JP4120559B2 - 排気ガス浄化用触媒 - Google Patents
排気ガス浄化用触媒 Download PDFInfo
- Publication number
- JP4120559B2 JP4120559B2 JP2003364852A JP2003364852A JP4120559B2 JP 4120559 B2 JP4120559 B2 JP 4120559B2 JP 2003364852 A JP2003364852 A JP 2003364852A JP 2003364852 A JP2003364852 A JP 2003364852A JP 4120559 B2 JP4120559 B2 JP 4120559B2
- Authority
- JP
- Japan
- Prior art keywords
- exhaust gas
- zirconium
- purification catalyst
- gas purification
- temperature
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images



























Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D53/00—Separation of gases or vapours; Recovering vapours of volatile solvents from gases; Chemical or biological purification of waste gases, e.g. engine exhaust gases, smoke, fumes, flue gases, aerosols
- B01D53/34—Chemical or biological purification of waste gases
- B01D53/92—Chemical or biological purification of waste gases of engine exhaust gases
- B01D53/94—Chemical or biological purification of waste gases of engine exhaust gases by catalytic processes
- B01D53/9445—Simultaneously removing carbon monoxide, hydrocarbons or nitrogen oxides making use of three-way catalysts [TWC] or four-way-catalysts [FWC]
- B01D53/945—Simultaneously removing carbon monoxide, hydrocarbons or nitrogen oxides making use of three-way catalysts [TWC] or four-way-catalysts [FWC] characterised by a specific catalyst
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/002—Mixed oxides other than spinels, e.g. perovskite
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/56—Platinum group metals
- B01J23/58—Platinum group metals with alkali- or alkaline earth metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/56—Platinum group metals
- B01J23/63—Platinum group metals with rare earths or actinides
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02T—CLIMATE CHANGE MITIGATION TECHNOLOGIES RELATED TO TRANSPORTATION
- Y02T10/00—Road transport of goods or passengers
- Y02T10/10—Internal combustion engine [ICE] based vehicles
- Y02T10/12—Improving ICE efficiencies
Description
リーン空燃比のもとで燃焼が行われているときには排気ガス中に過剰酸素O2が含まれている。従って超強塩基性点に吸着された一酸化窒素N−O- この過剰酸素O2によって酸化され、それによって硝酸イオンNO3 -となる。即ち、排気ガス中の酸素濃度が高いときには反応が硝酸イオンNO3 -を生成する方向に進み、斯くしてリーン空燃比のもとで燃焼が行われているときには一部の超強塩基性点に硝酸イオンNO3 -が生成、保持される。なお、硝酸イオンNO3 -は一酸化窒素NOが結晶を構成している酸素イオンO2-と結合することによっても生成され、また生成された硝酸イオンNO3 -が結晶を構成しているジルコニウムZr4+に吸着された状態で排気ガス浄化用触媒20上に保持される場合もある。
図8を参照すると、まず初めにステップ100において図6に示すマップから一酸化窒素量Q(NO)が算出される。次いでステップ101ではΣQにQ(NO)を加算することによって積算量ΣQが算出される。次いでステップ102では積算量ΣQが設定量QXを越えたか否かが判別され、ΣQ>QXになったときにはステップ103に進んで付与すべきエネルギ量が算出される。次いでステップ104ではエネルギを付与する処理が行われ、次いでステップ105ではΣQがクリアされる。
即ち、図9に示されるように、排気ガス浄化用触媒20上に保持されている酸素イオンO-と一酸化窒素NOの合計積算量ΣQが設定量QXを越えると燃焼室5内又は排気ガス中に還元剤が供給されて燃焼室5内又は排気ガスの空燃比A/Fがスパイク状にリッチにされ、それによって排気ガス浄化用触媒20に保持されている酸素イオンO-がパージされる。
図13を参照すると、まず初めにステップ200において排気ガス浄化用触媒20の温度TCが基準温度Tsよりも高いか否かが判別される。TC>Tsのときにはステップ201に進んで排気ガス浄化用触媒20に保持されている酸素のパージ作用が行われる。即ちステップ201では図6に示すマップから一酸化窒素量Q(NO)が算出される。次いでステップ203ではΣQにQ(NO)を加算することによって積算量ΣQが算出される。次いでステップ204では積算量ΣQが設定量QXを越えたか否かが判別され、ΣQ>QXになったときにはステップ205に進んで供給すべき還元剤量が算出される。次いでステップ206では還元剤を供給することにより空燃比をリッチにする処理が行われ、次いでステップ207ではΣQがクリアされる。
図16を参照すると、まず初めにステップ220において排気ガス浄化用触媒20の温度TCが基準温度Tsよりも高いか否かが判別される。TC>Tsのときにはステップ221に進んで前回の処理サイクルから今回の処理サイクルまでの時間ΔtをΣtに加算し、それによって経過時間Σtが算出される。次いでステップ222では図13から目標とすべき経過時間tXが算出される。次いでステップ223では経過時間Σtが目標経過時間tXを越えたか否かが判別され、Σt>tXになったときにはステップ224に進んで供給すべき還元剤量が算出される。次いでステップ225では還元剤を供給することにより空燃比をリッチにする処理が行われ、次いでステップ226ではΣtがクリアされる。
一方、ステップ220においてTC≦Tsであると判断されたときにはステップ208に進んで図14に示すNO還元処理が実行される。
図18を参照すると、まず初めにステップ230において排気ガス浄化用触媒20から流出した排気ガス中のNOx濃度DeがNOx濃度センサ44により検出される。次いでステップ231ではNOx濃度センサ44により検出されたNOx濃度Deが許容値DXよりも大きくなったか否かが判別される。De≦DXのときには処理サイクルを完了する。これに対してDe>DXになるとステップ232に進んで排気ガス浄化用触媒20の温度TCが基準温度Tsよりも高いか否かが判別される。TC>Tsのときにはステップ233に進んで供給すべき還元剤量が算出される。次いでステップ234では還元剤を供給することにより空燃比をリッチにする処理が行われる。このとき供給される還元剤の量は当量比=1よりも少ない。
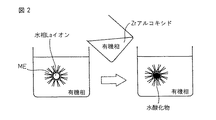
容積3Lのビーカーに界面活性剤溶液をつくり、これに硝酸ランタン0.03モルを蒸留水140部に溶解をした水溶液を滴下して攪拌し、マイクロエマルジョン液を調製した。次に、ジルコニウムブトキシド0.12モルをシクロヘキサン200部に溶解した溶液を滴下し、ジルコニウムブトキシドの加水分解を行った。直ちに白色の曇りが生じた。その後、沈殿の凝集を制御するために、アンモニア水でpHを8.5に調整した。その後、1時間攪拌を続け、生成物の熟成を行った。母液を濾別し、得られた沈殿をエタノールで3回洗浄し、80℃で一晩乾燥後、大気中600℃で2時間焼成して、ランタンとジルコニウムを含む複合酸化物(ランタンジルコニア)を得た。この複合酸化物のLa/Zrモル比は1/4であった。
次に、実施例1において製造したランタンジルコニアについて、常法によってモノリス基材にコートし、白金を1wt%担持させ、さらにアルカリ金属としてセシウムをランタンと同モル数担持させて、本発明の排気ガス浄化用触媒を得た。また、比較として、共沈法及びアルコキシド法により得られたランタンジルコニアを用い、同様にして白金及びセシウムを担持させた。
6…燃料噴射弁
7…点火栓
9…吸気ポート
11…排気ポート
20…排気ガス浄化用触媒
Claims (9)
- 結晶性のジルコニウム複合酸化物にアルカリ金属と貴金属を担持させた排気ガス浄化用触媒において、前記ジルコニウム複合酸化物が、3価の希土類金属より選ばれる少なくとも1種の元素によってジルコニウムの一部が置換されており、この元素置換による結晶格子の伸びが略理論値となっていることを特徴とする排気ガス浄化触媒。
- 前記3価の希土類金属より選ばれる少なくとも1種の元素が、ジルコニウム複合酸化物中の全金属元素の全モル数を基準として5〜50モル%存在する、請求項1記載の排気ガス浄化用触媒。
- ジルコニウムの一部がランタンにより置換されている、請求項1記載の排気ガス浄化用触媒。
- ジルコニウム複合酸化物に担持される前記アルカリ金属がセシウムである、請求項1記載の排気ガス浄化用触媒。
- ジルコニウム複合酸化物に担持される前記貴金属が白金である、請求項1記載の排気ガス浄化用触媒。
- 結晶性のジルコニウム複合酸化物にアルカリ金属と貴金属を担持させた排気ガス浄化用触媒の製造方法において、加水分解してジルコニウムの水酸化物を生成する有機化合物を溶解した有機相と、3価の希土類金属より選ばれる第2の元素をイオンとして含む水相とを接触させることにより、それらの界面におけるジルコニウム有機化合物の加水分解反応によりジルコニウムの水酸化物を生成させる過程でこの生成物中に第2の元素を取り込み、得られる複合水酸化物を焼成してジルコニウム及び第2の元素の複合酸化物を得、さらにアルカリ金属と貴金属を担持させることを特徴とする排気ガス浄化用触媒の製造方法。
- 前記加水分解してジルコニウムの水酸化物を生成する有機化合物が、ジルコニウムアルコキシド、アセチルアセトンジルコニウム錯体から選ばれる1種である、請求項6記載の排気ガス浄化用触媒の製造方法。
- 前記加水分解してジルコニウムの水酸化物を生成する有機化合物が、ジルコニウムブトキシドである、請求項6記載の排気ガス浄化用触媒の製造方法。
- 前記第2の元素がランタンである、請求項6記載の排気ガス浄化用触媒の製造方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003364852A JP4120559B2 (ja) | 2003-10-24 | 2003-10-24 | 排気ガス浄化用触媒 |
| EP04792725A EP1681096B1 (en) | 2003-10-24 | 2004-10-14 | Catalyst for exhaust gas cleaning |
| CN200480034776A CN100586559C (zh) | 2003-10-24 | 2004-10-14 | 废气净化用催化剂 |
| US10/576,025 US20070066479A1 (en) | 2003-10-24 | 2004-10-14 | Exhaust gas purifying catalyst |
| PCT/JP2004/015575 WO2005039759A1 (ja) | 2003-10-24 | 2004-10-14 | 排気ガス浄化用触媒 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003364852A JP4120559B2 (ja) | 2003-10-24 | 2003-10-24 | 排気ガス浄化用触媒 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2005125254A JP2005125254A (ja) | 2005-05-19 |
| JP2005125254A5 JP2005125254A5 (ja) | 2006-08-31 |
| JP4120559B2 true JP4120559B2 (ja) | 2008-07-16 |
Family
ID=34510134
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003364852A Expired - Fee Related JP4120559B2 (ja) | 2003-10-24 | 2003-10-24 | 排気ガス浄化用触媒 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20070066479A1 (ja) |
| EP (1) | EP1681096B1 (ja) |
| JP (1) | JP4120559B2 (ja) |
| CN (1) | CN100586559C (ja) |
| WO (1) | WO2005039759A1 (ja) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4325648B2 (ja) * | 2005-10-24 | 2009-09-02 | トヨタ自動車株式会社 | 触媒担体及び排ガス浄化用触媒 |
| JP4881758B2 (ja) | 2006-04-28 | 2012-02-22 | 日産自動車株式会社 | 排気ガス浄化用触媒及びその製造方法 |
| EP1941945B1 (en) | 2007-01-05 | 2012-01-18 | Kabushiki Kaisha Toyota Chuo Kenkyusho | Catalyst for removing particulate matter and method using the same for removing particulate matter |
| US20090264283A1 (en) * | 2008-04-16 | 2009-10-22 | Basf Catalysts Llc | Stabilized Iridium and Ruthenium Catalysts |
| FR2939695B1 (fr) * | 2008-12-17 | 2011-12-30 | Saint Gobain Ct Recherches | Structure de purification incorporant un systeme de catalyse supporte par une zircone a l'etat reduit. |
| US8003567B2 (en) * | 2009-08-17 | 2011-08-23 | Honda Motor Co., Ltd. | Nanocomposite support materials |
| KR102454125B1 (ko) * | 2015-03-20 | 2022-10-14 | 토프쉐 에이/에스 | 촉매화된 세라믹 캔들 필터 및 공정 오프가스 또는 배기가스의 정화 방법 |
| WO2018035434A1 (en) * | 2016-08-19 | 2018-02-22 | Kohler Co. | System and method for low co emission engine |
Family Cites Families (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02175602A (ja) | 1988-12-28 | 1990-07-06 | Ricoh Co Ltd | 超微粒子状金属酸化物組成物の製法及びそれによって得られた超微粒子状酸化ジルコニウム組成物 |
| US5747410A (en) * | 1992-07-03 | 1998-05-05 | Kabushiki Kaisha Riken | Exhaust gas cleaner and method of cleaning exhaust gas |
| JPH06178937A (ja) * | 1992-10-15 | 1994-06-28 | Riken Corp | 窒素酸化物除去触媒及び除去方法 |
| JP4098835B2 (ja) * | 1993-12-07 | 2008-06-11 | トヨタ自動車株式会社 | 排気ガス浄化用触媒 |
| JPH08281106A (ja) * | 1995-04-11 | 1996-10-29 | Nissan Motor Co Ltd | 排気ガス浄化用触媒及びその製造方法 |
| JPH0924274A (ja) * | 1995-05-09 | 1997-01-28 | Hitachi Ltd | 排ガス浄化触媒及び排ガス浄化システム |
| JP3498453B2 (ja) * | 1995-11-27 | 2004-02-16 | 日産自動車株式会社 | 排気ガス浄化用触媒及びその製造方法 |
| JP4053623B2 (ja) * | 1996-12-27 | 2008-02-27 | 阿南化成株式会社 | ジルコニウム−セリウム系複合酸化物及びその製造方法 |
| JP3466856B2 (ja) * | 1997-02-05 | 2003-11-17 | トヨタ自動車株式会社 | 排ガス浄化触媒およびその製造方法 |
| JP2001170487A (ja) * | 1999-12-15 | 2001-06-26 | Toyota Central Res & Dev Lab Inc | 排ガス浄化用触媒および排ガス浄化方法 |
| EP1020223A3 (en) * | 1999-01-12 | 2001-09-12 | Kabushiki Kaisha Toyota Chuo Kenkyusho | Porous material and production process thereof, catalyst comprising the porous material and process for purifying exhaust gas |
| EP1172139B1 (en) * | 2000-07-14 | 2006-10-11 | Kabushiki Kaisha Toyota Chuo Kenkyusho | Catalyst for purifying exhaust gas |
| JP3758487B2 (ja) * | 2000-09-08 | 2006-03-22 | トヨタ自動車株式会社 | 吸収還元型nox浄化用触媒 |
| WO2002094716A1 (en) | 2001-05-23 | 2002-11-28 | Svenska Rymdaktiebolaget | Sintering resistant catalyst material and a method for the preparation thereof |
| JP3845274B2 (ja) * | 2001-06-26 | 2006-11-15 | ダイハツ工業株式会社 | 排ガス浄化用触媒 |
| JP4228278B2 (ja) * | 2002-03-19 | 2009-02-25 | トヨタ自動車株式会社 | 排ガス浄化用触媒 |
| JP3758601B2 (ja) * | 2002-05-15 | 2006-03-22 | トヨタ自動車株式会社 | 吸蔵還元型NOx浄化用触媒 |
| JP4812233B2 (ja) * | 2003-02-28 | 2011-11-09 | トヨタ自動車株式会社 | 複合酸化物の製造方法 |
| DE602004007187T3 (de) * | 2003-05-21 | 2017-09-28 | Toyota Jidosha Kabushiki Kaisha | Verfahren zur Herstellung eines porösen Mischoxids |
-
2003
- 2003-10-24 JP JP2003364852A patent/JP4120559B2/ja not_active Expired - Fee Related
-
2004
- 2004-10-14 CN CN200480034776A patent/CN100586559C/zh not_active Expired - Fee Related
- 2004-10-14 WO PCT/JP2004/015575 patent/WO2005039759A1/ja active Search and Examination
- 2004-10-14 US US10/576,025 patent/US20070066479A1/en not_active Abandoned
- 2004-10-14 EP EP04792725A patent/EP1681096B1/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| EP1681096B1 (en) | 2012-11-21 |
| EP1681096A4 (en) | 2010-09-15 |
| US20070066479A1 (en) | 2007-03-22 |
| CN1886194A (zh) | 2006-12-27 |
| WO2005039759A1 (ja) | 2005-05-06 |
| JP2005125254A (ja) | 2005-05-19 |
| EP1681096A1 (en) | 2006-07-19 |
| CN100586559C (zh) | 2010-02-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8752367B2 (en) | Exhaust system for lean burn IC engine including particulate filter and NOx absorbent | |
| RU2480592C1 (ru) | Система очистки выхлопных газов двигателя внутреннего сгорания | |
| JP5959147B2 (ja) | 内燃機関の排気ガスを清浄化する方法 | |
| US8413426B2 (en) | Method of exhaust cleaning for internal combustion engine and exhaust cleaner | |
| JP5919286B2 (ja) | NOx貯蔵成分 | |
| JP2006242020A (ja) | 排気浄化装置 | |
| WO2000035564A1 (fr) | Systeme de regulation de gaz d'echappement pour moteurs a combustion interne, procede de regulation de gaz d'echappement et catalyseur de regulation de gaz d'echappement | |
| JP4120559B2 (ja) | 排気ガス浄化用触媒 | |
| JP3912294B2 (ja) | 内燃機関の排気浄化方法および排気浄化装置 | |
| JP2006291847A (ja) | ディーゼル排ガス浄化装置及びディーゼル排ガス浄化用触媒 | |
| JP2003193822A (ja) | 内燃機関の排気浄化装置 | |
| JP4196573B2 (ja) | 内燃機関の排気浄化方法及び浄化装置 | |
| JP2005169357A (ja) | 排ガス浄化システム | |
| JP2011136278A (ja) | 排ガス処理触媒およびそれを用いた排ガス浄化方法ならびに排ガス浄化装置 | |
| JP2007084391A (ja) | 自動車用排ガス浄化装置及び水素製造触媒 | |
| JP4374797B2 (ja) | 内燃機関の排気浄化方法 | |
| JP2007278212A (ja) | 排ガス浄化装置及び排ガス浄化方法 | |
| JP2010106799A (ja) | 排気浄化システム | |
| JP2003278536A (ja) | 内燃機関の排気浄化装置 | |
| JP2005330887A (ja) | 排ガス浄化装置及び排ガス浄化方法 | |
| JP2001271629A (ja) | 排気ガス浄化方法および排気ガス浄化装置 | |
| JP2004092433A (ja) | 微粒子酸化除去方法および内燃機関の排気浄化装置 | |
| JP2014001737A (ja) | 排ガス浄化方法および排ガス浄化装置 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20060713 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20060713 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20080401 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20080414 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110509 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110509 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110509 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120509 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130509 Year of fee payment: 5 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140509 Year of fee payment: 6 |
|
| LAPS | Cancellation because of no payment of annual fees |