WO2011078298A1 - ポリアクリル酸系吸水性樹脂粉末及びその製造方法 - Google Patents
ポリアクリル酸系吸水性樹脂粉末及びその製造方法 Download PDFInfo
- Publication number
- WO2011078298A1 WO2011078298A1 PCT/JP2010/073254 JP2010073254W WO2011078298A1 WO 2011078298 A1 WO2011078298 A1 WO 2011078298A1 JP 2010073254 W JP2010073254 W JP 2010073254W WO 2011078298 A1 WO2011078298 A1 WO 2011078298A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- water
- resin powder
- absorbent resin
- polymerization
- aqueous solution
- Prior art date
Links
Images











Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/12—Powdering or granulating
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L15/00—Chemical aspects of, or use of materials for, bandages, dressings or absorbent pads
- A61L15/16—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons
- A61L15/42—Use of materials characterised by their function or physical properties
- A61L15/48—Surfactants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L15/00—Chemical aspects of, or use of materials for, bandages, dressings or absorbent pads
- A61L15/16—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons
- A61L15/42—Use of materials characterised by their function or physical properties
- A61L15/60—Liquid-swellable gel-forming materials, e.g. super-absorbents
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/26—Synthetic macromolecular compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/001—Multistage polymerisation processes characterised by a change in reactor conditions without deactivating the intermediate polymer
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/12—Polymerisation in non-solvents
- C08F2/16—Aqueous medium
- C08F2/22—Emulsion polymerisation
- C08F2/24—Emulsion polymerisation with the aid of emulsifying agents
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/12—Polymerisation in non-solvents
- C08F2/16—Aqueous medium
- C08F2/22—Emulsion polymerisation
- C08F2/24—Emulsion polymerisation with the aid of emulsifying agents
- C08F2/30—Emulsion polymerisation with the aid of emulsifying agents non-ionic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F20/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride, ester, amide, imide or nitrile thereof
- C08F20/02—Monocarboxylic acids having less than ten carbon atoms, Derivatives thereof
- C08F20/04—Acids, Metal salts or ammonium salts thereof
- C08F20/06—Acrylic acid; Methacrylic acid; Metal salts or ammonium salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/04—Acids; Metal salts or ammonium salts thereof
- C08F220/06—Acrylic acid; Methacrylic acid; Metal salts or ammonium salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F6/00—Post-polymerisation treatments
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F6/00—Post-polymerisation treatments
- C08F6/26—Treatment of polymers prepared in bulk also solid polymers or polymer melts
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/24—Crosslinking, e.g. vulcanising, of macromolecules
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2810/00—Chemical modification of a polymer
- C08F2810/20—Chemical modification of a polymer leading to a crosslinking, either explicitly or inherently
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2333/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers
- C08J2333/04—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers esters
- C08J2333/06—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers esters of esters containing only carbon, hydrogen, and oxygen, the oxygen atom being present only as part of the carboxyl radical
- C08J2333/08—Homopolymers or copolymers of acrylic acid esters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2333/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers
- C08J2333/04—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers esters
- C08J2333/06—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers esters of esters containing only carbon, hydrogen, and oxygen, the oxygen atom being present only as part of the carboxyl radical
- C08J2333/10—Homopolymers or copolymers of methacrylic acid esters
Definitions
- the present invention relates to a polyacrylic acid-based water absorbent resin powder and a method for producing the same. More particularly, the present invention relates to a water-absorbing resin powder used for sanitary goods such as paper diapers and sanitary napkins, and has excellent water absorption performance (especially high water absorption speed). It is about.
- Water-absorbing resin (SAP / Super Absorbent Polymer) is a water-swellable, water-insoluble polymer gelling agent, absorbent articles such as paper diapers and sanitary napkins, water retaining agents for agriculture and horticulture, and industrial waterstops. As a material etc., it is frequently used mainly for disposable use.
- a water-absorbing resin many monomers and hydrophilic polymers have been proposed as raw materials, and in particular, a polyacrylic acid-based water-absorbing material using acrylic acid and / or a salt thereof as a monomer. Resins are most commonly used industrially due to their high water absorption performance (Non-patent Document 1).
- Such a water-absorbent resin is produced through a polymerization process, a drying process, if necessary, a removal process of undried material, a pulverization process, a classification process, a surface crosslinking process, and the like (Patent Documents 1 to 5).
- water-absorbing resins are also required to have many functions. Specifically, the gel strength, water-soluble content, water absorption speed, water absorption capacity under pressure, liquid permeability, particle size distribution, urine resistance, antibacterial properties, impact resistance (resistance to resistance) Many physical properties such as damage property), powder flowability, deodorant property, coloring resistance (whiteness), and low dust are required for the water-absorbent resin. For this reason, many proposals have been made in the above and below patent documents, such as many surface cross-linking techniques, additives, and manufacturing process changes.
- liquid permeability has been seen as a more important factor as the amount of water-absorbent resin used in paper diapers increases in recent years (for example, 50% by weight or more).
- improvement methods and improvement techniques for under-load liquid permeability and under-load liquid permeability such as SFC (Saline Flow Conductivity / Patent Document 6) and GBP (Gel Bed Permeability / Patent Documents 7 to 9) have been proposed. Yes.
- the water absorption rate is also an important basic physical property of the water-absorbent resin, and as a method for improving the water absorption rate, a technique for improving the water absorption rate by increasing the specific surface area is known.
- a technique for finely controlling the particle diameter Patent Document 10
- a technique for granulating fine particles having a large surface area Patent Documents 11 to 13
- a technique for freeze-drying a hydrous gel to make it porous Patent Document 14
- techniques for surface cross-linking of particles simultaneously with granulation Patent Documents 15 to 17
- techniques for foam polymerization Patent Documents 18 to 35
- techniques for foaming and cross-linking after polymerization Patent Document 36
- Patent Documents 10 to 36 the water absorption speed is improved to some extent by increasing the surface area of the water absorbent resin, but it still does not show a sufficient effect, and a special apparatus or an expensive raw material (a large amount of surfactant or Foaming agent) was required. Furthermore, problems such as deterioration of other physical properties such as liquid permeability (Patent Documents 6 to 9), impact resistance (Patent Document 37), bulk specific gravity (Patent Documents 38 and 39) and the like, which are required in recent years. Had.
- the water-absorbing resin is used in absorbent articles such as paper diapers and sanitary napkins, and is often combined with white pulp.
- Many color improvement techniques have been proposed for the whiteness of patents (Patent Documents 40 to 42).
- Patent Documents 40 to 42 Many color improvement techniques have been proposed for the whiteness of patents (Patent Documents 40 to 42).
- the present situation is that these are not sufficient in terms of the cost and safety of the coloring inhibitor, the complexity of the process, and the effect.
- the problem to be solved by the present invention is to maintain the other physical properties of the water-absorbent resin (liquid permeability, bulk specific gravity, surface tension, water absorption capacity under pressure, impact resistance, etc.) or more expensive raw materials. It is to provide a white water-absorbent resin powder having an improved water absorption speed (for example, FSR) without using any device. In particular, it is to provide a white water-absorbent resin powder having both high liquid permeability (for example, SFC) and water absorption speed (for example, FSR).
- SFC high liquid permeability
- FSR water absorption speed
- the present inventor paid attention to a method of dispersing bubbles in monomers during polymerization, and solved the above problems by using a specific method, and reduced bulk specific gravity and surface tension. Further, it was found that a water-absorbing resin powder excellent in whiteness, liquid permeability, and impact resistance (also known as damage resistance) was obtained, and the present invention was completed.
- a water-absorbing resin powder excellent in whiteness, liquid permeability, and impact resistance also known as damage resistance
- the present invention was completed.
- techniques for increasing the surface area, foaming polymerization, and making porous to improve the water absorption rate have been proposed in the above-mentioned patent documents, but the present invention is the first to use closed cells (also known as internal bubbles) that have not been focused on. Attention has been paid to find out that the above problems can be solved by controlling the internal cell ratio (Closed-Cell Rate) of the water-absorbent resin within a specific range, and the present invention has been completed.
- the method for producing a water-absorbent resin of the present invention (first method) is obtained by polymerizing an aqueous acrylic acid monomer solution containing bubbles and the polymerization step.
- a method for producing a polyacrylic acid-based water-absorbent resin powder comprising a step of drying a hydrogel crosslinked polymer, wherein the solubility of dissolved gas in an aqueous monomer solution in the presence of a surfactant and / or a dispersant It is characterized by including a bubble generation and containing step of generating and containing bubbles in the aqueous acrylic acid monomer solution by reducing.
- the method for producing a water-absorbent resin of the present invention (second method) is obtained by polymerizing an aqueous acrylic acid monomer solution containing bubbles and the polymerization step.
- a method for producing a polyacrylic acid-based water-absorbing resin powder comprising a step of drying a hydrogel crosslinked polymer, wherein the temperature of the acrylic acid-based monomer aqueous solution is increased in the presence of a surfactant and / or a dispersant. It is characterized by including the process to make.
- the manufacturing method (3rd method) of the water absorbing resin of this invention is obtained by the process of superposing
- a method for producing a polyacrylic acid-based water-absorbent resin powder comprising a step of drying a water-containing gel-like cross-linked polymer, which is water-soluble in an acrylic acid-based monomer aqueous solution in the presence of a surfactant and / or a dispersant. It includes a step of mixing organic substances.
- the water absorbent resin of the present invention has a particle size of 850 to 150 ⁇ m with a ratio of 95 weight.
- % of polyacrylic acid-based water-absorbent resin powder characterized in that the internal cell ratio defined by the following formula is 2.8 to 6.6%.
- the water-absorbent resin (second water-absorbent resin) of the present invention is a polyacrylic acid-based water-absorbent containing a surfactant and / or a dispersant therein.
- the resin powder is characterized in that the surface tension is 60 [mN / m] or more and the powder surface is coated with a surfactant.
- the method for producing a water-absorbent resin powder according to the present invention a water-absorbent resin powder having a high water absorption rate can be produced efficiently and efficiently without using a large amount of a surfactant.
- the water-absorbent resin powder of the present invention obtained by the production method of the present invention is a novel water-absorbent resin that has both water absorption speed and liquid permeability and excellent impact resistance. It is a powder.
- FIG. 1 is a perspective view showing an example of an apparatus used in a continuous temperature raising method by heating an acrylic acid monomer aqueous solution to which the first method and the second method of the present invention are applied.
- FIG. 2 is a flow diagram showing an outline of a method of raising temperature and generating bubbles by neutralization of an acrylic acid monomer aqueous solution to which the first method and the second method of the present invention are applied.
- FIG. 3 is an electron micrograph (50 times magnification) of the water-absorbent resin powder obtained in Example 2 and Comparative Example 2. In Examples and Comparative Examples, which will be described later, after the polymerization and drying, the water-absorbent resin powder is in an irregularly crushed shape as shown in the photograph.
- FIG. 1 is a perspective view showing an example of an apparatus used in a continuous temperature raising method by heating an acrylic acid monomer aqueous solution to which the first method and the second method of the present invention are applied.
- FIG. 2 is a flow diagram showing an outline of a method of
- FIG. 4 is a flowchart showing an outline of a method for raising temperature and generating bubbles by neutralization of an aqueous acrylic acid monomer solution in the presence of a surfactant in Example 2.
- FIG. 5 is a flowchart showing an outline of a temperature raising method by neutralization of an acrylic acid monomer aqueous solution in Comparative Example 3.
- FIG. 6 shows a preferred embodiment of the present invention that can be applied to the first method and the second method of the present invention.
- an inert gas for example, nitrogen
- FIG. 7 shows a preferred embodiment of the present invention that can be applied to the first method and the second method of the present invention.
- an inert gas for example, nitrogen
- FIG. 8 shows a preferred embodiment of the present invention that can be applied to the first method and the second method of the present invention.
- an inert gas for example, nitrogen It is a flowchart which shows the outline of the embodiment which deoxygenates before superposition
- FIG. 9 shows a preferred embodiment of the present invention that can be applied to the first method and the second method of the present invention.
- an inert gas for example, nitrogen It is a flowchart which shows the outline of the embodiment which deoxygenates before superposition
- an inert gas for example, nitrogen It is a flowchart which shows the outline of the embodiment which deoxygenates before superposition
- FIG. 10 is a flowchart showing an outline of gas solubility reduction and bubble generation by mixing a water-soluble organic compound in an acrylic acid monomer aqueous solution, which can be applied to the first method and the third method of the present invention. is there.
- FIG. 11 is a flowchart showing an outline of gas solubility reduction and bubble generation by mixing a water-soluble organic compound in an acrylic acid monomer aqueous solution, which can be applied to the first method and the third method of the present invention. is there.
- FIG. 12 is a cross-sectional view schematically showing closed cells (Opened Cell) and open cells (Closed Cell) in the water absorbent resin powder.
- the water-absorbent resin (described later) of the present invention has a characteristic of having an internal cell ratio (also known as closed cells) controlled within a specific range.
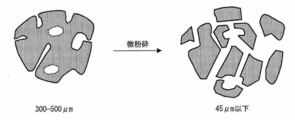
- FIG. 13 is a cross-sectional view schematically showing an operation for finely pulverizing a water-absorbent resin powder (for example, the ratio of the particle size of 850 to 150 ⁇ m is 95% by weight or more) for measuring the true density of the present invention to less than 45 ⁇ m. is there.
- the true density [g / cm 3 ] of the water-absorbent resin powder is measured by performing dry density measurement with helium gas after the closed cells are substantially broken or formed into open cells by finely pulverizing the water-absorbent resin powder. it can.
- FIG. 14 shows a preferred embodiment of the present invention that can be applied to the first method and the second method of the present invention.
- a hydrated gel-like cross-linked polymer is charged into a screw extruder using a belt polymerizer to obtain a crushed gel. It is sectional drawing which shows the outline of the process to obtain.
- FIG. 15 shows a preferred embodiment of the present invention that can be applied to the first method and the second method of the present invention.
- an inert gas for example, nitrogen
- the present invention is not limited to the following embodiments, and various modifications are possible within the scope of the claims, and technical means disclosed in different embodiments are appropriately combined. Embodiments obtained in this manner are also included in the technical scope of the present invention.
- Water absorbent resin powder The “water-absorbent resin powder” in the present invention means a water-swellable water-insoluble polymer gelling agent. “Water swellability” means that the CRC (water absorption capacity under no pressure) specified by ERT441.2-02 is 5 [g / g] or more, and “water-insoluble” means ERT470. Ext (water soluble content) specified in 2-02 is 0 to 50% by weight.
- the water-absorbent resin powder can be designed as appropriate according to its use, and is not particularly limited, and is not limited to a form in which the total amount (100% by weight) is a polymer, and maintains the above performance. Within the range, an additive or the like may be contained, and the water absorbent resin composition containing a small amount of the additive is also collectively referred to as a water absorbent resin powder in the present invention. Moreover, the presence or absence of surface cross-linking does not matter.
- the shape of the water-absorbing resin include a sheet shape, a fiber shape, a film shape, a gel shape, and the like, preferably a powder shape, and particularly preferably a powdered water-absorbing resin having a particle size and moisture content described below.
- the “polyacrylic acid-based water-absorbent resin powder” in the present invention optionally contains a graft component, and contains, as a repeating unit, acrylic acid and / or a salt thereof (hereinafter referred to as acrylic acid (salt)) as a main component. It means a polymer.
- polyacrylate type (neutralization type) polymers are also collectively referred to as polyacrylic acid.
- CRC is an abbreviation for Centrifugation Retention Capacity (centrifuge retention capacity), and means water absorption capacity without pressure (hereinafter also referred to as “water absorption capacity”). Specifically, the water absorption capacity (unit: [g / g] after 30 minutes of free swelling of 0.200 g of the water-absorbing resin in the nonwoven fabric with respect to a 0.9 wt% sodium chloride aqueous solution and further draining with a centrifuge ).
- AAP is an abbreviation for Absorption against Pressure, which means water absorption capacity under pressure. Specifically, it is the water absorption capacity (unit: [g / g]) after swelling under a load of 2.06 kPa for 1 hour with respect to 0.900 g of a 0.9 wt% aqueous sodium chloride solution.
- ERT442.2-02 “Absorption Under Pressure” is described, but the contents are substantially the same. Further, the load condition may be changed to 4.83 kPa (0.7 psi) for measurement.
- Ext is an abbreviation for Extractables and means a water-soluble component (water-soluble component amount). Specifically, it is a value (unit:% by weight) measured by pH titration of 1 g of water absorbent resin powder for 16 hours with respect to 200 g of 0.9 wt% sodium chloride aqueous solution for 16 hours.
- PSD is an abbreviation for Particle Size Distribution and means a particle size distribution measured by sieve classification.
- the weight average particle size (D50) and the particle size distribution width are measured by the same method as “(1) Average Particle Diameter and Distribution of Particle Diameter” described in US Pat. No. 2006-204755.
- liquid permeability The flow of liquid flowing between particles of the swollen water-absorbent resin powder under load or no load is referred to as “liquid permeability”.
- Typical measurement methods for this “liquid permeability” include SFC (Saline Flow Conductivity / Saline Flow Inductivity) and GBP (Gel Bed Permeability / Gel Bed Permeability).
- SFC saline flow inductivity
- GBP liquid permeability of a 0.69 wt% sodium chloride aqueous solution to the water absorbent resin powder under load or free expansion. It is measured according to the GBP test method described in International Publication No. 2005/016393 pamphlet.
- X to Y indicating a range means “X or more and Y or less”.
- t (ton) which is a unit of weight means “Metric ton”
- ppm means “ppm by weight” unless otherwise noted.
- weight and “mass”, “wt%” and “mass%”, “part by weight” and “part by mass” are treated as synonyms.
- ⁇ acid (salt) means “ ⁇ acid and / or salt thereof”
- (meth) acryl means “acryl and / or methacryl”.
- the manufacturing method of the water-absorbing resin powder according to the present invention is a method of foaming in the method of foam polymerization for improving the water absorption speed. Has characteristics.
- air bubbles are uniformly dispersed in the monomer aqueous solution before polymerization, not only the water absorption speed of the resulting water absorbent resin powder is improved, but also the whiteness is improved.
- other expensive physical properties and equipment can be maintained without maintaining or almost damaging other physical properties (liquid permeability, bulk specific gravity, surface tension, water absorption capacity under pressure, impact resistance (damage resistance), etc.) of the water absorbent resin powder. There is no need to use it.
- the production method (first method) of the present invention comprises a polymerization step of an aqueous acrylic acid monomer solution containing air bubbles, and if necessary, gel refinement of a hydrogel crosslinked polymer during or after polymerization.
- the method for producing a polyacrylic acid-based water-absorbent resin powder comprising a step and a drying step of a hydrogel crosslinked polymer, in the presence of a surfactant and / or a dispersant, It includes a bubble generation and containing step of generating and containing bubbles by lowering the solubility of dissolved gas.
- the surfactant may be added before the bubble generation-containing step, it may be performed after the polymerization step, but is preferably performed before the polymerization step.
- the aqueous acrylic acid monomer solution during or after adjustment before the polymerization step contains a surfactant and / or a dispersant, and the aqueous acrylic acid monomer solution
- a method for reducing the solubility of the dissolved gas for example, a method of increasing the temperature of an acrylic acid-based monomer aqueous solution and / or a method of mixing a water-soluble organic substance in an acrylic acid-based monomer aqueous solution. Is mentioned.
- the bubble generation and containing step may be performed after the polymerization step is started because it may be performed until the polymerization step is completed, but it is preferably performed before the polymerization step.
- the method for producing the water-absorbent resin powder of the present invention preferably comprises a step of polymerizing an aqueous acrylic acid monomer solution containing bubbles and, if necessary, a gel of a hydrogel crosslinked polymer after or after polymerization.
- a method for producing a polyacrylic acid-based water-absorbent resin powder comprising a fine graining step and a drying step of a hydrogel crosslinked polymer, wherein an acrylic acid monomer aqueous solution during or after adjustment before the polymerization step Contains a surfactant and / or a dispersant, and includes a step of raising the temperature of the aqueous acrylic acid monomer solution before the polymerization step, and / or a polymerization step A step of mixing a water-soluble organic substance into the acrylic acid monomer aqueous solution before the polymerization step, wherein the acrylic acid monomer aqueous solution before or after adjustment contains a surfactant and / or a dispersant. Including a manufacturing method (third manufacturing method).
- the water-soluble organic substance is acrylic acid, particularly unneutralized acrylic acid, and its water content is preferably 20% by weight or less, more preferably 2% by weight or less, and still more preferably 0. .5% by weight or less.
- the mixing ratio of the aqueous acrylic acid monomer solution containing the surfactant and / or dispersant and the water-soluble organic substance is appropriately determined, and is preferably 1: 9 to 9: 1 by weight, and preferably 2: 8 to 8: 2 is more preferable, and 3: 7 to 7: 3 is particularly preferable.
- the step of reducing the solubility of dissolved gas to generate bubbles, the step of raising the temperature of the acrylic acid monomer aqueous solution, and the step of mixing the water-soluble organic matter are under pressure and under reduced pressure, respectively.
- the atmosphere is substantially atmospheric pressure because of the simplicity of operation, processes and equipment.
- substantially normal pressure means a state of ⁇ 10% with respect to atmospheric pressure, preferably ⁇ 5%, more preferably ⁇ 2%, still more preferably ⁇ 1%, particularly preferably 0. % (Same as atmospheric pressure).
- the range is substantially 0%.
- bubbles are contained by these methods, but preferably, before or after the step of reducing the solubility of the dissolved gas, a step of mixing the inert gas into the acrylic acid monomer aqueous solution is performed. Including.
- the time from the end of the step of reducing the solubility of dissolved gas (the step of generating bubbles) to the start of polymerization in the polymerization step exceeds 0 due to the stability of the generated bubbles. It is preferably within 300 seconds, more preferably within 120 seconds, still more preferably within 60 seconds, and particularly preferably within 30 seconds.
- the above time exceeds 300 seconds, the generated bubbles are united or disappeared before polymerization, and the effect of improving the water absorption rate tends to be small, which is not preferable.
- the “acrylic acid monomer aqueous solution” is an aqueous solution of a monomer mainly composed of acrylic acid and / or a salt thereof described later, and if necessary, a crosslinking agent or other graft component described later, This refers to a composition in which constituents of a water-absorbent resin such as components (chelating agent, surfactant, dispersant) and the like are prepared. In this state, a polymerization initiator is added to be used for polymerization.
- the acrylic acid may be unneutralized or may be a salt type (completely neutralized type or partially neutralized type).
- the monomer aqueous solution may exceed the saturation concentration, and so-called supersaturated aqueous solution of acrylic acid (salt) and aqueous slurry solution (aqueous dispersion) are also included in the category of the acrylic acid-based monomer aqueous solution in the present invention.
- an acrylic acid monomer aqueous solution having a saturated concentration or less is preferably used from the viewpoint of the physical properties of the resulting water-absorbent resin powder.
- the monomer polymerization solvent is water, and the acrylic acid monomer is an aqueous solution.
- the aqueous solution is not limited when 100% by weight of the solvent is water, preferably Even if another water-soluble organic solvent (for example, alcohol) is used in combination with 0 to 30% by weight, more preferably 0 to 5% by weight, it is referred to as an aqueous solution in the present invention.
- the solubility of the gas is reduced (first method), preferably the temperature is increased (second method) or the water-soluble organic substance is mixed (third method) with respect to the aqueous acrylic acid solution.
- a basic aqueous solution for example, caustic soda aqueous solution
- an aqueous acrylic acid solution (acrylic acid + NaOH aqueous solution) is generated at the start of mixing, 100% by weight acrylic acid (water
- neutralization is carried out for no acrylic acid or acrylic acid containing a small amount of water (for example, acrylic acid having a water content of 2% by weight or less) is also included in the first to third methods of the present invention. .
- the adjusted acrylic acid-based monomer aqueous solution before the polymerization step is an acrylic acid-based monomer aqueous solution before being introduced into the polymerization apparatus or acrylic acid before being introduced into the polymerization apparatus and starting polymerization. Refers to an aqueous monomer solution.
- the acrylic acid-based monomer aqueous solution under adjustment (also known as adjustment in the present invention) in the present invention is a mixture of all components in the above-mentioned monomer aqueous solution mainly composed of acrylic acid and / or a salt thereof. It refers to an aqueous solution of the previous acrylic acid or a salt thereof, typically an aqueous solution of an acrylic acid aqueous solution, a partially neutralized or completely neutralized acrylate, or the like.
- the final acrylic acid monomer aqueous solution can be further neutralized, mixed with water as a solvent, or mixed with the above-mentioned trace components with respect to the acrylic acid monomer aqueous solution being adjusted. It is said.
- the acrylic acid monomer aqueous solution in the step of reducing the solubility of dissolved gas to generate and contain bubbles, that is, the step of raising the temperature of the acrylic acid monomer aqueous solution, the acrylic acid monomer aqueous solution
- the solubility of the gas decreases as the temperature rises.
- the temperature increase width is preferably + 5 ° C. or more, more preferably +10 to + 100 ° C., still more preferably +20 to + 90 ° C., and particularly preferably +30 to + 80 ° C. from the amount of bubbles generated.
- the temperature of the monomer aqueous solution before the temperature rise is preferably 0 to 60 ° C., more preferably 20 to 50 ° C.
- the temperature of the aqueous monomer solution after the temperature rise (width) after the temperature rise is preferably 40 to 100 ° C., more preferably in the range described later.
- FIG. 2 and FIG. 4 are schematic flow charts showing temperature rise and bubble generation by typical heat of neutralization.
- the heat of neutralization of acrylic acid is 13.9 [kcal / mol] (25 ° C.)
- the specific heat of water is 1 [cal / ° C./g] (25 ° C.)
- the specific heat of acrylic acid is 0.66 [cal / mol].
- ° C / g] 25 ° C
- the temperature of the acrylic acid aqueous solution is preferably raised by the heat of neutralization of the acrylic acid.
- the range of temperature rise can also be predicted from the heat of neutralization and specific heat.
- the temperature when the temperature is raised by the heat of neutralization of acrylic acid 13.9 [kcal / mol] (25 ° C.), it may be appropriately heated or cooled during the neutralization reaction in order to control the range of the temperature rise.
- the reaction system may be insulated from the neutralization reaction.
- the solubility of the gas is reduced by the temperature rise, and bubbles are generated in the aqueous acrylic acid solution. Bubbles generated by such a mechanism (not particularly limiting the present invention) are very fine compared to the conventional foaming methods described in Patent Documents 18 to 35 and the like, and are further increased by a surfactant and / or a dispersant. It is presumed that the problem of the present invention is solved by stabilization.
- thermo raising method other than the temperature raising method utilizing the heat of neutralization, there is a method of raising the temperature by heating an acrylic acid monomer aqueous solution. What is necessary is just to heat.
- FIG. 1 shows an apparatus diagram (schematic diagram) of a continuous temperature raising method by heating an acrylic acid monomer aqueous solution to which the first method and the second method of the present invention can be applied.
- the apparatus shown in FIG. 1 is an apparatus that can be used in one method for generating bubbles by raising the temperature of an aqueous acrylic acid monomer solution.
- FIG. 6 to FIG. 9 are flowcharts showing the outline of the embodiment of the gas solubility decrease and bubble generation included in the embodiment of the present invention due to the temperature rise. These temperature raising methods may be used in combination, or may be other methods.
- the method for producing a water-absorbent resin powder of the present invention provides a water-absorbent resin powder that improves the water absorption rate (for example, FSR) and maintains liquid permeability (for example, SFC). Therefore, it is preferably provided in a method for producing a water-absorbent resin powder excellent in FSR and SFC, which will be described later, and there is no decrease in other physical properties. Although preferable physical properties are in the ranges described later, the water-absorbing resin powder has a saline flow conductivity (SFC) of 20 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ sec ⁇ g ⁇ 1 ] or more and a water absorption rate (FSR).
- SFC saline flow conductivity
- FSR and SFC are those described in “(3-3) SFC (saline flow inductivity)” and “(3-5) FSR (water absorption rate)” described later.
- the acrylic acid-based monomer aqueous solution adjusting step is a step of obtaining an acrylic acid-based monomer aqueous solution in which a gas is dispersed. This will be specifically described below.
- the acrylic acid monomer used in the present invention is not particularly limited as long as acrylic acid is used and can be made into a water absorbent resin powder by polymerization.
- Anionic unsaturated monomers such as sulfonic acid, 2- (meth) acryloylethanesulfonic acid, 2- (meth) acryloylpropanesulfonic acid, 2-hydroxyethyl (meth) acryloyl phosphate and salts thereof; Saturated monomers; phenolic hydroxyl group-containing unsaturated monomers; amide group-containing unsaturated monomers such
- the amount of acrylic acid and / or its salt used is 50 mol% or more, preferably 70 mol% or more, based on the total monomer components (excluding internal cross-linking agents described later). More preferably, it is 80 mol% or more, More preferably, it is 90 mol% or more, Most preferably, it is 95 mol% or more (an upper limit is 100 mol%).
- polyacrylic acid is a concept including polyacrylate (especially monovalent salt).
- the neutralization rate of the monomer or polymer thereof is not particularly limited, but the polymerization gel may be neutralized after polymerization, if necessary. In applications that may come into contact with the human body, such as sanitary products, neutralization after polymerization is not required.
- the neutralization rate is preferably 40 to 90 mol%, more preferably 50 to 80 mol%, and still more preferably 60 to 74 mol%. Also from the viewpoint of solving the problems of the present invention, when the neutralization rate is low, the water absorption rate (for example, FSR) tends to decrease, and when the neutralization rate is high, the reactivity of the surface cross-linking agent is decreased.
- an acid group monomer such as acrylic acid or a polymer thereof may be partially or entirely salt type in terms of water absorption capacity without load (CRC) or water absorption rate (FSR), such as sodium salt, lithium salt, Monovalent salts such as potassium salts, ammonium salts and amines, especially alkali metal salts, sodium salts and / or potassium salts are preferred, and sodium salts are more preferred from the viewpoint of cost and physical properties.
- CRC water absorption capacity without load
- FSR water absorption rate
- Monovalent salts such as potassium salts, ammonium salts and amines, especially alkali metal salts, sodium salts and / or potassium salts are preferred, and sodium salts are more preferred from the viewpoint of cost and physical properties.
- an internal cross-linking agent is used as necessary.
- an internal crosslinking agent a conventionally known internal crosslinking agent can be used. Specifically, for example, N, N′-methylenebis (meth) acrylamide, (poly) ethylene glycol di (meth) acrylate, (poly) propylene glycol di (meth) acrylate, trimethylolpropane tri (meth) acrylate, glycerin Tri (meth) acrylate, glycerin acrylate methacrylate, ethylene oxide modified trimethylolpropane tri (meth) acrylate, pentaerythritol hexa (meth) acrylate, triallyl cyanurate, triallyl isocyanurate, triallyl phosphate, triallylamine, poly (meta ) Allyloxyalkane, (Poly) ethylene glycol diglycidyl ether, glycerol diglycidyl ether,
- the amount of the internal cross-linking agent used can be appropriately determined depending on the desired properties of the water-absorbent resin, but is usually preferably 0.001 to 5 mol% with respect to the total amount of the acrylic acid-based monomer. 0.005 to 2 mol% is more preferable, and 0.01 to 1 mol% is still more preferable.
- the amount of the internal cross-linking agent used is less than 0.001 mol%, the water-soluble content of the resulting water-absorbent resin powder is increased, so that there is a possibility that a sufficient amount of water absorption cannot be secured under pressure.
- the amount of the internal cross-linking agent used exceeds 5 mol%, the cross-linking density becomes high, and the water absorption amount of the resulting water-absorbent resin powder may be insufficient.
- the internal crosslinking agent may be added all at once to the reaction system or may be added in portions.
- Bubble dispersion In the method of dispersing bubbles in an aqueous acrylic acid monomer solution, dissolved gas contained in the aqueous solution is generated in the presence of a surfactant and / or a dispersant, and these are effectively made into fine bubbles (microbubbles). Bubbles or nanobubbles).
- a gas for example, an inert gas
- At least one of the following methods (a) and (b) is used as a method for dispersing bubbles in the acrylic acid-based monomer aqueous solution.
- a method for dispersing bubbles in an acrylic acid monomer aqueous solution the temperature of the acrylic acid monomer aqueous solution prepared by mixing the monomer and / or salt thereof, and an internal cross-linking agent and water as required is increased.
- a method of decreasing the solubility of the gas in the aqueous solution by raising the temperature in the adjustment step of the acrylic monomer aqueous solution.
- the temperature of the heated monomer aqueous solution is preferably a high temperature at which the solubility of the gas is lowered, specifically, 40 ° C. to the boiling point of the aqueous solution, more preferably 50 to 100 ° C., and still more preferably 60 ° C. ⁇ 98 ° C, most preferably 70-95 ° C.
- the time required for the temperature rise is preferably 60 seconds or less, more preferably 30 seconds or less, and even more preferably 10 seconds or less, and it is preferable to rapidly warm the monomer aqueous solution in order to generate as many bubbles as possible.
- the heat of neutralization (13.9 [kcal / mol] (25 ° C.)) in the step of increasing the degree of neutralization of the monomer (neutralization) And the like may be used in one-step neutralization or multiple steps (two-step neutralization).
- neutralization may be performed continuously or in batch. Further, it may be performed in one stage up to a predetermined neutralization rate, or may be performed in multiple stages (for example, two stages).
- Two-step neutralization involves adding a base in two steps, and is shown in FIG. 4, Example 2 and the like.
- the gas may be dissolved or dispersed in advance in the aqueous monomer solution before the temperature increase.
- a method for dispersing bubbles in an aqueous acrylic acid monomer solution when the monomer and / or salt thereof, and if necessary, an internal cross-linking agent and water are mixed, the gas is not dissolved, or Examples include a method for reducing the solubility of gas by mixing water-soluble organic substances that are hardly dissolved, aqueous acrylic acid monomers to be mixed, or water-soluble organic substances that do not dissolve in gas compared to water. It is done.
- a compound is used.
- a gas-free monomer solution for example, acrylic acid
- a gas-containing (dissolved) acrylic acid-based monomer aqueous solution a gas that cannot be dissolved in the mixed aqueous solution is generated. It can be dispersed in the aqueous solution as fine bubbles.
- the number average diameter (volume average particle diameter) of the bubbles introduced into the acrylic acid monomer aqueous solution by the method (a) or (b) is preferably 50 ⁇ m or less, and preferably 50 nm (more preferably 10 ⁇ m) to 500 ⁇ m. Is more preferable, and 100 nm (more preferably 10 ⁇ m) to 100 ⁇ m is even more preferable.
- the average diameter of the bubbles is less than 50 nm, the surface area does not increase, so that the water absorption rate may be inferior. Moreover, when an average diameter exceeds 500 micrometers, there exists a possibility that the intensity
- the solubility of gas in water is determined by the type and temperature of the gas. For example, for water at 25 ° C., carbon dioxide (1.05 [ml / ml]), oxygen (0.0285 [ml / ml]) ), Nitrogen (0.0147 [ml / ml]), and the solubility of these gases is reduced by increasing the temperature or mixing with a water-soluble organic substance (preferably acrylic acid). What is necessary is just to make it disperse
- the amount of bubbles is appropriately determined depending on the type of gas and the method of decreasing the solubility (temperature rise range and mixing ratio of water-soluble organic substances).
- the volume of the monomer aqueous solution is preferably 1.01 to 1. It is preferable to disperse the bubbles in the acrylic acid monomer aqueous solution so as to be 1 time, more preferably 1.02 to 1.08 times.
- the bubbles are dispersed by lowering the solubility of the dissolved gas in the aqueous acrylic acid monomer solution. It may be dispersed. That is, the bubbles may be dispersed in the aqueous acrylic acid monomer solution by bubbles dispersed with a reduced solubility, or bubbles dispersed with a gas introduced from the outside if necessary.
- examples of the gas constituting the bubbles dispersed in the aqueous acrylic acid monomer solution include oxygen, air, nitrogen, carbon dioxide, ozone, and mixtures thereof. Preferably, nitrogen, carbon dioxide, etc. An inert gas is used.
- air and nitrogen are particularly preferable in view of polymerizability and cost.
- the pressure at the time of introducing the gas or after the introduction is appropriately determined by normal pressure, pressurization, and decompression.
- a preferable introduction method when introducing gas from the outside is the method described in Japanese Patent Application No. 2009-292318 (application date: December 24, 2009) and its priority application PCT / JP2010 / 001004. In the following, “(2-1-4) Gas introduction method”.
- such gas dissolution and / or dispersion may be performed before or after the bubble generation and inclusion process described above.
- bubbles can be introduced more stably through a step of reducing the solubility of the present invention.
- the method of introducing microbubbles includes (a) a monomer aqueous solution and gas pressurization method, (b) monomer aqueous solution and gas swirl flow formation method, (c) Examples thereof include at least one method selected from a method of mixing a gas through a pore with a monomer aqueous solution.
- a monomer aqueous solution and gas pressurization method includes (a) a monomer aqueous solution and gas pressurization method, (b) monomer aqueous solution and gas swirl flow formation method, (c) Examples thereof include at least one method selected from a method of mixing a gas through a pore with a monomer aqueous solution.
- each method will be described.
- (A) Pressurization of monomer aqueous solution and gas As a method for introducing microbubbles, a pressure dissolution method by pressurization of the monomer aqueous solution and gas is preferably used.
- the gas is preferably pressurized and dissolved in the liquid at an absolute pressure of about 100 to 1000 kPa (absolute pressure), more preferably about 200 to 400 kPa, and particularly preferably about 250 to 350 kPa.
- absolute pressure absolute pressure
- p HC
- a shearing force to the monomer aqueous solution and gas mixture before or during pressurization, if necessary.
- a high speed rotating pump or the like is used to apply the shearing force.
- the gas is finely dispersed by a shearing force and is preferably further pressurized. After applying the shearing force to the monomer aqueous solution and the gas, the gas is pressurized to 0.1 to 1 MPa, and further released under pressure as described later. .
- the concentration of the gas component in the acrylic acid monomer aqueous solution in which the gas is dissolved and / or dispersed as described above is 1.01 to 10 times the saturation solubility of the gas at a predetermined temperature. Preferably, it is 1.05 to 5 times, more preferably 1.06 to 3 times.
- (B) Formation of monomer aqueous solution and gas swirl flow As another method for introducing microbubbles, formation of monomer aqueous solution and gas swirl flow is preferable.
- This method is a method in which gas-liquid two-phase fluid is swirled and bubbles are dispersed at the outlet (discharge port of the mixer), and the ratio of gas flow rate to liquid flow rate is preferably 1/7 to 1/15.
- the speed is preferably 10 to 10,000 revolutions per second, more preferably 100 to 1,000 revolutions.
- swirling microbubble generator examples include, for example, International Publication No. 00/69550, Japanese Patent Publication “JP 2003-205228”, “JP 2000-447”, and “JP 2006-”. No. 116365 ”etc., but is not particularly limited.
- (C) Mixing of gas through pores into monomer aqueous solution
- a method of introducing microbubbles a method of generating bubbles from pores of various porous materials, membranes, filters, etc. 2 O—CaO—Al 2 O 3 —B 2 O 3 —SiO 2 -based glass) or the like is used, and a surfactant in the range described later such as higher than 0 and 0.03% by weight or less is preferably used.
- the said method can be performed using Kinoshita Rika Kogyo Co., Ltd. Kinoshita type
- Microbubble generator In order to introduce microbubbles, a microbubble generator having a function of pressurizing an aqueous monomer solution and an inert gas or generating a swirling flow may be used. By this operation, the generated microbubbles can be suspended and held in the monomer aqueous solution until the start of polymerization.
- microbubble generator that can be used in the method according to the present invention is not particularly limited, and commercially available devices can be used. An example of a commercial product is illustrated below.
- OHR line mixer OHR Fluid Engineering Laboratory Co., Ltd.
- M-type microbubble generator Na planet Research Laboratories
- Microbubble generator for business use SMB-450 type Ishimaru Trading Co., Ltd.
- Microbubble generator Mbelife Kansai Autome Equipment Co., Ltd.
- Sphere built-in type bubble generator MBG type Nishida Tekko Co., Ltd.
- Pomparator Teikoku Electric Manufacturing Co., Ltd.
- the microbubble generator has a water inlet and a water outlet. When liquid (water or monomer) flows into the water inlet at a certain pressure or higher, the gas mixed inside the water Are collected in the center due to the density difference, and a gas axis is formed.
- the number average diameter of the microbubbles generated by the microbubble generator is preferably 50 nm (more preferably 10 ⁇ m) to 500 ⁇ m, more preferably 100 nm (more preferably 10 ⁇ m) to 100 ⁇ m.
- the average diameter of the bubbles is less than 50 nm, the surface area does not increase, so that the water absorption rate may be inferior.
- an average diameter exceeds 500 micrometers there exists a possibility that the intensity
- the throughput of the microbubble generator can be set as appropriate depending on the desired properties of the water-absorbent resin powder, but it is desirable to increase the flow rate of the aqueous monomer solution.
- the flow rate of the monomer aqueous solution is preferably 500 [kg / hr], more preferably 1000 [kg / hr], and still more preferably 2000 [kg / hr].
- the production amount per hour is not limited to the use of the microbubble generator, and the production method of the present invention is generally suitably applicable for industrial huge scale production. Although an upper limit is determined suitably, Preferably it is the said range (for example, 300 ton / hr or less). Therefore, the production method of the present invention is preferably applicable to continuous production, particularly continuous production of the production amount.
- Static mixer system Static mixer that has no moving parts and is mixed when fluid passes through an element fixed inside the tube, or a mushroom-like shape attached to the spiral flow guide and tube inside the circular tube
- An OHR mixer in which microbubbles are generated by crushing a gas-liquid two-phase flow that flows in a swirling manner by the protrusions of the above-mentioned projections.
- Cavitation method There is a method of generating microbubbles by deforming the flow path so that cavitation is intentionally generated in the gas distributor.
- Venturi method There is a method in which, when gas and liquid are simultaneously flowed through the straw section (throttle), large bubbles are blasted by shock waves generated by a sudden change in the liquid flow velocity, and microbubbles are generated.
- Rotating type A method in which a stirring blade is rotated at a high speed, gas is self-supplied, and microbubbles are generated can be mentioned.
- Ultrasonic method A method of generating microbubbles by appropriately setting an ultrasonic frequency, a pressure amplitude and the like can be mentioned.
- Electrolytic decomposition method There is a method of generating micro-order bubbles by electrolysis of water.
- the gas-liquid consisting of the monomer aqueous solution and gas is sheared, and the shearing method includes (3) centrifugal pump And a static mixer having a combination of shearing and swirling flow represented by an OHR mixer.
- one or more of the methods (a) to (c) and (1) to (8) can be applied, preferably (a) or (b), more preferably (a). If necessary, a shearing force generated by a microbubble generator may be applied.
- the gas and the monomer aqueous solution are pressurized beyond the atmospheric pressure (preferably, the range described in (a) above, the absolute pressure).
- the amount and size of the bubbles are controlled by opening to atmospheric pressure (or reduced pressure, in particular, slightly reduced pressure within ⁇ 10 mmHg). It is preferable to control the bubbles by controlling the pressure, temperature and opening time, particularly the opening time. With such control, it is possible to obtain a water-absorbing resin having high liquid permeability and high impact resistance at a target high water absorption speed.
- the above-mentioned (a) to (c) and various microbubbles may be used when preparing an aqueous monomer solution by mixing the acrylic acid or a salt thereof, a solvent, a crosslinking agent, and a polymerization initiator. What is necessary is just to set it as a pressurized state in a generator, ie, the pressure which exceeds atmospheric pressure in piping or an apparatus, and supply the said monomer aqueous solution to a superposition
- T1 is the time from the release of atmospheric pressure to the start of polymerization.
- T is the time from when atmospheric pressure is released until the kaolin turbidity returns to the value before the gas is dissolved or dispersed in the monomer aqueous solution.
- T1 is defined by the white turbidity of the monomer aqueous solution (derived from the formation of the polymer) or the temperature rise (1 ° C. or more) due to the heat of polymerization, and T is the initiator using a 100 ml graduated cylinder as described later. It can be defined by the time (T) until bubbles are dispersed in an aqueous monomer solution not contained and allowed to stand at room temperature under atmospheric pressure until the cloudiness derived from the bubbles disappears.
- the white turbidity is usually derived from a large bubble having a wavelength longer than that of visible light, and the fact that the inclusion of nanobubbles does not substantially affect the white turbidity is also described in the following turbidity (kaolin turbidity).
- the lower limit of T1 is appropriately determined, but is preferably a defoaming time described later (preferably 5 seconds or more, more preferably 10 to 3600 seconds), and the upper limit is a monomer composition including the amount of surfactant. Determined by the method of bubble inclusion.
- a gas can be introduced from the outside into the acrylic acid monomer aqueous solution as necessary.
- the present invention is not limited to the step of reducing the solubility of the gas. It is important that the dissolved gas becomes bubbles.
- the solubility of the gas is lowered by raising the temperature of the acrylic monomer aqueous solution or mixing the water-soluble organic substance, and bubbles are dispersed or dissolved in the monomer aqueous solution.
- the amount of dissolved gas before the operation for reducing the solubility of the gas is large.
- the dissolved gas in the aqueous monomer solution is preferably more than 1 ppm before the operation for reducing the solubility of the gas, more preferably 2 to 50,000 ppm, more preferably 3 to 30,000 ppm, most preferably 3 to 10,000 ppm.
- the amount of these gases is appropriately determined depending on the type of gas, temperature, composition of the acrylic monomer aqueous solution, and the like.
- bubbles can be stably suspended by using a surfactant and / or a dispersant.
- the water absorbing resin powder which has a desired physical property can be obtained by adjusting suitably the kind and quantity of surfactant and / or a dispersing agent.
- the surfactant is a non-polymer surfactant and the dispersant is a polymer dispersant.
- the amount of the surfactant and / or dispersant used is appropriately determined depending on the type, but preferably the surface tension of the water-absorbent resin powder obtained is 60 [mN / m] or more, and further described in “(3 The amount is preferably within the range indicated by “-7) surface tension”.
- the surface tension of the water-absorbent resin powder obtained is less than 60 [mN / m], the amount of return tends to increase when used with paper diapers, which is not preferable.
- reactive or polymerizable surfactants such as water-absorbent resin powder and its monomers, such as unsaturated polymerizable groups (particularly ⁇ , ⁇ -unsaturated double bonds) and reactions
- a surfactant having a functional group hydroxyl group or amino group
- a hydrophilic surfactant having high solubility in water for example, HLB is 1 to 18, particularly preferably 8 to 15 is also suitable.
- the type of the surfactant used in the present invention is not particularly limited.
- anionic surfactants, nonionic surfactants, cationic surfactants, amphoteric surfactants, fluorine-based surfactants examples thereof include organic metal surfactants, and specific examples include surfactants described in Patent Document 28 (International Publication No. WO 97/017397) or Patent Document 30 (US Pat. No. 6,107,358).
- the amount of these surfactants used is typically greater than 0 based on the monomers used, although it depends on the type of surfactant used and the desired physical properties (especially water absorption rate and surface tension). 2% by weight or less, preferably 0.03% by weight or less, more preferably more than 0 and 0.015% by weight, more preferably more than 0 and 0.01% by weight or less, most preferably more than 0 and 0.008% by weight % Or less.
- the amount of the surfactant used can be applied to the water-absorbent resin powder, and if necessary, as a final product obtained after coating with the surfactant described in “(2-7) Surface coating step” described later.
- the present invention can also be applied to water absorbent resin powder.
- the surfactant is too much, it may be difficult to control foaming, and the surface tension of the resulting water-absorbent resin powder will be excessively lowered, resulting in an increase in the amount of return. It is not preferred for actual use in diapers.
- a very small amount of surfactant improves the transportability and damage resistance of the resulting water-absorbent resin powder, and as a result, improves the physical properties of the water-absorbent resin powder after surface cross-linking and after powder transport.
- the surfactant is preferably used in an amount of more than 0 ppm, more preferably 0.1 ppm or more, and even more preferably 1 ppm or more.
- usable surfactants are not particularly limited including those exemplified in Patent Document 28 and Patent Document 30, but nonionic surfactants, anionic surfactants, and cationic surfactants.
- Various agents and amphoteric surfactants can be used, and they may have a polymerizable or reactive group with the monomer of the water absorbent resin powder.
- Nonionic surfactants include, for example, polyoxyalkylene alkyl ethers such as polyoxyethylene lauryl ether, polyoxyethylene cetyl ether, polyoxyethylene stearyl ether, polyoxyethylene oleyl ether; polyoxyethylene octyl phenyl ether, polyoxy Polyoxyalkylene alkyl phenyl ethers such as ethylene nonyl phenyl ether; polyoxyalkylene alkyl amino ethers such as polyoxyethylene lauryl amino ether and polyoxyethylene stearyl amino ether; sorbitan monolaurate, sorbitan monopalmitate, sorbitan monostearate, Sorbitan fatty acid esters such as sorbitan monooleate; polyoxyethylene sorbitan monolaurate Polyoxyalkylene sorbitan monopalmitate, polyoxyethylene sorbitan monostearate, polyoxyalkylene sorbitan fatty acid ester such as polyoxyethylene sorbitan monoole
- anionic surfactant examples include sodium polyoxyethylene lauryl ether sulfate, sodium polyoxyethylene octylphenyl ether sulfate, sodium polyoxyethylene nonylphenyl ether sulfate, triethanolamine lauryl sulfate, sodium lauryl sulfate, potassium lauryl sulfate.
- Sulfate salts such as ammonium lauryl sulfate; sulfonates such as sodium dodecylbenzene sulfonate, sodium alkylnaphthalene sulfonate and sodium dialkylsulfosuccinate; anionic surfactants represented by phosphate ester salts such as potassium alkyl phosphate Agents.
- cationic surfactant examples include cationic surfactants represented by quaternary ammonium salts such as lauryltrimethylammonium chloride, stearyltrimethylammonium chloride, cetyltrimethylammonium chloride, and stearyltrimethylammonium chloride. .
- silicone surfactants include anionic, nonionic, and cationic silicone surfactants, and polyoxyalkylene-modified silicone surfactants. Specifically, modified with polyoxyethylene-modified dimethylpolysiloxane, polyoxyethylene / polyoxypropylene block or random copolymer-modified dimesyl polysiloxane, and polyoxyethylene having an alkyl group having 1 to 12 carbon atoms at the terminal Dimethylpolysiloxane modified, dimethylpolysiloxane modified with block or random copolymer of polyoxyethylene / polyoxypropylene having an alkyl group having 1 to 12 carbon atoms at the end, end of dimethylpolysiloxane and / or inside of molecule And the above-mentioned polyoxyalkylene-modified products of dimethylpolysiloxane derivatives having amino groups, epoxy groups and the like.
- Polyoxyethylene-modified dimethylpolysiloxane, polyoxyethylene / polyoxypropylene block or random copolymer-modified dimesyl polysiloxane are preferable, and polyoxyethylene-modified dimethylpolysiloxane is more preferable because it can be obtained industrially at low cost. Oxyethylene-modified dimethylpolysiloxane.
- surfactants may be used alone or in combination of two or more thereof, or may be used in combination with a dispersant described below (particularly, a polymer dispersant).
- a dispersant described below (particularly, a polymer dispersant).
- anionic surfactants, nonionic surfactants or silicone surfactants, and further, nonionic surfactants or silicone surfactants may be used from the viewpoint of effects. preferable.
- the acrylic monomer aqueous solution during or after adjustment before the polymerization step includes a dispersant, and the dispersant is a hydrophilic polymer dispersant exhibiting water absorption. More preferably, it is a water-soluble polymer dispersant, and its weight average molecular weight is appropriately determined depending on the type of the dispersant, but is preferably 500 to 10 million, more preferably 5,000 to 500. 10,000, particularly preferably about 10,000 to 3,000,000.
- dispersants are not particularly limited.
- starch, starch derivatives, cellulose, cellulose derivatives, polyvinyl alcohol, carboxymethyl cellulose (sodium), hydroxyethyl cellulose, polyacrylic acid (salt), polyacrylic acid (Salt) Hydrophilic polymers such as cross-linked products are exemplified, and among these, a water-soluble polymer dispersant selected from starch, cellulose, and PVA is preferable from the viewpoint of the effect of the present invention.
- the amount of the dispersant used is preferably more than 0 parts by weight and 50 parts by weight or less, more preferably 0.01 to 20 parts by weight, still more preferably 0.05 to 10 parts by weight with respect to 100 parts by weight of the monomer. Parts, most preferably 0.1 to 5 parts by weight.
- the amount of the dispersant is similarly applied to the water-absorbent resin powder as a hydrophilic polymer dispersant used in place of the water-soluble polymer. If the amount of the dispersant is too large, it may be difficult to control foaming, and the absorbent capacity of the resulting water-absorbent resin powder is lowered, which is not preferable for actual use in a paper diaper.
- a polymerization inhibitor is preferably contained during the polymerization.
- the polymerization inhibitor include N-oxyxyl compounds, manganese compounds and substituted phenol compounds exemplified in International Publication No. 2008/096713, preferably substituted phenols, particularly methoxyphenols.
- methoxyphenols that can be preferably used include o, m, p-methoxyphenol and methoxy having one or more substituents such as a methyl group, a t-butyl group, and a hydroxyl group.
- Phenols are exemplified, and in the present invention, p-methoxyphenol is particularly preferable.
- the content of methoxyphenols may be 10 to 200 ppm, preferably 5 to 160 ppm, more preferably 10 to 160 ppm, still more preferably 10 to 100 ppm, particularly preferably 10 to 80 ppm, and most preferably 10 to 70 ppm. It is.
- the monomer aqueous solution preferably contains 10 to 200 ppm, more preferably p-methoxyphenol within the above range. It is preferable.
- bubbles generated by such a mechanism are very fine, and further stabilized by a surfactant and / or a dispersing agent, so that the liquid can be further passed at a high water absorption rate. It is possible to provide a white water-absorbing resin powder having excellent properties.
- the degassing (deoxygenation) of the aqueous monomer solution described in Patent Documents 1 and 3 with a simple inert gas does not stabilize the bubbles during polymerization, and does not solve the problem of the present invention.
- reducing the solubility of a gas refers to an operation for reducing the solubility of the same gas, and is a different concept from a degassing operation for removing dissolved oxygen (substituting oxygen with an inert gas).
- the aqueous solution in the presence of a surfactant and / or a dispersant, the aqueous solution is heated or mixed with a poor solvent (preferably acrylic acid) in the aqueous solution to lower the solubility of the dissolved gas and generate bubbles to disperse. This is a completely new foaming method.
- degassing substitution of dissolved oxygen and inert gas
- the dissolved oxygen is preferably 1 ppm or less, more preferably 0.5 ppm or less.
- a gas introduction step shown in “(2-1-3) Gas” may be provided separately.
- polymerization with a monomer slurry is a physical property (water absorption capacity, water-soluble content). , Residual monomer, etc.) may decrease. Therefore, preferably, when the monomer is an acid group-containing monomer, the neutralization rate is such that the neutralized salt does not precipitate in the aqueous monomer solution. That is, not an aqueous dispersion of an acrylic acid monomer, but preferably an aqueous acrylic acid monomer solution is polymerized in the present invention. Precipitation of the neutralized salt is determined by the solubility of the neutralized salt in water, such as the monomer concentration, neutralization rate, temperature, pressure, and neutralizing base. It depends on the conditions and is appropriately determined.
- the present invention preferably further includes a defoaming step.
- a defoaming step By including the defoaming step, large bubbles are sequentially removed from the monomer, and excessive foaming and a decrease in bulk specific gravity are suppressed.
- the defoaming time is preferably 5 seconds or more, more preferably 10 seconds to 60 minutes, further preferably 30 seconds to 30 minutes, and particularly preferably 60 seconds to 20 minutes. It is adjusted so as to leave the desired fine bubbles in the monomer aqueous solution.
- the size of bubbles in the aqueous monomer solution after the defoaming step is preferably 100 ⁇ m or less, more preferably 50 ⁇ m or less, still more preferably 20 ⁇ m or less, and particularly preferably 5 ⁇ m or less in terms of volume average diameter.
- the expansion ratio of the monomer aqueous solution after the defoaming step relative to the monomer aqueous solution before the bubble dispersion step is preferably 1.1 times or less, more preferably 1.05 or less, and still more preferably 1.02 or less. Particularly preferably, it is 1.01 or less, and usually the lower limit exceeds 1.
- the defoaming step used in the present invention may be a known technique, such as the method described in US Pat. No. 6,667,372 or the method described in Techno Systems Publishing Co., Ltd. “Foam Engineering First Edition”, pages 759-774. Etc.
- the defoaming step is preferably performed by circulating a monomer aqueous solution containing a circulating air flow to the circulation tank.
- the upper space of the circulation tank preferably contains 1% by volume or more of oxygen.
- at least a part of the monomer aqueous solution is supplied to the polymerization step through the neutralization step or the neutralization step from the step of circulating the monomer aqueous solution containing the air flow and the circulation line, and is polymerized.
- the aqueous monomer solution may be degassed by accumulating bubbles by allowing the monomer aqueous solution to stand by for a certain period of time in the pipe or in the polymerization apparatus until the start of polymerization.
- the monomer aqueous solution preferentially containing fine bubbles obtained
- a polymerization initiator or irradiation with ultraviolet rays may be performed.
- the resulting water-absorbent resin powder is scale-like or the bulk specific gravity is excessively reduced (for example, 0.5 [g / cm 3 ] or less).
- liquid permeability and impact resistance are also reduced.
- any of these defoaming methods may be used, but the purpose is to obtain an aqueous monomer solution preferentially containing fine bubbles after defoaming mainly on large bubbles. Not intended. In this way, the water-absorbent resin powder obtained through the defoaming step becomes a porous polymer having fine and uniform pores.
- microbubbles (fine bubbles) generated through the defoaming step are performed by the concentrated ions acting on the bubble interface, generating a repulsive force of static electricity, and preventing the gas from escaping.
- Nanobubbles may be generated by crushing microbubbles by a self-pressurizing effect or an adiabatic compression effect.
- the rising speed is 5400 [ ⁇ m / s] and the internal pressure is 1.04 ⁇ 10 5 Pa.
- the rising speed is 54 [ ⁇ m / s]
- the internal pressure is 1.31 ⁇ 10 5 Pa
- the rising speed is 0.54 [ ⁇ m / s] and the internal pressure is 3.95 ⁇ 10 5 Pa.
- the expansion ratio of the volume of the aqueous monomer solution in the polymerization step relative to the volume of the aqueous monomer solution before the bubble generation-containing step is preferably 1.1 times or less, more preferably 1.05 times or less, and still more preferably 1. It is 02 times or less, particularly preferably 1.01 times or less, and most preferably 1.00 times (preferably more than 1).
- a method for polymerizing by dispersing a large amount of bubbles has been known.
- polymerization is performed without excessively dispersing bubbles by using the above technique, so that there is almost no decrease in bulk specific gravity.
- microbubbles or nanobubbles having a volume average diameter of 100 ⁇ m or less are contained in the monomer aqueous solution in the polymerization step.
- the volume average diameter of the contained microbubbles or nanobubbles is preferably 100 ⁇ m or less, more preferably 50 ⁇ m or less, still more preferably 20 ⁇ m or less, and particularly preferably 5 ⁇ m or less.
- laser diffraction scattering method also known as static light scattering method
- dynamic light scattering method also known as dynamic light scattering method
- electrical detection band method common name: Coulter canter
- particle counter method light scattering method, light blocking method
- visualization method by camera photographing
- interference image method by laser light and CCD camera, and the like.
- the number can be measured by (c) the electrical detection band method or (d) the particle counter method, and the nano-order measurement can be performed by (b) dynamic light scattering method, (a) laser diffraction scattering method (also known as a different name). A static light scattering method). These measuring methods are used as appropriate, but preferably, a light scattering method, particularly a dynamic light scattering method is used.
- (2-3) Polymerization Step Polymerization is carried out at normal pressure, reduced pressure, or increased pressure, preferably at normal pressure (or in the vicinity thereof, usually ⁇ 10 mmHg). Also, in order to promote polymerization and improve physical properties, a degassing step of dissolved oxygen (for example, a substitution step with an inert gas) is provided if necessary at the time of polymerization based on the schematic flow charts shown in FIGS. May be.
- a degassing step of dissolved oxygen for example, a substitution step with an inert gas
- the polymerization initiator used in the present invention is appropriately selected depending on the polymerization form, and is not particularly limited. Examples thereof include a photodegradable polymerization initiator, a thermal decomposition polymerization initiator, and a redox polymerization initiator. it can.
- Examples of the photodegradable polymerization initiator include benzoin derivatives, benzyl derivatives, acetophenone derivatives, benzophenone derivatives, azo compounds, and the like.
- Examples of the thermal decomposition type polymerization initiator include persulfates (sodium persulfate, potassium persulfate, ammonium persulfate), peroxides (hydrogen peroxide, t-butyl peroxide, methyl ethyl ketone peroxide), azo Compounds (2,2′-azobis (2-amidinopropane) dihydrochloride, 2,2′-azobis [2- (2-imidazolin-2-yl) propane] dihydrochloride, etc.) and the like.
- examples of the redox polymerization initiator include a system in which a reducing compound such as L-ascorbic acid or sodium bisulfite is used in combination with the persulfate or peroxide, and the both are combined. Moreover, it can also be mentioned as a preferable aspect to use together the said photodegradable initiator and a thermal decomposable polymerization initiator.
- an azo polymerization initiator that generates N 2 by thermal decomposition may be used to promote foaming.
- the amount of these polymerization initiators used is preferably 0.0001 to 1 mol%, more preferably 0.0005 to 0.5 mol%, based on the monomer.
- the usage-amount of a polymerization initiator exceeds 1 mol%, since there exists a possibility of having a bad influence on the color tone of a water absorbing resin, it is unpreferable.
- the usage-amount of a polymerization initiator is less than 0.0001 mol%, since there exists a possibility of increasing the amount of residual monomers, it is unpreferable.
- a chain transfer agent such as hypophosphorous acid (salt), a chelating agent, etc. may be added to the reaction system before or during the polymerization.
- aqueous solution polymerization when polymerizing the monomer aqueous solution, aqueous solution polymerization is usually employed from the viewpoint of the properties of the water absorbent resin such as liquid permeability and water absorption speed of the resulting water absorbent resin and ease of polymerization control.
- kneader polymerization or belt polymerization more preferably continuous aqueous solution polymerization, still more preferably high concentration continuous aqueous solution polymerization, particularly preferably high concentration high temperature starting continuous aqueous solution polymerization.
- the above polymerization method is preferably employed in a production apparatus on a huge scale with a large production amount of water-absorbing resin per line, that is, continuous polymerization and continuous production (drying step to surface cross-linking step).
- the production amount is preferably 0.5 [t / hr], more preferably 1 [t / hr], still more preferably 5 [t / hr], and particularly preferably 10 [t / hr].
- aqueous solution polymerization examples include continuous belt polymerization (disclosed in US Pat. Nos. 4,893,999 and 6,241,928, US Patent Application Publication No. 2005/215734, etc.), continuous kneader polymerization, batch kneader polymerization (US Pat. No. 6,987,151). , And the like are disclosed in No. 6710141). In these aqueous solution polymerizations, a water-absorbing resin can be produced with high productivity.
- the polymerization initiation temperature is 0 ° C. or higher, preferably 30 ° C. or higher, more preferably 35 ° C. or higher, further preferably 40 ° C. or higher, particularly preferably 50 ° C. or higher (the upper limit is the boiling point).
- Initiation continuous aqueous solution polymerization high concentration continuous aqueous solution polymerization with a monomer concentration of preferably 35% by weight or more, more preferably 40% by weight or more, and even more preferably 45% by weight or more (the upper limit is a saturated concentration), or these
- a combined high concentration / high temperature starting continuous aqueous solution polymerization can be exemplified as a preferred example.
- the present invention provides a water-absorbing resin having excellent monomer stability and high whiteness.
- Such high temperature initiation polymerization is exemplified in U.S. Pat. Nos. 6,906,159 and 7,091,253, etc., but the method of the present invention is excellent in the stability of the monomer before polymerization, so that it is on an industrial scale. Is easy to produce.
- the polymerization start time (the time from the addition of the polymerization initiator to the start of polymerization) is preferably more than 0 and within 300 seconds from the viewpoint of suppressing the reduction of bubbles in the aqueous monomer solution. 1 to 240 seconds is more preferable. If the polymerization start time exceeds 300 seconds, the amount of bubbles introduced into the water-absorbent resin powder decreases, and the effects of the present invention may not be exhibited.
- Particularly suitable foaming polymerization conditions As the polymerization method, spray polymerization, droplet polymerization, aqueous solution polymerization, reverse phase suspension polymerization, etc. can be widely applied. Particularly suitable polymerization methods for solving the problems include aqueous solution polymerization, especially continuous belt polymerization or continuous polymerization. An example is kneader polymerization.
- the aqueous solution polymerization is preferably performed at the following temperature and concentration.
- the polymerization is preferably initiated at a high temperature.
- the polymerization initiation temperature in the polymerization step is preferably 40 ° C or higher, more preferably 50 ° C or higher, still more preferably 60 ° C or higher, and particularly preferably 70. More than 80 degreeC, Most preferably, it is 80 degreeC or more.
- the maximum temperature reached during polymerization is high.
- the maximum temperature reached during polymerization in the polymerization step is preferably 100 ° C. or higher, more preferably 100 to 130 ° C. More preferably, it is 105 to 120 ° C.
- the concentration of the aqueous monomer solution at the time of polymerization is not particularly limited, but is preferably in the range of 20% by weight to the saturated concentration, more preferably in the range of 25 to 80% by weight, and still more preferably in the range of 30 to 70% by weight. .
- productivity is lowered, which is not preferable.
- polymerization with a monomer slurry (aqueous dispersion of acrylate) as described in Patent Document 35 Japanese Patent Laid-Open No. 1-318021
- Patent Document 35 Japanese Patent Laid-Open No. 1-318021
- the concentration of the acrylic acid monomer aqueous solution is preferably as high as possible in the polymerization step. Specifically, it is preferably 40% by weight or more, more preferably 45% by weight or more, and more preferably 50% by weight or more. More preferably (the upper limit is usually 90% by weight or less, preferably 80% by weight or less, more preferably 70% by weight or less). This solid content is also applied to the hydrogel crosslinked polymer after polymerization. In addition, when the monomer concentration during the polymerization is 40% by weight or more, and further 45% by weight or more, the stability of fine bubbles is enhanced, so that the present invention is particularly advantageous.
- the gel is preferably atomized (also referred to as gel pulverization) during or after polymerization.
- gel refining particularly by kneading, it is possible to achieve both water absorption speed and liquid permeability, and further improve impact resistance.
- aqueous solution polymerization particularly belt polymerization or kneader polymerization is performed, during polymerization (particularly kneader polymerization) or after polymerization (particularly It is preferable to perform gel pulverization in belt polymerization, and if necessary, kneader polymerization.
- the gel crushing apparatus that can be used in the present invention is not particularly limited, and is a gel crusher equipped with a plurality of rotary stirring blades, such as a batch type or continuous double arm kneader, a single screw extruder, and a twin screw. Examples thereof include an extruder and a meat chopper. Among these, a screw-type extruder having a perforated plate at the tip is preferable. For example, a screw-type extruder disclosed in Japanese Laid-Open Patent Publication No. 2000-63527 is cited.
- the gel particle diameter after fine graining is preferably in the range of 0.5 to 3 mm, more preferably in the range of 0.6 to 2 mm, and still more preferably in the range of 0.8 to 3 in terms of weight average particle diameter (specified by sieve classification).
- the range is 1.5 mm.
- the content of the coarse gel of 5 mm or more is preferably 10% by weight or less, more preferably 5% by weight or less, and still more preferably 1% by weight or less.
- the temperature of the hydrogel before gel grinding is preferably 60 to 120 ° C., more preferably 65 to 110 ° C., from the viewpoints of particle size control and physical properties.
- gel temperature is less than 60 ° C., the hardness increases due to the characteristics of the hydrogel, and therefore it may be difficult to control the particle shape and particle size distribution during pulverization.
- the gel temperature exceeds 120 ° C., the softness of the hydrogel increases, which may make it difficult to control the particle shape and particle size distribution.
- the method for controlling the gel temperature can be appropriately controlled by the polymerization temperature, heating or cooling after polymerization, or the like. That is, in the present invention, the polymerization step is preferably carried out by continuous kneader polymerization, and the hydrogel crosslinked polymer is finely divided during the polymerization. In the present invention, the polymerization step is preferably carried out by continuous belt polymerization, and the hydrogel crosslinked polymer is finely divided after the polymerization.
- gel pulverization described in Japanese Patent Application “Japanese Patent Application No. 2010-088993” application date; April 7, 2010
- GGE gel pulverization energy
- the gel grinding energy (GGE) for grinding the hydrogel is preferably 60 [J / g] or less, more preferably 50 [J / g] or less, and 40 [J / g] as the upper limit. g] or less is more preferable.
- gel grinding energy in Japanese Patent Application No. 2010-088993 refers to the energy per unit weight required by the gel grinding device when the hydrogel crosslinked polymer is gel ground.
- GGE is an abbreviation for Gel Grinding Energy. GGE is calculated by the following equation 1 when the gel crusher is driven by three-phase AC power.
- the unit of voltage is [V]
- the unit of current is [A]
- the weight of the hydrogel crosslinked polymer charged into the gel crusher for 1 second is [g / s].
- the “power factor” and “motor efficiency” are values unique to the apparatus that vary depending on the operating conditions of the apparatus, and take values from 0 to 1. These values can be obtained by inquiries to the device manufacturer.
- control with gel grinding energy (GGE) 18 to 60 [J / g] is one of the means for achieving the above.
- the amount of increase in the water-soluble content of the hydrogel crosslinked polymer is preferably 5% by weight or less, more preferably 4% by weight or less, and still more preferably 3% by weight or less.
- the water-absorbent resin powder of the object of the present invention may not be obtained, so the water-containing gel is fragmented during or after polymerization.
- the hydrogel after fragmentation is subjected to a drying step, and is preferably subjected to a surface crosslinking step. Since sheet-like water-absorbing resin foams such as Patent Documents 19 and 22 do not solve the problem of the present application, in the present invention, a powdery water-absorbing resin is obtained by pulverization before or after drying.
- the water-containing gel-like crosslinked polymer is dried to form a dry polymer.
- the resin solid content determined from the loss on drying (1 g of powder or particles is heated at 180 ° C. for 3 hours) is preferably 80% by weight or more, more preferably 85 to 99% by weight, still more preferably 90 to 98% by weight as a powder. Particularly preferably, the content is adjusted in the range of 92 to 97% by weight to obtain a dry polymer.
- the drying temperature is not particularly limited, but is preferably 100 to 300 ° C, more preferably 150 to 250 ° C.
- the aggregate obtained in the drying step may be supplied to the pulverization step as it is.
- a hydrated gel containing bubbles (particularly closed cells) is obtained in the polymerization step, and the hydrated gel containing such bubbles is preferable because foaming is further promoted during high-temperature drying.
- the drying time and the drying apparatus are appropriately determined.
- the drying time is preferably 1 minute to 5 hours, and more preferably 5 minutes to 1 hour.
- One or two or more of aeration band drying, stirring drying, and drying by azeotropic dehydration can be used.
- the weight average particle diameter (D50) of the water-absorbent resin powder before surface crosslinking is preferably 200 to 600 ⁇ m as a powder from the viewpoint of water absorption speed, liquid permeability, and water absorption magnification under pressure in order to solve the problems of the present invention. More preferably, it is adjusted to 200 to 550 ⁇ m, further preferably 250 to 500 ⁇ m, and particularly preferably 350 to 450 ⁇ m. Further, from the viewpoint of liquid permeability and the like, it is better that the number of particles less than 150 ⁇ m is smaller with a JIS standard sieve, and it is usually adjusted to 0 to 5% by weight, preferably 0 to 3% by weight, particularly preferably 0 to 1% by weight. Such particle size control can be performed at the time of polymerization, gel pulverization or pulverization after drying, and classification, but it is particularly preferable to perform it during classification after drying.
- the shape of the water-absorbent resin powder may be a spherical shape or an aggregate thereof, or may be an irregularly crushed shape obtained through a pulverization process on a polymer gel or a dried polymer (for example, FIG. 3, FIG. 12, FIG. 13). However, from the viewpoint of water absorption speed, it is preferably an irregularly crushed shape or a granulated product thereof.
- the ratio of water absorption rate, liquid permeability, and water absorption magnification under pressure is preferably 850 ⁇ m (passage) to 150 ⁇ m, more preferably 710 ⁇ m (passage).
- the particle size before surface crosslinking is preferably applied to the water-absorbent resin particles (or the water-absorbent resin powder of the present invention described later), which is preferably the final product, after surface crosslinking. Applied.
- surface cross-linking step In order to further improve the water absorption rate (and liquid permeability), it is preferable to further include a step of surface cross-linking the polyacrylic acid water-absorbing resin powder after drying.
- the surface cross-linking may be performed with a surface cross-linking agent described later, the monomer may be polymerized on the surface of the water-absorbent resin, and a radical polymerization initiator such as persulfate or UV initiator is added, followed by heating or ultraviolet irradiation. You may go by.
- the surface cross-linking step in the present invention is preferably carried out with a surface cross-linking agent, and further a covalent surface cross-linking agent, particularly in the present invention, a covalent surface cross-linking agent is used in combination.
- the production method of the present invention is applied to a method for producing a water-absorbent resin powder having a high water absorption capacity (AAP) and liquid permeability (SFC) under high pressure and continuous production at a huge scale (particularly 1 [t / hr]).
- AAP water absorption capacity
- SFC liquid permeability
- it is suitably applied to water-absorbing resin powders at high temperature surface crosslinking.
- Covalent bonding surface cross-linking agent examples include various organic or inorganic cross-linking agents, but organic surface cross-linking agents can be preferably used.
- the surface crosslinking agent is preferably a polyhydric alcohol compound, an epoxy compound, a polyvalent amine compound or a condensate thereof with a haloepoxy compound, an oxazoline compound, a (mono, di, or poly) oxazolidinone compound, or an alkylene carbonate compound.
- a dehydration-reactive cross-linking agent composed of a polyhydric alcohol compound, an alkylene carbonate compound, or an oxazolidinone compound that requires a reaction at a high temperature can be used.
- a dehydration-reactive crosslinking agent When a dehydration-reactive crosslinking agent is not used, more specifically, compounds exemplified in US Pat. Nos. 6,228,930, 6071976, 6254990, and the like can be mentioned.
- liquid permeability and the like may be improved by using a polyamine polymer or a polyvalent metal salt as an ion-bonding surface cross-linking agent.
- a polyamine polymer or a polyvalent metal salt as an ion-bonding surface cross-linking agent.
- Such an ion-binding surface cross-linking agent is preferable as an electrostatic spacer for the swollen gel particles, which contributes to an improvement in liquid permeability.
- liquid permeability in the case of liquid permeability, in particular, when SFC is 20 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ sec ⁇ g ⁇ 1 ] or more, and further in the range described later, an organic surface crosslinking agent (covalent surface crosslinking) and It is preferable that liquid permeability is improved by using an ion-binding surface cross-linking agent or water-insoluble fine particles described later in combination.
- the polyvalent metal salt (inorganic surface crosslinking agent) used is a divalent or higher, preferably trivalent or tetravalent polyvalent metal salt (organic salt or inorganic salt) or hydroxide, that is, a polyvalent metal cation. Can be illustrated.
- the polyvalent metal that can be used include aluminum and zirconium.
- Preferred examples of the polyvalent metal salt include aluminum cations such as aluminum lactate and aluminum sulfate.
- polyamine polymer to be used examples include polyerylenimine, polyvinylamine, polyallylamine and the like, and the weight average molecular weight is appropriately determined from 1,000 to 5,000,000 and 10,000 to 1,000,000.
- water-insoluble fine particles used include inorganic fine powders such as silicon oxide, aluminum oxide, clay and kaolin, and organic fine powders such as aluminum lactate, calcium lactate and metal soap (polyvalent metal salt of long chain fatty acid).
- the volume average particle diameter is preferably 10 ⁇ m or less, more preferably 1 ⁇ m or less.
- the water-absorbent resin powder further contains a liquid permeability improver selected from the polyvalent metal cation, polyamine polymer and water-insoluble fine particles exemplified in the above (2-6) in the surface crosslinking.
- a liquid permeability improver selected from the polyvalent metal cation, polyamine polymer and water-insoluble fine particles exemplified in the above (2-6) in the surface crosslinking.
- the amount of the organic surface cross-linking agent (covalent surface cross-linking agent) used is preferably in the range of 0.001 to 10 parts by weight, more preferably 0.01 to 5 parts by weight with respect to 100 parts by weight of the water absorbent resin powder. It is determined as appropriate within the range.
- the amount of the liquid permeability improver selected from polyvalent metal cations, polyamine polymers, and water-insoluble fine particles is preferably 0 to 5 parts by weight, more preferably 0.001 to 3 parts by weight, and 0.01 to 2 parts by weight. Part is more preferable, and 0.05 to 1 part by weight is particularly preferable.
- water in combination with the surface cross-linking agent, preferably water can be used.
- the amount of water used is preferably in the range of 0.5 to 20 parts by weight, more preferably 0.5 to 10 parts by weight with respect to 100 parts by weight of the water absorbent resin powder.
- the amount is preferably 0.001 to 10 parts by weight, more preferably 0.01 to 5 parts by weight with respect to 100 parts by weight of the water absorbent resin powder.
- a hydrophilic organic solvent may be used, and the amount thereof is preferably more than 0 parts by weight and 10 parts by weight or less, more preferably 0 parts by weight with respect to 100 parts by weight of the water absorbent resin powder. It is in the range of more than 5 parts by weight.
- the range does not hinder the effect of the present invention, for example, more than 0 parts by weight and 10 parts by weight, preferably more than 0 parts by weight and 5 parts by weight, more preferably Is more than 0 part by weight and 1 part by weight or less, and a water-insoluble fine particle powder or a surfactant may coexist.
- the surfactant used and the amount of use thereof are exemplified in US Pat. No. 7,473,739.
- the water-absorbent resin powder after mixing the surface cross-linking agent is subjected to a heat treatment and, if necessary, a cooling treatment.
- the heating temperature is preferably 70 to 300 ° C, more preferably 120 to 250 ° C, still more preferably 150 to 250 ° C, and the heating time is preferably in the range of 1 minute to 2 hours.
- the water absorption capacity under pressure (AAP) described later is in the range described below, preferably 20 [g / g] or more, more preferably 23 to 30 [g / g].
- AAP water absorption capacity under pressure
- the SFC is improved to the following range (for example, 20 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ sec ⁇ g ⁇ 1 ] or more, more preferably the range described later)
- the CRC is preferably 15 to 45 [ g / g], more preferably 20 to 40 [g / g], still more preferably 25 to 35 [g / g], and particularly preferably 28 to 33 [g / g].
- the liquid permeability can be further improved by further including a liquid permeability improver selected from polyvalent metal cations, polyamine polymers, and water-insoluble fine particles.
- This step is a step of coating the surface of the water-absorbent resin powder with a surfactant in order to obtain a water-absorbent resin powder having a high water absorption rate and high liquid permeability.
- the water-absorbent resin powder of the present invention is a foam, it tends to be weak in impact resistance as a powder, and the physical properties may be deteriorated due to breakage at the time of surface crosslinking or after surface crosslinking. This tendency becomes more conspicuous as the scale during production (production amount per hour) is larger.
- the production amount per line is preferably 0.5 [t / hr] or more, and hereinafter, 1 [t / hr in order. ], 5 [t / hr] or more, and 10 [t / hr] or more, the more prominent.
- the surface of the water-absorbing resin powder is preferably further cross-linked after drying, particularly during continuous production on the huge scale. And a step of coating the surface with a surfactant at the same time or separately from the surface crosslinking.
- the type and amount of the surfactant are appropriately determined, but the amount used is preferably 2% by weight or less, and in the following order 0.03% by weight or less, 0.015% by weight or less, 0.01% by weight or less. 0.008% by weight or less is preferable, and the lower limit is preferably 0.1 ppm or more, and more preferably 1 ppm or more. Preferably, it is used in an amount and type that maintains the above surface tension (preferably 60 [mN / m] or more, more preferably a range indicated by “(3-7) Surface tension” described later). From the viewpoint of water absorption speed and impact resistance, water is preferably contained simultaneously with the surfactant. Water is preferably used or contained in the water-absorbent resin in an amount of 0.1 to 10% by weight, more preferably 1 to 8% by weight, and particularly preferably 2 to 7% by weight.
- the fine powder after the classification step is recycled (reused) in the step before the drying step. That is, the water-absorbent resin after the polymerization step, preferably after the heat drying step, is adjusted to the above particle size through a pulverization and classification step as necessary.
- coarse particles for example, 1 mm or more
- fine particles to be removed by classification for example, less than 150 ⁇ m or even less than 106 ⁇ m
- the production method of the present invention may preferably include a fine powder recycling step.
- the fine powder recycling process is the process of separating the fine powder generated in the drying process and, if necessary, the pulverization and classification processes (particularly, the fine powder containing 70% by weight or more of the powder having a particle diameter of 150 ⁇ m or less), as it is, or hydrated or It is granulated and recycled before the pulverization process, preferably the polymerization process, the process of refining the foamed polymer, or the process of recycling to the heat drying process.
- the particle size of the base polymer is controlled by recycling the fine powder.
- the water absorption rate can be further improved by adding fine powder.
- the fine powder may be fine powder before surface crosslinking or fine powder after surface crosslinking, and the amount of fine powder recycled is preferably 1 to 40% by weight of the dry polymer, and more preferably 5 to 30% by weight.
- the fine powder recycling method preferably used in the present invention is a method in which a water-absorbing resin fine powder, a hydrate or granulated product thereof, and, if necessary, inorganic fine particles are mixed with an aqueous monomer solution during polymerization or a hydrogel during polymerization. is there. Further, the fine powder to be recycled may be used to increase the viscosity of the monomer during polymerization to promote foaming.
- the fine powder recycling method to gel during polymerization is exemplified in International Publication Nos. 2007/074167, 2009/109563, 2009/153196, 2010/006937, and a monomer aqueous solution before polymerization.
- Examples of the method for recycling fine powders to US Pat. Nos. 92/001008 and 92/020723, and the method for recycling fine powders to dryers are exemplified in US Pat. No. 6,228,930. It is preferably applied.
- the reducing agent is consumed in the polymerization, Is preferably added after drying, especially after surface crosslinking.
- Chelating agents that can be used include chelating agents exemplified in US Pat. Nos. 6,599,989, 6,469,080, and European Patent No. 2,163,302, particularly non-polymeric chelating agents, organophosphorus chelating agents, and aminoalbonic acid chelating agents. Can be used.
- Examples of the ⁇ -hydroxycarboxylic acid include apple succinic acid, succinic acid, lactic acid and salts thereof (particularly monovalent salts) exemplified in US Patent Application Publication No. 2009/0312183.
- Examples of the inorganic or organic reducing agent (especially sulfur-based inorganic reducing agent) that can be used include sulfur-based reducing agents exemplified in U.S. Patent Application Publication No. 2010/0062252, particularly sulfites or hydrogen sulfites.
- a second classification step an evaporation monomer recycling step, a granulation step, a fine powder removal step, and the like may be provided as necessary.
- an additive may be used in the monomer or a polymer thereof for the purpose of color stability with time and prevention of gel degradation.
- the water-absorbent resin powder may be added to an oxidant, an antioxidant, water, a polyvalent metal compound, a water-insoluble inorganic or organic powder such as silica or metal soap, a deodorant, an antibacterial agent, pulp or heat.
- Plastic fibers and the like may be added to the water-absorbent resin powder in an amount of more than 0% by weight and 3% by weight or less, preferably more than 0% by weight and 1% by weight or less.
- a preferable amount of the surfactant in the water-absorbent resin powder is within the above range.
- the water absorbent resin powder obtained by the above production method of the present invention has a predetermined amount of voids inside.
- the present invention provides a novel water absorbent resin powder having a specific range of bubble content (bubble content inside the water absorbent resin particles, also referred to as porosity).
- the present invention is a polyacrylic acid-based water-absorbent resin powder, and has an internal cell ratio (also known as closed cell ratio) defined by the following formula of 2.8 to 6.6%.
- a water-absorbent resin powder is provided.
- (Internal bubble ratio [%]) ⁇ (true density [g / cm 3 ]) ⁇ (apparent density [g / cm 3 ]) ⁇ / (true density [g / cm 3 ]) ⁇ 100
- the true density [g / cm 3 ] of the water-absorbent resin powder sufficiently dried (moisture content of less than 1% by weight, preferably less than 0.5% by weight, more preferably less than 0.1% by weight).
- the polyacrylic acid-based water absorbent resin powder has a slight difference due to the neutralization rate, the type of the salt (for example, sodium polyacrylate having a neutralization rate of 75 mol%) and a trace amount of raw material. Indicates a constant value.
- the “apparent density” of the water-absorbent resin powder is a density that takes into account voids (other names; bubbles, particularly closed cells) inside the particles.
- voids other names; bubbles, particularly closed cells
- the water-absorbent resin obtained by foaming polymerization or the water-absorbent resin that has undergone the granulation process are not connected to the outside (closed pores; voids: independent). Bubbles).
- the density of the water-absorbent resin is measured by dry density measurement, since the introduced gas cannot reach the closed pores, the measured density is an apparent value obtained from the volume containing the closed pores (closed cells). It becomes density.
- the effective number of the measured value of the apparent density and the true density is appropriately determined by a measuring device or the like, and is, for example, 3 or 4 digits after the decimal point.
- Example 15 described below discloses 1.588 [g / cm 3 ] as an apparent density.
- the apparent density of the water-absorbent resin is described in p. 197 to 199 discloses wet measurement in which the apparent density of the water-absorbent resin after 40 to 60 mesh-Cut is determined by volume with methanol.
- the apparent density of the present invention is determined by the dry measurement described above for all particle sizes. It was found that the internal porosity defined by the apparent density is important for the water-absorbent resin.
- the density of the water-absorbing resin can be accurately determined by dry density measurement using a predetermined gas.
- the measurement principle of the solid dry density measurement is well known in the constant volume expansion method, which is a method for obtaining the solid volume with a specific gas. Specifically, when the volume V CELL of the sample chamber and the volume V EXP of the expansion chamber are known, if the pressures (gauge pressures) P 1 g and P 2 g are measured, the sample volume V SAMP can be obtained separately.
- the density can be obtained by measuring the mass of the material and dividing the mass by the volume (Reference: Shimadzu Corporation website; http://www.shimadzu.co.jp/powder/lecture/middle/m04.html) .
- the known value may be used as it is, and the known value may vary depending on the trace amount raw material of the water absorbent resin. If it is unknown, it may be determined by the method described later.
- the true density in the present invention is the dry density of the water-absorbent resin substantially free of closed cells after the closed cells in the powder are broken or made into open cells by pulverization by the method described later (see FIG. 13). It is obtained by measuring.
- the open cell is a bubble that communicates with the outside and is not counted as the volume of the powder when measuring the dry density of the powder. Therefore, the closed cell and the open cell can be easily discriminated by measuring the dry density of the powder.
- the internal cell ratio of the water-absorbent resin of the present invention (specified by the measurement method in the examples) is 2.8 to 6.6%, preferably 3.0 to 6.5%, more preferably 3.5 to It is 6.5%, particularly preferably 3.8 to 6.5%, and most preferably 4.0 to 6.5%.
- FSR water absorption rate
- SFC liquid permeability
- Patent Documents 18 to 35 and the like Conventionally, in order to improve the water absorption rate, foam polymerization of a water-absorbing resin is known in Patent Documents 18 to 35 and the like.
- Patent Document 31 US Pat. No. 6,100,358
- Comparative Example 8 of the present application
- excessive internal bubbles exceed 6.6%, or a large amount of surfactant (for example, 0.1 to 10% by weight) is used for foaming as in Patent Documents 28 and 29.
- the surface tension of the obtained water-absorbent resin powder is reduced (particularly less than 60 [mN / m], and further less than 55 [mN / m]), or fine powder is generated due to excessive foaming (particularly 10% by weight or more). Or had problems.
- p. 197 to 199 and Table 5.6 include commercially available (polyacrylic acid) water-absorbing resins (5 types), 40 to 60 mesh-Cut (corresponding to upper and lower limits of 425 to 250 ⁇ m powder) BET surface area, water absorption Disclose speed, water absorption, bulk specific gravity and apparent density.
- Non-Patent Document 1 as specific values of the apparent density by the methanol wet method, trade names Arasorb 720 (Arakawa Chemical) and Sanwet 1M-1000 (Sanyo Kasei) are 1.500 [g / cm 3 ], Aridall 1078 ( American Colloid) discloses 1.250 [g / cm 3 ], Aquakeep (Sumitomo Seika) and Drytech 510 (Dow Chemical) disclose 1.667 [g / cm 3 ]. That is, Non-Patent Document 1 discloses five types of commercially available water-absorbing resins having an apparent density of 1.250 to 1.667 [g / cm 3 ].
- Non-Patent Document 1 the apparent density (methanol wet method) of 40-60 mesh-Cut products is different from the dry density of the entire particle size of the present application, and there is no description of individual true density or chemical composition.
- Aquakeep reverse phase suspension polymerization / spherical particles
- Table 5.6 the internal cell ratio of the commercially available water-absorbent resin (Table 5.6) is 0% or close to 0% (Aquakeep, Drytech 510), and about 10 to 25% (Arasorb720, Sanwet 1M-1000, Aridall 1078) based on the above assumption.
- the present invention is characterized in that the internal cell ratio (2.8 to 6.6%) and the particle size are controlled to a specific narrow range (the ratio of the particle size of 850 to 150 ⁇ m is 95% by weight or
- Patent Document 31 (U.S. Pat. No. 5,856,370) uses an azo compound, and the density in a dry state exceeds 1.0 [g / cm 3 ], and the density in a swollen state (in a specific gravity bottle). The measurement) discloses a porous water-absorbent resin having 1.0 [g / cm 3 ], but does not disclose the specific internal cell ratio or particle size of the present invention.
- Such a water-absorbent resin powder (first water-absorbent resin) of the present invention has a high impact resistance and has a small amount of fine powder, and a ratio of a particle size of 850 to 150 ⁇ m is 95% by weight or more, despite being obtained by foam polymerization. Further, the upper and lower limits (850/710 ⁇ m to 150 ⁇ m) and the weight average particle diameter (D50) of the range (2-5) are shown. Conventionally, a technique for reducing the particle size as in Patent Document 10 is known to improve the water absorption speed, but such a method involves an increase in fine powder, but the present invention does not have such a problem. The more preferable particle size of the water absorbent resin powder of the present invention is as described in the above (2-5).
- the water absorbent resin powder (first water absorbent resin) of the present invention is obtained by foam polymerization, a large amount of surfactant (for example, 0.1 to 10% by weight) is not required, the surface tension is not reduced, the surface tension is 60 mN / m or more, and the surface tension is in the range of (3-7) described later.
- the surface tension can be adjusted by the type and amount of the surfactant used, but is preferably used within the range (2-1-6).
- the water-absorbent resin powder of the present invention using the production method of the present invention (reducing gas solubility) as an example of the production method has a particle size ratio of 850 to 150 ⁇ m of 95% by weight or more, preferably 98% by weight or more.
- the polyacrylic acid-based water-absorbent resin powder is 99% by weight or more
- the surface tension is 60 [mN / m] or more
- the internal cell ratio defined by the following formula is 2.8 to 6.6%. It is a water-absorbent resin powder characterized by being.
- Such a water-absorbent resin powder of the present invention is obtained, for example, by a production method including the above-described surface crosslinking, particularly surface crosslinking up to the above-mentioned CRC range, and preferably, the absorption capacity under pressure at a load of 50 [g / cm 2 ].
- This is a water-absorbent resin powder having (AAP) of 15 [g / g] or more.
- the preferred range of AAP is as follows. When AAP is low, when the water absorbent resin concentration is high in paper diapers, sufficient water absorption performance may not be exhibited.
- Such a water-absorbent resin powder of the present invention is obtained, for example, by a production method including the above-described surface crosslinking, particularly surface crosslinking up to the above-mentioned CRC range, and preferably has a saline flow conductivity (SFC) of 20 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ sec ⁇ g ⁇ 1 ] or more.
- SFC saline flow conductivity
- Such a water-absorbent resin powder preferably further contains a liquid permeability improver selected from polyvalent metal cations, polyamine polymers and water-insoluble fine particles exemplified in the above (2-6) in the surface crosslinking.
- a liquid permeability improver selected from polyvalent metal cations, polyamine polymers and water-insoluble fine particles exemplified in the above (2-6) in the surface crosslinking.
- the water-absorbent resin powder of the present invention preferably further contains a surfactant by the method exemplified in the above (2-6) or (2-1-6).
- a surfactant By containing the surfactant, it is possible to obtain a water-absorbing resin powder having more excellent impact resistance.
- a suitable use amount is in the above-mentioned range, and it is preferably added as an aqueous solution.
- Such a water-absorbent resin powder of the present invention preferably further contains a surfactant in the above range or the above range of surface tension by using a surfactant during polymerization or before and after surface crosslinking.
- Such a water-absorbent resin powder of the present invention preferably has 5 to 60 ppm, more preferably 5 to 40 ppm, and still more preferably 5 to 30 ppm p-methoxyphenol (MEHQ) as shown in (2-1-7) above. Furthermore, by including, it can be set as the water absorbing resin powder which was further excellent in weather resistance. When p-methoxyphenol is excessive, coloring may occur, and when it is small, weather resistance may be decreased.
- MEHQ p-methoxyphenol
- Non-Patent Document 1 In “2.5.3 Inhibition” (polymerization inhibitor) (p.39-44) of Non-Patent Document 1, p-methoxyphenol in commercially available water-absorbing resins (8 types) in Table 2.5 is 16 Although the fact that it is ⁇ 151 ppm is disclosed, Non-Patent Document 1 does not disclose the effects of the present application (preventing coloring and improving light resistance by controlling to a specific range).
- the amount of p-methoxyphenol can be adjusted in the final product water-absorbing resin according to the adjustment in the polymerization step and the drying step, for example, according to the method described in PCT / JP2010 / 067086.
- the neutralization step is performed with a basic substance having an iron content of 0 to 7 ppm
- the polymerization step includes 90 to 100 mol% of acrylic acid (salt), and a monomer concentration of 30 to A condition in which a 55 wt% aqueous monomer solution is used with a radical polymerization initiator of 0.001 to 1 mol%, the maximum temperature reached during polymerization is 130 ° C. or less, and the polymerization time is 0.5 minutes to 3 hours.
- the drying step is the drying temperature of the particulate hydrous gel-like crosslinked polymer obtained in the polymerization step (including the gel refinement step).
- Such a water-absorbent resin powder of the present invention preferably further contains an additive selected from a chelating agent, ⁇ -hydroxycarboxylic acid, inorganic or organic reducing agent, as shown in (2-9) above.
- an additive selected from a chelating agent, ⁇ -hydroxycarboxylic acid, inorganic or organic reducing agent, as shown in (2-9) above.
- the water absorbent resin is a polyacrylic acid water absorbent resin powder containing a surfactant and / or a dispersant therein, as in the second water absorbent resin described later.
- the surface tension is 60 [mN / m] or more and the powder surface is coated with a surfactant.
- SFC saline flow conductivity
- FSR water absorption rate
- the water absorbent resin (first water absorbent resin powder) preferably has a predetermined moisture content (specified in the examples) of 15% by weight or less, like the second water absorbent resin described later. 0.1 to 10% by weight is more preferable, and a water content of 1 to 8% by weight is more preferable.
- the water content is low, the water absorption rate (for example, FSR) and impact resistance are reduced, and conversely, when the water content is high, the water absorption capacity under no pressure (CRC) and the water absorption capacity under pressure (AAP) tend to decrease. It is in.
- the water content can be controlled by adjusting the heating temperature and time after polymerization, or by adding water.
- a surfactant and / or a dispersant is preferably used at the time of polymerization, and the surface of the water absorbent resin powder is preferably further coated with a surfactant.
- the water-absorbent resin powder obtained by the production method is a polyacrylic acid-based water-absorbent resin powder containing a surfactant and / or a dispersant substantially uniformly inside, and has a surface tension of 60 [mN / m] or more.
- a water absorbent resin powder, wherein the powder surface is further coated with a surfactant.
- the surface and internal surfactants can be discriminated by, for example, quantifying the presence or absence of a difference in the density of the surfactant in the thickness direction of the particles by polishing the surface of the particles or extracting the solvent only on the surface.
- the present invention is a polyacrylic acid-based water absorbent resin containing a surfactant and / or a dispersant as a novel water absorbent resin (second water absorbent resin powder), and has a surface tension of 60 [ mN / m] and a water-absorbent resin powder, the powder surface of which is further coated with a surfactant.
- Such a new water-absorbent resin maintains a high surface tension, so that the amount of return (Re-Wet) when using a paper diaper is small, and a very small amount of surfactant is disclosed in US Pat. No. 7,282,262.
- the insertion distance can be controlled high.
- Such a water absorbent resin powder (second water absorbent resin powder) preferably has an insertion distance (PID) of 13 [mm] or more.
- Insertion distance can be controlled by the use of surfactants inside the particles (during polymerization) or on the particle surface (especially surface cross-linking after drying), especially the use of surfactants inside and on the surface (see Examples and below) (See also Table 6).
- Such a water-absorbent resin has both liquid permeability and water absorption speed, and preferably has a saline flow conductivity (SFC) of 20 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ sec ⁇ g ⁇ 1 ] or more, A water-absorbing resin powder having a water absorption rate (FSR) of 0.25 [g / g / sec] or more.
- SFC saline flow conductivity
- FSR water absorption rate
- the water absorbent resin powder obtained by the production method of the present invention or the first or second water absorbent resin powder of the present invention preferably satisfies the following physical properties.
- the water absorbent resin powder obtained by the production method of the present invention or the first or second water absorbent resin powder of the present invention preferably satisfies the following physical properties.
- the following physical properties especially paper diapers, at least one of the following (3-1) to (3-7), and more than 2 including AAP, especially 3 Preferably it is controlled. If the following conditions are not satisfied, the high-density paper diaper described below may not exhibit sufficient performance.
- the production method of the present invention can be preferably applied to the following water-absorbent resin powder production method, but more preferably can be applied to control and improve liquid permeability (SFC) and water absorption rate (FSR).
- SFC liquid permeability
- FSR water absorption rate
- the polyacrylic acid water-absorbent resin powder according to the present invention preferably has a water absorption rate index of 90 or more and a bulk specific gravity of 0.58 to 0.8 [g / cm 3 ] defined by the following formula: More preferably, it is 6 to 0.8 [g / cm 3 ].
- Such a method for producing the water-absorbent resin powder of the present invention has a water absorption rate index defined by the following formula of 90 or more, a bulk specific gravity of 0.58 to 0.8 [g / cm 3 ], 6-0.8 [g / cm 3 ] polyacrylic acid water-absorbing resin powder is surface-crosslinked.
- a preferable method for surface crosslinking and a method for controlling the water absorption rate index can be carried out by the above-described methods, preferably surface crosslinking is performed, and preferably has a water absorption rate index in the above range.
- FSR indicates the water absorption rate at 20 times swelling in physiological saline.
- the water absorption rate index is preferably as high as 90, 95, 100, 105, 110, 115, 120 in order, and the upper limit is 150, and 140 is sufficient.
- Such a novel water-absorbent resin powder is excellent in liquid permeability and impact resistance and can be preferably used for absorbent articles such as paper diapers. When the water absorption rate index is low or too high, it tends to be unsuitable for actual use.
- Such a water-absorbent resin powder has a foam structure (also known as a porous structure), and the porous structure can be discriminated by confirming the particle surface with an electron micrograph.
- the average pore diameter on the particle surface is preferably 100 ⁇ m or less, more preferably 0.1 to 90 ⁇ m, and still more preferably 1 to 50 ⁇ m.
- the main component of each water absorbent resin powder is porous particles.
- AAP Water absorption capacity under pressure
- AAP Water absorption capacity under pressure
- the water absorption capacity (AAP) is preferably controlled to 20 [g / g] or more, more preferably 22 [g / g] or more, and further preferably 24 [g / g] or more.
- the upper limit of AAP is preferably as high as possible, but is usually 40 [g / g], more preferably 35 [g / g], particularly about 30 [g / g] in the case of a load of 4.8 kP, in view of balance with other physical properties. .
- the water absorption capacity (CRC) under no pressure is preferably 10 [g / g] or more, more preferably 20 [g / g] or more, still more preferably 25 [g / g] or more, particularly preferably 30 [g. / G] or more.
- CRC can be controlled by the amount of crosslinking agent and the like.
- Non-Patent Document 1 discloses the water absorption ratio of a commercially available water absorbent resin (there is no detailed description of the measurement method). Specifically, the fact that Aquakeep is 65.4 [g / g] and Sanwet 1M-1000 is 58.3 [g / g] is disclosed, but the subject of the present invention (coexistence of liquid permeability and water absorption speed) In the present invention, it is preferable to control the water absorption capacity (CRC) under no pressure within the above range.
- CRC water absorption capacity
- SFC is a well-known measurement method and can be defined by the method described in US Pat. No. 5,562,646, for example.
- the water-absorbent resin with controlled closed cell rate according to the present invention can achieve both high SFC and FSR.
- the upper limit of SFC is appropriately determined, but is preferably about 1000 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ sec ⁇ g ⁇ 1 ] in view of balance with other physical properties.
- the water-absorbent resin of the present invention (particularly the first water-absorbent resin with controlled closed cell ratio or the second water-absorbent resin powder containing a surfactant on the inside and on the surface) has an SFC of 50 [ ⁇ 10 ⁇ 7.
- the SFC drop width is preferably 15 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ sec ⁇ g ⁇ 1 ] or less, more preferably 10 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ sec ⁇ g ⁇ 1 ] or less, Particularly preferably, it can be 5 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ sec ⁇ g ⁇ 1 ] or less, and the damage resistance is excellent.
- the water-soluble content is preferably 35% by weight or less, more preferably 25% by weight or less, still more preferably 15% by weight or less, and particularly preferably 10% by weight or less.
- the water-absorbent resin of the present invention has a water absorption rate (FSR) of 1 g of water-absorbent resin powder with respect to 20 g of physiological saline, usually 0.05 [g / g / sec] or more, preferably 0.1 [g / g / sec] or more, more preferably 0.15 [g / g / sec] or more, still more preferably 0.20 [g / g / sec] or more, particularly preferably Is 0.25 [g / g / sec] or more.
- FSR water absorption rate
- the measurement method of FSR is defined in International Publication No. 2009/016055.
- the water-absorbent resin with controlled closed cell rate according to the present invention can achieve both high SFC and FRS.
- the bulk specific gravity of the water-absorbent resin powder is usually 0.58 to 0.8 (synonymous with 0.58 to 0.80) [g / cm 3 ], preferably 0. 0.6 to 0.8 (synonymous with 0.60 to 0.80) [g / cm 3 ], more preferably 0.63 to 0.77 [g / cm 3 ], still more preferably 0.8. 66 to 0.74 [g / cm 3 ].
- it has a foam structure (also called a porous structure), but has a high bulk specific gravity.
- Non-Patent Document 1 the bulk specific gravity of a commercially available water-absorbent resin (the measurement method is not described in detail) is disclosed. Specifically, the fact that Aquakeep is 0.4430 [g / cm 3 ], Sanwet 1M-1000 is 0.5624 [g / cm 3 ], and Drytech 510 is 0.8989 [g / cm 3 ] is disclosed. In the present invention, it is preferable to control within the above range. Bulk specific gravity can be controlled by the production method of the present invention.
- the surface tension (specified by the measurement method in the examples) is preferably 60 [mN / m] or more, more preferably 65 [mN / m] or more, and still more preferably 67 [mN / m]. As described above, it is particularly preferably 70 [mN / m] or more, most preferably 72 [mN / m] or more, and there is no substantial reduction in surface tension. The upper limit is usually 75 [mN / m].
- the use of the water-absorbent resin powder of the present invention is not particularly limited, but it can be preferably used for absorbent articles such as paper diapers, sanitary napkins and incontinence pads. Since the water-absorbent resin powder of the present invention has both liquid permeability and water absorption speed and is excellent in impact resistance and absorption capacity under pressure, it can be preferably used for absorbent articles having a high content of water-absorbent resin.
- the content (core concentration) of the water-absorbent resin powder in the absorbent body optionally containing other absorbent materials (pulp fibers, etc.) in this absorbent article is 30 to 100% by weight, preferably 40 to 100% by weight. More preferably, the effect of the present invention is exhibited at 50 to 100% by weight, further preferably 60 to 100% by weight, particularly preferably 70 to 100% by weight, and most preferably 75 to 95% by weight.
- the solid content was measured as follows.
- the bulk specific gravity was measured according to JIS K 3362 using a bulk specific gravity measuring instrument (manufactured by Kuramochi Scientific Instruments). After 100.0 g of the water-absorbing resin powder mixed sufficiently to eliminate the unevenness due to the particle size is put into the funnel with the damper closed, the damper is quickly opened, and the water-absorbing resin powder is received in the receiver (weight W9 [G]). After the water-absorbing resin powder swelled from the receiver is rubbed off with a glass rod, the weight (weight W10 [g]) of the receiver containing the water-absorbent resin powder is accurately weighed to 0.1 g, and the number is 5 Therefore, the bulk specific gravity was calculated.
- the temperature of the environment where the measurement was performed was 24.2 ° C.
- the relative humidity was 43% RH.
- the whiteness is an index indicating the whiteness of the powder, and is calculated using X, Y, Z values or L, a, b values.
- the WB value of the water-absorbent resin powder useful for comparing the degree of whiteness was measured using a spectral color difference meter.
- coloring (progress) during storage for a long period of time and coloring (progress) in a sanitary material are referred to as coloration with time.
- the true density in the present invention was determined by measuring the dry density of the water-absorbent resin powder in which the internal closed cells were broken or formed into open cells by finely pulverizing them into a 45 ⁇ m passage through a JIS standard sieve.
- the bubble diameter (closed cell) contained in the water-absorbent resin is usually 1 to 300 ⁇ m.
- the portion close to the bubbles is preferentially pulverized at the time of pulverization, and the water-absorbent resin powder pulverized to 45 ⁇ m or less contains almost no closed cells. Therefore, the water-absorbent resin pulverized to 45 ⁇ m or less
- the true density of the water absorbent resin powder was determined by measuring the dry density of the powder.
- the true density was measured using a water absorbent resin powder pulverized to less than 45 ⁇ m of JIS standard sieve. That is, in a ball mill pot (made by Terraoka Co., Ltd., magnetic ball mill pot model No. 90, inner dimension: diameter 80 mm, height 75 mm, outer dimension: diameter 90 mm, height 110 mm), 400 g of water-absorbing resin powder 15.0 g. A cylindrical magnetic ball (diameter 13 mm, length 13 mm) was placed and pulverized for 2 hours at 60 Hz using a ball mill. As a result, a water-absorbing resin powder in which 70% by weight or more of the water-absorbing resin powder passed through a JIS standard sieve of 45 ⁇ m was obtained.
- p-methoxyphenol vs. water-absorbing resin
- ERT470.2-02 stirred is 1 hour
- a metal halide lamp with a stirring blade having four flat blades having a distance from the center of the shaft to the blade tip of 3.0 cm and a width of 1.0 cm while stirring the 20-fold swollen gel in the separable flask ( Using an ultraviolet irradiation device (manufactured by Ushio Electric; UVL-1500M2-N1) (manufactured by Ushio; UV-152 / 1MNSC3-AA06), an ultraviolet ray with an irradiation intensity of 60 [mW / cm 2 ] was applied at room temperature for 1 minute. Irradiated to obtain a water-containing gel-like water-absorbing agent subjected to a weather resistance promotion test.
- UVL-1500M2-N1 manufactured by Ushio; UV-152 / 1MNSC3-AA06
- the deterioration rate can be calculated from the difference in soluble content between the water-containing gel water-absorbing agent (after deterioration) and the water-absorbent resin powder (before deterioration).
- the individual soluble components are not particularly described, but they were all in the range of 15% by weight or less (10% by weight or less).
- Example 1 In a polypropylene container with a capacity of 3 liters, 181.1 g of acrylic acid (containing p-methoxyphenol: 70 mg / L), 1727.0 g of 37 wt% aqueous sodium acrylate, polyethylene glycol diacrylate (molecular weight 523) as an internal cross-linking agent 4.38 g, 13.7 wt% polyoxyethylene (20) sorbitan monostearate (manufactured by Kao Co., Ltd.) aqueous solution 59.7 g as a surfactant was added and dissolved (mixed) to dissolve the monomer aqueous solution (1) The liquid temperature was adjusted to 24 ° C.
- the monomer aqueous solution (1) is placed in a stainless steel coil heat exchanger (self-standing coil exchanger, model number JC-S1; ASONE Corporation, Research Comprehensive Equipment Catalog) immersed in an oil bath at 100 ° C. Then, it was passed at 0.5 [L / min] using a metering pump, and the liquid temperature was raised to 98.5 ° C. At this time, the monomer aqueous solution (1) containing the surfactant was clouded by the introduction of very fine bubbles. This white turbidity phenomenon is caused by a decrease in gas solubility due to the temperature rise of the monomer aqueous solution (1).
- the monomer aqueous solution (1) was poured into a bat-type container and the polymerization reaction started 40 seconds later.
- the polymerization reaction proceeded by expanding and foaming up, down, left and right while generating water vapor, and then contracted to a size slightly larger than the bat-type container. This expansion and contraction was completed within about 1 minute.
- a hydrogel crosslinked polymer (hydrogel) was taken out.
- a hydrogel crosslinked polymer (hydrogel) obtained by the above polymerization reaction was used as a meat chopper (manufactured by Iizuka Kogyo Co., Ltd., MEAT-CHOPER TYPE: 12VR-400KSOX, die hole diameter: 6.4 mm, hole number: 38, die thickness. 8 mm) to obtain a finely divided hydrogel crosslinked polymer.
- the input amount of the hydrogel was 350 [g / min]
- pulverization was performed while adding deionized water adjusted to 90 ° C. at 80 [g / min] in parallel with the addition of the hydrogel.
- the finely divided hydrogel crosslinked polymer obtained by the above crushing operation was spread on a stainless steel wire mesh having an opening of 850 ⁇ m and dried with hot air at 180 ° C. for 30 minutes. Subsequently, the dried product obtained by the drying operation is pulverized using a roll mill (manufactured by Inoguchi Giken Co., Ltd., WML type roll pulverizer), and then classified using a JIS standard sieve having an opening of 850 ⁇ m and an opening of 45 ⁇ m. did.
- Example 2 In a polypropylene container having a capacity of 2 liters, 351.6 g of acrylic acid, 2.17 g of polyethylene glycol diacrylate (molecular weight 523) as an internal crosslinking agent, 94.6 g of 0.1 wt% diethylenetriaminepentaacetic acid / trisodium aqueous solution as a chelating agent, 144.9 g of 48.5% by weight sodium hydroxide aqueous solution, 6.45 g of 1.0% by weight polyoxyethylene (20) sorbitan monostearate (manufactured by Kao Corporation) as a surfactant, deionized water (ion-exchanged water) ) 236.0 g was charged and dissolved (mixed) to prepare an aqueous monomer solution (2 ′).
- the temperature of the aqueous solution (2 ′) was raised to 65 ° C. by the heat of neutralization in the first stage immediately after the production.
- the aqueous solution (2 ′) containing the surfactant was clouded with very fine bubbles introduced due to a decrease in the solubility of the gas accompanying the temperature increase.
- the monomer aqueous solution (2 ′) was cooled while stirring, and when the liquid temperature reached 53 ° C., 148.9 g of a 48.5 wt% sodium hydroxide aqueous solution adjusted to 30 ° C. was added.
- the monomer aqueous solution (2) was prepared by mixing. At this time, the temperature of the monomer aqueous solution (2) rose to 83.5 ° C. due to the heat of neutralization in the second stage immediately after the production.
- the monomer aqueous solution (2) containing the surfactant was clouded due to the introduction of very fine bubbles due to the decrease in gas solubility accompanying the temperature increase.
- the polymerization reaction started 15 seconds after the monomer aqueous solution (2) was poured into the vat-type container.
- the polymerization reaction proceeded by expanding and foaming up, down, left and right while generating water vapor, and then contracted to a size slightly larger than the bat-type container. This expansion and contraction was completed within about 1 minute.
- a hydrogel crosslinked polymer (hydrogel) was taken out. The series of operations were performed in an open air system, and the peak temperature during polymerization was 108 ° C.
- hydrogel crosslinked polymer hydrogel
- Example 3 In a polypropylene container with a capacity of 1 liter, 379.07 g of a 37 wt% sodium acrylate aqueous solution, 0.995 g of polyethylene glycol diacrylate (molecular weight 523) as an internal cross-linking agent, 176.31 g of deionized water (ion exchange water), surface activity As an agent, 0.04 g of polyoxyethylene (20) sorbitan monostearate (manufactured by Kao Corporation) was added and dissolved (mixed) to prepare a monomer aqueous solution (3). The monomer aqueous solution (3) was deaerated using nitrogen gas for 5 minutes while adjusting the temperature to 25 ° C.
- the hydrogel crosslinked polymer was taken out from the polypropylene container. Thereafter, in the same manner as in Example 1, the obtained hydrogel crosslinked polymer was subjected to operations of crushing, drying, crushing, and classification.
- Example 4 37 wt% sodium acrylate aqueous solution 595.4 [g / min], 48 wt% sodium hydroxide aqueous solution 198.6 [g / min], 100 wt% acrylic acid 300.1 [g / min], as an internal crosslinking agent Polyethylene glycol diacrylate (molecular weight 523) 2.71 [g / min], deionized water (ion-exchanged water) 204.7 [g / min], 31 wt% diethylenetriaminepentaacetic acid / trisodium aqueous solution 0.42 [g / min], 10 wt% polyoxyethylene (20) sorbitan monostearate (manufactured by Kao Corporation) aqueous solution 0.29 [g / min] as a surfactant is mixed by line mixing, and monomer aqueous solution (4) was made.
- Stainless steel coil heat exchanger self-standing coil type exchanger, model number JC-S1; ASONE Corporation, Research Comprehensive Equipment Catalog), in which the monomer aqueous solution (4) is continuously immersed in an oil bath at 95 ° C. ( (See FIG. 1).
- the monomer aqueous solution (4) containing a surfactant was clouded with very fine bubbles introduced therein.
- the jacket temperature of the polymerization apparatus was set to 95 ° C., and nitrogen gas was blown into the polymerization apparatus at 20 [L / min] (see FIG. 6).
- the monomer aqueous solution (4) started the polymerization reaction immediately after being supplied to the polymerization apparatus.
- the polymerization reaction and the shearing of the hydrogel crosslinked polymer were simultaneously performed, and the crushed hydrogel crosslinked polymer was continuously discharged from the polymerization apparatus. Thereafter, in the same manner as in Example 1, the obtained hydrogel crosslinked polymer was subjected to operations of crushing, drying, crushing, and classification.
- Example 5 Combined use with introduction of bubbles (for example, introduction of microbubbles)
- polyethylene glycol diacrylate molethacrylate
- 0 0.1 wt% diethylenetriaminepentaacetic acid / trisodium aqueous solution 113.5 g
- the temperature of the aqueous solution (5 ') was increased to 64 ° C by the first stage neutralization heat immediately after the production. While adjusting the temperature of this aqueous solution (5 ′) to 55 ° C., using a microbubble generator (Auratech Co., Ltd., model: OM4-GP-040) under an absolute pressure of 0.30 to 0.35 MPa. Then, microbubbles were introduced into the aqueous solution (5′A) for 1 minute using nitrogen gas as an introduction gas. The aqueous solution (5 ') containing the surfactant was clouded with very fine bubbles introduced by the microbubble generator.
- a microbubble generator Auratech Co., Ltd., model: OM4-GP-040
- the polymerization reaction started 10 seconds after the monomer aqueous solution (5) was poured into the vat-shaped container.
- the polymerization reaction proceeded by expanding and foaming up, down, left and right while generating water vapor, and then contracted to a size slightly larger than the bat-type container. This expansion and contraction was completed within about 1 minute.
- a hydrogel crosslinked polymer (hydrogel) was taken out. The series of operations was performed in an open air system, and the peak temperature during polymerization was 111 ° C.
- hydrogel hydrogel crosslinked polymer
- Example 1 an irregularly shaped water-absorbent resin powder (5) having a solid content of 97% by weight, a weight average particle size (D50) of 451 ⁇ m, and a logarithmic standard deviation ( ⁇ ) of 0.36 in the particle size distribution was obtained.
- Table 1 shows properties of the water absorbent resin powder (5).
- Example 6 Combined use with introduction of bubbles (for example, introduction of microbubbles) Addition of an aqueous solution of 10.0% by weight polyoxyethylene (20) sorbitan monostearate (manufactured by Kao Corporation) as a surfactant The same operation as in Example 5 was performed except that the change was made immediately after the introduction of.
- polyoxyethylene (20) sorbitan monostearate manufactured by Kao Corporation
- Example 7 In place of the 1.0 wt% polyoxyethylene (20) sorbitan monostearate (made by Kao Corporation) aqueous solution of Example 2 as the surfactant, a 1.0 wt% sorbitan monolaurate (made by Kao Corporation) aqueous solution A monomer aqueous solution (7) was prepared in the same manner as in Example 2 except that was used. The temperature of the aqueous monomer solution (7) rose to 63 ° C. by the heat of neutralization immediately after preparation.
- Example 2 Thereafter, the same operation as in Example 2 was performed, and an irregularly crushed water-absorbent resin powder having a solid content of 96% by weight, a weight average particle size (D50) of 439 ⁇ m, and a logarithmic standard deviation of particle size distribution ( ⁇ ) of 0.44 ( 7) was obtained.
- Table 1 shows properties of the water absorbent resin powder (7).
- Example 8 Instead of the 1.0 wt% polyoxyethylene (20) sorbitan monostearate (made by Kao Corporation) aqueous solution of Example 2 as a surfactant, 1.0 wt% polyether-modified silicone (side chain modified terminal OH type) ) (Toray Dow Corning Co., Ltd.) A monomer aqueous solution (8) was prepared in the same manner as in Example 2 except that an aqueous solution was used. The temperature of the aqueous monomer solution (8) was increased to 63 ° C. by the heat of neutralization immediately after preparation.
- Example 2 Thereafter, the same operation as in Example 2 was performed, and an irregularly crushed water-absorbent resin powder having a solid content of 97% by weight, a weight average particle size (D50) of 427 ⁇ m, and a logarithmic standard deviation ( ⁇ ) of 0.39 of the particle size distribution ( 8) was obtained.
- Table 1 shows properties of the water absorbent resin powder (8).
- Example 9 Instead of 6.45 g of the 1.0 wt% polyoxyethylene (20) sorbitan monostearate (made by Kao Corporation) aqueous solution of Example 2 as a surfactant, 3.0 wt% sodium carboxymethylcellulose (Sigma Aldrich Japan Co., Ltd.) A monomer aqueous solution (9) was prepared in the same manner as in Example 2 except that 14.3 g of an aqueous solution (manufactured by company) was used. The temperature of the aqueous monomer solution (9) rose to 64 ° C. due to the heat of neutralization immediately after preparation.
- Example 2 Thereafter, the same operation as in Example 2 was carried out, and an irregularly crushed water-absorbent resin powder having a solid content of 96% by weight, a weight average particle size (D50) of 463 ⁇ m, and a logarithmic standard deviation ( ⁇ ) of 0.39 in the particle size distribution ( 9) was obtained.
- Table 1 shows properties of the water absorbent resin powder (9).
- Example 1 As a surfactant, 59.7 g of the 13.7 wt% polyoxyethylene (20) sorbitan monostearate (produced by Kao Corporation) aqueous solution of Example 1 was added to 59.7 g of deionized water (ion-exchanged water) of the same weight. Except that it was replaced, the same operation as in Example 1 was performed, and the comparative water absorption in an irregularly crushed shape having a solid content of 97% by weight, a weight average particle size (D50) of 432 ⁇ m, and a logarithmic standard deviation ( ⁇ ) of 0.44 Resin powder (1) was obtained. Table 1 shows properties of the comparative water absorbent resin powder (1).
- the comparative monomer aqueous solution (1) prepared in Comparative Example 1 was heated to 98.5 ° C. by the heat exchanger (FIG. 1), which was almost the same as Example 1, but contained a surfactant.
- the generated bubbles were not stable, and as a result, fine bubbles were not introduced, and it was almost colorless and transparent.
- Example 2 As a surfactant, 6.45 g of a 1.0 wt% polyoxyethylene (20) sorbitan monostearate (manufactured by Kao Corporation) aqueous solution of Example 2 was added to 6.45 g of deionized water (ion exchange water) of the same weight. Except for the above, the same operation as in Example 2 was performed, and the comparatively water absorption in an irregularly crushed shape having a solid content of 96% by weight, a weight average particle size (D50) of 455 ⁇ m, and a logarithmic standard deviation of particle size distribution ( ⁇ ) of 0.37. Resin powder (2) was obtained. Table 1 shows properties of the comparative water absorbent resin powder (2) obtained. Table 2 shows the whiteness of the comparative water absorbent resin powder (2).
- the comparative monomer aqueous solution (2) prepared in Comparative Example 2 does not contain a surfactant, the generated bubbles are not stable, and fine bubbles are not introduced (white turbidity).
- the aqueous monomer solution after the temperature increase was almost colorless and transparent.
- Example 3 As a surfactant, the 1.0 wt% polyoxyethylene (20) sorbitan monostearate (produced by Kao Corporation) aqueous solution of Example 2 was not mixed with the monomer aqueous solution (2 ′), but the monomer aqueous solution. (2 ′) and a 48.5 wt% aqueous sodium hydroxide solution were mixed (83 ° C.), except that it was added to the monomer aqueous solution (2). An irregularly crushed comparative water-absorbent resin powder (2) having 97% by weight, a weight average particle diameter (D50) of 444 ⁇ m, and a logarithmic standard deviation ( ⁇ ) of 0.43 of the particle size distribution was obtained. Table 1 shows properties of the comparative water absorbent resin powder (3).
- Example 4 The same operation as in Example 3 was carried out except that 0.04 g of polyoxyethylene (20) sorbitan monostearate of Example 3 (manufactured by Kao Corporation) was not used as the surfactant, and the solid content was 96% by weight, An irregularly crushed comparative water-absorbent resin powder (4) having a weight average particle size (D50) of 458 ⁇ m and a logarithmic standard deviation ( ⁇ ) of 0.40 in particle size distribution was obtained. Table 1 shows properties of the comparative water absorbent resin powder (4) obtained.
- D50 weight average particle size
- ⁇ logarithmic standard deviation
- Example 5 The same operation as in Example 4 was carried out except that the 10% by weight polyoxyethylene (20) sorbitan monostearate (made by Kao Corporation) aqueous solution of Example 4 was not used as the surfactant, and the solid content was 97% by weight.
- Table 1 shows properties of the comparative water absorbent resin powder (5).
- foam polymerization was performed using carbonate. That is, in a polypropylene container having a capacity of 1 liter, acrylic acid 421.7 g, polyethylene glycol diacrylate (molecular weight 523) 2.754 g as an internal crosslinking agent, 0.1 wt% diethylenetriaminepentaacetic acid / trisodium aqueous solution 113.43 g, 48 A 0.4 wt% sodium hydroxide aqueous solution (140.4 g) and deionized water (ion exchange water) 292.3 g were added and dissolved (mixed) to prepare a comparative monomer aqueous solution (6 ′).
- a comparative monomer aqueous solution (6) is prepared by quickly adding 211.9 g of a 48.5% by weight sodium hydroxide aqueous solution adjusted to 40 ° C. to the comparative monomer aqueous solution (6 ′) and mixing them. did. At this time, the temperature of the comparative monomer aqueous solution (6) was 85 ° C.
- the vat-shaped container was heated using a hot plate (manufactured by Inoue Seieido Co., Ltd .; NEO HOTPLATE HI-1000) until the surface temperature reached 80 ° C.
- the polymerization reaction started soon after the comparative monomer aqueous solution (6) was poured into the vat-type container.
- the polymerization reaction proceeded by expanding and foaming up, down, left and right while generating water vapor, and then contracted to a size slightly larger than the bat-type container. This expansion and contraction was completed within about 1 minute.
- a hydrogel crosslinked polymer (hydrogel) was taken out.
- a hydrogel crosslinked polymer (hydrogel) obtained by the above polymerization reaction was used as a meat chopper (manufactured by Iizuka Kogyo Co., Ltd., MEAT-CHOPER TYPE: 12VR-400KSOX, die hole diameter: 6.4 mm, hole number: 38, die thickness. 8 mm) to obtain a finely divided hydrogel crosslinked polymer.
- the input amount of the hydrogel was 350 [g / min]
- pulverization was performed while adding deionized water adjusted to 90 ° C. at 80 [g / min] in parallel with the addition of the hydrogel.
- the finely divided hydrogel crosslinked polymer obtained by the above crushing operation was spread on a stainless steel wire mesh having an opening of 850 ⁇ m and dried with hot air at 180 ° C. for 30 minutes. Subsequently, the dried product obtained by the drying operation is pulverized using a roll mill (manufactured by Inoguchi Giken Co., Ltd., WML type roll pulverizer), and then classified using a JIS standard sieve having an opening of 850 ⁇ m and an opening of 45 ⁇ m. did.
- Comparative Example 7 Polymerization was carried out in the presence of a water-absorbent resin powder in accordance with Patent Document 34 (US Patent Publication No. 2007/0225422). That is, in Comparative Example 6, it was obtained in Reference Example 1 immediately after the addition of 17.55 g of a 4 wt% aqueous sodium persulfate solution without adding 5.2 g of sodium hydrogen carbonate (manufactured by Wako Pure Chemical Industries, Ltd.).
- Comparative Example 7 The same operations as in Comparative Example 7 were performed except that 25.8 g of the reference water-absorbent resin powder (1) was added, and the solid content was 97 wt%, the weight average particle size (D50) 446 ⁇ m, the logarithmic standard deviation of the particle size distribution ( A comparatively water-absorbent resin powder (7) having an irregularly crushed shape ( ⁇ ) 0.36 was obtained.
- Table 1 shows properties of the comparative water absorbent resin powder (7).
- Comparative Example 9 The comparative water-absorbent resin powder (2) obtained in Comparative Example 2 was classified using a JIS standard sieve having an opening of 600 ⁇ m, solid content was 97 wt%, weight average particle size (D50) 336 ⁇ m, logarithmic standard deviation of particle size distribution An irregular crushed comparative water-absorbent resin powder (9) with ( ⁇ ) 0.39 was obtained. Table 1 shows properties of the comparative water absorbent resin powder (9).
- Example 10 the reference water-absorbing resin powder (1) was continuously supplied at a rate of 57 [g / min] (fine powder) in the vicinity where the monomer aqueous solution (4) was supplied to the polymerization apparatus and polymerization started immediately. Except for being recycled), the same operation as in Example 4 was performed, and an irregularly crushed powder having a solid content of 96% by weight, a weight average particle size (D50) of 437 ⁇ m, and a logarithmic standard deviation of particle size distribution ( ⁇ ) of 0.43. Water-absorbent resin powder (10) was obtained. Table 1 shows properties of the water absorbent resin powder (10) obtained.
- the production method of the present invention does not impair other physical properties, does not require expensive auxiliary materials (a large amount of surfactant or foaming agent) or special equipment, and has improved the water absorption rate (FSR). It can be seen that a water absorbent resin powder is provided.
- the conventional foam polymerization shown in Comparative Examples 6 to 8 has insufficient improvement in water absorption rate (FSR) (Comparative Examples 6 and 7, 0.2 FSR), or bulk specific gravity is greatly reduced (Comparative Example 8).
- FSR water absorption rate
- Comparative Example 9 The method of reducing the average particle diameter as in Comparative Example 9 not only has an insufficient effect of improving the water absorption rate (FSR), but also the liquid permeability (for example, SFC) is greatly reduced by finer particles, or the amount of fine powder (For example, 150 ⁇ m passing material) greatly increases.
- the water-absorbent resin powder (2) of the present invention is used in Example 2 and Comparative Example 2 even when the same amount of chelating agent (acting as an anti-coloring agent or deterioration preventing agent) is used. It turns out that it is whiter (WB, X, Y, and Z are large).
- the water-absorbent resin powder of the present invention not only has a high water absorption rate (FSR), but can effectively improve whiteness even with the same amount of anti-coloring agent (chelating agent).
- Example 11 From 100 parts by weight of the water-absorbent resin powder (2) obtained in Example 2, from 0.48 parts by weight of 1,4-butanediol, 0.75 parts by weight of propylene glycol, and 4.0 parts by weight of deionized water. The resulting surface cross-linking agent solution was sprayed uniformly on the water-absorbent resin powder (2) and mixed. The water-absorbent resin particles mixed with the surface crosslinking agent solution were subjected to surface crosslinking treatment by heating with a hot air dryer (temperature: 180 ° C.) for 45 minutes. After the heat treatment, the obtained water-absorbent resin particles were pulverized until they passed through a JIS standard sieve having an opening of 850 ⁇ m, thereby obtaining water-absorbent resin particles whose surfaces were crosslinked.
- a hot air dryer temperature: 180 ° C.
- Example 12 With respect to 100 parts by weight of the water-absorbent resin powder (2) obtained in Example 2, 0.48 parts by weight of 1,4-butanediol, 0.75 parts by weight of propylene glycol, polyoxyethylene (20) sorbitan monostear A surface crosslinker solution consisting of 0.001 part by weight (made by Kao Corporation) (10 ppm with respect to the water absorbent resin powder) and 4.0 parts by weight of deionized water is uniformly applied to the water absorbent resin powder (2). A water absorbent resin powder (12) was obtained in the same manner as in Example 11 except for spraying. Table 3 shows properties of the water absorbent resin powder (12) obtained.
- the water-absorbent resin powder (12) obtained in Example 12 was obtained by further coating the surface of the water-absorbent resin powder (2) having 150 ppm of the surfactant substantially uniformly with 10 ppm of the surfactant.
- the surface tension was 67.4 [mN / m].
- Comparative Example 10 The same operation as in Example 11 was performed on the comparative water absorbent resin powder (2) obtained in Comparative Example 2 to obtain a comparative water absorbent resin powder (10).
- Table 3 shows properties of the comparative water absorbent resin powder (10).
- Example 11 The same operation as in Example 11 was performed on the comparative water absorbent resin powder (9) obtained in Comparative Example 9 to obtain a comparative water absorbent resin powder (11).
- Table 3 shows properties of the comparative water absorbent resin powder (11).
- Comparative Example 12 The same operation as in Example 11 was performed on the comparative water absorbent resin powder (8) obtained in Comparative Example 8 to obtain a comparative water absorbent resin powder (12).
- Table 3 shows properties of the comparative water absorbent resin powder (12).
- Example 13 The water-absorbent resin powder (11) obtained in Example 11 (internal cell ratio: 3.93%) was subjected to the damage resistance test described in “(5-9) Damage resistance test” above, and Water-absorbent resin powder (13) was obtained.
- Table 4 shows properties of the water absorbent resin powder (13) before and after the damage resistance test.
- PS test means “damage resistance test”.
- Example 14 The water-absorbent resin powder (12) obtained in Example 12 (internal cell ratio: 6.42%) was subjected to the damage resistance test described in “(5-9) Damage resistance test” above, and the damage was determined. Water-absorbent resin powder (14) was obtained. Table 4 shows properties of the water absorbent resin powder (14) before and after the damage resistance test.
- Comparative Example 13 The comparative water-absorbent resin powder (10) obtained in Comparative Example 10 (internal cell ratio 2.60%) was subjected to the damage resistance test described in the above “(5-9) Damage resistance test”. A comparative water absorbent resin powder (13) to which damage was given was obtained. Table 4 shows various physical properties of the comparative water absorbent resin powder (13) before and after the damage resistance test.
- Comparative Example 14 The comparative water absorbent resin powder (12) obtained in Comparative Example 12 (internal cell ratio 6.83%) was subjected to the damage resistance test described in the above “(5-9) Damage resistance test”. A comparative water absorbent resin powder (14) to which damage was given was obtained. Table 4 shows various physical properties of the comparative water absorbent resin powder (14) before and after the damage resistance test.
- the water-absorbent resin powder of the present invention has little deterioration in physical properties (SFC and FSR, particularly SFC) and is excellent in damage resistance. It can also be seen that the damage resistance is excellent by controlling the internal cell ratio of the water-absorbent resin powder to 2.8 to 6.6%. Such a water-absorbent resin powder does not deteriorate physical properties due to damage during pneumatic transportation or paper diaper production, and maintains high physical properties even after diaper production, particularly after high-concentration diaper production.
- Examples 15 and 16 With respect to the water absorbent resin powders (11) and (12) obtained in Examples 11 and 12, the apparent densities described in the above “(5-10) Apparent density” and “(5-11) True density”. Then, the true density was measured, and the internal bubble rate described in “(5-12) Internal bubble rate” was calculated. The results are shown in Table 5.
- Non-Patent Document 1 a commercially available paper diaper is purchased and used for the water-absorbent resin marketed at the time of filing in 2010, in order to examine the internal cell ratio, and the water-absorbent resin powder used. was taken out and analyzed.
- the true density of the water-absorbent resin fine particles after drying at 180 ° C. for 3 hours or more and finely pulverized to less than 45 ⁇ m is almost the same (1.652 to 1.656) regardless of the measurement sample.
- the true density is uniquely determined by the chemical composition of the water-absorbent resin (polymer repeating units and other trace raw materials), and it can be seen that the true density of the water-absorbent resin powder was accurately measured by this measurement method. .
- Non-Patent Document 1 As described in [3] Physical Properties of Polyacrylic Acid-Based Water Absorbent Resin Powder, Table 5.6 (p. 197 to 199) of Non-Patent Document 1 (issued 1998) includes a commercially available water absorbent resin (5 As for the type), the BET surface area, water absorption rate, water absorption rate, bulk specific gravity and apparent density of 40 to 60 mesh-Cut (corresponding to upper and lower limit 425 to 250 ⁇ m powder) products are disclosed. However, since Non-Patent Document 1 does not disclose a detailed measurement method, it cannot be directly compared with the product of the present invention. However, Patent Document 1 discloses a water-absorbent resin powder that satisfies the internal cell ratio and particle size of the present invention. Does not suggest.
- Non-Patent Document 1 For example, regarding the commercially available (polyacrylic acid) water absorbent resin disclosed in Table 5.6 of Non-Patent Document 1, five types of commercially available water absorbents having an apparent density of 1.250 to 1.667 [g / cm 3 ] are used. A resin is disclosed. Assuming that the true density (chemical composition) of the water-absorbent resin is all the same, the internal cell ratio is 0% or close to 0% (the apparent density after 40 to 60 mesh-Cut is 1.667 [g / cm 3 ] Aquakeep, Drytech 510) and about 10-25% of types (apparent density 1.500 [g / cm 3 ] Arasorb 720, Sanwet 1M-1000, 1.250 [g / cm 3 ] Aridall 1078 ). In addition, as above-mentioned, the five types of commercially available water-absorbing resins are different from the water-absorbing resin of the present invention in terms of bulk specific gravity and water absorption ratio.
- the water-absorbent resin of the present invention is a novel polyacrylic acid (2.8% to 6.6%, more preferably 3.0% to 6.5%) having an internal cell ratio ( Salt) water-absorbent resin powder, and it can be seen that the conventional problems (compatibility between liquid permeability and water absorption speed and damage resistance) are solved.
- Such a water-absorbing resin can keep both the FSR and SFC, which are normally in a reciprocal relationship, high even after being subjected to process damage during pneumatic transportation or paper diaper manufacturing.
- Example 17 For the water-absorbent resin powder (11) obtained in Example 11 (internal cell ratio: 3.93%) (using 150 ppm of surfactant as the monomer), the above-mentioned “(5-13) Maximum insertion load (PIL) and The maximum insertion load (PIL) and insertion distance (PID) described in “Insertion distance (PID)” were measured. The results are shown in Table 6.
- Example 18 The water-absorbing resin powder (2) obtained in Example 2 was subjected to the same surface cross-linking operation (using 10 ppm of surfactant) as in Example 12 to obtain a water-absorbing resin powder (18).
- the water-absorbent resin powder (18) further provided the surface of the water-absorbent resin powder (2) having a surfactant of 150 ppm substantially uniformly inside. It was coated with 10 ppm of a surfactant, and its surface tension was 67.4 [mN / m].
- Comparative Example 19 With respect to the comparative water absorbent resin powder (10) obtained in Comparative Example 10 (internal cell ratio: 2.60%) (no surfactant used), the maximum insertion load (PIL) and the insertion distance (PID) were measured. The results are shown in Table 6.
- Comparative Example 20 The comparative water absorbent resin powder (2) obtained in Comparative Example 2 was subjected to the same surface crosslinking operation as in Example 12 to obtain a comparative water absorbent resin powder (20).
- the comparative water-absorbent resin powder (20) is one in which the inside of the particle (central part) does not contain a surfactant and the particle surface is coated with 10 ppm of the surfactant.
- About the comparative water-absorbent resin powder (20), Maximum insertion load (PIL) and insertion distance (PID) were measured. The results are shown in Table 6.
- the surfactant is uniformly contained inside, and the surface of the water-absorbent resin is coated with the surfactant (Example 18), so that the insertion distance (PID) does not contain the surfactant. Or it turns out that it becomes overwhelmingly large compared with the case where it is contained in either the inside or the surface. Since the surfactant is uniformly contained inside and the surface of the water-absorbent resin is coated with the surfactant, the amount of the surfactant used can be reduced (in order to obtain the same effect). In particular, it is possible to obtain a water-absorbing resin having no reduction in surface tension, high slipperiness, and excellent handleability and damage resistance.
- Examples 19 to 22 show the effects of p-methoxyphenol in solving the problems of the present invention (particularly coloring and damage resistance).
- Example 19 With respect to the water absorbent resin powder (1) obtained in Example 1, the amount of p-methoxyphenol was measured and found to be 12 ppm.
- Example 20 In Example 1, polymerization was carried out with a p-methoxyphenol amount of 1 ppm.
- the p-methoxyphenol in the obtained water absorbent resin powder (20) was measured and found to be ND (less than 1 ppm).
- the weather resistance (photodegradation) described in the above (5-15) of the water-absorbent resin powder (20) having p-methoxyphenol of ND (less than 1 ppm) is the water-absorbent resin powder (1) of p-methoxyphenol of 12 ppm. ) About 10% worse. It can be seen that p-methoxyphenol improves the weather resistance of the water-absorbent resin.
- the amount of p-methoxyphenol in the monomer and / or the water-absorbent resin is important in weather resistance (damage resistance to light).
- Example 21 In Example 1, polymerization was carried out with a p-methoxyphenol amount of 230 ppm.
- the p-methoxyphenol in the obtained water absorbent resin powder (21) was measured and found to be 82 ppm.
- the initial coloration (coloring immediately after production) of the water-absorbent resin powder (21) having 82 ppm of p-methoxyphenol was worse than the water-absorbent resin powder (1) having 12 ppm of p-methoxyphenol, and had an L value of 88.3, a The value was -1.8 and the b value was 10.2. It can be seen that the amount of p-methoxyphenol in the monomer and / or the water-absorbent resin is important in coloring.
- Example 22 In Example 2, the polymerization was carried out with a p-methoxyphenol amount of 70 ppm. Furthermore, surface cross-linking was carried out in the same manner as in Example 11 to obtain a water absorbent resin powder (22) having the same physical properties as in Example 11. When p-methoxyphenol was measured, it was 10 ppm.
- Example 23 37 wt% sodium acrylate aqueous solution 595.4 [g / min], 48 wt% sodium hydroxide aqueous solution 198.6 [g / min], 100 wt% acrylic acid 300.1 [g / min], as an internal crosslinking agent Polyethylene glycol diacrylate (molecular weight 523) 2.71 [g / min], deionized water (ion exchange water) 203.9 [g / min], 31 wt% diethylenetriaminepentaacetic acid / trisodium aqueous solution 0.42 [g / min], 10% by weight polyoxyethylene (20) sorbitan monostearate (manufactured by Kao Corporation) aqueous solution 0.46 [g / min] as a surfactant is continuously mixed using a disperser, and the disperser 3% by weight sodium persulfate aqueous solution 26.0 [g / min] was line-mixed with the monomer aqueous solution after passing through It was fed
- the belt polymerization machine includes an endless belt having a length of 3.8 m and a width of 60 cm, the surface of which is coated with a fluororesin, and the bottom side of the belt and the circumference of the polymerization machine are heated and kept at about 90 ° C. It has an intake pipe for collecting evaporated water.
- the temperature of the aqueous monomer solution supplied onto the belt was controlled by passing water through a disperser so that the temperature was 92 ° C. (See Figure 14)
- the temperature of the aqueous monomer solution (23) supplied to the polymerization apparatus was 92 ° C., and the amount of dissolved oxygen was 4.30 [ml / l].
- the monomer aqueous solution (23) containing a surfactant was clouded with very fine bubbles introduced, and was continuously supplied to the belt polymerization machine. Thereafter, the polymerization reaction started immediately, and polymerization was carried out for about 2 minutes in the polymerization machine to continuously obtain a band-like hydrogel polymer (hydrogel) from the outlet of the polymerization machine.
- the obtained gel had a water-soluble content of 3.2% by weight, a solid content of 53% by weight, and a weight-average molecular weight of 228521 [Da].
- the obtained hydrogel was crushed by a screw extruder (meet chopper) having the following specifications into a length of 200 mm.
- the screw extruder was provided with a porous plate at the tip, and the diameter of the porous plate was 100 mm, the hole diameter was 7.5 mm, the number of holes was 55, and the thickness was 6 mm.
- the supply rate of the hydrogel was set to 1600 [g / min], and hot water at 90 ° C. (supply rate; 50 [g / min]) and water vapor (supply rate; 70 [g] / Min]) was simultaneously supplied to the meat chopper, and the screw shaft rotation speed was 412 rpm.
- the temperature of the hydrogel before gel grinding was 94 ° C.
- the temperature of the hydrogel after gel grinding (hereinafter referred to as “grinding gel”) was 103 ° C.
- the weight average particle diameter (D50) of the obtained ground gel (23) was 897 ⁇ m, the logarithmic standard deviation ( ⁇ ) of the particle size distribution was 0.98, the water-soluble content was 3.8% by weight, and the solid content was 49.4.
- the weight average molecular weight of the weight%, water-soluble component was 263330 [Da].
- Example 24 gel pulverization conditions were as follows: the hole diameter of the perforated plate was 19.0 mm, the number of holes was 10, and the thickness was 10.5 mm.
- the feed rate of the hydrogel was set to 1600 [g / min], and 90 ° C. warm water (feed rate; 63 [g / min]) and water vapor (feed rate; 95 [g / min]) were simultaneously applied to the meat chopper.
- a crushed gel (24) was obtained in the same manner as in Example 23 except that the screw shaft rotation speed was 257 rpm.
- the weight average particle diameter (D50) of the obtained crushed gel (24) was 1232 ⁇ m, the logarithmic standard deviation ( ⁇ ) of the particle size distribution was 1.88, the water-soluble content was 3.8% by weight, and the solid content was 51.1.
- the weight average molecular weight of the weight% water-soluble component was 229121 [Da].
- Example 25 In Example 23, in a 37 wt% sodium acrylate aqueous solution and deionized water (ion exchange water) used as an aqueous monomer solution, a microbubble generator (manufactured by Auratech Co., Ltd., model: OM4-GP-040) was used. The gel was obtained in the same manner as in Example 23 except that nitrogen gas was introduced as the introduction gas. The obtained gel had a water-soluble content of 3.0% by weight, a solid content of 53% by weight, and a weight-average molecular weight of 236521 [Da].
- a microbubble generator manufactured by Auratech Co., Ltd., model: OM4-GP-040
- This gel was pulverized under the same conditions as in Example 23.
- the weight average particle diameter (D50) of the obtained crushed gel (25) was 879 ⁇ m
- the logarithmic standard deviation ( ⁇ ) of the particle size distribution was 0.97
- the water-soluble content was 3.6% by weight
- the solid content was 48.8.
- the weight average molecular weight of the weight%, water-soluble component was 269981 [Da].
- the water-absorbent resin powder obtained according to the present invention When used for sanitary materials such as paper diapers, it has both liquid permeability and water absorption speed, and is excellent in impact resistance (damage resistance) and whiteness. Absorption performance (water absorption rate) superior to sanitary materials can be provided.
Abstract
Description
(内部気泡率[%])={(真密度[g/cm3])-(見かけ密度[g/cm3])}/(真密度[g/cm3])×100
更に、上記課題(特に耐ダメージ性)を解決するために、本発明の吸水性樹脂(第2の吸水性樹脂)は、界面活性剤及び/又は分散剤を内部に含むポリアクリル酸系吸水性樹脂粉末であって、表面張力が60[mN/m]以上で、かつ、粉末表面が界面活性剤で被覆されてなることを特徴としている。
(1-1)「吸水性樹脂粉末」
本発明における「吸水性樹脂粉末」とは、水膨潤性水不溶性の高分子ゲル化剤を意味する。尚、「水膨潤性」とは、ERT441.2-02で規定するCRC(無加圧下吸水倍率)が5[g/g]以上であることをいい、又、「水不溶性」とは、ERT470.2-02で規定するExt(水可溶分)が0~50重量%であることをいう。
本発明における「ポリアクリル酸系吸水性樹脂粉末」とは、任意にグラフト成分を含み、繰り返し単位として、アクリル酸及び/又はその塩(以下、アクリル酸(塩)と称する)を主成分とする重合体を意味する。
「EDANA」は、欧州不織布工業会(European Disposables and Nonwovens Assoiations)の略称であり、「ERT」は、欧州標準(ほぼ世界標準)である吸水性樹脂の測定方法(EDANA Recommended Test Metods)の略称である。なお、本発明においては、特に断りのない限り、ERT原本(公知文献:2002年改定)に準拠して、吸水性樹脂粉末の物性を測定する。
「CRC」は、Centrifuge Retention Capacity(遠心分離機保持容量)の略称であり、無加圧下吸水倍率(以下、「吸水倍率」と称することもある)を意味する。具体的には、不織布中の吸水性樹脂0.200gの0.9重量%塩化ナトリウム水溶液に対する30分間の自由膨潤後、更に遠心分離機で水切りした後の吸水倍率(単位;[g/g])である。
「AAP」は、Absorption Against Pressureの略称であり、加圧下吸水倍率を意味する。具体的には、吸水性樹脂0.900gの0.9重量%塩化ナトリウム水溶液に対する1時間、2.06kPaでの荷重下膨潤後の吸水倍率(単位;[g/g])である。尚、ERT442.2-02では、Absorption Under Pressureと表記されているが、実質的に同一内容である。又、荷重条件を4.83kPa(0.7psi)に変更して測定することもある。
「Ext」は、Extractablesの略称であり、水可溶分(水可溶成分量)を意味する。具体的には、0.9重量%塩化ナトリウム水溶液200gに対して、吸水性樹脂粉末1gを16時間攪拌した後、溶解したポリマー量をpH滴定で測定した値(単位;重量%)である。
「PSD」とは、Particle Size Disributionの略称であり、篩分級により測定される粒度分布を意味する。尚、重量平均粒子径(D50)及び粒子径分布幅は米国特許2006-204755号に記載された「(1) Average Particle Diameter and Distribution of Particle Diameter」と同様の方法で測定する。
荷重下又は無荷重下における膨潤した吸水性樹脂粉末の粒子間を流れる液の流れを「通液性」という。この「通液性」の代表的な測定方法として、SFC(Saline Flow Conductivity/生理食塩水流れ誘導性)や、GBP(Gel Bed Permeability/ゲル床透過性)がある。
本明細書において、範囲を示す「X~Y」は、「X以上Y以下」であることを意味する。又、重量の単位である「t(トン)」は、「Metric ton(メトリック トン)」であることを意味し、更に、特に注釈のない限り、「ppm」は「重量ppm」を意味する。又、「重量」と「質量」、「重量%」と「質量%」、「重量部」と「質量部」は同義語として扱う。更に、「~酸(塩)」は「~酸及び/又はその塩」を意味し、「(メタ)アクリル」は「アクリル及び/又はメタクリル」を意味する。
本発明の吸水性樹脂粉末の製造方法は、吸水速度の向上のために発泡重合する方法において、上記特許文献に対して気泡の含有方法に特徴を有する。かかる手法によって、重合前の単量体水溶液に気泡が均一に分散するため、得られる吸水性樹脂粉末の吸水速度が向上するだけでなく、更に白色度も向上する。更に吸水性樹脂粉末の他の物性(通液性、嵩比重、表面張力、加圧下吸水倍率、耐衝撃性(耐ダメージ性)等)を維持又は殆ど損なうことなく、更に高価な原料や装置を用いる必要もない。
本発明で「アクリル酸系単量体水溶液」とは、後述のアクリル酸及び/又はその塩を主成分とする単量体の水溶液であって、必要により架橋剤やその他後述のグラフト成分、微量成分(キレート剤、界面活性剤、分散剤)等、吸水性樹脂の構成成分を調合したものを指し、このままの状態で重合開始剤を添加して重合に供せるものを指す。
本発明の第1又は第2の方法において、溶存気体の溶解度を低下させて気泡を発生含有させる工程、つまり、アクリル酸系単量体水溶液を昇温する工程では、アクリル酸系単量体水溶液の昇温によって気体の溶解度が低下する。昇温幅は、気泡の発生量から好ましくは、+5℃以上、より好ましくは+10~+100℃、更に好ましくは+20~+90℃、特に好ましくは+30~+80℃である。
アクリル酸系単量体水溶液調整工程は、気体を分散させたアクリル酸系単量体水溶液を得る工程である。以下、具体的に説明する。
本発明で用いられるアクリル酸系単量体としては、アクリル酸が使用され、重合により吸水性樹脂粉末となり得るものであれば特に限定されないが、以下に示すようなものが挙げられる。例えば、(メタ)アクリル酸、(無水)マレイン酸、イタコン酸、ケイ皮酸、ビニルスルホン酸、アリルトルエンスルホン酸、ビニルトルエンスルホン酸、スチレンスルホン酸、2-(メタ)アクリルアミド-2-メチルプロパンスルホン酸、2-(メタ)アクリロイルエタンスルホン酸、2-(メタ)アクリロイルプロパンスルホン酸、2-ヒドロキシエチル(メタ)アクリロイルフォスフェート等のアニオン性不飽和単量体及びその塩;メルカプト基含有不飽和単量体;フェノール性水酸基含有不飽和単量体;(メタ)アクリルアミド、N-エチル(メタ)アクリルアミド、N,N-ジメチル(メタ)アクリルアミド等のアミド基含有不飽和単量体;N,N-ジメチルアミノエチル(メタ)アクリレート、N,N-ジメチルアミノプロピル(メタ)アクリレート、N,N-ジメチルアミノプロピル(メタ)アクリルアミド等のアミノ基含有不飽和単量体等が挙げられる。
アクリル酸系単量体水溶液への気泡の分散方法は、界面活性剤及び/又は分散剤の存在下で、水溶液中に含まれる溶存ガスを発生させて、それらを効果的に微細な気泡(マイクロバブル又はナノバブル)として分散させる方法である。ここで、水溶液中にガスを溶存させるために、予め水溶液中にガス(例えば、不活性ガス)を導入してもよいし、しなくてもよい。
アクリル酸系単量体水溶液への気泡の分散方法として、単量体及び/又はその塩、必要に応じて内部架橋剤及び水を混合して調製されたアクリル酸系単量体水溶液を昇温する方法、又は、アクリル酸系単量体水溶液の調整段階で昇温して水溶液中の気体の溶解度を低下させる方法が挙げられる。
アクリル酸系単量体水溶液への気泡の分散方法として、単量体及び/又はその塩、必要に応じて内部架橋剤及び水を混合して調製する際に、気体が溶存していない、又はほとんど溶存していない水溶性有機物、あるいは混合対象であるアクリル酸系単量体水溶液や、水に比べて気体が溶解していない水溶性有機物の混合を行い、気体の溶解度を低下させる方法が挙げられる。上記水溶性有機物としては、酸素の溶解度が好ましくは0.02[ml/ml]以下、より好ましくは0.01[ml/ml]以下、特に好ましくは0.005[ml/ml]以下の有機化合物が使用される。例えば、気体を含む(溶存する)アクリル酸系単量体水溶液に気体を含まない単量体(例えばアクリル酸)を混合することで、混合後の水溶液に溶存できない気体が発生し、その気体を微細な気泡として水溶液中に分散せしめることができる。
本発明に係る製造方法では、アクリル酸系単量体水溶液中の溶存気体の溶解度を低下させて気泡を分散させるが、別途、外部から気体を導入して気泡を分散させてもよい。即ち、溶解度を低下させて分散させる気泡や、更に必要により外部から導入する気体で分散させる気泡によって、アクリル酸系単量体水溶液に気泡を分散させればよい。その際、アクリル酸系単量体水溶液へ分散させる気泡を構成する気体としては、酸素、空気、窒素、炭酸ガス、オゾンやそれらの混合物等が挙げられるが、好ましくは、窒素、炭酸ガス等の不活性ガスが使用される。更に好ましくは、重合性やコスト面から空気、窒素が特に好ましい。気体を導入する際又は導入後の圧力は常圧、加圧、減圧で適宜決定される。又、気体を外部から導入する場合の好ましい導入方法は、特願2009-292318号(出願日;2009年12月24日)及びその優先権出願PCT/JP2010/001004号に記載された方法であり、下記「(2-1-4)気体の導入方法」で示す。
本発明に係る製造方法では、アクリル酸系単量体水溶液中の溶存気体の溶解度を低下させて気泡を分散させるが、別途、外部からの気体導入を併用してもよい。この場合、外部から導入する気体とアクリル酸系単量体水溶液とを混合すればよく、アクリル酸系単量体水溶液へ気体を導入させる方法としては、スタティックミキサー方式、キャビテーション方式、ベンチュリー方式等の公知の方法を適宜利用することができ、それらの方法を併用してもよい。更に、気体の導入量を多くできるマイクロバブル(又はナノバブル)の導入が好適である。即ち、後述の実施例5、6のように、マイクロバブル又はナノバブルの導入を併用することが好ましい。
マイクロバブルの導入方法として、好ましくは、単量体水溶液及び気体の加圧による加圧溶解方法が用いられる。具体的には、液中に気体を好ましくは100~1000kPa(絶対圧)、より好ましくは200~400kPa、特に好ましくは250~350kPa程度に絶対圧として加圧して溶解させ、減圧弁を通して液中にフラッシュ操作すると、減圧され過飽和となった気体が液中からマイクロバブルとなり放出される。気体の液中への溶解度はヘンリーの法則(p=HC)に従い、温度と圧力で決定される。かかる加圧によって、一旦溶解させた気泡を経て、分散させた気泡が得られる。
上記加圧溶解方法の一例として、アクリル酸系単量体水溶液を得る工程において、単量体水溶液に、気体を過飽和で含有させる方法が挙げられる。従って、上記して得られる、気体を溶解及び/又は分散させたアクリル酸系単量体水溶液における該気体成分の濃度は、該気体の所定温度における飽和溶解度に対して1.01~10倍が好ましく、1.05~5倍がより好ましく、1.06~3倍が更に好ましい。
別のマイクロバブルの導入方法として、好ましくは、単量体水溶液及び気体の旋回流の形成が挙げられる。該方法は、気液二相流体を旋回させて出口(混合機の吐出口)で気泡を分散させる方法であり、ガス流量と液流量との比は1/7~1/15が好ましく、旋回速度は毎秒10~10000回転が好ましく、100~1000回転が更に好ましい。
マイクロバブルの導入方法として、各種多孔質物質、膜、フィルター等の細孔から気泡を生成させる方法であり、多孔質ガラス(Na2O-CaO-Al2O3-B2O3-SiO2系ガラス)等が使用され、好ましくは、0より高く0.03重量%以下等後述の範囲の界面活性剤が使用される。上記方法は、例えば、木下理化工業株式会社製木下式ガラスボールフィルター(フィルター粒子No.4)を用いて行うことができる。
マイクロバブルを導入するために、モノマー水溶液と不活性ガスとを加圧や旋回流を発生させる機能を有するマイクロバブル発生装置を用いてもよい。この操作により、発生したマイクロバブルを、重合開始時までモノマー水溶液内に懸濁、保持させておくことができる。
M型マイクロバブル発生装置(株式会社ナノプラネット研究所)
業務用マイクロバブル発生装置SMB-450型(石丸商行有限会社)
マイクロバブル発生装置Mbelife(関西オートメ機器株式会社)
球体内蔵型気泡発生装置MBG型(西田鉄工株式会社)
ポンパレーター(株式会社帝国電機製作所)
マイクロバブルの発生器には入水口と出水口があり、この入水口に、ある一定以上の圧力で液体(水や単量体)を流入させた場合、内部では水の中に混ざっている気体が密度差により中心部に集められ、気体軸が形成される。これによってマイクロバブル発生器の内部には外周と中心部の間で圧力勾配が生じる。この時、気体軸の中心部はほぼ真空状態となり、一方では加圧され噴出しようとする水と、真空状態(超負圧の状態)の気体軸へと流入しようとする水とが衝突し、また旋回しながら気体軸がこの間を通り抜ける時に気体はせん断され微細化してマイクロバブルとなるのである。
本発明に係る製造方法では、上述した方法(a)~(c)やマイクロバブル発生装置に加えて、下記(1)~(8)の方法を使用ないし併用することができる。
可動部分がなく、流体が、管内部に固定されたエレメントを通過する際に混合されるスタティックミキサーや、円管内部に螺旋流誘導部と管内部に取り付けられたキノコ状の突起により旋回状に流れる気液2相流を破砕してマイクロバブルが発生されるOHRミキサーが挙げられる。
ガス分散器内に意図的にキャビテーションが発生するように流路を変形させてマイクロバブルを発生させる方法が挙げられる。
ポンプによる渦流攪拌作用とポンプでの昇圧により、液中に気体を加圧溶解させ、溶解しきれない気体を旋回流式マイクロバブル発生器でマイクロ化させる方法が挙げられる。
ストロー部(絞り)に気液を同時に流すと液流速の急激な変化により生成した衝撃波により大気泡が発破させ、マイクロバブルが発生する方法が挙げられる。
攪拌翼を高速回転され、ガスを自給させマイクロバブルを発生させる方法が挙げられる。
超音波周波数、圧力振幅等を適宜設定してマイクロバブルを発生させる方法が挙げられる。
気体(窒素ガス)と水蒸気との混合ガスを液中に細いノズルから吹き込むと、水蒸気が凝集し、凝集しない気体(窒素ガス)の気泡が残る。
水の電気分解でマイクロオーダーの気泡を発生させる方法が挙げられる。
気泡含有方法として上記(a)の加圧溶解方法やマイクロバブル発生装置では、気体と単量体水溶液とは大気圧を超えて加圧(好ましくは、上記(a)に記載の範囲、絶対圧として0.1~1MPa)された後、大気圧(或いは減圧、特に-10mmHg以内の微減圧)に開放されることで、気泡の量や大きさが制御される。かかる圧力、温度や開放時間、特に開放時間を制御することで、気泡を制御することが好ましい。かかる制御で目的とする高吸水速度で通液性や耐衝撃性の高い吸水性樹脂を得ることができる。
本発明においては、アクリル酸系単量体水溶液に対して、必要により外部から気体を導入することができるが、本発明は、気体の溶解度を低下させる工程によって溶存気体が気泡となることが重要である。
本発明において、界面活性剤及び/又は分散剤を用いることで気泡を安定的に懸濁させることができる。又、界面活性剤及び/又は分散剤の種類や量を適宜調整することにより、所望の物性を有する吸水性樹脂粉末を得ることができる。ここで、好ましくは、界面活性剤は非高分子界面活性剤であり、分散剤は高分子分散剤である。
本発明において用いられる界面活性剤としては、その種類は特に限定されないが、例えば、アニオン性界面活性剤、ノニオン性界面活性剤、カチオン性界面活性剤、両性界面活性剤、フッ素系界面活性剤、有機金属界面活性剤等が挙げられ、具体的には、前記特許文献28(国際公開第97/017397号)又は特許文献30(米国特許第6107358号)に記載の界面活性剤が挙げられる。
本発明に係る製造方法では、重合工程前の調整中又は調整後のアクリル酸系単量体水溶液が分散剤を含むことが好ましく、分散剤は、吸水性を示す親水性高分子分散剤であることが好ましく、更に好ましくは水溶性高分子分散剤であり、その重量平均分子量は、分散剤の種類で適宜決定されるが、好ましくは500~1,000万、更に好ましくは5,000~500万、特に好ましくは1万~300万程度である。
本発明に係る方法では、重合時に好ましくは重合禁止剤を含む。重合禁止剤としては、国際公開第2008/096713号に例示のN-オキシキシル化合物、マンガン化合物、置換フェノール化合物が挙げられ、好ましくは置換フェノール類、特にメトキシフェノール類が挙げられる。
本発明の第1~第3の方法では、かかる気体の溶解度の低下、具体的には、昇温、水溶性有機物の混合によって気体の溶解度が低下して、アクリル酸系単量体水溶液中に気泡が発生する。しかしながら、上記特許文献18~35等に記載の従来の発泡方法と比べて、本発明では、高価な原料(発泡剤や多量の界面活性剤)や特殊な装置もあえて必要としない。又、後述の「〔3〕ポリアクリル酸系吸水性樹脂粉末の物性」でも記載しているが、本発明で得られる吸水性樹脂粉末は、非特許文献1のTable5.6で開示されている市販の吸水性樹脂(詳細な製造方法は開示されておらず不明)のように、過度の嵩比重や見かけ密度の低下がない。
本発明では好ましくは脱泡工程を更に含む。脱泡工程を含むことで、単量体から大きな気泡が順次除去され、過度の発泡や嵩比重の低下を抑制する。脱泡時間としては5秒以上が好ましく、10秒~60分がより好ましく、30秒~30分が更に好ましく、60秒~20分が特に好ましい。目的とする細かい泡を単量体水溶液に残すように調整される。
重合は、常圧、減圧、又は加圧下で行われ、好ましくは常圧(ないしその近傍、通常±10mmHg)で行われる。又、重合を促進し物性を向上させるため、図7~図11に示した概略フロー図に基づいて、重合時に必要により溶存酸素の脱気工程(例えば、不活性ガスでの置換工程)を設けてもよい。
本発明において使用される重合開始剤は、重合形態によって適宜選択され、特に限定されないが、例えば、光分解型重合開始剤、熱分解型重合開始剤、レドックス系重合開始剤等を例示することができる。
上記重合に際しては、更に必要に応じて、重合前又は重合途中の反応系に、次亜燐酸(塩)等の連鎖移動剤、キレート剤等を添加してもよい。
本発明においては、上記単量体水溶液を重合するに際して、得られる吸水性樹脂の通液性や吸水速度といった吸水性樹脂の物性や重合制御の容易性等の観点から、通常、水溶液重合が採用され、好ましくはニーダー重合又はベルト重合、より好ましくは連続水溶液重合、更に好ましくは高濃度連続水溶液重合、特に好ましくは高濃度高温開始連続水溶液重合が採用される。
重合方法としては、噴霧重合、液滴重合、水溶液重合又は逆相懸濁重合等広く適用することができるが、課題の解決に特に好適な重合方法としては、水溶液重合、中でも連続ベルト重合又は連続ニーダー重合が挙げられる。
本発明では、好ましくは重合時又は重合後にゲルが細粒化(別称;ゲル粉砕)される。上記の発泡ゲルをゲル細粒化、特に混練によってゲル細粒化することで、吸水速度と通液性の両立が図れ、更に耐衝撃性も向上する。即ち、本発明の課題解決のため、ゲル粉砕が不要な逆相懸濁重合を採用するよりも、水溶液重合、特にベルト重合やニーダー重合を行い、重合中(特にニーダー重合)又は重合後(特にベルト重合、更には必要によりニーダー重合)にゲル粉砕を行うことが好ましい。
上記で得られた含水ゲル状架橋重合体は、重合時又は重合後に細分化される。重合時の細分化にはニーダーや逆相懸濁重合が用いられ、重合後の細粒化にはミートチョパー等が使用される。
表面架橋前の吸水性樹脂粉末の重量平均粒子径(D50)は、本発明の課題解決のため、吸水速度や通液性、加圧下吸水倍率の面からも、粉末として好ましくは200~600μm、より好ましくは200~550μm、更に好ましくは250~500μm、特に好ましくは350~450μmに調整される。又、通液性等の面からJIS標準篩で150μm未満の粒子が少ないほどよく、通常0~5重量%、好ましくは0~3重量%、特に好ましくは0~1重量%に調整される。かかる粒度制御は、重合時又はゲル粉砕や乾燥後の粉砕、分級時に行えるが、特に乾燥後の分級時に行うことが好ましい。
吸水速度(や通液性)をより向上させるため、乾燥後に、ポリアクリル酸系吸水性樹脂粉末を表面架橋する工程を更に含むことが好ましい。表面架橋は後述の表面架橋剤で行ってよく、吸水性樹脂表面で単量体を重合させてもよく、過硫酸塩やUV開始剤等のラジカル重合開始剤を加えて加熱又は紫外線照射することによって行ってもよい。
本発明では乾燥後の表面架橋工程を更に含むことが好ましい。本発明の製造方法では、高い加圧下吸水倍率(AAP)及び通液性(SFC)の吸水性樹脂粉末の製造方法や巨大スケール(特に1[t/hr])での連続生産に適用され、特に高温表面架橋での吸水性樹脂粉末に好適に適用される。
本発明で用いることができる表面架橋剤としては、種々の有機又は無機架橋剤を例示できるが、有機表面架橋剤が好ましく使用できる。
また、上記有機表面架橋剤(共有結合性)以外にイオン結合性表面架橋剤としてポリアミンポリマーや多価金属塩を使用して通液性等を向上させてもよい。かかるイオン結合性表面架橋剤は膨潤ゲル粒子の静電的にスペーサーとして通液性の向上に寄与して好ましい。本発明で特に通液性、特にSFCを20[×10-7・cm3・sec・g-1]以上、さらには後述の範囲にする場合、有機表面架橋剤(共有結合性表面架橋)とイオン結合性表面架橋剤又は後述の水不溶性微粒子が併用されて通液性が向上されることが好ましい。
有機表面架橋剤(共有結合性表面架橋剤)の使用量は、吸水性樹脂粉末100重量部に対して、好ましくは0.001~10重量部の範囲、より好ましくは0.01~5重量部の範囲で適宜決定される。又、多価金属カチオン、ポリアミンポリマー、水不溶性微粒子から選ばれる通液性向上剤の使用量は0~5重量部が好ましく、0.001~3重量部がより好ましく、0.01~2重量部が更に好ましく、0.05~1重量部が特に好ましい。
本工程は、高吸水速度及び高通液性の吸水性樹脂粉末を得るために、界面活性剤で吸水性樹脂粉末の表面を被覆する工程である。
本発明の課題をより解決するために、乾燥工程後に好ましくは、分級工程を含み、分級工程後の微粉が乾燥工程以前の工程においてリサイクル(再利用)される。即ち、重合工程後、好ましくは加熱乾燥工程後の吸水性樹脂は必要により粉砕、分級工程を経て、上記粒度に調整される。又、分級で除去される粗大粒子(例えば1mm以上)は必要により粉砕してもよく、又、分級で除去される微粒子(例えば150μm未満、更には106μm未満)は廃棄してもよく、他の用途に使用してもよく、微粉リサイクルしてもよい。微粉が除去されることでより通液性(例えばSFC)が向上することが見いだされた。更に除去後の微粉がリサクイルされることでより吸水速度(例えばFSR)が向上することが見いだされた。
一般的には、表面積の大きな吸水性樹脂は着色や劣化しやすい傾向もあるため、本発明では着色防止や劣化防止のために、キレート剤(特に有機リン系キレート剤、アミノアルボン酸系キレート剤)、α-ヒドロキシカルボン酸(特に乳酸又はその塩)、無機又は有機還元剤(特に硫黄系無機還元剤)から選ばれる着色防止剤又は耐尿性(耐候性)向上剤を更に含むことが好ましい。これらの使用量は吸水性樹脂100重量部に対して0~3重量部が好ましく、0.001~1重量部がより好ましく、0.05~0.5重量部が特に好ましい。これらは単量体や含水ゲル、乾燥重合体や粉末等に添加され、添加工程は重合工程以降に適宜決定されるが、これらの中で還元剤は重合で消費されるため、重合後、更には乾燥後に特に表面架橋後に添加するこが好ましい。
上記以外に、必要により、第2の分級工程、蒸発モノマーのリサイクル工程、造粒工程、微粉除去工程等を設けてもよい。更には、経時色安定性効果やゲル劣化防止等のために、添加剤を単量体あるいはその重合物に使用してもよい。
(新規な第1の吸水性樹脂粉末(図12,13参照))
本発明の上記製造方法で得られた吸水性樹脂粉末は、その内部に所定量の空隙を有する。本発明は特定範囲の気泡含有率(吸水性樹脂粒子内部での気泡含有率、別称;空隙率)を有する新規な吸水性樹脂粉末を提供する。
(内部気泡率[%])={(真密度[g/cm3])-(見かけ密度[g/cm3])}/(真密度[g/cm3])×100
ここで十分に乾燥(含水率1重量%未満が好ましく、0.5重量%未満がより好ましく、0.1重量%未満が特に好ましい)した吸水性樹脂粉末についての真密度[g/cm3]は、化学組成(高分子の繰り返し単位やその他、架橋剤等の微量原料や、任意に使用されるグラフト成分)によって一義的に決定される。従って、ポリアクリル酸系吸水性樹脂粉末においては、その中和率、その塩の種類(例えば、中和率75モル%のポリアクリル酸ナトリウム)や微量原料由来による若干の差を有するが、ほぼ一定値を示す。
(内部気泡率[%])={(真密度[g/cm3])-(見かけ密度[g/cm3])}/(真密度[g/cm3])×100
かかる本発明の吸水性樹脂粉末は、例えば、上記表面架橋、特に上記CRCの範囲までの表面架橋を含む製造方法によって得られ、好ましくは、荷重50[g/cm2]での加圧下吸収倍率(AAP)が15[g/g]以上である吸水性樹脂粉末である。AAPの好ましい範囲は下記であり、AAPが低い場合、紙オムツで吸水性樹脂濃度が高い場合、十分な吸水性能を発揮しないこともある。
上記の吸水性樹脂粉末の製造方法において、重合時に界面活性剤及び/又は分散剤を使用した上に、好ましくは、吸水性樹脂粉末の表面が界面活性剤で更に被覆される工程を含み、かかる製造方法で得られた吸水性樹脂粉末は、界面活性剤及び/又は分散剤を内部に実質均一に含むポリアクリル酸系吸水性樹脂粉末であって、表面張力が60[mN/m]以上で、かつ粉末表面が更に界面活性剤で更に被覆されてなる、吸水性樹脂粉末を提供する。
又、本発明の製造方法で得られる吸水性樹脂粉末又は上記本発明の第1又は第2の吸水性樹脂粉末は、好ましくは下記物性を満たしてなる。衛生材料、特に紙オムツを目的とする場合、上記重合や表面架橋をもって、下記(3-1)~(3-7)の少なくとも1つ、更にはAAPを含め2つ以上、特に3つ以上に制御されることが好ましい。下記を満たさない場合、後述の高濃度紙オムツでは十分な性能を発揮しないことがある。
紙オムツでのモレを防止するため、上記重合後の表面架橋を達成手段の一例として、1.9kPaの加圧下、更には4.8kPaの加圧下での0.9重量%の塩化ナトリウム水溶液に対する吸水倍率(AAP)が好ましくは20[g/g]以上、より好ましくは22[g/g]以上、更に好ましくは24[g/g]以上に制御される。AAPの上限は高いほど好ましいが、他の物性とのバランスから通常40[g/g]、更には35[g/g]、特に荷重4.8kPの場合は30[g/g]程度が好ましい。
無加圧下吸水倍率(CRC)は、好ましくは10[g/g]以上であり、より好ましくは20[g/g]以上、更に好ましくは25[g/g]以上、特に好ましくは30[g/g]以上に制御される。CRCは高いほど好ましく上限値は特に限定されないが、他の物性(特に通液性)のバランスから、好ましくは50[g/g]以下、より好ましくは45[g/g]以下、更に好ましくは40[g/g]以下である。CRCは架橋剤量等で制御できる。尚、上記非特許文献1のTable5.6には、市販の吸水性樹脂の吸水倍率(測定方法の詳細な記載はない)について開示されている。具体的には、Aquakeepが65.4[g/g]、Sanwet 1M-1000が58.3[g/g]である事実を開示するが、本発明の課題(通液性と吸水速度の両立)をより解決するため、本発明では上記範囲内に無加圧下吸水倍率(CRC)を制御することが好ましい。
紙オムツでのモレを防止するため、上記重合及びその粒度制御した表面架橋を達成手段の一例として、加圧下での液の通液特性である0.69重量%生理食塩水流れ誘導性(SFC)は1[×10-7・cm3・sec・g-1]以上であり、以下順に20[×10-7・cm3・sec・g-1]以上、50[×10-7・cm3・sec・g-1]以上、70[×10-7・cm3・sec・g-1]以上、100[×10-7・cm3・sec・g-1]以上、120[×10-7・cm3・sec・g-1]以上が好ましく、特に140[×10-7・cm3・sec・g-1]以上に制御される。SFCは周知の測定法であり、例えば、米国特許第5562646号に記載方法で規定できる。本発明の独立気泡率を制御した吸水性樹脂は、SFC及びFSRを高く両立することができる。SFCの上限については適宜決定されるが、他の物性とのバランスから好ましくは1000[×10-7・cm3・sec・g-1]程度である。
水可溶分は、好ましくは35重量%以下、より好ましくは25重量%以下であり、更に好ましくは15重量%以下、特に好ましくは10重量%以下である。
上記重合(発泡重合)を達成手段の一例として、本発明の吸水性樹脂は、20gの生理食塩水に対する吸水性樹脂粉末1gでの吸水速度(FSR)は、通常0.05[g/g/sec]以上、好ましくは0.1[g/g/sec]以上、より好ましくは0.15[g/g/sec]以上、更に好ましくは0.20[g/g/sec]以上、特に好ましくは0.25[g/g/sec]以上である。上限としては、好ましくは0.50[g/g/sec]以下、より好ましくは1.0[g/g/sec]以下である。FSRの測定法は国際公開第2009/016055号パンフレットで規定される。本発明の独立気泡率を制御した吸水性樹脂は、SFC及びFRSを高く両立することができる。
吸水性樹脂粉末の嵩比重は、通常、0.58~0.8(0.58~0.80と同義である)[g/cm3]であり、好ましくは0.6~0.8(0.60~0.80と同義である)[g/cm3]であり、より好ましくは0.63~0.77[g/cm3]、更に好ましくは0.66~0.74[g/cm3]である。本発明では発泡構造(別称;多孔質構造)であるが、高い嵩比重を有する。
表面張力(実施例の測定法で規定)は、好ましくは60[mN/m]以上、より好ましくは65[mN/m]以上、更に好ましくは67[mN/m]以上、特に好ましくは70[mN/m]以上、最も好ましくは72[mN/m]以上であり、実質的な表面張力の低下もない。上限は通常75[mN/m]で十分である。
本発明の吸水性樹脂粉末の用途は特に限定されないが、好ましくは、紙オムツ、生理ナプキン、失禁パット等の吸収性物品に使用され得る。本発明の吸水性樹脂粉末は通液性と吸水速度とを両立し、更に耐衝撃性や加圧下吸収倍率にも優れるため、吸水性樹脂の含有率が高い吸収物品に好ましく使用できる。この吸収性物品中の、任意に他の吸収性材料(パルプ繊維等)を含む吸収体における吸水性樹脂粉末の含有量(コア濃度)は、30~100重量%、好ましくは40~100重量%、より好ましくは50~100重量%、更に好ましくは60~100重量%、特に好ましくは70~100重量%、最も好ましくは75~95重量%で本発明の効果が発揮される。
以下、実施例に従って発明を説明するが、本発明は実施例に限定され解釈させるものではない。又、本発明の特許請求の範囲や実施例に記載の諸物性は、以下の測定法(5-1)~(5-15)に従って求めた。尚、特に断りのない限り、各実施例での各工程は実質常圧(大気圧の±5%、更に好ましくは1%以内)で行なわれ、同一工程では意図的な加圧又は減圧による圧力変化は加えずに実施した。
米国特許出願公開第2006/204755号に準じて、標準篩で分級して重量平均粒子径(D50)及び粒度分布の対数標準偏差(σζ)を求めた。
ERT441.2-0.2に従い、0.90重量%塩化ナトリウム水溶液(生理食塩水とも称する)に対する無加圧下で30分の吸水倍率(CRC)を求めた。
吸水性樹脂粉末において、180℃で揮発しない成分が占める割合を表す。含水率との関係は、{固形分=100-含水率}となる。
吸水性樹脂粉末1.00gを25mlガラス製ビーカー(直径32~34mm、高さ50mm)に入れた。この際、ビーカーに入れた吸水性樹脂粉末の上面が水平となるようにした(必要により、慎重にビーカーをたたく等の処置を行うことで吸水性樹脂粉末表面を水平にしてもよい。)。
嵩比重測定器(蔵持科学機器製作所製)を用い、JIS K 3362に準じて測定した。粒度による偏りを無くすため十分に混合された吸水性樹脂粉末100.0gを、ダンパーを閉めた漏斗に入れた後、速やかにダンパーを開け、吸水性樹脂粉末を内容量100mlの受器(重量W9[g])に落とした。受器から盛り上がった吸水性樹脂粉末は、ガラス棒ですり落とした後、吸水性樹脂粉末の入った受器の重さ(重量W10[g])を0.1gまで正確に量り、数5にしたがって嵩比重を算出した。
十分に洗浄された100mlのビーカーに20℃に調整された生理食塩水50mlを入れ、まず、生理食塩水の表面張力を表面張力計(KRUSS社製のK11自動表面張力計)を用いて測定した。この測定において表面張力の値が71~75[mN/m]の範囲でなくてはならない。
SFCは周知の測定法であり、米国特許第5562646号に記載の手法にて測定を行った。
初期着色(吸水性樹脂の製造直後の着色)として、白色度とは粉体の白さを示す指標であり、X、Y、Z値又はL、a、b値を用いて算出される。中でも白さ度合いを比較するために有用な吸水性樹脂粉末のWB値を、分光色差計を用いて測定した。尚、初期着色に対して、長期間保存中での着色(進行)や衛生材料中での着色(進行)を経時着色という。
粉体仕込みセル:φ35mm、高さ15mm
(5-9)耐ダメージ性試験
特許文献38(米国特許6562879号)及びその対応特許である日本国公開特許公報「特開2000-302876号」(12頁[0001]、[0002])に記載の(機械的ダメージ試験)方法にて振動時間を10分間として、吸水性樹脂粉末にダメージを与えた。
吸水性樹脂粉末の水分を更に除き、(粉末内部の独立気泡も考慮した)粉末の見かけ密度を乾式密度測定(所定重量の吸水性樹脂粉末体積での乾式測定)で行った。
JIS標準篩で45μm通過物に微粉砕することで内部の独立気泡を破壊又は連続気泡化した吸水性樹脂粉末について、その乾式密度を測定することで、本発明での真密度を求めた。
上記「(5-10)見かけ密度」に記載した方法で測定した見かけ密度(密度ρ1[g/cm3]、及び上記「(5-11)真密度」に記載した方法で測定した真密度(密度ρ2[g/cm3])を用いて、吸水性樹脂粉末の内部気泡率を下記数6に従って算出した。
米国特許第7282262号に開示された最大挿入荷重(PIL)及び挿入距離(PID)の測定方法に基づいて、上記2項目を測定した。
ERT470.2-02の可溶分測定に準じて、0.9重量%塩化ナトリウム水溶液200mlに、吸水性樹脂1.000gを添加し、1時間攪拌(攪拌時間は16時間から1時間に変更)した後の濾液について、分析することで求められる。
PCT/JP2010/067086号に記載された方法に準して規定される。
容量3リットルのポリプロピレン製容器に、アクリル酸(p-メトキシフェノール:70mg/L含有品)181.1g、37重量%アクリル酸ナトリウム水溶液1727.0g、内部架橋剤としてポリエチレングリコールジアクリレート(分子量523)4.38g、界面活性剤として13.7重量%ポリオキシエチレン(20)ソルビタンモノステアレート(花王株式会社製)水溶液59.7gを投入し、溶解(混合)させて単量体水溶液(1)を作製し、液温を24℃に調整した。
容量2リットルのポリプロピレン製容器に、アクリル酸351.6g、内部架橋剤としてポリエチレングリコールジアクリレート(分子量523)2.17g、キレート剤として0.1重量%ジエチレントリアミン5酢酸・3ナトリウム水溶液94.6g、48.5重量%水酸化ナトリウム水溶液144.9g、界面活性剤として1.0重量%ポリオキシエチレン(20)ソルビタンモノステアレート(花王株式会社製)水溶液6.45g、脱イオン水(イオン交換水)236.0gを投入し、溶解(混合)させて単量体水溶液(2’)を作製した。該水溶液(2’)の温度は、作製直後の1段階目の中和熱によって65℃まで上昇した。この温度上昇に伴う気体の溶解度低下によって、界面活性剤を含んだ該水溶液(2’)は、非常に細かい気泡が導入されて白濁していた。
容量1リットルのポリプロピレン製容器に、37重量%アクリル酸ナトリウム水溶液379.07g、内部架橋剤としてポリエチレングリコールジアクリレート(分子量523)0.995g、脱イオン水(イオン交換水)176.31g、界面活性剤としてポリオキシエチレン(20)ソルビタンモノステアレート(花王株式会社製)0.04gを投入し、溶解(混合)させて単量体水溶液(3)を作製した。該単量体水溶液(3)を25℃に調温しながら、窒素ガスを用いて5分間脱気処理を行った。
37重量%アクリル酸ナトリウム水溶液595.4[g/min]、48重量%水酸化ナトリウム水溶液198.6[g/min]、100重量%アクリル酸300.1[g/min]、内部架橋剤としてポリエチレングリコールジアクリレート(分子量523)2.71[g/min]、脱イオン水(イオン交換水)204.7[g/min]、31重量%ジエチレントリアミン5酢酸・3ナトリウム水溶液0.42[g/min]、界面活性剤として10重量%ポリオキシエチレン(20)ソルビタンモノステアレート(花王株式会社製)水溶液0.29[g/min]をラインミキシングにて混合し、単量体水溶液(4)を作製した。連続して上記単量体水溶液(4)を、95℃のオイルバスに浸漬したステンレス製のコイル式熱交換器(自立コイル式交換機、型番JC-S1;アズワン株式会社、研究総合機器カタログ)(図1参照)に通過させた。
容量2リットルのポリプロピレン製容器に、アクリル酸421.9g、内部架橋剤としてポリエチレングリコールジアクリレート(分子量523)2.60g、0.1重量%ジエチレントリアミン5酢酸・3ナトリウム水溶液113.5g、48.5重量%水酸化ナトリウム水溶液173.8g、界面活性剤として10.0重量%ポリオキシエチレン(20)ソルビタンモノステアレート(花王株式会社製)水溶液0.44g、脱イオン水(イオン交換水)290.4gを投入し、溶解(混合)させて単量体水溶液(5’)を作製した。
界面活性剤として10.0重量%ポリオキシエチレン(20)ソルビタンモノステアレート(花王株式会社製)水溶液の添加を、マイクロバブルの導入直後に変更したこと以外は実施例5と同様の操作を行った。
界面活性剤として実施例2の1.0重量%ポリオキシエチレン(20)ソルビタンモノステアレート(花王株式会社製)水溶液に代えて、1.0重量%ソルビタンモノラウレート(花王株式会社製)水溶液を用いたこと以外は実施例2と同様の操作を行い、単量体水溶液(7)を作製した。該単量体水溶液(7)の温度は、作成直後の中和熱によって63℃まで上昇した。
界面活性剤として実施例2の1.0重量%ポリオキシエチレン(20)ソルビタンモノステアレート(花王株式会社製)水溶液に代えて、1.0重量%ポリエーテル変性シリコーン(側鎖変性末端OH型)(東レ・ダウコーニング株式会社製)水溶液を用いたこと以外は実施例2と同様の操作を行い、単量体水溶液(8)を作製した。該単量体水溶液(8)の温度は、作製直後の中和熱によって63℃まで上昇した。
界面活性剤として実施例2の1.0重量%ポリオキシエチレン(20)ソルビタンモノステアレート(花王株式会社製)水溶液6.45gに代えて、3.0重量%カルボキシメチルセルロースナトリウム(シグマアルドリッチジャパン株式会社製)水溶液14.3gを用いたこと以外は実施例2と同様の操作を行い、単量体水溶液(9)を作製した。該単量体水溶液(9)の温度は、作成直後の中和熱によって64℃まで上昇した。
界面活性剤として実施例1の13.7重量%ポリオキシエチレン(20)ソルビタンモノステアレート(花王株式会社製)水溶液59.7gを、同重量の脱イオン水(イオン交換水)59.7gに代えたこと以外は実施例1と同様の操作を行い、固形分97重量%、重量平均粒子径(D50)432μm、粒度分布の対数標準偏差(σζ)0.44の不定形破砕状の比較吸水性樹脂粉末(1)を得た。得られた比較吸水性樹脂粉末(1)の諸物性を表1に示す。
界面活性剤として実施例2の1.0重量%ポリオキシエチレン(20)ソルビタンモノステアレート(花王株式会社製)水溶液6.45gを、同重量の脱イオン水(イオン交換水)6.45gに代えたこと以外は実施例2と同様の操作を行い、固形分96重量%、重量平均粒子径(D50)455μm、粒度分布の対数標準偏差(σζ)0.37の不定形破砕状の比較吸水性樹脂粉末(2)を得た。得られた比較吸水性樹脂粉末(2)の諸物性を表1に示す。また比較吸水性樹脂粉末(2)の白色度を表2に示す。
界面活性剤として実施例2の1.0重量%ポリオキシエチレン(20)ソルビタンモノステアレート(花王株式会社製)水溶液を単量体水溶液(2’)には混合せず、該単量体水溶液(2’)と48.5重量%水酸化ナトリウム水溶液とを混合した後(83℃)の、単量体水溶液(2)に添加したこと以外は実施例2と同様の操作を行い、固形分97重量%、重量平均粒子径(D50)444μm、粒度分布の対数標準偏差(σζ)0.43の不定形破砕状の比較吸水性樹脂粉末(2)を得た。得られた比較吸水性樹脂粉末(3)の諸物性を表1に示す。
界面活性剤として実施例3のポリオキシエチレン(20)ソルビタンモノステアレート(花王株式会社製)0.04gを用いなかったこと以外は実施例3と同様の操作を行い、固形分96重量%、重量平均粒子径(D50)458μm、粒度分布の対数標準偏差(σζ)0.40の不定形破砕状の比較吸水性樹脂粉末(4)を得た。得られた比較吸水性樹脂粉末(4)の諸物性を表1に示す。
界面活性剤として実施例4の10重量%ポリオキシエチレン(20)ソルビタンモノステアレート(花王株式会社製)水溶液を用いなかったこと以外は実施例4と同様の操作を行い、固形分97重量%、重量平均粒子径(D50)450μm、粒度分布の対数標準偏差(σζ)0.37の不定形破砕状の比較吸水性樹脂粉末(5)を得た。得られた比較吸水性樹脂粉末(5)の諸物性を表1に示す。
上記特許文献18~25に準じて炭酸塩を用いて発泡重合を行った。すなわち、容量1リットルのポリプロピレン製容器に、アクリル酸421.7g、内部架橋剤としてポリエチレングリコールジアクリレート(分子量523)2.754g、0.1重量%ジエチレントリアミン5酢酸・3ナトリウム水溶液113.43g、48.5重量%水酸化ナトリウム水溶液140.4g、脱イオン水(イオン交換水)292.3gを投入し、溶解(混合)させて比較単量体水溶液(6’)を作製した。
比較例2で得られた吸水性樹脂粉末(2)を目開き150μm及び目開き45μmのJIS標準篩を用いて分級し、150μmを通過し45μmを通過しない粒子92重量%と45μmを通過した粒子8重量%からなる参考吸水性樹脂粉末(1)を得た。
上記特許文献34(米国特許公開2007/0225422号)に準じて、吸水性樹脂粉末の存在下で重合を行った。すなわち、比較例6において、炭酸水素ナトリウム(和光純薬工業株式会社製)5.2gを添加せずに、4重量%の過硫酸ナトリウム水溶液17.55gを添加した直後に参考例1で得られた参考吸水性樹脂粉末(1)25.8gを添加したこと以外は比較例7と同様の操作を行い、固形分97重量%、重量平均粒子径(D50)446μm、粒度分布の対数標準偏差(σζ)0.36の不定形破砕状の比較吸水性樹脂粉末(7)を得た。得られた比較吸水性樹脂粉末(7)の諸物性を表1に示す。
上記特許文献30(米国特許6107358号)に準じて、気泡分散による発泡重合を行った。すなわち、上記発泡重合により得られた含水ゲル状架橋重合体をミートチョッパー(飯塚工業株式会社製、MEAT-CHOPPER TYPE:12VR-400KSOX、ダイ孔径:6.4mm、孔数:38、ダイ厚み8mm)により解砕し、乾燥、粉砕、分級を行い、固形分95重量%、重量平均粒子径(D50)450μm、粒度分布の対数標準偏差(σζ)0.39の不定形破砕状の比較吸水性樹脂粉末(8)を得た。得られた比較吸水性樹脂粉末(8)の諸物性を表1に示す。なお、単量体水溶液に気泡は分散されていたがその気泡径は非常に大きいものであった。
比較例2で得られた比較吸水性樹脂粉末(2)を目開き600μmのJIS標準篩を用いて分級し、固形分97重量%、重量平均粒子径(D50)336μm、粒度分布の対数標準偏差(σζ)0.39の不定形破砕状の比較吸水性樹脂粉末(9)を得た。得られた比較吸水性樹脂粉末(9)の諸物性を表1に示す。
実施例4において、単量体水溶液(4)が重合装置に供給されて直ぐに重合が開始される付近に、参考吸水性樹脂粉末(1)を57[g/min]で連続的に供給(微粉リサイクル)したこと以外は、実施例4と同様の操作を行い、固形分96重量%、重量平均粒子径(D50)437μm、粒度分布の対数標準偏差(σζ)0.43の不定形破砕状の吸水性樹脂粉末(10)を得た。得られた吸水性樹脂粉末(10)の諸物性を表1に示す。


表1より、本発明の製造方法は他の物性を損なうことなく、高価な副原料(多量の界面活性剤や発泡剤)や特殊な設備はあえて必要とせず、吸水速度(FSR)の向上した吸水性樹脂粉末を提供することがわかる。
実施例2で得られた吸水性樹脂粉末(2)100重量部に対して、1,4-ブタンジオール0.48重量部、プロピレングリコール0.75重量部、脱イオン水4.0重量部からなる表面架橋剤溶液を、該吸水性樹脂粉末(2)に均一にスプレーし、混合した。表面架橋剤溶液を混合した吸水性樹脂粒子を熱風乾燥機(温度:180℃)で45分間加熱表面架橋処理した。加熱処理後、得られた吸水性樹脂粒子を目開き850μmのJIS標準篩を通過するまで粉砕することで、表面が架橋された吸水性樹脂粒子を得た。
実施例2で得られた吸水性樹脂粉末(2)100重量部に対して、1,4-ブタンジオール0.48重量部、プロピレングリコール0.75重量部、ポリオキシエチレン(20)ソルビタンモノステアレート(花王株式会社製)0.001重量部(吸水性樹脂粉末に対して10ppm)、脱イオン水4.0重量部からなる表面架橋剤溶液を、該吸水性樹脂粉末(2)に均一にスプレーしたこと以外は、実施例11と同様にして吸水性樹脂粉末(12)を得た。得られた吸水性樹脂粉末(12)の諸物性を表3に示す。
比較例2で得られた比較吸水性樹脂粉末(2)に対して、実施例11と同様の操作を行い、比較吸水性樹脂粉末(10)を得た。得られた比較吸水性樹脂粉末(10)の諸物性を表3に示す。
比較例9で得られた比較吸水性樹脂粉末(9)に対して、実施例11と同様の操作を行い、比較吸水性樹脂粉末(11)を得た。得られた比較吸水性樹脂粉末(11)の諸物性を表3に示す。
比較例8で得られた比較吸水性樹脂粉末(8)に対して、実施例11と同様の操作を行い、比較吸水性樹脂粉末(12)を得た。得られた比較吸水性樹脂粉末(12)の諸物性を表3に示す。

表3より、本発明の製造方法によって得られた吸水性樹脂粉末では、共に表面積に依存するため相反する、吸水速度(FSR)と通液性(SFC)とを高い値で両立することができる。また、吸水性樹脂粉末の内部気泡率(詳細は後述の実施例15等と及び表5)を2.8~6.6%に制御することで、吸水速度(FSR)と通液性(SFC)とを高い値で両立することができる。また、通液性向上剤として作用する多価金属カチオンの使用によって、SFCが大きく向上していることも判る。
実施例11で得られた吸水性樹脂粉末(11)(内部気泡率3.93%)に対して、上記「(5-9)耐ダメージ性試験」に記載した耐ダメージ性試験を行い、ダメージが与えられた吸水性樹脂粉末(13)を得た。耐ダメージ性試験前後の吸水性樹脂粉末(13)の諸物性を表4に示す。なお、表4中「PS試験」は、「耐ダメージ性試験」を意味する。
実施例12で得られた吸水性樹脂粉末(12)(内部気泡率6.42%)に対して、上記「(5-9)耐ダメージ性試験」に記載した耐ダメージ性試験を行い、ダメージが与えられた吸水性樹脂粉末(14)を得た。耐ダメージ性試験前後の吸水性樹脂粉末(14)の諸物性を表4に示す。
比較例10で得られた比較吸水性樹脂粉末(10)(内部気泡率2.60%)に対して、上記「(5-9)耐ダメージ性試験」に記載した耐ダメージ性試験を行い、ダメージが与えられた比較吸水性樹脂粉末(13)を得た。耐ダメージ性試験前後の比較吸水性樹脂粉末(13)の諸物性を表4に示す。
比較例12で得られた比較吸水性樹脂粉末(12)(内部気泡率6.83%)に対して、上記「(5-9)耐ダメージ性試験」に記載した耐ダメージ性試験を行い、ダメージが与えられた比較吸水性樹脂粉末(14)を得た。耐ダメージ性試験前後の比較吸水性樹脂粉末(14)の諸物性を表4に示す。
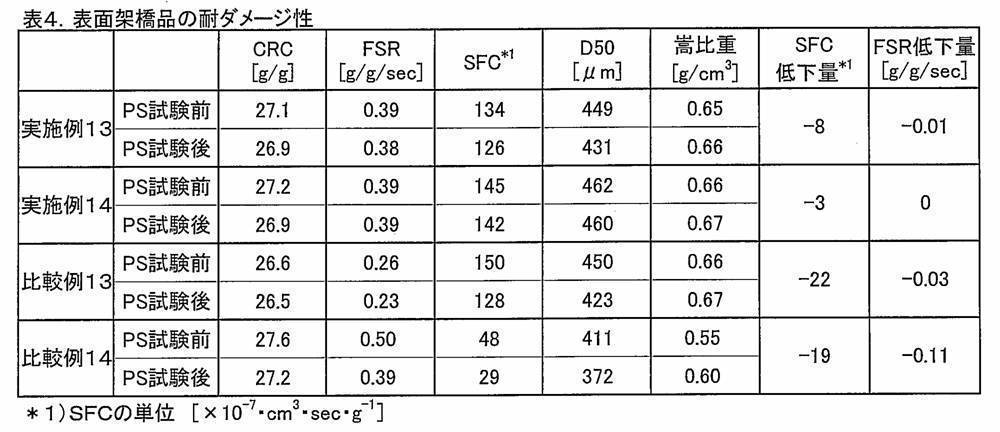
表4より、本発明の吸水性樹脂粉末は、物性の低下(SFCやFSR、特にSFC)が少なく耐ダメージ性に優れていることが分かる。また、吸水性樹脂粉末の内部気泡率を2.8~6.6%に制御することで、耐ダメージ性に優れることが分かる。かかる吸水性樹脂粉末は、空気輸送時や紙オムツ製造時におけるダメージによる物性低下がなく、おむつ製造後、特に高濃度おむつ製造後であっても高物性を維持する。
上記実施例11、12で得られた吸水性樹脂粉末(11)及び(12)に対して、上記「(5-10)見かけ密度」及び「(5-11)真密度」に記載した見かけ密度及び真密度の測定を行い、上記「(5-12)内部気泡率」に記載した内部気泡率を算出した。その結果を表5に示す。
上記比較例10、11で得られた比較吸水性樹脂粉末(10)及び(11)に対して、上記「(5-10)見かけ密度」及び「(5-11)真密度」に記載した見かけ密度及び真密度の測定を行い、上記「(5-12)内部気泡率」に記載した内部気泡率を算出した。その結果を表5に示す。
非特許文献1(1998年)に加えて、2010年出願時点に市販されている吸水性樹脂について、その内部気泡率を調べるため、市販の紙オムツを購入し、使用されている吸水性樹脂粉末を取り出して分析した。

表5より、180℃で3時間以上乾燥しさらに45μm未満に微粉砕後の吸水性樹脂微粒子について、その真密度は測定サンプルに関わらず、ほぼ同じ測定値(1.652~1.656)を示すことから、吸水性樹脂の化学組成(高分子の繰り返し単位やその他微量原料)で真密度は一義的に決まり、この測定法により吸水性樹脂粉末の真密度が正確に測定されたことが分かる。
実施例11で得られた吸水性樹脂粉末(11)(内部気泡率3.93%)(単量体に界面活性剤150ppm使用)について、上記「(5-13)最大挿入荷重(PIL)及び挿入距離(PID)」に記載した最大挿入荷重(PIL)及び挿入距離(PID)を測定した。結果を表6に示す。
実施例2で得られた吸水性樹脂粉末(2)に対して、実施例12と同様の表面架橋操作(界面活性剤10ppm使用)を行い、吸水性樹脂粉末(18)を得た。
比較例10で得られた比較吸水性樹脂粉末(10)(内部気泡率2.60%)(界面活性剤を未使用)について、最大挿入荷重(PIL)及び挿入距離(PID)を測定した。結果を表6に示す。
比較例2で得られた比較吸水性樹脂粉末(2)に対して、実施例12と同様の表面架橋操作を行い、比較吸水性樹脂粉末(20)を得た。かかる比較吸水性樹脂粉末(20)は粒子内部(中心部)は界面活性剤を含まず、粒子表面が界面活性剤10ppmで被覆されたものであり、かかる比較吸水性樹脂粉末(20)について、最大挿入荷重(PIL)及び挿入距離(PID)を測定した。結果を表6に示す。

表6より、界面活性剤を内部に均一に含み、かつ、吸水性樹脂表面が界面活性剤で被覆される(実施例18)ことで、挿入距離(PID)が、界面活性剤を含まない、又は内部若しくは表面のどちらか一方に含まれる場合と比較して、圧倒的に大きくなることが分かる。界面活性剤を内部に均一に含み、かつ、吸水性樹脂表面が界面活性剤で被覆されることで、(同じ効果を得るために)界面活性剤の使用量を低減させることができるため、実質的に表面張力の低下もなく、滑り性が高く、取り扱い性及び耐ダメージ性に優れた吸水性樹脂を得ることができる。
実施例1で得られた吸水性樹脂粉末(1)について、そのp-メトキシフェノール量を測定したところ、12ppmであった。
実施例1において、p-メトキシフェノール量1ppmで重合した。得られた吸水性樹脂粉末(20)のp-メトキシフェノールを測定したところ、ND(1ppm未満)であった。p-メトキシフェノールがND(1ppm未満)の吸水性樹脂粉末(20)の上記(5-15)に記載の耐候性(光劣化)は、p-メトキシフェノールが12ppmである吸水性樹脂粉末(1)より約10%悪かった。p-メトキシフェノールが吸水性樹脂の耐候性を向上させることがわかる。以上、耐候性(光に対する耐ダメージ性)において、単量体及び/又は吸水性樹脂中のp-メトキシフェノール量が重要であることが分かる。
実施例1において、p-メトキシフェノール量230ppmで重合した。得られた吸水性樹脂粉末(21)のp-メトキシフェノールを測定したところ、82ppmであった。p-メトキシフェノールが82ppmの吸水性樹脂粉末(21)の初期着色(製造直後の着色)は、p-メトキシフェノールが12ppmである吸水性樹脂粉末(1)より悪く、L値88.3、a値-1.8、b値10.2であった。着色において、単量体及び/又は吸水性樹脂中のp-メトキシフェノール量が重要であることが分かる。
実施例2において、p-メトキシフェノール量70ppmで重合した。さらに、実施例11と同様に表面架橋を行い、実施例11と同様の物性の吸水性樹脂粉末(22)を得た。p-メトキシフェノールを測定したところ、10ppmであった。
37重量%アクリル酸ナトリウム水溶液595.4[g/min]、48重量%水酸化ナトリウム水溶液198.6[g/min]、100重量%アクリル酸300.1[g/min]、内部架橋剤としてポリエチレングリコールジアクリレート(分子量523)2.71[g/min]、脱イオン水(イオン交換水)203.9[g/min]、31重量%ジエチレントリアミン5酢酸・3ナトリウム水溶液0.42[g/min]、界面活性剤として10重量%ポリオキシエチレン(20)ソルビタンモノステアレート(花王株式会社製)水溶液0.46[g/min]を連続的に分散機を用いて混合し、該分散機を通過した後の単量体水溶液に、3重量%過硫酸ナトリウム水溶液26.0[g/min]をラインミキシングし、ベルト重合機に供給した。該ベルト重合機は、表面がフッ素樹脂コーティングされた長さ3.8m、幅60cmのエンドレスベルトを備え、該ベルトの底面側および重合機の周囲が約90℃に加熱且つ保温され、中央部に蒸発水を回収する為の吸気配管を備えている。又、ベルト上に供給する単量体水溶液の温度は92℃となるように分散機中に通水してコントロールした。(図14参照)
該重合装置に供給される単量体水溶液(23)の温度は92℃であり、溶存酸素量は4.30[ml/l]であった。
実施例23において、ゲル粉砕の条件として、多孔板の孔径19.0mm、孔数10個、厚み10.5mmであった。含水ゲルの供給速度を1600[g/min]に設定し、更に90℃の温水(供給速度;63[g/min])、及び水蒸気(供給速度;95[g/min])を同時にミートチョッパーに供給し、スクリュー軸回転数を257rpmとしたこと以外は実施例23と同様にして粉砕ゲル(24)を得た。
実施例23において、単量体水溶液として用いられる37重量%アクリル酸ナトリウム水溶液及び脱イオン水(イオン交換水)中に、マイクロバブル発生装置(株式会社オーラテック社製、型式:OM4-GP-040)を用いて、窒素ガスを導入気体として導入したこと以外は実施例23と同様にしてゲルを得た。得られたゲルの水可溶分は3.0重量%、固形分は53重量%、水可溶分の重量平均分子量は236521[Da]であった。

Claims (43)
- 気泡を含有したアクリル酸系単量体水溶液を重合する工程と、当該重合工程で得られた含水ゲル状架橋重合体を乾燥する工程とを含むポリアクリル酸系吸水性樹脂粉末の製造方法であって、
界面活性剤及び/又は分散剤存在下で単量体水溶液中の溶存気体の溶解度を低下させることによって、該アクリル酸系単量体水溶液中に気泡を発生させ含有させる気泡発生含有工程を含むことを特徴とする製造方法。 - 上記気泡発生含有工程における、溶存気体の溶解度低下は、界面活性剤及び/又は分散剤を含有するアクリル酸系単量体水溶液を昇温することによって行う、請求項1に記載の製造方法。
- 上記気泡発生含有工程における、溶存気体の溶解度低下は、界面活性剤及び/又は分散剤を含有するアクリル酸系単量体水溶液に水溶性有機物を混合することによって行う、請求項1に記載の製造方法。
- 上記気泡発生含有工程を上記重合工程前に行う、請求項1~3の何れか1項に記載の製造方法。
- 気泡を含有したアクリル酸系単量体水溶液を重合する工程と、当該重合工程で得られた含水ゲル状架橋重合体を乾燥する工程とを含むポリアクリル酸系吸水性樹脂粉末の製造方法であって、
界面活性剤及び/又は分散剤存在下でアクリル酸系単量体水溶液を昇温させる工程を含むことを特徴とする製造方法。 - 気泡を含有したアクリル酸系単量体水溶液を重合する工程と、当該重合工程で得られた含水ゲル状架橋重合体を乾燥する工程とを含むポリアクリル酸系吸水性樹脂粉末の製造方法であって、
界面活性剤及び/又は分散剤存在下でアクリル酸系単量体水溶液に水溶性有機物を混合させる工程を含むことを特徴とする製造方法。 - 重合工程前に、界面活性剤及び/又は分散剤をアクリル酸系単量体水溶液へ添加する、請求項1~6に記載の製造方法。
- 上記水溶性有機物がアクリル酸である、請求項3又は6に記載の製造方法。
- 上記気泡発生含有工程が実質的に常圧で行われる、請求項1~4の何れか1項に記載の製造方法。
- 上記気泡発生含有工程の前又は後に、アクリル酸系単量体水溶液に不活性気体を混合する工程を含む、請求項1又は4に記載の製造方法。
- 上記気泡発生含有工程の終了時から、重合工程での重合の開始時までの時間が300秒以内である、請求項1~4の何れか1項に記載の製造方法。
- アクリル酸系単量体水溶液の上記昇温では、5℃以上昇温させる、請求項2又は5に記載の製造方法。
- 上記昇温後のアクリル酸系単量体水溶液の温度が40~100℃である、請求項2、5又は12に記載の製造方法。
- 上記昇温が、界面活性剤を含有するアクリル酸系単量体水溶液中のアクリル酸の中和熱を利用して行われる、請求項2、5、12又は13に記載の製造方法。
- 上記昇温が、アクリル酸系単量体水溶液を加熱することによって行われる、請求項2、5、12又は13に記載の製造方法。
- 上記界面活性剤が、ノニオン系界面活性剤又はシリコーン系界面活性剤である、請求項1~15の何れか1項に記載の製造方法。
- 上記分散剤が水溶性高分子である、請求項1~16の何れか1項に記載の製造方法。
- 重合時の最高到達温度が100℃以上である、請求項1~17の何れか1項に記載の製造方法。
- 上記単量体水溶液を重合する工程において、単量体水溶液におけるアクリル酸系単量体の濃度が40重量%以上である、請求項1~18の何れか1項に記載の製造方法。
- 含水ゲル状架橋重合体を乾燥する上記工程後に、ポリアクリル酸系吸水性樹脂粉末を表面架橋する工程をさらに含む、請求項1~19の何れか1項に記載の製造方法。
- 単量体水溶液における重合禁止剤メトキシフェノールの含有量が10~200ppmである、請求項1~20の何れか1項に記載の製造方法。
- 重合工程が連続ニーダー重合で行われ、重合中にゲル粉砕が行われ、更に必要により重合後にゲル粉砕が行われる、請求項1~21の何れか1項に記載の製造方法。
- 重合工程が連続ベルト重合で行われ、重合後にゲル粉砕が行われる、請求項1~21の何れか1項に記載の製造方法。
- 含水ゲル状架橋重合体を乾燥する上記工程後に、分級する工程をさらに含み、
分級工程後の微粉を、乾燥工程以前の工程において再利用する、請求項1~23の何れか1項に記載の製造方法。 - 得られる吸水性樹脂の表面張力が60[mN/m]以上を示す範囲で、アクリル酸系単量体水溶液が界面活性剤及び/又は分散剤を含有する、請求項1~24の何れか1項に記載の製造方法。
- 含水ゲル状架橋重合体を乾燥する上記工程後に、得られたポリアクリル酸系吸水性樹脂粉末を表面架橋する工程と、該表面架橋する工程と同時又は別途、ポリアクリル酸系吸水性樹脂粉末の表面をさらに界面活性剤で被覆する工程とをさらに含む、請求項1~25の何れか1項に記載の製造方法。
- 得られたポリアクリル酸系吸水性樹脂粉末の生理食塩水流れ誘導性が20[×10-7・cm3・sec・g-1]以上であり、吸水速度(FSR)が0.25[g/g/sec]以上である、請求項1~26の何れか1項に記載の製造方法。
- 気泡発生含有工程前における単量体水溶液の体積に対して、気泡発生含有工程後における単量体水溶液の体積が1.01~1.1倍となっている、請求項1~27の何れか1項に記載の製造方法。
- 気泡の体積平均粒子径が50μm以下である、請求項1~28の何れか1項に記載の製造方法。
- アクリル酸系単量体水溶液に水溶性有機物を混合させる工程、アクリル酸系単量体水溶液の昇温工程、或いは気泡発生工程含有工程の前又は後に気体を導入する工程を更に含む、請求項1~29の何れか1項に記載の製造方法。
- 重合中又は重合後に、ゲル粉砕エネルギー(GGE)18~60[J/g]でのゲル粉砕及び/又は水可溶分の重量平均分子量を10,000~500,000[Da]増加させるゲル粉砕が行われる、請求項1~30の何れか1項に記載の製造方法。
- 粒度850~150μmの割合が95重量%以上であるポリアクリル酸系吸水性樹脂粉末であって、下記式で規定される内部気泡率が2.8~6.6%であることを特徴とする、吸水性樹脂粉末。
(内部気泡率[%])={(真密度[g/cm3])-(見かけ密度[g/cm3])}/(真密度[g/cm3])×100 - 界面活性剤及び/又は分散剤を内部に含むポリアクリル酸系吸水性樹脂粉末であって、
表面張力が60[mN/m]以上で、かつ、粉末表面が界面活性剤で被覆されてなることを特徴とする、吸水性樹脂粉末。 - 生理食塩水流れ誘導性が20[×10-7・cm3・sec・g-1]以上で、吸水速度(FSR)が0.25[g/g/sec]以上である、請求項32又は33に記載の吸水性樹脂粉末。
- 無荷重下吸水倍率(CRC)が25[g/g]以上であり、荷重50[g/cm2]での加圧下吸収倍率(AAP)が15[g/g]以上である、請求項32~34の何れか1項に記載の吸水性樹脂粉末。
- 無荷重下吸水倍率(CRC)が25[g/g]以上であり、生理食塩水流れ誘導性(SFC)が20[×10-7・cm3・sec・g-1]以上である、請求項32~35の何れか1項に記載の吸水性樹脂粉末。
- 吸水性樹脂粉末が多価金属カチオン、ポリアミンポリマー及び水不溶性微粒子からなる群から選ばれる1種以上の通液性向上剤をさらに含む、請求項32~36の何れか1項に記載の吸水性樹脂粉末。
- 界面活性剤をさらに含む、請求項32に記載の吸水性樹脂粉末。
- p-メトキシフェノールを5~60ppmさらに含む、請求項32~38の何れか1項に記載の吸水性樹脂粉末。
- キレート剤、α-ヒドロキシカルボン酸、及び無機又は有機還元剤からなる群から選ばれる1種以上の添加剤をさらに含む、請求項32~39の何れか1項に記載の吸水性樹脂粉末。
- 下記式
(吸水速度指数)=(FSR[g/g/sec])×(嵩比重[g/cm3])×(重量平均粒子径[μm])
(ただし、FSRは生理食塩水への20倍膨潤での吸水速度を示す。)
で規定される吸水速度指数が90以上で、かつ、嵩比重が0.6~0.8[g/cm3]である、請求項32~40の何れか1項に記載の吸水性樹脂粉末。 - 表面張力が60[mN/m]以上である、請求項32~41の何れか1項に記載の吸水性樹脂粉末。
- 嵩比重が0.58~0.80[g/cm3]である、請求項32~42の何れか1項に記載の吸水性樹脂粉末。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020127019375A KR101895624B1 (ko) | 2009-12-24 | 2010-12-23 | 폴리아크릴산계 흡수성 수지분말 및 그 제조방법 |
| EP10839528.6A EP2518092B1 (en) | 2009-12-24 | 2010-12-23 | Water-absorbable polyacrylic acid resin powder, and process for production thereof |
| US13/518,438 US9334376B2 (en) | 2009-12-24 | 2010-12-23 | Water-absorbable polyacrylic acid resin powder, and process for production thereof |
| SG2012046322A SG181879A1 (en) | 2009-12-24 | 2010-12-23 | Water-absorbable polyacrylic acid resin powder, and process for production thereof |
| JP2011547617A JP5647625B2 (ja) | 2009-12-24 | 2010-12-23 | ポリアクリル酸系吸水性樹脂粉末及びその製造方法 |
| CN201080058964.1A CN102712712B (zh) | 2009-12-24 | 2010-12-23 | 聚丙烯酸系吸水性树脂粉末及其制造方法 |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009292318 | 2009-12-24 | ||
| JP2009-292318 | 2009-12-24 | ||
| JP2010088993 | 2010-04-07 | ||
| JP2010-088993 | 2010-04-07 | ||
| JP2010-149907 | 2010-06-30 | ||
| JP2010149907 | 2010-06-30 | ||
| JP2010-179515 | 2010-08-10 | ||
| JP2010179515 | 2010-08-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011078298A1 true WO2011078298A1 (ja) | 2011-06-30 |
Family
ID=44195823
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/073254 WO2011078298A1 (ja) | 2009-12-24 | 2010-12-23 | ポリアクリル酸系吸水性樹脂粉末及びその製造方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US9334376B2 (ja) |
| EP (1) | EP2518092B1 (ja) |
| JP (2) | JP5647625B2 (ja) |
| KR (1) | KR101895624B1 (ja) |
| CN (1) | CN102712712B (ja) |
| SG (1) | SG181879A1 (ja) |
| WO (1) | WO2011078298A1 (ja) |
Cited By (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012102407A1 (ja) | 2011-01-28 | 2012-08-02 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂粉末の製造方法 |
| WO2013002387A1 (ja) | 2011-06-29 | 2013-01-03 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂粉末及びその製造方法 |
| WO2013018571A1 (ja) * | 2011-08-03 | 2013-02-07 | 住友精化株式会社 | 吸水性樹脂粒子、吸水性樹脂粒子を製造する方法、吸収体、吸収性物品及び止水材 |
| WO2013073682A1 (ja) * | 2011-11-16 | 2013-05-23 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂の製造方法 |
| JPWO2011126079A1 (ja) * | 2010-04-07 | 2013-07-11 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂粉末の製造方法及びポリアクリル酸(塩)系吸水性樹脂粉末 |
| WO2014041968A1 (ja) | 2012-09-11 | 2014-03-20 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水剤の製造方法及びその吸水剤 |
| WO2014041969A1 (ja) | 2012-09-11 | 2014-03-20 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水剤の製造方法及びその吸水剤 |
| WO2014054731A1 (ja) | 2012-10-03 | 2014-04-10 | 株式会社日本触媒 | 吸水剤及びその製造方法 |
| EP2731975A1 (de) | 2011-07-14 | 2014-05-21 | Basf Se | Verfahren zur herstellung wasserabsorbierender polymerpartikel mit hoher anquellgeschwindigkeit |
| EP2739660A1 (de) | 2011-11-17 | 2014-06-11 | Evonik Degussa GmbH | Superabsorbierende polymere mit schnellen absorptionseigenschaften sowie verfahren zu dessen herstellung |
| WO2014119553A1 (ja) | 2013-01-29 | 2014-08-07 | 株式会社日本触媒 | 吸水性樹脂材料及びその製造方法 |
| JP2014533530A (ja) * | 2011-11-17 | 2014-12-15 | エボニック デグサ ゲーエムベーハーEvonik Degussa GmbH | 高充填あるいは繊維フリーの衛生物品用の高吸収性ポリマー |
| EP2814854A1 (de) | 2012-02-15 | 2014-12-24 | Basf Se | Wasserabsorbierende polymerpartikel mit hoher quellgeschwindigkeit und hoher permeabilität |
| WO2015030129A1 (ja) | 2013-08-28 | 2015-03-05 | 株式会社日本触媒 | ゲル粉砕装置、及びポリアクリル酸(塩)系吸水性樹脂粉末の製造方法、並びに吸水性樹脂粉末 |
| WO2015046604A1 (ja) | 2013-09-30 | 2015-04-02 | 株式会社日本触媒 | 粒子状吸水剤の充填方法および粒子状吸水剤充填物のサンプリング方法 |
| WO2015093594A1 (ja) | 2013-12-20 | 2015-06-25 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水剤及びその製造方法 |
| KR20160127742A (ko) | 2014-02-28 | 2016-11-04 | 가부시키가이샤 닛폰 쇼쿠바이 | 폴리(메트)아크릴산(염)계 입자상 흡수제 및 제조 방법 |
| KR20160128350A (ko) | 2014-03-03 | 2016-11-07 | 가부시키가이샤 닛폰 쇼쿠바이 | 폴리아크릴산(염)계 흡수성 수지의 제조 방법 |
| WO2016204302A1 (ja) | 2015-06-19 | 2016-12-22 | 株式会社日本触媒 | ポリ(メタ)アクリル酸(塩)系粒子状吸水剤及び製造方法 |
| JP2017502108A (ja) * | 2013-12-10 | 2017-01-19 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
| US9587081B2 (en) | 2012-02-15 | 2017-03-07 | Basf Se | Water-absorbing polymer particles with high free swell rate and high permeability |
| CN106661166A (zh) * | 2014-05-08 | 2017-05-10 | 巴斯夫欧洲公司 | 吸水性聚合物颗粒 |
| KR20170063818A (ko) | 2014-09-29 | 2017-06-08 | 가부시키가이샤 닛폰 쇼쿠바이 | 흡수성 수지 분말 및 흡수성 수지 분말의 탄성률의 측정 방법 |
| KR20170103849A (ko) | 2015-01-07 | 2017-09-13 | 가부시키가이샤 닛폰 쇼쿠바이 | 흡수제 및 그의 제조 방법, 그리고 평가 방법 및 측정 방법 |
| US9950306B2 (en) | 2011-07-14 | 2018-04-24 | Basf Se | Process for producing water-absorbing polymer particles with high free swell rate |
| JP2018203997A (ja) * | 2017-05-30 | 2018-12-27 | Sdpグローバル株式会社 | 吸水性樹脂粒子及びその製造方法 |
| US10307732B2 (en) | 2013-04-10 | 2019-06-04 | Evonik Corporation | Particulate superabsorbent polymer composition having improved stability and fast absorption |
| JP2019104903A (ja) * | 2017-12-12 | 2019-06-27 | 三洋化成工業株式会社 | 水性(共)重合体組成物 |
| EP3520978A1 (en) | 2013-08-28 | 2019-08-07 | Nippon Shokubai Co., Ltd. | Polyacrylic acid (polyacrylate) superabsorbent polymer powder |
| JPWO2018159803A1 (ja) * | 2017-03-02 | 2019-12-26 | 住友精化株式会社 | 吸水性樹脂及び土嚢 |
| JP2020500985A (ja) * | 2017-10-27 | 2020-01-16 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
| US10682625B2 (en) | 2015-10-02 | 2020-06-16 | Sdp Global Co., Ltd. | Absorbent resin composition and method for producing same |
| WO2020145383A1 (ja) | 2019-01-11 | 2020-07-16 | 株式会社日本触媒 | 吸水剤、及び吸水剤の製造方法 |
| KR20200128969A (ko) * | 2019-05-07 | 2020-11-17 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 및 고흡수성 수지 |
| US11718694B2 (en) | 2019-01-07 | 2023-08-08 | Lg Chem, Ltd. | Super absorbent polymer and preparation method thereof |
Families Citing this family (60)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009130915A1 (ja) * | 2008-04-25 | 2009-10-29 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂およびその製造方法 |
| JP6053109B2 (ja) * | 2012-07-05 | 2016-12-27 | 花王株式会社 | 水不溶性高分子ビルダー |
| US9550843B2 (en) | 2012-11-27 | 2017-01-24 | Nippon Shokubai Co., Ltd. | Method for producing polyacrylic acid (salt)-based water absorbent resin |
| KR101595037B1 (ko) | 2013-01-15 | 2016-02-17 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| US9375507B2 (en) * | 2013-04-10 | 2016-06-28 | Evonik Corporation | Particulate superabsorbent polymer composition having improved stability |
| KR20140134219A (ko) * | 2013-05-13 | 2014-11-21 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| DE102013208942A1 (de) * | 2013-05-15 | 2014-11-20 | Evonik Industries Ag | Superabsorbierende Polymere mit schnellen Absorptionseigenschaften sowie Verfahren zu dessen Herstellung |
| DE102013209023A1 (de) * | 2013-05-15 | 2014-11-20 | Evonik Industries Ag | Superabsorbierende Polymere mit schnellen Absorptionseigenschaften sowie Verfahren zu dessen Herstellung |
| CN105392805B (zh) | 2013-08-01 | 2017-07-25 | 株式会社Lg化学 | 超吸收性聚合物 |
| KR101718942B1 (ko) | 2013-09-17 | 2017-03-22 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| WO2015163517A1 (en) * | 2014-04-25 | 2015-10-29 | Songwon Industrial Co., Ltd. | Release of polymer gel from polymerization belt in production of water-absorbent polymer particles |
| WO2016056866A1 (ko) * | 2014-10-08 | 2016-04-14 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| KR20160061743A (ko) | 2014-11-24 | 2016-06-01 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| KR101769100B1 (ko) | 2014-11-27 | 2017-08-30 | 주식회사 엘지화학 | 가압하 흡수 속도가 빠른 고흡수성 수지 및 이의 제조 방법 |
| CN107428949B (zh) | 2015-03-31 | 2020-03-17 | 株式会社日本触媒 | 聚丙烯酸(盐)系吸水性树脂粉末及其制造方法、以及其评价方法 |
| KR101871968B1 (ko) | 2015-06-01 | 2018-06-27 | 주식회사 엘지화학 | 고흡수성 수지 |
| KR101949454B1 (ko) * | 2015-06-15 | 2019-02-18 | 주식회사 엘지화학 | 고흡수성 수지 |
| KR101949995B1 (ko) * | 2015-07-06 | 2019-02-19 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 및 이로부터 제조된 고흡수성 수지 |
| KR101855351B1 (ko) | 2015-08-13 | 2018-05-04 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| EP3167859B1 (en) | 2015-11-16 | 2020-05-06 | The Procter and Gamble Company | Absorbent cores having material free areas |
| US10604636B2 (en) * | 2015-12-17 | 2020-03-31 | Case Western Reserve University | Elastomeric and viscoelastic materials formed from poly(acrylic acid) gels |
| KR102119813B1 (ko) | 2015-12-23 | 2020-06-05 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| DE102016200324A1 (de) * | 2016-01-14 | 2017-07-20 | MTU Aero Engines AG | Verfahren zum Ermitteln einer Konzentration wenigstens eines Werkstoffs in einem Pulver für ein additives Herstellverfahren |
| WO2017146347A1 (ko) | 2016-02-25 | 2017-08-31 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| KR101949458B1 (ko) * | 2016-03-11 | 2019-02-18 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 및 고흡수성 수지 |
| KR101704789B1 (ko) | 2016-03-23 | 2017-02-08 | 주식회사 엘지화학 | 고흡수성 수지 |
| EP3437729B1 (en) | 2016-03-28 | 2023-12-13 | Nippon Shokubai Co., Ltd. | Water-absorbing agent and method for producing same, and absorbent article produced using water-absorbing agent |
| WO2017170605A1 (ja) * | 2016-03-28 | 2017-10-05 | 株式会社日本触媒 | 粒子状吸水剤 |
| CN105859947B (zh) * | 2016-04-27 | 2018-05-15 | 东华大学 | 一种聚甲基丙烯酸纳米水凝胶的制备方法 |
| CN106491279A (zh) * | 2016-10-21 | 2017-03-15 | 青岛首创嘉业工贸有限公司 | 一种活性卫生巾及其制备方法 |
| KR102075733B1 (ko) | 2016-12-13 | 2020-02-10 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| KR102093352B1 (ko) | 2016-12-19 | 2020-03-25 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| KR102102459B1 (ko) | 2016-12-20 | 2020-04-20 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| KR102086050B1 (ko) * | 2016-12-20 | 2020-03-06 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| KR102193459B1 (ko) | 2016-12-20 | 2020-12-21 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| EP3558629B1 (de) | 2016-12-21 | 2023-11-15 | Basf Se | Einwellenextruder und verwendung eines einwellenextruders sowie verfahren zum ändern einer morphologie eines superabsorbierenden polymergels mit einem einwellenextruder |
| WO2018117548A1 (ko) | 2016-12-23 | 2018-06-28 | 주식회사 엘지화학 | 다공성 고흡수성 수지의 제조 방법 및 다공성 고흡수성 수지 |
| EP3590981B1 (en) * | 2017-03-02 | 2023-09-13 | Sumitomo Seika Chemicals Co., Ltd. | Water-absorbing resin, and absorbent article |
| KR20190120219A (ko) | 2017-03-02 | 2019-10-23 | 스미토모 세이카 가부시키가이샤 | 흡수성 수지, 토양 보수재, 및 농원예 재료 |
| KR102526286B1 (ko) * | 2017-03-02 | 2023-04-27 | 스미토모 세이카 가부시키가이샤 | 흡수성 수지 및 흡수성 물품 |
| KR102487977B1 (ko) * | 2017-07-28 | 2023-01-11 | 주식회사 엘지화학 | 고흡수성 수지 및 그 제조 방법 |
| US11078343B2 (en) | 2017-10-06 | 2021-08-03 | Evonik Operations Gmbh | Absorbent polymeric foam for shoe insoles |
| EP3696532A4 (en) | 2017-10-12 | 2021-12-15 | Nippon Shokubai Co., Ltd. | METHOD OF MEASURING PROPERTIES OF PARTICULAR ABSORBENT AGENT, AND PARTICULAR ABSORBENT AGENT |
| KR102566440B1 (ko) * | 2017-12-15 | 2023-08-14 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| WO2019124536A1 (ja) | 2017-12-21 | 2019-06-27 | 株式会社日本触媒 | 発熱体組成物用吸水性樹脂粉末、及び発熱体組成物 |
| KR101940692B1 (ko) * | 2018-07-10 | 2019-01-21 | (주) 대영하이텍 | 화재용 안전 마스크 |
| WO2020020675A1 (de) | 2018-07-24 | 2020-01-30 | Basf Se | Verfahren zur herstellung von superabsorbern |
| CN109111544B (zh) * | 2018-07-25 | 2020-09-01 | 安徽富瑞雪化工科技股份有限公司 | 一种高倍数耐盐性高吸水树脂 |
| KR102620072B1 (ko) * | 2018-10-12 | 2023-12-29 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 및 고흡수성 수지 |
| CN109776741B (zh) * | 2019-01-23 | 2021-05-07 | 上海华谊新材料有限公司 | 高吸水性树脂的制造方法 |
| US20220080386A1 (en) | 2019-01-24 | 2022-03-17 | Basf Se | Method for producing superabsorbent particles |
| CN113811337A (zh) * | 2019-05-07 | 2021-12-17 | 金伯利-克拉克环球有限公司 | 吸收制品 |
| KR102614036B1 (ko) * | 2019-05-07 | 2023-12-13 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 및 고흡수성 수지 |
| CN114787245A (zh) | 2019-12-13 | 2022-07-22 | 住友精化株式会社 | 吸水性树脂粒子及吸收体 |
| WO2022025122A1 (ja) * | 2020-07-31 | 2022-02-03 | 住友精化株式会社 | 吸水性樹脂粒子を製造する方法 |
| KR20220050584A (ko) | 2020-10-16 | 2022-04-25 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| KR20220054051A (ko) * | 2020-10-23 | 2022-05-02 | 주식회사 엘지화학 | 고흡수성 수지 제조용 중합 반응기 |
| WO2022131838A1 (ko) * | 2020-12-18 | 2022-06-23 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| JP2023540289A (ja) * | 2020-12-18 | 2023-09-22 | エルジー・ケム・リミテッド | 高吸水性樹脂およびその製造方法 |
| TWI777661B (zh) * | 2021-07-08 | 2022-09-11 | 臺灣塑膠工業股份有限公司 | 吸水性樹脂及其製造方法 |
Citations (75)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4703067A (en) | 1981-10-26 | 1987-10-27 | American Colloid Company | Process for preparing dry solid water absorbing polyacrylate resin |
| JPH01318021A (ja) | 1988-06-17 | 1989-12-22 | Kazuo Saotome | 吸水性樹脂成形物の製造方法 |
| US4893999A (en) | 1985-12-18 | 1990-01-16 | Chemische Fabrik Stockhausen Gmbh | Apparatus for the continuous production of polymers and copolymers of water-soluble monomers |
| US5002986A (en) | 1989-02-28 | 1991-03-26 | Hoechst Celanese Corporation | Fluid absorbent compositions and process for their preparation |
| JPH03115313A (ja) * | 1989-09-28 | 1991-05-16 | Kazuo Saotome | 吸水性樹脂の製造方法 |
| WO1991015368A1 (en) | 1990-04-02 | 1991-10-17 | The Procter & Gamble Company | Particulate, absorbent, polymeric compositions containing interparticle crosslinked aggregates |
| WO1992001008A1 (de) | 1990-07-09 | 1992-01-23 | Chemische Fabrik Stockhausen Gmbh | Verfahren zur herstellung wasserquellbarer produkte unter verwendung von feinstanteilen wasserquellbarer polymerer |
| US5124188A (en) | 1990-04-02 | 1992-06-23 | The Procter & Gamble Company | Porous, absorbent, polymeric macrostructures and methods of making the same |
| US5154713A (en) | 1991-10-22 | 1992-10-13 | Nalco Chemical Company | Enhancing absorption rates of superabsorbents by incorporating a blowing agent |
| WO1992018171A1 (en) | 1991-04-12 | 1992-10-29 | The Procter & Gamble Company | Absorbent structures containing specific particle size distributions of superabsorbent hydrogel-forming materials |
| WO1992020723A1 (en) | 1991-05-16 | 1992-11-26 | The Dow Chemical Company | Process for recycling aqueous fluid absorbent fines to a polymerizer |
| US5314420A (en) | 1993-09-17 | 1994-05-24 | Nalco Chemical Company | Superabsorbent polymer having improved absorption rate and absorption under pressure |
| WO1994022502A1 (en) | 1993-03-26 | 1994-10-13 | The Procter & Gamble Company | Superabsorbent polymer foam |
| WO1995002002A1 (de) | 1993-07-09 | 1995-01-19 | Chemische Fabrik Stockhausen Gmbh | Pulverförmige, vernetzte, wässrige flüssigkeiten sowie körperflüssigkeiten absorbierende polymere, verfahren zu ihrer herstellung und ihre anwendung |
| US5451613A (en) | 1993-09-17 | 1995-09-19 | Nalco Chemical Company | Superabsorbent polymer having improved absorption rate and absorption under pressure |
| US5562646A (en) | 1994-03-29 | 1996-10-08 | The Proctor & Gamble Company | Absorbent members for body fluids having good wet integrity and relatively high concentrations of hydrogel-forming absorbent polymer having high porosity |
| US5624967A (en) | 1994-06-08 | 1997-04-29 | Nippon Shokubai Co., Ltd. | Water-absorbing resin and process for producing same |
| WO1997017397A1 (de) | 1995-11-03 | 1997-05-15 | Basf Aktiengesellschaft | Wasserabsorbierende, schaumförmige, vernetzte hydrogel-polymere |
| JPH10114801A (ja) * | 1996-08-23 | 1998-05-06 | Nippon Shokubai Co Ltd | 高吸水速度吸水性樹脂およびその製造方法 |
| JPH10168129A (ja) * | 1996-12-16 | 1998-06-23 | Nippon Shokubai Co Ltd | 吸水性樹脂の製造方法 |
| US5856370A (en) | 1993-12-23 | 1999-01-05 | Stockhausen Gmbh & Co. Kg | Cross-linked synthetic polymers having a porous structure, a high absorption rate for water, aqueous solutions and body fluids, a process for their production and their use in the absorption and/or retention of water and/or aqueous liquids |
| EP0450922B1 (en) | 1990-04-02 | 1999-07-07 | Nippon Shokubai Kagaku Kogyo Co. Ltd. | Method for production of fluid stable aggregate |
| US5985944A (en) | 1994-12-08 | 1999-11-16 | Nippon Shokubai Co., Ltd. | Water-absorbent resin, process for production thereof, and water-absorbent resin composition |
| JP2000000447A (ja) | 1997-12-30 | 2000-01-07 | Hirobumi Onari | 旋回式微細気泡発生装置 |
| JP2000063527A (ja) | 1998-08-12 | 2000-02-29 | Nippon Shokubai Co Ltd | 含水ゲル状架橋重合体の細粒化方法 |
| JP2000506911A (ja) * | 1996-02-28 | 2000-06-06 | ビーエーエスエフ アクチェンゲゼルシャフト | 吸水性のフォーム状の架橋ポリマー |
| US6071976A (en) | 1995-12-27 | 2000-06-06 | Nippon Shokubai Co., Ltd. | Water absorbing agent, manufacturing method thereof, and manufacturing machine thereof |
| US6107358A (en) | 1996-08-23 | 2000-08-22 | Nippon Shokubai Co., Ltd. | Water-absorbent resin and method for production thereof |
| WO2000052087A1 (de) | 1999-03-03 | 2000-09-08 | Basf Aktiengesellschaft | Wasserabsorbierende, schaumförmige, vernetzte polymerisate mit verbesserter verteilungswirkung, verfahren zu ihrer herstellung und ihre verwendung |
| JP2000302876A (ja) | 1999-02-15 | 2000-10-31 | Nippon Shokubai Co Ltd | 吸水性樹脂粉末の製造方法、吸水性樹脂粉末、およびその用途 |
| WO2000069550A1 (fr) | 1999-05-15 | 2000-11-23 | Hirofumi Ohnari | Generateur oscillant de microbulles d'air |
| US6228930B1 (en) | 1997-06-18 | 2001-05-08 | Nippon Shokubai Co., Ltd. | Water-absorbent resin granule-containing composition and production process for water-absorbent resin granule |
| US6241928B1 (en) | 1998-04-28 | 2001-06-05 | Nippon Shokubai Co., Ltd. | Method for production of shaped hydrogel of absorbent resin |
| US6254990B1 (en) | 1998-02-18 | 2001-07-03 | Nippon Shokubai Co., Ltd. | Surface-crosslinking process for water-absorbent resin |
| US6359049B1 (en) | 1999-03-12 | 2002-03-19 | Basf Aktiengesellschaft | Color-stable superabsorbent polymer composition |
| US6414214B1 (en) | 1999-10-04 | 2002-07-02 | Basf Aktiengesellschaft | Mechanically stable hydrogel-forming polymers |
| US6469080B2 (en) | 1999-12-15 | 2002-10-22 | Nippon Shokubai Co., Ltd. | Water-absorbent resin composition |
| US6562879B1 (en) | 1999-02-15 | 2003-05-13 | Nippon Shokubai Co., Ltd. | Water-absorbent resin powder and its production process and use |
| JP2003205228A (ja) | 1997-12-30 | 2003-07-22 | Hirobumi Onari | 旋回式微細気泡発生装置 |
| US6599989B2 (en) | 1998-03-03 | 2003-07-29 | Nippon Skokubai Co., Ltd. | Water-absorbent agents containing polycarboxylic amine chelating agents |
| US6667372B1 (en) | 1999-11-02 | 2003-12-23 | Nippon Shokubai Co., Ltd. | Continuous manufacturing method of water-absorbent polymer |
| US6710141B1 (en) | 1999-11-20 | 2004-03-23 | Basf Aktiengesellschaft | Method for continuously producing cross-linked fine-particle geleous polymerizates |
| WO2004052949A1 (de) | 2002-12-09 | 2004-06-24 | Basf Aktiengesellschaft | Verfahren zur herstellung geruchsarmer hydrogel-bildender polymerisate |
| WO2004069915A2 (en) | 2003-02-10 | 2004-08-19 | Nippon Shokubai Co., Ltd. | Particulate water-absorbing agent |
| WO2005012406A1 (de) | 2003-07-25 | 2005-02-10 | Stockhausen Gmbh | Verfahren zur agglomeration von superabsorberfeinteilchen |
| WO2005016393A1 (en) | 2003-07-31 | 2005-02-24 | Kimberly-Clark Worldwide, Inc. | Absorbent materials and articles |
| US6906159B2 (en) | 2000-08-03 | 2005-06-14 | Nippon Shokubai Co., Ltd. | Water-absorbent resin, hydropolymer, process for producing them, and uses of them |
| JP2005162834A (ja) | 2003-12-01 | 2005-06-23 | Nippon Shokubai Co Ltd | 吸水性樹脂の製法 |
| WO2005063313A1 (en) | 2003-12-19 | 2005-07-14 | Stockhausen, Inc. | Superabsorbent polymer having increased rate of water absorption |
| US6939914B2 (en) | 2002-11-08 | 2005-09-06 | Kimberly-Clark Worldwide, Inc. | High stiffness absorbent polymers having improved absorbency rates and method for making the same |
| US20050215734A1 (en) | 2004-03-24 | 2005-09-29 | Yorimichi Dairoku | Method for continuous production of water-absorbent resin |
| US20050256469A1 (en) | 2003-04-25 | 2005-11-17 | Kimberly-Clark Worldwide Inc. | Absorbent structure with superabsorbent material |
| US6987151B2 (en) | 2001-09-12 | 2006-01-17 | Dow Global Technologies Inc. | Continuous polymerization process for the manufacture of superabsorbent polymers |
| WO2006008905A1 (ja) | 2004-07-15 | 2006-01-26 | Sumitomo Seika Chemicals Co., Ltd. | 吸水性樹脂の製造方法 |
| JP2006116365A (ja) | 2004-09-27 | 2006-05-11 | Nanoplanet Kenkyusho:Kk | 旋回式微細気泡発生装置及び同気泡発生方法 |
| US7169843B2 (en) | 2003-04-25 | 2007-01-30 | Stockhausen, Inc. | Superabsorbent polymer with high permeability |
| US7173086B2 (en) | 2003-10-31 | 2007-02-06 | Stockhausen, Inc. | Superabsorbent polymer with high permeability |
| WO2007074167A2 (de) | 2005-12-29 | 2007-07-05 | Basf Se | Herstellung eines wasserabsorbierenden harzes unter einmischen eines teilchenförmigen additivs |
| US7265190B2 (en) | 2002-11-07 | 2007-09-04 | Nippon Shokubai Co., Ltd. | Process and apparatus for production of water-absorbent resin |
| US20070225422A1 (en) | 2006-03-24 | 2007-09-27 | Nippon Shokubai Co., Ltd. | Water-absorbing resin and method for manufacturing the same |
| US7282262B2 (en) | 2003-02-10 | 2007-10-16 | Nippon Shokubai Co., Ltd. | Particulate water absorbent containing water absorbent resin as a main component |
| JP2007284675A (ja) * | 2006-03-24 | 2007-11-01 | Nippon Shokubai Co Ltd | 吸水性樹脂およびその製造方法 |
| EP1521601B1 (de) | 2002-07-11 | 2008-05-07 | Stockhausen GmbH | Wasserabsorbierende, schaumf rmige polymergebilde |
| WO2008096713A1 (ja) | 2007-02-05 | 2008-08-14 | Nippon Shokubai Co., Ltd. | 粒子状吸水剤およびその製造方法 |
| US7473739B2 (en) | 2004-02-05 | 2009-01-06 | Nippon Shokubai Co., Ltd. | Particulate water absorbent agent and production method thereof, and water absorbent article |
| WO2009016055A2 (en) | 2007-07-27 | 2009-02-05 | Basf Se | Water-absorbing polymeric particles and method for the production thereof |
| WO2009048145A1 (ja) * | 2007-10-10 | 2009-04-16 | Nippon Shokubai Co., Ltd. | 吸水性樹脂組成物およびその製造方法 |
| WO2009062902A2 (de) | 2007-11-15 | 2009-05-22 | Basf Se | Superabsorbierender schaum mit grafischen zeichen an der oberfläche |
| WO2009109563A1 (en) | 2008-03-05 | 2009-09-11 | Basf Se | Process for producing superabsorbents |
| US20090312183A1 (en) | 2006-08-31 | 2009-12-17 | Hirotama Fujimaru | Particulate water absorbing agent and production method thereof |
| WO2009153196A1 (de) | 2008-06-19 | 2009-12-23 | Basf Se | Verfahren zur kontinuierlichen herstellung wasserabsorbierender polymerpartikel |
| WO2010006937A1 (de) | 2008-07-15 | 2010-01-21 | Basf Se | Verfahren zur herstellung wasserabsorbierender polymerpartikel |
| EP1957188B1 (en) | 2005-11-30 | 2010-02-24 | Basf Se | A method of drying an absorbent polymer with a surfactant |
| US20100062252A1 (en) | 2005-02-15 | 2010-03-11 | Nippon Shokubai Co., Ltd | Water absorbing agent, water absorbing article and method for production of water absorbing agent |
| EP2163302A1 (en) | 2007-07-04 | 2010-03-17 | Nippon Shokubai Co., Ltd. | Particulate water-absorbing agent and method for producing the same |
Family Cites Families (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5734101A (en) | 1980-08-11 | 1982-02-24 | Nippon Shokubai Kagaku Kogyo Co Ltd | Novel polymerization process |
| US4985518A (en) | 1981-10-26 | 1991-01-15 | American Colloid Company | Process for preparing water-absorbing resins |
| JPS61126103A (ja) | 1984-11-21 | 1986-06-13 | Nippon Shokubai Kagaku Kogyo Co Ltd | 重合体の製造法 |
| US4820773A (en) | 1986-04-21 | 1989-04-11 | American Colloid Company | Water absorbent resins prepared by polymerization in the presence of styrene-maleic anhydride copolymers |
| KR0130652B1 (ko) | 1987-08-14 | 1998-04-07 | 존 휴즈 | 수분 흡수성 수지의 제조 방법 |
| JPH0696619B2 (ja) | 1989-09-28 | 1994-11-30 | 三洋化成工業株式会社 | 高吸水性ポリマー組成物及びその製造方法ならびにそれからなる高吸水性物品 |
| US5145906A (en) | 1989-09-28 | 1992-09-08 | Hoechst Celanese Corporation | Super-absorbent polymer having improved absorbency properties |
| JPH04175319A (ja) | 1990-07-17 | 1992-06-23 | Sanyo Chem Ind Ltd | 吸水性樹脂の製造法 |
| DE69108804T2 (de) | 1990-07-17 | 1995-08-24 | Sanyo Chemical Ind Ltd | Verfahren zur Herstellung von Wasser absorbierenden Harzen. |
| JPH04236203A (ja) | 1991-01-19 | 1992-08-25 | Sanyo Chem Ind Ltd | 吸水性ヒドロゲル成形物の製造法 |
| US5118719A (en) | 1991-10-22 | 1992-06-02 | Nalco Chemical Company | Enhancing absorption rates of superabsorbents by incorporating a blowing agent |
| JP3103754B2 (ja) | 1995-10-31 | 2000-10-30 | 三洋化成工業株式会社 | 改質された吸水性樹脂粒子およびその製法 |
| DE19646484C2 (de) | 1995-11-21 | 2000-10-19 | Stockhausen Chem Fab Gmbh | Flüssigkeitsabsorbierende Polymere, Verfahren zu deren Herstellung und deren Verwendung |
| JP3895422B2 (ja) | 1997-03-17 | 2007-03-22 | 株式会社日本触媒 | 親水性重合体の製造方法 |
| JPH1057805A (ja) | 1996-08-23 | 1998-03-03 | Nippon Shokubai Co Ltd | 高吸水速度吸水性樹脂組成物 |
| JP4286335B2 (ja) | 1996-10-24 | 2009-06-24 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| JPH10130324A (ja) * | 1996-10-24 | 1998-05-19 | Nippon Shokubai Co Ltd | 吸水性樹脂の製造方法 |
| JP4180134B2 (ja) | 1997-07-25 | 2008-11-12 | 株式会社日本触媒 | 含水吸水性架橋重合体の切断方法 |
| WO1998017453A1 (fr) | 1996-10-24 | 1998-04-30 | Nippon Shokubai Co., Ltd. | Procede de production d'une resine absorbant l'eau |
| JP3913867B2 (ja) | 1997-11-14 | 2007-05-09 | 株式会社日本触媒 | 吸水剤およびその製造方法 |
| TW200504099A (en) * | 2003-07-18 | 2005-02-01 | Nippon Catalytic Chem Ind | Method for production of water-soluble porous polymer and water-soluble porous polymer |
| TW200635969A (en) | 2005-04-06 | 2006-10-16 | Nippon Catalytic Chem Ind | Particulate water absorbing agent, water-absorbent core and absorbing article |
| DE102005042038A1 (de) | 2005-09-02 | 2007-03-08 | Basf Ag | Verfahren zur Herstellung wasserabsorbierender Polymere |
| TWI394789B (zh) * | 2005-12-22 | 2013-05-01 | Nippon Catalytic Chem Ind | 吸水性樹脂組成物及其製造方法、吸收性物品 |
| JP5078131B2 (ja) | 2007-03-27 | 2012-11-21 | 旭化成ケミカルズ株式会社 | 超高速吸収能力をもつ吸収性複合体 |
| EP2238180B1 (de) | 2008-01-23 | 2012-04-04 | Basf Se | Schäumbare copolymere auf basis nachwachsender rohstoffe |
| JP5600670B2 (ja) | 2009-02-17 | 2014-10-01 | 株式会社日本触媒 | ポリアクリル酸系吸水性樹脂粉末およびその製造方法 |
-
2010
- 2010-12-23 JP JP2011547617A patent/JP5647625B2/ja active Active
- 2010-12-23 WO PCT/JP2010/073254 patent/WO2011078298A1/ja active Application Filing
- 2010-12-23 SG SG2012046322A patent/SG181879A1/en unknown
- 2010-12-23 US US13/518,438 patent/US9334376B2/en active Active
- 2010-12-23 CN CN201080058964.1A patent/CN102712712B/zh active Active
- 2010-12-23 KR KR1020127019375A patent/KR101895624B1/ko active IP Right Grant
- 2010-12-23 EP EP10839528.6A patent/EP2518092B1/en active Active
-
2014
- 2014-03-06 JP JP2014044309A patent/JP5889349B2/ja active Active
Patent Citations (82)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4703067A (en) | 1981-10-26 | 1987-10-27 | American Colloid Company | Process for preparing dry solid water absorbing polyacrylate resin |
| US4893999A (en) | 1985-12-18 | 1990-01-16 | Chemische Fabrik Stockhausen Gmbh | Apparatus for the continuous production of polymers and copolymers of water-soluble monomers |
| JPH01318021A (ja) | 1988-06-17 | 1989-12-22 | Kazuo Saotome | 吸水性樹脂成形物の製造方法 |
| US5002986A (en) | 1989-02-28 | 1991-03-26 | Hoechst Celanese Corporation | Fluid absorbent compositions and process for their preparation |
| JPH03115313A (ja) * | 1989-09-28 | 1991-05-16 | Kazuo Saotome | 吸水性樹脂の製造方法 |
| WO1991015368A1 (en) | 1990-04-02 | 1991-10-17 | The Procter & Gamble Company | Particulate, absorbent, polymeric compositions containing interparticle crosslinked aggregates |
| US5124188A (en) | 1990-04-02 | 1992-06-23 | The Procter & Gamble Company | Porous, absorbent, polymeric macrostructures and methods of making the same |
| EP0450922B1 (en) | 1990-04-02 | 1999-07-07 | Nippon Shokubai Kagaku Kogyo Co. Ltd. | Method for production of fluid stable aggregate |
| EP0595803B1 (en) | 1990-04-02 | 2002-02-13 | The Procter & Gamble Company | Porous, absorbent, polymeric macrostructures and methods of making the same |
| WO1992001008A1 (de) | 1990-07-09 | 1992-01-23 | Chemische Fabrik Stockhausen Gmbh | Verfahren zur herstellung wasserquellbarer produkte unter verwendung von feinstanteilen wasserquellbarer polymerer |
| WO1992018171A1 (en) | 1991-04-12 | 1992-10-29 | The Procter & Gamble Company | Absorbent structures containing specific particle size distributions of superabsorbent hydrogel-forming materials |
| WO1992020723A1 (en) | 1991-05-16 | 1992-11-26 | The Dow Chemical Company | Process for recycling aqueous fluid absorbent fines to a polymerizer |
| US5154713A (en) | 1991-10-22 | 1992-10-13 | Nalco Chemical Company | Enhancing absorption rates of superabsorbents by incorporating a blowing agent |
| WO1994022502A1 (en) | 1993-03-26 | 1994-10-13 | The Procter & Gamble Company | Superabsorbent polymer foam |
| WO1995002002A1 (de) | 1993-07-09 | 1995-01-19 | Chemische Fabrik Stockhausen Gmbh | Pulverförmige, vernetzte, wässrige flüssigkeiten sowie körperflüssigkeiten absorbierende polymere, verfahren zu ihrer herstellung und ihre anwendung |
| US5399591A (en) | 1993-09-17 | 1995-03-21 | Nalco Chemical Company | Superabsorbent polymer having improved absorption rate and absorption under pressure |
| US5451613A (en) | 1993-09-17 | 1995-09-19 | Nalco Chemical Company | Superabsorbent polymer having improved absorption rate and absorption under pressure |
| US5462972A (en) | 1993-09-17 | 1995-10-31 | Nalco Chemical Company | Superabsorbent polymer having improved absorption rate and absorption under pressure |
| US5314420A (en) | 1993-09-17 | 1994-05-24 | Nalco Chemical Company | Superabsorbent polymer having improved absorption rate and absorption under pressure |
| US5856370A (en) | 1993-12-23 | 1999-01-05 | Stockhausen Gmbh & Co. Kg | Cross-linked synthetic polymers having a porous structure, a high absorption rate for water, aqueous solutions and body fluids, a process for their production and their use in the absorption and/or retention of water and/or aqueous liquids |
| US5669894A (en) | 1994-03-29 | 1997-09-23 | The Procter & Gamble Company | Absorbent members for body fluids having good wet integrity and relatively high concentrations of hydrogel-forming absorbent polymer |
| US5562646A (en) | 1994-03-29 | 1996-10-08 | The Proctor & Gamble Company | Absorbent members for body fluids having good wet integrity and relatively high concentrations of hydrogel-forming absorbent polymer having high porosity |
| US5624967A (en) | 1994-06-08 | 1997-04-29 | Nippon Shokubai Co., Ltd. | Water-absorbing resin and process for producing same |
| US5985944A (en) | 1994-12-08 | 1999-11-16 | Nippon Shokubai Co., Ltd. | Water-absorbent resin, process for production thereof, and water-absorbent resin composition |
| WO1997017397A1 (de) | 1995-11-03 | 1997-05-15 | Basf Aktiengesellschaft | Wasserabsorbierende, schaumförmige, vernetzte hydrogel-polymere |
| US6071976A (en) | 1995-12-27 | 2000-06-06 | Nippon Shokubai Co., Ltd. | Water absorbing agent, manufacturing method thereof, and manufacturing machine thereof |
| JP2000506911A (ja) * | 1996-02-28 | 2000-06-06 | ビーエーエスエフ アクチェンゲゼルシャフト | 吸水性のフォーム状の架橋ポリマー |
| US6107358A (en) | 1996-08-23 | 2000-08-22 | Nippon Shokubai Co., Ltd. | Water-absorbent resin and method for production thereof |
| JPH10114801A (ja) * | 1996-08-23 | 1998-05-06 | Nippon Shokubai Co Ltd | 高吸水速度吸水性樹脂およびその製造方法 |
| JPH10168129A (ja) * | 1996-12-16 | 1998-06-23 | Nippon Shokubai Co Ltd | 吸水性樹脂の製造方法 |
| US6228930B1 (en) | 1997-06-18 | 2001-05-08 | Nippon Shokubai Co., Ltd. | Water-absorbent resin granule-containing composition and production process for water-absorbent resin granule |
| JP2003205228A (ja) | 1997-12-30 | 2003-07-22 | Hirobumi Onari | 旋回式微細気泡発生装置 |
| JP2000000447A (ja) | 1997-12-30 | 2000-01-07 | Hirobumi Onari | 旋回式微細気泡発生装置 |
| US6254990B1 (en) | 1998-02-18 | 2001-07-03 | Nippon Shokubai Co., Ltd. | Surface-crosslinking process for water-absorbent resin |
| US6599989B2 (en) | 1998-03-03 | 2003-07-29 | Nippon Skokubai Co., Ltd. | Water-absorbent agents containing polycarboxylic amine chelating agents |
| US6241928B1 (en) | 1998-04-28 | 2001-06-05 | Nippon Shokubai Co., Ltd. | Method for production of shaped hydrogel of absorbent resin |
| JP2000063527A (ja) | 1998-08-12 | 2000-02-29 | Nippon Shokubai Co Ltd | 含水ゲル状架橋重合体の細粒化方法 |
| JP2000302876A (ja) | 1999-02-15 | 2000-10-31 | Nippon Shokubai Co Ltd | 吸水性樹脂粉末の製造方法、吸水性樹脂粉末、およびその用途 |
| US6562879B1 (en) | 1999-02-15 | 2003-05-13 | Nippon Shokubai Co., Ltd. | Water-absorbent resin powder and its production process and use |
| WO2000052087A1 (de) | 1999-03-03 | 2000-09-08 | Basf Aktiengesellschaft | Wasserabsorbierende, schaumförmige, vernetzte polymerisate mit verbesserter verteilungswirkung, verfahren zu ihrer herstellung und ihre verwendung |
| US6359049B1 (en) | 1999-03-12 | 2002-03-19 | Basf Aktiengesellschaft | Color-stable superabsorbent polymer composition |
| WO2000069550A1 (fr) | 1999-05-15 | 2000-11-23 | Hirofumi Ohnari | Generateur oscillant de microbulles d'air |
| US6414214B1 (en) | 1999-10-04 | 2002-07-02 | Basf Aktiengesellschaft | Mechanically stable hydrogel-forming polymers |
| US6667372B1 (en) | 1999-11-02 | 2003-12-23 | Nippon Shokubai Co., Ltd. | Continuous manufacturing method of water-absorbent polymer |
| US6710141B1 (en) | 1999-11-20 | 2004-03-23 | Basf Aktiengesellschaft | Method for continuously producing cross-linked fine-particle geleous polymerizates |
| US6469080B2 (en) | 1999-12-15 | 2002-10-22 | Nippon Shokubai Co., Ltd. | Water-absorbent resin composition |
| US7091253B2 (en) | 2000-08-03 | 2006-08-15 | Nippon Shokubai Co., Ltd. | Water-absorbent resin, hydropolymer, process for producing them, and uses of them |
| US6906159B2 (en) | 2000-08-03 | 2005-06-14 | Nippon Shokubai Co., Ltd. | Water-absorbent resin, hydropolymer, process for producing them, and uses of them |
| US6987151B2 (en) | 2001-09-12 | 2006-01-17 | Dow Global Technologies Inc. | Continuous polymerization process for the manufacture of superabsorbent polymers |
| EP1521601B1 (de) | 2002-07-11 | 2008-05-07 | Stockhausen GmbH | Wasserabsorbierende, schaumf rmige polymergebilde |
| US7265190B2 (en) | 2002-11-07 | 2007-09-04 | Nippon Shokubai Co., Ltd. | Process and apparatus for production of water-absorbent resin |
| US6939914B2 (en) | 2002-11-08 | 2005-09-06 | Kimberly-Clark Worldwide, Inc. | High stiffness absorbent polymers having improved absorbency rates and method for making the same |
| WO2004052949A1 (de) | 2002-12-09 | 2004-06-24 | Basf Aktiengesellschaft | Verfahren zur herstellung geruchsarmer hydrogel-bildender polymerisate |
| WO2004069915A2 (en) | 2003-02-10 | 2004-08-19 | Nippon Shokubai Co., Ltd. | Particulate water-absorbing agent |
| US20060204755A1 (en) | 2003-02-10 | 2006-09-14 | Kazushi Torii | Walter-absorbing agent |
| US7282262B2 (en) | 2003-02-10 | 2007-10-16 | Nippon Shokubai Co., Ltd. | Particulate water absorbent containing water absorbent resin as a main component |
| US20050256469A1 (en) | 2003-04-25 | 2005-11-17 | Kimberly-Clark Worldwide Inc. | Absorbent structure with superabsorbent material |
| US7169843B2 (en) | 2003-04-25 | 2007-01-30 | Stockhausen, Inc. | Superabsorbent polymer with high permeability |
| WO2005012406A1 (de) | 2003-07-25 | 2005-02-10 | Stockhausen Gmbh | Verfahren zur agglomeration von superabsorberfeinteilchen |
| WO2005016393A1 (en) | 2003-07-31 | 2005-02-24 | Kimberly-Clark Worldwide, Inc. | Absorbent materials and articles |
| US7173086B2 (en) | 2003-10-31 | 2007-02-06 | Stockhausen, Inc. | Superabsorbent polymer with high permeability |
| JP2005162834A (ja) | 2003-12-01 | 2005-06-23 | Nippon Shokubai Co Ltd | 吸水性樹脂の製法 |
| WO2005063313A1 (en) | 2003-12-19 | 2005-07-14 | Stockhausen, Inc. | Superabsorbent polymer having increased rate of water absorption |
| US7582705B2 (en) | 2004-02-05 | 2009-09-01 | Nippon Shokubai Co., Ltd. | Particulate water absorbent agent and production method thereof, and water absorbent article |
| US7473739B2 (en) | 2004-02-05 | 2009-01-06 | Nippon Shokubai Co., Ltd. | Particulate water absorbent agent and production method thereof, and water absorbent article |
| US20050215734A1 (en) | 2004-03-24 | 2005-09-29 | Yorimichi Dairoku | Method for continuous production of water-absorbent resin |
| WO2006008905A1 (ja) | 2004-07-15 | 2006-01-26 | Sumitomo Seika Chemicals Co., Ltd. | 吸水性樹脂の製造方法 |
| JP2006116365A (ja) | 2004-09-27 | 2006-05-11 | Nanoplanet Kenkyusho:Kk | 旋回式微細気泡発生装置及び同気泡発生方法 |
| US20100062252A1 (en) | 2005-02-15 | 2010-03-11 | Nippon Shokubai Co., Ltd | Water absorbing agent, water absorbing article and method for production of water absorbing agent |
| EP1957188B1 (en) | 2005-11-30 | 2010-02-24 | Basf Se | A method of drying an absorbent polymer with a surfactant |
| WO2007074167A2 (de) | 2005-12-29 | 2007-07-05 | Basf Se | Herstellung eines wasserabsorbierenden harzes unter einmischen eines teilchenförmigen additivs |
| US20070225422A1 (en) | 2006-03-24 | 2007-09-27 | Nippon Shokubai Co., Ltd. | Water-absorbing resin and method for manufacturing the same |
| JP2007284675A (ja) * | 2006-03-24 | 2007-11-01 | Nippon Shokubai Co Ltd | 吸水性樹脂およびその製造方法 |
| US20090312183A1 (en) | 2006-08-31 | 2009-12-17 | Hirotama Fujimaru | Particulate water absorbing agent and production method thereof |
| WO2008096713A1 (ja) | 2007-02-05 | 2008-08-14 | Nippon Shokubai Co., Ltd. | 粒子状吸水剤およびその製造方法 |
| EP2163302A1 (en) | 2007-07-04 | 2010-03-17 | Nippon Shokubai Co., Ltd. | Particulate water-absorbing agent and method for producing the same |
| WO2009016055A2 (en) | 2007-07-27 | 2009-02-05 | Basf Se | Water-absorbing polymeric particles and method for the production thereof |
| WO2009048145A1 (ja) * | 2007-10-10 | 2009-04-16 | Nippon Shokubai Co., Ltd. | 吸水性樹脂組成物およびその製造方法 |
| WO2009062902A2 (de) | 2007-11-15 | 2009-05-22 | Basf Se | Superabsorbierender schaum mit grafischen zeichen an der oberfläche |
| WO2009109563A1 (en) | 2008-03-05 | 2009-09-11 | Basf Se | Process for producing superabsorbents |
| WO2009153196A1 (de) | 2008-06-19 | 2009-12-23 | Basf Se | Verfahren zur kontinuierlichen herstellung wasserabsorbierender polymerpartikel |
| WO2010006937A1 (de) | 2008-07-15 | 2010-01-21 | Basf Se | Verfahren zur herstellung wasserabsorbierender polymerpartikel |
Non-Patent Citations (3)
| Title |
|---|
| "Foaming engineering", TECHNOSYSTEM CO. LTD., pages: 759 - 774 |
| "Modern Superabsorbent Polymer Technology", 1998, pages 39-44 - 197-99 |
| See also references of EP2518092A4 * |
Cited By (79)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10434495B2 (en) | 2010-04-07 | 2019-10-08 | Nippon Shokubai Co., Ltd. | Method for producing water absorbent polyacrylic acid (salt) resin powder, and water absorbent polyacrylic acid (salt) resin powder |
| KR101908142B1 (ko) | 2010-04-07 | 2018-10-15 | 가부시키가이샤 닛폰 쇼쿠바이 | 폴리아크릴산(염)계 흡수성 수지분말의 제조방법 및 폴리아크릴산(염)계 흡수성 수지분말 |
| JP5676572B2 (ja) * | 2010-04-07 | 2015-02-25 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂粉末の製造方法及びポリアクリル酸(塩)系吸水性樹脂粉末 |
| US9447203B2 (en) | 2010-04-07 | 2016-09-20 | Nippom Shokubai Co., Ltd. | Method for producing water absorbent polyacrylic acid (salt) resin powder, and water absorbent polyacrylic acid (salt) resin powder |
| JPWO2011126079A1 (ja) * | 2010-04-07 | 2013-07-11 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂粉末の製造方法及びポリアクリル酸(塩)系吸水性樹脂粉末 |
| KR101946227B1 (ko) | 2010-04-07 | 2019-02-08 | 가부시키가이샤 닛폰 쇼쿠바이 | 폴리아크릴산(염)계 흡수성 수지분말의 제조방법 및 폴리아크릴산(염)계 흡수성 수지분말 |
| WO2012102407A1 (ja) | 2011-01-28 | 2012-08-02 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂粉末の製造方法 |
| US20140031473A1 (en) * | 2011-01-28 | 2014-01-30 | Nippon Shokubai Co., Ltd. | Method for producing polyacrylic acid (salt)-based water absorbent resin powder |
| US9044525B2 (en) | 2011-06-29 | 2015-06-02 | Nippon Shokubai Co., Ltd. | Polyacrylic acid (salt)-based water absorbent resin powder and method for producing the same |
| JPWO2013002387A1 (ja) * | 2011-06-29 | 2015-02-23 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂粉末及びその製造方法 |
| US20140193641A1 (en) * | 2011-06-29 | 2014-07-10 | Nippon Shokubai Co., Ltd. | Polyacrylic Acid (Salt)-Based Water Absorbent Resin Powder and Method for Producing the Same |
| JP5599513B2 (ja) * | 2011-06-29 | 2014-10-01 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂粉末及びその製造方法 |
| WO2013002387A1 (ja) | 2011-06-29 | 2013-01-03 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂粉末及びその製造方法 |
| EP2731975A1 (de) | 2011-07-14 | 2014-05-21 | Basf Se | Verfahren zur herstellung wasserabsorbierender polymerpartikel mit hoher anquellgeschwindigkeit |
| US9950306B2 (en) | 2011-07-14 | 2018-04-24 | Basf Se | Process for producing water-absorbing polymer particles with high free swell rate |
| US9199218B2 (en) | 2011-08-03 | 2015-12-01 | Sumitomo Seika Chemicals Co., Ltd. | Water absorbing resin particles, method for manufacturing water absorbing resin particles, absorption body, absorptive article, and water-sealing material |
| WO2013018571A1 (ja) * | 2011-08-03 | 2013-02-07 | 住友精化株式会社 | 吸水性樹脂粒子、吸水性樹脂粒子を製造する方法、吸収体、吸収性物品及び止水材 |
| JP5551836B2 (ja) * | 2011-11-16 | 2014-07-16 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂の製造方法 |
| US9012356B2 (en) | 2011-11-16 | 2015-04-21 | Nippon Shokubai Co., Ltd. | Method for producing polyacrylic acid (salt)-based water absorbent resin |
| WO2013073682A1 (ja) * | 2011-11-16 | 2013-05-23 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂の製造方法 |
| JP2014533530A (ja) * | 2011-11-17 | 2014-12-15 | エボニック デグサ ゲーエムベーハーEvonik Degussa GmbH | 高充填あるいは繊維フリーの衛生物品用の高吸収性ポリマー |
| EP2739660A1 (de) | 2011-11-17 | 2014-06-11 | Evonik Degussa GmbH | Superabsorbierende polymere mit schnellen absorptionseigenschaften sowie verfahren zu dessen herstellung |
| US10391195B2 (en) | 2011-11-17 | 2019-08-27 | Evonik Degussa Gmbh | Super-absorbing polymers with rapid absorption properties and method for producing the same |
| US9555148B2 (en) | 2011-11-17 | 2017-01-31 | Evonik Degussa Gmbh | Superabsorbing polymers with rapid absorption properties and method for producing the same |
| EP2773691B1 (de) | 2011-11-17 | 2020-07-15 | Evonik Operations GmbH | Superabsorbierende polymere für hochgefüllte oder faserfreie hygieneartikel |
| JP2015508836A (ja) * | 2012-02-15 | 2015-03-23 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | 高い膨潤速度および高い透過率を有する吸水性ポリマー粒子 |
| US9587081B2 (en) | 2012-02-15 | 2017-03-07 | Basf Se | Water-absorbing polymer particles with high free swell rate and high permeability |
| US9738769B2 (en) | 2012-02-15 | 2017-08-22 | Basf Se | Water-absorbing polymer particles with high free swell rate and high permeability |
| EP2814854A1 (de) | 2012-02-15 | 2014-12-24 | Basf Se | Wasserabsorbierende polymerpartikel mit hoher quellgeschwindigkeit und hoher permeabilität |
| EP2814854B1 (de) * | 2012-02-15 | 2019-01-23 | Basf Se | Wasserabsorbierende polymerpartikel mit hoher quellgeschwindigkeit und hoher permeabilität |
| WO2014041968A1 (ja) | 2012-09-11 | 2014-03-20 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水剤の製造方法及びその吸水剤 |
| WO2014041969A1 (ja) | 2012-09-11 | 2014-03-20 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水剤の製造方法及びその吸水剤 |
| KR20150067218A (ko) | 2012-10-03 | 2015-06-17 | 가부시키가이샤 닛폰 쇼쿠바이 | 흡수제 및 그의 제조 방법 |
| EP3369480A1 (en) | 2012-10-03 | 2018-09-05 | Nippon Shokubai Co., Ltd. | Water absorbing agent |
| WO2014054731A1 (ja) | 2012-10-03 | 2014-04-10 | 株式会社日本触媒 | 吸水剤及びその製造方法 |
| US10046304B2 (en) | 2012-10-03 | 2018-08-14 | Nippon Shokubai Co., Ltd. | Water absorbing agent and method for producing the same |
| WO2014119553A1 (ja) | 2013-01-29 | 2014-08-07 | 株式会社日本触媒 | 吸水性樹脂材料及びその製造方法 |
| US10307732B2 (en) | 2013-04-10 | 2019-06-04 | Evonik Corporation | Particulate superabsorbent polymer composition having improved stability and fast absorption |
| WO2015030129A1 (ja) | 2013-08-28 | 2015-03-05 | 株式会社日本触媒 | ゲル粉砕装置、及びポリアクリル酸(塩)系吸水性樹脂粉末の製造方法、並びに吸水性樹脂粉末 |
| EP3401070A1 (en) | 2013-08-28 | 2018-11-14 | Nippon Shokubai Co., Ltd. | A water absorbent resin powder and an absorbent body |
| EP3520978A1 (en) | 2013-08-28 | 2019-08-07 | Nippon Shokubai Co., Ltd. | Polyacrylic acid (polyacrylate) superabsorbent polymer powder |
| WO2015046604A1 (ja) | 2013-09-30 | 2015-04-02 | 株式会社日本触媒 | 粒子状吸水剤の充填方法および粒子状吸水剤充填物のサンプリング方法 |
| US10577135B2 (en) | 2013-09-30 | 2020-03-03 | Nippon Shokubai Co., Ltd. | Method for filling particulate water absorbing agent and method for sampling filled particulate water absorbing agent |
| US10934031B2 (en) | 2013-09-30 | 2021-03-02 | Nippon Shokubai Co., Ltd. | Method for filling particulate water absorbing agent and method for sampling filled particulate water absorbing agent |
| EP4159307A1 (en) | 2013-09-30 | 2023-04-05 | Nippon Shokubai Co., Ltd. | Method for filling particulate water absorbing agent and method for sampling filled particulate water absorbing agent |
| KR20160064113A (ko) | 2013-09-30 | 2016-06-07 | 가부시키가이샤 닛폰 쇼쿠바이 | 입자상 흡수제의 충전 방법 및 입자상 흡수제 충전물의 샘플링 방법 |
| JP2017502108A (ja) * | 2013-12-10 | 2017-01-19 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
| WO2015093594A1 (ja) | 2013-12-20 | 2015-06-25 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水剤及びその製造方法 |
| EP4252728A2 (en) | 2013-12-20 | 2023-10-04 | Nippon Shokubai Co., Ltd. | Water absorbing agent based on polyacrylic acid and/or a salt thereof |
| KR20160102217A (ko) | 2013-12-20 | 2016-08-29 | 가부시키가이샤 닛폰 쇼쿠바이 | 폴리아크릴산(염)계 흡수제 및 그의 제조 방법 |
| US10646612B2 (en) | 2013-12-20 | 2020-05-12 | Nippon Shokubai Co., Ltd. | Polyacrylic acid (salt) water absorbent, and method for producing same |
| JPWO2015129917A1 (ja) * | 2014-02-28 | 2017-03-30 | 株式会社日本触媒 | ポリ(メタ)アクリル酸(塩)系粒子状吸水剤及び製造方法 |
| US10207250B2 (en) | 2014-02-28 | 2019-02-19 | Nippon Shokubai Co., Ltd. | Poly(meth)acrylic acid (salt)-based particulate absorbent |
| KR20160127742A (ko) | 2014-02-28 | 2016-11-04 | 가부시키가이샤 닛폰 쇼쿠바이 | 폴리(메트)아크릴산(염)계 입자상 흡수제 및 제조 방법 |
| KR20160128350A (ko) | 2014-03-03 | 2016-11-07 | 가부시키가이샤 닛폰 쇼쿠바이 | 폴리아크릴산(염)계 흡수성 수지의 제조 방법 |
| US9896529B2 (en) | 2014-03-03 | 2018-02-20 | Nippon Shokubai Co., Ltd. | Method for producing polyacrylic acid (salt)-based water-absorbable resin |
| CN106661166B (zh) * | 2014-05-08 | 2019-08-13 | 巴斯夫欧洲公司 | 吸水性聚合物颗粒 |
| CN106661166A (zh) * | 2014-05-08 | 2017-05-10 | 巴斯夫欧洲公司 | 吸水性聚合物颗粒 |
| KR20170063818A (ko) | 2014-09-29 | 2017-06-08 | 가부시키가이샤 닛폰 쇼쿠바이 | 흡수성 수지 분말 및 흡수성 수지 분말의 탄성률의 측정 방법 |
| US10300458B2 (en) | 2014-09-29 | 2019-05-28 | Nippon Shokubai Co., Ltd. | Water-absorbable resin powder, and method for determining elastic modulus of water-absorbable resin powder |
| EP3984633A1 (en) | 2015-01-07 | 2022-04-20 | Nippon Shokubai Co., Ltd. | Water absorbent agent |
| US10695746B2 (en) | 2015-01-07 | 2020-06-30 | Nippon Shokubai Co., Ltd. | Water absorbent agent |
| KR20170103849A (ko) | 2015-01-07 | 2017-09-13 | 가부시키가이샤 닛폰 쇼쿠바이 | 흡수제 및 그의 제조 방법, 그리고 평가 방법 및 측정 방법 |
| JPWO2016111223A1 (ja) * | 2015-01-07 | 2017-12-07 | 株式会社日本触媒 | 吸水剤及びその製造方法、並びに評価方法及び測定方法 |
| US11958921B2 (en) | 2015-06-19 | 2024-04-16 | Nippon Shokubai Co., Ltd. | Poly (meth) acrylic acid (salt)-based particulate water-absorbing agent and production method therefor |
| WO2016204302A1 (ja) | 2015-06-19 | 2016-12-22 | 株式会社日本触媒 | ポリ(メタ)アクリル酸(塩)系粒子状吸水剤及び製造方法 |
| US11535689B2 (en) | 2015-06-19 | 2022-12-27 | Nippon Shokubai Co., Ltd. | Poly (meth) acrylic acid (salt)-based particulate water-absorbing agent and production method therefor |
| US10682625B2 (en) | 2015-10-02 | 2020-06-16 | Sdp Global Co., Ltd. | Absorbent resin composition and method for producing same |
| JPWO2018159803A1 (ja) * | 2017-03-02 | 2019-12-26 | 住友精化株式会社 | 吸水性樹脂及び土嚢 |
| JP7239276B2 (ja) | 2017-05-30 | 2023-03-14 | Sdpグローバル株式会社 | 吸水性樹脂粒子及びその製造方法 |
| JP2018203997A (ja) * | 2017-05-30 | 2018-12-27 | Sdpグローバル株式会社 | 吸水性樹脂粒子及びその製造方法 |
| US11407848B2 (en) | 2017-10-27 | 2022-08-09 | Lg Chem, Ltd. | Method for preparing super absorbent polymer |
| JP2020500985A (ja) * | 2017-10-27 | 2020-01-16 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
| JP7181775B2 (ja) | 2017-12-12 | 2022-12-01 | 三洋化成工業株式会社 | 水性(共)重合体組成物 |
| JP2019104903A (ja) * | 2017-12-12 | 2019-06-27 | 三洋化成工業株式会社 | 水性(共)重合体組成物 |
| US11718694B2 (en) | 2019-01-07 | 2023-08-08 | Lg Chem, Ltd. | Super absorbent polymer and preparation method thereof |
| WO2020145383A1 (ja) | 2019-01-11 | 2020-07-16 | 株式会社日本触媒 | 吸水剤、及び吸水剤の製造方法 |
| KR102316433B1 (ko) * | 2019-05-07 | 2021-10-21 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 및 고흡수성 수지 |
| KR20200128969A (ko) * | 2019-05-07 | 2020-11-17 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 및 고흡수성 수지 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2518092A4 (en) | 2013-05-22 |
| JP5889349B2 (ja) | 2016-03-22 |
| JPWO2011078298A1 (ja) | 2013-05-09 |
| US9334376B2 (en) | 2016-05-10 |
| JP2014098172A (ja) | 2014-05-29 |
| EP2518092A1 (en) | 2012-10-31 |
| CN102712712B (zh) | 2015-05-06 |
| SG181879A1 (en) | 2012-07-30 |
| CN102712712A (zh) | 2012-10-03 |
| KR20120132475A (ko) | 2012-12-05 |
| KR101895624B1 (ko) | 2018-09-05 |
| JP5647625B2 (ja) | 2015-01-07 |
| EP2518092B1 (en) | 2017-03-15 |
| US20120258851A1 (en) | 2012-10-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5889349B2 (ja) | ポリアクリル酸系吸水性樹脂粉末及びその製造方法 | |
| JP6359600B2 (ja) | ポリアクリル酸系吸水性樹脂粉末の製造方法 | |
| JP6157853B2 (ja) | ポリアクリル酸系吸水性樹脂及びその製造方法 | |
| JP5616437B2 (ja) | ポリアクリル酸(塩)系吸水性樹脂粉末の製造方法 | |
| JP6441894B2 (ja) | ポリ(メタ)アクリル酸(塩)系粒子状吸水剤及び製造方法 | |
| JP5591448B2 (ja) | 吸水剤およびその製造方法 | |
| JP6029800B2 (ja) | 吸水性樹脂粒子 | |
| JPWO2015093594A1 (ja) | ポリアクリル酸(塩)系吸水剤及びその製造方法 | |
| JP2007056071A (ja) | 改質された吸水性樹脂の製法 | |
| JP7100742B1 (ja) | ポリ(メタ)アクリル酸(塩)系吸水性樹脂、及び吸収体 | |
| EP4338832A1 (en) | Poly(meth)acrylic acid (salt) water-absorbing resin and absorbent article | |
| JP2022175088A (ja) | ポリ(メタ)アクリル酸(塩)系吸水性樹脂、及び吸収体 | |
| JP2023540766A (ja) | 高吸水性樹脂およびその製造方法 | |
| WO2022196763A1 (ja) | 吸水性樹脂の製造方法 | |
| JP2023088497A (ja) | 表面架橋された(メタ)アクリル酸(塩)系吸水性樹脂を含む吸水剤および(メタ)アクリル酸(塩)系吸水性樹脂の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080058964.1 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10839528 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011547617 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13518438 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2010839528 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010839528 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20127019375 Country of ref document: KR Kind code of ref document: A |