WO2003104334A1 - μ−オキソ架橋型異種金属フタロシアニン化合物及びその選択的製造方法 - Google Patents
μ−オキソ架橋型異種金属フタロシアニン化合物及びその選択的製造方法 Download PDFInfo
- Publication number
- WO2003104334A1 WO2003104334A1 PCT/JP2003/007240 JP0307240W WO03104334A1 WO 2003104334 A1 WO2003104334 A1 WO 2003104334A1 JP 0307240 W JP0307240 W JP 0307240W WO 03104334 A1 WO03104334 A1 WO 03104334A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phthalocyanine
- group
- metal
- oxo
- heterometallic
- Prior art date
Links
Classifications
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G5/00—Recording members for original recording by exposure, e.g. to light, to heat, to electrons; Manufacture thereof; Selection of materials therefor
- G03G5/02—Charge-receiving layers
- G03G5/04—Photoconductive layers; Charge-generation layers or charge-transporting layers; Additives therefor; Binders therefor
- G03G5/06—Photoconductive layers; Charge-generation layers or charge-transporting layers; Additives therefor; Binders therefor characterised by the photoconductive material being organic
- G03G5/0664—Dyes
- G03G5/0696—Phthalocyanines
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B47/00—Porphines; Azaporphines
- C09B47/04—Phthalocyanines abbreviation: Pc
- C09B47/08—Preparation from other phthalocyanine compounds, e.g. cobaltphthalocyanineamine complex
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B47/00—Porphines; Azaporphines
- C09B47/04—Phthalocyanines abbreviation: Pc
- C09B47/32—Cationic phthalocyanine dyes
-
- G—PHYSICS
- G11—INFORMATION STORAGE
- G11B—INFORMATION STORAGE BASED ON RELATIVE MOVEMENT BETWEEN RECORD CARRIER AND TRANSDUCER
- G11B7/00—Recording or reproducing by optical means, e.g. recording using a thermal beam of optical radiation by modifying optical properties or the physical structure, reproducing using an optical beam at lower power by sensing optical properties; Record carriers therefor
- G11B7/24—Record carriers characterised by shape, structure or physical properties, or by the selection of the material
- G11B7/241—Record carriers characterised by shape, structure or physical properties, or by the selection of the material characterised by the selection of the material
- G11B7/242—Record carriers characterised by shape, structure or physical properties, or by the selection of the material characterised by the selection of the material of recording layers
- G11B7/244—Record carriers characterised by shape, structure or physical properties, or by the selection of the material characterised by the selection of the material of recording layers comprising organic materials only
- G11B7/246—Record carriers characterised by shape, structure or physical properties, or by the selection of the material characterised by the selection of the material of recording layers comprising organic materials only containing dyes
- G11B7/248—Record carriers characterised by shape, structure or physical properties, or by the selection of the material characterised by the selection of the material of recording layers comprising organic materials only containing dyes porphines; azaporphines, e.g. phthalocyanines
Definitions
- the present invention relates to a ⁇ -oxo bridged heterometallic phthalocyanine compound and a method for selectively producing the compound.
- the present invention relates to a novel oxo-bridged heterometallic phthalocyanine compound useful for a charge generating material such as an organic photoreceptor, a photoconductive material, an optical recording material, an organic solar cell material, a nonlinear optical material, and the like. It relates to the manufacturing method.
- Metal phthalocyanine-based compounds are attracting attention as organic photoconductive substances having a sensitivity around 800 ⁇ m, which is the wavelength range of transmission of semiconductor lasers.
- Many organic photoconductors (OPCs) containing such an organic photoconductive substance as an active ingredient have been proposed.
- OPCs organic photoconductors
- an organic photoconductor using a titaerphthalocyanine compound as a charge generation material has been put to practical use. .
- a phthalocyanine (P c) compound generates electric charge by light irradiation, and exhibits various electric characteristics depending on its crystal transformation, presence / absence and type of a central metal, and the like.
- P c phthalocyanine
- Japanese Patent Application Laid-Open No. 2-272720 discloses that a metal-free phthalocyanine has the same amount or less as the metal-free phthalocyanine.
- Japanese Patent Application Laid-Open No. Hei 4 (1993) 713 describes a mixed crystal comprising oxytitanium phthalocyanine and at least one type of hydroxymetal phthalocyanine.
- Japanese Patent Application Laid-Open No. 4-184452 describes a coating solution containing tita; ⁇ phthalocyanine and a multilayer phthalocyanine derivative, which is used for a photoreceptor.
- Hei 8-677829 discloses that at least two or more phthalocyanine compounds are dissolved in an acid, and this is added to a mixed solution of water and an organic solvent having a dielectric constant of 20 or less.
- Japanese Patent Application Laid-Open No. 2002-127790 describes a mixed crystal composed of at least three kinds of phthalocyanines having different central substances.
- Japanese Patent Application Laid-Open No. 9-210720 describes a novel oxo-aluminum phthalocyanine dimer having a novel crystal modification
- Japanese Patent Application Laid-Open No. 10-88023 discloses / i- An oxo monogallium phthalocyanine dimer is described.
- Japanese Patent Publication No. 7-29595 describes an alkoxy-bridged metal phthalocyanine dimer.
- JP-A-6-145550 describes a method of reprecipitating a phthalocyanine mixture containing titanyl phthalocyanine and a metal halide phthalocyanine having a trivalent central metal in an acid paste method in an acid pasting method. I have. However, the above-mentioned literature does not describe a ⁇ -oxo cross-linked compound, nor does it describe selectively obtaining them.
- Japanese Patent Application Laid-Open No. 2000-219817 discloses a monooxoaluminum / gallium phthalocyanine dimer.
- the phthalocyanine dimer described here is necessarily obtained stochastically as a mixture of three types of ⁇ -oxo aluminum phthalocyanine dimer and ⁇ -oxo gallium phthalocyanine dimer. It is.
- U.S. Patent No. 4 9 0 0 8 1 7 No. for example, (HO) G e P c -OS i P c OS i (C 6 H x 3) polycyclic phthalocyanine compound such that 3 Is described.
- the center metal is tetravalent Si or Ge.
- the method is based on a dehydration reaction of a hydroxy-substituted metal (IV) phthalocyanine in an organic solvent.
- An object of the present invention is to provide a novel / Z-oxo crosslinked heterometallic phthalocyanine compound which can impart a variety of photosensitivity and electrical characteristics to an organic photoreceptor as a charge generation material.
- Another object of the present invention is to provide a method for producing the ⁇ -oxo-bridged heterometallic phthalocyanine compound in a simple and selective manner with a high yield.
- the present invention provides a formula
- M l represents a metal atom capable of taking a valence of up to 3 (excluding indium)
- M 2 represents a metal atom capable of taking a valence of 4
- R represents Independently represents one or more substituents and / or substituted atoms
- (A m ⁇ ) represents m-valent anion A
- n / m represents the number of anion
- n is an atom of M 2 Represents an integer selected from 0 or 1 to 3 corresponding to the value
- m represents 1 or 2.
- the present invention provides a ⁇ -oxo bridged heterometallic phthalocyanine compound represented by the formula (1), whereby the above object can be achieved.
- the present invention provides a ⁇ -oxo crosslinked heterometallic compound comprising a step of reacting phthalocyanine whose central metal is a metal halide (III) and phthalocyanine whose central metal is an oxymetal (IV) in equimolar amounts.
- An object of the present invention is to provide a method for producing a phthalocyanine compound, whereby the above object can be achieved.
- the present invention provides a step of reacting phthalocyanine whose central metal is a metal halide (III) with phthalocyanine whose central metal is an oxymetal (IV) in equimolar amounts, and using the obtained compound with aqueous ammonia.
- An object of the present invention is to provide a method for producing a muoxo-crosslinked type heterometallic phthalocyanine compound, which comprises a washing step, whereby the above object can be achieved.
- FIG. 1 is an XRD spectrum of the first embodiment.
- FIG. 2 is a TOF-MS spectrum of the first embodiment.
- FIG. 3 is an XRD spectrum of Example 2.
- FIG. 4 is a TOF-MS spectrum of Example 2.
- FIG. 5 is an XRD spectrum of Example 4.
- FIG. 6 is a TOF-MS spectrum of Example 4.
- FIG. 7 is a TOF-MS spectrum of Example 6.
- FIG. 8 is a TOF-MS spectrum of Example 7.
- FIG. 9 is a 1 H-NMR spectrum of Example 7.
- Figure 10 is a 1 3 C-NMR spectrum of Example 7.
- FIG. 11 shows an IR spectrum of the seventh embodiment.
- FIG. 12 is a near-ultraviolet-visible-near-infrared absorption spectrum of Example 7.
- FIG. 13 is a TOF-MS spectrum of Comparative Example 2.
- the ⁇ _oxo-bridged heterometallic phthalocyanine compound of the present invention is a compound having a structure in which central metal atoms (Ml, M2) of a metal phthalocyanine containing Ml as a central metal and a metal phthalocyanine containing M2 as a central metal are oxo-bridged.
- Ml means a metal atom that can take up to three valencies.
- metal atoms of Group 3A (eg, Sc, Y) or Group 3B (eg, A1, Ga, In, T1) of the periodic table are included in Ml.
- M2 means a metal atom capable of taking the valence of 4.
- metal atoms of groups 4A to 7A, 8 and 4B to 6B of the periodic table are included in M2.
- Metal atoms of Group 3A or 3B of the Periodic Table (eg A1, Ga) are not included in M2. It should be noted that, in the state of being included in the structure of the ⁇ -oxo crosslinked heterometallic phthalocyanine compound, ⁇ 2 may exist in a trivalent form.
- the ⁇ -oxo-bridged heterometallic phthalocyanine compound of the present invention may have one or more substituents and / or substituent atoms (R) on their aromatic rings.
- the type of the substituent or the substituent atom is not particularly limited as long as it is stably present in the compound.
- an alkyl group for example, a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, a tert-butyl group, an isoamyl group
- an alkoxy group for example, a methoxy group, an ethoxy group, an isopropoxy group, Butoxy group
- phenoxy group aryl group (for example, phenyl group, tolyl group), aralkyl group (for example, benzyl group), aryl group, alkenyl group, cyano group, halogen atom (Eg, Cl, Br, I, F, etc.), carboxylate ester group, sulfonic ester group, nitro group, amino group and the like.
- the ⁇ -oxo-bridged heterometallic phthalocyanine compound of the present invention may have a positive charge (11 +) corresponding to the valence of the central metal atom ( ⁇ 2). Therefore, they usually exist in a solution with an appropriate anion ( ⁇ ).
- halogen ions e.g., C 1-
- it is a hydroxy ion (OH—) when washed with ammonia water after the reaction.
- the above ⁇ _oxo-bridged heterometallic phthalocyanine compound includes a phthalocyanine whose central metal is a halogenated metal ( ⁇ ) (hereinafter, referred to as a metal halide phthalocyanine) and a phthalocyanine whose central metal is an oxymetal (IV) ( Hereinafter, it is referred to as oxymetal (IV) phthalocyanine.)
- ⁇ halogenated metal
- IV oxymetal
- the metal (III) halide phthalocyanine is, for example, of the formula
- R and M 1 are as defined above, and X represents a halogen atom].
- the metal halide (III) phthalocyanine can be obtained by using a known method. For example, it can be obtained by reacting phthalonitrile, 3-diiminoisoindoline, or a derivative thereof with a haptic compound of a trivalent metal atom in a high boiling organic solvent such as 1-chloronaphthalene or quinoline. it can. Further, the product can be washed with hot DMF or DMF after hot filtration.
- the halogen atom of the halide is fluorine, chlorine, bromine or iodine, Preferably it is chlorine.
- Preferred examples of the central metal atom M 1 of the metal halide (III) phthalocyanine described above include, for example, aluminum [A 1 (111)] and gallium [G a ( ⁇ )]. However, it is preferable not to use indium [I n (III)] as M 1. In the case of metal (III) phthalocyanine whose central metal is indium, In is eliminated from the phthalocyanine ring by sulfuric acid treatment, and metal-free phthalocyanine is by-produced, so that the reaction of the present invention is less likely to occur. .
- the halogenated metal (III) phthalocyanine preferably includes chlorogallium phthalocyanine and chloroaluminum phthalocyanine.
- oxymetal (IV) phthalocyanine is, for example, of the formula
- the oxymetal (IV) phthalocyanine can be obtained using a known method.
- a high-boiling organic solvent such as 1-chloronaphthalene or quinoline
- phthalonitrile, 1,3-diiminoisoindoline, or a derivative thereof is converted to a halide of a trivalent to hexavalent metal atom (for example, titanium chloride).
- Vanadyl chloride and molybdenum chloride Vanadyl chloride and molybdenum chloride
- Hydrolysis can occur by washing with DMF or the like, but is preferably performed in dilute hydrochloric acid if necessary.
- Examples of the central metal atom M 2 of the above oxymetal (IV) phthalocyanine include titanium (T i), vanadium (V), molybdenum (Mo), palladium (P d), and the like.
- vanadi phthalocyanine [0 VPc]
- oxymolybdene phthalocyanine Nin [0 MoPc].
- titanyl phthalocyanine is preferable.
- titanyl phthalocyanine is generally reacted with phthalonitrile or 1,3-diiminoisoindoline with a titanium chloride (eg, titanium tetrachloride) in a high boiling organic solvent such as 1-chloronaphthalene or quinoline.
- a titanium chloride eg, titanium tetrachloride
- a high boiling organic solvent such as 1-chloronaphthalene or quinoline.
- Cloguchi titanium phthalocyanine can be obtained by hydrolyzing in dilute hydrochloric acid water.
- phthalonitrile and titanium tetrachloride are mixed with a proton-accepting type reaction accelerator (for example, 1,8-diazabicyclo [5.4.0] indene-7-cene (DBU) or 1,5) in an alcohol solvent.
- DBU 1,8-diazabicyclo [5.4.0] indene-7-cene
- the ⁇ -oxo-bridged heterometallic phthalocyanine compound of the present invention can be produced by reacting the above-mentioned metal halide (III) phthalocyanine with oxymetal (IV) phthalocyanine.
- the mixing molar ratio of the halogenated metal (III) phthalocyanine and the oxymetal (IV) phthalocyanine is 1: 1 and equimolar, and is preferable. This is because the reaction at this mixing ratio can selectively obtain the desired / oxo-bridged heterometallic phthalocyanine in high yield.
- a metal (III) phthalocyanine halide and an oxy metal (IV) phthalocyanine are mixed and reacted in an equimolar amount in the presence of concentrated sulfuric acid. It is preferable to use sulfuric acid having a concentration of 95% or more as concentrated sulfuric acid.
- the metal (III) phthalocyanine and the oxy metal (IV) phthalocyanine can be dissolved in concentrated sulfuric acid under cooling (for example, at 5 ° C. or lower) and reacted for 2 to 3 hours. Further, after this reaction, the compound can be precipitated by pouring the reaction compound into a large amount of water-Z ice. By this operation, the compound can be refined and purified.
- the process of dissolving the reactants and the like in concentrated sulfuric acid and pouring the solution into water / ice to precipitate solids, which is then refined and purified is called “acid pasting treatment”.
- a so-called acid pasting treatment is used to react a metal halide (III) phthalocyanine with an oxymetal (IV) phthalocyanine to form a ⁇ -oxo metal.
- a so-bridged heterometallic phthalocyanine compound is obtained.
- the obtained compound is further washed with aqueous ammonia, whereby the acid radical can be easily removed from the reaction compound.
- the compound can be easily purified by adding the reaction compound to water and an ammonia solution, and then sufficiently washing the filtered compound with water and ion-exchanged water and drying.
- the preferred aqueous ammonia is one having a concentration of 1% or more, preferably 5 to 50%, and more preferably 25% aqueous ammonia. According to this method, a ⁇ -oxo crosslinked heterometallic phthalocyanine compound can be easily and selectively produced in high yield.
- a metal halide (III) phthalocyanine / oxymetal (IV) phthalocyanine which is preferable for the production of the / oxo-bridged heterometallic phthalocyanine compound of the present invention, is preferably a gallium phthalocyanine / titanole Phthalocyanine, chlorogallium phthalocyanine Vanazino phthalocyanine, chlorogallium phthalocyanine / oxymolybdenum phthalocyanine, chloroanoreminium phthalocyanine / titania phthalocyanine, chloroanolemin phthalocyanine / oxymolypdenphthalocyanine Chlorogallium phthalocyanine; and zoxypalladium phthalocyanine.
- these metal (in) phthalocyanine halides and zinc metal (IV) phthalocyanines may have one or more substituents and / or substituent atoms on their aromatic rings.
- the ⁇ -oxo crosslinked heterometallic phthalocyanine of the present invention thus obtained is useful as a charge generation material such as an organic photoreceptor, a photoconductive material, an optical recording material, an organic solar cell material, a nonlinear optical material, and the like. It is.
- D—BiII type dye (donor dye (D) and Axceptor dye (A), which had been conventionally obtained separately by separation and purification from a mixed system due to difficulty in selective production) Is a dye connected by a ⁇ bond.
- An oxo-bridged heterometallic phthalocyanine compound can be easily obtained.
- the ⁇ -oxo crosslinked heterometallic phthalocyanine compound is useful for a charge generation material such as an organic photoreceptor, a photoconductive material, an optical recording material, an organic solar cell material, a nonlinear optical material, and the like.
- the present invention Is capable of unambiguously inducing the polarization of the intramolecular electronic state by reacting a metal halide (III) phthalocyanine with an oxymetal (IV) phthalocyanine. It can be obtained simply and selectively in high yield.
- the obtained dichlorotitanium phthalocyanine was dispersed in 3% aqueous hydrochloric acid (100 Om1) and washed with water until the pH reached 6 or more. Next, this water-moist cake was put into about 50 Om 1 of DMF previously heated to 100 to 120 ° C., stirred at this temperature for about 1 hour, and filtered while hot. The obtained DMF wet cake was replaced with methanol (10 Om1) and dried to obtain 29.1 g of titanium phthalocyanine [O2TiPc].
- a four-necked flask was charged with 145.5 g (1.13 mo1) of phthalonitrile, 680 ml of 1-chloronaphthalene and 50. Og (0.284mo1) of gallium (III) chloride and heated. The mixture was stirred under reflux at 255 ° C for 12 hours. Thereafter, the reflux was stopped, the mixture was allowed to cool to about 130 ° C, filtered while hot, and sprinkled and washed with 2000 ml of hot dimethylformamide (100 ° C DMF) and 1000 ml of DMF.
- the obtained cake was dispersed again in 1500 ml of DMF, stirred and refluxed for 3 hours, filtered while hot at 110 ° C, and washed by sprinkling with 1000 ml of hot DMF (110 ° C) and 1000 ml of DMF. After repeating this operation twice, the obtained cake was washed with 1000 ml of methanol and 1000 ml of water, dried at 70 ° C., and treated with chlorogallium phthalocyanine [C 1 G a P c] 128.8 g ( Yield 73.5%).
- a four-necked flask was charged with 180. Og (1.4 lmo 1) of phthaloetrinole, 90 Om 1 of naphthalene i-monochrome and 47. Og (0.353 mol 1) of Shirai-Dai Aluminum (III), and heated. The mixture was stirred at 240 ° C for 6 hours under reflux. After that, stop the reflux Stop, cool to about 130 ° C, filter while hot, shake with hot toluene (100 ° C) 180 Oml, Tonolen 80m1, Acetone 900m1, and wash with Tonolen 100m Replaced by l.
- the obtained cake was stirred and refluxed in 75 Oml of toluene for 3 hours, filtered while hot at 100 ° C, washed with 1800 ml of hot toluene (100 ° C), 180 ml of toluene, 900 ml of acetone, and washed with water. The solvent was replaced with 400 ml.
- the obtained cake was added to 4500 ml of water, and heated and dispersed at 70 ° C for 1 hour. After filtration while hot, the mixture was washed with 900 ml of acetone and 1000 ml of water, and dried at 70 ° C. to obtain 187.6 g (yield 92.5%) of chloroanoreminium phthalocyanine [C 1 A 1 Pc].
- the chlorogallium phthalocyanine obtained in Synthesis Example 4 was dissolved in concentrated sulfuric acid, and subjected to acid pasting. As described in the Chemical Society of Japan 12, 878, 1997, the products thus obtained were identified as hydroxygallium phthalocyanine (H OGaP c) and ⁇ -oxogallium phthalocyanine dimer (P cGa—O— G a P c).
- Tetrakis tert-butyl-lutitanyl phthalocyanine [0 Ti P c (t-Bu)] in the same manner as in Synthesis Example 1 except that the starting material (phthalonitrinole) in Synthesis Example 1 was changed to tert-butynolephate nitrile. 4 ] was synthesized.
- Table 1 shows the result of elemental analysis of this compound (PcGa-0-TiPc: molecular weight: 75.64).
- Fig. 1 shows the X-ray diffraction spectrum (XRD spectrum) of this compound.
- Fig. 2 shows the mass spectrometry spectrum of this compound by TOF-MS (time-of-flight mass spectrometry).
- the TOF-MS was measured using “KOMPACT MALD I III” in the detection mode “POS I TIVE”, the extraction voltage “LOW (5 KV)”, and the flight mode “REFLECT I ON”.
- Table 2 shows the result of elemental analysis of this compound (PcGa-O-VPc: molecular weight: 78.72).
- Figure 3 shows the XRD spectrum of this compound.
- Fig. 4 shows the spectrum of this compound by TOF-MS.
- Table 3 shows the results of elemental analysis of this compound (PcGa—O—MoPc: molecular weight 1223.72).
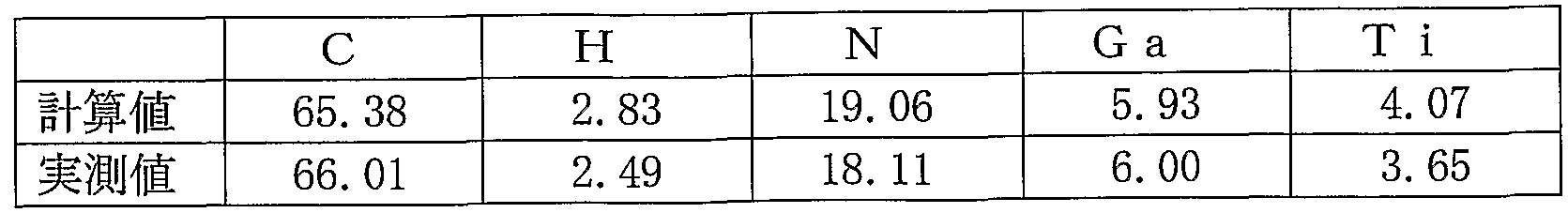
- Table 4 shows the results of elemental analysis of this compound (PcAl ——— Tipc: molecular weight: 1 132.90).
- Figure 5 shows the XRD spectrum of this compound.
- Fig. 6 shows the spectrum of this compound by TOF-MS.
- a wet cake, 0.4 L of water and 0.2 L of 25% aqueous ammonia were charged into a 1 L separab / reflask and dispersed for 6 hours. After filtration under reduced pressure, hot water The sample was sprinkled and washed with 1.5 L and ion-exchanged water 1.5 L. The wet cake was dried at 70 ° C. to obtain 10.2 g (yield: 89.8%) of a blue solid [Chemical Formula 5].
- Table 5 shows the results of elemental analysis of this compound (PcA1-0-VPc: molecular weight 1 135.98).
- Table 6 shows the results of elemental analysis of this compound (PcGa-O-TiPc (tert-Bu)) having a molecular weight of 14007.
- Fig. 7 shows the mass spectrum of this compound by TOF-MS.
- Table 7 shows the result of elemental analysis of this compound (molecular weight: 1624.5).
- FIG. 8 shows the mass spectrum of this compound measured by T ⁇ ⁇ ⁇ F-MS.
- Figure 1 shows the 1 H-NMR spectrum
- Figure 13 shows the 13 C-NMR spectrum
- Figure 11 shows the IR spectrum. See Figure 12.
- the cake After washing with 200 Om 1 of water, the cake was dispersed in 180 Om 1 of water and suction filtered. The cake was washed with 800 ml of water. The washed cake was added to 550 ml of warm water and 66 ml of 25% aqueous ammonia, and dispersed under reflux for 6 hours. After filtration, the cake was washed with 6 ml of hot water (60 ° C) and 1650 ml of ion exchange water (I EW). When the pH and the conductivity of the filtrate reached the ion-exchanged water level, the filtrate was dried at 70 ° C. to obtain 10.5 g (yield: 89.8%) of a blue solid.
- I EW ion exchange water
- Table 8 shows the results of elemental analysis of this sample.
- FIG. 13 shows a mass spectrometry spectrum of this compound by TOF-MS.
- the titanyl phthalocyanine obtained in Synthesis Example 1 and the product known to be a mixture of the hydroxygallium phthalocyanine and ⁇ - oxogallium phthalocyanine dimer of Synthesis Example 6 are simply mixed in equal amounts.
- a sample was prepared. Under the following conditions, for 10 samples prepared was Tsu mass analysis by TOF MS 0 [Table 9]
- Example 5 ClAlPc VOPc Described in Example 5
- ClGaPc chlorogallium phthalocyanine
- VOP c Vanadyl Phthalocyan
- GPL —Oxo-gallium phthalocyanine dimer
- M + and [M_ ⁇ H] + indicate the parent peak (PP) of the heterometallic phthalocyanine dimer in TOF-MS analysis.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Physics & Mathematics (AREA)
- General Physics & Mathematics (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Photoreceptors In Electrophotography (AREA)
Abstract
Description


















Claims



Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020047020043A KR100946448B1 (ko) | 2002-06-10 | 2003-06-09 | μ-옥소 가교형 이종 금속 프탈로시아닌 화합물 및 그의선택적 제조방법 |
| EP03730862A EP1514904B1 (en) | 2002-06-10 | 2003-06-09 | µ-OXO CROSSLINKED DISSIMILAR METAL PHTHALOCYANINE COMPOUND AND PROCESS FOR SELECTIVELY PRODUCING THE SAME |
| US10/516,884 US7462711B2 (en) | 2002-06-10 | 2003-06-09 | U-oxo crosslinked dissimilar metal phthalocyanine compound and process for selectively producing the same |
| DE60319081T DE60319081T2 (de) | 2002-06-10 | 2003-06-09 | µ-OXO-VERNETZTE PHTHALOCYANINVERBINDUNG MIT UNGLEICHEN METALLEN UND EIN VERFAHREN ZU DEREN SELEKTIVER HERSTELLUNG |
| JP2004511398A JP4461011B2 (ja) | 2002-06-10 | 2003-06-09 | μ−オキソ架橋型異種金属フタロシアニン化合物及びその選択的製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002168580 | 2002-06-10 | ||
| JP2002-168580 | 2002-06-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2003104334A1 true WO2003104334A1 (ja) | 2003-12-18 |
Family
ID=29727690
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2003/007240 WO2003104334A1 (ja) | 2002-06-10 | 2003-06-09 | μ−オキソ架橋型異種金属フタロシアニン化合物及びその選択的製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US7462711B2 (ja) |
| EP (1) | EP1514904B1 (ja) |
| JP (1) | JP4461011B2 (ja) |
| KR (1) | KR100946448B1 (ja) |
| DE (1) | DE60319081T2 (ja) |
| WO (1) | WO2003104334A1 (ja) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR100695531B1 (ko) * | 2005-10-11 | 2007-03-16 | 호서대학교 산학협력단 | 말단에 관능기를 갖는 옥소 가교형 프탈로시아닌 화합물 및그 제조방법 |
| EP2285907A4 (en) * | 2008-04-01 | 2012-01-04 | New Jersey Tech Inst | MICROWAVE-ASSISTED SYNTHESIS OF PERFLUOROPHTHALOCYANINE MOLECULES |
| JP2010163473A (ja) * | 2009-01-13 | 2010-07-29 | Konica Minolta Business Technologies Inc | インクジェット記録用シアンインク |
| JP6141145B2 (ja) * | 2012-08-22 | 2017-06-07 | キヤノン株式会社 | 色素化合物、並びに該色素化合物を含有するインク、カラーフィルター用レジスト組成物、及び感熱転写記録用インクシート |
| WO2014140829A1 (en) | 2013-03-11 | 2014-09-18 | Saudi Basic Industries Corporation | Aryloxy-phthalocyanines of group iii metals |
| KR101679999B1 (ko) | 2013-03-11 | 2016-11-25 | 사우디 베이식 인더스트리즈 코포레이션 | 태양전지에 사용하기 위한 ⅳ 족 금속의 아릴옥시-프탈로시아닌 |
| KR101577693B1 (ko) * | 2013-12-27 | 2015-12-15 | 나노씨엠에스(주) | 폴리 옥소가교형 프탈로시아닌 화합물, 이의 제조방법 및 이를 이용한 근적외선 흡수 및 반사 조성물 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06145550A (ja) * | 1992-11-02 | 1994-05-24 | Hitachi Chem Co Ltd | フタロシアニン組成物、その製造法およびそれを用いた電子写真感光体ならびに電荷発生層用塗液 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2754739B2 (ja) * | 1989-06-06 | 1998-05-20 | 日本電気株式会社 | フタロシアニン結晶とその製造方法及びこれを用いた電子写真感光体 |
| EP0798346A3 (en) | 1996-03-29 | 1997-12-10 | Xerox Corporation | Hydroxygallium phthalocyanine and alkoxygallium phthalocyanine dimers |
| US6093514A (en) | 1998-11-26 | 2000-07-25 | Orient Chemical Industries, Ltd. | (μ)-oxo-aluminum/gallium phthalocyanine dimer |
| JP4430862B2 (ja) * | 2002-11-08 | 2010-03-10 | オリヱント化学工業株式会社 | μ−オキソ架橋型異種金属化合物及びその選択的製造方法 |
| JP4146245B2 (ja) * | 2003-01-06 | 2008-09-10 | オリヱント化学工業株式会社 | 電子写真有機感光体 |
-
2003
- 2003-06-09 EP EP03730862A patent/EP1514904B1/en not_active Expired - Lifetime
- 2003-06-09 DE DE60319081T patent/DE60319081T2/de not_active Expired - Lifetime
- 2003-06-09 JP JP2004511398A patent/JP4461011B2/ja not_active Expired - Fee Related
- 2003-06-09 US US10/516,884 patent/US7462711B2/en not_active Expired - Fee Related
- 2003-06-09 WO PCT/JP2003/007240 patent/WO2003104334A1/ja active IP Right Grant
- 2003-06-09 KR KR1020047020043A patent/KR100946448B1/ko not_active IP Right Cessation
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06145550A (ja) * | 1992-11-02 | 1994-05-24 | Hitachi Chem Co Ltd | フタロシアニン組成物、その製造法およびそれを用いた電子写真感光体ならびに電荷発生層用塗液 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1514904A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1514904A4 (en) | 2005-11-09 |
| US20060020129A1 (en) | 2006-01-26 |
| JPWO2003104334A1 (ja) | 2005-10-06 |
| EP1514904A1 (en) | 2005-03-16 |
| DE60319081D1 (de) | 2008-03-27 |
| KR20050020832A (ko) | 2005-03-04 |
| DE60319081T2 (de) | 2009-02-05 |
| US7462711B2 (en) | 2008-12-09 |
| JP4461011B2 (ja) | 2010-05-12 |
| KR100946448B1 (ko) | 2010-03-10 |
| EP1514904B1 (en) | 2008-02-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3764506B2 (ja) | アルコキシ橋かけ金属フタロシアニン二量体の製造方法 | |
| JPH08259835A (ja) | ヒドロキシガリウムフタロシアニンの製造方法及び積層型光導電性画像形成部材 | |
| JPS61171771A (ja) | 電子写真用感光体の製造方法 | |
| JPS63366A (ja) | 結晶型オキシチタニウムフタロシアニン及びその製造方法 | |
| JPH075846B2 (ja) | A型オキシチタニウムフタロシアニンの製造方法 | |
| WO2003104334A1 (ja) | μ−オキソ架橋型異種金属フタロシアニン化合物及びその選択的製造方法 | |
| JPH0598174A (ja) | チタンフタロシアニンの製造法 | |
| JPH07292268A (ja) | アルコキシ橋かけ金属フタロシアニン二量体 | |
| JPH06122833A (ja) | ヒドロキシメタルフタロシアニン顔料の顔料化方法 | |
| JPH06293769A (ja) | 金属フタロシアニン類の製造方法および電子写真感光体 | |
| JP4430862B2 (ja) | μ−オキソ架橋型異種金属化合物及びその選択的製造方法 | |
| JP3665362B2 (ja) | Iv型オキシチタンフタロシアニンの製造方法及びiv型チタニルフタロシアニンの製造方法 | |
| JP4146245B2 (ja) | 電子写真有機感光体 | |
| JP4159125B2 (ja) | x型無金属フタロシアニンの製造方法 | |
| JP5064813B2 (ja) | Y型チタニルフタロシアニン結晶の製造方法 | |
| JP3141157B2 (ja) | 結晶変換によるチタニルフタロシアニンの製造方法 | |
| JPH02169671A (ja) | 結晶型オキシチタニウムフタロシアニンおよびそれを含む電子写真感光体の製造方法 | |
| JP2765399B2 (ja) | ヒドロキシガリウムフタロシアニン結晶の製造方法 | |
| JPS63141982A (ja) | フタロシアニンヘキサデカフルオリド類化合物 | |
| JPH0586302A (ja) | チタニルフタロシアニンの精製方法 | |
| JP5052217B2 (ja) | 分散液、及び電子写真感光体の製造方法 | |
| JPH07207171A (ja) | クロロガリウムフタロシアニンの製造方法 | |
| JP5052216B2 (ja) | チタニルフタロシアニン結晶、及びその製造方法 | |
| JP3646383B2 (ja) | ジハロゲノチタニウムフタロシアニンの製造方法およびこれを原料としたオキシチタニウムフタロシアニンならびにそれを用いた電子写真感光体 | |
| JPH0321669A (ja) | β型チタニルフタロシアニンの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): JP KR US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2004511398 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 2006020129 Country of ref document: US Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10516884 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003730862 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020047020043 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020047020043 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003730862 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10516884 Country of ref document: US |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2003730862 Country of ref document: EP |