JP4325648B2 - 触媒担体及び排ガス浄化用触媒 - Google Patents
触媒担体及び排ガス浄化用触媒 Download PDFInfo
- Publication number
- JP4325648B2 JP4325648B2 JP2006194548A JP2006194548A JP4325648B2 JP 4325648 B2 JP4325648 B2 JP 4325648B2 JP 2006194548 A JP2006194548 A JP 2006194548A JP 2006194548 A JP2006194548 A JP 2006194548A JP 4325648 B2 JP4325648 B2 JP 4325648B2
- Authority
- JP
- Japan
- Prior art keywords
- catalyst
- oxide
- electron
- particles
- noble metal
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 239000003054 catalyst Substances 0.000 title claims description 176
- 238000000746 purification Methods 0.000 title claims description 24
- 239000002131 composite material Substances 0.000 claims description 86
- 239000002245 particle Substances 0.000 claims description 79
- 229910000510 noble metal Inorganic materials 0.000 claims description 70
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 claims description 67
- 229910052746 lanthanum Inorganic materials 0.000 claims description 57
- FZLIPJUXYLNCLC-UHFFFAOYSA-N lanthanum atom Chemical compound [La] FZLIPJUXYLNCLC-UHFFFAOYSA-N 0.000 claims description 57
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 44
- 229910052779 Neodymium Inorganic materials 0.000 claims description 41
- QEFYFXOXNSNQGX-UHFFFAOYSA-N neodymium atom Chemical compound [Nd] QEFYFXOXNSNQGX-UHFFFAOYSA-N 0.000 claims description 41
- 229910052697 platinum Inorganic materials 0.000 claims description 32
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 claims description 30
- 229910052710 silicon Inorganic materials 0.000 claims description 30
- 239000010703 silicon Substances 0.000 claims description 30
- 229910052763 palladium Inorganic materials 0.000 claims description 21
- 229910052727 yttrium Inorganic materials 0.000 claims description 21
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical compound [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 claims description 21
- 238000006479 redox reaction Methods 0.000 claims description 5
- 230000003647 oxidation Effects 0.000 claims description 3
- 238000007254 oxidation reaction Methods 0.000 claims description 3
- 230000003197 catalytic effect Effects 0.000 claims description 2
- 238000005245 sintering Methods 0.000 description 45
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 38
- 239000000377 silicon dioxide Substances 0.000 description 34
- 239000007789 gas Substances 0.000 description 30
- 239000000243 solution Substances 0.000 description 29
- MCMNRKCIXSYSNV-UHFFFAOYSA-N Zirconium dioxide Chemical compound O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 description 24
- 239000002923 metal particle Substances 0.000 description 23
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 21
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 21
- 239000004094 surface-active agent Substances 0.000 description 20
- 239000004530 micro-emulsion Substances 0.000 description 17
- 239000008346 aqueous phase Substances 0.000 description 16
- 239000011777 magnesium Substances 0.000 description 15
- 229910044991 metal oxide Inorganic materials 0.000 description 15
- 239000012702 metal oxide precursor Substances 0.000 description 15
- 150000004706 metal oxides Chemical class 0.000 description 15
- -1 silicon (Si) Chemical class 0.000 description 15
- 150000003839 salts Chemical class 0.000 description 14
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 13
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 13
- 229910052749 magnesium Inorganic materials 0.000 description 13
- 238000000034 method Methods 0.000 description 13
- 239000000203 mixture Substances 0.000 description 13
- 239000010936 titanium Substances 0.000 description 13
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 12
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 12
- 235000011114 ammonium hydroxide Nutrition 0.000 description 12
- 239000012298 atmosphere Substances 0.000 description 12
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 12
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 description 11
- 150000004703 alkoxides Chemical class 0.000 description 11
- 239000012071 phase Substances 0.000 description 11
- 239000010948 rhodium Substances 0.000 description 11
- 230000001629 suppression Effects 0.000 description 11
- 229910052726 zirconium Inorganic materials 0.000 description 11
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 10
- 230000000694 effects Effects 0.000 description 10
- 229910052751 metal Inorganic materials 0.000 description 10
- 229910052760 oxygen Inorganic materials 0.000 description 10
- 239000001301 oxygen Substances 0.000 description 10
- 229910052703 rhodium Inorganic materials 0.000 description 10
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 10
- 239000011163 secondary particle Substances 0.000 description 10
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 9
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 9
- 239000006185 dispersion Substances 0.000 description 9
- 239000007788 liquid Substances 0.000 description 9
- 238000002156 mixing Methods 0.000 description 9
- 239000011164 primary particle Substances 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- 229910052719 titanium Inorganic materials 0.000 description 9
- 239000012153 distilled water Substances 0.000 description 8
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 7
- BOTDANWDWHJENH-UHFFFAOYSA-N Tetraethyl orthosilicate Chemical compound CCO[Si](OCC)(OCC)OCC BOTDANWDWHJENH-UHFFFAOYSA-N 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- CETPSERCERDGAM-UHFFFAOYSA-N ceric oxide Chemical compound O=[Ce]=O CETPSERCERDGAM-UHFFFAOYSA-N 0.000 description 7
- 229910000422 cerium(IV) oxide Inorganic materials 0.000 description 7
- 239000002184 metal Substances 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- 239000007864 aqueous solution Substances 0.000 description 6
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 6
- 125000000217 alkyl group Chemical group 0.000 description 5
- 229910052782 aluminium Inorganic materials 0.000 description 5
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 5
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 5
- 238000001354 calcination Methods 0.000 description 5
- 238000007796 conventional method Methods 0.000 description 5
- 238000001035 drying Methods 0.000 description 5
- 229930195733 hydrocarbon Natural products 0.000 description 5
- 150000002430 hydrocarbons Chemical class 0.000 description 5
- 230000002209 hydrophobic effect Effects 0.000 description 5
- 238000005259 measurement Methods 0.000 description 5
- 238000000593 microemulsion method Methods 0.000 description 5
- 230000007935 neutral effect Effects 0.000 description 5
- 230000001590 oxidative effect Effects 0.000 description 5
- 238000011156 evaluation Methods 0.000 description 4
- 230000003993 interaction Effects 0.000 description 4
- 239000003960 organic solvent Substances 0.000 description 4
- 239000002243 precursor Substances 0.000 description 4
- 229910052761 rare earth metal Inorganic materials 0.000 description 4
- FDCJDKXCCYFOCV-UHFFFAOYSA-N 1-hexadecoxyhexadecane Chemical compound CCCCCCCCCCCCCCCCOCCCCCCCCCCCCCCCC FDCJDKXCCYFOCV-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 229910018072 Al 2 O 3 Inorganic materials 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 3
- 150000001342 alkaline earth metals Chemical class 0.000 description 3
- 230000000052 comparative effect Effects 0.000 description 3
- 238000010586 diagram Methods 0.000 description 3
- 239000000446 fuel Substances 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- FYDKNKUEBJQCCN-UHFFFAOYSA-N lanthanum(3+);trinitrate Chemical compound [La+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O FYDKNKUEBJQCCN-UHFFFAOYSA-N 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 238000002844 melting Methods 0.000 description 3
- 230000008018 melting Effects 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 238000006722 reduction reaction Methods 0.000 description 3
- 230000000087 stabilizing effect Effects 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- PAWQVTBBRAZDMG-UHFFFAOYSA-N 2-(3-bromo-2-fluorophenyl)acetic acid Chemical compound OC(=O)CC1=CC=CC(Br)=C1F PAWQVTBBRAZDMG-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- 229910017625 MgSiO Inorganic materials 0.000 description 2
- 229910002651 NO3 Inorganic materials 0.000 description 2
- 238000002441 X-ray diffraction Methods 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000003945 anionic surfactant Substances 0.000 description 2
- QVQLCTNNEUAWMS-UHFFFAOYSA-N barium oxide Chemical compound [Ba]=O QVQLCTNNEUAWMS-UHFFFAOYSA-N 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- ZRHOFLXFAAEXEE-UHFFFAOYSA-J butanoate;titanium(4+) Chemical compound [Ti+4].CCCC([O-])=O.CCCC([O-])=O.CCCC([O-])=O.CCCC([O-])=O ZRHOFLXFAAEXEE-UHFFFAOYSA-J 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 239000003093 cationic surfactant Substances 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 238000010304 firing Methods 0.000 description 2
- ZSIAUFGUXNUGDI-UHFFFAOYSA-N hexan-1-ol Chemical compound CCCCCCO ZSIAUFGUXNUGDI-UHFFFAOYSA-N 0.000 description 2
- 230000003301 hydrolyzing effect Effects 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- YIXJRHPUWRPCBB-UHFFFAOYSA-N magnesium nitrate Chemical compound [Mg+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O YIXJRHPUWRPCBB-UHFFFAOYSA-N 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- 239000000693 micelle Substances 0.000 description 2
- 150000002823 nitrates Chemical class 0.000 description 2
- 239000002736 nonionic surfactant Substances 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 239000010970 precious metal Substances 0.000 description 2
- 150000002910 rare earth metals Chemical class 0.000 description 2
- VSZWPYCFIRKVQL-UHFFFAOYSA-N selanylidenegallium;selenium Chemical compound [Se].[Se]=[Ga].[Se]=[Ga] VSZWPYCFIRKVQL-UHFFFAOYSA-N 0.000 description 2
- 239000001763 2-hydroxyethyl(trimethyl)azanium Substances 0.000 description 1
- NGDQQLAVJWUYSF-UHFFFAOYSA-N 4-methyl-2-phenyl-1,3-thiazole-5-sulfonyl chloride Chemical compound S1C(S(Cl)(=O)=O)=C(C)N=C1C1=CC=CC=C1 NGDQQLAVJWUYSF-UHFFFAOYSA-N 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- 235000019743 Choline chloride Nutrition 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 229910021193 La 2 O 3 Inorganic materials 0.000 description 1
- 229910004298 SiO 2 Inorganic materials 0.000 description 1
- 229910010413 TiO 2 Inorganic materials 0.000 description 1
- 229910001361 White metal Inorganic materials 0.000 description 1
- GEIAQOFPUVMAGM-UHFFFAOYSA-N ZrO Inorganic materials [Zr]=O GEIAQOFPUVMAGM-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000004931 aggregating effect Effects 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 125000005210 alkyl ammonium group Chemical group 0.000 description 1
- 150000005215 alkyl ethers Chemical class 0.000 description 1
- 229910001038 basic metal oxide Inorganic materials 0.000 description 1
- 239000003637 basic solution Substances 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- WOWHHFRSBJGXCM-UHFFFAOYSA-M cetyltrimethylammonium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCC[N+](C)(C)C WOWHHFRSBJGXCM-UHFFFAOYSA-M 0.000 description 1
- RLGQACBPNDBWTB-UHFFFAOYSA-N cetyltrimethylammonium ion Chemical compound CCCCCCCCCCCCCCCC[N+](C)(C)C RLGQACBPNDBWTB-UHFFFAOYSA-N 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 150000003841 chloride salts Chemical class 0.000 description 1
- SGMZJAMFUVOLNK-UHFFFAOYSA-M choline chloride Chemical compound [Cl-].C[N+](C)(C)CCO SGMZJAMFUVOLNK-UHFFFAOYSA-M 0.000 description 1
- 229960003178 choline chloride Drugs 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 238000007598 dipping method Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000005470 impregnation Methods 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 150000003893 lactate salts Chemical class 0.000 description 1
- MRELNEQAGSRDBK-UHFFFAOYSA-N lanthanum(3+);oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[La+3].[La+3] MRELNEQAGSRDBK-UHFFFAOYSA-N 0.000 description 1
- 238000004768 lowest unoccupied molecular orbital Methods 0.000 description 1
- CFYGEIAZMVFFDE-UHFFFAOYSA-N neodymium(3+);trinitrate Chemical compound [Nd+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O CFYGEIAZMVFFDE-UHFFFAOYSA-N 0.000 description 1
- UJVRJBAUJYZFIX-UHFFFAOYSA-N nitric acid;oxozirconium Chemical compound [Zr]=O.O[N+]([O-])=O.O[N+]([O-])=O UJVRJBAUJYZFIX-UHFFFAOYSA-N 0.000 description 1
- GSGDTSDELPUTKU-UHFFFAOYSA-N nonoxybenzene Chemical compound CCCCCCCCCOC1=CC=CC=C1 GSGDTSDELPUTKU-UHFFFAOYSA-N 0.000 description 1
- ZPIRTVJRHUMMOI-UHFFFAOYSA-N octoxybenzene Chemical compound CCCCCCCCOC1=CC=CC=C1 ZPIRTVJRHUMMOI-UHFFFAOYSA-N 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- DGMKFQYCZXERLX-UHFFFAOYSA-N proglumide Chemical compound CCCN(CCC)C(=O)C(CCC(O)=O)NC(=O)C1=CC=CC=C1 DGMKFQYCZXERLX-UHFFFAOYSA-N 0.000 description 1
- 229960003857 proglumide Drugs 0.000 description 1
- 229910001404 rare earth metal oxide Inorganic materials 0.000 description 1
- 230000005070 ripening Effects 0.000 description 1
- 238000007613 slurry method Methods 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 239000006104 solid solution Substances 0.000 description 1
- 229910002076 stabilized zirconia Inorganic materials 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 239000010969 white metal Substances 0.000 description 1
Images










Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D53/00—Separation of gases or vapours; Recovering vapours of volatile solvents from gases; Chemical or biological purification of waste gases, e.g. engine exhaust gases, smoke, fumes, flue gases, aerosols
- B01D53/34—Chemical or biological purification of waste gases
- B01D53/92—Chemical or biological purification of waste gases of engine exhaust gases
- B01D53/94—Chemical or biological purification of waste gases of engine exhaust gases by catalytic processes
- B01D53/9445—Simultaneously removing carbon monoxide, hydrocarbons or nitrogen oxides making use of three-way catalysts [TWC] or four-way-catalysts [FWC]
- B01D53/945—Simultaneously removing carbon monoxide, hydrocarbons or nitrogen oxides making use of three-way catalysts [TWC] or four-way-catalysts [FWC] characterised by a specific catalyst
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/002—Mixed oxides other than spinels, e.g. perovskite
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/10—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of rare earths
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/56—Platinum group metals
- B01J23/63—Platinum group metals with rare earths or actinides
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/50—Catalysts, in general, characterised by their form or physical properties characterised by their shape or configuration
- B01J35/56—Foraminous structures having flow-through passages or channels, e.g. grids or three-dimensional monoliths
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/024—Multiple impregnation or coating
- B01J37/0242—Coating followed by impregnation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/03—Precipitation; Co-precipitation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2255/00—Catalysts
- B01D2255/10—Noble metals or compounds thereof
- B01D2255/102—Platinum group metals
- B01D2255/1021—Platinum
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2255/00—Catalysts
- B01D2255/10—Noble metals or compounds thereof
- B01D2255/102—Platinum group metals
- B01D2255/1023—Palladium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2255/00—Catalysts
- B01D2255/20—Metals or compounds thereof
- B01D2255/206—Rare earth metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/30—Catalysts, in general, characterised by their form or physical properties characterised by their physical properties
- B01J35/391—Physical properties of the active metal ingredient
- B01J35/393—Metal or metal oxide crystallite size
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/60—Catalysts, in general, characterised by their form or physical properties characterised by their surface properties or porosity
- B01J35/61—Surface area
- B01J35/613—10-100 m2/g
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02T—CLIMATE CHANGE MITIGATION TECHNOLOGIES RELATED TO TRANSPORTATION
- Y02T10/00—Road transport of goods or passengers
- Y02T10/10—Internal combustion engine [ICE] based vehicles
- Y02T10/12—Improving ICE efficiencies
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Combustion & Propulsion (AREA)
- Biomedical Technology (AREA)
- Environmental & Geological Engineering (AREA)
- Analytical Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Catalysts (AREA)
- Exhaust Gas Treatment By Means Of Catalyst (AREA)
- Exhaust Gas After Treatment (AREA)
Description
本発明の触媒担体を製造する本発明の方法は、疎水性溶媒相中に水性相が分散している分散液を提供すること;分散液中に分散している水性相中において、電子受容性元素の塩及び他の元素の塩を加水分解して、金属酸化物前駆体を析出させ、この金属酸化物前駆体を凝集させること;並びに凝集させた金属酸化物前駆体を乾燥及び焼成することを含む。
{1.90(ケイ素の電気陰性度)×1+3.44(酸素の電気陰性度)×2}/3
≒2.93

電子受容性元素は、他の元素と共に複合酸化物を構成している。この電子受容性元素は、他の元素と組み合わせて複合酸化物としたときに、貴金属触媒粒子が接近若しくは接触することによりその貴金属元素から電子を受容する電子受容性があり且つ酸化還元反応で原子価の変化がない元素として選択できる。
電子受容性元素は単独で用いずに、他の元素、特に他の金属元素との複合酸化物として用いられる。これは、耐熱性を向上させることに加えて、電子受容性元素の塩基性を弱めて、複合酸化物が全体として中性若しくは中性に近くなるようにするためである。従って、当該他の元素は、従来、排ガス用触媒の担体若しくは基材として用いられている金属元素で良く、具体的には、ケイ素(Si)、アルミニウム(Al)、ジルコニウム(Zr)、チタン(Ti)及びそれらの組み合わせからなる群より選択できる。
本発明の触媒担体を構成している複合酸化物は、電気陰性度が、好ましくは2.55〜2.80、より好ましくは2.60〜2.73である。この電気陰性度によれば、適度な酸塩基性によって、触媒活性を良好な状態に維持することができる。
ネオジム、ランタン等の元素をジルコニウム等の他の元素の酸化物に添加する場合、ネオジム等の元素と他の元素との合計に対する電子受容性元素のモル比が0.01〜0.3程度のときには、一般に、ネオジム等の元素によって他の元素の酸化物が安定化され、触媒担体としての耐熱性が向上することが知られている。
本発明の触媒担体では、電子受容性元素と他の金属との複合酸化物を、粒子状に形成して用いることができる。
上記の担体に貴金属触媒を担持することにより、本発明の排ガス浄化用触媒が得られる。この貴金属触媒は、具体的には白金(Pt)、ロジウム(ロジウム)、パラジウム(Pd)及びそれらの組み合わせからなる群より選択される貴金属を挙げることができ、特に排ガス浄化触媒としての使用の間に比較的シンタリングする傾向が大きい白金粒子、パラジウム及びそれらの組合せからなる群より選択される貴金属を挙げることができる。貴金属の担持のためには、従来知られている含浸法(スラリー法)、浸漬法等を用いることができる。
本発明の触媒担体を製造する本発明の方法は、疎水性溶媒相中に水性相が分散している分散液を提供すること;分散液中に分散している水性相中において、電子受容性元素の塩及び他の元素の塩を加水分解して、金属酸化物前駆体を析出させ、この金属酸化物前駆体を凝集させること;並びに凝集させた金属酸化物前駆体を乾燥及び焼成することを含む。
下記に様にしてマイクロエマルション法によって、La10Si6O27の組成の酸化ランタン−シリカ複合酸化物触媒担体を合成した。
下記に様にしてマイクロエマルション法によって、NdZrO3.5の組成の酸化ネオジウム−ジルコニア複合酸化物触媒担体を合成した。
下記に様にしてマイクロエマルション法によって、Y10Si6O27の組成の酸化イットリウム−シリカ複合酸化物触媒担体20gを合成した。
下記に様にしてマイクロエマルション法によって、MgSiO3の組成の酸化マグネシウム−シリカ複合酸化物触媒担体30gを合成した。
下記に様にしてマイクロエマルション法によって、LaTiO3.5の組成のパイロクロア構造酸化ランタン−チタニア複合酸化物触媒担体30gを合成した。
上記と同様にしてマイクロエマルション法によって、本発明の酸化ネオジウム−シリカ複合酸化物(Nd10Si6O27)触媒担体、酸化ランタン−アルミナ複合酸化物(LaAlO3)触媒担体、酸化ネオジウム−アルミナ複合酸化物(NdAlO3)触媒担体、酸化ランタン−ジルコニア複合酸化物(LaZrO3.5)触媒担体、酸化マグネシウム−アルミナ複合酸化物(MgAlO2.5)触媒担体、酸化イットリウム−ジルコニア複合酸化物(YZrO3.5)触媒担体、酸化ネオジム−チタニア複合酸化物(NdTiO3.5)触媒担体、及び酸化イットリウム−チタニア複合酸化物(YTiO3.5)触媒担体を合成した。
従来技術の触媒担体として、La2O3、Al2O3、ZrO2、TiO2、SiO2を得た。
本発明及び従来技術の触媒担体について電気陰性度を求めた。またこれらの触媒担体に1wt%の白金粒子を定法に従って担持し、空気中において800℃で2時間にわたって焼成した後の白金粒子の粒径を測定した。これらの測定結果を、下記の表3に示す。
1/Spr n=1/So n+kt …(1)
(Sprは貴金属粒子の表面積、Soは初期の貴金属粒子表面積、k及びnは任意の定数、tは時間である)。
1/Spr 2=1/So 2+kt …(2)
1/Spr∝d …(3)
∴1/Spr 2∝d2 …(4)
(dは貴金属粒子の粒子径、Sprは貴金属粒子の表面積)。
t=tr/Ssup 1/2 …(5)
t∝1/Ssup 1/2 …(6)
d2=k’/Ssup 1/2+Q …(7)
(dは貴金属粒子の粒子径、Ssupは担体表面積、k’及びQは定数)
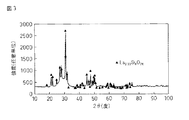
2 酸化ランタン−シリカ複合酸化物
Claims (7)
- 貴金属触媒粒子の貴金属元素が接近若しくは接触することによりその貴金属元素から電子を受容する電子受容性があり且つ酸化還元反応で原子価の変化がない電子受容性元素、及び他の元素、の複合酸化物から構成されており、
前記電子受容性元素が、ランタン、ネオジム、イットリウム及びそれらの組み合わせからなる群より選択され、
前記他の元素が、ケイ素であり、且つ
前記電子受容性元素と前記他の元素との合計に対する前記電子受容性元素のモル比が、0.5〜0.7である、
触媒担体。 - 前記電子受容性元素がランタンであり、前記他の元素がケイ素であり、且つランタンとケイ素との合計に対するランタンのモル比(La/(La+Si))が、0.5〜0.7である、請求項1に記載の触媒担体。
- 前記電子受容性元素がネオジムであり、前記他の元素がケイ素であり、且つネオジムとケイ素との合計に対するネオジムのモル比(Nd/(Nd+Si))が、0.5〜0.7である、請求項1に記載の触媒担体。
- 前記電子受容性元素がイットリウムであり、前記他の元素がケイ素であり、且つイットリウムとケイ素との合計に対するイットリウムのモル比(Y/(Y+Si))が、0.5〜0.7である、請求項1に記載の触媒担体。
- 前記複合酸化物の電気陰性度が、2.55〜2.80である、請求項1〜4のいずれかに記載の触媒担体。
- 請求項1〜5のいずれかに記載の触媒担体に、貴金属触媒粒子が担持されてなる、排ガス浄化用触媒。
- 前記貴金属触媒粒子が、白金、パラジウム及びそれらの組み合わせからなる群より選択される、請求項6に記載の排ガス浄化用触媒。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006194548A JP4325648B2 (ja) | 2005-10-24 | 2006-07-14 | 触媒担体及び排ガス浄化用触媒 |
| EP06832403A EP1946836B1 (en) | 2005-10-24 | 2006-10-24 | Catalyst support and catalyst for exhaust-gas purification |
| PCT/JP2006/321589 WO2007049778A1 (ja) | 2005-10-24 | 2006-10-24 | 触媒担体及び排ガス浄化用触媒 |
| CN2006800394261A CN101291731B (zh) | 2005-10-24 | 2006-10-24 | 催化剂载体和排气净化用催化剂 |
| US12/084,050 US7776783B2 (en) | 2005-10-24 | 2006-10-24 | Catalyst carrier and exhaust gas purification catalyst |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005308552 | 2005-10-24 | ||
| JP2006194548A JP4325648B2 (ja) | 2005-10-24 | 2006-07-14 | 触媒担体及び排ガス浄化用触媒 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009080138A Division JP4770959B2 (ja) | 2005-10-24 | 2009-03-27 | 触媒担体及び排ガス浄化用触媒 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2007144393A JP2007144393A (ja) | 2007-06-14 |
| JP2007144393A5 JP2007144393A5 (ja) | 2008-06-26 |
| JP4325648B2 true JP4325648B2 (ja) | 2009-09-02 |
Family
ID=37967884
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2006194548A Expired - Fee Related JP4325648B2 (ja) | 2005-10-24 | 2006-07-14 | 触媒担体及び排ガス浄化用触媒 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US7776783B2 (ja) |
| EP (1) | EP1946836B1 (ja) |
| JP (1) | JP4325648B2 (ja) |
| WO (1) | WO2007049778A1 (ja) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5078062B2 (ja) * | 2005-10-26 | 2012-11-21 | 三井金属鉱業株式会社 | 排ガス浄化用触媒 |
| JP5140987B2 (ja) * | 2006-10-24 | 2013-02-13 | トヨタ自動車株式会社 | 触媒担体及びその製造方法、並びに排ガス浄化触媒 |
| JP5205999B2 (ja) * | 2008-02-07 | 2013-06-05 | トヨタ自動車株式会社 | 排ガス浄化触媒 |
| JP5131053B2 (ja) * | 2008-06-24 | 2013-01-30 | トヨタ自動車株式会社 | 貴金属担持触媒及び触媒装置 |
| US8507403B2 (en) * | 2008-06-27 | 2013-08-13 | Cabot Corporation | Process for producing exhaust treatment catalyst powders, and their use |
| JP5227363B2 (ja) * | 2009-06-08 | 2013-07-03 | 三井金属鉱業株式会社 | 排ガス浄化用触媒 |
| CN104968430B (zh) * | 2013-01-31 | 2018-05-08 | 优美科触媒日本有限公司 | 废气净化用催化剂以及使用该催化剂的废气净化方法 |
| WO2014156676A1 (ja) * | 2013-03-29 | 2014-10-02 | 三井金属鉱業株式会社 | 排気ガス処理用触媒構造体 |
| WO2016117240A1 (ja) * | 2015-01-19 | 2016-07-28 | 三井金属鉱業株式会社 | 排ガス浄化触媒用担体及び排ガス浄化触媒 |
| JP6714989B2 (ja) | 2015-01-19 | 2020-07-01 | 国立大学法人秋田大学 | 排ガス浄化触媒用担体及び排ガス浄化触媒 |
| CN106179396B (zh) * | 2016-07-08 | 2019-02-15 | 宁波钛安新材料科技有限公司 | 一种分解臭氧的复合催化剂及其制备方法 |
| US10857520B2 (en) | 2016-11-11 | 2020-12-08 | N.E. Chemcat Corporation | Exhaust gas-purifying three-way catalyst and method for producing the same, and exhaust gas-purifying catalytic converter |
| JP6769839B2 (ja) * | 2016-11-11 | 2020-10-14 | エヌ・イーケムキャット株式会社 | 排ガス浄化用三元触媒及びその製造方法、並びに排ガス浄化用触媒コンバータ |
| JP6769862B2 (ja) * | 2016-12-26 | 2020-10-14 | エヌ・イーケムキャット株式会社 | 排ガス浄化用三元触媒及びその製造方法、並びに排ガス浄化用触媒コンバータ |
Family Cites Families (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1369745A (en) | 1971-07-20 | 1974-10-09 | Grace W R & Co | Process of converting noxious components in the exhaust gas of and internal combustion engine to less harmful entities |
| US3905918A (en) * | 1972-09-16 | 1975-09-16 | Heraeus Gmbh W C | Catalyst for purifying exhaust gases |
| JPS6135851A (ja) | 1984-07-30 | 1986-02-20 | Hitachi Ltd | 高温で安定な触媒用担体およびその調製方法 |
| JPS63264150A (ja) | 1987-04-20 | 1988-11-01 | Hitachi Ltd | 触媒製造方法 |
| JPH01168343A (ja) * | 1987-12-22 | 1989-07-03 | Toyota Central Res & Dev Lab Inc | 排気ガス用浄化触媒 |
| JPH02175602A (ja) * | 1988-12-28 | 1990-07-06 | Ricoh Co Ltd | 超微粒子状金属酸化物組成物の製法及びそれによって得られた超微粒子状酸化ジルコニウム組成物 |
| JP2930975B2 (ja) | 1989-07-17 | 1999-08-09 | バブコツク日立株式会社 | 燃焼用触媒の製造方法 |
| US5023224A (en) * | 1989-08-31 | 1991-06-11 | Shell Oil Company | Alkoxylation process catalyzed by lanthanum silicates and metasilicates |
| JPH0388800A (ja) * | 1989-08-31 | 1991-04-15 | Tokin Corp | レーザ用酸化物単結晶 |
| JPH04180835A (ja) | 1990-11-13 | 1992-06-29 | Toyota Motor Corp | 排気ガス浄化用触媒の製造方法 |
| JP2628798B2 (ja) | 1991-03-14 | 1997-07-09 | エヌ・イーケムキャット株式会社 | 耐熱性に優れた排気ガス浄化用触媒及びその製造方法 |
| JPH04298235A (ja) | 1991-03-27 | 1992-10-22 | Sekiyu Sangyo Kasseika Center | 窒素酸化物接触還元用触媒 |
| JP3212429B2 (ja) | 1993-11-17 | 2001-09-25 | 三菱重工業株式会社 | 排気ガス処理方法 |
| JP3436427B2 (ja) * | 1994-10-21 | 2003-08-11 | 株式会社豊田中央研究所 | 排ガス浄化用触媒及び排ガス浄化方法 |
| JP3498453B2 (ja) | 1995-11-27 | 2004-02-16 | 日産自動車株式会社 | 排気ガス浄化用触媒及びその製造方法 |
| JPH09313938A (ja) * | 1996-06-03 | 1997-12-09 | Nissan Motor Co Ltd | 排気ガス浄化用触媒 |
| JPH11169728A (ja) | 1997-12-15 | 1999-06-29 | Toho Gas Co Ltd | メタン酸化触媒 |
| JP2001252563A (ja) | 2000-03-10 | 2001-09-18 | Toyota Motor Corp | 排ガス浄化用触媒及び排ガス浄化装置 |
| JP2001314763A (ja) | 2000-05-10 | 2001-11-13 | Johnson Matthey Japan Inc | NOx吸蔵還元型触媒用支持材とそれを用いたNOx吸蔵還元型触媒 |
| JP2002001120A (ja) * | 2000-06-16 | 2002-01-08 | Toyota Central Res & Dev Lab Inc | 排ガス浄化用触媒 |
| DE60031258T2 (de) * | 2000-07-14 | 2007-05-03 | Kabushiki Kaisha Toyota Chuo Kenkyusho | Katalysator zum Reinigen von Abgas |
| JP3858625B2 (ja) | 2000-07-27 | 2006-12-20 | 株式会社豊田中央研究所 | 複合酸化物とその製造方法及び排ガス浄化用触媒とその製造方法 |
| JP4284847B2 (ja) | 2000-08-31 | 2009-06-24 | パナソニック株式会社 | 便座・便蓋電動開閉装置 |
| JP2002079094A (ja) | 2000-09-08 | 2002-03-19 | Toyota Central Res & Dev Lab Inc | 高温NOx吸蔵還元型触媒 |
| JP2002087896A (ja) * | 2000-09-12 | 2002-03-27 | Mitsubishi Heavy Ind Ltd | 自己修復性高耐熱耐酸化性皮膜及び積層体 |
| JP2002233755A (ja) | 2001-02-08 | 2002-08-20 | Toyota Central Res & Dev Lab Inc | 飽和炭化水素酸化用触媒 |
| JP2002346387A (ja) | 2001-05-23 | 2002-12-03 | Isuzu Motors Ltd | 排気ガス浄化触媒 |
| US7371352B2 (en) * | 2001-09-26 | 2008-05-13 | Siemens Power Generation, Inc. | Catalyst element having a thermal barrier coating as the catalyst substrate |
| JP4180835B2 (ja) | 2002-05-07 | 2008-11-12 | 坂本工業株式会社 | 管端の加工装置 |
| US6677064B1 (en) * | 2002-05-29 | 2004-01-13 | Siemens Westinghouse Power Corporation | In-situ formation of multiphase deposited thermal barrier coatings |
| JP4298235B2 (ja) | 2002-07-30 | 2009-07-15 | 古河機械金属株式会社 | 空気二次電池 |
| US20040024071A1 (en) * | 2002-08-01 | 2004-02-05 | Meier Paul F. | Perovskite compositions and method of making and process of using such compositions |
| JP4812233B2 (ja) * | 2003-02-28 | 2011-11-09 | トヨタ自動車株式会社 | 複合酸化物の製造方法 |
| JP3843102B2 (ja) | 2003-08-06 | 2006-11-08 | 本田技研工業株式会社 | 排ガス浄化触媒及びその製造方法、並びに排ガス浄化触媒装置 |
| JP4120559B2 (ja) * | 2003-10-24 | 2008-07-16 | トヨタ自動車株式会社 | 排気ガス浄化用触媒 |
| CN1657139A (zh) | 2004-02-16 | 2005-08-24 | 中国科学院大连化学物理研究所 | 一种氮氧化物存储-还原催化剂制备及存储-还原消除氮氧化物的方法 |
| JP2006035153A (ja) | 2004-07-29 | 2006-02-09 | Honda Motor Co Ltd | 排ガス浄化触媒 |
| JP4157514B2 (ja) | 2004-10-21 | 2008-10-01 | 本田技研工業株式会社 | 排ガス浄化触媒及びそれを備えた車用排ガス浄化装置 |
| JP2006131678A (ja) * | 2004-11-02 | 2006-05-25 | Lintec Corp | 開環重合用固体酸触媒及び開環重合方法 |
| JP2006137651A (ja) | 2004-11-15 | 2006-06-01 | Toyota Central Res & Dev Lab Inc | 複合酸化物及び排ガス浄化用触媒 |
| CN101180125B (zh) * | 2005-03-24 | 2014-09-10 | 里贾纳大学 | 用于生产氢的催化剂 |
| JP4720270B2 (ja) | 2005-04-18 | 2011-07-13 | トヨタ自動車株式会社 | 排ガス浄化用触媒およびその製造方法 |
| JP2007283207A (ja) * | 2006-04-17 | 2007-11-01 | Toyota Central Res & Dev Lab Inc | 排ガス浄化用触媒及びその製造方法 |
-
2006
- 2006-07-14 JP JP2006194548A patent/JP4325648B2/ja not_active Expired - Fee Related
- 2006-10-24 WO PCT/JP2006/321589 patent/WO2007049778A1/ja active Application Filing
- 2006-10-24 US US12/084,050 patent/US7776783B2/en not_active Expired - Fee Related
- 2006-10-24 EP EP06832403A patent/EP1946836B1/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| US7776783B2 (en) | 2010-08-17 |
| WO2007049778A1 (ja) | 2007-05-03 |
| EP1946836A4 (en) | 2010-09-15 |
| EP1946836B1 (en) | 2013-01-16 |
| US20090131249A1 (en) | 2009-05-21 |
| EP1946836A1 (en) | 2008-07-23 |
| JP2007144393A (ja) | 2007-06-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4325648B2 (ja) | 触媒担体及び排ガス浄化用触媒 | |
| JP5147687B2 (ja) | 排ガス浄化触媒及びその製造方法 | |
| JP5194397B2 (ja) | 排ガス浄化触媒及びその製造方法 | |
| RU2423177C1 (ru) | Структура ядро-оболочка, способ ее получения и катализатор очистки выхлопных газов, содержащий структуру ядро-оболочка | |
| JP5076377B2 (ja) | 排ガス浄化触媒 | |
| KR100881300B1 (ko) | 금속 산화물 입자의 제조 공정 및 배기 가스 정화용 촉매 | |
| JP5465842B2 (ja) | コアシェル構造体及び当該コアシェル構造体を含む排ガス浄化用触媒 | |
| US7820136B2 (en) | Process for production of compound oxides | |
| JP2005314134A (ja) | 金属酸化物粒子及びその製造方法、並びに排ガス浄化触媒 | |
| JP2006263550A (ja) | 触媒担体粉末及び排ガス浄化触媒 | |
| JP4710744B2 (ja) | 複合金属酸化物の製造方法 | |
| WO2017163916A1 (ja) | 排ガス浄化用触媒及びその製造方法並びにそれを用いた排ガス浄化装置 | |
| JP4770959B2 (ja) | 触媒担体及び排ガス浄化用触媒 | |
| JP4165419B2 (ja) | 金属酸化物粒子及び排ガス浄化触媒の製造方法 | |
| JP2007105632A (ja) | 排ガス浄化触媒 | |
| JP2006320797A (ja) | 触媒及びその製造方法 | |
| JP5019019B2 (ja) | 排ガス浄化用触媒担体、それを用いた排ガス浄化用触媒及び排ガス浄化方法 | |
| JP7262975B2 (ja) | セリア・ジルコニア系複合酸化物酸素吸収放出材料および排ガス浄化触媒 | |
| JP5140987B2 (ja) | 触媒担体及びその製造方法、並びに排ガス浄化触媒 | |
| JP2001232199A (ja) | 排ガス浄化用触媒 | |
| JP2009279546A (ja) | コアシェル構造体の製造方法及びそれにより製造されたコアシェル構造体を含む排ガス浄化用触媒 | |
| WO2013098987A1 (ja) | 排気ガス浄化用触媒のための担体、排気ガス浄化用触媒及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080508 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20080508 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090203 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090327 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20090519 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20090601 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120619 Year of fee payment: 3 |
|
| R151 | Written notification of patent or utility model registration |
Ref document number: 4325648 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R151 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120619 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120619 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130619 Year of fee payment: 4 |
|
| LAPS | Cancellation because of no payment of annual fees |