RU2379289C2 - Соединение индолина и способ его получения - Google Patents
Соединение индолина и способ его получения Download PDFInfo
- Publication number
- RU2379289C2 RU2379289C2 RU2007115888/04A RU2007115888A RU2379289C2 RU 2379289 C2 RU2379289 C2 RU 2379289C2 RU 2007115888/04 A RU2007115888/04 A RU 2007115888/04A RU 2007115888 A RU2007115888 A RU 2007115888A RU 2379289 C2 RU2379289 C2 RU 2379289C2
- Authority
- RU
- Russia
- Prior art keywords
- propyl
- dihydro
- ethyl
- trifluoroethoxy
- phenoxy
- Prior art date
Links
- 0 C[C@](Cc1cc(C#N)c2N(CCCO)CCc2c1)NCCOc1c(*)cccc1 Chemical compound C[C@](Cc1cc(C#N)c2N(CCCO)CCc2c1)NCCOc1c(*)cccc1 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
- C07D209/42—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/08—Indoles; Hydrogenated indoles with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to carbon atoms of the hetero ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
Abstract
Изобретение относится к улучшенному способу получения силодозина формулы ! ! используемого при расстройстве мочеиспускания. Способ заключается в смешении нового соединения 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино]пропил]-2,3-дигидро-1Н-индол-1-ил}пропилбензоата с щавелевой кислотой, с получением соответствующего оксалата, с последующим его гидролизом, приводящим к образованию нового соединения 1-(3-гидроксипропил)-5-[(2R-)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино]пропил]-2,3-дигидро-1H-индол-7-карбонитрила, и его гидролизом с получением целевого соединения. Описаны способы получения новых промежуточных соединений. 8 н. и 4 з.п. ф-лы.
Description
Область техники
Настоящее изобретение относится к способу получения соединения индолина, пригодного в качестве лекарственного средства, а также получению его промежуточных соединений. В особенности настоящее изобретение относится к способу получения соединения индолина (общее название: силодозин), представленного следующей структурной формулой:
[Соед.1]

которое полезно в качестве терапевтического агента при расстройстве мочеиспускания, связанного с доброкачественной гиперплазией простаты, а также к получению промежуточных соединений для использования в его производстве.
Уровень техники
Силодозин обладает селективным ингибирующим влиянием против сокращения гладкой мускулатуры уретры и снижает внутреннее давление уретры без большого влияния на кровяное давление. Более того, силодозин селективно влияет на α1A подтип адренорецептора и чрезвычайно полезен в качестве терапевтического агента при расстройстве мочеиспускания, связанного с доброкачественной гиперплазией простаты и тому подобного (см. патентные ссылки 1 и 2).
В качестве эффективного и рационального способа получения силодозина предлагали или сообщали о том, что оптически активное соединение амина, представляемое следующей общей формулой:
[Соед. 2]

в которой R1 представляет собой атом водорода или защитную группу для гидроксила, вводили в реакцию с соединением феноксиэтана, представленного следующей общей формулой:
[Соед.3]

в которой X представляет собой уходящую группу, и необязательно снимали защиту, и цианогруппу превращали в карбамоильную группу (см. патентные ссылки 3 и 4).
Однако в вышеупомянутых способах получения иногда из-за реакции одной молекулы оптически активного соединения амина и двух молекул соединения феноксиэтана в качестве побочного продукта образовывалось диалкильное соединение (C), представленное следующей общей формулой:
[Соед.4]

в которой R1 представляет собой атом водорода или защитную группу для гидроксила. Поскольку представляется сложным удалить побочный продукт методом очистки, используемым в обычном промышленном производстве, таким как перекристаллизация и тому подобное, то для удаления побочного продукта требуется использовать такой метод очистки, как колоночная хроматография или тому подобное. Таким образом, усложняясь, процесс очистки перестает удовлетворять способу промышленного производства. Таким образом, требуется разработка способа очистки, более подходящего для промышленного производства.
Патентная ссылка 1: Японская патентная публикация H6-220015;
Патентная ссылка 2: Японская патентная публикация 2000-247998;
Патентная ссылка 3: Японская патентная публикация 2001-199956;
Патентная ссылка 4: Японская патентная публикация 2002-265444.
Раскрытие изобретения
Задачи, которые должно решить изобретение
Цель настоящего изобретения заключается в обеспечении способа промышленного получения силодозина.
Способы решения задачи
Для достижения вышеупомянутой цели изобретатели настоятельно изучили и обнаружили, что путем превращения 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоата, представленного следующей структурной формулой:
[Соед.5]

в оксалат и его выделения при помощи перекристаллизации может быть удален побочный продукт (C-a), представленный формулой
[Соед.6]

что формирует таким образом основу настоящего изобретения.
То есть, настоящее изобретение относится к способу получения 1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-7-карбоксамида, представленного структурной формулой (3):
[Соед.9]

который включает смешивание 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоата, представленного следующей формулой (1):
[Соед.7]
с щавелевой кислотой с получением 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоат монооксалата, последующий гидролиз оксалата с получением 1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]-этил}амино)пропил]-2,3-дигидро-1H-индол-7-карбонитрила, представленного структурной формулой (2):
[Соед.8]

и дальнейший гидролиз соединения, представленного общей формулой (2), а также получение промежуточных соединений, используемых в способе получения.
Эффект от изобретения
3-{7-Циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)-фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоат монооксалат, получаемый в качестве промежуточного соединения согласно способу получения настоящего изобретения, хорошо кристаллизуется, легко отделим от побочного продукта (C-a) и прост в обращении. Таким образом, данный оксалат становится отличнейшим промежуточным продуктом в способе промышленного производства.
Наилучший вариант воплощения изобретения
Способ получения согласно настоящему изобретению включает 4 стадии, как объясняется ниже.
(Стадия 1)
Получение 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоата
3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоат, используемый в способе получения согласно настоящему изобретению, может быть получен способом, аналогичным тому, который описан в Патентной ссылке 3, по реакции 3-{7-циано-5-[(2R)-2-аминопропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоата, представленного структурной формулой(A):
[Соед.10]

или его соли с соединением феноксиэтана, представленным общей формулой (B):
[Соед.11]

в которой X представляет собой уходящую группу,
в органическом растворителе и предпочтительно в присутствии основания.
В качестве уходящей группы Х в общей формуле (B) могут быть приведены примеры атомов хлора, брома и йода, низшей алкилсульфонилоксигруппы, такой как метансульфонилоксигруппа и ей подобные, арилсульфонилоксигруппы, такой как бензолсульфонилоксигруппа или толуолсульфонилоксигруппа и им подобные. Среди них низшая алкилсульфонилоксигруппа является предпочтительной.
В качестве органического растворителя, используемого как растворитель для реакции, может быть использован любой органический растворитель, если только он не ингибирует реакцию. В качестве примера можно привести низший спирт, такой как метанол, этанол, пропанол, изопропиловый спирт, трет-бутанол и им подобные; апротонный полярный растворитель, такой как диметилформамид, диметилсульфоксид, ацетонитрил и им подобные, или смеси растворителей, выбираемых из них же. Среди них низший спирт является предпочтительным, в особенности трет-бутанол является наиболее предпочтительным.
В качестве основания может быть приведен пример неорганического основания, такого как гидроксид щелочного металла, такой как гидроксид натрия, гидроксид калия и им подобные, и карбонат щелочного металла, такой как карбонат натрия, карбонат калия, карбонат цезия и им подобные, а также органическое основание, такое как амин низшего алкила, такой как триэтиламин, диизопропиламин и им подобные. Среди них неорганическое основание, особенно карбонат щелочного металла, является предпочтительным, и карбонат натрия является особенно предпочтительным.
Реакцию обычно проводят при температуре от комнатной до точки кипения органического растворителя, используемого в реакции в течение времени от 30 минут до 48 часов.
После реакции 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2- трифторэтокси)фенокси]этил}амино)пропил-2,3-дигидро-1H-индол-1-ил}
пропилбензоат может быть получен с использованием обычного метода. Вышеупомянутый побочный продукт (C-a) обычно входит в состав продукта в количестве приблизительно от 5 до 20%, хотя его количество отличается в зависимости от условий реакции. Количество содержащегося побочного продукта может быть вычислено по отношению площади, измеренной методом высокоэффективной жидкостной хроматографии в следующих условиях.
Условия измерения
Колонка: Inertsil ODS-2
Длина волны: 254 нм
Подвижная фаза: метанол:0,01 моль/л фосфатный буфер (pH 7,6) = 7:3
(Стадия 2)
Получение 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино]пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоат монооксалата.
Кристаллический 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2- трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоат монооксалат может быть выделен путем растворения почти эквимолярных количеств 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоата и щавелевой кислоты в подходящем растворителе и, необязательно, при нагревании раствора до образования 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоат монооксалата и его кристаллизации. В качестве растворителя могут быть приведены примеры низшего спирта, такого как метанол, этанол, пропанол, изопропиловый спирт и им подобные, или вышеупомянутого спирта, содержащего воду, смесь растворителей, выбираемых из них же, и тому подобное. Среди них низший спирт является предпочтительным, особенно предпочтительным является этанол, изопропиловый спирт и смешанный растворитель, состоящий из воды и изопропилового спирта.
Хотя это может зависеть от растворителя, предпочтительное количество щавелевой кислоты, которое следует использовать, представляет собой обычно от 0,7 до 1,5 эквивалентов по отношению к 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоату.
Кристаллический оксалат может быть выкристаллизован, если оставить стоять вышеупомянутый раствор оксалата. В то же время могут быть использованы необязательные затравочные кристаллы оксалата или охлаждение. Более того, оксалат также может быть выкристаллизован при концентрировании раствора оксалата или при добавлении к раствору оксалата плохого растворителя.
Путем вышеупомянутого способа количество содержащегося побочного продукта (C-a) может быть понижено до 1% или меньше при помощи 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоат монооксалата. Таким образом, полученный оксалат может быть непосредственно использован в следующей реакции.
(Стадия 3)
Получение 1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-7-карбонитрила
1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-7-карбонитрил может быть получен путем гидролиза 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоат монооксалата в подходящем растворителе.
Реакция гидролиза может быть проведена с использованием щелочи, такой как гидроксид щелочного металла, такого как гидроксид натрия, гидроксид калия или им подобные; карбоната щелочного металла, такого как карбонат натрия, карбонат калия, карбонат цезия и им подобные, или используя кислоту, такую как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота или им подобные. Среди них щелочь является предпочтительной, особенно предпочтительным является гидроксид щелочного металла.
В качестве растворителя, используемого для гидролиза, можно привести примеры воды, низшего спирта, такого как метанол, этанол, пропанол, изопропиловый спирт и им подобные; водорастворимые органические растворители, такие как ацетон, тетрагидрофуран, диоксан и им подобные, а также смеси растворителей, выбираемых из них же. Среди них смешанный растворитель из воды и низшего спирта является предпочтительным.
Реакция гидролиза обычно может быть проведена при температуре от 0°С до точки кипения используемого растворителя в течение времени от 30 минут до 48 часов, и тогда 1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-7-карбонитрил может быть получен с использованием обычного метода. Полученное соединение может быть использовано для следующей реакции непосредственно или необязательно после дополнительной очистки.
(Стадия 4)
Получение 1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-7-карбоксамида
1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-7-карбоксамид может быть получен при гидролизе 1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-7-карбонитрила в подходящем растворителе.
Реакция гидролиза может быть проведена, используя щелочь, такую как гидроксид щелочного металла, такой как гидроксид натрия, гидроксид калия или им подобные; карбонат щелочного металла, такой как карбонат натрия, карбонат калия, карбонат цезия или им подобные, или используя кислоту, такую как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота или им подобные. Среди них щелочь является предпочтительной, особенно предпочтительным является гидроксид щелочного металла. В дополнение, предпочтительным является, чтобы реакцию гидролиза проводили в присутствии окисляющего агента, такого как пероксид водорода или ему подобного.
В качестве растворителя, используемого для гидролиза, можно привести примеры воды, низшего спирта, такого как метанол, этанол, пропанол, изопропиловый спирт и им подобные; водорастворимые органические растворители, такие как ацетон, тетрагидрофуран, диоксан, диметилсульфоксид и им подобные, а также смеси растворителей, выбираемых из тех же самых и им подобных. Среди них смешанный растворитель из воды и диметилсульфоксида является предпочтительным.
Реакция гидролиза может быть проведена при температуре от 0°С до 100°С в течение времени от 30 минут до 48 часов, и тогда 1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-7-карбоксамид может быть получен с использованием обычного метода.
ПРИМЕРЫ
Настоящее изобретение далее иллюстрируется более детально при помощи следующих примеров, однако изобретение этим не ограничивается.
Пример 1
3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил)-2,3-дигидро-1H-индол-1-ил)пропилбензоат
К смеси этилацетата (50 мл) и водного раствора (50 мл) карбоната калия (13,5 г) понемногу добавляли 3-[5-((2R)-2-аминопропил)-7-циано-2,3-дигидро-1H-индол-1-ил]пропилбензоат(2R,3R)монотартрат (5,0 г) и смесь перемешивали при комнатной температуре 2 часа. Слой этилацетата отделяли и водный слой экстрагировали раствором этилацетата (50 мл). Объединенный слой этилацетата промывали водным раствором карбоната калия и сушили над безводным сульфатом натрия. Фильтрат концентрировали при пониженном давлении. Полученное масло растворяли в безводном трет-бутаноле (25 мл) и к раствору добавляли 2-[2-(2,2,2-трифторэтокси)фенокси]этилметансульфонат (3,67 г) и карбонат натрия (1,08 г). Смесь кипятили с обратным холодильником в течение 24 часов. После этого реакционной смеси давали остыть и затем добавляли водный раствор бикарбоната натрия (50 мл). Смесь дважды экстрагировали этилацетатом (50 мл). Объединенный слой этилацетата промывали водным раствором бикарбоната натрия, водой и солевым раствором и сушили над безводным сульфатом натрия. Фильтрат концентрировали при пониженном давлении, получая 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоат (6,40 г). На данном этапе содержание побочного продукта (C-a) в полученном продукте составляло 13,6%. Продукт был использован для следующей реакции. Структура полученного 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоата была подтверждена ЯМР-анализом при использовании небольшого количества очищенного продукта.
1H-ЯМР (CDCl3) δppm: 1,06 (3H, д, J=6,4 Гц), 2,15 (2H, м), 2,44 (1H, дд, J=6,9, 13,8 Гц), 2,61 (1H, дд, J=6,3, 13,8 Гц), 2,85-3,10 (5H, м), 3,57 (2H, т, J=8,6 Гц), 3,74 (2H, т, J=7,2 Гц), 4,05-4,15 (2H, м), 4,32 (2H, кв., J=8,4 Гц), 4,47 (2H, т, J=6,4 Гц), 6,89-7,06 (6H, м), 7,44 (2H, т, J=7,8 Гц), 7,55 (1H, т, J=7,5 Гц), 8,06 (2H, д, J=8,4 Гц).
Пример 2
3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоат монооксалат
Изопропиловый спирт (50 мл) и дигидрат щавелевой кислоты (1,20 г) добавляли к 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоату (6,40 г), который был получен в примере 1, и смесь растворяли при нагревании. После введения затравки целевого соединения смесь оставляли на ночь. Осажденные кристаллы отделяли фильтрованием и промывали небольшим количеством охлажденного изопропилового спирта и сушили под вакуумом до получения 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоат монооксалата (5,43 г). На данном этапе содержание побочного продукта (C-a) в получаемом продукте составляло 0,9%.
1H-ЯМР (DMSO-d6) δppm: 1,13 (3H, д, J=6,2 Гц), 2,08 (2H, м), 2,45-2,57 (1H, м), 2,88-3,05 (3H, м), 3,35-3,50 (3H, м), 3,60 (1H, т, J=8,6 Гц), 3,70 (2H, т, J=7,1 Гц), 4,29 (2H, шир.с), 4,39 (2H, т, J=6,1 Гц), 4,71 (2H, кв., J=8,9 Гц), 6,95-7,16 (6H, м), 7,51 (2H, т, J=7,7 Гц), 7,65 (1H, т, J=7,4 Гц), 7,99 (2H, д, J=7,4 Гц).
Пример 3
1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-7-карбонитрил
3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-1-ил}пропилбензоат монооксалат (10,0 г) растворяли в метаноле (40 мл), затем понемногу добавляли водный раствор гидроксида калия, приготовленный из гидроксида калия (2,93 г) и воды (10 мл), и смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли воду (150 мл) и последовательно экстрагировали этилацетатом (150 мл и 50 мл). Объединенный слой этилацетата промывали насыщенным водным раствором бикарбоната натрия и солевым раствором и сушили над безводным сульфатом натрия. Фильтрат концентрировали при пониженном давлении, получая 1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-7-карбонитрил (7,86 г).
1H-ЯМР (CDCl3) δppm: 1,05 (3H, д, J=6,1 Гц), 1,85-1,95 (2H, м), 2,43 (1H, дд, J=13,5, 6,8 Гц), 2,60 (1H, дд, J=13,7, 6,3 Гц), 2,80-3,10 (5H, м), 3,57 (2H, т, J=8,8 Гц), 3,67 (2H, т, J=7,2 Гц), 3,80 (2H, т, J=6,0 Гц), 4,05-4,15 (2H, м), 4,32 (2H, кв., J=8,4 Гц), 6,85-7,05 (5H, м).
Пример 4
1-(3-Гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-7-карбоксамид
1-(3-Гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-7-карбонитрил (6,00 г) растворяли в диметилсульфоксиде (75 мл) и к этому раствору добавляли 5 моль/л водный раствор гидроксида натрия (4,50 мл). К реакционной смеси понемногу добавляли 30% пероксид водорода (2,63 мл) при температуре не выше 25°C. Реакционную смесь перемешивали при температуре от 20 до 25°C в течение 5 часов. К реакционной смеси осторожно добавляли водный раствор сульфита натрия, приготовленный из сульфита натрия (2,1 г), растворенного в воде (150 мл). Реакционную смесь дважды экстрагировали этилацетатом (50 мл). Объединенный слой этилацетата дважды экстрагировали 2 моль/л соляной кислоты. Экстрагированный водный раствор соляной кислоты нейтрализовали бикарбонатом натрия и дважды экстрагировали этилацетатом (50 мл). Объединенный слой этилацетата промывали насыщенным водным раствором бикарбоната натрия и солевым раствором и сушили над безводным сульфатом натрия. Фильтрат концентрировали при пониженном давлении и остаток растворяли в этилацетате. Раствор охлаждали до получения 1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1H-индол-7-карбоксамида (4,49 г).
1H-ЯМР (CDCl3) δppm: 1,08 (3H, д, J=6,2 Гц), 1,75-1,85 (2H, м), 2,53 (1H, дд, J=13,6, 6,7 Гц), 2,68 (1H, дд, J=13,6, 6,6 Гц), 2,90-3,10 (5H, м), 3,19 (2H, т, J=6,7 Гц), 3,41 (2H, т, J=8,5 Гц), 3,75 (2H, т, J=5,6 Гц), 4,05-4,15 (2H, м), 4,30 (2H, кв., J=8,4), 5,79 (1H, шир.с), 6,65 (1H, шир.с), 6,85-7,05 (5H, м), 7,16 (1H, с).
Claims (12)
1. Способ получения 1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-7-карбоксамида, представленного структурной формулой (3):
[Соединение 3]

включающий смешивание 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-1-ил}пропилбензоата, представленного структурной формулой (1):
[Соединение 1]
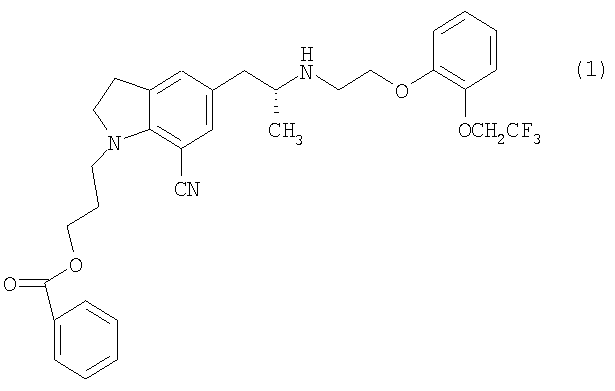
с щавелевой кислотой с образованием 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-1-ил}пропилбензоата монооксалата с последующим гидролизом оксалата с образованием 1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-7-карбонитрила, представленного структурной формулой (2):
[Соединение 2]

и гидролиз соединения, представленного структурной формулой (2).
[Соединение 3]
включающий смешивание 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-1-ил}пропилбензоата, представленного структурной формулой (1):
[Соединение 1]
с щавелевой кислотой с образованием 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-1-ил}пропилбензоата монооксалата с последующим гидролизом оксалата с образованием 1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-7-карбонитрила, представленного структурной формулой (2):
[Соединение 2]
и гидролиз соединения, представленного структурной формулой (2).
2. Способ получения по п.1, включающий выделение 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-1-ил}пропилбензоата монооксалата.
3. Способ получения по п.1 или 2, включающий гидролиз 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-1-ил}пропилбензоата монооксалата с гидроксидом щелочного металла.
4. Способ получения по п.1, в котором 1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-7-карбонитрил гидролизуют в присутствии окисляющего агента.
5. Способ получения по п.4, в котором окисляющим агентом является пероксид водорода.
6. Способ получения 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-1-ил}пропилбензоата, представленного структурной формулой (1):
[Соединение 6]
включающий взаимодействие 3-{7-циано-5-[(2R)-2-аминопропил]-2,3-дигидро-1Н-индол-1-ил}пропилбензоата, представленного структурной формулой (А):
[Соединение 4]

с соединением феноксиэтана, представленным общей формулой (В):
[Соединение 5]

где X представляет собой уходящую группу.
[Соединение 6]
включающий взаимодействие 3-{7-циано-5-[(2R)-2-аминопропил]-2,3-дигидро-1Н-индол-1-ил}пропилбензоата, представленного структурной формулой (А):
[Соединение 4]
с соединением феноксиэтана, представленным общей формулой (В):
[Соединение 5]
где X представляет собой уходящую группу.
7. Способ получения 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-1-ил}пропилбензоата монооксалата, включающий смешивание 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-1-ил}пропилбензоата и щавелевой кислоты.
8. Способ получения 1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-7-карбонитрила, представленного структурной формулой (2):
[Соединение 7]
включающий гидролиз 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-1-ил}пропилбензоата монооксалата.
[Соединение 7]
включающий гидролиз 3-{7-циано-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-1-ил}пропилбензоата монооксалата.
9. Способ получения 1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-7-карбоксамида, представленного структурной формулой (3):
[Соединение 8]
включающий гидролиз 1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-7-карбонитрила.
[Соединение 8]
включающий гидролиз 1-(3-гидроксипропил)-5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-7-карбонитрила.
10. 3-{7-Циано-
5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-1-ил)пропилбензоат.
5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-1-ил)пропилбензоат.
11. 3-{7-Циано-
5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-1-ил}пропилбензоат монооксалат.
5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-1-ил}пропилбензоат монооксалат.
12. 1-(3-Гидроксипропил)-
5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-7-карбонитрил.
5-[(2R)-2-({2-[2-(2,2,2-трифторэтокси)фенокси]этил}амино)пропил]-2,3-дигидро-1Н-индол-7-карбонитрил.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004313040 | 2004-10-27 | ||
| JP2004-313040 | 2004-10-27 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2007115888A RU2007115888A (ru) | 2008-11-10 |
| RU2379289C2 true RU2379289C2 (ru) | 2010-01-20 |
Family
ID=36227738
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2007115888/04A RU2379289C2 (ru) | 2004-10-27 | 2005-10-24 | Соединение индолина и способ его получения |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US7834193B2 (ru) |
| EP (1) | EP1806340B1 (ru) |
| JP (1) | JP5049013B2 (ru) |
| KR (1) | KR101249865B1 (ru) |
| CN (1) | CN101048376B (ru) |
| AT (1) | ATE508113T1 (ru) |
| AU (1) | AU2005298078B2 (ru) |
| BR (1) | BRPI0517515B8 (ru) |
| CA (1) | CA2583660C (ru) |
| DE (1) | DE602005027881D1 (ru) |
| ES (1) | ES2362788T3 (ru) |
| HK (1) | HK1113364A1 (ru) |
| IL (1) | IL182642A (ru) |
| MX (1) | MX2007005126A (ru) |
| RU (1) | RU2379289C2 (ru) |
| TW (1) | TW200621709A (ru) |
| WO (1) | WO2006046499A1 (ru) |
| ZA (1) | ZA200703996B (ru) |
Families Citing this family (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| UA78854C2 (en) * | 2002-09-06 | 2007-04-25 | Kissei Pharmaceutical | Crystal for an oral solid drug and oral solid drug for dysuria treatment containing the same |
| US8013007B2 (en) | 2007-02-26 | 2011-09-06 | Concert Pharmaceuticals, Inc. | Alpha 1A-adrenoceptor antagonists |
| MX2009009230A (es) * | 2007-02-28 | 2009-11-26 | Kissei Pharmaceutical | Uso de silodosina en una administracion diaria para tratar hiperplasia prostatica benigna. |
| CN101585798B (zh) * | 2008-05-20 | 2011-06-15 | 浙江华海药业股份有限公司 | 光学活性化合物1-(3-苯甲酰氧基丙基)-5-(2-(1-苯基乙基胺)丙基)-7-氰基吲哚啉及其制备方法和用途 |
| CN101993405B (zh) * | 2009-08-27 | 2014-01-15 | 浙江华海药业股份有限公司 | 吲哚啉衍生物、及其制备方法和用途 |
| CN101993407B (zh) * | 2009-08-27 | 2014-01-29 | 浙江华海药业股份有限公司 | 用于制备西洛多辛的吲哚啉化合物及其制备方法 |
| CN101993406B (zh) * | 2009-08-27 | 2014-01-15 | 浙江华海药业股份有限公司 | 光学活性的吲哚啉化合物及其制备方法 |
| JP5661773B2 (ja) * | 2009-09-12 | 2015-01-28 | サンド・アクチエンゲゼルシヤフト | インドリン誘導体の調製方法およびこれらの中間体 |
| CN101768056B (zh) * | 2010-01-19 | 2013-10-16 | 扬子江药业集团有限公司 | 一种取代的苯基甲基醚的制备方法 |
| WO2011101864A1 (en) | 2010-02-17 | 2011-08-25 | Panacea Biotec Ltd | Novel process for the synthesis of phenoxyethyl derivatives |
| WO2011124704A1 (en) | 2010-04-09 | 2011-10-13 | Ratiopharm Gmbh | Process for preparing an intermediate for silodosin |
| CN102311319A (zh) * | 2010-07-05 | 2012-01-11 | 浙江华海药业股份有限公司 | 一种制备2-(2,2,2-三氟乙基)苯甲醚的方法 |
| WO2012014186A1 (en) * | 2010-07-30 | 2012-02-02 | Ranbaxy Laboratories Limited | Process for the preparation of silodosin and its novel intermediates |
| CN102229558B (zh) * | 2010-08-05 | 2013-05-08 | 南京海纳医药科技有限公司 | 西洛多辛的晶型δ、它的制备方法和包含它的药物组合物 |
| CN102229557B (zh) * | 2010-08-05 | 2013-03-27 | 南京海纳医药科技有限公司 | 西洛多辛的半水合物晶体、制备方法和包含它的药物组合物 |
| CZ303061B6 (cs) * | 2010-11-12 | 2012-03-14 | Zentiva, K.S. | Zpusob výroby (-)-1-(3-hydroxypropyl)-5-[(2R)-2-({2,2,2-trifluorethoxy)fenoxyethyl}amino)propyl]-2,3-dihydro-1H-indol-7-karboxamidu |
| CZ2010836A3 (cs) * | 2010-11-12 | 2012-03-14 | Zentiva, K.S. | Zpusob výroby (-)-1-(3-hydroxypropyl)-5-[(2R)-2-({2,2,2-trifluorethoxy)fenoxyethyl}amino)propyl]-2,3-dihydro-1H-indol-7-karboxamidu |
| WO2012062229A1 (en) | 2010-11-12 | 2012-05-18 | Zentiva, K.S. | A method of manufacturing (-)-l-(3-hydroxypropyl)-5-[(2r)-2-({2,2,2-trifluoroethoxy)- phenoxyethyl}amino)propyl]-2,3-dihydro-lh-indole-7-carboxamide |
| WO2012131710A2 (en) * | 2011-03-30 | 2012-10-04 | Panacea Biotec Ltd | Novel process for the synthesis of indoline derivatives |
| WO2012147019A1 (en) * | 2011-04-26 | 2012-11-01 | Orchid Chemicals And Pharamceuticals Limited | An improved process for the preparation of silodosin |
| WO2012147107A2 (en) * | 2011-04-29 | 2012-11-01 | Msn Laboratories Limited | Novel & improved processes for the preparation of indoline derivatives and its pharmaceutical composition |
| CN102382029B (zh) * | 2011-07-26 | 2016-06-29 | 浙江华海药业股份有限公司 | 一种西洛多辛中间体成盐制备方法 |
| WO2013072935A2 (en) | 2011-10-10 | 2013-05-23 | Cadila Healthcare Limited | Process for the preparation of silodosin |
| IN2014KN01030A (ru) | 2011-10-21 | 2015-10-09 | Sandoz Ag | |
| CN102675182B (zh) * | 2012-05-24 | 2014-03-26 | 临海天宇药业有限公司 | 1-(3-苯甲酰氧基丙基)-5-(2-羰基丙基)-7-腈基吲哚啉的制备方法 |
| WO2014118606A2 (en) * | 2013-01-29 | 2014-08-07 | Alembic Pharmaceuticals Limited | A novel process for the preparation of silodosin |
| CN103159664B (zh) * | 2013-03-29 | 2016-03-23 | 深圳市海滨制药有限公司 | 一种赛洛多辛原料药及其制备方法、药物组合物 |
| CN104140389A (zh) * | 2013-05-06 | 2014-11-12 | 昆明积大制药股份有限公司 | 一种赛洛多辛及其中间体的制备方法 |
| CN103420893B (zh) * | 2013-08-02 | 2015-11-25 | 江苏和成新材料有限公司 | 制备西洛多辛中间体的方法 |
| WO2015119057A1 (ja) * | 2014-02-06 | 2015-08-13 | 宇部興産株式会社 | インドリン化合物の製造方法 |
| KR20150098565A (ko) * | 2014-02-20 | 2015-08-28 | 한미정밀화학주식회사 | 실로도신 제조에 사용되는 신규 중간체, 이의 제조방법 및 이를 이용한 실로도신의 제조방법 |
| KR20150127996A (ko) * | 2014-05-08 | 2015-11-18 | 한미정밀화학주식회사 | 실로도신 감마형 결정의 제조방법 |
| JP6242298B2 (ja) * | 2014-06-13 | 2017-12-06 | 株式会社トクヤマ | 1−(3−ベンゾイルオキシプロピル)−7−シアノ−5−[(2r)−2−({2−[2−(2,2,2−トリフルオロエトキシ)フェノキシ]エチル}アミノ)プロピル]インドリンまたはその塩を製造する方法 |
| KR101628946B1 (ko) * | 2014-07-09 | 2016-06-09 | 동방에프티엘(주) | 실로도신의 개선된 제조방법 |
| KR101725393B1 (ko) | 2014-07-24 | 2017-04-27 | 주식회사 경보제약 | 실로도신의 제조 방법 및 신규 중간체 |
| WO2016042441A1 (en) | 2014-09-18 | 2016-03-24 | Mankind Research Centre | A novel process for the preparation of considerably pure silodosin |
| JP2016088847A (ja) * | 2014-10-30 | 2016-05-23 | 株式会社トクヤマ | (‐)‐1‐(3‐ヒドロキシプロピル)‐5‐[[(2r)‐2‐({2‐[2‐(2,2,2‐トリフルオロエトキシ)フェノキシ]エチル}アミノ)プロピル]‐2,3‐ジヒドロ‐1h‐インドール‐7‐カルボキサミド])の製造方法 |
| CN104478784B (zh) * | 2014-11-17 | 2017-02-22 | 武汉工程大学 | 一种西洛多辛草酸盐的晶型及其制备方法 |
| KR101694262B1 (ko) * | 2014-12-18 | 2017-01-10 | (주)다산메디켐 | 실로도신의 결정형의 제조방법 |
| US9356405B1 (en) | 2015-03-05 | 2016-05-31 | Sony Corporation | Connector tongue element for an electrical connector plug receptacle and a method for producing the same |
| RU2750898C2 (ru) | 2015-09-23 | 2021-07-05 | Биокон Лимитед | Кристаллические формы промежуточного соединения позаконазола и способ получения аморфного позаконазола |
| JP2018528233A (ja) * | 2015-09-23 | 2018-09-27 | バイオコン・リミテッドBiocon Limited | インドリン化合物および新規インドリン塩を製造するプロセス |
| ES2607639B1 (es) | 2015-09-30 | 2018-02-28 | Urquima, S.A | Sal de ácido maleico de un intermedio de silodosina |
| CN106995399A (zh) * | 2016-01-25 | 2017-08-01 | 北京天泰恒华医药技术有限公司 | 一种制备赛洛多辛的方法 |
| KR102365411B1 (ko) * | 2017-08-08 | 2022-02-21 | (주)헥사파마텍 | 신규의 설폰아마이드 중간체 및 이를 사용한 실로도신의 제조방법 |
| KR102163068B1 (ko) * | 2018-07-04 | 2020-10-07 | 주식회사 가피바이오 | 실로도신 합성용 중간체의 제조 방법 및 이를 이용한 실로도신의 제조 방법 |
| CN111217735B (zh) * | 2018-11-27 | 2023-03-14 | 上海汇伦医药股份有限公司 | 赛洛多辛中间体的制备方法 |
| WO2020237645A1 (zh) * | 2019-05-31 | 2020-12-03 | 上海汇伦生命科技有限公司 | 赛洛多辛中间体的制备方法 |
| CN110452149B (zh) * | 2019-06-13 | 2021-11-02 | 北京鑫开元医药科技有限公司 | 二氢吲哚化合物的制备方法、二氢吲哚化合物及用途 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0600675B1 (en) * | 1992-12-02 | 1998-07-08 | Kissei Pharmaceutical Co., Ltd. | Indoline compounds for the treatment of dysuria |
| JP3331047B2 (ja) * | 1994-06-01 | 2002-10-07 | キッセイ薬品工業株式会社 | インドリン誘導体 |
| JP2526811B2 (ja) * | 1994-07-22 | 1996-08-21 | 田辺製薬株式会社 | フェネチルアミン誘導体及びその製法 |
| ES2220043T3 (es) * | 1998-02-27 | 2004-12-01 | Kissei Pharmaceutical Co., Ltd. | Derivados del indol y composiciones medicinales que los contienen. |
| JP4634560B2 (ja) * | 2000-01-14 | 2011-02-16 | キッセイ薬品工業株式会社 | 光学活性なインドリン誘導体の製造方法およびその製造中間体 |
| JP4921646B2 (ja) * | 2001-03-08 | 2012-04-25 | キッセイ薬品工業株式会社 | 1−(3−ベンジルオキシプロピル)−5−(2−置換プロピル)インドリン誘導体およびその使用方法 |
| JP2006188470A (ja) * | 2005-01-07 | 2006-07-20 | Kissei Pharmaceut Co Ltd | インドリン誘導体およびその製造方法 |
-
2005
- 2005-10-24 CN CN2005800370402A patent/CN101048376B/zh active Active
- 2005-10-24 JP JP2006543120A patent/JP5049013B2/ja active Active
- 2005-10-24 AU AU2005298078A patent/AU2005298078B2/en not_active Ceased
- 2005-10-24 EP EP05795703A patent/EP1806340B1/en not_active Not-in-force
- 2005-10-24 RU RU2007115888/04A patent/RU2379289C2/ru active
- 2005-10-24 MX MX2007005126A patent/MX2007005126A/es active IP Right Grant
- 2005-10-24 KR KR1020077011689A patent/KR101249865B1/ko active IP Right Grant
- 2005-10-24 ES ES05795703T patent/ES2362788T3/es active Active
- 2005-10-24 AT AT05795703T patent/ATE508113T1/de not_active IP Right Cessation
- 2005-10-24 WO PCT/JP2005/019478 patent/WO2006046499A1/ja active Application Filing
- 2005-10-24 DE DE602005027881T patent/DE602005027881D1/de active Active
- 2005-10-24 CA CA2583660A patent/CA2583660C/en active Active
- 2005-10-24 ZA ZA200703996A patent/ZA200703996B/xx unknown
- 2005-10-24 BR BRPI0517515A patent/BRPI0517515B8/pt active IP Right Grant
- 2005-10-26 TW TW094137403A patent/TW200621709A/zh unknown
-
2007
- 2007-04-16 US US11/787,312 patent/US7834193B2/en active Active
- 2007-04-18 IL IL182642A patent/IL182642A/en active IP Right Grant
-
2008
- 2008-03-28 HK HK08103492.9A patent/HK1113364A1/xx unknown
Also Published As
| Publication number | Publication date |
|---|---|
| IL182642A (en) | 2012-10-31 |
| JPWO2006046499A1 (ja) | 2008-05-22 |
| ZA200703996B (en) | 2008-09-25 |
| RU2007115888A (ru) | 2008-11-10 |
| CA2583660A1 (en) | 2006-05-04 |
| KR20070084499A (ko) | 2007-08-24 |
| EP1806340B1 (en) | 2011-05-04 |
| CN101048376B (zh) | 2011-03-23 |
| EP1806340A4 (en) | 2009-08-12 |
| BRPI0517515B8 (pt) | 2021-05-25 |
| MX2007005126A (es) | 2007-07-04 |
| AU2005298078A1 (en) | 2006-05-04 |
| ES2362788T3 (es) | 2011-07-13 |
| TWI356819B (ru) | 2012-01-21 |
| AU2005298078B2 (en) | 2011-03-31 |
| EP1806340A1 (en) | 2007-07-11 |
| CA2583660C (en) | 2013-02-12 |
| US20070197627A1 (en) | 2007-08-23 |
| CN101048376A (zh) | 2007-10-03 |
| WO2006046499A1 (ja) | 2006-05-04 |
| US7834193B2 (en) | 2010-11-16 |
| BRPI0517515A (pt) | 2008-10-14 |
| BRPI0517515B1 (pt) | 2021-03-09 |
| ATE508113T1 (de) | 2011-05-15 |
| JP5049013B2 (ja) | 2012-10-17 |
| IL182642A0 (en) | 2007-07-24 |
| KR101249865B1 (ko) | 2013-04-02 |
| HK1113364A1 (en) | 2008-10-03 |
| TW200621709A (en) | 2006-07-01 |
| DE602005027881D1 (de) | 2011-06-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2379289C2 (ru) | Соединение индолина и способ его получения | |
| KR102187608B1 (ko) | Ssao 억제제 | |
| EP2970123B1 (en) | Salt of omecamtiv mecarbil and process for preparing salt | |
| JP2013532164A (ja) | トロンビン特異的インヒビターを調製する方法 | |
| MX2010010157A (es) | Moduladores del receptor de prostaciclina (pg12) utiles para el tratamiento de trastornos relacionados con este. | |
| JP6832946B2 (ja) | キナーゼ阻害剤およびその中間体の調製方法 | |
| JP2019523273A (ja) | ベリノスタットの多形形態、およびその調製のためのプロセス | |
| TW200831090A (en) | Factor Xa inhibitor | |
| CA2627724A1 (en) | Process for the preparation of betamimetic benzoxazinone derivatives | |
| CA2538315C (en) | Matrix metalloproteinase inhibitors | |
| US20180339964A1 (en) | The process of preparing indoline compounds and a novel indolinesalt | |
| TR201802657T4 (tr) | 3-sübstitüe (indol-1-il)-asetik asit esterlerinin hazırlanmasına yönelik proses. | |
| JP2007522181A (ja) | シクロオキシゲナーゼ−1及びシクロオキシゲナーゼ−2阻害剤としての置換アゼチジン化合物、ならびにそれらの調製、および薬剤としての使用 | |
| JP5305593B2 (ja) | 高化学的r−5−(2−(2−エトキシフェノキシエチルアミノ)プロピル)−2−メトキシベンゼンスルホンアミド塩酸塩の調製 | |
| JP4323718B2 (ja) | 置換アルキルアミン又はその塩の製造方法 | |
| DK170095B1 (da) | Fremgangsmåde til fremstilling af octahydroindolderivater samt mellemprodukter til brug derved | |
| WO2019163731A1 (ja) | オキサゾリジノン化合物の製造方法 | |
| US9169236B2 (en) | Efficient process for the preparation of lapatinib and salts thereof by means of new intermediates | |
| JP4501015B2 (ja) | アミノピリミジン化合物の製造方法 | |
| KR100612779B1 (ko) | 키랄 글리시딜프탈이미드를 고광학순도로 제조하는 방법 | |
| WO2012026529A1 (ja) | イソキノリン誘導体又はその塩の新規製造方法 | |
| JP4886948B2 (ja) | ビフェニルエチルアミン誘導体およびその製造方法 | |
| WO2004065368A1 (ja) | トランドラプリル合成中間体の製造方法 | |
| JPWO2003050077A1 (ja) | 置換フェニルプロピオン酸誘導体の新規な安定結晶とその製法 | |
| JP2002047269A (ja) | ヘテロ環化合物の製造方法およびその中間体 |