to act and then a reducing agent is allowed to act; a reducing agent such as dithiothreitol, 2-mercaptoethanol, tris (2-carboxyethyl) phosphine hydrochloride (TCEP) is allowed to act on the antibody, and the hinge part in the antibody The disulfide bond is reduced to form a sulfhydryl group; however, it is not limited thereto.
- TCEP as a reducing agent is used in an amount of 0.3 to 3 molar equivalents per one intra-antibody hinge disulfide, and reacted with the antibody in a buffer containing a chelating agent, whereby the intra-antibody hinge Antibodies in which partial disulfides are partially or completely reduced can be obtained.
- the chelating agent include ethylenediaminetetraacetic acid (EDTA) and diethylenetriaminepentaacetic acid (DTPA). These may be used at a concentration of 1 mM to 20 mM.
- EDTA ethylenediaminetetraacetic acid
- DTPA diethylenetriaminepentaacetic acid
- the buffer solution sodium phosphate, sodium borate, sodium acetate solution and the like can be used.
- the antibody (3a) having a partially or completely reduced sulfhydryl group can be obtained by reacting the antibody with TCEP at 4 ° C. to 37 ° C. for 1 to 4 hours.
- the drug-linker moiety can be bound by a thioether bond.
- an antibody-drug conjugate (1) in which 2 to 8 drugs are bound per antibody is obtained. Can be manufactured.
- a solution in which the compound (2) is dissolved may be added to a buffer solution containing the antibody (3a) having a sulfhydryl group and reacted.
- a buffer solution a sodium acetate solution, sodium phosphate, sodium borate, or the like may be used.
- the pH during the reaction is 5 to 9, and more preferably, the reaction may be performed in the vicinity of pH 7.
- an organic solvent such as dimethylsulfoxide (DMSO), dimethylformamide (DMF), dimethylacetamide (DMA), N-methyl-2-pyridone (NMP) can be used.
- the organic solvent solution in which the compound (2) is dissolved may be reacted by adding 1 to 20% v / v to a buffer solution containing the antibody (3a) having a sulfhydryl group.
- the reaction temperature is 0 to 37 ° C., more preferably 10 to 25 ° C., and the reaction time is 0.5 to 2 hours.
- the reaction can be terminated by deactivating the reactivity of the unreacted compound (2) with a thiol-containing reagent.
- Thiol-containing reagents are, for example, cysteine or N-acetyl-L-cysteine (NAC).
- reaction can be completed by adding 1 to 2 molar equivalents of NAC to the compound (2) used and incubating at room temperature for 10 to 30 minutes.
- the produced antibody-drug conjugate (1) was concentrated, buffer exchanged, purified, antibody concentration and the average number of drugs per antibody molecule were measured by the following common operations. Identification can be performed.
- Common operation A Concentration of antibody or antibody-drug conjugate aqueous solution Place the antibody or antibody-drug conjugate solution in a container of Amicon Ultra (50,000 MWCO, Millipore Corporation) and centrifuge (Allegra X-15R, Beckman Coulter). The antibody or antibody-drug conjugate solution was concentrated by centrifugation using 2000 G to 3800 G for 5 to 20 minutes.
- Common operation B Measurement of antibody concentration Using a UV measuring device (Nanodrop 1000, Thermo Fisher Scientific Inc.), antibody concentration was measured according to the method prescribed by the manufacturer.
- This fractionated fraction is placed on the NAP-25 column again, and the gel filtration purification procedure for elution with a buffer solution is repeated 2 to 3 times in total, so that unbound drug linkers and low molecular compounds (Tris (2-carboxyethyl) Antibody-drug conjugates without phosphine hydrochloride (TCEP), N-acetyl-L-cysteine (NAC), dimethyl sulfoxide) were obtained.
- Tris (2-carboxyethyl) Antibody-drug conjugates without phosphine hydrochloride (TCEP), N-acetyl-L-cysteine (NAC), dimethyl sulfoxide were obtained.
- the bound drug concentration in the antibody-drug conjugate can be calculated by measuring the UV absorbance at two wavelengths of 280 nm and 370 nm of the antibody-drug conjugate aqueous solution and performing the following calculation. Since the total absorbance at a certain wavelength is equal to the sum of the absorbances of all the absorbing species present in the system (addition of absorbance), the molar extinction coefficient of the antibody and drug changes before and after conjugation of the antibody and drug. Assuming that there is no antibody, the antibody concentration and the drug concentration in the antibody-drug conjugate are expressed by the following relational expression.
- a 280 represents the absorbance of the antibody-drug conjugate aqueous solution at 280 nm
- a 370 represents the absorbance of the antibody-drug conjugate aqueous solution at 370 nm
- a A, 280 represents the absorbance of the antibody at 280 nm
- a A , 370 indicates the absorbance of the antibody at 370 nm
- AD, 280 indicates the absorbance of the conjugate precursor at 280 nm
- AD, 370 indicates the absorbance of the conjugate precursor at 370 nm
- ⁇ A, 280 is at 280 nm.
- ⁇ A, 370 shows the molar extinction coefficient of the antibody at 370 nm
- ⁇ D, 280 shows the molar extinction coefficient of the conjugate precursor at 280 nm
- ⁇ D, 370 shows the conjugate at 370 nm.
- C a antibody - indicates antibody concentration in drug conjugates
- C D antibodies It shows the drug concentration in the drug conjugate.
- ⁇ A, 280 can be estimated from the amino acid sequence of an antibody by a known calculation method (Protein Science, 1995, vol. 4, 2411-2423).
- ⁇ A, 370 is usually zero.
- C A and C D can be obtained by measuring A 280 and A 370 of the antibody-drug conjugate aqueous solution and substituting these values into equations (I) and (II) to solve the simultaneous equations.
- C D can be drug average binding per antibody determined by the dividing in C A.
- Common operation F Measurement of the average number of drugs per antibody molecule in antibody-drug conjugate (2)
- the average number of drugs per antibody molecule in the antibody-drug conjugate can be determined by high performance liquid chromatography (HPLC) analysis using the following method in addition to the common operation E described above.
- HPLC high performance liquid chromatography
- HPLC analysis HPLC analysis is performed under the following measurement conditions.
- HPLC system Agilent 1290 HPLC system (Agilent Technologies)
- Detector UV absorbance meter (measurement wavelength: 280 nm)
- Column PLRP-S (2.1 ⁇ 50 mm, 8 ⁇ m, 1000 mm; Agilent Technologies, P / N PL 1912-1802)
- Mobile phase A 0.04% trifluoroacetic acid (TFA) aqueous solution
- Mobile phase B acetonitrile solution containing 0.04% TFA
- Gradient program 29% -36% (0 min-12.5 min), 36% -42 % (12.5-15 minutes), 42% -29% (15 minutes-15.1 minutes), 29% -29% (15.1 minutes-25 minutes)
- Sample injection volume 15 ⁇ L [F-3.
- the detection peak can be assigned to any one of L 0 , L 1 , H 0 , H 1 , H 2 , and H 3 by comparing the holding times with L 0 and H 0 .
- the peak area value is corrected according to the following formula using the molar extinction coefficient of the L chain, H chain, and drug linker according to the number of bonds of the drug linker. .
- the molar extinction coefficient (280 nm) of the L chain and H chain in each antibody is determined by the known calculation method (Protein Science, 1995, vol.4, 2411-2423). Values deduced from the sequence can be used. In the case of hTINA, 34690 was used as the L chain molar extinction coefficient and 95000 as the estimated value of the H chain molar extinction coefficient according to the amino acid sequence.
- the molar extinction coefficient (280 nm) of the drug linker is the measured molar extinction coefficient (280 nm) of the compound obtained by reacting each drug linker with mercaptoethanol or N-acetylcysteine and converting the maleimide group to succinimide thioether. It was. [F-3-3]
- the ratio (%) of each chain peak area to the total peak area correction value is calculated according to the following formula.
- n 3 represents an integer from 2 to 8
- L 2 represents —NH— (CH 2 CH 2 —O) n 4 —CH 2 CH 2 —C ( ⁇ O) — or a single bond
- n 4 represents an integer of 1 to 6
- L P represents a peptide residue composed of 2 to 7 amino acids selected from phenylalanine, glycine, valine, lysine, citrulline, serine, glutamic acid, aspartic acid
- n 1 represents an integer of 0 to 6
- n 2 represents an integer of 0 to 5
- L a represents -O- or a single bond
- L 2 is a single bond or, when —NH— (CH 2 CH 2 —O) n 4 —CH 2 CH 2 —C ( ⁇ O) —, n 4 is an integer from 2 to 4.
- the peptide residue of L P phenylalanine, glycine, valine, lysine, citrulline, serine, glutamic acid, compound is a peptide residue consisting of the amino acid selected from aspartic acid is preferred as a production intermediate.
- a compound in which L P is a peptide residue composed of 4 amino acids is preferred as a production intermediate. More specifically, the compound L P is a tetrapeptide residue of -GGFG- is preferred as a production intermediate.
- -NH- (CH 2) n 1 -L a - (CH 2) n 2 - The, -NH-CH 2 CH 2 - , - NH-CH 2 CH 2 CH 2 -, - NH-CH 2 CH 2 CH 2 CH 2 -, - NH-CH 2 CH 2 CH 2 CH 2 CH 2 -, - NH-CH 2 -O-CH 2 -, or -NH-CH 2 CH 2 -O- CH 2 - with Certain compounds are preferred as production intermediates, and more preferably as —NH—CH 2 CH 2 CH 2 —, —NH—CH 2 —O—CH 2 —, or —NH—CH 2 CH 2 —O—CH 2 It is a certain compound.
- n 3 is an integer of 2 to 5
- L 2 is a single bond
- —NH— (CH 2 ) n 1 -L a — (CH 2 ) n 2 - is, -NH-CH 2 CH 2 - , - NH-CH 2 CH 2 CH 2 -, - NH-CH 2 CH 2 CH 2 CH 2 -, - NH-CH 2 CH 2 CH 2 CH 2 CH 2 -, —NH—CH 2 —O—CH 2 —, or —NH—CH 2 CH 2 —O—CH 2 — is preferred as a production intermediate.
- -NH- (CH 2) n 1 -L a - (CH 2) n 2 - is, -NH-CH 2 CH 2 - , - NH-CH 2 CH 2 CH 2 -, - NH-CH 2 —O—CH 2 — or —NH—CH 2 CH 2 —O—CH 2 —.
- the compound whose n ⁇ 3 > is the integer 2 or 5 is preferable.
- n 3 is an integer of 2 to 5
- L 2 is —NH— (CH 2 CH 2 —O) n 4 —CH 2 CH 2 —C ( ⁇ O).
- n 4 is an integer from 2 to 4 and -NH- (CH 2 ) n 1 -L a- (CH 2 ) n 2 -is -NH-CH 2 CH 2- , -NH- CH 2 CH 2 CH 2- , -NH-CH 2 CH 2 CH 2 CH 2- , -NH-CH 2 CH 2 CH 2 CH 2- , -NH-CH 2 CH 2 CH 2 CH 2- , -NH-CH 2 CH 2 CH 2 CH 2- , -NH-CH 2 -O-CH 2- , or- A compound that is NH—CH 2 CH 2 —O—CH 2 — is preferred as a production intermediate.
- n 4 is an integer of 2 or 4.
- -NH- (CH 2 ) n 1 -L a- (CH 2 ) n 2 - is substituted with -NH-CH 2 CH 2 CH 2- , -NH-CH 2 -O-CH 2- , or -NH Compounds that are —CH 2 CH 2 —O—CH 2 — are preferred.
- a drug-linker compound selected from the group of production intermediate compounds described above is reacted with an anti-TROP2 antibody or a reactive derivative thereof to form a thioether bond at a disulfide bond portion present in the hinge part of the anti-TROP2 antibody.
- Inventive anti-TROP2 antibody-drug conjugates can be prepared.
- a reactive derivative of the anti-TROP2 antibody is preferably used, and a reactive derivative obtained by reducing the anti-TROP2 antibody is particularly preferable.
- conjugates for example, about ⁇ 1 having the same average number of drugs prepared under the same conditions into a new lot. it can. In that case, the average number of drugs falls within the average number of drugs before mixing.
- L 1 ′ represents a terminal maleimidyl group
- P 1 , P 2 and P 3 represent protecting groups.
- the compound (6) is obtained by derivatizing the carboxylic acid (5) into an active ester, mixed acid anhydride, acid halide or the like and reacting with NH 2 -DX (4) or a pharmacologically acceptable salt thereof in the presence of a base.
- NH 2 -DX (4) is exatecan (chemical name: (1S, 9S) -1-amino-9-ethyl-5-fluoro-2,3-dihydro-9-hydroxy-4-methyl-1H, 12H- Benzo [de] pyrano [3 ′, 4 ′: 6,7] indolidino [1,2-b] quinoline-10,13 (9H, 15H) -dione).
- reaction reagents and conditions usually used for peptide synthesis may be applied mutatis mutandis.
- active esters There are various types of active esters.
- phenols such as p-nitrophenol, N-hydroxybenzotriazole or N-hydroxysuccinimide, and carboxylic acid (5) are mixed with N, N′-dicyclohexylcarbodiimide or 1 It can be produced by reacting with a condensing agent such as -ethyl-3- (3-dimethylaminopropyl) carbodiimide / hydrochloride.
- the active ester is a reaction of carboxylic acid (5) with pentafluorophenyl trifluoroacetate or the like; a reaction of carboxylic acid (5) with 1-benzotriazolyloxytripyrrolidinophosphonium hexafluorophosphite; Reaction of (5) with diethyl cyanophosphonate (salt insertion method); Reaction of carboxylic acid (5) with triphenylphosphine and 2,2′-dipyridyl disulfide (Mukayama method); Carboxylic acid (5) and 4- ( It can also be produced by a reaction with a triazine derivative such as 4,6-dimethoxy-1,3,5-triazin-2-yl) -4-methylmorpholinium chloride (DMTMM); The reaction can also be performed by an acid halide method or the like that can be produced by treating carboxylic acid (5) with an acid halide such as thionyl chloride or oxalyl chloride in
- the base used in each of the above steps include alkali metals or alkalis such as sodium carbonate, potassium carbonate, sodium ethoxide, potassium butoxide, sodium hydroxide, potassium hydroxide, sodium hydride, potassium hydride and the like.
- inert solvent used in this reaction examples include halogenated hydrocarbon solvents such as dichloromethane, chloroform and carbon tetrachloride; ether solvents such as tetrahydrofuran, 1,2-dimethoxyethane and dioxane; aromatics such as benzene and toluene.
- halogenated hydrocarbon solvents such as dichloromethane, chloroform and carbon tetrachloride
- ether solvents such as tetrahydrofuran, 1,2-dimethoxyethane and dioxane
- aromatics such as benzene and toluene.
- Hydrocarbon solvents such as N, N-dimethylformamide, N, N-dimethylacetamide, N-methylpyrrolidin-2-one; and in addition to these, dimethyl sulfoxide, sulfolane It is also possible to use sulfoxide solvents such as; ketone solvents such as acetone and methyl ethyl ketone; alcohol solvents such as methanol and ethanol. Furthermore, these can also be mixed and used.
- Examples of the protecting group P 1 for the terminal amino group of the compound (6) include tert-butyloxycarbonyl group, 9-fluorenylmethyloxycarbonyl group, benzyloxycarbonyl group, and other amino groups usually used for peptide synthesis. Protecting groups can be used.
- protecting groups for amino groups include alkanoyl groups such as acetyl groups; alkoxycarbonyl groups such as methoxycarbonyl groups and ethoxycarbonyl groups; aryls such as paramethoxybenzyloxycarbonyl groups and para (or ortho) nitrobenzyloxycarbonyl groups Methoxycarbonyl group; arylmethyl group such as benzyl group and triphenylmethyl group; aroyl group such as benzoyl group; arylsulfonyl group such as 2,4-dinitrobenzenesulfonyl group and orthonitrobenzenesulfonyl group;
- the protecting group P 1 may be selected according to the properties of the compound protecting the amino group.
- Compound (7) can be produced by deprotecting the protecting group P 1 of the terminal amino group of the obtained compound (6). For this deprotection, a reagent and conditions corresponding to the protecting group may be selected.
- the compound (9) can be produced by inducing the peptide carboxylic acid (8) whose N-terminal is protected with P 2 to an active ester, mixed acid anhydride or the like and reacting with the resulting compound (7).
- Reaction conditions, reagents, bases and inert solvents for forming peptide bonds between peptide carboxylic acid (8) and compound (7) may be appropriately selected from those described in the synthesis of compound (6). .
- the protecting group P 2 may be appropriately selected from those described for the protecting group of the compound (6) and used depending on the properties of the compound protecting the amino group. Further, as is usually used in peptide synthesis, the amino acid or peptide constituting peptide carboxylic acid (8) can be sequentially subjected to reaction and deprotection and extended to produce compound (9).
- Compound (10) can be produced by deprotecting amino-protecting group P 2 of the resulting compound (9). For this deprotection, a reagent and conditions corresponding to the protecting group may be selected.
- Compound (2) or can be produced by derivatizing carboxylic acid (11) into an active ester, mixed acid anhydride, acid halide or the like and reacting with the resulting compound (10). The reaction conditions, reagents, base, and inert solvent for forming a peptide bond between the carboxylic acid (11) and the compound (10) may be appropriately selected from those described in the synthesis of the compound (6).
- Compound (9) can also be produced, for example, by the following method.
- Peptide carboxylic acid (8) whose N-terminal is protected with P 2 is derived into an active ester, mixed acid anhydride, etc., and is reacted with an amine compound (12) whose carboxy group is protected with P 3 in the presence of a base. (13) can be manufactured.
- Reaction conditions, reagents, bases, and inert solvents for forming peptide bonds between peptide carboxylic acid (8) and compound (12) may be appropriately selected from those described in the synthesis of compound (6). .
- the amino-protecting group P 2 of the compound (13) may be protected with a commonly used protecting group.
- hydroxyl-protecting groups include alkoxymethyl groups such as methoxymethyl groups; arylmethyl groups such as benzyl groups, 4-methoxybenzyl groups and triphenylmethyl groups; alkanoyl groups such as acetyl groups; benzoyl groups and the like An aroyl group; a silyl group such as a tert-butyldiphenylsilyl group; and the like.
- the carboxy group can be protected as an ester with an alkyl group such as a methyl group, an ethyl group, or a tert-butyl group, an allyl group, or an arylmethyl group such as a benzyl group.
- the amino group is an alkyloxycarbonyl group such as tert-butyloxycarbonyl group, methoxycarbonyl group, ethoxycarbonyl group; allyloxycarbonyl group, or 9-fluorenylmethyloxycarbonyl group, benzyloxycarbonyl group, paramethoxybenzyloxy Arylmethoxycarbonyl group such as carbonyl group, para (or ortho) nitrobenzyloxycarbonyl group; alkanoyl group such as acetyl group; arylmethyl group such as benzyl group and triphenylmethyl group; aroyl group such as benzoyl group; And arylsulfonyl groups such as 2,4-dinitrobenzenesulfonyl group and orthonitrobenzenesulfonyl group.
- alkyloxycarbonyl group such as tert-butyloxycarbonyl group, methoxycarbonyl group, ethoxycarbonyl group; ally
- a protective group usually used as a protective group for the carboxy group in organic synthetic chemistry, particularly peptide synthesis may be used.
- a methyl group, an ethyl group, or a tert-butyl group may be used.
- alkyl esters, allyl esters, benzyl esters, etc. which may be selected from the above protecting groups.
- the protecting group for the amino group and the protecting group for the carboxy group can be removed by different methods or conditions.
- a typical combination includes a combination in which P 2 is a tert-butyloxycarbonyl group and P 3 is a benzyl group.
- protecting groups may be selected from those described above according to the properties of the compounds protecting amino groups and carboxy groups, and when cleaving these protecting groups, reagents and conditions corresponding to the protecting groups may be selected. Good.
- the compound (14) can be produced by deprotecting the protecting group P 3 of the carboxy group of the obtained compound (13). For this deprotection, a reagent and conditions corresponding to the protecting group may be selected.
- the compound (9) can be produced by derivatizing the obtained compound (14) into an active ester, mixed acid anhydride, acid halide or the like and reacting with the compound (4) in the presence of a base.
- reaction reagents and conditions usually used for peptide synthesis may be applied mutatis mutandis, and reaction conditions, reagents, bases, and inert solvents may be appropriately selected from those described in the synthesis of compound (6). .
- Compound (2) can also be produced, for example, by the following method.
- Compound (15) can be produced by deprotecting amino-protecting group P 2 of compound (13). For this deprotection, a reagent and conditions corresponding to the protecting group may be selected.
- Compound (16) can be produced by derivatizing carboxylic acid derivative (11) into an active ester, mixed acid anhydride, acid halide or the like and reacting with the obtained compound (15) in the presence of a base. .
- the reaction conditions, reagents, base, and inert solvent for forming an amide bond between peptide carboxylic acid (11) and compound (15) may be appropriately selected from those described in the synthesis of compound (6). .
- the compound (17) can be produced by deprotecting the protecting group of the carboxy group of the obtained compound (16). This deprotection can be carried out in the same manner as the deprotection of the carboxy group in the production of compound (14).
- Compound (2) can be produced by inducing compound (17) to an active ester, mixed acid anhydride, acid halide or the like and reacting with compound (4) in the presence of a base. For this reaction, reaction reagents and conditions usually used for peptide synthesis may be applied mutatis mutandis, and reaction conditions, reagents, bases, and inert solvents may be appropriately selected from those described in the synthesis of compound (6). .
- L 1 ' is the L 1 terminal is converted into maleimidyl group structure
- P 4 is a protecting group
- Compound (19) can be produced by inducing compound (11) to an active ester, mixed acid anhydride, etc., and reacting with peptide carboxylic acid (18) whose C-terminal is protected with P 4 in the presence of a base.
- Reaction conditions, reagents, bases and inert solvents for forming peptide bonds between peptide carboxylic acid (18) and compound (11) may be appropriately selected from those described in the synthesis of compound (6).
- the protecting group P 4 for the carboxy group of the compound (18) may be appropriately selected from the protecting groups described above.
- the compound (20) can be produced by deprotecting the protecting group of the carboxy group of the obtained compound (19).
- This deprotection can be carried out in the same manner as the deprotection of the carboxy group in the production of compound (14).
- the compound (2) can be produced by inducing the obtained compound (20) to an active ester or a mixed acid anhydride and reacting with the compound (7).
- reaction reagents and conditions usually used for peptide synthesis may be applied mutatis mutandis, and reaction conditions, reagents, bases, and inert solvents may be appropriately selected from those described in the synthesis of compound (6). .
- L P represents the same as described above, L represents an acyl group, an alkanoyl group such as an acetyl group or an aroyl group such as a benzoyl group, or a hydrogen atom, and X and Y are An oligopeptide consisting of 1 to 3 amino acids, P 5 and P 7 are amino-protecting groups, and P 6 is a carboxy-protecting group.
- the compound represented by the formula (21) is a method described in JP-A No. 2002-60351, a method described in literature (J. Org. Chem., 51, 3196, 1986), or an application of the method. And it can manufacture by removing a protective group and functional group conversion as needed. In addition, it can be obtained by treating an amino acid having a protected terminal amino group or an acid amide of an oligopeptide having a protected amino group with an aldehyde or a ketone.
- Compound (23) can be produced by reacting compound (21) with compound (22) having a hydroxyl group in an inert solvent in the presence of an acid or a base under cooling and at room temperature.
- acids examples include inorganic acids such as hydrofluoric acid, hydrochloric acid, sulfuric acid, nitric acid, phosphoric acid, and boric acid; organic acids such as acetic acid, citric acid, paratoluenesulfonic acid, and methanesulfonic acid; tetrafluoro And Lewis acids such as borate, zinc chloride, tin chloride, aluminum chloride, and iron chloride.
- inorganic acids such as hydrofluoric acid, hydrochloric acid, sulfuric acid, nitric acid, phosphoric acid, and boric acid
- organic acids such as acetic acid, citric acid, paratoluenesulfonic acid, and methanesulfonic acid
- tetrafluoro And Lewis acids such as borate, zinc chloride, tin chloride, aluminum chloride, and iron chloride.
- sulfonic acids particularly paratoluenesulfonic acid, are preferred.
- the base may be appropriately selected from the above-mentioned bases, particularly alkali metal alkoxides such as potassium tert-butoxide; alkali metal hydroxides such as sodium hydroxide and potassium hydroxide; hydrogenation Preferred are alkali metal hydrides such as sodium and potassium hydride; organometallic bases typified by dialkylaminolithium such as lithium diisopropylamide; organometallic bases of bissilylamines such as lithium bis (trimethylsilyl) amide; As the solvent used in the reaction, ether solvents such as tetrahydrofuran and 1,4-dioxane; aromatic hydrocarbon solvents such as benzene and toluene; The above solvent may be a mixture with water.
- alkali metal alkoxides such as potassium tert-butoxide
- alkali metal hydroxides such as sodium hydroxide and potassium hydroxide
- the amino protecting group exemplified in P 5 is not particularly limited as long as it is a group usually used for protecting an amino group.
- the amino-protecting groups described in Production Method 2 can be mentioned, but the amino-protecting groups exemplified in P 5 may be cleaved during this reaction. In that case, the protecting group may be introduced again by reacting with an appropriate amino group protecting reagent as needed.
- Compound (24) can be produced by removing protecting group P 6 of compound (23).
- protecting group for the carboxy group exemplified as P 6 those representative in Production Method 2 are described, and can be appropriately selected therefrom.
- protecting group P 6 of the protecting group P 5 and the carboxy group of the amino group is a protecting group which can be removed in different ways, or condition.
- a representative combination is a combination in which P 5 is a 9-fluorenylmethyloxycarbonyl group and P 6 is a benzyl group.
- Those protecting groups may be selected according to the properties of the compound protecting the amino group and carboxy group, and reagents and conditions corresponding to the protecting groups may be selected when removing these protecting groups.
- the compound (25) is produced by derivatizing the carboxylic acid (24) into an active ester, mixed acid anhydride, acid halide or the like and reacting with the compound (4) or a pharmacologically acceptable salt thereof in the presence of a base. Then, the compound (26) can be produced by removing the protecting group P 5 of the obtained compound (25). In the reaction between the compound (4) and the carboxylic acid (24) and the reaction for removing the protecting group P 6 , the same reagents and reaction conditions as described in Production Method 2 may be used.
- Compound (9b) is produced by reacting compound (26) with an amino acid with a terminal amino group protected or an oligopeptide (27) with an amino group protected, and protecting group P 7 of compound (9b) obtained is reacted.
- Compound (10b) can be produced by removing.
- the amino protecting group shown as P 7 is not particularly limited as long as it is usually a group used for protecting an amino group, and representative examples include the amino protecting group described in Production Method 2.
- a reagent and conditions corresponding to the protecting group may be selected.
- reaction reagents and conditions usually used for peptide synthesis may be applied mutatis mutandis.
- the compound (10b) produced by the above method can be led to the compound (1) of the present invention according to the above production method.
- the anti-TROP2 antibody-drug conjugate of the present invention may be left in the atmosphere or purified by recrystallization or the like to absorb water, adsorbed water, or become a hydrate. Yes, such water-containing compounds and salts are also encompassed by the present invention.
- the present invention also includes compounds labeled with various radioactive or non-radioactive isotopes.
- One or more of the atoms making up the antibody-drug conjugate of the present invention may also contain unnatural proportions of atomic isotopes. Examples of atomic isotopes include deuterium ( 2 H), tritium ( 3 H), iodine-125 ( 125 I), carbon-14 ( 14 C), and the like.
- the compound of the present invention can be radiolabeled with a radioisotope such as tritium ( 3 H), iodine-125 ( 125 I) or carbon-14 ( 14 C).
- Radiolabeled compounds are useful as therapeutic or prophylactic agents, research reagents such as assay reagents, and diagnostic agents such as in vivo diagnostic imaging agents. All isotope variants of the antibody-drug conjugates of the present invention, whether radioactive or not, are encompassed within the scope of the present invention.
- the anti-TROP2 antibody-drug conjugate of the present invention exhibits cytotoxic activity against cancer cells, it can be used as a pharmaceutical, particularly as a therapeutic and / or prophylactic agent for cancer. That is, the anti-TROP2 antibody-drug conjugate of the present invention can be selected and used as a drug for chemotherapy, which is a main therapeutic method for cancer treatment, and as a result, cancer cell growth is delayed and Can be suppressed, and cancer cells can be destroyed.
- chemotherapy which is a main therapeutic method for cancer treatment
- cancer cell growth is delayed and Can be suppressed, and cancer cells can be destroyed.
- the anti-TROP2 antibody-drug conjugate of the present invention can be used not only as a drug alone but also as a drug combined with other therapies in adjuvant therapy, and can be combined with surgery, radiation therapy, hormone therapy and the like. Furthermore, it can also be used as a drug for pharmacotherapy in neoadjuvant therapy. In addition to the therapeutic use as described above, it can be expected that the antibody binds to a finely metastasized cancer cell by binding to an antigen, suppresses the growth of the cancer cell, and further destroys it.
- suppression of cancer metastasis and prevention can be expected by administering the anti-TROP2 antibody-drug conjugate of the present invention.
- an effect of suppressing and destroying cancer cells in a body fluid during a metastasis process and an effect of suppressing and destroying fine cancer cells immediately after implantation in any tissue can be expected.
- suppression of cancer metastasis, especially after surgical removal of cancer can be expected, and these can be expected to suppress cancer metastasis.
- the anti-TROP2 antibody-drug conjugate of the present invention can be administered to a patient systemically as a systemic therapy or locally to a cancer tissue to expect a therapeutic effect.
- the types of cancer to which the anti-TROP2 antibody-drug conjugate of the present invention is applied include lung cancer, renal cancer, urothelial cancer, colon cancer, prostate cancer, glioblastoma multiforme, ovarian cancer, pancreatic cancer, breast cancer, Examples include melanoma, liver cancer, bladder cancer, gastric cancer, cervical cancer, head and neck cancer, esophageal cancer, etc., but a protein capable of recognizing the antibody in the antibody-drug conjugate is expressed in cancer cells to be treated. It is not limited to these as long as they are cancer cells.
- the anti-TROP2 antibody-drug conjugate of the present invention can be suitably administered to a mammal, but is more preferably a human.
- the substance used in the pharmaceutical composition containing the anti-TROP2 antibody-drug conjugate of the present invention is appropriately selected and applied from the formulation additives and the like commonly used in this field at the dosage and concentration. Can do.
- the anti-TROP2 antibody-drug conjugate of the present invention can be administered as a pharmaceutical composition comprising one or more pharmaceutically compatible ingredients.
- the pharmaceutical composition typically includes one or more pharmaceutical carriers (eg, sterile liquids).
- Liquids here include, for example, water and oils (oils of petroleum, animal origin, plant origin or synthetic origin).
- the oil may be, for example, peanut oil, soybean oil, mineral oil, sesame oil and the like.
- Water is a more typical carrier when the pharmaceutical composition is administered intravenously.
- Saline solutions and aqueous dextrose and glycerol solutions can also be employed as liquid carriers, particularly for injectable solutions. Suitable pharmaceutical excipients are known in the art.
- composition can also contain minor amounts of wetting or emulsifying agents, or pH buffering agents, if desired.
- suitable pharmaceutical carriers are E. W. It is described in “Remington's Pharmaceutical Sciences” by Martin. The formulation corresponds to the mode of administration.
- Introduction methods can include, but are not limited to, intradermal, intramuscular, intraperitoneal, intravenous, and subcutaneous routes. Administration can be, for example, by infusion or bolus injection. In certain preferred embodiments, administration of the ligand drug conjugate is by infusion. Parenteral administration is the preferred route of administration.
- the pharmaceutical composition is formulated according to routine procedures as a pharmaceutical composition adapted for intravenous administration to humans.
- compositions for intravenous administration are solutions in sterile isotonic aqueous buffer.
- the medicament may also include a solubilizing agent and a local anesthetic (eg, lignocaine) to relieve pain at the site of injection.
- a local anesthetic eg, lignocaine
- the ingredients are either separately or together in a unit dosage form, for example, as a dry lyophilized powder or anhydrous concentrate in a hermetically sealed container such as an ampoule or sachet indicating the amount of active agent Mixed and supplied either.
- the medicament is in a form to be administered by infusion, it can be dispensed, for example, with an infusion bottle containing sterile pharmaceutical grade water or saline.
- an ampule of sterile water for injection or saline can be provided, for example, so that the ingredients can be mixed prior to administration.
- the pharmaceutical composition of the present invention may be a pharmaceutical composition containing only the anti-TROP2 antibody-drug conjugate of the present application, or a pharmaceutical containing the anti-TROP2 antibody-drug conjugate and at least one other cancer therapeutic agent. It may be a composition.
- the anti-TROP2 antibody-drug conjugate of the present invention can also be administered together with other cancer therapeutic agents, thereby enhancing the anticancer effect.
- Other anticancer agents used for such a purpose may be administered to an individual simultaneously or separately with the antibody-drug conjugate, or may be administered at different intervals. .
- cancer therapeutic agents abraxane, paclitaxel, cisplatin, gemcitabine, irinotecan (CPT-11), paclitaxel, pemetrexed, sorafenib, vinblastin, or an analog described in International Publication No. 2003/038043 H Prorelin, goserelin, etc.), estramustine phosphate, estrogen antagonists (tamoxifen, raloxifene, etc.), aromatase inhibitors (anastrozole, letrozole, exemestane, etc.), etc. There is no limitation as long as the drug has.
- Such a pharmaceutical composition may be formulated as a freeze-dried preparation or a liquid preparation as a preparation having the selected composition and the required purity.
- a pharmaceutical composition When formulated as a lyophilized formulation, it may be a formulation containing appropriate formulation additives used in this field.
- liquid preparations can be formulated as liquid preparations containing various preparation additives used in this field.
- the anti-TROP2 antibody-drug conjugate contained in the pharmaceutical composition of the present invention has the affinity of the antibody-drug conjugate for the antigen, that is, dissociation for the antigen.
- Kd value the higher the affinity (the lower the Kd value), the more effective the drug can be exerted even with a small dose. Therefore, in determining the dose of the antibody-drug conjugate, the dose can be set based on the affinity state between the antibody-drug conjugate and the antigen.
- the antibody-drug conjugate of the present invention is administered to humans, for example, about 0.001 to 100 mg / kg may be administered once or several times at intervals of 1 to 180 days. .
- Example 1 Immunization of mice and acquisition of hybridoma
- 5 ⁇ 10 6 NCI-H322 cells human non-small cell lung cancer cell line, ATCC CRL-5806; ATCC: American Type Culture Collection
- RPMI-1640 Roswell Park Memorial
- PBS phosphate buffered saline
- PBS phosphate buffered saline
- mice BALB / c mice (6 weeks old) were immunized intraperitoneally with NCI-H322 cells (1 ⁇ 10 7 cells) for the first immunization.
- the second to fifth rounds were immunized intraperitoneally with 1 ⁇ 10 6 NCI-H322 cells at weekly intervals.
- NCI-H322 cells were immunized with 1 ⁇ 10 6 cells / 200 ⁇ l PBS into the tail vein and the abdominal cavity, respectively. Spleen cells were removed 3 days after the final immunization.
- RPMI 1640FBS (+) medium (10 ml) was added to the cell suspension, passed through a cell strainer, and centrifuged at 1500 rpm for 5 minutes at room temperature. The supernatant was discarded and the spleen cells were resuspended in RPMI 1640 FBS ( ⁇ ) medium (10 ml).
- P3U1 cells mouse myeloma cell line
- EDTA (0.02%) solution 10 ml
- the P3U1 cell suspension was centrifuged at 1500 rpm for 5 minutes at room temperature.
- the supernatant was discarded and resuspended in RPMI 1640 FBS ( ⁇ ) medium (10 ml).
- NCI-H322 cells treated with ⁇ -Gal reporter gene assay using Galacto-Light Plus Reporter Gene Assay System were washed with 200 ⁇ l / well of PBS, and Lysis Solution was added at 50 ⁇ l / well. Allowed to stand for 10 minutes.
- This cell lysate (10 ⁇ L) was diluted 100-fold with Galacton-Plus Galacto Reaction Buffer Diluent, added to White microwell SH 96 well plate (Nunc), and reacted at room temperature for 1 hour.
- Accelerator II was added at 150 ⁇ l / well, chemiluminescence was measured for 5 seconds using a multi-label counter Wallac 1420ARVOsx (Perkin Elmer), and the average value per second was determined as RLU (luminescence) to NCI-H322 cells. The amount of virus infection was expressed.
- RLU measured value
- clones having a measured value (RLU) of 5000 RLU or more were selected from the entire group (minimum value 1383 RLU, average value 10914 RLU, maximum value 78746 RLU).
- 81 positive wells were selected from 960 well hybridoma wells obtained by one cell fusion.
- the assay was performed in duplicate by the same method as in the primary screening, and both wells with measured values of 5000 RLU or more were made positive, and 81 to 52 wells obtained in the primary screening. Positive wells were selected. The selected clones were subcloned 2 to 4 times to establish 44 monoclonal hybridoma cell lines.
- Example 2 Purification of antibody from hybridoma
- pristane Pristane; 2,6,10,14-tetramethylpentadecane; 0.5 ml
- Monoclonal antibody-producing hybridomas were injected intraperitoneally.
- ascites was collected after 10 to 21 days after the hybridoma was changed to ascites tumor.
- the resulting ascites was centrifuged to remove solids, salted out with 40-50% ammonium sulfate, purified by caprylic acid precipitation method, DEAE-Sepharose column, protein G-column, and IgG or IgM fraction was obtained. Collected and used as a purified monoclonal antibody.
- Example 3 Identification of antigen bound by antibody produced by hybridoma
- TINA1 is one of the antibodies produced by the hybridoma prepared in Example 2.
- the washed cells were resuspended in lysis buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.6, 1% NP-40 + Protease inhibitor, Complete EDTA free (Roche) 1 tablet / 50 ml; 2 ml) at 4 ° C. For 30 minutes.
- Protein G Sepharose / Protein G Sepharose / lysis buffer (50% slurry; 30 ⁇ l) obtained by substituting the buffer of Protein G Sepharose (Protein G Sepharose 4 Fast Flow (GE Healthcare)) with the lysis buffer was added to the cell lysis solution at 4 ° C. And then centrifuged at 4 ° C. for 5 minutes, and the supernatant was collected.
- TINA1 antibody (3 ⁇ g) was added to the supernatant, and the mixture was rotated at 4 ° C. for 1 hour, and then Protein G Sepharose / lysis buffer (50% slurry; 60 ⁇ l) was added and rotated at 4 ° C. for 2 hours.
- Protein G Sepharose 6 was was was rotated at 4 ° C. for 1 hour, and then Protein G Sepharose / lysis buffer (50% slurry; 60 ⁇ l) was added and rotated at 4 ° C. for 2 hours.
- lysis buffer 1 ml
- 1 ⁇ SDS sample buffer / 5% 2-ME (2-mercaptoethanol) buffer (62.5 mM Tris-HCl (pH 6.8 at 25 ° C.), 2 % (W / v) SDS, 10% glycerol and 0.01% (w / v) phenol red).
- SDS-PAGE polyacrylamide gel electrophoresis
- the membrane was washed 4 times for 10 minutes with PBS-T, and then the target band was detected using ECL Western blotting detection reagents (Amersham) and Hyperfilm ECL (Amersham).
- ECL Western blotting detection reagents Amersham
- Hyperfilm ECL Hyperfilm ECL
- An immunoprecipitate obtained by immunoprecipitation of NCI-H322 cells labeled with biotin according to the procedure of Example 3-1) with KCI7A3 antibody and TINA1 antibody whose antigen was already determined to be TROP2 by mass spectrometry was obtained. Analysis was performed by SDS-PAGE and Western blotting with or without DTT added, respectively.
- TINA1 antibody Since it was predicted from the band pattern that the antigen of TINA1 antibody was TROP2, forced expression analysis by gene introduction of cDNA was performed without mass spectrometry. As a result of FACS analysis, TINA1 antibody showed a strong positive reaction in human TROP2-expressing CHOK1 cells, indicating that the antigen of TINA1 antibody is TROP2.
- lung cancer cell line PC14 lung cancer cell line NCI-H322, lung cancer cell line NCI-H2122, lung cancer cell line LCAM1, lung cancer cell line LC2 / ad, pancreatic cancer cell line MIAPaCa2, pancreatic cancer cell line PK-1, prostate cancer cell line PC3
- colon cancer cell line HCT116 melanoma cell line A375
- FACS analysis was performed. All the lung cancer cell lines examined were TROP2-positive, and PC3, PK1, and SKOV3 were positive in cell lines other than lung cancer. On the other hand, all normal blood cells were negative.
- Antibody internalization activity evaluation system DT3C a recombinant complex protein, was prepared for the purpose of antibody internalization and measurement of immunotoxin activity.
- This DT3C is a protein having three catalytic domains of diphtheria toxin (DT) and three antibody binding domains of protein G.
- DT3C specifically binds to the Fc part of the antibody and is stable, and when it is taken into cells, it induces cell death by inhibiting protein synthesis.
- antibody internalization and cytocidal effect by immunotoxin can be observed simultaneously (Yamaguchi, M., Hamada, H., et al., Biochemical and Biophysical Research Communications 454 (2014) 600- 603).
- NCI-H322 cells 50 ⁇ L of 2 ⁇ 10 4 cells / mL (RPMI1640 medium supplemented with 20% Low IgG FBS) were seeded, incubated at room temperature for 30 minutes, and then cultured in a 37 ° C. CO 2 incubator for 3 days. The supernatant after culturing, 10% WST-10% FBS -RPMI1640 the (100 [mu] L) was added, 37 ° C. CO 2 1 hour incubation in an incubator, the number of viable cells microplate reader (OD 450 ⁇ OD 640, infinite200 , Tecan Japan Co., Ltd.). In the culture supernatant of the evaluated hybridoma cells, strong internalization and immunotoxin activity were observed with antibodies against CD59, CD71, EGFR, EpCAM, and TROP2 (FIG. 10).
- Example 4 Difference in internalization and immunotoxin activity of anti-TROP2 antibody in each clone Example 1 or TINA1 (immunogen: lung cancer strain NCI-H322), KCL7A3, and KCL2D6 (immunogen: Pancreatic cancer strain KCL-MOH1), Pr1E11 and Pr8H10 (immunogen: prostate cancer strain Pc-1), NY16 and NY17 (immunogen: pancreatic cancer strain PK-1) and commercially available 77220 (R & D System) anti-TROP2 antibodies was evaluated in the same manner as in Example 4-3). As a result, the TINA1 antibody had the strongest activity among the eight anti-TROP2 antibodies (FIG. 12).
- Example 5 Determination of nucleotide sequence of cDNA encoding variable region of TINA1 antibody gene and production of chimeric TINA1 (hereinafter, cTINA1) antibody] 5-1) Determination of nucleotide sequence of cDNA encoding variable region of TINA1 antibody gene 5-1-1) Preparation of mRNA from TINA1 antibody-producing hybridoma Production of TINA1 antibody to amplify cDNA containing variable region of TINA1 antibody MRNA was prepared from the hybridoma using mRNA Isolation kit (Roche applied science).
- a cDNA containing a heavy chain variable region amplified by 5'-RACE PCR was purified using MinElute PCR Purification Kit (QIAGEN), cloned using Zero Blunt TOPO PCR Cloning Kit (Invitrogen), and cloned Sequence analysis of the nucleotide sequence of the cDNA containing the variable region of the strand was performed.
- sequence primer the above-mentioned primer mG2aVR2 designed from the sequence of the constant region of the mouse heavy chain in the database and NUP (Nested Universal Primer A: attached to SMARTer RACE cDNA Amplification Kit) were used.
- sequence analysis was performed using a gene sequence analyzer (“ABI PRISM 3700 DNA Analyzer” or “Applied Biosystems 3730xl Analyzer”, Applied Biosystems), and the sequence reaction was performed using Gene Amp 9ed (Apli, Inc.).
- the nucleotide sequence of the cDNA encoding the variable region of the determined heavy chain of TINA1 antibody is shown in SEQ ID NO: 1 in the sequence listing, and the amino acid sequence is shown in SEQ ID NO: 2.
- Amplification of cDNA containing the light chain variable region of TINA1 antibody by 5'-RACE PCR using this primer combination and the cDNA synthesized in Example 5-1-2) (5'-RACE-Ready cDNA) as a template did.
- PCR was performed using a touchdown PCR program according to the manual of SMARTER RACE cDNA Amplification Kit (CLONTECH) using KOD-plus- (TOYOBO) as Polymerase.
- a cDNA containing a light chain variable region amplified by 5′-RACE PCR was purified using MinElute PCR Purification Kit (QIAGEN), cloned using Zero Blunt TOPO PCR Cloning Kit (Invitrogen), and cloned.
- Sequence analysis of the nucleotide sequence of the cDNA containing the variable region of the strand was performed.
- the primers mKVR2 and NUP designed from the sequence of the constant region of the mouse light chain in the database were used as sequence primers.
- the above-mentioned apparatus was used for sequence analysis and sequence reaction.
- the determined nucleotide sequence of cDNA encoding the light chain variable region of the TINA1 antibody is shown in SEQ ID NO: 3 in the sequence listing, and the amino acid sequence is shown in SEQ ID NO: 4.
- PCR was carried out with the following primer set, and the obtained 3.8 kb fragment was phosphorylated and self-ligated to downstream of the CMV promoter to establish a signal sequence, cloning site, and human kappa chain.
- Chimeric and humanized antibody light chain expression vectors pCMA-LK with normal regions were constructed.
- Primer set 5'-tataccgtcgacccttagtagtaggctttggc-3 '(SEQ ID NO: 35: primer 3.3-F1) 5′-gctatggcaggggcctgccgcccccgacgtgt-3 ′ (SEQ ID NO: 36: primer 3.3-R1)
- chimeric and humanized antibody IgG1-type heavy chain expression vector pCMA-G1 A DNA fragment obtained by digesting pCMA-LK with XbaI and PmeI to remove the ⁇ chain secretion signal and the human ⁇ chain constant region; A DNA fragment comprising a DNA sequence encoding the amino acid sequence of the human heavy chain signal sequence shown in SEQ ID NO: 6 and the human IgG1 constant region was ligated using an In-Fusion Advantage PCR cloning kit (CLONTECH), downstream of the CMV promoter. A chimeric and humanized antibody IgG1-type heavy chain expression vector pCMA-G1 having a signal sequence, a cloning site, a human IgG1 heavy chain constant region was constructed.
- cTINA1 antibody heavy chain expression vector Using cDNA containing the variable region of the heavy chain of TINA1 antibody obtained in Example 5-1-3) as a template, KOD-Plus- (TOYOBO) and In-Fusion HD PCR cloning at the site where the DNA fragment containing cDNA encoding the variable region of the heavy chain was amplified with the following primer set and the chimeric and humanized IgG1-type heavy chain expression vector pCMA-G1 was cleaved with the restriction enzyme BlpI
- a cTINA1 antibody heavy chain expression vector was constructed by insertion using a kit (CLONTECH). The obtained expression vector was designated as “pCMA-G1 / cTINA1”.
- the nucleotide sequence of the cTINA1 antibody heavy chain is shown in SEQ ID NO: 7, and the amino acid sequence is shown in SEQ ID NO: 8.
- the nucleotide sequence of SEQ ID NO: 7 and the amino acid sequence of SEQ ID NO: 8 are also shown in FIG.
- cTINA1 antibody heavy chain primer set 5′-CCAGATGGGTGCTGAGCCAGATCCAGTGGGTGCAGTCTGGACCTGAG-3 ′
- SEQ ID NO: 37 primer TINA1H-F
- SEQ ID NO: 38: primer TINA1H-R primer TINA1H-R
- the light chain nucleotide sequence of the cTINA1 antibody is shown in SEQ ID NO: 9 in the sequence listing, and the amino acid sequence is shown in SEQ ID NO: 10.
- the nucleotide sequence of SEQ ID NO: 9 and the amino acid sequence of SEQ ID NO: 10 are also shown in FIG.
- cTINA1 antibody light chain primer set 5′-ATCTCCGGGCCGTACGGCGGACATGTGATGACCCAGTCTCCAAAATC-3 ′
- SEQ ID NO: 39: primer TINA1L-F 5′-GGAGGGGGCGGCCCACAGCCCGTTTCAGCTCCAGCTTGGTCCCAGC-3 ′
- SEQ ID NO: 40 primer TINA1L-R
- FreeStyle 293F cells Small-scale production of cTINA1 antibody FreeStyle 293F cells (Invitrogen) were subcultured and cultured according to the manual. 1 ⁇ 10 7 FreeStyle 293F cells (Invitrogen) in logarithmic growth phase were diluted to 9.6 mL with FreeStyle 293 expression medium (Invitrogen), then seeded at 30 mL Square Storage Bottle, 37 ° C., Nalgen 8 The culture was shaken at 90 rpm for 1 hour in a% CO 2 incubator.
- Polyethyleneimine (Polyscience # 24765; 30 ⁇ g) was dissolved in Opti-Pro SFM (Invitrogen; 200 ⁇ L), and then a light chain expression vector (6 ⁇ g) and a light chain expression vector prepared using PureLink HiPure Plasmid kit (Invitrogen) (4 ⁇ g) was added to Opti-Pro SFM (Invitrogen; 200 ⁇ L).
- the expression vector / Opti-Pro SFM mixed solution (200 ⁇ L) was added to the Polyethyleneimine / Opti-Pro SFM mixed solution (200 ⁇ L), stirred gently, allowed to stand for another 5 minutes, and then added to FreeStyle 293F cells.
- a culture supernatant obtained by shaking culture at 90 rpm in a 37 ° C., 8% CO 2 incubator for 7 days was filtered through a Minisart-Plus filter (Sartorius) to obtain a sample for evaluation.
- the human chimeric TINA1 antibody obtained by the combination of pCMA-G1 / cTINA1 and pCMA-LK / cTINA1 was named “cTINA1 antibody”.
- Example 6 Design of humanized antibody of mouse anti-TROP2 monoclonal antibody] 6-1) Design of humanized version of TINA1 6-1-1) Molecular modeling of variable region of TINA1 Molecular modeling of the variable region of TINA1 is a method known as homology modeling (Methods in Enzymology, 203, 121-153). (1991)). Primary sequence of variable region of human immunoglobulin registered in Protein Data Bank (Nuc. Acid Res. 35, D301-D303 (2007)) (three-dimensional structure derived from X-ray crystal structure is available) Comparison was made with the previously determined variable region of TINA1.
- 1ZEA was selected as having the highest sequence homology among antibodies that are similarly defective in the framework to the variable region of the heavy chain of TINA1.
- 3IU4 was also selected as having the highest sequence homology to the variable region of the light chain of TINA1.
- the three-dimensional structure of the framework region was created by combining the coordinates of 1ZEA and 3IU4 corresponding to the heavy and light chains of TINA1 to obtain a “framework model”. A representative conformation for each CDR was then incorporated into the framework model.
- an energy calculation was performed to eliminate adverse interatomic contacts. The above procedure was performed using a commercially available protein tertiary structure analysis program Discovery Studio (Accelrys, Inc.).
- the amino acid residues in the framework region for HuPR1A3 were aligned with the amino acid residues for TINA1, and the positions where different amino acids were used were identified. The positions of these residues are analyzed using the three-dimensional model of TINA1 constructed above, and the donor residues to be graphed on the acceptor are described in Queen et. al. (Proc. Natl. Acad. Sci. USA 86, 1000029-10033 (1989)).
- the humanized TINA1 sequence was constructed as described in the examples below by transferring several selected donor residues into the acceptor antibody.
- TINA1 heavy chain 6-2-1) hTINA1-H1 type heavy chain The amino acid number 21 (isoleucine) of the TINA1 heavy chain shown in SEQ ID NO: 8 in the sequence listing is valine, amino acid number 28 (proline) is alanine, amino acid number 30 (leucine) is valine, and amino acid number 35 (glutamic acid).
- amino acid number 36 to serine
- amino acid number 38 arginine to lysine
- amino acid number 39 isoleucine
- amino acid number 57 (glutamine) to arginine
- amino acid number 58 lysine
- Glutamine amino acid number 59 (methionine) to alanine
- amino acid number 62 to glutamine
- amino acid number 65 (lysine) to glutamic acid
- amino acid number 67 isoleucine
- amino acid number 87 phenylalanine
- amino acid number 89 (phenylalanine) to isoleucine, amino acid number 91 (leucine) to alanine, amino acid number 92 (glutamic acid) to aspartic acid, amino acid number 95 (alanine) to threonine, Amino acid number 102 (isoleucine) to leucine, amino acid number 104 (asparagine) to serine, amino acid number 107 (asparagine) to serine, amino acid number 111 (threonine) to alanine, amino acid number 112 (threonine) to valine,
- the humanized TINA1 heavy chain designed with the replacement of amino acid number 114 (phenylalanine) with tyrosine, amino acid number 132 (alanine) with glutamine, and amino acid number 135 (alanine) with leucine is designated as “hTINA1”.
- H1 was designated as type heavy chain ".
- the amino acid sequence of the hTINA1-H1 type heavy chain is set forth in SEQ ID NO: 12 in the Sequence Listing.
- the sequence consisting of amino acid residues 1 to 19 of the amino acid sequence of SEQ ID NO: 12, the sequence consisting of amino acid residues 20 to 140, and the sequence consisting of amino acid residues 141 to 470 are a signal sequence and a heavy chain, respectively. It corresponds to the variable region and heavy chain constant region.
- the nucleotide sequence encoding the amino acid sequence of SEQ ID NO: 12 is described in SEQ ID NO: 11 of the sequence listing.
- the sequence consisting of the 1st to 57th nucleotides, the sequence consisting of the 58th to 420th nucleotides, and the sequence consisting of the 421th to 1410th nucleotides of the nucleotide sequence of SEQ ID NO: 11, respectively, are a signal sequence, a heavy chain variable region sequence, and a heavy chain constant.
- the normal region sequence is encoded.
- the nucleotide sequence of SEQ ID NO: 11 and the amino acid sequence of SEQ ID NO: 12 are also shown in FIG.
- hTINA1-H2 type heavy chain The amino acid number 21 (isoleucine) of the TINA1 heavy chain shown in SEQ ID NO: 8 in the sequence listing is valine, amino acid number 28 (proline) is alanine, amino acid number 30 (leucine) is valine, and amino acid number 35 (glutamic acid).
- amino acid number 36 to serine
- amino acid number 38 arginine to lysine
- amino acid number 39 isoleucine
- amino acid number 57 (glutamine) to arginine
- amino acid number 58 lysine
- Glutamine amino acid number 59 (methionine) to alanine
- amino acid number 62 to glutamine
- amino acid number 65 (lysine) to glutamic acid
- amino acid number 67 isoleucine
- amino acid number 87 phenylalanine
- the amino acid sequence of the hTINA1-H2 type heavy chain is set forth in SEQ ID NO: 14 in the sequence listing.
- the sequence consisting of amino acid residues 1 to 19 of the amino acid sequence of SEQ ID NO: 14, the sequence consisting of amino acid residues 20 to 140, and the sequence consisting of amino acid residues 141 to 470 are a signal sequence and a heavy chain, respectively. It corresponds to the variable region and heavy chain constant region.
- the nucleotide sequence encoding the amino acid sequence of SEQ ID NO: 14 is described in SEQ ID NO: 13 of the sequence listing.
- the sequence consisting of the 1st to 57th nucleotides, the sequence consisting of the 58th to 420th nucleotides, and the sequence consisting of the 421th to 1410th nucleotides of the nucleotide sequence of SEQ ID NO: 13 are a signal sequence, a heavy chain variable region sequence, and a heavy chain constant, respectively.
- the normal region sequence is encoded.
- the nucleotide sequence of SEQ ID NO: 13 and the amino acid sequence of SEQ ID NO: 14 are also shown in FIG.
- hTINA1-H3 type heavy chain Amino acid number 28 (proline) of TINA1 heavy chain shown in SEQ ID NO: 8 in the sequence listing is alanine, amino acid number 30 (leucine) is valine, amino acid number 36 (threonine) is serine, and amino acid number 38 (arginine) is In lysine, amino acid number 39 (isoleucine) is valine, amino acid number 58 (lysine) is glutamine, amino acid number 65 (lysine) is glutamic acid, amino acid number 67 (isoleucine) is methionine, and amino acid number 87 (phenylalanine).
- the amino acid sequence of the hTINA1-H3 type heavy chain is set forth in SEQ ID NO: 16 in the Sequence Listing.
- the sequence consisting of the 1st to 19th amino acid residues, the sequence consisting of the 20th to 140th amino acid residues, and the sequence consisting of the 141st to 470th amino acid residues of the amino acid sequence of SEQ ID NO: 16 are respectively a signal sequence and a heavy chain It corresponds to the variable region and heavy chain constant region.
- the nucleotide sequence encoding the amino acid sequence of SEQ ID NO: 16 is described in SEQ ID NO: 15 of the sequence listing.
- the sequence consisting of the 1st to 57th nucleotides, the sequence consisting of the 58th to 420th nucleotides, and the sequence consisting of the 421th to 1410th nucleotides of the nucleotide sequence of SEQ ID NO: 15 are a signal sequence, a heavy chain variable region sequence, and a heavy chain constant, respectively.
- the normal region sequence is encoded.
- the nucleotide sequence of SEQ ID NO: 15 and the amino acid sequence of SEQ ID NO: 16 are also shown in FIG.
- TINA1 light chain 6-3-1) Humanization of TINA1 light chain 6-3-1) hTINA1-L1 type light chain: The amino acid number 23 (valine) of the TINA1 light chain shown in SEQ ID NO: 10 in the sequence listing is glutamine, amino acid number 28 (histidine) is proline, amino acid number 29 (lysine) is serine, and amino acid number 30 (phenylalanine).
- amino acid number 31 (methionine) to leucine, amino acid number 33 (threonine) to alanine, amino acid number 40 (serine) to threonine, amino acid number 62 (glutamine) to lysine, amino acid number 63 (serine) Alanine, amino acid number 80 (aspartic acid) to serine, amino acid number 83 (threonine) to serine, amino acid number 90 (alanine) to aspartic acid, amino acid number 93 (phenylalanine) to leucine, amino acid number 98 (valine) ) To leucine, amino acid number Replacing 100 (alanine) with proline, amino acid number 103 (leucine) with phenylalanine, amino acid number 120 (alanine) with glutamine, amino acid number 126 (leucine) with isoleucine, and amino acid number 129 (alanine) with threonine.
- the humanized TINA1 light chain designed with was named “hTINA1-L1 type light chain”.
- the amino acid sequence of the hTINA1-L1 type light chain is set forth in SEQ ID NO: 18 in the sequence listing.
- a sequence consisting of amino acid residues 1 to 20 in the amino acid sequence of SEQ ID NO: 18, a sequence consisting of amino acid residues 21 to 129, and a sequence consisting of amino acid residues 130 to 234 are a signal sequence and a light chain, respectively. It corresponds to the variable region and the light chain constant region.
- the nucleotide sequence encoding the amino acid sequence of SEQ ID NO: 18 is described in SEQ ID NO: 17 of the sequence listing.
- the sequence consisting of the 1st to 60th nucleotides of the nucleotide sequence of SEQ ID NO: 17, the sequence consisting of the 61st to 387th nucleotides, the sequence consisting of the 388th to 702th nucleotides are respectively a signal sequence, a light chain variable region sequence and a light chain constant.
- the normal region sequence is encoded.
- the nucleotide sequence of SEQ ID NO: 17 and the amino acid sequence of SEQ ID NO: 18 are also shown in FIG.
- hTINA1-L2 type light chain The amino acid number 28 (histidine) of the TINA1 light chain shown in SEQ ID NO: 10 in the sequence listing is proline, amino acid number 29 (lysine) is serine, amino acid number 30 (phenylalanine) is serine, and amino acid number 31 (methionine) is Leucine, amino acid number 33 (threonine) to alanine, amino acid number 40 (serine) to threonine, amino acid number 62 (glutamine) to lysine, amino acid number 63 (serine) to alanine, amino acid number 80 (aspartic acid) Amino acid number 83 (threonine) as serine, amino acid number 90 (alanine) as aspartic acid, amino acid number 93 (phenylalanine) as leucine, amino acid number 98 (valine) as leucine, amino acid number 100 (alanine) ) To proline, amino acid No.
- hTINA1-L2 type light chain The amino acid sequence of the hTINA1-L2 type light chain is set forth in SEQ ID NO: 20 in the sequence listing.
- a sequence consisting of amino acid residues 1 to 20 of the amino acid sequence of SEQ ID NO: 20, a sequence consisting of amino acid residues 21 to 129, and a sequence consisting of amino acid residues 130 to 234 are a signal sequence and a light chain, respectively.
- the nucleotide sequence encoding the amino acid sequence of SEQ ID NO: 20 is described in SEQ ID NO: 19 in the sequence listing.
- the sequence consisting of the 1st to 60th nucleotides of the nucleotide sequence of SEQ ID NO: 19, the sequence consisting of the 61st to 387th nucleotides, the sequence consisting of the 388th to 702th nucleotides are respectively a signal sequence, a light chain variable region sequence and a light chain constant.
- the normal region sequence is encoded.
- the nucleotide sequence of SEQ ID NO: 19 and the amino acid sequence of SEQ ID NO: 20 are also shown in FIG.
- hTINA1-L3 type light chain The amino acid number 28 (histidine) of the TINA1 light chain shown in SEQ ID NO: 10 in the sequence listing is proline, amino acid number 29 (lysine) is serine, amino acid number 30 (phenylalanine) is serine, and amino acid number 31 (methionine) is Leucine, amino acid number 33 (threonine) to alanine, amino acid number 40 (serine) to threonine, amino acid number 62 (glutamine) to lysine, amino acid number 63 (serine) to glutamine, amino acid number 80 (aspartic acid) Amino acid number 83 (threonine) as serine, amino acid number 90 (alanine) as aspartic acid, amino acid number 93 (phenylalanine) as leucine, amino acid number 98 (valine) as leucine, amino acid number 100 (alanine) ) To proline, amino Humanized TINA1 light designed to replace number
- the chain was named “hTINA1-L3 type light chain”.
- the amino acid sequence of the hTINA1-L3 type light chain is set forth in SEQ ID NO: 22 in the sequence listing.
- the sequence consisting of amino acid residues 1 to 20 in the amino acid sequence of SEQ ID NO: 22, the sequence consisting of amino acid residues 21 to 129, and the sequence consisting of amino acid residues 130 to 234 are respectively a signal sequence and a light chain It corresponds to the variable region and the light chain constant region.
- the nucleotide sequence encoding the amino acid sequence of SEQ ID NO: 22 is described in SEQ ID NO: 21 of the Sequence Listing.
- the sequence consisting of the 1st to 60th nucleotides of the nucleotide sequence of SEQ ID NO: 21, the sequence consisting of the 61st to 387th nucleotides, the sequence consisting of the 388th to 702th nucleotides are respectively a signal sequence, a light chain variable region sequence and a light chain constant.
- the normal region sequence is encoded.
- the nucleotide sequence of SEQ ID NO: 21 and the amino acid sequence of SEQ ID NO: 22 are also shown in FIG.
- Example 7 Construction of hTINA1 antibody expression vector and production of antibody
- 7-1) Construction of hTINA1 heavy chain expression vector 7-1-1)
- a DNA fragment containing a DNA sequence encoding the variable region of H1 was synthesized (Geneart Artificial Gene Synthesis Service).
- hTINA1-H1 expression vector was constructed by inserting the expression vector pCMA-G1 into the site cleaved with the restriction enzyme BlpI using an In-Fusion HD PCR cloning kit (CLONTECH). The obtained expression vector was designated as “pCMA-G1 / hTINA1-H1”.
- hTINA1-H2 Expression Vector DNA comprising a DNA sequence encoding the variable region of hTINA1-H2 shown in nucleotide numbers 36 to 437 of the nucleotide sequence of hTINA1-H2 shown in SEQ ID NO: 13 of the Sequence Listing Fragments were synthesized (GENEART Artificial Gene Synthesis Service), and an hTINA1-H2 expression vector was constructed in the same manner as in Example 7-1-1). The resulting expression vector was named “pCMA-G1 / hTINA1-H2”.
- hTINA1-L1 expression vector was constructed by inserting pCMA-LK into a site cut with the restriction enzyme BsiWI using an In-Fusion HD PCR cloning kit (CLONTECH). The obtained expression vector was designated as “pCMA-LK / hTINA1-L1”.
- hTINA1-L2 expression vector DNA comprising a DNA sequence encoding the variable region of hTINA1-L2 shown in nucleotide numbers 38 to 402 of the nucleotide sequence of hTINA1-L2 shown in SEQ ID NO: 19 of the Sequence Listing Fragments were synthesized (GENEART Artificial Gene Synthesis Service), and an hTINA1-L2 expression vector was constructed in the same manner as in Example 7-2-1). The obtained expression vector was designated as “pCMA-LK / hTINA1-L2”.
- hTINA1 antibody 7-3-1) Production and purification of hTINA1 antibody 7-3-1) Small-scale production of hTINA1 antibody Production was carried out in the same manner as in Example 5-2-5).
- the hTINA1 antibody obtained by the combination of pCMA-G1 / hTINA1-H1 and pCMA-LK / hTINA1-L1 is represented by “hTINA1-H1L1”, the combination of pCMA-G1 / hTINA1-H2 and pCMA-LK / hTINA1-L1.
- the obtained hTINA1 antibody was designated as “hTINA1-H2L1,” and pCMA-G1 / hTINA1-H2 and the combination of pCMA-LK / hTINA1-L2 were designated as hTINA1 antibody as “hTINA1-H2L2,” and pCMA-G1 / hTINA1-
- the hTINA1 antibody obtained by the combination of H3 and pCMA-LK / hTINA1-L3 was named “hTINA1-H3L3”.
- hTINA1 antibody hTINA1-H1L1, hTINA1-H2L1, hTINA1-H2L2, and hTINA1-H3L3 were produced by the following method.
- FreeStyle 293F cells (Invitrogen) were subcultured and cultured according to the manual. 1.2 ⁇ 10 9 cells of FreeStyle 293F cells in the logarithmic growth phase (Invitrogen) were seeded in 3L Fernbach Erlenmeyer Flask (CORNING Inc.), FreeStyle293 expression medium was diluted with (Invitrogen, Inc.) 1.0 ⁇ 10 6 cells / Ml, and cultured with shaking at 90 rpm in an 8% CO 2 incubator at 37 ° C.
- hRS7 antibody light chain expression vector The DNA sequence encoding the variable region of the hRS7 antibody light chain shown in nucleotide numbers 38 to 402 of the nucleotide sequence of the hRS7 antibody light chain shown in SEQ ID NO: 31 in the sequence listing is included. A DNA fragment was synthesized (GENEART, an artificial gene synthesis service), and an hRS7 antibody light chain expression vector was constructed in the same manner as in Example 7-2-1). The obtained expression vector was named “pCMA-LK / hRS7”. The amino acid sequence of the hRS7 antibody heavy chain is shown in SEQ ID NO: 32 in the Sequence Listing.
- hRS7 antibody 1-3-1 Production and purification of hRS7 antibody 1-3-1) Production of hRS7 antibody
- the hRS7 antibody is obtained by combining pCMA-G1 / hRS7 and pCMA-LK / hRS7 in the same manner as in Example 7-3-2). Produced in.
- Example 8 Measurement of antigen-binding ability of hTINA1 antibody and hRS7 antibody
- 8-1) Measurement of antigen binding ability using small-scale production antibody (culture supernatant) Dissociation constant measurement of antibody and antigen (Recombinant Human TROP-2 Fc chimera) was performed using Biacore 3000 (GE Healthcare Bioscience ( The antibody was captured (captured) with an immobilized anti-human IgG (Fab) antibody as a ligand, and the antigen was measured as an analyte.
- Biacore 3000 GE Healthcare Bioscience
- An anti-human IgG (Fab) antibody (Human Fab capture kit, GE Healthcare Biosciences) was covalently bound to a sensor chip CM5 (BIAcore, Inc.) by an amine coupling method at about 2000 RU.
- the reference cell was similarly fixed.
- HBS-EP + (10 mM HEPES pH 7.4, 0.15 M NaCl, 3 mM EDTA, 0.05% Surfactant P20) was used as a running buffer. After adding the culture supernatant containing the antibody for about 80 seconds on the chip on which the anti-human IgG (Fab) antibody is immobilized, a dilution series solution of the antigen (1-1000 nM) is added for 300 seconds at a flow rate of 30 ⁇ l / min.
- the dissociation phase for 600 seconds was monitored.
- 10 mM Gly-HCl pH 1.5 containing 20% DMSO was added at a flow rate of 10 ⁇ l / min for 60 seconds.
- HBS-EP + (10 mM HEPES pH 7.4, 0.15 M NaCl, 3 mM EDTA, 0.05% Surfactant P20) was used as a running buffer.
- a dilution series solution (1-1000 nM) of the antigen is added at a flow rate of 30 ⁇ l / min for 300 seconds, and then dissociated for 600 seconds. The phase was monitored.
- 25 mM NaOH diluted with a running buffer was added twice for 3 seconds at a flow rate of 100 ⁇ l / min. Data analysis was performed by the above method.
- Step 1 tert-butyl (4- ⁇ [(1S, 9S) -9-ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2,3,9,10,13,15 -Hexahydro-1H, 12H-benzo [de] pyrano [3 ', 4': 6,7] indolidino [1,2-b] quinolin-1-yl] amino ⁇ -4-oxobutyl) carbamate 4- (tert- Butoxycarbonylamino) butanoic acid (0.237 g, 1.13 mmol) was dissolved in dichloromethane (10 mL), N-hydroxysuccinimide (0.130 g, 1.13 mmol), and 1-ethyl-3- (3-dimethylamino).
- Step 2 4-amino-N-[(1S, 9S) -9-ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2,3,9,10,13,15- Hexahydro-1H, 12H-benzo [de] pyrano [3 ′, 4 ′: 6,7] indolidino [1,2-b] quinolin-1-yl] butanamide trifluoroacetate
- Compound obtained in Step 1 above 0.388 g, 0.61 mmol
- dichloromethane 9 mL
- Trifluoroacetic acid 9 mL
- Step 3 N- (tert-Butoxycarbonyl) glycylglycyl-L-phenylalanyl-N- (4- ⁇ [(1S, 9S) -9-ethyl-5-fluoro-9-hydroxy-4-methyl-10, 13-Dioxo-2,3,9,10,13,15-hexahydro-1H, 12H-benzo [de] pyrano [3 ′, 4 ′: 6,7] indolidino [1,2-b] quinoline-1- Yl] amino ⁇ -4-oxobutyl) glycinamide N- (tert-butoxycarbonyl) glycylglycyl-L-phenylalanylglycine (0.081 g, 0.19 mmol) was dissolved in dichloromethane (3 mL) to give N-hydroxysuccinimide ( 0.021 g, 0.19 mol) and 1-ethyl-3- (3-dimethylamino
- Step 4 Glycylglycyl-L-phenylalanyl-N- (4- ⁇ [(1S, 9S) -9-ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2,3 9,10,13,15-hexahydro-1H, 12H-benzo [de] pyrano [3 ′, 4 ′: 6,7] indolizino [1,2-b] quinolin-1-yl] amino ⁇ -4-oxobutyl ) Glycinamide trifluoroacetate
- the compound obtained in Step 3 above (1.97 g, 2.10 mmol) was dissolved in dichloromethane (7 mL).
- Trifluoroacetic acid (7 mL) was added and stirred for 1 hour.
- the title compound (1.97 g, 99%) was obtained.
- Step 5 N- ⁇ 3- [2- (2- ⁇ [3- (2,5-Dioxo-2,5-dihydro-1H-pyrrol-1-yl) propanoyl] amino ⁇ ethoxy) ethoxy] propanoyl ⁇ glycylglycyl -L-phenylalanyl-N- (4- ⁇ [(1S, 9S) -9-ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2,3,9,10, 13,15-hexahydro-1H, 12H-benzo [de] pyrano [3 ′, 4 ′: 6,7] indolizino [1,2-b] quinolin-1-yl] amino ⁇ -4-oxobutyl) glycinamide To a solution of the compound obtained in Step 4 (100 mg, 0.119 mmol) in N, N-dimethylformamide (1.20 mL), diisopropylethyl
- Step 6 Antibody-drug conjugate (1)
- Antibody reduction 10 mg of hTINA1-H1L1 prepared in Example 7 in PBS 6.0 / EDTA using common procedure B (using 1.54 as the 280 nm extinction coefficient) and C described in Production Method 1 / ML.
- This solution (10.0 mL) was collected in a 50 mL tube, and a 10 mM TCEP (Tokyo Chemical Industry Co., Ltd.) aqueous solution (0.317 mL; 4.6 equivalents per antibody molecule) and a 1 M dipotassium hydrogen phosphate aqueous solution (Nacalai) Tesque, Inc .; 0.500 mL) was added.
- TCEP Tokyo Chemical Industry Co., Ltd.
- Step 1 Antibody-drug conjugate (2)
- Antibody reduction 10 mg of hTINA1-H1L1 prepared in Example 7 in PBS 6.0 / EDTA using common procedure B (using 1.54 as the 280 nm extinction coefficient) and C described in Production Method 1 / ML.
- This solution (2.00 mL) was collected in a 4 mL tube, and a 10 mM TCEP (Tokyo Chemical Industry Co., Ltd.) aqueous solution (0.0690 mL; 5.0 equivalents for one antibody molecule) and a 1 M dipotassium hydrogen phosphate aqueous solution (Nacalai) Tesque, Inc .; 0.100 mL) was added.
- TCEP Tokyo Chemical Industry Co., Ltd.
- Step 1 Antibody-drug conjugate (3)
- Antibody reduction 10 mg of hTINA1-H1L1 prepared in Example 7 in PBS 6.0 / EDTA using common procedure B (using 1.54 as the 280 nm extinction coefficient) and C described in Production Method 1 / ML.
- This solution 5.0 mL was collected in a 15 mL container, and a 1 M aqueous solution of dipotassium hydrogen phosphate (Nacalai Tesque, Inc .; 0.0813 mL) was added with stirring, followed by stirring at 37 ° C. for 10 minutes.
- Step 1 Antibody-drug conjugate (4) Antibody reduction:
- the hTINA1-H1L1 prepared in Example 7 was treated with PBS 6.0 / EDTA using the common procedure B described in Production Method 1 (using 1.54 as the 280 nm extinction coefficient) and C.
- This solution (5.00 mL) was collected in a 15 mL container, and 1 M aqueous solution of dipotassium hydrogen phosphate (Nacalai Tesque, Inc .; 0.0813 mL) was added with stirring, followed by stirring at 37 ° C. for 10 minutes.
- Step 1 Antibody-drug conjugate (5)
- TCEP Tokyo Chemical Industry Co., Ltd.
- Step 1 ( ⁇ N-[(9H-fluoren-9-ylmethoxy) carbonyl] glycyl ⁇ amino) methyl acetate N-9-fluorenylmethoxycarbonylglycylglycine (4.33 g, 12.2 mmol), tetrahydrofuran (120 ml ) And toluene (40.0 ml) were added pyridine (1.16 ml, 14.7 mmol and lead tetraacetate (6.84 g, 14.7 mmol), and the mixture was heated to reflux for 5 hours.
- Step 2 Benzyl [( ⁇ N-[(9H-fluoren-9-ylmethoxy) carbonyl] glycyl ⁇ amino) methoxy] acetate
- the compound obtained in Step 1 above (3.68 g, 10.0 mmol) and benzyl glycolate (4
- potassium tert-butoxide (2.24 g, 20.0 mmol) was added to the reaction solution at 0 ° C., and the mixture was extracted with ethyl acetate and chloroform.
- Step 3 [( ⁇ N-[(9H-Fluoren-9-ylmethoxy) carbonyl] glycyl ⁇ amino) methoxy] acetic acid
- the compound obtained in Step 2 above (1.88 g, 3.96 mmol) was ethanol (40.0 mL). , Dissolved in ethyl acetate (20.0 ml). Palladium carbon catalyst (376 mg) was added, and the mixture was stirred at room temperature for 2 hours under a hydrogen atmosphere. Insoluble materials were removed by Celite filtration, and the solvent was distilled off under reduced pressure to obtain the title compound (1.52 g, quantitative) as a colorless solid.
- Step 4 9H-fluoren-9-ylmethyl (2- ⁇ [(2- ⁇ [(1S, 9S) -9-ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2, 3,9,10,13,15-hexahydro-1H, 12H-benzo [de] pyrano [3 ′, 4 ′: 6,7] indolidino [1,2-b] quinolin-1-yl] amino ⁇ -2 -Oxoethoxy) methyl] amino ⁇ -2-oxoethyl) carbamate Under ice-cooling, exatecan mesylate (0.283 g, 0.533 mmol), N-hydroxysuccinimide (61.4 mg, 0.533 mmol), and the above steps To a solution of the compound obtained in 3 (0.205 g, 0.533 mmol) in N, N-dimethylformamide (10.0 mL), N, N-diisopropylethylamine (92
- Step 5 N-[(2- ⁇ [(1S, 9S) -9-ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2,3,9,10,13,15 -Hexahydro-1H, 12H-benzo [de] pyrano [3 ', 4': 6,7] indolizino [1,2-b] quinolin-1-yl] amino ⁇ -2-oxoethoxy) methyl] glycinamide Piperidine (1.1 mL) was added to a solution of the compound obtained in step 4 (0.881 g, 1.10 mmol) in N, N-dimethylformamide (11.0 mL), and the mixture was stirred at room temperature for 2 hours. The solvent was distilled off under reduced pressure to obtain a mixture containing the title compound. This mixture was used in the next reaction without further purification.
- Step 6 N-[(9H-Fluoren-9-ylmethoxy) carbonyl] glycylglycyl-L-phenylalanyl-N-[(2- ⁇ [(1S, 9S) -9-ethyl-5-fluoro-9-hydroxy -4-Methyl-10,13-dioxo-2,3,9,10,13,15-hexahydro-1H, 12H-benzo [de] pyrano [3 ′, 4 ′: 6,7] indolidino [1,2 -B] quinolin-1-yl] amino ⁇ -2-oxoethoxy) methyl] glycinamide Under ice-cooling, the mixture obtained in Step 5 above (0.439 mmol), N-hydroxysuccinimide (0.101 g, 0.878 mmol) ), And N-[(9H-fluoren-9-ylmethoxy) carbonyl] glycylglycyl
- Step 7 Glycylglycyl-L-phenylalanyl-N-[(2- ⁇ [(1S, 9S) -9-ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2,3 , 9,10,13,15-hexahydro-1H, 12H-benzo [de] pyrano [3 ′, 4 ′: 6,7] indolizino [1,2-b] quinolin-1-yl] amino ⁇ -2- Oxoethoxy) methyl] glycinamide Piperidine (0.251 mL, 2.53 mmol) was added to a solution of the compound obtained in Step 6 (0.269 g, 0.253 mmol) in N, N-dimethylformamide (4.00 mL). And stirred at room temperature for 2 hours. The solvent was distilled off under reduced pressure to obtain a mixture containing the title compound. This mixture was used in the next reaction without further purification.
- Step 8 N- [6- (2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl) hexanoyl] glycylglycyl-L-phenylalanyl-N-[(2- ⁇ [(1S, 9S) -9-Ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2,3,9,10,13,15-hexahydro-1H, 12H-benzo [de] pyrano [3 ', 4': 6,7] Indolizino [1,2-b] quinolin-1-yl] amino ⁇ -2-oxoethoxy) methyl] glycinamide N of the compound obtained in Step 7 above (0.253 mmol) To a solution of N-dimethylformamide (10.0 mL) was added N-succinimidyl 6-maleimidohexanoate (0.156 g, 0.506 mmol), and the
- Step 9 Antibody-drug conjugate (6)
- Antibody reduction 10 mg of hTINA1-H1L1 prepared in Example 7 in PBS 6.0 / EDTA using common procedure B (using 1.54 as the 280 nm extinction coefficient) and C described in Production Method 1 / ML.
- This solution (10.0 mL) was collected in a 50 mL tube, and a 10 mM TCEP (Tokyo Chemical Industry Co., Ltd.) aqueous solution (0.317 mL; 4.6 equivalents per antibody molecule) and a 1 M dipotassium hydrogen phosphate aqueous solution (Nacalai) Tesque, Inc .; 0.500 mL) was added.
- TCEP Tokyo Chemical Industry Co., Ltd.
- Step 1 Antibody-drug conjugate (7)
- Antibody reduction 10 mg of hTINA1-H1L1 prepared in Example 7 in PBS 6.0 / EDTA using common procedure B (using 1.54 as the 280 nm extinction coefficient) and C described in Production Method 1 / ML.
- This solution (2.0 mL) was collected in a 4 mL tube, and a 10 mM TCEP (Tokyo Chemical Industry Co., Ltd.) aqueous solution (0.0690 mL; 5.0 equivalents per antibody molecule) and a 1 M dipotassium hydrogen phosphate aqueous solution (Nacalai) Tesque, Inc .; 0.0299 mL) was added.
- TCEP Tokyo Chemical Industry Co., Ltd.
- Step 1 Antibody-drug conjugate (8)
- Antibody reduction 10 mg of hTINA1-H1L1 prepared in Example 7 in PBS 6.0 / EDTA using common procedure B (using 1.54 as the 280 nm extinction coefficient) and C described in Production Method 1 / ML.
- This solution (30.0 mL) was collected in a 100 mL container, and 1 M aqueous solution of dipotassium hydrogen phosphate (Nacalai Tesque, Inc .; 0.4875 mL) was added with stirring, followed by stirring at 37 ° C. for 10 minutes.
- Step 1 Antibody-drug conjugate (9)
- Antibody reduction 10 mg of hTINA1-H1L1 prepared in Example 7 in PBS 6.0 / EDTA using common procedure B (using 1.54 as the 280 nm extinction coefficient) and C described in Production Method 1 / ML.
- This solution 5.0 mL was collected in a 15 mL container, and a 1 M aqueous solution of dipotassium hydrogen phosphate (Nacalai Tesque, Inc .; 0.0813 mL) was added with stirring, followed by stirring at 37 ° C. for 10 minutes.
- Step 1 Antibody-drug conjugate (10)
- Antibody reduction hRS7 prepared in Reference Example 1 was prepared at 10 mg / mL in PBS 6.0 / EDTA using the common procedure B (using 1.56 as the 280 nm extinction coefficient) and C described in Production Method 1.
- This solution (2.0 mL) was collected in a 4 mL tube, and a 10 mM TCEP (Tokyo Chemical Industry Co., Ltd.) aqueous solution (0.0690 mL; 5.0 equivalents per antibody molecule) and a 1 M dipotassium hydrogen phosphate aqueous solution (Nacalai) Tesque, Inc .; 0.0299 mL) was added.
- TCEP Tokyo Chemical Industry Co., Ltd.
- Step 1 tert-butyl [2- (2- ⁇ [(1S, 9S) -9-ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2,3,9,10, 13,15-hexahydro-1H, 12H-benzo [de] pyrano [3 ′, 4 ′: 6,7] indolizino [1,2-b] quinolin-1-yl] amino ⁇ -2-oxoethoxy) ethyl] Carbamate Exatecan Mesylate (3.10 g, 5.47 mol) was replaced with 4- (tert-butoxycarbonylamino) butanoic acid instead of ⁇ 2-[(tert-butoxycarbonyl) amino] ethoxy ⁇ acetic acid (J.
- Step 2 2- (2-Aminoethoxy) -N-[(1S, 9S) -9-ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2,3,9,10 , 13,15-Hexahydro-1H, 12H-benzo [de] pyrano [3 ′, 4 ′: 6,7] indolizino [1,2-b] quinolin-1-yl] acetamide trifluoroacetate salt
- the obtained compound (1.50 g, 2.36 mol) was reacted in the same manner as in Step 2 of Example 1 to obtain the title compound (1.50 g, quantitative) as a pale yellow solid.
- Step 3 N- (tert-Butoxycarbonyl) glycylglycyl-L-phenylalanyl-N- [2- (2- ⁇ [(1S, 9S) -9-ethyl-5-fluoro-9-hydroxy-4-methyl -10,13-dioxo-2,3,9,10,13,15-hexahydro-1H, 12H-benzo [de] pyrano [3 ', 4': 6,7] indolidino [1,2-b] quinoline -1-yl] amino ⁇ -2-oxoethoxy) ethyl] glycinamide
- the compound obtained in Step 2 above (554 mg, 0.85 mmol) was reacted in the same manner as in Step 3 of Example 1 to give the title compound (775 mg, 95%).
- Step 4 Glycylglycyl-L-phenylalanyl-N- [2- (2- ⁇ [(1S, 9S) -9-ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2 , 3,9,10,13,15-hexahydro-1H, 12H-benzo [de] pyrano [3 ′, 4 ′: 6,7] indolizino [1,2-b] quinolin-1-yl] amino ⁇ - 2-Oxoethoxy) ethyl] glycinamide trifluoroacetate
- the compound obtained in Step 3 (630 mg, 0.659 mmol) was reacted in the same manner as in Step 4 of Example 1 to give the title compound (588 mg, 92%).
- Step 5 N- [6- (2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl) hexanoyl] glycylglycyl-L-phenylalanyl-N- [2- (2- ⁇ [( 1S, 9S) -9-ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2,3,9,10,13,15-hexahydro-1H, 12H-benzo [de] pyrano [3 ′, 4 ′: 6,7] Indolizino [1,2-b] quinolin-1-yl] amino ⁇ -2-oxoethoxy) ethyl] glycinamide Compound obtained in Step 4 above (240 mg, 0.247 mmol ), Triethylamine (31.4 ⁇ L, 0.22 mmol) instead of diisopropylethylamine, 3- (2- (2- (3-maleimidopropanamid
- Step 6 Antibody-drug conjugate (11)
- Antibody reduction 10 mg of hTINA1-H1L1 prepared in Example 7 in PBS 6.0 / EDTA using common procedure B (using 1.54 as the 280 nm extinction coefficient) and C described in Production Method 1 / ML.
- This solution (3.0 mL) was collected in a 15 mL container, and 1 M aqueous solution of dipotassium hydrogen phosphate (Nacalai Tesque, Inc .; 0.0488 mL) was added with stirring, followed by stirring at 37 ° C. for 10 minutes.
- Antibody concentration 1.93 mg / mL, antibody yield: 27.0 mg (90%), drug average binding number per antibody molecule (n) measured in common operation E: 7.1; in common operation F The average number of drugs bound per molecule of antibody measured (n): 7.0.
- Step 1 Antibody-drug conjugate (12)
- This solution (10.0 mL) was collected in a 50 mL tube, and a 10 mM TCEP (Tokyo Chemical Industry Co., Ltd.) aqueous solution (0.317 mL; 4.6 equivalents per antibody molecule) and a 1 M dipotassium hydrogen phosphate aqueous solution (Nacalai) Tesque, Inc .; 0.500 mL) was added.
- TCEP Tokyo Chemical Industry Co., Ltd.
- Step 1 Antibody-drug conjugate (13)
- Antibody reduction Using hTINA1-H1L1 prepared in Example 7 with the common procedure B (using 1.54 as the 280 nm extinction coefficient) and C described in Production Method 1, the medium was changed to PBS 6.0 / EDTA. Substitution was made to an antibody concentration of 10 mg / mL. This solution (100 mL) was put into a polycarbonate 250 mL Erlenmeyer flask container, and 1 M aqueous solution of dipotassium hydrogenphosphate (1.4 mL) was added at room temperature with stirring with a magnetic stirrer, and then 10 mM TCEP aqueous solution (1.62 mL; 2.5 equivalents to molecule).
- an ultrafiltration membrane Merck, Pellicon XL Cassette, Ultracell 30 KDa
- a tube pump US Coal Palmer Master Flex Pump model 77521-40, a pump head model 7518-00
- a tube US
- Ultrafiltration was performed using an ultrafiltration apparatus composed of Cole Palmer Master Flex Tube L / S16). That is, by performing ultrafiltration purification while adding ABS (a total of 1600 mL) as a purification buffer to the reaction solution, unbound drug linkers and other low molecular weight reagents are removed and the buffer solution is replaced with ABS. And further concentrated.
- All antibody-drug conjugates were diluted with physiological saline (Otsuka Pharmaceutical Factory) and administered at a volume of 10 mL / kg into the tail vein.
- 2 ⁇ 10 6 cells in which human colon cancer cell line COLO205 purchased from ATCC is suspended in physiological saline are subcutaneously implanted into the right flank of female BALB / c-nu / nu mice (Day 0), and randomly grouped on Day 7 Divided.
- Antibody-drug conjugates (1), (6), and (12) were administered into Day 7, 14, and 21 via the tail vein at a dose of 10 mg / kg.
- hTINA1-H1L1 antibody and hRS7 antibody to which no drug was bound were similarly administered at a dose of 25 mg / kg.
- ADC antitumor effect (2) Human pancreatic adenocarcinoma cell line Bx-PC3 purchased from ATCC was transplanted into female BALB / c-nu / nu mice, and solid passage tumor pieces were subcutaneously transplanted into the right flank of female BALB / c-nu / nu mice. (Day 0), Day 16 was randomly grouped. Antibody-drug conjugates (1), (6), and (12) were all administered to Day 16, 23, 30 at a dose of 10 mg / kg via the tail vein. As negative controls, hTINA1-H1L1 antibody and hRS7 antibody to which no drug was bound were similarly administered at a dose of 25 mg / kg. The administration of the antibody-drug conjugates (1) and (6) significantly reduced the tumor volume compared to the administration of the antibody-drug conjugate (12), and both exhibited a tumor growth inhibitory effect (FIG. 14).
- Example 21-d ADC antitumor effect (4)
- COLO205 was subcutaneously transplanted into female BALB / c-nu / nu mice (Day 0), and random grouping was performed on Day 11.
- antibody-drug conjugates (2) and (5) were all administered at 10 mg / kg
- antibody-drug conjugates (7) and (10) were all administered at 3 mg / kg via the tail vein. All of the administrations of antibody-drug conjugates (2), (5), (7), and (10) exhibited a tumor growth inhibitory effect (FIG. 16).
- Example 21-e Antitumor effect of ADC (5)
- Bx-PC3 was subcutaneously transplanted into female BALB / c-nu / nu mice (Day 0), and randomized grouping was performed on Day 25.
- Antibody-drug conjugates (2), (5), (7), and (10) were all administered to Day 25 and 32 via the tail vein at a dose of 3 mg / kg. All of the administrations of antibody-drug conjugates (2), (5), (7), and (10) exhibited a tumor growth inhibitory effect (FIG. 17).
- Example 21-g ADC antitumor effect (7)
- Bx-PC3 was subcutaneously transplanted into female BALB / c-nu / nu mice (Day 0), and Day 21 was randomly grouped.
- antibody-drug conjugates (3), (4), (8), (9) were all administered into the tail vein at a dose of 3 mg / kg. All of the antibody-drug conjugates (3), (4), (8), and (9) exhibited a tumor growth inhibitory effect (FIG. 19).
- ADC antitumor effect 9
- Human gastric cancer cell line NCI-N87 1 ⁇ 10 7 cells purchased from ATCC was suspended in physiological saline, subcutaneously implanted into female BALB / c-nu / nu mice (Day 0), and randomly grouped on Day 6 did.
- Antibody-drug conjugates (3), (4), (8), and (9) were all administered to Day 6 via the tail vein at a dose of 3 mg / kg. All of the administrations of antibody-drug conjugates (3), (4), (8), and (9) exhibited a tumor growth inhibitory effect (FIG. 21).
- ADC antitumor effect (11) Human pharyngeal carcinoma cell line FaDu 3 ⁇ 10 6 cells purchased from ATCC was suspended in physiological saline, subcutaneously implanted into female BALB / c-nu / nu mice (Day 0), and randomized to Day 11 .
- Antibody-drug conjugates (3), (4), (8), and (9) were all administered to Day 11 via the tail vein at a dose of 3 mg / kg. All of the antibody-drug conjugates (3), (4), (8), and (9) exhibited a tumor growth inhibitory effect (FIG. 23).
- Example 21-m Antitumor effect of ADC (13)
- CFPAC-1 was subcutaneously transplanted into female BALB / c-nu / nu mice (Day 0), and grouping was performed randomly on Day 14.
- antibody-drug conjugates (8) and (13) were all administered into the tail vein at a dose of 1 mg / kg. Both administrations of antibody-drug conjugates (8) and (13) exhibited a tumor growth inhibitory effect (FIG. 25).
- BxPC3 and NCI-H292 are RPMI 1640 Medium (Gibco) containing 10% fetal bovine serum (Moregate), NIH: OVCAR-3 is RPMI 1640 Medium containing 20% fetal bovine serum and 0.01 mg / mL Insulin (Invitrogen), CFPAC-1 is Iscove's Modified Dulbecco's Medium (Gibco) containing 10% fetal calf serum, and FaDu, Calu-3 and Calu-6 are Eagle's Minimum Essential Medium (ATCC) containing 10% fetal bovine serum.
- Gibco fetal bovine serum
- Ad Eagle's Minimum Essential Medium
- CaOV3 and A375 are Dulbecco's Modified Eagle Medium (Gibco) containing 10% fetal bovine serum, each 2.2 ⁇ 1 It was prepared in 6 cells / mL, and seeded each 90 ⁇ L microplate for 96-well cell culture. Add 10 ⁇ L each of RPMI1640 Medium to antibody-drug conjugate (4), (8) diluted for 100 nM, 20 nM, 4 nM, 0.8 nM, 0.16 nM, 0.032 nM, 0.0064 nM with RPMI1640 Medium or for comparison. Then, the cells were cultured for 6 days at 37 ° C. and 5% CO 2 .
- Cell growth inhibition rate (%) a ⁇ b ⁇ 100 a: Average value of sample-added wells after 6 days of culture-Average value of wells without sample added at the start of culture b: Average value of wells added with medium after 6 days of culture-Average value of wells without sample added at the start of culture
- the GI 50 value was calculated by the following formula.
- GI 50 (nM) antilog ((50 ⁇ f) ⁇ (LOG 10 (d) ⁇ LOG 10 (c)) ⁇ (fe) + LOG 10 (d)) c: specimen concentration c d: specimen concentration d e: Cell growth inhibition rate at sample concentration c f: Cell growth inhibition rate c at sample concentration d, d is c> d at two points sandwiching 50% cell growth inhibition rate.
- Antibody-drug conjugates (4) and (8) have a GI 50 against TROP2 antigen positive cell lines BxPC3, NCI-H292, NIH: OVCAR-3, CFPAC-1, FaDu, Calu-3, CaOV3. An anti-cell effect of ⁇ 1 (nM) was shown. On the other hand, no anti-cell effect was shown against Calu-6, A375, which is a TROP2 antigen negative cell line (> 100 (nM)).
- SEQ ID NO: 1 Nucleotide sequence of cDNA encoding variable region of heavy chain of TINA1 antibody
- SEQ ID NO: 2 Amino acid sequence of variable region of heavy chain of TINA1 antibody
- SEQ ID NO: 3 cDNA encoding variable region of light chain of TINA1 antibody
- Nucleotide sequence SEQ ID NO: 4 amino acid sequence of the light chain variable region of TINA1 antibody
- SEQ ID NO: 5 nucleotide sequence encoding human ⁇ chain secretion signal and human ⁇ chain constant region
- SEQ ID NO: 6 human heavy chain secretion signal and human IgG1
- SEQ ID NO: 9 cTINA1 antibody light chain nucleotide sequence
- SEQ ID NO: 10 cTINA1 antibody light chain amino acid
- SEQ ID NO: 11 hTINA
Abstract
Description

hTROP2遺伝子は約50%の相同性を持つヒトTrop-1(EpCAM,EGP-2,TACSTD1)と共にTACSTD遺伝子ファミリーを構成している(非特許文献21)。hTROP2蛋白質は、N末端26アミノ酸残基から成るシグナル配列、248アミノ酸残基から成る細胞外ドメイン、23アミノ酸残基から成る細胞膜貫通ドメイン、26アミノ酸残基から成る細胞内ドメインで構成されている。細胞外ドメインには4つのN結合型糖鎖付加部位があり、見かけ上の分子量は理論計算値35キロダルトンより10キロダルトン前後増加することが知られている(非特許文献19)。
これまでhTROP2の生理学的リガンドは同定されておらず、分子機能は明らかにされていないが、腫瘍細胞においてカルシウムシグナルを伝達することが示され(非特許文献20)、またCa2+依存性キナーゼであるプロテインキナーゼCにより細胞内セリン303残基がリン酸化されること(非特許文献18)や、細胞内ドメインにPIP2結合配列を有することから、腫瘍細胞におけるシグナル伝達機能が示唆されている(非特許文献22)。
臨床検体を用いた免疫組織化学解析により、hTROP2は種々の上皮細胞癌種において過剰発現していること、かつ、正常組織においてはいくつかの組織の上皮細胞での発現に限られ、その発現量も腫瘍組織と比較して低いことが示されている(非特許文献23~27)。またhTrop2の発現は大腸癌(非特許文献23)、胃癌(非特許文献24)、膵臓癌(非特許文献25)、口腔癌(非特許文献26)、グリオーマ(非特許文献27)において予後不良と相関することも報告されている。
さらに大腸癌細胞を用いたモデルから、hTROP2の発現が腫瘍細胞の足場非依存的細胞増殖及び免疫不全マウスでの腫瘍形成に関与していることが報告されている(非特許文献28)。
このために本発明者らは特定の構造のリンカーを創出し、このリンカーを介して抗TROP2抗体とエキサテカンとを結合させた抗体-薬物コンジュゲートを獲得することに成功し、同コンジュゲートが優れた抗腫瘍効果を発揮することを見出して本発明を完成させたのである。
[1]次式

-L1-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-
で示される構造のリンカーを介して、抗TROP2抗体のヒンジ部に存在するジスルフィド結合部分において形成させたチオエーテル結合によって結合させたことを特徴とする抗体-薬物コンジュゲートに関するものである。
式中、n1は、0から6の整数を示し、
n2は、0から5の整数を示し、
L1は、-(Succinimid-3-yl-N)-(CH2)n3-C(=O)-を示し、
ここで、n3は、2から8の整数を示し、
L2は、-NH-(CH2CH2-O)n4-CH2CH2-C(=O)-又は単結合を示し、
ここで、n4は、1から6の整数を示し、
LPは、2から7個のアミノ酸で構成されるペプチド残基を示し、
Laは、-O-又は単結合を示し、
-(Succinimid-3-yl-N)-は次式:

[2]LPのペプチド残基が、フェニルアラニン、グリシン、バリン、リシン、シトルリン、セリン、グルタミン酸、アスパラギン酸から選ばれるアミノ酸からなるペプチド残基である[1]に記載の抗体-薬物コンジュゲート。
[3]LPが、以下の群から選ばれるペプチド残基である[1]又は[2]に記載の抗体-薬物コンジュゲート:
-GGF-、
-DGGF-、
-(D-)D-GGF-、
-EGGF-、
-GGFG-、
-SGGF-、
-KGGF-、
-DGGFG-、
-GGFGG-、
-DDGGFG-、
-KDGGFG-、及び
-GGFGGGF-;
ここで『(D-)D』はD-アスパラギン酸を示す。
[4]LPが、4個のアミノ酸で構成されるペプチド残基である[1]又は[2]に記載の抗体-薬物コンジュゲート。
[5]LPが、テトラペプチド残基の-GGFG-である[1]から[4]のいずれか一項に記載の抗体-薬物コンジュゲート。
[7]n3が2から5の整数であって、L2が-NH-(CH2CH2-O)n4-CH2CH2-C(=O)-であり、n4が2又は4である[1]から[5]のいずれか一項に記載の抗体-薬物コンジュゲート。
[8]-NH-(CH2)n1-La-(CH2)n2-C(=O)-が、4から7原子の鎖長を有する部分構造である[1]から[7]のいずれか一項に記載の抗体-薬物コンジュゲート。
[9]-NH-(CH2)n1-La-(CH2)n2-C(=O)-が、5又は6原子の鎖長を有する部分構造である[1]から[7]のいずれか一項に記載の抗体-薬物コンジュゲート。
[10]-NH-(CH2)n1-La-(CH2)n2-C(=O)-が、
-NH-CH2CH2-C(=O)-、
-NH-CH2CH2CH2-C(=O)-、
-NH-CH2CH2CH2CH2-C(=O)-、
-NH-CH2CH2CH2CH2CH2-C(=O)-、
-NH-CH2-O-CH2-C(=O)-、又は
-NH-CH2CH2-O-CH2-C(=O)-である[1]から[9]のいずれか一項に記載の抗体-薬物コンジュゲート。
[11]-NH-(CH2)n1-La-(CH2)n2-C(=O)-が、
-NH-CH2CH2CH2-C(=O)-、
-NH-CH2-O-CH2-C(=O)-、又は
-NH-CH2CH2-O-CH2-C(=O)-
である[1]から[9]のいずれか一項に記載の抗体-薬物コンジュゲート。
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)。

-(NH-DX)は次式:

-GGFG-は、-Gly-Gly-Phe-Gly-のテトラペプチド残基を示す。
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)。

-L1-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-
で示される構造のリンカーを介して、抗TROP2抗体のヒンジ部に存在するジスルフィド結合部分において形成させたチオエーテル結合を介して結合させたことを特徴とする抗体-薬物コンジュゲート。
ここで、抗TROP2抗体はL1の末端において結合し、抗腫瘍性化合物は-(CH2)n2-C(=O)-部分のカルボニル基に結合する。
式中、n1は、0から6の整数を示し、
n2は、0から5の整数を示し、
L1は、-(Succinimid-3-yl-N)-(CH2)n3-C(=O)-を示し、
ここで、n3は、2から8の整数を示し、
L2は、-NH-(CH2CH2-O)n4-CH2CH2-C(=O)-又は単結合を示し、
ここで、n4は、1から6の整数を示し、
LPは、-GGFG-のテトラペプチド残基を示し、
Laは、-O-又は単結合を示し、
-(Succinimid-3-yl-N)-は次式:

n1が1であり、n2が1であり、n3が5であり、L2が単結合であり、Laが-O-であるか、又は
n1が2であり、n2が1であり、n3が5であり、L2が単結合であり、Laが-O-である[14]に記載の抗体-薬物コンジュゲート。
[16]n3が2又は5であって、L2が単結合である[14]又は[15]に記載の抗体-薬物コンジュゲート。
[17]n3が2又は5であって、L2が-NH-(CH2CH2-O)n4-CH2CH2-C(=O)-であり、n4が2又は4である[14]又は[15]に記載の抗体-薬物コンジュゲート。
[18]-NH-(CH2)n1-La-(CH2)n2-C(=O)-が、
-NH-CH2CH2CH2-C(=O)-、
-NH-CH2-O-CH2-C(=O)-、又は
-NH-CH2CH2-O-CH2-C(=O)-
である[14]から[17]のいずれか一項に記載の抗体-薬物コンジュゲート。
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX);

-(NH-DX)は次式:

-GGFG-は、-Gly-Gly-Phe-Gly-のテトラペプチド残基を示す。
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)。
ここで、-(Succinimid-3-yl-N)-、-(NH-DX)及び-GGFG-は、上記の通りである。
[22]選択された1種の薬物-リンカー構造の1抗体あたりの平均結合数が2から8個の範囲である[1]から[20]のいずれか一項に記載の抗体-薬物コンジュゲート。
[23]選択された1種の薬物-リンカー構造の1抗体あたりの平均結合数が3から8個の範囲である[1]から[20]のいずれか一に記載の抗体-薬物コンジュゲート。
[25][1]から[23]のいずれか一項に記載の抗体-薬物コンジュゲート、その塩、又はそれらの水和物を含有する抗腫瘍薬及び/又は抗癌薬。
[26]肺癌、腎癌、尿路上皮癌、大腸癌、前立腺癌、多形神経膠芽腫、卵巣癌、膵癌、乳癌、メラノーマ、肝癌、膀胱癌、胃癌、子宮頸癌、頭頸部癌、又は食道癌に適用するための[25]に記載の抗腫瘍薬及び/又は抗癌薬。
[27][1]から[23]のいずれか一項に記載の抗体-薬物コンジュゲート、その塩、又はそれらの水和物を活性成分とし、薬学的に許容される製剤成分とを含有する医薬組成物。
[28]肺癌、腎癌、尿路上皮癌、大腸癌、前立腺癌、多形神経膠芽腫、卵巣癌、膵癌、乳癌、メラノーマ、肝癌、膀胱癌、胃癌、子宮頸癌、頭頸部癌、又は食道癌に適用するための[27]に記載の医薬組成物。
[29][1]から[23]のいずれか一項に記載の抗体-薬物コンジュゲート、その塩、又はそれらの水和物を投与することを特徴とする腫瘍及び/又は癌の治療方法。
(maleimid-N-yl)-(CH2)n3-C(=O)-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-(NH-DX)
を抗TROP2抗体又はその反応性誘導体と反応させ、該抗体のヒンジ部に存在するジスルフィド結合部分においてチオエーテル結合を形成させる方法によって薬物-リンカー部分を該抗体に結合させることを特徴とする抗体-薬物コンジュゲートの製造方法。
L2は、-NH-(CH2CH2-O)n4-CH2CH2-C(=O)-又は単結合を示し、
ここで、n4は、1から6の整数を示し、
LPは、フェニルアラニン、グリシン、バリン、リシン、シトルリン、セリン、グルタミン酸、アスパラギン酸から選ばれる2から7個のアミノ酸で構成されるペプチド残基を示し、
n1は、0から6の整数を示し、
n2は、0から5の整数を示し、
Laは、-O-又は単結合を示し、
(maleimid-N-yl)-は、次式

-(NH-DX)は、次式

[33]選択された1種の薬物-リンカー構造の1抗体あたりの平均結合数が2から8個の範囲である[30]又は[31]に記載の製造方法。
[34]選択された1種の薬物-リンカー構造の1抗体あたりの平均結合数が3から8個の範囲である[30]又は[31]に記載の製造方法。
[35][30]から[34]のいずれかの製造方法によって得られる抗体-薬物コンジュゲート。
(maleimid-N-yl)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、及び
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)。

-(NH-DX)は、次式

-GGFG-は、-Gly-Gly-Phe-Gly-のテトラペプチド残基を示す。
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、及び
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)。
ここで、(maleimid-N-yl)-、-(NH-DX)、及び-GGFG-は、上記の通りである。
[39]選択された1種の薬物-リンカー構造の1抗体あたりの平均結合数が2から8個の範囲である[36]又は[37]に記載の抗体-薬物コンジュゲート。
[40]選択された1種の薬物-リンカー構造の1抗体あたりの平均結合数が3から8個の範囲である[36]又は[37]に記載の抗体-薬物コンジュゲート。
本発明の抗TROP2抗体-薬物コンジュゲートに使用される抗TROP2抗体は、いずれの種に由来してもよいが、好ましくは、ヒト、ラット、マウス、及びウサギを例示できる。ヒト以外の種に由来する場合は、周知の技術を用いて、キメラ化又はヒト化することが好ましい。本発明の抗体は、ポリクローナル抗体であっても、モノクローナル抗体であってもよいが、モノクローナル抗体が好ましい。
抗TROP2抗体は腫瘍細胞を標的にできる抗体であり、すなわち腫瘍細胞を認識できる特性、腫瘍細胞に結合できる特性、そして腫瘍細胞内に取り込まれて内在化する特性等を備えており、抗腫瘍活性を有する化合物を、リンカーを介して結合させて抗体-薬物コンジュゲートとすることができる。
抗体の腫瘍細胞への結合性は、フローサイトメトリーを用いて確認することができる。腫瘍細胞内への抗体の取り込みの確認方法としては、(1)治療抗体に結合する二次抗体(蛍光標識)を用いて細胞内に取り込まれた抗体を蛍光顕微鏡で可視化するアッセイ(Cell Death and Differentiation (2008) 15, 751-761)、(2)治療抗体に結合する二次抗体(蛍光標識)を用いて細胞内に取り込まれた蛍光量を測定するアッセイ(Molecular Biology of the Cell Vol. 15, 5268-5282, December 2004)、又は(3)治療抗体に結合するイムノトキシンを用いて、細胞内に取り込まれると毒素が放出されて細胞増殖が抑制されるというMab-ZAPアッセイ(Bio Techniques 28:162-165, January 2000)等を挙げることができる。なお、イムノトキシンとしては、ジフテテリア毒素の触媒領域とプロテインGとのリコンビナント複合蛋白質も使用可能である。
抗体-薬物コンジュゲートは抗腫瘍効果を発揮する化合物を結合させてあるので、抗体自体が抗腫瘍効果を有することは、好ましいが、必須ではない。抗腫瘍性化合物の細胞障害性を腫瘍細胞において特異的・選択的に発揮させる目的からは、抗体が内在化して腫瘍細胞内に移行する性質のあることが重要であり、好ましい。
また、公知の方法(例えば、Kohler and Milstein, Nature(1975)256, p.495-497;Kennet, R.ed., Monoclonal Antibodies, p.365-367, Plenum Press, N.Y.(1980))に従って、抗原に対する抗体を産生する抗体産生細胞とミエローマ細胞とを融合させることによってハイブリドーマを樹立し、モノクローナル抗体を得ることもできる。
なお、抗原は抗原蛋白質をコードする遺伝子を遺伝子操作によって宿主細胞に産生させることによって得ることができる。具体的には、抗原遺伝子を発現可能なベクターを作製し、これを宿主細胞に導入して該遺伝子を発現させ、発現した抗原を精製すればよい。上記の遺伝子操作による抗原発現細胞、或は抗原を発現している細胞株、を動物に免疫する方法を用いることによっても抗体を取得できる。
抗TROP2抗体は、公知の手段によって取得することができる。
(1)以下の特性を有することを特徴とする抗体;
(a)TROP2に特異的に結合する
(b)TROP2と結合することによってTROP2発現細胞に内在化する活性を有する
(2)TROP2がヒトTROP2である上記(1)に記載の抗体。
(3)重鎖における相補性決定領域として配列番号23に記載のアミノ酸配列からなるCDRH1、配列番号24に記載のアミノ酸配列からなるCDRH2、及び配列番号25に記載のアミノ酸配列からなるCDRH3、並びに軽鎖における相補性決定領域として配列番号26に記載のアミノ酸配列からなるCDRL1、配列番号27に記載のアミノ酸配列からなるCDRL2、及び配列番号28に記載のアミノ酸配列からなるCDRL3を有する上記(1)又は(2)に記載の抗体。
(4)定常領域がヒト由来定常領域である上記(1)乃至(3)のいずれかに記載の抗体。
(5)ヒト化されている上記(1)乃至(4)のいずれかに記載の抗体。
(6)(a)配列番号12においてアミノ酸番号20乃至140に記載のアミノ酸配列、(b)配列番号14においてアミノ酸番号20乃至140に記載のアミノ酸配列、(c)配列番号16においてアミノ酸番号20乃至140に記載のアミノ酸配列、(d)(a)乃至(c)の配列に対して少なくとも95%以上の相同性を有するアミノ酸配列、及び(e)(a)乃至(c)の配列において1又は数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列からなる群から選択されたアミノ酸配列からなる重鎖の可変領域、並びに(f)配列番号18においてアミノ酸番号21乃至129に記載のアミノ酸配列、(g)配列番号20においてアミノ酸番号21乃至129に記載のアミノ酸配列、(h)配列番号22においてアミノ酸番号21乃至129に記載のアミノ酸配列、(i)(f)乃至(h)の配列に対して少なくとも95%以上の相同性を有するアミノ酸配列、及び(j)(f)乃至(h)の配列において1又は数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列からなる群から選択されたアミノ酸配列からなる軽鎖の可変領域、を有する上記(5)に記載の抗体。
(7)配列番号12においてアミノ酸番号20乃至140に記載のアミノ酸配列からなる重鎖の可変領域及び配列番号18においてアミノ酸番号21乃至129に記載のアミノ酸配列からなる軽鎖の可変領域、配列番号12においてアミノ酸番号20乃至140に記載のアミノ酸配列からなる重鎖の可変領域及び配列番号20においてアミノ酸番号21乃至129に記載のアミノ酸配列からなる軽鎖の可変領域、配列番号12においてアミノ酸番号20乃至140に記載のアミノ酸配列からなる重鎖の可変領域及び配列番号22においてアミノ酸番号21乃至129に記載のアミノ酸配列からなる軽鎖の可変領域、配列番号14においてアミノ酸番号20乃至140に記載のアミノ酸配列からなる重鎖の可変領域及び配列番号18においてアミノ酸番号21乃至129に記載のアミノ酸配列からなる軽鎖の可変領域、配列番号14においてアミノ酸番号20乃至140に記載のアミノ酸配列からなる重鎖の可変領域及び配列番号20においてアミノ酸番号21乃至129に記載のアミノ酸配列からなる軽鎖の可変領域、配列番号14においてアミノ酸番号20乃至140に記載のアミノ酸配列からなる重鎖の可変領域及び配列番号22においてアミノ酸番号21乃至129に記載のアミノ酸配列からなる軽鎖の可変領域、配列番号16においてアミノ酸番号20乃至140に記載のアミノ酸配列からなる重鎖の可変領域及び配列番号18においてアミノ酸番号21乃至129に記載のアミノ酸配列からなる軽鎖の可変領域、配列番号16においてアミノ酸番号20乃至140に記載のアミノ酸配列からなる重鎖の可変領域及び配列番号20においてアミノ酸番号21乃至129に記載のアミノ酸配列からなる軽鎖の可変領域、並びに配列番号16においてアミノ酸番号20乃至140に記載のアミノ酸配列からなる重鎖の可変領域及び配列番号22においてアミノ酸番号21乃至129に記載のアミノ酸配列からなる軽鎖の可変領域からなる群から選択される重鎖の可変領域及び軽鎖の可変領域を有する上記(6)に記載の抗体。
(8)配列番号12においてアミノ酸番号20乃至140に記載のアミノ酸配列からなる重鎖の可変領域及び配列番号18においてアミノ酸番号21乃至129に記載のアミノ酸配列からなる軽鎖の可変領域、配列番号14においてアミノ酸番号20乃至140に記載のアミノ酸配列からなる重鎖の可変領域及び配列番号18においてアミノ酸番号21乃至129に記載のアミノ酸配列からなる軽鎖の可変領域、配列番号14においてアミノ酸番号20乃至140に記載のアミノ酸配列からなる重鎖の可変領域及び配列番号20においてアミノ酸番号21乃至129に記載のアミノ酸配列からなる軽鎖の可変領域、並びに配列番号16においてアミノ酸番号20乃至140に記載のアミノ酸配列からなる重鎖の可変領域及び配列番号22においてアミノ酸番号21乃至129に記載のアミノ酸配列からなる軽鎖の可変領域からなる群から選択される重鎖の可変領域及び軽鎖の可変領域を有する上記(7)に記載の抗体。
(9)配列番号12においてアミノ酸番号20乃至470に記載のアミノ酸配列からなる重鎖及び配列番号18においてアミノ酸番号21乃至234に記載のアミノ酸配列からなる軽鎖、配列番号12においてアミノ酸番号20乃至470に記載のアミノ酸配列からなる重鎖及び配列番号20においてアミノ酸番号21乃至234に記載のアミノ酸配列からなる軽鎖、配列番号12においてアミノ酸番号20乃至470に記載のアミノ酸配列からなる重鎖及び配列番号22においてアミノ酸番号21乃至234に記載のアミノ酸配列からなる軽鎖、配列番号14においてアミノ酸番号20乃至470に記載のアミノ酸配列からなる重鎖及び配列番号18においてアミノ酸番号21乃至234に記載のアミノ酸配列からなる軽鎖、配列番号14においてアミノ酸番号20乃至470に記載のアミノ酸配列からなる重鎖及び配列番号20においてアミノ酸番号21乃至234に記載のアミノ酸配列からなる軽鎖、配列番号14においてアミノ酸番号20乃至470に記載のアミノ酸配列からなる重鎖及び配列番号22においてアミノ酸番号21乃至234に記載のアミノ酸配列からなる軽鎖、配列番号16においてアミノ酸番号20乃至470に記載のアミノ酸配列からなる重鎖及び配列番号18においてアミノ酸番号21乃至234に記載のアミノ酸配列からなる軽鎖、配列番号16においてアミノ酸番号20乃至470に記載のアミノ酸配列からなる重鎖及び配列番号20においてアミノ酸番号21乃至234に記載のアミノ酸配列からなる軽鎖、並びに配列番号16においてアミノ酸番号20乃至470に記載のアミノ酸配列からなる重鎖及び配列番号22においてアミノ酸番号21乃至234に記載のアミノ酸配列からなる軽鎖からなる群から選択される重鎖及び軽鎖からなる上記(6)又は(7)に記載の抗体。
(10)配列番号12に記載のアミノ酸配列からなる重鎖及び配列番号18に記載のアミノ酸配列からなる軽鎖、配列番号12に記載のアミノ酸配列からなる重鎖及び配列番号20に記載のアミノ酸配列からなる軽鎖、配列番号12に記載のアミノ酸配列からなる重鎖及び配列番号22に記載のアミノ酸配列からなる軽鎖、配列番号14に記載のアミノ酸配列からなる重鎖及び配列番号18に記載のアミノ酸配列からなる軽鎖、配列番号14に記載のアミノ酸配列からなる重鎖及び配列番号20に記載のアミノ酸配列からなる軽鎖、配列番号14に記載のアミノ酸配列からなる重鎖及び配列番号22に記載のアミノ酸配列からなる軽鎖、配列番号16に記載のアミノ酸配列からなる重鎖及び配列番号18に記載のアミノ酸配列からなる軽鎖、配列番号16に記載のアミノ酸配列からなる重鎖及び配列番号20に記載のアミノ酸配列からなる軽鎖、並びに配列番号16に記載のアミノ酸配列からなる重鎖及び配列番号22に記載のアミノ酸配列からなる軽鎖からなる群から選択される重鎖及び軽鎖からなる上記(6)又は(7)に記載の抗体。
(11)配列番号12においてアミノ酸番号20乃至470に記載のアミノ酸配列からなる重鎖及び配列番号18においてアミノ酸番号21乃至234に記載のアミノ酸配列からなる軽鎖、配列番号14においてアミノ酸番号20乃至470に記載のアミノ酸配列からなる重鎖及び配列番号18においてアミノ酸番号21乃至234に記載のアミノ酸配列からなる軽鎖、配列番号14においてアミノ酸番号20乃至470に記載のアミノ酸配列からなる重鎖及び配列番号20においてアミノ酸番号21乃至234に記載のアミノ酸配列からなる軽鎖、並びに配列番号16においてアミノ酸番号20乃至470に記載のアミノ酸配列からなる重鎖及び配列番号22においてアミノ酸番号21乃至234に記載のアミノ酸配列からなる軽鎖からなる群から選択される重鎖及び軽鎖からなる上記(8)に記載の抗体。
(12)重鎖カルボキシル末端のリシン残基が欠失している上記(1)乃至(11)のいずれかに記載の抗体。
(13)上記(1)乃至(12)のいずれかに記載の抗体をコードするポリヌクレオチドを含有する発現ベクターによって形質転換された宿主細胞を培養する工程及び当該工程で得られた培養物から目的の抗体を採取する工程を含む当該抗体の製造方法によって得られる抗体。
本明細書において、「癌」と「腫瘍」は同じ意味に用いている。
本明細書において、「遺伝子」という語には、DNAのみならずそのmRNA、cDNA、及びそのcRNAも含まれる。
本明細書において、「ポリヌクレオチド」という語は核酸と同じ意味で用いており、DNA、RNA、プローブ、オリゴヌクレオチド、及びプライマーも含まれる。
本明細においては、「ポリペプチド」と「蛋白質」は区別せずに用いている。
本明細書において、「細胞」には、動物個体内の細胞、培養細胞も含んでいる。
本明細書において、「TROP2」は、TROP2蛋白質と同じ意味で用いている。
本明細書における「CDR」とは、相補性決定領域(CDR:Complemetarity deterring region)を意味する。抗体分子の重鎖及び軽鎖にはそれぞれ3箇所のCDRがあることが知られている。CDRは、超可変領域(hypervariable domain)とも呼ばれ、抗体の重鎖及び軽鎖の可変領域内にあって、一次構造の変異性が特に高い部位であり、重鎖及び軽鎖のポリペプチド鎖の一次構造上において、それぞれ3ヶ所に分離している。本明細書中においては、抗体のCDRについて、重鎖のCDRを重鎖アミノ酸配列のアミノ末端側からCDRH1、CDRH2、CDRH3と表記し、軽鎖のCDRを軽鎖アミノ酸配列のアミノ末端側からCDRL1、CDRL2、CDRL3と表記する。これらの部位は立体構造の上で相互に近接し、結合する抗原に対する特異性を決定している。
本発明において、「ストリンジェントな条件下でハイブリダイズする」とは、市販のハイブリダイゼーション溶液ExpressHyb Hybridization Solution(クロンテック社製)中、68℃でハイブリダイズすること、又はDNAを固定したフィルターを用いて0.7-1.0MのNaCl存在下、68℃でハイブリダイゼーションを行った後、0.1-2倍濃度のSSC溶液(1倍濃度SSCとは150mM NaCl、15mMクエン酸ナトリウムからなる)を用い、68℃で洗浄することによって同定することができる条件又はそれと同等の条件でハイブリダイズすることをいう。
TROP2は、ヒト栄養膜細胞(trophoblasts)に発現するTACSTDファミリーのひとつであり、ヒト栄養膜細胞と癌細胞に共通する免疫抵抗性に関与する、1回膜貫通型の1型細胞膜蛋白質である。
本発明で用いるTROP2蛋白質は、ヒト、非ヒト哺乳動物(ラット、マウス等)のTROP2発現細胞から直接精製して使用するか、或は当該細胞の細胞膜画分を調製して使用することができ、また、TROP2をin vitroにて合成する、或は遺伝子操作によって宿主細胞に産生させることによって得ることができる。遺伝子操作では、具体的には、TROP2 cDNAを発現可能なベクターに組み込んだ後、転写と翻訳に必要な酵素、基質及びエネルギー物質を含む溶液中で合成する、或は他の原核生物、又は真核生物の宿主細胞を形質転換させることによってTROP2を発現させることによって、該蛋白質を得ることが出来る。また、前記の遺伝子操作によるTROP2発現細胞、或はTROP2を発現している細胞株をTROP2蛋白質として使用することも可能である。
TROP2のDNA配列及びアミノ酸配列は公的データベース上に公開されており、例えばNM_002353,NP_002344(NCBI)等のアクセッション番号により参照可能である。
また、上記TROP2のアミノ酸配列において、1又は数個のアミノ酸が置換、欠失及び/又は付加されたアミノ酸配列からなり、当該蛋白質と同等の生物活性を有する蛋白質もTROP2に含まれる。
ヒトTROP2蛋白質は、N末端26アミノ酸残基から成るシグナル配列、248アミノ酸残基から成る細胞外ドメイン、23アミノ酸残基から成る細胞膜貫通ドメイン、26アミノ酸残基から成る細胞内ドメインで構成されている。
本発明のTROP2に対する抗体は、この分野で通常実施される方法を用いて、TROP2又はTROP2のアミノ酸配列から選択される任意のポリペプチドを動物に免疫し、生体内に産生される抗体を採取、精製することによって得ることができる。抗原となるTROP2の生物種はヒトに限定されず、マウス、ラット等のヒト以外の動物に由来するTROP2を動物に免疫することもできる。この場合には、取得された異種TROP2に結合する抗体とヒトTROP2との交差性を試験することによって、ヒトの疾患に適用可能な抗体を選別できる。
また、公知の方法(例えば、Kohler and Milstein,Nature(1975)256,p.495-497;Kennet,R.ed.,Monoclonal Antibodies,p.365-367,Plenum Press,N.Y.(1980))に従って、TROP2に対する抗体を産生する抗体産生細胞とミエローマ細胞とを融合させることによってハイブリドーマを樹立し、モノクローナル抗体を得ることもできる。
なお、抗原となるTROP2はTROP2遺伝子を遺伝子操作によって宿主細胞に発現させることによって得ることができる。
具体的には、TROP2遺伝子を発現可能なベクターを作製し、これを宿主細胞に導入して該遺伝子を発現させ、発現したTROP2を精製すればよい。
また、上記の遺伝子操作によるTROP2発現細胞、或はTROP2を発現している細胞株をTROP2蛋白質として使用することも可能である。以下、具体的にTROP2に対する抗体の取得方法を説明する。
抗TROP2抗体を作製するための抗原としては、TROP2又はその少なくとも6個の連続した部分アミノ酸配列からなるポリペプチド、或はこれらに任意のアミノ酸配列や担体が付加された誘導体を挙げることができる。
TROP2は、ヒトの腫瘍組織或は腫瘍細胞から直接精製して使用することができ、また、TROP2をin vitroにて合成する、或は遺伝子操作によって宿主細胞に産生させることによって得ることができる。
遺伝子操作では、具体的には、TROP2のcDNAを発現可能なベクターに組み込んだ後、転写と翻訳に必要な酵素、基質及びエネルギー物質を含む溶液中で合成する、或は他の原核生物又は真核生物の宿主細胞を形質転換してTROP2を発現させることによって、抗原を得ることができる。
また、膜蛋白質であるTROP2の細胞外領域と抗体の定常領域とを連結した融合蛋白質を適切な宿主・ベクター系において発現させることによって、分泌蛋白質として抗原を得ることも可能である。
TROP2のcDNAは例えば、TROP2のcDNAを発現しているcDNAライブラリーを鋳型として、TROP2 cDNAを特異的に増幅するプライマーを用いてポリメラーゼ連鎖反応(以下「PCR」という;Saiki,R. K.,et al.,Science(1988)239,p.487-489 参照)を行う、いわゆるPCR法によって取得することができる。
ポリペプチドのin vitro合成としては、例えばロシュ・ダイアグノスティックス社製のラピッドトランスレーションシステム(RTS)を挙げることができるが、これに限定されることはない。
原核細胞の宿主としては、例えば、大腸菌(Escherichia coli)や枯草菌(Bacillus subtilis)等を挙げることができる。目的の遺伝子をこれらの宿主細胞内で形質転換させるには、宿主と適合し得る種由来のレプリコンすなわち複製起点と、調節配列を含んでいるプラスミドベクターで宿主細胞を形質転換させる。また、ベクターとしては、形質転換細胞に表現形質(表現型)の選択性を付与することができる配列を有するものが好ましい。
真核細胞の宿主細胞には、脊椎動物、昆虫、酵母等の細胞が含まれ、脊椎動物細胞としては、例えば、サルの細胞であるCOS細胞(Gluzman,Y.Cell(1981)23,p.175-182、ATCC CRL-1650;ATCC:American Type Culture Collection)、マウス線維芽細胞NIH3T3(ATCC No.CRL-1658)やチャイニーズ・ハムスター卵巣細胞(CHO細胞、ATCC CCL-61)のジヒドロ葉酸還元酵素欠損株(Urlaub,G. and Chasin,L.A.Proc.Natl.Acad.Sci.USA(1980)77,p.4126-4220)等がよく用いられているが、これらに限定されない。
上記の様にして得られる形質転換体は、この分野で通常実施される方法に従って培養することができ、該培養によって細胞内又は細胞外に目的のポリペプチドが産生される。
該培養に用いられる培地は、当業者であれば採用した宿主細胞に応じて慣用される各種のものを適宜選択することができ、大腸菌であれば、例えば、LB培地に必要に応じて、アンピシリン等の抗生物質やIPMGを添加して用いることができる。
上記培養によって、形質転換体の細胞内又は細胞外に産生される組換え蛋白質は、該蛋白質の物理的性質や化学的性質等を利用した各種の公知の分離操作法によって分離・精製することができる。
該方法としては、具体的には例えば、通常の蛋白質沈殿剤による処理、限外濾過、分子ふるいクロマトグラフィー(ゲル濾過)、吸着クロマトグラフィー、イオン交換クロマトグラフィー、アフィニティークロマトグラフィー等の各種液体クロマトグラフィー、透析法、これらの組合せ等を例示できる。
また、発現させる組換え蛋白質に6残基からなるヒスチジンタグを繋げることによって、ニッケルアフィニティーカラムで効率的に精製することができる。或は、発現させる組換え蛋白質にIgGのFc領域を繋げることによって、プロテインAカラムで効率的に精製することができる。
上記方法を組合せることによって容易に高収率、高純度で目的とするポリペプチドを大量に製造できる。
上記に述べた形質転換体自体を抗原として使用することも可能である。また、TROP2を発現する細胞株を抗原として使用することも可能である。この様な細胞株としては、ヒト化肺癌株NCI-H322、PC14、NCIH-H2122、又はLCAM1、ヒト前立腺癌株PC3、ヒト膵臓癌株BxPC-3、Capan-1、又はPK-1、ヒト卵巣癌株SKOV3並びにヒト大腸癌株COLO205を挙げることができるが、TROP2を発現する限り、これらの細胞株に限定されることはない。
TROP2と特異的に結合する抗体の例として、TROP2と特異的に結合するモノクローナル抗体を挙げることができるが、その取得方法は、以下に記載する通りである。
モノクローナル抗体の製造にあたっては、一般に下記の様な作業工程が必要である。
すなわち、
(a)抗原として使用する生体高分子の精製、又は抗原発現細胞の調製、
(b)抗原を動物に注射することによって免疫した後に血液を採取し、その抗体価を検定して脾臓摘出の時期を決定してから、抗体産生細胞を調製する工程、
(c)骨髄腫細胞(以下「ミエローマ」という)の調製、
(d)抗体産生細胞とミエローマとの細胞融合、
(e)目的とする抗体を産生するハイブリドーマ群の選別、
(f)単一細胞クローンへの分割(クローニング)、
(g)場合によっては、モノクローナル抗体を大量に製造するためのハイブリドーマの培養、又はハイブリドーマを移植した動物の飼育、
(h)この様にして製造されたモノクローナル抗体の生理活性、及びその結合特異性の検討、或は標識試薬としての特性の検定
等である。
以下、モノクローナル抗体の作製法を上記工程に沿って詳述するが、該抗体の作製法はこれに制限されず、例えば脾細胞以外の抗体産生細胞及びミエローマを使用することもできる。
抗原としては、前記した様な方法で調製したTROP2又はその一部を使用することができる。
また、TROP2発現組換え体細胞によって調製した膜画分、又はTROP2発現組換え体細胞自身、さらに、当業者に周知の方法を用いて化学合成した本発明の蛋白質の部分ペプチドを抗原として使用することもできる。
さらに、TROP2発現細胞株を抗原として使用することもできる。
工程(a)で得られた抗原と、フロインドの完全又は不完全アジュバント、或はカリミョウバンの様な助剤とを混合し、免疫原として実験動物に免疫する。この他に、抗原発現細胞を免疫原として実験動物に免疫する方法もある。実験動物は公知のハイブリドーマ作製法で用いられる動物を支障なく使用することができる。具体的には、例えばマウス、ラット、ヤギ、ヒツジ、ウシ、ウマ等を使用することができる。ただし、摘出した抗体産生細胞と融合させるミエローマ細胞の入手容易性等の観点から、マウス又はラットを被免疫動物とするのが好ましい。
また、実際に使用するマウス及びラットの系統には特に制限はなく、マウスの場合には、例えば各系統A、AKR、BALB/c、BDP、BA、CE、C3H、57BL、C57BL、C57L、DBA、FL、HTH、HT1、LP、NZB、NZW、RF、R III、SJL、SWR、WB、129等が、またラットの場合には、例えば、Wistar、Low、Lewis、Sprague、Dawley、ACI、BN、Fischer等を用いることができる。
これらのマウス及びラットは例えば日本クレア株式会社、日本チャ-ルス・リバー株式会社等の実験動物飼育販売業者より入手することができる。
被免疫動物としては、後述のミエローマ細胞との融合適合性を勘案すれば、マウスではBALB/c系統が、ラットではWistar及びLow系統が特に好ましい。
また、抗原のヒトとマウスでの相同性を考慮し、自己抗体を除去する生体機構を低下させたマウス、すなわち自己免疫疾患マウスを用いることも好ましい。
なお、これらのマウス又はラットの免疫時の週齢は、好ましくは5~12週齢、さらに好ましくは6~8週齢である。
TROP2又はこの組換え体によって動物を免疫するには、例えば、Weir,D.M.,Handbook of Experimental Immunology Vol.I.II.III.,Blackwell Scientific Publications,Oxford(1987);Kabat,E.A.and Mayer,M.M.,Experimental Immunochemistry,Charles C Thomas Publisher Springfield,Illinois(1964)等に詳しく記載されている公知の方法を用いることができる。
これらの免疫法のうち、本発明において好適な方法を具体的に示せば、例えば以下の通りである。
すなわち、まず、抗原である膜蛋白質画分、又は抗原を発現させた細胞を動物の皮内又は腹腔内に投与する。ただし、免疫効率を高めるためには両者の併用が好ましく、前半は皮内投与を行い、後半又は最終回のみ腹腔内投与を行うと、特に免疫効率を高めることができる。
抗原の投与スケジュールは、被免疫動物の種類、個体差等によって異なるが、一般には、抗原投与回数3~6回、投与間隔2~6週間が好ましく、投与回数3~4回、投与間隔2~4週間がさらに好ましい。
また、抗原の投与量は、動物の種類、個体差等によって異なるが、一般には0.05~5mg、好ましくは0.1~0.5mg程度とする。
追加免疫は、以上の通りの抗原投与の1~6週間後、好ましくは1~4週間後、さらに好ましくは1~3週間後に行う。免疫原が細胞の場合には、1×106乃至1×107個の細胞を使用する。
なお、追加免疫を行う際の抗原投与量は、動物の種類、大きさ等によって異なるが、一般に、例えばマウスの場合には0.05~5mg、好ましくは0.1~0.5mg、さらに好ましくは0.1~0.2mg程度とする。免疫原が細胞の場合には、1×106乃至1×107個の細胞を使用する。
上記追加免疫から1~10日後、好ましくは2~5日後、さらに好ましくは2~3日後に被免疫動物から抗体産生細胞を含む脾臓細胞又はリンパ球を無菌的に取り出す。その際に抗体価を測定し、抗体価が十分高くなった動物を抗体産生細胞の供給源として用いれば、以後の操作の効率を高めることができる。
ここで用いられる抗体価の測定法としては、例えば、RIA法又はELISA法を挙げることができるがこれらの方法に制限されない。本発明における抗体価の測定は、例えばELISA法によれば、以下に記載する様な手順によって行うことができる。
まず、精製又は部分精製した抗原をELISA用96穴プレート等の固相表面に吸着させ、さらに抗原が吸着していない固相表面を抗原と無関係な蛋白質、例えばウシ血清アルブミン(BSA)によって覆い、該表面を洗浄後、第一抗体として段階希釈した試料(例えばマウス血清)に接触させ、上記抗原に試料中の抗体を結合させる。
さらに第二抗体として酵素標識されたマウス抗体に対する抗体を加えてマウス抗体に結合させ、洗浄後該酵素の基質を加え、基質分解に基づく発色による吸光度の変化等を測定することによって、抗体価を算出する。
被免疫動物の脾臓細胞又はリンパ球からの抗体産生細胞の分離は、公知の方法(例えば、Kohler et al.,Nature(1975)256,p.495;Kohler et al.,Eur.J.Immunol.(1977)6,p.511;Milstein et al.,Nature(1977),266,p.550;Walsh,Nature,(1977)266,p.495)に従って行うことができる。例えば、脾臓細胞の場合には、脾臓を細切して細胞をステンレスメッシュで濾過した後、イーグル最小必須培地(MEM)に浮遊させて抗体産生細胞を分離する一般的方法を採用することができる。
細胞融合に用いるミエローマ細胞には特段の制限はなく、公知の細胞株から適宜選択して用いることができる。ただし、融合細胞からハイブリドーマを選択する際の利便性を考慮して、その選択手続が確立しているHGPRT(Hypoxanthine-guanine phosphoribosyl transferase)欠損株を用いるのが好ましい。
すなわち、マウス由来のX63-Ag8(X63)、NS1-ANS/1(NS1)、P3X63-Ag8.U1(P3U1)、X63-Ag8.653(X63.653)、SP2/0-Ag14(SP2/0)、MPC11-45.6TG1.7(45.6TG)、FO、S149/5XXO、BU.1等、ラット由来の210.RSY3.Ag.1.2.3(Y3)等、ヒト由来のU266AR(SKO-007)、GM1500・GTG-A12(GM1500)、UC729-6、LICR-LOW-HMy2(HMy2)、8226AR/NIP4-1(NP41)等である。これらのHGPRT欠損株は例えば、ATCC等から入手することができる。
これらの細胞株は、適当な培地、例えば8-アザグアニン培地(RPMI-1640培地にグルタミン、2-メルカプトエタノール、ゲンタマイシン及びウシ胎児血清(以下「FBS」という)を加えた培地に8-アザグアニンを加えた培地)、イスコフ改変ダルベッコ培地(Iscove’s Modified Dulbecco’s Medium;IMDM)、又はダルベッコ改変イーグル培地(Dulbecco’s Modified Eagle Medium;DMEM)で継代培養するが、細胞融合の3乃至4日前に正常培地(例えば、10% FCSを含むASF104培地(味の素株式会社製))で継代培養し、融合当日に2×107以上の細胞数を確保しておく。
抗体産生細胞とミエローマ細胞との融合は、公知の方法(Weir,D.M.,Handbookof Experimental Immunology Vol.I.II.III.,Blackwell Scientific Publications,Oxford(1987);Kabat,E.A.and Mayer,M.M.,Experimental Immunochemistry,Charles C Thomas Publisher Springfield,Illinois(1964)等)に従い、細胞の生存率を極度に低下させない程度の条件下で適宜実施することができる。
その様な方法として、例えば、ポリエチレングリコール等の高濃度ポリマー溶液中で抗体産生細胞とミエローマ細胞とを混合する化学的方法、電気的刺激を利用する物理的方法等を用いることができる。このうち、上記化学的方法の具体例を示せば以下の通りである。
すなわち、高濃度ポリマー溶液としてポリエチレングリコールを用いる場合には、分子量1500~6000、好ましくは2000~4000のポリエチレングリコール溶液中で、30~40℃、好ましくは35~38℃の温度で抗体産生細胞とミエローマ細胞とを1~10分間、好ましくは5~8分間混合する。
上記細胞融合によって得られるハイブリドーマの選択方法は特に制限はないが、通常HAT(ヒポキサンチン・アミノプテリン・チミジン)選択法(Kohler et al.,Nature(1975)256,p.495;Milstein et al.,Nature(1977)266,p.550)が用いられる。
この方法は、アミノプテリンで生存し得ないHGPRT欠損株のミエローマ細胞を用いてハイブリドーマを得る場合に有効である。すなわち、未融合細胞及びハイブリドーマをHAT培地で培養することによって、アミノプテリンに対する耐性を持ち合わせたハイブリドーマのみを選択的に残存させ、かつ増殖させることができる。
ハイブリドーマのクローニング法としては、例えばメチルセルロース法、軟アガロース法、限界希釈法等の公知の方法を用いることができる(例えばBarbara, B.M.and Stanley,M.S.:Selected Methods in Cellular Immunology,W.H.Freeman and Company,San Francisco(1980)参照)。これらの方法のうち、特にメチルセルロース法等の三次元培養法が好適である。例えば、細胞融合によって形成されたハイブリドーマ群をClonaCell-HY Selection Medium D(StemCell Technologies社製 #03804)等のメチルセルロース培地に懸濁して培養し、形成されたハイブリドーマコロニーを回収することでモノクローンハイブリドーマの取得が可能である。回収された各ハイブリドーマコロニーを培養し、得られたハイブリドーマ培養上清中に安定して抗体価の認められたものをTROP2モノクローナル抗体産生ハイブリドーマ株として選択する。
TINA1抗体の重鎖可変領域は、配列表の配列番号2に示されるアミノ酸配列を有する。また、TINA1抗体の軽鎖可変領域は、配列表の配列番号4に示されるアミノ酸配列を有する。
この様にして選択されたハイブリドーマは、これを培養することによって、モノクローナル抗体を効率よく得ることができるが、培養に先立ち、目的とするモノクローナル抗体を産生するハイブリドーマをスクリーニングすることが望ましい。
このスクリーニングにはそれ自体既知の方法が採用できる。
本発明における抗体価の測定は、例えば上記(b)の項目で説明したELISA法によって行うことができる。
以上の方法によって得たハイブリドーマは、液体窒素中又は-80℃以下の冷凍庫中に凍結状態で保存することができる。
クローニングを完了したハイブリドーマは、培地をHT培地から正常培地に換えて培養される。
大量培養は、大型培養瓶を用いた回転培養、或はスピナー培養で行われる。この大量培養における上清から、ゲル濾過等、当業者に周知の方法を用いて精製することによって、本発明の蛋白質に特異的に結合するモノクローナル抗体を得ることができる。
また、同系統のマウス(例えば、上記のBALB/c)、或はNu/Nuマウスの腹腔内にハイブリドーマを注射し、該ハイブリド-マを増殖させることによって、本発明のモノクローナル抗体を大量に含む腹水を得ることができる。
腹腔内に投与する場合には、事前(3~7日前)に2,6,10,14-テトラメチルペンタデカン(2,6,10,14-tetramethylpentadecane;プリスタン)等の鉱物油を投与すると、より多量の腹水が得られる。
例えば、ハイブリドーマと同系統のマウスの腹腔内に予め免疫抑制剤を注射し、T細胞を不活性化した後、20日後に106~107個のハイブリドーマ・クローン細胞を、血清を含まない培地中に浮遊(0.5ml)させて腹腔内に投与し、通常腹部が膨満し、腹水がたまったところでマウスより腹水を採取する。この方法によって、培養液中に比べて約100倍以上の濃度のモノクローナル抗体が得られる。
上記方法によって得たモノクローナル抗体は、例えばWeir,D.M.:Handbook of Experimental Immunology,Vol.I,II,III,Blackwell Scientific Publications,Oxford(1978)に記載されている方法で精製することができる。
かくして得られるモノクローナル抗体は、TROP2に対して高い抗原特異性を有する。
かくして得られたモノクローナル抗体のアイソタイプ及びサブクラスの決定は以下の様に行うことができる。
まず、同定法としてはオクテルロニー(Ouchterlony)法、ELISA法、又はRIA法を挙げることができる。
オクテルロニー法は簡便ではあるが、モノクローナル抗体の濃度が低い場合には濃縮操作が必要である。
一方、ELISA法又はRIA法を用いた場合は、培養上清をそのまま抗原吸着固相と反応させ、さらに第二次抗体として各種イムノグロブリンアイソタイプ、サブクラスに対応する抗体を用いることによって、モノクローナル抗体のアイソタイプ、サブクラスを同定することが可能である。
また、さらに簡便な方法として、市販の同定用のキット(例えば、マウスタイパーキット;バイオラッド社製)等を利用することもできる。
さらに、蛋白質の定量は、フォーリンロウリー法及び280nmにおける吸光度(1.4(OD280)=イムノグロブリン1mg/ml)より算出する方法によって行うことができる。
さらに、(2)の(a)乃至(h)の工程を再度実施して別途に独立してモノクローナル抗体を取得した場合においても、TINA1抗体と同等の細胞傷害活性を有する抗体を取得することが可能である。この様な抗体の一例として、TINA1抗体と同一のエピトープに結合する抗体を挙げることができる。新たに作製されたモノクローナル抗体が、TINA1抗体の結合する部分ペプチド又は部分立体構造に結合すれば、該モノクローナル抗体がTINA1抗体と同一のエピトープに結合すると判定することができる。また、TINA1抗体のTROP2に対する結合に対して該モノクローナル抗体が競合する(即ち、該モノクローナル抗体がTINA1抗体とTROP2の結合を妨げる)ことを確認することによって、具体的なエピトープの配列又は構造が決定されていなくても、該モノクローナル抗体が抗TROP2抗体と同一のエピトープに結合すると判定することができる。エピトープが同一であることが確認された場合、該モノクローナル抗体がTINA1抗体と同等の抗原結合能又は生物活性を有していることが強く期待される。
本発明の抗体には、上記TROP2に対するモノクローナル抗体に加え、ヒトに対する異種抗原性を低下させること等を目的として人為的に改変した遺伝子組換え型抗体、例えば、キメラ(Chimeric)抗体、ヒト化(Humanized)抗体、ヒト抗体等も含まれる。これらの抗体は、既知の方法を用いて製造することができる。
キメラ抗体としては、抗体の可変領域と定常領域が互いに異種である抗体、例えばマウス又はラット由来抗体の可変領域をヒト由来の定常領域に接合したキメラ抗体を挙げることができる(Proc.Natl.Acad.Sci.U.S.A.,81,6851-6855,(1984)参照)。
ヒト化抗体としては、相補性決定領域(CDR)のみをヒト由来の抗体に組み込んだ抗体(Nature(1986)321,p.522-525参照)、CDR移植法によって、CDRの配列に加えて一部のフレームワークのアミノ酸残基もヒト抗体に移植した抗体(国際公開第90/07861号)を挙げることができる。
但し、TINA1抗体由来のヒト化抗体としては、TINA1抗体の6種全てのCDR配列を保持する限り、特定のヒト化抗体に限定されることはない。なお、TINA1抗体の重鎖可変領域は、配列表の配列番号23に示されるアミノ酸配列からなるCDRH1(TAGMQ)、配列番号24に示されるアミノ酸配列からなるCDRH2(WINTHSGVPKYAEDFKG)、及び配列番号25に示されるアミノ酸配列からなるCDRH3(SGFGSSYWYFDV)を保有している。また、TINA1抗体の軽鎖可変領域は、配列表の配列番号26に示されるアミノ酸配列からなるCDRL1(KASQDVSTAVA)、配列番号27に示されるアミノ酸配列からなるCDRL2(SASYRYT)、及び配列番号28に示されるアミノ酸配列からなるCDRL3(QQHYITPLT)を保有している。
なお、本明細書における「数個」とは、1乃至10個、1乃至9個、1乃至8個、1乃至7個、1乃至6個、1乃至5個、1乃至4個、1乃至3個、又は1若しくは2個を意味する。
また、配列表の配列番号18、20、又は22に示される軽鎖アミノ酸配列中で、1乃至20番目のアミノ酸残基からなるアミノ酸配列はシグナル配列であり、21乃至129番目のアミノ酸残基からなるアミノ酸配列は可変領域であり、130乃至234番目のアミノ酸残基からなるアミノ酸配列は定常領域である。配列番号18の配列は図6に、配列番号20の配列は図7に、配列番号22の配列は図8に各々記載されている。
また、遺伝子組換え技術によって、その様なヒト抗体の重鎖及び軽鎖の各々をコードするcDNA、好ましくは該cDNAを含むベクターによって真核細胞を形質転換し、遺伝子組換えヒトモノクローナル抗体を産生する形質転換細胞を培養することによって、この抗体を培養上清中から得ることもできる。
ここで、宿主としては例えば真核細胞、好ましくはCHO細胞、リンパ球やミエローマ等の哺乳動物細胞を用いることができる。
例えば、ヒト抗体の可変領域を一本鎖抗体(scFv)としてファージ表面に発現させて、抗原に結合するファージを選択するファージディスプレイ法(Nature Biotechnology(2005),23,(9),p.1105-1116)を用いることができる。
抗原に結合することで選択されたファージの遺伝子を解析することによって、抗原に結合するヒト抗体の可変領域をコードするDNA配列を決定することができる。
抗原に結合するscFvのDNA配列が明らかになれば、当該配列を有する発現ベクターを作製し、適当な宿主に導入して発現させることによってヒト抗体を取得することができる(国際公開第92/01047号、同92/20791号、同93/06213号、同93/11236号、同93/19172号、同95/01438号、同95/15388号;Annu.Rev.Immunol(1994)12,p.433-455;Nature Biotechnology(2005)23(9),p.1105-1116)。
新たに作製されたヒト抗体が、TINA1抗体の結合する部分ペプチド又は部分立体構造に結合すれば、該ヒト抗体がTINA1抗体と同一のエピトープに結合すると判定することができる。また、TINA1抗体のTROP2に対する結合に対して該ヒト抗体が競合する(すなわち、該ヒト抗体が、TINA1抗体とTROP2の結合を妨げる)ことを確認することによって、具体的なエピトープの配列又は構造が決定されていなくても、該ヒト抗体がTINA1抗体と同一のエピトープに結合すると判定することができる。エピトープが同一であることが確認された場合、該ヒト抗体がTINA1抗体と同等の生物活性を有していることが強く期待される。
以上の方法によって得られたキメラ抗体、ヒト化抗体、又はヒト抗体は、公知の方法等によって抗原に対する結合性を評価し、好適な抗体を選抜することができる。
抗体遺伝子を一旦単離した後、適当な宿主に導入して抗体を作製する場合には、適当な宿主と発現ベクターの組み合わせを使用することができる。抗体遺伝子の具体例としては、本明細書に記載された抗体の重鎖配列をコードする遺伝子、及び軽鎖配列をコードする遺伝子を組み合わせたものを挙げることができる。宿主細胞を形質転換する際には、重鎖配列遺伝子と軽鎖配列遺伝子は、同一の発現ベクターに挿入されていることが可能であり、又別々の発現ベクターに挿入されていることも可能である。
真核細胞を宿主として使用する場合、動物細胞、植物細胞や、真核微生物を用いることができる。特に動物細胞としては、哺乳類細胞、例えば、サルの細胞であるCOS細胞(Gluzman,Y.Cell(1981)23,p.175-182、ATCC CRL-1650)、マウス線維芽細胞NIH3T3(ATCC No.CRL-1658)やチャイニーズ・ハムスター卵巣細胞(CHO細胞、ATCC CCL-61)のジヒドロ葉酸還元酵素欠損株(Urlaub,G.and Chasin,L.A.Proc.Natl.Acad.Sci.U.S.A.(1980)77,p.4126-4220)を挙げることができる。
原核細胞を使用する場合は、例えば、大腸菌、枯草菌を挙げることができる。
これらの細胞に目的とする抗体遺伝子を形質転換によって導入し、形質転換された細胞をin vitroで培養することによって抗体が得られる。当該培養においては抗体の配列によって収量が異なる場合があり、同等な結合活性を持つ抗体の中から収量を指標に医薬としての生産が容易なものを選別することが可能である。よって、本発明の抗体には、上記形質転換された宿主細胞を培養する工程、及び当該工程で得られた培養物から目的の抗体又は当該抗体の機能性断片を採取する工程を含むことを特徴とする当該抗体の製造方法によって得られる抗体も含まれる。
クロマトグラフィーとしては、アフィニティークロマトグラフィー、イオン交換クロマトグラフィー、疎水性クロマトグラフィー、ゲル濾過クロマトグラフィー、逆相クロマトグラフィー、吸着クロマトグラフィー等を挙げることができる。
これらのクロマトグラフィーは、HPLCやFPLC等の液体クロマトグラフィーを用いて行うことができる。
アフィニティークロマトグラフィーに用いるカラムとしては、プロテインAカラム、プロテインGカラムを挙げることができる。例えばプロテインAカラムを用いたカラムとして、Hyper D,POROS,Sepharose F.F.(ファルマシア)等を挙げることができる。
また抗原を固定化した担体を用いて、抗原への結合性を利用して抗体を精製することも可能である。
本発明の抗TROP2抗体-薬物コンジュゲートに結合される抗腫瘍性化合物について述べる。本発明で使用される抗腫瘍性化合物としては、抗腫瘍効果を有する化合物であって、リンカー構造に結合できる置換基、部分構造を有するものであれば特に制限はない。抗腫瘍性化合物は、リンカーの一部又は全部が腫瘍細胞内で切断されて抗腫瘍性化合物部分が遊離して抗腫瘍効果が発現される。リンカーが薬物との結合部分で切断されれば抗腫瘍性化合物が未修飾の構造で遊離され、その本来の抗腫瘍効果が発揮される。
本発明で使用される抗腫瘍性化合物として、カンプトテシン誘導体であるエキサテカン((1S,9S)-1-アミノ-9-エチル-5-フルオロ-2,3-ジヒドロ-9-ヒドロキシ-4-メチル-1H,12H-ベンゾ[de]ピラノ[3',4':6,7]インドリジノ[1,2-b]キノリン-10,13(9H,15H)-ジオン;次式:)

エキサテカンはカンプトテシン構造を有するので、酸性水性媒体中(例えばpH3程度)ではラクトン環が形成された構造(閉環体)に平衡が偏り、一方、塩基性水性媒体中(例えばpH10程度)ではラクトン環が開環した構造(開環体)に平衡が偏ることが知られている。この様な閉環構造及び開環構造に対応するエキサテカン残基を導入した薬物コンジュゲートであっても同等の抗腫瘍効果が期待され、いずれの状態のものも本発明の範囲に包含されることはいうまでもない。
本発明の抗TROP2抗体-薬物コンジュゲートにおいて抗腫瘍性化合物を抗TROP2抗体に結合させるリンカー構造について述べる。当該リンカーは、次式:
-L1-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-
の構造を有しており、抗体はL1の末端(L2が結合するのとは反対側の末端)で結合し、抗腫瘍性化合物は-La-(CH2)n2-C(=O)-部分のカルボニル基で結合する。
n1は、0から6の整数を示すが、好ましくは1から5の整数であり、より好ましくは1から3である。
L1は、
-(Succinimid-3-yl-N)-(CH2)n3-C(=O)-
の構造で示される。
ここで、n3は、2から8の整数であり、『-(Succinimid-3-yl-N)-』は、次式


-(Succinimid-3-yl-N)-CH2CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-
等を挙げることができる。
L2は、
-NH-(CH2CH2-O)n4-CH2CH2-C(=O)-
で示される構造であるが、L2は存在しなくともよく、この場合L2は単結合となる。また、n4は、1から6の整数であり、好ましくは2から4である。L2は末端のアミノ基でL1に結合し、反対の末端のカルボニル基でLPと結合する。
-NH-CH2CH2-O-CH2CH2-C(=O)-、
-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-、
-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-、
-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-、
-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-、
-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-
等を挙げることができる。
LPは、2から7個のアミノ酸で構成されるペプチド残基である。すなわち、2から7個のアミノ酸がペプチド結合したオリゴペプチドの残基によって構成される。LPは、N末端においてL2に結合し、C末端においてリンカーの-NH-(CH2)n1-La-(CH2)n2-C(=O)-部分のアミノ基に結合する。
LPのアミノ酸配列は、特に限定されないが、構成するアミノ酸として、フェニルアラニン(Phe;F)、チロシン(Tyr;Y)、ロイシン(Leu;L)、グリシン(Gly;G)、アラニン(Ala;A)、バリン(Val;V)、リシン(Lys;K)、シトルリン(Cit)、セリン(Ser;S)、グルタミン酸(Glu;E)、アスパラギン酸(Asp;D)等を挙げることができる。
これらのうちで好ましくは、フェニルアラニン、グリシン、バリン、リシン、シトルリン、セリン、グルタミン酸、アスパラギン酸を挙げることができる。アミノ酸の種類によって、薬物遊離のパターンをコントロールすることができる。アミノ酸の数は、2から7個でよい。
-GGF-、
-DGGF-、
-(D-)D-GGF-、
-EGGF-、
-GGFG-、
-SGGF-、
-KGGF-、
-DGGFG-、
-GGFGG-、
-DDGGFG-、
-KDGGFG-、
-GGFGGGF-
を挙げることができる。上記の『(D-)D』はD-アスパラギン酸を意味する。本発明の抗体-薬物コンジュゲートの特に好ましいLPとして、-GGFG-のテトラペプチド残基を挙げることができる。
La-(CH2)n2-C(=O)-におけるLaは、-O-の構造であるか、又は単結合である。n2は、0から5の整数であるが、好ましくは0から3であり、より好ましくは0または1である。
La-(CH2)n2-C(=O)-としては以下の構造のものを挙げることができる。
-O-CH2-C(=O)-、
-O-CH2CH2-C(=O)-、
-O-CH2CH2CH2-C(=O)-、
-O-CH2CH2CH2CH2-C(=O)-、
-O-CH2CH2CH2CH2CH2-C(=O)-、
-CH2-C(=O)-、
-CH2CH2-C(=O)-、
-CH2CH2CH2-C(=O)-、
-CH2CH2CH2CH2-C(=O)-、
-CH2CH2CH2CH2CH2-C(=O)-。
これらのうちでは、
-O-CH2-C(=O)-、
-O-CH2CH2-C(=O)-
である場合か、Laが単結合でn2が0である場合が好ましい。
-NH-CH2-C(=O)-、
-NH-CH2CH2-C(=O)-、
-NH-CH2-O-CH2-C(=O)-、
-NH-CH2CH2-O-C(=O)-、
-NH-CH2CH2-O-CH2-C(=O)-、
-NH-CH2CH2CH2-C(=O)-、
-NH-CH2CH2CH2CH2-C(=O)-、
-NH-CH2CH2CH2CH2CH2-C(=O)-、
等を挙げることができる。
-NH-CH2CH2CH2-C(=O)-、
-NH-CH2-O-CH2-C(=O)-、
-NH-CH2CH2-O-C(=O)-
である。
NH2-CH2CH2-C(=O)-(NH-DX)、
NH2-CH2CH2CH2-C(=O)-(NH-DX)、
NH2-CH2-O-CH2-C(=O)-(NH-DX)、
NH2-CH2CH2-O-CH2-C(=O)-(NH-DX)。
なお、NH2-CH2-O-CH2-C(=O)-(NH-DX)の場合は同分子内にあるアミナール構造が不安定であるため、さらに自己分解して
HO-CH2-C(=O)-(NH-DX)
が遊離されることが確認された。これらの化合物は本発明の抗体-薬物コンジュゲートの製造中間体としても好適に用いることができる。
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)。
これらのうちでより好ましくは、次のものである。
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)。
さらに、好ましくは、次のものである。
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)。
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-。
これらのうちでより好ましくは、次のものである。
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-。
さらに、好ましくは、次のものを挙げることができる。
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-。
次に、本発明の抗体-薬物コンジュゲート或はその製造中間体の代表的な製造方法について説明する。なお、以下において、化合物を示すために、各反応式中に示される化合物の番号を用いる。すなわち、『式(1)の化合物』、『化合物(1)』等と称する。またこれ以外の番号の化合物についても同様に記載する。
式(1)で示される、チオエーテルを介して抗体と薬物-リンカー構造が結合している抗体-薬物コンジュゲートは、例えば下記の方法によって製造することができる。



スルフヒドリル基を有する抗体(3a)は、当業者周知の方法で得ることができる(Hermanson, G.T, Bioconjugate Techniques, pp.56-136, pp.456-493, Academic Press(1996))。例えば、Traut’s試薬を抗体のアミノ基に作用させる;N-サクシンイミジル S-アセチルチオアルカノエート類を抗体のアミノ基に作用させた後、ヒドロキシルアミンを作用させる;N-サクシンイミジル 3-(ピリジルジチオ)プロピオネートを作用させた後、還元剤を作用させる;ジチオトレイトール、2-メルカプトエタノール、トリス(2-カルボキシエチル)ホスフィン塩酸塩(TCEP)等の還元剤を抗体に作用させて抗体内ヒンジ部のジスルフィド結合を還元してスルフヒドリル基を生成させる;等の方法を挙げることができるがこれらに限定されることはない。
具体的には、還元剤としてTCEPを、抗体内ヒンジ部ジスルフィド1個当たりに対して0.3乃至3モル当量用い、キレート剤を含む緩衝液中で、抗体と反応させることで、抗体内ヒンジ部ジスルフィドが部分的又は完全に還元された抗体を得ることができる。キレート剤としては、例えばエチレンジアミン四酢酸(EDTA)やジエチレントリアミン5酢酸(DTPA)等を挙げることができる。これらを1mM乃至20mMの濃度で用いればよい。緩衝液としては、リン酸ナトリウムやホウ酸ナトリウム、酢酸ナトリウム溶液等を用いることができる。具体的には、抗体は4℃乃至37℃で1乃至4時間TCEPと反応させることで部分的又は完全に還元されたスルフヒドリル基を有する抗体(3a)を得ることが出来る。
ここでスルフヒドリル基を薬物-リンカー部分に付加させる反応を実施することでチオエーテル結合によって薬物-リンカー部分を結合させることができる。
スルフヒドリル基を有する抗体(3a)1個あたり、2乃至20モル当量の化合物(2)を使用して、抗体1個当たり2個乃至8個の薬物が結合した抗体―薬物コンジュゲート(1)を製造することができる。具体的には、スルフヒドリル基を有する抗体(3a)を含む緩衝液に、化合物(2)を溶解させた溶液を加えて反応させればよい。ここで、緩衝液としては、酢酸ナトリウム溶液、リン酸ナトリウムやホウ酸ナトリウム等を用いればよい。反応時のpHは5乃至9であり、より好適にはpH7付近で反応させればよい。化合物(2)を溶解させる溶媒としては、ジメチルスルホキシド(DMSO)、ジメチルホルムアミド(DMF)、ジメチルアセトアミド(DMA)、N-メチル-2-ピリドン(NMP)等の有機溶媒を用いることができる。
化合物(2)を溶解させた有機溶媒溶液を、スルフヒドリル基を有する抗体(3a)を含む緩衝液に1乃至20%v/vを加えて反応させればよい。反応温度は、0乃至37℃、より好適には10乃至25℃であり、反応時間は、0.5乃至2時間である。反応は、未反応の化合物(2)の反応性をチオール含有試薬によって失活させることによって終了できる。チオール含有試薬は例えば、システインまたはN-アセチル-L-システイン(NAC)である。より具体的には、NACを、用いた化合物(2)に対して、1乃至2モル当量加え、室温で10乃至30分インキュベートすることにより反応を終了できる。
製造した抗体-薬物コンジュゲート(1)は、以下の共通操作によって濃縮、バッファー交換、精製、抗体濃度及び抗体一分子あたりの薬物平均結合数の測定を行い、抗体-薬物コンジュゲート(1)の同定を行うことができる。
Amicon Ultra(50,000 MWCO,Millipore Corporation)の容器内に抗体又は抗体-薬物コンジュゲート溶液を入れ、遠心機(Allegra X-15R,Beckman Coulter,Inc.)を用いた遠心操作(2000G乃至3800Gで5乃至20分間遠心)にて、抗体又は抗体-薬物コンジュゲート溶液を濃縮した。
共通操作B:抗体の濃度測定
UV測定器(Nanodrop 1000,Thermo Fisher Scientific Inc.)を用いて、メーカー規定の方法に従い、抗体濃度の測定を行った。その際に、抗体ごとに異なる280nm吸光係数(1.3mLmg-1cm-1乃至1.8mLmg-1cm-1)を用いた。
共通操作C-1:抗体のバッファー交換
Sephadex G-25担体を使用したNAP-25カラム(Cat.No.17-0852-02,GE Healthcare Japan Corporation)を、メーカー規定の方法に従い、塩化ナトリウム(137mM)及びエチレンジアミン四酢酸(EDTA,5mM)を含むリン酸緩衝液(10mM,pH6.0;本明細書でPBS6.0/EDTAと称する)にて平衡化させた。このNAP-25カラム一本につき、抗体水溶液2.5mLをのせた後、PBS6.0/EDTA3.5mLで溶出させた画分(3.5mL)を分取した。この画分を共通操作Aによって濃縮し、共通操作Bを用いて抗体濃度の測定を行った後に、PBS6.0/EDTAを用いて10mg/mLに抗体濃度を調整した。
共通操作C-2:抗体のバッファー交換
Sephadex G-25担体を使用したNAP-25カラム(Cat.No.17-0852-02,GE Healthcare Japan Corporation)を、メーカー規定の方法に従い、塩化ナトリウム(50mM)及びEDTA(2mM)を含むリン酸緩衝液(50mM,pH6.5;本明細書でPBS6.5/EDTAと称する)にて平衡化させた。このNAP-25カラム一本につき、抗体水溶液2.5mLをのせた後、PBS6.5/EDTA3.5mLで溶出させた画分(3.5mL)を分取した。この画分を共通操作Aによって濃縮し、共通操作Bを用いて抗体濃度の測定を行った後に、PBS6.5/EDTAを用いて20mg/mLに抗体濃度を調整した。
共通操作D:抗体-薬物コンジュゲートの精製
市販のリン酸緩衝液(PBS7.4,Cat.No.10010-023,Invitrogen)、塩化ナトリウム(137mM)を含むリン酸ナトリウム緩衝液(10mM,pH6.0;本明細書でPBS6.0と称する)又はSorbitol(5%)を含む酢酸緩衝液(10mM,pH5.5;本明細書でABSと称する)のいずれかの緩衝液でNAP-25カラムを平衡化させた。このNAP-25カラムに、抗体-薬物コンジュゲート反応水溶液(約1.5mL)をのせ、メーカー規定の量の緩衝液で溶出させることで、抗体画分を分取した。この分取画分を再びNAP-25カラムにのせ、緩衝液で溶出させるゲルろ過精製操作を計2乃至3回繰り返すことで、未結合の薬物リンカーや低分子化合物(トリス(2-カルボキシエチル)ホスフィン塩酸塩(TCEP)、N-アセチル-L-システイン(NAC)、ジメチルスルホキシド)を除いた抗体-薬物コンジュゲートを得た。
共通操作E:抗体-薬物コンジュゲートにおける抗体濃度及び抗体一分子あたりの薬物平均結合数の測定(1)
抗体-薬物コンジュゲートにおける結合薬物濃度は、抗体-薬物コンジュゲート水溶液の280nm及び370nmの二波長におけるUV吸光度を測定した後に下記の計算を行うことで、算出することができる。
ある波長における全吸光度は系内に存在する全ての吸収化学種の吸光度の和に等しい(吸光度の加成性)ことから、抗体と薬物のコンジュゲーション前後において、抗体及び薬物のモル吸光係数に変化がないと仮定すると、抗体-薬物コンジュゲートにおける抗体濃度及び薬物濃度は、下記の関係式で示される。
A280=AD,280+AA,280=εD,280CD+εA,280CA 式(I)
A370=AD,370+AA,370=εD,370CD+εA,370CA 式(II)
ここで、A280は280nmにおける抗体-薬物コンジュゲート水溶液の吸光度を示し、A370は370nmにおける抗体-薬物コンジュゲート水溶液の吸光度を示し、AA,280は280nmにおける抗体の吸光度を示し、AA,370は370nmにおける抗体の吸光度を示し、AD,280は280nmにおけるコンジュゲート前駆体の吸光度を示し、AD,370は370nmにおけるコンジュゲート前駆体の吸光度を示し、εA,280は280nmにおける抗体のモル吸光係数を示し、εA,370は370nmにおける抗体のモル吸光係数を示し、εD,280は280nmにおけるコンジュゲート前駆体のモル吸光係数を示し、εD,370は370nmにおけるコンジュゲート前駆体のモル吸光係数を示し、CAは抗体-薬物コンジュゲートにおける抗体濃度を示し、CDは抗体-薬物コンジュゲートにおける薬物濃度を示す。
ここで、εA,280、εA,370、εD,280、εD,370は、事前に用意した値(計算推定値又は化合物のUV測定から得られた実測値)が用いられる。例えば、εA,280は、抗体のアミノ酸配列から、既知の計算方法(Protein Science, 1995, vol.4, 2411-2423)によって推定することが出来る。εA,370は、通常、ゼロである。εD,280及びεD,370は、用いるコンジュゲート前駆体をあるモル濃度に溶解させた溶液の吸光度を測定することで、ランベルト・ベールの法則(吸光度=モル濃度×モル吸光係数×セル光路長)によって、得ることができる。抗体-薬物コンジュゲート水溶液のA280及びA370を測定し、これらの値を式(I)及び(II)に代入して連立方程式を解くことによって、CA及びCDを求めることができる。さらにCDをCAで除することで1抗体あたりの薬物平均結合数が求めることができる。
抗体-薬物コンジュゲートにおける抗体一分子あたりの薬物平均結合数は、前述の共通操作Eに加え、以下の方法を用いる高速液体クロマトグラフィー(HPLC)分析によっても求めることができる。
[F-1.HPLC分析用サンプルの調製(抗体-薬物コンジュゲートの還元)]
抗体-薬物コンジュゲート溶液(約1mg/mL、60μL)をジチオトレイトール(DTT)水溶液(100mM、15μL)と混合する。混合物を37℃で30分インキュベートすることで、抗体-薬物コンジュゲートのL鎖及びH鎖間のジスルフィド結合を切断したサンプルを、HPLC分析に用いる。
[F-2.HPLC分析]
HPLC分析を、下記の測定条件にて行う。
HPLCシステム:Agilent 1290 HPLCシステム(Agilent Technologies)
検出器:紫外吸光度計(測定波長:280nm)
カラム:PLRP-S(2.1×50mm、8μm、1000Å;Agilent Technologies、P/N PL1912-1802)
カラム温度:80℃
移動相A:0.04%トリフルオロ酢酸(TFA)水溶液
移動相B:0.04%TFAを含むアセトニトリル溶液
グラジエントプログラム:29%-36%(0分-12.5分)、36%-42%(12.5-15分)、42%-29%(15分-15.1分)、29%-29%(15.1分-25分)
サンプル注入量:15μL
[F-3.データ解析]
〔F-3-1〕 薬物の結合していない抗体のL鎖(L0)及びH鎖(H0)に対して、薬物の結合したL鎖(薬物が一つ結合したL鎖:L1)及びH鎖(薬物が一つ結合したH鎖:H1、薬物が二つ結合したH鎖:H2、薬物が三つ結合したH鎖:H3)は、結合した薬物の数に比例して疎水性が増して保持時間が大きくなることから、L0、L1、H0、H1、H2、H3の順に溶出される。L0及びH0との保持時間比較により検出ピークをL0、L1、H0、H1、H2、H3のいずれかに割り当てることができる。
〔F-3-2〕 薬物リンカーにUV吸収があるため、薬物リンカーの結合数に応じて、L鎖、H鎖及び薬物リンカーのモル吸光係数を用いて下式に従ってピーク面積値の補正を行う。


〔F-3-3〕 ピーク面積補正値合計に対する各鎖ピーク面積比(%)を下式に従って計算する。

薬物平均結合数=(L0ピーク面積比×0+L0ピーク面積比×1+H0ピーク面積比×0+H1ピーク面積比×1+H2ピーク面積比×2+H3ピーク面積比×3)/100×2
(maleimid-N-yl)-(CH2)n3-C(=O)-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-(NH-DX)
で示される化合物である。
式中、
n3は、整数の2から8を示し、
L2は、-NH-(CH2CH2-O)n4-CH2CH2-C(=O)-又は単結合を示し、
ここで、n4は、1から6の整数を示し、
LPは、フェニルアラニン、グリシン、バリン、リシン、シトルリン、セリン、グルタミン酸、アスパラギン酸から選ばれる2から7個のアミノ酸で構成されるペプチド残基を示し、
n1は、0から6の整数を示し、
n2は、0から5の整数を示し、
Laは、-O-又は単結合を示し、
(maleimid-N-yl)-は、次式

-(NH-DX)は、次式

LPのペプチド残基としては、フェニルアラニン、グリシン、バリン、リシン、シトルリン、セリン、グルタミン酸、アスパラギン酸から選ばれるアミノ酸からなるペプチド残基である化合物が製造中間体として好ましい。この様なペプチド残基のうち、LPが4個のアミノ酸で構成されるペプチド残基である化合物が製造中間体として好ましい。より具体的には、LPが-GGFG-のテトラペプチド残基である化合物が製造中間体として好ましい。
(maleimid-N-yl)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)。
(maleimid-N-yl)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)。
(maleimid-N-yl)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、又は
(maleimid-N-yl)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
で示される化合物がさらに好ましい化合物である。
先の製造方法で使用した中間体である式(2)で示される化合物及びそれらの薬理上許容される塩は例えば下記の方法によって製造することができる。

この反応は、ペプチド合成に通常用いる反応試薬や条件を準用すればよい。活性エステルには各種のものがあるが、例えばp-ニトロフェノール等のフェノール類、N-ヒドロキシベンゾトリアゾール或はN-ヒドロキシスクシンイミド等とカルボン酸(5)をN,N’-ジシクロヘキシルカルボジイミド或は1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド・塩酸塩等の縮合剤を用いて反応させれば製造できる。また、活性エステルは、カルボン酸(5)とペンタフルオロフェニルトリフルオロアセテート等との反応;カルボン酸(5)と1-ベンゾトリアゾリルオキシトリピロリジノホスホニウムヘキサフルオロホスファイトとの反応;カルボン酸(5)とシアノホスホン酸ジエチルとの反応(塩入法);カルボン酸(5)とトリフェニルホスフィン及び2,2’-ジピリジルジスルフィドとの反応(向山法);カルボン酸(5)と4-(4,6-ジメトキシ-1,3,5-トリアジン-2-イル)-4-メチルモルホリニウムクロライド(DMTMM)等のトリアジン誘導体との反応;等によっても製造することができる。また、カルボン酸(5)を塩基存在下に塩化チオニル、オキザリルクロリド等の酸ハロゲン化物で処理することによって製造できる酸ハライド法等によって反応を行うこともできる。
上記の様に得たカルボン酸(5)の活性エステル、混合酸無水物、又は酸ハロゲン化物を化合物(4)と適当な塩基存在下に不活性な溶媒中で-78℃~150℃の反応温度で反応させることによって化合物(6)を製造することができる。なお、「不活性な溶媒」とはその溶媒が採用された反応において実施される目的とされた反応を阻害することのない溶媒を意味する。
得られた化合物(6)の末端アミノ基の保護基P1を脱保護させることによって化合物(7)を製造することができる。この脱保護は、その保護基に応じた試薬や条件を選択すればよい。
N末端をP2で保護したペプチドカルボン酸(8)を活性エステル、混合酸無水物等に誘導し、得られた化合物(7)に反応させることによって、化合物(9)を製造することができる。ペプチドカルボン酸(8)と化合物(7)のペプチド結合を形成する反応条件や試薬、塩基、及び不活性な溶媒は、化合物(6)の合成で述べたものから適宜選択して使用すればよい。保護基P2は、化合物(6)の保護基で述べたものから適宜選択して使用すればよく、アミノ基を保護する化合物の性質等に応じて選択すればよい。また、ペプチド合成に通常用いられている様に、ペプチドカルボン酸(8)を構成するアミノ酸又はペプチドを順次反応と脱保護を繰り返し、伸長させて化合物(9)を製造することもできる。
得られた化合物(9)のアミノ基の保護基P2を脱保護させることによって化合物(10)を製造することができる。この脱保護は、その保護基に応じた試薬や条件を選択すればよい。
カルボン酸(11)を活性エステル、混合酸無水物、又は酸ハロゲン化物等に誘導し、得られた化合物(10)に反応させることによって、化合物(2)又はを製造することができる。カルボン酸(11)と化合物(10)のペプチド結合を形成する反応条件や試薬、塩基、及び不活性溶媒は、化合物(6)の合成で述べたものから適宜選択して使用すればよい。
N末端をP2で保護したペプチドカルボン酸(8)を活性エステル、混合酸無水物等に誘導し、塩基存在下、カルボキシ基をP3で保護したアミン化合物(12)と反応させることによって化合物(13)を製造することができる。ペプチドカルボン酸(8)と化合物(12)のペプチド結合を形成する反応条件や試薬、塩基、及び不活性な溶媒は、化合物(6)の合成で述べたものから適宜選択して使用すればよい。
化合物(13)のアミノ基の保護基P2は、通常用いられる保護基で保護されていてよい。
具体的には水酸基の保護基としては、メトキシメチル基等のアルコキシメチル基;ベンジル基、4-メトキシベンジル基、トリフェニルメチル基等のアリールメチル基;アセチル基等のアルカノイル基;ベンゾイル基等のアロイル基;tert-ブチルジフェニルシリル基等のシリル基;等を挙げることができる。カルボキシ基は、メチル基、エチル基、tert-ブチル基等のアルキル基、アリル基、又はベンジル基等のアリールメチル基とのエステル等として保護することができる。アミノ基は、tert-ブチルオキシカルボニル基、メトキシカルボニル基、エトキシカルボニル基等のアルキルオキシカルボニル基;アリルオキシカルボニル基、又は9-フルオレニルメチルオキシカルボニル基、ベンジルオキシカルボニル基、パラメトキシベンジルオキシカルボニル基、パラ(又はオルト)ニトロベンジルオキシカルボニル基等のアリールメトキシカルボニル基;のほか、アセチル基等のアルカノイル基;ベンジル基、トリフェニルメチル基等のアリールメチル基;ベンゾイル基等のアロイル基;2,4-ジニトロベンゼンスルホニル基、オルトニトロベンゼンスルホニル基等のアリールスルホニル基;等を挙げることができる。
カルボキシ基の保護基P3としては、有機合成化学、中でもペプチド合成においてカルボキシ基の保護基として通常用いられている保護基を使用すればよく、具体的にはメチル基、エチル基、tert-ブチル等のアルキルエステル、アリルエステル、ベンジルエステル等であり上記の保護基から適宜選択して使用すればよい。この場合に、アミノ基の保護基とカルボキシ基の保護基が異なる方法又は条件で除去できることが好ましい。例えばP2がtert-ブチルオキシカルボニル基であり、P3がベンジル基である組み合わせ等を代表的なものとして挙げることができる。それらの保護基はアミノ基とカルボキシ基を保護する化合物の性質等に応じて上述したものから選択すればよく、それらの保護基の切断に際してもその保護基に応じた試薬や条件を選択すればよい。
得られた化合物(13)のカルボキシ基の保護基P3を脱保護させることによって化合物(14)を製造することができる。この脱保護は、その保護基に応じた試薬や条件を選択すればよい。
得られた化合物(14)を活性エステル、混合酸無水物、又は酸ハロゲン化物等に誘導し、塩基存在下、化合物(4)と反応させることによって化合物(9)を製造することができる。この反応はペプチド合成に通常用いる反応試薬や条件を準用すればよく、反応条件や試薬、塩基、及び不活性な溶媒は化合物(6)の合成で述べたものから適宜選択して使用すればよい。
化合物(13)のアミノ基の保護基P2を脱保護させることによって化合物(15)を製造することができる。この脱保護は、その保護基に応じた試薬や条件を選択すればよい。
カルボン酸誘導体(11)を活性エステル、混合酸無水物、又は酸ハロゲン化物等に誘導し、塩基存在下、得られた化合物(15)と反応させることによって化合物(16)を製造することができる。ペプチドカルボン酸(11)と化合物(15)のアミド結合を形成する反応条件や試薬、塩基、及び不活性な溶媒は、化合物(6)の合成で述べたものから適宜選択して使用すればよい。
得られた化合物(16)のカルボキシ基の保護基を脱保護させることによって化合物(17)を製造することができる。この脱保護は、化合物(14)の製造におけるカルボキシ基の脱保護と同様に行うことができる。
化合物(17)を活性エステル、混合酸無水物、又は酸ハロゲン化物等に誘導し、塩基存在下、化合物(4)と反応させることによって化合物(2)を製造することができる。この反応はペプチド合成に通常用いる反応試薬や条件を準用すればよく、反応条件や試薬、塩基、及び不活性な溶媒は化合物(6)の合成で述べたものから適宜選択して使用すればよい。
中間体の式(2)で示される化合物は下記の方法によっても製造することもできる。

得られた化合物(19)のカルボキシ基の保護基を脱保護させることによって化合物(20)を製造することができる。この脱保護は、化合物(14)の製造におけるカルボキシ基の脱保護と同様に行うことができる。
得られた化合物(20)を活性エステル、又は混合酸無水物等に誘導し、化合物(7)に反応させることによって、化合物(2)を製造することができる。この反応はペプチド合成に通常用いる反応試薬や条件を準用すればよく、反応条件や試薬、塩基、及び不活性な溶媒は化合物(6)の合成で述べたものから適宜選択して使用すればよい。
以下に、製造方法2に記載の製造中間体(10)のうち、n1=1、La=Oの化合物(10b)の製造方法について詳述する。式(10b)で示される化合物、その塩又はそれらの溶媒和物は、例えば下記の方法で製造することができる。

化合物(21)を、不活性な溶媒中、酸または塩基存在下で冷却下から室温の温度条件で水酸基を有する化合物(22)と反応させることによって、化合物(23)を製造することができる。
ここで使用できる酸としては例えば、フッ化水素酸、塩酸、硫酸、硝酸、リン酸、ホウ酸等の無機酸;酢酸、クエン酸、パラトルエンスルホン酸、メタンスルホン酸等の有機酸;テトラフルオロボレート、塩化亜鉛、塩化スズ、塩化アルミニウム、塩化鉄等のルイス酸;等を挙げることができる。これらのうちではスルホン酸類、特にパラトルエンスルホン酸が好ましい。さらに塩基としては、既に述べられた塩基の中から適宜選択して使用すればよく、特にカリウム tert-ブトキシド等のアルカリ金属アルコキシド;水酸化ナトリウム、水酸化カリウム等のアルカリ金属水酸化物;水素化ナトリウム、水素化カリウム等のアルカリ金属水素化物;リチウム ジイソプロピルアミド等のジアルキルアミノリチウムに代表される有機金属塩基;リチウム ビス(トリメチルシリル)アミド等のビスシリルアミンの有機金属塩基;等が好ましい。反応に用いる溶媒としては、テトラヒドロフラン、1,4-ジオキサン等のエーテル系溶媒;ベンゼン、トルエン等の芳香族炭化水素系溶媒;等が用いられる。上記の溶媒は水との混合物としてもよい。また、P5に例示されるアミノ基の保護基としては、通常、アミノ基の保護に用いられる基であれば特に制限はない。代表的なものとして製造方法2で記載したアミノ基の保護基を挙げることができるが、本反応中においてP5に例示されるアミノ基の保護基が切断される場合がある。その場合には、必要に応じて適当なアミノ基の保護試薬と適宜反応させて再度保護基を導入すればよい。
化合物(24)は、化合物(23)の保護基P6を除去することによって製造することができる。ここで、P6として例示されるカルボキシ基の保護基としては、製造方法2に代表的なものが記載されており、ここから適宜選択することができる。化合物(23)においては、アミノ基の保護基P5とカルボキシ基の保護基P6が異なる方法又は条件で除去できる保護基であることが望ましい。例えば、P5が9-フルオレニルメチルオキシカルボニル基であり、P6がベンジル基である組み合わせ等を代表的なものとして挙げることができる。それらの保護基は、アミノ基及びカルボキシ基を保護する化合物の性質等に応じて選択すればよく、それらの保護基の除去に際してもその保護基に応じた試薬や条件を選択すればよい。
カルボン酸(24)を活性エステル、混合酸無水物、又は酸ハロゲン化物等に誘導し、塩基存在下、化合物(4)又はその薬理上許容される塩と反応させることによって化合物(25)を製造し、得られた化合物(25)の保護基P5を除去することによって化合物(26)を製造することができる。化合物(4)とカルボン酸(24)との反応及び保護基P6を除去する反応では、製造方法2で述べた試薬や反応条件と同様のものを用いればよい。
化合物(26)と末端アミノ基が保護されたアミノ酸又はアミノ基が保護されたオリゴペプチド(27)を反応させることによって化合物(9b)を製造し、得られた化合物(9b)の保護基P7を除去することによって化合物(10b)を製造することができる。P7として示されるアミノ基の保護基としては、通常、アミノ基の保護に用いられる基であれば特に制限はなく、代表的なものとして製造方法2で記載したアミノ基の保護基を挙げることができ、その除去に際しても保護基に応じた試薬や条件を選択すればよい。化合物(26)と化合物(27)との反応では、ペプチド合成に通常使用される反応試薬や条件を準用すればよい。上記の方法で製造した化合物(10b)は、上述の製造方法に従って本発明化合物(1)に導くことができる。
また、本発明には、種々の放射性または非放射性同位体でラベルされた化合物も包含される。本発明の抗体-薬物コンジュゲートを構成する原子の1以上に、原子同位体の非天然割合も含有し得る。原子同位体としては、例えば、重水素(2H)、トリチウム(3H)、ヨウ素-125(125I)または炭素-14(14C)等を挙げることができる。また、本発明化合物は、例えば、トリチウム(3H)、ヨウ素-125(125I)または炭素-14(14C)等の放射性同位体で放射性標識され得る。放射性標識された化合物は、治療または予防剤、研究試薬、例えば、アッセイ試薬、及び診断剤、例えば、インビボ画像診断剤として有用である。本発明の抗体-薬物コンジュゲートの全ての同位体変異種は、放射性であると否とを問わず、本発明の範囲に包含される。
本発明の抗TROP2抗体-薬物コンジュゲートは、癌細胞に対して細胞傷害活性を示すことから、医薬として、特に癌に対する治療剤及び/又は予防剤として使用することができる。
すなわち本発明の抗TROP2抗体-薬物コンジュゲートは、癌治療の主要な治療法である化学療法のための薬剤として選択して使用することができ、その結果として、癌細胞の成長を遅らせ、増殖を抑え、さらには癌細胞を破壊することができる。これ等によって、癌患者において、癌による症状からの解放や、QOLの改善を達成でき、癌患者の生命を保って治療効果が達成される。癌細胞の破壊には至らない場合であっても、癌細胞の増殖の抑制やコントロールによって癌患者においてより高いQOLを達成しつつより長期の生存を達成させることができる。
本発明の抗TROP2抗体-薬物コンジュゲートは、薬物単独での使用の他、アジュバント療法において他の療法と組み合わせる薬剤としても使用でき、外科手術や、放射線療法、ホルモン療法等と組み合わせることができる。さらにはネオアジュバント療法における薬物療法の薬剤として使用することもできる。
以上のような治療的使用の他、抗体の抗原への結合性によって微細な転移癌細胞に結合して癌細胞の増殖を押さえ、さらには破壊する効果も期待することができる。すなわち、特に原発性の癌細胞においてTROP2の発現が確認されたときに本発明の抗TROP2抗体-薬物コンジュゲートを投与することよって癌転移の抑制や、予防効果を期待することができる。例えば、転移過程で体液中にある癌細胞を抑制、破壊する効果や、いずれかの組織に着床した直後の微細な癌細胞に対する抑制、破壊等の効果が期待できる。また、特に外科的な癌の除去後においての癌転移の抑制、予防効果が期待でき、これらによって癌の転移抑制効果が期待できる。
本発明の抗TROP2抗体-薬物コンジュゲートは、患者に対しては全身療法として全身的に投与する他、癌組織に局所的に投与して治療効果を期待することができる。
1-1)マウス免疫に用いる細胞の準備
5×106個のNCI-H322細胞(ヒト非小細胞肺癌細胞株、ATCC CRL-5806;ATCC:American Type Culture Collection)をRPMI-1640(Roswell Park Memorial Institute-1640)培地(10ml)で5日間培養後に回収し、PBS(リン酸緩衝生理食塩水)で2回洗浄し、PBS(500μl)中に再懸濁した。
BALB/cマウス(6週齢)に対し、1回目の免疫はNCI-H322細胞(1×107個)を腹腔内へ免疫した。2~5回目は1週間間隔で1×106個のNCI-H322細胞を腹腔内へ免疫した。6回目の最終免疫では、NCI-H322細胞を1×106細胞/200μl PBSで尾静脈ならびに腹腔内へそれぞれ免疫した。脾臓細胞は最終免疫から3日後に摘出した。
免疫済みマウスの脾臓を摘出し、すりつぶしてRPMI 1640 10%FBS(ウシ胎児血清)(+)培地に懸濁した。細胞懸濁液をセルストレイナー(100μm、BD Falcon社)に通した後、1500rpmで5分間、室温にて遠心して上清を廃棄した。Tris-NH4Cl溶液(20mM Tris-HCl pH7.5、0.83%NH4Cl;10mL)を加え、室温で5分間処理した。細胞懸濁液にRPMI 1640FBS(+)培地(10ml)を加え、セルストレイナーに通した後、1500rpmで5分間、室温にて遠心した。上清を廃棄し、脾臓細胞をRPMI 1640 FBS(-)培地(10ml)で再懸濁した。
P3U1細胞(マウスミエローマ細胞株)を回収し、1500rpmで5分間、室温にて遠心した。P3U1細胞にEDTA(0.02%)溶液(10ml)を加え、37℃で5分間処理した。P3U1細胞懸濁液を1500rpmで5分間、室温にて遠心した。上清を廃棄し、RPMI 1640 FBS(-)培地(10ml)で再懸濁した。
脾臓細胞とミエローマ細胞を5:1になるように混合し、遠心分離(1200rpm、5分間)した。得られた沈殿画分の細胞群をよくほぐした後、攪拌しながら、ポリエチレングリコール-4000(PEG-4000;1mL)を約1分かけて徐々に加えた。その後該細胞液に1分毎にRPMI培地(1mL)を数回加えた後、RPMI培地を加えて全量を50mLとした。該細胞懸濁液を遠心分離(900rpm、5分間)し、得られた沈殿画分の細胞をゆるやかにほぐした後、HAT培地(10%ウシ胎児血清及びHAT Media Supplementを加えた MI1640培地;100mL)中にゆるやかに懸濁した。該懸濁液を96ウェル培養用プレートに200μL/ウェルずつ分注し、37℃、5%CO2のインキュベーター中で、50%コンフルエントとなるまで培養した。
NCI-H322細胞を、96ウェルプレートに5×103細胞/ウェルで播種し、37℃で48時間培養した。細胞を150μl/ウェルのPBSで2回洗浄し、各ウェルに対して、各ハイブリドーマ培養上清(50μl)を加え4℃で1時間反応させた。150μl/ウェルのPBSで2回洗浄し、アデノウイルスAx3CAZ3-FZ33(抗体に結合するようにZ33ファイバーが改変されたβ-ガラクトシダーゼ発現アデノウイルス(米国特許出願公開第2012/0237518号明細書を参照))を3×106vp/100μl(1×103vp/細胞)の濃度になるようにRPMI1640(-)培地で希釈し、この希釈溶液を100μl/ウェルで加えた。4℃で1時間反応後、150μl/ウェルのPBSで2回洗浄した。RPMI1640 FBS(+)培地を100μl/ウェルで加えて37℃で24時間培養した。Galacto-Light Plus Reporter Gene Assay System(Applied Biosystems社)を使用したβ-Galレポーター遺伝子アッセイで処理したNCI-H322細胞を200μl/ウェルのPBSで洗浄し、Lysis Solutionを50μl/ウェルで加えて室温で10分間静置した。この細胞溶解液(10μL)をGalacton-Plus Galacto Reaction Buffer Diluentで100倍に希釈し、White microwell SH 96 well plate(Nunc社)に加えて室温で1時間反応させた。Accelerator IIを150μl/ウェルで加え、マルチラベルカウンターWallac 1420ARVOsx(Perkin Elmer社)を用いて化学発光を5秒間測定し、1秒あたりの平均値をRLU(発光量)として、NCI-H322細胞へのウイルス感染量を表した。この様に行ったハイブリドーマ群のスクリーニングにおいて、測定値(RLU)が群全体(最小値1383RLU、平均値10914RLU、最大値78746RLU)の中で、5000RLU以上のクローンを選抜した。まず、1次スクリーニングとして、一回の細胞融合で得られた960ウェルのハイブリドーマウェルのうち81ウェルの陽性ウェルを選抜した。さらに、確認スクリーニングとして、1次スクリーニングと同様の手法により、デュプリケート(duplicate)でアッセイを行って、両方の測定値が5000RLU以上のウェルを陽性とし、1次スクリーニングで得られた81ウェルから52ウェルの陽性ウェルを選抜した。選抜したクローンは2~4回サブクローニングを行い、モノクローナルなハイブリドーマ細胞株44株を樹立した。
プリスタン(Pristane;2,6,10,14-テトラメチルペンタデカン;0.5ml)を腹腔内投与して予め2週間飼育した8~10週齢のマウス又はヌードマウスに、実施例1で得られたモノクローナル抗体産生ハイブリドーマを腹腔内に注射した。さらに10~21日してハイブリドーマを腹水癌化させた後に腹水を採取した。得られた腹水を遠心分離して固形分を除去後、40~50%硫酸アンモニウムで塩析し、カプリル酸沈殿法、DEAE-セファロースカラム、プロテインG-カラムによる精製を行い、IgG又はIgM画分を集め、精製モノクローナル抗体とした。
実施例2において調製されたハイブリドーマの産生する抗体の一つであるTINA1について抗原の同定を行った。
5×106個のNCI-H322細胞を回収し、PBSで3回洗浄した。EZ-Link Sulfo-NHS-Biotin(PIERCE社)を0.1mg/mlの濃度でPBSに懸濁した。NCI-H322細胞をBiotin/PBS溶液中、室温で30分間ローテートした後に100mMグリシン/PBS溶液(25ml)で2回洗浄し、その後、PBS(25ml)で3回洗浄した。洗浄後の細胞を溶解バッファー(150mM NaCl、50mM Tris-HCl pH7.6、1%NP-40 + Protease inhibitor、Complete EDTA free(Roche社)1粒/50ml;2ml)中に再懸濁し、4℃で30分間処理した。Protein G Sepharose(Protein G Sepharose 4 Fast Flow(GE Healthcare社))のバッファーを溶解バッファーに置換して得られたProtein G Sepharose/溶解バッファー(50%スラリー;30μl)を細胞溶解液に加え、4℃で1時間ローテートした後に4℃で5分間遠心し、上清を回収した。この上清にTINA1抗体(3μg)を加え、4℃で1時間ローテートした後にProtein G Sepharose/溶解バッファー(50%スラリー;60μl)を加え、4℃で2時間ローテートした。溶解バッファー(1ml)でProtein G Sepharoseを6回洗浄した後に1×SDSサンプルバッファー/5% 2-ME(2-メルカプトエタノール)buffer(62.5mM Tris-HCl(25℃でpH6.8)、2%(w/v)SDS、10% glycerol及び0.01%(w/v)phenol red)に再懸濁した。100℃で5分間懸濁液を処理した後、溶液を回収し、SDS-PAGE(ポリアクリルアミドゲル電気泳動)のためのサンプルとした。
3-1)で調製したSDS-PAGEサンプルをReady Gels J 5-20%(BioRad社)を用いて20mAで泳動した後、ゲルよりメンブレンへ0.1mA/cm2でブロッティングした。メンブレンをPBS-T(PBS(-)-0.05% Tween20)で5分間洗浄した後、1時間ブロッキングを行った。メンブレンをPBS-Tで5分間3回洗浄した後にStreptavidin-horseradish peroxidase conjugate(Amersham社;PBS-Tで2000倍に希釈して使用)と1時間反応させた。メンブレンをPBS-Tで10分間4回洗浄した後に、ECL western blotting detection reagents(Amersham社)及びHyperfilm ECL(Amersham社)を用いて目的のバンドを検出した。実施例3-1)の手順によりビオチンラベルしたNCI-H322細胞を、既に質量分析によって抗原がTROP2であることが確定しているKCI7A3抗体及びTINA1抗体で免疫沈降して得られた免疫沈降物を、それぞれDTT非添加或はDTT添加でSDS-PAGE及びウエスタンブロッティングにより解析した。KCI7A3抗体とTINA1抗体を用いたいずれの場合においても、DTT非添加では分子量46kDaのバンドが、DTTを添加したサンプルでは分子量37kDaのバンドが検出された。
バンドパターンからTINA1抗体の抗原がTROP2であることが予測できたため、質量分析を行わずにcDNAの遺伝子導入による強制発現解析を行った。FACS解析の結果、ヒトTROP2発現CHOK1細胞ではTINA1抗体は強陽性反応を示したため、TINA1抗体の抗原はTROP2であることが示された。また、肺癌細胞株PC14、肺癌細胞株NCI-H322、肺癌細胞株NCI-H2122、肺癌細胞株LCAM1、肺癌細胞株LC2/ad、膵癌細胞株MIAPaCa2、膵癌細胞株PK-1、前立腺癌細胞株PC3、大腸癌細胞株HCT116、メラノーマ細胞株A375、卵巣癌細胞株SKOV3、造血系腫瘍細胞株RPMI8226、造血系腫瘍細胞株K562、PBMC(ヒト末梢血単核細胞)及びヒト血小板を使用して同様なFACS解析を行った。調査した肺癌細胞株はいずれもTROP2陽性であり、肺癌以外の細胞株ではPC3、PK1、SKOV3が陽性であった。一方、正常血液細胞はいずれも陰性であった。
4-1)抗体内在化活性評価系
抗体の内在化及びイムノトキシン活性の測定を目的にリコンビナント複合蛋白質であるDT3Cを作製した。このDT3Cは、ジフテリア毒素(DT)の触媒領域とプロテインGの抗体結合領域を3個兼ね備えた蛋白質である。DT3Cは抗体のFc部分に特異的に結合し安定で、細胞内へ取り込まれると蛋白質合成阻害により細胞死を誘導する。この系を用いることで抗体の内在化とイムノトキシンによる殺細胞効果を同時に観察することができる(Yamaguchi, M., Hamada, H., et al., Biochemical and Biophysical Research Communications 454 (2014) 600-603)。
96wll Plateに4μg/mLのDT3C(25μL)を加え、さらに実施例1又はこれに準じた方法で取得された11種のハイブリドーマの培養上清(25μL)を加え、室温で30分インキュベーションした。なお、TINA1抗体産生ハイブリドーマ以外のハイブリドーマが産生する抗体の認識する抗原は、CD9、CD46、CD55、CD59、CD71、CD73、CD147、CD276、EpCAM、又はEGFRであることを予め確認した。2×104個/mL(20% Low IgG FBS加RPMI1640培地)のNCI-H322細胞(50μL)を播種し、室温で30分インキュベーションした後、37℃ CO2インキュベータで3日間培養した。培養後に上清を除き、10%WST-10%FBS-RPMI1640(100μL)を加え、37℃ CO2インキュベータ内で1時間インキュベーションし、生細胞数をマイクロプレートリーダー(OD450~OD640、infinite200、テカンジャパン株式会社)で測定した。評価したハイブリドーマ細胞の培養上清の中で、CD59、CD71、EGFR、EpCAM、及びTROP2に対する抗体で強い内在化ならびにイムノトキシン活性が認められた(図10)。
96well platにDT3C(0、0.004、0.04、0.4、4、40μg/mL)希釈溶液(25μL)を加え、各抗体(40μg/mL;25μL)を加え、室温で30分インキュベーションした。さらに、2×104個/mL(20%LowIgGFBS加RPMI1640培地)のNCI-H322細胞(50μL)を播種し、室温で30分インキュベーションした後、37℃ CO2インキュベータで3日間培養した。培養後に上清を除き、10%WST-1加10%FBS-RPMI1640(100μL)を加え、37℃ CO2インキュベータ内で1時間インキュベーションし、生細胞数をプレートリーダー(OD450~OD640)で測定した。評価した抗体の中で、TROP2に対する抗体であるTINA1の内在化ならびにイムノトキシン活性が最も強かった(図11)。
実施例1又はこれに準じて取得されたTINA1(免疫原:肺癌株NCI-H322)、KCL7A3、及びKCL2D6(免疫原:膵臓癌株KCL-MOH1)、Pr1E11及びPr8H10(免疫原:前立腺癌株Pc-1)、NY16及びNY17(免疫原:膵臓癌株PK-1)並びに市販の77220(R&D System)の各抗TROP2抗体の内在化ならびにイムノトキシン活性について実施例4-3)と同様に評価した。その結果、8種の抗TROP2抗体の中でTINA1抗体が最も強い活性を有していた(図12)。
5-1)TINA1抗体遺伝子の可変領域をコードするcDNAのヌクレオチド配列の決定
5-1-1)TINA1抗体産生ハイブリドーマからのmRNAの調製
TINA1抗体の可変領域を含むcDNAを増幅するため、TINA1抗体産生ハイブリドーマよりmRNA Isolation kit(Roche applied science社)を用いてmRNAを調製した。
cDNA(5’-RACE-Ready cDNA)の合成は実施例5-1-1)で調製したmRNA(100ng)とSMARTer RACE cDNA Amplification Kit(CLONTECH社)を用いて実施した。
重鎖遺伝子の可変領域のcDNAをPCRで増幅するためのプライマーとして、UPM(Universal Primer A Mix:SMARTer RACE cDNA Amplification Kitに付属)、及び5’-AGAGTTCCAGGTCAAGGTCACTGGCTCAGG-3’(配列番号33:プライマー mG2aVR2)の配列を有するオリゴヌクレオチドを用いた。UPMはSMARTer RACE cDNA Amplification Kit(CLONTECH社)に付属のものを使用し、mG2aVR2はデータベースのマウス重鎖(IgG2a)の定常領域の配列から設計した。
このプライマーの組み合わせと、実施例5-1-2)で合成したcDNA(5’-RACE-Ready cDNA)を鋳型とした5’-RACE PCRによりTINA1抗体の重鎖の可変領域を含むcDNAを増幅した。PCRは、PolymeraseとしてKOD-plus(TOYOBO社)を用い、SMARTer RACE cDNA Amplification Kit(CLONTECH社)のマニュアルに従い、タッチダウンPCRプログラムで実施した。
5’-RACE PCRで増幅した重鎖の可変領域を含むcDNAをMinElute PCR Purification Kit(QIAGEN社)を用いて精製後、Zero Blunt TOPO PCR Cloning Kit(Invitrogen社)を用いてクローニングし、クローニングした重鎖の可変領域を含むcDNAのヌクレオチド配列のシークエンス解析を実施した。シークエンスプライマーとして、データベースのマウス重鎖の定常領域の配列から設計した上記プライマー mG2aVR2、及びNUP(Nested Universal Primer A:SMARTer RACE cDNA Amplification Kitに付属)を用いた。
シークエンス解析は遺伝子配列解析装置(「ABI PRISM 3700 DNA Analyzer」或は「Applied Biosystems 3730xl Analyzer」、Applied Biosystems社)を用いて実施し、シークエンス反応は、Gene Amp 9700(Applied Biosystems社)を用いた。
決定されたTINA1抗体の重鎖の可変領域をコードするcDNAのヌクレオチド配列を配列表の配列番号1に示し、アミノ酸配列を配列番号2に示した。
TINA1抗体の軽鎖遺伝子の可変領域のcDNAをPCRで増幅するためのプライマーとして、UPM(Universal Primer A Mix:SMARTer RACE cDNA Amplification Kitに付属)及び5’-AGTCCAACTGTTCAGGACGCCATTTTGTCG-3’(配列番号34:プライマー mKVR2)の配列を有するオリゴヌクレオチドを用いた。UPMはSMARTer RACE cDNA Amplification Kit(CLONTECH社)に付属のものを使用し、mKVR2はデータベースのマウス軽鎖の定常領域の配列から設計した。
このプライマーの組み合わせと、実施例5-1-2)で合成したcDNA(5’-RACE-Ready cDNA)を鋳型とした5’-RACE PCRによりTINA1抗体の軽鎖の可変領域を含むcDNAを増幅した。PCRは、PolymeraseとしてKOD-plus-(TOYOBO社)を用い、SMARTer RACE cDNA Amplification Kit(CLONTECH社)のマニュアルに従い、タッチダウンPCRプログラムで実施した。
5’-RACE PCRで増幅した軽鎖の可変領域を含むcDNAをMinElute PCR Purification Kit(QIAGEN社)を用いて精製後、Zero Blunt TOPO PCR Cloning Kit(Invitrogen社)を用いてクローニングし、クローニングした軽鎖の可変領域を含むcDNAのヌクレオチド配列のシークエンス解析を実施した。
シークエンスプライマーとして、データベースのマウス軽鎖の定常領域の配列から設計した上記プライマーmKVR2及びNUPを用いた。
シークエンス解析及びシークエンス反応には、上述の装置を用いた。
決定されたTINA1抗体の軽鎖の可変領域をコードするcDNAのヌクレオチド配列を配列表の配列番号3に示し、アミノ酸配列を配列番号4に示した。
5-2-1)キメラ及びヒト化抗体軽鎖発現ベクターpCMA-LKの構築
プラスミドpcDNA3.3-TOPO/LacZ(Invitrogen社)を制限酵素XbaI及びPmeIで消化して得られる約5.4kbのフラグメントと、配列番号5に示すヒトκ鎖分泌シグナル及びヒトκ鎖定常領域をコードするDNA配列を含むDNA断片を、In-Fusion Advantage PCRクローニングキット(CLONTECH社)を用いて結合して、pcDNA3.3/LKを作製した。
pcDNA3.3/LKを鋳型として、下記プライマーセットでPCRを行い、得られた約3.8kbのフラグメントをリン酸化後セルフライゲーションすることによりCMVプロモーターの下流にシグナル配列、クローニングサイト、及びヒトκ鎖定常領域を持つ、キメラ及びヒト化抗体軽鎖発現ベクターpCMA-LKを構築した。
プライマーセット
5’-tataccgtcgacctctagctagagcttggc-3’(配列番号35:プライマー 3.3-F1)
5’-gctatggcagggcctgccgccccgacgttg-3’(配列番号36:プライマー 3.3-R1)
pCMA-LKをXbaI及びPmeIで消化してκ鎖分泌シグナル及びヒトκ鎖定常領域を取り除いたDNA断片と、配列番号6に示すヒト重鎖シグナル配列及びヒトIgG1定常領域のアミノ酸をコードするDNA配列を含むDNA断片を、In-Fusion Advantage PCRクローニングキット(CLONTECH社)を用いて結合して、CMVプロモーターの下流にシグナル配列、クローニングサイト、ヒトIgG1重鎖定常領域をもつキメラ及びヒト化抗体IgG1タイプ重鎖発現ベクターpCMA-G1を構築した。
実施例5-1-3)で得られたTINA1抗体の重鎖の可変領域を含むcDNAをテンプレートとして、KOD-Plus-(TOYOBO社)と下記のプライマーセットで重鎖の可変領域をコードするcDNAを含むDNA断片を増幅し、キメラ及びヒト化IgG1タイプ重鎖発現ベクターpCMA-G1を制限酵素BlpIで切断した箇所にIn-Fusion HD PCRクローニングキット(CLONTECH社)を用いて挿入することにより、cTINA1抗体重鎖発現ベクターを構築した。得られた発現ベクターを「pCMA-G1/cTINA1」と命名した。cTINA1抗体重鎖のヌクレオチド配列を配列番号7に示し、アミノ酸配列を配列番号8に示した。配列番号7のヌクレオチド配列及び配列番号8のアミノ酸配列は、図1にも記載されている。
cTINA1抗体重鎖用プライマーセット
5’-CCAGATGGGTGCTGAGCCAGATCCAGTTGGTGCAGTCTGGACCTGAG-3’(配列番号37:プライマー TINA1H-F)
5’-CTTGGTGGAGGCTGAGCTGACGGTGACCGCGGTCCCTGCGCCCCAGAC-3’(配列番号38:プライマー TINA1H-R)
実施例5-1-4)で得られたTINA1抗体の軽鎖の可変領域を含むcDNAをテンプレートとして、KOD-Plus-(TOYOBO社)と下記のプライマーセットで軽鎖の可変領域をコードするcDNAを含むDAN断片を増幅し、キメラ及びヒト化抗体軽鎖発現汎用ベクターpCMA-LKを制限酵素BsiWIで切断した箇所にIn-Fusion HD PCRクローニングキット(CLONTECH社)を用いて挿入することにより、cTINA1抗体の軽鎖発現ベクターを構築した。得られた発現ベクターを「pCMA-LK/cTINA1」と命名した。cTINA1抗体の軽鎖のヌクレオチド配列を配列表の配列番号9に示し、アミノ酸配列を配列番号10に示した。配列番号9のヌクレオチド配列及び配列番号10のアミノ酸配列は、図2にも記載されている。
cTINA1抗体軽鎖用プライマーセット
5’-ATCTCCGGCGCGTACGGCGACATTGTGATGACCCAGTCTCACAAATTC-3’(配列番号39:プライマー TINA1L-F)
5’-GGAGGGGGCGGCCACAGCCCGTTTCAGCTCCAGCTTGGTCCCAGC-3’(配列番号40:プライマー TINA1L-R)
FreeStyle 293F細胞(Invitrogen社)はマニュアルに従い、継代、培養をおこなった。
対数増殖期の1×107個のFreeStyle 293F細胞(Invitrogen社)をFreeStyle293 expression medium (Invitrogen社)で9.6mLに希釈した後に、30mL Square Storage Bottle(Nalgene社)に播種し、37℃、8%CO2インキュベーター内で90rpmで一時間振とう培養した。Polyethyleneimine(Polyscience #24765;30μg)をOpti-Pro SFM(Invitrogen社;200μL)に溶解し、次にPureLink HiPure Plasmidキット(Invitrogen社)を用いて調製した軽鎖発現ベクター(6μg)及び重鎖発現ベクター(4μg)をOpti-Pro SFM(Invitrogen社;200μL)に添加した。Polyethyleneimine/Opti-Pro SFM混合液(200μL)に、発現ベクター/Opti-Pro SFM混合液(200μL)を加え穏やかに攪拌し、さらに5分間放置した後にFreeStyle 293F細胞に添加した。37℃、8%CO2インキュベーターで7日間、90rpmで振とう培養して得られた培養上清をMinisart-Plus filter(Sartorius社)でろ過して評価用のサンプルとした。
pCMA-G1/cTINA1とpCMA-LK/cTINA1との組合せによって取得されたヒトキメラTINA1抗体を「cTINA1抗体」と命名した。
6-1)TINA1のヒト化バージョンの設計
6-1-1)TINA1の可変領域の分子モデリング
TINA1の可変領域の分子モデリングは、相同性モデリングとして公知の方法(Methods in Enzymology,203,121-153(1991))によって実行された。Protein Data Bank(Nuc. Acid Res.35,D301-D303(2007))に登録されるヒト免疫グロブリンの可変領域の1次配列(X線結晶構造から誘導される三次元構造が入手可能)を、先に決定されたTINA1の可変領域と比較した。結果として、1ZEAが、TINA1の重鎖の可変領域に対して同様にフレームワーク中に欠損がある抗体の中で、最も高い配列相同性を有するとして選択された。また、3IU4が、TINA1の軽鎖の可変領域に対して最も高い配列相同性を有するとして選択された。フレームワーク領域の三次元構造は、TINA1の重鎖及び軽鎖に対応する1ZEA及び3IU4の座標を組み合わせて、「フレームワークモデル」を得ることによって作製された。次いで、それぞれのCDRについての代表的なコンホメーションがフレームワークモデルに組み込まれた。
最後に、エネルギーの点でTINA1の可変領域の可能性のある分子モデルを得るために、不利な原子間接触を除くためのエネルギー計算を行った。上記手順を、市販の蛋白質立体構造解析プログラムDiscovery Studio(Accelrys,Inc.)を用いて行った。
ヒト化TINA1抗体の構築を、CDRグラフティング(Proc.Natl.Acad.Sci.USA 86,10029-10033(1989))として公知の方法によって行った。アクセプター抗体は、フレームワーク領域内のアミノ酸相同性に基づいて選択された。TINA1のフレームワーク領域の配列を、抗体のアミノ酸配列のKabatデータベース(Nuc.Acid Res.29,205-206(2001))の全てのヒトフレームワークと比較し、結果として、HuPR1A3抗体がフレームワーク領域についての74%の配列相同性に起因して、アクセプターとして選択された。HuPR1A3についてのフレームワーク領域のアミノ酸残基を、TINA1についてのアミノ酸残基と整列させ、異なるアミノ酸が使用される位置を同定した。これらの残基の位置は、上で構築されたTINA1の三次元モデルを使用して分析され、そしてアクセプター上にグラフティングされるべきドナー残基が、Queen et. al.,(Proc.Natl.Acad.Sci.USA 86,10029-10033(1989))によって与えられる基準によって選択された。選択されたいくつかのドナー残基をアクセプター抗体に移入することによって、ヒト化TINA1配列を以下の実施例に記載される様に構築した。
6-2-1)hTINA1-H1タイプ重鎖:
配列表の配列番号8に示されるTINA1重鎖のアミノ酸番号21(イソロイシン)をバリンに、アミノ酸番号28(プロリン)をアラニンに、アミノ酸番号30(ロイシン)をバリンに、アミノ酸番号35(グルタミン酸)をアラニンに、アミノ酸番号36(スレオニン)をセリンに、アミノ酸番号38(アルギニン)をリシンに、アミノ酸番号39(イソロイシン)をバリンに、アミノ酸番号57(グルタミン)をアルギニンに、アミノ酸番号58(リシン)をグルタミンに、アミノ酸番号59(メチオニン)をアラニンに、アミノ酸番号62(リシン)をグルタミンに、アミノ酸番号65(リシン)をグルタミン酸に、アミノ酸番号67(イソロイシン)をメチオニンに、アミノ酸番号87(フェニルアラニン)をバリンに、アミノ酸番号88(アラニン)をスレオニンに、アミノ酸番号89(フェニルアラニン)をイソロイシンに、アミノ酸番号91(ロイシン)をアラニンに、アミノ酸番号92(グルタミン酸)をアスパラギン酸に、アミノ酸番号95(アラニン)をスレオニンに、アミノ酸番号102(イソロイシン)をロイシンに、アミノ酸番号104(アスパラギン)をセリンに、アミノ酸番号107(アスパラギン)をセリンに、アミノ酸番号111(スレオニン)をアラニンに、アミノ酸番号112(スレオニン)をバリンに、アミノ酸番号114(フェニルアラニン)をチロシンに、アミノ酸番号132(アラニン)をグルタミンに、アミノ酸番号135(アラニン)をロイシンに、置き換えることを伴い設計されたヒト化TINA1重鎖を「hTINA1-H1タイプ重鎖」と命名した。
hTINA1-H1タイプ重鎖のアミノ酸配列は、配列表の配列番号12に記載されている。配列番号12のアミノ酸配列の1乃至19番目のアミノ残基からなる配列、20乃至140番目のアミノ酸残基からなる配列、141乃至470番目のアミノ酸残基からなる配列が、それぞれシグナル配列、重鎖可変領域、重鎖定常領域に相当する。配列番号12のアミノ酸配列をコードするヌクレオチド配列は、配列表の配列番号11に記載されている。配列番号11のヌクレオチド配列の1乃至57番目のヌクレオチドからなる配列、58乃至420番目のヌクレオチドからなる配列、421乃至1410番目のヌクレオチドからなる配列が、それぞれシグナル配列、重鎖可変領域配列、重鎖定常領域配列をコードしている。配列番号11のヌクレオチド配列及び配列番号12のアミノ酸配列は、図3にも記載されている。
配列表の配列番号8に示されるTINA1重鎖のアミノ酸番号21(イソロイシン)をバリンに、アミノ酸番号28(プロリン)をアラニンに、アミノ酸番号30(ロイシン)をバリンに、アミノ酸番号35(グルタミン酸)をアラニンに、アミノ酸番号36(スレオニン)をセリンに、アミノ酸番号38(アルギニン)をリシンに、アミノ酸番号39(イソロイシン)をバリンに、アミノ酸番号57(グルタミン)をアルギニンに、アミノ酸番号58(リシン)をグルタミンに、アミノ酸番号59(メチオニン)をアラニンに、アミノ酸番号62(リシン)をグルタミンに、アミノ酸番号65(リシン)をグルタミン酸に、アミノ酸番号67(イソロイシン)をメチオニンに、アミノ酸番号87(フェニルアラニン)をバリンに、アミノ酸番号88(アラニン)をスレオニンに、アミノ酸番号89(フェニルアラニン)をイソロイシンに、アミノ酸番号92(グルタミン酸)をアスパラギン酸に、アミノ酸番号95(アラニン)をスレオニンに、アミノ酸番号102(イソロイシン)をロイシンに、アミノ酸番号104(アスパラギン)をセリンに、アミノ酸番号107(アスパラギン)をセリンに、アミノ酸番号111(スレオニン)をアラニンに、アミノ酸番号112(スレオニン)をバリンに、アミノ酸番号114(フェニルアラニン)をチロシンに、アミノ酸番号132(アラニン)をグルタミンに、アミノ酸番号135(アラニン)をロイシンに、置き換えることを伴い設計されたヒト化TINA1重鎖を「hTINA1-H2タイプ重鎖」と命名した。
hTINA1-H2タイプ重鎖のアミノ酸配列は、配列表の配列番号14に記載されている。配列番号14のアミノ酸配列の1乃至19番目のアミノ残基からなる配列、20乃至140番目のアミノ酸残基からなる配列、141乃至470番目のアミノ酸残基からなる配列が、それぞれシグナル配列、重鎖可変領域、重鎖定常領域に相当する。配列番号14のアミノ酸配列をコードするヌクレオチド配列は、配列表の配列番号13に記載されている。配列番号13のヌクレオチド配列の1乃至57番目のヌクレオチドからなる配列、58乃至420番目のヌクレオチドからなる配列、421乃至1410番目のヌクレオチドからなる配列が、それぞれシグナル配列、重鎖可変領域配列、重鎖定常領域配列をコードしている。配列番号13のヌクレオチド配列及び配列番号14のアミノ酸配列は、図4にも記載されている。
配列表の配列番号8に示されるTINA1重鎖のアミノ酸番号28(プロリン)をアラニンに、アミノ酸番号30(ロイシン)をバリンに、アミノ酸番号36(スレオニン)をセリンに、アミノ酸番号38(アルギニン)をリシンに、アミノ酸番号39(イソロイシン)をバリンに、アミノ酸番号58(リシン)をグルタミンに、アミノ酸番号65(リシン)をグルタミン酸に、アミノ酸番号67(イソロイシン)をメチオニンに、アミノ酸番号87(フェニルアラニン)をバリンに、アミノ酸番号88(アラニン)をスレオニンに、アミノ酸番号92(グルタミン酸)をアスパラギン酸に、アミノ酸番号95(アラニン)をスレオニンに、アミノ酸番号102(イソロイシン)をロイシンに、アミノ酸番号104(アスパラギン)をセリンに、アミノ酸番号107(アスパラギン)をセリンに、アミノ酸番号111(スレオニン)をアラニンに、アミノ酸番号112(スレオニン)をバリンに、アミノ酸番号114(フェニルアラニン)をチロシンに、アミノ酸番号132(アラニン)をグルタミンに、アミノ酸番号135(アラニン)をロイシンに、置き換えることを伴い設計されたヒト化TINA1重鎖を「hTINA1-H3タイプ重鎖」と命名した。
hTINA1-H3タイプ重鎖のアミノ酸配列は、配列表の配列番号16に記載されている。配列番号16のアミノ酸配列の1乃至19番目のアミノ残基からなる配列、20乃至140番目のアミノ酸残基からなる配列、141乃至470番目のアミノ酸残基からなる配列が、それぞれシグナル配列、重鎖可変領域、重鎖定常領域に相当する。配列番号16のアミノ酸配列をコードするヌクレオチド配列は、配列表の配列番号15に記載されている。配列番号15のヌクレオチド配列の1乃至57番目のヌクレオチドからなる配列、58乃至420番目のヌクレオチドからなる配列、421乃至1410番目のヌクレオチドからなる配列が、それぞれシグナル配列、重鎖可変領域配列、重鎖定常領域配列をコードしている。配列番号15のヌクレオチド配列及び配列番号16のアミノ酸配列は、図5にも記載されている。
6-3-1)hTINA1-L1タイプ軽鎖:
配列表の配列番号10に示されるTINA1軽鎖のアミノ酸番号23(バリン)をグルタミンに、アミノ酸番号28(ヒスチジン)をプロリンに、アミノ酸番号29(リシン)をセリンに、アミノ酸番号30(フェニルアラニン)をセリンに、アミノ酸番号31(メチオニン)をロイシンに、アミノ酸番号33(スレオニン)をアラニンに、アミノ酸番号40(セリン)をスレオニンに、アミノ酸番号62(グルタミン)をリシンに、アミノ酸番号63(セリン)をアラニンに、アミノ酸番号80(アスパラギン酸)をセリンに、アミノ酸番号83(スレオニン)をセリンに、アミノ酸番号90(アラニン)をアスパラギン酸に、アミノ酸番号93(フェニルアラニン)をロイシンに、アミノ酸番号98(バリン)をロイシンに、アミノ酸番号100(アラニン)をプロリンに、アミノ酸番号103(ロイシン)をフェニルアラニンに、アミノ酸番号120(アラニン)をグルタミンに、アミノ酸番号126(ロイシン)をイソロイシンに、アミノ酸番号129(アラニン)をスレオニンに、置き換えることを伴い設計されたヒト化TINA1軽鎖を「hTINA1-L1タイプ軽鎖」と命名した。
hTINA1-L1タイプ軽鎖のアミノ酸配列は、配列表の配列番号18に記載されている。配列番号18のアミノ酸配列の1乃至20番目のアミノ残基からなる配列、21乃至129番目のアミノ酸残基からなる配列、130乃至234番目のアミノ酸残基からなる配列が、それぞれシグナル配列、軽鎖可変領域、軽鎖定常領域に相当する。配列番号18のアミノ酸配列をコードするヌクレオチド配列は、配列表の配列番号17に記載されている。配列番号17のヌクレオチド配列の1乃至60番目のヌクレオチドからなる配列、61乃至387番目のヌクレオチドからなる配列、388乃至702番目のヌクレオチドからなる配列が、それぞれシグナル配列、軽鎖可変領域配列、軽鎖定常領域配列をコードしている。配列番号17のヌクレオチド配列及び配列番号18のアミノ酸配列は、図6にも記載されている。
配列表の配列番号10に示されるTINA1軽鎖のアミノ酸番号28(ヒスチジン)をプロリンに、アミノ酸番号29(リシン)をセリンに、アミノ酸番号30(フェニルアラニン)をセリンに、アミノ酸番号31(メチオニン)をロイシンに、アミノ酸番号33(スレオニン)をアラニンに、アミノ酸番号40(セリン)をスレオニンに、アミノ酸番号62(グルタミン)をリシンに、アミノ酸番号63(セリン)をアラニンに、アミノ酸番号80(アスパラギン酸)をセリンに、アミノ酸番号83(スレオニン)をセリンに、アミノ酸番号90(アラニン)をアスパラギン酸に、アミノ酸番号93(フェニルアラニン)をロイシンに、アミノ酸番号98(バリン)をロイシンに、アミノ酸番号100(アラニン)をプロリンに、アミノ酸番号103(ロイシン)をフェニルアラニンに、アミノ酸番号120(アラニン)をグルタミンに、アミノ酸番号126(ロイシン)をイソロイシンに、アミノ酸番号129(アラニン)をスレオニンに、置き換えることを伴い設計されたヒト化TINA1軽鎖を「hTINA1-L2タイプ軽鎖」と命名した。
hTINA1-L2タイプ軽鎖のアミノ酸配列は、配列表の配列番号20に記載されている。配列番号20のアミノ酸配列の1乃至20番目のアミノ残基からなる配列、21乃至129番目のアミノ酸残基からなる配列、130乃至234番目のアミノ酸残基からなる配列が、それぞれシグナル配列、軽鎖可変領域、軽鎖定常領域に相当する。配列番号20のアミノ酸配列をコードするヌクレオチド配列は、配列表の配列番号19に記載されている。配列番号19のヌクレオチド配列の1乃至60番目のヌクレオチドからなる配列、61乃至387番目のヌクレオチドからなる配列、388乃至702番目のヌクレオチドからなる配列が、それぞれシグナル配列、軽鎖可変領域配列、軽鎖定常領域配列をコードしている。配列番号19のヌクレオチド配列及び配列番号20のアミノ酸配列は、図7にも記載されている。
配列表の配列番号10に示されるTINA1軽鎖のアミノ酸番号28(ヒスチジン)をプロリンに、アミノ酸番号29(リシン)をセリンに、アミノ酸番号30(フェニルアラニン)をセリンに、アミノ酸番号31(メチオニン)をロイシンに、アミノ酸番号33(スレオニン)をアラニンに、アミノ酸番号40(セリン)をスレオニンに、アミノ酸番号62(グルタミン)をリシンに、アミノ酸番号63(セリン)をグルタミンに、アミノ酸番号80(アスパラギン酸)をセリンに、アミノ酸番号83(スレオニン)をセリンに、アミノ酸番号90(アラニン)をアスパラギン酸に、アミノ酸番号93(フェニルアラニン)をロイシンに、アミノ酸番号98(バリン)をロイシンに、アミノ酸番号100(アラニン)をプロリンに、アミノ酸番号103(ロイシン)をフェニルアラニンに、アミノ酸番号120(アラニン)をグルタミンに、アミノ酸番号126(ロイシン)をイソロイシンに、アミノ酸番号129(アラニン)をスレオニンに、置き換えることを伴い設計されたヒト化TINA1軽鎖を「hTINA1-L3タイプ軽鎖」と命名した。
hTINA1-L3タイプ軽鎖のアミノ酸配列は、配列表の配列番号22に記載されている。配列番号22のアミノ酸配列の1乃至20番目のアミノ残基からなる配列、21乃至129番目のアミノ酸残基からなる配列、130乃至234番目のアミノ酸残基からなる配列が、それぞれシグナル配列、軽鎖可変領域、軽鎖定常領域に相当する。配列番号22のアミノ酸配列をコードするヌクレオチド配列は、配列表の配列番号21に記載されている。配列番号21のヌクレオチド配列の1乃至60番目のヌクレオチドからなる配列、61乃至387番目のヌクレオチドからなる配列、388乃至702番目のヌクレオチドからなる配列が、それぞれシグナル配列、軽鎖可変領域配列、軽鎖定常領域配列をコードしている。配列番号21のヌクレオチド配列及び配列番号22のアミノ酸配列は、図8にも記載されている。
7-1)hTINA1の重鎖発現ベクターの構築
7-1-1)hTINA1-H1発現ベクターの構築
配列表の配列番号11に示すhTINA1-H1のヌクレオチド配列のヌクレオチド番号36乃至437に示されるhTINA1-H1の可変領域をコードするDNA配列を含むDNA断片を合成した(GENEART社 人工遺伝子合成サービス)。合成したDNA断片をテンプレートとして、KOD-Plus-(TOYOBO社)と下記のプライマーセットでhTINA1-H1の可変領域をコードするDNA配列を含むDNA断片を増幅し、キメラ及びヒト化抗体IgG1タイプ重鎖発現ベクターpCMA-G1を制限酵素BlpIで切断した箇所にIn-Fusion HD PCRクローニングキット(CLONTECH社)を用いて挿入することによりhTINA1-H1発現ベクターを構築した。得られた発現ベクターを「pCMA-G1/hTINA1-H1」と命名した。
プライマーセット
5’-agctcccagatgggtgctgagc-3’(配列番号41:プライマー EG-Inf-F)
5’-gggcccttggtggaggctgagc-3’(配列番号42:プライマー EG1-Inf-R)
配列表の配列番号13に示すhTINA1-H2のヌクレオチド配列のヌクレオチド番号36乃至437に示されるhTINA1-H2の可変領域をコードするDNA配列を含むDNA断片を合成し(GENEART社 人工遺伝子合成サービス)、実施例7-1-1)と同様の方法でhTINA1-H2発現ベクターを構築した。得られた発現ベクターを「pCMA-G1/hTINA1-H2」と命名した。
配列表の配列番号15に示すhTINA1-H3のヌクレオチド配列のヌクレオチド番号36乃至437に示されるhTINA1-H3の可変領域をコードするDNA配列を含むDNA断片を合成し(GENEART社 人工遺伝子合成サービス)、実施例7-1-1)と同様の方法でhTINA1-H3発現ベクターを構築した。得られた発現ベクターを「pCMA-G1/hTINA1-H3」と命名した。
7-2-1)hTINA1-L1発現ベクターの構築
配列表の配列番号17に示すhTINA1-L1のヌクレオチド配列のヌクレオチド番号38乃至402に示されるhTINA1-L1の可変領域をコードするDNA配列を含むDNA断片を合成した(GENEART社 人工遺伝子合成サービス)。合成したDNA断片をテンプレートとして、KOD-Plus-(TOYOBO社)と下記のプライマーセットでhTINA1-L1の可変領域をコードするDNA配列を含むDNA断片を増幅し、キメラ及びヒト化抗体軽鎖発現ベクターpCMA-LKを制限酵素BsiWIで切断した箇所にIn-Fusion HD PCRクローニングキット(CLONTECH社)を用いて挿入することによりhTINA1-L1発現ベクターを構築した。得られた発現ベクターを「pCMA-LK/hTINA1-L1」と命名した。
プライマーセット
5’-ctgtggatctccggcgcgtacggc-3’(配列番号43:プライマー CM-LKF)
5’-ggagggggcggccaccgtacg-3’(配列番号44:プライマー
KCL-Inf-R)
配列表の配列番号19に示すhTINA1-L2のヌクレオチド配列のヌクレオチド番号38乃至402に示されるhTINA1-L2の可変領域をコードするDNA配列を含むDNA断片を合成し(GENEART社 人工遺伝子合成サービス)、実施例7-2-1)と同様の方法でhTINA1-L2発現ベクターを構築した。得られた発現ベクターを「pCMA-LK/hTINA1-L2」と命名した。
配列表の配列番号21に示すhTINA1-L3のヌクレオチド配列のヌクレオチド番号38乃至402に示されるhTINA1-L3の可変領域をコードするDNA配列を含むDNA断片を合成し(GENEART社 人工遺伝子合成サービス)、実施例7-2-1)と同様の方法でhTINA1-L3発現ベクターを構築した。得られた発現ベクターを「pCMA-LK/hTINA1-L3」と命名した。
7-3-1)hTINA1抗体の小スケール生産
実施例5-2-5)と同様の方法で生産した。
pCMA-G1/hTINA1-H1とpCMA-LK/hTINA1-L1との組合せによって取得されたhTINA1抗体を「hTINA1-H1L1」、pCMA-G1/hTINA1-H2とpCMA-LK/hTINA1-L1との組合せによって取得されたhTINA1抗体を「hTINA1-H2L1」、pCMA-G1/hTINA1-H2とpCMA-LK/hTINA1-L2との組合せによって取得されたhTINA1抗体を「hTINA1-H2L2」、及びpCMA-G1/hTINA1-H3とpCMA-LK/hTINA1-L3との組合せによって取得されたhTINA1抗体を「hTINA1-H3L3」と命名した。
hTINA1-H1L1、hTINA1-H2L1、hTINA1-H2L2、及びhTINA1-H3L3を以下の方法で生産した。
FreeStyle 293F細胞(Invitrogen社)はマニュアルに従い、継代、培養をおこなった。対数増殖期の1.2×109個のFreeStyle 293F細胞(Invitrogen社)を3L Fernbach Erlenmeyer Flask(CORNING社)に播種し、FreeStyle293 expression medium(Invitrogen社)で希釈して1.0×106細胞/mlに調製した後に、37℃、8%CO2インキュベーター内で90rpmで一時間振とう培養した。Polyethyleneimine(Polyscience #24765;3.6mg)をOpti-Pro SFM(Invitrogen社;20ml)に溶解し、次にPureLink HiPure Plasmidキット(Invitrogen社)を用いて調製した軽鎖発現ベクター(0.8mg)及び重鎖発現ベクター(0.4mg)をOpti-Pro SFM(Invitrogen社;20ml)に添加した。Polyethyleneimine/Opti-Pro SFM混合液(20ml)に、発現ベクター/Opti-Pro SFM混合液(20ml)を加えて穏やかに攪拌し、さらに5分間放置した後にFreeStyle 293F細胞に添加した。37℃、8%CO2インキュベーターで7日間、90rpmで振とう培養して得られた培養上清をDisposable Capsule Filter (ADVANTEC #CCS-045-E1H)でろ過した。
上記7-3-2)で得られた培養上清から抗体を、rProteinAアフィニティークロマトグラフィー(4-6℃)とセラミックハイドロキシアパタイト(室温)の2段階工程で精製した。rProteinAアフィニティークロマトグラフィー精製後とセラミックハイドロキシアパタイト精製後のバッファー置換工程は4-6℃で実施した。最初に、培養上清を、PBSで平衡化したMabSelectSuRe(GE Healthcare Bioscience社製、HiTrapカラム)にアプライした。培養上清がカラムに全て入った後、カラム容量2倍以上のPBSでカラムを洗浄した。次に2Mアルギニン塩酸塩溶液(pH4.0)で溶出し、抗体の含まれる画分を集めた。その画分を透析(Thermo Scientific社、Slide-A-Lyzer Dialysis Cassette)によりPBSに置換した後、5mMリン酸ナトリウム/50mM MES/pH7.0のバッファーで5倍希釈した抗体溶液を、5mM NaPi/50mM MES/30mM NaCl/pH7.0のバッファーで平衡化されたセラミックハイドロキシアパタイトカラム(日本バイオラッド、Bio-Scale CHTType‐I Hydroxyapatite Column)にアプライした。塩化ナトリウムによる直線的濃度勾配溶出を実施し、抗体の含まれる画分を集めた。その画分を透析(Thermo Scientific社、Slide-A-Lyzer Dialysis Cassette)によりHBSor(25mM ヒスチジン/5% ソルビトール、pH6.0)への液置換を行った。最後にCentrifugal UF Filter Device VIVASPIN20(分画分子量UF10K,Sartorius社,4℃)にて濃縮し、IgG濃度を20mg/ml以上に調製し精製サンプルとした。
hRS7抗体は、国際公開第2003/074566号に記載されている軽鎖、及び重鎖のアミノ酸配列をもとに作製した。
1-1)hRS7抗体重鎖発現ベクターの構築
配列表の配列番号29に示すhRS7抗体重鎖のヌクレオチド配列のヌクレオチド番号36乃至437に示されるhRS7抗体重鎖の可変領域をコードするDNA配列を含むDNA断片を合成し(GENEART社 人工遺伝子合成サービス)、実施例7-1-1)と同様の方法でhRS7抗体重鎖発現ベクターを構築した。得られた発現ベクターを「pCMA-G1/hRS7」と命名した。hRS7抗体重鎖のアミノ酸配列を配列表の配列番号30に示した。
配列表の配列番号31に示すhRS7抗体軽鎖のヌクレオチド配列のヌクレオチド番号38乃至402に示されるhRS7抗体軽鎖の可変領域をコードするDNA配列を含むDNA断片を合成し(GENEART社 人工遺伝子合成サービス)、実施例7-2-1)と同様の方法でhRS7抗体軽鎖発現ベクターを構築した。得られた発現ベクターを「pCMA-LK/hRS7」と命名した。hRS7抗体重鎖のアミノ酸配列を配列表の配列番号32に示した。
1-3-1)hRS7抗体の生産
hRS7抗体はpCMA-G1/hRS7とpCMA-LK/hRS7との組合せにより実施例7-3-2)と同様の方法で生産した。
上記1-3-1)で得られた培養上清から実施例7-3-3)と同様の方法で精製した。
8-1)小スケール生産の抗体(培養上清)を用いた抗原結合能の測定
抗体と抗原(Recombinant Human TROP-2 Fc chimera)との解離定数測定は、Biacore 3000(GEヘルスケアバイオサイエンス(株))を使用し、固定化した抗ヒトIgG(Fab)抗体に抗体をリガンドとして捕捉(キャプチャー)し、抗原をアナライトとして測定するキャプチャー法にて行った。抗ヒトIgG(Fab)抗体(Human Fab capture kit、GEヘルスケアバイオサイエンス(株))は、センサーチップCM5(BIAcore,Inc.)へアミンカップリング法にて約2000RU共有結合させた。リファレンスセルにも同様に固定化した。ランニングバッファーとしてHBS-EP+(10mM HEPES pH7.4、0.15M NaCl,3mM EDTA、0.05%Surfactant P20)を用いた。抗ヒトIgG(Fab)抗体を固定化したチップ上に、抗体を含む培養上清を約80秒間添加した後、抗原の希釈系列溶液(1-1000nM)を流速30μl/分で300秒間添加し、引き続き600秒間の解離相をモニターした。再生溶液として、20%DMSOを含む10mM Gly-HCl pH1.5を流速10μl/分で60秒間添加した。データの解析には、分析ソフトウェア(BIAevaluation software, version 4.1)のBivalent結合モデルを用いて、結合速度定数kon、解離速度定数koff及び解離定数(KD;KD=koff/kon)を算出した。

抗体と抗原(Recombinant Human TROP-2 Fc chimera)との解離定数測定は、Biacore 3000(GEヘルスケアバイオサイエンス(株))を使用し、固定化した抗ヒトIgG(Fab)抗体に抗体をリガンドとして捕捉(キャプチャー)し、抗原をアナライトとして測定するキャプチャー法にて行った。抗ヒトIgG(Fab)抗体(Human Fab capture kit、GEヘルスケアバイオサイエンス(株))は、センサーチップCM5(BIAcore,Inc.)へアミンカップリング法にて約2000RU共有結合させた。リファレンスセルにも同様に固定化した。ランニングバッファーとしてHBS-EP+(10mM HEPES pH7.4、0.15M NaCl,3mM EDTA、0.05%Surfactant P20)を用いた。抗ヒトIgG(Fab)抗体を固定化したチップ上に、抗体を約1分間添加した後、抗原の希釈系列溶液(1-1000nM)を流速30μl/分で300秒間添加し、引き続き600秒間の解離相をモニターした。再生溶液として、ランニングバッファーにて希釈した25mM NaOHを流速100μl/分で3秒間、2回添加した。データの解析は、上記方法により行った。


4-(tert-ブトキシカルボニルアミノ)ブタン酸(0.237g、1.13mmoL)をジクロロメタン(10mL)に溶解し、N-ヒドロキシスクシンイミド(0.130g、1.13mmoL)、及び1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(0.216g、1.13mmoL)を加えて1時間撹拌した。エキサテカンのメシル酸塩(0.500g、0.94mmoL)、及びトリエチルアミン(0.157mL、1.13mmoL)を加えたN,N-ジメチルホルムアミド溶液(10mL)に滴下し、室温にて一日間撹拌した。溶媒を減圧留去し、得られた残留物をシリカゲルカラムクロマトグラフィー[クロロホルム~クロロホルム:メタノ-ル=8:2(v/v)]にて精製し、標記化合物(0.595g、定量的)を得た。
1H-NMR(400MHz,DMSO-d6)δ:0.87(3H,t,J=7.2Hz),1.31(9H,s),1.58(1H,t,J=7.2Hz),1.66(2H,t,J=7.2Hz),1.89-1.82(2H,m),2.12-2.21(3H,m),2.39(3H,s),2.92(2H,t,J=6.5Hz),3.17(2H,s),5.16(1H,d,J=19.2Hz),5.24(1H,d,J=18.8Hz),5.42(2H,s),5.59-5.55(1H,m),6.53(1H,s),6.78(1H,t,J=6.3Hz),7.30(1H,s),7.79(1H,d,J=11.0Hz),8.40(1H,d,J=8.6Hz).
MS(APCI)m/z:621(M+H)+.
上記工程1で得た化合物(0.388g、0.61mmoL)をジクロロメタン(9mL)に溶解した。トリフルオロ酢酸(9mL)を加えて4時間撹拌した。溶媒を減圧留去し、得られた残留物をシリカゲルカラムクロマトグラフィー[クロロホルム~クロロホルム:メタノール:水=7:3:1(v/v/v)の分配有機層]にて精製し、標記化合物(0.343g、定量的)を得た。
1H-NMR(400MHz,DMSO-d6)δ:0.87(3H,t,J=7.2Hz),1.79-1.92(4H,m),2.10-2.17(2H,m),2.27(2H,t,J=7.0Hz),2.40(3H,s),2.80-2.86(2H,m),3.15-3.20(2H,m),5.15(1H,d,J=18.8Hz),5.26(1H,d,J=18.8Hz),5.42(2H,s),5.54-5.61(1H,m),6.55(1H,s),7.32(1H,s),7.72(3H,brs),7.82(1H,d,J=11.0Hz),8.54(1H,d,J=8.6Hz).
MS(APCI)m/z:521(M+H)+.
N-(tert-ブトキシカルボニル)グリシルグリシル-L-フェニルアラニルグリシン(0.081g、0.19mmoL)をジクロロメタン(3mL)に溶解し、N-ヒドロキシスクシンイミド(0.021g、0.19moL)及び、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(0.036g、0.19mmoL)を加えて3.5時間撹拌した。その反応溶液を上記工程2で得た化合物(0.080g、0.15mmoL)を加えたN,N-ジメチルホルムアミド溶液(1.5mL)に滴下し、室温にて4時間撹拌した。溶媒を減圧留去し、得られた残留物をシリカゲルカラムクロマトグラフィー[クロロホルム~クロロホルム:メタノ-ル=8:2(v/v)]にて精製し、標記化合物(0.106g、73%)を得た。
1H-NMR(400MHz,DMSO-d6)δ:0.87(3H,t,J=7.4Hz),1.36(9H,s),1.71(2H,m),1.86(2H,t,J=7.8Hz),2.15-2.19(4H,m),2.40(3H,s),2.77(1H,dd,J=12.7,8.8Hz),3.02(1H,dd,J=14.1,4.7Hz),3.08-3.11(2H,m),3.16-3.19(2H,m),3.54(2H,d,J=5.9Hz),3.57-3.77(4H,m),4.46-4.48(1H,m),5.16(1H,d,J=19.2Hz),5.25(1H,d,J=18.8Hz),5.42(2H,s),5.55-5.60(1H,m),6.53(1H,s),7.00(1H,t,J=6.3Hz),7.17-7.26(5H,m),7.31(1H,s),7.71(1H,t,J=5.7Hz),7.80(1H,d,J=11.0Hz),7.92(1H,t,J=5.7Hz),8.15(1H,d,J=8.2Hz),8.27(1H,t,J=5.5Hz),8.46(1H,d,J=8.2Hz).
MS(APCI)m/z:939(M+H)+.
上記工程3で得た化合物(1.97g、2.10mmoL)をジクロロメタン(7mL)に溶解した。トリフルオロ酢酸(7mL)を加えて1時間撹拌した。溶媒を減圧留去し、トルエンを加えて共沸し、得られた残留物をシリカゲルカラムクロマトグラフィー[クロロホルム~クロロホルム:メタノール:水=7:3:1(v/v/v)の分配有機層]にて精製し、標記化合物(1.97g、99%)を得た。
1H-NMR(400MHz,DMSO-d6)δ:0.87(3H,t,J=7.4Hz),1.71-1.73(2H,m),1.82-1.90(2H,m),2.12-2.20(4H,m),2.40(3H,s),2.75(1H,dd,J=13.7,9.4Hz),3.03-3.09(3H,m),3.18-3.19(2H,m),3.58-3.60(2H,m),3.64(1H,d,J=5.9Hz),3.69(1H,d,J=5.9Hz),3.72(1H,d,J=5.5Hz),3.87(1H,dd,J=16.8,5.9Hz),4.50-4.56(1H,m),5.16(1H,d,J=19.2Hz),5.25(1H,d,J=18.8Hz),5.42(2H,s),5.55-5.60(1H,m),7.17-7.27(5H,m),7.32(1H,s),7.78-7.81(2H,m),7.95-7.97(3H,m),8.33-8.35(2H,m),8.48-8.51(2H,m).
MS(APCI)m/z:839(M+H)+.
上記工程4で得た化合物(100mg,0.119mmoL)のN,N-ジメチルホルムアミド(1.20mL)溶液に、ジイソプロピルエチルアミン(20.8μL,0.119mmoL)、3-(2-(2-(3-マレインイミドプロパンアミド)エトキシ)エトキシ)プロパン酸N-スクシンイミジル(50.7mg,0.119mmoL)を加え、室温で1時間撹拌した。溶媒を減圧留去し、得られた残留物をシリカゲルカラムクロマトグラフィー[クロロホルム~クロロホルム:メタノール=5:1(v/v)]にて精製し、標記化合物(66.5mg,48%)を淡黄色固体として得た。
1H-NMR(400MHz,DMSO-d6)δ:0.85(3H,t,J=7.4Hz),1.65-1.74(2H,m),1.77-1.90(2H,m),2.07-2.19(4H,m),2.30(2H,t,J=7.2Hz),2.33-2.36(2H,m),2.38(3H,s),2.76(1H,dd,J=13.7,9.8Hz),2.96-3.18(9H,m),3.42-3.44(4H,m),3.53-3.76(10H,m),4.43(1H,td,J=8.6,4.7Hz),5.14(1H,d,J=18.8Hz),5.23(1H,d,J=18.8Hz),5.38(1H,d,J=17.2Hz),5.42(1H,d,J=17.2Hz),5.52-5.58(1H,m),6.52(1H,s),6.98(2H,s),7.12-7.17(1H,m),7.18-7.25(4H,m),7.29(1H,s),7.69(1H,t,J=5.5Hz),7.78(1H,d,J=11.3Hz),7.98-8.03(2H,m),8.11(1H,d,J=7.8Hz),8.16(1H,t,J=5.7Hz),8.23(1H,t,J=5.9Hz),8.44(1H,d,J=9.0Hz).
MS(APCI)m/z:1149(M+H)+.
抗体の還元:実施例7にて作製したhTINA1-H1L1を、製造方法1に記載した共通操作B(280nm吸光係数として1.54を使用)及びCを用いて、PBS6.0/EDTAにて10mg/mLに調製した。本溶液(10.0mL)を50mLチューブに採取し、10mM TCEP(東京化成工業株式会社)水溶液(0.317mL;抗体一分子に対して4.6当量)及び1Mリン酸水素二カリウム水溶液(Nacalai Tesque,Inc.;0.500mL)を加えた。本溶液のpHが7.4±0.1内であることを確認した後に、37℃で1時間インキュベートすることによって、抗体内ヒンジ部のジスルフィド結合を還元した。
抗体と薬物リンカーのコンジュゲーション:上記溶液を常温水浴で10分間インキュベートした後にジメチルスルホキシド(0.567mL)を添加した。次いで上記工程5で得た化合物の10mMジメチルスルホキシド溶液(0.635mL;抗体一分子に対して9.2当量)を加え、チューブローテーター(MTR-103、アズワン株式会社)を用いて室温で40分間撹拌し、薬物リンカーを抗体へ結合させた。次に、100mM NAC(Sigma-Aldrich Co.LLC)水溶液(0.127mL;抗体一分子に対して18.4当量)を加え、さらに室温にて20分間撹拌し、薬物リンカーの反応を停止させた。
精製:上記溶液を、製造方法1に記載した共通操作Dによる精製を行い、標記抗体-薬物コンジュゲートを含有する溶液を35.0mL得た。
特性評価:製造方法1に記載した共通操作E(εD,280=4964(実測値)、εD,370=18982(実測値)を使用)を使用して、下記の特性値を得た。
抗体濃度:2.70mg/mL,抗体収量:94.5mg(95%),共通操作Eにて測定された抗体一分子あたりの薬物平均結合数(n):6.6。

抗体の還元:実施例7にて作製したhTINA1-H1L1を、製造方法1に記載した共通操作B(280nm吸光係数として1.54を使用)及びCを用いて、PBS6.0/EDTAにて10mg/mLに調製した。本溶液(2.00mL)を4mLチューブに採取し、10mM TCEP(東京化成工業株式会社)水溶液(0.0690mL;抗体一分子に対して5.0当量)及び1Mリン酸水素二カリウム水溶液(Nacalai Tesque,Inc.;0.100mL)を加えた。本溶液のpHが7.4±0.1内であることを確認した後に、37℃で1時間インキュベートすることによって、抗体内ヒンジ部のジスルフィド結合を還元した。
抗体と薬物リンカーのコンジュゲーション:上記溶液を15℃水浴で10分間インキュベートした後に実施例9工程5で得た化合物の10mMジメチルスルホキシド溶液(0.127mL;抗体一分子に対して9.2当量)を加え、15℃水浴にて1時間インキュベートし、薬物リンカーを抗体へ結合させた。次に、100mM NAC(Sigma-Aldrich Co.LLC)水溶液(0.0190mL;抗体一分子に対して13.8当量)を加え、チューブローテーターを用いて室温で20分間撹拌し、薬物リンカーの反応を停止させた。
精製:上記溶液を、製造方法1に記載した共通操作Dによる精製を行い、標記抗体-薬物コンジュゲートを含有する溶液を9.00mL得た。
特性評価:製造方法1に記載した共通操作E(εD,280=4964(実測値)、εD,370=18982(実測値)を使用)を使用して、下記の特性値を得た。
抗体濃度:2.08mg/mL,抗体収量:18.7mg(94%),共通操作Eにて測定された抗体一分子あたりの薬物平均結合数(n):6.1。

抗体の還元:実施例7にて作製したhTINA1-H1L1を、製造方法1に記載した共通操作B(280nm吸光係数として1.54を使用)及びCを用いて、PBS6.0/EDTAにて10mg/mLに調製した。本溶液(5.0mL)を15mL容器に採取し、撹拌下1Mリン酸水素二カリウム水溶液(Nacalai Tesque,Inc.;0.0813mL)を加えた後、37℃で10分間撹拌した。撹拌下10mM TCEP(東京化成工業株式会社)水溶液(0.0745mL;抗体一分子に対して2.3当量)を加えた後、本溶液のpHが7.0±0.1内であることを確認し、37℃で1時間撹拌することによって、抗体内ヒンジ部のジスルフィド結合を還元した。
抗体と薬物リンカーのコンジュゲーション:上記溶液を15℃水浴で10分間撹拌した後に実施例9工程5で得た化合物の10mMジメチルスルホキシド溶液(0.162mL;抗体一分子に対して5.0当量)をゆっくり滴下しながら加え、15℃水浴にて1時間撹拌し、薬物リンカーを抗体へ結合させた。次に、100mM NAC(Sigma-Aldrich Co.LLC)水溶液(0.0418mL;抗体一分子に対して12.9当量)を加え、室温で20分間撹拌し、薬物リンカーの反応を停止させた。
精製:上記溶液を、製造方法1に記載した共通操作Dによる精製を行い、標記抗体-薬物コンジュゲートを含有する溶液を21.0mL得た。
特性評価:製造方法1に記載した共通操作E及びF(εD,280=4964(実測値)、εD,370=18982(実測値)を使用)を使用して、下記の特性値を得た。
抗体濃度:2.19mg/mL,抗体収量:46.0mg(92%),共通操作Eにて測定された抗体一分子あたりの薬物平均結合数(n):3.6;共通操作Fにて測定された抗体一分子あたりの薬物平均結合数(n):3.6。

抗体の還元:実施例7にて作製したhTINA1-H1L1を、製造方法1に記載した共通操作B(280nm吸光係数として1.54を使用)及びCを用いて、PBS6.0/EDTAにて10.0mg/mLに調製した。本溶液(5.00mL)を15mL容器に採取し、撹拌下1Mリン酸水素二カリウム水溶液(Nacalai Tesque,Inc.;0.0813mL)を加えた後、37℃で10分間撹拌した。撹拌下10mM TCEP(東京化成工業株式会社)水溶液(0.162mL;抗体一分子に対して5.0当量)を加えた後、本溶液のpHが7.0±0.1内であることを確認し、37℃で1時間撹拌することによって、抗体内ヒンジ部のジスルフィド結合を還元した。
抗体と薬物リンカーのコンジュゲーション:上記溶液を15℃水浴で10分間撹拌した後に実施例9工程5で得た化合物の10mMジメチルスルホキシド溶液(0.389mL;抗体一分子に対して12.0当量)をゆっくり滴下しながら加え、15℃水浴にて1時間撹拌し、薬物リンカーを抗体へ結合させた。次に、100mM NAC(Sigma-Aldrich Co.LLC)水溶液(0.0418mL;抗体一分子に対して12.9当量)を加え、室温で20分間撹拌し、薬物リンカーの反応を停止させた。
精製:上記溶液を、製造方法1に記載した共通操作Dによる精製を行い、標記抗体-薬物コンジュゲートを含有する溶液を21.0mL得た。
特性評価:製造方法1に記載した共通操作E及びF(εD,280=4964(実測値)、εD,370=18982(実測値)を使用)を使用して、下記の特性値を得た。
抗体濃度:2.19mg/mL,抗体収量:46.0mg(92%),共通操作Eにて測定された抗体一分子あたりの薬物平均結合数(n):7.0;共通操作Fにて測定された抗体一分子あたりの薬物平均結合数(n):7.0。

抗体の還元:参考例1にて作製したhRS7を、製造方法1に記載した共通操作B(280nm吸光係数として1.56を使用)及びCを用いて、PBS6.0/EDTAにて10mg/mLに調製した。本溶液(2.0mL)を4mLチューブに採取し、10mM TCEP(東京化成工業株式会社)水溶液(0.0690mL;抗体一分子に対して5.0当量)及び1Mリン酸水素二カリウム水溶液(Nacalai Tesque,Inc.;0.100mL)を加えた。本溶液のpHが7.4±0.1内であることを確認した後に、37℃で1時間インキュベートすることによって、抗体内ヒンジ部のジスルフィド結合を還元した。
抗体と薬物リンカーのコンジュゲーション:上記溶液を15℃水浴で10分間インキュベートした後に実施例9工程5で得た化合物の10mMジメチルスルホキシド溶液(0.127mL;抗体一分子に対して9.2当量)を加え、15℃水浴にて1時間インキュベートし、薬物リンカーを抗体へ結合させた。次に、100mM NAC(Sigma-Aldrich Co.LLC)水溶液(0.0190mL;抗体一分子に対して13.8当量)を加え、チューブローテーターを用いて室温で20分間撹拌し、薬物リンカーの反応を停止させた。
精製:上記溶液を、製造方法1に記載した共通操作Dによる精製を行い、標記抗体-薬物コンジュゲートを含有する溶液を9.00mL得た。
特性評価:製造方法1に記載した共通操作E(εD,280=4964(実測値)、εD,370=18982(実測値)を使用)を使用して、下記の特性値を得た。
抗体濃度:2.04mg/mL,抗体収量:18.4mg(92%),共通操作Eにて測定された抗体一分子あたりの薬物平均結合数(n):6.2。

N-9-フルオレニルメトキシカルボニルグリシルグリシン(4.33g,12.2mmol)、テトラヒドロフラン(120ml)、及びトルエン(40.0ml)からなる混合物に、ピリジン(1.16ml,14.7mmol及び四酢酸鉛(6.84g,14.7mmol)を加え、5時間加熱還流した。反応液を室温まで冷却した後、不溶物をセライト濾過によって除き、減圧下濃縮した。得られた残留物を酢酸エチルに溶解し、水及び飽和食塩水で洗浄後、有機層を無水硫酸マグネシウムで乾燥した。溶媒を減圧下で留去した後、得られた残留物をシリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=9:1(v/v)~酢酸エチル]にて精製し、標記化合物(3.00g,67%)を無色固体として得た。
1H-NMR(400MHz,CDCl3)δ:2.07(3H,s),3.90(2H,d,J=5.1Hz),4.23(1H,t,J=7.0Hz),4.46(2H,d,J=6.6Hz),5.26(2H,d,J=7.0Hz),5.32(1H,brs),6.96(1H,brs),7.32(2H,t,J=7.3Hz),7.41(2H,t,J=7.3Hz),7.59(2H,d,J=7.3Hz),7.77(2H,d,J=7.3Hz).
上記工程1で得た化合物(3.68g,10.0mmoL)及びベンジルグリコレート(4.99g,30.0mmoL)のテトラヒドロフラン(40.0mL)溶液に、カリウム tert-ブトキシド(2.24g,20.0mmoL)を0℃で加え、室温にて15分間撹拌した。反応溶液に酢酸エチル、水を0℃で加え、酢酸エチル、クロロホルムで抽出し、得られた有機層を硫酸ナトリウムで乾燥し、ろ過した。溶媒を減圧留去し、得られた残留物をジオキサン(40.0mL)、水(10.0mL)に溶解し、炭酸水素ナトリウム(1.01g,12.0mmoL)、クロロギ酸9-フルオレニルメチル(2.59g,10.0mmoL)を加え、室温で2時間撹拌した。反応溶液に水を加え、酢酸エチルで抽出し、得られた有機層を硫酸ナトリウムで乾燥し、ろ過した。溶媒を減圧留去し、得られた残留物をシリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=100:0(v/v)~0:100]にて精製し、無色油状の標記化合物(1.88g、40%)を得た。
1H-NMR(400MHz,CDCl3)δ:3.84(2H,d,J=5.5Hz),4.24(3H,t,J=6.5Hz),4.49(2H,d,J=6.7Hz),4.88(2H,d,J=6.7Hz),5.15-5.27(1H,m),5.19(2H,s),6.74(1H,brs),7.31-7.39(7H,m),7.43(2H,t,J=7.4Hz),7.61(2H,d,J=7.4Hz),7.79(2H,d,J=7.4Hz).
上記工程2で得た化合物(1.88g、3.96mmoL)をエタノール(40.0mL)、酢酸エチル(20.0ml)に溶解した。パラジウム炭素触媒(376mg)を加え、水素雰囲気下、室温にて2時間撹拌した。不溶物をセライト濾過によって除き、溶媒を減圧留去し、標記化合物(1.52g、定量的)を無色固体として得た。
1H-NMR(400MHz,DMSO-d6)δ:3.62(2H,d,J=6.3Hz),3.97(2H,s),4.18-4.32(3H,m),4.60(2H,d,J=6.7Hz),7.29-7.46(4H,m),7.58(1H,t,J=5.9Hz),7.72(2H,d,J=7.4Hz),7.90(2H,d,J=7.4Hz),8.71(1H,t,J=6.5Hz).
氷冷下、エキサテカンのメシル酸塩(0.283g,0.533mmoL)、N-ヒドロキシスクシンイミド(61.4mg,0.533mmoL)、及び上記工程3で得た化合物(0.205g,0.533mmoL)のN,N-ジメチルホルムアミド(10.0mL)溶液に、N,N-ジイソプロピルエチルアミン(92.9μL,0.533mmoL)及びN,N’-ジシクロヘキシルカルボジイミド(0.143g,0.693mmoL)を加え、室温にて3日間撹拌した。溶媒を減圧留去し、得られた残留物をシリカゲルカラムクロマトグラフィー[クロロホルム~クロロホルム:メタノール:水=7:3:1(v/v/v)の分配有機層]にて精製し、標記化合物(0.352g,82%)を淡茶色固体として得た。
1H-NMR(400MHz,DMSO-d6)δ:0.81(3H,t,J=7.4Hz),1.73-1.87(2H,m),2.06-2.20(2H,m),2.34(3H,s),3.01-3.23(2H,m),3.58(2H,d,J=6.7Hz),3.98(2H,s),4.13-4.25(3H,m),4.60(2H,d,J=6.7Hz),5.09-5.22(2H,m),5.32-5.42(2H,m),5.50-5.59(1H,m),6.49(1H,s),7.24-7.30(3H,m),7.36(2H,t,J=7.4Hz),7.53(1H,t,J=6.3Hz),7.66(2H,d,J=7.4Hz),7.75(1H,d,J=11.0Hz),7.84(2H,d,J=7.4Hz),8.47(1H,d,J=8.6Hz),8.77(1H,t,J=6.7Hz).
MS(ESI)m/z:802(M+H)+.
上記工程4で得た化合物(0.881g,1.10mmoL)のN,N-ジメチルホルムアミド(11.0mL)溶液に、ピペリジン(1.1mL)を加え、室温で2時間撹拌した。溶媒を減圧留去し、標記化合物を含む混合物を得た。本混合物は、これ以上の精製は行わずに次の反応に用いた。
氷冷下、上記工程5で得た混合物(0.439mmoL)、N-ヒドロキシスクシンイミド(0.101g,0.878mmoL)、及びN-[(9H-フルオレン-9-イルメトキシ)カルボニル]グリシルグリシル-L-フェニルアラニン(特開2002-60351号公報;0.440g,0.878mmoL)のN,N-ジメチルホルムアミド(50.0mL)溶液に、N,N’-ジシクロヘキシルカルボジイミド(0.181g,0.878mmoL)を加え、室温にて4日間撹拌した。溶媒を減圧留去し、得られた残留物をシリカゲルカラムクロマトグラフィー[クロロホルム~クロロホルム:メタノール=9:1(v/v)]にて精製し、標記化合物(0.269g,58%)を淡橙色固体として得た。
MS(ESI)m/z:1063(M+H)+.
上記工程6で得た化合物(0.269g,0.253mmoL)のN,N-ジメチルホルムアミド(4.00mL)溶液に、ピペリジン(0.251mL,2.53mmoL)を加え、室温で2時間撹拌した。溶媒を減圧留去し、標記化合物を含む混合物を得た。本混合物は、これ以上の精製は行わずに次の反応に用いた。
上記工程7で得た化合物(0.253mmoL)のN,N-ジメチルホルムアミド(10.0mL)溶液に、6-マレイミドヘキサン酸N-スクシンイミジル(0.156g,0.506mmoL)を加え、室温で3日間撹拌した。溶媒を減圧留去し、得られた残留物をシリカゲルカラムクロマトグラフィー[クロロホルム~クロロホルム:メタノール=9:1(v/v)]にて精製し、標記化合物(0.100g,38%)を淡黄色固体として得た。
1H-NMR(400MHz,DMSO-d6)δ:0.83(3H,t,J=7.2Hz),1.09-1.21(2H,m),1.33-1.47(4H,m),1.75-1.90(2H,m),2.00-2.23(4H,m),2.36(3H,s),2.69-2.81(1H,m),2.94-3.03(1H,m),3.06-3.22(2H,m),3.23-3.74(6H,m),3.98(2H,s),4.39-4.50(1H,m),4.60(2H,d,J=6.7Hz),5.17(2H,s),5.39(2H,s),5.53-5.61(1H,m),6.50(1H,s),6.96(2H,s),7.11-7.24(5H,m),7.28(1H,s),7.75(1H,d,J=11.0Hz),7.97(1H,t,J=5.7Hz),8.03(1H,t,J=5.9Hz),8.09(1H,d,J=7.8Hz),8.27(1H,t,J=6.5Hz),8.48(1H,d,J=9.0Hz),8.60(1H,t,J=6.5Hz).
MS(ESI)m/z:1034(M+H)+.
抗体の還元:実施例7にて作製したhTINA1-H1L1を、製造方法1に記載した共通操作B(280nm吸光係数として1.54を使用)及びCを用いて、PBS6.0/EDTAにて10mg/mLに調製した。本溶液(10.0mL)を50mLチューブに採取し、10mM TCEP(東京化成工業株式会社)水溶液(0.317mL;抗体一分子に対して4.6当量)及び1Mリン酸水素二カリウム水溶液(Nacalai Tesque,Inc.;0.500mL)を加えた。本溶液のpHが7.4±0.1内であることを確認した後に、37℃で1時間インキュベートすることによって、抗体内ヒンジ部のジスルフィド結合を還元した。
抗体と薬物リンカーのコンジュゲーション:上記溶液を常温水浴で10分間インキュベートした後にジメチルスルホキシド(0.567mL)を添加した。次いで上記工程8で得た化合物の10mMジメチルスルホキシド溶液(0.635mL;抗体一分子に対して9.2当量)を加え、チューブローテーターを用いて室温で40分間撹拌し、薬物リンカーを抗体へ結合させた。次に、100mM NAC(Sigma-Aldrich Co.LLC)水溶液(0.127mL;抗体一分子に対して18.4当量)を加え、さらに室温にて20分間撹拌し、薬物リンカーの反応を停止させた。
精製:上記溶液を、製造方法1に記載した共通操作Dによる精製を行い、標記抗体-薬物コンジュゲートを含有する溶液を35.0mL得た。
特性評価:製造方法1に記載した共通操作E(εD,280=5178(実測値)、εD,370=20217(実測値)を使用)を使用して、下記の特性値を得た。
抗体濃度:2.70mg/mL,抗体収量:94.5mg(95%),共通操作Eにて測定された抗体一分子あたりの薬物平均結合数(n):6.4。

抗体の還元:実施例7にて作製したhTINA1-H1L1を、製造方法1に記載した共通操作B(280nm吸光係数として1.54を使用)及びCを用いて、PBS6.0/EDTAにて10mg/mLに調製した。本溶液(2.0mL)を4mLチューブに採取し、10mM TCEP(東京化成工業株式会社)水溶液(0.0690mL;抗体一分子に対して5.0当量)及び1Mリン酸水素二カリウム水溶液(Nacalai Tesque,Inc.;0.0299mL)を加えた。本溶液のpHが7.0±0.1内であることを確認した後に、37℃で1時間インキュベートすることによって、抗体内ヒンジ部のジスルフィド結合を還元した。
抗体と薬物リンカーのコンジュゲーション:上記溶液を15℃水浴で10分間インキュベートした後に実施例14工程8で得た化合物の10mMジメチルスルホキシド溶液(0.127mL;抗体一分子に対して9.2当量)を加え、15℃水浴にて1時間インキュベートし、薬物リンカーを抗体へ結合させた。次に、100mM NAC(Sigma-Aldrich Co.LLC)水溶液(0.0190mL;抗体一分子に対して13.8当量)を加え、チューブローテーターを用いて室温で20分間撹拌し、薬物リンカーの反応を停止させた。
精製:上記溶液を、製造方法1に記載した共通操作Dによる精製を行い、標記抗体-薬物コンジュゲートを含有する溶液を9.00mL得た。
特性評価:製造方法1に記載した共通操作E(εD,280=5178(実測値)、εD,370=20217(実測値)を使用)を使用して、下記の特性値を得た。
抗体濃度:2.04mg/mL,抗体収量:18.4mg(92%),共通操作Eにて測定された抗体一分子あたりの薬物平均結合数(n):5.7。

抗体の還元:実施例7にて作製したhTINA1-H1L1を、製造方法1に記載した共通操作B(280nm吸光係数として1.54を使用)及びCを用いて、PBS6.0/EDTAにて10mg/mLに調製した。本溶液(30.0mL)を100mL容器に採取し、撹拌下1Mリン酸水素二カリウム水溶液(Nacalai Tesque,Inc.;0.4875mL)を加えた後、37℃で10分間撹拌した。撹拌下10mM TCEP(東京化成工業株式会社)水溶液(0.9721mL;抗体一分子に対して5.0当量)を加えた後、本溶液のpHが7.0±0.1内であることを確認し、37℃で1時間撹拌することによって、抗体内ヒンジ部のジスルフィド結合を還元した。
抗体と薬物リンカーのコンジュゲーション:上記溶液を15℃水浴で10分間撹拌した後に実施例14工程8で得た化合物の10mMジメチルスルホキシド溶液(2.33mL;抗体一分子に対して12.0当量)をゆっくり滴下しながら加え、15℃水浴にて1時間撹拌し、薬物リンカーを抗体へ結合させた。次に、100mM NAC(Sigma-Aldrich Co.LLC)水溶液(0.251mL;抗体一分子に対して12.9当量)を加え、室温で20分間撹拌し、薬物リンカーの反応を停止させた。
精製:上記溶液を、製造方法1に記載した共通操作Dによる精製を行い、標記抗体-薬物コンジュゲートを含有する溶液を98.0mL得た。その後、製造方法1に記載した共通操作Aによる濃縮操作を行い、標記抗体-薬物コンジュゲートを含有する溶液を17.5mL得た。
特性評価:製造方法1に記載した共通操作E及びF(εD,280=5178(実測値)、εD,370=20217(実測値)を使用)を使用して、下記の特性値を得た。
抗体濃度:14.6mg/mL,抗体収量:256mg(85%),共通操作Eにて測定された抗体一分子あたりの薬物平均結合数(n):6.7;共通操作Fにて測定された抗体一分子あたりの薬物平均結合数(n):7.0。

抗体の還元:実施例7にて作製したhTINA1-H1L1を、製造方法1に記載した共通操作B(280nm吸光係数として1.54を使用)及びCを用いて、PBS6.0/EDTAにて10mg/mLに調製した。本溶液(5.0mL)を15mL容器に採取し、撹拌下1Mリン酸水素二カリウム水溶液(Nacalai Tesque,Inc.;0.0813mL)を加えた後、37℃で10分間撹拌した。撹拌下10mM TCEP(東京化成工業株式会社)水溶液(0.0778mL;抗体一分子に対して2.4当量)を加えた後、本溶液のpHが7.0±0.1内であることを確認し、37℃で1時間撹拌することによって、抗体内ヒンジ部のジスルフィド結合を還元した。
抗体と薬物リンカーのコンジュゲーション:上記溶液を15℃水浴で10分間撹拌した後に実施例14工程8で得た化合物の10mMジメチルスルホキシド溶液(0.162mL;抗体一分子に対して5.0当量)をゆっくり滴下しながら加え、15℃水浴にて1時間撹拌し、薬物リンカーを抗体へ結合させた。次に、100mM NAC(Sigma-Aldrich Co.LLC)水溶液(0.0418mL;抗体一分子に対して12.9当量)を加え、室温で20分間撹拌し、薬物リンカーの反応を停止させた。
精製:上記溶液を、製造方法1に記載した共通操作Dによる精製を行い、標記抗体-薬物コンジュゲートを含有する溶液を21.0mL得た。
特性評価:製造方法1に記載した共通操作E及びF(εD,280=5178(実測値)、εD,370=20217(実測値)を使用)を使用して、下記の特性値を得た。
抗体濃度:2.26mg/mL,抗体収量:47.5mg(95%),共通操作Eにて測定された抗体一分子あたりの薬物平均結合数(n):3.5;共通操作Fにて測定された抗体一分子あたりの薬物平均結合数(n):3.6。

抗体の還元:参考例1にて作製したhRS7を、製造方法1に記載した共通操作B(280nm吸光係数として1.56を使用)及びCを用いて、PBS6.0/EDTAにて10mg/mLに調製した。本溶液(2.0mL)を4mLチューブに採取し、10mM TCEP(東京化成工業株式会社)水溶液(0.0690mL;抗体一分子に対して5.0当量)及び1Mリン酸水素二カリウム水溶液(Nacalai Tesque,Inc.;0.0299mL)を加えた。本溶液のpHが7.0±0.1内であることを確認した後に、37℃で1時間インキュベートすることによって、抗体内ヒンジ部のジスルフィド結合を還元した。
抗体と薬物リンカーのコンジュゲーション:上記溶液を15℃水浴で10分間インキュベートした後に実施例14工程8で得た化合物の10mMジメチルスルホキシド溶液(0.1269mL;抗体一分子に対して9.2当量)を加え、15℃水浴にて1時間インキュベートし、薬物リンカーを抗体へ結合させた。次に、100mM NAC(Sigma-Aldrich Co.LLC)水溶液(0.0190mL;抗体一分子に対して13.8当量)を加え、チューブローテーターを用いて室温で20分間撹拌し、薬物リンカーの反応を停止させた。
精製:上記溶液を、製造方法1に記載した共通操作Dによる精製を行い、標記抗体-薬物コンジュゲートを含有する溶液を9.00mL得た。
特性評価:製造方法1に記載した共通操作E(εD,280=5178(実測値)、εD,370=20217(実測値)を使用)を使用して、下記の特性値を得た。
抗体濃度:2.07mg/mL,抗体収量:18.6mg(93%),共通操作Eにて測定された抗体一分子あたりの薬物平均結合数(n):5.6。

エキサテカンのメシル酸塩(3.10g、5.47moL)を、4-(tert-ブトキシカルボニルアミノ)ブタン酸の代わりに{2-[(tert-ブトキシカルボニル)アミノ]エトキシ}酢酸(J.Med.Chem.,1992年,35巻,2928頁;1.55g,6.01mmol)を用いて、実施例1の工程1と同様に反応させ、標記化合物(2.56g,73%)を淡黄色固体として得た。
1H-NMR(400MHz,DMSO-d6)δ:0.87(3H,t,J=7.3Hz),1.26(9H,s),1.81-1.91(2H,m),2.13-2.22(2H,m),2.40(3H,s),3.08-3.26(4H,m),3.43-3.53(2H,m),4.00(1H,d,J=15.1Hz),4.05(1H,d,J=15.1Hz),5.14(1H,d,J=18.7Hz),5.22(1H,d,J=18.7Hz),5.40(1H,d,J=16.6Hz),5.44(1H,d,J=16.6Hz),5.59-5.66(1H,m),6.53(1H,s),6.86(1H,t,J=5.4Hz),7.31(1H,s),7.79(1H,d,J=10.9Hz),8.49(1H,d,J=9.1Hz).
MS(APCI)m/z:637(M+H)+.
上記工程1で得た化合物(1.50g,2.36mol)を、実施例1の工程2と同様に反応させ、標記化合物(1.50g,定量的)を淡黄色固体として得た。
1H-NMR(400MHz,DMSO-d6)δ:0.87(3H,t,J=7.5Hz),1.81-1.92(2H,m),2.15-2.23(2H,m),2.41(3H,s),3.05(2H,t,J=5.1Hz),3.15-3.23(2H,m),3.71(2H,t,J=5.1Hz),4.10(2H,s),5.19(1H,d,J=18.7Hz),5.24(1H,d,J=18.7Hz),5.43(2H,s),5.58-5.66(1H,m),6.55(1H,s),7.33(1H,s),7.73-7.84(4H,m),8.55(1H,d,J=9.1Hz).
MS(APCI)m/z:537(M+H)+.
上記工程2で得た化合物(554mg,0.85mmol)を、実施例1の工程3と同様に反応させ、標記化合物(775mg,95%)を得た。
1H-NMR(400MHz,DMSO-d6)δ:0.85(3H,t,J=7.3Hz),1.36(9H,s),1.78-1.89(2H,m),2.13-2.22(2H,m),2.39(3H,s),2.71(1H,dd,J=13.4,9.8Hz),2.95(1H,dd,J=13.4,4.3Hz),3.09-3.23(1H,m),3.23-3.32(2H,m),3.40-3.62(8H,m),3.73(1H,dd,J=16.5,5.5Hz),4.03(2H,s),4.39-4.47(1H,m),5.17(1H,d,J=18.9Hz),5.25(1H,d,J=18.9Hz),5.41(1H,d,J=16.8Hz),5.45(1H,d,J=16.8Hz),5.57-5.64(1H,m),6.54(1H,s),6.99(1H,t,J=5.8Hz),7.13-7.26(5H,m),7.31(1H,s),7.76-7.82(2H,m),7.90(1H,t,J=5.2Hz),8.13(1H,d,J=7.9Hz),8.27(1H,t,J=5.8Hz),8.49(1H,d,J=8.5Hz).
MS(APCI)m/z:955(M+H)+.
上記工程3で得た化合物(630mg,0.659mmol)を、実施例1の工程4と同様に反応させ、標記化合物(588mg,92%)を得た。
1H-NMR(400MHz,DMSO-d6)δ:0.86(3H,t,J=7.3Hz),1.79-1.90(2H,m),2.13-2.22(2H,m),2.39(3H,s),2.71(1H,dd,J=13.4,10.1Hz),2.99(1H,dd,J=13.4,4.3Hz),3.09-3.23(1H,m),3.24-3.32(3H,m),3.41-3.71(7H,m),3.86(1H,dd,J=16.8,5.8Hz),4.04(2H,s),4.52(1H,td,J=9.0,4.1Hz),5.17(1H,d,J=18.9Hz),5.25(1H,d,J=18.9Hz),5.41(1H,d,J=16.5Hz),5.45(1H,d,J=16.5Hz),5.56-5.65(1H,m),6.55(1H,s),7.13-7.26(5H,m),7.32(1H,s),7.80(1H,d,J=11.0Hz),7.87-8.01(4H,m),8.29-8.36(2H,m),8.46-8.55(2H,m).
MS(APCI)m/z:855(M+H)+.
上記工程4で得た化合物(240mg,0.247mmol)を、ジイソプロピルエチルアミンの代わりにトリエチルアミン(31.4μL,0.22mmoL)を、3-(2-(2-(3-マレインイミドプロパンアミド)エトキシ)エトキシ)プロパン酸N-スクシンイミジルの代わりに6-マレイミドヘキサン酸N-スクシンイミジル(95.3mg,0.31mmoL)を用いて、実施例1の工程5と同様に反応させ、標記化合物(162mg,62%)を得た。
1H-NMR(400MHz,DMSO-d6)δ:0.86(3H,t,J=7.6Hz),1.13-1.22(2H,m),1.40-1.51(4H,m),1.78-1.90(2H,m),2.09(2H,t,J=7.6Hz),2.14-2.21(2H,m),2.39(3H,s),2.74(1H,dd,J=13.6,9.7Hz),2.96(1H,dd,J=13.6,4.5Hz),3.08-3.24(1H,m),3.24-3.30(1H,m),3.33-3.40(4H,m),3.47-3.68(7H,m),3.72(1H,dd,J=16.6,5.7Hz),4.03(2H,s),4.42(1H,td,J=8.6,4.2Hz),5.17(1H,d,J=18.7Hz),5.25(1H,d,J=18.7Hz),5.40(1H,d,J=17.2Hz),5.44(1H,d,J=17.2Hz),5.57-5.64(1H,m),6.52(1H,s),6.99(2H,s),7.13-7.25(5H,m),7.31(1H,s),7.74-7.81(2H,m),7.99(1H,t,J=5.7Hz),8.03-8.11(2H,m),8.22(1H,t,J=5.7Hz),8.47(1H,d,J=9.1Hz).
MS(APCI)m/z:1048(M+H)+.
抗体の還元:実施例7にて作製したhTINA1-H1L1を、製造方法1に記載した共通操作B(280nm吸光係数として1.54を使用)及びCを用いて、PBS6.0/EDTAにて10mg/mLに調製した。本溶液(3.0mL)を15mL容器に採取し、撹拌下1Mリン酸水素二カリウム水溶液(Nacalai Tesque,Inc.;0.0488mL)を加えた後、37℃で10分間撹拌した。撹拌下10mM TCEP(東京化成工業株式会社)水溶液(0.0972mL;抗体一分子に対して5.0当量)を加えた後、本溶液のpHが7.0±0.1内であることを確認し、37℃で1時間撹拌することによって、抗体内ヒンジ部のジスルフィド結合を還元した。
抗体と薬物リンカーのコンジュゲーション:上記溶液を15℃水浴で10分間撹拌した後に実施例11a工程8で得た化合物の10mMジメチルスルホキシド溶液(0.2333mL;抗体一分子に対して12.0当量)をゆっくり滴下しながら加え、15℃水浴にて1時間撹拌し、薬物リンカーを抗体へ結合させた。次に、100mM NAC(Sigma-Aldrich Co.LLC)水溶液(0.0251mL;抗体一分子に対して12.9当量)を加え、室温で20分間撹拌し、薬物リンカーの反応を停止させた。
精製:上記溶液を、製造方法1に記載した共通操作Dによる精製を行い、標記抗体-薬物コンジュゲートを含有する溶液を14mL得た。
特性評価:製造方法1に記載した共通操作E及びF(εD,280=5193(実測値)、εD,370=20347(実測値)を使用)を使用して、下記の特性値を得た。
抗体濃度:1.93mg/mL,抗体収量:27.0mg(90%),共通操作Eにて測定された抗体一分子あたりの薬物平均結合数(n):7.1;共通操作Fにて測定された抗体一分子あたりの薬物平均結合数(n):7.0。

抗体の還元:参考例1にて作製したhRS7を、製造方法1に記載した共通操作B(280nm吸光係数として1.54を使用)及びCを用いて、PBS6.0/EDTAにて10mg/mLに調製した。本溶液(10.0mL)を50mLチューブに採取し、10mM TCEP(東京化成工業株式会社)水溶液(0.317mL;抗体一分子に対して4.6当量)及び1Mリン酸水素二カリウム水溶液(Nacalai Tesque,Inc.;0.500mL)を加えた。本溶液のpHが7.4±0.1内であることを確認した後に、37℃で1時間インキュベートすることによって、抗体内ヒンジ部のジスルフィド結合を還元した。
抗体と薬物リンカーのコンジュゲーション:上記溶液を常温水浴で10分間インキュベートした後にジメチルスルホキシド(0.567mL)を添加した。次いで、米国特許公開第2011/0293513号明細書に従って合成したCL2A-SN38の10mMジメチルスルホキシド溶液(0.635mL;抗体一分子に対して9.2当量)を加え、チューブローテーターを用いて室温で40分間撹拌し、薬物リンカーを抗体へ結合させた。次に、100mM NAC(Sigma-Aldrich Co.LLC)水溶液(0.127mL;抗体一分子に対して18.4当量)を加え、さらに室温にて20分間撹拌し、薬物リンカーの反応を停止させた。
精製:上記反応溶液について、製造方法1-共通操作Dに記載したゲルろ過精製操作を2回繰り返し、次いでポリソルベート80(0.01%)を含有する25mMトレハロース溶液にて同様にNAP-25カラムによるゲルろ過精製操作を1回行った後に得られる溶出液(35mL)を凍結乾燥操した。
特性評価:凍結乾燥前の溶出液について製造方法1に記載した共通操作Eを使用して、下記の特性値を得た。
抗体濃度:2.78mg/mL,抗体収量:97.3mg(97%),抗体一分子あたりの薬物平均結合数(n):5.6

抗体の還元:実施例7にて作製したhTINA1-H1L1を、製造方法1に記載した共通操作B(280nm吸光係数として1.54を使用)及びCを用いて、媒体をPBS6.0/EDTAに置換し、10mg/mLの抗体濃度に調製した。本溶液(100mL)をポリカーボネート製の250mL三角フラスコ容器に入れ、マグネティックスターラー撹拌下、室温で1Mリン酸水素二カリウム水溶液(1.4mL)を加えた後、10mM TCEP水溶液(1.62mL;抗体一分子に対して2.5当量)を加えた。本溶液のpHが7.0±0.1内であることを確認した後に、撹拌を停止し、37℃で2時間インキュベートすることにより、抗体内ヒンジ部のジスルフィド結合を還元した。
抗体と薬物リンカーのコンジュゲーション:上記溶液を15℃に冷却後、撹拌下、DMSO(3.24mL)をゆっくり滴下して加えた。次いで、実施例14工程8の化合物を10mM含むDMSO溶液(1.76mL;抗体一分子に対して5.0当量)をゆっくり滴下して加えた。この溶液を15℃にて、1時間撹拌し、薬物リンカーを抗体へ結合させた。次に、撹拌下、100mM NAC水溶液(0.324mL;抗体一分子に対して5.0当量)を加え、さらに室温で20分間インキュベートし、未反応の薬物リンカーの反応性を停止させた。
精製:撹拌下、上記溶液に20%酢酸水(約0.52mL)とABS(100mL)をゆっくり加え、本溶液のpHを5.5±0.1にした。この溶液を精密ろ過(0.45μm、PVDF膜)することにより白濁物を除いて、ろ液を約200mL得た。このろ液に対して、限外ろ過膜(メルク株式会社、Pellicon XL Cassette、Ultracell 30KDa)、チューブポンプ(米国コールパーマー社マスターフレックスポンプ model 77521-40、ポンプヘッド model 7518-00)及びチューブ(米国コールパーマー社マスターフレックスチューブ L/S16)で構成された限外ろ過装置を用い、限外ろ過精製を行った。すなわち、反応液に精製緩衝液としてABS(計1600mL)を滴下しながら、限外ろ過精製を行うことで、未結合の薬物リンカー及び他の低分子量試薬を除去するとともに緩衝液をABSへ置換し、さらに濃縮まで行った。得られた精製溶液に対して、精密ろ過(0.22μm、PVDF膜)を行い、標記抗体-薬物コンジュゲートを含有する溶液を88mL得た。
特性評価:共通操作Eと共通操作F(εD,280=5178、εD,370=20217を使用)を使用して、下記の特性値を得た。
抗体濃度:9.96mg/mL,抗体収量:876mg(88%),共通操作Eにて測定された抗体一分子あたりの薬物平均結合数(n):3.8;共通操作Fにて測定された抗体一分子あたりの薬物平均結合数(n):3.8。
21-a)ADCの抗腫瘍効果(1)
マウス:5-6週齢の雌BALB/c-nu/nuマウス(日本チャールス・リバー株式会社)を実験使用前にSPF条件下で4-7日間馴化した。マウスには滅菌した固形飼料(FR-2,Funabashi Farms Co.,Ltd)を給餌し、滅菌した水道水(5-15ppm次亜塩素酸ナトリウム溶液を添加して調製)を与えた。
測定、計算式:全ての研究において、腫瘍の長径及び短径を電子式デジタルキャリパー(CD-15C,Mitutoyo Corp.)で1週間に2回測定し、腫瘍体積(mm3)を計算した。計算式は以下に示す通り。
腫瘍体積(mm3)=1/2×長径(mm)×[短径(mm)]2
ATCCから購入したヒト膵臓腺癌細胞株Bx-PC3を雌BALB/c-nu/nuマウスに移植し、更に固形継代した腫瘍片を雌BALB/c-nu/nuマウス右側腹部に皮下移植して(Day0)、Day16に無作為に群分けを実施した。抗体-薬物コンジュゲート(1)、(6)、(12)をDay16、23、30に全て10mg/kgの用量で尾静脈内投与した。陰性対照として薬物を結合していないhTINA1-H1L1抗体及びhRS7抗体を25mg/kgの用量で同様に投与した。抗体-薬物コンジュゲート(1)、(6)の投与によって腫瘍体積が抗体-薬物コンジュゲート(12)の投与に比べて著しく減少し、いずれも腫瘍増殖抑制効果を発揮した(図14)。
ATCCから購入したヒト膵臓腺癌細胞株Capan-1を雌BALB/c-nu/nuマウスに移植し、更に固形継代した腫瘍片を雌BALB/c-nu/nuマウス右側腹部に皮下移植して(Day0)、Day18に無作為に群分けを実施した。抗体-薬物コンジュゲート(1)、(6)、(12)をDay18、25、32に全て10mg/kgの用量で尾静脈内投与した。陰性対照として薬物を結合していないhTINA1-H1L1抗体及びhRS7抗体を25mg/kgの用量で同様に投与した。抗体-薬物コンジュゲート(1)、(6)の投与によって腫瘍体積が抗体-薬物コンジュゲート(12)の投与に比べて著しく減少し、いずれも腫瘍増殖抑制効果を発揮した(図15)。
実施例21-a)と同様にCOLO205を雌BALB/c-nu/nuマウスに皮下移植し(Day0)、Day11に無作為に群分けを実施した。Day11、18、25に抗体-薬物コンジュゲート(2)、(5)を全て10mg/kg、抗体-薬物コンジュゲート(7)、(10)を全て3mg/kgの用量で尾静脈内投与した。抗体-薬物コンジュゲート(2)、(5)、(7)、(10)の投与によっていずれも腫瘍増殖抑制効果を発揮した(図16)。
実施例21-b)と同様にBx-PC3を雌BALB/c-nu/nuマウスに皮下移植し(Day0)、Day25に無作為に群分けを実施した。抗体-薬物コンジュゲート(2)、(5)、(7)、(10)をDay25、32に全て3mg/kgの用量で尾静脈内投与した。抗体-薬物コンジュゲート(2)、(5)、(7)、(10)の投与によっていずれも腫瘍増殖抑制効果を発揮した(図17)。
実施例21-a)と同様にCOLO205を雌BALB/c-nu/nuマウスに皮下移植し(Day0)、Day9に無作為に群分けを実施した。Day9、16に抗体-薬物コンジュゲート(3)、(4)、(8)、(9)を全て10mg/kgの用量で尾静脈内投与した。抗体-薬物コンジュゲート(3)、(4)、(8)、(9)の投与によっていずれも腫瘍増殖抑制効果を発揮した(図18)。
実施例21-b)と同様にBx-PC3を雌BALB/c-nu/nuマウスに皮下移植し(Day0)、Day21に無作為に群分けを実施した。Day21、28に抗体-薬物コンジュゲート(3)、(4)、(8)、(9)を全て3mg/kgの用量で尾静脈内投与した。抗体-薬物コンジュゲート(3)、(4)、(8)、(9)の投与によっていずれも腫瘍増殖抑制効果を発揮した(図19)。
ATCCから購入したヒト卵巣癌細胞株NIH:OVCAR-3 8×106cellsをMatrigel(Becton,Dickinson and Company)に懸濁し、雌BALB/c-nu/nuマウスに皮下移植し(Day0)、Day25に無作為に群分けを実施した。Day25に抗体-薬物コンジュゲート(3)、(4)、(8)、(9)を全て3mg/kgの用量で尾静脈内投与した。抗体-薬物コンジュゲート(3)、(4)、(8)、(9)の投与によっていずれも腫瘍増殖抑制効果を発揮した(図20)。
ATCCから購入したヒト胃癌細胞株NCI-N87 1×107cellsを生理食塩水に懸濁し、雌BALB/c-nu/nuマウスに皮下移植し(Day0)、Day6に無作為に群分けを実施した。Day6に抗体-薬物コンジュゲート(3)、(4)、(8)、(9)を全て3mg/kgの用量で尾静脈内投与した。抗体-薬物コンジュゲート(3)、(4)、(8)、(9)の投与によっていずれも腫瘍増殖抑制効果を発揮した(図21)。
ATCCから購入したヒト肺癌細胞株NCI-H292 5×106cellsを生理食塩水に懸濁し、雌BALB/c-nu/nuマウスに皮下移植し(Day0)、Day9に無作為に群分けを実施した。Day9に抗体-薬物コンジュゲート(3)、(4)、(8)、(9)を全て3mg/kgの用量で尾静脈内投与した。抗体-薬物コンジュゲート(3)、(4)、(8)、(9)の投与によっていずれも腫瘍増殖抑制効果を発揮した(図22)。
ATCCから購入したヒト咽頭癌細胞株FaDu 3×106cellsを生理食塩水に懸濁し、雌BALB/c-nu/nuマウスに皮下移植し(Day0)、Day11に無作為に群分けを実施した。Day11に抗体-薬物コンジュゲート(3)、(4)、(8)、(9)を全て3mg/kgの用量で尾静脈内投与した。抗体-薬物コンジュゲート(3)、(4)、(8)、(9)の投与によっていずれも腫瘍増殖抑制効果を発揮した(図23)。
ATCCから購入したヒト膵臓腺癌細胞株CFPAC-1 4×106cellsを生理食塩水に懸濁し、雌BALB/c-nu/nuマウスに皮下移植し(Day0)、Day14に無作為に群分けを実施した。Day14に抗体-薬物コンジュゲート(3)、(4)、(8)、(9)を全て3mg/kgの用量で尾静脈内投与した。抗体-薬物コンジュゲート(3)、(4)、(8)、(9)の投与によっていずれも腫瘍増殖抑制効果を発揮した(図24)。
実施例21-lと同様に、CFPAC-1を雌BALB/c-nu/nuマウスに皮下移植し(Day0)、Day14に無作為に群分けを実施した。Day14に抗体-薬物コンジュゲート(8)、(13)を全て1mg/kgの用量で尾静脈内投与した。抗体-薬物コンジュゲート(8)、(13)の投与によっていずれも腫瘍増殖抑制効果を発揮した(図25)。
ATCCから購入したヒト膵臓腺癌細胞株HPAC 3×106cellsを生理食塩水に懸濁して雌BALB/c-nu/nuマウスに皮下移植し(Day0)、Day12に無作為に群分けを実施した。Day12に抗体-薬物コンジュゲート(8)、(13)を全て3mg/kgの用量で尾静脈内投与した。抗体-薬物コンジュゲート(8)、(13)の投与によっていずれも腫瘍増殖抑制効果を発揮した(図26)。
公益財団法人ヒューマンサイエンス振興財団から入手したヒト食道癌組織をNOGマウス(公益財団法人 実験動物中央研究所)の皮下に移植し増殖させて得た腫瘍片を、さらに雌NOD-scidマウス(日本チャールス・リバー株式会社)に皮下移植し(Day0)、Day27に無作為に群分けを実施した。Day27に抗体-薬物コンジュゲート(8)、(13)を全て3mg/kgの用量で尾静脈内投与した。抗体-薬物コンジュゲート(8)、(13)の投与によっていずれも腫瘍増殖抑制効果を発揮した(図27)。
TROP2抗原陽性細胞株であるBxPC3、NCI-H292、NIH:OVCAR-3、CFPAC-1、FaDu、ヒト肺腺癌細胞株Calu-3(ATCC)、ヒト卵巣癌細胞株CaOV3(ATCC)、及びTROP2抗原陰性細胞株であるヒト肺癌細胞株Calu-6(ATCC)、ヒト皮膚黒色腫細胞株A375(ATCC)を各ADCの抗細胞効果の評価に使用した。BxPC3及びNCI-H292は10%ウシ胎児血清(Moregate)を含むRPMI1640 Medium(Gibco)で、NIH:OVCAR-3は20%ウシ胎児血清及び0.01mg/mL Insulin(Invitrogen)を含むRPMI1640 Mediumで、CFPAC-1は10%ウシ胎児血清を含むIscove‘s Modified Dulbecco’s Medium(Gibco)で、FaDu、Calu-3及びCalu-6は10%ウシ胎児血清を含むEagle’s Minimum Essential Medium(ATCC)で、CaOV3及びA375は10%ウシ胎児血清を含むDulbecco’s Modified Eagle Medium(Gibco)で、それぞれ2.2×106cells/mLに調製し、96穴細胞培養用マイクロプレートに90μLずつ播種した。更にRPMI1640 Mediumで100nM、20nM、4nM、0.8nM、0.16nM、0.032nM、0.0064nMに希釈した抗体-薬物コンジュゲート(4)、(8)もしくは比較のためRPMI1640 Mediumを10μLずつ添加して、37℃、5%CO2下で6日間培養した。培養後、マイクロプレートをインキュベーターから取り出し、室温で30分間静置した。培養液と等量のCellTiter-Glo Luminescent Cell Viability Assay(Promega)を添加し、プレートミキサーで10分間攪拌して細胞を溶解後にプレートリーダーで発光量を計測した。
6日間培養後の細胞増殖阻害率は次式で算出した。
細胞増殖阻害率(%)=a÷b×100
a:6日間培養後の検体添加ウェルの平均値-培養開始時の検体未添加ウェルの平均値
b:6日間培養後の培地添加ウェルの平均値-培養開始時の検体未添加ウェルの平均値
またGI50値は次式で算出した。
GI50(nM)=antilog((50-f)×(LOG10(d)-LOG10(c))÷(f-e)+LOG10(d))
c:検体濃度c
d:検体濃度d
e:検体濃度cにおける細胞増殖阻害率
f:検体濃度dにおける細胞増殖阻害率
c、dは細胞増殖阻害率50%を挟む2点でc>d。
配列番号2:TINA1抗体の重鎖の可変領域のアミノ酸配列
配列番号3:TINA1抗体の軽鎖の可変領域をコードするcDNAのヌクレオチド配列
配列番号4:TINA1抗体の軽鎖の可変領域のアミノ酸配列
配列番号5:ヒトκ鎖分泌シグナル及びヒトκ鎖定常領域をコードするヌクレオチド配列
配列番号6:ヒト重鎖分泌シグナル及びヒトIgG1定常領域をコードするヌクレオチド配列
配列番号7:cTINA1抗体重鎖のヌクレオチド配列
配列番号8:cTINA1抗体重鎖のアミノ酸配列
配列番号9:cTINA1抗体軽鎖のヌクレオチド配列
配列番号10:cTINA1抗体軽鎖のアミノ酸配列
配列番号11:hTINA1-H1のヌクレオチド配列
配列番号12:hTINA1-H1のアミノ酸配列
配列番号13:hTINA1-H2のヌクレオチド配列
配列番号14:hTINA1-H2のアミノ酸配列
配列番号15:hTINA1-H3のヌクレオチド配列
配列番号16:hTINA1-H3のアミノ酸配列
配列番号17:hTINA1-L1のヌクレオチド配列
配列番号18:hTINA1-L1のアミノ酸配列
配列番号19:hTINA1-L2のヌクレオチド配列
配列番号20:hTINA1-L2のアミノ酸配列
配列番号21:hTINA1-L3のヌクレオチド配列
配列番号22:hTINA1-L3のアミノ酸配列
配列番号23:TINA1抗体のCDRH1のアミノ酸配列
配列番号24:TINA1抗体のCDRH2のアミノ酸配列
配列番号25:TINA1抗体のCDRH3のアミノ酸配列
配列番号26:TINA1抗体のCDRL1のアミノ酸配列
配列番号27:TINA1抗体のCDRL2のアミノ酸配列
配列番号28:TINA1抗体のCDRL3のアミノ酸配列
配列番号29:hRS7抗体重鎖のヌクレオチド配列
配列番号30:hRS7抗体重鎖のアミノ酸配列
配列番号31:hRS7抗体軽鎖のヌクレオチド配列
配列番号32:hRS7抗体軽鎖のアミノ酸配列
配列番号33:プライマー mG2aVR2
配列番号34:プライマー mKVR2
配列番号35:プライマー 3.3-F1
配列番号36:プライマー 3.3-R1
配列番号37:プライマー TINA1H-F
配列番号38:プライマー TINA1H-R
配列番号39:プライマー TINA1L-F
配列番号40:プライマー TINA1L-R
配列番号41:プライマー EG-Inf-F
配列番号42:プライマー EG1-Inf-R
配列番号43:プライマー CM-LKF
配列番号44:プライマー KCL-Inf-R
Claims (27)
- 次式

-L1-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-
で示される構造のリンカーを介して、抗TROP2抗体のヒンジ部に存在するジスルフィド結合部分において形成させたチオエーテル結合によって結合させたことを特徴とする抗体-薬物コンジュゲート。
(ここで、抗TROP2抗体は、L1の末端において結合する。抗腫瘍性化合物は、1位のアミノ基の窒素原子を結合部位として、-(CH2)n2-C(=O)-部分のカルボニル基に結合する。
式中、n1は、0から6の整数を示し、
n2は、0から5の整数を示し、
L1は、-(Succinimid-3-yl-N)-(CH2)n3-C(=O)-を示し、
ここで、n3は、2から8の整数を示し、
L2は、-NH-(CH2CH2-O)n4-CH2CH2-C(=O)-又は単結合を示し、
ここで、n4は、1から6の整数を示し
LPは、2から7個のアミノ酸で構成されるペプチド残基を示し、
Laは、-O-又は単結合を示し、
-(Succinimid-3-yl-N)-は次式:

- LPのペプチド残基が、フェニルアラニン、グリシン、バリン、リシン、シトルリン、セリン、グルタミン酸、アスパラギン酸から選ばれるアミノ酸からなるペプチド残基である請求項1記載の抗体-薬物コンジュゲート。
- LPが、以下の群から選ばれるペプチド残基である請求項1又は2に記載の抗体-薬物コンジュゲート。
-GGF-、
-DGGF-、
-(D-)D-GGF-、
-EGGF-、
-GGFG-、
-SGGF-、
-KGGF-、
-DGGFG-、
-GGFGG-、
-DDGGFG-、
-KDGGFG-、
-GGFGGGF-;
(ここで『(D-)D』はD-アスパラギン酸を示す。) - LPが、4個のアミノ酸で構成されるペプチド残基である請求項1又は2に記載の抗体-薬物コンジュゲート。
- LPが、テトラペプチド残基の-GGFG-である請求項1~4のいずれか一項に記載の抗体-薬物コンジュゲート。
- n3が2から5の整数であって、L2が単結合である請求項1~5のいずれか一項に記載の抗体-薬物コンジュゲート。
- n3が2から5の整数であって、L2が-NH-(CH2CH2-O)n4-CH2CH2-C(=O)-であり、n4が2又は4である請求項1~5のいずれか一項に記載の抗体-薬物コンジュゲート。
- -NH-(CH2)n1-La-(CH2)n2-C(=O)-が、4から7原子の鎖長を有する部分構造である請求項1~7のいずれか一項に記載の抗体-薬物コンジュゲート。
- -NH-(CH2)n1-La-(CH2)n2-C(=O)-が、5又は6原子の鎖長を有する部分構造である請求項1~7のいずれか一項に記載の抗体-薬物コンジュゲート。
- -NH-(CH2)n1-La-(CH2)n2-C(=O)-が、以下のいずれかである請求項1~9のいずれか一項に記載の抗体-薬物コンジュゲート。
-NH-CH2CH2-C(=O)-、
-NH-CH2CH2CH2-C(=O)-、
-NH-CH2CH2CH2CH2-C(=O)-、
-NH-CH2CH2CH2CH2CH2-C(=O)-、
-NH-CH2-O-CH2-C(=O)-、
-NH-CH2CH2-O-CH2-C(=O)-。 - -NH-(CH2)n1-La-(CH2)n2-C(=O)-が、以下のいずれかである請求項10に記載の抗体-薬物コンジュゲート。
-NH-CH2CH2CH2-C(=O)-、
-NH-CH2-O-CH2-C(=O)-、
-NH-CH2CH2-O-CH2-C(=O)-。 - -L1-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-に薬物を結合させた薬物-リンカー構造部分が、次の群から選ばれる1種の薬物-リンカー構造である請求項1~9のいずれか一項に記載の抗体-薬物コンジュゲート。
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX);
(ここで、-(Succinimid-3-yl-N)-は次式:

-(NH-DX)は次式:

-GGFG-は、-Gly-Gly-Phe-Gly-のテトラペプチド残基を示す。) - 前記薬物-リンカー構造部分が、次の群から選ばれる1種の薬物-リンカー構造である請求項12に記載の抗体-薬物コンジュゲート。
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)、
-(Succinimid-3-yl-N)-CH2CH2-C(=O)-NH-CH2CH2-O-CH2CH2-O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)。 - 次式

-L1-L2-LP-NH-(CH2)n1-La-(CH2)n2-C(=O)-
で示される構造のリンカーを介して、抗TROP2抗体のヒンジ部に存在するジスルフィド結合部分において形成させたチオエーテル結合を介して結合させたことを特徴とする抗体-薬物コンジュゲート。
(ここで、抗TROP2抗体はL1の末端において結合する。抗腫瘍性化合物は-(CH2)n2-C(=O)-部分のカルボニル基に結合する。
式中、n1は、0から6の整数を示し、
n2は、0から5の整数を示し、
L1は、-(Succinimid-3-yl-N)-(CH2)n3-C(=O)-を示し、
ここで、n3は、2から8の整数を示し、
L2は、-NH-(CH2CH2-O)n4-CH2CH2-C(=O)-又は単結合を示し、
ここで、n4は、1から6の整数を示し、
LPは、-GGFG-のテトラペプチド残基を示し、
Laは、-O-又は単結合を示し、
-(Succinimid-3-yl-N)-は次式:

- n1が3であり、n2が0であり、n3が2であり、L2が-NH-(CH2CH2-O)n4-CH2CH2-C(=O)-であって、n4が2であり、Laが単結合であるか、
n1が1であり、n2が1であり、n3が5であり、L2が単結合であり、Laが-O-であるか、又は
n1が2であり、n2が1であり、n3が5であり、L2が単結合であり、Laが-O-である、請求項14に記載の抗体-薬物コンジュゲート。 - n3が2又は5であって、L2が単結合である請求項14又は15に記載の抗体-薬物コンジュゲート。
- n3が2又は5であって、L2が-NH-(CH2CH2-O)n4-CH2CH2-C(=O)-であり、n4が2又は4である請求項14又は15に記載の抗体-薬物コンジュゲート。
- -NH-(CH2)n1-La-(CH2)n2-C(=O)-が、以下のいずれかである請求項14~17のいずれか一項に記載の抗体-薬物コンジュゲート。
-NH-CH2CH2CH2-C(=O)-、
-NH-CH2-O-CH2-C(=O)-、
-NH-CH2CH2-O-CH2-C(=O)-。 - 選択された1種の薬物-リンカー構造の1抗体あたりの平均結合数が1から10個の範囲である請求項1~18のいずれか一項に記載の抗体-薬物コンジュゲート。
- 選択された1種の薬物-リンカー構造の1抗体あたりの平均結合数が2から8個の範囲である請求項1~18のいずれか一項に記載の抗体-薬物コンジュゲート。
- 選択された1種の薬物-リンカー構造の1抗体あたりの平均結合数が3から8個の範囲である請求項1~18のいずれか一項に記載の抗体-薬物コンジュゲート。
- 請求項1~21のいずれか一項に記載の抗体-薬物コンジュゲート、その塩、又はそれらの水和物を含有する医薬。
- 請求項1~21のいずれか一項に記載の抗体-薬物コンジュゲート、その塩、又はそれらの水和物を含有する抗腫瘍薬及び/又は抗癌薬。
- 肺癌、腎癌、尿路上皮癌、大腸癌、前立腺癌、多形神経膠芽腫、卵巣癌、膵癌、乳癌、メラノーマ、肝癌、膀胱癌、胃癌、子宮頸癌、頭頸部癌、又は食道癌に適用するための請求項23記載の抗腫瘍薬及び/又は抗癌薬。
- 請求項1~21のいずれか一項に記載の抗体-薬物コンジュゲート、その塩、又はそれらの水和物を活性成分とし、薬学的に許容される製剤成分とを含有する医薬組成物。
- 肺癌、腎癌、尿路上皮癌、大腸癌、前立腺癌、多形神経膠芽腫、卵巣癌、膵癌、乳癌、メラノーマ、肝癌、膀胱癌、胃癌、子宮頸癌、頭頸部癌、又は食道癌に適用するための請求項25記載の医薬組成物。
- 請求項1~21のいずれか一項に記載の抗体-薬物コンジュゲート、その塩、又はそれらの水和物を投与することを特徴とする腫瘍及び/又は癌の治療方法。
Priority Applications (42)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020227031871A KR102491710B1 (ko) | 2013-12-25 | 2014-12-24 | 항 trop2 항체-약물 컨쥬게이트 |
| KR1020167015563A KR101941758B1 (ko) | 2013-12-25 | 2014-12-24 | 항 trop2 항체-약물 컨쥬게이트 |
| KR1020237002152A KR102535900B1 (ko) | 2013-12-25 | 2014-12-24 | 항 trop2 항체-약물 컨쥬게이트 |
| ES14874745T ES2703903T3 (es) | 2013-12-25 | 2014-12-24 | Conjugado de fármaco-anticuerpo anti-trop2 |
| RS20181597A RS58173B1 (sr) | 2013-12-25 | 2014-12-24 | Konjugat anti-trop2 antitelo-lek |
| SG11201605215YA SG11201605215YA (en) | 2013-12-25 | 2014-12-24 | Anti-trop2 antibody-drug conjugate |
| KR1020207006601A KR102237803B1 (ko) | 2013-12-25 | 2014-12-24 | 항 trop2 항체-약물 컨쥬게이트 |
| DK14874745.4T DK3088419T3 (en) | 2013-12-25 | 2014-12-24 | ANTI-TROP2 ANTIBODY-MEDICINE CONJUGATE |
| MYPI2016001186A MY195162A (en) | 2013-12-25 | 2014-12-24 | Anti-Trop2 Antibody-Drug Conjugate |
| KR1020197000700A KR102088169B1 (ko) | 2013-12-25 | 2014-12-24 | 항 trop2 항체-약물 컨쥬게이트 |
| JP2015554565A JP6130517B2 (ja) | 2013-12-25 | 2014-12-24 | 抗trop2抗体−薬物コンジュゲート |
| KR1020227015978A KR102444529B1 (ko) | 2013-12-25 | 2014-12-24 | 항 trop2 항체-약물 컨쥬게이트 |
| NZ721213A NZ721213A (en) | 2013-12-25 | 2014-12-24 | Anti-trop2 antibody-drug conjugate |
| BR112016013704-3A BR112016013704B1 (pt) | 2013-12-25 | 2014-12-24 | Conjugado de anticorpo-fármaco anti-trop2,fármaco antitumor e/ou anticâncer,composição farmacêutica e uso dos mesmos. |
| RU2016130095A RU2705367C2 (ru) | 2013-12-25 | 2014-12-24 | Конъюгат анти-trop2 антитело-лекарственное средство |
| EP18187671.5A EP3424955A1 (en) | 2013-12-25 | 2014-12-24 | Anti-trop2 antibody-drug conjugate |
| KR1020217009822A KR102288093B1 (ko) | 2013-12-25 | 2014-12-24 | 항 trop2 항체-약물 컨쥬게이트 |
| KR1020227000880A KR102399141B1 (ko) | 2013-12-25 | 2014-12-24 | 항 trop2 항체-약물 컨쥬게이트 |
| AU2014371934A AU2014371934B2 (en) | 2013-12-25 | 2014-12-24 | Anti-TROP2 antibody-drug conjugate |
| PL14874745T PL3088419T3 (pl) | 2013-12-25 | 2014-12-24 | Koniugat lek-przeciwciało anty-trop2 |
| LTEP14874745.4T LT3088419T (lt) | 2013-12-25 | 2014-12-24 | Anti-trop2 antikūno-vaisto konjugatas |
| MX2016008192A MX2016008192A (es) | 2013-12-25 | 2014-12-24 | Conjugado anticuerpo anti-trop2-farmaco. |
| CN202310800520.XA CN116987193A (zh) | 2013-12-25 | 2014-12-24 | 抗trop2抗体-药物偶联物 |
| EP14874745.4A EP3088419B1 (en) | 2013-12-25 | 2014-12-24 | Anti-trop2 antibody-drug conjugate |
| KR1020237016957A KR20230078820A (ko) | 2013-12-25 | 2014-12-24 | 항 trop2 항체-약물 컨쥬게이트 |
| CN201480071134.0A CN105849126B (zh) | 2013-12-25 | 2014-12-24 | 抗trop2抗体-药物偶联物 |
| SI201430973T SI3088419T1 (sl) | 2013-12-25 | 2014-12-24 | Zdravilni konjugat anti trop2 protitelesa |
| CA2933666A CA2933666C (en) | 2013-12-25 | 2014-12-24 | Anti-trop2 antibody-drug conjugate |
| KR1020217024615A KR102351164B1 (ko) | 2013-12-25 | 2014-12-24 | 항 trop2 항체-약물 컨쥬게이트 |
| US15/187,179 US9850312B2 (en) | 2013-12-25 | 2016-06-20 | Anti-TROP2 antibody-drug conjugate |
| IL246361A IL246361B (en) | 2013-12-25 | 2016-06-20 | Antibody conjugates - antidote - trop 2, preparations containing them and their uses |
| PH12016501233A PH12016501233A1 (en) | 2013-12-25 | 2016-06-22 | Anti-trop2 antibody-drug conjugate |
| US15/821,662 US10227417B2 (en) | 2013-12-25 | 2017-11-22 | Anti-TROP2 antibody-drug conjugate |
| CY20191100002T CY1121246T1 (el) | 2013-12-25 | 2019-01-02 | Συζευγμα αντισωματος αντι-trop2-φαρμακου |
| HRP20190056TT HRP20190056T1 (hr) | 2013-12-25 | 2019-01-09 | Konjugat anti-trop2 antitijelo-lijek |
| US16/256,715 US11008398B2 (en) | 2013-12-25 | 2019-01-24 | Anti-TROP2 antibody-drug conjugate |
| IL267618A IL267618B (en) | 2013-12-25 | 2019-06-24 | Methods of producing antibody-drug conjugates |
| AU2020202305A AU2020202305B2 (en) | 2013-12-25 | 2020-03-31 | Anti-trop2 antibody-drug conjugate |
| PH12020551943A PH12020551943A1 (en) | 2013-12-25 | 2020-11-13 | Anti-trop2 antibody-drug conjugate |
| US17/230,543 US20210238303A1 (en) | 2013-12-25 | 2021-04-14 | Anti-trop2 antibody-drug conjugate |
| IL291446A IL291446A (en) | 2013-12-25 | 2022-03-16 | Anti-trop2 antibodies and methods for their production |
| AU2023200883A AU2023200883A1 (en) | 2013-12-25 | 2023-02-16 | Anti-trop2 antibody-drug conjugate |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013-267548 | 2013-12-25 | ||
| JP2013267548 | 2013-12-25 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/187,179 Continuation US9850312B2 (en) | 2013-12-25 | 2016-06-20 | Anti-TROP2 antibody-drug conjugate |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015098099A1 true WO2015098099A1 (ja) | 2015-07-02 |
Family
ID=53477995
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/006421 WO2015098099A1 (ja) | 2013-12-25 | 2014-12-24 | 抗trop2抗体-薬物コンジュゲート |
Country Status (27)
| Country | Link |
|---|---|
| US (4) | US9850312B2 (ja) |
| EP (2) | EP3424955A1 (ja) |
| JP (6) | JP6130517B2 (ja) |
| KR (10) | KR102491710B1 (ja) |
| CN (3) | CN105849126B (ja) |
| AU (3) | AU2014371934B2 (ja) |
| CA (1) | CA2933666C (ja) |
| CY (1) | CY1121246T1 (ja) |
| DK (1) | DK3088419T3 (ja) |
| ES (1) | ES2703903T3 (ja) |
| HR (1) | HRP20190056T1 (ja) |
| HU (1) | HUE042512T2 (ja) |
| IL (3) | IL246361B (ja) |
| LT (1) | LT3088419T (ja) |
| MX (3) | MX2016008192A (ja) |
| MY (1) | MY195162A (ja) |
| NZ (1) | NZ721213A (ja) |
| PH (2) | PH12016501233A1 (ja) |
| PL (1) | PL3088419T3 (ja) |
| PT (1) | PT3088419T (ja) |
| RS (1) | RS58173B1 (ja) |
| RU (2) | RU2743077C2 (ja) |
| SG (2) | SG11201605215YA (ja) |
| SI (1) | SI3088419T1 (ja) |
| TR (1) | TR201900176T4 (ja) |
| TW (5) | TWI812438B (ja) |
| WO (1) | WO2015098099A1 (ja) |
Cited By (61)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2015155976A1 (ja) * | 2014-04-10 | 2017-04-13 | 第一三共株式会社 | 抗her2抗体−薬物コンジュゲート |
| WO2018110515A1 (ja) | 2016-12-12 | 2018-06-21 | 第一三共株式会社 | 抗体-薬物コンジュゲートと免疫チェックポイント阻害剤の組み合わせ |
| WO2019039483A1 (ja) | 2017-08-23 | 2019-02-28 | 第一三共株式会社 | 抗体-薬物コンジュゲートの製剤及びその凍結乾燥方法 |
| WO2019044946A1 (ja) | 2017-08-31 | 2019-03-07 | 第一三共株式会社 | 抗体-薬物コンジュゲートの新規製造方法 |
| WO2019044947A1 (ja) | 2017-08-31 | 2019-03-07 | 第一三共株式会社 | 抗体-薬物コンジュゲートの改良製造方法 |
| WO2019065964A1 (ja) | 2017-09-29 | 2019-04-04 | 第一三共株式会社 | 抗体-ピロロベンゾジアゼピン誘導体コンジュゲート |
| JP2020503243A (ja) * | 2017-08-11 | 2020-01-30 | バイオ − テラ ソリューションズ、リミテッド | Trop2陽性疾患の治療のための化合物及び方法 |
| WO2020022363A1 (ja) | 2018-07-25 | 2020-01-30 | 第一三共株式会社 | 抗体-薬物コンジュゲートの効果的な製造方法 |
| WO2020022475A1 (ja) | 2018-07-27 | 2020-01-30 | 第一三共株式会社 | 抗体-薬物コンジュゲートの薬物部位を認識する蛋白質 |
| WO2020027100A1 (ja) | 2018-07-31 | 2020-02-06 | 第一三共株式会社 | 抗体-薬物コンジュゲート投与による転移性脳腫瘍の治療 |
| WO2020031936A1 (ja) | 2018-08-06 | 2020-02-13 | 第一三共株式会社 | 抗体-薬物コンジュゲートとチューブリン阻害剤の組み合わせ |
| WO2020040245A1 (ja) | 2018-08-23 | 2020-02-27 | 第一三共株式会社 | 抗体薬物複合体の感受性マーカー |
| WO2020094670A1 (en) | 2018-11-05 | 2020-05-14 | Synaffix B.V. | Antibody-conjugates for targeting of tumours expressing trop-2 |
| WO2020122034A1 (ja) | 2018-12-11 | 2020-06-18 | 第一三共株式会社 | 抗体-薬物コンジュゲートとparp阻害剤の組み合わせ |
| WO2020130125A1 (ja) | 2018-12-21 | 2020-06-25 | 第一三共株式会社 | 抗体-薬物コンジュゲートとキナーゼ阻害剤の組み合わせ |
| WO2020196474A1 (ja) | 2019-03-25 | 2020-10-01 | 第一三共株式会社 | 抗体-ピロロベンゾジアゼピン誘導体コンジュゲート |
| WO2020196712A1 (ja) | 2019-03-27 | 2020-10-01 | 第一三共株式会社 | 抗体-ピロロベンゾジアゼピン誘導体コンジュゲートとparp阻害剤の組み合わせ |
| WO2020240467A1 (en) | 2019-05-29 | 2020-12-03 | Daiichi Sankyo Company, Limited | Dosage of an antibody-drug conjugate |
| WO2020249063A1 (en) * | 2019-06-13 | 2020-12-17 | Bio-Thera Solutions, Ltd. | Methods for the treatment of trop2 positive diseases |
| JP2021059599A (ja) * | 2017-05-15 | 2021-04-15 | 第一三共株式会社 | 抗cdh6抗体及び抗cdh6抗体−薬物コンジュゲート |
| WO2021147993A1 (zh) | 2020-01-22 | 2021-07-29 | 江苏恒瑞医药股份有限公司 | 抗trop-2抗体-依喜替康类似物偶联物及其医药用途 |
| WO2021177438A1 (ja) | 2020-03-06 | 2021-09-10 | 第一三共株式会社 | 新規環状ジヌクレオチド誘導体を含む抗体薬物コンジュゲート |
| WO2021226440A1 (en) | 2020-05-08 | 2021-11-11 | Bolt Biotherapeutics, Inc. | Elastase-substrate, peptide linker immunoconjugates, and uses thereof |
| US11173213B2 (en) | 2015-06-29 | 2021-11-16 | Daiichi Sankyo Company, Limited | Method for selectively manufacturing antibody-drug conjugate |
| WO2021260579A1 (en) | 2020-06-24 | 2021-12-30 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and atr inhibitor |
| WO2021260578A1 (en) | 2020-06-24 | 2021-12-30 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and cdk9 inhibitor |
| WO2021260582A1 (en) | 2020-06-24 | 2021-12-30 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and aurora b inhibitor |
| WO2021260580A1 (en) | 2020-06-24 | 2021-12-30 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and atm inhibitor |
| WO2021260583A1 (en) | 2020-06-24 | 2021-12-30 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and dna-pk inhibitor |
| WO2022014698A1 (ja) | 2020-07-17 | 2022-01-20 | 第一三共株式会社 | 抗体-薬物コンジュゲートの製造方法 |
| WO2022036101A1 (en) | 2020-08-13 | 2022-02-17 | Bolt Biotherapeutics, Inc. | Pyrazoloazepine immunoconjugates, and uses thereof |
| WO2022074617A1 (en) | 2020-10-09 | 2022-04-14 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and parp1 selective inhibitor |
| WO2022102695A1 (ja) | 2020-11-12 | 2022-05-19 | 第一三共株式会社 | 抗b7-h3抗体-薬物コンジュゲート投与による中皮腫の治療 |
| WO2022102634A1 (ja) | 2020-11-11 | 2022-05-19 | 第一三共株式会社 | 抗体-薬物コンジュゲートと抗SIRPα抗体の組み合わせ |
| WO2022163846A1 (ja) | 2021-02-01 | 2022-08-04 | 第一三共株式会社 | 抗体-免疫賦活化剤コンジュゲートの新規製造方法 |
| WO2022191313A1 (ja) | 2021-03-12 | 2022-09-15 | 第一三共株式会社 | 糖鎖及び糖鎖を含む医薬品の製造方法 |
| WO2022204528A1 (en) | 2021-03-26 | 2022-09-29 | Bolt Biotherapeutics, Inc. | 2-amino-4-carboxamide-benzazepine immunoconjugates, and uses thereof |
| WO2022204536A1 (en) | 2021-03-26 | 2022-09-29 | Bolt Biotherapeutics, Inc. | 2-amino-4-carboxamide-benzazepine immunoconjugates, and uses thereof |
| WO2022261301A1 (en) | 2021-06-11 | 2022-12-15 | Gilead Sciences, Inc. | Combination mcl-1 inhibitors with anti-cancer agents |
| WO2022261310A1 (en) | 2021-06-11 | 2022-12-15 | Gilead Sciences, Inc. | Combination mcl-1 inhibitors with anti-body drug conjugates |
| WO2023274365A1 (zh) * | 2021-07-02 | 2023-01-05 | 和迈生物科技有限公司 | 抗trop2单域抗体及其应用 |
| WO2023001248A1 (zh) | 2021-07-21 | 2023-01-26 | 江苏恒瑞医药股份有限公司 | 一种含抗trop2抗体药物偶联物的药物组合物及其用途 |
| WO2023077030A1 (en) | 2021-10-29 | 2023-05-04 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2023076599A1 (en) | 2021-10-29 | 2023-05-04 | Bolt Biotherapeutics, Inc. | Tlr agonist immunoconjugates with cysteine-mutant antibodies, and uses thereof |
| WO2023076983A1 (en) | 2021-10-28 | 2023-05-04 | Gilead Sciences, Inc. | Pyridizin-3(2h)-one derivatives |
| WO2023089527A1 (en) | 2021-11-18 | 2023-05-25 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and parp1 selective inhibitor |
| WO2023122581A2 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023122615A1 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023126823A1 (en) | 2021-12-28 | 2023-07-06 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and atr inhibitor |
| WO2023126822A1 (en) | 2021-12-28 | 2023-07-06 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and rasg12c inhibitor |
| WO2023147418A1 (en) | 2022-01-28 | 2023-08-03 | Gilead Sciences, Inc. | Parp7 inhibitors |
| EP4245756A1 (en) | 2022-03-17 | 2023-09-20 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023175483A1 (en) | 2022-03-16 | 2023-09-21 | Astrazeneca Uk Limited | A scoring method for an anti-trop2 antibody‑drug conjugate therapy |
| WO2023180485A1 (en) | 2022-03-23 | 2023-09-28 | Synaffix B.V. | Antibody-conjugates for targeting of tumours expressing trop-2 |
| WO2023201268A1 (en) | 2022-04-13 | 2023-10-19 | Gilead Sciences, Inc. | Combination therapy for treating tumor antigen expressing cancers |
| WO2023201267A1 (en) | 2022-04-13 | 2023-10-19 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
| WO2023205719A1 (en) | 2022-04-21 | 2023-10-26 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| WO2023209591A1 (en) | 2022-04-27 | 2023-11-02 | Daiichi Sankyo Company, Limited | Combination of antibody-drug conjugate with ezh1 and/or ezh2 inhibitor |
| WO2023218378A1 (en) | 2022-05-11 | 2023-11-16 | Daiichi Sankyo Company, Limited | Combination of an antibody specific for a tumor antigen and a cd47 inhibitor |
| WO2024006929A1 (en) | 2022-07-01 | 2024-01-04 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2024023750A1 (en) | 2022-07-28 | 2024-02-01 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and bispecific checkpoint inhibitor |
Families Citing this family (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SI3088419T1 (sl) | 2013-12-25 | 2019-01-31 | Daiichi Sankyo Company | Zdravilni konjugat anti trop2 protitelesa |
| KR20230162159A (ko) | 2014-01-31 | 2023-11-28 | 다이이찌 산쿄 가부시키가이샤 | 항-her2 항체-약물 접합체 |
| US20170151281A1 (en) | 2015-02-19 | 2017-06-01 | Batu Biologics, Inc. | Chimeric antigen receptor dendritic cell (car-dc) for treatment of cancer |
| AU2017292934B2 (en) | 2016-07-07 | 2024-04-04 | Bolt Biotherapeutics, Inc. | Antibody adjuvant conjugates |
| CN108066772B (zh) * | 2016-11-14 | 2021-07-13 | 中国科学院上海药物研究所 | 靶向tacstd2的抗体与药物偶联体(adc)分子 |
| BR112019012847A2 (pt) | 2017-01-17 | 2019-12-10 | Daiichi Sankyo Co Ltd | anticorpo ou fragmento funcional do anticorpo, polinucleotídeo, vetor de expressão, células hospedeiras, método para produção de um anticorpo de interesse ou de um fragmento funcional do anticorpo e para produção de um conjugado de anticorpo-fármaco, conjugado de anticorpo-fármaco, composição farmacêutica, fármaco antitumoral, e, método de tratamento de um tumor. |
| EP3420906A1 (en) | 2017-06-29 | 2019-01-02 | Koninklijke Philips N.V. | Image contrast enhancement of an x-ray image |
| KR102385495B1 (ko) * | 2017-10-14 | 2022-04-15 | 애브비 인코포레이티드 | 항-cd71 활성화 가능한 항체 약물 접합체 및 이의 사용 방법 |
| BR112020023373A2 (pt) | 2018-05-18 | 2021-02-09 | Daiichi Sankyo Company, Limited | conjugado, composição, e, uso de um conjugado ou de uma composição |
| CN114573699A (zh) * | 2018-07-09 | 2022-06-03 | 启德医药科技(苏州)有限公司 | 滋养层细胞表面抗原2(trop2)特异性抗体 |
| TW202031298A (zh) | 2018-11-14 | 2020-09-01 | 日商第一三共股份有限公司 | 抗cdh6抗體-吡咯并苯二氮呯衍生物結合物 |
| JP7232925B2 (ja) * | 2019-02-15 | 2023-03-03 | ウーシー バイオロジクス アイルランド リミテッド | 改善された均一性を有する抗体薬物コンジュゲートの調製するプロセス |
| WO2020190725A1 (en) | 2019-03-15 | 2020-09-24 | Bolt Biotherapeutics, Inc. | Immunoconjugates targeting her2 |
| WO2020191092A1 (en) * | 2019-03-19 | 2020-09-24 | Cspc Dophen Corporation | Anti-trophoblast cell surface antigen 2 (trop2) antibodies and antibody drug conjugates comprising same |
| WO2020200880A1 (en) | 2019-03-29 | 2020-10-08 | Medimmune Limited | Compounds and conjugates thereof |
| DK3958977T3 (en) * | 2019-04-26 | 2023-12-11 | Immunogen Inc | Camptothecinderivater |
| CN112390885B (zh) * | 2019-08-12 | 2024-01-19 | 凯惠科技发展(上海)有限公司 | 一种trop2抗体及其制备方法、其偶联物和应用 |
| CN112646038A (zh) * | 2019-10-11 | 2021-04-13 | 迈威(上海)生物科技股份有限公司 | 抗人Trop-2抗体及其应用 |
| US20230101266A1 (en) * | 2019-12-31 | 2023-03-30 | Genequantum Healthcare (Suzhou) Co., Ltd. | Anti-trop2 antibodies, antibody-drug conjugates, and application of the same |
| US20210316003A1 (en) | 2020-03-20 | 2021-10-14 | Immunomedics, Inc. | Biomarkers for sacituzumab govitecan therapy |
| WO2021190480A1 (zh) * | 2020-03-24 | 2021-09-30 | 上海翰森生物医药科技有限公司 | 抗体-药物偶联物及其医药用途 |
| CN113527497B (zh) * | 2020-04-16 | 2022-06-10 | 上海洛启生物医药技术有限公司 | 抗Trop2纳米抗体及其应用 |
| WO2022001864A1 (zh) * | 2020-06-28 | 2022-01-06 | 昆山新蕴达生物科技有限公司 | 一种抗体-药物偶联物,其制备方法及应用 |
| CN112321715B (zh) * | 2020-11-03 | 2022-05-10 | 博奥信生物技术(南京)有限公司 | 抗trop2纳米抗体及其制备方法和应用 |
| AU2021376354A1 (en) * | 2020-11-04 | 2023-06-22 | Myeloid Therapeutics, Inc. | Engineered chimeric fusion protein compositions and methods of use thereof |
| AU2020482223A1 (en) * | 2020-12-18 | 2023-07-27 | Shanghai Fudan-Zhangjiang Bio-Pharmaceutical Co., Ltd. | Trop2 targeting antibody-drug conjugate, and preparation method and use therefor |
| JP2024503658A (ja) | 2021-01-13 | 2024-01-26 | メモリアル スローン-ケタリング キャンサー センター | 抗dll3抗体-薬物コンジュゲート |
| CN116761824A (zh) * | 2021-01-18 | 2023-09-15 | 上海药明合联生物技术有限公司 | 工程化抗-trop2抗体及其抗体-药物偶联物 |
| AU2021423664A1 (en) | 2021-01-28 | 2023-09-07 | Genequantum Healthcare (Suzhou) Co., Ltd. | Ligase fusion proteins and applications thereof |
| CN117295524A (zh) | 2021-06-02 | 2023-12-26 | 百奥泰生物制药股份有限公司 | 药物偶联物及其用途 |
| TW202320861A (zh) | 2021-09-15 | 2023-06-01 | 日商第一三共股份有限公司 | 以抗體-藥物結合物治療耐化療的癌症之方法 |
| KR20230143869A (ko) * | 2022-04-06 | 2023-10-13 | 주식회사 레고켐 바이오사이언스 | 인간 trop2에 대한 항체를 포함하는 항체-약물 접합체 및 이의 용도 |
| WO2023228095A1 (en) | 2022-05-24 | 2023-11-30 | Daiichi Sankyo Company, Limited | Dosage regimen of an anti-cdh6 antibody-drug conjugate |
| WO2023240135A2 (en) | 2022-06-07 | 2023-12-14 | Actinium Pharmaceuticals, Inc. | Bifunctional chelators and conjugates |
| WO2024012566A2 (en) | 2022-07-15 | 2024-01-18 | Genequantum Healthcare (Suzhou) Co., Ltd. | Antibody, linkers, payload, conjugates and applications thereof |
| KR20240032207A (ko) | 2022-08-31 | 2024-03-12 | 경북대학교 산학협력단 | Trop2에 결합하는 펩타이드 및 이의 용도 |
Citations (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1990007861A1 (en) | 1988-12-28 | 1990-07-26 | Protein Design Labs, Inc. | CHIMERIC IMMUNOGLOBULINS SPECIFIC FOR p55 TAC PROTEIN OF THE IL-2 RECEPTOR |
| WO1992001047A1 (en) | 1990-07-10 | 1992-01-23 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1992020791A1 (en) | 1990-07-10 | 1992-11-26 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| JPH0559061A (ja) | 1991-01-16 | 1993-03-09 | Dai Ichi Seiyaku Co Ltd | 六環性化合物 |
| WO1993006213A1 (en) | 1991-09-23 | 1993-04-01 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| WO1993011236A1 (en) | 1991-12-02 | 1993-06-10 | Medical Research Council | Production of anti-self antibodies from antibody segment repertoires and displayed on phage |
| WO1993019172A1 (en) | 1992-03-24 | 1993-09-30 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| JPH0687746A (ja) | 1992-07-16 | 1994-03-29 | Dai Ichi Seiyaku Co Ltd | 抗腫瘍剤 |
| WO1995001438A1 (en) | 1993-06-30 | 1995-01-12 | Medical Research Council | Sbp members with a chemical moiety covalently bound within the binding site; production and selection thereof |
| WO1995015388A1 (en) | 1993-12-03 | 1995-06-08 | Medical Research Council | Recombinant binding proteins and peptides |
| JPH08337584A (ja) | 1995-04-10 | 1996-12-24 | Dai Ichi Seiyaku Co Ltd | 縮合六環式アミノ化合物、これを含有する医薬及びその製法 |
| WO1997046260A1 (en) | 1996-06-06 | 1997-12-11 | Daiichi Pharmaceutical Co., Ltd. | Drug complexes |
| WO1999054342A1 (en) | 1998-04-20 | 1999-10-28 | Pablo Umana | Glycosylation engineering of antibodies for improving antibody-dependent cellular cytotoxicity |
| WO2000025825A1 (fr) | 1998-10-30 | 2000-05-11 | Daiichi Pharmaceutical Co., Ltd. | Composes dds et procede de dosage de ces composes |
| WO2000061739A1 (en) | 1999-04-09 | 2000-10-19 | Kyowa Hakko Kogyo Co., Ltd. | Method for controlling the activity of immunologically functional molecule |
| JP2002060351A (ja) | 2000-03-22 | 2002-02-26 | Dai Ichi Seiyaku Co Ltd | 水酸基を有する薬物を含むdds化合物 |
| WO2002031140A1 (fr) | 2000-10-06 | 2002-04-18 | Kyowa Hakko Kogyo Co., Ltd. | Cellules produisant des compositions d'anticorps |
| WO2003038043A2 (en) | 2001-11-01 | 2003-05-08 | Uab Research Foundation | Combinations of dr5 antibody and other therapeutic agents |
| WO2003074566A2 (en) | 2002-03-01 | 2003-09-12 | Immunomedics, Inc. | Rs7 antibodies |
| JP2005511627A (ja) * | 2001-11-20 | 2005-04-28 | シアトル ジェネティクス,インコーポレーテッド | 抗cd30抗体を使用する免疫学的疾患の治療 |
| JP2008521828A (ja) * | 2004-11-29 | 2008-06-26 | シアトル ジェネティックス, インコーポレイテッド | 操作された抗体およびイムノコンジュゲート |
| WO2008144891A1 (en) | 2007-05-30 | 2008-12-04 | F. Hoffmann-La Roche Ag | Humanized and chimeric anti-trop-2 antibodies that mediate cancer cell cytotoxicity |
| JP2009538629A (ja) * | 2006-05-30 | 2009-11-12 | ジェネンテック・インコーポレーテッド | 抗体およびイムノコンジュゲートとこれらの使用方法 |
| WO2011068845A1 (en) | 2009-12-02 | 2011-06-09 | Immunomedics, Inc. | Combining radioimmunotherapy and antibody-drug conjugates for improved cancer therapy |
| US7999083B2 (en) | 2002-12-13 | 2011-08-16 | Immunomedics, Inc. | Immunoconjugates with an intracellularly-cleavable linkage |
| WO2011145744A1 (ja) | 2010-05-17 | 2011-11-24 | 株式会社リブテック | in vivoで抗腫瘍活性を有する抗ヒトTROP-2抗体 |
| WO2011155579A1 (ja) | 2010-06-10 | 2011-12-15 | 北海道公立大学法人札幌医科大学 | 抗Trop-2抗体 |
| JP2012100671A (ja) * | 2005-04-22 | 2012-05-31 | Amgen | 延長された血液半減期を有するトキシンペプチド |
| WO2013068946A2 (en) | 2011-11-11 | 2013-05-16 | Rinat Neuroscience Corp. | Antibodies specific for trop-2 and their uses |
| WO2013077458A1 (ja) | 2011-11-22 | 2013-05-30 | 株式会社リブテック | in vivoで抗腫瘍活性を有する抗ヒトTROP-2抗体 |
| JP2013534906A (ja) * | 2010-05-13 | 2013-09-09 | インディアナ ユニバーシティー リサーチ アンド テクノロジー コーポレーション | 核内ホルモン受容体の活性を示すグルカゴンスーパーファミリーのペプチド |
| WO2014061277A1 (ja) * | 2012-10-19 | 2014-04-24 | 第一三共株式会社 | 親水性構造を含むリンカーで結合させた抗体-薬物コンジュゲート |
Family Cites Families (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3040121B2 (ja) | 1988-01-12 | 2000-05-08 | ジェネンテク,インコーポレイテッド | 増殖因子レセプターの機能を阻害することにより腫瘍細胞を処置する方法 |
| US5658920A (en) | 1991-01-16 | 1997-08-19 | Daiichi Pharmaceutical Co., Ltd. | Substituted 1H,12H-benz-[DE]pyrano[3',4':6,7] indolizino[1,2-B]quinoline-10,13(9H,15H)-dione compound |
| LU91067I2 (fr) | 1991-06-14 | 2004-04-02 | Genentech Inc | Trastuzumab et ses variantes et dérivés immuno chimiques y compris les immotoxines |
| US6214345B1 (en) | 1993-05-14 | 2001-04-10 | Bristol-Myers Squibb Co. | Lysosomal enzyme-cleavable antitumor drug conjugates |
| US6504029B1 (en) | 1995-04-10 | 2003-01-07 | Daiichi Pharmaceutical Co., Ltd. | Condensed-hexacyclic compounds and a process therefor |
| SG50747A1 (en) | 1995-08-02 | 1998-07-20 | Tanabe Seiyaku Co | Comptothecin derivatives |
| CA2192725C (en) | 1995-12-28 | 2004-04-20 | Kenji Tsujihara | Camptothecin derivatives |
| JPH1095802A (ja) | 1995-12-28 | 1998-04-14 | Tanabe Seiyaku Co Ltd | カンプトテシン誘導体 |
| TW409058B (en) | 1996-06-06 | 2000-10-21 | Daiichi Seiyaku Co | Method for preparation of a drug complex |
| JPH1171280A (ja) | 1997-06-26 | 1999-03-16 | Tanabe Seiyaku Co Ltd | 医薬組成物 |
| JPH1192405A (ja) | 1997-09-19 | 1999-04-06 | Dai Ichi Seiyaku Co Ltd | 薬物複合体 |
| TWI253935B (en) | 1998-05-22 | 2006-05-01 | Daiichi Seiyaku Co | Drug complex |
| SI2803367T1 (en) | 1999-06-25 | 2018-04-30 | Immunogen, Inc. | Treatment methods with maytansinoid conjugated antibody anti-ERBB |
| KR20030031502A (ko) | 2000-06-29 | 2003-04-21 | 다이이찌 세이야꾸 가부시기가이샤 | 디디에스 화합물 및 그의 제조방법 |
| BR0112417A (pt) | 2000-07-13 | 2003-07-01 | Daiichi Seiyaku Co | Composições farmacêuticas contendo composto dds |
| KR100573565B1 (ko) | 2001-08-29 | 2006-04-25 | 주식회사 포스코 | 요철부가 형성된 실린더 링과 이 실린더 링이 결합된 맨드릴 |
| US8877901B2 (en) | 2002-12-13 | 2014-11-04 | Immunomedics, Inc. | Camptothecin-binding moiety conjugates |
| US7585491B2 (en) | 2002-12-13 | 2009-09-08 | Immunomedics, Inc. | Immunoconjugates with an intracellularly-cleavable linkage |
| US9745380B2 (en) * | 2002-03-01 | 2017-08-29 | Immunomedics, Inc. | RS7 antibodies |
| DK1483294T3 (da) * | 2002-03-01 | 2010-11-08 | Immunomedics Inc | Internaliserende anti-CD74-antistoffer og fremgangsmåder til deres anvendelse |
| US20040202666A1 (en) * | 2003-01-24 | 2004-10-14 | Immunomedics, Inc. | Anti-cancer anthracycline drug-antibody conjugates |
| EP1725586B1 (en) | 2004-03-02 | 2015-01-14 | Seattle Genetics, Inc. | Partially loaded antibodies and methods of their conjugation |
| NZ550934A (en) | 2004-05-19 | 2010-05-28 | Medarex Inc | Chemical linkers and conjugates thereof |
| KR20120064120A (ko) | 2004-06-01 | 2012-06-18 | 제넨테크, 인크. | 항체 약물 접합체 및 방법 |
| US7560111B2 (en) | 2004-07-22 | 2009-07-14 | Genentech, Inc. | HER2 antibody composition |
| US8802820B2 (en) | 2004-11-12 | 2014-08-12 | Xencor, Inc. | Fc variants with altered binding to FcRn |
| CN102580084B (zh) | 2005-01-21 | 2016-11-23 | 健泰科生物技术公司 | Her抗体的固定剂量给药 |
| DE102005009099A1 (de) | 2005-02-28 | 2006-08-31 | Ktb Tumorforschungsgesellschaft Mbh | Proteinbindende Camptothecin-Peptid-Derivate und diese enthaltende Arzneimittel |
| DE102005009084A1 (de) | 2005-02-28 | 2006-08-31 | Ktb Tumorforschungsgesellschaft Mbh | Proteinbindende Anthrazyklin-Peptid-Derivate und diese enthaltende Arzneimittel |
| ZA200809776B (en) | 2006-05-30 | 2010-03-31 | Genentech Inc | Antibodies and immunoconjugates and uses therefor |
| US7780340B2 (en) | 2006-11-30 | 2010-08-24 | Advanced Process Technologies, Inc. | Cheese vat having fluid accessible seal assembly |
| JP5769616B2 (ja) | 2008-04-30 | 2015-08-26 | イミュノジェン・インコーポレーテッド | クロスリンカーおよびそれらの使用 |
| ES2544971T3 (es) | 2008-05-13 | 2015-09-07 | Genentech, Inc. | Análisis de conjugados de fármacos y anticuerpos mediante espectrometría de masas con captura por afinidad basada en esferas |
| KR20110112301A (ko) | 2008-11-18 | 2011-10-12 | 메리맥 파마슈티컬즈, 인크. | 인간 혈청 알부민 링커 및 그 콘쥬게이트 |
| US20120171201A1 (en) | 2009-07-22 | 2012-07-05 | Enzon Pharmaceuticals, Inc. | Methods of treating her2 positive cancer with her2 receptor antagonist in combination with multi-arm polymeric conjugates of 7-ethyl-10-hydroxycamptothecin |
| US20110042149A1 (en) | 2009-08-18 | 2011-02-24 | Baker Hughes Incorporated | Methods of forming polycrystalline diamond elements, polycrystalline diamond elements, and earth-boring tools carrying such polycrystalline diamond elements |
| WO2011038159A2 (en) | 2009-09-24 | 2011-03-31 | Seattle Genetics, Inc. | Dr5 ligand drug conjugates |
| CA2799202C (en) | 2010-05-18 | 2016-07-05 | Cerulean Pharma Inc. | Compositions and methods for treatment of autoimmune and other diseases |
| CA2804185C (en) | 2010-07-12 | 2017-03-21 | Covx Technologies Ireland Limited | Multifunctional antibody conjugates |
| ES2544608T3 (es) * | 2010-11-17 | 2015-09-02 | Genentech, Inc. | Conjugados de anticuerpo y de alaninil-maitansinol |
| MX2014007121A (es) | 2011-12-14 | 2014-09-04 | Seattle Genetics Inc | Nuevos conjugados de principio activo-ligante (adc) y su uso. |
| US20130280282A1 (en) | 2012-04-24 | 2013-10-24 | Daiichi Sankyo Co., Ltd. | Dr5 ligand drug conjugates |
| KR20150023729A (ko) | 2012-06-14 | 2015-03-05 | 암브룩스, 인코포레이티드 | 핵 수용체 리간드 폴리펩티드에 접합된 항-psma 항체 |
| EP3632471A1 (en) | 2012-10-11 | 2020-04-08 | Daiichi Sankyo Company, Limited | Antibody-drug conjugate |
| CN105051032B (zh) | 2013-01-03 | 2017-08-15 | 赛特瑞恩股份有限公司 | 抗体‑连接子‑药物偶联物、其制法及包含其的抗癌药物组合物 |
| SI3088419T1 (sl) * | 2013-12-25 | 2019-01-31 | Daiichi Sankyo Company | Zdravilni konjugat anti trop2 protitelesa |
| KR20230162159A (ko) * | 2014-01-31 | 2023-11-28 | 다이이찌 산쿄 가부시키가이샤 | 항-her2 항체-약물 접합체 |
| IL300540B1 (en) * | 2014-04-10 | 2024-04-01 | Daiichi Sankyo Co Ltd | Preparation method for drug conjugates - antibody against - HER3 |
-
2014
- 2014-12-24 SI SI201430973T patent/SI3088419T1/sl unknown
- 2014-12-24 DK DK14874745.4T patent/DK3088419T3/en active
- 2014-12-24 WO PCT/JP2014/006421 patent/WO2015098099A1/ja active Application Filing
- 2014-12-24 NZ NZ721213A patent/NZ721213A/en unknown
- 2014-12-24 KR KR1020227031871A patent/KR102491710B1/ko active IP Right Grant
- 2014-12-24 KR KR1020207006601A patent/KR102237803B1/ko active IP Right Grant
- 2014-12-24 RU RU2019134399A patent/RU2743077C2/ru active
- 2014-12-24 KR KR1020227015978A patent/KR102444529B1/ko active IP Right Grant
- 2014-12-24 PL PL14874745T patent/PL3088419T3/pl unknown
- 2014-12-24 MX MX2016008192A patent/MX2016008192A/es active IP Right Grant
- 2014-12-24 RU RU2016130095A patent/RU2705367C2/ru active
- 2014-12-24 AU AU2014371934A patent/AU2014371934B2/en active Active
- 2014-12-24 CN CN201480071134.0A patent/CN105849126B/zh active Active
- 2014-12-24 JP JP2015554565A patent/JP6130517B2/ja active Active
- 2014-12-24 RS RS20181597A patent/RS58173B1/sr unknown
- 2014-12-24 KR KR1020167015563A patent/KR101941758B1/ko active IP Right Grant
- 2014-12-24 KR KR1020217009822A patent/KR102288093B1/ko active IP Right Grant
- 2014-12-24 HU HUE14874745A patent/HUE042512T2/hu unknown
- 2014-12-24 KR KR1020227000880A patent/KR102399141B1/ko active IP Right Grant
- 2014-12-24 TR TR2019/00176T patent/TR201900176T4/tr unknown
- 2014-12-24 SG SG11201605215YA patent/SG11201605215YA/en unknown
- 2014-12-24 ES ES14874745T patent/ES2703903T3/es active Active
- 2014-12-24 KR KR1020237002152A patent/KR102535900B1/ko active IP Right Grant
- 2014-12-24 KR KR1020217024615A patent/KR102351164B1/ko active IP Right Grant
- 2014-12-24 SG SG10201902571VA patent/SG10201902571VA/en unknown
- 2014-12-24 PT PT14874745T patent/PT3088419T/pt unknown
- 2014-12-24 EP EP18187671.5A patent/EP3424955A1/en active Pending
- 2014-12-24 KR KR1020197000700A patent/KR102088169B1/ko active IP Right Grant
- 2014-12-24 KR KR1020237016957A patent/KR20230078820A/ko not_active Application Discontinuation
- 2014-12-24 MY MYPI2016001186A patent/MY195162A/en unknown
- 2014-12-24 EP EP14874745.4A patent/EP3088419B1/en active Active
- 2014-12-24 CN CN202010034409.0A patent/CN111228510A/zh active Pending
- 2014-12-24 LT LTEP14874745.4T patent/LT3088419T/lt unknown
- 2014-12-24 CN CN202310800520.XA patent/CN116987193A/zh active Pending
- 2014-12-24 CA CA2933666A patent/CA2933666C/en active Active
- 2014-12-25 TW TW111132669A patent/TWI812438B/zh active
- 2014-12-25 TW TW112127350A patent/TW202344273A/zh unknown
- 2014-12-25 TW TW103145458A patent/TWI633893B/zh active
- 2014-12-25 TW TW107124674A patent/TWI712423B/zh active
- 2014-12-25 TW TW109138661A patent/TWI779386B/zh active
-
2016
- 2016-06-20 MX MX2020009971A patent/MX2020009971A/es unknown
- 2016-06-20 MX MX2020009970A patent/MX2020009970A/es unknown
- 2016-06-20 US US15/187,179 patent/US9850312B2/en active Active
- 2016-06-20 IL IL246361A patent/IL246361B/en active IP Right Grant
- 2016-06-22 PH PH12016501233A patent/PH12016501233A1/en unknown
-
2017
- 2017-04-13 JP JP2017079392A patent/JP6449366B2/ja active Active
- 2017-11-22 US US15/821,662 patent/US10227417B2/en active Active
-
2018
- 2018-12-05 JP JP2018227733A patent/JP6832907B2/ja active Active
-
2019
- 2019-01-02 CY CY20191100002T patent/CY1121246T1/el unknown
- 2019-01-09 HR HRP20190056TT patent/HRP20190056T1/hr unknown
- 2019-01-24 US US16/256,715 patent/US11008398B2/en active Active
- 2019-06-24 IL IL267618A patent/IL267618B/en unknown
-
2020
- 2020-03-31 AU AU2020202305A patent/AU2020202305B2/en active Active
- 2020-08-03 JP JP2020131505A patent/JP7030909B2/ja active Active
- 2020-11-13 PH PH12020551943A patent/PH12020551943A1/en unknown
-
2021
- 2021-04-14 US US17/230,543 patent/US20210238303A1/en active Pending
-
2022
- 2022-02-22 JP JP2022025259A patent/JP7259104B2/ja active Active
- 2022-03-16 IL IL291446A patent/IL291446A/en unknown
-
2023
- 2023-02-16 AU AU2023200883A patent/AU2023200883A1/en active Pending
- 2023-04-05 JP JP2023061539A patent/JP2023085465A/ja active Pending
Patent Citations (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1990007861A1 (en) | 1988-12-28 | 1990-07-26 | Protein Design Labs, Inc. | CHIMERIC IMMUNOGLOBULINS SPECIFIC FOR p55 TAC PROTEIN OF THE IL-2 RECEPTOR |
| WO1992001047A1 (en) | 1990-07-10 | 1992-01-23 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1992020791A1 (en) | 1990-07-10 | 1992-11-26 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| JPH0559061A (ja) | 1991-01-16 | 1993-03-09 | Dai Ichi Seiyaku Co Ltd | 六環性化合物 |
| WO1993006213A1 (en) | 1991-09-23 | 1993-04-01 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| WO1993011236A1 (en) | 1991-12-02 | 1993-06-10 | Medical Research Council | Production of anti-self antibodies from antibody segment repertoires and displayed on phage |
| WO1993019172A1 (en) | 1992-03-24 | 1993-09-30 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| JPH0687746A (ja) | 1992-07-16 | 1994-03-29 | Dai Ichi Seiyaku Co Ltd | 抗腫瘍剤 |
| WO1995001438A1 (en) | 1993-06-30 | 1995-01-12 | Medical Research Council | Sbp members with a chemical moiety covalently bound within the binding site; production and selection thereof |
| WO1995015388A1 (en) | 1993-12-03 | 1995-06-08 | Medical Research Council | Recombinant binding proteins and peptides |
| JPH08337584A (ja) | 1995-04-10 | 1996-12-24 | Dai Ichi Seiyaku Co Ltd | 縮合六環式アミノ化合物、これを含有する医薬及びその製法 |
| WO1997046260A1 (en) | 1996-06-06 | 1997-12-11 | Daiichi Pharmaceutical Co., Ltd. | Drug complexes |
| WO1999054342A1 (en) | 1998-04-20 | 1999-10-28 | Pablo Umana | Glycosylation engineering of antibodies for improving antibody-dependent cellular cytotoxicity |
| WO2000025825A1 (fr) | 1998-10-30 | 2000-05-11 | Daiichi Pharmaceutical Co., Ltd. | Composes dds et procede de dosage de ces composes |
| WO2000061739A1 (en) | 1999-04-09 | 2000-10-19 | Kyowa Hakko Kogyo Co., Ltd. | Method for controlling the activity of immunologically functional molecule |
| JP2002060351A (ja) | 2000-03-22 | 2002-02-26 | Dai Ichi Seiyaku Co Ltd | 水酸基を有する薬物を含むdds化合物 |
| WO2002031140A1 (fr) | 2000-10-06 | 2002-04-18 | Kyowa Hakko Kogyo Co., Ltd. | Cellules produisant des compositions d'anticorps |
| WO2003038043A2 (en) | 2001-11-01 | 2003-05-08 | Uab Research Foundation | Combinations of dr5 antibody and other therapeutic agents |
| JP2005511627A (ja) * | 2001-11-20 | 2005-04-28 | シアトル ジェネティクス,インコーポレーテッド | 抗cd30抗体を使用する免疫学的疾患の治療 |
| WO2003074566A2 (en) | 2002-03-01 | 2003-09-12 | Immunomedics, Inc. | Rs7 antibodies |
| US7999083B2 (en) | 2002-12-13 | 2011-08-16 | Immunomedics, Inc. | Immunoconjugates with an intracellularly-cleavable linkage |
| US20110293513A1 (en) | 2002-12-13 | 2011-12-01 | Immunomedics, Inc. | Immunoconjugates with an Intracellularly-Cleavable Linkage |
| JP2008521828A (ja) * | 2004-11-29 | 2008-06-26 | シアトル ジェネティックス, インコーポレイテッド | 操作された抗体およびイムノコンジュゲート |
| JP2012100671A (ja) * | 2005-04-22 | 2012-05-31 | Amgen | 延長された血液半減期を有するトキシンペプチド |
| JP2009538629A (ja) * | 2006-05-30 | 2009-11-12 | ジェネンテック・インコーポレーテッド | 抗体およびイムノコンジュゲートとこれらの使用方法 |
| WO2008144891A1 (en) | 2007-05-30 | 2008-12-04 | F. Hoffmann-La Roche Ag | Humanized and chimeric anti-trop-2 antibodies that mediate cancer cell cytotoxicity |
| WO2011068845A1 (en) | 2009-12-02 | 2011-06-09 | Immunomedics, Inc. | Combining radioimmunotherapy and antibody-drug conjugates for improved cancer therapy |
| JP2013534906A (ja) * | 2010-05-13 | 2013-09-09 | インディアナ ユニバーシティー リサーチ アンド テクノロジー コーポレーション | 核内ホルモン受容体の活性を示すグルカゴンスーパーファミリーのペプチド |
| WO2011145744A1 (ja) | 2010-05-17 | 2011-11-24 | 株式会社リブテック | in vivoで抗腫瘍活性を有する抗ヒトTROP-2抗体 |
| US20120237518A1 (en) | 2010-06-10 | 2012-09-20 | Kyowa Hakko Kirin Co., Ltd | Anti-trop-2 antibody |
| WO2011155579A1 (ja) | 2010-06-10 | 2011-12-15 | 北海道公立大学法人札幌医科大学 | 抗Trop-2抗体 |
| WO2013068946A2 (en) | 2011-11-11 | 2013-05-16 | Rinat Neuroscience Corp. | Antibodies specific for trop-2 and their uses |
| WO2013077458A1 (ja) | 2011-11-22 | 2013-05-30 | 株式会社リブテック | in vivoで抗腫瘍活性を有する抗ヒトTROP-2抗体 |
| WO2014061277A1 (ja) * | 2012-10-19 | 2014-04-24 | 第一三共株式会社 | 親水性構造を含むリンカーで結合させた抗体-薬物コンジュゲート |
Non-Patent Citations (74)
| Title |
|---|
| "Antibodies: A Laboratory Manual", 1988, COLD SPRING HARBOR LABORATORY |
| "Monoclonal Antibodies", 1980, PLENUM PRESS, pages: 365 - 367 |
| "Strategies for Protein Purification and Characterization: A Laboratory Course Manual", 1996, COLD SPRING HARBOR LABORATORY PRESS |
| ALLEY, S. C. ET AL., CURRENT OPINION IN CHEMICAL BIOLOGY, vol. 14, 2010, pages 529 - 537 |
| ALTSCHUL, STEPHEN F.; THOMAS L. MADDEN; ALEJANDRO A. SCHAEFFER; JINGHUI ZHANG; ZHENG ZHANG; WEBB MILLER; DAVID J. LIPMAN: "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs", NUCLEIC ACIDS RES., vol. 25, 1997, pages 3389 - 3402, XP002905950, DOI: doi:10.1093/nar/25.17.3389 |
| ANALYTICAL BIOCHEMISTRY, vol. 360, 2007, pages 75 - 83 |
| ANNU. REV. IMMUNOL., vol. 12, 1994, pages 433 - 455 |
| BARBARA, B. M.; STANLEY, M. S.: "Selected Methods in Cellular Immunology", 1980, W. H. FREEMAN AND COMPANY |
| BASU A ET AL., INT. J. CANCER, vol. 62, no. 4, 1995, pages 472 - 479 |
| BIO TECHNIQUES, vol. 28, January 2000 (2000-01-01), pages 162 - 165 |
| CALABRESE G ET AL., CYTOGENET. CELL GENET., vol. 92, no. 1-2, 2001, pages 164 - 165 |
| CARMEN, S. ET AL., BRIEFINGS IN FUNCTIONAL GENOMICS AND PROTEOMICS, vol. 1, no. 2, 2002, pages 189 - 203 |
| CELL DEATH AND DIFFERENTIATION, vol. 15, 2008, pages 751 - 761 |
| DAMLE N.K., EXPERT OPIN. BIOL. THER., vol. 4, 2004, pages 1445 - 1452 |
| DE JAGER, R. ET AL., ANN N Y ACAD SCI, vol. 922, 2000, pages 260 - 273 |
| DUCRY, L. ET AL., BIOCONJUGATE CHEM., vol. 21, 2010, pages 5 - 13 |
| EL SEWEDY T ET AL., INT. J. CANCER, vol. 75, no. 2, 1998, pages 324 - 330 |
| FAULK WP ET AL., PROC. NATL. ACAD. SCI., vol. 75, no. 4, 1978, pages 1947 - 1951 |
| FONG D ET AL., BR. J. CANCER, vol. 99, no. 8, 2008, pages 1290 - 1295 |
| FONG D ET AL., MOD. PATHOL, vol. 21, no. 2, 2008, pages 186 - 191 |
| FORNARO M ET AL., INT. J. CANCER, vol. 62, no. 5, 1995, pages 610 - 618 |
| GLUZMAN, Y., CELL, vol. 23, 1981, pages 175 - 182 |
| HERMANSON, G.T: "Bioconjugate Techniques", 1996, ACADEMIC PRESS, pages: 56 - 136,456- |
| IKUO MITSUI ET AL.: "A New Water-soluble Camptothecin Derivative, DX-8951f, Exhibits Potent Antitumor Activity against Human Tumors in vitro and in vivo", JPN. J. CANCER RES., vol. 86, 1995, pages 776 - 782, XP009059567 * |
| INOUE, K. ET AL.: "Polymer Drugs in the Clinical Stage", 2003, pages: 145 - 153 |
| J. MED. CHEM., vol. 35, 1992, pages 2928 |
| J. ORG. CHEM, vol. 51, 1986, pages 3196 |
| JOTO, N. ET AL., INT J CANCER, vol. 72, 1997, pages 680 - 686 |
| JOURNAL OF CHROMATOGRAPHY A, vol. 705, 1995, pages 129 - 134 |
| KABAT, E. A.; MAYER, M. M.: "Experimental Immunochemistry", 1964, CHARLES C THOMAS PUBLISHER |
| KABAT, E. A.; MAYER, M. M.: "Experimental Immunochemistry", 1964, CHARLES C THOMAS PUBLISHER SPRINGFIELD |
| KOHLER ET AL., EUR. J. IMMUNOL., vol. 6, 1977, pages 511 |
| KOHLER ET AL., NATURE, vol. 256, 1975, pages 495 |
| KOHLER; MILSTEIN, NATURE, vol. 256, 1975, pages 495 - 497 |
| KUMAZAWA, E. ET AL., CANCER CHEMOTHER. PHARMACOL., vol. 42, 1998, pages 210 - 220 |
| KUMAZAWA, E. ET AL., CANCER SCI, vol. 95, 2004, pages 168 - 175 |
| KUMAZAWA, E.; TOHGO, A., EXP. OPIN. INVEST. DRUGS, vol. 7, 1998, pages 625 - 632 |
| KUROIWA, Y. ET AL., NUCL. ACIDS RES., vol. 26, 1998, pages 3447 - 3448 |
| LINNENBACH A J ET AL., PROC. NATL. ACAD. SCI., vol. 86, no. 1, 1989, pages 27 - 31 |
| LIPINSKI M ET AL., PROC. NATL. ACAD. SCI., vol. 78, no. 8, 1981, pages 5147 - 5150 |
| LORI BURTON, PHARMACEUTICAL DEVELOPMENT AND TECHNOLOGY, vol. 12, 2007, pages 265 - 273 |
| METHODS IN ENZYMOLOGY, vol. 203, 1991, pages 121 - 153 |
| MILSTEIN ET AL., NATURE, vol. 266, 1977, pages 550 |
| MITSUI, I. ET AL., JPN J. CANCER RES, vol. 86, 1995, pages 776 - 782 |
| MOLECULAR BIOLOGY OF THE CELL, vol. 15, December 2004 (2004-12-01), pages 5268 - 5282 |
| MUHLMANN G ET AL., J. CLIN. PATHOL., vol. 62, no. 2, 2009, pages 152 - 158 |
| NATURE BIOTECHNOLOGY, vol. 23, no. 9, 2005, pages 1105 - 1116 |
| NATURE, vol. 321, 1986, pages 522 - 525 |
| NING S ET AL., NEUROL. SCI., vol. 34, no. 10, 2013, pages 1745 - 1750 |
| NUC. ACID RES., vol. 29, 2001, pages 205 - 206 |
| NUC. ACID RES., vol. 35, 2007, pages D301 - D303 |
| OHMACHI T ET AL., CLIN. CANCER RES., vol. 12, no. 10, 2006, pages 3057 - 3063 |
| PROC. NATL. ACAD. SCI. USA, vol. 81, 1984, pages 6851 - 6855 |
| PROC. NATL. ACAD. SCI. USA, vol. 86, 1989, pages 10029 - 10033 |
| PROTEIN SCIENCE, vol. 4, 1995, pages 2411 - 2423 |
| QUEEN ET AL., PROC. NATL. ACAD. SCI. USA, vol. 86, 1989, pages 10029 - 10033 |
| RIPANI E ET AL., INT. J. CANCER, vol. 76, no. 5, 1998, pages 671 - 676 |
| SAIKI, R. K. ET AL., SCIENCE, vol. 239, 1988, pages 487 - 489 |
| SENTER P. D. ET AL., NATURE BIOTECHNOLOGY, vol. 30, 2012, pages 631 - 637 |
| SIRIWARDENA, D. ET AL., OPHTHALMOLOGY, vol. 109, no. 3, 2002, pages 427 - 431 |
| SOEPENBERG, 0. ET AL., CLINICAL CANCER RESEARCH, vol. 11, 2005, pages 703 - 711 |
| TAKIGUCHI, S. ET AL., JPN J. CANCER RES., vol. 88, 1997, pages 760 - 769 |
| TOMIZUKA, K. ET AL., NATURE GENETICS, vol. 16, 1997, pages 133 - 143 |
| TOMIZUKA, K. ET AL., PROC. NATL. ACAD. SCI. USA, vol. 97, 2000, pages 722 - 727 |
| URLAUB, G.; CHASIN, L. A., PROC. NATL. ACAD. SCI. USA, vol. 77, 1980, pages 4126 - 4220 |
| WALSH, NATURE, vol. 266, 1977, pages 495 |
| WANG J ET AL., MOL. CANCER THER, vol. 7, no. 2, 2008, pages 280 - 285 |
| WEIR, D. M.: "Handbook of Experimental Immunology", vol. I. II. I, 1987, BLACKWELL SCIENTIFIC PUBLICATION |
| WEIR, D. M.: "Handbook of Experimental Immunology", vol. I. II. I, 1987, BLACKWELL SCIENTIFIC PUBLICATIONS |
| WEIR, D. M.: "Handbook of Experimental Immunology", vol. VOL. I,, 1978, BLACKWELL SCIENTIFIC PUBLICATIONS |
| WENTE M. N. ET AL., INVESTIGATIONAL NEW DRUGS, vol. 23, 2005, pages 339 - 347 |
| WORMSTONE, I. M. ET AL., INVESTIGATIVE OPHTHALMOLOGY VISUAL SCIENCE, vol. 43, no. 7, 2002, pages 2301 - 2308 |
| YAMAGUCHI, M.; HAMADA, H. ET AL., BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS, vol. 454, 2014, pages 600 - 603 |
| YOSHIDA, H. ET AL.: "Animal Cell Technology: Basic and Applied Aspects", vol. 10, 1999, KLUWER ACADEMIC PUBLISHERS, pages: 69 - 73 |
Cited By (89)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2015155976A1 (ja) * | 2014-04-10 | 2017-04-13 | 第一三共株式会社 | 抗her2抗体−薬物コンジュゲート |
| JP2020079237A (ja) * | 2014-04-10 | 2020-05-28 | 第一三共株式会社 | 抗her2抗体−薬物コンジュゲート |
| US11173213B2 (en) | 2015-06-29 | 2021-11-16 | Daiichi Sankyo Company, Limited | Method for selectively manufacturing antibody-drug conjugate |
| JPWO2018110515A1 (ja) * | 2016-12-12 | 2019-10-24 | 第一三共株式会社 | 抗体−薬物コンジュゲートと免疫チェックポイント阻害剤の組み合わせ |
| WO2018110515A1 (ja) | 2016-12-12 | 2018-06-21 | 第一三共株式会社 | 抗体-薬物コンジュゲートと免疫チェックポイント阻害剤の組み合わせ |
| US11273155B2 (en) | 2016-12-12 | 2022-03-15 | Daiichi Sankyo Company, Limited | Combination of antibody-drug conjugate and immune checkpoint inhibitor |
| KR20190095280A (ko) | 2016-12-12 | 2019-08-14 | 다이이찌 산쿄 가부시키가이샤 | 항체-약물 콘주게이트와 면역 체크 포인트 저해제의 조합 |
| JP7064629B2 (ja) | 2017-05-15 | 2022-05-10 | 第一三共株式会社 | 抗cdh6抗体及び抗cdh6抗体-薬物コンジュゲート |
| JP2021059599A (ja) * | 2017-05-15 | 2021-04-15 | 第一三共株式会社 | 抗cdh6抗体及び抗cdh6抗体−薬物コンジュゲート |
| US11446386B2 (en) | 2017-05-15 | 2022-09-20 | Daiichi Sankyo Company, Limited | Anti-CDH6 antibody and method of producing an anti-CDH6 antibody-drug conjugate |
| JP2020503243A (ja) * | 2017-08-11 | 2020-01-30 | バイオ − テラ ソリューションズ、リミテッド | Trop2陽性疾患の治療のための化合物及び方法 |
| KR20200044044A (ko) | 2017-08-23 | 2020-04-28 | 다이이찌 산쿄 가부시키가이샤 | 항체-약물 콘주게이트의 제제 및 그 동결 건조 방법 |
| WO2019039483A1 (ja) | 2017-08-23 | 2019-02-28 | 第一三共株式会社 | 抗体-薬物コンジュゲートの製剤及びその凍結乾燥方法 |
| JPWO2019044947A1 (ja) * | 2017-08-31 | 2020-10-15 | 第一三共株式会社 | 抗体−薬物コンジュゲートの改良製造方法 |
| WO2019044946A1 (ja) | 2017-08-31 | 2019-03-07 | 第一三共株式会社 | 抗体-薬物コンジュゲートの新規製造方法 |
| KR20200041993A (ko) | 2017-08-31 | 2020-04-22 | 다이이찌 산쿄 가부시키가이샤 | 항체-약물 콘주게이트의 개량 제조 방법 |
| AU2018327171B2 (en) * | 2017-08-31 | 2023-03-09 | Daiichi Sankyo Company, Limited | Improved method for producing antibody-drug conjugate |
| KR20220104292A (ko) | 2017-08-31 | 2022-07-26 | 다이이찌 산쿄 가부시키가이샤 | 항체-약물 콘주게이트의 신규 제조 방법 |
| KR20200033949A (ko) | 2017-08-31 | 2020-03-30 | 다이이찌 산쿄 가부시키가이샤 | 항체-약물 콘주게이트의 신규 제조 방법 |
| JP7366745B2 (ja) | 2017-08-31 | 2023-10-23 | 第一三共株式会社 | 抗体-薬物コンジュゲートの改良製造方法 |
| KR20240018674A (ko) | 2017-08-31 | 2024-02-13 | 다이이찌 산쿄 가부시키가이샤 | 항체-약물 콘주게이트의 신규 제조 방법 |
| WO2019044947A1 (ja) | 2017-08-31 | 2019-03-07 | 第一三共株式会社 | 抗体-薬物コンジュゲートの改良製造方法 |
| US11318212B2 (en) | 2017-08-31 | 2022-05-03 | Daiichi Sankyo Company, Limited | Method for producing antibody-drug conjugate |
| US11945882B2 (en) | 2017-08-31 | 2024-04-02 | Daiichi Sankyo Company, Limited | Method for producing antibody-drug conjugate |
| WO2019065964A1 (ja) | 2017-09-29 | 2019-04-04 | 第一三共株式会社 | 抗体-ピロロベンゾジアゼピン誘導体コンジュゲート |
| KR20210038904A (ko) | 2018-07-25 | 2021-04-08 | 다이이찌 산쿄 가부시키가이샤 | 항체-약물 콘쥬게이트의 효과적인 제조 방법 |
| WO2020022363A1 (ja) | 2018-07-25 | 2020-01-30 | 第一三共株式会社 | 抗体-薬物コンジュゲートの効果的な製造方法 |
| KR20210040059A (ko) | 2018-07-27 | 2021-04-12 | 다이이찌 산쿄 가부시키가이샤 | 항체-약물 콘주게이트의 약물 부위를 인식하는 단백질 |
| WO2020022475A1 (ja) | 2018-07-27 | 2020-01-30 | 第一三共株式会社 | 抗体-薬物コンジュゲートの薬物部位を認識する蛋白質 |
| WO2020027100A1 (ja) | 2018-07-31 | 2020-02-06 | 第一三共株式会社 | 抗体-薬物コンジュゲート投与による転移性脳腫瘍の治療 |
| KR20210038625A (ko) | 2018-07-31 | 2021-04-07 | 다이이찌 산쿄 가부시키가이샤 | 항체-약물 콘주게이트 투여에 의한 전이성 뇌종양의 치료 |
| WO2020031936A1 (ja) | 2018-08-06 | 2020-02-13 | 第一三共株式会社 | 抗体-薬物コンジュゲートとチューブリン阻害剤の組み合わせ |
| KR20210042120A (ko) | 2018-08-06 | 2021-04-16 | 다이이찌 산쿄 가부시키가이샤 | 항체-약물 콘주게이트와 튜불린 저해제의 조합 |
| JP7458981B2 (ja) | 2018-08-06 | 2024-04-01 | 第一三共株式会社 | 抗体-薬物コンジュゲートとチューブリン阻害剤の組み合わせ |
| WO2020040245A1 (ja) | 2018-08-23 | 2020-02-27 | 第一三共株式会社 | 抗体薬物複合体の感受性マーカー |
| WO2020094670A1 (en) | 2018-11-05 | 2020-05-14 | Synaffix B.V. | Antibody-conjugates for targeting of tumours expressing trop-2 |
| WO2020122034A1 (ja) | 2018-12-11 | 2020-06-18 | 第一三共株式会社 | 抗体-薬物コンジュゲートとparp阻害剤の組み合わせ |
| CN113271942A (zh) * | 2018-12-11 | 2021-08-17 | 第一三共株式会社 | 抗体-药物缀合物与parp抑制剂的组合 |
| KR20210102341A (ko) | 2018-12-11 | 2021-08-19 | 다이이찌 산쿄 가부시키가이샤 | 항체-약물 컨쥬게이트와 parp 저해제의 조합 |
| WO2020130125A1 (ja) | 2018-12-21 | 2020-06-25 | 第一三共株式会社 | 抗体-薬物コンジュゲートとキナーゼ阻害剤の組み合わせ |
| KR20210107069A (ko) | 2018-12-21 | 2021-08-31 | 다이이찌 산쿄 가부시키가이샤 | 항체-약물 컨쥬게이트와 키나아제 저해제의 조합 |
| WO2020196474A1 (ja) | 2019-03-25 | 2020-10-01 | 第一三共株式会社 | 抗体-ピロロベンゾジアゼピン誘導体コンジュゲート |
| WO2020196712A1 (ja) | 2019-03-27 | 2020-10-01 | 第一三共株式会社 | 抗体-ピロロベンゾジアゼピン誘導体コンジュゲートとparp阻害剤の組み合わせ |
| WO2020240467A1 (en) | 2019-05-29 | 2020-12-03 | Daiichi Sankyo Company, Limited | Dosage of an antibody-drug conjugate |
| WO2020249063A1 (en) * | 2019-06-13 | 2020-12-17 | Bio-Thera Solutions, Ltd. | Methods for the treatment of trop2 positive diseases |
| WO2021147993A1 (zh) | 2020-01-22 | 2021-07-29 | 江苏恒瑞医药股份有限公司 | 抗trop-2抗体-依喜替康类似物偶联物及其医药用途 |
| WO2021177438A1 (ja) | 2020-03-06 | 2021-09-10 | 第一三共株式会社 | 新規環状ジヌクレオチド誘導体を含む抗体薬物コンジュゲート |
| WO2021226440A1 (en) | 2020-05-08 | 2021-11-11 | Bolt Biotherapeutics, Inc. | Elastase-substrate, peptide linker immunoconjugates, and uses thereof |
| WO2021260583A1 (en) | 2020-06-24 | 2021-12-30 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and dna-pk inhibitor |
| WO2021260580A1 (en) | 2020-06-24 | 2021-12-30 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and atm inhibitor |
| WO2021260582A1 (en) | 2020-06-24 | 2021-12-30 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and aurora b inhibitor |
| WO2021260578A1 (en) | 2020-06-24 | 2021-12-30 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and cdk9 inhibitor |
| WO2021260579A1 (en) | 2020-06-24 | 2021-12-30 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and atr inhibitor |
| WO2022014698A1 (ja) | 2020-07-17 | 2022-01-20 | 第一三共株式会社 | 抗体-薬物コンジュゲートの製造方法 |
| WO2022036101A1 (en) | 2020-08-13 | 2022-02-17 | Bolt Biotherapeutics, Inc. | Pyrazoloazepine immunoconjugates, and uses thereof |
| WO2022074617A1 (en) | 2020-10-09 | 2022-04-14 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and parp1 selective inhibitor |
| WO2022102634A1 (ja) | 2020-11-11 | 2022-05-19 | 第一三共株式会社 | 抗体-薬物コンジュゲートと抗SIRPα抗体の組み合わせ |
| KR20230106645A (ko) | 2020-11-11 | 2023-07-13 | 다이이찌 산쿄 가부시키가이샤 | 항체-약물 콘주게이트와 항 SIRPα 항체의 조합 |
| WO2022102695A1 (ja) | 2020-11-12 | 2022-05-19 | 第一三共株式会社 | 抗b7-h3抗体-薬物コンジュゲート投与による中皮腫の治療 |
| WO2022163846A1 (ja) | 2021-02-01 | 2022-08-04 | 第一三共株式会社 | 抗体-免疫賦活化剤コンジュゲートの新規製造方法 |
| WO2022191313A1 (ja) | 2021-03-12 | 2022-09-15 | 第一三共株式会社 | 糖鎖及び糖鎖を含む医薬品の製造方法 |
| WO2022204536A1 (en) | 2021-03-26 | 2022-09-29 | Bolt Biotherapeutics, Inc. | 2-amino-4-carboxamide-benzazepine immunoconjugates, and uses thereof |
| WO2022204528A1 (en) | 2021-03-26 | 2022-09-29 | Bolt Biotherapeutics, Inc. | 2-amino-4-carboxamide-benzazepine immunoconjugates, and uses thereof |
| WO2022261301A1 (en) | 2021-06-11 | 2022-12-15 | Gilead Sciences, Inc. | Combination mcl-1 inhibitors with anti-cancer agents |
| US11931424B2 (en) | 2021-06-11 | 2024-03-19 | Gilead Sciences, Inc. | Combination MCL-1 inhibitors with anti-body drug conjugates |
| US11957693B2 (en) | 2021-06-11 | 2024-04-16 | Gilead Sciences, Inc. | Combination MCL-1 inhibitors with anti-cancer agents |
| WO2022261310A1 (en) | 2021-06-11 | 2022-12-15 | Gilead Sciences, Inc. | Combination mcl-1 inhibitors with anti-body drug conjugates |
| WO2023274365A1 (zh) * | 2021-07-02 | 2023-01-05 | 和迈生物科技有限公司 | 抗trop2单域抗体及其应用 |
| WO2023001248A1 (zh) | 2021-07-21 | 2023-01-26 | 江苏恒瑞医药股份有限公司 | 一种含抗trop2抗体药物偶联物的药物组合物及其用途 |
| WO2023076983A1 (en) | 2021-10-28 | 2023-05-04 | Gilead Sciences, Inc. | Pyridizin-3(2h)-one derivatives |
| WO2023077030A1 (en) | 2021-10-29 | 2023-05-04 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2023076599A1 (en) | 2021-10-29 | 2023-05-04 | Bolt Biotherapeutics, Inc. | Tlr agonist immunoconjugates with cysteine-mutant antibodies, and uses thereof |
| WO2023089527A1 (en) | 2021-11-18 | 2023-05-25 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and parp1 selective inhibitor |
| WO2023122581A2 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023122615A1 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023126823A1 (en) | 2021-12-28 | 2023-07-06 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and atr inhibitor |
| WO2023126822A1 (en) | 2021-12-28 | 2023-07-06 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and rasg12c inhibitor |
| WO2023147418A1 (en) | 2022-01-28 | 2023-08-03 | Gilead Sciences, Inc. | Parp7 inhibitors |
| WO2023175483A1 (en) | 2022-03-16 | 2023-09-21 | Astrazeneca Uk Limited | A scoring method for an anti-trop2 antibody‑drug conjugate therapy |
| EP4245756A1 (en) | 2022-03-17 | 2023-09-20 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023178181A1 (en) | 2022-03-17 | 2023-09-21 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023180485A1 (en) | 2022-03-23 | 2023-09-28 | Synaffix B.V. | Antibody-conjugates for targeting of tumours expressing trop-2 |
| WO2023201267A1 (en) | 2022-04-13 | 2023-10-19 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
| WO2023201268A1 (en) | 2022-04-13 | 2023-10-19 | Gilead Sciences, Inc. | Combination therapy for treating tumor antigen expressing cancers |
| WO2023205719A1 (en) | 2022-04-21 | 2023-10-26 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| WO2023209591A1 (en) | 2022-04-27 | 2023-11-02 | Daiichi Sankyo Company, Limited | Combination of antibody-drug conjugate with ezh1 and/or ezh2 inhibitor |
| WO2023218378A1 (en) | 2022-05-11 | 2023-11-16 | Daiichi Sankyo Company, Limited | Combination of an antibody specific for a tumor antigen and a cd47 inhibitor |
| WO2024006929A1 (en) | 2022-07-01 | 2024-01-04 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2024023750A1 (en) | 2022-07-28 | 2024-02-01 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and bispecific checkpoint inhibitor |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7259104B2 (ja) | 抗trop2抗体-薬物コンジュゲート | |
| JP7146031B2 (ja) | 抗her2抗体-薬物コンジュゲート | |
| JP5953378B2 (ja) | 抗体−薬物コンジュゲート | |
| WO2015146132A1 (ja) | 抗cd98抗体-薬物コンジュゲート | |
| BR112016013704B1 (pt) | Conjugado de anticorpo-fármaco anti-trop2,fármaco antitumor e/ou anticâncer,composição farmacêutica e uso dos mesmos. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14874745 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015554565 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20167015563 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2933666 Country of ref document: CA |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014874745 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014874745 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 246361 Country of ref document: IL Ref document number: MX/A/2016/008192 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12016501233 Country of ref document: PH |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112016013704 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2014371934 Country of ref document: AU Date of ref document: 20141224 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: NC2016/0000099 Country of ref document: CO |
|
| ENP | Entry into the national phase |
Ref document number: 2016130095 Country of ref document: RU Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 112016013704 Country of ref document: BR Kind code of ref document: A2 Effective date: 20160614 |