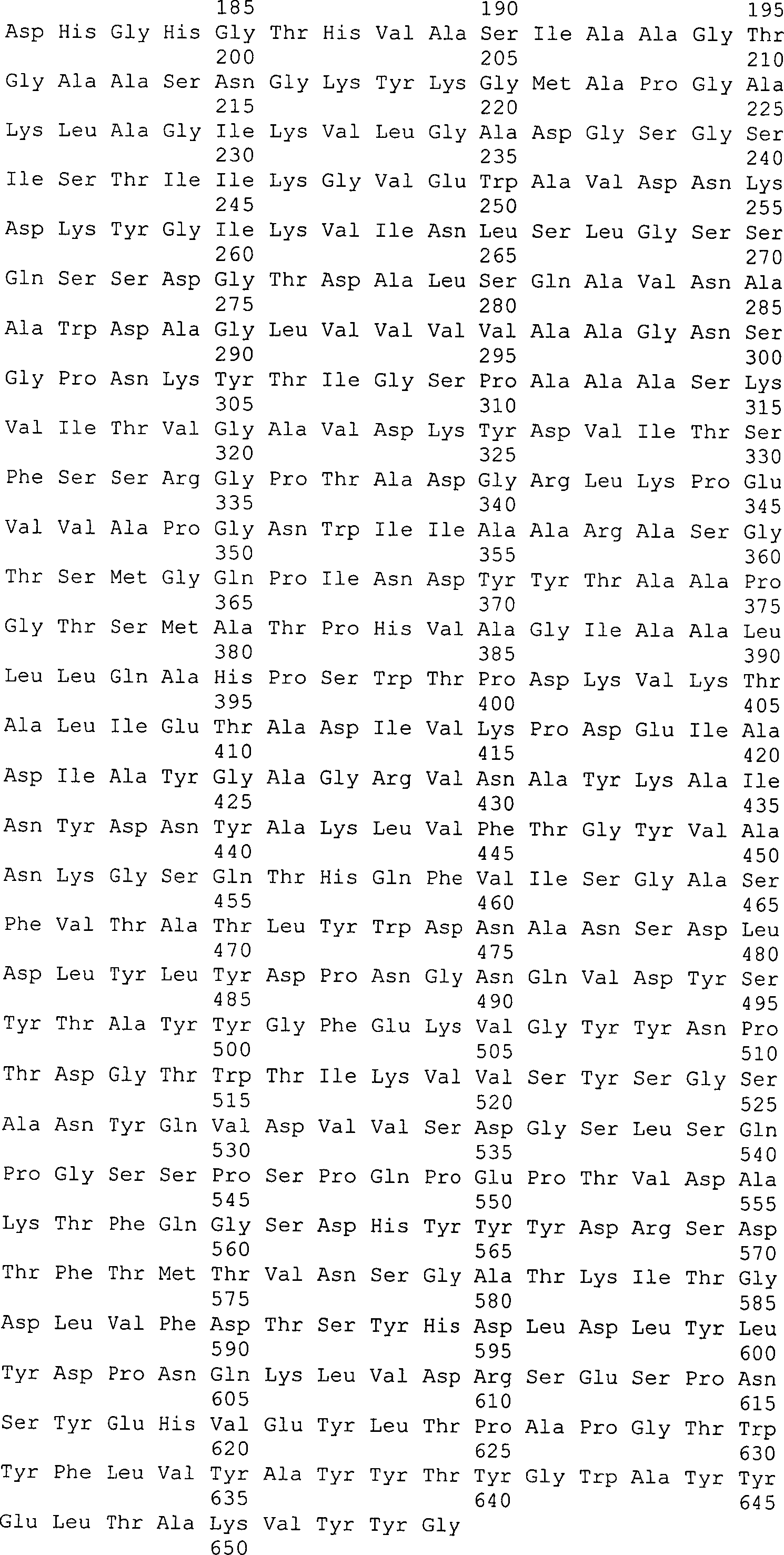-
Technisches
Gebiet
-
Die
vorliegende Erfindung betrifft eine hyperthermostabile Protease,
die als ein Enzym zur industriellen Verwendung geeignet ist, ein
Gen, das diese kodiert und ein Verfahren zum Herstellen des Enzyms
durch Gentechnik.
-
Technischer
Hintergrund
-
Eine
Protease ist ein Enzym, das Peptidbindungen in Proteinen spaltet.
Eine Anzahl an solchen Enzymen ist in Tieren, Pflanzen und Mikroorganismen
gefunden worden. Die Protease wird als ein Reagenz sowohl zur Verwendung
im Labor als auch als ein Pharmazeutikum in industriellen Gebieten,
zum Beispiel als ein Zusatz zu einem Detergenz, zur Lebensmittelverarbeitung
und zur chemischen Synthese unter Verwendung einer reversen Reaktion
verwendet. Daher kann gesagt werden, dass die Protease für die Industrie
ein außerordentlich
wichtiges Enzym ist. Da hohe physikalische und chemische Stabilität für eine Protease
benötigt
wird, die in industriellen Gebieten verwendet wird, wird bevorzugt
ein thermostabiles Enzym gegenüber
anderen verwendet. Da durch Bakterien der Gattung Bacillus hergestellte
Proteasen relativ hohe Thermostabilität aufweisen, werden sie hauptsächlich als
Proteasen für
industrielle Verwendung verwendet. Jedoch wurden bei der Suche eines
besseren Enzyms Versuche unternommen, ein Enzym von einem bei hohen
Temperaturen wachsenden Organismus zu erhalten, z.B. einem thermophilen
oder einem hyperthermophilen Bakterium der Gattung Bacillus.
-
Zum
Beispiel ist das hyperthermophile Pyrococcus furiosus für die Herstellung
einer Protease bekannt (Appl. Environ. Microbiol., 56:1992-1998
(1990); FEMS Microbiol. Letters, 71:17-20 (1990); J. Gen. Microbiol., 137:1193-1199
(1991)).
-
Zusätzlich wurde
von dem hyperthermophilen Pyrococcus sp.-Stamm KOD1 berichtet, dass
er eine Thiol-Protease (eine Cystein-Protease) herstellt (Appl.
Environ. Microbiol., 60:4559-4566 (1994)). Hyperthermophile der
Gattungen Thermococcus, Staphylothermus und Thermobacteroides sind
auch für
die Herstellung von Proteasen bekannt (Appl. Microbiol. Biotechnol.,
34:715-719 (1991)).
-
Die
Proteasen der Hyperthermophilen wie vorstehend beschrieben haben
eine hohe Thermostabilität. Daher
wird davon ausgegangen, dass sie anstelle der momentan verwendeten
thermostabilen Proteasen oder in einem Gebiet verwendet werden können, in
dem die Verwendung einer Protease nicht erwägt worden ist.
-
Jedoch
wachsen die meisten der diese Enzyme herstellenden Mikroorganismen
nur bei hoher Temperatur. Zum Beispiel muss Pyrococcus furiosus
bei 90-100°C
kultiviert werden. Kultivierung bei solch hoher Temperatur ist im
Hinblick auf Energiekosten nachteilig. Des Weiteren sind die Leistungsfähigkeiten
der Proteasen von den Hyperthermophilen geringer als die Leistungsfähigkeiten
der herkömmlichen
mikrobiellen Proteasen. Daher haben die Verfahren zur industriellen
Herstellung der Proteasen von Hyperthermophilen Nachteile.
-
Im Übrigen ist
die Herstellung eines Enzyms durch Gentechnik, durch Isolierung
des Gens für
das Enzym von Interesse und sein Einbringen in einen Wirtsorganismus,
der leicht kultiviert werden kann, momentan in der Technik üblich. Jedoch
wird das in den Wirt eingebrachte Gen des Enzyms nicht immer so
effizient exprimiert wie erwartet. Es wird angenommen, dass der
Hauptgrund dafür
ist, dass der GC-Gehalt oder die Codon-Verwendung des eingebrachten
Gens von denen der Gene des Wirtes unterschiedlich ist. Daher ist
es notwendig, das Expressionsverfahren für jedes einzubringende Gen
und/oder jeden Wirt zu optimieren, um eine geeignete Leistungsfähigkeit
eines Enzyms für
die beabsichtigte Verwendung zu erreichen.
-
Aufgaben der
Erfindung
-
Die
Aufgaben der vorliegenden Erfindung sind die Bereitstellung einer
Protease von einem Hyperthermophilen, die vorteilhaft für die industrielle
Verwendung ist, die Isolierung eines Gens, das die Protease des Hyperthermophilen
kodiert, und die Bereitstellung eines Verfahrens zur Herstellung
der hyperthermostabilen Protease unter Verwendung des Gens durch
Gentechnik, um die vorstehend beschriebenen Probleme zu lösen.
-
Zusammenfassung
der Erfindung
-
Unter
den durch Hyperthermophile hergestellten Proteasen können einige
als Subtilisin-Typ der alkalischen Proteasen basierend auf der Aminosäure-Sequenzhomologie
klassifiziert werden. Wenn ein Gen für solch eine Protease in Bacillus
subtilis eingeführt
wird, das im Allgemeinen zur Herstellung durch Gentechnik verwendet
wird, ist die Leistungsfähigkeit
dieses Enzyms deutlich geringer als die eines von Natur aus von
Bacillus subtilis hergestellten Proteins.
-
Die
Erfinder haben nach intensiven Studien herausgefunden, dass durch
Platzieren eines für
ein Signalpeptid kodierenden Gens (Signalsequenz), abgeleitet von
einem Subtilisin, upstream eines zu exprimierenden Protease-Gens,
abgeleitet von einem Hyperthermophilen, und durch Modifizieren der
Aminosäure-Sequenz
um die Spaltstelle, das Gen von Interesse in Bacillus subtilis mit
hoher Effizienz exprimiert wird. Des Weiteren ist herausgefunden
worden, dass der Expressionsgrad des Enzyms durch Entfernen eines
Teils, der nicht essentiell für
die enzymatische Aktivität
ist, im Protease-Gen, abgeleitet vom Hyperthermophilen von Interesse,
erhöht
werden kann. Folglich ist die vorliegende Erfindung vollständig ausgeführt worden.
-
Die
vorliegende Erfindung wird im Folgenden umrissen.
-
Der
erste Gegenstand der Erfindung ist eine Protease, die aus einer
Aminosäuresequenz
besteht, in der ein oder mehrere Aminosäurereste vom C-Terminus der
Aminosäuresequenz,
dargestellt durch die SEQ ID NO:4 des Sequenzprotokolls, entfernt
worden sind, und die eine thermostabile Protease-Aktivität aufweist.
-
In
einer Ausführungsform
des ersten Gegenstands der Erfindung besteht die Protease aus der
Aminosäuresequenz,
dargestellt durch die SEQ ID NO:1 des Sequenzprotokolls.
-
Der
zweite Gegenstand der Erfindung ist ein Gen, das für ein Protein
kodiert, das aus einer Aminosäuresequenz
besteht, in der ein oder mehrere Aminosäurereste vom C-Terminus der
Aminosäuresequenz,
dargestellt durch die SEQ ID NO:4 des Sequenzprotokolls, entfernt
worden sind, und das eine thermostabile Protease-Aktivität aufweist.
-
In
einer Ausführungsform
des zweiten Gegenstands der Erfindung kodiert das Protease-Gen die
Aminosäuresequenz,
dargestellt durch die SEQ ID NO:1 des Sequenzprotokolls.
-
In
einer zweiten Ausführungsform
besteht das Protease-Gen aus der Basensequenz, dargestellt durch die
SEQ ID NO:2 des Sequenzprotokolls.
-
Der
dritte Gegenstand der Erfindung ist ein Protease-Gen, das mit dem
Protease-Gen gemäß der zweiten
Ausführungsform
des zweiten Gegenstands der Erfindung unter Bedingungen hybridisiert,
die durch 6×SSC
mit 0,5% SDS, 0,1% Rinderserumalbumin (BSA), 0,1% Polyvinylpyrrolidon,
0,1% Ficoll 400, 0,01% denaturierte Lachs-Sperma-DNA bei 50°C definiert
sind, und das ein Protein mit einer thermostabilen Protease-Aktivität kodiert.
-
Der
vierte Gegenstand der Erfindung ist ein Gen, das eine Aminosäuresequenz
kodiert, dargestellt durch Formel I: SIG-Ala-Gly-Gly-Asn-PRO, wobei
SIG eine Aminosäuresequenz
eines Signalpeptids, abgeleitet von einem Subtilisin, darstellt
und PRO die Aminosäuresequenz
der Protease gemäß des ersten
Gegenstands der Erfindung umfasst.
-
Der
fünfte
Gegenstand der Erfindung ist ein Plasmid-Vektor, der das Gen gemäß dem vierten
Gegenstand der Erfindung umfasst.
-
In
einer Ausführungsform
des fünften
Gegenstands der Erfindung ist der Plasmid-Vektor pSPO124ΔC (FERM BP-6294).
-
Der
sechste Gegenstand der Erfindung ist ein Verfahren zur Herstellung
eines Proteins, das die Kultivierung eines Bakteriums der Gattung
Bacillus, in welches der Plasmid-Vektor gemäß des fünften Gegenstands der Erfindung
eingeführt
ist, und die Isolierung des Proteins von Interesse aus der Kultur
umfasst.
-
In
einer Ausführungsform
des sechsten Gegenstands der Erfindung ist das Bakterium der Gattung
Bacillus Bacillus subtilis.
-
In
einer weiteren Ausführungsform
wird das Plasmid pSPO124 Δ C
(FERM BP-6294) in das Bakterium der Gattung Bacillus eingeführt.
-
In
noch einer weiteren Ausführungsform
umfasst das Verfahren die Kultivierung von Bacillus subtilis DB104/pSPO124 Δ C FERM BP-6294
und das Isolieren des Proteins von Interesse aus der Kultur.
-
Eine
Mutation wie Deletion, Substitution, Insertion oder Addition von
einem bis einigen Aminosäureresten
in einer Aminosäuresequenz
kann in einem natürlich
vorkommenden Protein einschließlich
des durch die vorliegende Erfindung offenbarten Proteins gebildet
werden. Solch eine Mutation kann aufgrund eines Polymorphismus oder
einer Mutation des das Protein kodierenden Gens gebildet werden
oder sie kann aufgrund einer Modifikation des Proteins in vivo oder
während
Reinigung nach der Synthese geschehen. Dennoch ist es bekannt, dass
solch ein mutiertes Protein physiologische und biologische Aktivitäten aufweisen
kann, die äquivalent
zu denen eines Proteins ohne eine Mutation sind. Dies ist auf ein
Protein anwendbar, in das solch eine Mutation künstlich in seine Aminosäuresequenz
eingeführt
wird. In diesem Fall ist es möglich,
eine große Vielfalt
an Mutationen zu bilden. Zum Beispiel ist bekannt, dass ein Polypeptid,
in dem ein Cystein-Rest in der Aminosäuresequenz des humanen Interleukin-2
(IL-2) durch einen Serin-Rest ersetzt wird, eine Interleukin-2-Aktivität beibehält (Science,
224:1431 (1984)). Folglich kann eine Protease mit einer Aminosäuresequenz,
in der ein oder mehrere Aminosäurereste
in der Aminosäuresequenz,
offenbart durch die vorliegende Erfindung, deletiert, substituiert,
insertiert oder addiert sind, und mit einer Protease-Aktivität, die äquivalent
zu der der erfindungsgemäßen Protease
ist, gebildet werden.
-
Soweit
hierin verwendet ist "ein
Gen, das (mit einem bestimmten Gen) hybridisiert", ein Gen mit einer Basensequenz ähnlich zu
der des bestimmten Gens. Es ist wahrscheinlich, dass ein Gen mit
einer Basensequenz ähnlich
zu der eines bestimmten Gens ein Protein kodiert mit einer Aminosäuresequenz
und einer Funktion ähnlich
zu denen des durch das bestimmte Gen kodierten Proteins. Ähnlichkeiten
von Basensequenzen von Genen können
durch die Bestimmung untersucht werden, ob die Gene oder Teile davon
ein Hybrid miteinander unter stringenten Bedingungen bilden (hybridisieren)
oder nicht. Durch Ausnutzung dieses Verfahrens kann ein Gen erhalten
werden, das ein Protein kodiert mit einer ähnlichen Funktion wie der des
durch das bestimmte Gen kodierten Proteins. Das heißt, dass
ein Gen mit einer zu der des Gens der vorliegenden Erfindung ähnlichen
Basensequenz durch Verwendung des durch die vorliegende Erfindung
erhaltenen Gens oder einem Teil davon als eine Sonde zum Ausführen einer
Hybridisierung gemäß eines
bekannten Verfah rens erhalten werden kann. Hybridisierung kann gemäß des Verfahrens
ausgeführt
werden, z.B. wie beschrieben in T. Maniatis et al., Hrsg., Molecular
Cloning: A Laboratory Manual, 2. Auflage, veröffentlicht von Cold Spring
Harbor Laboratory, 1989. Insbesondere kann Hybridisierung unter
den folgenden Bedingungen ausgeführt
werden. Kurz zusammengefasst wird eine Membran, auf die DNAs immobilisiert
worden sind, in 6×SSC
(1×SSC
bedeutet 0,15 M NaCl, 0,015 M Natriumcitrat, pH 7,0) mit 0,5% SDS,
0,1% Rinderserumalbumin (BSA), 0,1% Polyvinylpyrrolidon, 0,1% Ficoll
400, 0,01% denaturierter Lachs-Sperma-DNA bei 50°C für 12-20 Stunden mit einer Sonde
inkubiert. Nach Inkubation wird die Membran gewaschen, bis die Signale
für die
immobilisierten DNAs vom Hintergrund unterschieden werden können, angefangen
mit Waschen in 2×SSC
mit 0,5% SDS bei 37°C
unter Verringerung der SSC-Konzentration bis 0,1x und Erhöhung der
Temperatur bis 50°C.
-
Alternativ
kann anstatt der Hybridisierung ein Gen-Vervielfältigungsverfahren (z.B. ein
PCR-Verfahren) verwendet werden, das Teile der Basensequenz des
durch die vorliegende Erfindung erhaltenen Gens als Primer einsetzt.
Ob das dadurch erhaltene Gen ein Protein mit der Funktion von Interesse
kodiert oder nicht, kann durch Expression des Gens unter Verwendung
eines geeigneten Wirts und eines geeigneten Expressionssystems und
Untersuchung der Aktivität
des erhaltenen Proteins bestimmt werden.
-
Kurze Beschreibung
der Zeichnungen
-
1 ist
die Restriktionsenzym-Karte des Plasmids pSTC3.
-
2 vergleicht
die Aminosäuresequenzen
der Protease PFUS, der Protease TCES und eines Subtilisins.
-
3 vergleicht
die Aminosäuresequenzen
der Protease PFUS, der Protease TCES und eines Subtilisins.
-
4 vergleicht
die Aminosäuresequenzen
der Protease PFUS, der Protease TCES und eines Subtilisins.
-
5 vergleicht
die Aminosäuresequenzen
der Protease PFUS, der Protease TCES und eines Subtilisins.
-
6 ist
die Restriktionsenzym-Karte des Plasmids pSNP1.
-
7 ist
die Restriktionsenzym-Karte des Plasmids pPS1.
-
8 ist
die Restriktionsenzym-Karte des Plasmids pNAPS1.
-
Genaue Beschreibung
der Erfindung
-
Die
erfindungsgemäße hyperthermostabile
Protease umfasst Proteasen von verschiedenen Hyperthermophilen.
Zum Beispiel beschreibt WO 95/34645 Proteasen von Pyrococcus furiosus
und Thermococcus celer.
-
Ein
Protease-Gen von Pyrococcus furiosus DSM3638 wurde aus einer genomischen
DNA-Bibliothek des Stamms basierend auf der Expression einer thermostabilen
Protease-Aktivität
isoliert. Ein dieses Gen enthaltendes Plasmid wird als Plasmid pTPR12
bezeichnet. Mit diesem Plasmid transformiertes Escherichia coli JM109
wird bezeichnet und gekennzeichnet als Escherichia coli JM109/pTPR12
und wurde am 24. Mai 1994 (Datum der ursprünglichen Hinterlegung) gemäß dem Budapester
Vertrag am Nationalen Institut für
Biowissenschaft und Humantechnologie, Amt für industrielle Wissenschaft
und Technologie, Ministerium für
internationalen Handel und Industrie, 1-3, Higashi 1-chome, Tsukuba-shi,
Ibaraki-ken, Japan unter der Zugangsnummer FERM BP-5103 hinterlegt.
-
Diese
Protease wird hierin nachstehend als Protease PFUL bezeichnet. Protease
PFUL ist eine Protease mit hoher Thermostabilität und weist eine Protease-Aktivität sogar
bei 95°C
auf.
-
Die
Basensequenz des von Pyrococcus furiosus abgeleiteten DNA-Fragments,
eingefügt
in das Plasmid pTPR12, ist bestimmt worden. Die Basensequenz des
etwa 4,8 kb großen,
von zwei DraI-Schnittstellen flankierten Teils des in das Plasmid
pTPR12 eingefügten
DNA-Fragments ist in der SEQ ID NO:5 des Sequenzprotokolls gezeigt.
Des Weiteren ist die Aminosäuresequenz
des von dieser Basensequenz abgeleiteten Genprodukts in der SEQ
ID NO:6 des Sequenzprotokolls gezeigt. Mit anderen Worten ist die
in der SEQ ID NO:6 des Sequenzprotokolls gezeigte Aminosäuresequenz,
die Aminosäuresequenz
der Protease PFUL. Wie in der Sequenz gezeigt besteht die Protease
PFUL aus 1398 Aminosäureresten
und ist eine Protease mit einem hohen Molekulargewicht von über 150
000.
-
Ein
Vergleich der Aminosäuresequenz
der Protease PFUL, wie gezeigt in SEQ ID NO:6 des Sequenzprotokolls,
mit bekannten Aminosäuresequenzen
von Proteasen von Mikroorganismen hat gezeigt, dass die Aminosäuresequenz
der ersten Hälfte
der Protease PFUL homolog zu denen einer Reihe von alkalischen Serin-Proteasen
ist, dargestellt durch ein Subtilisin (Protein Engineering, 4:719-737
(1991)). Es gibt eine extrem hohe Homologie um die vier Aminosäurereste,
von denen angenommen wird, dass sie wichtig für die katalytische Aktivität der Protease
sind.
-
Wie
vorstehend beschrieben ist herausgefunden worden, dass eine unter
Proteasen abgeleitet von Mesophilen gemeinsame Region in der Aminosäuresequenz
der Protease PFUL, hergestellt durch ein hyperthermophiles Pyrococcus
furiosus, konserviert ist. Daher wird davon ausgegangen, dass eine
durch ein anderes Hyperthermophil als Pyrococcus furiosus hergestellte
Protease ebenfalls diese Region aufweist.
-
Zum
Beispiel kann ein Gen für
eine hyperthermostabile Protease mittels Durchführung einer PCR unter Verwendung
einer chromosomalen DNA von verschiedenen Hyperthermophilen als
ein Template und der Oligonukleotide PRO-1F, PRO-2F, PRO-2R und
PRO-4R in Kombination als Primer gescreent werden. Diese Oligonukleotide
werden basierend auf der Basensequenz im Gen der Protease PFUL synthetisiert,
die eine Region in der Aminosäuresequenz
der Protease PFUL kodiert, die hohe Homologie mit Subtilisinen oder
dergleichen aufweist. Die Basensequenzen der Oligonukleotide PRO-1F,
PRO-2F, PRO-2R und PRO-4R sind in den SEQ ID NOs: 7, 8, 9 bzw. 10
des Sequenzprotokolls gezeigt.
-
Als
ein Hyperthermophil, von dem die erfindungsgemäße Protease abgeleitet ist,
kann ein Bakterium verwendet werden, das zur Gattung Pyrococcus,
Thermococcus, Staphylothermus, Thermobacteroides und dergleichen
gehört.
Als ein Bakterium, das zur Gattung Thermococcus gehört, kann
z.B. Thermococcus celer DSM2476 verwendet werden. Dieser Stamm ist
von der Deutschen Sammlung von Mikroorganismen und Zellkulturen
GmbH erhältlich.
Bei Durchführung
einer PCR unter Verwendung einer chromosomalen DNA von Thermococcus
celer DSM2476 als ein Template und einer Kombination der Oligonukleotide
PRO-1F und PRO-2R oder der Oligonukleotide PRO-2F und PRO-4R als
Primer werden spezifische DNA-Fragmente amplifiziert, was die Anwesenheit
eines Protease-Gens anzeigt. Des weiteren können durch Herstellung rekombinanter
Plasmide, in denen die DNA-Fragmente in einen geeigneten Plasmid-Vektor
eingefügt
sind, und Bestimmung der Basensequenzen der eingefügten DNA-Fragmente
durch ein Didesoxy-Verfahren, die durch die Fragmente kodierten
Aminosäuresequenzen
abgeleitet werden. Als Ergebnis ist es erwiesen, dass solche DNA-Fragmente
eine Aminosäuresequenz
kodieren, die homolog zu den Aminosäuresequenzen der Protease PFUL
und alkalischer Serinproteasen von verschiedenen Mikroorganismen
ist, und dass die PCR-amplifizierten DNA-Fragmente von einem Protease-Gen
als ein Template amplifiziert wurden.
-
Als
nächstes
kann ein Gen für
eine hyperthermostabile Protease (zum Beispiel ein Gen für eine hyperthermostabile
Protease hergestellt durch Thermococcus celer) durch Screenen einer
Gen-Bibliothek von einem Hyperthermophilen unter Verwendung des
PCR-amplifizierten DNA-Fragments oder des Oligonukleotids wie vorstehend
beschrieben als eine Sonde erhalten werden.
-
Zum
Beispiel kann ein das Gen von Interesse enthaltender Phagen-Klon
mittels Durchführen
einer Plaque-Hybridisierung gegen eine Bibliothek unter Verwendung
des PCR-amplifizierten DNA-Fragments als eine Sonde erhalten werden.
Solch eine Bibliothek wird durch Ligation von lambda-GEM-11-Vektor
(Promega) und DNA-Fragmenten,
die aus einem teilweisen Verdau der chromosomalen DNA von Thermococcus
celer DSM2476 mit dem Restriktionsenzym Sau3AI erhalten wurden,
und dann durch Verpackung von diesen in lambda-Phagenpartikel mittels
eines in vitro-Verpackungsverfahrens
gebildet.
-
Es
ist durch Analyse eines DNA-Fragments, das in einem so erhaltenen
Phagen-Klons enthalten
ist, herausgefunden worden, dass ein Protease-Gen in einem SacI-Fragment von etwa
1,9 kb enthalten ist. Des weiteren ist durch Bestimmung seiner Basensequenz
herausgefunden worden, dass diesem Fragment die 5'-Region des Protease-Gens
fehlt. Die 5'-Region
kann durch PCR unter Verwendung einer Kassette und von Kassettenprimern
erhalten werden (Takara Shuzo Gene Technology Product Guide, 1994-1995,
Seiten 250-251). Folglich kann ein DNA-Fragment erhalten werden,
das die 5'-Region
des Gens der hypothermostabilen Protease umfasst, die in dem Plasmid
pTCS6 nicht vorhanden ist. Des weiteren kann die Basensequenz des
gesamten Gens der hyperthermostabilen Protease abgeleitet von Thermococcus
celer aus den Basensequenzen der zwei DNA-Fragmente bestimmt werden.
-
Die
Basensequenz eines in der bestimmten Basensequenz gefundenen offenen
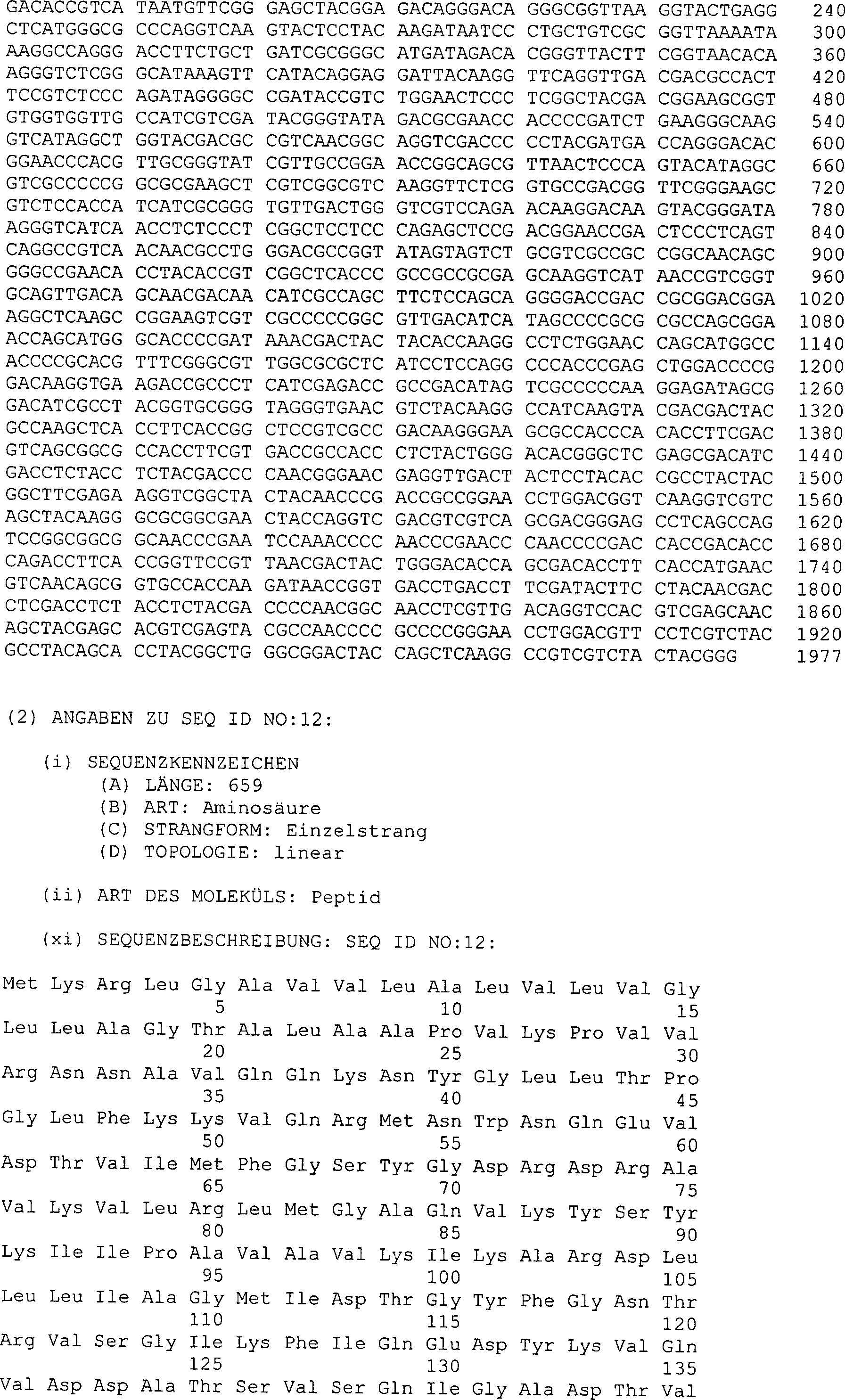
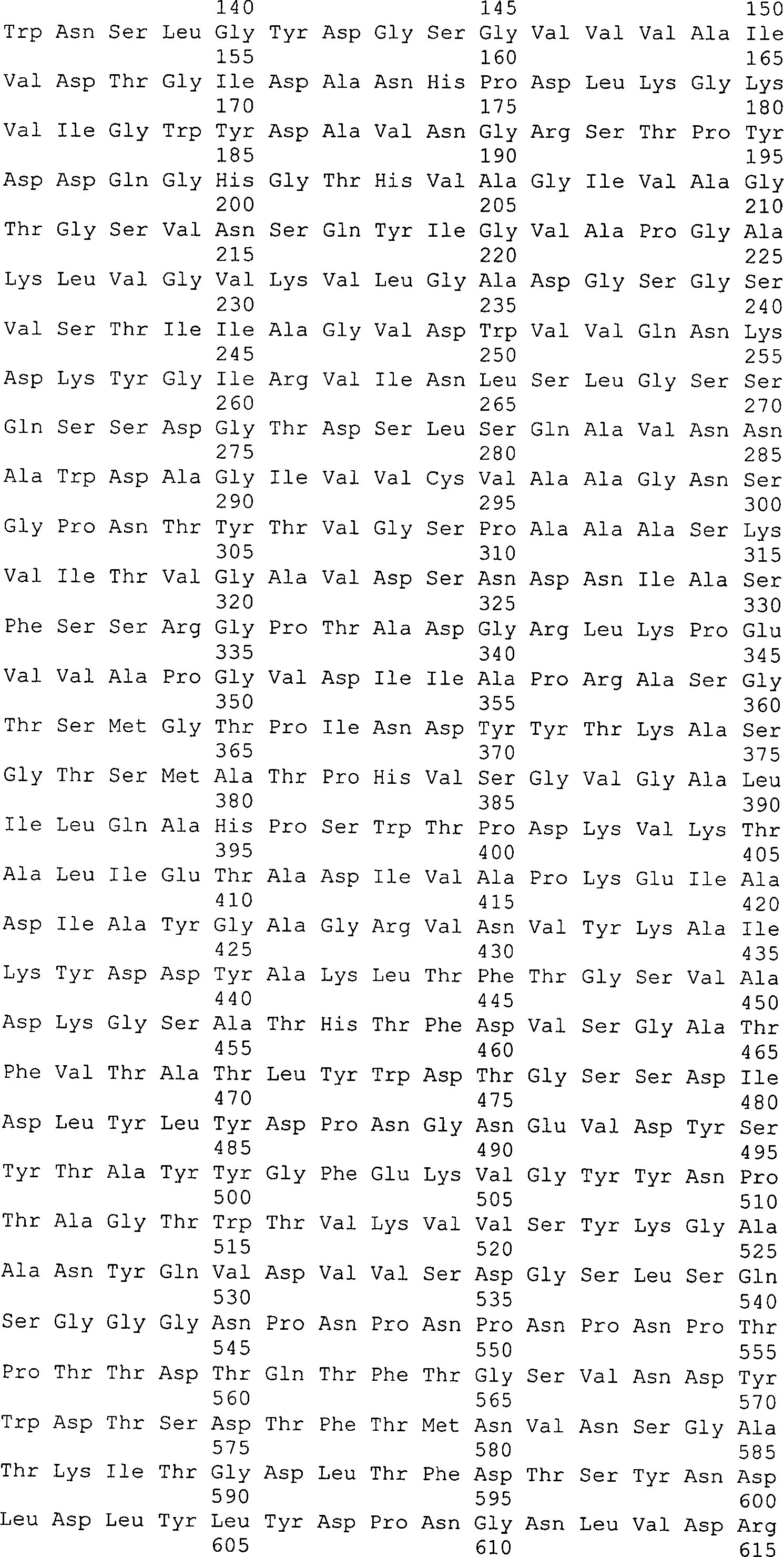
Leserahmens ist in der SEQ ID NO:11 des Sequenzprotokolls gezeigt,
und die von die ser Basensequenz abgeleitete Aminosäuresequenz
ist in der SEQ ID NO:12 des Sequenzprotokolls gezeigt. Die Basensequenz
des Gens, das die hyperthermostabile Protease von Thermococcus celer
kodiert, und die Aminosäuresequenz
der Protease wurden daher bestimmt. Diese Protease wird als Protease
TCES bezeichnet.
-
Ein
Expressionsvektor, in dem das gesamte Gen der Protease TCES durch
Kombinieren der zwei DNA-Fragmente wiederhergestellt worden ist,
kann konstruiert werden. Als jedoch Escherichia coli als Wirt verwendet
wurde, wurden keine Transformanten erhalten, in die das Expressionsplasmid
von Interesse eingeführt
worden war, wahrscheinlich weil die Bildung des von dem Gen exprimierten
Produkts in Zellen schädlich oder
tödlich
für Escherichia
coli sein kann. In solch einem Fall ist es z.B. möglich, Bacillus
subtilis als Wirt für eine
extrazelluläre
Sekretion der Protease zu verwenden und die Aktivität zu bestimmen.
-
Als
ein Bacillus subtilis-Stamm kann Bacillus subtilis DB 104 verwendet
werden, der ein bekannter Stamm ist wie beschrieben in Gene, 83:215-233
(1989). Als ein Klonierungsvektor kann das Plasmid pUB 18-P43 verwendet
werden, das ein Geschenk von Dr. Sui-Lam Wong, University of Calgary,
ist. Das Plasmid enthält
ein Kanamycin-Resistenzgen als einen selektierbaren Marker.
-
Ein
rekombinantes Plasmid, in dem das Gen der Protease TCES downstream
des Promoters P43 in dem Plasmid-Vektor pUB 18-P43 eingefügt ist,
wird als Plasmid pSTC3 bezeichnet. Mit diesem Plasmid transformiertes
Bacillus subtilis DB 104 wird bezeichnet und gekennzeichnet als
Bacillus subtilis DB 104/pSTC3 und wurde am 1. Dezember 1995 (Datum
der ursprünglichen
Hinterlegung) gemäß dem Budapester
Vertag am Nationalen Institut für
Biowissenschaft und Humantechnologie, Amt für Industrielle Wissenschaft
und Technologie, Ministerium für
Internationalen Handel und Industrie, 1-3, Higashi 1-chome, Tsukuba-shi,
Ibaraki-ken, Japan unter der Zugangsnummer FERM BP-5635 hinterlegt.
-
Die
Restriktionsenzym-Karte des Plasmids pSTC3 ist in 1 gezeigt.
In 1 kennzeichnet die breite Linie das DNA-Fragment,
das in den Plasmid-Vektor pUB 18-P43 eingefügt worden ist.
-
Eine
thermostabile Protease-Aktivität
ist sowohl im Überstand
als auch im Zellextrakt der Kultur von Bacillus subtilis DB104/pSTC3
gefunden worden.
-
Die
Haupteigenschaften einer ungereinigten Enzym-Präparation der Protease erhalten
von der Kultur des Transformanten sind wie folgt.
-
(1) Aktivität:
-
Baut
Casein und Gelatine unter Bildung kurzkettiger Polypeptide ab.
-
Hydrolysiert
Succinyl-L-leucyl-L-leucyl-L-valyl-L-tyrosin-4-methylcoumarin-7-amid (Suc-Leu-Leu-Val-Tyr-MCA)
unter Bildung einer fluoreszierenden Substanz (7-Amino-4-methylcoumarin).
-
Hydrolysiert
Succinyl-L-alanyl-L-alanyl-L-prolyl-L-phenylalanin-p-nitroanilid (Suc-Ala-Ala-Pro-Phe-p-NA)
unter Bildung einer gelben Substanz (p-Nitroanilin).
-
(2) Optimale Temperatur:
-
Weist
eine enzymatische Aktivität
bei 37-95°C
mit der optimalen Temperatur bei 70-80°C auf.
-
(3) Optimaler pH:
-
Weist
eine enzymatische Aktivität
bei pH 5,5-9 mit dem optimalen pH bei pH 7-8 auf.
-
(4) Thermostabilität:
-
Behält 90% oder
mehr von ihrer enzymatischen Aktivität nach Behandlung bei 80°C für 3 Stunden.
-
Beim
Abgleich der Aminosäuresequenzen
der Protease PFUL, der Protease TCES und eines Subtilisins (Subtilisin
BNP'; Nucl. Acids
Res., 11:7911-7925 (1983)), so dass homologe Regionen sich zueinander anordnen
wie gezeigt in 2-5, wurde
herausgefunden, dass am C-Terminus und zwischen den homologen Regionen
der Protease PFUL Sequenzen liegen, die nicht in der Protease TCES
oder dem Subtilisin gefunden werden. Nach diesen Ergebnissen kann
eine Protease mit einem Molekulargewicht geringer als das der Protease
PFUL und ähnlich
zu dem der Protease TCES oder von Subtilisinen in Pyrococcus furiosus
zusätzlich
zur Protease PFUL vorkommen.
-
Daraufhin
wurde eine Southern-Hybridisierung gegen eine chromosomale DNA hergestellt
aus Pyrococcus furiosus unter Verwendung einer DNA-Sonde aus der
homologen Region durchgeführt.
Es wurde ein Signal beobachtet, das unterschiedlich von dem für das Gen
der Protease PFUL war, was auf die Existenz eines anderen Protease-Gens
hinweist.
-
Dieses
neue Protease-Gen kann durch das folgende Verfahren isoliert werden.
-
Zum
Beispiel wird ein DNA-Fragment, das ein für die neue Protease kodierendes
Gen enthält,
durch Verdau einer chromosomalen DNA von Pyrococcus furiosus mit
einem geeigneten Restriktionsenzym und Durchführung einer Southern-Hybridisierung
gegen die verdaute DNA wie vorstehend beschrieben erhalten. Die
Basensequenz des DNA-Fragments wird bestimmt, um zu bestätigen, dass
die Basensequenz eine Aminosäuresequenz
kodiert, die homolog zu der vorstehend genannten Protease ist. Wenn
das DNA-Fragment nicht das gesamte Gen von Interesse enthält, wird
der restliche Teil des weiteren durch ein inverses PCR-Verfahren
oder dergleichen erhalten.
-
Wenn
zum Beispiel eine chromosomale DNA von Pyrococcus furiosus mit den
Restriktionsenzymen SacI und SpeI (Takara Shuzo) verdaut und für eine Southern-Hybridisierung verwendet
wird, wird ein Signal mit einer Größe von etwa 0,6 kb beobachtet.
DNA-Fragmente dieser Größe werden
isoliert und zwischen die SpeI-SacI-Stellen in den Plasmid-Vektor pBluescript
SK(–)
(Stratagene) eingefügt,
und Escherichia coli JM 109 wird mit den erhaltenen rekombinanten
Plasmiden transformiert. Ein Klon, in den das Fragment von Interesse eingebaut
worden ist, kann von den Transformanten durch Kolonie-Hybridisierung
unter Verwendung der gleichen Sonde wie die, die für die Southern-Hybridisierung
wie vorstehend beschrieben verwendet wurde, erhalten werden. Ob
das in den erhaltenen Klonen enthaltene Plasmid die Sequenz hat,
die die Protease kodiert oder nicht, kann durch Bestimmung der Basensequenz
des DNA-Fragments, das in das Plasmid eingefügt worden ist, bestätigt werden.
Die Anwesenheit des Protease-Gens in dem Plasmid wurde dadurch bestätigt. Dieses
Plasmid wird als das Plasmid pSS3 bezeichnet.
-
Es
ist herausgefunden worden, dass die Aminosäuresequenz, die von der Basensequenz
des in das Plasmid pSS3 eingefügten
DNA-Fragments abgeleitet worden ist, Homologie mit Sequenzen von
Subtilisinen, der Protease PFUL, der Protease TCES und dergleichen
hat. Das Produkt des von dem Gen der Protease PFUL unterschied lichen
Protease-Gens, von dem ein Teil neu von Pyrococcus furiosus wie
vorstehend beschrieben erhalten worden war, wird als Protease PFUS
bezeichnet. Die Regionen, die die N-terminalen und C-terminalen
Regionen der Protease kodieren, können durch ein inverses PCR-Verfahren
erhalten werden.
-
Für eine inverse
PCR verwendete Primer können
basierend auf der Basensequenz des DNA-Fragments, das in das Plasmid
pSS3 eingefügt
worden ist, hergestellt werden. Eine chromosomale DNA von Pyrococcus
furiosus wird mit einem geeigneten Restriktionsenzym verdaut und
die erhaltenen DNA-Fragmente werden dann einer intramolekularen
Ligationsreaktion unterworfen. Durch Durchführung einer PCR unter Verwendung
des Reaktionsgemisches als Template und der vorstehend genannten
Primer können
DNA-Fragmente erhalten werden, die den Regionen entsprechen, die
das Fragment des Protease-Gens flankieren, das in dem Plasmid pSS3
enthalten ist. Die Aminosäuresequenz
des durch diese Regionen kodierten Enzymproteins kann durch Analyse
der Basensequenzen der so erhaltenen DNA-Fragmente abgeleitet werden. Des weiteren
können
Primer hergestellt werden, die zur Amplifikation des gesamten Gens
der Protease PFUS unter Verwendung einer chromosomalen DNA von Pyrococcus
furiosus als Template imstande sind. Die Primer NPF-4 und NPR-4
können
entworfen werden. Der Primer NPF-4 hat die Basensequenz, die direkt
upstream des Anfangscodons des Gens der Protease PFUS liegt, und
kann eine BamHI-Schnittstelle 5' zu
der Sequenz einfügen.
Der Primer NPR-4 hat eine Sequenz, die komplementär zu dem
3'-Teil des Gens
der Protease PFUS ist, und kann eine SphI-Schnittstelle 5' zu der Sequenz einfügen.
-
Die
Basensequenzen der Primer NPF-4 und NPR-4 sind in den SEQ ID NOs:
13 und 14 des Sequenzprotokolls gezeigt. Diese zwei Primer können zur
Amplifikation des gesamten Gens der Protease PFUS unter Verwendung
einer chromosomalen DNA von Pyrococcus furiosus als Template verwendet
werden.
-
Wie
die Protease TCES kann die Protease PFUS in Bacillus subtilis als
Wirt exprimiert werden. Ein Plasmid zur Expression der Protease
PFUS kann basierend auf dem Expressionsplasmid für die Protease TCES, pSTC3,
hergestellt werden. Insbesondere kann ein Plasmid zur Expression
der Protease PFUS durch Ersetzen des Gens der Protease TCES im Plasmid
pSTC3 mit dem DNA-Fragment, das das gesamte Gen der Protease PFUS
enthält
und durch PCR mit den Primern wie vorstehend beschrieben amplifiziert
worden ist, hergestellt werden. Das so hergestellte Expressionsplasmid
wird als das Plasmid pSNP1 bezeichnet. Mit diesem Plasmid transformiertes
Bacillus subtilis DB 104 wird bezeichnet und gekennzeichnet als
Bacillus subtilis DB104/pSNP1 und wurde am 1. Dezember 1995 (Datum
der ursprünglichen
Hinterlegung) gemäß dem Budapester
Vertrag am Nationalen Institut für
Biowissenschaft und Humantechnologie, Amt für Industrielle Wissenschaft
und Technologie, Ministerium für
Internationalen Handel und Industrie, 1-3, Higashi 1-chome, Tsukuba-shi,
Ibaraki-ken, Japan unter der Zugangsnummer FERM BP-5634 hinterlegt.
Die Restriktionsenzym-Karte des Plasmids pSNP1 ist in 6 gezeigt.
-
Die
Basensequenz, die einem offenen Leserahmen in dem für die Protease
PFUS kodierenden Gen entspricht, und die Aminosäuresequenz der Protease PFUS,
die von der Basensequenz abgeleitet ist, sind in den SEQ ID NOs:
15 bzw. 16 des Sequenzprotokolls bezeichnet.
-
Eine
thermostabile Protease-Aktivität
wurde sowohl in dem Überstand
als auch in dem Zellextrakt der Kultur von Bacillus subtilis DB104/pSNP1
gefunden. Das heißt,
dass ein Teil der exprimierten Protease PFUS in den Überstand
der Kultur sekretiert wird.
-
Die
Haupteigenschaften der Protease, die aus der Kultur des Transformanten
erhalten worden ist, sind wie folgt.
-
(1) Aktivität:
-
Baut
Casein und Gelatine unter Bildung kurzkettiger Polypeptide ab.
-
Hydrolysiert
Succinyl-L-leucyl-L-leucyl-L-valyl-L-tyrosin-4-methylcoumarin-7-amid (Suc-Leu-Leu-Val-Tyr-MCA)
unter Bildung einer fluoreszierenden Substanz (7-Amino-4-methylcoumarin).
-
Hydrolysiert
Succinyl-L-alanyl-L-alanyl-L-prolyl-L-phenylalanin-p-nitroanild (Suc-Ala-Ala-Pro-Phe-p-NA)
unter Bildung einer gelben Substanz (p-Nitroanilin).
-
(2) Optimale Temperatur:
-
Weist
eine enzymatische Aktivität
bei 40-110°C
mit der optimalen Temperatur von 80-95°C auf.
-
(3) Optimaler pH:
-
Weist
eine enzymatische Aktivität
bei pH 5-10 mit dem optimalen pH bei pH 6-8 auf.
-
(4) Thermostabilität:
-
Behält 90% oder
mehr von ihrer enzymatischen Aktivität nach Behandlung bei 95°C für 8 Stunden.
-
(5) pH-Stabilität:
-
Behält 95% oder
mehr von ihrer Aktivität
nach Behandlung bei pH 5-11 bei 95°C für 60 Minuten.
-
(6) Molekulargewicht:
-
Weist
ein Molekulargewicht von etwa 45 kDa auf einer SDS-PAGE auf.
-
Protease-Gene,
die homolog zu dem Gen der Protease TCES und dem Gen der Protease
PFUS sind, können
aus anderen Hyperthermophilen als Pyrococcus furiosus und Thermococcus
celer unter Verwendung eines Verfahrens erhalten werden, das ähnlich zu
dem ist, das zum Erhalten des Gens der Protease TCES und des Gens
der Protease PFUS verwendet worden ist.
-
Ein
DNA-Fragment von etwa 1 kb, das eine Sequenz von dem Rest an Position
323 zu dem Rest an Position 650 der Aminosäure-Sequenz der Protease PFUL
wie in SEQ ID NO:6 des Sequenzprotokolls gezeigt kodiert, kann hergestellt
werden und als eine Sonde für
eine genomische Southern-Hybridisierung gegen chromosomale DNAs
von Staphylothermus marinus DSM3639 und Thermobacteroides proteoliticus
DSM 5265 verwendet werden. Als ein Ergebnis werden Signale an der
Position von etwa 4,8 kb für
die mit PstI (Takara Shuzo) verdaute chromosomale DNA von Staphylothermus
marinus und an der Position von etwa 3,5 kb für die mit XbaI verdaute chromosomale
DNA von Thermobacteroides proteoliticus beobachtet.
-
Mit
diesen Ergebnissen wurde bewiesen, dass es Sequenzen auf den chromosomalen
DNAs von Staphylothermus marinus und Thermobacteroides proteoliticus
gibt, die homolog zu denen der Gene der Protease PFUL, der Protease
PFUS, der Protease TCES und dergleichen sind. Die die hyperthermostabilen
Proteasen kodierenden Gene in Staphylothermus marinus und Thermobacteroides
proteoliticus können
von den so detektierten DNA-Fragmenten durch Verwenden eines Verfahrens
isoliert und identifiziert werden, das zur Isolierung und Identifizierung
der die Protease TCES und die Protease PFUS kodierenden Gene verwendet
wurde.
-
Im
Allgemeinen wird davon ausgegangen, dass die Verwendung eines Promotors,
der wirksam in einem Wirt arbeitet, vorteilhaft gegenüber einem
Promotor, der inhärent
mit dem das Protein von Interesse kodierenden Gen assoziiert ist,
zur Herstellung eines Proteins in einer großen Menge durch Gentechnik
sein würde.
Obwohl der P43-Promotor, der zur Herstellung der Expressionssysteme
für die
Protease TCES und die Protease PFUS verwendet wurde, ein von Bacillus
subtilis abgeleiteter Promotor ist, war er für die Expression der zwei Proteasen
nicht ausreichend wirksam.
-
Daher
kann ein Gen, das zu einem hohen Grad in Bacillus subtilis exprimiert
wird, insbesondere ein Gen für
ein sekretiertes Protein, zur Erhöhung des Expressionsgrads verwendet
werden. Gene für α-Amylase oder
verschiedene extrazelluläre
Proteasen können
verwendet werden. Zum Beispiel wird erwartet, dass die Verwendung
eines Promotors und einer Signalpeptid-kodierenden Region eines
Subtilisin-Gens
den Expressionsgrad der Protease PFUS erhöhen kann.
-
Insbesondere
kann die Protease PFUS als ein Fusionsprotein unter Kontrolle des
Promotors des Subtilisin-Gens durch Platzieren des gesamten Gens
der Protease PFUS downstream der Region, die das Signalpeptid des
Subtilisin-Gens kodiert, einschließlich der Promotorregion exprimiert
werden, so dass die Translationsrahmen der zwei Gene übereinstimmen.
-
Zum
Beispiel kann das Gen, das Subtilisin E kodiert, als das erfindungsgemäß verwendete
Subtilisin-Gen verwendet werden. Der Promotor und die Signalpeptid-kodierende
Region des Subtilisin E-Gens, die in das Plasmid pKWZ wie beschrieben
in J. Bacteriol., 171:2657-2665 (1989) eingefügt worden sind, können verwendet
werden. Die Basensequenz der 5'-upstream-Region
einschließlich
der Promotorsequenz ist in der Referenz (supra) beschrieben und
die Basensequenz der Region, die Subtilisin kodiert, ist in J. Bacteriol., 158:411-418
(1984) beschrieben.
-
Basierend
auf diesen Sequenzen werden der Primer SUB4 zum Einfügen einer
EcoRI-Schnittstelle upstream von der Promotorsequenz des Gens und
der Primer BmR1 zum Einfügen
einer BamHI-Schnittstelle downstream der das Signalpeptid von Subtilisin
E kodierenden Region synthetisiert. Die Basensequenzen der Primer
SUB4 und BmR1 sind in den SEQ ID NOs:17 bzw. 18 des Sequenzprotokolls
gezeigt. Die Primer SUB4 und BmR1 können zur Amplifikation eines
DNA-Fragments von etwa 0,3 kb, das den Promotor und die Signalpeptid-kodierende
Region des Subtilisin E-Gens enthält, mittels PCR unter Verwendung
des Plasmids pKWZ als Template verwendet werden.
-
Das
Gen der Protease PFUS, das downstream des DNA-Fragments eingefügt werden
soll, kann von einer chromosomalen DNA von Pyrococcus furiosus durch
ein PCR-Verfahren
erhalten werden. Der Primer NPF-4 kann als ein Primer verwendet
werden, der mit der 5'-Region
des Gens hybridisiert. Der Primer NPM-1, der basierend auf der Basensequenz
downstream des Terminator-Codons des Gens entwickelt wurde und eine
SphI-Schnittstelle hat, kann als ein Primer verwendet werden, der
mit der 3'-Region
des Gens hybridisiert. Diese Sequenz des Primers NPM-1 ist in der
SEQ ID NO:19 des Sequenzprotokolls gezeigt.
-
Eine
im Gen vorhandene BamHI-Schnittstelle würde ein Problem für ein Verfahren
darstellen, in dem eine BamHI-Schnittstelle zum Verbinden des Gens
der Protease PFUS mit dem 0,3 kb-DNA-Fragment verwendet wird. Die
Primer mutRR und mutFR zum Entfernen der BamHI-Schnittstelle durch
ein PCR-Mutageneseverfahren können
basierend auf der Basensequenz des Gens der Protease PFUS wie gezeigt
in der SEQ ID NO:15 des Sequenzprotokolls hergestellt werden. Die
Basensequenzen der Primer mutRR und mutFR sind in den SEQ ID NOs:20
bzw. 21 des Sequenzprotokolls gezeigt. Wenn diese Primer zum Entfernen
der BamHI-Schnittstelle verwendet werden, wird der durch diese Stelle
kodierte Aminosäurerest
(d.h. Glycin an Position 560 in der Aminosäuresequenz der Protease PFUS
wie gezeigt in der SEQ ID NO:16 des Sequenzprotokolls), aufgrund
der in diese Stelle eingefügten
Basensubstitution durch Valin ersetzt.
-
Das
Gen der Protease PFUS, das mit dem Promotor und der Signalpeptid-kodierenden
Region des Subtilisin E-Gens verbunden werden soll, kann durch Verwendung dieser
Primer erhalten werden. Insbesondere werden zwei PCRs unter Verwendung
einer chromosomalen DNA von Pyrococcus furiosus als Template und
des Primerpaares mutRR und NPF-4 oder des Primerpaares mutFR und
NPM-1 durchgeführt.
Zusätzlich wird
eine zweite PCR unter Verwendung eines Heteroduplexes, der durch
Vermischen der entsprechenden PCR-amplifizierten DNA-Fragmente gebildet
wird, als Template und der Primer NPF-4 und NPM-1 durchgeführt. Folglich
kann das gesamte Gen der Protease PFUS mit etwa 2,4 kb amplifiziert
werden, ohne dass es eine interne BamHI-Schnittstelle enthält.
-
Ein
DNA-Fragment mit etwa 2,4 kb, das durch Verdau des PCR-amplifizierten
DNA-Fragments mit BamHI
und SphI erhalten wird, wird isoliert und verwendet, um ein BamHI-SphI-Fragment
in dem Plasmid pSNP1 zu ersetzen, das das Gen der Protease PFUS
enthält.
Ein so hergestellter Expressionsvektor wird als das Plasmid pPS1
bezeichnet. Mit diesem Plasmid transformiertes Bacillus subtilis
DB 104 wird als Bacillus subtilis DB104/pPS1 bezeichnet. Eine Protease-Aktivität wird in
sowohl dem Überstand
als auch dem Zellextrakt der Kultur dieses Transformanten gefunden,
die ähnlich
ist zu der, die für
den das Plasmid pSNP1 beinhaltenden Transformanten beobachtet wurde.
Dies zeigt, dass die Aminosäure-Substitution
die enzymatische Aktivität
nicht beeinflusst. Die Restriktionsenzym-Karte des Plasmids pPS1
ist in 7 gezeigt.
-
Das
DNA-Fragment von etwa 0,3 kb, das den Promotor und die Signalpeptid-kodierende
Region des Subtilisin E-Gens enthält, wird mit EcoRI und BamHI
verdaut und verwendet, um das EcoRI-BamHI-Fragment, das den Promotor
P43 und eine Ribosom-Bindestelle enthält, in dem Plasmid pPS1 zu
ersetzen. Ein so hergestelltes Expressionsplasmid wird als pNAPS1
bezeichnet. Mit diesem Plasmid transformiertes Bacillus subtilis
DB 104 wird als Bacillus subtilis DB 104/pNAPS1 bezeichnet. Eine
thermostabile Protease-Aktivität
wird sowohl im Überstand
als auch im Zellextrakt der Kultur des Transformanten gefunden,
wobei der Expressionsgrad verglichen mit dem von Bacillus subtilis
DB104/pSNP1 erhöht
ist. Die Restriktionsenzym-Karte des Plasmids pNAPS1 ist in 8 gezeigt.
-
Die
von dem Transformanten exprimierte Protease weist enzymatische Eigenschaften
auf, die äquivalent
zu denen der durch Bacillus subtilis DB104/pSNP1 exprimierten Protease
wie vorstehend beschrieben sind. Die durch den Transformanten exprimierte
Protease wurde gereinigt. Die Analyse der N-terminalen Aminosäuresequenz
der gereinigten Protease lieferte die Aminosäuresequenz wie gezeigt in der SEQ
ID NO:22 des Sequenzprotokolls. Diese Sequenz ist identisch mit
der Sequenz von Position 133 bis Position 144 der Aminosäuresequenz
der Protease PFUS wie gezeigt in der SEQ ID NO:15 des Sequenzprotokolls,
was zeigt, dass die vollprozessierte Protease PFUS ein Enzym ist,
das aus einem Polypeptid besteht, das an dieser Position startet.
Die aufgrund dieser Ergebnisse angenommene Aminosäuresequenz
der vollprozessierten Protease PFUS ist in der SEQ ID NO:4 des Sequenzprotokolls
gezeigt.
-
Obwohl
die Menge an durch Bacillus subtilis DB 104/pNAPS1 gebildeter Protease
im Vergleich mit der Menge der durch Bacillus subtilis DB104/pSNP1
(FERM BP-5634) gebildeten Protease erhöht ist, ist eine höhere Produktivität wünschenswert.
Es wird erwartet, dass der Expressionsgrad der Protease durch Modifizieren
der Verbindung in dem durch pNAPS1 kodierten fusionierten Peptid
zwischen dem Signalpeptid des Subtilisins und der Protease PFUS
zur Steigerung der Effizienz beim Entfernen des Signalpeptids erhöht wird. Dem
Plasmid pNAPS1 wird ein Peptid, das aus den drei Aminosäureresten
Ala-Gly-Ser besteht, zwischen den C-terminalen Aminosäurerest
des Signalpeptids von Subtilisin E wie gezeigt in der SEQ ID NO:3
des Sequenzprotokolls (Ala) und dem N-terminalen Aminosäurerest
der Protease PFUS (Met) eingefügt.
Ein Transformant mit erhöhtem
Expressionsgrad der Protease kann durch Einführen einer Mutation in die
dieses Peptid kodierende DNA in das Plasmid pNAPS1 und durch Untersuchen
der Protease-Produktivität
des Transformanten, in den das mutierte Plasmid eingefügt wurde,
erhalten werden.
-
Zuerst
wird ein mutiertes Plasmid hergestellt, indem der Ser-kodierende
Teil in dem 3-Aminosäurepeptid
in dem Gen, das das Fusionsprotein, Subtilisin E-Protease PFUS,
kodiert, in dem Plasmid pNAPS1 so modifiziert wird, dass die Basensequenz
des Teils zwei zufällige
Aminosäurereste
kodiert. Solch ein mutiertes Plasmid kann mittels PCR hergestellt
werden. Zum Beispiel können
die Primer SPOF0 und SPOR0 mit Sequenzen, in denen das Ser-kodierende
Codon (TCC) durch sechs zufällige
Basen substituiert ist (die Basensequenzen der Primer SPOF0 und
SPOR0 sind in den SEQ ID NOs:24 bzw. 25 des Sequenzprotokolls gezeigt),
und die Primer SUB3 und NPR-10, die basierend auf der Basensequenz
um diese Region herum hergestellt worden sind (die Basensequenzen
der Primer SUB3 und NPR-10 sind in den SEQ ID NOs:26 bzw. 27 des
Sequenzprotokolls gezeigt), zur Durchführung einer PCR zum Erhalten
eines DNA-Fragments verwendet werden, in das die beabsichtigte Mutation
an der dem Ser-kodierenden Codon (TCC) entsprechenden Stelle eingefügt wurde.
Ein mutiertes Plasmid, das das Protease-Gen mit der eingefügten Mutation enthält, kann durch
Ersetzen der entsprechenden Region in dem Plasmid pNAPS1 mit dem
erhaltenen Fragment erhalten werden.
-
Ein
Transformant mit erhöhtem
Expressionsgrad kann dann durch Einführen der so erhaltenen mutierten
Plasmide in einen geeigneten Wirt, z.B. Bacillus subtilis DB 104,
und durch Bestimmen der Menge der durch die Transformanten exprimierten
Protease erhalten werden. Der Expressionsgrad der Protease kann durch
Bestimmen der Aktivität
in der unabhängigen
Kultur des isolierten Transformanten bestätigt werden. Alternativ kann
ein Transformant mit erhöhtem
Expressionsgrad leicht durch Verwenden einer Agarplatte ausgewählt werden,
die ein Substrat enthält.
-
Insbesondere
werden die Transformanten, in die die mutierten Plasmide eingefügt worden
sind, auf Agarplatten wachsen gelassen, die Magermilch enthalten.
Danach werden die Platten bei einer Temperatur inkubiert, bei der
die Protease PFUS ihre Aktivität
zeigt, z.B. bei 70°C.
Magermilch um eine Kolonie eines Transformanten, der eine Protease
exprimiert, wird abgebaut und dadurch klar. Der Expressionsgrad
der Protease kann aus der Größe des klaren
Bereichs abgeschätzt
werden.
-
Einer
der so erhaltenen Transformanten, der einen hohen Grad an Protease-Aktivität verglichen
mit Bacillus subtilis DB104/pNAPS1 exprimiert, wird als Bacillus
subtilis DB 104/pSPO124 bezeichnet. Das in diesem Transformanten
enthaltene Plasmid wurde präpariert
(dieses Plasmid wird als pSPO124 bezeichnet). Eine Analyse der Basensequenz
des Plasmids zeigte, dass der Ser-kodierende Teil in die Basensequenz GGGAAT
geändert
wurde, d.h. dass ein Protein durch das Plasmid kodiert wurde, indem
Ser in Gly-Asn geändert
wurde.
-
Folglich
wurde bewiesen, dass der Expressionsgrad des Proteins von Interesse
in einem Bakterium der Gattung Bacillus als Wirt dadurch erhöht werden
kann, dass ein Peptid, das aus den vier Aminosäureresten Ala-Gly-Gly-Asn besteht,
downstream des Signalpeptids eines Subtilisins platziert wird, es
an den N-Terminus des Proteins von Interesse fusioniert wird und
das fusionierte Protein exprimiert wird. Zusätzlich zu Subtilisin E (von
Bacillus subtilis), das in der vorliegenden Erfindung verwendet
wird, sind Subtilisin BPN' von
Bacillus amyloliquefaciens (Nucl. Acids Res., 11:7911-7925 (1983)),
Subtilisin Carlsberg von Bacillus licheniformis (Nucl. Acids Res.,
13:8913-8926 (1985)) und dergleichen als Subtilisine bekannt, die
durch Bakterien der Gattung Bacillus hergestellt werden. Die Signalpeptide
von ih nen können
vorzugsweise für
die vorliegende Erfindung verwendet werden, obwohl ihre Aminosäuresequenzen
voneinander leicht variieren. Verschiedene Promotoren, die in einem
Bakterium der Gattung Bacillus wirken, können anstatt des Promotors
des Subtilisin E-Gens verwendet werden, das in der vorliegenden
Erfindung zur Expressionskontrolle verwendet wird.
-
Das
zu exprimierende Protein ist nicht eingeschränkt. Es ist möglich, ein
Protein durch Gentechnik durch Anwenden der vorliegenden Erfindung
stark zu exprimieren, solange das Gen für das Protein verfügbar ist.
Es ist offensichtlich, dass die vorliegende Erfindung zur Expression
eines Proteins verwendet werden kann, das von einem anderen Organismus
als dem Wirt abgeleitet ist, da ein Protein abgeleitet von Pyrococcus
furiosus, das taxonomisch unterschiedlich von Bakterien der Gattung
Bacillus ist, stark exprimiert wird. Die vorliegende Erfindung wird
bevorzugt zur Herstellung der Protease PFUL, der Protease TCES sowie
von Proteasen von Staphylothermus marinus und Thermobacteroides
proteoliticus, die strukturell ähnlich
zur Protease PFUS sind, mittels Gentechnik verwendet.
-
Basierend
auf der Homologie mit Subtilisinen wird vermutet, dass die Protease
PFUS als ein Vorläufer-Protein
mit einem Signalpeptid und einem Pro-Peptid exprimiert wird und
dann zur Bildung eines gereiften Enzyms prozessiert wird. Des Weiteren
kann basierend auf den Ergebnissen der N-terminalen Aminosäuresequenzanalyse
des ausgereiften Protease PFUS-Enzyms angenommen werden, dass das
ausgereifte Enzym ein Enzym ist, das aus der Aminosäuresequenz
wie in der SEQ ID NO:4 des Sequenzprotokolls gezeigt besteht. Jedoch
liegt das Molekulargewicht der gereinigten ausgereiften Protease
PFUS bei etwa 45 kDa, was geringer als das für die Aminosäuresequenz
berechnete ist. Dies legt nahe, dass die als ein Vorläufer exprimierte
Protease PFUS in eine ausgereifte Protease umgewandelt wird, nachdem
auch ihr C-terminales Peptid prozessiert wurde.
-
Wenn
das durch die Prozessierung entfernte C-terminale Peptid nicht essentiell
für die
enzymatische Aktivität
oder für
die Faltung des Enzymproteins in die richtige Struktur ist, wird
erwartet, dass der Expressionsgrad der Protease PFUS auch durch
Entfernen der diesen Teil kodierenden Region vom Gen und Exprimieren
der Protease erhöht
werden kann.
-
Das
Molekulargewicht der ausgereiften, von Bacillus subtilis DB104/pNAPS1
erhaltenen Protease PFUS kann präzise,
z.B. durch Verwendung eines Massenspektrometers, gemessen werden.
Durch das gemessene Molekulargewicht und die N-terminale Aminosäuresequenz
der ausgereiften Protease PFUS, die wie vorstehend beschrieben bestimmt
wurden, wurde herausgefunden, dass die Protease ein Polypeptid ist,
das der Aminosäuresequenz
wie gezeigt in der SEQ ID NO:15 des Sequenzprotokolls von Ala bei
Position 133 bis Thr bei Position 552 entspricht. Des Weiteren kann
ein Plasmid, das die Protease PFUS ohne ein Polypeptid exprimiert,
das nicht essentiell für
ihre enzymatische Aktivität
ist, durch Einführung
eines Terminationscodons in der Nähe des Teils hergestellt werden,
der Thr an Position 552 im Gen der Protease PFUS enthalten im Plasmid
pNAPS1 kodiert. Insbesondere kann ein DNA-Fragment mit einer Basensequenz,
in die das beabsichtigte Terminationscodon eingeführt worden
ist, mittels PCR unter Verwendung des Primers NPR544, der ein Terminationscodon
(TGA) an der C-terminalen Seite des den 544. Aminosäurerest
kodierenden Codons aus den vom Initiationscodon im Gen der Protease
PFUS im Plasmid pNAPS1 (Ser) einführen kann (die Basensequenz des
Primers NPR544 ist in der SEQ ID NO:28 des Sequenzprotokolls gezeigt)
und des Primers NPFE81 erhalten werden, der die Basensequenz des
Gebiets upstream der NspV-Schnittstelle
im Gen aufweist (die Basensequenz des Primers NPFE81 ist in der
SEQ ID NO:29 des Sequenzprotokolls gezeigt). Ein mutiertes Plasmid,
das das Protease-Gen enthält,
in das die Mutation von Interesse eingeführt worden ist, kann durch
Ersetzen der entsprechenden Region in dem Plasmid pNAPS1 durch das
Fragment erhalten werden. Dieses Plasmid wird als das Plasmid pNAPSΔC bezeichnet.
Mit diesem Plasmid transformiertes Bacillus subtilis DB 104 wird
als Bacillus subtilis DB104/pNAPSΔC
bezeichnet.
-
Dieser
Transformant exprimiert eine Protease-Aktivität mit Eigenschaften äquivalent
zu denen der Protease PFUS und mit einem höheren Expressionsgrad als dem
von Bacillus subtilis DB104/pNAPS1.
-
Folglich
wurde herausgefunden, dass das im Plasmid pNAPSΔC enthaltene Gen der Protease
PFUS eine Region umfasst, die ausreichend für die Expression der Aktivität des Enzyms
ist. Die Basensequenz der Region, die die in dem Plasmid vorhandene
Protease PFUS kodiert, ist in der SEQ ID NO:2 des Sequenzprotokolls
gezeigt. Die durch die Basensequenz kodierte Aminosäuresequenz
ist in SEQ ID NO:1 des Sequenzprotokolls gezeigt.
-
Des
Weiteren kann die Protease PFUS ohne ihr C-terminales Peptid durch
Einführung
einer Mutation ähnlich
zu der im Plasmid pNAPSΔC
in das Gen der Protease PFUS im Plasmid pSP0124 exprimiert werden.
-
Insbesondere
kann das Plasmid von Interesse durch Mischen und Ligieren eines
DNA-Fragments von etwa 13 kb erhalten durch Verdau des Plasmids
pNAPSΔC
mit NspV und SphI mit dem Plasmid pSP0124 hergestellt werden, das
mit NspV und SphI verdaut worden ist. Dieses Plasmid wird als das
Plasmid pSO124ΔC bezeichnet.
Mit diesem Plasmid transformiertes Bacillus subtilis DB 104 wird
bezeichnet und gekennzeichnet als Bacillus subtilis DB 104/pSO124ΔC und wurde
am 16. Mai 1997 (Datum der ursprünglichen
Hinterlegung) gemäß dem Budapester
Vertrag am Nationalen Institut für
Biowissenschaften und Humantechnologie, Amt für Industrielle Wissenschaft
und Technologie, Ministerium für
Internationalen Handel und Industrie, 1-3, Higashi 1-chome, Tsukuba-shi,
Ibaraki-ken, Japan unter der Zugriffsnummer FERM BP-6294 hinterlegt.
Der Expressionsgrad der Protease ist in dieser Transformante verglichen
mit dem von Bacillus subtilis DB104/pNAPS1 erhöht.
-
Die
enzymatischen Eigenschaften sowie die physikalischen und chemischen
Eigenschaften der durch die Transformanten Bacillus subtilis DB104/pNAPSΔC und Bacillus
subtilis DB 104/pSPO124ΔC
hergestellten Proteasen scheinen identisch mit denen der durch Bacillus
subtilis DB104/pSNP1 hergestellten Protease zu sein. Die Haupteigenschaften
der von den Kulturen der zwei Transformanten erhaltenen Proteasen
sind wie folgt:
-
(1) Aktivität:
-
Baut
Casein und Gelatine unter Bildung kurzkettiger Polypeptide ab.
-
Hydrolysiert
Succinyl-L-leucyl-L-leucyl-L-valyl-L-tyrosin-4-methylcoumarin-7-amid (Suc-Leu-Leu-Val-Tyr-MCA)
unter Bildung einer fluoreszierenden Substanz (7-Amino-4-methylcoumarin).
-
Hydrolysiert
Succinyl-L-alanyl-L-alanyl-L-prolyl-L-phenylalanin-p-nitroanild (Suc-Ala-Ala-Pro-Phe-p-NA)
unter Bildung einer gelben Substanz (p-Nitroanilin).
-
(2) Optimale Temperatur:
-
Weist
eine enzymatische Aktivität
bei 40-110°C
mit einer optimalen Temperatur bei 80-95°C auf.
-
(3) Optimaler pH-Wert:
-
Weist
eine enzymatische Aktivität
bei pH 5-10 mit einem optimalen pH-Wert bei 6-8 auf.
-
(4) Thermostabilität:
-
Behält 90% oder
mehr ihrer enzymatischen Aktivität
nach Behandlung bei 95°C
für 8 Stunden.
-
(5) pH-Stabilität:
-
Behält 95% oder
mehr ihrer Aktivität
nach Behandlung bei pH 5-11 bei 95°C für 60 Minuten.
-
(6) Molekulargewicht:
-
Weist
ein Molekulargewicht von etwa 45 kDa auf einer SDS-PAGE auf.
-
Folglich
werden Proteasen mit hoher Thermostabilität und Gene davon bereitgestellt.
Auch wird durch die vorliegende Erfindung ein neues System zur Expression
eines Proteins offenbart, das die Expression der Protease in hohen
Mengen ermöglicht.
Das Expressionssystem ist für
die Herstellung der erfindungsgemäßen Protease sowie von verschiedenen
Proteinen durch Gentechnik geeignet.
-
Die
folgenden Beispiele veranschaulichen die vorliegende Erfindung genauer,
sind aber nicht beschränkend
aufzufassen.
-
Beispiel 1
-
(1) Herstellung einer
chromosomalen DNA von Pyrococcus furiosus
-
Pyrococcus
furiosus DSM3638 wurde wie folgt kultiviert.
-
Ein
Medium mit 1% Trypton, 0,5% Hefeextrakt, 1% löslicher Stärke, 3,5% Jamarine S Feststoff
(Jamarine Laboratory), 0,5% Jamarine S Flüssigkeit (Jamarine Laboratory),
0,003% MgSO4, 0,001% NaCl, 0,0001% FeSO4·7H2O, 0,0001% CoSO4,
0,0001% CaCl2·7H2O,
0,0001% ZnSO4, 0,1 ppm CuSO4·5H2O, 0,1 ppm H3BO3, 0,1 ppm KAl (SO4)2, 0,1 ppm Na2MoO4·2H2O und 0,25 ppm NiCl2·H2O wurde in eine 2 1-Mediumflasche gegeben
und bei 120°C
für 20
Minuten sterilisiert. Stickstoffgas wurde zur Entfernung von gelöstem Sauerstoff
hindurchgeleitet und dann wurde das Medium mit dem Bakterienstamm
angeimpft und bei 95°C
für 16
Stunden ohne Schütteln
kultiviert. Nach dem Kultivieren wurden die Zellen durch Zentrifugieren
gesammelt.
-
Die
erhaltenen Zellen wurden dann in 4 ml 50 mM Tris-HCl (pH 8,0) mit
25% Sucrose aufgenommen. 2 ml 0,2 M EDTA und 0,8 ml Lysozym (5 mg/ml)
wurden zu der Suspension zugegeben. Das Gemisch wurde eine Stunde
bei 20°C
inkubiert. 24 ml SET-Lösung
(150 mM NaCl, 1 mM EDTA, 20 mM Tris-HCl, pH 8,0), 4 ml 5% SDS und
400 μl Proteinase
K (10 mg/ml) wurden dem Gemisch zugeführt. Dieses wurde für eine weitere Stunde
bei 37°C
inkubiert. Die Reaktion wurde durch Extrahieren des Gemisches mit
Phenol-Chloroform beendet. Dann wurde eine Ethanolfällung zum
Erhalten von etwa 3,2 mg der chromosomalen DNA durchgeführt.
-
Beispiel 2
-
(1) Synthese der Primer
zum Herstellen des Plasmids pNSP1
-
Zur
Synthese von Primern, die zur Amplifizierung des gesamten Gens der
Protease PFUS verwendet werden, wurde das Plasmid pSNP1, das das
gesamte Gen enthält,
von Bacillus subtilis DB104/pSNP1 (FERM BP-5634) isoliert und die
Basensequenz der benötigten
Region wurde bestimmt. Basierend auf der Basensequenz wurden der
Primer NPF-4 zum Einfügen
einer BamHI-Schnittstelle direkt upstream des Initiationscodons
des Gens der Protease PFUS und der Primer NPM-1 synthetisiert, der
mit der 3'-Region
des Gens hybridisiert und eine Erkennungsstelle für SphI enthält. Die
Basensequenzen der Primer NPF-4 und NPM-1 sind in den SEQ ID NOs:13
bzw. 19 des Sequenzprotokolls gezeigt.
-
Die
Primer mutRR und mutFR zum Entfernen der etwa 1,7 kb downstream
des Initiationscodons im Gen der Protease PFUS vorhandenen BamHI-Schnittstelle
wur den auch synthetisiert. Die Basensequenzen der Primer mutRR und
mutFR sind in den SEQ ID NOs:20 bzw. 21 des Sequenzprotokolls gezeigt.
-
(2) Herstellung des Plasmids
pPS1
-
Zwei
Sätze der
LA-PCR-Reaktionsgemische, von denen jedes eine chromosomale DNA
von Pyrococcus furiosus als Template und eine Kombination der Primer
NPF-4 und mutRR oder eine Kombination der Primer mutFR und NPM-1
enthält,
wurden hergestellt und 30 Reaktionszyklen von 94°C für 30 Sekunden-55°C für 1 Minute-68°C für 3 Minuten
unterworfen. LA-PCR-Kit Ver. 2 (Takara Shuzo) wurde zum Herstellen
der LA-PCR-Reaktionsgemische verwendet. Aliquots der Reaktionsgemische
wurden einer Agarose-Gel-Elektrophorese unterworfen und die Amplifizierung
eines DNA-Fragments von etwa 1,8 kb mit den Primern NPF-4 und mutRR
bzw. eines DNA-Fragments von etwa 0,6 kb mit den Primern mutFR und
NPM-1 wurde beobachtet.
-
Die
Primer wurden von den zwei PCR-Reaktionsgemischen unter Verwendung
von SUPREC-02 (Takara Shuzo) zur Herstellung amplifizierter DNA-Fragmente
entfernt. Ein LA-PCR-Reaktionsgemisch, das diese zwei amplifizierten
DNA-Fragmente aber nicht die Primer oder LA-Taq enthielt, wurde
hergestellt, 10 Minuten bei 94°C
Hitzedenaturiert, innerhalb von 30 Minuten auf 30°C abgekühlt und
dann 15 Minuten bei 30°C
zur Bildung eines Heterokomplexes inkubiert. Danach wurde LA-Taq
(Takara Shuzo) zu dem Reaktionsgemisch für eine Reaktion bei 72°C für 30 Minuten
zugegeben. Die Primer NPF-4 und NPM-1 wurden dann zu dem Reaktionsgemisch
zugegeben, das dann 25 Reaktionszyklen bei 94°C für 30 Sekunden-55°C für 1 Minute-68°C für 3 Minuten
unterworfen wurde. Amplifizierung eines DNA-Fragments von etwa 2,4
kb wurde in dem Reaktionsgemisch beobachtet.
-
Das
DNA-Fragment von etwa 2,4 kb wurde mit BamHI und SphI (beide vom
Takara Shuzo) verdaut. Das Fragment wurde mit dem Plasmid pSNP1,
das zur Entfernung des gesamten Gens der Protease PFUS mit BamHI
und SphI verdaut worden ist, gemischt und ligiert und dann in Bacillus
subtilis DB 104 eingeführt. Plasmide
wurden von erhaltenen Kanamycin-resistenten Transformanten präpariert.
Ein Plasmid, in das nur ein Molekül des Fragments von etwa 2,4
kb insertiert wurde, wurde ausgewählt und als das Plasmid pPS1
bezeichnet. Mit diesem Plasmid pPS1 transformiertes Bacillus subtilis
DB104 wurde als Bacillus subtilis DB104/pPS1 bezeichnet.
-
Die
Restriktionsenzym-Karte des Plasmids pPS1 ist in 7 gezeigt.
-
(3) Amplifizierung eines
DNA-Fragments für
die Promotor-Signalpeptid-kodierende Region des Gens von Subtilisin
E
-
Primer
zum Erhalten der Promotor-Signalpeptid-kodierenden Region des Gens
von Subtilisin E wurden synthetisiert. Zuerst wurde der Primer SUB4
basierend auf der Basensequenz der Promotor-Region des Gens von
Subtilisin E wie beschrieben in J. Bacteriol., 171:2657-2665 (1989)
synthetisiert, der mit der Sequenz upstream dieser Region hybridisiert
und eine EcoRI-Schnittstelle enthält (die Basensequenz des Primers SUB4
ist in der SEQ ID NO:17 des Sequenzprotokolls gezeigt). Der Primer
BmR1, der eine BamHI-Schnittstelle direkt downstream der Signalpeptid-kodierenden
Region einfügen
kann, wurde basierend auf der Basensequenz des Gens von Subtilisin
E wie beschrieben in J. Bacteriol., 158:411-418 (1984) synthetisiert
(die Basensequenz des Primers BmR1 ist in der SEQ ID NO:18 des Sequenzprotokolls
gezeigt).
-
Ein
PCR-Reaktionsgemisch, das das Plasmid pKWZ, das das Gen von Subtilisin
E wie beschrieben in J. Bacteriol., 171:2657-2665 enthält, als
Template und die Primer SUB4 und BmR1 enthält, wurde hergestellt und 30
Reaktionszyklen von 94°C
für 30
Sekunden-55°C
für 1 Minute-68°C für 2 Minuten
unterworfen. Ein Aliquot des Reaktionsgemisches wurde einer Agarose-Gel-Elektrophorese
unterworfen und die Amplifikation eines DNA-Fragments von etwa 0,3
kb wurde beobachtet.
-
(4) Herstellung des Protease-Expressionsplasmids
pNAPS1
-
Das
DNA-Fragment von etwa 0,3 kb wie vorstehend beschrieben wurde mit
EcoRI (Takara Shuzo) und BamHI verdaut, mit dem in Beispiel 3 beschriebenen
Plasmid pPS1, das mit EcoRI und BamHI verdaut wurde, gemischt und
ligiert und dann in Bacillus subtilis DB 104 eingeführt. Von
den erhaltenen Kanamycin-resistenten Transformanten wurden Plasmide
präpariert.
Ein Plasmid, in das nur ein Molekül des Fragments von etwa 0,3
kb insertiert war, wurde ausgewählt
und als das Plasmid pNAPS1 bezeichnet. Mit dem Plasmid pNAPS1 transformiertes
Bacillus subtilis DB 104 wurde als Bacillus subtilis DB 104/pNAPS1
bezeichnet.
-
Die
Restriktionsenzym-Karte des Plasmids pNAPS1 ist in 8 gezeigt.
-
(5) Herstellung des Plasmids
pSNP2
-
Der
Primer SUB17R zur Einführung
einer BamHI-Schnittstelle upstream der Signalpeptid-kodierenden Region
des Gens von Subtilisin E in dem vorstehend genannten Plasmid pNAPS1
wurde synthetisiert (die Basensequenz des Primers SUB17R ist in
der SEQ ID NO:23 des Sequenzprotokolls gezeigt). Ein PCR-Reaktionsgemisch,
das das Plasmid pNAPS1 als Template und die Primer SUB17R und SUB4
enthält,
wurde hergestellt und 25 Rektionszyklen von 94°C für 30 Sekunden-55°C für 1 Minute-72°C für 1 Minute
unterworfen. Das amplifizierte DNA-Fragment von etwa 0,21 kb wurde
mit EcoRI und BamHI zum Erhalten eines DNA-Fragments von etwa 0,2
kb verdaut, das den Promotor und die SD-Sequenz des Gens von Subtilisin
E enthält.
Dieses Fragment wurde mit dem Plasmid pNAPS1, das mit EcoRI und
BamHI verdaut worden war, gemischt und ligiert. Das Reaktionsgemisch
wurde zur Transformierung von Bacillus subtilis DB 104 verwendet.
Von erhaltenen Kanamycin-resistenten Transformanten wurden Plasmide
präpariert.
Ein Plasmid, in das das DNA-Fragment von etwa 0,2 kb insertiert
war, wurde ausgewählt
und als das Plasmid pSNP2 bezeichnet.
-
(6) Bildung eines mutierten
Plasmids, das eine Protease stark exprimiert
-
Die
Primer SPOF0 und SPOR0 zum Ersetzen der Sequenz, die den Aminosäurerest
Ser (Basensequenz: TCC) an der Verbindung zwischen der Signalpeptid-kodierenden
Region des Gens von Subtilisin E in dem Plasmid pNAPS1 und dem Initiationscodon
des Gens der Protease PFUS kodiert, mit einer Sequenz für zwei zufällige Aminosäurereste,
wurden synthetisiert (die Basensequenzen der Primer SPOF0 und SPOR0 sind
in den SEQ ID NOs: 24 bzw. 25 des Sequenzprotokolls gezeigt). Der
Primer SUB3 zum Einführen
einer BamHI-Schnittstelle direkt upstream der Signalpeptid-kodierenden
Region im Gen von Subtilisin E im Plasmid pNAPS1 und der Primer
NPR-10, der eine SpeI-Schnittstelle in der Protease PFUS-kodierenden
Region enthält,
wurden synthetisiert (die Basensequenzen der Primer SUB3 und NPR-10 sind in den SEQ
ID NOs: 26 bzw. 27 des Sequenzprotokolls gezeigt).
-
PCR-Reaktionsgemische,
von denen jedes das Plasmid pNAPS1 als Template und eine Kombination der
Primer SPOF0 und NPR-10 oder eine Kombination der Primer SUB3 und
SPOR0 enthielt, wurden hergestellt und 20 Reaktionszyklen von 94°C für 30 Sekunden-50°C für 1 Minute-72°C für 1 Minute
unterworfen. Die in den zwei Reaktionsgemischen amplifizierten DNA-Fragmente
von etwa 0,13 kb und etwa 0,35 kb wurden zusammengemischt, 10 Minuten
bei 94°C
denaturiert und schrittweise auf 37°C zum Bilden eines Heteroduplexes
abgekühlt.
Von den Heteroduplexen wurde dann eine doppelsträngige DNA mittels Taq-Polymerase (Takara
Shuzo) gebildet. Ein PCR-Reaktionsgemisch, das die so erhaltene
doppelsträngige
DNA als Template und die Primer SUB3 und NPR-10 enthielt, wurde
hergestellt und 25 Reaktionszyklen von 94°C für 30 Sekunden-50°C für 1 Minute-72°C für 1 Minute
unterworfen. Ein DNA-Fragment, das durch Verdau des amplifizierten DNA-Fragments
von etwa 0,43 kb mit BamHI und SpeI (Takara Shuzo) erhalten wurde,
wurde mit dem Plasmid pSNP2, das mit BamHI und SpeI verdaut worden
war, gemischt und ligiert. Das Reaktionsgemisch wurde zum Transformieren
von Bacillus subtilis DB 104 verwendet.
-
Erhaltene
Kanamycin-resistente Transformanten wurden auf Magermilchplatten
(LB-Agarmedium zur Hochtemperatur-Kultivierung mit 10 μg/ml Kanamycin
und 1% Magermilch) zur Bildung von Kolonien überimpft. Danach wurden die
Platten bei 70°C
inkubiert und die durch die entsprechenden Transformanten exprimierten
Protease-Aktivitäten
wurden basierend auf dem Grad des Abbaus der Magermilch um die Kolonien untersucht.
Als ein Ergebnis wurde ein Klon isoliert, der eine besonders hohe
Aktivität
aufwies, und ein Plasmid, das als das Plasmid pSP0124 bezeichnet
wurde, wurde von diesem Klon präpariert.
Mit diesem Plasmid transformiertes Bacillus subtilis DB104 wurde
als Bacillus subtilis DB104/pSPO124 bezeichnet. Die Basensequenz
des Plasmids pSP0124 wurde analysiert und es wurde herausgefunden,
dass die Basensequenz, die Ser in dem Plasmid pNAPS1 kodiert, durch
die Basensequenz GGGAAT ausgetauscht wurde, d.h. dass ein Protein
kodiert wurde, indem Ser durch die beiden Aminosäurereste Gly-Asn ausgetauscht
worden war. Zusätzlich
zeigte sich, dass das Pro (CCA) entsprechende 25. Codon ab dem Initiationscodon
des Gens der Protease PFUS in ein Leu (CTA)-kodierendes Codon gleichzeitig
mit der vorstehend beschriebenen Mutation geändert wurde.
-
(7) Herstellung des Protease-Expressionsplasmids
pNAPSΔC
-
Ein
Terminationscodon wurde auf der C-terminalen Seite des 544. Aminosäurerestes
ab dem Initiationscodon des Gens der Protease PFUS in dem Plasmid
pNAPS1 zur Herstellung eines Plasmids eingeführt, das eine Protease exprimiert,
der der Teil downstream dieser Stelle fehlt. Der Primer NPR544 wurde
synthetisiert, der ein Terminationscodon (Basensequenz: TGA) an
der C-terminalen Seite des den 544. Aminosäurerest kodierenden Codons
in das Gen einführt
und eine SphI-Schnittstelle hat (die Basensequenz des Primers NPR544
ist in der SEQ ID NO:28 des Sequenzprotokolls gezeigt). Zusätzlich wurde
der Primer NPFE81 basierend auf der Basensequenz des Teils upstream
der NspV-Schnittstelle im Gen synthetisiert (die Basensequenz des
Primers NPFE81 ist in der SEQ ID NO:29 des Sequenzprotokolls gezeigt).
-
Ein
PCR-Reaktionsgemisch, das das Plasmid pNAPS1 als Template und die
Primer NPFE81 und NPR544 enthielt, wurde hergestellt und 20 Reaktionszyklen
von 94°C
für 30
Sekunden-50°C
für 1 Minute-72°C für 1 Minute
unterworfen. Das amplifizierte DNA-Fragment von etwa 0,61 kb wurde
mit NspV (Takara Shuzo) und SpeI zum Erhalt eines das Terminationscodon
enthaltenden DNA-Fragments von etwa 0,13 kb verdaut. Dieses DNA-Fragment
wurde mit dem Plasmid pNAPS1, das mit den Restriktionsenzymen NspV
und SphI verdaut worden war, gemischt und ligiert. Das Reaktionsgemisch
wurde zum Transformieren von Bacillus subtilis DB 104 verwendet.
Von den erhaltenen Kanamycin-resistenten Transformanten wurden Plasmide
präpariert. Ein
Plasmid, in das das DNA-Fragment von etwa 0,13 kb insertiert war,
wurde ausgewählt
und als das Plasmid pNAPSΔC
bezeichnet. Mit dem Plasmid pNAPSΔC
transformiertes Bacillus subtilis DB 104 wurde als Bacillus subtilis
DB104/pNAPSΔC
bezeichnet.
-
(8) Herstellung des Protease-Expressionsplasmids
pSPO124ΔC
-
Ein
DNA-Fragment von etwa 1,3 kb, das durch Verdau des Plasmids pNAPSΔC mit NspV
und SphI erhalten wurde, wurde isoliert und dann mit dem Plasmid
pSP0124, das mit NspV und SphI verdaut worden war, vermischt und
ligiert. Das Reaktionsgemisch wurde zum Transformieren von Bacillus
subtilis DB 104 verwendet. Von den erhaltenen Kanamycin-resistenten
Transformanten wurden Plasmide präpariert. Ein Plasmid, in das
das DNA-Fragment von etwa 1,3 kb insertiert war, wurde ausgewählt und
als das Plasmid pSPO124ΔC bezeichnet.
Mit dem Plasmid pSPO124ΔC
transformiertes Bacillus subtilis DB 104 wurde als Bacillus subtilis DB
104/pSPO124ΔC
bezeichnet.
-
Beispiel 3
-
(1) Kultivieren von mit
einem das Gen der Protease PFUS enthaltenden Plasmids transformiertem
Bacillus subtilis und Herstellen einer ungereinigten Enzymlösung
-
Bacillus
subtilis DB104/pNAPS1, welches Bacillus subtilis DB104 ist, in das
das das Gen der Protease PFUS enthaltene Plasmid pNAPS1 wie beschrieben
in Beispiel 2 eingefügt
wurde, wurde 24 Stunden bei 37°C in
2 ml LB-Medium (Trypton 10 g/l, Hefeextrakt 5 g/l, NaCl 5 g/l, pH
7,2) mit 10 μg/ml
Kanamycin kultiviert. Die Kultur wurde zum Erhalten eines Kulturüberstandes
(Präparat
1-S) und von Zellen zentrifugiert.
-
Die
Zellen wurden in 100 μl
50 mM Tris-HCl, pH 7,5 aufgenommen und 45 Minuten bei 37°C nach Zugabe
von 2 mg Lysozym (Sigma) verdaut. Die verdaute Probe wurde 10 Minuten
bei 95°C
Hitze-behandelt und dann wurde ein Überstand durch Zentrifugation
zum Erhalten eines zellfreien Extrakts (Präparat 1-L) gesammelt.
-
In ähnlicher
Weise wurden Kulturüberstände und
zellfreie Extrakte von das Plasmid pSP0124 enthaltendem Bacillus
subtilis DB104/pSPO124, das Plasmid pNAPSΔC enthaltendem Bacillus subtilis DB104/pNAPSΔC oder das
Plasmid pSPO124ΔC
enthaltendem Bacillus subtilis DB104/pSPO124ΔC erhalten. Der Kulturüberstand
und der zellfreie Extrakt von Bacillus subtilis DB104/pSPO124 wurden
als 124-S bzw. 124-L bezeichnet. Der Kulturüberstand und der zellfreie
Extrakt von Bacillus subtilis DB104/pNAPSΔC wurden als ΔC-S bzw. ΔC-L bezeichnet.
Der Kulturüberstand
und der zellfreie Extrakt von Bacillus subtilis DB 104/pSPO124ΔC wurden
als 124ΔC-S
bzw. 124ΔC-L
bezeichnet. Protease-Aktivitäten
wurden mit diesen Präparaten
bestimmt und die Konzentration der in jedem Präparat enthaltenen Protease
wurde bestimmt.
-
(2) Vergleich der Protease-Produktivitäten
-
Die
Aktivität
der Protease PFUS wurde durch spektroskopisches Messen der Menge
an p-Nitroanilin bestimmt, das in einer enzymatischen Hydrolysereaktion
unter Verwendung von Suc-Ala-Ala-Pro-Phe-p-NA (Sigma) als Substrat
gebildet wurde. Kurz zusammengefasst wurde ein auf seine enzymatische
Aktivität
zu messendes Enzympräparat
geeignet verdünnt.
50 μl 1
mM Suc-Ala-Ala-Pro-Phe-p-NA-Lösung
in 100 mM Phosphatpuffer, pH 7,0, wurden zu 50 μl der verdünnten Probenlösung gegeben.
Dann erfolgte die Reaktion für
30 Minuten bei 95°C.
Nach Beendigung der Rektion durch Abkühlen auf Eis wurde die Absorption
bei 405 nm zum Berechnen der Menge an gebildetem p-Nitroanilin gemessen.
Eine Unit des Enzyms wurde als die Menge des Enzyms definiert, die
1 μmol p-Nitroanilin
pro 1 Minute bei 95°C
bildete. Die Menge des in dem Kulturüberstand oder den Zellen exprimierten
Enzymproteins wurde basierend auf der gemessenen enzymatischen Aktivität unter
der Annahme berechnet, dass die spezifische Aktivität bei 9,5
Units/mg Protein der Protease PFUS liegt.
-
Die
Protease-Aktivität
von jedem im Beispiel 3-(1) hergestellten Enzympräparat wurde
gemessen. Die aus den Messungen berechnete Produktivität an Protease
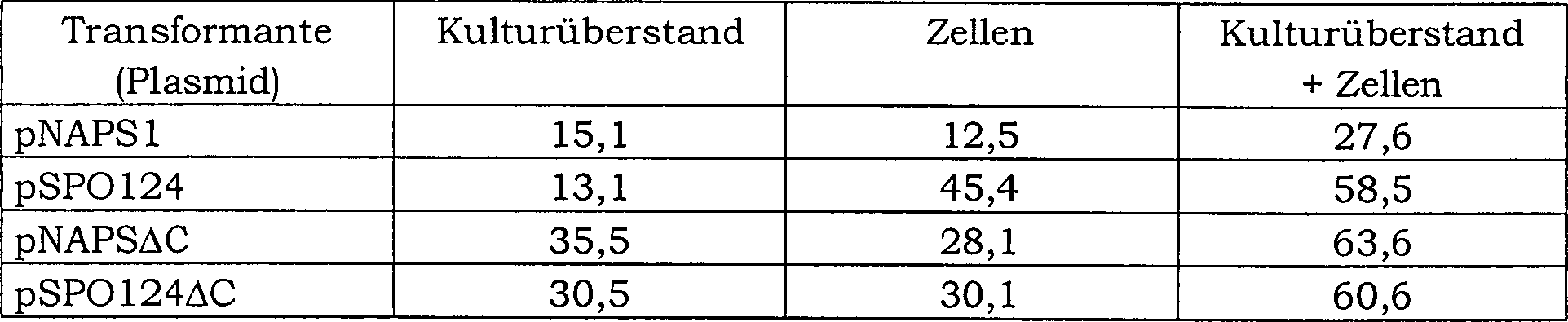
PFUS pro 1 l Kultur jedes Transformanten ist in Tabelle 1 gezeigt.
-
Im
Bacillus subtilis DB 104/pSPO124 erhöhte sich die Produktivität an Protease
PFUS in den Zellen um 3,6-fach verglichen mit der von Bacillus subtilis
DB104/pNAPS1. In Bacillus subtilis DB104/pNAPSΔC erhöhte sich die Produktivität an Protease
PFUS im Zellüberstand
um 2,4-fach bzw. in den Zellen um 2,2-fach. Auch in Bacillus subtilis
DB104/pSPO124ΔC
erhöhte
sich die Produktivität
an Protease PFUS im Kulturüberstand
um 2-fach bzw. in den Zellen um 2,4-fach. Die Produktivität pro Zelle
erhöhte
sich auch.
-
Die
Gesamtmenge an im Zellüberstand
und den Zellen hergestellter Protease PFUS erhöhte sich um 2,1-fach bei Bacillus
subtilis DB 104/pSPO124, um 2,1-fach bei Bacillus subtilis DB104/pNAPSΔC bzw. um 2,2-fach
bei Bacillus subtilis DB104/pSPO124ΔC verglichen zu der von Bacillus
subtilis DB104/pNAPS1. Tabelle
1 Die
Produktivität
an Protease PFUS (mg/l der Kultur)
-
Beispiel 4
-
(1) Herstellen von gereinigtem
Enzympräparat
der ausgereiften Protease PFUS
-
Bacillus
subtilis DB104/pNAPS1 und Bacillus subtilis DB104/pSPO124ΔC, die beide
Bacillus subtilis DB 104 sind, in die das Gen der erfindungsgemäßen hyperthermostabilen
Protease wie beschrieben in Beispiel 2 eingeführt wurde, wurden getrennt
voneinander in 5 ml LB-Medium mit 10 μg/ml Kanamycin überimpft und
unter Schütteln
7 Stunden bei 37°C
kultiviert. Die 5 ml-Kulturen wurden in 500 ml TM-Medium (Sojabohnenpulver
5 g/l, Polypepton 10 g/l, Fleischextrakt 5 g/l, Hefeextrakt 2 g/l,
Glucose 10 g/l, FeSO4·7H2O
10 mg/l, MnSO4·4H2O
10 mg/l, ZnSO4·7H2O
1 mg/l, pH 7,0) mit 10 μg/ml
Kanamycin in 5 1-Erlenmeyerkolben überimpft und unter Schütteln 3
Tage bei 30°C
kultiviert. Die erhaltenen Kulturen wurden mit Ultraschall aufgeschlossen, 30
Minuten bei 95°C
Hitze-behandelt und dann zum Sammeln des Überstands zentrifugiert. Zu
den Überständen wurde
Ammoniumsulfat bis 25% Sättigung
gegeben. Die durch nachfolgende Zentrifugation erhaltenen Überstände wurden
dann auf Micro-Prep Methyl-HIC-Säulen
(Bio-Rad) aufgetragen, die mit 25 mM Tris-HCl-Puffer (pH 7,6) mit
25% gesättigtem
Ammoniumsulfat äquilibriert
waren. Nach Waschen des Gels mit dem gleichen Puffer wurde die an
die Säulen
adsorbierte Protease PFUS durch schrittweise Elution unter Verwendung
von 25 mM Tris-HCl-Puffer (pH 7,6) mit 40% Ethanol eluiert. Die
so erhaltenen, die Protease PFUS enthaltenen Fraktionen wurden einer
Gelfiltration unter Verwendung von NAP-25-Säulen (Pharmacia), äquilibriert
mit 0,05% Trifluoressigsäure
mit 20% Acetonitril, unterworfen und während Denaturieren der Protease PFUS
entsalzt. Dann wurden gereinigte Präparate der Protease PFUS erhalten.
Die von Bacillus subtilis DB104/pNAPS1 und Bacillus subtilis DB
104/pSPO124ΔC
erhaltenen Präparate
wurden als NAPS-1 bzw. SPO-124ΔC
bezeichnet.
-
Eine
Elektrophorese von beiden gereinigten Enzympräparaten auf einem 0,1% SDS-10% Polyacrylamidgel
mit nachfolgender Anfärbung
mit Coomassie Brilliant Blue R-250
zeigte einzelne Banden für
die beiden gereinigten Enzympräparate
NAPS-1 und SPO-124ΔC
mit einem abgeschätzten
Molekulargewicht von etwa 45 kDa.
-
(2) Analyse der N-terminalen
Aminosäuresequenz
der ausgereiften Protease PFUS
-
N-terminale
Aminosäuresequenzen
der gereinigten Enzympräparate
NAPS-1 und SPO-124ΔC
wurden durch ein automatisiertes Edman-Verfahren unter Verwendung
eines G 1000A-Proteinsequenzers (Hewlett-Packard) analysiert. Beide
N-terminalen Aminosäuresequenzen
der zwei gereinigten Enzympräparate
waren wie in der SEQ ID NO:22 des Sequenzprotokolls gezeigt. Diese
Sequenz stimmt mit der Sequenz N-terminate Aminosäuresequenzen
der gereinigten Enzympräparate
NAPS-1 und SPO-124ΔC
wurden durch ein automatisiertes Edman-Verfahren unter Verwendung
eines G 1000A-Proteinsequenzers (Hewlett-Packard) analysiert. Beide
N-terminalen Aminosäuresequenzen
der zwei gereinigten Enzympräparate
waren wie in der SEQ ID NO:22 des Sequenzprotokolls gezeigt. Diese
Sequenz stimmt mit der Sequenz von Position 133 bis Position 144
der Aminosäuresequenz
der Protease PFUS wie in der SEQ ID NO:15 des Sequenzprotokolls
gezeigt überein.
Dies zeigt, dass sowohl NAPS-1 als auch SPO-124ΔC Enzyme sind, die aus einem
von diesem Teil startenden Polypeptid bestehen.
-
(3) Massenspektrometrische
Analyse der ausgereiften Protease PFUS
-