ES2945932T3 - Pirrolobenzodiazepinas y conjugados dirigidos - Google Patents
Pirrolobenzodiazepinas y conjugados dirigidos Download PDFInfo
- Publication number
- ES2945932T3 ES2945932T3 ES18168997T ES18168997T ES2945932T3 ES 2945932 T3 ES2945932 T3 ES 2945932T3 ES 18168997 T ES18168997 T ES 18168997T ES 18168997 T ES18168997 T ES 18168997T ES 2945932 T3 ES2945932 T3 ES 2945932T3
- Authority
- ES
- Spain
- Prior art keywords
- point
- cancer
- binding
- group
- attachment
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
- A61K31/5513—1,4-Benzodiazepines, e.g. diazepam or clozapine
- A61K31/5517—1,4-Benzodiazepines, e.g. diazepam or clozapine condensed with five-membered rings having nitrogen as a ring hetero atom, e.g. imidazobenzodiazepines, triazolam
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/55—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound the modifying agent being also a pharmacologically or therapeutically active agent, i.e. the entire conjugate being a codrug
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68035—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a pyrrolobenzodiazepine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Immunology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Cell Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Peptides Or Proteins (AREA)
- Saccharide Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Un Conjugado que tiene la fórmula IV: L - (LU-D) P (IV) o una de sus sales o solvatos farmacéuticamente aceptables; en la que L es una unidad de ligando seleccionada del grupo que consiste en un anticuerpo y un fragmento de unión a antígeno de un anticuerpo, LU es una unidad enlazadora, p es de 1 a 20; y D se selecciona del grupo que consta de D1 a D6 que tiene las estructuras definidas en la reivindicación 1 y donde LU es: donde el asterisco indica el punto de unión a D, la línea ondulada indica el punto de unión a la Unidad de Ligando, Y es NH y E es ácido glucurónico; y A1 es una Unidad de Camilla. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Pirrolobenzodiazepinas y conjugados dirigidos
La presente invención se refiere a pirrolobenzodiazepinas (PBD), en particular a dímeros de pirrolobenzodiazepina que tienen un doble enlace C2-C3 y un grupo arilo en la posición C2 en cada unidad monomérica, y su inclusión en conjugados dirigidos.
Antecedentes de la invención
Algunas pirrolobenzodiazepinas (PBD) tienen la capacidad de reconocer y unirse a secuencias específicas de ADN; la secuencia preferente es PuGPu. El primer antibiótico antitumoral de PBD, antramicina, se descubrió en 1965 (Leimgruber, et al., J. Am. Chem. Soc., 87,5793-5795 (1965); Leimgruber, et al., J. Am. Chem. Soc., 87, 5791-5793 (1965)). Desde entonces, se han presentado numerosos PBD de origen natural, y se han desarrollado numerosas rutas de síntesis para obtener varios análogos (Thurston, et al., Chem. Rev. 1994, 433-465 (1994); Antonow, D. y Thurston. DE, Chem. Rev. 2011 111 (4), 2815-2864). Los miembros de la familia incluyen abeimicina (Hochlowski, et al., J. Antibiotics, 40, 145-148 (1987)), quicamicina (Konishi, et al., J. Antibiotics, 37, 200-206 (1984)), DC-81 (Patente japonesa 58-180 487; Thurston, et al., Chem. Brit., 26.767-772 (1990); Bose, et al., Tetrahedron, 48, 751 758 (1992)), mazetramicina (Kuminoto, et al., J. Antibiotics, 33, 665-667 (1980)), neotrramicinas A y B (Takeuchi, et al., J. Antibiotics, 29, 93-96 (1976)), porotramicina (Tsunakawa, et al., J. Antibiotics, 41, 1366-1373 (1988)), protracarcina (Shimizu, et al, J. Antibiotics, 29, 2492-2503 (1982); Langley y Thurston, J. Org. Chem., 52, 91-97 (1987)), sibanomicina (DC-102) (Hara, et al., J. Antibiotics, 41, 702-704 (1988); Itoh, et al., J. Antibiotics, 41, 1281 1284 (1988)), sibiromicina (Leber, et al., J. Am. Chem. Soc., 110, 2992-2993 (1988)) y tomamicina (Arima, et al., J. Antibiotics, 25, 437-444 (1972)). Las PBD tienen la estructura general:
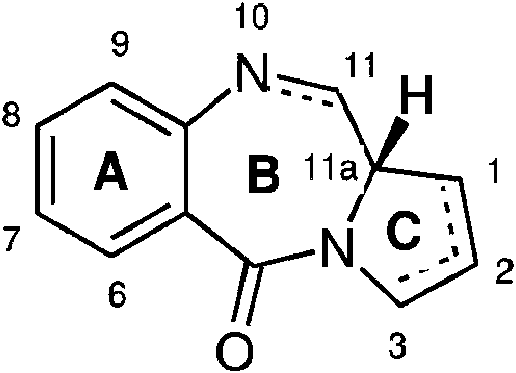
Se diferencian en el número, tipo y posición de los sustituyentes, tanto en sus anillos A aromáticos como en los anillos C de pirrolo, y en el grado de saturación del anillo C. En el anillo B hay una imina (N=C), una carbinolamina (NH-CH(OH)), o un metil éter de carbinolamina (NH-CH(OMe)) en la posición N10-C11 que es el centro electrófilo responsable de alquilar el ADN. Todos los productos naturales conocidos tienen una configuración (S) en la posición quiral C11a que les proporciona un giro levógiro cuando se ven desde el anillo C hacia el anillo A. Esto les da la forma tridimensional adecuada para la isohelicidad con el surco menor del ADN de la forma B, lo que conduce a un ajuste ceñido en el sitio de unión (Kohn, en Antibiotics III. Springer-Verlag, Nueva York, págs. 3-11 (1975); Hurley y Needham-VanDevanter, Acc. Chem. Res., 19, 230-237 (1986)). Su capacidad para formar un aducto en el surco menor les permite interferir con el procesamiento del ADN, de ahí su uso como agentes antitumorales.
Se ha divulgado anteriormente que la actividad biológica de estas moléculas se puede potenciar uniendo dos unidades PBD entre sí a través de sus funcionalidades C8/C'-hidroxilo a través de un enlazador de alquileno flexible (Bose, DS, et al., J. Am. Chem. Soc., 114, 4939-4941 (1992); Thurston, DE, et al., J. Org. Chem., 61, 8141-8147 (1996)). Se cree que los dímeros de PBD forman lesiones en el ADN selectivas de la secuencia tal como el enlace cruzado 5'-Pu-GATC-Py-3' palindrómico entre cadenas (Smellie, M., et al., Biochemistry, 42, 8232-8239 (2003); Martin, C., et al., Biochemistry, 44, 4135-4147) que se cree que es el principal responsable de su actividad biológica. Un ejemplo de un dímero de PBD, SG2000 (s JG-136):

ha entrado recientemente en ensayos clínicos de fase II en el área de oncología (Gregson, S., et al., J. Med. Chem., 44, 737-748 (2001); Alley, MC, et al., Cancer Research, 64, 6700-6706 (2004); Hartley, JA, et al., Cancer Research, 64, 6693-6699 (2004)).
Más recientemente, los presentes inventores han divulgado previamente, en el documento WO 2005/085251, compuestos de PBD dímeros que tienen sustituyentes de arilo en C2, tales como SG2202 (ZC-207):

y en el documento WO2006/111759, bisulfitos de dichos compuestos de PBD, por ejemplo SG2285 (ZC-423):
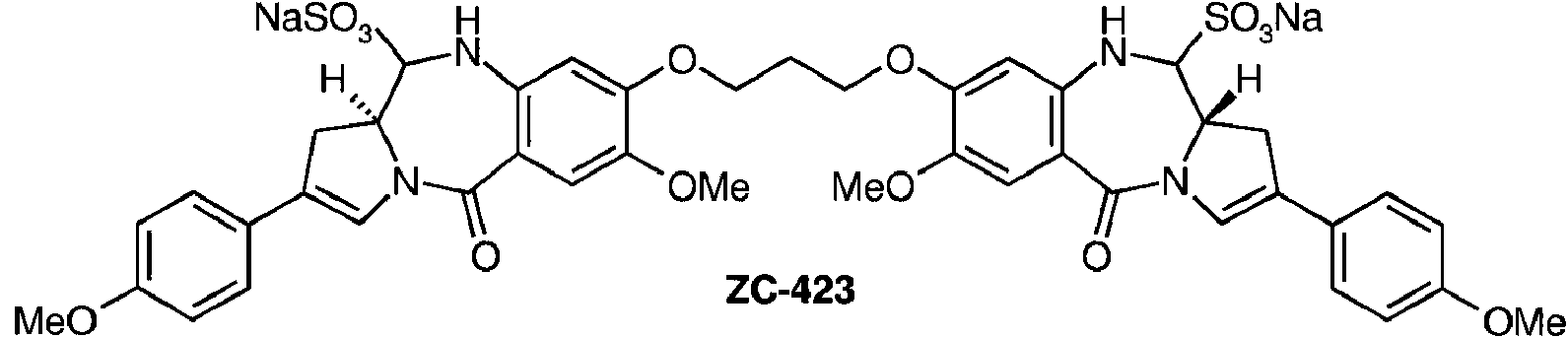
Se ha demostrado que estos compuestos son agentes citotóxicos altamente útiles (Howard, PW, et al., Bioorg. Med. Chem. (2009), 19 (22), 6463-6466, doi: 10.1016/j.bmcl.2009.09.012).
Debido a la forma en que estos compuestos altamente potentes actúan en la reticulación del ADN, estas moléculas se han fabricado simétricamente. Esto proporciona una síntesis directa, bien construyendo los restos de PBD simultáneamente que ya han formado el enlace del dímero, o haciendo reaccionar restos de PBD ya construidos con el grupo de unión del dímero.
El documento WO 2010/043880 divulga un compuesto de PBD dimérico que tiene grupos arilo en la posición C2 de cada monómero, donde uno de estos grupos arilo tiene un sustituyente diseñado para proporcionar un anclaje para unir el compuesto a otro resto. La solicitud internacional PCT/US2011/032664, en trámite junto con la presente, presentada el 15 de abril de 2011, divulga la inclusión de estos compuestos dímeros de PBD en conjugados dirigidos.
Divulgación de la invención
Los presentes inventores han desarrollado compuestos de PBD dímeros no asimétricos adicionales específicos para su inclusión en conjugados dirigidos. Estos compuestos pueden tener ventajas en su preparación y uso, particularmente en sus propiedades biológicas y la síntesis de conjugados, y las propiedades biológicas de estos conjugados.
La presente invención comprende un conjugado que tiene la fórmula IV:

o una sal farmacéuticamente aceptable o solvato del mismo;
en donde L es una unidad de ligando seleccionada entre el grupo que consiste en un anticuerpo y un fragmento de unión a antígeno de un anticuerpo,
LU es una unidad de enlazador,
p es 1 a 20 y
D se selecciona entre el grupo que consiste en D1 a D6, que tienen las estructuras:

continuación

en donde
(a) R10 es H, y R11 es OH, ORA, en donde RA es alquilo C1-4 saturado; o
(b) R10 y R11 forman un doble enlace nitrógeno-carbono entre los átomos de nitrógeno y carbono a los que están unidos; o
(c) R10 es H y R11 es SOzM, en donde z es 2 o 3 y M es un catión monovalente farmacéuticamente aceptable o ambos M juntos son un catión divalente farmacéuticamente aceptable y
el asterisco indica el punto de unión a la unidad de enlazador (LU), en donde LU es:

en donde el asterisco indica el punto de unión a D, la línea ondulada indica el punto de unión a la unidad de ligando, Y esH N y E e s ácido glucurónico y A 1 es una unidad de extensión.
Un segundo aspecto de la presente invención proporciona un conjugado del primer aspecto de la invención para su uso en el tratamiento de una enfermedad proliferativa o autoinmunitaria.
Un experto en la técnica puede determinar fácilmente si un conjugado candidato trata o no una afección proliferativa para cualquier tipo de célula particular. Por ejemplo, los ensayos que se pueden usar de manera práctica para evaluar la actividad ofrecida por un compuesto particular se describen en los ejemplos siguientes.
Los compuestos diméricos de PBD asimétricos de la presente invención se preparan según estrategias diferentes a las anteriormente utilizadas para fabricar compuesto diméricos de PBD simétricos. En particular, los presentes inventores han desarrollado un método que implica añadir cada sustituyente de C2 a un núcleo de PBD dimérico en etapas del proceso independientes.
Los Conjugados tienen la siguiente fórmula IV:
L -(LU-D)p (IV)
o una sal o solvato farmacéuticamente aceptable del mismo, en la que L es una unidad de ligando (es decir, un agente de direccionamiento), LU es una unidad de enlazador y D es una unidad de fármaco que es un dímero de PBD (véase a continuación). El subíndice p es de 1 a 20. Por consiguiente, los conjugados comprenden una unidad de ligando covalentemente unida a al menos una unidad de fármaco mediante una unidad de enlazador. La unidad de ligando, descrita con más detalle a continuación, es un agente de direccionamiento que se une a un resto diana. La unidad de ligando puede, por ejemplo, unirse específicamente a un componente celular (un agente de unión a célula) o a otras moléculas diana de interés. Por consiguiente, la presente invención también proporciona un conjugado para su uso en el tratamiento de una enfermedad proliferativa o una enfermedad autoinmunitaria. La unidad de ligando se selecciona entre un anticuerpo y un fragmento de unión a antígeno de un anticuerpo.
En los conjugados de la presente invención, el dímero de PBD, D, se selecciona entre el grupo que consiste en:


o una sal o solvato farmacéuticamente aceptable del mismo, donde R10 y R11 son como se han definido en el primer aspecto, y el asterisco indica el punto de unión a la unidad de enlazador (LU) en donde LU es:

en donde el asterisco indica el punto de unión a D, la línea ondulada indica el punto de unión a la unidad de ligando, Y es NH y E es ácido glucurónico; y A1 es una unidad de extensión.
La carga del fármaco está representada por p, el número de moléculas de fármaco por unidad de ligando (por ejemplo, un anticuerpo). La carga de fármaco puede oscilar entre 1 y 20 unidades de fármaco (D) por unidad de ligando (por ejemplo, Ab o mAb). Para las composiciones, p representa la carga promedio de fármaco de los conjugados en la composición y p oscila entre 1 y 20.
En algunas realizaciones, p es de aproximadamente 1 a aproximadamente 8 unidades de fármaco por unidad de ligando. En algunas realizaciones, p es 1. En algunas realizaciones, p es 2. En algunas realizaciones, p es de aproximadamente 2 a aproximadamente 8 unidades de fármaco por unidad de ligando. En algunas realizaciones, p es de aproximadamente 2 a aproximadamente 6, 2 a aproximadamente 5, o de 2 a aproximadamente 4 unidades de fármaco por unidad de ligando. En algunas realizaciones, p es aproximadamente 2, aproximadamente 4, aproximadamente 6 o aproximadamente 8 unidades de fármaco por unidad de ligando.
El número promedio de unidades de fármaco por unidad de ligando en una preparación procedente de una reacción de conjugación puede caracterizarse por medios convencionales tales como espectroscopia de masas, ensayo ELISA y HPLC. También se puede determinar la distribución cuantitativa de los conjugados en función de p. En algunos casos, la separación, purificación y caracterización de conjugados homogéneos, donde p tiene un valor determinado, a partir de conjugados con otras cargas de fármaco se puede conseguir por medios tales como HPLC de fase inversa o electroforesis.
En un tercer aspecto, la presente invención se refiere a compuestos de enlazador-fármaco (es decir, enlazadores de fármaco) que comprenden dímeros de PBD (véase anteriormente) unidos a una unidad de enlace. Estos enlazadores de fármaco pueden usarse como intermedios para la síntesis de conjugados que comprenden dímeros de PBD unidos a un agente de direccionamiento.
Estos enlazadores de fármaco tienen la siguiente formula V:
LU-D (V)
o una sal o solvato farmacéuticamente aceptable del mismo, en la que LU es una unidad de enlazador y D es una unidad de fármaco que es un dímero de PBD, como se define en el primer aspecto de la invención.
Figura
La Figura 1 muestra el efecto sobre el volumen tumoral medio después del tratamiento con dos conjugados no reivindicados.
Definiciones
Cationes farmacéuticamente aceptables
Ejemplos de cationes monovalentes y divalentes farmacéuticamente aceptables se tratan en Berge, et al., J. Pharm. Sci., 66, 1-19 (1977).
El catión farmacéuticamente aceptable puede ser inorgánico u orgánico.
Ejemplos de cationes monovalentes inorgánicos farmacéuticamente aceptables incluyen, pero sin limitación, iones de metales alcalinos tales como Na+ y K+. Ejemplos de cationes divalentes inorgánicos farmacéuticamente aceptables incluyen, pero sin limitación, cationes alcalinotérreos tales como Ca2+ y Mg2+. Ejemplos de cationes orgánicos farmacéuticamente aceptables incluyen, pero sin limitación, ion amonio (es decir NH4+) e iones de amonio sustituidos (por ejemplo, NH3R+, NH2R2+, NHR3+, NR4+). Ejemplos de algunos iones amonio sustituido adecuados son los procedentes de: etilamina, dietilamina, diciclohexilamina, trietilamina, butilamina, etilendiamina, etanolamina, dietanolamina, piperazina, bencilamina, fenilbencilamina, colina, meglumina y trometamina, así como aminoácidos, tales como lisina y arginina. Un ejemplo de un ion de amonio cuaternario común es N(CH3)4+.
Grupos
El término “alquilo C1-4 saturado", como se usa en el presente documento, se refiere a un resto monovalente obtenido mediante la eliminación de un átomo de hidrógeno de un átomo de carbono de un compuesto hidrocarburo que tiene de 1 a 4 átomos de carbono, que puede ser alifático o alicíclico. De manera similar, el término “alquilo C1-2 saturado", como se usa en el presente documento, se refiere a un resto monovalente obtenido mediante la eliminación de un átomo de hidrógeno de un átomo de carbono de un compuesto hidrocarburo que tiene de 1 a 2 átomos de carbono, es decir metilo o etilo.
Ejemplos de grupos alquilo saturados incluyen, pero sin limitación, metilo (C1), etilo (C2), propilo (C3), y butilo (C4).
Ejemplos de grupos alquilo lineales saturados incluyen, pero sin limitación, metilo (C1), etilo (C2), n-propilo (C3) y nbutilo (C4).
Ejemplos de grupos alquilo ramificados saturados incluyen iso-propilo (C3), iso-butilo (C4), sec-butilo (C4) y terc-butilo
(C4).
Oxo (ceto, -ona): =O.
Grupo protector de oxígeno: el término “grupo protector de oxígeno” se refiere a un resto que enmascara un grupo hidroxi, y estos son bien conocidos en la técnica. Un importante número de grupos adecuados se describe en las páginas 23 a 200 de Greene, T.W. y Wuts, G.M., Protective Groups in Organic Synthesis, 3a Edición, John Wiley & Sons, Inc., 1999. Las clases de especial interés incluyen éteres de sililo (por ejemplo, TMS, TBDMS), éteres de metilo (por ejemplo, THP) y ésteres de metilo (por ejemplo, acetato) sustituidos.
Éter: -OR, en la que R es un sustituyente éter, por ejemplo, un grupo alquilo C1-7 (también denominado un grupo alcoxi C1-7, descrito más adelante), un grupo de heterociclilo C3-20 (también denominado un grupo heterocicliloxi C3-20) o un grupo arilo C5-20 (también denominado un grupo ariloxi C5-20 ), preferentemente un grupo alquilo C1-7.
Carboxi (ácido carboxílico): -C(=O)OH.
Éster (carboxilato, éster de ácido carboxílico, oxicarbonilo): -C(=O)OR, en la que R es un sustituyente éster, por ejemplo, un grupo alquilo C1-7, un grupo heterociclilo C3-20, o un grupo arilo C5-20, preferentemente un grupo alquilo C1-7. Ejemplos de grupos éster incluyen, pero sin limitación, -C(=O)OCH3, -C(=O)OCH2CH3 , -C(=O)OC(CH3)3 y -C(=O)OPh.
Sulfonato (éster de ácido sulfónico): -S(=O)2OR, en la que R es un sustituyente sulfonato, por ejemplo, un grupo alquilo C1-7, un grupo heterociclilo C3-20, o un grupo arilo C5-20, preferentemente un grupo alquilo C1-7. Ejemplos de grupos sulfonato incluyen, pero sin limitación, -S(=O)2OCH3 (metoxisulfonilo; metil sulfonato) y -S(=O)2OCH2CH3 (etoxisulfonilo; sulfonato de etilo).
Conjugados
La presente invención proporciona conjugados que comprenden un dímero de PBD conectado a una unidad de ligando a través de una unidad de enlazador. La unidad de enlazador está unida por un extremo a la unidad de ligando (L) y por el otro extremo al compuesto PBD dimérico (D).
Preferencias
Las siguientes preferencias pueden aplicarse a todos los aspectos de la invención como se ha descrito anteriormente, o pueden referirse a un único aspecto. Las preferencias se pueden combinar juntas en cualquier combinación.
El conjugado tiene la fórmula:

o una sal o solvato farmacéuticamente aceptable del mismo, en la que L, A1, L1, L2, D y p son como se describen a continuación.
L1 y L2 junto con -OC(=O)- representan:
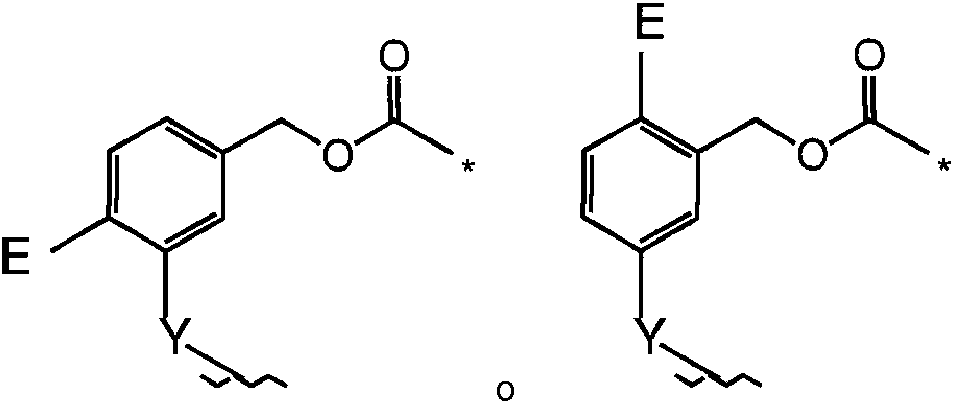
donde el asterisco indica el punto de unión a la unidad de fármaco, la línea ondulada indica el punto de unión a A1, Y es NH y E es ácido glucurónico (por ejemplo, ácido p-glucurónico).
E se selecciona de modo que el grupo sea susceptible a escisión, por ejemplo, con luz o mediante la acción de una enzima. E es ácido glucurónico (por ejemplo, ácido p-glucurónico). El ácido glucurónico puede ser susceptible a la acción de una p-glucuronidasa.
En una realización, el grupo A1 es:

donde el asterisco indica el punto de unión a Y, la línea ondulada indica el punto de unión a la unidad de ligando y n es 0 a 6. En una realización, n es 5.
En una realización, el grupo A1 es:

donde el asterisco indica el punto de unión a Y, la línea ondulada indica el punto de unión a la unidad de ligando y n es 0 a 6. En una realización, n es 5.
En una realización, el grupo A1 es:

donde el asterisco indica el punto de unión a Y, la línea ondulada indica el punto de unión a la unidad de ligando, n es 0 o 1 y m es 0 a 30. En una realización preferida, n es 1 y m es 0 a 10, 1 a 8, preferentemente 4 a 8, lo más preferente 4 u 8.
En una realización, el grupo A1 es:

donde el asterisco indica el punto de unión a Y, la línea ondulada indica el punto de unión a la unidad de ligando, n es 0 o 1 y m es 0 a 30. En una realización preferida, n es 1 y m es 0 a 10, 1 a 8, preferentemente 4 a 8, lo más preferente 4 u 8.
En una realización, la conexión entre la unidad de ligando y A1 es a través de un resto tiol de la unidad de ligando y un grupo maleimida de A1.
En una realización, la conexión entre la unidad de ligando y A1 es:

donde el asterisco indica el punto de unión a la porción restante de A1, y la línea ondulada indica el punto de unión de la porción restante de la unidad de ligando. En esta realización, el átomo de S normalmente procede de la unidad de ligando.
En una realización, los grupos A1-L1 son:

donde
el asterisco indica el punto de unión a L2, la línea ondulada indica el punto de unión a la unidad de ligando y n es 0 a 6. En una realización, n es 5.
En una realización, los grupos A1-L1 son:

en los que el asterisco indica el punto de unión a L2, la línea ondulada indica el punto de unión a la unidad de ligando y n es 0 a 6. En una realización, n es 5.
En una realización, los grupos A1-L1 son:

en los que el asterisco indica el punto de unión a L2, la línea ondulada indica el punto de unión a la unidad de ligando, n es 0 o 1 y m es 0 a 30. En una realización preferida, n es 1 y m es 0 a 10, 1 a 8, preferentemente 4 a 8, lo más preferente 4 u 8.
En una realización, los grupos A1-L1 son:

en los que el asterisco indica el punto de unión a L2, la línea ondulada indica el punto de unión a la unidad de ligando, n es 0 o 1 y m es 0 a 30. En una realización preferida, n es 1 y m es 0 a 10, 1 a 7, preferentemente 3 a 7, lo más preferente 3 u 7.
En una realización, los grupos L-A1-L1 son:

en los que el asterisco indica el punto de unión a L2, S es un grupo de azufre de la unidad de ligando, la línea ondulada indica el punto de unión al resto de la unidad ligando y n es 0 a 6. En una realización, n es 5.
En una realización, los grupos L-A1-L1 son:

en los que el asterisco indica el punto de unión a L2, S es un grupo de azufre de la unidad de ligando, la línea ondulada indica el punto de unión al resto de la unidad de ligando y n es 0 a 6. En una realización, n es 5.
En una realización, los grupos L-A1-L1 son:

en los que el asterisco indica el punto de unión a L2, S es un grupo de azufre de la unidad de ligando, la línea ondulada indica el punto de unión al resto de la unidad de ligando, n es 0 o 1 y m es 0 a 30. En una realización preferida, n es 1 y m es 0 a 10, 1 a 8, preferentemente 4 a 8, lo más preferente 4 u 8.
En una realización, los grupos L-A1-L1 son:

en los que el asterisco indica el punto de unión a L2, la línea ondulada indica el punto de unión a la unidad de ligando, n es 0 o 1 y m es 0 a 30. En una realización preferida, n es 1 y m es 0 a 10, 1 a 7, preferentemente 4 a 8, lo más preferente 4 u 8.
Enlazador-fármacos
En otras realizaciones, los compuestos de enlazador-fármaco se proporcionan para su conjugación a una unidad de ligando. En una realización, los compuestos de enlazador-fármaco están diseñados para la conexión a un agente de unión a célula.
El compuesto enlazador a fármaco tiene la fórmula:
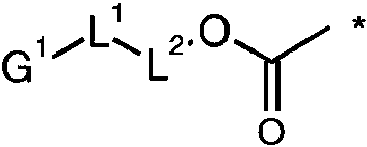
donde el asterisco indica el punto de unión a la unidad de fármaco (D, como se ha definido anteriormente), G1 es un grupo de extensión (A1) para formar una conexión a una unidad de ligando L1 y L2 son como se han definido anteriormente en donde L1 y L2 junto con -OC(=O)- representan:

donde el asterisco indica el punto de unión a la unidad de fármaco, la línea ondulada indica el punto de unión a G1 (A1), Y es NH y E es ácido glucurónico (por ejemplo, ácido p-glucurónico). Las referencias a la conexión a A1 se pueden interpretar aquí como referencias a una conexión a G1.
El grupo funcional G1 forma un grupo de conexión tras la reacción con una unidad de ligando (por ejemplo, un agente de unión a la célula).
En una realización, el grupo funcional G1 es o comprende un grupo maleimida para reaccionar con un grupo apropiado en la unidad de ligando. En una realización preferida, G1 comprende un grupo maleimida.
En una realización, el grupo G1 es un grupo alquilmaleimida. Este grupo es adecuado para su reacción con grupos tiol, particularmente grupos tiol de cisteína, presente en el agente de unión a la célula, por ejemplo presente en un anticuerpo.
En una realización, el grupo G1 es:

en el que el asterisco indica el punto de unión a Y y n es 0 a 6. En una realización n es 5.
En una realización, el grupo G1 es:

en el que el asterisco indica el punto de unión a Y y n es 0 a 6. En una realización, n es 5.
En una realización, el grupo G1 es:

donde el asterisco indica el punto de unión a Y, n es 0 o 1 y m es 0 a 30. En una realización preferida, n es 1 y m es 0 a 10, 1 a 2, preferentemente 4 a 8, y lo más preferentemente 4 u 8.
En una realización, el grupo G1 es:

donde el asterisco indica el punto de unión a Y, n es 0 o 1 y m es 0 a 30. En una realización preferida, n es 1 y m es 0 a 10, 1 a 8, preferentemente 4 a 8, y lo más preferentemente 4 u 8.
Unidad de ligando
La unidad de ligando se une específicamente a una molécula diana y puede ser un anticuerpo o un fragmento de un anticuerpo. La unidad de ligando también se denomina en el presente documento "agente de unión" o "agente de direccionamiento".
Los términos "se une específicamente" y "unión específica" se refieren a la unión de un anticuerpo u otra proteína, polipéptido o péptido a una molécula predeterminada (por ejemplo, un antígeno). Normalmente, el anticuerpo u otra molécula se une con una afinidad de al menos aproximadamente 1x107 M-1, y se une a la molécula predeterminada con una afinidad que es al menos dos veces mayor que su afinidad por la unión a una molécula no específica (por ejemplo, ASB, caseína) distinta de la molécula predeterminada o una molécula estrechamente relacionada.
Ejemplos de unidades de ligando incluyen los agentes descritos para su uso en el documento WO 2007/085930.
En algunas realizaciones, la unidad de ligando es un agente de unión a célula que se une a una diana extracelular de una célula. El agente de unión a célula también es un anticuerpo o un fragmento de unión al antígeno de un anticuerpo. Por tanto, en una realización, la presente invención proporciona un conjugado anticuerpo-fármaco (CAF).
En una realización, el anticuerpo es un anticuerpo monoclonal; un anticuerpo quimérico; un anticuerpo humanizado; un anticuerpo completamente humano; o un anticuerpo de cadena simple. Una realización del anticuerpo es un fragmento de uno de estos anticuerpos que tiene actividad biológica. Ejemplos de dichos fragmentos incluyen fragmentos Fab, Fab', F(ab')2 y Fv.
El anticuerpo puede ser un diacuerpo, un anticuerpo de dominio (ACD) o un anticuerpo monocatenario.
En una realización, el anticuerpo es un anticuerpo monoclonal.
Los anticuerpos para su uso en la presente invención incluyen los anticuerpos descritos en el documento WO 2005/082023. Son especialmente preferidos los anticuerpos contra antígenos asociados a tumores. Ejemplos de esos antígenos conocidos en la técnica incluyen, pero sin limitación, dichos antígenos asociados a tumores definidos en el documento WO 2005/082023. Véanse, por ejemplo, las páginas 41-55.
En algunas realizaciones, los conjugados están diseñados para dirigirse a células tumorales a través de los antígenos de su superficie celular. Los antígenos pueden ser antígenos de superficie celular que están expresados en exceso, o que se expresan en momentos o tipos celulares anómalos. Preferentemente, el antígeno diana se expresa solo en células proliferativas (preferentemente células tumorales); sin embargo, esto rara vez se observa en la práctica. Como resultado, los antígenos diana normalmente se seleccionan sobre la base de la expresión diferencial entre tejido proliferativo y sano.
Se han sensibilizado anticuerpos para dirigirse a antígenos específicos relacionados con tumores, que incluyen:
Cripto, CD19, CD20, CD22, CD30, CD33, a la glicoproteína NMB, CanAg, Her2 (ErbB2/Neu), CD56 (NCAM), CD70, CD79, CD138, al PSCA, al PSMA (antígeno prostático específico de membrana), al BCMA, a la E-selectina, EphB2, a la melanotransferina, Muc16 y TMEFF2. En cualquiera de las realizaciones proporcionadas en el presente documento, la unidad de ligando puede ser un anticuerpo monoclonal que se une específicamente al antígeno Cripto, al antígeno CD19, al antígeno CD20, al antígeno CD22, al antígeno CD30, al antígeno CD33, a la glicoproteína NMB, al antígeno CanAg, al antígeno Her2 (ErbB2/Neu ), al antígeno CD56 (NCAM), al antígeno CD70, al antígeno CD79, al antígeno CD138, al PSCA, al PSMA (antígeno prostático específico de membrana), al BCMA, a la E-selectina, EphB2, a la melanotransferina, al antígeno Muc16 o al antígeno TMEFF2.
La unidad de ligando está conectada a la unidad de enlazador. En una realización, la unidad de ligando está conectada a A1 de la unidad de enlazador.
En una realización, la conexión entre la unidad de ligando y la unidad de enlazador es a través de un enlace tioéter.
En una realización, la conexión entre la unidad de ligando y la unidad de enlazador es a través de un enlace disulfuro.
En una realización, la conexión entre la unidad de ligando y la unidad de enlazador es a través de un enlace amida.
En una realización, la conexión entre la unidad de ligando y la unidad de enlazador es a través de un enlace éster.
En una realización, la conexión entre la unidad de ligando y el enlazador se forma entre un grupo tiol de un resto de cisteína de la unidad de ligando y un grupo maleimida de la unidad de enlazador.
Los restos de cisteína de la unidad de ligando pueden estar disponibles para su reacción con el grupo funcional de la unidad de enlazador para formar una conexión. En otras realizaciones, por ejemplo, cuando la unidad de ligando es un anticuerpo, los grupos tiol del anticuerpo pueden participar en enlaces disulfuro intercatenarios. Estos enlaces intercatenarios se pueden convertir en grupos tiol libres, por ejemplo, mediante el tratamiento del anticuerpo con DTT antes de la reacción con el grupo funcional de la unidad del enlazador.
En algunas realizaciones, el resto de cisteína se introduce en la cadena pesada o ligera de un anticuerpo. Las posiciones para la inserción de cisteína por sustitución en cadenas pesadas o ligeras de anticuerpos incluyen las
descritas en la Solicitud Publicada de EE.UU. n.° 2007-0092940 y la publicación de la Patente Internacional WO2008/070593.
Métodos de tratamiento
Los compuestos y conjugados de la presente invención se pueden usar en un método de terapia. Dicho método de tratamiento puede comprender administrar a un sujeto que necesita tratamiento una cantidad terapéuticamente eficaz de un compuesto o conjugado divulgado en el presente documento. El término "cantidad terapéuticamente eficaz" es una cantidad suficiente para mostrar un beneficio a un paciente. Dicho beneficio puede ser al menos la mejora de al menos un síntoma. La cantidad real administrada, y la velocidad y el ciclo de tiempo de administración, dependerán de la naturaleza y la gravedad de lo que se está tratando. La prescripción del tratamiento, por ejemplo, las decisiones sobre la dosificación, es de la responsabilidad de los facultativos generales y otros médicos.
Un compuesto o conjugado se puede administrar solo o en combinación con otros tratamientos, de forma simultánea o secuencial dependiendo de la afección a tratar. Ejemplos de tratamientos y terapias incluyen, pero sin limitación, quimioterapia (la administración de agentes activos, incluidos, por ejemplo, fármacos); cirugía; y radioterapia.
Las composiciones farmacéuticas de acuerdo con la presente invención, y para su uso de acuerdo con la presente invención, pueden comprender, además del principio activo, es decir un compuesto o conjugado divulgado en el presente documento, un excipiente, transportador, tampón, estabilizador farmacéuticamente aceptables u otros materiales bien conocidos por los expertos en la materia. Dichos materiales deben ser no tóxicos y no deben interferir con la eficacia del principio activo. La naturaleza precisa del transportador u otro material dependerá de la vía de administración, que puede ser oral, o por inyección, por ejemplo, cutánea, subcutánea o intravenosa.
Las composiciones farmacéuticas para la administración oral pueden estar en forma de comprimido, cápsula, polvo o líquido. Un comprimido puede comprender un transportador sólido o un adyuvante. Las composiciones farmacéuticas líquidas generalmente comprenden un transportador líquido tal como agua, petróleo, aceites animales o vegetales, aceite mineral o aceite sintético. Puede incluirse solución salina fisiológica, dextrosa u otra solución de sacáridos o glicoles tales como etilenglicol, propilenglicol o polietilenglicol. Una cápsula puede comprender un vehículo sólido tal como gelatina.
Para la inyección intravenosa, cutánea o subcutánea, o inyección en el sitio de aflicción, el principio activo estará en forma de una solución acuosa parenteralmente aceptable, que no tenga pirógenos y que tenga un pH, isotonicidad y estabilidad adecuados. Los expertos en la materia serán bien capaces de preparar soluciones adecuadas usando, por ejemplo, vehículos isotónicos tales como inyección de cloruro de sodio, inyección de Ringer, inyección de Ringer lactato. Pueden incluirse conservantes, estabilizantes, tampones, antioxidantes y/u otros aditivos, según sea necesario.
Los compuestos y conjugados pueden usarse para tratar una enfermedad proliferativa y una enfermedad autoinmunitaria. El término "enfermedad proliferativa" se refiere a una proliferación celular no deseada o incontrolada de células excesivas o anómalas que no es deseada, tales como, crecimiento neoplásico o hiperplásico, tanto in vitro como in vivo.
Ejemplos de afecciones proliferativas incluyen, pero sin limitación, proliferación celular benigna, premaligna y maligna, incluyendo, aunque sin limitación, neoplasias y tumores (por ejemplo, histiocitoma, glioma, astrocitoma, osteoma), cánceres (por ejemplo, cáncer de pulmón, cáncer de pulmón de células pequeñas, cáncer gastrointestinal, cáncer de intestino, cáncer de colon, carcinoma de mama, carcinoma de ovario, cáncer de próstata, cáncer de testículos, cáncer de hígado, cáncer de riñón, cáncer de vejiga, cáncer pancreático, cáncer de cerebro, sarcoma, osteosarcoma, sarcoma de Kaposi, melanoma), leucemias, psoriasis, enfermedades óseas, trastornos fibroproliferativos (por ejemplo, de tejidos conectivos) y aterosclerosis. Otros cánceres de interés incluyen, pero sin limitación, hematológicos; neoplasias tales como leucemias y linfomas, tales como linfoma no Hodgkin, y subtipos tales como LDCGB, zona marginal, zona del manto, y folicular, linfoma de Hodgkin, LMA y otros cánceres de origen en linfocitos B o T.
Ejemplos de enfermedades autoinmunitarias incluyen las siguientes: artritis reumatoide, enfermedades desmielinizantes autoinmunitarias (por ejemplo, esclerosis múltiple, encefalomielitis alérgica), artritis psoriásica, oftalmopatía endocrina, uveorretinitis, lupus eritematoso sistémico, miastenia grave, enfermedad de Graves, glomerulonefritis, trastorno hepatológico autoinmunitario, enfermedad inflamatoria del intestino (por ejemplo, enfermedad de Crohn), anafilaxia, reacción alérgica, síndrome de Sjogren, diabetes mellitus de tipo I, cirrosis biliar primaria, granulomatosis de Wegener, fibromialgia, polimiositis, dermatomiositis, insuficiencia endocrina múltiple, síndrome de Schmidt, uveítis autoinmunitaria, enfermedad de Addison, adrenalitis, tiroiditis, tiroiditis de Hashimoto, enfermedad tiroidea autoinmunitaria, anemia perniciosa, atrofia gástrica, hepatitis crónica, hepatitis lupoide, ateroesclerosis, lupus eritematoso cutáneo subagudo, hipoparatiroidismo, síndrome de Dressler, trombocitopenia autoinmunitaria, púrpura trombocitopénica idiopática, anemia hemolítica, pénfigo vulgar, pénfigo, dermatitis herpetiforme, alopecia areata, penfigoide, esclerodermia, esclerosis sistémica progresiva, síndrome CREST (calcinosis, fenómeno de Raynaud, dismotilidad esofágica, esclerodactilia y telangiectasia), infertilidad
autoinmunitaria masculina y femenina, espondilitis anquilosante, colitis ulcerosa, enfermedad del tejido conectivo mixto, poliarteritis nodosa, vasculitis necrosante sistémica, dermatitis atópica, rinitis atópica, síndrome de Goodpasture, enfermedad de Chagas, sarcoidosis, fiebre reumática, asma, aborto recurrente, síndrome antifosfolípido, pulmón del granjero, eritema multiforme, síndrome poscardiotomía, síndrome de Cushing, hepatitis crónica activa autoinmunitaria, pulmón del cuidador de aves, necrolisis epidérmica tóxica, síndrome de Alport, alveolitis, alveolitis alérgica, alveolitis fibrosante, enfermedad pulmonar intersticial, eritema nudoso, pioderma gangrenoso, reacción a la transfusión, arteritis de Takayasu, polimialgia reumática, arteritis temporal, esquistosomiasis, arteritis de células gigantes, ascariasis, aspergilosis, síndrome de Sampter, eccema, granulomatosis linfomatoide, enfermedad de Behcet, síndrome de Caplan, enfermedad de Kawasaki, dengue, encefalomielitis, endocarditis, fibrosis endomiocardial, endoftalmitis, eritema elevado y diutino, psoriasis, eritroblastosis fetal, fascitis eosinofílica, síndrome de Shulman, síndrome de Felty, filariasis, ciclitis, ciclitis crónica, ciclitis heterocrónica, ciclitis de Fuch, nefropatía IgA, púrpura de Henoch-Schonlein, enfermedad de injerto frente a huésped, rechazo de trasplantes, cardiomiopatía, síndrome de Eaton-Lambert, policondritis recidivante, crioglobulinemia, macroglobulinemia de Waldenstrom, síndrome de Evan, e insuficiencia gonadal autoinmunitaria.
En algunas realizaciones, la enfermedad autoinmunitaria es un trastorno de los linfocitos B (por ejemplo, lupus eritematoso sistémico, síndrome de Goodpasture, artritis reumatoide y diabetes tipo I), linfocitos Th1 (por ejemplo, artritis reumatoide, esclerosis múltiple, psoriasis, síndrome de Sjogren, tiroiditis de Hashimoto, enfermedad de Graves, cirrosis biliar primaria, granulomatosis de Wegener, tuberculosis, o enfermedad de injerto frente a hospedador) o linfocitos Th2 (por ejemplo, dermatitis atópica, lupus eritematoso sistémico, asma atópica, rinoconjuntivitis, rinitis alérgica, síndrome de Omenn, esclerosis sistémica, o enfermedad crónica de injerto frente a hospedador). En general, los trastornos que implican células dendríticas implican trastornos de linfocitos Th1 o linfocitos Th2. En algunas realizaciones, el trastorno autoinmunitario es un trastorno inmunológico mediado por linfocitos T.
En algunas realizaciones, la cantidad del conjugado administrado oscila entre aproximadamente 0,01 y aproximadamente 10 mg/kg por dosis. En algunas realizaciones, la cantidad del conjugado administrado oscila entre aproximadamente 0,01 y aproximadamente 5 mg/kg por dosis. En algunas realizaciones, la cantidad del conjugado administrado oscila entre aproximadamente 0,05 y aproximadamente 5 mg/kg por dosis. En algunas realizaciones, la cantidad del conjugado administrado oscila entre aproximadamente 0,1 y aproximadamente 5 mg/kg por dosis. En algunas realizaciones, la cantidad del conjugado administrado oscila entre aproximadamente 0,1 y aproximadamente 4 mg/kg por dosis. En algunas realizaciones, la cantidad del conjugado administrado oscila entre aproximadamente 0,05 y aproximadamente 3 mg/kg por dosis. En algunas realizaciones, la cantidad del conjugado administrado oscila entre aproximadamente 0,1 y aproximadamente 3 mg/kg por dosis. En algunas realizaciones, la cantidad del conjugado administrado oscila entre aproximadamente 0,1 y aproximadamente 2 mg/kg por dosis.
Incluye otras formas
Salvo que se especifique otra cosa, se incluyen en lo anterior las formas iónicas, sal, solvatos y protegidas bien conocidas de estos sustituyentes. Por ejemplo, una referencia al ácido carboxílico (-COOH) también incluye la forma (carboxilato) aniónica (-COO), una sal o un solvato de la misma, así como las formas protegidas convencionales. De manera similar, una referencia a un grupo amino incluye la forma protonada (-N+HR1R2), una sal o un solvato del grupo amino, por ejemplo, una sal de clorhidrato, así como las formas protegidas convencionales de un grupo amino. De manera similar, una referencia a un grupo hidroxilo también incluye la forma aniónica (-O'), una sal o un solvato de la misma, así como las formas protegidas convencionales.
Sales
Puede ser conveniente o deseable preparar, purificar y/o manipular una sal correspondiente del compuesto activo, por ejemplo, una sal farmacéuticamente aceptable. Ejemplos de sales farmacéuticamente aceptables se tratan en Berge, et at., J. Pharm. Sci., 66, 1-19 (1977).
Por ejemplo, si el compuesto es aniónico, o tiene un grupo funcional que puede ser aniónico (por ejemplo, -COOH puede ser -COO), a continuación, la sal se puede formar con un catión adecuado. Ejemplos de cationes inorgánicos adecuados incluyen, pero sin limitación, iones de metal alcalino tales como Na+ y K+, cationes alcalinotérreos tales como Ca2+ y Mg2+, y otros cationes tales como Al+3. Ejemplos de cationes orgánicos adecuados incluyen, pero sin limitación, ion amonio (es decir NH4+) e iones de amonio sustituidos (por ejemplo, NH3R+, NH2R2+, NHR3+, NR4+). Ejemplos de algunos iones amonio sustituido adecuados son los procedentes de: etilamina, dietilamina, diciclohexilamina, trietilamina, butilamina, etilendiamina, etanolamina, dietanolamina, piperazina, bencilamina, fenilbencilamina, colina, meglumina y trometamina, así como aminoácidos, tales como lisina y arginina. Un ejemplo de un ion de amonio cuaternario común es N(CH3)4+.
Si el compuesto es catiónico, o tiene un grupo funcional que puede ser catiónico (por ejemplo, -NH2 puede ser -NH3+), entonces se puede formar una sal con un anión adecuado. Ejemplos de aniones inorgánicos adecuados incluyen, pero sin limitación, los obtenidos a partir de los siguientes ácidos inorgánicos: clorhídrico, bromhídrico, yodhídrico, sulfúrico, sulfuroso, nítrico, nitroso, fosfórico y fosforoso.
Ejemplos de aniones orgánicos adecuados incluyen, pero sin limitación, los obtenidos a partir de los siguientes ácidos orgánicos: 2-acetioxibenzoico, acético, ascórbico, aspártico, benzoico, alcanforsulfónico, cinámico, cítrico, edético, etanodisulfónico, etanosulfónico, fumárico, gluqueptónico, glucónico, glutámico, glicólico, hidroximaleico, hidroxinaftalenocarboxílico, isetiónico, láctico, lactobiónico, láurico, maleico, málico, metanosulfónico, múcico, oleico, oxálico, palmítico, pamoico, pantoténico, fenilacético, fenilsulfónico, propiónico, pirúvico, salicílico, esteárico, succínico, sulfanílico, tartárico, toluenosulfónico y valérico. Ejemplos de aniones orgánicos poliméricos adecuados incluyen, pero sin limitación, los obtenidos a partir de los siguientes ácidos poliméricos: ácido tánico, carboximetilcelulosa.
Solvatos
Puede ser conveniente o deseable preparar, purificar y/o manejar un solvato correspondiente del compuesto activo. El término "solvato" se usa en el presente documento en el sentido convencional para referirse a un complejo de soluto (por ejemplo, principio activo, sal de principio activo) y disolvente. Si el disolvente es agua, el solvato se puede definir convenientemente como un hidrato, por ejemplo, un monohidrato, un dihidrato, un trihidrato, etc.
Carbinolaminas
La invención incluye compuestos en los que un disolvente se añade a través del enlace imina del resto PBD, que se ilustra a continuación donde el disolvente es agua o un alcohol (RAOH, donde RA es alquilo C1-4):

Estas formas se pueden llamar denominar formas de carbinolamina y éter de carbinolamina de PBD. El resto de estos equilibrios depende de las condiciones en las que se encuentran los compuestos, así como de la naturaleza del resto en sí.
Estos compuestos especiales se pueden aislar en forma sólida, por ejemplo, por liofilización.
Isómeros
Determinados compuestos pueden existir en una o más formas geométricas, ópticas, enantioméricas, diastereoméricas, epiméricas, atrópicas, estereoisoméricas, tautoméricas, conformacionales o anoméricas especiales, incluyendo, aunque sin limitación, formas cis y trans; formas E y Z; formas c, t, y r; formas endo y exo; formas R, S y meso; formas D y L; formas d y l; formas (+) y (-); formas ceto, enol y enolato; formas sin y anti; formas sinclinal y anticlinal; formas a y p; formas axiales y ecuatoriales; formas de bote, silla, torsionadas, de sobre y media silla; y combinaciones de las mismas, colectivamente denominadas en lo sucesivo "isómeros" (o "formas isoméricas").
Cabe destacar que, salvo en los descrito a continuación para formas tautómeras, específicamente excluidas del término "isómeros", como se usa en el presente documento, son isómeros estructurales (o constitucionales) (es decir, isómeros que difieren en las conexiones entre átomos en lugar de simplemente por la posición de los átomos en el espacio). Por ejemplo, una referencia a un grupo metoxi, -OCH3 , no debe interpretarse como una referencia a su isómero estructural, un grupo hidroximetilo, -CH2OH. De manera similar, una referencia al ortoclorofenilo no debe interpretarse como una referencia a su isómero estructural, el metaclorofenilo. Sin embargo, una referencia a una clase de estructuras bien puede incluir formas estructuralmente isoméricas comprendidas en dicha clase (por ejemplo, alquilo C1-7 incluye n-propilo e iso-propilo; butilo incluye n-, iso-, sec-, y terc-butilo; metoxifenilo incluye orto-, meta- y para-metoxifenilo).
La exclusión anterior no concierne a las formas tautoméricas, por ejemplo, formas ceto, enol y enolato, como en, por ejemplo, los siguientes pares tautoméricos: ceto/enol (ilustrado a continuación), imina/enamina, amida/iminoalcohol, amidina/amidina, nitroso/oxima, tiocetona/enetiol, N-nitroso/hidroxiazo y nitro/aci-nitro.

ceto enol enolato
Se debe tener en cuenta que en el término "isómero" se incluyen específicamente compuestos con una o más sustituciones isotópicas. Por ejemplo, H puede estar en cualquier forma isotópica, incluyendo 1H, 2H (D) y 3H (T); C puede estar en cualquier forma isotópica, incluyendo 12C, 13C, y 14C; O puede estar en cualquier forma isotópica, incluyendo 16O y 18O; y similares.
Salvo que se especifique otra cosa, una referencia a un compuesto particular incluye todas las formas isoméricas, incluyendo (total o parcialmente) formas racémicas y otras mezclas de las mismas. Los métodos de preparación (por ejemplo, síntesis asimétrica) y separación (por ejemplo, cristalización fraccionada y medios cromatográficos) de dichas formas isoméricas se conocen en la técnica o se obtienen fácilmente adaptando los métodos enseñados en el presente documento, o los métodos conocidos, de una manera conocida.
Rutas de síntesis generales
La síntesis de los compuestos de PBD se trata extensamente en las siguientes referencias:
a) documento WO 00/12508 (páginas 14 a 30);
b) documento WO 2005/023814 (páginas 3 a 10);
c) documento WO 2004/043963 (páginas 28 a 29); y
d) documento WO 2005/085251 (páginas 30 a 39).
Ruta de síntesis
Los compuestos de la presente invención, en los que R10 y R11 forman un doble enlace nitrógeno-carbono entre los átomos de nitrógeno y de carbono a los que están unidos, pueden sintetizarse a partir de un compuesto de Fórmula 2:

donde n es 0 o 1, y R2 y R12 representan los grupos aromáticos C2 de los compuestos de la presente invención, como se muestra en la tabla siguiente:

continuación

ProtN es un grupo protector de nitrógeno para la síntesis y ProtO es un grupo de oxígeno protegido para la síntesis o un grupo oxo, desprotegiendo el enlace imina por métodos convencionales.
El compuesto producido puede estar en su forma de carbinolamina o éter de carbinolamina dependiendo de los disolventes usados. Por ejemplo, si ProtN es Troc y ProtO es un grupo protector de oxígeno para la síntesis, entonces la desprotección se lleva a cabo usando un par Cd/Pb para producir el compuesto de la presente invención. Si ProtN es SEM o un grupo análogo, y ProtO es un grupo oxo, entonces el grupo oxo puede eliminarse por reducción, lo que conduce a un intermedio de carbinolamina protegida, que luego puede tratarse para eliminar el grupo protector SEM, seguido de la eliminación del agua. La reducción del compuesto de Fórmula 2 se puede realizar, por ejemplo, con superhidruro o tetraborohidruro de litio, mientras que un medio adecuado para eliminar el grupo protector SEM es el tratamiento con gel de sílice.
Los compuestos de fórmula 2 se pueden sintetizar a partir de un compuesto de fórmula 3a:

donde R2, ProtN y ProtO son como se han definido para los compuestos de fórmula 2, mediante el acoplamiento de un derivado organometálico que comprende R12, tal como un derivado de organoboro. El derivado de organoboro puede ser un boronato o un ácido borónico.
Los compuestos de fórmula 2 se pueden sintetizar a partir de un compuesto de fórmula 3b:

donde R12, ProtN y ProtO son como se han definido para los compuestos de fórmula 2, mediante el acoplamiento de un derivado organometálico que comprende R2, tal como un derivado de organoboro. El derivado de organoboro puede ser un boronato o un ácido borónico.
Los compuestos de fórmula 3a 3b pueden sintetizarse a partir de un compuesto de fórmula 4:

donde ProtN y ProtO son como se han definido para los compuestos de fórmula 2, mediante el acoplamiento de aproximadamente un único equivalente (por ejemplo, 0,9 o 1 a 1,1 o 1,2) de un derivado organometálico, tal como un derivado de organoboro, que comprende R2 o R12.
Los acoplamientos descritos anteriormente se llevan a cabo normalmente en presencia de un catalizador de paladio, por ejemplo Pd(PPh3)4, Pd(OCOCH3)2, PdCl2 , Pd2(dba)3. El acoplamiento se puede llevar a cabo en condiciones convencionales, o también se puede llevar a cabo en condiciones de microondas.
Las dos etapas de acoplamiento normalmente se llevan a cabo secuencialmente. Pueden llevarse a cabo con o sin purificación entre las dos etapas. Si no se lleva a cabo la purificación, entonces las dos etapas se pueden llevar a cabo en el mismo recipiente de reacción. La purificación normalmente se requiere después de la segunda etapa de acoplamiento. La purificación del compuesto a partir de los subproductos no deseados puede llevarse a cabo por cromatografía en columna o separación por intercambio iónico.
La síntesis de compuestos de fórmula 4 en los que ProtO es un grupo oxo y ProtN es SEM se describen en detalle en el documento WO 00/12508. En particular, se hace referencia al esquema 7 de la página 24, donde el compuesto anterior se designa como intermedio P. Este método de síntesis también se describe en el documento WO 2004/043963. También se hace referencia adicional a la síntesis de los compuestos 8a y 8b en el documento WO 2010/043880 (páginas 36 a 45).
La síntesis de compuestos de fórmula 4 en la que ProtO es un grupo de oxígeno protegido para la síntesis se describe en el documento WO 2005/085251.
Los compuestos de la presente invención donde R10 y R10’ son H y R11 y R11’ son SOzM, se pueden sintetizar a partir de los compuestos de la presente invención donde R10 y R11 forman un enlace doble nitrógeno-carbono entre los átomos de nitrógeno y carbono a los están unidos, mediante la adición de la sal de bisulfito o sulfinato adecuada, seguida por una etapa de purificación necesaria. En el documento GB 2053894 se describen otros métodos.
En algunas realizaciones de la invención, puede resultar que los compuestos de Fórmula 2 se usen en la síntesis de los compuestos enlazadores de fármaco. En estas realizaciones, la eliminación de los grupos protectores N10/C11 se puede realizar durante la síntesis de los compuestos enlazadores de fármaco.
Grupos protectores de nitrógeno para síntesis
Los grupos protectores de nitrógeno para síntesis son bien conocidos en la técnica. En la presente invención, los grupos protectores de especial interés son los grupos carbamato como protectores de nitrógeno y los grupos protectores hemiaminales para el nitrógeno.
Los grupos protectores especialmente preferidos incluyen Troc, Teoc, Fmoc, BOC, Doc, Hoc, TcBOC, 1-Adoc y 2-Adoc.
Otros posibles grupos son nitrobenciloxicarbonilo (por ejemplo, 4-nitrobenciloxicarbonilo) y 2-(fenilsulfonil)etoxicarbonilo.
Estos grupos protectores que se pueden eliminar mediante catálisis de paladio no son preferidos, por ejemplo Alloc. Grupo de oxígeno protegido para síntesis
Los grupos de oxígeno protegido para síntesis son bien conocidos en la técnica. Un importante número de grupos protectores de oxígeno adecuados se describe en las páginas 23 a 200 de Greene, T.W. y Wuts, G.M., Protective Groups in Organic Synthesis, 3a Edición, John Wiley & Sons, Inc., 1999.
Las clases de especial interés incluyen éteres de sililo, éteres de metilo, éteres de alquilo, éteres de bencilo, ésteres, acetatos, benzoatos, carbonatos, y sulfonatos.
Los grupos protectores de oxígeno preferidos incluyen acetatos, TBS y THP.
Síntesis de conjugados de fármacos
Los conjugados que comprenden dímeros de PBD como se describe en el presente documento pueden prepararse usando el conocimiento del experto en la técnica en combinación con las enseñanzas proporcionadas en el presente documento. Por ejemplo, los enlazadores se describen en la Patente de EE.UU. n.° 6.214.345, Patente de EE.UU. n.° 7.498.298 así como en el documento WO 2009/0117531. Se pueden preparar otros enlazadores de acuerdo con las referencias citadas en el presente documento o conocidas de los expertos en la técnica.
Los compuestos de enlazador-fármaco se pueden preparar de acuerdo con los métodos conocidos en la técnica en combinación con las enseñanzas proporcionadas en el presente documento. Por ejemplo, el enlace de sustituyentes de amina (de la unidad de dímero de PBD del fármaco) a los grupos activos de las unidades del enlazador se puede realizar de acuerdo con los métodos descritos en general en las Patentes de EE.UU. números 6.214.345 y
7.498.298; y el documento WO 2009-0117531, o como conocen de otra manera los expertos en la técnica.
Los anticuerpos pueden conjugarse con compuestos de enlazador-fármaco como se describe en Doronina et al., Nature Biotechnology, 2003, 21, 778-784). En resumen, los anticuerpos (4-5 mg/ml) en PBS que contiene borato de sodio 50 mM a pH 7,4 se reducen con clorhidrato de tris(carboxietil)fosfina (TCEP) a 37 °C. El progreso de la reacción, que reduce los disulfuros intercatenarios, se controla por reacción con 5,5'-ditiobis(ácido 2-nitrobenzoico) y se deja avanzar hasta que se consigue el nivel deseado de tioles/mAb. El anticuerpo reducido se enfría luego a 0 °C y se alquila con 1,5 equivalentes de enlazador de fármaco maleimida por tiol de anticuerpo. Después de 1 hora, la reacción se interrumpe mediante la adición de 5 equivalentes de N-acetil cisteína. El enlazador de fármaco inactivado se elimina por filtración en gel sobre una columna PD-10. El CAF se filtra luego en condiciones estériles a través de un filtro de jeringa de 0,22 μm. La concentración de proteína puede determinarse por análisis espectral a 280 nm y 329 nm, respectivamente, con corrección por la contribución de la absorbancia del fármaco a 280 nm. La cromatografía de exclusión por tamaño se puede usar para determinar la extensión de la agregación de anticuerpos, y la RP-HPLC se puede usar para determinar los niveles del enlazador de fármaco inactivado con NAC restantes.
Los anticuerpos con restos de cisteína introducidos pueden conjugarse con compuestos de enlace-fármaco como se describe en la publicación de Patente Internacional WO2008/070593. Los anticuerpos que contienen un resto de cisteína introducido en la cadena pesada se reducen completamente añadiendo 10 equivalentes de TCEP y EDTA 1 mM y ajustando el pH a 7,4 con tampón Tris 1 M (pH 9,0). Después de una incubación de 1 hora a 37 °C, la reacción se enfría a 22°C y se añaden 30 equivalentes de ácido deshidroascórbico para reoxidar selectivamente los disulfuros nativos, mientras que deja la cisteína introducida en el estado reducido. El pH se ajusta a 6,5 con tampón Tris 1 M (pH 3,7) y se deja que la reacción avance durante 1 hora a 22 °C. El pH de la solución se eleva de nuevo a 7,4 mediante la adición de tampón Tris 1 M (pH 9,0). Se colocan 3,5 equivalentes del enlazador de fármaco PBD en DMSO en un recipiente adecuado para la dilución con propilenglicol antes de la adición a la reacción. Para mantener la solubilidad del enlazador de fármaco PBD, el propio anticuerpo primero se diluye con propilenglicol hasta una concentración final de 33 % (por ejemplo, si la solución de anticuerpo estaba en un volumen de reacción de 60 ml, se añadieron 30 ml de propilenglicol). Este mismo volumen de propilenglicol (30 ml en este ejemplo) se añade al enlazador de fármaco PBD como diluyente. Después de la mezcla, la solución de enlazador de fármaco PBD en propilenglicol se añade a la solución de anticuerpo para efectuar la conjugación; la concentración final de propilenglicol es del 50 %. La reacción se deja avanzar durante 30 minutos y luego se inactiva mediante la adición de 5 equivalentes de N-acetil cisteína. El CAF se purifica por ultrafiltración a través de una membrana de 30 kD. (Se debe tener en cuenta que la concentración de propilenglicol usada en la reacción puede reducirse para cualquier PBD especial, ya que su único fin es mantener la solubilidad del enlazador de fármaco en el medio acuoso).
Esquemas ilustrativos para la síntesis de enlazadores de fármaco
El siguiente esquema es ilustrativo de las rutas para sintetizar enlazadores de fármaco.
Esquema A

R2' representa la parte de R2 (como se ha definido anteriormente) que une el núcleo de PBD con el grupo NH2 (para el compuesto C1, el grupo NH2 está sustituido por NHMe). n es como se ha definido anteriormente.
El enlazador de glucurónido intermedio S1 (referencia: Jeffrey et al., Bioconjugate Chemistry, 2006, 17, 831-840) se puede tratar con difosgeno en diclrorometano a -78 °C para proporcionar el cloroformiato de glucurónico, que a continuación se hace reaccionar con el PBD dimérico S2 disuelto en CH2CI2 mediante adición gota a gota. El calentamiento de la reacción a 0° C durante 2 horas seguido de extracción proporcionará el compuesto S3. El tratamiento de una solución de S3 en una mezcla de igual cantidad de disolventes MeOH, tetrahidrofurano, y agua (enfriada a 0 °C) con hidróxido de litio monohidrato durante 4 horas, seguido de reacción con ácido acético glacial proporcionará el compuesto S4. La adición de éster de maleidocaproil NHS a una solución de S4 en DMF, seguido de diisopropiletilamina y agitación a temperatura ambiente en una atmósfera de nitrógeno durante 2 horas proporcionará el enlazador de fármaco deseado S5.
Los métodos de los Ejemplos 2, 3, 4, 6, 7 y 8 se podrían adaptar a todos los compuestos de PBD de la presente invención.
Otras preferencias
Las siguientes preferencias pueden aplicarse a todos los aspectos de la invención como se ha descrito anteriormente, o pueden referirse a un único aspecto. Las preferencias se pueden combinar juntas en cualquier combinación.
Conjugados
Los conjugados de la presente invención incluyen, por ejemplo, los de fórmula:
IV:
L -(LU-D)p (IV)
o una sal farmacéuticamente aceptable o solvato de los mismos;
en donde L es una unidad de ligando seleccionada entre el grupo que consiste en un anticuerpo y un fragmento de unión a antígeno de un anticuerpo,
LU es una unidad de enlazador,
P es 1 a 20 y
D se selecciona entre el grupo que consiste en D1 a D6 que tienen las estructuras:


en donde
(a) R10 es H y R11 es OH, ORA, en donde RA es alquilo C1-4 saturado o
(b) R10 y R11 forman un doble enlace nitrógeno-carbono entre los átomos de nitrógeno y de carbono a los cuales están unidos o
(c) R10 es H y R11 es SOzM, en donde z es 2 o 3 y M es un catión monovalente farmacéuticamente aceptable o ambos M juntos son un catión divalente farmacéuticamente aceptable y
el asterisco indica el punto de unión a la unidad de enlazador (LU), en donde LU es:

Y es NH y E es ácido glucurónico
A1 es una unidad de extensión.
Los conjugados preferidos de la presente invención incluyen aquellos en donde A1 es

en la que la línea ondulada indica el punto de unión a la unidad de ligando (por ejemplo, anticuerpo) y el asterisco indica el punto de unión a D.
En una realización particularmente preferida, para todos los conjugados, la conexión entre el anticuerpo y la unidad de enlazador se forma entre un grupo tiol de un resto de cisteína del anticuerpo y un grupo maleimida de la unidad del enlazador.
En una realización particularmente preferida, para todos los conjugados preferidos, el anticuerpo es un anticuerpo monoclonal que se une específicamente al antígeno Cripto, al antígeno CD19, al antígeno CD20, al antígeno CD22, al antígeno CD30, al antígeno CD33, a la glicoproteína NMB, al antígeno CanAg, al antígeno Her2 (ErbB2/Neu ), al antígeno CD56 (NCAM), al antígeno CD70, al antígeno CD79, al antígeno CD138, al PSCA, al Ps MA (antígeno prostático específico de membrana), al BCMA, a la E-selectina, EphB2, a la melanotransferina, al antígeno Muc16 o al antígeno TMEFF2.
Ejemplos
Métodos experimentales generales para el Ejemplo 1
Las rotaciones ópticas se midieron en un polarímetro ADP 220 (Belitngham Stanley Ltd.) y las concentraciones (c) se dan en g/100 ml. Los puntos de fusión se midieron usando un aparato digital de punto de fusión (electrotérmico). Los
espectros IR se registraron en un espectrómetro Perkin-Elmer Spectrum 1000 FT IR. Los espectros de RMN de 1H y 13C se adquirieron a 300 K usando un espectrómetro de RMN Bruker Avance a 400 y 100 MHz, respectivamente. Los desplazamientos químicos se presentan relativos a TMS ( 6 = 0,0 ppm), y las señales se designan como s (singlete), d (doblete), t (triplete), dt (doblete triplete), dd (doblete de dobletes), ddd (doble doblete de dobletes) o m (multiplete), con constantes de acoplamiento dadas en hercios (Hz). Los datos de la espectroscopia de masas (EM) se recogieron usando un instrumento Waters Micromass ZQ acoplado a una HPLC Waters 2695 con un Waters 2996 PDA. Los parámetros del Waters Micromass ZQ usados fueron: capilar (kV), 3,38; cono (V), 35; extractor (V), 3,0; temperatura de la fuente (°C), 100; temperatura de desolvatación (°C). 200; caudal del cono (L/h), 50; caudal de desolvatación (L/h), 250. Se registraron datos de espectroscopia de masas de alta resolución (EMAR) en un Waters Micromass QTOF Global en modo W positivo usando puntas de vidrio de borosilicato recubierto de metal para introducir las muestras en el instrumento. La cromatografía en capa fina (CCF) se realizó sobre placas de aluminio de gel de sílice (Merck 60, F254), y la cromatografía ultrarrápida utilizó gel de sílice (Merck 60, malla 230-400 ASTM). Excepto por el HOBt (Novabiochem) y los reactivos soportados en sólido (Argonaut), el resto de productos químicos y disolventes se adquirieron de Sigma-Aldrich y se usaron tal como se suministran sin purificación adicional. Los disolventes anhidros se prepararon por destilación bajo una atmósfera de nitrógeno seco en presencia de un agente de secado adecuado y se almacenaron sobre tamices moleculares de 4 A o alambre de sodio. El éter de petróleo se refiere a la fracción con punto de ebullición en 40-60 °C.
Condiciones generales de CL/EM: La HPLC (Waters Alliance 2695) se llevó a cabo usando una fase móvil de agua (A) (ácido fórmico al 0,1 %) y acetonitrilo (B) (ácido fórmico al 0,1 %). Gradiente: composición inicial 5% de B durante 1,0 min y después 5 % de B hasta 95 % de B durante 3 min. La composición se mantuvo durante 0,5 min a 95 % de B, y luego regresó a 5 % de B en 0,3 minutos. El ciclo total en gradiente es de 5 min. Caudal 3,0 ml/min, 400 μl se dividió con un accesorio en forma de T de volumen cero que se introduce en el espectrómetro de masas. Intervalo de detección de la longitud de onda: 220 a 400 nm. Tipo de función: matriz de diodos (535 exploraciones). Columna: Phenomenex® Onyx Monolithic C1850 x 4,60 mm
Ejemplo 1 no reivindicado

(a) (S)-2-(4-metoxifenil)-7-metoxi-8-(3-((S)-7-metoxi-2-(trifluorometilsulfonil)-5,11 -dioxo- 10-((2-(trimetilsilil)etoxi)metil)-5,10,11,11a-tetrahidro-1H-pirrolo[2,1-c][1,4]benzodiazepin-8-iloxi)pentoxioxi)-10-((2-(trimetilsilil)etoxi)metil)-1H-pirrolo[2,1-c] [1,4]benzodiazepina-5,11(10H, 11aH)-diona (2)
Un complejo de tetraquis-tris-fenilfosfina paladio (38 mg, 3,23 X 10-5 mol, 0,02 equiv.) se añadió a una mezcla desgasificada en agitación de 1,1-[[(pentano-1,5-diil)dioxi]bis(11aS)-7-metoxi-2-[[(trifluorometil)sulfonil]oxi]-10-((2-(trimetilsilil)etoxi)metil)-1,10,11,11a-tetrahidro-5H-pirrolo[2,1 -c][1,4]-benzodiazepin-5,11 -diona] (1) (Compuesto 8 b del documento WO 2010/043880) (185 mg, 1,62 mmol, 1,0 equiv.), ácido 4-metoxifenilborónico (234 mg, 1,54 mmol, 0,95 equiv.) y Na2CO3 (274 mg, 2,59 mmol, 1,6 equiv.) en tolueno/etanol/agua (10 ml/5 ml/5 ml). La mezcla de reacción se dejó en agitación a temperatura ambiente en una atmósfera de argón durante 3 horas. La mezcla de reacción se diluyó con acetato de etilo y la porción acuosa se separó. La porción orgánica se lavó con agua, solución
acuosa saturada de salmuera, se secó (MgSO4) y se evaporó a presión reducida. La purificación por cromatografía en columna ultrarrápida [elución en gradiente, etilacetato 30 %/n-hexano 70 % a etilacetato 80 %/n-hexano 20 %] dio como resultado el producto en forma de una espuma de color amarillo (0,7 g, 39 %). Datos analíticos: TR 3,97 min; EM (ES+) m/z (intensidad relativa) 1103 ([M + H]+, 100).
(b) (S)-2-(4-(aminometil)fenil)-7-metoxi-8-((5-(((S)-7-metoxi-2-(4-metoxifenil)-5,11-dioxo-10-((2-(trimetilsilil)etoxi)metil)-5,10,11,11a-tetrahidro-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-8-il)oxi)pentil)oxi)-10-((2-(trimetilsilil)etoxi)metil)-1H-benzo[e]pirmlo[1,2-a][1,4]diazepina-5,11(WH, 11aH)-diona (3)
Un complejo de tetraquis-tris-fenilfosfina paladio (30 mg, 2,5 X 10-5 mol, 0,04 equiv.) se añadió a una mezcla desgasificada del metoxitriflato (2) (700 mg, 0,63 mmol, 1,0 equiv.), ácido 4-aminometilfenilborónico (190 mg, 1.015 mmol, 1,6 equiv.) y Na2CO3 (303 mg, 2,85 mmol, 4,5 equiv.) en tolueno/etanol/agua (20 ml/10 ml/10 ml). La mezcla de reacción se calentó a 75 °C en una atmósfera de argón durante 3 horas. La mezcla de reacción se diluyó con acetato de etilo y la porción acuosa se separó. La porción orgánica se lavó con agua, solución acuosa saturada de salmuera, se secó (MgSO4) y se evaporó a presión reducida para dar el proporcionó el producto en forma de una espuma de color marrón. La purificación por cromatografía en columna ultrarrápida [elución en gradiente, etilacetato 50 %/n-hexano 50 % a etilacetato 100 %] dio como resultado el producto (0,55 g, 82 %). Datos analíticos: TR 3,48 min; EM (ES+) m/z (intensidad relativa) 1060 ([M+H]+, 100).
Métodos experimentales generales para el Ejemplo 2
Todos los disolventes anhidros disponibles en el mercado se usaron sin purificación adicional. La cromatografía de capa fina analítica se realizó sobre láminas de aluminio de gel de sílice 60 F254 (EMD Chemicals, Gibbstown, NJ). La cromatografía radial se realizó en un aparato Cromatotrón (Harris Research, Palo Alto, CA). La HPLC analítica se realizó en un sistema de entrega de disolvente Varian ProStar 210 configurado con un detector Varian ProStar 330 PDA. Las muestras se eluyeron en una columna C12 Phenomenex Synergi 2,0 x 150 mm, 4 μm, 80 A de fase inversa. La fase móvil ácida consistió en acetonitrilo y agua, conteniendo ambos bien ácido trifluoroacético al 0,05 % o ácido fórmico al 0,1 % (denotado para cada compuesto). Los compuestos se eluyeron con un gradiente lineal de acetonitrilo ácido desde el 5 % a 1 min después de la inyección, a 95 % a 11 min, seguido de acetonitrilo isocrática al 95% a 15 minutos (caudal = 1,0 ml/min). La CLEM se realizó en un espectrómetro de masas ZMD Micromass interconectado con un instrumento HP Agilent 1100 HPLC equipado con una columna C12 Phenomenex Synergi 2,0 x 150 mm, 4 μm, 80 A de fase inversa. El eluyente ácido consistía en un gradiente lineal de acetonitrilo del 5 % al 95% en ácido fórmico acuoso al 0,1 % durante 10 min, seguido de acetonitrilo isocrático al 95% durante 5 min (caudal = 0,4 ml/min). La HPLC preparativa se llevó a cabo en un sistema de entrega de disolvente Varian ProStar 210 configurado con un detector Varian ProStar 330 PDA. Los productos se purificaron sobre una columna C12 Phenomenex Synergi 10,0 x 250 mm, 4 μm, 80 A de fase inversa eluyendo con ácido fórmico al 0,1 % en agua (disolvente A) y ácido fórmico al 0,1 % en acetonitrilo (disolvente B). El método de purificación consistió en el siguiente gradiente de disolvente A a disolvente B: 90:10 de 0 a 5 min; 90:10 a 10:90 de 5 min a 80 min; seguido de isocrático 10:90 durante 5 min. El caudal fue de 4,6 ml/min con seguimiento a 254 nm. Los datos de los espectros de RMN se recogieron en un espectrómetro Varian Mercury 400 MHz. Las constantes de acoplamiento (J) se notifican en hertzios.
Ejemplo 2 no reivindicado

(a) ((S)-1-(((S)-1-((4-((S)-7-Metoxi-8-((5-(((S)-7-metoxi-2-(4-metoxifenil)-5,11-dioxo-10-((2-(tnmetilsilil)etoxi)metil)-5,10,11,11a-tetrahidro-lH-benzo[e]pirrolo[1,2-a][1,4]diazepin-8-il)oxi)pentil)oxi)-5,11-dioxo-10-((2-(trimetilsilil)etoxi)metil)-5,10,11,11a-tetrahidro-lH-benzo[e]pirrolo[1,2-a][1,4]diazepin-2-il)bencil)amino)-1-oxopropan-2-il)amino)-3-metil-1-oxobutan-2-il)carbamato de alilo(4)
Un matraz de 10 ml se cargó con alloc-Val-Ala (15 mg, 57 pmol), EEDQ (17 mg, 69 μmol) y 0,72 ml de CH2CI2 anhidro. Se añadió metanol (40 μl) para facilitar la disolución y la mezcla se agitó en una atmósfera de nitrógeno durante 15 minutos. Se añadió SEM-dilactamabencilamina 3 (40 mg, 38 μmol) a continuación y la reacción se agitó a temperatura ambiente durante 4 horas, momento en el cual la CL-EM reveló la conversión en producto. La reacción se concentró, se disolvió en una cantidad mínima de CH2CI2, y se purificó por cromatografía radial en una placa de cromatotrón de 1 mm eluida con mezclas CH2Ch/MeOH (100:0 a 90:10 CH2Cl2/MeOH) para proporcionar 4 (42 mg, 85 %). CL-EM: t* 15,50 min, m/z (ES+) observado 1315,1 (M+H)+.
(b) ((S)-1-(((S)-1-((4-((S)-7-Metoxi- 8-((5-(((S)- 7-metoxi-2-(4-metoxifenil)-5-oxo-5,11a-dihidro- 1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-8-il)oxi)pentil)oxi)-5-oxo-5,11a-dihidro-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-2-il)bencil)amino)-1-oxopropan-2-il)amino)-3-metil-1-oxobutan-2-il)carbamato de alilo (5)
Un matraz de 10 ml secado a la llama se cargó con 4 (40 mg, 30 pmol) y THF anhidro (0,6 ml) y se enfrió a -78 °C. Se añadió trietilborohidruro de litio (60 μl de una solución 1 M en t Hf ) gota a gota y la reacción se agitó en atmósfera de nitrógeno a -78 °C durante 2 h, momento en el cual la CL-EM reveló aproximadamente una conversión del 50 %. A continuación se añadieron 30 μl del reductor, y la agitación se continuó durante dos horas más, momento en el que la reacción se había completado. Después la reacción se detuvo mediante la adición de 1 ml de agua y se calentar hasta temperatura ambiente, después se diluyó con 25 ml de salmuera y se extrajo tres veces con 25 ml de diclorometano. Los extractos orgánicos combinados se lavaron con salmuera, se secaron con sulfato sódico, y se concentraron a sequedad. El residuo obtenido se disolvió en cloroformo (0,75 ml), etanol (2 ml, y agua (0,3 ml), y se añadieron 800 mg de gel de sílice, proporcionando una suspensión espesa que se cerró herméticamente y se agitó a temperatura ambiente durante cuatro días. En ese momento, el análisis por CCF reveló
la conversión a la imina 5. El gel de sílice se filtró a continuación y se lavó varias veces con metanol al 10 % en cloroformo hasta que no se observó más absorbancia del PBD en el filtrado. A continuación los extractos orgánicos combinados se lavaron con salmuera, se secaron con sulfato sódico, y se concentraron a sequedad. El residuo se disolvió a continuación en la cantidad mínima de CH2Ch, y se purificó por cromatografía radial en una placa de cromatotrón de 1 mm eluida con mezclas de CH2Ch/MeOH (100:0 a 90:10 CH2Ch/MeOH) para proporcionar 3 (19 mg, 61 %). HPLC analítica: tR 11,99 min. CL-EM: tR 12,76 min, m/z (ES+) observado 1022,4 (M+H)+.
(c) 6-(2,5-Dioxo-2,5-dihidro-1H-pirrol-1-il)-N-((S)-1-(((S)-1-((4-((S)-7-metoxi-8-((5-(((S)- 7-metoxi-2-(4-metoxifenil)-5-oxo-5,11a-dihidro-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-8-il)oxi)pentil)oxi)-5-oxo-5, 11a-dihidro-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-2-il)bencil)amino)-1-oxopropan-2-il)amino)-3-metil-1-oxobutan-2-il)hexanamida (6)
La diimina de PMD 5 protegida con Alloc (19 mg, 19 μmol) se añadió a un matraz secado con llama y se disolvió en diclorometano anhidro (1,9 ml). Se añadieron trifenilfosfina (0,25 mg, 1 pmol), pirrolidina (3,1 μl, 38 μl), y tetraquis(trifenilfosfina)paladio (0) (0,5 mg, 0,5 μmol) se añadieron, la reacción se agitó a continuación en una atmósfera de nitrógeno durante 30 minutos a temperatura ambiente, momento en el cual la CL-EM reveló la desprotección completa de alloc. La reacción se cargó directamente sobre una placa de cromatotrón de 1 mm eluida con mezclas de C ^C^/M eO H (100:0 a 80:20 CH2Ch/MeOH) para proporcionar la amina libre (14 mg, 79 %). HPLC analítica: tR 9,32 min. CL-EM: tR 11,61 min, m/z (ES+) observado 938,5 (M+H)+. La amina libre se disolvió a continuación en DMF anhidra (0,37 ml) y se añadió éster de maleimidocaproil NHS (6,9 mg, 22 pmol), seguido de diisopropiletilamina (13 μl, 75 μl). La reacción se agitó a temperatura ambiente durante tres horas, momento en el cual la CL-EM reveló el consumo completo del material de partida. La mezcla de reacción se diluyó con diclorometano y se purificó por cromatografía radial en una placa de cromatotrón de 1 mm eluida con mezclas de CH2Ch/MeOH (100:0 a 90:10 CH2Ch/MeOH) para proporcionar 4 (15 mg, 89%). HPLC analítica: tR 11,63 min. CLEM: tR 12,73 min, m/z (ES+) observado 1132,1 (M+H)+.
Métodos experimentales generales para los Ejemplos 3-7. Todos los disolventes anhidros disponibles en el mercado se usaron sin purificación adicional. La cromatografía de capa fina analítica se realizó sobre láminas de aluminio de gel de sílice 60 F254 (EMD Chemicals, Gibbstown, NJ). La cromatografía radial se realizó en un aparato Cromatotrón (Harris Research, Palo Alto, CA). La HPLC analítica se realizó en un sistema de entrega de disolvente Varian ProStar 210 configurado con un detector Varian ProStar 330 PDA. Las muestras se eluyeron en una columna C12 Phenomenex Synergi 2,0 x 150 mm, 4 μm, 80 A de fase inversa. La fase móvil ácida consistió en acetonitrilo y agua, conteniendo ambos bien ácido trifluoroacético al 0,05% o ácido fórmico al 0,1% (denotado para cada compuesto). Los compuestos se eluyeron con un gradiente lineal de acetonitrilo ácido desde el 5 % a 1 min después de la inyección, a 95 % a 11 min, seguido de acetonitrilo isocrático al 95 % a 15 minutos (caudal = 1,0 ml/min). La CLEM se realizó en un espectrómetro de masas ZMD Micromass interconectado con un instrumento HP Agilent 1100 HPLC equipado con una columna C12 Phenomenex Synergi 2,0 x 150 mm, 4 μm, 80 A de fase inversa. El eluyente ácido consistía en un gradiente lineal de acetonitrilo del 5% al 95% en ácido fórmico acuoso al 0,1 % durante 10 min, seguido de acetonitrilo isocrático al 95 % durante 5 min (caudal = 0,4 ml/min). La HPLC preparativa se llevó a cabo en un sistema de entrega de disolvente Varian ProStar 210 configurado con un detector Varian ProStar 330 PDA. Los productos se purificaron sobre una columna C12 Phenomenex Synergi 10,0 x 250 mm, 4 μm, 80 A de fase inversa eluyendo con ácido fórmico al 0,1 % en agua (disolvente A) y ácido fórmico al 0,1 % en acetonitrilo (disolvente B). El método de purificación consistió en el siguiente gradiente de disolvente A a disolvente B: 90:10 de 0 a 5 min; 90:10 a 10:90 de 5 min a 80 min; seguido de isocrático 10:90 durante 5 min. El caudal fue de 4,6 ml/min con seguimiento a 254 nm. Los datos de los espectros de RMN se recogieron en un espectrómetro Varian Mercury 400 MHz. Las constantes de acoplamiento (J) se notifican en hertzios.
Ejemplo 3 no reivindicado

(a) (S)-2-(4-(aminometil)fenil) - 7-metoxi- 8-((5-(((S)-7-metoxi-2-(4-metoxifenil) -5-oxo-5,11a-dihidro- 1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-8-il)oxi)pentil)oxi)-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-5( 11aH)-ona (7)
Un matraz secado a la llama se cargó con dilactama SEM 3 (100 mg, 94 |jmol, 1 eq) disuelta en tetrahidrofurano anhidro (THF, 1,9 ml), y se enfrió a -78 °C. Se añadió gota a gota trietilborohidruro de litio (0,19 ml de una solución 1 M en THF, 188 jmol, 2 equiv.) se añadió gota a gota y la reacción se agitó en una atmósfera de nitrógeno durante 1 hora, momento en el cual la CL reveló una conversión incompleta en producto. Se añadieron 0,1 ml más del reductor y la reacción se agitó durante una hora más. La reacción se interrumpió por adición de agua (3 ml) y se dejó calentar a temperatura ambiente, después se diluyó con salmuera (25 ml) y se extrajo tres veces con diclorometano (25 ml). Los extractos orgánicos combinados se lavaron con salmuera (25 ml), se secaron con sulfato de sodio, y se evaporaron a sequedad. El residuo se disolvió en una mezcla de diclorometano (2,4 ml), etanol (6,2 ml), y agua (0,9 ml), y se añadió gel de sílice (2,4 g). La suspensión resultante se agitó a temperatura ambiente durante 3 días. El análisis por CCF reveló la conversión a la imina 7, momento en el cual la suspensión se filtró sobre un embudo de vidrio sinterizado y la torta de gel de sílice se lavó con metanol al 10 % en cloroformo hasta que no se observó más absorbancia de PBD en el filtrado. La concentración del filtrado proporcionó 70 mg del dímero de imina 7 en bruto, que se dividió, y 40 mg se tomaron para su purificación. El material se disolvió en una cantidad mínima de diclorometano y se purificó por cromatografía radial en una placa de cromatotrón de 1 mm eluida con mezclas de CH2Cl2/MeOH (100:0 a 80:20) para proporcionar 7 (11 mg, 27 % para el material dividido). CCF: Rf = 0,21, MeOH al 20 % en CH2CI2. CL-EM: t* 11,30 min, m/z (ES+) observado 768,3 (M+H)+.
(b) 4-((S)-7-metoxi-8- ((5-(((S)-7-metoxi-2-(4-metoxifenil) -5-oxo-5,11a-dihidro- 1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-8-il)oxi)pentil)oxi)-5-oxo-5,11a-dihidro- 1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-2-il)bencilcarbamato de 4-((S)-2-((S)-2-(6-(2,5-dioxo-2,5-dihidro- 1H-pirrol-1 -il)hexanamido)-3-metilbutanamido)-5-ureidopentanamido)bencilo (9)
Un matraz secado a la llama se cargó con bencilamina 7 (7,3 mg, 9,5 jmol, 1 equiv.) disuelta en dimetilformamida anhidra (0,2 ml). Se añadió maleimidocaproil-Valina-Citrulina-PAB-OCO-pNP (Dubowchik et al., Bioconjugate Chemistry, 2002, 13, 855-869) (7 mg, 9,5 jmol, 1 equiv.), seguido de diisopropiletilamina (16,5 jl, 95 jmol, 10 equiv.), la reacción se agitó a continuación a temperatura ambiente en una atmósfera de nitrógeno. La CL reveló la conversión del producto después de 1,5 horas; la reacción se diluyó con diclorometano y se cargó directamente sobre una placa de cromatotrón de 1 mm eluida con mezclas de CH2Cb/MeOH mixtures (100:0 a 80:20) para proporcionar en enlazador de fármaco 9 purificado (9,3 mg, 72 %). CCF: Rf = 0,24, MeOH al 10 % en CH2CI2. CLEM: t* 12,61 min, m/z (ES+) observado 1366,8 (M+H)+.
Ejemplo 4

(a) Triacetato de (2S,3R,4S,5R,6R)-2-(2-(3-((((9H-fluoren-9-il)metoxi)carbonil)amino)propanamido)-4-((((4-((S)-7-metoxi-8-((5-(((S)-7-metoxi-2-(4-metoxifenil)-5-oxo-5,11a-dihidro- 1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-8-il)oxi)pentil)oxi)-5-oxo-5,11a-dihidro- 1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-2-il)bencil)carbamoil)oxi)metil)fenoxi)-6-metiltetrahidro-2H-piran-3,4,5-triilo (12)
Un matraz secado a la llama se cargó con dilactama SEM 3 (40 mg, 38 |jmol, 1 eq) disuelta en tetrahidrofurano anhidro (0,8 ml), y se enfrió a -78 °C. Se añadió gota a gota trietilborohidruro de litio (80 j l de una solución 1 M en THF, 76 jmol, 2 eq.) y la reacción se agitó bajo una atmósfera de nitrógeno durante 1,5 horas, momento en el cual la CL reveló una conversión incompleta en producto. Se añadieron 40 j l del reductor y la reacción se agitó durante una hora más. La reacción se interrumpió por adición de agua (1 ml) y se dejó calentar a temperatura ambiente, después se diluyó con salmuera (25 ml) y se extrajo tres veces con diclorometano (25 ml). Los extractos orgánicos combinados se lavaron con salmuera (25 ml), se secaron con sulfato de sodio, y se evaporaron a sequedad. La SEM carbinolamina 10 (39 mg) se llevó a la siguiente etapa sin purificación adicional. Un enlazador de glucurónico 11 activado (Jeffrey et at., Bioconjugate Chemistry, 2006, 17, 831-840) (38 mg, 42 jmol, 1,1 equiv.) se disolvió en dimetilformamida anhidra (0,6 ml) y se añadió a un matraz que contenía 10 (39 mg, 37 jmol, 1 equiv.). Se añadió diisopropiletilamina (13 jl, 74 jmol, 2 equiv.) y la reacción se agitó a temperatura ambiente bajo atmósfera de nitrógeno; después de 1,5 horas, la CL-EM reveló la conversión al producto acoplado. La reacción se diluyó con salmuera (25 ml) y se extrajo tres veces con diclorometano (25 ml). Los extractos orgánicos combinados se lavaron con salmuera (25 ml), se secaron con sulfato de sodio, y se evaporaron a sequedad. El residuo obtenido se disolvió en una mezcla de cloroformo (0,7 ml), etanol (1,25 ml), y agua (0,17 ml), y gel de sílice (1 g) se añadió. La suspensión resultante se agitó a temperatura ambiente durante 3 días. El análisis por CCF reveló la conversión a la imina 12, momento en el cual la suspensión se filtró sobre un embudo de vidrio sinterizado y la torta de gel de sílice se lavó con metanol al 10% en cloroformo hasta que no se observó más absorbancia de PBD en el filtrado. La concentración del filtrado proporcionó el producto 12 en bruto. El material se disolvió en una cantidad mínima de diclorometano y se purificó por cromatografía radial en una placa de cromatotrón de 1 mm eluida con mezclas de CH2Cl2/MeOH (100:0 a 80:20) para proporcionar 12 (25 mg, 36 %). CL-EM: m/z (ES+) observado 1542,9 (M+H)+.
(b) ácido (2S,3S,4S,5R,6S)-6-(2-(3-aminopropanamido)-4-((((4-((S)-7-metoxi-8-((5-(((S)-7-metoxi-2-(4-metoxifenil)-5-oxo-5,11a-dihidro-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-8-il)oxi)pentil)oxi)-5-oxo-5,11a-dihidro-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-2-il)bencil)carbamoil)oxi)metil)fenoxi)-3,4,5-trihidroxitetrahidro-2H-piran-2-carboxílico (13)
El enlazador de glucurónido protegido 12 (25 mg, 16 pmol, 1 equiv.) se disolvió en metanol (0,5 ml) y tetrahidrofurano (0,5 ml), y se enfrió a 0 °C. Hidróxido de litio monohidrato (4 mg, 96 pmol, 6 equiv.) se disolvió en agua (0,5 ml) y se añadió gota a gota a la reacción, que se dejó calentar hasta la temperatura ambiente después y se controló mediante CL-EM. Se añadió más cantidad de LiOH (3,2 mg, 76 pmol, 4,8 equiv.) en 0,4 ml de agua a la reacción después de 2 h para impulsar adicionalmente la conversión al producto. Se añadió ácido acético glacial (11 μl, 195 pmol, 12 equiv.), seguido de 1 ml de dimetilsulfóxido, a continuación, los disolventes volátiles se eliminaron por evaporación rotatoria. El producto en bruto se purificó por HPLC preparativa para proporcionar el enlazador de glucurónido desprotegido 13 (2 mg, 11 %). CL-EM: tR 11,54 min, m/z (ES+) observado 1180,0 (M+H)+.
(c) ácido (2S, 3S, 4S, 5R, 6S)-6-(2-(3-(6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanamido)propanamido)-4-((((4-((S)-7-metoxi- 8-((5-(((S)- 7-metoxi-2-(4-metoxifenil)-5-oxo-5,11a-dihidro- 1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-8-H)oxi)pentil)oxi)-5-oxo-5,11a-dihidro-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-2-il)bendl)carbamoil)oxi)metil)fenoxi)-3-4,5-trihidroxitetrahidro-2H-piran-2-carboxílico (14)
Un matraz secado a la llama se cargó con enlazador de glucurónido 13 (2 mg, 1,7 pmol, 1 equiv.), N-hidroxisuccinimida éster del ácido maleimidocaproico (0,8 mg, 2,6 pmol, 1,5 equiv.), y dimetilformamida anhidra (85 μl). Se añadió diisopropiletilamina (1,5 μl, 8,5 pmol, 5 equiv.), la reacción se agitó a continuación a temperatura ambiente en una atmósfera de nitrógeno. Después de 2 horas, la HPLC reveló la conversión al producto. La reacción se diluyó en dimetilsulfóxido y se purificó por HPLC preparativa para proporcionar el enlazador de glucurónido PBD 14 (1,4 mg, 61 %). CL-EM: tR 12,30 min, m/z (ES+) observado 1373,7 (M+H)+.
Ejemplo 5 no reivindicado

(a) trifluorometanosulfonato de (S)-8-((5-(((S)-2-(4-aminofenil)-7-metoxi-5,11-dioxo-10-((2-(trimetilsiIil)etoxi)metil)-5,10,11,11a-tetrahidro-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin -8-il)oxi)pentil)oxi)-7-metoxi-5,11-dioxo-10-((2-(trimetilsilil)etoxi)metil)-5,10,11,11a-tetrahidro-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-2-ilo (15)
Un matraz se cargó con bistriflato 1 (500 mg, 437 pmol, 1 equiv.) disuelto en tolueno (6,5 ml), etanol (3,2 ml), y agua (3,2 ml). A la solución agitada se añadió éster de pinacol del ácido 4-aminofenilborónico (87 mg, 398 pmol, 0,91
equiv.), carbonato sódico (213 mg, 2,0 mmol, 4,6 equiv.), y tetraquis(trifenilfosfina)paladio (0) (20 mg, 17,5 |jmol, 0,04 equiv.), la reacción se agitó vigorosamente a temperatura ambiente bajo atmósfera de nitrógeno con seguimiento mediante CL-EM. Después de tres horas la reacción había avanzado hasta aproximadamente una conversión del 50 % hasta el producto. La reacción se concentró y después se repartió entre acetato de etilo (100 ml) y agua (100 ml). A continuación, la capa orgánica se lavó con agua (100 ml) y salmuera (100 ml), se secó sobre sulfato de sodio y se concentró a sequedad para proporcionar anilina triflato 15 bruta. El producto bruto se purificó por cromatografía ultrarrápida, eluyendo con mezclas de hexanos:acetato de etilo (60:40 a 30:70), para proporcionar anilina triflato 15 pura (118 mg, 25 %). CCF: Fr = 0,43, 25 % hexanos en acetato de etilo. CLEM: t« 8,30 min, m/z (ES+) observado 1088,2 (M+H)+.
(b) (S)-2-(4-aminofenil)-8-((5-(((S)-2-(4-(3-dimetilamino)propoxi)fenil)-7-metoxi-5,11-dioxo-10-((2-(trimetilsilil)etoxi)metil)-5,10,11,11a-tetrahidro-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-8-il)oxi)pentil)oxi)-7-metoxi-10-((2- (trimetilsilil)etoxi)metil)-1H-benzo[e]pirrolo[1,2-a][1,4]diazepina-5,11(10H, 11aH)-diona (16)
Un matraz se cargó con anilina triflato 15 (118 mg, 109 jmol, 1 equiv.) disuelta en tolueno (0,7 ml), etanol (2,3 ml), y agua (0,3 ml). A la solución agitada se añadió éster de pinacol del ácido 4-[3-(dimetilamino)propiloxi]fenilborónico (43 mg, 142 jmol, 1,3 equiv.), carbonato sódico (53 mg, 0,5 mmol, 4,6 equiv.), y tetraquis(trifenilfosfina)paladio (0) (5 mg, 4,4 jmol, 0,04 equiv.), la reacción se agitó vigorosamente a temperatura ambiente bajo atmósfera de nitrógeno con seguimiento mediante CL-EM. Después de cuatro horas la reacción había llegado a la finalización. La reacción se concentró y después se repartió entre acetato de etilo (25 ml) y agua (25 ml). La capa acuosa se extrajo dos veces con acetato de etilo (25 ml). A continuación, la capa orgánica se lavó con agua (50 ml) y salmuera (50 ml), se secó sobre sulfato de sodio y se concentró a sequedad para proporcionar dilactama SEM 16 bruta. El producto bruto se purificó por cromatografía ultrarrápida, eluyendo con mezclas de hexanos:acetato de etilo (50:50 a 0:100), para proporcionar el producto puro 16 (78 mg, 64%). CCF: Fr = 0,38, metanol al 20% en CH2CI2. CL-EM: m/z (ES+) observado 1117,8 (M+H)+. RMN 1H (d7-DMF) 8 (ppm) 0,00 (s, 18H), 0,90 (m, 4H), 1,74 (m, 3H), 1,96 (m, 6H), 2,21 (S, 6H), 2,44 (t, J = 7,2 Hz, 2H), 3,25 (m, 2H), 3,62 (m, 4H), 3,80 (m, 2H), 3,96 (s, 5H), 4,11 (t, J = 6,4 Hz, 2H), 4,20 (m, 3H), 4,92 (m, 2H), 5,31 (dd, J = 6, 10 Hz, 2H), 5,48 (m, 4H), 6,73 (t, J = 8,4 Hz, 2H), 7,01 (t, J = 8,8 Hz, 2H), 7,29 (m, 3H), 7,40 (m, 4H), 7,47 (m, 1H), 7,56 (t, J = 8,4 Hz, 2H).
(c) (S)-2-(4-aminofenil)-8-((5-(((S)-2-(4-(3-(dimetilamino)propoxi)fenil)-7-metoxi-5-oxo-5,11a-dihidro-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-8-il)oxi)pentil)oxi)-7-metoxi-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-5(11aH)-ona (17)
Un matraz secado a la llama se cargó con dilactama SEM 16 (70 mg, 63 jmol, 1 eq) disuelta en tetrahidrofurano anhidro (1,3 ml), y se enfrió a -78 °C. Se añadió gota a gota trietilborohidruro de litio (0,13 ml de una solución 1 M en THF, 126 jmol, 2 equiv.) se añadió gota a gota y la reacción se agitó en una atmósfera de nitrógeno durante 1,5 h, momento en el cual la CL reveló una conversión incompleta en producto. Se añadieron 65 j l del reductor y la reacción se agitó durante una hora más. La reacción se interrumpió por adición de agua (1 ml) y se dejó calentar a temperatura ambiente, después se diluyó con salmuera (25 ml) y se extrajo tres veces con diclorometano (25 ml). Los extractos orgánicos combinados se lavaron con salmuera (25 ml), se secaron con sulfato de sodio, y se evaporaron a sequedad. El residuo se disolvió en una mezcla de diclorometano (1,2 ml), etanol (3,2 ml), y agua (0,5 ml), y se añadió gel de sílice (1,6 g). La suspensión resultante se agitó a temperatura ambiente durante 4 días. El análisis de CCF reveló la conversión a dímero de imina 17, momento en el cual la suspensión se filtró sobre un embudo de vidrio sinterizado y la torta de gel de sílice se lavó con metanol al 10 % en cloroformo hasta que no se observó más absorbancia de PBD en el filtrado. La concentración del filtrado proporcionó el dímero imina bruto 17. El material se disolvió en diclorometano mínimo y se purificó por cromatografía radial sobre una placa de cromatotrón de 1 mm eluída con mezclas CH2Ch/MeOH (100:0 a 60:40) para proporcionar 17 (31 mg, 60%). CL-EM: tR 11,14 min, m/z (ES*) observado 825,4 (M+H)+.
Ejemplo 6 no reivindicado

N-(4-((S)-8-((5-(((S)-2-(4-(3-(dimetilamino)propoxi)fenil) - 7-metoxi-5-oxo-5,11a-dihidro- 1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-8-il)oxi)pentil)oxi)- 7-metoxi-5-oxo-5,11a-dihidro- 1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-2-il)fenil)-6-(2,5-dioxo-2,5-dihidro- 1H-pirrol- 1-il)hexanamida (18)
Un matraz secado a la llama se cargó con ácido maleimidocaproico (5,2 mg, 25 pmol, 1,5 equiv.) disuelto en 0,33 ml de metanol al 5 % en diclorometano anhidro. El ácido fue activado previamente mediante la adición de N-etoxicarbonil-2-etoxi-1,2-dihidroquinolina (7,3 mg, 30 pmol 1,8 eq), seguido de agitación a temperatura ambiente bajo atmósfera de nitrógeno durante 15 minutos. A continuación, se añadió el ácido activado a un matraz secado a la llama que contiene el dímero PBD 17 (13,5 mg, 16 pmol, 1 eq). La reacción se agitó durante 4 h a temperatura ambiente en una atmósfera de nitrógeno, momento en el cual la CL-EM reveló la conversión en producto. El material se diluyó con diclorometano y se purificó por cromatografía radial en una placa de cromatotrón de 1 mm eluida con mezclas de CH2Ch/MeOH (100:0 a 80:20) para proporcionar 18 (7,3 mg, 44%). CL-EM: t* 9,09 min, m/z (ES+) observado 1018,3 (M+H)+.
Ejemplo 7 no reivindicado

N-((S)-1-(((S)-1-((4-((S)-8-((5-(((S)-2-(4-(3-(dimetilamino)propoxi)fenil)-7-metoxi-5-oxo-5,11a-dihidro-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-8-il)oxi)pentil)oxi)-7-metoxi-5-oxo-5,11a-dihidro-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-2-il)fenil)amino)-1-oxopropan-2-il)amino)-3-metil-1-oxobutan-2-il)-6-(2,5-dioxo-2,5-dihidro- 1H-pirrol-1-il)hexanamida (19)
Un matraz secado a la llama se cargó con enlazador de maleimidocaproil-valina-alanina (compuesto 36 del Ejemplo 13 del documento WO 2011/130613 A1) (9 mg, 24 pmol, 1,5 eq) disuelto en 0,33 ml de metanol al 5% en diclorometano anhidro. El ácido fue activado previamente mediante la adición de N-etoxicarbonil-2-etoxi-1,2-dihidroquinolina (7,1 mg, 29 pmol 1,8 eq), seguido de agitación a temperatura ambiente bajo atmósfera de nitrógeno durante 15 minutos. A continuación, se añadió el ácido activado a un matraz secado a la llama que contiene el dímero PBD 17 (13 mg, 16 pmol, 1 eq). La reacción se agitó durante 7 h a temperatura ambiente en una atmósfera de nitrógeno, momento en el cual la CL-EM reveló la conversión en producto. El material se diluyó en diclorometano y se purificó por cromatografía radial sobre una placa de cromatotrón de 1 mm eluida con mezclas CH2Cl2/MeOH (100:0 a 80:20) para proporcionar 19 (5,1 mg, 27 %). CL-EM: t* 9,09 min, m/z (ES+) observado 1188,4 (M+H)+.
Ejemplo 8 no reivindicado

(a) 2-(4-(2,5,8,11-tetraoxatrídecan-13-iloxi)fenil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (21)
A una mezcla de éster de pinacol del ácido 4-hidroxifenilborónico (880 mg, 4 mmol), bromometiltetraetilenglicol (1,6 g, 6 mmol) y DMF (10 ml) se añadió Cs2CO3 (1,5 g; 8 mmol). La mezcla de reacción se agitó durante -65 h, y se vertió en acetato de etilo (100 ml). La mezcla se lavó con HCl 0,1 N (200 ml), agua (3 x 100 ml) y salmuera (50 ml) y la fase orgánica se secó con Na2SO4. La decantación y la concentración proporcionaron un aceite de color pardo que se purificó en una placa de cromatotrón radial de 2 mm eluida con acetato de etilo al 50 % en hexanos seguido de acetato de etilo al 100 % para dar 1,21 g (74 %): RMN (d6-DMSO, 400 MHz) 7,59 (d, J = 8,6 Hz, 2H), 6,93 (d, J = 8,6 Hz, 2H), 4,11 (t, J = 4,7 Hz, 2H), 3,73 (m, 2H), 3,60-3,45 (m, 14H), 3,41 (m, 2H), 3,23 (s, 3H), 1,27 (s, 12H); CL-EM: m/z (ES+) observado 433,64 (M+Na)+.
(b) (R)-2-(4-(2,5,8,11-tetraoxatridecan-13-iloxi)fenil)-8-((5-(((R)-2-(4-aminofenil)-7-metoxi-5,11-dioxo-10-((2-(trimetilsilil)etoxi)metil)-5,10,11,11a-tetrahidro-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-8-il) oxi)pentil)oxi)-7-metoxi-10-((2-(trimetilsilil)etoxi)metil)-1H-benzo[e]pirrolo[1,2-a][1,4]diazepina-5,11(10H, 11aH)-diona (22)
A una mezcla del monotriflato 15 (200 mg, 0,18 mmol) y éster de boronato TEG (111 mg, 0,27 mmol) en una mezcla de tolueno (2 ml) y etanol (1 ml) se añadió Na2CO32 M (0,5 ml) y tetraquis Pd (6 mg, 0,054 mmol). Después de agitar a temperatura ambiente durante 3 h, la mezcla de reacción se vertió en acetato de etilo, se lavó con agua (3 x 50 ml) y salmuera (50 ml) y se secó con Na2SO4. La solución se decantó, se concentró y después se purificó en una placa de cromatotrón radial de 2 mm eluida con metanol al 5 % en diclorometano. Esto produjo 207 mg (94 %) en forma de un sólido de color amarillo: RMN 1H (CDCI3 , 400 MHz) □ 7,39-7,34 (m, 7H), 7,26 (m, 2H), 6,89 (d, J = 9,0 Hz, 2H), 6,66 (d, J = 8,6 Hz, 2H), 4,70 (dd, J = 10,2, 2,0 Hz, 2H). 4,18-4,03 (m, 8H), 3,93 (s, 3H), 3,90-3,62 (m, 21 H), 3,55 (dd, J = 5,1, 3,2 Hz, 2H) 3,38 (s, 3H), 3,12 (pent, J = 5,0H, 2H), 2,05 (m, 6H), 1,72 (m, 2H), 1,0 (m, 4H), 0,3 (s, 18 H); CLEM: m/z (ES+) observado 1222,98 (M+H)+.
(c) (R)-2-(4-(2,5,8,11-tetraoxatridecan-13-iloxi)fenil)-8-((5-(((R)-2-(4-aminofenil)-7-metoxi-5-oxo-5,11a-dihidro-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-8-il)oxi)pentil)oxi)-7-metoxi-1H-benzo[e]pirrolo[1,2-a)[1,4]diazepin-5(11aH)-ona (23)
A una mezcla de la dilactama SEM (207 mg, 0,17 mmol) en THF (10 ml) a -78 °C se añadió Superhidruro (LiHBEt4 en forma de una solución 1 N en THF. 0,34 ml, 0,34 mmol). La mezcla de reacción se agitó durante aproximadamente 2 h, momento en el que el análisis por CL-EM de la mezcla de reacción reveló una conversión de aproximadamente el 50 % al intermedio de SEM-carbinoiamina completamente reducida, como material monorreducido aún presente. Se añadió una alícuota adicional de Superhidruro (0,34 ml, 0,34 mmol) y la reacción se agitó a -78 °C durante una hora más. La inspección por CL-EM reveló que la reacción aún no había progresado hasta su finalización, por tanto, se añadió una tercera alícuota de Superhidruro (0,34 ml, 0,34 mmol) y la mezcla de
reacción se introdujo en un congelador a -80 °C durante 16 horas. La mezcla de reacción se inactivó después con agua (5 ml), se dejó calentar a temperatura ambiente y se vertió en acetato de etilo (100 ml). Después de lavar con salmuera, la mezcla se secó con Na2SO4. La fase orgánica se decantó, se concentró a presión reducida, y posteriormente se trató con una mezcla de etanol (14 ml) diclorometano (5 ml), agua (7 ml) y gel de sílice (5 g) durante 72 horas. La mezcla se filtró a través de un embudo de vidrio con placa fritada y se lavó varias veces metanol al 10 % metanol en diclorometano, antes de concentrar la solución a presión reducida. La mezcla se purificó en una placa de cromatotrón radial de 2 mm eluida con metanol al 5 % metanol en diclorometano para dar 46 mg (29 %): CLEM: m/z (ES+) observado 930,85 (M+H)+.
(d) N-((S)-1-(((S)-1-((4-((R)-8-((5-(((R)-2-(4-(2,5,8,11-tetraoxatridecan-13-iloxi)fenil)-7-metoxi-5-oxo-5,11a-dihidro-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-8-il)oxi)pentil)oxi)-7-metoxi-5-oxo-5,11a-dihidro-1H-benzo[e]pirrolo[1,2-a][1,4]diazepin-2-il)fenil)amino)-1-oxopropan-2-il)amino)-3-metil-1-oxobutan-2-il)-6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanamida (24)
A una mezcla del mc-val-ala-OH (50 mg, 0,129 mmol) en metanol al 5 % en diclorometano (1 ml) a 0 °C se añadió EEDQ (32 mg, 0,129 mmol). La mezcla se agitó durante 15 min, y a continuación se añadió una solución de la anilina (44 mg, 0,044 mmol) en metanol al 5 % en diclorometano (1 ml). La mezcla de reacción se dejó en agitación durante 3 horas, se diluyó con diclorometano (2 ml) y se aspiró directamente sobre una placa de cromatotrón radial de 2 mm. El producto se eluyó con un gradiente de metanol 2,5 % al 5 % en diclorometano para dar 22,5 mg (40 %): CL-EM: m/z (ES+) observado 1294 (M+H)+.
Ejemplo 9 - Preparación de conjugados de dímeros de PBD
Anticuerpos con cisteínas introducidas: Los anticuerpos frente a CD70 que contienen un resto de cisteína en la posición 239 de la cadena pesada se redujeron totalmente mediante la adición de 10 equivalentes de TCEP y EDTA 1 mM y ajustando el pH a 7,4 con tampón Tris 1 M (pH 9,0). Después de una incubación de 1 hora a 37 °C, la reacción se enfrió a 22 °C y se añadieron 30 equivalentes de ácido deshidroascórbico para reoxidar selectivamente los disulfuros naturales, mientras que deja la cisteína 239 en el estado reducido. El pH se ajustó a 6,5 con tampón Tris 1 M (pH 3,7) y la reacción se dejó avanzar durante 1 hora a 22 °C. El pH de la solución se elevó entonces de nuevo a 7,4 mediante la adición de tampón Tris 1 M (pH 9,0). 3,5 equivalentes del enlazador de fármaco PBD en DMSO se colocaron en un recipiente adecuado para la dilución con propilenglicol antes de la adición a la reacción. Para mantener la solubilidad del enlazador de fármaco PBD, el propio anticuerpo se diluyó primero con propilenglicol hasta una concentración final del 33 % (por ejemplo, si la solución de anticuerpo estaba en un volumen de reacción de 60 ml, se añadieron 30 ml de propilenglicol). A continuación se añadió este mismo volumen de propilenglicol (30 ml en este ejemplo) al enlazador de fármaco PBD como diluyente. Después de la mezcla, se añadió la solución del enlazador de fármaco PBD en propilenglicol a la solución de anticuerpo para efectuar la conjugación; la concentración final de propilenglicol es del 50 %. La reacción se dejó avanzar durante 30 minutos y después se inactivó mediante la adición de 5 equivalentes de N-acetil cisteína. El CAF se purificó después por ultrafiltración a través de una membrana de 30 kD. (Se debe tener en cuenta que la concentración de propilenglicol usada en la reacción puede reducirse para cualquier PBD especial, ya que su único fin es mantener la solubilidad del enlazador de fármaco en el medio acuoso).
Ejemplo 10: Determinación de la actividad in vitro de conjugados seleccionados
La actividad citotóxica in vitro del conjugado de anticuerpo-fármaco seleccionado se evaluó usando un ensayo de reducción de resazurina (Sigma, St. Louis, Mo., EE.UU.) (Referencia: Doronina et al., Nature Biotechnology, 2003, 21, 778-784). El conjugado anticuerpo-fármaco se preparó como se ha descrito anteriormente.
Para el ensayo de 96 horas, se sembraron células cultivadas en crecimiento en fase logarítmica durante 24 horas en placas de 96 pocillos que contenían 150 μl de RPMI 1640 suplementado con 20 % de SFB. Se prepararon diluciones en serie de CAF en medio de cultivo de células en concentración de trabajo 4x; se añadieron 50 μl de cada dilución a las placas de 96 pocillos. Después de la adición de CAF, las células se incubaron con artículos de ensayo durante 4 días a 37 °C. La resazurina se añadió entonces a cada pocillo para conseguir una concentración final de 50 μM, y las placas se incubaron durante 4 horas adicionales a 37 °C. Las placas se leyeron entonces para determinar la extensión de la reducción del colorante en un lector de placa de fusión HT (Packard Instruments, Meridien, CT, EE.UU.) con longitudes de onda de excitación y emisión de 530 y 590 nm, respectivamente. El valor de CI50, determinado por triplicado, se define en este caso como la concentración que da como resultado una reducción del 50 % en el crecimiento celular relativo a los controles no tratados.
Haciendo referencia a la tabla siguiente, se muestra la citotoxicidad in vitro de los CAF usando el ensayo de 96 horas. Los CAF se ensayaron de nuevo frente a líneas celulares de antígeno positivas y negativas, h1F6 es el anticuerpo humanizado anti-CD70 descrito a continuación.
Actividad in vitro
T l 1 - I n M n r mi n 4 h r

T l 2 - I n M n r mi n h r

T l - I n M n r mi n h r

Ejemplo 11 no reivindicado: Determinación de la citotoxicidad in vivo de conjugados seleccionados
Se llevaron a cabo todos los estudios de acuerdo con el Animal Care and Use Committee en una instalación que está totalmente acreditada por la Association for Assessment and Accreditation of Laboratory Animal Care. La tolerabilidad del CAF se evaluó primero para asegurar que los conjugados eran tolerados en las dosis seleccionadas para los experimentos de xenoinjerto. Los ratones BALB/c fueron tratados con dosis crecientes de CAF formulado en PBS con arginina 0,5 M y 0,01 % de Tween 20. Los ratones se controlaron para la pérdida de peso y los signos externos de morbilidad después del tratamiento; los que experimentaron una pérdida de peso mayor que 20 % o mostraron signos de morbilidad fueron sacrificados. El anticuerpo usado era un anticuerpo CD70, una lgG1 de h1F6 humanizada (WO2006/113909), con una mutación puntual que sustituye la cisteína por la serina en la posición 239. La conjugación con la unidad de fármaco se realiza a través de la cisteína introducida en la posición 239. Un promedio de 2 fármacos se carga por anticuerpo.
Los experimentos de terapia in vivo se llevaron a cabo en modelos de xenoinjerto en ratones portadores de carcinoma de células renales CD70+. Los fragmentos del tumor (Caki-1) se implantaron en el flanco derecho de ratones atímicos. Los ratones se aleatorizaron a los grupos de estudio (n=5 (786-0) donde cada grupo promedia aproximadamente 100 mm3. El CAF o los controles se dosificaron ip de acuerdo con el calendario indicado. El volumen del tumor en función del tiempo se determinó usando la fórmula (L x W2)/2. Los animales se sacrificaron cuando el volumen del tumor alcanzó 1000 mm3. Los ratones que muestran regresiones duraderas se sacrificaron alrededor del día 100 después del implante.
En referencia a la Figura 1, se muestran los resultados de un estudio de tratamiento usando un conjugado h1F6-compuesto 18 y un conjugado h1F6-compuesto 19 en un modelo de carcinoma de células renales CD70+. La dosificación se realizó a q7d x 2. En la figura, * es no tratado, ■ es tratamiento con h1F63c-18 at 0,1 mg/kg, □ es tratamiento con h1F63c-18 a 0,3 mg/kg, ▲ es tratamiento con h1F63c-19 a 1 mg/kg, A es tratamiento con h1F63c-19 a 3 mg/kg.
Los resultados de un experimento de tolerabilidad en ratones con un conjugado h1F6-compuesto 6 demostró que una sola dosis de CAF a 1 mg/kg era bien tolerada sin signos de pérdida de peso ni morbilidad al cabo de 30 días. La administración de una dosis más alta (2,5 mg/kg) dio como resultado la pérdida de peso.
Claims (9)
1. Un conjugado que tiene la fórmula IV:

o una sal o un solvato del mismo farmacéuticamente aceptables;
en la que L es una unidad de ligando seleccionada de entre el grupo que consiste en un anticuerpo y un fragmento de unión a antígeno de un anticuerpo,
Lu es una unidad de enlazador,
p es de 1 a 20 y
D se selecciona de entre el grupo que consiste en D1 a D6, que tienen las estructuras:


en las que
(a) R10 es H y R11 es OH, ORA, en donde RA es alquilo C1-4 saturado o
(b) R10 y R11 forman un doble enlace nitrógeno-carbono entre los átomos de nitrógeno y de carbono a los que están unidos; o
(c) R10 es H y R11 es SOzM, en donde z es 2 o 3 y M es un catión monovalente farmacéuticamente aceptable, o ambos M conjuntamente son un catión divalente farmacéuticamente aceptable y
el asterisco indica el punto de unión de la unidad de enlazador (LU), en donde LU es:

en las que el asterisco indica el punto de unión a D, la línea ondulada indica el punto de unión a la unidad de ligando, Y es n H y E es ácido glucurónico; y
A1 es una unidad de extensión.
2. El conjugado de la reivindicación 1, en donde A1 se selecciona de entre el grupo que consiste en:

en la que el asterisco indica el punto de unión a Y, la línea ondulada indica el punto de unión a la unidad de ligando y n es de 0 a 6 ;
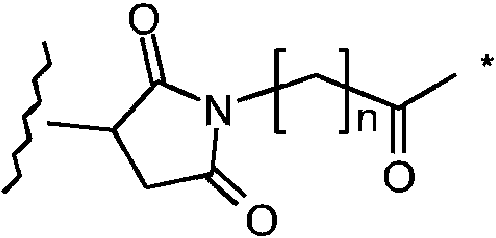
en la que el asterisco indica el punto de unión a Y, la línea ondulada indica el punto de unión a la unidad de ligando y n es de 0 a 6 ;

en la que el asterisco indica el punto de unión a Y, la línea ondulada indica el punto de unión a la unidad de ligando,
n es 0 o 1 y m es de 0 a 30 y

en la que el asterisco indica el punto de unión a Y, la línea ondulada indica el punto de unión a la unidad de ligando, n es 0 o 1 y m es de 0 a 30.
3. El conjugado de la reivindicación 1, en donde A1 es:
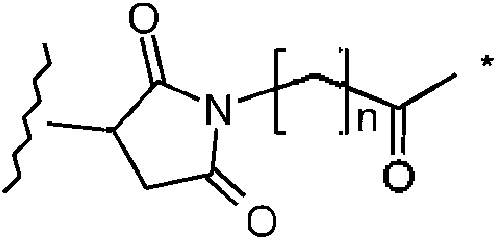
en la que el asterisco indica el punto de unión a Y, la línea ondulada indica el punto de unión a la unidad de ligando y n es de 0 a 6.
4. El conjugado de la reivindicación 3, en donde n es 5.
5. Un conjugado de acuerdo con una cualquiera de las reivindicaciones 1 a 4 para su uso en el tratamiento de una enfermedad proliferativa o una enfermedad autoinmunitaria.
6. El conjugado de la reivindicación 5 en donde la enfermedad proliferativa es un cáncer y el cáncer es una neoplasia hematológica, especialmente leucemias y linfomas, especialmente linfoma no de Hodgkin, y subtipos tales como LDCGB, zona marginal, zona del manto y folicular, linfoma de Hodgkin, LMA y otros cánceres de origen en linfocitos B o de origen en linfocitos T.
7. El conjugado de la reivindicación 5 en donde la enfermedad proliferativa es un cáncer y el cáncer es cáncer de pulmón, cáncer microcítico de pulmón, cáncer gastrointestinal, cáncer de intestino, cáncer de colon, carcinoma de mama, carcinoma de ovario, cáncer de próstata, cáncer de testículos, cáncer de hígado, cáncer de riñón, cáncer de vejiga, cáncer pancreático, cáncer de cerebro, sarcoma, osteosarcoma, sarcoma de Kaposi o melanoma.
8. Un enlazador de fármaco de fórmula V:
LU-D (V)
o una sal o un solvato farmacéuticamente aceptables del mismo, en donde LU es una unidad de enlazador y D se selecciona del grupo que consiste en D1 a D6, que tienen las estructuras:


en las que
a) R10 es H y R11 es OH, ORA, donde RA es alquilo C1-4 saturado o
b) R10 y R11 forman un doble enlace nitrógeno-carbono entre los átomos de nitrógeno y de carbono a los que están unidos o
c) R10 es H y R11 es SOzM, donde z es 2 o 3 y M es un catión monovalente farmacéuticamente aceptable o ambos M juntos son un catión divalente farmacéuticamente aceptable;
el asterisco indica el punto de unión a la unidad de ligando (LU), en donde LU es:

donde el asterisco indica el punto de unión a D, Y es NH y E es ácido glucurónico;
G1 es una unidad de extensión para formar una conexión a una unidad de ligando.
9. El enlazador a fármaco de la reivindicación 8, en donde G1 se selecciona de entre el grupo que consiste en:

en la que el asterisco indica el punto de unión a Y y n es de 0 a 6;

en la que el asterisco indica el punto de unión a Y y n es de 0 a 6;

en la que el asterisco indica el punto de unión a Y, n es 0 o 1 y m es de 0 a 30 y
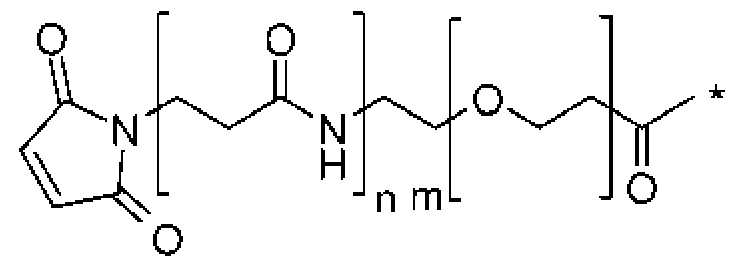
en la que el asterisco indica el punto de unión a Y, n es 0 o 1 y m es 0 a 30.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161547192P | 2011-10-14 | 2011-10-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2945932T3 true ES2945932T3 (es) | 2023-07-10 |
Family
ID=48082465
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18168997T Active ES2945932T3 (es) | 2011-10-14 | 2012-10-12 | Pirrolobenzodiazepinas y conjugados dirigidos |
| ES12840661.8T Active ES2687246T3 (es) | 2011-10-14 | 2012-10-12 | Pirrolobenzodiazepinas y conjugados dirigidos |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES12840661.8T Active ES2687246T3 (es) | 2011-10-14 | 2012-10-12 | Pirrolobenzodiazepinas y conjugados dirigidos |
Country Status (7)
| Country | Link |
|---|---|
| US (3) | US9526798B2 (es) |
| EP (2) | EP2755642B1 (es) |
| JP (2) | JP6393617B2 (es) |
| CN (2) | CN108164551A (es) |
| CA (1) | CA2850373C (es) |
| ES (2) | ES2945932T3 (es) |
| WO (1) | WO2013055990A1 (es) |
Families Citing this family (168)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0819095D0 (en) | 2008-10-17 | 2008-11-26 | Spirogen Ltd | Pyrrolobenzodiazepines |
| KR101772354B1 (ko) | 2010-04-15 | 2017-08-28 | 시애틀 지네틱스, 인크. | 표적화된 피롤로벤조디아제핀 접합체 |
| ES2623057T3 (es) | 2010-04-15 | 2017-07-10 | Medimmune Limited | Pirrolobenzodiazepinas usadas para tratar enfermedades proliferativas |
| WO2013041606A1 (en) | 2011-09-20 | 2013-03-28 | Spirogen Sàrl | Pyrrolobenzodiazepines as unsymmetrical dimeric pbd compounds for inclusion in targeted conjugates |
| US9388187B2 (en) | 2011-10-14 | 2016-07-12 | Medimmune Limited | Pyrrolobenzodiazepines |
| MX358757B (es) | 2011-10-14 | 2018-09-03 | Seattle Genetics Inc | Pirrolobenzodiazepinas y conjugados dirigidos. |
| US9526798B2 (en) | 2011-10-14 | 2016-12-27 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| BR112014008888A2 (pt) | 2011-10-14 | 2017-04-18 | Seattle Genetics Inc | pirrolobenzodiazepinas |
| HRP20181646T2 (hr) | 2012-10-12 | 2019-08-09 | Adc Therapeutics Sa | Konjugati pirolobenzodiazepin - anti-psma protutijela |
| WO2014057120A1 (en) | 2012-10-12 | 2014-04-17 | Adc Therapeutics Sàrl | Pyrrolobenzodiazepine-antibody conjugates |
| MX364328B (es) | 2012-10-12 | 2019-04-23 | Medimmune Ltd | Conjugados del anticuerpo pirrolobenzodiazepina. |
| WO2014057117A1 (en) | 2012-10-12 | 2014-04-17 | Adc Therapeutics Sàrl | Pyrrolobenzodiazepine-antibody conjugates |
| KR101645905B1 (ko) | 2012-10-12 | 2016-08-04 | 스피로즌 살 | 피롤로벤조디아제핀 및 그의 컨주게이트 |
| HRP20190366T1 (hr) | 2012-10-12 | 2019-04-19 | Medimmune Limited | Pirolobenzodiazepini i njihovi konjugati |
| ES2680153T3 (es) | 2012-10-12 | 2018-09-04 | Adc Therapeutics Sa | Conjugados de anticuerpos anti-PSMA-pirrolobenzodiazepinas |
| AU2013328673B2 (en) | 2012-10-12 | 2017-07-13 | Medimmune Limited | Synthesis and intermediates of pyrrolobenzodiazepine derivatives for conjugation |
| PL2906251T3 (pl) | 2012-10-12 | 2018-02-28 | Adc Therapeutics Sa | Koniugaty pirolobenzodiazepina-przeciwciało anty-CD22 |
| US9353150B2 (en) | 2012-12-04 | 2016-05-31 | Massachusetts Institute Of Technology | Substituted pyrazino[1′,2′:1 ,5]pyrrolo[2,3-b]-indole-1,4-diones for cancer treatment |
| AU2013366493B2 (en) | 2012-12-21 | 2017-08-24 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| CN105246894A (zh) | 2012-12-21 | 2016-01-13 | 斯皮罗根有限公司 | 用于治疗增殖性和自身免疫疾病的非对称吡咯并苯并二氮杂卓二聚物 |
| EP3441072B1 (en) | 2013-03-13 | 2020-12-30 | Seagen Inc. | Activated carbon filtration for purification of benzodiazepine adcs |
| WO2014140862A2 (en) | 2013-03-13 | 2014-09-18 | Spirogen Sarl | Pyrrolobenzodiazepines and conjugates thereof |
| MX385713B (es) | 2013-03-13 | 2025-03-18 | Seagen Inc | Ciclodextrina y formulaciones de conjugados anticuerpo-fármaco. |
| JP6445519B2 (ja) | 2013-03-13 | 2018-12-26 | メドイミューン・リミテッドMedImmune Limited | ピロロベンゾジアゼピン及びそのコンジュゲート |
| AU2014233385C1 (en) | 2013-03-15 | 2019-09-19 | Regeneron Pharmaceuticals, Inc. | Biologically active molecules, conjugates thereof, and therapeutic uses |
| US10208125B2 (en) | 2013-07-15 | 2019-02-19 | University of Pittsburgh—of the Commonwealth System of Higher Education | Anti-mucin 1 binding agents and uses thereof |
| KR102252925B1 (ko) | 2013-08-26 | 2021-05-18 | 리제너론 파마슈티칼스 인코포레이티드 | 마크롤라이드 디아스테레오머를 포함하는 약제학적 조성물, 이의 합성 방법 및 치료학적 용도 |
| US10010624B2 (en) | 2013-10-11 | 2018-07-03 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| US9950078B2 (en) | 2013-10-11 | 2018-04-24 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| EP3054986B1 (en) | 2013-10-11 | 2019-03-20 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| GB201317982D0 (en) | 2013-10-11 | 2013-11-27 | Spirogen Sarl | Pyrrolobenzodiazepines and conjugates thereof |
| GB201317981D0 (en) | 2013-10-11 | 2013-11-27 | Spirogen Sarl | Pyrrolobenzodiazepines and conjugates thereof |
| CA3187392A1 (en) | 2013-10-15 | 2015-04-23 | Seagen Inc. | Pegylated drug-linkers for improved ligand-drug conjugate pharmacokinetics |
| MX381017B (es) | 2013-12-16 | 2025-03-12 | Genentech Inc | Compuestos peptidomiméticos y conjugados anticuerpo-fármaco de estos. |
| TWI727919B (zh) | 2013-12-19 | 2021-05-21 | 美商西雅圖遺傳學公司 | 與標的-藥物結合物併用之亞甲基胺基甲酸酯連接物 |
| RS59077B1 (sr) | 2014-03-11 | 2019-09-30 | Regeneron Pharma | Anti-egfrviii antitela i njihova primena |
| WO2015157595A1 (en) | 2014-04-11 | 2015-10-15 | Medimmune, Llc | Conjugated compounds comprising cysteine-engineered antibodies |
| KR101628872B1 (ko) * | 2014-05-28 | 2016-06-09 | 주식회사 레고켐 바이오사이언스 | 자가-희생 기를 포함하는 화합물 |
| WO2016037644A1 (en) | 2014-09-10 | 2016-03-17 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| GB201416112D0 (en) | 2014-09-12 | 2014-10-29 | Medimmune Ltd | Pyrrolobenzodiazepines and conjugates thereof |
| AU2015314954B2 (en) | 2014-09-12 | 2021-05-13 | Genentech, Inc. | Anti-HER2 antibodies and immunoconjugates |
| US10780096B2 (en) | 2014-11-25 | 2020-09-22 | Adc Therapeutics Sa | Pyrrolobenzodiazepine-antibody conjugates |
| WO2016115191A1 (en) | 2015-01-14 | 2016-07-21 | Bristol-Myers Squibb Company | Benzodiazepine dimers, conjugates thereof, and methods of making and using |
| ES2747386T3 (es) | 2015-01-14 | 2020-03-10 | Bristol Myers Squibb Co | Dímeros de benzodiacepina unidos por heteroarileno, conjugados de los mismos y métodos de preparación y uso |
| KR20240142591A (ko) | 2015-03-27 | 2024-09-30 | 리제너론 파아마슈티컬스, 인크. | 메이탄시노이드 유도체, 이의 컨주게이트, 및 사용 방법 |
| GB201506402D0 (en) | 2015-04-15 | 2015-05-27 | Berkel Patricius H C Van And Howard Philip W | Site-specific antibody-drug conjugates |
| GB201506411D0 (en) | 2015-04-15 | 2015-05-27 | Bergenbio As | Humanized anti-axl antibodies |
| EA201792516A1 (ru) | 2015-06-23 | 2018-05-31 | Бристол-Маерс Сквибб Компани | Макроциклические димеры бензодиазепина, их конъюгаты, получение и применение |
| US11191844B2 (en) | 2015-07-06 | 2021-12-07 | Regeneran Pharmaceuticals, Inc. | Multispecific antigen-binding molecules and uses thereof |
| NZ741261A (en) | 2015-10-02 | 2019-11-29 | Genentech Inc | Pyrrolobenzodiazepine antibody drug conjugates and methods of use |
| EP3165532B1 (en) | 2015-11-03 | 2018-12-19 | Industrial Technology Research Institute | Auristatin derivatives, linker-drugs and ligand-drug conjugates |
| EP3380126A4 (en) | 2015-11-25 | 2019-07-24 | LegoChem Biosciences, Inc. | ANTIBODY-ACTIVE CONJUGATES WITH BRANCHED LINKERS AND ASSOCIATED METHODS |
| CA3006242A1 (en) | 2015-11-25 | 2017-06-01 | Legochem Biosciences, Inc. | Conjugates comprising self-immolative groups and methods related thereto |
| CA3006247A1 (en) | 2015-11-25 | 2017-06-01 | Legochem Biosciences, Inc. | Conjugates comprising peptide groups and methods related thereto |
| US11229708B2 (en) | 2015-12-04 | 2022-01-25 | Seagen Inc. | Conjugates of quaternized tubulysin compounds |
| US11793880B2 (en) | 2015-12-04 | 2023-10-24 | Seagen Inc. | Conjugates of quaternized tubulysin compounds |
| GB201521709D0 (en) * | 2015-12-09 | 2016-01-20 | Kings College London And Sec Dep For Health The | PBD Antibacterial agents |
| MA43416A (fr) | 2015-12-11 | 2018-10-17 | Regeneron Pharma | Méthodes pour ralentir ou empêcher la croissance de tumeurs résistantes au blocage de l'egfr et/ou d'erbb3 |
| KR102885186B1 (ko) | 2016-01-25 | 2025-11-17 | 리제너론 파아마슈티컬스, 인크. | 메이탄시노이드 유도체, 그의 접합체, 및 사용 방법 |
| GB201601431D0 (en) | 2016-01-26 | 2016-03-09 | Medimmune Ltd | Pyrrolobenzodiazepines |
| GB201602356D0 (en) | 2016-02-10 | 2016-03-23 | Medimmune Ltd | Pyrrolobenzodiazepine Conjugates |
| GB201602359D0 (en) | 2016-02-10 | 2016-03-23 | Medimmune Ltd | Pyrrolobenzodiazepine Conjugates |
| MA43835A (fr) | 2016-03-25 | 2018-11-28 | Seattle Genetics Inc | Procédé de préparation de lieurs de médicaments pégylés et leurs intermédiaires |
| KR102775461B1 (ko) | 2016-04-01 | 2025-02-28 | 어비디티 바이오사이언시스 인크. | 핵산-폴리펩타이드 조성물 및 이의 용도 |
| EP3448891A1 (en) | 2016-04-28 | 2019-03-06 | Regeneron Pharmaceuticals, Inc. | Methods of making multispecific antigen-binding molecules |
| GB201607478D0 (en) | 2016-04-29 | 2016-06-15 | Medimmune Ltd | Pyrrolobenzodiazepine Conjugates |
| WO2017197045A1 (en) | 2016-05-11 | 2017-11-16 | Movassaghi Mohammad | Convergent and enantioselective total synthesis of communesin analogs |
| US10143695B2 (en) | 2016-05-18 | 2018-12-04 | Mersana Therapeutics, Inc. | Pyrrolobenzodiazepines and conjugates thereof |
| JP7049276B2 (ja) | 2016-06-24 | 2022-04-06 | メルサナ セラピューティクス インコーポレイテッド | ピロロベンゾジアゼピンおよびその結合体 |
| WO2018002902A1 (en) | 2016-07-01 | 2018-01-04 | Glaxosmithkline Intellectual Property (No.2) Limited | Antibody-drug conjugates and therapeutic methods using the same |
| HRP20221202T1 (hr) | 2016-09-23 | 2022-12-09 | Regeneron Pharmaceuticals, Inc. | Anti-muc16 (mucin 16) antitijela |
| WO2018058001A1 (en) | 2016-09-23 | 2018-03-29 | Regeneron Pharmaceuticals, Inc. | Anti-steap2 antibodies, antibody-drug conjugates, and bispecific antigen-binding molecules that bind steap2 and cd3, and uses thereof |
| WO2018071455A1 (en) | 2016-10-10 | 2018-04-19 | Cellerant Therapeutics, Inc. | Isoquinolidinobenzodiazepine (iqb)-1(chloromethyl)-2,3-dihydro-1h-benzo[e]indole (cbi) dimers |
| GB201617466D0 (en) * | 2016-10-14 | 2016-11-30 | Medimmune Ltd | Pyrrolobenzodiazepine conjugates |
| KR20190074310A (ko) | 2016-11-08 | 2019-06-27 | 리제너론 파마슈티칼스 인코포레이티드 | 스테로이드 및 이의 단백질-접합체 |
| TWI782930B (zh) | 2016-11-16 | 2022-11-11 | 美商再生元醫藥公司 | 抗met抗體,結合met之雙特異性抗原結合分子及其使用方法 |
| GB201619490D0 (en) * | 2016-11-17 | 2017-01-04 | Medimmune Ltd | Pyrrolobenzodiazepine conjugates |
| JP7350313B2 (ja) | 2016-12-16 | 2023-09-26 | ブルーフィン バイオメディシン, インコーポレイテッド | 抗cubドメイン含有タンパク質1(cdcp1)抗体、抗体薬物コンジュゲート、およびその使用方法 |
| SG10202107429WA (en) | 2017-01-06 | 2021-08-30 | Avidity Biosciences Inc | Nucleic acid-polypeptide compositions and methods of inducing exon skipping |
| UA125198C2 (uk) | 2017-02-08 | 2022-01-26 | Ейдісі Терапьютікс Са | Кон'югати піролобензодіазепін-антитіло |
| GB201702031D0 (en) | 2017-02-08 | 2017-03-22 | Medlmmune Ltd | Pyrrolobenzodiazepine-antibody conjugates |
| KR102697394B1 (ko) | 2017-02-28 | 2024-08-20 | 씨젠 인크. | 컨쥬게이션을 위한 시스테인 변이된 항체 |
| KR102648564B1 (ko) | 2017-03-24 | 2024-03-19 | 씨젠 인크. | 글루쿠로니드 약물-링커의 제조 공정 및 그 중간물 |
| AU2018246806B2 (en) | 2017-03-29 | 2022-05-12 | Ligachem Biosciences Inc. | Pyrrolobenzodiazepine dimer precursor and ligand-linker conjugate compound thereof |
| SI3612537T1 (sl) | 2017-04-18 | 2022-10-28 | Medimmune Limited | Konjugati pirolobenzodiazepina |
| WO2018193102A1 (en) | 2017-04-20 | 2018-10-25 | Adc Therapeutics Sa | Combination therapy with an anti-axl antibody-drug conjugate |
| WO2018209239A1 (en) | 2017-05-11 | 2018-11-15 | Massachusetts Institute Of Technology | Potent agelastatin derivatives as modulators for cancer invasion and metastasis |
| CA3063871A1 (en) | 2017-05-18 | 2018-11-22 | Regeneron Pharmaceuticals, Inc. | Cyclodextrin protein drug conjugates |
| CN111065622A (zh) | 2017-05-18 | 2020-04-24 | 里珍纳龙药品有限公司 | 双八氢菲羧酰胺类化合物及其蛋白质偶联物 |
| EP3635009B1 (en) | 2017-06-07 | 2026-02-25 | Regeneron Pharmaceuticals, Inc. | Compositions and methods for internalizing enzymes |
| JP7145891B2 (ja) | 2017-06-14 | 2022-10-03 | アーデーセー セラピューティクス ソシエテ アノニム | 抗cd19 adcを投与するための投与レジメ |
| AU2018289581C1 (en) | 2017-06-23 | 2025-01-30 | VelosBio Inc. | ROR1 antibody immunoconjugates |
| GB201711809D0 (en) | 2017-07-21 | 2017-09-06 | Governors Of The Univ Of Alberta | Antisense oligonucleotide |
| JP7368856B2 (ja) | 2017-07-25 | 2023-10-25 | トゥルーバインディング,インコーポレイテッド | Tim-3とそのリガンドとの相互作用の遮断によるがん治療 |
| BR112020003003A2 (pt) | 2017-08-18 | 2020-08-11 | Medimmune Limited | conjugados de pirrolobenzodiazepina |
| GB201714115D0 (en) | 2017-09-04 | 2017-10-18 | Femtogenix Ltd | Cytotoxic agents |
| CN117003875A (zh) | 2017-09-29 | 2023-11-07 | 第一三共株式会社 | 抗体或其功能性片段、多核苷酸、表达载体、宿主细胞、糖链重构抗体、药物组合物、应用 |
| CN111655268B (zh) | 2017-10-04 | 2024-03-26 | 艾维迪提生物科学公司 | 核酸-多肽组合物及其用途 |
| US10640508B2 (en) | 2017-10-13 | 2020-05-05 | Massachusetts Institute Of Technology | Diazene directed modular synthesis of compounds with quaternary carbon centers |
| JP7426931B2 (ja) | 2017-11-07 | 2024-02-02 | レゲネロン ファーマシューティカルス,インコーポレーテッド | 抗体薬物コンジュゲートのための親水性リンカー |
| CN111417409B (zh) | 2017-11-14 | 2022-07-08 | 麦迪穆有限责任公司 | 吡咯并苯并二氮杂䓬缀合物 |
| US11638760B2 (en) | 2017-11-27 | 2023-05-02 | Mersana Therapeutics, Inc. | Pyrrolobenzodiazepine antibody conjugates |
| CN118638787A (zh) | 2017-12-06 | 2024-09-13 | 艾维迪提生物科学公司 | 治疗肌萎缩和强直性肌营养不良的组合物和方法 |
| JP2021506883A (ja) | 2017-12-21 | 2021-02-22 | メルサナ セラピューティクス インコーポレイテッド | ピロロベンゾジアゼピン抗体結合体 |
| BR112020013492A2 (pt) | 2018-01-08 | 2020-12-08 | Regeneron Pharmaceuticals, Inc. | Esteroides e conjugados de anticorpo dos mesmos |
| GB201803342D0 (en) | 2018-03-01 | 2018-04-18 | Medimmune Ltd | Methods |
| MX2020010110A (es) | 2018-03-28 | 2020-11-06 | Mitsubishi Tanabe Pharma Corp | Conjugados de farmacos de agentes de union monoclonales de cmet, y usos de los mismos. |
| GB201806022D0 (en) | 2018-04-12 | 2018-05-30 | Medimmune Ltd | Pyrrolobenzodiazepines and conjugates thereof |
| IL278244B2 (en) | 2018-04-30 | 2026-01-01 | Regeneron Pharma | Antibodies and bispecific antigen-binding molecules that bind HER2 and/or APLP2, conjugates and uses thereof |
| CA3099680A1 (en) | 2018-05-09 | 2019-11-14 | Legochem Biosciences, Inc. | Compositions and methods related to anti-cd19 antibody drug conjugates |
| KR20210008008A (ko) | 2018-05-09 | 2021-01-20 | 리제너론 파마슈티칼스 인코포레이티드 | 항-msr1 항체 및 이의 사용 방법 |
| MA52626A (fr) | 2018-05-17 | 2021-03-24 | Regeneron Pharma | Anticorps anti-cd63, conjugués et leurs utilisations |
| EP4428142A3 (en) | 2018-11-20 | 2024-11-20 | Regeneron Pharmaceuticals, Inc. | Bis-octahydrophenanthrene carboxamide derivatives and protein conjugates thereof |
| IL319265A (en) | 2018-12-21 | 2025-04-01 | Avidity Biosciences Inc | Anti-transferrin receptor antibodies and their uses |
| WO2020132483A1 (en) | 2018-12-21 | 2020-06-25 | Regeneron Pharmaceuticals, Inc. | Rifamycin analogs and antibody-drug conjugates thereof |
| MY208067A (en) | 2018-12-21 | 2025-04-11 | Regeneron Pharma | Tubulysins and protein-tubulysin conjugates |
| KR20200084802A (ko) | 2019-01-03 | 2020-07-13 | 주식회사 레고켐 바이오사이언스 | 안전성이 향상된 피롤로벤조디아제핀 이량체 화합물 및 이의 용도 |
| JP2022517767A (ja) | 2019-01-08 | 2022-03-10 | レゲネロン ファーマシューティカルス,インコーポレーテッド | 無痕跡型リンカー及びそのタンパク質-コンジュゲート |
| GB201901197D0 (en) | 2019-01-29 | 2019-03-20 | Femtogenix Ltd | G-A Crosslinking cytotoxic agents |
| CN120058944A (zh) | 2019-01-30 | 2025-05-30 | 真和制药有限公司 | 抗gal3抗体及其用途 |
| SG11202108525VA (en) | 2019-02-21 | 2021-09-29 | Regeneron Pharma | Methods of treating ocular cancer using anti-met antibodies and bispecific antigen binding molecules that bind met |
| MX2021010477A (es) | 2019-03-15 | 2021-10-01 | Medimmune Ltd | Dimeros de azetidobenzodiazepina y conjugados que los comprenden para uso en el tratamiento de cancer. |
| EP3959319A4 (en) | 2019-04-25 | 2023-06-07 | Avidity Biosciences, Inc. | NUCLEIC ACID COMPOSITIONS AND METHODS FOR MULTI-EXON SKIPPING |
| WO2020247054A1 (en) | 2019-06-05 | 2020-12-10 | Massachusetts Institute Of Technology | Compounds, conjugates, and compositions of epipolythiodiketopiperazines and polythiodiketopiperazines and uses thereof |
| JP7581252B2 (ja) | 2019-06-06 | 2024-11-12 | アビディティー バイオサイエンシーズ,インク. | Unaアミダイトおよびその使用 |
| US11578090B2 (en) | 2019-06-06 | 2023-02-14 | Avidity Biosciences, Inc. | Nucleic acid-polypeptide compositions and uses thereof |
| KR20210028544A (ko) | 2019-09-04 | 2021-03-12 | 주식회사 레고켐 바이오사이언스 | 인간 ror1에 대한 항체를 포함하는 항체 약물 접합체 및 이의 용도 |
| CN114340684B (zh) | 2019-09-16 | 2025-09-12 | 瑞泽恩制药公司 | 用于免疫pet成像的放射性标记的met结合蛋白 |
| US11814428B2 (en) | 2019-09-19 | 2023-11-14 | Regeneron Pharmaceuticals, Inc. | Anti-PTCRA antibody-drug conjugates and uses thereof |
| US20240123081A1 (en) | 2019-10-25 | 2024-04-18 | Medimmune, Llc | Branched moiety for use in conjugates |
| CA3164060A1 (en) | 2019-12-06 | 2021-06-10 | Truebinding, Inc. | Antibodies that disrupt the interaction of gal3 and insulin receptor or integrins and methods of use thereof |
| IL294989A (en) | 2020-01-24 | 2022-09-01 | Regeneron Pharma | Conjugated protein-antiviral compounds |
| MX2022010599A (es) | 2020-02-28 | 2022-09-09 | Regeneron Pharma | Moleculas biespecificas de union al antigeno que se unen a her2, y metodos de uso de los mismos. |
| AU2021237465A1 (en) | 2020-03-19 | 2022-10-13 | Avidity Biosciences, Inc. | Compositions and methods of treating Facioscapulohumeral muscular dystrophy |
| IL296393B1 (en) | 2020-03-27 | 2025-12-01 | Avidity Biosciences Inc | Compositions and methods of treating muscle dystrophy |
| US11701427B2 (en) | 2020-04-16 | 2023-07-18 | Regeneron Pharmaceuticals, Inc. | Diels-alder conjugation methods |
| JP2023528797A (ja) | 2020-05-26 | 2023-07-06 | トゥルーバインディング,インコーポレイテッド | ガレクチン-3を遮断することにより炎症性疾患を処置する方法 |
| CN115867563B (zh) | 2020-06-24 | 2025-06-27 | 里珍纳龙药品有限公司 | 微管溶素及蛋白质-微管溶素偶联物 |
| KR20240038138A (ko) | 2020-07-13 | 2024-03-22 | 리제너론 파마슈티칼스 인코포레이티드 | 단백질에서 글루타민 잔기에 접합된 캄토테신 유사체 및 그의 용도 |
| WO2022056494A1 (en) | 2020-09-14 | 2022-03-17 | Regeneron Pharmaceuticals, Inc. | Antibody-drug conjugates comprising glp1 peptidomimetics and uses thereof |
| NZ797493A (en) | 2020-10-22 | 2024-05-31 | Regeneron Pharma | Anti-fgfr2 antibodies and methods of use thereof |
| WO2022182415A1 (en) | 2021-02-24 | 2022-09-01 | Massachusetts Institute Of Technology | Himastatin derivatives, and processes of preparation thereof, and uses thereof |
| US20250026838A1 (en) | 2021-07-13 | 2025-01-23 | Truebinding, Inc. | Methods of preventing protein aggregation |
| CA3220081A1 (en) | 2021-07-28 | 2023-02-02 | Thomas Nittoli | Protein-antiviral compound conjugates |
| CN118265544A (zh) | 2021-09-16 | 2024-06-28 | 艾维迪提生物科学公司 | 治疗面肩肱型肌营养不良的组合物和方法 |
| CN118524852A (zh) | 2021-11-09 | 2024-08-20 | 真和制药有限公司 | 治疗或抑制心血管疾病的方法 |
| CN118660722A (zh) | 2021-12-23 | 2024-09-17 | 米雷楚来有限公司 | 用于递送多核苷酸的组合物 |
| US20230414775A1 (en) | 2021-12-29 | 2023-12-28 | Regeneron Pharmaceuticals, Inc. | Tubulysins and protein-tubulysin conjugates |
| MX2024008640A (es) | 2022-01-12 | 2024-07-24 | Regeneron Pharma | Analogos de camptotecina conjugados a un residuo de glutamina en una proteina y su uso. |
| AU2023208050A1 (en) | 2022-01-14 | 2024-07-04 | Regeneron Pharmaceuticals, Inc. | Verrucarin a derivatives and antibody drug conjugates thereof |
| EP4489855A1 (en) | 2022-03-11 | 2025-01-15 | Regeneron Pharmaceuticals, Inc. | Anti-glp1r antibody-drug conjugates comprising glp1 peptidomimetics and uses thereof |
| US12071621B2 (en) | 2022-04-05 | 2024-08-27 | Avidity Biosciences, Inc. | Anti-transferrin receptor antibody-PMO conjugates for inducing DMD exon 44 skipping |
| US20260000782A1 (en) | 2022-04-07 | 2026-01-01 | Twinpig Biolab Inc. | Itgb2-mediated drug delivery system |
| US20250367307A1 (en) | 2022-06-30 | 2025-12-04 | Toray Industries, Inc. | Pharmaceutical composition for cancer treatment and/or prevention |
| CN120239705A (zh) | 2022-07-21 | 2025-07-01 | 萤火虫生物股份有限公司 | 糖皮质激素受体激动剂和其缀合物 |
| AU2023314808A1 (en) | 2022-07-29 | 2025-03-20 | Regeneron Pharmaceuticals, Inc. | Compositions and methods for transferrin receptor (tfr)-mediated delivery to the brain and muscle |
| WO2024107765A2 (en) | 2022-11-14 | 2024-05-23 | Regeneron Pharmaceuticals, Inc. | Compositions and methods for fibroblast growth factor receptor 3-mediated delivery to astrocytes |
| EP4626553A2 (en) | 2022-11-30 | 2025-10-08 | Regeneron Pharmaceuticals, Inc. | Tlr7 agonists and antibody-drug-conjugates thereof |
| WO2024138000A1 (en) | 2022-12-21 | 2024-06-27 | Regeneron Pharmaceuticals, Inc. | Prodrugs of topoisomerase i inhibitor for adc conjugations and methods of use thereof |
| EP4661914A1 (en) | 2023-02-09 | 2025-12-17 | Regeneron Pharmaceuticals, Inc. | Antibody-drug conjugates via inverse electron demand diels-alder reactions |
| CN121240890A (zh) | 2023-04-18 | 2025-12-30 | 阿斯利康(瑞典)有限公司 | 包含可裂解接头的缀合物 |
| AU2024265507A1 (en) | 2023-05-02 | 2025-10-30 | Regeneron Pharmaceuticals, Inc. | Anti-human m-cadherin (cdh15) antibodies, conjugates, and uses thereof for delivery of genetic payloads to muscle cells |
| WO2025006639A2 (en) | 2023-06-27 | 2025-01-02 | Avidity Biosciences, Inc. | Compositions and methods of using prkag2-targeting antibody-oligonucleotide conjugates abstract |
| US12409230B2 (en) | 2023-06-30 | 2025-09-09 | Avidity Biosciences, Inc. | Compositions comprising PLN-targeting anti-transferrin receptor antibody-polynucleotides and methods of use thereof to treat cardiomyopathy |
| CN121568720A (zh) | 2023-07-10 | 2026-02-24 | 瑞泽恩制药公司 | 抗人类cacng1抗体-药物缀合物及其用途 |
| US20250171516A1 (en) | 2023-11-03 | 2025-05-29 | Regeneron Pharmaceuticals, Inc. | Peptide acids as a glp1r agonist and antibody-drug conjugates thereof |
| WO2025117727A1 (en) | 2023-11-29 | 2025-06-05 | Regeneron Pharmaceuticals, Inc. | Analogs of quinoxaline/quinoline cytotoxins, linker- payloads, protein-drug conjugates, and uses thereof |
| WO2026020031A2 (en) | 2024-07-18 | 2026-01-22 | Novarock Biotherapeutics, Ltd. | Cdh17 antibodies and uses thereof |
Family Cites Families (118)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3361742A (en) | 1964-12-07 | 1968-01-02 | Hoffmann La Roche | 5-oxo-1h-pyrrolo-[2, 1-c][1, 4]-benzodiazepin-2-crylamides |
| US3523941A (en) | 1967-03-06 | 1970-08-11 | Hoffmann La Roche | Benzodiazepine compounds and process for their preparation |
| US3524849A (en) | 1967-10-27 | 1970-08-18 | Hoffmann La Roche | Process for the preparation of pyrrolo-benzodiazepine acrylamides and intermediates useful therein |
| JPS4843755B1 (es) | 1969-06-26 | 1973-12-20 | ||
| DE1965304A1 (de) | 1968-12-30 | 1970-07-23 | Fujisawa Pharmaceutical Co | Benzdiazepinon-Verbindungen und Verfahren zu ihrer Herstellung |
| IL33558A (en) | 1968-12-30 | 1973-10-25 | Fujisawa Pharmaceutical Co | Antibiotic pyrrolo-benzodiazepine compound,its derivatives and processes for their production |
| JPS5382792U (es) | 1976-12-10 | 1978-07-08 | ||
| JPS6053033B2 (ja) | 1976-12-28 | 1985-11-22 | 財団法人微生物化学研究会 | 新制癌抗生物質マゼスラマイシン及びその製造方法 |
| JPS585916B2 (ja) | 1977-12-27 | 1983-02-02 | 株式会社ミドリ十字 | 新規ベンゾジアゼピン系化合物 |
| JPS5615289A (en) | 1979-07-17 | 1981-02-14 | Green Cross Corp:The | Novel benzodiazepinnbased compound 3 |
| JPS57131791A (en) | 1980-12-31 | 1982-08-14 | Fujisawa Pharmaceut Co Ltd | Benzodiazepine derivative and its preparation |
| JPH0353356Y2 (es) | 1981-02-06 | 1991-11-21 | ||
| CA1184175A (en) | 1981-02-27 | 1985-03-19 | Walter Hunkeler | Imidazodiazepines |
| CA1185602A (en) | 1981-02-27 | 1985-04-16 | Emilio Kyburz | Imidazodiazepines |
| CA1173441A (en) | 1981-02-27 | 1984-08-28 | Hoffmann-La Roche Limited | Imidazodiazepines |
| JPS58180487A (ja) | 1982-04-16 | 1983-10-21 | Kyowa Hakko Kogyo Co Ltd | 抗生物質dc−81およびその製造法 |
| JPS58180487U (ja) | 1982-05-28 | 1983-12-02 | 松下電工株式会社 | 光線式報知器の組立体 |
| US4427588A (en) | 1982-11-08 | 1984-01-24 | Bristol-Myers Company | Process for conversion of oxotomaymycin to tomaymycin |
| US4427587A (en) | 1982-11-10 | 1984-01-24 | Bristol-Myers Company | Total synthesis of antitumor antibiotics BBM-2040A and BBM-2040B |
| JPS59152329A (ja) | 1983-02-17 | 1984-08-31 | Green Cross Corp:The | 局所障害抑制剤 |
| FR2586683B1 (fr) | 1985-08-29 | 1988-07-01 | Centre Nat Rech Scient | Nouveaux derives de neothramycine, leur procede de preparation et leur application en tant que medicaments |
| JP2660201B2 (ja) | 1988-08-05 | 1997-10-08 | 塩野義製薬株式会社 | 新規ピロロ[1,4]ベンゾジアゼピン誘導体および老人性痴呆薬 |
| FR2676230B1 (fr) | 1991-05-07 | 1993-08-27 | Centre Nat Rech Scient | Nouveaux derives de pyrrolo [1,4]-benzodiazepines, leur procede de preparation et medicaments les contenant. |
| GB9205051D0 (en) | 1992-03-09 | 1992-04-22 | Cancer Res Campaign Tech | Pyrrolobenzodiazepine derivatives,their preparation,and compositions containing them |
| FR2696176B1 (fr) | 1992-09-28 | 1994-11-10 | Synthelabo | Dérivés de pipéridine, leur préparation et leur application en thérapeutique. |
| US6214345B1 (en) | 1993-05-14 | 2001-04-10 | Bristol-Myers Squibb Co. | Lysosomal enzyme-cleavable antitumor drug conjugates |
| GB9316162D0 (en) | 1993-08-04 | 1993-09-22 | Zeneca Ltd | Fungicides |
| EP1754995B1 (en) | 1998-07-08 | 2012-04-04 | E Ink Corporation | Methods for achieving improved color in microencapsulted electrophoretic devices |
| GB9818732D0 (en) | 1998-08-27 | 1998-10-21 | Univ Portsmouth | Collection of compounds |
| CA2341471C (en) | 1998-08-27 | 2009-04-07 | Spirogen Limited | Pyrrolbenzodiazepines |
| GB9818731D0 (en) | 1998-08-27 | 1998-10-21 | Univ Portsmouth | Compounds |
| GB9818730D0 (en) | 1998-08-27 | 1998-10-21 | Univ Portsmouth | Collections of compounds |
| US6909006B1 (en) | 1999-08-27 | 2005-06-21 | Spirogen Limited | Cyclopropylindole derivatives |
| CN1315805C (zh) | 2000-09-19 | 2007-05-16 | 斯皮罗根公司 | Cc-1065和多卡米新的非手性类似物的组合物及其使用方法 |
| US6362331B1 (en) | 2001-03-30 | 2002-03-26 | Council Of Scientific And Industrial Research | Process for the preparation of antitumor agents |
| US6660856B2 (en) | 2002-03-08 | 2003-12-09 | Kaohsiung Medical University | Synthesis of pyrrolo[2,1-c][1,4]benzodiazepine analogues |
| DK1545613T3 (da) | 2002-07-31 | 2011-11-14 | Seattle Genetics Inc | Auristatinkonjugater og deres anvendelse til behandling af cancer, en autoimmun sygdom eller en infektiøs sygdom |
| US20040138269A1 (en) | 2002-10-11 | 2004-07-15 | Sugen, Inc. | Substituted pyrroles as kinase inhibitors |
| GB0226593D0 (en) | 2002-11-14 | 2002-12-24 | Consultants Ltd | Compounds |
| ATE421967T1 (de) | 2003-03-31 | 2009-02-15 | Council Scient Ind Res | Nichtvernetzende pyrroloä2,1-cüä1, 4übenzodiazepine als potentielle antitumor- agentien und ihre herstellung |
| GB0321295D0 (en) | 2003-09-11 | 2003-10-15 | Spirogen Ltd | Synthesis of protected pyrrolobenzodiazepines |
| GB0416511D0 (en) | 2003-10-22 | 2004-08-25 | Spirogen Ltd | Pyrrolobenzodiazepines |
| WO2005040170A2 (en) | 2003-10-22 | 2005-05-06 | Government Of The United States Of America, Represented By The Secretary, Department Of Health And Human Services | Pyrrolobenzodiazepine derivatives, compositions comprising the same and methods related thereto |
| CN107213469A (zh) | 2003-11-06 | 2017-09-29 | 西雅图基因公司 | 能够与配体偶联的单甲基缬氨酸化合物 |
| EP1718667B1 (en) | 2004-02-23 | 2013-01-09 | Genentech, Inc. | Heterocyclic self-immolative linkers and conjugates |
| GB0404577D0 (en) | 2004-03-01 | 2004-04-07 | Spirogen Ltd | Pyrrolobenzodiazepines |
| GB0404578D0 (en) | 2004-03-01 | 2004-04-07 | Spirogen Ltd | Pyrrolobenzodiazepines |
| GB0404574D0 (en) | 2004-03-01 | 2004-04-07 | Spirogen Ltd | Amino acids |
| CA2558195C (en) | 2004-03-01 | 2012-11-06 | Spirogen Limited | 11-hydroxy-5h-pyrrolo[2,1-c][1,4]benzodiazepin-5-one derivatives as key intermediates for the preparation of c2 substituted pyrrolobenzodiazepines |
| DE102004010943A1 (de) | 2004-03-03 | 2005-09-29 | Degussa Ag | Verfahren zur Herstellung von N-geschützten 4-Ketprolinderivaten |
| US7528126B2 (en) | 2004-03-09 | 2009-05-05 | Spirogen Limited | Pyrrolobenzodiazepines |
| FR2869231B1 (fr) | 2004-04-27 | 2008-03-14 | Sod Conseils Rech Applic | Composition therapeutique contenant au moins un derive de la pyrrolobenzodiazepine et la fludarabine |
| GB0410725D0 (en) | 2004-05-13 | 2004-06-16 | Spirogen Ltd | Pyrrolobenzodiazepine therapeutic agents |
| KR101270829B1 (ko) | 2004-09-23 | 2013-06-07 | 제넨테크, 인크. | 시스테인 유전자조작 항체 및 접합체 |
| EP1831418A2 (en) | 2004-12-24 | 2007-09-12 | Showa Denko Kabushiki Kaisha | Production method of thermoelectric semiconductor alloy, thermoelectric conversion module and thermoelectric power generating device |
| CN101203241B (zh) | 2005-04-19 | 2012-02-22 | 西雅图基因公司 | 人源化抗-cd70结合物和其应用 |
| NZ563136A (en) * | 2005-04-21 | 2009-11-27 | Spirogen Ltd | Pyrrolobenzodiazepines |
| US20070154906A1 (en) | 2005-10-05 | 2007-07-05 | Spirogen Ltd. | Methods to identify therapeutic candidates |
| US8637664B2 (en) | 2005-10-05 | 2014-01-28 | Spirogen Sarl | Alkyl 4- [4- (5-oxo-2,3,5, 11a-tetrahydo-5H-pyrrolo [2, 1-c] [1,4] benzodiazepine-8-yloxy)-butyrylamino]-1H-pyrrole-2-carboxylate derivatives and related compounds for the treatment of a proliferative disease |
| SI1813614T1 (sl) | 2006-01-25 | 2012-01-31 | Sanofi 174 | Citotoksična sredstva, ki obsegajo nove tomajmicinske derivate |
| NZ574215A (en) | 2006-07-18 | 2012-07-27 | Sanofi Aventis | Antagonist antibody against epha2 for the treatment of cancer |
| EP1914242A1 (en) | 2006-10-19 | 2008-04-23 | Sanofi-Aventis | Novel anti-CD38 antibodies for the treatment of cancer |
| WO2008050140A2 (en) | 2006-10-27 | 2008-05-02 | Spirogen Limited | Compounds for treatment of parasitic infection |
| DK2099823T4 (da) | 2006-12-01 | 2022-05-09 | Seagen Inc | Målbindingsmiddelvarianter og anvendelser deraf |
| SI2019104T1 (sl) | 2007-07-19 | 2013-12-31 | Sanofi | Citotoksična sredstva, ki obsegajo nove tomaimicinske derivate, in njihova terapevtska uporaba |
| GB0722088D0 (en) | 2007-11-09 | 2007-12-19 | Spirogen Ltd | Pyrrolobenzodiazepines |
| GB0722087D0 (en) | 2007-11-09 | 2007-12-19 | Spirogen Ltd | Polyamides |
| DK2265283T3 (da) | 2008-03-18 | 2014-10-20 | Seattle Genetics Inc | Auristatin-lægemiddel-linker-konjugater |
| GB0813432D0 (en) | 2008-07-22 | 2008-08-27 | Spirogen Ltd | Pyrrolobenzodiazepines |
| GB0819095D0 (en) | 2008-10-17 | 2008-11-26 | Spirogen Ltd | Pyrrolobenzodiazepines |
| GB0819097D0 (en) | 2008-10-17 | 2008-11-26 | Spirogen Ltd | Pyrrolobenzodiazepines |
| SG173152A1 (en) | 2009-02-05 | 2011-08-29 | Immunogen Inc | Novel benzodiazepine derivatives |
| FR2949469A1 (fr) | 2009-08-25 | 2011-03-04 | Sanofi Aventis | Derives anticancereux, leur preparation et leur application en therapeutique |
| EP2480230A4 (en) | 2009-09-24 | 2015-06-10 | Seattle Genetics Inc | DR5 Ligand-HEILMITTELKONJUGATE |
| US9040526B2 (en) | 2010-02-09 | 2015-05-26 | Bristol-Myers Squibb Company | Benzylpyrrolidinone derivatives as modulators of chemokine receptor activity |
| GB201006340D0 (en) | 2010-04-15 | 2010-06-02 | Spirogen Ltd | Synthesis method and intermediates |
| ES2623057T3 (es) | 2010-04-15 | 2017-07-10 | Medimmune Limited | Pirrolobenzodiazepinas usadas para tratar enfermedades proliferativas |
| KR101772354B1 (ko) | 2010-04-15 | 2017-08-28 | 시애틀 지네틱스, 인크. | 표적화된 피롤로벤조디아제핀 접합체 |
| WO2011130598A1 (en) | 2010-04-15 | 2011-10-20 | Spirogen Limited | Pyrrolobenzodiazepines and conjugates thereof |
| KR20190089048A (ko) | 2011-02-15 | 2019-07-29 | 이뮤노젠 아이엔씨 | 컨쥬게이트의 제조방법 |
| WO2013041606A1 (en) | 2011-09-20 | 2013-03-28 | Spirogen Sàrl | Pyrrolobenzodiazepines as unsymmetrical dimeric pbd compounds for inclusion in targeted conjugates |
| EP2751076B1 (en) | 2011-10-14 | 2018-04-25 | MedImmune Limited | Synthesis method and intermediates useful in the preparation of pyrrolobenzodiazepines |
| US9388187B2 (en) | 2011-10-14 | 2016-07-12 | Medimmune Limited | Pyrrolobenzodiazepines |
| BR112014008888A2 (pt) | 2011-10-14 | 2017-04-18 | Seattle Genetics Inc | pirrolobenzodiazepinas |
| US9526798B2 (en) | 2011-10-14 | 2016-12-27 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| WO2013055987A1 (en) | 2011-10-14 | 2013-04-18 | Spirogen Sàrl | Pyrrolobenzodiazepines and conjugates thereof |
| MX358757B (es) | 2011-10-14 | 2018-09-03 | Seattle Genetics Inc | Pirrolobenzodiazepinas y conjugados dirigidos. |
| KR101960130B1 (ko) | 2012-04-30 | 2019-03-19 | 메디뮨 리미티드 | 피롤로벤조디아제핀 |
| NZ701290A (en) | 2012-04-30 | 2016-08-26 | Medimmune Ltd | Pyrrolobenzodiazepines |
| TW201406785A (zh) | 2012-07-09 | 2014-02-16 | Genentech Inc | 抗cd22抗體及免疫結合物 |
| IN2014DN10652A (es) | 2012-07-09 | 2015-09-11 | Genentech Inc | |
| BR112015002193A2 (pt) | 2012-08-02 | 2017-07-04 | Genentech Inc | anticorpos anti-etbr e imunoconjugados |
| WO2014057118A1 (en) | 2012-10-12 | 2014-04-17 | Adc Therapeutics Sarl | Pyrrolobenzodiazepine-anti-cd22 antibody conjugates |
| MX364328B (es) | 2012-10-12 | 2019-04-23 | Medimmune Ltd | Conjugados del anticuerpo pirrolobenzodiazepina. |
| HRP20171264T1 (hr) | 2012-10-12 | 2017-10-20 | Adc Therapeutics Sa | Konjugati protutijela anti-her2-pirolobenzodiazepina |
| WO2014057117A1 (en) | 2012-10-12 | 2014-04-17 | Adc Therapeutics Sàrl | Pyrrolobenzodiazepine-antibody conjugates |
| ES2680153T3 (es) | 2012-10-12 | 2018-09-04 | Adc Therapeutics Sa | Conjugados de anticuerpos anti-PSMA-pirrolobenzodiazepinas |
| HRP20190366T1 (hr) | 2012-10-12 | 2019-04-19 | Medimmune Limited | Pirolobenzodiazepini i njihovi konjugati |
| KR101645905B1 (ko) | 2012-10-12 | 2016-08-04 | 스피로즌 살 | 피롤로벤조디아제핀 및 그의 컨주게이트 |
| HRP20181646T2 (hr) | 2012-10-12 | 2019-08-09 | Adc Therapeutics Sa | Konjugati pirolobenzodiazepin - anti-psma protutijela |
| AU2013328673B2 (en) | 2012-10-12 | 2017-07-13 | Medimmune Limited | Synthesis and intermediates of pyrrolobenzodiazepine derivatives for conjugation |
| PL2906251T3 (pl) | 2012-10-12 | 2018-02-28 | Adc Therapeutics Sa | Koniugaty pirolobenzodiazepina-przeciwciało anty-CD22 |
| WO2014057120A1 (en) | 2012-10-12 | 2014-04-17 | Adc Therapeutics Sàrl | Pyrrolobenzodiazepine-antibody conjugates |
| CN105246894A (zh) | 2012-12-21 | 2016-01-13 | 斯皮罗根有限公司 | 用于治疗增殖性和自身免疫疾病的非对称吡咯并苯并二氮杂卓二聚物 |
| AU2013366493B2 (en) | 2012-12-21 | 2017-08-24 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| RU2019109456A (ru) | 2013-02-22 | 2019-04-10 | ЭББВИ СТЕМСЕНТРКС ЭлЭлСи | Новые конъюгаты антител и их применения |
| EP2968585B1 (en) | 2013-03-13 | 2018-07-18 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| JP6445519B2 (ja) | 2013-03-13 | 2018-12-26 | メドイミューン・リミテッドMedImmune Limited | ピロロベンゾジアゼピン及びそのコンジュゲート |
| WO2014140862A2 (en) | 2013-03-13 | 2014-09-18 | Spirogen Sarl | Pyrrolobenzodiazepines and conjugates thereof |
| WO2014174111A1 (en) | 2013-04-26 | 2014-10-30 | Pierre Fabre Medicament | Axl antibody-drug conjugate and its use for the treatment of cancer |
| GB201317982D0 (en) | 2013-10-11 | 2013-11-27 | Spirogen Sarl | Pyrrolobenzodiazepines and conjugates thereof |
| US9950078B2 (en) | 2013-10-11 | 2018-04-24 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| EP3054986B1 (en) | 2013-10-11 | 2019-03-20 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| EP3054984A1 (en) | 2013-10-11 | 2016-08-17 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| GB201317981D0 (en) | 2013-10-11 | 2013-11-27 | Spirogen Sarl | Pyrrolobenzodiazepines and conjugates thereof |
| US10010624B2 (en) | 2013-10-11 | 2018-07-03 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| MX381017B (es) | 2013-12-16 | 2025-03-12 | Genentech Inc | Compuestos peptidomiméticos y conjugados anticuerpo-fármaco de estos. |
| GB201406767D0 (en) | 2014-04-15 | 2014-05-28 | Cancer Rec Tech Ltd | Humanized anti-Tn-MUC1 antibodies anf their conjugates |
-
2012
- 2012-10-12 US US14/351,168 patent/US9526798B2/en active Active
- 2012-10-12 ES ES18168997T patent/ES2945932T3/es active Active
- 2012-10-12 CN CN201810169400.3A patent/CN108164551A/zh active Pending
- 2012-10-12 CN CN201280062295.4A patent/CN103987384A/zh active Pending
- 2012-10-12 CA CA2850373A patent/CA2850373C/en active Active
- 2012-10-12 EP EP12840661.8A patent/EP2755642B1/en active Active
- 2012-10-12 JP JP2014535897A patent/JP6393617B2/ja active Active
- 2012-10-12 WO PCT/US2012/059867 patent/WO2013055990A1/en not_active Ceased
- 2012-10-12 ES ES12840661.8T patent/ES2687246T3/es active Active
- 2012-10-12 EP EP18168997.7A patent/EP3388435B1/en active Active
-
2016
- 2016-06-14 US US15/181,900 patent/US9713647B2/en active Active
-
2017
- 2017-06-27 US US15/635,063 patent/US10328084B2/en active Active
-
2018
- 2018-06-04 JP JP2018107170A patent/JP7123635B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| ES2687246T3 (es) | 2018-10-24 |
| HK1256557A1 (en) | 2019-09-27 |
| CA2850373A1 (en) | 2013-04-18 |
| CN103987384A (zh) | 2014-08-13 |
| JP6393617B2 (ja) | 2018-09-19 |
| EP2755642B1 (en) | 2018-07-18 |
| CN108164551A (zh) | 2018-06-15 |
| EP2755642A4 (en) | 2015-06-24 |
| JP2018150371A (ja) | 2018-09-27 |
| US9713647B2 (en) | 2017-07-25 |
| US20140286970A1 (en) | 2014-09-25 |
| CA2850373C (en) | 2019-07-16 |
| EP3388435B1 (en) | 2023-05-03 |
| WO2013055990A1 (en) | 2013-04-18 |
| US10328084B2 (en) | 2019-06-25 |
| EP3388435A1 (en) | 2018-10-17 |
| HK1200091A1 (en) | 2015-07-31 |
| JP7123635B2 (ja) | 2022-08-23 |
| US20170290924A1 (en) | 2017-10-12 |
| US20160289239A1 (en) | 2016-10-06 |
| EP2755642A1 (en) | 2014-07-23 |
| JP2014528480A (ja) | 2014-10-27 |
| US9526798B2 (en) | 2016-12-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2945932T3 (es) | Pirrolobenzodiazepinas y conjugados dirigidos | |
| ES2660233T3 (es) | Pirrolobenzodiazepinas y conjugados dirigidos | |
| US10561739B2 (en) | Targeted pyrrolobenzodiazapine conjugates | |
| JP5875083B2 (ja) | 増殖性疾患治療用ピロロベンゾジアゼピン | |
| JP6117801B2 (ja) | ピロロベンゾジアゼピン | |
| HK1256557B (en) | Pyrrolobenzodiazepines and targeted conjugates | |
| HK1194923B (en) | Pyrrolobenzodiazepines and targeted conjugates | |
| HK1194923A (en) | Pyrrolobenzodiazepines and targeted conjugates |