CN105008518B - 在空气-液体界面处培养人胚胎干细胞以用于分化成胰腺内分泌细胞 - Google Patents
在空气-液体界面处培养人胚胎干细胞以用于分化成胰腺内分泌细胞 Download PDFInfo
- Publication number
- CN105008518B CN105008518B CN201380074040.4A CN201380074040A CN105008518B CN 105008518 B CN105008518 B CN 105008518B CN 201380074040 A CN201380074040 A CN 201380074040A CN 105008518 B CN105008518 B CN 105008518B
- Authority
- CN
- China
- Prior art keywords
- cells
- stage
- medium
- inhibitor
- tgf
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 230000004069 differentiation Effects 0.000 title claims abstract description 78
- 230000009996 pancreatic endocrine effect Effects 0.000 title claims abstract description 64
- 210000003890 endocrine cell Anatomy 0.000 title claims abstract description 41
- 239000007788 liquid Substances 0.000 title abstract description 152
- 238000012258 culturing Methods 0.000 title abstract description 76
- 210000001671 embryonic stem cell Anatomy 0.000 title description 71
- 210000004027 cell Anatomy 0.000 claims abstract description 835
- 210000001778 pluripotent stem cell Anatomy 0.000 claims abstract description 88
- 239000003112 inhibitor Substances 0.000 claims description 227
- 239000002609 medium Substances 0.000 claims description 204
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 claims description 138
- FOORCIAZMIWALX-ULJHMMPZSA-N (z)-n-(4-benzylpiperazin-1-yl)-1-(3,5-dimethyl-1-phenylpyrazol-4-yl)methanimine Chemical compound CC1=NN(C=2C=CC=CC=2)C(C)=C1\C=N/N(CC1)CCN1CC1=CC=CC=C1 FOORCIAZMIWALX-ULJHMMPZSA-N 0.000 claims description 69
- 235000010323 ascorbic acid Nutrition 0.000 claims description 67
- 229960005070 ascorbic acid Drugs 0.000 claims description 67
- 239000011668 ascorbic acid Substances 0.000 claims description 67
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 claims description 64
- 229930002330 retinoic acid Natural products 0.000 claims description 64
- 229960001727 tretinoin Drugs 0.000 claims description 64
- 210000001900 endoderm Anatomy 0.000 claims description 55
- 210000004039 endoderm cell Anatomy 0.000 claims description 52
- 239000002243 precursor Substances 0.000 claims description 50
- 239000001963 growth medium Substances 0.000 claims description 43
- 108091005735 TGF-beta receptors Proteins 0.000 claims description 39
- 102000016715 Transforming Growth Factor beta Receptors Human genes 0.000 claims description 39
- 210000002438 upper gastrointestinal tract Anatomy 0.000 claims description 38
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 claims description 32
- 229960002897 heparin Drugs 0.000 claims description 32
- 229920000669 heparin Polymers 0.000 claims description 32
- 239000005557 antagonist Substances 0.000 claims description 25
- 102000001893 Bone Morphogenetic Protein Receptors Human genes 0.000 claims description 22
- 108010040422 Bone Morphogenetic Protein Receptors Proteins 0.000 claims description 22
- 239000000203 mixture Substances 0.000 claims description 19
- 229910000368 zinc sulfate Inorganic materials 0.000 claims description 19
- -1 L Y2109761 Chemical compound 0.000 claims description 18
- 102000045246 noggin Human genes 0.000 claims description 18
- 108700007229 noggin Proteins 0.000 claims description 18
- NWONKYPBYAMBJT-UHFFFAOYSA-L zinc sulfate Chemical compound [Zn+2].[O-]S([O-])(=O)=O NWONKYPBYAMBJT-UHFFFAOYSA-L 0.000 claims description 17
- AUYYCJSJGJYCDS-LBPRGKRZSA-N Thyrolar Chemical class IC1=CC(C[C@H](N)C(O)=O)=CC(I)=C1OC1=CC=C(O)C(I)=C1 AUYYCJSJGJYCDS-LBPRGKRZSA-N 0.000 claims description 16
- 108091006082 receptor inhibitors Proteins 0.000 claims description 14
- 102000013380 Smoothened Receptor Human genes 0.000 claims description 13
- 230000008410 smoothened signaling pathway Effects 0.000 claims description 13
- ZXSWZQSYZYMZKS-UHFFFAOYSA-N 2-methoxyethyl 4-(3-hydroxyphenyl)-7-(2-methoxyphenyl)-2-methyl-5-oxo-4,6,7,8-tetrahydro-1h-quinoline-3-carboxylate Chemical compound COCCOC(=O)C1=C(C)NC(CC(CC2=O)C=3C(=CC=CC=3)OC)=C2C1C1=CC=CC(O)=C1 ZXSWZQSYZYMZKS-UHFFFAOYSA-N 0.000 claims description 10
- 239000005495 thyroid hormone Substances 0.000 claims description 8
- 229940036555 thyroid hormone Drugs 0.000 claims description 8
- XUIIKFGFIJCVMT-LBPRGKRZSA-N L-thyroxine Chemical compound IC1=CC(C[C@H]([NH3+])C([O-])=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-LBPRGKRZSA-N 0.000 claims description 7
- 229950008325 levothyroxine Drugs 0.000 claims description 7
- XUIIKFGFIJCVMT-UHFFFAOYSA-N thyroxine-binding globulin Natural products IC1=CC(CC([NH3+])C([O-])=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-UHFFFAOYSA-N 0.000 claims description 7
- 229960001763 zinc sulfate Drugs 0.000 claims description 7
- QASFUMOKHFSJGL-LAFRSMQTSA-N Cyclopamine Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H](CC2=C3C)[C@@H]1[C@@H]2CC[C@@]13O[C@@H]2C[C@H](C)CN[C@H]2[C@H]1C QASFUMOKHFSJGL-LAFRSMQTSA-N 0.000 claims description 6
- 102000006533 chordin Human genes 0.000 claims description 6
- 108010008846 chordin Proteins 0.000 claims description 6
- QASFUMOKHFSJGL-UHFFFAOYSA-N cyclopamine Natural products C1C=C2CC(O)CCC2(C)C(CC2=C3C)C1C2CCC13OC2CC(C)CNC2C1C QASFUMOKHFSJGL-UHFFFAOYSA-N 0.000 claims description 6
- 229940035722 triiodothyronine Drugs 0.000 claims description 6
- ULFUJLFTRWWLPO-UHFFFAOYSA-N ethyl 2,7,7-trimethyl-5-oxo-4-(4-phenylphenyl)-1,4,6,8-tetrahydroquinoline-3-carboxylate Chemical compound CCOC(=O)C1=C(C)NC(CC(C)(C)CC2=O)=C2C1C(C=C1)=CC=C1C1=CC=CC=C1 ULFUJLFTRWWLPO-UHFFFAOYSA-N 0.000 claims description 5
- 102100021796 Sonic hedgehog protein Human genes 0.000 claims description 4
- 230000024245 cell differentiation Effects 0.000 claims description 4
- 230000001939 inductive effect Effects 0.000 claims description 4
- 108010090739 Smoothened Receptor Proteins 0.000 claims description 3
- 230000019491 signal transduction Effects 0.000 claims description 3
- 239000013589 supplement Substances 0.000 claims description 3
- 208000032740 autosomal recessive 10 intellectual disability Diseases 0.000 claims description 2
- 210000004263 induced pluripotent stem cell Anatomy 0.000 claims 2
- 102100028096 Homeobox protein Nkx-6.2 Human genes 0.000 abstract description 97
- 101000578254 Homo sapiens Homeobox protein Nkx-6.1 Proteins 0.000 abstract description 97
- 101000578258 Homo sapiens Homeobox protein Nkx-6.2 Proteins 0.000 abstract description 97
- 238000000034 method Methods 0.000 abstract description 97
- 102100041030 Pancreas/duodenum homeobox protein 1 Human genes 0.000 abstract description 58
- 101710183548 Pyridoxal 5'-phosphate synthase subunit PdxS Proteins 0.000 abstract description 58
- 238000004113 cell culture Methods 0.000 abstract description 24
- 238000001727 in vivo Methods 0.000 abstract description 12
- 230000035800 maturation Effects 0.000 abstract description 10
- 101000576323 Homo sapiens Motor neuron and pancreas homeobox protein 1 Proteins 0.000 abstract description 4
- 102100025170 Motor neuron and pancreas homeobox protein 1 Human genes 0.000 abstract description 4
- 230000001737 promoting effect Effects 0.000 abstract 1
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 170
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 100
- 230000014509 gene expression Effects 0.000 description 94
- 238000010790 dilution Methods 0.000 description 86
- 239000012895 dilution Substances 0.000 description 86
- 102000004877 Insulin Human genes 0.000 description 85
- 108090001061 Insulin Proteins 0.000 description 85
- 229940125396 insulin Drugs 0.000 description 85
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 81
- 239000008103 glucose Substances 0.000 description 51
- 102100028071 Fibroblast growth factor 7 Human genes 0.000 description 44
- 101001060261 Homo sapiens Fibroblast growth factor 7 Proteins 0.000 description 44
- 102000010792 Chromogranin A Human genes 0.000 description 40
- 108010038447 Chromogranin A Proteins 0.000 description 40
- 210000000130 stem cell Anatomy 0.000 description 38
- 150000001875 compounds Chemical class 0.000 description 37
- 235000014113 dietary fatty acids Nutrition 0.000 description 35
- 229930195729 fatty acid Natural products 0.000 description 35
- 239000000194 fatty acid Substances 0.000 description 35
- 150000004665 fatty acids Chemical class 0.000 description 35
- 102100039939 Growth/differentiation factor 8 Human genes 0.000 description 33
- 239000002953 phosphate buffered saline Substances 0.000 description 33
- 102000051325 Glucagon Human genes 0.000 description 32
- 108060003199 Glucagon Proteins 0.000 description 32
- 101000886562 Homo sapiens Growth/differentiation factor 8 Proteins 0.000 description 32
- 229960004666 glucagon Drugs 0.000 description 32
- MASNOZXLGMXCHN-ZLPAWPGGSA-N glucagon Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 MASNOZXLGMXCHN-ZLPAWPGGSA-N 0.000 description 32
- HJCMDXDYPOUFDY-WHFBIAKZSA-N Ala-Gln Chemical compound C[C@H](N)C(=O)N[C@H](C(O)=O)CCC(N)=O HJCMDXDYPOUFDY-WHFBIAKZSA-N 0.000 description 30
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 30
- 230000002124 endocrine Effects 0.000 description 29
- 239000011575 calcium Substances 0.000 description 26
- 108090000623 proteins and genes Proteins 0.000 description 26
- 238000003753 real-time PCR Methods 0.000 description 26
- IYOZTVGMEWJPKR-VOMCLLRMSA-N 4-[(1R)-1-aminoethyl]-N-pyridin-4-yl-1-cyclohexanecarboxamide Chemical compound C1CC([C@H](N)C)CCC1C(=O)NC1=CC=NC=C1 IYOZTVGMEWJPKR-VOMCLLRMSA-N 0.000 description 23
- 238000004458 analytical method Methods 0.000 description 23
- 101000603702 Homo sapiens Neurogenin-3 Proteins 0.000 description 22
- 102100038553 Neurogenin-3 Human genes 0.000 description 22
- 108700014808 Homeobox Protein Nkx-2.2 Proteins 0.000 description 21
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 21
- 102100024645 ATP-binding cassette sub-family C member 8 Human genes 0.000 description 20
- 101100518002 Danio rerio nkx2.2a gene Proteins 0.000 description 20
- 102100027886 Homeobox protein Nkx-2.2 Human genes 0.000 description 20
- 101000760570 Homo sapiens ATP-binding cassette sub-family C member 8 Proteins 0.000 description 20
- 101100460496 Homo sapiens NKX2-2 gene Proteins 0.000 description 20
- 239000003550 marker Substances 0.000 description 20
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 18
- 210000001035 gastrointestinal tract Anatomy 0.000 description 18
- 102000036770 Islet Amyloid Polypeptide Human genes 0.000 description 17
- 108010041872 Islet Amyloid Polypeptide Proteins 0.000 description 17
- 102000003923 Protein Kinase C Human genes 0.000 description 16
- 108090000315 Protein Kinase C Proteins 0.000 description 16
- 239000007640 basal medium Substances 0.000 description 16
- 102000005962 receptors Human genes 0.000 description 16
- 108020003175 receptors Proteins 0.000 description 16
- 239000011435 rock Substances 0.000 description 16
- 239000000243 solution Substances 0.000 description 16
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 15
- 239000012190 activator Substances 0.000 description 15
- 229940088597 hormone Drugs 0.000 description 15
- 239000005556 hormone Substances 0.000 description 15
- 238000011081 inoculation Methods 0.000 description 15
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 15
- 239000006285 cell suspension Substances 0.000 description 14
- 235000017557 sodium bicarbonate Nutrition 0.000 description 14
- 239000000126 substance Substances 0.000 description 14
- 238000002054 transplantation Methods 0.000 description 14
- VOUAQYXWVJDEQY-QENPJCQMSA-N 33017-11-7 Chemical compound OC(=O)CC[C@H](N)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)NCC(=O)NCC(=O)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N1[C@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O)CCC1 VOUAQYXWVJDEQY-QENPJCQMSA-N 0.000 description 13
- 230000000694 effects Effects 0.000 description 13
- 101150004578 gdf-8 gene Proteins 0.000 description 13
- 230000001965 increasing effect Effects 0.000 description 13
- 238000004519 manufacturing process Methods 0.000 description 13
- 229940088594 vitamin Drugs 0.000 description 13
- 229930003231 vitamin Natural products 0.000 description 13
- 235000013343 vitamin Nutrition 0.000 description 13
- 239000011782 vitamin Substances 0.000 description 13
- 150000003722 vitamin derivatives Chemical class 0.000 description 13
- 108010083123 CDX2 Transcription Factor Proteins 0.000 description 12
- 102000004190 Enzymes Human genes 0.000 description 12
- 108090000790 Enzymes Proteins 0.000 description 12
- 102000003974 Fibroblast growth factor 2 Human genes 0.000 description 12
- 102100031671 Homeobox protein CDX-2 Human genes 0.000 description 12
- 101000971533 Homo sapiens Killer cell lectin-like receptor subfamily G member 1 Proteins 0.000 description 12
- 101000979205 Homo sapiens Transcription factor MafA Proteins 0.000 description 12
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 12
- 102000004218 Insulin-Like Growth Factor I Human genes 0.000 description 12
- 102100021457 Killer cell lectin-like receptor subfamily G member 1 Human genes 0.000 description 12
- 108010032788 PAX6 Transcription Factor Proteins 0.000 description 12
- 229910052791 calcium Inorganic materials 0.000 description 12
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 12
- 229940088598 enzyme Drugs 0.000 description 12
- OGQSCIYDJSNCMY-UHFFFAOYSA-H iron(3+);methyl-dioxido-oxo-$l^{5}-arsane Chemical compound [Fe+3].[Fe+3].C[As]([O-])([O-])=O.C[As]([O-])([O-])=O.C[As]([O-])([O-])=O OGQSCIYDJSNCMY-UHFFFAOYSA-H 0.000 description 12
- 239000000758 substrate Substances 0.000 description 12
- 239000011686 zinc sulphate Substances 0.000 description 12
- 230000012010 growth Effects 0.000 description 11
- 239000010410 layer Substances 0.000 description 11
- 102000004169 proteins and genes Human genes 0.000 description 11
- 210000001519 tissue Anatomy 0.000 description 11
- 102000018233 Fibroblast Growth Factor Human genes 0.000 description 10
- 108050007372 Fibroblast Growth Factor Proteins 0.000 description 10
- 241001465754 Metazoa Species 0.000 description 10
- 102100037506 Paired box protein Pax-6 Human genes 0.000 description 10
- 102000005157 Somatostatin Human genes 0.000 description 10
- 108010056088 Somatostatin Proteins 0.000 description 10
- 238000005516 engineering process Methods 0.000 description 10
- 229940126864 fibroblast growth factor Drugs 0.000 description 10
- 239000011159 matrix material Substances 0.000 description 10
- 229960000553 somatostatin Drugs 0.000 description 10
- NHXLMOGPVYXJNR-ATOGVRKGSA-N somatostatin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N1)[C@@H](C)O)NC(=O)CNC(=O)[C@H](C)N)C(O)=O)=O)[C@H](O)C)C1=CC=CC=C1 NHXLMOGPVYXJNR-ATOGVRKGSA-N 0.000 description 10
- 101001128694 Homo sapiens Neuroendocrine convertase 1 Proteins 0.000 description 9
- 101000613495 Homo sapiens Paired box protein Pax-4 Proteins 0.000 description 9
- 102100032132 Neuroendocrine convertase 1 Human genes 0.000 description 9
- 102100040909 Paired box protein Pax-4 Human genes 0.000 description 9
- 102000052651 Pancreatic hormone Human genes 0.000 description 9
- 238000000338 in vitro Methods 0.000 description 9
- 239000004025 pancreas hormone Substances 0.000 description 9
- 229940032957 pancreatic hormone Drugs 0.000 description 9
- 239000011148 porous material Substances 0.000 description 9
- 230000009261 transgenic effect Effects 0.000 description 9
- 108010075254 C-Peptide Proteins 0.000 description 8
- 101000687905 Homo sapiens Transcription factor SOX-2 Proteins 0.000 description 8
- 241000288906 Primates Species 0.000 description 8
- 102100024270 Transcription factor SOX-2 Human genes 0.000 description 8
- 230000015572 biosynthetic process Effects 0.000 description 8
- 206010012601 diabetes mellitus Diseases 0.000 description 8
- 238000010899 nucleation Methods 0.000 description 8
- 230000008569 process Effects 0.000 description 8
- 108010059616 Activins Proteins 0.000 description 7
- 102000005606 Activins Human genes 0.000 description 7
- 102100029284 Hepatocyte nuclear factor 3-beta Human genes 0.000 description 7
- 101001062347 Homo sapiens Hepatocyte nuclear factor 3-beta Proteins 0.000 description 7
- 102100032063 Neurogenic differentiation factor 1 Human genes 0.000 description 7
- 108050000588 Neurogenic differentiation factor 1 Proteins 0.000 description 7
- 101800001268 Pancreatic hormone Proteins 0.000 description 7
- 239000000488 activin Substances 0.000 description 7
- 210000002950 fibroblast Anatomy 0.000 description 7
- 210000001654 germ layer Anatomy 0.000 description 7
- 239000003102 growth factor Substances 0.000 description 7
- 210000000496 pancreas Anatomy 0.000 description 7
- 238000010186 staining Methods 0.000 description 7
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 6
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 6
- 101800001586 Ghrelin Proteins 0.000 description 6
- 102400000442 Ghrelin-28 Human genes 0.000 description 6
- 101000711846 Homo sapiens Transcription factor SOX-9 Proteins 0.000 description 6
- 102100034204 Transcription factor SOX-9 Human genes 0.000 description 6
- PLOPBXQQPZYQFA-AXPWDRQUSA-N amlintide Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H]1NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)CCCCN)CSSC1)[C@@H](C)O)C(C)C)C1=CC=CC=C1 PLOPBXQQPZYQFA-AXPWDRQUSA-N 0.000 description 6
- 239000006143 cell culture medium Substances 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 230000002354 daily effect Effects 0.000 description 6
- GNKDKYIHGQKHHM-RJKLHVOGSA-N ghrelin Chemical compound C([C@H](NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)CN)COC(=O)CCCCCCC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C1=CC=CC=C1 GNKDKYIHGQKHHM-RJKLHVOGSA-N 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 102100028412 Fibroblast growth factor 10 Human genes 0.000 description 5
- 102100022054 Hepatocyte nuclear factor 4-alpha Human genes 0.000 description 5
- 101000917237 Homo sapiens Fibroblast growth factor 10 Proteins 0.000 description 5
- 101001045740 Homo sapiens Hepatocyte nuclear factor 4-alpha Proteins 0.000 description 5
- 241000699670 Mus sp. Species 0.000 description 5
- 229920002472 Starch Polymers 0.000 description 5
- 230000004186 co-expression Effects 0.000 description 5
- 238000011161 development Methods 0.000 description 5
- 230000018109 developmental process Effects 0.000 description 5
- 238000012744 immunostaining Methods 0.000 description 5
- 239000003446 ligand Substances 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 230000037361 pathway Effects 0.000 description 5
- 210000002966 serum Anatomy 0.000 description 5
- 235000019698 starch Nutrition 0.000 description 5
- 239000008107 starch Substances 0.000 description 5
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 4
- 101100257359 Caenorhabditis elegans sox-2 gene Proteins 0.000 description 4
- 241000124008 Mammalia Species 0.000 description 4
- 241000699666 Mus <mouse, genus> Species 0.000 description 4
- 101100257363 Mus musculus Sox2 gene Proteins 0.000 description 4
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 4
- 102100035423 POU domain, class 5, transcription factor 1 Human genes 0.000 description 4
- 239000013614 RNA sample Substances 0.000 description 4
- 102000040945 Transcription factor Human genes 0.000 description 4
- 108091023040 Transcription factor Proteins 0.000 description 4
- 102000013814 Wnt Human genes 0.000 description 4
- 108050003627 Wnt Proteins 0.000 description 4
- 102000044880 Wnt3A Human genes 0.000 description 4
- 108700013515 Wnt3A Proteins 0.000 description 4
- 230000007423 decrease Effects 0.000 description 4
- 239000012091 fetal bovine serum Substances 0.000 description 4
- 238000012606 in vitro cell culture Methods 0.000 description 4
- 230000000968 intestinal effect Effects 0.000 description 4
- 210000004185 liver Anatomy 0.000 description 4
- 210000003716 mesoderm Anatomy 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 230000001105 regulatory effect Effects 0.000 description 4
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 3
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 3
- 101150095289 FGF7 gene Proteins 0.000 description 3
- 102100024785 Fibroblast growth factor 2 Human genes 0.000 description 3
- 101000976622 Homo sapiens Zinc finger protein 42 homolog Proteins 0.000 description 3
- 102000007354 PAX6 Transcription Factor Human genes 0.000 description 3
- 102100023550 Zinc finger protein 42 homolog Human genes 0.000 description 3
- 108010023082 activin A Proteins 0.000 description 3
- 210000002469 basement membrane Anatomy 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 230000022131 cell cycle Effects 0.000 description 3
- 239000003636 conditioned culture medium Substances 0.000 description 3
- 238000009826 distribution Methods 0.000 description 3
- 210000003981 ectoderm Anatomy 0.000 description 3
- 210000002744 extracellular matrix Anatomy 0.000 description 3
- 230000007045 gastrulation Effects 0.000 description 3
- 210000000936 intestine Anatomy 0.000 description 3
- 238000004264 monolayer culture Methods 0.000 description 3
- 102000039446 nucleic acids Human genes 0.000 description 3
- 108020004707 nucleic acids Proteins 0.000 description 3
- 150000007523 nucleic acids Chemical class 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 230000035755 proliferation Effects 0.000 description 3
- 239000002464 receptor antagonist Substances 0.000 description 3
- 229940044551 receptor antagonist Drugs 0.000 description 3
- 238000003757 reverse transcription PCR Methods 0.000 description 3
- 230000003248 secreting effect Effects 0.000 description 3
- 230000011664 signaling Effects 0.000 description 3
- 239000002356 single layer Substances 0.000 description 3
- 210000001685 thyroid gland Anatomy 0.000 description 3
- 235000019154 vitamin C Nutrition 0.000 description 3
- 239000011718 vitamin C Substances 0.000 description 3
- 101150068520 wnt3a gene Proteins 0.000 description 3
- 235000009529 zinc sulphate Nutrition 0.000 description 3
- WOLVEMPZUIFSII-IHHOKICGSA-N (2e,4e)-n-[(2s,5s)-5-(hydroxymethyl)-1-methyl-3-oxo-2-propan-2-yl-2,4,5,6-tetrahydro-1,4-benzodiazocin-8-yl]-5-[4-(trifluoromethyl)phenyl]penta-2,4-dienamide Chemical compound CN([C@H](C(N[C@H](CO)CC1=C2)=O)C(C)C)C1=CC=C2NC(=O)\C=C\C=C\C1=CC=C(C(F)(F)F)C=C1 WOLVEMPZUIFSII-IHHOKICGSA-N 0.000 description 2
- QSHGISMANBKLQL-OWJWWREXSA-N (2s)-2-[[2-(3,5-difluorophenyl)acetyl]amino]-n-[(7s)-5-methyl-6-oxo-7h-benzo[d][1]benzazepin-7-yl]propanamide Chemical compound N([C@@H](C)C(=O)N[C@@H]1C(N(C)C2=CC=CC=C2C2=CC=CC=C21)=O)C(=O)CC1=CC(F)=CC(F)=C1 QSHGISMANBKLQL-OWJWWREXSA-N 0.000 description 2
- CDOVNWNANFFLFJ-UHFFFAOYSA-N 4-[6-[4-(1-piperazinyl)phenyl]-3-pyrazolo[1,5-a]pyrimidinyl]quinoline Chemical compound C1CNCCN1C1=CC=C(C2=CN3N=CC(=C3N=C2)C=2C3=CC=CC=C3N=CC=2)C=C1 CDOVNWNANFFLFJ-UHFFFAOYSA-N 0.000 description 2
- HFDKKNHCYWNNNQ-YOGANYHLSA-N 75976-10-2 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@@H](NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)N)C(C)C)[C@@H](C)O)C1=CC=C(O)C=C1 HFDKKNHCYWNNNQ-YOGANYHLSA-N 0.000 description 2
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 2
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 2
- 102100022595 Broad substrate specificity ATP-binding cassette transporter ABCG2 Human genes 0.000 description 2
- 102100031650 C-X-C chemokine receptor type 4 Human genes 0.000 description 2
- 102000007345 Chromogranins Human genes 0.000 description 2
- 108010007718 Chromogranins Proteins 0.000 description 2
- PHEDXBVPIONUQT-UHFFFAOYSA-N Cocarcinogen A1 Natural products CCCCCCCCCCCCCC(=O)OC1C(C)C2(O)C3C=C(C)C(=O)C3(O)CC(CO)=CC2C2C1(OC(C)=O)C2(C)C PHEDXBVPIONUQT-UHFFFAOYSA-N 0.000 description 2
- 108010069241 Connexin 43 Proteins 0.000 description 2
- 102000001045 Connexin 43 Human genes 0.000 description 2
- ZZZCUOFIHGPKAK-UHFFFAOYSA-N D-erythro-ascorbic acid Natural products OCC1OC(=O)C(O)=C1O ZZZCUOFIHGPKAK-UHFFFAOYSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 102100037060 Forkhead box protein D3 Human genes 0.000 description 2
- 102100039290 Gap junction gamma-1 protein Human genes 0.000 description 2
- 102400000921 Gastrin Human genes 0.000 description 2
- 108010052343 Gastrins Proteins 0.000 description 2
- 108060003393 Granulin Proteins 0.000 description 2
- 108010051696 Growth Hormone Proteins 0.000 description 2
- 229940121827 Hedgehog pathway inhibitor Drugs 0.000 description 2
- 102100029087 Hepatocyte nuclear factor 6 Human genes 0.000 description 2
- 101000922348 Homo sapiens C-X-C chemokine receptor type 4 Proteins 0.000 description 2
- 101001029308 Homo sapiens Forkhead box protein D3 Proteins 0.000 description 2
- 101000988619 Homo sapiens Hepatocyte nuclear factor 6 Proteins 0.000 description 2
- 101001069749 Homo sapiens Prospero homeobox protein 1 Proteins 0.000 description 2
- 101000652324 Homo sapiens Transcription factor SOX-17 Proteins 0.000 description 2
- 101000777245 Homo sapiens Undifferentiated embryonic cell transcription factor 1 Proteins 0.000 description 2
- 101000976653 Homo sapiens Zinc finger protein ZIC 1 Proteins 0.000 description 2
- 108010090306 Member 2 Subfamily G ATP Binding Cassette Transporter Proteins 0.000 description 2
- 241001529936 Murinae Species 0.000 description 2
- 101100369076 Mus musculus Tdgf1 gene Proteins 0.000 description 2
- 101710126211 POU domain, class 5, transcription factor 1 Proteins 0.000 description 2
- 102000018886 Pancreatic Polypeptide Human genes 0.000 description 2
- 108700020479 Pancreatic hormone Proteins 0.000 description 2
- 229930040373 Paraformaldehyde Natural products 0.000 description 2
- 102100033880 Prospero homeobox protein 1 Human genes 0.000 description 2
- 239000012980 RPMI-1640 medium Substances 0.000 description 2
- 238000011579 SCID mouse model Methods 0.000 description 2
- 102100038803 Somatotropin Human genes 0.000 description 2
- 101000983124 Sus scrofa Pancreatic prohormone precursor Proteins 0.000 description 2
- 102100030243 Transcription factor SOX-17 Human genes 0.000 description 2
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 2
- 102100031278 Undifferentiated embryonic cell transcription factor 1 Human genes 0.000 description 2
- 229930003268 Vitamin C Natural products 0.000 description 2
- 102100023497 Zinc finger protein ZIC 1 Human genes 0.000 description 2
- 108010076089 accutase Proteins 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 210000002459 blastocyst Anatomy 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 230000011748 cell maturation Effects 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- AOXOCDRNSPFDPE-UKEONUMOSA-N chembl413654 Chemical compound C([C@H](C(=O)NCC(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](C)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)CNC(=O)[C@@H](N)CCC(O)=O)C1=CC=C(O)C=C1 AOXOCDRNSPFDPE-UKEONUMOSA-N 0.000 description 2
- 108010015426 connexin 45 Proteins 0.000 description 2
- 108010007093 dispase Proteins 0.000 description 2
- 210000002242 embryoid body Anatomy 0.000 description 2
- 210000000981 epithelium Anatomy 0.000 description 2
- 230000001605 fetal effect Effects 0.000 description 2
- 210000003953 foreskin Anatomy 0.000 description 2
- 239000003540 gamma secretase inhibitor Substances 0.000 description 2
- 239000000122 growth hormone Substances 0.000 description 2
- 210000003494 hepatocyte Anatomy 0.000 description 2
- 238000010842 high-capacity cDNA reverse transcription kit Methods 0.000 description 2
- 210000005260 human cell Anatomy 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 229910052500 inorganic mineral Inorganic materials 0.000 description 2
- 210000004153 islets of langerhan Anatomy 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 108020004999 messenger RNA Proteins 0.000 description 2
- 235000010755 mineral Nutrition 0.000 description 2
- 239000011707 mineral Substances 0.000 description 2
- 229960003966 nicotinamide Drugs 0.000 description 2
- 235000005152 nicotinamide Nutrition 0.000 description 2
- 239000011570 nicotinamide Substances 0.000 description 2
- 229920002866 paraformaldehyde Polymers 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- 102000014187 peptide receptors Human genes 0.000 description 2
- 108010011903 peptide receptors Proteins 0.000 description 2
- BQJRUJTZSGYBEZ-YVQNUNKESA-N phorbol 12,13-dibutanoate Chemical compound C([C@]1(O)C(=O)C(C)=C[C@H]1[C@@]1(O)[C@H](C)[C@H]2OC(=O)CCC)C(CO)=C[C@H]1[C@H]1[C@]2(OC(=O)CCC)C1(C)C BQJRUJTZSGYBEZ-YVQNUNKESA-N 0.000 description 2
- PHEDXBVPIONUQT-RGYGYFBISA-N phorbol 13-acetate 12-myristate Chemical compound C([C@]1(O)C(=O)C(C)=C[C@H]1[C@@]1(O)[C@H](C)[C@H]2OC(=O)CCCCCCCCCCCCC)C(CO)=C[C@H]1[C@H]1[C@]2(OC(C)=O)C1(C)C PHEDXBVPIONUQT-RGYGYFBISA-N 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 239000012474 protein marker Substances 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- 101150015916 smg1 gene Proteins 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 210000001082 somatic cell Anatomy 0.000 description 2
- 210000000278 spinal cord Anatomy 0.000 description 2
- 210000002784 stomach Anatomy 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 230000003827 upregulation Effects 0.000 description 2
- 102000009310 vitamin D receptors Human genes 0.000 description 2
- 108050000156 vitamin D receptors Proteins 0.000 description 2
- LUZOFMGZMUZSSK-LRDDRELGSA-N (-)-indolactam V Chemical compound C1[C@@H](CO)NC(=O)[C@H](C(C)C)N(C)C2=CC=CC3=C2C1=CN3 LUZOFMGZMUZSSK-LRDDRELGSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- VHZOKTILAXIWHP-UHFFFAOYSA-N 1,4-diazepane;dihydrochloride Chemical compound Cl.Cl.C1CNCCNC1 VHZOKTILAXIWHP-UHFFFAOYSA-N 0.000 description 1
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- 125000004204 2-methoxyphenyl group Chemical group [H]C1=C([H])C(*)=C(OC([H])([H])[H])C([H])=C1[H] 0.000 description 1
- WONYMNWUJVKVII-UHFFFAOYSA-N 3,5-diiodothyropropionic acid Chemical compound IC1=CC(CCC(=O)O)=CC(I)=C1OC1=CC=C(O)C=C1 WONYMNWUJVKVII-UHFFFAOYSA-N 0.000 description 1
- ZDYIYHVFVNXRLD-UHFFFAOYSA-N 3-(3,5-dimethyl-1-phenylpyrazol-4-yl)-2-methylidenepiperazin-1-amine Chemical compound CC1=NN(C(=C1C1C(N(CCN1)N)=C)C)C1=CC=CC=C1 ZDYIYHVFVNXRLD-UHFFFAOYSA-N 0.000 description 1
- 125000004208 3-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C([H])C(*)=C1[H] 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000004939 6-pyridyl group Chemical group N1=CC=CC=C1* 0.000 description 1
- 102000018918 Activin Receptors Human genes 0.000 description 1
- 108010052946 Activin Receptors Proteins 0.000 description 1
- 102100027647 Activin receptor type-2B Human genes 0.000 description 1
- 241000272814 Anser sp. Species 0.000 description 1
- 101150018431 Arx gene Proteins 0.000 description 1
- 210000002237 B-cell of pancreatic islet Anatomy 0.000 description 1
- 102400001242 Betacellulin Human genes 0.000 description 1
- 101800001382 Betacellulin Proteins 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 102000007350 Bone Morphogenetic Proteins Human genes 0.000 description 1
- 108010007726 Bone Morphogenetic Proteins Proteins 0.000 description 1
- 102100025422 Bone morphogenetic protein receptor type-2 Human genes 0.000 description 1
- 108050008407 Bone morphogenetic protein receptor type-2 Proteins 0.000 description 1
- 102100037904 CD9 antigen Human genes 0.000 description 1
- 102000024905 CD99 Human genes 0.000 description 1
- 108060001253 CD99 Proteins 0.000 description 1
- 241000202252 Cerberus Species 0.000 description 1
- 108060005980 Collagenase Proteins 0.000 description 1
- 102000029816 Collagenase Human genes 0.000 description 1
- 206010010099 Combined immunodeficiency Diseases 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- SNPLKNRPJHDVJA-ZETCQYMHSA-N D-panthenol Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCCO SNPLKNRPJHDVJA-ZETCQYMHSA-N 0.000 description 1
- 101000923091 Danio rerio Aristaless-related homeobox protein Proteins 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- 102000018711 Facilitative Glucose Transport Proteins Human genes 0.000 description 1
- 102000003971 Fibroblast Growth Factor 1 Human genes 0.000 description 1
- 108090000386 Fibroblast Growth Factor 1 Proteins 0.000 description 1
- 102100028072 Fibroblast growth factor 4 Human genes 0.000 description 1
- 102100037362 Fibronectin Human genes 0.000 description 1
- 108010067306 Fibronectins Proteins 0.000 description 1
- 102000001267 GSK3 Human genes 0.000 description 1
- 108060006662 GSK3 Proteins 0.000 description 1
- 229940125373 Gamma-Secretase Inhibitor Drugs 0.000 description 1
- 108091052347 Glucose transporter family Proteins 0.000 description 1
- 108010051975 Glycogen Synthase Kinase 3 beta Proteins 0.000 description 1
- 102100038104 Glycogen synthase kinase-3 beta Human genes 0.000 description 1
- 101150097704 HHEX gene Proteins 0.000 description 1
- 102000003693 Hedgehog Proteins Human genes 0.000 description 1
- 108090000031 Hedgehog Proteins Proteins 0.000 description 1
- 102100035961 Hematopoietically-expressed homeobox protein HHEX Human genes 0.000 description 1
- 229920002971 Heparan sulfate Polymers 0.000 description 1
- 108010087745 Hepatocyte Nuclear Factor 3-beta Proteins 0.000 description 1
- 102000009094 Hepatocyte Nuclear Factor 3-beta Human genes 0.000 description 1
- 102100022057 Hepatocyte nuclear factor 1-alpha Human genes 0.000 description 1
- 108700005087 Homeobox Genes Proteins 0.000 description 1
- 102100031470 Homeobox protein ARX Human genes 0.000 description 1
- 102100031672 Homeobox protein CDX-1 Human genes 0.000 description 1
- 102100031670 Homeobox protein CDX-4 Human genes 0.000 description 1
- 102100030634 Homeobox protein OTX2 Human genes 0.000 description 1
- 101000738354 Homo sapiens CD9 antigen Proteins 0.000 description 1
- 101001060274 Homo sapiens Fibroblast growth factor 4 Proteins 0.000 description 1
- 101001021503 Homo sapiens Hematopoietically-expressed homeobox protein HHEX Proteins 0.000 description 1
- 101001045751 Homo sapiens Hepatocyte nuclear factor 1-alpha Proteins 0.000 description 1
- 101000923090 Homo sapiens Homeobox protein ARX Proteins 0.000 description 1
- 101000777808 Homo sapiens Homeobox protein CDX-1 Proteins 0.000 description 1
- 101000777790 Homo sapiens Homeobox protein CDX-4 Proteins 0.000 description 1
- 101000584400 Homo sapiens Homeobox protein OTX2 Proteins 0.000 description 1
- 101000738523 Homo sapiens Pancreas transcription factor 1 subunit alpha Proteins 0.000 description 1
- 101000655352 Homo sapiens Telomerase reverse transcriptase Proteins 0.000 description 1
- 101000819074 Homo sapiens Transcription factor GATA-4 Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- LUZOFMGZMUZSSK-UHFFFAOYSA-N Indolactam-V Natural products C1C(CO)NC(=O)C(C(C)C)N(C)C2=CC=CC3=C2C1=CN3 LUZOFMGZMUZSSK-UHFFFAOYSA-N 0.000 description 1
- 102000011845 Iodide peroxidase Human genes 0.000 description 1
- 108010036012 Iodide peroxidase Proteins 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 229930182816 L-glutamine Natural products 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 108010085895 Laminin Proteins 0.000 description 1
- 102000007547 Laminin Human genes 0.000 description 1
- 102100027754 Mast/stem cell growth factor receptor Kit Human genes 0.000 description 1
- 102100025169 Max-binding protein MNT Human genes 0.000 description 1
- 101100460498 Mus musculus Nkx2-2 gene Proteins 0.000 description 1
- 101100096242 Mus musculus Sox9 gene Proteins 0.000 description 1
- 108010056852 Myostatin Proteins 0.000 description 1
- 238000011789 NOD SCID mouse Methods 0.000 description 1
- 101100519293 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pdx-1 gene Proteins 0.000 description 1
- 102100037369 Nidogen-1 Human genes 0.000 description 1
- 238000000636 Northern blotting Methods 0.000 description 1
- 102000007399 Nuclear hormone receptor Human genes 0.000 description 1
- 108020005497 Nuclear hormone receptor Proteins 0.000 description 1
- 101150081664 PAX6 gene Proteins 0.000 description 1
- 238000010222 PCR analysis Methods 0.000 description 1
- 102100037878 Pancreas transcription factor 1 subunit alpha Human genes 0.000 description 1
- 101150075928 Pax4 gene Proteins 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 102100033237 Pro-epidermal growth factor Human genes 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 108010067787 Proteoglycans Proteins 0.000 description 1
- 102000016611 Proteoglycans Human genes 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 description 1
- 239000012979 RPMI medium Substances 0.000 description 1
- 238000011529 RT qPCR Methods 0.000 description 1
- 101100437153 Rattus norvegicus Acvr2b gene Proteins 0.000 description 1
- 102000034527 Retinoid X Receptors Human genes 0.000 description 1
- 108010038912 Retinoid X Receptors Proteins 0.000 description 1
- 206010043276 Teratoma Diseases 0.000 description 1
- 108010071769 Thyroid Hormone Receptors beta Proteins 0.000 description 1
- 102100021380 Transcription factor GATA-4 Human genes 0.000 description 1
- 101710195626 Transcriptional activator protein Proteins 0.000 description 1
- 102000004338 Transferrin Human genes 0.000 description 1
- 108090000901 Transferrin Proteins 0.000 description 1
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 1
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 1
- 108091008605 VEGF receptors Proteins 0.000 description 1
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- 229930003316 Vitamin D Natural products 0.000 description 1
- QYSXJUFSXHHAJI-XFEUOLMDSA-N Vitamin D3 Natural products C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@H](C)CCCC(C)C)=C/C=C1\C[C@@H](O)CCC1=C QYSXJUFSXHHAJI-XFEUOLMDSA-N 0.000 description 1
- CIUQDSCDWFSTQR-UHFFFAOYSA-N [C]1=CC=CC=C1 Chemical compound [C]1=CC=CC=C1 CIUQDSCDWFSTQR-UHFFFAOYSA-N 0.000 description 1
- 238000004115 adherent culture Methods 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 210000000577 adipose tissue Anatomy 0.000 description 1
- 238000011166 aliquoting Methods 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 238000003491 array Methods 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 210000003445 biliary tract Anatomy 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 229940112869 bone morphogenetic protein Drugs 0.000 description 1
- 239000003715 calcium chelating agent Substances 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229960002424 collagenase Drugs 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 230000001143 conditioned effect Effects 0.000 description 1
- 230000003750 conditioning effect Effects 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 239000000430 cytokine receptor antagonist Substances 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000032459 dedifferentiation Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 230000023011 digestive tract development Effects 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 229910001882 dioxygen Inorganic materials 0.000 description 1
- KAKKHKRHCKCAGH-UHFFFAOYSA-L disodium;(4-nitrophenyl) phosphate;hexahydrate Chemical compound O.O.O.O.O.O.[Na+].[Na+].[O-][N+](=O)C1=CC=C(OP([O-])([O-])=O)C=C1 KAKKHKRHCKCAGH-UHFFFAOYSA-L 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 229940121647 egfr inhibitor Drugs 0.000 description 1
- 230000003028 elevating effect Effects 0.000 description 1
- 230000013020 embryo development Effects 0.000 description 1
- 210000002308 embryonic cell Anatomy 0.000 description 1
- 239000002158 endotoxin Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000006862 enzymatic digestion Effects 0.000 description 1
- 108060002566 ephrin Proteins 0.000 description 1
- 102000012803 ephrin Human genes 0.000 description 1
- 239000003797 essential amino acid Substances 0.000 description 1
- 235000020776 essential amino acid Nutrition 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000010195 expression analysis Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000008246 gaseous mixture Substances 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 210000001647 gastrula Anatomy 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 238000003633 gene expression assay Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 210000004907 gland Anatomy 0.000 description 1
- 230000000762 glandular Effects 0.000 description 1
- 101150090422 gsk-3 gene Proteins 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- 230000005745 host immune response Effects 0.000 description 1
- 210000003917 human chromosome Anatomy 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 238000010166 immunofluorescence Methods 0.000 description 1
- 238000002991 immunohistochemical analysis Methods 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000007901 in situ hybridization Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 230000003914 insulin secretion Effects 0.000 description 1
- 210000002660 insulin-secreting cell Anatomy 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 108010082117 matrigel Proteins 0.000 description 1
- 210000004379 membrane Anatomy 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 229940029985 mineral supplement Drugs 0.000 description 1
- 235000020786 mineral supplement Nutrition 0.000 description 1
- 210000000663 muscle cell Anatomy 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- KVQVEZQDNHMQJV-UHFFFAOYSA-N n-[(3-benzamidophenyl)carbamothioyl]-3,4,5-trimethoxybenzamide Chemical compound COC1=C(OC)C(OC)=CC(C(=O)NC(=S)NC=2C=C(NC(=O)C=3C=CC=CC=3)C=CC=2)=C1 KVQVEZQDNHMQJV-UHFFFAOYSA-N 0.000 description 1
- NFVJNJQRWPQVOA-UHFFFAOYSA-N n-[2-chloro-5-(trifluoromethyl)phenyl]-2-[3-(4-ethyl-5-ethylsulfanyl-1,2,4-triazol-3-yl)piperidin-1-yl]acetamide Chemical compound CCN1C(SCC)=NN=C1C1CN(CC(=O)NC=2C(=CC=C(C=2)C(F)(F)F)Cl)CCC1 NFVJNJQRWPQVOA-UHFFFAOYSA-N 0.000 description 1
- YOVNFNXUCOWYSG-UHFFFAOYSA-N n-[3-[2-(4-amino-1,2,5-oxadiazol-3-yl)-1-ethylimidazo[4,5-c]pyridin-6-yl]oxyphenyl]-4-(2-morpholin-4-ylethoxy)benzamide Chemical compound C1=C2N(CC)C(C=3C(=NON=3)N)=NC2=CN=C1OC(C=1)=CC=CC=1NC(=O)C(C=C1)=CC=C1OCCN1CCOCC1 YOVNFNXUCOWYSG-UHFFFAOYSA-N 0.000 description 1
- 210000003061 neural cell Anatomy 0.000 description 1
- 108010008217 nidogen Proteins 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 125000003835 nucleoside group Chemical group 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- 210000002747 omentum Anatomy 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 230000005305 organ development Effects 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 230000015031 pancreas development Effects 0.000 description 1
- 210000003577 pancreatic endocrine progenitor Anatomy 0.000 description 1
- 210000004923 pancreatic tissue Anatomy 0.000 description 1
- 210000004303 peritoneum Anatomy 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 210000002826 placenta Anatomy 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 210000003240 portal vein Anatomy 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 210000001811 primitive streak Anatomy 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 108010028138 prohibitin Proteins 0.000 description 1
- 102000016670 prohibitin Human genes 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 150000003195 pteridines Chemical class 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000007261 regionalization Effects 0.000 description 1
- 238000009256 replacement therapy Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 102000000568 rho-Associated Kinases Human genes 0.000 description 1
- 108010041788 rho-Associated Kinases Proteins 0.000 description 1
- 210000003752 saphenous vein Anatomy 0.000 description 1
- 238000007790 scraping Methods 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000012679 serum free medium Substances 0.000 description 1
- MFBOGIVSZKQAPD-UHFFFAOYSA-M sodium butyrate Chemical compound [Na+].CCCC([O-])=O MFBOGIVSZKQAPD-UHFFFAOYSA-M 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 125000002813 thiocarbonyl group Chemical group *C(*)=S 0.000 description 1
- 210000001541 thymus gland Anatomy 0.000 description 1
- 102000004217 thyroid hormone receptors Human genes 0.000 description 1
- 108090000721 thyroid hormone receptors Proteins 0.000 description 1
- 230000036962 time dependent Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 108091006107 transcriptional repressors Proteins 0.000 description 1
- 239000012581 transferrin Substances 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 235000019166 vitamin D Nutrition 0.000 description 1
- 239000011710 vitamin D Substances 0.000 description 1
- 150000003710 vitamin D derivatives Chemical class 0.000 description 1
- 229940046008 vitamin d Drugs 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
Images































































































































































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0603—Embryonic cells ; Embryoid bodies
- C12N5/0606—Pluripotent embryonic cells, e.g. embryonic stem cells [ES]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0676—Pancreatic cells
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5044—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics involving specific cell types
- G01N33/507—Pancreatic cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/05—Inorganic components
- C12N2500/10—Metals; Metal chelators
- C12N2500/20—Transition metals
- C12N2500/24—Iron; Fe chelators; Transferrin
- C12N2500/25—Insulin-transferrin; Insulin-transferrin-selenium
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/105—Insulin-like growth factors [IGF]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/115—Basic fibroblast growth factor (bFGF, FGF-2)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/117—Keratinocyte growth factors (KGF-1, i.e. FGF-7; KGF-2, i.e. FGF-12)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/15—Transforming growth factor beta (TGF-β)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/30—Hormones
- C12N2501/38—Hormones with nuclear receptors
- C12N2501/385—Hormones with nuclear receptors of the family of the retinoic acid recptor, e.g. RAR, RXR; Peroxisome proliferator-activated receptor [PPAR]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/30—Hormones
- C12N2501/38—Hormones with nuclear receptors
- C12N2501/395—Thyroid hormones
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/40—Regulators of development
- C12N2501/415—Wnt; Frizzeled
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/70—Enzymes
- C12N2501/72—Transferases [EC 2.]
- C12N2501/727—Kinases (EC 2.7.)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/999—Small molecules not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/02—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from embryonic cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2533/00—Supports or coatings for cell culture, characterised by material
- C12N2533/30—Synthetic polymers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2533/00—Supports or coatings for cell culture, characterised by material
- C12N2533/90—Substrates of biological origin, e.g. extracellular matrix, decellularised tissue
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Chemical & Material Sciences (AREA)
- Biotechnology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Cell Biology (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Organic Chemistry (AREA)
- Genetics & Genomics (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Immunology (AREA)
- General Engineering & Computer Science (AREA)
- Urology & Nephrology (AREA)
- Molecular Biology (AREA)
- Hematology (AREA)
- Reproductive Health (AREA)
- Gynecology & Obstetrics (AREA)
- Developmental Biology & Embryology (AREA)
- Physics & Mathematics (AREA)
- Toxicology (AREA)
- Food Science & Technology (AREA)
- Medicinal Chemistry (AREA)
- Tropical Medicine & Parasitology (AREA)
- Analytical Chemistry (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- User Interface Of Digital Computer (AREA)
Abstract
本发明提供了通过在培养容器中的空气‑液体界面处培养,来促进多能干细胞分化成表达PDX1、NKX6.1和HB9的胰腺内分泌细胞的方法、细胞培养物和分化培养基。本发明还提供了在所述空气‑液体界面处培养的细胞的体内成熟。
Description
相关申请的交叉引用
本申请要求美国临时申请61/747,662(提交于2012年12月31日)的 优先权,所述美国临时申请全文以引用的方式并入。
技术领域
本发明属于细胞分化领域。更具体地,本发明提供用于通过在空气-液 体界面处培养细胞来从人多能干细胞生成胰腺内胚层细胞、胰腺内分泌前 体细胞和单激素胰腺内分泌细胞的方法、细胞培养物和培养基。
背景技术
用于Ⅰ型糖尿病的细胞替代疗法的进展以及可移植胰岛的缺乏已使得 人们把注意力集中在开发适于移植的胰岛素分泌细胞(即β细胞)的来源 上。一种方法是从多能干细胞诸如胚胎干细胞生成功能性β细胞。
在脊椎动物胚胎发育中,多能细胞可在称为原肠胚形成的过程中产生 包括三个胚层(外胚层、中胚层和内胚层)的细胞群。组织诸如甲状腺、 胸腺、胰腺、肠和肝脏将从内胚层经由中间阶段发育而来。该过程中的中 间阶段是定形内胚层的形成。
到原肠胚形成为止,内胚层划分成可通过一组因子的表达来识别的前- 后域,所述因子唯一地标记内胚层的前、中和后区。例如,HHEX和SOX2 识别内胚层的前区,而CDX1、CDX2和CDX4识别内胚层的后区。
内胚层组织的迁移使内胚层与有助于肠管的区域化的不同的中胚层组 织紧密接近。这通过多种分泌因子诸如FGF、WNT、TGF-β、视黄酸(RA) 和BMP配体及其拮抗剂来完成。例如,FGF4和BMP促进假定后肠内胚层 中的CDX2的表达并阻遏前基因Hhex和SOX2的表达(2000 Development, 127:1563–1567)。WNT信号转导也已显示与FGF信号转导平行工作,以促 进后肠发育并抑制前肠命运(2007 Development,134:2207–2217)。最后,由 间充质分泌的视黄酸调节前肠-后肠边界(2002 CurrBiol,12:1215-1220)。
特异性转录因子的表达水平可用于指定组织的身份。在定形内胚层转 化成原肠管期间,肠管变得区域化成广泛域,所述广泛域可通过限制性基 因表达模式在分子水平上进行观察。该肠管中的区域化胰腺域显示PDX1 的极高表达以及CDX2和SOX2的极低表达。PDX1、NKX6.1、PTFlA和 NKX2.2在胰腺组织中高度表达;而CDX2在肠组织中高度表达。
定形内胚层分化成胰内胚层从而形成胰腺。背侧和腹侧胰腺域产生自 前肠上皮。前肠也产生食道、气管、肺、甲状腺、胃、肝脏和胆管系统。
胰腺内胚层的细胞表达胰-十二指肠同源盒基因PDX1。在不存在 PDX1时,胰腺形成腹胰芽和背胰芽后不再发育。因而,PDX1表达标志着 胰腺器官形成的一个关键步骤。成熟的胰腺包括胰腺内胚层分化成的外分 泌组织和内分泌组织。
D’Amour等人描述了在高浓度活化素和低血清的存在下,产生人胚胎 干细胞衍生的定形内胚层的富集培养物(Nature Biotechnology 2005, 23:1534-1541;美国专利7,704,738)。将这些细胞移植在小鼠的肾包膜 下,据报道导致分化成具有内胚层组织特征的更成熟的细胞(美国专利 7,704,738)。在添加FGF10和视黄酸后,人胚胎干细胞衍生的定形内胚层 细胞可进一步分化成PDX1阳性细胞(美国专利申请公布 2005/0266554)。这些胰腺前体细胞在免疫缺陷小鼠的脂肪垫中的后续移植 导致在3-4个月成熟期后形成功能胰腺内分泌细胞(美国专利7,993,920和 美国专利7,534,608)。
Fisk等人报道用于由人胚胎干细胞产生胰岛细胞的系统(美国专利 7,033,831)。在这种情况下,分化途径分成三个阶段。人胚胎干细胞首先 使用丁酸钠和活化素A的组合而分化成内胚层(美国专利7,326,572)。然 后将细胞和与EGF或β细胞素(betacellulin)结合的BMP拮抗剂诸如头蛋白 (Noggin)一起培养,以生成PDX1阳性细胞。通过烟酰胺诱导终末分化。
小分子抑制剂也已用于诱导胰腺内分泌前体细胞。例如,TGF-β受体 和BMP受体的小分子抑制剂(Development 2011,138:861-871;Diabetes 2011, 60:239-247)已用于显著提高胰腺内分泌细胞的数目。另外,小分子活化剂 也已用于生成定形内胚层细胞或胰腺前体细胞(Curr Opin Cell Biol 2009, 21:727-732;Nature Chem Biol 2009,5:258-265)。
HB9(也称为HlXB9和MNX1)是一种在胰腺发育早期表达的BHLH 转录活化因子蛋白,该表达始于大约胚胎期第8天。HB9也在脊索和脊髓 中表达。在以PDX1和NKX6.1表达细胞表达的胰腺上皮中,HB9的表达 是短暂的并且在大约第10.5天达到峰值。在大约第12.5天时,HB9表达下 降,并且在后期阶段其表达仅局限于β细胞。在HB9的无效突变的纯合子小鼠中,胰腺的背瓣无法发育(Nat Genet 23:67-70,1999;Nat Genet 23:71-75, 1999)。HB9-/-β-细胞表达低水平的葡萄糖转运蛋白、GLUT2和NKX6.1。 此外,HB9-/-胰腺显示出胰岛素阳性细胞数目显著降低,但是并未显著影 响其他胰腺激素的表达。因此,对HB9的时间控制对于正常的β细胞发育 和功能而言是至关重要的。虽然对关于调控β细胞中HB9表达的因子知之 甚少,但是对斑马鱼的近期研究表明视黄酸可正向调控HB9的表达(Development,138,4597-4608,2011)。
甲状腺激素,即四碘甲状腺原氨酸(“T4”)和三碘甲状腺原氨酸 (“T3”)是由甲状腺腺体产生的基于酪氨酸的激素,并且主要负责代谢调 控。甲状腺激素在血液中的主要形式是T4,T4相较于T3具有更长的半衰 期。释放到血液中的T4与T3的比率是大致20比1。在细胞中由脱碘酶将 T4转变成活性更强的T3(活性是T4的三至四倍)。
T3结合至甲状腺激素受体TRα1和TRβ1(TR)。TR是一种核激素受 体,该受体与类视黄醇X受体形成异源二聚体。当不存在配体时,所述二 聚体结合至四碘甲状腺原氨酸应答元件(TRE),并且充当转录抑制子。T3 结合至TR降低了对TRE依赖性基因的抑制并且引发各种靶基因的表达。 虽然多项研究已显示T3对增进β细胞增殖、减少细胞凋亡以及改善胰岛素 分泌的作用,但是T3对细胞分化的作用尚不明确。
转化生长因子β(“TGF-β”)是多种生物学过程中所涉及的多效性细胞 因子大家族的一员,所述生物学过程包括生长控制、分化、迁移、细胞存 活、纤维变性和发育命运的特化。TGF-β超家族成员通过包括II型受体和I 型受体在内的受体复合物传导信号。TGF-B配体(诸如活化素和生长分化 因子(“GDF”))使II型受体与I型受体结合在一起。所述II型受体磷酸化 并活化所述复合物中的I型受体。哺乳动物有五种II型受体:TβR-II、ActR-II、ActR-IIB、BMPR-II和AMHR-II,以及七种I型受体(ALK 1–7)。 活化素和相关的配体经由ActR-II或ActR-IIB与ALK4或ALK5的组合来传 导信号,并且BMP通过ALK2、ALK3和ALK6与ActR-II、ActR-IIB或 BMPR-II的组合来传导信号。AMH通过AMHR-II与ALK6的复合物来传导信号,并且Nodal已经显示为通过ActR-IIB和ALK7的复合物来传导信号 (Cell.2003,113(6):685-700)。在TGF-β配体结合至适当的受体之后,所产生 的信号主要通过Smads复合物的活化来转导至细胞核。在活化时,所述I型 受体磷酸化由受体调控的Smads亚族的成员。所述磷酸化使所述成员活化 并且使得它们能够与常见的中介体Smad(Smad4)一起形成复合物。 Smad1、Smad5和Smad8是ALK1、ALK2、ALK3和ALK6的底物,而 Smad2和Smad3是ALK4、ALK5和ALK7的底物(FASEB J 13:2105–2124)。 活化的Smad复合物在细胞核中积聚,在细胞核中所述复合物直接参与通常 与其他特异性DNA结合转录因子相关联的靶基因的转录。选择性抑制 TGF-β受体的化合物已被开发用于治疗应用,以及在从各种干细胞群重新编 程和分化的情形中用于调控细胞命运。具体地,ALK5抑制剂已经早就用于 将胚胎干细胞的分化引导至内分泌命运(Diabetes,2011,60(1):239-47)。
一般来讲,祖细胞分化成功能性β细胞的过程经历多个阶段;并且对 从祖细胞诸如人多能干细胞生成胰腺细胞的方案的改善已经取得了显著进 展。虽然有这些研究进展,但是祖细胞分化过程中的每一步骤均提出独特 的挑战。因此,仍然存在针对形成功能性内分泌细胞的方案的需要,并且 具体地功能性β细胞。
附图说明
图1A至图1H示出使用实例1中所述的方法在空气-液体界面处培养的 细胞在以下时间点的相位对比图像:第1天(图1A);第5天(图1B); 第6天(图1C);第7天(图1D);第9天(图1E);第13天(图 1F);第16天(图1G);以及第21天(图1H)。
图2A至图2K示出使用在实例1中所述的方法在空气-液体界面处分化 一周并且针对以下物质进行免疫染色的细胞的图像:DAPI(图2A);胰 岛素(图2B);HB9(图2C);DAPI(图2D);胰高血糖素(图 2E);胰岛素(图2F);DAPI(图2G);胰岛素(图2H);生长抑素(图2I);NKX6.1(图2J);以及胰岛素(图2K)。实验组A-C、D-F、 G-I和J-K取自相同的视野。
图3A至图3H示出使用在实例1中所述的方法在空气-液体界面处分化 两周并且针对以下物质进行免疫染色的细胞的图像:胰岛素(图3A);胰 高血糖素(图3B);胰岛素(图3C);生长抑素(图3D);胰岛素(图 3E);NKX6.1(图3F);HB9(图3G);以及NKX6.1(图2H)。实验组A-B、C-D、E-F和G-H取自相同的视野。
图4A至图4D示出使用在实例1中所述的方法在空气-液体界面处分化 三周并且针对以下物质进行免疫染色的细胞的图像:胰岛素(图4A),胰 高血糖素(图4B),胰岛素(图4C),以及生长抑素(图4D)。实验组 A-B和C-D取自相同的视野。
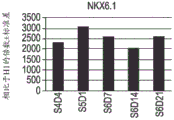
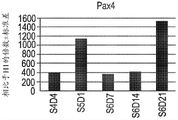
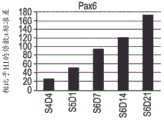
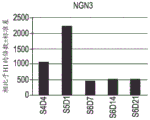
图5A至图5R示出了如实例1中所概述分化的人胚胎干细胞系H1的 细胞中下列基因的表达的实时PCR分析的数据:PDX1(图5A);NKX6.1 (图5B);PAX4(图5C);PAX6(图5D);NGN3(图5E);NKX2.2 (图5F);ABCC8(图5G);嗜铬粒蛋白-A(图5H);PCSK1(图 5I);IAPP(图5J);胰岛素(图5K);胰高血糖素(图5L);生长抑 素(图5M);生长素释放肽(图5N);PTFlA(图5O);ZIC1(图 5P);CDX2(图5Q);以及SOX9(图5R)。细胞是在第5阶段(S5) 之后在空气-液体界面处培养的。
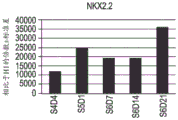
图6A至图6L示出了如实例2中所概述分化的人胚胎干细胞系H1的 细胞中下列基因的表达的实时PCR分析的数据:PDX1(图6A);NKX6.1 (图6B);PAX4(图6C);PAX6(图6D);NGN3(图6E);NKX2.2 (图6F);ABCC8(图6G);嗜铬粒蛋白-A(图6H);PCSK1(图 6I);IAPP(图6J);胰岛素(图6K);以及胰高血糖素(图6L)。
图7A至图7L示出了如实例3中所概述分化的人胚胎干细胞系H1的 细胞中下列基因的表达的实时PCR分析的数据:PDX1(图7A);NKX6.1 (图7B);PAX4(图7C);PAX6(图7D);NGN3(图7E);NKX2.2 (图7F);ABCC8(图7G);嗜铬粒蛋白-A(图7H);PCSK1(图 7I);IAPP(图7J);胰岛素(图7K);以及胰高血糖素(图7L)。
图8A至图8H示出了如实例4中所概述分化的人胚胎干细胞系H1的 细胞中下列基因的表达的实时PCR分析的数据:PDX1(图8A);NKX6.1 (图8B);NGN3(图8C);ABCC8(图8D);PCSK1(图8E);生长 素释放肽(图8F);胰高血糖素(图8G);以及胰岛素(图8H)。
图9A至图9F示出了如实例4中所概述分化的人胚胎干细胞系H1的 细胞中下列基因的表达的实时PCR分析的数据:PDX1(图9A);NKX6.1 (图9B);NGN3(图9C);ABCC8(图9D);胰高血糖素(图9E); 以及胰岛素(图9F)。
图10A至图10B示出对根据实例4在空气-液体界面处培养并用1微摩 尔SD208抑制剂(图10A)或1微摩尔ALK5抑制剂II(图10B)处理并 且针对嗜铬粒蛋白-A(胰腺内分泌标志物)和NKX6.1(胰腺前体标志物和 β细胞特异性标志物)进行染色的第6阶段细胞的免疫染色结果。
图11A至图11H示出了如实例6中所述分化的人胚胎干细胞系H1的 细胞中下列基因的表达的实时PCR分析的数据:ABCC8(图11A);胰高 血糖素(图11B);淀粉不溶素(图11C);胰岛素(图11D);NGN3 (图11E);NKX2.2(图11F);NKX6.1(图11G);以及PDX1(图 11H)。所述数据示出为相对于未分化的H1细胞系成倍增长。
图12A至图12H示出了如实例7中所概述分化的并且在ALI处培养的 人胚胎干细胞系H1的细胞中下列基因的表达的实时PCR分析的数据: ABCC8(图12A);胰高血糖素(图12B);淀粉不溶素(图12C);胰 岛素(图12D);NGN3(图12E);NKX2.2(图12F);NKX6.1(图12G);以及PDX1(图12H)。
图13A至图13H示出了如实例8中所概述分化的并且在ALI处培养的 人胚胎干细胞系H1的细胞中下列基因的表达的实时PCR分析的数据: ABCC8(图13A);胰高血糖素(图13B);淀粉不溶素(图13C);胰 岛素(图13D);NGN3(图13E);NKX2.2(图13F);NKX6.1(图13G);以及PDX1(图13H)。
图14A至图14H示出了如实例9中所概述分化的并且在ALI处培养的 人胚胎干细胞系H1的细胞中下列基因的表达的实时PCR分析的数据: ABCC8(图14A);胰高血糖素(图14B);淀粉不溶素(图14C);胰 岛素(图14D);ISL-1(图14E);MNX1(图14F);NKX6.1(图14G);以及SLC30A8(图14H)。
图15A至图15J示出根据实例10分化并且针对下列基因进行染色的第 5阶段第3天(S5D3)的细胞的FACS分布图:同种型对照(图15A); NKX6.1(图15B);NKX2.2(图15C);NKX6.1(Y轴)与胰岛素(X 轴)共染色(图15D);PDX1(X轴)与KI-67(Y轴)共染色(图 15E);PAX6(图15F);ISL-1(图15G);FOXA2(图15H);NeuroD (图15I);以及胰高血糖素(Y轴)与胰岛素(X轴)共染色(图 15J)。
图16A至图16I示出根据实例10分化并且针对下列基因进行染色的第 6阶段第5天的细胞的FACS分布图:同种型对照(图16A);NKX6.1(Y 轴)与嗜铬粒蛋白-A(X轴)共染色(图16B);NKX2.2(Y轴)与嗜铬 粒蛋白-A(X轴)共染色(图16C);NKX6.1(Y轴)与胰岛素(X轴) 共染色(图16D);PDX1(X轴)与KI-67(Y轴)共染色(图16E); PAX6(图16F);ISL-1(图16G);FOXA2(图16H);以及NeuroD (图16I)。
图17A至图17I示出根据实例10分化并且针对下列基因进行染色的第 6阶段第15天的FACS(细胞的荧光活化细胞分选)(Fluorescence-activated cell sorting;FACS)分布图:同种型对照(图17A);NKX6.1(Y轴)与嗜 铬粒蛋白-A(X轴)共染色(图17B);NKX2.2(Y轴)与嗜铬粒蛋白-A (X轴)共染色(图17C);胰高血糖素(Y轴)与胰岛素(X轴)共染色 (图17D);NKX6.1(Y轴)与胰岛素(X轴)共染色(图17E);PDX1 (X轴)与KI-67(Y轴)共染色(图17F);ISL-1(图17G);FOXA2 (图17H);以及NeuroD(图17I)。
图18A至图18C示出根据实例1分化并且针对下列基因进行染色的第 4阶段第4天(S4D4)的FACS(细胞的荧光活化细胞分选)分布图: NKX6.1(Y轴)与嗜铬粒蛋白-A(X轴)共染色(图18A);PDX1(X 轴)与KI-67(Y轴)共染色(图18B);以及NKX6.1(Y轴)与胰岛素 (X轴)共染色(图18C)。
图19A至图19C示出根据实例11分化并且针对下列基因进行染色的 第6阶段第6天的FACS(细胞的荧光活化细胞分选)分布图:NKX6.1(Y 轴)与嗜铬粒蛋白-A(X轴)共染色(图19A);PDX1(X轴)与KI-67 (Y轴)共染色(图19B);以及NKX6.1(Y轴)与胰岛素(X轴)共染 色(图19C)。
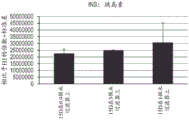
图20示出如实例11所述移植有各种细胞群的NOD-SCID小鼠中的人 C肽制备的体内动力学。
图21A至图21F示出如实例12中所概述分化的人胚胎干细胞系H1的 细胞中下列基因的表达的实时PCR分析的数据:淀粉不溶素(图21A); 胰岛素(图21B);MAFA(图21C);NKX6.1(图21D);PTF1a(图 21E);以及SOX9(图21F)。
图22A至图22D示出如实例13中所概述分化的人胚胎干细胞系H1的 细胞中下列基因的实时PCR数据:MAFA(图22A);胰岛素(图 22B);淀粉不溶素(图22C);以及NKX6.1(图22D)。
图23A至图23F示出如实例5中所概述的分化的人胚胎干细胞系H1 的细胞中下列基因的表达的实时PCR分析的数据:PDX1(图23A); NKX6.1(图23B);NGN3(图23C);ABCC8(图23D);胰高血糖素 (图23E);以及胰岛素(图23F)。
具体实施方式
在结合附图阅读以下对本发明的详细说明时能够更好地进行理解。提 供附图是出于说明本发明的某些实施例的目的。然而,本发明不限于示出 的精确布置方式、实例和工具。为了清晰说明本公开内容,以非限制性方 式将本发明的具体实施方式分成描述或阐明本发明某些特征、实施例或应 用的小节。
本发明涉及通过以下方法使内胚层祖细胞诸如多能干细胞分化成表现 出胰腺内分泌细胞特征的细胞:至少部分地在存在于开放培养容器中或部 分填充有培养基的培养容器中的空气-液体界面处培养所述祖细胞。虽然为 了方便起见而在本文中称作“空气”,但是本发明不限于在周围环境中存 在的气体和组合物的混合物。本发明具体地设想和包括具有与周围环境不 同的组合物的气态混合物,包括例如富含特定组分或其中特定组分已经被 耗尽或消除的混合物。
另外,本发明提供用于使多能干细胞分化成表现出胰腺内分泌细胞特 征的细胞的细胞培养物,以及引发和促进此类分化的分化培养基。有利 地,这些细胞培养物和分化培养基可与空气-液体界面处的分化结合使用来 提供先前未达到的表达胰腺内分泌细胞的特征性标志物的细胞的收率。
培养可在从多能干细胞至胰腺内分泌细胞的分化途径中所涉及的所有 阶段在空气-液体界面处发生,或培养可涉及在分化的早期阶段在浸没于培 养基中的平坦培养物上培养,而在分化的后期阶段在空气-液体界面处培 养。优选地,本发明的过程涉及早期阶段期间在浸没于培养基中的支撑表 面上培养多能干细胞和随后在分化的后期阶段在空气-液体界面处培养的组 合。在此类实施例中,可将所述细胞初始接种在固体表面上以进行浸没培 养,随后将细胞从所述固体载体去除并且重新接种到多孔支承体上以进行 在空气-液体界面处的培养。作为另一种选择,可将所述细胞初始接种在多 孔支承体上,随后在分化的早期阶段将该支承体浸没于培养基中,并且随 后在分化的后期阶段将该支承体定位在空气-液体界面处。相较于在整个过 程中以浸没状态培养细胞,于分化的后期阶段在空气-液体界面处培养显著 提升了分泌性标志物的表达,指示更大百分比的细胞已分化成胰腺内分泌 细胞。
在一个实施例中,本发明涉及在部分填充有培养基的培养容器的空气- 液体界面处使内胚层祖细胞分化成对于NKX6.1、PDX1和HB9呈阳性的胰 腺内胚层祖细胞。本发明部分地基于以下发现:在空气-液体界面处培养显 著提升了内分泌标志物的表达。此外,已经发现胰腺内分泌前体细胞可容 易地在空气-液体界面处生成,从而使得主要生成单激素胰岛素阳性细胞。 已发现空气-液体界面处的单细胞接种改善了胰岛素制备的一致性。
定义
干细胞是通过其在单细胞水平上既自我更新又分化的能力来定义的未 分化细胞。干细胞可产生子代细胞,包括自我更新祖细胞、非更新祖细胞 和终末分化细胞。干细胞的特征还在于其在体外分化成来自多个胚层(内 胚层、中胚层和外胚层)的多种细胞谱系的功能细胞的能力。干细胞还在 移植后产生多种胚层的组织,并且在注射到胚泡内之后,促成基本上至大 部分(如果不是所有的话)组织。
干细胞是根据其发育潜能分类的。多能干细胞能够产生所有胚胎细胞 类型。
分化是这样一个过程,通过该过程,非特化的(“非定向的”)或少 特化的细胞可获得特化细胞(例如,神经细胞或肌细胞)的特征。分化细 胞是已在细胞的谱系内占据更特化的(“定向的”)位置的细胞。术语 “定向的”当应用于分化过程时,指在分化途径中已经进行到这么一种程 度的细胞:在正常环境下,它会继续分化成特定的细胞类型或细胞类型亚群,且在正常环境下不能分化成另一细胞类型或退回到分化程度较低的细 胞类型。“去分化”指细胞退回到细胞的谱系内特化(或定向)程度较低 的地位的过程。本文所用的“细胞的谱系”限定细胞的遗传,即它来自哪 些细胞和它能产生什么细胞。细胞的谱系将细胞定位于发育和分化的遗传 方案内。谱系特异性标志物指与感兴趣谱系的细胞的表型明确相关的特 征,并且可用来评估未定向细胞向感兴趣的谱系的分化。
本文所用的“标志物”是在感兴趣的细胞中差异表达的核酸或多肽分 子。在该语境中,差异表达意指与未分化细胞相比针对阳性标志物的升高 的水平以及针对阴性标志物的下降的水平。标志物核酸或多肽在感兴趣的 细胞中的可检测水平充分地高于或低于在其他细胞中的那些,使得可使用 本领域已知的多种方法中的任何一种将感兴趣的细胞与其他细胞鉴别和区 分开来。
如本文所用,当在细胞中充分地检测到特定标志物时,细胞“对于特 定标志物是阳性的”或是“阳性的”。相似地,当在细胞中未充分检测到 特定标志物时,细胞“对于特定标志物是阴性的”或是“阴性的”。具体 地,用FACS检测到的阳性通常大于2%,而用FACS检测到的阴性阈值通 常小于1%。用PCR检测到的阳性通常小于34个循环(Cts);而用PCR检测到的阴性通常大于34.5个循环。
为了尝试在静态体外细胞培养物中将多能干细胞的分化复制到功能性 胰腺内分泌细胞中,所述分化过程通常被视为通过多个连续阶段进化。具 体地,所述分化过程常常被视为通过六个阶段进化。在该逐步进化中, “第1阶段”是指该分化过程的第一步骤,其中使多能干细胞分化成表达 定形内胚层细胞的特征性标志物的细胞(在下文被替代地称为“第1阶段 细胞”)。“第2阶段”是指第二步骤,其中使表达定形内胚层细胞的特 征性标志物的细胞分化成表达肠管细胞的特征性标志物的细胞(在下文被 替代地称为“第2阶段细胞”)。“第3阶段”是指第三步骤,其中使表 达肠管细胞的特征性标志物的细胞分化成表达前肠内胚层细胞的特征性标 志物的细胞(在下文被替代地称为“第3阶段细胞”)。“第4阶段”是 指第四步骤,其中使表达前肠内胚层细胞的特征性标志物的细胞分化成表 达胰腺前肠前体细胞的特征性标志物的细胞(在下文被替代地称为“第4 阶段细胞”)。“第5阶段”是指第五步骤,其中使表达胰腺前肠前体细 胞的特征性标志物的细胞分化成表达胰腺内胚层细胞和/或胰腺内分泌前体 细胞的特征性标志物的细胞(在下文被统称为“胰腺内胚层/内分泌前体细 胞”或被替代地称为“第5阶段细胞”)。“第6阶段”是指使表达胰腺 内胚层/内分泌前体细胞的特征性标志物的细胞分化成表达胰腺内分泌细胞 的特征性标志物的细胞(在下文被替代地称为“第6阶段细胞”)。
然而应当指出的是,并非具体群体中的所有细胞都以相同速率通过这 些阶段进化。因此,在体外细胞培养物中检测到存在比群体中存在的大部 分细胞沿分化途径进化更少或更多的细胞的情况并不少见,具体地在后期 分化阶段。例如,在第5阶段的细胞培养期间观察到胰腺内分泌细胞的特 征性标志物的出现的情况并不少见。出于说明本发明的目的,与上文识别 的各阶段相关联的各种细胞类型的特性如本文所述。
本文所用的“定形内胚层细胞”指具有在原肠胚形成期间从上胚层产 生的细胞的特性并形成胃肠道及其衍生物的细胞。定形内胚层细胞表达下 述标志物中的至少一种:FOXA2(也被称为肝细胞核因子 3β(“HNF3β”))、GATA4、SOX17、CXCR4、Brachyury、Cerberus、OTX2、goosecoid、C-Kit、CD99和MIXL1。定形内胚层细胞的特征性标志 物包括CXCR4、FOXA2和SOX17。因此,定形内胚层细胞可通过其对 CXCR4、FOXA2和SOX17的表达来表征。此外,取决于允许细胞保持在 第1阶段的时长,可观察到HNF4α的增长。
如本文所用的“肠管细胞”是指源于定形内胚层的细胞,所述细胞能 够产生所有的内胚层器官,诸如肺、肝、胰腺、胃和肠。肠管细胞可通过 相较于由定形内胚层细胞对HNF4α的表达,对HNF4α的基本上增长的表 达来表征。例如,在第2阶段期间可观察到HNF4α的mRNA表达的十到四 十倍的增长。
如本文所用的“前肠内胚层细胞”指产生食道、肺、胃、肝、胰腺、 胆囊和一部分十二指肠的内胚层细胞。前肠内胚层细胞表达下述标志物中 的至少一种:PDX1、FOXA2、CDX2、SOX2和HNF4α。前肠内胚层细胞 可通过其相较于肠管细胞,对PDX1的表达的增长来表征。例如,通常第3 阶段培养物中大于百分之五十的细胞表达PDX1。
如本文所用,“胰腺前肠前体细胞”是指表达下述标志物中的至少一 种的细胞:PDX1、NKX6.1、HNF6、NGN3、SOX9、PAX4、PAX6、 ISL1、胃泌素、FOXA2、PTF1a、PROX1和HNF4α。胰腺前肠前体细胞可 通过对PDX1、NKX6.1和SOX9的表达为阳性来表征。
如本文所用,“胰腺内胚层细胞”是指表达下述标志物中的至少一种 的细胞:PDX1、NKX6.1、HNF1β、PTF1α、HNF6、HNF4α、SOX9、 NGN3;胃泌素;HB9或PROX1。胰腺内胚层细胞可通过其缺乏对CDX2 或SOX2的基本的表达来表征。
如本文所用,“胰腺内分泌前体细胞”是指能够变成胰腺激素表达细 胞的胰腺内胚层细胞。胰腺内分泌前体细胞表达下述标志物中的至少一 种:NGN3;NKX2.2;NeuroD1;ISL1;PAX4;PAX6;或ARX。胰腺内 分泌前体细胞可通过其对NKX2.2和NeuroD1的表达来表征。
如本文所用,“胰腺内分泌细胞”指能够表达至少一种下列激素的细 胞:胰岛素、胰高血糖素、生长抑素、生长素释放肽和胰多肽。除了这些 激素以外,胰腺内分泌细胞的特征性标志物还包括以下一种或多种标志 物:NGN3、NeuroD1、ISL1、PDX1、NKX6.1、PAX4、ARX、NKX2.2和 PAX6。表达β细胞的特征性标志物的胰腺内分泌细胞可通过其对胰岛素以 及至少一种下列转录因子的表达来表征:PDX1、NKX2.2、NKX6.1、 NeuroD1、ISL1、HNF3β、MAFA和PAX6。
在本文中可互换使用的是“d1”、“1d”和“第1天”;“d2”、 “2d”和“第2天”等等。这些数字字母组合是指在本专利申请的逐步分 化方案过程中的不同阶段中温育的具体天数。
在本文使用的“葡萄糖”是指右旋糖,在自然界中通常发现的糖。
本文所用的“NeuroD1”识别在胰腺内分泌祖细胞中表达的蛋白质及 其编码基因。
“LDN-193189”是指((6-(4-(2-(哌啶-1-基)乙氧基)苯基)-3-(吡啶-4-基) 吡唑并[1,5-a]嘧啶,盐酸盐;DM-3189)),一种可以商标STEMOLECULETM购自Stemgent,Inc.,Cambridge,MA,USA的BMP受体抑 制剂。
多能干细胞的表征、来源、扩增和培养
A.多能干细胞的表征
多能干细胞可表达一种或多种指定的TRA-1-60和TRA-1-81抗体 (Thomson等人,1998,Science 282:1145-1147)。多能干细胞在体外的分 化导致TRA-1-60和TRA-1-81丧失表达。未分化的多能干细胞通常具有碱 性磷酸酶活性,所述碱性磷酸酶活性可通过用4%多聚甲醛固定细胞,然后 用以商标 Red出售的碱性磷酸酶底物试剂盒显色来检测,如由制 造商(Vector Laboratories,CA,USA)描述的。未分化的多能干细胞通常也表达OCT4和TERT,如通过RT-PCR检测。
Red出售的碱性磷酸酶底物试剂盒显色来检测,如由制 造商(Vector Laboratories,CA,USA)描述的。未分化的多能干细胞通常也表达OCT4和TERT,如通过RT-PCR检测。
增殖的多能干细胞的另一理想表型是分化成所有三个胚层(内胚层、 中胚层和外胚层)的细胞的潜能。可以例如通过如下方式来确认干细胞的 多能性:将细胞注射入重症联合免疫缺陷(SCID)小鼠,使用4%的多聚甲醛 固定形成的畸胎瘤,然后针对来自该三个胚层的细胞类型的证据进行组织 学检测。作为另一种选择,可以通过产生胚状体并评估胚状体中与三个胚 层相关的标志物的存在来确定多能性。
增殖的多能干细胞系可用标准G显带技术进行核型分析并与公开的相 应灵长类物种的核型相比较。希望获得具有“正常核型”(其意指细胞是 整倍体的)的细胞,其中所有人染色体都存在并且无明显的改变。
B.多能干细胞的来源
可使用的多能干细胞的示例性类型包括建立的多能细胞系。非限制性例子是建立的人胚胎干细胞系或人胚胎干细胞,诸如人胚胎干细胞系H1、H7和H9 (WiCell ResearchInstitute, Madison, WI, USA)。另外合适的是取自已在不存在饲养细胞的情况下培养的多能干细胞群的细胞。还可使用诱导多能细胞(IPS)或重新编程的多能细胞,其可使用多种多能相关的转录因子的被迫表达而来源于成人体细胞,所述转录因子诸如OCT4、NANOG、SOX2、KLF4和ZFP42 (Annu Rev Genomics Hum Genet 2011, 12:165-185);还参见IPS,Cell, 126(4):663-676)。在本发明的方法中使用的人胚胎干细胞也可如由Thomson等人描述的进行制备(美国专利5,843,780;Science, 1998, 282:1145-1147;Curr Top Dev Biol1998, 38:133-165;Proc Natl Acad Sci U.S.A. 1995, 92:7844-7848)。也可使用突变的人胚胎干细胞系,诸如BG01v (BresaGen, Athens, Ga.);或衍生自成人体细胞的细胞,诸如在Takahashi等人,Cell 131:1-12 (2007)中公开的细胞。在某些实施例中,适用于本发明的多能干细胞可以根据在以下文献中描述的方法来获得:Li等人(Cell Stem Cell 4:16-19, 2009);Maherali等人(Cell Stem Cell 1:55-70, 2007);Stadtfeld等人(Cell Stem Cell 2:230-240);Nakagawa等人(Nature Biotechnol 26:101-106, 2008);Takahashi等人(Cell 131:861-872, 2007);以及美国专利申请公布2011/0104805的正式申请。在某些实施例中,所述多能干细胞可为非胚胎来源的。所有这些参考文献、专利和专利申请全文以引用方式并入本文中,尤其当它们与多能细胞的分离、培养、扩增和分化有关时。
C.多能干细胞的扩增和培养
通常在饲养细胞层上培养多能干细胞,饲养细胞可以多种方式支持多 能干细胞。作为另一种选择,可在培养系统中培养多能干细胞,所述培养 系统基本上不含饲养细胞,但支持多能干细胞的增殖而不会经历基本的分 化。往往使用通过此前培养另一细胞类型而调理过的培养基来支持多能干 细胞在无饲养细胞的培养物中生长而不分化。作为另一种选择,可用化学 限定的培养基来支持多能干细胞在无饲养细胞的培养物中生长而不分化。
多能细胞在培养物中可使用多种饲养层或通过使用基质蛋白质涂布的 容器容易地扩增。作为另一种选择,与限定的培养基诸如以商标 出售的培养基(StemCellTechnologies,Vancouver,Canada)组合的化学限定的 表面可用于细胞的常规扩增。多能细胞可使用酶促消化、机械分离或使用多 种钙螯合剂诸如乙二胺四乙酸(EDTA)容易地从培养皿中取出。作为另一种 选择,多能细胞可在不存在任何基质蛋白质或饲养层的情况下悬浮扩增。
出售的培养基(StemCellTechnologies,Vancouver,Canada)组合的化学限定的 表面可用于细胞的常规扩增。多能细胞可使用酶促消化、机械分离或使用多 种钙螯合剂诸如乙二胺四乙酸(EDTA)容易地从培养皿中取出。作为另一种 选择,多能细胞可在不存在任何基质蛋白质或饲养层的情况下悬浮扩增。
在受权利要求保护的本发明中可以使用多种不同的扩增和培养多能干 细胞的方法。例如,本发明的方法可使用Reubinoff等人、Thompson等 人、Richard等人和美国专利申请公布2002/0072117的正式申请。Reubinoff 等人(Nature Biotechnology 18:399-404(2000))和Thompson等人(Science 282:1145-1147(1998))公开了用小鼠胚胎成纤维细胞饲养细胞层来培养来自 人胚泡的多能干细胞系。Richards等人(Stem Cells 21:546-556,2003)评估了 一组十一种不同的成人、胎儿和新生儿饲养细胞层在支持人多能干细胞培养方面的能力,注意到在成人皮肤成纤维饲养细胞上培养的人胚胎干细胞 系保持人胚胎干细胞形态并且保留多能性。美国专利申请公布 2002/0072117公开了产生支持灵长类多能干细胞在无饲养细胞的培养物中 生长的培养基的细胞系。所采用的细胞系是从胚胎组织获得或从胚胎干细 胞分化而来的间质细胞系和成纤维细胞样细胞系。美国专利申请公布 2002/072117还公开了所述细胞系作为原代饲养细胞层的用途。
扩增和培养多能干细胞的其他合适方法公开于例如Wang等人、 Stojkovic等人、Miyamoo等人和Amit等人的文献中。Wang等人(Stem Cells 23:1221-1227,2005)公开了用于在衍生自人胚胎干细胞的饲养细胞层上长期 生长的人多能干细胞的方法。Stojkovic等人(Stem Cells 200523:306-314, 2005)公开了一种衍生自人胚胎干细胞自发分化的饲养细胞系统。Miyamoto 等人(Stem Cells 22:433-440,2004)公开了从人胎盘获得的饲养细胞源。Amit 等人(Biol.Reprod 68:2150-2156,2003)公开了衍生自人包皮的饲养细胞层。
扩增和培养多能干细胞的其他合适方法公开于例如Inzunza等人的文 献、美国专利6,642,048、WO 2005/014799、Xu等人的文献和美国专利申 请公布2007/0010011的正式申请。Inzunza等人(Stem Cells 23:544-549,2005) 公开了来自人出生后包皮成纤维细胞的饲养细胞层。美国专利6,642,048公 开了可支持灵长类多能干细胞在无饲养细胞的培养物中生长的培养基以及 可用于产生此类培养基的细胞系。美国专利6,642,048报道从胚胎组织获得 或从胚胎干细胞分化成的间质细胞系和成纤维样细胞系,以及获得此类细 胞系、处理培养基以及使用此类培养基培育干细胞的方法。WO 2005/014799公开了一种用于哺乳动物细胞的维持、增殖和分化的条件培养 基。WO 2005/014799报道,通过鼠细胞(那些分化并永生化的转基因肝细 胞,称为MMH(Met鼠肝细胞))的细胞分泌活性对根据本发明制备的培 养基进行调理。Xu等人(Stem Cells 22:972-980,2004)公开了一种从人胚胎干 细胞衍生物获得的条件培养基,所述人胚胎干细胞衍生物已被基因修饰而过 表达人端粒酶逆转录酶。美国专利申请公布2007/0010011公开了一种用于 维持多能干细胞的化学限定的培养基。
一种替代的培养系统采用补充有能促进胚胎干细胞增殖的生长因子的 无血清培养基。此类培养系统的例子包括但不限于Cheon等人、Levenstein 等人的文献和美国专利申请公布2005/0148070。Cheon等人(BioReprod DOI:10.1095/biolreprod.105.046870,2005年10月19日)公开了一种无饲养 细胞的无血清培养系统,其中胚胎干细胞维持在补充有能引发胚胎干细胞自 我更新的不同生长因子的未经调理的血清替代(SR)培养基中。Levenstein等 人(Stem Cells 24:568-574,2006)公开了使用补充有bFGF的培养基,在不存 在成纤维细胞或条件培养基的情况下长期培养人胚胎干细胞的方法。美国 专利申请公布2005/0148070公开了一种在无血清且无成纤维细胞饲养细胞 的限定的培养基中培养人胚胎干细胞的方法,该方法包括:在含有白蛋白、 氨基酸、维生素、矿物质、至少一种转铁蛋白或转铁蛋白替代品、至少一种 胰岛素或胰岛素替代品的培养基中培养干细胞,该培养基基本上无哺乳动物 胎血清且含有至少约100ng/ml的能够对成纤维细胞生长因子信号转导受体 进行活化的成纤维细胞生长因子,其中该生长因子的供给来源不是仅为成 纤维细胞饲养层,该培养基支持干细胞在无饲养细胞或条件培养基的情况 下以未分化状态增殖。
培养和扩增多能干细胞的其他合适的方法公开于美国专利申请公布 2005/0233446、美国专利6,800,480、美国专利申请公布2005/0244962和 WO 2005/065354中。美国专利申请公布2005/0233446公开了一种可用于培 养干细胞的限定的培养基,所述干细胞包括未分化的灵长类原始干细胞。在 溶液中,此培养基与培养中的干细胞相比基本上是等渗的。在给定的培养物 中,特定的培养基包含基础培养基和各为一定量的bFGF、胰岛素和抗坏血 酸,所述一定量的bFGF、胰岛素和抗坏血酸为支持原始干细胞进行基本上 非分化性生长所必需的。美国专利6,800,480报道,提供了用于使灵长类来 源的原始干细胞以基本上未分化状态生长的细胞培养基,其包括低渗透压、 低内毒素基础培养基,此培养基对于支持灵长类来源的原始干细胞的生长是 有效的。美国专利6,800,480的公开内容还报道,该基础培养基混合了有效 支持灵长类来源的原始干细胞生长的营养血清以及选自饲养细胞和衍生自饲 养细胞的胞外基质组分的基质。还应注意该培养基包括非必需氨基酸、抗氧 化剂和选自核苷和丙酮酸盐的第一生长因子。美国专利申请公布 2005/0244962报道,本公开的一个方面提供一种培养灵长类胚胎干细胞的 方法,并且该培养物中的干细胞基本上不含哺乳动物胎儿血清(优选地也 基本上不含任何动物血清)并且处于存在从除成纤维细胞饲养细胞层以外 的来源供应的成纤维细胞生长因子的情况下。
WO 2005/065354公开了一种限定的等渗培养基,该培养基基本上不含 饲养细胞且不含血清,包含:基础培养基、bFGF、胰岛素和抗坏血酸,所 述基础培养基、bFGF、胰岛素和抗坏血酸的量足以支持基本上未分化的哺 乳动物干细胞生长。此外,WO 2005/086845公开了一种维持未分化的干细 胞的方法,所述方法包括使干细胞暴露于转化生长因子-β(TGF-β)蛋白家族 的成员、成纤维细胞生长因子(FGF)蛋白家族的成员或烟酰胺(NIC),所述 成员或烟酰胺的量足以维持所述细胞处于未分化状态达足以实现所需结果 的一段时间。
可将多能干细胞接种至合适的培养基质上。在一个实施例中,合适的 培养基质是细胞外基质组分,例如衍生自基底膜的组分,或可形成粘着分 子受体-配体偶联物的一部分的组分。一种合适的培养基质是以商标 MATRIGELTM(BD Biosciences,Franklin Lakes,NJ)出售的重构基底膜。 MATRIGELTM是来自Engelbreth-Holm Swarm肿瘤细胞的可溶性制剂,其在 室温下胶凝而形成重构的基底膜。
在本领域中已知的其他细胞外基质组分和组分混合物适合作为替代 物。取决于所扩增的细胞类型,这可包括层粘连蛋白、纤连蛋白、蛋白聚 糖、巢蛋白、硫酸乙酰肝素等,单独的或呈各种组合。
可在存在可促进细胞存活、增殖和保持理想特性的培养基存在的情况 下,以合适的分布将多能干细胞接种于所述基质上。所有这些特性可得益 于对接种分布的认真考虑并可易于由本领域技术人员确定。合适的培养基 可用以下组分制备:Dulbecco改良的Eagle培养基(DMEM),由Life Technologies Corporation,Grand Island,NY以商标GIBCOTM(货号11965- 092)出售;敲除的Dulbecco改良的Eagle培养基(KO DMEM),由LifeTechnologies Corporation,Grand Island,NY以商标GIBCOTM(货号10829- 018)出售;Ham的F12/50%DMEM基础培养基;200mM的L-谷氨酰胺, 由Life Technologies Corporation,Grand Island,NY以商标GIBCOTM(货号 15039-027)出售;非必需氨基酸溶液,由LifeTechnologies Corporation, Grand Island,NY以商标GIBCOTM(货号11140-050)出售;β-巯基乙醇, Sigma-Aldrich Company,LLC Saint Louis,MO,(货号M7522);人重组碱 性成纤维细胞生长因子(bFGF),由Life Technologies Corporation,Grand Island,NY以商标GIBCOTM(货号13256-029)出售。
多能干细胞的分化
当多能细胞向β细胞分化时,所述细胞通过多个阶段分化,每一阶段 可通过特定标志物的存在或不存在来表征。细胞分化至这些阶段是通过特 定培养条件实现的,所述培养条件包括存在和缺乏添加至培养基中的某些 因子。一般来讲,这种分化可涉及多能干细胞分化成定形内胚层细胞。这 些定形内胚层细胞可随后进一步分化成肠管细胞,所述肠管细胞可继而分 化成前肠内胚层细胞。前肠内胚层细胞可分化成胰腺前肠前体细胞,该胰腺前肠前体细胞可继而进一步分化成胰腺内胚层细胞、胰腺内分泌前体细 胞,或胰腺内胚层细胞和胰腺内分泌前体细胞两者。这些细胞可随后分化 成产生胰腺激素的细胞(诸如β细胞)。本发明通过以下步骤提供多能干 细胞向胰腺内分泌细胞的分阶段分化:在部分填充有培养基的培养容器中 存在的空气-液体界面处培养细胞,具体地在空气-液体界面处培养第4阶段 至第6阶段的细胞。
多能干细胞向表达胰腺内分泌细胞的特征性标志物的细胞的分化
多能干细胞的特征是本领域技术人员熟知的,并且多能干细胞的其他 特征有待继续识别。多能干细胞标志物包括例如一种或多种下列标志物的 表达:ABCG2、cripto、FOXD3、CONNEXIN43、CONNEXIN45、OCT4、 SOX2、NANOG、hTERT、UTF1、ZFP42、SSEA-3、SSEA-4、TRA-1- 60、TRA-1-81。
示例性的多能干细胞包括人胚胎干细胞系H9(NIH代码:WA09)、人 胚胎干细胞系H1(NIH代码:WA01)、人胚胎干细胞系H7(NIH代码: WA07),以及人胚胎干细胞系SA002(Cellartis,Sweden)。表达下述多能细胞 特征性标志物中的至少一种的细胞也是合适的:ABCG2、cripto、CD9、 FOXD3、CONNEXIN43、CONNEXIN45、OCT4、SOX2、NANOG、 hTERT、UTF1、ZFP42、SSEA-3、SSEA-4、TRA-1-60和TRA-1-81。
还适用于本发明的是表达定形内胚层谱系的特征性标志物中的至少一 种的细胞。在本发明的一个实施例中,表达定形内胚层谱系的特征性标志 物的细胞为原条前体细胞。在一个替代实施例中,表达定形内胚层谱系的 特征性标志物的细胞是中内胚层细胞。在一个替代实施例中,表达定形内 胚层谱系的特征性标志物的细胞是定形内胚层细胞。
还适用于本发明的是表达胰腺内胚层谱系的特征性标志物中的至少一 种的细胞。在本发明的一个实施例中,表达胰腺内胚层谱系的特征性标志 物的细胞是胰腺内胚层细胞,其中PDX1和NKX6.1的表达基本上高于 CDX2和SOX2的表达。在某些实施例中,大于百分之三十的细胞表达 PDX1和NKX6.1,并且小于百分之三十的细胞表达CDX2或SOX2,如用FACS所测量的。特别有用的是其中PDX1和NKX6.1的表达比CDX2或 SOX2的表达至少高两倍的细胞。
还适用于本发明的是表达胰腺内分泌谱系的特征性标志物中的至少一 种的细胞。在本发明的一个实施例中,表达胰腺内分泌谱系的特征性标志 物的细胞为胰腺内分泌细胞。在一个实施例中,胰腺内分泌细胞能够表达 以下激素中的至少一种:胰岛素、胰高血糖素、生长抑素或胰多肽。在一 个优选的实施例中,所述胰腺内分泌细胞是产生胰岛素的β细胞。
在本发明的某些实施例中,为了得到表达胰腺内分泌细胞的特征性标 志物的细胞,利用从多能干细胞或诱导多能细胞开始的方案,优选地从多 能干细胞开始的方案。该方案包括以下阶段:
第1阶段:将从细胞培养系获得的多能干细胞,诸如胚胎干细胞,用 合适的因子处理以诱导分化成表达定形内胚层细胞的特征性标志物的细胞。
第2阶段:将从第1阶段获得的细胞用合适的因子处理以进一步诱导 分化成表达肠管细胞的特征性标志物的细胞。
第3阶段:将从第2阶段获得的细胞用合适的因子处理以进一步诱导 分化成表达前肠内胚层细胞的特征性标志物的细胞。
第4阶段:将从第3阶段获得的细胞用合适的因子处理以进一步诱导 分化成表达胰腺前肠前体细胞的特征性标志物的细胞。将所述细胞任选地 在第4阶段后期时在空气-液体界面处培养。
第5阶段:将从第4阶段获得的细胞用合适的因子处理并且在空气-液 体界面处培养以进一步诱导分化成表达胰腺内胚层/内分泌前体细胞的特征 性标志物的细胞。
第6阶段:将从第5阶段获得的细胞用合适的因子处理并且在空气-液 体界面处培养以进一步诱导分化成表达胰腺内分泌细胞的特征性标志物的 细胞。
虽然在某些实施例中本发明涵盖使多能干细胞(例如,第1阶段之前 的细胞)分化成第6阶段细胞,但是本发明也涵盖使其他中间阶段的细胞 向第6阶段分化。具体地,本发明涵盖第4阶段细胞至第6阶段细胞的分 化。此外,虽然该过程被描述为离散的阶段,但是处理以及细胞经历分化 过程的进程可为顺序的或连续的。
第1阶段:使多能干细胞分化成表达定形内胚层细胞的特征性标志物的细胞
可通过本领域已知的任何方法或通过本文提出的任何方法,使多能干 细胞分化成表达定形内胚层细胞的特征性标志物的细胞。可用于使多能干 细胞分化成表达定形内胚层细胞的特征性标志物的细胞的方法公开于:美 国专利申请公布2007/0254359;美国专利申请公布2009/0170198;美国专 利申请公布2009/0170198;美国专利申请公布2011/0091971;美国专利申 请公布2010/0015711;美国专利申请公布2010/0015711;美国专利申请公 布2012/0190111;美国专利申请公布2012/0190112;美国专利申请公布 2012/0196365;美国专利申请公布20100015711;美国专利申请公布 2012/0190111;美国专利申请公布2012/0190112;美国专利申请公布 2012/0196365;美国专利申请公布20100015711;美国专利申请公布 2012/0190111;美国专利申请公布2012/0190112;美国专利申请公布2012/0196365;美国临时专利申请61/076,900;美国临时专利申请 61/076,908;以及美国临时专利申请61/076,915,所述专利申请全文以引用 方式并入,因为它们涉及多能干细胞并且涉及使多能干细胞分化成表达定 形内胚层谱系的特征性标志物的细胞。
在本发明的一个实施例中,用补充有活化素A和WNT3A的培养基处 理多能干细胞,以导致生成表达定形内胚层细胞的特征性标志物的细胞。 处理可涉及使多能干细胞与包含约50ng/ml至约150ng/ml、或约75ng/ml至 约125ng/ml、或约100ng/ml的活化素A的培养基接触。所述处理还可涉及 使所述细胞与约10ng/ml至约50ng/ml、或约15ng/ml至约30ng/ml、或约 20ng/ml的WNT3A接触。可培养所述多能细胞约两至五天,优选地约三 天,以促进所述多能细胞分化成表达定形内胚层细胞的特征性标志物的细 胞。在一个实施例中,在活化素A和WNT3A的存在下培养细胞一天,接 着在活化素A(不存在WNT3A)的存在下培养细胞剩余的时间。
在本发明的另一个实施例中,用补充有生长分化因子8(“GDF8”)和 糖原合成酶激酶-3β(“GSK3β”)抑制剂(诸如在美国专利申请公布 2010/0015711中公开的环苯胺-吡啶三嗪化合物;该专利全文以引用方式并 入本文)的培养基处理多能干细胞,以诱导分化成表达定形内胚层细胞的 特征性标志物的细胞。优选的GSK3β抑制剂是14-丙-2-烯-1-基- 3,5,7,14,17,23,27-七氮杂四环[19.3.1.1~2,6~.1~8,12~]二十七- 1(25),2(27),3,5,8(26),9,11,21,23-壬-16-酮,在本文中被称作(“MCX化合 物”)。处理可涉及使多能干细胞与补充有约50ng/ml至约150ng/ml、或 约75ng/ml至约125ng/ml、或约100ng/ml的GDF8的培养基接触。所述处 理还可涉及使所述细胞与约0.1μM至5μM、或约0.5μM至约2.5μM,优选 地约1μM的MCX化合物接触。可培养所述多能细胞约两至五天,优选地 约两至三天,以促进所述多能细胞分化成表达定形内胚层细胞的特征性标 志物的细胞。
在一个实施例中,在GDF8和MCX化合物的存在下培养细胞一天, 接着在GDF8和较低浓度的MCX化合物的存在下培养细胞一天,接着在存 在GDF8并且缺乏MCX化合物的情况下培养细胞一天。具体地,在GDF8 和约1μM的MCX化合物的存在下培养细胞一天,接着在GDF8和约 0.1μM的MCX化合物的存在下培养细胞一天,接着在存在GDF8并且缺乏 MCX化合物的情况下培养细胞一天。在一个替代实施例中,在GDF8和约 1μM的MCX化合物的存在下培养细胞一天,接着在GDF8和约0.1μM的 MCX化合物的存在下培养细胞一天。
表达定形内胚层细胞的特征性标志物的细胞的生成可通过在进行特定 方案之前或之后检测该标志物的存在来确定。多能干细胞通常不表达这类 标志物。因此,当细胞开始表达定形内胚层细胞的特征性标志物时,可检 测到多能细胞的分化。用于评估蛋白质标志物和核酸标志物在培养的或分 离的细胞中的表达的方法是本领域的标准方法。这些方法包括定量反转录 聚合酶链式反应(RT-PCR)、Northern印迹、原位杂交(参见,例如,Current Protocols in Molecular Biology(Ausubel等人编辑,2001增刊),以 及免疫测定(诸如切片材料的免疫组织化学分析)、Western印迹和在完整 细胞中可接近的标志物的流式细胞术分析(FACS)(参见,例如,Harlow和 Lane,Using Antibodies:A LaboratoryManual,New York:Cold Spring Harbor Laboratory Press(1998))。
另外,可通过将处理过的细胞群暴露于可特异性识别由感兴趣的分化 细胞表达的蛋白质标志物的试剂(诸如抗体)来确定分化效率。
还可将分化的细胞进一步纯化。例如,在用本发明方法处理多能干细 胞后,可通过使处理过的细胞群暴露于特异性识别由经纯化的分化细胞特 征性表达的蛋白质标志物的试剂(诸如抗体)来纯化分化的细胞。
第2阶段:使表达定形内胚层细胞的特征性标志物的细胞分化成表达肠管细胞的
特征性标志物的细胞
表达定形内胚层细胞的特征性标志物的细胞可进一步分化成表达肠管 细胞的特征性标志物的细胞。在一个实施例中,表达肠管细胞的特征性标 志物的细胞的形成包括:用包含成纤维细胞生长因子(“FGF”)7或FGF10 的培养基来培养表达定形内胚层细胞的特征性标志物的细胞,从而分化这 些细胞。例如,该培养基可包含从约25ng/ml至约75ng/ml、或从约 30ng/ml至约60ng/ml、或约50ng/ml的FGF7或FGF10,优选地是FGF7。 可在这些条件下培养细胞约两至三天,优选地约两天。
在另一个实施例中,分化成表达肠管细胞的特征性标志物的细胞包 括:用FGF7或FGF10和抗坏血酸(维生素C)培养表达定形内胚层细胞 的特征性标志物的细胞。该培养基可包括从约0.1mM至约0.5mM的抗坏血 酸,或从约0.2mM至约0.4mM,或约0.25mM的抗坏血酸。该培养基还可 包括从约10ng/ml至约35ng/ml,或从约15ng/ml至约30ng/ml,或约25ng/ml的FGF7或FGF10,优选地FGF7。例如,该培养基可包括约 0.25mM的抗坏血酸和约25ng/ml的FGF7。在一个实施例中,用FGF7和抗 坏血酸处理第1阶段细胞2天。
第3阶段:使表达肠管细胞的特征性标志物的细胞分化成表达前肠内胚层细胞的
特征性标志物的细胞
表达肠管细胞的特征性标志物的细胞可进一步分化成表达前肠内胚层 细胞的特征性标志物的细胞。在一个实施例中,第2阶段细胞通过以下方 式而进一步分化成第3阶段细胞:在补充有Smoothened(“SMO”)受体抑 制剂(诸如“MRT10”(N-[[[3-苯甲酰基氨基)苯基]氨基]硫代甲酰]-3,4,5-三 甲氧基苯甲酰胺))或环杷明)或Sonic Hedgehog(“SHH”)信号转导途径拮 抗剂(诸如Smoothened拮抗剂1(“SANT-1”)((E)-4-苯甲基-N-((3,5-二甲基-1-苯基-1H-吡唑-4-基)亚甲基-哌嗪-1-胺)),或Hedgehog途径抑制剂1 (“HPI-1”)(2-甲氧基乙基1,4,5,6,7,8-六氢化-4-(3-羟基苯基)-7-(2-甲氧基苯 基)-2-甲基-5-氧代-3-喹啉羧酸酯))、视黄酸、以及头蛋白的培养基中培养 这些细胞。作为另一种选择,第2阶段细胞可通过以下步骤分化成第3阶 段细胞:在补充有SMO受体抑制剂、SHH信号转导途径拮抗剂、视黄 酸、以及头蛋白的培养基中培养这些细胞。可培养细胞约两至四天,优选 地约两天。在一个实施例中,该培养基补充有从约0.1μM至约0.3μM的 SANT-1、从约0.5μM至约3μM的视黄酸、以及从约75ng/ml至约 125ng/ml的头蛋白。在另一个实施例中,该培养基补充有约0.25μM的 SANT-1、约2μM的视黄酸和约100ng/ml的头蛋白。
在一个替代实施例中,第2阶段细胞通过以下方式进一步分化成第3 阶段细胞:用补充有FGF7或FGF10、视黄酸、SMO受体抑制剂(诸如 MRT10或环杷明)或SHH信号转导途径拮抗剂(诸如SANT-1或HPI- 1)、蛋白激酶C(“PKC”)活化剂(诸如((2S,5S)-(E,E)-8-(5-(4-(三氟甲基) 苯基)-2,4-戊二烯酰氨基)苯并内酰胺(“TPB”)),EMD Chemicals,Inc.,Gibbstown,NJ)、12,13-二丁酸佛波醇(“PDBu”)、佛波醇-12-肉豆蔻酸酯- 13-乙酸酯(“PMA”)或吲哚内酰胺V(“ILV”))、骨形态发生蛋白 (“BMP”)抑制剂(诸如LDN-193189、头蛋白或脊索发生素)、以及抗坏血 酸的培养基处理第2阶段细胞。在另一个实施例中,该培养基可补充有 FGF7或FGF10、视黄酸、SMO受体抑制剂、SHH信号转导途径拮抗剂 (诸如SANT-1)、PKC活化剂(诸如TPB)、BMP抑制剂(诸如LDN- 193189)、以及抗坏血酸。可在存在这些生长因子、小分子激动剂和拮抗 剂的情况下培养细胞约两至四天,优选地约两至三天。
在另一个实施例中,该培养基补充有从约15ng/ml至约35ng/ml的 FGF7、从约0.5μM至约2μM的视黄酸、从约0.1μM至约0.4μM的SANT- 1、从约100至约300nM的TPB、从约50nM至约200nM的LDN-193189、 以及从约0.15mM至约0.35mM的抗坏血酸。在另一个实施例中,该培养基 补充有约25ng/ml的FGF7、约1μM的视黄酸、约0.25μM的SANT-1、约 200nM的TPB、约100nM的LDN-193189、以及约0.25mM的抗坏血酸。
通过在空气-液体界面处培养产生第4阶段细胞至第6阶段细胞
虽然本发明设想在从多能细胞至胰腺内分泌细胞的途径中的所有阶段 中在空气-液体界面处培养,但是本发明优选地提供在浸没的培养物中形成 第1阶段至第3阶段细胞,以及通过在空气-液体界面处培养细胞形成第4 阶段至第6阶段细胞。因此在某些实施例中,本发明提供使多能细胞逐步 分化的方法,所述方法包括在第4阶段至第6阶段期间在空气-液体界面处 培养。在某些实施例中,可在整个第4阶段至第6阶段期间在空气-液体界面处培养细胞。在其他实施例中,仅第4阶段后期至第6阶段,或仅第5 阶段和第6阶段,或仅第4阶段和第5阶段,或仅第4阶段和第6阶段包括 在空气-液体界面处培养。
当在空气-液体界面(空气-液体界面)处培养细胞时,可在多孔基质 上培养细胞,使得所述细胞的顶侧与空气接触,并且底侧与细胞培养基接 触。例如,可将足够体积的培养基添加至包含所述多孔基质(例如,滤器 芯子(filter insert))的培养容器的底部,使得培养基接触存在于基质上的细 胞的底表面但是不包封或浸没它们。合适的多孔基质可由将不会对细胞的 生长和分化产生不利影响的任意材料形成。示例性多孔基质是由聚合物制 成的,诸如聚对苯二甲酸乙二酯(PET)、聚酯或聚碳酸酯。合适的多孔基质 可经涂覆或未经涂覆。在一个实施例中,多孔基质可涂覆有 MATRIGELTM。在本发明的一个实施例中,多孔基质是可涂覆有 MATRIGELTM的多孔滤器芯子。然而优选地,多孔基质是未经涂覆的滤器芯 子。基质的孔隙率应当足以维持细胞活力并且促进细胞的分化。合适的基 质包括具有以下孔尺寸:从约0.3至约3.0μm、约0.3至约2.0μm、约0.3 至约1.0μm、约0.3至约0.8μm、约0.3至约0.6μm、约0.3至约0.5μm、约 0.5至约3.0μm、约0.6至约3.0μm、约0.8至约3.0μm、约1.0至约 3.0μm、约2.0μm至约3.0μm,优选地约0.4μm;以及以下孔密度:从约50 至约120百万个孔/cm2、约60至约110百万个孔/cm2、约70至约100百万 个孔/cm2,优选地约80至约100百万个孔/cm2、约90至约100百万个孔 /cm2,更优选地约100百万个孔/cm2的滤器芯子。
该培养基可有利地每天或每隔一天替换或更换。在多孔基质顶部上生 长的细胞一般不是单个细胞,相反它们是以片形式或以细胞团簇形式存在 的。相较于浸没在培养基中的细胞,在空气-液体界面处培养的细胞可经历 更高的氧张力。
本发明因而涵盖在空气-液体界面处产生第4阶段至第6阶段细胞,优 选地第5阶段和第6阶段细胞。可完全在空气-液体界面处完全在平面的培 养物中培养第4阶段细胞,或可在第4阶段的早期部分期间在浸没的平面 培养物中培养细胞并且随后在第4阶段的后期部分期间在空气-液体界面处 培养细胞。这些细胞可通过使多能干细胞分化产生,或通过进一步分化来 源于其他装置的第3阶段、第4阶段或第5阶段细胞而产生。
在一个实施例中,本发明提供了一种用于从多能干细胞产生表达胰腺 内分泌细胞的特征性标志物的细胞,优选地是β细胞的方法,所述方法包 括培养多能干细胞,使所述多能干细胞分化成表达前肠内胚层细胞的特征 性标志物的细胞;以及通过在空气-液体界面处培养来使表达前肠内胚层细 胞的特征性标志物的细胞分化成表达胰腺内分泌/β细胞的特征性标志物的 细胞。
在另一个实施例中,本发明提供了一种用于从多能干细胞产生表达胰 腺内分泌细胞(优选地是β细胞)的特征性标志物的细胞的方法,所述方 法包括培养多能干细胞,使所述多能干细胞分化成表达胰腺前肠前体细胞 的特征性标志物的细胞;以及通过在空气-液体界面处培养来使表达胰腺前 肠前体细胞的特征性标志物的细胞分化成表达胰腺内分泌细胞的特征性标 志物的细胞。
该方法可包括用补充有三碘甲状腺原氨酸(T3)、四碘甲状腺原氨酸 (T4)、T3或T4的类似物、或它们的混合物(在下文被统称为 “T3/T4”)、或活化素受体样激酶(“ALK”)5抑制剂、或T3/T4和 ALK5抑制剂两者的培养基进行处理。合适的甲状腺激素类似物可包括:得自R&D Systems,Inc.的GC-1(Sobertirome),目录号4554;DITPA(3,5-二 碘甲状腺丙酸);KB-141,在J.Steroid Biochem.Mol.Biol.2008,111:262- 267和Proc.Natl.Acad.Sci.US2003,100:10067-10072中有所论述; MB07344,在Proc.Natl.Acad.Sci.US 2007,104:15490-15495中有所论述; T0681,在PLoS One,2010,5e8722和J.Lipid Res.2009,50:938-944中有所 论述;以及GC-24,在PLoS One,2010e8722和Endocr.Pract.2012, 18(6):954-964中有所论述,所述文献的公开内容全文并入本文。可用的 ALK5抑制剂包括:ALK5抑制剂II(Enzo,Farmingdale,NY); ALK5i(Axxora,San Diego,CA);SD208(R&D systems(MN));TGF-B抑 制剂SB431542(Xcess Biosciences(San Diego,CA));ITD-1(Xcess Biosciences(San Diego,CA));LY2109761(Xcess Biosciences(San Diego, CA));A83-01(XcessBiosciences(San Diego,CA));LY2157299(Xcess Biosciences(San Diego,CA));TGF-β受体抑制剂V(EMD Chemicals, Gibstown,NJ));TGF-β受体抑制剂I(EMD Chemicals,Gibstown,NJ)); TGF-β受体抑制剂IV(EMD Chemicals,Gibstown,NJ));TGF-β受体抑制剂VII(EMD Chemicals,Gibstown,NJ));TGF-β受体抑制剂VIII(EMD Chemicals,Gibstown,NJ));TGF-β受体抑制剂II(EMD Chemicals,Gibstown, NJ));TGF-β受体抑制剂VI(EMDChemicals,Gibstown,NJ));TGF-β受体 抑制剂III(EMD Chemicals,Gibstown,NJ))。该方法可包括通过用补充有 T3/T4或ALK5抑制剂的培养基处理并且在平面培养物中培养,来使表达前 肠内胚层细胞的特征性标志物的细胞分化成表达胰腺前肠前体细胞的特征 性标志物的细胞。该方法还可包括通过用补充有T3/T4、或ALK5抑制剂、 或T3/T4和ALK5抑制剂两者的培养基处理并且在空气-液体界面处培养, 来使表达胰腺前肠前体细胞的特征性标志物的细胞分化成表达β细胞的特 征性标志物的细胞。
在一个实施例中,该方法包括用补充有T3/T4和ALK5抑制剂的培养 基处理。在其他实施例中,该方法包括用补充有T3/T4或ALK5抑制剂的 培养基处理第3阶段细胞。该方法还可包括用补充有T3/T4和ALK5抑制 剂的培养基处理表达胰腺内胚层/内分泌前体细胞的特征性标志物的细胞。
本发明的一个实施例是一种形成表达β细胞的特征性标志物的细胞的 方法,所述方法包括通过在空气-液体界面处培养,来使表达前肠内胚层细 胞的特征性标志物的细胞分化成表达β细胞的特征性标志物的细胞。表达β 细胞的特征性标志物的细胞表达胰岛素以及至少一种下列转录因子: PDX1、NKX2.2、NKX6.1、NeuroD1、1SL1、HNF3β、MAFA、PAX4和PAX6。在一个实施例中,本发明的方法导致形成对于NKX6.1、PDX1和 HB9呈阳性的细胞。因此,本发明提供了一种通过在足以诱导PDX1、NKX6.1和HB9在人类细胞中表达的条件下在空气-液体界面处培养胰腺内 胚层细胞来诱导此类表达的方法。本发明还提供了一种通过在空气-液体界 面处培养胰腺内胚层细胞来诱导PDX1、NKX6.1和NGN3在人类细胞中表 达的方法。该方法可包括用补充有T3、ALK5抑制剂,或T3和ALK5抑制 剂两者的培养基处理。因此,在一个实施例中,培养基可补充有T3,而在 另一个实施例中,培养基可补充有ALK5抑制剂。在另一个实施例中,培 养基可补充有T3和ALK5抑制剂两者。第6阶段细胞可为对NKX6.1、 PDX1和HB9呈阳性的细胞。在其他实施例中,第6阶段细胞是单激素阳 性细胞。例如,第6阶段细胞可为(a)共表达NKX6.1和嗜铬粒蛋白-A或(b) 共表达NKX6.1和胰岛素的细胞。
在空气-液体界面处培养细胞包括将细胞接种到多孔基质诸如多孔滤器 芯子上。在某些实施例中,基质的孔尺寸范围可为从约0.4至约3微米, 或本文所提及的任意孔尺寸。接种可通过以下方式实现:将细胞作为单细 胞或细胞簇从单层培养物释放到悬浮液中,随后将所述细胞悬浮液等分到 定位于空气-液体界面处的多孔基质上。可从悬浮液中将所述细胞接种到多 孔基质上,所述悬浮液包含约1000个细胞/μl至约100,000个细胞/μl、约 1000个细胞/μl至约90,000个细胞/μl、约1000个细胞/μl至约80,000个细 胞/μl、约1000个细胞/μl至约70,000个细胞/μl、约1000个细胞/μl至约 60,000个细胞/μl、约1000个细胞/μl至约50,000个细胞/μl、约1000个细 胞/μl至约40,000个细胞/μl、约1000个细胞/μl至约30,000个细胞/μl、约 1000个细胞/μl至约20,000个细胞/μl、约1000个细胞/μl至约10,000个细 胞/μl、约1000个细胞/μl至约5000个细胞/μl、约5000个细胞/μl至约 100,000个细胞/μl、约10,000个细胞/μl至约100,000个细胞/μl、约20,000 个细胞/μl至约100,000个细胞/μl、约30,000个细胞/μl至约100,000个细胞 /μl、约40,000个细胞/μl至约100,000个细胞/μl、约50,000个细胞/μl至约 100,000个细胞/μl、约60,000个细胞/μl至约100,000个细胞/μl、约20,000 个细胞/μl至约80,000个细胞/μl、约30,000个细胞/μl至约70,000个细胞 /μl、约40,000个细胞/μl至约60,000个细胞/μl,优选地约50,000个细胞 /μl。所述细胞可作为包含个别细胞或细胞团的细胞悬浮液的液滴而接种。 所得的细胞沉积物可包含从约5×106至约5×107个细胞/cm2、约6×106至约 5×107个细胞/cm2、约7×106至约5×107个细胞/cm2、约8×106至约5×107个 细胞/cm2、约9×106至约5×107个细胞/cm2、约1×107至约5×107个细胞 /cm2、约2×107至约5×107个细胞/cm2、约2×107至约5×107个细胞/cm2、约3×107至约5×107个细胞/cm2、约3×107至约5×107个细胞/cm2、约4×107至约5×107个细胞/cm2、约5×106至约4×107个细胞/cm2、约5×106至约 3×107个细胞/cm2、约5×106至约2×107个细胞/cm2、约5×106至约1×107个 细胞/cm2、约5×106至约9×106个细胞/cm2、约5×106至约8×106个细胞 /cm2、约5×106至约7×106个细胞/cm2、约5×106至约6×106个细胞/cm2、 约7×106至约4×107个细胞/cm2、约8×106至约3×107个细胞/cm2、约9×106至约2×107个细胞/cm2,优选地大约或约1×107个细胞/cm2。
在一个实施例中,本发明涉及一种通过在多孔基质上的空气-液体界面 处培养并且使PDX1和NKX6.1共阳性的胰腺内胚层前体细胞群分化成 PDX1和NKX6.1共阳性的胰腺内分泌细胞来提升HB9蛋白的表达的方 法。作为另一种选择,可以通过在空气-液体界面处培养并且使主要由 PDX1阳性细胞构成的前肠内胚层细胞群分化来诱导HB9蛋白表达。在一些实施例中,胰腺内胚层细胞群是通过使多能细胞至少部分地在空气-液体 界面处逐步分化而获得的。
在另一个实施例中,本发明提供一种通过在空气-液体界面处培养和分 化PDX1和NKX6.1共表达细胞群来增加单激素阳性细胞(例如,共表达 NKX6.1和胰岛素的细胞,或产生NKX6.1和嗜铬粒蛋白-A的细胞)的数目 的方法。在另一个实施例中,通过用选自以下的化合物处理,将在空气-液 体界面处培养的胰腺内胚层细胞进一步分化成胰腺内分泌细胞:ALK5抑制 剂、BMP抑制剂、γ-分泌酶抑制剂、Ephrin配体、EphB抑制剂、PKC抑制 剂、EGFr抑制剂、视黄酸、维生素C、T3/T4、葡萄糖、细胞周期调节因 子、WNT调节因子、SHH抑制剂、或它们的组合。
在一些实施例中,在空气-液体界面处培养的胰腺内胚层细胞进一步分 化成胰腺内分泌前体细胞和表达胰腺激素的细胞。在一个替代实施例中, 本发明涵盖用本发明的方法制备的表达胰岛素但不表达NKX6.1的细胞。 在一些实施例中,将在空气-液体界面处生成的胰腺内胚层群体移植到糖尿 病动物体内,以在体内进一步成熟为功能性胰腺内分泌细胞。
第4阶段:使表达前肠内胚层细胞的特征性标志物的细胞分化成表达胰腺前肠前
体细胞的特征性标志物的细胞
在一个实施例中,本发明的方法包括用包含补充有以下一种或多种物 质的生长培养基的分化培养基处理第3阶段细胞:(a)ALK5抑制剂,选 自:TGF-β受体抑制剂V、TGF-β受体抑制剂I、TGF-β受体抑制剂IV、 TGF-β受体抑制剂VII、TGF-β受体抑制剂VIII、TGF-β受体抑制剂II、 TGF-β受体抑制剂VI、TGF-β受体抑制剂III、TGF-B抑制剂SB431542、 SD208、ITD-1、LY2109761、A83-01、LY2157299、ALK5i和ALK5抑制 剂II;(b)甲状腺激素,选自:T3、T4、T3的类似物、T4的类似物、以及 它们的混合物;(c)smoothened受体抑制剂,选自MRT10或环杷明; (d)SHH信号转导途径拮抗剂,选自SANT-1或HPI-1;(e)BMP受体抑制 剂,选自LDN-193189、头蛋白或脊索发生素;(f)PKC活化剂,选自 TPB、PDBu、PMA和ILV;(g)成纤维细胞生长因子,选自FGF7或 FGF10;(h)视黄酸;(i)抗坏血酸;(j)肝素;以及(k)硫酸锌。例如,生长培 养基,诸如MCDB131或BLAR,可补充有SMO抑制剂(诸如MRT10或 环杷明)或SHH信号转导途径拮抗剂(诸如SANT-1或HPI-1)、BMP抑 制剂(诸如LDN-193189、头蛋白或脊索发生素)、抗坏血酸,以及PKC 活化剂(诸如TPB、PDBu、PMA或ILV),以提供可用的分化培养基。 在此类培养基中培养第3阶段细胞约两至四天,优选地约三天,往往足以 使第3阶段细胞分化成第4阶段细胞。在另一个实施例中,培养基可补充 有SMO抑制剂和SHH信号转导途径拮抗剂。在一个优选的实施例中,可 用补充有约0.25μM的SANT-1;约100nM的视黄酸;约2ng/ml的FGF7; 约100nM的LDN-193189;约0.25mM的抗坏血酸;以及约100nM的TPB 的培养基处理第3阶段细胞三天。在另一个实施例中,培养基还补充有 T3,诸如从约5nM至约25nM、或约10nM的T3。
在第4阶段中,可在平面培养物上或在空气-液体界面处培养细胞。具 体地,本发明提供了一种用于使衍生自多能干细胞的细胞在空气-液体界面 处分化的体外细胞培养物,所述体外细胞培养物包括:(a)培养容器;(b)在 所述容器内一定体积的生长培养基,所述体积足以填充所述容器的体积的 仅一部分;(c)所述容器内的空气,所述空气填充所述容器邻接所述培养基 的部分;(d)多孔基质,所述多孔基质定位在所述培养基和所述空气之间的 界面处;以及(e)衍生自多能干细胞的细胞,所述细胞设置在所述基质的表 面上,使得所述培养基接触所述细胞表面的仅一部分。作为另一种选择, 通过用如上所述经补充的培养基在平面培养物中处理,表达前肠内胚层细 胞的特征性标志物的细胞可分化成表达胰腺前肠前体细胞的特征性标志物 的细胞。
在另一个实施例中,在第4阶段结束时可用Rho相关激酶(“ROCK”) 抑制剂处理细胞,所述抑制剂诸如Y27632((1R,4r)-4-((R)-1-氨基乙基)-N- (吡啶-4-基)环己甲酰胺)、GSK269962(N-[3-[[2-(4-氨基-1,2,5- 二唑-3?- 基)-1-乙基-1H-咪唑并[4,5-c]吡啶-6-基]氧]苯基]-4-[2-(4-吗啉基)乙氧基]苯甲 酰胺)、H1152((S)-(+)-2-甲基-1-[(4-甲基-5-异喹啉基)磺酰]高哌嗪二盐酸 盐)和SR3677(N-[2-[2-(二甲氨基)乙氧基]-4-(1H-吡唑-4-基)苯基-2,3-二氢- 1,4-苯并二氧杂苯-2-甲酰胺二盐酸盐)。在某些实施例中可使用约10μM的 ROCK抑制剂。
二唑-3?- 基)-1-乙基-1H-咪唑并[4,5-c]吡啶-6-基]氧]苯基]-4-[2-(4-吗啉基)乙氧基]苯甲 酰胺)、H1152((S)-(+)-2-甲基-1-[(4-甲基-5-异喹啉基)磺酰]高哌嗪二盐酸 盐)和SR3677(N-[2-[2-(二甲氨基)乙氧基]-4-(1H-吡唑-4-基)苯基-2,3-二氢- 1,4-苯并二氧杂苯-2-甲酰胺二盐酸盐)。在某些实施例中可使用约10μM的 ROCK抑制剂。
在某些实施例中,仅在第4阶段的后期在空气-液体界面处培养细胞。 在一个实施例中,仅在第4阶段的后期在空气-液体界面处培养用ROCK抑 制剂处理过的细胞。在某些实施例中,在空气-液体界面处培养之前,可用 细胞脱附溶液处理细胞,所述细胞脱附溶液为诸如包含蛋白水解酶和胶原 蛋白酶的溶液。
在一个替代实施例中,可用包含补充有ALK5抑制剂、头蛋白和PKC 活化剂(诸如TPB)的生长培养基的分化培养基处理第3阶段细胞。在某 些实施例中,培养基可补充有约0.1μM的ALK5抑制剂、约100ng/ml的头 蛋白、以及约500nM的TPB。细胞培养物可为单层形式。处理可持续总共 约三天。在某些实施例中,可处理所述细胞两天,随后在最后一天可用蛋 白水解酶、胶原蛋白酶,或蛋白水解酶和胶原蛋白酶两者,诸如分散酶处 理细胞,使细胞分散成直径小于约100微米的细胞簇,接着在ALK5抑制 剂和LDN-193189的存在下培养。在某些实施例中,可在补充有约200nM 的ALK5抑制剂和约100nM的LDN-193189的培养基中培养直径小于约 100微米的细胞簇。
第5阶段:使表达胰腺前肠前体细胞的特征性标志物的细胞分化成表达胰腺内胚
层/内分泌前体细胞的特征性标志物的细胞
在一个实施例中,本发明的方法包括用包含补充有以下一种或多种物 质的生长培养基的分化培养基处理第4阶段细胞:(a)ALK5抑制剂,选 自:TGF-β受体抑制剂V、TGF-β受体抑制剂I、TGF-β受体抑制剂IV、TGF-β受体抑制剂VII、TGF-β受体抑制剂VIII、TGF-β受体抑制剂II、 TGF-β受体抑制剂VI、TGF-β受体抑制剂III、TGF-B抑制剂SB431542、 SD208、ITD-1、LY2109761、A83-01、LY2157299、ALK5i和ALK5抑制 剂II;(b)甲状腺激素,选自:T3、T4、T3的类似物、T4的类似物、以及 它们的混合物;(c)smoothened受体抑制剂,选自MRT10或环杷明;(d) SHH信号转导途径拮抗剂,选自SANT-1或HPI-1;(e)BMP受体抑制剂, 选自LDN-193189、头蛋白或脊索发生素;(f)PKC活化剂,选自TPB、 PDBu、PMA和ILV;(g)成纤维细胞生长因子,选自FGF7或FGF10;(h) 视黄酸;(i)抗坏血酸;(j)肝素;以及(k)硫酸锌;以及在空气-液体界面处培 养细胞约两至四天,优选地约三天,以使所述细胞分化成第5阶段细胞。 在另一个实施例中,生长培养基补充有SMO抑制剂(诸如MRT10或环杷 明)或SHH信号转导途径拮抗剂(诸如SANT-1或HPI-1)、视黄酸、 T3、抗坏血酸、BMP受体抑制剂(诸如LDN-193189、头蛋白或脊索发生 素)、以及ALK5抑制剂。在另一个实施例中,本发明的方法包括:用补 充有SMO抑制剂、SHH信号转导途径拮抗剂、视黄酸、T3、抗坏血酸、 BMP受体抑制剂和ALK5抑制剂的培养基处理第4阶段细胞,以及在空气- 液体界面处培养细胞约两至四天,优选地约三天,以使所述细胞分化成第5 阶段细胞。在一个实施例中,通过用补充有约0.25μM的SANT-1、约 50nM的视黄酸、约0.25mM的抗坏血酸、约50nM的LDN-193189、约 10nM的T3和约1000nM的ALK5抑制剂的培养基处理第4阶段细胞,来 使第4阶段细胞分化成第5阶段细胞。在某些实施例中,ALK5抑制剂是 SD208((2-(5-氯-2-氟苯基)-N-4-吡啶基-4-蝶啶胺)。在一个实施例中,培 养基补充有约1000nM的SD208。
在另一个实施例中,本发明的方法包括:用补充有肝素、SMO抑制剂 或SHH信号转导途径拮抗剂、视黄酸、BMP受体抑制剂和ALK5抑制剂的 培养基处理第4阶段细胞,以及在空气-液体界面处培养细胞约两至四天, 优选地约三天,以使所述细胞分化成第5阶段细胞。在一个替代实施例 中,培养基可补充有SMO抑制剂和SHH信号转导途径拮抗剂两者,以及 视黄酸、BMP受体抑制剂和ALK5抑制剂。
培养基还可补充有ZnSO4。例如,可添加约10μM的ZnSO4。因此, 在一个实施例中,可通过用补充有肝素、ZnSO4、SMO抑制剂或SHH信号 转导途径拮抗剂、视黄酸、LDN-193189和ALK5抑制剂II的培养基处理第 4阶段细胞,来使第4阶段细胞分化成第5阶段细胞。在一个替代实施例 中,培养基可补充有SMO抑制剂和SHH信号转导途径拮抗剂两者。在一 个实施例中,通过用补充有约10μg/ml的肝素、约0.25μM的SANT-1、约 50nM的视黄酸、约50nM的LDN-193189、约10nM的T3和约1000nM的 ALK5抑制剂的培养基处理第4阶段细胞,来使第4阶段细胞分化成第5阶 段细胞。合适的ALK5抑制剂包括但不限于SD208、ALK5抑制剂II、 TGF-β受体抑制剂V、TGF-β受体抑制剂I、TGF-β受体抑制剂IV、TGF-β 受体抑制剂VII、TGF-β受体抑制剂VIII、TGF-β受体抑制剂II、TGF-β受 体抑制剂VI、TGF-β受体抑制剂III,以及它们的组合。
在一个实施例中,ALK5抑制剂是ALK5抑制剂II。在另一个实施例 中,使用约1000nM的ALK5抑制剂II。在一个替代实施例中,用补充有 约10μg/ml的肝素、约0.25μM的SANT-1、约50nM的视黄酸、约100nM 的LDN-193189,以及约10000nM的ALK5抑制剂II的培养基处理第4阶 段细胞。
在另一个另选实施例中,本发明的方法包括:用补充有SMO抑制剂或 SHH信号转导途径拮抗剂、视黄酸以及ALK5抑制剂的培养基处理第4阶 段细胞,以及在空气-液体界面处培养细胞约2天,以使所述细胞分化成第 5阶段细胞。在一个替代实施例中,培养基可补充有SMO抑制剂和SHH信 号转导途径拮抗剂两者。在一个实施例中,通过用补充有约0.25μM的SANT-1、约50nM的视黄酸、约50nM的LDN-193189,以及约1000nM的 ALK5抑制剂(诸如SD208或ALK5抑制剂II)的培养基处理第4阶段细 胞,来使第4阶段细胞分化成第5阶段细胞。在某些实施例中,培养基可 为MCDB-131(Life Technologies Corporation,Grand Island,NY)。
用于在空气-液体界面处培养的接种细胞量可变化。例如,为了在空 气-液体界面处培养所述细胞,可将包含从约2×105个细胞/μl至约6×105个 细胞/μl、3×105个细胞/μl至约6×105个细胞/μl、4×105个细胞/μl至约6×105个细胞/μl、5×105个细胞/μl至约6×105个细胞/μl、2×105个细胞/μl至约 5×105个细胞/μl、2×105个细胞/μl至约4×105个细胞/μl,或约3×105个细胞 /μl的细胞悬浮液的液滴接种到多孔基质上,诸如定位在空气-液体界面处的 过滤器。在一些实施例中,将包含从约0.5×105个细胞/μl至约0.75×105个细胞/μl、约0.6×105个细胞/μl至约0.75×105个细胞/μl,或约0.5×105个细胞 /μl至约0.6×105个细胞/μl的细胞悬浮液的液滴接种到多孔支承体上,以在 空气-液体界面处培养。
在另一个实施例中,本发明的方法包括:用补充有BMP受体抑制剂 (例如,LDN-193189、头蛋白或脊索发生素)和ALK5抑制剂的培养基处 理第4阶段细胞约1天,以使第4阶段细胞分化成第5阶段细胞。例如,培 养基可补充有约100nM的LDN-193189和约200nM的ALK5抑制剂。优选 地,该实施例还包括用分散酶预处理所述细胞。所述细胞可为细胞簇形式。在某些实施例中,在空气-液体界面处培养之前,可用细胞脱附溶液处 理细胞,所述细胞脱附溶液为诸如包含蛋白水解酶和胶原蛋白酶的溶液。 在一个实施例中,利用根据本发明的实施例培养的第4阶段细胞并且使所 述细胞分化成第5阶段细胞,而在其他实施例中可利用根据其他方案培养 的第4阶段细胞。
根据前述方法,本发明还提供了一种用于使表达胰腺前肠前体细胞的 特征性标志物的细胞分化成表达胰腺内胚层/胰腺内分泌前体细胞的特征性 标志物的细胞的细胞培养物,所述细胞培养物包括:(a)培养容器;(b)在所 述容器内一定体积的生长培养基,所述体积足以填充所述容器的体积的仅 一部分;(c)所述容器内的空气,所述空气填充所述容器邻接所述培养基的 部分;(d)多孔基质,所述多孔基质定位在所述培养基和所述空气之间的界 面处;以及(e)衍生自多能干细胞的表达胰腺前肠前体细胞的特征性标志物 的细胞,所述细胞设置在所述基质的表面上,使得所述培养基接触所述细 胞表面的仅一部分。
在某些实施例中,在第5阶段中在空气-液体界面处培养细胞可提升胰 腺激素的表达。因此,本发明还提供通过在空气-液体界面处培养细胞来提 升胰腺激素的表达的方法。在一些实施例中,第5阶段细胞可如本文所述 并且如在下表VIII至XIII中所示出地处理。在某些实施例中,该方法还可 减少PTF1a、SOX9、CDX2(肠标志物)、ZIC1(外胚层标志物和SOX2 (前内胚层标志物)的表达。
在一个实施例中,该方法包括通过用补充有T3/T4,或ALK5抑制 剂,或T3/T4和ALK5抑制剂两者的培养基处理并且在空气-液体界面处培 养,来使表达胰腺前肠前体细胞的特征性标志物的细胞分化成表达胰腺内 分泌细胞的特征性标志物的细胞。
第6阶段:使表达胰腺内胚层/胰腺内分泌前体细胞的特征性标志物的细胞分化成
表达胰腺内分泌细胞的特征性标志物的细胞
在本发明的一个实施例中,该方法包括:用包含补充有以下一种或多 种物质的生长培养基的分化培养基处理第5阶段细胞:(a)ALK5抑制剂, 选自:TGF-β受体抑制剂V、TGF-β受体抑制剂I、TGF-β受体抑制剂IV、 TGF-β受体抑制剂VII、TGF-β受体抑制剂VIII、TGF-β受体抑制剂II、 TGF-β受体抑制剂VI、TGF-β受体抑制剂III、TGF-B抑制剂SB431542、SD208、ITD-1、LY2109761、A83-01、LY2157299、ALK5i和ALK5抑制 剂II;(b)甲状腺激素,选自:T3、T4、T3的类似物、T4的类似物、以及 它们的混合物;(c)smoothened受体抑制剂,选自MRT10或环杷明;(d) SHH信号转导途径拮抗剂,选自SANT-1或HPI-1;(e)BMP受体抑制剂,选自LDN-193189、头蛋白或脊索发生素;(f)PKC活化剂,选自TPB、 PDBu、PMA和ILV;(g)成纤维细胞生长因子,选自FGF7或FGF10;(h) 视黄酸;(i)抗坏血酸;(j)肝素;以及(k)硫酸锌;以及在空气-液体界面处培 养第5阶段细胞约两至四天,优选地约三天,以使第5阶段细胞分化成第6 阶段细胞。在一个实施例中,生长培养基补充有SMO抑制剂(诸如 MRT10或环杷明)或SHH信号转导途径拮抗剂(诸如SANT-1或HPI- 1)、视黄酸、抗坏血酸、T3/T4,以及ALK5抑制剂。在一个替代实施例 中,培养基可补充有SMO抑制剂和SHH信号转导途径拮抗剂两者。可通 过用补充有约0.25μM的SANT-1、约50nM的RA、约0.25mM的抗坏血 酸、约500mM的ALK5抑制剂,以及约0.1nM的T3的培养基处理第5阶 段细胞约三天,来使第5阶段细胞分化成第6阶段细胞。作为另一种选 择,可通过用补充有约0.25μM的SANT-1、约50nM的视黄酸、约 0.25mM的抗坏血酸、约500nM的ALK5抑制剂和10nM的T3的培养基处 理第5阶段细胞约三天,来使第5阶段细胞分化成第6阶段细胞。可根据 需要,将细胞在此类培养基中再培养两天或以上。
作为另一种选择,可通过用补充有肝素、SMO抑制剂或SHH信号转 导途径拮抗剂、BMP抑制剂、T3/T4和ALK5抑制剂的培养基处理并且在 空气-液体界面处培养第5阶段细胞约六至十四天、或约6天、或约7天、 或约8天、或约9天、或约10天、或约11天、或约12天、或约13天,以 及或约14天,来使第5阶段细胞分化成第6阶段细胞。在一个替代实施例 中,培养基可补充有SMO抑制剂和SHH信号转导途径拮抗剂两者。例 如,可在补充有约10μg/ml的肝素;约0.25μM的SANT-1;约100nM的 LDN-193189;约1000nM的T3和约500至约10,000nM、约1000至约 10,000nM、约5000至约10,000nM、约600至约5000nM、约700至约 5000nM、约800至约5000nM、约900至约5000nM、约1000nM至约 5000nM、约600至约1000nM、约700至约1000nM、约800至约 1000nM、约600至约1200nM、约700至约1200nM、约800至约 1200nM、约900至约1200nM、或约500nM、或约1000mM,以及或约 10,000nM的ALK5抑制剂的培养基中培养所述细胞。
合适的ALK5抑制剂包括但不限于SD208、ALK5抑制剂II、TGF-β受 体抑制剂V、TGF-β受体抑制剂I、TGF-β受体抑制剂IV、TGF-β受体抑制 剂VII、TGF-β受体抑制剂VIII、TGF-β受体抑制剂II、TGF-β受体抑制剂 VI、TGF-β受体抑制剂III,以及它们的组合。
在一个实施例中,ALK5抑制剂是ALK5抑制剂II。在另一个实施例 中,使用约1000nM的ALK5抑制剂II。因此,在一个实施例中,可通过用 补充有肝素、SMO抑制剂或SHH信号转导途径拮抗剂、BMP抑制剂、 T3/T4,以及ALK5抑制剂的培养基处理并且在空气-液体界面处培养第5阶 段细胞约六天来使第5阶段细胞分化成第6阶段细胞。在一个替代实施例中,培养基可补充有SMO抑制剂和SHH信号转导途径拮抗剂两者。在某 些实施例中,在空气-液体界面处培养之前,可用细胞脱附溶液处理细胞, 所述细胞脱附溶液为诸如包含蛋白水解酶和胶原蛋白酶的溶液。
在另一个实施例中,可通过用补充有肝素、SMO抑制剂或SHH信号 转导途径拮抗剂、BMP抑制剂、T3,以及ALK5抑制剂II的培养基处理并 且在空气-液体界面处培养第5阶段细胞约5天至约15天、约6天至约14 天、约7天至约13天、约8天至约12天、约9天至约11天、约5天至约 10天、约10天至约15天、或约5天、或约6天、或约7天、或约8天、 或约9天、或约10天、或约11天、或约12天、或约13天、或约14天, 或约15天,使第5阶段细胞分化成第6阶段细胞。在一个实施例中,在空 气-液体界面处培养细胞5天或以上、6天或以上、7天或以上、8天或以 上、9天或以上、10天或以上、11天或以上、12天或以上、13天或以上、 14天或以上、15天或以上。在一个实施例中,在空气-液体界面处培养细胞 15天或以下、14天或以下、13天或以下、12天或以下、11天或以下、10 天或以下、9天或以下、8天或以下、7天或以下、6天或以下、5天或以 下。在一个实施例中,将细胞在空气-液体界面处培养约10天。在另一个实施例中,将细胞在空气-液体界面处培养约11天。在一个替代实施例中,将 细胞在空气-液体界面处培养约12天。在另一个实施例中,将细胞在空气- 液体界面处培养约15天。在这些实施例中,培养基可补充有约10μg/ml的 肝素、约0.25μM的SANT-1、约100nM的LDN-193189、约1000nM的 T3、以及约10,000nM的ALK5抑制剂II。在某些实施例中,培养基还可补 充有硫酸锌(ZnSO4)。例如,培养基还可补充有约10μM的ZnSO4。在一个 替代实施例中,培养基可补充有SMO抑制剂和SHH信号转导途径拮抗剂 两者。
根据前述方法,本发明还提供了一种用于使表达胰腺内胚层/胰腺内分 泌前体细胞的特征性标志物的细胞分化成表达胰腺内分泌细胞的特征性标 志物的细胞的细胞培养物,所述细胞培养物包括:(a)培养容器;(b)在所述 容器内一定体积的生长培养基,所述体积足以填充所述容器的体积的仅一 部分;(c)所述容器内的空气,所述空气填充所述容器邻接所述培养基的部 分;(d)多孔基质,所述多孔基质定位在所述培养基和所述空气之间的界面 处;以及(e)衍生自多能干细胞的表达胰腺内胚层/胰腺内分泌前体细胞的特 征性标志物的细胞,所述细胞设置在所述基质的表面上,使得所述培养基 接触所述细胞表面的仅一部分。
在一个实施例中,利用根据本发明的实施例培养的第5阶段细胞并且 使所述细胞分化成第6阶段细胞,而在其他实施例中可利用根据其他方案 培养的第5阶段细胞。
在另一个实施例中,本发明的方法导致第6阶段细胞的产生,所述第 6阶段细胞是单激素阳性的。因此,在一个实施例中,本发明的方法产生共 表达NKX6.1和嗜铬粒蛋白-A的第6阶段细胞。在另一个实施例中,本发 明的方法产生共表达NKX6.1和胰岛素的第6阶段细胞。
在本发明的某些实施例中,该方法在第4阶段至第6阶段采用BLAR 定制培养基(参见表II)。培养基可优选地每天更换或另选地每隔一天更 换。在本发明的某些实施例中,所述方法包括:用指定组分按照在本文表 VIII至XIII中示出的量处理第4阶段至第6阶段细胞。
在另一个实施例中,本发明涉及一种用于产生共表达NKX6.1和嗜铬 粒蛋白-A的第6阶段细胞的方法,该方法包括在第4至第6阶段中,优选 地在第5阶段和第6阶段在空气-液体界面处培养细胞。在另一个实施例 中,本发明涉及一种通过在第4至第6阶段中,优选地在第5阶段和第6阶 段在空气-液体界面处培养细胞,来产生表达NKX6.1细胞的单激素胰岛素 阳性细胞的方法。
在第4阶段期间或之后在空气-液体界面处培养细胞可显著提升胰腺内 胚层标志物和内分泌相关标志物的表达。因此,本发明提供通过在第4阶 段期间或之后在空气-液体界面处培养细胞来提升胰腺内胚层标志物和内分 泌相关标志物的表达的方法。
在另一个实施例中,本发明还提供通过在存在ALK5抑制剂的情况 下,在空气-液体界面处培养第4阶段细胞和后续细胞,来增加共表达胰岛 素、嗜铬粒蛋白-A、或嗜铬粒蛋白-A和胰岛素两者的NKX6.1阳性细胞的 收率的方法。在一个实施例中,ALK5抑制剂是ALK5抑制剂II。其他合适 的ALK5抑制剂包括但不限于TGF-β受体抑制剂V、TGF-β受体抑制剂I、TGF-β受体抑制剂IV、TGF-β受体抑制剂VII、TGF-β受体抑制剂VIII、 TGF-β受体抑制剂II、TGF-β受体抑制剂VI、TGF-β受体抑制剂III、以及 它们的组合。在一些实施例中,除了ALK5抑制剂之外,还可如下表VIII 至XIII所述地处理细胞。
在一个实施例中,本发明提供通过在存在ALK5抑制剂II的情况下在 第5阶段期间在空气-液体界面处培养细胞,来增加共表达胰岛素、嗜铬粒 蛋白-A、或嗜铬粒蛋白-A和胰岛素两者的NKX6.1阳性细胞的方法。在一 个实施例中,该方法还包括在第5阶段期间在ALK5抑制剂II和T3的存在 下培养细胞。
第6阶段细胞的体内成熟
在本发明的某些实施例中,根据本发明的方法制备的第6阶段细胞可 进一步在体内成熟。在一个实施例中,这些细胞可通过移植到哺乳动物的 体内而进一步成熟。例如,可将细胞移植到小鼠肾包膜下方。在一个实施 例中,在体内进一步成熟的第6阶段细胞是共表达NXK6.1和胰岛素的细 胞。在另一个实施例中,在体内进一步成熟的第6阶段细胞是共表达 NXK6.1和嗜铬粒蛋白的细胞。在一个替代实施例中,(a)共表达NXK6.1和 胰岛素的细胞或(b)共表达NXK6.1和嗜铬粒蛋白的细胞的体内成熟导致早 期的C肽产生。在某些实施例中,从移植约3百万个第6阶段细胞产生的 C-肽水平类似于通过移植约3,000个人类胰岛产生的C-肽量。
根据本文所述的方法在空气-液体界面处培养还适用于根据化合物对胰 腺激素和内分泌标志物的分泌的影响来筛选化合物。具体地,在空气-液体 界面处培养的第4阶段至第6阶段细胞可以多种培养形式使用,包括从384 至6孔形式,以评估以多种剂量和时间间隔包含多种小分子制剂或生物制 剂对胰腺内胚层、胰腺内分泌前体、胰腺内分泌标志物和胰腺β细胞标志 物的后续表达的影响。此类评估可通过以下方式实现:用PCR测量基因表达,用FACS、免疫染色测量蛋白质表达,或用ELISA来测量受到小分子 制剂或生物制剂的添加影响的细胞的细胞因子分泌。
可使用本发明的方法获得的细胞
本发明还提供通过本发明的方法可获得的细胞或细胞群。本发明还提 供已通过本发明的方法获得的细胞或细胞群。
本发明还提供优选地表达胰腺内分泌细胞的特征性标志物的细胞或细 胞群,所述细胞或细胞群通过显著共表达NKX6.1和嗜铬粒蛋白-A来表 征。本发明还提供一种优选地表达胰腺内分泌细胞的特征性标志物的胰岛 素阳性细胞或胰岛素阳性细胞群,所述细胞或细胞群通过NKX6.1表达 (任选地>30%)来表征。这些细胞或细胞群是先前未描述的细胞群,如在 实例10中所解释的。
治疗方法
本发明提供治疗方法。具体地,本发明提供了一种用于治疗患有糖尿 病,或有发展糖尿病风险的患者的方法。
本发明还提供可通过本发明的方法获得或已通过本发明的方法获得以 用于治疗方法的细胞或细胞群。具体地,本发明提供可通过本发明的方法 获得或已通过本发明的方法获得以用于治疗患有糖尿病,或有发展糖尿病 风险的患者的方法的细胞或细胞群。
糖尿病可为1型糖尿病或2型糖尿病。
在一个实施例中,治疗方法包括将已通过本发明的方法获得或可通过 本发明的方法获得的细胞植入患者。
在一个实施例中,治疗方法包括:
例如如本文所述,使多能干细胞体外分化成第1阶段、第2阶 段、第3阶段、第4阶段、第5阶段或第6阶段细胞,
以及将分化的细胞植入患者。
在一个实施例中,该方法还包括在使多能干细胞分化的步骤之前,例 如如本文所述的培养多能干细胞的步骤。
在一个实施例中,该方法还包括在植入步骤之后,使细胞体内分化的 步骤。
在一个实施例中,患者是哺乳动物,优选地是人类。
在一个实施例中,可将这些细胞可作为分散的细胞植入,或可使这些 细胞形成簇,可将这些细胞簇输注进肝门静脉内。作为另一种选择,可将 细胞设置在生物相容性的可降解聚合物型支持物中、多孔的非可降解性装 置中或可进行封装以保护其免受宿主免疫应答的破坏。可将细胞植入到受 体中的适当位置内。植入位点包括例如肝脏、天然的胰腺、肾包膜下空 间、网膜、腹膜、浆膜下空间、肠、胃或皮下袋。
为了增强植入细胞在体内的进一步分化、存活率或活性,可在施用细 胞之前、与之同时或之后施用额外的因子,例如生长因子、抗氧化剂或抗 炎剂。这些因子可由内源性细胞分泌并原位暴露于所施用的细胞。可通过 本领域已知的内源施用的生长因子或外源施用的生长因子的任意组合来诱 导植入细胞进行分化。
移植中所用的细胞量取决于多种因素,包括患者的状况和对该疗法的 响应,并且可由本领域技术人员确定。
在一个实施例中,治疗方法还包括在移植前将所述细胞结合到三维支 持物中。在移植进患者前,可将该细胞体外保持在该支持物上。作为另一 种选择,可将包含该细胞的支持物直接植入到患者中而无需进行额外的体 外培养。可任选将至少一种有利于植入细胞的存活和功能的药剂掺入该支 持物中。
实例
实例1
在空气-液体界面处培养胰腺内分泌前体细胞
该实例检查并证实胰腺内分泌前体细胞(第5阶段细胞)当在空气-液 体界面处培养时可进一步成熟。为了在空气-液体界面处培养胰腺内分泌前 体细胞,基于如下所论述的方案将胚胎干细胞分化成胰腺内分泌前体细胞。
将人胚胎干细胞系H1的细胞作为单细胞以1×105个细胞/cm2接种在 MATRIGELTM(1:30的稀释度;BD Biosciences,Franklin Lakes,NJ)涂覆的 培养皿上的 培养基(StemCell Technologies,Vancouver,Canada) 中,所述培养基补充有10μM的Y27632(Rock抑制剂,目录号Y0503, Sigma-Aldrich,St.Louis,MO)。接种后四十八小时,将培养物在不完全 PBS(不含Mg或Ca的磷酸盐缓冲盐水溶液)中洗涤。随后根据以下方案 使所述细胞分化:
培养基(StemCell Technologies,Vancouver,Canada) 中,所述培养基补充有10μM的Y27632(Rock抑制剂,目录号Y0503, Sigma-Aldrich,St.Louis,MO)。接种后四十八小时,将培养物在不完全 PBS(不含Mg或Ca的磷酸盐缓冲盐水溶液)中洗涤。随后根据以下方案 使所述细胞分化:
a)第1阶段:(3天):使接种在用1:30的MATRIGELTM涂覆的表 面上的未分化H1细胞的60-70%汇合贴壁培养物暴露于 RPMI 1640培养基(Life TechnologiesCorporation,Grand Island,NY) 仅一天,所述
RPMI 1640培养基(Life TechnologiesCorporation,Grand Island,NY) 仅一天,所述 RPMI 1640培养基补充有0.2%的胎牛血清 (FBS)(Hyclone,Utah)、100ng/ml的活化素-A(AA;Pepro-tech; Rocky Hill,NJ)以及20ng/ml的Wnt3A(R&D Systems,Inc., Minneapolis,MN)。在接下来的两天,在补充有0.5%的FBS和 100ng/ml的AA的
RPMI 1640培养基补充有0.2%的胎牛血清 (FBS)(Hyclone,Utah)、100ng/ml的活化素-A(AA;Pepro-tech; Rocky Hill,NJ)以及20ng/ml的Wnt3A(R&D Systems,Inc., Minneapolis,MN)。在接下来的两天,在补充有0.5%的FBS和 100ng/ml的AA的 RPMI培养基中培养细胞。
RPMI培养基中培养细胞。
b)第2阶段:(3天):随后使第1阶段细胞暴露于Dulbecco改良的 eagle培养基(DMEM-F12)(Life Technologies Corporation,NY)三 天,所述培养基补充有2%的FBS和50ng/ml的FGF7(Pepro- tech)。
c)第3阶段:(4天):第2阶段细胞随后在DMEM-HG培养基 (Life TechnologiesCorporation,Grand Island,NY)中培养四天,所述 DMEM-HG培养基补充有0.25μM的SANT-1(Sigma-Aldrich;St. Louis,MO)、2μM的视黄酸(Sigma-Aldrich)、100ng/ml的头蛋白 (R&DSystems),以及由Life Technologies Corporation,Grand Island, NY以商标 出售的1%(v/v)的补充剂(目录号:17504044)。
出售的1%(v/v)的补充剂(目录号:17504044)。
d)第4阶段:(3天):第3阶段细胞随后可在单层形式的DMEM- HG培养基中培养三天,所述DMEM-HG培养基补充有0.1μM的ALK5抑制剂(ALK5i;Axxora,San Diego,CA)、100ng/ml的头蛋 白、500nM的TPB((2S,5S)-(E,E)-8-(5-(4-(三氟甲基)苯基)-2,4-戊 二烯酰氨基)苯并内酰胺;EMD Chemicals Inc,Gibbstown NJ)和 1%的B27。在培养的最后一天,用5mg/ml的分散酶(Becton Dickinson,Bedford,MA,#354235)处理细胞5分钟,接着温和地吹打混匀细胞并使细胞分散成细胞簇(<100微米)。将所述细胞簇 移入一次性的125ml聚苯乙烯转瓶(Corning),并且使所述细胞在 含补充有200nM的ALK5抑制剂、100nM的LDN-193189(Stemgent,CA)和1%的B27的DMEM-HG培养基的悬浮液中以80 至100rpm旋转过夜。
e)第5阶段:(1天):第4阶段细胞随后用5mg/ml的分散酶处理 5分钟,接着温和地吹打混匀细胞并使细胞分散成细胞簇(<100 微米)。将所述细胞簇移入一次性的125ml聚苯乙烯转瓶(Corning, NY),并且使所述细胞在含补充有200nM的ALK5抑制剂、 100nM的LDN-193189(Stemgent,CA)和1%的B27的DMEM-HG 的悬浮液中以80至100rpm旋转过夜。
将第5阶段第1天(S5D1)的细胞簇接种到6孔板中的0.4微米多孔 细胞培养物滤器芯子(BD Biosciences,PET膜,#353493)上(在10微升 等分试样中包含约1百万个细胞),以及通过在滤芯的底部添加1.5ml的 DMEM-HG+1%的B27并且在所述滤芯上方无培养基来在空气-液体界面处 培养所述细胞簇3周。图1A至图1H示出在空气-液体界面处在各个接种后时间点的细胞簇的相位对比图像。图2A至图2K示出在细胞簇接种到过滤 器上后一周时下列蛋白的免疫染色结果:DAPI(图2A);胰岛素(图 2B);HB9(图2C);DAPI(图2D);胰高血糖素(图2E);胰岛素 (图2F);DAPI(图2G);胰岛素(图2H);生长抑素(图2I); NKX6.1(图2J);以及胰岛素(图2K)。而图3A至图3H示出了在接种 到过滤器上后2周时下列蛋白的免疫染色结果:胰岛素(图3A);胰高血 糖素(图3B);胰岛素(图3C);生长抑素(图3D);胰岛素(图 3E);NKX6.1(图3F);HB9(图3G);以及NKX6.1(图3H)。在图 2中,实验组A-C、D-F、G-I和J-K取自相同的视野。在图3中,实验组 A-B、C-D、E-F和G-H分别取自相同的视野。图4A至图4D示出了在接种 到过滤器上后3周时下列蛋白的免疫染色结果:胰岛素(图4A);胰高血 糖素(图4B);胰岛素(图4C);以及生长抑素(图4D)。在图4中, 实验组A-B和C-D分别取自相同的视野。
在第4阶段和随后的培养中,采集mRNA以进行对相关胰腺内胚层/内 分泌基因的PCR分析。用 小量提取试剂盒(
小量提取试剂盒( Mini Kit) (Qiagen;Valencia,CA)提取总RNA,并且用高容量cDNA逆转录试剂盒 (High Capacity cDNA Reverse TranscriptionKit)(Applied Biosystems,Foster City,CA)根据制造商的使用说明逆转录所述总RNA。cDNA使用Taqman 通用预混液(Taqman Universal Master Mix)和预加载到定制的Taqman阵列 (Taqman Arrays)(Applied Biosystems)上的Taqman基因表达分析(Taqman GeneExpression Assays)进行扩增。所述数据使用序列检测软件(Applied Biosystems)进行分析,并且使用ΔΔCt方法(即,用内部对照校准的qPCR 结果(ΔΔCt=ΔCtsample-ΔCtreference))对未分化的人胚胎干(hES)细胞进行归一 化。所有引物均购自AppliedBiosystems。FACS和免疫荧光分析是如先前 所描述地进行的(Diabetes,61,20126,2012)。
Mini Kit) (Qiagen;Valencia,CA)提取总RNA,并且用高容量cDNA逆转录试剂盒 (High Capacity cDNA Reverse TranscriptionKit)(Applied Biosystems,Foster City,CA)根据制造商的使用说明逆转录所述总RNA。cDNA使用Taqman 通用预混液(Taqman Universal Master Mix)和预加载到定制的Taqman阵列 (Taqman Arrays)(Applied Biosystems)上的Taqman基因表达分析(Taqman GeneExpression Assays)进行扩增。所述数据使用序列检测软件(Applied Biosystems)进行分析,并且使用ΔΔCt方法(即,用内部对照校准的qPCR 结果(ΔΔCt=ΔCtsample-ΔCtreference))对未分化的人胚胎干(hES)细胞进行归一 化。所有引物均购自AppliedBiosystems。FACS和免疫荧光分析是如先前 所描述地进行的(Diabetes,61,20126,2012)。
图5A至图5R示出了如实例1中所概述分化的人胚胎干细胞系H1的 细胞中下列基因的表达的实时PCR分析的数据:PDX1(图5A);NKX6.1 (图5B);Pax4(图5C);Pax6(图5D);NGN3(图5E);NKX2.2 (图5F);ABCC8(图5G);嗜铬粒蛋白-A(图5H);PCSK1(图 5I);IAPP(图5J);胰岛素(图5K);胰高血糖素(图5L);生长抑 素(图5M);生长素释放肽(图5N);Ptf1a(图5O);Zic1(图 5P);CDX2(图5Q);以及SOX9(图5R)。在空气-液体界面处培养3 周之后,与内分泌细胞的成熟相关联的标志物诸如ABCC8、IAPP(淀粉不 溶素)和PCSK1的表达存在显著的时间依赖性增加。存在PTF1a和SOX9 表达的显著下降和非常低的CDX2(肠标志物)、ZIC1(外胚层标志物) 和SOX2(前内胚层标志物)表达,而NKX6.1和PDX1的表达维持在非常 高的水平。所有胰腺激素的表达通过在空气-液体界面处培养3周而得到显 著的提升。
实例2
使用各种滤器芯子在空气-液体界面处培养胰腺内分泌前体细胞
该实例检查在胰腺内胚层细胞在空气-液体界面处的分化中滤器芯子的 类型和孔隙率。为了检查滤器芯子的类型和孔隙率的影响,使用以下论述 的方案来使胚胎干细胞分化。
将人胚胎干细胞系H1的细胞(传代40代)作为单细胞以1×105个细 胞/cm2接种在MATRIGELTM(1:30的稀释度;BD Biosciences,NJ)涂覆的 培养皿上或MATRIGELTM涂覆的滤器芯子(Millipore PIHT 30R 48)上的培养 基中,所述培养基包含DMEM-F12(Invitrogen,Ca)、GlutaMaxTM(1:100的 稀释度,Invitrogen)、0.25mM的抗坏血酸(Sigma,MO)、100ng/ml的FGF2 (R&D systems,MN)、1ng/ml的TGF-β(R&D systems)、ITS-X(1:100的 稀释度)、2%无脂肪酸BSA(Lampire,PA),以及20ng/ml的IGF-1(R&D systems),所述培养基补充有10μM的Y27632(Rock抑制剂,目录号 Y0503,Sigma-Aldrich,St.Louis,MO)。接种后四十八小时,将培养物在 不完全PBS(不含Mg或Ca的磷酸盐缓冲盐水溶液)中洗涤。随后根据以 下方案使所述细胞分化:
a.第1阶段(3天):细胞在MCDB-131培养基(Invitrogen,目录 号10372-019)中培养一天,所述MCDB-131培养基补充有2%的 无脂肪酸BSA(Proliant,目录号68700)、0.0012g/ml的碳酸氢钠 (Sigma-Aldrich,目录号S3187)、1X的GlutaMaxTM (Invitrogen,目录号35050-079)、4.5mM的D-葡萄糖(Sigma- Aldrich,目录号G8769)、100ng/ml的GDF8(R&DSystems)和 1μM的MCX化合物。细胞随后在MCDB-131培养基中再培养一 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、100ng/ml的GDF8和0.1μM的MCX化合物。接着,细胞随后 在MCDB-131培养基中再培养一天,所述MCDB-131培养基补充 有2%的无脂肪酸BSA、0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄糖和100ng/ml的GDF8。
b.第2阶段(2天):第1阶段细胞随后用MCDB-131培养基处理两 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、0.25mM的抗坏血酸(Sigma,MO)和25ng/ml的FGF7(R&D Systems,MN)。
c.第3阶段(2天):第2阶段细胞随后用MCDB-131培养基处理两 天,所述MCDB-131培养基补充有1:200稀释度的ITS-X (Invitrogen,CA)、4.5mM的葡萄糖、1X的GlutaMaxTM、0.0017g/ml 的碳酸氢钠、2%的无脂肪酸BSA、0.25μM的SANT-1(Sigma, MO)、1μM的RA(Sigma,MO)、25ng/ml的FGF7、0.25mM的抗 坏血酸、200nM的TPB(PKC活化剂;目录号565740;EMD Chemicals,Gibbstown,NJ),以及100nM的LDN-193189(BMP 受体抑制剂;目录号04-0019;Stemgent)。
d.第4阶段(3天):第3阶段细胞随后用MCDB-131培养基处理三 天,所述MCDB-131培养基补充有1:200稀释度的ITS-X、4.5mM 的葡萄糖、1X的GlutaMaxTM、0.0017g/ml的碳酸氢钠、2%的无脂 肪酸BSA、0.25μM的SANT-1、100nM的RA、2ng/ml的FGF7、 100nM的LDN-193189、0.25mM的抗坏血酸、10nM的T3(T6397, Sigma)和100nM的TPB。
e.第5阶段(3天):第4阶段细胞随后用MCDB-131培养基处理三 天,所述MCDB-131培养基补充有1:200稀释度的ITS-X、4.5mM 的葡萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢钠、2%的无脂 肪酸BSA、0.25μM的SANT-1、50nM的RA、0.25mM的抗坏血 酸、10nM的T3、50nM的LDN-193189、1000nM的ALK5抑制剂 SD208。SD208((2-(5-氯-2-氟苯基)-N-4-吡啶基-4-蝶啶胺)是一种 2,4-二取代的蝶啶,是TGF-βR I激酶的ATP-竞争性抑制剂,公开 于Molecular Pharmacology 2007,72:152-161中并且具有如式I中 所示出的结构。
式1:

f.第6阶段(5天):第5阶段细胞随后用MCDB-131培养基处理三 天,所述MCDB-131培养基补充有1:200稀释度的ITS-X、4.5mM 的葡萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢钠、2%的无脂 肪酸BSA、0.25μM的SANT-1、50nM的RA、0.25mM的抗坏血 酸、500nM的ALK5抑制剂、0.1nM的T3。
在一些培养物中,在第3阶段至第6阶段,在平皿上培养的细胞在室 温下用1X的ACCUTASETM(StemCell Tech,Vancouver)处理1-3分钟,之后 去除所述酶并且用细胞刮棒刮擦所述细胞。将所得的细胞悬浮液以约2- 6×106个细胞的密度接种到6孔板中的0.4微米多孔细胞培养物滤器芯子 上,所述滤器芯子具有约4.2cm2的表面积。所使用的各种滤器芯子在表I 中示出。将1.5ml的培养基添加至每一滤芯的底部,并且不另外添加培养基 至过滤器的顶上侧。在一些培养物中,将过滤器用MATRIGELTM(1:30的 稀释度)在室温下涂覆1小时,并将所述细胞接种到在空气-液体界面处的 经涂覆滤芯的顶部上,或在所述滤芯的顶端部上有培养基。培养基在研究 持续期间每隔一天更换。
图6示出了如实例2中所概述分化的人胚胎干细胞系H1的细胞中下列 基因的表达的实时PCR分析的数据:PDX1(图6A);NKX6.1(图 6B);PAX4(图6C);PAX6(图6D);NGN3(图6E);NKX2.2(图 6F);ABCC8(图6G);嗜铬粒蛋白-A(图6H);PCSK1(图6I); IAPP(图6J);胰岛素(图6K);以及胰高血糖素(图5L)。在滤器芯 子(Corning,3412;BD,353493;以及Millipore PIHT 30R 48)上培养第 4阶段(S4)细胞显著增加胰腺内胚层标志物以及内分泌相关的标志物。用 MATRIGELTM涂覆过滤器显著地减弱在空气-液体界面处的过滤器上培养的 益处。此外,在低孔隙密度的滤器芯子(BD 353090,Corning 3452)上培养的 细胞显示出较少的存活率和分化。

实例3
相较于平面培养物或维持在液体-液体界面处的过滤器上的培养物,在空气-液体
界面处培养的胰腺内胚层细胞显示出提升的内分泌标志物表达
该实例涉及在平面基质上培养的胰腺内胚层细胞相较于在空气-液体界 面处培养的那些胰腺内胚层细胞的分化倾向差异。此外,空气-液体界面的 效果通过在滤芯上使细胞分化但是将培养基同时添加至所述滤芯的顶部和 底部而进一步突出。
将人胚胎干细胞系H1的细胞(传代40代)作为单细胞以1×105个细 胞/cm2接种在MATRIGELTM(1:30的稀释度;BD Biosciences,NJ)涂覆的 培养皿上或MATRIGELTM涂覆的滤器芯子(Millipore PIHT 30R 48)上的培养 基中,所述培养基包含DMEM-F12(Invitrogen,Ca)、GlutaMaxTM(1:100的 稀释度,Invitrogen)、0.25mM的抗坏血酸(Sigma,MO)、100ng/ml的FGF2 (R&D systems,MN)、1ng/ml的TGF-β(R&D systems)、ITS-X(1:100的 稀释度)、2%无脂肪酸BSA(Lampire,PA),以及20ng/ml的IGF-1(R&D systems)),所述培养基补充有10μM的Y27632(Rock抑制剂,目录号 Y0503,Sigma-Aldrich,St.Louis,MO)。接种后四十八小时,将培养物在 不完全PBS(不含Mg或Ca的磷酸盐缓冲盐水溶液)中洗涤。对于在滤器 芯子上培养的细胞,在第1阶段开始时,在一些培养物中仅将培养基添加 至滤芯的底部,而滤芯的顶部则保持在空气-液体界面处,而在其他培养中 还将培养基添加至滤器芯子的顶部以及滤芯的底部。随后根据以下方案使 所述细胞分化:
a)第1阶段(3天):细胞在MCDB-131培养基(Invitrogen,目录 号10372-019)中培养一天,所述MCDB-131培养基补充有2%的 无脂肪酸BSA(Proliant,目录号68700)、0.0012g/ml的碳酸氢钠 (Sigma-Aldrich,目录号S3187)、1X的GlutaMaxTM (Invitrogen,目录号35050-079)、4.5mM的D-葡萄糖(Sigma- Aldrich,目录号G8769)、100ng/ml的GDF8(R&DSystems)和 1μM的MCX化合物。细胞随后在MCDB-131培养基中再培养一 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、100ng/ml的GDF8和0.1μM的MCX化合物。接着,细胞在 MCDB-131培养基中再培养一天,所述MCDB-131培养基补充有 2%的无脂肪酸BSA、0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、 4.5mM的D--葡萄糖和100ng/ml的GDF8。
b)第2阶段(2天):第1阶段细胞随后用MCDB-131培养基处理两 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、0.25mM的抗坏血酸(Sigma,MO)和25ng/ml的FGF7(R&D Systems,MN)。
c)第3阶段(2天):第2阶段细胞随后用BLAR定制培养基(由 Invitrogen制备,BLAR培养基的组分请参见表II)处理两天,所 述BLAR定制培养基补充有1:200稀释度的ITS-X(Invitrogen, CA)、4.5mM的葡萄糖、1X的GlutaMaxTM、0.0017g/ml的碳酸氢 钠、2%的无脂肪酸BSA、0.25μM的SANT-1(Sigma,MO)、1μM 的RA(Sigma,MO)、25ng/ml的FGF7、0.25mM的抗坏血酸、 200nM的TPB(PKC活化剂;目录号565740;EMD Chemicals, Gibbstown,NJ),以及100nM的LDN-193189(BMP受体抑制 剂;目录号04-0019;Stemgent)。
d)第4阶段(3天):第3阶段细胞随后用BLAR培养基处理三天, 所述BLAR培养基补充有1:200稀释度的ITS-X、4.5mM的葡萄 糖、1X的GlutaMaxTM、0.0017g/ml的碳酸氢钠、2%的无脂肪酸 BSA、0.25μM的SANT-1、100nM的RA、2ng/ml的FGF7、100nM的LDN-193189、0.25mM的抗坏血酸、10nM的T3(T6397, Sigma)和100nM的TPB。
e)第5阶段(3天):第4阶段细胞用BLAR培养基处理三天,所述 BLAR培养基补充有1:200稀释度的ITS-X、4.5mM的葡萄糖、1X 的GlutaMaxTM、0.0015g/ml的碳酸氢钠、2%的无脂肪酸BSA、 0.25μM的SANT-1、50nM的RA、0.25mM的抗坏血酸、10nM的 T3、50nM的LDN-193189、1000nM的ALK5抑制剂(SD208)。
f)第6阶段(5天):第5阶段细胞随后用BLAR培养基处理三天, 所述BLAR培养基补充有1:200稀释度的ITS-X、4.5mM的葡萄 糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢钠、2%的无脂肪酸 BSA、0.25μM的SANT-1、50nM的RA、0.025mM的抗坏血酸、 500nM的ALK5抑制剂、0.1nM的T3。
在一些培养物中,在第3阶段结束时,在平皿上培养的细胞在室温下 用1X的ACCUTASETM(StemCell Tech,Vancouver)处理1-3分钟,之后去除 所述酶并且用细胞刮棒刮擦所述细胞。将所得的细胞悬浮液以2-6×106个细 胞的密度(在50-100μl的等分试样中)接种到6孔板中的0.4微米多孔细胞 培养物滤器芯子(BD 353493)上。将1.5ml的培养基添加至每一滤芯的底 部,并且不另外添加培养基至过滤器的顶上侧。培养基在研究持续期间每 隔一天更换。
图7示出了如实例3中所概述分化的人胚胎干细胞系H1的细胞中下列 基因的表达的实时PCR分析的数据:PDX1(图7A);NKX6.1(图 7B);PAX4(图7C);PAX6(图7D);NGN3(图7E);NKX2.2(图 7F);ABCC8(图7G);嗜铬粒蛋白-A(图7H);PCSK1(图7I); IAPP(图7J);胰岛素(图7K);以及胰高血糖素(图7L)。
在第4阶段至第6阶段中在空气-液体界面处的滤器芯子上培养细胞显 著提升了与胰腺内分泌相关的标志物。此外,相较于在平面培养物上或在 空气-液体界面处培养的细胞,在滤器芯子的顶部和底部上都有培养基的情 况下从第1阶段开始时在MATRIGELTM涂覆的滤器芯子上培养和分化的细 胞确实显示出较低水平的胰腺内胚层和内分泌表达。此外,相较于所有测 试的构型,在空气-液体界面处的滤器芯子上培养的胰腺内胚层细胞显示出 最高的胰腺内分泌细胞表达。

这一页的其余部分故意保留空白
实例4
在空气-液体界面处培养并且用ALK5抑制剂II处理的胰腺内胚层细胞显示出显著
更多数量的共表达嗜铬粒蛋白-A和胰岛素的NKX6.1阳性细胞
进行该实例以显示ALK5抑制剂II独特地在空气-液体界面处产生表达 NKX6.1和胰岛素或嗜铬粒蛋白-A的大量细胞群。此外,这种观察结果是 在空气-液体界面处的培养物所特有的。在单层平面培养物中的浸没培养未 能显示出表达胰岛素或嗜铬粒蛋白-A的大量NKX6.1细胞。
将人胚胎干细胞系H1的细胞(传代40代)作为单细胞以1×105个细 胞/cm2接种在MATRIGELTM(1:30的稀释度;BD Biosciences,NJ)涂覆的 培养皿上或Matrigel涂覆的滤器芯子(Millipore PIHT 30R 48)上的培养基 中,所述培养基包含DMEM-F12(Invitrogen,Ca)、GlutaMaxTM(1:100的稀 释度,Invitrogen)、0.25mM的抗坏血酸(Sigma,MO)、100ng/ml的FGF2 (R&D systems,MN)、1ng/ml的TGF-β(R&D systems)、ITS-X(1:100的 稀释度)、2%的无脂肪酸BSA(Lampire,PA),以及20ng/ml的IGF-1(R& D systems),所述培养基补充有10μM的Y27632(Rock抑制剂,目录号 Y0503,Sigma-Aldrich,St.Louis,MO)。接种后四十八小时,将培养物在 不完全PBS(不含Mg或Ca的磷酸盐缓冲盐水溶液)中洗涤。随后根据以 下方案使所述细胞分化:
a)第1阶段(3天):细胞在MCDB-131培养基(Invitrogen,目录 号10372-019)中培养一天,所述MCDB-131培养基补充有2%的 无脂肪酸BSA(Proliant,目录号68700)、0.0012g/ml的碳酸氢钠 (SigmaAldrich,目录号S3187)、1X的GlutaMaxTM (Invitrogen,目录号35050-079)、4.5mM的D-葡萄糖 (SigmaAldrich,目录号G8769)、100ng/ml的GDF8(R&DSystems)和1μM的MCX化合物。细胞随后在MCDB-131培养基 中再培养一天,所述MCDB-131培养基补充有2%的无脂肪酸 BSA、0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄糖、100ng/ml的GDF8和0.1μM的MCX化合物。细胞随后在 MCDB-131培养基中再培养一天,所述MCDB-131培养基补充有 2%的无脂肪酸BSA、0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄糖和100ng/ml的GDF8。
b)第2阶段(2天):第1阶段细胞随后用MCDB-131培养基处理两 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、0.25mM的抗坏血酸(Sigma,MO)和25ng/ml的FGF7(R&D Systems,MN)。
c)第3阶段(2天):第2阶段细胞随后用BLAR定制培养基 (Invitrogen)处理两天,所述BLAR定制培养基补充有1:200稀释度 的ITS-X(Invitrogen,Ca)、4.5mM的葡萄糖、1X的GlutaMaxTM、 0.0017g/ml的碳酸氢钠、2%的无脂肪酸BSA、0.25μM的SANT-1 (Sigma,MO)、1μM的RA(Sigma,MO)、25ng/ml的FGF7、 0.25mM的抗坏血酸、200nM的TPB(PKC活化剂;目录号565740;EMD Chemicals,Gibbstown,NJ),以及100nM的LDN- 193189(BMP受体抑制剂;目录号04-0019;Stemgent)。
d)第4阶段(3天):第3阶段细胞随后用BLAR培养基处理三天, 所述BLAR培养基补充有1:200稀释度的ITS-X、4.5mM的葡萄 糖、1X的GlutaMaxTM、0.0017g/ml的碳酸氢钠、2%的无脂肪酸 BSA、0.25μM的SANT-1、100nM的RA、2ng/ml的FGF7、 100nM的LDN-193189、0.25mM的抗坏血酸和100nM的TPB。
e)第5阶段(-3天):第4阶段细胞随后用BLAR培养基处理三 天,所述BLAR培养基补充有1:200稀释度的ITS-X、20mM的葡 萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢钠、2%的无脂肪酸 BSA、0.25μM的SANT-1、50nM的RA、50nM的LDN、500- 1000nM的各种ALK5抑制剂(关于所使用的抑制剂的列表请参见 表III)。
f)第6阶段(7天):第5阶段细胞随后用BLAR培养基处理,所述 BLAR培养基补充有1:200稀释度的ITS-X、20mM的葡萄糖、1X 的GlutaMaxTM、0.0015g/ml的碳酸氢钠、2%的无脂肪酸BSA、 0.25μM的SANT-1、50nM的RA、500-1000nM的ALK5抑制剂 (关于所测试的抑制剂的列表请参见表III)。
在一些培养物中,在第4阶段的第1天结束时,在平皿上培养的细胞 在室温下用1X的ACCUTASETM(StemCell Tech,Vancouver)处理1-3分钟, 之后去除所述酶并且用细胞刮棒刮擦所述细胞。将所得的细胞悬浮液以2- 4×106个细胞的密度(在25-50μl的等分试样中)接种到6孔板中的0.4微米 多孔细胞培养物滤器芯子(BD 353493)上。将1.5ml的培养基添加至每一滤 芯的底部,并且不另外添加培养基至过滤器的顶上侧。培养基在研究持续 期间每隔一天更换。
图8示出了如在实例4中所概述的在空气-液体界面处分化和培养的人 胚胎干细胞系H1的细胞在空气-液体界面处的第4阶段第1天(S4D1)、 第5阶段第3天(S5D3)和第6阶段第6天(S6D6)之后,其中下列基因 的表达的实时PCR分析的数据:PDX1(图8A)、NKX6.1(图8B)、 NGN3(图8C)、ABCC8(图8D)、PCSK1(图8E)、生长素释放肽 (图8F)、胰高血糖素(图8G),以及胰岛素(图8H)。
图9示出了如在实例4中所概述分化并且在第5阶段第3天(S5D3) 和第6阶段第4天(S6D4)在平面单层培养物中培养的人胚胎干细胞系H1 的细胞中下列基因的表达的实时PCR分析的数据:PDX1(图9A)、 NKX6.1(图9B)、NGN3(图9C)、ABCC8(图9D)、PCSK1(图 9E)、生长素释放肽(图9F)、胰高血糖素(图9G),以及胰岛素(图 9H)。图8和图9的比较显示出在第5阶段和第6阶段用ALK5抑制剂II 处理平面培养物导致相较于第5阶段,第6阶段的胰岛素表达下降。然 而,用ALK5抑制剂对在空气-液体界面处的培养物的处理导致相较于第5 阶段,第6阶段的胰岛素表达提升。相较于单层培养物,相同的模式还应 用于在空气-液体界面处的培养物中的NGN3和NKX6.1表达。
图10示出了在空气-液体界面处在用1微摩尔SD208抑制剂(实验组 9A)或1微摩尔ALK5抑制剂II(实验组10B)处理过的培养基中培养并且 针对嗜铬粒蛋白-A(胰腺内分泌标志物)和NKX6.1(胰腺前体标志物和β 细胞特异性标志物)进行染色的第6阶段细胞的免疫染色结果。用ALK5抑 制剂II处理过的培养物导致NKX6.1和嗜铬粒蛋白-A的共表达。然而,用 SD208处理过的培养物显示出非常低的NKX6.1和嗜铬粒蛋白-A共表达。
在空气-液体界面处的滤器芯子上培养胰腺前肠前体细胞与ALK5抑制 剂II组合显著提升了共表达胰岛素或嗜铬粒蛋白-A的NKX6.1阳性细胞的 数目。此外,应用于在传统单层培养物上培养的细胞的相同方案未能显示 出大量的共表达胰岛素或嗜铬粒蛋白-A的NKX6.1阳性细胞。最后,用除ALK5抑制剂II之外的ALK5抑制剂对在空气-液体界面处培养的细胞处理 未能显示出大量的共表达胰岛素或嗜铬粒蛋白-A的NKX6.1阳性细胞。这 些结果表明在空气-液体界面处培养和用补充有ALK5抑制剂II的培养基培 养的独特组合导致胰岛素/嗜铬粒蛋白-A和NKX6.1的共表达。

实例5
在空气-液体界面处培养的细胞的第5阶段和第6阶段中各种ALK5抑制剂的比较
该实例显示ALK5抑制剂II独特地在空气-液体界面处产生表达 NKX6.1和胰岛素或嗜铬粒蛋白-A的大量细胞群。另外的TGF-β抑制剂如 表IV中所列出的进行测试。
将人胚胎干细胞系H1的细胞(传代40代)作为单细胞以1×105个细 胞/cm2接种在MATRIGELTM(1:30的稀释度;BD Biosciences,NJ)涂覆的 培养皿上的培养基中,所述培养基包含DMEM-F12(Invitrogen,Ca)、 GlutaMaxTM(1:100的稀释度,Invitrogen)、0.25mM的抗坏血酸(Sigma, MO)、100ng/ml的FGF2(R&D systems,MN)、1ng/ml的TGF-β(R&D systems)、ITS-X(1:100的稀释度)、2%的无脂肪酸BSA(Lampire,PA), 以及20ng/ml的IGF-1(R&Dsystems),所述培养基补充有10μM的 Y27632(Rock抑制剂,目录号Y0503,Sigma-Aldrich)。接种后四十八小 时,将培养物在不完全PBS(不含Mg或Ca的磷酸盐缓冲盐水溶液)中洗 涤。随后根据以下方案使所述细胞分化:
a.第1阶段(3天):细胞在MCDB-131培养基(Invitrogen,目录 号10372-019)中培养一天,所述MCDB-131培养基补充有2%的 无脂肪酸BSA(Proliant,目录号68700)、0.0012g/ml的碳酸氢钠 (Sigma-Aldrich,目录号S3187)、1X的GlutaMaxTM (Invitrogen,目录号35050-079)、4.5mM的D-葡萄糖(Sigma- Aldrich,目录号G8769)、100ng/ml的GDF8(R&DSystems)和 1μM的MCX化合物。细胞随后在MCDB-131培养基中再培养一 天,所述MCDB-131培养基补充有:2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、100ng/ml的GDF8和0.1μM的MCX化合物。细胞随后在 MCDB-131培养基中再培养一天,所述MCDB-131培养基补充有 2%的无脂肪酸BSA、0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、 4.5mM的D--葡萄糖和100ng/ml的GDF8。
b.第2阶段(2天):第1阶段细胞随后用MCDB-131培养基处理两 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、0.25mM的抗坏血酸(Sigma,MO)和25ng/ml的FGF7(R&D Systems,MN)。
c.第3阶段(2天):第2阶段细胞随后用BLAR定制培养基 (Invitrogen)处理两天,所述BLAR定制培养基补充有1:200稀释度 的ITS-X(Invitrogen,CA)、4.5mM的葡萄糖、1X的GlutaMaxTM、 0.0017g/ml的碳酸氢钠、2%的无脂肪酸BSA、0.25μM的SANT-1 (Sigma,MO)、1μM的RA(Sigma,MO)、25ng/ml的FGF7、 0.25mM的抗坏血酸、200nM的TPB(PKC活化剂;目录号565740;EMD Chemicals,Gibbstown,NJ),以及100nM的LDN- 193189(BMP受体抑制剂;目录号04-0019;Stemgent)。
d.第4阶段(2天):第3阶段细胞随后用BLAR培养基处理两天, 所述BLAR培养基补充有1:200稀释度的ITS-X、4.5mM的葡萄 糖、1X的GlutaMaxTM、0.0017g/ml的碳酸氢钠、2%的无脂肪酸 BSA、0.25μM的SANT-1、100nM的RA、2ng/ml的FGF7、 100nM的LDN-193189、0.25mM的抗坏血酸和100nM的TPB;随 后在第4阶段结束时,在平皿上培养的细胞用10μM的Y27632处 理4小时,用PBS冲洗,然后在室温下用1X的TrypLETM (Invitrogen)处理5分钟,之后去除所述酶并且用基础培养基冲洗, 然后用细胞刮棒刮擦所述细胞。将所得的细胞悬浮液以0.5- 0.75×105个细胞的密度(在10μl的等分试样中)接种到6孔板中 的0.4微米多孔细胞培养物滤器芯子(BD 353493)上,或接种到 10cm培养皿中的10cm滤器芯子(Corning,#3419)上。将1.5ml的培 养基添加至6孔板中每一滤芯的底部,以及将7.5ml的培养基添加到每一10cm滤芯的底部。不另外添加培养基至过滤器的顶上侧。 培养基在研究持续期间每天更换。
e.第5阶段(3天):第4阶段细胞随后在空气-液体界面处在BLAR 培养基中培养三天,所述BLAR培养基补充有1:200稀释度的 ITS-X、20mM的葡萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢 钠、2%的无脂肪酸BSA、10μg/ml的肝素(Sigma,#H3149)、 0.25μM的SANT-1、50nM的RA、100nM的LDN-193189、 1000nM的各种ALK5抑制剂(关于所使用的抑制剂的列表请参见 表IV)。
f.第6阶段(6天):第5阶段细胞随后用BLAR培养基处理六天, 所述BLAR培养基补充有1:200稀释度的ITS-X、20mM的葡萄 糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢钠、2%的无脂肪酸 BSA、10μg/ml的肝素(Sigma,#H3149)、0.25μM的SANT-1; 100nM的LDN-193189、1000nM的T3、1000nM的ALK5抑制剂 (关于所测试的抑制剂的列表请参见表IV)。
图23示出了如在实例5中所概述的在空气-液体界面处分化和培养的 人胚胎干细胞系H1的细胞在第5阶段第4天和第6阶段第6天之后,其中 下列基因的表达的实时PCR分析的数据:PDX1(图23A)、NKX6.1(图 23B)、NGN3(图23C)、ABCC8(图23D)、胰高血糖素(图23E),以及胰岛素(图23F)。类似于实例4的结果,在空气-液体界面处的滤器 芯子上培养胰腺前肠前体细胞和用ALK5抑制剂II处理显著提升了胰岛素 和胰高血糖素的表达。用其他ALK5抑制剂处理胰腺前肠前体细胞并未导 致胰岛素或胰高血糖素的显著表达。

实例6
在空气-液体界面处的接种细胞密度对后续分化成内分泌细胞的影响
该实例识别在空气-液体界面的接种密度范围以及所得的内分泌标志物 的表达。为了进行该实例中的研究,使用以下论述的方案来使胚胎干细胞 分化。
将人胚胎干细胞系H1的细胞(传代40代)作为单细胞以1×105个细 胞/cm2接种在MATRIGELTM(1:30的稀释度;BD Biosciences,NJ)涂覆的 培养皿上的培养基中,所述培养基包含DMEM-F12(Invitrogen,CA)、 GlutaMaxTM(1:100的稀释度,Invitrogen)、0.25mM的抗坏血酸(Sigma, MO)、100ng/ml的FGF2(R&D systems,MN)、1ng/ml的TGF-β(R&D systems)、ITS-X(1:100的稀释度)、2%的无脂肪酸BSA(Lampire,PA), 以及20ng/ml的IGF-1(R&Dsystems),所述培养基补充有10μM的 Y27632(Rock抑制剂,目录号Y0503,Sigma)。接种后四十八小时,将 培养物在不完全PBS(不含Mg或Ca的磷酸盐缓冲盐水溶液)中洗涤。随 后根据以下方案使所述细胞分化:
a)第1阶段(3天):细胞在MCDB-131培养基(Invitrogen,目录 号10372-019)中培养一天,所述MCDB-131培养基补充有2%的 无脂肪酸BSA(Proliant,目录号68700)、0.0012g/ml的碳酸氢钠 (Sigma-Aldrich,目录号S3187)、1X的GlutaMaxTM (Invitrogen,目录号35050-079)、4.5mM的D-葡萄糖(Sigma- Aldrich,目录号G8769)、100ng/ml的GDF8(R&DSystems)和1μM的MCX化合物。细胞随后在MCDB-131培养基中再培养一 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄糖、100ng/ml的GDF8和0.1μM的MCX化合物。接着,细胞在 MCDB-131培养基中再培养一天,所述MCDB-131培养基补充有 2%的无脂肪酸BSA、0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄糖和100ng/ml的GDF8。
b)第2阶段(2天):第1阶段细胞随后用MCDB-131培养基处理两 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、0.25mM的抗坏血酸(Sigma,MO)和25ng/ml的FGF7(R&D Systems,MN)。
c)第3阶段(2天):第2阶段细胞随后用BLAR定制培养基 (Invitrogen)处理两天,所述BLAR定制培养基补充有1:200稀释度 的ITS-X(Invitrogen,CA)、4.5mM的葡萄糖、1X的GlutaMaxTM、 0.0017g/ml的碳酸氢钠、2%的无脂肪酸BSA、0.25μM的SANT-1 (Sigma,MO)、1μM的RA(Sigma,MO)、25ng/ml的FGF7、 0.25mM的抗坏血酸、200nM的TPB(PKC活化剂;目录号565740;EMD Chemicals,Gibbstown,NJ),以及100nM的LDN- 193189(BMP受体抑制剂;目录号04-0019;Stemgent)。随后, 在第3阶段结束时,在平皿上培养的细胞用10μM的Y27632处理 4小时,用PBS冲洗,然后在室温下用1X的TrypLETM(Invitrogen) 处理5分钟,之后去除所述酶并且用基础培养基冲洗,然后用细 胞刮棒刮擦所述细胞。将所得的细胞悬浮液以0.1、0.5、1和 5×106个细胞的密度(在10μl的等分试样中)接种到6孔板中的 0.4微米多孔细胞培养物滤器芯子(BD 353493)上。将1.5ml的培养 基添加至每一滤芯的底部,并且不另外添加培养基至过滤器的顶 上侧。培养基在研究持续期间每天更换。
d)第4阶段(2天):第3阶段细胞随后在空气-液体界面处在BLAR 培养基中培养两天,所述BLAR培养基补充有1:200稀释度的 ITS-X、4.5mM的葡萄糖、1X的GlutaMaxTM、0.0017g/ml的碳酸氢 钠、2%的无脂肪酸BSA、10μg/ml的肝素(Sigma,#H3149)、 0.25μM的SANT-1、100nM的RA、2ng/mlFGF7、100nM的LDN- 193189、0.25mM的抗坏血酸,以及100nM的TPB。
e第5阶段(3天):第4阶段细胞随后用BLAR培养基处理三天, 所述BLAR培养基补充有1:200稀释度的ITS-X、20mM的葡萄 糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢钠、2%的无脂肪酸 BSA、10μg/ml的肝素(Sigma,#H3149)、0.25μM的SANT-1、 50nM的RA、100nM的LDN-193189、10000nM的ALK5抑制剂 II。
f)第6阶段(14天):第5阶段细胞随后用BLAR培养基处理十四 天,所述BLAR培养基补充有1:200稀释度的ITS-X、20mM的葡 萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢钠、2%的无脂肪酸 BSA、10μg/ml的肝素(Sigma,#H3149)、0.25μM的SANT-1、 10000nM的ALK5抑制剂、100nM的LDN-193189,以及1000nM 的T3。
采集第4阶段、第5阶段和第6阶段的RNA样本,并且通过实时PCR 进行分析。图11示出了如实例6中所概述分化的并且在空气-液体界面处培 养的人胚胎干细胞系H1的细胞中下列基因的表达的实时PCR分析的数 据:ABCC8(图11A);胰高血糖素(图11B);淀粉不溶素(图 11C);胰岛素(图11D);NGN3(图11E);NKX2.2(图11F); NKX6.1(图11G);以及PDX1(图11H)。在0.1-1×106个细胞/10μl的范 围内的接种密度导致在第5阶段和第6阶段处相似的胰腺内胚层标志物和 内分泌标志物表达。在空气-液体界面处最高的测试接种密度(5×106个细 胞/10μl)处,存在内分泌标志物表达的下降。
实例7
0.4微米、1微米和3微米孔尺寸的滤器芯子的比较
该实例比较过滤器孔尺寸对在空气-液体界面处的随后分化的影响。为 了进行该实例中的研究,使用以下论述的方案来使胚胎干细胞分化。
将人胚胎干细胞系H1的细胞(传代40代)作为单细胞以1×105个细 胞/cm2接种在MATRIGELTM(1:30的稀释度;BD Biosciences,NJ)涂覆的 培养皿上的培养基中,所述培养基包含DMEM-F12(Invitrogen,CA)、GlutaMaxTM(1:100的稀释度,Invitrogen)、0.25mM的抗坏血酸(Sigma, MO)、100ng/ml的FGF2(R&D systems,MN)、1ng/ml的TGF-β(R&D systems)、ITS-X(1:100的稀释度)、2%的无脂肪酸BSA(Lampire,PA), 以及20ng/ml的IGF-1(R&Dsystems),所述培养基补充有10μM的 Y27632(Rock抑制剂,目录号Y0503,Sigma)。接种后四十八小时,将 培养物在不完全PBS(不含Mg或Ca的磷酸盐缓冲盐水溶液)中洗涤。随 后根据以下方案使所述细胞分化:
a)第1阶段(3天):细胞在MCDB-131培养基(Invitrogen,目录 号10372-019)中培养一天,所述MCDB-131培养基补充有2%的 无脂肪酸BSA(Proliant,目录号68700)、0.0012g/ml的碳酸氢钠 (Sigma-Aldrich,目录号S3187)、1X的GlutaMaxTM (Invitrogen,目录号35050-079)、4.5mM的D-葡萄糖(Sigma- Aldrich,目录号G8769)、100ng/ml的GDF8(R&DSystems)和 1μM的MCX。细胞随后在MCDB-131培养基中再培养一天,所 述MCDB-131培养基补充有2%的无脂肪酸BSA、0.0012g/ml的碳 酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄糖、100ng/ml的 GDF8和0.1μM的MCX化合物。细胞随后在MCDB-131培养基中 再培养一天,所述MCDB-131培养基补充有2%的无脂肪酸 BSA、0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡 萄糖和100ng/ml的GDF8。
b)第2阶段(2天):第1阶段细胞随后用MCDB-131培养基处理两 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、0.25mM的抗坏血酸(Sigma,MO)和25ng/ml的FGF7(R&D Systems,MN)。
c)第3阶段(2天):第2阶段细胞随后用BLAR定制培养基 (Invitrogen)处理两天,所述BLAR定制培养基补充有1:200稀释度 的ITS-X(Invitrogen,CA)、4.5mM的葡萄糖、1X的GlutaMaxTM、 0.0017g/ml的碳酸氢钠、2%的无脂肪酸BSA、0.25μM的SANT-1 (Sigma,MO)、1μM的RA(Sigma,MO)、25ng/ml的FGF7、 0.25mM的抗坏血酸、200nM的TPB(PKC活化剂;目录号565740;EMD Chemicals,Gibbstown,NJ),以及100nM的LDN- 193189(BMP受体抑制剂;目录号04-0019;Stemgent)。
d)第4阶段(2天):第3阶段细胞随后用BLAR培养基处理三天, 所述BLAR培养基补充有1:200稀释度的ITS-X、4.5mM的葡萄 糖、1X的GlutaMaxTM、0.0017g/ml的碳酸氢钠、2%的无脂肪酸 BSA、0.25μM的SANT-1、100nM的RA、2ng/ml的FGF7、 100nM的LDN-193189、0.25mM的抗坏血酸和100nM的TPB。随 后,在第4阶段结束时,在平皿上培养的细胞用10μM的Y27632 处理4小时,用PBS冲洗,然后在室温下用1X的TrypLETM (Invitrogen)处理5分钟,之后去除所述酶并且用基础培养基冲洗, 然后用细胞刮棒刮擦所述细胞。将所得的细胞悬浮液以0.5- 0.75×106个细胞的密度(在10μl的等分试样中)接种到6孔板中 的0.4微米、1微米或3微米多孔细胞培养物滤器芯子上。将 1.5ml的培养基添加至每一滤芯的底部,并且不另外添加培养基至 过滤器的顶上侧。培养基在研究持续期间每天更换。
e)第5阶段(3天):第4阶段细胞随后在空气-液体界面处在BLAR 培养基中培养三天,所述BLAR培养基补充有1:200稀释度的 ITS-X、20mM的葡萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢 钠、2%的无脂肪酸BSA、10μg/ml的肝素(Sigma,#H3149)、 0.25μM的SANT-1、50nM的RA、100nM的LDN-193189、 10000nM的ALK5抑制剂II。
f)第6阶段(15天):第5阶段细胞随后用BLAR培养基处理十五 天,所述BLAR培养基补充有1:200稀释度的ITS-X、20mM的葡 萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢钠、2%的无脂肪酸 BSA、10μg/ml的肝素(Sigma,#H3149)、0.25μM的SANT-1、100nM 的LDN-193189、1000nM的T3、10000nM的ALK5抑制剂II。
采集第6阶段的RNA样本,并且通过实时PCR进行分析。图12示出 了如本实例中所概述分化的并且在空气-液体界面处培养的人胚胎干细胞系 H1的细胞中下列基因的表达的实时PCR分析的数据:ABCC8(图 12A);胰高血糖素(图12B);淀粉不溶素(图12C);胰岛素(图 12D);NGN3(图12E);NKX2.2(图12F);NKX6.1(图12G);以 及PDX1(图12H)。在从0.4微米至3微米范围内的滤器芯子孔尺寸并未 显著影响在空气-液体界面处的胰腺内胚层标志物或内分泌标志物的表达。
实例8
胰腺前肠前体细胞在空气-液体界面处的滤器芯子上与在液体-液体(L/L)界面处
的滤器芯子上的分化比较
该实例比较在空气-液体界面处培养与在液体-液体界面处培养对滤器 芯子上的胰腺前肠前体细胞分化的影响。为了进行该实例中的研究,使用 以下论述的方案来使胚胎干细胞分化。
将人胚胎干细胞系H1的细胞(传代40代)作为单细胞以1×105个细 胞/cm2接种在MATRIGELTM(1:30的稀释度;BD Biosciences,NJ)涂覆的 培养皿上的培养基中,所述培养基包含DMEM-F12(Invitrogen,CA)、 GlutaMaxTM(1:100的稀释度,Invitrogen)、0.25mM的抗坏血酸(Sigma, MO)、100ng/ml的FGF2(R&D systems,MN)、1ng/ml的TGF-β(R&D systems)、ITS-X(1:100的稀释度)、2%的无脂肪酸BSA(Lampire,PA), 以及20ng/ml的IGF-1(R&Dsystems),所述培养基补充有10μM的 Y27632(Rock抑制剂,目录号Y0503,Sigma-Aldrich)。接种后四十八小 时,将培养物在不完全PBS(不含Mg或Ca的磷酸盐缓冲盐水溶液)中洗 涤。随后根据以下方案使所述细胞分化:
a)第1阶段(3天):细胞在MCDB-131培养基(Invitrogen,目录 号10372-019)中培养一天,所述MCDB-131培养基补充有2%的 无脂肪酸BSA(Proliant,目录号68700)、0.0012g/ml的碳酸氢钠 (Sigma-Aldrich,目录号S3187)、1X的GlutaMaxTM (Invitrogen,目录号35050-079)、4.5mM的D-葡萄糖(Sigma- Aldrich,目录号G8769)、100ng/ml的GDF8(R&DSystems)和 1μM的MCX化合物。细胞随后在MCDB-131培养基中再培养一 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、100ng/ml的GDF8和0.1μM的MCX化合物。细胞随后在 MCDB-131培养基中再培养一天,所述MCDB-131培养基补充有 2%的无脂肪酸BSA、0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄糖和100ng/ml的GDF8。
b)第2阶段(2天):第1阶段细胞随后用MCDB-131培养基处理两 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、0.25mM的抗坏血酸(Sigma,MO)和25ng/ml的FGF7(R&D Systems,MN)。
c)第3阶段(2天):第2阶段细胞随后用BLAR定制培养基 (Invitrogen)处理两天,所述BLAR定制培养基补充有1:200稀释度 的ITS-X(Invitrogen,CA)、4.5mM的葡萄糖、1X的GlutaMaxTM、 0.0017g/ml的碳酸氢钠、2%的无脂肪酸BSA、0.25μM的SANT-1 (Sigma,MO)、1μM的RA(Sigma,MO)、25ng/ml的FGF7、 0.25mM的抗坏血酸、200nM的TPB(PKC活化剂;目录号565740;EMD Chemicals,Gibbstown,NJ),以及100nM的LDN- 193189(BMP受体抑制剂;目录号04-0019;Stemgent)。
d)第4阶段(2天):第3阶段细胞随后用BLAR培养基处理两天, 所述BLAR培养基补充有1:200稀释度的ITS-X、4.5mM的葡萄 糖、1X的GlutaMaxTM、0.0017g/ml的碳酸氢钠、2%的无脂肪酸 BSA、0.25μM的SANT-1、100nM的RA、2ng/ml的FGF7、 100nM的LDN-193189、0.25mM的抗坏血酸和100nM的TPB;随 后在第4阶段结束时,在平皿上培养的细胞用10μM的Y27632处 理4小时,用PBS冲洗,然后在室温下用1X的TrypLETM (Invitrogen)处理5分钟,之后去除所述酶并且用基础培养基冲洗, 然后用细胞刮棒刮擦所述细胞。将所得的细胞悬浮液以0.5- 0.75×106个细胞的密度(在10μl的等分试样中)接种到6孔板中 经MATRIGELTM涂覆的0.4微米多孔细胞培养物滤器芯子上。将 1.5ml的培养基添加至每一滤芯的底部,并且不另外添加培养基至 过滤器的顶上侧。对于L/L条件,还将培养基添加至滤器芯子的 顶部上,以产生液体-液体界面。培养基在研究持续期间每天更 换。
e)第5阶段(3天):第4阶段细胞随后在空气-液体界面处在BLAR 培养基中培养三天,所述BLAR培养基补充有1:200稀释度的 ITS-X、20mM的葡萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢 钠、2%的无脂肪酸BSA、10μg/ml的肝素(Sigma,#H3149)、 0.25μM的SANT-1、50nM的RA、100nM的LDN-193189、 10000nM的各种ALK5抑制剂II。
f)第6阶段(10天):第5阶段细胞随后用BLAR培养基处理十 天,所述BLAR培养基补充有1:200稀释度的ITS-X、20mM的葡 萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢钠、2%的无脂肪酸 BSA、10μg/ml的肝素(Sigma,#H3149)、0.25μM的SANT-1、 100nM的LDN-193189、1000nM的T3、10000nM的ALK5抑制剂 II。
采集第5阶段和第6阶段的RNA样本,并且通过实时PCR进行分 析。图13示出了如实例8中所概述分化的并且在空气-液体界面处培养的人 胚胎干细胞系H1的细胞中下列基因的表达的实时PCR分析的数据: ABCC8(图13A);胰高血糖素(图13B);淀粉不溶素(图13C);胰 岛素(图13D);NGN3(图13E);NKX2.2(图13F);NKX6.1(图 13G);以及PDX1(图13H)。相较于空气-液体界面条件,在L/L条件下 观测到的最显著差异为胰高血糖素的显著上调(7X)。
实例9
在空气-液体界面处培养的胰腺内胚层/内分泌前体细胞可用于筛选化合物库
该实例检查了空气-液体界面培养物的筛选化合物库的用途。为此,使 用以下论述的方案来使胚胎干细胞分化。
将人胚胎干细胞系H1的细胞(传代40代)作为单细胞以1×105个细 胞/cm2接种在MATRIGELTM(1:30的稀释度;BD Biosciences,NJ)涂覆的 培养皿上的培养基中,所述培养基包含DMEM-F12(Invitrogen,Ca)、 GlutaMaxTM(1:100的稀释度,Invitrogen)、0.25mM的抗坏血酸(Sigma, MO)、100ng/ml的FGF2(R&D systems,MN)、1ng/ml的TGF-β(R&D systems)、ITS-X(1:100的稀释度)、2%的无脂肪酸BSA(Lampire,PA), 以及20ng/ml的IGF-1(R&Dsystems),所述培养基补充有10μM的 Y27632(Rock抑制剂,目录号Y0503,Sigma)。接种后四十八小时,将 培养物在不完全PBS(不含Mg或Ca的磷酸盐缓冲盐水溶液)中洗涤。随 后根据以下方案使所述细胞分化:
a)第1阶段(3天):细胞在MCDB-131培养基(Invitrogen,目录 号10372-019)中培养一天,所述MCDB-131培养基补充有2%的 无脂肪酸BSA(Proliant,目录号68700)、0.0012g/ml的碳酸氢钠 (Sigma-Aldrich,目录号S3187)、1X的GlutaMaxTM (Invitrogen,目录号35050-079)、4.5mM的D-葡萄糖(Sigma- Aldrich,目录号G8769)、100ng/ml的GDF8(R&DSystems)和 1μM的MCX化合物。细胞随后在MCDB-131培养基中再培养一 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、100ng/ml的GDF8和0.1μM的MCX化合物。细胞随后在 MCDB-131培养基中再培养一天,所述MCDB-131培养基补充有 2%的无脂肪酸BSA、0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄糖和100ng/ml的GDF8。
b)第2阶段(2天):第1阶段细胞随后用MCDB-131培养基处理两 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、0.25mM的抗坏血酸(Sigma,MO)和25ng/ml的FGF7(R&D Systems,MN)。
c)第3阶段(2天):第2阶段细胞随后用BLAR定制培养基 (Invitrogen)处理两天,所述BLAR定制培养基补充有1:200稀释度 的ITS-X(Invitrogen,CA)、4.5mM的葡萄糖、1X的GlutaMaxTM、 0.0017g/ml的碳酸氢钠、2%的无脂肪酸BSA、0.25μM的SANT-1 (Sigma,MO)、1μM的RA(Sigma,MO)、25ng/ml的FGF7、 0.25mM的抗坏血酸、200nM的TPB(PKC活化剂;目录号565740;EMD Chemicals,Gibbstown,NJ),以及100nM的LDN- 193189(BMP受体抑制剂;目录号04-0019;Stemgent)。
d)第4阶段(2天):第3阶段细胞随后用BLAR培养基处理两天, 所述BLAR培养基补充有1:200稀释度的ITS-X、4.5mM的葡萄 糖、1X的GlutaMaxTM、0.0017g/ml的碳酸氢钠、2%的无脂肪酸 BSA、0.25μM的SANT-1、100nM的RA、2ng/ml的FGF7、 100nM的LDN-193189、0.25mM的抗坏血酸和200nM的TPB;随 后在第4阶段结束时,在平皿上培养的细胞用10μM的Y27632处 理4小时,用PBS冲洗,然后在室温下用1X的TrypLETM (Invitrogen)处理5分钟,之后去除所述酶并且用基础培养基冲洗, 然后用细胞刮棒刮擦所述细胞。将所得的细胞悬浮液以0.5- 0.75×106个细胞的密度(在10μl的等分试样中)接种到6孔板中 经MATRIGELTM涂覆的0.4微米多孔细胞培养物滤器芯子上。将 1.5ml的培养基添加至每一滤芯的底部,并且不另外添加培养基至 过滤器的顶上侧。
e)第5阶段(3天):第4阶段细胞随后在空气-液体界面处在BLAR 培养基中培养三天,所述BLAR培养基补充有1:200稀释度的 ITS-X、20mM的葡萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢 钠、2%的无脂肪酸BSA、10μg/ml的肝素(Sigma,#H3149)、10μM 的ZnSO4(Sigma,Z0251)、0.25μM的SANT-1、50nM的RA、 100nM的LDN-193189、10000nM的ALK5抑制剂II。
f)第6阶段(12天):第5阶段细胞随后用BLAR培养基处理十二 天,所述BLAR培养基补充有1:200稀释度的ITS-X、20mM的葡 萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢钠、2%的无脂肪酸 BSA、10μg/ml的肝素(Sigma,#H3149)、10μM的ZnSO4(Sigma, Z0251)、0.25μM的SANT-1、100nM的LDN-193189、1000nM的 T3、10000nM的ALK5抑制剂II。在该阶段,筛选在表V中列出 的化合物以识别影响内胚层标志物和内分泌标志物的潜在化合物。


采集第6阶段的RNA样本,并且通过实时PCR进行分析。图14示出 了如实例9中所概述分化的并且在空气-液体界面处培养的人胚胎干细胞系 H1的细胞中下列基因的表达的实时PCR分析的数据:ABCC8(图 14A);胰高血糖素(图14B);淀粉不溶素(图14C);胰岛素(图14D);ISL-1(图14E);MNX1(图14F);NKX6.1(图14G);以及 SLC30A8(图14H)。该实例显示在空气-液体界面处培养的细胞作为筛选 工具的潜能。
实例10
在空气-液体界面处培养的第5阶段和第6阶段细胞的FACS分布图
该实例研究在空气-液体界面处的第5阶段和第6阶段培养物的组成。 为了进行该实例中的研究,使用以下论述的方案来使胚胎干细胞分化成第5 阶段和第6阶段培养物。
将人胚胎干细胞系H1的细胞(传代40代)作为单细胞以1×105个细 胞/cm2接种在MATRIGELTM(1:30的稀释度;BD Biosciences,NJ)涂覆的 培养皿上的培养基中,所述培养基包含DMEM-F12(Invitrogen,Ca)、 GlutaMaxTM(1:100的稀释度,Invitrogen)、0.25mM的抗坏血酸(Sigma, MO)、100ng/ml的FGF2(R&D systems,MN)、1ng/ml的TGF-β(R&D systems)、ITS-X(1:100的稀释度)、2%的无脂肪酸BSA(Lampire,PA), 以及20ng/ml的IGF-1(R&Dsystems),所述培养基补充有10μM的 Y27632(Rock抑制剂,目录号Y0503,Sigma)。接种后四十八小时,将 培养物在不完全PBS(不含Mg或Ca的磷酸盐缓冲盐水溶液)中洗涤。随 后根据以下方案使所述细胞分化:
a)第1阶段(3天):细胞在MCDB-131培养基(Invitrogen,目录 号10372-019)中培养一天,所述MCDB-131培养基补充有2%的 无脂肪酸BSA(Proliant,目录号68700)、0.0012g/ml的碳酸氢钠 (Sigma-Aldrich,目录号S3187)、1X的GlutaMaxTM (Invitrogen,目录号35050-079)、4.5mM的D-葡萄糖(Sigma- Aldrich,目录号G8769)、100ng/ml的GDF8(R&DSystems)和 1μM的MCX化合物。细胞随后在MCDB-131培养基中再培养一 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、100ng/ml的GDF8和0.1μM的MCX化合物。细胞随后在 MCDB-131培养基中再培养一天,所述MCDB-131培养基补充有 2%的无脂肪酸BSA、0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄糖和100ng/ml的GDF8。
b)第2阶段(2天):第1阶段细胞随后用MCDB-131培养基处理两 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、0.25mM的抗坏血酸(Sigma,MO)和25ng/ml的FGF7(R&D Systems,MN)。
c)第3阶段(2天):第2阶段细胞随后用BLAR定制培养基 (Invitrogen)处理两天,所述BLAR定制培养基补充有1:200稀释度 的ITS-X(Invitrogen,Ca)、4.5mM的葡萄糖、1X的GlutaMaxTM、 0.0017g/ml的碳酸氢钠、2%的无脂肪酸BSA、0.25μM的SANT-1 (Sigma,MO)、1μM的RA(Sigma,MO)、25ng/ml的FGF7、 0.25mM的抗坏血酸、200nM的TPB(PKC活化剂;目录号565740;EMD Chemicals,Gibbstown,NJ),以及100nM的LDN- 193189(BMP受体抑制剂;目录号04-0019;Stemgent)。
d)第4阶段(3天):第3阶段细胞随后用BLAR培养基处理三天, 所述BLAR培养基补充有1:200稀释度的ITS-X、4.5mM的葡萄 糖、1X的GlutaMaxTM、0.0017g/ml的碳酸氢钠、2%的无脂肪酸 BSA、0.25μM的SANT-1、100nM的RA、2ng/ml的FGF7、 100nM的LDN-193189、0.25mM的抗坏血酸和200nM的TPB;随 后在第4阶段结束时,在平皿上培养的细胞用10μM的Y27632处 理4小时,用PBS冲洗,然后在室温下用1X的TrypLETM (Invitrogen)处理5分钟,之后去除所述酶并且用基础培养基冲洗, 然后用细胞刮棒刮擦所述细胞。将所得的细胞悬浮液以0.5- 0.75×106个细胞的密度(在10μl的等分试样中)接种到6孔板中 经MATRIGELTM涂覆的0.4微米多孔细胞培养物滤器芯子上。将 1.5ml的培养基添加至每一滤芯的底部,并且不另外添加培养基至 过滤器的顶上侧。
e)第5阶段(3天):第4阶段细胞随后在空气-液体界面处在BLAR 培养基中培养三天,所述BLAR培养基补充有1:200稀释度的 ITS-X、20mM的葡萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢 钠、2%的无脂肪酸BSA、10μg/ml的肝素(Sigma,#H3149)、10μM 的ZnSO4(Sigma,Z0251)、0.25μM的SANT-1、50nM的RA、 100nM的LDN-193189、10000nM的各种ALK5抑制剂II。
f)第6阶段(15天):第5阶段细胞随后用BLAR培养基处理五至 十五天,所述BLAR培养基补充有1:200稀释度的ITS-X、20mM 的葡萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢钠、2%的无脂 肪酸BSA、10μg/ml的肝素(Sigma,#H3149)、10μM的ZnSO4 (Sigma,Z0251)、0.25μM的SANT-1、100nM的LDN-193189、 1000nM的T3、10000nM的ALK5抑制剂II。
收集第5阶段和在第6阶段各时间点的细胞,并且通过FACS进行分 析。如先前所述地(Diabetes,61,2016,2012)并且使用表VI中列出的抗体进 行FACS染色。图15示出了在第5阶段收集的细胞的FACS分布图。图16 示出了在空气-液体界面处培养的第6阶段第5天细胞的FACS分布图。最 后,图17示出了在空气-液体界面处培养的第6阶段第15天细胞的分布图。如图15所示,在第5阶段,在活跃细胞周期中存在少量共表达胰岛素 和NKX6.1的细胞(约1%)和大量的PDX1阳性细胞,如通过PDX1和 KI-67的共表达所测量的(约23%;KI-67指示细胞处于活跃细胞周期 中)。然而,到第6阶段第5天为止(图16),存在显著的PDX1+细胞增 殖下降(8%),同时存在共表达嗜铬粒蛋白-A的NKX6.1+细胞的数目的显著 增长(51%;嗜铬粒蛋白-A是胰腺内分泌标志物)或共表达胰岛素的 NKX6.1+细胞的数目的显著增长(14%)。此外,存在表达内分泌前体标志物 ISL-1、NeuroD和NKX2.2的细胞的显著增多。这表明第6阶段的独特培养 物允许细胞从增殖祖细胞命运脱离而快速成熟为早熟内分泌细胞。此外, 通过延长第6阶段至15天,观测到了共表达胰岛素和NXK6.1的细胞百分 比的增长(33%)(图17)。此外,还存在细胞周期中PDX1阳性细胞的百分 比的下降(1%),并且还存在ISL-1和NeuroD的百分比的增长。最后,大部 分激素阳性细胞是单激素胰岛素阳性细胞(34%的单激素胰岛素阳性细 胞,7%的单激素胰高血糖素阳性细胞,以及8%的多激素细胞)。NKX6.1 和嗜铬粒蛋白-A,以及表达NKX6.1(>30%)的单激素胰岛素阳性细胞的显 著共表达突出了先前未描述的细胞群。

实例11
NKX6.1+嗜铬粒蛋白-A+胰岛素+细胞、NKX6.1+嗜铬粒蛋白-A-胰岛素-和共表达
PDX1和NKX6.1的胰岛素祖细胞相对于人类胰岛在SCID小鼠中的体内成熟
该实例突出了分化的细胞的体外组成及其对体内细胞性能的影响。具 体地,将根据实例1在平面培养物上制备的5百万个第4阶段第4天 (PDX1+NKX6.1+)胰腺前肠前体细胞、在空气-液体界面处培养的5百万个 NKX6.1+嗜铬粒蛋白-A阴性细胞,以及根据实例10在空气-液体界面处制 备的3百万个NKX6.1+嗜铬粒蛋白-A阳性细胞移植到未患糖尿病的SCID 小鼠的肾包膜中,如在(Diabetes 2012,61(8):2016-29)中所述的。用循环的人 类C-肽作为对细胞的成熟状态随时间变化的量度来追踪小鼠。此外,在独 立的小鼠群组中,将1500-4000个人类尸体胰岛(PRODO labs,Irvine,CA)当 量作为阳性对照移植。
NKX6.1+嗜铬粒蛋白-A阴性细胞群如下所述制备:
将人胚胎干细胞系H1的细胞(传代40代)作为单细胞以1×105个细 胞/cm2接种在MATRIGELTM(1:30的稀释度;BD Biosciences,NJ)涂覆的 培养皿上的培养基中,所述培养基包含DMEM-F12(Invitrogen,Ca)、 GlutaMaxTM(1:100的稀释度,Invitrogen)、0.25mM的抗坏血酸(Sigma, MO)、100ng/ml的FGF2(R&D systems,MN)、1ng/ml的TGF-β(R&D systems)、ITS-X(1:100的稀释度)、2%的无脂肪酸BSA(Lampire,PA), 以及20ng/ml的IGF-1(R&Dsystems),所述培养基补充有10μM的 Y27632(Rock抑制剂,目录号Y0503,Sigma)。接种后四十八小时,将 培养物在不完全PBS(不含Mg或Ca的磷酸盐缓冲盐水溶液)中洗涤。随 后根据以下方案使所述细胞分化:
a.第1阶段(3天):细胞在MCDB-131培养基(Invitrogen,目录 号10372-019)中培养一天,所述MCDB-131培养基补充有2%的 无脂肪酸BSA(Proliant,目录号68700)、0.0012g/ml的碳酸氢钠 (Sigma-Aldrich,目录号S3187)、1X的GlutaMaxTM (Invitrogen,目录号35050-079)、4.5mM的D-葡萄糖(Sigma- Aldrich,目录号G8769)、100ng/ml的GDF8(R&DSystems)和 1μM的MCX化合物。细胞随后在MCDB-131培养基中再培养一 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、100ng/ml的GDF8和0.1μM的MCX化合物。细胞随后在 MCDB-131培养基中再培养一天,所述MCDB-131培养基补充有 2%的无脂肪酸BSA、0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄糖和100ng/ml的GDF8。
b.第2阶段(2天):第1阶段细胞随后用MCDB-131培养基处理两 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、0.25mM的抗坏血酸(Sigma,MO)和25ng/ml的FGF7(R&D Systems,MN)。
c.第3阶段(2天):第2阶段细胞随后用MCDB-131培养基处理两 天,所述MCDB-131培养基补充有1:200稀释度的ITS-X (Invitrogen,CA)、4.5mM的葡萄糖、1X的GlutaMaxTM、0.0017g/ml 的碳酸氢钠、2%的无脂肪酸BSA、0.25μM的SANT-1(Sigma, MO)、1μM的RA(Sigma,MO)、25ng/ml的FGF7、0.25mM的抗 坏血酸、200nM的TPB(PKC活化剂;目录号565740;EMD Chemicals,Gibbstown,NJ),以及100nM的LDN-193189(BMP 受体抑制剂;目录号04-0019;Stemgent)。第3阶段细胞随后在 室温下用1X的ACCUTASETM处理1-3分钟,之后去除所述酶并且 用细胞刮棒刮擦所述细胞。将所得的细胞悬浮液以约2×106个细胞 /10μl的密度接种到0.4微米多孔细胞培养物滤器芯子上。将1.5ml 的培养基添加至每一滤芯的底部,并且不另外添加培养基至过滤 器的顶上侧。
d.第4阶段(2天):第3阶段细胞随后在空气-液体界面处在 MCDB-131培养基中培养两天,所述MCDB-131培养基补充有 1:200稀释度的ITS-X、4.5mM的葡萄糖、1X的GlutaMaxTM、 0.0017g/ml的碳酸氢钠、2%的无脂肪酸BSA、0.25μM的SANT- 1、100nM的RA、2ng/ml的FGF7、100nM的LDN-193189、 0.25mM的抗坏血酸、100nM的T3(T6397,Sigma)和100nM的 TPB。
e.第5阶段(2天):第4阶段细胞随后用MCDB-131培养基处理两 天,所述MCDB-131培养基补充有1:200稀释度的ITS-X、20mM 的葡萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢钠、2%的无脂 肪酸BSA、0.25μM的SANT-1、50nM的RA、50nM的LDN- 193189、500nM的ALK5抑制剂(SD208)。
f.第6阶段(6天):第5阶段细胞随后用MCDB-131培养基处理六 天,所述MCDB-131培养基补充有1:200稀释度的ITS-X、20mM 的葡萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢钠、2%的无脂 肪酸BSA;0.25μM的SANT-1、50nM的RA、500nM的ALK5抑 制剂。
下表VII突出了三种人胚胎干细胞来源的细胞群的各种胰腺内胚层标 志物和内分泌标志物的表达水平。具体地,表VII比较了下列的结果:(1) 根据实例1(第4阶段第4天)产生的第4阶段第4天的细胞群;(2)根据实 例11产生的胰腺内胚层/内分泌前体细胞群(胰腺内胚层/内分泌);以及 (3)根据实例10(NKX6.1+嗜铬粒蛋白-A+胰岛素+)产生的NKX6.1+嗜铬 粒蛋白-A+胰岛素+细胞群。每种细胞群的FACS分布图信息示出在图17至 图19中。具体地,图17示出了NKX6.1+嗜铬粒蛋白-A+细胞群的FACS分 布图;图18示出了第4阶段第4天的细胞的FACS分布图;以及图19示出 了根据实例11产生的第6阶段第6天胰腺内分泌细胞的FACS分布图。

在移植之后,周期性地测试小鼠中循环的人类C-肽的浓度。循环的人 类C-肽是通过经由隐静脉采血来测试的。将血浆保存在-20℃下,随后使用 人类C-肽通过ELISA试剂盒(Alpco Diagnostics,Salem,NH)来进行测定。这 些结果也在图20中以图线示出。
图20示出了三种ES来源的细胞群相比于多种剂量的人类胰岛的产生 C-肽的动力学图。表达基本上共表达的NKX6.1和嗜铬粒蛋白-A以及 NKX6.1和胰岛素的细胞群导致显著地早产生C-肽。在第12周时,C-肽产 生的水平类似于移植约4000个人类胰岛。然而,到移植后第16周为止, 人类C-肽的水平已经几乎是通过移植4000个人类胰岛实现的C-肽量值的 两倍。
然而,移植表达PDX1和NKX6.1的祖细胞或胰腺前体细胞和多激素 细胞(嗜铬粒蛋白-A+NKX6.1-)的混合细胞群需要显著更长时期来分泌与 4000个人类胰岛等效水平的C-肽。
实例12
γ分泌酶抑制剂XX的添加进一步扩增在空气-液体界面处培养的第6阶段细胞的
成熟标志物
该实例突出NOTCH抑制剂,诸如γ分泌酶抑制剂,进一步增加β细 胞的成熟标志物,同时维持NKX6.1的表达。将人胚胎干细胞系H1的细胞 (传代42代)作为单细胞以1×105个细胞/cm2接种在MATRIGELTM(1:30 的稀释度;BD Biosciences,NJ)涂覆的培养皿上的培养基中,所述培养基 包含DMEM-F12(Invitrogen,Ca)、GlutaMaxTM(1:100的稀释度,Invitrogen)、0.25mM的抗坏血酸(Sigma,MO)、100ng/ml的FGF2(R&D systems,MN)、1ng/ml的TGF-β(R&D systems)、ITS-X(1:100的稀释 度)、2%的无脂肪酸BSA(Lampire,PA),以及20ng/ml的IGF-1(R&D systems),所述培养基补充有10μM的Y27632(Rock抑制剂,目录号Y0503,Sigma)。接种后四十八小时,将培养物在不完全PBS(不含Mg 或Ca的磷酸盐缓冲盐水溶液)中洗涤。随后根据以下方案使所述细胞分 化:
a)第1阶段(3天):细胞在MCDB-131培养基(Invitrogen,目录 号10372-019)中培养一天,所述MCDB-131培养基补充有2%的 无脂肪酸BSA(Proliant,目录号68700)、0.0012g/ml的碳酸氢钠 (Sigma-Aldrich,目录号S3187)、1X的GlutaMaxTM (Invitrogen,目录号35050-079)、4.5mM的D-葡萄糖(Sigma- Aldrich,目录号G8769)、100ng/ml的GDF8(R&DSystems)和 1μM的MCX化合物。细胞随后在MCDB-131培养基中再培养一 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、100ng/ml的GDF8和0.1μM的MCX化合物。细胞随后在 MCDB-131培养基中再培养一天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄糖和100ng/ml的GDF8。
b)第2阶段(2天):第1阶段细胞随后用MCDB-131培养基处理两 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、0.25mM的抗坏血酸(Sigma,MO)和25ng/ml的FGF7(R&D Systems,MN)。
c)第3阶段(2天):第2阶段细胞随后用BLAR定制培养基 (Invitrogen)处理两天,所述BLAR定制培养基补充有1:200稀释度 的ITS-X(Invitrogen,Ca)、4.5mM的葡萄糖、1X的GlutaMaxTM、 0.0017g/ml的碳酸氢钠、2%的无脂肪酸BSA、0.25μM的SANT-1 (Sigma,MO)、1μM的RA(Sigma,MO)、25ng/ml的FGF7、 0.25mM的抗坏血酸、200nM的TPB(PKC活化剂;目录号565740;EMD Chemicals,Gibbstown,NJ),以及100nM的LDN- 193189(BMP受体抑制剂;目录号04-0019;Stemgent)。
d)第4阶段(3天):第3阶段细胞随后用BLAR培养基处理三天, 所述BLAR培养基补充有1:200稀释度的ITS-X、4.5mM的葡萄 糖、1X的GlutaMaxTM、0.0017g/ml的碳酸氢钠、2%的无脂肪酸 BSA、0.25μM的SANT-1、100nM的RA、2ng/ml的FGF7、 100nM的LDN-193189、0.25mM的抗坏血酸和200nM的TPB;随 后在第4阶段结束时,在平皿上培养的细胞用10μM的Y27632处 理4小时,用PBS冲洗,然后在室温下用1X的TrypLETM (Invitrogen)处理5分钟,之后去除所述酶并且用基础培养基冲洗, 然后用细胞刮棒刮擦所述细胞。将所得的细胞悬浮液以0.5- 0.75×106个细胞的密度(在10μl的等分试样中)接种到6孔板中 经MATRIGELTM涂覆的0.4微米多孔细胞培养物滤器芯子上。将 1.5ml的培养基添加至每一滤芯的底部,并且不另外添加培养基至 过滤器的顶上侧。
e)第5阶段(3天):第4阶段细胞随后在空气-液体界面处在BLAR 培养基中培养三天,所述BLAR培养基补充有1:200稀释度的 ITS-X、20mM的葡萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢 钠、2%的无脂肪酸BSA、10μg/ml的肝素(Sigma,#H3149)、10μM 的ZnSO4(Sigma,Z0251)、0.25μM的SANT-1、50nM的RA、 100nM的LDN-193189、10000nM的各种ALK5抑制剂II。
f)第6阶段(14天):第5阶段细胞随后用BLAR培养基处理十四 天,所述BLAR培养基补充有1:200稀释度的ITS-X、20mM的葡 萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢钠、2%的无脂肪酸 BSA、10μg/ml的肝素(Sigma,#H3149)、10μM的ZnSO4(Sigma, Z0251)、0.25μM的SANT-1、100nM的LDN-193189、1000nM的 T3、10000nM的ALK5抑制剂II。
在第6阶段,测试各种剂量(100nM至5000nM)的γ分泌酶抑制剂 XX(EMD,#565789)。采集第6阶段第4天和第6阶段第8天的mRNA。图 21示出了关键的β细胞成熟标志物以及胰腺祖细胞标志物的PCR数据。如 图21所示,成熟标志物,诸如淀粉不溶素(实验组21A)、胰岛素(实验 组21B)和MAFA(实验组21C)显著上调,而NKX6.1(实验组21D)的 表达则未受到显著影响。然而,胰腺前体标志物,诸如PTF1a(实验组 21E)和SOX9(实验组21F)显著下调。
实例13
ALK5抑制剂的存在对于MAFA的上调至关重要,并且进一步添加T3还提升MAFA表达
该实例突出了ALK5抑制剂II添加上调MAFA表达的能力,以及T3 与ALK5抑制剂和LDN-193189的添加进一步提升MAFA表达的能力。
将人胚胎干细胞系H1的细胞(传代42代)作为单细胞以1×105个细 胞/cm2接种在MATRIGELTM(1:30的稀释度;BD Biosciences,NJ)涂覆的 培养皿上的培养基中,所述培养基包含DMEM-F12(Invitrogen,Ca)、 GlutaMaxTM(1:100的稀释度,Invitrogen)、0.25mM的抗坏血酸(Sigma, MO)、100ng/ml的FGF2(R&D systems,MN)、1ng/ml的TGF-β(R&D systems)、ITS-X(1:100的稀释度)、2%的无脂肪酸BSA(Lampire,PA), 以及20ng/ml的IGF-1(R&Dsystems),所述培养基补充有10μM的 Y27632(Rock抑制剂,目录号Y0503,Sigma)。接种后四十八小时,将 培养物在不完全PBS(不含Mg或Ca的磷酸盐缓冲盐水溶液)中洗涤。随 后根据以下方案使所述细胞分化:
a)第1阶段(3天):细胞在MCDB-131培养基(Invitrogen,目录 号10372-019)中培养一天,所述MCDB-131培养基补充有2%的 无脂肪酸BSA(Proliant,目录号68700)、0.0012g/ml的碳酸氢钠 (Sigma-Aldrich,目录号S3187)、1X的GlutaMaxTM (Invitrogen,目录号35050-079)、4.5mM的D-葡萄糖(Sigma- Aldrich,目录号G8769)、100ng/ml的GDF8(R&DSystems)和 1μM的MCX化合物。细胞随后在MCDB-131培养基中再培养一 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、100ng/ml的GDF8和0.1μM的MCX化合物。细胞随后在 MCDB-131培养基中再培养一天,所述MCDB-131培养基补充有 2%的无脂肪酸BSA、0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄糖和100ng/ml的GDF8。
b)第2阶段(2天):第1阶段细胞随后用MCDB-131培养基处理两 天,所述MCDB-131培养基补充有2%的无脂肪酸BSA、 0.0012g/ml的碳酸氢钠、1X的GlutaMaxTM、4.5mM的D--葡萄 糖、0.25mM的抗坏血酸(Sigma,MO)和25ng/ml的FGF7(R&D Systems,MN)。
c)第3阶段(2天):第2阶段细胞随后用BLAR定制培养基 (Invitrogen)处理两天,所述BLAR定制培养基补充有1:200稀释度 的ITS-X(Invitrogen,Ca)、4.5mM的葡萄糖、1X的GlutaMaxTM、 0.0017g/ml的碳酸氢钠、2%的无脂肪酸BSA、0.25μM的SANT-1 (Sigma,MO)、1μM的RA(Sigma,MO)、25ng/ml的FGF7、 0.25mM的抗坏血酸、200nM的TPB(PKC活化剂;目录号565740;EMD Chemicals,Gibbstown,NJ),以及100nM的LDN- 193189(BMP受体抑制剂;目录号04-0019;Stemgent)。
d)第4阶段(3天):第3阶段细胞随后用BLAR培养基处理三天, 所述BLAR培养基补充有1:200稀释度的ITS-X、4.5mM的葡萄 糖、1X的GlutaMaxTM、0.0017g/ml的碳酸氢钠、2%的无脂肪酸 BSA、0.25μM的SANT-1、100nM的RA、2ng/ml的FGF7、 100nM的LDN-193189、0.25mM的抗坏血酸和200nM的TPB;随 后在第4阶段结束时,在平皿上培养的细胞用10μM的Y27632处 理4小时,用PBS冲洗,然后在室温下用1X的TrypLETM (Invitrogen)处理5分钟,之后去除所述酶并且用基础培养基冲洗, 然后用细胞刮棒刮擦所述细胞。将所得的细胞悬浮液以0.5- 0.75×106个细胞的密度(在10μl的等分试样中)接种到6孔板中 经MATRIGELTM涂覆的0.4微米多孔细胞培养物滤器芯子上。将 1.5ml的培养基添加至每一滤芯的底部,并且不另外添加培养基至 过滤器的顶上侧。
e)第5阶段(3天):第4阶段细胞随后在空气-液体界面处在BLAR 培养基中培养三天,所述BLAR培养基补充有1:200稀释度的 ITS-X、20mM的葡萄糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢 钠、2%的无脂肪酸BSA、10μg/ml的肝素(Sigma,#H3149)、10μM 的ZnSO4(Sigma,Z0251)、0.25μM的SANT-1、50nM的RA、 100nM的LDN-193189、10000nM的ALK5抑制剂II。
f)第6阶段(8天):第5阶段细胞随后用BLAR培养基处理八天, 所述BLAR培养基补充有1:200稀释度的ITS-X、20mM的葡萄 糖、1X的GlutaMaxTM、0.0015g/ml的碳酸氢钠、2%的无脂肪酸 BSA、10μg/ml的肝素(Sigma,#H3149)、10μM的ZnSO4(Sigma, Z0251)、0.25μM的SANT-1、100nM的LDN-193189、1000nM的 T3、10000nM的ALK5抑制剂II。
在第6阶段,按照各种组合去除ALK5抑制剂、T3或LDN,以测试每 种因子对NKX6.1、胰岛素和MAFA的表达的影响。采集第6阶段第5天 和第6阶段第8天的mRNA。图22示出了关键的β细胞成熟标志物以及胰 腺祖细胞标志物的PCR数据。如图22所示,在第6阶段ALK5抑制剂的去 除导致MAFA的表达显著下降。而ALK5抑制剂、LDN-183189(BMP受 体抑制剂)和T3的组合显著提升以下标志物的表达:MAFA(图22A)、 胰岛素(图22B)、淀粉不溶素(图22C),并且适度地改善了NKX6.1的表 达(图22D)。
实例14
用于在空气-液体界面处培养第6阶段细胞的其他方案
该实例公开了用于在空气-液体界面处培养第6阶段细胞的其他材料和 方法。
所使用的材料包括以下物质:得自Corning的10cm滤器芯子(目录号 3419,0.4微米聚碳酸酯膜);MCDB-131培养基(Invitrogen,目录号 10372-019)或BLAR定制培养基(由Invitrogen制备);ITS-X(Invitrogen, Ca);甲状腺激素(T3):Sigma ALK5抑制剂II-ENZO(目录号ALX-27- 445);LDN-193189-StemGent(#04-0074);肝素(Sigma,H3149);以及无脂肪酸BSA(Proliant/Lampire,7500804)。
第6阶段的基础培养基的制备:添加1.5克/升的碳酸氢钠至MCDB- 131培养基,加上2%的BSA,加上1:200X的ITS-X,加上另外15mM的 葡萄糖。
第6阶段的分化培养基的制备:向第6阶段的基础培养基中添加10微 摩尔的ALK5抑制剂II、100nM的LDN-193189,以及1微摩尔的T3。
方法
添加7.5ml的第6阶段分化培养基至10cm滤器芯子的底部。添加少量 (20-30μl)的细胞簇至滤器芯子的顶部。通常,每10cm的滤芯上放置约50 个细胞簇。在接种时,每个细胞簇包含约0.5M的细胞。
培养基优选地每天更换。细胞可通过单独地去除细胞团(诸如通过使 用广口移液管头)来从滤器芯子去除,或所有的细胞团可通过用基础培养 基冲洗过滤器的顶部来一次性去除。所述细胞团松散地贴附在滤芯上。
除了在先前实例中描述的具体培养条件以外,用于使多能细胞或它们 的子代分化成胰腺内分泌细胞的其他合适的培养条件示出于表VIII至XIII 中。如在这些表中所使用的,“ALK5 inh.”是ALK5抑制剂,“RA”是视 黄酸,“Vit.C”是抗坏血酸,“inh.”是抑制剂,“act.”是活化剂, ZnSO4是硫酸锌,“MCX”是MCX化合物,以及“AA”是活化素A。在 某些实施例中,在一个阶段(例如,第4阶段)中的任一处理剂可与另一 阶段(例如,第5阶段)中的任一处理剂组合。




这一页故意保留空白

这一页故意保留空白
如在本发明的方法中使用、在表X示出的组分的示例性范围如下所 示:

如下所示的表XI示出了适用于本发明方法的实施例的另选的示例性培 养条件。
这一页故意保留空白


如下所示的表XII示出了适用于本发明方法的实施例的另选的示例性 培养条件。


如上文所详述的,本发明特别提供了一种形成表达β细胞的特征性标 志物的细胞的方法,所述方法包括:通过用补充有T3/T4,或ALK5抑制 剂、或T3/T4和ALK5抑制剂两者的培养基处理并且在空气-液体界面处培 养表达前肠内胚层细胞的特征性标志物的细胞,来使所述细胞分化成表达β 细胞的特征性标志物的细胞。在一个实施例中,仅在空气-液体界面处培养 第4阶段至第6阶段细胞。第6阶段细胞可对NKX6.1、PDX1和HB9呈阳 性。因而,本发明还提供了一种诱导衍生自多能干细胞的细胞中的PDX1、 NKX6.1和HB9表达的方法,所述方法包括:(a)培养多能干细胞;(b)使所 述多能干细胞分化成表达前肠内胚层细胞的特征性标志物的细胞;以及(c) 通过用补充有T3/T4,或ALK5抑制剂,或T3/T4和ALK5抑制剂两者的培 养基处理并且在空气-液体界面处培养表达前肠内胚层细胞的特征性标志物 的细胞,来使所述细胞分化成表达PDX1、NKX6.1和HB9的细胞。此外, 所得的第6阶段细胞可为单激素阳性细胞。在一个实施例中,第6阶段细 胞共表达NKX6.1和嗜铬粒蛋白-A。在另一个实施例中,第6阶段细胞共 表达NKX6.1和胰岛素。
在某些实施例中,所述方法包括用补充有T3/T4和ALK5抑制剂(诸 如ALK5抑制剂II)的培养基处理第5阶段细胞。在这些实施例中,培养 基可有利地还补充有一种或多种以下物质:视黄酸、抗坏血酸、SANT-1或 LDN-139189。
尽管已结合各种具体材料、程序和实例描述和例示了本发明,然而应 当理解,本发明并不限于所选材料和程序的特定组合。本领域的技术人员 将理解,可对这些细节作出多种变型。说明书和示例应被认为仅为示例性 的,本发明的真实范围和实质由以下权利要求书限定。本申请中所提到的 所有参考文献、专利和专利申请均全文以引用方式并入本文中。
Claims (16)
1.一种可用于诱导衍生自多能干细胞的细胞的分化的培养基,所述培养基包括生长培养基,所述生长培养基补充有:
a. ALK5抑制剂,所述ALK5抑制剂选自:ALK5抑制剂II、ALK5i、SD208、TGF-B抑制剂SB431542、ITD-1、LY2109761、A83-01、LY2157299、TGF-β受体抑制剂V、TGF-β受体抑制剂I、TGF-β受体抑制剂IV、TGF-β受体抑制剂VII、TGF-β受体抑制剂VIII、TGF-β受体抑制剂II、TGF-β受体抑制剂VI、TGF-β受体抑制剂III;
b. BMP受体抑制剂,所述BMP受体抑制剂选自LDN-193189、头蛋白或脊索发生素;以及
c. 甲状腺激素,所述甲状腺激素选自三碘甲状腺原氨酸、四碘甲状腺原氨酸、三碘甲状腺原氨酸的类似物、四碘甲状腺原氨酸的类似物、以及它们的混合物;
其中所述多能干细胞是非胚胎来源的多能干细胞,H1、H7、H9建立的细胞系的多能干细胞,或诱导的多能干细胞。
2.根据权利要求1所述的培养基,所述培养基还包含选自SANT-1或HPI-1的SHH信号转导途径拮抗剂。
3.根据权利要求2所述的培养基,所述培养基还包含视黄酸。
4.根据权利要求3所述的培养基,所述培养基包含ALK5抑制剂II、SANT-1、LDN-193189和视黄酸。
5.根据权利要求4所述的培养基,所述培养基还包含一种或多种补充剂,所述一种或多种补充剂选自:
a. smoothened受体抑制剂,所述smoothened受体抑制剂选自MRT10或环杷明;
b. 抗坏血酸;
c. 肝素;以及
d. 硫酸锌。
6.根据权利要求4所述的分化培养基,其中所述生长培养基选自MCDB-131培养基和BLAR培养基。
7.根据权利要求5所述的分化培养基,其中所述生长培养基补充有ALK5抑制剂II、SANT-1、LDN-193189、肝素、硫酸锌和视黄酸。
8.一种可用于诱导多能干细胞的分化的培养基,所述培养基包括生长培养基,所述生长培养基补充有:
a. ALK5抑制剂,所述ALK5抑制剂选自:ALK5抑制剂II、ALK5i、SD208、TGF-B抑制剂SB431542、ITD-1、LY2109761、A83-01、LY2157299、TGF-β受体抑制剂V、TGF-β受体抑制剂I、TGF-β受体抑制剂IV、TGF-β受体抑制剂VII、TGF-β受体抑制剂VIII、TGF-β受体抑制剂II、TGF-β受体抑制剂VI、TGF-β受体抑制剂III;
b. 甲状腺激素,所述甲状腺激素选自三碘甲状腺原氨酸、四碘甲状腺原氨酸、三碘甲状腺原氨酸的类似物、四碘甲状腺原氨酸的类似物、以及它们的混合物;和
c. SHH信号转导途径拮抗剂,所述SHH信号转导途径拮抗剂选自SANT-1或HPI-1;
其中所述多能干细胞是非胚胎来源的多能干细胞,H1、H7、H9建立的细胞系的多能干细胞,或诱导的多能干细胞。
9.根据权利要求8所述的培养基,还包含视黄酸。
10.根据权利要求9所述的培养基,还包含抗坏血酸。
11.根据权利要求8所述的培养基,还包含一种或多种补充剂,所述一种或多种补充剂选自:
a. BMP受体抑制剂,所述BMP受体抑制剂选自LDN-193189、头蛋白或脊索发生素;
b. 肝素;以及
c. 硫酸锌。
12.根据权利要求11所述的分化培养基,其中所述生长培养基选自MCDB-131培养基和BLAR培养基。
13.根据权利要求12所述的分化培养基,其中所述生长培养基补充有ALK5抑制剂II、LDN-139189、硫酸锌、三碘甲状腺原氨酸、SANT-1和肝素。
14.根据权利要求1所述的分化培养基,其中所述衍生自多能干细胞的细胞是前肠内胚层细胞、胰腺前肠前体细胞或胰腺内胚层/胰腺内分泌细胞。
15.根据权利要求1所述的分化培养基,其中所述衍生自多能干细胞的细胞是胰腺内胚层/胰腺内分泌细胞。
16.根据权利要求1所述的培养基,其中所述衍生自多能干细胞的细胞是前肠内胚层细胞。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261747662P | 2012-12-31 | 2012-12-31 | |
| US61/747662 | 2012-12-31 | ||
| PCT/US2013/075939 WO2014105543A1 (en) | 2012-12-31 | 2013-12-18 | Culturing of human embryonic stem cells at the air-liquid interface for differentiation into pancreatic endocrine cells |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN105008518A CN105008518A (zh) | 2015-10-28 |
| CN105008518B true CN105008518B (zh) | 2020-08-07 |
Family
ID=51017432
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201380074040.4A Active CN105008518B (zh) | 2012-12-31 | 2013-12-18 | 在空气-液体界面处培养人胚胎干细胞以用于分化成胰腺内分泌细胞 |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US10344264B2 (zh) |
| EP (1) | EP2938724B1 (zh) |
| JP (1) | JP6557146B2 (zh) |
| KR (1) | KR102084561B1 (zh) |
| CN (1) | CN105008518B (zh) |
| AR (1) | AR094347A1 (zh) |
| AU (4) | AU2013368221B2 (zh) |
| BR (1) | BR112015015770A2 (zh) |
| CA (1) | CA2896655C (zh) |
| DK (1) | DK2938724T3 (zh) |
| ES (1) | ES2837763T3 (zh) |
| HK (1) | HK1217110A1 (zh) |
| MX (1) | MX2015008577A (zh) |
| PH (1) | PH12015501465A1 (zh) |
| RU (3) | RU2768963C2 (zh) |
| SG (2) | SG10201709338RA (zh) |
| WO (1) | WO2014105543A1 (zh) |
Families Citing this family (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101617243B1 (ko) | 2007-07-31 | 2016-05-02 | 라이프스캔, 인코포레이티드 | 인간 배아 줄기 세포의 분화 |
| EP2229434B1 (en) | 2007-11-27 | 2011-09-07 | Lifescan, Inc. | Differentiation of human embryonic stem cells |
| EP2456862A4 (en) | 2009-07-20 | 2013-02-27 | Janssen Biotech Inc | DIFFERENTIATION OF HUMAN EMBRYONIC STEM CELLS |
| KR102203056B1 (ko) | 2011-12-22 | 2021-01-14 | 얀센 바이오테크 인코포레이티드 | 인간 배아 줄기 세포의 단일 인슐린 호르몬 양성 세포로의 분화 |
| US8859286B2 (en) | 2013-03-14 | 2014-10-14 | Viacyte, Inc. | In vitro differentiation of pluripotent stem cells to pancreatic endoderm cells (PEC) and endocrine cells |
| MX2015017103A (es) | 2013-06-11 | 2016-11-07 | Harvard College | Celulas beta derivadas de células madre ( sc-beta ) y composiciones y métodos para generarlas. |
| EP3143127B1 (en) | 2014-05-16 | 2021-07-14 | Janssen Biotech, Inc. | Use of small molecules to enhance mafa expression in pancreatic endocrine cells |
| WO2016100898A1 (en) | 2014-12-18 | 2016-06-23 | President And Fellows Of Harvard College | Serum-free in vitro directed differentiation protocol for generating stem cell-derived b cells and uses thereof |
| EP3234110B1 (en) | 2014-12-18 | 2024-02-28 | President and Fellows of Harvard College | METHODS FOR GENERATING STEM CELL-DERIVED ß CELLS AND USES THEREOF |
| WO2016100930A1 (en) | 2014-12-18 | 2016-06-23 | President And Fellows Of Harvard College | Methods for generating stem cell-derived b cells and methods of use thereof |
| US20160175363A1 (en) * | 2014-12-18 | 2016-06-23 | President And Fellows Of Harvard College | Methods for generating stem cell-derived beta cells and uses thereof |
| MA45329A (fr) * | 2016-04-08 | 2019-02-13 | Univ California | Production de cellules bêta matures pleinement fonctionnelles à partir de progénitrices pancréatiques humaines |
| KR101870240B1 (ko) * | 2016-04-12 | 2018-06-22 | 서울대학교산학협력단 | 지방줄기세포에서 신경줄기세포, 신경세포 및 가바성 신경세포로의 분화 유도 방법 |
| MA45502A (fr) | 2016-06-21 | 2019-04-24 | Janssen Biotech Inc | Génération de cellules bêta fonctionnelles dérivées de cellules souches pluripotentes humaines ayant une respiration mitochondriale glucose-dépendante et une réponse en sécrétion d'insuline en deux phases |
| EP3500276A4 (en) * | 2016-08-19 | 2020-01-15 | Memorial Sloan-Kettering Cancer Center | METHODS OF DIFFERENTIATING STEM CELLS IN ENDODERM |
| WO2018038042A1 (ja) * | 2016-08-24 | 2018-03-01 | 学校法人慶應義塾 | ヒト下痢症ウイルスの感染・増殖培養用2dオルガノイド及びその使用 |
| FR3061206B1 (fr) * | 2016-12-23 | 2020-06-26 | Laboratoires Clarins | Modele de peau incluant epiderme, derme et hypoderme, son procede de preparation et son utilisation |
| US10767164B2 (en) | 2017-03-30 | 2020-09-08 | The Research Foundation For The State University Of New York | Microenvironments for self-assembly of islet organoids from stem cells differentiation |
| IL305391B2 (en) | 2017-11-15 | 2024-09-01 | Vertex Pharma | Preparations for the production of islet cells and methods of use |
| EP3833365A4 (en) | 2018-08-10 | 2022-05-11 | Vertex Pharmaceuticals Incorporated | ISLE DIFFERENTIATION DERIVED FROM STEM CELLS |
| US10724052B2 (en) | 2018-09-07 | 2020-07-28 | Crispr Therapeutics Ag | Universal donor cells |
| EP3856205A4 (en) * | 2018-09-25 | 2022-06-15 | The Regents of the University of California | PRODUCTION AND ACCUMULATION OF PANCREATIC ENDOCRIN PROGENIOR CELLS |
| EP4386081A3 (en) * | 2019-05-21 | 2024-10-16 | President and Fellows of Harvard College | Endocrine differentiation-inducing molecule |
| AU2020282355B2 (en) | 2019-05-31 | 2023-11-02 | Viacyte, Inc. | A biocompatible membrane composite |
| EP3975926A1 (en) | 2019-05-31 | 2022-04-06 | W.L. Gore & Associates, Inc. | A biocompatible membrane composite |
| CN114401752B (zh) | 2019-05-31 | 2023-04-04 | W.L.戈尔及同仁股份有限公司 | 具有受控氧扩散距离的细胞封装装置 |
| CA3139590C (en) | 2019-05-31 | 2024-01-23 | W. L. Gore & Associates, Inc. | A biocompatible membrane composite |
| KR20220058579A (ko) | 2019-09-05 | 2022-05-09 | 크리스퍼 테라퓨틱스 아게 | 보편적 공여자 세포 |
| CA3150235A1 (en) | 2019-09-05 | 2021-03-11 | Alireza Rezania | UNIVERSAL DONOR CELLS |
| AU2021414617A1 (en) | 2020-12-31 | 2023-08-10 | Crispr Therapeutics Ag | Universal donor cells |
| CN112961823B (zh) * | 2021-03-19 | 2024-01-23 | 上海爱萨尔生物科技有限公司 | 一种诱导多能干细胞定向分化制备胰岛β细胞的培养液 |
| CN113046299B (zh) * | 2021-03-19 | 2024-06-28 | 上海爱萨尔生物科技有限公司 | 一种诱导多能干细胞定向分化制备胰岛β细胞的添加剂 |
| CN112980774B (zh) * | 2021-03-19 | 2022-04-01 | 上海爱萨尔生物科技有限公司 | 一种诱导多能干细胞定向分化制备胰岛β细胞的培养方法 |
| JP2024536496A (ja) * | 2021-10-15 | 2024-10-04 | サイミューン セラピューティクス インコーポレイテッド | 胸腺細胞及びその製造方法 |
| US20230377685A1 (en) | 2022-04-15 | 2023-11-23 | Aspen Neuroscience, Inc. | Methods of classifying the differentiation state of cells and related compositions of differentiated cells |
| CN115011544B (zh) * | 2022-05-30 | 2023-11-17 | 广州国家实验室 | 体外诱导获得胰岛δ细胞的方法及其应用 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090004152A1 (en) * | 2006-03-02 | 2009-01-01 | Cythera, Inc. | Methods of producing pancreatic hormones |
| US20110281355A1 (en) * | 2010-05-12 | 2011-11-17 | Centocor Ortho Biotech Inc. | Differentiation of Human Embryonic Stem Cells |
| WO2012030540A2 (en) * | 2010-08-31 | 2012-03-08 | Janssen Biotech, Inc. | Differentiation of pluripotent stem cells |
| CN102741395A (zh) * | 2009-12-23 | 2012-10-17 | 森托科尔奥索生物科技公司 | 人胚胎干细胞的分化 |
| US20120264209A1 (en) * | 2006-05-02 | 2012-10-18 | Jon Odorico | Methods and devices for differentiating pluripotent stem cells into cells of the pancreatic lineage |
Family Cites Families (265)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3209652A (en) | 1961-03-30 | 1965-10-05 | Burgsmueller Karl | Thread whirling method |
| AT326803B (de) | 1968-08-26 | 1975-12-29 | Binder Fa G | Maschenware sowie verfahren zur herstellung derselben |
| US3935067A (en) | 1974-11-22 | 1976-01-27 | Wyo-Ben Products, Inc. | Inorganic support for culture media |
| CA1201400A (en) | 1982-04-16 | 1986-03-04 | Joel L. Williams | Chemically specific surfaces for influencing cell activity during culture |
| US4499802A (en) | 1982-09-29 | 1985-02-19 | Container Graphics Corporation | Rotary cutting die with scrap ejection |
| US4537773A (en) | 1983-12-05 | 1985-08-27 | E. I. Du Pont De Nemours And Company | α-Aminoboronic acid derivatives |
| US4557264A (en) | 1984-04-09 | 1985-12-10 | Ethicon Inc. | Surgical filament from polypropylene blended with polyethylene |
| US5215893A (en) | 1985-10-03 | 1993-06-01 | Genentech, Inc. | Nucleic acid encoding the ba chain prodomains of inhibin and method for synthesizing polypeptides using such nucleic acid |
| US5089396A (en) | 1985-10-03 | 1992-02-18 | Genentech, Inc. | Nucleic acid encoding β chain prodomains of inhibin and method for synthesizing polypeptides using such nucleic acid |
| US4737578A (en) | 1986-02-10 | 1988-04-12 | The Salk Institute For Biological Studies | Human inhibin |
| US5863531A (en) | 1986-04-18 | 1999-01-26 | Advanced Tissue Sciences, Inc. | In vitro preparation of tubular tissue structures by stromal cell culture on a three-dimensional framework |
| US5567612A (en) | 1986-11-20 | 1996-10-22 | Massachusetts Institute Of Technology | Genitourinary cell-matrix structure for implantation into a human and a method of making |
| US5759830A (en) | 1986-11-20 | 1998-06-02 | Massachusetts Institute Of Technology | Three-dimensional fibrous scaffold containing attached cells for producing vascularized tissue in vivo |
| CA1340581C (en) | 1986-11-20 | 1999-06-08 | Joseph P. Vacanti | Chimeric neomorphogenesis of organs by controlled cellular implantation using artificial matrices |
| NZ229354A (en) | 1988-07-01 | 1990-09-26 | Becton Dickinson Co | Treating polymer surfaces with a gas plasma and then applying a layer of endothelial cells to the surface |
| EP0363125A3 (en) | 1988-10-03 | 1990-08-16 | Hana Biologics Inc. | Proliferated pancreatic endocrine cell product and process |
| US5837539A (en) | 1990-11-16 | 1998-11-17 | Osiris Therapeutics, Inc. | Monoclonal antibodies for human mesenchymal stem cells |
| DK0628639T3 (da) | 1991-04-25 | 2000-01-24 | Chugai Pharmaceutical Co Ltd | Rekonstitueret humant antistof mod human interleukin-6-receptor |
| US5449383A (en) | 1992-03-18 | 1995-09-12 | Chatelier; Ronald C. | Cell growth substrates |
| GB9206861D0 (en) | 1992-03-28 | 1992-05-13 | Univ Manchester | Wound healing and treatment of fibrotic disorders |
| CA2114282A1 (en) | 1993-01-28 | 1994-07-29 | Lothar Schilder | Multi-layered implant |
| JP3525221B2 (ja) | 1993-02-17 | 2004-05-10 | 味の素株式会社 | 免疫抑制剤 |
| JP2813467B2 (ja) | 1993-04-08 | 1998-10-22 | ヒューマン・セル・カルチャーズ・インコーポレーテッド | 細胞培養法および培地 |
| US5523226A (en) * | 1993-05-14 | 1996-06-04 | Biotechnology Research And Development Corp. | Transgenic swine compositions and methods |
| GB9310557D0 (en) | 1993-05-21 | 1993-07-07 | Smithkline Beecham Plc | Novel process and apparatus |
| TW257671B (zh) | 1993-11-19 | 1995-09-21 | Ciba Geigy | |
| US5834308A (en) | 1994-04-28 | 1998-11-10 | University Of Florida Research Foundation, Inc. | In vitro growth of functional islets of Langerhans |
| US6703017B1 (en) | 1994-04-28 | 2004-03-09 | Ixion Biotechnology, Inc. | Reversal of insulin-dependent diabetes by islet-producing stem cells, islet progenitor cells and islet-like structures |
| US6001647A (en) | 1994-04-28 | 1999-12-14 | Ixion Biotechnology, Inc. | In vitro growth of functional islets of Langerhans and in vivo uses thereof |
| US6083903A (en) | 1994-10-28 | 2000-07-04 | Leukosite, Inc. | Boronic ester and acid compounds, synthesis and uses |
| JP4079461B2 (ja) | 1994-12-29 | 2008-04-23 | 中外製薬株式会社 | Il−6アンタゴニストを含んでなる抗腫瘍剤の作用増強剤 |
| US5843780A (en) | 1995-01-20 | 1998-12-01 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| US5718922A (en) | 1995-05-31 | 1998-02-17 | Schepens Eye Research Institute, Inc. | Intravitreal microsphere drug delivery and method of preparation |
| US5908782A (en) | 1995-06-05 | 1999-06-01 | Osiris Therapeutics, Inc. | Chemically defined medium for human mesenchymal stem cells |
| US5681561A (en) | 1995-06-07 | 1997-10-28 | Life Medical Sciences, Inc. | Compositions and methods for improving autologous fat grafting |
| UA65572C2 (en) | 1997-04-24 | 2004-04-15 | Ortho Mcneil Pharm Inc | Substituted imidazoles, intermediate compounds for the preparation thereof, a method for the preparation of substituted imidazoles and a method for the treatment of inflammatory diseases |
| DK1028737T3 (da) | 1997-07-03 | 2007-08-13 | Osiris Therapeutics Inc | Humane mesenchymale stamceller fra perifert blod |
| WO1999014318A1 (en) | 1997-09-16 | 1999-03-25 | Board Of Regents, The University Of Texas System | Method for the complete chemical synthesis and assembly of genes and genomes |
| US6670127B2 (en) | 1997-09-16 | 2003-12-30 | Egea Biosciences, Inc. | Method for assembly of a polynucleotide encoding a target polypeptide |
| JP3880795B2 (ja) | 1997-10-23 | 2007-02-14 | ジェロン・コーポレーション | フィーダー細胞を含まない培養物中で、霊長類由来始原幹細胞を増殖させるための方法 |
| AR014195A1 (es) | 1997-12-29 | 2001-02-07 | Ortho Mcneil Pharm Inc | Compuestos de trifenilpropanamida utiles para el tratamiento de procesos inflamatorios, composiciones anti-inflamatorias que los comprenden, ymetodos para prepararlos |
| US6328960B1 (en) | 1998-03-18 | 2001-12-11 | Osiris Therapeutics, Inc. | Mesenchymal stem cells for prevention and treatment of immune responses in transplantation |
| MY132496A (en) | 1998-05-11 | 2007-10-31 | Vertex Pharma | Inhibitors of p38 |
| US6413773B1 (en) | 1998-06-01 | 2002-07-02 | The Regents Of The University Of California | Phosphatidylinositol 3-kinase inhibitors as stimulators of endocrine differentiation |
| US6667176B1 (en) | 2000-01-11 | 2003-12-23 | Geron Corporation | cDNA libraries reflecting gene expression during growth and differentiation of human pluripotent stem cells |
| US7410798B2 (en) | 2001-01-10 | 2008-08-12 | Geron Corporation | Culture system for rapid expansion of human embryonic stem cells |
| US6610540B1 (en) | 1998-11-18 | 2003-08-26 | California Institute Of Technology | Low oxygen culturing of central nervous system progenitor cells |
| US6413556B1 (en) | 1999-01-08 | 2002-07-02 | Sky High, Llc | Aqueous anti-apoptotic compositions |
| EP1144597A2 (en) | 1999-01-21 | 2001-10-17 | Vitro Diagnostics, Inc. | Immortalized cell lines and methods of making the same |
| JP2002536012A (ja) | 1999-02-11 | 2002-10-29 | ハン、ジャエ・ヨン | 鳥類の多能性胚性生殖細胞株 |
| US6815203B1 (en) | 1999-06-23 | 2004-11-09 | Joslin Diabetes Center, Inc. | Methods of making pancreatic islet cells |
| US6333029B1 (en) | 1999-06-30 | 2001-12-25 | Ethicon, Inc. | Porous tissue scaffoldings for the repair of regeneration of tissue |
| US6306424B1 (en) | 1999-06-30 | 2001-10-23 | Ethicon, Inc. | Foam composite for the repair or regeneration of tissue |
| EP1224259A4 (en) | 1999-09-27 | 2005-04-27 | Univ Florida | INVERSION OF INSULIN DEPENDENT DIABETES BY ISOLATED STEM CELLS, PROGENITOR ISLANDIC CELLS, AND INSULAR TYPE STRUCTURES |
| US6685936B2 (en) | 1999-10-12 | 2004-02-03 | Osiris Therapeutics, Inc. | Suppressor cells induced by culture with mesenchymal stem cells for treatment of immune responses in transplantation |
| US20030082155A1 (en) | 1999-12-06 | 2003-05-01 | Habener Joel F. | Stem cells of the islets of langerhans and their use in treating diabetes mellitus |
| US6753153B2 (en) | 1999-12-13 | 2004-06-22 | The Scripps Research Institute | Markers for identification and isolation of pancreatic islet α and β progenitors |
| US7005252B1 (en) | 2000-03-09 | 2006-02-28 | Wisconsin Alumni Research Foundation | Serum free cultivation of primate embryonic stem cells |
| US7439064B2 (en) | 2000-03-09 | 2008-10-21 | Wicell Research Institute, Inc. | Cultivation of human embryonic stem cells in the absence of feeder cells or without conditioned medium |
| US6436704B1 (en) | 2000-04-10 | 2002-08-20 | Raven Biotechnologies, Inc. | Human pancreatic epithelial progenitor cells and methods of isolation and use thereof |
| US6458589B1 (en) | 2000-04-27 | 2002-10-01 | Geron Corporation | Hepatocyte lineage cells derived from pluripotent stem cells |
| KR100947577B1 (ko) | 2000-06-26 | 2010-03-15 | 엔씨 메디컬 리서치 가부시키가이샤 | 신경계 세포로 분화할 수 있는 세포를 포함하는 세포분획 |
| DK1333833T3 (da) | 2000-10-23 | 2011-12-12 | Glaxosmithkline Llc | Ny trisubstitueret 8H-pyridol[2,3-d]pyrimidin-7-on-forbindelse til behandling af CSBP/RK/p38-kinnasemedierede sygdomme |
| ES2263681T3 (es) | 2000-12-08 | 2006-12-16 | Ortho-Mcneil Pharmaceutical, Inc. | Compuestos de pirrolina indazolil-substituidos como inhibidores de la kinasa. |
| AU2737102A (en) | 2000-12-08 | 2002-06-18 | Ortho Mcneil Pharm Inc | Macroheterocylic compounds useful as kinase inhibitors |
| US6599323B2 (en) | 2000-12-21 | 2003-07-29 | Ethicon, Inc. | Reinforced tissue implants and methods of manufacture and use |
| CA2435826A1 (en) | 2001-01-24 | 2002-08-01 | The Government Of The United States Of America | Differentiation of stem cells to pancreatic endocrine cells |
| CA2435124A1 (en) | 2001-01-25 | 2002-08-01 | Millennium Pharmaceuticals, Inc. | Formulation of boronic acid compounds |
| US6656488B2 (en) | 2001-04-11 | 2003-12-02 | Ethicon Endo-Surgery, Inc. | Bioabsorbable bag containing bioabsorbable materials of different bioabsorption rates for tissue engineering |
| EP1379626A2 (en) | 2001-04-19 | 2004-01-14 | DeveloGen Aktiengesellschaft für entwicklungsbiologische Forschung | A method for differentiating stem cells into insulin-producing cells |
| WO2002088335A1 (fr) | 2001-04-24 | 2002-11-07 | Ajinomoto Co., Inc. | Cellules souches et procede d'extraction de ces cellules |
| CA2447015A1 (en) | 2001-05-15 | 2002-11-21 | Rappaport Family Institute For Research In The Medical Sciences | Insulin producing cells derived from human embryonic stem cells |
| US6626950B2 (en) | 2001-06-28 | 2003-09-30 | Ethicon, Inc. | Composite scaffold with post anchor for the repair and regeneration of tissue |
| KR100418195B1 (ko) | 2001-07-05 | 2004-02-11 | 주식회사 우리기술 | 전력케이블의 다중절연진단장치 및 그 방법 |
| GB0117583D0 (en) | 2001-07-19 | 2001-09-12 | Astrazeneca Ab | Novel compounds |
| WO2003014313A2 (en) | 2001-08-06 | 2003-02-20 | Bresagen, Ltd. | Alternative compositions and methods for the culture of stem cells |
| US6617152B2 (en) | 2001-09-04 | 2003-09-09 | Corning Inc | Method for creating a cell growth surface on a polymeric substrate |
| EP1298201A1 (en) | 2001-09-27 | 2003-04-02 | Cardion AG | Process for the production of cells exhibiting an islet-beta-cell-like state |
| US20030138951A1 (en) | 2001-10-18 | 2003-07-24 | Li Yin | Conversion of liver stem and progenitor cells to pancreatic functional cells |
| JP4330995B2 (ja) | 2001-11-15 | 2009-09-16 | チルドレンズ メディカル センター コーポレーション | 絨毛膜絨毛、羊水、および胎盤からの胎児性幹細胞を単離、増殖、および分化させる方法、ならびにその治療的使用方法 |
| KR100930139B1 (ko) | 2001-12-07 | 2009-12-07 | 사이토리 테라퓨틱스, 인크. | 처리된 리포애스퍼레이트 세포로 환자를 치료하기 위한시스템 및 방법 |
| WO2003050249A2 (en) | 2001-12-07 | 2003-06-19 | Geron Corporation | Islet cells from human embryonic stem cells |
| WO2003054169A1 (en) | 2001-12-21 | 2003-07-03 | Thromb-X Nv | Compositions for the in vitro derivation and culture of embryonic stem (es) cell lines with germline transmission capability |
| EP1461421A2 (en) | 2001-12-28 | 2004-09-29 | Cellartis AB | A method for the establishment of a pluripotent human blastocyst-derived stem cell line |
| US20030162290A1 (en) | 2002-01-25 | 2003-08-28 | Kazutomo Inoue | Method for inducing differentiation of embryonic stem cells into functioning cells |
| US20030180268A1 (en) | 2002-02-05 | 2003-09-25 | Anthony Atala | Tissue engineered construct for supplementing or replacing a damaged organ |
| JPWO2003087349A1 (ja) | 2002-04-17 | 2005-08-18 | 大塚製薬株式会社 | 間葉系細胞から膵β細胞を形成する方法 |
| US20040161419A1 (en) | 2002-04-19 | 2004-08-19 | Strom Stephen C. | Placental stem cells and uses thereof |
| EP1506192B1 (en) | 2002-05-08 | 2008-02-27 | Janssen Pharmaceutica N.V. | Substituted pyrroline kinase inhibitors |
| US20060003446A1 (en) * | 2002-05-17 | 2006-01-05 | Gordon Keller | Mesoderm and definitive endoderm cell populations |
| BR0311360A (pt) | 2002-05-28 | 2006-06-06 | Becton Dickinson Co | métodos para expansão e transdiferenciação in vitro de células acinares pancreáticas humanas em células produtoras de insulina |
| JP2005531609A (ja) | 2002-06-05 | 2005-10-20 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | キナーゼ阻害剤としてのシスインドリル−マレイミド誘導体 |
| GB0212976D0 (en) | 2002-06-06 | 2002-07-17 | Tonejet Corp Pty Ltd | Ejection method and apparatus |
| CN1171991C (zh) | 2002-07-08 | 2004-10-20 | 徐如祥 | 人神经干细胞的培养方法 |
| US6877147B2 (en) | 2002-07-22 | 2005-04-05 | Broadcom Corporation | Technique to assess timing delay by use of layout quality analyzer comparison |
| US7838290B2 (en) | 2002-07-25 | 2010-11-23 | The Scripps Research Institute | Hematopoietic stem cells and methods of treatment of neovascular eye diseases therewith |
| CA2494040A1 (en) | 2002-07-29 | 2004-02-05 | Es Cell International Pte Ltd. | Multi-step method for the differentiation of insulin positive, glucose |
| AU2003262628A1 (en) | 2002-08-14 | 2004-03-03 | University Of Florida | Bone marrow cell differentiation |
| AU2003268534A1 (en) | 2002-09-06 | 2004-03-29 | Amcyte Inc. | Cd56 positive human adult pancreatic endocrine progenitor cells |
| US9969977B2 (en) | 2002-09-20 | 2018-05-15 | Garnet Biotherapeutics | Cell populations which co-express CD49c and CD90 |
| US20040062753A1 (en) | 2002-09-27 | 2004-04-01 | Alireza Rezania | Composite scaffolds seeded with mammalian cells |
| AU2003285172A1 (en) | 2002-11-08 | 2004-06-03 | The Johns Hopkins University | Human embryonic stem cell cultures, and compositions and methods for growing same |
| US7144999B2 (en) | 2002-11-23 | 2006-12-05 | Isis Pharmaceuticals, Inc. | Modulation of hypoxia-inducible factor 1 alpha expression |
| WO2004050827A2 (en) | 2002-12-05 | 2004-06-17 | Technion Research & Development Foundation Ltd. | Cultured human pancreatic islets, and uses thereof |
| JP4613069B2 (ja) | 2002-12-16 | 2011-01-12 | テクニオン リサーチ アンド ディベロップメント ファウンデーション リミテッド | 支持細胞非含有、異種非含有のヒト胚性幹細胞の調製方法およびこれらを使用して調製された幹細胞培養物 |
| US20050118148A1 (en) | 2002-12-20 | 2005-06-02 | Roland Stein | Compositions and methods related to mammalian Maf-A |
| RU2359671C2 (ru) | 2003-01-29 | 2009-06-27 | Такеда Фармасьютикал Компани Лимитед | Способ получения препарата с покрытием |
| EP1588708A4 (en) | 2003-01-29 | 2006-03-01 | Takeda Pharmaceutical | METHOD FOR PRODUCING COATED PREPARATION |
| WO2004073633A2 (en) | 2003-02-14 | 2004-09-02 | The Board Of Trustees Of The Leland Stanford Junior University | Methods and compositions for modulating the development of stem cells |
| US20070154981A1 (en) | 2003-02-14 | 2007-07-05 | The Board Of Trustees Of The Leland Stanford Junior University | Insulin-producing cells derived from stem cells |
| CA2520861A1 (en) | 2003-03-27 | 2004-10-14 | Ixion Biotechnology, Inc. | Method for transdifferentiation of non-pancreatic stem cells to the pancreatic pathway |
| WO2004090110A2 (en) | 2003-03-31 | 2004-10-21 | Bresagen Inc. | Compositions and methods for the control, differentiation and/or manipulation of pluripotent cells through a gamma-secretase signaling pathway |
| US20090203141A1 (en) | 2003-05-15 | 2009-08-13 | Shi-Lung Lin | Generation of tumor-free embryonic stem-like pluripotent cells using inducible recombinant RNA agents |
| PL1641914T3 (pl) | 2003-06-27 | 2017-01-31 | DePuy Synthes Products, Inc. | Komórki pochodzące z poporodowej tkanki łożyska oraz sposoby uzyskiwania i zastosowania tych komórek |
| IL161903A0 (en) | 2003-07-17 | 2005-11-20 | Gamida Cell Ltd | Ex vivo progenitor and stem cell expansion for usein the treatment of disease of endodermally- deri ved organs |
| ITRM20030395A1 (it) | 2003-08-12 | 2005-02-13 | Istituto Naz Per Le Malattie Infettive Lazz | Terreno di coltura per il mantenimento, la proliferazione e il differenziamento di cellule di mammifero. |
| US20050042595A1 (en) | 2003-08-14 | 2005-02-24 | Martin Haas | Banking of multipotent amniotic fetal stem cells |
| US7157275B2 (en) | 2003-08-15 | 2007-01-02 | Becton, Dickinson And Company | Peptides for enhanced cell attachment and growth |
| US20060205072A1 (en) | 2003-08-27 | 2006-09-14 | Nobuko Uchida | Enriched pancreatic stem cell and progenitor cell populations, and methods for identifying, isolating and enriching for such populations |
| US6939633B2 (en) | 2003-09-17 | 2005-09-06 | General Motors Corporation | Fuel cell shutdown and startup using a cathode recycle loop |
| EP1696899A1 (en) | 2003-12-17 | 2006-09-06 | Allergan, Inc. | Methods for treating retinoid responsive disorders using selective inhibitors of cyp26a and cyp26b |
| US20060030042A1 (en) | 2003-12-19 | 2006-02-09 | Ali Brivanlou | Maintenance of embryonic stem cells by the GSK-3 inhibitor 6-bromoindirubin-3'-oxime |
| GB0329498D0 (en) | 2003-12-19 | 2004-01-28 | Novartis Ag | Organic compounds |
| CN103898045B (zh) | 2003-12-23 | 2019-02-01 | 维亚希特公司 | 定形内胚层 |
| US7541185B2 (en) | 2003-12-23 | 2009-06-02 | Cythera, Inc. | Methods for identifying factors for differentiating definitive endoderm |
| US7625753B2 (en) | 2003-12-23 | 2009-12-01 | Cythera, Inc. | Expansion of definitive endoderm cells |
| CN109628371B (zh) | 2003-12-23 | 2021-02-19 | 维亚希特公司 | 定形内胚层 |
| US20050266554A1 (en) | 2004-04-27 | 2005-12-01 | D Amour Kevin A | PDX1 expressing endoderm |
| TWI334443B (en) | 2003-12-31 | 2010-12-11 | Ind Tech Res Inst | Method of single cell culture of undifferentiated human embryonic stem cells |
| WO2005065354A2 (en) | 2003-12-31 | 2005-07-21 | The Burnham Institute | Defined media for pluripotent stem cell culture |
| US7794704B2 (en) | 2004-01-23 | 2010-09-14 | Advanced Cell Technology, Inc. | Methods for producing enriched populations of human retinal pigment epithelium cells for treatment of retinal degeneration |
| WO2005071066A1 (en) | 2004-01-23 | 2005-08-04 | Board Of Regents, The University Of Texas System | Methods and compositions for preparing pancreatic insulin secreting cells |
| US20070298453A1 (en) | 2004-02-12 | 2007-12-27 | University Of Newcastle Upon Tyne | Stem Cells |
| WO2005080598A1 (ja) | 2004-02-19 | 2005-09-01 | Dainippon Sumitomo Pharma Co., Ltd. | 体細胞核初期化物質のスクリーニング方法 |
| US20060281174A1 (en) | 2004-03-09 | 2006-12-14 | Gang Xu | Methods for generating insulin-producing cells |
| EP1730261A4 (en) | 2004-03-10 | 2007-11-28 | Univ California | COMPOSITIONS AND METHODS FOR GROWING EMBRYONIC STEM CELLS |
| CN1934245B (zh) | 2004-03-23 | 2012-07-04 | 第一三共株式会社 | 多能干细胞的增殖方法 |
| WO2005097980A2 (en) | 2004-03-26 | 2005-10-20 | Geron Corporation | New protocols for making hepatocytes from embryonic stem cells |
| AU2005230832B2 (en) | 2004-04-01 | 2010-11-11 | Wisconsin Alumni Research Foundation | Differentiation of stem cells to endoderm and pancreatic lineage |
| DK2377922T3 (da) | 2004-04-27 | 2020-05-04 | Viacyte Inc | PDX1-eksprimerende endoderm |
| US7690808B2 (en) | 2004-06-08 | 2010-04-06 | Il Shik Yoon | Prefabricated light reflecting system mounted to ceiling of elevator cage |
| CN102925406B (zh) | 2004-07-09 | 2019-11-22 | 维亚希特公司 | 鉴定用于分化定型内胚层的因子的方法 |
| WO2006020919A2 (en) | 2004-08-13 | 2006-02-23 | University Of Georgia Research Foundation, Inc. | Compositions and methods for self-renewal and differentiation in human embryonic stem cells |
| US20080268533A1 (en) | 2004-08-25 | 2008-10-30 | University Of Georgia Research Foundation, Inc. | Methods and Compositions Utilizing Myc and Gsk3Beta to Manipulate the Pluripotency of Embryonic Stem Cells |
| DE102004043256B4 (de) | 2004-09-07 | 2013-09-19 | Rheinische Friedrich-Wilhelms-Universität Bonn | Skalierbarer Prozess zur Kultivierung undifferenzierter Stammzellen in Suspension |
| JP2006075022A (ja) * | 2004-09-07 | 2006-03-23 | Foundation For Biomedical Research & Innovation | 膵臓ホルモン産生細胞取得方法 |
| KR20070058584A (ko) | 2004-09-08 | 2007-06-08 | 위스콘신 얼럼나이 리서어치 화운데이션 | 인간 배아 줄기 세포의 배양 |
| JP5420837B2 (ja) | 2004-09-08 | 2014-02-19 | ウィスコンシン アラムニ リサーチ ファンデーション | 胚幹細胞の培地及び培養 |
| WO2006079854A1 (en) | 2005-01-28 | 2006-08-03 | Novathera Ltd | Methods for embryonic stem cell culture |
| JP2008528038A (ja) | 2005-01-31 | 2008-07-31 | エス セル インターナショナル ピーティーイー リミテッド | 胚性幹細胞の指示された分化及びその利用 |
| WO2006088867A2 (en) | 2005-02-15 | 2006-08-24 | Medistem Laboratories, Incorporated | Method for expansion of stem cells |
| ES2627419T3 (es) | 2005-03-04 | 2017-07-28 | Lifescan, Inc. | Células estromales adultas derivadas del páncreas |
| GB0505970D0 (en) | 2005-03-23 | 2005-04-27 | Univ Edinburgh | Culture medium containing kinase inhibitor, and uses thereof |
| WO2006113470A2 (en) | 2005-04-15 | 2006-10-26 | Geron Corporation | Cancer treatment by combined inhibition of proteasome and telomerase activities |
| CN100425694C (zh) | 2005-04-15 | 2008-10-15 | 北京大学 | 诱导胚胎干细胞向胰腺细胞分化的方法 |
| US20080208351A1 (en) | 2005-04-26 | 2008-08-28 | Aarhus Universitet | Biocompatible Material for Surgical Implants and Cell Guiding Tissue Culture Surfaces |
| JP4557797B2 (ja) | 2005-05-20 | 2010-10-06 | 株式会社日立製作所 | 光ディスク装置 |
| JP5092124B2 (ja) | 2005-05-24 | 2012-12-05 | 国立大学法人 熊本大学 | Es細胞の分化誘導方法 |
| AU2006202209B2 (en) | 2005-05-27 | 2011-04-14 | Lifescan, Inc. | Amniotic fluid derived cells |
| US20100234400A1 (en) | 2005-06-10 | 2010-09-16 | Irm Llc | Compounds that maintain pluripotency of embryonic stem cells |
| WO2006138433A2 (en) | 2005-06-14 | 2006-12-28 | The Regents Of The University Of California | Induction of cell differentiation by class i bhlh polypeptides |
| EP1931764A1 (en) | 2005-06-21 | 2008-06-18 | GE Healthcare Bio-Sciences AB | Method for cell culture |
| CN101233226B (zh) | 2005-06-22 | 2017-08-11 | 阿斯特利亚斯生物治疗股份公司 | 人胚胎干细胞的悬浮培养物 |
| NZ564179A (en) | 2005-06-30 | 2010-09-30 | Janssen Pharmaceutica Nv | Cyclic anilino - pyridinotriazines as GSK-3 inhibitors |
| US20080194021A1 (en) | 2005-07-29 | 2008-08-14 | Mays Robert W | Use of a Gsk-3 Inhibitor to Maintain Potency of Culture Cells |
| US20090087907A1 (en) | 2005-07-29 | 2009-04-02 | Alice Pebay | Compositions and Methods for Growth of Pluripotent Cells |
| WO2007025234A2 (en) | 2005-08-26 | 2007-03-01 | The Trustees Of Columbia University In The City Of New York | Generation of pancreatic endocrine cells from primary duct cell cultures and methods of use for treatment of diabetes |
| EP3354723B1 (en) | 2005-08-29 | 2023-12-13 | Technion Research & Development Foundation Ltd. | Media for culturing stem cells |
| KR20080056182A (ko) | 2005-09-02 | 2008-06-20 | 에이전시 포 사이언스, 테크놀로지 앤드 리서치 | 간엽 줄기 세포의 유도 방법 |
| GB2444686B (en) | 2005-09-12 | 2010-08-25 | Es Cell Int Pte Ltd | Differentiation of pluripotent stem cells using p38 MAPK inhibitors or prostaglandins |
| WO2007039986A1 (ja) | 2005-10-05 | 2007-04-12 | Osaka University | 脂肪組織由来細胞から膵内分泌細胞を得る方法 |
| EP1941032A2 (en) | 2005-10-14 | 2008-07-09 | Regents Of The University Of Minnesota | Differentiation of non-embryonic stem cells to cells having a pancreatic phenotype |
| US7732202B2 (en) | 2005-10-21 | 2010-06-08 | International Stem Cell Corporation | Oxygen tension for the parthenogenic activation of human oocytes for the production of human embryonic stem cells |
| EP1957636B1 (en) | 2005-10-27 | 2018-07-04 | Viacyte, Inc. | Pdx1-expressing dorsal and ventral foregut endoderm |
| EP3418297B1 (en) | 2005-12-13 | 2023-04-05 | Kyoto University | Nuclear reprogramming factor |
| WO2007082963A1 (es) | 2006-01-18 | 2007-07-26 | Fundación Instituto Valenciano De Infertilidad | Líneas de células madre embrionarias humanas y métodos para usar las mismas |
| SG170021A1 (en) | 2006-02-23 | 2011-04-29 | Novocell Inc | Compositions and methods useful for culturing differentiable cells |
| EP2650359B1 (en) | 2006-03-02 | 2022-05-04 | Viacyte, Inc. | Endocrine precursor cells, pancreatic hormone-expressing cells and methods of production |
| US8741643B2 (en) | 2006-04-28 | 2014-06-03 | Lifescan, Inc. | Differentiation of pluripotent stem cells to definitive endoderm lineage |
| EP4438720A2 (en) | 2006-04-28 | 2024-10-02 | Janssen Biotech, Inc. | Differentiation of human embryonic stem cells |
| US20070259423A1 (en) | 2006-05-02 | 2007-11-08 | Jon Odorico | Method of differentiating stem cells into cells of the endoderm and pancreatic lineage |
| US7964402B2 (en) | 2006-05-25 | 2011-06-21 | Sanford-Burnham Medical Research Institute | Methods for culture and production of single cell populations of human embryonic stem cells |
| CN101541953A (zh) | 2006-06-02 | 2009-09-23 | 佐治亚大学研究基金会 | 通过从人胚胎干细胞获得的定形内胚层细胞的分化得到胰和肝内胚层细胞及组织 |
| WO2007143193A1 (en) | 2006-06-02 | 2007-12-13 | University Of Georgia Research Foundation, Inc. | Pancreatic and liver endoderm cells and tissue by differentiation of definitive endoderm cells obtained from human embryonic stems |
| US8415153B2 (en) | 2006-06-19 | 2013-04-09 | Geron Corporation | Differentiation and enrichment of islet-like cells from human pluripotent stem cells |
| CN100494359C (zh) | 2006-06-23 | 2009-06-03 | 中日友好医院 | 神经干细胞三维立体培养体外扩增的方法 |
| US20080003676A1 (en) | 2006-06-26 | 2008-01-03 | Millipore Corporation | Growth of embryonic stem cells |
| WO2008036447A2 (en) | 2006-06-26 | 2008-03-27 | Lifescan, Inc. | Pluripotent stem cell culture |
| WO2008004990A2 (en) | 2006-07-06 | 2008-01-10 | Es Cell International Pte Ltd | Method for stem cell culture and cells derived therefrom |
| WO2008013664A2 (en) | 2006-07-26 | 2008-01-31 | Cythera, Inc. | Methods of producing pancreatic hormones |
| EP2733203B1 (en) | 2006-08-02 | 2018-10-10 | Technion Research & Development Foundation Ltd. | Methods of expanding embryonic stem cells in a suspension culture |
| KR101331510B1 (ko) | 2006-08-30 | 2013-11-20 | 재단법인서울대학교산학협력재단 | 저농도의 포도당을 함유하는 인간 배아줄기세포용 배지조성물 및 이를 이용한 인간 배아 줄기세포로부터 인슐린생산 세포 또는 세포괴로 분화시키는 방법, 그리고그로부터 유도된 인슐린 생산 세포 또는 세포괴 |
| JP2008099662A (ja) | 2006-09-22 | 2008-05-01 | Institute Of Physical & Chemical Research | 幹細胞の培養方法 |
| WO2008039521A2 (en) | 2006-09-26 | 2008-04-03 | Nmt Medical, Inc. | Method for modifying a medical implant surface for promoting tissue growth |
| WO2008048647A1 (en) | 2006-10-17 | 2008-04-24 | Cythera, Inc. | Modulation of the phosphatidylinositol-3-kinase pathway in the differentiation of human embryonic stem cells |
| BRPI0718310A2 (pt) | 2006-10-17 | 2013-11-19 | Stiefel Laboratories | Metabólitos de talarozol |
| CA2666789C (en) | 2006-10-18 | 2016-11-22 | Yong Zhao | Embryonic-like stem cells derived from adult human peripheral blood and methods of use |
| EP2088190A4 (en) | 2006-11-09 | 2011-01-05 | Japan Government | METHOD FOR THE CULTURE AND PASSAGE OF PRIMATE EMBRYONIC STRAIN CELL, AND METHOD FOR INDUCING DIFFERENTIATION OF EMBRYONIC STEM CELL |
| US8217027B2 (en) | 2006-12-21 | 2012-07-10 | Abbott Laboratories | Sphingosine-1-phosphate receptor agonist and antagonist compounds |
| WO2008086005A1 (en) | 2007-01-09 | 2008-07-17 | University Of South Florida | Compositions including triciribine and bortezomib and derivatives thereof and methods of use thereof |
| WO2008094597A2 (en) | 2007-01-30 | 2008-08-07 | University Of Georgia Research Foundation, Inc. | Early mesoderm cells, a stable population of mesendoderm cells that has utility for generation of endoderm and mesoderm lineages and multipotent migratory cells (mmc) |
| GB0703188D0 (en) | 2007-02-19 | 2007-03-28 | Roger Land Building | Large scale production of stem cells |
| US20090053182A1 (en) | 2007-05-25 | 2009-02-26 | Medistem Laboratories, Inc. | Endometrial stem cells and methods of making and using same |
| JP5991796B2 (ja) | 2007-06-29 | 2016-09-14 | セルラー ダイナミクス インターナショナル, インコーポレイテッド | 胚性幹細胞培養のための自動化された方法および装置 |
| EP3192865B1 (en) | 2007-07-01 | 2021-04-21 | Janssen Biotech, Inc. | Single pluripotent stem cell culture |
| BRPI0814425A2 (pt) * | 2007-07-18 | 2014-10-21 | Lifescan Inc | Diferenciação de células-tronco embrionárias humanas |
| KR101617243B1 (ko) * | 2007-07-31 | 2016-05-02 | 라이프스캔, 인코포레이티드 | 인간 배아 줄기 세포의 분화 |
| ES2665434T3 (es) | 2007-07-31 | 2018-04-25 | Lifescan, Inc. | Diferenciación de células madre pluripotentes usando células alimentadoras humanas |
| KR101544498B1 (ko) | 2007-08-24 | 2015-08-17 | 스티칭 허트 네덜란드 칸커 인스티튜트 | 종양성 질환의 치료를 위한 조성물 |
| WO2009061442A1 (en) | 2007-11-06 | 2009-05-14 | Children's Medical Center Corporation | Method to produce induced pluripotent stem (ips) cells form non-embryonic human cells |
| EP2229434B1 (en) * | 2007-11-27 | 2011-09-07 | Lifescan, Inc. | Differentiation of human embryonic stem cells |
| SG154367A1 (en) | 2008-01-31 | 2009-08-28 | Es Cell Int Pte Ltd | Method of differentiating stem cells |
| WO2009096049A1 (ja) | 2008-02-01 | 2009-08-06 | Kyoto University | 人工多能性幹細胞由来分化細胞 |
| EP2250252A2 (en) | 2008-02-11 | 2010-11-17 | Cambridge Enterprise Limited | Improved reprogramming of mammalian cells, and the cells obtained |
| CN102046779A (zh) | 2008-02-21 | 2011-05-04 | 森托科尔奥索生物科技公司 | 用于细胞粘附、培养和分离的方法、表面改性培养板和组合物 |
| JPWO2009110215A1 (ja) | 2008-03-03 | 2011-07-14 | 独立行政法人科学技術振興機構 | 繊毛細胞の分化誘導方法 |
| SG188918A1 (zh) | 2008-03-17 | 2013-04-30 | Agency Science Tech & Res | |
| RU2359030C1 (ru) | 2008-03-19 | 2009-06-20 | Общество С Ограниченной Ответственностью "Лаборатория Клеточных Технологий" | Способ получения эндотелиальных клеток из эмбриональных стволовых клеток человека (варианты) |
| WO2009131568A1 (en) | 2008-04-21 | 2009-10-29 | Cythera, Inc. | Methods for purifying endoderm and pancreatic endoderm cells derived from human embryonic stem cells |
| US8338170B2 (en) | 2008-04-21 | 2012-12-25 | Viacyte, Inc. | Methods for purifying endoderm and pancreatic endoderm cells derived from human embryonic stem cells |
| WO2009132083A2 (en) | 2008-04-22 | 2009-10-29 | President And Fellows Of Harvard College | Compositions and methods for promoting the generation of pdx1+ pancreatic cells |
| US8623648B2 (en) | 2008-04-24 | 2014-01-07 | Janssen Biotech, Inc. | Treatment of pluripotent cells |
| US7939322B2 (en) | 2008-04-24 | 2011-05-10 | Centocor Ortho Biotech Inc. | Cells expressing pluripotency markers and expressing markers characteristic of the definitive endoderm |
| EP2993226B1 (en) | 2008-06-03 | 2020-12-16 | Viacyte, Inc. | Growth factors for production of definitive endoderm |
| US20090298178A1 (en) | 2008-06-03 | 2009-12-03 | D Amour Kevin Allen | Growth factors for production of definitive endoderm |
| DE102008032236A1 (de) | 2008-06-30 | 2010-04-01 | Eberhard-Karls-Universität Tübingen | Isolierung und/oder Identifizierung von Stammzellen mit adipozytärem, chondrozytärem und pankreatischem Differenzierungspotential |
| RU2011103183A (ru) | 2008-06-30 | 2012-08-10 | Сентокор Орто Байотек Инк. (Us) | Дифференцирование плюрипотентных стволовых клеток |
| PL2310492T3 (pl) | 2008-06-30 | 2015-12-31 | Janssen Biotech Inc | Różnocowanie pluripotencjalnych komórek macierzystych |
| US20100028307A1 (en) | 2008-07-31 | 2010-02-04 | O'neil John J | Pluripotent stem cell differentiation |
| US9683215B2 (en) | 2008-08-22 | 2017-06-20 | President And Fellows Of Harvard College | Methods of reprogramming cells |
| KR102025158B1 (ko) | 2008-10-31 | 2019-09-25 | 얀센 바이오테크 인코포레이티드 | 인간 배아 줄기 세포의 췌장 내분비 계통으로의 분화 |
| AU2009308967C1 (en) | 2008-10-31 | 2017-04-20 | Janssen Biotech, Inc. | Differentiation of human embryonic stem cells to the pancreatic endocrine lineage |
| CN102361970B (zh) | 2008-11-04 | 2018-03-27 | 韦尔赛特公司 | 干细胞聚集体悬浮组合物及其分化方法 |
| US8008075B2 (en) | 2008-11-04 | 2011-08-30 | Viacyte, Inc. | Stem cell aggregate suspension compositions and methods of differentiation thereof |
| EP4176888A1 (en) | 2008-11-14 | 2023-05-10 | ViaCyte, Inc. | Encapsulation of pancreatic cells derived from human pluripotent stem cells |
| MX356756B (es) | 2008-11-20 | 2018-06-11 | Centocor Ortho Biotech Inc | Células madre pluripotentes en microportadores. |
| WO2010063848A1 (en) | 2008-12-05 | 2010-06-10 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and medium for neural differentiation of pluripotent cells |
| SG177416A1 (en) | 2009-07-20 | 2012-02-28 | Janssen Biotech Inc | Differentiation of human embryonic stem cells |
| AU2010276440B2 (en) | 2009-07-20 | 2014-07-03 | Janssen Biotech Inc. | Differentiation of human embryonic stem cells |
| EP2456862A4 (en) | 2009-07-20 | 2013-02-27 | Janssen Biotech Inc | DIFFERENTIATION OF HUMAN EMBRYONIC STEM CELLS |
| EP2464723B1 (en) | 2009-08-12 | 2016-06-22 | Kyoto University | Method for inducing differentiation of pluripotent stem cells into neural precursor cells |
| CN107058389A (zh) | 2009-10-29 | 2017-08-18 | 詹森生物科技公司 | 多能干细胞 |
| ES2779048T3 (es) | 2009-11-12 | 2020-08-13 | Technion Res & Dev Foundation | Medios de cultivo, cultivos celulares y métodos de cultivo de células madre pluripotentes en un estado indiferenciado |
| FI20096288A0 (fi) | 2009-12-04 | 2009-12-04 | Kristiina Rajala | Formulations and methods for culturing stem cells |
| CN102712902B (zh) | 2009-12-23 | 2019-01-08 | 詹森生物科技公司 | 人胚胎干细胞的分化 |
| US8932853B2 (en) | 2009-12-29 | 2015-01-13 | Takeda Pharmaceutical Company Limited | Method for manufacturing pancreatic-hormone-producing cells |
| CN102858958A (zh) | 2010-02-03 | 2013-01-02 | 日本国立癌症研究中心 | 诱导肝干细胞及其制造方法、以及该细胞的用途 |
| CA2791846A1 (en) | 2010-03-02 | 2011-09-09 | National University Of Singapore | Culture additives to boost stem cell proliferation and differentiation response |
| JP5909482B2 (ja) | 2010-03-31 | 2016-04-26 | ザ スクリプス リサーチ インスティテュート | 細胞の再プログラム |
| EP2563908B1 (en) | 2010-04-25 | 2019-01-09 | Icahn School of Medicine at Mount Sinai | Generation of anterior foregut endoderm from pluripotent cells |
| US8932857B2 (en) | 2010-06-15 | 2015-01-13 | Kyoto University | Method for selecting reduced differentiation resistance human induced pluripotent stem cells |
| US9085757B2 (en) | 2010-06-17 | 2015-07-21 | Regents Of The University Of Minnesota | Production of insulin producing cells |
| CA2807418C (en) | 2010-08-05 | 2017-04-04 | Wisconsin Alumni Research Foundation | Simplified basic media for human pluripotent cell culture |
| MY177150A (en) | 2011-02-28 | 2020-09-08 | Stempeutics Res Malaysia Sdn Bhd | Isolation and expansion of adult stem cells, their therapeutic composition and uses thereof |
| US20130274184A1 (en) | 2011-10-11 | 2013-10-17 | The Trustees Of Columbia University In The City Of New York | Er stress relievers in beta cell protection |
| WO2013056072A1 (en) | 2011-10-13 | 2013-04-18 | Wisconsin Alumni Research Foundation | Generation of cardiomyocytes from human pluripotent stem cells |
| EP2766474B1 (en) | 2011-10-14 | 2020-10-07 | Children's Medical Center Corporation | Inhibition and enhancement of reprogramming by chromatin modifying enzymes |
| KR102203056B1 (ko) | 2011-12-22 | 2021-01-14 | 얀센 바이오테크 인코포레이티드 | 인간 배아 줄기 세포의 단일 인슐린 호르몬 양성 세포로의 분화 |
| US10519422B2 (en) | 2012-02-29 | 2019-12-31 | Riken | Method of producing human retinal pigment epithelial cells |
| CN108034633B (zh) * | 2012-06-08 | 2022-08-02 | 詹森生物科技公司 | 人胚胎干细胞向胰腺内分泌细胞的分化 |
| US20150247123A1 (en) | 2012-09-03 | 2015-09-03 | Novo Nordisk A/S | Generation of pancreatic endoderm from Pluripotent Stem cells using small molecules |
| AU2013368224B2 (en) | 2012-12-31 | 2018-09-27 | Janssen Biotech, Inc. | Differentiation of human embryonic stem cells into pancreatic endocrine cells using HB9 regulators |
| US8987471B2 (en) | 2013-02-14 | 2015-03-24 | Allergan, Inc. | Substituted dihydropyrazoles as sphingosine receptor modulators |
| US8859286B2 (en) | 2013-03-14 | 2014-10-14 | Viacyte, Inc. | In vitro differentiation of pluripotent stem cells to pancreatic endoderm cells (PEC) and endocrine cells |
| EP2970892A1 (en) * | 2013-03-15 | 2016-01-20 | The Jackson Laboratory | Isolation of non-embryonic stem cells and uses thereof |
-
2013
- 2013-12-18 EP EP13869259.5A patent/EP2938724B1/en active Active
- 2013-12-18 KR KR1020157020562A patent/KR102084561B1/ko active IP Right Grant
- 2013-12-18 RU RU2018116648A patent/RU2768963C2/ru active
- 2013-12-18 RU RU2018116647A patent/RU2018116647A/ru unknown
- 2013-12-18 SG SG10201709338RA patent/SG10201709338RA/en unknown
- 2013-12-18 ES ES13869259T patent/ES2837763T3/es active Active
- 2013-12-18 SG SG11201505128SA patent/SG11201505128SA/en unknown
- 2013-12-18 US US13/998,884 patent/US10344264B2/en active Active
- 2013-12-18 RU RU2015131835A patent/RU2658488C2/ru active
- 2013-12-18 WO PCT/US2013/075939 patent/WO2014105543A1/en active Application Filing
- 2013-12-18 MX MX2015008577A patent/MX2015008577A/es unknown
- 2013-12-18 BR BR112015015770A patent/BR112015015770A2/pt not_active Application Discontinuation
- 2013-12-18 DK DK13869259.5T patent/DK2938724T3/da active
- 2013-12-18 CA CA2896655A patent/CA2896655C/en active Active
- 2013-12-18 JP JP2015550478A patent/JP6557146B2/ja active Active
- 2013-12-18 CN CN201380074040.4A patent/CN105008518B/zh active Active
- 2013-12-18 AU AU2013368221A patent/AU2013368221B2/en active Active
-
2014
- 2014-01-03 AR ARP140100011A patent/AR094347A1/es unknown
-
2015
- 2015-06-25 PH PH12015501465A patent/PH12015501465A1/en unknown
-
2016
- 2016-05-04 HK HK16105105.3A patent/HK1217110A1/zh unknown
-
2018
- 2018-12-14 AU AU2018279009A patent/AU2018279009B2/en active Active
-
2021
- 2021-10-09 AU AU2021245259A patent/AU2021245259B2/en active Active
-
2023
- 2023-12-19 AU AU2023285724A patent/AU2023285724A1/en not_active Abandoned
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090004152A1 (en) * | 2006-03-02 | 2009-01-01 | Cythera, Inc. | Methods of producing pancreatic hormones |
| US20120264209A1 (en) * | 2006-05-02 | 2012-10-18 | Jon Odorico | Methods and devices for differentiating pluripotent stem cells into cells of the pancreatic lineage |
| CN102741395A (zh) * | 2009-12-23 | 2012-10-17 | 森托科尔奥索生物科技公司 | 人胚胎干细胞的分化 |
| US20110281355A1 (en) * | 2010-05-12 | 2011-11-17 | Centocor Ortho Biotech Inc. | Differentiation of Human Embryonic Stem Cells |
| WO2012030540A2 (en) * | 2010-08-31 | 2012-03-08 | Janssen Biotech, Inc. | Differentiation of pluripotent stem cells |
Non-Patent Citations (1)
| Title |
|---|
| Maturation of Human Embryonic Stem Cell–Derived Pancreatic Progenitors Into Functional Islets Capable of Treating Pre-existing Diabetes in Mice;Alireza Rezania et al;《DIABETES》;20120831;第61卷;全文 * |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN105008518B (zh) | 在空气-液体界面处培养人胚胎干细胞以用于分化成胰腺内分泌细胞 | |
| JP7278990B2 (ja) | Hb9調節物を用いたヒト胚性幹細胞の膵臓内分泌細胞への分化 | |
| US11890304B2 (en) | Pancreatic endocrine cells and methods thereof | |
| KR102607813B1 (ko) | 글루코스-의존성 미토콘드리아 호흡 및 2-상 인슐린 분비 반응을 나타내는 인간 만능 줄기 세포 유래 기능성 베타 세포의 생성 | |
| KR20170002630A (ko) | 췌장 내분비 세포에서 mafa 발현을 향상시키기 위한 소분자의 용도 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |