-
Die
Erfindung beansprucht die Priorität der US 09/062 328 vom 17.
April 1998.
-
Die
Erfindung betrifft neue substituierte tricyclische organische Verbindungen,
die zur Hemmung der sPLA2 vermittelten Freisetzung
von Fettsäuren
für Zustände, wie
dem septischen Schock, brauchbar sind.
-
Die
Struktur und die physikalischen Eigenschaften der humanen nicht
aus dem Pankreas stammenden sekretorischen Phospholipase A2 (hierin "sPLA2" genannt) wurden
ausführlich
in zwei Artikeln beschrieben, nämlich "Cloning and Recombinant
Expression of Phospholipase A2 Present in
Rheumatoid Arthritic Synovial Fluid", von Jeffrey J. Seilhammer, Waldemar
Pruzanski, Peter Vadas, Shelley Plant, Judy A. Miller, Jean Kloss
und Lorin K. Johnson, The Journal of Biological Chemistry, Band
264, Nr. 10, Ausgabe vom 5. April, Seiten 5335–5338, 1989 und "Structure and Properties
of a Human Non-pancreatic Phospholipase A2" von Ruth M. Kramer,
Catherine Hession, Berit Johansen, Gretchen Hayes, Paula McGray,
Pingchang E. Chaow, Richard Tizard und R. Blake Pepinsky, The Journal
of Biological Chemistry, Band 264, Nr. 10, Ausgabe vom 5. April Seiten
5768–5775,
1989, deren Beschreibungen hiermit eingeführt sind.
-
Man
glaubt, daß die
sPLA2 ein geschwindigkeitslimitierendes
Enzym in der Arachidonsäurekaskade ist,
die Membranphospholipide hydrolysiert. Daher ist es wichtig, Verbindungen
zu entwickeln, die die sPLA2 vermittelte
Freisetzung von Fettsäuren
(beispielsweise Arachidonsäure)
hemmen. Solche Verbindungen wären
bei der allgemeinen Behandlung von Zuständen brauchbar, die durch die Überproduktion
von sPLA2 hervorgerufen und/oder aufrechterhalten
werden, wie septischer Schock, Atemnotsyndrom beim Erwachsenen, Pankreatitis,
durch Trauma induzierter Schock, Bronchialasthma, allergische Rhinitis,
rheumatoide Arthritis und dergleichen.
-
Es
ist erwünscht,
neue Verbindungen und Behandlungen für Erkrankungen zu entwickeln,
die durch sPLA2 induziert sind.
-
Die
von der Erfindung umfassten Verbindungen sind aus der Gruppe ausgewählt, die
besteht aus
[9-Benzyl-5-carbamoyl-1-fluorcarbazol-4-yl]oxyessigsäure,
{9-[(Phenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-[(3-Fluorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-[(3-Chlorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-[(3-Trifluormethylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-[(2-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-[(3-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-[(3-Trifluormethoxyphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
[9-Benzyl-5-carbamoyl-1-chlorcarbazol-4-yl]oxyessigsäure,
[9-(Cyclohexyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäure und
[9-[(Cyclopentyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäure oder
einem pharmazeutisch annehmbaren Razemat, Solvat, Tautomer, optischen
Isomer, Prodrugderivat, oder ein Salz hiervon.
-
Die
Erfindung betrifft auch eine pharmazeutische Formulierung, die eine
Verbindung, welche aus der Gruppe ausgewählt ist, die besteht aus
[9-Benzyl-5-carbamoyl-1-fluorcarbazol-4-yl]oxyessigsäure,
{9-[(Phenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-[(3-Fluorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-[(3-Chlorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-[(3-Trifluormethylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-[(2-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-[(3-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-[(3-Trifluormethoxyphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
[9-Benzyl-5-carbamoyl-1-chlorcarbazol-4-yl]oxyessigsäure,
[9-(Cyclohexyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäure und
[9-[(Cyclopentyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäure zusammen
mit einem oder mehreren pharmazeutisch annehmbaren Verdünnungsmitteln,
Trägern
und Hilfsstoffen umfasst.
-
Die
Erfindung betrifft auch die Verwendung einer Verbindung, die aus
der Gruppe ausgewählt
ist, welche besteht aus
[9-Benzyl-5-carbamoyl-1-fluorcarbazol-4-yl]oxyessigsäure,
{9-[(Phenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-[(3-Fluorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-[(3-Chlorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-((3-Trifluormethylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-[(2-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-((3-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-[(3-Trifluormethoxyphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
[9-Benzyl-5-carbamoyl-1-chlorcarbazol-4-yl]oxyessigsäure,
[9-(Cyclohexyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäure und
[9-[(Cyclopentyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäure zur
Herstellung eines Arzneimittels zur Hemmung der sPLA2 bei
einem Säuger.
-
Gemäß einem
weiteren Aspekt der Erfindung wird die Verwendung einer Verbindung,
die aus der Gruppe ausgewählt
ist, welche besteht aus
[9-Benzyl-5-carbamoyl-1-fluorcarbazol-4-yl]oxyessigsäure,
{9-[(Phenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-[(3-Fluorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-[(3-Chlorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-[(3-Trifluormethylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-[(2-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-[(3-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-[(3-Trifluormethoxyphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
(9-Benzyl-5-carbamoyl-1-chlorcarbazol-4-yl]oxyessigsäure,
[9-(Cyclohexyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäure und
[9-[(Cyclopentyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäure zur
Herstellung eines Arzneimittels zur selektiven Hemmung der sPLA2 bei einem Säuger bereitgestellt.
-
Die
Erfindung liefert ferner eine Verbindung, die aus der Gruppe ausgewählt ist,
welche besteht aus
[9-Benzyl-5-carbamoyl-1-fluorcarbazol-4-yl]oxyessigsäure,
{9-[(Phenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-[(3-Fluorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-[(3-Chlorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-[(3-Trifluormethylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-[(2-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-[(3-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-[(3-Trifluormethoxyphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
[9-Benzyl-5-carbamoyl-1-chlorcarbazol-4-yl]oxyessigsäure,
[9-(Cyclohexyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäure und
[9-[(Cyclopentyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäure zur
Verwendung als Arzneimittel zur Behandlung von entzündlichen
Erkrankungen, wie septischem Schock, Atemstresssyndrom beim Erwachsenen, Pankreatitis,
Trauma induziertem Schock, Bronchialasthma, allergischer Rhinitis,
rheumatoider Arthritis, cystischer Fibrose, Schlaganfall, akuter
Bronchitis, chronischer Bronchitis, akuter Broncheolitis, chronischer
Broncheolitis, Osteoarthritis, Gicht, Spondylarthropathie, ankylosierender
Spondylitis, Reiter Syndrom, psoriatischer Arthropathie, enterapathischer
Spondylitis, juveniler Arthropathie oder juveniler alkylosierender
Spondylitis, reaktiver Arthropathie, infektiöser oder postinfektiöser Arthritis,
Gonokokkenarthritis, Tuberkulosearthritis, viraler Arthritis, pilzbedingter
Arthritis, Syphilis-bedingter Arthritis, Lyme-Erkankung, Arthritis,
die mit "vaskulitischen
Syndromen" assoziiert
ist, Polyarteriitis nodosa, hyperempfindlicher Vaskulitis, Luegenec
Granulomatose, Polymyalgia rheumatica, Gelenkszellarteritis, Arthropathie
durch Calciumkristallablagerung, Pseudogicht, nicht-artikulärem Rheuma,
Bursitis, Tenosynovitis, Epicondylitis (Tennisellenbogen), Carpaltunnelsyndrom,
Verletzung durch wiederkehrende Tätigkeit (Tippen), verschiedenen
Formen der Arthritis, neuropatischer Gelenkserkrankung (Charcotgelenk
und Gelenke), Hämarthrose
(hämarthrotisch),
Purpura Schönlein-Hennoch, hypertropher
Osteoarthropathie, multizentrischer Reticulohistiocytose, Arthritis,
die mit bestimmten Erkrankungen assoziiert ist, Surcoilose, Hämochromatose,
Sichelzellerkrankung und anderen Hämoglobinopathien, Hyperlipoproteinämie, Hypogammaglobulinämie, Hyperparathyreoidismus,
Akromegalie, familiärem
Mittelmeerfieber, Behat Erkrankung, systemischem Lupus erythrematodes
oder Polychondritisrückfall
und verwandten Erkrankungen, das die Verabreichung einer therapeutisch
wirksamen Menge der Verbindung der Formel 1 in einer Menge an einen
behandlungsbedürftigen
Säuger
umfasst, die zur Hemmung der durch sPLA2 vermittelten
Freisetzung von Fettsäuren
und hierbei zur Hemmung der Prävention
der Arachidonsäurekaskade
und ihrer schädlichen
Produkte ausreicht.
-
Andere
Ziele, Merkmale und Vorteile der vorliegenden Erfindung werden aus
der anschließenden
Beschreibung und den Patentansprüchen
deutlich.
-
Definitionen:
-
Die
pharmazeutisch annehmbaren Salze der obigen Tricyclen sind ein weiterer
Aspekt der Erfindung. In den Fällen,
in denen die erfindungsgemäßen Verbindungen
funktionelle saure Gruppen besitzen, können verschiedene Salze gebildet
werden, die wasserlöslicher
und physiologisch geeigneter sind als die Ausgangsverbindung. Repräsentative
pharmazeutisch annehmbare Salze sind unter anderem die Alkali- und
Erdalkalimetallsalze, wie Lithium, Natrium, Kalium, Calcium, Magnesium,
Aluminium und dergleichen. Salze werden bequem durch die Behandlung
der Säure
in Lösung
mit einer Base oder durch Aussetzen der Säure gegenüber einem Ionenaustauscherharz
aus der freien Säure
hergestellt.
-
In
der Definition der pharmazeutisch annehmbaren Salze eingeschlossen
sind die relativ untoxischen, anorganischen und organischen Basenadditionssalze
der erfindungsgemäßen Verbindungen,
beispielsweise Ammonium-, quarternäre Ammonium- und Aminkationen,
die von stickstoffhaltigen Basen mit einer ausreichenden Basizität zur Bildung
von Salzen mit den erfindungsgemäßen Verbindungen
stammen (siehe beispielsweise S.M. Berge et al., "Pharmaceutical Salts", J. Phar. Sci.,
66: 1–19
(1977)).
-
Die
erfindungsgemäßen Verbindungen
können
chirale Zentren aufweisen und daher in optisch aktiven Formen vorkommen.
Die R- und S-Isomere und razemischen Gemische sind von der Erfindung
abgedeckt. Ein bestimmtes Stereoisomer kann durch in der Technik
gut bekannte Verfahren durch die Verwendung von stereospezifischen
Reaktionen mit Ausgangsmaterialien hergestellt werden, die die asymmetrischen
Zentren enthalten und bereits getrennt sind, oder alternativ dazu
durch Verfahren, die zu Gemischen der Stereoisomeren führen und
die anschließende
Trennung durch bekannte Verfahren.
-
Prodrugs
sind Derivate der erfindungsgemäßen Verbindungen,
die chemisch oder metabolisch spaltbare Gruppen aufweisen und durch
Solvolyse oder unter physiologischen Bedingungen zu den erfindungsgemäßen Verbindungen
werden, die in vivo pharmazeutisch wirksam sind. Derivate der erfindungsgemäßen Verbindungen
haben sowohl in ihren Säure-
als auch Basenderivatformen eine Aktivität, aber die Säurederivatform
bietet oft Vorteile hinsichtlich Löslichkeit, Gewebekompatibilität oder verzögerter Freisetzung
in einem Säugerorganismus
(siehe H. Bundgard, Design of Prodrugs, Seiten 7–9, 21–24, Elsevier, Amsterdam 1985). Prodrugs
umfassen dem Fachmann gut bekannte Säurederivate, wie beispielsweise
Ester, die durch die Umsetzung der sauren Ausgangsverbindung mit
einem geeigneten Alkohol hergestellt werden. Einfache aliphatische
Ester (beispielsweise Methyl, Ethyl, Propyl, Isopropyl, Butyl, sek-Butyl,
tert-Butyl) oder aromatische Ester, die von sauren Gruppen stammen,
die in den erfindungsgemäßen Verbindungen
vorkommen, sind bevorzugte Prodrugs. Andere bevorzugte Ester umfassen
Morpholinoethyloxy, Diethylglycolamid und Diethylaminocarbonylmethoxy.
-
Die
folgende Liste an Abkürzungen
wird in den Beispielen und Präparationen
verwendet:
HCl = Chlorwasserstoffsäure
EtOAc = Ethylacetat
DMF
= Dimethylformamid
THF = Tetrahydrofuran
Et2O
= Diethylether
H2O = Wasser
NaOH
= Natriumhydroxid
EtOH = Ethanol
Na2SO4 = Natriumsulfat
NaHCO3 =
Natriumbicarbonat
Celite = Diatomäenerde
CH2Cl2 = Methylenchlorid
H2SO4 = Schwefelsäure
McOH = Methanol
Rh/Al2O3 = Rhodium auf
Aluminiumoxid
DDQ = 2,3-Dichlor-5,6-dicyano-1,4-benzochinon
TLC
= Dünnschichtchromatographie
NaH
= Natriumhydrid
NH4OH = Ammoniumhydroxid
LiOH
= Lithiumhydroxid
NH3 = Ammoniak
Cs2CO3 = Cäsiumcarbonat
NH4OAc = Ammoniumacetat
-
Die
folgenden Präparationen
der Zwischenprodukte und die Beispiele der Endprodukte erläutern weiter
die Herstellung der erfindungsgemäßen Verbindungen. Die Beispiele
sind nur erläuternd
und sollen den Schutzumfang der Erfindung in keiner Weise beschränken.
-
Prägaration 1
-
Herstellung von 5-Carbomethoxy-1,2-dihydro-9H-carbazol-4(3H)-on
aus 2-Brom-3-nitrobenzoesäure
-
-
a) Methyl-2-brom-3-nitrobenzoat
-
Eine
Lösung
aus 2-Brom-3-nitrobenzoesäure
(28,4 g, 115,0 mM), Iodmethan (18,0 g, 127 mM) und Kaliumcarbonat
(19,0 g, 137,4 mM) in 100 ml DMF wird bei Raumtemperatur für 72 Stunden
gerührt.
Das Gemisch wird in 1,5 Liter H2O gegossen.
Der entstehende Niederschlag wird durch Filtration gesammelt und
unter Bildung von 28,79 g (96%) an Methyl-2-brom-3-nitrobenzoat
als weißer
Feststoff getrocknet.
1H NMR (DMSO-d6) δ 8,3
(dd, 1H, J = 1 und 8 Hz), 7,9 (dd, 1H, J = 1 und 8 Hz), 7,7 (t,
1H, J = 8 Hz) und 3,9 (s, 3H).
IR (KBr, cm–1)
2950, 1738, 1541, 1435, 1364, 1298 und 1142.
MS (FD) m/e 259,
261.
-
Elementaranalyse
für C8H6NO4Br:
Berechnet: C 36,95, H 2,33, N 5,39. Gefunden: C 37,14, H 2,37, N 5,45.
-
b) Methyl-2-brom-3-aminobenzoat
-
Chlorwasserstoffgas
wird durch eine Lösung
aus Methyl-2-brom-3-nitrobenzoat (0,20 g, 0,77 mM) und 0,1 g an
3% sulfidiertem Platin auf Kohle in 25 ml Ethylacetat für 24 Stunden
bei Raumtemperatur gegeben. Der Katalysator wird durch Filtration
durch Celite entfernt. Eine Konzentration des Filtrats ergibt 0,175
g (99%) an Methyl-2-brom-3-aminobenzoat als gelbes Öl.
1H NMR (CDCl3) δ 7,15 (t,
1H, J = 8 Hz), 7,1 (dd, 1H, J = 1 und 8 Hz), 6,8 (dd, 1H, J = 1
und 8 Hz) und 3,95 (s, 3H).
IR (CHCl3,
cm–1)
3550, 3380, 2980, 2900, 1729, 1613, 1465, 1451, 1434, 1324, 1266
und 1025.
MS (FD) m/e 230, 232.
Elementaranalyse für C8H8NO2Br:
Berechnet:
C 41,77, H 3,51, N 6,09. Gefunden: C 42,01, H 3,29, N 6,00.
-
b') In einem alternativen
Verfahren kann Methyl-2-brom-3-aminobenzoat folgendermaßen hergestellt werden:
Eine
Lösung
aus Zinn-(II)-chlorid (15,0 g, 76,1 mM) in 30 ml konzentrierter
Chlorwasserstoffsäure
wird langsam zu einer Lösung
aus Methyl-2-brom-3-nitrobenzoat (4,0 g, 15,4 mM) in 90 ml Ethanol
bei 15–30°C über 1 Stunde
gegeben. Das Gemisch wird dann auf 50–60°C für 15 Minuten erhitzt. Das Gemisch
wird auf Raumtemperatur gekühlt
und durch langsame Zugabe von festem Natriumhydroxid alkalisch gemacht,
wobei die Temperatur auf 30–35°C gehalten
wird. Das entstehende Gemisch wird dreimal mit Chloroform extrahiert.
Die Extrakte werden mit Kochsalzlösung gewaschen, über Natriumsulfat
getrocknet, filtriert und unter Bildung von 3,51 g (99%) an Methyl-2-brom-3-aminobenzoat
als gelbes Öl
konzentriert, das in allen Aspekten zu dem durch katalytische Hydrierung
abgeleiteten Material identisch ist, wie es oben beschrieben ist.
-
c) 3-(3-Carbomethoxy-2-bromanilin)cyclohex-2-en-1-on
-
Ein
Gemisch aus Methyl-2-brom-3-aminobenzoat (13,2 g, 60,0 mM) und 1,3-Cyclohexandion
(8,4 g, 75 mM) wird bei 125°C
unter einem Stickstoffstrom für
4 h erhitzt. Der entstehende Feststoff wird durch HPLC auf Silicagel
(Elution mit Methylenchlorid/Ethylacetat) unter Bildung von 17,2
g (88%) an 3-(3-Carbomethoxy-2-bromanilin)cyclohex-2-en-1-on
als hellbrauner Schaum gereinigt.
1H
NMR (DMSO-d6) δ 8,75 (s, 1H), 7,6-7,4 (m, 3H),
4,65 (s, 1H), 3,85 (s, 3H), 2,6 (t, 2H, J = 6 Hz), 2,15 (t, 2H, J
= 6 Hz) und 1,9 (m, 2H).
IR (CHCl3,
cm–1)
3400, 3004, 2954, 1732, 1607, 1588, 1573, 1513, 1464, 1436, 1412,
1308, 1249, 1177 und 1144.
MS (ES) m/e 322, 324, 326.
Elementaranalyse
für C14H14NO3Br:
Berechnet: C 51,85, H 4,32, N 4,32. Gefunden: C 53,60, H 4,73, N
4,09.
-
d) 5-Carbomethoxy-1,2-dihydro-9H-carbazol-4(3H)-on
-
Eine
Suspension aus 3-(3-Carbomethoxy-2-bromanilin)cyclohex-2-en-1-on
(15,8 g, 48,8 mM), Palladiumacetat (1,12 g, 5,0 mM), Tri-o-tolylphosphin
(3,1 g, 10,0 mM) und Triethylamin (6,3 g, 62,0 mM) in 120 ml Acetonitril
wird am Rückfluss
für 8 Stunden
erhitzt. Das Lösemittel
wird im Vakuum entfernt. Der Rückstand wird
in Methylenchlorid gelöst,
zweimal mit 1 N HCl, zweimal mit H2O und
einmal mit gesättigter
Kochsalzlösung
gewaschen, über
wasserfreiem Magnesiumsulfat getrocknet, filtriert und unter Bildung
von 17 g eines hellbraunen Schaums konzentriert. Eine Reinigung
durch HPLC auf Silicagel (Elution mit einem Gradienten Methylenchlorid/Ethylacetat)
ergibt 9,2 g (78%) an 5-Carbomethoxy-1,2-dihydro-9H-carbazol-4(3H)-on als gelben
Feststoff, der identisch zu dem Material ist, das aus 3-(3-Carbomethoxy-2-chloranilin)cyclohex-2-en-1-on abgeleitet
ist, wie dies oben beschrieben ist.
1H
NMR (DMSO-d6) δ 7,5 (d, 1H, J = 8 Hz), 7,25-7,1
(m, 2H), 5,7 (s, 1H), 3,8 (s, 3H), 2,95 (t, 2H, J = 6 Hz), 2,4 (t,
2H, J = 6 Hz) und 2,1 (m, 2H).
MS (ES) m/e 242,244.
-
Beispiel 1
-
Herstellung von {9-[(Phenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäurenatriumsalz
-
-
A. 9-[(Phenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
-
Eine
Suspension aus 5-Carbomethoxy-1,2-dihydro-9H-carbazol-4(3H)-on (300
mg, 1,23 mM), Benzylbromid (210 mg, 1,23 mM) und Kaliumcarbonat
(170 mg, 1,23 mM) in 15 ml DMF wird bei Raumtemperatur für 6 Stunden
gerührt.
Das Gemisch wird mit 80 ml H2O verdünnt und
in einen Kühlschrank
gegeben. Der entstehende weiße
Niederschlag wird durch Filtration gesammelt, mit H2O
gewaschen und im Vakuum unter Bildung von 325 mg (79%) an 9-[(Phenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on als weißer Feststoff getrocknet.
1N NMR (DMSO-d6) δ 7,7 (dd,
1H, J = 1 und 8 Hz), 7,45-7,0 (m, 7H), 5,6 (s, 2H), 3,8 (s, 3H),
3,05 (t, 2H, J = 6 Hz), 2,5 (t, 2H, J = 6 Hz) und 2,2 (m, 2H).
IR
(KBr, cm–1)
3421, 1726, 1676, 1636, 1473, 1450, 1435, 1288, 1122, 764, 745 und
706. MS (ES) m/e 334.
Elementaranalyse für C21H19NO3: Berechnet:
C 75,68, H 5,71, N 4,20. Gefunden: 70,85, H 5,53, N 4,49.
-
B. 9-[(Phenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol
-
(a)
Eine Lösung
aus 9-[(Phenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
(1,5 g, 4,5 mM) und 2,3-Dichlor-5,6-dicyano-1,4-benzochinon (1,12
g, 5,0 mM) in 25 ml Toluol wird zwischen 80–90°C für 6 h gerührt. Das Gemisch wird direkt
durch Säulenchromatographie
auf Silicagel (Elution mit Methylenchlorid/Ethylacetat) unter Bildung
von 420 mg (28%) an 9-[(Phenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol
als gelber Feststoff gereinigt.
1H
NMR (DMSO-d6) δ 10,25 (s, 1H), 7,7 (d, 1H,
J = 8 Hz), 7,4 (t, 1H, J = 8 Hz), 7,4-7,0 (m, 8H), 6,6 (d, 1H, J
= 8 Hz), 5,6 (s, 2H) und 3,8 (s, 3H).
IR (CHCl3,
cm–1)
1723, 1685, 1621, 1597, 1568, 1496, 1453, 1442, 1392, 1286, 1267,
1156 und 1138.
MS (ES) m/e 330, 332.
Elementaranalyse
für C21H17NO3:
Berechnet: C 76,13, H 5,14, N 4,23. Gefunden: C 75,90, H 5,20, N
4,46.
-
(b)
Zu einer Lösung
aus 9-[(Phenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
(2,87 g, 8,61 mM) in 29 ml Dioxan wird 60% Natriumhydrid in Mineralöl (0,79
g, 19,8 mM) gegeben. Die Reaktion wird für 8 Minuten gerührt und
dann wird Methylbenzolsulfinat (1,80 ml, 13,8 mM) zugegeben. Die
Reaktion wird für weitere
1,5 h gerührt
und dann mit 43 ml Dioxan und 1,13 ml Essigsäure verdünnt. Das Gemisch wird für 1 h am
Rückfluss
erhitzt, mit Ethylacetat verdünnt
und mit gesättigtem
NaHCO3 zweimal und dann mit Kochsalzlösung extrahiert.
Nach dem Trocknen (NaSO4) ergibt eine Eindampfung
im Vakuum 4,90 g. Das Gemisch wird durch Säulenchromatographie auf Silicagel
(Elution mit Toluol/Methylenchlorid) unter Bildung von 2,31 g (81%) an
9-[(Phenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol gereinigt.
1H NMR (DMSO-d6) δ 10,25 (s,
1H), 7,7 (d, 1H, J = 8 Hz), 7,4 (t, 1H, J = 8 Hz), 7,4-7,0 (m, 8H),
6,6 (d, 1H, J = 8 Hz), 5,6 (s, 2H) und 3,8 (s, 3H).
IR (CHCl3, cm–1) 1723, 1685, 1621,
1597, 1568, 1496, 1453, 1442, 1392, 1286, 1267, 1156 und 1138.
MS
(ES) m/e 330, 332.
Elementaranalyse für C21H17NO3: Berechnet:
C 76,13, H 5,14, N 4,23. Gefunden: C 75,90, H 5,20, N 4,46.
-
C. 9-[(Phenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
-
Eine
Lösung
aus 9-[(Phenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol (200 mg,
0,6 mM) in 4 ml McOH und 40 ml konzentriertem wässrigem Ammoniumhydroxid wird
für 30
h bei 40–50°C ultrabeschallt.
Das Gemisch wird mit Ethylacetat verdünnt und mit 5 N HCl auf pH
1 angesäuert.
Die wässrige
Phase wird dreimal mit Ethylacetat extrahiert. Die vereinigten organischen
Extrakte werden mit gesättigter
Kochsalzlösung
gewaschen, über
Magnesiumsulfat getrocknet, filtriert und konzentriert. Der Rückstand
wird durch Säulenchromatographie
auf Silicagel (Elution mit einem Gradienten Methylenchlorid/Ethylacetat)
unter Bildung von 50 mg (26%) an 9-[(Phenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
als weißer
Feststoff gereinigt.
1H NMR (DMSO-d6) δ 10,5
(s, 1H), 8,8 (br s, 1H), 8,4 (br s, 1H), 7,85 (dd, 1H, J = 1 und
8 Hz), 7,5-7,1 (m, 9H), 6,6 (d, 1H, J = 8 Hz) und 5,8 (s, 2H).
IR
(KBr, cm–1)
3428, 3198, 3063, 1631, 1599, 1579, 1562, 1496, 1442, 1330, 1261,
1215, 775 und 697.
MS (ES) m/e 315, 317.
Elementaranalyse
für C20H16N2O2. Berechnet: C 75,95, H 5,06, N 8,86. Gefunden:
C 74,88, H 5,40, N 7,78.
-
D. {9-[(Phenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäuremethylester
-
40%
Methanolisches Triton B (0,11 ml, 0,24 mM) wird zu einer Lösung von
9-[(Phenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
(70 mg, 0,22 mM) in 20 ml DMF bei 0°C gegeben. Nach 15 Minuten wird
Methylbromacetat (70 mg, 0,44 mM) zugegeben und das entstehende
Gemisch wird bei Raumtemperatur für 5 h gerührt. Das Gemisch wird mit Ethylacetat
verdünnt,
mit 1 N HCl, H2O und gesättigter Kochsalzlösung gewaschen, über Magnesiumsulfat
getrocknet, filtriert und konzentriert. Der Rückstand wird mit dem rohen
Material vereinigt, das aus einem ähnlichen Lauf unter Verwendung
von 45 mg (0,14 mM [0,36 mM gesamt]) an 9-[(Phenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
stammt. Die vereinigten Rückstände werden
durch Säulenchromatographie
auf Silicagel (Elution mit Ethylacetat) unter Bildung von 76 mg
(54%) an {9-[(Phenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäuremethylester
als weißer
Feststoff gereinigt.
1H NMR (DMSO-d6) δ 7,65
(d, 1H, J = 8 Hz), 7,5 (br s, 1H), 7,4-7,15 (m, 9H), 7,1 (d, 1H,
J = 8 Hz), 6,6 (d, 1H, J = 8 Hz), 5,7 (s, 2H), 4,9 (s, 2H) und 3,75
(s, 3H).
IR (KBr, cm–1) 3367, 3200, 1760,
1643, 1579, 1496, 1452, 1427, 1216, 1157, 772 und 716.
MS (FD)
m/e 388.
Elementaranalyse für
C23H20N2O4: Berechnet: C 71,13, H 5,15, N 7,22. Gefunden:
C 70,77, H 5,49, N 6,79.
-
E. {9-[(Phenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäurenatriumsalz
-
Eine
Lösung
aus {9-[(Phenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäuremethylester
(10,1 mg, 0,025 mM) und 0,025 ml (0,025 mM) an 1 N NaOH in 3 ml
Ethanol wird für
16 h bei 25°C
gerührt.
Der entstehende weiße
Niederschlag wird durch Filtration gesammelt, mit einer kleinen
Menge an EtOH gewaschen und dann unter Bildung von 7,1 mg (70%)
an {9-[(Phenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäurenatriumsalz als weißes Pulver
gewaschen.
1H NMR (DMSO-d6) δ 7,6 (d,
1H, J = 8 Hz), 7,5-7,05 (m, 11H), 6,55 (d, 1H, J = 8 Hz), 5,75 (s,
2H) und 4,3 (s, 2H).
IR (KBr, cm–1)
3471, 1657, 1615, 1591, 1496, 1453, 1412, 1330, 1272 und 1151.
MS
(ES) m/e 373, 375, 397.
Elementaranalyse für C22H17N2O4Na:
C 66,67, H 4,29, N 7,07. Gefunden: C 66,75, H 4,55, N 6,83.
-
Beispiel 2
-
Herstellung von [9-Benzyl-4-carbamoyl-8-fluor-1,2,3,4-tetrahydrocarbazol-5-yl]oxyessigsäure
-
A. Herstellung von (2-Chlor-4-fluorphenyl)-ethylcarbonat
-
Eine
Lösung
aus 19,16 g an 2-Chlor-4-fluorphenol in 65,4 ml an 2 N wässriger
Natriumhydroxidlösung wird
in einem Eisbad gekühlt
und tropfenweise mit 16,3 ml Ethylchlorformiat behandelt. Nach dem
Rühren
bei Raumtemperatur über
Nacht wird das Zweiphasenreaktionsgemisch mit 100 ml Wasser verdünnt und
mit 300 ml eines 1:1 Pentan/Ethergemisches extrahiert. Der Extrakt
wird dreimal mit 0,02 N Natriumhydroxidlösung, Wasser und dann Kochsalzlösung gewaschen.
Nach dem Trocknen und Eindampfung werden 27,63 g (97%) der Untertitelverbindung
erhalten.
NMR (300 MHz, CDCl3): δ 7,23-7,18
(m, 2H), 7,00 (dt, J = 8,4, 2,7, 1H), 4,35 (q, J = 7,1, 2H), 1,40
(t, J = 7,1, 3H).
-
B. Herstellung von (2-Chlor-4-fluor-5-nitrophenyl)-ethylcarbonat
-
Eine
Lösung
aus 27,63 g an (2-Chlor-4-fluorphenyl)-ethylcarbonat in 60 ml Dichlormethan
wird in einem Eisbad gekühlt
und tropfenweise mit 31,86 g eines 1:2 Gemisches aus rauchender
Salpetersäure
(90%) und konzentrierter Schwefelsäure behandelt. Die Reaktion
wird für
2 Stunden bei Raumtemperatur gerührt
und dann mit Eis gekühlt
und mit weiteren 4,5 g desselben Nitrierungsgemisches behandelt.
Die Reaktion wird über Nacht
bei Raumtemperatur gerührt,
in 200 ml Eis und Wasser gegossen und zweimal mit Dichlormethan
extrahiert. Die Extrakte werden mit Wasser und dann mit Kochsalzlösung gewaschen, über Magnesiumsulfat
getrocknet und unter Bildung von 33,01 g (99%) der Untertitelverbindung
konzentriert.
Smp. 50–51°C.
Elementaranalyse:
Berechnet: C 41,01, H 2,68, N 5,31, Cl 13,45. Gefunden: C 41,03,
H 2,59, N 5,38, Cl 13,71.
-
C. Herstellung von 2-Chlor-4-fluor-5-nitroanisol
-
Eine
Lösung
aus 15,0 g an (2-Chlor-4-fluor-5-nitrophenyl)-ethylcarbonat in 100
ml Dimethylformamid wird mit 18,6 g Cäsiumcarbonat, 7,1 ml Iodmethan
und 7 ml Methanol behandelt und über
Nacht bei Raumtemperatur gerührt.
Das Reaktionsgemisch wird in Wasser gegossen und zweimal mit Ether
extrahiert. Die Extrakte werden zweimal mit Wasser und dann mit
Kochsalzlösung
gewaschen, über
Magnesiumsulfat getrocknet und unter Bildung von 11,4 g der Untertitelverbindung
konzentriert.
Smp. 69–70°C. Bsp. 57,
C.
Elementaranalyse: Berechnet: C 40,90, H 2,45, N 6,81, Cl
17,25. Gefunden: C 41,20, H 2,48, N 6,70, Cl 17,44.
-
D. Herstellung von 2-Fluor-5-methoxyanilin
-
Eine
Lösung
aus 5,63 g an 2-Chlor-4-fluor-5-nitroanisol in 90 ml Ethanol und
5 ml Triethylamin wird bei Raumtemperatur unter 60 psi mit 1,0 g
an 5% Palladium auf Kohle für
vier Stunden hydriert. Der Katalysator wird abfiltriert und das
Lösemittel
wird verdampft. Der Rückstand
wird in Chloroform aufgeschlämmt
und durch ein Kissen aus Silicagel filtriert und dann eingedampft.
Dieser Rückstand
wird auf Silicagel mittels eines Hexan/Chloroformgemisches unter
Bildung von 2,77 g (72%) der Untertitelverbindung chromatographiert.
Smp.
253–254°C.
NMR
(300 MHz, CDCl3): δ 6,88 (dd, J = 10,6, 8,9, 1H),
6,32 (dd, J = 7,4, 3,0, 1H), 6,20 (dt, J = 8,9, 3,2, 1H), 3,73 (s,
3H), 3,72 (br, 2H).
-
E. Herstellung von N-Benzyl-2-fluor-5-methoxyanilin
-
Dieses
Verfahren wird gemäß dem von
Tietze und Grote, Chem. Ber. 126 (12), 2733 (1993) ausgeführt. Eine
Lösung
aus 2,73 g an 2-Fluor-5-methoxyanilin und 2,67 g Benzaldehyd in
48 ml Methanol wird mit 3,43 g Zinkchlorid behandelt und dann in
einem Eisbad gekühlt.
Natriumcyanoborhydrid (1,58 g) wird in kleinen Portionen bei Raumtemperatur über 30 Minuten
zugegeben und die Reaktion wird für fünf Stunden bei Raumtemperatur
gerührt.
Nach der Verdampfung des Lösemittels
wird der Rückstand
in 40 ml an 1 N Natriumhydroxidlösung
aufgeschlämmt
und dann zweimal mit Ether extrahiert. Die Extrakte werden mit Wasser
und dann mit Kochsalzlösung
gewaschen, über
Magnesiumsulfat getrocknet und konzentriert. Der Rückstand
wird aus Hexan unter Bildung von 2,61 g umkristallisiert und die
Mutterlaugen werden auf Silicagel unter Verwendung von 20:1 Hexan/Ether
unter Bildung von weiteren 1,4 g der Untertitelverbindung (90%)
chromatographiert.
Smp. 56–58°C.
Elementaranalyse:
Berechnet: C 72,71, H 6,10; N 6,06. Gefunden: C 72,51, H 6,06, N
5,99.
-
F. Herstellung von Ethyl-9-benzyl-5-methoxy-8-fluor-1,2,3,4-tetrahydrocarbazol-4-carboxylat
-
Eine
Lösung
aus 0,62 g an N-Benzyl-2-fluor-5-methoxyanilin in 20 ml trockenem
Tetrahydrofuran wird in einem Eisbad gekühlt und mit 11,3 ml an 0,5
M Kaliumbis(trimethylsilyl)amid in Toluol behandelt. Nach dem Rühren für 30 Minuten
werden 0,74 g an 2-Carboethoxy-6-bromcyclohexanon (Sheehan und Mumaw,
JACS, 72, 2127 (1950)) in 4 ml Tetrahydrofuran zugegeben und die
Reaktion kann sich langsam über
2 Stunden auf Raumtemperatur erwärmen.
Die Reaktion wird mit gesättigter
Ammoniumchloridlö sung
gestoppt und zweimal mit Ether extrahiert. Die Extrakte werden mit
Wasser und dann mit Kochsalzlösung
gewaschen, über
Magnesiumsulfat getrocknet und konzentriert. Dieser Rückstand
wird auf Silicagel mittels eines Hexan/Ethergemisches unter Bildung
von 0,796 g (74%) an N-alkylierten Zwischenproduktdiastereomeren
chromatographiert. Dieses Gemisch wird in 20 ml Benzol mit 0,99
g Zinkchlorid über
Nacht am Rückfluss
erhitzt. Das Lösemittel wird
verdampft und der Rückstand
wird zwischen 25 ml an 1 N HCl und 25 ml Ethylacetat aufgeteilt
und dann erneut mit Ethylacetat extrahiert. Die organischen Phasen
werden mit Wasser und dann Kochsalzlösung gewaschen, über Magnesiumsulfat
getrocknet und unter Bildung von 0,734 g (96%) der Untertitelverbindung
konzentriert.
ESIMS m/e 382 (M++1).
Elementaranalyse:
Berechnet: C 72,42, H 6,34, N 3,67. Gefunden: C 72,20, H 6,26, N
3,70.
-
G. Herstellung von 9-Benzyl-5-methoxy-8-fluor-1,2,3,4-tetrahydrocarbazol-4-carboxamid
-
Ethyl-9-benzyl-5-methoxy-8-fluor-1,2,3,4-tetrahydrocarbazol-4-carboxylat
(0,722 g) wird ähnlich
wie in Beispiel 49, Teil C, beschrieben, behandelt und auf Silicagel
mittels 1% Methanol in Dichlormethan unter Bildung von 0,482 g (72%)
der Untertitelverbindung chromatographiert.
ESIMS m/e 353 (M++1).
Elementaranalyse: Berechnet: C
71,57, H 6,01, N 7,95. Gefunden: C 71,42, H 5,83, N 7,75.
-
H. Herstellung von [9-Benzyl-4-carbamoyl-8-fluor-1,2,3,4-tetrahydrocarbazol-5-yl]oxyessigsäuremethylester
-
9-Benzyl-5-methoxy-8-Fluor-1,2,3,4-tetrahydrocarbazol-4-carboxamid
(0,170 g) wird ähnlich
wie in Beispiel 49, Teil D, beschrieben, umgewandelt und auf Silicagel
mittels Methanol/0–1%
in Dichlormethan unter Bildung von 85 mg (50%) der Untertitelverbindung
chromatographiert.
Smp. 183–185°C.
Elementaranalyse: Berechnet:
C 67,31, H 5,65, N 6,82. Gefunden: C 67,58, H 5,48, N 6,95.
-
1. Herstellung von [9-Benzyl-4-carbamoyl-8-Fluor-1,2,3,4-tetrahydrocarbazol-5-yl]oxyessigsäure
-
9-Benzyl-4-carbamoyl-8-fluor-1,2,3,4-tetrahydrocarbazol-5-yl]oxyessigsäuremethylester
(71 mg) wird ähnlich
wie in Beispiel 50, Teil D, beschrieben, unter Bildung von 65 mg
der Titelverbindung hydrolysiert.
ESIMS m/e 397 (M++1),
395 (M+-1).
NMR (300 MHz, d6-DMSO): δ 13,03
(br, 1H), 7,31-7,19 (m, 3H), 6,97 (d, J = 7,4, 2H), 6,95 (br, 1H),
6,70 (d, J = 3,8, 1H), 6,67 (dd, J = 12,4, 3,9, 1H), 6,28 (dd, J
= 8,5, 2,6, 1H), 5,39 (ABq, 2H), 4,64 (s, 2H), 3,92 (br, 1H), 2,71
(m, 1H), 2,44 (m, 1H), 2,02 (m, 2H), 1,76 (m, 2H).
-
Beispiel 3
-
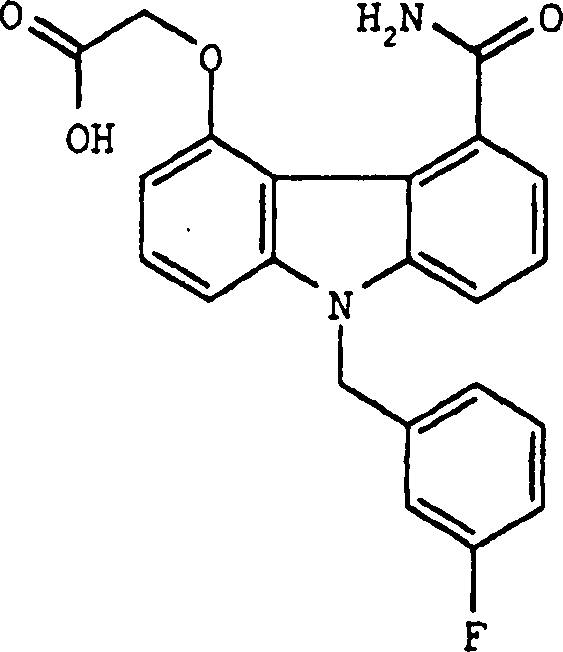
Herstellung von [9-Benzyl-5-carbamoyl-1-fluorcarbazol-4-yl]oxyessigsäure
-
A. Herstellung von 9-Benzyl-5-carbamoyl-4-methoxy-1-fluorcarbazol
-
Eine
Lösung
aus 0,458 g an 9-Benzyl-5-methoxy-8-fluor-1,2,3,4-tetrahydrocarbazol-4-carboxamid
in 13 ml trockenem Dioxan unter Stickstoff wird mit 0,59 g an 2,3-Dichlor-5,6-dicyano-1,4-benzochinon
behandelt und am Rückfluss
für eine
Stunde erhitzt. Das Reaktionsgemisch wird abgekühlt und filtriert und der Niederschlag
wird mit 15 ml Dioxan gewaschen. Das Filtrat und die Waschlösung werden
in gesättigte
Natriumbicarbonatlösung
gegossen und dreimal mit Ethylacetat extrahiert. Die Extrakte werden
mit gesättigtem
Natriumbicarbonat, Wasser und dann Kochsalzlösung gewaschen, über Magnesiumsulfat
getrocknet und konzentriert. Dieser Rückstand wird auf Silicagel
mittels Dichlormethan/0–2%
Methanol unter Bildung von 0,45 g der Untertitelverbindung chromatographiert.
ESIMS
m/e 349 (M++1).
Elementaranalyse: Berechnet:
C 72,42, H 4,92, N 8,04. Gefunden: C 72,35, H 4,81, N 7,88.
-
B. Herstellung von [9-Benzyl-5-carbamoyl-1-fluorcarbazol-4-yl]oxyessigsäuremethylester
-
Eine
Lösung
aus 0,45 g an 9-Benzyl-5-carbamoyl-4-methoxy-1-fluorcarbazol in
25 ml Dichlormethan wird in einem Eisbad gekühlt und tropfenweise mit 12
ml an 1,0 M Bortribromidlösung
in Dichlormethan behandelt. Die Reaktion kann sich langsam über 2 Stunden
auf Raumtemperatur erwärmen
und wird dann durch Gießen
in Eis und die Zugabe von 50 ml an 1 N HCl gestoppt. Das Gemisch
wird mit Dichlormethan extrahiert (3 × 200 ml) und die Extrakte
werden über
Magnesiumsulfat getrocknet und unter Bildung von 0,35 g (78%) des demethylierten
Zwischenprodukts konzentriert. Dieses Zwischenprodukt (0,215 g)
wird alkyliert und ähnlich
zu Beispiel GH1, Teil D unter Bildung von 0,166 g (64%) der Untertitelverbindung
gereinigt.
Smp. 190–191°C
Elementaranalyse:
Berechnet: C 67,97, H 4,71, N 6,89. Gefunden: C 67,81, H 4,94, N
6,96.
-
C. Herstellung von [9-Benzyl-5-carbamoyl-1-fluorcarbazol-4-yl]oxyessigsäure
-
[9-Benzyl-5-carbamoyl-1-fluorcarbazol-4-yl]oxyessigsäuremethylester
(56 mg) wird hydrolysiert und ähnlich
wie in Beispiel 50, Teil D, unter Bildung von 54 mg der Titelverbindung
isoliert.
FDMS m/e 392 (M+)
ESIMS
m/e 393 (M++1), 391 (M+-1).
NMR
(300 MHz, d6-DMSO): δ 12,92 (br, 1H), 7,70 (m, 2H),
7,45 (t, J = 7,5, 1H), 7,39 (br, 1H), 7,28-7,17 (m, 4H), 7,12 (d,
J = 7,2, 1H), 7,07 (d, J = 7,0, 2H), 6,51 (dd, J = 8,8, 2,7, 1H),
5,77 (s, 2H), 4,80 (s, 2H).
Elementaranalyse: Berechnet: C
67,34, H 4,37, N 7,14. Gefunden: C 66,92, H 4,49, N 6,77.
-
Beispiel 4
-
Herstellung von {9-[(3-Fluorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure
-
-
A. 9-[(3-Fluorphenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
-
Es
wird 40% methanolisches Triton B (2,06 ml, 4,53 mM) langsam tropfenweise
zu einer Lösung
aus 5-Carbomethoxy-1,2-dihydro-9H-carbazol-4(3H)-on (930,0 mg, 3,82
mM) in 5 ml DMF bei 0°C
gegeben. Nach 5 Minuten wird 3-Fluorbenzylchlorid (664,0 mg, 4,59
mM) zugegeben und das entstehende Gemisch wird bei 0°C für 3 h und
dann bei Raumtemperatur für
20 Stunden gerührt.
Das Gemisch wird mit Ethylacetat verdünnt, dreimal mit 1 N HCl, dreimal
mit H2O und einmal mit gesättigter
Kochsalzlösung
gewaschen, über
wasserfreiem Magnesiumsulfat getrocknet, filtriert und konzentriert.
Der Rückstand
wird durch Säulenchromatographie auf
Silicagel (Elution mit einem Gradienten aus Methylenchlorid / Ethylacetat)
unter Bildung von 502,3 mg (37%) an 9-[(3-Fluorphenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
als gelber Schaum gereinigt.
1H NMR
(CDCl3) δ 7,4-7,2
(m, 4H), 6,9 (m, 1H), 6,7 (m, 2H), 5,35 (s, 2H), 4,05 (s, 3H), 2,9
(t, 2H, J = 6 Hz), 2,65 (t, 2H, J = 6 Hz) und 2,3 (m, 2H).
IR
(CHCl3, cm–1)
3050, 2950, 1725, 1654, 1464, 1451, 1440, 1288 und 1119. MS (ES)
m/e 350, 352.
Elementaranalyse für C21H18NO3F: Berechnet:
C 71,78, H 5,16, N 3,99. Gefunden: C 72,00, H 4,95, N 4,11.
-
B. 9-((3-Fluorphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol
-
Eine
Lösung
aus 9-[(3-Fluorphenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
(434,0 mg, 1,23 mM) und 2,3-Dichlor-5,6-dicyano-1,4-benzochinon
(324,0 mg, 1,42 mM) in 20 ml Toluol wird zwischen 70–80°C für 5 h gerührt. Das
Gemisch wird direkt durch Säulenchromatographie
auf Silicagel (Elution mit Methylenchlorid) unter Bildung von 137,0
mg (32%) an 9-[(3-Fluorphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol als gelber Schaum
gereinigt.
1H NMR (DMSO-d6) δ 10,2 (s,
1H), 7,7 (d, 1H, J = 8 Hz), 7,4 (t, 1H, J = 8 Hz), 7,3 (m, 2H),
7,2 (d, 1H, J = 8 Hz), 7,1 (d, 1H, J = 8 Hz), 7,05-6,85 (m, 3H),
6,6 (d, 1H, J = 8 Hz), 5,65 (s, 2H) und 3,85 (s, 3H).
IR (CNCl3, cm–1) 3200 (br), 1687,
1597, 1452, 1442, 1285 und 1267.
MS (ES) m/e 348, 350.
Elementaranalyse
für C21H16NO3F:
Berechnet: C 72,20, H 4,62, N 4,01. Gefunden: C 72,30, H 4,66, N
4,04.
-
C. 9-[(3-Fluorphenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
-
Eine
Lösung
aus 9-[(3-Fluorphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol (130,8
mg, 0,37 mM) in 5 ml THF und 20 ml konzentriertes wässriges
Ammoniumhydroxid wird für
5 h bei 40–50°C ultrabeschallt. Das
Gemisch wird mit Ethylacetat verdünnt und mit 5 N HCl auf pH
1 angesäuert.
Die wässrige
Phase wird zweimal mit Ethylacetat extrahiert. Die vereinigten organischen
Extrakte werden mit gesättigter
Kochsalzlösung
gewaschen, über
Magnesiumsulfat getrocknet, filtriert und konzentriert. Der Rückstand
wird durch Säulenchromatographie
auf Silicagel (Elution mit einem Gradienten aus Methylenchlorid/Ethylacetat)
unter Bildung von 57,4 mg (45%) an 9-[(3-Fluorphenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
als weißer
Feststoff gereinigt.
1H NMR (DMSO-d6) δ 10,5
(s, 1H), 8,8 (br s, 1H), 8,4 (br s, 1H), 7,8 (dd, 1H, J = 1 und
8 Hz), 7,5 (m, 2H), 7,3 (m, 2H), 7,15-7,0 (m, 2H), 6,95 (d, 1H,
J = 8 Hz), 6,85 (d, 1H, J = 8 Hz), 6,6 (d, 1H, J = 8 Hz) und 5,7
(s, 2H).
IR (CHCl3, cm–1)
3431, 3200 (br), 1628, 1614, 1600, 1580, 1546, 1488, 1448, 1329,
1261 und 776.
MS (ES) m/e 333, 335.
Elementaranalyse für C20H15N2O2F: Berechnet: C 71,85, H 4,52, N 8,38. Gefunden:
C 74,45, H 6,01, N 8,48.
-
D. {9-[(3-Fluorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-tert-butylester
-
Es
wird 40% methanolisches Triton B (0,086 ml, 0,19 mM) zu einer Lösung aus
9-[(3-Fluorphenyl)methyl]-4-hydroxy-5-carbamoylcarbazol (51,9 mg,
0,155 mM) in 3 ml DMF bei Raumtemperatur gegeben. Nach 3 Minuten
wird t-Butylbromacetat (87,8 mg, 0,44 mM) zugegeben und das entstehende
Gemisch wird bei Raumtemperatur für 5 Stunden gerührt. Das
Gemisch wird mit Ethylacetat verdünnt, viermal mit H2O
und gesättigter
Kochsalzlösung
gewaschen, über
Magnesiumsulfat getrocknet, filtriert und konzentriert. Der Rückstand
wird durch Säulenchromatographie
auf Silicagel (Elution mit einem Gradienten aus Methylenchlorid/Ethylacetat)
unter Bildung von 44,0 mg (63%) an {9-[(3-Fluorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-tert-butylester
als weißer
Feststoff gereinigt.
1H NMR (DMSO-d6) δ 7,6
(d, 1H, J = 8 Hz), 7,5-6,8 (m, 10H), 6,55 (d, 1H, J = 8 Hz), 5,7
(s, 2H), 4,8 (s, 2H) und 1,45 (s, 9H).
IR (CHCl3,
cm–1)
3450, 3400, 1746, 1674, 1592, 1457, 1369 und 1151.
MS (FD)
m/e 448.
Elementaranalyse für
C26H25N2O4F: Berechnet: C 69,63, H 5,62, N 6,25. Gefunden:
C 69,35, H 5,44, N 6,23.
-
E. {9-[(3-Fluorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure
-
Eine
Lösung
aus {9-[(3-Fluorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-tert-butylester (40,0
mg, 0,089 mM) in 2 ml Trifluoressigsäure wird bei Raumtemperatur
für 5 Stunden
gerührt.
Das Lösemittel wird
im Vakuum entfernt. Der Rückstand
wird mit Ethylether behandelt und dann im Vakuum unter Bildung von 35,0
mg (100%) an {9-[(3-Fluorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure als
weißes
Pulver getrocknet.
1H NMR (DMSO-d6) δ 13,0
(br s, 1H), 7,75 (s, 1H), 7,6 (d, 1H, J = 8 Hz), 7,5-7,25 (m, 5H),
7,2-6,8 (m, 4H), 6,6 (d, 1H, J = 8 Hz), 5,7 (s, 2H) und 4,8 (s,
2H).
IR (KBr, cm–1) 3423, 3400, 1736,
1637, 1615, 1589, 1499, 1487, 1450, 1436, 1331, 1250 und 1156.
MS
(ES) m/e 391, 393.
Elementaranalyse für C22H17N2O4F:
Berechnet: C 67,34, H 4,37, N 7,14. Gefunden: C 67,63, H 4,22, N
7,35.
-
Beispiel 5
-
Herstellung von {9-[(3-Chlorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure
-
-
A. 9-[(3-Chlorphenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
-
Eine
Suspension aus 5-Carbomethoxy-1,2-dihydro-9H-carbazol-4(3H)-on (527,0
mg, 2,17 mM), 3-Chlorbenzylbromid (802,2 mg, 3,90 mM), eine katalytische
Menge Natriumiodid (ca. 1 mg) und Kaliumcarbonat (500,0 mg, 3,62
mM) wird bei Raumtemperatur für
150 Stunden gerührt.
Das Gemisch wird mit Ethylacetat verdünnt, fünfmal mit H2O
und einmal mit gesättigter
Kochsalzlösung
gewaschen, über
wasserfreiem Magnesiumsulfat getrocknet, filtriert und konzentriert.
Der Rückstand
wird durch Säulenchromatographie
auf Silicagel (Elution mit einem Gradienten aus Methylenchlorid/Ethylacetat)
unter Bildung 537,1 mg (67%) an 9-[(3-Chlorphenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on als gelber Schaum
gereinigt.
1H NMR (CDCl3) δ 7,5-7,2
(m, 5H), 7,1 (s, 1H), 6,85 (m, 1H), 5,35 (s, 2H), 4,05 (s, 3H),
2,9 (t, 2H, J = 6 Hz), 2,65 (t, 2H, J = 6 Hz) und 2,3 (m, 2H).
IR
(CNCl3, cm–1)
3050, 2950, 1725, 1654, 1464, 1444, 1432, 1288 und 1120.
MS
(ES) m/e 366, 368, 370.
Elementaranalyse für C21H18NO3Cl: Berechnet:
C 68,57, H 4,93, N 3,81. Gefunden: C 68,61, H 4,92, N 3,70.
-
B. 9-[(3-Chlorphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol
-
Eine
Lösung
aus 9-[(3-Chlorphenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
(480,5 mg, 1,31 mM) und 2,3-Dichlor-5,6-dicyano-1,4-benzochinon
(325,7 mg, 1,43 mM) in 50 ml Toluol wird zwischen 70–80°C für 3 Stunden
gerührt.
Das Gemisch wird direkt durch Säulenchromatographie
auf Silicagel (Elution mit Methylenchlorid) unter Bildung von 172,6
mg (36%) an 9-[(3-Chlorphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol als gelber
Schaum gereinigt.
1H NMR (CDCl3) δ 10,4
(s, 1H), 8,05 (d, 1H, J = 8 Hz), 7,6 (d, 1H, J = 8 Hz), 7,4 (m,
2H), 7,3-7,1 (m, 3H), 6,9-6,7 (m, 3H), 5,55 (s, 2H) und 4,15 (s,
3H).
IR (CHCl3, cm–1)
3200 (br), 1684, 1598, 1442, 1428, 1331, 1285 und 1267.
MS
(ES) m/e 364, 366, 368.
Elementaranalyse für C21H16NO3Cl: Berechnet:
C 68,95, H 4,41, N 3,83. Gefunden: C 69,23, H 4,52, N 3,88.
-
C. 9-[(3-Chlorphenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
-
Eine
Lösung
aus 9-[(3-Chlorphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol (156,2
mg, 0,43 mM) in 5 ml THF und 20 ml konzentriertes wässriges
Ammoniumhydroxid wird für
5 Stunden bei 40–50°C ultrabeschallt.
Das Gemisch wird mit Ethylacetat verdünnt und mit 5 N HCl auf pH
1 angesäuert.
Die wässrige
Phase wird zweimal mit Ethylacetat extrahiert. Die vereinigten organischen
Extrakte werden mit gesättigter
Kochsalzlösung
gewaschen, über
Magnesiumsulfat getrocknet, filtriert und konzentriert. Der Rückstand
wird durch Säulenchromatographie
auf Silicagel (Elution mit einem Gradienten aus Methylenchlorid/Ethylacetat)
unter Bildung von 69,7 mg (47%) an 9-[(3-Chlorphenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
als weißer
Feststoff gereinigt.
1H NMR (DMSO-d6) δ 10,5
(s, 1H), 8,8 (br s, 1H), 8,4 (br s, 1H), 7,8 (dd, 1H, J = 1 und
8 Hz), 7,45 (m, 2H), 7,3 (m, 3H), 7,2 (s, 1H), 7,1 (d, 1H, J = 8
Hz), 6,95 (s, 1H), 6,6 (d, 1H, J = 8 Hz) und 5,7 (s, 2H).
IR
(CHCl3, cm–1)
3433, 3202 (br), 1630, 1600, 1580, 1564, 1433, 1330, 1261 und 776.
MS (ES) m/e 349, 351, 353.
Elementaranalyse für C20H15N2O2Cl: Berechnet: C 68,48, H 4,31, N 7,99.
Gefunden: C 68,64, H 4,55, N 7,93.
-
D. {9-[(3-Chlorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-tert-butylester
-
Es
wird 40% methanolisches Triton B (0,053 ml, 0,12 mM) zu einer Lösung aus
9-[(3-Chlorphenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
(33,2 mg, 0,12 mM) in 2 ml DMF bei Raumtemperatur gegeben. Nach
3 Minuten wird t-Butylbromacetat (53,8 mg, 0,27 mM) zugegeben und
das entstehende Gemisch wird bei Raumtemperatur für 20 Stunden
gerührt.
Das Gemisch wird mit Ethylacetat verdünnt, viermal mit H2O
und einmal mit gesättigter
Kochsalzlösung
gewaschen, über
Magnesiumsulfat getrocknet, filtriert und konzentriert. Der Rückstand
wird durch Säulenchromatographie
auf Silicagel (Elution mit einem Gradienten aus Methylenchlorid/Ethylacetat)
unter Bildung von 42,1 mg (95%) an {9-[(3-Chlorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-tert-butylester
als weißer
Feststoff gereinigt.
1H NMR (DMSO-d6) δ 7,6
(d, 1H, J = 8 Hz), 7,5-6,8 (m, 10H), 6,55 (d, 1H, J = 8 Hz), 5,7
(s, 2H), 4,8 (s, 2H) und 1,45 (s, 9H).
IR (CHCl3,
cm–1)
3450, 3400, 1744, 1676, 1591, 1457, 1369 und 1150.
MS (FD)
m/e 464,466.
Elementaranalyse für C26H25N2O4Cl:
Berechnet: C 67,17, H 5,42, N 6,03. Gefunden: C 67,17, H 5,65, N
5,97.
-
E. {9-[(3-Chlorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure
-
Eine
Lösung
aus {9-[(3-Chlorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-tert-butylester (35,6
mg, 0,077 mM) in 2 ml Trifluoressigsäure wird bei Raumtemperatur
für 6 Stunden
gerührt.
Das Lösemittel wird
im Vakuum entfernt. Der Rückstand
wird mit Ethylacetat behandelt und dann im Vakuum unter Bildung von
31,4 mg (100%) an {9-[(3-Chlorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure als
weißes
Pulver getrocknet.
1H NMR (DMSO-d6) δ 13,0
(br s, 1H), 7,75 (s, 1H), 7,6 (d, 1H, J = 8 Hz), 7,4-7,25 (m, 7H),
7,2 (d, 1H, J = 8 Hz), 7,0 (br t, 1H), 6,6 (d, 1H, J = 8 Hz), 5,7
(s, 2H) und 4,8 (s, 2H).
IR (KBr, cm–1)
3456, 3416, 3335, 1735, 1638, 1617, 1580, 1499, 1452, 1431, 1431,
1329, 1255, 1157, 772, 764 und 717.
MS (ES) m/e 407, 409, 411.
Elementaranalyse
für C22H17N2O4Cl: Berechnet: C 64,63, H 4,19, N 6,85.
Gefunden: C 64,55, H 4,12, N 6,74.
-
Beispiel 6
-
Herstellung von {9-[(3-Trifluormethylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäurenatriumsalz
-
-
A. 9-[(3-Trifluormethylphenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
-
Es
wird 40% methanolisches Triton B (2,18 ml, 4,8 mM) langsam tropfenweise
zu einer Lösung
aus 5-Carbomethoxy-1,2-dihydro-9H-carbazol-4(3H)-on (973 mg, 4,0
mM) in 10 ml DMF bei –10°C gegeben.
Nach 30 Minuten wird 3-(Trifluormethyl)benzylchlorid (1,53 g, 6,0
mM) und Natriumiodid (900 mg, 6,0 mM) zugegeben und das entstehende
Gemisch wird bei Raumtemperatur für 25 Stunden gerührt. Das
Gemisch wird mit Ethylacetat verdünnt, fünfmal mit H2O,
1 N HCl, H2O und gesättigtem NaHCO3 und
gesättigter
Kochsalzlösung gewaschen, über wasserfreiem
Magnesiumsulfat getrocknet, filtriert, konzentriert und im Vakuum
getrocknet. Der Rückstand
wird mittels Säulenchromatographie
auf Silicagel (Elution mit einem Gradienten aus Methylenchlorid/Ethylacetat)
unter Bildung von 1,02 g (63%) an 9-[(3-Trifluormethylphenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
als hellbrauner Feststoff gereinigt.
1H
NMR (CDCl3) δ 7,6 (d, 1H, J = 8 Hz), 7,45–7,2 (m,
5H), 7,0 (d, 1H, J = 8 Hz), 5,4 (s, 2H), 4,05 (s, 3H), 2,85 (t,
2H, J = 6 Hz), 2,6 (t, 2H, J = 6 Hz) und 2,2 (m, 2H).
IR (KBr,
cm–1)
1727 und 1652.
MS (ES) m/e 400, 402.
Elementaranalyse
für C22H18NO3F3: Berechnet: C 65,83, H 4,52, N 3,49, F
14,20. Gefunden: C 65,63, H 4,58, N 3,39, F 14,14.
-
B. 9-[(3-Trifluormethylphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol
-
Eine
Lösung
aus 9-[(3-Trifluormethylphenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on (1,21 g, 3,00
mM) und 2,3-Dichlor-5,6-dicyano-1,4-benzochinon (764 mg, 3,3 mM)
in 25 ml Toluol wird zwischen 80–90°C für 7 Stunden gerührt. Das
Gemisch wird direkt durch Säulenchromatographie
auf Silicagel (Elution mit Methylenchlorid) unter Bildung von 340,0
mg (28%) an 9-[(3-Trifluormethylphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol
als gelber Feststoff gereinigt.
1H
NMR (CDCl3) δ 10,35 (s, 1H), 8,0 (d, 1H,
J = 8 Hz), 7,6-7,3 (m, 6H), 7,05 (d, 1H, J = 8 Hz), 6,85 (m, 2H), 5,6
(s, 2H) und 4,1 (s, 3H).
IR (CHCl3,
cm–1)
3378 und 1712.
MS (ES) m/e 398, 400.
Elementaranalyse
für C22H16NO3F3: Berechnet: C 66,17, H 4,04, N 3,51. Gefunden:
C 66,99, H 4,12, N 3,53.
-
C. 9-[(3-Trifluormethylphenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
-
Eine
Lösung
aus 9-[(3-Trifluormethylphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol
(250 mg, 0,625 mM) in 5 ml THF und 20 ml konzentriertes, wässriges
Ammoniumhydroxid wird für
30 h bei 40–50°C ultrabeschallt.
Das Gemisch wird mit Ethylacetat verdünnt und mit 5 N HCl auf pH
1 angesäuert.
Die wässrige Phase
wird dreimal mit Ethylacetat extrahiert. Die vereinigten organischen
Extrakte werden mit gesättigter Kochsalzlösung gewaschen, über Magnesiumsulfat
getrocknet, filtriert und konzentriert. Der Rückstand wird durch Säulenchromatographie
auf Silicagel (Elution mit einem Gradienten aus Methylenchlorid/Ethylacetat) unter
Bildung von 120 mg (50%) an 9-[(3-Trifluormethylphenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
als weißer
Feststoff gereinigt.
1H NMR (DMSO-d6) δ 10,5
(s, 1H), 8,8 (br s, 1H), 8,4 (br s, 1H), 7,8 (d, 1H, J = 8 Hz),
7,6-7,5 (m, 5H), 7,3 (t, 1H, J = 8 Hz), 7,15 (d, 1H, J = 8 Hz),
7,1 (d, 1H, J = 8 Hz), 6,6 (d, 1H, J = 8 Hz) und 5,8 (s, 2H).
IR
(KBr, cm–1)
3429, 3206 und 1630.
MS (ES) m/e 383, 385.
Elementaranalyse
für C21H15N2O2F3: Berechnet: C
65,62, H 3,93, N 7,29. Gefunden: C 67,50, H 4,00, N 7,19.
-
D. {9-[(3-Trifluormethylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäuremethylester
-
Es
wird 40% methanolisches Triton B (0,18 ml, 0,4 mM) zu einer Lösung aus
9-[(3-Trifluormethylphenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
(115 mg, 0,3 mM) in 5 ml DMF bei Raumtemperatur gegeben. Nach 15
Minuten wird Methylbromacetat (95 mg, 0,6 mM) zugegeben und das
entstehende Gemisch wird bei Raumtemperatur für 22 Stunden gerührt. Das
Gemisch wird mit Ethylacetat verdünnt, viermal mit H2O,
1 N HCl, H2O, gesättigtem NaHCO3 und
gesättigter
Kochsalzlösung
gewaschen, über
Magnesiumsulfat getrocknet, filtriert und konzentriert. Der Rückstand
wird mittels Säulenchromatographie auf
Silicagel (Elution mit Ethylacetat) unter Bildung von 120 mg (88%)
an {9-[(3-Trifluormethylphenyl)-methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäuremethylester
als weißer
Feststoff gereinigt.
1H NMR (CDCl3) δ 7,5-7,2
(m, 7H), 7,1 (d, 1H, J = 8 Hz), 7,0 (d, 1H, J = 8 Hz), 6,6 (d, 1H,
J = 8 Hz), 6,4 (br s, 1H), 6,0 (br s, 1H), 5,55 (s, 2H), 4,9 (s,
2H) und 3,9 (s, 3H).
IR (KBr, cm–1)
1763 und 1673.
MS (ES) m/e 457.
Elementaranalyse für C24H19N2O4F3: Berechnet: C
63,16, H 4,20, N 6,14. Gefunden: C 61,37, H 4,19, N 5,77.
-
E. {9-[(3-Trifluormethylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäurenatriumsalz
-
Eine
Lösung
aus {9-[(3-Trifluormethylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäuremethylester
(91 mg, 0,153 mM) und 0,22 ml (0,22 mM) an 1 N NaOH in 8 ml Ethanol
wird für
17 h bei 25°C
gerührt. Das
Ethanol wird im Vakuum entfernt. Der entstehende weiße Niederschlag
wird durch Filtration gesammelt, mit kleinen Mengen an EtOH und
Diethylether gewaschen und im Vakuum unter Bildung von 75 mg (81%)
an {9-[(3-Trifluormethylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäurenatriumsalz
als weißer
Pulver getrocknet.
1H NMR (DMSO-d6) δ 7,65
(s, 1H), 7,6 (m, 4H), 7,45 (t, 1H, J = 8 Hz), 7,35 (t, 1H, J = 8
Hz), 7,3 (t, 1H, J = 8 Hz), 7,2 (d, 1H, J = 8 Hz), 7,1 (d, 1H, J
= 8 Hz), 7,05 (d, 1H, J = 8 Hz), 6,5 (d, 1H, J = 8 Hz), 5,75 (s,
2H) und 4,3 (s, 2H).
IR (KBr, cm–1)
1665 und 1618.
MS (ES) m/e 441, 443.
Elementaranalyse
für C23H16N2O4F3Na: Berechnet:
C 59,49, H 3,47, N 6,03. Gefunden: C 60,69, H 3,78, N 5,75.
-
Beispiel 7
-
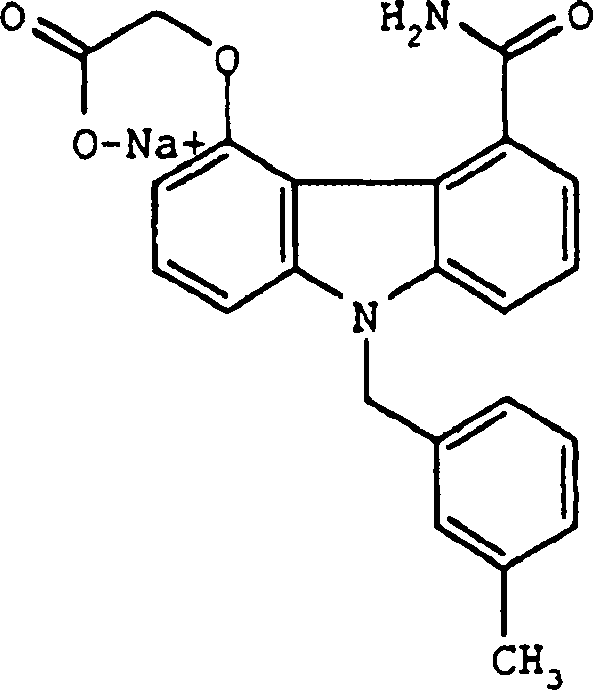
Herstellung von {9-[(2-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäurenatriumsalz
-
-
A. 9-[(2-Methylphenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4-(3H)-on
-
Eine
Suspension aus 5-Carbomethoxy-1,2-dihydro-9H-carbazol-4(3H)-on (870
mg, 3,58 mM), α-Brom-o-xylol (662
mg, 3,58 mM) und Kaliumcarbonat (500 mg, 3,61 mM) in 20 ml DMF wird
bei Raumtemperatur für
20 Stunden gerührt.
Das Gemisch wird mit Ethylacetat verdünnt, mit H2O
und gesättigter
Kochsalzlösung
gewaschen, über
wasserfreiem Magnesiumsulfat getrocknet, filtriert und unter Bildung von
1,21 g (98%) an 9-[(2-Methylphenyl)methyl]-5-carbomethoxy-l,2-dihydrocarbazol-4-(3H)-on
als dunkles Öl
konzentriert.
1H NMR (DMSO-d6) δ 7,5-7,2
(m, 4H), 7,15 (t, 1H, J = 8 Hz), 7,0 (t, 1H, J = 8 Hz), 6,15 (d,
1H, J = 8 Hz), 5,55 (s, 2H), 3,85 (s, 3H), 2,6 (m, 2H), 2,4 (m,
2H), 2,4 (s, 3H) und 2,1 (m, 2H).
IR (CHCl3,
cm–1)
3010, 2952, 1724, 1671, 1653, 1604, 1460, 1444, 1290, 1174 und 1122.
MS
(ES) m/e 348,5.
Elementaranalyse für C22H21NO3: Berechnet:
C 76,08, H 6,05, N 4,03. Gefunden: C 73,33, H 6,36, N 4,30.
-
B. 9-[(2-Methylphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol
-
Eine
Lösung
aus 9-[(2-Methylphenyl)methyl]-5-carbomethoxy-l,2-dihydrocarbazol-4-(3H)-on
(1 ,2 g, 3,5 mM) und 2,3-Dichlor-5,6-dicyano-1,4-benzochinon (800
mg, 3,6 mM) in 70 ml Toluol wird bei 80–90°C für 5 Stunden gerührt. Das
Gemisch wird direkt durch Säulenchromatographie
auf Silicagel (Elution mit Methylenchlorid) unter Bildung von 260
mg (22%) an 9-[(2-Methylphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol als
gelber Feststoff gereinigt.
1H NMR
(DMSO-d6) δ 10,25 (s, 1H), 7,5 (d, 1H,
J = 8 Hz), 7,4 (t, 1H, J = 8 Hz), 7,3-7,1 (m, 4H), 6,9 (m, 2H), 6,6
(d, 1H, J = 8 Hz), 6,1 (d, 1H, J = 8 Hz), 5,65 (s, 2H), 3,8 (s,
3H) und 2,5 (s, 3H).
IR (KBr, cm–1)
3200, 1672, 1440, 1426, 1332, 1302, 1265, 1216, 1141, 761, 749 und
718.
MS (ES) m/e 344, 346.
Elementaranalyse für C22H19NO3:
Berechnet: C 76,52, H 5,51, N 4,06. Gefunden: C 76,44, H 5,66, N
3,94.
-
C. 9-[(2-Methylphenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
-
Eine
Lösung
aus 9-[(2-Methylphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol
(260 mg, 0,75 mM) in 10 ml THF und 30 ml konzentriertem wässrigem
Ammoniumhydroxid wird für
5 Stunden bei 40–50°C ultrabeschallt.
Das Gemisch wird mit Ethylacetat verdünnt und mit 5 N HCl auf pH
1 angesäuert.
Die wässrige
Phase wird dreimal mit Ethylacetat extrahiert. Die vereinigten organischen
Extrakte werden mit H2O und gesättigter Kochsalzlösung gewaschen, über Magnesiumsulfat
getrocknet, filtriert und konzentriert. Der Rückstand wird durch Säulenchromatographie
auf Silicagel (Elution mit einem Gradienten aus Hexan/Ethylacetat)
unter Bildung von 90 mg (36%) an 9-[(2-Methylphenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
als hellbrauner Feststoff gereinigt.
1H
NMR (DMSO-d6) δ 10,5 (s, 1H), 8,8 (br s, 1H),
8,4 (br s, 1H), 7,7 (m, 1H), 7,5 (m, 2H), 7,3 (m, 2H), 7,1 (t, 1H,
J = 8 Hz), 6,95 (d, 1H, J = 8 Hz), 6,85 (t, 1H, J = 8 Hz), 6,6 (d,
1H, J = 8 Hz), 5,95 (d, 1H, J = 8 Hz), 5,7 (s, 2H) und 2,5 (s, 3H).
IR
(KBr, cm–1)
3451, 3191, 1627, 1600, 1584, 1562, 1435, 1329, 1322, 1263 und 774.
MS
(ES) m/e 329, 331.
Elementaranalyse für C21H18N2O2:
Berechnet: C 76,36, H 5,45, N 8,48. Gefunden: C 75,66, H 5,79, N
8,07.
-
D. {9-[(2-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäuremethylester
-
Es
wird 40% methanolisches Triton B (0,45 ml, 0,99 mM) zu einer Lösung aus
9-[(2-Methylphenyl)methyl]-4-hydroxy-5-carbamoylcarbazol (80 mg,
0,24 mM) in 8 ml DMF bei Raumtemperatur gegeben. Nach 3 Minuten
wird Methylbromacetat (115 mg, 0,72 mM) zugegeben und das entstehende
Gemisch wird bei Raumtemperatur für 48 Stunden gerührt. Das
Gemisch wird mit Ethylacetat verdünnt, mit H2O,
1 N HCl, H2O und gesättigter Kochsalzlösung gewaschen, über Magnesiumsulfat
getrocknet, filtriert und konzentriert. Der Rückstand wird durch Säulenchromatographie
auf Silicagel (Elution mit Ethylacetat) unter Bildung von 80 mg
(82%) an {9-[(2-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäuremethylester
als weißer
Feststoff gereinigt.
1H NMR (DMSO-d6) δ 7,56
(br s, 1H), 7,5-7,1 (m, 9H), 6,9 (t, 1H, J = 8 Hz), 6,6 (d, 1H,
J = 8 Hz), 5,65 (s, 2H), 4,9 (s, 2H), 3,8 (s, 3H) und 2,5 (s, 3H).
IR
(KBr, cm–1)
3367, 3153, 1760, 1740, 1672, 1644, 1619, 1591, 1578, 1498, 1456,
1425, 1327, 1200, 1153, 1109, 1100 und 777.
MS (FD) m/e 402.
Elementaranalyse
für C24H22N2O4: Berechnet: C 71,64, H 5,47, N 6,96. Gefunden:
C 71,51, H 5,56, N 6,67.
-
E. {9-[(2-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäurenatriumsalz
-
Eine
Suspension aus {9-[(2-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäuremethylester
(15,5 mg, 0,039 mM) und 0,04 ml (0,04 mM) an 1 N NaOH in 5 ml Ethanol
wird für
24 Stunden bei 25°C gerührt. Der
entstehende weiße
Niederschlag wird durch Filtration gesammelt, mit einer kleinen
Menge an EtOH gewaschen und dann im Vakuum unter Bildung von 10
mg (63%) an {9-[(2-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäurenatriumsalz
als weißes
Pulver getrocknet.
1H NMR (DMSO-d6) δ 7,55
(br s, 1H), 7,5-7,0 (m, 7H), 6,9 (d, 1H, J = 8 Hz), 6,85 (t, 1H,
J = 8 Hz), 6,6 (d, 1H, J = 8 Hz), 6,2 (d, 1H, J = 8 Hz), 5,6 (s,
2H), 4,35 (s, 2H) und 2,5 (s, 3H).
IR (KBr, cm–1)
3390, 1656, 1613, 1595, 1573, 1498, 1455, 1408, 1325, 1332 und 719.
MS
(ES) m/e 387, 389.
Elementaranalyse für C23H19N2O4:
Berechnet: C 67,32, H 4,63, N 6,83. Gefunden: C 64,72, H 4,44, N
6,40.
-
Beispiel 8
-
Herstellung von {9-[(3-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäurenatriumsalz
-
-
A. {9-[(3-Methylphenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
-
Eine
Suspension aus 5-Carbomethoxy-1,2-dihydro-9H-carbazol-4(3H)-on (870
mg, 3,58 mM), α-Brom-m-xylol (662
mg, 3,58 mM) und Kaliumcarbonat (500 mg, 3,61 mM) in 20 ml DMF wird
bei Raumtemperatur für
16 Stunden gerührt.
Das Gemisch wird mit Ethylacetat verdünnt, mit H2O
und gesättigter
Kochsalzlösung
gewaschen, über
wasserfreiem Magnesiumsulfat getrocknet, filtriert und unter Bildung
von 1,18 g (95%) an 9-[(3-Methylphenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4-(3H)-on
als dunkles Öl
konzentriert.
1H NMR (DMSO-d6) δ 7,65
(dd, 1H, J = 1 und 8 Hz), 7,3-7,1 (m, 3H), 7,05 (d, 1H, J = 8 Hz),
7,0 (s, 1H), 6,85 (d, 1H, J = 8 Hz), 5,5 (s, 2H), 3,8 (s, 3H), 3,0
(m, 2H), 2,45 (m, 2H), 2,3 (s, 3H) und 2,1 (m, 2H).
IR (CHCl3, cm–1) 3010, 2953, 1724,
1652, 1605, 1465, 1442, 1288, 1174 und 1119.
MS (ES) m/e 348,5.
Elementaranalyse
für C22H21NO3:
Berechnet: C 76,08, H 6,05, N 4,03. Gefunden: C 74,53, H 6,03, N
3,68.
-
B. 9-[(3-Methylphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol
-
Eine
Lösung
aus 9-[(3-Methylphenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
(1,18 g, 3,4 mM) und 2,3-Dichlor-5,6-dicyano-1,4-benzochinon (800
mg, 3,6 mM) in 70 ml Toluol wird bei 80–90°C für 6 Stunden gerührt. Das
Gemisch wird direkt durch Säulenchromatographie
auf Silicagel (Elution mit Methylenchlorid) unter Bildung von 300
mg (26%) an 9-[(3-Methylphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol als
gelber Feststoff gereinigt.
1H NMR
(DMSO-d6) δ 10,2 (s, 1H), 7,65 (d, 1H,
J = 8 Hz), 7,35 (t, 1H, J = 8 Hz), 7,25 (t, 1H, J = 8 Hz), 7,2-7,0 (m,
4H), 6,9 (m, 2H), 6,6 (d, 1H, J = 8 Hz), 5,6 (s, 2H), 3,85 (s, 3H)
und 2,2 (s, 3H).
IR (KBr, cm–1)
3200, 1673, 1596, 1440, 1426, 1394, 1265, 1216, 1152, 750, 711 und
694.
MS (ES) m/e 344, 346.
Elementaranalyse für C22H19NO3:
Berechnet: C 76,52, H 5,51, N 4,06. Gefunden: C 76,22, H 5,55, N
3,97.
-
C. 9-[(3-Methylphenyl)methylj-4-hydroxy-5-carbamoylcarbazol
-
Eine
Lösung
aus 9-[(3-Methylphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol
(300 mg, 0,87 mM) in 10 ml THF und 30 ml konzentiertem wässrigem
Ammoniumhydroxid wird für
5 Stunden bei 40–50°C ultrabeschallt.
Das Gemisch wird mit Ethylacetat verdünnt und mit 5 N HCl auf pH
1 angesäuert.
Die wässrige
Phase wird dreimal mit Ethylacetat extrahiert. Die vereinigten organischen
Extrakte werden mit H2O und gesättigter Kochsalzlösung gewaschen, über Magnesiumsulfat
getrocknet, filtriert und konzentriert. Der Rückstand wird durch Säulenchromatographie
auf Silicagel (Elution mit einem Gradienten aus Hexan/Ethylacetat)
unter Bildung von 114 mg (40%) an 9-[(3-Methylphenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
als nicht ganz weißer Feststoff
gereinigt.
1H NMR (DMSO-d6) δ 10,5 (s,
1H), 8,8 (br s, 1H), 8,4 (br s, 1H), 7,8 (dd, 1H, J = 1 und 8 Hz),
7,4 (m, 2H), 7,3 (t, 1H, J = 8 Hz), 7,15-7,0 (m, 3H), 6,85 (d, 1H,
J = 8 Hz), 6,6 (d, 1H, J = 8 Hz), 5,95 (d, 1H, J = 8 Hz), 5,65 (s, 2H)
und 2,25 (s, 3H).
IR (KBr, cm–1)
3434, 3203, 1629, 1599, 1579, 1552, 1443, 1330, 1262, 1214 und 776.
MS
(ES) m/e 329, 331.
Elementaranalyse für C21H18N2O2:
Berechnet: C 76,36, H 5,45, N 8,48. Gefunden: C 77,56, H 5,67, N
8,26.
-
D. {9-[(3-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäuremethylester
-
Es
wird 40% methanolisches Triton B (0,45 ml, 0,99 mM) zu einer Lösung aus
9-[(3-Methylphenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
(100 mg, 0,30 mM) in 8 ml DMF bei Raumtemperatur gegeben. Nach 3 Minuten
wird Methylbromacetat (115 mg, 0,72 mM) zugegeben und das entstehende
Gemisch wird bei Raumtemperatur für 24 Stunden gerührt. Das
Gemisch wird mit Ethylacetat verdünnt, mit H2O
und gesättigter
Kochsalzlösung
gewaschen, über
Magnesiumsulfat getrocknet, filtriert und konzentriert. Der Rückstand
wird durch Säulenchromatographie
auf Silicagel (Elution mit Ethylacetat) unter Bildung von 80 mg
(66%) an {9-[(3-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäuremethylester
als weißer
Feststoff gereinigt.
1H NMR (DMSO-d6) δ 7,6
(d, 1H, J = 8 Hz), 7,55 (br s, 1H), 7,45-7,0 (m, 8H), 6,9 (d, 1H,
J = 8 Hz), 6,6 (d, 1H, J = 8 Hz), 5,65 (s, 2H), 4,9 (s, 2H), 3,75
(s, 3H) und 2,2 (s, 3H).
IR (KBr, cm–1)
3367, 3157, 1760, 1642, 1589, 1499, 1455, 1424, 1328, 1216, 1151,
1102, 772 und 714.
MS (FD) m/e 402.
Elementaranalyse für C24H22N2O4: Berechnet: C 71,64, H 5,47, N 6,96. Gefunden:
C 71,01, H 5,60, N 6,66.
-
E. {9-[(3-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäurenatriumsalz
-
Eine
Suspension aus {9-[(3-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäuremethylester
(15,8 mg, 0,039 mM) und 0,04 ml (0,04 mM) an 1 N NaOH in 5 ml Ethanol
wird für
24 Stunden bei 25°C gerührt. Der
entstehende weiße
Niederschlag wird durch Filtration gesammelt, mit einer kleinen
Menge an EtOH gewaschen und dann im Vakuum unter Bildung von 10
mg (62%) an {9-[(3-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäurenatriumsalz
als weißes
Pulver getrocknet.
1H NMR (DMSO-d6) δ 7,55
(d, 1H, J = 8 Hz), 7,5-7,0 (m, 9H), 6,85 (d, 1H, J = 8 Hz), 6,55
(d, 1H, J = 8 Hz), 5,6 (s, 2H), 4,35 (s, 2H) und 2,2 (s, 3H).
IR
(KBr, cm–1)
3390, 1656, 1613, 1595, 1573, 1498, 1455, 1408, 1325, 1332 und 719.
MS
(ES) m/e 387, 389.
Elementaranalyse für C23H19N2O4Na:
Berechnet: C 67,32, H 4,63, N 6,83. Gefunden: C 61,20, H 4,64, N
6, 06.
-
Beispiel 9
-
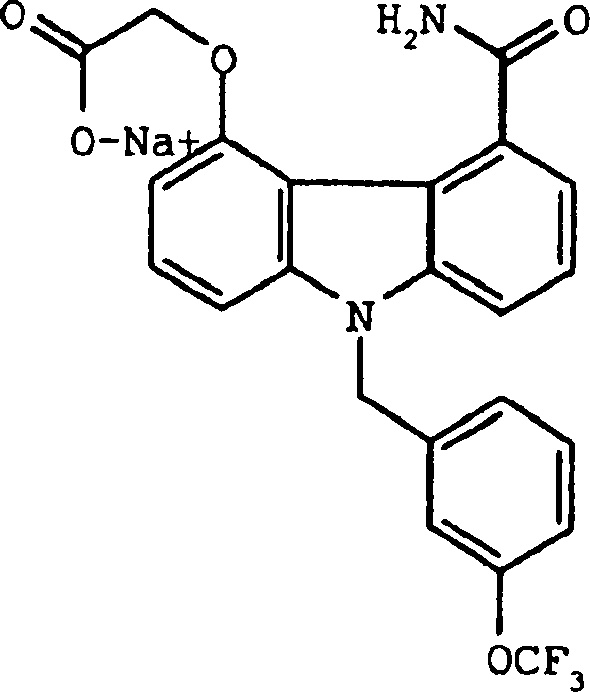
Herstellung von {9-[(3-Trifluormethoxyphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäurenatriumsalz
-
-
A. {9-[(3-Trifluormethoxyphenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
-
Eine
Suspension aus 5-Carbomethoxy-1,2-dihydro-9H-carbazol-4(3H)-on (935
mg, 3,85 mM), 3-Trifluormethoxybenzylbromid (1,0 g, 3,93 mM) und
Kaliumcarbonat (531 mg, 3,85 mM) in 20 ml DMF wird bei Raumtemperatur
für 17
Stunden gerührt.
Das Gemisch wird mit Ethylacetat verdünnt, mit H2O
und gesättigter
Kochsalzlösung
gewaschen, über
wasserfreiem Magnesiumsulfat getrocknet, filtriert und unter Bildung
von 1,6 g (100%) an 9-[(3-Trifluormethoxyphenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4-(3H)-on
als Schaum konzentriert.
1H NMR (DMSO-d6) δ 7,7
(dd, 1H, J = 1 und 8 Hz), 7,45 (t, 1H, J = 8 Hz), 7,3-7,1 (m, 4H),
7,05 (d, 1H, J = 8 Hz), 5,6 (s, 2H), 3,8 (s, 3H), 3,0 (m, 2H), 2,45
(m, 2H) und 2,1 (m, 2H).
IR (CNCl3,
cm–1)
1729, 1647, 1439, 1259, 1176 und 1116.
MS (ES) m/e 418.
Elementaranalyse
für C22H18NO4F3: Berechnet: C 63,31, H 4,32, N 3,36. Gefunden:
C 63,12, H 4,35, N 3,31.
-
B. 9-[(3-Trifluormethoxyphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol
-
Eine
Lösung
aus 9-[(3-Trifluormethoxyphenyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4-(3H)-on (0,75 g,
1,8 mM) und 2,3-Dichlor-5,6-dicyano-1,4-benzochinon (490 mg, 2,16
mM) in 70 ml Toluol wird am Rückfluss
für 6 Stunden
gerührt.
Das Gemisch wird direkt durch Säulenchromatographie
auf Silicagel (Elution mit Methylenchlorid) unter Bildung von 300
mg (40%) an 9-[(3-Trifluormethoxyphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol
als gelber Feststoff gereinigt.
1H
NMR (DMSO-d6) δ 10,25 (s, 1H), 7,7 (d, 1H,
J = 8 Hz), 7,5-7,0 (m, 8H), 6,6 (d, 1H, J = 8 Hz), 5,7 (s, 2H) und 3,85
(s, 3H).
IR (KBr, cm–1) 3200, 1673, 1441,
1268, 1217, 1173 und 753.
MS (ES) m/e 414, 416.
Elementaranalyse
für C22H16NO3F3: Berechnet: C 63,61, H 3,86, N 3,37. Gefunden:
C 63,40, H 3,99, N 3,43.
-
C. 9-[(3-Trifluormethoxyphenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
-
Eine
Lösung
aus 9-[(3-Trifluormethoxyphenyl)methyl]-4-hydroxy-5-carbomethoxycarbazol
(260 mg, 0,62 mM) in 10 ml THF und 30 ml konzentiertem wässrigem
Ammoniumhydroxid wird für
132 Stunden kräftig gerührt. Das
Gemisch wird mit Ethylacetat verdünnt und mit 5 N HCl auf pH
1 angesäuert.
Die wässrige
Phase wird dreimal mit Ethylacetat extrahiert. Die vereinigten organischen
Extrakte werden mit H2O und gesättigter Kochsalzlösung gewaschen, über Magnesiumsulfat
getrocknet, filtriert und konzentriert. Der Rückstand wird durch Säulenchromatographie
auf Silicagel (Elution mit einem Gradienten aus Hexan/Ethylacetat)
unter Bildung von 150 mg (60%) an 9-[(3-Trifluormethoxyphenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
als nicht ganz weißer
Feststoff gereinigt.
1H NMR (DMSO-d6) δ 10,5
(s, 1H), 8,8 (br s, 1H), 8,4 (br s, 1H), 7,85 (dd, 1H, J = 1 und
8 Hz), 7,5-7,15 (m, 5H), 7,1 (d, 1H, J = 8 Hz), 7,0 (d, 1H, J =
8 Hz), 6,6 (d, 1H, J = 8 Hz), 5,95 (d, 1H, J = 8 Hz) und 5,65 (s,
2H).
IR (KBr, cm–1) 3431, 3203, 1629,
1601, 1580, 1548, 1446, 1330, 1261, 1215 und 777.
MS (ES) m/e
399, 401.
Elementaranalyse für C21H15N2O2F3: Berechnet: C 63,00, H 3,75, N 7,0. Gefunden:
C 63,15, H 4,07, N 6,84.
-
D. {9-[(3-Trifluormethoxyphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäuremethylester
-
Es
wird 40% methanolisches Triton B (0,15 ml, 0,34 mM) zu einer Lösung aus
9-[(3-Trifluormethoxyphenyl)methyl]-4-hydroxy-5-carbamoylcarbazol
(115 mg, 0,28 mM) in 8 ml DMF bei Raumtemperatur gegeben. Nach 3
Minuten wird Methylbromacetat (65 mg, 0,41 mM) zugegeben und das
entstehende Gemisch wird bei Raumtemperatur für 23 Stunden gerührt. Das
Gemisch wird mit Ethylacetat verdünnt, mit H2O
und gesättigter
Kochsalzlösung
gewaschen, über
Magnesiumsulfat getrocknet, filtriert und konzentriert. Der Rückstand wird
durch Säulenchromatographie
auf Silicagel (Elution mit Ethylacetat) unter Bildung von 112 mg
(83%) an {9-[(3-Trifluormethoxyphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäuremethylester
als weißer
Feststoff gereinigt.
1H NMR (DMSO-d6) δ 7,6
(d, 1H, J = 8 Hz), 7,55 (br s, 1H), 7,5-7,0 (m, 9H), 6,6 (d, 1H,
J = 8 Hz), 5,7 (s, 2H), 4,9 (s, 2H) und 3,75 (s, 3H).
IR (KBr,
cm–1)
3488, 3141, 1763, 1674, 1501, 1444, 1269, 1215, 1178, 1102, 772
und 714.
MS (FD) m/e 472.
Elementaranalyse für C24H19N2O5F3: Berechnet: C
61,02, H 4,03, N 5,93. Gefunden: C 61,05, H 4,17, N 5,81.
-
E. {9-[(3-Trifluormethoxyphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäurenatriumsalz
-
Eine
Suspension aus {9-[(3-Trifluormethoxyphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäuremethylester
(22,4 mg, 0,047 mM) und 0,065 ml (0,065 mM) an 1 N NaOH in 5 ml
Ethanol wird für
24 Stunden bei 25°C
gerührt.
Das Lösemittel
wird im Vakuum entfernt und der Rückstand wird in EtOH suspendiert.
Der entstehende weiße
Niederschlag wird durch Filtration gesammelt, mit einer kleinen
Menge an EtOH gewaschen und dann im Vakuum unter Bildung von 9 mg
(41%) an {9-[(3-Trifluormethoxyphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäurenatriumsalz
als weißes
Pulver getrocknet.
MS (ES) m/e 457, 459.
-
Beispiel 10
-
Herstellung von [9-Benzyl-5-carbamoyl-1-methylcarbazol-4-yl]oxyessigsäure
-
A. Herstellung von 5-Carbamoyl-4-methoxy-1-methylcarbazol
-
Eine
Lösung
aus 0,805 g an 9-Benzyl-5-methoxy-8-methyl-1,2,3,4-tetrahydrocarbazol-4-carboxamid in
24 ml Carbitol wird mit 1,1 g an 5% Palladium auf Kohle behandelt
und am Rückfluss
für 6 Stunden
mit Luftzufuhr erhitzt. Nach dem Abkühlen wird die Lösung durch
ein Kissen aus Celite filtriert und das Kissen wird mit Ethylacetat
gewaschen. Die Filtrate werden mit Ether verdünnt und viermal mit Wasser
gewaschen und über Magnesiumsulfat
getrocknet und konzentriert. Der Rückstand wird auf Silicagel
mittels Methanol/0–4%
in Dichlormethan unter Bildung von 0,166 g (28%) an debenzyliertem
Carbazol chromatographiert.
ESIMS m/e 255 (M++1),
253 (M+-1)
NMR (300 MHz, CDCl3): δ 8,13
(br, 1H), 7,51 (d, J = 8,1, 1H), 7,40 (t, J = 7,6, 1H), 7,32 (d,
J = 7,2, 1H), 7,18 (d, J = 7,8, 1H), 6,60 (d, J = 8,0, 1H), 5,68
(br, 2H), 3,99 (s, 3H), 2,50 (s, 3H).
-
B. Herstellung von 9-Benzyl-5-carbamoyl-4-methoxy-1-methylcarbazol
-
Eine
Lösung
aus 0,148 g an 5-Carbamoyl-4-methoxy-1-methylcarbazol in 1,1 ml
Dimethylformamid wird zu 0,026 g Natriumhydrid (60% in Mineralöl) in 0,4
ml Dimethylformamid gegeben und für 60 Minuten bei Raumtemperatur
gerührt.
Benzylbromid (0,076 ml) wird dann zugegeben und die Reaktion wird über Nacht
gerührt.
Das Reaktionsgemisch wird in 20 ml gesättigte Ammoniumchloridlösung gegossen
und dann zweimal mit Ethylacetat extrahiert. Die Extrakte werden
mit Wasser und dann mit Kochsalzlösung gewaschen, über Magnesiumsulfat
getrocknet und konzentriert. Der Rückstand wird mit Hexan gespült und in
Dichlormethan gelöst, filtriert
und unter Bildung von 0,21 g der Untertitelverbindung konzentriert.
FDMS
m/e 344 (M+).
Elementaranalyse: Berechnet:
C 76,72, H 5,85, N 8,13. Gefunden: C 75,20, H 6,19, N 7,54.
-
C. Herstellung von [9-Benzyl-5-carbamoyl-1-methylcarbazol-4-yl]oxyessigsäuremethylester
-
Eine
Lösung
aus 0,23 g an 9-Benzyl-5-carbamoyl-4-methoxy-1-methylcarbazol in
4 ml Dimethylformamid wird zu einer 1 ml Lösung aus Natriumethanthiolat
(hergestellt aus 0,116 g Natriumhydrid 60% Dispersion und 0,22 ml
Ethanthiol unter Stickstoff) gegeben und bei 110°C für 15 Stunden erhitzt. Das Reaktionsgemisch wird
gekühlt,
in 20 ml an 1 N HCl gegossen und zweimal mit Ethylacetat extrahiert.
Die Extrakte werden zweimal mit Wasser und dann mit Kochsalzlösung gewaschen, über Magnesiumsulfat
getrocknet und konzentriert. Der Rückstand wird auf Silicagel
unter Verwendung von Methanol/0–1%
in Dichlormethan unter Bildung von 0,146 g (66%) des demethylierten
Zwischenprodukts gegeben. Eine Lösung
aus 0,146 g dieses Zwischenprodukts in 1,5 ml Dimethylfomamid wird
zu 0,021 g Natriumhydrid (60% in Mineralöl) in 0,5 ml Dimethylformamid gegeben.
Nach dem Rühren
für 10
Minuten bei Raumtemperatur werden 0,054 ml Methylbromacetat zugegeben.
Nach dem Rühren
für 5 Stunden
bei Raumtemperatur wird das Reaktionsgemisch in Wasser gegossen und
zweimal mit Ethylacetat extrahiert. Die Extrakte werden mit Wasser
und dann mit Kochsalzlösung
gewaschen, über
Magnesiumsulfat getrocknet und konzentriert. Der Rückstand
wird auf Silicagel unter Verwendung von Methanol/0–2% in Dichlormethan
unter Bildung von 0,10 g (56%) der Untertitelverbindung chromatographiert.
Smp.
228–230°C.
ESIMS
m/e 403 (M++1).
Elementaranalyse: Berechnet:
C 71,63, H 5,51, N 6,96. Gefunden: C 71,34, H 5,60, N 6,70.
-
D. Herstellung von [9-Benzyl-5-carbamoyl-1-methylcarbazol-4-yl]oxyessigsäure
-
Eine
Aufschlämmung
aus 32 mg (0,0795 mmol) an [9-Benzyl-5-carbamoyl-1-methylcarbazol-4-yl]oxyessigsäuremethylester
in 1 ml Tetrahydrofuran und 3,5 ml Methanol wird mit 0,3 ml wässriger
2 N Natriumhydroxidlösung
behandelt und über
Nacht bei Raumtemperatur gerührt.
Das Lösemittel
wird verdampft und der Rückstand
wird zwischen 1:1 Ethylacetat/Tetrahydrofuran und 0,2 N HCl Lösung aufgeteilt.
Nach einer weiteren Extraktion mit 1:1 Ethylacetat/Tetrahydrofuran
werden die Extrakte mit Kochsalzlösung gewaschen, über Magnesiumsulfat
getrocknet und unter Bildung der Titelverbindung (27 mg) konzentriert.
Smp.
253–254°C.
ESIMS
m/e 389 (M++1), 387 (M+-1).
NMR
(300 MHz, d6-DMSO): δ 12,83 (br, 1H), 7,75 (br, 1H),
7,53 (d, J = 8,2, 1H), 7,41-7,34 (m, 2H), 7,28-7,17 (m, 3H), 7,07 (m, 2H), 6,90 (d,
J = 7,2, 2H), 6,49 (d, J = 8,1, 1H), 5,89 (s, 2H), 4,79 (s, 2H),
2,52 (s, 3H).
-
Beispiel 11
-
Herstellung von [9-Benzyl-4-carbamoyl-8-fluor-1,2,3,4-tetrahydrocarbazol-5-yl]oxyessigsäure
-
A. Herstellung von (2-Chlor-4-fluorphenyl)-ethylcarbonat
-
Eine
Lösung
aus 19,16 g an 2-Chlor-4-fluorphenol in 65,4 ml an 2 N wässriger
Natriumhydroxidlösung wird
in einem Eisbad gekühlt
und tropfenweise mit 16,3 ml Ethylchlorformiat behandelt. Nach dem
Rühren
bei Raumtemperatur über
Nacht wird das Zweiphasenreaktionsgemisch mit 100 ml Wasser verdünnt und
mit 300 ml eines 1:1 Pentan/Ethergemisches extrahiert. Der Extrakt
wird dreimal mit 0,02 N Natriumhydroxidlösung, Wasser und dann Kochsalzlösung gewaschen.
Nach dem Trocknen und Eindampfung werden 27,63 g (97%) der Untertitelverbindung
erhalten.
NMR (300 MHz, CDCl3): δ 7,23-7,18
(m, 2H), 7,00 (dt, J = 8,4, 2,7, 1H), 4,35 (q, J = 7,1, 2H), 1,40
(t, J = 7,1, 3H).
-
B. Herstellung von (2-Chlor-4-fluor-5-nitrophenyl)-ethylcarbonat
-
Eine
Lösung
aus 27,63 g an (2-Chlor-4-fluorphenyl)-ethylcarbonat in 60 ml Dichlormethan
wird in einem Eisbad gekühlt
und tropfenweise mit 31,86 g eines 1:2 Gemisches aus rauchender
Salpetersäure
(90%) und konzentrierter Schwefelsäure behandelt. Die Reaktion
wird für
2 Stunden bei Raumtemperatur gerührt
und dann mit Eis gekühlt
und mit weiteren 4,5 g desselben Nitrierungsgemisches behandelt.
Die Reaktion wird über Nacht
bei Raumtemperatur gerührt,
in 200 ml Eis und Wasser gegossen und zweimal mit Dichlormethan
extrahiert. Die Extrakte werden mit Wasser und dann mit Kochsalzlösung gewaschen, über Magnesiumsulfat
getrocknet und unter Bildung von 33,01 g (99%) der Untertitelverbindung
konzentriert.
Smp. 50–51°C.
Elementaranalyse:
Berechnet: C 41,01, H 2,68, N 5,31, Cl 13,45. Gefunden: C 41,03,
H 2,59, N 5,38, Cl 13,71.
-
C. Herstellung von 2-Chlor-4-fuor-5-nitroanisol
-
Eine
Lösung
aus 15,0 g an (2-Chlor-4-fluor-5-nitrophenyl)-ethylcarbonat in 100
ml Dimethylformamid wird mit 18,6 g Cäsiumcarbonat, 7,1 ml Iodmethan
und 7 ml Methanol behandelt und über
Nacht bei Raumtemperatur gerührt.
Das Reaktionsgemisch wird in Wasser gegossen und zweimal mit Ether
extrahiert. Die Extrakte werden zweimal mit Wasser und dann mit
Kochsalzlösung
gewaschen, über
Magnesiumsulfat getrocknet und unter Bildung von 11,4 g der Untertitelverbindung
konzentriert.
Smp. 69–70°C. Bsp. 57,
C.
Elementaranalyse: Berechnet: C 40,90, H 2,45, N 6,81, Cl
17,25. Gefunden: C 41,20, H 2,48, N 6,70, Cl 17,44.
-
D. Herstellung von 2-Fluor-5-methoxyanilin
-
Eine
Lösung
aus 5,63 g an 2-Chlor-4-fluor-5-nitroanisol in 90 ml Ethanol und
5 ml Triethylamin wird bei Raumtemperatur unter 60 psi mit 1,0 g
an 5% Palladium auf Kohle für
vier Stunden hydriert. Der Katalysator wird abfiltriert und das
Lösemittel
wird verdampft. Der Rückstand
wird in Chloroform aufgeschlämmt
und durch ein Kissen aus Silicagel filtriert und dann eingedampft.
Dieser Rückstand
wird auf Silicagel mittels eines Hexan/Chloroformgemisches unter
Bildung von 2,77 g (72%) der Untertitelverbindung chromatographiert.
Smp.
253–254°C.
NMR
(300 MHz, CDCl3): δ 6,88 (dd, J = 10,6, 8,9, 1H),
6,32 (dd, J = 7,4, 3,0, 1H), 6,20 (dt, J = 8,9, 3,2, 1H), 3,73 (s,
3H), 3,72 (br, 2H).
-
E. Herstellung von N-Benzyl-2-Fuor-5-methoxyanilin
-
Dieses
Verfahren wird gemäß dem von
Tietze und Grote, Chem. Ber. 126 (12), 2733 (1993) ausgeführt. Eine
Lösung
aus 2,73 g an 2-Fluor-5-methoxyanilin und 2,67 g Benzaldehyd in
48 ml Methanol wird mit 3,43 g Zinkchlorid behandelt und dann in
einem Eisbad gekühlt.
Natriumcyanoborhydrid (1,58 g) wird in kleinen Portionen bei Raumtemperatur über 30 Minuten
zugegeben und die Reaktion wird für fünf Stunden bei Raumtemperatur
gerührt.
Nach der Verdampfung des Lösemittels
wird der Rückstand
in 40 ml an 1 N Natriumhydroxidlösung
aufgeschlämmt
und dann zweimal mit Ether extrahiert. Die Extrakte werden mit Wasser
und dann mit Kochsalzlösung
gewaschen, über
Magnesiumsulfat getrocknet und konzentriert. Der Rückstand
wird aus Hexan unter Bildung von 2,61 g kristallisiert und die Mutterlaugen
werden auf Silicagel unter Verwendung von 20:1 Hexan/Ether unter
Bildung von weiteren 1,4 g der Untertitelverbindung (90%) chromatographiert.
Smp.
56–58°C.
Elementaranalyse:
Berechnet: C 72,71, H 6,10; N 6,06. Gefunden: C 72,51, H 6,06, N
5,99.
-
F. Herstellung von Ethyl-9-benzyl-5-methoxy-8-Fluor-1,2,3,4-tetrahydrocarbazol-4-carboxylat
-
Eine
Lösung
aus 0,62 g an N-Benzyl-2-fluor-5-methoxyanilin in 20 ml trockenem
Tetrahydrofuran wird in einem Eisbad gekühlt und mit 11,3 ml an 0,5
M Kaliumbis(trimethylsilyl)amid in Toluol behandelt. Nach dem Rühren für 30 Minuten
werden 0,74 g an 2-Carboethoxy-6-bromcyclohexanon (Sheehan und Mumaw,
JACS, 72, 2127 (1950)) in 4 ml Tetrahydrofuran zugegeben und die
Reaktion kann sich langsam über
2 Stunden auf Raumtemperatur erwärmen.
Die Reaktion wird mit gesättigter
Ammoniumchloridlösung
gestoppt und zweimal mit Ether extrahiert. Die Extrakte werden mit
Wasser und dann mit Kochsalzlösung
gewaschen, über
Magnesiumsulfat getrocknet und konzentriert. Dieser Rückstand
wird auf Silicagel mittels eines Hexan/Ethergemisches unter Bildung
von 0,796 g (74%) an N-alkylierten Zwischenproduktdiastereomeren
chromatographiert. Dieses Gemisch wird in 20 ml Benzol mit 0,99
g Zinkchlorid über
Nacht am Rückfluss
erhitzt. Das Lösemittel wird
verdampft und der Rückstand
wird zwischen 25 ml an 1 N HCl und 25 ml Ethylacetat aufgeteilt
und dann erneut mit Ethylacetat extrahiert. Die organischen Phasen
werden mit Wasser und dann Kochsalzlösung gewaschen, über Magnesiumsulfat
getrocknet und unter Bildung von 0,734 g (96%) der Untertitelverbindung
konzentriert.
ESIMS m/e 382 (M++1).
Elementaranalyse:
Berechnet: C 72,42, H 6,34, N 3,67. Gefunden: C 72,20, H 6,26, N
3,70.
-
G. Herstellung von 9-Benzyl-5-methoxy-8-fluor-1,2,3,4-tetrahydrocarbazol-4-carboxamid
-
Ethyl-9-benzyl-5-methoxy-8-fluor-1,2,3,4-tetrahydrocarbazol-4-carboxylat
(0,722 g) wird ähnlich
wie in Beispiel 49, Teil C, beschrieben, behandelt und auf Silicagel
mittels 1% Methanol in Dichlormethan unter Bildung von 0,482 g (72%)
der Untertitelverbindung chromatographiert.
ESIMS m/e 353 (M++1).
Elementaranalyse: Berechnet: C
71,57, H 6,01, N 7,95. Gefunden: C 71,42, H 5,83, N 7,75.
-
H. Herstellung von [9-Benzyl-4-carbamoyl-8-fluor-1,2,3,4-tetrahydrocarbazol-5-yl]oxyessigsäuremethylester
-
9-Benzyl-5-methoxy-8-fluor-1,2,3,4-tetrahydrocarbazol-4-carboxamid
(0,170 g) wird ähnlich
wie in Beispiel 49, Teil D, beschrieben, umgewandelt und auf Silicagel
mittels Methanol/0–1%
in Dichlormethan unter Bildung von 85 mg (50%) der Untertitelverbindung
chromatographiert.
Smp. 183–185°C.
Elementaranalyse: Berechnet:
C 67,31, H 5,65, N 6,82. Gefunden: C 67,58, H 5,48, N 6,95.
-
1. Herstellung von [9-Benzyl-4-carbamoyl-8-fluor-1,2,3,4-tetrahydrocarbazol-5-yl]oxyessigsäure
-
9-Benzyl-4-carbamoyl-8-Fluor-1,2,3,4-tetrahydrocarbazol-5-yl]oxyessigsäuremethylester
(71 mg) wird ähnlich
wie in Beispiel 50, Teil D, beschrieben, unter Bildung von 65 mg
der Titelverbindung hydrolysiert.
ESIMS m/e 397 (M++1),
395 (M+-1).
NMR (300 MHz, d6-DMSO): δ 13,03
(br, 1H), 7,31-7,19 (m, 3H), 6,97 (d, J = 7,4, 2H), 6,95 (br, 1H),
6,70 (d, J = 3,8, 1H), 6,67 (dd, J = 12,4, 3,9, 1H), 6,28 (dd, J
= 8,5, 2,6, 1H), 5,39 (ABq, 2H), 4,64 (s, 2H), 3,92 (br, 1H), 2,71
(m, 1H), 2,44 (m, 1H), 2,02 (m, 2H), 1,76 (m, 2H).
-
Beispiel 12
-
Herstellung von [9-Benzyl-5-carbamoyl-1-chlorcarbazol-4-yl]oxyessigsäure
-
A. Herstellung von 9-Benzyl-5-carbamoyl-4-methoxy-1-chlorcarbazol
-
Eine
Lösung
von 1,0 g an 9-Benzyl-5-methoxy-8-methyl-1,2,3,4-tetrahydrocarbazol-4-carboxamid wird ähnlich zu
Beispiel 51 Teil A oxidiert und auf Silicagel mittels Dichlormethan/0–1% Methanol
unter Bildung von 0,66 g (67%) der Untertitelverbindung chromatographiert.
FDMS m/e 364 (M+).
Elementaranalyse
Berechnet:
C 69,14, H 4,70, N 7,68, Cl 9,72
Gefunden: C 69,40, H 4,64,
N 7,49, Cl 9,98
-
B. Herstellung von 5-Carbamoyl-4-hydroxy-1-chlorcarbazol
-
Eine
Lösung
aus 0,66 g an 9-Benzyl-5-carbamoyl-4-methoxy-1-chlorcarbazol in
40 ml Dichlormethan wird in einem Eisbad gekühlt und tropfenweise mit 14
ml an 1,0 M Bortribromidlösung
in Dichlormethan behandelt. Die Reaktion kann sich langsam über 2 Stunden
auf Raumtemperatur erwärmen
und wird dann durch Gießen
auf Eis und der anschließenden
Zugabe von 50 ml an 1 N HCl gestoppt. Das Gemisch wird mit Dichlormethan
(3 × 200
ml) extrahiert und die Extrakte werden mit Kochsalzlösung gewaschen,
mit Magnesiumsufat getrocknet und konzentriert. Die wässrigen
Phasen zeigen einen Niederschlag und werden dann zweimal mit Ethylacetat
extrahiert, mit Kochsalzlösung
gewaschen, mit Magnesiumsulfat getrocknet und unter Bildung von 0,287
g der Untertitelverbindung konzentriert. Der erste Rückstand
wird auf Silicagel mittel 0,5% Methanol in Dichlormethan unter Bildung
von weiteren 93 mg der Untertitelverbindung chromatographiert. (Gesamtausbeute
80%).
ESIMS m/e 259 (M+-1),
NMR
(300 MHz, d6-DMSO): δ 11,79 (s, 1H), 10,76 (s, 1H),
8,87 (br s, 1H), 8,41 (br s, 1H), 7,77 (t, J = 4,6, 1H), 7,48 (d,
J = 4,2, 2H), 7,34 (d, J = 8,5, 1H), 6,54 (d, J = 8,5, 1H).
-
C. Herstellung von [5-Carbamoyl-1-chlorcarbazol-4-yl]oxyessigsäuremethylester
-
Eine
Lösung
aus 0,28 g an 5-Carbamoyl-4-hydroxy-1-chlorcarbazol in 6 ml Tetrahydrofuran
wird zu 0,043 g an Natriumhydrid (60% in Mineralöl) in 1 ml Tetrahydrofuran
gegeben und für
60 Minuten bei Raumtemperatur gerührt. Methylbromacetat (0,11
ml) wird dann zugegeben und die Reaktion wird über Nacht gerührt. Das
Reaktionsgemisch wird in 20 ml gesättigte Ammoniumchloridlösung gegossen
und dann zweimal mit Ethylacetat extrahiert. Die Extrakte werden
mit Wasser und dann mit Kochsalzlösung gewaschen, über Magnesiumsulfat
getrocknet und konzentriert. Der Rückstand wird dann auf Silicagel
chromatographiert, wobei mit Chloroform und dann mit 2:1 Chloroform/Ethylacetat
unter Bildung von 0,16 g (45%) der Untertitelverbindung eluiert
wird.
ESIMS m/e 333 (M++1), 335 (M++3), 331 (M+-1).
NMR
(300 MHz, d6-DMSO): δ 11,73 (s, 1H), 7,56 (d, J =
8,1, 1H), 7,50 (br s, 1H), 7,43-7,35 (m, 2H), 7,18 (br s, 1H), 7,06
(d, J = 7,8, 1H), 6,56 (d, J = 8,6, 1H), 4,90 (s, 2H), 3,70 (s,
3H).
-
D. Herstellung von [9-Benzyl-5-carbamoyl-1-chlorcarbazol-4-yl]oxyessigsäuremethylester
-
Eine
Lösung
aus 78 mg an [5-Carbamoyl-1-chlorcarbazol-4-yl]oxyessigsäuremethylester
in 0,8 ml trockenem Dimethylformamid wird dann zu 10 mg Natriumhydrid
(60% in Mineralöl)
in 0,2 ml Dimethylformamid gegeben und für 15 Minuten gerührt. Benzylbromid
(0,031 ml) wird dann zugegeben und die Reaktion wird über Nacht
gerührt.
Das Reaktionsgemisch wird in Wasser gegossen und mit 1 ml an 1 N
HCl Lösung
angesäuert
und zweimal mit Ethylacetat extrahiert. Die Extrakte werden mit
Wasser (3×)
und dann mit Kochsalzlösung
gewaschen, über
Magnesiumsulfat getrocknet und konzentriert. Der Rückstand
wird auf Silicagel unter Bildung von 40 mg der Untertitelverbindung
chromatographiert, wobei mit Methanol/0–2% in Dichlormethan eluiert
wird.
ESIMS m/e 423 (M++1), 425 (M++3).
NMR (300 MHz, CDCl3): δ 7,43-7,22
(m, 7H), 7,06 (d, J = 7,3, 2H), 6,51 (d, J = 8,6, 1H), 6,05 (s,
2H), 5,80 (br, 2H), 4,88 (s, 2H), 3,83 (s, 3H).
-
E. Herstellung von [9-Benzyl-5-carbamoyl-1-chlorcarbazol-4-yl]oxyessigsäure
-
[9-Benzyl-5-carbamoyl-1-chlorcarbazol-4-yl]oxyessigsäuremethylester
(15 mg) wird ähnlich
zur Beschreibung von Beispiel 50, Teil D unter Bildung von 14 mg
der Titelverbindung hydrolysiert.
Smp. 240–242°C.
ESIMS m/e 409 (M++1), 411 (M++3),
407 (M+-1).
NMR (300 MHz, d6-DMSO): δ 12,94
(br, 1H), 7,70 (br, 1H), 7,61 (d, J = 8,3, 1H), 7,43 (t, J = 7,8,
1H), 7,36 (m, 2H), 7,28-7,19 (m, 3H), 7,13 (d, J = 7,2, 1H), 6,99
(d, J = 7,4, 2H), 6,63 (d, J = 8,6, 1H), 6,08 (s, 2H), 4,83 (s, 2H).
-
Beispiel 13
-
Herstellung von [9-(Cyclohexyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäure
-
-
A. 9-[(Cyclohexyl)methyi]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
-
Eine
0°C Suspension
aus 5-Carbomethoxy-1,2-dihydro-9H-carbazol-4(3H)-on (1,0 g, 4,11
mmol) einer katalytischen Menge NaI (etwa 10 mg) und K2CO3 (1,1 g, 8,2 mmol) in 10 ml DMF wird mit
Cyclohexylmethylbromid (0,631 ml, 4,52 mmol) behandelt. Nach dem
Rühren über Nacht
bei Umgebungstemperatur werden zusätzliche 0,63 ml Cyclohexylmethylbromid
zugegeben und das entstehende Gemisch wird bei 60°C für 3 Stunden
erhitzt. Das Gemisch wird in H2O (30 ml)
gegossen und mit EtOAc (2 × 25
ml) extrahiert. Die vereinigten organischen Phasen werden mit H2O (4 × 50
ml) gewaschen, über
wasser freiem Na2SO4 getrocknet,
filtriert und im Vakuum konzentriert. Der Rückstand wird durch Radialchromatographie
auf Silicagel (Elution mit einem Gradienten aus 20% bis 40% EtOAc/Hexan)
unter Bildung von 1,36 g (4,01 mmol, 97%) an 9-[(Cyclohexyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
als weißer
Schaum gereinigt.
IR (CHCl3, cm–1)
3011, 2932, 2857, 1725, 1649, 1469, 1446, 1288 und 1120.
MS
(ES) m/e 340 (M+1), 453 (M+AcO–).
FAB HRMS m/e,
Berechnet für
C21H26NO3: 340,1913. Gefunden: 340,1916 (M+1).
Elementaranalyse
für C21H25NO3:
Berechnet: C 74,31, H 7,42, N 4,13. Gefunden: C 72,65, H 7,39, N
4,70.
-
B. 9-[(Cyclohexyl)methyl]-4-hydroxy-5-carbomethoxycarbazol
-
Eine
Lösung
aus 9-[(Cyclohexyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
(1,16 g, 3,42 mmol) und 2,3-Dichlor-5,6-dicyano-1,4-benzochinon
(853 mg, 3,76 mmol) in 20 ml Toluol wird für 3 Stunden auf 80°C erhitzt.
Das Gemisch wird direkt durch Säulenchromatographie
auf Silicagel (Elution mit CH2Cl2) unter Bildung von 259 mg (0,768 mmol,
22%) an 9-[(Cyclohexyl)methyl]-4-hydroxy-5-carbomethoxycarbazol als gelbes Öl gereinigt,
das sich langsam verfestigt.
MS (ES) m/e 338 (M+1), 336 (M-1).
Elementaranalyse
für C21H23NO3:
Berechnet: C 74,75, H 6,87, N 4,15. Gefunden: C 74,95, H 6,99, N
4,42.
-
C. 9-[(Cyclohexyl)methyl]-4-hydroxy-5-carbamoylcarbazol
-
Eine
Lösung
aus 9-[(Cyclohexyl)methyl]-4-hydroxy-5-carbomethoxycarbazol (205
mg, 0,608 mmol) in 5 ml THF und 20 ml konzentriertem, wässrigem
Ammoniumhydroxid wird mit einem Strom aus NH3 Gas
behandelt, um die Sättigung
sicherzustellen. Das Reaktionsgefäß wird verschlossen und das
Gemisch wird bei 35°C
unter Rühren
erhitzt, bis die TLC den vollständigen
Verbrauch des Ausgangsmaterials anzeigt (20 Stunden). Das THF wird
verdampft und die wässrige
Phase wird filtriert. Der grüne
Feststoffniederschlag wird in THF gelöst und durch Radialchromatographie
auf Silicagel (Elution mit CH2Cl2) gereinigt. Der entstehende Schaum wird
mit Ether unter Bildung von 138 mg (70%) der Titelverbindung als
nicht ganz weißer
Feststoff behandelt.
IR (KBr, cm–1)
3418, 3200, 3131, 1629, 1600, 1443, 1261, 778.
FAB HRMS m/e
Berechnet für
C20H23N2O2: 323,1760. Gefunden: 323,1760. (M+1)
-
D. [9-(Cyclohexyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäuremethylester
-
Ein
Gemisch aus 9-[(Cyclohexyl)methyl]-4-hydroxy-5-carbamoylcarbazol
(60 mg, 0,186 mmol) und Cs2CO3 (150
mg, 0,460 mmol) in 2 ml DMF wird mit Methylbromacetat (0,023 ml,
0,242 mmol) behandelt. Die Reaktion wird für 2 Stunden bei Umgebungstemperatur
gerührt
und dann wird sie mit EtOAc und H2O (jeweils 10
ml) verdünnt.
Die wässrige
Phase wird mit festem NaCl gesättigt
und mit EtOAc (2 × 10
ml) extrahiert. Die vereinigten organischen Phasen werden mit H2O (2 × 25
ml) gewaschen, über
wasserfreiem Na2SO4 getrocknet,
filtriert und im Vakuum konzentriert. Eine Reinigung des rohen Rückstands
durch Blitzchromatographie auf Silicagel (Elution mit einem Gradienten
aus 0% bis 90% EtOAc/He xan), gefolgt von einer Behandlung mit Et2O/EtOAc ergibt 45 mg (0,114 mmol, 61%) der
Titelverbindung als nicht ganz weißen Feststoff.
MS (ES)
m/e 395 (M+1), 378 (M+H-NH3), 453 (M+AcO–).
Elementaranalyse
für C23H26N2O4 × 0,3H2O: Berechnet: C 69,08, H 6,71, N 7,01. Gefunden:
C 69,13, H 6,71, N 7,09.
-
E. 9-[(Cyclohexyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäure
-
Eine
Aufschlämmung
aus [9-[(Cyclohexyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäuremethylester
(20 mg, 0,051 mmol) in 0,3 ml THF und 0,1 ml McOH wird mit 0,1 ml
an 1 N wässrigem
LiOH (0,1 mmol) behandelt und das Gemisch wird bei Raumtemperatur
für 2 Stunden
gerührt.
Die Reaktion wird mit 0,2 N HCl angesäuert und die organischen Anteile
werden im Vakuum entfernt. Der weiße Niederschlag wird aus der wässrigen
Phase abfiltriert und unter Bildung von 16 mg (0,042 mmol, 83%)
der Titelsäure
als weißes
Pulver mit Et2O gewaschen.
MS (ES)
m/e 381 (M+1), 364 (M+H-NH3), 379 (M-1).
Elementaranalysen
für C22H24N2O4: Berechnet: C 69,46, H 6,36, N 7,36. Gefunden:
C 69,34, H 6,35, N 7,29.
-
Beispiel 14
-
Herstellung von [9-[(Cyclopentyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäure
-
-
A. 9-[(Cyclopentyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
-
Eine
Suspension aus 5-Carbomethoxy-1,2-dihydro-9N-carbazol-4(3H)-on (820
mg, 3,37 mmol), einer katalytischen Menge NaI (etwa 10 mg) und K2CO3 (930 mg, 6,74
mmol) in 6 ml DMF wird mit Cyclopentylmethylchlorid (JOC, 1964,
29, 421–423,
400 mg, 3,37 mmol) behandelt. Nach dem Rühren über Nacht bei Umgebungstemperatur
werden zusätzliche
800 mg Cyclopentylmethylchlorid und 1 g NaI zugegeben und das entstehende
Gemisch wird bei 80°C über Nacht
erhitzt. Es werden zusätzliche
800 mg an Cyclopentylmethylchlorid und 2,2 g Cs2CO3 zugegeben und das Reaktionsgemisch wird
für 24
Stunden auf 80°C
erhitzt. Es werden zusätzliche
1,6 g Cyclopentylmethylchlorid zugegeben und das Reaktionsgemisch
wird bei 80°C
für 3 Tage
erhitzt. Das Gemisch wird in H2O (30 ml)
gegossen und mit EtOAc (3 × 10
ml) extrahiert. Die vereinigten organischen Phasen werden über wasserfreiem
Na2SO4 getrocknet,
filtriert und im Vakuum konzentriert. Der Rückstand wird durch Radialchromatographie
auf Silicagel (Elution mit einem Gradienten aus 10% bis 40% EtOAc/Hexan)
unter Bildung von 775 mg (2,38 mmol, 71%) an 9-[(Cyclopentyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
als brauner Schaum gereinigt.
MS (ES) m/e 326 (M+1), 384 (M+AcO–).
Elementaranalyse
für C20H23NO3:
Berechnet: C 73,82, H 7,12, N 4,30. Gefunden: C 74,12, H 7,21, N
4,45.
-
B. 9-[(Cyclopentyl)methyl]-4-hydroxy-5-carbomethoxycarbazol
-
Eine
Lösung
aus 9-[(Cyclopentyl)methyl]-5-carbomethoxy-1,2-dihydrocarbazol-4(3H)-on
(730 mg, 2,24 mmol) und 2,3-Dichlor-5,6-dicyano-1,4-benzochinon
(560 mg, 2,47 mmol) in 20 ml Toluol wird für 3 Stunden auf 80°C erhitzt.
Das Gemisch wird direkt durch Säulenchromatographie
auf Silicagel (Elution mit CH2Cl2) unter Bildung von 140 mg (0,433 mmol,
19%) an 9-[(Cyclopentyl)methyl]-4-hydroxy-5-carbomethoxycarbazol als gelbes Öl gereinigt,
das sich langsam verfestigt.
MS (ES) m/e 324 (M+1), 322 (M-1).
Elementaranalyse
für C20H21NO3 × 0,3H2O: Berechnet: C 73,06, H 6,62, N 4,26. Gefunden:
C 73,19, H 6,44, N 4,40.
-
C. 9-[(Cyclopentyl)methyl]-4-hydroxy-5-carbamoylcarbazol
-
Eine
Lösung
aus 9-[(Cyclopentyl)methyl]-4-hydroxy-5-carbomethoxycarbazol (110
mg, 0,34 mmol) in 3 ml THF und 20 ml konzentriertem, wässrigem
Ammoniumhydroxid wird mit einem Strom aus NH3 Gas
behandelt, um die Sättigung
sicherzustellen. Das Reaktionsgefäß wird verschlossen und das
Gemisch wird bei 35°C
unter Rühren
erhitzt, bis die TLC den vollständigen
Verbrauch des Ausgangsmaterials anzeigt (20 Stunden). Das THF wird
verdampft und die wässrige
Phase wird filtriert. Der entstehende Feststoff wird mit Ether unter
Bildung von 50 mg (0,162 mmol, 48%) der Titelverbindung als grünlich-weißer Feststoff
behandelt.
IR (KBr, cm–1) 3416, 3199, 3126,
1630, 1599, 1442, 1262, 778.
FAB HRMS m/e Berechnet für C20H21N2O2: 309,1603. Gefunden: 309,1607 (M+1).
-
D. [9-(Cyclopentyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäuremethylester
-
Ein
Gemisch aus 9-[(Cyclopentyl)methylJ-4-hydroxy-5-carbamoylcarbazol
(45 mg, 0,146 mmol) und Cs2CO3 (120
mg, 0,365 mmol) in 2 ml DMF wird mit Methylbromacetat (0,018 ml,
0,19 mmol) behandelt. Die Reaktion wird für 2 Stunden bei Umgebungstemperatur
gerührt
und dann wird sie mit EtOAc und H2O (jeweils 10
ml) verdünnt.
Die wässrige
Phase wird mit festem NaCl gesättigt
und mit EtOAc (2 × 10
ml) extrahiert. Die vereinigten organischen Phasen werden mit H2O (2 × 25
ml) gewaschen, über
wasserfreiem Na2SO4 getrocknet,
filtriert und im Vakuum konzentriert. Eine Reinigung des rohen Rückstands
durch Blitzchromatographie auf Silicagel (Elution mit einem Gradienten
aus 0% bis 100% EtOAc/Hexan), gefolgt von einer Behandlung mit Et2O/EtOAc ergibt 26 mg (0,0683 mmol, 47%)
der Titelverbindung als hellbraunen Feststoff. MS (ES) m/e 381 (M+1),
364 (M+H-NH3), 439 (M+AcO).
Elementaranalyse
für C23H26N2O4 × 0,1H2O:
Berechnet: C 69,13, H 6,38, N 7,33.
Gefunden:
C 68,99, H 6,39, N 7,41.
-
E. 9-[(Cyclopentyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäure
-
Eine
Aufschlämmung
aus [9-[(Cyclopentyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäuremethylester
(20 mg, 0,065 mmol) in 0,3 ml THF und 0,1 ml MeOH wird mit 0,1 ml
an 1 N wässrigem
LiOH (0,1 mmol) behandelt und das Gemisch wird bei Raumtemperatur
für 2 Stunden
gerührt.
Die Reaktion wird mit 0,2 N HCl angesäuert und die organischen Anteile
werden im Vakuum entfernt. Der weiße Niederschlag wird aus der wässrigen
Phase abfiltriert und unter Bildung von 15 mg (0,0409 mmol, 63%)
der Titelverbindung als weißes Pulver
mit Et2O gewaschen.
MS (ES) m/e 367
(M+1), 350 (M+H-NH3), 365 (M-1).
Elementaranalyse
für C21H22N2O4 × 0,3H2O. Berechnet: C 67,84, H 6,13, N 7,53. Gefunden:
C 67,73, H 5,97, N 7,70.
-
Die
hierin beschriebenen Verbindungen dürften ihre nützliche
therapeutische Wirkung im Prinzip durch die direkte Hemmung der
humanen sPLA2 erreichen, und nicht, indem
sie als Antagonisten für
Arachidonsäure wirken
und auch nicht für
andere wirksame Mittel unter der Arachidonsäure in der Arachidonsäurekaskade,
wie 5-Lipoxygenasen, Cyclooxygenasen usw. Das Verfahren zur Hemmung
der durch sPLA2 vermittelten Freisetzung
von Fettsäuren
umfasst das Zusammenbringen von sPLA2 mit
einer therapeutisch wirksamen Menge einer Verbindung der Formel
(I), die aus der Gruppe ausgewählt
ist, welche besteht aus [9-Benzyl-5-carbamoyl-1-fluorcarbazol-4-yl]oxyessigsäure,
{9-[(Phenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-[(3-Fluorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-[(3-Chlorphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure,
{9-[(3-Trifluormethylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-[(2-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-[(3-Methylphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
{9-[(3-Trifluormethoxyphenyl)methyl]-5-carbamoylcarbazol-4-yl}oxyessigsäure-Natriumsalz,
[9-Benzyl-5-carbamoyl-1-chlorcarbazol-4-yl]oxyessigsäure,
[9-(Cyclohexyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäure und
[9-[(Cyclopentyl)methyl]-5-carbamoylcarbazol-4-yl]oxyessigsäure oder
einem pharmazeutisch annehmbaren Salz hiervon.
-
Die
erfindungsgemäßen Verbindungen
können
in einem Verfahren zur Behandlung eines Säugers (beispielsweise eines
Menschen) zur Linderung der pathologischen Wirkungen von septischem
Schock, Atemstresssyndrom beim Erwachsenen, Pankreatitis, Trauma,
Bronchialasthma, allergischer Rhinitis und rheumatoider Arthritis
verwendet werden, wobei das Verfahren die Verabreichung einer Verbindung
der Formel (I) in einer therapeutisch wirksamen Menge an den Säuger umfasst.
Eine "therapeutisch
wirksame Menge" ist
eine Menge, die zur Hemmung der durch sPLA2 vermittelten
Freisetzung von Fettsäuren
und somit zur Hemmung oder Prävention
der Arachidonsäurekaskade
und ihrer schädlichen
Produkte ausreichend ist. Die therapeutische Menge einer erfindungsgemäßen Verbindung,
die zur Hemmung von sPLA2 erforderlich ist,
kann leicht durch die Entnahme einer Körperflüssigkeitsprobe und der Bestimmung
des sPLA2 Gehalts durch herkömmliche
Verfahren bestimmt werden.
-
In
diesem Dokument wird die Person oder das Tier, die zu behandeln
sind, als "Säuger" beschrieben und
es soll so verstanden werden, dass der am meisten bevorzugte Patient
ein Mensch ist. Jedoch muss erwähnt
werden, dass die Untersuchung von . gestörten Zuständen des zentralen Nervensystems
bei Tieren jetzt erst beginnt und dass in einigen Fällen solche
Behandlungen angewendet werden. Demnach ist die Verwendung der vorliegenden
Verbindungen bei Tieren ebenfalls umfasst. Es ist verständlich,
dass sich die Dosisbereiche für
Tiere notwendigerweise von den dem Menschen verabreichten Dosen
ziemlich unterscheiden und dass demnach die beschriebenen Dosisbereiche
erneut berechnet werden müssen.
Beispielsweise kann ein kleiner Hund nur 1/10 der typischen Größe eines
Menschen aufweisen und es ist daher notwendig, eine viel kleinere
Dosis zu verwenden. Die Bestimmung der wirksamen Menge für ein bestimmtes
Tier wird auf dieselbe Weise ausgeführt, wie sie später im Fall
des Menschen beschrieben ist und Tierärzte sind mit solchen Bestimmungen
gut vertraut.
-
Wie
vorher erwähnt
sind die erfindungsgemäßen Verbindungen
zur Hemmung der durch die sPLA2 vermittelten
Freisetzung von Fettsäuren
brauchbar, wie Arachidonsäure.
Durch den Ausdruck "hemmend" ist die Prävention
oder therapeutisch signifikante Verringerung der durch die sPLA2 hervorgerufenen Freisetzung von Fettsäuren durch
die erfindungsgemäßen Verbindungen
gemeint. Mit "pharmazeutisch
annehmbar" ist gemeint,
dass der Träger,
das Verdünnungsmittel
oder der Hilfsstoff mit den anderen Bestandteilen der Formulierung
kompatibel sein muss und für
den Empfänger
hiervon nicht schädlich
sein darf.
-
Im
allgemeinen werden die erfindungsgemäßen Verbindungen am liebsten
in einer Dosis verab reicht, die im allgemeinen wirksame Ergebnisse
erzielt, ohne schwere Nebenwirkungen zu verursachen und kann entweder
als Einheitsdosis verabreicht werden oder, falls erwünscht, kann
die Dosis in bequeme Untereinheiten aufgeteilt werden, die zu geeigneten
Zeiten über
den Tag verabreicht werden.
-
Die
bestimmte Dosis einer gemäß der Erfindung
verabreichten Verbindung, um therapeutische oder prophylaktische
Wirkungen zu erzielen, wird natürlich
von den besonderen Umständen
bestimmt, die den Fall umgeben, einschließlich beispielsweise, dem Verabreichungsweg,
dem Alter, Gewicht und der Reaktion des einzelnen Patienten, dem
bestimmten zu behandelnden Zustand und der Schwere der Symptome
des Patienten. Typische Tagesdosen enthalten eine nicht-toxische
Dosismenge von etwa 0,01 mg/kg bis etwa 50 mg/kg Körpergewicht
des erfindungsgemäßen Wirkstoffs.
-
Vorzugsweise
ist die pharmazeutische Formulierung eine Einheitsdosierungsform.
Die Einheitsdosierungsform kann eine Kapsel oder eine Tablette oder
die geeignete Anzahl dieser sein. Die Menge an Wirkstoff in einer
Einheitsdosierungsformzusammensetzung kann von etwa 0,1 bis etwa
1000 Milligramm oder mehr gemäß der bestimmten
involvierten Behandlung variiert oder eingestellt werden. Es ist
verständlich,
dass Routinevariationen bezüglich
der Dosis in Abhängigkeit
des Alters und des Zustands des Patienten notwendig sind. Die Dosis
hängt auch
vom Verabreichungsweg ab.
-
Ein "chronischer" Zustand steht für einen
störenden
Zustand mit langsamem Fortschritt und langer Dauer. Daher wird dieser
bei Diagnose behandelt und während
dem Verlauf der Krankheit fortgesetzt. Ein "akuter" Zustand ist ein schlechter Zustand
von kurzer Dauer, dem eine Besserungsperiode folgt. In einem akuten
Fall wird die Verbindung beim Einsetzen der Symptome verabreicht
und abgesetzt, wenn die Symptome verschwinden.
-
Pankreatitis,
Trauma-induzierter Schock, Bronchialasthma, allergische Rhinitis
und rheumatoide Arthritis können
als akuter Vorfall oder als chronischer Vorfall auftreten. Daher
umfasst die Behandlung dieser Zustände sowohl die akuten als auch
die chronischen Formen. Septischer Schock und Atemstresssyndrom
beim Erwachsenen sind auf der anderen Seite akute Zustände, die
bei Diagnose behandelt werden.
-
Die
Verbindungen können
auf eine Vielzahl an Arten verabreicht werden, einschließlich oral,
durch Aerosol, rektal, transdermal, subkutan, intravenös, intramuskulär und intranasal.
-
Pharmazeutische
Formulierungen der Erfindung werden hergestellt durch Kombination
(beispielsweise Mischen) einer therapeutisch wirksamen Menge der
erfindungsgemäßen Verbindungen
mit einem pharmazeutisch annehmbaren Träger oder Verdünnungsmittel
hierfür.
Die vorliegenden pharmazeutischen Formulierungen werden durch gut
bekannte Verfahren mittels gut bekannter und leicht verfügbarer Inhaltsstoffe
hergestellt.
-
Bei
der Herstellung der erfindungsgemäßen Zusammensetzungen wird
der Wirkstoff gewöhnlich
mit einem Träger
gemischt oder mit einem Träger
verdünnt
oder in einem Träger
eingeschlossen, der in Form einer Kapsel, eines Sachets, eines Papiers
oder eines anderen Behälters
vorliegen kann. Wenn der Träger
als Verdünnungsmittel
dient, kann dies ein festes, halbfestes oder flüssiges Material sein, das als
Vehikel dient, oder kann in Form von Tabletten, Pillen, Pulvern,
Lonzetten, Elixieren, Suspensionen, Emulsionen, Lösungen,
Sirupen, Aerosolen (als Feststoff oder in einem flüssigen Medium)
oder Salben vorliegen, die beispielsweise bis zu 10 Gewichtsprozent
des Wirkstoffs enthalten. Die erfindungsgemäßen Verbindungen werden vorzugsweise vor
der Verabreichung formuliert.
-
Für die pharmazeutischen
Formulierungen kann jeder in der Technik bekannte Träger verwendet
werden. In einer solchen Formulierung kann der Träger ein
Feststoff, eine Flüssigkeit
oder ein Gemisch eines Feststoffs und einer Flüssigkeit sein. Formulierungen
in fester Form umfassen Pulver, Tabletten und Kapseln. Ein fester
Träger
kann eine oder mehrere Substanzen umfassen, die auch als Geschmacksmittel,
Gleitmittel, Löslichkeitsvermittler,
Suspendiermittel, Bindemittel, Tablettenzerfallshilfsmittel und
verkapselndes Material wirken können.
-
Tabletten
zur oralen Verabreichung können
geeignete Hilfsstoffe enthalten, wie Calciumcarbonat, Natriumcarbonat,
Lactose, Calciumphosphat, zusammen mit Zerfallshilfsstoffen, wie
Maisstärke
oder Alginsäure und/oder
Bindemittel, beispielsweise Gelatine oder Akaziengummi, und Gleitmittel,
wie Magnesiumstearat, Stearinsäure
oder Talkum.
-
In
Pulvern ist der Träger
ein fein verteilter Feststoff, der mit dem fein verteilten Wirkstoff
gemischt ist. In Tabletten wird der Wirkstoff mit einem Träger in geeigneten
Anteilen gemischt, der die notwendigen Bindungseigenschaften aufweist,
und in der gewünschten
Form und Größe verpresst.
Die Pulver und Tabletten enthalten vorzugsweise etwa 1 bis etwa
99 Gewichtsprozent des Wirkstoffs, der die neue Verbindung der Erfindung
ist. Geeignete feste Träger
sind Magnesiumcarbonat, Magnesiumstearat, Talkum, Zucker, Lactose, Pektin,
Dextrin, Stärke,
Gelatine, Tragacanth, Methylcellulose, Natriumcarboxymethylcellulose,
niedrig schmelzende Wachse und Kakaobutter.
-
Sterile
Formulierungen in flüssiger
Form umfassen Suspensionen, Emulsionen, Sirupe und Elixiere.
-
Der
Wirkstoff kann in einem pharmazeutisch annehmbaren Träger gelöst oder
suspendiert werden, wie sterilem Wasser, sterilem organischem Lösemittel
oder einem Gemisch aus beidem. Der Wirkstoff kann oft in einem geeigneten
organischen Lösemittel
gelöst
werden, beispielsweise wässrigem
Propylenglycol. Andere Zusammensetzungen können durch Dispergieren des
fein verteilten Wirkstoffs in wässriger
Stärke
oder Natriumcarboxymethylcelluloselösung oder in einem geeigneten Öl hergestellt
werden.
-
Die
folgenden pharmazeutischen Formulierungen 1 bis 8 sind nur erläuternd und
sollen den Schutzumfang der Erfindung in keiner Weise beschränken. "Wirkstoff" steht für eine Verbindung
der Formel (I) oder ein pharmazeutisch annehmbares Salz, Solvat
oder Prodrug hiervon.
-
Formulierung 1
-
Hartgelatinekapseln
werden unter Verwendung folgender Inhaltsstoffe hergestellt:
| | Menge |
| | (mg/Kapsel) |
| Verbindung
von Beispiel 1 | 250 |
| Stärke, getrocknet | 200 |
| Magnesiumstearat | 10 |
| Gesamt | 460
mg |
-
Formulierung 2
-
Eine
Tablette wird unter Verwendung der folgenden Inhaltsstoffe hergestellt:
| | Menge |
| | (mg/Tablette) |
| Verbindung
von Beispiel 2 | 250 |
| mikrokristalline
Cellulose | 400 |
| pyrogen
hergestelltes Siliciumdioxid | 10 |
| Stearinsäure | 5 |
| Gesamt | 665
mg |
-
Die
Bestandteile werden vermischt und unter Bildung von Tabletten gepresst,
wobei jede 665 mg wiegt.
-
Formulierung 3
-
Eine
Aerosollösung,
die die folgenden Bestandteile enthält, wird hergestellt:
| | Gewicht |
| Verbindung
von Beispiel 3 | 0,25 |
| Ethanol | 25,75 |
| Propellant
22 (Chlordifluormethan) | 74,00 |
| Gesamt | 100,00 |
-
Der
Wirkstoff wird mit Ethanol gemischt und das Gemisch wird zu einem
Teil Propellant 22 gegeben, auf –30°C abgekühlt und in ein Abfüllgerät gegeben.
Die erforderliche Menge wird anschließend in einen Edelstahlbehälter gefüllt und
mit dem Rest des Propellants verdünnt. Die Ventileinheiten werden
anschließend
am Behälter
angebracht.
-
Formulierung 4
-
Tabletten,
die jeweils 60 mg des Wirkstoffs enthalten, werden folgendermaßen hergestellt:
| Verbindung
von Beispiel 4 | 60
mg |
| Stärke | 45
mg |
| Mikrokristalline
Cellulose | 35
mg |
| Polyvinylpyrrolidon
(als 10% Lösung
in Wasser) | 4
mg |
| Natriumcarboxymethylstärke | 4,5
mg |
| Magnesiumstearat | 0,5
mg |
| Talkum | 1
mg |
| Gesamt | 150
mg |
-
Der
Wirkstoff, die Stärke
und die Cellulose werden durch ein Nr. 45 Mesh U.S. Sieb gegeben
und sorgfältig
vermischt. Die wässrige
Lösung,
die Polyvinylpyrrolidon enthält,
wird mit dem entstehenden Pulver vermischt und das Gemisch wird
anschließend
durch ein Nr. 14 Mesh U.S. Sieb gegeben. Die so hergestellten Granula
werden bei 50°C
getrocknet und durch ein Nr. 18 Mesh U.S. Sieb gegeben. Die Natriumcarboxymethylstärke, das
Magnesiumstearat und das Talkum werden, nachdem sie vorher durch
ein Nr. 60 Mesh U.S. Sieb gegeben wurden, zu den Granula gegeben
und nach dem Mischen in einer Tablettenmaschine unter Bildung von
Tabletten gepresst, die jeweils 150 mg wiegen.
-
Formulierung 5
-
Kapseln,
die jeweils 80 mg des Wirkstoffs enthalten, werden folgendermaßen hergestellt:
| Verbindung
von Beispiel 5 | 80
mg |
| Stärke | 59
mg |
| Mikrokristalline
Cellulose | 59
mg |
| Magnesiumstearat | 2
mg |
| Gesamt | 200
mg |
-
Der
Wirkstoff, die Cellulose, die Stärke
und das Magnesiumstearat werden gemischt, durch ein Nr. 45 Mesh
U.S. Sieb gegeben und in Hartgelatinekapseln in 200 mg Mengen abgefüllt.
-
Formulierung 6
-
Zäpfchen,
die jeweils 225 mg des Wirkstoffs enthalten, werden folgendermaßen hergestellt:
| Verbindung
von Beispiel 6 | 225
mg |
| Gesättigte Fettsäureglyceride | 2000
mg |
| Gesamt | 2225
mg |
-
Der
Wirkstoff wird durch ein Nr. 60 Mesh U.S. Sieb gegeben und in den
gesättigten
Fettsäureglyceriden
suspendiert, die vorher bei möglichst
geringer Hitze geschmolzen werden. Das Gemisch wird anschließend in
eine Zäpfchenform
mit einer nominalen Kapazität
von 2 g gegossen und abgekühlt.
-
Formulierung 7
-
Suspensionen,
die jeweils 50 mg des Wirkstoffs pro 5 ml Dosis enthalten, werden
folgendermaßen hergestellt:
| Verbindung
von Beispiel 7 | 50
mg |
| Natriumcarboxymethylcellulose | 50
mg |
| Sirup | 1,25
ml |
| Benzoesäurelösung | 0,10
ml |
| Geschmacksstoff | q.v. |
| Farbstoff | q.v. |
| Gereinigtes
Wasser auf gesamt | 5
ml |
-
Der
Wirkstoff wird durch ein Nr.45 Mesh U.S. Sieb gegeben und mit Natriumcarboxymethylcellulose und
Sirup vermischt, um eine glatte Paste zu erhalten. Die Benzoesäurelösung, der
Geschmacksstoff und der Farbstoff werden mit einem Anteil Wasser
vermischt, und unter Rühren
zugegeben. Anschließend
wird ausreichend Wasser zugegeben, um das erforderliche Volumen
zu erhalten.
-
Formulierung 8
-
Eine
intravenöse
Formulierung kann folgendermaßen
hergestellt werden:
| Verbindung
von Beispiel 8 | 100
mg |
| Isotonische
Kochsalzlösung | 1000
ml |
-
Die
Lösung
der obigen Inhaltsstoffe wird im allgemeinen einem Patienten mit
einer Geschwindigkeit von 1 ml pro Minute intravenös verabreicht.
-
Testexperimente
-
Testbeispiel 1
-
Das
folgende chromogene Testverfahren wird zur Identifizierung und Evaluierung
von Inhibitoren der rekombinanten humanen sekretierten Phospholipase
A2 verwendet. Der hierin beschriebene Test
wurde mittels Mikrotiterplatten mit 96 Vertiefungen für ein Screening
mit hohem Durchsatz angepasst. Eine allgemeine Beschreibung des
Testverfahrens findet man in dem Artikel "Analysis of Human Synovial Fluid Phospholipase A2 on Short Chain Phosphatidylcholin-Mixed
Micelles: Development of a Spectrophotometric Assay Suitable for
a Microtiterplate Reader",
von Laure J. Reynolds, Lori L. Hughes und Edward A. Dennis, Analytical
Biochemistry, 204, Seiten 190–197,
1992 (wobei die Beschreibung hiermit eingeführt ist).
-
Reagenzien:
-
- Reaktionspuffer:
CaCl2·2H2O (1,47 g/l)
KCl (7,455 g/l)
Rinderserumalbumin
(fettsäurefrei)
(1 g/l) (Sigma A-7030, Produkt von Sigma Chemical Co. St. Louis,
Mo, USA)
Tris HCl (3,94 g/l)
pH 7,5 (eingestellt mit NaOH)
- Enzympuffer
0,05 NaOAc·3 H2O,
pH 4,5
0,2 NaCl
pH eingestellt mit Essigsäure auf
4,5
- DTNB – 5,5'-Dithiobis-2-nitrobenzoesäure
- Razemisches Diheptanoyl Thio-PC
razemisches 1,2-Bis(heptanoylthio)-1,2-didesoxy-sn-glycero-3-phosphorylcholin
Triton
X-100® angesetzt
mit 6,249 mg/ml in Reaktionspuffer entspricht 10 μM
Triton
X-100® ist
ein nicht-ionisches Polyoxyethylendetergenz, das von Pierce Chemical
Company,
3747 N. Meridian Road, Rockford, Illinois 61101 vertrieben
wird.
- Reaktionsgemisch
Ein gemessenes Volumen an razemischem
Diheptanoylthio-PC in Chloroform mit einer Konzentration von 100 mg/ml
wird eingedampft und in 10 mM rückgelöst
Triton
X-100® wässrige Lösung des
nichtionischen Detergenzes
Man gibt Reaktionspuffer zur Lösung und
dann DTNB, um das Reaktionsgemisch zu erhalten.
Das so erhaltene
Reaktionsgemisch enthält
1 mM Diheptanoylthio-PC Substrat, 0,29 mM Triton X-100® Detergenz
und 0,12 mM DTNB in einer gepufferten wässrigen Lösung bei pH 7,5
-
Testverfahren
-
- 1. Zugabe von 0,2 ml Reaktionsgemisch zu allen
Vertiefungen.
- 2. Zugabe von 10 μl
Testverbindung (oder Lösemittelkontrolle)
zu geeigneten Vertiefungen, 20 Sekunden Mischen.
- 3. Zugabe von 50 Nanogramm sPLA2 (10 μl) zu geeigneten
Vertiefungen.
- 4. Inkubation der Platte bei 40°C für 30 Minuten.
- 5. Ablesen der Absorption der Vertiefungen bei 405 nm mit einem
automatischen Mikrotiterphotometer.
-
Alle
Verbindungen werden dreifach getestet. Typische Verbindungen werden
in einer Endkonzentration von 5 μg/ml
getestet. Verbindungen werden als wirksam betrachtet, wenn sie eine
Hemmung von 40% oder mehr verglichen zu den ungehemmten Kontrollreaktionen
aufweisen, wenn sie bei 405 nm gemessen werden. Das Fehlen der Farbentwicklung
bei 405 nm zeigt eine Hemmung an. Verbindungen, die anfangs als
aktiv befunden werden, werden erneut getestet, um ihre Aktivität zu bestätigen, und
wenn sie ausreichend aktiv sind, werden die HK50 Werte
bestimmt. Typischerweise werden die HK50 Werte
durch serielle Zweifachverdünnung der
Testverbindung bestimmt, so dass die Endkonzentration in der Reaktion
von 45 μg/ml
bis 0,35 μg/ml
reicht. Stärkere
Inhibitoren erfordern eine signifikant stärkere Verdünnung. In allen Fällen wird
die prozentuale Hemmung bei 405 nm gemessen, die durch die Enzymreaktionen
hervorgerufen wird, die Inhibitoren enthalten, relativ zu nicht-gehemmten
Kontrollreaktionen. Jede Probe wird dreifach titriert und die Ergebnisse
werden für die
Auftragung und die Berechnung der HK50 Werte
gemittelt. Die HK50 Werte werden durch Auftragung
der logarithmischen Konzentrationen gegen die Hemmwerte im Bereich
von 10–90%
Hemmung bestimmt.
-
Die
erfindungsgemäßen Verbindungen
werden in Testbeispiel 1 getestet und sind bei Konzentrationen von
weniger als 100 μM
wirksam.
-
Testbeispiel 2
-
Verfahren:
-
Männliche
Hartley-Meerschweinchen (500–700
g) werden durch cervikale Dislokation getötet und ihre Herzen und Lungen
werden intakt entnommen und in einen belüfteten (95% O2 :
5% CO2) Krebs Puffer gegeben. Dorsale Pleurastreifen
(4 × 1 × 25 mm)
werden von intakten Parenchymsegmenten (8 × 4 × 25 mm) herausgeschnitten,
die parallel zur äußeren Spitze
der unteren Lungenlappen geschnitten wurden. Zwei benachbarte Pleurastreifen,
die von einem einzelnen Lappen erhalten wurden und eine einzelne
Gewebeprobe darstellen, werden an beiden Enden angebunden und unabhängig an
einen Metallträgerstab
angebracht. Ein Stab wird an einen Grass Kraftübertragungsumwandler (Modell
FTO3C, Produkt von Grass Medical Instruments Co., Quincy, MA, USA)
angebracht. Veränderungen
in der isometrischen Spannung werden auf einen Monitor und einem
Thermoschreiber (Produkt von Modular Instruments, Malvern, PA) angezeigt.
Alle Gewebe werden in ummantelte 10 ml Gewebebäder gegeben und auf 37°C gehalten.
Die Gewebebäder
werden kontinuierlich belüftet
und enthalten eine modifizierte Krebslösung der folgenden Zusammensetzung
(mM): NaCl 118,2, KCl 4,6, CaCl2·2H2O 2,5, MgSO4·7H2O 1,2, NaHCO3 24,8,
KH2PO4 1,0, und
Dextrose 10,0. Pleurastreifen von den gegenüberliegenden Lungenlappen werden
für gepaarte
Experimente verwendet. Vorläufige
Daten, die aus den Spannungs-/Antwortkurven erzeugt werden, zeigen,
dass die Ruhespannung von 800 mg optimal ist. Die Gewebe können sich
für 45
Minuten äquilibrieren,
wenn die Badflüssigkeit
periodisch ausgetauscht wurde.
-
Kumulative Konzentrations-Antwortkurven
-
Anfänglich werden
die Gewebe dreimal mit KCl (40 mM) provoziert, um die Gewebelebensfähigkeit
zu testen und eine konsistente Antwort zu erhalten. Nach der Aufzeichnung
der maximalen Reaktion gegenüber KCl
werden die Gewebe gewaschen und können vor der nächsten Provokation
auf die Grundlinie zurückkehren.
Man erhält
kumulative Konzentrations-Antwortkurven aus den Pleurastreifen,
indem man die Agonistkonzentration (sPLA2)
im Gewebebad durch Zunahmen um halbe log10 erhöht, während die vorhergehende Konzentration
mit den Geweben in Kontakt bleibt (siehe obige Referenz 1). Die
Agonistkonzentration wird erhöht, nachdem
die durch die vorangehende Konzentration ausgelöste Kontraktion ein Plateau
erreicht hat. Man erhält
eine Konzentrations-Antwortkurve aus jedem Gewebe. Um die Variabilität zwischen
Geweben zu minimieren, die von verschiedenen Tieren erhalten wurden,
werden die Kontraktionsreaktionen als Prozentsatz der maximalen
Reaktion ausgedrückt,
die mit der letzten KCl Provokation erhalten wurde. Wenn man die
Effekte verschiedener Arzneimittel auf die kontraktilen Effekte
der sPLA2 untersucht, werden die Verbindungen
und ihre jeweiligen Träger
30 Minuten vor dem Start der sPLA2 Konzentrations-Antwortkurven
zugegeben.
-
Statistische Analyse
-
Daten
von verschiedenen Experimenten werden vereinigt und als Prozentsatz
der maximalen KCl Reaktionen dargestellt (Mittel ± Standardabweichung).
Um die durch die Arzneimittel hervorgerufenen Verschiebungen nach
rechts in den Konzentrations-Antwortkurven abzuschätzen, werden
die Kurven simultan mittels statistischer nichtlinearer Modellmethoden
analysiert, die den von Waud (1976), Gleichung 26, Seite 163 (Referenz
2) beschriebenen ähnlich
sind. Das Modell umfasst vier Parameter: Die maximale Gewebereaktion,
die für
jede Kurve als gleich angenommen wird, die ED50 für die Kontrollkurve,
die Steilheit der Kurven und die pA2, die
Konzentration an Agonist, die eine zweifache Erhöhung des Agonisten erfordert,
um eine äquivalente
Reaktion zu erreichen. Die Schild Steigung wird mittels statistischer
nichtlinearer Modellmethoden mit 1 bestimmt, die zu den von Waud
(1976), Gleichung 27, Seite 164 beschriebenen ähnlich sind (Referenz 2). Die
dem Wert 1 entsprechende Schild-Steigung zeigt, dass das Modell
mit den Annahmen eines kompetitiven Antagonisten konsistent ist,
und daher die pA2 als scheinbare KB interpretiert werden kann, die Dissoziationskonstante
des Inhibitors.
-
Um
die Arzneimittel-induzierte Unterdrückung der Maximalreaktionen
abzuschätzen,
werden die sPLA2 Reaktionen (10 μg/ml) in
Abwesenheit und Anwesenheit des Arzneimittels bestimmt und die prozentuale
Unterdrückung
wird für
jedes Gewebepaar berechnet. Repräsentative
Beispiele der Hemmaktivitäten
sind in der Tabelle 2 unten gezeigt.
Referenz 1 – van, J.M.:
Cumulative dose-response curves. II.
Technique for the making
of dose-response curves in isolated organs and the evaluation of
drug parameters. Arch. Int. Pharmacodyn. Ther. 143: 299–330, 1963.
Referenz
2 – D.
Waud: Analysis of dose-response relationships, in Advances in General
and Cellular Pharmacology, Herausgeber Narahashi, Bianchi 1: 145–178, 1976.
-
Die
erfindungsgemäßen Verbindungen
werden im Testbeispiel 2 getestet und sind bei Konzentrationen unter
20 μM wirksam.
-
Testbeispiel 3
-
sPLA2 Test
in transgenen Mäusen
-
Materialien und Methoden
-
Die
in diesen Studien verwendeten Mäuse
sind reife, 6–8
Monate alte, mit ZnSO4 stimulierte, transgene
Mäuse der
hemizygoten Linie 2608a (Fox et al., 1996).
Die transgenen Mäuse
dieser Linie exprimieren humane sPLA2 in
der Leber und anderen Geweben und erreichen typischerweise Spiegel
von humaner sPLA2 in ihrem Kreislauf von
etwa 173 ± 10
ng/ml, wenn sie maximal mit ZnSO4 stimuliert
werden (Fox et al., 1996). Die Mäuse
werden unter konstanter Luftfeuchtigkeit und Temperatur gehalten
und erhalten freien Zugang zu Futter und Wasser. Die Beleuchtung
des Tierraums wird in einem 12 stündigen Hell-Dunkel-Zyklus gehalten
und alle Experimente werden zur selben Zeit des Tages während der
Lichtperiode am frühen
Morgen ausgeführt.
-
Für intravenöse Tests
werden die Verbindungen oder der Träger als i.v. Bolus über die
Schwanzvene in einem Volumen von 0,15 ml verabreicht. Der Träger besteht
aus 1–5%
Dimethylsulfoxid, 1–5%
Ethanol und 10–30%
Polyethylenglycol 300 in H2O, wobei die
Konzentrationen dieser Bestandteile gemäß der Löslichkeit der Verbindung angepasst
werden. Die Mäuse
lässt man
retro-orbital vor der Verabreichung des Arzneimittels oder des Trägers und
30 Minuten, 2 und 4 Stunden danach bluten. 3 bis 6 Mäuse werden
für jede
Dosis verwendet. Die katalytische PLA2 Aktivität im Serum
wird mit einem modifi zierten gemischten Mizelltest aus Phosphatidylcholin/-desoxycholin
(Fox et al. 1996, Schadlich et al. 1987) unter Verwendung von 3
mM Natriumdesoxycholat und 1 mM 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholin getestet.
-
Für einen
oralen Test werden die Verbindungen in 1–5% Ethanol/10–30% Polyethylenglycol
300 in H2O gelöst oder in 5% Dextrose in H2O suspendiert und durch orale Gabe verabreicht.
Das Serum wird aus retro-orbitalem Blut gewonnen und wie oben auf
eine katalytische PLA2 Aktivität getestet.
-
Literaturangaben:
-
- N. Fox, M. Song, J. Schrementi, J.D. Sharp, D.L. White,
D.W. Snyder, L.W. Hartley, D.G. Carlson, N.J. Bach, R.D. Dillard,
S.E. Draheim, J. L. Bobbit, L. Fisher und E.D. Mihelich, 1996, Eur.
J. Pharmacol. 308: 195.
- H.R. Schadlich, M. Buchler und H.G. Reger, 1987, J. Clin. Chem.
Clin Biochem. 25, 505.
-
Die
Verbindungen der vorliegenden Erfindung werden in Testbeispiel 3
getestet und als wirksam befunden.
-
Obwohl
die vorliegende Erfindung durch bestimmte Ausführungsformen erläutert wurde,
ist es nicht beabsichtigt, dass diese bestimmten Beispiele den Schutzumfang
der Erfindung beschränken,
wie er in den Patentansprüchen
beschrieben ist.