WO2014157149A1 - ポリカーボネート樹脂積層体 - Google Patents
ポリカーボネート樹脂積層体 Download PDFInfo
- Publication number
- WO2014157149A1 WO2014157149A1 PCT/JP2014/058200 JP2014058200W WO2014157149A1 WO 2014157149 A1 WO2014157149 A1 WO 2014157149A1 JP 2014058200 W JP2014058200 W JP 2014058200W WO 2014157149 A1 WO2014157149 A1 WO 2014157149A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- layer
- solid solution
- core
- titanium oxide
- mass
- Prior art date
Links
Images
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/36—Layered products comprising a layer of synthetic resin comprising polyesters
- B32B27/365—Layered products comprising a layer of synthetic resin comprising polyesters comprising polycarbonates
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B25/00—Layered products comprising a layer of natural or synthetic rubber
- B32B25/04—Layered products comprising a layer of natural or synthetic rubber comprising rubber as the main or only constituent of a layer, which is next to another layer of the same or of a different material
- B32B25/08—Layered products comprising a layer of natural or synthetic rubber comprising rubber as the main or only constituent of a layer, which is next to another layer of the same or of a different material of synthetic resin
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/06—Layered products comprising a layer of synthetic resin as the main or only constituent of a layer, which is next to another layer of the same or of a different material
- B32B27/08—Layered products comprising a layer of synthetic resin as the main or only constituent of a layer, which is next to another layer of the same or of a different material of synthetic resin
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/18—Layered products comprising a layer of synthetic resin characterised by the use of special additives
- B32B27/20—Layered products comprising a layer of synthetic resin characterised by the use of special additives using fillers, pigments, thixotroping agents
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/30—Layered products comprising a layer of synthetic resin comprising vinyl (co)polymers; comprising acrylic (co)polymers
- B32B27/308—Layered products comprising a layer of synthetic resin comprising vinyl (co)polymers; comprising acrylic (co)polymers comprising acrylic (co)polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B5/00—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts
- B32B5/16—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by features of a layer formed of particles, e.g. chips, powder or granules
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B7/00—Layered products characterised by the relation between layers; Layered products characterised by the relative orientation of features between layers, or by the relative values of a measurable parameter between layers, i.e. products comprising layers having different physical, chemical or physicochemical properties; Layered products characterised by the interconnection of layers
- B32B7/04—Interconnection of layers
- B32B7/12—Interconnection of layers using interposed adhesives or interposed materials with bonding properties
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G23/00—Compounds of titanium
- C01G23/04—Oxides; Hydroxides
- C01G23/047—Titanium dioxide
- C01G23/053—Producing by wet processes, e.g. hydrolysing titanium salts
- C01G23/0536—Producing by wet processes, e.g. hydrolysing titanium salts by hydrolysing chloride-containing salts
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09C—TREATMENT OF INORGANIC MATERIALS, OTHER THAN FIBROUS FILLERS, TO ENHANCE THEIR PIGMENTING OR FILLING PROPERTIES ; PREPARATION OF CARBON BLACK ; PREPARATION OF INORGANIC MATERIALS WHICH ARE NO SINGLE CHEMICAL COMPOUNDS AND WHICH ARE MAINLY USED AS PIGMENTS OR FILLERS
- C09C1/00—Treatment of specific inorganic materials other than fibrous fillers; Preparation of carbon black
- C09C1/04—Compounds of zinc
- C09C1/043—Zinc oxide
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09C—TREATMENT OF INORGANIC MATERIALS, OTHER THAN FIBROUS FILLERS, TO ENHANCE THEIR PIGMENTING OR FILLING PROPERTIES ; PREPARATION OF CARBON BLACK ; PREPARATION OF INORGANIC MATERIALS WHICH ARE NO SINGLE CHEMICAL COMPOUNDS AND WHICH ARE MAINLY USED AS PIGMENTS OR FILLERS
- C09C1/00—Treatment of specific inorganic materials other than fibrous fillers; Preparation of carbon black
- C09C1/36—Compounds of titanium
- C09C1/3607—Titanium dioxide
- C09C1/3653—Treatment with inorganic compounds
- C09C1/3661—Coating
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09C—TREATMENT OF INORGANIC MATERIALS, OTHER THAN FIBROUS FILLERS, TO ENHANCE THEIR PIGMENTING OR FILLING PROPERTIES ; PREPARATION OF CARBON BLACK ; PREPARATION OF INORGANIC MATERIALS WHICH ARE NO SINGLE CHEMICAL COMPOUNDS AND WHICH ARE MAINLY USED AS PIGMENTS OR FILLERS
- C09C1/00—Treatment of specific inorganic materials other than fibrous fillers; Preparation of carbon black
- C09C1/36—Compounds of titanium
- C09C1/3607—Titanium dioxide
- C09C1/3684—Treatment with organo-silicon compounds
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09C—TREATMENT OF INORGANIC MATERIALS, OTHER THAN FIBROUS FILLERS, TO ENHANCE THEIR PIGMENTING OR FILLING PROPERTIES ; PREPARATION OF CARBON BLACK ; PREPARATION OF INORGANIC MATERIALS WHICH ARE NO SINGLE CHEMICAL COMPOUNDS AND WHICH ARE MAINLY USED AS PIGMENTS OR FILLERS
- C09C1/00—Treatment of specific inorganic materials other than fibrous fillers; Preparation of carbon black
- C09C1/36—Compounds of titanium
- C09C1/3692—Combinations of treatments provided for in groups C09C1/3615 - C09C1/3684
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D151/00—Coating compositions based on graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Coating compositions based on derivatives of such polymers
- C09D151/06—Coating compositions based on graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Coating compositions based on derivatives of such polymers grafted on to homopolymers or copolymers of aliphatic hydrocarbons containing only one carbon-to-carbon double bond
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D183/00—Coating compositions based on macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing silicon, with or without sulfur, nitrogen, oxygen, or carbon only; Coating compositions based on derivatives of such polymers
- C09D183/04—Polysiloxanes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2255/00—Coating on the layer surface
- B32B2255/10—Coating on the layer surface on synthetic resin layer or on natural or synthetic rubber layer
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2255/00—Coating on the layer surface
- B32B2255/26—Polymeric coating
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/40—Properties of the layers or laminate having particular optical properties
- B32B2307/412—Transparent
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/40—Properties of the layers or laminate having particular optical properties
- B32B2307/42—Polarizing, birefringent, filtering
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/50—Properties of the layers or laminate having particular mechanical properties
- B32B2307/558—Impact strength, toughness
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/50—Properties of the layers or laminate having particular mechanical properties
- B32B2307/584—Scratch resistance
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/70—Other properties
- B32B2307/71—Resistive to light or to UV
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/70—Other properties
- B32B2307/712—Weather resistant
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/50—Solid solutions
- C01P2002/52—Solid solutions containing elements as dopants
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/50—Solid solutions
- C01P2002/52—Solid solutions containing elements as dopants
- C01P2002/54—Solid solutions containing elements as dopants one element only
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/80—Crystal-structural characteristics defined by measured data other than those specified in group C01P2002/70
- C01P2002/84—Crystal-structural characteristics defined by measured data other than those specified in group C01P2002/70 by UV- or VIS- data
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/64—Nanometer sized, i.e. from 1-100 nanometer
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/18—Oxygen-containing compounds, e.g. metal carbonyls
- C08K3/20—Oxides; Hydroxides
- C08K3/22—Oxides; Hydroxides of metals
- C08K2003/2237—Oxides; Hydroxides of metals of titanium
- C08K2003/2241—Titanium dioxide
Definitions
- the present invention relates to a laminate in which a specific hard coat layer is formed on a laminate substrate in which a thermoplastic resin layer having ultraviolet absorbing performance is molded and laminated on one side or both sides of a polycarbonate resin layer. More specifically, the hard coat layer has tetragonal titanium oxide fine particles in which one or more atoms of the group consisting of cobalt, tin, and manganese are dissolved, and has a shell of silicon oxide or aluminum oxide outside the nucleus.
- a core-shell type tetragonal titanium oxide solid solution in which a core-shell type tetragonal titanium oxide solid solution is dispersed in one or more dispersion media of the group consisting of water, alcohol, ether, ester, and ketone.
- the volume-based 50% cumulative distribution diameter (D 50 ) of the nuclear fine particles measured by the dynamic light scattering method was 50 nm or less, and the volume-based 50% cumulative distribution diameter (D 50 ) of the core-shell solid solution was 100 nm or less.
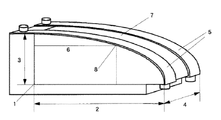
- a silicone hard coat layer formed by coating and curing a silicone hard coat composition it is excellent in scratch resistance, ultraviolet shielding properties, and outdoor durability, and more preferably, a three-point bending test in accordance with JIS K7171. When implemented, this is a polycarbonate resin laminate having a radius of curvature R of 100 or less.
- Polycarbonate resin has excellent transparency, impact resistance, and high heat distortion temperature, and has excellent dimensional stability and processability. , Glasses, goggles, sound insulation walls, traffic light lenses, curve mirrors, windshields, nameplates, etc. However, since the polycarbonate resin has insufficient weather resistance, conventionally, a method of coating the polycarbonate resin layer with an acrylic resin containing an ultraviolet absorber by lamination, coating, coextrusion method or the like is known.
- Patent Document 1 discloses obtaining a laminate having excellent pencil hardness in addition to abrasion resistance and chemical resistance. When used for a long time, there arises a problem that peeling occurs at the interface between the polycarbonate resin layer and the acrylic resin film.
- Patent Documents 2 to 9 disclose a method of adding an ultraviolet absorber to a primer layer, a method of introducing an ultraviolet-absorbing organic substituent into a organic resin constituting the primer via a chemical bond, and colloidal silica. There has been proposed a method of adding ultraviolet-shielding inorganic oxide fine particles to the polysiloxane paint containing.
- ultraviolet absorber and “ultraviolet-absorbing organic substituent” as used herein refers to substituents such as benzophenone, benzotriazole, and triazine, and organic compounds containing them.
- the ultraviolet shielding inorganic oxide fine particles refer to, for example, zinc oxide or titanium oxide fine particles.
- a primer composition containing an ultraviolet absorber component must be separately manufactured, and in the primer coating process, a coating environment and a heating equipment for curing that prevent contamination of dust, dust, and dust.
- the process is complicated and multistage, and has the disadvantage of being inferior in economic efficiency.
- the addition of a large amount of UV absorber to the primer coating may deteriorate the adhesion with the protective coating by the colloidal silica-containing polysiloxane coating applied to the upper surface of the substrate or the undercoat layer, or during the heat curing process, For example, if it is removed from the composition by volatilization, or if it is used outdoors for a long period of time, the ultraviolet absorber gradually bleeds out, causing cracks, whitening or yellowing. It was. Further, the protective coating layer made of colloidal silica-containing polysiloxane on the upper surface has a problem that a large amount of an ultraviolet absorber cannot be added from the viewpoint of scratch resistance.
- a method for improving the above-mentioned problems, particularly process simplification and economic efficiency while achieving weather resistance and scratch resistance a method of silicone hard coating on a laminated substrate such as a co-extrusion method is also disclosed.
- adhesion and crack resistance are determined by defining the linear expansion coefficient difference between the base material and the acrylic resin layer and the linear expansion coefficient difference between the acrylic resin layer and the organosiloxane resin layer. An excellent laminate is disclosed.
- Patent Document 11 there is an example of a laminate in which a hard coat layer containing zinc oxide fine particles as an ultraviolet cut agent is provided on a base material formed by coextrusion with a polycarbonate substrate and an acrylic layer.
- a hard coat layer containing zinc oxide fine particles as an ultraviolet cut agent is provided on a base material formed by coextrusion with a polycarbonate substrate and an acrylic layer.
- the ultraviolet shielding property and outdoor durability are not yet sufficient, and there is no description about bending workability.
- An object of the present invention is to provide a polycarbonate resin laminate excellent in scratch resistance, ultraviolet shielding properties, outdoor durability, and bending workability.
- a core-shell type tetragonal titanium oxide solid solution having tetragonal titanium oxide fine particles with solid atoms dissolved therein and a silicon oxide or aluminum oxide shell outside the nucleus is composed of water, alcohol, ether, ester, and ketone.
- a core-shell type tetragonal titanium oxide solid solution dispersion dispersed in one or more dispersion mediums of a group, and a volume-based 50% cumulative distribution diameter of the above-mentioned core particles measured by a dynamic light scattering method using laser light in (D 50) is 50nm or less, the 50% cumulative distribution diameter on a volume basis of the core-shell solid solution (D 50) is at 100nm or less, the cobalt, tin, manganese
- the silicone hard coat layer (C) is obtained by coating and curing a silicone hard coat composition containing a solid solution dispersion having a total solid solution amount (M) of 10 to 1,000 in terms of molar ratio with titanium (Ti / M).
- the laminated base material is a polycarbonate resin layer (A layer) and a thermoplastic resin layer (B layer) having ultraviolet absorbing performance by any one of a co-extrusion method, a thermocompression bonding method, or an insert molding method.
- the thermoplastic resin layer (B layer) having ultraviolet absorbing performance is composed of a thermoplastic resin containing benzotriazoles or triazines as an ultraviolet absorber, [1] or [2] The polycarbonate resin laminate described in 1.
- the thermoplastic resin layer (B layer) having ultraviolet absorbing performance is formed by using a (meth) acrylic resin composition.
- the hard coat layer (C layer) is a cured film of a silicone hard coat composition containing the following components (c-1) to (c-5): The polycarbonate resin laminate according to any one of the above.
- (C-1) Core-shell type tetragonal crystal having tetragonal titanium oxide fine particles in which one or more atoms of the group consisting of cobalt, tin, and manganese are solid-solved as a nucleus, and a shell of silicon oxide or aluminum oxide outside the nucleus.
- the amount of the colloidal silica as the component (c-5) is 5 to 100 parts by mass with respect to 100 parts by mass of the solid content of the silicone resin as the component (c-2).
- a specific hard coat layer is formed on a laminate base material obtained by molding and laminating a thermoplastic resin layer (B layer) having ultraviolet absorbing performance on a layer (A layer) made of a polycarbonate resin.
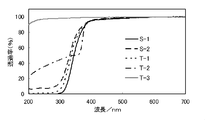
- tool for a quarter ellipse test method for implementing limit stress property evaluation is shown. It is a graph which shows the transmittance
- Polycarbonate resin layer As the polycarbonate resin used in the present invention, a bisphenol A type polycarbonate is suitably used. Besides, various polycarbonate resins polymerized by using other dihydric phenols and having a high heat resistance or a low water absorption rate, and fats. Various heat-resistant polycarbonate resins polymerized using a group diol may be used. The polycarbonate resin may be produced by any production method, and in the case of interfacial polycondensation, a terminal stopper of a monohydric phenol is usually used.
- the polycarbonate resin may be a branched polycarbonate resin obtained by polymerizing trifunctional phenols, and further a copolymer polycarbonate obtained by copolymerizing an aliphatic dicarboxylic acid, an aromatic dicarboxylic acid, or a divalent aliphatic or alicyclic alcohol. It may be.
- the viscosity average molecular weight of the polycarbonate resin is in the range of 13,000 to 40,000, it can be applied to a wide range of fields.
- the viscosity average molecular weight is less than 20,000, the fluidity is excellent, and among the resin windows for vehicles, it is suitable for complex shapes and large resin molded products (for example, back door windows), and the viscosity average molecular weight is 20,000 or more. If it is, it will be excellent in strength and suitable for general resin windows for vehicles.
- the resin window for vehicles which is a preferred application of the present invention, it is necessary to select a molecular weight in accordance with the intended molded product. Since the resin plate of the present invention is thick, distortion at the time of molding is within an allowable limit even at a relatively high molecular weight.
- the upper limit of the viscosity average molecular weight is more preferably 35,000, still more preferably 30,000 from the viewpoint of versatility.
- the viscosity average molecular weight should just be satisfied as the whole polycarbonate resin, and the thing satisfying this range is included by the mixture of 2 or more types from which molecular weight differs.
- mixing of a polycarbonate having a viscosity average molecular weight exceeding 50,000 (more preferably 80,000 or more, and further preferably 100,000 or more) may be advantageous in terms of increasing entropy elasticity at the time of melting.
- the present invention is effective in suppressing jetting.
- the effect of improving the entropy elasticity becomes more prominent as the molecular weight of the polycarbonate is higher, but the upper limit of the molecular weight is practically 2 million, preferably 300,000, more preferably 200,000.
- a predetermined effect can be obtained without particularly impairing moldability.
- polycarbonate resins polymerized using other dihydric phenols and having high heat resistance or low water absorption include the following.
- BPM 4,4 ′-(m-phenylenediisopropylidene) diphenol
- BCF 9,9-bis (4-hydroxy-3-methylphenyl) fluorene
- the bisphenol A component is 10 to 95 mol% (more preferably 50 to 90 mol%, more preferably 60 to 85 mol%), And a copolycarbonate having a BCF component content of 5 to 90 mol% (more preferably 10 to 50 mol%, still more preferably 15 to 40 mol%).
- the BPM component is 20 to 80 mol% (more preferably 40 to 75 mol%, more preferably 45 to 65 mol%) in 100 mol% of the dihydric phenol component constituting the polycarbonate, and
- the 1,1-bis (4-hydroxyphenyl) -3,3,5-trimethylcyclohexane component is 20 to 80 mol% (more preferably 25 to 60 mol%, more preferably 35 to 55 mol%).
- bisphenol C 2,2-bis (4-hydroxy-3-methylphenyl) propane
- various heat-resistant polycarbonate resins polymerized using an aliphatic diol include polycarbonates in which the aliphatic diol constituting the polycarbonate is isosorbide, isomannide, or isoidide.
- isosorbide (1,4; 3,6-dianhydro-D-sorbitol) is particularly preferable because of its ease of production and excellent heat resistance.
- These special polycarbonates may be used alone or in combination of two or more. Moreover, these can also be mixed and used for the bisphenol A type polycarbonate generally used.
- the above polycarbonate resin can contain various conventionally known additives as long as the transparency is not impaired.
- additives include heat stabilizers, antioxidants, ultraviolet absorbers, light stabilizers, colorants, mold release agents, sliding agents, infrared absorbers, light diffusing agents, fluorescent whitening agents, and antistatic agents.
- examples include agents, flame retardants, flame retardant aids, plasticizers, reinforcing fillers, impact modifiers, photocatalytic antifouling agents, and photochromic agents.
- the heat stabilizer, antioxidant, ultraviolet absorber, light stabilizer, colorant, mold release agent, and the like can be blended with known appropriate amounts in conventional polycarbonate resins.
- thermoplastic resin layer (B layer)
- the thermoplastic resin used in the present invention is not particularly limited, polyolefin resin such as polyethylene and polypropylene, amorphous polyolefin resin such as polydicyclopentadiene, acrylic resin such as polycarbonate resin and polymethyl methacrylate, polyethylene terephthalate, polybutylene terephthalate, poly Examples thereof include polyester resins such as (ethylene-2,6-naphthalate), polystyrene, polyarylate, polyethersulfone, polyetheretherketone, polyimide, phenolic resin, and urea resin.
- polycarbonate resins having excellent transparency acrylic resins such as polymethyl methacrylate, polyester resins such as polyethylene terephthalate, polybutylene terephthalate, poly (ethylene-2,6-naphthalate), polystyrene, polypropylene, polyarylate, polyethersulfone Is preferred.
- acrylic resins such as polymethyl methacrylate
- polyester resins such as polyethylene terephthalate, polybutylene terephthalate, poly (ethylene-2,6-naphthalate), polystyrene, polypropylene, polyarylate, polyethersulfone Is preferred.
- the thickness of the thermoplastic resin layer is preferably 50 ⁇ m or more and 200 ⁇ m or less, and more preferably 60 ⁇ m or more and 180 ⁇ m or less. Above the lower limit, it is preferable because sufficient weather resistance and scratch resistance can be obtained, and below the upper limit, the environmental resistance decreases due to the moisture absorption rate of the thermoplastic resin layer, and further, the impact resistance does not decrease, and bending This is preferable because cracks are less likely to occur in the substrate.
- the thermoplastic resin layer used in the present invention is formed using a thermoplastic resin composition having ultraviolet absorption performance.
- the organic ultraviolet absorber is preferable for improving weather resistance, and among them, a benzotriazole-based or triazine-based ultraviolet absorber having a low photolysis rate and excellent durability is preferable. Of these, triazine-based ultraviolet absorbers are more preferable.
- the amount of the organic ultraviolet absorber contained in the thermoplastic resin is preferably 0.3 to 35.0 parts by mass, and 1.0 to 25.0 parts by mass with respect to 100 parts by mass of the thermoplastic resin. More preferably, it is 2.0 to 15.0 parts by mass.
- the inorganic ultraviolet absorber is preferably metal oxide fine particles.
- at least one metal oxide fine particle selected from titanium oxide fine particles, cerium oxide fine particles, and zinc oxide fine particles has a relatively small band gap and absorption.
- the light energy is preferably converted to energy that does not deteriorate the organic resin, and zinc oxide fine particles are particularly preferable because of high transparency of the coating film and excellent dispersibility in a polar solvent.
- the metal oxide fine particles are preferably contained in an amount of 0.1 to 16.0 parts by mass, more preferably 1.0 to 8.0 parts by mass, and still more preferably 2 to 100 parts by mass of the thermoplastic resin. It is preferably contained in an amount of 0.0 to 6.0 parts by mass. If it is at least the lower limit, good weather resistance can be obtained, and it is preferable to be at most the upper limit, since the adhesion between the thermoplastic resin layer and the organosiloxane resin layer is good.
- the ultraviolet absorbing group is bonded to a part of the thermoplastic resin skeleton.
- Specific examples include a polymer obtained by copolymerizing a (meth) acryl monomer having an ultraviolet absorbing group and another copolymerizable (meth) acryl monomer.
- the (meth) acrylic monomer having an ultraviolet absorbing group is preferably a benzophenone type, benzotriazole type, or benzotriazine type monomer, and more preferably a benzotriazole type monomer.
- the amount of the (meth) acrylic monomer having an ultraviolet absorbing group is preferably 1 to 40% by mass, particularly 3 to 25% by mass, based on the copolymer composition. If it is less than 1% by mass, good UV absorption ability cannot be obtained, and if it exceeds 40% by mass, the glass transition temperature of the (meth) acrylic copolymer is lowered, and the silicone hard coat layer (C layer) on the surface. In some cases, cracks are likely to occur, and whitening may occur during moisture resistance.
- the thermoplastic resin layer used in the present invention may contain a (meth) acrylic rubber component for the purpose of imparting impact resistance and bending workability.
- (Meth) acryl refers to methacryl or acrylic.
- the (meth) acrylic rubber component is a polymer having a (meth) acrylic acid ester monomer unit and a polyfunctional monomer unit as essential components.
- the (meth) acrylic acid ester monomer is preferably a (meth) acrylic acid alkyl ester having an alkyl group having 1 to 12 carbon atoms.
- methyl (meth) acrylate, ethyl (meth) acrylate, propyl (meth) acrylate, (n-butyl) (meth) acrylate, isobutyl (meth) acrylate, (t-butyl) (meth) acrylate, (Meth) acrylic acid-2-ethylhexyl and the like can be used, and those having an alkyl group with 1 to 8 carbon atoms are particularly preferred. These can be used alone or in combination of two or more.
- the content of the acrylate monomer unit in 100% by mass of the (meth) acrylic rubber component is preferably 75% by mass or more, more preferably 85% by mass or more, and particularly preferably 95% by mass or more.
- the content of the (meth) acrylic acid ester monomer unit is less than the above lower limit, the weather resistance, impact resistance, rigidity of the (meth) acrylic rubber rubber component and the thermoplastic resin layer (B layer) to be obtained. Any of the appearances may be degraded.
- the total content of the polyfunctional monomer units is 0.3 to 3 parts by mass with respect to 100 parts by mass of the (meth) acrylic acid ester monomer units, 2 parts by mass or less is preferable, 1.5 parts by mass or less is particularly preferable, and 0.4 parts by mass or more is preferable and 0.5 parts by mass or more is particularly preferable.
- the polyfunctional monomer unit content exceeds the above upper limit, the impact resistance of the obtained (meth) acrylic rubber component and the thermoplastic resin layer (B layer) may be lowered, and when the content is less than the above lower limit. The appearance may be degraded.
- the polyfunctional monomer unit may have a plurality of unsaturated bonds, but a polyfunctional monomer unit having two unsaturated bonds and a polyfunctional monomer having three unsaturated bonds. It is preferable that it contains an ionic monomer unit. When a polyfunctional monomer having 4 or more unsaturated bonds is present, the resulting (meth) acrylic rubber component may be deteriorated in appearance or gelled.
- Polyfunctional monomers include allyl (meth) acrylate, ethylene glycol di (meth) acrylate, 1,3-butanediol di (meth) acrylate, 1,6-hexanediol di (meth)
- examples thereof include di (meth) acrylic acid esters of diols such as acrylic acid esters, 2-propenyl (meth) acrylate, divinylbenzene and the like.
- allyl (meth) acrylate having an allyl group, 2- (meth) acrylic acid 2- Propenyl and the like are preferred, and allyl (meth) acrylate is particularly preferred from the viewpoint of improving the physical properties of the resulting resin composition.
- Examples of the polyfunctional monomer having 3 or more unsaturated bonds include triallyl isocyanurate having an aromatic ring, triallyl cyanurate, triallyl trimetate, and the like. Among them, triallyl isocyanurate having a triazine ring, Triallyl cyanurate is preferred, and triallyl isocyanurate is particularly preferred from the viewpoint of polymerization stability.
- polyfunctional monomers having two unsaturated bonds and the polyfunctional monomers having three unsaturated bonds may be used alone or in combination of two or more. Also good.
- (meth) acrylic rubber component in addition to the acrylic acid ester monomer and the polyfunctional monomer, other monomers that can be copolymerized with the acrylic acid ester monomer are used if necessary. be able to.
- (meth) acrylic acid ester monomers include aromatic vinyl monomers such as styrene, ⁇ -methylstyrene, p-methylstyrene, acrylonitrile, methacrylonitrile, etc. And unsaturated nitrile monomers. These can be used alone or in combination of two or more.
- the (meth) acrylic rubber component is preferably produced by emulsion polymerization of the above monomer mixture.
- the emulsifier used in the emulsion polymerization is not limited, but an anionic emulsifier is preferred from the viewpoint of excellent stability of the latex during emulsion polymerization and an increase in the polymerization rate.
- An emulsifier may be used individually by 1 type, and may mix and use 2 or more types.
- the volume average particle diameter of the (meth) acrylic rubber component is preferably 100 nm or more, and more preferably 200 nm or more. When the volume average particle diameter is less than the lower limit, the impact resistance of the obtained (meth) acrylic rubber-based fine particles and the thermoplastic resin layer (B layer) may be lowered. Further, the upper limit of the volume average particle diameter is preferably 900 nm or less, and more preferably 600 nm or less. When the volume average particle diameter exceeds the above upper limit, the appearance of the resulting (meth) acrylic rubber component and the thermoplastic resin layer (B layer) may deteriorate.
- the (meth) acrylic rubber component may be graft polymerized with a vinyl monomer.
- the vinyl monomer used for the graft polymerization preferably includes an unsaturated nitrile monomer and an aromatic vinyl monomer, and other monomers as required.
- unsaturated nitrile monomers examples include acrylonitrile and methacrylonitrile. These can be used alone or in combination of two or more.
- aromatic vinyl monomer examples include styrene, ⁇ -methylstyrene, vinyltoluene and the like. These can be used alone or in combination of two or more.
- Other monomers are monomers copolymerizable with unsaturated nitrile monomers and aromatic vinyl monomers, and unsaturated nitrile monomers and aromatic vinyl monomers are Exclude monomer.
- Other monomers include methyl (meth) acrylate, ethyl (meth) acrylate, n-butyl (meth) acrylate, 2-ethylhexyl (meth) acrylate, glycidyl (meth) acrylate, (meth) Examples thereof include N, N-dimethylaminoethyl acrylate, (meth) acrylamide, maleic anhydride, and N-substituted maleimide. Also about another monomer, 1 type may be used independently and 2 or more types may be used together.
- an aromatic vinyl monomer such as styrene and an unsaturated nitrile monomer such as acrylonitrile are used because the resulting molded article has excellent impact resistance.
- a monomer mixture with a monomer is preferable, and a mixture of styrene and acrylonitrile is particularly preferable.
- the addition amount of the (meth) acrylic rubber component is preferably 0 to 30 parts by mass, more preferably 0.1 to 20 parts by mass with respect to 100 parts by mass of the resin component of the thermoplastic resin. If the addition amount of the (meth) acrylic rubber component in the thermoplastic resin is not more than the upper limit, good appearance and fluidity of the resulting thermoplastic resin layer (B layer) can be maintained.
- thermoplastic resin may contain other components such as organic dyes, inorganic dyes, pigments, antioxidants, antistatic agents, and surfactants as necessary.
- the hard coat layer used in the present invention is formed by coating and curing a specific silicone hard coat composition.
- the silicone hard coat composition is at least one selected from (c-1) a core-shell type tetragonal titanium oxide solid solution dispersion, (c-2) a specific alkoxysilane and a partial hydrolysis condensate thereof.
- (C-3) a silicone resin obtained by (co) hydrolysis / condensation, (c-3) a curing catalyst, (c-4) a solvent, and, if necessary, (c-5) colloidal silica, and (c-1)
- the solid content of the core-shell type tetragonal titanium oxide solid solution dispersion of component (1) is preferably 1 to 30% by mass with respect to the solid content of the silicone resin of component (c-2).
- Component (c-1) The component (c-1) is composed of tetragonal titanium oxide fine particles in which one or more atoms of the group consisting of cobalt, tin, and manganese are solid-solubilized, and silicon oxide is formed outside the nucleus.
- the volume-based 50% cumulative distribution diameter (D 50 ) of the core particles measured by dynamic light scattering using light is 50 nm or less, and the volume-based 50% cumulative distribution diameter (D 50 ) of the core-shell type fine particles.
- tin and manganese are preferred as the solid solution atoms, and the solid solution amounts of tin (Ti / Sn) are 10 to 1,000 and manganese (Ti / Mn) 10 to 1 in terms of molar ratio with titanium, respectively. 1,000 is preferred. More preferably, tin (Ti / Sn) is 20 to 200, and manganese (Ti / Mn) is 20 to 200.
- the solid solution mode of cobalt, tin, and manganese may be a substitution type or an interstitial type.
- the substitution type is a solid solution mode formed by substitution of solid solution atoms at the titanium (IV) ion sites of titanium oxide
- the interstitial type is between the crystal lattices of titanium oxide. This is a solid solution mode formed by the presence of solid solution atoms.
- an F center that causes coloration is easily formed, and since the symmetry around the metal ion is poor, the Frank-Condon factor of the vibronic transition in the metal ion is increased, and visible light is easily absorbed. Therefore, the substitution type is preferable.
- the shell of silicon oxide formed outside the core of tetragonal titanium oxide fine particles in which cobalt, tin, and manganese are dissolved contains silicon oxide as the main component and contains other components such as zirconium, tin, and aluminum. It may be formed by any method.
- the silicon oxide shell can be formed by hydrolytic condensation of tetraalkoxysilane.
- tetraalkoxysilane those usually available such as tetramethoxysilane, tetraethoxysilane, tetra (n-propoxy) silane, tetra (i-propoxy) silane, tetra (n-butoxy) silane may be used.
- Tetraethoxysilane is preferably used from the viewpoint of reactivity and safety.
- commercially available “KBE-04” manufactured by Shin-Etsu Chemical Co., Ltd.
- the hydrolysis and condensation of tetraalkoxysilane may be performed in water, and a condensation catalyst such as ammonia, aluminum salt, organoaluminum, tin salt, or organotin may be used as appropriate.
- Ammonia is used as a dispersant for the core fine particles. It is particularly preferable because it also has the above action.
- the volume-based 50% cumulative distribution diameter (D 50 ) of the core-shell type tetragonal titanium oxide solid solution measured by a dynamic light scattering method using laser light is 100 nm or less, More preferably, it is 50 nm or less.
- D 50 value of the core fine particles and the core-shell type solid solution exceeds the upper limit, the dispersion is not preferable because it is opaque.
- D 50 volume-based 50% cumulative distribution diameter
- average particle diameter for example, Nanotrac UPA-EX150 (manufactured by Nikkiso Co., Ltd.) ), LA-910 (manufactured by HORIBA, Ltd.) and the like.
- Examples of the dispersion medium for dispersing the core-shell type tetragonal titanium oxide solid solution include one or more dispersion media of the group consisting of water, alcohol, ether, ester, and ketone. Of these, water, alcohol, and a mixed solvent thereof are preferable. As water, for example, deionized water (ion exchange water), distilled water, pure water and the like are preferable. As the alcohol, for example, lower alcohols such as methanol, ethanol and isopropanol are preferable. Among these, deionized water and pure water are most preferable from the viewpoints of productivity and cost.
- the concentration of the core-shell type tetragonal titanium oxide solid solution is 0.1 to 30% by mass. Preferably, it is 5 to 15% by mass.
- This dispersion medium is allowed to contain a basic substance (dispersant) used in the production process of the core-shell type tetragonal titanium oxide solid solution.
- the basic substance has properties as a pH adjuster and a dispersant, it may be used in an aqueous solution having an appropriate concentration together with the dispersion medium.
- the core-shell type tetragonal titanium oxide solid solution dispersion does not contain a dispersant other than the basic substance, for example, a polymer dispersant.
- a dispersant other than the basic substance for example, a polymer dispersant.
- a polymer dispersing agent that has been conventionally used as a dispersing agent for titanium oxide fine particles by containing the basic substance.
- Examples of basic substances (dispersants) in such a core-shell type tetragonal titanium oxide solid solution dispersion include ammonia, alkali metal hydroxides, phosphate compounds, hydrogen phosphate compounds, carbonate compounds and bicarbonate compounds. is there.
- the core-shell type tetragonal titanium oxide solid solution dispersion having such a configuration has high transparency.
- the transmittance of light having a wavelength of 550 nm passing through is usually 80% or more, more preferably 85% or more, and still more preferably 90% or more. Such transmittance can be easily obtained by measuring an ultraviolet-visible transmission spectrum.
- Such a core-shell type tetragonal titanium oxide solid solution dispersion (c-1) component is one in which methylene blue is added and no fading is observed after irradiation with black light, and the photocatalytic activity is blocked.
- magenta blue fading test 0.5 mass% core-shell type tetragonal titanium oxide solid solution dispersion added to methylene blue to 0.01 mmol / L was placed in a borosilicate glass bottle and black light (UV Irradiation intensity: 0.5 mW / cm 2 ) can be performed by colorimetry after irradiation for 24 hours, and can be confirmed by a decrease rate of absorbance at 653 nm.
- the rate of decrease in absorbance after this fading test is preferably within 10%. If it exceeds 10%, in the weather resistance test, the surface of the cured film (C layer) formed from the silicone hard coat composition is partially decomposed due to photocatalytic activity, and causes curing shrinkage. As a result, cracks are likely to occur early.
- the solid content of the core-shell type tetragonal titanium oxide solid solution dispersion as the component (c-1) is 1 to 30% by mass, preferably 3 to 3%, based on the solid content of the silicone resin as the component (c-2) described later. It is blended so as to be 20% by mass, more preferably 5 to 15% by mass. If it is less than 1% by mass, it is insufficient for imparting ultraviolet shielding ability to the composition, and if it exceeds 30% by mass, the coating film formed from the composition tends to undergo curing shrinkage, causing cracks. Therefore, it is not preferable.
- a method for producing a core-shell type tetragonal titanium oxide solid solution dispersion in which one or more atoms of the group consisting of cobalt, tin, and manganese having the above-described structure are formed as a solid solution includes the following steps (A) to (D). .
- ⁇ Process (A) In this step, first, an aqueous dispersion of tetragonal titanium oxide solid solution fine particles in which one or more atoms of the group consisting of cobalt, tin, and manganese are dissolved in tetragonal titanium oxide is prepared.
- the method for obtaining this aqueous dispersion is not particularly limited, but the raw material titanium compound, cobalt compound, tin compound, manganese compound, basic substance and hydrogen peroxide are reacted in an aqueous dispersion medium to obtain cobalt, After obtaining a peroxotitanic acid solution containing one or more atoms of the group consisting of tin and manganese, this is hydrothermally treated to form a tetragonal system containing one or more atoms of the group consisting of cobalt, tin and manganese. A method of obtaining a titanium oxide fine particle dispersion is preferred.
- the reaction until obtaining a peroxotitanic acid solution containing one or more atoms of the group consisting of cobalt, tin, and manganese in the preceding stage is performed by adding a basic substance to a raw material titanium compound in an aqueous dispersion medium to form titanium hydroxide.
- the raw material titanium compound for example, inorganic acid salts such as titanium hydrochloride, nitrate and sulfate, organic acid salts such as formic acid, citric acid, succinic acid, lactic acid and glycolic acid, alkalis are added to these aqueous solutions.
- inorganic acid salts such as titanium hydrochloride, nitrate and sulfate
- organic acid salts such as formic acid, citric acid, succinic acid, lactic acid and glycolic acid, alkalis are added to these aqueous solutions.
- examples thereof include titanium hydroxide precipitated by hydrolysis by addition, and one or more of these may be used in combination.
- Hydrogen peroxide is used to convert the above raw material titanium compound or titanium hydroxide into peroxotitanium, that is, a titanium oxide compound containing a Ti—O—O—Ti bond. used.
- the amount of hydrogen peroxide added is preferably 1.5 to 5 times the total number of moles of Ti, Co, Sn, and Mn.
- the reaction temperature in the reaction of adding hydrogen peroxide to convert the raw material titanium compound or titanium hydroxide to peroxotitanic acid is preferably 5 to 60 ° C., and the reaction time is 30 minutes to 24 hours. It is preferable.
- the peroxotitanic acid solution thus obtained may contain a basic substance or an acidic substance for pH adjustment or the like.
- the basic substance herein include the above-described ammonia
- the acidic substance include inorganic acids such as sulfuric acid, nitric acid, hydrochloric acid, carbonic acid, phosphoric acid, and hydrogen peroxide, and formic acid, Organic acids such as citric acid, succinic acid, lactic acid and glycolic acid can be mentioned.
- the pH of the obtained peroxotitanic acid solution is preferably 1 to 7, particularly 4 to 7, from the viewpoint of handling safety.
- the reaction until obtaining a tetragonal titanium oxide fine particle dispersion in which one or more atoms of the group consisting of cobalt, tin, and manganese are dissolved in the latter stage is carried out by using the peroxotitanic acid solution at a pressure of 0.01-4. It is subjected to a hydrothermal reaction under the conditions of 0.5 MPa, preferably 0.15 to 4.5 MPa, temperature 80 to 250 ° C., preferably 120 to 250 ° C., and reaction time 1 minute to 24 hours. As a result, peroxotitanic acid is converted into tetragonal titanium oxide fine particles in which one or more atoms of the group consisting of cobalt, tin, and manganese are dissolved.
- the tetraalkoxy titanium oxide fine particle dispersion in which one or more atoms of the group consisting of cobalt, tin, and manganese thus obtained are solid-dissolved is added to tetraalkoxy such as monohydric alcohol, ammonia, and tetraethoxysilane. Add silane.
- the monohydric alcohol methanol, ethanol, propanol, isopropyl alcohol, and any mixture thereof are used, and ethanol is particularly preferably used.
- a monohydric alcohol is used in an amount of 100 parts by mass or less, preferably 50 parts by mass or less, based on 100 parts by mass of the titanium oxide fine particle dispersion.
- by changing the blending amount of the monohydric alcohol it becomes possible to control the thickness of the silicon oxide shell formed on the outside of the nucleus composed of the solid tetragonal titanium oxide fine particles in the next step.
- the solubility of the silicon reactant such as tetraalkoxysilane in the reaction system is increased while the dispersion state of titanium oxide is not adversely affected.
- the core-shell type tetragonal titanium oxide solid solution dispersion in which at least one atom of the group consisting of cobalt, tin, and manganese obtained in the next step is dissolved is a mechanical unit operation such as pulverization and classification in the manufacturing step. In spite of not having passed, it can be set as the range of the said specific cumulative particle size distribution diameter, and the transparency in a visible part can be provided.
- Ammonia is ammonia water, and may be replaced by addition of ammonia water by blowing ammonia gas into a solid solution of tetragonal titanium oxide fine particle dispersion, and a reactant capable of generating ammonia in the dispersion. It may replace with the addition of ammonia water by adding.
- the concentration of the ammonia water is not particularly limited, and any commercially available ammonia water may be used. In the process of the present invention, for example, the pH of the solid solution of tetragonal titanium oxide fine particles dispersed with 28% by mass of concentrated aqueous ammonia is 9 to 12, more preferably 9.5 to 11.5. It is preferable to add ammonia water up to the amount.
- tetraalkoxysilane those described above can be used, but tetraethoxysilane is preferable.
- tetraethoxysilane (partial) hydrolyzate can also be used as tetraethoxysilane.
- Such a tetraethoxysilane or a (partial) hydrolyzate of tetraethoxysilane may be any commercially available product.
- trade name “KBE-04” tetraethoxysilane: manufactured by Shin-Etsu Chemical Co., Ltd.
- trade names “silicate 35", “silicate 45” tetrahydrosilane partial hydrolysis condensate: manufactured by Tama Chemical Industry Co., Ltd.
- trade names "ESI40”, “ESI48” tetraethoxysilane partial hydrolysis) Condensate: Colcoat Co., Ltd.
- These tetraethoxysilanes or the like may be used alone or in combination.
- the compounding amount of tetraalkoxysilane is 20 to 50% by mass, preferably 25 to 45% by mass, more preferably 30 to 40% by mass with respect to titanium oxide containing silicon oxide after hydrolysis.
- the amount is less than 20% by mass, the formation of the shell is insufficient.
- the amount is more than 50% by mass, the aggregation of the particles is promoted and the dispersion may become opaque.
- the method of adding monohydric alcohol, ammonia and tetraalkoxysilane such as tetraethoxysilane to the solid solution of the tetragonal titanium oxide fine particle dispersion and mixing them may be carried out by any method, for example, magnetic Stirring, mechanical stirring, shaking stirring, etc. can be used.
- the method for rapidly heating the mixture obtained in the step (A) may be any existing method, such as microwave heating, a microreactor capable of achieving high heat exchange efficiency, and an external having a large heat capacity. Heat exchange with a heat source or the like can be used. In particular, a heating method using a microwave is preferable because it allows uniform and rapid heating. In addition, the process of irradiating and heating microwaves may be a batch process or a continuous process.
- the time from reaching room temperature to just below the boiling point of the dispersion medium is within 10 minutes. This is because when the heating method exceeds 10 minutes, the particles are aggregated, which is not preferable.
- the frequency can be appropriately selected from electromagnetic waves having a frequency of 300 MHz to 3 THz.
- the microwave frequency band that can be normally used is determined by the Radio Law as 2.45 GHz, 5.8 GHz, 24 GHz, etc.
- 2.45 GHz is often used for consumer use. Therefore, the magnetron for oscillation of this frequency is advantageous in terms of equipment cost.
- this standard depends on the laws and economic conditions of specific countries and regions, and technically does not limit the frequency.
- Any commercially available apparatus may be used as long as the microwave output has a rating of 100 W to 24 kW, preferably 100 W to 20 kW.
- ⁇ ReactorEx manufactured by Shikoku Keiki Kogyo Co., Ltd.
- Advanced manufactured by Biotage Co., Ltd.
- the output of the microwave is adjusted, or the reaction liquid volume is adjusted appropriately for batch reactions and the reaction flow rate is adjusted for continuous reactions. Can be done.
- ⁇ Process (C) water is removed from the core-shell type tetragonal system titanium oxide solid solution dispersion obtained in the step (B) and concentrated.
- the concentration method may be any existing method, for example, atmospheric concentration, vacuum concentration, azeotropic dehydration, ultrafiltration, reverse osmosis, lyophilization, etc. It is easy to dry, and if the temperature is low, the dispersion may freeze. In the inorganic fine particle dispersion, the state change is not always reversible, and the state change or contact with the solvent may lead to the quality of the dispersion. From this point, it is preferable to use at least one selected from normal pressure concentration, reduced pressure concentration, and ultrafiltration, and in particular, reduced pressure concentration under a pressure of 50 mmHg or less is preferable because of mild conditions.
- the concentration of the core-shell type tetragonal titanium oxide solid solution (solid content) in the core-shell type tetragonal titanium oxide solid solution dispersion is 5 to 20% by mass, preferably 8 to 17% by mass, more preferably 10%. Concentrate to ⁇ 15 wt%.
- the amount is less than 5% by mass, the amount of water becomes too large and the composition cannot be balanced, which is not suitable.
- the amount is more than 20% by mass, the stability of the dispersion deteriorates and gels with time. This is not preferable because there is a possibility.
- ammonia is further removed from the core-shell type tetragonal titanium oxide solid solution dispersion obtained in the step (C).
- Any existing method may be used for removing ammonia.
- ion exchange, adsorption or the like is used, and removal of ammonia by a cation exchange resin is particularly preferable.
- Any commercially available cation exchange resin may be used.
- Amberlite IR120B manufactured by Organo Corporation
- Amberlite 200CT manufactured by Organo Corporation
- Amberlite IR124 organo Corporation
- Amberlite FPC3500 manufactured by Organo Corporation
- Amberlite IRC76 manufactured by Organo Corporation
- Diaion SK104 manufactured by Mitsubishi Chemical Corporation
- Diaion PK208 manufactured by Mitsubishi Chemical Corporation
- the cation exchange resin used in the step of removing ammonia is removed by filtration.
- Filtration is sufficient if the purpose of separating the ion exchange resin and the core shell dispersion is achieved. Filtration is usually treated as a classification operation in the mechanical unit operation, but the filtration in this case is not involved in the classification of the core-shell particles. Therefore, any type of mesh or qualitative filter paper may be used as long as it is coarse and efficiently allows the core-shell dispersion to pass through.
- the concentration of ammonia in the core-shell type tetragonal titanium oxide solid solution dispersion is 0.1% by mass or less, preferably 0.05% by mass or less, more preferably 0.01% by mass or less. Remove.
- the amount is more than 0.1% by mass, the action of silicone as a condensation catalyst becomes remarkable in the composition, and as a result, cracks are caused in the silicone hard coat film, which is not preferable.
- Component (c-2) is represented by the following general formula (1): (R 1 ) m (R 2 ) n Si (OR 3 ) 4-mn (1) (Wherein R 1 and R 2 are each independently a hydrogen atom or a substituted or unsubstituted monovalent hydrocarbon group, R 3 is an alkyl group having 1 to 3 carbon atoms, m, n Are each independently 0 or 1, and m + n is 0, 1 or 2.) (Co) hydrolysis of at least one selected from alkoxysilanes and partial hydrolysis condensates thereof -A silicone resin obtained by condensation.
- R 1 and R 2 are a hydrogen atom or a substituted or unsubstituted monovalent hydrocarbon group, preferably having 1 to 12 carbon atoms, particularly 1 to 8 carbon atoms, such as a hydrogen atom; a methyl group Alkyl group such as ethyl group, propyl group, butyl group, pentyl group, hexyl group, heptyl group, octyl group; cycloalkyl group such as cyclopentyl group, cyclohexyl group; alkenyl group such as vinyl group, allyl group; phenyl group, etc.
- a halogen-substituted hydrocarbon group such as a chloromethyl group, ⁇ -chloropropyl group, 3,3 ′, 3 ′′ -trifluoropropyl group; ⁇ -methacryloxypropyl group, ⁇ -glycidoxypropyl group, Substitution of (meth) acryloxy, epoxy, mercapto, amino group such as 3,4-epoxycyclohexylethyl group, ⁇ -mercaptopropyl group, ⁇ -aminopropyl group, etc. And the like can be exemplified hydrocarbon groups.
- an alkyl group is preferable when used for applications requiring particularly scratch resistance and weather resistance
- epoxy and (meth) acryloxy-substituted hydrocarbon groups are preferable when toughness and dyeability are required. .
- R 3 is an alkyl group having 1 to 3 carbon atoms, and examples thereof include a methyl group, an ethyl group, an n-propyl group, and an i-propyl group. Among these, a methyl group and an ethyl group are preferable in consideration of the high reactivity of the hydrolysis condensation and the high vapor pressure of the alcohol R 3 OH to be produced and the ease of distilling off.
- At least one selected from the alkoxysilane of the general formula (1) and its partial hydrolysis-condensation product can be (co) hydrolyzed and condensed by a known method. That's fine.
- an alkoxysilane or a partially hydrolyzed condensate thereof alone or in a mixture is (co) hydrolyzed with water having a pH of 1 to 7.5, preferably 2 to 7.
- a material in which fine metal oxide particles such as silica sol are dispersed in water may be used.
- metal oxide fine particles such as silica sol dispersed in water or an organic solvent may coexist at the time of hydrolysis.
- the hydrolysis / condensation reaction may be carried out by mixing water, an acidic hydrolysis catalyst, and an alkoxysilane in the presence of the component (c-1).
- the component (c-1) may be partially reacted with the hydrolysis condensate of (c-2) and / or (c-2). Is more preferable. That is, by adding (c-1) during the reaction step, as a result, (c-1) is surface-treated by the binder component itself, and the dispersibility is further improved.
- the storage stability may be insufficient even though temporary dispersion stability of the inorganic particles can be secured.
- the silicone used for the coating and the silicone used for the binder may differ in composition (primary structure), and even if the composition is the same, the degree of condensation, molecular weight distribution, etc. are strictly defined (secondary structure) This is because it is common that the silicone is not the same in (1).
- the secondary structure is the same, so the compatibility is improved and the dispersibility is also improved.
- the amount of water used may be in the range of 20 parts by weight to 3,000 parts by weight of water with respect to 100 parts by weight of the total of alkoxysilane and / or its partial hydrolysis condensate.
- the amount of water used may be in the range of 20 parts by weight to 3,000 parts by weight of water with respect to 100 parts by weight of the total of alkoxysilane and / or its partial hydrolysis condensate.
- Hydrolysis may be performed by dropping or adding water into alkoxysilane or a partial hydrolysis condensate thereof, or conversely dropping or adding alkoxysilane or a partial hydrolysis condensate thereof into water.
- an organic solvent may be contained.
- condensation may be performed continuously following hydrolysis, and is usually performed under heating at a liquid temperature of room temperature or 100 ° C. Gelation may occur at temperatures higher than 100 ° C. Furthermore, condensation can be accelerated
- an organic solvent may be added for the purpose of adjusting the progress and concentration of the condensation, and the metal oxide fine particles such as silica sol dispersed in water or an organic solvent, Component (c-1) may be added.
- silicone resin undergoes condensation, increases in molecular weight, and decreases in solubility in water and generated alcohol. Therefore, as an organic solvent to be added, silicone resin is well dissolved and has a boiling point of 80 ° C. or higher.
- the organic solvent having a relatively high polarity is preferred.
- organic solvents include alcohols such as isopropyl alcohol, n-butanol, isobutanol, t-butanol and diacetone alcohol; methylpropyl ketone, diethyl ketone, methyl isobutyl ketone, cyclohexanone, diacetone alcohol and the like.
- Ketones such as dipropyl ether, dibutyl ether, anisole, dioxane, ethylene glycol monoethyl ether, ethylene glycol monobutyl ether, propylene glycol monomethyl ether, propylene glycol monomethyl ether acetate; propyl acetate, butyl acetate, cyclohexyl acetate, etc.
- ethers such as dipropyl ether, dibutyl ether, anisole, dioxane, ethylene glycol monoethyl ether, ethylene glycol monobutyl ether, propylene glycol monomethyl ether, propylene glycol monomethyl ether acetate; propyl acetate, butyl acetate, cyclohexyl acetate, etc.
- the blending amount of the organic solvent may be an amount sufficient to dissolve the silicone resin, but it is often in the range of 100 to 1,000% by mass with respect to the solid content of the silicone resin.
- the coating composition contains This is not preferable because the resin concentration as an active ingredient becomes thin and it becomes difficult to form a good film.
- the component (c-2) When the (silane) component (c-2) is obtained by (co) hydrolytic condensation of the alkoxysilane of formula (1) and / or its partial hydrolysis-condensation product, the component (c-1)
- the core-shell type tetragonal titanium oxide solid solution dispersion may be (co) hydrolyzed by adding to the alkoxysilane and / or its partially hydrolyzed condensate, and the (c-5) component colloidal silica described later may be used. When used, this colloidal silica may be added to the (co) hydrolytic condensation system.
- Component (c-3) is a curing catalyst that accelerates the reaction of condensable groups such as silanol groups and alkoxy groups contained in the silicone resin of component (c-2).
- condensable groups such as silanol groups and alkoxy groups contained in the silicone resin of component (c-2).
- sodium propionate, sodium acetate, sodium formate, trimethylbenzylammonium hydroxide, tetramethylammonium hydroxide, tris (acetylacetonato) aluminum, and diisopropoxy (ethylacetoacetate) aluminum are particularly preferable.
- the amount of component (c-3) is not particularly limited as long as it is an amount effective for curing the silicone resin of component (c-2).
- the content is preferably 0.0001 to 30% by mass, more preferably 0.001 to 10% by mass, based on the solid content. If it is less than 0.0001% by mass, curing may be insufficient and the hardness may decrease, and if it is more than 30% by mass, cracks may easily occur in the coating film, and water resistance may decrease.
- the component (c-4) is a solvent and is not particularly limited as long as it dissolves or disperses the components (c-1) to (c-3).
- a highly polar organic solvent is preferably the main solvent.
- the organic solvent include alcohols such as methanol, ethanol, isopropyl alcohol, n-butanol, isobutanol, t-butanol, diacetone alcohol; methyl propyl ketone, diethyl ketone, methyl isobutyl ketone, cyclohexanone, diacetone alcohol Ketones such as dipropyl ether, dibutyl ether, anisole, dioxane, ethylene glycol monoethyl ether, ethylene glycol monobutyl ether, propylene glycol monomethyl ether, propylene glycol monomethyl ether acetate, etc .; ethyl acetate, propyl acetate, butyl acetate And esters such as
- the addition amount of the component (c-4) it is preferable to use an amount that makes the solid content concentration of the silicone hard coat composition 1 to 30% by mass, particularly 5 to 25% by mass. Outside this range, defects may occur in the coating film coated and cured with the composition. For example, if the concentration is less than the above range, sagging, twisting, and spotting tend to occur in the coating film, and the desired hardness and scratch resistance may not be obtained. On the other hand, if the concentration exceeds the above range, the coating film There is a risk that brushing, whitening, and cracks are likely to occur.
- the component (c-5) is colloidal silica, and can be added in an appropriate amount when it is desired to particularly improve the hardness and scratch resistance of the coating film.
- Nanosized silica having a particle size of about 5 to 50 nm is colloidally dispersed in a medium of water or an organic solvent, and a commercially available water dispersion or organic dispersion type can be used. Specific examples include Snowtex-O, OS, OL, methanol silica sol manufactured by Nissan Chemical Industries, Ltd.
- the amount of colloidal silica added is 0 to 100 parts by weight, preferably 5 to 100 parts by weight, particularly 5 to 50 parts by weight, based on 100 parts by weight of the silicone resin solid content of component (Q).
- silicone hard coat compositions pH adjusters, leveling agents, thickeners, pigments, dyes, metal oxide fine particles, metal powders, antioxidants, UV absorbers, UV stabilizers as necessary , Heat ray reflection / absorption imparting agent, flexibility imparting agent, antistatic agent, antifouling property imparting agent, water repellency imparting agent and the like can be added within a range that does not adversely affect the purpose and effect of the present invention. .
- the silicone hard coat composition can be obtained by mixing the above components (c-1) to (c-5) and, if necessary, other components according to a conventional method.
- a normal application method can be used, for example, various application methods such as brush coating, spraying, dipping, flow coating, roll coating, curtain coating, spin coating, knife coating and the like. You can choose.
- the curing after applying the silicone hard coat composition may be left in the air and air-dried, or may be heated.
- the curing temperature and the curing time are not limited, it is preferable to heat at a temperature lower than the heat resistant temperature of the substrate for 10 minutes to 2 hours. Specifically, heating at 80 to 135 ° C. for 30 minutes to 2 hours is more preferable.
- the thickness of the hard coat layer (C layer) is 0.5 to 30 ⁇ m, preferably 1 to 25 ⁇ m, particularly 1.5 to 20 ⁇ m. If it is less than 0.5 ⁇ m, good scratch resistance cannot be obtained, and if it exceeds 30 ⁇ m, cracks may be generated or bending resistance may be lowered.
- thermoplastic resin layer (B layer) on the surface of the polycarbonate resin layer (A layer)
- a method by molding rather than coating may be used.
- the insert molding method or the coextrusion method is preferably performed, and the coextrusion method is more preferable.
- the specific example of a laminated body manufacturing process is described below.
- thermocompression bonding method any method can be adopted as the thermocompression bonding method.
- a method of thermocompression bonding an acrylic resin film and a polycarbonate resin layer with a laminating machine or a press machine, or a method of thermocompression bonding the acrylic resin film to the polycarbonate resin layer immediately after extrusion In particular, a method of continuously thermocompression bonding to a polycarbonate resin layer immediately after extrusion is industrially advantageous.
- the thermocompression bonding conditions in this case vary depending on the thickness of the polycarbonate resin layer or the acrylic resin film, the state of the crimping surface, etc., and cannot be specified unconditionally.
- a linear pressure of 0.05 kg / cm or more, preferably about 1 to 10 kg / cm is applied at a glass transition point of ⁇ 10 ° C. to a glass transition point + 150 ° C., preferably at a glass transition point of ⁇ 5 ° C. to a glass transition point + 100 ° C.
- Thermocompression bonding is possible.
- thermoplastic resin film is inserted between male and female molds for injection molding, and a molten resin is injected from one side of the mold for injection. At the same time that the molded body is formed, the above-mentioned film is bonded to the molded body, or a thermoplastic resin film is pre-shaped by vacuum molding or pressure molding, etc., and then inserted into an injection mold.
- a molten resin is injected and integrally molded with a thermoplastic resin film.
- an extrusion apparatus used for the production of a laminate generally one main extruder for extruding a polycarbonate resin constituting a substrate layer and one or more sub-extrusions for extruding a thermoplastic resin constituting a coating layer.
- the sub-extruder is usually smaller than the main extruder.
- the temperature of the sub-extruder is appropriately set depending on the type of thermoplastic resin.
- the temperature condition of the main extruder is usually 230 to 290 ° C., preferably 240 to 280 ° C.
- the temperature condition of the sub-extruder is usually 220 to 270 ° C., preferably 230 to 260 ° C.
- a known method such as a feed block method or a multi-manifold method can be used.
- the molten resin laminated in the feed block is guided to a sheet forming die such as a T die, and after being formed into a sheet shape, the molten resin flows into a forming roll (polishing roll) whose surface is mirror-finished. Form.
- This sheet-like molded product is mirror-finished and cooled while passing through the molding roll, thereby forming a laminate.
- the molten resin laminated in the die is similarly formed into a sheet shape inside the die, and then surface finish and cooling are performed with a forming roll to form a laminated plate.
- the die temperature is usually 220 to 280 ° C., preferably 230 to 270 ° C.
- the forming roll temperature is usually 100 to 190 ° C., preferably 110 to 180 ° C.
- a vertical roll or a horizontal roll can be appropriately used.
- the thickness of the laminate of the polycarbonate resin layer (A layer) and the thermoplastic resin layer (B layer) obtained by coextrusion is 0.5 mm to 20.0 mm, more preferably 1.0 mm to 15.0 mm. 1.5 mm to 10.0 mm is particularly preferable. Above the lower limit, the bending with respect to the load from the outside becomes small, which is preferable for outdoor use, and below the upper limit, it is preferable because the outside can be seen without distortion without inhibiting the reduction in thickness and weight.
- the polycarbonate resin laminate of the present invention may have a printing layer in addition to the above steps.
- a printed layer is formed at the peripheral edge in glazing, and has a blinding function for the adhesive and the structural member formed at the peripheral edge. It also has design and design functions for building materials.
- the printing layer may be formed on any one or both sides of the A layer or the B layer.
- various methods such as various printing methods, spray coating, and brush coating can be applied.
- the printing method is not particularly limited, and printing can be performed on a flat or curved sheet surface by a conventionally known method. For example, methods such as spray printing, offset printing, flexographic printing, gravure printing, screen printing, and ink jet printing are exemplified, and among these, screen printing is most preferably applied.
- the polycarbonate resin laminate of the present invention can be trimmed, drilled, and attached with peripheral members in addition to the above steps to form a final part or final product as a resin glazing.
- peripheral member include a frame, a pin, a screw, a fastener, a cushioning material, a seal material, a hinge, and a lock mechanism.
- Such peripheral members are attached using fixing means such as adhesion, adhesion, screwing, welding, fitting, ultrasonic welding, and laser welding.
- the polycarbonate resin laminate of the present invention can be attached to a final product such as a vehicle body using various fixing means, similarly to the attachment of the peripheral member described above. In the case of glazing, gluing is applied as the most preferred means for such attachment.
- gluing is applied as the most preferred means for such attachment.
- any of hard adhesive, semi-rigid adhesive, and elastic adhesive can be used, but in the present invention, an elastic adhesive that is excellent as a structural adhesive is preferable, and in particular, a urethane-based elastic adhesive is used.
- the sealing performance, strength and cost are preferable.
- the adhesive layer may be formed on the polycarbonate resin layer, on the thermoplastic resin layer having ultraviolet absorption performance, on the printed layer, or on the hard coat layer.
- the layer once laminated may be removed by various chemical treatments, blasting, polishing, and cutting methods to form an adhesive layer.
- the surface layer of the portion where the adhesive layer is provided can be made a predetermined layer by filling the plate without printing and in the formation of the hard coat layer mainly by masking.
- a urethane-based elastic adhesive which is a suitable immobilizing means
- a primer for urethane adhesives preferably has a polyisocyanate compound as a main component on the base material layer and the printed layer, and is usually a commercially available product called a body primer or a coating film primer. Available to: On the other hand, on the hard coat layer, those containing a polyisocyanate compound and a silane compound as main components are preferable, and what is usually called a glass primer can be suitably used.
- the urethane adhesive primer is generally mixed with a solvent, a filler, a catalyst, a desiccant, a resin component, and optionally other compounds.
- the polyisocyanate compound preferably contains a polyisocyanate containing an aromatic ring as a main component. Such polyisocyanate compounds are excellent in reactivity. More preferably, at least one polyisocyanate compound selected from the group consisting of MDI, TDI, triphenylmethane-4,4,4-triisocyanate, and tris (p-isocyanatephenyl) thiophosphate is used for adhesion. In 100 mol% of the polyisocyanate compound in the primer, preferably 50 mol% or more, more preferably 55 to 90 mol%.
- aliphatic polyisocyanate compounds and alicyclic polyisocyanate compounds can be used.
- examples of such compounds include isocyanurate-modified products of TDI and HDI, adduct-modified products of HDI and trimethylolpropane, isocyanurate-modified products of HDI, and isocyanurate-modified products of IPDI.
- Isocyanurate modified products are preferred because the reactivity can be easily adjusted.
- the primer applied on the resin base material layer and the printing ink layer is preferably a combination of MDI, tris (p-isocyanatephenyl) thiophosphate, and an isocyanurate modified product of TDI and HDI. It is preferable that (p-isocyanatophenyl) thiophosphate is in the range of 50 to 70 mol% in a total of 100 mol% of these three.
- a silane coupling agent and a reaction product of a silane compound and a polyisocyanate compound are preferably used as main components.
- the silane coupling agent various conventionally known agents can be used.
- epoxy group-containing silane coupling agents such as ⁇ -glycidoxypropyltrimethoxysilane, and N- ( ⁇ -aminoethyl) - ⁇ -An amino group-containing silane coupling agent such as aminopropylmethyldimethoxysilane is preferably used in combination.
- a vinyl group-containing silane coupling agent such as vinyltrimethoxysilane as necessary.
- reaction product of the silane compound and the polyisocyanate compound a reaction product of the above various polyisocyanate compounds and a mercapto group-containing alkoxysilane compound such as ⁇ -mercaptopropyltrimethoxysilane can be used.
- polyisocyanate compounds include aliphatic polyisocyanate compounds represented by HDI, alicyclic polyisocyanate compounds represented by IPDI, and adduct modified products, isocyanurate modified products, and biuret modified products.
- the primer layer for adhesion is formed by applying a primer composition using various applicators and drying at normal temperature.
- a coating method for example, a brush coating method, a spray coating method, a wire bar method, a blade method, and a roll coating method can be used.
- the thickness of the adhesion primer layer is preferably in the range of 2 to 40 ⁇ m, more preferably 3 to 30 ⁇ m, and more preferably 5 to 20 ⁇ m.
- urethane adhesive in the present invention either a moisture curable one-component urethane adhesive or a two-component urethane adhesive can be used, but the moisture curable one-component urethane adhesive is particularly excellent in production efficiency. This is preferable.
- Moisture-curable one-component urethane adhesives are usually composed mainly of isocyanate group-containing compounds, especially isocyanate group-terminated urethane prepolymers (hereinafter referred to as NCO-terminated prepolymers), and plasticizers, fillers, and catalysts. , And optionally other compounds.
- the other compounds are intended to impart desired properties to the composition, and include, for example, adhesives such as polyisocyanate compounds and silane coupling agents such as ⁇ -mercaptopropyltrimethoxysilane, heat resistance, and the like. It includes a (meth) acrylate copolymer for imparting adhesiveness, and a foaming agent or microballoon for imparting lightness, vibration damping and soundproofing.
- the content of the prepolymer is usually preferably 15 to 50% by mass, more preferably 20 to 45% by mass, and further preferably 30 to 45% by mass in the total amount of the urethane adhesive composition. Is done.
- Representative examples of preferred embodiments of the urethane adhesive composition include various adhesives for direct glazing such as WS-222 manufactured by Yokohama Rubber Co., Ltd. and # 560 manufactured by Sunstar Giken Co., Ltd. Is done.
- bending is applied as the most effective means in such attachment.
- the tolerance for bending of the polycarbonate laminate is larger because it can cope with a final product having a complicated shape.
- the tolerance for bending of the laminate is preferably 100 or less, more preferably R90 or less, and still more preferably 80 or less, when a three-point bending test is performed in accordance with JIS K7171. It is preferable that The lower limit is preferably as small as possible, and is not particularly limited, but is usually 50 or more.
- the limit stress after being held at 80 ° C. for 114 hours is preferably 18 MPa or more, more preferably 20 MPa or more, and further preferably 22 MPa or more.
- the critical stress after being held at 110 ° C. for 21 hours is preferably 9 MPa or more, more preferably 10 MPa or more, and still more preferably 11 MPa or more.
- the upper limit is preferably as large as possible and is not particularly limited, but is usually 54 MPa or less.
- the polycarbonate resin laminate can be bent forcibly at room temperature according to the shape of the final product frame, the flexibility of design of the outdoor structure including the roof and its roof is possible. Therefore, it can be used for general purposes and suitable for outdoor structures.
- ⁇ Adhesion of hard coat layer> After making 100 squares with 11 cuts at 1 mm intervals horizontally and vertically with a cutter on the hard coat layer, and attaching cello tape (registered trademark) (adhesive tape made by Nichiban Co., Ltd.) to this eye, It peeled at a stretch in the direction of 90 °. The hard coat layer did not peel off, and the number of remaining squares was counted.
- ⁇ Weather resistance test> The test piece was exposed to the super xenon test (UV irradiation intensity 180 W / m 2 , black panel temperature 63 ° C.) for 1000 hours and 6000 hours, and the test piece was taken out to determine the yellowness (YI), crack property, adhesion, and appearance. evaluated.
- YI yellowness
- ⁇ Yellowness> In accordance with JIS Z8722, measurement was performed with an integrating sphere spectrophotometer CE-7000A (manufactured by X-Rite), and the yellowing difference ( ⁇ YI) before and after the weather resistance test was measured.
- ⁇ Weather crack resistance> The appearance after the weather resistance test (the presence or absence of cracks) was evaluated according to the following criteria.
- 1, 1 is the center of the ellipse
- 2 is the major axis radius of the ellipse (10 cm)
- 3 is the minor axis radius of the ellipse (4 cm)
- 4 is the width of the jig (4 cm)
- 5 is the presser fitting (each 1 cm wide)
- 6 is the horizontal distance (cm) from the center of the ellipse to the portion where the crack is generated with the least strain
- 7 is the sample (polycarbonate resin laminate)
- 8 is the portion where the crack is generated with the least strain.
- thermoplastic resin used for thermoplastic resin layer (B layer) III-1) Manufacture of acrylic resin-B1, B3, B5 Commercially available acrylic resin (Mitsubishi Rayon Co., Ltd. polymethyl methacrylate resin (VH001) ))It was used. Further, an acrylic resin film (B5) having a thickness of 100 ⁇ m was obtained by extrusion through a T-shaped die having a set temperature of 230 ° C.
- AIBN azobisisobutyronitrile
- Precipitation of titanium hydroxide containing tin and manganese by gradually adding 300 parts by mass of 5% by mass of ammonia water (manufactured by Wako Pure Chemical Industries, Ltd.) to this metal salt aqueous solution mixture, neutralizing and hydrolyzing. I got a thing.
- the pH of the titanium hydroxide slurry at this time was 8.
- the resulting precipitate of titanium hydroxide was deionized by repeatedly adding ion-exchanged water and decanting. 100 parts by mass of 30% by mass hydrogen peroxide (manufactured by Wako Pure Chemical Industries, Ltd.) is gradually added to the titanium hydroxide precipitate containing tin and manganese after the deionization treatment, and then at 60 ° C. for 3 hours.
- the average particle diameter of this titanium oxide dispersion (i) is 50% cumulative on a volume basis measured by a dynamic light scattering method using a laser beam (manufactured by Nikkiso Co., Ltd., apparatus name “Nanotrack UPA-EX150”).
- the particle size distribution diameter (D 50 ) was 14 nm.
- the reaction vessel after heating was cooled to room temperature with a water bath.
- the content liquid was taken out into a round bottom flask and concentrated by vacuum batch distillation. After concentration, it was brought into contact with 10 parts by mass of Amberlite 200CT (manufactured by Organo Corporation) for 3 hours.
- This mixture was filtered with a filter paper (Advantec 2B) to separate the ion exchange resin, and a core-shell type titanium oxide solid solution aqueous dispersion (CS-i) was obtained as a filtrate.
- a certain amount of dispersion (CS-i) is precisely weighed (using AUX-220 manufactured by Shimadzu Corporation) and treated in an oven at 105 ° C (using a perfect oven manufactured by Espec) for 3 hours to volatilize the dispersion medium. As a result, it was revealed that the solid content was 15% by mass. After the dispersion (CS-i) was diluted so that the solid content was 1% by mass, the average particle size (D 50 ) was measured in the same manner as the titanium oxide dispersion (i). It turns out that.
- KP-341 manufactured by Shin-Etsu Chemical Co., Ltd.
- acetic acid as a pH adjuster
- tetrabutylammonium hydroxide concentration 10% by weight aqueous solution
- Wako Pure Chemical Industries, Ltd., special grade 2.5 parts by mass was added, stirred at room temperature, filtered through a filter paper, and a silicone resin hard coat composition having a non-volatile content of 20.9% by mass (S1) Got.
- V-2 Preparation of a silicone resin hard coat composition (S2) containing a core-shell type titanium oxide solid solution in which cobalt atoms are dissolved (4 mol% of cobalt with respect to 100 mol% of titanium).
- S2 silicone resin hard coat composition
- a silicone resin hard coat composition S2 containing a core-shell type titanium oxide solid solution in which cobalt atoms are dissolved (4 mol% of cobalt with respect to 100 mol% of titanium.
- Snowtex O manufactured by Nissan Chemical Industries, Ltd .: water-dispersed silica sol, average particle size of 15 to 20 nm, SiO 2 containing 20% by mass Article
- 100 parts by mass and 44.2 parts by mass of a 0.25N acetic acid aqueous solution were dropped, and hydrolysis was carried out while cooling so that the internal temperature did not exceed 40 ° C.
- RTTDNB15WT% -E88 manufactured by CIK Nanotech Co., Ltd .: a coating containing alumina and zirconia with titanium oxide fine particles (cobalt 4 mol% with respect to 100 mol% titanium) prepared by direct current arc plasma method.
- the average particle diameter (D 50 ) of the titanium oxide dispersion (RTTDNB15WT% -E88) and the core-shell type titanium oxide dispersion before the silicon oxide coating on the surface was measured in the same manner as (V-1). However, they were 40.0 nm and 82.0 nm, respectively.
- the RTTDNB15WT% -E88 used at this time was diluted with methanol so that the solid content was 1% by mass, the UV-visible transmittance spectrum was measured, and the transmittance at 550 nm was 90%, maintaining good transparency. It was.
- methylene blue (Wako special grade) was added to the dispersion after dilution with methanol so that the solid content was 0.5% by mass, and the resulting dispersion was added to 0.01 mmol / L. (Intensity: 0.5 mW / cm 2 , measured using Iwasaki Electric Co., Ltd., eye ultraviolet illuminance meter UVP365-1) for 24 hours after color irradiation, the rate of decrease in absorbance was 8%. It was.
- V-3 Preparation of Silicone Resin Hard Coat Composition (T1) Containing Core-Shell Type Titanium Oxide Solid Solution (Titanium 100 mol%) that does not Dissolve Other Atoms Tin (IV) Chloride Pentahydrate and Chloride Without using manganese (II) tetrahydrate, the same operation as in the above (V-1) was performed to obtain a core-shell type titanium oxide aqueous dispersion (CS-ii) in which other atoms were not dissolved. .
- a certain amount of dispersion (CS-ii) is precisely weighed (using AUX-220 manufactured by Shimadzu Corporation) and treated in an oven at 105 ° C (using a perfect oven manufactured by ESPEC) for 3 hours to volatilize the dispersion medium. As a result, it was revealed that the solid content was 15% by mass.
- the average particle diameter (D 50 ) is measured by the same method as (V-1). As a result, they were 20 nm and 34.6 nm, respectively. Further, when the dispersion (CS-ii) was diluted so that the solid content was 1% by mass and then the ultraviolet-visible transmittance spectrum was measured, the transmittance at 550 nm was maintained at 90%, which was good transparency. Further, methylene blue (Wako special grade) was added to a 0.5 mass% core-shell type titanium oxide aqueous dispersion so as to have a concentration of 0.01 mmol / L.
- methyltrimethoxysilane 136 parts by mass of methyltrimethoxysilane was charged into a 2 L flask, cooled to a liquid temperature of about 10 ° C., then Snowtex O (manufactured by Nissan Chemical Industries, Ltd .: water-dispersed silica sol, average particle size of 15 to 20 nm, 20 mass% SiO 2 containing product) 100 parts by mass, core shell type titanium oxide dispersion (CS-ii) 44.2 parts by mass was added. When the mixed solution was added, self-heating due to hydrolysis was observed, and the internal temperature rose to 50 ° C. After completion of the addition, the mixture was stirred at 60 ° C. for 3 hours to complete the hydrolysis.
- Snowtex O manufactured by Nissan Chemical Industries, Ltd .: water-dispersed silica sol, average particle size of 15 to 20 nm, 20 mass% SiO 2 containing product
- CS-ii core shell type titanium oxide dispersion
- KP-341 manufactured by Shin-Etsu Chemical Co., Ltd.
- acetic acid as a pH adjuster
- tetrabutylammonium hydroxide concentration 10% by weight aqueous solution
- Wako Pure Chemical Industries, Ltd. special grade
- the average particle diameter (D 50 ) of ZNTAB15WT% -E16 (2) measured by the same method as in (V-1) was 45.0 nm and 105.0 nm, respectively, and the decrease rate of methylene blue colorimetric absorbance was 24%. there were.
- KP-341 manufactured by Shin-Etsu Chemical Co., Ltd.
- acetic acid as a pH adjuster
- tetramethylammonium hydroxide concentration 20% by weight aqueous solution
- Wako Pure Chemical Industries, Ltd., special grade 1.8 parts by mass was added, stirred at room temperature, filtered through a filter paper, and a silicone resin hard coat composition (T3) having a nonvolatile content concentration of 19.4% by mass Got.
- UV curable acrylate hard coat agent (T4) 100 parts by mass of polyfunctional acrylate oligomer (Shin Nakamura Chemical Co., Ltd. U-15HA), phenyl-1-hydroxycyclohexyl ketone (BASF Co., Ltd.) Irgacure 184) 5 parts by mass, 1-methoxy-2-propanol 250 parts by mass, 2-propanol 100 parts by mass, organic solvent-dispersed colloidal silica (Snowtex O-40, solid content concentration 40 mass%, manufactured by Nissan Chemical Industries, Ltd.) 29 2 parts by mass were mixed to obtain an ultraviolet curable acrylate hard coat agent (T4).
- the ratio of colloidal silica and / or alkoxysilane hydrolysis condensate was 30% by mass.
- melamine resin hard coat agent (T5) Methylated methylolmelamine [Nippon Cytec Industries, Ltd. Cymel 301] 100 parts by mass, 1.6-hexanediol 70 parts by mass, maleic acid 5 parts by mass, 150 parts by mass of isopropyl alcohol, 320 parts by mass of isobutyl alcohol, and 25 parts by mass of ethylene glycol monobutyl ether were mixed to obtain a melamine resin hard coat agent (T5).
- the ratio of colloidal silica and / or alkoxysilane hydrolysis condensate was 0% by mass.
- Example 1 On the B layer of the coextruded base material prepared by using (A1) and (B1) described in (II) and (III) above, the hard coat agent (S1) prepared in (V) is subjected to thermosetting. It was applied by a flow coating method so that the film thickness was 4.0 ⁇ m, allowed to stand at 25 ° C. for 15 minutes and then thermally cured at 120 ° C. for 1 hour to obtain a PC resin laminate. The results are shown in Table 2.
- Example 2 On the B layer of the coextruded base material prepared using (A1) and (B1) described in (II) and (III) above, the hard coat agent (S2) prepared in (V) is subjected to heat curing. It was applied by a flow coating method so that the film thickness was 4.0 ⁇ m, allowed to stand at 25 ° C. for 15 minutes and then thermally cured at 120 ° C. for 1 hour to obtain a PC resin laminate. The results are shown in Table 2.
- Example 3 On the B layer of the coextruded base material prepared using (A1) and (B2) described in (II) and (III) above, the hard coating agent (S1) prepared in (V) is subjected to heat curing. It was applied by a flow coating method so that the film thickness was 4.0 ⁇ m, allowed to stand at 25 ° C. for 15 minutes, and then thermally cured at 120 ° C. for 1 hour to obtain a PC resin laminate. The results are shown in Table 2.
- Example 4 On the B layer of the coextruded base material prepared using (A1) and (B3) described in (II) and (III) above, the hard coating agent (S1) prepared in (V) is subjected to heat curing. It was applied by a flow coating method so that the film thickness was 4.0 ⁇ m, allowed to stand at 25 ° C. for 15 minutes and then thermally cured at 120 ° C. for 1 hour to obtain a PC resin laminate. The results are shown in Table 2.
- Example 5 The base material layer prepared in (IV-1) above is set in the lower chamber of a vacuum hot press molding machine (MP-22S manufactured by Meiki Seisakusho), and the acrylic resin film (B5) prepared in (III) above is placed in the upper chamber. Set. The temperature of the lower chamber and the upper chamber was set to 180 ° C. and 120 ° C., respectively, and when the base material layer and the acrylic resin film were sufficiently heated, pressurization was performed at 4 MPa for 90 seconds to obtain a laminated base material. The hard coating agent (S1) prepared in (V) is applied on the layer B of the laminated base material by a flow coating method so that the film thickness after thermosetting becomes 4.0 ⁇ m, and left at 25 ° C. for 15 minutes. Then, it heat-cured at 120 degreeC for 1 hour, and obtained the PC resin laminated body. The results are shown in Table 2.
- S1 and S2 which have good ultraviolet shielding properties
- S1 and S2 have no problem even in a weather resistance test of 6,000 hours as shown in Tables 2 and 3, but in T1, they are solid as component (c-1). Since an unsolubilized core-shell type tetragonal titanium oxide dispersion is used, cracks and peeling occur at 6,000 hours. From this, it was found that the effect of improving the weather resistance by the core-shell type tetragonal titanium oxide solid solution dispersion as the component (c-1) was remarkable.
- the laminate of the present invention is excellent in scratch resistance, ultraviolet shielding properties, outdoor durability, and bending workability, so it is used outdoors such as vehicles, airplane windows, windshields, building windows, road sound insulation walls, etc. It can be used for a wide range of applications such as applications. Therefore, the industrial effect of the outdoor application of the present invention is exceptional.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Wood Science & Technology (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Environmental & Geological Engineering (AREA)
- General Life Sciences & Earth Sciences (AREA)
- Geology (AREA)
- Inorganic Chemistry (AREA)
- Laminated Bodies (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Description
[1]ポリカーボネート樹脂層(A層)の片面又は両面に紫外線吸収性性能を有する熱可塑性樹脂層(B層)を成形積層した積層基材に、コバルト、スズ、マンガンからなる群の1以上の原子を固溶した正方晶系酸化チタン微粒子を核とし、該核の外側に酸化ケイ素もしくは酸化アルミニウムの殻を有するコアシェル型正方晶系酸化チタン固溶体を、水、アルコール、エーテル、エステル、ケトンからなる群の1以上の分散媒中に分散したコアシェル型正方晶系酸化チタン固溶体分散液であって、レーザー光を用いた動的光散乱法で測定した上記核微粒子の体積基準の50%累積分布径(D50)が50nm以下で、上記コアシェル固溶体の体積基準の50%累積分布径(D50)が100nm以下であり、前記コバルト、スズ、マンガンからなる群の全固溶量(M)が、チタンとのモル比(Ti/M)で10~1,000の固溶体分散液を含むシリコーンハードコート組成物を塗布硬化してなるシリコーンハードコート層(C層)を形成したポリカーボネート樹脂積層体。
[2]積層基材が、ポリカーボネート樹脂層(A層)及び紫外線吸収性性能を有する熱可塑性樹脂層(B層)を共押出法、熱圧着法、又はインサートモールド成形法のいずれかの方法により成形積層したものである上記[1]に記載のポリカーボネート樹脂積層体。
[3]紫外線吸収性性能を有する熱可塑性樹脂層(B層)は、紫外線吸収剤としてベンゾトリアゾール類又はトリアジン類を含む熱可塑性系樹脂からなることを特徴とする上記[1]又は[2]に記載のポリカーボネート樹脂積層体。
[4]紫外線吸収性性能を有する熱可塑性樹脂層(B層)は、(メタ)アクリル系樹脂組成物を用いて形成されたものであることを特徴とする上記[1]~[3]のいずれかに記載のポリカーボネート樹脂積層体。
[5]紫外線吸収性性能を有する熱可塑性樹脂層(B層)は、(メタ)アクリル系ゴム成分を含むことを特徴とする上記[1]~[4]のいずれかに記載のポリカーボネート樹脂積層体。
[6]ハードコート層(C層)は、下記成分(c-1)~(c-5)を含むシリコーンハードコート組成物の硬化被膜であること特徴とする上記[1]~[5]のいずれかに記載のポリカーボネート樹脂積層体。
(c-1)コバルト、スズ、マンガンからなる群の1以上の原子を固溶した正方晶系酸化チタン微粒子を核とし、該核の外側に酸化ケイ素もしくは酸化アルミニウムの殻を有するコアシェル型正方晶系酸化チタン固溶体を、水、アルコール、エーテル、エステル、ケトンからなる群の1以上の分散媒中に分散したコアシェル型正方晶系酸化チタン固溶体分散液であって、レーザー光を用いた動的光散乱法で測定した上記核微粒子の体積基準の50%累積分布径(D50)が50nm以下で、上記コアシェル固溶体の体積基準の50%累積分布径(D50)が100nm以下であり、前記コバルト、スズ、マンガンからなる群の全固溶量(M)が、チタンとのモル比(Ti/M)で10~1,000の固溶体分散液、
(c-2)下記一般式(1):
(R1)m(R2)nSi(OR3)4-m-n (1)
(式中、R1及びR2は、各々独立に、水素原子、又は置換もしくは非置換の一価炭化水素基であり、置換基同士が相互に結合していてもよく、R3は、炭素数1~3のアルキル基であり、m,nは、各々独立に、0又は1であり、且つm+nは、0,1又は2である。)
で表されるアルコキシシラン及びその部分加水分解縮合物から選ばれる少なくとも1種を(共)加水分解・縮合することにより得られたシリコーンレジン、
(c-3)硬化触媒、
(c-4)溶剤、
(c-5)必要によりコロイダルシリカ
を含み、且つ(c-1)成分のコアシェル型正方晶系酸化チタン固溶体分散液の固形分が、(c-2)成分のシリコーンレジンの固形分に対して、1~30質量%である。
[7](c-5)成分のコロイダルシリカの配合量が、(c-2)成分のシリコーンレジン固形分100質量部に対し5~100質量部であることを特徴とする上記[6]に記載のポリカーボネート樹脂積層体。
[8](c-1)成分が、スズ及びマンガンを固溶した正方晶系酸化チタン微粒子を核とし、該核の外側に酸化ケイ素の殻を有するコアシェル型正方晶系酸化チタン固溶体を、水、アルコールからなる群の1以上の分散媒中に分散したコアシェル型正方晶系酸化チタン固溶体分散液であって、レーザー光を用いた動的光散乱法で測定した上記核微粒子の体積基準の50%累積分布径(D50)が30nm以下で、上記コアシェル固溶体の体積基準の50%累積分布径(D50)が50nm以下であり、前記スズ成分の固溶量が、チタンとのモル比(Ti/Sn)で10~1,000、前記マンガン成分の固溶量が、チタンとのモル比(Ti/Mn)で10~1,000である固溶体分散液であって、下記工程(A)から(D)を経て得られたものであることを特徴とする上記[6]又は[7]に記載のポリカーボネート樹脂積層体。
(A)スズ及びマンガンを固溶した正方晶系酸化チタン微粒子分散液に、一価アルコール、アンモニア、及びテトラアルコキシシランを配合する工程、
(B)前記(A)の工程で得られた混合物を急速加熱することにより、スズ及びマンガンを固溶した正方晶系酸化チタン微粒子を核とし該核の外側に酸化ケイ素の殻を有するコアシェル型正方晶系酸化チタン固溶体を形成させる工程、
(C)前記(B)の工程で得られたコアシェル型正方晶系酸化チタン固溶体水分散液から水を除去して濃縮する工程、及び
(D)アンモニアを除去する工程
[9]JIS K7171に準拠して3点曲げ試験を実施した際に、クラックが入る曲率半径Rが100mm以下である上記[1]~[8]のいずれかに記載のポリカーボネート樹脂積層体。
[10]上記[1]~[9]のいずれかに記載のポリカーボネート樹脂積層体の熱可塑性樹脂層(B層)又はハードコート層(C層)上に接着用プライマー層及び弾性接着剤層を順次介して構造部材が接着取り付けられてなる接着構成体。
(i)ポリカーボネート樹脂層(A層)
本発明に用いるポリカーボネート樹脂は、ビスフェノールA型ポリカーボネートが好適に用いられ、またそれ以外にも、他の二価フェノールを用いて重合された高耐熱性又は低吸水率の各種のポリカーボネート樹脂や、脂肪族ジオールを用いて重合された高耐熱性の各種のポリカーボネート樹脂であってもよい。ポリカーボネート樹脂はいかなる製造方法によって製造されたものでもよく、界面重縮合の場合は通常一価フェノール類の末端停止剤が使用される。ポリカーボネート樹脂は3官能フェノール類を重合させた分岐ポリカーボネート樹脂であってもよく、更に脂肪族ジカルボン酸や芳香族ジカルボン酸、又は二価の脂肪族又は脂環族アルコールを共重合させた共重合ポリカーボネートであってもよい。ポリカーボネート樹脂の粘度平均分子量は13,000~40,000の範囲であると、幅広い分野に適用可能となる。粘度平均分子量が20,000未満であると流動性に優れ、車両用樹脂窓の中でも、複雑な形状や大型の樹脂成型品(例えばバックドアウィンドウ)に好適となり、粘度平均分子量が20,000以上であると強度に優れ、車両用樹脂窓全般に好適となる。本発明の好適な用途である車両用樹脂窓においては、目的とする成型品に合わせて分子量の選択が必要である。本発明の樹脂板は厚肉であるため、比較的高い分子量においても成形時の歪みは許容限度内となる。粘度平均分子量の上限は、汎用性の点からより好ましくは35,000、更に好ましくは30,000である。
ηSP/c=[η]+0.45×[η]2c(但し[η]は極限粘度)
[η]=1.23×10-4M0.83
c=0.7
本発明に用いる熱可塑性樹脂は、特に制限はなく、ポリエチレン、ポリプロピレン等のポリオレフィン樹脂、ポリジシクロペンタジエン等のアモルファスポリオレフィン樹脂、ポリカーボネート樹脂、ポリメチルメタクリレート等のアクリル樹脂、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリ(エチレン-2,6-ナフタレート)等のポリエステル樹脂、ポリスチレン、ポリアリレート、ポリエーテルスルホン、ポリエーテルエーテルケトン、ポリイミド、フェノール樹脂、尿素樹脂などが挙げられる。中でも優れた透明性を有するポリカーボネート樹脂、ポリメチルメタクリレート等のアクリル樹脂、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリ(エチレン-2,6-ナフタレート)等のポリエステル樹脂、ポリスチレン、ポリプロピレン、ポリアリレート、ポリエーテルスルホンが好ましい。具体的には 三菱レイヨン(株)製「VH001」、「IRG304」、ダイセル・エボニック(株)製「hw55」、アルケマ(株)製「HT121」、クラレ製「パラペットHR-L」、「パラペットGR01240」、「パラペットGR01270」、旭化成ケミカルズ(株)製「PM120N」等の市販品を用いることができる。
中でもトリアジン系紫外線吸収剤がより好ましい。熱可塑性樹脂中に含有する有機系紫外線吸収剤の量は、熱可塑性樹脂100質量部に対して0.3~35.0質量部であることが好ましく、1.0~25.0質量部であることがより好ましく、2.0~15.0質量部であることが更に好ましい。
金属酸化物微粒子は、熱可塑性樹脂100質量部に対して0.1~16.0質量部含有することが好ましく、1.0~8.0質量部含有することがより好ましく、更に好ましくは2.0~6.0質量部含有することが好ましい。下限以上であると、良好な耐候性が得られるため好ましく、上限以下であると、熱可塑性樹脂層とオルガノシロキサン樹脂層の密着性が良好であるためより好ましい。
(メタ)アクリル系ゴム成分は、上記のような単量体混合物を乳化重合することにより製造されることが好ましい。
本発明に用いるハードコート層は、特定のシリコーンハードコート組成物を塗布硬化してなる。具体的には、シリコーンハードコート組成物は、(c-1)コアシェル型正方晶系酸化チタン固溶体分散液、(c-2)特定のアルコキシシラン及びその部分加水分解縮合物から選ばれる少なくとも1種を(共)加水分解・縮合することにより得られるシリコーンレジン、(c-3)硬化触媒、(c-4)溶剤、及び必要により(c-5)コロイダルシリカを含有し、且つ(c-1)成分のコアシェル型正方晶系酸化チタン固溶体分散液の固形分が、(c-2)成分のシリコーンレジンの固形分に対して、1~30質量%であるものを用いることが好ましい。
(c-1)成分は、コバルト、スズ、及びマンガンからなる群の1以上の原子を固溶した正方晶系酸化チタン微粒子を核とし、該核の外側に酸化ケイ素の殻を有するコアシェル型正方晶系酸化チタン微粒子を、水、アルコール、エーテル、エステル、ケトンからなる群の1以上の分散媒中に分散したコアシェル型正方晶系酸化チタン固溶体分散液であって、レーザー光を用いた動的光散乱法で測定した該核微粒子の体積基準の50%累積分布径(D50)が50nm以下で、該コアシェル型微粒子の体積基準の50%累積分布径(D50)が100nm以下であり、前記コバルト、スズ、及びマンガン各成分の合計固溶量(M)が、チタンとのモル比(Ti/M)で10~1,000であるコアシェル型正方晶系酸化チタン固溶体分散液である。
この工程では、先ず、コバルト、スズ、及びマンガンからなる群の1以上の原子が正方晶系酸化チタンに固溶している正方晶系酸化チタン固溶体微粒子の水分散体を用意する。この水分散体を得る方法は、特に限定されないが、原料となるチタン化合物、コバルト化合物、スズ化合物、マンガン化合物、塩基性物質及び過酸化水素を水性分散媒中で反応させて、一旦、コバルト、スズ、及びマンガンからなる群の1以上の原子を含有したペルオキソチタン酸溶液を得た後、これを水熱処理してコバルト、スズ、及びマンガンからなる群の1以上の原子を含有した正方晶系酸化チタン微粒子分散液を得る方法が好ましい。
ここでは、上記(A)の工程で得られた混合物を急速加熱することにより、固溶化した正方晶系酸化チタン微粒子を核とし、該核の外側に酸化ケイ素の殻を有するコアシェル型正方晶系酸化チタン固溶体の微粒子を形成させる。
ここでは、上記(B)の工程で得られたコアシェル型正方晶系酸化チタン固溶体分散液から水を除去して濃縮する。
濃縮方法は、既存のどのような方法でもよく、例えば、常圧濃縮、減圧濃縮、共沸脱水、限外ろ過、逆浸透、凍結乾燥等が用いられるが、濃縮時に高温であれば分散液が乾固しやすく、また低温であれば分散液が凍結するおそれがある。無機微粒子分散液では、状態変化が必ずしも可逆的ではなく、状態変化や溶媒との接触は、分散液の変質につながることがある。この点から、常圧濃縮、減圧濃縮、及び限外ろ過から選ばれる少なくとも1種を用いることが好ましく、特には、温和な条件である点から、50mmHg以下の圧力下での減圧濃縮が好ましい。
ここでは、上記(C)工程で得られたコアシェル型正方晶系酸化チタン固溶体分散液から、更にアンモニアを除去する。
アンモニアを除去する方法は、既存のどのような方法でもよく、例えば、イオン交換、吸着等が用いられ、特に、陽イオン交換樹脂によるアンモニアの除去が好ましい。
(c-2)成分は、下記一般式(1):
(R1)m(R2)nSi(OR3)4-m-n (1)
(式中、R1及びR2は、各々独立に、水素原子、又は置換もしくは非置換の一価炭化水素基であり、R3は、炭素数1~3のアルキル基であり、m,nは、各々独立に、0又は1であり、且つm+nは、0,1又は2である。)で表されるアルコキシシラン及びその部分加水分解縮合物から選ばれる少なくとも1種を(共)加水分解・縮合することにより得られるシリコーンレジンである。
(c-3)成分は、(c-2)成分のシリコーンレジン中に含まれる、シラノール基、アルコキシ基等の縮合可能な基が縮合する反応を促進する硬化触媒であり、例えば、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、ナトリウムメチラート、プロピオン酸ナトリウム、プロピオン酸カリウム、酢酸ナトリウム、酢酸カリウム、ギ酸ナトリウム、ギ酸カリウム、トリメチルベンジルアンモニウムヒドロキサイド、テトラメチルアンモニウムヒドロキサイド、テトラメチルアンモニウムアセテート、n-ヘキシルアミン、トリブチルアミン、ジアザビシクロウンデセン(DBU)、ジシアンジアミド等の塩基性化合物類;テトライソプロピルチタネート、テトラブチルチタネート、チタンアセチルアセトナート、アルミニウムトリイソブトキシド、アルミニウムトリイソプロポキシド、トリス(アセチルアセトナート)アルミニウム、ジイソプロポキシ(エチルアセトアセテート)アルミニウム、過塩素酸アルミニウム、塩化アルミニウム、コバルトオクチレート、コバルトアセチルアセトナート、鉄アセチルアセトナート、スズアセチルアセトナート、ジブチルスズオクチレート、ジブチルスズラウレート等の含金属化合物類;p-トルエンスルホン酸、トリクロル酢酸等の酸性化合物類などが挙げられる。この中で特にプロピオン酸ナトリウム、酢酸ナトリウム、ギ酸ナトリウム、トリメチルベンジルアンモニウムヒドロキサイド、テトラメチルアンモニウムヒドロキサイド、トリス(アセチルアセトナート)アルミニウム、ジイソプロポキシ(エチルアセトアセテート)アルミニウムが好ましい。
(c-4)成分は、溶剤であり、上記(c-1)~(c-3)成分を溶解する又は分散するものであれば特に限定されるものではないが、極性の高い有機溶剤が主溶剤であることが好ましい。有機溶剤の具体例としては、メタノール、エタノール、イソプロピルアルコール、n-ブタノール、イソブタノール、t-ブタノール、ジアセトンアルコール等のアルコール類;メチルプロピルケトン、ジエチルケトン、メチルイソブチルケトン、シクロヘキサノン、ジアセトンアルコール等のケトン類;ジプロピルエーテル、ジブチルエーテル、アニソール、ジオキサン、エチレングリコールモノエチルエーテル、エチレングリコールモノブチルエーテル、プロピレングリコールモノメチルエーテル、プロピレングリコールモノメチルエーテルアセテート等のエーテル類;酢酸エチル、酢酸プロピル、酢酸ブチル、酢酸シクロヘキシル等のエステル類などを挙げることができ、これらからなる群より選ばれた1種もしくは2種以上の混合物を使用することができる。
(c-5)成分は、コロイダルシリカであり、塗膜の硬度、耐擦傷性を特に高めたい場合、適量添加することができる。粒子径5~50nm程度のナノサイズのシリカが水や有機溶剤の媒体にコロイド分散している形態であり、市販されている水分散、有機分散タイプが使用可能である。具体的には、日産化学工業(株)製スノーテックス-O、OS、OL、メタノールシリカゾル等が挙げられる。コロイダルシリカの添加量は、(Q)成分のシリコーンレジン固形分100質量部に対し、0~100質量部、好ましくは5~100質量部、特に5~50質量部がよい。
シリコーンハードコート組成物には、必要に応じて、pH調整剤、レベリング剤、増粘剤、顔料、染料、金属酸化物微粒子、金属粉、酸化防止剤、紫外線吸収剤、紫外線安定剤、熱線反射・吸収性付与剤、可撓性付与剤、帯電防止剤、防汚性付与剤、撥水性付与剤等を本発明の目的や効果に悪影響を与えない範囲内で添加することができる。
ポリカーボネート樹脂層(A層)表面に熱可塑性樹脂層(B層)を積層するにはコーティングではなく、成形による方法を用いればよく、特に熱圧着法、インサートモールド成形法又は共押出法で行うことが好ましく、共押出法がより好ましい。積層体製造工程の具体例を以下に記す。
本発明のポリカーボネート樹脂積層体は上記の工程以外に印刷層を有していてもよい。かかる印刷層はグレージングにおいては周縁部に形成され、周縁部に形成される接着剤や構造部材の目隠し機能を有する。また建材用途などにおいては意匠、デザインの機能を有する。印刷層は、A層又はB層において、いずれか1面又は両面に形成されても構わない。印刷層をインキ塗工で形成する場合、かかる形成方法としては、各種の印刷方法、スプレー塗装、及び刷毛塗りなどの各種の方法が適用できる。印刷方法は特に限定されず、従来公知の方法で、平板のもしくは湾曲したシート表面に印刷できる。例えば、スプレー印刷、オフセット印刷、フレキソ印刷、グラビア印刷、スクリーン印刷、及びインクジェット印刷などの方法が例示され、これらの中でもスクリーン印刷が最も好ましく適用できる。
本発明のポリカーボネート樹脂積層体は、上述の周辺部材の取り付けと同様に、各種の固定化手段を用いて車体の如き最終製品に取り付けることができる。グレージングの場合かかる取り付けにおいては、接着が最も好適な手段として適用される。かかる接着方法には、硬質接着剤、半硬質接着剤、及び弾性接着剤のいずれも利用できるが、本発明では構造接着剤として優れている弾性接着剤が好ましく、特にウレタン系弾性接着剤が、そのシーリング性能、強度及びコストなどの点から好ましい。かかる接着層の形成は、ポリカーボネート樹脂層上、紫外線吸収性能を有する熱可塑性樹脂層上、印刷層上、及びハードコート層上のいずれに形成されてもよい。更に、一旦積層された層を各種化学処理、ブラスト、研磨、及び切削などの方法により除去し、接着層を形成してもよい。印刷層の形成においては版を埋めて印刷しないことにより、またハードコート層の形成では主としてマスキングを施すことにより、接着層を設ける部位の表面層を所定の層にすることができる。
また、限界応力性試験を実施した際は、80℃で114時間保持した後の限界応力が18MPa以上であることが好ましく、より好ましくは20MPa以上、更に好ましくは22MPa以上であることが好ましい。110℃で21時間保持した後の限界応力が9MPa以上であることが好ましく、より好ましくは10MPa以上、更に好ましくは11MPa以上であることが好ましい。なお、その上限は大きければ大きいほどよく、特に限定されるものではないが、通常54MPa以下である。
<透明性>
実施例で得られた樹脂積層体を、JIS K7361-1、JIS K7136に準拠して、全光線透過率値(Tt)、ヘイズ値(Haze)を評価した。
<耐擦傷性>
ASTM1044に準拠し、テーバー磨耗試験機にて摩耗輪CS-10Fを装着、荷重500g下での500回転後のヘイズ値(Haze)を濁度計(NDH2000、日本電色工業製)を用いて測定し、試験後と試験前のヘイズ値差(ΔHz)を測定した。
ハードコート層にカッターで1mm間隔に縦横に各11本の切れ目を入れて100個のマス目を作り、この目にセロテープ(登録商標)(ニチバン(株)製粘着テープ)を貼り付けた後、90°の方向に一気に剥した。ハードコート層が剥離せず、残ったマス目の数を数えた。
<沸水試験後の密着性>
試験片を沸騰水中に3時間浸漬した後に、目視にて外観観察、及び前記初期密着性と同様にして密着性試験を行った。
試験片をスーパーキセノンテスト(UV照射強度180W/m2、ブラックパネル温度63℃)にて1000時間、6000時間暴露し、試験片を取り出して黄色度(YI)、クラック性、密着性、外観を評価した。
<黄変度>
JIS Z8722に準拠し、積分球分光光度計CE-7000A(X-Rite社製)にて測定し、耐候性試験後と試験前の黄変度差(ΔYI)を測定した。
<耐候クラック性>
耐候性試験後の外観(クラックの有無)を下記の基準で評価した。
○:異常なし
△:僅かにクラックあり
×:塗膜全体にクラックあり
<耐候密着性>
耐候性試験後に、前記初期密着性と同様にして密着性試験を行った。
<耐候剥離>
耐候性試験後の外観(剥離の有無)を下記の基準で評価した。
○:異常なし
△:一部剥離あり
×:全面剥離
<曲げ加工性評価>
JIS K7171に準拠し、3点曲げ試験を実施し、クラックが発生した曲率半径(R)を求めた。曲率半径は次式より求める。
R=(a2+502)/(2a) (2)
a:3点曲げ治具がサンプルを押した距離(mm)
ベル・テレフォン社が開発した1/4楕円法に従って、図1に示すように、厚み1.0mm、幅40.0mm、長さ120.0mmのサンプルを1/4楕円法治具にセットし、23℃で24時間後のクラック発生点までの長さと試料厚さより限界歪みと限界応力を求めた。クラック発生点の歪みは次式より求める。なお、図1中、1は楕円中心、2は楕円の長軸半径(10cm)、3は楕円の短軸半径(4cm)、4はジグの幅(4cm)、5は押え金具(それぞれ幅1cm)、6は楕円中心から最も少ない歪みでクラックが生じている部分までの水平距離(cm)、7は試料(ポリカーボネート樹脂積層体)、8は最も少ない歪みでクラックが生じている部分を示す。
ε=〔0.02(1-0.0084X2)-3/2〕t (3)
ε:歪み(%)
t:試料厚さ(cm)
X:楕円の中心からX座標軸方向でクラック発生限界点までの距離(cm)
ベル・テレフォン社が開発した1/4楕円法に従って、厚み1.0mm、幅40.0mm、長さ120.0mmのサンプルを1/4楕円法治具にセットし、80℃の耐熱乾燥機にて114時間経過した後と110℃の耐熱乾燥機にて21時間経過した後の限界歪みを求める。クラック発生点の歪みと限界応力は次式より求める(クラック発生点の歪みは上式3と同様)。
限界応力=εE(MPa) (4)
E:PC曲げ弾性率(2000)
<紫外線遮蔽性>
石英板((株)藤原製作所製、長さ40mm×幅10mm×厚み1mm)の長さ40mm×幅10mmの片面に、シリコーンハードコート組成物を硬化膜厚が4μmとなるようフローコート、静置した。15分間の静置後、120℃で1時間加熱し、塗膜を硬化させた。得られた石英板付き硬化膜は紫外可視分光光度計(島津製作所(株)製)で透過率を測定した。
常法によりビスフェノールAとホスゲンを界面重合法で重合して得た粘度平均分子量23,900のポリカーボネート樹脂粉粒体(帝人化成(株)製:パンライトL-1250WP)を使用した。
(III-1)アクリル樹脂-B1、B3、B5の製造
市販のアクリル樹脂(三菱レイヨン(株)製ポリメチルメタクリレート樹脂(VH001))を使用した。
また、設定温度230℃のT型ダイスを介して押出することで、厚み100μmのアクリル樹脂フィルム(B5)を得た。
市販のアクリル樹脂(三菱レイヨン(株)製ポリメチルメタクリレート樹脂(VH001))と市販(メタ)アクリル系ゴム成分(三菱レイヨン(株)製ポリメチルメタクリレート樹脂(IRG304))をVH001/IRG304(100/18)の割合で混合した樹脂を使用した。
還流冷却器及び撹拌装置を備え、窒素置換したフラスコ中にエチルメタクリレート(以下EMAと省略する)74.2質量部、シクロヘキシルメタクリレート(以下CHMAと省略する)33.6質量部、2-ヒドロキシエチルメタクリレート(以下HEMAと省略する)13.0質量部、LA-82(ADEKA(株)製ヒンダードアミン系光安定性基含有メタクリレート;1,2,2,6,6-ペンタメチル-4-ピペリジルメタクリレート12.0質量部、メチルイソブチルケトン(以下MIBKと省略する)132.8質量部及び2-ブタノール(以下2-BuOHと省略する)66.4質量部を添加混合した。混合物に窒素ガスを15分間通気して脱酸素した後、窒素ガス気流下にて70℃に昇温し、アゾビスイソブチロニトリル(以下AIBNと省略する)0.33質量部を加え、窒素ガス気流中、70℃で5時間撹拌下に反応させた。更にAIBN:0.08質量部を加えて80℃に昇温し、3時間反応させ、不揮発分濃度が39.7質量%のアクリル共重合体溶液を得た。アクリル共重合体の重量平均分子量はGPCの測定(カラム;Shodex GPCA-804、溶離液;THF)からポリスチレン換算で115,000であった。アクリル共重合体溶液100質量部に、MIBK:68.6質量部、2-BuOH:34.2質量部、1-メトキシ-2-プロパノール(以下PMAと省略する):133質量部を加えて混合し、チヌビン400(チバ・スペシャルティ・ケミカルズ社製トリアジン系紫外線吸収剤)4.24質量部、及びチヌビン479(チバ・スペシャルティ・ケミカルズ社製トリアジン系紫外線吸収剤)1.06質量部、アクリル共重合体溶液中のアクリル共重合体のヒドロキシ基1当量に対してイソシアネート基が1.0当量になるようにVESTANAT B1358/100(デグサ・ジャパン(株)製ブロック化されたポリイソシアネート化合物)10.1質量部を添加し、更にジメチルチンジネオデカノエート:0.015質量部を加えて25℃で1時間撹拌し、アクリル樹脂塗料(B4:アクリル樹脂塗料)を得た。
(IV-1)単層シートの製造
ポリカーボネート樹脂-A1を用いて、粘度平均分子量23,900のポリカーボネート樹脂シート[帝人化成(株)パンライトシートPC-1151]を得た。なお、限界応力性評価は厚み1.0mm、その他の評価は厚み3.5mmにて実施した。
(IV-2)共押出シートの製造
上記ポリカーボネート樹脂層を構成するポリカーボネート樹脂-A1はスクリュー径40mmの単軸押出機で、また、上記熱可塑性樹脂層を形成するアクリル樹脂-Bはスクリュー径30mmの単軸押出機でそれぞれ溶融させ、フィードブロック法にて2層に積層させ、設定温度280℃のT型ダイスを介して押出し、得られるシートを鏡面仕上げされたロールにて冷却し、ポリカーボネート樹脂(帝人化成(株)製、商品名:パンライトL-1250WP、粘度平均分子量23,900)シートの片面に、表1記載の市販のアクリル樹脂層100質量部に対しベンゾトリアゾール型紫外線吸収剤としてアデカスタブLA-31(ADEKA(株)社製)を2.0質量部添加し、それぞれ積層した樹脂積層体を得た。なお、限界応力性評価は厚み1.0mm、その他の評価は厚み3.5mmにて実施し、アクリル樹脂層の種類と厚みは表1に示した通りである。

(V-1)スズ及びマンガン原子を固溶したコアシェル型酸化チタン固溶体(チタン100モル%に対し、スズ6モル%、マンガン2モル%)を含有するシリコーン樹脂系ハードコート組成物(S1)の調製
36質量%の塩化チタン(IV)水溶液(石原産業(株)製、製品名:TC-36)66質量部に塩化スズ(IV)五水和物(和光純薬工業(株)製)2.6質量部、塩化マンガン(II)四水和物(和光純薬工業(株)製)0.5質量部を添加し、よく混合した後、これをイオン交換水1,000質量部で希釈した。この金属塩水溶液混合物に5質量%のアンモニア水(和光純薬工業(株)製)300質量部を徐々に添加して中和、加水分解することによりスズ、マンガンを含有する水酸化チタンの沈殿物を得た。このときの水酸化チタンスラリーのpHは8であった。得られた水酸化チタンの沈殿物を、イオン交換水の添加とデカンテーションを繰り返して脱イオン処理した。この脱イオン処理後のスズ、マンガンを含有する水酸化チタン沈殿物に30質量%過酸化水素水(和光純薬工業(株)製)100質量部を徐々に添加し、その後60℃で3時間撹拌して十分に反応させた。その後、純水を添加して濃度調整を行うことにより、褐色透明のスズ、マンガン含有ペルオキソチタン酸溶液(固形分濃度1質量%)を得た。容積500mLのオートクレーブ(耐圧硝子工業(株)製、製品名:TEM-D500)に、上記のように合成したペルオキソチタン酸溶液350mLを仕込み、これを200℃、1.5MPaの条件下、240分間水熱処理した。その後、オートクレーブ内の反応混合物を、サンプリング管を経由して、25℃の水浴中に保持した容器に排出し、急速に冷却することで反応を停止させ、酸化チタン分散液(i)を得た。この酸化チタン分散液(i)の平均粒子径は、レーザー光を用いた動的光散乱法(日機装(株)製、装置名「ナノトラックUPA-EX150」)によって測定した体積基準の50%累積粒度分布径(D50)で14nmであった。
2Lのフラスコに、メチルトリメトキシシラン136質量部を仕込み、液温が約10℃になるよう冷却後、スノーテックスO(日産化学工業(株)製:水分散シリカゾル、平均粒子径15~20nm、SiO2 20質量%含有品)100質量部、0.25Nの酢酸水溶液44.2質量部を滴下し、内温が40℃を超えないように冷却しながら加水分解を行った。滴下終了後、40℃以下で1時間、次いで、60℃にて3時間撹拌し、加水分解を完結させた。その後、シクロヘキサノン142質量部を投入し、加水分解で生成したメタノールを、常圧にて液温が92℃になるまで加熱留去すると共に、縮合させた後、希釈剤としてイソブタノール189.3質量部、レベリング剤としてKP-341(信越化学工業(株)製)0.1質量部、pH調整剤として酢酸6.5質量部、及び硬化触媒として水酸化テトラメチルアンモニウム(濃度20質量%水溶液、和光純薬工業(株)、特級)1.8質量部を加え、室温にて撹拌した。更に、RTTDNB15WT%-E88(CIKナノテック(株)製:直流アークプラズマ法で製造したコバルト原子を固溶化した酸化チタン微粒子(チタン100モル%に対し、コバルト4モル%)をアルミナ及びジルコニアを含む被膜で被覆した後、メチルトリメトキシシランで表面処理してから、高分子分散剤を用いて、混合アルコールに分散したコバルト原子固溶化コアシェル型酸化チタン固溶体分散液、固形分濃度15質量%)65.4質量部を添加した後、撹拌、濾紙濾過を行い、不揮発分濃度19.2質量%のシリコーン樹脂系ハードコート組成物(S2)を得た。
塩化スズ(IV)五水和物及び塩化マンガン(II)四水和物を使用せずに、上記(V-1)と同様な操作を行い、他原子を固溶していないコアシェル型酸化チタン水分散液(CS-ii)を得た。分散液(CS-ii)の一定量を精密秤量(島津製作所(株)製AUX-220使用)し、105℃のオーブン(エスペック社製パーフェクトオーブン使用)で3時間処理して分散媒を揮発させたところ、固形分が15質量%であることが明らかとなった。
(V-2)で使用したRTTDNB15WT%-E67の代わりに、ZNTAB15WT%-E16(2)(CIKナノテック(株)製:直流アークプラズマ法で製造した酸化亜鉛微粒子をシリカ被覆した後、メチルトリメトキシシランで表面処理してから、高分子分散剤を用いて、混合アルコールに分散した分散体、固形分濃度15%)を用いて、(V-2)と同様な方法で、不揮発分濃度19.8質量%のシリコーン樹脂系ハードコート組成物(T2)を得た。
なお(V-1)と同様な方法で測定したZNTAB15WT%-E16(2)における平均粒子径(D50)はそれぞれ、45.0nm、105.0nm、メチレンブルー比色吸光度の減少率は24%であった。
2Lのフラスコに、メチルトリメトキシシラン136質量部を仕込み、液温が約10℃になるよう冷却後、スノーテックスO(日産化学工業(株)製:水分散シリカゾル、平均粒子径15~20nm、SiO2 20質量%含有品)100質量部、0.25N酢酸水溶液44.2質量部の混合液を加えた。混合液を加えると加水分解に伴う自己発熱が見られ内部温度が50℃に上昇した。添加終了後、60℃にて3時間撹拌し、加水分解を完結させた。その後、シクロヘキサノン142質量部を投入し、加水分解で生成したメタノールを、常圧にて液温が92℃になるまで加熱留去すると共に、縮合させた後、希釈剤としてイソブタノール189.3質量部、レベリング剤としてKP-341(信越化学工業(株)製)0.1質量部、pH調整剤として酢酸1.6質量部、及び硬化触媒として水酸化テトラメチルアンモニム(濃度20質量%水溶液、和光純薬工業(株)、特級)1.8質量部を加え、室温にて撹拌した後、濾紙濾過を行い、不揮発分濃度19.4質量%のシリコーン樹脂系ハードコート組成物(T3)を得た。
多官能アクリレートオリゴマー(新中村化学(株)製U-15HA)100質量部、フェニル-1-ヒドロキシシクロヘキシルケトン(BASF(株)製Irgacure184)5質量部、1-メトキシ-2-プロパノール250質量部、2-プロパノール100質量部、有機溶剤分散コロイダルシリカ(日産化学工業(株)製スノーテックスO-40 固形分濃度40質量%)29.2質量部を混合して紫外線硬化型アクリレートハードコート剤(T4)を得た。
コロイダルシリカ及び/又はアルコキシシラン加水分解縮合物の割合は30質量%であった。
メチル化メチロールメラミン[日本サイテックインダストリーズ(株)製サイメル301]100質量部、1.6-ヘキサンジオール70質量部、マレイン酸5質量部、イソプロピルアルコール150質量部、イソブチルアルコール320質量部、エチレングリコールモノブチルエーテル25質量部混合してメラミン樹脂ハードコート剤(T5)を得た。
コロイダルシリカ及び/又はアルコキシシラン加水分解縮合物の割合は0質量%であった。
上記(II)、(III)に記載の(A1)、(B1)を用いて作成した共押出基材のB層上に、(V)で作成したハードコート剤(S1)を熱硬化後の膜厚が4.0μmとなるようにフローコート法で塗布し、25℃で15分間静置後、120℃で1時間熱硬化させPC樹脂積層体を得た。結果を表2に示した。
上記(II)、(III)に記載の(A1)、(B1)を用いて作成した共押出基材のB層上に、(V)で作成したハードコート剤(S2)を熱硬化後の膜厚が4.0μmとなるようにフローコート法で塗布し、25℃で15分間静置後、120℃で1時間熱硬化させPC樹脂積層体を得た。結果を表2に示した。
上記(II)、(III)に記載の(A1)、(B2)を用いて作成した共押出基材のB層上に、(V)で作成したハードコート剤(S1)を熱硬化後の膜厚が4.0μmになるようにフローコート法で塗布し、25℃で15分間静置後、120℃で1時間熱硬化させPC樹脂積層体を得た。結果を表2に示した。
上記(II)、(III)に記載の(A1)、(B3)を用いて作成した共押出基材のB層上に、(V)で作成したハードコート剤(S1)を熱硬化後の膜厚が4.0μmになるようにフローコート法で塗布し、25℃で15分間静置後、120℃で1時間熱硬化させPC樹脂積層体を得た。結果を表2に示した。
上記(IV-1)で作成した基材層を真空熱プレス成形機(名機製作所製MP-22S)の下チャンバーにセットし、上記(III)で作成したアクリル樹脂フィルム(B5)を上チャンバーにセットした。下チャンバーと上チャンバーの温度をそれぞれ180℃と120℃に設定し、基材層とアクリル樹脂フィルムが十分に加熱されたところで、4MPaで90秒間加圧して積層基材を得た。この積層基材のB層上に(V)で作成したハードコート剤(S1)を熱硬化後の膜厚が4.0μmになるようにフローコート法で塗布し、25℃で15分間静置後、120℃で1時間熱硬化させPC樹脂積層体を得た。結果を表2に示した。
上記(II)、(III)に記載の(A1)、(B1)を用いて作成した共押出基材に、(V)で作成したハードコート剤(T1)を熱硬化後の膜厚が4.0μmとなるように、B層上にフローコート法で塗布し、25℃で15分間静置後、120℃で1時間熱硬化させPC樹脂積層体を得た。結果を表3に示した。
上記(II)、(III)に記載の(A1)、(B1)を用いて作成した共押出基材に、(V)で作成したハードコート剤(T2)を熱硬化後の膜厚が4.0μmとなるように、B層上にフローコート法で塗布し、25℃で15分間静置後、120℃で1時間熱硬化させPC樹脂積層体を得た。結果を表3に示した。
上記(IV-1)で作成した基材層に、上記(III)で得られたアクリル樹脂塗料(B4)を用いて、熱硬化後の膜厚が8.0μmになるようにフローコート法によって片面塗布し、25℃で20分間静置後、130℃で1時間熱硬化させた。
次いで(V)で作成したハードコート剤(S1)を熱硬化後の膜厚が4.0μmになるようにフローコート法で塗布し、25℃で20分間静置後、120℃で1時間熱硬化させた。結果を表3に示した。
上記(IV-1)で作成した基材層に、上記(III)で得られたアクリル樹脂塗料(B4)を用いて、熱硬化後の膜厚が8.0μmになるようにフローコート法によって片面塗布し、25℃で20分間静置後、130℃で1時間熱硬化させた。
次いで(V)で作成したハードコート剤(T3)を熱硬化後の膜厚が4.0μmになるようにフローコート法で塗布し、25℃で20分間静置後、120℃で1時間熱硬化させた。結果を表3に示した。
上記(IV-1)で作成した基材層に、上記(V)で作成したハードコート剤(T4)を紫外線硬化後の膜厚が4.0μmになるようにフローコート法で片面塗布し、25℃で1分間、80℃で1分間静置後、積算照度が600mJ/cm2になるように高圧水銀ランプで紫外線を照射して硬化させた。結果を表2に示した。
上記(IV-1)で作成した基材層に、上記(V)で作成したハードコート剤(T5)を熱硬化後の膜厚が4.0μmになるようにフローコート法で片面塗布し、25℃で15分間静置後、130℃で1時間熱硬化させPC樹脂積層体を得た。結果を表3に示した。
上記(II)、(III)に記載の(A1)、(B1)を用いて作成した共押出基材に、(V)で作成したハードコート剤(T3)を熱硬化後の膜厚が4.0μmとなるようB層上にフローコート法で塗布し、25℃で15分間静置後、120℃で1時間熱硬化させPC樹脂積層体を得た。結果を表3に示した。
上記(V)で作成したシリコーンハードコート組成物(S1、S2、T1、T2、及びT3)の硬化膜について、紫外可視分光光度計(島津製作所(株)製)にて透過率を測定した。結果を図2に示した。図2から、S1、S2、及びT1の硬化膜は、可視領域(400~700nm)における透明性と紫外領域(200~400nm)における遮蔽性に優れている。一方、T2の硬化膜では、紫外領域における遮蔽能は半減し、T3ではほとんどないことが明らかである。この結果から、(c-1)成分のコアシェル型正方晶系酸化チタン固溶体分散液による紫外線遮蔽効果は顕著であることが判った。また紫外線遮蔽性のよいS1、S2、T1のうち、S1及びS2では、表2,3の通り、耐候性試験6,000時間でも問題みられないが、T1では(c-1)成分として固溶化していないコアシェル型正方晶系酸化チタン分散液を用いているため、6,000時間ではクラック、剥離が発生している。このことから、(c-1)成分としてコアシェル型正方晶系酸化チタン固溶体分散液による耐候性向上効果は顕著であることが判った。

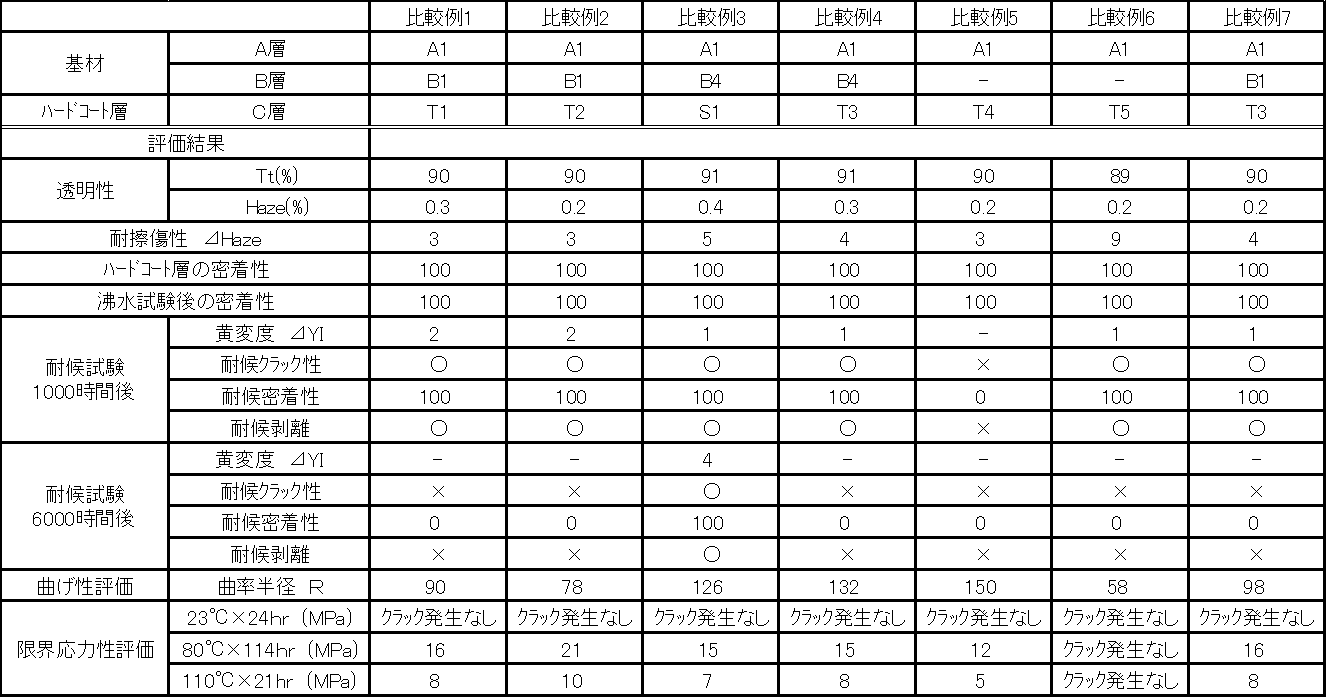
2 楕円の長軸半径(10cm)
3 楕円の短軸半径(4cm)
4 ジグの幅(4cm)
5 押え金具(それぞれ幅1cm)
6 楕円中心から最も少ない歪みでクラックが生じている部分までの水平距離(cm)
7 ポリカーボネート樹脂積層体
8 最も少ない歪みでクラックが生じている部分
Claims (10)
- ポリカーボネート樹脂層(A層)の片面又は両面に紫外線吸収性性能を有する熱可塑性樹脂層(B層)を成形積層した積層基材に、コバルト、スズ、マンガンからなる群の1以上の原子を固溶した正方晶系酸化チタン微粒子を核とし、該核の外側に酸化ケイ素もしくは酸化アルミニウムの殻を有するコアシェル型正方晶系酸化チタン固溶体を、水、アルコール、エーテル、エステル、ケトンからなる群の1以上の分散媒中に分散したコアシェル型正方晶系酸化チタン固溶体分散液であって、レーザー光を用いた動的光散乱法で測定した上記核微粒子の体積基準の50%累積分布径(D50)が50nm以下で、上記コアシェル固溶体の体積基準の50%累積分布径(D50)が100nm以下であり、前記コバルト、スズ、マンガンからなる群の全固溶量(M)が、チタンとのモル比(Ti/M)で10~1,000の固溶体分散液を含むシリコーンハードコート組成物を塗布硬化してなるシリコーンハードコート層(C層)を形成したポリカーボネート樹脂積層体。
- 積層基材が、ポリカーボネート樹脂層(A層)及び紫外線吸収性性能を有する熱可塑性樹脂層(B層)を共押出法、熱圧着法、又はインサートモールド成形法のいずれかの方法により成形積層したものである請求項1に記載のポリカーボネート樹脂積層体。
- 紫外線吸収性性能を有する熱可塑性樹脂層(B層)は、紫外線吸収剤としてベンゾトリアゾール類又はトリアジン類を含む熱可塑性樹脂からなることを特徴とする請求項1又は2に記載のポリカーボネート樹脂積層体。
- 紫外線吸収性性能を有する熱可塑性樹脂層(B層)は、(メタ)アクリル系樹脂組成物を用いて形成されたものであることを特徴とする請求項1~3のいずれか1項に記載のポリカーボネート樹脂積層体。
- 紫外線吸収性性能を有する熱可塑性樹脂層(B層)は、(メタ)アクリル系ゴム成分を含むことを特徴とする請求項1~4のいずれか1項に記載のポリカーボネート樹脂積層体。
- ハードコート層(C層)は、下記成分(c-1)~(c-5)を含むシリコーンハードコート組成物の硬化被膜であること特徴とする請求項1~5のいずれか1項に記載のポリカーボネート樹脂積層体。
(c-1)コバルト、スズ、マンガンからなる群の1以上の原子を固溶した正方晶系酸化チタン微粒子を核とし、該核の外側に酸化ケイ素もしくは酸化アルミニウムの殻を有するコアシェル型正方晶系酸化チタン固溶体を、水、アルコール、エーテル、エステル、ケトンからなる群の1以上の分散媒中に分散したコアシェル型正方晶系酸化チタン固溶体分散液であって、レーザー光を用いた動的光散乱法で測定した上記核微粒子の体積基準の50%累積分布径(D50)が50nm以下で、上記コアシェル固溶体の体積基準の50%累積分布径(D50)が100nm以下であり、前記コバルト、スズ、マンガンからなる群の全固溶量(M)が、チタンとのモル比(Ti/M)で10~1,000の固溶体分散液、
(c-2)下記一般式(1):
(R1)m(R2)nSi(OR3)4-m-n (1)
(式中、R1及びR2は、各々独立に、水素原子、又は置換若しくは非置換の一価炭化水素基であり、置換基同士が相互に結合していてもよく、R3は、炭素数1~3のアルキル基であり、m,nは、各々独立に、0又は1であり、且つm+nは、0,1又は2である。)
で表されるアルコキシシラン及びその部分加水分解縮合物から選ばれる少なくとも1種を(共)加水分解・縮合することにより得られたシリコーンレジン、
(c-3)硬化触媒、
(c-4)溶剤、
(c-5)必要によりコロイダルシリカ
を含み、且つ(c-1)成分のコアシェル型正方晶系酸化チタン固溶体分散液の固形分が、(c-2)成分のシリコーンレジンの固形分に対して、1~30質量%である。 - (c-5)成分のコロイダルシリカの配合量が、(c-2)成分のシリコーンレジン固形分100質量部に対し5~100質量部であることを特徴とする請求項6に記載のポリカーボネート樹脂積層体。
- (c-1)成分が、スズ及びマンガンを固溶した正方晶系酸化チタン微粒子を核とし、該核の外側に酸化ケイ素の殻を有するコアシェル型正方晶系酸化チタン固溶体を、水、アルコールからなる群の1以上の分散媒中に分散したコアシェル型正方晶系酸化チタン固溶体分散液であって、レーザー光を用いた動的光散乱法で測定した上記核微粒子の体積基準の50%累積分布径(D50)が30nm以下で、上記コアシェル固溶体の体積基準の50%累積分布径(D50)が50nm以下であり、前記スズ成分の固溶量が、チタンとのモル比(Ti/Sn)で10~1,000、前記マンガン成分の固溶量が、チタンとのモル比(Ti/Mn)で10~1,000である固溶体分散液であって、下記工程(A)から(D)を経て得られたものであることを特徴とする請求項6又は7に記載のポリカーボネート樹脂積層体。
(A)スズ及びマンガンを固溶した正方晶系酸化チタン微粒子分散液に、一価アルコール、アンモニア、及びテトラアルコキシシランを配合する工程、
(B)前記(A)の工程で得られた混合物を急速加熱することにより、スズ及びマンガンを固溶した正方晶系酸化チタン微粒子を核とし該核の外側に酸化ケイ素の殻を有するコアシェル型正方晶系酸化チタン固溶体を形成させる工程、
(C)前記(B)の工程で得られたコアシェル型正方晶系酸化チタン固溶体水分散液から水を除去して濃縮する工程、及び
(D)アンモニアを除去する工程 - JIS K7171に準拠して3点曲げ試験を実施した際に、クラックが入る曲率半径Rが100mm以下である請求項1~8のいずれか1項に記載のポリカーボネート樹脂積層体。
- 請求項1~9のいずれか1項に記載のポリカーボネート樹脂積層体の熱可塑性樹脂層(B層)又はハードコート層(C層)上に接着用プライマー層及び弾性接着剤層を順次介して構造部材が接着取り付けられてなる接着構成体。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/777,707 US20160271922A1 (en) | 2013-03-26 | 2014-03-25 | Polycarbonate resin laminate |
| EP14773481.8A EP2979862B1 (en) | 2013-03-26 | 2014-03-25 | Polycarbonate resin laminate |
| CN201480018126.XA CN105050810B (zh) | 2013-03-26 | 2014-03-25 | 聚碳酸酯树脂层叠体 |
| JP2015508519A JP5967293B2 (ja) | 2013-03-26 | 2014-03-25 | ポリカーボネート樹脂積層体 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013-063721 | 2013-03-26 | ||
| JP2013063721 | 2013-03-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014157149A1 true WO2014157149A1 (ja) | 2014-10-02 |
Family
ID=51624120
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/058200 WO2014157149A1 (ja) | 2013-03-26 | 2014-03-25 | ポリカーボネート樹脂積層体 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20160271922A1 (ja) |
| EP (1) | EP2979862B1 (ja) |
| JP (1) | JP5967293B2 (ja) |
| CN (1) | CN105050810B (ja) |
| WO (1) | WO2014157149A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3009485A1 (en) * | 2014-10-17 | 2016-04-20 | Shin-Etsu Chemical Co., Ltd. | Activation energy radiation-curable silicone coating composition and coated article |
| WO2021070632A1 (ja) * | 2019-10-09 | 2021-04-15 | 帝人株式会社 | 湾曲部材の製造方法、及び、熱曲げ用ハードコート層付ポリカーボネート樹脂積層体 |
| JP2022524984A (ja) * | 2019-03-04 | 2022-05-11 | モメンティブ パフォーマンス マテリアルズ インコーポレイテッド | 保護コーティング組成物およびそれを含むコーティングされた金属基材 |
| WO2023286634A1 (ja) * | 2021-07-15 | 2023-01-19 | 帝人株式会社 | 積層体 |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20170031525A1 (en) | 2010-05-14 | 2017-02-02 | Racing Optics, Inc. | Touch screen shield |
| US9295297B2 (en) | 2014-06-17 | 2016-03-29 | Racing Optics, Inc. | Adhesive mountable stack of removable layers |
| KR101746460B1 (ko) * | 2015-11-20 | 2017-06-13 | 현대자동차주식회사 | 폴리카보네이트 글레이징용 코팅도료 조성물로 코팅된 도장품 |
| WO2017115819A1 (ja) * | 2015-12-28 | 2017-07-06 | 信越化学工業株式会社 | 積層体の製造方法 |
| JP6810792B2 (ja) * | 2017-03-31 | 2021-01-06 | 三井化学株式会社 | 積層体の製造方法、積層体およびハードコート液 |
| EP3473497B1 (en) | 2017-10-17 | 2021-11-03 | SABIC Global Technologies B.V. | Front end panel for an electric vehicle |
| JP7188003B2 (ja) * | 2018-11-14 | 2022-12-13 | 大日本印刷株式会社 | ハードコート基材接合体、及び、その製造方法 |
| US11524493B2 (en) | 2019-02-01 | 2022-12-13 | Racing Optics, Inc. | Thermoform windshield stack with integrated formable mold |
| US11846788B2 (en) | 2019-02-01 | 2023-12-19 | Racing Optics, Inc. | Thermoform windshield stack with integrated formable mold |
| US11364715B2 (en) | 2019-05-21 | 2022-06-21 | Racing Optics, Inc. | Polymer safety glazing for vehicles |
| US11648723B2 (en) | 2019-12-03 | 2023-05-16 | Racing Optics, Inc. | Method and apparatus for reducing non-normal incidence distortion in glazing films |
| US20230086403A1 (en) * | 2020-03-04 | 2023-03-23 | Asahi Kasei Kabushiki Kaisha | Laminate, hard coating film, and coating material composition |
| US11548356B2 (en) | 2020-03-10 | 2023-01-10 | Racing Optics, Inc. | Protective barrier for safety glazing |
| US11490667B1 (en) | 2021-06-08 | 2022-11-08 | Racing Optics, Inc. | Low haze UV blocking removable lens stack |
| CN113429922A (zh) * | 2021-06-18 | 2021-09-24 | 山西中科正泰机械制造有限公司 | 一种3dp打印机打印用pmma蜡粉的粘合剂及其制备方法和应用 |
| US11307329B1 (en) | 2021-07-27 | 2022-04-19 | Racing Optics, Inc. | Low reflectance removable lens stack |
| US11709296B2 (en) | 2021-07-27 | 2023-07-25 | Racing Optics, Inc. | Low reflectance removable lens stack |
| US11933943B2 (en) | 2022-06-06 | 2024-03-19 | Laminated Film Llc | Stack of sterile peelable lenses with low creep |
| US11808952B1 (en) | 2022-09-26 | 2023-11-07 | Racing Optics, Inc. | Low static optical removable lens stack |
Citations (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5692059A (en) | 1979-12-21 | 1981-07-25 | Gen Electric | Wearrproof silicon coated polycarbonate article |
| JPS5931130A (ja) | 1982-08-17 | 1984-02-20 | Dainippon Printing Co Ltd | 射出成形同時絵付装置 |
| JPS62196113A (ja) | 1986-02-21 | 1987-08-29 | Dainippon Printing Co Ltd | 射出成形同時絵付方法および装置 |
| JPH01149878A (ja) | 1987-12-04 | 1989-06-12 | Shin Etsu Chem Co Ltd | プライマー組成物とその用法 |
| JPH04106161A (ja) | 1990-08-24 | 1992-04-08 | Toray Ind Inc | プライマ及び複層コーティング物品 |
| JPH06172508A (ja) | 1992-12-03 | 1994-06-21 | Teijin Chem Ltd | 芳香族ポリカーボネート共重合体 |
| JPH079484A (ja) | 1993-06-22 | 1995-01-13 | Dainippon Printing Co Ltd | 射出成形同時絵付け方法及びそれに用いる絵付けフィルム |
| JPH0827370A (ja) | 1994-07-12 | 1996-01-30 | Teijin Chem Ltd | 芳香族ポリカーボネート組成物 |
| JP3102696B2 (ja) | 1990-09-10 | 2000-10-23 | 日本エーアールシー株式会社 | 被覆組成物および該組成物を用いる被覆樹脂成形品 |
| JP2001047574A (ja) | 1999-08-11 | 2001-02-20 | Nippon Shokubai Co Ltd | 紫外線吸収性樹脂部材 |
| JP2001055435A (ja) | 1999-08-19 | 2001-02-27 | Mitsubishi Gas Chem Co Inc | 光記録媒体用成形材料 |
| JP2001198947A (ja) | 2000-01-21 | 2001-07-24 | Teijin Ltd | ポリカーボネート樹脂積層成形体およびその製造方法 |
| JP2002117580A (ja) | 2000-10-03 | 2002-04-19 | Teijin Chem Ltd | 光ディスク基板および光ディスク |
| JP2002129003A (ja) | 2000-10-26 | 2002-05-09 | Teijin Chem Ltd | 電磁波遮蔽性樹脂組成物 |
| JP2004001393A (ja) | 2002-04-22 | 2004-01-08 | Mitsubishi Gas Chem Co Inc | 透明な樹脂積層体及びそれを用いた成形品 |
| JP3841141B2 (ja) | 2000-02-04 | 2006-11-01 | 信越化学工業株式会社 | 下塗り剤組成物及びコーティング方法 |
| JP2009279806A (ja) * | 2008-05-21 | 2009-12-03 | Nippon Shokubai Co Ltd | 積層板、積層板の製造方法および、表示装置用前面板 |
| JP2010202731A (ja) | 2009-03-02 | 2010-09-16 | Shin-Etsu Chemical Co Ltd | 紫外線遮蔽性シリコーンコーティング組成物及び被覆物品 |
| JP2011183758A (ja) | 2010-03-11 | 2011-09-22 | Shin-Etsu Chemical Co Ltd | ポリカーボネート樹脂積層体 |
| JP2012077267A (ja) | 2010-10-06 | 2012-04-19 | Shin-Etsu Chemical Co Ltd | 紫外線遮蔽性シリコーンコーティング組成物及び被覆物品 |
| JP2012111142A (ja) * | 2010-11-25 | 2012-06-14 | Teijin Chem Ltd | 色調安定性に優れた耐候性樹脂グレージング |
| JP2014019611A (ja) * | 2012-07-19 | 2014-02-03 | Shin Etsu Chem Co Ltd | コアシェル型正方晶系酸化チタン固溶体水分散液、その製造方法、紫外線遮蔽性シリコーンコーティング組成物、及び被覆物品 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4326679B2 (ja) * | 2000-07-19 | 2009-09-09 | リンテック株式会社 | ハードコートフィルム |
| JPWO2008047940A1 (ja) * | 2006-10-20 | 2010-02-25 | Mgcフィルシート株式会社 | ポリカーボネート樹脂を含む積層体 |
| GB0705614D0 (en) * | 2007-03-23 | 2007-05-02 | Croda Int Plc | Particulate titanium dioxide |
| JP5557662B2 (ja) * | 2010-09-10 | 2014-07-23 | 日揮触媒化成株式会社 | コアシェル型無機酸化物微粒子の分散液、その製造方法および該分散液を含む塗料組成物 |
-
2014
- 2014-03-25 EP EP14773481.8A patent/EP2979862B1/en active Active
- 2014-03-25 JP JP2015508519A patent/JP5967293B2/ja not_active Expired - Fee Related
- 2014-03-25 US US14/777,707 patent/US20160271922A1/en not_active Abandoned
- 2014-03-25 CN CN201480018126.XA patent/CN105050810B/zh active Active
- 2014-03-25 WO PCT/JP2014/058200 patent/WO2014157149A1/ja active Application Filing
Patent Citations (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5692059A (en) | 1979-12-21 | 1981-07-25 | Gen Electric | Wearrproof silicon coated polycarbonate article |
| JPS5931130A (ja) | 1982-08-17 | 1984-02-20 | Dainippon Printing Co Ltd | 射出成形同時絵付装置 |
| JPS62196113A (ja) | 1986-02-21 | 1987-08-29 | Dainippon Printing Co Ltd | 射出成形同時絵付方法および装置 |
| JPH01149878A (ja) | 1987-12-04 | 1989-06-12 | Shin Etsu Chem Co Ltd | プライマー組成物とその用法 |
| JPH04106161A (ja) | 1990-08-24 | 1992-04-08 | Toray Ind Inc | プライマ及び複層コーティング物品 |
| JP3102696B2 (ja) | 1990-09-10 | 2000-10-23 | 日本エーアールシー株式会社 | 被覆組成物および該組成物を用いる被覆樹脂成形品 |
| JPH06172508A (ja) | 1992-12-03 | 1994-06-21 | Teijin Chem Ltd | 芳香族ポリカーボネート共重合体 |
| JPH079484A (ja) | 1993-06-22 | 1995-01-13 | Dainippon Printing Co Ltd | 射出成形同時絵付け方法及びそれに用いる絵付けフィルム |
| JPH0827370A (ja) | 1994-07-12 | 1996-01-30 | Teijin Chem Ltd | 芳香族ポリカーボネート組成物 |
| JP2001047574A (ja) | 1999-08-11 | 2001-02-20 | Nippon Shokubai Co Ltd | 紫外線吸収性樹脂部材 |
| JP2001055435A (ja) | 1999-08-19 | 2001-02-27 | Mitsubishi Gas Chem Co Inc | 光記録媒体用成形材料 |
| JP2001198947A (ja) | 2000-01-21 | 2001-07-24 | Teijin Ltd | ポリカーボネート樹脂積層成形体およびその製造方法 |
| JP3841141B2 (ja) | 2000-02-04 | 2006-11-01 | 信越化学工業株式会社 | 下塗り剤組成物及びコーティング方法 |
| JP2002117580A (ja) | 2000-10-03 | 2002-04-19 | Teijin Chem Ltd | 光ディスク基板および光ディスク |
| JP2002129003A (ja) | 2000-10-26 | 2002-05-09 | Teijin Chem Ltd | 電磁波遮蔽性樹脂組成物 |
| JP2004001393A (ja) | 2002-04-22 | 2004-01-08 | Mitsubishi Gas Chem Co Inc | 透明な樹脂積層体及びそれを用いた成形品 |
| JP2009279806A (ja) * | 2008-05-21 | 2009-12-03 | Nippon Shokubai Co Ltd | 積層板、積層板の製造方法および、表示装置用前面板 |
| JP2010202731A (ja) | 2009-03-02 | 2010-09-16 | Shin-Etsu Chemical Co Ltd | 紫外線遮蔽性シリコーンコーティング組成物及び被覆物品 |
| JP2011183758A (ja) | 2010-03-11 | 2011-09-22 | Shin-Etsu Chemical Co Ltd | ポリカーボネート樹脂積層体 |
| JP2012077267A (ja) | 2010-10-06 | 2012-04-19 | Shin-Etsu Chemical Co Ltd | 紫外線遮蔽性シリコーンコーティング組成物及び被覆物品 |
| JP2012111142A (ja) * | 2010-11-25 | 2012-06-14 | Teijin Chem Ltd | 色調安定性に優れた耐候性樹脂グレージング |
| JP2014019611A (ja) * | 2012-07-19 | 2014-02-03 | Shin Etsu Chem Co Ltd | コアシェル型正方晶系酸化チタン固溶体水分散液、その製造方法、紫外線遮蔽性シリコーンコーティング組成物、及び被覆物品 |
Non-Patent Citations (1)
| Title |
|---|
| "Adhesives and Sealants: General Knowledge, Application Techniques", 2006, ELSEVIER SCIENCE LTD., pages: 385 |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3009485A1 (en) * | 2014-10-17 | 2016-04-20 | Shin-Etsu Chemical Co., Ltd. | Activation energy radiation-curable silicone coating composition and coated article |
| CN105524549A (zh) * | 2014-10-17 | 2016-04-27 | 信越化学工业株式会社 | 活性能量辐射可固化的有机硅涂料组合物和涂覆制品 |
| JP2016079395A (ja) * | 2014-10-17 | 2016-05-16 | 信越化学工業株式会社 | 活性エネルギー線硬化型シリコーンコーティング組成物及び被覆物品 |
| JP2022524984A (ja) * | 2019-03-04 | 2022-05-11 | モメンティブ パフォーマンス マテリアルズ インコーポレイテッド | 保護コーティング組成物およびそれを含むコーティングされた金属基材 |
| JP7499782B2 (ja) | 2019-03-04 | 2024-06-14 | モメンティブ パフォーマンス マテリアルズ インコーポレイテッド | 保護コーティング組成物およびそれを含むコーティングされた金属基材 |
| WO2021070632A1 (ja) * | 2019-10-09 | 2021-04-15 | 帝人株式会社 | 湾曲部材の製造方法、及び、熱曲げ用ハードコート層付ポリカーボネート樹脂積層体 |
| JPWO2021070632A1 (ja) * | 2019-10-09 | 2021-04-15 | ||
| JP7236554B2 (ja) | 2019-10-09 | 2023-03-09 | 帝人株式会社 | 湾曲部材の製造方法、及び、熱曲げ用ハードコート層付ポリカーボネート樹脂積層体 |
| WO2023286634A1 (ja) * | 2021-07-15 | 2023-01-19 | 帝人株式会社 | 積層体 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2979862B1 (en) | 2018-07-04 |
| CN105050810B (zh) | 2018-01-23 |
| US20160271922A1 (en) | 2016-09-22 |
| EP2979862A4 (en) | 2017-01-25 |
| CN105050810A (zh) | 2015-11-11 |
| JP5967293B2 (ja) | 2016-08-10 |
| EP2979862A1 (en) | 2016-02-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5967293B2 (ja) | ポリカーボネート樹脂積層体 | |
| JPWO2014157149A1 (ja) | ポリカーボネート樹脂積層体 | |
| EP2708513B1 (en) | Core/shell type tetragonal titanium oxide particle water dispersion, making method, uv-shielding silicone coating composition and coated article | |
| EP2226352B1 (en) | UV-shielding silicone coating composition and coated article | |
| JP5267488B2 (ja) | ポリカーボネート樹脂積層体 | |
| EP2436736B1 (en) | Organosiloxane resin composition and laminate comprising same | |
| EP1925631B1 (en) | Composite polymer, thermosetting coating composition, and molded article | |
| KR102406819B1 (ko) | 광촉매 적층체 | |
| US20210032429A1 (en) | Organic resin laminate | |
| JP5877086B2 (ja) | プラスチック積層体およびその製造方法 | |
| WO2019172313A1 (ja) | ガラス積層体 | |
| JP2002254560A (ja) | 紫外線遮蔽性積層体および紫外線遮蔽層形成用樹脂組成物 | |
| JP6130163B2 (ja) | ポリカーボネート樹脂積層体 | |
| JP5919027B2 (ja) | 鱗片状金属酸化物微粒子硬化用プラスチック積層体の製造方法 | |
| TWI617458B (zh) | a laminate having a top coat composed of scaly metal oxide fine particles | |
| KR20150041404A (ko) | 폴리카보네이트 글래이징 및 그 제조방법 | |
| JP5893985B2 (ja) | アクリル樹脂塗料およびその積層体 | |
| WO2023286634A1 (ja) | 積層体 | |
| JP2017025251A (ja) | 樹脂組成物およびそれからなる成形品 | |
| JP2017025252A (ja) | 樹脂組成物およびそれからなる成形品 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201480018126.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14773481 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015508519 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014773481 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14777707 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |