WO2011157339A1 - Metallkomplexe - Google Patents
Metallkomplexe Download PDFInfo
- Publication number
- WO2011157339A1 WO2011157339A1 PCT/EP2011/002467 EP2011002467W WO2011157339A1 WO 2011157339 A1 WO2011157339 A1 WO 2011157339A1 EP 2011002467 W EP2011002467 W EP 2011002467W WO 2011157339 A1 WO2011157339 A1 WO 2011157339A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- mmol
- group
- atoms
- radicals
- Prior art date
Links
- 0 *OC1C(CC2)CC2C1 Chemical compound *OC1C(CC2)CC2C1 0.000 description 13
- GHIBFRBZYLTPOA-UHFFFAOYSA-N CC(C)(C)c([n]1c2cc(C)c3C)cc(nc(C(C)(C)C)cc4)c4c1nc2c3Br Chemical compound CC(C)(C)c([n]1c2cc(C)c3C)cc(nc(C(C)(C)C)cc4)c4c1nc2c3Br GHIBFRBZYLTPOA-UHFFFAOYSA-N 0.000 description 1
- ZILQCFXCEHFMCZ-UHFFFAOYSA-N CC(C)(C)c1ncc2[nH]c(-c3c(N)nc(C(C)(C)C)nc3)nc2c1 Chemical compound CC(C)(C)c1ncc2[nH]c(-c3c(N)nc(C(C)(C)C)nc3)nc2c1 ZILQCFXCEHFMCZ-UHFFFAOYSA-N 0.000 description 1
- RIYARDQGQOVCPK-UHFFFAOYSA-N COC1(CC2)CCC2CC1 Chemical compound COC1(CC2)CCC2CC1 RIYARDQGQOVCPK-UHFFFAOYSA-N 0.000 description 1
- XTLGIZVLOGKJKT-UHFFFAOYSA-N COC1C2C(C3)CC1CC3C2 Chemical compound COC1C2C(C3)CC1CC3C2 XTLGIZVLOGKJKT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/10—Organic polymers or oligomers
- H10K85/111—Organic polymers or oligomers comprising aromatic, heteroaromatic, or aryl chains, e.g. polyaniline, polyphenylene or polyphenylene vinylene
- H10K85/113—Heteroaromatic compounds comprising sulfur or selene, e.g. polythiophene
- H10K85/1135—Polyethylene dioxythiophene [PEDOT]; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C15/00—Cyclic hydrocarbons containing only six-membered aromatic rings as cyclic parts
- C07C15/20—Polycyclic condensed hydrocarbons
- C07C15/27—Polycyclic condensed hydrocarbons containing three rings
- C07C15/30—Phenanthrenes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C15/00—Cyclic hydrocarbons containing only six-membered aromatic rings as cyclic parts
- C07C15/20—Polycyclic condensed hydrocarbons
- C07C15/38—Polycyclic condensed hydrocarbons containing four rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F15/00—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic System
- C07F15/0006—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic System compounds of the platinum group
- C07F15/0033—Iridium compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F15/00—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic System
- C07F15/0006—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic System compounds of the platinum group
- C07F15/0086—Platinum compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F19/00—Metal compounds according to more than one of main groups C07F1/00 - C07F17/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic System
- C07F5/02—Boron compounds
- C07F5/025—Boronic and borinic acid compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic System
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0803—Compounds with Si-C or Si-Si linkages
- C07F7/081—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te
- C07F7/0812—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te comprising a heterocyclic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6564—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms
- C07F9/6568—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus atoms as the only ring hetero atoms
- C07F9/65683—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus atoms as the only ring hetero atoms the ring phosphorus atom being part of a phosphine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6564—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms
- C07F9/6568—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus atoms as the only ring hetero atoms
- C07F9/65685—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus atoms as the only ring hetero atoms the ring phosphorus atom being part of a phosphine oxide or thioxide
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6564—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms
- C07F9/6581—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus and nitrogen atoms with or without oxygen or sulfur atoms, as ring hetero atoms
- C07F9/6584—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus and nitrogen atoms with or without oxygen or sulfur atoms, as ring hetero atoms having one phosphorus atom as ring hetero atom
- C07F9/65848—Cyclic amide derivatives of acids of phosphorus, in which two nitrogen atoms belong to the ring
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D139/00—Coating compositions based on homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a single or double bond to nitrogen or by a heterocyclic ring containing nitrogen; Coating compositions based on derivatives of such polymers
- C09D139/04—Homopolymers or copolymers of monomers containing heterocyclic rings having nitrogen as ring member
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K11/00—Luminescent, e.g. electroluminescent, chemiluminescent materials
- C09K11/06—Luminescent, e.g. electroluminescent, chemiluminescent materials containing organic luminescent materials
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05B—ELECTRIC HEATING; ELECTRIC LIGHT SOURCES NOT OTHERWISE PROVIDED FOR; CIRCUIT ARRANGEMENTS FOR ELECTRIC LIGHT SOURCES, IN GENERAL
- H05B33/00—Electroluminescent light sources
- H05B33/12—Light sources with substantially two-dimensional radiating surfaces
- H05B33/14—Light sources with substantially two-dimensional radiating surfaces characterised by the chemical or physical composition or the arrangement of the electroluminescent material, or by the simultaneous addition of the electroluminescent material in or onto the light source
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
- H10K50/10—OLEDs or polymer light-emitting diodes [PLED]
- H10K50/11—OLEDs or polymer light-emitting diodes [PLED] characterised by the electroluminescent [EL] layers
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/30—Coordination compounds
- H10K85/341—Transition metal complexes, e.g. Ru(II)polypyridine complexes
- H10K85/342—Transition metal complexes, e.g. Ru(II)polypyridine complexes comprising iridium
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/615—Polycyclic condensed aromatic hydrocarbons, e.g. anthracene
- H10K85/622—Polycyclic condensed aromatic hydrocarbons, e.g. anthracene containing four rings, e.g. pyrene
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/615—Polycyclic condensed aromatic hydrocarbons, e.g. anthracene
- H10K85/626—Polycyclic condensed aromatic hydrocarbons, e.g. anthracene containing more than one polycyclic condensed aromatic rings, e.g. bis-anthracene
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/649—Aromatic compounds comprising a hetero atom
- H10K85/656—Aromatic compounds comprising a hetero atom comprising two or more different heteroatoms per ring
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/649—Aromatic compounds comprising a hetero atom
- H10K85/657—Polycyclic condensed heteroaromatic hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/02—Ortho- or ortho- and peri-condensed systems
- C07C2603/04—Ortho- or ortho- and peri-condensed systems containing three rings
- C07C2603/22—Ortho- or ortho- and peri-condensed systems containing three rings containing only six-membered rings
- C07C2603/24—Anthracenes; Hydrogenated anthracenes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/02—Ortho- or ortho- and peri-condensed systems
- C07C2603/40—Ortho- or ortho- and peri-condensed systems containing four condensed rings
- C07C2603/42—Ortho- or ortho- and peri-condensed systems containing four condensed rings containing only six-membered rings
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1003—Carbocyclic compounds
- C09K2211/1007—Non-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1003—Carbocyclic compounds
- C09K2211/1011—Condensed systems
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1018—Heterocyclic compounds
- C09K2211/1022—Heterocyclic compounds bridged by heteroatoms, e.g. N, P, Si or B
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1018—Heterocyclic compounds
- C09K2211/1025—Heterocyclic compounds characterised by ligands
- C09K2211/1029—Heterocyclic compounds characterised by ligands containing one nitrogen atom as the heteroatom
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1018—Heterocyclic compounds
- C09K2211/1025—Heterocyclic compounds characterised by ligands
- C09K2211/1044—Heterocyclic compounds characterised by ligands containing two nitrogen atoms as heteroatoms
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1018—Heterocyclic compounds
- C09K2211/1025—Heterocyclic compounds characterised by ligands
- C09K2211/1059—Heterocyclic compounds characterised by ligands containing three nitrogen atoms as heteroatoms
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1018—Heterocyclic compounds
- C09K2211/1025—Heterocyclic compounds characterised by ligands
- C09K2211/1088—Heterocyclic compounds characterised by ligands containing oxygen as the only heteroatom
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1018—Heterocyclic compounds
- C09K2211/1025—Heterocyclic compounds characterised by ligands
- C09K2211/1092—Heterocyclic compounds characterised by ligands containing sulfur as the only heteroatom
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1018—Heterocyclic compounds
- C09K2211/1025—Heterocyclic compounds characterised by ligands
- C09K2211/1096—Heterocyclic compounds characterised by ligands containing other heteroatoms
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/18—Metal complexes
- C09K2211/185—Metal complexes of the platinum group, i.e. Os, Ir, Pt, Ru, Rh or Pd
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K2101/00—Properties of the organic materials covered by group H10K85/00
- H10K2101/10—Triplet emission
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K2102/00—Constructional details relating to the organic devices covered by this subclass
- H10K2102/10—Transparent electrodes, e.g. using graphene
- H10K2102/101—Transparent electrodes, e.g. using graphene comprising transparent conductive oxides [TCO]
- H10K2102/103—Transparent electrodes, e.g. using graphene comprising transparent conductive oxides [TCO] comprising indium oxides, e.g. ITO
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/615—Polycyclic condensed aromatic hydrocarbons, e.g. anthracene
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E10/00—Energy generation through renewable energy sources
- Y02E10/50—Photovoltaic [PV] energy
- Y02E10/549—Organic PV cells
Definitions
- the present invention relates to metal complexes which are suitable for use as emitters in organic electroluminescent devices.
- OLEDs organic electroluminescent devices
- OLEDs organic electroluminescent devices
- organometallic complexes which exhibit phosphorescence instead of fluorescence are increasingly being used as emitting materials (M.A. Baldo et al., Appl. Phys. Lett. 1999, 75, 4-6).
- organometallic compounds for quantum mechanical reasons, up to four times energy and power efficiency is possible using organometallic compounds as phosphorescence emitters.
- OLEDs which in the shorter wavelength
- iridium complexes are used in phosphorescent OLEDs as triplet emitters in particular.
- the object of the present invention is therefore to provide new metal complexes which are suitable as emitters for use in OLEDs.
- the object is to provide emitters, which are suitable for blue phosphorescent OLEDs, and which thereby
- the invention thus relates to a compound according to formula (1),
- R 3 is the same or different at each occurrence, H, D, F or a
- aliphatic, aromatic and / or heteroaromatic hydrocarbon radical having 1 to 20 C atoms, in which also one or more H atoms may be replaced by F; two or more substituents R 3 may also together form a mono- or polycyclic aliphatic ring system;
- a substituent R or R 1 additionally coordinate to the metal; with the proviso that R 1 is a branched or cyclic alkyl group having 4 to 20 C atoms, each with one or more R 2 radicals may be substituted with one or more non-adjacent CH 2 groups not directly attached to the aromatic carbon of the
- the indices n and m are chosen such that the coordination number on the metal M in total, depending on the metal, corresponds to the usual coordination number for this metal. This is usually the coordination number 4, 5 or 6 for transition metals depending on the metal. It is generally known that metal coordination compounds have different coordination numbers depending on the metal and on the oxidation state of the metal, ie bind a different number of ligands.
- An aryl group for the purposes of this invention contains 6 to 40 carbon atoms; a heteroaryl group in the context of this invention contains 2 to 40 carbon atoms and at least one heteroatom, with the proviso that the sum of
- an aryl group or heteroaryl group is either a simple aromatic cycle, ie benzene, or a simple heteroaromatic cycle, for example pyridine, pyrimidine, thiophene, etc., or a fused aryl or heteroaryl group, for example naphthalene, anthracene, phenanthrene, quinoline, isoquinoline, etc. understood.
- An aromatic ring system in the sense of this invention contains 6 to 60 carbon atoms in the ring system.
- a heteroaromatic ring system in the sense of this invention contains 1 to 60 C atoms and at least one heteroatom in the ring system, with the proviso that the sum of C atoms and heteroatoms gives at least 5.
- the heteroatoms are preferably selected from N, O and / or S.
- An aromatic or heteroaromatic ring system in the sense of this invention is to be understood as meaning a system which does not necessarily contain only aryl or heteroaryl groups, but in which several aryls are also present - or heteroaryl groups by a non-aromatic unit (preferably less than 10% of the atoms other than H), such as.
- N or O atom or a carbonyl group may be interrupted.
- systems such as 9,9'-spirobifluorene, 9,9-diarylfluorene, triarylamine, diaryl ethers, stilbene, etc. are to be understood as aromatic ring systems in the context of this invention, and also systems in which two or more aryl groups, for example by a linear or cyclic alkyl group or interrupted by a silyl group.
- systems in which two or more aryl or heteroaryl groups are bonded directly to each other, such as.
- biphenyl or terphenyl also be understood as an aromatic or heteroaromatic ring system.
- a cyclic alkyl, alkoxy or thioalkoxy group is understood as meaning a monocyclic, a bicyclic or a polycyclic group.
- a C 1 to C 40 -alkyl group in which individual H atoms or CH 2 groups may also be substituted by the abovementioned groups, for example the radicals methyl, ethyl, n-propyl, i-propyl , Cyclopropyl, n-butyl, i-butyl, s-butyl, t-butyl, cyclobutyl, 2-methylbutyl, n-pentyl, s-pentyl, tert-pentyl, 2-pentyl, neo-pentyl, cyclopentyl, n-hexyl , s-hexyl, tert -hexyl, 2-hexyl, 3-hexyl, neo-hexyl, cyclohexyl, 1-methylcyclopentyl, 2-methylpentyl, n-heptyl, 2-heptyl
- Pentenyl cyclopentenyl, hexenyl, cyclohexenyl, heptenyl, cycloheptenyl, octenyl, cyclooctenyl or cyclooctadienyl understood.
- alkynyl group is meant, for example, ethynyl, propynyl, butynyl, pentynyl, hexynyl, heptynyl or octynyl.
- a C to C 40 alkoxy group for example, methoxy, trifluoromethoxy, ethoxy, n-
- aromatic or heteroaromatic ring system having 5-60 aromatic ring atoms, which may be substituted in each case with the abovementioned radicals R and which may be linked via any position on the aromatic or heteroaromatic, are understood, for example, groups which are derived from benzene, Naphthalene, anthracene, benzanthracene, phenanthrene, benzophenanthrene, pyrene, chrysene, perylene, fluoranthene, benzfluoranthene, naphthacene, pentacene, benzpyrene, biphenyl, biphenylene, terphenyl, terphenylene, fluorene, spirobifluorene, dihydrophenanthrene, dihydropyrene, tetrahydropyrene, cis- or trans indenofluorene, cis or trans monobenzoindenofluorene, cis or trans dibenzoindene
- M is a transition metal or a main group metal. If M for one
- Main group metal is, then it is preferably a metal of the third, fourth or fifth main group, in particular for tin.
- M is a transition metal, with lanthanides and actinides being excluded, in particular being a tetracoordinate, a pentacoordinate or a hexacoordinate transition metal, more preferably selected from the group consisting of chromium, molybdenum, Tungsten, rhenium,
- the metals can be present in different oxidation states.
- M is a tetracoordinate metal
- the subscript n is 1 or 2.
- a preferred tetracoordinate metal is Pt (II).
- M is a hexacoordinated metal
- a preferred hexacoordinated metal is Ir (III).
- the ligand L are preferably one, two, three or four groups X, more preferably one, two or three groups X, most preferably one or two groups X for N.
- each X which stands for N at least one X adjacent to this N is CR 1 .
- preferred substructures of the formula (2) are the substructures of the following formulas (7) to (13),
- preferred substructures of the formula (2) are the substructures of the following formulas (14) to (27),
- preferred substructures of the formula (2) are the substructures of the following formulas (28) to (36),
- preferred substructures of the formula (2) are the substructures of the following formulas (37) and (38),
- R 1 is attached as a substituent adjacent to at least one X, which is nitrogen.
- a group R 1 is attached as a substituent adjacent to each X which is nitrogen.
- R 1 is a group selected from CF 3 , OCF 3 , branched or cyclic alkyl or alkoxy groups having at least 3 C atoms, triply substituted silyl groups, aromatic or heteroaromatic ring systems or aralkyl or heteroaralkyl. These groups are sterically demanding groups.
- R 1 is an alkyl group
- this alkyl group preferably has 4 to 10 C atoms. It is furthermore preferably a secondary or tertiary alkyl group in which the secondary or tertiary carbon atom is bonded either directly to the ligand or via a
- CH2 group is bound to the ligand.
- This alkyl group is particularly preferably selected from the structures of the following formulas (R 1 -1) to (R 1 -33), wherein in each case the attachment of these groups to the ligand is also shown:
- R 1 is an alkoxy group
- this alkoxy group preferably has 3 to 10 C atoms.
- this alkoxy group is selected from the structures of the following formulas (R 1 -34) to (R 1 -47), wherein in each case the attachment of these groups to the ligand is also shown:
- R 1 is a dialkylamino group
- each of these alkyl groups preferably has 1 to 8 C atoms, more preferably 1 to 6 C atoms.
- suitable alkyl groups are methyl, ethyl or the structures listed above as groups (R 1 -1) to (R 1 -33).
- the dialkylamino group is particularly preferably selected from the structures of the following formulas (R 1 -48) to (R 1 -55), where in each case the attachment of these groups to the ligand is also shown:
- Lig denotes the attachment of the alkyl group to the ligand.
- R 1 is an aralkyl group
- this aralkyl group is preferably selected from the structures of the following formulas (R 1 -56) to (R 1 -69), wherein in each case the attachment of these groups to the ligand is also shown:
- Lig denotes the attachment of the aralkyl group to the ligand and the phenyl groups can each be substituted by one or more radicals R 2 .
- the alkyl, alkoxy, dialkylamino and aralkyl groups may, depending on the exact structure, also have one or more stereocenters. Since the basic structure of the complex can also be a chiral structure, it is possible to form diastereomers, especially if there are several such alkyl, alkoxy, dialkylamino and aralkyl groups with stereocenters.
- the complexes according to the invention then comprise both the mixtures of the different diastereomers or the corresponding racemates as well as the individual isolated diastereomers or enantiomers.
- R is an aromatic or heteroaromatic ring system
- this aromatic or heteroaromatic ring system preferably has 5 to 30 aromatic ring atoms, more preferably 5 to 24 aromatic ring atoms. Furthermore, this contains aromatic or
- heteroaromatic ring system does not prefer aryl or heteroaryl groups in which more than two aromatic six-membered rings are condensed directly to each other.
- the aromatic or heteroaromatic ring system contains no condensed aryl or heteroaryl groups at all, and most preferably it contains only phenyl groups.
- the aromatic ring system is preferably selected from the structures of the following formulas (R 1 -70) to (R 1 -84), where in each case the attachment of these groups to the ligand is shown:
- Lig denotes the attachment of the aromatic or heteroaromatic ring system to the ligand and the phenyl groups may each be substituted by one or more radicals R 2 .
- heteroaromatic ring system is preferably selected from the structures of the following formulas (R 1 -85) to (R 1 -112), wherein in each case the attachment of these groups to the ligand is also shown:
- Lig denotes the attachment of the aromatic or heteroaromatic ring system to the ligand and the aromatic and heteroaromatic groups may each be substituted by one or more radicals R 2 .
- radicals R at each occurrence are identically or differently selected from the group consisting of H, D, F, N (R 2 ) 2 , a straight-chain alkyl group having 1 to 6 C atoms or a branched or cyclic alkyl group having 3 to 10 carbon atoms, wherein one or more H atoms may be replaced by D or F, or an aromatic or heteroaromatic ring system having 5 to 24 aromatic ring atoms, each by one or several radicals R 2 may be substituted; in this case, two adjacent radicals R or R with R 1 can also form a mono- or polycyclic aliphatic ring system with one another.
- substituents which have a + M effect and which substituents have an M effect is known to one skilled in the art of organic chemistry.
- substituents which have a + M effect are F, Cl, Br, I, amines, alkoxy groups, OH, N (R 2 ) 2 or thioalkoxy groups.
- substituents having an M effect are CN, NO 2 , ketones, aldehydes, phosphine oxide groups, sulfoxide groups or sulfones.
- the substituent R which is bonded in the ortho position to the metal coordination, represents a group which also coordinates or binds to the metal M.
- Groups R are aryl or heteroaryl groups, for example phenyl or pyridyl, aryl or alkyl cyanides, aryl or alkyl isocyanides, amines or amides, alcohols or alcoholates, thio alcohols or thioalcoholates,
- the substructures ML of the following formulas (39) to (44) are accessible:
- Formula (43) wherein the symbols and indices used have the same meanings as described above, X 1 is the same or different at each occurrence of C or N and W is the same or different at each occurrence for S, O or NR 2 stands.
- a bridging unit V may also be present which links this ligand L with one or more further ligands L or L '.
- a bridging unit V is present, so that the ligands tridentate or polydentate or polypodal Character.
- Preferred structures with polydentate ligands or with polydentate ligands are the metal complexes of the following formulas (45) to (50),
- Formula (49) Formula (50) where the symbols used have the abovementioned meanings, V preferably representing a single bond or a bridging unit containing 1 to 80 atoms from the third, fourth, fifth and / or sixth main group (group 13, 14, 15 or 16 according to IUPAC) or a 3- to 6-membered homo- or heterocycle, which covalently connects the partial ligands L with each other or L with L 'with each other.
- the bridging unit V can also be constructed asymmetrically, ie the combination of V to L or L 'does not have to be identical.
- the bridging unit V may be neutral, single, double or triple negative or single, double or triple positively charged.
- V is preferably neutral or simply negative or simply positively charged, more preferably neutral.
- the charge of V is preferably chosen so that a total of a neutral complex is formed.
- the preferences mentioned above for the substructure ML n apply to the ligands and n is preferably at least 2.
- V 1 is CR 2 , NR, O or S.
- group V has no significant influence on the electronic properties of the complex, as the task of this group is essentially to: by bridging L with each other or with L 'to increase the chemical and thermal stability of the complexes.
- V is a trivalent group, ie three ligands L are bridged with one another or two ligands L with L 'or one ligand L with two ligands L', V is preferably the same or different at each occurrence selected from the group consisting of B, B ( R 2 ) -. B (C (R 2 ) 2 ) 3,
- N (C O) 3> N (C (R 2 ) 2 C (R 2 ) 2 ) 3 , (R 2 ) N (C (R 2 ) 2 C (R) 2 ) + , P, P (R 2 ) + , PO, PS, PSe, PTe, P (O) 3 , PO (O) 3 , P (OC (R 2 ) 2 ) 3 , PO (OC (R 2 ) 2 ) 3 , P (C ( R 2 ) 2 ) 3 ,
- the other symbols used have the meanings given above.
- V stands for a group CR 2
- the two radicals R can also be linked to one another so that structures such as, for example, 9,9-fluorene are suitable groups V.
- V is a divalent group, ie two ligands L bridged to each other or a ligand L with L ', V is preferably the same or different at each occurrence selected from the group consisting of BR 2 , B (R 2 ) 2 ⁇
- Formula (63) wherein the dashed bonds in each case indicate the bond to the partial ligands L or L ', Y in each occurrence is identical or different for C (R 2 ) 2 , N (R 2 ), O or S. and the other symbols used in each case have the meanings given above.
- preferred ligands U are described as they occur in formula (1).
- the ligand groups L ' may also be selected if these are bonded to L via a bridging unit V, as indicated in formulas (45) to (50).
- the ligands L ' are preferably neutral, monoanionic, dianionic or trianionic ligands, particularly preferably neutral or monoanionic ligands.
- the ligands L ' may also be bonded to L via a bridging group V.
- Preferred neutral, monodentate ligands U are selected from the group consisting of carbon monoxide, nitrogen monoxide, alkyl cyanides, such as.
- alkyl cyanides such as.
- amines such as.
- Trifluorophosphine trimethylphosphine, tricyclohexylphosphine, tri-ieri-butylphosphine, triphenylphosphine, tris (pentafluorophenyl) phosphine, dimethylphenylphosphine, methyldiphenylphosphine, bis (tert-butyl) phenylphosphine, phosphites, such as. For example, trimethyl phosphite, triethyl phosphite, arsines, such as.
- Trifluorarsine trimethylarsine, tricyclohexylarsine, tri-tert-butylarsine, triphenylarsine, tris (pentafluorophenyl) -arsine, stibines, such as. Trifluorostibine, trimethylstibine, tricyclohexylstibine, tri-ferf-butylstibine, triphenylstibin, tris (pentafluorophenyl) stibine, nitrogen-containing heterocycles, such as. As pyridine, pyridazine, pyrazine, pyrimidine, triazine, and carbenes, in particular Arduengo carbenes.
- Preferred monoanionic, monodentate ligands L ' are selected from hydride, pointing, the halides F ", Cl ⁇ , Br ⁇ and ⁇ , Alkylacetyliden such.
- Propanolate, / so-propanolate, terf-butylate, phenolate, aliphatic or aromatic thioalcoholates such.
- Carboxylates such as. Acetate, trifluoroacetate, propionate, benzoate,
- Aryl groups such as. Phenyl, naphthyl, and anionic nitrogen-containing heterocycles such as pyrrolidine, imidazolide, pyrazolide.
- the alkyl groups in these groups are preferably C 1 -C 6 -alkyl groups, more preferably C 1 -C 10 -alkyl groups, very particularly preferably C 1 -C 4 -alkyl groups.
- An aryl group is also understood to mean heteroaryl groups. These groups are as defined above.
- Preferred neutral or mono- or dianionic, bidentate or higher-dentate ligands L ' are selected from diamines, such as.
- Example ethylene diamine, ⁇ , ⁇ , ⁇ ', ⁇ ' tetramethylethylenediamine, propylenediamine, ⁇ , ⁇ , ⁇ ', ⁇ ' - tetramethylpropylenediamine, cis- or trans-diaminocyclohexane, cis- or trans-N, N, N ' , N'-tetramethyldiaminocyclohexane, imines, such as. B.
- diphosphines such as.
- acetylacetone benzoylacetone
- 1,1-diketone 1,
- 3-keto esters such.
- ethyl acetoacetate carboxylates derived from aminocarboxylic acids, such as.
- pyridine-2-carboxylic acid quinoline-2-carboxylic acid, glycine, ⁇ , ⁇ -dimethylglycine, alanine, N, N-dimethylamino-alanine
- salicyliminates derived from salicylimines, such as.
- methylsalicylimine, ethylsalicylimine, phenylsalicylimine dialcoholates derived from dialcohols, such as.
- ethylene glycol, 3-propylene glycol and dithiolates derived from dithiols, such as. B. 1, 2-ethylenedithiol, 1, 3-propylenedithiol.
- Preferred tridentate ligands are borates of nitrogen-containing heterocycles, such as. As tetrakis (1-imidazolyl) borate and tetrakis (1-pyrazolyl) borate.
- bidentate monoanionic, neutral or dianionic ligands L ' in particular monoanionic ligands which have with the metal a cyclometallated five-membered ring or six-membered ring with at least one metal-carbon bond, in particular a cyclometallated five-membered ring.
- ligands such as are generally used in the field of phosphorescent metal complexes for organic electroluminescent devices, ie, phenylpyridine, naphthylpyridine, phenylquinoline, phenylisoquinoline, etc. ligands, each of which may be substituted by one or more R radicals.
- phosphorescent metal complexes for organic electroluminescent devices, ie, phenylpyridine, naphthylpyridine, phenylquinoline, phenylisoquinoline, etc.
- Electroluminescent devices a plurality of such ligands is known, and he can without further inventive step other such
- ligand L for compounds according to formula (1).
- the combination of two groups as represented by the following formulas (65) to (92), is particularly suitable, one group preferably bonding via a neutral nitrogen atom or a carbene carbon atom and the other group preferably via a negatively charged Carbon atom or a negatively charged nitrogen atom binds.
- the ligand L 'can then be formed from the groups of the formulas (65) to (92) by virtue of these groups being identified in each case by the to draw the marked position together. The position at which the groups coordinate to the metal are indicated by * .
- These groups can also be bound to the ligand L via one or two bridging units V.
- X is the same or different CR or N on each occurrence, and R has the same meaning as described above.
- a maximum of three symbols X in each group represent N, more preferably, at most two symbols X in each group represent N, most preferably, at most one symbol X in each group represents N. More preferably, all symbols X stand for CR.
- formulas (76) to (80) may also contain oxygen instead of sulfur.
- ligands L ' are 1,3,5-cis, cis-cyclohexane derivatives, in particular of the formula (93), 1,1,1-tri (methylene) methane derivatives, in particular of the formula (94) and 1, 1, 1- trisubstituted methanes, in particular of the formula (95) and (96),
- Formula (94) wherein in the formulas in each case the coordination to the metal M is shown, R has the abovementioned meaning and A, identically or differently on each occurrence, stands for 0 ⁇ , S ⁇ , ⁇ COO, PR 2 or NR 2.
- H atoms may be replaced by D or F, or an aromatic or heteroaromatic ring system having 5 to 14 aromatic ring atoms, each of which may be substituted by one or more radicals R 2 ; Two or more adjacent radicals R may also form a mono- or polycyclic, aliphatic, aromatic and / or benzo-fused ring system with one another.
- radicals R at each occurrence are identically or differently selected from the group consisting of H, D, F, Br, CN, B (OR 2 ) 2 , a straight-chain alkyl group having 1 to 5 C atoms, in particular methyl, or a branched or cyclic alkyl group having 3 to 5 C-atoms, in particular iso-propyl or tert-butyl, wherein one or more H atoms may be replaced by D or F, or an aromatic or heteroaromatic ring system having 5 to 12 aromatic ring atoms, the each may be substituted by one or more radicals R 2 ; two or more radicals R may also together form a mono- or polycyclic, aliphatic, aromatic and / or benzoannulated ring system.
- the complexes according to the invention can be facial or pseudofacial, or they can be meridional or pseudomeridional.
- the abovementioned preferred embodiments can be combined with one another as desired. In a particularly preferred embodiment of the invention, the abovementioned preferred embodiments apply simultaneously.
- the metal complexes according to the invention can in principle be prepared by various methods. However, the methods described below have been found to be particularly suitable.
- Another object of the present invention is a process for preparing the metal complex compounds of formula (1) by reacting the corresponding free ligands with metal alkoxides of the formula (97), with metal ketoketonates of the formula (98), with metal halides of the formula ( 99) or with dimeric metal complexes of the formula (100),
- metal compounds in particular iridium compounds, which carry both alcoholate and / or halide and / or hydroxyl and also ketoketonate radicals. These connections can also be loaded.
- iridium compounds which are particularly suitable as starting materials are disclosed in WO 2004/085449.
- [IrCl 2 (acac) 2 r for example Na [IrCl 2 (acac) 2 ]
- metal complexes with acetylacetonate derivatives as ligands for example Ir (acac) 3 or tris (2,2,6,6-tetramethylheptane). 3,5-dionato) iridium, and lrCl 3 xH 2 O, where x is usually a number between 2 and 4.
- Suitable platinum starting materials are, for example, PtCl 2 , K 2 [PtCl 4 ],
- Heteroleptic complexes can also be used, for example, according to WO
- melt means that the ligand is melted and the metal precursor is dissolved or suspended in this melt.
- the compounds of the invention according to formula (1) can be obtained in high purity, preferably more than 99% (determined by means of 1 H-NMR and / or HPLC).
- the compounds according to the invention can also be made soluble by suitable substitution, for example by longer alkyl groups (about 4 to 20 C atoms), in particular branched alkyl groups, or optionally substituted aryl groups, for example xylyl, mesityl or branched terphenyl or quaterphenyl groups.
- suitable substitution for example by longer alkyl groups (about 4 to 20 C atoms), in particular branched alkyl groups, or optionally substituted aryl groups, for example xylyl, mesityl or branched terphenyl or quaterphenyl groups.
- Such compounds are then soluble in common organic solvents, such as toluene or xylene at room temperature in sufficient concentration to process the complexes from solution can.
- These soluble compounds are particularly suitable for processing from solution, for example by printing processes.
- An electronic device is understood to mean a device which contains anode, cathode and at least one layer, this layer containing at least one organic or organometallic compound.
- the electronic device according to the invention thus contains anode, cathode and at least one layer which contains at least one compound of the above-mentioned formula (1).
- organic electroluminescent devices OLEDs, PLEDs
- organic integrated circuits O-ICs
- organic field effect transistors O-FETs
- organic thin-film transistors O-TFTs
- organic light-emitting transistors O -LETs
- organic solar cells O-SCs
- organic optical detectors organic photoreceptors
- organic field quench devices O-FQDs
- light-emitting electrochemical cells LOCs
- O-lasers organic laser diodes
- Active components are generally the organic or inorganic materials incorporated between the anode and cathode, for example, charge injection, charge transport or charge blocking materials, but especially emission materials and matrix materials.
- the compounds according to the invention exhibit particularly good properties as emission material in organic electroluminescent devices.
- a preferred embodiment of the invention are therefore organic electroluminescent devices.
- the organic electroluminescent device includes cathode, anode and at least one emitting layer. In addition to these layers, they may also contain further layers, for example one or more hole injection layers, hole transport layers, hole blocking layers, electron transport layers, electron injection layers, exciton blocking layers, electron blocking layers, charge generation layers and / or organic or inorganic p / n junctions.
- interlayers may be introduced between two emitting layers which, for example, have an exciton-blocking function and / or control the charge balance in the electroluminescent device. It should be noted, however, that not necessarily each of these layers must be present.
- the organic electroluminescent device can be any organic electroluminescent device.
- the organic electroluminescent device can be any organic electroluminescent device.
- emissive layers may include multiple emissive layers. If there are multiple emission layers, these preferably have a total of several emission maxima between 380 nm and 750 nm, so that a total of white emission results, ie in the emitting layers different emitting compounds are used, which can fluoresce or phosphoresce. Particular preference is given to three-layer systems, the three layers exhibiting blue, green and orange or red emission (for the basic structure see, for example, WO 2005/01 1013) or systems having more than three emitting layers. It may also be a hybrid system wherein one or more layers fluoresce and one or more other layers phosphoresce.
- Organic electroluminescent device the compound according to formula (1) or the above-mentioned preferred embodiments as an emitting compound in one or more emitting layers.
- the compound of the formula (1) is used as an emitting compound in an emitting layer, it is preferably used in U.S.P.
- the mixture of the compound according to formula (1) and the matrix material contains between 0.1 and 99% by volume, preferably between 1 and 90% by volume, more preferably between 3 and 40% by volume, in particular between 5 and 15% by volume .-% of the compound according to formula (1) based on the total mixture of emitter and matrix material. Accordingly, the mixture contains between 99.9 and 1% by volume, preferably between 99 and 10% by volume, more preferably between 97 and 60% by volume, in particular between 95 and 85% by volume of the matrix material, based on the total mixture Emitter and matrix material.
- the triplet level of the matrix material is higher than the triplet level of the emitter.
- Suitable matrix materials for the compounds according to the invention are ketones, phosphine oxides, sulfoxides and sulfones, for. B. according to WO 2004/013080, WO 2004/093207, WO 2006/005627 or WO
- a plurality of different matrix materials as a mixture, in particular at least one electron-conducting matrix material and at least one hole-conducting matrix material.
- a preferred combination is, for example, the use of an aromatic ketone, a triazine derivative or a phosphine oxide derivative with a triarylamine derivative or a carbazole derivative as a mixed matrix for the metal complex according to the invention. Also preferred is the use of a mixture of a charge transporting
- Matrix material and an electrically inert matrix material which is not or not significantly involved in charge transport, such.
- the triplet emitter with the shorter-wave emission spectrum serves as a co-matrix for the triplet emitter with the longer-wave emission spectrum.
- the complexes of the formula (1) according to the invention as Co-matrix for longer-wave emitting triplet emitter for example, green or red emitting triplet emitter, are used.
- the compounds according to the invention can also be used in other functions in the electronic device, for example as hole transport material in a hole injection or transport layer, as charge generation material or as electron blocking material.
- the complexes according to the invention can be used as matrix material for other phosphorescent metal complexes in an emitting layer.
- low work function metals, metal alloys or multilayer structures of various metals are preferable, such as alkaline earth metals, alkali metals, main group metals or lanthanides (eg, Ca, Ba, Mg, Al, In, Mg, Yb, Sm, etc.).
- alkaline earth metals alkali metals, main group metals or lanthanides (eg, Ca, Ba, Mg, Al, In, Mg, Yb, Sm, etc.).
- alloys of an alkali or alkaline earth metal and silver for example an alloy of magnesium and silver.
- further metals which have a relatively high work function such as, for example, B. Ag, which then usually combinations of metals, such as Mg / Ag, Ca / Ag or Ba / Ag are used.
- a metallic cathode and the organic semiconductor may also be preferred to introduce between a metallic cathode and the organic semiconductor a thin intermediate layer of a material with a high dielectric constant.
- a metallic cathode and the organic semiconductor may also be preferred to introduce between a metallic cathode and the organic semiconductor a thin intermediate layer of a material with a high dielectric constant.
- a material with a high dielectric constant for example, alkali metal or alkaline earth metal fluorides, but also the corresponding oxides or carbonates in question (eg., LiF, Li 2 O, BaF 2 ,
- organic alkali metal complexes for.
- the layer thickness of this layer is preferably between 0.5 and 5 nm.
- materials with a high work function are preferred.
- the anode has a work function greater than 4.5 eV. Vacuum up.
- metals with a high redox potential such as Ag, Pt or Au, are suitable for this purpose.
- metal / metal oxide electrodes eg Al / Ni / NiO x , Al / PtO x ) may also be preferred.
- At least one of the electrodes must be transparent or partially be transparent to allow either the irradiation of the organic material (O-SC) or the extraction of light (OLED / PLED, O-LASER).
- Preferred anode materials are conductive mixed metal oxides. Particularly preferred are indium tin oxide (ITO) or indium zinc oxide (IZO). Also preferred are conductive, doped organic materials, in particular conductive doped polymers, for. B. PEDOT, PANI or derivatives of these polymers.
- the device is structured according to the application), contacted and finally hermetically sealed because the life of such devices drastically shortened in the presence of water and / or air.
- an organic electroluminescent device characterized in that one or more layers are coated with a sublimation process.
- an organic electroluminescent device characterized in that one or more layers are coated with the OVPD (Organic Vapor Phase Deposition) method or with the aid of a carrier gas sublimation. The materials are applied at a pressure between 10 "applied 5 mbar and 1 bar.
- OVPD Organic Vapor Phase Deposition
- a special case of this method is the OVJP (organic vapor jet printing) method in which the materials are applied directly through a nozzle and patterned (eg. BMS Arnold et al., Appl. Phys. Lett., 2008, 92, 053301).
- an organic electroluminescent device characterized in that one or more layers of solution, such. B. by spin coating, or with any printing process, such.
- Suitable substitution As screen printing, flexographic printing, offset printing or Nozzle printing, but more preferably LITI (Light Induced Thermal Imaging, thermal transfer printing) or ink-jet printing (inkjet printing), are produced.
- LITI Light Induced Thermal Imaging, thermal transfer printing
- ink-jet printing inkjet printing
- soluble compounds are necessary, which are obtained for example by suitable substitution.
- the organic electroluminescent device can also be manufactured as a hybrid system by applying one or more layers of solution and depositing one or more other layers.
- a hybrid system by applying one or more layers of solution and depositing one or more other layers.
- the electronic devices according to the invention are distinguished by the following surprising advantages over the prior art:
- Compounds according to formula (1) as emitting materials have excellent efficiency. 3.
- the metal complexes according to the invention make it possible to obtain organic electroluminescent devices which phosphoresce in the blue color range. In particular, blue phosphorescence according to the prior art is very difficult to realize with good efficiencies and lifetimes.
- the metal complexes of the invention are synthetically good and accessible in high yield.
- the following syntheses are carried out under an inert gas atmosphere in dried solvents.
- the metal complexes are additionally handled in the absence of light.
- the solvents and reagents may e.g. from Sigma-ALDRICH or ABCR.
- the reaction mixture is stirred into a solution of 40 g of potassium carbonate in 2000 ml of water, stirred for 15 min. After, extracted with three 300 ml portions of dichloromethane, the organic phase is washed twice with 300 ml of water, once with 500 ml of sat. Saline and dried over sodium sulfate. The dichloromethane solution is filtered through silica gel and the dichloromethane is removed in vacuo. The yellow residue is taken up in 500 ml of methanol, rendered inert with nitrogen while stirring, admixed with 3 g of 10% Pd / C and hydrogenated at room temperature in an autoclave at 2 bar hydrogen pressure.
- a solution of 100 mmol of the aldehyde and 110 mmol of 1, 2-diaminobenzene in a mixture of 150 ml of DMF and 5 ml of water is placed in a cold water bath (about 1000 ml at about 10 ° C) and then with stirring in portions with 33.8 g (55 mmol) of oxone [70693-62-8] added so that the temperature does not rise above 35 ° C. After the exothermic reaction has subsided, it is stirred at room temperature until complete conversion of the aldehyde (1-6 h). The reaction mixture is stirred into a solution of 40 g of potassium carbonate in 2000 ml of water and stirred for 15 min. to.
- Precipitated solids are filtered off, washed three times with 100 ml of water and then sucked dry. Oils are extracted with three 300 ml portions of dichloromethane, the organic phase is washed twice with 300 ml of water and once with 500 ml of sat. Washed brine, and dried over sodium sulfate. The dichloromethane solution is filtered through a short silica gel column, the dichloromethane is removed in vacuo and the residue recrystallized from ethyl acetate / ether or ethanol / water.
- a solution of 100 mmol of the aldehyde and 110 mmol of 1, 2-diaminobenzene in 100 ml of ethanol is placed in an apparatus consisting of a 500 ml flask with water separator and reflux condenser, and 30 min. stirred at room temperature. Then, 40 ml of nitrobenzene are added and the reaction mixture is heated to a gentle reflux (oil bath temperature about 220 ° C.), whereby the ethanol and
- reaction mixture is stirred into a solution of 40 g of potassium carbonate solution in 2000 ml of water, stirred 15 minute after, sucks off the solid formed, washed three times with 100 ml of water and dried in vacuo. Dissolve the solid in about 50 ml of hot ethyl acetate and added on cooling diethyl ether to light turbidity, stirred for 12 h, filtered from the crystals, washed once with 50 ml of diethyl ether and dried in vacuo. Yield: 16.6 g (66 mmol), 66%. Purity:> 95% after 1 H NMR. 8-tert-butyl-quinazoline, S49
- a well-stirred mixture of 100 mmol of the 2- (2-amino-phenyl) -benzimidazole derivative, 350 mmol of the carboxylic acid chloride and 300 mmol of the carboxylic acid is refluxed for 24 to 100 hours with carboxylic acid chlorides boiling below 150 ° C. or at 150 ° C to 180 ° C for carboxylic acid chlorides boiling above 150 ° C, heated until the 2- (2-amino-phenyl) - benzimidazole derivative is reacted. After cooling, the reaction mixture is taken up in ethanol or ethyl acetate (50-200 ml).
- the Reaction mixture is added with good stirring in a mixture of 500 g of ice and 500 ml of aqueous conc. Ammonia stirred. If the product precipitates as a solid, it is filtered off with suction, washed with water and sucked dry. If the product precipitates as an oil, this is extracted with three portions of 300 ml of ethyl acetate. The organic phase is separated, washed with 500 ml of water and concentrated in vacuo. The crude product is taken up in ethyl acetate or dichloromethane, filtered through a short column of Alox, basic, activity grade 1 or silica gel to remove brown impurities. After two recrystallizations
- a mixture of 20.9 g (100 mmol) of 2- (2-aminophenyl) benzimidazole, 42.2 g (350 mmol) of pivaloyl chloride and 30.6 g (300 mmol) of pivalic acid is refluxed for 50 h.
- the reaction mixture is allowed to cool to about 60.degree. C., 100 ml of ethanol are added, the mixture thus obtained is stirred into a mixture of 500 g of ice and 500 ml of conc. Ammonia, stirred for 5 min. after, then sucks from the precipitated solid, washed twice with 100 ml of water and sucks this dry.
- a mixture of 20.9 g (100 mmol) of 2- (2-aminophenyl) benzimidazole, 47.1 g (350 mmol) of 3,3-dimethylbutyric acid chloride and 34.8 g (300 mmol) of 3,3-dimethylbutyric acid is refluxed for 20 h.
- the reaction mixture is allowed to cool to about 60.degree. C., 100 ml of ethanol are added, the mixture thus obtained is stirred into a mixture of 500 g of ice and 500 ml of conc. Ammonia, stirred for 15 min. After, then sucks from the precipitated solid, washed three times with each 00 ml of water and sucks it dry.
- a well-stirred mixture of 100 mmol of the 2- (2- (N-alkylamido) -phenyl) -benzimidazole derivative and 350 mmol of the carbonyl chloride is refluxed for 24 to 100 h - with carboxylic acid chlorides which are present under Boil 50 ° C - or at 150 ° C to 180 ° C - for carboxylic acid chlorides boiling above 150 ° C - heated until the 2- (2- (N-alkylamido) phenyl) - benzimidazole derivative is implemented. If the reaction mixture is too mushy, with an inert, in the boiling point adapted to the carboxylic acid chloride solvent used, for.
- dioxane or diethylene glycol dimethyl ether As dioxane or diethylene glycol dimethyl ether, diluted. After cooling, the reaction mixture is taken up in dioxane (50-200 ml). The reaction mixture is added with good stirring to a mixture of 500 g of ice and 500 ml of aqueous conc. Ammonia stirred. If the product precipitates as a solid, it is filtered off with suction, washed with water and sucked dry. If the product is an oil, it is extracted with three portions of 300 ml of ethyl acetate each time. The organic phase is separated, washed with water and concentrated in vacuo.
- the crude product is taken up in ethyl acetate or dichloromethane, filtered through a short column of Alox, basic, activity grade 1 or silica gel to remove brown impurities. After two recrystallization of the product thus obtained (methanol, ethanol, acetone, dioxane, etc.) this is by Kugelrohr distillation or sublimation (p about 1 x 10 ⁇ 5 mbar, T about 150 - 230 ° C) of low boilers and brown minor components freed. Purity by 1 H-NMR typically> 99.5%.
- a mixture of 25.4 g (100 mmol) of 6-chloro-benzo [4,5] imidazo [1,2-c] quinazoline, S61 and 200 mmol of the sodium alkoxide is refluxed in 200 ml of the corresponding alcohol, until the 6-chloro-benzo [4,5] imidazo [1,2-c] quinazoline is reacted (2-12 h).
- the alcohol or solvent is distilled off, the solid is taken up in 300 ml of water and stirred. After filtration with suction, the solid is washed twice with 100 ml of water and once with 30 ml of cold methanol and then dried in vacuo.
- a solution of 500 mmol of pyridine-3-carboxaldehyde and 550 mmol of 1,2-diaminobenzene in 1000 ml of nitrobenzene is placed in an apparatus consisting of a 2000 ml one-necked flask with tap piece and attached distillation bridge and stirred for 2 h at 200 ° C, the formed Distilled off water. Then raise the temperature to about 215 ° C and distilled from the nitrobenzene in the argon stream. At the end of the distillation, a vacuum of about 100 mbar is applied to remove the last residues of nitrobenzene, then the reaction mixture is allowed to cool.
- a mixture of 100 mmol of the 2- (2-amino-pyridin-3-yl) -benzimidazole derivative (used as an equimolar mixture with the corresponding 2-tert-butylbenzimidazole derivative as obtained from Synthesis 16) and 1 mol of the corresponding carboxylic acid chloride is refluxed for 8 to 40 hours with carbonyl chlorides boiling below 150 ° C, or at 150 ° C to 180 ° C with carboxylic acid chlorides boiling above 150 ° C, until the 2- (2-amino -pyridin-3-yl) benzimidazole derivative is reacted.
- reaction mixture is allowed to cool to 80 ° C, optionally diluted with 100 ml of dioxane, then stirred into a mixture of 500 ml of conc. Ammonia solution and 500 g of ice and stirred for 3 h.
- Example 2- (2-Amino carboxylic acid product Auspyridin-3-yl) chloride prey benzimidazole
- reaction mixture is allowed to cool to 80 ° C, optionally diluted with 100 ml of dioxane, then stirred into a mixture of 500 ml of conc. Ammonia solution and 500 g of ice and stirred for 3 h. It is then filtered off from the solid, washed twice with 100 ml of water and dried in vacuo. The solid is taken up in 1000 ml of ethyl acetate, filtered through a short silica gel column, washed with 500 ml of ethyl acetate, the ethyl acetate is removed in vacuo and the brown residue is recrystallized from methanol.
- the solids thus obtained are ( "180 p approximately 1 x 10 -5 mbar, T - 220 ° C) by sublimation freed of low boilers and non-volatile secondary components purity according to 1 H-NMR is typically> 99.5% pure..
- Example 2- (4-Amino carboxylic acid product auspyridin-3-yl) chloride prey benzimidazole
- Formula (24) Formula (25) Formula (26) Formula (27) A well-stirred mixture of 100 mmol of the 4-chloro-quinazoline derivative, 29.8 g (130 mmol) of 3-bromo-4-amino-6-tert-butylpyridine (S40), 35.0 g (250 mmol) of potassium carbonate, 200 g glass beads (3 mm diameter), 2.6 g (10 mmol) of triphenylphosphine and 450 mg (2 mmol) of palladium (II) acetate in 500 ml of o-xylene is heated under reflux until the 4-chloro-quinazoline derivative is consumed ( typically 16 h).
- Solids are freed from low-boiling components and non-volatile secondary components by sublimation (p ca. 1 ⁇ 10 -5 mbar, T ca. 200-240 ° C.). Purity by 1 H-NMR typically> 99.5%.
- the solids thus obtained are ( "200 p approximately 1 x 10 -5 mbar, T - 240 ° C) by sublimation freed of low boilers and non-volatile secondary components purity according to 1 H-NMR is typically> 99.5% pure..
- the solid is taken up in 100 ml of diethylene glycol, 4 ml of hydrazine hydrate are added and the mixture is heated slowly to 190 ° C. on a water separator. After 16 h, allowed to cool to room temperature, diluted with 50 ml of methanol, sucks from the precipitated crystals, washed three times with 30 ml of methanol and crystallized twice from DMF. The solid is through
- Variant A tris-acetylacetonato-iridium (III) as iridium starting material
- a mixture of 10 mmol Tris-acetylacetonato iridium (III) [15635-87-7] and 60 mmol of the ligand L will be under vacuum ( "5 mbar 10) melted in a 50 ml glass vial.
- the vial is on for the specified time
- the ampoule is opened, the sinter cake is made with 100 g glass beads (3 mm diameter) in 100
- the fine suspension is decanted from the glass spheres, the solid is filtered off with suction and dried in vacuo.
- the dry solid is suspended in a hot extractor on a 10 cm high Alox bed (Alox.
- Basic activity level 1 Basic activity level 1 and then with the specified extractant (original amount about 500 ml) extra- hiert. After completion of the extraction, the extractant is concentrated in vacuo to about 100 ml. Metal complexes which have too good solubility in the extractant are brought to crystallization by the dropwise addition of 200 ml of methanol. The solid of the suspensions thus obtained is filtered off, washed once with about 50 ml of methanol and
- the metal complex is tempered or sublimed.
- the annealing is carried out in a high vacuum (p about 10 ⁇ 6 mbar) in the temperature range of 200 - 300 ° C.
- the sublimation takes place in a high vacuum (p 10 ⁇ 6 mbar) in the temperature range of about 320 to about 400 ° C, wherein the sublimation is preferably carried out in the form of a fractional sublimation.
- the derived metal complexes are obtained as a diastereomeric mixture.
- Variant B tris- (2,2,6,6-tetramethyl-3,5-heptanedionato) iridium as
- lr (L180) 3 L180 lr (L180) 3 such as lr (L162) 29% lr (L181) 3 L181 lr (L181) 3 as Example lr (L62) 36% lr (L182) 3 L182 lr (L182 ) 3 as exemplified by Ir (L162) 44% Ir (L183) 3 L183 Ir (L183) 3 as Ex. Ir (L162) 38% Ir (L184) 3 L184 Ir (L184) 3 as Ex. Ir (L162) 37 % lr (L185) 3 L185 lr (L185) 3 as Ex.
- Step 1
- Step 2 the ampoule is opened, the sinter cake is mixed with 100 g glass balls (3 mm diameter) The mixture is then stirred in 100 ml of the suspension medium for 3 hours while mechanically digested, and the fine suspension is decanted from the glass beads, the solid is filtered off with suction and dried in vacuo.
- the resulting crude chloro-dimer of the formula [Ir (L) 2 Cl] 2 is suspended in a mixture of 75 ml of 2-ethoxyethanol and 25 ml of water, with 13 mmol of the co-ligand CL or of the co-ligand compound CL and 5 mmol sodium carbonate added. After 20 h under reflux, another 75 ml of water are added dropwise, filtered off with suction from the solid, washed three times with 50 ml of water and three times with 50 ml of methanol and dried in vacuo. The dry solid is placed in a hot extractor on a 10 cm high Alox bed (Alox, basic activity level 1) and then extracted with the specified extractant (amount of about 500 ml).
- the extractant After completion of the extraction, the extractant is concentrated in vacuo to about 100 ml. Metal complexes which have too good solubility in the extractant are brought to crystallization by the dropwise addition of 200 ml of methanol. The solid of the suspensions thus obtained is filtered off with suction, washed once with about 50 ml of methanol and dried. After drying, the purity of the metal complex is determined by NMR and / or HPLC. If the purity is below 99.5%, the hot extraction step is repeated, if a purity of 99.5 - 99.9% is reached, the metal complex is tempered or sublimed.
- the annealing is carried out in high vacuum (p about 0 "6 mbar) in the temperature range 200-300 ° C
- the sublimation is carried out in high vacuum (p about 10 -6 mbar) in the temperature range of about 300 to about 390 ° C. wherein the sublimation is preferably carried out in the form of a fractional sublimation.
- Step 1
- the crude chloro-dimer of the formula [Ir (L) 2 Cl] 2 is according to WO 2007/065523, Example 5 in the presence of 80 mmol of the co-ligand CL and 75 mmol of ⁇ , ⁇ -dimethylglycine in 1000 ml of a dioxane-water Mixed (1: 1, vv) further implemented.
- the solid thus obtained is placed in a hot extractor on a 10 cm high Alox bed (Alox, basic activity level 1) and then with the specified extractant (amount of approx.
- the resulting suspensions are filtered off with suction, washed once with about 50 ml of methanol and dried. After drying, the purity of the metal complex is determined by NMR and / or HPLC. If the purity is below 99.5%, the hot extraction step is repeated, if a purity of 99.5 - 99.9% is reached, the metal complex is tempered or sublimed. Annealing is carried out in a high vacuum (p approx. 10 "6 mbar) in
- Step 1
- the resulting crude chloro-dimer of the formula [Ir (L) 2 Cl] 2 is suspended in 00 ml of THF, the suspension is admixed with 40 mmol of the co-ligand CL, 20 mmol of silver (I) trifluoroacetate and 80 mmol of potassium carbonate and Heated under reflux for 24 h. After cooling, the THF is removed in vacuo. The residue is dissolved in 200 ml of a mixture of ethanol and conc. Ammonia solution (1: 1, vv) was added. The suspension is stirred for 1 h at room temperature, the solid is filtered off with suction, twice with 50 ml of a mixture of ethanol and conc.
- Step 1
- the resulting crude chloro-dimer of the formula [Ir (L) 2 Cl] 2 is suspended in 1000 ml of dichloromethane and 150 ml of ethanol, the suspension is mixed with 40 mmol of silver (I) trifluoromethanesulfonate and stirred at room temperature for 24 h. It sucks from the precipitated solid (AgCl) over a short bed of Celite and the filtrate concentrated to dryness in vacuo. The resulting solid is taken up in 100 ml of ethanol, mixed with 30 mmol of the co-ligand CL and then heated under reflux for 30 h.
- the mixture is filtered off with suction from the solid, washed twice with 50 ml of ethanol each time and dried in vacuo.
- the solid thus obtained is placed in a hot extractor on a 10 cm high Alox bed (Alox, basic activity level 1) and then with the indicated Extracting agent (amount of template about 500 ml) extracted. After completion of the extraction, the extractant is concentrated in vacuo to about 100 ml. Metal complexes which have too good solubility in the extractant are brought to crystallization by the dropwise addition of 200 ml of methanol.
- the solid of the suspensions thus obtained is filtered off with suction, washed once with about 50 ml of methanol and dried.
- the purity of the metal complex is determined by NMR and / or HPLC. If the purity is below 99.5%, the hot extraction step is repeated, if a purity of 99.5 - 99.9% is reached, the metal complex is tempered or sublimed.
- the annealing is carried out in a high vacuum (p about 10 ⁇ 6 mbar) in the temperature range of 200 - 300 ° C.
- the sublimation is carried out in a high vacuum (p about 10 ⁇ 6 mbar) in the temperature range of about 300 to about 390 ° C, wherein the sublimation is preferably carried out in the form of a fractional sublimation.
- Alox is replaced by celite in the hot extraction step.
- a mixture of 10 mmol of Ir complex Ir (L) 2 (CL), and 30 mmol of the ligand L will be under vacuum ( "5 mbar 10) melted in a 50 ml glass vial.
- the vial is for the indicated time at the indicated temperature
- the ampoule is opened, the sinter cake is mixed with 100 g glass beads (3 mm diameter) in 100 ml of the suspension medium 3
- the fine suspension is decanted from the glass beads, the solid is filtered off with suction and dried under reduced pressure, and the dry solid is placed in a hot extractor on a 10 cm high Alox bed (Alox, basic activity level 1) 500 ml)
- the extractant is concentrated in vacuo to about 100 ml ie in the extraction medium have too good solubility, are brought by the dropwise addition of 200 ml of methanol to crystallize.
- the solid of the suspensions thus obtained is filtered off with suction, washed once with about 50 ml of methanol and dried. After drying, the purity of the metal complex is determined by NMR and / or HPLC. If the purity is below 99.5%, the heat repeated extraction step, is reached a purity of 99.5 - 99.9%, the metal complex is tempered or sublimed.
- the annealing is carried out in high vacuum (p about 10 "6 mbar) in the temperature range 200-300 ° C, the sublimation is carried out in high vacuum (p about 10". 6 mbar) in the temperature range of about 340 to about 400 ° C, wherein the sublimation is preferably carried out in the form of a fractional sublimation.
- Step 1
- Ligand L 60 ml of 2-ethoxyethanol and 30 ml of water is refluxed for 160 h. After cooling, the product is filtered off with suction from the solid, washed once with 20 ml of a mixture of ethanol and water (1: 1, v) and three times with 10 ml of ethanol each time and dried in vacuo.
- the crude chloro-dimer of the formula [Pt (L) Cl] 2 thus obtained is suspended in a mixture of 60 ml of 2-ethoxyethanol and 20 ml of water and treated with 12 mmol of the co-ligand CL or of the co-ligand compound CL and 12 mmol of sodium carbonate. After 20 h under reflux, another 100 ml of water are added dropwise, filtered off with suction from the solid, washed three times with 50 ml of water and three times with 50 ml of methanol and dried in vacuo. The solid thus obtained is placed in a hot extractor on a 10 cm high bed of celite (Alox, basic activity level 1) and then extracted with the specified extractant (initial amount about 500 ml).
- the Extractant concentrated in vacuo to about 100 ml.
- Metal complexes which have too good solubility in the extraction medium are brought to crystallization by dropwise addition of 200 ml of methanol.
- the solid of the suspensions thus obtained is filtered off with suction, washed once with about 50 ml of methanol and dried. After drying, the purity of the metal complex is determined by NMR and / or HPLC. If the purity is below 99.5%, the hot extraction step is repeated, if a purity of 99.5 - 99.9% is reached, the metal complex is tempered or sublimed.
- the annealing is carried out in high vacuum (p about 10 "6 mbar) in the temperature range 200-300 ° C
- the sublimation is carried out in high vacuum (p about 10 -6 mbar) in the temperature range of about 300 to about 390 °. C, wherein the sublimation preferably in the form of a fractionated
- Ligand L 50 mmol of lithium acetate, anhydrous in 100 ml of glacial acetic acid is refluxed for 60 h. After dropwise addition of 100 ml
- the purity of the metal complex is determined by NMR and / or HPLC. If the purity is below 99.5%, the hot extraction step is repeated; is reached a purity of 99.5 - 99.9%, the Pt complex is sublimated. The sublimation takes place under high vacuum (p ca. 10 "6 mbar) in
- inventive OLEDs and OLEDs according to the prior art is carried out according to a general method according to WO 2004/058911, based on the circumstances described here
- the OLEDs have the following layer structure: substrate / optional hole injection layer (HIL) / hole transport layer (HTL) /
- Electron injection layer EIL
- cathode is formed by a 100 nm thick aluminum layer
- the emission layer always consists of at least one matrix material (host material, host material) and an emitting dopant (dopant, emitter), which is admixed to the matrix material or the matrix materials by co-evaporation in a specific volume fraction.
- the electron transport layer may consist of a mixture of two materials.
- the exact structure of the OLEDs is shown in Table 1.
- the materials used to make the OLEDs are shown in Table 3.
- the OLEDs are characterized by default.
- the electroluminescence spectra, the current efficiency (measured in cd / A) and the voltage (measured at 1000 cd / m 2 in V) are determined from current-voltage-brightness characteristics (IUL characteristic curves).
- IUL characteristic curves current-voltage-brightness characteristics
- LD50 means that the said lifetime is the time at which the luminance has dropped to 50% of the starting luminance, ie from 4000 cd / m 2 to 2000 cd / m 2 .
- the values for the lifetime can be converted to an indication for other starting luminous densities with the aid of conversion formulas known to the person skilled in the art.
- the life for a starting luminous flux of 1000 cd / m 2 is a common statement.
- the compounds according to the invention can be used inter alia as phosphorescent emitter materials in the emission layer in OLEDs.
- the metal complexes with the central atoms Ir and Pt are used.
- the compounds lr (ref) 3 is used.
- the results of the OLEDs are in
- Table 2 summarized. The OLEDs show here that the materials according to the invention lead to efficient blue and green emitting OLEDs.
- HTM EBM2 M4 M3: Ir (L108) 3 ETM1 LiQ nm 5nm 15nm (85% ⁇ .10% .5%) 30nm 2nm
- HTM EBM2 M4.M3 Ir (L134 ETM1 LiQ nm 5nm 15nm (85%: 10%: 5%) 30nm 2nm
- HTM EBM2 M4 M3: Ir (L135) 3 ETM1 LiQ nm 5nm 15nm (85%: 10%: 5%) 30nm 2nm
- HTM EBM1 M2 M3: Ir (L1) 2 (CL1) ETM1 LiQ nm 5nm 15nm (88%: 12%) 30nm 2nm
- HTM EBM2 M4 M3: Ir (L1) 2 (CL7) ETM1 LiQ nm 5nm 15nm (85%: 10%: 5%) 30nm 2nm
- HTM EBM2 M4 M3: Ir (L91 (CL7) ETM1 LiQ nm 5nm 15nm (85%: 10%: 5%) 30nm 2nm
- HIM HTM EBM2 M5 M2: Ir (L165) 3 M5 ETM1 LiQ 20 nm 5 nm 15 nm (10%: 85%: 5%) 10 nm 30 nm 2 nm
- HIM HTM EBM2 M5 M2: Ir (L167) 3 M5 ETM1 LiQ 20 nm 5 nm 15 nm (10%: 85%: 5%) 10 nm 30 nm 2 nm
Abstract
Description















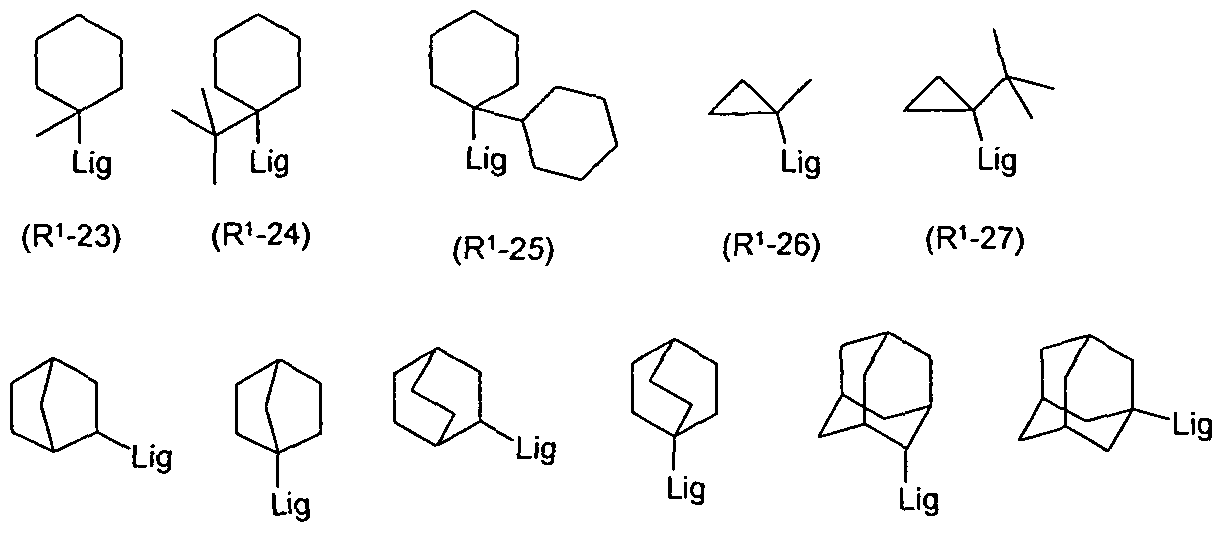




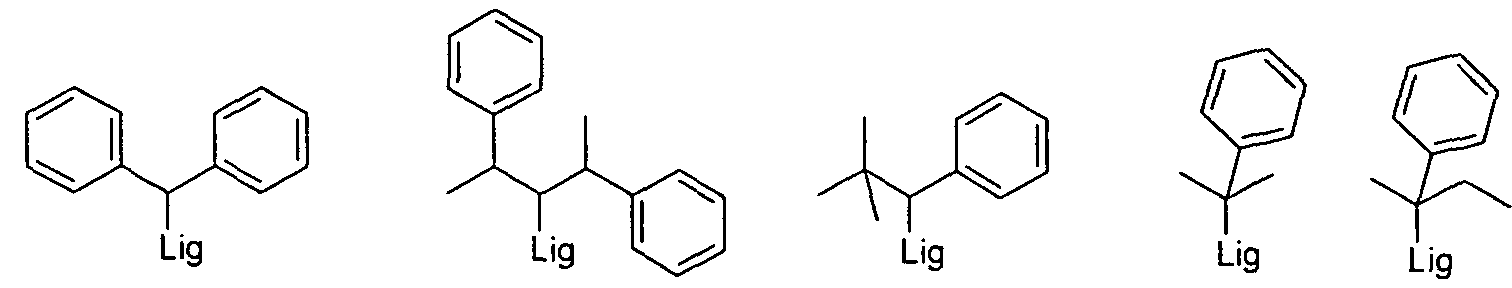
























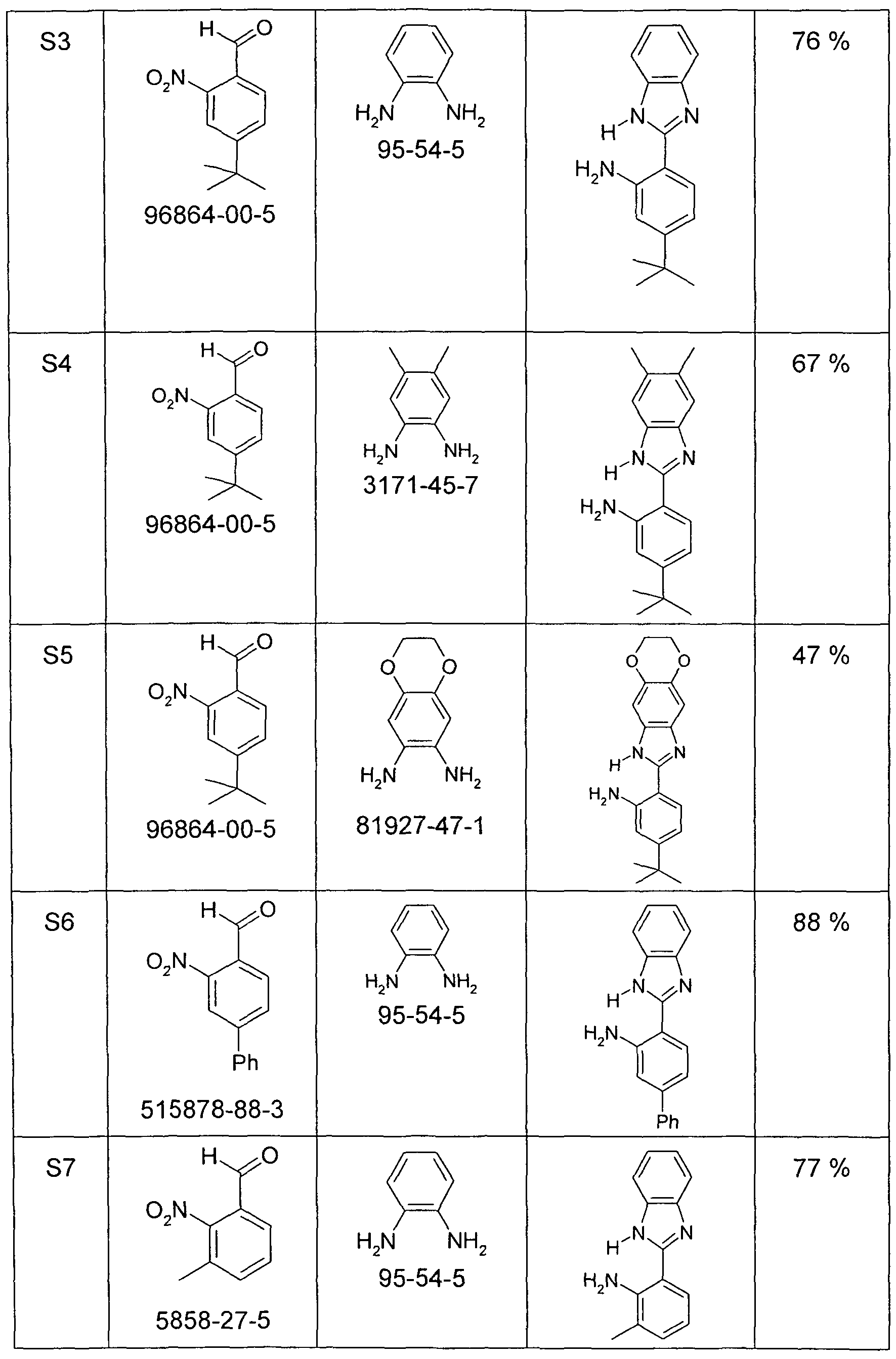




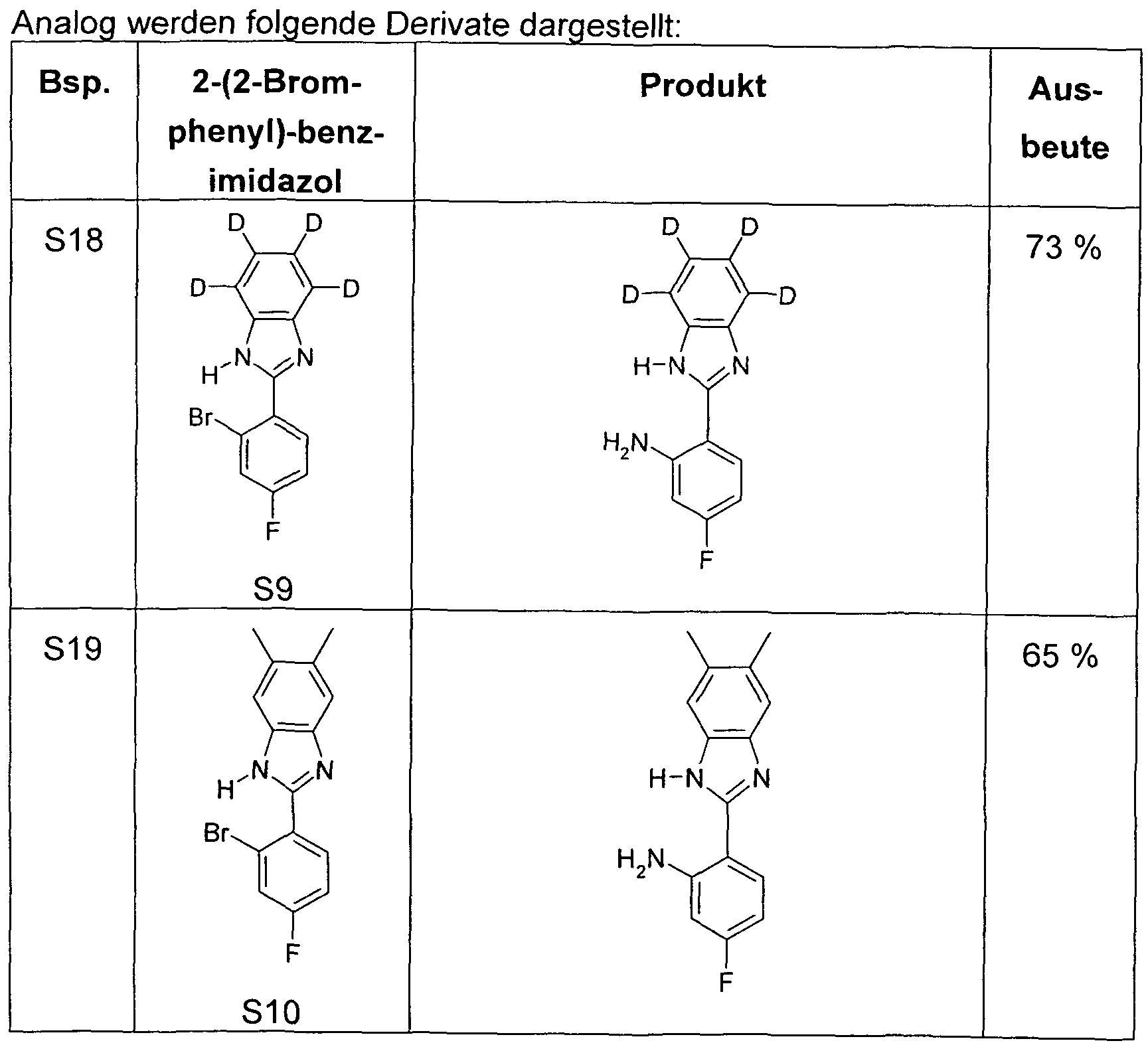
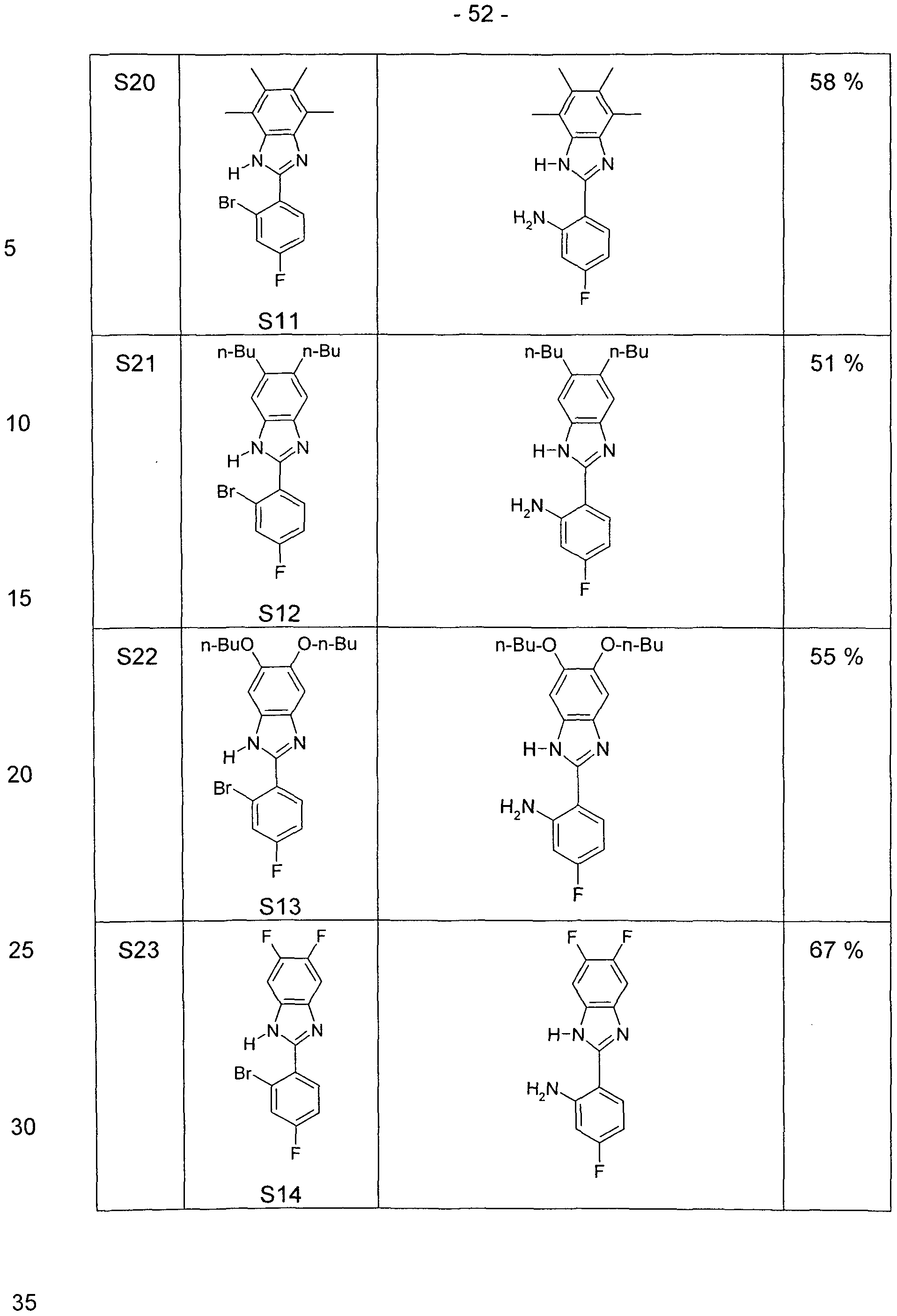
















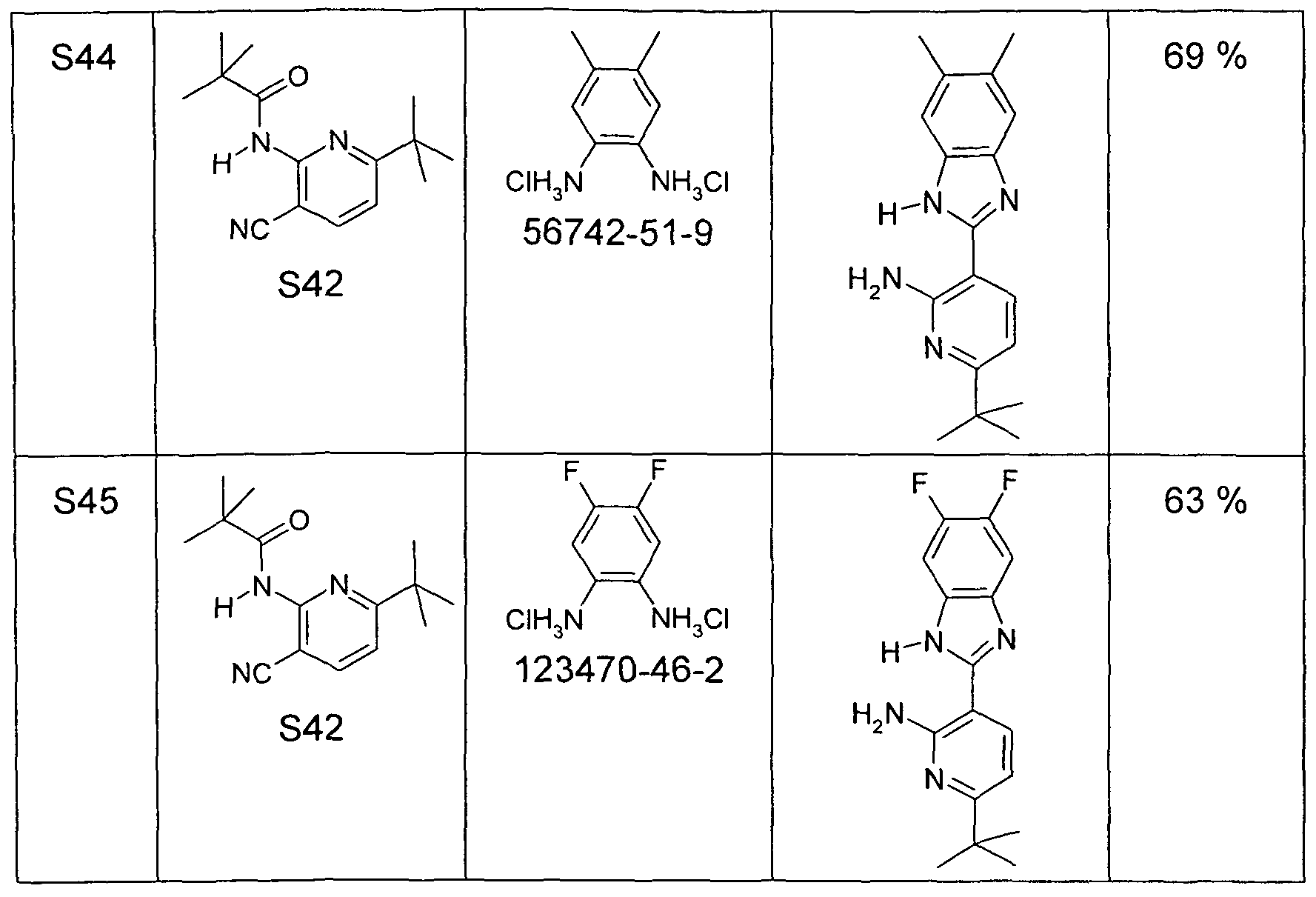




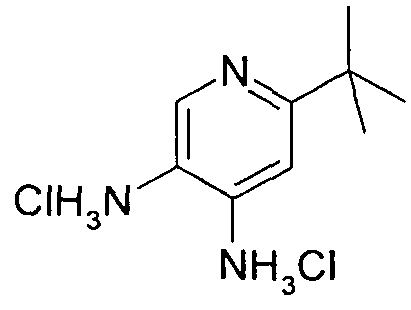










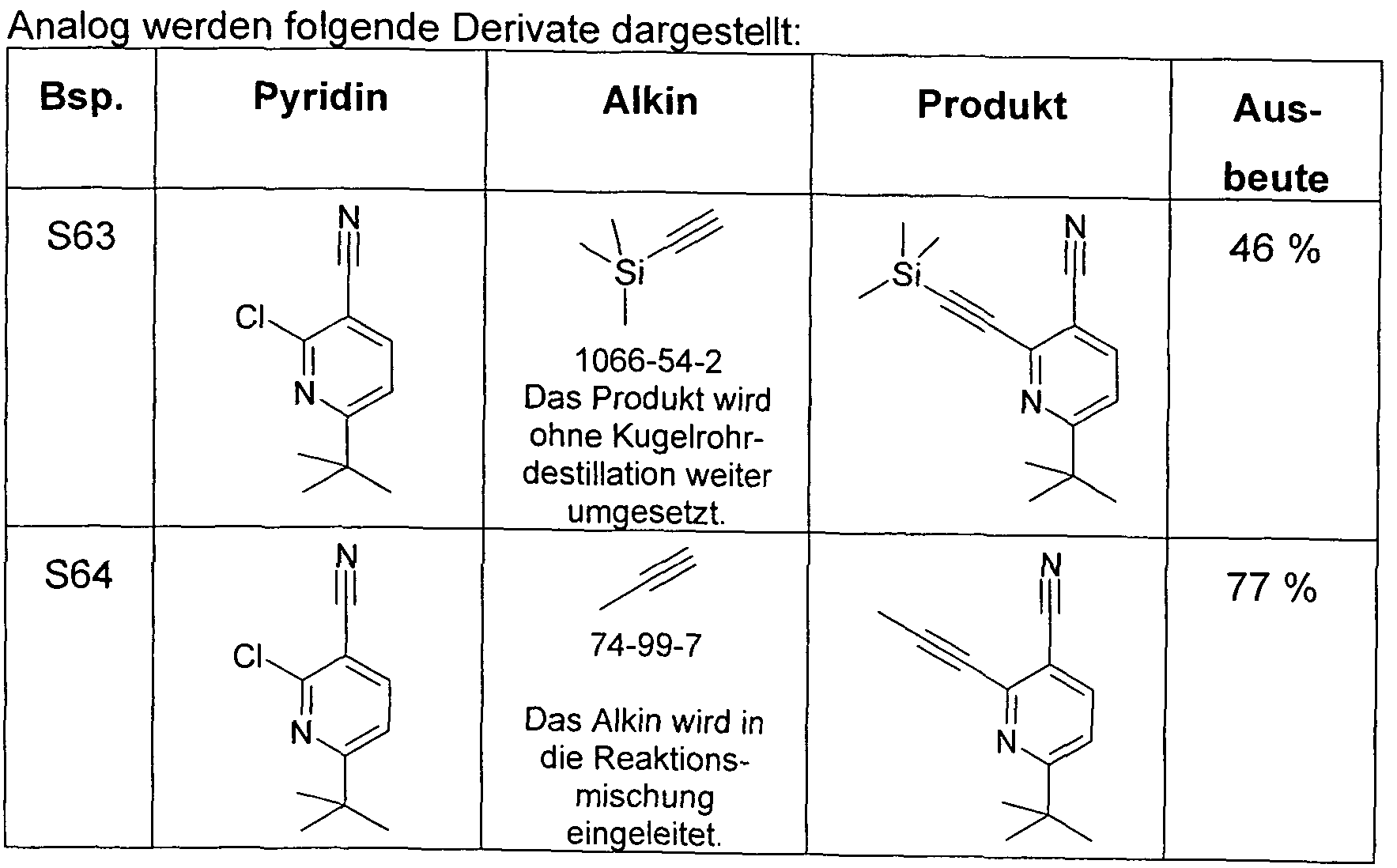











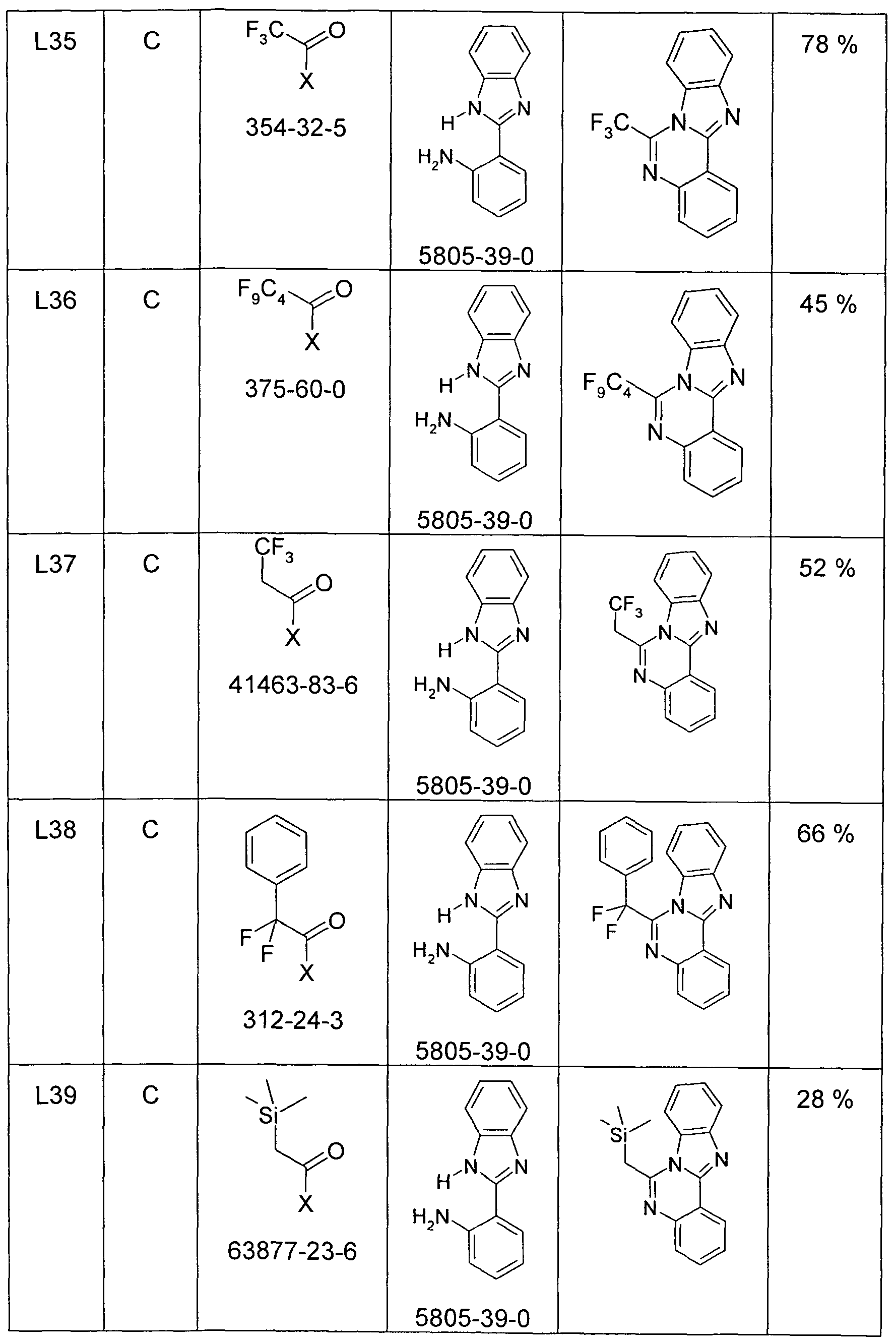































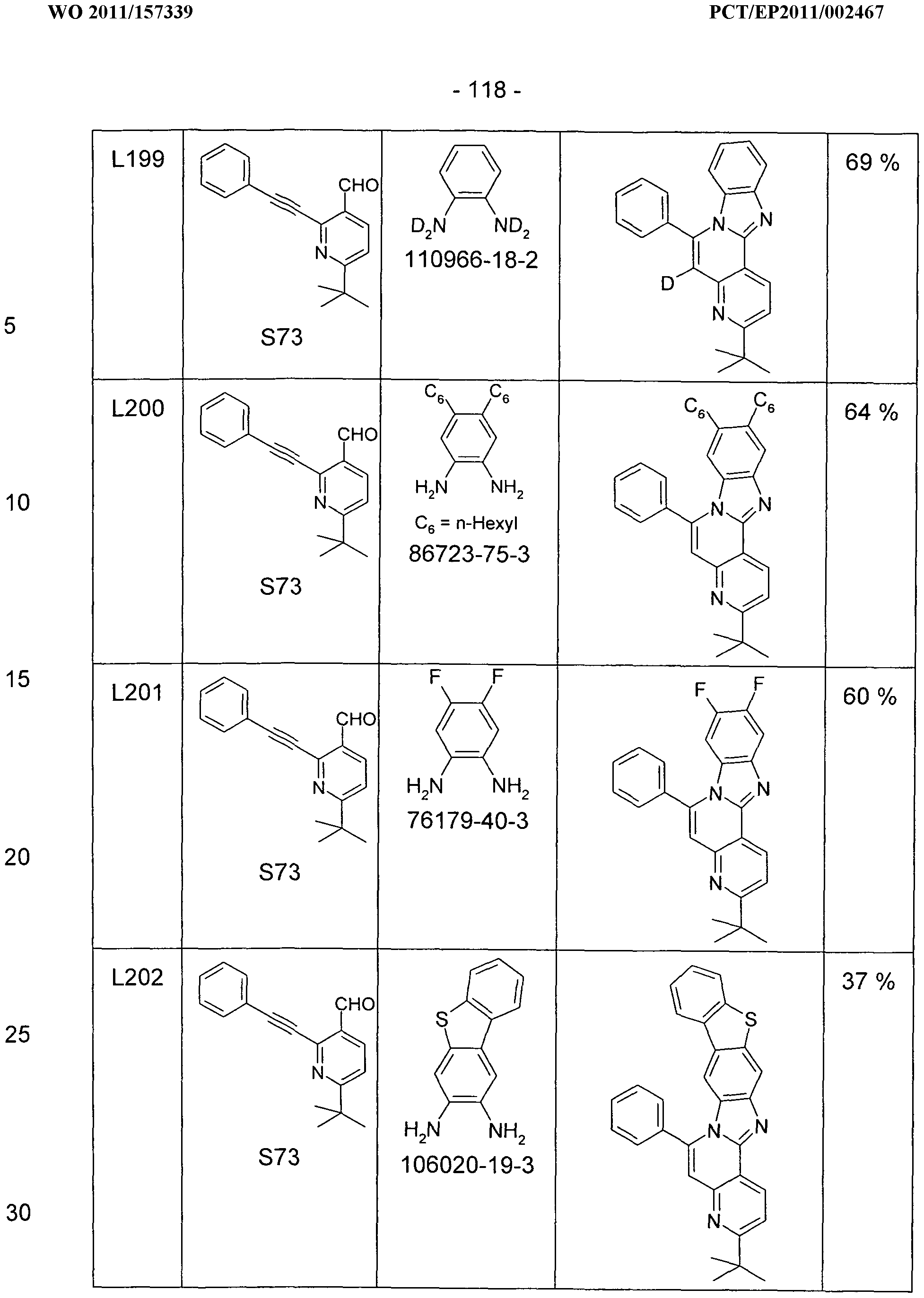






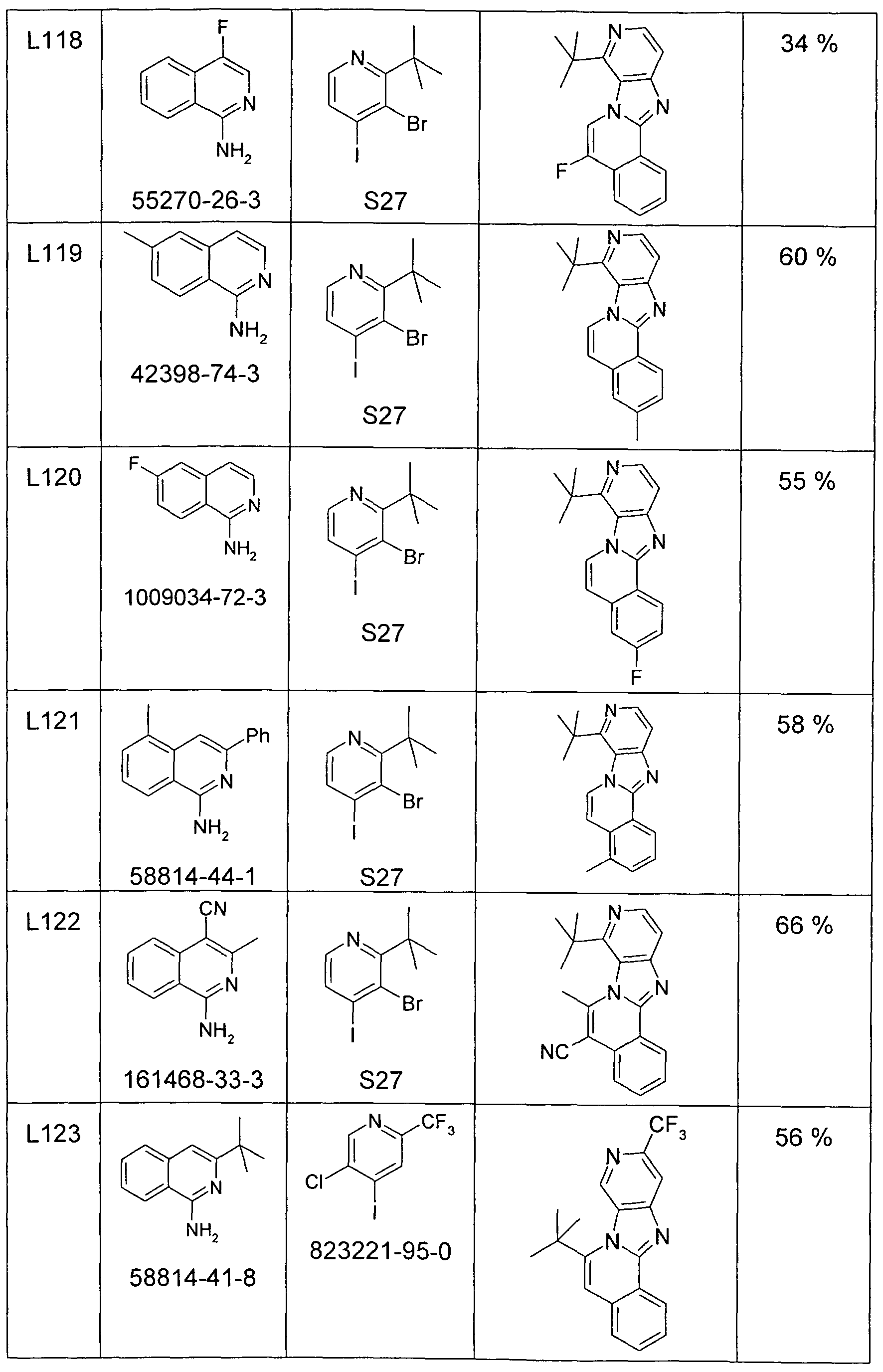












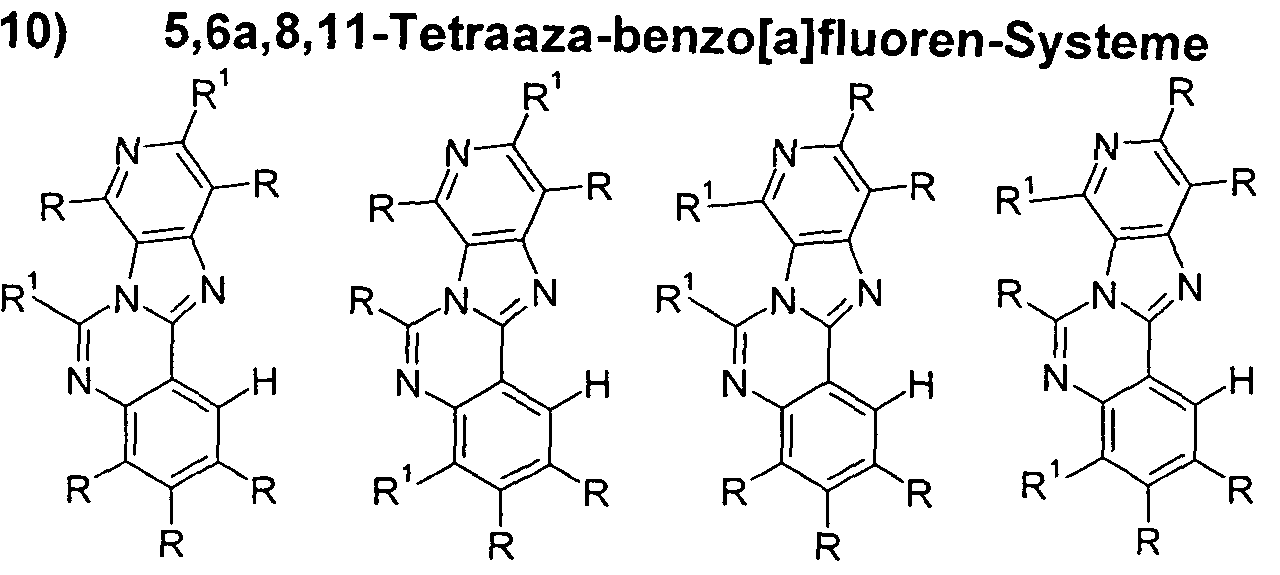









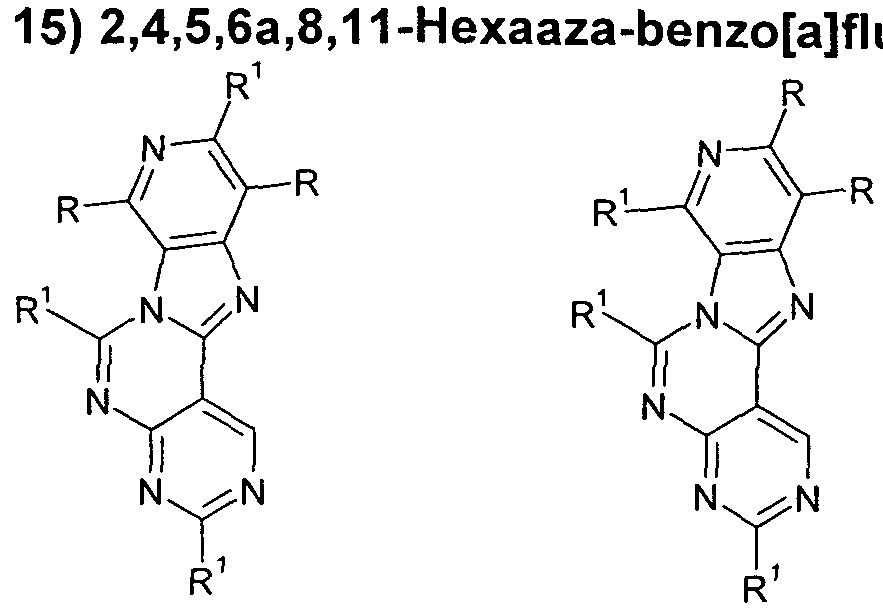




















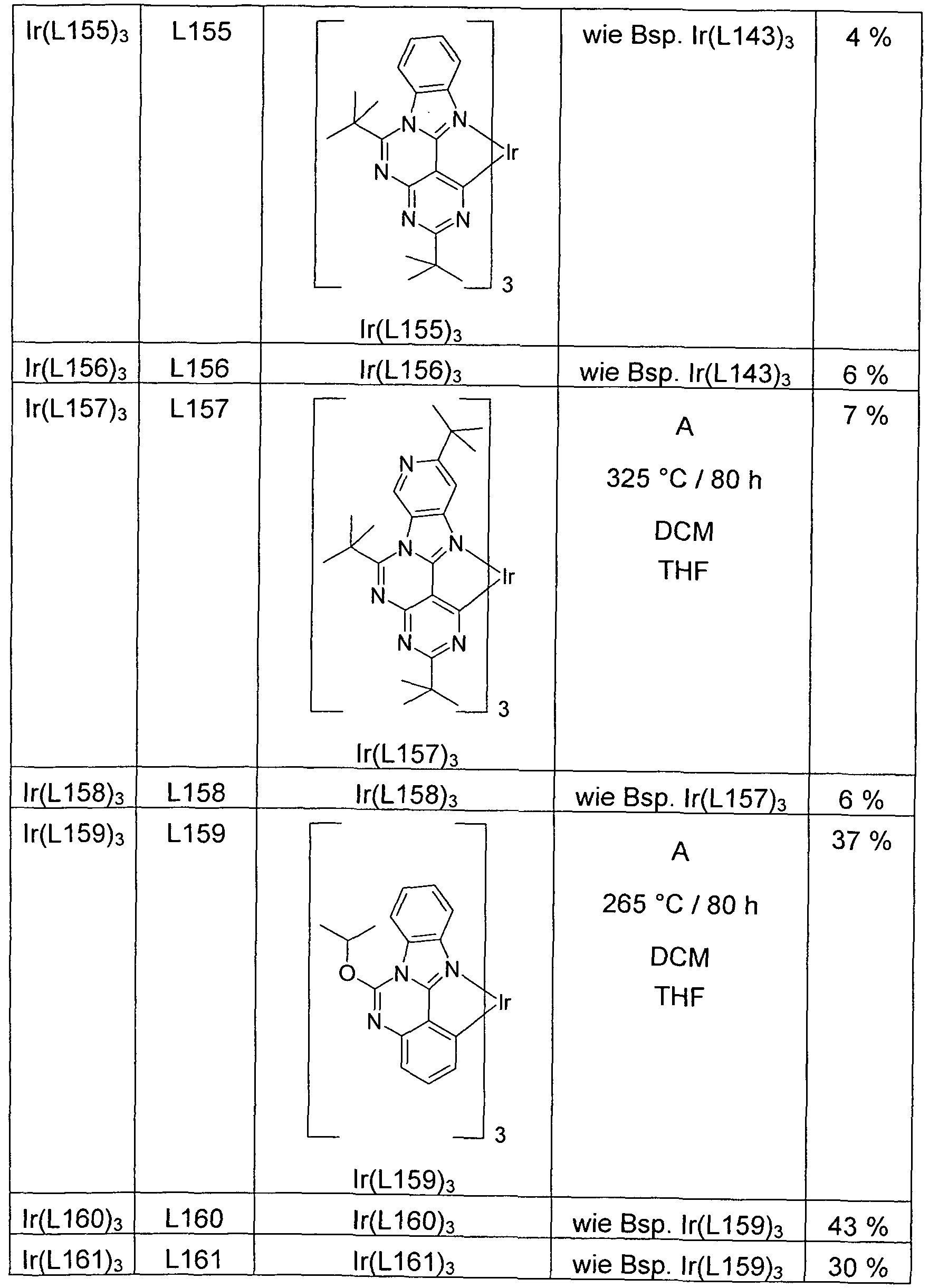




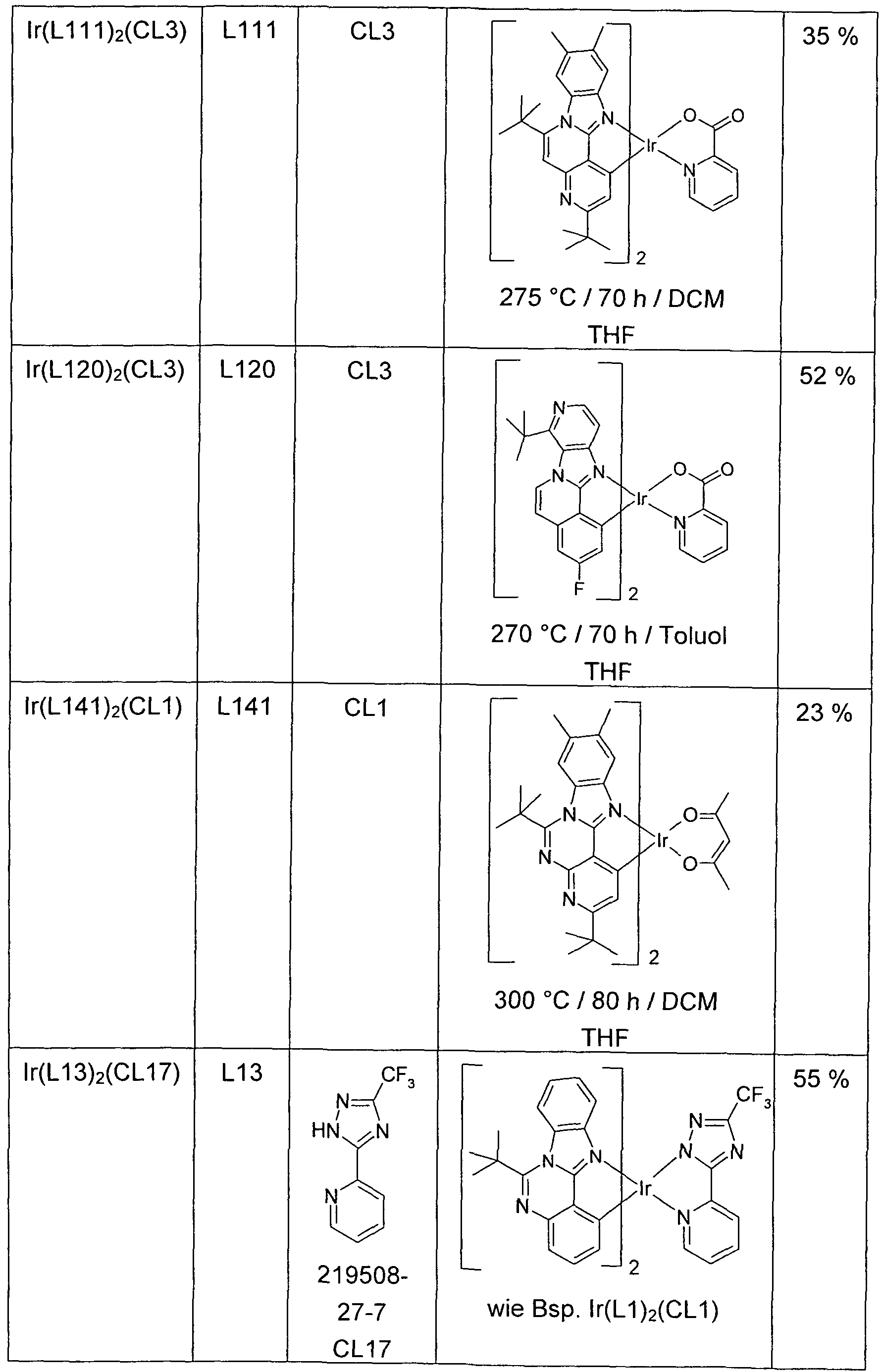

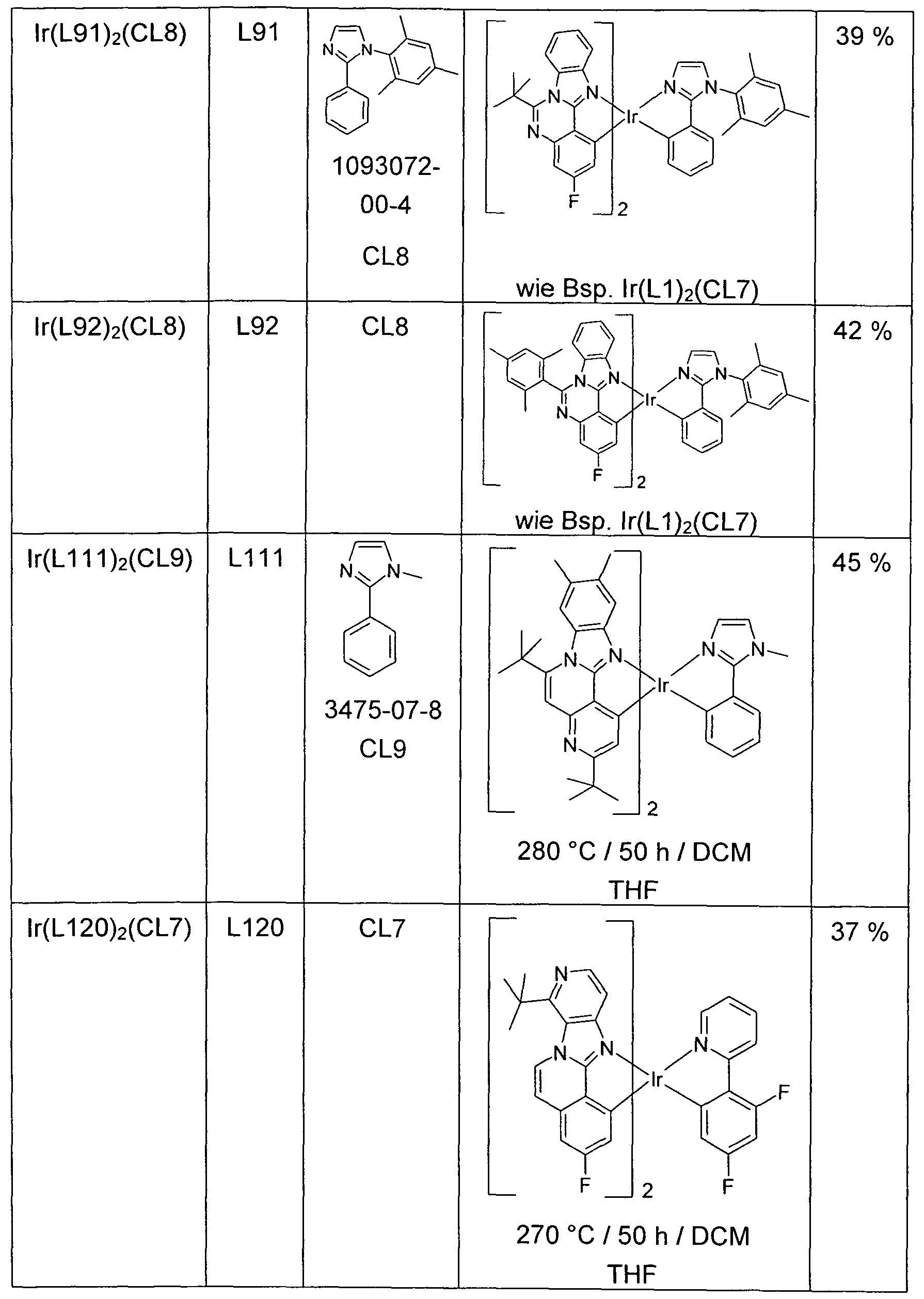
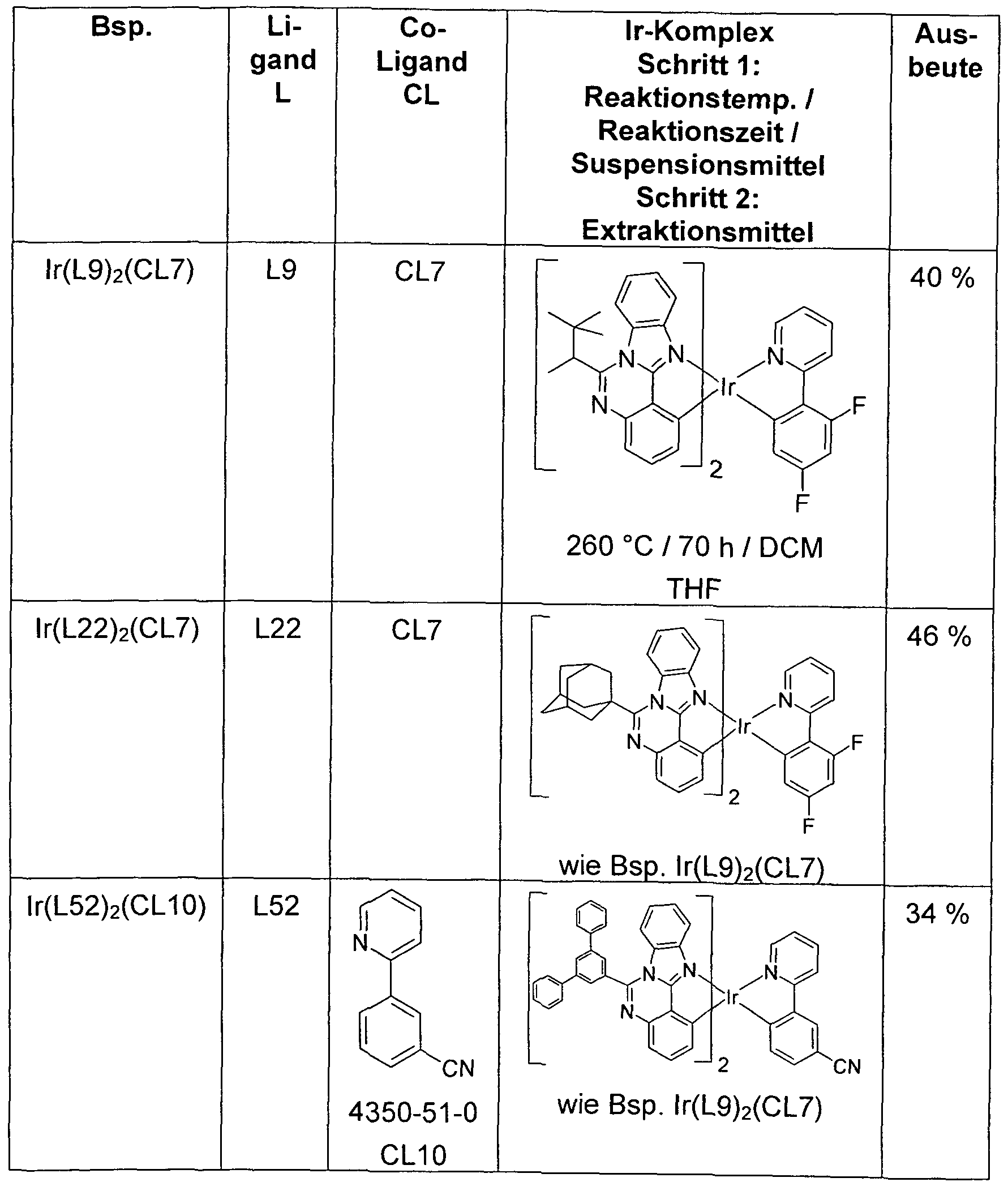




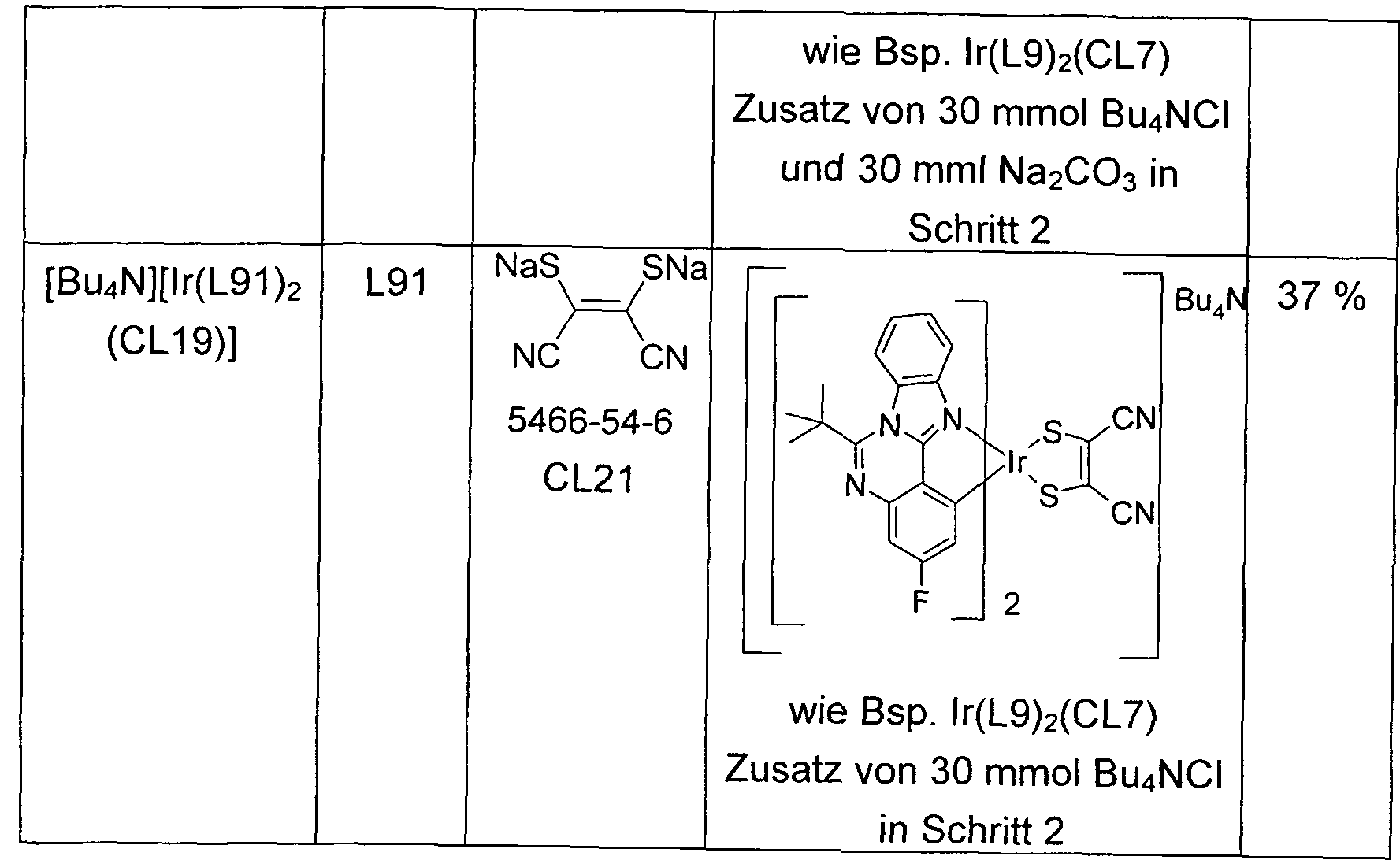
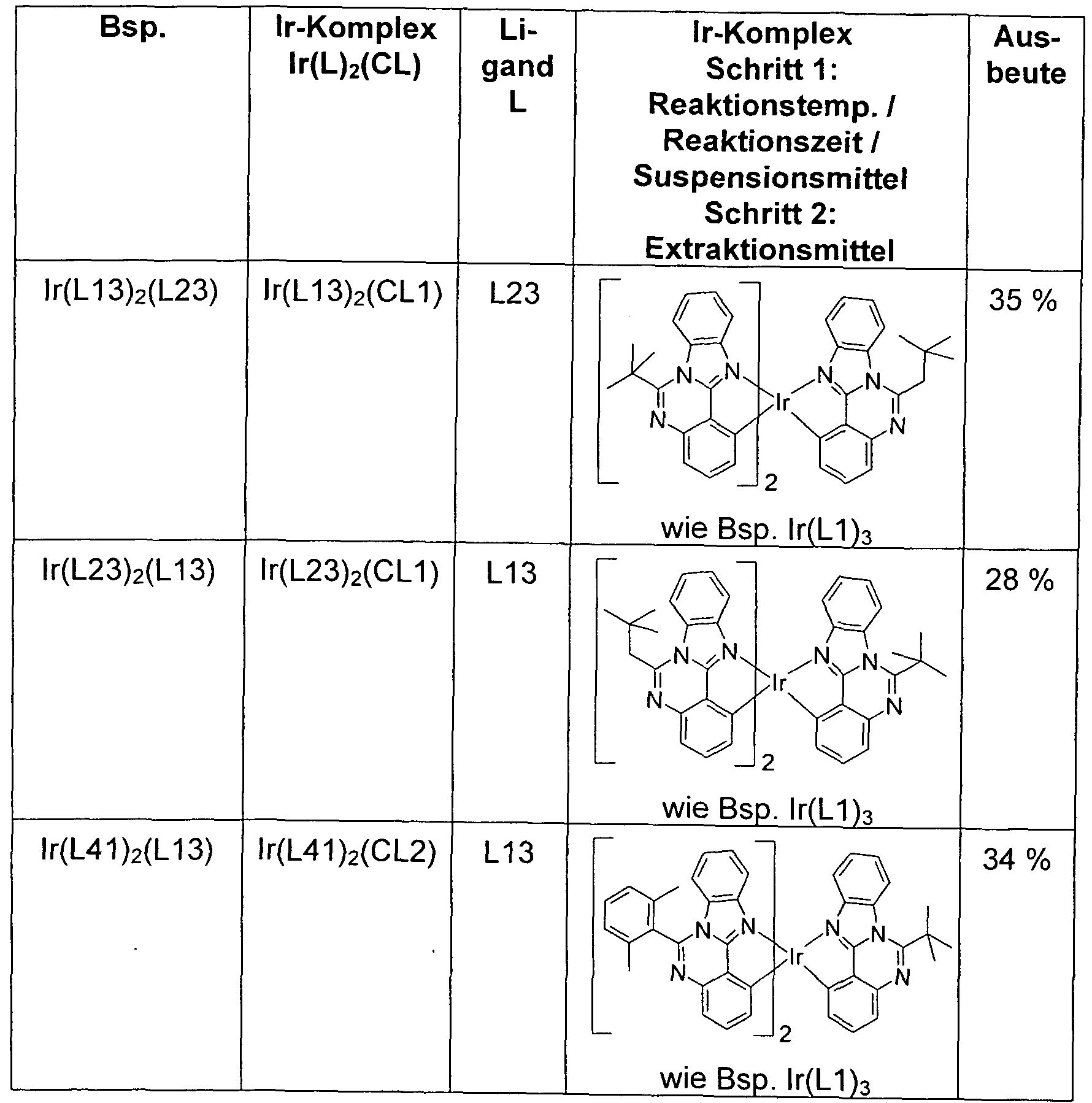



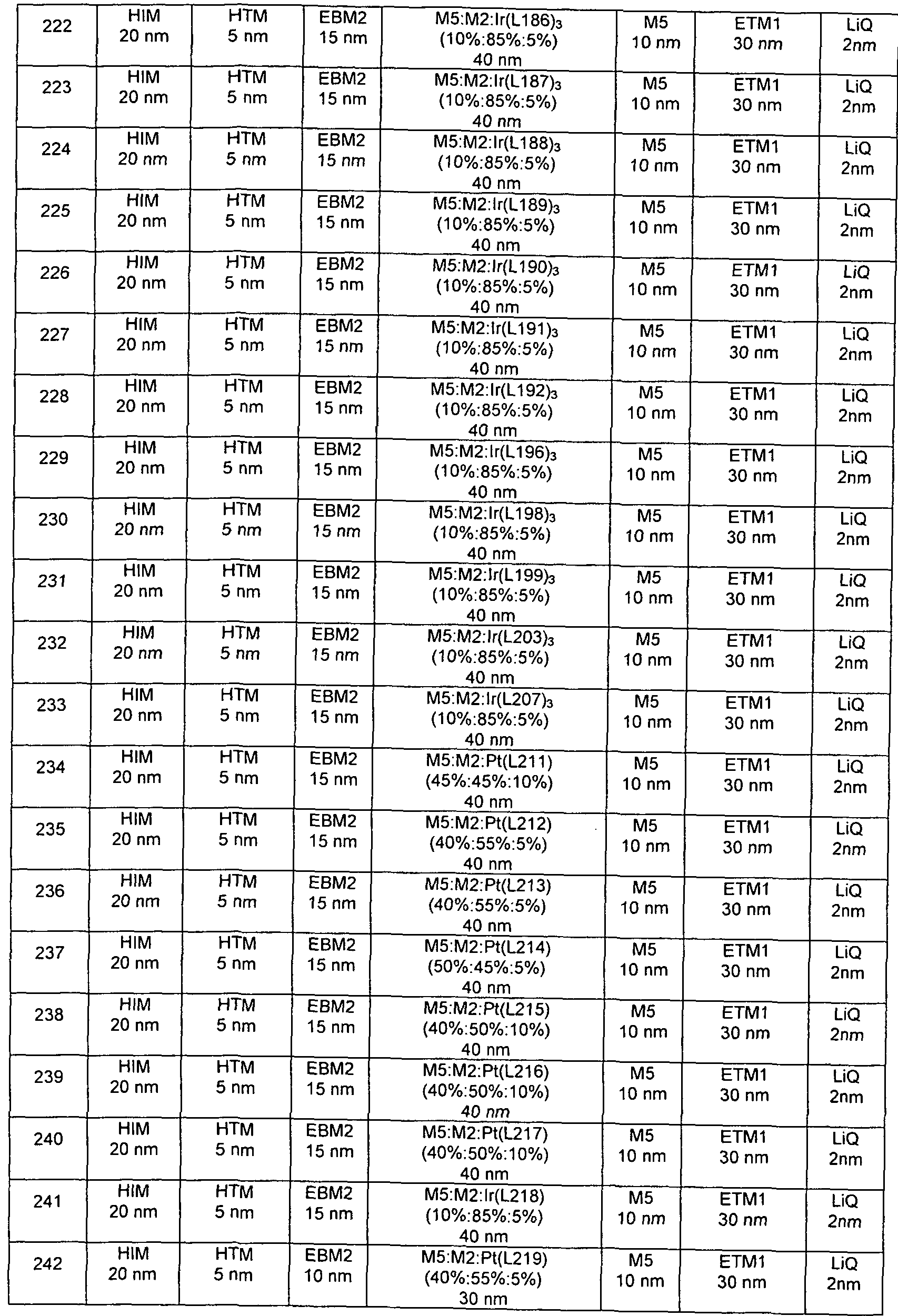





Claims





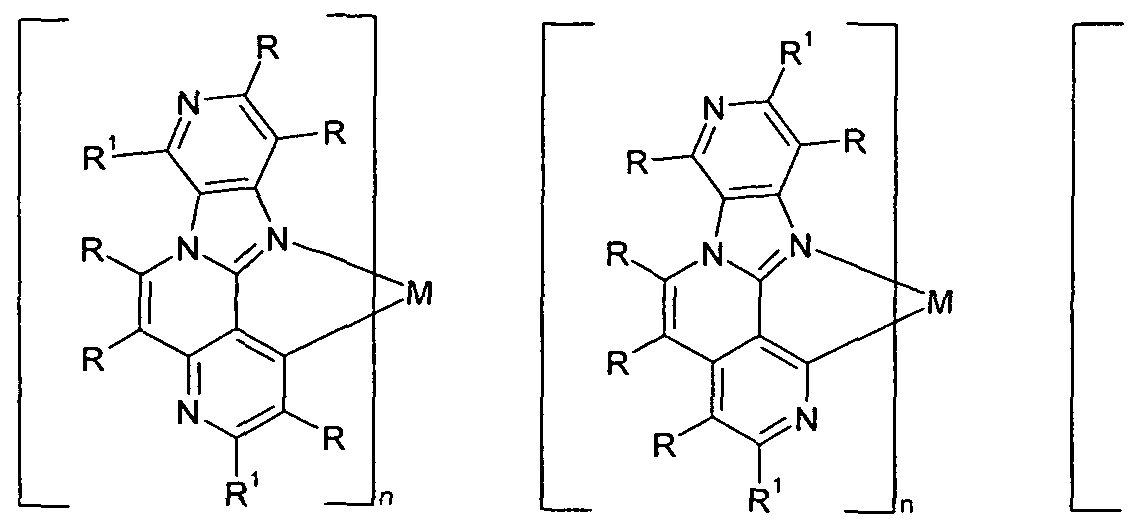


















Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020137000877A KR20130087499A (ko) | 2010-06-15 | 2011-05-18 | 금속 착물 |
| US13/703,683 US9273080B2 (en) | 2010-06-15 | 2011-05-18 | Metal complexes |
| JP2013514565A JP6054290B2 (ja) | 2010-06-15 | 2011-05-18 | 金属錯体 |
| CN201180029938.0A CN102939296B (zh) | 2010-06-15 | 2011-05-18 | 金属络合物 |
| DE112011102008.2T DE112011102008B4 (de) | 2010-06-15 | 2011-05-18 | Metallkomplexe |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10006208.2 | 2010-06-15 | ||
| EP10006208 | 2010-06-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011157339A1 true WO2011157339A1 (de) | 2011-12-22 |
Family
ID=44263181
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2011/002467 WO2011157339A1 (de) | 2010-06-15 | 2011-05-18 | Metallkomplexe |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9273080B2 (de) |
| JP (1) | JP6054290B2 (de) |
| KR (1) | KR20130087499A (de) |
| CN (1) | CN102939296B (de) |
| DE (1) | DE112011102008B4 (de) |
| WO (1) | WO2011157339A1 (de) |
Cited By (200)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012163465A1 (de) | 2011-06-03 | 2012-12-06 | Merck Patent Gmbh | Organische elektrolumineszenzvorrichtung |
| WO2013041176A1 (de) | 2011-09-21 | 2013-03-28 | Merck Patent Gmbh | Carbazolderivate für organische elektrolumineszenzvorrichtungen |
| WO2013083216A1 (de) | 2011-11-17 | 2013-06-13 | Merck Patent Gmbh | Spiro -dihydroacridinderivate und ihre verwendung als materialien für organische elektrolumineszenzvorrichtungen |
| EP2623508A1 (de) | 2012-02-02 | 2013-08-07 | Konica Minolta Advanced Layers, Inc. | Iridiumkomplexverbindung, Material aus organischem elektrolumineszentem Material, organisches elektrolumineszentes Material, Beleuchtungsvorrichtung und Anzeigevorrichtung |
| WO2013120577A1 (en) | 2012-02-14 | 2013-08-22 | Merck Patent Gmbh | Spirobifluorene compounds for organic electroluminescent devices |
| WO2013139431A1 (de) | 2012-03-23 | 2013-09-26 | Merck Patent Gmbh | 9,9'-spirobixanthenderivate für elektrolumineszenzvorrichtungen |
| WO2014008982A1 (de) | 2012-07-13 | 2014-01-16 | Merck Patent Gmbh | Metallkomplexe |
| WO2014008967A2 (de) | 2012-07-10 | 2014-01-16 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2014015931A1 (de) | 2012-07-23 | 2014-01-30 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2014023388A1 (de) | 2012-08-10 | 2014-02-13 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2014030666A1 (ja) | 2012-08-24 | 2014-02-27 | コニカミノルタ株式会社 | 透明電極、電子デバイス、および透明電極の製造方法 |
| JP2014045101A (ja) * | 2012-08-28 | 2014-03-13 | Konica Minolta Inc | 有機エレクトロルミネッセンス素子、照明装置および表示装置 |
| WO2014147134A1 (en) | 2013-03-20 | 2014-09-25 | Basf Se | Azabenzimidazole carbene complexes as efficiency booster in oleds |
| WO2014157494A1 (ja) | 2013-03-29 | 2014-10-02 | コニカミノルタ株式会社 | 有機エレクトロルミネッセンス素子用材料、有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| WO2014157618A1 (ja) | 2013-03-29 | 2014-10-02 | コニカミノルタ株式会社 | 有機エレクトロルミネッセンス素子、それを具備した照明装置及び表示装置 |
| WO2015000955A1 (en) | 2013-07-02 | 2015-01-08 | Basf Se | Monosubstituted diazabenzimidazole carbene metal complexes for use in organic light emitting diodes |
| WO2015104045A1 (de) * | 2014-01-13 | 2015-07-16 | Merck Patent Gmbh | Metallkomplexe |
| WO2015117718A1 (de) * | 2014-02-05 | 2015-08-13 | Merck Patent Gmbh | Metallkomplexe |
| KR20150096520A (ko) * | 2012-12-21 | 2015-08-24 | 메르크 파텐트 게엠베하 | 금속 착물 |
| KR20150096805A (ko) * | 2012-12-21 | 2015-08-25 | 메르크 파텐트 게엠베하 | 금속 착물 |
| KR20150100842A (ko) * | 2012-12-21 | 2015-09-02 | 메르크 파텐트 게엠베하 | 금속 착물 |
| WO2015169412A1 (de) | 2014-05-05 | 2015-11-12 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| KR101576570B1 (ko) | 2013-02-18 | 2015-12-10 | 주식회사 두산 | 유기 화합물 및 이를 포함하는 유기 전계 발광 소자 |
| WO2015192941A1 (de) | 2014-06-18 | 2015-12-23 | Merck Patent Gmbh | Zusammensetzungen für elektronische vorrichtungen |
| WO2015197156A1 (de) | 2014-06-25 | 2015-12-30 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| US9382253B2 (en) | 2010-07-16 | 2016-07-05 | Merck Patent Gmbh | Metal complexes |
| WO2016198144A1 (en) | 2015-06-10 | 2016-12-15 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2017012687A1 (en) | 2015-07-22 | 2017-01-26 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2017016630A1 (en) | 2015-07-30 | 2017-02-02 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2017025166A1 (en) | 2015-08-13 | 2017-02-16 | Merck Patent Gmbh | Hexamethylindanes |
| WO2017071791A1 (en) | 2015-10-27 | 2017-05-04 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| EP3200255A2 (de) | 2016-01-06 | 2017-08-02 | Konica Minolta, Inc. | Organisches elektrolumineszenzelement, verfahren zur herstellung eines organischen elektrolumineszenzelements, sowie anzeige und beleuchtungsvorrichtung |
| WO2017148565A1 (de) | 2016-03-03 | 2017-09-08 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2017157983A1 (de) | 2016-03-17 | 2017-09-21 | Merck Patent Gmbh | Verbindungen mit spirobifluoren-strukturen |
| WO2017178311A1 (de) | 2016-04-11 | 2017-10-19 | Merck Patent Gmbh | Heterocyclische verbindungen mit dibenzofuran- und/oder dibenzothiophen-strukturen |
| EP3239161A1 (de) | 2013-07-31 | 2017-11-01 | UDC Ireland Limited | Lumineszente diazabenzimidazol-carben-metall-komplexe |
| WO2017186760A1 (en) | 2016-04-29 | 2017-11-02 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2017207596A1 (en) | 2016-06-03 | 2017-12-07 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| US9862739B2 (en) | 2014-03-31 | 2018-01-09 | Udc Ireland Limited | Metal complexes, comprising carbene ligands having an O-substituted non-cyclometalated aryl group and their use in organic light emitting diodes |
| WO2018050584A1 (de) | 2016-09-14 | 2018-03-22 | Merck Patent Gmbh | Verbindungen mit spirobifluoren-strukturen |
| WO2018050583A1 (de) | 2016-09-14 | 2018-03-22 | Merck Patent Gmbh | Verbindungen mit carbazol-strukturen |
| WO2018060218A1 (de) | 2016-09-30 | 2018-04-05 | Merck Patent Gmbh | Carbazole mit diazadibenzofuran- oder diazadibenzothiophen-strukturen |
| WO2018060307A1 (de) | 2016-09-30 | 2018-04-05 | Merck Patent Gmbh | Verbindungen mit diazadibenzofuran- oder diazadibenzothiophen-strukturen |
| WO2018087020A1 (en) | 2016-11-08 | 2018-05-17 | Merck Patent Gmbh | Compounds for electronic devices |
| WO2018087346A1 (de) | 2016-11-14 | 2018-05-17 | Merck Patent Gmbh | Verbindungen mit einer akzeptor- und einer donorgruppe |
| WO2018087022A1 (de) | 2016-11-09 | 2018-05-17 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2018091435A1 (en) | 2016-11-17 | 2018-05-24 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2018095395A1 (zh) | 2016-11-23 | 2018-05-31 | 广州华睿光电材料有限公司 | 高聚物、包含其的混合物、组合物和有机电子器件以及用于聚合的单体 |
| WO2018095392A1 (zh) | 2016-11-23 | 2018-05-31 | 广州华睿光电材料有限公司 | 有机混合物、组合物以及有机电子器件 |
| WO2018095381A1 (zh) | 2016-11-23 | 2018-05-31 | 广州华睿光电材料有限公司 | 印刷油墨组合物及其制备方法和用途 |
| WO2018099846A1 (de) | 2016-11-30 | 2018-06-07 | Merck Patent Gmbh | Verbindungen mit valerolaktam-strukturen |
| WO2018103744A1 (zh) | 2016-12-08 | 2018-06-14 | 广州华睿光电材料有限公司 | 混合物、组合物及有机电子器件 |
| WO2018104193A1 (de) | 2016-12-05 | 2018-06-14 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2018104195A1 (de) | 2016-12-05 | 2018-06-14 | Merck Patent Gmbh | Stickstoffhaltige heterocyclen zur verwendung in oleds |
| WO2018104194A1 (de) | 2016-12-05 | 2018-06-14 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2018114883A1 (de) | 2016-12-22 | 2018-06-28 | Merck Patent Gmbh | Mischungen umfassend mindestens zwei organisch funktionelle verbindungen |
| WO2018113785A1 (zh) | 2016-12-22 | 2018-06-28 | 广州华睿光电材料有限公司 | 含呋喃交联基团的聚合物及其应用 |
| WO2018127465A1 (de) | 2017-01-04 | 2018-07-12 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2018138306A1 (de) | 2017-01-30 | 2018-08-02 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2018138039A1 (de) | 2017-01-25 | 2018-08-02 | Merck Patent Gmbh | Carbazolderivate |
| WO2018149769A1 (de) | 2017-02-14 | 2018-08-23 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2018166934A1 (de) | 2017-03-15 | 2018-09-20 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2018166932A1 (de) | 2017-03-13 | 2018-09-20 | Merck Patent Gmbh | Verbindungen mit arylamin-strukturen |
| WO2018189134A1 (de) | 2017-04-13 | 2018-10-18 | Merck Patent Gmbh | Zusammensetzung für organische elektronische vorrichtungen |
| CN108780849A (zh) * | 2016-03-15 | 2018-11-09 | 新日铁住金化学株式会社 | 有机电致发光元件 |
| WO2018206537A1 (en) | 2017-05-11 | 2018-11-15 | Merck Patent Gmbh | Carbazole-based bodipys for organic electroluminescent devices |
| WO2018206526A1 (en) | 2017-05-11 | 2018-11-15 | Merck Patent Gmbh | Organoboron complexes for organic electroluminescent devices |
| WO2018234346A1 (en) | 2017-06-23 | 2018-12-27 | Merck Patent Gmbh | MATERIALS FOR ORGANIC ELECTROLUMINESCENT DEVICES |
| WO2019007867A1 (de) | 2017-07-05 | 2019-01-10 | Merck Patent Gmbh | Zusammensetzung für organische elektronische vorrichtungen |
| WO2019007866A1 (de) | 2017-07-05 | 2019-01-10 | Merck Patent Gmbh | Zusammensetzung für organische elektronische vorrichtungen |
| WO2019052933A1 (de) | 2017-09-12 | 2019-03-21 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2019068679A1 (en) | 2017-10-06 | 2019-04-11 | Merck Patent Gmbh | MATERIALS FOR ORGANIC ELECTROLUMINESCENT DEVICES |
| WO2019081391A1 (de) | 2017-10-24 | 2019-05-02 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2019096717A2 (de) | 2017-11-14 | 2019-05-23 | Merck Patent Gmbh | Zusammensetzung für organische elektronische vorrichtungen |
| US10323180B2 (en) | 2014-12-04 | 2019-06-18 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Deuterated organic compound, mixture and composition containing said compound, and organic electronic device |
| WO2019121483A1 (en) | 2017-12-20 | 2019-06-27 | Merck Patent Gmbh | Heteroaromatic compounds |
| WO2019121458A1 (de) | 2017-12-19 | 2019-06-27 | Merck Patent Gmbh | Heterocyclische verbindung zur verwendung in electronischen vorrichtungen |
| US10364316B2 (en) | 2015-01-13 | 2019-07-30 | Guangzhou Chinaray Optoelectronics Materials Ltd. | Conjugated polymer containing ethynyl crosslinking group, mixture, formulation, organic electronic device containing the same and application therof |
| WO2019145316A1 (de) | 2018-01-25 | 2019-08-01 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2019175149A1 (en) | 2018-03-16 | 2019-09-19 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2019229011A1 (de) | 2018-05-30 | 2019-12-05 | Merck Patent Gmbh | Zusammensetzung für organische elektronische vorrichtungen |
| WO2019233904A1 (de) | 2018-06-07 | 2019-12-12 | Merck Patent Gmbh | Organische elektrolumineszenzvorrichtungen |
| US10510967B2 (en) | 2014-12-11 | 2019-12-17 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Organic compound, and mixture, formulation and organic device comprising the same |
| WO2020011686A1 (de) | 2018-07-09 | 2020-01-16 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2020016264A1 (en) | 2018-07-20 | 2020-01-23 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2020053150A1 (en) | 2018-09-12 | 2020-03-19 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2020053314A1 (de) | 2018-09-12 | 2020-03-19 | Merck Patent Gmbh | Elektrolumineszierende vorrichtungen |
| WO2020053315A1 (de) | 2018-09-12 | 2020-03-19 | Merck Patent Gmbh | Elektrolumineszierende vorrichtungen |
| WO2020064666A1 (de) | 2018-09-27 | 2020-04-02 | Merck Patent Gmbh | Verbindungen, die in einer organischen elektronischen vorrichtung als aktive verbindungen einsetzbar sind |
| WO2020064662A2 (de) | 2018-09-27 | 2020-04-02 | Merck Patent Gmbh | Verfahren zur herstellung von sterisch gehinderten stickstoffhaltigen heteroaromatischen verbindungen |
| WO2020094542A1 (de) | 2018-11-06 | 2020-05-14 | Merck Patent Gmbh | 5,6-diphenyl-5,6-dihydro-dibenz[c,e][1,2]azaphosphorin- und 6-phenyl-6h-dibenzo[c,e][1,2]thiazin-5,5-dioxid-derivate und ähnliche verbindungen als organische elektrolumineszenzmaterialien für oleds |
| WO2020094539A1 (de) | 2018-11-05 | 2020-05-14 | Merck Patent Gmbh | In einer organischen elektronischen vorrichtung einsetzbare verbindungen |
| WO2020099349A1 (de) | 2018-11-14 | 2020-05-22 | Merck Patent Gmbh | Zur herstellung einer organischen elektronischen vorrichtung einsetzbare verbindungen |
| WO2020099307A1 (de) | 2018-11-15 | 2020-05-22 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2020127165A1 (de) | 2018-12-19 | 2020-06-25 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2020148243A1 (en) | 2019-01-16 | 2020-07-23 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2020148303A1 (de) | 2019-01-17 | 2020-07-23 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2020169241A1 (de) | 2019-02-18 | 2020-08-27 | Merck Patent Gmbh | Zusammensetzung für organische elektronische vorrichtungen |
| WO2020178230A1 (en) | 2019-03-04 | 2020-09-10 | Merck Patent Gmbh | Ligands for nano-sized materials |
| WO2020182779A1 (de) | 2019-03-12 | 2020-09-17 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2020187865A1 (de) | 2019-03-20 | 2020-09-24 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2020193447A1 (de) | 2019-03-25 | 2020-10-01 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| US10804470B2 (en) | 2016-11-23 | 2020-10-13 | Guangzhou Chinaray Optoelectronic Materials Ltd | Organic compound |
| WO2020208051A1 (en) | 2019-04-11 | 2020-10-15 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2020212296A1 (de) | 2019-04-15 | 2020-10-22 | Merck Patent Gmbh | Metallkomplexe |
| US10840450B2 (en) | 2014-12-04 | 2020-11-17 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Polymer, and mixture or formulation, and organic electronic device containing same, and monomer thereof |
| WO2021013775A1 (de) | 2019-07-22 | 2021-01-28 | Merck Patent Gmbh | Verfahren zur herstellung ortho-metallierter metallverbindungen |
| WO2021037401A1 (de) | 2019-08-26 | 2021-03-04 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2021043703A1 (de) | 2019-09-02 | 2021-03-11 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2021043755A1 (de) | 2019-09-03 | 2021-03-11 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2021052921A1 (de) | 2019-09-19 | 2021-03-25 | Merck Patent Gmbh | Mischung von zwei hostmaterialien und organische elektrolumineszierende vorrichtung damit |
| WO2021053046A1 (de) | 2019-09-20 | 2021-03-25 | Merck Patent Gmbh | Peri-kondensierte heterozyklische verbindungen als materialien für elektronische vorrichtungen |
| WO2021052924A1 (en) | 2019-09-16 | 2021-03-25 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| US10968243B2 (en) | 2015-12-04 | 2021-04-06 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Organometallic complex and application thereof in electronic devices |
| WO2021078710A1 (en) | 2019-10-22 | 2021-04-29 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2021078831A1 (de) | 2019-10-25 | 2021-04-29 | Merck Patent Gmbh | In einer organischen elektronischen vorrichtung einsetzbare verbindungen |
| WO2021089447A1 (de) | 2019-11-04 | 2021-05-14 | Merck Patent Gmbh | Organische elektrolumineszierende vorrichtung |
| WO2021089450A1 (en) | 2019-11-04 | 2021-05-14 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2021094269A1 (en) | 2019-11-12 | 2021-05-20 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2021110741A1 (en) | 2019-12-04 | 2021-06-10 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2021122740A1 (de) | 2019-12-19 | 2021-06-24 | Merck Patent Gmbh | Polycyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2021122535A1 (de) | 2019-12-17 | 2021-06-24 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2021122538A1 (de) | 2019-12-18 | 2021-06-24 | Merck Patent Gmbh | Aromatische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2021151922A1 (de) | 2020-01-29 | 2021-08-05 | Merck Patent Gmbh | Benzimidazol-derivate |
| WO2021170522A1 (de) | 2020-02-25 | 2021-09-02 | Merck Patent Gmbh | Verwendung von heterocyclischen verbindungen in einer organischen elektronischen vorrichtung |
| WO2021175706A1 (de) | 2020-03-02 | 2021-09-10 | Merck Patent Gmbh | Verwendung von sulfonverbindungen in einer organischen elektronischen vorrichtung |
| WO2021180614A1 (de) | 2020-03-11 | 2021-09-16 | Merck Patent Gmbh | Organische elektrolumineszierende vorrichtung |
| WO2021180625A1 (de) | 2020-03-11 | 2021-09-16 | Merck Patent Gmbh | Organische elektrolumineszierende vorrichtung |
| WO2021185829A1 (de) | 2020-03-17 | 2021-09-23 | Merck Patent Gmbh | Heterocyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2021185712A1 (de) | 2020-03-17 | 2021-09-23 | Merck Patent Gmbh | Heteroaromatische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2021191117A1 (de) | 2020-03-24 | 2021-09-30 | Merck Patent Gmbh | Materialien für elektronische vorrichtungen |
| WO2021191058A1 (en) | 2020-03-23 | 2021-09-30 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2021191183A1 (de) | 2020-03-26 | 2021-09-30 | Merck Patent Gmbh | Cyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2021198213A1 (de) | 2020-04-02 | 2021-10-07 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2021204646A1 (de) | 2020-04-06 | 2021-10-14 | Merck Patent Gmbh | Polycyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| US11161933B2 (en) | 2016-12-13 | 2021-11-02 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Conjugated polymer and use thereof in organic electronic device |
| WO2021239772A1 (de) | 2020-05-29 | 2021-12-02 | Merck Patent Gmbh | Organische elektrolumineszierende vorrichtung |
| WO2021254984A1 (de) | 2020-06-18 | 2021-12-23 | Merck Patent Gmbh | Indenoazanaphthaline |
| WO2021259824A1 (de) | 2020-06-23 | 2021-12-30 | Merck Patent Gmbh | Verfahren zur herstellung einer mischung |
| WO2022002772A1 (de) | 2020-06-29 | 2022-01-06 | Merck Patent Gmbh | Heteroaromatische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2022002771A1 (de) | 2020-06-29 | 2022-01-06 | Merck Patent Gmbh | Heterocyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| US11239428B2 (en) | 2016-11-23 | 2022-02-01 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Boron-containing organic compound and applications thereof, organic mixture, and organic electronic device |
| WO2022029096A1 (de) | 2020-08-06 | 2022-02-10 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2022034046A1 (de) | 2020-08-13 | 2022-02-17 | Merck Patent Gmbh | Metallkomplexe |
| WO2022038066A1 (de) | 2020-08-19 | 2022-02-24 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2022038065A1 (de) | 2020-08-18 | 2022-02-24 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| US11292875B2 (en) | 2016-12-22 | 2022-04-05 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Cross-linkable polymer based on Diels-Alder reaction and use thereof in organic electronic device |
| WO2022069421A1 (de) | 2020-09-30 | 2022-04-07 | Merck Patent Gmbh | Zur strukturierung von funktionalen schichten organischer elektrolumineszenzvorrichtungen einsetzbare verbindungen |
| WO2022069422A1 (de) | 2020-09-30 | 2022-04-07 | Merck Patent Gmbh | Verbindungen zur strukturierung von funktionalen schichten organischer elektrolumineszenzvorrichtungen |
| WO2022079067A1 (de) | 2020-10-16 | 2022-04-21 | Merck Patent Gmbh | Verbindungen mit heteroatomen für organische elektrolumineszenzvorrichtungen |
| WO2022079068A1 (de) | 2020-10-16 | 2022-04-21 | Merck Patent Gmbh | Heterocyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2022101171A1 (de) | 2020-11-10 | 2022-05-19 | Merck Patent Gmbh | Schwefelhaltige verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2022117473A1 (de) | 2020-12-02 | 2022-06-09 | Merck Patent Gmbh | Heterocyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2022122682A2 (de) | 2020-12-10 | 2022-06-16 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2022129114A1 (de) | 2020-12-18 | 2022-06-23 | Merck Patent Gmbh | Stickstoffhaltige verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2022129113A1 (de) | 2020-12-18 | 2022-06-23 | Merck Patent Gmbh | Stickstoffhaltige heteroaromaten für organische elektrolumineszenzvorrichtungen |
| WO2022129116A1 (de) | 2020-12-18 | 2022-06-23 | Merck Patent Gmbh | Indolo[3.2.1-jk]carbazole-6-carbonitril-derivate als blau fluoreszierende emitter zur verwendung in oleds |
| WO2022148717A1 (de) | 2021-01-05 | 2022-07-14 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2022157343A1 (de) | 2021-01-25 | 2022-07-28 | Merck Patent Gmbh | Stickstoffhaltige verbindungen für organische elektrolumineszenzvorrichtungen |
| US11404651B2 (en) | 2017-12-14 | 2022-08-02 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Transition metal complex material and application thereof in electronic devices |
| WO2022184601A1 (de) | 2021-03-02 | 2022-09-09 | Merck Patent Gmbh | Verbindungen für organische elektrolumineszenzvorrichtungen |
| US11447496B2 (en) | 2016-11-23 | 2022-09-20 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Nitrogen-containing fused heterocyclic ring compound and application thereof |
| WO2022194799A1 (de) | 2021-03-18 | 2022-09-22 | Merck Patent Gmbh | Heteroaromatische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2022200638A1 (de) | 2021-07-06 | 2022-09-29 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2022229126A1 (de) | 2021-04-29 | 2022-11-03 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2022229298A1 (de) | 2021-04-29 | 2022-11-03 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2022229234A1 (de) | 2021-04-30 | 2022-11-03 | Merck Patent Gmbh | Stickstoffhaltige, heterocyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2022243403A1 (de) | 2021-05-21 | 2022-11-24 | Merck Patent Gmbh | Verfahren zur kontinuierlichen aufreinigung von mindestens einem funktionalen material und vorrichtung zur kontinuierlichen aufreinigung von mindestens einem funktionalen material |
| US11555128B2 (en) | 2015-11-12 | 2023-01-17 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Printing composition, electronic device comprising same and preparation method for functional material thin film |
| US11594690B2 (en) | 2017-12-14 | 2023-02-28 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Organometallic complex, and polymer, mixture and formulation comprising same, and use thereof in electronic device |
| WO2023036976A1 (en) | 2021-09-13 | 2023-03-16 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2023041454A1 (de) | 2021-09-14 | 2023-03-23 | Merck Patent Gmbh | Borhaltige, heterocyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2023052313A1 (de) | 2021-09-28 | 2023-04-06 | Merck Patent Gmbh | Materialien für elektronische vorrichtungen |
| WO2023052314A1 (de) | 2021-09-28 | 2023-04-06 | Merck Patent Gmbh | Materialien für elektronische vorrichtungen |
| WO2023052275A1 (de) | 2021-09-28 | 2023-04-06 | Merck Patent Gmbh | Materialien für elektronische vorrichtungen |
| WO2023052272A1 (de) | 2021-09-28 | 2023-04-06 | Merck Patent Gmbh | Materialien für elektronische vorrichtungen |
| US11634444B2 (en) | 2016-11-23 | 2023-04-25 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Metal organic complex, high polymer, composition, and organic electronic component |
| WO2023072799A1 (de) | 2021-10-27 | 2023-05-04 | Merck Patent Gmbh | Bor- und stickstoffhaltige, heterocyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2023094412A1 (de) | 2021-11-25 | 2023-06-01 | Merck Patent Gmbh | Materialien für elektronische vorrichtungen |
| WO2023099543A1 (en) | 2021-11-30 | 2023-06-08 | Merck Patent Gmbh | Compounds having fluorene structures |
| US11680059B2 (en) | 2017-12-21 | 2023-06-20 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Organic mixture and application thereof in organic electronic devices |
| WO2023110742A1 (de) | 2021-12-13 | 2023-06-22 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2023117837A1 (de) | 2021-12-21 | 2023-06-29 | Merck Patent Gmbh | Verfahren zur herstellung von deuterierten organischen verbindungen |
| WO2023117836A1 (en) | 2021-12-21 | 2023-06-29 | Merck Patent Gmbh | Electronic devices |
| WO2023117835A1 (en) | 2021-12-21 | 2023-06-29 | Merck Patent Gmbh | Electronic devices |
| WO2023152346A1 (de) | 2022-02-14 | 2023-08-17 | Merck Patent Gmbh | Materialien für elektronische vorrichtungen |
| WO2023152063A1 (de) | 2022-02-09 | 2023-08-17 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| EP4236652A2 (de) | 2015-07-29 | 2023-08-30 | Merck Patent GmbH | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2023161168A1 (de) | 2022-02-23 | 2023-08-31 | Merck Patent Gmbh | Aromatische heterocyclen für organische elektrolumineszenzvorrichtungen |
| WO2023161167A1 (de) | 2022-02-23 | 2023-08-31 | Merck Patent Gmbh | Stickstoffhaltige heterocyclen für organische elektrolumineszenzvorrichtungen |
| EP4271160A2 (de) | 2015-02-13 | 2023-11-01 | Merck Patent GmbH | Aromatisches heterocyclisches derivat und organisches elektrolumineszenzelement, beleuchtungsvorrichtung und anzeigevorrichtung mit dem aromatischen heterocyclischen derivat |
| WO2023213837A1 (de) | 2022-05-06 | 2023-11-09 | Merck Patent Gmbh | Cyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2023222559A1 (de) | 2022-05-18 | 2023-11-23 | Merck Patent Gmbh | Verfahren zur herstellung von deuterierten organischen verbindungen |
| WO2023247663A1 (de) | 2022-06-24 | 2023-12-28 | Merck Patent Gmbh | Zusammensetzung für organische elektronische vorrichtungen |
| WO2023247662A1 (de) | 2022-06-24 | 2023-12-28 | Merck Patent Gmbh | Zusammensetzung für organische elektronische vorrichtungen |
| WO2024013004A1 (de) | 2022-07-11 | 2024-01-18 | Merck Patent Gmbh | Materialien für elektronische vorrichtungen |
| WO2024033282A1 (en) | 2022-08-09 | 2024-02-15 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| US11932659B2 (en) | 2016-07-25 | 2024-03-19 | Udc Ireland Limited | Metal complexes for use as emitters in organic electroluminescence devices |
| WO2024061942A1 (de) | 2022-09-22 | 2024-03-28 | Merck Patent Gmbh | Stickstoffenthaltende verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2024061948A1 (de) | 2022-09-22 | 2024-03-28 | Merck Patent Gmbh | Stickstoffenthaltende heterocyclen für organische elektrolumineszenzvorrichtungen |
Families Citing this family (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4551480B1 (ja) * | 2009-08-31 | 2010-09-29 | 富士フイルム株式会社 | 有機電界発光素子 |
| KR102134951B1 (ko) | 2011-02-16 | 2020-07-16 | 가부시키가이샤 한도오따이 에네루기 켄큐쇼 | 발광 소자 |
| KR20190014600A (ko) | 2011-03-23 | 2019-02-12 | 가부시키가이샤 한도오따이 에네루기 켄큐쇼 | 발광 소자 |
| KR102069497B1 (ko) | 2012-01-13 | 2020-01-23 | 미쯔비시 케미컬 주식회사 | 이리듐 착물 화합물 그리고 그 화합물을 함유하는 용액 조성물, 유기 전계 발광 소자, 표시 장치 및 조명 장치 |
| EP2960240B1 (de) | 2013-02-07 | 2017-05-03 | LG Chem, Ltd. | Heterocyclische verbindung und organisches lichtemittierendes element damit |
| KR101711917B1 (ko) | 2013-12-09 | 2017-03-13 | 제일모직 주식회사 | 화합물, 이를 포함하는 유기광전자소자 및 표시장치 |
| US9397302B2 (en) | 2014-10-08 | 2016-07-19 | Universal Display Corporation | Organic electroluminescent materials and devices |
| US10854826B2 (en) | 2014-10-08 | 2020-12-01 | Universal Display Corporation | Organic electroluminescent compounds, compositions and devices |
| JP6655873B2 (ja) * | 2014-11-04 | 2020-03-04 | 株式会社半導体エネルギー研究所 | イリジウム錯体、発光素子、発光装置、電子機器、及び照明装置 |
| JP6595752B2 (ja) * | 2014-11-04 | 2019-10-23 | 株式会社半導体エネルギー研究所 | 発光素子、発光装置、電子機器、及び照明装置 |
| KR101778446B1 (ko) * | 2015-06-05 | 2017-09-14 | 주식회사 엘지화학 | 화합물 및 이를 포함하는 유기 전자 소자 |
| US10825997B2 (en) | 2015-06-25 | 2020-11-03 | Universal Display Corporation | Organic electroluminescent materials and devices |
| US10672996B2 (en) | 2015-09-03 | 2020-06-02 | Universal Display Corporation | Organic electroluminescent materials and devices |
| US10608186B2 (en) * | 2016-09-14 | 2020-03-31 | Universal Display Corporation | Organic electroluminescent materials and devices |
| US11239432B2 (en) * | 2016-10-14 | 2022-02-01 | Universal Display Corporation | Organic electroluminescent materials and devices |
| US11545636B2 (en) * | 2016-12-15 | 2023-01-03 | Universal Display Corporation | Organic electroluminescent materials and devices |
| US11925103B2 (en) * | 2018-06-05 | 2024-03-05 | Universal Display Corporation | Organic electroluminescent materials and devices |
| US11228004B2 (en) | 2018-06-22 | 2022-01-18 | Universal Display Corporation | Organic electroluminescent materials and devices |
| US11370809B2 (en) * | 2019-02-08 | 2022-06-28 | Universal Display Corporation | Organic electroluminescent materials and devices |
| JP7298187B2 (ja) * | 2019-02-28 | 2023-06-27 | 住友化学株式会社 | 金属錯体及び前記金属錯体を含む組成物 |
| CN111848642B (zh) * | 2020-07-31 | 2023-01-24 | 武汉天马微电子有限公司 | 化合物、显示面板以及显示装置 |
Citations (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4539507A (en) | 1983-03-25 | 1985-09-03 | Eastman Kodak Company | Organic electroluminescent devices having improved power conversion efficiencies |
| US5151629A (en) | 1991-08-01 | 1992-09-29 | Eastman Kodak Company | Blue emitting internal junction organic electroluminescent device (I) |
| EP0652273A1 (de) | 1993-11-09 | 1995-05-10 | Shinko Electric Industries Co. Ltd. | Organisches Material für elektrolumineszente Vorrichtung und elektrolumineszente Vorrichtung |
| EP0676461A2 (de) | 1994-04-07 | 1995-10-11 | Hoechst Aktiengesellschaft | Spiroverbindungen und ihre Verwendung als Elektrolumineszenzmaterialien |
| WO1998027136A1 (de) | 1996-12-16 | 1998-06-25 | Aventis Research & Technologies Gmbh & Co Kg | ARYLSUBSTITUIERTE POLY(p-ARYLENVINYLENE), VERFAHREN ZUR HERSTELLUNG UND DEREN VERWENDUNG IN ELEKTROLUMINESZENZBAUELEMENTEN |
| EP1205527A1 (de) | 2000-03-27 | 2002-05-15 | Idemitsu Kosan Co., Ltd. | Organische elektrolumineszierende vorrichtung |
| WO2002060910A1 (de) | 2001-02-01 | 2002-08-08 | Covion Organic Semiconductors Gmbh | Verfahren zur herstellung von hochreinen, tris-ortho-metallierten organo-iridium-verbindungen |
| WO2004013080A1 (en) | 2002-08-01 | 2004-02-12 | Covion Organic Semiconductors Gmbh | Spirobifluorene derivatives, their preparation and uses thereof |
| WO2004037887A2 (de) | 2002-10-25 | 2004-05-06 | Covion Organic Semiconductors Gmbh | Arylamin-einheiten enthaltende konjugierte polymere, deren darstellung und verwendung |
| WO2004058911A2 (de) | 2002-12-23 | 2004-07-15 | Covion Organic Semiconductors Gmbh | Organisches elektrolumineszenzelement |
| WO2004081017A1 (de) | 2003-03-11 | 2004-09-23 | Covion Organic Semiconductors Gmbh | Metallkomplexe |
| WO2004085449A1 (de) | 2003-03-27 | 2004-10-07 | Covion Organic Semiconductors Gmbh | Verfahren zur herstellung von hochreinen organo-iridium-verbindungen |
| JP2004288381A (ja) | 2003-03-19 | 2004-10-14 | Konica Minolta Holdings Inc | 有機エレクトロルミネッセンス素子 |
| WO2004093207A2 (de) | 2003-04-15 | 2004-10-28 | Covion Organic Semiconductors Gmbh | Mischungen von organischen zur emission befähigten halbleitern und matrixmaterialien, deren verwendung und elektronikbauteile enthaltend diese mischungen |
| WO2005011013A1 (de) | 2003-07-21 | 2005-02-03 | Covion Organic Semiconductors Gmbh | Organisches elektrolumineszenzelement |
| US20050069729A1 (en) | 2003-09-30 | 2005-03-31 | Konica Minolta Holdings, Inc. | Organic electroluminescent element, illuminator, display and compound |
| WO2005042548A1 (de) | 2003-10-30 | 2005-05-12 | Merck Patent Gmbh | Verfahren zur herstellung von heteroleptischer, ortho-metallierter organometall-verbindungen |
| WO2005111172A2 (de) | 2004-05-11 | 2005-11-24 | Merck Patent Gmbh | Neue materialmischungen für die elektrolumineszenz |
| WO2005113563A1 (de) | 2004-05-19 | 2005-12-01 | Merck Patent Gmbh | Metallkomplexe |
| JP2005347160A (ja) | 2004-06-04 | 2005-12-15 | Konica Minolta Holdings Inc | 有機エレクトロルミネッセンス素子、照明装置及び表示装置 |
| EP1617711A1 (de) | 2003-04-23 | 2006-01-18 | Konica Minolta Holdings, Inc. | Organisches elektrolumineszenzbauelement und anzeige |
| WO2006005627A1 (en) | 2004-07-15 | 2006-01-19 | Merck Patent Gmbh | Oligomeric derivatives of spirobifluorene, their preparation and use |
| WO2006008069A1 (de) | 2004-07-16 | 2006-01-26 | Merck Patent Gmbh | Metallkomplexe |
| WO2006117052A1 (de) | 2005-05-03 | 2006-11-09 | Merck Patent Gmbh | Organische elektrolumineszenzvorrichtung und in deren herstellung verwendete boronsäure- und borinsäure-derivate |
| EP1731584A1 (de) | 2004-03-31 | 2006-12-13 | Konica Minolta Holdings, Inc. | Organischer elektrolumineszenzvorrichtungsstoff, organische elektrolumineszenzvorrichtung, display und beleuchtungsvorrichtung |
| WO2007063754A1 (ja) | 2005-12-01 | 2007-06-07 | Nippon Steel Chemical Co., Ltd. | 有機電界発光素子用化合物及び有機電界発光素子 |
| WO2007065523A1 (de) | 2005-12-05 | 2007-06-14 | Merck Patent Gmbh | Verfahren zur herstellung ortho-metallierter metallverbindungen |
| WO2007073303A2 (en) | 2005-12-23 | 2007-06-28 | Astrazeneca Ab | Novel benzimidazole derivatives as vanilloid receptor 1 (vrl) inhibitors |
| WO2007095118A2 (en) | 2006-02-10 | 2007-08-23 | Universal Display Corporation | METAL COMPLEXES OF CYCLOMETALLATED IMIDAZO[1,2-f]PHENANTHRIDINE AND DIIMIDAZO[1,2-A:1',2'-C]QUINAZOLINE LIGANDS AND ISOELECTRONIC AND BENZANNULATED ANALOGS THEREOF |
| WO2007137725A1 (de) | 2006-05-31 | 2007-12-06 | Merck Patent Gmbh | Neue materialien für organische elektrolumineszenzvorrichtungen |
| WO2008056746A1 (fr) | 2006-11-09 | 2008-05-15 | Nippon Steel Chemical Co., Ltd. | Composé pour un dispositif électroluminescent organique et dispositif électroluminescent organique |
| WO2008086851A1 (de) | 2007-01-18 | 2008-07-24 | Merck Patent Gmbh | Carbazol-derivate für organische elektrolumineszenzvorrichtungen |
| WO2009062578A1 (de) | 2007-11-12 | 2009-05-22 | Merck Patent Gmbh | Organische elektrolumineszenzvorrichtungen enthaltend azomethin-metall-komplexe |
| US20090136779A1 (en) | 2007-11-26 | 2009-05-28 | Chien-Hong Cheng | Conjugated compounds containing hydroindoloacridine structural elements, and their use |
| US20090134784A1 (en) | 2004-10-21 | 2009-05-28 | Universal Display Corporation | Carbazole-containing materials in phosphorescent light emitting diodes |
| WO2009148015A1 (ja) | 2008-06-05 | 2009-12-10 | 出光興産株式会社 | ハロゲン化合物、多環系化合物及びそれを用いた有機エレクトロルミネッセンス素子 |
| WO2010006680A1 (de) | 2008-07-18 | 2010-01-21 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2010015306A1 (de) | 2008-08-08 | 2010-02-11 | Merck Patent Gmbh, | Organische elektrolumineszenzvorrichtung |
| WO2010050778A1 (en) | 2008-10-31 | 2010-05-06 | Gracel Display Inc. | Novel compounds for organic electronic material and organic electronic device using the same |
| WO2010054730A1 (de) | 2008-11-11 | 2010-05-20 | Merck Patent Gmbh | Organische elektrolumineszenzvorrichtungen |
| WO2010054729A2 (de) | 2008-11-11 | 2010-05-20 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2010086089A1 (de) | 2009-02-02 | 2010-08-05 | Merck Patent Gmbh | Metallkomplexe |
| WO2010108579A1 (de) | 2009-03-23 | 2010-09-30 | Merck Patent Gmbh | Organische elektrolumineszenzvorrichtung |
| WO2010136109A1 (de) | 2009-05-29 | 2010-12-02 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2011000455A1 (de) | 2009-06-30 | 2011-01-06 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| DE102009048791A1 (de) | 2009-10-08 | 2011-04-14 | Merck Patent Gmbh | Materialien für organische Elektrolumineszenzvorrichtungen |
| DE102010005697A1 (de) | 2010-01-25 | 2011-07-28 | Merck Patent GmbH, 64293 | Verbindungen für elektronische Vorrichtungen |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6014304B2 (ja) * | 2010-01-15 | 2016-10-25 | ユー・ディー・シー アイルランド リミテッド | 有機電界発光素子 |
-
2011
- 2011-05-18 DE DE112011102008.2T patent/DE112011102008B4/de active Active
- 2011-05-18 US US13/703,683 patent/US9273080B2/en active Active
- 2011-05-18 CN CN201180029938.0A patent/CN102939296B/zh active Active
- 2011-05-18 KR KR1020137000877A patent/KR20130087499A/ko not_active Application Discontinuation
- 2011-05-18 WO PCT/EP2011/002467 patent/WO2011157339A1/de active Application Filing
- 2011-05-18 JP JP2013514565A patent/JP6054290B2/ja active Active
Patent Citations (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4539507A (en) | 1983-03-25 | 1985-09-03 | Eastman Kodak Company | Organic electroluminescent devices having improved power conversion efficiencies |
| US5151629A (en) | 1991-08-01 | 1992-09-29 | Eastman Kodak Company | Blue emitting internal junction organic electroluminescent device (I) |
| EP0652273A1 (de) | 1993-11-09 | 1995-05-10 | Shinko Electric Industries Co. Ltd. | Organisches Material für elektrolumineszente Vorrichtung und elektrolumineszente Vorrichtung |
| EP0676461A2 (de) | 1994-04-07 | 1995-10-11 | Hoechst Aktiengesellschaft | Spiroverbindungen und ihre Verwendung als Elektrolumineszenzmaterialien |
| WO1998027136A1 (de) | 1996-12-16 | 1998-06-25 | Aventis Research & Technologies Gmbh & Co Kg | ARYLSUBSTITUIERTE POLY(p-ARYLENVINYLENE), VERFAHREN ZUR HERSTELLUNG UND DEREN VERWENDUNG IN ELEKTROLUMINESZENZBAUELEMENTEN |
| EP1205527A1 (de) | 2000-03-27 | 2002-05-15 | Idemitsu Kosan Co., Ltd. | Organische elektrolumineszierende vorrichtung |
| WO2002060910A1 (de) | 2001-02-01 | 2002-08-08 | Covion Organic Semiconductors Gmbh | Verfahren zur herstellung von hochreinen, tris-ortho-metallierten organo-iridium-verbindungen |
| WO2004013080A1 (en) | 2002-08-01 | 2004-02-12 | Covion Organic Semiconductors Gmbh | Spirobifluorene derivatives, their preparation and uses thereof |
| WO2004037887A2 (de) | 2002-10-25 | 2004-05-06 | Covion Organic Semiconductors Gmbh | Arylamin-einheiten enthaltende konjugierte polymere, deren darstellung und verwendung |
| WO2004058911A2 (de) | 2002-12-23 | 2004-07-15 | Covion Organic Semiconductors Gmbh | Organisches elektrolumineszenzelement |
| WO2004081017A1 (de) | 2003-03-11 | 2004-09-23 | Covion Organic Semiconductors Gmbh | Metallkomplexe |
| JP2004288381A (ja) | 2003-03-19 | 2004-10-14 | Konica Minolta Holdings Inc | 有機エレクトロルミネッセンス素子 |
| WO2004085449A1 (de) | 2003-03-27 | 2004-10-07 | Covion Organic Semiconductors Gmbh | Verfahren zur herstellung von hochreinen organo-iridium-verbindungen |
| WO2004093207A2 (de) | 2003-04-15 | 2004-10-28 | Covion Organic Semiconductors Gmbh | Mischungen von organischen zur emission befähigten halbleitern und matrixmaterialien, deren verwendung und elektronikbauteile enthaltend diese mischungen |
| EP1617711A1 (de) | 2003-04-23 | 2006-01-18 | Konica Minolta Holdings, Inc. | Organisches elektrolumineszenzbauelement und anzeige |
| EP1617710A1 (de) | 2003-04-23 | 2006-01-18 | Konica Minolta Holdings, Inc. | Material für ein organisches elektrolumineszenzgerät, organisches elektrolumineszenzgerät, beleuchtungsvorrichtung und anzeige |
| WO2005011013A1 (de) | 2003-07-21 | 2005-02-03 | Covion Organic Semiconductors Gmbh | Organisches elektrolumineszenzelement |
| US20050069729A1 (en) | 2003-09-30 | 2005-03-31 | Konica Minolta Holdings, Inc. | Organic electroluminescent element, illuminator, display and compound |
| WO2005039246A1 (ja) | 2003-09-30 | 2005-04-28 | Konica Minolta Holdings, Inc. | 有機エレクトロルミネッセンス素子、照明装置、表示装置 |
| WO2005042548A1 (de) | 2003-10-30 | 2005-05-12 | Merck Patent Gmbh | Verfahren zur herstellung von heteroleptischer, ortho-metallierter organometall-verbindungen |
| EP1731584A1 (de) | 2004-03-31 | 2006-12-13 | Konica Minolta Holdings, Inc. | Organischer elektrolumineszenzvorrichtungsstoff, organische elektrolumineszenzvorrichtung, display und beleuchtungsvorrichtung |
| WO2005111172A2 (de) | 2004-05-11 | 2005-11-24 | Merck Patent Gmbh | Neue materialmischungen für die elektrolumineszenz |
| WO2005113563A1 (de) | 2004-05-19 | 2005-12-01 | Merck Patent Gmbh | Metallkomplexe |
| JP2005347160A (ja) | 2004-06-04 | 2005-12-15 | Konica Minolta Holdings Inc | 有機エレクトロルミネッセンス素子、照明装置及び表示装置 |
| WO2006005627A1 (en) | 2004-07-15 | 2006-01-19 | Merck Patent Gmbh | Oligomeric derivatives of spirobifluorene, their preparation and use |
| WO2006008069A1 (de) | 2004-07-16 | 2006-01-26 | Merck Patent Gmbh | Metallkomplexe |
| US20090134784A1 (en) | 2004-10-21 | 2009-05-28 | Universal Display Corporation | Carbazole-containing materials in phosphorescent light emitting diodes |
| WO2006117052A1 (de) | 2005-05-03 | 2006-11-09 | Merck Patent Gmbh | Organische elektrolumineszenzvorrichtung und in deren herstellung verwendete boronsäure- und borinsäure-derivate |
| WO2007063754A1 (ja) | 2005-12-01 | 2007-06-07 | Nippon Steel Chemical Co., Ltd. | 有機電界発光素子用化合物及び有機電界発光素子 |
| WO2007065523A1 (de) | 2005-12-05 | 2007-06-14 | Merck Patent Gmbh | Verfahren zur herstellung ortho-metallierter metallverbindungen |
| WO2007073303A2 (en) | 2005-12-23 | 2007-06-28 | Astrazeneca Ab | Novel benzimidazole derivatives as vanilloid receptor 1 (vrl) inhibitors |
| WO2007095118A2 (en) | 2006-02-10 | 2007-08-23 | Universal Display Corporation | METAL COMPLEXES OF CYCLOMETALLATED IMIDAZO[1,2-f]PHENANTHRIDINE AND DIIMIDAZO[1,2-A:1',2'-C]QUINAZOLINE LIGANDS AND ISOELECTRONIC AND BENZANNULATED ANALOGS THEREOF |
| WO2007137725A1 (de) | 2006-05-31 | 2007-12-06 | Merck Patent Gmbh | Neue materialien für organische elektrolumineszenzvorrichtungen |
| WO2008056746A1 (fr) | 2006-11-09 | 2008-05-15 | Nippon Steel Chemical Co., Ltd. | Composé pour un dispositif électroluminescent organique et dispositif électroluminescent organique |
| WO2008086851A1 (de) | 2007-01-18 | 2008-07-24 | Merck Patent Gmbh | Carbazol-derivate für organische elektrolumineszenzvorrichtungen |
| WO2009062578A1 (de) | 2007-11-12 | 2009-05-22 | Merck Patent Gmbh | Organische elektrolumineszenzvorrichtungen enthaltend azomethin-metall-komplexe |
| US20090136779A1 (en) | 2007-11-26 | 2009-05-28 | Chien-Hong Cheng | Conjugated compounds containing hydroindoloacridine structural elements, and their use |
| WO2009148015A1 (ja) | 2008-06-05 | 2009-12-10 | 出光興産株式会社 | ハロゲン化合物、多環系化合物及びそれを用いた有機エレクトロルミネッセンス素子 |
| WO2010006680A1 (de) | 2008-07-18 | 2010-01-21 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2010015306A1 (de) | 2008-08-08 | 2010-02-11 | Merck Patent Gmbh, | Organische elektrolumineszenzvorrichtung |
| WO2010050778A1 (en) | 2008-10-31 | 2010-05-06 | Gracel Display Inc. | Novel compounds for organic electronic material and organic electronic device using the same |
| WO2010054730A1 (de) | 2008-11-11 | 2010-05-20 | Merck Patent Gmbh | Organische elektrolumineszenzvorrichtungen |
| WO2010054729A2 (de) | 2008-11-11 | 2010-05-20 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2010086089A1 (de) | 2009-02-02 | 2010-08-05 | Merck Patent Gmbh | Metallkomplexe |
| WO2010108579A1 (de) | 2009-03-23 | 2010-09-30 | Merck Patent Gmbh | Organische elektrolumineszenzvorrichtung |
| WO2010136109A1 (de) | 2009-05-29 | 2010-12-02 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2011000455A1 (de) | 2009-06-30 | 2011-01-06 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| DE102009048791A1 (de) | 2009-10-08 | 2011-04-14 | Merck Patent Gmbh | Materialien für organische Elektrolumineszenzvorrichtungen |
| DE102010005697A1 (de) | 2010-01-25 | 2011-07-28 | Merck Patent GmbH, 64293 | Verbindungen für elektronische Vorrichtungen |
Non-Patent Citations (13)
| Title |
|---|
| "Darstellung nach Organikum", 1965, VEB DEUTSCHER VERLAG DER WISSENSCHAFTEN, pages: 409 |
| A. ZHANG ET AL., J. COMBI. CHEM., vol. 9, no. 6, 2007, pages 916 |
| D. JERCHEL ET AL., ANN. CHEM., vol. 575, 1952, pages 162 |
| K. T. J. LOONES ET AL., TETRAHEDRON, vol. 63, 2007, pages 3818 |
| M. A. BALDO, APPL. PHYS. LETT., vol. 75, 1999, pages 4 - 6 |
| M. BERG ET AL., CHEM. MED. CHEM., vol. 4, no. 2, 2009, pages 249 |
| M. S. ARNOLD ET AL., APPL. PHYS. LETT., vol. 92, 2008, pages 053301 |
| N. UMEDA ET AL., ANGEW. CHEM INT. ED., vol. 47, 2008, pages 4019 |
| N. XIUA ET AL., ANGEW. CHEM. INT. ED., vol. 48, 2009, pages 337 |
| P. N. W. BAXTER, CHEM. EUR. J., vol. 9, 2003, pages 2531 |
| SAHA ET AL., SYNTH. COMMUN, vol. 37, 2007, pages 3455 |
| SAHA ET AL., SYNTH. COMMUN., vol. 37, no. 19, 2007, pages 3455 |
| V. CANIBANO ET AL., SYNTHESIS, vol. 14, 2001, pages 2175 |
Cited By (240)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9382253B2 (en) | 2010-07-16 | 2016-07-05 | Merck Patent Gmbh | Metal complexes |
| US9118022B2 (en) * | 2011-06-03 | 2015-08-25 | Merck Patent Gmbh | Organic electroluminescent device |
| US20140088305A1 (en) * | 2011-06-03 | 2014-03-27 | Merck Patent Gmbh | Organic electroluminescent device |
| WO2012163465A1 (de) | 2011-06-03 | 2012-12-06 | Merck Patent Gmbh | Organische elektrolumineszenzvorrichtung |
| CN103619986A (zh) * | 2011-06-03 | 2014-03-05 | 默克专利有限公司 | 有机电致发光器件 |
| WO2013041176A1 (de) | 2011-09-21 | 2013-03-28 | Merck Patent Gmbh | Carbazolderivate für organische elektrolumineszenzvorrichtungen |
| WO2013083216A1 (de) | 2011-11-17 | 2013-06-13 | Merck Patent Gmbh | Spiro -dihydroacridinderivate und ihre verwendung als materialien für organische elektrolumineszenzvorrichtungen |
| EP2623508A1 (de) | 2012-02-02 | 2013-08-07 | Konica Minolta Advanced Layers, Inc. | Iridiumkomplexverbindung, Material aus organischem elektrolumineszentem Material, organisches elektrolumineszentes Material, Beleuchtungsvorrichtung und Anzeigevorrichtung |
| WO2013120577A1 (en) | 2012-02-14 | 2013-08-22 | Merck Patent Gmbh | Spirobifluorene compounds for organic electroluminescent devices |
| EP3101088A1 (de) | 2012-02-14 | 2016-12-07 | Merck Patent GmbH | Materialien für organische elektrolumineszenzvorrichtungen |
| EP3235892A1 (de) | 2012-02-14 | 2017-10-25 | Merck Patent GmbH | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2013139431A1 (de) | 2012-03-23 | 2013-09-26 | Merck Patent Gmbh | 9,9'-spirobixanthenderivate für elektrolumineszenzvorrichtungen |
| WO2014008967A2 (de) | 2012-07-10 | 2014-01-16 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| CN104428392A (zh) * | 2012-07-13 | 2015-03-18 | 默克专利有限公司 | 金属络合物 |
| TWI671305B (zh) * | 2012-07-13 | 2019-09-11 | 德商麥克專利有限公司 | 金屬錯合物類 |
| CN104428392B (zh) * | 2012-07-13 | 2017-05-31 | 默克专利有限公司 | 金属络合物 |
| US9837622B2 (en) | 2012-07-13 | 2017-12-05 | Merck Patent Gmbh | Metal complexes |
| KR102076481B1 (ko) | 2012-07-13 | 2020-02-12 | 메르크 파텐트 게엠베하 | 금속 착물 |
| KR20150036612A (ko) * | 2012-07-13 | 2015-04-07 | 메르크 파텐트 게엠베하 | 금속 착물 |
| WO2014008982A1 (de) | 2012-07-13 | 2014-01-16 | Merck Patent Gmbh | Metallkomplexe |
| WO2014015931A1 (de) | 2012-07-23 | 2014-01-30 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2014023388A1 (de) | 2012-08-10 | 2014-02-13 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2014030666A1 (ja) | 2012-08-24 | 2014-02-27 | コニカミノルタ株式会社 | 透明電極、電子デバイス、および透明電極の製造方法 |
| JP2014045101A (ja) * | 2012-08-28 | 2014-03-13 | Konica Minolta Inc | 有機エレクトロルミネッセンス素子、照明装置および表示装置 |
| KR20150096520A (ko) * | 2012-12-21 | 2015-08-24 | 메르크 파텐트 게엠베하 | 금속 착물 |
| JP2016508127A (ja) * | 2012-12-21 | 2016-03-17 | メルク パテント ゲーエムベーハー | 金属錯体 |
| KR20150096805A (ko) * | 2012-12-21 | 2015-08-25 | 메르크 파텐트 게엠베하 | 금속 착물 |
| KR102188212B1 (ko) * | 2012-12-21 | 2020-12-08 | 메르크 파텐트 게엠베하 | 금속 착물 |
| KR102146232B1 (ko) * | 2012-12-21 | 2020-08-20 | 메르크 파텐트 게엠베하 | 금속 착물 |
| KR102188214B1 (ko) * | 2012-12-21 | 2020-12-08 | 메르크 파텐트 게엠베하 | 금속 착물 |
| KR20150100842A (ko) * | 2012-12-21 | 2015-09-02 | 메르크 파텐트 게엠베하 | 금속 착물 |
| JP2016507491A (ja) * | 2012-12-21 | 2016-03-10 | メルク パテント ゲーエムベーハー | 金属錯体 |
| JP2016507490A (ja) * | 2012-12-21 | 2016-03-10 | メルク パテント ゲーエムベーハー | 金属錯体 |
| KR101576570B1 (ko) | 2013-02-18 | 2015-12-10 | 주식회사 두산 | 유기 화합물 및 이를 포함하는 유기 전계 발광 소자 |
| WO2014147134A1 (en) | 2013-03-20 | 2014-09-25 | Basf Se | Azabenzimidazole carbene complexes as efficiency booster in oleds |
| WO2014157618A1 (ja) | 2013-03-29 | 2014-10-02 | コニカミノルタ株式会社 | 有機エレクトロルミネッセンス素子、それを具備した照明装置及び表示装置 |
| WO2014157494A1 (ja) | 2013-03-29 | 2014-10-02 | コニカミノルタ株式会社 | 有機エレクトロルミネッセンス素子用材料、有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| EP3266789A1 (de) | 2013-07-02 | 2018-01-10 | UDC Ireland Limited | Monosubstituierte diazabenzimidazolcarben-metall-komplexe zur verwendung in organischen leuchtdioden |
| EP3608329A1 (de) | 2013-07-02 | 2020-02-12 | UDC Ireland Limited | Monosubstituierte diazabenzimidazolcarben-metall-komplexe zur verwendung in organischen leuchtdioden |
| WO2015000955A1 (en) | 2013-07-02 | 2015-01-08 | Basf Se | Monosubstituted diazabenzimidazole carbene metal complexes for use in organic light emitting diodes |
| EP3239161A1 (de) | 2013-07-31 | 2017-11-01 | UDC Ireland Limited | Lumineszente diazabenzimidazol-carben-metall-komplexe |
| KR20160107305A (ko) * | 2014-01-13 | 2016-09-13 | 메르크 파텐트 게엠베하 | 금속 착물 |
| US11005050B2 (en) | 2014-01-13 | 2021-05-11 | Merck Patent Gmbh | Metal complexes |
| KR102378657B1 (ko) | 2014-01-13 | 2022-03-24 | 메르크 파텐트 게엠베하 | 금속 착물 |
| WO2015104045A1 (de) * | 2014-01-13 | 2015-07-16 | Merck Patent Gmbh | Metallkomplexe |
| US11393988B2 (en) | 2014-02-05 | 2022-07-19 | Merck Patent Gmbh | Metal complexes |
| WO2015117718A1 (de) * | 2014-02-05 | 2015-08-13 | Merck Patent Gmbh | Metallkomplexe |
| US10370396B2 (en) | 2014-03-31 | 2019-08-06 | Udc Ireland Limited | Metal complexes, comprising carbene ligands having an O-substituted non-cyclometallated aryl group and their use in organic light emitting diodes |
| US10118939B2 (en) | 2014-03-31 | 2018-11-06 | Udc Ireland Limited | Metal complexes, comprising carbene ligands having an o-substituted non-cyclometalated aryl group and their use in organic light emitting diodes |
| US9862739B2 (en) | 2014-03-31 | 2018-01-09 | Udc Ireland Limited | Metal complexes, comprising carbene ligands having an O-substituted non-cyclometalated aryl group and their use in organic light emitting diodes |
| WO2015169412A1 (de) | 2014-05-05 | 2015-11-12 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| DE102014008722A1 (de) | 2014-06-18 | 2015-12-24 | Merck Patent Gmbh | Zusammensetzungen für elektronische Vorrichtungen |
| WO2015192941A1 (de) | 2014-06-18 | 2015-12-23 | Merck Patent Gmbh | Zusammensetzungen für elektronische vorrichtungen |
| WO2015197156A1 (de) | 2014-06-25 | 2015-12-30 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| US10323180B2 (en) | 2014-12-04 | 2019-06-18 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Deuterated organic compound, mixture and composition containing said compound, and organic electronic device |
| US10840450B2 (en) | 2014-12-04 | 2020-11-17 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Polymer, and mixture or formulation, and organic electronic device containing same, and monomer thereof |
| US10510967B2 (en) | 2014-12-11 | 2019-12-17 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Organic compound, and mixture, formulation and organic device comprising the same |
| US10364316B2 (en) | 2015-01-13 | 2019-07-30 | Guangzhou Chinaray Optoelectronics Materials Ltd. | Conjugated polymer containing ethynyl crosslinking group, mixture, formulation, organic electronic device containing the same and application therof |
| EP4271160A2 (de) | 2015-02-13 | 2023-11-01 | Merck Patent GmbH | Aromatisches heterocyclisches derivat und organisches elektrolumineszenzelement, beleuchtungsvorrichtung und anzeigevorrichtung mit dem aromatischen heterocyclischen derivat |
| WO2016198144A1 (en) | 2015-06-10 | 2016-12-15 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2017012687A1 (en) | 2015-07-22 | 2017-01-26 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| EP4236652A2 (de) | 2015-07-29 | 2023-08-30 | Merck Patent GmbH | Materialien für organische elektrolumineszenzvorrichtungen |
| EP4301110A2 (de) | 2015-07-30 | 2024-01-03 | Merck Patent GmbH | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2017016630A1 (en) | 2015-07-30 | 2017-02-02 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2017025166A1 (en) | 2015-08-13 | 2017-02-16 | Merck Patent Gmbh | Hexamethylindanes |
| WO2017071791A1 (en) | 2015-10-27 | 2017-05-04 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| US11555128B2 (en) | 2015-11-12 | 2023-01-17 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Printing composition, electronic device comprising same and preparation method for functional material thin film |
| US10968243B2 (en) | 2015-12-04 | 2021-04-06 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Organometallic complex and application thereof in electronic devices |
| EP3200255A2 (de) | 2016-01-06 | 2017-08-02 | Konica Minolta, Inc. | Organisches elektrolumineszenzelement, verfahren zur herstellung eines organischen elektrolumineszenzelements, sowie anzeige und beleuchtungsvorrichtung |
| WO2017148564A1 (de) | 2016-03-03 | 2017-09-08 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2017148565A1 (de) | 2016-03-03 | 2017-09-08 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| CN108780849A (zh) * | 2016-03-15 | 2018-11-09 | 新日铁住金化学株式会社 | 有机电致发光元件 |
| CN108780849B (zh) * | 2016-03-15 | 2020-10-27 | 日铁化学材料株式会社 | 有机电致发光元件 |
| WO2017157983A1 (de) | 2016-03-17 | 2017-09-21 | Merck Patent Gmbh | Verbindungen mit spirobifluoren-strukturen |
| WO2017178311A1 (de) | 2016-04-11 | 2017-10-19 | Merck Patent Gmbh | Heterocyclische verbindungen mit dibenzofuran- und/oder dibenzothiophen-strukturen |
| WO2017186760A1 (en) | 2016-04-29 | 2017-11-02 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2017207596A1 (en) | 2016-06-03 | 2017-12-07 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| EP3978477A2 (de) | 2016-06-03 | 2022-04-06 | Merck Patent GmbH | Materialien für organische elektrolumineszente vorrichtungen |
| US11932659B2 (en) | 2016-07-25 | 2024-03-19 | Udc Ireland Limited | Metal complexes for use as emitters in organic electroluminescence devices |
| WO2018050583A1 (de) | 2016-09-14 | 2018-03-22 | Merck Patent Gmbh | Verbindungen mit carbazol-strukturen |
| WO2018050584A1 (de) | 2016-09-14 | 2018-03-22 | Merck Patent Gmbh | Verbindungen mit spirobifluoren-strukturen |
| WO2018060218A1 (de) | 2016-09-30 | 2018-04-05 | Merck Patent Gmbh | Carbazole mit diazadibenzofuran- oder diazadibenzothiophen-strukturen |
| WO2018060307A1 (de) | 2016-09-30 | 2018-04-05 | Merck Patent Gmbh | Verbindungen mit diazadibenzofuran- oder diazadibenzothiophen-strukturen |
| WO2018087020A1 (en) | 2016-11-08 | 2018-05-17 | Merck Patent Gmbh | Compounds for electronic devices |
| WO2018087022A1 (de) | 2016-11-09 | 2018-05-17 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2018087346A1 (de) | 2016-11-14 | 2018-05-17 | Merck Patent Gmbh | Verbindungen mit einer akzeptor- und einer donorgruppe |
| EP4271163A2 (de) | 2016-11-14 | 2023-11-01 | Merck Patent GmbH | Verbindungen mit einer akzeptor- und einer donorgruppe |
| WO2018091435A1 (en) | 2016-11-17 | 2018-05-24 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| US11447496B2 (en) | 2016-11-23 | 2022-09-20 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Nitrogen-containing fused heterocyclic ring compound and application thereof |
| US11453745B2 (en) | 2016-11-23 | 2022-09-27 | Guangzhou Chinaray Optoelectronic Materials Ltd. | High polymer, mixture containing same, composition, organic electronic component, and monomer for polymerization |
| US11239428B2 (en) | 2016-11-23 | 2022-02-01 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Boron-containing organic compound and applications thereof, organic mixture, and organic electronic device |
| US10804470B2 (en) | 2016-11-23 | 2020-10-13 | Guangzhou Chinaray Optoelectronic Materials Ltd | Organic compound |
| WO2018095395A1 (zh) | 2016-11-23 | 2018-05-31 | 广州华睿光电材料有限公司 | 高聚物、包含其的混合物、组合物和有机电子器件以及用于聚合的单体 |
| WO2018095392A1 (zh) | 2016-11-23 | 2018-05-31 | 广州华睿光电材料有限公司 | 有机混合物、组合物以及有机电子器件 |
| US11634444B2 (en) | 2016-11-23 | 2023-04-25 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Metal organic complex, high polymer, composition, and organic electronic component |
| US11248138B2 (en) | 2016-11-23 | 2022-02-15 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Printing ink formulations, preparation methods and uses thereof |
| WO2018095381A1 (zh) | 2016-11-23 | 2018-05-31 | 广州华睿光电材料有限公司 | 印刷油墨组合物及其制备方法和用途 |
| WO2018099846A1 (de) | 2016-11-30 | 2018-06-07 | Merck Patent Gmbh | Verbindungen mit valerolaktam-strukturen |
| WO2018104195A1 (de) | 2016-12-05 | 2018-06-14 | Merck Patent Gmbh | Stickstoffhaltige heterocyclen zur verwendung in oleds |
| EP3978491A1 (de) | 2016-12-05 | 2022-04-06 | Merck Patent GmbH | Stickstoffhaltige heterocyclen zur verwendung in oleds |
| WO2018104193A1 (de) | 2016-12-05 | 2018-06-14 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2018104194A1 (de) | 2016-12-05 | 2018-06-14 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2018103744A1 (zh) | 2016-12-08 | 2018-06-14 | 广州华睿光电材料有限公司 | 混合物、组合物及有机电子器件 |
| US10978642B2 (en) | 2016-12-08 | 2021-04-13 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Mixture, composition and organic electronic device |
| US11161933B2 (en) | 2016-12-13 | 2021-11-02 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Conjugated polymer and use thereof in organic electronic device |
| WO2018114883A1 (de) | 2016-12-22 | 2018-06-28 | Merck Patent Gmbh | Mischungen umfassend mindestens zwei organisch funktionelle verbindungen |
| WO2018113785A1 (zh) | 2016-12-22 | 2018-06-28 | 广州华睿光电材料有限公司 | 含呋喃交联基团的聚合物及其应用 |
| US11292875B2 (en) | 2016-12-22 | 2022-04-05 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Cross-linkable polymer based on Diels-Alder reaction and use thereof in organic electronic device |
| US11289654B2 (en) | 2016-12-22 | 2022-03-29 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Polymers containing furanyl crosslinkable groups and uses thereof |
| WO2018127465A1 (de) | 2017-01-04 | 2018-07-12 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2018138039A1 (de) | 2017-01-25 | 2018-08-02 | Merck Patent Gmbh | Carbazolderivate |
| WO2018138306A1 (de) | 2017-01-30 | 2018-08-02 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2018149769A1 (de) | 2017-02-14 | 2018-08-23 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2018166932A1 (de) | 2017-03-13 | 2018-09-20 | Merck Patent Gmbh | Verbindungen mit arylamin-strukturen |
| WO2018166934A1 (de) | 2017-03-15 | 2018-09-20 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2018189134A1 (de) | 2017-04-13 | 2018-10-18 | Merck Patent Gmbh | Zusammensetzung für organische elektronische vorrichtungen |
| WO2018206537A1 (en) | 2017-05-11 | 2018-11-15 | Merck Patent Gmbh | Carbazole-based bodipys for organic electroluminescent devices |
| WO2018206526A1 (en) | 2017-05-11 | 2018-11-15 | Merck Patent Gmbh | Organoboron complexes for organic electroluminescent devices |
| WO2018234346A1 (en) | 2017-06-23 | 2018-12-27 | Merck Patent Gmbh | MATERIALS FOR ORGANIC ELECTROLUMINESCENT DEVICES |
| WO2019007867A1 (de) | 2017-07-05 | 2019-01-10 | Merck Patent Gmbh | Zusammensetzung für organische elektronische vorrichtungen |
| WO2019007866A1 (de) | 2017-07-05 | 2019-01-10 | Merck Patent Gmbh | Zusammensetzung für organische elektronische vorrichtungen |
| EP4186898A1 (de) | 2017-07-05 | 2023-05-31 | Merck Patent GmbH | Zusammensetzung für organische elektronische verbindungen |
| WO2019052933A1 (de) | 2017-09-12 | 2019-03-21 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2019068679A1 (en) | 2017-10-06 | 2019-04-11 | Merck Patent Gmbh | MATERIALS FOR ORGANIC ELECTROLUMINESCENT DEVICES |
| WO2019081391A1 (de) | 2017-10-24 | 2019-05-02 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2019096717A2 (de) | 2017-11-14 | 2019-05-23 | Merck Patent Gmbh | Zusammensetzung für organische elektronische vorrichtungen |
| US11594690B2 (en) | 2017-12-14 | 2023-02-28 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Organometallic complex, and polymer, mixture and formulation comprising same, and use thereof in electronic device |
| US11404651B2 (en) | 2017-12-14 | 2022-08-02 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Transition metal complex material and application thereof in electronic devices |
| WO2019121458A1 (de) | 2017-12-19 | 2019-06-27 | Merck Patent Gmbh | Heterocyclische verbindung zur verwendung in electronischen vorrichtungen |
| WO2019121483A1 (en) | 2017-12-20 | 2019-06-27 | Merck Patent Gmbh | Heteroaromatic compounds |
| US11680059B2 (en) | 2017-12-21 | 2023-06-20 | Guangzhou Chinaray Optoelectronic Materials Ltd. | Organic mixture and application thereof in organic electronic devices |
| WO2019145316A1 (de) | 2018-01-25 | 2019-08-01 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2019175149A1 (en) | 2018-03-16 | 2019-09-19 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2019229011A1 (de) | 2018-05-30 | 2019-12-05 | Merck Patent Gmbh | Zusammensetzung für organische elektronische vorrichtungen |
| WO2019233904A1 (de) | 2018-06-07 | 2019-12-12 | Merck Patent Gmbh | Organische elektrolumineszenzvorrichtungen |
| WO2020011686A1 (de) | 2018-07-09 | 2020-01-16 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2020016264A1 (en) | 2018-07-20 | 2020-01-23 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2020053315A1 (de) | 2018-09-12 | 2020-03-19 | Merck Patent Gmbh | Elektrolumineszierende vorrichtungen |
| WO2020053314A1 (de) | 2018-09-12 | 2020-03-19 | Merck Patent Gmbh | Elektrolumineszierende vorrichtungen |
| DE202019005924U1 (de) | 2018-09-12 | 2023-05-10 | MERCK Patent Gesellschaft mit beschränkter Haftung | Elektrolumineszierende Vorrichtungen |
| DE202019005923U1 (de) | 2018-09-12 | 2023-06-27 | MERCK Patent Gesellschaft mit beschränkter Haftung | Elektrolumineszierende Vorrichtungen |
| WO2020053150A1 (en) | 2018-09-12 | 2020-03-19 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2020064666A1 (de) | 2018-09-27 | 2020-04-02 | Merck Patent Gmbh | Verbindungen, die in einer organischen elektronischen vorrichtung als aktive verbindungen einsetzbar sind |
| EP4190880A1 (de) | 2018-09-27 | 2023-06-07 | Merck Patent GmbH | Verbindungen, die in einer organischen elektronischen vorrichtung als aktive verbindungen einsetzbar sind |
| WO2020064662A2 (de) | 2018-09-27 | 2020-04-02 | Merck Patent Gmbh | Verfahren zur herstellung von sterisch gehinderten stickstoffhaltigen heteroaromatischen verbindungen |
| WO2020094539A1 (de) | 2018-11-05 | 2020-05-14 | Merck Patent Gmbh | In einer organischen elektronischen vorrichtung einsetzbare verbindungen |
| WO2020094542A1 (de) | 2018-11-06 | 2020-05-14 | Merck Patent Gmbh | 5,6-diphenyl-5,6-dihydro-dibenz[c,e][1,2]azaphosphorin- und 6-phenyl-6h-dibenzo[c,e][1,2]thiazin-5,5-dioxid-derivate und ähnliche verbindungen als organische elektrolumineszenzmaterialien für oleds |
| WO2020099349A1 (de) | 2018-11-14 | 2020-05-22 | Merck Patent Gmbh | Zur herstellung einer organischen elektronischen vorrichtung einsetzbare verbindungen |
| WO2020099307A1 (de) | 2018-11-15 | 2020-05-22 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2020127165A1 (de) | 2018-12-19 | 2020-06-25 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2020148243A1 (en) | 2019-01-16 | 2020-07-23 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2020148303A1 (de) | 2019-01-17 | 2020-07-23 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2020169241A1 (de) | 2019-02-18 | 2020-08-27 | Merck Patent Gmbh | Zusammensetzung für organische elektronische vorrichtungen |
| WO2020178230A1 (en) | 2019-03-04 | 2020-09-10 | Merck Patent Gmbh | Ligands for nano-sized materials |
| WO2020182779A1 (de) | 2019-03-12 | 2020-09-17 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2020187865A1 (de) | 2019-03-20 | 2020-09-24 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2020193447A1 (de) | 2019-03-25 | 2020-10-01 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2020208051A1 (en) | 2019-04-11 | 2020-10-15 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2020212296A1 (de) | 2019-04-15 | 2020-10-22 | Merck Patent Gmbh | Metallkomplexe |
| WO2021013775A1 (de) | 2019-07-22 | 2021-01-28 | Merck Patent Gmbh | Verfahren zur herstellung ortho-metallierter metallverbindungen |
| WO2021037401A1 (de) | 2019-08-26 | 2021-03-04 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2021043703A1 (de) | 2019-09-02 | 2021-03-11 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2021043755A1 (de) | 2019-09-03 | 2021-03-11 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2021052924A1 (en) | 2019-09-16 | 2021-03-25 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2021052921A1 (de) | 2019-09-19 | 2021-03-25 | Merck Patent Gmbh | Mischung von zwei hostmaterialien und organische elektrolumineszierende vorrichtung damit |
| WO2021053046A1 (de) | 2019-09-20 | 2021-03-25 | Merck Patent Gmbh | Peri-kondensierte heterozyklische verbindungen als materialien für elektronische vorrichtungen |
| WO2021078710A1 (en) | 2019-10-22 | 2021-04-29 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2021078831A1 (de) | 2019-10-25 | 2021-04-29 | Merck Patent Gmbh | In einer organischen elektronischen vorrichtung einsetzbare verbindungen |
| WO2021089447A1 (de) | 2019-11-04 | 2021-05-14 | Merck Patent Gmbh | Organische elektrolumineszierende vorrichtung |
| WO2021089450A1 (en) | 2019-11-04 | 2021-05-14 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2021094269A1 (en) | 2019-11-12 | 2021-05-20 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2021110741A1 (en) | 2019-12-04 | 2021-06-10 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2021122535A1 (de) | 2019-12-17 | 2021-06-24 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2021122538A1 (de) | 2019-12-18 | 2021-06-24 | Merck Patent Gmbh | Aromatische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2021122740A1 (de) | 2019-12-19 | 2021-06-24 | Merck Patent Gmbh | Polycyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2021151922A1 (de) | 2020-01-29 | 2021-08-05 | Merck Patent Gmbh | Benzimidazol-derivate |
| WO2021170522A1 (de) | 2020-02-25 | 2021-09-02 | Merck Patent Gmbh | Verwendung von heterocyclischen verbindungen in einer organischen elektronischen vorrichtung |
| WO2021175706A1 (de) | 2020-03-02 | 2021-09-10 | Merck Patent Gmbh | Verwendung von sulfonverbindungen in einer organischen elektronischen vorrichtung |
| WO2021180625A1 (de) | 2020-03-11 | 2021-09-16 | Merck Patent Gmbh | Organische elektrolumineszierende vorrichtung |
| WO2021180614A1 (de) | 2020-03-11 | 2021-09-16 | Merck Patent Gmbh | Organische elektrolumineszierende vorrichtung |
| WO2021185712A1 (de) | 2020-03-17 | 2021-09-23 | Merck Patent Gmbh | Heteroaromatische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2021185829A1 (de) | 2020-03-17 | 2021-09-23 | Merck Patent Gmbh | Heterocyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2021191058A1 (en) | 2020-03-23 | 2021-09-30 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2021191117A1 (de) | 2020-03-24 | 2021-09-30 | Merck Patent Gmbh | Materialien für elektronische vorrichtungen |
| WO2021191183A1 (de) | 2020-03-26 | 2021-09-30 | Merck Patent Gmbh | Cyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2021198213A1 (de) | 2020-04-02 | 2021-10-07 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2021204646A1 (de) | 2020-04-06 | 2021-10-14 | Merck Patent Gmbh | Polycyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2021239772A1 (de) | 2020-05-29 | 2021-12-02 | Merck Patent Gmbh | Organische elektrolumineszierende vorrichtung |
| WO2021254984A1 (de) | 2020-06-18 | 2021-12-23 | Merck Patent Gmbh | Indenoazanaphthaline |
| WO2021259824A1 (de) | 2020-06-23 | 2021-12-30 | Merck Patent Gmbh | Verfahren zur herstellung einer mischung |
| WO2022002772A1 (de) | 2020-06-29 | 2022-01-06 | Merck Patent Gmbh | Heteroaromatische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2022002771A1 (de) | 2020-06-29 | 2022-01-06 | Merck Patent Gmbh | Heterocyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2022029096A1 (de) | 2020-08-06 | 2022-02-10 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2022034046A1 (de) | 2020-08-13 | 2022-02-17 | Merck Patent Gmbh | Metallkomplexe |
| WO2022038065A1 (de) | 2020-08-18 | 2022-02-24 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2022038066A1 (de) | 2020-08-19 | 2022-02-24 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2022069421A1 (de) | 2020-09-30 | 2022-04-07 | Merck Patent Gmbh | Zur strukturierung von funktionalen schichten organischer elektrolumineszenzvorrichtungen einsetzbare verbindungen |
| WO2022069422A1 (de) | 2020-09-30 | 2022-04-07 | Merck Patent Gmbh | Verbindungen zur strukturierung von funktionalen schichten organischer elektrolumineszenzvorrichtungen |
| WO2022079068A1 (de) | 2020-10-16 | 2022-04-21 | Merck Patent Gmbh | Heterocyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2022079067A1 (de) | 2020-10-16 | 2022-04-21 | Merck Patent Gmbh | Verbindungen mit heteroatomen für organische elektrolumineszenzvorrichtungen |
| WO2022101171A1 (de) | 2020-11-10 | 2022-05-19 | Merck Patent Gmbh | Schwefelhaltige verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2022117473A1 (de) | 2020-12-02 | 2022-06-09 | Merck Patent Gmbh | Heterocyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2022122682A2 (de) | 2020-12-10 | 2022-06-16 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2022129114A1 (de) | 2020-12-18 | 2022-06-23 | Merck Patent Gmbh | Stickstoffhaltige verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2022129113A1 (de) | 2020-12-18 | 2022-06-23 | Merck Patent Gmbh | Stickstoffhaltige heteroaromaten für organische elektrolumineszenzvorrichtungen |
| WO2022129116A1 (de) | 2020-12-18 | 2022-06-23 | Merck Patent Gmbh | Indolo[3.2.1-jk]carbazole-6-carbonitril-derivate als blau fluoreszierende emitter zur verwendung in oleds |
| WO2022148717A1 (de) | 2021-01-05 | 2022-07-14 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2022157343A1 (de) | 2021-01-25 | 2022-07-28 | Merck Patent Gmbh | Stickstoffhaltige verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2022184601A1 (de) | 2021-03-02 | 2022-09-09 | Merck Patent Gmbh | Verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2022194799A1 (de) | 2021-03-18 | 2022-09-22 | Merck Patent Gmbh | Heteroaromatische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2022229126A1 (de) | 2021-04-29 | 2022-11-03 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2022229298A1 (de) | 2021-04-29 | 2022-11-03 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2022229234A1 (de) | 2021-04-30 | 2022-11-03 | Merck Patent Gmbh | Stickstoffhaltige, heterocyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2022243403A1 (de) | 2021-05-21 | 2022-11-24 | Merck Patent Gmbh | Verfahren zur kontinuierlichen aufreinigung von mindestens einem funktionalen material und vorrichtung zur kontinuierlichen aufreinigung von mindestens einem funktionalen material |
| WO2022200638A1 (de) | 2021-07-06 | 2022-09-29 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2023036976A1 (en) | 2021-09-13 | 2023-03-16 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2023041454A1 (de) | 2021-09-14 | 2023-03-23 | Merck Patent Gmbh | Borhaltige, heterocyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2023052314A1 (de) | 2021-09-28 | 2023-04-06 | Merck Patent Gmbh | Materialien für elektronische vorrichtungen |
| WO2023052313A1 (de) | 2021-09-28 | 2023-04-06 | Merck Patent Gmbh | Materialien für elektronische vorrichtungen |
| WO2023052272A1 (de) | 2021-09-28 | 2023-04-06 | Merck Patent Gmbh | Materialien für elektronische vorrichtungen |
| WO2023052275A1 (de) | 2021-09-28 | 2023-04-06 | Merck Patent Gmbh | Materialien für elektronische vorrichtungen |
| WO2023072799A1 (de) | 2021-10-27 | 2023-05-04 | Merck Patent Gmbh | Bor- und stickstoffhaltige, heterocyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2023094412A1 (de) | 2021-11-25 | 2023-06-01 | Merck Patent Gmbh | Materialien für elektronische vorrichtungen |
| WO2023099543A1 (en) | 2021-11-30 | 2023-06-08 | Merck Patent Gmbh | Compounds having fluorene structures |
| WO2023110742A1 (de) | 2021-12-13 | 2023-06-22 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2023117835A1 (en) | 2021-12-21 | 2023-06-29 | Merck Patent Gmbh | Electronic devices |
| WO2023117836A1 (en) | 2021-12-21 | 2023-06-29 | Merck Patent Gmbh | Electronic devices |
| WO2023117837A1 (de) | 2021-12-21 | 2023-06-29 | Merck Patent Gmbh | Verfahren zur herstellung von deuterierten organischen verbindungen |
| WO2023152063A1 (de) | 2022-02-09 | 2023-08-17 | Merck Patent Gmbh | Materialien für organische elektrolumineszenzvorrichtungen |
| WO2023152346A1 (de) | 2022-02-14 | 2023-08-17 | Merck Patent Gmbh | Materialien für elektronische vorrichtungen |
| WO2023161167A1 (de) | 2022-02-23 | 2023-08-31 | Merck Patent Gmbh | Stickstoffhaltige heterocyclen für organische elektrolumineszenzvorrichtungen |
| WO2023161168A1 (de) | 2022-02-23 | 2023-08-31 | Merck Patent Gmbh | Aromatische heterocyclen für organische elektrolumineszenzvorrichtungen |
| WO2023213837A1 (de) | 2022-05-06 | 2023-11-09 | Merck Patent Gmbh | Cyclische verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2023222559A1 (de) | 2022-05-18 | 2023-11-23 | Merck Patent Gmbh | Verfahren zur herstellung von deuterierten organischen verbindungen |
| WO2023247663A1 (de) | 2022-06-24 | 2023-12-28 | Merck Patent Gmbh | Zusammensetzung für organische elektronische vorrichtungen |
| WO2023247662A1 (de) | 2022-06-24 | 2023-12-28 | Merck Patent Gmbh | Zusammensetzung für organische elektronische vorrichtungen |
| WO2024013004A1 (de) | 2022-07-11 | 2024-01-18 | Merck Patent Gmbh | Materialien für elektronische vorrichtungen |
| WO2024033282A1 (en) | 2022-08-09 | 2024-02-15 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
| WO2024061942A1 (de) | 2022-09-22 | 2024-03-28 | Merck Patent Gmbh | Stickstoffenthaltende verbindungen für organische elektrolumineszenzvorrichtungen |
| WO2024061948A1 (de) | 2022-09-22 | 2024-03-28 | Merck Patent Gmbh | Stickstoffenthaltende heterocyclen für organische elektrolumineszenzvorrichtungen |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102939296A (zh) | 2013-02-20 |
| US9273080B2 (en) | 2016-03-01 |
| JP6054290B2 (ja) | 2016-12-27 |
| US20130082209A1 (en) | 2013-04-04 |
| CN102939296B (zh) | 2016-02-10 |
| DE112011102008B4 (de) | 2022-04-21 |
| JP2013531652A (ja) | 2013-08-08 |
| KR20130087499A (ko) | 2013-08-06 |
| DE112011102008A5 (de) | 2013-04-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE112011102008B4 (de) | Metallkomplexe | |
| EP3174890B1 (de) | Metallkomplexe | |
| EP2872590B1 (de) | Metallkomplexe | |
| EP3160976B1 (de) | Metallkomplexe | |
| EP2742054B1 (de) | Metallkomplexe | |
| EP2726490B1 (de) | Metallkomplexe | |
| DE112010004049B4 (de) | Metallkomplexe | |
| EP2714704B1 (de) | Metallkomplexe | |
| EP2906575B1 (de) | Metallkomplexe | |
| EP2935292B1 (de) | Metallkomplexe | |
| EP3526228A1 (de) | Metallkomplexe | |
| WO2012007086A1 (de) | Metallkomplexe | |
| EP3044284A1 (de) | Metallkomplexe | |
| WO2014023377A2 (de) | Metallkomplexe | |
| WO2010086089A1 (de) | Metallkomplexe | |
| EP3046927A1 (de) | Polycyclische phenyl-pyridin iridiumkomplexe und derivate davon für oled | |
| WO2009146770A2 (de) | Elektronische vorrichtung enthaltend metallkomplexe | |
| WO2012007088A1 (de) | Metallkomplexe | |
| WO2014044347A1 (de) | Metallkomplexe | |
| EP2984093A1 (de) | Metallkomplexe und ihre verwendung in elektronischen vorrichtungen | |
| EP3956338A1 (de) | Metallkomplexe |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180029938.0 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11720396 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13703683 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2013514565 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 112011102008 Country of ref document: DE Ref document number: 1120111020082 Country of ref document: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20137000877 Country of ref document: KR Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: R225 Ref document number: 112011102008 Country of ref document: DE Effective date: 20130404 |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11720396 Country of ref document: EP Kind code of ref document: A1 |