CN100586559C - 废气净化用催化剂 - Google Patents
废气净化用催化剂 Download PDFInfo
- Publication number
- CN100586559C CN100586559C CN200480034776A CN200480034776A CN100586559C CN 100586559 C CN100586559 C CN 100586559C CN 200480034776 A CN200480034776 A CN 200480034776A CN 200480034776 A CN200480034776 A CN 200480034776A CN 100586559 C CN100586559 C CN 100586559C
- Authority
- CN
- China
- Prior art keywords
- exhaust gas
- purification catalyst
- gas purification
- zirconium
- temperature
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 239000003054 catalyst Substances 0.000 title claims abstract description 258
- 238000004140 cleaning Methods 0.000 title abstract 3
- 229910052726 zirconium Inorganic materials 0.000 claims abstract description 66
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 claims abstract description 59
- 229910052751 metal Inorganic materials 0.000 claims abstract description 30
- 239000002131 composite material Substances 0.000 claims abstract description 20
- 229910052783 alkali metal Inorganic materials 0.000 claims abstract description 14
- 150000001340 alkali metals Chemical class 0.000 claims abstract description 14
- 229910052761 rare earth metal Inorganic materials 0.000 claims abstract description 13
- 150000002910 rare earth metals Chemical class 0.000 claims abstract description 13
- 238000000746 purification Methods 0.000 claims description 230
- -1 zirconium organic compound Chemical class 0.000 claims description 86
- 238000000034 method Methods 0.000 claims description 28
- 208000035126 Facies Diseases 0.000 claims description 25
- 238000006460 hydrolysis reaction Methods 0.000 claims description 24
- 230000007062 hydrolysis Effects 0.000 claims description 23
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 claims description 21
- YRKCREAYFQTBPV-UHFFFAOYSA-N acetylacetone Chemical compound CC(=O)CC(C)=O YRKCREAYFQTBPV-UHFFFAOYSA-N 0.000 claims description 18
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 18
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical group [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 claims description 15
- 150000002500 ions Chemical class 0.000 claims description 13
- 229910000510 noble metal Inorganic materials 0.000 claims description 13
- 229910052746 lanthanum Inorganic materials 0.000 claims description 12
- FZLIPJUXYLNCLC-UHFFFAOYSA-N lanthanum atom Chemical compound [La] FZLIPJUXYLNCLC-UHFFFAOYSA-N 0.000 claims description 12
- 238000004519 manufacturing process Methods 0.000 claims description 12
- 229910052792 caesium Inorganic materials 0.000 claims description 7
- 229910052697 platinum Inorganic materials 0.000 claims description 7
- 230000008569 process Effects 0.000 claims description 7
- 238000006467 substitution reaction Methods 0.000 claims description 7
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical group [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 claims description 6
- 150000002894 organic compounds Chemical class 0.000 claims description 5
- 125000006606 n-butoxy group Chemical group 0.000 claims description 2
- 239000002184 metal Substances 0.000 abstract description 14
- 229910052784 alkaline earth metal Inorganic materials 0.000 abstract description 11
- 150000001342 alkaline earth metals Chemical class 0.000 abstract description 11
- 238000006073 displacement reaction Methods 0.000 abstract description 4
- 239000013078 crystal Substances 0.000 abstract description 3
- RVTZCBVAJQQJTK-UHFFFAOYSA-N oxygen(2-);zirconium(4+) Chemical compound [O-2].[O-2].[Zr+4] RVTZCBVAJQQJTK-UHFFFAOYSA-N 0.000 abstract description 3
- 229910001928 zirconium oxide Inorganic materials 0.000 abstract description 3
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 493
- 239000007789 gas Substances 0.000 description 275
- 239000000446 fuel Substances 0.000 description 137
- 239000002912 waste gas Substances 0.000 description 85
- 229960003753 nitric oxide Drugs 0.000 description 78
- 238000002485 combustion reaction Methods 0.000 description 73
- 230000009467 reduction Effects 0.000 description 49
- 239000003638 chemical reducing agent Substances 0.000 description 48
- 239000001301 oxygen Substances 0.000 description 44
- 229910052760 oxygen Inorganic materials 0.000 description 44
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 41
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 30
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 21
- 239000008346 aqueous phase Substances 0.000 description 16
- 150000004703 alkoxides Chemical class 0.000 description 15
- 239000000203 mixture Substances 0.000 description 15
- 239000004094 surface-active agent Substances 0.000 description 14
- 230000006835 compression Effects 0.000 description 13
- 238000007906 compression Methods 0.000 description 13
- 150000002902 organometallic compounds Chemical class 0.000 description 13
- 238000001816 cooling Methods 0.000 description 11
- 239000004530 micro-emulsion Substances 0.000 description 11
- 239000000047 product Substances 0.000 description 11
- 238000000354 decomposition reaction Methods 0.000 description 10
- 229910052757 nitrogen Inorganic materials 0.000 description 10
- SLODBEHWNYQCRC-UHFFFAOYSA-N [La+3].[O-2].[Zr+4] Chemical compound [La+3].[O-2].[Zr+4] SLODBEHWNYQCRC-UHFFFAOYSA-N 0.000 description 9
- 230000001186 cumulative effect Effects 0.000 description 9
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 8
- 229910002091 carbon monoxide Inorganic materials 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 8
- 238000007254 oxidation reaction Methods 0.000 description 8
- 238000010521 absorption reaction Methods 0.000 description 7
- 239000012298 atmosphere Substances 0.000 description 7
- 229910021645 metal ion Inorganic materials 0.000 description 7
- 238000012545 processing Methods 0.000 description 7
- PNEYBMLMFCGWSK-UHFFFAOYSA-N Alumina Chemical compound [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- 230000009102 absorption Effects 0.000 description 6
- 230000003647 oxidation Effects 0.000 description 6
- 230000000630 rising effect Effects 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 5
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 5
- 239000002202 Polyethylene glycol Substances 0.000 description 5
- 239000011358 absorbing material Substances 0.000 description 5
- 239000003245 coal Substances 0.000 description 5
- 230000007547 defect Effects 0.000 description 5
- 238000010586 diagram Methods 0.000 description 5
- 229930195733 hydrocarbon Natural products 0.000 description 5
- 150000002430 hydrocarbons Chemical class 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 229920001223 polyethylene glycol Polymers 0.000 description 5
- 239000000779 smoke Substances 0.000 description 5
- 239000004215 Carbon black (E152) Substances 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- MCMNRKCIXSYSNV-UHFFFAOYSA-N Zirconium dioxide Chemical compound O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 description 4
- 238000000975 co-precipitation Methods 0.000 description 4
- 238000010304 firing Methods 0.000 description 4
- ZSIAUFGUXNUGDI-UHFFFAOYSA-N hexan-1-ol Chemical compound CCCCCCO ZSIAUFGUXNUGDI-UHFFFAOYSA-N 0.000 description 4
- 230000007246 mechanism Effects 0.000 description 4
- 239000003595 mist Substances 0.000 description 4
- 239000003960 organic solvent Substances 0.000 description 4
- 230000002000 scavenging effect Effects 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 239000007762 w/o emulsion Substances 0.000 description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 238000004364 calculation method Methods 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 239000012467 final product Substances 0.000 description 3
- 229910044991 metal oxide Inorganic materials 0.000 description 3
- 150000004706 metal oxides Chemical class 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 238000011084 recovery Methods 0.000 description 3
- 239000010948 rhodium Substances 0.000 description 3
- 239000000243 solution Substances 0.000 description 3
- 239000007921 spray Substances 0.000 description 3
- 238000003860 storage Methods 0.000 description 3
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 2
- 229910002651 NO3 Inorganic materials 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- 239000002250 absorbent Substances 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 239000003945 anionic surfactant Substances 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 125000002091 cationic group Chemical group 0.000 description 2
- 210000004027 cell Anatomy 0.000 description 2
- 238000009841 combustion method Methods 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 239000000498 cooling water Substances 0.000 description 2
- 239000003599 detergent Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 230000012447 hatching Effects 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 238000011068 loading method Methods 0.000 description 2
- 150000004692 metal hydroxides Chemical class 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- JCXJVPUVTGWSNB-UHFFFAOYSA-N nitrogen dioxide Inorganic materials O=[N]=O JCXJVPUVTGWSNB-UHFFFAOYSA-N 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 229910052703 rhodium Inorganic materials 0.000 description 2
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 2
- 230000001052 transient effect Effects 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- FDCJDKXCCYFOCV-UHFFFAOYSA-N 1-hexadecoxyhexadecane Chemical compound CCCCCCCCCCCCCCCCOCCCCCCCCCCCCCCCC FDCJDKXCCYFOCV-UHFFFAOYSA-N 0.000 description 1
- MGWGWNFMUOTEHG-UHFFFAOYSA-N 4-(3,5-dimethylphenyl)-1,3-thiazol-2-amine Chemical compound CC1=CC(C)=CC(C=2N=C(N)SC=2)=C1 MGWGWNFMUOTEHG-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- YQEVIZPKEOELNL-UHFFFAOYSA-N CCCCO[Zr] Chemical compound CCCCO[Zr] YQEVIZPKEOELNL-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 229910052684 Cerium Inorganic materials 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 229910052692 Dysprosium Inorganic materials 0.000 description 1
- 229910052691 Erbium Inorganic materials 0.000 description 1
- 229910052693 Europium Inorganic materials 0.000 description 1
- 229910001111 Fine metal Inorganic materials 0.000 description 1
- 229910052688 Gadolinium Inorganic materials 0.000 description 1
- GYHNNYVSQQEPJS-UHFFFAOYSA-N Gallium Chemical compound [Ga] GYHNNYVSQQEPJS-UHFFFAOYSA-N 0.000 description 1
- 240000004859 Gamochaeta purpurea Species 0.000 description 1
- 229910052689 Holmium Inorganic materials 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 229910052765 Lutetium Inorganic materials 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 229910052779 Neodymium Inorganic materials 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 229910052773 Promethium Inorganic materials 0.000 description 1
- 229910052772 Samarium Inorganic materials 0.000 description 1
- 229910052775 Thulium Inorganic materials 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- 229910052769 Ytterbium Inorganic materials 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 150000005215 alkyl ethers Chemical class 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 239000004411 aluminium Substances 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 235000011114 ammonium hydroxide Nutrition 0.000 description 1
- GETQZCLCWQTVFV-UHFFFAOYSA-N anhydrous trimethylamine Natural products CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 229910052788 barium Inorganic materials 0.000 description 1
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 229910052790 beryllium Inorganic materials 0.000 description 1
- ATBAMAFKBVZNFJ-UHFFFAOYSA-N beryllium atom Chemical compound [Be] ATBAMAFKBVZNFJ-UHFFFAOYSA-N 0.000 description 1
- 230000002457 bidirectional effect Effects 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- 150000001663 caesium Chemical class 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000035568 catharsis Effects 0.000 description 1
- 210000003850 cellular structure Anatomy 0.000 description 1
- GWXLDORMOJMVQZ-UHFFFAOYSA-N cerium Chemical compound [Ce] GWXLDORMOJMVQZ-UHFFFAOYSA-N 0.000 description 1
- RLGQACBPNDBWTB-UHFFFAOYSA-N cetyltrimethylammonium ion Chemical compound CCCCCCCCCCCCCCCC[N+](C)(C)C RLGQACBPNDBWTB-UHFFFAOYSA-N 0.000 description 1
- 238000005660 chlorination reaction Methods 0.000 description 1
- 229910052878 cordierite Inorganic materials 0.000 description 1
- 150000001934 cyclohexanes Chemical class 0.000 description 1
- JSKIRARMQDRGJZ-UHFFFAOYSA-N dimagnesium dioxido-bis[(1-oxido-3-oxo-2,4,6,8,9-pentaoxa-1,3-disila-5,7-dialuminabicyclo[3.3.1]nonan-7-yl)oxy]silane Chemical compound [Mg++].[Mg++].[O-][Si]([O-])(O[Al]1O[Al]2O[Si](=O)O[Si]([O-])(O1)O2)O[Al]1O[Al]2O[Si](=O)O[Si]([O-])(O1)O2 JSKIRARMQDRGJZ-UHFFFAOYSA-N 0.000 description 1
- 238000007599 discharging Methods 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- KBQHZAAAGSGFKK-UHFFFAOYSA-N dysprosium atom Chemical compound [Dy] KBQHZAAAGSGFKK-UHFFFAOYSA-N 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- UYAHIZSMUZPPFV-UHFFFAOYSA-N erbium Chemical compound [Er] UYAHIZSMUZPPFV-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- OGPBJKLSAFTDLK-UHFFFAOYSA-N europium atom Chemical compound [Eu] OGPBJKLSAFTDLK-UHFFFAOYSA-N 0.000 description 1
- 229910052730 francium Inorganic materials 0.000 description 1
- KLMCZVJOEAUDNE-UHFFFAOYSA-N francium atom Chemical compound [Fr] KLMCZVJOEAUDNE-UHFFFAOYSA-N 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 239000002828 fuel tank Substances 0.000 description 1
- UIWYJDYFSGRHKR-UHFFFAOYSA-N gadolinium atom Chemical compound [Gd] UIWYJDYFSGRHKR-UHFFFAOYSA-N 0.000 description 1
- 229910052733 gallium Inorganic materials 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- KJZYNXUDTRRSPN-UHFFFAOYSA-N holmium atom Chemical compound [Ho] KJZYNXUDTRRSPN-UHFFFAOYSA-N 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 1
- 230000001535 kindling effect Effects 0.000 description 1
- FYDKNKUEBJQCCN-UHFFFAOYSA-N lanthanum(3+);trinitrate Chemical compound [La+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O FYDKNKUEBJQCCN-UHFFFAOYSA-N 0.000 description 1
- XQBXQQNSKADUDV-UHFFFAOYSA-N lanthanum;nitric acid Chemical compound [La].O[N+]([O-])=O XQBXQQNSKADUDV-UHFFFAOYSA-N 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- OHSVLFRHMCKCQY-UHFFFAOYSA-N lutetium atom Chemical compound [Lu] OHSVLFRHMCKCQY-UHFFFAOYSA-N 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 235000013372 meat Nutrition 0.000 description 1
- 229910000000 metal hydroxide Inorganic materials 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 239000000693 micelle Substances 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- QEFYFXOXNSNQGX-UHFFFAOYSA-N neodymium atom Chemical compound [Nd] QEFYFXOXNSNQGX-UHFFFAOYSA-N 0.000 description 1
- ZPIRTVJRHUMMOI-UHFFFAOYSA-N octoxybenzene Chemical compound CCCCCCCCOC1=CC=CC=C1 ZPIRTVJRHUMMOI-UHFFFAOYSA-N 0.000 description 1
- 229920002114 octoxynol-9 Polymers 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000005501 phase interface Effects 0.000 description 1
- 239000003495 polar organic solvent Substances 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- VQMWBBYLQSCNPO-UHFFFAOYSA-N promethium atom Chemical compound [Pm] VQMWBBYLQSCNPO-UHFFFAOYSA-N 0.000 description 1
- 229910052701 rubidium Inorganic materials 0.000 description 1
- IGLNJRXAVVLDKE-UHFFFAOYSA-N rubidium atom Chemical compound [Rb] IGLNJRXAVVLDKE-UHFFFAOYSA-N 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- KZUNJOHGWZRPMI-UHFFFAOYSA-N samarium atom Chemical compound [Sm] KZUNJOHGWZRPMI-UHFFFAOYSA-N 0.000 description 1
- 229910052706 scandium Inorganic materials 0.000 description 1
- SIXSYDAISGFNSX-UHFFFAOYSA-N scandium atom Chemical compound [Sc] SIXSYDAISGFNSX-UHFFFAOYSA-N 0.000 description 1
- 239000013049 sediment Substances 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000006104 solid solution Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 229910052712 strontium Inorganic materials 0.000 description 1
- CIOAGBVUUVVLOB-UHFFFAOYSA-N strontium atom Chemical compound [Sr] CIOAGBVUUVVLOB-UHFFFAOYSA-N 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 229910052716 thallium Inorganic materials 0.000 description 1
- BKVIYDNLLOSFOA-UHFFFAOYSA-N thallium Chemical compound [Tl] BKVIYDNLLOSFOA-UHFFFAOYSA-N 0.000 description 1
- NAWDYIZEMPQZHO-UHFFFAOYSA-N ytterbium Chemical compound [Yb] NAWDYIZEMPQZHO-UHFFFAOYSA-N 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical compound [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 description 1
Images


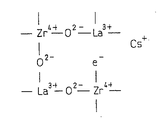




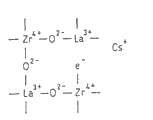
























Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D53/00—Separation of gases or vapours; Recovering vapours of volatile solvents from gases; Chemical or biological purification of waste gases, e.g. engine exhaust gases, smoke, fumes, flue gases, aerosols
- B01D53/34—Chemical or biological purification of waste gases
- B01D53/92—Chemical or biological purification of waste gases of engine exhaust gases
- B01D53/94—Chemical or biological purification of waste gases of engine exhaust gases by catalytic processes
- B01D53/9445—Simultaneously removing carbon monoxide, hydrocarbons or nitrogen oxides making use of three-way catalysts [TWC] or four-way-catalysts [FWC]
- B01D53/945—Simultaneously removing carbon monoxide, hydrocarbons or nitrogen oxides making use of three-way catalysts [TWC] or four-way-catalysts [FWC] characterised by a specific catalyst
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/002—Mixed oxides other than spinels, e.g. perovskite
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/56—Platinum group metals
- B01J23/58—Platinum group metals with alkali- or alkaline earth metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/56—Platinum group metals
- B01J23/63—Platinum group metals with rare earths or actinides
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02T—CLIMATE CHANGE MITIGATION TECHNOLOGIES RELATED TO TRANSPORTATION
- Y02T10/00—Road transport of goods or passengers
- Y02T10/10—Internal combustion engine [ICE] based vehicles
- Y02T10/12—Improving ICE efficiencies
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Combustion & Propulsion (AREA)
- Biomedical Technology (AREA)
- Environmental & Geological Engineering (AREA)
- Analytical Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Catalysts (AREA)
- Exhaust Gas Treatment By Means Of Catalyst (AREA)
- Exhaust Gas After Treatment (AREA)
Abstract
提供一种在高温也能发挥净化性能的废气净化用催化剂。该催化剂在结晶性的锆复合氧化物上载持碱金属和贵金属,上述锆复合氧化物被选自碱土金属、稀土金属以及IIIB族元素中的至少一种元素置换一部分锆,通过该元素置换,晶格的延伸大致变为理论值。
Description
技术领域
本发明涉及一种用于净化汽车等的内燃机废气的废气净化用催化剂。
背景技术
一般地,从汽车用发动机等内燃机排出的废气中含有碳氢化合物(以下称为HC)、一氧化碳(CO)、氮氧化合物(NOx)等物质(排放物)。为了减少这些物质的排出量,除了使排放物的空燃比等的燃烧条件最适化之外,一般还使用利用废气净化用催化剂去除废气中含有的物质的方法。
作为该废气净化用催化剂,一般有在氧化铝等多孔质金属氧化物载体中载持了铂(Pt)、铑(Rh)、钯(Pd)等贵金属的所谓三元催化剂。公知的是,该三元催化剂氧化CO和HC,并且具有将NOx还原成N2的能力。
另一方面,近年来,从地球环境保护的观点出发,抑制从汽车用发动机等内燃机排出的二氧化碳(CO2)的产生量已经成为课题,作为其对策,开发了在氧过剩(稀混合气体)氛围内进行稀薄燃烧的稀薄混合气发动机。在该稀薄混合气发动机中,降低了燃料的使用量,其结果是能够抑制作为燃烧废气的CO2的产生。
对此,以往的三元催化剂是,空燃比为理论空燃比(化学计量比)时,废气中的HC、CO、NOx同时进行氧化/还原,在上述稀混合气燃烧时废气的氧过剩氛围中,可以氧化去除HC和CO,但是对于NOx的还原去除没有显示出充分的净化性能。
因此,开发了一种在稀混合气体燃烧中,经常在氧过剩的稀混合气体条件下进行燃烧,在短时间内从化学计量比~浓条件,由此使废气作为还原气氛还原净化NOx的系统。并且,开发了一种最适于该系统的、利用在稀混合气体氛围中吸收NOx、在化学计量比~浓混合氛围中放出被吸收了的NOx的NOx吸收材料的NOx吸收还原型废气净化用催化剂。
该NOx吸收还原型的废气净化用催化剂构成为,在氧化铝等的多孔质金属氧化物的载体上形成碱金属、碱土金属或稀土金属构成的NOx吸收材料的层,并将铂等的贵金属催化剂载持在载体表面上。在该催化剂中,废气的空燃比为稀混合气体时,废气中含有的NOx通过贵金属催化剂被氧化,以硝酸盐的形势被NOx吸收材料吸收。其次,废气的空燃比在短时间内变浓时,该期间内放出吸收在NOx吸收材料中的NOx,并与HC或CO的还原性成分反应而被净化,其次再将废气的空燃比返回稀混合气体时,开始向NOx吸收材料的NOx吸收,即使是来自稀薄混合气发动机的废气也能够高效率地净化NOx。
但是,该NOx吸收还原性的废气净化用催化剂有废气温度特别是在500℃以上的高温时,NOx净化能力大幅下降的问题。因此,提案了一种在高温区域也具有较高NOx净化能力、由钙钛矿型复合氧化物构成的废气净化用催化剂。(例如参照特开2002-143684号公报)
在该废气净化用催化剂中,具有钙钛矿性复合氧化物促进的NOx直接分解作用,因此与以往相比,在更广的温度范围内具有较高的NOx净化性能,但是在大概700℃时NOx吸收达到极限,在更高的温度时无法保持NOx。并且,在实用条件下不能达到充分的NOx净化率。
因此,本发明的目的在于提供一种废气净化用催化剂,作为催化剂载体,利用在结晶结构内具有特定元素的锆复合氧化物,由此能够在高达1000℃的高温达到NOx净化。
发明内容
为了解决上述问题点,根据本发明的第一方式,一种废气净化用催化剂,在结晶性的锆复合氧化物上载持碱金属和贵金属,上述锆复合氧化物被选自碱土金属、稀土金属以及IIIB族元素中的至少一种元素置换一部分锆,通过该元素置换,晶格的延伸大致变为理论值。
根据第二方式,在第一方式中,上述选自碱土金属、稀土金属以及IIIB族元素中的至少一种元素,以锆复合氧化物中的所有金属元素的总摩尔数为基准,存在5~50摩尔%。
根据第三方式,在第一方式中,一部分锆被镧置换。
根据第四方式,在第一方式中,锆复合氧化物上载持的上述碱金属是铯。
根据第五方式,在第一方式中,锆复合氧化物上载持的上述贵金属是铂。
附图说明
图1是用于说明发现本发明的催化剂的超强碱性点的情况的图,图1(A)表示氧化锆的结晶结构,图1(B)表示用镧置换了氧化锆的一部分锆的结晶结构,图1(C)表示本发明的锆复合氧化物的结晶结构。
图2是用于说明本发明的催化剂的制造工序的图。
图3是火花点火式内燃机的整体图。
图4是用于说明一氧化氮的吸附和分解的情况的图,图4(A)和图4(B)表示在具有超强碱性点的载体上吸附了一氧化氮的状态,图4(C)表示氮分解了的状态,图4(D)表示氧脱离了的状态。
图5是表示应提供的能量的量和废气净化用催化剂的温度的关系的图。
图6是表示废气中的一氧化氮量的变换的图。
图7是表示提供的能量的量的图。
图8是用于控制能量的提供的流程图。
图9是表示空燃比的加浓控制的图。
图10是表示氧浓度与NOx浓度的变化的时间表。
图11是表示应供给的还原剂量与废气净化用催化剂的温度的关系的图。
图12是表示空燃比的加浓控制的图。
图13是用于控制还原剂的供给的流程图。
图14是用于进行硝酸离子和一氧化氮的还原处理的流程图。
图15是表示经过时间的图。
图16是用于控制还原剂供给的流程图。
图17是表示火花点火式内燃机的另一实施例的整体图。
图18是用于控制还原剂供给的流程图。
图19是表示火花点火式内燃机的又一实施例的整体图。
图20是表示火花点火式内燃机的又一实施例的整体图。
图21是表示压燃式内燃机的整体图。
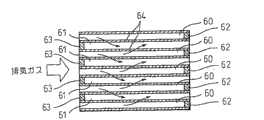
图22是表示微粒过滤器的图,图22(A)是主视图,图22(B)是侧剖视图。
图23是表示烟雾产生量的图。
图24(A)是表示燃烧室内的平均气温和曲轴转角的关系的图表,图24(B)是表示燃料及其周围的气温与曲轴转角的关系的图表。
图25是表示运转区域I、II的图。
图26是表示空燃比A/F的图。
图27是表示节气门开度等的变化的图。
图28是表示镧锆氧化物的晶格间隔的图表。
图29是表示催化剂的高温NOx吸收性能的图表。
具体实施方式
本发明的废气净化用催化剂是在结晶性的锆复合氧化物载体上载持碱金属和贵金属的催化剂。作为该碱金属,可以使用锂、钠、钾、铷、铯、钫,作为贵金属,可以使用铂、钯、铑等。优选的是,碱金属的载持量为0.05~0.3摩尔/升,贵金属的载持量为1~5克/升。
本发明的废气净化用催化剂的载体,如图1(A)所示,基本上具有氧化锆的结晶结构,该结晶结构中的锆的一部分被选自碱土金属、稀土金属以及IIIB族元素中的至少一种元素置换的锆复合氧化物。在此,作为碱土金属,可以使用铍、镁、钙、锶、钡。作为稀土金属,可以使用钪、钇、镧、钕、钷、钐、铕、钆、镝、钬、铒、铥、镱、镥。作为IIIB族元素,可以使用硼、铝、镓、铟、铊。选自该碱土金属、稀土金属以及IIIB族元素中的至少一种元素的含量,以锆复合氧化物中的所有金属元素的总摩尔数为基准,存在5~50摩尔%。
构成结晶性氧化锆的氧化锆为4价,因此,其被2价的碱土金属、3价的稀土金属或IIIB族元素,例如镧置换时,如图1(B)所示,在晶格内形成不存在氧的氧缺陷。
并且,在该复合氧化物中,如上所述,碱金属例如铯被载持,通过该铯,如图1(C)所示,电子e-被提供到氧缺陷中。被提供了该电子的氧缺陷显示出非常强的碱性,因此,被提供了电子的氧缺陷以下称为超强碱性点。
在本发明的废气净化用催化剂中,作为载体的锆复合氧化物在整体都具有图1(C)所示结晶结构,并且,整体上均匀分布无数超强碱性点。利用通过镧等置换了氧化锆中的一部分锆的载体的以往的催化剂,是由共沉淀法或者醇盐法这种以往的复合氧化物的制造方法制造的,不能充分使镧置换锆,没有足够的超强碱性点,并且也不能使这些超强碱性点均匀分布。对此,本发明的催化剂的载体中,通过使用规定方法,能够使足够量的镧等与锆置换,使足够量的超强碱性点均匀分布。该足够量的镧等的置换可以由元素置换引起的锆氧化物的晶格的延伸大致为理论值这一点反映。
由此,通过选自碱土金属、稀土金属以及IIIB族元素中的至少一种金属来置换一部分锆、该元素置换引起的晶格的延伸大致为理论值的锆复合氧化物可以利用以下方法进行制造。
即,通过使水解而生成锆的氢氧化物的有机化合物溶解后的有机相、与将选自碱土金属、稀±金属以及IIIB族元素中的第二元素作为离子包括在内的水相接触,在通过其界面的锆有机化合物的水解反应来生成锆的氢氧化物的过程中,向该生成物中引入第二元素,烧成得到的复合氢氧化物(前驱物),得到锆和第二元素的复合氧化物。
在此,水解生成锆的氢氧化物的有机化合物是公知的,在本发明中可以使用其中任意一个。例如,可以列举出锆醇盐、乙酰丙酮锆络合物。
锆Zr(OR)4的水解反应也是公知的,形式上可以表达为:Zr(OR)4+4H2O→Zr(OH)4+4ROH,其次,Zr(OH)4→ZrO2+2H2O。
乙酰丙酮锆络合物(CH3COCH2COCH3)4Zr的水解反应也是公知的,可以表示为(CH3COCH2COCH3)4Zr+4ROH→4CH3COCH2C(OH)CH3+Zr(OH)4,其次,Zr(OH)4→ZrO2+2H2O。
如果从极性有机溶剂、无极性有机溶剂等之中选择适当的溶剂,则锆醇盐或乙酰丙酮锆络合物等的有机金属化合物能够相对容易地溶解。作为有机溶剂的例子,有环己烷、苯等的碳氢化合物、己醇等直链醇、丙酮等的酮类。作为有机溶剂的选择标准,有形成表面活性剂的溶解度的其他微乳剂的区域的大小(水/表面活性剂的摩尔比较大)等。
公知的是,在溶解了这种水解生成氢氧化物的有机金属化合物的有机相中添加水时,有机金属化合物的水解反应开始,并进行。一般地,在溶解了有机金属化合物的有机相中添加水,并搅拌,能够得到金属氢氧化物。
并且,公知的是,在有机相(油相)中形成用表面活性剂使水相微细地分散的油中水型乳剂、微乳剂,在该有机相(油相)中添加有机金属化合物(将有机金属化合物溶解到有机溶剂中的溶液),并进行搅拌,生成微细的金属氢氧化物或氧化物。虽然并不仅限于此,但可以考虑由表面活性剂围住的水相所构成的的许多胶粒表面变为反应核,或者使表面活性剂生成的氢氧化物的微粒子稳定,由此得到微细的生成物粒子。
公知的是,如上所述,在水解反应中,多个水解性有机金属化合物溶解到有机相中,由此,使其与水接触时,该多个有机金属化合物水解,多个金属的氢氧化物同时生成。
在本发明中,其特征在于,使该水解性有机金属化合物中的一种(含锆的化合物)存在于有机相中,该有机相与水相接触时,从碱土金属、稀土金属以及IIIB族元素所构成的群中选出第二金属元素,并且第三以后的金属元素不是像以往那样存在于有机相中,而是在水相中作为离子存在。
使其在水相中作为离子存在,可以使用水溶性金属盐,特别是,硝酸盐、氯化物等无机酸盐,还有乙酸盐、乳酸盐、草酸盐等有机酸盐。存在于水相中的第二元素的离子除了可以是金属的单体离子之外,还可以是包括第二元素的络离子。第三以后的元素的离子也是同样的。
使有机相与水相接触时,有机相中的有机锆化合物与水接触,由此引起水解反应,生成锆的氢氧化物或氧化物,但此时,根据本发明,可以发现存在于水相中的金属离子被吸入作为水解生成物的锆的氢氧化物(或氧化物)中。该现象是以往不知道的。虽然没有充分理解水相中的离子即使不做特别的沉淀操作也能被吸入氢氧化物中的理由,但以有机锆化合物为醇盐的情形为例进行说明时,可以理解为,醇盐被水解时,水相中的第二金属离子诱导醇盐,进行水解,或者醇盐进行了水解的微小的氢氧化物捕获水相中规定量的金属离子,并凝集而成。
根据本发明,尤其是在本新制造方法中,存在于水相中的第二金属元素的离子被吸入到有机相中的锆的有机锆化合物水解得到的氢氧化物中,可以获得得到的氢氧化物中的锆和第二金属元素非常均匀地分散了的氢氧化物,可以发现与以往的醇盐法、即有机相中存在多个金属醇盐的情况相比,能够得到显著优秀的均匀度。即使在比较低的烧成温度下,也能得到烧成后的复合氧化物的锆和第二金属元素以原子水平理想混合的复合氧化物(固溶体)。这是以往的金属醇盐法无法达到的。在以往的金属醇盐法中,由于根据金属醇盐的种类不同,稳定性也不同,第一金属元素和第二金属元素之间只能得到不均匀的生成物。
本发明中所用的复合氧化物中的锆及第二金属元素的相对比可以根据有机相中的锆的量和水相中的第二金属元素的量的比进行调整。
在本发明中,优选的是,反应体系是油中水型乳剂类或者微乳剂类。这种情况下,可以考虑为,第一,微乳剂直径小至数nm~十数nm,油相-水相界面非常宽大(直径为10nm的情况下为8000m2/升左右)而引起的水解速度的高速化,第二,水相被分壳化,每个仅含有极少量的金属离子(大概100个左右)而引起的均匀化的效果。
在这种目的下,微乳剂的水相的直径为2~40nm,优选的是2~15nm,更优选的是2~10nm。
形成油中水型乳剂类或微乳剂类的方法是公知的。作为有机相介质,可以使用环己烷、苯等碳氢化合物、己醇等直链醇、丙酮等的酮类等与上述有机溶剂同样的物质。在本方案中使用的表面活性剂可以是非离子型表面活性剂、阴离子型表面活性剂、阳离子型表面活性剂等多种活性剂根据用途与有机相(油相)成分组合使用。
作为非离子型表面活性剂,可以使用以聚氧化乙烯(n=5)壬基苯基醚为代表的聚氧化乙烯壬基苯基醚类,聚氧化乙烯(n=10)辛基苯基醚为代表的聚氧化乙烯辛基苯基醚类,聚氧化乙烯(n=7)十六烷基醚等为代表的聚氧化乙烯烷基醚类表面活性剂,聚氧化乙烯山梨糖醇三油酸酯为代表的聚氧化乙烯山梨糖醇类表面活性剂等。
作为阴离子型表面活性剂,可以使用双-2-亚乙基己基硫代琥珀酸钠等,阳离子型表面活性剂可以使用氯化十六烷基三甲基胺或溴化十六烷基三甲基胺等。
优选的是,使用油中水型乳剂类或微乳剂类,但也可以使用水中油型乳剂类。
在本发明中,在制造三种以上元素的复合氧化物的情况下,使第三以后的元素存在于水相中。这是由于使有机相中存在多个水解性有机金属化合物时,有机相中的水解性有机金属化合物间的稳定性存在差异,因而产生不均匀的生成物。但是,虽然锆和第二金属元属间必须均匀,但锆和第三金属元素间均匀性不重要时,有机相中也可以存在第三元素的有机金属化合物。
如上所述,使有机相和水相接触并进行水解反应时,一般生成氢氧化物(前驱物)。根据本发明,总之在生成物干燥后进行烧成,制造复合氧化物。生成物的分离、干燥方法与以往同样即可。
烧成条件也可以与以往同样地,烧成温度、烧成氛围等根据特定的复合氧化物的种类选择即可。但是,一般来说,可以以与以往相比更低的温度烧成。可以看作是,由于金属元素预先均匀分散,因此,使得金属元素在固体中扩散的能量少也是可以的。
图2以利用微乳剂合成镧锆氧化物为例示意性地表示了上述锆复合氧化物的制造方法。参照图2,微乳剂ME的水相中溶解有硝酸镧等,向其中添加并混合锆醇盐,由此合成镧锆氧化物。即,向微乳剂的有机相中仅添加一种金属醇盐,即锆醇盐。由于有机相中存在多个金属醇盐时其稳定性有差异,因此,相对于有机相与水接触时不能得到均匀的水解生成物,根据本发方法能够解决该问题。因此,在合成三种以上的金属元素的复合氧化物时,第三元素以后添加到水相中。
水解性有机金属化合物的水解反应是公知的。可以看出,根据本发明,使有机相中的水解性有机锆化合物与水相接触,进行水解反应时,在水相中,第二元素作为离子存在时,第二元素被吸入作为水解反应生成物的氢氧化物中。该反应是:水相中的第二元素离子被表面活性剂的亲水基电吸引,有机锆、化合物被水解时同时被吸入,由此变为含有第二元素的复合氧化物。并且,可以看出,由于该反应,有机锆化合物中含有的锆和水相中的第二元素可以均匀地分散、混合在水解反应生成物、甚至复合氧化物中。
将这样得到锆复合氧化物作为载体使用,通过使用与以往同样的方法,将碱金属和贵金属载持在该载体上,得到本发明的废气净化用催化剂。
对于利用这样得到的本发明的废气净化用催化剂进行的、以稀空燃比进行燃烧时净化NOx的作用进行说明。图3表示将本发明的废气净化用催化剂应用于火花点火式内燃机的情况。另外,本发明也可以应用于压燃式内燃机。
参照图3,1是内燃机主体,2是汽缸体、3是汽缸盖、4是活塞、5是燃烧室、6是电控式燃料喷射阀、7是火花塞、8是进气阀、9是进气口、10是废气阀、11是废气口。进气口9通过所对应的进气支管12与稳压箱13连接,稳压箱13通过进气管道14与空气过滤器15连接。在进气管道14内配置了由步进电动机16驱动的节气门17、还在进气管道14内配置了用于检测吸入空气的质量流量的吸入空气量传感器18。另一方面,废气口11通过废气歧管19与内置有本发明的废气净化用催化剂20的催化转化器21连接。
废气歧管19和稳压箱13通过废气再循环(下文称为EGR)通道22彼此连接,在EGR通道22内配置了电控式EGR控制阀23。此外,还在EGR通道22周围配置了用于冷却在EGR通道22内流动的EGR气体的冷却装置24。在图1所示的实施例中,内燃机冷却水被引入冷却装置24内,利用内燃机冷却水冷却EGR气体。另一方面,各燃料喷射阀6通过燃料供给管25与油箱、即所谓的共轨26连接。由排出量可变的电控式燃料泵27向该共轨26内提供燃料,提供给共轨26内的燃料通过各燃料供给管25提供给燃料喷射阀6。在共轨26上安装了用于检测共轨26内的燃料压力的燃料压力传感器28,根据燃料压力传感器28的输出信号控制燃料泵27的排出量,以便使共轨26内的燃料压力达到目标燃料压力。
由于电子控制单元30由数字计算机构成,具有通过双向总线31彼此连接的ROM(只读存储器)32、RAM(随机存取存储器)33、CPU(微处理机)34、输入口35以及输出口36。吸入空气量传感器18和燃料压力传感器28的输出信号通过所对应的AD变换器37输入到输入口35。此外,在加速踏板40上连接着产生与加速踏板40的下踏量L成比例的输出电压的负载传感器41,负载传感器41的输出电压通过所对应的AD变换器37输入到输入口35。输入口35上还连接着曲轴转角传感器42,例如曲轴每转动30°即产生输出脉冲。另外,输出口36通过所对应的驱动电路38而与燃料喷射阀6、火花塞7、用于驱动节气门的步进电动机16、EGR控制阀23以及燃料泵27连接。
在活塞4的顶面上形成有空腔43,当内燃机低负载运转时,从燃料喷射阀6朝空腔43内喷射燃料F。该燃料F被空腔43的底壁面引导而朝向火花塞7,从而在火花塞7的周围形成混合气体。接着该混合气体被火花塞7点燃,进行成层燃烧。此时燃烧室5内的平均空燃比变得稀薄,因而废气的空燃比也变得稀薄。
当内燃机中等负载运转时,分为进气行程初期和压缩行程末期两次喷射燃料。通过进气行程初期的燃料喷射可在燃烧室5内形成扩散到整个燃烧室5内的稀薄混合气体,通过压缩行程末期的燃料喷射可在火花塞7的周围形成构成火种的混合气体。此时燃烧室5内的平均空燃比仍然稀薄,因而废气的空燃比自然也稀薄。
另一方面,当内燃机高负载运转时,在进气行程初期喷射燃料,由此可以在燃烧室5内形成均匀的混合气体。这时,燃烧室5内的空燃比可以是稀空燃比、理论空燃比或浓空燃比中的任何一种。由于内燃机通常在低负载或中负载状态下运转,因而通常可在稀空燃比状态下持续进行燃烧。
当在稀空燃比状态下进行燃烧时,利用废气净化用催化剂20净化从燃烧室5排出的NOx,但关于该废气净化用催化剂20的NOx净化作用的机理虽然尚不十分明了,但至今为止分析的结果可以认为是通过下述机理来净化NOx的。
即,当在稀空燃比状态下进行燃烧时,废气中含有一氧化氮NO以及二氧化氮NO2等氮氧化物NOx和过剩的氧O2。在此情况下,废气含有的大部分氮氧化物NOx是一氧化氮NO,因此下文以此为例对一氧化氮NO的净化机理加以说明。
如上所述,本发明中的废气净化用催化剂20具有超强碱性点。一旦存在此种超强碱性点,则本身呈酸性的一氧化氮NO不论废气净化用催化剂20的温度是高是低均可被超强碱性点吸引,其结果是一氧化氮NO以图4A或图4B所示的某种形式被废气净化用催化剂20的超强碱性点所捕获。在此情况下,由于上述废气净化用催化剂20的整个载体上均匀布满无数个超强碱性点,因而废气净化用催化剂20可吸附极大数量的一氧化氮NO。
一氧化氮(NO)一被超强碱性点吸附,即产生一氧化氮(NO)分解作用和一氧化氮(NO)的氧化反应。因此首先对一氧化氮(NO)的分解作用加以说明。
如上所述,废气中的一氧化氮(NO)受废气净化用催化剂20上的超强碱性点的吸引而被超强碱性点吸附并捕获。这时可向一氧化氮(NO)提供电子(e-)。当向一氧化氮(NO)提供的电子(e-)时,则一氧化氮(NO)的N-O键产生分解,在此情况下,废气净化用催化剂20的温度越高,该N-O键越容易断裂。事实上,一氧化氮(NO)一吸附到超强碱性点上,该N-O键很快就会断裂,并分解为氮(N)和氧(O),这时,如图4(C)所示,氧以氧离子(O-)的形式继续保持在超强碱性点上,而氮(N)则脱离超强碱性点在废气净化用催化剂20上移动。
在废气净化用催化剂20上移动的氮(N)与被废气净化用催化剂20的其它超强碱性点吸附的一氧化氮(NO)的氮(N)或在废气净化用催化剂20上移动的其它氮(N)结合后生成氮分子(N2),脱离废气净化用催化剂20。这样即可净化NOx。
然而,由于一氧化氮(NO)一吸附到超强碱性点上,该一氧化氮(NO)很快就分解。氧(O)以氧离子(O-)的形式被超强碱性点捕获,因而存在于废气净化用催化剂20上的超强碱性点会逐渐被氧离子(O-)掩埋。如上所述,超强碱性点一旦被氧离子(O-)掩埋,则废气中的一氧化氮(NO)即与吸附到超强碱性点上的一氧化氮(NO)中的氮(N)结合,结果生成N2O。
下面对废气净化用催化剂20上的一氧化氮(NO)的氧化反应加以说明。
当在稀空燃比状态下进行燃烧时,废气中含有过剩的氧(O2)。因此,被吸附到超强碱性点上的一氧化氮(N-O-)被该过剩氧(O2)所氧化,并因此形成硝酸根离子(NO3 -)。即,当废气中的氧浓度高时,反应朝生成硝酸根离子(NO3 -)的方向进行,因此在稀空燃比状态下进行燃烧时,在部分超强碱性点上生成并保持硝酸根离子(NO3 -)。而硝酸根离子(NO3 -)也可通过一氧化氮(NO)与构成晶体的氧离子(O2-)结合而生成,此外,所生成的硝酸根离子(NO3 -)有时也以吸附到构成晶体的锆(Zr4+)上的形式保持在废气净化用催化剂20之上。
但该硝酸根离子(NO3 -)一旦温度升高即分解,变为一氧化氮(NO)释放出来。因此,废气净化用催化剂20的温度一升高,则在废气净化用催化剂20上几乎不存在硝酸根离子(NO3 -)。因此,若把废气净化用催化剂20上几乎不存在硝酸根离子(NO3 -)时的废气净化用催化剂20的下限温度称为基准温度,则该基准温度由废气净化用催化剂20决定,在本发明的废气净化用催化剂20的情况下,该基准温度大体为600℃,即,当废气净化用催化剂20的温度低于该基准温度时,废气净化用催化剂20上就会生成硝酸根离子(NO3 -),当废气净化用催化剂20的温度高于该基准温度时,则废气净化用催化剂20上几乎不存在硝酸根离子(NO3 -)。
另一方面,当在稀空燃比状态下进行燃烧时,载持在废气净化用催化剂20上的铈(Ce)之类的金属被废气中含有的过剩氧(O2)氧化(Ce2O3+1/2O2→2CeO2),因而氧可储存在废气净化用催化剂20上。由于该储存的氧稳定进入了晶构之内,因而即使废气净化用催化剂20的温度上升,该储存的氧也不会从废气净化用催化剂20上脱离。
综上所述,当在稀空燃比状态下进行燃烧且废气净化用催化剂20的温度高于基准温度时,废气净化用催化剂20上的超强碱性点上保持着氧离子(O-)或尚未分解的一氧化氮(NO),此外,在废气净化用催化剂20上还保持着所储存的氧。但此时,在废气净化用催化剂20上几乎不存在硝酸根离子(NO3 -)。
相对于此,即使在稀空燃比状态下进行燃烧且废气净化用催化剂20的温度低于基准温度时,在废气净化用催化剂20上的超强碱性点上仍保持着氧离子(O-)或尚未分解的一氧化氮(NO),此外,废气净化用催化剂20上还保持着所储存的氧。但此时,在废气净化用催化剂20上生成了大量的硝酸根离子(NO3 -)。
即,换言之,当废气净化用催化剂20的温度低于基准温度时,废气中的一氧化氮(NO)在废气净化用催化剂20上变为硝酸根离子(NO3 -),因此,虽然这时在废气净化用催化剂20上存在大量的硝酸根离子(NO3 -),但是保持在废气净化用催化剂20上的氧离子(O-)相对较少。
与之相反,当废气净化用催化剂20的温度低于基准温度时,即便生成硝酸根离子(NO3 -)也会马上分解,因而废气净化用催化剂20上几乎不存在硝酸根离子(NO3 -)。此外,由于这时吸附到废气净化用催化剂20的超强碱性点上的一氧化氮(NO)分解作用活跃,因而被超强碱性点捕获的氧离子(O-)的量逐渐增加。
下面对废气净化用催化剂20的NOx净化性能的恢复处理过程加以说明。由于该恢复处理随着废气净化用催化剂20的温度而变化,因而首先对废气净化用催化剂20的温度高于基准温度的情况加以说明。
当以稀空燃比状态进行燃烧且废气净化用催化剂20的温度高于基准温度时,如上所述,在废气净化用催化剂20的超强碱性点上可保持被分解的氧离子(O-)。因此如果在稀空燃比状态下持续燃烧,则废气净化用催化剂20的超强碱性点就会逐渐被氧离子(O-)掩埋,这样一氧化氮(NO)的超强碱性点的个数逐渐减少。其结果是NOx的净化率逐渐下降。
这时,如果使保持在超强碱性点上的氧离子(O-)脱离,即清除保持在超强碱性点上的氧离子(O-),则废气净化用催化剂20就会变为如图4(D)所示向氧缺陷提供电子(e-)的原始形式,这样可以获得很高的NOx净化率。
但是从图4(C)可知,超强碱性点位于电性为正的金属离子之间,因而电性为负的氧离子(O-)很容易保持在这些金属离子之间。但由于该氧离子(O-)和金属离子间的结合力很弱,因而氧离子(O-)处于极不稳定的状态。因此,如果将保持在超强碱性点上的氧离子中的一部分氧离子(O-)从超强碱性点上清除,则受该清除作用的激发,保持在超强碱性点上的其余氧离子(O-)也可被一并清除。但此时并不能清除储存在废气净化用催化剂20上的氧。
如上所述,虽然对于在部分氧离子的清除作用激发下一并清除其余的氧离子(O-)的机理尚不明了,但可以认为是利用被清除的部分氧离子形成稳定的氧分子时所释放出的能量将其余的氧离子(O-)一并清除掉的。事实上,已通过实验证明了,通过向废气净化用催化剂20提供将保持在废气净化用催化剂20上的氧离子(O-)从废气净化用催化剂20上清除所需的能量,从而将保持在废气净化用催化剂20上的部分氧离子从废气净化用催化剂20上清除,则在该清除作用激发下将保持在废气净化用催化剂20上的其余氧离子(O-)一并从废气净化用催化剂20上清除掉。而一旦提供该能量,则促进超强碱性点上的一氧化氮(NO)的分解作用,因而也可将从吸附的一氧化氮(NO)中分解出来的氧离子(O-)清除。
即,为了清除保持在废气净化用催化剂20上的所有氧离子(O-),由于并不需要提供清除所有氧离子(O-)所需的能量,而只需要提供清除所有氧离子(O-)中的一部分氧离子(O-)所需的能量即可,因而优点在于,可减少用于清除氧离子(O-)的能量。
另外,作为应提供的能量有各种能量。例如若提高废气的温度或废气净化用催化剂20的温度,则可以清除保持在废气净化用催化剂20上的氧离子(O-)。因此,作为可提供的能量,可使用热能。
废气净化用催化剂20的温度越高,保持在废气净化用催化剂20上的氧离子(O-)越容易脱离。因而如图5所示,将保持在废气净化用催化剂20上的一部分氧离子(O-)从废气净化用催化剂20上清除所需的能量E,随着废气净化用催化剂20的温度TC升高而减少。
如上所述,当废气净化用催化剂20的温度高于基准温度时,若在稀空燃比状态下继续燃烧,则废气净化用催化剂20上的超强碱性点会逐渐被氧离子(O-)掩埋,这样一来一氧化氮(NO)可吸附的超强碱性点的数量会逐渐减少。其结果是,NOx的净化率逐渐下降。因此,周期性地向废气净化用催化剂20提供能量,以便在废气净化用催化剂20被氧离子(O-)掩埋之前将保持在废气净化用催化剂20上的氧离子(O-)从废气净化用催化剂20上清除掉。
在此情况下,可每隔一定时间或内燃机的转速的累计值每超过设定值时,或车辆的行驶距离每超过一定距离时即提供能量。此外,在此情况下,可以随着废气净化用催化剂20的温度升高而加大向废气净化用催化剂20提高能量之后到下次提供能量间的时间间隔。
此外,还可推算出保持在废气净化用催化剂20上的氧离子(O-)和一氧化氮(NO)的总量,当该推算总量超过设定量时提供能量。即,废气中包含的一氧化氮(NO)以原有形式或分解后的氧离子(O-)的形式保持在废气净化用催化剂20上。因此,保持在废气净化用催化剂20上的氧离子(O-)和一氧化氮(NO)的总量为废气中所含的一氧化氮(NO)的累计量。而废气中所含的一氧化氮(NO)的量取决于内燃机的运转状态,在图6中作为内燃机负载L和内燃机转速N的函数,以图像形式表示出通过实验求出的每单位时间内由内燃机排出的一氧化氮量Q(NO)。
当使用上述图像的情况下,保持在废气净化用催化剂20上的氧离子(O-)和一氧化氮(NO)的总量可根据图6所示的一氧化氮量Q(NO)的累计值推算出。因此在本发明中,把图6所示的一氧化氮量Q(NO)的累计值作为保持在废气净化用催化剂20上的氧离子(O-)和一氧化氮(NO)的推算总量使用。
图7示出废气净化用催化剂20的温度高于基准温度时的图6所示的Q(NO)的累计值∑Q、废气净化用催化剂20的温度TC以及所提供的能量之间的关系。
从图7可知,当保持在废气净化用催化剂20上的氧离子(O-)和一氧化氮(NO)的累计量∑Q超过设定量QX时则提供能量。这时即可将保持在废气净化用催化剂20上的氧离子(O-)清除掉。此外,一提供能量,则促进吸附在废气净化用催化剂20上的一氧化氮(NO)的分解作用,这时分解后的氧离子(O-)也可被清除掉。此外如上所述,由于废气净化用催化剂20的温度越高,提供能量时氧离子(O-)越容易被清除,因此当保持在废气净化用催化剂20上的氧离子(O-)的数量相同的情况下,废气净化用催化剂20的温度越高,清除所有氧离子(O-)所需的能量越少。因此如图7所示,提供给废气净化用催化剂20的能量的量,随着废气净化用催化剂20的温度TC升高而减少。
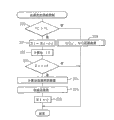
图8示出能量的供给控制过程。
从图8可知,首先在步骤100中根据图6所示的图像计算出一氧化氮量Q(NO)。接着在步骤101中通过在∑Q上累加Q(NO)而计算出累计量∑Q。接着在步骤102中判断累计量∑Q是否超过设定量QX,当∑Q>QX时进入步骤103,计算出应提供的能量。接着在步骤104中进行提供能量的处理,接着在步骤105中将∑Q清零。
下面对第2实施例加以说明,其中,利用提供给燃烧室5内或废气中的还原剂生成应提供的能量,在稀空燃比状态下进行燃烧,且废气净化用催化剂20温度高于由废气净化用催化剂20决定的基准温度时,在从废气净化用催化剂20上清除保持在废气净化用催化剂20上的氧离子(O-)时,通过向燃烧置5内或废气中提供还原剂,以尖峰状加浓燃烧室5内的空燃比或废气的空燃比。
在此情况下,可以通过提供还原剂,以周期性加浓燃烧室5内的空燃比或废气的空燃比,例如每隔一定时间,或内燃机的转速的累计值每超过设定值时,或车辆的行驶距离每超过一定距离时即加浓空燃比。
另外,在该第2实施例中也可根据保持在废气净化用催化剂20上的氧离子(O-)和一氧化氮(NO)的累计总量进行空燃比的加浓控制。
图9示出进行此种加浓控制时的情况。
即,如图9所示,当保持在废气净化用催化剂20上的氧离子(O-)和一氧化氮(NO)的累加总量∑Q超过设定量QX时,则通过向燃烧室5内或废气中提供还原剂而以尖峰状加浓燃烧室5内或废气的空燃比A/F,这样即可清除保持在废气净化用催化剂20上的氧离子(O-)。
在该实施例中,作为还原剂使用了含碳氢化合物等的燃料,在此情况下,作为还原剂起作用的燃料是相对于理论空燃比过剩的燃料部分。即,就图9而言,以剖面线表示的浓于理论空燃比一侧的部分表示还原剂量Qr。该还原剂可以通过增加燃料喷射阀6的喷射量而提供到燃烧室5内,也可以提供给从燃烧室5排出的废气中。
当在稀空燃比状态下进行燃烧,且废气净化用催化剂20的温度高于由废气净化用催化剂20决定的基准温度时,若向废气净化用催化剂20提供使保持在废气净化用催化剂20上的部分氧脱离废气净化用催化剂20所需的还原剂,则可将保持在废气净化用催化剂20上的其余氧从废气净化用催化剂20上清除掉。参照图10更为详细地对此时的现象进行说明。
图10示出当把流入废气净化用催化剂20的废气的空燃比A/F维持在稀空燃比状态下时和以尖峰状加浓时从废气净化用催化剂20流出的废气中的氧浓度(%)的变化和NOx浓度(p.p.m.)的变化。
如上所述,当通过在稀空燃比状态下进行燃烧,废气净化用催化剂20的温度高于基准温度时,废气净化用催化剂20上保持着氧离子(O-)和一氧化氮(NO),此外,废气净化用催化剂20上还保持着所储存的氧。但在废气净化用催化剂20上却几乎不存在硝酸根离子(NO3 -)。
若在此状态下将空燃比由稀切换为浓,则保持在废气净化用催化剂20上的一部分氧离子(O-)就会脱离超强碱性点,在这些氧离子(O-)的脱离作用的激发下,其余的氧离子(O-)也会一并脱离。一般而言,虽然在空燃比A/F浓的情况下废气中仍含有未燃氧,但是如果忽略此种未燃氧,则空燃比A/F由稀切换为浓时,如果是通常的催化剂,则从催化剂中流出的废气的氧浓度为零。
然而本发明中所使用的废气净化用催化剂20,由于将空燃比A/F由稀切换为浓时,保持在废气净化用催化剂20上的氧离子(O-)脱离,因而此时从废气净化用催化剂20流出的废气中的氧浓度如图10所示,受脱离的氧离子(O-)的影响并不为零。即,当将空燃比A/F由稀切换为浓时,虽然脱离的一部分氧离子(O-)被还原,但脱离的大部分氧离子(O-)并未被还原剂还原,而是以氧分子(O2)的形式从废气净化用催化剂20中排出,因而如图10所示,当把空燃比A/F从稀切换为浓时,从废气净化用催化剂20中流出的废气中的氧浓度为一定量。接着,由于脱离的氧离子(O-)的量随着时间变少,其氧浓度如图10所示逐渐减少到零,一旦减少到零,在之后的空燃比A/F处于浓空燃比的期间内,氧浓度维持在零上。
另一方面,当将空燃比A/F由稀切换为浓时,保持在废气净化用催化剂20的超强碱性点上的部分一氧化氮(NO)被分解,分解后的氧离子(O-)脱离。此外,此时其余的一氧化氮(NO)被还原剂所还原,分解为氮和二氧化碳,另外,储存在废气净化用催化剂20中的氧(O2 -)也被还原剂还原。因而如图10所示,在空燃比处于浓空燃比的期间内,从废气净化用催化剂20中流出的废气中的NOx浓度为零。
如上所述,一提供还原剂即可将一部分氧离子(O-)从废气净化用催化剂20上清除,在该清除作用的激发下,也可将保持在废气净化用催化剂20上的其余氧离子(O-)从废气净化用催化剂20上一并清除。此外,一提供还原剂,即可将吸附在废气净化用催化剂20上的一氧化氮(NO)还原。因而可以说利用还原剂生成应提供的能量极其理想。
图11表示以当量比表示的还原剂量Qr与废气净化用催化剂20的温度TC之间的关系,用于为了恢复废气净化用催化剂20的净化性能而加浓空燃比时。其中,在利用还原剂还原一氧化氮(NO)时,将还原从暂时加浓燃烧室5内的空燃比或废气的空燃比后到下次加浓燃烧室5内的空燃比或废气的空燃比的期间内所生成的一氧化氮(NO)所需的还原剂量,称为还原剂/NO的当量比为1的还原剂量Qr。换言之,假设当废气中的一氧化氮(NO)全部以硝酸根离子(NO3 -)的形式被废气净化用催化剂20所吸收时,将理论上还原该被吸收的硝酸根离子(NO3 -)所需的还原剂的量Qr称为当量比=1的还原剂量。
从图11可知,当废气净化用催化剂20的温度TC高于基准温度Ts时,还原剂量的当量比小于1.0。换言之,当废气净化用催化剂20的温度TC高于基准温度Ts时,为了清除保持在废气净化用催化剂20上的氧离子(O-)而加浓燃烧室内5内的空燃比或废气的空燃比时的还原剂量Qr少于还原从上次加浓燃烧室5内的空燃比或废气的空燃比到此次加浓燃烧室5内的空燃比或废气的空燃比的期间内所生成的一氧化氮(NO)所需的还原剂量,即少于当量比为1.0的还原剂量。
在使用本发明的废气净化用催化剂的实施例中,可在废气净化用催化剂20的温度TC为1000℃左右的高温下净化NOx,当在废气净化用催化剂20的温度TC为1000℃左右的高温下加浓空燃比时,提供当量比为1.0以下的量的还原剂即可恢复废气净化用催化剂20的净化性能。即,通过提供少于还原进入废气净化用催化剂20中的一氧化氮(NO)所需量的还原剂即可恢复废气净化用催化剂20的NOx净化性能,因而可减少用于恢复NOx净化性能的燃料消耗量。
因此,从图11可知,加浓空燃比时应提供的还原剂的量Qr,当废气净化用催化剂20的温度TC为800℃左右时,只需要还原流入废气净化用催化剂20的废气中所含的一氧化氮(NO)所需的还原剂量的三分之一左右,即当量比为1.0的还原剂量的三分之一左右,当废气净化用催化剂20的温度TC为900℃左右时只需要还原流入废气净化用催化剂20的废气中所含的一氧化氮(NO)所需还原剂量的四分之一左右,当废气净化用催化剂20的温度TC为1000℃左右时,只需要还原流入吸收分解催化剂20的废气中所含的一氧化氮(NO)所需还原剂量的六分之一左右。即,从图9及图11可知,为了清除保持在废气净化用催化剂20上的氧离子(O-)而提供的还原剂量Qr可随着废气净化用催化剂20的温度TC升高而减少。
另一方面,当废气净化用催化剂20的温度TC低于基准温度Ts时,如图10所示加浓空燃比时应提供的还原剂的量Qr为当量比为1.0以上的还原剂量。即,如上文所述,当在稀空燃比状态下进行燃烧且废气净化用催化剂20的温度TC低于基准温度Ts时,在废气净化用催化剂20的超强碱性点上仍保持着氧离子(O-)和一氧化氮(NO),此外,在废气净化用催化剂20上仍保持着储存的氧。不过,这时废气中的一氧化氮(NO)在废气净化用催化剂20上变成硝酸根离子(NO3 -),因此,废气净化用催化剂20上有大量硝酸根离子(NO3 -),而保持在废气净化用催化剂20上的氧离子(O-)和一氧化氮(NO)很少。即,当废气净化用催化剂20的温度TC低于基准温度Ts时,废气中的一氧化氮(NO)中的大部分以硝酸根离子(NO3 -)的形式被废气净化用催化剂20所吸收,因而,可净化废气中的NOx。
这时若加浓空燃比,则可将废气净化用催化剂20所吸收的硝酸根离子(NO3 -)以及一氧化氮(NO)还原。然而由于此时采用还原剂进行的硝酸根离子(NO3 -)的还原效率并非100%,因而为了还原废气净化用催化剂20所吸收的硝酸根离子(NO3 -)需要比还原废气净化用催化剂20吸收的硝酸根离子(NO3 -)以及一氧化氮(NO)所需的还原剂量还要多的还原剂。因而如上所述,加浓空燃比时提供的还原剂量Qr可设定为当量比为1.0以上的还原剂量。
即使在废气净化用催化剂20的温度TC低于基准温度Ts时仍可累加根据图6所示的图像计算出的一氧化氮量Q(NO),如图12所示,当该累计量∑Q(NO)超出允许量MAX时,可暂时加浓空燃比A/F。这样即可恢复废气净化用催化剂20的净化性能。比较图12及图9可知,此时的还原剂量Qr与图9所示的情况相比增多了许多。此外,还可看出,此时的还原剂Qr并不依赖于废气净化用催化剂20的温度TC。
图13表示还原剂的供给控制过程。
参照图13,首先在步骤200中判断废气净化用催化剂20上的温度TC是否高于基准温度Ts。当TC>Ts时则进入步骤201,清除保持在废气净化用催化剂20上的氧。即在步骤201中根据图6所示的图像计算出一氧化氮量Q(NO)。接着在步骤203中通过将Q(NO)累加到∑Q上而计算出累计量∑Q。接着在步骤204中判断累计量∑Q是否超过设定量QX,当∑Q>QX时则进入步骤205,计算出应提供的还原剂量。接着在步骤206中通过提供还原剂进行处理以加浓空燃比,然后在步骤207中将∑Q清零。
另一方面,当在步骤200中判断为TC≤Ts时,则进入步骤209,进行NO还原处理,以还原废气净化用催化剂20所吸收的硝酸根离子(NO3 -)以及一氧化氮(NO)。该NO还原处理如图14所示。参照图14,首先在步骤210中根据图6所示的图像累加一氧化氮量Q(NO),接着在步骤211中通过将Q(NO)累加到∑Q(NO)上而计算出累计量∑Q(NO)。接着在步骤212中判断累计量∑Q(NO)是否超过允许量(MAX),当∑Q(NO)>MAX时进入步骤213,计算出应提供的还原剂量。接着,通过在步骤214中供给还原剂来进行加浓空燃比处理,接着在步骤215中将∑Q(NO)清零。
如上文所述,当废气净化用催化剂20的温度TC高于基准温度Ts时,可根据废气净化用催化剂20的温度TC的升高程度相应减少加浓空燃比时的还原剂量Qr。这意味着,在还原剂量Qr大体恒定的情况下,可根据废气净化用催化剂20的温度TC的升高程度相应延长加浓空燃比之后到下次加浓空燃比为止的时间间隔。
因而如图15所示,在采用本发明的废气净化用催化剂的第3实施例中,将为了清除保持在废气净化用催化剂20上的氧离子(O-)而加浓燃烧室5内的空燃比或废气的空燃比之后到下次加浓燃烧室5内的空燃比或废气的空燃比的时间间隔tX随着废气净化用催化剂20的温度TC的升高程度相应增大。
图16表示用于实行第3实施例的还原剂的供给控制过程。
参照图16,首先在步骤220中判断废气净化用催化剂20的温度TC是否高于基准温度Ts。当TC>Ts时进入步骤221,将从上次处理循环到此次处理循环的时间Δt累加到∑t上,由此计算出经过时间∑t。接着在步骤222中根据图13计算出目标经过时间tX。接着在步骤223中判断经过时间∑t是否超过目标经过时间tX,当∑t>tX时进入步骤224,计算出应提供的还原剂量。接着在步骤225中通过提供还原剂进行加浓空燃比的处理,接着在步骤226中将∑t清零。
另一方面,当在步骤220中判断为TC≤Ts时,进入步骤208,实施图14所示的NO还原处理。
图17示出第4实施例。如图17所示,在该实施例中,在处于废气净化用催化剂20下游的排气管43内配置用于检测废气净化用催化剂20的废气中的NOx浓度的NOx浓度传感器44。
在废气净化用催化剂20的超强碱性点尚未被氧离子(O-)掩埋的期间内,由于废气中所含的NOx被废气净化用催化剂20捕获,因而从废气净化用催化剂20中流出的废气中几乎不含NOx。然而,一旦废气净化用催化剂20的超强碱性点中有相当一部分被氧离子(O-)掩埋,则未被废气净化用催化剂20捕获,而直接通过废气净化用催化剂20的NOx量会逐渐增大。因而在该第4实施例中,当从废气净化用催化剂20中流出的废气中的NOx浓度超过允许值时,则判断为超强碱性点有相当一部分己被氧离子(O-)掩埋,使流入废气净化用催化剂20的废气的空燃比以尖峰状由稀变浓。
图18表示用于实施该第4实施例的还原剂的供给控制过程。
参照图18,首先在步骤230中利用NOx浓度传感器44检测出从废气净化用催化剂20中流出的废气中的NOx浓度De。接着在步骤231中判断NOx浓度传感器44所检测出的NOx浓度De是否大于允许值DX。当De≤DX时结束该处理循环。与之相反,当De>DX时,则进入步骤232,判断废气净化用催化剂20的温度TC是否高于基准温度Ts。当TC>Ts时进入步骤233,计算出应提供的还原剂量。接着在步骤234中通过提供还原剂进行加浓空燃比的处理。此时提供的还原剂的量少于当量比=1时。
另一方面,当在步骤232中判断出TC≤Ts时,则进入步骤235,计算出应提供的还原剂量。接着,在步骤236中通过提供还原剂进行加浓空燃比的处理。此时提供的还原剂的量多于当量比=1时。
图19示出另一实施例。在本实施例中,如虚线所示,将废气净化用催化剂50载持在汽缸盖3的内壁面以及活塞4的顶面等燃烧室5的内壁面上,或将废气净化用催化剂51载持在废气口11的内壁面以及废气歧管19的内壁面等废气通道的内壁面上。当将废气净化用催化剂50载持在燃烧室5的内壁面上时,燃烧室5内的燃烧气体或已燃气体与废气净化用催化剂50接触,上述燃烧气体或已燃气体中所含的氮氧化物,主要是一氧化氮NO,吸附到废气净化用催化剂50上之后分解为氮(N)和氧(O),当将废气净化用催化剂51载持于废气通道的内壁面上时,从燃烧室5排出的废气与废气净化用催化剂51接触,该废气中所含的一氧化氮(NO)吸附到废气净化用催化剂51上之后分解为氮(N)和氧(O)。
在图20所示的实施例中,在处于废气净化用催化剂20上游的废气歧管19内配置还原剂供给阀52,当需要加浓废气的空燃比时,由该还原剂供给阀52向废气提供还原剂。
图21表示将本发明应用于压燃式内燃机时的情况。在图21中,以相同标号表示与图3所示的火花点火式内燃机相同的构成。在图21中,1是内燃机主体,5是各汽缸的燃烧室,6是用于分别向各个燃烧室5内喷射燃料的电控式燃料喷射阀,13a是进气歧管,19是废气歧管。进气歧管13a经由进气管道14与废气涡轮增压器53的压缩机53a的出口连接,压缩机53a的入口与空气过滤器15连接。在进气管道14内配置节气门17,并在进气管道14周围配置用于冷却在进气管道14内流动的吸入空气的冷却装置54。另一方面,废气歧管19与废气涡轮增压器53的废气涡轮53b的入口连接,废气涡轮53b的出口与内置有废气净化用催化剂20的催化转化器21连接。在废气歧管19的集合部出口上配置还原剂供给阀55,为了加浓废气的空燃比而提供由碳氢化合物之类构成的还原剂。废气歧管19和进气歧管13a经由EGR通道22彼此连接,在EGR通道22内配置电控式EGR控制阀23。而且,在EGR通道22周围配置用于冷却在EGR通道22内流动的EGR气体的冷却装置24。另外,各燃料喷射阀6经由燃料供给管25与共轨26连接,由排出量可变的电控式燃料泵27向该共轨26内提供燃料。
在该压燃式内燃机中,在稀空燃比状态下持续进行燃烧,当为了恢复废气净化用催化剂20的净化性能而使废气的空燃比以周期性尖峰状加浓时,由还原剂供给阀55向废气中提供还原剂。
而在该压燃式内燃机中,当废气净化用催化剂20的温度TC高于由废气净化用催化剂20决定的基准温度Ts时,周期性提供的还原剂的量少于还原从上次提供还原剂之后到此次提供还原剂的期间内流入废气净化用催化剂20的NOx所需的还原剂量;当废气净化用催化剂20的温度TC低于由废气净化用催化剂20决定的基准温度Ts时,周期性提供的还原剂的量多于还原从上次提供还原剂之后到此次提供还原剂的期间内流入废气净化用催化剂20的NOx所需的还原剂量。
下面对图21所示的实施例加以说明,其中,配置微粒过滤器以代替废气净化用催化剂20,在该微粒过滤器上形成废气净化用催化剂层。
图22(A)及图22(B)示出该微粒过滤器的结构。图22(A)是微粒过滤器的主视图,图22(B)是微粒过滤器的侧剖视图。如图21A及图21B所示,微粒过滤器具有蜂窝式结构,具有彼此平行延伸的多个废气通道60、61。这些废气通道包括:下游端被旋塞62封闭的废气流入通道60和上游端被旋塞63封闭的废气流出通道61。图22(A)中带剖面线的部分表示旋塞63。因此废气流入通道60和废气流出通道61彼此通过薄壁的隔板64交替配置。换言之,配置废气流入通道60和废气流出通道61,使得各个废气流入通道60被4个废气流出通道61包围,各个废气流出通道61同样被4个废气流入通道60包围。
微粒过滤器例如由堇青石之类的多孔质材料形成,因此,如图22(B)中箭头所示,流入到废气流入通道60内的废气穿过周围的隔板64后,从相邻的废气流出通道61中流出。在该实施例中,在各废气流入通道60以及各废气流出通道61的周围壁面,即各隔板64的两侧表面上以及隔板64内的微孔内壁面上均形成废气净化用催化剂层。
在该实施例中,当需要恢复废气净化用催化剂的NOx净化性能时,同样可加浓废气的空燃比。而且,在该实施例中,废气中所含的微粒被微粒过滤器捕获,可利用废气的热量使被捕获的微粒依次燃烧。当有大量的微粒堆积在微粒过滤器上时,则提供还原剂并使废气升温,由此使堆积的微粒点火燃烧。
下面说明一种低温燃烧法,该低温燃烧法应用于在压燃式内燃机中为了恢复废气净化用催化剂的NOx净化性能而加浓燃烧室内的空燃比时。
在图21所示的压燃式内燃机中,若增大EGR率(EGR气体量/(EGR气体量+吸入空气量)),则烟雾的发生量逐渐增大并达到峰值,若进一步提高EGR率,则烟雾的发生量反而急剧下降。参照图23进行说明,图23表示EGR气体的冷却程度改变时的EGR率与烟雾之间的关系。而图23中的曲线A表示强力冷却EGR气体并将EGR气体温度维持在90℃左右时的情况,曲线B表示利用小型冷却装置来冷却EGR气体时的情况,曲线C表示未强制冷却EGR气体时的情况。
如图23的曲线A所示,在强力冷却EGR气体的情况下,在EGR率稍微低于50%的位置上,烟雾发生量达到峰值;这时若将EGR率大致设定在55%以上,则几乎不产生烟雾。另外,如图23的曲线B所示,在稍微冷却EGR气体的情况下,在EGR率稍高于50%的位置上烟雾发生量达到峰值;这时若将EGR率大致设定在65%以上,则几乎不产生烟雾。此外,如图23的曲线C所示,在未强制冷却EGR气体的情况下,在EGR率为55%的位置附近,烟雾发生量达到峰值;此时若将EGR率大致设定在70%以上,则几乎不产生烟雾。
如上所述,若将EGR率设定为55%以上则几乎不产生烟雾,其原因在于,由于EGR气体的吸热作用,燃烧时的燃料及周围的气体温度并不太高,即,进行低温燃烧,其结果是,碳氢化合物未能生成煤烟。
该低温燃烧具有下述特征:与空燃比无关,可以抑制烟雾产生,并可以减少NOx的产生量。即,当加浓空燃比时虽然燃料过剩,但由于将燃烧温度控制在低温,因而过剩的燃料不产生煤烟,所以不会产生烟雾。而且,这时,NOx的产生量也极少。另外,当平均空燃比稀薄时,或者当空燃比为理论空燃比时,虽然随着燃烧温度的升高而产生少量的煤烟,但是由于在低温燃烧下可以将燃烧温度控制在较低温度,因而完全不产生烟雾,NOx也产生极少。
另外,当进行该低温燃烧时,虽然燃料及其周围的气体温度变低,但是废气的温度却升高。参照图24(A)及图24(B)对此进行说明。
图24(A)的实线表示进行低温燃烧时的燃烧室5内的平均气体温度Tg与曲轴转角之间的关系;图24(A)的虚线表示进行正常燃烧时的燃烧室5内的平均气体温度Tg与曲轴转角之间的关系。并且,图24(B)的实线表示进行低温燃烧时的燃料及其周围的气体温度Tf与曲轴转角之间的关系;图24(B)的虚线表示进行正常燃烧时的燃料及其周围的气体温度Tf与曲轴转角之间的关系。
进行低温燃烧时与进行正常燃烧时相比,EGR气体量大,因此如图24(A)所示,在压缩上止点之前,即压缩行程期间,以实线表示的低温燃烧时的平均气体温度Tg高于以虚线表示的正常燃烧时的平均气体温度Tg。而此时,如图24(B)所示,燃料及其周围的气体温度Tf与平均气体温度Tg大体相同。
接着在压缩上止点附近开始燃烧,当在此情况下进行低温燃烧时,如图24(B)的实线所示,由于EGR气体的吸热作用,燃料及其周围的气体温度Tf并不太高。与之相反,在进行正常燃烧的情况下,由于燃料周围存在大量氧,所以如图24(B)的虚线所示,燃料及其周围的气体温度Tf极高。如上所述,虽然正常燃烧时燃料及其周围的气体温度Tf比低温燃烧时高出许多,但占大部分的其余气体的温度反而是正常燃烧时低于低温燃烧时,因此如图24(A)所示,压缩上止点附近的燃烧室5内的平均气体温度Tg,在低温燃烧时高于正常燃烧时。其结果是,如图24(A)所示,燃烧结束后的燃烧室5内的已燃气体温度,在低温燃烧时高于正常燃烧时,因而在进行低温燃烧时废气温度上升。
不过,若内燃机的要求转矩TQ变高,即燃料喷射量增加,则由于燃烧时的燃料及周围的气体温度升高,因而很难进行低温燃烧。即,低温燃烧仅限于燃烧产生的发热量较少的内燃机中低负载运转时。
在图25中,区域I表示可以进行第1燃烧即低温燃烧的运转区域,其中所述第1燃烧为,燃烧室5的非活性气体量多于煤烟产生量达到峰值时的非活性气体量;区域II表示只能进行第2燃烧即正常燃烧的运转区域,其中所述第2燃烧为,燃烧室的非活性气体量少于煤烟产生量达到峰值时的非活性气体量。
图26示出在运转区域I中进行低温燃烧时的目标空燃比A/F,图27示出在运转区域I中进行低温燃烧时与要求转矩TQ对应的节气门17的开度、EGR控制阀23的开度、EGR率、空燃比、喷射开始时间θS、喷射结束时间θE以及喷射量。另外,在图27中还一并表示出在运转区域II内进行正常燃烧时的节气门17的开度等。
从图26及图27可知,当在运转区域I内进行低温燃烧时,可以将EGR率设定在55%以上,空燃比A/F可设定为15.5到18左右的稀空燃比。如上述,在运转区域I内进行低温燃烧时,即使加浓空燃比也几乎不产生烟雾。
如上所述,当进行低温燃烧时,可以加浓空燃而几乎不产生烟雾。因此,当为恢复废气净化用催化剂的NOx净化作用而需要加浓空燃比时,可进行低温燃烧,从而在低温燃烧状态下加浓空燃比。
此外,如上所述,当进行低温燃烧时,则废气温度上升。因而,当为了使堆积的微粒点火燃烧而需要升高废气温度时,也可以进行低温燃烧。
实施例
实施例1
在容积为3L的烧杯中制作表面活性剂溶液,在其中滴下将0.03摩尔硝酸镧溶解到蒸馏水140份中的水溶液,并进行搅拌,调制微乳剂液。接着,滴下将0.12摩尔四正丁氧基锆溶解到环己烷200份中的溶液,进行四正丁氧基锆的水解。立刻生成白色的沉淀物。其后,为了控制沉淀的凝集,利用氨水将pH值调节到8.5。其后连续搅拌1小时,进行生成物的熟化。过滤母液,将得到的沉淀在乙醇中清洗三次,在80℃的状态下干燥一晚后,在空气中以200℃烧成2小时,得到包括镧和锆的复合氧化物(镧锆氧化物)。该复合氧化物的La/Zr的摩尔比是1/4。
通过X线衍射法测量这样得到的镧锆氧化物,求出(111)面的面间隔。其结果如图28所示。并且,为了进行比较,图28还是出了以往方法的共沉淀法和醇盐法所制造的同样的镧锆氧化物的对应数据。
在图28中,实线所示直线是连接在ZrO2(La含有率为0)和LaZrO3.5(La含有率50%)的组成中的理论晶格的(111)面间隔的值之间的直线,表示各组成(La含有率)的计算上的面间隔。共沉淀法或以往的醇盐法得到的是比理论值短的晶格常数,表示大部分La没有置换到ZrO2晶格。对此,本发明的镧锆氧化物是完全按照理论值的面间隔,La3+离子几乎完全置换到ZrO2晶格。
实施例2
其次,对于实施例1中制造的镧锆氧化物,利用普通方法涂布在单片基板上,并使其载持1重量%的铂,并载持与镧同摩尔数的铯作为碱金属,得到本发明的废气净化用催化剂。并且,作为比较,使用通过共沉淀法和醇盐法得到的镧锆氧化物,并同样使其载持铂和铯。
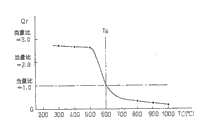
对于这些催化剂的高温NOX吸收性能进行讨论。实验是在600℃的还原气氛中还原催化剂后,在NO714ppm+O23%/N2平衡导通下,以20℃/分的速度从750℃降温到100℃,测量NOX的降低率。结果如图29所示。
如图29所示,很明显,利用以往的催化剂时,在700℃以上时NOx的保持能力突然消失,相对于此,利用本发明的催化剂时,即使在1000℃的高温也能保持NOx。
Claims (9)
1.一种废气净化用催化剂,在结晶性的锆复合氧化物上载持碱金属和贵金属,其特征在于,上述锆复合氧化物被选自三价的稀土金属的至少一种元素置换一部分锆,通过该元素置换,晶格的延伸大致变为理论值。
2.如权利要求1所述的废气净化用催化剂,所述选自三价的稀土金属的至少一种元素,以锆复合氧化物中的所有金属元素的总摩尔数为基准,存在5~50摩尔%。
3.如权利要求1所述的废气净化用催化剂,一部分锆被镧置换。
4.如权利要求1所述的废气净化用催化剂,锆复合氧化物上载持的所述碱金属是铯。
5.如权利要求1所述的废气净化用催化剂,锆复合氧化物上载持的所述贵金属是铂。
6.一种权利要求1所述的废气净化用催化剂的制造方法,制造在结晶性的锆复合氧化物上载持碱金属和贵金属的废气净化用催化剂,其特征在于,通过使水解而生成锆的氢氧化物的有机化合物溶解后的有机相,与将选自三价的稀土金属的第二元素作为离子包括在内的水相接触,在通过其界面的锆有机化合物的水解反应来生成锆的氢氧化物的过程中,向该生成物中引入第二元素,烧成得到的复合氢氧化物,得到锆和第二元素的复合氧化物,进而使其载持碱金属和贵金属。
7.根据权利要求6所述的废气净化用催化剂的制造方法,所述水解而生成锆的氢氧化物的有机化合物是选自锆醇盐、乙酰丙酮锆络合物中的一种。
8.根据权利要求6所述的废气净化用催化剂的制造方法,所述水解而生成锆的氢氧化物的有机化合物是四正丁氧基锆。
9.根据权利要求6所述的废气净化用催化剂的制造方法,所述第二元素为镧。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP364852/2003 | 2003-10-24 | ||
| JP2003364852A JP4120559B2 (ja) | 2003-10-24 | 2003-10-24 | 排気ガス浄化用触媒 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1886194A CN1886194A (zh) | 2006-12-27 |
| CN100586559C true CN100586559C (zh) | 2010-02-03 |
Family
ID=34510134
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN200480034776A Expired - Fee Related CN100586559C (zh) | 2003-10-24 | 2004-10-14 | 废气净化用催化剂 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20070066479A1 (zh) |
| EP (1) | EP1681096B1 (zh) |
| JP (1) | JP4120559B2 (zh) |
| CN (1) | CN100586559C (zh) |
| WO (1) | WO2005039759A1 (zh) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4325648B2 (ja) * | 2005-10-24 | 2009-09-02 | トヨタ自動車株式会社 | 触媒担体及び排ガス浄化用触媒 |
| JP4881758B2 (ja) | 2006-04-28 | 2012-02-22 | 日産自動車株式会社 | 排気ガス浄化用触媒及びその製造方法 |
| EP1941945B1 (en) | 2007-01-05 | 2012-01-18 | Kabushiki Kaisha Toyota Chuo Kenkyusho | Catalyst for removing particulate matter and method using the same for removing particulate matter |
| US20090264283A1 (en) * | 2008-04-16 | 2009-10-22 | Basf Catalysts Llc | Stabilized Iridium and Ruthenium Catalysts |
| FR2939695B1 (fr) * | 2008-12-17 | 2011-12-30 | Saint Gobain Ct Recherches | Structure de purification incorporant un systeme de catalyse supporte par une zircone a l'etat reduit. |
| US8003567B2 (en) * | 2009-08-17 | 2011-08-23 | Honda Motor Co., Ltd. | Nanocomposite support materials |
| KR102454125B1 (ko) * | 2015-03-20 | 2022-10-14 | 토프쉐 에이/에스 | 촉매화된 세라믹 캔들 필터 및 공정 오프가스 또는 배기가스의 정화 방법 |
| US11149617B2 (en) * | 2016-08-19 | 2021-10-19 | Kohler Co. | System and method for low CO emission engine |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003001109A (ja) * | 2001-06-26 | 2003-01-07 | Daihatsu Motor Co Ltd | 排ガス浄化用触媒 |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02175602A (ja) | 1988-12-28 | 1990-07-06 | Ricoh Co Ltd | 超微粒子状金属酸化物組成物の製法及びそれによって得られた超微粒子状酸化ジルコニウム組成物 |
| US5747410A (en) * | 1992-07-03 | 1998-05-05 | Kabushiki Kaisha Riken | Exhaust gas cleaner and method of cleaning exhaust gas |
| JPH06178937A (ja) * | 1992-10-15 | 1994-06-28 | Riken Corp | 窒素酸化物除去触媒及び除去方法 |
| JP4098835B2 (ja) * | 1993-12-07 | 2008-06-11 | トヨタ自動車株式会社 | 排気ガス浄化用触媒 |
| JPH08281106A (ja) * | 1995-04-11 | 1996-10-29 | Nissan Motor Co Ltd | 排気ガス浄化用触媒及びその製造方法 |
| JPH0924274A (ja) * | 1995-05-09 | 1997-01-28 | Hitachi Ltd | 排ガス浄化触媒及び排ガス浄化システム |
| JP3498453B2 (ja) * | 1995-11-27 | 2004-02-16 | 日産自動車株式会社 | 排気ガス浄化用触媒及びその製造方法 |
| JP4053623B2 (ja) | 1996-12-27 | 2008-02-27 | 阿南化成株式会社 | ジルコニウム−セリウム系複合酸化物及びその製造方法 |
| JP3466856B2 (ja) * | 1997-02-05 | 2003-11-17 | トヨタ自動車株式会社 | 排ガス浄化触媒およびその製造方法 |
| JP2001170487A (ja) * | 1999-12-15 | 2001-06-26 | Toyota Central Res & Dev Lab Inc | 排ガス浄化用触媒および排ガス浄化方法 |
| EP1020223A3 (en) * | 1999-01-12 | 2001-09-12 | Kabushiki Kaisha Toyota Chuo Kenkyusho | Porous material and production process thereof, catalyst comprising the porous material and process for purifying exhaust gas |
| EP1172139B1 (en) * | 2000-07-14 | 2006-10-11 | Kabushiki Kaisha Toyota Chuo Kenkyusho | Catalyst for purifying exhaust gas |
| JP3758487B2 (ja) * | 2000-09-08 | 2006-03-22 | トヨタ自動車株式会社 | 吸収還元型nox浄化用触媒 |
| WO2002094716A1 (en) * | 2001-05-23 | 2002-11-28 | Svenska Rymdaktiebolaget | Sintering resistant catalyst material and a method for the preparation thereof |
| JP4228278B2 (ja) * | 2002-03-19 | 2009-02-25 | トヨタ自動車株式会社 | 排ガス浄化用触媒 |
| JP3758601B2 (ja) * | 2002-05-15 | 2006-03-22 | トヨタ自動車株式会社 | 吸蔵還元型NOx浄化用触媒 |
| JP4812233B2 (ja) * | 2003-02-28 | 2011-11-09 | トヨタ自動車株式会社 | 複合酸化物の製造方法 |
| DE602004007187T3 (de) * | 2003-05-21 | 2017-09-28 | Toyota Jidosha Kabushiki Kaisha | Verfahren zur Herstellung eines porösen Mischoxids |
-
2003
- 2003-10-24 JP JP2003364852A patent/JP4120559B2/ja not_active Expired - Fee Related
-
2004
- 2004-10-14 WO PCT/JP2004/015575 patent/WO2005039759A1/ja active Search and Examination
- 2004-10-14 US US10/576,025 patent/US20070066479A1/en not_active Abandoned
- 2004-10-14 CN CN200480034776A patent/CN100586559C/zh not_active Expired - Fee Related
- 2004-10-14 EP EP04792725A patent/EP1681096B1/en not_active Expired - Lifetime
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003001109A (ja) * | 2001-06-26 | 2003-01-07 | Daihatsu Motor Co Ltd | 排ガス浄化用触媒 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2005125254A (ja) | 2005-05-19 |
| EP1681096A1 (en) | 2006-07-19 |
| EP1681096B1 (en) | 2012-11-21 |
| US20070066479A1 (en) | 2007-03-22 |
| JP4120559B2 (ja) | 2008-07-16 |
| CN1886194A (zh) | 2006-12-27 |
| WO2005039759A1 (ja) | 2005-05-06 |
| EP1681096A4 (en) | 2010-09-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101292077B (zh) | 内燃机的排气净化装置 | |
| CN100398789C (zh) | 排气净化方法和排气净化装置 | |
| CN100420829C (zh) | 压缩点火式内燃机的废气净化装置 | |
| RU2480592C1 (ru) | Система очистки выхлопных газов двигателя внутреннего сгорания | |
| US7375054B2 (en) | Exhaust gas treatment catalyst and exhaust gas treatment method | |
| JP4270224B2 (ja) | 内燃機関の排気浄化装置 | |
| CN1676893A (zh) | 废气净化设备 | |
| CN101784764B (zh) | 用于内燃机的废气净化装置及其控制方法 | |
| CN101542085A (zh) | 内燃机的排气净化装置 | |
| CN102859136A (zh) | 内燃机的排气净化装置 | |
| KR100841427B1 (ko) | 내연기관의 배기정화시스템 | |
| KR20020024595A (ko) | 내연 기관의 배기 가스 정화장치 | |
| CN100586559C (zh) | 废气净化用催化剂 | |
| CN100449123C (zh) | 内燃机的排气净化方法及排气净化装置 | |
| CN100582454C (zh) | 内燃机的排气净化装置 | |
| US11007480B2 (en) | Exhaust gas purification system | |
| JP2003193822A (ja) | 内燃機関の排気浄化装置 | |
| JP2001336414A (ja) | 排気ガス浄化方法および排気ガス浄化装置 | |
| CN101006252A (zh) | 包括NOx吸收剂的排气系统中的还原剂的添加 | |
| CN103154454B (zh) | 内燃机的排气净化装置 | |
| CN101542086B (zh) | 内燃机的排气净化装置 | |
| JP2003049631A (ja) | 排気浄化装置 | |
| JP5142086B2 (ja) | 排気浄化システム | |
| JP2002364339A (ja) | 排気ガス浄化装置、および排気ガスの浄化方法 | |
| JP2003278529A (ja) | 内燃機関の排気浄化方法及び浄化装置 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20100203 Termination date: 20171014 |