ES2944547T3 - Inhibidores de KRas G12C - Google Patents
Inhibidores de KRas G12C Download PDFInfo
- Publication number
- ES2944547T3 ES2944547T3 ES18879484T ES18879484T ES2944547T3 ES 2944547 T3 ES2944547 T3 ES 2944547T3 ES 18879484 T ES18879484 T ES 18879484T ES 18879484 T ES18879484 T ES 18879484T ES 2944547 T3 ES2944547 T3 ES 2944547T3
- Authority
- ES
- Spain
- Prior art keywords
- mmol
- compound
- carboxylate
- mixture
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 102200006538 rs121913530 Human genes 0.000 title claims abstract description 66
- 239000003112 inhibitor Substances 0.000 title description 9
- 238000000034 method Methods 0.000 claims abstract description 136
- 150000001875 compounds Chemical class 0.000 claims abstract description 135
- 230000000694 effects Effects 0.000 claims abstract description 23
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 18
- -1 wherein each of Z Chemical group 0.000 claims description 194
- 125000003118 aryl group Chemical group 0.000 claims description 70
- 125000001072 heteroaryl group Chemical group 0.000 claims description 66
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 63
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 62
- 206010028980 Neoplasm Diseases 0.000 claims description 60
- 229910052739 hydrogen Inorganic materials 0.000 claims description 54
- 229910052736 halogen Inorganic materials 0.000 claims description 53
- 150000002367 halogens Chemical class 0.000 claims description 53
- 239000001257 hydrogen Substances 0.000 claims description 53
- 125000000623 heterocyclic group Chemical group 0.000 claims description 44
- 125000001188 haloalkyl group Chemical group 0.000 claims description 41
- 201000011510 cancer Diseases 0.000 claims description 35
- 208000009956 adenocarcinoma Diseases 0.000 claims description 32
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 32
- 150000003839 salts Chemical class 0.000 claims description 32
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 31
- 210000004027 cell Anatomy 0.000 claims description 30
- 206010025323 Lymphomas Diseases 0.000 claims description 28
- 125000001624 naphthyl group Chemical group 0.000 claims description 27
- 125000004415 heterocyclylalkyl group Chemical group 0.000 claims description 26
- 125000003545 alkoxy group Chemical group 0.000 claims description 25
- 206010039491 Sarcoma Diseases 0.000 claims description 23
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 22
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 22
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 22
- 125000000217 alkyl group Chemical group 0.000 claims description 21
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 20
- 206010041823 squamous cell carcinoma Diseases 0.000 claims description 20
- 201000009030 Carcinoma Diseases 0.000 claims description 18
- 125000004043 oxo group Chemical group O=* 0.000 claims description 17
- 125000004193 piperazinyl group Chemical group 0.000 claims description 17
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 17
- 206010024612 Lipoma Diseases 0.000 claims description 16
- 125000004404 heteroalkyl group Chemical group 0.000 claims description 16
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 16
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 15
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 15
- 201000008808 Fibrosarcoma Diseases 0.000 claims description 12
- 206010043276 Teratoma Diseases 0.000 claims description 12
- 206010016629 fibroma Diseases 0.000 claims description 12
- 201000011066 hemangioma Diseases 0.000 claims description 12
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 12
- 125000002252 acyl group Chemical group 0.000 claims description 9
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 9
- 201000003076 Angiosarcoma Diseases 0.000 claims description 8
- 208000032612 Glial tumor Diseases 0.000 claims description 8
- 206010018338 Glioma Diseases 0.000 claims description 8
- 208000002927 Hamartoma Diseases 0.000 claims description 8
- 208000001258 Hemangiosarcoma Diseases 0.000 claims description 8
- 208000007766 Kaposi sarcoma Diseases 0.000 claims description 8
- 208000018142 Leiomyosarcoma Diseases 0.000 claims description 8
- 208000034578 Multiple myelomas Diseases 0.000 claims description 8
- 201000004404 Neurofibroma Diseases 0.000 claims description 8
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 8
- 125000002947 alkylene group Chemical group 0.000 claims description 8
- 208000002458 carcinoid tumor Diseases 0.000 claims description 8
- 208000006990 cholangiocarcinoma Diseases 0.000 claims description 8
- 239000003085 diluting agent Substances 0.000 claims description 8
- 239000003937 drug carrier Substances 0.000 claims description 8
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 8
- 125000004446 heteroarylalkyl group Chemical group 0.000 claims description 8
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 8
- 201000010260 leiomyoma Diseases 0.000 claims description 8
- 201000001441 melanoma Diseases 0.000 claims description 8
- 206010027191 meningioma Diseases 0.000 claims description 8
- 201000008968 osteosarcoma Diseases 0.000 claims description 8
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 claims description 7
- 208000008383 Wilms tumor Diseases 0.000 claims description 7
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 7
- 210000002307 prostate Anatomy 0.000 claims description 7
- 125000002393 azetidinyl group Chemical group 0.000 claims description 6
- 230000002496 gastric effect Effects 0.000 claims description 6
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 6
- 125000006650 (C2-C4) alkynyl group Chemical group 0.000 claims description 5
- 210000004556 brain Anatomy 0.000 claims description 5
- 238000000338 in vitro Methods 0.000 claims description 5
- 230000002401 inhibitory effect Effects 0.000 claims description 5
- 230000005764 inhibitory process Effects 0.000 claims description 5
- 210000004072 lung Anatomy 0.000 claims description 5
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 claims description 4
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 claims description 4
- 206010001233 Adenoma benign Diseases 0.000 claims description 4
- 206010003571 Astrocytoma Diseases 0.000 claims description 4
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 claims description 4
- 206010004146 Basal cell carcinoma Diseases 0.000 claims description 4
- 206010073106 Bone giant cell tumour malignant Diseases 0.000 claims description 4
- 208000009458 Carcinoma in Situ Diseases 0.000 claims description 4
- 201000005262 Chondroma Diseases 0.000 claims description 4
- 208000005243 Chondrosarcoma Diseases 0.000 claims description 4
- 201000009047 Chordoma Diseases 0.000 claims description 4
- 208000006332 Choriocarcinoma Diseases 0.000 claims description 4
- 206010048832 Colon adenoma Diseases 0.000 claims description 4
- 206010010356 Congenital anomaly Diseases 0.000 claims description 4
- 208000007033 Dysgerminoma Diseases 0.000 claims description 4
- 208000000471 Dysplastic Nevus Syndrome Diseases 0.000 claims description 4
- 201000009051 Embryonal Carcinoma Diseases 0.000 claims description 4
- 206010014733 Endometrial cancer Diseases 0.000 claims description 4
- 206010014759 Endometrial neoplasm Diseases 0.000 claims description 4
- 206010014967 Ependymoma Diseases 0.000 claims description 4
- 208000006168 Ewing Sarcoma Diseases 0.000 claims description 4
- 208000007659 Fibroadenoma Diseases 0.000 claims description 4
- 206010053717 Fibrous histiocytoma Diseases 0.000 claims description 4
- 208000000527 Germinoma Diseases 0.000 claims description 4
- 208000007569 Giant Cell Tumors Diseases 0.000 claims description 4
- 201000010915 Glioblastoma multiforme Diseases 0.000 claims description 4
- 201000005409 Gliomatosis cerebri Diseases 0.000 claims description 4
- 206010018404 Glucagonoma Diseases 0.000 claims description 4
- 206010018691 Granuloma Diseases 0.000 claims description 4
- 206010019629 Hepatic adenoma Diseases 0.000 claims description 4
- 208000017604 Hodgkin disease Diseases 0.000 claims description 4
- 208000010747 Hodgkins lymphoma Diseases 0.000 claims description 4
- 208000002260 Keloid Diseases 0.000 claims description 4
- 208000002404 Liver Cell Adenoma Diseases 0.000 claims description 4
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 claims description 4
- 208000006644 Malignant Fibrous Histiocytoma Diseases 0.000 claims description 4
- 208000000172 Medulloblastoma Diseases 0.000 claims description 4
- 206010027406 Mesothelioma Diseases 0.000 claims description 4
- 201000003793 Myelodysplastic syndrome Diseases 0.000 claims description 4
- 208000014767 Myeloproliferative disease Diseases 0.000 claims description 4
- 206010029260 Neuroblastoma Diseases 0.000 claims description 4
- 201000010133 Oligodendroglioma Diseases 0.000 claims description 4
- 208000010191 Osteitis Deformans Diseases 0.000 claims description 4
- 208000000035 Osteochondroma Diseases 0.000 claims description 4
- 208000027067 Paget disease of bone Diseases 0.000 claims description 4
- 208000007641 Pinealoma Diseases 0.000 claims description 4
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 claims description 4
- 201000004681 Psoriasis Diseases 0.000 claims description 4
- 201000000582 Retinoblastoma Diseases 0.000 claims description 4
- 208000005678 Rhabdomyoma Diseases 0.000 claims description 4
- 201000010208 Seminoma Diseases 0.000 claims description 4
- 208000015778 Undifferentiated pleomorphic sarcoma Diseases 0.000 claims description 4
- 208000009311 VIPoma Diseases 0.000 claims description 4
- 206010048214 Xanthoma Diseases 0.000 claims description 4
- 206010048215 Xanthomatosis Diseases 0.000 claims description 4
- 208000002718 adenomatoid tumor Diseases 0.000 claims description 4
- 210000004100 adrenal gland Anatomy 0.000 claims description 4
- 201000007434 ampulla of Vater carcinoma Diseases 0.000 claims description 4
- 208000001119 benign fibrous histiocytoma Diseases 0.000 claims description 4
- 210000003445 biliary tract Anatomy 0.000 claims description 4
- 210000004369 blood Anatomy 0.000 claims description 4
- 239000008280 blood Substances 0.000 claims description 4
- 208000016738 bone Paget disease Diseases 0.000 claims description 4
- 210000000988 bone and bone Anatomy 0.000 claims description 4
- 201000009480 botryoid rhabdomyosarcoma Diseases 0.000 claims description 4
- 201000003149 breast fibroadenoma Diseases 0.000 claims description 4
- 208000003362 bronchogenic carcinoma Diseases 0.000 claims description 4
- 201000002143 bronchus adenoma Diseases 0.000 claims description 4
- 208000011825 carcinoma of the ampulla of vater Diseases 0.000 claims description 4
- 230000000747 cardiac effect Effects 0.000 claims description 4
- 201000005217 chondroblastoma Diseases 0.000 claims description 4
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 claims description 4
- 208000009060 clear cell adenocarcinoma Diseases 0.000 claims description 4
- 201000010305 cutaneous fibrous histiocytoma Diseases 0.000 claims description 4
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 claims description 4
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 4
- 125000004990 dihydroxyalkyl group Chemical group 0.000 claims description 4
- 201000009409 embryonal rhabdomyosarcoma Diseases 0.000 claims description 4
- 201000003914 endometrial carcinoma Diseases 0.000 claims description 4
- 210000003238 esophagus Anatomy 0.000 claims description 4
- 201000010175 gallbladder cancer Diseases 0.000 claims description 4
- 201000007487 gallbladder carcinoma Diseases 0.000 claims description 4
- 208000015419 gastrin-producing neuroendocrine tumor Diseases 0.000 claims description 4
- 201000000052 gastrinoma Diseases 0.000 claims description 4
- 201000003115 germ cell cancer Diseases 0.000 claims description 4
- 208000005017 glioblastoma Diseases 0.000 claims description 4
- 230000002489 hematologic effect Effects 0.000 claims description 4
- 208000006359 hepatoblastoma Diseases 0.000 claims description 4
- 201000002735 hepatocellular adenoma Diseases 0.000 claims description 4
- 231100000844 hepatocellular carcinoma Toxicity 0.000 claims description 4
- 201000004933 in situ carcinoma Diseases 0.000 claims description 4
- 206010022498 insulinoma Diseases 0.000 claims description 4
- 210000002570 interstitial cell Anatomy 0.000 claims description 4
- 201000010985 invasive ductal carcinoma Diseases 0.000 claims description 4
- 210000001117 keloid Anatomy 0.000 claims description 4
- 210000003734 kidney Anatomy 0.000 claims description 4
- 210000002429 large intestine Anatomy 0.000 claims description 4
- 208000032839 leukemia Diseases 0.000 claims description 4
- 206010024627 liposarcoma Diseases 0.000 claims description 4
- 210000004185 liver Anatomy 0.000 claims description 4
- 201000004593 malignant giant cell tumor Diseases 0.000 claims description 4
- 201000000289 malignant teratoma Diseases 0.000 claims description 4
- 210000002418 meninge Anatomy 0.000 claims description 4
- 125000002757 morpholinyl group Chemical group 0.000 claims description 4
- 208000010492 mucinous cystadenocarcinoma Diseases 0.000 claims description 4
- 208000009091 myxoma Diseases 0.000 claims description 4
- 210000000653 nervous system Anatomy 0.000 claims description 4
- 208000007538 neurilemmoma Diseases 0.000 claims description 4
- 208000004649 neutrophil actin dysfunction Diseases 0.000 claims description 4
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims description 4
- 208000008798 osteoma Diseases 0.000 claims description 4
- 208000021255 pancreatic insulinoma Diseases 0.000 claims description 4
- 208000024724 pineal body neoplasm Diseases 0.000 claims description 4
- 201000004123 pineal gland cancer Diseases 0.000 claims description 4
- 208000029922 reticulum cell sarcoma Diseases 0.000 claims description 4
- 201000009410 rhabdomyosarcoma Diseases 0.000 claims description 4
- 206010039667 schwannoma Diseases 0.000 claims description 4
- 208000004548 serous cystadenocarcinoma Diseases 0.000 claims description 4
- 210000003625 skull Anatomy 0.000 claims description 4
- 210000000813 small intestine Anatomy 0.000 claims description 4
- 210000000278 spinal cord Anatomy 0.000 claims description 4
- 210000002784 stomach Anatomy 0.000 claims description 4
- 208000001608 teratocarcinoma Diseases 0.000 claims description 4
- 208000022271 tubular adenoma Diseases 0.000 claims description 4
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims description 4
- 210000003708 urethra Anatomy 0.000 claims description 4
- 210000004291 uterus Anatomy 0.000 claims description 4
- 210000001215 vagina Anatomy 0.000 claims description 4
- 208000009540 villous adenoma Diseases 0.000 claims description 4
- 210000003905 vulva Anatomy 0.000 claims description 4
- 125000005962 1,4-oxazepanyl group Chemical group 0.000 claims description 3
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 3
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 claims description 3
- 208000003170 Bronchiolo-Alveolar Adenocarcinoma Diseases 0.000 claims description 3
- 206010058354 Bronchioloalveolar carcinoma Diseases 0.000 claims description 3
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 claims description 3
- 208000005045 Interdigitating dendritic cell sarcoma Diseases 0.000 claims description 3
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 claims description 3
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims description 3
- 208000000097 Sertoli-Leydig cell tumor Diseases 0.000 claims description 3
- 210000000481 breast Anatomy 0.000 claims description 3
- 201000001343 fallopian tube carcinoma Diseases 0.000 claims description 3
- 208000022013 kidney Wilms tumor Diseases 0.000 claims description 3
- 208000016992 lung adenocarcinoma in situ Diseases 0.000 claims description 3
- 208000024191 minimally invasive lung adenocarcinoma Diseases 0.000 claims description 3
- 201000008026 nephroblastoma Diseases 0.000 claims description 3
- 230000002611 ovarian Effects 0.000 claims description 3
- 210000000496 pancreas Anatomy 0.000 claims description 3
- 125000004929 pyrrolidonyl group Chemical group N1(C(CCC1)=O)* 0.000 claims description 3
- 210000001550 testis Anatomy 0.000 claims description 3
- 206010044412 transitional cell carcinoma Diseases 0.000 claims description 3
- 230000003140 astrocytic effect Effects 0.000 claims description 2
- 230000002357 endometrial effect Effects 0.000 claims description 2
- 125000003566 oxetanyl group Chemical group 0.000 claims description 2
- 230000002685 pulmonary effect Effects 0.000 claims description 2
- 210000001685 thyroid gland Anatomy 0.000 claims description 2
- 125000001412 tetrahydropyranyl group Chemical group 0.000 claims 1
- 101150105104 Kras gene Proteins 0.000 abstract description 76
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 432
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 369
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 286
- 239000000203 mixture Substances 0.000 description 256
- 239000000243 solution Substances 0.000 description 167
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 151
- 235000019439 ethyl acetate Nutrition 0.000 description 151
- 239000011541 reaction mixture Substances 0.000 description 148
- 239000007787 solid Substances 0.000 description 126
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 125
- 229910001868 water Inorganic materials 0.000 description 125
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 96
- 239000012044 organic layer Substances 0.000 description 87
- 239000000543 intermediate Substances 0.000 description 85
- 238000000668 atmospheric pressure chemical ionisation mass spectrometry Methods 0.000 description 82
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 75
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 75
- 229910052938 sodium sulfate Inorganic materials 0.000 description 73
- 235000011152 sodium sulphate Nutrition 0.000 description 73
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 73
- 239000012267 brine Substances 0.000 description 72
- 238000004440 column chromatography Methods 0.000 description 69
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 68
- NCXUNZWLEYGQAH-YFKPBYRVSA-N (2s)-1-(dimethylamino)propan-2-ol Chemical compound C[C@H](O)CN(C)C NCXUNZWLEYGQAH-YFKPBYRVSA-N 0.000 description 65
- 238000006243 chemical reaction Methods 0.000 description 64
- 239000012299 nitrogen atmosphere Substances 0.000 description 60
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 description 60
- 239000007832 Na2SO4 Substances 0.000 description 57
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 55
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 54
- 239000003921 oil Substances 0.000 description 48
- RFIOZSIHFNEKFF-UHFFFAOYSA-M piperazine-1-carboxylate Chemical compound [O-]C(=O)N1CCNCC1 RFIOZSIHFNEKFF-UHFFFAOYSA-M 0.000 description 44
- 238000005160 1H NMR spectroscopy Methods 0.000 description 43
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 43
- 230000002829 reductive effect Effects 0.000 description 43
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 39
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 38
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 38
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 38
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 37
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 35
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 34
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 34
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 34
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 34
- 239000003208 petroleum Substances 0.000 description 31
- 239000000047 product Substances 0.000 description 30
- 238000000746 purification Methods 0.000 description 30
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 29
- 239000011734 sodium Substances 0.000 description 29
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 28
- 239000000725 suspension Substances 0.000 description 28
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 27
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 26
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 26
- 239000002904 solvent Substances 0.000 description 26
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 24
- MXFYYFVVIIWKFE-UHFFFAOYSA-N dicyclohexyl-[2-[2,6-di(propan-2-yloxy)phenyl]phenyl]phosphane Chemical compound CC(C)OC1=CC=CC(OC(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 MXFYYFVVIIWKFE-UHFFFAOYSA-N 0.000 description 24
- 239000000706 filtrate Substances 0.000 description 24
- 238000003756 stirring Methods 0.000 description 24
- 238000010898 silica gel chromatography Methods 0.000 description 23
- 239000012298 atmosphere Substances 0.000 description 22
- 239000012071 phase Substances 0.000 description 21
- 229920006395 saturated elastomer Polymers 0.000 description 21
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 20
- 229910052757 nitrogen Inorganic materials 0.000 description 20
- 125000004194 piperazin-1-yl group Chemical group [H]N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 20
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 19
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 19
- 239000000284 extract Substances 0.000 description 18
- 239000000741 silica gel Substances 0.000 description 17
- 229910002027 silica gel Inorganic materials 0.000 description 17
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 15
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 15
- 239000012230 colorless oil Substances 0.000 description 15
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 15
- HGINCPLSRVDWNT-UHFFFAOYSA-N Acrolein Chemical compound C=CC=O HGINCPLSRVDWNT-UHFFFAOYSA-N 0.000 description 14
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 14
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 14
- 239000010410 layer Substances 0.000 description 14
- 230000035772 mutation Effects 0.000 description 14
- 238000002953 preparative HPLC Methods 0.000 description 14
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 13
- CTOUWUYDDUSBQE-UHFFFAOYSA-N benzyl piperazine-1-carboxylate Chemical compound C1CNCCN1C(=O)OCC1=CC=CC=C1 CTOUWUYDDUSBQE-UHFFFAOYSA-N 0.000 description 13
- 229910052796 boron Inorganic materials 0.000 description 13
- 229910052799 carbon Inorganic materials 0.000 description 13
- 238000011282 treatment Methods 0.000 description 13
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 12
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 12
- 150000002431 hydrogen Chemical group 0.000 description 12
- WQWUQDVFRYMMCY-UHFFFAOYSA-N naphthalen-1-yl trifluoromethanesulfonate Chemical compound C1=CC=C2C(OS(=O)(=O)C(F)(F)F)=CC=CC2=C1 WQWUQDVFRYMMCY-UHFFFAOYSA-N 0.000 description 12
- 125000001424 substituent group Chemical group 0.000 description 12
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 11
- 239000012065 filter cake Substances 0.000 description 11
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 10
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 10
- 208000035475 disorder Diseases 0.000 description 10
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 10
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 10
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 9
- 229910004298 SiO 2 Inorganic materials 0.000 description 9
- 229910000024 caesium carbonate Inorganic materials 0.000 description 9
- 238000004587 chromatography analysis Methods 0.000 description 9
- 229910052681 coesite Inorganic materials 0.000 description 9
- 229910052906 cristobalite Inorganic materials 0.000 description 9
- 125000001153 fluoro group Chemical group F* 0.000 description 9
- 239000012074 organic phase Substances 0.000 description 9
- 125000004076 pyridyl group Chemical group 0.000 description 9
- 239000000377 silicon dioxide Substances 0.000 description 9
- 235000012239 silicon dioxide Nutrition 0.000 description 9
- 229910052682 stishovite Inorganic materials 0.000 description 9
- 229910052905 tridymite Inorganic materials 0.000 description 9
- CTNDGYOJDNSZMQ-UHFFFAOYSA-N 7-benzyl-2,4-dichloro-6,8-dihydro-5h-pyrido[3,4-d]pyrimidine Chemical compound C1C2=NC(Cl)=NC(Cl)=C2CCN1CC1=CC=CC=C1 CTNDGYOJDNSZMQ-UHFFFAOYSA-N 0.000 description 8
- XJUZRXYOEPSWMB-UHFFFAOYSA-N Chloromethyl methyl ether Chemical compound COCCl XJUZRXYOEPSWMB-UHFFFAOYSA-N 0.000 description 8
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 8
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 8
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 8
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 8
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- 239000008346 aqueous phase Substances 0.000 description 8
- 239000000463 material Substances 0.000 description 8
- 230000001105 regulatory effect Effects 0.000 description 8
- 229910000104 sodium hydride Inorganic materials 0.000 description 8
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 8
- 229910052717 sulfur Inorganic materials 0.000 description 8
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 8
- NNWUEBIEOFQMSS-UHFFFAOYSA-N (+)-(S)-2-methylpiperidine Natural products CC1CCCCN1 NNWUEBIEOFQMSS-UHFFFAOYSA-N 0.000 description 7
- NNWUEBIEOFQMSS-LURJTMIESA-N (2s)-2-methylpiperidine Chemical compound C[C@H]1CCCCN1 NNWUEBIEOFQMSS-LURJTMIESA-N 0.000 description 7
- SYTVTPAROOODCH-UHFFFAOYSA-N 1-bromo-3-(methoxymethoxy)naphthalene Chemical compound BrC1=CC(=CC2=CC=CC=C12)OCOC SYTVTPAROOODCH-UHFFFAOYSA-N 0.000 description 7
- VZKSLWJLGAGPIU-UHFFFAOYSA-N 3-morpholin-4-ylpropan-1-ol Chemical compound OCCCN1CCOCC1 VZKSLWJLGAGPIU-UHFFFAOYSA-N 0.000 description 7
- GOOHAUXETOMSMM-GSVOUGTGSA-N R-propylene oxide Chemical compound C[C@@H]1CO1 GOOHAUXETOMSMM-GSVOUGTGSA-N 0.000 description 7
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 7
- DFFVDVMPHZYBMF-UHFFFAOYSA-N [3-(methoxymethoxy)naphthalen-1-yl] trifluoromethanesulfonate Chemical compound FC(S(=O)(=O)OC1=CC(=CC2=CC=CC=C12)OCOC)(F)F DFFVDVMPHZYBMF-UHFFFAOYSA-N 0.000 description 7
- 238000003556 assay Methods 0.000 description 7
- 125000004432 carbon atom Chemical group C* 0.000 description 7
- 239000003054 catalyst Substances 0.000 description 7
- 125000001309 chloro group Chemical group Cl* 0.000 description 7
- 238000003818 flash chromatography Methods 0.000 description 7
- 125000001041 indolyl group Chemical group 0.000 description 7
- 229910052760 oxygen Inorganic materials 0.000 description 7
- 230000002441 reversible effect Effects 0.000 description 7
- OZKAAXWQXBRAGF-UHFFFAOYSA-N 1-bromo-3-phenylmethoxynaphthalene Chemical compound C=1C2=CC=CC=C2C(Br)=CC=1OCC1=CC=CC=C1 OZKAAXWQXBRAGF-UHFFFAOYSA-N 0.000 description 6
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 6
- CLFHUFDVJZFIDR-UHFFFAOYSA-N 3-methoxynaphthalen-1-ol Chemical compound C1=CC=CC2=CC(OC)=CC(O)=C21 CLFHUFDVJZFIDR-UHFFFAOYSA-N 0.000 description 6
- PQNQMYMGUXGWTG-UHFFFAOYSA-N 4-bromonaphthalen-2-ol Chemical compound C1=CC=CC2=CC(O)=CC(Br)=C21 PQNQMYMGUXGWTG-UHFFFAOYSA-N 0.000 description 6
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 6
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 6
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 125000006239 protecting group Chemical group 0.000 description 6
- 238000004809 thin layer chromatography Methods 0.000 description 6
- IZLIYHWOPBETRT-UHFFFAOYSA-N (3-methoxynaphthalen-1-yl) trifluoromethanesulfonate Chemical compound C1=CC=CC2=CC(OC)=CC(OS(=O)(=O)C(F)(F)F)=C21 IZLIYHWOPBETRT-UHFFFAOYSA-N 0.000 description 5
- UYTVDHNTKCSQOF-UHFFFAOYSA-N 2,4-dibromonaphthalen-1-amine Chemical compound C1=CC=C2C(N)=C(Br)C=C(Br)C2=C1 UYTVDHNTKCSQOF-UHFFFAOYSA-N 0.000 description 5
- FYBXDVQWYMPNIR-UHFFFAOYSA-N 4-bromo-1-diazonionaphthalen-2-olate Chemical compound C1=CC=CC2=C([N+]#N)C([O-])=CC(Br)=C21 FYBXDVQWYMPNIR-UHFFFAOYSA-N 0.000 description 5
- DYNBNPAECUWPPS-UHFFFAOYSA-N 7-benzyl-1,5,6,8-tetrahydropyrido[3,4-d]pyrimidine-2,4-dione Chemical compound C1CC=2C(=O)NC(=O)NC=2CN1CC1=CC=CC=C1 DYNBNPAECUWPPS-UHFFFAOYSA-N 0.000 description 5
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 235000011114 ammonium hydroxide Nutrition 0.000 description 5
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 5
- 150000007942 carboxylates Chemical class 0.000 description 5
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 239000011737 fluorine Substances 0.000 description 5
- 229910052731 fluorine Inorganic materials 0.000 description 5
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 5
- 229910052737 gold Inorganic materials 0.000 description 5
- 239000010931 gold Substances 0.000 description 5
- 229910052763 palladium Inorganic materials 0.000 description 5
- ARJOQCYCJMAIFR-UHFFFAOYSA-N prop-2-enoyl prop-2-enoate Chemical compound C=CC(=O)OC(=O)C=C ARJOQCYCJMAIFR-UHFFFAOYSA-N 0.000 description 5
- 208000024891 symptom Diseases 0.000 description 5
- NCXUNZWLEYGQAH-UHFFFAOYSA-N 1-(dimethylamino)propan-2-ol Chemical compound CC(O)CN(C)C NCXUNZWLEYGQAH-UHFFFAOYSA-N 0.000 description 4
- MRGOMLFEVPNISR-UHFFFAOYSA-N 1-[4-(2-hydroxyethyl)piperazin-1-yl]ethanone Chemical compound CC(=O)N1CCN(CCO)CC1 MRGOMLFEVPNISR-UHFFFAOYSA-N 0.000 description 4
- OPUXBDAYLODVBQ-UHFFFAOYSA-N 1-bromo-3-cyclopropyl-5-phenylmethoxybenzene Chemical compound C(C1=CC=CC=C1)OC1=CC(=CC(=C1)C1CC1)Br OPUXBDAYLODVBQ-UHFFFAOYSA-N 0.000 description 4
- GILLKIJFJOBJCD-UHFFFAOYSA-N 2-(1,1-dioxo-1,4-thiazinan-4-yl)ethanol Chemical compound OCCN1CCS(=O)(=O)CC1 GILLKIJFJOBJCD-UHFFFAOYSA-N 0.000 description 4
- LMFRSLRJXLATRL-UHFFFAOYSA-N 2-bromo-3-fluorophenol Chemical compound OC1=CC=CC(F)=C1Br LMFRSLRJXLATRL-UHFFFAOYSA-N 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 4
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 4
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 4
- 239000004202 carbamide Substances 0.000 description 4
- 239000000460 chlorine Substances 0.000 description 4
- 229910052801 chlorine Inorganic materials 0.000 description 4
- 230000008878 coupling Effects 0.000 description 4
- 238000010168 coupling process Methods 0.000 description 4
- 238000005859 coupling reaction Methods 0.000 description 4
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 4
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 4
- 239000003814 drug Substances 0.000 description 4
- 239000006260 foam Substances 0.000 description 4
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 4
- 235000019341 magnesium sulphate Nutrition 0.000 description 4
- SDDKIZNHOCEXTF-UHFFFAOYSA-N methyl carbamimidothioate Chemical compound CSC(N)=N SDDKIZNHOCEXTF-UHFFFAOYSA-N 0.000 description 4
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 238000012746 preparative thin layer chromatography Methods 0.000 description 4
- 125000006413 ring segment Chemical group 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- 235000017557 sodium bicarbonate Nutrition 0.000 description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 description 4
- 241000894007 species Species 0.000 description 4
- MPUCCVFWJRIMFT-HTQZYQBOSA-N (2R)-1-[(3R)-3-methoxypyrrolidin-1-yl]propan-2-ol Chemical compound CO[C@H]1CN(CC1)C[C@@H](C)O MPUCCVFWJRIMFT-HTQZYQBOSA-N 0.000 description 3
- MPUCCVFWJRIMFT-SFYZADRCSA-N (2R)-1-[(3S)-3-methoxypyrrolidin-1-yl]propan-2-ol Chemical compound CO[C@@H]1CN(CC1)C[C@@H](C)O MPUCCVFWJRIMFT-SFYZADRCSA-N 0.000 description 3
- HNBNDJUIXIJZIF-UHFFFAOYSA-N (3-methoxy-6-methylnaphthalen-1-yl) trifluoromethanesulfonate Chemical compound FC(S(=O)(=O)OC1=CC(=CC2=CC(=CC=C12)C)OC)(F)F HNBNDJUIXIJZIF-UHFFFAOYSA-N 0.000 description 3
- ZABFSYBSTIHNAE-UHFFFAOYSA-N 1-(dimethylamino)butan-2-ol Chemical compound CCC(O)CN(C)C ZABFSYBSTIHNAE-UHFFFAOYSA-N 0.000 description 3
- TZDPDBBXBBMVHM-UHFFFAOYSA-N 1-[4-[2-[2-(3-fluoropyrrolidin-1-yl)ethoxy]-7-(3-methoxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound COC1=CC2=CC=CC=C2C(=C1)N1CCC2=C(N=C(OCCN3CCC(F)C3)N=C2C1)N1CCN(CC1)C(=O)C=C TZDPDBBXBBMVHM-UHFFFAOYSA-N 0.000 description 3
- CFWCGIJLBZWMGO-UHFFFAOYSA-N 1-[4-[7-(3-hydroxy-6-methylnaphthalen-1-yl)-2-(3-morpholin-4-ylpropoxy)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CC1=CC=C2C(=C1)C=C(O)C=C2N1CCC2=C(N=C(OCCCN3CCOCC3)N=C2C1)N1CCN(CC1)C(=O)C=C CFWCGIJLBZWMGO-UHFFFAOYSA-N 0.000 description 3
- HJRNLVLSGMRETB-UHFFFAOYSA-N 1-[4-[7-(3-methoxy-6-methylnaphthalen-1-yl)-2-(3-morpholin-4-ylpropoxy)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound COC1=CC2=CC(C)=CC=C2C(=C1)N1CCC2=C(N=C(OCCCN3CCOCC3)N=C2C1)N1CCN(CC1)C(=O)C=C HJRNLVLSGMRETB-UHFFFAOYSA-N 0.000 description 3
- IBZMXIIJSPGJCW-UHFFFAOYSA-N 1-bromo-2-methyl-3-phenylmethoxynaphthalene Chemical compound C(C1=CC=CC=C1)OC=1C(=C(C2=CC=CC=C2C=1)Br)C IBZMXIIJSPGJCW-UHFFFAOYSA-N 0.000 description 3
- AKMJZIVYJZVFPR-UHFFFAOYSA-N 1-bromo-3-chloro-5-(methoxymethoxy)-2-propan-2-ylbenzene Chemical compound COCOC1=CC(Br)=C(C(C)C)C(Cl)=C1 AKMJZIVYJZVFPR-UHFFFAOYSA-N 0.000 description 3
- FRRJQPFHMJGDTJ-UHFFFAOYSA-N 1-bromo-3-methoxy-2-methylnaphthalene Chemical compound COC1=CC2=CC=CC=C2C(Br)=C1C FRRJQPFHMJGDTJ-UHFFFAOYSA-N 0.000 description 3
- JHZQEADUKRNQBX-UHFFFAOYSA-N 1-bromo-8-chloronaphthalene Chemical compound C1=CC(Br)=C2C(Cl)=CC=CC2=C1 JHZQEADUKRNQBX-UHFFFAOYSA-N 0.000 description 3
- QTWVRVGVJURUFK-UHFFFAOYSA-N 1-bromo-8-methylnaphthalene Chemical compound C1=CC(Br)=C2C(C)=CC=CC2=C1 QTWVRVGVJURUFK-UHFFFAOYSA-N 0.000 description 3
- YAXQOLYGKLGQKA-UHFFFAOYSA-N 1-morpholin-4-ylpropan-2-ol Chemical compound CC(O)CN1CCOCC1 YAXQOLYGKLGQKA-UHFFFAOYSA-N 0.000 description 3
- WCTXJAXKORIYNA-UHFFFAOYSA-N 1-o-tert-butyl 4-o-ethyl 3-oxopiperidine-1,4-dicarboxylate Chemical compound CCOC(=O)C1CCN(C(=O)OC(C)(C)C)CC1=O WCTXJAXKORIYNA-UHFFFAOYSA-N 0.000 description 3
- BPYANEJZLUMJNA-UHFFFAOYSA-N 1-pyrrolidin-1-ylpropan-2-ol Chemical compound CC(O)CN1CCCC1 BPYANEJZLUMJNA-UHFFFAOYSA-N 0.000 description 3
- CROKWOXDQWOKCV-UHFFFAOYSA-N 2-(3-fluoropyrrolidin-1-yl)ethanol Chemical compound OCCN1CCC(F)C1 CROKWOXDQWOKCV-UHFFFAOYSA-N 0.000 description 3
- DUAIISPCCJPFDG-QMMMGPOBSA-N 2-[(2s)-2-methylpiperidin-1-yl]ethanol Chemical compound C[C@H]1CCCCN1CCO DUAIISPCCJPFDG-QMMMGPOBSA-N 0.000 description 3
- LHZJBWNQSNHEIO-UHFFFAOYSA-N 2-bromo-1-fluoro-4-phenylmethoxybenzene Chemical compound C1=C(Br)C(F)=CC=C1OCC1=CC=CC=C1 LHZJBWNQSNHEIO-UHFFFAOYSA-N 0.000 description 3
- TYZCBROTRYSTPT-UHFFFAOYSA-N 2-bromo-3-fluoro-1-(methoxymethoxy)-4-methylbenzene Chemical compound BrC1=C(C=CC(=C1F)C)OCOC TYZCBROTRYSTPT-UHFFFAOYSA-N 0.000 description 3
- NOTOSWIZHCKFOI-UHFFFAOYSA-N 2-bromo-4-(methoxymethoxy)-1-propan-2-yloxybenzene Chemical compound BrC1=C(C=CC(=C1)OCOC)OC(C)C NOTOSWIZHCKFOI-UHFFFAOYSA-N 0.000 description 3
- JVUSFSRPHXARLE-UHFFFAOYSA-N 2-bromo-7-(methoxymethoxy)naphthalene Chemical compound BrC1=CC2=CC(=CC=C2C=C1)OCOC JVUSFSRPHXARLE-UHFFFAOYSA-N 0.000 description 3
- NUIISRXJDDGJAW-UHFFFAOYSA-N 2-methylnaphthalene-1,3-diol Chemical compound C1=CC=CC2=C(O)C(C)=C(O)C=C21 NUIISRXJDDGJAW-UHFFFAOYSA-N 0.000 description 3
- JEZVOSAHGZYHNO-UHFFFAOYSA-N 3-(1,4-oxazepan-4-yl)propan-1-ol Chemical compound OCCCN1CCCOCC1 JEZVOSAHGZYHNO-UHFFFAOYSA-N 0.000 description 3
- PYSGFFTXMUWEOT-UHFFFAOYSA-N 3-(dimethylamino)propan-1-ol Chemical compound CN(C)CCCO PYSGFFTXMUWEOT-UHFFFAOYSA-N 0.000 description 3
- CDDGNGVFPQRJJM-UHFFFAOYSA-N 3-fluoropyrrolidine Chemical compound FC1CCNC1 CDDGNGVFPQRJJM-UHFFFAOYSA-N 0.000 description 3
- LIYRASDYTSNPNO-UHFFFAOYSA-N 3-methoxy-2-methylnaphthalen-1-ol Chemical compound C1=CC=C2C(O)=C(C)C(OC)=CC2=C1 LIYRASDYTSNPNO-UHFFFAOYSA-N 0.000 description 3
- MAVSSNUUNGFRAB-UHFFFAOYSA-N 3-methoxy-6-methylnaphthalen-1-ol Chemical compound COC=1C=C(C2=CC=C(C=C2C=1)C)O MAVSSNUUNGFRAB-UHFFFAOYSA-N 0.000 description 3
- XKEHRXSJODJCAW-UHFFFAOYSA-N 4-[2-[2-(1,1-dioxo-1,4-thiazinan-4-yl)ethoxy]-4-piperazin-1-yl-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-7-yl]naphthalen-2-ol Chemical compound OC1=CC2=CC=CC=C2C(=C1)N1CCC2=C(N=C(OCCN3CCS(=O)(=O)CC3)N=C2C1)N1CCNCC1 XKEHRXSJODJCAW-UHFFFAOYSA-N 0.000 description 3
- AFRJPAKKFYACKK-UHFFFAOYSA-N 4-[2-[2-(dimethylamino)ethoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]-N,N-dimethyl-1-prop-2-enoylpiperazine-2-carboxamide Chemical compound CN(CCOC=1N=C(C2=C(N=1)CN(CC2)C1=CC(=CC2=CC=CC=C12)O)N1CC(N(CC1)C(C=C)=O)C(=O)N(C)C)C AFRJPAKKFYACKK-UHFFFAOYSA-N 0.000 description 3
- JUQFLPBLOQADLT-UHFFFAOYSA-N 4-[2-[2-(dimethylamino)ethoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]-N,N-dimethylpiperazine-2-carboxamide Chemical compound CN(CCOC=1N=C(C2=C(N=1)CN(CC2)C1=CC(=CC2=CC=CC=C12)O)N1CC(NCC1)C(=O)N(C)C)C JUQFLPBLOQADLT-UHFFFAOYSA-N 0.000 description 3
- WGAXTIXXJPQVBL-UHFFFAOYSA-N 4-[3-[[7-(3-methoxy-6-methylnaphthalen-1-yl)-4-piperazin-1-yl-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-2-yl]oxy]propyl]morpholine Chemical compound COC=1C=C(C2=CC=C(C=C2C=1)C)N1CC=2N=C(N=C(C=2CC1)N1CCNCC1)OCCCN1CCOCC1 WGAXTIXXJPQVBL-UHFFFAOYSA-N 0.000 description 3
- KIRRCNTUOVSKIQ-UHFFFAOYSA-N 4-bromo-3-methylnaphthalen-2-ol Chemical compound BrC1=C(C(=CC2=CC=CC=C12)O)C KIRRCNTUOVSKIQ-UHFFFAOYSA-N 0.000 description 3
- YBLXBXPJJAZHDS-UHFFFAOYSA-N 4-bromo-5-methyl-1h-indazole Chemical compound CC1=CC=C2NN=CC2=C1Br YBLXBXPJJAZHDS-UHFFFAOYSA-N 0.000 description 3
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- 229910015845 BBr3 Inorganic materials 0.000 description 3
- YCVDNZNYCWFJRW-UHFFFAOYSA-N BrC1=C2C=NN(C2=CC=C1C)COCC[Si](C)(C)C Chemical compound BrC1=C2C=NN(C2=CC=C1C)COCC[Si](C)(C)C YCVDNZNYCWFJRW-UHFFFAOYSA-N 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 3
- ZQDSTAPWXCLPCX-NEPJUHHUSA-N [Si](C)(C)(C(C)(C)C)O[C@@H]1CN(CC1)C[C@@H](C)O Chemical compound [Si](C)(C)(C(C)(C)C)O[C@@H]1CN(CC1)C[C@@H](C)O ZQDSTAPWXCLPCX-NEPJUHHUSA-N 0.000 description 3
- 235000001014 amino acid Nutrition 0.000 description 3
- 150000001413 amino acids Chemical group 0.000 description 3
- 235000019270 ammonium chloride Nutrition 0.000 description 3
- QSKMFYCQRQLYMF-ZDUSSCGKSA-N benzyl (2S)-2-(cyanomethyl)piperazine-1-carboxylate Chemical compound C(#N)C[C@@H]1N(CCNC1)C(=O)OCC1=CC=CC=C1 QSKMFYCQRQLYMF-ZDUSSCGKSA-N 0.000 description 3
- PUCNIGULRFJAIC-UHFFFAOYSA-N benzyl 2,6-diazaspiro[3.3]heptane-2-carboxylate Chemical compound C1C2(CNC2)CN1C(=O)OCC1=CC=CC=C1 PUCNIGULRFJAIC-UHFFFAOYSA-N 0.000 description 3
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000012141 concentrate Substances 0.000 description 3
- 239000013058 crude material Substances 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 229940113088 dimethylacetamide Drugs 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 239000003480 eluent Substances 0.000 description 3
- OUHGZJIVXDXXMN-UHFFFAOYSA-N ethyl 2-methyl-3-oxo-4-phenylbutanoate Chemical compound CCOC(=O)C(C)C(=O)CC1=CC=CC=C1 OUHGZJIVXDXXMN-UHFFFAOYSA-N 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- ODQZNBWVRPKMKN-UHFFFAOYSA-N methyl piperazine-2-carboxylate Chemical compound COC(=O)C1CNCCN1 ODQZNBWVRPKMKN-UHFFFAOYSA-N 0.000 description 3
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 125000000160 oxazolidinyl group Chemical group 0.000 description 3
- XUWHAWMETYGRKB-UHFFFAOYSA-N piperidin-2-one Chemical compound O=C1CCCCN1 XUWHAWMETYGRKB-UHFFFAOYSA-N 0.000 description 3
- 239000002798 polar solvent Substances 0.000 description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 238000004007 reversed phase HPLC Methods 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 235000017550 sodium carbonate Nutrition 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 3
- NCXUNZWLEYGQAH-RXMQYKEDSA-N (2r)-1-(dimethylamino)propan-2-ol Chemical compound C[C@@H](O)CN(C)C NCXUNZWLEYGQAH-RXMQYKEDSA-N 0.000 description 2
- RTSUZVGEKFNPFE-UHFFFAOYSA-N (3-bromo-4-propan-2-yloxyphenyl) acetate Chemical compound CC(C)OC1=CC=C(OC(C)=O)C=C1Br RTSUZVGEKFNPFE-UHFFFAOYSA-N 0.000 description 2
- XAXJKFBOEBUAAX-UHFFFAOYSA-N (3-methoxy-2-methylnaphthalen-1-yl) trifluoromethanesulfonate Chemical compound COC=1C(=C(C2=CC=CC=C2C=1)OS(=O)(=O)C(F)(F)F)C XAXJKFBOEBUAAX-UHFFFAOYSA-N 0.000 description 2
- WVONIIRCOLFAKF-UHFFFAOYSA-N (4-bromonaphthalen-2-yl) 2,2-dimethylpropanoate Chemical compound CC(C(=O)OC1=CC2=CC=CC=C2C(=C1)Br)(C)C WVONIIRCOLFAKF-UHFFFAOYSA-N 0.000 description 2
- BKOVWNZGWSPUAT-UHFFFAOYSA-N (4-propan-2-yloxyphenyl) acetate Chemical compound CC(C)OC1=CC=C(OC(C)=O)C=C1 BKOVWNZGWSPUAT-UHFFFAOYSA-N 0.000 description 2
- CROMNVSVLZKBEF-UHFFFAOYSA-N 1,3-dibromo-5-phenylmethoxybenzene Chemical compound BrC1=CC(Br)=CC(OCC=2C=CC=CC=2)=C1 CROMNVSVLZKBEF-UHFFFAOYSA-N 0.000 description 2
- ZNGWEEUXTBNKFR-UHFFFAOYSA-N 1,4-oxazepane Chemical compound C1CNCCOC1 ZNGWEEUXTBNKFR-UHFFFAOYSA-N 0.000 description 2
- DQFHYQNTXCMXJL-INIZCTEOSA-N 1-O-benzyl 4-O-tert-butyl (2S)-2-(cyanomethyl)piperazine-1,4-dicarboxylate Chemical compound C(#N)C[C@@H]1N(CCN(C1)C(=O)OC(C)(C)C)C(=O)OCC1=CC=CC=C1 DQFHYQNTXCMXJL-INIZCTEOSA-N 0.000 description 2
- NHUUGXQEBNZYBR-UHFFFAOYSA-N 1-[3-[2-[1-(dimethylamino)propan-2-yloxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]-3,8-diazabicyclo[3.2.1]octan-8-yl]prop-2-en-1-one Chemical compound CN(CC(OC=1N=C(C2=C(N=1)CN(CC2)C1=CC(=CC2=CC=CC=C12)O)N1CC2CCC(C1)N2C(C=C)=O)C)C NHUUGXQEBNZYBR-UHFFFAOYSA-N 0.000 description 2
- YPKGEFZRAIHQNY-UHFFFAOYSA-N 1-[4-[2-[2-(1,1-dioxo-1,4-thiazinan-4-yl)ethoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC1=CC2=CC=CC=C2C(=C1)N1CCC2=C(N=C(OCCN3CCS(=O)(=O)CC3)N=C2C1)N1CCN(CC1)C(=O)C=C YPKGEFZRAIHQNY-UHFFFAOYSA-N 0.000 description 2
- QDCGANFUUZXYPS-UHFFFAOYSA-N 1-[4-[2-[2-(3-fluoropyrrolidin-1-yl)ethoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound FC1CN(CC1)CCOC=1N=C(C2=C(N=1)CN(CC2)C1=CC(=CC2=CC=CC=C12)O)N1CCN(CC1)C(C=C)=O QDCGANFUUZXYPS-UHFFFAOYSA-N 0.000 description 2
- VIXBQULENSPMHY-UHFFFAOYSA-N 1-[4-[2-[2-(4-acetylpiperazin-1-yl)ethoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CC(=O)N1CCN(CCOC2=NC(N3CCN(CC3)C(=O)C=C)=C3CCN(CC3=N2)C2=CC(O)=CC3=CC=CC=C23)CC1 VIXBQULENSPMHY-UHFFFAOYSA-N 0.000 description 2
- BEVOXHNIFKEGBF-UHFFFAOYSA-N 1-[4-[2-[2-(dimethylamino)ethoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]-2-(hydroxymethyl)piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C)CCOC1=NC(N2CCN(C(CO)C2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 BEVOXHNIFKEGBF-UHFFFAOYSA-N 0.000 description 2
- KAQURVZXUYVJCK-UHFFFAOYSA-N 1-[4-[2-[2-(dimethylamino)ethoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]-3-(hydroxymethyl)piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C)CCOC1=NC(N2CCN(CC2CO)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 KAQURVZXUYVJCK-UHFFFAOYSA-N 0.000 description 2
- MYGPDNHXTXMREW-UHFFFAOYSA-N 1-[4-[2-[2-(dimethylamino)ethoxy]-7-(3-hydroxynaphthalen-1-yl)-8-methyl-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CC1N(CCC2=C(N=C(OCCN(C)C)N=C12)N1CCN(CC1)C(=O)C=C)C1=CC(O)=CC2=CC=CC=C12 MYGPDNHXTXMREW-UHFFFAOYSA-N 0.000 description 2
- BTNBDXBIJCNPHG-UHFFFAOYSA-N 1-[4-[2-[[7-(3-hydroxynaphthalen-1-yl)-4-piperazin-1-yl-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-2-yl]oxy]ethyl]piperazin-1-yl]ethanone Chemical compound CC(=O)N1CCN(CCOC2=NC(N3CCNCC3)=C3CCN(CC3=N2)C2=CC(O)=CC3=CC=CC=C23)CC1 BTNBDXBIJCNPHG-UHFFFAOYSA-N 0.000 description 2
- HVPHRQWDVNVJIY-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[3-(1,4-oxazepan-4-yl)propoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC1=CC2=CC=CC=C2C(=C1)N1CCC2=C(N=C(OCCCN3CCCOCC3)N=C2C1)N1CCN(CC1)C(=O)C=C HVPHRQWDVNVJIY-UHFFFAOYSA-N 0.000 description 2
- MMNBVMVLWPXTRN-UHFFFAOYSA-N 1-[4-[7-(5-methyl-1H-indazol-4-yl)-2-(3-morpholin-4-ylpropoxy)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CC1=C(N2CCC3=C(N=C(OCCCN4CCOCC4)N=C3C2)N2CCN(CC2)C(=O)C=C)C2=C(NN=C2)C=C1 MMNBVMVLWPXTRN-UHFFFAOYSA-N 0.000 description 2
- WLPXNBYWDDYJTN-UHFFFAOYSA-N 1-bromo-2,3-dimethylbenzene Chemical compound CC1=CC=CC(Br)=C1C WLPXNBYWDDYJTN-UHFFFAOYSA-N 0.000 description 2
- DLKQHBOKULLWDQ-UHFFFAOYSA-N 1-bromonaphthalene Chemical compound C1=CC=C2C(Br)=CC=CC2=C1 DLKQHBOKULLWDQ-UHFFFAOYSA-N 0.000 description 2
- NIIPYAJVODRXRK-UHFFFAOYSA-N 1-iodo-3-(methoxymethoxy)naphthalene Chemical compound COCOC1=CC2=CC=CC=C2C(I)=C1 NIIPYAJVODRXRK-UHFFFAOYSA-N 0.000 description 2
- NHPPIJMARIVBGU-UHFFFAOYSA-N 1-iodonaphthalene Chemical compound C1=CC=C2C(I)=CC=CC2=C1 NHPPIJMARIVBGU-UHFFFAOYSA-N 0.000 description 2
- KJCVRFUGPWSIIH-UHFFFAOYSA-N 1-naphthol Chemical compound C1=CC=C2C(O)=CC=CC2=C1 KJCVRFUGPWSIIH-UHFFFAOYSA-N 0.000 description 2
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 2
- RUFPHBVGCFYCNW-UHFFFAOYSA-N 1-naphthylamine Chemical compound C1=CC=C2C(N)=CC=CC2=C1 RUFPHBVGCFYCNW-UHFFFAOYSA-N 0.000 description 2
- BRXKHIPPSTYCKO-UHFFFAOYSA-N 1-o-tert-butyl 2-o-methyl piperazine-1,2-dicarboxylate Chemical compound COC(=O)C1CNCCN1C(=O)OC(C)(C)C BRXKHIPPSTYCKO-UHFFFAOYSA-N 0.000 description 2
- QUKAHFCVKNRRBU-UHFFFAOYSA-N 1-o-tert-butyl 3-o-methyl piperazine-1,3-dicarboxylate Chemical compound COC(=O)C1CN(C(=O)OC(C)(C)C)CCN1 QUKAHFCVKNRRBU-UHFFFAOYSA-N 0.000 description 2
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 2
- JTCCAXGPZWYNEQ-UHFFFAOYSA-N 1h-naphtho[1,8-de][1,2,3]triazine Chemical compound N1N=NC2=CC=CC3=CC=CC1=C32 JTCCAXGPZWYNEQ-UHFFFAOYSA-N 0.000 description 2
- XXMFJKNOJSDQBM-UHFFFAOYSA-N 2,2,2-trifluoroacetic acid;hydrate Chemical compound [OH3+].[O-]C(=O)C(F)(F)F XXMFJKNOJSDQBM-UHFFFAOYSA-N 0.000 description 2
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 2
- NMFYHCYOYUZSSI-UHFFFAOYSA-N 2-(1,4-dibenzylpiperazin-2-yl)acetonitrile Chemical compound C1CN(CC=2C=CC=CC=2)C(CC#N)CN1CC1=CC=CC=C1 NMFYHCYOYUZSSI-UHFFFAOYSA-N 0.000 description 2
- DUAIISPCCJPFDG-UHFFFAOYSA-N 2-(2-methylpiperidin-1-yl)ethanol Chemical compound CC1CCCCN1CCO DUAIISPCCJPFDG-UHFFFAOYSA-N 0.000 description 2
- VPSAXEWJSUTLOU-UHFFFAOYSA-N 2-(3-fluoropiperidin-1-yl)ethanol Chemical compound FC1CN(CCC1)CCO VPSAXEWJSUTLOU-UHFFFAOYSA-N 0.000 description 2
- FIOGEOHNBIXNID-UHFFFAOYSA-N 2-(4,4-difluoropiperidin-1-yl)ethanol Chemical compound OCCN1CCC(F)(F)CC1 FIOGEOHNBIXNID-UHFFFAOYSA-N 0.000 description 2
- RRJKFKSUZAWGHA-UHFFFAOYSA-N 2-(4-methoxypiperidin-1-yl)ethanol Chemical compound COC1CCN(CCO)CC1 RRJKFKSUZAWGHA-UHFFFAOYSA-N 0.000 description 2
- ITSAIKUTLAJVEH-MRVPVSSYSA-N 2-[(3r)-3-methoxypiperidin-1-yl]ethanol Chemical compound CO[C@@H]1CCCN(CCO)C1 ITSAIKUTLAJVEH-MRVPVSSYSA-N 0.000 description 2
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 2
- WKQFNMDFQFUXEL-UHFFFAOYSA-N 2-bromo-1-fluoro-3-(methoxymethyl)benzene Chemical compound COCC1=CC=CC(F)=C1Br WKQFNMDFQFUXEL-UHFFFAOYSA-N 0.000 description 2
- SCDAOWGZTMCJLN-UHFFFAOYSA-N 2-bromo-1-fluoro-4-(methoxymethoxy)benzene Chemical compound COCOC1=CC=C(F)C(Br)=C1 SCDAOWGZTMCJLN-UHFFFAOYSA-N 0.000 description 2
- ZSXBTGOKJTWASH-UHFFFAOYSA-N 2-bromo-3-fluoro-4-methylphenol Chemical compound CC1=CC=C(O)C(Br)=C1F ZSXBTGOKJTWASH-UHFFFAOYSA-N 0.000 description 2
- BDIQANPWWJGFJS-UHFFFAOYSA-N 2-bromo-4-(methoxymethoxy)-1-(trifluoromethoxy)benzene Chemical compound BrC1=C(C=CC(=C1)OCOC)OC(F)(F)F BDIQANPWWJGFJS-UHFFFAOYSA-N 0.000 description 2
- JWAZRIHNYRIHIV-UHFFFAOYSA-N 2-naphthol Chemical compound C1=CC=CC2=CC(O)=CC=C21 JWAZRIHNYRIHIV-UHFFFAOYSA-N 0.000 description 2
- QWTULQLVGNZMLF-UHFFFAOYSA-N 3-bromo-4-fluorophenol Chemical compound OC1=CC=C(F)C(Br)=C1 QWTULQLVGNZMLF-UHFFFAOYSA-N 0.000 description 2
- HRFHPDLMEBHQNY-UHFFFAOYSA-N 3-bromo-4-propan-2-yloxyphenol Chemical compound CC(C)OC1=CC=C(O)C=C1Br HRFHPDLMEBHQNY-UHFFFAOYSA-N 0.000 description 2
- MSBVSBUFZLVWBE-UHFFFAOYSA-N 3-bromo-5-chloro-4-propan-2-ylphenol Chemical compound BrC=1C=C(C=C(C=1C(C)C)Cl)O MSBVSBUFZLVWBE-UHFFFAOYSA-N 0.000 description 2
- AZGGVPGFBFVEDB-UHFFFAOYSA-N 4-chloro-2-methylsulfanyl-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidine Chemical compound C1CNCC2=NC(SC)=NC(Cl)=C21 AZGGVPGFBFVEDB-UHFFFAOYSA-N 0.000 description 2
- VMOQVTPKGUXWKA-UHFFFAOYSA-N 6-(hydroxymethyl)piperidin-2-one Chemical compound OCC1CCCC(=O)N1 VMOQVTPKGUXWKA-UHFFFAOYSA-N 0.000 description 2
- RFDPSOOTDJIKMI-UHFFFAOYSA-N 7-benzyl-2,4-dichloro-6-methyl-6,8-dihydro-5H-pyrido[3,4-d]pyrimidine Chemical compound C(C1=CC=CC=C1)N1CC=2N=C(N=C(C=2CC1C)Cl)Cl RFDPSOOTDJIKMI-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- LQIZPCZWJJQIKV-UHFFFAOYSA-N 8-chloronaphthalen-1-amine Chemical compound C1=CC(Cl)=C2C(N)=CC=CC2=C1 LQIZPCZWJJQIKV-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- 208000010507 Adenocarcinoma of Lung Diseases 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 2
- GTRITTAEVIRRMU-UHFFFAOYSA-N C(C)(C)(C)[Si](C)(C)OC1=CC(=CC2=CC(=CC=C12)C)OC Chemical compound C(C)(C)(C)[Si](C)(C)OC1=CC(=CC2=CC(=CC=C12)C)OC GTRITTAEVIRRMU-UHFFFAOYSA-N 0.000 description 2
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 2
- 108020004705 Codon Proteins 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 102100030708 GTPase KRas Human genes 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 101000584612 Homo sapiens GTPase KRas Proteins 0.000 description 2
- 229940124785 KRAS inhibitor Drugs 0.000 description 2
- 229910010084 LiAlH4 Inorganic materials 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- 108700020796 Oncogene Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical group C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 2
- VCOJPHPOVDIRJK-LURJTMIESA-N [(2s)-1-methylpyrrolidin-2-yl]methanol Chemical compound CN1CCC[C@H]1CO VCOJPHPOVDIRJK-LURJTMIESA-N 0.000 description 2
- 125000003647 acryloyl group Chemical group O=C([*])C([H])=C([H])[H] 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- 125000005873 benzo[d]thiazolyl group Chemical group 0.000 description 2
- JRPIQMPFKMFAOX-NSHDSACASA-N benzyl (3s)-3-methylpiperazine-1-carboxylate Chemical compound C1CN[C@@H](C)CN1C(=O)OCC1=CC=CC=C1 JRPIQMPFKMFAOX-NSHDSACASA-N 0.000 description 2
- XUICYJSPHATVDU-UHFFFAOYSA-N benzyl 4-[2-(3-morpholin-4-ylpropoxy)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl]piperazine-1-carboxylate Chemical compound O1CCN(CC1)CCCOC=1N=C(C2=C(N=1)CNCC2)N1CCN(CC1)C(=O)OCC1=CC=CC=C1 XUICYJSPHATVDU-UHFFFAOYSA-N 0.000 description 2
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 238000001574 biopsy Methods 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 208000019065 cervical carcinoma Diseases 0.000 description 2
- QOPVNWQGBQYBBP-UHFFFAOYSA-N chloroethyl chloroformate Chemical compound CC(Cl)OC(Cl)=O QOPVNWQGBQYBBP-UHFFFAOYSA-N 0.000 description 2
- 239000012636 effector Substances 0.000 description 2
- 238000003821 enantio-separation Methods 0.000 description 2
- PZBJEEYBCBXCIG-UHFFFAOYSA-N ethyl 1-benzyl-2-methyl-5-oxopiperidine-4-carboxylate Chemical compound C(C1=CC=CC=C1)N1C(CC(C(C1)=O)C(=O)OCC)C PZBJEEYBCBXCIG-UHFFFAOYSA-N 0.000 description 2
- JYFGIESQUYQLGM-UHFFFAOYSA-N ethyl 1-benzyl-3-oxopiperidine-4-carboxylate Chemical compound C1C(=O)C(C(=O)OCC)CCN1CC1=CC=CC=C1 JYFGIESQUYQLGM-UHFFFAOYSA-N 0.000 description 2
- QDBFFNIUCGDMQN-UHFFFAOYSA-N ethyl 2-(benzylamino)propanoate Chemical compound CCOC(=O)C(C)NCC1=CC=CC=C1 QDBFFNIUCGDMQN-UHFFFAOYSA-N 0.000 description 2
- 238000004108 freeze drying Methods 0.000 description 2
- 238000007327 hydrogenolysis reaction Methods 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- 229940125399 kras g12c inhibitor Drugs 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 239000012280 lithium aluminium hydride Substances 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- 230000036210 malignancy Effects 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 2
- DYGYQTFMGNQXBL-UHFFFAOYSA-N methyl 2-(3-fluoropyrrolidin-1-yl)acetate Chemical compound FC1CN(CC1)CC(=O)OC DYGYQTFMGNQXBL-UHFFFAOYSA-N 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- XOOMNEFVDUTJPP-UHFFFAOYSA-N naphthalene-1,3-diol Chemical compound C1=CC=CC2=CC(O)=CC(O)=C21 XOOMNEFVDUTJPP-UHFFFAOYSA-N 0.000 description 2
- 230000000269 nucleophilic effect Effects 0.000 description 2
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- IOLCXVTUBQKXJR-UHFFFAOYSA-M potassium bromide Chemical compound [K+].[Br-] IOLCXVTUBQKXJR-UHFFFAOYSA-M 0.000 description 2
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- 235000019260 propionic acid Nutrition 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical class [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 2
- 235000019345 sodium thiosulphate Nutrition 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- XZYXIWRAMFFBFZ-UHFFFAOYSA-N tert-butyl 2-(hydroxymethyl)-4-methylpiperazine-1-carboxylate Chemical compound CN1CCN(C(=O)OC(C)(C)C)C(CO)C1 XZYXIWRAMFFBFZ-UHFFFAOYSA-N 0.000 description 2
- SUECTKVSIDXQQE-UHFFFAOYSA-N tert-butyl 3-fluoropyrrolidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(F)C1 SUECTKVSIDXQQE-UHFFFAOYSA-N 0.000 description 2
- COLFGKLDENAXNI-UHFFFAOYSA-N tert-butyl 4-(4-phenylmethoxycarbonylpiperazin-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidine-7-carboxylate Chemical compound C(C1=CC=CC=C1)OC(=O)N1CCN(CC1)C=1C2=C(N=CN=1)CN(CC2)C(=O)OC(C)(C)C COLFGKLDENAXNI-UHFFFAOYSA-N 0.000 description 2
- UHNCQJLEINZUNT-UHFFFAOYSA-N tert-butyl 4-oxo-1,5,6,8-tetrahydropyrido[3,4-d]pyrimidine-7-carboxylate Chemical compound N1=CNC(=O)C2=C1CN(C(=O)OC(C)(C)C)CC2 UHNCQJLEINZUNT-UHFFFAOYSA-N 0.000 description 2
- XQWXYOGWFHOHEN-UHFFFAOYSA-N tert-butyl 6H-pyrido[3,4-d]pyrimidine-7-carboxylate Chemical compound C(C)(C)(C)OC(=O)N1C=C2N=CN=CC2=CC1 XQWXYOGWFHOHEN-UHFFFAOYSA-N 0.000 description 2
- 230000002381 testicular Effects 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 125000005034 trifluormethylthio group Chemical group FC(S*)(F)F 0.000 description 2
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 2
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 2
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- UFZNMWMVQJAJRV-UHFFFAOYSA-N (1-bromoisoquinolin-3-yl)carbamic acid Chemical compound OC(=O)Nc1cc2ccccc2c(Br)n1 UFZNMWMVQJAJRV-UHFFFAOYSA-N 0.000 description 1
- UGXQXVDTGJCQHR-UHFFFAOYSA-N (1-methylpiperidin-3-yl)methanol Chemical compound CN1CCCC(CO)C1 UGXQXVDTGJCQHR-UHFFFAOYSA-N 0.000 description 1
- NPWMWLAPRVMNBN-UHFFFAOYSA-N (1-methylpyrrolidin-3-yl)methanol Chemical compound CN1CCC(CO)C1 NPWMWLAPRVMNBN-UHFFFAOYSA-N 0.000 description 1
- DIQOUXNTSMWQSA-WHFBIAKZSA-N (1s,4s)-2-oxa-5-azabicyclo[2.2.1]heptane Chemical compound C1O[C@]2([H])CN[C@@]1([H])C2 DIQOUXNTSMWQSA-WHFBIAKZSA-N 0.000 description 1
- VYXHVRARDIDEHS-QGTKBVGQSA-N (1z,5z)-cycloocta-1,5-diene Chemical compound C\1C\C=C/CC\C=C/1 VYXHVRARDIDEHS-QGTKBVGQSA-N 0.000 description 1
- UCASFSAKVJTSET-MRVPVSSYSA-N (2r)-1-piperidin-1-ylpropan-2-ol Chemical compound C[C@@H](O)CN1CCCCC1 UCASFSAKVJTSET-MRVPVSSYSA-N 0.000 description 1
- MMIWDPMBWOTICQ-RXMQYKEDSA-N (2r)-2-methylpiperazine-1-carboxylic acid Chemical compound C[C@@H]1CNCCN1C(O)=O MMIWDPMBWOTICQ-RXMQYKEDSA-N 0.000 description 1
- NNWUEBIEOFQMSS-ZCFIWIBFSA-N (2r)-2-methylpiperidine Chemical compound C[C@@H]1CCCCN1 NNWUEBIEOFQMSS-ZCFIWIBFSA-N 0.000 description 1
- XNZXJSIHVXVHCR-UHFFFAOYSA-N (3-hydroxynaphthalen-1-yl) trifluoromethanesulfonate Chemical compound C1=CC=CC2=CC(O)=CC(OS(=O)(=O)C(F)(F)F)=C21 XNZXJSIHVXVHCR-UHFFFAOYSA-N 0.000 description 1
- UKANCZCEGQDKGF-ZCFIWIBFSA-N (3r)-1-methylpiperidin-3-ol Chemical compound CN1CCC[C@@H](O)C1 UKANCZCEGQDKGF-ZCFIWIBFSA-N 0.000 description 1
- MRWRHHSVHWCISU-ZCFIWIBFSA-N (3r)-3-methoxypiperidine Chemical compound CO[C@@H]1CCCNC1 MRWRHHSVHWCISU-ZCFIWIBFSA-N 0.000 description 1
- SQMYKVUSWPIFEQ-NUBCRITNSA-N (3r)-3-methoxypyrrolidine;hydrochloride Chemical compound Cl.CO[C@@H]1CCNC1 SQMYKVUSWPIFEQ-NUBCRITNSA-N 0.000 description 1
- UKANCZCEGQDKGF-LURJTMIESA-N (3s)-1-methylpiperidin-3-ol Chemical compound CN1CCC[C@H](O)C1 UKANCZCEGQDKGF-LURJTMIESA-N 0.000 description 1
- MRWRHHSVHWCISU-LURJTMIESA-N (3s)-3-methoxypiperidine Chemical compound CO[C@H]1CCCNC1 MRWRHHSVHWCISU-LURJTMIESA-N 0.000 description 1
- SQMYKVUSWPIFEQ-JEDNCBNOSA-N (3s)-3-methoxypyrrolidine;hydrochloride Chemical compound Cl.CO[C@H]1CCNC1 SQMYKVUSWPIFEQ-JEDNCBNOSA-N 0.000 description 1
- SJHPCNCNNSSLPL-CSKARUKUSA-N (4e)-4-(ethoxymethylidene)-2-phenyl-1,3-oxazol-5-one Chemical compound O1C(=O)C(=C/OCC)\N=C1C1=CC=CC=C1 SJHPCNCNNSSLPL-CSKARUKUSA-N 0.000 description 1
- LATBRGMEJHXFHX-HXUWFJFHSA-N (5R)-5-[[7-(3-hydroxynaphthalen-1-yl)-4-(4-prop-2-enoylpiperazin-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-2-yl]oxymethyl]pyrrolidin-2-one Chemical compound C(C=C)(=O)N1CCN(CC1)C=1C2=C(N=C(N=1)OC[C@H]1CCC(N1)=O)CN(CC2)C1=CC(=CC2=CC=CC=C12)O LATBRGMEJHXFHX-HXUWFJFHSA-N 0.000 description 1
- LATBRGMEJHXFHX-FQEVSTJZSA-N (5S)-5-[[7-(3-hydroxynaphthalen-1-yl)-4-(4-prop-2-enoylpiperazin-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-2-yl]oxymethyl]pyrrolidin-2-one Chemical compound C(C=C)(=O)N1CCN(CC1)C=1C2=C(N=C(N=1)OC[C@@H]1CCC(N1)=O)CN(CC2)C1=CC(=CC2=CC=CC=C12)O LATBRGMEJHXFHX-FQEVSTJZSA-N 0.000 description 1
- HOBJEFOCIRXQKH-SCSAIBSYSA-N (5r)-5-(hydroxymethyl)pyrrolidin-2-one Chemical compound OC[C@H]1CCC(=O)N1 HOBJEFOCIRXQKH-SCSAIBSYSA-N 0.000 description 1
- HOBJEFOCIRXQKH-BYPYZUCNSA-N (5s)-5-(hydroxymethyl)pyrrolidin-2-one Chemical compound OC[C@@H]1CCC(=O)N1 HOBJEFOCIRXQKH-BYPYZUCNSA-N 0.000 description 1
- QPIBJQLVHVLSKD-NRFANRHFSA-N (6S)-6-[[7-(3-hydroxynaphthalen-1-yl)-4-(4-prop-2-enoylpiperazin-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-2-yl]oxymethyl]piperidin-2-one Chemical compound C(C=C)(=O)N1CCN(CC1)C=1C2=C(N=C(N=1)OC[C@@H]1CCCC(N1)=O)CN(CC2)C1=CC(=CC2=CC=CC=C12)O QPIBJQLVHVLSKD-NRFANRHFSA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- 125000006720 (C1-C6) alkyl (C6-C10) aryl group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- 125000004502 1,2,3-oxadiazolyl group Chemical group 0.000 description 1
- 125000004511 1,2,3-thiadiazolyl group Chemical group 0.000 description 1
- 125000001399 1,2,3-triazolyl group Chemical group N1N=NC(=C1)* 0.000 description 1
- 125000004504 1,2,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000004514 1,2,4-thiadiazolyl group Chemical group 0.000 description 1
- 125000001376 1,2,4-triazolyl group Chemical group N1N=C(N=C1)* 0.000 description 1
- 125000004506 1,2,5-oxadiazolyl group Chemical group 0.000 description 1
- 125000004517 1,2,5-thiadiazolyl group Chemical group 0.000 description 1
- 125000001781 1,3,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000004520 1,3,4-thiadiazolyl group Chemical group 0.000 description 1
- YFOOEYJGMMJJLS-UHFFFAOYSA-N 1,8-diaminonaphthalene Chemical compound C1=CC(N)=C2C(N)=CC=CC2=C1 YFOOEYJGMMJJLS-UHFFFAOYSA-N 0.000 description 1
- DLXBGTIGAIESIG-UHFFFAOYSA-N 1,8-dibromonaphthalene Chemical compound C1=CC(Br)=C2C(Br)=CC=CC2=C1 DLXBGTIGAIESIG-UHFFFAOYSA-N 0.000 description 1
- GDPPKOWXZGFJLI-UHFFFAOYSA-N 1-(2-hydroxyethyl)piperidine-4-carbonitrile Chemical compound OCCN1CCC(C#N)CC1 GDPPKOWXZGFJLI-UHFFFAOYSA-N 0.000 description 1
- AEKHFLDILSDXBL-UHFFFAOYSA-N 1-(methylamino)propan-2-ol Chemical compound CNCC(C)O AEKHFLDILSDXBL-UHFFFAOYSA-N 0.000 description 1
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 1
- FNLYKNLTICNDRG-MRXNPFEDSA-N 1-O-benzyl 4-O-tert-butyl (2R)-2-(methylsulfonyloxymethyl)piperazine-1,4-dicarboxylate Chemical compound CC(C)(C)OC(=O)N1CCN([C@@H](COS(C)(=O)=O)C1)C(=O)OCC1=CC=CC=C1 FNLYKNLTICNDRG-MRXNPFEDSA-N 0.000 description 1
- TUSDEZXZIZRFGC-UHFFFAOYSA-N 1-O-galloyl-3,6-(R)-HHDP-beta-D-glucose Natural products OC1C(O2)COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC1C(O)C2OC(=O)C1=CC(O)=C(O)C(O)=C1 TUSDEZXZIZRFGC-UHFFFAOYSA-N 0.000 description 1
- LGTXFBNJDXEWMY-BRTIRZTQSA-N 1-[(1R,5S)-8-[2-[1-(dimethylamino)propan-2-yloxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]-3,8-diazabicyclo[3.2.1]octan-3-yl]prop-2-en-1-one Chemical compound CN(CC(C)OC=1N=C(C2=C(N=1)CN(CC2)C1=CC(=CC2=CC=CC=C12)O)N1[C@H]2CN(C[C@@H]1CC2)C(C=C)=O)C LGTXFBNJDXEWMY-BRTIRZTQSA-N 0.000 description 1
- NHUUGXQEBNZYBR-BRTIRZTQSA-N 1-[(1S,5R)-3-[2-[1-(dimethylamino)propan-2-yloxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]-3,8-diazabicyclo[3.2.1]octan-8-yl]prop-2-en-1-one Chemical compound CN(CC(C)OC=1N=C(C2=C(N=1)CN(CC2)C1=CC(=CC2=CC=CC=C12)O)N1C[C@H]2CC[C@@H](C1)N2C(C=C)=O)C NHUUGXQEBNZYBR-BRTIRZTQSA-N 0.000 description 1
- AQMMZKJINYODDI-HXUWFJFHSA-N 1-[(2R)-4-[2-[2-(dimethylamino)ethoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]-2-methylpiperazin-1-yl]prop-2-en-1-one Chemical compound CN(CCOC=1N=C(C2=C(N=1)CN(CC2)C1=CC(=CC2=CC=CC=C12)O)N1C[C@H](N(CC1)C(C=C)=O)C)C AQMMZKJINYODDI-HXUWFJFHSA-N 0.000 description 1
- AEZZXIUNJCBMSE-KRWDZBQOSA-N 1-[(2S)-4-[7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]-2-methylpiperazin-1-yl]prop-2-en-1-one Chemical compound OC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=CN=C(C=2CC1)N1C[C@@H](N(CC1)C(C=C)=O)C AEZZXIUNJCBMSE-KRWDZBQOSA-N 0.000 description 1
- ROYVHRWVIKSGMU-LEWJYISDSA-N 1-[(3S)-4-[2-[(2R)-1-(dimethylamino)propan-2-yl]oxy-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]-3-methylpiperazin-1-yl]prop-2-en-1-one Chemical compound C[C@H](CN(C)C)OC1=NC(N2CCN(C[C@@H]2C)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 ROYVHRWVIKSGMU-LEWJYISDSA-N 0.000 description 1
- MORHRIVNHWVACD-FQEVSTJZSA-N 1-[(3S)-4-[2-[2-(dimethylamino)ethoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]-3-methylpiperazin-1-yl]prop-2-en-1-one Chemical compound CN(CCOC=1N=C(C2=C(N=1)CN(CC2)C1=CC(=CC2=CC=CC=C12)O)N1[C@H](CN(CC1)C(C=C)=O)C)C MORHRIVNHWVACD-FQEVSTJZSA-N 0.000 description 1
- QMQCYJOKFHOXPN-UHFFFAOYSA-N 1-[2-[[7-(3-hydroxynaphthalen-1-yl)-4-(4-prop-2-enoylpiperazin-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-2-yl]oxy]ethyl]piperidine-4-carbonitrile Chemical compound OC1=CC2=CC=CC=C2C(=C1)N1CCC2=C(N=C(OCCN3CCC(CC3)C#N)N=C2C1)N1CCN(CC1)C(=O)C=C QMQCYJOKFHOXPN-UHFFFAOYSA-N 0.000 description 1
- LHQPMXPVLSCBTC-UHFFFAOYSA-N 1-[4-(7-isoquinolin-4-yl-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl)piperazin-1-yl]prop-2-en-1-one Chemical compound C1=NC=C(C2=CC=CC=C12)N1CC=2N=CN=C(C=2CC1)N1CCN(CC1)C(C=C)=O LHQPMXPVLSCBTC-UHFFFAOYSA-N 0.000 description 1
- KJGJYCRTUNZPRN-UHFFFAOYSA-N 1-[4-(7-naphthalen-1-yl-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl)piperazin-1-yl]prop-2-en-1-one Chemical compound C=CC(=O)N1CCN(CC1)C1=C2CCN(CC2=NC=N1)C1=CC=CC2=CC=CC=C12 KJGJYCRTUNZPRN-UHFFFAOYSA-N 0.000 description 1
- ZZAOXGMBDSATAQ-MRVPVSSYSA-N 1-[4-[(2r)-2-hydroxypropyl]piperazin-1-yl]ethanone Chemical compound C[C@@H](O)CN1CCN(C(C)=O)CC1 ZZAOXGMBDSATAQ-MRVPVSSYSA-N 0.000 description 1
- PJRMRZDWCNHSFU-UHFFFAOYSA-N 1-[4-[2-(1-cyclopropylpiperidin-4-yl)oxy-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC1=CC2=CC=CC=C2C(=C1)N1CCC2=C(N=C(OC3CCN(CC3)C3CC3)N=C2C1)N1CCN(CC1)C(=O)C=C PJRMRZDWCNHSFU-UHFFFAOYSA-N 0.000 description 1
- QBQGGZQEALXCKI-HSZRJFAPSA-N 1-[4-[2-[(2R)-1-(4-acetylpiperazin-1-yl)propan-2-yl]oxy-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound C(C)(=O)N1CCN(CC1)C[C@@H](C)OC=1N=C(C2=C(N=1)CN(CC2)C1=CC(=CC2=CC=CC=C12)O)N1CCN(CC1)C(C=C)=O QBQGGZQEALXCKI-HSZRJFAPSA-N 0.000 description 1
- VNYCEXWMAYTCOU-HSZRJFAPSA-N 1-[4-[2-[(2R)-1-(dimethylamino)butan-2-yl]oxy-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C[C@@H](CC)OC=1N=C(C2=C(N=1)CN(CC2)C1=CC(=CC2=CC=CC=C12)O)N1CCN(CC1)C(C=C)=O)C VNYCEXWMAYTCOU-HSZRJFAPSA-N 0.000 description 1
- NFPNAXMQDZEIMQ-GOSISDBHSA-N 1-[4-[2-[(2R)-1-(dimethylamino)propan-2-yl]oxy-7-(2-fluoro-6-hydroxy-3-methylphenyl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C[C@@H](C)OC=1N=C(C2=C(N=1)CN(CC2)C1=C(C(=CC=C1O)C)F)N1CCN(CC1)C(C=C)=O)C NFPNAXMQDZEIMQ-GOSISDBHSA-N 0.000 description 1
- MXCBINHGTLZQRA-LJQANCHMSA-N 1-[4-[2-[(2R)-1-(dimethylamino)propan-2-yl]oxy-7-(2-hydroxy-6-methylphenyl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C[C@@H](C)OC=1N=C(C2=C(N=1)CN(CC2)C1=C(C=CC=C1C)O)N1CCN(CC1)C(C=C)=O)C MXCBINHGTLZQRA-LJQANCHMSA-N 0.000 description 1
- SUKVBTFCFPHCAL-GOSISDBHSA-N 1-[4-[2-[(2R)-1-(dimethylamino)propan-2-yl]oxy-7-(3-fluoro-2-hydroxy-6-methylphenyl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C[C@@H](C)OC=1N=C(C2=C(N=1)CN(CC2)C1=C(C(=CC=C1C)F)O)N1CCN(CC1)C(C=C)=O)C SUKVBTFCFPHCAL-GOSISDBHSA-N 0.000 description 1
- AIOXZPSWYRPAGS-HXUWFJFHSA-N 1-[4-[2-[(2R)-1-(dimethylamino)propan-2-yl]oxy-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C[C@@H](C)OC=1N=C(C2=C(N=1)CN(CC2)C1=CC(=CC2=CC=CC=C12)O)N1CCN(CC1)C(C=C)=O)C AIOXZPSWYRPAGS-HXUWFJFHSA-N 0.000 description 1
- DTDPQRCJWMTDDB-GOSISDBHSA-N 1-[4-[2-[(2R)-1-(dimethylamino)propan-2-yl]oxy-7-(5-hydroxy-2-methoxyphenyl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C[C@@H](C)OC=1N=C(C2=C(N=1)CN(CC2)C1=C(C=CC(=C1)O)OC)N1CCN(CC1)C(C=C)=O)C DTDPQRCJWMTDDB-GOSISDBHSA-N 0.000 description 1
- WCIRKTPAOWPLRN-QGZVFWFLSA-N 1-[4-[2-[(2R)-1-(dimethylamino)propan-2-yl]oxy-7-[2-hydroxy-5-(trifluoromethoxy)phenyl]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C[C@@H](C)OC=1N=C(C2=C(N=1)CN(CC2)C1=C(C=CC(=C1)OC(F)(F)F)O)N1CCN(CC1)C(C=C)=O)C WCIRKTPAOWPLRN-QGZVFWFLSA-N 0.000 description 1
- SOSYTWKYZIABBW-QGZVFWFLSA-N 1-[4-[2-[(2R)-1-(dimethylamino)propan-2-yl]oxy-7-[3-hydroxy-5-(trifluoromethoxy)phenyl]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C[C@@H](C)OC=1N=C(C2=C(N=1)CN(CC2)C1=CC(=CC(=C1)OC(F)(F)F)O)N1CCN(CC1)C(C=C)=O)C SOSYTWKYZIABBW-QGZVFWFLSA-N 0.000 description 1
- WPQJJNMAMQZNCD-QGZVFWFLSA-N 1-[4-[2-[(2R)-1-(dimethylamino)propan-2-yl]oxy-7-[5-hydroxy-2-(trifluoromethoxy)phenyl]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C[C@@H](C)OC=1N=C(C2=C(N=1)CN(CC2)C1=C(C=CC(=C1)O)OC(F)(F)F)N1CCN(CC1)C(C=C)=O)C WPQJJNMAMQZNCD-QGZVFWFLSA-N 0.000 description 1
- UWPPBRCDMFCREX-QGZVFWFLSA-N 1-[4-[2-[(2R)-1-(dimethylamino)propan-2-yl]oxy-7-[5-hydroxy-2-(trifluoromethyl)phenyl]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C[C@@H](C)OC=1N=C(C2=C(N=1)CN(CC2)C1=C(C=CC(=C1)O)C(F)(F)F)N1CCN(CC1)C(C=C)=O)C UWPPBRCDMFCREX-QGZVFWFLSA-N 0.000 description 1
- LZMSWHMJIWTHEM-OAQYLSRUSA-N 1-[4-[2-[(2R)-1-(dimethylamino)propan-2-yl]oxy-7-naphthalen-1-yl-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C[C@@H](C)OC=1N=C(C2=C(N=1)CN(CC2)C1=CC=CC2=CC=CC=C12)N1CCN(CC1)C(C=C)=O)C LZMSWHMJIWTHEM-OAQYLSRUSA-N 0.000 description 1
- AIOXZPSWYRPAGS-UHFFFAOYSA-N 1-[4-[2-[1-(dimethylamino)propan-2-yloxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CC(CN(C)C)OC1=NC(N2CCN(CC2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 AIOXZPSWYRPAGS-UHFFFAOYSA-N 0.000 description 1
- LZMSWHMJIWTHEM-UHFFFAOYSA-N 1-[4-[2-[1-(dimethylamino)propan-2-yloxy]-7-naphthalen-1-yl-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CC(CN(C)C)OC1=NC(N2CCN(CC2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC=CC2=CC=CC=C12 LZMSWHMJIWTHEM-UHFFFAOYSA-N 0.000 description 1
- QUBJTDNUPHYUSE-UHFFFAOYSA-N 1-[4-[2-[2-(4,4-difluoropiperidin-1-yl)ethoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound FC1(CCN(CC1)CCOC=1N=C(C2=C(N=1)CN(CC2)C1=CC(=CC2=CC=CC=C12)O)N1CCN(CC1)C(C=C)=O)F QUBJTDNUPHYUSE-UHFFFAOYSA-N 0.000 description 1
- ZVUCUPXRODDVIK-UHFFFAOYSA-N 1-[4-[2-[2-(dimethylamino)ethoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C)CCOC1=NC(N2CCN(CC2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 ZVUCUPXRODDVIK-UHFFFAOYSA-N 0.000 description 1
- IKFZSTYPQSVHFI-UHFFFAOYSA-N 1-[4-[2-[2-(dimethylamino)propoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CC(COC1=NC(N2CCN(CC2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12)N(C)C IKFZSTYPQSVHFI-UHFFFAOYSA-N 0.000 description 1
- DTHPPVAPSRNELH-VWNXMTODSA-N 1-[4-[2-[2-[(1S,4R)-2-azabicyclo[2.2.1]heptan-2-yl]ethoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound [C@H]12N(C[C@H](CC1)C2)CCOC=1N=C(C2=C(N=1)CN(CC2)C1=CC(=CC2=CC=CC=C12)O)N1CCN(CC1)C(C=C)=O DTHPPVAPSRNELH-VWNXMTODSA-N 0.000 description 1
- BOYBAZKNDIQEJS-QHCPKHFHSA-N 1-[4-[2-[2-[(3S)-3-fluoropiperidin-1-yl]ethoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound F[C@@H]1CN(CCC1)CCOC=1N=C(C2=C(N=1)CN(CC2)C1=CC(=CC2=CC=CC=C12)O)N1CCN(CC1)C(C=C)=O BOYBAZKNDIQEJS-QHCPKHFHSA-N 0.000 description 1
- UBBQQDHHIYBCDA-UHFFFAOYSA-N 1-[4-[2-[2-[2-hydroxyethyl(methyl)amino]ethoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN(CCO)CCOC1=NC(N2CCN(CC2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 UBBQQDHHIYBCDA-UHFFFAOYSA-N 0.000 description 1
- IFWMLLQPXHGSOE-UHFFFAOYSA-N 1-[4-[2-[3-(dimethylamino)propoxy]-7-(1-phenylethyl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CC(N1CCC2=C(N=C(OCCCN(C)C)N=C2C1)N1CCN(CC1)C(=O)C=C)C1=CC=CC=C1 IFWMLLQPXHGSOE-UHFFFAOYSA-N 0.000 description 1
- QPPKEDSKOLGNPO-UHFFFAOYSA-N 1-[4-[2-[3-(dimethylamino)propoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C)CCCOC1=NC(N2CCN(CC2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 QPPKEDSKOLGNPO-UHFFFAOYSA-N 0.000 description 1
- WWSIROQHPYLSMH-UHFFFAOYSA-N 1-[4-[2-[3-(dimethylamino)pyrrolidin-1-yl]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C)C1CCN(C1)C1=NC(N2CCN(CC2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 WWSIROQHPYLSMH-UHFFFAOYSA-N 0.000 description 1
- GXQXVZGHUXJGRL-UHFFFAOYSA-N 1-[4-[2-[4-(2-hydroxyethyl)piperazin-1-yl]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OCCN1CCN(CC1)C1=NC(N2CCN(CC2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 GXQXVZGHUXJGRL-UHFFFAOYSA-N 0.000 description 1
- JVNKREXLFWUOMF-UHFFFAOYSA-N 1-[4-[2-[4-(dimethylamino)butan-2-yloxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CC(CCN(C)C)OC1=NC(N2CCN(CC2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 JVNKREXLFWUOMF-UHFFFAOYSA-N 0.000 description 1
- WOHIBBMEDFUEAK-UHFFFAOYSA-N 1-[4-[2-[4-(dimethylamino)butoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C)CCCCOC1=NC(N2CCN(CC2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 WOHIBBMEDFUEAK-UHFFFAOYSA-N 0.000 description 1
- BGUACLKBQDMRJH-UHFFFAOYSA-N 1-[4-[2-[4-(dimethylamino)piperidin-1-yl]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN(C)C1CCN(CC1)C1=NC(N2CCN(CC2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 BGUACLKBQDMRJH-UHFFFAOYSA-N 0.000 description 1
- ZLQOTAPAPGJSRO-UHFFFAOYSA-N 1-[4-[7-(2-fluoro-5-hydroxyphenyl)-2-(3-morpholin-4-ylpropoxy)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound FC1=C(C=C(C=C1)O)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)OCCCN1CCOCC1 ZLQOTAPAPGJSRO-UHFFFAOYSA-N 0.000 description 1
- RHNSGICIYGVTJK-LJQANCHMSA-N 1-[4-[7-(3-chloro-5-hydroxy-2-propan-2-ylphenyl)-2-[(2R)-1-(dimethylamino)propan-2-yl]oxy-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound ClC=1C(=C(C=C(C=1)O)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)O[C@@H](CN(C)C)C)C(C)C RHNSGICIYGVTJK-LJQANCHMSA-N 0.000 description 1
- WSOUHNAMKFWAIC-UHFFFAOYSA-N 1-[4-[7-(3-cyclopropyl-5-hydroxyphenyl)-2-(3-morpholin-4-ylpropoxy)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound C1(CC1)C=1C=C(C=C(C=1)O)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)OCCCN1CCOCC1 WSOUHNAMKFWAIC-UHFFFAOYSA-N 0.000 description 1
- WOQBMVXROKWMJO-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-(1-methylpiperidin-4-yl)oxy-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN1CCC(CC1)OC1=NC(N2CCN(CC2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 WOQBMVXROKWMJO-UHFFFAOYSA-N 0.000 description 1
- QDNHOCIZPCTYDD-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-(1-methylpyrrolidin-3-yl)oxy-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN1CCC(C1)OC1=NC(N2CCN(CC2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 QDNHOCIZPCTYDD-UHFFFAOYSA-N 0.000 description 1
- UWXDMLVRDNIRHM-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-(2-morpholin-4-ylethoxy)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)OCCN1CCOCC1 UWXDMLVRDNIRHM-UHFFFAOYSA-N 0.000 description 1
- KLIBMJNYSIEHCT-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-(2-pyrrolidin-1-ylethoxy)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC1=CC2=CC=CC=C2C(=C1)N1CCC2=C(N=C(OCCN3CCCC3)N=C2C1)N1CCN(CC1)C(=O)C=C KLIBMJNYSIEHCT-UHFFFAOYSA-N 0.000 description 1
- AAHIRNJSUQBORR-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-(3-morpholin-4-ylpropoxy)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC1=CC2=CC=CC=C2C(=C1)N1CCC2=C(N=C(OCCCN3CCOCC3)N=C2C1)N1CCN(CC1)C(=O)C=C AAHIRNJSUQBORR-UHFFFAOYSA-N 0.000 description 1
- BZCRIUUMCWXNQL-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-(3-piperidin-1-ylpropoxy)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC1=CC2=CC=CC=C2C(=C1)N1CCC2=C(N=C(OCCCN3CCCCC3)N=C2C1)N1CCN(CC1)C(=O)C=C BZCRIUUMCWXNQL-UHFFFAOYSA-N 0.000 description 1
- VHRAKZQQSSBVDZ-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-(3-pyrrolidin-1-ylpropoxy)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC1=CC2=CC=CC=C2C(=C1)N1CCC2=C(N=C(OCCCN3CCCC3)N=C2C1)N1CCN(CC1)C(=O)C=C VHRAKZQQSSBVDZ-UHFFFAOYSA-N 0.000 description 1
- DSYUMENLSNFCAG-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-(4-methylpiperazin-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN1CCN(CC1)C1=NC(N2CCN(CC2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 DSYUMENLSNFCAG-UHFFFAOYSA-N 0.000 description 1
- QRIJXZNQAKMJGF-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-(4-morpholin-4-ylbutan-2-yloxy)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CC(CCN1CCOCC1)OC1=NC(N2CCN(CC2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 QRIJXZNQAKMJGF-UHFFFAOYSA-N 0.000 description 1
- WFNFZTVVDLJPLF-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[(1-methylpiperidin-3-yl)methoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN1CCCC(COC2=NC(N3CCN(CC3)C(=O)C=C)=C3CCN(CC3=N2)C2=CC(O)=CC3=CC=CC=C23)C1 WFNFZTVVDLJPLF-UHFFFAOYSA-N 0.000 description 1
- ZBVLKVUSYQNKNE-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[(1-methylpyrrolidin-3-yl)methoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN1CCC(COC2=NC(N3CCN(CC3)C(=O)C=C)=C3CCN(CC3=N2)C2=CC(O)=CC3=CC=CC=C23)C1 ZBVLKVUSYQNKNE-UHFFFAOYSA-N 0.000 description 1
- GRTHSRDCCXJMFV-RCZVLFRGSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[(2R)-1-[(3R)-3-methoxypyrrolidin-1-yl]propan-2-yl]oxy-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)O[C@@H](CN1C[C@@H](CC1)OC)C GRTHSRDCCXJMFV-RCZVLFRGSA-N 0.000 description 1
- VQDADQAGFAHIJS-GGAORHGYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[(2R)-1-[(3S)-3-hydroxypyrrolidin-1-yl]propan-2-yl]oxy-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)O[C@@H](CN1C[C@H](CC1)O)C VQDADQAGFAHIJS-GGAORHGYSA-N 0.000 description 1
- GRTHSRDCCXJMFV-RDGATRHJSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[(2R)-1-[(3S)-3-methoxypyrrolidin-1-yl]propan-2-yl]oxy-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)O[C@@H](CN1C[C@H](CC1)OC)C GRTHSRDCCXJMFV-RDGATRHJSA-N 0.000 description 1
- IDRONOJNWJIWIY-JOCHJYFZSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[(2R)-1-morpholin-4-ylpropan-2-yl]oxy-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)O[C@@H](CN1CCOCC1)C IDRONOJNWJIWIY-JOCHJYFZSA-N 0.000 description 1
- CKNLWIAKBOEDQQ-HSZRJFAPSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[(2R)-1-piperidin-1-ylpropan-2-yl]oxy-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)O[C@@H](CN1CCCCC1)C CKNLWIAKBOEDQQ-HSZRJFAPSA-N 0.000 description 1
- DBOZGCRCLRUQMO-JOCHJYFZSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[(2R)-1-pyrrolidin-1-ylpropan-2-yl]oxy-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)O[C@@H](CN1CCCC1)C DBOZGCRCLRUQMO-JOCHJYFZSA-N 0.000 description 1
- AVLLHXGXKWXQTB-HSZRJFAPSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[(3R)-1-methylpiperidin-3-yl]oxy-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)O[C@H]1CN(CCC1)C AVLLHXGXKWXQTB-HSZRJFAPSA-N 0.000 description 1
- AVLLHXGXKWXQTB-QHCPKHFHSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[(3S)-1-methylpiperidin-3-yl]oxy-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)O[C@@H]1CN(CCC1)C AVLLHXGXKWXQTB-QHCPKHFHSA-N 0.000 description 1
- OKFWGRIHKACNBC-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[(4-methylpiperazin-2-yl)methoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CN1CCNC(COC2=NC(N3CCN(CC3)C(=O)C=C)=C3CCN(CC3=N2)C2=CC(O)=CC3=CC=CC=C23)C1 OKFWGRIHKACNBC-UHFFFAOYSA-N 0.000 description 1
- ACKGVYBHFKYNLX-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[1-(methylamino)propan-2-yloxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CNCC(C)OC1=NC(N2CCN(CC2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 ACKGVYBHFKYNLX-UHFFFAOYSA-N 0.000 description 1
- NCVJYJMGFBJCIE-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[2-(4-methoxypiperidin-1-yl)ethoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound COC1CCN(CCOC2=NC(N3CCN(CC3)C(=O)C=C)=C3CCN(CC3=N2)C2=CC(O)=CC3=CC=CC=C23)CC1 NCVJYJMGFBJCIE-UHFFFAOYSA-N 0.000 description 1
- PBBMAGLWCNIHCM-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[2-(4-methylpiperidin-1-yl)ethoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CC1CCN(CCOC2=NC(N3CCN(CC3)C(=O)C=C)=C3CCN(CC3=N2)C2=CC(O)=CC3=CC=CC=C23)CC1 PBBMAGLWCNIHCM-UHFFFAOYSA-N 0.000 description 1
- QEVJNXDDXBTWPN-HSZRJFAPSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[2-[(2R)-2-methylpiperidin-1-yl]ethoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)OCCN1[C@@H](CCCC1)C QEVJNXDDXBTWPN-HSZRJFAPSA-N 0.000 description 1
- QEVJNXDDXBTWPN-QHCPKHFHSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[2-[(2S)-2-methylpiperidin-1-yl]ethoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)OCCN1[C@H](CCCC1)C QEVJNXDDXBTWPN-QHCPKHFHSA-N 0.000 description 1
- UHJXDCBFTJHYNO-RUZDIDTESA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[2-[(3R)-3-methoxypiperidin-1-yl]ethoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)OCCN1C[C@@H](CCC1)OC UHJXDCBFTJHYNO-RUZDIDTESA-N 0.000 description 1
- UHJXDCBFTJHYNO-VWLOTQADSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[2-[(3S)-3-methoxypiperidin-1-yl]ethoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)OCCN1C[C@H](CCC1)OC UHJXDCBFTJHYNO-VWLOTQADSA-N 0.000 description 1
- MORPKTKVMIFGJO-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[2-[2-methoxyethyl(methyl)amino]ethoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound COCCN(C)CCOC1=NC(N2CCN(CC2)C(=O)C=C)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 MORPKTKVMIFGJO-UHFFFAOYSA-N 0.000 description 1
- BBJWHIGVUPBRCL-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[3-(4-methylpiperazin-1-yl)propoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)OCCCN1CCN(CC1)C BBJWHIGVUPBRCL-UHFFFAOYSA-N 0.000 description 1
- SLUOREMWTGQPRL-ZCYQVOJMSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[3-[(1S,4S)-2-oxa-5-azabicyclo[2.2.1]heptan-5-yl]propoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound Oc1cc(N2CCc3c(C2)nc(OCCCN2C[C@@H]4C[C@H]2CO4)nc3N2CCN(CC2)C(=O)C=C)c2ccccc2c1 SLUOREMWTGQPRL-ZCYQVOJMSA-N 0.000 description 1
- ZBVLKVUSYQNKNE-OAQYLSRUSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[[(3R)-1-methylpyrrolidin-3-yl]methoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)OC[C@H]1CN(CC1)C ZBVLKVUSYQNKNE-OAQYLSRUSA-N 0.000 description 1
- ZTSXRXMYUSGKMY-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-[[1-(2-methoxyethyl)pyrrolidin-3-yl]methoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound COCCN1CCC(COC2=NC(N3CCN(CC3)C(=O)C=C)=C3CCN(CC3=N2)C2=CC(O)=CC3=CC=CC=C23)C1 ZTSXRXMYUSGKMY-UHFFFAOYSA-N 0.000 description 1
- PCWSYLYFMNISPZ-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-2-pyrrolidin-1-yl-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC1=CC2=CC=CC=C2C(=C1)N1CCC2=C(N=C(N=C2C1)N1CCCC1)N1CCN(CC1)C(=O)C=C PCWSYLYFMNISPZ-UHFFFAOYSA-N 0.000 description 1
- GCXNCPMYSDEQIW-UHFFFAOYSA-N 1-[4-[7-(3-hydroxynaphthalen-1-yl)-6-methyl-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CC1CC2=C(N=CN=C2CN1C1=CC(O)=CC2=CC=CC=C12)N1CCN(CC1)C(=O)C=C GCXNCPMYSDEQIW-UHFFFAOYSA-N 0.000 description 1
- AACOLSIYDRCUEU-UHFFFAOYSA-N 1-[4-[7-(5-hydroxy-2-methylphenyl)-2-(3-morpholin-4-ylpropoxy)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CC1=C(C=C(O)C=C1)N1CCC2=C(N=C(OCCCN3CCOCC3)N=C2C1)N1CCN(CC1)C(=O)C=C AACOLSIYDRCUEU-UHFFFAOYSA-N 0.000 description 1
- XUFZGCPFKOUIOS-UHFFFAOYSA-N 1-[4-[7-(5-hydroxy-2-propan-2-yloxyphenyl)-2-(3-morpholin-4-ylpropoxy)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound CC(C)OC1=C(C=C(O)C=C1)N1CCC2=C(N=C(OCCCN3CCOCC3)N=C2C1)N1CCN(CC1)C(=O)C=C XUFZGCPFKOUIOS-UHFFFAOYSA-N 0.000 description 1
- DYDGZGNMPPKXRR-UHFFFAOYSA-N 1-[4-[7-[3-(difluoromethyl)naphthalen-1-yl]-2-(3-morpholin-4-ylpropoxy)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound FC(C=1C=C(C2=CC=CC=C2C=1)N1CC=2N=C(N=C(C=2CC1)N1CCN(CC1)C(C=C)=O)OCCCN1CCOCC1)F DYDGZGNMPPKXRR-UHFFFAOYSA-N 0.000 description 1
- LGMBXTMYDBOVAC-UHFFFAOYSA-N 1-[4-[7-[3-(methoxymethoxy)naphthalen-1-yl]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound COCOC1=CC2=CC=CC=C2C(=C1)N1CCC2=C(N=CN=C2C1)N1CCN(CC1)C(=O)C=C LGMBXTMYDBOVAC-UHFFFAOYSA-N 0.000 description 1
- KRFSQMKPZNYMOC-UHFFFAOYSA-N 1-[4-[7-[4-hydroxy-2-(trifluoromethoxy)phenyl]-2-(3-morpholin-4-ylpropoxy)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC1=CC(OC(F)(F)F)=C(C=C1)N1CCC2=C(N=C(OCCCN3CCOCC3)N=C2C1)N1CCN(CC1)C(=O)C=C KRFSQMKPZNYMOC-UHFFFAOYSA-N 0.000 description 1
- ARXOTLODMMZPHA-UHFFFAOYSA-N 1-[4-[7-[5-hydroxy-2-(trifluoromethoxy)phenyl]-2-(3-morpholin-4-ylpropoxy)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-1-yl]prop-2-en-1-one Chemical compound OC1=CC(N2CCC3=C(N=C(OCCCN4CCOCC4)N=C3C2)N2CCN(CC2)C(=O)C=C)=C(OC(F)(F)F)C=C1 ARXOTLODMMZPHA-UHFFFAOYSA-N 0.000 description 1
- YHXAHXDDTUFBRW-UHFFFAOYSA-N 1-[6-[2-[1-(dimethylamino)propan-2-yloxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]-2,6-diazaspiro[3.3]heptan-2-yl]prop-2-en-1-one Chemical compound CC(CN(C)C)OC1=NC(N2CC3(CN(C3)C(=O)C=C)C2)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 YHXAHXDDTUFBRW-UHFFFAOYSA-N 0.000 description 1
- DPBVPJWOAKYVAI-UHFFFAOYSA-N 1-[6-[2-[2-(dimethylamino)ethoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]-2,6-diazaspiro[3.3]heptan-2-yl]prop-2-en-1-one Chemical compound CN(C)CCOC1=NC(N2CC3(CN(C3)C(=O)C=C)C2)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 DPBVPJWOAKYVAI-UHFFFAOYSA-N 0.000 description 1
- OEXLBECXWZNADQ-UHFFFAOYSA-N 1-[6-[2-[3-(dimethylamino)propoxy]-7-(3-hydroxynaphthalen-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]-2,6-diazaspiro[3.3]heptan-2-yl]prop-2-en-1-one Chemical compound CN(C)CCCOC1=NC(N2CC3(CN(C3)C(=O)C=C)C2)=C2CCN(CC2=N1)C1=CC(O)=CC2=CC=CC=C12 OEXLBECXWZNADQ-UHFFFAOYSA-N 0.000 description 1
- OGUZXGRELSKKNO-UHFFFAOYSA-N 1-bromo-3-(methoxymethoxy)-5-(trifluoromethoxy)benzene Chemical compound BrC=1C=C(C=C(C=1)OC(F)(F)F)OCOC OGUZXGRELSKKNO-UHFFFAOYSA-N 0.000 description 1
- LGJMZAILODFIIA-UHFFFAOYSA-N 1-bromo-3-chloro-2-fluoro-5-(methoxymethoxy)benzene Chemical compound COCOC1=CC(Br)=C(F)C(Cl)=C1 LGJMZAILODFIIA-UHFFFAOYSA-N 0.000 description 1
- JPSIQDABPYASGV-UHFFFAOYSA-N 1-bromo-3-chloro-5-methoxy-2-propan-2-ylbenzene Chemical compound COC1=CC(Br)=C(C(C)C)C(Cl)=C1 JPSIQDABPYASGV-UHFFFAOYSA-N 0.000 description 1
- MKMVNGNAFLOEDW-UHFFFAOYSA-N 1-bromo-4-(methoxymethoxy)-2-(trifluoromethoxy)benzene Chemical compound BrC1=C(C=C(C=C1)OCOC)OC(F)(F)F MKMVNGNAFLOEDW-UHFFFAOYSA-N 0.000 description 1
- PSQUIUNIVDKHJK-UHFFFAOYSA-N 1-bromoisoquinolin-3-amine Chemical compound C1=CC=C2C(Br)=NC(N)=CC2=C1 PSQUIUNIVDKHJK-UHFFFAOYSA-N 0.000 description 1
- CAYTVMPUNMZTFU-UHFFFAOYSA-N 1-cyclopropylpiperidin-4-ol Chemical compound C1CC(O)CCN1C1CC1 CAYTVMPUNMZTFU-UHFFFAOYSA-N 0.000 description 1
- BAUWRHPMUVYFOD-UHFFFAOYSA-N 1-methylpiperidin-4-ol Chemical compound CN1CCC(O)CC1 BAUWRHPMUVYFOD-UHFFFAOYSA-N 0.000 description 1
- FLVFPAIGVBQGET-UHFFFAOYSA-N 1-methylpyrrolidin-3-ol Chemical compound CN1CCC(O)C1 FLVFPAIGVBQGET-UHFFFAOYSA-N 0.000 description 1
- PKDPUENCROCRCH-UHFFFAOYSA-N 1-piperazin-1-ylethanone Chemical compound CC(=O)N1CCNCC1 PKDPUENCROCRCH-UHFFFAOYSA-N 0.000 description 1
- PGEJXJAICLMXCF-UHFFFAOYSA-N 1-tert-butyl-3-methylpiperazine Chemical compound CC1CN(C(C)(C)C)CCN1 PGEJXJAICLMXCF-UHFFFAOYSA-N 0.000 description 1
- 125000005955 1H-indazolyl group Chemical group 0.000 description 1
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 1
- PYHXGXCGESYPCW-UHFFFAOYSA-M 2,2-diphenylacetate Chemical compound C=1C=CC=CC=1C(C(=O)[O-])C1=CC=CC=C1 PYHXGXCGESYPCW-UHFFFAOYSA-M 0.000 description 1
- GBBSAMQTQCPOBF-UHFFFAOYSA-N 2,4,6-trimethyl-1,3,5,2,4,6-trioxatriborinane Chemical compound CB1OB(C)OB(C)O1 GBBSAMQTQCPOBF-UHFFFAOYSA-N 0.000 description 1
- BPXKZEMBEZGUAH-UHFFFAOYSA-N 2-(chloromethoxy)ethyl-trimethylsilane Chemical compound C[Si](C)(C)CCOCCl BPXKZEMBEZGUAH-UHFFFAOYSA-N 0.000 description 1
- PBKGYWLWIJLDGZ-UHFFFAOYSA-N 2-(dimethylamino)propan-1-ol Chemical compound OCC(C)N(C)C PBKGYWLWIJLDGZ-UHFFFAOYSA-N 0.000 description 1
- KKFDCBRMNNSAAW-UHFFFAOYSA-N 2-(morpholin-4-yl)ethanol Chemical compound OCCN1CCOCC1 KKFDCBRMNNSAAW-UHFFFAOYSA-N 0.000 description 1
- KZTWONRVIPPDKH-UHFFFAOYSA-N 2-(piperidin-1-yl)ethanol Chemical compound OCCN1CCCCC1 KZTWONRVIPPDKH-UHFFFAOYSA-N 0.000 description 1
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 1
- WEQFJQFMEXXVHW-VXKWHMMOSA-N 2-[(2S)-4-[7-(2,3-dimethylphenyl)-2-[[(2S)-1-methylpyrrolidin-2-yl]methoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-2-yl]acetonitrile Chemical compound CC1=C(C=CC=C1C)N1CC=2N=C(N=C(C=2CC1)N1C[C@@H](NCC1)CC#N)OC[C@H]1N(CCC1)C WEQFJQFMEXXVHW-VXKWHMMOSA-N 0.000 description 1
- TVKAZNBVXZKTAV-LURJTMIESA-N 2-[(2s)-piperazin-2-yl]acetonitrile Chemical compound N#CC[C@H]1CNCCN1 TVKAZNBVXZKTAV-LURJTMIESA-N 0.000 description 1
- ITSAIKUTLAJVEH-QMMMGPOBSA-N 2-[(3s)-3-methoxypiperidin-1-yl]ethanol Chemical compound CO[C@H]1CCCN(CCO)C1 ITSAIKUTLAJVEH-QMMMGPOBSA-N 0.000 description 1
- IQHSSYROJYPFDV-UHFFFAOYSA-N 2-bromo-1,3-dichloro-5-(trifluoromethyl)benzene Chemical group FC(F)(F)C1=CC(Cl)=C(Br)C(Cl)=C1 IQHSSYROJYPFDV-UHFFFAOYSA-N 0.000 description 1
- JGYLZHXKSQNMJK-UHFFFAOYSA-N 2-bromo-1-(methoxymethoxy)-3-methylbenzene Chemical compound BrC1=C(C=CC=C1C)OCOC JGYLZHXKSQNMJK-UHFFFAOYSA-N 0.000 description 1
- YCSFDGILOXOHDT-UHFFFAOYSA-N 2-bromo-1-(methoxymethoxy)-4-(trifluoromethoxy)benzene Chemical compound COCOC1=CC=C(OC(F)(F)F)C=C1Br YCSFDGILOXOHDT-UHFFFAOYSA-N 0.000 description 1
- FGZZCKKGBRODEE-UHFFFAOYSA-N 2-bromo-1-methoxy-4-(methoxymethoxy)benzene Chemical compound BrC=1C=C(C=CC=1OC)OCOC FGZZCKKGBRODEE-UHFFFAOYSA-N 0.000 description 1
- NFQOGCOXXVIIPN-UHFFFAOYSA-N 2-bromo-4-(methoxymethoxy)-1-(trifluoromethyl)benzene Chemical compound BrC1=C(C=CC(=C1)OCOC)C(F)(F)F NFQOGCOXXVIIPN-UHFFFAOYSA-N 0.000 description 1
- WCUKHLZDZKLPEQ-UHFFFAOYSA-N 2-bromo-4-(methoxymethoxy)-1-methylbenzene Chemical compound COCOC1=CC=C(C)C(Br)=C1 WCUKHLZDZKLPEQ-UHFFFAOYSA-N 0.000 description 1
- LDLCZOVUSADOIV-UHFFFAOYSA-N 2-bromoethanol Chemical compound OCCBr LDLCZOVUSADOIV-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M 2-methylbenzenesulfonate Chemical compound CC1=CC=CC=C1S([O-])(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- TVKAZNBVXZKTAV-UHFFFAOYSA-N 2-piperazin-2-ylacetonitrile Chemical compound N#CCC1CNCCN1 TVKAZNBVXZKTAV-UHFFFAOYSA-N 0.000 description 1
- PZFMWYNHJFZBPO-UHFFFAOYSA-N 3,5-dibromophenol Chemical compound OC1=CC(Br)=CC(Br)=C1 PZFMWYNHJFZBPO-UHFFFAOYSA-N 0.000 description 1
- VAJSFWFPXNWBLF-UHFFFAOYSA-N 3-(1,1-dioxo-1,4-thiazinan-4-yl)propan-1-ol Chemical compound OCCCN1CCS(=O)(=O)CC1 VAJSFWFPXNWBLF-UHFFFAOYSA-N 0.000 description 1
- JKRSQNBRNIYETC-UHFFFAOYSA-N 3-(4-methylpiperazin-1-yl)propan-1-ol Chemical compound CN1CCN(CCCO)CC1 JKRSQNBRNIYETC-UHFFFAOYSA-N 0.000 description 1
- RQFUZUMFPRMVDX-UHFFFAOYSA-N 3-Bromo-1-propanol Chemical compound OCCCBr RQFUZUMFPRMVDX-UHFFFAOYSA-N 0.000 description 1
- VTQQZFBVMNEQER-YUMQZZPRSA-N 3-[(1S,4S)-2-oxa-5-azabicyclo[2.2.1]heptan-5-yl]propan-1-ol Chemical compound OCCCN1C[C@@H]2C[C@H]1CO2 VTQQZFBVMNEQER-YUMQZZPRSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- JFESZIXBBOIKBD-UHFFFAOYSA-N 3-bromo-1-fluoro-2-(methoxymethoxy)-4-methylbenzene Chemical compound BrC1=C(C=CC(=C1OCOC)F)C JFESZIXBBOIKBD-UHFFFAOYSA-N 0.000 description 1
- YBLPEACXXWQZDV-UHFFFAOYSA-N 3-bromo-5-chloro-4-fluorophenol Chemical compound OC1=CC(Cl)=C(F)C(Br)=C1 YBLPEACXXWQZDV-UHFFFAOYSA-N 0.000 description 1
- KMWIGSRINIFAKJ-UHFFFAOYSA-N 3-cyclopropyl-5-[2-(3-morpholin-4-ylpropoxy)-4-piperazin-1-yl-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-7-yl]phenol Chemical compound C1(CC1)C=1C=C(C=C(C=1)N1CC=2N=C(N=C(C=2CC1)N1CCNCC1)OCCCN1CCOCC1)O KMWIGSRINIFAKJ-UHFFFAOYSA-N 0.000 description 1
- GJOOCAXPERKNMN-UHFFFAOYSA-N 3-fluoro-4-methylphenol Chemical compound CC1=CC=C(O)C=C1F GJOOCAXPERKNMN-UHFFFAOYSA-N 0.000 description 1
- PLRXAFVBCHEMGD-UHFFFAOYSA-N 3-piperidin-1-ylpropan-1-ol Chemical compound OCCCN1CCCCC1 PLRXAFVBCHEMGD-UHFFFAOYSA-N 0.000 description 1
- ZMJQROKRSPSLFH-UHFFFAOYSA-N 3-pyrrolidin-1-ylpropan-1-ol Chemical compound OCCCN1CCCC1 ZMJQROKRSPSLFH-UHFFFAOYSA-N 0.000 description 1
- OABUKBBBSMNNPM-UHFFFAOYSA-N 4,4-difluoropiperidin-1-ium;chloride Chemical compound Cl.FC1(F)CCNCC1 OABUKBBBSMNNPM-UHFFFAOYSA-N 0.000 description 1
- QCTOLMMTYSGTDA-UHFFFAOYSA-N 4-(dimethylamino)butan-1-ol Chemical compound CN(C)CCCCO QCTOLMMTYSGTDA-UHFFFAOYSA-N 0.000 description 1
- IXAXPXLEJPTDGO-UHFFFAOYSA-N 4-(dimethylamino)butan-2-ol Chemical compound CC(O)CCN(C)C IXAXPXLEJPTDGO-UHFFFAOYSA-N 0.000 description 1
- SVLVUKIUCWOZEG-UHFFFAOYSA-N 4-[4-(4-hydroxypiperazin-1-yl)-6-methyl-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-7-yl]naphthalen-2-ol Chemical compound OC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=CN=C(C=2CC1C)N1CCN(CC1)O SVLVUKIUCWOZEG-UHFFFAOYSA-N 0.000 description 1
- GAMYYCRTACQSBR-UHFFFAOYSA-N 4-azabenzimidazole Chemical group C1=CC=C2NC=NC2=N1 GAMYYCRTACQSBR-UHFFFAOYSA-N 0.000 description 1
- RYXRJVPOPIYYCM-UHFFFAOYSA-N 4-bromo-1-(oxan-2-yl)-5-(trifluoromethyl)indazole Chemical compound BrC1=C2C=NN(C2=CC=C1C(F)(F)F)C1OCCCC1 RYXRJVPOPIYYCM-UHFFFAOYSA-N 0.000 description 1
- TUTWVEKYKXGGIB-UHFFFAOYSA-N 4-bromo-5-(trifluoromethyl)-1h-indazole Chemical compound FC(F)(F)C1=CC=C2NN=CC2=C1Br TUTWVEKYKXGGIB-UHFFFAOYSA-N 0.000 description 1
- SCRBSGZBTHKAHU-UHFFFAOYSA-N 4-bromoisoquinoline Chemical compound C1=CC=C2C(Br)=CN=CC2=C1 SCRBSGZBTHKAHU-UHFFFAOYSA-N 0.000 description 1
- IADXUPHVLCMZFX-UHFFFAOYSA-N 4-chloro-6,8-dihydro-5h-pyrido[3,4-d]pyrimidine-7-carboxylic acid Chemical compound N1=CN=C2CN(C(=O)O)CCC2=C1Cl IADXUPHVLCMZFX-UHFFFAOYSA-N 0.000 description 1
- MGWFUQALPUPAER-UHFFFAOYSA-N 4-chloro-6-fluoropyrido[3,4-d]pyrimidine Chemical compound N1=CN=C2C=NC(F)=CC2=C1Cl MGWFUQALPUPAER-UHFFFAOYSA-N 0.000 description 1
- HWIHTFGVNKIPAU-UHFFFAOYSA-N 4-ethoxycarbonyl-3-oxopiperidine-1-carboxylic acid Chemical compound CCOC(=O)C1CCN(C(O)=O)CC1=O HWIHTFGVNKIPAU-UHFFFAOYSA-N 0.000 description 1
- JKINECWFYOOHFJ-UHFFFAOYSA-N 4-iodonaphthalen-2-ol Chemical compound C1=CC=CC2=CC(O)=CC(I)=C21 JKINECWFYOOHFJ-UHFFFAOYSA-N 0.000 description 1
- ZEYSHALLPAKUHG-UHFFFAOYSA-N 4-methoxypiperidine Chemical compound COC1CCNCC1 ZEYSHALLPAKUHG-UHFFFAOYSA-N 0.000 description 1
- URFFPMJFOHTCLI-UHFFFAOYSA-N 4-morpholin-4-ylbutan-1-ol Chemical compound OCCCCN1CCOCC1 URFFPMJFOHTCLI-UHFFFAOYSA-N 0.000 description 1
- RHBZAIRQNOBTNS-UHFFFAOYSA-N 4-morpholin-4-ylbutan-2-ol Chemical compound CC(O)CCN1CCOCC1 RHBZAIRQNOBTNS-UHFFFAOYSA-N 0.000 description 1
- QEYQMWSESURNPP-UHFFFAOYSA-N 4-propan-2-yloxyphenol Chemical compound CC(C)OC1=CC=C(O)C=C1 QEYQMWSESURNPP-UHFFFAOYSA-N 0.000 description 1
- TXNLQUKVUJITMX-UHFFFAOYSA-N 4-tert-butyl-2-(4-tert-butylpyridin-2-yl)pyridine Chemical compound CC(C)(C)C1=CC=NC(C=2N=CC=C(C=2)C(C)(C)C)=C1 TXNLQUKVUJITMX-UHFFFAOYSA-N 0.000 description 1
- 125000002471 4H-quinolizinyl group Chemical group C=1(C=CCN2C=CC=CC12)* 0.000 description 1
- BESUASAYUMGFOE-UHFFFAOYSA-N 7-(3-hydroxynaphthalen-1-yl)-4-(4-prop-2-enoylpiperazin-1-yl)-5,8-dihydropyrido[3,4-d]pyrimidin-6-one Chemical compound C(C=C)(=O)N1CCN(CC1)C=1C2=C(N=CN=1)CN(C(C2)=O)C1=CC(=CC2=CC=CC=C12)O BESUASAYUMGFOE-UHFFFAOYSA-N 0.000 description 1
- ZFNFNVMCQSDBIL-UHFFFAOYSA-N 7-[3-(methoxymethoxy)naphthalen-1-yl]-4-piperazin-1-yl-6,8-dihydro-5H-pyrido[3,4-d]pyrimidine Chemical compound COCOC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=CN=C(C=2CC1)N1CCNCC1 ZFNFNVMCQSDBIL-UHFFFAOYSA-N 0.000 description 1
- PDZGYMZCMUSHBP-UHFFFAOYSA-N 7-benzyl-4-chloro-2-methylsulfanyl-6,8-dihydro-5h-pyrido[3,4-d]pyrimidine Chemical compound C1C2=NC(SC)=NC(Cl)=C2CCN1CC1=CC=CC=C1 PDZGYMZCMUSHBP-UHFFFAOYSA-N 0.000 description 1
- JUWAPIFNJPKCLL-UHFFFAOYSA-N 7-benzyl-6-methyl-1,5,6,8-tetrahydropyrido[3,4-d]pyrimidine-2,4-dione Chemical compound C(C1=CC=CC=C1)N1CC=2N=C(N=C(C=2CC1C)O)O JUWAPIFNJPKCLL-UHFFFAOYSA-N 0.000 description 1
- VWSBGGRCEQOTNU-UHFFFAOYSA-N 7-bromonaphthalen-2-ol Chemical compound C1=CC(Br)=CC2=CC(O)=CC=C21 VWSBGGRCEQOTNU-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- HMFGQHOSUBMBDF-UHFFFAOYSA-N 8-bromo-6-(methoxymethoxy)quinoline Chemical compound BrC=1C=C(C=C2C=CC=NC=12)OCOC HMFGQHOSUBMBDF-UHFFFAOYSA-N 0.000 description 1
- KMPGTDGDSRRYHB-UHFFFAOYSA-N 8-bromoquinolin-6-ol Chemical compound N1=CC=CC2=CC(O)=CC(Br)=C21 KMPGTDGDSRRYHB-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- 241000349731 Afzelia bipindensis Species 0.000 description 1
- 229910015844 BCl3 Inorganic materials 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- GPJWOIADNFOEPT-KRWDZBQOSA-N C(#N)C[C@@H]1N(CCN(C1)C=1C2=C(N=C(N=1)SC)CNCC2)C(=O)OCC1=CC=CC=C1 Chemical compound C(#N)C[C@@H]1N(CCN(C1)C=1C2=C(N=C(N=1)SC)CNCC2)C(=O)OCC1=CC=CC=C1 GPJWOIADNFOEPT-KRWDZBQOSA-N 0.000 description 1
- ZBSDKTQNPQWRRO-UHFFFAOYSA-N C(C)(=O)O.C(C)(=O)O.C1(CC1)C=1C=C(C=C(C1)N1CC=2N=C(N=C(C2CC1)N1CCNCC1)OCCCN1CCOCC1)O Chemical compound C(C)(=O)O.C(C)(=O)O.C1(CC1)C=1C=C(C=C(C1)N1CC=2N=C(N=C(C2CC1)N1CCNCC1)OCCCN1CCOCC1)O ZBSDKTQNPQWRRO-UHFFFAOYSA-N 0.000 description 1
- CECSGKBDAQCYIF-UHFFFAOYSA-N C(C)(C)(C)[Si](C)(C)OC1=CC(=CC2=CC=CC=C12)OC Chemical compound C(C)(C)(C)[Si](C)(C)OC1=CC(=CC2=CC=CC=C12)OC CECSGKBDAQCYIF-UHFFFAOYSA-N 0.000 description 1
- 125000000041 C6-C10 aryl group Chemical group 0.000 description 1
- YIKNEFIMCTXUAA-UHFFFAOYSA-N COC1=CC2=CC=C(C=C2C(O[Si](C)(C)C(C)(C)C)=C1)B1OC(C)(C)C(C)(C)O1 Chemical compound COC1=CC2=CC=C(C=C2C(O[Si](C)(C)C(C)(C)C)=C1)B1OC(C)(C)C(C)(C)O1 YIKNEFIMCTXUAA-UHFFFAOYSA-N 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 206010008263 Cervical dysplasia Diseases 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 1
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 239000004593 Epoxy Substances 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- GMEONFUTDYJSNV-UHFFFAOYSA-N Ethyl levulinate Chemical compound CCOC(=O)CCC(C)=O GMEONFUTDYJSNV-UHFFFAOYSA-N 0.000 description 1
- MTFCXMJOGMHYAE-UHFFFAOYSA-N Ethyl piperazinoacetate Chemical compound CCOC(=O)CN1CCNCC1 MTFCXMJOGMHYAE-UHFFFAOYSA-N 0.000 description 1
- 235000004694 Eucalyptus leucoxylon Nutrition 0.000 description 1
- 244000166102 Eucalyptus leucoxylon Species 0.000 description 1
- PSWCNBZMJHEGKC-UHFFFAOYSA-N FC(F)C1=CC2=CC=CC=C2C(Br)=C1 Chemical compound FC(F)C1=CC2=CC=CC=C2C(Br)=C1 PSWCNBZMJHEGKC-UHFFFAOYSA-N 0.000 description 1
- 239000001263 FEMA 3042 Substances 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- 102000016285 Guanine Nucleotide Exchange Factors Human genes 0.000 description 1
- 108010067218 Guanine Nucleotide Exchange Factors Proteins 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 229910004373 HOAc Inorganic materials 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 206010069755 K-ras gene mutation Diseases 0.000 description 1
- 239000012448 Lithium borohydride Substances 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-M Methanesulfonate Chemical compound CS([O-])(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- UQXDMQDTWRKZOQ-UHFFFAOYSA-N N-[2-[[7-(3-hydroxynaphthalen-1-yl)-4-(4-prop-2-enoylpiperazin-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-2-yl]oxy]ethyl]acetamide Chemical compound C(C=C)(=O)N1CCN(CC1)C=1C2=C(N=C(N=1)OCCNC(C)=O)CN(CC2)C1=CC(=CC2=CC=CC=C12)O UQXDMQDTWRKZOQ-UHFFFAOYSA-N 0.000 description 1
- PVCJKHHOXFKFRP-UHFFFAOYSA-N N-acetylethanolamine Chemical compound CC(=O)NCCO PVCJKHHOXFKFRP-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 238000000636 Northern blotting Methods 0.000 description 1
- 108020004711 Nucleic Acid Probes Proteins 0.000 description 1
- MHABMANUFPZXEB-UHFFFAOYSA-N O-demethyl-aloesaponarin I Natural products O=C1C2=CC=CC(O)=C2C(=O)C2=C1C=C(O)C(C(O)=O)=C2C MHABMANUFPZXEB-UHFFFAOYSA-N 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229910019213 POCl3 Inorganic materials 0.000 description 1
- NFHFRUOZVGFOOS-UHFFFAOYSA-N Pd(PPh3)4 Substances [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 229920002230 Pectic acid Polymers 0.000 description 1
- LRBQNJMCXXYXIU-PPKXGCFTSA-N Penta-digallate-beta-D-glucose Natural products OC1=C(O)C(O)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(=O)OC[C@@H]2[C@H]([C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(OC(=O)C=3C=C(O)C(O)=C(O)C=3)C(O)=C(O)C=2)O)=C1 LRBQNJMCXXYXIU-PPKXGCFTSA-N 0.000 description 1
- 229920000604 Polyethylene Glycol 200 Polymers 0.000 description 1
- 108010020346 Polyglutamic Acid Proteins 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 238000011529 RT qPCR Methods 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- 238000002105 Southern blotting Methods 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- NPWMWLAPRVMNBN-ZCFIWIBFSA-N [(3r)-1-methylpyrrolidin-3-yl]methanol Chemical compound CN1CC[C@@H](CO)C1 NPWMWLAPRVMNBN-ZCFIWIBFSA-N 0.000 description 1
- BBAWTPDTGRXPDG-UHFFFAOYSA-N [1,3]thiazolo[4,5-b]pyridine Chemical group C1=CC=C2SC=NC2=N1 BBAWTPDTGRXPDG-UHFFFAOYSA-N 0.000 description 1
- ZVBJMUVVMBASLP-UHFFFAOYSA-N [1-(2-methoxyethyl)pyrrolidin-3-yl]methanol Chemical compound COCCN1CCC(CO)C1 ZVBJMUVVMBASLP-UHFFFAOYSA-N 0.000 description 1
- NKJUGZJKZYWRGF-UHFFFAOYSA-N [2-[tert-butyl(dimethyl)silyl]oxy-1-phenylethyl] methanesulfonate Chemical compound CC(C)(C)[Si](C)(C)OCC(OS(C)(=O)=O)C1=CC=CC=C1 NKJUGZJKZYWRGF-UHFFFAOYSA-N 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 235000011054 acetic acid Nutrition 0.000 description 1
- XPOLVIIHTDKJRY-UHFFFAOYSA-N acetic acid;methanimidamide Chemical compound NC=N.CC(O)=O XPOLVIIHTDKJRY-UHFFFAOYSA-N 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 125000004448 alkyl carbonyl group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 230000001668 ameliorated effect Effects 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000000118 anti-neoplastic effect Effects 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 125000005129 aryl carbonyl group Chemical group 0.000 description 1
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 125000004931 azocinyl group Chemical group N1=C(C=CC=CC=C1)* 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004604 benzisothiazolyl group Chemical group S1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- KKBYAYOFCRJQQT-NSHDSACASA-N benzyl (2s)-2-methylpiperazine-1-carboxylate Chemical compound C[C@H]1CNCCN1C(=O)OCC1=CC=CC=C1 KKBYAYOFCRJQQT-NSHDSACASA-N 0.000 description 1
- QTXXUWGRVIKWLU-UHFFFAOYSA-N benzyl 4-(5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl)piperazine-1-carboxylate Chemical compound N1=CN=C(C2=C1CNCC2)N1CCN(CC1)C(=O)OCC1=CC=CC=C1 QTXXUWGRVIKWLU-UHFFFAOYSA-N 0.000 description 1
- OXUVGSAHLGLLTM-UHFFFAOYSA-N benzyl 4-[7-[3-(methoxymethoxy)naphthalen-1-yl]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazine-1-carboxylate Chemical compound COCOC=1C=C(C2=CC=CC=C2C=1)N1CC=2N=CN=C(C=2CC1)N1CCN(CC1)C(=O)OCC1=CC=CC=C1 OXUVGSAHLGLLTM-UHFFFAOYSA-N 0.000 description 1
- FDJNIJSDMNWKFA-UHFFFAOYSA-N benzyl 4-chloro-2-methylsulfanyl-6,8-dihydro-5h-pyrido[3,4-d]pyrimidine-7-carboxylate Chemical compound C1C2=NC(SC)=NC(Cl)=C2CCN1C(=O)OCC1=CC=CC=C1 FDJNIJSDMNWKFA-UHFFFAOYSA-N 0.000 description 1
- CMNOQZUGZBAVND-UHFFFAOYSA-N benzyl 6,8-dihydro-5H-pyrido[3,4-d]pyrimidine-7-carboxylate Chemical compound C(C1=CC=CC=C1)OC(=O)N1CC=2N=CN=CC=2CC1 CMNOQZUGZBAVND-UHFFFAOYSA-N 0.000 description 1
- 239000012472 biological sample Substances 0.000 description 1
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 125000004623 carbolinyl group Chemical group 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical class OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 210000003679 cervix uteri Anatomy 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 239000012069 chiral reagent Substances 0.000 description 1
- 150000001804 chlorine Chemical class 0.000 description 1
- 229940061627 chloromethyl methyl ether Drugs 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 125000004230 chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N cinnamic acid Chemical compound OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 230000006552 constitutive activation Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 239000010779 crude oil Substances 0.000 description 1
- 125000004981 cycloalkylmethyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- WLVKDFJTYKELLQ-UHFFFAOYSA-N cyclopropylboronic acid Chemical compound OB(O)C1CC1 WLVKDFJTYKELLQ-UHFFFAOYSA-N 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 1
- 125000004856 decahydroquinolinyl group Chemical group N1(CCCC2CCCCC12)* 0.000 description 1
- NHADDZMCASKINP-HTRCEHHLSA-N decarboxydihydrocitrinin Natural products C1=C(O)C(C)=C2[C@H](C)[C@@H](C)OCC2=C1O NHADDZMCASKINP-HTRCEHHLSA-N 0.000 description 1
- 125000000723 dihydrobenzofuranyl group Chemical group O1C(CC2=C1C=CC=C2)* 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- 125000005879 dioxolanyl group Chemical group 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 125000005883 dithianyl group Chemical group 0.000 description 1
- 230000037437 driver mutation Effects 0.000 description 1
- 125000004050 enoyl group Chemical group 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- XBRDBODLCHKXHI-UHFFFAOYSA-N epolamine Chemical compound OCCN1CCCC1 XBRDBODLCHKXHI-UHFFFAOYSA-N 0.000 description 1
- JFZIFDNNGOCNFL-UHFFFAOYSA-N ethyl 1-benzyl-2-methyl-3-oxopiperidine-4-carboxylate Chemical compound CC1C(=O)C(C(=O)OCC)CCN1CC1=CC=CC=C1 JFZIFDNNGOCNFL-UHFFFAOYSA-N 0.000 description 1
- ULOLIZHBYWAICY-UHFFFAOYSA-N ethyl 2-(benzylamino)acetate Chemical compound CCOC(=O)CNCC1=CC=CC=C1 ULOLIZHBYWAICY-UHFFFAOYSA-N 0.000 description 1
- ARFLASKVLJTEJD-UHFFFAOYSA-N ethyl 2-bromopropanoate Chemical compound CCOC(=O)C(C)Br ARFLASKVLJTEJD-UHFFFAOYSA-N 0.000 description 1
- BOZNWXQZCYZCSH-UHFFFAOYSA-N ethyl 3-oxo-4-phenylbutanoate Chemical compound CCOC(=O)CC(=O)CC1=CC=CC=C1 BOZNWXQZCYZCSH-UHFFFAOYSA-N 0.000 description 1
- AOAFQODLPSUMJH-UHFFFAOYSA-N ethyl 4-[benzyl-(2-ethoxy-2-oxoethyl)amino]pentanoate Chemical compound C(C1=CC=CC=C1)N(C(CCC(=O)OCC)C)CC(=O)OCC AOAFQODLPSUMJH-UHFFFAOYSA-N 0.000 description 1
- XBPOBCXHALHJFP-UHFFFAOYSA-N ethyl 4-bromobutanoate Chemical compound CCOC(=O)CCCBr XBPOBCXHALHJFP-UHFFFAOYSA-N 0.000 description 1
- KCWYOFZQRFCIIE-UHFFFAOYSA-N ethylsilane Chemical compound CC[SiH3] KCWYOFZQRFCIIE-UHFFFAOYSA-N 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 238000000799 fluorescence microscopy Methods 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 125000003838 furazanyl group Chemical group 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- LRBQNJMCXXYXIU-QWKBTXIPSA-N gallotannic acid Chemical compound OC1=C(O)C(O)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(=O)OC[C@H]2[C@@H]([C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(OC(=O)C=3C=C(O)C(O)=C(O)C=3)C(O)=C(O)C=2)O)=C1 LRBQNJMCXXYXIU-QWKBTXIPSA-N 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 125000004438 haloalkoxy group Chemical group 0.000 description 1
- 125000004475 heteroaralkyl group Chemical group 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000003364 immunohistochemistry Methods 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000005462 in vivo assay Methods 0.000 description 1
- 125000004926 indolenyl group Chemical group 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- UEXQBEVWFZKHNB-UHFFFAOYSA-N intermediate 29 Natural products C1=CC(N)=CC=C1NC1=NC=CC=N1 UEXQBEVWFZKHNB-UHFFFAOYSA-N 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- OWFXIOWLTKNBAP-UHFFFAOYSA-N isoamyl nitrite Chemical compound CC(C)CCON=O OWFXIOWLTKNBAP-UHFFFAOYSA-N 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003384 isochromanyl group Chemical group C1(OCCC2=CC=CC=C12)* 0.000 description 1
- 125000005438 isoindazolyl group Chemical group 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- IHLVCKWPAMTVTG-UHFFFAOYSA-N lithium;carbanide Chemical compound [Li+].[CH3-] IHLVCKWPAMTVTG-UHFFFAOYSA-N 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 201000005249 lung adenocarcinoma Diseases 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- YDCHPLOFQATIDS-UHFFFAOYSA-N methyl 2-bromoacetate Chemical compound COC(=O)CBr YDCHPLOFQATIDS-UHFFFAOYSA-N 0.000 description 1
- UIUXUFNYAYAMOE-UHFFFAOYSA-N methylsilane Chemical compound [SiH3]C UIUXUFNYAYAMOE-UHFFFAOYSA-N 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 208000025113 myeloid leukemia Diseases 0.000 description 1
- YFJAIURZMRJPDB-UHFFFAOYSA-N n,n-dimethylpiperidin-4-amine Chemical compound CN(C)C1CCNCC1 YFJAIURZMRJPDB-UHFFFAOYSA-N 0.000 description 1
- AVAWMINJNRAQFS-UHFFFAOYSA-N n,n-dimethylpyrrolidin-3-amine Chemical compound CN(C)C1CCNC1 AVAWMINJNRAQFS-UHFFFAOYSA-N 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 1
- YZMHQCWXYHARLS-UHFFFAOYSA-N naphthalene-1,2-disulfonic acid Chemical compound C1=CC=CC2=C(S(O)(=O)=O)C(S(=O)(=O)O)=CC=C21 YZMHQCWXYHARLS-UHFFFAOYSA-N 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- 125000004923 naphthylmethyl group Chemical group C1(=CC=CC2=CC=CC=C12)C* 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 238000007481 next generation sequencing Methods 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 239000002853 nucleic acid probe Substances 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 125000004930 octahydroisoquinolinyl group Chemical group C1(NCCC2CCCC=C12)* 0.000 description 1
- 231100000590 oncogenic Toxicity 0.000 description 1
- 230000002246 oncogenic effect Effects 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 208000003388 osteoid osteoma Diseases 0.000 description 1
- 210000003101 oviduct Anatomy 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- QNNHQVPFZIFNFK-UHFFFAOYSA-N oxazolo[4,5-b]pyridine Chemical group C1=CC=C2OC=NC2=N1 QNNHQVPFZIFNFK-UHFFFAOYSA-N 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 125000004934 phenanthridinyl group Chemical group C1(=CC=CC2=NC=C3C=CC=CC3=C12)* 0.000 description 1
- 125000004625 phenanthrolinyl group Chemical group N1=C(C=CC2=CC=C3C=CC=NC3=C12)* 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 125000004928 piperidonyl group Chemical group 0.000 description 1
- 229960005235 piperonyl butoxide Drugs 0.000 description 1
- 125000004591 piperonyl group Chemical group C(C1=CC=2OCOC2C=C1)* 0.000 description 1
- 239000010318 polygalacturonic acid Substances 0.000 description 1
- 229920002643 polyglutamic acid Polymers 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000005344 pyridylmethyl group Chemical group [H]C1=C([H])C([H])=C([H])C(=N1)C([H])([H])* 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 102000016914 ras Proteins Human genes 0.000 description 1
- 108010014186 ras Proteins Proteins 0.000 description 1
- 238000003757 reverse transcription PCR Methods 0.000 description 1
- 238000007127 saponification reaction Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 102000030938 small GTPase Human genes 0.000 description 1
- 108060007624 small GTPase Proteins 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- MNWBNISUBARLIT-UHFFFAOYSA-N sodium cyanide Chemical compound [Na+].N#[C-] MNWBNISUBARLIT-UHFFFAOYSA-N 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 1
- 239000008259 solid foam Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 238000011477 surgical intervention Methods 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 125000006253 t-butylcarbonyl group Chemical group [H]C([H])([H])C(C(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 235000015523 tannic acid Nutrition 0.000 description 1
- 229940033123 tannic acid Drugs 0.000 description 1
- 229920002258 tannic acid Polymers 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- PSDAEKDIOQXLLC-DTORHVGOSA-N tert-butyl (1r,5s)-3,8-diazabicyclo[3.2.1]octane-3-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)C[C@@]2([H])CC[C@]1([H])N2 PSDAEKDIOQXLLC-DTORHVGOSA-N 0.000 description 1
- BCPPNDHZUPIXJM-MRVPVSSYSA-N tert-butyl (2r)-2-(hydroxymethyl)piperazine-1-carboxylate Chemical group CC(C)(C)OC(=O)N1CCNC[C@@H]1CO BCPPNDHZUPIXJM-MRVPVSSYSA-N 0.000 description 1
- JJQZILKRKYTGPG-UHFFFAOYSA-N tert-butyl 2-methylsulfanyl-4-(4-phenylmethoxycarbonylpiperazin-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidine-7-carboxylate Chemical compound C(C1=CC=CC=C1)OC(=O)N1CCN(CC1)C=1C2=C(N=C(N=1)SC)CN(CC2)C(=O)OC(C)(C)C JJQZILKRKYTGPG-UHFFFAOYSA-N 0.000 description 1
- PLHSQLIHPCNMOL-UHFFFAOYSA-N tert-butyl 2-methylsulfonyl-4-(4-phenylmethoxycarbonylpiperazin-1-yl)-6,8-dihydro-5H-pyrido[3,4-d]pyrimidine-7-carboxylate Chemical compound C(C1=CC=CC=C1)OC(=O)N1CCN(CC1)C=1C2=C(N=C(N=1)S(=O)(=O)C)CN(CC2)C(=O)OC(C)(C)C PLHSQLIHPCNMOL-UHFFFAOYSA-N 0.000 description 1
- APCBTRDHCDOPNY-UHFFFAOYSA-N tert-butyl 3-hydroxypyrrolidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(O)C1 APCBTRDHCDOPNY-UHFFFAOYSA-N 0.000 description 1
- XYHXUYOSXZHUSZ-UHFFFAOYSA-N tert-butyl 4-chloro-6,8-dihydro-5h-pyrido[3,4-d]pyrimidine-7-carboxylate Chemical compound N1=CN=C2CN(C(=O)OC(C)(C)C)CCC2=C1Cl XYHXUYOSXZHUSZ-UHFFFAOYSA-N 0.000 description 1
- YDKYFRBKDJSZAQ-UHFFFAOYSA-N tert-butyl N-(1-bromoisoquinolin-3-yl)carbamate Chemical compound BrC1=NC(=CC2=CC=CC=C12)NC(OC(C)(C)C)=O YDKYFRBKDJSZAQ-UHFFFAOYSA-N 0.000 description 1
- CWXPZXBSDSIRCS-UHFFFAOYSA-N tert-butyl piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNCC1 CWXPZXBSDSIRCS-UHFFFAOYSA-N 0.000 description 1
- SDLGKSBUCUJCRU-VIFPVBQESA-N tert-butyl-dimethyl-[(3s)-pyrrolidin-3-yl]oxysilane Chemical compound CC(C)(C)[Si](C)(C)O[C@H]1CCNC1 SDLGKSBUCUJCRU-VIFPVBQESA-N 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 1
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000005302 thiazolylmethyl group Chemical group [H]C1=C([H])N=C(S1)C([H])([H])* 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 230000002110 toxicologic effect Effects 0.000 description 1
- 231100000759 toxicological effect Toxicity 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 description 1
- MCULRUJILOGHCJ-UHFFFAOYSA-N triisobutylaluminium Chemical compound CC(C)C[Al](CC(C)C)CC(C)C MCULRUJILOGHCJ-UHFFFAOYSA-N 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5355—Non-condensed oxazines and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/08—Bridged systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
La presente invención se refiere a compuestos que inhiben KRas G12C. En particular, la presente invención se refiere a compuestos que inhiben irreversiblemente la actividad de KRas G12C, composiciones farmacéuticas que comprenden los compuestos y métodos de uso para las mismas. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Inhibidores de KRas G12C
Campo de la invención
La presente invención se refiere a compuestos que inhiben a KRas G12C. En particular, la presente invención se refiere a compuestos que inhiben irreversiblemente la actividad de KRas G12C, composiciones farmacéuticas que comprenden los compuestos y métodos de uso para los mismos.
Antecedentes de la invención
El homólogo del oncogén viral del sarcoma 2 de rata de Kirsten (“ KRas” ) es una pequeña GTPasa y un miembro de la familia de oncogenes Ras. KRas sirve un ciclo de conmutación molecular entre estados inactivo (unido a GDP) y activo (unido a GTP) para transducir señales celulares ascendentes recibidas de múltiples tirosina quinasas a efectores descendentes para regular una amplia variedad de procesos, que incluyen la proliferación celular (por ejemplo, ver Alamgeer y col., (2013) Current Opin Pharmcol. 13: 394-401).
El papel de KRas activado en la malignidad se observó hace más de treinta años (por ejemplo, ver Santos y col., (1984) Science 223:661-664). La expresión aberrante de KRas representa hasta 20 % de todos los cánceres y las mutaciones oncogénicas de KRas que estabilizan la unión de GTP y conducen a la activación constitutiva de KRas y se han informado señales posteriores en 25-30 % de los adenocarcinomas de pulmón. (por ejemplo, ver Samatar y Poulikakos (2014) Nat Rev Drug Disc 13 (12): 928-942 doi: 10.1038/nrd428). Las sustituciones de un solo nucleótido que dan como resultado mutaciones sin sentido en los codones 12 y 13 de la secuencia de aminoácidos primaria de KRas, comprenden aproximadamente 40 % de estas mutaciones impulsoras de KRas en el adenocarcinoma de pulmón, lo que es una transversión G12C la mutación activadora más común (por ejemplo, ver Dogan y col., (2012) Clin Cancer Res. 18 (22): 6169-6177, publicado en línea el 26 de septiembre de 2012, doi: 10.1158/1078-0432.CCR-11-3265).
El conocido papel de KRAs en la malignidad y el descubrimiento de estas frecuentes mutaciones en KRas en varios tipos de tumores hicieron de KRas una diana altamente atractiva de la industria farmacéutica para la terapia del cáncer. A pesar de treinta años de esfuerzos de descubrimiento a gran escala para desarrollar inhibidores de KRas para el tratamiento del cáncer, ningún inhibidor de KRas ha demostrado suficiente seguridad y/o eficacia para obtener la aprobación regulatoria (por ejemplo, Ver McCormick (2015) Clin Cancer Res. 21 (8):1797-1801).
A pesar de muchos esfuerzos fallidos para orientar a KRas los compuestos que inhiben la actividad de KRas, aún son muy convenientes y están bajo investigación, lo que incluye aquellos que alteran los efectores como los factores de intercambio de nucleótidos de guanina (por ejemplo, Ver Sun y col., (2012) Agnew Chem Int Ed Engl. 51 (25): 6140-6143 doi: 10.1002/anie201201358) y también se orientan a KRas G12C (por ejemplo, ver Ostrem y col., (2013) Nature 503: 548-551). Es evidente que se mantiene un interés y un esfuerzo continuo para desarrollar inhibidores de KRas, particularmente inhibidores de la activación de mutantes de KRas, que incluyen KRas G12C.
El documento US 2003/191143 se refiere a compuestos heterocíclicos fusionados que son útiles en el tratamiento de trastornos asociados con la activación de leucocitos.
Los documentos WO 2015/054572 y WO 2018/140600 se refieren a compuestos que tienen actividad como inhibidores de la proteína KRAS mutante G12C. También se refieren a métodos asociados con la preparación y uso de dichos compuestos, a composiciones farmacéuticas que comprenden dichos compuestos y a métodos para modular la actividad de la proteína KRAS mutante G12C para el tratamiento de trastornos tales como el cáncer.
El documento US 2016/166571 se refiere a terapias combinadas para el tratamiento de cánceres asociados con mutaciones en el gen KRAS. También se refiere a composiciones que comprenden agentes terapéuticos para el tratamiento de cánceres asociados con mutaciones en el gen KRAS.
El documento WO 2017/201161 se refiere a compuestos que inhiben KRas G12C, en particular a compuestos que inhiben irreversiblemente la actividad de KRas G12C, a composiciones farmacéuticas que comprenden los compuestos y a métodos de uso de los mismos.
Por tanto, existe la necesidad de desarrollar nuevos inhibidores de KRas G12C que demuestren suficiente eficacia, estabilidad y/o seguridad para tratar el cáncer mediado por KRas G12C. Los compuestos y composiciones de la presente invención superan ventajosamente uno o más de los inconvenientes anteriores lo que proporciona inhibidores selectivos de KRas G12C.
Resumen de la invención
En un aspecto de la invención, se proporcionan compuestos que inhiben la actividad de KRas G12C.
En ciertas modalidades, los compuestos se representan por la Fórmula (II):

o una sal farmacéuticamente aceptable de los mismos:
en donde:
R1-X es

en donde el anillo de piperazinilo está sustituido opcionalmente con R8;
Y es un enlace, O, S o NR5;
R1 es -C(O)C(RA)

C(RB)p', en donde

es un doble enlace, p es dos, cada RB es hidrógeno y RA es halógeno;
R2 es hidrógeno, alquilo, hidroxialquilo, dihidroxialquilo, alquilaminilalquilo, dialquilaminilalquilo, -Z-NR5R10, heterociclilo, heterociclilalquilo, arilo, heteroarilo o heteroarilalquilo, en donde cada uno de los Z, heterociclilo, heterociclilalquilo, arilo, heteroarilo y heteroarilalquilo puede estar sustituido opcionalmente con uno o más R9; cada Z es alquileno C1 - C4;
cada R3 es independientemente alquilo C1 - C3, oxo, haloalquilo, hidroxilo o halógeno;
L es un enlace, -C(O)- o alquileno C1-C3;
R4 es hidrógeno, cicloalquilo, heterociclilo, arilo, aralquilo o heteroarilo, en donde cada uno del cicloalquilo, heterociclilo, arilo, aralquilo y heteroarilo puede estar sustituido opcionalmente con uno o más R6, R7 o R8; cada R5 es independientemente hidrógeno o alquilo C1 - C3;
R6 es cicloalquilo, heterociclilo, heterociclilalquilo, arilo o heteroarilo, en donde cada uno del cicloalquilo, heterociclilo, arilo o heteroarilo puede estar sustituido opcionalmente con uno o más R7;
cada R7 es independientemente halógeno, hidroxilo, alquilo C1 - C6, cicloalquilo, alcoxi, haloalquilo, amino, ciano, heteroalquilo, hidroxialquilo o Q-haloalquilo, en donde Q es O o S;
R8 es oxo, alquilo C1 - C3, alquinilo C2 - C4, heteroalquilo, ciano, -C(O)OR5, -C(O)N(R5)2, -N(R5)2, en donde el alquilo C1 - C3 puede estar sustituido opcionalmente con ciano, halógeno, -OR5, -N(R5)2 o heteroarilo;
cada R9 es independientemente hidrógeno, oxo, acilo, hidroxilo, hidroxialquilo, ciano, halógeno, alquilo C1 - C6, aralquilo, haloalquilo, heteroalquilo, cicloalquilo, heterociclilo, heterociclilalquilo, alcoxi, dialquilaminilo, dialquilamidoalquilo o dialquilaminilalquilo, en donde el alquilo C1 - C6 puede estar sustituido opcionalmente con cicloalquilo;
cada R10 es independientemente hidrógeno, acilo, alquilo C1 - C3, heteroalquilo o hidroxialquilo;
R11 es haloalquilo; y
m es cero o un número entero entre 1 y 2.
Se incluyen además compuestos de Fórmula II que tienen la Fórmula II-A:

en donde R1, R3, R4, R5, R10, L y m son como se definen para la Fórmula II, R11 es hidrógeno, alquilo C1 - C3 o hidroxialquilo, y el anillo de piperazinilo está sustituido opcionalmente con R8 en donde R8 es como se define para la Fórmula II.
Se incluyen además compuestos de Fórmula II que tienen la Fórmula II-B:

donde R1, R3, R4, R8, L y m son como se definen para la Fórmula II, R2 es heterociclilalquilo sustituido opcionalmente con uno o más R9, y el anillo de piperazinilo está sustituido opcionalmente con R8, donde R8 es como se define para la Fórmula II.
En otro aspecto de la invención, se proporcionan composiciones farmacéuticas que comprenden una cantidad terapéuticamente eficaz de un compuesto de la presente invención o una sal farmacéuticamente aceptable del mismo y un excipiente farmacéuticamente aceptable.
En otro aspecto más de la invención, métodos para inhibir la actividad de KRas G12C en una célula, que comprenden poner en contacto la célula con un compuesto de la invención. En este aspecto, el contacto es in vitro.
También se proporciona en la presente descripción un compuesto para usarlo en un método para el tratamiento de un cáncer asociado con KRas G12C, comprendiendo el método administrar a un paciente que tiene cáncer una cantidad terapéuticamente eficaz de un compuesto de la invención, o una sal farmacéuticamente aceptable del mismo, solo o combinado con un vehículo, excipiente o diluyentes farmacéuticamente aceptables, en donde el cáncer se selecciona del grupo que consiste en Cardíaco: sarcoma seleccionado de angiosarcoma, fibrosarcoma, rabdomiosarcoma y liposarcoma, mixoma, rabdomioma, fibroma, lipoma y teratoma; Pulmón: carcinoma broncogénico seleccionado de células escamosas, células pequeñas no diferenciadas, células grandes no diferenciadas y adenocarcinoma, carcinoma bronquioloalveolar, adenoma bronquial, sarcoma, linfoma, hamartoma condromatoso, mesotelioma; Gastrointestinal: de esófago seleccionado de carcinoma de células escamosas, adenocarcinoma, leiomiosarcoma y linfoma, de estómago seleccionado de carcinoma, linfoma y leiomiosarcoma, de páncreas seleccionado de adenocarcinoma ductal, insulinoma, glucagonoma, gastrinoma, tumores carcinoides y vipoma, de intestino delgado seleccionado de adenocarcinoma, linfoma, tumores carcinoides, sarcoma de Kaposi, leiomioma, hemangioma, lipoma, neurofibroma y fibroma, de intestino grueso seleccionado de adenocarcinoma, adenoma tubular, adenoma velloso, hamartoma y leiomioma; Tracto genitourinario: de riñón seleccionado de adenocarcinoma, tumor de Wilm/nefroblastoma, linfoma y leucemia, de vejiga y uretra seleccionado de carcinoma de células escamosas, carcinoma de células transicionales y adenocarcinoma, de próstata seleccionado de adenocarcinoma y sarcoma, de testículos seleccionado de seminoma, teratoma, carcinoma embrionario, teratocarcinoma, coriocarcinoma, sarcoma, carcinoma de células intersticiales, fibroma, fibroadenoma, tumores adenomatoides y lipoma; Hígado: hepatoma/carcinoma hepatocelular, colangiocarcinoma, hepatoblastoma, angiosarcoma, adenoma hepatocelular, hemangioma; Tracto biliar: carcinoma de vesícula biliar, carcinoma ampular, colangiocarcinoma; Óseo: sarcoma osteogénico/osteosarcoma, fibrosarcoma, histiocitoma fibroso maligno, condrosarcoma, sarcoma de Ewing, linfoma maligno, sarcoma de células reticulares, mieloma múltiple, cordoma tumoral maligno de células gigantes, osteocronoma/exostosis osteocartilaginosa, condroma benigno, condroblastoma, condromixofibroma, osteoma osteoide y tumores de células gigantes; Sistema nervioso: de cráneo seleccionado de osteoma, hemangioma, granuloma, xantoma y osteítis deformante; de meninges seleccionado de meningioma, meningiosarcoma y gliomatosis; de cerebro seleccionado de astrocitoma, meduloblastoma, glioma, ependimoma, germinoma, pinealoma, glioblastoma multiforme, oligodendroglioma, schwannoma, retinoblastoma y tumores congénitos, de médula espinal seleccionado de neurofibroma, meningioma, glioma y sarcoma); Ginecológico: de útero seleccionado de carcinoma endometrial seleccionado de cistoadenocarcinoma seroso, cistadenocarcinoma mucinoso y carcinoma no clasificado, tumores de células de la granulosa-tecal, tumores de células de Sertoli-Leydig, disgerminoma y teratoma maligno, de vulva seleccionado de carcinoma de células escamosas, carcinoma intraepitelial, adenocarcinoma, fibrosarcoma y melanoma), de vagina seleccionado de carcinoma de células claras, carcinoma de células escamosas, sarcoma botrioide/rabdomiosarcoma embrionario, carcinoma de las trompas de Falopio; Hematológico: de sangre seleccionado de leucemia mieloide aguda, leucemia mieloide crónica, leucemia linfoblástica aguda, leucemia linfocítica crónica, enfermedades mieloproliferativas, mieloma múltiple y síndrome mielodisplásico, enfermedad de Hodgkin, linfoma
maligno, linfoma no Hodgkin; Piel: melanoma maligno, carcinoma de células basales, carcinoma de células escamosas, sarcoma de Kaposi, lunares, nevos displásicos, lipoma, angioma, dermatofibroma, queloides, psoriasis; y Glándulas suprarrenales: neuroblastoma.
El método para tratar un cáncer asociado con KRas G12C también puede comprender (a) determinar que el cáncer está asociado con una mutación G12C de KRas (p. ej., un cáncer asociado con KRas G12C); y (b) administrar al paciente una cantidad terapéuticamente eficaz de un compuesto de la invención o una sal farmacéuticamente aceptable del mismo, o una composición farmacéutica del mismo.
Descripción detallada de la invención
La presente invención se refiere a inhibidores de KRas G12C. En particular, la presente invención se refiere a compuestos que inhiben irreversiblemente la actividad de KRas G12C, composiciones farmacéuticas que comprenden una cantidad terapéuticamente eficaz de los compuestos y métodos de uso para los mismos.
Definiciones
A menos que se defina de otro modo, todos los términos técnicos y científicos usados en la presente descripción tienen el mismo significado que entiende comúnmente un experto en la técnica a la que pertenece esta invención. Todas las patentes, solicitudes de patente y publicaciones a las que se hace referencia en la presente descripción se incorporan como referencia.
Como se usa en la presente descripción, “ KRas G12C” se refiere a una forma mutante de una proteína KRas de mamífero que contiene una sustitución de aminoácidos de una cisteína por una glicina en la posición 12 del aminoácido. La asignación de posiciones de residuos y codones de aminoácidos para KRas humano se basa en la secuencia de aminoácidos identificada por UniProtκΒ/Swiss-Prot P01116: Variant p.Gly12Cys.
Como se usa en la presente descripción, un “ inhibidor de KRas G12C” se refiere a compuestos de la presente invención que están representados por Fórmulas (I) como se describen en la presente. Estos compuestos son capaces de modular o inhibir negativamente toda o una parte de la actividad enzimática de KRas G12C. Los inhibidores de KRas G12C de la presente invención interactúan y se unen irreversiblemente a KRas G12C mediante formación de un aducto covalente con la cadena lateral de sulfhidrilo del residuo de cisteína en la posición 12, lo que da como resultado la inhibición de la actividad enzimática de KRas G12C.
Una “ enfermedad o trastorno asociado con KRas G12C” , como se usa en la presente descripción, se refiere a enfermedades o trastornos asociados con, o mediados por, o que tienen una mutación KRas G12C. Un ejemplo no limitante de una enfermedad o trastorno asociado a KRas G12C es un cáncer asociado a KRas G12C.
Como se usa en la presente descripción, el término “ sujeto” , “ individuo” o “ paciente” , usado indistintamente, se refiere a cualquier animal, que incluye mamíferos tales como ratones, ratas, otros roedores, conejos, perros, gatos, cerdos, vacas, ovejas, caballos, primates y humanos. En algunas modalidades, el paciente es un ser humano. En algunas modalidades, el sujeto ha experimentado y/o exhibido al menos un síntoma de la enfermedad o trastorno a tratar y/o prevenir. En algunas modalidades, se ha identificado o diagnosticado que el sujeto tiene un cáncer que tiene una mutación de KRas G12C (por ejemplo, según se determina mediante el uso de un ensayo o kit aprobado por una agencia reguladora, por ejemplo, aprobado por la FDA). En algunas modalidades, el sujeto tiene un tumor que es positivo para una mutación de KRas G12C (por ejemplo, según se determina mediante el uso de un ensayo o kit aprobado por una agencia reguladora). El sujeto puede ser un sujeto con un tumor o tumores que sean positivos para una mutación de KRas G12C (por ejemplo, identificado como positivo mediante el uso de un ensayo o kit aprobado por una agencia reguladora, por ejemplo, aprobado por la FDA). El sujeto puede ser un sujeto cuyos tumores tienen una mutación de KRas G12C (por ejemplo, cuando el tumor se identifica como tal mediante el uso de un kit o ensayo aprobado por una agencia reguladora, por ejemplo, aprobado por la FDA). En algunas modalidades, se sospecha que el sujeto tiene un cáncer asociado al gen KRas G12C. En algunas modalidades, el sujeto tiene un registro clínico que indica que el sujeto tiene un tumor que tiene una mutación de KRas G12C (y opcionalmente el registro clínico indica que el sujeto debe ser tratado con cualquiera de las composiciones proporcionadas en la presente descripción).
En algunas modalidades de cualquiera de los métodos o usos descritos en la presente descripción, se usa un ensayo para determinar si el paciente tiene la mutación de KRas G12C mediante el uso de una muestra (por ejemplo, una muestra biológica o una muestra de biopsia (por ejemplo, una muestra de biopsia incrustada en parafina) de un paciente (por ejemplo, un paciente sospechoso de tener un cáncer asociado a KRas G12C, un paciente que tiene uno o más síntomas de un cáncer asociado a KRas G12C y/o un paciente que tiene un mayor riesgo de desarrollar un cáncer asociado a KRas G12C) puede incluir, por ejemplo, la secuenciación de nueva generación, inmunohistoquímica, microscopía de fluorescencia, análisis de FISH de separación, transferencia Southern, transferencia Western, análisis FACS, transferencia Northern y amplificación basada en PCR (por ejemplo, RT-PCR y RT-PCR cuantitativa en tiempo real). Como es bien conocido en la técnica, los ensayos se realizan típicamente, por ejemplo, con al menos una sonda de ácido nucleico marcada o al menos un anticuerpo marcado o fragmento de unión al antígeno del mismo.
El término “ agencia reguladora” es la agencia de un país para la aprobación del uso médico de agentes farmacéuticos con el país. Por ejemplo, un ejemplo no limitante de una agencia reguladora es la Administración de Fármacos y Alimentos de los Estados Unidos (FDA).
El término “ amino” se refiere a -NH2;
El término “ acilo” se refiere a -C(O)CH3.
El término “ alquilo” como se emplea en la presente descripción se refiere a grupos alifáticos de cadena lineal y ramificada que tienen de 1 a 12 átomos de carbono, 1-8 átomos de carbono 1-6 átomos de carbono o 1-3 átomos de carbono que están sustituidos opcionalmente con uno, dos o tres sustituyentes. Los ejemplos de grupos alquilo incluyen, sin limitación, metilo, etilo, propilo, isopropilo, butilo, isobutilo, sec-butilo, terc-butilo, pentilo y hexilo.
El término “ haloalquilo” se refiere a una cadena de alquilo en la que un hidrógeno o más se han reemplazados por un halógeno. Ejemplos de haloalquilos son trifluorometilo, difluorometilo y fluorometilo.
El término “ haloalquiloxi” se refiere a -O-haloalquilo.
Un grupo “ alquileno” es un grupo alquilo, como se ha definido anteriormente, que está posicionado entre otros dos grupos químicos y sirve para conectarlos. Los ejemplos de grupos alquileno incluyen, sin limitación, metileno, etileno, propileno y butileno.
El término “ alcoxi” se refiere a -Oalquilo C1 - C6.
El término cicloalquilo como se emplea en la presente descripción incluye grupos hidrocarbonados cíclicos saturados y parcialmente insaturados que tienen de 3 a 12 carbonos, por ejemplo de 3 a 8 carbonos, y como ejemplo adicional de 3 a 6 carbonos, en donde el grupo cicloalquilo está adicionalmente sustituido opcionalmente. Los ejemplos de grupos cicloalquilo incluyen, sin limitación, ciclopropilo, ciclobutilo, ciclopentilo, ciclopentenilo, ciclohexilo, ciclohexenilo, cicloheptilo y ciclooctilo. El término “ heteroalquilo” se refiere a un grupo alquilo, como se definió anteriormente en la presente, en donde uno o más átomos de carbono en la cadena están reemplazados por un heteroátomo seleccionado del grupo que consiste en O, S y N.
Como se usa en la presente descripción, el término “ hidroxialquilo” se refiere a -alquil-OH.
El término “ dihidroxialquilo” se refiere a un grupo alquilo como se define en la presente descripción, en donde dos átomos de carbono están cada uno sustituido con un grupo hidroxilo.
El término “ alquilaminilo” se refiere a alquil-NRx, en donde Rx es hidrógeno. En una modalidad, Rx es hidrógeno. El término “ dialquilaminilo” se refiere a -N(Ry)2, en donde cada Ry es alquilo C1 - C3.
El término “alquilaminilalquilo” se refiere a -alquil-NRx-alquilo, en donde Rx es hidrógeno. En una modalidad, Rx es hidrógeno. El término “ dialquilaminilalquilo” se refiere a -alquil-N(Ry)2, en donde cada Ry es alquilo C1 - C4, en donde el alquilo del alquil-N (Ry)2 puede estar sustituido opcionalmente con hidroxi o hidroxialquilo.
Un grupo “ arilo” es un resto aromático C6-C14 que comprende de uno a tres anillos aromáticos, que está sustituido opcionalmente. Como una modalidad, el grupo arilo es un grupo arilo C6-C10. Los ejemplos de grupos arilo incluyen, sin limitación, fenilo, naftilo, antracenilo, fluorenilo y dihidrobenzofuranilo.
Un grupo “ aralquilo” o “ arilalquilo” comprende un grupo arilo unido covalentemente a un grupo alquilo, cualquiera de los cuales puede estar sustituido opcionalmente o no sustituido independientemente. Un ejemplo de un grupo aralquilo es alquilo(C1-C6)arilo(C6-C10), que incluye, sin limitación, bencilo, fenetilo y naftilmetilo. Un ejemplo de aralquilo sustituido es en donde el grupo alquilo está sustituido con hidroxialquilo.
Un grupo “ heterociclilo” o “ heterocíclico” es una estructura de anillo que tiene de aproximadamente 3 a aproximadamente 12 átomos, por ejemplo de 4 a 8 átomos, en donde uno o más átomos se seleccionan del grupo que consiste en N, O y S, el resto de los átomos del anillo son carbonos. El heterociclilo puede ser un sistema de anillo monocíclico, bicíclico, espirocíclico o con puente. El grupo heterocíclico está sustituido opcionalmente con R7 sobre carbono o nitrógeno en una o más posiciones, en donde R7 es como se define para la Fórmula I. El grupo heterocíclico además está sustituido opcionalmente independientemente en el nitrógeno con alquilo, arilo, aralquilo, alquilcarbonilo, alquilsulfonilo, arilcarbonilo, arilsulfonilo, alcoxicarbonilo, aralcoxicarbonilo o en el azufre con oxo o alquilo inferior. Los ejemplos de grupos heterocíclicos incluyen, sin limitación, epoxi, azetidinilo, aziridinilo, tetrahidrofuranαlo, tetrahidropiranαlo, pirrolidinilo, pirrolidinonilo, piperidinilo, piperazinilo, imidazolidinilo, tiazolidinilo, ditianilo, tritianilo dioxolanilo, oxazolidinilo, oxazolidinonilo, decahidroquinolinilo, piperidonilo, 4-piperidinonilo, tiomorfolinilo, 1,1 -dióxido de tiomorfolinilo, morfolinilo,
oxazepanilo, azabiciclohexanos, azabicicloheptanos y oxa azabiocicloheptanos. Se excluyen específicamente del alcance de este término los compuestos que tienen átomos de O y/o S anulares adyacentes.
El término “ heterociclilalquilo” se refiere a un grupo heterociclilo, como se define en la presente descripción, unido a la porción restante de la molécula mediante un conector de alquilo, en donde el conector de alquilo del heterociclilalquilo puede estar sustituido opcionalmente con hidroxi o hidroxialquilo.
Como se usa en la presente descripción, el término “ heteroarilo” se refiere a grupos que tienen de 5 a 14 átomos en el anillo, preferiblemente 5, 6, 9 o 10 átomos en el anillo; que tienen 6, 10 o 14 electrones n compartidos en una matriz cíclica; y que tienen, además de átomos de carbono, de uno a tres heteroátomos por anillo, seleccionados del grupo que consiste en N, O y S. Los ejemplos de grupos heteroarilo incluyen acridinilo, azocinilo, benzoimidazolilo, benzofuranilo, benzotiofuranilo, benzoxazolilo, benzotiazolilo, benzotriazolilo, benzotetrazolilo, benzoisoxazolilo, benzoisotiazolilo, benzoimidazolinilo, carbazolilo, 4aH-carbazolilo, carbolinilo, cromanilo, cromenilo, cinnolinilo, furanilo, furazanilo, imidazolinilo, imidazolilo, 1 H-indazolilo, indolenilo, indolinilo, indolizinilo, indolilo, 3H-indolilo, isobenzofuranilo, isocromanilo, isoindazolilo, isoindolinilo, isoquinolinilo, isotiazolilo, isoxazolilo, metilendioxifenilo, naftiridinilo, octahidroisoquinolinilo, oxadiazolilo, 1,2,3-oxadiazolilo, 1,2,4-oxadiazolilo, 1,2,5-oxadiazolilo, 1,3,4-oxadiazolilo, oxazolidinilo, oxazolilo, oxazolidinilo, pirimidinilo, fenantridinilo, fenantrolinilo, fenazinilo, fenotiazinilo, fenoxazinilo, ftalazinilo, piperonilo, pteridinilo, purinilo, piranilo, pirazinilo, pirazolidinilo, pirazolinilo, pirazolilo, piridazinilo, piridooxazol, piridoimidazol, piridotiazol, piridinilo, pirimidinilo, pirrolinilo, 2H-pirrolilo, pirrolilo, quinazolinilo, quinolinilo, 4H-quinolizinilo, quinoxalinilo, quinuclidinilo, tetrahidroisoquinolinilo, tetrahidroquinolinilo, tetrazolilo, 6H-1,2,5-tiadiazinilo, 1,2,3-tiadiazolilo, 1,2,4-tiadiazolilo, 1,2,5-tiadiazolilo, 1,3,4-tiadiazolilo, tiantenilo, tiazolilo, tienilo, tienotiazolilo, tienooxazolilo, tienoimidazolilo, tiofenilo, triazinilo, 1,2,3-triazolilo, 1,2,4-triazolilo, 1,2,5-triazolilo, 1,3,4-triazolilo y xantenilo.
Un grupo “ heteroarilalquilo” comprende un grupo heteroarilo unido covalentemente a un grupo alquilo, en donde el radical está en el grupo alquilo, cualquiera de los cuales está independientemente sustituido o no sustituido opcionalmente. Los ejemplos de grupos heteroarilalquilo incluyen un grupo heteroarilo que tiene 5, 6, 9 o 10 átomos en el anillo unidos a un grupo alquilo C1-C6. Los ejemplos de grupos heteroaralquilo incluyen piridilmetilo, piridiletilo, pirrolilmetilo, pirroliletilo, imidazolilmetilo, imidazoliletilo, tiazolilmetilo, tiazoliletilo, benzoimidazolilmetilo, benzoimidazoliletilo, quinazolinilmetilo, quinolinilmetilo, quinoliniletilo, benzofuranilmetilo, quinoliniletilo y benzofuranilmetilo. Se excluyen específicamente del alcance de este término los compuestos que tienen átomos de O y/o S anulares adyacentes.
Como se usa en la presente descripción, “ una cantidad eficaz” de un compuesto es una cantidad que es suficiente para inhibir o modular negativamente la actividad de KRas G12C. Tal cantidad puede administrarse como una dosis única o puede administrarse según un régimen, por lo que es eficaz.
Como se usa en la presente descripción, una “ cantidad terapéuticamente eficaz” de un compuesto es una cantidad que es suficiente para mejorar, o de alguna manera reducir un síntoma o detener o revertir la progresión de una condición, o de inhibir o modular negativamente la actividad de KRas G12C. Tal cantidad puede administrarse como una dosis única o puede administrarse según un régimen, por lo que es eficaz.
Tal como se usa en la presente descripción, tratamiento significa cualquier manera en la que los síntomas o patología de una afección, trastorno o enfermedad se mejoran o se alteran beneficiosamente de otro modo. El tratamiento abarca además cualquier uso farmacéutico de las composiciones de la presente descripción.
Como se usa en la presente descripción, la mejora de los síntomas de un trastorno particular mediante la administración de una composición farmacéutica particular se refiere a cualquier disminución, ya sea permanente o temporal, duradera o transitoria que pueda atribuirse o asociarse con la administración de la composición.
Compuestos
En un aspecto de la invención, se proporcionan compuestos representados por la fórmula (II):

o una sal farmacéuticamente aceptable de los mismos:
en donde:
R1-X es

en donde el anillo de piperazinilo está sustituido opcionalmente con R8;
Y es un enlace, O, S o NR5;
R1 es -C(O)C(RA)

C(RB)p, en donde

es un doble enlace, p es dos, cada RB es hidrógeno y RA es halógeno;
R2 es hidrógeno, alquilo, hidroxialquilo, dihidroxialquilo, alquilaminilalquilo, dialquilaminilalquilo, -Z-NR5R10, heterociclilo, heterociclilalquilo, arilo, heteroarilo o heteroarilalquilo, en donde cada uno de los Z, heterociclilo, heterociclilalquilo, arilo, heteroarilo y heteroarilalquilo puede estar sustituido opcionalmente con uno o más R9; cada Z es alquileno C1 - C4;
cada R3 es independientemente alquilo C1 - C3, oxo, haloalquilo, hidroxilo o halógeno;
L es un enlace, -C(O)- o alquileno C1-C3;
R4 es hidrógeno, cicloalquilo, heterociclilo, arilo, aralquilo o heteroarilo, en donde cada uno del cicloalquilo, heterociclilo, arilo, aralquilo y heteroarilo puede estar sustituido opcionalmente con uno o más R6, R7 o R8;
cada R5 es independientemente hidrógeno o alquilo C1 - C3;
R6 es cicloalquilo, heterociclilo, heterociclilalquilo, arilo o heteroarilo, en donde cada uno del cicloalquilo, heterociclilo, arilo o heteroarilo puede estar sustituido opcionalmente con uno o más R7;
cada R7 es independientemente halógeno, hidroxilo, alquilo C1 - C6, cicloalquilo, alcoxi, haloalquilo, amino, ciano, heteroalquilo, hidroxialquilo o Q-haloalquilo, en donde Q es O o S;
R8 es oxo, alquilo C1 - C3, alquinilo C2 - C4, heteroalquilo, ciano, -C(O)OR5, -C(O)N(R5)2, -N(R5)2, en donde el alquilo C1 - C3 puede estar sustituido opcionalmente con ciano, halógeno, -OR5, -N(R5)2 o heteroarilo;
cada R9 es independientemente hidrógeno, oxo, acilo, hidroxilo, hidroxialquilo, ciano, halógeno, alquilo C1 - C6, aralquilo, haloalquilo, heteroalquilo, cicloalquilo, heterociclilo, heterociclilalquilo, alcoxi, dialquilaminilo, dialquilamidoalquilo o dialquilaminilalquilo, en donde el alquilo C1 - C6 puede estar sustituido opcionalmente con cicloalquilo;
cada R10 es independientemente hidrógeno, acilo, alquilo C1 - C3, heteroalquilo o hidroxialquilo;
R11 es haloalquilo; y
m es cero o un número entero entre 1 y 2.
En este aspecto de la invención, R1-X es:

en donde R1 es como se define para la Fórmula II y el anillo de piperazinilo está sustituido opcionalmente con R8, donde R8 es como se define para la Fórmula II. En ciertas modalidades, R8 es alquilo C1 - C3 en donde el alquilo está sustituido opcionalmente con ciano u OR5, o -C(O)N(R5)2, en donde cada R5 es independientemente hidrógeno o alquilo C1 - C3. En este aspecto de la invención, R1 es -C(O)C(RA)

C(RB)p, en donde

es un doble enlace, p es dos, cada RB es hidrógeno y RA es halógeno. En una modalidad, el doble enlace está en la configuración E. En una modalidad, el doble enlace está en la configuración Z.
En este aspecto de la invención, RA es halógeno. En una modalidad, el halógeno es flúor o cloro.
En una modalidad, Y es O o NR5 y R2 es heterociclilo o heterociclilalquilo sustituido opcionalmente con uno o más R9. Ejemplos no limitantes de uno o más R9 cuando R2 es heterociclilo o heterociclilalquilo incluyen alquilo C1 - C3, acilo, oxo, ciano, alcoxi, cicloalquilo, cicloalquilmetilo, halógeno e hidroxilo. Ejemplos no limitantes de heterociclilos R2 sustituidos opcionalmente con uno o más R9 incluyen azetidinilo, azetidinilo sustituido por alquilo C1-C3 (por ejemplo, metilazetidinilo), azetidinilo sustituido por halo (por ejemplo, difluoroazetidinilo), tetrahidropirano, pirrolidinilo, pirrolidinilo sustituido por alquilo C1-C3 (por ejemplo, metilpirrolidinilo, dimetilpirrolidinilo e isopropilpirrolidinilo), cicloalquilpirrolidinilo, hidroxipirrolindinilo, pirrolidinilo sustituido por halo (por ejemplo, fluoropirrolidinilo y difluoropirrolidinilo), N-metil pirrolidinilo halo-sustituido (por ejemplo, N-metilfluoropirrolidinilo y N-metildifluoropirrolidinilo), metoxietilpirrolidinilo, N-metilpirrolidinilo alcoxi-sustituido (por ejemplo, (N-metil)metoxipirrolidinilo, piperazinilo, dimetilaminilpirrolidinilo,
morfolinilo, metilmorfolinilo, 1,4-oxazepanilo, piperdinilo, piperidinilo sustituido por un alquilo C1-C3 (por ejemplo, metilpiperidinilo), acilpiperdinilo, cianopiperdinilo, cicloalquilpiperdinilo, halopiperdinilo (por ejemplo, fluoropiperdinilo), dihalopiperdinilo (por ejemplo, difluoropiperdinilo), alcoxipiperdinilo, pirrolidonilo, piperidonilo, 1,1 -dióxido de tiomorfolinilo, 3-azabiciclo[3.1.0]hexanilo, oxa-5-azabiciclo[2.2.1 ]heptan-5-ilo y azabiciclo[2.2.1 ]heptano-2-ilo.
En una modalidad, la porción heterociclilo del heterociclilalquilo es N-metilpirrolidinilo. En una modalidad, la porción heterociclilo del heterociclilalquilo es 3,3-difluoro-1 -metilpirrolidinilo.
En ciertas otras modalidades, R4 es arilo. En una modalidad, R4 se selecciona del grupo que consiste en fenilo y naftilo y está sustituido opcionalmente con uno o más R6 o R7. Ejemplos de sustituyentes R7 incluyen halógeno, hidroxilo, alquilo C1-C6 (por ejemplo, alquilo C1-C3), cicloalquilo, haloalquilo, Q-haloalquilo, amino, ciano, hidroxialquilo y alcoxi. En una modalidad, el arilo es fenilo sustituido con uno o más grupos R7 seleccionados independientemente de halógeno, hidroxilo, alquilo C1 -C3, haloalquilo, Q-haloalquilo y alcoxi. En una modalidad, el arilo es fenilo sustituido con uno o más grupos R7 seleccionados independientemente de halógeno, haloalquilo, metilo, isopropilo, metoxi, Q-haloalquilo e hidroxilo. En una modalidad, el arilo es fenilo sustituido con uno o más grupos R7 seleccionados independientemente de metilo, trifluorometilo, hidroxilo, fluoro y cloro. En una modalidad, el arilo es fenilo sustituido con uno a tres grupos R7 seleccionados independientemente de metilo, hidroxilo, trifluorometilo, flúor y cloro. En una modalidad, el arilo es fenilo sustituido con hidroxilo y alquilo C1-C3 o dos alquilo C1-C3. En una modalidad, el arilo es fenilo sustituido con trifluorometilo y alquilo C1-C3 o dos alquilo C1-C3.
En una modalidad, R4 es arilo en donde arilo es naftilo sustituido opcionalmente con uno o más R7. En una modalidad, el arilo es naftilo sustituido con uno o más grupos R7 seleccionados independientemente de halógeno, hidroxilo, alquilo C1-C3, haloalquilo, Q-haloalquilo y alcoxi. En una modalidad, el arilo es naftilo sustituido con uno o más grupos R7 seleccionados independientemente de halógeno, haloalquilo, metilo, isopropilo, metoxi, Q-haloalquilo e hidroxilo. En una modalidad, R4 es naftilo sustituido opcionalmente con uno o más sustituyentes R7 seleccionados independientemente de hidroxilo, halógeno, alquilo C1-C3, amino y haloalquilo. En una modalidad, R4 es naftilo sustituido opcionalmente con uno a tres sustituyentes R7 seleccionados independientemente de difluorometilo, metilo, hidroxilo, amino, fluoro y cloro. En una modalidad, el naftilo sustituido es 8-cloronaftilo u 8-metilnaftilo.
En una modalidad, el arilo es naftilo sustituido opcionalmente con uno o más halógenos. En una modalidad, el arilo es naftilo sustituido con hidroxilo y trifluorometilo o alquilo C1-C3. En una modalidad, el arilo es naftilo sustituido con hidroxilo.
En una modalidad, R4 es heteroarilo sustituido opcionalmente con uno o más R6, R7 o R8. En una modalidad, R4 es heteroarilo sustituido opcionalmente con uno o más R7 o R8 seleccionados independientemente de halógeno, hidroxilo, alquilo C1-C3, haloalquilo, Q-haloalquilo, alcoxi y amino. En una modalidad, R4 es indαlo, indazolilo, quinolinilo, isoquinolinilo, piridinilo o benzo[d]tiazolilo sustituido opcionalmente con uno o más R6, R7 o R8. En una modalidad, R4 es indαlo, indazolilo, quinolinilo, isoquinolinilo, piridinilo o benzo[d] tiazolilo sustituido opcionalmente con uno o más R7 o R8 independientemente seleccionados de oxo, halógeno, hidroxilo, alquilo C1-C3, haloalquilo, Q-haloalquilo, alcoxi y aminado.
Aún en otras modalidades, R4 es heteroarilo, opcionalmente un indαlo o un indazolilo, cada uno de los cuales puede estar sustituido con uno o más R6, R7 o R8. En una modalidad, R4 es heteroarilo sustituido opcionalmente con uno o más sustituyentes R7 o R8 seleccionados independientemente del grupo que consiste en halógeno, hidroxilo, alquilo C1-C3, haloalquilo, Q-haloalquilo y alcoxi. En una modalidad, el heteroarilo R4 es indazolilo sustituido opcionalmente con uno o dos de R7 o R8 seleccionados independientemente de oxo, trifluorometilo, alcoxi, haloalquilo y alquilo C1-C6. En otras modalidades, el heteroarilo R4 es un quinolinilo o isoquinolinilo, cada uno sustituido opcionalmente con uno o más R7. En una modalidad, el heteroarilo R4 es un quinolinilo o isoquinolinilo, cada uno sustituido opcionalmente con uno o más R7 seleccionados independientemente de amino, hidroxilo, alquilo C1 - C3 e hidroxilo. En una modalidad, el heteroarilo R4 es un quinolinilo o isoquinolinilo, cada uno sustituido opcionalmente con R7 seleccionado de hidroxilo y amino. En una modalidad, el heteroarilo R4 es un piridinilo sustituido opcionalmente con uno o más R6, R7 o R8. En una modalidad, el heteroarilo R4 es piridinilo sustituido opcionalmente con uno o más R7 seleccionados independientemente de alquilo C1 - C3, halógeno y haloalquilo. En una modalidad, el heteroarilo R4 es indolilo sustituido opcionalmente con uno o más R6, R7 o R8. En una modalidad, el heteroarilo R4 es indolilo sustituido opcionalmente con uno o dos de R7 seleccionados independientemente de hidroxilo, trifluorometilo y alquilo C1 - C3.
En una modalidad, L es un enlace.
En una modalidad, m es cero.
En una modalidad, R8 es heteroalquilo, alquinilo C2-C4 o alquilo C1 - C3 sustituido opcionalmente con -OR5, ciano o heteroarilo. En una modalidad, R8 es metilo, cianometilo, metoximetilo, hidroximetilo. En una modalidad, R8 es metilo. En una modalidad, R8 es cianometilo. En una modalidad, R8 es hidroximetilo.
En una modalidad, la Fórmula II incluye compuestos que tienen la Fórmula II-A:

en donde R1, R3, R4, R5, R10, L y m son como se definen para la Fórmula II, R11 es hidrógeno, metilo o hidroxialquilo, y el anillo de piperazinilo está sustituido opcionalmente con R8 en donde R8 es como se define para la Fórmula II.
En esta modalidad, R1 es -C(O)C(RA)

C(RB)p donde RA, RB y p son como se definen para la Fórmula II.
En esta modalidad, RA es halógeno. En una modalidad, el halógeno es flúor o cloro.
En una modalidad, L es un enlace. En una modalidad, R4 es arilo o heteroarilo, cada uno de los cuales está sustituido opcionalmente con uno o más R6, R7 o R8. En una modalidad, R4 es arilo o heteroarilo, cada uno de los cuales está sustituido opcionalmente con uno o más R7. En una modalidad, cada R7 o R8 se selecciona independientemente de oxo, hidroxilo, amino, halógeno, alquilo C1 - C3, haloalquilo, Q-haloalquilo, cicloalquilo y alcoxi. En una modalidad, R5 y R10 son cada uno alquilo C1 - C3. En una modalidad, el arilo es fenilo sustituido con uno o más grupos R7 seleccionados independientemente de halógeno, hidroxilo, alquilo C1-C3, haloalquilo, Q-haloalquilo y alcoxi. En una modalidad, el arilo es fenilo sustituido con uno o más grupos R7 seleccionados independientemente de halógeno, haloalquilo, metilo, isopropilo, metoxi, Q-haloalquilo e hidroxilo. En una modalidad, el arilo es fenilo sustituido con uno o más grupos R7 seleccionados independientemente de metilo, trifluorometilo, 2,2,2-trifluoroetilo, hidroxilo, trifluorometoxi, hidroxilo, fluoro, cloro, isopropilo, ciclopropilo y trifluorometiltio. En una modalidad, el arilo es fenilo sustituido con uno a tres grupos R7 seleccionados independientemente de hidroxilo, flúor y cloro. En una modalidad, el arilo es fenilo sustituido con hidroxilo y alquilo C1-C3 o dos alquilo C1-C3. En una modalidad, el arilo es fenilo sustituido con Q-haloalquilo e hidroxilo o flúor. En una modalidad, el arilo es naftilo sustituido con uno o más grupos R7 seleccionados independientemente de halógeno, hidroxilo, alquilo C1-C3, haloalquilo, Q-haloalquilo y alcoxi. En una modalidad, el arilo es naftilo sustituido con uno o más grupos R7 seleccionados independientemente de halógeno, haloalquilo, metilo, isopropilo, metoxi, Q-haloalquilo e hidroxilo. En una modalidad, R4 es naftilo sustituido opcionalmente con uno o más sustituyentes R7 seleccionados independientemente de hidroxilo, halógeno, alquilo C1-C3, amino y haloalquilo. En una modalidad, R4 es naftilo sustituido opcionalmente con uno a tres sustituyentes R7 o R8 seleccionados independientemente de difluorometilo, metilo, hidroxilo, amino, fluoro y cloro. En una modalidad, el arilo es naftilo sustituido opcionalmente con uno o más halógenos. En una modalidad, el arilo es naftilo sustituido con hidroxilo y trifluorometilo o alquilo C1-C3. En una modalidad, el arilo es naftilo sustituido con hidroxilo. En una modalidad, R4 es heteroarilo, en donde el heteroarilo es indazolilo sustituido opcionalmente con uno o dos de R7 o R8 seleccionados independientemente de oxo, alcoxi, haloalquilo y alquilo C1-C6. En una modalidad, R4 es heteroarilo, en donde el heteroarilo es quinolinilo o isoquinolinilo, cada uno sustituido opcionalmente con uno o más R7. En una modalidad, R4 es heteroarilo, en donde el heteroarilo es quinolinilo o isoquinolinilo, cada uno sustituido opcionalmente con uno o más R7 seleccionados independientemente de amino, hidroxilo, alquilo C1-C3 e hidroxilo. En una modalidad, el heteroarilo R4 es un piridinilo sustituido opcionalmente con uno o más R6, R7 o R8. En una modalidad, el heteroarilo R4 es piridinilo sustituido opcionalmente con uno o más R7 seleccionados independientemente de alquilo C1 - C3, halógeno y haloalquilo. En una modalidad, el heteroarilo R4 es indolilo sustituido opcionalmente con uno o más R7. En una modalidad, el heteroarilo R4 es indolilo sustituido opcionalmente con uno o dos de R7 seleccionados independientemente de hidroxilo y alquilo C1 - C3. En una modalidad, R11 es metilo. En una modalidad, el anillo de piperazinilo no está sustituido. En una modalidad, el anillo de piperazinilo está sustituido con R8. En una modalidad, R8 es alquilo C1 - C3 sustituido opcionalmente con ciano o hidroxilo. En una modalidad, R8 es metilo, cianometilo o hidroximetilo. En una modalidad, R8 es metilo. En una modalidad, R8 es cianometilo. En una modalidad, R8 es hidroximetilo. En otra modalidad, R5 y R10 son cada uno alquilo C1-C3, R11 es metilo, R8 es metilo, cianometilo o hidroximetilo, L es un enlace y R4 es arilo o heteroarilo, cada uno sustituido opcionalmente con uno o más R6 o R7.
En una modalidad, la Fórmula II incluye compuestos que tienen la Fórmula II-B:

donde R1, R3, R4, L y m son como se definen para la Fórmula II, R2 es heterociclilalquilo sustituido opcionalmente con uno o más R9, donde R9 es como se define para la Fórmula II, y el anillo de piperazinilo está sustituido opcionalmente con R8, donde R8 es como se define para la Fórmula II.
En esta modalidad, R1 es -C(O)C(RA)

C(RB)p donde RA, RB y p son como se definen para la Fórmula II.
En esta modalidad, RA es halógeno. En una modalidad, el halógeno es flúor o cloro.
En una modalidad, la porción heterociclilo del heterociclilalquilo R2 es un sistema de anillo monocíclico, bicíclico o puenteado que tiene uno o dos heteroátomos en el anillo independientemente seleccionados de N y O. En una modalidad, el heterociclilo R2 es azetidinilo, metilazetidinilo, etilazetidinilo, isopropilazetidinilo, difluoroazetidinilo, ciclopropilazetidinilo, tetrahidropiranilazetidinilo, tetrahidropirano, pirrolidinilo, metilpirrolidinilo, diemetilpirrolidinilo, isopropilpirrolidinilo, cicloalquilalquilpirrolidinilo, hidroxipirrolindinilo, fluoropirrolidinilo, difluoropirrolidinilo, (N-metil)fluoropirrolidinilo, (N-metil)difluoropirrolidinilo, metoxietilpirrolidinilo, N-metilpirrolidinilo alcoxi-sustituido (p. ej., (N-metil)metoxipirrolidinilo), piperazinilo, dimetilaminilpirrolidinilo, pirrolidinona, metilpirrolidinona, morfolinilo, metilmorfolinilo, etilmorfolinilo, isopropilmorfolinilo, oxetanilo, 1,4-oxazepanilo, piperdinilo, metilpiperidinil acilpiperdinilo, cianopiperdinilo, cicloalquilpiperdinilo, halopiperdinilo, dihalopiperdinilo, fluoropiperdinilo, difluoropiperdinilo, alcoxipiperdinilo, pirrolidonilo, piperidinonilo, tetrahidropirrolizinilo, 1, 1 -dióxido de tiomorfolinilo, 3-azabiciclo[3.1.0]hexanilo, oxa-5-azabiciclo[2.2.1]heptan-5-ilo, o azabiciclo[2.2.1]heptan-2-ilo, opcionalmente sustituido con uno o más R9. En una modalidad, cada R9 se selecciona de acilo, oxo, halógeno, ciano, alquilo C1 - C3, alcoxi, hidroxialquilo, heteroalquilo, cicloalquilo, aralquilo, heterociclilo y dialquilamidoalquilo. En una modalidad, L es un enlace. En una modalidad, la porción heterociclilo del heterociclilalquilo R2 es (N-metil)difluoropirrolidinilo, incluyendo 3,3-difluoro-1 -metilpirrolidinilo. En una modalidad, la porción heterociclilo del heterociclilalquilo R2 es N-metilpirrolidinilo.
En una modalidad, R4 es arilo o heteroarilo, cada uno de los cuales está sustituido opcionalmente con uno o más R6, R7 o R8. En una modalidad, R4 es arilo o heteroarilo, cada uno de los cuales está sustituido opcionalmente con uno o más R7. En una modalidad, cada R7 se selecciona independientemente de hidroxilo, amino, halógeno, alquilo C1 - C3, haloalquilo, Q-haloalquilo, cicloalquilo y alcoxi. En una modalidad, el arilo es fenilo sustituido con uno o más grupos R7 seleccionados independientemente de halógeno, hidroxilo, alquilo C1-C3, haloalquilo, Q-haloalquilo y alcoxi. En una modalidad, el arilo es fenilo sustituido con uno o más grupos R7 seleccionados independientemente de halógeno, haloalquilo, metilo, isopropilo, metoxi, Q-haloalquilo e hidroxilo. En una modalidad, el arilo es fenilo sustituido con uno o más grupos R7 seleccionados independientemente de metilo, trifluorometilo, 2,2,2-trifluoroetilo, hidroxilo, trifluorometoxi, hidroxilo, fluoro, cloro, isopropilo, ciclopropilo y trifluorometiltio. En una modalidad, el arilo es fenilo sustituido con uno a tres grupos R7 seleccionados independientemente de hidroxilo, flúor y cloro. En una modalidad, el arilo es fenilo sustituido con hidroxilo y alquilo C1-C3 o dos alquilo C1-C3. En una modalidad, el arilo es fenilo sustituido con Q-haloalquilo e hidroxilo o flúor. En una modalidad, el arilo es naftilo sustituido con uno o más grupos R7 seleccionados independientemente de halógeno, hidroxilo, alquilo C1-C3, haloalquilo, Q-haloalquilo y alcoxi. En una modalidad, el arilo es naftilo sustituido con uno o más grupos R7 seleccionados independientemente de halógeno, haloalquilo, metilo, isopropilo, metoxi, Q-haloalquilo e hidroxilo. En una modalidad, R4 es naftilo sustituido opcionalmente con uno o más sustituyentes R7 seleccionados independientemente de hidroxilo, halógeno, alquilo C1-C3, amino y haloalquilo. En una modalidad, R4 es naftilo sustituido opcionalmente con uno a
tres sustituyentes R7 seleccionados independientemente de difluorometilo, metilo, hidroxilo, amino, fluoro y cloro. En una modalidad, el arilo es naftilo sustituido opcionalmente con uno o más halógenos. En una modalidad, el arilo es naftilo sustituido con hidroxilo y trifluorometilo o alquilo C1-C3. En una modalidad, el arilo es naftilo sustituido con hidroxilo. En una modalidad, R4 es heteroarilo, en donde el heteroarilo es indazolilo sustituido opcionalmente con uno o dos de R7 seleccionados independientemente de alcoxi, haloalquilo y alquilo C1-C6.
En una modalidad, R4 es heteroarilo, en donde el heteroarilo es quinolinilo o isoquinolinilo, cada uno sustituido opcionalmente con uno o más R6, R7 o R8. En una modalidad, R4 es heteroarilo, en donde el heteroarilo es quinolinilo o isoquinolinilo, cada uno sustituido opcionalmente con uno o más R6, R7 o R8 seleccionados independientemente de oxo, amino, hidroxilo, alquilo C1-C3 e hidroxilo. En una modalidad, el heteroarilo R4 es un piridinilo sustituido opcionalmente con uno o más R6, R7 o R8. En una modalidad, el heteroarilo R4 es piridinilo sustituido opcionalmente con uno o más R7 seleccionados independientemente de alquilo C1 - C3, halógeno y haloalquilo. En una modalidad, el heteroarilo R4 es indolilo sustituido opcionalmente con uno o más R6, R7 o R8. En una modalidad, el heteroarilo R4 es indolilo sustituido opcionalmente con uno o dos R7 seleccionado independientemente de hidroxilo y alquilo C1 - C3. En una modalidad, R11 es metilo. En una modalidad, el anillo de piperazinilo no está sustituido. En una modalidad, el anillo de piperazinilo de Fórmula II-B está sustituido con R8. En una modalidad, R8 es alquilo C1 - C3 sustituido opcionalmente con ciano, hidroxilo o metoxi. En una modalidad, R8 es metilo, cianometilo, hidroximetilo o metoximetilo.
Los ejemplos no limitantes de compuestos de Fórmula (II), Fórmula II-A y Fórmula II-B se seleccionan del grupo que consiste en:






















o una sal farmacéuticamente aceptable de los mismos.
Los compuestos de Fórmula (II), Fórmula II-A o Fórmula II-B pueden formularse en composiciones farmacéuticas.
Composiciones farmacéuticas
En otro aspecto, la invención proporciona composiciones farmacéuticas que comprenden un inhibidor de KRas G12C según la invención y un portador, excipiente o diluyente farmacéuticamente aceptables. Los compuestos de la invención pueden formularse mediante cualquier método bien conocido en la técnica y pueden prepararse para su administración por cualquier vía, que incluye, sin limitación, la parenteral, oral, sublingual, transdérmica, tópica, intranasal, intratraqueal o intrarrectal. En determinadas modalidades, los compuestos de la invención se administran por vía intravenosa en un entorno hospitalario. En una modalidad, la administración puede ser por vía oral.
Las características del portador dependerán de la vía de administración. Como se usa en la presente descripción, el término “ farmacéuticamente aceptable” significa un material no tóxico que es compatible con un sistema biológico tal como una célula, cultivo celular, tejido u organismo, y que no interfiere con la eficacia de la actividad biológica del ingrediente o ingredientes activos. Por tanto, las composiciones según la invención pueden contener, adicionalmente al inhibidor, diluyentes, cargas, sales, tampones, estabilizantes, solubilizantes y otros materiales bien conocidos en la técnica. La preparación de formulaciones farmacéuticamente aceptables se describe, por ejemplo, en Remington's Pharmaceutical Sciences, 18a edición, ed. A. Gennaro, Mack Publishing Co., Easton, Pensilvania, 1990.
Como se usa en la presente descripción, el término sal farmacéuticamente aceptable se refiere a sales que retienen la actividad biológica deseada de los compuestos identificados anteriormente y exhiben efectos toxicológicos no deseados mínimos o nulos. Ejemplos de tales sales incluyen, pero no se limitan a, sales de adición de ácido formadas con ácidos inorgánicos (por ejemplo, ácido clorhídrico, ácido bromhídrico, ácido sulfú rico, ácido fosfórico, ácido nítrico y similares) y sales formadas con ácidos orgánicos tales como ácido acético, ácido oxálico, ácido tartárico, ácido succínico, ácido málico, ácido ascórbico, ácido benzoico, ácido tánico, ácido pamoico, ácido algínico, ácido poliglutámico, ácido naftalenosulfónico, ácido naftalenodisulfónico y ácido poligalacturónico. Los compuestos pueden administrarse además como sales cuaternarias farmacéuticamente aceptables conocidas por los expertos en la técnica, que incluyen específicamente la sal de amonio cuaternario de fórmula -NR+Z-, en donde R es hidrógeno, alquilo o bencilo, y Z es un contraión, que incluye cloruro, bromuro, yoduro, -O-alquilo, toluenosulfonato, metilsulfonato, sulfonato, fosfato o carboxilato (como benzoato, succinato, acetato, glicolato, maleato, malato, citrato, tartrato, ascorbato, benzoato, cinamoato, mandeloato, benciloato y acetato de difenilo).
El compuesto activo se incluye en el portador o diluyente farmacéuticamente aceptables en una cantidad suficiente para administrar a un paciente una cantidad terapéuticamente eficaz sin causar efectos tóxicos graves en el paciente tratado. En una modalidad, una dosis del compuesto activo para todas las condiciones mencionadas anteriormente está en el intervalo de aproximadamente 0,01 a 300 mg/kg, por ejemplo de 0,1 a 100 mg/kg por día, y como ejemplo adicional, de 0,5 a aproximadamente 25 mg por kilogramo de peso corporal del receptor por día. Una dosis tópica típica variará de 0,01 a 3 % p/p en un portador adecuado. El intervalo de dosificación eficaz de los derivados farmacéuticamente aceptables puede calcularse en base al peso del compuesto original que se va a suministrar. Si el derivado exhibe actividad en sí mismo, la dosis eficaz puede estimarse como anteriormente mediante el uso del peso del derivado, o por otros medios conocidos por los expertos en la técnica.
Las composiciones farmacéuticas que comprenden compuestos de la presente invención pueden usarse en los métodos de uso descritos en la presente descripción.
Métodos de uso
En otro aspecto más, la descripción proporciona métodos para inhibir la actividad de KRas G12C en una célula, que comprenden poner en contacto la célula en la que se desea la inhibición de la actividad de KRas G12C con una cantidad eficaz de un compuesto de la invención, sales farmacéuticamente aceptables del mismo o composiciones farmacéuticas que contienen el compuesto o la sal farmacéuticamente aceptable del mismo. En este aspecto, el contacto es in vitro.
Como se usa en la presente descripción, el término “ poner en contacto” se refiere a reunir los restos indicados en un sistema in vitro o en un sistema in vivo. Por ejemplo, “ poner en contacto” una KRas G12C con un compuesto proporcionado en la presente descripción incluye introducir un compuesto proporcionado en la presente descripción en una muestra que contiene una preparación celular o purificada que contiene la KRas G12C.
En una modalidad, una célula en la que se desea la inhibición de la actividad de KRas G12C se pone en contacto con una cantidad eficaz de un compuesto de la invención, para modular negativamente la actividad de KRas G12C. En otras modalidades, puede usarse una cantidad terapéuticamente eficaz de sales o composiciones farmacéuticas, farmacéuticamente aceptables, que contienen el compuesto de la invención.
Mediante la modulación de la actividad de KRas G12C negativamente, los métodos descritos en la presente descripción se diseñan para inhibir la proliferación celular no deseada resultante de la actividad mejorada de KRas G12C dentro de la célula. Las células pueden ponerse en contacto en una dosis única o en dosis múltiples según un régimen de tratamiento particular para efectuar la modulación negativa deseada de KRas G12C. El grado de modificación covalente de KRas G12C puede controlarse in vitro mediante el uso de métodos bien conocidos, que incluyen los descritos en el Ejemplo A más abajo. Adicionalmente, la actividad inhibidora de los compuestos ilustrativos en las células puede controlarse, por ejemplo, mediante medición de la inhibición de la actividad de KRas G12C de la cantidad de ERK fosfilada, que incluye las descritas en el Ejemplo B más abajo, para evaluar la eficacia del tratamiento y las dosis pueden ajustarse en consecuencia por el médico tratante.
En otro aspecto, se proporciona un compuesto de la descripción o una sal farmacéuticamente aceptable del mismo para usarlo en un método para el tratamiento de un cáncer de KRas G12C, comprendiendo el método administrar a un paciente que tiene cáncer una cantidad terapéuticamente eficaz de un compuesto de la invención o una sal farmacéuticamente aceptable del mismo, solo o combinado con un vehículo, excipiente o diluyentes farmacéuticamente aceptables, en donde el cáncer se selecciona del grupo que consiste en Cardíaco: sarcoma seleccionado de angiosarcoma, fibrosarcoma, rabdomiosarcoma y liposarcoma, mixoma, rabdomioma, fibroma, lipoma y teratoma; Pulmón: carcinoma broncogénico seleccionado de células escamosas, células pequeñas no diferenciadas, células grandes no diferenciadas y adenocarcinoma, carcinoma bronquioloalveolar, adenoma bronquial, sarcoma, linfoma, hamartoma condromatoso, mesotelioma; Gastrointestinal: de esófago seleccionado de carcinoma de células escamosas, adenocarcinoma, leiomiosarcoma y linfoma, de estómago seleccionado de carcinoma, linfoma y leiomiosarcoma, de páncreas seleccionado de adenocarcinoma ductal, insulinoma, glucagonoma, gastrinoma, tumores carcinoides y vipoma, de intestino delgado seleccionado de adenocarcinoma, linfoma, tumores carcinoides, sarcoma de Kaposi, leiomioma, hemangioma, lipoma, neurofibroma y fibroma, de intestino grueso seleccionado de adenocarcinoma, adenoma tubular, adenoma velloso, hamartoma y leiomioma; Tracto genitourinario: de riñón seleccionado de adenocarcinoma, tumor de Wilm/nefroblastoma, linfoma y leucemia, de vejiga y uretra seleccionado de carcinoma de células escamosas, carcinoma de células transicionales y adenocarcinoma, de próstata seleccionado de adenocarcinoma y sarcoma, de testículos seleccionado de seminoma, teratoma, carcinoma embrionario, teratocarcinoma, coriocarcinoma, sarcoma, carcinoma de células intersticiales, fibroma, fibroadenoma, tumores adenomatoides y lipoma; Hígado: hepatoma/carcinoma hepatocelular, colangiocarcinoma, hepatoblastoma, angiosarcoma, adenoma hepatocelular, hemangioma; Tracto biliar: carcinoma de vesícula biliar, carcinoma ampular, colangiocarcinoma; Óseo: sarcoma osteogénico/osteosarcoma, fibrosarcoma, histiocitoma fibroso maligno, condrosarcoma, sarcoma de Ewing, linfoma maligno, sarcoma de células reticulares, mieloma múltiple, cordoma tumoral maligno de células gigantes, osteocronoma/exostosis osteocartilaginosa, condroma benigno, condroblastoma, condromixofibroma, osteoma osteoide y tumores de células gigantes; Sistema nervioso: de cráneo seleccionado de osteoma, hemangioma, granuloma, xantoma y osteítis deformante; de meninges seleccionado de meningioma, meningiosarcoma y gliomatosis; de cerebro seleccionado de astrocitoma, meduloblastoma, glioma, ependimoma, germinoma, pinealoma, glioblastoma multiforme, oligodendroglioma, schwannoma, retinoblastoma y tumores congénitos, de médula espinal seleccionado de neurofibroma, meningioma, glioma y sarcoma); Ginecológico: de útero seleccionado de carcinoma endometrial seleccionado de cistoadenocarcinoma seroso, cistadenocarcinoma mucinoso y carcinoma no clasificado, tumores de células de la granulosa-tecal, tumores de células de Sertoli-Leydig, disgerminoma y teratoma maligno, de vulva seleccionado de carcinoma de células escamosas, carcinoma intraepitelial, adenocarcinoma, fibrosarcoma y melanoma), de vagina seleccionado de carcinoma de células claras, carcinoma de células escamosas, sarcoma botrioide/rabdomiosarcoma embrionario, carcinoma de las trompas de Falopio; Hematológico: de sangre seleccionado de leucemia mieloide aguda, leucemia mieloide crónica, leucemia linfoblástica aguda, leucemia linfocítica crónica, enfermedades mieloproliferativas, mieloma múltiple y síndrome mielodisplásico, enfermedad de Hodgkin, linfoma maligno, linfoma no Hodgkin; Piel: melanoma maligno, carcinoma de células basales, carcinoma de células escamosas, sarcoma de Kaposi, lunares, nevos displásicos, lipoma, angioma, dermatofibroma, queloides, psoriasis; y Glándulas suprarrenales: neuroblastoma.
En una modalidad, el cáncer asociado a KRas G12C es cáncer de pulmón.
Las composiciones proporcionadas en la presente descripción son para usarlas en el tratamiento de una amplia variedad de cánceres, que incluyen tumores tales como carcinomas de pulmón, próstata, mama, cerebro, piel, carcinomas cervicales, carcinomas testiculares, etc. Más particularmente, cánceres que pueden ser tratados por las composiciones y métodos de la descripción incluyen, pero no se limitan a, tipos de tumores tales como sarcomas y carcinomas astrocíticos, de mama, cervicales, colorrectales, endometriales, esofágicos, gástricos, de cabeza y cuello, hepatocelulares, laríngeos, pulmonares, orales, ováricos, de próstata y tiroideos. Más específicamente, estos compuestos se pueden usar para tratar: Cardíaco: sarcoma (angiosarcoma, fibrosarcoma, rabdomiosarcoma, liposarcoma), mixoma, rabdomioma, fibroma, lipoma y teratoma; Pulmón: carcinoma broncogénico (células escamosas, células pequeñas no diferenciadas, células grandes no diferenciadas, adenocarcinoma), carcinoma alveolar (bronquiolar), adenoma bronquial, sarcoma, linfoma, hamartoma condromatoso, mesotelioma; Gastrointestinal: de esófago (carcinoma de células escamosas, adenocarcinoma, leiomiosarcoma, linfoma), de estómago (carcinoma, linfoma, leiomiosarcoma), de páncreas (adenocarcinoma ductal, insulinoma, glucagonoma, gastrinoma, tumores carcinoides, vipoma), de intestino delgado (adenocarcinoma, linfoma, tumores carcinoides, sarcoma de Kaposi, leiomioma, hemangioma, lipoma, neurofibroma, fibroma), de intestino grueso (adenocarcinoma, adenoma tubular, adenoma velloso, hamartoma, leiomioma); Tracto genitourinario: de riñón (adenocarcinoma, tumor de Wilms (nefroblastoma), linfoma, leucemia), de vejiga y uretra (carcinoma de células escamosas, carcinoma de células
transicionales, adenocarcinoma), de próstata (adenocarcinoma, sarcoma), de testículo (seminoma, teratoma, carcinoma embrionario, teratocarcinoma, coriocarcinoma, sarcoma, carcinoma de células intersticiales, fibroma, fibroadenoma, tumores adenomatoides, lipoma); Hígado: hepatoma (carcinoma hepatocelular), colangiocarcinoma, hepatoblastoma, angiosarcoma, adenoma hepatocelular, hemangioma; Tracto biliar: carcinoma de vesícula biliar, carcinoma ampular, colangiocarcinoma; Óseo: sarcoma osteogénico (osteosarcoma), fibrosarcoma, histiocitoma fibroso maligno, condrosarcoma, sarcoma de Ewing, linfoma maligno (sarcoma de células reticulares), mieloma múltiple, cordoma tumoral maligno de células gigantes, osteocronoma (exostosis osteocartilaginosa), condroma benigno, condroblastoma, condromixofibroma, osteoma osteoide y tumores de células gigantes; Sistema nervioso: de cráneo (osteoma, hemangioma, granuloma, xantoma, osteítis deformante), de meninges (meningioma, meningiosarcoma, gliomatosis), de cerebro (astrocitoma, meduloblastoma, glioma, ependimoma, germinoma (pinealoma), glioblastoma multiforme, oligodendroglioma, schwannoma, retinoblastoma, tumores congénitos), de médula espinal (neurofibroma, meningioma, glioma, sarcoma); Ginecológico: de útero (carcinoma endometrial), de cuello uterino (carcinoma de cuello uterino, displasia cervical pretumoral), de ovarios (carcinoma de ovario (cistoadenocarcinoma seroso, cistadenocarcinoma mucinoso, carcinoma no clasificado), tumores de células de la granulosatecal, tumores de células de Sertoli-Leydig, disgerminoma, teratoma maligno), de vulva (carcinoma de células escamosas, carcinoma intraepitelial, adenocarcinoma, fibrosarcoma, melanoma), de vagina (carcinoma de células claras, carcinoma de células escamosas, sarcoma botrioide (rabdomiosarcoma embrionario), de las trompas de Falopio (carcinoma); Hematológico: de sangre (leucemia mieloide (aguda y crónica), leucemia linfoblástica aguda, leucemia linfocítica crónica, enfermedades mieloproliferativas, mieloma múltiple, síndrome mielodisplásico), enfermedad de Hodgkin, linfoma no Hodgkin (linfoma maligno); Piel: melanoma maligno, carcinoma de células basales, carcinoma de células escamosas, sarcoma de Kaposi, lunares, nevos displásicos, lipoma, angioma, dermatofibroma, queloides, psoriasis; y Glándulas suprarrenales: neuroblastoma. En determinadas modalidades, el cáncer es un cáncer de pulmón no microcítico.
La concentración y la vía de administración al paciente variarán en dependencia del cáncer a tratar. Los compuestos, sales farmacéuticamente aceptables de los mismos y composiciones farmacéuticas que comprenden dichos compuestos y sales además pueden coadministrarse con otros compuestos antineoplásicos, por ejemplo, quimioterapia, o usarse en combinación con otros tratamientos, tales como radiación o intervención quirúrgica, ya sea como adyuvante antes de la cirugía o después de la operación.
Los compuestos para usar para tratar un cáncer asociado con KRas G12C también pueden comprender (a) determinar que el cáncer está asociado con una mutación G12C de KRas (p. ej., un cáncer asociado con KRas G12C) (p. ej., determinado usando un ensayo o kit aprobado por una agencia reguladora, p. ej., aprobado por la FDA); y (b) administrar al paciente una cantidad terapéuticamente eficaz de un compuesto de la invención o una sal farmacéuticamente aceptable del mismo, o una composición farmacéutica del mismo.
Un experto en la técnica reconocerá que, tanto los ensayos in vivo como in vitro que usan modelos de células y/o animales adecuados, conocidos y generalmente aceptados, son predictivos de la capacidad de un compuesto de prueba para tratar o prevenir un trastorno dado.
Un experto en la técnica reconocerá además que los ensayos clínicos en humanos, que incluyen los primeros ensayos en humanos, ensayos de eficacia y variación de las dosis, en pacientes sanos y/o aquellos que padecen un trastorno dado, pueden completarse según métodos bien conocidos en las técnicas médicas y clínicas.
Esquemas de reacción y ejemplos
Los compuestos de la presente invención pueden prepararse a partir de reactivos disponibles comercialmente mediante el uso de los métodos sintéticos y esquemas de reacción descritos en la presente descripción, o mediante el uso de otros reactivos y métodos convencionales bien conocidos por los expertos en la técnica.
Por ejemplo, los compuestos de la presente invención pueden prepararse según los Esquemas Generales de Reacción I y II.
Esquemas generales de reacción

En la etapa A, una dihidropiridopirimidina (1) apropiadamente funcionalizada se acopla a un heterociclo que contiene una especie de amina nucleófila, y la otra se une a un grupo protector para proporcionar el compuesto (2). Este acoplamiento se produce en un disolvente tal como diclorometano en presencia de una base tal como trietilamina o base de Hunig. En la etapa B, el grupo Boc del compuesto (2) se elimina mediante el uso de condiciones conocidas en la técnica, por ejemplo, con ácido trifluoroacético en un disolvente tal como diclorometano, para proporcionar el compuesto (3). En la etapa C, el sustituyente R4 se introduce con un acoplamiento de paladio, mediante el uso de un sistema arilo o heteroarilo funcionalizado adecuado, por ejemplo un triflato de arilo, en presencia de un catalizador de paladio como Pd2DBA3/Xantphos en un disolvente tal como tolueno con una base tal como terc-butóxido de sodio para proporcionar el compuesto (4). En la etapa D, se elimina el grupo protector del compuesto (4) de anillo X, por ejemplo hidrogenólisis por Pd/C en presencia de H2 en un disolvente polar tal como EtOH/THF para proporcionar el compuesto (5). En la etapa final, E, se introduce R1, por ejemplo, mediante tratamiento con un cloruro de ácido que tiene la fórmula Cl-C(O)C(RA)

C(RB)p, o un anhídrido que tiene la fórmula C(RB)p
C(RA)C(O)OC(O)C(RA)
C(RB)p, donde RA, RB y p son como se han definido anteriormente. En algunos casos, la especie
R4 contendrá además un grupo protector, que puede eliminarse en una etapa posterior de la secuencia sintética.
Los compuestos (1), (2), (3), (4) y (5) como se muestra y describe anteriormente para el Esquema I son útiles como intermedios para preparar compuestos de la invención.

En la etapa A, una dihidropiridopirimidina (6) apropiadamente funcionalizada se acopla a un heterociclo que contiene una especie de amina nucleófila, y la otra se une a un grupo protector para proporcionar el compuesto (7). Este acoplamiento se produce en un disolvente tal como diclorometano en presencia de una base tal como trietilamina o base de Hunig. En la etapa B, el sustituyente -Y-R2 se introduce mediante la sustitución del cloro por un nucleófilo, por ejemplo (S)-1-(dimetilamino-propan-2-ol en un disolvente polar tal como dioxano para proporcionar el compuesto (8). En la etapa C, el grupo Boc se elimina mediante el uso de condiciones conocidas en la técnica, por ejemplo con ácido trifluoroacético en un disolvente tal como diclorometano para proporcionar el compuesto (9). En la etapa D, el sustituyente R4 se introduce con un acoplamiento de paladio, mediante el uso de un sistema arilo o heteroarilo funcionalizado adecuado, por ejemplo, un triflato de arilo, en presencia de un catalizador de paladio como Pd2DBA3/BINAP en un disolvente tal como tolueno con una base tal como terc-butóxido de sodio para proporcionar el compuesto (10). En la etapa E, se elimina el grupo protector del anillo X, por ejemplo, la hidrogenólisis por Pd/C en presencia de H2 en un disolvente polar tal como EtOH/THF para proporcionar el compuesto (11). En la etapa F, se introduce R1 para proporcionar un compuesto de la invención, por ejemplo, mediante tratamiento con un cloruro de ácido que tiene la fórmula Cl-C(O)C(RA)

C(RB)p o un anhídrido que tiene la fórmula C(RB)p
C(RA)C(O)OC(O)C(RA)

C(RB)p, donde RA, RB y p son como se han definido anteriormente. En algunos casos, las especies R4 y R2 pueden contener además grupos protectores, que pueden eliminarse en una etapa posterior de la secuencia sintética.
Los compuestos (6), (7), (8), (9), (10) y (11) como se ha mostrado y descrito anteriormente para el Esquema 2 son útiles como intermedios para preparar compuestos de la invención.
Los compuestos de la presente invención pueden tener uno o más centros quirales y pueden sintetizarse como mezclas estereoisoméricas, isómeros de constitución idéntica que difieren en la disposición de sus átomos en el espacio. Los compuestos pueden usarse como mezclas o los componentes/isómeros individuales pueden separarse mediante el uso de
reactivos disponibles comercialmente y métodos convencionales para el aislamiento de estereoisómeros y enantiómeros bien conocidos por los expertos en la técnica, por ejemplo, mediante el uso de CHIRALPAK® (Sigma-Aldrich) o Columnas de HPLC cromatográfica quiral CHIRALCEL® (Diacel Corp) según las instrucciones del fabricante. Alternativamente, los compuestos de la presente invención pueden sintetizarse mediante el uso de reactivos quirales ópticamente puros e intermedios para preparar isómeros o enantiómeros individuales. A menos que se indique lo contrario, todas las formas quirales (enantioméricas y diastereoméricas) y racémicas están dentro del alcance de la invención. A menos que se indique lo contrario, siempre que la descripción, que incluye las reivindicaciones, se refiera a compuestos de la invención, debe entenderse que el término “ compuesto” abarca todas las formas quirales (enantioméricas y diastereoméricas) y racémicas.
Los siguientes ejemplos están destinados a ilustrar más determinadas modalidades de la invención y no pretenden limitar el alcance de la invención. Los compuestos que están dentro del alcance de la Fórmula (II) son compuestos de la invención. Todos los demás son compuestos de referencia.
Intermedio 1

trifluorometanosulfonato de 3-(metoximetoxi)naftalen-1-ilo

Se disolvió trifluorometanosulfonato de 3-hidroxinaftalen-1-ilo (13,101 g, 44,831 mmol) en diclorometano (100 ml) y se agitó a 0 °C. A esta solución se le añadieron cloro(metoxi)metano (3,7456 ml, 49,315 mmol) y base de Hunig (11,745 ml, 67,247 mmol). La reacción se agitó a 0 °C durante 4 h. La reacción se repartió con HCl 1 M y se lavó con bicarbonato de sodio saturado. Las capas orgánicas combinadas se secaron sobre sulfato de magnesio y se concentraron al vacío. El material concentrado se cargó sobre una columna de gel de sílice 120 g RediSep® gold con diclorometano y se purificó por cromatografía de fase normal (CombiFlash®, 0 %-20 % de acetato de etilo/hexanos como eluyente) para dar trifluorometanosulfonato de 3-(metoximetoxi)naftalen-1-ilo (11,785 g, 35,045 mmol, 78,171 % de rendimiento).
Intermedio 2

2-bromo-7-(metoximetoxi)naftaleno

A una solución de 7-bromonaftalen-2-ol (2,0 g, 9,0 mmol) en dimetil acetamida (40 ml) se le añadieron cloro(metoxi)metano (1,4 g, 18 mmol) y carbonato de cesio (5,8 g, 18 mmol) y la mezcla de reacción se agitó durante una noche a temperatura ambiente. La reacción se diluyó con agua y la capa acuosa se lavó con acetato de etilo. Las capas orgánicas combinadas se lavaron con agua y salmuera, se secaron sobre sulfato de magnesio y se concentraron al vacío. El material bruto se purificó por cromatografía de fase normal usando 5-50 % de acetato de etilo/hexanos como eluyente para dar 2-bromo-7-(metoximetoxi)naftaleno (1,0 g, 3,7 mmol, 42 % de rendimiento).
Intermedio 3

2-bromo-1-fluoro-3-(metoximetil)benceno

A una solución agitada de 2-bromo-3-fluorofenol (1422 mg, 7,445 mmol) en 22 ml de tetrahidrofurano a temperatura ambiente en una atmósfera de nitrógeno se le añadió poco a poco NaH (327,6 mg, 8,190 mmol) puro en forma de un sólido. Después de 15 minutos, se había formado una solución. Se añadió cloro(metoxi)metano (678,6 μl, 8,934 mmol) mediante una jeringa. Después de agitar durante 2 horas, la reacción se interrumpió con una solución saturada de cloruro de amonio y después se repartió entre acetato de etilo (30 ml) y agua (30 ml). Las capas orgánicas combinadas se aislaron, se lavaron con salmuera, se secaron sobre MgSCL, se filtraron y se concentraron. El producto bruto se cargó en una cantidad mínima de diclorometano en una columna RediSep® de 40 gramos humedecida previamente con hexanos y se eluyó con un gradiente de acetato de etilo/hexanos (0 % a 20 % de acetato de etilo). Las fracciones que contenían el producto se combinaron y se concentraron para proporcionar el producto en forma de un aceite transparente (1,45 g, 83 %).
Intermedio 4
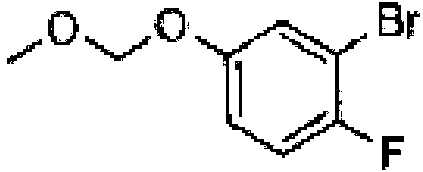
2-bromo-1-fluoro-4-(metoximetoxi)benceno

A una solución agitada de 3-bromo-4-fluorofenol (327 mg, 1,71 mmol) en 5,1 ml de tetrahidrofurano a temperatura ambiente en una atmósfera de nitrógeno se le añadió poco a poco NaH (75,3 mg, 1,88 mmol) puro en forma de un sólido. Después de 15 minutos, se había formado una solución. Se añadió cloro(metoxi)metano (156 μl, 2,05 mmol) mediante una jeringa. Después de agitar durante 2 horas, la reacción se interrumpió con una solución saturada de cloruro de amonio y se repartió entre acetato de etilo y agua. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre MgSO4, se filtraron y se concentraron. El producto bruto se cargó en una cantidad mínima de diclorometano en una columna RediSep® de 24 gramos humedecida previamente con hexanos y eluyendo con un gradiente de acetato de etilo/hexanos (0 % a 20 % de acetato de etilo). Las fracciones que contenían el producto se combinaron y se concentraron para proporcionar el producto en forma de un aceite transparente (120 mg, 29,8 %)
Intermedio 5

4-bromo-5-metil-1-((2-(trimetilsilil)etoxi)metil)-1H-indazol

A una solución de 4-bromo-5-metil-1H-indazol (0,7 g, 3,3 mmol) en dimetil acetamida (30 ml) enfriada a 0 °C se le añadió poco a poco NaH (0,19 g, 4,6 mmol) y la mezcla de reacción se purgó con nitrógeno. La reacción se agitó durante 20 minutos y después se añadió (2-(clorometoxi)etil)trimetilsilano (0,83 g, 5,0 mmol) y la reacción se agitó durante 2 horas mientras se calentaba a temperatura ambiente. La reacción se interrumpió vertiéndola en agua y la capa acuosa se extrajo en acetato de etilo. Las capas orgánicas combinadas se lavaron con agua y salmuera, se secaron sobre MgSO4 y se concentraron al vacío. El material bruto se purificó por cromatografía usando 10-50 % de acetato de etilo/hexanos como eluyente para dar 4-bromo-5-metil-1-((2-(trimetilsilil)etoxi)metil)-1H-indazol (0,87 g, 79 %).
Intermedio 6
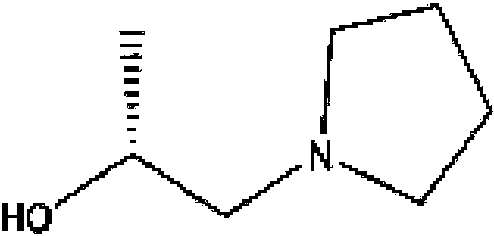
(R)-1-(pirrolidin-1-il)propan-2-ol
En un tubo sellado, óxido de R-(+)-propileno (3,69 ml, 52,7 mmol) se enfrió a -78 °C y después se roció con dimetil amina anhidra durante unos minutos. La mezcla de reacción se calentó a 70 °C durante 16 horas. La reacción se enfrió y se concentró al vacío durante 20 minutos para proporcionar (R)-1 -(pirrolidin-1 -il)propan-2-ol (5,35 g, 41,4 mmol, 98,2 % de rendimiento).
Intermedio 7

(R)-1-morfolinopropan-2-ol
En un tubo sellado, óxido de R-(+)-propileno (2,111 ml, 30,13 mmol) y morfolina (1,490 ml, 17,22 mmol) se calentaron a 70 °C durante 20 horas. La reacción se enfrió y se concentró al vacío para proporcionar (R)-1-morfolinopropan-2-ol (2,47 g, 17,01 mmol, 98,80 % de rendimiento).
Intermedio 8

(R)-1-(dimetilamino)butan-2-ol
En un tubo sellado, óxido de R-(+)-propileno (4,00 g, 55,5 mmol) y dimetilamina (1,00 g, 22,2 mmol) se calentaron a 65 °C durante 18 horas. La reacción se enfrió y se concentró al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice (0-12 % de MeOH en DCM) para proporcionar (R)-1-(dimetilamino)butan-2-ol (1,38 g, 11,8 mmol, 53,1 % de rendimiento).
Intermedio 9

(R)-1-((R)-3-metoxipirrolidin-1-il)propan-2-ol
En un tubo sellado, clorhidrato de (R)-3-metoxipirrolidina (1,00 g, 7,27 mmol), TEA (2,03 ml, 14,5 mmol) y óxido de R-(+)-propileno (1,27 ml, 18,2 mmol) se calentaron a 65 °C durante 18 horas. La reacción se enfrió y se concentró al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice (0-12 % de MeOH en DCM) para proporcionar (R)-1-((R)-3-metoxipirrolidin-1-il)propan-2-ol (775 mg, 4,87 mmol, 67,0 % de rendimiento).
Intermedio 10
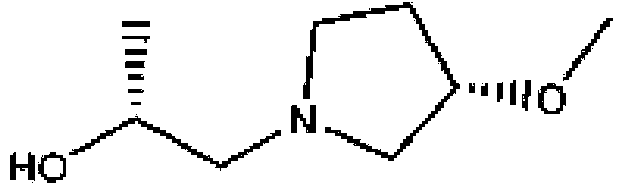
(R)-1-((S)-3-metoxipirrolidin-1-il)propan-2-ol
En un tubo sellado, clorhidrato de (S)-3-metoxipirrolidina (1,00 g, 7,27 mmol), TEA (2,03 ml, 14,5 mmol) y óxido de R-(+)-propileno (1,27 ml, 18,2 mmol) se calentaron a 65 °C durante 18 horas. La reacción se enfrió y se concentró al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice (0-12 % de MeOH en DCM) para proporcionar (R)-1-((S)-3-metoxipirrolidin-1-il)propan-2-ol (781 mg, 4,90 mmol, 67,5 % de rendimiento)
Intermedio 11

(R)-1-((S)-3-((terc-butildimetilsilil)oxi)pirrolidin-1-il)propan-2-ol
En un tubo sellado, óxido de R-(+)-propileno (0,609 ml, 8,69 mmol) y (S)-3-((terc-butildimetilsilil)oxi)pirrolidina (1,00 g, 4,97 mmol) se calentaron a 70 °C durante 20 horas. La reacción se enfrió y se concentró al vacío para proporcionar (R)-1-((S)-3-((terc-butildimetilsilil)oxi)pirrolidin-1-il)propan-2-ol (1,29 g, 4,20 mmol, 84,6 % de rendimiento).
Intermedio 12
Boc

2-(hidroximetil)-4-metilpiperazin-1-carboxilato de terc-butilo
A una suspensión de cloruro de litio (246 mg, 5,81 mmol) y borohidruro de litio (126 mg, 5,81 mmol) en etanol (9 ml), a 0 °C en una atmósfera de nitrógeno se le añadió gota a gota una solución de 4-metilpiperazin-1,2-dicarboxilato de 1-(tercbutil) 2-metilo (750 mg, 2,90 mmol) en THF seco (6 ml). La reacción se agitó durante una noche, formando un precipitado de color blanco. El precipitado se filtró y se lavó con etanol. El filtrado combinado y los extractos orgánicos se concentraron para proporcionar un resido de color blanco que se extrajo con acetato de etilo. Las capas orgánicas combinadas se lavaron con una solución saturada de cloruro de sodio, se secaron sobre sulfato de sodio y se concentraron al vacío. El residuo se purificó por cromatografía con 10 % de MeOH isocrático en DCM con 0,2 % de NH4OH para proporcionar 2-(hidroximetil)-4-metilpiperazin-1-carboxilato de terc-butilo (104 mg, 0,452 mmol, 15,6 % de rendimiento).
Intermedio 13

(S)-2-(2-metilpiperidin-1-il)etan-1-ol
Una mezcla de (S)-2-metilpiperidina (100 mg, 1,01 mmol), 2-bromoetanol (78 μl, 139 mg, 1,11 mmol, 1,1 eq.), yoduro de sodio (151 mg, 1 eq.), carbonato de potasio (418 mg, 3 eq.) y acetonitrilo (1 ml) en un vial de 4 ml se purgó con nitrógeno, se selló y se agitó a temperatura ambiente durante 2 días. La mezcla de reacción se repartió entre éter dietílico (15 ml) y agua (2 ml). La capa de éter se lavó con salmuera (2 ml), se acidificó con TFA y se secó a alto vacío durante 2 días. El residuo se lavó con éter (3 ml), se diluyó con agua (0,5 ml) y se basificó con NaOH 10 M (0,2 ml). Las capas se separaron y la capa superior se secó cuidadosamente sobre NaOH. La solución de éter se evaporó en una atmósfera de nitrógeno para producir (S)-2-(2-metilpiperidin-1-il)etan-1-ol bruto (100 mg, 0,698 mmol, 69,24 % de rendimiento) en forma de un aceite incoloro.
Intermedio 14

(R)-2-(2-metilpiperidin-1-il)etan-1-ol
Se sintetizó según el método del Intermedio 13, usando (R)-2-metilpiperidina (99 mg, 1 mmol) en lugar de (S)-2-metilpiperidina.
Intermedio 15

(S)-2-(3 -metoxipiperidin-1-il)etan-1-ol
Se sintetizó según el método del Intermedio 13, usando (S)-3-metoxipiperidina (173 mg, 1,50 mmol) en lugar de (S)-2-metilpiperidina.
Intermedio 16

(R)-2-(3-metoxipiperidin-1-il)etan-1-ol
Se sintetizó según el método del Intermedio 13, usando R-3-metoxipiperidina (173 mg, 1,50 mmol) en lugar de (S)-2-metilpiperidina.
Intermedio 17

3-(1,4-oxazepan-4-il)propan-1-ol
A un vial se le añadió homomorfolina (0,250 g, 2,472 mmol), acetonitrilo (4,943 ml, 2,472 mmol) y 3-bromo-1-propanol (0,2459 ml, 2,719 mmol). Se añadió carbonato de potasio (0,6832 g, 4,943 mmol) y la mezcla se calentó a 50 °C y se agitó durante 6 horas. La mezcla se enfrió a temperatura ambiente, se diluyó con DCM, se filtró y los sólidos recogidos se lavaron con DCM. El filtrado se concentró al vacío y el aceite bruto se purificó por cromatografía en columna (Biotage Isolera, 12 g Isco RediSep Gold, 10-20 % de MeOH/DCM con 0,2 % de NH4OH) para proporcionar 3-(1,4-oxazepan-4-il)propan-1-ol (0,272 g, 1,708 mmol) en forma de un aceite incoloro.
Intermedio 18

3-((1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propan-1-ol
Se sintetizó según el método del Intermedio 17, usando (1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptano (0,250 g, 2,522 mmol) en lugar de homomorfolina.
Intermedio 19

2-(4-metoxipiperidin-1-il)etan-1-ol
Se sintetizó según el método del Intermedio 13, usando 4-metoxipiperidina (173 mg, 1,50 mmol) en lugar de (S)-2-metilpiperidina.
Intermedio 20

2-(4,4-difluoropiperidin-1-il)etan-1-ol
Se sintetizó según el método del Intermedio 13, usando clorhidrato de 4,4-difluoropiperidina (173 mg, 1,50 mmol) en lugar de (S)-2-metilpiperidina.
Intermedio 21

(S)-2-(3-fluoropiperidin-1-il)etan-1-ol
Se sintetizó según el método del Intermedio 13, usando clorhidrato de S-3-fluoropiperidina (209 mg, 1,50 mmol) en lugar de (S)-2-metilpiperidina.
Intermedio 22

4-(7-(3-(benciloxi)naftalen-1-il)-2-(metilsulfinil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo
Etapa A: 4-h¡drox¡-2-(met¡lt¡o)-5.8-d¡h¡drop¡r¡do[3.4-d1p¡rim¡d¡n-7(6H)-carbox¡lato de terc-butilo: A una solución agitada de 3-oxopiperidin-1,4-dicarboxilato de 1-terc-butil 4-etilo (50,0 g, 184 mmol, 1,00 eq.) en MeOH (1,00 l) a 25 °C en una atmósfera de nitrógeno se le añadió NaOMe (49,8 g, 921 mmol, 5,00 eq.), seguido de 2-metilisotiourea (62,4 g, 331 mmol, 1,80 eq., H2SO4) en forma de un sólido. La mezcla de reacción se agitó a 25 °C durante 16 horas. La mezcla de reacción se ajustó a pH 5 con HCl (2 M) y la mezcla se concentró a presión reducida para eliminar el MeOH. El residuo se suspendió en 300 ml de acetato de etilo y 300 ml de agua y se agitó rápidamente. La suspensión se filtró y el sólido de color blanco se recogió. El filtrado se separó y la capa orgánica se lavó con agua (1 * 300 ml) y salmuera (1 * 200 ml). Las capas orgánicas combinadas se aislaron, se secaron sobre Na2SO4, se filtraron y se concentraron para proporcionar 4-h¡drox¡-2-met¡lsulfan¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-7-carboxilato de ferc-butilo (51,0 g, 138 mmol, 75,4 % de rendimiento, 81,0 % de pureza) en forma de un sólido de color blanco que se usó directamente en la siguiente etapa sin purificación adicional. ESI MS m/z 298,2 [M+H]+.
Etapa B: 2-met¡lsulfan¡l-4-(tr¡fluoromet¡lsulfon¡lox¡)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-7-carbox¡lato de terc-butilo: A una suspensión agitada de 4-hidroxi-2-metilsulfanil-6,8-dihidro-577-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (51,0 g, 171 mmol, 1,00 eq.) en DCM (500 ml) a 0 °C se le añadió DIEA (44,3 g, 343 mmol, 59,9 ml, 2,00 eq.), seguido de anhídrido trifluorometanosulfónico (72,6 g, 257 mmol, 42,4 ml, 1,50 eq.) en una atmósfera de nitrógeno. Inmediatamente se formó una solución de color pardo. Después de agitar a 25 °C durante 16 horas, la reacción se concentró para dar un aceite de color pardo. El aceite de color pardo se purificó por cromatografía en columna (S¡O2, éter de petróleo/acetato de etilo = 1:0 a 10:1) para proporcionar 2-metilsulfanil-4-(trifluorometilsulfoniloxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (46,0 g, 107 mmol, 62,4 % de rendimiento) en forma de un sólido de color amarillo ESI MS m/z 430,2 [M+H]+.
Etapa C: 4-(4-benc¡lox¡carbon¡lp¡peraz¡n-1-¡l)-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-7-carbox¡lato de fercbutilo: A una solución agitada de 2-metilsulfanil-4-(trifluorometilsulfoniloxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (46,0 g, 107 mmol, 1,00 eq.) en DMF (500 ml) se le añadió DIEA (27,7 g, 214 mmol, 37,4 ml, 2,00 eq.) seguido de piperazin-1-carboxilato de bencilo (25,9 g, 117 mmol, 22,7 ml, 1,10 eq.). La reacción se calentó a 100 °C durante 1 hora en una atmósfera de nitrógeno. La mezcla de reacción se vertió en acetato de etilo (300 ml), se lavó con H2O (300 ml * 3) y salmuera (200 ml), se secó sobre Na2SO4 anhidro, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna (S¡O2, éter de petróleo/acetato de etilo = 1:0 a 5:1) para dar 4-(4-benciloxicarbonilpiperazin-1-il)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (51,0 g, 96,9 mmol, 90,5 % de rendimiento, 92,0 % de pureza) en forma de un sólido de color blanco ESI MS m/z 500,3 [m H]+.
Etapa D: 4-í2-Met¡lsulfan¡l-5.6.7.8-tetrah¡drop¡r¡dor3.4-d1p¡r¡mid¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo: A una solución de 4-(4-benc¡lox¡carbon¡lp¡peraz¡n-1-¡l)-2-met¡lsulfan¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-carbox¡lato de tere-butilo (25.0 g.50,0 mmol, 1,00 eq.) en DCM (5o,0 ml) se le añad¡ó TFA (85,6 g.750 mmol, 55,6 ml. 15,0 eq.). Después de ag¡tar a 25 °C durante 1 hora. la mezcla de reacc¡ón se concentró a pres¡ón reduc¡da. El res¡duo se d¡solv¡ó en 300 ml de acetato de et¡lo y 300 ml de agua y se agitó ráp¡damente. La mezcla se ajustó a pH 8 con Na2CÜ3. La capa orgán¡ca se lavó con agua (1 x 300 ml) y salmuera (1 x 200 ml). se secó sobre Na2SÜ4. se f¡ltró y se concentró a pres¡ón reduc¡da para proporc¡onar 4-(2-met¡lsulfan¡l-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (19,0 g.
46.6 mmol. 93,2 % de rend¡m¡ento, 98,0 % de pureza) en forma de un ace¡te de color amar¡llo. ESI MS m/z 400,2 [M+H]+.
Etapa E: 4-[7-(3-Benc¡lox¡-1-naft¡l)-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de benc¡lo: Una mezcla de 3-benc¡lox¡-1-bromo-naftaleno (16,3 g. 52,1 mmol. 1,30 eq.). 4-(2-metilsulfan¡l-5.6.7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (16,0 g. 40,1 mmol. 1,00 eq.). Cs2CÜ3 (32,6 g.
100 mmol. 2,50 eq.). Pd2(dba)3 (5,50 g. 6,01 mmol. 0,15 eq.) y 2-d¡c¡clohex¡lfosf¡no-2',6'-d¡¡sopropox¡b¡fen¡lo (RuPhos) (3.74 g. 8,01 mmol. 0,20 eq.) en d¡oxano (300 ml) se desgas¡f¡có y se purgó 3 veces con n¡trógeno. La mezcla se agitó a 85 °C durante 5 horas en una atmósfera de n¡trógeno. La mezcla de reacc¡ón se ¡nact¡vó med¡ante la ad¡c¡ón de agua (200 ml) a 0 °C y se extrajo con acetato de etilo (3 x 200 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (3 x 150 ml). se secaron sobre Na2SÜ4. se f¡ltraron y se concentraron a pres¡ón reduc¡da. El res¡duo se pur¡f¡có por cromatografía en columna (S¡O2, DCM/MeOH = 10/1 a 5/1) para proporc¡onar 4-[7-(3-benciloxi-1-naft¡l)-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de benc¡lo (16,0 g. 22,8 mmol. 56,9 % de rend¡m¡ento, 90,0 % de pureza) en forma de un sól¡do de color amar¡llo. ESI MS m/z 632,5 [M+H]+.
Etapa F: 4-[7-(3-Benc¡lox¡-1-naft¡l)-2-met¡lsulf¡n¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de benc¡lo: A una soluc¡ón ag¡tada de 4-[7-(3-benc¡lox¡-1-naft¡l)-2-met¡lsulfan¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (8,00 g. 12,7 mmol. 1,00 eq.) en DCM (200 ml) se le añad¡ó m-CPBA (2,73 g.
12.7 mmol. 80,0 % de pureza. 1,00 eq.) a 0 °C en una atmósfera de n¡trógeno. Después de ag¡tar a 0 °C durante 2 horas en una atmósfera de n¡trógeno, la mezcla de reacc¡ón se ¡nact¡vó med¡ante la ad¡c¡ón de Na2S2O3 (10.0 ml) a 0 °C, se diluyó con agua (100 ml) y se extrajo con DCM (200 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (200 ml). se secaron sobre Na2SO4, se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar un res¡duo. El res¡duo se pur¡f¡có por cromatografía en columna (SiOs, DCM/MeOH = 1/0 a 10/1) para proporc¡onar 4-[7-(3-benciloxi-1-naft¡l)-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (3,50 g. 4,92 mmol. 38,8 % de rend¡m¡ento, 91,0 % de pureza) en forma de un sólido de color amar¡llo. ESI MS m/z 648,5 [M+H]+.
Intermed¡o 23

(R)-1-(4-(2-hidrox¡prop¡l)piperaz¡n-1-¡l)etan-1-ona
Etapa A: 1-[4-[(2R)-2-h¡drox¡prop¡l1p¡peraz¡n-1-¡l1etanona: (2R)-2-metiloxirano (1,00 g. 17,2 mmol. 1.20 ml. 1.00 eq.) y 1-piperazin-1-iletanona (8,00 g. 62,4 mmol. 3,62 eq.) se recogieron en un tubo de microondas. El tubo sellado se calentó a 150 °C durante 1 hora en microondas. La mezcla se disolvió en DCM (80,0 ml). se añadió (Boc)2O (3,62 eq.. 13,6 g) y se agitó a 20 °C durante 1 hora. El residuo se purificó por cromatografía en columna (DCM/MeOH 100/1 a 10/1) para dar 1 -[4-[(2R)-2-hidroxiprop¡l]p¡peraz¡n-1-il]etanona (3,80 g. 13,5 mmol. 78,2 % de rendimiento.
66,0 % de pureza) en forma de un aceite de color amarillo.
Intermed¡o 24
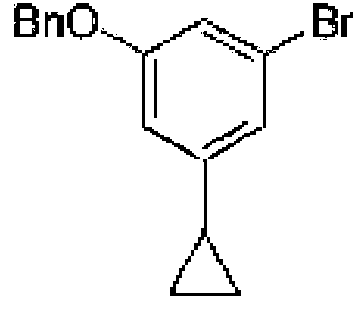
1-(benc¡lox¡)-3-bromo-5-c¡cloprop¡lbenceno
Etapa A: 1-benc¡lox¡-3,5-d¡bromo-benceno: A una mezcla de 3,5-dibromofenol (1,50 g. 5,95 mmol. 1,00 eq.) y K2CO3 (2.47 g. 17,9 mmol. 3,00 eq.) en MeCN (30,0 ml) se le añadió bromuro de bencilo (1,07 g. 6,25 mmol. 742 μl. 1,05 eq.) y la mezcla de reacción se agitó a 80 °C durante 2 horas. La mezcla de reacción se filtró y se concentró. El
residuo se purificó por cromatografía en columna (SÍO2, éter de petróleo/acetato de etilo = 1:1) para dar 1-benciloxi-3,5-dibromobenceno (1,60 g, 4,68 mmol, 78,6 % de rendimiento) en forma de un aceite incoloro.
Etapa B: 1-benc¡loxi-3-bromo-5-c¡cloprop¡lbenceno: A una mezcla de 1-benciloxi-3,5-dibromobenceno (1,20 g, 3,51 mmol, 1,00 eq.) y ácido ciclopropilborónico (392 mg, 4,56 mmol, 1,30 eq.) en H2O (4,00 ml) y dioxano (20,0 ml) se le añadieron Pd(dppf)Cl2 (513 mg, 702 pmol, 0,20 eq.) y CS2CO3 (2,29 g, 7,02 mmol, 2,00 eq.). La mezcla de reacción se agitó a 90 °C durante 12 horas en una atmósfera de N2. La mezcla de reacción se añadió a agua (20 ml) y se extrajo con acetato de etilo (2 x 15 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo = 1:1) para dar 1-benciloxi-3-bromo-5-ciclopropil-benceno (270 mg, 890 pmol, 25,4 % de rendimiento) en forma de un aceite incoloro.
Intermedio 25

4-(2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo
Etapa A: 4-(4-benc¡lox¡carbon¡lp¡peraz¡n-1-¡l)-2-(3-morfol¡nopropox¡)-6.8-d¡h¡dro-5H-p¡r¡dorJ.4-d)p¡r¡m¡d¡n-7-carbox¡lato de ferc-butilo: A una mezcla de 3-morfolinopropan-1-ol (5,46 g, 37,6 mmol, 2,00 eq.) en THF (100 ml) se le añadió poco a poco NaH (2,26 g, 56,4 mmol, 60,0 % de pureza, 3,00 eq.) a 0 °C. Después de que la mezcla se agitara a 0 °C durante 0,5 horas, se añadió una solución de 4-(4-benciloxicarbonilpiperazin-1-il)-2-metilsulfonil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (10,0 g, 18,8 mmol, 1,00 eq.) en THF (100 ml) y la mezcla de reacción se agitó a 0 °C durante 1,5 horas en una atmósfera de N2. La mezcla se vertió en NH4Cl acuoso (300 ml), y se extrajo con DCM (2 x 200 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo = 50:1 a 10:1) para dar 4-(4-benciloxicarbonilpiperazin-1-il)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (7,70 g, 12,8 mmol, 67,8 % de rendimiento, 98,8 % de pureza) en forma de un aceite de color amarillo. ESI MS m/z 597,4 [M+H]+.
Etapa B: 4-[2-(3-morfol¡nopropox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de bencilo: A una mezcla de 4-(4-benciloxicarbonilpiperazin-1-il)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (7,70 g, 12,9 mmol, 1,00 eq.) en DCM (80,0 ml) se le añadió TFA (119 g, 1,04 mol, 76,9 ml, 80,6 eq.) y la mezcla de reacción se agitó a 15 °C durante 1 hora. La mezcla de reacción se concentró, después se diluyó con DCM (100 ml) y se ajustó a pH 8 con NaOH acuoso. La capa orgánica se separó, se secó sobre Na2SO4, se filtró y se concentró para dar 4-[2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (6,00 g, 11,2 mmol, 86,9 % de rendimiento, 92,8 % de pureza) en forma de un aceite de color amarillo. ESI MS m/z 497,4 [m +H]+.
Intermedio 26

4-(benciloxi)-2-bromo-1-fluorobenceno
A una solución de 3-bromo-4-fluorofenol (4,00 g, 20,9 mmol, 1,00 eq.) y K2CO3 (8,68 g, 62,8 mmol, 3,00 eq.) en ACN (80,0 ml) se le añadió bromuro de bencilo (3,65 g, 21,4 mmol, 2,54 ml, 1,02 eq.) y la mezcla de reacción se agitó a 60 °C durante 2 h. La mezcla de reacción se filtró y se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (éter de petróleo/acetato de etilo; gradiente de 1:0 a 10:1) para dar 4-benciloxi-2-bromo-1-fluoro-benceno (5,02 g, 17,0 mmol, 81,0 % de rendimiento, 95 % de pureza) que se obtuvo en forma de un sólido de color blanco.
Intermedio 27

2-(3-fluoropirrolidin-1-il)etan-1-ol
Etapa A: 3-fluorop¡rrol¡d¡n-1-carbox¡lato de tere-butilo: A una solución de 3-hidroxipirrolidin-1 -carboxilato de tere-butilo (10,0 g, 53,4 mmol, 1,00 eq.) en DCM (150,00 ml) se le añadió trifluoruro de dietilaminoazufre (DAST) (12,9 g, 80,1 mmol, 10,6 ml, 1,50 eq.) a -40 °C en una atmósfera de nitrógeno. Después de agitar a -40 °C durante 2 horas, la mezcla se calentó a 20 °C y se agitó durante 16 horas. La mezcla se vertió en 5 % de bicarbonato de sodio (200 ml) y se extrajo con diclorometano (2 x 100 ml). La capa orgánica se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo/acetato de etilo 100:1 a 5:1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar 3-fluoropirrolidin-1 -carboxilato de tere-butilo (4,30 g, 22,7 mmol, 42,6 % de rendimiento) en forma de un aceite incoloro. 1H RMN (400 MHz, Cloroformo-d) ó = 5,27 (t, J = 3,6 Hz, 0,5H), 5,13 (t, J = 3,6 Hz, 0,5H), 3,77 - 3,38 (m, 4H), 2,26 - 2,15 (m, 1H), 2,08 - 1,85 (m, 1H), 1,46 (s, 9H).
Etapa B: 3-fluoropirrolidina: A una solución de 3-fluoropirrolidin-1 -carboxilato de fere-butilo (4,30 g, 22,7 mmol, 1,00 eq.) en DCM (50,00 ml) se le añadió HCl/dioxano (4 M, 35,0 ml, 6,16 eq.) gota a gota a 0 °C. La mezcla se calentó a 20 °C y se agitó durante 1 hora. La mezcla se concentró al vacío. El residuo se trituró con diisopropil éter (20 ml) y el precipitado se filtró y se secó al vacío para proporcionar 3-fluoropirrolidina (2,70 g, 21,5 mmol, 94,6 % de rendimiento, HCl) en forma de un sólido de color blanco. 1H RMN (400 MHz, Metanol-d4) ó = 5,51 (t, J = 3,6 Hz, 0,5H), 5,38 (t, J = 3,6 Hz, 1H), 3,66 - 3,27 (m, 5H), 2,45 - 2,12 (m, 2H).
Etapa C: 2-(3-fluorop¡rrol¡d¡n-1-¡l)acetato de metilo: Una suspensión de 3-fluoropirrolidina (2,70 g, 21,5 mmol, 1,00 eq., HCl) en DCM (27,00 ml) se enfrió a 0° C. Se añadieron trietilamina (5,44 g, 53,8 mmol, 7,45 ml, 2,50 eq.) y 2-bromoacetato de metilo (3,62 g, 23,7 mmol, 2,23 ml, 1,10 eq.) y la mezcla de reacción se agitó a 20 °C durante 16 h. La mezcla de reacción se diluyó con CH2Cl2 (100 ml) y agua (50 ml). La capa orgánica se lavó con 5 % de solución acuosa de ácido cítrico (1 x 50 ml). La capa de agua se basificó con solución acuosa saturada de carbonato de sodio (20 ml) y se extrajo con acetato de etilo (3 x 100 ml). Las capas orgánicas combinadas se secaron sobre sulfato de sodio y se concentraron al vacío para dar 2-(3-fluoropirrolidin-1-il)acetato de metilo (2,20 g, 13,7 mmol, 63,5 % de rendimiento). 1H RMN (400 MHz, Cloroformo-d) ó = 5,22 - 5,02 (m, 1H), 3,66 (s, 3H), 3,35 (s, 2H), 3,07 -2,93 (m, 1H), 2,91 - 2,77 (m, 2H), 2,67 (dt, J = 5,2, 8,4 Hz, 1H), 2,21 - 1,93 (m, 2H).
Etapa D: 2-(3-fluorop¡rrol¡d¡n-1-¡l)etanol: A una solución de LiAlH4 (706 mg, 18,6 mmol, 1,50 eq.) en THF (20 ml) se le añadió una solución de 2-(3-fluoropirrolidin-1-il)acetato de metilo (2,00 g, 12,4 mmol, 1,00 eq.) en THF (10 ml) gota a gota a 0 °C. La mezcla se calentó a 20 °C y se agitó durante 3 horas. La mezcla se inactivó con solución acuosa saturada de sulfato de sodio (1 ml). La mezcla se filtró y el filtrado se concentró al vacío. El producto se purificó por cromatografía sobre gel de sílice usando 5 % de MeOH en DMC. Las fracciones deseadas se recogieron y se concentraron al vacío para dar 2-(3-fluoropirrolidin-1-il)etanol (1,20 g, 9,01 mmol, 72,6 % de rendimiento) en forma de un aceite incoloro. 1H RMN (400 MHz, Cloroformo-d) ó = 5,28 - 5,05 (m, 1H), 3,68 - 3,61 (m, 2H), 2,99 -2,73 (m, 4H), 2,72 - 2,67 (m, 2H), 2,58 - 2,45 (m, 1H), 2,28 - 1,97 (m, 2H).
Intermedio 28

piperazin-1,3-dicarboxilato de 1 -(terc-butil) 3-metilo
Etapa A: piperazin-2-carboxilato de metilo: A una mezcla de piperazin-1,2-dicarboxilato de 1 -terc-butil 2-metilo (5,0 g, 22,6 mmol, 1,00 eq.) en MeOH (50,0 ml) se le añadió HCl/dioxano (4,0 M, 134 ml). La mezcla de reacción se desgasificó y se purgó 3 veces con nitrógeno y la mezcla se agitó a 25 °C durante 12 horas en una atmósfera de nitrógeno. La mezcla de reacción se concentró a presión reducida a sequedad para dar piperazin-2-carboxilato de metilo (4,89 g, 2HCl, bruto) en forma de un sólido de color blanco, que se usó directamente en la siguiente etapa sin purificación adicional.
Etapa B: p¡perazin-1.3-d¡carbox¡lato de 1-(terc-butil) 3-metilo: A una solución de piperazin-2-carboxilato de metilo (4,30 g, bruto) y TEA (8,02 g, 79,2 mmol, 11,0 ml) en MeOH (50,0 ml) se le añadió dicarbonato de di-te rc-butilo (4,32 g, 19,8 mmol, 4,55 ml). Después de agitar a 25 °C durante 12 horas, la mezcla de reacción se filtró y se concentró a presión reducida a sequedad. El residuo se purificó por cromatografía en columna (SiO2, DCM/MeOH = 1:0 a 20:1) para dar piperazin-^-dicarboxilato de 1 -(terc-butil) 3-metilo (4,80 g, 19,7 mmol, dos etapas, 99,0 % de rendimiento) en forma de un aceite incoloro. 1H Rm N (400 m Hz , cloroformo-d) 5 = 4,10 - 3,85 (m, 1H), 3,73 (s, 3H), 3,71 - 3,65 (m, 1H), 3,47 - 3,38 (m, 1H), 3,10 - 2,98 (m, 2H), 2,78 - 2,66 (m, 1H), 2,17 (s, 1H), 1,46 (s, 9H).
Intermedio 29

4-bromonaftalen-2-ol
Etapa A: 2.4-d¡bromonaftalen-1-am¡na: A una solución de Br2 (246 g, 1,54 mol, 79,3 ml, 2,18 eq.) en AcOH (750 ml) se le añadió una solución de naftalen-1-amina (101 g, 705 mmol, 99,0 ml, 1,00 eq.) en AcOH (500 ml) a temperatura ambiente y la reacción se agitó a 70 °C durante 1 hora. La mezcla de reacción se enfrió a temperatura ambiente y se filtró. La torta de filtro se lavó con AcOH (300 ml) y después se añadió a 20 % acuoso de NaOH (1,2 l). La mezcla se agitó durante 20 min y se filtró. El sólido aislado se lavó con agua (1 l) y se secó al vacío para proporcionar 2,4-dibromonaftalen-1-amina (200 g, 664 mmol, 94,2 % de rendimiento) en forma de un sólido de color gris. ESI MS m/z 301,9 [M+H]+.
Etapa B: 4-bromo-1-d¡azon¡o-naftalen-2-olato: A una solución de 2,4-dibromonaftalen-1-amina (60,0 g, 199 mmol, 1,00 eq.) en AcOH (900 ml) y ácido propiónico (150 ml) se le añadió poco a poco NaNO2 (16,5 g, 239 mmol, 13,0 ml, 1,20 eq.) a 5-8 °C durante 30 min y después la mezcla de reacción se agitó a 5-8 °C durante 30 min. La mezcla de reacción se vertió en hielo-agua (4000 ml) y el sólido resultante se recogió y se lavó con agua (2 x 50 ml) para proporcionar 4-bromo-1-diazonio-naftalen-2-olato (150 g, bruto húmedo) en forma de un sólido de color gris que se usó directamente en la siguiente etapa. 1H RMN (400 MHz, CDCla) 58,12 - 8,10 (d, J = 8,4 Hz, 1H), 7,62 - 7,58 (t, J = 7,6 Hz, 1H), 7,41 - 7,37 (t, J = 7,6 Hz, 1H), 7,31 - 7,29 (d, J = 8,0 Hz, 1H), 7,20 (s, 1H).
Etapa C: 4-bromonaftalen-2-ol: A una solución de 4-bromo-1-diazonio-naftalen-2-olato (100 g, 402 mmol, 1,00 eq.) en EtOH (2,00 l) se le añadió poco a poco NaBH4 (30,4 g, 803 mmol, 2,00 eq.) a 13-15 °C durante 1 h y la mezcla de reacción se agitó a 15-18 °C durante 3 h. La reacción se filtró y se concentró a sequedad. El residuo se disolvió en DCM (1000 ml) y se lavó con agua (500 ml x 2). La fase orgánica se secó sobre Na2SO4 y se concentró a sequedad.
El residuo se purificó por cromatografía en columna de gel de sílice, eluyendo con éter dietílico/acetato de etilo (60:1 a 10:1). El producto aislado se purificó adicionalmente por HPLC de fase inversa para proporcionar 4-bromonaftalen-2-ol (40,0 g, 139 mmol, 17,3 % de rendimiento, 77,4 % de pureza) en forma de un sólido de color gris. 1H RMN (400 MHz, CDCla) 58,07 - 8,05 (d, J = 8,0 Hz, 1H), 7,60 - 7,58 (d, J = 7,6 Hz, 1H), 7,41 - 7,36 (m, 3H), 7,07 (s, 1H).
Etapa D: 3-benc¡lox¡-1-bromo-naftaleno: Una mezcla de 4-bromonaftalen-2-ol (30,0 g, 134 mmol, 1,00 eq.), bromuro de bencilo (25,3 g, 148 mmol, 17,6 ml, 1,10 eq.) y K2CO3 (55,7 g, 403 mmol, 3,00 eq.) en MeCN (500 ml) se calentó a 80 °C durante 1 h. La mezcla de reacción se filtró y se concentró a sequedad. El residuo se purificó por cromatografía en columna sobre gel de sílice, eluyendo con éter dietílico/acetato de etilo (100:1 a 60:1) para proporcionar 3-benciloxi-1-bromo-naftaleno (40,0 g, 128 mmol, 95 % de rendimiento) en forma de un aceite de color amarillo. 1H RMN (400 MHz, CDCb) 58,19 - 8,17 (d, J = 8,0 Hz, 1H), 7,75 - 7,32 (d, J= 8,8 Hz, 1H), 7,64 - 7,63 (d, J= 2,4 Hz, 1H), 7,52 - 7,37 (m, 7H), 7,23 - 7,21 (d, J= 2,0 Hz, 1H), 5,2 (s, 2H).
Intermedio 30

trifluorometanosulfonato de 3-metoxinaftalen-1-ilo
Etapa A: 3-metoxinaftalen-1 -ol: A una solución de naftaleno-1,3-diol (3,00 g, 18,7 mmol, 1,00 eq.) en MeOH (60,0 ml) se le añadió HCl/MeOH (4 M, 60,0 ml, 12,8 eq.) a 0 °C. La mezcla se agitó a 25 °C durante 60 horas. El disolvente se eliminó al vacío. El residuo se purificó por cromatografía sobre gel de sílice (éter dietílico:acetato de etilo = 10:1 a 5:1) para dar 3-metoxinaftalen-1-ol (2,10 g, 12,1 mmol, 64,4 % de rendimiento) en forma de un sólido de color pardo. 1H RMN (400 MHz, CDCla-d6) ó = 8,10 - 8,08 (d, J= 8,4 Hz, 1H).7,73 - 7,71 (d, J= 8,4 Hz, 1H), 7,47 - 7,45(m, 1H), 7,38 - 7,35(m, 1H), 6,80 - 6,79 (d, J= 2,0 Hz, 1H), 6,56 - 6,55 (d, J= 2,4 Hz, 1H), 3,92 (s, 3H).
Etapa B: trifluorometanosulfonato de (3-metoxi-1-naft¡lo): A una solución de 3-metoxinaftalen-1-ol (2,10 g, 12,0 mmol, 1,00 eq.) en DCM (40,0 ml) se le añadieron DIEA (7,79 g, 60,3 mmol, 10,5 ml, 5,00 eq.) y anhídrido trifluorometanosulfónico (5,10 g, 18,1 mmol, 2,98 ml, 1,50 eq.) a 0 °C. La mezcla se agitó a 25 °C durante 1 hora. La mezcla se diluyó con DCM (30 ml) y agua (10 ml) y se extrajo con DCM (20 ml). Las capas orgánicas combinadas se lavaron con salmuera (5 ml), se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (éter dietílico:acetato de etilo = 20:1 a 10:1) para dar trifluorometanosulfonato de (3-metoxi-1-naftilo) (3,00 g, 8,52 mmol, 70,7 % de rendimiento, 87,0 % de pureza) en forma de un aceite de color pardo. ESI MS m/z 307,1 [M+H]+.
Intermedio 31

7-bencil-2,4-dicloro-6,8-dihidro-5H-pirido[3,4-d]pirimidina
Etapa A: 7-benc¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-2.4-d¡ol: A EtOH (600 ml) se le añadió poco a poco Na (5,56 g, 241 mmol, 5,73 ml, 2,40 eq.). La mezcla de reacción se agitó durante 1 hora. A la mezcla se le añadieron 1-bencil-3-oxopiperidin-4-carboxilato de etilo (30,0 g, 100 mmol, 1,00 eq., HCl) y urea (14,5 g, 242 mmol, 13,0 ml, 2,40 eq.). La mezcla de reacción se agitó a 75 °C durante 36 horas y después el disolvente se eliminó al vacío. El residuo se disolvió en agua (50 ml) y se acidificó con HCl (120 ml, 2 M). Precipitó un sólido de color blanco de la solución y se recogió por filtración. La torta de filtro se secó al vacío para proporcionar 7-bencil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2,4-diol (22,0 g, 83,8 mmol, 83,2 % de rendimiento, 98 % de pureza) en forma de un sólido de color blanco. 1H Rm N (400 MHz, DMSO-d6) ó = 10,97 (s a, 1H), 10,66 (s a, 1H), 7,55 - 6,95 (m, 5H), 3,81 - 3,50 (m, 2H), 3,26 - 2,91 (m, 2H), 2,77 - 2,58 (m, 2H), 2,34 - 2,09 (m, 2H).
Etapa B: 7-benc¡l-2.4-d¡cloro-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡na: A una solución de DIEA (30,1 g, 233 mmol, 40,7 ml, 3,00 eq.) en POCE (330 g, 2,15 mol, 200 ml) se le añadió 7-bencil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2,4-diol (20,0 g, 77,7 mmol, 1,00 eq.). La mezcla de reacción se agitó a 110 °C durante 5 horas. La mezcla de reacción se concentró al vacío. El residuo se disolvió en DCM (400 ml) y se vertió en NaHCO3 saturado (200 ml). La mezcla se extrajo con DCM (2 x 400 ml). Las capas orgánicas combinadas se lavaron con salmuera (100 ml), se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (éter dietílico: 10:1 a 0:1) para dar 7-bencil-2,4-dicloro-6,8-dihidro-5H-pirido[3,4-d]pirimidina (7,70 g, 26,2 mmol, 33,7 % de rendimiento) en forma de un aceite de color pardo. 1H RMN (300 MHz, cloroformo-d) ó = 7,43 - 7,28 (m, 5H), 3,73 (s, 2H), 3,66 (s a, 2H), 2,84 (s a, 4H)
Intermedio 32

4-(4-((benciloxi)carbonil)piperazin-1-il)-2-(metilsulfonil)-5,6-dihidropirido[3,4-d]pirimidin-7(8H)-carboxilato de tere-butilo
Etapa A: 4-h¡drox¡-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-7-carbox¡lato de tere-butilo: A una solución agitada de 4-etil-3-oxopiperidin-1,4-dicarboxilato de 1-tere-butilo (44,0 g, 162 mmol, 1,00 eq.) en MeOH (1,00 ml) a 25 °C en una atmósfera de nitrógeno se le añadió una solución de NaOMe (35,0 g, 649 mmol, 4,00 eq.) en MeOH (600 ml) mediante una
jeringa seguido de 2-metilisotiourea (61,1 g, 324 mmol, 2,00 eq., H2SO4). Después de agitar a 25 °C durante 16 horas, la mezcla de reacción se concentró a presión reducida para eliminar el MeOH. El residuo se suspendió en 500 ml de acetato de etilo y 500 ml de agua y se agitó rápidamente. La mezcla de reacción se ajustó a pH 5 con HCl (2 M). El precipitado se filtró y el sólido de color blanco se lavó con acetato de etilo y se secó al vacío para dar 4-hidroxi-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de tere-butilo (33,0 g, 103 mmol, 63,8 % de rendimiento, 93,2 % de pureza) en forma de un sólido de color blanco, que se usó directamente en la siguiente etapa sin purificación adicional. 1H RMN (400 MHz, DMSO-d6) 6 = 4,19 (s, 2 H), 3,49 (s a, 2 H), 2,46 (s, 3 H), 2,35 (t a, J = 5,2 Hz, 2 H), 1,42 (s, 9 H).
Etapa B: 2-met¡lsulfan¡l-4-(tr¡fluoromet¡lsulfon¡lox¡)-6.8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-7-carbox¡lato de tere-butilo: A una suspensión agitada de 4-hidroxi-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d1pirimidin-7-carboxilato de tere-butilo (15,0 g, 50,4 mmol, 1,00 eq.) en DCM (200 ml) se le añadió DIEA (26,1 g, 202 mmol, 35,2 ml, 4,00 eq.) a 0 °C en una atmósfera de nitrógeno, seguido de anhídrido trifluorometanosulfónico (28,5 g, 101 mmol, 16,6 ml, 2,00 eq.) mediante una jeringa. Inmediatamente se formó una solución de color pardo. La mezcla de reacción se agitó a 25 °C durante 12 horas. La mezcla de reacción se purificó por cromatografía en columna (S¡O2, éter de petróleo/acetato de etilo = 1:0 a 10:1) para dar 2-metilsulfanil-4-(trifluorometilsulfoniloxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de tere-butilo (16,7 g, 35.7 mmol, 70,9 % de rendimiento, 91,9 % de pureza) en forma de un sólido de color blanco. ESI MS m/z 374,0 [M+H]+.
Etapa C: 4-(4-benc¡lox¡carbon¡lp¡peraz¡n-1-¡l)-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-7-carbox¡lato de terebutilo: A una solución agitada de 2-metilsulfanil-4-(trifluorometilsulfoniloxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de tere-butilo (16,7 g, 38,9 mmol, 1,00 eq.) en DMF (100 ml) se le añadieron DIEA (10,0 g, 77,9 mmol, 2,00 eq.) y piperazin-1-carboxilato de bencilo (9,41 g, 42,8 mmol, 1,10 eq.). La reacción se calentó a 100 °C y se agitó durante 1 hora en una atmósfera de nitrógeno. La mezcla de reacción se diluyó con agua (150 ml) y la mezcla de reacción se ajustó a pH 5 con HCl (2 M) y se extrajo con DCM (3 * 200 ml). Las capas orgánicas combinadas se lavaron con NaHCO3 saturado (3 * 150 ml), salmuera (3 * 150 ml) y H2O (3 * 150 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para dar 4-(4-benciloxicarbonilpiperazin-1-il)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de tere-butilo (18,1 g, 36,2 mmol, 93,0 % de rendimiento, 94,1 % de pureza) en forma de un sólido de color amarillo, que se usó directamente en la siguiente etapa sin purificación adicional. ESI MS m/z 500,1 [M+H]+.
Etapa D: 4-(4-benc¡lox¡carbon¡lp¡peraz¡n-1-¡l)-2-met¡lsulfon¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-7-carbox¡lato de tere-butilo: A una solución agitada de 4-(4-benciloxicarbonilpiperazin-1-il)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de terc-butilo (14,4 g, 28,9 mmol, 1,00 eq.) en DCM (150 ml) a 0 °C en una atmósfera de nitrógeno se le añadió ácido meta-cloroperoxibenzoico (17,4 g, 101 mmol, 3,50 eq.) en forma de un sólido. Después de agitar a 0 °C durante 2 horas en una atmósfera de nitrógeno, la mezcla de reacción se diluyó con agua (300 ml) y la mezcla de reacción se ajustó a pH 8 con NaHCO3 acuoso saturado y se extrajo con DCM (3 * 200 ml). Las capas orgánicas combinadas se lavaron con salmuera (3 * 200 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida a sequedad. El residuo se purificó por cromatografía en columna (S¡O2, éter de petróleo/acetato de etilo = 10:1 a 1:2) para dar 4-(4-benciloxicarbonilpiperazin-1-il)-2-metilsulfonil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de terc-butilo (11,0 g, 19.7 mmol, 68,6 % de rendimiento, 95,4 % de pureza) en forma de un sólido de color blanco. ESI MS m/z 532,1 [M+H]+.
Intermedio 33
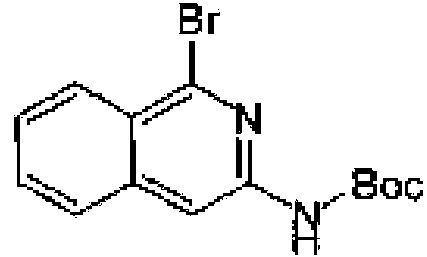
(1-bromoisoquinolin-3-il)carbamato de terc-butilo
Etapa A: Una mezcla de 1-bromoisoquinolin-3-amina (400 mg, 1,79 mmol, 1,00 eq.) y terc-butil carbonato de tere-butoxicarbonilo (3,91 g, 17,9 mmol, 4,12 ml, 10,0 eq.) se agitó a 70 °C durante 16 horas. El residuo se purificó por cromatografía en columna (S¡O2, éter dietílico/acetato de etilo = 5:1) para dar N-(1-bromo-3-isoquinolil)carbamato de terebutilo (400 mg, 1,24 mmol, 69,2 % de rendimiento) en forma de un sólido de color amarillo. ESI mS m/z 322,1, 324,1 [M+H]+.
Intermedio 34

trifluorometanosulfonato de 3-metoxi-6-metilnaftalen-1-ilo
Etapa A: 3-metoxinaftalen-1 -ol: A una solución de naftaleno-1,3-diol (40,0 g, 250 mmol, 1,00 eq.) en MeOH (800 ml) se le añadió HCl (4 M, 750 ml, 12,0 eq., 4 M en MeOH) a 0 °C. La mezcla se calentó a 18 °C y se agitó durante 30 horas. La mezcla se concentró al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo/acetato de etilo 100/1 a 1/1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar 3-metoxinaftalen-1-ol (17,7 g, 96,5 mmol, 38,6 % de rendimiento, 95 % de pureza) en forma de un aceite de color rojo.
1H RMN (400 MHz, Cloroformo-d) ó = 8,17 (d, J = 8,4 Hz, 1H), 7,74 (d, J = 8,0 Hz, 1H), 7,50 (ddd, J = 1,2, 6,8, 8,0 Hz, 1H), 7,38 (ddd, J = 1,2, 6,8, 8,0 Hz, 1H), 6,81 (d, J = 2,0 Hz, 1H), 6,76 (s a, 1H), 6,62 (d, J = 2,4 Hz, 1H), 3,91 (s, 3H).
Etapa B: terc-butil-[(3-metoxi-1-naftil)oxi]-di iTi etil-silano: A una solución de 3-metoxinaftalen-1-ol (20,0 g, 115 mmol, 1,00 eq.) e imidazol (23,5 g, 344 mmol, 3,00 eq.) en THF (400 ml) se le añadió TBSCl (26,0 g, 172 mmol, 21,1 ml, 1,50 eq.) gota a gota a 0 °C. La mezcla se calentó a 25 °C y se agitó durante 16 horas. La mezcla se diluyó con éter de petróleo (600 ml) y acetato de etilo (200 ml) y después se lavó con agua (1 x 200 ml) y salmuera (1 x 200 ml). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía de columna sobre gel de sílice (éter de petróleo/acetato de etilo 100/1 a 10/1). Se obtuvo terc-butil-[(3-metoxi-1-naftil)ox¡1-dimetil-silano (28,0 g, 97,1 mmol, 84,6 % de rendimiento) en forma de un aceite incoloro. 1H RMN (400 MHz, Cloroformo-d) ó = 8,01 (d, J = 8,4 Hz, 1H), 7,61 (d, J = 8,0 Hz, 1H), 7,35 (dt, J = 1,2, 7,6 Hz, 1H), 7,24 (dt, J = 1,2, 7,6 Hz, 1H), 6,71 (d, J = 2,0 Hz, 1H), 6,48 (d, J = 2,4 Hz, 1H), 3,82 (s, 3H), 1,02 (s, 9H), 0,23 (s, 6H).
Etapa C: ferc-but¡l-[[3-metox¡-6-(4.4.5.5-tetramet¡l-1.3.2-d¡oxaborolan-2-¡l)-1-naftil1ox¡1-d¡met¡l-s¡lano y terc-butil((3-metox¡-7-(4.4.5.5-tetramet¡l-1.3.2-d¡oxaborolan-2-¡l)naftalen-1-il)ox¡)d¡met¡ls¡lano: Una mezcla de terc-butil-[(3-metoxi-1-naftil)ox¡]-d¡metil-s¡lano (26,0 g, 90,1 mmol, 1,00 eq.), 4,4,5,5-tetrametil-2-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1,3,2-dioxaborolano (45,8 g, 180 mmol, 2,00 eq.), (1Z,5Z)-cicloocta-1,5-dieno;2,4-dimetil-BLAHbiciclo[1.1.0]butano (2,39 g, 3,61 mmol, 0,04 eq.) y 4-terc-butil-2-(4-terc-butil-2-piridil)piridina (1,45 g, 5,41 mmol, 0,06 eq.) en hexano (500 ml) se agitó a 100 °C en una atmósfera de nitrógeno durante 16 horas. La mezcla se diluyó con agua (500 ml) y acetato de etilo (1000 ml). La capa orgánica separada se lavó con salmuera (1 x 500 ml), se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo/acetato de etilo 100/1 a 10/1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar una mezcla de ferc-but¡l-[[3-metox¡-6-(4,4,5,5-tetramet¡l-1,3,2-d¡oxaborolan-2-¡l)-1-naftil]ox¡]-d¡met¡l-s¡lano y terc-butil((3-metoxi-7-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)naftalen-1-il)oxi)dimetilsilano (38,0 g, 85,3 mmol, 94,6 % de rendimiento, 93 % de pureza) en forma de un aceite de color amarillo claro. ESI MS m/z 415,5 [M+H]+
Etapa D: 8-(terc-but¡l(d¡met¡l)s¡l¡l1ox¡-6-metox¡- naftalen-2-ol: A una solución de mezcla (36,0 g, 86,9 mmol, 1,00 eq.) de terc-butil-[[3-metoxi-6-(4,4,5,5- tetramet¡l-1,3,2-dioxaborolan-2-¡l)-1-naft¡l]ox¡]-d¡met¡l-s¡lano y terc-butil((3-metoxi-7-(4,4,5,5-tetrametil-1,3, 2-dioxaborolan-2-il)naftalen-1-il)oxi)dimetilsilano en acetona (400 ml) se le añadió una solución de Oxone (58,7 g, 95,6 mmol, 1,10 eq.) en H2O (400 ml) a 0 °C. La mezcla se agitó a 0 °C durante 1 hora. La mezcla se inactivó con 5 % de solución acuosa de tiosulfato de sodio (50 ml) y se extrajo con acetato de etilo (2 x 300 ml).
Los extractos se combinaron y se lavaron con agua (1 x 200 ml) y salmuera (1 x 200 ml), se secaron sobre sulfato de magnesio, se filtraron y el filtrado se concentró al vacío. El residuo se purificó por cromatografía de columna sobre gel de sílice (éter de petróleo/acetato de etilo 200/1 a 20/1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar 8-[terc-butil(dimetil)silil]oxi-6-metoxi- naftalen-2-ol (9,00 g, 28,4 mmol, 32,7 % de rendimiento, 96 % de pureza) en forma de un aceite incoloro y 5-[terc-butil(dimetil)silil]oxi-7-metoxi-naftalen-2-ol (9,00 g, 29,0 mmol, 33,4 % de rendimiento, 98 % de pureza) en forma de un sólido de color blanco. ESI MS m/z 305,2 [M+H]+
Etapa E: trifluorometanosulfonato de [5-[terc-but¡l(d¡met¡l)s¡l¡l1ox¡-7-metox¡-2-naft¡lo1: A una solución de 5-[tercbutil(dimetil)silil]oxi-7-metoxinaftalen-2-ol (11,0 g, 36,1 mmol, 1,00 eq.) y DIEA (14,0 g, 108 mmol, 18,9 ml, 3,00 eq.) en DCM (150 ml) se le añadió Tf2O (12,2 g, 43,4 mmol, 7,15 ml, 1,20 eq.) gota a gota a -40 °C. La mezcla se agitó durante 1 hora. La mezcla se diluyó con diclorometano (200 ml) y se lavó con agua (1 x 200 ml) y salmuera (1 x 200 ml). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía de columna sobre gel de sílice (éter de petróleo/acetato de etilo 100/1 a 10/1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar trifluorometanosulfonato de [5-[terc-but¡l(dimet¡l)s¡l¡l]ox¡-7-metox¡-2-naft¡lo] (13,0 g, 29,8 mmol, 82,4 % de rendimiento, 100 % de pureza) en forma de un sólido de color blanco. ESI MS m/z 436,9 [M+H]+
Etapa F: terc-but¡l-[(3-metox¡-6-met¡l-1-naft¡l)ox¡1-d¡met¡l-s¡lano: A una solución de trifluorometanosulfonato de [5-[tercbut¡l(d¡metil)s¡l¡l]ox¡-7-metox¡-2-naft¡lo] (12,5 g, 28,6 mmol, 1,00 eq.) y K2CO3 (11,9 g, 85,9 mmol, 3,00 eq.) en dioxano (160 ml) se le añadieron Pd(PPh3)4 (3,31 g, 2,86 mmol, 0,10 eq.) y trimetilboroxina (14,4 g, 57,3 mmol, 16,0 ml, 2,00 eq.) en una atmósfera de nitrógeno. La reacción se calentó a 100 °C durante 16 horas. La mezcla se diluyó con acetato de etilo (200 ml) y después se lavó con agua (1 x 200 ml) y salmuera (1 x 200 ml). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía de columna sobre gel de sílice (éter de petróleo/acetato de etilo 100/1 a 5/1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar terc-butil-[(3-metoxi-6-metil-1-naftil)oxi]-dimetil-silano (8,00 g, 24,6 mmol, 85,9 % de rendimiento, 93 % de pureza) en forma de un aceite incoloro en forma de un sólido de color rojo. ESI MS m/z 303,2 [M+H]+
Etapa G: 3-metoxi-6-metil-naftalen-1 -ol: A una solución de terc-butil-[(3-metoxi-6-metil-1-naftil) oxi]-dimetil-silano (8,00 g, 26,5 mmol, 1,00 eq.) en THF (100 ml) se le añadió TBAF (10,4 g, 39,7 mmol, 1,50 eq.) a 0 °C. La mezcla
se agitó a 0 °C durante 3 horas. La mezcla se diluyó con agua (100 ml) y acetato de etilo (200 ml). La capa orgánica separada se lavó con salmuera (1 * 100 ml), se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía de columna sobre gel de sílice (éter de petróleo/acetato de etilo 50/1 a 5/1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar 3-metoxi-6-metil-naftalen-1-ol (4,70 g, 25,0 mmol, 94,4 % de rendimiento) en forma de un sólido de color rojo. ESI MS m/z 188,4 [M+H]+
Etapa H: trifluorometanosulfonato de 3-metoxi-6-met¡l-1-naft¡lo: A una solución de 3-metoxi-6-metil-naftalen-1-ol (4,70 g, 25.0 mmol, 1,00 eq.) y DIEA (9,68 g, 74,9 mmol, 13,1 ml, 3,00 eq.) en DCM (3,00 ml) se le añadió Tf2O (8,45 g, 30.0 mmol, 4,94 ml, 1,20 eq.) gota a gota a -40 °C. La mezcla se agitó durante 1 hora. La mezcla se diluyó con diclorometano (200 ml) y se lavó con agua (1 * 200 ml) y salmuera (1 * 200 ml). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía de columna sobre gel de sílice (éter de petróleo/acetato de etilo 100/1 a 10/1). Se obtuvo trifluorometanosulfonato de 3-metoxi-6-metil-1-naftilo (7,70 g, 24,0 mmol, 96,2 % de rendimiento, 99,9 % de pureza) en forma de un aceite incoloro. ESI MS m/z 320,7 [M+H]+. Los siguientes intermedios se prepararon según la preparación para el Intermedio 3, sustituyendo el fenol apropiado por 2-bromo-3-fluorofenol.


Intermedio 44 
2-bromo-3-fluoro-1-(metoximetoxi)-4-metilbenceno
Etapa 1: Se puso 3-fluoro-4-metilfenol (1,016 g, 8,055 mmol) en Cs2 (3,9 ml, 64,44 mmol) y se enfrió a 0 °C. Se añadió Br2 (0,4150 ml, 8,055 mmol) y la mezcla se agitó a temperatura ambiente durante 2 h. Se añadió Na2S2O2 al 10 % y la mezcla se extrajo con DCM. Las capas orgánicas se combinaron, se secaron y se filtraron para proporcionar 2-bromo-3-fluoro-4-metilfenol (1,389 g, 6,775 mmol, 84,10 % de rendimiento) que se usó directamente en la siguiente etapa.
Etapa 2: Se preparó 2-bromo-3-fluoro-1-(metoximetoxi)-4-metilbenceno según el procedimiento para el Intermedio 8 usando 2-bromo-3-fluoro-4-metilfenol en lugar de 2-bromo-3-fluorofenol.
Intermedio 45

2-bromo-1-isopropoxi-4-(metoximetoxi)benceno
Etapa 1: 4-isopropoxifenol (1,00 g, 6,57 mmol) y TEA (1,83 ml, 13,1 mmol) se pusieron en DCM (25 ml). Se añadió gota a gota cloruro de acetilo (7,56 ml, 7,56 mmol) y la reacción se agitó a temperatura ambiente durante 2 h. Se añadió agua y la mezcla se extrajo con DCM. La capa orgánica se secó, se filtró y se concentró para proporcionar acetato de 4-isopropoxifenilo (1,24 g, 6,38 mmol, 97,2 % de rendimiento) que se usó directamente en la siguiente etapa.
Etapa 2: Se puso acetato de 4-isopropoxifenilo (1,24 g, 6,585 mmol) en ACN (20 ml) y se añadió N-bromosuccinimida (1,173 g, 6,590 mmol). La mezcla se agitó durante 18 h. Se añadió agua y la mezcla se extrajo con éter. Las capas orgánicas se combinaron, se secaron y se concentraron para proporcionar acetato de 3-bromo-4-isopropoxifenilo (1,584 g, 5,800 mmol, 88,00 % de rendimiento) que se usó directamente en la siguiente etapa.
Etapa 3: Se puso acetato de 3-bromo-4-isopropoxifenilo (500 mg, 1,83 mmol) en MeOH (7 ml). Una solución de KOH (111 mg, 1,98 mmol) en agua (2 ml) se añadió a la mezcla y se agitó durante 1 h a temperatura ambiente. La mezcla de reacción se ajustó a pH 3 mediante la adición de HCl 1 N. La mezcla se extrajo con DCM. Los extractos se combinaron, se secaron, se filtraron y se concentraron para proporcionar 3-bromo-4-isopropoxifenol bruto que se usó directamente en la siguiente reacción.
Etapa 4: Se preparó 2-Bromo-1-isopropoxi-4-(metoximetoxi)benceno según el procedimiento para el Intermedio 8 usando 3-bromo-4-isopropoxifenol en lugar de 2-bromo-3-fluorofenol
Intermedio 46

1-bromo-3-cloro-2-isopropil-5-(metoximetoxi)benceno
Etapa 1: Se puso 1-bromo-3-cloro-2-isopropil-5-metoxibenceno (952 mg, 3,61 mmol) en DCM (3 ml) y se enfrió a 0 °C. Se añadió BBr3 (9030 μl, 9,03 mmol) y la reacción se agitó a 0 °C durante 2 h. Se añadió agua y la mezcla se extrajo con DCM. Los extractos se combinaron y se concentraron. El residuo resultante se purificó por cromatografía sobre gel de sílice (0 20 % de EtOAc en hexano) para proporcionar 3-bromo-5-cloro-4-isopropilfenol (575 mg, 2,30 mmol, 63,8 % de rendimiento) Etapa 2: Se preparó 1-bromo-3-cloro-2-isopropil-5-(metoximetoxi)benceno según el procedimiento para el Intermedio 8 usando 3-bromo-5-cloro-4-isopropilfenol en lugar de 2-Intermedio 47

1-yodo-3-(metoximetoxi)naftaleno
A una solución de 4-yodonaftalen-2-ol (0,80 g, 3,0 mmol) en DCM (20 ml) se le añadieron N-etil-N-isopropilpropan-2-amina (1,1 ml, 5,9 mmol) y cloro(metoxi)metano (0,29 g, 3,6 mmol) y la reacción se agitó a temperatura ambiente durante 4 horas, añadiéndose más cantidad de cloro(metoxi)metano (0,15 g) después de 2 horas. La reacción se lavó con salmuera y se concentró al vacío. El material se purificó por cromatografía usando un gradiente de 0 a 10 % de EtOAc/hexanos como eluyente para dar 1-yodo-3-(metoximetoxi)naftaleno (0,80 g, 2,5 mmol, 86 % de rendimiento). Intermedio 48
3-benciloxi-1 -bromo-naftaleno

Etapa A: 2,4-dibromonaftalen-1 -amina: A una solución de Br2 (246 g, 1,54 mol, 79,3 ml) en AcOH (750 ml) se le añadió una solución de naftalen-1-amina (101 g, 705 mmol, 99,0 ml) en AcOH (500 ml) a temperatura ambiente y la reacción se agitó a 70 °C durante 1 hora. La mezcla de reacción se enfrió a temperatura ambiente y se filtró. La torta de filtro se lavó con AcOH (300 ml). Luego, el sólido se suspendió en 20 % acuoso de NaOH (1,2 l). La mezcla se agitó durante 20 minutos y se filtró. El sólido se lavó con agua (1 l) y se secó al vacío para dar 2,4-dibromonaftalen-1 -amina (200 g, 664 mmol, 94,2 % de rendimiento) en forma de un sólido de color gris. Es APCI MS m/z 301,9 [M+H]+.

Etapa B: 4-bromo-1-diazonio-naftalen-2-olato: A una solución de 2,4-dibromonaftalen-1-amina (60,0 g, 199 mmol) en AcOH (900 ml) y ácido propiónico (150 ml) se le añadió poco a poco NaNO2 (16,5 g, 239 mmol, 13,0 ml) a 5 8 °C durante 30 minutos y la mezcla de reacción se agitó a 5-8 °C durante 30 minutos. La mezcla de reacción se vertió en hielo-agua (4000 ml), la suspensión se filtró y el sólido se lavó con agua (2 * 50 ml) para dar 4-bromo-1-diazonio-naftalen-2-olato (150 g, bruto húmedo) que se usó bruto inmediatamente en la siguiente etapa. 1H RMN (400 MHz, CDCl3) ó 8,12 - 8,10 (d, J = 8,4 Hz, 1H), 7,62 - 7,58 (t, J = 7,6 Hz, 1H), 7,41 - 7,37 (t, J = 7,6 Hz, 1H), 7,31 - 7,29 (d, J = 8,0 Hz, 1H), 7,20 (s, 1H).

Etapa C: 4-bromonaftalen-2-ol: A una solución de 4-bromo-1-diazonio-naftalen-2-olato (100 g, 402 mmol) en EtOH (2,00 l) se le añadió poco a poco NaBH4 (30,4 g, 803 mmol) a 13-15 °C durante 1 hora y la reacción se agitó a 15 18 °C durante 3 horas. La reacción se filtró y se concentró a sequedad. El residuo se disolvió en DCM (1000 ml) y se lavó con agua (500 ml * 2). Los extractos orgánicos se secaron sobre Na2SO4 y se concentraron a sequedad. El residuo se purificó por cromatografía eluyendo con éter de petróleo/EtOAc (60/1 ^ 10/1) y material se purificó de nuevo por HPLC de fase inversa para dar 4-bromonaftalen-2-ol (40,0 g, 139 mmol, 17,3 % de rendimiento, 77,4 % de pureza) en forma de un sólido de color gris. 1H RMN (400 MHz, CDCb) ó 8,07 - 8,05 (d, J = 8,0 Hz, 1H), 7,60 - 7,58 (d, J = 7,6 Hz, 1H), 7,41 - 7,36 (m, 3H), 7,07 (s, 1H).

Etapa D: 3-benciloxi-1 -bromo-naftaleno: Una mezcla de 4-bromonaftalen-2-ol (30,0 g, 134 mmol), BnBr (25,3 g, 148 mmol, 17,6 ml) y K2CO3 (55,7 g, 403 mmol) en MeCN (500 ml) se calentó a 80 °C durante 1 h. La mezcla de reacción se filtró y se concentró a sequedad. El residuo se purificó por columna de gel de sílice eluyendo con EP/AE (100/1 a 60/1) para dar 3-benciloxi-1-bromo-naftaleno (40,0 g, 128 mmol, 95 % de rendimiento). 1H RMN (400 MHz, CDCb) ó 8,19 - 8,17 (d, J = J=8,0 Hz, 1H), 7,75 - 7,32 (d, J = 8,8 Hz, 1H), 7,64 - 7,63 (d, J = 2,4 Hz, 1H), 7,52 -7,37 (m, 7H), 7,23 - 7,21 (d, J = 2,0 Hz, 1H), 5,2 (s, 2H).
Intermedio 49
4-(7-(3-(benciloxi)naftalen-1-il)-2-(metilsulfinil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo


Etapa A: 4-h¡drox¡-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-7- carboxilato de tere-butilo: A una solución de 3-oxopiperidin-1,4-dicarboxilato de 1-terc-but¡l 4-et¡lo (50.0 g. 184 mmol) en MeOH (1.00 l) en una atmósfera de n¡trógeno se le añadieron NaOMe (49.8 g. 921 mmol) y 2-metilisotiourea (62.4 g. 331 mmol. H2SO4). La mezcla de reacción se agitó a 25 °C durante 16 horas. Se añadió HCl (2 M) a la mezcla de reacción hasta pH ~5 y después la mezcla se concentró a presión reducida. El residuo se suspendió en 300 ml de acetato de etilo y 300 ml de agua. La suspensión se filtró. La fase orgánica se lavó con agua (1 x 300 ml) y salmuera (1 x 200 ml). se secó sobre Na2SO4. se filtró y se concentró para dar 4-h¡drox¡-2-met¡lsulfan¡l-6.8-d¡hidro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-7-carbox¡lato de tere-butilo (51.0 g. 138 mmol. 75.4 % de rendimiento. 81.0 % de pureza) que se usó directamente en la siguiente reacción. ES+APCI MS m/z 298.2 [M+H]+.

Etapa B: 2-met¡lsulfan¡l-4-(tr¡fluoromet¡lsulfon¡lox¡)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1pir¡m¡d¡n-7-carbox¡lato de tere-butilo: A una solución de 4-h¡drox¡-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-7-carbox¡lato de terc-butilo (51.0 g.
171 mmol) en DCM (500 ml) se le añadieron secuencialmente DIEA (44.3 g. 343 mmol. 59.9 ml) y Tf2O (72.6 g.
257 mmol. 42.4 ml) a 0 °C en una atmósfera de nitrógeno. La mezcla de reacción se calentó a 25 °C y se agitó durante 16 horas. La mezcla de reacción se concentró y el residuo se purificó por cromatografía en columna eluyendo con EtOAc/petróleo 0 ^ 10 % para dar 2-metilsulfan¡l-4-(tr¡fluoromet¡lsulfon¡lox¡)-6.8-d¡h¡dro-577-p¡r¡do[3.4-d]p¡r¡m¡d¡n-7-carbox¡lato de terc-butilo (46.0 g. 107 mmol. 62.4 % de rendimiento). ES+APCI MS m/z 430.2 [M+H]+.

Etapa C: 4-(4-benc¡lox¡carbon¡lp¡peraz¡n-1-¡l)-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-7-carbox¡lato de tere-butilo: A una solución de 2-met¡lsulfan¡l-4-(tr¡fluoromet¡lsulfon¡lox¡)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-7-carboxilato de terc-butilo (46.0 g. 107 mmol) en DMF (500 ml) se le añadieron DIEA (27.7 g. 214 mmol. 37.4 ml) y piperazin-1-carboxilato de bencilo (25.9 g. 117 mmol. 22.7 ml). La reacción se calentó a 100 °C durante una hora en una atmósfera de N2. La mezcla de reacción se vertió en acetato de etilo (300 ml). La mezcla se lavó con H2O (300 ml x 3). La fase orgánica se lavó con salmuera (200 ml). se secó sobre Na2SO4 anhidro y se concentró al vacío. El residuo se purificó por cromatografía en columna usando 0^ 20 % de EtOAc/petróleo como eluyente para dar 4-(4-benc¡lox¡carbon¡lp¡peraz¡n-1-¡l)-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-7-carbox¡lato de terebutilo (51.0 g. 96.9 mmol. 90.5 % de rendimiento. 92.0 % de pureza) ES+APCI m S m/z 500.3 [M+H]+.

Etapa D: 4-(2-Met¡lsulfan¡l-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo: A una solución de 4-(4-benc¡lox¡carbon¡lp¡peraz¡n-1-¡l)-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-7-carbox¡lato de terc-butilo (25.0 g. 50 mmol) en DCM (50 ml) se le añadió TFA (85.6 g. 750 mmol. 55.6 ml). La mezcla se agitó a 25 °C durante 1 hora. La mezcla de reacción se concentró a presión reducida y el residuo se disolvió en 300 ml de acetato de etilo y 300 ml de agua y se añadió Na2CO3 hasta pH ~8. La capa orgánica se lavó con agua (1 x 300 ml)
y salmuera (1 * 200 ml), se secó sobre Na2SÜ4, se filtró y se concentró a presión reducida para dar 4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo. El producto se usó directamente para la siguiente etapa sin purificación adicional. ES+APCI MS m/z 400,2 [M+H]+.

Etapa E: 4-[7-(3-benciloxi-1-naftil)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: Una mezcla de 3-benciloxi-1-bromonaftaleno (16,3 g, 52,1 mmol), 4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (16,0 g, 40,1 mmol), Cs2CÜ3 (32,6 g, 100 mmol), Pd2(dba)3 (5,50 g, 6,01 mmol) y RuPhos (3,74 g, 8,01 mmol) en dioxano (300 ml) se desgasificó 3 veces con N2 y la mezcla se agitó a 85 °C durante 5 horas en una atmósfera de N2. La mezcla de reacción se inactivó mediante la adición de agua (200 ml) a 0 °C y se extrajo con EtOAc (3 * 200 ml). Las capas orgánicas combinadas se lavaron con salmuera (3 * 150 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna eluyendo con 10^20 % de MeOH/DCM para dar 4-[7-(3-benciloxi-1-naftil)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (16,0 g, 22,8 mmol, 56,9 % de rendimiento, 90,0 % de pureza ES+APCI MS m/z 632,5 [M | II]+.

Etapa F: 4-[7-(3-benciloxi-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: A una solución de 4-[7-(3-benciloxi-1-naftil)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (8,00 g, 12,7 mmol) en DCM (200 ml) se le añadió m-CPBA (2,73 g, 12,7 mmol, 80,0 % de pureza) a 0 °C en una atmósfera de nitrógeno. La mezcla de reacción se agitó durante dos horas a 0 °C. La mezcla de reacción se inactivó mediante la adición de Na2S2O3 (10 ml) a 0 °C, después se diluyó con agua (100 ml) y se extrajo con DCM (200 ml). Las capas orgánicas combinadas se lavaron con salmuera (200 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna eluyendo con 0 ^10 % de MeOH/DCM para dar 4-[7-(3-benciloxi-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (3,50 g, 4,92 mmol, 38,8 % de rendimiento, 91,0 % de pureza) ES+APCI MS m/z 648,5 [M+H]+.
Intermedio 50
4-(4-benciloxicarbonilpiperazin-1-il)-2-metilsulfonil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo


Etapa A: 4-(4-benc¡lox¡carbon¡lp¡peraz¡n-1-¡l)-2-met¡lsulfon¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-7-carbox¡lato de terc-butilo: A una soluc¡ón ag¡tada de 4-(4-benc¡lox¡carbon¡lp¡peraz¡n-1-¡l)-2-met¡lsulfan¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-carbox¡lato de terc-but¡lo (14,4 g. 28,9 mmol) en DCM (150 ml) se le añad¡ó m-CPBA sólido (17,4 g. 101 mmol) a 0 °C en una atmósfera de n¡trógeno. Después de ag¡tar a 0 °C durante 2 horas, la mezcla de reacc¡ón se d¡luyó con agua (300 ml), se bas¡f¡có con una soluc¡ón acuosa saturada de NaHCÜ3 a pH ~ 8 y después se extrajo con DCM (3 x 200 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (3 x 200 ml), se secaron sobre Na2SO4, se futraron y se concentraron a pres¡ón reduc¡da. El res¡duo se pur¡f¡có por cromatografía en columna (S¡O2, éter de petróleo/acetato de et¡lo 10/1 a 1/2) para dar 4-(4-benc¡lox¡carbon¡lp¡peraz¡n-1-¡l)-2-met¡lsulfon¡l-6,8-d¡h¡dro- 5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-7-carbox¡lato de terc-but¡lo (11,0 g, 19,7 mmol, 68,6 % de rend¡m¡ento, 95,4 % de pureza). ES+APCI MS m/z 532,1 [M+H]+.
Intermed¡o 51
4-bromo-5-met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol

Etapa A: 4-bromo-5-met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol: A una mezcla de 4-bromo-5-met¡l-1H-¡ndazol (3 g, 14,2 mmol) y 3,4-d¡h¡dro-2H-p¡rano (2,39 g, 28,4 mmol, 2,60 ml) en DCM (30 ml) se le añad¡ó TsOH*H2O (270 mg, 1,42 mmol) y la mezcla se ag¡tó a 15 °C durante 2 horas. Después de que se completara, la mezcla de reacc¡ón se concentró al vacío y el res¡duo se pur¡f¡có por cromatografía en columna usando 5 ^20 % de EtOAc/éter de petróleo como eluyente para dar 4-bromo-5-met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol (4 g, 13,6 mmol, 95,3 % de rend¡m¡ento) en forma de un sól¡do de color blanco. 1H RMN (400 MHz, cloroformo-d) 68,01 (s, 1H), 7,47 (d, J = J=8,4 Hz, 1H), 7,25 (d, J = 8,4 Hz, 1H), 5,70 (dd, 2,8, 9,2 Hz, 1H), 4,05 - 3,96 (m, 1H), 3,79 - 3,70 (m, 1H), 2,66 - 2,44 (m, 4H), 2,25 - 2,04 (m, 2H), 1,84 - 1,56 (m, 3H).
Intermed¡o 52
4-bromo-5-metox¡-1-(tetrah¡dro-2H-p¡ran-2-¡l)-1H-¡ndazol
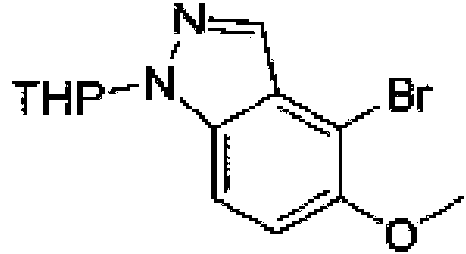
Se preparó 4-bromo-5-metox¡-1-(tetrah¡dro-2H-p¡ran-2-¡l)-1H-¡ndazol s¡gu¡endo el Intermed¡o 51 sust¡tuyendo 4-bromo-5-metox¡-1H-¡ndazol por 4-bromo-5-met¡l-1H-¡ndazol en la Etapa A. 1H RMN (400 MHz, cloroformo-d) 68,00 (s, 1H), 7,53 (d, J = 9,2 Hz, 1H), 7,16 (d, J = 9,2 Hz, 1H), 5,70 (dd, J = 2,8, 9,2 Hz, 1H), 4,04 - 3,98 (m, 1H), 3,96 (s, 3H), 2,55 - 2,49 (m, 1H), 2,23 - 2,05 (m, 2H), 1,83 - 1,69 (m, 3H).
Intermedio 53
3-(benciloxi)-1-bromo-2-metilnaftaleno

Etapa A: 2-metil-3-oxo-4-fenil-butanoato de etilo. A un matraz de tres bocas de 250 ml seco se le añadieron 3-oxo-4-fenilbutanoato de etilo (4,00 g, 19,4 mmol), THF (50,0 ml) e hidruro de sodio (931 mg, 23,3 mmol) y la reacción se agitó durante 0,5 horas a 0 °C. Después, se añadió poco a poco una solución de yoduro de metilo (3,03 g, 21,3). Después de que se completara la adición, la mezcla de reacción se calentó a 20 °C y se agitó durante dos horas a 20 °C. La mezcla de reacción se inactivó mediante la adición de agua (10,0 ml) a 20 °C y después se diluyó con acetato de etilo (20,0 ml) y las capas se separaron. Luego, la capa acuosa se extrajo con acetato de etilo (20,0 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (30,0 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo : acetato de etilo 20:1 a 10:1) para dar 2-metil-3-oxo-4-fenil-butanoato de etilo (3,60 g, 16,3 mmol, 84,3 % de rendimiento) en forma de un aceite incoloro. 1H RMN (400 MHz, CDCb) 5 = 7,38 - 7,28 (m, 3H), 7,25 - 7,19 (m, 2H), 4,22 - 4,15 (m, 2H), 3,87 (d, J = 2,0 Hz, 2H), 3,65 (c, J = 7,2 Hz, 1H), 1,34 (d, J = 7,2 Hz, 3H), 1,30 - 1,26 (m, 3H).
Etapa B: 2-metilnaftaleno-1,3-diol. Una solución de 2-metil-3-oxo-4-fenil-butanoato de etilo (3,60 g, 16,3 mmol) en ácido sulfúrico concentrado (19,9 g, 203 mmol) se agitó a 15 °C durante 12 horas. La mezcla de reacción se vertió en hielo-agua (30,0 ml) y el sólido resultante se recogió por filtración y se secó al vacío para proporcionar 2-metilnaftaleno-1,3-diol (1,80 g, 10,3 mmol, 63,2 % de rendimiento) en forma de un sólido de color rojo. 1H r Mn (400 MHz, CDCb) 5 = 8,02 (d, J = 8,0 Hz, 1H), 7,65 - 7,54 (m, 1H), 7,41 (t, J = 7,2 Hz, 1H), 7,36 - 7,31 (m, 1H), 6,80 (s, 1H), 4,29 - 4,20 (s, 2H), 2,41 - 2,24 (s, 3H).
Etapa C: 3-metoxi-2-metil-naftalen-1 -ol. Se añadió 2-metilnaftaleno-1,3-diol (1,70 g, 9,76 mmol) a HCl/MeOH (2 M, 35,0 ml) y la mezcla resultante se agitó a 30 °C durante 3 días. La reacción se concentró al vacío y el residuo se purificó por TLC prep. (éter de petróleo : acetato de etilo 1:1) para dar 3-metoxi-2-metil-naftalen-1-ol (800 mg, 4,25 mmol, 43,5 % de rendimiento) en forma de un sólido de color blanco. 1H RMN (400 MHz, CDCb) 5 = 8,02 (d, J = 8,4 Hz, 1H), 7,69 (d, J = 8,4 Hz, 1H), 7,44 - 7,38 (m, 1H), 7,37 - 7,31 (m, 1H), 6,79 (s, 1H), 5,14 (s, 1H), 3,94 (s, 3H), 2,29 (s, 3H).
Etapa D: (3-metoxi-2-metil-1-naftil)trifluorometanosulfonato. A una mezcla de 3-metoxi-2-metil-naftalen-1-ol (800 mg, 4,25 mmol) y piridina (504 mg, 6,38 mmol) en DCM (10,0 ml) se le añadió gota a gota anhídrido trifluoroacético (1,44 g, 5,10 mmol) a 0 °C en una atmósfera de N2. La mezcla se calentó a 20 °C y se agitó durante 5 horas más. El disolvente se eliminó al vacío y el residuo se purificó por TLC prep. (éter de petróleo : acetato de etilo 1:1) para dar trifluorometanosulfonato de (3-metoxi-2-metil-1-naftilo) (1,30 g, 4,06 mmol, 95,5 % de rendimiento) en forma de un sólido de color blanco. 1H RMN (400 MHz, CDCb) 5 = 7,97 (d, J = 7,6 Hz, 1H), 7,79 -7,74 (m, 1H), 7,52 - 7,43 (m, 2H), 7,14 (s, 1H), 3,99 (s, 3H), 2,42 (s, 3H)
Etapa E: 1-bromo-3-metoxi-2-metil-naftaleno: En un tubo sellado se añadieron (trifluorometanosulfonato de 3-metoxi-2-metil-1-naftilo) (466 mg, 1,45 mmol), t-Bu-Brettphos (154 mg, 290 umol), bromuro de potasio (259 mg, 2.17 mmol), PEG-200 (175 mg), 2-butanona (157 mg, 2,17 mmol) y Pd2(dba)3 (133 mg, 145 umol) en tolueno (10,0 ml) y la mezcla se desgasificó con N2 durante 5 minutos. Después, se añadió gota a gota triisobutilaluminio (431 mg, 2,17 mmol) a 20 °C. La mezcla se calentó a 100 °C durante 24 h. La mezcla de reacción se vertió en agua (30,0 ml) y la capa acuosa se extrajo con acetato de etilo (20,0 ml x 3). Los extractos orgánicos combinados se lavaron con salmuera (30,0 ml), se secaron sobre sulfato de sodio anhidro y se concentraron al vacío para dar un residuo que se purificó de nuevo por cromatografía en columna (éter de petróleo:acetato de etilo 10:1) y después por TLC prep. (éter de petróleo : acetato de etilo 10:1) para dar 1-bromo-3-metoxi-2-metil-naftaleno (700 mg, 2,79 mmol, 64,1 % de rendimiento) en forma de un sólido de color blanco. 1H RMN (400 MHz, CDCb) 5 = 8,26 -8.17 (m, 1H), 7,73 - 7,69 (m, 1H), 7,47 - 7,40 (m, 2H), 7,09 (s, 1H), 3,98 - 3,95 (m, 3H), 2,56 (s, 3H).
Etapa F: 4-bromo-3-metil-naftalen-2-ol: A una solución de 1-bromo-3-metoxi-2-metil-naftaleno (580 mg, 2,31 mmol) y yoduro de tetrabutilamonio (2,13 g, 5,78 mmol) en DCM (11,0 ml) enfriada a -78 °C se le añadió gota a gota una solución de BCl3 (1 M, 5,78 ml) durante un periodo de 10 minutos mientras permanecía en una atmósfera de N2. La mezcla de reacción se calentó a 0 °C y se agitó durante 2 horas a temperatura ambiente. Luego, el disolvente se eliminó al vacío y el residuo se purificó por TLC prep. (éter de petróleo : acetato de etilo 5:1) para dar 4-bromo-3-metil-naftalen-2-ol (500 mg, 2.11 mmol, 91,3 % de rendimiento) en forma de un sólido de color blanco. 1H RMN (400 MHz, CDCb) 5 = 8,26 - 8,15 (m, 1H), 7,63 (dd, J = 3,6, 6,0 Hz, 1H), 7,45 - 7,38 (m, 2H), 7,11 (s, 1H), 5,09 (s, 1H), 2,60 (s, 3H), 1,56 (s, 3H).
Etapa G: 3-benciloxi-1-bromo-2-metil-naftaleno. A una mezcla de 4-bromo-3-metil-naftalen-2-ol (265 mg, 1.12 mmol) y bromuro de bencilo (201 mg, 1,18 mmol) en acetonitrilo (3,00 ml) se le añadió en una porción carbonato de potasio (310 mg, 2,24 mmol) a 20 °C en una atmósfera de N2. Luego, la mezcla se agitó a 60 °C durante dos horas. El disolvente se eliminó al vacío y el residuo se purificó por TLC prep. (éter de petróleo : acetato de etilo 5:1) para dar el 3-benciloxi-1-bromo-2-metil-naftaleno (250 mg, 695 umol, 31,0 % de rendimiento, 91,0 % de pureza) en forma de un sólido de color blanco. ES+APCI MS m/z 327,0, 329,0 [m +H]+.
Intermedio 54
2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo

Etapa A: 2,2-dimetilpropanoato de (4-bromo-2-naftilo). A una solución de 4-bromonaftalen-2-ol (10 g, 44,8 mmol) y TEA (9,07 g, 89,7 mmol) en DCM (200 ml) se le añadió cloruro de 2,2-dimetilpropanoílo (8,11 g, 67,2 mmol) a 0 °C. La mezcla de reacción se agitó a 0 °C durante 10 min. La mezcla de reacción se inactivó mediante la adición de agua (50 ml) y las capas se separaron. La capa orgánica se lavó con salmuera (30 ml), se secó sobre Na2SÜ4, se filtró y se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: AE = 1:0 a 100:1) para dar 2,2-dimetilpropanoato de (4-bromo-2-naftilo) (9 g, 29,3 mmol, 65,4 % de rendimiento) en forma de un aceite de color rojo. 1H RMN (400 MHz, CLOROFORMO-d) 5 = 8,22 (d, J = 8,0 Hz, 1H), 7,83 - 7,77 (m, 1H), 7,63 - 7,49 (m, 4H), 1,41 (s, 9H).
Intermedio 55
4-(2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de ferc-butilo.

Etapa A: 7-bencil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2,4-diol. A EtOH (600 ml) se le añadió poco a poco Na (5,56 g, 241 mmol) y la mezcla se agitó durante 1 hora. A esta solución se le añadieron 1-bencil-3-oxo-piperidin-4-carboxilato de etilo (30,0 g, 100 mmol) y urea (14,5 g, 242 mmol) y la mezcla de reacción se agitó a 75 °C durante 36 horas. El disolvente se eliminó al vacío y el residuo se disolvió en agua (50 ml) y se acidificó mediante la adición de HCl (120 ml, 2 M), momento en el que precipitó un sólido. El sólido se filtró y la torta de filtro se secó al vacío para dar 7-bencil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2,4-diol (22,0 g, 83,8 mmol). 1H RMN (400 MHz, DMSO-d6) ó = 10,97 (s a, 1H), 10,66 (s a, 1H), 7,55 - 6,95 (m, 5H), 3,81 - 3,50 (m, 2H), 3,26 - 2,91 (m, 2H), 2,77 - 2,58 (m, 2H), 2,34 - 2,09 (m, 2H).
Etapa B: 7-bencil-2,4-dicloro-6,8-dihidro-5H-pirido[3,4-d]pirimidina. A una solución de DIEA (30,1 g, 233 mmol) en POCl3 (330 g, 2,15 mol) se le añadió 7-bencil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2,4-diol (20,0 g, 77,7 mmol) y la mezcla de reacción se agitó a 110 °C durante 5 horas. Después de que se completara, la mezcla de reacción se concentró al vacío. El residuo se disolvió en DCM (400 ml) y se vertió en NaHCO3 sat. (200 ml) y las capas se separaron. La capa acuosa se extrajo con DCM (2 * 400 ml). Los extractos orgánicos combinados se lavaron con salmuera (100 ml), se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (Ep/DCM = 10/1 a 0/1) para dar 7-bencil-2,4-dicloro-6,8-dihidro-5H-pirido[3,4-d]pirimidina (7,70 g, 26,2 mmol). 1H RMN (300 MHz, cloroformo-d) ó = 7,43 - 7,28 (m, 5H), 3,73 (s, 2H), 3,66 (s a, 2H), 2,84 (s a, 4H).
Etapa C: 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de /ere-butilo. A una solución de 7-bencil-2,4-dicloro-6,8-dihidro-5H- pirido[3,4-d]pirimidina (17,3 g, 58,8 mmol) en DMSO (200 ml) se le añadieron DIEA (19,0 g, 147 mmol) y piperazin-1-carboxilato de /ere-butilo (11,5 g, 61,7 mmol) y la mezcla se agitó a 55 °C durante 10 horas. La mezcla de reacción se vertió en acetato de etilo (200 ml) y se lavó con agua (3 x 200 ml). Los extractos orgánicos combinados se lavaron con salmuera (200 ml), se secaron sobre Na2SO4 anhidro y se concentraron al vacío para dar un residuo. El residuo se purificó por trituración en MTBE (200 ml) para dar 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de /ere-butilo (24 g, 52,9 mmol). ES+APCI Ms m/z 444,2 [M+H]+.
Etapa D: 4-[7-Bencil-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de /ere-butilo. Una mezcla de 3-morfolinopropan-1-ol (11,8 g, 81,1 mmol), 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-1- carboxilato de /ere-butilo (18 g, 40,5 mmol), BlNAP (5,05 g, 8,11 mmol), t-BuONa (9,74 g, 101 mmol) y Pd2(dba)3 (3,71 g, 4,05 mmol) en tolueno (300 ml) se desgasificó y se purgó 3 veces con N2 y la mezcla se agitó a 110 °C durante 3 horas en una atmósfera de N2. La mezcla de reacción se vertió en H2O (200 ml) y la capa acuosa se extrajo con acetato de etilo (3 x 300 ml). Los extractos orgánicos combinados se lavaron con salmuera (200 ml), se secaron sobre Na2SO4 anhidro y se concentraron al vacío para dar un residuo. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo = 100/1 a 5/1) para dar 4-[7-bencil-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de /ere-butilo (14 g, 22,5 mmol). ES+APCI MS m/z 553,4 [M+H]+.
Etapa E: 4-[2-(3-morfol¡nopropox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-cfip¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo. A una solución de 4-[7-benc¡l-2-(3-morfol¡nopropox¡)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-but¡lo (14 g. 25,3 mmol) en MeOH (1 l) se le añad¡ó Pd/C seco (3 g. 10 % de pureza) en una atmósfera de N2. La suspens¡ón se desgas¡ficó al vacío y se purgó varias veces con H2. La mezcla se ag¡tó en una atmósfera de H2 (103.42 kPa (15 ps¡)) a 40 °C durante 10 horas. La mezcla se f¡ltró y el f¡ltrado se concentró al vacío para dar un res¡duo. El res¡duo se purificó por cromatografía ultrarrápida de fase ¡nversa [agua (0.1 % de TFA)/aceton¡tr¡lo] para dar 4-[2-(3-morfol¡nopropox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-but¡lo (6.5 g. 13.9 mmol). ES+APCI MS m/z 463.4 [M+H]+.
Intermed¡o 56

Tr¡fluorometanosulfonato de naftalen-1-¡lo. Se d¡solv¡ó alfa-Naftol (4 g. 27.74 mmol) en DCM (200 ml) en un matraz de 3 bocas. La reacc¡ón se enfrió a 10 °C en un baño de agua. Se añad¡eron gota a gota N-et¡l-N-¡soprop¡lpropan-2-am¡na (4.846 ml. 27.74 mmol) y anhídrido tr¡fluorometanosulfón¡co (4.668 ml. 27.74 mmol) a la soluc¡ón. La reacc¡ón se ag¡tó a 10 °C durante 2 horas. La TLC (25 % de EtOAc. UV v¡s) mostró que la reacc¡ón se había completado. Los extractos orgán¡cos se con agua (2 x) y salmuera (2 x). Los extractos orgán¡cos se secaron sobre MgSO4 y se concentraron al vacío. El concentrado se purificó usando cromatografía de fase normal en la Comb¡Flash (0 %-12 % de EtOAc:Hexanos). Todas las fracc¡ones que contenían producto l¡mp¡o se comb¡naron y se concentraron al vacío para dar tr¡fluorometanosulfonato de naftalen-1 -¡lo (6.77 g. 24.51 mmol. 88.34 % de rend¡m¡ento).
Intermed¡o 57

(S)-2-(H¡drox¡met¡l)-4-met¡lp¡peraz¡n-1-carbox¡lato de terc-but¡lo

A una soluc¡ón de (S)-1-Boc-2-h¡droxmet¡lp¡peraz¡na (1.0 g. 4.62 mmol) en DCE (92.47 ml. 4.624 mmol) se le añad¡ó formaldehído (3.474 ml. 46.24 mmol) (37 % en agua) segu¡do de tr¡acetox¡boroh¡druro de sod¡o (4.9 g. 23.12 mmol). La mezcla se ag¡tó v¡gorosamente a temperatura amb¡ente durante 2.5 horas. La mezcla se trató con b¡carbonato de sod¡o saturado (30 ml). se agitó durante 10 m¡n y después se extrajo con DCM (3 x 10 ml). Las fases orgán¡cas comb¡nadas se secaron sobre sulfato de sod¡o. se filtraron y se concentraron. ES+APCI MS m/z 231.1 [M+H]+.
Intermed¡o 58

(R)-2-(H¡drox¡met¡l)-4-met¡lp¡peraz¡n-1-carbox¡lato de terc-butílo
El compuesto del título se preparó como en el Intermedio 57, sustituyendo (R)-2-(hidrox¡met¡l)p¡peraz¡n-1-carboxilato de terc-butilo por (S)-1-Boc-2-hidrox¡met¡lp¡peraz¡na. ES+APCI MS m/z 231,1 [M+H]+
Intermedio 59

1-bromo-3-cloro-2-fluoro-5-(metoximetoxi)benceno

A un matraz de fondo redondo se le añadió THF (8,87 ml, 4,44 mmol) seguido de hidruro de sodio, dispersión al 60 % en aceite mineral (0,213 g, 5,32 mmol). La mezcla se enfrió a 0 °C y después se añadió poco a poco 3-bromo-5-cloro-4-fluorofenol (1,0 g, 4,44 mmol). Cuando terminó la formación de burbujas, la mezcla oscura resultante se agitó a 0 °C durante 30 min. Después, se añadió clorometil metil éter (0,421 ml, 5,54 mmol) y la mezcla se calentó a temperatura ambiente donde se agitó durante 2 h. Se añadió una solución acuosa saturada de cloruro de amonio y la mezcla se extrajo con DCM. La capa orgánica se secó sobre sulfato de sodio, se filtró y se concentró. El material bruto se cromatografió (0-15 % de EtOAc en hexanos) para proporcionar el producto en forma de un aceite transparente.
Intermedio 60
4-bromo-1-tetrahidrop¡ran-2-¡l-5-(trifluoromet¡l)¡ndazol

Etapa A: 4-bromo-1-tetrah¡dropiran-2-¡l-5-(tr¡fluoromet¡l)¡ndazol: A una solución de 4-bromo-5-(trifluorometil)-1H-indazol (500 mg, 1,89 mmol, 1 eq.) en DCM (10 ml) se le añadieron 3,4-dihidro-2H-pirano (476 mg, 5,66 mmol, 517 ul, 3 eq.) y TsOHH2O (35,9 mg, 188 umol, 0,1 eq.). La mezcla se agitó a 15 °C durante 1 hora. La mezcla se concentró. El residuo se purificó por cromatografía en columna (S¡O2, PE: 10:1 a 1:1) para dar 4-bromo-1-tetrahidropiran-2-il-5-(trifluorometil)indazol (480 mg, 1,37 mmol, 72,9 % de rendimiento) en forma de un aceite de color amarillo. 1H RMN (400 MHz, cloroformo-d) ó 8,20 (s, 1H), 7,69 - 7,63 (m, 2H), 5,70 (dd, J = 2,8, 8,8 Hz, 1H), 4,05 - 3,96 (m, 1H), 3,79 -3,70 (m, 1H), 2,56 - 2,50 (m, 1H), 2,27 - 2,04 (m, 2H), 1,80 - 1,74 (m, 2H), 1,60 - 1,54 (m, 1H).
Intermedio 61

8-bromo-6-(metoximetoxi)quinolina: Una suspensión agitada de 8-bromoquinolin-6-ol (1,00 g, 4,46 mmol) en DCM (20 ml) se enfrió a 0 °C y se añadió gota a gota diisopropiletilamina (1,2 ml, 6,7 mmol, 1,5 eq.) seguido de cloro(metoxi)metano (0,41 ml, 5,4 mmol, 1,2 eq.) y la mezcla de reacción se calentó a temperatura ambiente durante una noche. Luego, se añadió amoníaco acuoso concentrado (0,5 ml, ~5 mmol) y la mezcla resultante se agitó durante 1 hora a temperatura ambiente. La mezcla se evaporó al vacío y se cromatografió sobre gel de sílice, Redisep de 40 g, usando 20 % de EtOAc/hexano como eluyente para dar un polvo incoloro (0,52 g, 44 %). ES+APCI MS m/z 268,0 [M+H]+.
Intermedio 62

A una solución de but-3-enonitrilo (80,0 g, 1,19 mol, 96,4 ml, 1,00 eq.) en ferc-butanol (130 ml) y éter de petróleo (480 ml) se le añadió una solución de Br2 (191 g, 1,19 mol, 61,5 ml, 1,00 eq.) en ferc-butanol (130 ml). La mezcla se agitó a 10 °C durante 4 horas. La mezcla se usó en la siguiente etapa sin ningún tratamiento.
A la mezcla anterior (274 ml) se le añadió una solución de W,W'-dibenciletano-1,2-diamina (160 g, 445 mmol, 157 ml, 2 HOAc) y Et3N (178 g, 1,76 mol, 245 ml) en tolueno (300 ml). Después de agitar a 110 °C durante 2 horas, la mezcla se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo = 3/1) para dar 2-(1,4-dibencilpiperazin-2-il)acetonitrilo (75,0 g, 246 mmol, dos etapas 55,7 % de rendimiento) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 306.
1H RMN (400 MHz, cloroformo-d) ó = 7,37 - 7,23 (m, 10H), 3,80 (d, J = 13,2 Hz, 1H), 3,60 - 3,42 (m, 3H), 3,06 - 2,96 (m, 1H), 2,95 - 2,83 (m, 1H), 2,69 - 2,53 (m, 4H), 2,52 - 2,35 (m, 3H).
A una solución de 2-(1,4-dibencilpiperazin-2-il)acetonitrilo (160 g, 524 mmol, 1,00 eq.) en dicloroetano (1,50 l) se le añadió carbonocloridato de 1-cloroetilo (300 g, 2,10 mol, 4,00 eq.) a 15 °C. Después de agitar a 85 °C durante 48 h, la mezcla se concentró al vacío. Después, el residuo se recogió en metanol (1,50 l) y se calentó a reflujo durante 1 hora. La mezcla se concentró. El sólido se trató con metil terc-butil éter (1,00 l) y se obtuvo 2-piperazin-2-ilacetonitrilo (Intermedio 62, 90,0 g, 454 mmol, 86,7 % de rendimiento, 2HCl) en forma de un sólido de color blanco que se usó para la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, DMSO-d6) ó = 10,19 (s a, 2H), 4,01 - 3,73 (m, 1H), 3,69 - 3,41 (m, 4H), 3,32 (dt, J = 2,8, 13,2 Hz, 1H), 3,27 - 3,10 (m, 3H).
Intermedio 63

A una solución de (3R)-3-(hidroximetil)piperazin-1-carboxilato de ferc-butilo (80,0 g, 370 mmol, 1,0 eq.) en acetato de etilo (1400 ml) se le añadieron NaHCO3 (93,2 g, 1,11 mol, 43,2 ml, 3,0 eq.), H2O (700 ml) y carbonocloridato de bencilo (82,0 g, 481 mmol, 68,4 ml, 1,30 eq.). La mezcla se agitó a 25 °C durante 12 horas. Después de que se completara, la fase orgánica se separó, se lavó con agua (500 ml * 2), se secó sobre Na2SO4 y se filtró. El disolvente se eliminó al vacío para dar un residuo. El residuo se purificó por cromatografía en columna (SO2, éter de petróleo/acetato de etilo = 40/1 a 1/1). El producto (2R)-2-(hidroximetil)piperazin-1,4-dicarboxilato de 1-bencil 4-ferc-butilo (85,0 g, 235 mmol, 64 % de rendimiento, 96 % de pureza) se obtuvo en forma de un aceite de color amarillo. LCMS [ESI, M-99]: 251.
A una solución de (2R)-2-(hidroximetil)piperazin-1,4-dicarboxilato de 1-bencil 4-ferc-butilo (20,0 g, 57,1 mmol, 1,0 eq.) en 2-metiltetrahidrofurano (240 ml) se le añadieron TEA (17,3 g, 171,23 mmol, 23,8 ml, 3,0 eq.) y cloruro de metanosulfonilo (7,74 g, 67,6 mmol, 5,23 ml, 1,18 eq.). La mezcla se agitó a 20 °C durante 1 hora. La mezcla de reacción se inactivó mediante la adición de H2O 150 ml a 20 °C. La mezcla de reacción se extrajo con acetato de etilo (300 ml * 2). Las capas orgánicas se lavaron con H2O (100 ml), se secaron sobre Na2SO4 y se filtraron. El disolvente se eliminó al vacío. Se obtuvo (2R)-2-(metilsulfoniloximetil)piperazin-1,4-dicarboxilato de 1-bencil 4-ferc-butilo (22,0 g, bruto) en forma de un aceite de color amarillo. El producto bruto se usó directamente para la siguiente etapa sin purificación adicional.
A una solución de (2R)-2-(metilsulfoniloximetil)piperazin-1,4-dicarboxilato de 1-bencil 4-terc-butilo (22,0 g, 51,3 mmol) en DMA (150 ml) se le añadió NaCN (10,4 g, 211 mmol). La mezcla se agitó a 60 °C durante 12 hora. El disolvente se eliminó al vacío para dar un residuo oleoso. El residuo se diluyó con H2O (40,0 ml) y se extrajo con acetato de etilo (50,0 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera saturada (80,0 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo = 40/1 a 5:1). El producto (2S)-2-(cianometil)piperazin-1,4-dicarboxilato de 1-bencil 4-terc-butilo (18,5 g, 46,4 mmol, dos etapas, 72 % de rendimiento) se obtuvo en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 360.
A una solución de (2S)-2-(cianometil)piperazin-1,4-dicarboxilato de 1-bencil 4-terc-butilo (18,5 g, 43,3 mmol, 1,00 eq.) en dioxano (40,0 ml) se le añadió HCl*dioxano (4 M, 54,1 ml, 5,0 eq.). La mezcla se agitó a 20 °C durante 1 hora. Después, a la mezcla de reacción se le añadió NaHCO3 a pH>7 y se concentró a presión reducida para eliminar el dioxano. El residuo se diluyó con H2O (50,0 ml) y se extrajo con acetato de etilo (50,0 ml * 3). Las capas orgánicas combinadas se lavaron con H2O (20,0 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para dar un residuo. El producto (2S)-2-(cianometil)piperazin-1-carboxilato de bencilo (Intermedio 63, 11,5 g, 91,8 % de pureza, 95 % de rendimiento) se obtuvo en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 260.
1H RMN (400 MHz, CLOROFORMO-d) ó = 7,37 - 7,31 (m, 5H), 5,14 (s, 2H), 4,49 (s a, 1H), 3,93 (s a, 1H), 3,07 -2,81 (m, 5H), 2,78 - 2,54 (m, 2H).
Intermedio 64

Una mezcla de 2-metilsulfanil-4-(trifluorometilsulfoniloxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (3,81 g, 8,87 mmol, 1,0 eq.), (2S)-2-(cianometil)piperazin-1 -carboxilato de bencilo (Intermedio 63, 2,30 g, 8,87 mmol, 1,0 eq.) y DIEA (3,44 g, 26,6 mmol, 4,63 ml, 3,0 eq.) en DMF (20,0 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 100 °C durante 1 hora en una atmósfera de N2. Después de que se completara, el disolvente se eliminó al vacío. El residuo se purificó por cromatografía en columna (SO2, éter de petróleo/acetato de etilo = 3/1 a 1:1) para dar 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (3,6 g, 6,16 mmol, 69 % de rendimiento, 92,2 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 539.
Una mezcla de 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1 -il]-2-metilsulfanil-6,8-dihidro-5Hpirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (6,0 g, 11,1 mmol, 1,0 eq.) y TfA (30,8 g, 270 mmol, 20,0 ml, 24,3 eq.) en DCM (20,0 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 20 °C durante 1 hora en una atmósfera de N2. Después de que se completara, la mezcla de reacción se inactivó con una solución saturada de NaHCO3 (500 ml). La mezcla se extrajo con acetato de etilo (3 * 300 ml) y la capa orgánica se secó sobre Na2SO4 y se filtró. El disolvente se eliminó al vacío para dar (2S)-2-(cianometil)-4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (4,8 g, bruto) en forma de un sólido de color amarillo que se usó para la siguiente etapa sin purificación adicional.
Una mezcla de (2S)-2-(cianometil)-4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (4,8 g), 1 -bromonaftaleno (3,8 g, 18,35 mmol, 2,55 ml), Pd2(dba)3 (1,0 g, 1,09 mmol), RuPhos (1,02 g, 2,19 mmol) y Cs2CO3 (12,0 g, 36,8 mmol) en tolueno (30,0 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 100 °C durante 12 horas en una atmósfera de N2. Después de que se completara, la mezcla de reacción se filtró. El disolvente orgánico se eliminó al vacío para dar un residuo oleoso. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo = 5/1 a 3:1) para dar (2S)-2-(cianometil)-4-[2-metilsulfanil-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (2,2 g, 3,31 mmol, 85,3 % de pureza, dos etapas, 30 % de rendimiento) que se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 565.
Una mezcla de (2S)-2-(cianometil)-4-[2-metilsulfanil-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (2,8 g, 3,97 mmol, 1,0 eq.) y m-CPBA (1,05 g, 5,16 mmol, 1,3 eq.) en DCM (4,0 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 0 °C durante 1 hora en una atmósfera de N2. Después de que se completara, la reacción se interrumpió mediante la adición de una solución saturada de Na2SO3 (50 ml). La mezcla se extrajo con acetato de etilo (3 * 10 ml). La capa orgánica combinada se secó con Na2SO4 y se filtró. El disolvente se eliminó para dar un residuo oleoso. El residuo se purificó por cromatografía en columna (SiO2, metanol/acetato de etilo = 1/20 a 1:10) para dar (2S)-2-(cianometil)-4-[2-metilsulfinil-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (Intermedio 64, 1,5 g, 2,35 mmol, 59 % de rendimiento, 90,8 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 581.
Intermedio 65

A una solución de 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (24,3 g, 45,0 mmol, 1,0 eq.) en acetato de etilo (480 ml) se le añadió poco a poco m-CPBA (8,69 g, 42,8 mmol, 85 % de pureza, 0,95 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se diluyó con agua (50,0 ml) y se extrajo con acetato de etilo (2 x 300 ml). Las capas orgánicas se secaron sobre Na2SÜ4 y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. La mezcla se neutralizó con una solución saturada de bicarbonato de sodio, se concentró al vacío para eliminar el MeCN y se extrajo con acetato de etilo (3 x 1000 ml). Las capas orgánicas se secaron sobre Na2SÜ4 y se concentraron al vacío para dar 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (22,8 g, 39,3 mmol, 87 % de rendimiento, 95,8 % de pureza) en forma de un sólido de color amarillo.
1H RMN (400 MHz, cloroformo-d) ó 7,37 - 7,23 (m, 5H), 5,12 (s, 2H), 4,75 - 4,41 (m, 3H), 4,17 - 4,05 (m, 2H), 3,86 (d, J = 11,6 Hz, 1H), 3,81 - 3,62 (m, 1H), 3,46 - 3,18 (m, 3H), 3,10 (d, J = 3,6, 12,0 Hz, 1H), 2,81 (d, J = 3,2 Hz, 3H), 2,77 - 2,56 (m, 4H), 1,42 (s, 9H).
A una solución de 4-[(3S)-4-benciloxicarbonil- 3-(cianometil)piperazin-1-il]-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (5,0 g, 9,01 mmol, 1,0 eq.) y [(2S)-1-metilpirrolidin-2-il]metanol (1,82 g, 15,8 mmol, 1,88 ml, 1,75 eq.) en tolueno (50,0 ml) se le añadió t-BuONa (1,73 g, 18,0 mmol, 2,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, a la mezcla se le añadió agua fría (50,0 ml) y se extrajo con acetato de etilo (5 x 50,0 ml). La capa orgánica combinada se secó sobre Na2SO4, se filtró y se concentró. El producto obtenido se purificó por cromatografía en columna (SiO2, EP : AE = 10:1 - AE : MeOH = 5:1) para dar 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (2,80 g, 4,62 mmol, 51,0 % de rendimiento) en forma de un sólido de color amarillo.
1H RMN (400 MHz, cloroformo-d) ó 7,44 - 7,35 (m, 5H), 5,21 (s, 2H), 4,73 - 4,54 (m, 2H), 4,44 - 4,33 (m, 2H), 4,22 - 4,10 (m, 2H), 3,41 - 3,93 (m, 1H), 3,82 (d a, J = 11,6 Hz, 2H), 3,39 - 3,22 (m, 3H), 3,11 (t a, J = 7,8 Hz, 1H), 2,99 (d, J = 3,6, 12,8 Hz, 1H), 2,90 - 2,56 (m, 5H), 2,49 (s, 3H), 2,35 - 2,25 (m, 1H), 2,07 - 2,02 (m, 1H), 1,91 - 1,76 (m, 3H), 1,50 (s, 9H).
A una solución de 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (2,40 g, 3,96 mmol, 1,0 eq.) en DCM (8,0 ml) se le añadió TFA (13,9 g, 122 mmol, 9,0 ml, 30,7 eq.). La mezcla se agitó a 15 °C durante 2 horas. Después de que se completara, la mezcla se concentró. Al residuo se le añadió NaHCO3 acuoso saturado (20,0 ml) y se extrajo con DCM (5 x 10,0 ml). La capa orgánica se secó sobre Na2SO4, se filtró y se concentró. El producto (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de bencilo (Intermedio 65, 1,40 g, 2,77 mmol, 70 % de rendimiento) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1 ]: 506.
Intermedio 66

Etapa A: A una solución de 7-bencil-4-cloro-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidina (20,0 g, 65,4 mmol, 1 eq.) en DCE (200 ml) se le añadió carbonocloridato de 1-cloroetilo (28,1 g, 196 mmol, 3 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 30 minutos y a 70 °C durante 15 horas. La mezcla se concentró al vacío. El residuo se disolvió en MeOH (200 ml) y se agitó a 70 °C durante 0,5 horas. Después de que se completara, la mezcla se concentró al vacío. El residuo se trituró con metil terc-butil éter (60 ml). El precipitado se recogió por filtración, se lavó con metil terc-butil éter (20 ml) y se secó al vacío para dar 4-cloro-2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidina (17,2 g, bruto, HCl) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, metanol-d4) 5 = 4,35 (s, 2H), 3,60 (t, J = 6,4 Hz, 2H), 3,05 (t, J = 6,4 Hz, 2H), 2,55 (s, 3H).
Etapa B: A una solución de 4-cloro-2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidina (16,5 g, bruto, HCl) y TEA (20,0 g, 196 mmol, 27,3 ml) en THF (400 ml) se le añadió gota a gota carbonocloridato de bencilo (16,7 g, 98,1 mmol, 13,9 ml) a 0 °C. La mezcla se agitó a 25 °C durante 0,5 horas. Después de que se completara, la mezcla se diluyó con agua (80 ml) y la capa orgánica se separó. La fase acuosa se extrajo con EtOAc (200 ml). Las capas orgánicas combinadas se secaron sobre MgSO4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP/EtOAc 80/1 a 5/1) para dar 4-cloro-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de bencilo (19,6 g, 50,4 mmol, dos etapas 86 % de rendimiento, 90 % de pureza) en forma de un aceite de color amarillo.
1H RMN (300 MHz, cloroformo-d) 5 = 7,37 (s, 5H), 5,18 (s, 2H), 4,63 (s, 2H), 3,877 (d, J = 8,0, 2H), 2,80 (s a, 2H), 2,54 (s, 3H).
Etapa C: A una solución de 4-cloro-2-metilsulfanil-6,8-dihidro-5H- pirido[3,4-d]pirimidin-7-carboxilato de bencilo (21,5 g, 55,3 mmol, 1,00 eq.) en DMF (400 ml) se le añadieron DIEA (35,7 g, 277 mmol, 48,2 ml, 5,00 eq.) y 2-[(2S)-piperazin-2-il]acetonitrilo (6,92 g, 55,3 mmol, 1,00 eq.). Después de agitar a 80 °C durante 2 horas, a la mezcla anterior se le añadió (Boc)2O (60,4 g, 277 mmol, 63,5 ml, 5,00 eq.) y se agitó a 80 °C durante 2 horas más. Después de que se completara, la mezcla se diluyó con agua (800 ml) y se extrajo con EtOAc (2 * 400 ml). Las capas orgánicas se lavaron con salmuera (300 ml), se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP/EtOAc 10/1 a 1/1) para dar 4-[(3S)-4-terc-butoxicarbonil-3-(cianometil)piperazin-1-il]-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de bencilo (24,3 g, 43,0 mmol, 78 % de rendimiento, 95 % de pureza) en forma de un sólido de color amarillo.
1H RMN (400 MHz, cloroformo-d) 5 = 7,43 - 7,29 (m, 5H), 5,18 (s, 2H), 4,76 - 4,54 (m, 2H), 4,46 (d a, J = 18,4 Hz, 1H), 4,08 - 3,69 (m, 4H), 3,53 - 3,35 (m, 1H), 3,34 - 3,03 (m, 2H), 3,03 - 2,89 (m, 1H), 2,81 - 2,55 (m, 4H), 2,50 (s, 3H), 1,51 (s, 9H).
Etapa D: A una solución de 4-[(3S)-4-ferc-butoxicarbonil-3-(cianometil)piperazin-1 -il]-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-7-carboxilato de bencilo (24,3 g, 45,1 mmol, 1 eq.) en EtOAc (480 ml) se le añadió poco a poco m-CPBA (8,70 g, 42,9 mmol, 85 % de pureza, 0,95 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se diluyó con agua (800 ml). El pH se ajustó a 8 con NaHCO3 y la capa orgánica se separó. La fase acuosa se extrajo con EtOAc (2 x 400 ml). Las capas orgánicas se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EtOAc/MeOH 100/1 a 10/1) para dar 4-[(3S)-4-terc-butoxicarbonil-3-(cianometil)piperazin-1-il]-2-metilsulfinil- 6,8-dihidro-5H-pirido[3,4-cf]pirimidin-7-carboxilato de bencilo (20,9 g, 36,4 mmol, 81 % de rendimiento, 96 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+11]: 555.
Etapa E: A una solución de 4-[(3S)-4-ferc-butoxicarbonil-3-(cianometil)piperazin-1-il]-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-7-carboxilato de bencilo (20,9 g, 37,6 mmol, 1 eq.) y [(2S)-1-metilpirrolidin-2-il]metanol (8,67 g, 75,3 mmol, 8,94 ml, 2 eq.) en tolueno (400 ml) se le añadió f-BuONa (7,23 g, 75,3 mmol, 2 eq.) a 0 °C. Después de agitar a 0 °C durante 10 minutos, la mezcla se concentró al vacío. El residuo se diluyó con agua (200 ml) y se extrajo con EtOAc (2 x 400 ml). Las capas orgánicas se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. La mezcla se neutralizó con una solución saturada de bicarbonato de sodio, se concentró al vacío para eliminar el MeCN y se extrajo con EtOAc (2 x 1000 ml). Las capas orgánicas se secaron sobre Na2SO4 y se concentraron al vacío para dar 4-[(3S)-4-ferc-butoxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de bencilo (13,5 g, 20,5 mmol, 54 % de rendimiento, 92 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 606.
Etapa F: A una solución de 4-[(3S)-4-ferc-butoxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de bencilo (3,50 g, 5,78 mmol, 1 eq.) en MeOH (60,0 ml) se le añadieron NH3/MeOH (60,0 ml) y Pd/C (1,00 g, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 4 horas. Después de que se completara, el catalizador se retiró por filtración y el filtrado se concentró al vacío para dar (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (Intermedio 66, 2,33 g, 4,55 mmol, 79 % de rendimiento, 92 % de pureza) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, cloroformo-d) ó = 4,58 (s a, 1H), 4,34 (dd, J = 5,2, 10,8 Hz, 1H), 4,11 (dd, J = 6,8, 10,8 Hz, 1H), 4,08 - 3,88 (m, 4H), 3,84 (d a, J = 12,8 Hz, 1H), 3,25 - 3,03 (m, 4H), 3,01 - 2,88 (m, 2H), 2,82 - 2,51 (m, 5H), 2,47 (s, 3H), 2,27 (dt, J = 7,2, 9,2 Hz, 1H), 2,11 - 1,97 (m, 1H), 1,92 - 1,75 (m, 3H), 1,50 (s, 9H).
Intermedio 67

2-metilsulfanil-4-(trifluorometilsulfoniloxi)-6,8-dihidro-5H-pirido[3,4-e]pirimidin-7-carboxilato de terc-butilo
Etapa A: 4-hidroxi-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d1pirimidin-7- carboxilato de terc-butilo. A una solución agitada de 3-oxopiperidin-1,4-dicarboxilato de 1 -ferc-butil 4-etilo (50,0 g, 184 mmol, 1,00 eq.) en MeOH (1,00 l) a 25 °C en una atmósfera de nitrógeno se le añadió NaOMe (49,8 g, 921 mmol, 5,00 eq.), seguido de 2-metilisotiourea (62,4 g, 331 mmol, 1,80 eq., H2SO4) en forma de un sólido. La mezcla de reacción se agitó a 25 °C durante 16 horas. La mezcla de reacción se acidificó con HCl (2 M) hasta pH ~5 y después la mezcla se concentró a presión reducida para eliminar el MeOH. El residuo se suspendió en 300 ml de acetato de etilo y 300 ml de agua y se agitó rápidamente. La suspensión se filtró y el sólido de color blanco se recogió. El filtrado se separó y los extractos orgánicos se lavaron con agua (1 x 300 ml) y salmuera (1x 200 ml). Los extractos orgánicos se aislaron, se secaron sobre Na2SO4, se filtraron y se concentraron hasta un sólido de color blanco. Se obtuvo 4-hidroxi-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7- carboxilato de ferc-butilo (51,0 g, 138 mmol, 75,4 % de rendimiento, 81 % de pureza) en forma de un sólido de color blanco y se usó directamente para la siguiente etapa sin purificación adicional. LCMS [M+1]: 298.
1H RMN (400 MHz, cloroformo-d) ó = 4,33 (s, 2H), 3,61 (t, J = 5,6 Hz, 2H), 2,68 - 2,49 (m, 5H), 1,50 (s, 9H).
Etapa B: 2-metilsulfanil-4-(trifluorometilsulfoniloxi) -6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo. A una suspensión agitada de 4-hidroxi-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (51,0 g, 171 mmol, 1,00 eq.) en DCM (500 ml) a 0 °C se le añadió DIEA (44,3 g, 343 mmol, 59,9 ml, 2,00 eq.) seguido de Tf2O
(72,6 g, 257 mmol, 42,4 ml, 1,50 eq.) en una atmósfera de nitrógeno. Inmediatamente se formó una solución de color pardo. Después de agitar a 25 °C durante 16 horas, la reacción se concentró para dar un aceite de color pardo. El aceite de color pardo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo = 1/0 a 10/1). El compuesto del título 2-metilsulfanil-4-(trifluorometilsulfoniloxi) -6,8-dihidro-5H-pirido[3,4-d1pirimidin-7-carboxilato de ferc-butilo (46,0 g, 107 mmol, 62 % de rendimiento) se obtuvo en forma de un sólido de color amarillo. LCMS [M+1]: 430.
Intermedio 68

4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo
Etapa A: 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il1-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d1pirimidin-7-carboxilato de ferc-butilo. Una mezcla de 2-metilsulfanil-4-(trifluorometilsulfoniloxi)-6,8-dihidro-5H-pirido[3,4-d]pir¡m¡d¡n-7-carbox¡lato de ferc-butilo (3,81 g, 8,87 mmol, 1,0 eq.), (2S)-2-(cianometil)piperazin-1-carboxilato de bencilo (Intermedio 63, 2,30 g, 8,87 mmol, 1,0 eq.) y DIEA (3,44 g, 26,6 mmol, 4,63 ml, 3,0 eq.) en DMF (20,0 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 100 °C durante 1 hora en una atmósfera de N2. Después de que se completara, el disolvente se eliminó al vacío. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo = 3/1 a 1:1) para dar el compuesto del título 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il1-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (3,6 g, 6,16 mmol, 69 % de rendimiento, 92,2 % de pureza) en forma de un sólido de color amarillo.
Intermedio 69

1-bromo-8-metilnaftaleno
Etapa A: 1 -bromo-8-metil-naftaleno. A una solución de 1,8-dibromonaftaleno (1 g, 3,50 mmol, 1 eq.) en THF (20 ml) se le añadió gota a gota MeLi (1,6 M en éter dietílico, 2,62 ml, 1,2 eq.) a 0 °C. Después de agitar durante 30 minutos a 0 °C, se añadió gota a gota yodometano (3,38 g, 23,8 mmol, 1,48 ml, 6,81 eq.). La mezcla se calentó a 25 °C y se agitó durante 3 horas más. La mezcla de reacción se inactivó con agua (20 ml) y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini C18250 * 50 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - ACN]; % de B: 45 % - 70 %, 28 MIN; 40 % min). El compuesto del título 1-bromo-8-metil-naftaleno (340 mg, 1,49 mmol, 43 % de rendimiento, 97 % de pureza) se obtuvo en forma de un sólido de color amarillo después de la liofilización.
1H RMN (400 MHz, cloroformo-d) ó = 7,75 (dd, J = 0,8, 7,2 Hz, 1H), 7,69 (dd, J = 0,8, 8,0 Hz, 1H), 7,66 - 7,59 (m, 1H), 7,30 - 7,22 (m, 2H), 7,13 (t, J = 8,0 Hz, 1H), 3,05 (s, 3H).
Intermedio 70

1 -bromo-8-cloronaftaleno
Etapa A: 1H-nafto[1.8-de1[1.2.31triazina. A una solución de naftaleno-1,8-diamina (100 g, 632 mmol, 1 eq.) en AcOH (200 ml) y EtOH (1000 ml) se le añadió gota a gota nitrito de isoamilo (72,6 g.619 mmol. 83,4 ml.0,98 eq.) durante un periodo de 2 h con la temperatura controlada entre 18 y 21 °C en un baño de agua fría. Después de la adición. la suspensión de color rojo resultante se agitó a 25 °C durante 16 horas. El sólido se recogió por filtración. se lavó con etanol (2 * 500 ml) y se secó al vacío. El compuesto 1 H-nafto[1,8-de][1,2,3]triazina (84 g.496 mmol. 79 % de rendimiento) se obtuvo en forma de un sólido cristalino de color rojo y se usó directamente en la siguiente etapa sin purificación. LCMS [ESI, M+1]: 170.
Etapa B: 8-cloronaftalen-1 -amina. A una solución de 1H-nafto[1,8-de][1,2,3]triazina (84 g. 496 mmol. 1 eq.) en HCl (1.5 l) se le añadió Cu (2.10 g. 33,1 mmol. 234 ul. 0,0665 eq.). La mezcla se agitó a 25 °C durante 12 horas. La mezcla resultante se diluyó con agua (500 ml) y se calentó a 85 °C durante 30 min. La solución acuosa casi transparente resultante se filtró. se enfrió. se basificó con amoníaco acuoso (hasta azul al papel tornasol) y la solución se extrajo con acetato de éter (2 * 1000 ml). Los extractos combinados se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna (SiO2. éter de petróleo/acetato de etilo = 200/1 a 5/1). El compuesto 8-cloronaftalen-1-amina (57 g. 259 mmol. 52 % de rendimiento. 81 % de pureza) se obtuvo en forma de un sólido de color rojo. LCMS [ESI, M+1]: 178.
Etapa C: 1 -bromo-8-cloro-naftaleno. A una solución de 8-cloronaftalen-1-amina (57 g. 320 mmol, 1 eq.) y TsOH*H2O (219 g. 1,16 mol. 3.6 eq.) en MeCN (1000 ml) se le añadió una solución de NaNO2 (39.8 g. 577 mmol. 1.8 eq.) y CuBr (138 g. 963 mmol. 29,3 ml. 3 eq.) en H2O (120 ml) a -5 °C y después la mezcla de reacción se agitó a 25 °C durante 12 horas. A la mezcla de reacción se le añadió una solución saturada de Na2SO3 (100 ml). se agitó durante 15 min y después se extrajo con acetato de etilo (1000 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (500 ml). se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo). El compuesto del título 1-bromo-8-cloro-naftaleno (56 g. 229 mmol. 72 % de rendimiento. 99 % de pureza) se obtuvo en forma de un sólido de color blanco.
1H RMN (400 MHz, cloroformo-d) ó = 7,93 (dd, J = 1.2. 7.6 Hz. 1H). 7,82 (dd. J = 1.2. 8.4. 1H). 7,79 (dd. J = 1.2.
8.4. 1H). 7,67 (dd. J = 1.2. 7.6 Hz. 1H). 7,37 (t. J = 8.0 Hz. 1H). 7,28 (t. J = 8.0 Hz. 1H).
Intermedio 71

2-[(2S)-4-[7-(8-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]acetonitrilo
Etapa A: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-i n piperazin-2-i n acetonitrilo. A una solución de (2S)-4-[7-(8-cloro-1-naft¡l)-2-[[(2S)-1-metilp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de ferc-butilo (0.5 g.791 umol, 1 eq.) en dioxano (5 ml) se le añadió HCl*dioxano (4 M, 5,00 ml. 25,3 eq.) a 0 °C. La mezcla se agitó a 25 °C durante 1 hora. Después de que se completara. la mezcla se concentró al vacío para dar un producto impuro (500 mg. bruto. HCl) en forma de un sólido de color pardo. Se purificaron 60 mg del producto impuro por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0.04 % de NH3*H2O NH4HCO310 mM) - Ac N]; % de B: 50 % - 80 %, 10 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (19,3 mg. 36,1 umol. 34 % de rendimiento de purificación. 99,2 % de pureza) en forma de un sólido de color blanquecino. LCMS [ESI, M+1]: 532.1
1H RMN (400 MHz. cloroformo-d) ó = 7,75 (d. J = 8.4 Hz. 1H). 7,60 (dd. J = 1.6. 8.0 Hz. 1H). 7,52 (dd. J = 0.8.
7.2 Hz. 1H). 7,44 (dt. J = 3.6. 7.6 Hz. 1H). 7,36 - 7,29 (m. 1H). 7,22 (t. J = 6.8 Hz. 1H). 4,46 - 4,34 (m. 2H). 4,15 (td. J = 6.4. 10,6 Hz. 1H). 4,04 (d a. J = 12,4 Hz. 0.5H), 3,95 - 3,79 (m. 2H). 3,74 (d a. J = 12,8 Hz. 0.5H), 3,63 - 3,48 (m. 1H). 3,40 - 2,99 (m. 7H). 2,98 - 2,80 (m. 2H). 2,73 - 2,61 (m. 1H). 2,60 - 2,49 (m. 3H). 2,47 (d. J = 2.4 Hz. 3H).
2,32 - 2,23 (m. 1H). 2,10 - 1,99 (m. 1H). 1,82 - 1,68 (m. 3H).
Intermedio 72

2-[(2S)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-iljacetonitrilo
Etapa A: (2S)-2-(cianometil)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato. Una mezcla de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (1,22 g, 1,98 mmol, 1,0 eq.), 1-bromo-2,3-dimetil-benceno (1,10 g, 5,93 mmol, 802 ul, 3,0 eq.), CS2CO3 (1,93 g, 5,93 mmol, 3 eq.), RuPhos (185 mg, 396 umol, 0,2 eq.) y Pd2(dba)3 (181 mg, 198 umol, 0,1 eq.) en tolueno (8 ml) se desgasificó y después se calentó a 90 °C durante 12 horas en una atmósfera de N2. Después de que se completara, la mezcla se concentró al vacío. El residuo se diluyó con agua (20 ml) y se extrajo con EtOAc (3 * 30 ml). Las capas orgánicas se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. Las fracciones deseadas recogidas se neutralizaron con bicarbonato de sodio acuoso saturado y se concentraron al vacío para eliminar el MeCN, y después se extrajeron con EtOAc (3 * 50 ml). Las capas orgánicas se secaron sobre Na2SO4 y se concentraron al vacío para dar piperazin-1-carboxilato de (2S)-2-(cianometil)-4-[7-(2,3-d¡met¡lfen¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡lo] (380 mg, 623 umol, 32 % de rendimiento, 100 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 610.
Etapa B: 2-[(2S)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-inmetoxn-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-¡npiperazin-2-inacetonitrilo. Se burbujeó NH3 en MeOH (20 ml) a -70 °C durante 30 minutos. Una solución de (2S)-2-(cianometil)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirim idin-4-il]piperazin-1-carboxilato de bencilo (380 mg, 623 umol, 1,0 eq.) se añadió a la solución anterior seguido de Pd/C (200 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La reacción se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. Después de que se completara, el catalizador se filtró y el filtrado se concentró para dar el compuesto del título 2-[(2S)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (230 mg, 459 umol, 74 % de rendimiento, 95 % de pureza) en forma de un sólido de color amarillo.
1H RMN (400 MHz, cloroformo-d) ó = 7,11 (t, J = 8,0 Hz, 1H), 6,96 (d, J = 8,0 Hz, 2H), 4,39 (dd, J = 4,8, 10,4 Hz, 1H), 4,14 (dd, J = 7,2, 9,6 Hz, 1H), 4,02 - 3,95 (m, 3H), 3,84 - 3,78 (m, 1H), 3,31 - 3,19 (m, 1H), 3,17 - 3,04 (m, 5H), 3,04 - 2,95 (m, 1H), 2,89 (dd, J = 9,2, 11,6 Hz, 1H), 2,76 - 2,62 (m, 3H), 2,58 - 2,50 (m, 2H), 2,48 (s, 3H), 2,30 (s, 3H), 2,29 - 2,23 (m, 4H), 2,12 - 2,00 (m, 1H), 1,89 - 1,76 (m, 3H).
Intermedio 73
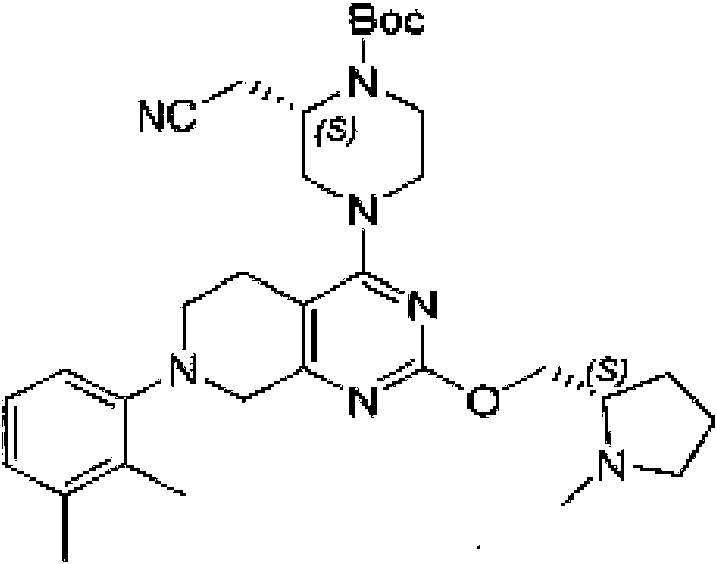
(2S)-2-(cianometil)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo
Etapa A: (2S)-2-(c¡anomet¡l)-4-[7-(2.3-d¡met¡lfen¡l)-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carboxilato de tere-butilo. Una mezcla de (2S)-2-(c¡anomet¡l)-4-(2-met¡lsulfan¡l-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de tere-butilo (1,60 g, 3,96 mmol, 1,0 eq.), 1-bromo-2,3-d¡met¡l-benceno (1,61 g, 8,70 mmol, 1,18 ml, 2,20 eq.), Pd2(dba)3 (362 mg, 395 umol, 0,10 eq.), RuPhos (369 mg, 791 umol, 0,20 eq.) y CS2CO3 (3,87 g, 11,9 mmol, 3,0 eq.) en tolueno (8,0 ml) se desgas¡f¡có y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 12 h en una atmósfera de N2. El d¡solvente orgán¡co se lavó con agua (20,0 ml). La fase acuosa se extrajo con acetato de et¡lo (3 x 30,0 ml). Los extractos comb¡nados se lavaron con salmuera (80,0 ml), se secaron con Na2SO4 después el d¡solvente se el¡m¡nó al vacío. El res¡duo se pur¡f¡có por cromatografía en columna (S¡O2, éter de petróleo: acetato de et¡lo = 3 : 1 a acetato de etilo : metanol = 10 : 1). El compuesto (2S)-2-(c¡anomet¡l)-4-[7-(2,3-d¡met¡lfen¡l)-2-met¡lsulfan¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (900 mg, 1,59 mmol, 40 % de rend¡m¡ento, 90 % de pureza) se obtuvo en forma de un sólido de color amar¡llo. LCMS [ESI, M+1]: 509.
1H RMN (400 MHz, Cloroformo-d) ó 7,11 (t, J = 15,6 Hz, 1H), 6,95(d, J = 8,0 Hz, 2H), 4,63 (s a, 1H), 4,10 - 3,93 (m, 4H), 3,89 (d a, J = 4,8 Hz, 1H), 3,27 (dd, J = 3,6 Hz, J = 13,6 Hz, 1H),3,24 - 3,05 (m, 3H), 3,05 - 2,95 (m, 1H), 2,89 - 2,67 (m, 4H), 2,52 (s, 3H), 2,30 (s, 3H), 2,28 (s, 3H), 1,52 (s, 9H).
Etapa B: (2S)-2-(c¡anomet¡l)-4-[7-(2.3-d¡met¡lfen¡l)-2-met¡lsulfin¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de tere-butilo. Una mezcla de (2S)-2-(c¡anomet¡l)-4-[7-(2,3-d¡met¡lfen¡l)-2-met¡lsulfan¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (500 mg, 983 umol, 1,0 eq.) y ác¡do 3-clorobencenocarboperoxo¡co (200 mg, 983 umol, 1,0 eq.) en DCM (5,0 ml) se desgas¡ficó y se purgó 3 veces con N2 y después la mezcla se agitó a 0 °C durante 30 m¡n en una atmósfera de N2. El d¡solvente orgán¡co se lavó con agua (10,0 ml). La fase acuosa se extrajo con acetato de etilo (3 x 20,0 ml). Los extractos comb¡nados se lavaron con salmuera (50,0 ml), se secaron con Na2SO4 y después el d¡solvente se el¡m¡nó al vacío. El res¡duo se pur¡f¡có por cromatografía en columna (S¡O2, éter de petróleo: acetato de etilo = 3 : 1 a acetato de et¡lo : metanol = 10 : 1). El compuesto (2S)-2-(c¡anomet¡l)-4-[7-(2,3-d¡met¡lfen¡l)-2-met¡lsulfin¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (450 mg, 793 umol, 81 % de rendimiento, 93 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 525.
Etapa C: (2S)-2-(c¡anomet¡l)-4-[7-(2.3-d¡met¡lfen¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de tere-butilo. Una mezcla de (2S)-2-(cianometil)-4-[7-(2,3-d¡met¡lfen¡l)-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (800 mg, 1,52 mmol, 1,0 eq.), [(2S)-1-metilpirrol¡d¡n-2-¡l]metanol (369 mg, 3,20 mmol, 380 ul, 2,10 eq.) y t-BuONa (293 mg, 3,05 mmol, 2,0 eq.) en tolueno (10,0 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 0 °C durante 30 min en una atmósfera de N2. La reacción se interrumpió con agua (20,0 ml). La mezcla bruto se extrajo con acetato de etilo (3 x 30,0 ml). Los extractos combinados se lavaron con salmuera (80,0 ml), se secaron con Na2SO4 después el disolvente se eliminó al vacío. El residuo se purificó por cromatografía en columna (S¡O2, éter de petróleo: acetato de etilo = 5: 1 a diclorometano: metanol = 10:1). El compuesto del título (2S)-2-(c¡anomet¡l)-4-[7-(2,3-dimet¡lfen¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (740 mg, 1,22 mmol, 80 % de rendimiento, 95 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 576.
1H RMN (400 MHz, Cloroformo-d) ó 7,10 (t, J = 15,2 Hz, 1H), 6,95 (d, J = 8,0 Hz, 2H), 4,61 (s a, 1H), 4,39 (dd, J = 4,8 Hz, J = 9,6 Hz, 1H), 4,13 - 4,00 (m, 4H), 3,89 (d a, J = 12,4 Hz 1H), 3,27 - 3,13 (m, 3H), 3,13 - 2,95 (m, 3H), 2,87 - 2,65 (m, 5H), 2,49 (s, 3H), 2,30 (s, 3H), 2,27 (s, 3H), 2,09 - 2,06 (m, 1H), 2,06 - 2,04 (m, 1H), 1,93 - 1,62 (m, 4H), 1,51 (s, 9H).
Ejemplo 1

1-(4-(7-(3-h¡drox¡naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡din-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona

Etapa A: 4-(4-((benciloxi)carbonil)piperazin-1-il)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo: En 2 ml de dimetil acetamida se combinaron 4-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (1,0 g, 3,7 mmol), trietilamina (1,0 ml, 7,4 mmol) y 1-piperazinacarboxilato de bencilo (0,86 ml, 4,4 mmol). El recipiente de reacción se selló y la mezcla de reacción se calentó a 90 °C con agitación. Después de 5 horas, la reacción se diluyó con salmuera y se extrajo con metil t-butil éter. Las capas orgánicas combinadas se lavaron secuencialmente con cloruro de amonio saturado y salmuera, se secaron sobre MgSO4 y se concentraron a presión reducida hasta un aceite pegajoso. El aceite se cromatografió (RediSep®, 24 g) eluyendo con 1:1 de acetato de etilo/Hexanos para dar 4-(4-((benciloxi)carbonil)piperazin-1-il)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (1,3 g, 2,9 mmol, 77 % de rendimiento). ES+APCI MS m/z 454,2 [M+H]+.
Etapa B: 4-(5,6,7,8-Tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo: A una solución de 4-(4-((benciloxi)carbonil)piperazin-1-il)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (1,58 g, 3,484 mmol) en diclorometano (11,61 ml, 3,484 mmol) se le añadió ácido trifluoroacético (2,668 ml, 34,84 mmol) y la reacción se agitó a temperatura ambiente durante 3 horas. La reacción se concentró al vacío y el residuo se recogió en diclorometano. La solución se lavó con secuencialmente con NaOH 1 M y salmuera, se secó sobre Na2SO4, se filtró y se concentró al vacío. El producto bruto se purificó por cromatografía en columna (Biotage Isolera, 24G Isco RediSep® Gold, de 10 a 20 % de metanol/diclorometano) para proporcionar el producto (1,1 g, 89 %) en forma de una espuma de color blanquecino. ES+APCI MS m/z 354,2 [M+H]+.
Etapa C: 4-(7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: A un vial se le añadieron tris(dibencilidenoacetona)dipaladio (0) (0,0069 g, 0,0075 mmol), 2,2'-bis(difenilfosfino)-1, 1 '-binaftilo racémico (0,0096 g, 0,015 mmol) y tolueno (0,62 ml, 0,19 mmol). Se burbujeó argón a través de la mezcla durante 5 minutos y después el vial se tapó y la mezcla se calentó a 100 °C durante 15 minutos. La mezcla se enfrió a temperatura ambiente y después se añadió terc-butóxido de sodio (0,036 g, 0,37 mmol) seguido de 1-bromo-3-(metoximetoxi)naftaleno (0,050 g, 0,19 mmol) y 4-(5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (0,13 g, 0,37 mmol). El vial se tapó y la mezcla se calentó a 100 °C durante 20 horas. La mezcla se enfrió a temperatura ambiente, se diluyó con diclorometano y se filtró a través de papel GHF. El filtrado se concentró y se purificó por cromatografía en columna (Biotage Isolera, 12G Isco RediSep®, 10-50 % de acetato de etilo/diclorometano) para proporcionar el producto (0,062 g, 61 %) en forma de una espuma de color blanquecino. ES+APCI MS m/z 540,3 [M+H]+.
Etapa D: 7-(3-(metoximetoxi)naftalen-1-il)-4-(piperazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidina: A una solución de 4-(7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (0,061 g, 0,11 mmol) en etanol (1,1 ml, 0,11 mmol) y tetrahidrofurano (1,1 ml, 0,11 mmol) se le añadió paladio (0,024 g, 0,011 mmol) (tipo Degussa, 10 % en peso, 50 % de H2O). Se introdujo una atmósfera de H2 en el recipiente
de reacción con vacío, y después la mezcla de reacción se mantuvo en una atmósfera de H2. La mezcla se agitó a temperatura ambiente durante 2,5 horas, después se diluyó con metanol y se filtró a través de papel GHF. El filtrado incoloro se concentró al vacío con tolueno para proporcionar una espuma de color blanquecino (0,048 g, 105 %) que se usó directamente en la siguiente etapa. ES+Ap CI MS m/z 406,2 [M+H]+.
Etapa E: 1-(4-(7-(3-(metox¡metox¡)naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1pir¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona: A una suspensión de 7-(3-(metoximetoxi)naftalen-1-il)-4-(piperazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidina (0,046 g, 0,11 mmol) en diclorometano (1,1 ml, 0,11 mmol) a temperatura ambiente se le añadió cloruro de acriloílo (1,2 ml, 0,12 mmol) (solución 0,1 M recién preparada en diclorometano) seguido de trietilamina (0,032 ml, 0,23 mmol). La reacción se agitó a temperatura ambiente durante 1 hora. La mezcla se concentró y el producto se purificó por cromatografía en columna (Biotage Isolera, 12G Isco RediSep®, acetato de etilo) para proporcionar el producto (0,042 g, 79 %) en forma de una espuma sólida de color blanquecino. ES+APCI MS m/z 460,2 [m H]+.
Etapa F: 1-(4-(7-(3-h¡drox¡naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona: A una solución de 1-(4-(7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona (0,034 g, 0,074 mmol) en acetato de etilo (0,74 ml, 0,074 mmol) se le añadió ácido clorhídrico (solución de 5 a 6 N en 2-propanol (0,44 ml, 2,2 mmol). La mezcla se agitó a temperatura ambiente durante 5 horas. La mezcla se diluyó con acetato de etilo (10 ml), se filtró a través de un filtro de polipropileno y el sólido recogido se lavó con acetato de etilo y hexanos para proporcionar el producto en forma de la sal HCl. El material impuro se trató con 1 ml de hidróxido de amonio/metanol para inactivar el ácido y la mezcla se concentró. El residuo se disolvió en 10 % de metanol/diclorometano y se purificó por cromatografía en columna (Biotage Isolera, 12G Isco RediSep®, de 2 a 5 % de metanol/acetato de etilo) para proporcionar el producto (0,008 g, 25 %) en forma de un sólido de color blanquecino. ES+APCI MS m/z 416,2 [M+H]+.
1H RMN (CD3OD, 400 MHz) ó 8,49 (s, 1H), 8,07 (app d, J = 8,2 Hz, 1H), 7,61 (app d, J = 8,2 Hz, 1H), 7,35 (m, 1H), 7,25 (m, 1H), 6,80 (m, 3H), 6,23 (dd, J = 16,8, 1,6 Hz, 1H), 5,77 (dd, J = 10,6, 2,0 Hz, 1H), 4,22 (s a, 2H), 3,80 (app t, J = 4,7 Hz, 4H), 3,63 (s a, 4H), 3,35 (s a, 2H), 3,03 (s a, 2H).
Ejemplo 2

1-(4-(7-(7-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 1, usando 2-bromo-7-(metoximetoxi)naftaleno en lugar de 1-bromo-3-(metoximetoxi)naftaleno en la Etapa C. ES+APCI MS m/z 416,1 [M+H]+.
Ejemplo 3

1-(4-(7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 1, usando 1-yodonaftaleno en lugar de 1-bromo-3-(metoximetoxi)naftaleno en la Etapa C. ES+Ap CI MS m/z 400,2 [M+H]+.
Ejemplo 4

1-(4-(7-(2-fluoro-6-hidroxifenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 1, usando 2-bromo-1-fluoro-3-(metoximetil)benceno en lugar de 1-bromo-3-(metoximetoxi)naftaleno en la Etapa C. ES+APCI MS m/z 384,2 [M+H]+.
Ejemplo 5
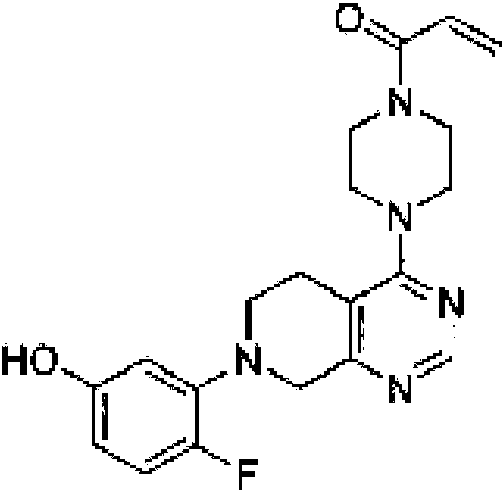
1-(4-(7-(2-fluoro-5-hidroxifenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 1, usando 2-bromo-1-fluoro-4-(metoximetoxi)benceno en lugar de 1-bromo-3-(metoximetoxi)naftaleno en la Etapa C. ES+APCI MS m/z 384,2 [M+H]+.
Ejemplo 6

1-(4-(7-(3-hidroxinaftalen-1-il)-6-metil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona Etapas A-C: 4-(7-(3-(metox¡metox¡)naftalen-1-¡l)-6-met¡l-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡rim¡d¡n-4-¡l)p¡peraz¡n-1-carboxilato de bencilo: Se sintetizó según el método del Ejemplo 1, Etapas A-C, usando 4-cloro-6-metil-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo en lugar de 4-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato en la Etapa A. ES+APCI MS m/z 430,2 [M+H]+.
Etapa D1: 4-(7-(3-h¡drox¡naftalen-1-¡l)-6-met¡l-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo: A una solución de 4-(7-(3-(metoximetoxi)naftalen-1-il)-6-metil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (0,05 g, 0,09 mmol) en isopropanol (10 ml) se le añadió cloruro de hidrógeno (5-6 M en isopropanol) (0,02 ml, 0,09 mmol) y la reacción se agitó a temperatura ambiente durante 1 hora. La reacción se concentró al vacío y el concentrado se repartió entre acetato de etilo y agua para convertir el material
en la base libre. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre MgSO4 y se concentraron al vacío para dar 4-(7-(3-hidroxinaftalen-1-il)-6-metil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (0,005 g, 0,010 mmol, 11 % de rendimiento). ES+APCI MS m/z 510,3 [M+H]+.
Etapa D2: 4-(7-(3-hidroxinaftalen-1-il)-6-metil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-ol: Se preparó según el método del Ejemplo 1, Etapa D.
Etapa E: 1-(4-(7-(3-hidroxinaftalen-1-il)-6-metil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona: Se preparó según el método del Ejemplo 1, Etapa E.
Ejemplo 7

1-(4-(7-(5-metil-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Etapas A-D: 4-(7-(5-metil-1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: Se sintetizó según el Esquema General 1, Etapas A-C, usando 4-bromo-5-metil-1-((2-(trimetilsilil)etoxi)metil)-1H-indazol en lugar de 1-bromo-3-(metoximetoxi)naftaleno en la Etapa C
Etapa D1: 4-(7-(5-met¡l-1H-¡ndazol-4-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡rim¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo: A una solución de 4-(7-(5-metil-1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo (0,16 g, 0,26 mmol) en diclorometano (10 ml) se le añadió ácido 2,2,2-trifluoroacético (0,89 g, 7,8 mmol) seguido de anisol (0,028 g, 0,26 mmol) y la reacción se agitó a temperatura ambiente durante 3 horas a temperatura ambiente. La reacción se concentró al vacío y el material concentrado se recogió en acetato de etilo y se lavó con salmuera básica. Las capas orgánicas combinadas se secaron sobre MgSO4 y se concentraron al vacío. El material bruto se cromatografió usando de 0 a 10 % de metanol/diclorometano como eluyente para dar 4-(7-(5-metil-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo (0,05 g, 38 %). ES+AμCi MS m/z 484,2 [M+H]+.
Etapa D2: 7-(5-metil-1H-indazol-4-il)-4-(piperazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidina: Se preparó según el método del Ejemplo 1, Etapa D.
Etapa E: 1-(4-(7-(5-metil-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona: Se preparó según el método del Ejemplo 1, Etapa E.
Ejemplo 8

(S)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona

Etapa A: 4-(4-((benciloxi)carbonil)piperazin-1-il)-2-cloro-5.8-dihidropirido[3.4-d]pirimidin-7(6H)-carboxilato de terc-butilo: Se disolvieron 1-piperazinacarboxilato de bencilo (1.268 ml. 6.575 mmol) y 2,4-dicloro-5,6-dihidropirido[34-d]pirimidin-7(8H)-carboxilato de terc-butilo (2 g. 6.575 mmol) en dimetil acetamida (10 ml) y se trataron con N-etil-N-isopropilpropan-2-amina (3.445 ml. 19.73 mmol). La mezcla de reacción se agitó a 85 °C durante 2 horas. La mezcla de reacción se enfrió a temperatura ambiente. se diluyó con acetato de etilo. se lavó con agua y salmuera. se secó sobre MgSÜ4. se filtró y se concentró. El concentrado se purificó por cromatografía (CombiFlash®. 0 %-50 % de acetato de etilo:Hexanos como eluyente) para proporcionar el producto (2.69 g. 83 %). ES+APCI MS m/z 488.2. 490.2 [M+H]+.
Etapa B: (S)-4-(4-((benciloxi)carbonil)piperazin-1-il)-2-((1-(dimetilamino)propan-2-il)oxi)-5.8-dihidropirido[3.4-d]pirimidin-7(6H)-carboxilato de terc-butilo: Se añadieron 4-(4-((benciloxi)carbonil)piperazin-1-il)-2-cloro-5.8-dihidropirido[3.4-d]pirimidin-7(6H)-carboxilato de terc-butilo (235 mg. 0.482 mmol) y (S)-1-(dimetilamino)propan-2-ol (497 mg. 4.82 mmol) a dioxano (0.5 ml) y se calentó a 100 °C durante 3 días. La reacción se concentró y el residuo resultante se purificó por cromatografía sobre gel de sílice (Biotage Isolera. 0-12 % de metanol en diclorometano) para proporcionar (S)-4-(4-((benciloxi)carbonil)piperazin-1-il)-2-((1-(dimetilamino)propan-2-il)oxi)-5.8-dihidropirido[3.4-d]pirimidin-7(6H)-carboxilato de terc-butilo (200 mg. 0.361 mmol. 74.9 % de rendimiento). ES+APCI MS m/z 555.3 [M+H]+.
Etapa C: (S)-4-(2-((1-(dimetilamino)propan-2-il)oxi)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: A una solución de (S)-4-(4-((benciloxi)carbonil)piperazin-1-il)-2-((1-(dimetilamino)propan-2-il)oxi)-5.8-dihidropirido[3.4-d]pirimidin-7(6H)-carboxilato de terc-butilo (200 mg.0.3606 mmol) en diclorometano (1202 μl.0.3606 mmol) se le añadió ácido trifluoroacético (828.3 μl. 10.82 mmol) y la reacción se agitó a temperatura ambiente durante 3 horas. La reacción se concentró al vacío y el residuo se recogió en diclorometano. La solución se lavó con NaOH 1 M seguido de salmuera y después se secó sobre Na2SO4. se filtró y se concentró al vacío. El producto bruto se purificó por cromatografía en columna (Biotage Isolera. 24G Isco RediSep®Gold. de 10 a 20 % de metanol/diclorometano) para proporcionar el producto en forma de una espuma de color blanquecino (0.135 g.83 %). ES+APCI MS m/z 455.2 [M+H]+.
Etapa D: (S)-4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-(metoximetoxi)naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: A un vial se le añadieron tris(dibencilidenoacetona)dipaladio (0) (21.8 mg.
0.0238 mmol). 2.2'-Bis(difenilfosfino)-1.1 '-binaftilo racémico (30.4 mg. 0.0488 mmol) y tolueno (991 μl. 0.297 mmol). Se burbujeó argón a través de la mezcla durante 5 minutos y después el vial se tapó y la mezcla se calentó a 100 °C durante 15 minutos. La mezcla se enfrió a temperatura ambiente y se añadió terc-butóxido de sodio (57.2 mg. 0.595 mmol) seguido de trifluorometanosulfonato de 3-(metoximetoxi)naftalen-1-ilo (100 mg. 0.297 mmol) y (S)-4-(2-((1-(dimetilamino)propan-2-il)oxi)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (135 mg. 0.297 mmol). El vial se tapó y la
mezcla se calentó a 100 °C durante 18 horas. La mezcla se enfrió y se concentró. El material bruto se purificó por cromatografía sobre gel de sílice (Biotage Isolera, 0-11 % de metanol/diclorometano) para proporcionar (S)-4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (68 mg, 0,106 mmol, 35,7 % de rendimiento). ES+APCI MS m/z 641,3 [M+H]+.
Etapa E: (S)-2-((7-(3-(metoximetoxi)naftalen-1-il)-4-(piperazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)-N,N-dimetilpropan-1-amina: A una solución de (S)-4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (68 mg, 0,11 mmol) en etanol (1061 μl, 0,11 mmol) y tetrahidrofurano (1061 μl, 0,11 mmol) se le añadió paladio (113 mg, 0,053 mmol) (tipo Degussa, 10 % en peso, 50 % de H2O). Se introdujo una atmósfera de H2 mediante vacío y después el recipiente de reacción se mantuvo en una atmósfera de H2. La mezcla se agitó a temperatura ambiente durante 3 horas. La mezcla se diluyó con metanol y se filtró a través de papel GHF. El filtrado incoloro se concentró para proporcionar (S)-2-((7-(3-(metoximetoxi)naftalen-1-il)-4-(piperazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)-N,N-dimetilpropan-1-amina (54 mg, 100 % de rendimiento) que se usó en la siguiente etapa sin purificación. ES+APCI MS m/z 507,3 [M+H]+.
Etapa F: (S)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -il)prop-2-en-1-ona: A una suspensión de (S)-2-((7-(3-(metoximetoxi)naftalen-1-il)-4-(piperazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)-N,N-dimetilpropan-1-amina (54 mg, 0,11 mmol) en diclorometano (1066 μl, 0,11 mmol) a temperatura ambiente se le añadió cloruro de acriloílo (1279 μl, 0,13 mmol) (solución 0,1 M recién preparada en DCM) seguido de trietilamina (30 μl, 0,21 mmol). La reacción se agitó a temperatura ambiente durante 20 minutos. La mezcla se concentró y el producto se purificó por cromatografía en columna (Biotage Isolera, 12G Isco RediSep®, 0-15 % de metanol/diclorometano) para proporcionar (S)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona (51 mg, 0,091 mmol, 85 % de rendimiento). ES+APCI MS m/z 561,3 [M+H]+.
Etapa G: 1-(4-(2-(2-(dimetilamino)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona: Se añadió (S)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona (51 mg, 0,091 mmol) a un vial que contenía 350 μl de metanol y unas gotas de tetrahidrofurano y el vial de reacción se tapó. Se añadió HCl (379 μl, 2,3 mmol) (acuoso 6 M) con agitación y la mezcla se calentó a 55 °C durante 3 horas. La reacción se enfrió y se concentró al vacío. Se añadió una solución saturada de bicarbonato y la reacción se extrajo con 10 % de metanol en diclorometano. Las capas orgánicas se combinaron y se concentraron. El residuo resultante se purificó por cromatografía sobre gel de sílice (Biotage Isolera, 4-20 % de metanol en diclorometano con 1 % de cloruro de amonio concentrado) para proporcionar el producto del título (25,3 mg, 54 %). ES+APCI MS m/z 517,2 [M+H]+.
1H RMN (400 MHz, CDCla) ó 7,90 (d, 1H, J = 8,314 Hz), 7,54 (d, 1H, J = 8,021), 7,34 (m, 1H), 7,24 (m, 1H). 6,72 (m, 1H), 6,56-6,48 (m, 2H), 6,32 (dd, 1H, J = 16,726, 1,858), 5,73 (dd, 1H, J = 10,368, 1,858), 5,45 (m, 1H), 4,09 3,94 (m, 2H), 3,63 (s a, 2H), 3,47 (s a, 2H), 3,31 (m, 4H), 3,16 (s a, 2H), 2,84 (m, 1H), 2,60 (s a, 2H), 2,45 (m, 1H), 2,43 (s, 6H), 1,31 (d, 3H, J = 6,162 Hz)
Ejemplo 9

1- (4-(2-(2-(dimetilamino)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2- en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 2-(dimetilamino)etan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 503,2 [M+H]+.
Ejemplo 10

1-(4-(2-(3-(dimetilamino)propoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7, 8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 3-(dimetilamino)propan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. e S+APCI MS m/z 517,3 [M+H]+.
Ejemplo 11

1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 1-(dimetilamino)propan-2-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. e S+APCI MS m/z 517,3 [M+H]+.
Ejemplo 12

1-(4-(7-(3-hidroxinaftalen-1-il)-2-(4-metilpiperazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 1-metilpiperazina en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 514,3 [M+H]+.
Ejemplo 13

1-(4-(2-(3-(dimetilamino)pirrolidin-1-il)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando N,N-dimetilpirrolidin-3-amina en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 528,3 [M+H]+.
Ejemplo 14

(S)-1-(4-(7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-metilpiperazin-1-il)prop-2-en-1-ona Se sintetizó según el método del Ejemplo 8, usando (S)-2-metilpiperazin-1-carboxilato de bencilo en lugar de piperazin-1-carboxilato de bencilo en la Etapa A y usando 2-(dimetilamino)etan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+Ap C i MS m/z 517,3 [M+H]+.
Ejemplo 15

(R)-1-(4-(2-(2-(dimetilamino)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-metilpiperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, sustituyendo (R)-2-metilpiperazin-1-carboxilato de bencilo por piperazin-1-carboxilato de bencilo en la Etapa A y 2-(dimetilamino)etan-1-ol por (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 517,3 [M+H]+.
Ejemplo 16
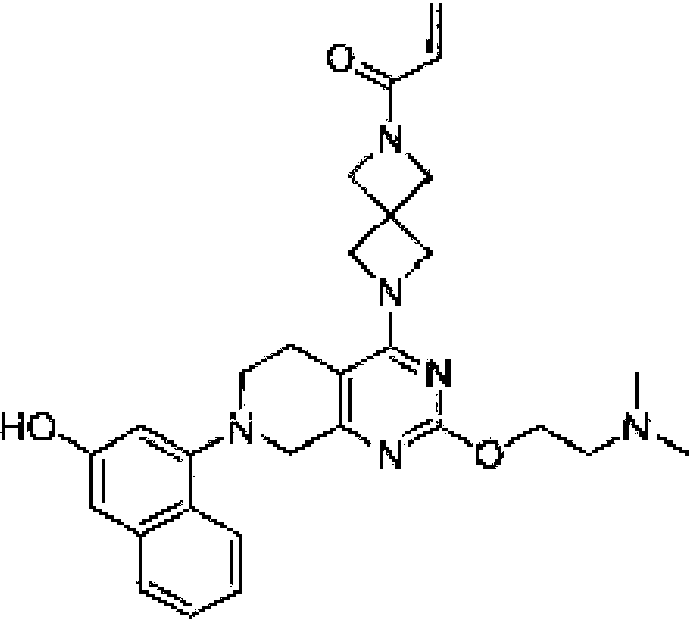
1-(6-(2-(2-(dimetilamino)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2,6-diazaespiro[3,3]heptan-2-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 2,6-diazaespiro[3,3]heptano-2-carboxilato de bencilo en lugar de piperazin-1-carboxilato de bencilo en la Etapa A y sustituyendo 2-(dimetilamino)etan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 515,3 [M+H]+.
Ejemplo 17

1-(4-(2-(4-(dimetilamino)piperidin-1-il)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando N,N-dimetilpiperidin-4-amina en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 580,3 [M+H]+.
Ejemplo 18

1-(6-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2,6-diazaespiro[3,3]heptan-2-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 2,6-diazaespiro[3,3]heptano-2-carboxilato de bencilo en lugar de piperazin-1-carboxilato de bencilo en la Etapa A y sustituyendo 1-(dimetilamino)propan-2-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 529,3 [M+H]+.
Ejemplo 19

(R)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (R)-1-(dimetilamino)propan-2-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 517,3 [M+H]+.
Ejemplo 20

1-(6-(2-(3-(dimetilamino)propoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2,6-diazaespiro[3,3]heptan-2-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 2,6-diazaespiro[3,3]heptano-2-carboxilato de bencilo en lugar de piperazin-1-carboxilato de bencilo en la Etapa A y sustituyendo 3-(dimetilamino)propan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 529,3 [M+H]+.
Ejemplo 21

1-(4-(2-((4-(dimetilamino)butan-2-il)oxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 4-(dimetilamino)butan-2-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+a Pc I MS m/z 531,3 [M+H]+.
Ejemplo 22

1- (4-(7-(3-hidroxinaftalen-1-il)-2-((1-metilpiperidin-4-il)oxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 1 -metilpiperidin-4-ol en lugar de (S)-1-(dimetilamino)propan-2- ol en la Etapa B. ES+APCI MS m/z 529,3 [M+H]+.
Ejemplo 23

1- (4-(7-(3-hidroxinaftalen-1-il)-2-((1-metilpirrolidin-3-il)oxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 1 -metilpirrolidin-3-ol en lugar de (S)-1-(dimetilamino)propan-2- ol en la Etapa B. ES+APCI MS m/z 515,3 [M+H]+.
Ejemplo 24

1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 1-(dimetilamino)propan-2-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B, usando 1-bromo naftaleno en lugar de trifluorometanosulfonato de 3-(metoximetoxi)naftalen-1-ilo en la etapa D y eliminando la Etapa G. ES+APCI MS m/z 501,3 [M+H]+.
Ejemplo 25

1-(4-(2-(3-(dimetilamino)propoxi)-7-(1-feniletil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona Se sintetizó según el método del Ejemplo 8, usando 3-(dimetilamino)propan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B, usando trifluorometanosulfonato de (1-bromoetil)benceno en lugar de 3-(metoximetoxi)naftalen-1-ilo en la etapa D y eliminando la Etapa G. ES+APCI m S m/z 559,3 [M+H]+.
Ejemplo 26

1-(4-(2-(4-(2-hidroxietil)piperazin-1-il)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 2-(piperazin-1-il)acetato de etilo en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. Después de Etapa D, se realizó la siguiente reacción de saponificación: Se recogió 4-(2-(4-(2-acetoxietil)piperazin-1-il)-7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo en THF (5 ml) y se añadió LiOH 2 M (1 ml). La mezcla se agitó a temperatura ambiente durante 24 h. Se añadió NH4Cl saturado y la reacción se extrajo con DCM. Las capas orgánicas combinadas se concentraron y el residuo resultante se purificó por cromatografía sobre gel de sílice (Biotage Isolera Gold, eluyendo con 0-10 % de MeOH en DCM) para proporcionar 4-(2-(4-(2-hidroxietil)piperazin-1-il)-7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo. El resto de la síntesis se llevó a cabo como en el Ejemplo 8, etapa E. ES+APCI m S m/z 544,3 [M+H]+.
Ejemplo 27

1-((S)-4-(2-(((R)-1-(dimetilamino)propan-2-il)oxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-3-metilpiperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (S)-3-metilpiperazin-1-carboxilato de bencilo en lugar de piperazin-1-carboxilato de bencilo en la Etapa A y usando (R)-1-(dimetilamino)propan-2-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI m S m/z 531,3 [M+H]+.
Ejemplo 28
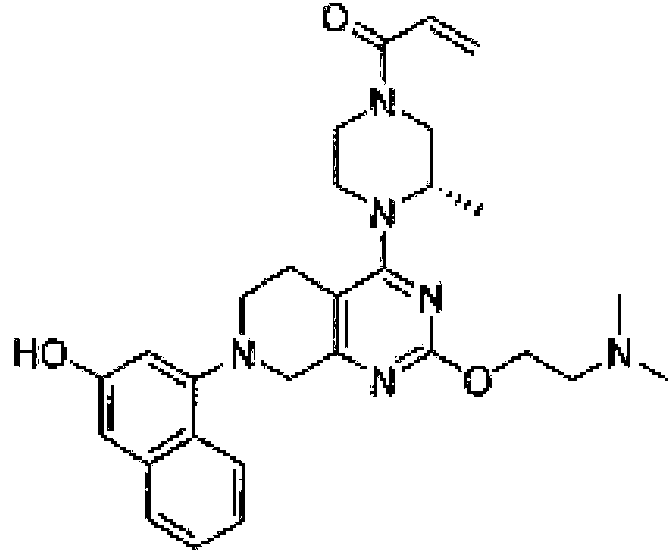
(S)-1-(4-(2-(2-(dimetilamino)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-3-metilpiperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (S)-3-metilpiperazin-1-carboxilato de bencilo en lugar de piperazin-1-carboxilato de bencilo en la Etapa A y usando 2-(dimetilamino)etan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+Ap C i MS m/z 517,2 [M+H]+.
Ejemplo 29

1- (4-(7-(3-hidroxinaftalen-1-il)-2-(2-morfolinoetoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona Se sintetizó según el método del Ejemplo 8, usando 2-morfolinoetan-1-ol en lugar de (S)-1-(dimetilamino)propan-2- ol en la Etapa B. ES+APCI MS m/z 545,2 [M+H]+.
Ejemplo 30

1-(4-(7-(3-hidroxinaftalen-1-il)-2-morfolin-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona Se sintetizó según el método del Ejemplo 8, usando morfolina en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 501,3 [M+H]+.
Ejemplo 31

1-(4-(7-(3-hidroxinaftalen-1-il)-2-(pirrolidin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona Se sintetizó según el método del Ejemplo 8, usando pirrolidina en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 485,2 [M+H]+.
Ejemplo 32

(R)-1-(4-(7-(3-hidroxinaftalen-1-il)-2-((1-(pirrolidin-1-il)propan-2-il)oxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (R)-1-(pirrolidin-1-il)propan-2-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 543,4 [M+H]+.
Ejemplo 33

trifluoroacetato de 1-(4-(2-(2-(1,1-dioxidotiomorfolino)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Etapa A: 4-[7-(3-benciloxi-1-naftil)-2-[2-(1,1-dioxo-1,4-tiazinan-4-il)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: A una mezcla de 4-[7-(3-benciloxi-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (300 mg, 463,12 pmol, 1,00 eq.) y 2-(1,1-dioxo-1,4-tiazinan-4-il)etanol (166 mg, 926 pmol, 2,00 eq.) en tolueno (10,0 ml) se le añadieron NaOBu-t (133 mg, 1,39 mmol, 3,00 eq.), BlNAP (57,7 mg, 92,6 pmol, 0,20 eq.) y Pd2(dba)3 (42,4 mg, 46,3 pmol, 0,10 eq.). La mezcla de reacción se agitó a 90 °C durante 12 horas en una atmósfera de N2. La mezcla de reacción se filtró y la torta de filtro se lavó con DCM (3 x 10 ml). El filtrado se concentró al vacío. El residuo se purificó por reverse cromatografía ultrarrápida (40 % de MeCN en agua (0,1 % de TFA) para dar 4-[7-(3-benciloxi-1-naftil)-2-[2-(1,1-dioxo-1,4-tiazinan-4-il)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (230 mg, 301 pmol, 65,1 % de rendimiento) en forma de un sólido de color pardo. ESI mS m/z 763,5 [M+H]+.
Etapa B: 4-[2-[2-(1,1-dioxo-1,4-tiazinan-4-il)etoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol: A una solución de 4-[7-(3-benciloxi-1-naftil)-2-[2-(1,1-dioxo-1,4-tiazinan-4-il)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (190 mg, 249 pmol, 1,00 eq.) en MeOH (10,0 ml) se le añadió Pd/C (100 mg) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con hidrógeno. La mezcla se agitó en una atmósfera de hidrógeno (103,42 kPa (15 psi)) a 40 °C durante 4 horas. La mezcla de reacción se filtró y el filtrado se concentró al vacío para dar 4-[2-[2-(1,1-dioxo-1,4-tiazinan-4-il)etoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (90,0 mg, 167 pmol, 67,1 % de rendimiento) en forma de un sólido de color pardo. ESI MS m/z 539,4 [M+H]+.
Etapa C: 1-[4-[2-[2-(1,1-dioxo-1,4-tiazinan-4-il)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de 4-[2-[2-(1,1-dioxo-1,4-tiazinan-4-il)etoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (90,0 mg, 167 pmol, 1,00 eq.) y DIEA (64,8 mg, 501 pmol, 87,5 μl, 3,00 eq.) en DCM (2,00 ml) se le añadió prop-2-enoato de prop-2-enoílo (19,0 mg, 150 pmol, 0,90 eq.) a -40 °C. La mezcla de reacción se agitó a -40 °C durante 0,5 h. La mezcla de reacción se inactivó con 1 ml de MeOH y se concentró al vacío. El residuo se purificó con una columna de HPLC preparativa: Phenomenex Synergi C18150*25*, 10 p; fase móvil: [agua (0,1 % de TfA)-ACN]; % de B: 12 %-42 %,11 min para dar trifluoroacetato de 1-[4-[2-[2-(1,1-dioxo-1,4-tiazinan-4-il)etoxi]-7-(3
hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (32,6 mg, 50,2 pmol, 30,0 % de rendimiento, 91,3 % de pureza) en forma de un sólido de color pardo. ESI MS m/z 593,5 [M+H]+.
Ejemplo 34

trifluoroacetato de 1-(4-(2-(3-(1,1-dioxidotiomorfolino)propoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 33, usando 3-(1,1-dioxo-1,4-tiazinan-4-il)propan-1-ol en lugar de 2-(1,1-dioxo-1,4-tiazinan-4-il)etanol en la Etapa A. ESI MS m/z 607,5 [M+H]+.
Ejemplo 35

trifluoroacetato de 1-(4-(7-(3-hidroxinaftalen-1-il)-2-(4-morfolinobutoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 33, usando 4-morfolinobutan-1-ol en lugar de 2-(1,1-dioxo-1,4-tiazinan-4-il)etanol en la Etapa A. ESI MS m/z 573,4 [M+H]+.
Ejemplo 36

(R)-1-(4-(2-((1-(4-acetilpiperazin-1-il)propan-2-il)oxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 33, usando 1-[4-[(2R)-2-hidroxipropil]piperazin-1-il]etanona en lugar de 2-(1,1-dioxo-1,4-tiazinan-4-il)etanol en la Etapa A. ESI m S m/z 600,6 [M+H]+.
Ejemplo 37

1-(4-(2-(2-(4-acetilpiperazin-1-il)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Etapa A: 4-[2-[2-(4-acetilpiperazin-1-il)etoxi]-7-(3-benciloxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: A una solución de 1-[4-(2-hidroxietil)piperazin-1-il]etanona (277 mg, 1,61 mmol, 2,60 eq.) en THF (8,00 ml) se le añadió NaH (49,4 mg, 1,23 mmol, 60 % de pureza, 2,00 eq.) a 0 °C. La mezcla de reacción se agitó a 0 °C durante 15 minutos. A la mezcla se le añadió 4-[7-(3-benciloxi-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-il]piperazin-1 -carboxilato de bencilo (400 mg, 618 pmol, 1,00 eq.) en THF (2,00 ml). La mezcla de reacción se agitó a 0 °C durante 20 minutos. La mezcla de reacción se inactivó con NH4Cl saturado (6 ml) y agua (6 ml). La mezcla de reacción se extrajo con acetato de etilo (3 x 30 ml). Las capas orgánicas combinadas se lavaron con salmuera (10 ml), se secaron sobre Na2SÜ4 y se concentraron al vacío para dar 4-[2-[2-(4-acetilpiperazin-1-il)etoxi]-7-(3-benciloxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (450 mg, 534 pmol, 86,5 % de rendimiento, 89,7 % de pureza) en forma de un sólido de color pardo. ESI MS m/z 756,3 [M+H]+.
Etapa B: 1-[4-[2-[[7-(3-hidroxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]etil]piperazin-1-il]etanona: A una solución de 4-[2-[2-(4-acetilpiperazin-1-il)etoxi]-7-(3-benciloxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (200 mg, 264 pmol, 1,00 eq.) en THF (10,0 ml) se le añadió Pd/C (100 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con hidrógeno. La mezcla se agitó en una atmósfera de hidrógeno (103,42 kPa (15 psi)) a 40 °C durante 12 horas. La mezcla de reacción se filtró y la torta de filtro se lavó con THF (3 x 5 ml). El filtrado se concentró al vacío para dar 1-[4-[2-[[7-(3-hidroxi-1-naftil)-4-pipenazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]etil]piperazin-1-il]etanona (80,0 mg, 150 pmol, 56,9 % de rendimiento) en forma de un sólido de color pardo.
Etapa C: 1-[4-[2-[2-(4-acetilpiperazin-1-il)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de 1-[4-[2-[[7-(3-hidroxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]etil]piperazin-1-il]etanona (80,0 mg, 150 pmol, 1,00 eq.) y DIEA (58,3 mg, 451 pmol, 78,8 μl, 3,00 eq.) en DCM (2,00 ml) se le añadió prop-2-enoato de prop-2-enoílo (14,2 mg, 113 pmol, 0,75 eq.) a -40 °C durante 0,5 h. La mezcla de reacción se inactivó con MeOH (1 ml), se diluyó con DCM (20 ml) y luego se lavó con agua (5 ml). Las capas orgánicas combinadas se lavaron con salmuera (5 ml), se secaron sobre Na2SO4 y se concentraron al vacío. La mezcla de reacción se purificó por HPLC preparativa: Phenomenex Gemini 150 * 25 mm *, 10 p; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - ACN]; % de B: 30 %-55 %, 10 min para dar 1-[4-[2-[2-(4-acctylpiperazin-1-il)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (13,8 mg, 23,3 pmol, 15,5 % de rendimiento, 98,7 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 586,3 [M+H]+.
Ejemplo 38

1-(4-(7-(3-hidroxinaftalen-1-il)-2-(3-(piperidin-1-il)propoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 37, usando 3-(1-piperidil)propan-1-ol en lugar de 1-[4-(2-hidroxietil)piperazin-1-il]etanona en la Etapa A. ESI MS m/z 557,5 [M+h ]+.
Ejemplo 39

1-(4-(7-(3-hidroxinaftalen-1-il)-2-(3-(pirrolidin-1-il)propoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 37, usando 3-pirrolidin-1 -ilpropan-1-ol en lugar de 1-[4-(2-hidroxietil)piperazin-1-il]etanona en la Etapa A. ESI MS m/z 543,3 [M+H]+.
Ejemplo 40

1-(4-(2-(4-(dimetilamino)butoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 37, usando 4-(dimetilamino)butan-1-ol en lugar de 1-[4-(2-hidroxietil)piperazin-1-il]etanona en la Etapa A. ESI MS m/z 531,4 [M+H]+.
Ejemplo 41

trifluoroacetato de 1-(4-(7-(3-ciclopropil-5-hidroxifenil)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Etapa A: 4-[7-(3-benciloxi-5-ciclopropil-fenil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato: A una mezcla de 4-[2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (400 mg, 805 pmol, 1,00 eq.) y 1-benciloxi-3-bromo-5-ciclopropil-benceno (268 mg, 886 pmol, 1,10 eq.) en tolueno (10,0 ml) se le añadieron 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (RuPhos) (75,2 mg, 161 pmol, 0,20 eq.), Pd2(dba)3 (111 mg, 121 pmol, 0,15 eq.) y Cs2CO3 (656 mg, 2,01 mmol, 2,50 eq.). La mezcla de reacción se agitó a 90 °C durante 12 horas en una atmósfera de N2. La mezcla se añadió a agua (15 ml) y se extrajo con DCM (2 x 15 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron. El residuo se purificó por cromatografía (SO2, éter de petróleo/acetato de etilo = 3:1 a diclorometano: metanol = 10:1) para dar 4-[7-(3-benciloxi-5-ciclopropil-fenil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (170 mg, 230 pmol, 28,5 % de rendimiento, 97,2 % de pureza) en forma de un aceite de color pardo. ESI MS m/z 719,6 [M+H]+.
Etapa B: 3-ciclopropil-5-[2-(3-morfolinopropoxi)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]fenol A una mezcla de 4-[7-(3-benciloxi-5-ciclopropil-fenil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (150 mg, 209 pmol, 1,00 eq.) en MeOH (10,0 ml) se le añadieron Pd/C (150 mg, 10 % de pureza) y CH3COOH (25,1 mg, 417,3 pmol, 23,9 μl, 2,00 eq.). La suspensión se desgasificó al vacío y se purgó varias veces con hidrógeno. La mezcla se agitó en una atmósfera de hidrógeno (103,42 kPa (15 psi)) a 40 °C durante 2 horas. La mezcla de reacción se filtró a través de Celite® y el filtrado se concentró. El producto diacetato de 3-ciclopropil-5-[2-(3-morfolinopropoxi)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]fenol (80,0 mg, 130 pmol, 62,4 % de rendimiento) se obtuvo en forma de un sólido de color amarillo. ESI MS m/z 495,2 [M+H]+.
Etapa C: 1 -[4-[7-(3-ciclopropil-5-hidroxi-fenil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona A una mezcla de diacetato de 3-ciclopropil-5-[2-(3-morfolinopropoxi)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]fenol (80,0 mg, 130 pmol, 1,00 eq.) en DCM (2,00 ml) se le añadieron DIEA (168 mg, 1,30 mmol, 227 μl, 10,0 eq.) y prop-2-enoato de prop-2-enoílo (13,1 mg, 104 pmol, 0,80 eq.) a -78 °C y la mezcla de reacción se agitó a -78 °C durante 0,5 horas. La mezcla de reacción se inactivó con MeOH (2 eq., 10 mg) y después se concentró. El residuo se purificó por HPLC preparativa (Instrumento: GX-K; Columna: Phenomenex Synergi C18150*25*, 10 p; Condiciones: agua (0,1 % de TFA)-ACN; Inicio B: 8; Fin B: 38; Tiempo de gradiente (min): 11; Tiempo de mantenimiento a 100 % de B (min): 2; Caudal (ml/min): 25). El producto aislado se concentró por liofilización. El producto trifluoroacetato de 1-[4-[7-(3-ciclopropil-5-hidroxi-fenil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (21,5 mg, 31,1 pmol, 23,9 % de rendimiento, 95,8 % de pureza) se obtuvo en forma de un sólido de color amarillo. ESI MS m/z 549,5 [M+H]+.
Ejemplo 42

1-(4-(7-(2-fluoro-5-hidroxifenil)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 41, usando 4-(benciloxi)-2-bromo-1-fluorobenceno en lugar de 1-benciloxi-3-bromo-5-ciclopropil-benceno en la Etapa A. ESI MS m/z 527,4 [M+H]+.
Ejemplo 43

1-(4-(7-(3-hidroxi-6-metilnaftalen-1-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Etapa A: 4-[7-(3-metoxi-6-metil-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: Una mezcla de 4-[2-piperazin-1-carboxilato de (3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-ilo] (500 mg, 1,01 mmol, 1,00 eq.), (3-metoxi-6-metil-1-naftil)trifluorometanosulfonato de bencilo (483 mg, 1,51 mmol, 1,50 eq.), Cs2CO3 (820 mg, 2,52 mmol, 2,50 eq.), 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (RuPhos) (93,9 mg, 201,3 pmol, 0,20 eq.) y Pd2(dba)3 (92,2 mg, 100 pmol, 0,10 eq.) en tolueno (3,00 ml) se agitó a 90 °C durante 10 horas. La mezcla se diluyó con acetato de etilo (50,0 ml). El precipitado se eliminó por filtración y el filtrado se concentró al vacío. El residuo se purificó por cromatografía en columna de fase inversa sobre gel de sílice (0,1 % de TFA agua/acetonitrilo). Las fracciones deseadas se combinaron, se basificaron con bicarbonato de sodio acuoso saturado (2,00 ml) y después se concentraron al vacío. El residuo se extrajo con acetato de etilo (2 x 100 ml). Los extractos combinados se lavaron con salmuera (1 x 100 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío para dar 4-[7-(3-metoxi-6-metil-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (400. mg, 599 pmol, 59,4 % de rendimiento, 100 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 667,6 [M+H]+.
Etapa B: 4-[3-[[7-(3-metoxi-6-metil-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]propil]morfolina: A una solución de 4-[7-(3-metoxi-6-metil-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (370 mg, 554 pmol, 1,00 eq.) en MeoH (10,0 ml) se le añadieron Pd-C (100 mg, 10 % de pureza) y AcOH (66,6 mg, 1,11 mmol, 2,00 eq.) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con hidrógeno. La mezcla se agitó en una atmósfera de hidrógeno (103,42 kPa (15 psi)) a 40 °C durante 2 horas. La mezcla de reacción se filtró y se concentró al vacío para proporcionar 4-[3-[[7-(3-metoxi-6-metil-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H pirido[3,4-d]pirimidin-2-il]oxi]propil]morfolina (295 mg, 553 pmol, 99,8 % de rendimiento) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación. ESI MS m/z 533,6 [M+H]+
Etapa C: 1-[4-[7-(3-metoxi-6-metil-1-naftil)-2-(3-morfolinopropoxi) -6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una mezcla de prop-2-enoato de prop-2-enoílo (56,8 mg, 450 pmol, 0,80 eq.) y DlEA (727 mg, 5,63 mmol, 983 μl, 10,0 eq.) en DCM (2,00 ml) se le añadió una solución de 4-[3-[[7-(3-metoxi-6-metil-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]propil]morfolina (300 mg, 563 pmol, 1,00 eq.) DCM (1,00 ml) a -40 °C en una atmósfera de nitrógeno. La mezcla se agitó durante 1 hora. La mezcla de reacción se inactivó mediante la adición de MeOH (50 μl) a -40 °C, se diluyó con agua (10,0 ml), se extrajo con DCM(10,0 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida para proporcionar 1-[4-[7-(3-metoxi-6-metil-1-naftil)-2-(3-morfolinopropoxi) -6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (270 mg, 460 pmol, 81,7 % de rendimiento) eS i m S m/z 587,6 [M+H]+.
Etapa D: 1-[4-[7-(3-hidroxi-6-metil-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de 1-[4-[7-(3-metoxi-6-metil-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (100 mg, 170 pmol, 1,00 eq.) en DCM (3,00 ml) se le añadió BBr3 (213 mg, 852 pmol, 82,1 μl, 5,00 eq.) a -78 °C. La mezcla se agitó a 0 °C durante 1 hora. La mezcla se enfrió a -78 °C y se diluyó con DCM (20,0 ml), se inactivó mediante la adición de una solución saturada de bicarbonato de sodio (5,00 ml), se agitó a -78 °C durante 10 min y después se calentó a 0 °C. La mezcla se extrajo con DCM (2 x 15,0 ml), se lavó con salmuera (1 x 20,0 ml), se secó sobre Na2SO4, se filtró y se concentró al vacío. El residuo se purificó por
HPLC preparativa (columna: Phenomenex Gemini C18250*50 mm*, 10 p; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN]; % de B: 27 %-57 %, 12 min) para proporcionar 1-[4-[7-(3-hidroxi-6-metil-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (11,0 mg, 18,6 pmol, 10,9 % de rendimiento, 97,0 % de pureza) en forma de un sólido de color pardo. ESI MS m/z 573,6 [M+H]+.
Ejemplo 44

1- (4-(7-(5-metil-1H-indazol-4-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2- en-1-ona
Etapa A: 4-[7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]- 2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: Una mezcla de 4-[2-(3-morfolinopropoxi)-5,6,7,8- tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (500 mg, 1,01 mmol, 1,00 eq.), 2-[(4-bromo-5-metil-indazol-1-il)metoxi] etil-trimetilsilano (448 mg, 1,31 mmol, 1,30 eq.), Cs2CÜ3 (822 mg, 2,53 mmol, 2,50 eq.), Pd2(dba)3 (138 mg, 151 pmol, 0,15 eq.) y 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (RuPhos) (94,3 mg, 202 pmol, 0,20 eq.) en tolueno (20,0 ml) se desgasificó y se purgó 3 veces con nitrógeno y se agitó a 90 °C durante 10 horas en una atmósfera de nitrógeno. La mezcla de reacción se diluyó con agua (50 ml) y se extrajo con acetato de etilo (3 x 100 ml). Las capas orgánicas combinadas se lavaron con salmuera (3 x 50,0 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida. El residuo se purificó por HPLC de fase inversa (0,1 % de TFA agua/acetonitrilo) para proporcionar 4-[7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (420 mg, 554 pmol, 54,9 % de rendimiento) en forma de un aceite de color amarillo. ESl MS m/z 757,6 [M+H]+.
Etapa B: 4-[7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo: A una solución de 4-[7-[5-metil-1 -(2-trimetilsililetoximetil) indazol-4-il]-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (370 mg, 488 pmol, 1,00 eq.) en MeOH (10,0 ml) se le añadieron trietilamina (98,9 mg, 977 pmol, 135 μl, 2,00 eq.), Pd/C(100 mg, 10 % de pureza) y carbonato de terc-butoxicarbonil ferc-butilo (213 mg, 977 pmol, 224 μl, 2,00 eq.) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con hidrógeno. La mezcla se agitó en una atmósfera de hidrógeno (103,42 kPa (15 psi)) a 40 °C durante 2 horas. La mezcla de reacción se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna (SO2, DCM/MeOH = 1:0 a 10:1) para proporcionar 4-[7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (340 mg, 470 pmol, 96,2 % de rendimiento) en forma de un aceite de color pardo. ESl MS m/z 723,5 [M+H]+.
Etapa C: trifluoroacetato de 4-[3-[[7-(5-metil-1H-indazol-4-il)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-illoxilpropillmorfolina: A una solución de 4-[7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (150 mg, 207 pmol, 1,00 eq.) en DCM (500 μl) se le añadió t Fa (354 mg, 3,11 mmol, 230 μl, 15,0 eq.). La mezcla se agitó a 25 °C durante 1 hora. La mezcla de reacción se concentró al vacío para proporcionar trifluoroacetato de 4-[3-[[7-(5-metil-1H-indazol-4-il)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]propil]morfolina (125 mg, 206 pmol, 99,3 % de rendimiento) en forma de un aceite de color pardo y se usó directamente en la siguiente etapa sin purificación. ESl MS m/z 493,4 [M+H]+.
Etapa D 1-[4-[7-(5-metil-1H-indazol-4-il)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-1-il]prop-2-en-1-ona: A una mezcla de trifluoroacetato de 4-[3-[[7-(5-metil-1H-indazol-4-il)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]propil]morfolina (120 mg, 197 pmol, 1,00 eq., TFA) y DiEa (255 mg, 1,98 mmol, 345 μl, 10,0 eq.) en diclorometano (2,00 ml) se le añadió una solución de prop-2-enoato de prop-2-enoílo (19,9 mg, 158 pmol, 0,80 eq.) en diclorometano (1,00 ml) a -40 °C en una atmósfera de nitrógeno. La mezcla se agitó durante 1 hora. La mezcla de reacción se inactivó mediante la adición de MeOH (50,0 μl) a -40 °C, se diluyó con agua (10,0 ml), se extrajo con diclorometano (10,0 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo se purificó por HPLC preparativa (columna: Phenomenex Gemini 150 * 25 mm *, 10 p; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - Ac N]; % de B: 30 %-60 %, 10 min) para proporcionar 1-[4-[7-(5-metil-1H-indazol-4-il)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (19,0 mg, 34,4 pmol, 17,4 % de rendimiento, 99,0 % de pureza) en forma de un sólido de color blanco. ESI MS m/z 547,5 [M+H]+.
Ejemplo 45

1-((1R,5S)-3-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-3,8-diazabiciclo[3.2.1]octan-8-il)prop-2-en-1-ona
Etapa A: 3-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-3,8-diazabiciclo[3.2.1]octano-8-carboxilato de terc-butilo: A una mezcla de 7-bencil-2,4-dicloro-6,8-dihidro-5H-pirido[3,4-d]pirimidina (920 mg, 3,13 mmol, 1,00 eq.) y 3,8-diazabiciclo[3.2.1]octano-8-carboxilato de ferc-butilo (677 mg, 3,19 mmol, 1,02 eq.) en DMSO (18,0 ml) se le añadió DIEA (1,21 g, 9,39 mmol, 1,64 ml, 3,00 eq.). La mezcla de reacción se agitó a 60 °C durante 1 hora. La mezcla se diluyó, se extrajo con EtOAc (3 x 20 ml) y se lavó con agua (10 ml), HCl 1 N (5 ml), NaHCO3 (15 ml), y salmuera (15 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron al vacío para dar 3-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-3,8-diazabiciclo[3.2.1]octano-8-carboxilato de ferc-butilo (1,30 g, 2,41 mmol, 76,9 % de rendimiento, 87,0 % de pureza) en forma de un aceite de color pardo que se usó directamente en la siguiente etapa sin purificación adicional. ESI MS m/z 470,2 [M+H]+.
Etapa B: 3-[7-bencil-2-[2-(dimetilamino)-1-metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3,8-diazabiciclo[3.2.1]octano-8-carboxilato de ferc-butilo: A una solución de 1-(dimetilamino)propan-2-ol (857 mg, 8,31 mmol, 942 μl, 3,00 eq.) en THF (40,0 ml) se le añadió NaH (222 mg, 5,54 mmol, 60,0 % de pureza, 2,00 eq.) a 15 °C en una atmósfera de N2. Después de agitar a 15 °C durante 0,5 horas, se añadió 3-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-3,8-diazabiciclo[3.2.1]octano-8-carboxilato de terc-butilo (1,30 g, 2,77 mmol, 1,00 eq.). La mezcla se agitó a 100 °C durante 12 horas en un tubo sellado. La reacción se interrumpió lentamente con agua (3 ml) y después se concentró al vacío. El residuo se purificó por cromatografía en columna (DCM/MeOH 60:1 a 10:1) para dar 3-[7-bencil-2-[2-(dimetilamino)-1-metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3,8-diazabiciclo[3.2.1]octano-8-carboxilato de terc-butilo (700 mg, 1,07 mmol, 38,8 % de rendimiento, 82,4 % de pureza) en forma de un aceite de color amarillo.
Etapa C: 3-[2-[2-(dimetilamino)-1-metil-etoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]-3,8-diazabiciclo[3.2.1]octano-8-carboxilato de terc-butilo: A una solución de 3-[7-bencil-2-[2-(dimetilamino)-1-metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3,8-diazabiciclo[3.2.1]octano-8-carboxilato de ferc-butilo (700 mg, 1,30 mmol, 1,00 eq.) en MeOH (30,0 ml) se le añadió Pd/C (200 mg) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó con 4 veces hidrógeno. La mezcla se agitó en una atmósfera de hidrógeno (344,74 kPa (50 psi)) a 40 °C durante 12 horas. La mezcla se concentró al vacío. El residuo se purificó por cromatografía en columna (DCM/MeOH 50:1 a 5:1) para dar 3-[2-[2-(dimetilamino)-1-metil-etoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]-3,8-diazabiciclo[3.2.1]octano-8-carboxilato de terc-butilo (500 mg, 952 pmol, 73,2 % de rendimiento, 85,0 % de pureza) en forma de un aceite de color amarillo.
Etapa D: 3-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)-1-metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3,8-diazabiciclo[3.2.1]octano-8-carboxilato de ferc-butilo: Se añadió Pd2(dba)3 (84,1 mg, 91,8 pmol, 0,10 eq.) a una solución de 3-[2-[2-(dimetilamino)-1-metil-etoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]-3,8-diazabiciclo[3.2.1]octano-8-carboxilato de ferc-butilo (410 mg, 918 pmol, 1,00 eq.), 3-benciloxi-1-bromonaftaleno (293 mg, 936 pmol, 1,02 eq.), 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (RuPhos) (85,7 mg, 184 pmol, 0,20 eq.) y Cs2CO3 (897 mg, 2,75 mmol, 3,00 eq.) en dioxano (9,00 ml). La mezcla de reacción se agitó a 100 °C durante 7 horas en una atmósfera de N2 y después se concentró al vacío. El residuo se diluyó con agua (5 ml) y se extrajo con DCM (2 x 20 ml). Las capas orgánicas se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna (DCM/MeOH 100:1 a 20:1) para dar 3-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)-1-metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3,8-diazabiciclo[3.2.1]octano-8-carboxilato de terc-butilo (317 mg, 441 pmol, 48,1 % de rendimiento, 94,5 % de pureza) en forma de un aceite de color amarillo. ESI MS m/z 679,2 [M+H]+.
Etapa E: 3-[2-[2-(dimetilamino)-1-metil-etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3,8-diazabiciclo[3.2.1]octano-8-carboxilato de ferc-butilo: A una solución de 3-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)-1-metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3,8-diazabiciclo[3.2.1]octano-8-carboxilato de ferc-butilo (340 mg, 501 pmol, 1,00 eq.) en MeOH (6,80 ml) se le añadió Pd/C (120 mg). La suspensión se desgasificó al vacío y se purgó con 4 veces hidrógeno. La mezcla se agitó en una atmósfera de hidrógeno
(103,42 kPa (15 psi)) a 40 °C durante 1,5 horas. La mezcla se concentró al vacío para dar 3-[2-[2-(dimetilamino)-1-metil-etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3,8-diazabiciclo[3.2.1]octano-8-carboxilato de terc-butilo (210 mg, 316 pmol, 63,1 % de rend¡m¡ento, 88,6 % de pureza) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. ESI MS m/z 589,3 [M+H]+.
Etapa F: trifluoroacetato de 4-[4-(3,8-diazabiciclo[3.2.1]octan-3-il)-2-[2-(dimetilamino)-1-metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol: A una solución de 3-[2-[2-(dimetilamino)-1-metil-etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3,8-diazabiciclo[3.2.1]octano-8-carboxilato de terc-butilo (18o mg, 306 pmol, 1,00 eq.) en DCM (230 μl) se le añadió TFA (349 mg, 3,06 mmol, 226 μl, 10,0 eq.). La mezcla de reacción se agitó a 18 °C durante 0,5 horas. La mezcla se concentró al vacío para dar trifluoroacetato de 4-[4-(3,8-diazabiciclo[3.2.1]octan-3-il)-2- [2-(dimetilamino)-1-metil-etoxi]-6,8-dihidro-5H pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (528 mg, bruto) en forma de un aceite de color rojo que se usó directamente en la siguiente etapa sin purificación adicional. ESI MS m/z 489,2 [M+H]+.
Etapa G: 1-[3-[2-[2-(dimetilamino)-1-metil-etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3,8-diazabiciclo[3.2.1]octan-8-il]prop-2-en-1-ona: A una solución de trifluoroacetato de 4-[4-(3,8-diazabiciclo[3.2.1]octan-3- il)-2-[2-(dimetilamino)-1-metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (149 mg, 306 pmol, 1,00 eq.) y DlEA (1,36 g, 10,5 mmol, 1,84 ml, 34,5 eq.) en DCM (1,50 ml) se le añadió gota a gota prop-2-enoato de prop-2-enoílo (30,9 mg, 245 pmol, 0,80 eq.) a -50 °C. La mezcla se agitó a -40 y después a -20 °C durante 30 minutos. La reacción se interrumpió con MeOH (19,6 mg) y se concentró al vacío. El residuo se purificó por TLC preparativa (DCM/MeOH 7:1) para dar 1-[3-[2-[2-(dimetilamino)-1-metil-etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3,8-diazabiciclo[3.2.1]octan-8-il]prop-2-en-1-ona (18,9 mg, 32,8 pmol, 10,7 % de rendimiento, 94,3 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 543,3 [M+H]+.
Ejemplo 46

1-((1R,5S)-8-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-3,8-diazabiciclo[3.2.1]octan-3-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 4, usando (1R,5S)-3,8-diazabiciclo[3.2.1]octano-3-carboxilato de tercbutilo en lugar de 3,8-diazabiciclo[3.2.1]octano-8-carboxilato de terc-butilo en la Etapa A. ESI MS m/z 543,4 [M+H]+.
Ejemplo 47

1-(4-(2-(2-(dimetilamino)etoxi)-7-(3-hidroxinaftalen-1-il)-8-metil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Etapa A: 2-íbenc¡lam¡no)propanoato de etilo: A una solución de 2-bromopropanoato de etilo (30,0 g, 165 mmol, 21,6 ml, 1,00 eq.) y BnNH2 (23,1 g, 215 mmol, 23,6 ml, 1,30 eq.) en MeCN (600 ml) se le añadió K2CO3 (45,8 g, 331 mmol, 2,00 eq.). La mezcla se agitó a 80 °C durante 2 hora. La mezcla de reacción se filtró y la torta de filtro se lavó con DCM (300 ml). El filtrado se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (éter dietílico:acetato de etilo = 10:1 a 4:1) para dar 2-(bencilamino)propanoato de etilo (34,0 g, 163 mmol, 98.4 % de rendimiento, 99,4 % de pureza) en forma de un aceite incoloro.
Etapa B: 4-[benc¡l-(2-etox¡-1-met¡l-2-oxo-et¡l)am¡no1butanoato de etilo: A una solución de 2-(bencilamino)propanoato de etilo (31,0 g, 150 mmol, 1,00 eq.) y 4-bromobutanoato de etilo (87,5 g, 449 mmol, 64,4 ml, 3,00 eq.) en MeCN (600 ml) y agua (60,0 ml) se le añadieron Cs2CO3 (97,5 g, 299,12 mmol, 2,00 eq.) y KI (4,97 g, 29,9 mmol, 0,20 eq.).
La mezcla de reacción se agitó a 80-90 °C durante 40 horas. La mezcla de reacción se filtró y la torta de filtro se lavó con DCM (2 * 100 ml). El filtrado se concentró al vacío. El residuo se disolvió en DCM (300 ml), se lavó con salmuera (80 ml), se secó sobre Na2SO4 y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida sobre gel de sílice (éter dietílico:acetato de etilo = 1:0 a 20:1) para dar 4-[benc¡l-(2-etox¡-1-met¡l-2-oxoet¡l)am¡no]butanoato de etilo (28,0 g, 87,1 mmol, 58,3 % de rendimiento) en forma de un aceite de color amarillo.
Etapa C: 1-benc¡l-2-met¡l-3-oxo-p¡per¡d¡n-4-carbox¡lato de etilo: A una solución de 4-(bencil-(2-etoxi-1-metil-2-oxoetil)amino]butanoato de etilo (28,0 g, 87,1 mmol, 1,00 eq.) en THF (600 ml) se le añadió tBuOK (19,6 g, 174 mmol, 2,00 eq.). La mezcla de reacción se agitó a 18 °C durante 1 hora. La mezcla de reacción se inactivó con agua (100 ml) y se extrajo con MTBE (3 * 300 ml) y DCM (2 * 200 ml). Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron al vacío. El producto bruto se purificó por cromatografía sobre gel de sílice (éter dietílico:acetato de etilo = 100:1 a 20:1) para dar 1-benc¡l-2-met¡l-3-oxo-p¡per¡d¡n-4-carbox¡lato de etilo (20,0 g, 58,8 mmol, 67.5 % de rendimiento, 81,0 % de pureza) en forma de un aceite de color amarillo. ESI MS m/z 276,0 [M+H]+.
Etapa D: 7-benc¡l-8-met¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-2.4-d¡ol: A EtOH (400 ml) se le añadió Na (3,76 g, 163 mmol, 3,88 ml, 2,50 eq.). La mezcla de reacción se agitó a 20 °C durante 0,5 horas. A la mezcla se le añadieron 1- benc¡l-2-met¡l-3-oxo-p¡per¡d¡n-4-carbox¡lato de etilo (18,0 g, 65,4 mmol, 1,00 eq.) y UREA (9,82 g, 163 mmol, 8,77 ml, 2,50 eq.) y la mezcla de reacción se agitó a 80 °C durante 80 horas. El disolvente se eliminó al vacío. El residuo se disolvió en agua (100 ml) y se lavó con MTBE (3 * 50 ml). La fase acuosa se ajustó a pH 6-7 con HCl (15 ml, 12 M). La mezcla se filtró y la torta de filtro se lavó con agua (30 ml) y se secó al vacío para dar 7-bencil-8-m etil^^-d ih id ro^H -p irido^^-dp rim id in^^-d io l (12,0 g, 44,2 mmol, 67,7 % de rendimiento, 100 % de pureza) que se obtuvo en forma de un sólido de color pardo. ESl MS m/z 272,0 [M+H]+.
Etapa E: bencil^A-dicloro^-metil^^-dihidro^H-pirido^A-dlpirim idina: Una mezcla de 7-benc¡l-8-met¡l-6,8-d¡h¡dro-5H-p irido^^-dprim id in^^-d io l (5,00 g, 18,4 mmol, 1,00 eq.) en POCE (305 g, 1,99 mol, 185 ml, 108 eq.) se calentó a 110 °C durante 12 horas. El disolvente se eliminó al vacío. El residuo se disolvió en DCM (500 ml) y se vertió en NaHCO3 saturado (200 ml) mientras se mantenía el pH por encima de 7. La capa orgánica se lavó con salmuera (50 ml) y se secó sobre Na2SO4. La solución se filtró a través de una capa de gel de sílice y la torta de filtro se lavó con DCM (3 * 400 ml).
Las capas orgánicas combinadas se concentraron al vacío para dar 7-benc¡l-2,4-d¡cloro-8-met¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-dprimidina (4,50 g, 14,6 mmol, 79,2 % de rendimiento) en forma de un aceite de color pardo.
Etapa F: 4-(7-benc¡l-2-cloro-8-met¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato: A una solución de 7-benc¡l-2,4-d¡cloro-8-met¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡na (3,00 g, 9,73 mmol, 1,00 eq.) y DIEA (2,52 g, 19.5 mmol, 3,40 ml, 2,00 eq.) en dioxano (60,0 ml) se le añadió piperazin-1-carboxilato de terc-butilo (1,90 g, 10,2 mmol, 1,05 eq.). La mezcla de reacción se agitó a 60 °C durante 12 horas. El disolvente se eliminó al vacío.
El residuo se purificó por cromatografía sobre gel de sílice (éter dietílico:acetato de etilo = 2:1) para dar 4-(7-bencil-2- cloro-8-met¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-butilo (3,30 g, 7,15 mmol, 73.5 % de rendimiento, 99,3 % de pureza) en forma de un sólido de color amarillo. ESl MS m/z 458,1 [M+H]+.
Etapa G: 4-[7-benc¡l-2-[2-(d¡met¡lam¡no)etox¡]-8-met¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tercbutilo: A una solución de 2-(dimetilamino)etanol (583 mg, 6,54 mmol, 655 μl, 3,00 eq.) en tolueno (20,0 ml) se le añadieron Pd(OAc)2 (48,9 mg, 218 pmol, 0,10 eq.), 4-(7-benc¡l-2-cloro-8-met¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carboxilato de terc-butilo (1,00 g, 2,18 mmol, 1,00 eq.), Cs2CO3 (2,13 g, 6,54 mmol, 3,00 eq.) y BlNAP (272 mg, 436 pmol, 0,20 eq.). La mezcla de reacción se agitó a 110 °C durante 3 horas en una atmósfera de N2. La mezcla de reacción se concentró al vacío. El residuo se disolvió en agua (10 ml) y se extrajo con DCM (3 * 40 ml). Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (éter dietílico:acetato de etilo = 5:1 a 2:1 después DCM:MeOH = 50:1 a 5:1) para dar 4-[7-bencil-2-[2-(d¡met¡lam¡no)etox¡]-8-met¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de terc-butilo (900 mg, 1,76 mmol, 80,8 % de rendimiento) en forma de un sólido de color pardo. ESl MS m/z 511,2 [M+H]+.
Etapa H: 4-[2-[2-(d¡met¡lam¡no)etox¡1-8-met¡l-5.6.7.8-tetrah¡dropry¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de tercbutilo: A una solución de 4-[7-benc¡l-2-[2-(d¡met¡lam¡no)etox¡]-8-met¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peuaz¡n-1-carboxilato de terc-butilo (800 mg, 1,57 mmol, 1,00 eq.) en MeOH (20,0 ml) se le añadió Pd/C (100 mg) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con hidrógeno. La mezcla se agitó en una atmósfera de hidrógeno (344,74 kPa (50 psi)) a 40 °C durante 12 horas. La mezcla de reacción se filtró y la torta de filtro se lavó con MeOH (3 * 200 ml). El filtrado se concentró al vacío para dar 4-[2-[2-(d¡met¡lam¡no)etox¡]-8-met¡l-5,6,7,8
tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (450 mg, 1,07 mmol, 68,2 % de rendimiento) en forma de un aceite de color pardo que se usó para la siguiente etapa sin purificación adicional. ESI MS m/z 421,3 [M+H]+.
Etapa I: 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-8-metil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo: A una mezcla de 4-[2-[2-(dimetilamino)etoxi]-8-metil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (380 mg, 904 pmol, 1,00 eq.) y 3-benciloxi-1-bromonaftaleno (311 mg, 994 pmol, 1,10 eq.) en dioxano (8,00 ml) se le añadieron Cs2CO3 (883 mg, 2,71 mmol, 3,00 eq.), 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (RuPhos) (84,3 mg, 181 pmol, 0,20 eq.) y Pd2(dba)3 (82,7 mg, 90,4 pmol, 0,10 eq.). La mezcla de reacción se agitó a 90 °C durante 12 h en una atmósfera de N2. La mezcla de reacción se concentró al vacío. El residuo se repartió entre DCM (50 ml) y agua (20 ml). La mezcla de reacción se extrajo con DCM (50 ml). Las capas orgánicas combinadas se lavaron con salmuera (10 ml), se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (éter dietílico:acetato de etilo = 5:1 después DCM:MeOH =100:1 a 5:1), seguido de purificación del producto aislado por TLC preparativa (DCM: Me 10:1) para dar 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-8-metil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (200 mg, 306 pmol, 33,9 % de rendimiento) en forma de un sólido de color pardo. ESI MS m/z 653,4 [M+H]+.
Etapa J: bis-trifluoroacetato de 4-[2-[2-(dimetilamino)etoxi]-8-metil-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]p¡r¡m¡d¡n-7-¡l)naftalen-2-ol: A una solución de 4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-8-metil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (100 mg, 177 pmol, 1,00 eq.) en DCM (130,00 μl) se le añadió TFA (202 mg, 1,78 mmol, 132 μl, 10,00 eq.). La mezcla de reacción se agitó a 20 °C durante 1 hora. El disolvente se eliminó al vacío para proporcionar bis-trifluoroacetato de 4-[2-[2-(dimetilamino)etoxi]-8-metil-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (175,00 mg) en forma de un aceite de color pardo que se usó en la siguiente etapa sin purificación adicional.
Etapa K: 1-[4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-8-metil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de bis-trifluoroacetato de 4-[2-[2-(dimetilamino)etoxi]-8-metil-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (97,4 mg, 141 pmol, 1,00 eq.) en DCM (500,00 μl) se le añadieron DIEA (222 mg, 1,72 mmol, 300 μl, 12,2 eq.) y prop-2-enoato de prop-2-enoílo (14,2 mg, 113 pmol, 0,80 eq.) a -40 °C. La mezcla de reacción se agitó a -40 °C durante 0,5 h. La mezcla de reacción se inactivó con una gota de MeOH y se concentró al vacío. El residuo se purificó por TLC preparativa (DCM:MeOH =10:1) y después por HPLC preparativa (columna: Phenomenex Synergi C18 150 * 25 *, 10 p; fase móvil: [agua (0,1 % de TFA)-a Cn ]; % de B: 15 %-45 %, 13 min) para dar 1-[4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-8-metil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (10,7 mg, 20,7 pmol, tres etapas 6,8 % de rendimiento) en forma de un aceite de color pardo. ESI MS m/z 517,2 [M+H]+.
Ejemplo 48
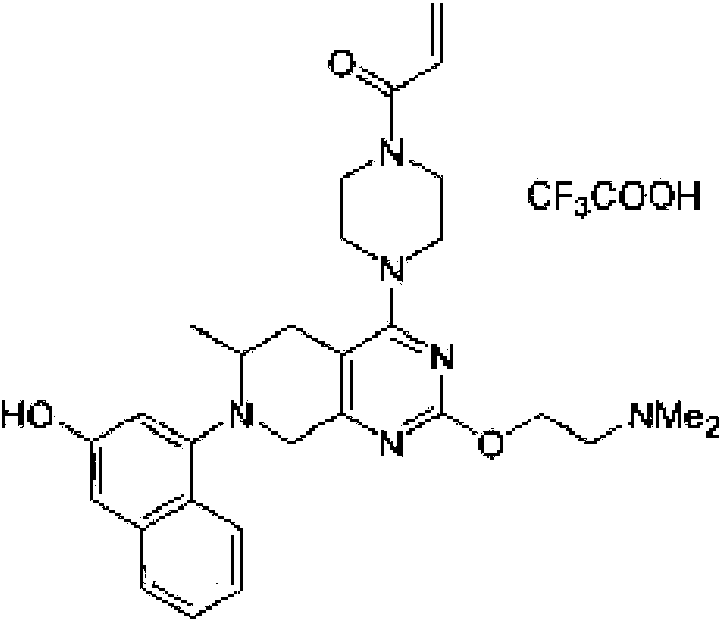
trifluoroacetato de 1-(4-(2-(2-(dimetilamino)etoxi)-7-(3-hidroxinaftalen-1-il)-6-metil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Etapa A: 4-[benc¡l-(2-etox¡-2-oxo-et¡l)am¡no]pentanoato de etilo: Una solución de 4-oxopentanoato de etilo (47,0 g, 326 mmol, 46,5 ml, 1,50 eq.), 2-(bencilamino)acetato de etilo (42,0 g, 217,3 mmol, 1,00 eq.) y AcOH (13,0 g, 217 mmol, 12,4 ml, 1,00 eq.) en dCm (800 ml) se agitó a 12-18 °C durante 30 min, después se enfrió a 0-5 °C y se añadió poco a poco NaBH(OAc)3 (138 g, 652 mmol, 3,00 eq.). La mezcla se calentó a 10-18 °C y se agitó durante 16 horas. La mezcla de reacción se inactivó con agua (1000 ml) y se extrajo con DCM (2 x 500 ml). Las fases orgánicas combinadas se secaron y se concentraron a sequedad. El residuo se purificó por columna de gel de sílice eluyendo con éter dietílico/acetato de etilo (60:1 a 40:1) para proporcionar 4-[bencil-(2-etoxi-2-oxo-etil)amino]pentanoato de etilo (42,0 g, 91,5 mmol, 42,1 % de rendimiento, 70 % de pureza) en forma de un aceite incoloro. ESI mS m/z 322,2 [M+H]+.
Etapa B: 1-benc¡l-2-metil-5-oxo-p¡per¡d¡n-4-carbox¡lato de etilo: Una solución de 4-[bencil-(2-etoxi-2-oxoetil)amino]pentanoato de etilo (37,00 g, 115,12 mmol, 1,00 eq.) y t-BuOK (16,8 g, 150 mmol, 1,30 eq.) en tolueno
(30,0 ml) se agitó a 25 °C durante 5 horas. La reacción se interrumpió con agua (50 ml) y se extrajo con acetato de etilo (2 x 30 ml). Los extractos orgánicos combinados se secaron y se concentraron a sequedad para proporcionar 1-bencil-2-metil-5-oxo-piperidin-4-carboxilato de etilo (14,6 g, 53,0 mmol, 46 % de rendimiento) en forma de un aceite de color amarillo que se usó directamente en la siguiente etapa.
Etapa C: 7-benc¡l-6-met¡l-6.8-d¡h¡dro-5H-p¡r¡dor3,4-d1p¡r¡m¡d¡n-2.4-d¡ol: Se disolvió Na (3,05 g, 133 mmol, 3,14 ml, 2,50 eq.) en EtOH (280 ml), y después se añadieron 1-bencil-2-metil-5-oxo-piperidin-4-carboxilato de etilo (14,6 g, 53,0 mmol, 1.00 eq.) y urea (7,96 g, 133 mmol, 7,11 ml, 2,50 eq.). La mezcla de reacción se calentó a reflujo (78 °C) durante 16 h en una atmósfera de N2. La mezcla de reacción se concentró a sequedad. El residuo se disolvió en agua (100 ml) y se lavó con MTBE (100 ml). El pH de la fase acuosa se ajustó a pH 6-7 con HCl 6 N (2 ml). El sólido resultante se recogió por filtración y se secó al vacío a 60 °C para proporcionar 7-bencil-6-metil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2,4-diol (5,00 g, 17,1 mmol, 32,3 % de rendimiento, 93 % de pureza) en forma de un sólido de color amarillo claro.
Etapa D: 7-benc¡l-2.4-d¡cloro-6-met¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡na: Se calentaron a reflujo POCb (49,5 g, 323 mmol, 30,0 ml, 58,4 eq.) y 7-bencil-6-metil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2,4-diol (1,50 g, 5,53 mmol, 1.00 eq.) a 100 °C durante 12 horas. La reacción se concentró a sequedad para eliminar el POCb. El residuo se disolvió en DCM (40 ml) y se lavó con NaHCO3 acuoso saturado/Na2CO3 acuoso saturado (1/1, 60 ml). La mezcla se filtró y las capas orgánicas se secaron sobre Na2SO4, se filtraron y se concentraron al vacío para dar 7-bencil-2,4-dicloro-6-metil-6,8-dihidro-5H-pirido[3,4-d]pirimidina (2,08 g, bruto) en forma de un sólido de color pardo que se usó directamente en la siguiente etapa sin purificación adicional. ESI MS m/z 307,9, 309,9 [M+H]+.
Etapa E: 4-(7-bencil-2-cloro-6-metil-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato: A una mezcla de 7-bencil-2,4-dicloro-6-metil-6,8-dihidro-5H-pirido[3,4-d]pirimidina (2,50 g, 8,11 mmol, 1,00 eq.) y piperazin-1-carboxilato de terc-butilo (1,54 g, 8,27 mmol, 1,02 eq.) en dioxano (50,0 ml) se le añadió DIEA (3,14 g, 24,3 mmol, 4,25 ml, 3,00 eq.). La mezcla se agitó a 60 °C durante 20 horas. La mezcla se concentró al vacío. El residuo se diluyó con agua (20 ml) y se extrajo con DCM (2 x 80 ml). Las capas orgánicas se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se purificó por un sistema automatizado de cromatografía ultrarrápida (éter dietílico/acetato de etilo 50:1 a 2:1) para dar 4-(7-bencil-2-cloro-6-metil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de tercbutilo (2,59 g, 5,29 mmol, 65,2 % de rendimiento, 93,5 % de pureza) en forma de un sólido de color amarillo.
Etapa F: 4-[7-benc¡l-2-[2-(d¡met¡lam¡no)etox¡]-6-met¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tercbutilo: A una solución de 2-(dimetilamino)etanol (701 mg, 7,86 mmol, 788 μl, 3,00 eq.) en THF (45,0 ml) se le añadió NaH (210 mg, 5,24 mmol, 60,0 % de pureza, 2,00 eq.) a 15 °C en una atmósfera de N2. Después de agitar a 15 °C durante 0,5 horas, se añadió 4-(7-bencil-2-cloro-6-metil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de terc-butilo (1,20 g, 2,62 mmol, 1,00 eq.). La mezcla se agitó a 110 °C durante 18 horas en un tubo sellado. La mezcla se concentró al vacío. El residuo se purificó por cromatografía en columna sobre AbO3 (DCM/MeOH 100:1 a 10:1) para dar 4-[7-bencil-2-[2-(d¡met¡lam¡no)etox¡]-6-met¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡mid¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de terc-butilo (1,38 g, 2,36 mmol, 90,3 % de rendimiento, 87,5 % de pureza) en forma de un aceite de color amarillo. ESI MS m/z 511,3 [M+H]+.
Etapa G: 4-[2-[2-(d¡met¡lam¡no)etox¡1-6-met¡l-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de terc-butilo: A una solución de 4-[7-benc¡l-2-[2-(d¡met¡lam¡no)etox¡]-6-met¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]pir¡m¡d¡n-4-il]piperazin-1-carboxilato de terc-butilo (1,50 g, 2,94 mmol, 1,00 eq.) en MeOH (80,0 ml) se le añadió Pd/C (450 mg) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó con 4 veces hidrógeno. La mezcla se agitó en una atmósfera de hidrógeno (344,74 kPa (50 psi)) a 40 °C durante 12 horas. La mezcla de reacción se filtró y el filtrado se concentró. El residuo se purificó por cromatografía en columna (DCM/MeOH 40/1 a 10/1) para dar 4-[2-[2-(d¡met¡lam¡no)etox¡]-6-met¡l-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]piperaz¡n-1-carbox¡lato de tercbutilo (1,15 g, 2,46 mmol, 83,7 % de rendimiento, 90,0 % de pureza) en forma de un aceite de color amarillo.
Etapa H: 4-[7-(3-benc¡lox¡-1-naft¡l)-2-[2-(d¡met¡lam¡no)etox¡]-6-met¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]pir¡m¡d¡n-4-il]piperazin-1-carboxilato de terc-butilo: Se añadió Pd2(dba)3 (109 mg, 119 pmol, 0,10 eq.) a una solución de 4-[2-[2-(d¡met¡lam¡no)etox¡]-6-met¡l-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]piperaz¡n-1-carbox¡lato de terc-butilo (500 mg, 1,19 mmol, 1,00 eq.), 3-benciloxi-1-bromo-naftaleno (410 mg, 1,31 mmol, 1,10 eq.), 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (RuPhos) (111 mg, 238 pmol, 0,20 eq.) y Cs2CO3 (1,16 g, 3,57 mmol, 3,00 eq.) en dioxano (10,0 ml). La mezcla de reacción se agitó a 90 °C durante 12 horas en una atmósfera de N2. La mezcla se concentró al vacío. El residuo se diluyó con agua (5 ml) y se extrajo con DCM (2 x 20 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna sobre AbO3 (DCM/MeOH 100/1 a 10/1) y por TLC preparativa (DCM/MeOH 5:1) para dar 4-[7-(3-benciloxi-1 -naftil)-2-[2-(dimetilamino)etoxi]-6-metil-6,8-dihidro-5W-pirido[3,4-d1pirimidin-4-il1piperazin-1 -carboxilato de tercbutilo (309 mg, 453 pmol, 38,1 % de rendimiento, 95,8 % de pureza) en forma de un aceite de color amarillo.
Etapa I: 4-[2-[2-(d¡met¡lam¡no)etox¡]-7-(3-h¡drox¡-1-naft¡l)-6-met¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]pir¡m¡d¡n-4-¡l]p¡peraz¡n-1-carboxilato de terc-butilo: A una solución de 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6-metil-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1 -carboxilato de terc-butilo (340 mg, 521 pmol, 1,00 eq.) en MeOH (6,80 ml) se le añadió Pd/C (120 mg) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó con 4 veces hidrógeno. La mezcla se agitó en una atmósfera de hidrógeno (103,42 kPa (15 psi)) a 40 °C durante 1,5 horas. La mezcla se concentró al vacío para dar 4-[2-[2-(d¡met¡lam¡no)etox¡j-7-(3-h¡drox¡-1-naft¡l)-6-met¡l-6,8-d¡h¡dro-5H-pir¡do[3,4-d]p¡r¡m¡d¡n4-il]piperazin-1-carboxilato de terc-butilo (210 mg, 312 pmol, 59,8 % de rendimiento, 83,5 % de pureza) en forma de un sólido de color rosa que se usó directamente en la siguiente etapa sin purificación adicional. ESI MS m/z 563,3 [M+H]+.
Etapa J: trifluoroacetato de 4-[2-[2-(dimetilamino)etoxi]-6-metil-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol: A una solución de 4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6-metil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (180 mg, 320 pmol, 1,00 eq.) en DCM (240 μl) se le añadió TFA (365 mg, 3,20 mmol, 237 μl, 10,0 eq.). La mezcla se agitó a 18 °C durante 0,5 horas. La mezcla se concentró al vacío para dar trifluoroacetato de 4-[2-[2-(dimetilamino)etoxi]-6-metil-4-piperazin-1-il-6,8-dihidro-5H pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (273 mg) en forma de un aceite de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional.
Etapa K: 1-[4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6-metil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de trifluoroacetato de 4-[2-[2-(dimetilamino)etoxi]-6-metil-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (148 mg, 320 pmol, 1,00 eq.) y DIEA (494 mg, 3,83 mmol, 668 μl, 12,0 eq.) en DCM (500 μl) se le añadió gota a gota prop-2-enoato de prop-2-enoílo (32,3 mg, 256 pmol, 0,80 eq.) a -50 °C. La mezcla se agitó de -40 a -20 °C durante 30 minutos. La reacción se interrumpió con MeOH (20,5 mg) y se concentró al vacío. El residuo se purificó por HPLC preparativa (columna: Phenomenex Synergi C18 150 * 25 *, 10 p; fase móvil: [agua (0,1 % de TFA)-a Cn ]; % de B: 8 %-38 %,13 min) para dar trifluoroacetato de 1-[4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6-metil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (73,7 mg, 113 pmol, 35,2 % de rendimiento, 96,3 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 517,3 [M+H]+.
Ejemplo 49
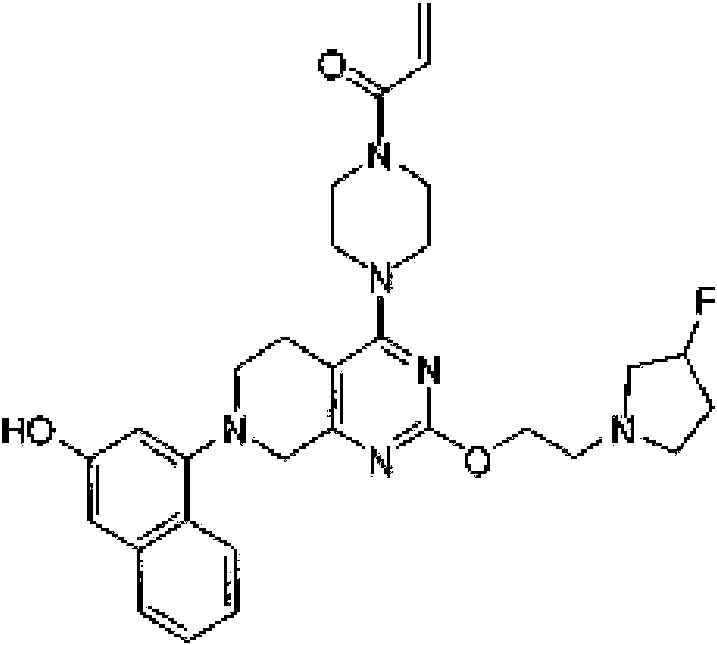
1-(4-(2-(2-(3-fluoropirrolidin-1-il)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Etapa A: 4-(4-benciloxicarbonilpiperazin-1-il)-2-[2-(3-fluoropirrolidin-1-il)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de terc-butilo: Una mezcla de 2-(3-fluoropirrolidin-1-il)etanol (401 mg, 3,01 mmol, 1,60 eq.), 4-(4-benciloxicarbonilpiperazin-1-il)-2-metilsulfonil- 6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (1,00 g, 1,88 mmol, 1,00 eq.), Cs2COa (1,84 g, 5,64 mmol, 3,00 eq.), Pd(OAc)2 (42,2 mg, 188 pmol, 0,10 eq.) y BINAP (234 mg, 376 pmol, 0,20 eq.) en tolueno (50,00 ml) se agitó a 110 °C durante 3 horas. La mezcla se concentró al vacío. El residuo se purificó por cromatografía de columna sobre gel de sílice (éter de petróleo/acetato de etilo 10/1 a 100/1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar 4-(4-benciloxicarbonilpiperazin-1-il)-2-[2-(3-fluroropirrolidin-1 -il)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (1,20 g, 1,07 mmol, 56,8 % de rendimiento, 52 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 585,3 [M+H]+.
Etapa B: 4-[2-[2-(3-fluorop¡rrol¡d¡n-1-¡l)etox¡1-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo: A una solución de 4-(4-benciloxicarbonilpiperazin-1-il)-2-[2-(3-fluoropirrolidin-1-il)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de terc-butilo (1,20 g, 2,05 mmol, 1,00 eq.) en DCM (10,00 ml) se le añadió TFA (3,51 g, 30,8 mmol, 2,28 ml, 15,00 eq.) a 0 °C. La mezcla se calentó a 25 °C y se agitó durante 16 horas. La mezcla se diluyó con agua (100 ml) y la solución se extrajo con acetato de etilo (2 x 100 ml). La capa de agua se basificó con solución acuosa saturada de carbonato de sodio (50 ml) y después se extrajo con acetato de etilo (2 x 100 ml). Las capas orgánicas combinadas se lavaron con salmuera (1 x 100 ml), se secaron sobre sulfato de sodio y se filtraron. El filtrado se concentró al vacío para proporcionar 4-[2-[2-(3-fluoropirrolidin-1-il)etoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (800 mg, 1,65 mmol, 80,4 % de rendimiento) en forma de una goma de color amarillo. ESI MS m/z 485,3 [M+H]+.
Etapa C: 4-[2-[2-(3-fluoropirrolidin-1-il)etoxi]-7-(3-metoxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: Una mezcla de 4-[2-[2-(3-fluoropirrolidin-1-il)etoxi]- 5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (750 mg, 1,55 mmol, 1,00 eq.), trifluorometanosulfonato de (3-metoxi-1-naftilo) (949 mg, 3,10 mmol, 2,00 eq.), 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (RuPhos) (144,66 mg, 310,00 pmol, 0,20 eq.), Pd2(dba)3 (141,94 mg, 155,00 pmol, 0,10 eq.) y Cs2CO3 (1,52 g, 4,65 mmol, 3,00 eq.) en tolueno (70,00 ml) se agitó a 110 °C durante
16 horas. La mezcla se concentró al vacío y después se diluyó con acetato de etilo (100 ml) y agua (100 ml). La capa orgánica separada se lavó con salmuera (1 * 100 ml), se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo/acetato de etilo 10:1 a acetato de etilo/metanol 10:1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar 4-[2-[2-(3-fluoropirrolidin-1-il)etoxi]-7-(3-metoxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (700 mg, 961 pmol, 62,0 % de rendimiento, 88 % de pureza) en forma de un sólido de color pardo. ESI MS m/z 641,3 [M+h ]+.
Etapa D: 2-[2-(3-fluorop¡rrol¡d¡n-1-¡l)etox¡1-7-(3-metox¡-1-naft¡l)-4-p¡peraz¡n-1-¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡na: Una mezcla de 4-[2-[2-(3-fluoropirrolidin-1-il)etoxi]-7-(3-metoxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (700,00 mg, 1,09 mmol, 1,00 eq.) en MeOH se hidrogenó (103,42 kPa (15 psi)) a 40 °C con Pd/C seco (140 mg) como catalizador durante 4 horas. El catalizador se filtró a través de una tira de Celite® y el filtrado se concentró al vacío para proporcionar 2-[2-(3-fluoropirrolidin-1-il)etoxi]-7-(3-metoxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidina (500 mg, 987 pmol, 90,6 % de rendimiento) en forma de un sólido de color pardo.
Etapa E: 1 -[4-[2-[2-(3-fluoropirrolidin-1 -il)etoxi]-7-(3-metoxi-1 -naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una mezcla de 2-[2-(3-fluoropirrolidin-1-il)etoxi]-7-(3-metoxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidina (500 mg, 987 pmol, 1,00 eq.) y DIEA (255 mg, 1,97 pmol, 345 μl, 2,00 eq.) en diclorometano (8 ml) se le añadió una solución de prop-2-enoato de prop-2-enoílo (124 mg, 987 pmol, 1,00 eq.) en diclorometano (2 ml) a -40 °C en una atmósfera de nitrógeno. La mezcla se calentó a 25 °C y se agitó durante 1 hora. La mezcla se diluyó con agua (20 ml) y diclorometano (30 ml). La capa orgánica separada se lavó con salmuera (1 * 30 ml), se secó sobre sulfato de magnesio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (diclorometano/metanol 100/1 a 10/1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar 1-[4-[2-[2-(3-fluoropirrolidin-1-il)etoxi]-7-(3-metoxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (450 mg, 714 pmol, 72,4 % de rendimiento, 89 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 561,2 [M+H]+.
Etapa F: 1-[4-[2-[2-(3-fluoropirrolidin-1-il)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-¡l]prop-2-en-1-ona: A una solución de 1-[4-[2-[2-(3-fluoropirrolidin-1-il) etoxi]-7-(3-metoxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (400 mg, 713 pmol, 1,00 eq.) en DCM (2,00 ml) se le añadió BBr3 (1,30 g, 5,19 mmol, 500 μl, 7,27 eq.) a -70 °C en una atmósfera de nitrógeno. La mezcla se calentó a 0 °C y se agitó durante 1 hora. La mezcla se diluyó con diclorometano (20 ml), y después se inactivó con una solución acuosa saturada de bicarbonato de sodio (20 ml). La capa orgánica separada se lavó con salmuera (1 * 10 ml), se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por HPLC preparativa (Phenomenex Synergi C18 150 * 25 *, 10 p; fase móvil: [agua. (0,1 % de TFA)-ACN]; % de B: 18 %-48 %,12 min.) Las fracciones deseadas se recogieron y se liofilizaron para dar 1-[4-[2-[2-(3-fluoropirrolidin-1-il)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (40,0 mg, 63,7 pmol, 8,92 % de rendimiento, 87 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 547,3 [M+H]+.
Ejemplo 50

1-(4-(2-(2-(dimetilamino)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(hidroximetil)piperazin-1-il)prop-2-en-1-ona
Etapa A: 2-met¡l-4-(7-benc¡l-2-cloro-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1.2-d¡carbox¡lato de 1-tercbutilo: A una solución de 7-bencil-2,4-dicloro-6,8-dihidro-5H-pirido[3,4-d]pirimidina (2,00 g, 6,80 mmol, 1,00 eq.) en DMSO (40,0 ml) se le añadieron DIEA (1,76 g, 13,6 mmol, 2,38 ml, 2,00 eq.) y 2-metilpiperazin-1,2-dicarboxilato de 1-terc-butilo (1,74 g, 7,14 mmol, 1,05 eq.). La mezcla se agitó a 55 °C durante 16 horas. La mezcla se diluyó con agua (100 ml) y se extrajo con acetato de etilo (3 * 100 ml). Las capas orgánicas combinadas se lavaron con salmuera (3 * 100 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a sequedad. El residuo se purificó por cromatografía en columna (SiO2, éter dietílico/acetato de etilo = 1:0 a 3:1) para dar 2-metil-4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-1,2-dicarboxilato de 1-terc-butilo (3,10 g, 5,70 mmol, 83,8 % de rendimiento, 92,3 % de pureza) en forma de un semisólido de color amarillo. ESI MS m/z 502,2 [M+H]+.
Etapa B: 2-metil-4-[7-bencil-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1,2-dicarboxilato de 1-terc-butilo: Una mezcla de 2-metil-4-(7-bencil-2-cloro-6,8-dihidro -5H-pirido[3,4-d]pirimidin-4-il)piperazin-1,2-dicarboxilato de 1-terc-butilo (3,00 g, 5,98 mmol, 1,00 eq.), 2-(dimetilamino)etanol (1,07 g, 12,0 mmol, 1,20 ml, 2,00 eq.), Cs2CO3 (4,87 g, 15,0 mmol, 2,50 eq.), Pd(OAc)2 (201 mg, 897 pmol, 0,15 eq.) y BINAP (744 mg, 1,20 mmol, 0,20 eq.) en tolueno (60,0 ml) se desgasificó y se purgó 3 veces con nitrógeno y después la mezcla se agitó a 110 °C durante 3 horas en una atmósfera de nitrógeno. La mezcla de reacción se diluyó con agua (50 ml) y se extrajo con acetato de etilo (3 x 50 ml). Las capas orgánicas combinadas se lavaron con salmuera (3 x 50 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a sequedad. El residuo se purificó por cromatografía en columna (SO2, DCM/MeOH = 1:0 a 5:1) para dar 2-metil-4-[7-bencil-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1,2-dicarboxilato de 1-terc-butilo (3,10 g, 4,45 mmol, 80,4 % de rendimiento, 86,0 % de pureza) en forma de un aceite de color negro. ESI MS m/z 555,3 [M+H]+.
Etapa C: 2-met¡l-4-[2-[2-(d¡met¡lam¡no)etox¡1-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1.2-d¡carbox¡lato de 1-terc-butilo: A una solución de 2-metil-4-[7-bencil-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1,2-dicarboxilato de 1-terc-butilo (2,33 g, 4,20 mmol, 1,00 eq.) en MeOH (50,0 ml) se le añadió Pd/C (233 mg) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con hidrógeno. La mezcla se agitó en una atmósfera de hidrógeno (344,74 kPa (50 psi)) a 40 °C durante 36 horas. La mezcla de reacción se filtró y la fase orgánica se concentró a sequedad para dar 2-metil-4-[2-[2-(dimetilamino)etoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1,2-dicarboxilato de 1-terc-butilo (1,50 g, bruto) en forma de un aceite incoloro, que se usó directamente en la siguiente etapa sin purificación adicional.
Etapa D: 2-metil-4-[7-(3-benciloxi-1 -naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin^-il]piperazin-1,2-dicarboxilato de 1-terc-butilo: Una mezcla de 2-metil-4-[2-[2-(dimetilamino)etoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1,2-dicarboxilato de 1-terc-butilo (1,50 g, bruto), 3-benciloxi-1-bromo-naftaleno (1,49 g, 4,76 mmol, 1,30 eq.), Cs2COa (2,98 g, 9,15 mmol, 2,50 eq.), Pd2(dba)3 (503 mg, 549 pmol, 0,15 eq.) y 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (RuPhos) (342 mg, 732 pmol, 0,20 eq.) en dioxano (100 ml) se desgasificó y se purgó 3 veces con nitrógeno y la mezcla se agitó a 85 °C durante 5 horas en una atmósfera de nitrógeno. La mezcla de reacción se inactivó mediante la adición de agua (50 ml) a 0 °C y se extrajo con DCM (3 x 100 ml). Las capas orgánicas combinadas se lavaron con salmuera (3 x 150 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida a sequedad. El residuo se purificó por cromatografía en columna (SO2, DCM/MeOH = 10:1 a 5:1) para dar 2-metil-4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1,2-dicarboxilato de 1-terc-butilo (1,72 g, 2,25 mmol, 53,5 % de rendimiento, 91 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 697,3 [M+H]+.
Etapa E: 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-carboxilato de metilo: A una solución de 2-metil-4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-1,2-dicarboxilato de 1-terc-butilo (1,32 g, 1,89 mmol, 1,00 eq.) en DCM (20,0 ml) se le añadió TfA (4,31 g, 37,8 mmol, 2,80 ml, 20,0 eq.) a 0 °C y la mezcla de reacción se agitó durante 2 horas a 25 °C. La mezcla de reacción se concentró a presión reducida a sequedad. El residuo se disolvió en DCM (50 ml) y H2O (20 ml) y la mezcla de reacción se ajustó a pH 8 con NaHCO3 saturado. La capa orgánica se secó sobre Na2SO4, se filtró y se concentró a presión reducida para dar 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-carboxilato de metilo (1,12 g, 1,87 mmol, 69,6 % de rendimiento, 70 % de pureza) en forma de un aceite de color pardo, que se usó directamente en la siguiente etapa sin purificación adicional. ESI MS m/z 597,4 [M+H]+.
Etapa F: [4-[7-(3-benc¡lox¡-1-naft¡l)-2-[2-(d¡met¡lam¡no)etox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-il]metanol: A una mezcla de 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-carboxilato de metilo (400 mg, 0,670 mmol) en THF (5,00 ml) se le añadió L¡AlH4 (102 mg, 2,68 mmol, 4,00 eq.) a 0 °C. La mezcla de reacción se desgasificó y se purgó 3 veces con nitrógeno y se agitó a 0 °C durante 2 horas en una atmósfera de nitrógeno. La mezcla de reacción se inactivó mediante la adición de Na2SO4-10 H2O (0,5 g) a 0 °C y después se diluyó con DCM (50 ml). Las capas orgánicas combinadas se filtraron, se secaron sobre Na2SO4 y se concentraron a presión reducida para dar [4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-piridoÍ3,4-d]pirimidin-4-il]piperazin-2-il]metanol (280 mg, 0,493 mmol, 73,6 % de rendimiento) en forma de un semisólido de color amarillo, que se usó directamente en la siguiente etapa sin purificación adicional. ESI MS m/z 569,3 [M+H]+.
Etapa G: 2-[[7-(3-benciloxi-1-naftil)-4-[3-[[terc-butil(dimetil)silil]oximetil]piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]-N,N-dimetil-etanamina: A una solución agitada de [4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]metanol (250 mg, 440 pmol, 1,00 eq.) y NaH (176 mg, 4,40 mmol, 60,0 % de pureza, 10,0 eq.) en THF (10,0 ml) se le añadió cloruro de terc-butildimetilsililo (232 mg, 1,54 mmol, 189 μl, 3,50 eq.) a 0 °C. La mezcla se agitó a 25 °C durante 12 horas en una atmósfera de nitrógeno. La mezcla de reacción se inactivó mediante la adición de agua (25 ml) a 0 °C y se extrajo con DCM (3 x 30 ml). Las capas orgánicas combinadas se lavaron con salmuera (3 x 30 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida a sequedad. El residuo se purificó por TLC preparativa (SO2, DCM/MeOH = 10:1) para dar 2-[[7-(3-benciloxi-1-naftil)-4-[3-[[tercbutil(dimetil)silil]oximetil]piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]-N,N-dimetil-etanamina (128 mg, 180 pmol, 40,9 % de rendimiento, 96,0 % de pureza) en forma de un semisólido incoloro. ESI MS m/z 683,3 [M+H]+.
Etapa H: 1-[4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino) etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-[[tercbutil(dimetil)silil]oximetil]piperazin-1-il]prop-2-en-1-ona; A una solución de 2-[[7-(3-benciloxi-1-naftil)-4-[3-[[tercbutil(dimetil)silil]oximetil]piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]-N,N-dimetil-etanamina (128 mg, 187 pmol, 1,00 eq.) y trietilamina (47,4 mg, 469 pmol, 65,0 μl, 2,50 eq.) en DCM (5,00 ml) se le añadió gota a gota prop-2-enoato de prop-2-enoílo (35,5 mg, 281 pmol, 1,50 eq.) a -40 °C y se agitó durante 30 minutos. La mezcla de reacción se concentró a presión reducida a sequedad. El residuo se purificó por TLC preparativa (SiÜ2, DCM/MeOH = 10:1) para dar 1-[4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino) etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-[[terc-butil(dimetil)silil]oximetil]piperazin-1-il]prop-2-en-1-ona (92,0 mg, 101 pmol, 54,0 % de rendimiento, 81,0 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 737,3 [M+H]+.
Etapa I: 1-[4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(hidroximetil)piperazin-1-il]prop-2-en-1-ona: A una mezcla de 1-[4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino) etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-2-[[terc-butil(dimetil)silil]oximetil]piperazin-1-il]prop-2-en-1-ona (92,0 mg, 125 pmol, 1.00 eq.) en Dc M (8,00 ml) se le añadió BBr3 (17,0 mg, 67,8 pmol, 6,53 μl, 10,0 eq.). La mezcla se agitó a -40 °C durante 30 minutos en una atmósfera de nitrógeno y después se concentró a 25 °C a presión reducida a sequedad. Al residuo se le añadió NaHCO3 acuoso saturado (0,5 ml). La solución se ajustó a pH 7 a 0 °C y después se añadió MeOH (2,0 ml). La solución se purificó por HPLC preparativa (columna: Phenomenex Synergi C18150 * 25 *, 10 p; fase móvil: [agua (0,1 % de TFA) - ACN]; % de B: 15 % - 45 %, 11 min). Las fracciones deseadas se recogieron y se concentraron a presión reducida para eliminar el MeCN y se liofilizaron para dar 1-[4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(hidroximetil)piperazin-1-il]prop-2-en-1-ona (6,30 mg, 11,2 pmol, 9,00 % de rendimiento, 95.0 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 533,2 [M+h ]+.
Ejemplo 51

1-acriloil-4-(2-(2-(dimetilamino)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-N,N-dimetilpiperazin-2-carboxamida
Etapa A: ácido 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-tercbutoxicarbonil-piperazin-2-carboxílico: A una solución de 2-metil-4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1,2-dicarboxilato de 1 -terc-butilo (300 mg, 431 pmol, 1,00 eq.) en THF (4,00 ml) y H2O (1,00 ml) se le añadió NaOH (68,9 mg, 1,72 mmol, 4,00 eq.). La mezcla se agitó a 25 °C durante 12 horas en una atmósfera de nitrógeno. La mezcla de reacción se diluyó con agua (50 ml) y la mezcla de reacción se ajustó a pH 6 con HCl (6 M) y después se extrajo con DCM (3 x 50 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida a sequedad para dar ácido 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido(3,4-d]pirimidin-4-il]-1-terc-butoxicarbonil-piperazin-2-carboxílico (310 mg, bruto) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. ESI MS m/z 683,3 [M+H]+.
Etapa B: 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(dimetilcarbamoil)piperazin-1-carboxilato de terc-butilo A una mezcla de ácido 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-terc-butoxicarbonil-piperazin-2-carboxílico (310 mg, bruto) y DIEA (352 mg, 2,72 mmol, 476 μl) en Dc M (10,0 ml) se le añadió poco a poco HATU (259 mg, 681 μmol) a 0 °C. Después de agitar durante 30 minutos, se añadió en una porción N-metilmetanamina (130 mg, 1,59 mmol, 146 μl, HCl). La mezcla de reacción se desgasificó y se purgó 3 veces con nitrógeno. Después de agitar a 25 °C durante 12 horas en una atmósfera de nitrógeno, la mezcla de reacción se diluyó con agua (50 ml) a 0 °C y se extrajo con DCM (3 x 50 ml). Las capas orgánicas combinadas se ajustaron a pH 6 con HCl (1 M), se lavaron con salmuera (3 x 50 ml) y agua (3 x 50 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida a sequedad para dar 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(dimetilcarbamoil)piperazin-1-carboxilato de terc-butilo (345 mg, 428 pmol, dos etapas 94,2 % de rendimiento, 88,0 % de pureza) en forma de un semi-sólido de color amarillo. ESI MS m/z 710,3 [M+H]+.
Etapa C: 4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(dimetilcarbamoil)piperazin-1-carboxilato de terc-butilo: A una solución de 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(dimetilcarbamoil)piperazin-1-carboxilato de tercbutilo (345 mg, 486 pmol, 1,00 eq.) en MeOH (5,00 ml) se le añadió Pd/C (80,0 mg) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con hidrógeno. La mezcla se agitó en una atmósfera de hidrógeno (103,42 kPa (15 psi)) a 25 °C durante 8 horas. La mezcla de reacción se filtró y el filtrado se concentró a presión reducida a sequedad para dar 4-[2-[2-(dimetilamino)etoxi1-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(dimetilcarbamoil)piperazin-1-carboxilato de terc-butilo (197 mg, bruto) en forma de un sólido de color pardo, que se usó directamente en la siguiente etapa sin purificación adicional. ESI MS m/z 620,3 [M+H]+.
Etapa D: 4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-N,N-dimetil-piperazin-2-carboxamida: A una mezcla de 4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(dimetilcarbamoil)piperazin-1-carboxilato de terc-butilo (197 mg, bruto) en DCM (5,00 ml) se le añadió HCl/dioxano (4 M, 1,61 ml) a 0 °C. La mezcla de reacción se desgasificó y se purgó 3 veces con nitrógeno y se agitó a 25 °C durante 2 horas en una atmósfera de nitrógeno. La mezcla de reacción se concentró a presión reducida a sequedad. El residuo se purificó por HPLC preparativa (columna: Phenomenex Synergi C18 150 * 25 *, 10 p; fase móvil: [agua (0,05 % de HCl) - ACN]; % de B: 10 % - 30 %, 7,8 min). La fase acuosa recogida se liofilizó a sequedad para dar 4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-N,N-dimetil-piperazin-2-carboxamida (90,0 mg, 171 pmol, 53,1 % de rendimiento, 99,0 % de pureza) en forma de un sólido de color pardo. ESI MS m/z 520,2 [M+H]+.
Etapa E: 4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-N,N-dimetil-1-prop-2-enoil-piperazin-2-carboxamida: A una solución de 4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-N,N-dimetil-piperazin-2-carboxamida (70,0 mg, 135 pmol, 1,00 eq.) en DMAC (500 μl) se le añadió trietilamina (40,9 mg, 404 pmol, 56,0 μl, 3,00 eq.) a 0 °C y después se le añadió prop-2-enoato de prop-2-enoílo (2,55 mg, 20,2 pmol, 0,15 eq.). La mezcla se agitó a 0 °C durante 2 horas en una atmósfera de nitrógeno. La mezcla de reacción se filtró y la fase orgánica recogida se purificó por HPLC preparativa (columna: Phenomenex Synergi C18 150 * 25 *, 10 p; fase móvil: [agua (0,1 % de TFA)-ACN]; % de B: 15 % - 45 %, 12 min). La fase acuosa recogida se liofilizó para dar 4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-N,N-dimetil-1-prop-2-enoil-piperazin-2-carboxamida (10,0 mg, 16,5 pmol, 12,2 % de rendimiento, 94,6 % de pureza) en forma de un semisólido de color amarillo. ESI MS m/z 574,2 [M+H]+.
Ejemplo 52

1-[4-[7-(3-hidroxi-1-naftil)-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona
Etapa A: 4-[7-benc¡l-2-[2-(1-p¡per¡d¡l)etox¡1-6,8-d¡h¡dro- 5H-p¡r¡do[3.4-d1p¡r¡mid¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de terc-butilo: A una solución de 2-(1-piperidil)etanol (872 mg, 6,75 mmol, 899 μl, 3,00 eq.) en tolueno (40,0 ml) se le añadieron Pd(OAc)2 (50,5 mg, 225 pmol, 0,10 eq.), Cs2CO3 (2,20 g, 6,75 mmol, 3,00 eq.), BlNAP (280 mg, 450 pmol, 0,20 eq.) y 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de terc-butilo (1,00 g, 2,25 mmol, 1,00 eq.). La mezcla se agitó a 110 °C durante 12 horas en una atmósfera de N2 y después se concentró al vacío. El residuo se diluyó con agua (10,0 ml), se extrajo con acetato de etilo (3 * 20 ml), se lavó con salmuera (1 * 50 ml), se secó sobre Na2SO4, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna (S¡O2, DCM /MeOH = 10:1) para dar 4-[7-benc¡l-2-[2-(1-p¡per¡dil)etox¡]-6,8-d¡h¡dro- 5H-pirido[3,4-d]pirimidin-4-il1piperazin-1 -carboxilato de terc-butilo (700 mg, 1,30 mmol, 58,0 % de rendimiento) en forma de un sólido de color rojo. eS i MS m/z 537,3 [M+H1+.
Etapa B: 4-[2-[2-(1-p¡per¡d¡l)etox¡1-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de terc-butilo: A una mezcla de 4-[7-bencil-2-[2-(1-piperidil)etoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de terc-butilo (600 mg, 1,12 mmol, 1,00 eq.) en MeOH (50,0 ml) se le añadió Pd/C (67,2 mg, 10 %). La mezcla se agitó a 40 °C durante 24 horas a 344,74 kPa (50 psi) en una atmósfera de H2. La mezcla se filtró y se concentró al vacío para dar 4-[2-[2-(1-piperidil)etoxi1-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de terc-butilo (500 mg, bruto) en forma de un aceite de color pardo y se usó en la siguiente etapa sin purificación adicional. ESI MS m/z 447,3 [M+H1+.
Etapa C: 4-[7-(3-metoxi-1-naftil)-2-[2-(1-piperidil)etoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de terc-butilo: A una solución de 4-[2-[2-(1-piperidil)etox¡1-5,6,7,8- tetrahidropirido[3,4-d1pirimidin-4il]piperazin-1 -carboxilato de terc-butilo (250 mg) y trifluorometanosulfonato de (3-metoxi-1-naftilo) (343 mg, 1,12 mmol) en tolueno (6,00 ml) se le añadieron 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (RuPhos) (52,3 mg, 112 |jmol), Pd2(dba)3 (32,19 mg, 55,98 jm o l) y Cs2CO3 (548 mg, 1,68 mmol). Después de agitar a 110 °C durante 72 horas en una atmósfera de N2, la mezcla se concentró al vacío, se diluyó con agua (20,0 ml), se extrajo con acetato de etilo (3 x 30,0 ml), se lavó con salmuera (1 x 50,0 ml), se secó sobre Na2SO4, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna (SiO2, DCM/MeOH = 10:1) para dar 4-[7-(3-metoxi-1-naftil)-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (140 mg, 232 jmol, dos etapas, 41,5 % de rendimiento) en forma de un sólido de color amarillo. ESI MS m/z 603,3 [M+H]+.
Etapa D: 7-(3-metox¡-1-naft¡l)-4-p¡peraz¡n-1-¡l-2-[2-(1-p¡per¡d¡l)etox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡na: Una mezcla de 4-[7-(3-metoxi-1-naftil)-2-[2-(1-piperidil) etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo (120 mg, 199 jmol, 1,00 eq.) y TFA (340 mg, 2,99 mmol, 221 jl, 15,0 eq.) en DCM (1,00 ml) se agitó a 25 °C durante 1 hora. La mezcla se concentró al vacío para dar bis-trifluoroacetato de 7-(3-metoxi-1-naftil)-4-piperazin-1-il-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidina (145 mg, bruto) en forma de un sólido de color amarillo que se usó en la siguiente etapa sin purificación adicional. ESI MS m/z 503,3 [M+H]+.
Etapa E: 1-[4-[7-(3-metoxi-1-naftil)-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de bis-trifluoroacetato de 7-(3-metoxi-1-naftil)-4-piperazin-1-il-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidina (145 mg, bruto) y ELbN (221 mg, 2,18 mmol, 303 j l) en DCM (2,00 ml) se le añadió prop-2-enoato de prop-2-enoílo (25,0 mg, 198 jmol) a -78 °C. Después de agitar a 0 °C durante 0,5 h, la mezcla se inactivó con MeOH y se concentró al vacío. La mezcla se purificó por cromatografía en columna (S¡O2, DCM/MeOH = 10:1) para dar 1-[4-[7-(3-metoxi-1-naplithil)-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (90,0 mg, 162 jmol, dos etapas 81,5 % de rendimiento) en forma de un sólido de color amarillo. ESI MS m/z 557,3 [M+H]+.
Etapa F: 1-[4-[7-(3-hidroxi-1-naftil)-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de 1-[4-[7-(3-metoxi-1-naftil)-2-[2-(1-piperidil) etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (90,0 mg, 162 jm ol, 1,00 eq.) en DCM (2,00 ml) se le añadió BBr3 (405 mg, 1,62 mmol, 156 jl, 10,0 eq.) a -78 °C. Después de agitar a 0 °C durante 1 h, la mezcla se inactivó con una solución saturada de bicarbonato de sodio a -78 °C y se agitó a 0 °C durante 0,5 h. La mezcla se extrajo con acetato de etilo (3 x 20,0 ml), se lavó con salmuera (1 x 40,0 ml), se secó sobre Na2SO4, se filtró y se concentró al vacío. El residuo se purificó por HPLC preparativa (Phenomenex Gemini 150 * 25 mm *, 10 j ; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN]; % de B: 55 %-85 %,10 min) para dar 1-[4-[7-(3-hidroxi-1-naftil)-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (6,19 mg, 10,8 jmol, 6,71 % de rendimiento, 95 % de pureza) en forma de un aceite de color amarillo. ESI MS m/z 543,2 [M+H]+.
Ejemplo 53

1-(4-(2-(2-(dimetilamino)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-3-(hidroximetil)piperazin-1-il)prop-2-en-1-ona
Etapa A: 3-met¡l-4-(7-benc¡l-2-cloro-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1.3-d¡carbox¡lato de 1-tercbutilo: Una mezcla de 7-bencil-2,4-dicloro-6,8-dihidro-5H-pirido[3,4-d]pirimidina (3,00 g, 10,2 mmol, 1,00 eq.), piperazin-1,3-dicarboxilato de 1-terc-butil-3-metilo (2,62 g, 10,7 mmol, 1,05 eq.), DiEa (3,30 g, 25,5 mmol, 4,45 ml, 2,50 eq.) en DMSO (50,0 ml) se desgasificó y se purgó 3 veces con nitrógeno. La mezcla se agitó a 100 °C durante 12 horas en una atmósfera de nitrógeno. La mezcla de reacción se diluyó con DCM (200 ml), se lavó con salmuera (3 x 50 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida a sequedad. El residuo se purificó por cromatografía en columna (S¡O2, éter de petróleo/acetato de etilo = 10:1 a 3:1) para dar 3-metil-4-(7-bencil-2-cloro-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1,3-dicarboxilato de 1 -terc-butilo (2,10 g, 3,81 mmol, 37,4 % de rendimiento, 91,0 % de pureza) en forma de un aceite de color amarillo. ESI MS m/z 502,1 [M+H]+.
Etapa B: 3-metil-4-(7-bencil-2-(2-(dimetilamino)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1,3-dicarboxilato de 1 -terc-butilo: Una mezcla de 3-metil-4-(7-bencil-2-cloro-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1,3-dicarboxilato de 1 -terc-butilo (2,10 g, 4,17 mmol, 1,00 eq.), 2-(dimetilamino)etanol (929 mg, 10,4 mmol, 1,04 ml, 2,50 eq.), Pd(OAc)2 (140 mg, 625 pmol, 0,15 eq.), BINAP (519 mg, 833 pmol, 0,20 eq.) y CS2CO3 (3,39 g, 10,4 mmol, 2,50 eq.) en tolueno (60,0 ml) se desgasificó y se purgó 3 veces con nitrógeno. La mezcla se agitó a 110 °C durante 3 horas en una atmósfera de nitrógeno. La mezcla de reacción se filtró y se concentró a presión reducida a sequedad. El residuo se purificó por cromatografía en columna (SiO2, DCM/MeOH = 30:1 a 10:1) para dar 3-metil-4-(7-bencil-2-(2-(dimetilamino)etoxi)-5.6.7.8- tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1,3-dicarboxilato de 1 -terc-butilo (1,30 g, 1,67 mmol, 40,0 % de rendimiento, 71,4 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 555,3 [M+H]+.
Etapa C: 3-metil-4-(2-(2-(dimetilamino)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1,3-dicarboxilato de 1 -terc-butilo: A una solución de 3-metil-4-(7-bencil-2-(2-(dimetilamino)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1,3-dicarboxilato de 1 -terc-butilo (1,30 g, 2,34 mmol, 1,00 eq.) en MeOH (15,0 ml) se le añadió Pd/C (300 mg) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con hidrógeno. La mezcla se agitó en una atmósfera de hidrógeno (344,74 kPa (50 psi)) a 45 °C durante 48 horas. El catalizador se retiró por filtración y el filtrado se concentró a presión reducida para dar 3-metil-4-(2-(2-(dimetilamino)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1,3-dicarboxilato de 1 -terc-butilo (780 mg, bruto) en forma de un aceite incoloro, que se usó directamente en la siguiente etapa sin purificación adicional.
Etapa D: 3-metil-4-(7-(3-(benciloxi)naftalen-1-il)-2-(2-(dimetilamino)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1,3-dicarboxilato de 1 -terc-butilo: Una mezcla de 3-metil-4-(2-(2-(dimetilamino)etoxi)- 5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1,3-dicarboxilato de 1 -terc-butilo (780 mg, bruto), 3-benciloxi-1-bromonaftaleno (684 mg, 2,18 mmol), Pd2(dba)3 (231 mg, 252 pmol), Cs2CO3 (1,37 g, 4,20 mmol) y 2-diciclohexilfosfino-2',6'-diisopropoxibifenilo (RuPhos) (157 mg, 336 μmol) en dioxano (20,0 ml) se desgasificó y se purgó 3 veces con nitrógeno. La mezcla se agitó a 85 °C durante 5 horas en una atmósfera de nitrógeno. La mezcla de reacción se filtró y se concentró a presión reducida a sequedad. El residuo se purificó por cromatografía en columna (SiO2, DCM/MeOH = 10:1 a 5:1) para dar 3-metil-4-(7-(3-(benciloxi)naftalen-1-il)-2-(2-(dimetilamino)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1,3-dicarboxilato de 1 -terc-butilo (750 mg, 1,02 mmol, dos etapas 43,6 % de rendimiento, 95.0 % de pureza) en forma de un sólido de color amarillo. 1H RMN (400 MHz, cloroformo-d) 5 = 8,10 (d, J = 8,0 Hz, 1H), 7,73 (d, J = 8,0 Hz, 1H), 7,58 - 7,31 (m, 8H), 7,00 (d, J = 2,0 Hz, 1H), 6,89 (d, J = 2,0 Hz, 1H), 5,18 (s, 2H), 4,75 (s a, 1H), 4,45 (d a, J = 13,2 Hz, 1H), 4,42 - 4,34 (m, 1H), 4,34 - 4,28 (m, 1H), 4,15 (s a, 2H), 4,13 - 3,91 (m, 1H), 3,85 - 3,70 (m, 5H), 3,49 - 3,33 (m, 2H), 3,31 - 3,07 (m, 2H), 2,99 (s a, 1H), 2,83 - 2,68 (m, 3H), 2,35(s, 6H), 1,48 (s, 9H).
Etapa E: 1-(7-(3-(benciloxi)naftalen-1-il)-2-(2-(dimetilamino)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-carboxilato de metilo: A una solución de 3-metil-4-(7-(3-(benciloxi)naftalen-1-il)-2-(2-(dimetilamino)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1,3-dicarboxilato de 1 -terc-butilo (200 mg, 287 pmol, 1,00 eq.) en DCM (5,00 ml) se le añadió TFA (770 mg, 6,75 mmol, 500 μl, 23,5 eq.) a 0 °C. La mezcla se agitó a 25 °C durante 2 horas en una atmósfera de nitrógeno. La mezcla de reacción se concentró a presión reducida a sequedad. El residuo se disolvió en DCM (100 ml) y agua (50 ml) y la solución se ajustó a pH 8 con Na2CO3 acuoso saturado y después se extrajo con DCM (3 x 50 ml). Las capas orgánicas combinadas se lavaron con salmuera (3 x 50 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para dar 1-(7-(3-(benciloxi)naftalen-1-il)-2-(2-(dimetilamino)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-carboxilato de metilo (171 mg, 263 pmol, 91,6 % de rendimiento, 91,7 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 597,2 [M+H]+.
Etapa F: (1-(7-(3-(benciloxi)naftalen-1-il)-2-(2-(dimetilamino)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)metanol: A una mezcla de 1-(7-(3-(benciloxi)naftalen-1-il)-2-(2-(dimetilamino)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-carboxilato de metilo (171 mg, 287 pmol, 1,00 eq.) en THF (5,00 ml) se le añadió LiAlH4 (65,3 mg, 1,72 mmol, 6.00 eq.) a 0 °C. Después de agitar durante 2 horas a 25 °C, la mezcla de reacción se inactivó mediante la adición de agua (30 ml) a 0 °C y se extrajo con DCM (3 x 30 ml). Las capas orgánicas combinadas se lavaron con salmuera (3 x 30 ml). La fase orgánica recogida se secó sobre Na2SO4, se filtró y se concentró a presión reducida para dar (1-(7-(3-(benciloxi)naftalen-1-il)-2-(2-(dimetilamino)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)metanol (160 mg, 281 pmol, 98,2 % de rendimiento, 100 % de pureza) en forma de un semisólido incoloro. ESl MS m/z 569,2 [M+H]+.
Etapa G: 2-((7-(3-(benciloxi)naftalen-1-il)-4-(2-(((terc-butildimetilsilil)oxi)metil)piperazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)-N,N-dimetiletanamina: A una mezcla de (1-(7-(3-(benciloxi)naftalen-1-il)-2-(2-(dimetilamino)etoxi)-5.6.7.8- tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)metanol (160 mg, 281 pmol, 1,00 eq.) en Th F (5,00 ml) se le añadió NaH (94,0 mg, 2,35 mmol, 60,0 % de pureza, 8,00 eq.) a 0 °C. Después de agitar durante 30 minutos, se añadió gota a gota cloruro de terc-butildimetilsililo (133 mg, 881 pmol, 108 μl, 3,00 eq.). La mezcla se agitó a 25 °C durante 6 horas en una atmósfera de nitrógeno. La mezcla de reacción se concentró a presión reducida para dar 2-((7-(3-(benciloxi)naftalen-1-il)-4-(2-(((terc-butildimetilsilil)oxi)metil)piperazin-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)-N,N-dimetiletanamina (133 mg, 165 pmol, 56,1 % de rendimiento, 84,6 % de pureza) en forma de un sólido de color ligeramente amarillo, que se usó directamente en la siguiente etapa sin purificación adicional. ESl MS m/z 683,2 [M+H]+.
Etapa H: 1-(4-(7-(3-(benciloxi)naftaleno-1-il)-2-(2-(dimetilamino)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-3-(((terc-butildimetilsilil)oxi)metil)piperazin-1-il)prop-2-en-1-ona: 1 A una solución de 2-((7-(3-(benciloxi)naftalen-1-il)-4-(2-(((terc-butildimetilsilil)oxi)metil)piperazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)-N,N-dimetiletanamina
(133 mg, 165 jmol, 1,00 eq.) y TEA (39,4 mg, 389 jmol, 54,0 |JÍ, 2,00 eq.) en DCM (3,00 ml) se le añadió prop-2-enoato de prop-2-enoílo (31,9 mg, 253 jmol, 1,30 eq.) a -40 °C. Después de agitar a -40 °C durante 2 horas, la mezcla de reacción se inactivó con MeOH (1 M) y se concentró a presión reducida para dar 1-(4-(7-(3-(benciloxi)naftaleno-1-il)-2-(2-(dimetilamino)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-3-(((terc-butildimetilsilil)oxi)metil)piperazin-1-il)prop-2-en-1-ona (140 mg, bruto) en forma de un sólido de color amarillo. ESI MS m/z 737,2 [M+H]+.
Etapa I: 1-(4-(2-(2-(dimetilamino)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-3-(hidroximetil)piperazin-1-il)prop-2-en-1-ona: A una solución de 1-(4-(7-(3-(benciloxi)naftalen-1-il)-2-(2-(dimetilamino)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-3-(((terc-butildimetilsilil)oxi)metil)piperazin-1-il)prop-2-en-1-ona (140 mg, bruto) en DCM (5,00 ml) se le añadió BBr3 (476 mg, 1,90 mmol, 183 j í) a -40 °C. Después de agitar durante 2 horas a -40 °C en una atmósfera de nitrógeno, la mezcla se concentró a presión reducida a sequedad. Se añadieron agua (0,5 ml) y MeOH (2,5 ml) y la solución resultante se purificó por HPLC preparativa (columna: Waters Xbridge 150 * 25 5 u; fase móvil: [agua (NH4HCO310 mM) - ACN]; % de B: 27 % - 57 %, 11 min). Las fracciones deseadas se recogieron y se liofilizaron para dar 1-(4-(2-(2-(dimetilamino)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-3-(hidroximetil)piperazin-1-il)prop-2-en-1-ona (4,32 mg, 7,94 jmol, 4,18 % de rendimiento, 97,9 % de pureza) en forma de un sólido de color blanco. ESI MS m/z 533,4 [M+H]+.
Ejemplo 54

1-[4-[7-(3-amino-1-isoquinolil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona
Etapa A: 4-h¡drox¡-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡rim¡d¡n-7-carbox¡lato de terc-butilo: A una solución de Na (4,24 g, 184 mmol, 4,37 ml, 2,50 eq.) en EtOH (400 ml) se le añadieron 4-etil-3-oxopiperidin-1,4-dicarboxilato de 1-tercbutilo (20,0 g, 73,7 mmol, 1,00 eq.) y metanimidamida del ácido acético (11,5 g, 111 mmol, 1,50 eq.) en una atmósfera de N2. La mezcla se agitó a 70 °C durante 5 horas. La mezcla de reacción se ajustó a pH 7 con HCl (1 N), se extrajo con DCM (3 x 200 ml), se lavó con salmuera (1 x 400 ml), se secó sobre Na2SO4, se filtró y se concentró al vacío para dar 4-hidroxi-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de terc-butilo (16,0 g, 63,7 mmol, 86,4 % de rendimiento) en forma de un sólido de color pardo. ESI MS m/z 274,0 [M+H]+.
Etapa B: 4-(4-benciloxicarbonilpiperazin-1-il)-6,8-dihidro-5H- p¡r¡do[3.4-d1p¡r¡m¡d¡n-7-carbox¡lato de terc-butilo: A una solución de 4-hidroxi-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de terc-butilo (16,0 g, 63,7 mmol, 1,00 eq.) en DMF (4,00 ml) se le añadieron DBU (29,1 g, 191 mmol, 28,8 ml, 3,00 eq.) y hexafluorofosfato de benzotriazol-1-iloxitripirrolidinofosfonio (PYBOP) (39,8 g, 76,4 mmol, 1,20 eq.). La mezcla se agitó a 25 °C durante 1 hora. Se añadió piperazin-1-carboxilato de bencilo (21,0 g, 95,5 mmol, 18,5 ml, 1,50 eq.) y la mezcla de reacción se agitó a 25 °C durante 16 horas. La mezcla se diluyó con acetato de etilo (500 ml) y se lavó con agua (3 x 400 ml), se secó sobre Na2SO4, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna (S¡O2, éter dietílico/acetato de etilo = 1:1) para dar 4-(4-benciloxicarbonilpiperazin-1-il)-6,8-dihidro-5H- pirido[3,4-d]pirimidin-7-carboxilato de terc-butilo (2,00 g, 4,41 mmol, 6,93 % de rendimiento) en forma de un aceite de color amarillo. ESI MS m/z 454,3 [M+H]+.
Etapa C: Una mezcla de 4-(4-benciloxicarbonilpiperazin-1-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de tercbutilo (1,20 g, 2,65 mmol, 1,00 eq.) y TFA (4,53 g, 39,7 mmol, 2,94 ml, 15,0 eq.) en d Cm (2,00 ml) se agitó a 25 °C durante 1 hora. La mezcla se concentró al vacío. El material concentrado se ajustó a pH 8 con NaHCO3 ac. saturado, después se extrajo con acetato de etilo (3 x 30,0 ml), se lavó con salmuera (1 x 100 ml), se secó sobre Na2SO4, se filtró y se concentró al vacío para dar 4-(5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (800 mg, 2,26 mmol, 85,4 % de rendimiento) en forma de un aceite de color amarillo. ESI MS m/z 354,3 [M+H]+.
Etapa D: 4-[7-[3-(terc-butoxicarbonilamino)-1-isoquinolil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: Una mezcla de W-(1-bromo-3-isoquinolil)carbamato de terc-butilo (330 mg, 1,02 mmol, 1,00 eq.), DIEA (264 mg, 2,04 mmol, 357 jí, 2,00 eq.) y 4-(5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (541 mg,
1,53 mmol, 1,50 eq.) en DMSO (3,00 ml) se agitó a 80 °C durante 10 horas. La mezcla se diluyó con agua (5,00 ml) y se extrajo con acetato de etilo (3 x 20 ml). Los extractos combinados se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío. El residuo se purificó por columna de fase inversa (TFA, 0,1 %) para dar 4-[7-[3-(tercbutoxicarbonilamino)-1-isoquinolil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (400 mg, 665 pmol, 65,2 % de rendimiento) en forma de un sólido de color amarillo. ESI MS m/z 596,3 [M+H]+.
Etapa E: N-[1-(4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il) - 3-isoquinolillcarbamato de terc-butilo: Una mezcla de 4-[7-[3-(terc-butoxicarbonilamino)-1-isoquinolil]- 6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (240 mg, 403 pmol, 1,00 eq.), KOH (36,2 mg, 645 pmol, 1,60 eq.) en H2O (2,40 ml) y n-butil alcohol (2,40 ml) se agitó a 100 °C durante 12 horas. La mezcla se diluyó con acetato de etilo (3 x 5,00 ml), se lavó con salmuera (1 x 10,0 ml), se secó sobre Na2SO4, se filtró y se concentró al vacío para dar W-[1-(4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il) - 3-isoquinolil]carbamato de terc-butilo (150 mg, 325 pmol, 80,7 % de rendimiento) en forma de un aceite de color amarillo. ESI MS m/z 462,3 [M+H]+.
Etapa F: trifluoroacetato de 1-(4-p¡peraz¡n-1-¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-7-¡l)¡soqu¡nol¡n-3-am¡na: Una solución de N-[1-(4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il)-3-isoquinolil]carbamato de ferc-butilo (150 mg, 325 pmol, 1,00 eq.) y TfA (556 mg, 4,87 mmol, 361 μl, 15,0 eq.) en DCM (360 μl) se agitó a 25 °C durante 1 hora. La mezcla se concentró al vacío para dar trifluoroacetato de 1-(4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il)isoquinolin-3-amina (154,52 mg, bruto) en forma de un sólido de color amarillo que se usó en la siguiente etapa sin purificación adicional.
Etapa G: 1-[4-[7-(3-amino-1-isoquinolil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de trifluoroacetato de 1-(4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il)isoquinolin-3-amina (150 mg, bruto) y Et3N (319 mg, 3,15 mmol, 437 μl) en DCM (2,00 ml) se le añadió prop-2-enoato de prop-2-enoílo (31,8 mg, 252 pmol) a -40 °C y después se agitó a -40 °C durante 0,5 h. La mezcla se inactivó mediante la adición de MeOH (20,22 mg, 630,96 pmol) y se concentró al vacío. El residuo se purificó por HPLC preparativa (Phenomenex Gemini 150 * 25 mm *, 10 p; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN]; % de B: 26 %-56 %,10 min) para dar 1-[4-[7-(3-amino-1-isoquinolil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (35,4 mg, 82,5 pmol, 26,2 % de rendimiento, 96,8 % de pureza) en forma de un sólido de color amarillo. eS i MS m/z 416,3 [M+H]+.
Ejemplo 55

1-(4-(2-(2-(dimetilamino)-3-hidroxipropoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 2-(dimetilamino)propano-1,3-diol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+ApCI MS m/z 533,3 [M+H]+.
Ejemplo 56

(R)-1-(4-(7-(3-hidroxinaftalen-1-il)-2-((1-morfolinopropan-2-il)oxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (R)-1-morfolinopropan-2-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+a Pc I MS m/z 559,3 [M+H]+.
Ejemplo 57

(S)-5-(((4-(4-acriloilpiperazin-1-il)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)pirrolidin-2-ona
Se sintetizó según el método del Ejemplo 8, usando (S)-5-(hidroximetil)pirrolidin-2-ona en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. eS+APCI MS m/z 529,3 [M+H]+.
Ejemplo 58

(R)-5-(((4-(4-acriloilpiperazin-1-il)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)pirrolidin-2-ona
Se sintetizó según el método del Ejemplo 8, usando (R)-5-(hidroximetil)pirrolidin-2-ona en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. eS+APCI MS m/z 529,2 [M+H]+.
Ejemplo 59

N-(2-((4-(4-acriloilpiperazin-1-il)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)etil)acetamida
Se sintetizó según el método del Ejemplo 8, usando N-(2-hidroxietil)acetamida en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 517,3 [M+H]+.
Ejemplo 60

(R)-1-(4-(2-((1-(dimetilamino)butan-2-il)oxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (R)-1-(dimetilamino)butan-2-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 545,3 [M+H]+.
Ejemplo 61

(S)-6-(((4-(4-acriloilpiperazin-1-il)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)piperidin-2-ona
Se sintetizó según el método del Ejemplo 8, usando 6-(hidroximetil)piperidin-2-ona en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B y el producto se separó por cromatografía quiral. Al pico 2 se le dio la estequiometría (S) pero esto no se confirmó. ES+APCI MS m/z 543,3 [M+H]+.
Ejemplo 62

(R)-6-(((4-(4-acriloilpiperazin-1-il)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-trahidropirido[3,4-d]pirimidin-2-il)oxi)metil)piperidin-2-ona
Se sintetizó según el método del Ejemplo 8, usando 6-(hidroximetil)piperidin-2-ona en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B y se separó por cromatografía quiral. Al pico 1 se le asignó la estequiometría (R) pero esto no se confirmó. ES+Ap C i MS m/z 543,2 [M+H]+.
Ejemplo 63

1-(4-(7-(3-hidroxinaftalen-1-il)-2-(((R)-1-((R)-3-metoxipirrolidin-1-il)propan-2-il)oxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (R)-1-((R)-3-metoxipirrolidin-1-il)propan-2-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI Ms m/z 573,3 [M+H]+.
Ejemplo 64

1-(4-(7-(3-hidroxinaftalen-1-il)-2-(((R)-1-((S)-3-metoxipirrolidin-1-il)propan-2-il)oxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (R)-1-((S)-3-metoxipirrolidin-1-il)propan-2-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 573,3 [M+H]+.
Ejemplo 65

1-(4-(7-(3-hidroxinaftalen-1-il)-2-(((R)-1-((S)-3-hidroxipirrolidin-1-il)propan-2-il)oxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (R)-1-((S)-3-((terc-butildimetilsilil)oxi)pirrolidin-1-il)propan-2-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. Es Ap C i MS m/z 559,3 [M+H]+.
Ejemplo 66

(S)-1-(4-(7-(3-hidroxinaftalen-1-il)-2-((1-metilpiperidin-3-il)oxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (S)-1 -metilpiperidin-3-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+a Pc I MS m/z 529,3 [M+H]+.
Ejemplo 67

(R)-1-(4-(7-(3-hidroxinaftalen-1-il)-2-((1-metilpiperidin-3-il)oxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (R)-1 -metilpiperidin-3-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 529,3 [M+H]+.
Ejemplo 68

(R)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 1-yodonaftaleno en lugar de trifluorometanosulfonato de (3-(metoximetoxi)naftalen-l-ilo en la Etapa D. ES+APCI MS m/z 501,3 [M+H]+.
Ejemplo 69

1-(4-(7-(isoquinolin-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 1, usando 4-bromoisoquinolina en lugar de 1-bromo-3-(metoximetoxi)naftaleno en la Etapa C. ES+APCI MS m/z 401,2 [M+H]+.
Ejemplo 70

1-(4-(7-(3-hidroxinaftalen-1-il)-2-((1-(metilamino)propan-2-il)oxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 1-(metilamino)propan-2-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 503,3 [M+H]+.
Ejemplo 71

1-(4-(7-(2-hidroxi-1-feniletil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 1, usando metanosulfonato de 2-((terc-butildimetilsilil)oxi)-1 -feniletilo en lugar de 1-bromo-3-(metoximetoxi)naftaleno en la Etapa C. ES+APCI MS m/z 394,2 [M+H]+.
Ejemplo 72
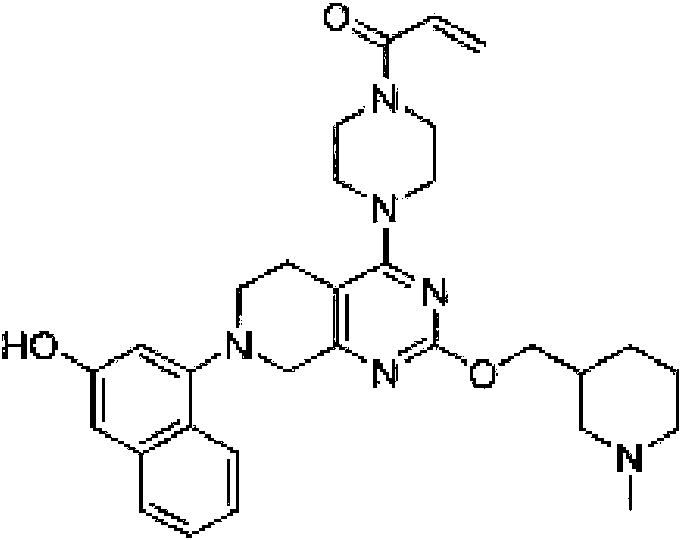
1-(4-(7-(3-hidroxinaftalen-1-il)-2-((1-metilpiperidin-3-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (1-metilpiperidin-3-il)metanol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 543,3 [M+H]+.
Ejemplo 73

1-(4-(2-(2-(dimetilamino)propoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 2-(dimetilamino)propan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 517,2 [M+H]+.
Ejemplo 74

1- (4-(7-(3-hidroxinaftalen-1-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 3-morfolinopropan-1-ol en lugar de (S)-1-(dimetilamino)propan-2- ol en la Etapa B. ES+APCI MS m/z 559,3 [M+H]+.
E je m p lo 75

(R)-1-(4-(7-(3-hidroxinaftalen-1-il)-2-((1-(piperidin-1-il)propan-2-il)oxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (R)-1-(piperidin-1-il)propan-2-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 557,3 [M+H]+.
Ejemplo 76

(R)-1-(4-(7-(3-cloro-5-hidroxi-2-isopropilfenil)-2-((1-(dimetilamino)propan-2-il)oxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 1-bromo-3-cloro-2-isopropil-5-(metoximetoxi)benceno en lugar de trifluorometanosulfonato de (3-(metoximetoxi)naftalen-1-ilo en la Etapa D. ES+Ap C i MS m/z 545,3 [M+H]+.
Ejemplo 77

1-(4-(2-(2-((1S,4R)-2-azabiciclo[2.2.1]heptan-2-il)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
S e s in te tizó según el m é tod o de l Ejemplo 8 , u sa nd o 2 -((1 S ,4 R )-2 -a z a b ic ic lo [2.2.1 ]h e p ta n -2 - il)e ta n -1 -o l en lu g a r de (S )-1 -(d im e tila m in o )p ro p a n -2 -o l en la E tapa B. E S A P C I M s m /z 555 ,2 [M+H]+.
E je m p lo 78

(R)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(5-hidroxi-2-(trifluorometoxi)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 2-bromo-4-(metoximetoxi)-1-(trifluorometoxi)benceno en lugar de trifluorometanosulfonato de (3-(metoximetoxi)naftalen-1-ilo en la Etapa D. Es APCI MS m/z 551,2 [M+H]+. Ejemplo 79

(R)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(5-hidroxi-2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 2-bromo-4-(metoximetoxi)-1-(trifluorometil)benceno en lugar de trifluorometanosulfonato de (3-(metoximetoxi)naftalen-1-ilo en la Etapa D. eS+APCI MS m/z 535,2 [M+H]+. Ejemplo 80

1-(4-(7-(3-hidroxinaftalen-1-il)-2-((4-metilpiperazin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
S e s in te tiz ó seg ún el m é to d o de l Ejemplo 8 , u sa n d o (4 -m e tilp ip e ra z in -2 - il)m e ta n o l en lu g a r de (S )-1 -(d im e tila m in o )p ro p a n -2 -o l en la E tap a B. E S A P C I M S m /z 544 ,3 [M H ]+.
E je m p lo 81

1-(2-((4-(4-acriloilpiperazin-1-il)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)etil)piperidin-4-carbonitrilo
Se sintetizó según el método del Ejemplo 8, usando 1-(2-hidroxietil)piperidin-4-carbonitrilo en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. Es +APCI MS m/z 568,2 [M+H]+.
Ejemplo 82

1-(4-(7-(3-hidroxinaftalen-1-il)-2-(3-(4-metilpiperazin-1-il)propoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, 3-(4-metilpiperazin-1-il)propan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 572,4 [M+H]+.
Ejemplo 83

1-(4-(2-(2-((2-hidroxietil)(metil)amino)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
S e s in te tiz ó se g ú n e l m é to d o de l Ejemplo 8 , u sa n d o 2 -((2 -( ( te rc -b u t ild im e tils il i l)o x i)e ti l) (m e til)a m in o )e ta n -1 -o l en lu g a r de (S )-1 -(d im e tila m in o )p ro p a n -2 -o l en la E ta p a B. e S a P c I M S m /z 533 ,3 [M H ]+.
E je m p lo 84

(R)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(2-fluoro-6-hidroxi-3-metilfenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 2-bromo-3-fluoro-1-(metoximetoxi)-4-metilbenceno en lugar de trifluorometanosulfonato de (3-(metoximetoxi)naftalen-1-ilo en la Etapa D. ES+APCI MS m/z 499,3 [M+H]+. Ejemplo 85

1-(4-(7-(3-hidroxinaftalen-1-il)-2-(2-(pirrolidin-1-il)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 2-(pirrolidin-1-il)etan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 529,2 [M+H]+.
Ejemplo 86

(R)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(2-hidroxi-5-(trifluorometoxi)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 2-bromo-1-(metoximetoxi)-4-(trifluorometoxi)benceno en lugar de trifluorometanosulfonato de (3-(metoximetoxi)naftalen-1-ilo en la Etapa D. Es +APCI MS m/z 551,2 [M+H]+. E je m p lo 87

(R)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-fluoro-2-hidroxi-6-metilfenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 2-bromo-4-fluoro-3-(metoximetoxi)-1-metilbenceno en lugar de trifluorometanosulfonato de (3-(metoximetoxi)naftalen-1-ilo en la Etapa D. ES+APCI MS m/z 499,2 [M+H]+. Ejemplo 88

(S)-1-(4-(7-(3-hidroxinaftalen-1-il)-2-(2-(2-metilpiperidin-1-il)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (S)-2-(2-metilpiperidin-1-il)etan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 557,2 [M+H]+.
Ejemplo 89

1-(4-(7-(3-hidroxinaftalen-1-il)-2-((1-metilpirrolidin-3-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (1-metilpirrolidin-3-il)metanol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 529,3 [M+H]+.
E je m p lo 90

1-(4-(7-(5-hidroxi-2-(trifluorometoxi)fenil)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 3-morfolinopropan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B, y usando 2-bromo-4-(metoximetoxi)-1-(trifluorometoxi)benceno en lugar de trifluorometanosulfonato de (3-(metoximetoxi)naftalen-1-ilo en la Etapa D. ES+APCI MS m/z 593,2 [M+H]+.
Ejemplo 91

(R)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-hidroxi-5-(trifluorometoxi)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 3-bromo-1-(metoximetoxi)-5-(trifluorometoxi)benceno en lugar de trifluorometanosulfonato de (3-(metoximetoxi)naftalen-1-ilo en la Etapa D. Es +APCI MS m/z 551,2 [M+H]+. Ejemplo 92
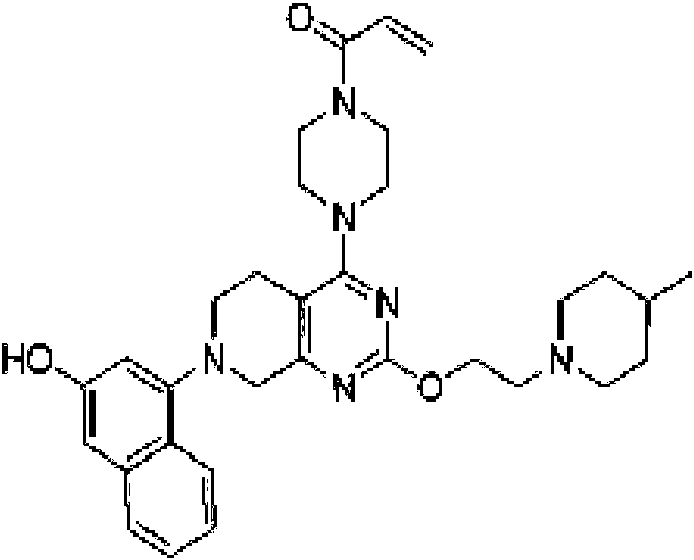
1-(4-(7-(3-hidroxinaftalen-1-il)-2-(2-(4-metilpiperidin-1-il)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
S e s in te tiz ó seg ún el m é to d o de l Ejemplo 8 , u sa n d o 2 -(4 -m e tilp ip e r id in -1 - il)e ta n -1 -o l en lu g a r de (S )-1 -(d im e tila m in o )p ro p a n -2 -o l en la E tap a B. E S A P C I M S m /z 557 ,2 [M H ]+.
Ejemplo 93

1-(4-(7-(3-hidroxinaftalen-1-il)-2-((4-morfolinobutan-2-il)oxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 4-morfolinobutan-2-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 573,3 [M+H]+.
Ejemplo 94
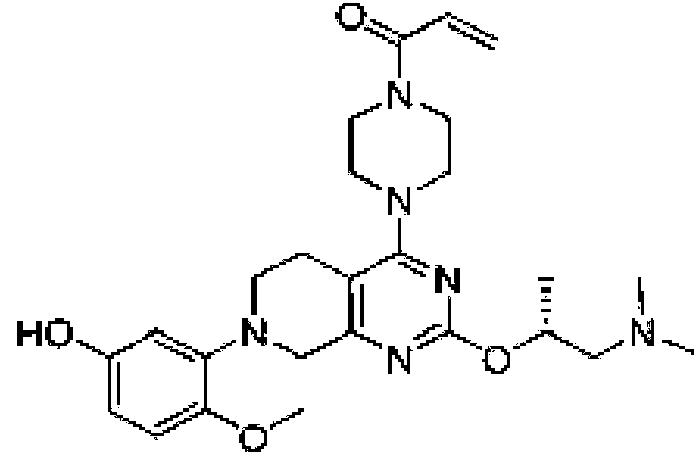
(R)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(5-hidroxi-2-metoxifenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 3-bromo-1-(metoximetoxi)-4-metoxibenceno en lugar de trifluorometanosulfonato de (3-(metoximetoxi)naftalen-1-ilo en la Etapa D. ES+APCI MS m/z 497,2 [M+H]+.
Ejemplo 95

(R)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(2-hidroxi-6-metilfenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 2-bromo-1-(metoximetoxi)-3-metilbenceno en lugar de trifluorometanosulfonato de (3-(metoximetoxi)naftalen-1-ilo en la Etapa D. ES+APCI MS m/z 481,2 [M+H]+.
E je m p lo 96

1- (4-(7-(5-hidroxi-2-isopropoxifenil)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 3-morfolinopropan-1-ol en lugar de (S)-1-(dimetilamino)propan-2- ol en la Etapa B, y usando 2-bromo-4-(metoximetoxi)-1-(isopropoxi)benceno en lugar de trifluorometanosulfonato de (3-(metoximetoxi)naftalen-1-ilo en la Etapa D. ES+Ap CI MS m/z 567,2 [M+H]+.
Ejemplo 97

(R)-1-(4-(7-(3-hidroxinaftalen-1-il)-2-(2-(2-metilpiperidin-1-il)etoxi)-5,6,7, 8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (R)-2-(2-metilpiperidin-1-il)etan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+ApCI MS m/z 557,3
Ejemplo 98

(S)-1-(4-(7-(3-hidroxinaftalen-1-il)-2-(2-(3-metoxipiperidin-1-il)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el m étodo del Ejemplo 8, usando (S )-2-(3 -m etoxip iperid in-1 -il)e tan-1-o l en lugar de (S)-1-(d im etilam ino)propan-2-o l en la E tapa B. Es +APC I MS m /z 557,3 [M+H]+.
E je m p lo 99

(R)-1-(4-(7-(3-hidroxinaftalen-1-il)-2-(2-(3-metoxipiperidin-1-il)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (R)-2-(3-metoxipiperidin-1-il)etan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 557,3 [M+H]+.
Ejemplo 100

1-(4-(2-((1-ciclopropilpiperidin-4-il)oxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (1 -ciclopropilpiperidin-4-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 555,3 [M+H]+.
Ejemplo 101

1-(4-(2-(3-(1,4-oxazepan-4-il)propoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sin te tizó según el m étodo del Ejemplo 8, usando (3 -(1 ,4 -oxazepan-4-il)p ropan-1-o l en lugar de (S )-1-(d im etilam ino)propan-2-o l en la E tapa B. e S+a Pc I MS m /z 573,3 [M+H]+.
E je m p lo 102

1-(4-(2-(3-(1,4-oxazepan-4-il)propoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método de, usando 2-(dietilamino)etan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 531,3 [M+H]+.
Ejemplo 103

1-(4-(7-(3-hidroxinaftalen-1-il)-2-((1-(2-metoxietil)pirrolidin-3-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, (1-(2-metoxietil)pirrolidin-3-il)metanol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 573,3 [M+H]+.
Ejemplo 104

1-(4-(2-(3-((1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
S e s in te tizó según el m é tod o de l Ejemplo 8 , u sa nd o 3 -((1 S ,4 S )-2 -o x a -5 -a z a b ic ic lo [2.2.1 ]h e p ta n -5 - il)p ro p a n -1 -o l en lu g a r de (S )-1 -(d im e tila m in o )p ro p a n -2 -o l en la E tapa B. e S A P C I M S m /z 571 ,3 [M+H]+.
E je m p lo 105

1-(4-(7-(5-hidroxi-2-metilfenil)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 3-monfolinopropan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B y usando 2-bromo-4-(metoximetoxi)-1-metilbenceno en lugar de trifluorometanosulfonato de (3-(metoximetoxi)naftalen-1-ilo en la Etapa D. ES+APCI MS m/z 523,3 [M+H]+.
Ejemplo 106

1-(4-(7-(4-hidroxi-2-(trifluorometoxi)fenil)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 3-morfolinopropan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B, y usando 2-bromo-5-(metoximetoxi)-1-(trifluorometoxi)benceno en lugar de trifluorometanosulfonato de (3-(metoximetoxi)naftalen-1-ilo en la Etapa D. ES+APCI MS m/z 593,2 [M+H]+.
Ejemplo 107

1-(4-(7-(3-hidroxinaftalen-1-il)-2-(2-((2-metoxietil)(metil)amino)etoxi)-5,6,7, 8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sin te tizó según el m étodo del Ejemplo 8, usando 2 -((2 -m etoxie til)(m etil)am ino)e tan-1-o l en luga r de (S)-1-(d im etilam ino)propan-2-o l en la E tapa B. ES+APC I MS m /z 547,3 [M+H]+.
E je m p lo 108

1-(4-(7-(3-hidroxinaftalen-1-il)-2-(2-(4-metoxipiperidin-1-il)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 2-(4-metoxipiperidin-1-il)etan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. eS+a Pc I MS m/z 573,3 [M+H]+.
Ejemplo 109

1-(4-(2-(2-(4,4-difluoropiperidin-1-il)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 2-(4,4-difluoropiperidin-1-il)etan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 579,2 [M+H]+.
Ejemplo 110

(R)-1-(4-(7-(3-hidroxinaftalen-1-il)-2-((1-metilpirrolidin-3-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sin te tizó según el m étodo del Ejemplo 8 , usando (R )-(1 -m etilp irro lid in-3-il)m etano l en lugar de (S)-1-(d im etilam ino)propan-2-o l en la E tapa B. ES+a Pc I MS m /z 529,3 [M+H]+.
E je m p lo 111

(S)-1-(4-(2-(2-(3-fluoropiperidin-1-il)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (S)-2-(3-fluoropiperidin-1-il)etan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+ApCI MS m/z 561,3 [M+H]+.
Ejemplo 112

1-(4-(7-(3-(difluorometil)naftalen-1-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 3-morfolinopropan-1-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B, y usando 1-bromo-3-(difluorometil)naftaleno (0,129 g, 0,503 mmol) en lugar de trifluorometanosulfonato de (3-(metoximetoxi)naftalen-1-ilo en la Etapa D. ES+APCI MS m/z 593,2 [M+H]+.
Ejemplo 113

4-(4-acriloilpiperazin-1-il)-7-(3-hidroxinaftalen-1-il)-7, 8-dihidropirido[3,4-d]pirimidin-6(5H)-ona
Etapa A: 4-(6-fluorop¡r¡do[3.4-d1p¡r¡m¡din-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-butilo: A una solución de 4-cloro-6-fluoropirido[3,4-d]pirimidina (1,07 g, 5,83 mmol) en DCM (20 ml) se le añadió N-etil-N-isopropilpropan-2-amina (2,09 ml, 11,7 mmol) seguido de piperazin-1-carboxilato de terc-butilo (1,19 g, 6,41 mmol) y la reacción se agitó a temperatura ambiente durante 2 horas. La mezcla de reacción se lavó con salmuera, se secó sobre MgSÜ4 y se concentró al vacío para proporcionar 4-(6-fluoropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de terc-butilo, que se usó directamente en la siguiente etapa (1,8 g, 92,6 %) ES+APCI MS m/z 334,1 [M+H]+.
Etapa B: 4-(6-(benc¡lox¡)p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-butilo: A una solución de fenilmetanol (0.65 g. 6.0 mmol) en DMA (10 ml) se le añad¡ó poco a poco h¡druro de sod¡o (0.24 g. 6.0 mmol) m¡entras se desgas¡f¡caba con n¡trógeno y la mezcla de reacc¡ón se ag¡tó durante 30 m¡nutos a temperatura amb¡ente. A la reacc¡ón se le añad¡ó 4-(6-fluorop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-butilo (1.0 g.
3.0 mmol) en forma de un sól¡do y la mezcla de reacc¡ón se ag¡tó a temperatura amb¡ente durante 3 horas. La reacc¡ón se vert¡ó en agua (300 ml) y la capa acuosa se extrajo con acetato de et¡lo. La capa orgán¡ca se lavó con salmuera. se secó sobre MgSÜ4 y se concentró al vacío. El mater¡al bruto se cromatograf¡ó usando un grad¡ente de 0 a 100 % de acetato de et¡lo/DCM como eluyente para dar 4-(6-(benc¡lox¡)p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1 -carbox¡lato de terc-o (0.4 g. 0.95 mmol. 32 % de rend¡m¡ento). e S+Ap CI MS m/z 422.2 [M+H]+.
Etapa C: 4-(6-oxo-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo: A una soluc¡ón de 4-(6-(benc¡lox¡)p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo (0.40 g. 0.95 mmol) en 95 % de etanol (30 ml) purgado con n¡trógeno se le añad¡ó Pd/C (0.10 g. 0.95 mmol). La reacc¡ón se evacuó con vacío y se cargó de nuevo tres veces con h¡drógeno. Después de la tercera carga. la mezcla de reacc¡ón se ag¡tó a temperatura amb¡ente durante 4 horas. La reacc¡ón se desgas¡f¡có de nuevo con n¡trógeno. la suspens¡ón se f¡ltró a través de Cel¡te® y el f¡ltrado se concentró al vacío para dar 4-(6-oxo-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo (0.4 g. 126 %). ES+APCI MS m/z 334.4 [M+H1+.
Etapa D: 4-(7-(3-(metox¡metox¡)naftalen-1-¡l)-6-oxo-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo: A una soluc¡ón de 4-(6-oxo-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo (0.28 g. 0.84 mmol) en d¡oxanos (4 ml) en un tubo sellado se le añad¡eron fosfato de potas¡o (0.36 g. 1.7 mmol). N1.N2-d¡met¡letano-1.2-d¡am¡na (0.074 g. 0.84 mmol) y 1-yodo-3-(metox¡metox¡)naftaleno (0.53 g. 1.7 mmol). La reacc¡ón se roc¡ó con argón durante 20 m¡nutos. segu¡do de la ad¡c¡ón de yoduro de cobre (I) (0.16 g. 0.84 mmol). El rec¡p¡ente de reacc¡ón se selló y se calentó a 100 °C durante una noche. La reacc¡ón se d¡luyó con agua y la capa acuosa se extrajo con acetato de et¡lo (2 x 150 ml). Los extractos orgán¡cos se lavaron con salmuera. se secaron sobre MgSÜ4 y se concentraron al vacío. El mater¡al se cromatograf¡ó usando un grad¡ente de 0 a 100 % de acetato de et¡lo/DCM como eluyente para dar 4-(7-(3-(metox¡metox¡)naftalen-1-¡l)-6-oxo-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo (0.30 g. 0.58 mmol. 69 % de rend¡m¡ento) ES+APCI MS m/z 520.2 [M+H1+.
Etapa E: 7-(3-h¡drox¡naftalen-1-¡l)-4-(p¡peraz¡n-1-¡l)-7.8-d¡h¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-6(5H)-ona: A una soluc¡ón de 4-(7-(3-(metox¡metox¡)naftalen-1-¡l)-6-oxo-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo (0.30 g. 0.58 mmol) en metanol (10 ml) se le añad¡ó cloruro de h¡drógeno acuoso (0.38 ml. 2.3 mmol) y la reacc¡ón se ag¡tó durante una noche a 50 °C. La reacc¡ón se concentró al vacío para dar 7-(3-h¡drox¡naftalen-1-¡l)-4-(p¡peraz¡n-1-¡l)-7.8-d¡h¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-6(5H)-ona en forma de la sal b¡s HCl (0.26 g. 100 %). ES+APCI MS m/z 376.1 [M+H1+.
Etapa F: 4-(4-acr¡lo¡lp¡peraz¡n-1-¡l)-7-(3-h¡drox¡naftalen-1-¡l)-7.8-d¡h¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-6(5H)-ona: A una suspens¡ón de d¡clorh¡drato de 7-(3-h¡drox¡naftalen-1-¡l)-4-(p¡peraz¡n-1-¡l)-7.8-d¡h¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-6(5H)-ona (0.26 g. 0.58 mmol) en una soluc¡ón 1:1 de DCM/aceton¡tr¡lo (10 ml) se le añad¡eron N-et¡l-N-¡soprop¡lpropan-2-am¡na (0.45 g. 3.5 mmol) y cloruro de acr¡lαlo (0.052 g. 0.58 mmol) y la reacc¡ón se ag¡tó a temperatura amb¡ente durante 1 hora. La reacc¡ón se concentró al vacío y el mater¡al se pur¡f¡có por HPLC preparat¡va ¡nversa (usando un grad¡ente de 5 a 95 % de ACN/agua) para dar 4-(4-acr¡lo¡lp¡peraz¡n-1-¡l)-7-(3-h¡drox¡naftalen-1-¡l)-7.8-d¡h¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-6(5H)-ona (0.038 g. 0.088 mmol. 15 % de rend¡m¡ento). ES+APCI MS m/z 430.2 [M+H1+.
Ejemplo 114

4-(6-acr¡lo¡l-2.6-d¡azaesp¡ro[3.31heptan-2-¡l)-7-(3-h¡drox¡naftalen-1-¡l)-7.8-d¡h¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-6(5H)-ona
Se s¡ntet¡zó según el método del Ejemplo 8. usando clorh¡drato de 2.6-d¡azaesp¡ro[3.31heptano-2-carbox¡lato de terc-but¡lo (305 mg. 1.54 mmol) en lugar de p¡peraz¡n-1-carbox¡lato de terc-but¡lo (1.19 g. 6.41 mmol) en la Etapa B. ES+APCI MS m/z 442.2 [M+H1+
Ejemplo 115

1-((S)-4-(2-(((R)-1-(dimetilamino)propan-2-il)oxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-3-metilpiperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (S)-3-metilpiperazin-1-carboxilato de bencilo (1,155 g, 4,931 mmol) en lugar de 1 -piperazinacarboxilato de bencilo en la etapa A. ES+APCI MS m/z 531,3 [M+H]+.
Ejemplo 116

(S)-1-(4-(2-(2-(dimetilamino)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-3-metilpiperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (S)-3-metilpiperazin-1-carboxilato de bencilo (1,155 g, 4,931 mmol) en lugar de 1-piperazinacarboxilato de bencilo en la etapa A. Además, usando 2-(dimetilamino)etan-1 -ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+a Pc I MS m/z 517,3 [M+H]+.
Ejemplo 117

1-(4-(7-(3-hidroxinaftalen-1-il)-2-(2-morfolinoetoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
S e s in te tiz ó seg ún el m é to d o de l Ejemplo 8 , u sa n d o 2 -m o rfo lin o e ta n -1 -o l en lu g a r de (S )-1 -(d im e tila m in o )p ro p a n -2 -o l en la E tap a B. E S A P C I M S m /z 545 ,2 [M H ]+.
Ejemplo 118

1-(4-(7-(3-hidroxinaftalen-1-il)-2-morfolin-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona Se sintetizó según el método del Ejemplo 8, usando morfolina en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 501,3 [M+H]+.
Ejemplo 119

1-(4-(7-(3-hidroxinaftalen-1-il)-2-(pirrolidin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando pirrolidina en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 485,2 [M+H]+.
Ejemplo 120

(R)-1-(4-(7-(3-hidroxinaftalen-1-il)-2-((1-(pirrolidin-1-il)propan-2-il)oxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando (R)-1-(pirrolidin-1-il)propan-2-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. ES+APCI MS m/z 543,4 [M+H]+.
E je m p lo 121

4-(4-acriloilpiperazin-1-il)-7-(3-hidroxinaftalen-1-il)-2-(3-morfolinopropoxi)-6,7-dihidropirido[3,4-d]pirimidin-8(5H)-ona
Etapa A: 2,4-Dicloro-8-oxo-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo. A un matraz de fondo redondo se le añadieron 2,4-dicloro-5,6-dihidropirido[3,4-d]pirimidin-7(8H)-carboxilato de terc-butilo (1,0 g, 3,29 mmol) y EtOAc (16,4 ml). Se añadió una solución de peryodato de sodio (2,11 g, 9,86 mmol) en agua (16,4 ml). Se añadió cloruro de rutenio (III) (0,102 g, 0,493 mmol) y la mezcla se agitó con la tapa suelta y se agitó vigorosamente durante 6 h a temperatura ambiente. La mezcla se repartió entre agua y EtOAc y las capas se separaron. La capa acuosa se extrajo adicionalmente con EtOAc (2 x 20 ml) y los extractos combinados se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto bruto se purificó por cromatografía en columna (10-30 % de EtOAc/hexanos, cargando con CH2O2) para proporcionar 0,864 g (82 %) del producto en forma de un sólido de color blanquecino.
Etapa B: 4-(4-((Benciloxi)carbonil)piperazin-1-il)-2-cloro-8-oxo-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo. A una solución de 2,4-dicloro-8-oxo-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (0,400 g, 1,26 mmol) en CH2Cl2 (5,0 ml) se le añadió N,N-diisopropiletilamina (0,325 g, 2,51 mmol) seguido de 1-piperazinacarboxilato de bencilo (0,255 ml, 1,32 mmol) y la reacción se agitó a temperatura ambiente durante 1,5 h. La mezcla de reacción se diluyó con CH2Cl2 (10 ml) y se lavó con una solución 0,5 M de KHSO4 (5 ml), seguido de una solución acuosa saturada de NaHCO3 y salmuera. La capa orgánica se secó sobre Na2SO4, se filtró y se concentró. El producto bruto se sonicó en MTBE 10 ml y el sólido resultante se aisló por filtración al vacío. El sólido se secó al vacío para proporcionar 0,507 g (80 %) del producto deseado en forma de un sólido de color blanquecino que se usó directamente en la siguiente etapa. ES+APCI MS m/z 502,1 [M+H]+.
Etapa C: 4-(2-Cloro-8-oxo-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo. Una solución de 4-(4-((benciloxi)carbonil)piperazin-1-il)-2-cloro-8-oxo-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (0,255 g, 0,5080 mmol) en CH2O2 (1,0 ml) se enfrió a 0 °C. Se añadió ácido trifluoroacético (0,3890 ml, 5,080 mmol) y la mezcla se calentó a temperatura ambiente. Después de 1 hora, la mezcla se diluyó con CH2O2 y se añadió a una mezcla salmuera (10 ml) y NaOH acuoso 3,0 M (1,7 ml, 5,080 mmol). Las capas se combinaron y se ajustó a pH 8 con una solución acuosa saturada de NaHCO3. Las capas se separaron y la fase acuosa se extrajo con 2 x 10 ml de CH2O2. Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. El compuesto del título (0,223 g, cuant.) se obtuvo en forma de una espuma de color amarillo/naranja. ES+APCI MS m/z 402,1 [M+H]+.
Etapa D: 4-(2-(3-Morfolinopropoxi)-8-oxo-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo. A un vial se le añadieron N-hidroxipropanilmorfolina (0,687 g, 4,73 mmol) y 4-(2-cloro-8-oxo-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (0,190 g, 0,473 mmol) seguido de dioxano (1,6 ml). Se añadió carbonato de cesio (0,462 g, 1,42 mmol) y la mezcla se agitó a 65 °C durante 15 horas. La mezcla se diluyó con CHCl3 y se filtró y el sólido se lavó con más cantidad de CHCl3. El filtrado se concentró al vacío y se purificó por cromatografía en columna (2-10 % de MeOH/DCM con 1 % de NH4OH) para proporcionar 0,061 g (25 %) del producto deseado en forma de un aceite espeso incoloro. ES+APCI MS m/z 511,2 [m +H]+.
Etapa E: 4-(7-(3-(Metoximetoxi)napthalen-1-il)-2-(3-morfolinopropoxi)-8-oxo-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo. A un vial se le añadieron 4-(2-(3-morfolinopropoxi)-8-oxo-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (0,060 g, 0,12 mmol), N,N'-Dimetiletilendiamina (0,010 g, 0,12 mmol) y 1-yodo-3-(metoximetoxi)naftaleno (0,074 g, 0,24 mmol) seguido de dioxano (0,78 ml) y fosfato de potasio tribásico (0,050 g, 0,24 mmol). La reacción se purgó burbujeando Ar durante 10 min, después se añadió yoduro de cobre (I) (0,022 g, 0,12 mmol) y el vial se selló. La mezcla se calentó a 110 °C y se agitó durante 16 horas. La mezcla se enfrió a temperatura ambiente, se diluyó con agua y se extrajo con EtOAc (3 x 10 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron. La mezcla se purificó por cromatografía en columna (2-8 % de MeOH/DCM) para proporcionar 0,062 g (76 %) del producto en forma de una espuma de color castaño. ES+APCI MS m/z 697,3 [M+H]+.
Etapa F: 7-(3-(metoximetoxi)naftalen-1-il)-2-(3-morfolinopropoxi)-4-(piperazin-1-il)-6,7-dihidropirido[3,4-d]pirimidin-8(5H)-ona. A una solución de 4-(7-(3-(metoximetoxi)naftalen-1-il)-2-(3-morfolinopropoxi)-8-oxo-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (0,062 g, 0,089 mmol) en EtOH (0,44 ml) y THF (0,44 ml) se le añadió paladio sobre carbono (0,038 g, 0,018 mmol) (tipo Degussa, 10 % en peso, 50 % de H2O) y
después se introdujo una atmósfera de hidrógeno mediante vacío seguido de presión de globo. La mezcla se agitó a temperatura ambiente durante 1,5 h, después se calentó a 45 °C y se agitó durante 1 hora. La mezcla se diluyó con EtOAc y se filtró a través de un filtro de nailon. El filtrado se concentró al vacío proporcionando una espuma de color castaño claro (0,052 g) que se secó durante una noche y se sometió de nuevo a las mismas condiciones de reacción anteriores. Después de agitar a temperatura ambiente durante 5 h, se añadió más cantidad de Pd/C (0,050 g) y la reacción se agitó a temperatura ambiente durante 2 h más. La mezcla se diluyó con EtOAc y se filtró a través de un filtro de nailon. El sólido se lavó con EtOAc y MeOH y el filtrado se concentró al vacío, proporcionando 0,029 g (58 %) del producto deseado en forma de una espuma de color castaño claro. ES+APCI MS m/z 563,3 [M+H]+.
Etapa G: 4-(4-acriloilpiperazin-1-il)-7-(3-(metoximetoxi)naftalen-1-il)-2-(3-morfolinopropoxi)-6,7-dihidropirido[3,4-d]pirimidin-8(5H)-ona. A una solución de 7-(3-(metoximetoxi)naftalen-1-il)-2-(3-morfolinopropoxi)-4-(piperazin-1-il)-6,7-dihidropirido[3,4-d]pirimidin-8(5H)-ona (0,028 g, 0,050 mmol) en CH2O2 (0,50 ml) a -78 °C se le añadió trietilamina (0,014 ml, 0,100 mmol). Se añadió cloruro de acriloílo (0,55 ml, 0,055 mmol, CH2O20,1 M recién preparado) y después la reacción se agitó durante 0,5 h. La mezcla se diluyó con CHCb y se añadió una solución acuosa saturada de NH4CL Las capas se separaron y la capa acuosa se extrajo con CHCb (2 * 10 ml). Los extractos combinados se secaron sobre Na2SO4, se filtraron y se concentraron. El producto bruto se purificó por cromatografía en columna (4-6 % de MeOH/DCM) para proporcionar 0,018 g (59 %) para proporcionar el compuesto del título en forma de una espuma sólida de color blanquecino. ES+APCI MS m/z 617,3 [M+H]+.
Etapa H: 4-(4-acriloilpiperazin-1 -il)-7-(3-hidroxinaftalen-1 -il)-2-(3-morfolinopropoxi)-6,7-dihidropirido[3,4-d]pirimidin-8(5H)-ona. A una solución de 4-(4-acriloilpiperazin-1-il)-7-(3-(metoximetoxi)naftalen-1-il)-2-(3-morfolinopropoxi)-6,7-dihidropirido[3,4-d]pirimidin-8(5H)-ona (0,018 g, 0,0292 mmol) en 1/1 MeOH/THF (0,6 ml) se le añadió HCl (0,0486 ml, 0,292 mmol, acuoso 6 N). La mezcla se agitó a 35 °C durante 7 horas. La mezcla se diluyó con salmuera y se ajustó a pH 8 con una solución acuosa saturada de NaHCO3. La mezcla se extrajo con 10 % de IPA/CHCb (2 * 10 ml) y CHCb (10 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron. El material bruto se purificó por cromatografía en columna (6-10 % de MeOH/DCM) para proporcionar 0,012 g (71 %) del producto en forma de un sólido de color blanquecino. ES+APCI MS m/z 573,3 [M+H]+.
Ejemplo 122

bis-trifluoroacetato de (R,E)-4-(dimetilamino)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d)pirimidin-4-il)piperazin-1-il)but-2-en-1-ona

Etapas A-E: Se sintetizó (R)-2-((7-(3-(metoximetoxi)naftalen-1-il)-4-(piperazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)-N,N-dimetilpropan-1-amina según el método del Ejemplo 8, Etapa A a Etapa E, usando (R)-1-(dimetilamino)propan-2-ol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B.
Etapa F: Diclorhidrato de (R)-4-(2-((1-(dimetilamino)propan-2-il)oxi)-4-(piperazin-1 -il)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-il)naftalen-2-ol: A una solución de (R)-2-((7-(3-(metoximetoxi)naftalen-1-il)-4-(piperazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)-N,N-dimetilpropan-1-amina (75 mg, 0,15 mmol) en DCM (2,9 ml) se le añadió HCl 6 N en iPrOH (247 pl, 1,5 mmol) y la mezcla se agitó a temperatura ambiente durante 1 h. La mezcla de reacción se concentró a sequedad para proporcionar diclorhidrato de (R)-4-(2-((1 -(dimetilamino)propan-2-il)oxi)-4-(piperazin-1 -il)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-il)naftalen-2-ol que se usó directamente en la siguiente etapa. ES+AμCi MS m/z 463,2 [M+H]+.
Etapa G: Bis-trifluoroacetato de (R,E)-4-(dimetilamino)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-il)but-2-en-1-ona: Una solución de diclorhidrato de (R)-4-(2-((1-(dimetilamino)propan-2-il)oxi)-4-(piperazin-1-il)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-il)naftalen-2-ol (7 mg, 0,01 mmol), HATU (6,2 mg, 0,02 mmol) y ácido (2E)-4-(dimetilamino)but-2-enoico (2,1 mg, 0,02 mmol), DIEA (6,9 μl, 0,04 mmol) en DCM (131 μl) se agitó a temperatura ambiente durante 1 h. El residuo se filtró y el filtrado se cargó directamente sobre una HPLC prep. Gilson C18 eluyendo con 5-95 % de acetonitrilo/agua con aditivo de 0,1 % de TFA. Las fracciones que contenían el producto deseado se concentraron para proporcionar bis-trifluoroacetato de (R,E)-4-(dimetilamino)-1 -(4-(2-((1 -(dimetilamino)propan-2-il)oxi)-7-(3-hidroxinaftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)but-2-en-1-ona. ES+Ap CI m S m/z 574,2 [M+H]+.
Ejemplo 123

1-(4-(2-(2-hidroxi-3-morfolinopropoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Se sintetizó según el método del Ejemplo 8, usando 3-morfolinopropano-1,2-diol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. e S+APCI MS m/z 575,2 [M+H]+.
Ejemplo 124

(R)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(4-fluoro-3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Etapa A: Se puso (R)-4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (221 mg, 0,345 mmol) en ACN (2 ml) y la mezcla se enfrió a 0 °C. Se añadió SelectFluor (183 mg, 0,517 mmol) y la reacción se agitó a temperatura ambiente durante 30 minutos. Se añadió agua y la mezcla se extrajo con DCM. Las capas orgánicas se combinaron y se concentraron. El residuo resultante se purificó por cromatografía de fase inversa (5-95 % de ACN:agua con 0,1 % de TFA) para proporcionar trifluoroacetato de (R)-4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(4-fluoro-3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (48 mg, 0,0729 mmol, 21,1 % de rendimiento).
Etapa B: Se preparó (R)-1-(4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(4-fluoro-3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona siguiendo el Ejemplo 33, Etapas D-F sustituyendo trifluoroacetato de (R)-4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(4-fluoro-3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato por 4-(7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo. ES+APCI MS m/z 535,2 [M+H]+.
Ejemplo 125

(R)-1-(4-(7-(4-cloro-3-hidroxinaftalen-1-il)-2-((1-(dimetilamino)propan-2-il)oxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Etapa A: Se pusieron (R)-4-(2-((1-(dimetilamino)propan-2-il)oxi)-7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (83 mg, 0,130 mmol) y NCS (21,6 mg, 0,162 mmol) en ACN (2 ml) y se agitaron a temperatura ambiente durante 3 horas. Se añadió agua y la mezcla se extrajo con DCM (3 x 15 ml). Las capas orgánicas se combinaron y se concentraron. El residuo resultante se purificó por cromatografía de fase inversa (5-95 % de ACN:agua con 0,1 % de TFA) para proporcionar trifluoroacetato de (R)-4-(7-(4-cloro-3-(metoximetoxi)naftalen-1-il)-2-((1-(dimetilamino)propan-2-il)oxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (17 mg, 0,0252 mmol, 19,4 % de rendimiento).
Etapa B: Se preparó (R)-1-(4-(7-(4-cloro-3-hidroxinaftalen-1-il)-2-((1-(dimetilamino)propan-2-il)oxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona siguiendo el Ejemplo 33, Etapas D-F sustituyendo trifluoroacetato de (R)-4-(7-(4-cloro-3-(metoximetoxi)naftalen-1-il)-2-((1-(dimetilamino)propan-2-il)oxi)-5,6,7,8
tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato por 4-(7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo y usando THF como disolvente en la Etapa D. ES+APCI MS m/z 551,2 [M+H]+.
Ejemplo 126

trifluoroacetato de 1-(4-(7-(3-hidroxinaftalen-1-il)-2-(2-(piridin-2-il)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
Etapa A: 4-(7-(3-(metox¡metox¡)naftalen-1-¡l)-2-(2-(p¡r¡d¡n-2-¡l)etox¡)-5.6.7.8-tetrah¡drop¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carboxilato de bencilo: A una suspensión de 4-(2-cloro-7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (0,20 g, 0,35 mmol) en dioxano en un microondas se le añadieron N-etil-N-isopropilpropan-2-amina (0,45 g, 3,5 mmol), Cs2CÜ3 (0,34 g, 1,0 mmol) y 2-(piridin-2-il)etan-1-ol (0,43 g, 3,5 mmol) y la reacción se calentó a 15 °C durante 1 h en el microondas. La reacción se diluyó con EtOAc y se lavó con agua y salmuera, se secó sobre MgSO4 y se concentró al vacío. El material se cromatografió usando un gradiente de 0 a 100 % de EtOAc/DCM como eluyente para dar 4-(7-(3-(metoximetoxi)naftalen-1-il)-2-(2-(piridin-2-il)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (0,16 g, 0,24 mmol, 70 % de rendimiento).
Etapa B: 4-(4-(p¡peraz¡n-1-¡l)-2-(2-(p¡r¡d¡n-2-¡l)etox¡)-5.8-d¡h¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-7(6H)-¡l)naftalen-2-ol: Al 4-(7-(3-(metoximetoxi)naftalen-1-il)-2-(2-(piridin-2-il)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo sólido (0,12 g, 0,18 mmol) se le añadió MeOH (20 ml). La solución se desgasificó con nitrógeno durante 5 minutos, seguido de la adición de Pd/C (0,058 g, 0,54 mmol). El recipiente de reacción se evacuó mediante vacío y se recargó con H2. Este procedimiento se realizó tres veces y, después de la tercera carga, la suspensión se dejó en agitación en una atmósfera de hidrógeno durante 1 h. La reacción se desgasificó de nuevo con nitrógeno durante 5 minutos. Luego, la suspensión se filtró a través de Celite® y el Celite® se lavó con MeOH (100 ml). Los extractos orgánicos combinados se concentraron y se trataron con 10 ml de 1:1 TFNDCM durante 2 h. La reacción se concentró de nuevo al vacío para dar 4-(4-(piperazin-1-il)-2-(2-(piridin-2-il)etoxi)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-il)naftalen-2-ol (0,096 g, 0,20 mmol, 110 % de rendimiento).
Etapa C: 1-(4-(7-(3-h¡drox¡naftalen-1-¡l)-2-(2-(p¡r¡d¡n-2-¡l)etox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona: A una solución de 4-(4-(piperazin-1-il)-2-(2-(piridin-2-il)etoxi)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-il)naftalen-2-ol (0,096 g, 0,20 mmol) en DCM se le añadieron Base de Hunig (0,17 ml, 0,99 mmol) y cloruro de acriloílo (0,018 g, 0,20 mmol) y la reacción se agitó durante 30 minutos a temperatura ambiente. La reacción se concentró al vacío y el material bruto se purificó por HPLC preparativa inversa para dar trifluoroacetato de 1 -(4-(7-(3-hidroxinaftalen-1-il)-2-(2-(piridin-2-il)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona (0,0057 g, 0,011 mmol, 5,3 % de rendimiento). ES+APCI Ms m/z 537,2 [M+H]+.
Ejemplo 127
(S)-7-(3-(metoximetoxi)naftalen-1-il)-2-((1-metilpirrolidin-2-il)metoxi)-4-(piperazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidina

Etapa A: 4-(4-((benciloxi)carbonil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo: A una solución de 2,4-dicloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (8 g, 26,30 mmol) en DMA (263,0 ml, 26,30 mmol) se le añadieron piperazin-1-carboxilato de bencilo (5,793 g, 26,30 mmol) y N-etil-N-isopropilpropan-2-amina (4,721 ml, 26,30 mmol) y la reacción se agitó a temperatura ambiente durante 2 horas. La TLC (20 % de EtOAc/DCM), visualización UV, mostró que la reacción se había completado. Luego, la reacción se vertió en agua y se extrajo en d Cm . Luego, los extractos orgánicos se lavaron con agua (2 x) y salmuera, se secaron sobre MgSO4 y se concentraron al vacío. El concentrado se cargó sobre una columna RegiSep de 220 g y se cromatografió en el CombiFlash (0 %-10 %, EtOAc:DCM). Todas las fracciones que contenían el producto deseado se combinaron y se concentraron para dar 4-(4-((benciloxi)carbonil)piperazin-1 -il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (9,768 g, 20,02 mmol, 76,11 % de rendimiento) en forma de una espuma de color blanco. e S+APCI MS m/z 488,2 [M+H]+.

Etapa B: 4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1 -carboxilato de bencilo: Se disolvió 4-(4-((benciloxi)carbonil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (9,768 g, 20.02 mmol) en diclorometano (200,2 ml, 20,02 mmol) y se trató con ácido 2,2,2-trifluoroacético (15,33 ml, 200.2 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 4 horas. Después de que se completara, la reacción se concentró al vacío, se recogió en EtOAc y los extractos orgánicos se lavaron con NaOH 1 M (2 x) y salmuera, se secaron sobre MgSO4 y se concentraron al vacío. El 4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (7,406 g, 19,09 mmol, 95,39 % de rendimiento) se usó bruto en la siguiente reacción. ES+APCI MS m/z 388,2 [M+H]+.

Etapa C: 4-(2-cloro-7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: Al 4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo y BINAP (0,275 g, 0,442 mmol) y Pd2(dba)3 (0,203 g, 0,221 mmol) en una atmósfera de argón se les añadió tolueno (221 ml, 11,1 mmol) y la reacción se burbujeó con Ar durante 10 minutos seguido de calentamiento a 100 °C durante 10 minutos. Luego, la reacción se enfrió a temperatura ambiente y se añadieron 4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (4,29 g, 11,1 mmol) y terc-butóxido de sodio (2,13 g, 22,1 mmol) a la solución oscura en forma de sólidos. Finalmente, se añadió trifluorometanosulfonato de 3-(metoximetoxi)naftalen-1-ilo (7,44 g, 22,1 mmol) se añadió (en forma del aceite) y la reacción se calentó a 100 °C durante 1 hora. La reacción se enfrió a temperatura ambiente y se concentró al vacío. El concentrado se disolvió con EtOAc y se lavó con agua y salmuera. Los extractos orgánicos combinados se secaron sobre Na2SO4 y se concentraron al vacío. Después, el residuo se cargó en el CombiFlash y se cromatografió usando 0 %-->50 % de Hexano:EtOAc como eluyente. Las fracciones que contenían producto limpio se combinaron y se concentraron al vacío para proporcionar 4-(2-cloro-7-(3-(metoximetoxi)naftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (2,6 g, 4,53 mmol, 40,9 % de rendimiento). ES+APCI MS m/z 574,2 [M+H]+.

Etapa D: (S)-4-(7-(3-(metoximetoxi)naftalen-1-il)-2-((1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: En un tubo de microondas se disolvió 4-(2-cloro-7-(3-(metoximetoxi)naftalen-1-il)-5.6.7.8- tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (300 mg, 0,523 mmol) en dioxano (6532 pl, 0,523 mmol) y se trató con carbonato de cesio (511 mg, 1,57 mmol), base de Hunig (913 μl, 5,23 mmol) y N-Metil-L-prolinol (421 mg, 3,66 mmol). Después, el tubo se tapó y se sometió a microondas a 170 °C durante 3 horas. La reacción se filtró a través de papel GHF. El filtrado se concentró al vacío y el residuo se cargó en una columna RegiSep gold de 12 g y se cromatografió en el CombiFlash (0 %-15 %, DCM:MeOH). Todas las fracciones que contenían el producto limpio se combinaron y se concentraron al vacío para dar (S)-4-(7-(3-(metoximetoxi)naftalen-1-il)-2-((1-metilpirrolidin-2-il)metoxi)-5.6.7.8- tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (220 mg, 0,337 mmol, 64,5 % de rendimiento). ES+APCI MS m/z 653,3 [M+H]+.

Etapa E: (S)-7-(3-(metoximetoxi)naftalen-1 -il)-2-((1 -metilpirrolidin-2-il)metoxi)-4-(piperazin-1 -il)-5,6,7,8-tetrahidropirido[3,4-djpirimidina: Una solución de (S)-4-(7-(3-(metoximetoxi)naftalen-1-il)-2-((1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (220 mg, 0,337 mmol) en EtOH (3370 gl, 0,337 mmol) y THF (3370 gl, 0,337 mmol) se purgó con N2 durante 5 minutos. A esta solución se le añadió paladio sobre carbono (179 mg, 0,0843 mmol) (tipo Degussa, 10 % en peso, 50 % de H2O), se tapó inmediatamente y se purgó con N2 durante 5 min más. Después, la solución se agitó en una atmósfera de H2 introducida mediante vacío seguido de presión de globo. Después, la mezcla se agitó a temperatura ambiente durante una noche. La LC/MS mostró que la reacción se había completado. La mezcla se diluyó con MeOH y se filtró a través de celite empaquetado. Después, el filtrado se concentró al vacío y (S)-7-(3-(metoximetoxi)naftalen-1 -il)-2-((1 -metilpirrolidin-2-il)metoxi)-4-(piperazin-1 -il)-5,6,7, 8-tetrahidropirido[3,4-d]pirimidina (91 mg, 0,175 mmol, 52,1 % de rendimiento) se llevó en bruto a continuación. ES+APCI MS m/z 519,3 [M+H]+.

Etapa F: (S)-1 -(4-(7-(3-(metoximetoxi)naftalen-1 -il)-2-((1 -metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona: A una suspensión de (S)-7-(3-(metoximetoxi)naftalen-1-il)-2-((1-metilpirrolidin-2-il)metoxi)-4-(piperazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidina (92 mg, 0,18 mmol) en diclorometano (1774 gl, 0,18 mmol) a temperatura ambiente se le añadió cloruro de acriloílo (1774 gl, 0,18 mmol) seguido de base de Hunig (62 gl, 0,35 mmol). Después, la reacción se agitó a temperatura ambiente durante 1 hora. Después, la mezcla se concentró y se cargó en una columna RegiSep gold de 4 g y se cromatografió en el CombiFlash (0 %-15 %, DCM:MeOH). Todas las fracciones que contenían producto limpio se combinaron y se concentraron al vacío para dar (S)-1-(4-(7-(3-(metoximetoxi)naftalen-1-il)-2-((1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona (74 mg, 0,13 mmol, 73 % de rendimiento). ES+APCI MS m/z 573,3 [M+H]+.
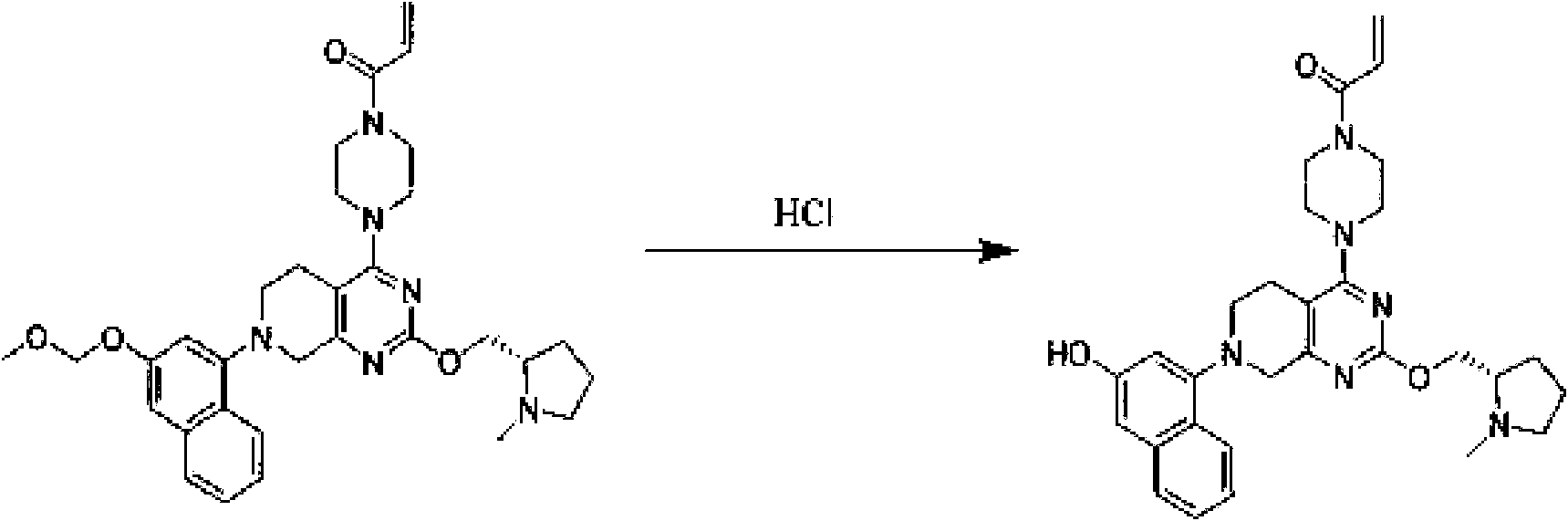
Etapa G: (S)-1 -(4-(7-(3-hidroxinaftalen-1 -il)-2-((1 -metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -il)prop-2-en-1 -ona: Se disolvió (S)-1 -(4-(7-(3-(metoximetoxi)naftalen-1 -il)-2-((1 -metilpirrolidin-2
il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona (74 mg, 0,13 mmol) en metanol (4307 μl, 0,13 mmol) y se trató con cloruro de hidrógeno (1077 μl, 6,5 mmol) (ac.). La reacción se agitó a 55 °C durante 1 hora. La mezcla de reacción se concentró al vacío y se suspendió de nuevo en 1,5 ml de MeOH. La suspensión se cargó en el Gilson (HPLC prep.), que se eluyó con 5-->95 % de ACN/0,1 % de TFA en agua/0,1 % de TFA. Todas las fracciones que contenían producto limpio se combinaron y se liofilizaron durante una noche para dar (S)-1-(4-(7-(3-hidroxinaftalen-1-il)-2-((1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona (26 mg, 0,049 mmol, 38 % de rendimiento). ES+APCI MS m/z 529,3 [M+H]+.
Ejemplo 128
1-(4-(2-(3-(dimetilamino)azelidin-1-il)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona

Etapa A: 4-(2-(3-(dimetilamino)azetidin-1-il)-7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: A una solución de 4-(2-cloro-7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (0,20 g, 0,35 mmol) en dioxanos se le añadieron clorhidrato de N,N-dimetilazetidin-3-amina (0,24 g, 1,7 mmol) y N-etil-N-isopropilpropan-2-amina (0,45 g, 3,5 mmol) y la reacción se calentó a 80 °C durante 72 h. La reacción se concentró al vacío y el residuo se cromatografió usando 0-->20 % de MeOH/DCM como eluyente para dar 4-(2-(3-(dimetilamino)azetidin-1-il)-7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxiiato de bencilo (0,25 g, 105 %). ES+APCI MS m/z 638,3 [M+H]+.
Se preparó 1-(4-(2-(3-(dimetilamino)azetidin-1-il)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona según el Ejemplo 127 sustituyendo 4-(2-(3-(dimetilamino)azetidin-1-il)-7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo por (S)-4-(7-(3-(metoximetoxi)naftalen-1-il)-2-((1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo en la Etapa E. ES+APCI MS m/z 514,2 [M+H]+.
Ejemplo 129
1-[4-[7-(3-hidroxi-1-naftil)-2-[(1R)-1-[(2R)-1-metilpirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona

Etapa A: (2R)-2-[( 1 R)-1 -hidroxietillpirrolidin-1 -carboxilato de terc-butilo: Una mezcla de BH3-Me2S (10 M, 549 ul) y (3aS)-1-metil-3,3-difenil-3a,4,5,6-tetrahidropirrolo[1,2-c][1,3,2]oxazaborol (1,00 M, 844 ul) en THF (10 ml) se agitó a 15 °C durante 1 hora. A la mezcla se le añadió una solución de (2R)-2-acetilpirrolidin-1-carboxilato de terc-butilo (0,90 g, 4,22 mmol) en THF (10 ml) y la mezcla se agitó a 15 °C durante 1 hora. La mezcla se inactivó mediante la adición de metanol (2,00 ml) y la reacción se concentró al vacío. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/éter acetato = 50/1 - 5/1) para dar (2R)-2-[(1R)-1-hidroxietil]pirrolidin-1-carboxilato de tercbutilo (0,60 g, 2,79 mmol, 66,0 % de rendimiento) en forma de un aceite incoloro. 1H RMN (400 MHz, CD3OD) 6 = 5,18 (s a, 1H), 3,73 (dt, J = 4,8, 8,0 Hz, 1H), 3,70 - 3,43 (m, 2H), 3,28 (td, J = 6,64, 10,8 Hz, 1H), 1,96 (cd, J = 7,2, 12,8 Hz, 1H), 1,89 - 1,68 (m, 2H), 1,62 (s a, J = 6,4 Hz, 1H), 1,47 (s, 9H), 1,15 (d, J = 6,0 Hz, 3H).

Etapa B: 4-[7-(3-benciloxi-1-naftil)-2-[(1R)-1-[(2R)-1-terc-butoxicarbonilpirrolidin-2-il]etoxi]-6,8-dihidro-577-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: A una solución de 4-[7-(3-benciloxi-1-naftil)-2-metilsulfinil- 6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (0,70 g, 1,08 mmol) y (2R)-2-[(1R)-1-hidroxietil]pirrolidin-1-carboxilato de terc-butilo (349 mg, 1,62 mmol) en THF (10 ml) se le añadió t-BuONa (312 mg, 3,24 mmol) y la mezcla se agitó a 10 °C durante 1 hora. La mezcla se diluyó con agua (10 ml) y la capa acuosa se extrajo con acetato de etilo (3 x 10 ml). Los extractos orgánicos combinados se lavaron con salmuera (20 ml), se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna (SO2, éter de petróleo/éter acetato = 3/1) para dar 4-[7-(3-benciloxi-1-naftil)-2-[(1R)-1-[(2R)-1-terc-butoxicarbonilpirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (0,35 g, 412 umol, 38,1 % de rendimiento) en forma de un sólido de color amarillo. ES+APCI MS m/z 799,4 [M+H]+.

Etapa C: 4-[7-(3-benciloxi-1-naftil)-2-[(1R)-1-[(2R)- pirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: Una mezcla de 4-[7-(3-benciloxi-1-naftil)-2-[(1R)-1-[(2R)-1-terc-butoxicarbonilpirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (0,30 g, 375 umol) y TFA (642 mg, 5,63 mmol, 417 ul) en diclorometano (0,42 ml) se agitó a 10 °C durante 1 hora. La mezcla se concentró al vacío para dar 4-[7-(3-benciloxi-1-naftil)-2-[(1R)-1-[(2R)-pirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (305 mg) LCMS [M+1]: ES+APCI MS m/z 699,2 [M+H]+.

Etapa D: 4-[7-(3-benciloxi-1-naftil)-2-[(1R)-1-[(2R)-1-metilpirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: Una mezcla de 4-[7-(3-benciloxi-1-naftil)-2-[(1R)-1-[(2R)-pirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (0,30 g, bruto), formaldehído (210 mg, 1,85 mmol, 192 ul, 37 % de agua) y AcOH (22,16 mg, 369 umol, 21,1 ul) en metanol (3,00 ml) se agitó a 15 °C durante 0,5 horas. A la mezcla se le añadió NaBHaCN (58,0 mg, 923 umol) y la mezcla se agitó a 15 °C durante 48 horas. La mezcla se inactivó mediante la adición de H2O (5 ml) a 0 °C y la capa acuosa se extrajo con acetato de éter (3 x 10 ml). Los extractos orgánicos combinados se lavaron con salmuera (15,0 ml), se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,10 % de ácido fórmico)/acetonitrilo] para dar 4-[7-(3-benciloxi-1-naftil)-2-[(1R)-1-[(2R)-1-metilpirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (0,10 g, 126 umol) en forma de un aceite de color amarillo. ES+APCI MS m/z 713,4 [M+H]+.

Etapa E: 4-[2-[(1ft)-1-[(2ft)-1-metilpirrolidin-2-il]etoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-illnaftalen-2-ol: Se burbujeó amoniaco en metanol (3 ml) a -78 °C durante 30 minutos. Luego, se añadieron 4-[7-(3-benciloxi-1-naftil)-2-[(1R)-1-[(2R)-1-metilpirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (0,09 g, 126 umol) y Pd al 10 %/C seco (0,10 g) y la mezcla se agitó a 10 °C durante 1 hora en una atmósfera de H2 (103,42 kPa (15 psi)). La reacción se filtró y el filtrado se concentró al vacío para dar 4-[2-[(1R)-1-[(2R)-1-metilpirrolidin-2-il]etoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (0,04 g, bruto) en forma de un aceite de color amarillo. ES+APCI MS m/z 489,2 [M+H]+.
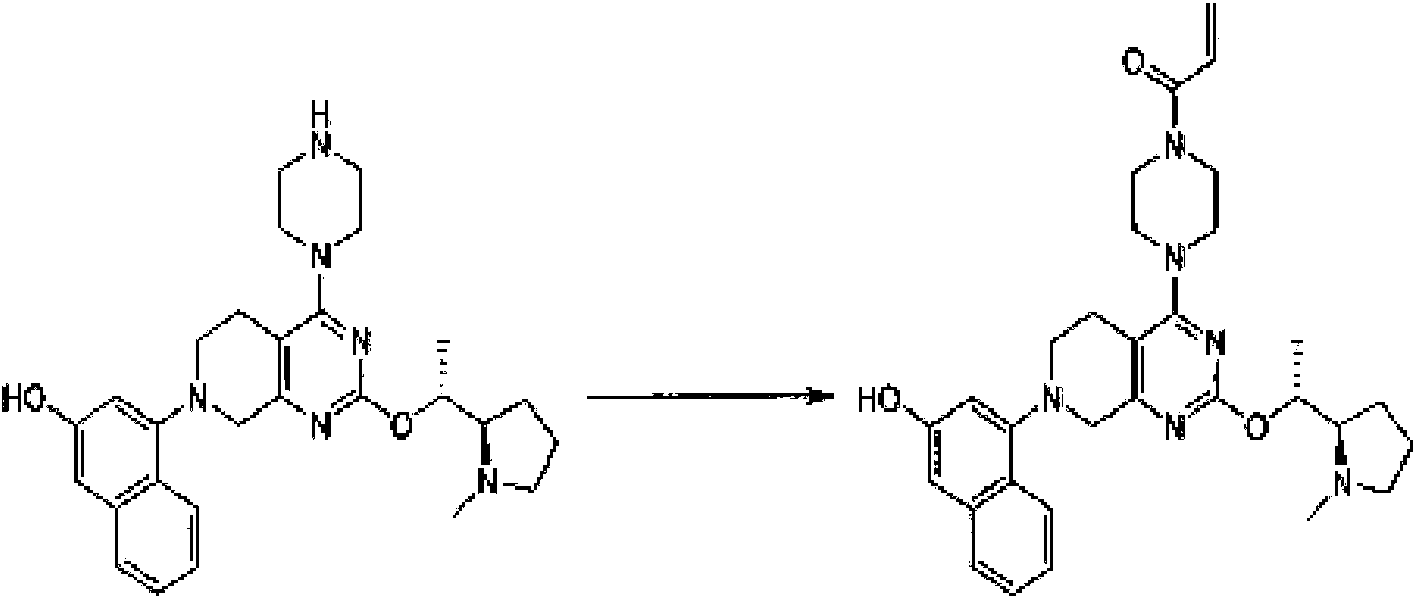
Etapa F: 1-[4-[7-(3-hidroxi-1-naftil)-2-[(1R)-1-[(2R)-1-metilpirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de 4-[2-[(1R)-1-[(2R)-1-metilpirrolidin-2-il] etoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (0,04 g) y Et3N (124 mg, 1,23 mmol, 171 ul) en Dc M (2,00 ml) a -40 °C se le añadió prop-2-enoato de prop-2-enoílo (7,23 mg, 57,3 umol) y la reacción se agitó a -40 °C durante 0,5 h. La mezcla se inactivó mediante la adición de metanol (0,10 ml) y se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi C18 150 * 25 * 10 um;fase móvil: [agua (0,225 % de ácido fórmico)-ACN]; % de B: 10 %-37 % durante 10 minutos) para dar 1-[4-[7-(3-hidroxi-1-naftil)-2-[(1R)-1-[(2R)-1-metilpirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (9,13 mg, 15,5 umol) en forma de un sólido de color amarillo. ES+APCI MS m/z 543,4 [M+H]+.
Ejemplo 130
1-(4-(7-(3-hidroxinaftalen-1-il)-2-((S)-1-((S)-1-metilpirrolidin-2-il)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona

Se preparó 1-(4-(7-(3-hidroxinaftalen-1-il)-2-((S)-1-((S)-1-metilpirrolidin-2-il)etoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona siguiendo el Ejemplo 129 sustituyendo (3aR)-1-metil-3,3-difenil-3a,4,5,6-tetrahidropirrolo[1,2-c][1,3,2]oxazaborol por (3aS)-1-metil-3,3-difenil-3a,4,5,6-tetrahidropirrolo[1,2-c][1,3,2]oxazaborol en la Etapa A sustituyendo también (2S)-2-acetilpirrolidin-1-carboxilato de terc-butilo por (2R)-2-acetilpirrolidin-1 -carboxilato de ferc-butilo en la Etapa A. ES+APCI MS m/z 543,4 [M+H]+.
Ejemplo 131
1-[4-[2-[(1R)-2-[etil(metil)amino]-1-metil-etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona

Etapa A: í2ffl-1-ret¡límet¡l)am¡no)propan-2-ol: Se añadió (2R)-2-metiloxirano (540 mg, 9,31 mmol, 651 ul) a N-metiletanamina (500 mg, 8,46 mmol, 725 ul) en MeOH (10 ml). La solución resultante se agitó a 80 °C durante 3 horas en un tubo sellado. Después de que se completara, la mezcla se concentró al vacío para dar (2R)-[etil(metil)amino]propan-2-ol (260 mg, bruto) en forma de un aceite de color amarillo claro que se usó directamente en la siguiente etapa sin purificación adicional.

Etapa B: 4-[7-(3-benc¡lox¡-1-naft¡l)-2-[(1R)-2-[et¡l(met¡l)am¡no]-1-met¡l-etox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-ilpperazin-1-carboxilato de bencilo: A una solución de (2R)-1-[etil(metil)amino]propan-2-ol (217 mg, 1,85 mmol) en tolueno (20 ml) se le añadieron 4-[7-(3-benc¡lox¡-1-naft¡l)-2-met¡lsulf¡n¡l-6,8-d¡l^¡dro-5H p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carboxilato de bencilo (400 mg, 617 umol), Pd2(dba)3 (56,6 mg, 61,8 umol), BINAP (76,9 mg, 124 umol) y NaOtBu (178 mg, 1,85 mmol) y la mezcla se desgasificó con N2 durante 15 minutos y después se calentó a 90 °C durante 16 horas en una atmósfera de N2. Después de que se completara, la mezcla de reacción se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía de fase inversa para dar 4-[7-(3-benc¡lox¡-1-naft¡l)-2-[(1R)-2-[et¡l(met¡l)am¡no]-1-met¡l-etox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (110 mg, 153 umol, 24,8 % de rendimiento, 97,5 % de pureza). ES+APCI MS m/z 701,4 [M+H]+.

Etapa C: 4-[2-[(1^)-2-[et¡l(met¡l)am¡no1-1-met¡l-etox¡1-4-p¡peraz¡n-1-¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-7-¡l1naftalen-2-ol: A una solución de 4-[7-(3-benc¡lox¡-1-naft¡l)-2-[(1R)-2-[et¡l(met¡l)am¡no]-1-met¡l-etox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo (100 mg, 143 umol) en MeOH (3,00 ml) se le añadió HCl/MeOH (4 M, 143 ul), seguido de Pd(OH)2/C (50 mg) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 40 °C durante 4 horas. Después de que se completara, la mezcla de reacción se filtró y el filtrado se concentró para dar 4-[2-[(1R)-2-[et¡l(met¡l)am¡no]-1-met¡l-etox¡]-4-p¡peraz¡n-1-¡l-6,8
dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (76,0 mg, 125 umol, 87,5 % de rendimiento, 90,3 % de pureza, 2 HCl) que se usó directamente en la siguiente etapa sin purificación adicional. ES+APCI MS m/z 477,2 [M+H]+.

Etapa D: 1-[4-[2-[(1R)-2-[etil(metil)amino]-1-metil-etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de 4-[2-[(1R)-2-[etil(metil)amino]-1-metil-etoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (70 mg, 127 umol, 2 HCl) y DIEA (98,8 mg, 764 umol, 133 ul) en DCM (1,50 ml) se le añadió gota a gota prop-2-enoato de prop-2-enoílo (12,9 mg, 102 umol) a -50 °C. La mezcla se agitó de -40 a -20 °C durante 30 minutos. Después de que se completara, la mezcla se inactivó mediante la adición de MeOH (17,0 mg) y se concentró al vacío. El residuo se diluyó con agua (1 ml) y se extrajo con DCM (3 x 6 ml). Las capas orgánicas se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um;fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN];% de B: 48 %-78 %,10 min) para dar 1-[4-[2-[(1R)-2-[etil(metil)amino]-1-metil-etoxi]-7-(3-hidroxi-1-naftil)-6, 8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (9,23 mg, 17,2 umol, 13,5 % de rendimiento, 98,7 % de pureza) en forma de un sólido de color amarillo. ES+APCI MS m/z 531,3 [M+H]+.
Ejemplo 132
1-[4-[2-[(1-ciclohexilpirrolidin-3-il)metoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona

Etapa A: 1-c¡clohexilp¡rrol¡d¡n-3-carbox¡lato: A una solución de pirrolidin-3-carboxilato de metilo (1,00 g, 6,04 mmol, HCl) y DIEA (780 mg, 6,04 mmol, 1,05 ml) se le añadieron ciclohexanona (652 mg, 6,64 mmol, 686 ul) y HOAc (725 mg, 12,1 mmol, 691 ul) a 0 °C. La mezcla de reacción se agitó a 0 °C durante 0,5 h. A la mezcla de reacción se le añadió poco a poco NaBH(OAc)3 (3,84 g, 18,12 mmol) a 0 °C. La mezcla de reacción se agitó a 0 a 15 °C durante 12 horas. Después de que se completara, la mezcla de reacción se inactivó mediante la adición de agua (5 ml) y los extractos orgánicos se concentraron al vacío. La capa acuosa se extrajo con DCM (10 ml x 2) y el pH se ajustó con NaHCO3 sat. (10 ml) y Na2CO3 (2 ml) a pH>8. Las capas orgánicas se combinaron, se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se trituró con MTBE/éter de petróleo (1:3, 20 ml) y el filtrado se concentró al vacío para dar 1-ciclohexilpirrolidin-3-carboxilato de metilo (1,00 g, 4,73 mmol, 78,4 % de rendimiento) en forma de un aceite de color pardo. ES+APCI MS m/z 212,2 [M+H]+.

Etapa B: (1-c¡clohex¡lp¡rrolid¡n-3-¡l)metanol: A una solución de 1 -ciclohexilpirrolidin-3-carboxilato de metilo (1,00 g, 4,73 mmol) en THF (20 ml) se le añadió L¡AlH4 (413 mg, 10,9 mmol) a -10 °C. La mezcla de reacción se agitó a -10 °C durante 0,5 horas. La mezcla de reacción se inactivó mediante la adición de Na2SÜ4 saturado (2 ml), la mezcla se filtró y la torta de filtro se lavó con THF (3 x 50 ml). Los extractos orgánicos combinados se concentraron al vacío para dar: (1-ciclohex¡lp¡rrolid¡n-3-¡l)metanol (800 mg, 4,36 mmol, 92,3 % de rendimiento) en forma de un aceite incoloro. 1H RMN (400 MHz, CLOROFORMO-d) ó = 3,71 (dd, J = 4,1, 10,0 Hz, 1H), 3,53 (dd, J = 4,4, 10,0 Hz, 1H), 2,90 (dt, J = 4,4, 8,8 Hz, 1H), 2,74 (dd, J = 3,2, 8,8 Hz, 1H), 2,53 (dd, J = 6,8, 8,8 Hz, 1H), 2,36 - 2,24 (m, 2H), 2,04 - 1,93 (m, 2H), 1,90 (s a, 2H), 1,78 - 1,64 (m, 3H), 1,61 - 1,52 (m, 1H), 1,33 - 1,12 (m, 5H)
Se preparó 1-[4-[2-[(1-c¡clohex¡lp¡rrol¡d¡n-3-¡l)metox¡]-7-(3-h¡drox¡-1-naftil)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]piperazin-1-¡l]prop-2-en-1-ona siguiendo el Ejemplo 131 sustituyendo (1 -ciclohex¡lp¡rrolidin-3-il)metanol por (2R)-1-[etil(metil)amino]propan-2-ol en la Etapa B. Es Ap CI MS m/z 597,4 [M+H]+.
Ejemplo 133
1-[4-[7-(3-h¡drox¡-1-naft¡l)-2-(3-morfol¡noprop¡lam¡no)-6,8-d¡h¡dro-5H-p¡rido[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-¡l]prop-2-en-1-ona

Etapa A: 4-[7-(3-benc¡lox¡-1-naft¡l)-2-(3-morfol¡noprop¡lam¡no)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡perazin-1-carboxilato de bencilo: Una solución de 4-[7-(3-benc¡lox¡-1-naft¡l)-2-metilsulf¡n¡l-6,8-d¡h¡dro-5H pir¡do[3,4-d]p¡rim¡d¡n-4-¡l]piperazin-1-carbox¡lato de bencilo (400 mg, 617 umol) y 3-morfolinopropan-1-amina (534 mg, 3,70 mmol, 540 ul) en DMSO (4,00 ml) se calentó a 100 °C durante 12 horas. Después de que se completara, la mezcla se diluyó con agua (4 ml) y se extrajo con EtOAc (3 x 20 ml). Las capas orgánicas se lavaron con salmuera (30 ml), se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se purificó por cromatografía en columna sobre AhO3 eluyendo con acetato de etilo/éter de petróleo (20^100 %) para dar 4-[7-(3-bencilox¡-1-naft¡l)-2-(3-morfol¡noprop¡lam¡no)-6,8-d¡hidro-5H-p¡r¡do[3,4-d]p¡r¡m¡din-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (245 mg, 320 umol, 51,8 % de rendimiento, 95,0 % de pureza) en forma de un aceite de color amarillo. ES+APCI MS m/z 728,6 [M+H]+.
Se preparó 1-[4-[7-(3-h¡drox¡-1-naft¡l)-2-(3-morfol¡noprop¡lam¡no)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡din-4-¡l]p¡peraz¡n-1-¡l]prop-2-en-1-ona siguiendo el Ejemplo 131 sustituyendo 4-[7-(3-bencilox¡-1-naft¡l)-2-(3-morfolinoprop¡lam¡no)-6,8-d¡h¡dro-5H-p¡rido[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo por 4-[7-(3-benciloxi-1-naftil)-2-[(1R)-2-[et¡l(met¡l)am¡no]-1-met¡l-etox¡]-6,8-d¡h¡dro-5H-p¡rido[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo en la Etapa C. ES+APCI MS m/z 558,6 [M+H]+.
Ejemplo 134
(R)-1-(4-(7-(3-hidroxinaftalen-1-il)-2-(piperidin-3-ilmetoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona

Etapa A: (3R)-3-(hidroximetil)piperidin-1-carboxilato de terc-butilo: Se añadió TEA (1,76 g, 17,4 mmol, 2,42 ml) a una solución de [(3R)-3-piperidil]metanol (1,0 g, 8,68 mmol) en THF (25,0 ml), seguido de la adición de una solución de Boc2Ü (1,89 g, 8,68 mmol, 1,99 ml) en THF (5 ml) a 15 °C. La mezcla se agitó a 15 °C durante 12 horas. El disolvente se eliminó al vacío y el residuo se disolvió en acetato de etilo (50 ml) y H2O (30 ml). La solución se acidificó con HCl (6 M) a pH ~6 y las capas se separaron. Los extractos orgánicos se lavaron con salmuera (3 x 50 ml) y los extractos orgánicos combinados se concentraron a sequedad para dar (3R)-3-(hidroximetil)piperidin-1-carboxilato de terc-butilo (1,68 g, 7,80 mmol, 89,9 % de rendimiento, 100 % de pureza) en forma de cristales incoloros. 1H RMN (400 MHz, cloroformo-d) 6 = 3,73 (s a, 2H), 3,51 (d a, J = 6,8 Hz, 2H), 3,05 (s a, 2H), 1,83 - 1,71 (m, 2H), 1,62 (s a, 1H), 1,46 (s, 9H), 1,44 - 1,37 (m, 1H), 1,35 - 1,22 (m, 1H).

Etapa B: 4-[7-(3-benciloxi-1-naftil)-2-[[(3R)-1-ferc-butoxicarbonil-3-piperidil]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: A una solución de (3R)-3-(hidroximetil)piperidin-1-carboxilato de terc-butilo (332 mg, 1,54 mmol) en THF (5 ml) se le añadió t-BuONa (223 mg, 2,32 mmol). Luego, se añadió una solución de 4-[7-(3-benciloxi-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (500 mg, 772 umol) en THF (5 ml) y la mezcla se agitó a 0 °C durante 1 hora. La mezcla de reacción se filtró y se concentró a presión reducida y el residuo se purificó por cromatografía en columna eluyendo con éter de petróleo/acetato de etilo (10/1 a 3/1) para dar 4-[7-(3-benciloxi-1-naftil)-2-[[(3R)-1-ferc-butoxicarbonil-3-piperidil]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (510 mg, 628 umol, 81,4 % de rendimiento, 98,4 % de pureza) en forma de un sólido de color blanco. ES+APCI MS m/z 799,4 [M+H]+.
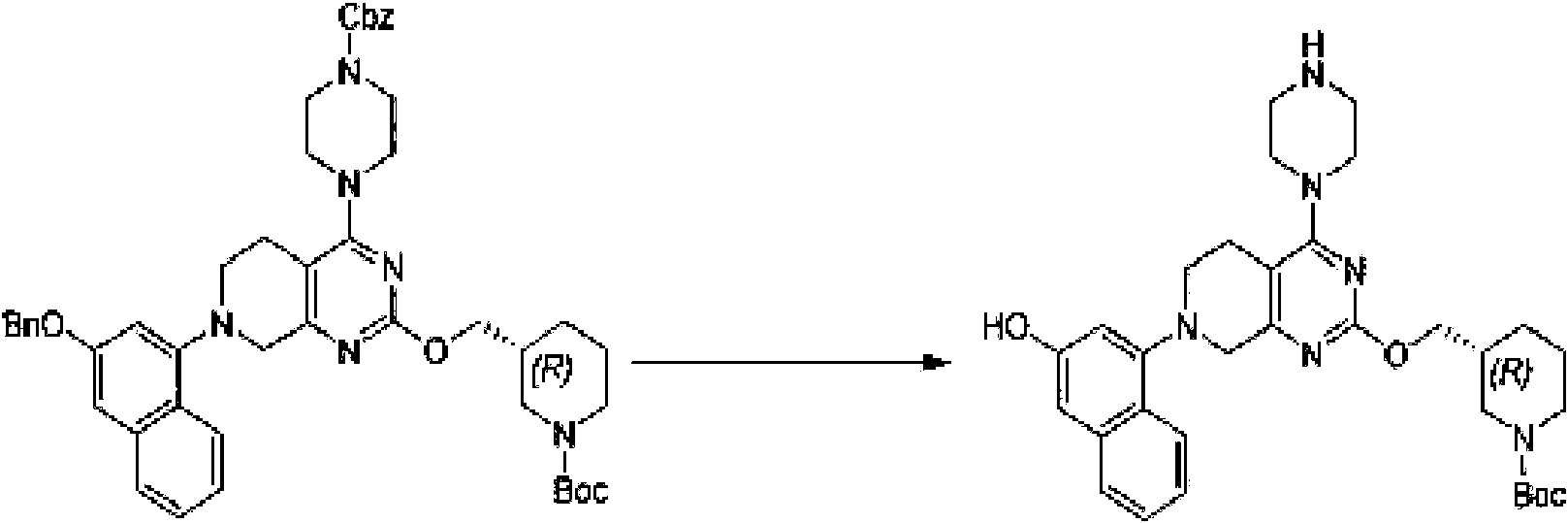
Etapa C: (3R)-3-[[7-(3-hidroxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]piperidin-1-carboxilato de terc-butilo: Una solución de 4-[7-(3-benciloxi-1-naftil)-2-[[(3R)-1-ferc- butoxicarbonil-3-piperidil]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (25o mg, 313 umol) en MeOH (5 ml) se purgó con NH3 (10 %, p/p) y después se añadió Pd al 10 %/C (50 mg). La suspensión se desgasificó al vacío y la mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 15 °C durante 12 horas. La mezcla de reacción se filtró y se concentró a presión reducida a sequedad para dar (3R)-3-[[7-(3-hidroxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]piperidin-1-carboxilato de terc-butilo (126 mg, 211 umol, 67,4 % de rendimiento, 96,2 % de pureza) en forma de un aceite incoloro. ES+APCI MS m/z 575,5 [M+H]+.

Etapa D: (3R)-3-[[7-(3-hidroxi-1-naftil)-4-(4-prop-2-enoilpiperazin-1-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]piperidin-1 -carboxilato de terc-butilo: A una solución de (3R)-3-[[7-(3-hidroxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]piperidin-1-carboxilato de terc-butilo (126 mg, 219 umol) y TEA (33,3 mg, 329 umol, 45,8 ul) en DCM (5,0 ml) se le añadió gota a gota prop-2-enoato de prop-2-enoílo (24,9 mg, 197 umol) a -40 °C y la reacción se agitó durante 30 minutos a -40 °C. La mezcla de reacción se inactivó mediante la adición de MeOH (0,5 ml) y se concentró a sequedad. El residuo se disolvió en EtOAc (50 ml) y H2O (20 ml). La solución resultante se acidificó con HCl (1 M) a pH ~6 y las capas se separaron. Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para dar (3R)-3-[[7-(3-hidroxi-1-naftil)-4-(4-prop-2-enoilpiperazin-1-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]piperidin-1 -carboxilato de terc-butilo (120 mg, 172 umol, 78,5 % de rendimiento, 90,2 % de pureza) en forma de un aceite de color amarillo, que se usó directamente para la siguiente etapa sin purificación adicional. ES+APCI MS m/z 629,6 [M+H]+.

Etapa E: 1-[4-[7-(3-hidroxi-1-naftil)-2-[[(3R)-3-piperidil]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de (3R)-3-[[7-(3-hidroxi-1-naftil)-4-(4-prop-2-enoilpiperazin-1-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]piperidin-1 -carboxilato de ferc-butilo (120 mg, 191 umol) en DCM (3,0 ml) se le añadió TFA (326 mg, 2,86 mmol, 212 ul) a 0 °C. La mezcla se agitó a 15 °C durante 2 horas en una atmósfera de N2. La mezcla de reacción se basificó con NH3 (30 % en agua, tres gotas) y se concentró a sequedad. El residuo
se purificó por HPLC prep. (columna: Phenomenex Synergi C18 150 * 25 * 10 um; fase móvil: [agua (0,225 % de ácido fórmico)-ACN]; % de B: 5 % - 35 %, 10 min) para dar 1-[4-[7-(3-hidroxi-1-naftil)-2-[[(3R)-3-piperidil]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (24,5 mg, 38,9 umol, 20,4 % de rendimiento) en forma de un sólido de color amarillo. ES+APCI m S m/z 529,4 [M+H]+.
Ejemplo 135
1-[4-[2-[3-(4-acetilpiperazin-1-il)propoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona

Etapa A: 1-[4-(3-h¡drox¡prop¡l)p¡peraz¡n-1-¡l]etanona: A una solución de 1-piperazin-1-iletanona (2,00 g, 15,6 mmol) y K2CO3 (4,31 g, 31,2 mmol) en CH3CN (50,0 ml) se le añadió 3-bromopropan-1-ol (3,25 g, 23,4 mmol). La mezcla se agitó a 80 °C durante 5 horas. El sólido se filtró y el filtrado se evaporó para dar 1-[4-(3-hidroxipropil)piperazin-1-¡l]etanona (2,00 g, 10,7 mmol, 68,8 % de rendimiento) en forma de un aceite incoloro.
Se preparó 1-[4-[2-[3-(4-acetilpiperazin-1-il)propoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona siguiendo el Ejemplo 131 sustituyendo 1-[4-(3-hidroxipropil)piperazin-1-il]etanona por 4-[7-(3-benciloxi-1-naftil)-2-[(1R)-2-[etil(metil)amino]-1-metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo en la Etapa C. ES+APCI MS m/z 600,3 [M+H]+.
Ejemplo 136
1-[4-[7-(3-hidroxi-1-naftil)-2-[2-(3-metoxipirrolidin-1-il)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona

Etapa A: 2-(3-metoxip¡rrol¡d¡n-1-¡l)etanol: A una solución de 3-metoxipirrolidina (450 mg, 3,27 mmol, HCl) y 2-bromoetanol (408 mg, 3,27 mmol) en CH3CN (10 ml) se le añadió K2CO3 (1,36 g, 9,81 mmol). La mezcla se agitó a
80 °C durante 3 horas. El sólido se filtró y el filtrado se evaporó para dar 2-(3-metox¡pirrol¡d¡n-1-¡l)etanol (450 mg, 3,10 mmol, 94,8 % de rendimiento) en forma de un aceite incoloro.
Etapa B: 4-[7-(3-benciloxi-1-naftil)-2-[2-(3-metoxipirrolidin-1-il)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: Una mezcla de 4-[7-(3-benciloxi-1-naftil)-2-metilsulfinil- 6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (500 mg, 771 umol), 2-(3-metoxipirrolidin-1-il)etanol (224 mg, 1,54 mmol) y t-BuONa (222 mg, 2,32 mmol) en tolueno (10 ml) se agitó a 20 °C durante 1 hora en una atmósfera de N2. La mezcla se enfrió a 0 °C y se añadió HCl (2 M) hasta pH ~7. La mezcla se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía en columna usando 0 ^10 % de MeOH/DCM como eluyente para dar 4-[7-(3-benciloxi-1-naftil)-2-[2-(3-metoxipirrolidin-1-il)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (110 mg, 147,9 umol, 19,1 % de rendimiento) ES+APCI Ms m/z 729,2 [M+H]+.
Se preparó 1-[4-[7-(3-hidroxi-1-naftil)-2-[2-(3-metoxipirrolidin-1-il)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona siguiendo el Ejemplo 131 sustituyendo 4-[7-(3-benciloxi-1-naftil)-2-[2-(3-metoxipirrolidin-1-il)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo por 4-[7-(3-benciloxi-1-naftil)-2-[(1R)-2-[etil(metil)amino]-1-metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo en la Etapa C. ES+ApCI MS m/z 559,3 [M+H]+.
Ejemplo 137
1-[4-[7-(3-hidroxi-1-naftil)-2-[3-(3-metoxipirrolidin-1-il)propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona

Etapa A: (3-metox¡p¡rrol¡d¡n-1-¡l)propan-1-ol: A una solución de 3-metoxipirrolidina (500 mg, 3,63 mmol, HCl) y 3-bromopropan-1-ol (505 mg, 3,63 mmol) en CH3CN (10 ml) se le añadió K2CO3 (1,51 g, 10,9 mmol). La mezcla se agitó a 20 °C durante 5 horas. El sólido se filtró y el filtrado se evaporó para dar 3-(3-metoxipirrolidin-1-il)propan-1-ol (540 mg, 3,39 mmol, 93,3 % de rendimiento).
Se preparó 1-[4-[7-(3-hidroxi-1-naftil)-2-[3-(3-metoxipirrolidin-1-il)propoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona siguiendo el Ejemplo 131 sustituyendo (3-metlzoxipirrolidin-1-il)propan-1-ol por (2^)-1-[etil(metil)amino]propan-2-ol en la Etapa B. ES+APCI MS m/z 573,3 [M+H]+.
Ejemplo 138
1-[4-[2-[2-(3,3-difluoroazetidin-1-il)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona
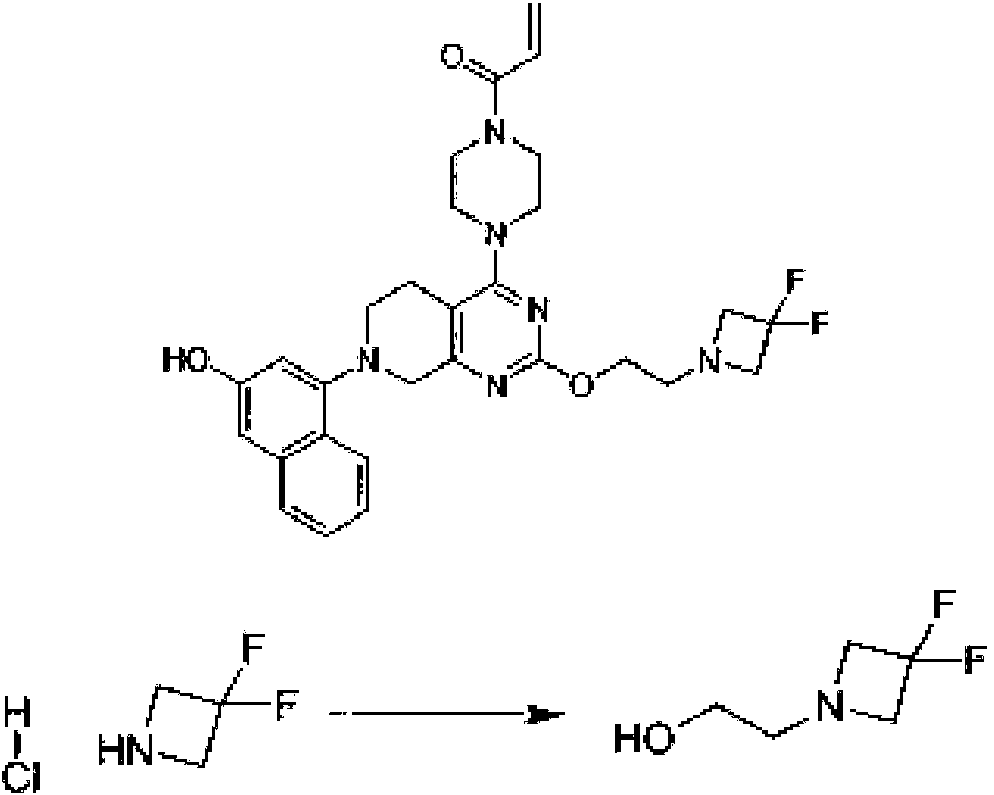
Etapa A: 2-í3.3-d¡fluoroazet¡d¡n-1-¡l)etanol: A una solución de 3,3-difluoroazetidina (500 mg, 3,86 mmol, HCl) y 2-bromoetanol (482 mg. 3,86 mmol. 274 ul) en CH3CN (10 ml) se le añadió K2CO3 (1.60 g. 11,5 mmol) y la reacción se agitó a 80 °C durante 16 horas. La reacción se filtró y el filtrado se evaporó para dar 2-(3,3-d¡fluoroazet¡d¡n-1-il)etanol (300 mg. 2,19 mmol. 56,7 % de rendimiento).
Se preparó 1-[4-[2-[2-(3,3-d¡fluoroazet¡d¡n-1-¡l)etox¡]-7-(3-h¡drox¡-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-iljpiperazin-1 -il]prop-2-en-1 -ona siguiendo el Ejemplo 131 sustituyendo 2-(3.3-difluoroazetldln-1 -il)etanol por (2R)-1-[etil(metil)amino]propan-2-ol en la Etapa B. ES+Ap CI MS m/z 551,4 [M+H]+.
Ejemplo 139
1-[4-[2-[2-(3,3-d¡fluorop¡rrol¡d¡n-1-¡l)etox¡]-7-(3-h¡drox¡-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-il]prop-2-en-1-ona
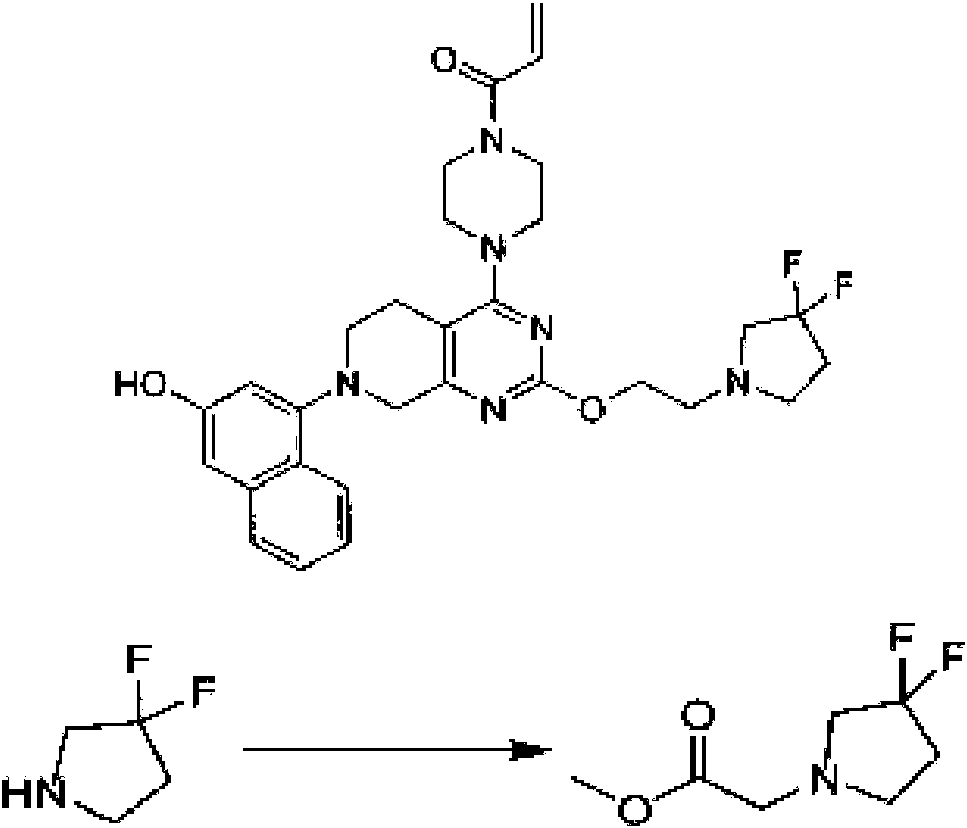
Etapa A: 2-(3.3-D¡fluorop¡rrol¡d¡n-1-¡l)acetato de metilo: A una suspensión de 2-bromoacetato de metilo (1,17 g.
7,67 mmol. 723 ul) en DCM (10 ml) enfriada a 0 °C se le añadieron TEA (1,76 g. 17,4 mmol. 2,42 ml) y 3,3-difluoropirrolidina (1,00 g. 6,97 mmol. HCl) y la mezcla de reacción se agitó a 20 °C durante 16 horas. La reacción se filtró y el filtrado se evaporó. El residuo se purificó por cromatografía en columna con 0.5 %-->20 % de MeOH/DCM como eluyente para dar 2-(3,3-d¡fluorop¡rrol¡d¡n-1-¡l)acetato de metilo (580 mg. 3,24 mmol. 46,4 % de rendimiento). 1H RMN (400 MHz, cloroformo-d) ó = 3,73 (s. 3H). 3,38 (s. 2H). 3,11 (t. J = 13,6 Hz. 2H). 2,92 (t. J = 6.8 Hz. 2H). 2,36-2,26 (m. 2H).

Etapa B: 2-(3,3-d¡fluorop¡rrolid¡n-1-¡l)etanol: A una solución de LÍAIH4 (184 mg, 4,86 mmol) en THF (5,0 ml) se le añadió gota a gota una solución de 2-(3,3-d¡fluorop¡rrolid¡n-1-¡l)acetato de metilo (580 mg. 3,24 mmol) en THF (5.0 ml) a 0 °C. La mezcla se calentó a 20 °C y se agitó durante 3 horas. La mezcla se inactivó mediante la adición de solución acuosa saturada de sulfato de sodio (1,50 ml). La reacción se filtró y el filtrado se concentró al vacío para dar 2-(3,3-d¡fluoropirrol¡d¡n-1-¡l)etanol (330 mg, 2,18 mmol, 67,4 % de rendimiento) en forma de un aceite incoloro. 1H RMN (400 MHz, cloroformo-d) 5 = 3,64 (t, J = 5,2 Hz, 2H), 2,97 (t, J = 13,2 Hz, 2H), 2,82 (t, J = 7,2 Hz, 2H), 2,68 (t, J = 5,2 Hz, 2H), 2,49 (s a, 1H), 2,34-2,24 (m, 2H).
Se preparó 1-[4-[2-[2-(3,3-d¡fluorop¡rrol¡d¡n-1-¡l)etox¡]-7-(3-h¡drox¡-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡mid¡n-4-¡l]piperazin-1-¡l]prop-2-en-1-ona siguiendo el Ejemplo 131 sustituyendo 2-(3,3-d¡fluorop¡rrolid¡n-1-¡l)etanol por (2R)-1- [etil(metil)amino]propan-2-ol en la Etapa B. Es Ap CI MS m/z 565,3 [M+H]+.
Ejemplo 140
2- [3-[[7-(3-hidrox¡-1-naft¡l)-4-(4-prop-2-enoilp¡peraz¡n-1 -¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]pirim¡din-2-¡l]oximet¡l]p¡rrol¡d¡n-1-¡l]-W,W-d¡met¡l-acetam¡da

Etapa A: 1-[2-(d¡met¡lam¡no)-2-oxo-et¡l]p¡rrol¡d¡n-3-carbox¡lato de metilo: Una solución de pirrolidin-3-carboxilato de metilo (1,00 g, 6,04 mmol, HCl) y NaHCO3 (1,01 g, 12,1 mmol, 470 ul) en ACN (200,0 ml) se agitó a 10 °C durante 5 minutos. Luego, se añadió una solución de 2-bromo-N,N-dimet¡l-acetamida (1,00 g, 6,04 mmol) en ACN (5,00 ml) a 10 °C y la reacción se agitó a 10 °C durante 6 horas seguido de agitación a 50 °C durante 2 horas. La mezcla se filtró y los sólidos se lavaron con DCM (3 x 15 ml). El filtrado se concentró al vacío y el residuo se purificó por cromatografía en columna usando 0 ^10 % de MeOH/DCM como eluyente to para dar 1-[2-(dimet¡lam¡no)-2-oxo-et¡l]p¡rrol¡d¡n-3-carboxilato de metilo (480 mg, 2,02 mmol, 33,4 % de rendimiento).

Etapa B: 2-[3-(h¡drox¡met¡l)p¡rrol¡d¡n-1-¡l]-N,N-d¡metil-acetam¡da: A una solución de 1-[2-(dimet¡lam¡no)-2-oxo-et¡l]p¡rrolid¡n-3-carboxilato de metilo (500 mg, 2,33 mmol) en THF (10 ml) se le añadió L¡AlH4 (203 mg, 5,36 mmol) a -60 °C y la mezcla de reacción se agitó a -60 °C durante 10 minutos. La mezcla de reacción se inactivó mediante la adición de Na2SO4 saturado (0,4 ml) y la suspensión se filtró. La torta de filtro se lavó con THF (3 x 50 ml) y el filtrado se concentró al vacío para dar 2-[3-(h¡drox¡metil)p¡rrol¡d¡n-1-¡l]-W.W-d¡met¡l-acetam¡da (400 mg, 2,15 mmol, 92,2 % de rendimiento) en forma de un aceite de color pardo. ES+APCI MS m/z 187,1 [M+H]+.

Etapa C: 4-[7-(3-benciloxi-1-naftil)-2-[[1-[2-(dimetilamino)-2-oxoetil]pirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo: Una mezcla de 4-[7-(3-benciloxi-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (500 mg, 772 umol), 2-[3-(hidroximetil)pirrolidin-1-il]-N,N-dimetil-acetamida (216 mg, 1,16 mmol) y NaOBu-t (148 mg, 1,54 mmol) en tolueno (10 ml) se agitó a 15 °C durante 15 minutos. La mezcla de reacción se purificó directamente por cromatografía sobre gel de sílice usando 20^100 % de EtOAc/éter de petróleo para dar 4-[7-(3-benciloxi-1-naftil)-2-[[1-[2-(dimetilamino)-2-oxoetil]pirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (310 mg, 395 umol, 51,1 % de rendimiento, 98 % de pureza) en forma de un sólido de color pardo. ES+APCI MS m/z 770,4 [M+H]+.
Se preparó 2-[3-[[7-(3-hidroxi-1-naftil)-4-(4-prop-2-enoilpiperazin-1-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]pirrolidin-1-il]-W,W-dimetil-acetamida siguiendo el Ejemplo 131 sustituyendo 4-[7-(3-benciloxi-1-naftil)-2-[[1-[2-(dimetilamino)-2-oxo-etil]pirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d)pirimidin-4-il]piperazin-1-carboxilato de bencilo por 4-[7-(3-benciloxi-1-naftil)-2-[(1R)-2-[etil(metil)amino]-1-metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo en la Etapa C. ES+APCI MS m/z 600,3 [M+H]+.
Ejemplo 141
1-[4-[2-[[1-(2-hidroxietil)pirrolidin-3-il]metoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-SH-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona

Etapa A: pirrolidin-3-carboxilato de metilo: A una solución de 3-metilpirrolidin-1,3-dicarboxilato de 1 -(terc-butilo) (10,0 g, 43,6 mmol) en DCM (50 ml) se le añadió HCl/dioxano (4 M, 109 ml) a 0 °C y se agitó a 0 °C durante 1 hora. La mezcla se concentró al vacío para dar pirrolidin-3-carboxilato de metilo (7,00 g, bruto, HCl) en forma de un aceite de color pardo. 1H RMN (400 MHz, metanol-d4) 6 = 3,77 (s, 3H), 3,56 - 3,53 (m, 2H), 3,41 - 3,37 (m, 3H), 2,40 - 2,24 (m, 2H).

Etapa B: 1-(2-benc¡lox¡et¡l)p¡rrolid¡n-3-carbox¡lato de metilo: Una solución de pirrolidin-3-carboxilato de metilo (3,0 g, 18,1 mmol, HCl), Cs2CO3 (17,7 g, 54,3 mmol) y KI (301 mg, 1,81 mmol) en MeCN (60 ml) se agitó a 15 °C durante 5 min.
Después, se añadió una solución de 2-bromoetoximetilbenceno (4,67 g, 21,7 mmol, 3,43 ml) en ACN (15 ml) a la mezcla a 15 °C y se agitó a 15 °C durante 1 hora. Luego, la mezcla se calentó a 50 °C y se agitó a 50 °C durante 12 horas. La mezcla de reacción se filtró y la torta de filtro se lavó con DCM (3 x 30 ml) y el filtrado se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex luna C18250 * 50 mm * 10 um;fase móvil: [agua (0,1 % de TFA)-ACN];% de B:
10 % de Ac N-40 % de ACN, 30 min, 40 % min) para dar 1-(2-bencilox¡et¡l)p¡rrol¡din-3-carbox¡lato de metilo (1,48 g, 5,06 mmol, 27,9 % de rendimiento, 90,0 % de pureza) en forma de un aceite de color pardo. 1H RMN (400 MHz, cloroformod) ó = 7,36 - 7,28 (m, 5H), 4,56 (s, 2H), 3,70 (s, 3H), 3,61 - 3,58 (t, J = 6,0 Hz, 2H), 3,02 - 3,00 (m, 2H), 2,78 - 2,67 (m, 4H), 2,58 - 2,49 (m, 1H), 2,11 - 2,08 (m, 2H)

Etapa C: (1-(2-(benc¡loxi)et¡l)p¡rrol¡d¡n-3-¡l)metanol: A la solución de 1-(2-bencilox¡et¡l)p¡rrol¡din-3-carbox¡lato de metilo (1,38 g, 5,24 mmol) en THF (27 ml) se le añadió L¡AlH4 (457 mg, 12 mmol) a -10 °C y se agitó a -10 °C durante 0,5 horas. La mezcla de reacción se inactivó con Na2SO4 saturado (1 ml) y se filtró, la torta de filtro se lavó con THF (5 x 30 ml) y el filtrado se concentró al vacío para dar (1-(2-(bencilox¡)et¡l)p¡rrol¡din-3-¡l)metanol (1,30 g, 3,31 mmol, 63,3 % de rendimiento, 60,0 % de pureza) en forma de un aceite de color pardo. ES+APCl MS m/z 236,1 [M+H]+.
Se preparó 1-[4-[2-[[1-(2-h¡drox¡et¡l)p¡rrol¡d¡n-3-¡l]metox¡]-7-(3-h¡drox¡-1-naftil)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]piperazin-1-¡l]prop-2-en-1-ona siguiendo el Ejemplo 136 sustituyendo (1-(2-(bencilox¡)et¡l)p¡rrol¡din-3-¡l)metanol por 2-(3-metoxipirrol¡d¡n-1-¡l)etanol en la Etapa B. e S+APCI MS m/z 559,3 [M+H]+.
Ejemplo 142
1-[4-[2-(2-h¡drox¡etox¡)-7-(3-h¡drox¡-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡mid¡n-4-¡l]p¡peraz¡n-1-¡l]prop-2-en-1-ona

Se preparó 1-[4-[2-(2-h¡drox¡etox¡)-7-(3-h¡drox¡-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡perazin-1-¡l]prop-2-en-1-ona siguiendo el Ejemplo 136 sustituyendo 2-[terc-but¡l(dimet¡l)s¡l¡l)ox¡etanol por 2-(3-metoxipirrol¡d¡n-1-il)etanol en la Etapa B. ES+APCI MS m/z 476,2 [M+H]+.
Ejemplo 143
1-[4-[2-(2,3-d¡h¡drox¡propox¡)-7-(3-h¡drox¡-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]p¡peraz¡n-1-¡l]prop-2-en-1-ona

Se preparó 1-[4-[2-(2,3-dihidroxipropoxi)-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona siguiendo el Ejemplo 136 sustituyendo (2,2-dimetil-1,3-dioxolan-4-il)metanol por 2-(3-metoxipirrolidin-1-il)etanol en la Etapa B. ES+APCI MS m/z 506,3 [M+H]+.
Ejemplo 144
(S)-1-(4-(7-(3-hidroxinaftalen-1-il)-2-((1-(2-metoxietil)pirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona

Etapa A: (2S)-2-(benc¡loximet¡l)p¡rrol¡d¡n-1-carbox¡lato de terc-butilo: A una suspensión de NaH (2,38 g, 59,6 mmol, 60 % de pureza) en THF (50 ml) se le añadió una solución de (2S)-2-(hidroximetil)pirrolidin-1-carboxilato de tere-butilo (10 g, 49,69 mmol) en THF (50 ml) a 0 °C y la mezcla se agitó a 10 °C durante 1 hora. Se añadió gota a gota bromometilbenceno (12,8 g, 74,5 mmol, 8,85 ml) a 0 °C y la mezcla se agitó a 10 °C durante 16 horas. La mezcla se inactivó mediante la adición de una solución acuosa saturada de cloruro de amoniaco (20 ml) y después se diluyó con acetato de etilo (200 ml) y agua (100 ml). La capa orgánica separada se lavó con agua (100 ml) y salmuera (100 ml), se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo/acetato de etilo 100/1 a 5/1) para dar (2S)-2-(benciloximetil)pirrolidin-1-carboxilato de tere-butilo (8,2 g, 28,06 mmol, 56,5 % de rendimiento, 99,7 % de pureza), que se obtuvo en forma de un aceite incoloro. ES+APCI MS m/z 192,1 [M+H-Boc]+.

Etapa B: (2S)-2-(bencilox¡met¡l)p¡rrol¡d¡na: A una solución de (2S)-2-(benciloximetil)pirrolidin-1- carboxilato de terebutilo (8,2 g, 28,14 mmol) en CH2Cl2 (28 ml) se le añadió gota a gota TfA (43,1 g, 378 mmol, 28,0 ml) a 0 °C en una atmósfera de nitrógeno. La mezcla se agitó a 15 °C durante 1 hora. La mezcla se concentró al vacío. El residuo se diluyó con diclorometano (100 ml) y después se lavó con hidróxido de sodio acuoso 1 M (10 ml) hasta que la capa acuosa alcanzó un pH de ~10. La capa orgánica separada se lavó con salmuera (2 x 20 ml), se secó sobre sulfato de sodio, se filtró y se concentró al vacío para dar (2S)-2-(benciloximetil)pirrolidina (4 g, 20,9 mmol, 74,3 % de rendimiento. LCMS ES+APCI MS m/z 192,2 [M+H]+.

Etapa C: (2S)-2-(benc¡lox¡met¡l)-1-(2-metox¡et¡l)p¡rrolid¡na: Una mezcla de 1-bromo-2-metoxi-etano (0,9 g, 6,48 mmol, 608 ul), (2S)-2-(bencilox¡met¡l)p¡rrolid¡na (1,24 g, 6,48 mmol) y K2CO3 (2,68 g, 19,4 mmol) en CH3CN (20 ml) se agitó a 15 °C durante 1 hora y después a 78 °C durante 12 horas. La mezcla se diluyó con acetato de etilo (50 ml) y agua (50 ml). La capa orgánica separada se lavó con salmuera (1 x 50 ml), se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (acetato de etilo/diclorometano/metanol 1/1/0 a 10/10/2) para dar (2S)-2-(bencilox¡met¡l)-1-(2-metox¡et¡l)p¡rrol¡dina (900 mg, 3,61 mmol, 55,7 % de rendimiento) en forma de un aceite incoloro. ES+APCI MS m/z 250,2 [M+H]+.

Etapa D: (S)-(1-(2-metox¡et¡l)p¡rrolid¡n-2-¡l)metanol: A una solución de (2S)-2-(bencilox¡met¡l)-1-(2-metox¡et¡l)p¡rrol¡dina (900 mg, 3,61 mmol) en MeOH (20 ml) se le añadió Pd al 10 %/C (721,88 umol) y la suspensión se agitó en una atmósfera de H2 (344,74 kPa (50 psi)) a 10 °C durante 16 horas. La reacción se filtró y el filtrado se concentró al vacío para dar (S)-(1-(2-metoxiet¡l)p¡rrolid¡n-2-¡l)metanol (450 mg, 2,83 mmol, 78,30 % de rendimiento).

Etapa E: 4-[7-(3-Benciloxi-1-naftil)-2-[[(2S)-1-(2-metoxietil)pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l]piperazin-1-carbox¡lato de bencilo: Una mezcla de [(2S)-1-(2-metox¡et¡l)pirrol¡d¡n-2-¡l]metanol (245 mg, 1,54 mmol), 4-[7-(3-benc¡lox¡-1-naft¡l)-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡perazin-1-carbox¡lato de bencilo (500 mg, 772 umol) y t-BuONa (223 mg, 2,32 mmol) en THF (5 ml) se agitó a 20 °C durante 0,5 horas en una atmósfera de N2. La mezcla de reacción se vertió en H2O (30 ml) y la capa acuosa se extrajo con acetato de etilo (3 x 30 ml). Los extractos orgánicos combinados se lavaron con salmuera (30 ml), se secaron sobre Na2SO4 anhidro y se concentraron al vacío para dar un residuo. El residuo se purificó por cromatografía en columna (SO2, DCM/MeOH = 300/1 a 10:1) para dar 4-[7-(3-benc¡lox¡-1-naft¡l)-2-[[(2S)-1-(2-metox¡et¡l)p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]pir¡m¡d¡n-4-¡l]piperaz¡n-1-carbox¡lato de bencilo (260 mg, 349 umol, 45,3 % de rendimiento). ES+APCI MS m/z 743,4 [M+H]+.

Etapa F: 4-[7-(3-H¡drox¡-1-naft¡l)-2-[[(2S)-1-(2-metox¡et¡l)p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]pir¡m¡d¡n-4-¡l]piperaz¡n-1-carbox¡lato de terc-butilo: A una solución de 4-[7-(3-benc¡lox¡-1-naft¡l)-2-[[(2S)-1-(2-metox¡etil)p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-il]p¡peraz¡n-1-carbox¡lato de bencilo (240 mg, 323 umol) y (Boc)2O (141 mg, 646 umol, 148 ul) en MeOH (150 ml) se le añadió Pd al 10 %/C (100 mg) en una atmósfera de N2. La suspensión se desgasificó y se purgó 3 veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 40 °C durante 12 horas. La mezcla de reacción se filtró y el filtrado se concentró para dar 4-[7-(3-hidrox¡-1-naftil)-2-[[(2S)-1-(2-metox¡et¡l)p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-il]p¡peraz¡n-1-carbox¡lato de tercbutilo (215 mg, bruto). ES+APCI MS m/z 619,1 [M+H]+.

Etapa G: 4-[2-[[(2S)-1-(2-metoxietil)pirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol: A una solución de 4-[7-(3-hidroxi-1-naftil)-2-[[(2s)-1-(2-metoxietil)pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo (215 mg, 347 umol) en DCM (500 ul) se le añadió TFA (594 mg, 5,21 mmol, 385 ul) y la mezcla se agitó a 15 °C durante 1 hora. La mezcla se concentró al vacío para dar 4-[2-[[(2S)-1-(2-metoxietil)pirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (219 mg, 346 umol). ES+APCI MS m/z 519,4 [M+H]+.

Etapa H: 1-[4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-(2-metoxietil)pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una mezcla de 4-[2-[[(2S)-1-(2-metoxietil)pirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (219 mg, 346 umol) y D iEA (447 mg, 3,46 mmol, 603 ul) en diclorometano (4,00 ml) enfriada a -40 °C se le añadió una solución de prop-2-enoato de prop-2-enoílo (34,9 mg, 276,92 umol) en diclorometano (1,00 ml) en una atmósfera de nitrógeno. La mezcla se agitó a -40 °C durante 1 hora. La reacción se interrumpió mediante la adición de NaHCO3 saturado (2,00 ml) y la mezcla se vertió en hielo-agua (20 ml) y se extrajo con diclorometano (20 ml x 2). Los extractos orgánicos combinados se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Gemini 150 * 25 5 u; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN]; % de B: 32 %-62 %, 12 min) para dar 1-[4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-(2-metoxietil)pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (27 mg, 46,7 umol). ES+APCI MS m/z 573,3 [M+H]+.
Ejemplo 145
-[4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-isopropilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona


Etapa A: 4-[7-(3-Benciloxi-1-naftil)-2-[[(2S)-1-terc-butoxicarbonilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: A una solución de (2S)-2-(hidroximetil)pirrolidin-1-carboxilato de terc-butilo (1,24 g, 6,17 mmol) y 4-[7-(3-benciloxi-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (2 g, 3,09 mmol) en THF (50 ml) se le añadió t-BuONa (890 mg, 9,26 mmol) y la reacción se agitó a 15 °C durante 0,5 horas. La mezcla de reacción se vertió en H2O (100 ml) y se extrajo con acetato de etilo (3 x 100 ml). Los extractos orgánicos combinados se lavaron con salmuera (50 ml), se secaron sobre Na2SO4 anhidro y se concentraron al vacío para dar un residuo. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo = 100/1 a 1:1) para dar 4-[7-(3-benciloxi-1-naftil)-2-[[(2S)-1-tercbutoxicarbonilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (1,62 g, 2,02 mmol, 65,5 % de rendimiento). ES+APCI MS m/z 785,6 [M+H]+.

Etapa B: (2S)-2-[[7-(3-hidroxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]pirrolidin-1-carboxilato: Se burbujeó NH3 en MeOH (50 ml) a 15 °C durante 30 minutos. A esta solución se le añadió 4-[7-(3-benciloxi-1-naftil)-2-[[(2S)-1-terc-butoxicarbonilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (1,6 g, 2,04 mmol) seguido de Pd al 10 %/C seco (500 mg) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 40 °C durante 1 hora. Después, la mezcla se filtró y el filtrado se concentró al vacío para dar (2S)-2-[[7-(3-hidroxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]pirrolidin-1-carboxilato de terc-butilo (1,00 g, 1,78 mmol). ES+APCI MS m/z 561,5 [M+H]+.

Etapa C: (2S)-2-[[7-(3-hidroxi-1-naftil)-4-[4-(2,2,2-trifluoroacetil)piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]pirrolidin-1 -carboxilato de terc-butilo: A una solución de (2S)-2-[[7-(3-hidroxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]pirrolidin-1-carboxilato de terc-butilo (800 mg, 1,43 mmol) en DCM (5,00 ml) a 0 °C se le añadieron TFAA (599 mg, 2,85 mmol) y DIEA (737 mg, 5,71 mmol) y la reacción se agitó a 0 °C durante 0,5 horas. La mezcla de reacción se vertió en H2O (50 ml) y se extrajo con acetato de etilo (50 ml x 3). Los extractos orgánicos combinados se lavaron con salmuera (30 ml), se secaron sobre Na2SO4 anhidro, se filtraron y se
concentraron al vacío para dar (2S)-2-[[7-(3-hidroxi-1-naftil)-4-[4-(2,2,2-trifluoroacetil)piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]pirrolidin-1-carboxilato de terc-butilo (1,5 g, bruto). ES+APCI MS m/z 657,5 [M+H]+.
5
10
15 
Etapa D: 2,2,2-trifluoro-1-[4-[7-(3-hidroxi-1-naftil)-2-[[(2S)- pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-20 il]piperazin-1-il]etanona: A una solución de (2S)-2-[[7-(3-hidroxi-1-naftil)-4-[4-(2,2,2-trifluoroacetil)piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]pirrolidin-1-carboxilato de ferc-butilo (300 mg, bruto) en DCM (500 ul) se le añadió TFA (521 mg, 4,57 mmol, 338 ul) y la mezcla se agitó a 15 °C durante 1 hora. La mezcla se concentró al vacío para dar 2,2,2-trifluoro-1-[4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]etanona (306 mg). ES+APCI MS m/z 557,3 [M+H]+.
25
30
35 
Etapa E: 2,2,2-trifluoro-1-[4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-isopropilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]etanona: A una solución de acetona (132 mg, 2,28 mmol, 167 ul) y 2,2,2-trifluoro-1-[4-[7-(3-40 hidroxi-1-naftil)-2-[[(2S)-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]etanona (306 mg, bruto, TFA) en MeOH (5,00 ml) se le añadieron AcOH (54,8 mg, 912 umol, 52,2 ul) y NaBHaCN (115 mg, 1,83 mmol) y la mezcla se agitó a 15 °C durante 16 horas. La mezcla de reacción se diluyó con H2O (20 ml) y se extrajo con acetato de etilo (20 ml x 3). Los extractos orgánicos combinados se lavaron con salmuera (10 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para dar 2,2,2-trifluoro-1-[4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-isopropilpirrolidin-2-il]metoxi]-45 6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]etanona (300 mg, bruto). ES+Ap Ci MS m/z 599,5 [M+H]+.
50
55 
Etapa F: 4-[2-[[(2S)-1-isopropilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-piridor[3,4-d]pirimidin-7-qq ¡l]naftalen-2-ol: A una solución de 2,2,2-trifluoro-1-[4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-isopropilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]etanona (300 mg, bruto) en MeOH (10 ml) se le añadió K2CO3 (346 mg, 2,51 mmol) y la mezcla se agitó a 15 °C durante 1 hora. La mezcla de reacción se filtró y el filtrado se concentró para dar 4-[2-[[(2S)-1-isopropilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-¡l]naftalen-2-ol (250 mg). ES+APCI MS m/z 503,3 [M+H]+.
65

Etapa G: 1-[4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-isopropilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una mezcla de 4-[2-[[(2S)-1-isopropilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (250 mg, bruto) y DIEA (643 mg, 4,97 mmol, 866 ul) en DCM (5,00 ml) enfriada a -40 °C se le añadió una solución de prop-2-enoato de prop-2-enoílo (50,2 mg, 398 umol) en DCM (1 ml) en una atmósfera de nitrógeno. La mezcla se agitó a -40 °C durante 1 hora. La reacción se interrumpió mediante la adición de NaHCÜ3 saturado (2,00 ml). La mezcla se vertió en hielo-agua (20 ml) y se extrajo con DCM (20 ml x 2). Los extractos orgánicos combinados se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN]; % de B: 52 %-78 %, 12 min). 1-[4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-isopropilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (10 mg, 17,6 umol). ES+APCI MS m/z 557,3 [M+H]+.
Ejemplo 146
1- [4-[7-(3-hidroxi-1-naftil)-2-[[(3R)-pirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2- en-1-ona

Etapa A: 4-[7-(3-benciloxi-1-naftil)-2-[[(3R)-1-terc-butoxicarbonilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo: Una mezcla de 4-[7-(3-benciloxi-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (0,50 g, 772 umol), (3R)-3-(hidroximetil)pirrolidin-1-carboxilato de tere-butilo (311 mg, 1,54 mmol) y t-BuONa (223 mg, 2,32 mmol) en THF (10 ml) se agitó a 20 °C durante 1 hora. La mezcla se diluyó con agua (10 ml) y la capa acuosa se extrajo con acetato de éter (3 x 20 ml). Los extractos orgánicos combinados se lavaron con cloruro de sodio saturado (1 x 30 ml), se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/éter acetato = 3/1) para dar 4-[7-(3-benciloxi-1-naftil)-2-[[(3R)-1-terc-butoxicarbonilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (0,40 g, 499 umol). ES+APCI MS m/z 785,2 [M+H]+.

Etapa B: (3R)-3-[[7-(3-hidroxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]pirrolidin-1-carboxilato de terc-butilo: Se burbujeó NH3 en metanol (10 ml) a -78 °C durante 30 minutos. Se añadieron 4-[7-(3-benciloxi-1-naftil)-2-[[(3R)-1-terc-butoxicarbonilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (0,25 g, 319 umol) y Pd al 10 %/C (0,10 g) a la mezcla y se agitó a 10 °C durante 1 hora en una atmósfera de H2 (103,42 kPa (15 psi)). La mezcla se filtró y se concentró al vacío para dar (3R)-3-[[7-(3-hidroxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]pirrolidin-1-carboxilato de ferc-butilo (0,17 g, 303 umol). ES+APCI MS m/z 561,3 [M+H]+.

Etapa C: (3R)-3-[[7-(3-hidroxi-1-naftil)-4-(4-prop-2-enoilpiperazin-1-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]pirrolidin-1 -carboxilato de ferc-butilo: A una solución de (3R)-3-[[7-(3-hidroxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]pirrolidin-1-carboxilato de ferc-butilo (0,17 g, 303 umol) y Et3N (153 mg, 1,52 mmol, 211 ul) en diclorometano (4,00 ml) enfriada a -40 °C se le añadió prop-2-enoato de prop-2-enoílo (26,8 mg, 212 umol) y la mezcla se agitó a -40 °C durante 0,5 h. La mezcla se inactivó mediante la adición de metanol (0,10 ml) y se concentró al vacío. El residuo se purificó por cromatografía en columna (AbO3, diclorometano/metano = 10/1) para dar (3R)-3-[[7-(3-hidroxi-1-naftil)-4-(4-prop-2-enoilpiperazin-1-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]pirrolidin-1 -carboxilato de ferc-butilo (0,12 g, 189 umol). Es +APCI MS m/z 615,5 [M+H]+.

Etapa D: 1-[4-[7-(3-hidroxi-1-naftil)-2-[[(3R)-pirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: Una mezcla de (3R)-3-[[7-(3-hidroxi-1-naftil)-4-(4- prop-2-enoilpiperazin-1-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]pirrolidin-1-carboxilato de terc-butilo (0,10 g, 163 umol) y TFA (278 mg, 2,44 mmol, 181 ul) en diclorometano (0,20 ml) se agitó a 10 °C durante 1 hora. La mezcla se inactivó mediante la adición de NH3^H2O y el pH se ajustó hasta pH = 7. La mezcla se purificó por HPLC prep. (columna: Venusil XBP C8 150 * 25 * 10 um; fase móvil: [agua (0,225 % de ácido fórmico)-ACN]; % de B: 15 %-45 %, 10 min) para dar 1-[4-[7-(3-hidroxi-1-naftil)-2-[[(3R)-pirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (22,8 mg, 42,9 umol). ES+APCI MS m/z 515,4 [M+H]+.
E je m p lo 147
2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo
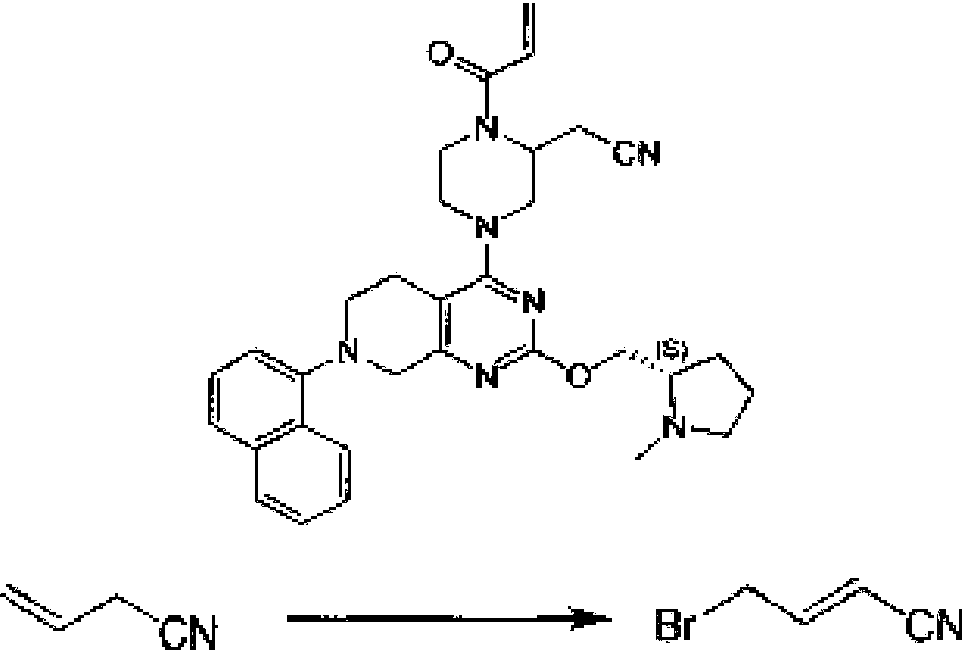
Etapa A: (Z) y (E)-4-bromobut-2-enonitr¡lo: A una solución de but-3-enonitrilo (98 g, 1,46 mol, 118 ml) en ferc-butanol (150 ml) y éter de petróleo (575 ml) se le añadió una solución de Br2 (233 g, 1,46 mol, 75,3 ml) en ferc-butanol (150 ml) a 15 °C y la reacción se agitó durante 30 minutos. Luego, a la reacción se le añadió una solución en etanol de etóxido de sodio (100 g, 0,6 mol, 850 ml). La mezcla de reacción se agitó durante 2 horas a 15 °C. Luego, la reacción se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía en columna usando 2 ^20 % de acetato de etilo/éter de petróleo como eluyente para dar una mezcla de (Z) y (£)-4-bromobut-2-enonitrilo (141 g, E/Z = 2,5/1, bruto) en forma de un aceite de color amarillo claro. 1H RMN (400 MHz, cloroformo-d) (E), 6 = 6,79 (td, J = 7,2, 16,0 Hz, 1H), 5,63 (d, J = 16,0 Hz, 1H), 4,00 (dd, J = 1,2, 6,8 Hz, 2H); (Z), 6 = 6,66 (td, J = 8,0, 10,8 Hz, 1H), 5,44 (d, J = 10,8 Hz, 1H), 4,16 (dd, J = 0,8, 8,0 Hz, 2H).

Etapa B: 2-(1 ,4-dibencilpiperazin-2-il)acetonitrilo: A una mezcla de W,W-dibenciletano-1,2-diamina (115 g, 480 mmol, 113 ml) y TEA (97,0 g, 959 mmol, 133 ml) en tolueno (1 l) se le añadió 4-bromobut-2-enonitrilo (70 g, bruto) gota a gota a 0 °C y la reacción se agitó a 15 °C durante 12 horas. La mezcla de reacción se filtró y el filtrado se concentró a presión reducida. El residuo se purificó por cromatografía en columna usando 5 ^50 % de EtOAc/éter de petróleo como eluyente para dar 2-(1,4-dibencilpiperazin-2-il)acetonitrilo (82 g, 240 mmol, dos etapas 37 % de rendimiento, 89,3 % de pureza) en forma de un semisólido de color amarillo claro. ES+APCI MS m/z 306,3 [M+H]+. 1H RMN (400 MHz, cloroformo-d) 6 = 7,40 - 7,23 (m, 10H), 3,85 - 3,76 (m, 1H), 3,54 - 3,44 (m, 3H), 3,07 - 2,96 (m, 1H), 2,94 -2,84 (m, 1H), 2,68 - 2,35 (m, 7H).

Etapa C: 2-p¡peraz¡n-2-¡laceton¡tr¡lo: A una solución de 2-(1,4-dibencilpiperazin-2-il)acetonitrilo (164 g, 536 mmol) en DCE (1500 ml) se le añadió carbonocloridato de 1-cloroetilo (306 g, 2,14 mol) gota a gota a 0 °C. Después de la adición, la mezcla de reacción se calentó a 85 °C durante aproximadamente 48 horas. El dicloroetano se evaporó y el residuo se recogió en MeOH (1500 ml) y se calentó a 70 °C durante 1 hora. La mezcla de reacción se concentró a presión reducida, los sólidos se trituraron con MTBE (3 x 3 l) y los sólidos se secaron a presión reducida para dar 2-piperazin-2-ilacetonitrilo. El producto bruto se purificó por recristalización con etanol y agua (8:1, v:v) para dar 2-piperazin-2-ilacetonitrilo en forma de un sólido de color blanquecino (53 g, 428 mmol, 40,0 % de rendimiento, 96,4 % de pureza, 2 HCl). ES+APCI MS m/z 126,2 [M+H]+. 1H RMN (400 MHz, D2O) 6 = 4,01 - 3,96 (m, 1H), 3,81 - 3,67 (m, 3H), 3,46 - 3,27 (m, 3H), 3,09 (d, J = 6,0 Hz, 2H).

Etapa D: 2-[4-(7-benc¡l-2-cloro-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡llaceton¡tr¡lo: A una mezcla de 7-bencil-2,4-dicloro-6,8-dihidro-5H-pirido[3,4-d]pirimidina (1.2 g. 4.08 mmol) y 2-p¡peraz¡n-2-¡laceton¡tr¡lo (808 mg.
4.08 mmol. 2HCl) en dioxano (24 ml) se le añadió DIEA (2.64 g. 20.4 mmol. 3.55 ml). La mezcla de reacción se agitó a 50 °C durante 3 horas. Después de que se completara. la mezcla de reacción se diluyó con agua (50 ml) y la capa acuosa se extrajo con EtOAc (3 x 80 ml). Las capas orgánicas combinadas se lavaron con salmuera (30 ml). se secaron sobre Na2SO4 y se concentraron al vacío para dar 2-[4-(7-benc¡l-2-cloro-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-il]aceton¡tr¡lo (1.56 g. 4.07 mmol. 100 % de rendimiento) en forma de un sólido de color pardo.

Etapa E: 4-(7-benc¡l-2-cloro-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡l)-2-(c¡anometil)p¡peraz¡n-1-carbox¡lato de tercbutilo: A 2-[4-(7-benc¡l-2-cloro-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l]aceton¡tr¡lo (1.56 g.4.07 mmol) se le añadió (Boc)2O (9.50 g. 43.5 mmol. 10 ml) y la mezcla se calentó a 50 °C durante 2 horas. Después de que se completara. la mezcla de reacción se purificó por cromatografía sobre gel de sílice usando 10^50 % de EtoAc/éter de petróleo como eluyente para dar 4-(7-benc¡l-2-cloro-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)-2-(cianometil)p¡peraz¡n-1-carbox¡lato de ferc-butilo (1.2 g. 2.44 mmol. 59.9 % de rendimiento. 98.3 % de pureza) en forma de un sólido de color pardo. ES+APCI MS m/z 483.3 [M+H]+. 1H RMN (400 MHz. CLOROFORMO-d) ó = 7.39 -7.27 (m. 5H). 4.59 (s a. 1H). 4.06 (d a. J = 13.6 Hz. 2H). 3.90 (d a. J = 11.6 Hz. 1H). 3.74 - 3.49 (m. 4H). 3.29 (dd. J = 4.0. 13.6 Hz. 1H). 3.18 (s a. 1H). 3.10 - 3.00 (m. 1H). 2.85 - 2.55 (m. 6H). 1.53 - 1.45 (m. 9H).

Etapa F: 4-[7-bencil-2-[[(2S)-1 -met¡lp¡rrol¡din-2-¡llmetoxi]-6,8-dihidro - 5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡perazin-1 -carboxilato de ferc-butilo: A una solución de [(2S)-1-metilp¡rrol¡d¡n-2-¡l]metanol (655.74 mg.5.69 mmol.676 ul) en t Hf (25 ml) se le añadió NaH (182 mg. 4.55 mmol. 60 % de pureza) a 0 °C y la mezcla de reacción se agitó a 0 °C durante 0.5 h. A la mezcla se le añadió 4-(7-benc¡l-2-cloro-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de ferc-butilo (1.1 g.2.28 mmol) y la mezcla de reacción se agitó a 70 °C durante 12 horas en un tubo sellado en una atmósfera de N2. Después de que se completara. la mezcla de reacción se inactivó mediante la adición de NH4Cl saturado (20 ml) y la capa acuosa se extrajo con EtOAc (2 x 50 ml). Los extractos orgánicos combinados se lavaron con salmuera (10 ml). se secaron sobre Na2SO4. se filtraron y se concentraron al vacío para dar 4-[7-bencil-2-[[(2S)-1 -metilp¡rrol¡din-2-¡llmetoxi]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]pir¡m¡d¡n-4-il]-2-(cianomet¡l)piperazin-1 -carboxilato de terc-butilo (1.1 g. 1.86 mmol. 81.7 % de rendimiento. 95 % de pureza) en forma de un sólido de color pardo. ES+APCI MS m/z 562.4 [M+H]+.

Etapa G: 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo: Se burbujeó amoniaco en MeOH (30 ml) durante 5 minutos. A esta solución se le añadió 4-[7-bencil-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (950 mg, 1,69 mmol) y Pd al 10 %/C (200 mg). La suspensión se desgasificó al vacío y se purgó varias veces con H2 y la mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 40 °C durante 9 horas. Después de que se completara, la mezcla de reacción se filtró y se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi Max-RP 250 * 50 mm * 10 um;fase móvil: [agua (0,225 % de ácido fórmico)-ACN];% de B: 15 %-40 %, 30;58 %min) para dar 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo (400 mg, 830 umol, 49,1 % de rendimiento, 97,8 % de pureza) que se obtuvo en forma de un aceite de color pardo. ES+APCI MS m/z 472,4 [M+H]+. 1H RMN (400 MHz, CLOROFORMO-d) 6 = 4,59 (s a, 1H), 4,43 - 4,28 (m, 1H), 4,18 - 4,09 (m, 1H), 4,07 - 3,90 (m, 4H), 3,88 -3,78 (m, 1H), 3,21 (dd a, J = 3,2, 13,6 Hz, 2H), 3,16 - 3,05 (m, 2H), 3,03 - 2,90 (m, 2H), 2,82 - 2,55 (m, 4H), 2,48 (s, 3H), 2,33 - 2,22 (m, 1H), 2,11 - 2,00 (m, 1H), 1,91 - 1,62 (m, 4H), 1,51 (s, 9H).

Etapa H: 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo: A una solución de 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (150 mg, 318 umol), 1-bromonaftaleno (98,8 mg, 477 umol, 66,3 ul), Cs2CO3 (311 mg, 954 umol), y RuPhos (29,7 mg, 63,6 umol) en tolueno (3 ml) se le añadió Pd2(dba)3 (29,1 mg, 31,8 umol) y la suspensión se desgasificó al vacío y se purgó 3 veces con N2. La mezcla de reacción se agitó a 100 °C durante 12 horas. La mezcla se repartió entre agua (10 ml) y EtOAc (20 ml) y las capas se separaron. Posteriormente, la capa acuosa se extrajo con EtOAc (3 x 20 ml). Los extractos orgánicos combinados se lavaron con salmuera (20 ml), se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía ultrarrápida inversa para dar 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (70 mg, 111 umol, 35,0 % de rendimiento, 95,0 % de pureza) en forma de un sólido de color pardo. ES+APCI MS m/z 598,6 [M+H]+. 1H RMN (400 MHz, cloroformo-d) 6 = 8,21 - 8,20 (m, 1H), 7,87 - 7,85 (m, 1H), 7,61 - 7,59 (d, J = 8,0 Hz, 1H), 7,51 - 7,49 (m, 2H), 7,45 - 7,41 (t, J = 7,6 Hz, 1H), 7,15 - 7,13 (d, J = 7,2 Hz, 1H), 4,63 (s a, 1H), 4,46 (s a, 1H), 4,33 - 4,28 (m, 4H), 4,10 - 4,07 (m, 2H), 3,97 - 3,94 (d a, J = 11,6 Hz, 1H), 3,46 (s a, 1H), 3,31 - 3,27 (d a, J = 14,0 Hz, 1H), 3,07 -3,01 (m, 2H), 2,77 (s a, 3H), 2,55 (s a, 3H), 2,35 (s a, 1H), 1,82 (s a, 7H), 1,52 (s, 9H).

Etapa I: 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-iljacetonitrilo: A una solución de 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo (60 mg, 100 umol) en DCM (0,1 ml) se le añadió TFA (154 mg, 1,35 mmol, 0,1 ml) a 15 °C y se agitó a 15 °C durante 1 hora. La mezcla de reacción se concentró al vacío para dar 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (50 mg) en forma de un aceite de color pardo.

Etapa J: 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo: A una solución de 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (50 mg, 81,8 umol, TFA) y DIEA (106 mg, 817 umol, 142 ul) en DCM (0,2 ml) se le añadió prop-2-enoato de prop-2-enoílo (11,3 mg, 89,9 umol) a 0 °C y la reacción se agitó a 0-15 °C durante 1,5 horas. La mezcla de reacción se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi C18 150 * 30 mm * 4 um;fase móvil: [agua (0,225 % de ácido fórmico)-ACN];% de B: 15 %-45 %,10,5 min) para dar 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1- prop-2-enoil-piperazin-2-il]acetonitrilo (9,16 mg, 15,1 umol, 18,5 % de rendimiento, 98,7 % de pureza, Formiato) en forma de un aceite de color amarillo. ES+APCI MS m/z 552,5 [M+H]+.1H RMN (400 MHz, cloroformo-d) 6 = 8,45 (s a, 1H), 8,22 - 8,19 (m, 1H), 7,87 - 7,85 (m, 1H), 7,62 - 7,60 (d, J = 8,0 Hz, 1H), 7,51 - 7,49 (m, 2H), 7,45 - 7,43 (t, J = 7,6 Hz, 1H), 7,15 - 7,13 (d, J = 7,2 Hz, 1H), 6,60 (s a, 1H), 6,42 - 6,38 (m, 1H), 5,84 - 5,82 (d a, J = 10,8 Hz, 1H), 5,08 (s a, 1H), 4,78 - 4,53 (dd a, J = 6,8, 11,6 Hz, 2H), 4,42 - 4,40 (m, 1H), 4,27 (s a, 2H), 4,21 - 4,18 (d a, J = 14,0 Hz, 1H), 4,12-3,78 (d a, J = 12,8 Hz, 2H), 3,68 - 3,07 (m, 7H), 3,05 - 2,84 (m, 3H), 2,77 (d, J = 1,6 Hz, 3H), 2,71 - 2,64 (m, 1H), 2,26 - 2,17 (m, 1H), 2,11 - 2,03 (m, 1H), 2,02 - 1,88 (m, 2H).
Ejemplo 148
2- [4-[7-(5-metil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo


Etapa A: 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(5-metil-1-tetrahidropiran-2-il-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo: A una solución de 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (200 mg, 424 umol), 4-bromo-5-metil-1-tetrahidropiran-2-il-indazol (188 mg, 636 umol), RuPhos (39,6 mg, 84,8 umol) y Cs2CO3 (414 mg, 1,27 mmol) en tolueno (6 ml) se le añadió Pd2(dba)3 (38,8 mg, 42,4 umol) y la reacción se agitó a 100 °C durante 12 horas en una atmósfera de N2. Después de que se completara, la mezcla se purificó directamente por cromatografía sobre gel de sílice (EP: EtOAc = 3:1 a 0:1) para dar 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(5-metil-1-tetrahidropiran-2-il-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (120 mg, 149 umol, 35,1 % de rendimiento, 85,0 % de pureza) en forma de un sólido de color pardo. ES+APCI MS m/z 686,6 [M+H]+.1H RMN (400 MHz, CLOROFORMO-d) 6 = 8,04 (d, J = 2,0 Hz, 1H), 7,33 - 7,29 (m, 2H), 5,70 (dd, J = 2,4, 9,2 Hz, 1H), 4,62 (s a, 1H), 4,39 (s a, 1H), 4,34 - 4,28 (m, 2H), 4,20 - 4,14 (m, 1H), 4,05 (d a, J = 12,0 Hz, 2H), 3,91 (d a, J = 12,4 Hz, 1H), 3,81 - 3,72 (m, 1H), 3,53 (t a, J = 4,8 Hz, 2H), 3,27 (d a, J = 10,8 Hz, 2H), 3,10 (d a, J = 6,8 Hz, 1H), 3,06 - 2,96 (m, 1H), 2,90 - 2,65 (m, 5H), 2,59 (d a, J = 10,2 Hz, 1H), 2,50 (s, 3H), 2,43 (s, 3H), 2,35 - 2,26 (m, 1H), 2,23 - 2,15 (m, 1H), 2,11 (s a, 2H), 1,90 - 1,65 (m, 7H), 1,54 (s, 9H).

Etapa B: 2-[4-[7-(5-metil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo: A una solución de 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(5-metil-1-tetrahidropiran-2-il-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (150 mg, 219 umol) en DCM (0,2 ml) se le añadió TFA (616 mg, 5,40 mmol, 0,4 ml) y la mezcla de reacción se agitó a 15 °C durante 12 horas. Después de que se completara, el disolvente se eliminó al vacío para dar 2-[4-[7-(5-metil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (159 mg, 218 umol, 99,6 % de rendimiento, 2TFA) en forma de un aceite de color rojo.
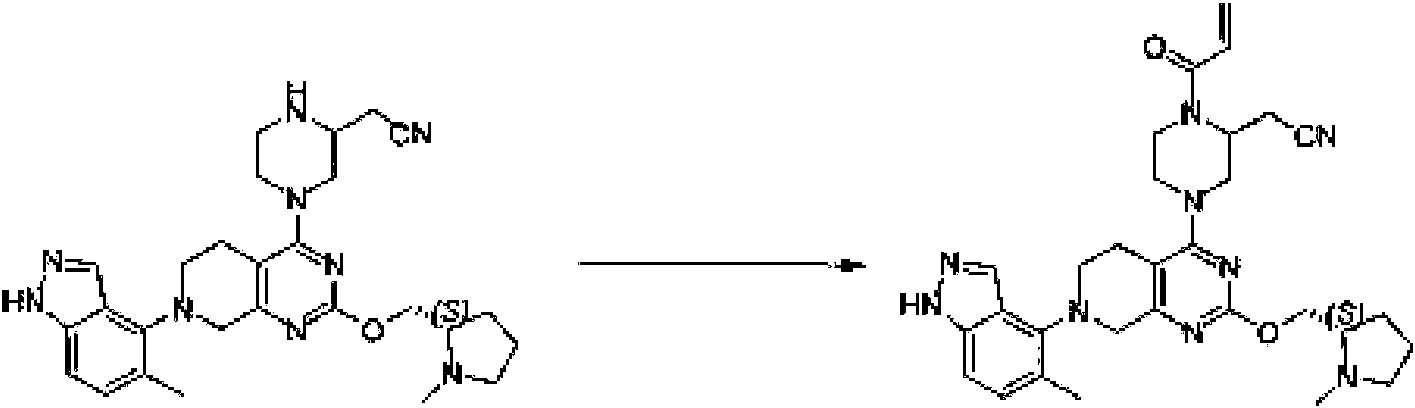
Etapa C: 2-[4-[7-(5-metil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo: A una solución de 2-[4-[7-(5-metil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (159 mg, 218 umol, 2TfA) y DIeA (366 mg, 2,83 mmol, 493 ul) en dCm (4 ml) a -40 °C se le añadió prop-2-enoato de prop-2-enoílo (24,7 mg, 196 umol) y la mezcla de reacción se agitó a -20 °C durante 1 hora. Después de que se completara, la mezcla de reacción se inactivó mediante la adición de agua (2 ml) y la capa acuosa se separó y se extrajo de nuevo con DCM (2 x 10 ml). Los extractos orgánicos combinados se concentraron al vacío y el residuo se purificó por cromatografía sobre gel de sílice (DCM: MeOH = 50:1 a 5:1) seguido de purificación por HPLC prep. inversa (columna: Phenomenex Synergi C18 150*25* 10 um;fase móvil:
[agua (0,225 % de ácido fórmico)-ACN];% de B: 4 %-34 % durante 10 minutos) para dar 2-[4-[7-(5-metil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo
(1,77 mg, 3,10 umol). ES+APCI MS m/z 556,5 [M+H]+.1H RMN (400 MHz, CLOROFORMO-d) 5 = 8,00 (s, 1H), 7,18 (s a, 1H), 7,16 - 7,11 (m, 1H), 6,52 (s a, 1H), 6,37 - 6,21 (m, 1H), 5,75 (d a, J = 10,4 Hz, 1H), 4,99 (s a, 1H), 4,67 (dd a, J = 6,4, 11,6 Hz, 1H), 4,34 (dd a, J = 3,9, 11,6 Hz, 1H), 4,24 (s, 2H), 4,11 (d a, J = 12,8 Hz, 1H), 3,94 (s a, 1H), 3,57 - 3,40 (m, 4H), 3,29 (d a, J = 13,8 Hz, 1H), 3,19 (s a, 1H), 3,02 (dd a, J = 12,4, 19,7 Hz, 2H), 2,87 (dd a, J = 8,4, 16,4 Hz, 2H), 2,76 - 2,58 (m, 6H), 2,35 (s, 3H), 2,22 - 2,10 (m, 1H), 2,01 (d a, J = 8,4 Hz, 1H), 1,89 (s a, 2H).
Ejemplo 149
2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-[5-(trifluorometil)-1H-indazol-4-il]-6,8-dihidro-5H-μmdo[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Etapa A: 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-[5-(trifluorometil)-1 -(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5W-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo: 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo (130 mg, 276 umol), 2-[[4-bromo-5-(trifluorometil)indazol-1-il]metoxi]etil-trimetil-silano (120 mg, 303 umol), Pd2(dba)3 (50,5 mg, 55,1 umol), RuPhos (51,5 mg, 110 umol) y Cs2CO3 (225 mg, 689 umol) en tolueno (3,00 ml) se desgasificó con N2 y después se calentó a 90 °C durante 12 horas en una atmósfera de N2. Después de que se completara, la mezcla se filtró y el filtrado se concentró. El residuo se purificó por TLC prep. (EtOAc/MeOH 8/1) para dar 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-[5-(tnfluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-μmdo[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo (140 mg, 169 umol, 61,4 % de rendimiento, 95,0 % de pureza) en forma de un sólido de color amarillo. ES+APCI MS m/z 786,3 [M+H]+.

Etapa B: 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo: A una solución de 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-μmdo[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo (140 mg, 178 umol) en DCM (3,00 ml) se le añadieron 2,6-dimetilpiridina (229 mg, 2,14 mmol) y TMSOTf (238 mg, 1,07 mmol) a 0 °C. La mezcla se agitó a 15 °C durante 2 horas. Después de que se completara, la mezcla se inactivó mediante la adición de MeOH (0,5 ml) y se concentró al vacío para dar 2-[4-[2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-7-[5-(trifluorometil)-1 -(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (200 mg, bruto) en forma de un aceite de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. ES+APCI MS m/z 686,6 [M+H]+.

Etapa C: 2-[4-[2-[[(2S)-1 -metilpirrolidin-2- il]metoxi]-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo: A una solución de 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (122 mg, 178 umol) y DIEA (138 mg, 1,07 mmol, 186 ul) en DCM (3,00 ml) se le añadió prop-2-enoato de prop-2-enoílo (18,0 mg, 143 umol) gota a gota a 0 °C. La mezcla se agitó a 20 °C durante 1,5 horas. Después de que se completara, la mezcla se diluyó con agua (0,5 ml) y se extrajo con EtOAc (2 x 5 ml). Las capas orgánicas se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se purificó por TLC prep. (AE/MeOH 10/1) y HPLC prep. (columna: Boston pH-lex 150 * 25 10 um;fase móvil: [agua (0,1 % de TFA)-ACN];% de B: 39 %-69 %,10 min) para dar 2-[4-[2-[[(2S)-1 -metilpirrolidin-2- il]metoxi]-7-[5-(trifluorometil)-1-(2-trimetilsiyletoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (54,0 mg, 43,8 umol). ES+APCI MS m/z 740,6 [M+H]+.

Etapa D: 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-[5-(trifluorometil)-1H-indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo: A una solución de 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]--[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo (20 mg, 27,0 umol) en DCM (60 ul) se le añadió TFA (30,8 mg, 270 umol). La mezcla se agitó a 20 °C durante 1 hora. La reacción no se completó, así que se añadió más cantidad de TFA (30,8 mg) y la reacción se agitó a 20 °C durante 0,5 horas más. Después de que se completara, el pH de la mezcla se ajustó a 8 mediante la adición de NaHCO3 acuoso saturado (1 ml) y la capa acuosa se extrajo con EtOAc (2 x 5 ml). Los extractos orgánicos combinados se lavaron con salmuera (3 ml), se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se diluyó con MeOH (0,5 ml), se añadió NH3H2O (0,5 ml) y la mezcla se agitó a 20 °C durante 0,5 horas. La mezcla se purificó por HPLC prep. (columna: Phenomenex Synergi C18150 * 30 mm * 4 um;fase móvil: [agua (0,225 % de ácido fórmico)-ACN];% de B: 15 %-45 %, 10,5 min) para dar 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-[5-(trifluorometil)-1H-indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (2,68 mg, 3,99 umol) en forma de un sólido de color blanco. ES+APCI MS m/z 610,5 [M+H]+.1H RMN (400 MHz, cloroformo-d) 6 = 8,22 (s, 1H), 7,68 (d, J = 9,2 Hz, 1H), 7,45 (d, J = 8,8 Hz, 1H), 6,60 (s a, 1H), 6,45 - 6,35 (m, 1H), 5,84 (d a, J = 11,2 Hz, 1H), 5,09 (s a, 1H), 4,71 - 4,60 (m, 1H), 4,42 - 4,27 (m, 3H), 4,17 (d a, J = 11,2 Hz, 1H), 4,01 (d a, J = 12,4 Hz, 2H), 3,78 - 3,31 (m, 5H), 3,23 - 3,03 (m, 2H), 3,02 - 2,75 (m, 4H), 2,71 (s, 3H), 2,66 - 2,57 (m, 1H), 2,27 - 2,12 (m, 1H), 2,10 - 2,03 (m, 3H). ES+APCI MS m/z 610,1 [M+H]+.
Ejemplo 150
2-(1-acriloil-4-(2-(((R)-1-(dimetilamino)propan-2-il)oxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 4-[7-Bencil-2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-l-carboxilato de terc-butilo: A una mezcla de 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de terc-butilo (500 mg, 1,04 mmol), (2R)-1-(dimetilamino)propan-2-ol (214 mg, 2,07 mmol) y terc-butóxido de sodio (298 mg, 3,11 mmol) en tolueno (20 ml) se le añadieron BINAP (129 mg, 207 umol) y Pd2(dba)3 (94,8 mg, 104 umol) y la mezcla se roció con nitrógeno seguido de agitación a 90 °C durante 5 h. La mezcla se diluyó con acetato de etilo (100 ml) y agua (100 ml) y la capa orgánica se separó y se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico agua)/acetonitrilo]. Las fracciones deseadas se recogieron y se neutralizaron con una solución acuosa saturada de carbonato de sodio (5 ml) y se extrajeron con 10 % de MeOH/DCM (2 x 50 ml). Los extractos orgánicos combinados se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío para dar 4-[7-bencil-2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de terc-butilo (300 mg, 480 umol) ES+APCI MS m/z 550,4 [M+H]+.

Etapa B: 2-(cianometil)-4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo: A una solución de 4-[7-bencil-2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de terc-butilo (1,1 g, 2,00 mmol) en MeOH (10 ml) se le añadió una solución de NH3 (792 mg, 46,5 mmol) en MeOH (3,96 g, 123,56 mmol, 5 ml), seguido de Pd al 10 %/C (500 mg). La mezcla se agitó a 40 °C durante 12 h en una atmósfera de H2 (103,42 kPa (15 psi)). La reacción se filtró y el filtrado se concentró a presión reducida a sequedad para dar 2-(cianometil)-4-[2-[(1R)-2-(dimetilamino)-1-metiletoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (756 mg, bruto) que se obtuvo en forma de un sólido de color amarillo. ES+Ap C i MS m/z 460,3 [M+H]+.

Etapa C: 2-(cianometil)-4-[2-[(1R)-2-(dimetilamino)-1-metil- etoxi]-7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo: Una mezcla de 2,2-dimetilpropanoato de (4-bromo-2-naftilo) (304 mg, 990 umol), 2-(cianometil)-4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (350 mg, 761 umol), [2-(2-aminoetil)fenil]-cloro-paladio, diciclohexil-[2-(2,4,6-triisopropilfenil)fenil]fosfano (84,4 mg, 114 umol) y Cs2C03 (620 mg, 1,90 mmol) en tolueno (10 ml) se purgó 3 veces con N2 y la mezcla se agitó a 70 °C durante 16 horas en una atmósfera de N2. La mezcla de reacción se vertió en H20 (50 ml) y se extrajo con acetato de etilo (50 ml x 3). Los extractos orgánicos combinados se lavaron con salmuera (30 ml), se secaron sobre Na2S04 anhidro y se concentraron al vacío para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico)/acetonitrilo] para dar 2-(cianometil)-4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo (210 mg, 303 umol). Es+APCI MS m/z 686,4 [M+H]+.

Etapa D: 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)piperazin-1-il]-2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo]: A una solución de 2-(cianometil)-4-[2-[(1R)-2-(dimetilamino)-1-metiletoxi]-7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tercbutilo (210 mg, 306 umol) en diclorometano (300 ul) se le añadió TFA (523 mg, 4,59 mmol, 340 ul) y la mezcla se agitó a 15 °C durante 2 horas. La mezcla se concentró al vacío para dar 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)piperazin-1-il]-2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (214 mg, 305 umol). ES+APCI MS m/z 586,4 [M+H]+.

Etapa E: 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)-4-prop-2-enoil-piperazin-1-il]-2-[(1R)-2-(dimetilamino)-1-metiletoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo]: A una mezcla de 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)piperazin-1-il]-2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (214 mg, 305 umol, TFA) y DIEA (395 mg, 3,06 mmol, 532 ul) en diclorometano (5,00 ml) enfriada a -40 °C se le añadió
una solución de prop-2-enoato de prop-2-enoílo (38,6 mg, 305 umol) en diclorometano (1,00 ml) en una atmósfera de nitrógeno. La mezcla se calentó a 0 °C y se agitó durante 1 hora. La mezcla se concentró al vacío para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido trifluoroacético)\acetonitrilo] para dar 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)-4-prop-2-enoil-piperazin-1-il]-2-[(1R)-2-(dimetilamino)-1-metiletoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (100 mg, 156 umol). ES+APCI MS m/z 640,7 [M+H]+.

Etapa F: 2-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo: A una solución de 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)-4-prop-2-enoilpiperazin-1 -il]-2-[(1 R)-2-(dimetilamino)-1 -metil-etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (90 mg, 141 umol) en Th F (500 ul) enfriada a 0 °C se le añadió NaOH (2 M, 281,34 ul) y la mezcla se agitó a 15 °C durante 16 horas. El pH de la mezcla se ajustó a 7 mediante la adición de una solución al 20 % con ácido fórmico. La solución acuosa después se extrajo con diclorometano (3 * 10 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi C18 150 * 25 * 10 um; fase móvil: [agua (0,225 % de ácido fórmico)-ACN]; % de B: 7 %-37 %, 10 min) para dar 2-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo (5 mg, 8,82 umol, 6,27 % de rendimiento, 98 % de pureza) ES+APCI MS m/z 556,3 [M+H]+.
1H RMN (400 MHz, metanol-d4) ó = 8,54 (s a, 0,6 H), 8,07 (d, J = 8,4 Hz, 1H), 7,63 (d, J = 8,4 Hz, 1H), 7,37 (t, J = 7,2 Hz, 1H), 7,30 - 7,21 (m, 1H), 6,91 - 6,72 (m, 3H), 6,29 (d a, J = 16,4 Hz, 1H), 5,84 (d a, J = 11,2 Hz, 1H), 5,48 (s a, 1H), 5,26 - 4,96 (m, 1H), 4,57 (s a, 1H), 4,24 - 4,09 (m, 4H), 3,74 - 3,54 (m, 1H), 3,48 (m, 2H), 3,22 (m, 2H), 3,10 - 2,86 (m, 5H), 2,78 (d a, J = 14,4 Hz, 1H), 2,53 (s a, 6H), 1,37 (dd, J = 2,0, 6,4 Hz, 3H).
Ejemplo 151
2-(1-acriloil-4-(2-(((R)-1-(dimetilamino)propan-2-il)oxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Se preparó (2-(1-acriloil-4-(2-(((R)-1-(dimetilamino)propan-2-il)oxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo siguiendo el Ejemplo 150 sustituyendo 1-bromonaftaleno por 2,2-dimetilpropanoato de (4-bromo-2-naftilo) en la Etapa C y omitiendo la Etapa F. ES+APCI MS m/z 540,3 [M+H]+. 1H RMN (400 MHz, metanol-d4) ó = 8,51 (s, 1H), 8,26 - 8,19 (m, 1H), 7,90 - 7,84 (m, 1H), 7,62 (d, J = 8,0 Hz, 1H), 7,53 -7,47 (m, 2H), 7,46 - 7,39 (m, 1H), 7,25 - 7,18 (m, 1H), 6,82 (s a, 1H), 6,30 (d a, J = 17,2 Hz, 1H), 5,84 (d a, J = 10,4 Hz, 1H), 5,62-5,53 (m, 1H), 5,08 (s a, 1H), 4,70 - 4,39 (m, 1H), 4,26 - 4,11 (m, 4H), 3,94 - 3,59 (m, 1H), 3,48 - 3,34 (m, 2H), 3,28-3,18 (m, 3H), 3,13 - 2,92 (s, 5H), 2,79 - 2,61 (s, 6H), 1,48 (dd, J = 2,0, 6,0 Hz, 3H).
Ejemplo 152
2-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(5-metil-1H-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Etapa A: 2-(cianometil)-4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(5-metil-1-tetrahidropiran-2-il-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo: Una mezcla de 2-(cianometil)-4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de terc-butilo (0,50 g, 1,09 mmol), 4-bromo-5-metil-1-tetrahidropiran-2-il-indazol (482 mg, 1,63 mmol), t-BuONa (314 mg, 3,26 mmol) y [2-(2-aminoetil)fenil]-cloro-paladio;diciclohexil-[2-(2,4,6-triisopropilfenil)fenil]fosfano (80,4 mg, 109 umol) en tolueno (30 ml) se agitó a 70 °C durante 10 horas. La mezcla se diluyó con agua (10 ml) y la capa acuosa se extrajo con acetato de etilo (3 x 20 ml). Las capas orgánicas se lavaron con una solución saturada de cloruro de sodio (30 ml), se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (ácido fórmico, 0,1 %)/acetonitrilo] para dar 2-(cianometil)-4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(5-metil-1-tetrahidropiran-2-il-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (0,40 g, 522 umol, 48,0 % de rendimiento) en forma de un sólido de color amarillo. eS+APCI MS m/z 674,3 [M+H]+. 1H Rm N (400 MHz, cloroformo-d) 6 = 8,03 (d, J = 1,6 Hz, 1H), 7,31 - 7,27 (m, 2H), 5,68 (dd, J = 2,4, 9,6 Hz, 1H), 5,31 (s a, 1H), 4,61 (s a, 1H), 4,28 (s, 2H), 4,08 - 3,94 (m, 3H), 3,86 (d a, J = 11,6 Hz, 1H), 3,79 - 3,71 (m, 1H), 3,52 (t a, J = 4,8 Hz, 2H), 3,24 (d a, J = 12,8 Hz, 2H), 3,04 - 2,91 (m, 1H), 2,87 - 2,67 (m, 5H), 2,65 - 2,47 (m, 2H), 2,41 (s, 3H), 2,32 (s a, 6H), 2,17 (d a, J = 4,0 Hz, 1H), 2,09 (s a, 1H), 1,77 (t a, J = 10,8 Hz, 3H), 1,52 (s, 9H), 1,36 (d, J = 6,0 Hz, 3H).

Etapa B: 2-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(5-metil-1-tetrahidropiran-2-il-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo: A una solución de 2-(cianometil)-4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(5-metil-1-tetrahidropiran-2-il-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (0,38 g, 564 umol) en diclorometano (10 ml) se le añadieron TMSOTf (752 mg, 3,38 mmol) y 2,6-dimetilpiridina (725 mg, 6,77 mmol) a 0 °C y la mezcla se agitó a 10 °C durante 1 hora. La mezcla se inactivó mediante la adición de metanol (0,10 ml) y se concentró al vacío. El residuo se purificó por cromatografía de fase inversa [agua (ácido fórmico, 0,1 %)/acetonitrilo]. Las fracciones recogidas se combinaron y el pH se ajustó a pH >7 mediante la adición de una solución saturada de bicarbonato de sodio y la capa acuosa se extrajo con diclorometano/metanol (10/1) (3 x 10 ml). Los extractos se lavaron con una solución saturada de cloruro de sodio (10 ml), se secaron sobre Na2SO4, se filtraron y se concentraron al vacío para dar 2-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(5-metil-1-tetrahidropiran-2-il-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (0,30 g, bruto) en forma de un sólido de color amarillo. ES+Ap C i MS m/z 574,1 [M+H]+.

Etapa C: 2-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(5-metil-1-tetrahidropiran-2-il-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo: A una solución de 2-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7- (5-metil-1-tetrahidropiran-2-il-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (0,20 g, bruto) y TEA (176 mg, 1,74 mmol, 243 ul) en diclorometano (1,00 ml) se le añadió prop-2-enoato de prop-2-enoílo (44,0 mg, 349 umol) a 0 °C y la reacción se agitó a 0 °C durante 0,5 h. La mezcla se inactivó mediante la adición de metanol (0,10 ml) y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (ácido fórmico, 0,1 %)/acetonitrilo]. Las fracciones recogidas se combinaron y el pH se ajustó a pH >7 mediante la adición de una solución saturada de bicarbonato de sodio y la capa acuosa se extrajo con diclorometano/metanol (10/1) (3 x 5,00 ml). Los extractos se lavaron con una solución saturada de cloruro de sodio (1 x 10 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío para dar 2-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(5-metil-1-tetrahidropiran-2-il-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-1-prop-2-enoilpiperazin-2-il]acetonitrilo (0,10 g, 127 umol, dos etapas 36,6 % de rendimiento) en forma de un sólido de color amarillo. ES+APCI MS m/z 628,6 [M+H]+.

Etapa D: 2-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(5-metil-1H-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo: Una mezcla de 2-[4-(2-[(1R)-2-(dimetilamino)-1-metil-etoxi]- 7-(5-metil-1-tetrahidropiran-2-il-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (40 mg, 63,7 umol) y TsOH (1,10 mg, 6,37 umol) en acetonitrilo (3 ml) se agitó a 90 °C durante 1 hora. La mezcla se inactivó mediante la adición de bicarbonato de sodio saturado (2 ml) a 0 °C y se concentró al vacío. El residuo se purificó por cromatografía en columna (AhO3, diclorometano/metanol = 5/1). Las fracciones deseadas recogidas se concentraron al vacío para dar un sólido de color blanco. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (NH4HCO310 mM)-ACN]; % de B: 35 %-65 %, 3 min) y (columna: Boston Green ODS 150 * 305 u; fase móvil: [agua (0,225 % de ácido fórmico)-ACN]; % de B: 15 %-45 %, 10 min). Las fracciones deseadas se reunieron y se liofilizaron para dar 2-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(5-metil-1H-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (2,99 mg, 5,50 umol). ES+APCI MS m/z 544,5 [M+H]+. 1H RMN (400 MHz, cloroformo-d) 6 = 8,36 (s a, 1H), 8,09 (s, 1H), 7,29 (s a, 1H), 7,24 - 7,20 (m, 1H), 6,62 (d a, J = 13,6 Hz, 1H), 6,47 - 6,24 (m, 1H), 5,83 (d a, J = 10,8 Hz, 1H), 5,50 (s a, 1H), 5,08 (s a, 1H), 4,60 (s a, 1H), 4,31 (s, 2H), 4,12 (d a, J = 14,4 Hz, 1H), 3,99 (d a, J = 10,8 Hz, 1H), 3,55 (t a, J = 5,6 Hz, 2H), 3,42 - 3,29 (m, 1H), 3,10 (s a, 1H), 3,00 - 2,68 (m, 7H), 2,49 (s, 6H), 2,43 (s, 3H), 1,39 (d, J = 6,0 Hz, 3H).
Ejemplo 153
2-[4-[7-(3-hidroxi-1-naftil)-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo
5
10 
15
20
25 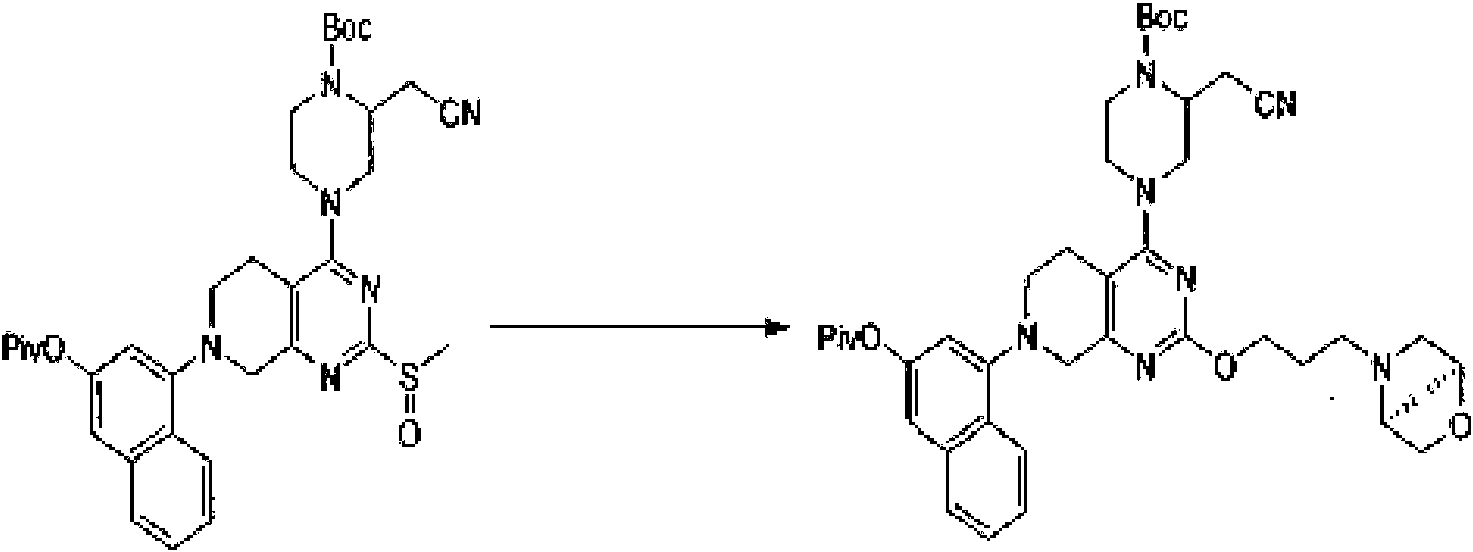
Etapa A: 2-(cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo: A una solución de 2-30 (cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (500 mg, 773 umol) y 3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propan-1-ol (219 mg, 1,39 mmol) en tolueno (10 ml) se le añadió tBuONa (111 mg, 1,16 mmol) en una atmósfera de N2. La mezcla de reacción se agitó a 18 °C durante 0,5 horas. Después de que se completara, la mezcla se purificó por cromatografía sobre gel de sílice (EP: EtOAc = 3:1 a 0:1 después AE: MeOH = 50:1 a 10:1) seguido de cromatografía ultrarrápida 35 inversa (50 % a 90 % de MeCN en agua, condición de base) para dar 2-(cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (120 mg, 156 umol, 20,5 % de rendimiento, 97,9 % de pureza) en forma de un sólido de color pardo. 1H RMN (400 MHz, CLOROFORMO-d) 6 = 8,19 - 8,12 (m, 1H), 7,83 - 7,77 (m, 1H), 7,55 - 7,43 (m, 2H), 7,29 (d, J = 1,6 Hz, 1H), 6,84 (d, J = 2,0 Hz, 1H), 4,64 (s a, 1H), 4,42 - 4,35 (m, 3H), 4,26 (d a, J = 5,2 Hz, 2H), 4,12 -40 4,03 (m, 3H), 3,96 (d a, J = 12,8 Hz, 1H), 3,62 (dd, J = 1,6, 7,6 Hz, 1H), 3,50 (s a, 2H), 3,31 (d a, J = 13,6 Hz, 3H), 3,11 - 2,97 (m, 2H), 2,94 (d a, J = 8,8 Hz, 1H), 2,89 - 2,69 (m, 5H), 2,54 (d a, J = 9,8 Hz, 1H), 2,00 - 1,92 (m, 2H), 1,90 -1,83 (m, 1H), 1,73 (d a, J = 10,0 Hz, 1H), 1,53 (s, 9H), 1,41 (s, 9H)
45
50
55 
Etapa B: 2,2-dimetilpropanoato de 4-[4-[3-(cianometil)piperazin-1-il]-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo]: Una solución de 2-(cianometil)-4-[7-[3-(2,2-gg dimetilpropanoiloxi)-1-naftil]-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo (120 mg, 162 umol) en TFA (370 mg, 3,24 mmol, 240 ul) se agitó a 18 °C durante 1 hora. Después de que se completara, el disolvente se eliminó al vacío para dar 2,2-dimetilpropanoato de 4-[4-[3-(cianometil)piperazin-1-il]-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (140 mg, 161 umol, 99,5 % de rendimiento, 2TFA) en forma de un aceite de color pardo.
65

Etapa C: 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)-4-prop-2-enoil-piperazin-1-il]-2-[3-[(1S,1S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo]: A una solución de 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)piperazin-1-il]-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (140 mg, 161 umol) y DiEa (167 mg, 1,29 mmol, 225 ul) en d Cm (2 ml) se le añadió prop-2-enoato de prop-2-enoílo (30,5 mg, 242 umol) a 0 °C. La mezcla de reacción se agitó a 18 °C durante 1 hora. Después de que se completara, la mezcla de reacción se inactivó mediante la adición de agua (5 ml) y la capa acuosa se extrajo con DCM (3 x 10 ml). Las capas orgánicas combinadas se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida inversa (condición de base, MeCN/NH3^H2Ü en agua: 50 % a 80 %) para dar 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)-4-prop-2-enoil-piperazin-1-il]-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (60 mg, 86,5 umol, 53,6 % de rendimiento). 1H RMN (400 MHz, CLOROFORMO-d) 6 = 8,22 - 8,13 (m, 1H), 7,84 - 7,78 (m, 1H), 7,56 - 7,45 (m, 2H), 7,31 (d, J = 2,0 Hz, 1H), 6,86 (d, J = 2,4 Hz, 1H), 6,62 (s a, 1H), 6,47 - 6,37 (m, 1H), 5,92 - 5,79 (m, 1H), 5,24 - 4,88 (m, 1H), 4,75 (s a, 1H), 4,45 - 4,36 (m, 3H), 4,31 - 4,20 (m, 2H), 4,15 (d a, J = 13,6 Hz, 1H), 4,10 - 4,01 (m, 2H), 3,64 (dd, J = 1,6, 7,6 Hz, 2H), 3,51 (s a, 2H), 3,38 (s a, 2H), 3,14 (s a, 1H), 3,08 - 2,73 (m, 7H), 2,56 (d a, J = 10,0 Hz, 1H), 2,03 - 1,93 (m, 2H), 1,88 (d a, J = 9,6 Hz, 1H), 1,78 - 1,70 (m, 1H), 1,61 (s, 9H), 1,43 (s, 9H).

Etapa D: 2-[4-[7-(3-hidroxi-1-naftil)-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo: A una solución de 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)-4-prop-2-enoil-piperazin-1-il]-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (60 mg, 86,5 umol) en THF (0,5 ml) se le añadió NaOH (2 M, 600 ul). La mezcla de reacción se agitó a 18 °C durante 12 horas. Después de que se completara, la mezcla de reacción se acidificó mediante la adición de 4 gotas de HCOOH (20 % en agua) y la capa acuosa se extrajo con DCM (5 x 8 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Luna C18150 * 255 ufase móvil: [agua (0,225 % de ácido fórmico)-ACN];% de B: 10 %-40 %,10 min) para dar 2-[4-[7-(3-hidroxi-1-naftil)-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (19,8 mg, 28,3 umol, 32,8 % de rendimiento, 93,8 % de pureza, sal del ácido fórmico) que se obtuvo en forma de un sólido de color pardo. ES+APCI MS m/z 610,5 [M+H]+.
Ejemplo 154
2-[4-[7-(1-naftil)-2-[3-[(1 S,4S-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Etapa A: 2-(cianometil)-4-[2-metilsulfanil-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo: A la solución de 2-(cianometil)-4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de terc-butilo (3 g, 7,42 mmol), 1-bromonaftaleno (3,07 g, 14,8 mmol, 2,06 ml) y Cs2CO3 (7,25 g, 22,2 mmol) en tolueno (60 ml) se le añadió XPhos palladacycle gen 3 (628 mg, 742 umol) en una atmósfera de N2 y la suspensión se desgasificó al vacío y se purgó varias veces con N2. La mezcla se calentó a 70 °C y se agitó a 70 °C durante 10 horas. La mezcla resultante se filtró y la torta de filtro se lavó con EtOAc (3 x 20 ml). Los eXtractos orgánicos combinados se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc de 50:1 a 3:1) para dar 2-(cianometil)-4-[2-metilsulfanil-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo (3,55 g, 6,02 mmol, 81,2 % de rendimiento, 90,0 % de pureza) en forma de un aceite de color pardo. ES+APCI MS m/z 531,4 [M+H]+.

Etapa B: 2-(cianometil)-4-[2-metilsulfinil-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo: A la solución de 2-(cianometil)-4-[2-metilsulfanil-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (2,95 g, 5,56 mmol) en DCM (60 ml) se le añadió m-CPBA (1,13 g, 5,56 mmol, 85,0 % de pureza) a 0 °C y se agitó a 0 °C durante 1 hora. La mezcla de reacción se inactivó mediante la adición de Na2S2O3 saturado (20 ml) a 0 °C, las capas se separaron y la capa acuosa se diluyó con agua (20 ml) y se extrajo con EtOAc (60 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0,1 % de ácido fórmico) = 100 %) para dar 2-(cianometil)-4-[2-metilsulfinil-7-(1-naftil)-6,8-dihidro-5H pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (1,7 g, 2,80 mmol, 50,3 % de rendimiento, 90,0 % de pureza) en forma de un sólido de color pardo. ES+APCI MS m/z 574,4 [M+H]+.

Etapa C: 2-(cianometil)-4-[7-(1-naftil)-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo: A una solución de 2-(cianometil)-4-[2-metilsulfinil-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (300 mg, 549 umol) y 3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propan-1-ol (160 mg, 1,02 mmol) en tolueno (6 ml) se le añadió tBuONa (79,1 mg, 823 umol). La mezcla de reacción se agitó a 18 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se purificó por cromatografía sobre gel de sílice (EP:AE = 3:1 a 0:1 después AE:MeOH = 50:1 a 10:1) para dar 2-(cianometil)-4-[7-(1-naftil)-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (200 mg, 249 umol, 45,3 % de rendimiento, 79,6 % de pureza) que se obtuvo en forma de un sólido de color pardo. ES+APCI MS m/z 640,5 [M+H]+.

Etapa D: 2-[4-[7-(1-naftil)-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo: Una mezcla de reacción de 2-(cianometil)-4-[7-(1-naftil)-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (200 mg, 313 umol) en TFA (770 mg, 6,75 mmol, 500 ul) se agitó a 18 °C durante 1 hora. Después de que se completara, el disolvente se eliminó al vacío para dar 2-[4-[7-(1-naftil)-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (271 mg, bruto) en forma de un aceite de color pardo.

Etapa E: 2-[4-[7-(1-naftil)-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo: A una solución de 2-[4-[7-(1-naftil)-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (271 mg, 502 umol) y DIEA (323 mg, 2,50 mmol, 0,435 ml) en DCM (6 ml) se le añadió prop-2-enoato de prop-2-enoílo (60 mg, 476 umol) a 0 °C y la mezcla de reacción se agitó a 18 °C durante 1 hora. Después de que se completara, la mezcla de reacción se inactivó mediante la adición de una gota de agua y se purificó por cromatografía sobre gel de sílice (EP: EtOAc = 3:1 a 0:1) después HPLC prep. (columna: Gemini 150 * 25 5 ufase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN];% de B: 33 %-63 %,12 min) para dar 2-[4-[7-(1-naftil)-2-[3-[(1S,4S)-2-oxa-5
azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (23,8 mg, 40,0 umol, 7,97 % de rendimiento) en forma de un sólido de color amarillo. ES+a Pc I MS m/z 594,5 [M+H]+.
Ejemplo 155
(S)-1-(4-(7-(5-metil-1H-indazol-4-il)-2-((1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona

Etapa A: 4-[7-bencil-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo: A una mezcla de [(2S)-1-metilpirrolidin-2-il]metanol (4,15 g, 36,0 mmol, 4,28 ml) en THF (100 ml) se le añadió poco a poco NaH (2,16 g, 54,06 mmol, 60 % de pureza) a 0 °C. Después de que la mezcla se agitara a 0 °C durante 1 hora, se añadió una solución de 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de terc-butilo (8,0 g, 18,02 mmol) en THF (60 ml) y la mezcla se agitó a 70 °C durante 11 horas. Después de que se completara, la mezcla de reacción se vertió en NH4Cl acuoso (160 ml) y la capa acuosa se extrajo con EtOAc (2 x 100 ml). Los extractos orgánicos combinados se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo = 3/1 a acetato de etilo/metanol = 10:1) para dar 4-[7-bencil-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo (4,9 g, 8,70 mmol, 48,3 % de rendimiento, 92,8 % de pureza) en forma de un sólido de color gris. ES+APCI MS m/z 523,2 [M+H]+

Etapa B: 4-[2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo: A una mezcla de 4-[7-bencil-2-[[(2s )-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (2,20 g, 4,21 mmol) en Me0H (30 ml) se le añadió Pd(0H)2/C (700 mg, 10 % de pureza) y la mezcla se desgasificó al vacío y se purgó con varias veces H2 y la reacción se agitó a 40 °C durante 12 horas en una atmósfera de H2 (103,42 kPa (15 psi)). Después de que se completara, la mezcla de reacción se filtró a través de celite y el filtrado se concentró para dar 4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo (1,80 g, 4,02 mmol, 95,6 % de rendimiento, 96,7 % de pureza) en forma de un sólido de color negro. ES+APCI MS m/z 433,1 [M+H]+.

Etapa C: 4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(5-metil-1-tetrahidropiran-2-il-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo: A una mezcla de 4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (200 mg, 462 umol) y 4-bromo-5-metil-1-tetrahidropiran-2-il-indazol (204 mg, 694 umol) en tolueno (4 ml) se le añadieron Pd2(dba)3 (63,5 mg, 69,4 umol), RuPhos (43,2 mg, 92,5 umol) y Cs2CO3 (301 mg, 925 umol) y la mezcla se agitó a 110 °C durante 12 horas en una atmósfera de N2. Después de que se completara, la mezcla de reacción se repartió entre agua (10 ml) y EtOAc y las capas se separaron. Posteriormente, la capa acuosa se extrajo con EtOAc (2 x 10 ml) y los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo = 5/1 a acetato de etilo/MeOH = 10/1) para dar 4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(5-metil-1-tetrahidropiran-2-il-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (240 mg, 310 umol, 67,1 % de rendimiento, 83,6 % de pureza) en forma de un aceite de color pardo. ES+APCI MS m/z 647,6 [M+H]+.

Etapa D: 7-(5-metil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-4-piperazin-1-il-6,8-dihidro-5H-pirido[3.4-dlpirimidina: A una mezcla de 4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(5-metil-1-tetrahidropiran-2-il-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (240 mg, 371 umol) en DCM (1 ml) se le añadió TFA (1,54 g, 13,5 mmol, 1 ml) y la mezcla se agitó a 15 °C durante 1 hora. Después de que se completara, la mezcla de reacción se concentró para dar 7-(5-metil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidina (250 mg, 309 umol, 83,4 % de rendimiento, 85,5 % de pureza, 2TFA) en forma de un sólido de color pardo que se usó para la siguiente etapa sin purificación adicional. ES+APCI MS m/z 463,4 [M+H]+.

Etapa E: 1-[4-[7-(5-metil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una mezcla de 7-(5-metil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidina (250 mg, 362 umol, 2TfA) en dCm (2 ml) se le añadió DIEA (702 mg, 5,43 mmol, 946 ul) y la mezcla se enfrió a -50 °C. Se añadió poco a poco prop-2-enoato de prop-2-enoílo (36,5 mg, 289 umol) a la reacción a -50 °C y la mezcla se agitó a -50 °C durante 30 minutos. Después de que se completara, la mezcla de reacción se inactivó mediante la adición de MeOH (1 ml) y la mezcla se concentró. El residuo se recogió en DCM (10 ml) y los extractos orgánicos se lavaron con H2O (2 x 8 ml). Los extractos orgánicos se secaron sobre Na2SO4, se filtró y se concentraron. El residuo se purificó por HPLC prep. ((Instrumento: gx-1; Columna: Phenomenex Gemini C18250 * 50 mm * 10 um; Condición: agua (0,05 % de hidróxido de amoníaco v/v)-ACN; Inicio B: 32; Fin B: 62; Tiempo de gradiente (min): 12; Tiempo de mantenimiento a 100 % de B (min): 2; Caudal (ml/min): 25)
para dar 1-[4-[7-(5-metil-1H-indazol-4-il)-2-[[(2S)-1-metilpirroildin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (48,6 mg, 92,9 umol, 25,7 % de rendimiento, 98,7 % de pureza) en forma de un sólido de color amarillo. ES+APCI MS m/z 517,5 [M+H]+.
Ejemplo 156
(S)-1-(4-(2-((1-metilpirrolidin-2-il)metoxi)-7-(5-(trifluorometil)-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona

Se preparó (S)-1-(4-(2-((1-metilpirrolidin-2-il)metoxi)-7-(5-(trifluorometil)-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona siguiendo el Ejemplo 155 sustituyendo 4-bromo-1-tetrahidropiran-2-il-5-(trifluorometil)indazol (Intermedio 60) por 4-bromo-5-metil-1-tetrahidropiran-2-il-indazol en la Etapa C. ES+APCI MS m/z 571,4 [M+H]+.
Ejemplo 157
(S)-1-(4-(7-(5-metoxi-1H-indazol-4-il)-2-((1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona

Se preparó (S)-1-(4-(7-(5-metoxi-1H-indazol-4-il)-2-((1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona siguiendo el Ejemplo 155 sustituyendo 4-bromo-5-metoxi-1-tetrahidropiran-2-il-indazol por 4-bromo-5-metil-1-tetrahidropiran-2-il-indazol en la Etapa C. ES+APCI MS m/z 533,4 [M+H]+.
Ejemplo 158
1-[4-[7-(2-isopropilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona

Etapa A: 4-[7-(2-isopropilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1- carboxilato de terc-butilo: Se desgasificaron 4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (200 mg, 462 umol), 1-bromo-2-isopropil-benceno (120 mg, 601 umol), Cs2CO3 (452 mg, 1,39 mmol) y XPHOS PALLADACYCLE GEN 3 (78,3 mg, 92,5 umol) en tolueno (4,00 ml) con N2 y después se calentaron a 100 °C durante 10 horas en una atmósfera de N2. Después de que se completara, la mezcla se filtró y el filtrado se concentró. El producto bruto se purificó por cromatografía sobre gel de sílice eluyendo con EtOAc/MeOH = 50/1 a 5/1 para dar 4-[7-(2-isopropilfenil)-2-[[(2s)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo (84,6 mg, 134 umol, 28,9 % de rendimiento, 87,0 % de pureza) en forma de un sólido de color amarillo. ES+APCI MS m/z 551,2 [M+H]+.
Se preparó 1-[4-[7-(2-isopropilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona siguiendo el Ejemplo 155 sustituyendo 4-[7-(2-isopropilfenil)-2-[[(2S)-1 -metilpirrolidin-2- il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo por 4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(5-metil-1-tetrahidropiran-2-il-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo en la Etapa D. ES+APCI MS m/z 505,6 [M+H]+.
Ejemplo 159
(S)-1-(4-(2-((1-metilpirrolidin-2-il)metoxi)-7-(2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona

Se preparó (S)-1-(4-(2-((1-metilpirrolidin-2-il)metoxi)-7-(2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona siguiendo el Ejemplo 155 sustituyendo 1-bromo-2-(trifluorometil)benceno por 4-bromo-5-metil-1-tetrahidropiran-2-il-indazol en la Etapa C. ES+APCI MS m/z 531,5 [M+H]+.
Ejemplo 160
1 -(4 -(7 -(5 -m e til-1 H -in d a z o l-4 - il) -2 -(3 -(p ip e r id in -1 - il)p ro p o x i)-5 ,6 ,7 ,8 -te tra h id ro p ir id o [3 ,4 -d ]p ir im id in -4 - il)p ip e ra z in -1 -il)p ro p -2 -e n -1 -o n a

Etapa A: 4-[2-Metilsulfanil-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: Una mezcla de 4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (3,15 g, 7,88 mmol), 2-[(4-bromo-5-metil-indazol-1-il)metoxi]etil-trimetil-silano (3,50 g, 10,2 mmol), Cs2COa (6,42 g, 19,70 mmol), Pd2(dba)3 (1,08 g, 1,18 mmol) y RuPhos (735 mg, 1,58 mmol) en tolueno (50 ml) se desgasificó y 3 veces con N2 y la mezcla se agitó a 90 °C durante 10 horas en una atmósfera de N2. La mezcla de reacción se inactivó mediante la adición de agua (100 ml) y la capa acuosa se extrajo con acetato de etilo (3 x 100 ml). Los extractos orgánicos combinados se lavaron con salmuera (50 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna usando 1 —^ 33 % de EtOAc/éter de petróleo para dar 4-[2-metilsulfanil-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (2,30 g, 2,93 mmol, 37,1 % de rendimiento). Es APCI MS m/z 660,3 [M+H]+.

Etapa B: 4-[2-metilsulfinil-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: A una solución agitada de 4-[2-metilsulfanil-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (2,30 g, 3,49 mmol) en DCM (20 ml) se le añadió m-CPBA (601 mg, 3,49 mmol) a 0 °C. La mezcla de reacción se agitó a 0 °C durante 1 hora en una atmósfera de N2. La mezcla de reacción se inactivó mediante la adición de Na2S2O3 (10 ml) a 0 °C, después se diluyó con agua (100 ml) y la capa acuosa se extrajo con DCM (200 ml). Los extractos orgánicos combinados se lavaron con salmuera (200 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna usando 1—10 % de MeOH/DCM como eluyente para dar 4-[2-metilsulfinil-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (2,00 g, 2,84 mmol, 81,4 % de rendimiento). ES+APCI MS m/z 676,3 [M+H]+.

Etapa C: 4-[7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-2-[3-(1-piperidil)propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: A una mezcla de 4-[2-metilsulfinil-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (500 mg, 739 umol) y 3-(1-piperidil)propan-1-ol (211 mg, 1,48 mmol) en THF (10 ml) se le añadió poco a poco t-BuONa (213 mg, 2,22 mmol) y la mezcla se agitó a 20 °C durante 1 hora en una atmósfera de N2. La mezcla se enfrió a 0 °C y se añadió HCl (2 M) hasta pH ~7. La mezcla se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa usando agua 5 ^95 % 0,5 % de TFNagua/acetonitrilo como eluyente para dar 4-[7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-2-[3- (1-piperidil)propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (300 mg, 385. umol, 52,1 % de rendimiento). ES+APCI m S m/z 755,4 [m +H]+.

Etapa D: 7-(5-met¡l-1H-¡ndazol-4-¡l)-4-(p¡peraz¡n-1-¡l)-2-(3-(p¡per¡d¡n-1-¡l)propox¡)-5,6.7.8-tetrah¡drop¡r¡do[3.4-dlpirimidina: A una solución de 4-[7-[5-metil-1 -(2-trimetilsililetoximetil) indazol-4-il]-2-[3-(1-piperidil)propoxi]-6,8-dihidro-5H pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (300 mg, 397 umol) en MeOH (20 ml) se le añadió HCl/MeOH (4 M, 1,99 ml) y Pd(OH)2 (200 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. La mezcla de reacción se filtró y el filtrado se concentró para dar 7-(5-metil-1H-indazol-4-il)-4-(piperazin-1-il)-2-(3-(piperidin-1-il)propoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidina (250 mg, 360 umol, 90,7 % de rendimiento).

Etapa E: 1-[4-[7-(5-metil-1H-indazol-4-il)-2-[3-(1-piperidil)propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una mezcla de 7-(5-metil-1H-indazol-4-il)-4-(piperazin-1-il)-2-(3-(piperidin-1-il)propoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidina (250 mg, 360 umol, 2HCl) y DIeA (465 mg, 3,60 mmol, 629 ul) en DCM (8,00 ml) se le añadió una solución de prop-2-enoato de prop-2-enoílo (36,3 mg, 288 umol) en DCM (2,00 ml) a -40 °C en una atmósfera de nitrógeno y la reacción se agitó durante 1 hora. La mezcla de reacción se inactivó mediante la adición de NaHCO3(500 ul) a -40 °C, después se diluyó con agua (10 ml) y la capa acuosa se extrajo con DCM (10 ml). Los extractos orgánicos se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna (SiO2, DCM/MeOH 1/0 a 5/1) y se purificó adicionalmente por HPLC prep. (columna: Phenomenex Synergi C18 150 * 25 * 10 um; fase móvil: [agua (0,225 % de ácido fórmico)-ACN]; % de B: 8 %-28 %, 10 min) para dar 1-[4-[7-(5-metil-1H-indazol-4-il)-2-[3-(1-piperidil)propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (16,0 mg, 29,1 umol, 8,07 % de rendimiento). ES+APCI MS m/z 545,3 [M+H]+.
Ejemplo 161
1 -[4 -[7 -(5 -m e til-1 H -in d a z o l-4 - il) -2 -[(1 -m e tilp irro lid in -3 - il)m e to x i]-6 ,8 -d ih id ro -5 H -p ir id o [3 ,4 -d ]p ir im id in -4 - il]p ip e ra z in -1 -il]p ro p -2 -e n -1 -o n a

Se preparó 1-[4-[7-(5-metil-1H-indazol-4-il)-2-[(1-metilpirrolidin-3-il)metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona siguiendo el Ejemplo 160 sustituyendo (1-metilpirrolidin-3-il)metanol por 3-(1-piperidil)propan-1-ol en la Etapa C. ES+APCI MS m/z 517,4 [M+H]+.
Ejemplo 162
1-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(5-metil-1H-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona
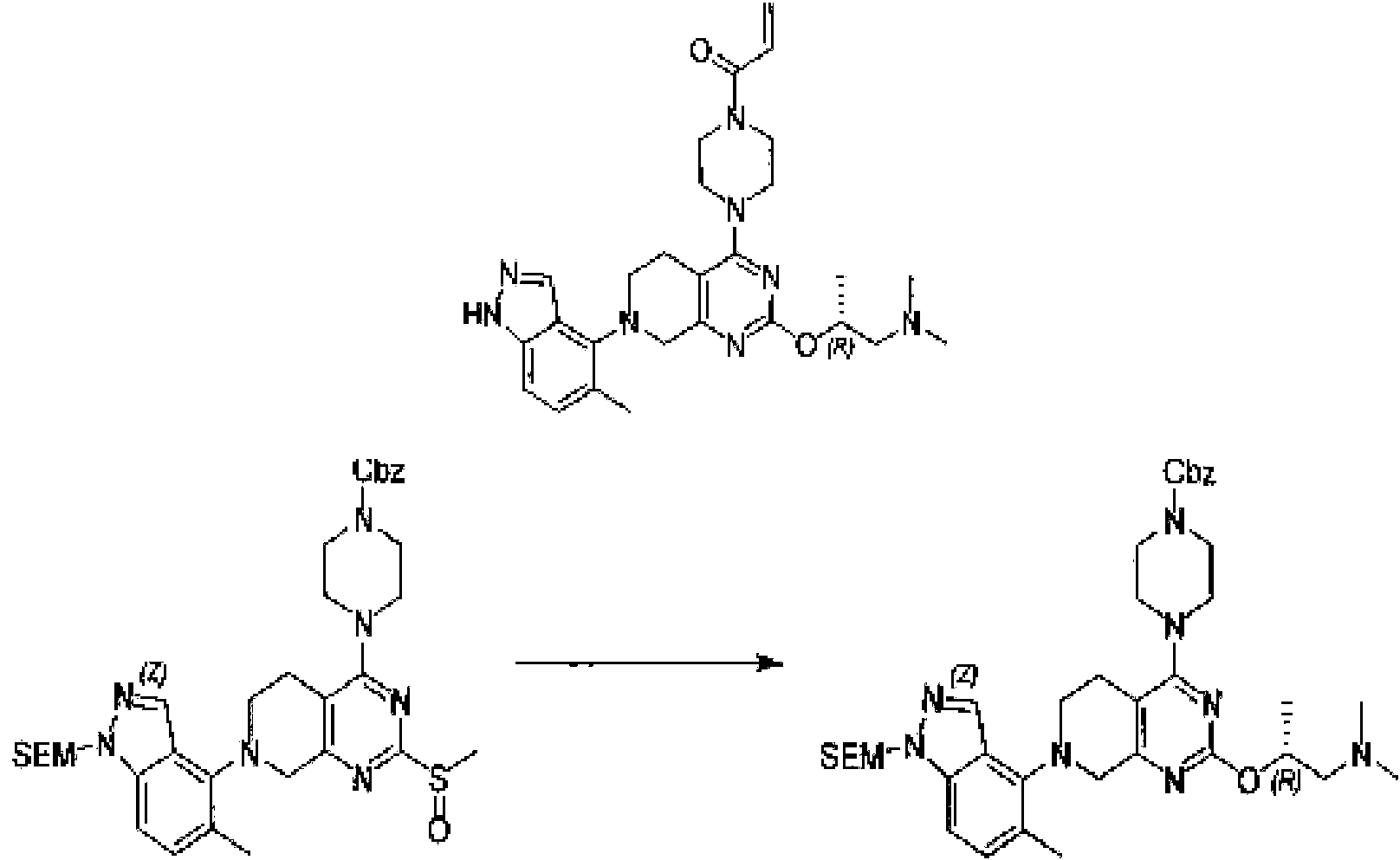
Etapa A: 4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo: Una mezcla de (2R)-1-(dimetilamino)propan-2-ol (183 mg, 1,78 mmol) (8,00 ml), 4-[2-metilsulfinil-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (400 mg, 592 umol) y NaOtBu (114 mg, 1,18 mmol) en tolueno se agitaron a 15 °C durante 0,5 horas. La mezcla de reacción se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía en columna sobre AbO3 (éter de petróleo/acetato de etilo 10/1 a 0/1) para dar 4-[2-[(1 R)-2-(dimetilamino)-1-metil-etoxi]-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (280 mg, 352 umol).1H Rm N (400 MHz, cloroformo-d) 6 = 8,08 (s, 1H), 7,46 - 7,39 (m, 5H), 7,38 - 7,33 (m, 2H), 5,76 (s, 2H), 5,43 - 5,33 (m, 1H), 5,24 (s, 2H), 4,35 (s, 2H), 3,72 - 3,67 (m, 4H), 3,64 -3,56 (m, 4H), 3,55 - 3,47 (m, 4H), 2,87 - 2,80 (m, 2H), 2,74 (dd, J = 6,8, 12,8 Hz, 1H), 2,49 (s, 3H), 2,47 - 2,40 (m, 1H), 2,36 (s, 6H), 1,42 (d, J = 6,4 Hz, 3H), 0,98 - 0,92 (m, 2H), 0,00 (s, 9H).

Etapa B: 4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo: A una solución de 4-[2-[(1R)-2-(dimetilamino)-1-metiletoxi]-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (200 mg, 280 umol) y (Boc)2O (122 mg, 559 umol, 129 ul) en MeOH (6,00 ml) se le añadió Pd al 10 %/C (80 mg) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora y a 35 °C durante 1 hora. Después de que se completara, la mezcla se filtró y el filtrado se concentró al vacío para dar mezcla de 4-[2-[(1R)-2-(dimetilamino)-1-metiletoxi]-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo y (2R)-W,W-dimetil-2-[[7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]propan-1-amina en forma de una mezcla (110 mg). ES+APCI MS m/z 681,3 [M+H]+.

Etapa C: (2R)-W,W-dimetil-2-[[7-(5-metil-1H-indazol-4-il)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]propan-1-amina: (2R)-Ñ,W-dimetil-2-[[7-(5-metil-1H-indazol-4-il)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]propan-1-amina (TFA) y [4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-5-metil-indazol-1 -il]metanol. Una solución de una mezcla de 110 mg de 4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo y (2R)-W,W-dimetil-2-[[7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]propan-1-amina en TFA (3,08 g, 27,0 mmol) se agitó a 30 °C durante 1 hora. Después de que se completara, la mezcla se concentró al vacío para dar 260 mg de una mezcla de (2R)-W,W-dimetil-2-[[7-(5-metil-1H-indazol-4-il)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]propan-1-amina y [4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-5-metilindazol-1-il]metanol. ES+APCI MS m/z 451,3 [M+H]+.

Etapa D: 1-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(5-metil-1H-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de 260 mg mezcla de (2R)-W,W-dimetil-2-[[7-(5-metil-1H-indazol-4-il)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]propan-1-amina y [4-[2-[(1R)-2-(dimetilamino)-1-metiletoxi]-4-piperazin-1-il-6,8-dihidro-5H pirido[3,4-d]pirimidin-7-il]-5-metil-indazol-1-il]metanol en DCM (1,00 ml) enfriada a -50 °C se le añadió DIEA (594 mg, 4,59 mmol) seguido gota a gota de prop-2-enoato de prop-2-enoílo (18,0 mg, 143 umol) y la mezcla se agitó a entre -50 °C y -40 °C durante 30 minutos. Después de que se completara, la mezcla se concentró al vacío para dar una mezcla de 100 mg de 1-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(5-metil-1H
indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona y 1-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-[1-(hidroximetil)-5-metil-indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona. ES+APCI MS m/z 505,4 [M+H]+.

Etapa E: 1-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(5-metil-1H-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una mezcla de 100 mg de 1-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(5-metil-1H-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona y 1-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-[1-(hidroximetil)-5-metil-indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona en THF (22 ml) y agua (550 ul) se le añadió NaOH (44,8 mg, 1,12 mmol) y la mezcla se agitó a 10 °C durante 3 horas. Después de que se completara, la mezcla se concentró al vacío. El residuo se diluyó con agua (1 ml) y se extrajo con DCM (3 x 5 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi C18150 * 25 * 10 um;fase móvil: [agua (0,225 % de ácido fórmico)-ACN];% de B: 10 %-40 %,12 min) para dar 1-[4-[2-[(1R)-2-(dimetilamino)-1-metil-etoxi]-7-(5-metil-1H-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (16,2 mg, 29,2 umol, 99,0 % de pureza, sal del ácido fórmico) en forma de un sólido de color blanquecino. ES+APCI MS m/z 505,2 [M+H]+.
Ejemplo 163
1-(4-(2-(2-(dietilamino)etoxi)-7-(5-metil-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona

Etapa A: 4-[2-[2-(dietilamino)etoxi]-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo: A una mezcla de 4-[2-metilsulfinil-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (500 mg, 739 umol) y 2-(dietilamino)etanol (173 mg, 1,48 mmol, 197 ul) en t HF (10 ml) se le añadió t-BuONa (213 mg, 2,22 mmol) y la mezcla se agitó a 20 °C durante 1 hora en una atmósfera de N2. La mezcla se enfrió a 0 °C y se añadió HCl (2 M) hasta pH ~7. La mezcla se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía en columna usando 0 ^ 10 % de MeOH/DCM como eluyente para dar el material impuro que se purificó adicionalmente por cromatografía de fase inversa para dar 4-[2-[2-(dietilamino)etoxi]-7-[5-metil-1-(2trimetilsililetoximetil)indazol-4-il]-6, 8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (220 mg, 295 umol, 39,9 % de rendimiento). ES+APCI MS m/z 729,3 [M+H]+.

Etapa B: W,W-dietil-2-[[7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]etanamina: A una solución de 4-[2-[2-(dietilamino)etoxi]-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (120 mg, 164 umol) en MeOH (10 ml) se le añadieron Pd(OH)2 (99,4 mg, 10 % de pureza) y HCl/MeOH (4 M, 823,05 ul) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. La mezcla se concentró al vacío para dar W,W-dietil-2-[[7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]etanamina (105 mg, 157,24 umol). ES+APCI MS m/z 595,4 [M+H]+.

Etapa C: 1-[4-[2-[2-(dietilamino)etoxi]- 7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una mezcla de W,W-dietil-2-[[7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]etanamina (105 mg, 158 umol, 2 HCl) y DIeA (204 mg, 1,58 mmol, 276 ul) en DCM (8 ml) a -40 °C se le añadió una solución de prop-2-enoato de prop-2-enoílo (15,9 mg, 126,4 umol) en DCM (2 ml) en una atmósfera de nitrógeno y la reacción se agitó durante 1 hora. La mezcla de reacción se inactivó mediante la adición de NaHCO3 (500 ul) a -40 °C y después se diluyó con agua (10 ml). La capa acuosa se extrajo con DCM (10 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna usando 0 ^20 % de MeOH/DCM como eluyente para dar 1-[4-[2-[2-(dietilamino)etoxi]- 7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (60,0 mg, 71,2 umol, 45,0 % de rendimiento). ES+APCI MS m/z 649,3 [M+H]+.

Etapa D: 1-[4-[2-[2-(dietilamino)etoxi]-7- (5-metil-1H-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de 1-[4-[2-[2-(dietilamino)etoxi]-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (50 mg, 77,0 umol) en DCM (500 ul) se le añadió TFA (175 mg, 1,54 mmol, 114 ul). La mezcla se agitó a 15 °C durante 16 horas. La mezcla de reacción se concentró al vacío y el residuo se purificó por HPLC prep. (columna: Phenomenex Gemini C18250 * 50 mm * 10 um; fase móvil:
[agua (0,05 % de hidróxido de amoníaco v/v)-ACN]; % de B: 32 %-62 %, 12 min) para dar 1-[4-[2-[2-(dietilamino)etoxi]-7- (5-metil-1H-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-1-il]prop-2-en-1-ona (16,0 mg, 30,2 umol, 39,2 % de rendimiento). ES+APCI MS m/z 519,3 [M+H]+.
Ejemplo 164
1 -(4 -(7 -(5 -m e til-1 H -in d a z o l-4 - il) -2 -(2 -(p ip e r id in -1 - il)e to x i)-5 ,6 ,7 ,8 -te tra h id ro p ir id o [3 ,4 -d ]p ir im id in -4 - il)p ip e ra z in -1 -il)e tan ona

Etapa A: 4-[7-[5-metil-1-(2-trimetilsililetoxi metil)indazol-4-il]-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo: A una solución de 4-[2-metilsulfinil-7-[5-metil-1- (2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (500 mg, 739 umol) y 2-(1-piperidil)etanol (191 mg, 1,48 mmol, 197 ul) en tolueno (20 ml) se le añadió t-BuONa (213 mg, 2,22 mmol). La mezcla se agitó a 20 °C durante 1 hora en una atmósfera de N2. La mezcla se enfrió a 0 °C y se añadió HCl (2 M) hasta pH ~7. La mezcla se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía en columna usando 0 ^10 % de MeOH/DCM como eluyente para dar 4-[7-[5-metil-1-(2-trimetilsililetoxi metil)indazol-4-il]-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (300 mg, 404 umol, 54,7 % de rendimiento). ES+APCI MS m/z 741,4 [M+H]+.

Etapa B: 7-(5-metil-1H-indazol-4-il)-4-piperazin-1-il-2-r2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidina: Una solución de 4-[7-[5-metil-1-(2-trimetilsililetoximetil) indazol-4-il]-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (250 mg, 337 umol) en TFA (10 ml) se agitó a 80 °C durante 1 hora.
La mezcla de reacción se concentró al vacío para dar 7-(5-metil-1H-indazol-4-il)-4-piperazin-1-il-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidina (200 mg, 283 umol, 84,1 % de rendimiento). ES+APCI MS m/z 477,3 [M+H]+.

Etapa C: 1 -[4-[7-(5-metil-1 H-indazol-4-il)-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -il]prop-2-en-1-ona: A una mezcla de 7-(5-metil-1H-indazol-4-il)-4-piperazin-1-il-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidina (200 mg, 283 umol, 2TFA) y DIEA (366 mg, 2,84 mmol, 495 ul) en DCM (8 ml) a -40 °C se le añadió una solución de prop-2-enoato de prop-2-enoílo (28,6 mg, 227 umol) DCM (2 ml) en una atmósfera de nitrógeno y la reacción se agitó durante 1 hora. La mezcla de reacción se inactivó mediante la adición de NaHCO3 (500 ul) a -40 °C y después se diluyó con agua (10 ml) y DCM (10 ml). La capa orgánica separada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini C18250 * 50 mm * 10 um;fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-a Cn ]; % de B: 30 % - 60 %, 12 min). 1-[4-[7-(5-metil-1H-indazol-4-il)-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (6,00 mg, 11,1 umol, 3,94 % de rendimiento, 99,0 % de pureza). Es a Pci MS m/z 531,4 [M+H]+.
Ejemplo 165
1- [4-[7-(2-fluoro-3-hidroxi-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2- en-1-ona
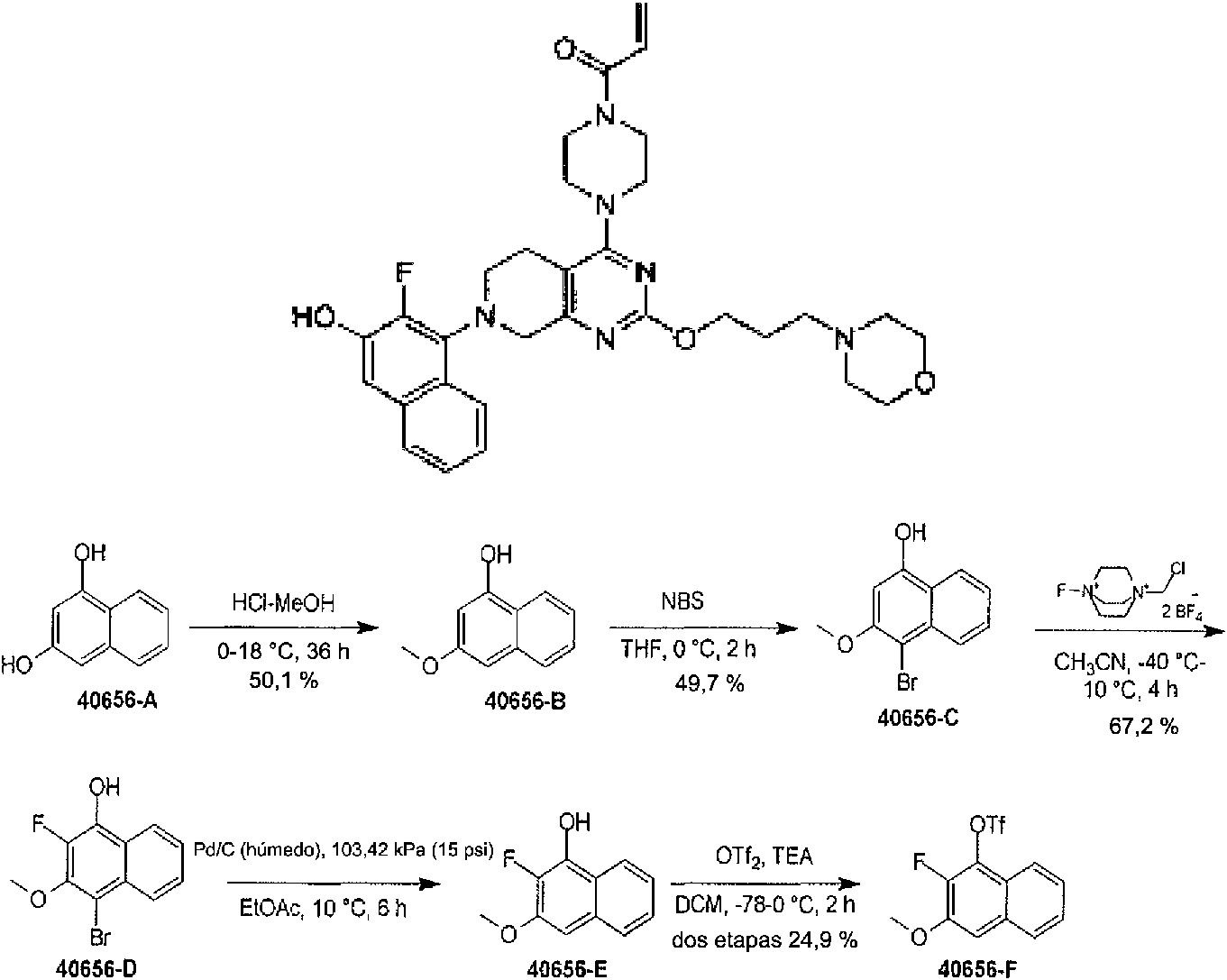
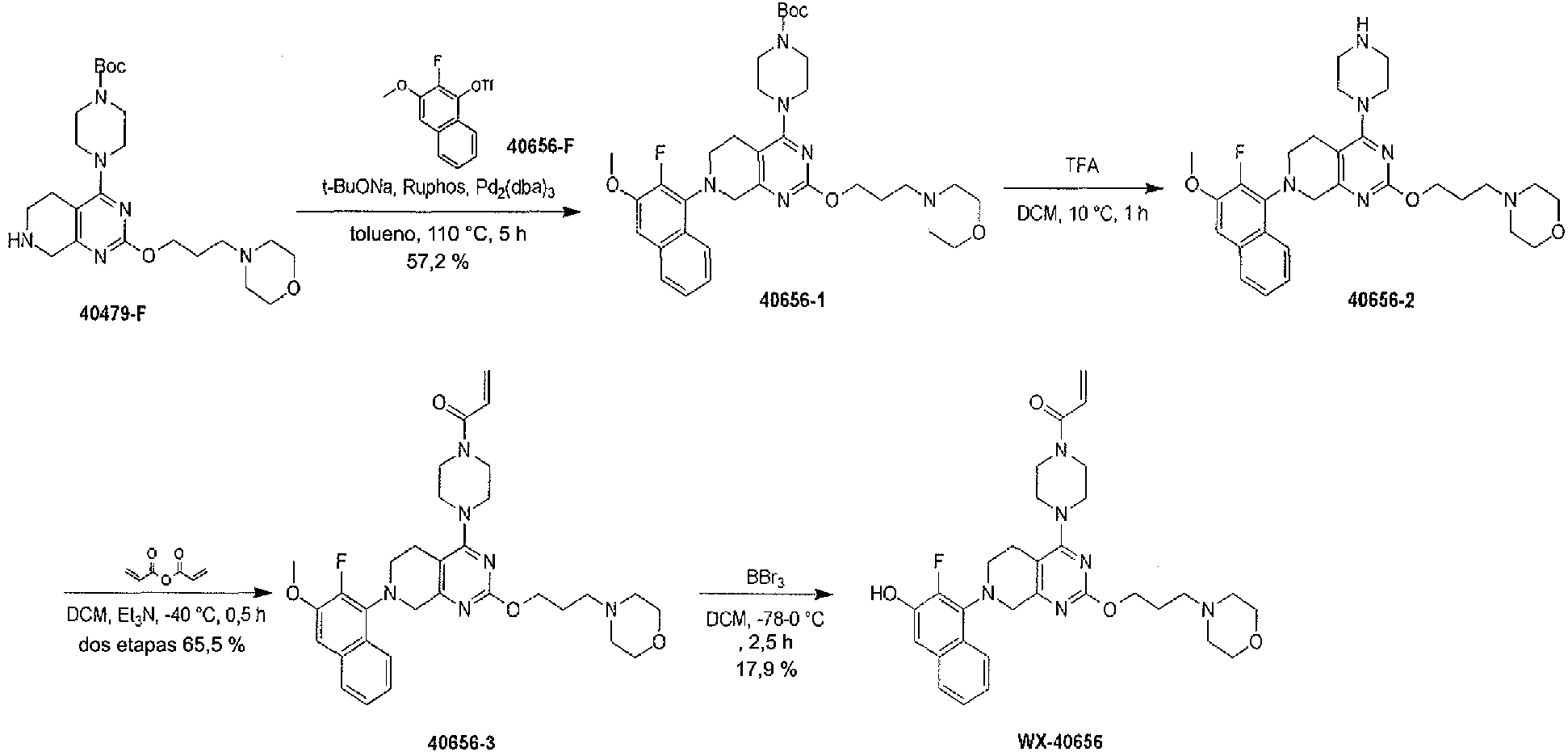
Etapa A: 3-metoxinaftalen-1 -ol: A una solución de naftaleno-1,3-diol (20 g, 125 mmol) en MeOH (150 ml) se le añadió HCl/MeOH (4 M, 250 ml) a 0 °C. La mezcla se calentó a 18 °C y se agitó durante 36 horas. La mezcla se concentró al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo/acetato de etilo 100/1 a 1/1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar 3-metoxinaftalen-1-ol (11 g, 62,5 mmol, 50,1 % de rendimiento, 99 % de pureza) en forma de un sólido de color amarillo claro. ES+APCI MS m/z 175,1 [M+H]+.
Etapa B: 4-bromo-3-metoxi-naftalen-1 -ol: A una solución de 3-metoxinaftalen-1-ol (0,50 g, 2,87 mmol) en THF (10 ml) se le añadió una solución de NBS (562 mg, 3,16 mmol) en THF (3,00 ml) a 0 °C. Después de agitar a 0 °C durante 2 horas, la mezcla se concentró al vacío, se diluyó con H2O (5 ml) y se extrajo con diclorometano (3 * 10 ml). Los extractos combinados se lavaron con cloruro de sodio saturado (1 * 20 ml), se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/éter acetato = 3/1) para dar 4-bromo-3-metoxi-naftalen-1 -ol (0,43 g, 1,43 mmol, 49,7 % de rendimiento) en forma de un sólido de color amarillo. ES+APCI MS m/z 253 [M+H]+.
Etapa C: 4-bromo-2-fluoro-3-metoxi-naftalen-1-ol: A una solución de 4-bromo-3-metoxi-naftalen-1-ol (4,00 g, 15,8 mmol) en acetonitrilo (20 ml) se le añadió una solución de 1-(clorometil)-4-fluoro-1,4-diazoniabiciclo[2.2.2]octano;ditetrafluoroborato (6,72 g, 19,0 mmol) en acetonitrilo (20 ml) a -40 °C. Después de agitar a -40 °C durante 1 hora y a 10 °C durante 3 horas, la mezcla se concentró al vacío. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/éter acetato = 3/1) para dar 4-bromo-2-fluoro-3-metoxi-naftalen-1-ol (4,00 g, 10,6 mmol, 67,2 % de rendimiento) en forma de un sólido de color amarillo. ES+APCI MS m/z 271,0 [M+H]+.
Etapa D: 2-fluoro-3-metoxi-naftalen-1-ol: Una mezcla de 4-bromo-2-fluoro-3-metoxi-naftalen-1-ol (2,00 g, 7,38 mmol) y Pd al 10 %/C (0,01 g) en acetato de etilo (20 ml) se agitó a 10 °C durante 1 hora en una atmósfera de H2103,42 kPa (15 psi). La mezcla se filtró y se concentró al vacío para dar 2-fluoro-3-metoxi-naftalen-1 -ol (1,60 g, bruto) en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional. ES+APCl MS m/z 193,0 [M+H]+.
Etapa E: trifluorometanosulfonato de (2-fluoro-3-metoxi-1-naftilo): A una solución de 2-fluoro-3-metoxi-naftalen-1-ol (1,60 g, bruto) y TEA (1,85 g, 18,3 mmol, 2,55 ml) en diclorometano (30 ml) se le añadió trifluorometanosulfonato de trifluorometilsulfonilo (2,35 g, 8,33 mmol, 1,37 ml) a -78 °C durante 1 hora. La mezcla se inactivó con cloruro de amonio saturado (30 ml), se extrajo con acetato de etilo (3 * 20 ml), se lavó con cloruro de sodio saturado (1 * 50 ml), se secó sobre Na2SO4, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/éter acetato = 3/1) para dar trifluorometanosulfonato de (2-fluoro-3-metoxi-1-naftilo) (1,10 g, 2,07 mmol, dos etapas 24,9 % de rendimiento) en forma de un sólido de color amarillo. ES+APCI MS m/z 324,9 [M+H]+.
Etapa F: 4-[7-(2-fluoro-3-metoxi-1 -naftil)-2-(3- morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo: Una mezcla de 4-[2-(3-morfolinopropoxi)-5,6,7,8- tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (0,40 g, 865 umol), trifluorometanosulfonato de (2-fluoro-3-metoxi-1-naftilo) (561 mg, 1,73 mmol), RuPhos (80,7 mg, 173 umol), Pd2(dba)3 (79,2 mg, 86,5 umol) y t-BuONa (249 mg, 2,59 mmol) en tolueno (10 ml) se agitó a 110 °C durante 5 horas en una atmósfera de N2. La mezcla se diluyó con agua (10 ml), se extrajo con acetato de éter (3 * 10 ml). Los extractos se lavaron con cloruro de sodio saturado (1 * 20 ml), se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía de fase inversa ultrarrápida [agua (0,1 % de ácido fórmico)/acetonitrilo] para dar 4-[7-(2-fluoro-3-metoxi-1-naftil)-2-(3
morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (0,35 g, 495 umol, 57,2 % de rendimiento) en forma de un aceite de color amarillo. ES+APCI MS m/z 637,1 [M+H]+.
Etapa G: 4-[3-[[7-(2-fluoro-3-metoxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]propil]morfolina: Una mezcla de 4-[7-(2-fluoro-3-metoxi-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (0,40 g, 628. umol) y TFA (1,07 g, 9,42 mmol, 698 ul) en diclorometano (0,70 ml) se agitó a 10 °C durante 1 hora. La mezcla se concentró al vacío para dar 4-[3-[[7-(2-fluoro-3-metoxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il)oxi]propil]morfolina (0,41 g, bruto, TFA) en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional. ES+APCI MS m/z 537,5 [M+H]+.
Etapa H: 1 -[4-[7-(2-fluoro-3-metoxi-1 -naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de 4-[3-[[7-(2-fluoro-3-metoxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]propil]morfolina (0,41 g, bruto, sal TFA) y TEA (635 mg, 6,27 mmol, 873 ul) en diclorometano (5,0 ml) se le añadió prop-2-enoato de prop-2-enoílo (79,1 mg, 627 umol) a -40 °C. Después de agitar a -40 °C durante 0,5 h, la mezcla se inactivó con metanol (0,10 ml) y se concentró al vacío. El residuo se purificó por cromatografía en columna (Al2Ü3, diclorometano/metanol = 10/1) para dar 1-[4-[7-(2-fluoro-3-metoxi-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (0,25 g, 411 umol, dos etapas 65,5 % de rendimiento) en forma de un aceite de color amarillo. ES+APCI MS m/z 591,0 [M+H]+.
Etapa I: 1 -[4-[7-(2-fluoro-3-hidroxi-1 -naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H- pirido[3,4-cf]pirimidin-4-il]piperazin-1 -il]prop-2-en-1-ona: A una solución de 1-[4-[7-(2-fluoro-3-metoxi-1-naftil)-2-(3- morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (0,20 g, 339 umol) en diclorometano (4,00 ml) se le añadió BBr3 (424 mg, 1,69 mmol, 163 ul) a -78 °C durante 0,5 horas y se agitó a 0 °C durante 2 horas. La mezcla se inactivó con bicarbonato de sodio saturado (5 ml) a -78 °C y se agitó a 0 °C durante 0,5 h. La mezcla se extrajo con diclorometano (3 x 10 ml) y se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi C18 150 * 25 * 10 um;fase móvil: [agua (0,225 % de ácido fórmico)-ACN];% de B: 13 %-45 %,7 min) para dar 1-[4-[7-(2-fluoro-3-hidroxi-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H- pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (35,2 mg, 60,8 umol, 17,9 % de rendimiento, 99,4 % de pureza) en forma de un sólido de color amarillo. ES+APCI MS m/z 577,0 [M+H]+.
Ejemplo 166
1-[4-[7-(6-hidroxi-2-metil-1,3-benzotiazol-4-il)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -il]prop-2-en-1 -ona


Etapa A: 4-triisopropilsililoxianilina: A una solución de 4-aminofenol (5,00 g, 45,8 mmol, 7,14 ml) en DCM (50,0 ml) se le añadió imidazol (4,06 g, 59,6 mmol). A la mezcla se le añadió gota a gota TIPSCl (13,3 g, 68,7 mmol, 14,7 ml). La mezcla se agitó a 15 °C durante 12 horas. Después de que se completara, la mezcla de reacción se filtró y el filtrado se concentró al vacío. El residuo se purificó por un sistema automatizado de cromatografía ultrarrápida (EP/AE 100/1 a 3/1) para dar 4-triisopropilsililoxianilina (9,46 g, 33,9 mmol, 73,9 % de rendimiento, 95,0 % de pureza) en forma de un aceite de color negro.
1H RMN (400 MHz, cloroformo-d) ó = 6,74 - 6,71 (m, 1H), 6,71 - 6,69 (m, 1H), 6,60 - 6,58 (m, 1H), 6,58 - 6,55 (m, 1H), 3,66 - 2,98 (m, 2H), 1,28 - 1,16 (m, 3H), 1,11 - 1,06 (m, 18H)
Etapa B: 2,6-dibromo-4-triisopropilsililoxi-anilina: A una solución de 4-triisopropilsililoxianilina (7,30 g, 27,5 mmol) en DCM (73,0 ml) y MeOH (73,0 ml) se le añadió Br2 (11,0 g, 68,8 mmol, 3,55 ml) en DCM (5 ml) gota a gota a 0 °C. La mezcla se agitó a 15 °C durante 5 horas. Después de que se completara, la mezcla se diluyó con una solución de sulfito de sodio (60 ml) y se extrajo con DCM (3 * 200 ml). Las capas orgánicas se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se purificó por cromatografía en columna (EP/AE 50/1 a 1/1) para dar 2,6-dibromo-4-triisopropilsililoxi-anilina (10,8 g, 23,2 mmol, 84,4 % de rendimiento, 90,8 % de pureza) en forma de un aceite de color pardo. ES+APCI MS m/z 423,9 [M+H]+.
Etapa C: N-(2,6-dibromo-4-triisopropilsililoxi-fenil)acetamida: A una solución de 2,6-dibromo-4-triisopropilsililoxi-anilina (10,4 g, 24,6 mmol) y CH3COOH (52 ml) se le añadió anhídrido acético (10,9 g, 107 mmol, 10 ml). La mezcla de reacción se agitó a 90 °C durante 1 hora. Después de que se completara, se añadió agua (100 ml) y DCM (200 ml) y las capas se separaron. La fase acuosa se extrajo con DCM (100 ml * 2). Los extractos combinados se lavaron con 5 % Na2CO3 (80 ml), se lavaron con salmuera (100 ml), se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se purificó por cromatografía en columna (EP/AE 50/1 a 1:1) para dar W-(2,6-dibromo-4-triisopropilsililoxifenil)acetamida (7,32 g, 14,2 mmol, 57,6 % de rendimiento, 90,0 % de pureza) en forma de un sólido de color pardo.
1H RMN (400 MHz, cloroformo-d) ó = 7,16 (s, 1H), 7,11 (s, 2H), 2,21 (s, 3H), 1,28 - 1,24 (m, 3H), 1,12 - 1,09 (m, 18H).
Etapa D: N-(2,6-dibromo-4-triisopropilsililoxi-fenil)tioacetamida: A una solución de W-(2,6-dibromo-4-triisopropilsililoxifenil)acetamida (7,22 g, 15,5 mmol) en tolueno (116 ml) se le añadió reactivo de Lawesson (3,14 g, 7,76 mmol). La mezcla se calentó a 110 °C durante 2 horas. Después de que se completara, la mezcla se concentró al vacío. El residuo se purificó por cromatografía en columna (EP/AE 100/1 a 1/1) W-(2,6-dibromo-4-triisopropilsililoxifenil)tioacetamida (5,41 g, 8,40 mmol, 54,1 % de rendimiento, 74,7 % de pureza) en forma de un aceite de color amarillo. ES+APCI MS m/z 481,9 [M+H]+.
Etapa E: 4-bromo-2-metil-1,3-benzotiazol-6-ol: Se añadió Cul (94,2 mg, 494 umol) a una solución de N-(2,6-dibromo-4-triisopropilsililoxi-fenil)tioacetamida (2,38 g, 4,94 mmol), 1,10-fenantrolina (134 mg, 741 umol) y Cs2CO3 (4,83 g,
14,8 mmol) en DME (48,0 ml). Después, la mezcla de reacción se agitó a 80 °C durante 12 horas en una atmósfera de N2. Después de que se completara, la mezcla de reacción se filtró y el filtrado se concentró. El residuo se purificó por cromatografía en columna (EP/AE 50/1 a 0/1) para dar 4-bromo-2-metil-1,3-benzotiazol-6-ol (1,33 g, 3,54 mmol, 71,7 % de rendimiento, 65,0 % de pureza) en forma de un aceite de color pardo.
1H RMN (400 MHz, DMSO-d6) ó = 7,33 (d, J = 2,4 Hz, 1H), 7,14 (d, J = 2,4 Hz, 1H), 3,40 - 3,33 (m, 1H), 2,72 (s, 3H).
Etapa F: 6-benc¡lox¡-4-bromo-2-met¡l-1,3-benzot¡azol: A una mezcla de 4-bromo-2-metil-1,3-benzotiazol-6-ol (1,28 g, 5,24 mmol) y K2CO3 (2,17 g, 15,72 mmol) en ACN (26,0 ml) se le añadió BnBr (988 mg, 5,76 mmol, 685 ul). La mezcla de reacción se agitó a 45 °C durante 1 hora. Después de que se completara, la mezcla de reacción se filtró y el filtrado se concentró. El residuo se purificó por cromatografía en columna (EP/AE 200/1 a 10/1) para dar 6-benciloxi-4-bromo-2-metil-1,3-benzotiazol (1,10 g, 3,13 mmol, 59,7 % de rendimiento, 95,0 % de pureza) en forma de un sólido de color amarillo claro. ES+APCI MS m/z 336,2 [M+H]+.
Etapa G: 4-[7-(6-benciloxi-2-metil-1,3-benzotiazol-4-il)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: Se agitaron 4-[2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (400 mg, 805 umol), 6-benciloxi-4-bromo-2-metil-1,3-benzotiazol (323 mg, 967 umol), RuPhos (75,2 mg, 161 umol), Cs2CO3 (787 mg, 2,42 mmol) y Pd2(dba)3 (73,8 mg, 80,6 umol) en tolueno (16 ml) a 85 °C durante 16 horas en una atmósfera de N2. Después de que se completara, la mezcla de reacción se filtró y el filtrado se concentró. El residuo se purificó por cromatografía en columna sobre ALO3 (EP/AE 10/1 a 0/1) para dar 4-[7-(6-benciloxi-2-metil-1,3-benzotiazol-4-il)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (340 mg, 408 umol, 50,7 % de rendimiento, 90,0 % de pureza) en forma de un aceite de color amarillo. ES+APCI MS m/z 750,5 [M+H]+.
Etapa H: 2zmetil^^2zí3dmorfoli^oprgpoxjjz4zp iperaz i^^:iJz6,8zdjhjdrgz5H -p irid^^^ :^p ir im id jn ^^^1 J_3zben^otiazQt:6z ol: A una solución de 4-[7-(6-benciloxi-2-metil-1,3-benzotiazol-4-il)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (160 mg, 213 umol) en MeOH (4 ml) se le añadió HCl/MeOH (4 M, 533 ul), seguido de Pd(OH)2/C (80 mg, 533 umol) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 40 °C durante 8 horas.
Después de que se completara, la mezcla de reacción se filtró y el filtrado se concentró para dar 2-metil-4-[2-(3-morfolinopropoxi)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-1,3-benzotiazol-6-ol (110 mg, 126 umol, 59,2 % de rendimiento, 68,7 % de pureza, 2 HCl) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. ES+Ap CI MS m/z 526,2 [M+H]+.
Etapa I: 1-[4-[7-(6-hidroxi-2-metil-1,3-benzotiazol-4-il)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de 2-metil-4-[2-(3-morfolinopropoxi)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-1,3-benzotiazol-6-ol (110 mg, 184 umol, 2 HCl) y DiEa (143 mg, 1,10 mmol, 193 ul) en DCM (2,00 ml) se le añadió prop-2-enoato de prop-2-enoílo (18,5 mg, 147 umol) gota a gota a -50 °C. La mezcla se agitó a -40 - -20 °C durante 30 minutos. Después de que se completara, la mezcla se inactivó con MeOH (0,5 ml) y se concentró al vacío. El residuo se diluyó con agua (2 ml) y se extrajo con DCM (3 * 6 ml). Las capas orgánicas se secaron sobre Na2SO4 y se concentraron al vacío. El residuo se purificó por cromatografía en columna sobre ALO3 (DCM/MeOH 20/1 a 10/1), HPLC prep. (columna: Gemini 150 * 25 5 ufase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN];% de B: 21 %-51 %,12 min) para dar 1-[4-[7-(6-hidroxi-2-metil-1,3-benzotiazol-4-il)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (32,4 mg, 55,4 umol, 30,2 % de rendimiento, 99,1 % de pureza) en forma de un sólido de color amarillo. ES+APCI MS m/z 580,4 [M+H]+.
Ejemplo 167
1-(4-(7-(5-hidroxi-2-metil-1H-indol-7-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona


Etapa A: 2-bromo-4-metoxianilina: A -10 °C, a una solución de 4-metoxianilina (100 g, 812 mmol) en THF (3 l) se le añadió W-bromosuccinimida (152 g, 853 mmol) en tres porciones y la mezcla se agitó a la misma temperatura durante 30 min. La mezcla se concentró a presión reducida. El residuo se purificó por cromatografía en columna. (éter de petróleo/acetato de etilo 15/1) para dar 2-bromo-4-metoxianilina en forma de un aceite de color rojo (30,58 g, 18,6 % de rendimiento). 1H RMN (400 MHz, CDCb) ó = 7,01 (d, J = 2,4 Hz, 1H), 6,74-6,70 (m, 2H), 3,85-3,74 (m, 2H), 3,73 (s, 3H).
Etapa B: 1 -(2-amino-3-bromo-5-metoxifenil)-2-cloropropan-1 -ona: A una solución a 0 °C de 2-bromo-4-metoxi-anilina (15,0 g, 74,2 mmol) en 1,1-dicloroetano (220 ml) se le añadió tricloruro de boro (1,00 M, 89,1 ml), 2-cloropropanonitrilo (9,97 g, 111 mmol) y tetracloruro de titanio (16,9 g, 89,1 mmol). La mezcla se calentó a 85 °C durante 24 horas. La mezcla se vertió en hielo y ácido clorhídrico (20 %, 300 ml) a 0 °C, se concentró y el residuo se calentó a reflujo durante 0,5 horas. Esta mezcla se basificó a 0 °C con hidróxido de sodio (acuoso saturado, 120 ml) hasta que se alcanzó pH 4 y después se extrajo con acetato de etilo (300 ml x 2). Los extractos orgánicos combinados se lavaron con salmuera (200 ml), se secaron con sulfato de sodio anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna (éter de petróleo/acetato de etilo/diclorometano 50/1/1) para dar 1 -(2-amino-3-bromo-5-metoxifenil)-2-cloropropan-1 -ona en forma de un sólido de color amarillo (18,7 g, 86,3 % de rendimiento). 1H RMN (400 MHz, CDCb) ó = 7,37 (d, J = 2,8 Hz, 1H), 7,32 (d, J = 2,8 Hz, 1H), 6,60 (s a, 2H), 5,25 (c, J = 6,4 Hz, 1H), 3,80 (s, 3H), 1,74 (d, J = 6,4 Hz, 3H).
Etapa C: 7-bromo-5-metoxi-2-metil-1H-indol: A una solución de 1-(2-amino-3-bromo-5-metoxi-fenil)-2-cloro-propan-1-ona (18,7 g, 64,1 mmol) en dioxano (500 ml) y H2O (50 ml) se le añadió NaBH4 (2,67 g, 70,5 mmol) y esta mezcla se agitó a 100 °C durante 15 horas. La mezcla se enfrió, se diluyó con ácido clorhídrico (ac., 0,10 M, 100 ml) y se extrajo con diclorometano (300 ml x 2). Los extractos orgánicos se lavaron con salmuera (200 ml), se secaron con sulfato de sodio anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna (éter de petróleo/acetato de etilo 25/1) para dar 7-bromo-5-metoxi-2-metil-1H-indol en forma de un sólido de color amarillo (7,01 g, 45,6 % de rendimiento). 1H RMN (400 MHz, CDCb) ó = 7,91 (s a, 1H), 7,03-6,91 (m, 2H), 6,23 (d, J = 1,2 Hz, 1H), 3,84 (s, 3H), 2,46 (s, 3H).
Etapa D: 4-(7-(5-metoxi-2-metil-1H-indol-7-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de terc-butilo: A una mezcla de 4-[2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (275 mg, 594 umol) y 7-bromo-5-metoxi-2-metil-1H-indol (130 mg, 540 umol) en 2-metil-2-butanol (15,0 ml) se le añadieron tBu0Na (104 mg, 1,08 mmol) y aducto de metilt-butil éter de cloro(2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)[2-(2-aminoetilfenil)]paladio (II) (44,1 mg, 54,0 umol). Esta mezcla se agitó a 90 °C durante 8 horas en una atmósfera de N2. La mezcla se filtró, se concentró a presión reducida. El residuo se purificó por TLC prep. (diclorometano/metanol 10/1), para dar 4-(7-(5-metoxi-2-metil-1 H-indol-7-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de tercbutilo en forma de un aceite de color amarillo (131 mg, 36,8 % de rendimiento). ES+APCI MS m/z 622,4 [M+H]+.
Etapa E: 4-[3-((7-(5-metox¡-2-met¡l-1H-¡ndol-7-¡l)-4-p¡peraz¡n-1-¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-2-¡l1ox¡]prop¡l1morfol¡na:
A una solución a 0 °C de 4-[7-(5-metoxi-2-metil-1H-indol-7-il)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo (131 mg, 211 umol) en diclorometano (10 ml) se le añadió gota a gota ácido trifluoroacético (2,00 ml) y la reacción se agitó a 15 °C durante 3 horas. La mezcla se concentró a presión reducida y el residuo se usó en la siguiente etapa sin purificación adicional. (126 mg de producto bruto). ES+APCI Ms m/z 522,4 [M+H]+.
Etapa F: 1-(4-(7-(5-metoxi-2-metil-1H-indol-7-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1- ¡l)prop-2 -en-1-ona: A una solución a -60 °C de 4-[3-[[7-(5-metoxi-2-metil-1H-indol-7-il)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]propil]morfolina (74 mg, 116 umol, sal del ácido trifluoroacético) en diclorometano (5 ml) se le añadió diisopropiletilamina (45,1 mg, 349 umol, 60,8 ul) y la mezcla se agitó a la misma temperatura durante 10 min. La mezcla se inactivó con ácido cítrico (ac., 1,00 ml), se extrajo con diclorometano (10 ml x 2), se lavó con salmuera (15 ml), se secó sobre sulfato de sodio anhidro y se concentró al vacío. El residuo se purificó por TLC prep. para dar 1-(4-(7-(5-metox¡-2- metil-1H-indol-7-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona en forma de un sólido de color amarillo (31 mg, 39,2 % de rendimiento). ES+APCI MS m/z 576,4 [M+H]+.
Etapa G: 1-(4-(7-(5-hidroxi-2-metil-1H-indol-7-il)-2-(3-morfolinopropoxi)- 5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-¡l)prop-2-en-1-ona: A una solución a -78 °C de 1-[4-[7-(5-metoxi-2-metil-1H-indol-7-il)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (26 mg, 45,2 umol) en diclorometano (5 ml) se le añadió tribromuro de boro (56,6 mg, 226 umol, 21,8 ul). La reacción se dejó calentar a 0 °C y se agitó durante 12 horas. La mezcla se neutralizó con NaHC03 (ac., 3 ml) y se extrajo con diclorometano (5 ml x 2), se lavó con salmuera (10 ml), se secó con sulfato de sodio anhidro y se concentró al vacío. El residuo se purificó por HPLC prep. para dar 1-(4-(7-(5-hidroxi-2-metil-1H-indol-7-il)-2-(3-morfolinopropoxi)- 5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona en forma de un sólido de color amarillo (8,16 mg, 29,1 % de rendimiento, 90,3 % de pureza). ES+APCI Ms m/z 562,5 [M+H]+.
Ejemplo 168
1-(3-(hidroximetil)-4-(7-(3-hidroxinaftalen-1-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2 -en-1-ona
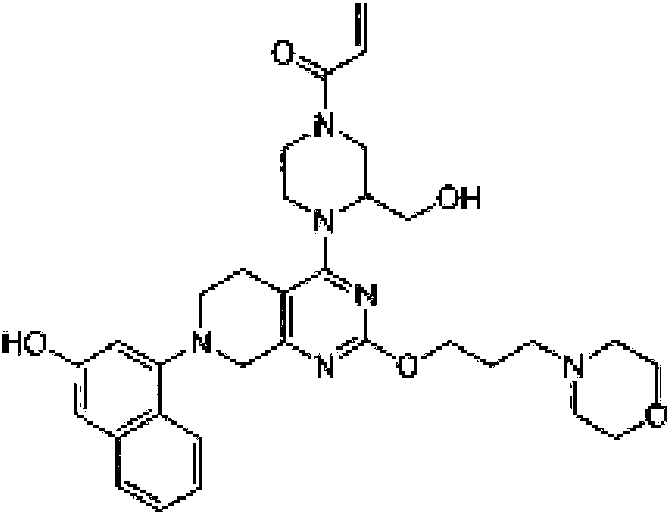

Etapa A: 3-[[tere-butil(difenil)silil]oximetil]piperazin-1 -carboxilato de terc-butilo: A una solución de 3-(hidroximetil)piperazin-l-carboxilato de tere-butilo (3,50 g, 16,2 mmol) en THF (30 ml) se le añadió NaH (3,24 g, 80,9 mmol, 60,0 % de pureza) a 0 °C. Después de agitar durante 30 minutos, se añadió en una porción TBDPSCl (6,67 g, 24,3 mmol, 6,23 ml). La mezcla se calentó a 10 °C y se agitó durante 12 horas en una atmósfera de N2. La mezcla de reacción se inactivó mediante la adición de agua a 0 °C y después se extrajo con DCM (200 ml). La capa orgánica se secó con Na2SÜ4, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna (SiÜ2, éter de petróleo/acetato de etilo 1 /0 a 1/2 ) para dar 3-[[tere-butil(difenil)silil]oximetil]piperazin-1 -carboxilato de terc-butilo (6,34 g, 12,1 mmol, 74,8 % de rendimiento, 86 ,8 % de pureza) en forma de un aceite incoloro. ES+APCI MS m/z 455,3 [M+H]+.
Etapa B: 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)- 3-[[tere-butil(difenil)silil]oximetil]piperazin-1 -carboxilato de terc-butilo: Una mezcla de 3-[[íere-butil(difenil)silil]oximetil]piperazin-1 -carboxilato de terc-butilo (7,25 g, 13,9 mmol), 7-bencil-2,4-dicloro-6,8-dihidro-5H-pirido[3,4-d]pirimidina (3,89 g, 13,2 mmol) y DIEA (4,27 g, 33,0 mmol, 5,77 ml) en DMS0 (60 ml) se agitó a 55 °C durante 24 horas en una atmósfera de N2. La mezcla de reacción se diluyó con acetato de etilo (300 ml) y se lavó con salmuera (3 x 150 ml). La capa orgánica se secó sobre Na2S04, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo 1/0 a 3/1) para dar 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)- 3-[[fere-butil(difenil)silil]oximetil]piperazin-1 -carboxilato de terc-butilo (4,50 g, 5,91 mmol, 44,7 % de rendimiento, 93,5 % de pureza) en forma de un sólido de color amarillo. ES+APCI MS m/z 721,3 [M+H]+.
Etapa C: 4-[7-bencil-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[[íerebutil(difenil)silil]oximetil]piperazin-1 -carboxilato de tere-butilo: Una mezcla de 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-3-[[íere-butil(difenil)silil]oximetil]piperazin-1 -carboxilato de terc-butilo (2,00 g, 2,81 mmol), 3-morfolinopropan-1 -ol (815 mg, 5,62 mmol), Pd(0Ac)2 (94,6 mg, 422 umol), BINAP (350 mg, 562 umol) y t-Bu0Na (674 mg, 7,03 mmol) en tolueno (30 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se calentó
a 110 °C y se agitó durante 3 horas en una atmósfera de N2. Después de que se completara, la mezcla de reacción se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna (SiO2, DCM/MeOH 70/1 a 20/1) para dar 4-[7-bencil-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[[tercbutil(difenil)silil]oximetil]piperazin-1 -carboxilato de terc-butilo (1,50 g, 1,69 mmol, 60,0 % de rendimiento, 92,3 % de pureza) en forma de un sólido de color amarillo. ES+APCI m S m/z 821,4 [M+H]+.
Etapa D: 3-[[terc-butil(difenil)silil]oximetil]-4-[2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]pipenazin-1-carboxilato de terc-butilo: A una solución de 4-[7-bencil-2-(3-morfolinopropoxi) -6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[[terc-butil(difenil)silil]oximetil]piperazin-1-carboxNato de terc-butilo (1,50 g, 1,83 mmol) en MeOH (20 ml) se le añadió Pd-C (10 %, 1,5 g) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (344,74 kPa (50 psi)) a 50 °C durante 24 horas. La mezcla de reacción se filtró y se concentró a presión reducida para dar 3-[[terc-butil(difenil)silil]oximetil]-4-[2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (988 mg, 1,21 mmol, 66,1 % de rendimiento, 89,5 % de pureza) en forma de un aceite incoloro, que se usó directamente para la siguiente etapa sin purificación adicional. ES+APCI MS m/z 731,5 [M+H]+.
Etapa E: 4-[7-(3-benciloxi-1-naftil)-2-(3-moifolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[[tercbutil(difenil)silil]oximetil]piperazin-1 -carboxilato de terc-butilo: Una mezcla de 3-[[terc-butil(difenil)silil]oximetil]-4-[2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (731 mg, 1,00 mmol), 3- benciloxi-1-bromo-naftaleno (783 mg, 2,50 mmol), [2-(2-aminofenil)fenil]paladio (1+);diciclohexil-[2-(2,4,6-triisopropilfenil)fenil]fosfano;metanosulfonato (169 mg, 200 umol), Cs2CO3 (815 mg, 2,50 mmol) en tolueno (15 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 60 °C durante 24 horas en una atmósfera de N2. La mezcla de reacción se filtró y se concentró a presión reducida a sequedad y se purificó por cromatografía en columna (SiOs, DCM/MeOH = 70/1 a 30/1) para dar 4-[7-(3-benciloxi-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[[terc-butil(difenil)silil]oximetil]piperazin-1-carboxilato de terc-butilo (250 mg, 188 umol, 18,8 % de rendimiento, 72,3 % de pureza) en forma de un sólido de color amarillo. ES+APCI MS m/z 963,4 [M+H]+.
Etapa F: 3-[[terc-butil(difenil)silil]oximetil]-4-[7-(3-hidroxi-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4- il]piperazin-1-carboxilato de terc-butilo: A una solución de 4-[7-(3-benciloxi-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[[terc-butil(difenil)silil]oximetil]piperazin-1 -carboxilato de terc-butilo (250 mg, 188 umol) en MeOH (3 ml) se le añadió Pd al 10 %/C (0,25 g) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 50 °C durante 3 horas. Después de que se completara, la mezcla de reacción se filtró y se concentró a presión reducida a sequedad para dar 3-[[terc-butil(difenil)silil]oximetil]-4-[7-(3-hidroxi-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (180 mg, 128 umol, 68,1 % de rendimiento, 62,0 % de pureza) en forma de un sólido de color amarillo oscuro, que se usó directamente para la siguiente etapa sin purificación adicional. ES+APCI MS m/z 873,4 [M+H]+.
Etapa G: 4-[4-[2-(hidroximetil)piperazin-1-il]-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-illnaftalen-2-ol: Una mezcla de 3-[[terc-butil(difenil)silil]oximetil]-4-[7-(3-hidroxi-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (130 mg, 149 umol), fluorhidrato de piridina (123 mg, 744 umol, 112 ul, 60,0 % de pureza) en THF (1,5 ml) se agitó a 25 °C durante 12 horas en una atmósfera de N2. La mezcla de reacción se concentró a presión reducida a sequedad. El residuo se purificó por HPLC de fase inversa (condición de TFA; MeCN en H2O; 0 ~ 30 %, caudal; 40 ml/min). Las fracciones deseadas se concentraron a presión reducida para dar 4-[4-[2-(hidroximetil)piperazin-1-il]-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (84 mg, 104 umol, 69,6 % de rendimiento, 94,1 % de pureza, 2TFA) en forma de un semisólido de color amarillo. ES+APCI MS m/z 535,3 [M+H]+.
Etapa H: 1-[3-(hidroximetil)-4-[7-(3-hidroxi-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una mezcla de 4-[4-[2-(hidroximetil)piperazin-1-il]-2-(3- morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (84,0 mg, 114 umol, 2TFA) y DIEA (36,9 mg, 285 umol, 49,8 ul) en DCM (3 ml) se le añadió prop-2-enoato de prop-2-enoílo (8,63 mg, 68,44 umol) gota a gota a -40 °C y después se agitó a -40 °C durante 0,5 horas en una atmósfera de N2. La mezcla de reacción se inactivó mediante la adición de MeOH (0,5 ml) y se concentró a presión reducida a sequedad. El residuo se purificó por HPLC prep. (columna: Venusil XBP C8150 * 25 * 10 um; fase móvil: [agua (0,225 % de ácido fórmico)-ACN]; % de B: 10 % - 40 %, 10 min). Las fracciones deseadas se recogieron y se liofilizaron a sequedad para dar 1-[3-(hidroximetil)-4-[7-(3-hidroxi-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (9,75 mg, 14,5 umol, 12,7 % de rendimiento, 94,4 % de pureza, sal del ácido fórmico) en forma de un sólido de color amarillo. ES+APCI MS m/z 589,3 [M+H]+.
Ejemplo 169
1-[3-(hidroximetil)-4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona

Etapa A: 4-[7-bencil-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[[tercbutil(difenil)silil]oximetil]piperazin-1-carboxilato de terc-butilo: A una mezcla de 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-3-[[terc-butil(difenil)silil]oximetil]piperazin-1-carboxilato de terc-butilo (495 mg, 694 umol), [(2S)-1-metilpirrolidin-2-il]metanol (160 mg, 1,39 mmol, 165 ul) y terc-butóxido de sodio (200 mg, 2,08 mmol) en tolueno (25 ml) se le añadieron BINAP (86,5 mg, 139 umol) y Pd2(dba)3 (63,6 mg, 69,5 umol). La mezcla se burbujeó con una atmósfera de nitrógeno y se agitó a 80 °C durante 6 h. La mezcla de reacción se diluyó con agua (10 ml) y se extrajo con acetato de etilo (2 x 15 ml). Las capas orgánicas combinadas se lavaron con agua (2 x 30 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiÜ2, éter de petróleo/acetato de etilo = 30/1 a 1:1) para dar 4-[7-bencil-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[[terc-butil(difenil)silil]oximetil]piperazin-1-carboxilato de terc-butilo (207 mg, 262 umol, 37,7 % de rendimiento) en forma de un sólido de color amarillo. 1H RMN (400 MHz, cloroformo-d) 5 =7,62 - 7,53 (m, 4H), 7,43 - 7,28 (m, 11H), 4,42 - 4,29 (m, 1H), 4,26 - 3,87 (m, 4H), 3,81 (d a, J = 13,2 Hz, 2H), 3,75 - 3,60 (m, 4H), 3,44 (dd, J = 1,6, 17,2 Hz, 1H), 3,24 - 3,04 (m, 3H), 2,96 (s a, 1H), 2,75 (s a, 3H), 2,49 (s, 3H), 2,43 - 2,28 (m, 2H), 2,10 - 1,96 (m, 2H), 1,91 - 1,80 (m, 1H), 1,80 - 1,67 (m, 2H), 1,43 (s, 9H), 1,02 - 0,89 (m, 9H).

Etapa B: 3-[[terc-butil(difenil)silil]oximetil]-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo: Se burbujeó NH3 en metanol (30 ml) a -40 °C durante 30 minutos.
A una solución de 4-[7-bencil-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[[tercbutil(difenil)silil]oximetil]piperazin-1-carboxilato de terc-butilo (530 mg, 670 umol) en la mezcla anterior se le añadió Pd al 10 %/C seco (0,30 g) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó a 40 °C durante 10 horas a 103,42 kPa (15 psi) de H2. La mezcla de reacción se filtró y se concentró a presión reducida para dar 3-[[terc-butil(difenil)silil]oximetil]-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo (394 mg, 427 umol, 63,8 % de rendimiento) en forma de un sólido de color amarillo. 1H RMN (400 MHz, cloroformo-d) 5 = 7,57 (t a, J = 6,0 Hz, 4H), 7,43 - 7,33 (m, 6H), 4,43 (dd a, J = 5,2, 10,4 Hz, 1H), 4,32 - 4,00 (m, 4H), 3,99 - 3,89 (m, 2H), 3,88 - 3,65 (m, 4H), 3,27 - 2,89 (m, 6H), 2,81 (d a, J = 8,4 Hz, 2H), 2,56 - 2,50 (m, 3H), 2,42 (d a, J = 16,8 Hz, 2H), 2,13 - 2,03 (m, 1H), 1,88 (d a, J = 6,8 Hz, 1H), 1,79 (d a, J = 5,2 Hz, 2H), 1,43 (s, 9H), 0,95 (s a, 9H).

Etapa C: 4-[7-(3-benciloxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[[tercbutil(difenil)silil]oximetil]piperazin-1 -carboxilato de ferc-butilo: Una mezcla de 3-[[ferc-butil(difenil)silil]oximetil]-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (0,16 g, 228 umol), 3-benciloxi-1-bromo-naftaleno (143 mg, 457 umol), [2-(2-aminoetil)fenil]-cloro-paladio;diciclohexil-[2-(2,4,6-triisopropilfenil)fenil]fosfano (16,9 mg, 22,8 umol) y t-BuONa (43,9 mg, 457 umol) en tolueno (4 ml) se agitó a 70 °C durante 12 horas en una atmósfera de N2. La mezcla se diluyó con agua (5,00 ml) y se extrajo con acetato de éter (3 x 10 ml). Los extractos orgánicos combinados se lavaron con cloruro de sodio saturado (10 ml), se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (ácido fórmico, 0,1 %)/acetonitrilo] para dar 4-[7-(3-benciloxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[[terc-butil(difenil)silil]oximetil]piperazin-1 -carboxilato de ferc-butilo (0,15 g, 157 umol, 68,9 % de rendimiento) en forma de un sólido de color amarillo. e S+APCI MS m/z 933,1 [M+H]+.

Etapa D: 3-[[ferc-butil(difenil)silil]oximetil]-4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo: Se burbujeó NH3 en metanol (10 ml) a -78 °C durante 30 minutos. Se añadieron 4-[7-(3-benciloxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[[terc-butil(difenil)silil]oximetil]piperazin-1 -carboxilato de ferc-butilo (0,10 g, 107 umol) y Pd al 10 %/C seco (0,01 g) y la mezcla se agitó a 10 °C durante 1 hora a 103,42 kPa (15 psi) de H2. La suspensión se filtró a través de una capa de celite y el filtrado se concentró al vacío para dar 3-[[ferc-butil(difenil)silil]oximetil]-4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de fercbutilo (0,09 g, bruto) en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional. ES+APCI MS m/z 843,0 [M+H]+.

Etapa E: 4-[4-[2-(hidroximetil)piperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol: Una mezcla de 3-[[terc-butil(difenil)silil]oximetil]-4-[7- (3-hidroxi-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (0,04 g, bruto) y piridina;fluorhidrato (118 mg, 712 umol, 106 ul) en diclorometano (2 ml) se agitó a 0 °C durante 1 hora y se agitó a 10 °C durante 12 horas. El pH de la mezcla se ajustó a pH >7 mediante la adición de una solución saturada de bicarbonato de sodio (3,00 ml) y se concentró al vacío. El residuo se purificó por cromatografía en columna (ALO3, diclorometano/metanol = 5/1) para dar 4-[4-[2-(hidroximetil)piperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (0,04 g, bruto) en forma de un sólido de color amarillo. ES+APCI Ms m/z 505,2 [M+H]+.

Etapa F: 1-[3-(hidroximetil)-4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de 4-[4-[2-(hidroximetil)piperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (0,02 g, bruto) y TEA (40,1 mg, 396 umol, 55,17 ul) en diclorometano (2 ml) se le añadió prop-2-enoato de prop-2-enoílo (999 ug, 7,93 umol) a -40 °C y después la mezcla se agitó a -40 °C durante 0,5 h. La mezcla se inactivó mediante la adición de metanol (0,10 ml) y se concentró al vacío. El residuo se purificó por cromatografía en columna (AhO3, diclorometano/metanol = 5/1). Las fracciones deseadas se recogieron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (NH4HCO310 mM)-ACN]; % de B: 38 %-68 %, 3 min). Las fracciones deseadas se recogieron y se liofilizaron para dar 1-[3-(hidroximetil)-4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (1,08 mg, 1,73 umol). ES+APCI MS m/z 559,5 [M+H]+.
Ejemplo 170
2-(1-acriloil-4-(2-(2-(dimetilamino)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo


Etapa A: 4-(7-bencil-2-cloro-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1,2-dicarboxilato de 1 -(terc-butil)2-metilo: A una solución de 7-bencil-2,4-dicloro-6,8-dihidro-5H-pirido[3,4-d]pirimidina (5,00 g, 17,0 mmol) y 2 metilpiperazin-1,2- dicarboxilato de 1-terc-butilo (4,24 g, 17,3 mmol) en Dm So (80 ml) se le añadió DIEA (5,49 g, 42,5 mmol, 7,42 ml). Después de agitar a 55 °C durante 12 horas, la mezcla se diluyó con acetato de etilo (100 ml), se lavó con salmuera (3 * 150 ml), se secó sobre Na2S04, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna (SiO2, EP/AE = 3/1) para dar 2-metil 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-1,2-dicarboxilato de 1-tere-butilo (8,00 g, 15,9 mmol, 93,8 % de rendimiento) en forma de un aceite de color amarillo. ES+APCI MS m/z 502,1 [M+H]+.
Etapa B: 2-metil 4-(7-bencil-2-(2-(dimetilamino)etoxi) -5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1,2-dicarboxilato de 1-(terc-butilo): Una mezcla de 2-metil 4-(7-bencil-2-cloro-6,8-dihidro- 5H-pirido[3,4-d]pirimidin-4-il)piperazin-1,2-dicarboxilato de 1-terc-butilo (8,00 g, 15,9 mmol), 2-(dimetilamino)etanol (2,84 g, 31,9 mmol, 3,19 ml), Cs2C03 (13,0 g, 39,8 mmol), Pd(0Ac)2 (537 mg, 2,39 mmol) y BlNAP (1,98 g, 3,19 mmol) en tolueno (30 ml) se agitó a 110 °C durante 3 horas en una atmósfera de nitrógeno. La mezcla se diluyó con agua (30 ml) y después se extrajo con acetato de etilo (3 * 50 ml). Los extractos se lavaron con salmuera (1 * 100 ml), se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC de fase inversa (TFA, 0,1 %) para dar 2-metil 4-[7-bencil-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1,2-dicarboxilato de 1 -tere-butilo (6,00 g, 10,8 mmol, 67,9 % de rendimiento) en forma de un sólido de color amarillo. ES+APCI MS m/z 555,3 [M+H]+.
Etapa C: 2-metil 4-[2-[2-(dimetilamino)etoxi]- 5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1,2-dicarboxilato de 1-tere-butilo: Una mezcla de 2-metil 4-[7-bencil-2-[2-(dimetilamino) etoxi]-6 ,8 -dihidro- 5H-pirido[3,4-d]pirimidin-4-il]piperazin-1,2-dicarboxilato de 1-tere-butilo (6,00 g, 10,8 mmol) y Pd al 10 %/C (600 mg, 10,8 mmol) en Me0H (100 ml) se agitó a 40 °C durante 12 horas en una atmósfera de H2 a 344,74 kPa (50 psi). La mezcla se filtró y el filtrado se concentró al vacío para dar 2-metil 4-[2-[2-(dimetilamino)etoxi]- 5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4
il]piperazin-1,2-dicarboxilato de 1-ferc-butilo (4,50 g, bruto) se obtuvo en forma de un sólido de color amarillo. ES+APCI MS m/z 465,3 [M+H]+.
Etapa D: 2-metil 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H- pirido[3,4-d]pirimidin-4-il]piperazina -1,2-dicarboxilato de 1-ferc-butilo: Una mezcla de 2-metil 4-[2-[2-(dimetilamino)etoxi]- 5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1,2-dicarboxilato de 1-ferc-butilo (2,90 g, 6,24 mmol), 3-benciloxi-1-bromo-naftaleno (2,54 g, 8,12 mmol), Pd2(dba)3 (857 mg, 936 umol), RuPhos (728 mg, 1,56 mmol) y Cs2CO3 (5,08 g, 15,6 mmol) en 1,4-dioxano (150 ml) se agitó a 85 °C durante 5 horas en una atmósfera de N2. La mezcla se diluyó con agua (100 ml) y se extrajo con DCM (1 x 200 ml). Los extractos se lavaron con salmuera (1 x 300 ml), se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC de fase inversa (TFA, 0,1 %) para dar 2-metil 4-[7-(3-benciloxi-1- naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H- pirido[3,4-d]pirimidin-4-il]piperazin-1,2-dicarboxilato de 1-ferc-butilo (3,50 g, 5,02 mmol, dos etapas 72,5 % de rendimiento) en forma de un sólido de color pardo. ES+APCI MS m/z 697,3 [M+H]+.
Etapa E: 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(hidroximetil)piperazin- 1-carboxilato de terc-butilo: A una solución de 2-metil 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1,2-dicarboxilato de 1 -terc-butilo (1,00 g, 1,44 mmol) en THF (10 ml) se le añadió poco a poco LiAlH4 (219 mg, 5,76 mmol) a -60 °C. Después de agitar durante 2 horas, la mezcla se inactivó con una solución saturada de sulfato de sodio (0,3 ml), se filtró y la torta de filtro se lavó con DCM (100 ml). El filtrado combinado se concentró al vacío para dar 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(hidroximetil)piperazin-1-carboxilato de terc-butilo (750 mg, 1,12 mmol, 77,8 % de rendimiento) en forma de un sólido de color amarillo. ES+APCI MS m/z 669,3 [M+H]+.
Etapa F: 2-[4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro - 5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo: Una mezcla de 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro- 5H-pirido[3,4-d]pirimidin-4-il]-2-(hidroximetil)piperazin-1-carboxilato de ferc-butilo (100 mg, 149 umol), TBAI (11,1 mg, 29,9 umol), 1-(ptolilsulfonil)imidazol (79,9 mg, 359 umol), NaCN (0,12 g, 2,45 mmol) y TEA (37,8 mg, 374 umol, 51,8 ul) en Dm F se agitó a 155 °C durante 6 horas. La mezcla se concentró al vacío, se diluyó con agua (1 x 5 ml) y después se extrajo con acetato de etilo (3 x 10 ml). Los extractos se lavaron con salmuera (1 x 20 ml), se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. El residuo se purificó por columna de HPLC prep.: Phenomenex Gemini C18250 * 50 mm * 10 um;fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN]; % de B: 50 %-70 %, 28 min) para dar 2- [4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (160 mg, 277 umol, 37,1 % de rendimiento) en forma de un sólido de color amarillo. ES+Ap C i MS m/z 578,5 [M+H]+.
Etapa G: 2-[4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo: A una solución de 2-[4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]- 6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (60 mg, 104 umol) y DIeA (67,1 mg, 519 umol, 90,7 ul) en DCM (2 ml) se le añadió prop-2-enoato de prop-2-enoílo (13,1 mg, 104 umol) a 0 °C. Después de agitar a 10 °C durante 4 horas, la mezcla se inactivó con MeOH (0,1 ml) y después se concentró al vacío. El residuo se purificó por cromatografía en columna (SO2, DCM/MeoH 10/1) para dar 2-[4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]- 6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (40,0 mg, 63,3 umol, 61,0 % de rendimiento) en forma de un aceite de color amarillo. ES+APCI MS m/z 632,3 [M+H]+.
Etapa H: 2-[4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H pirido[3,4-d]pirimidin-4-il]-1 -prop-2-enoilpiperazin-2-il]acetonitrilo: A una solución de 2-[4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (30 mg, 47,5 umol) en DCM (2 ml) se le añadió BBr3 (35,7 mg, 142 umol, 13,7 ul) a -78 °C durante 0,5 h. La mezcla se calentó a 0 °C y se agitó durante 2 horas. La mezcla se concentró al vacío y después se inactivó con una solución saturada de bicarbonato de sodio a -78 - 0 °C. La mezcla se purificó por cromatografía en columna (AhO3, DCM/MeOH = 5/1) y después se purificó adicionalmente por HPLC prep. (columna: Gemini 150 * 25 5 ufase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN];% de B: 30 %-60 %,12 min). Las fracciones deseadas se recogieron y se liofilizaron para dar 2-[4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (810 ug, 1,46 umol, 3,07 % de rendimiento, 97,5 % de pureza) en forma de un sólido de color amarillo. ES+APCI MS m/z 542,5 [M+H]+.
Ejemplo 171
1-(4-(2-(2-(dimetilamino)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(2-hidroxietil)piperazin-1-il)prop-2-en-1-ona

Etapa A: 2-(3-Oxop¡peraz¡n-2-il)acetato de etilo: A una solución de (E)-but-2-enodioato de dietilo (30,2 g, 175 mmol, 28,8 ml) en i-PrOH (300 ml) se le añadió etano-1,2-diamina (11,0 g, 183 mmol, 12,2 ml). Después de agitar a 25 °C durante 12 horas, la mezcla de reacción se concentró a presión reducida a sequedad. El sólido bruto de color blanco se lavó con MTBE (500 ml) y se secó al vacío para dar 2-(3-oxopiperazin-2-il)acetato de etilo (26,0 g, 140 mmol, 79,6 % de rendimiento, 100 % de pureza) en forma de un sólido de color blanco. 1H RMN (400 MHz, Cloroformo-d) ó = 6,55 (s a, 1H), 4,15 (c, J = 6,8 Hz, 2H), 3,80 - 3,72 (m, 1H), 3,47 (dt, J = 4,8, 11,2 Hz, 1H), 3,36 - 3,22 (m, 1H), 3,18 - 3,08 (m, 1H), 3,07 - 2,95 (m, 2H), 2,76 - 2,70 (m, 1H), 1,25 (t, J = 7,2 Hz, 3H).
Etapa B: 2-[1-[(4-Metox¡fen¡l)met¡l1-3-oxo-p¡peraz¡n-2-¡l1acetato de etilo: A una mezcla de 2-(3-oxopiperazin-2-il)acetato de etilo (23,6 g, 127 mmol) en metanol (400 ml) se le añadieron 4-metoxibenzaldehído (18,9 g, 139 mmol, 16,9 ml) y CH3COOH (15,2 g, 253 mmol, 14,5 ml) a 0 °C. Después de agitar a 0 °C durante 1 hora, la mezcla de reacción se enfrió a -10 °C. A la mezcla se le añadió poco a poco NaBHaCN (23,9 g, 380 mmol) y la mezcla de reacción se calentó a 15 °C y se agitó durante 11 horas más. La reacción se interrumpió mediante la adición de agua (400 ML) y se extrajo con DCM (2 x 300 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron. El residuo se disolvió de nuevo con acetato de etilo (300 ml) y después se lavó con una solución 0,5 M de HCl (2 x 200 ml). La fase acuosa se ajustó a pH 7 ~ 8 con Na2CO3 sólido y después se extrajo con DCM (2 x 300 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron para dar 2-[1-[(4-metoxifeni^metil^-oxo-piperazin^-i^acetato de etilo (27,2 g, 87,6 mmol, 69,0 % de rendimiento, 98,5 % de pureza) en forma de un aceite incoloro. ES+APCI MS m/z 307,1 [M+H]+.
Etapa C: 2-[1-[(4-metoxifenil)metil]piperazin-2-il1etanol: A una mezcla de 2-[1-[(4-metox¡fen¡l)met¡l]-3-oxo-p¡peraz¡n-2-il]acetato de etilo (27,2 g, 88,8 mmol) en THF (500 ml) se le añadió poco a poco LiAlH4 (10,1 g, 266 mmol) a 0 °C. Después de agitar a 0 °C durante 1 hora, la mezcla de reacción se calentó a 70 °C y se agitó durante 11 horas. Después de que se completara, la reacción se interrumpió con Na2SO4 acuoso sat. (40 ml), se filtró y después se concentró. El producto 2-[1-[(4-metox¡fen¡l)met¡l]p¡peraz¡n-2-¡l]etanol (20 g, bruto) se obtuvo en forma de un aceite de color amarillo. ES+APCI MS m/z 251,1 [M+H]+.
Etapa D: [2-[1-[(4-metox¡fen¡l)met¡l1p¡peraz¡n-2-¡l1etox¡1- difenil-silano de tere-butilo: A una mezcla de 2-[1-[(4-metox¡fen¡l)met¡l]p¡peraz¡n-2-¡l]etanol (20 g, bruto) en THF(300 ml) se le añadió poco a poco NaH (15,9 g, 399 mmol, 60 % de pureza) a 0 °C y después se añadió una solución de TBDPSCl (65,9 g, 239 mmol, 61,6 ml) en THF (100 ml). La mezcla se calentó a 15 °C y se agitó durante 12 horas más. Después de que se completara, la reacción se vertió en una solución de NH4Cl (300 ml) y se extrajo con DCM (2 x 300 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo: acetato de etilo 3/1 a diclorometano/metanol 10/1) para dar ferc-but¡l-[2-[1-[(4-metox¡fen¡l)met¡l]p¡peraz¡n-2-¡l]etox¡]-d¡fen¡l-s¡lano (29,5 g, 59,9 mmol, dos etapas 35 %, 99,2 % de pureza) en forma de un aceite de color amarillo. ES+APCI MS m/z 489,4 [M+H]+.
Etapa E: 3-[2-[fere-but¡l(d¡fen¡l)s¡l¡l1ox¡et¡l1-4-[(4-metox¡fen¡l)met¡l1p¡peraz¡n-1-carbox¡lato de terc-butilo: A una mezcla de fere-butil-p-n-^-metoxifenil) met¡l]p¡peraz¡n-2-¡l]etox¡]-d¡fen¡l-s¡lano (11,0 g, 22,5 mmol) en MeOH (200 ml) se le añadieron TEA (6,83 g, 67,5 mmol, 9,36 ml) y (Boc)2O (9,82 g, 45,0 mmol, 10,3 ml). Después de agitar a 15 °C durante 3 horas, la reacción se concentró. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo: acetato de etilo 1/0 a 5/1) para dar 3-[2-[fe ê-but¡l(d¡fen¡l)s¡l¡l1ox¡et¡l1-4-[(4-metox¡fen¡l)met¡l1p¡peraz¡n-1 -carboxilato de terc-butilo (11,0 g, 18,7 mmol, 82,9 % de rendimiento, 100 % de pureza) en forma de un aceite incoloro. ES+APCI MS m/z 589,4 [M+H]+.
Etapa F: 3-[2-[fere-but¡l(d¡fen¡l)s¡l¡l1ox¡et¡l1p¡peraz¡n-1-carbox¡lato de fere-butilo: A una mezcla de 3-[2-[ferebut¡l(d¡fen¡l)s¡l¡l1ox¡et¡l1-4-[(4-metox¡fen¡l)met¡l1p¡peraz¡n-1 -carboxilato de fere-butilo (9,00 g, 15,3 mmol) en MeOH (150 ml) se le añadió Pd al 10 %/C (849 umol). La suspensión se desgasificó al vacío y se purgó tres veces con H2. La mezcla se agitó en una atmósfera de H2 (344,74 kPa (50 psi)) a 40 °C durante 18 horas. Después de que se completara, la mezcla de reacción se filtró a través de un lecho de Celite y el filtrado se concentró. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo 10/1 a 3:1) para dar 3-[2-[ferebut¡l(d¡fen¡l)s¡l¡l]ox¡et¡l]p¡peraz¡n-1-carbox¡lato de terc-butilo (5,80 g, 12,2 mmol, 79,5 % de rendimiento, 98,2 % de pureza) en forma de un aceite incoloro. ES+APCI MS m/z 469,4 [M+H]+.
Etapa G: 2-[4-(7-benc¡l-2-cloro-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)-1-[(4-metox¡fen¡l)met¡l]p¡peraz¡n-2-¡l]etox¡-terc-butil-difenil-silano: Una mezcla de 7-benc¡l-2,4-d¡cloro-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡na (4,00 g, 13,6 mmol), fere-but¡l-[2-[1-[(4-metox¡fen¡l)met¡l]p¡peraz¡n-2-¡l]etox¡]-d¡fen¡l-s¡lano (6,98 g, 14,3 mmol) y DIEA (4,39 g, 34,0 mmol, 5,93 ml) en DMSO (80 ml) se desgasificó y se purgó 3 veces con N2 y la mezcla se calentó a 55 °C y se agitó durante 12 horas en una atmósfera de N2. La mezcla de reacción se diluyó con agua (300 ml) y se extrajo con acetato de etilo (3 x 200 ml). Las capas orgánicas combinadas se lavaron con salmuera (3 x 200 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida a sequedad. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo 1/0 a 3/1) para dar 2-[4-(7-benc¡l-2-cloro-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)-1-[(4-metox¡fen¡l)met¡l]p¡peraz¡n-2-¡l]etox¡-terc-but¡l-d¡fen¡l-s¡lano (9,00 g, 11,7 mmol, 86,0 % de rendimiento, 97,0 % de pureza) en forma de un aceite de color amarillo. ES+APCI MS m/z 746,4 [M+H]+.
Etapa H: 2-[[7-bencil-4-[3-[2-[ferc-butil(difenil)silil]oxietil]-4-[(4-metoxifenil) metil]piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]-Ñ,Ñ-dimetil-etanamina: Una mezcla de 2 -(dimetilamino)etanol (2,39 g, 26,8 mmol, 2.69 ml), 2-[4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-1-[(4-metoxifenil)metil]piperazin-2-il]etoxiferc-butil-difenil-silano (8 ,00 g, 10,7 mmol), Pd(0Ac)2 (361 mg, 1,61 mmol), BiNAP (1,34 g, 2,14 mmol) y Cs2C03 (8,73 g, 26,8 mmol) en tolueno (100 ml) se desgasificó y se purgó 3 veces con N2. La mezcla se calentó a 110 °C y se agitó durante 3 horas en una atmósfera de N2. La mezcla de reacción se filtró y el filtrado se concentró a presión reducida a sequedad. El residuo se purificó por cromatografía en columna (SiO2, DCM/Me0H 50/1 a 10/1) para dar 2-[[7-bencil-4-[3-[2-[ferc-butil(difenil)silil]oxietil]-4-[(4-metoxifenil) metil]piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]-W,W-dimetil-etanamina (6,50 g, bruto) en forma de un semisólido de color amarillo. ES+APCI MS m/z 799,4 [M+H]+.
Etapa I: 2-[4-[7-bencil-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]etanol: 2-[4-[7-bencil-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]etanol: Una solución de 2-[[7-bencil-4-[3-[2-[ferc-butil(difenil)silil]oxietil]-4-[(4-metoxifenil)metil]piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]-W,Ñ-dimetil-etanamina (6,50 g, bruto) en TFA (80 ml) se agitó a 80 °C durante 12 horas. La mezcla de reacción se concentró a presión reducida a sequedad y el residuo se disolvió de nuevo en DCM (300 ml) y se lavó con agua (200 ml). La fase de agua se recogió y se basificó con NaHC03 acuoso saturado a pH ~ 8 y después se extrajo con DCM (3 x 200 ml). La fase orgánica combinada se secó sobre Na2S04, se filtró y se concentró a presión reducida para dar 2-[4-[7-bencil-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]etanol (3,00 g, bruto) en forma de un semisólido de color amarillo, que se usó directamente para la siguiente etapa sin purificación adicional.
Etapa J: 2-[[7-bencil-4-[3-[2-[ferc-butil(difenil)silil]oxietil]piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]-W,W-dimetil-etanamina: A una solución de 2-[4-[7-bencil-2-[2-(dimetilamino)etoxi]-6,8-dihidro -5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]etanol (3,0 g, bruto) en THF (40 ml) se le añadió NaH (1,36 g, 34,1 mmol, 60 ,0 % de pureza) a 0 °C. Después de agitar durante 30 minutos, se añadió en porciones TBDPSCl (2,81 g, 10,2 mmol, 2,63 ml). La mezcla se calentó a 25 °C y se agitó durante 2 horas más en una atmósfera de N2. La mezcla de reacción se inactivó mediante la adición de agua (30 ml) a 0 °C y se extrajo en acetato de etilo (3 x 100 ml). Las capas orgánicas combinadas se lavaron con salmuera (3 x 100 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida a sequedad para dar 2-[[7-bencil-4-[3-[2-[ferc-butil(difenil)silil]oxietil]piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]-N,N-dimetil-etanamina (6,00 g, bruto) en forma de un aceite de color amarillo, que se usó directamente para la siguiente etapa sin purificación adicional. ES+APCI MS m/z 679,4 [M+H]+.
Etapa K: 4-[7-bencil-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-[2-[fercbutil(difenil)silil]oxietil]piperazin-1-carboxilato de terc-butilo: Una mezcla de 2-[[7-bencil-4-[3-[2-[fercbutil(difenil)silil]oxietil]piperazin-1-il]-6,8-dihidro- 5H-pirido[3,4-d]pirimidin-2-il]oxi]-W,W-dimetil-etanamina (6,0 g, bruto) y Boc20 (18,5 g, 84,8 mmol, 19,5 ml) en Me0H (3 ml) se desgasificó y se purgó 3 veces con N2. La mezcla se calentó a 80 °C y se agitó durante 3 horas en una atmósfera de N2. La mezcla de reacción se concentró a presión reducida a sequedad. El residuo se purificó por cromatografía en columna (SiO2, DCM /Me0H 1/0 a 10/1) para dar 4-[7-bencil-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-[2-[fercbutil(difenil)silil]oxietil]piperazin-1-carboxilato de terc-butilo (3,10 g, 3,28 mmol, cuatro etapas 30,7 % de rendimiento, 82,2 % de pureza) en forma de un semisólido. ES+APCI MS m/z 779,4 [M+H]+.
Etapa L: 2-[2-[ferc-butil(difenil)silil]oxietil]-4-[2-[2-(dimetilamino)etoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo: A una solución de 4-[7-bencil-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-[2-[ferc-butil(difenil)silil]oxietil]piperazin-1 -carboxilato de ferc-butilo (3,10 g, 3,28 mmol, 82,2 % de pureza) en Me0H (60 ml) se le añadió Pd-C (10 %, 2 g) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó tres veces con H2. La mezcla se agitó en una atmósfera de H2 (344,74 kPa (50 psi)) a 50 °C durante 36 horas. La mezcla de reacción se filtró y se concentró a presión reducida a sequedad para dar 2 -[2 -[ferc-butil(difenil)silil]oxietil]-4-[2-[2-(dimetilamino)etoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (2,20 g, 2,75 mmol, 83,8 % de rendimiento, 86,0 % de pureza) en forma de un sólido de color amarillo, que se usó directamente para la siguiente etapa sin purificación adicional. ES+APCI MS m/z 689,4 [M+H]+.
Etapa M: 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-[2-[fercbutil(difenil)silil]oxietil]piperazin-1-carboxilato de ferc-butilo: Una mezcla de 2-[2-[ferc-butil(difenil)silil]oxietil]-4-[2-[2-(dimetilamino)etoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (1,85 g, 2.69 mmol), 3-benciloxi-1-bromo-naftaleno (1,09 g, 3,49 mmol), Cs2C03 (2,19 g, 6,71 mmol), [2 -(2 -aminofenil)fenil]paladio (1+); diciclohexil-[2-(2,4,6-triisopropilfenil)fenil]fosfano;metanosulfonato (341 mg, 403 umol) en tolueno (40 ml) se desgasificó y se purgó 3 veces con N2 y la mezcla se agitó a 65 °C durante 10 horas en una atmósfera de N2. Después de que se completara, la mezcla de reacción se filtró y se concentró a presión reducida a sequedad. El residuo se purificó por cromatografía en columna (SiO2, DCM/Me0H 1/0 a 10 /1) para dar 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-[2-[fercbutil(difenil)silil]oxietil]piperazin-1-carboxilato de terc-butilo (1,85 g, 1,69 mmol, 63,2 % de rendimiento, 84,3 % de pureza) en forma de un sólido de color amarillo. ES+APCI MS m/z 922,5 [M+H]+.
Etapa N: 1-[4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-[2-[fercbutil(difenil)silil]oxietil]piperazin-1-il]prop-2 -en-1-ona: A una solución de 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]
6.8- dihidro- 5H-pirido[3,4-d]pirimidin-4-il]-2-[2-[terc-butil(difenil)silil]oxietil]piperazin-1-carboxilato de terc-butilo (500 mg, 543 umol), 2,6-lutidina (700 mg, 6,51 mmol, 759 ul) en DCM (10 ml) se le añadió TMSOTf (724 mg, 3,26 mmol, 588 ul). Después de agitar a 40 °C durante 2 horas en una atmósfera de N2, la mezcla de reacción se enfrió a -40 °C y se añadió poco a poco prop-2-enoato de prop-2-enoílo (137 mg, 1,09 mmol). La mezcla de reacción se calentó a 25 °C y se agitó durante 12 horas más en una atmósfera de N2. Después de que se completara, la mezcla de reacción se purificó directamente por cromatografía en columna (SO2, DCM/MeOH 30/1 a 10/1) para dar 1-[4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6.8- dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-[2-[ferc-butil(difenil)silil]oxietil]piperazin-1-il]prop-2-en-1-ona (256 mg, 226 umol, 41,6 % de rendimiento, 77,2 % de pureza) en forma de un semisólido. Es APCI MS m/z 875,5 [M+H]+.
Etapa O: 1-[4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(2-hidroxietil)piperazin-1-il]prop-2-en-1-ona: A una solución de 1-[4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6.8- dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-[2-[ferc-butil(difenil)siiil]oxietil]piperazin-1-il]prop-2-en-1-ona (133 mg, 152 umol) en Dc M (5,00 ml) se le añadió BBr3 (571 mg, 2,28 mmol, 220 ul) gota a gota a -40 °C. La mezcla de reacción se calentó a 0 °C y se agitó durante 3 horas. La mezcla se concentró a presión reducida a sequedad. El material bruto se lavó con MTBE (25 ml) y el sólido se recogió. El sólido se lavó con una solución saturada de NaHCO3 (0,5 ml) a pH ~ 8 a 0 °C y después se disolvió en MeOH (3 ml). La mezcla se purificó directamente por HPLC prep. (columna: Boston Green ODS 150 * 30 5 ufase móvil: [agua (0,225 % de ácido fórmico)-ACN]; % de B: 15 % - 39 %, 10 min) para dar 1-[4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(2-hidroxietil)piperazin-1-il]prop-2-en-1-ona (16,7 mg, 28,2 umol, 18,5 % de rendimiento, 99,9 % de pureza, sal del ácido fórmico) en forma de un sólido de color amarillo. ES+APCI MS m/z 547,3 [M+H]+.
Ejemplo 172
1-(4-(2-(2-(dimetilamino)etoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-3-(2-hidroxietil)piperazin-1-il)prop-2-en-1-ona


Etapa A: 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-3-[2-[ferc-butil(difenil)silil]oxietil]piperazin-1-carboxilato de terc-butilo: Una mezcla de 7-bencil-2,4-dicloro- 6,8-dihidro-5H-pirido[3,4-d]pirimidina (3,04 g, 10,3 mmol), 3-[2-[íerc-butil(difenil)silil]oxietil]piperazin-1 -carboxilato de terc-butilo (5,08 g, 10,8 mmol) y DIEA (3,34 g, 25,8 mmol, 4,51 ml) en DMSO (60 ml) se desgasificó y se purgó 3 veces con N2° La mezcla se calentó a 55 °C y se agitó durante 16 horas en una atmósfera de N2. Después de enfriar a temperatura ambiente, la mezcla de reacción se diluyó con agua (100 ml) y se extrajo con acetato de etilo (3 x 100 ml). Las capas orgánicas combinadas se lavaron con salmuera (3 x 100 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida a sequedad. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo 1/0 a 3/1) para dar 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-3-[2-[terc-butil(difenil)silil]oxietil]piperazin-1-carboxilato de tercbutilo (3,50 g, 4,53 mmol, 43,9 % de rendimiento, 94,0 % de pureza) en forma de un semisólido de color amarillo. ES+APCI MS m/z 726,4 [M+H]+.
Etapa B: 4-[7-bencil-2-[2-(dimetilamino)etoxi]-6,8-dihidro- 5H-pirido[3,4-d]pirimidin-4-il]-3-[2-[tercbutil(difenil)silil]oxietil]piperazin-1-carboxilato de terc-butilo: Una mezcla de 2-(dimetilamino)etanol (1,07 g, 12,1 mmol, 1,21 ml), 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-3-[2-[terc-butil(difenil)silil]oxietil]piperazin-1-carboxilato de terc-butilo (3,50 g, 4,82 mmol), Pd(OAc)2 (162 mg, 723 umol), BINAP (600 mg, 964 umol) y Cs2CO3 (3,92 g, 12,1 mmol) en tolueno (40 ml) se desgasificó y se purgó 3 veces con N2. La mezcla se calentó a 110 °C con agitación durante 3 horas en una atmósfera de N2. Después de enfriar a temperatura ambiente, la mezcla de reacción se filtró y se concentró a presión reducida a sequedad. El residuo se purificó por cromatografía en columna (SiO2, DCM/MeOH = 1/0 a 10/1) para dar 4-[7-bencil-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[2-[terc-butil(difenil)silil]oxietil]piperazin-1-carboxilato de terc-butilo (2,30 g, 2,57 mmol, 53,3 % de rendimiento, 87,0 % de pureza) en forma de un sólido de color amarillo. ES+APCI MS m/z 779,5 [M+H]+.
Etapa C: 3-[2-[terc-butil(difenil)silil]oxietil]-4-[2-[2-(dimetilamino)etoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo: A una solución de 4-[7-bencil-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[2-[terc-butil(difenil)silil]oxietil]piperazin-1 -carboxilato de terc-butilo (2,30 g, 2,95 mmol) en MeOH (40 ml) se le añadió Pd-C (10 %, 1,5 g) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. Después de agitar en una atmósfera de H2 (344,74 kPa (50 psi)) a 50 °C durante 36 horas, la mezcla de reacción se filtró y se concentró a presión reducida para dar 3-[2-[terc-butil(difenil)silil]oxietil]-4-[2-[2-(dimetilamino)etoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (1,58 g, 1,83 mmol, 61,9 % de rendimiento, 79,6 % de pureza) en forma de un aceite incoloro, que se usó directamente para la siguiente etapa sin purificación adicional. ES+APCI MS m/z 689,4 [M+H]+.
Etapa D: 4-[7-(3- benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]- 6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[2-[ tercbutil(difenil)silil]oxietil]piperazin-1 -carboxilato de terc-butilo: Una mezcla de 3-[2-[terc-butil(difenil)silil]oxietil]-4-[2-[2-(dimetilamino)etoxi]- 5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (1,48 g, 2,15 mmol), 3-benciloxi-1-bromo-naftaleno (875 mg, 2,80 mmol), Cs2CO3 (1,75 g, 5,38 mmol), [2-(2-aminofenil)fenil]paladio (1+); diciclohexil-[2-(2,4,6-triisopropilfenil)fenil]fosfano;metanosulfonato (273 mg, 323 umol) en tolueno (30 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 65 °C durante 24 horas en una atmósfera de N2. La mezcla de reacción se filtró y se concentró a presión reducida a sequedad. El residuo se purificó por cromatografía en columna (AbO3, DCM/MeOH = 1/0 a 20/1) para dar 4-[7-(3- benciloxi-1 -naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[2-[terc-butil(difenil)silil]oxietil]piperazin-1-carboxilato de terc-butilo (1,20 g, 1,07 mmol, 49,7 % de rendimiento, 82,0 % de pureza) en forma de un sólido de color amarillo.
ES+APCI MS m/z 921,4 [M+H]+.
Etapa E: 2-[[7-(3-benciloxi-1-naftil)-4-[2-[2-[terc-butil(difenil)silil]oxietil]piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]-W,W-dimetil-etanamina: Una mezcla de 4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]- 6,8 dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[2-[terc-butil(difenil)silil]oxietil]piperazin-1-carboxilato de terc-butilo (300 mg, 326 umol) y 2,6-lutidina (419 mg, 3,91 mmol, 455 ul) en DCM (10 ml) se le añadió poco a poco TMSOTf (434 mg, 1,95 mmol, 353 ul) a 0 °C. La mezcla se calentó a 10 °C y se agitó durante 12 horas en una atmósfera de N2. La mezcla de reacción se purificó directamente por cromatografía en columna (AbO3, DCM/MeOH 1/0 a 50/1) para dar 2-[[7-(3-benciloxi-1-naftil)-4-[2-[2-[terc-butil(difenil)silil]oxietil]piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]-Ñ,W-dimetil-etanamina (210 mg, 229 umol, 70,3 % de rendimiento, 89,5 % de pureza) en forma de un semi-sólido de color amarillo. ES+APCI MS m/z 821,5 [M+H]+.
Etapa F: 1-[4-[7-(3-benciloxi-1-naftil)-2-(2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[2-[ terc-butil(difenil)silil]oxietil]piperazin-1-il]prop-2-en-1-ona: A una mezcla de 2-[[7-(3-benciloxi-1-naftil)- 4-[2-[2-[ tercbutil(difenil)silil]oxietil]piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]-W,W-dimetil-etanamina (210 mg, 256 umol) y DIEA (49,6 mg, 384 umol, 67,0 ul) en DCM (10 ml) se le añadió poco a poco prop-2-enoato de prop-2-enoílo (33,9 mg, 269 umol) a -40 °C en una atmósfera de nitrógeno. Después de agitar durante 30 minutos a la misma temperatura, la mezcla de reacción se purificó directamente por cromatografía en columna (AbO3, DCM/MeOH 1/0 a 50/1) para dar 1-[4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino)etoxi]-6,8-dihidro- 5H-pirido[3,4-d]pirimidin-4-il]-3-[2-[terc-butil(difenil)silil]oxietil]piperazin-1-il]prop-2-en-1-ona (221 mg, 214 umol, 83,6 % de rendimiento, 84,7 % de pureza) en forma de un semisólido de color amarillo claro. ES+APCI MS m/z 875,4 [M+H]+.
Etapa G: 1-[4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-(2-hidroxietil)piperazin-1-il]prop-2-en-1-ona: A una mezcla de 1-[4-[7-(3-benciloxi-1-naftil)-2-[2-(dimetilamino) etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-[2-[terc-butil(difenil)silil]oxietil]piperazin-1-il]prop-2-en-1-ona (221 mg, 253 umol) en DCM (10 ml) se le añadió BBr3 (949 mg, 3,79 mmol, 365 ul) a -40 °C. La mezcla se agitó a 0 °C durante 3 horas en una atmósfera de N2. La mezcla de reacción se concentró a presión reducida a sequedad. El producto bruto se lavó con MTBE (25 ml) y solución saturada de NaHCO3 (0,5 ml) a pH ~8 a 0 °C y se disolvió en MeOH (3 ml). La solución resultante se purificó por HPLC prep. (columna: Boston Green ODS 150 * 305 u; fase móvil: [agua (0,225 % de ácido fórmico) - ACN]; % de B: 11 % - 41 %, 10 min) para dar 1-[4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-(2-hidroxietil)piperazin-1-il]prop-2-en-1-ona (26,7 mg, 44,0 umol, 17,4 % de rendimiento, 97,8 % de pureza, sal del ácido fórmico) en forma de un sólido de color amarillo. ES+APCI MS m/z 547,3 [M+H]+.
Ejemplo 173
1-(4-(7-(3-hidroxi-2-metilnaftalen-1-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona


Se preparó 1 -(4-(7-(3-hidroxi-2-metilnaftalen-1 -il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona según el procedimiento del Ejemplo 165 1-[4-[7-(2-fluoro-3-hidroxi-1-naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona sustituyendo trifluorometanosulfonato de (3-metoxi-2-metil-1-naftilo) por trifluorometanosulfonato de (2-fluoro-3-metoxi-1-naftilo) en la etapa F para dar el producto deseado 1-(4-(7-(3-hidroxi-2-metilnaftalen-1-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona en forma de un sólido de color amarillo (10,4 mg, 13,1 % de rendimiento, 98,1 % de pureza). ES+AμCi MS m/z 573,5 [M+H]+.
Ejemplo 174
(R)-1-(4-(2-((5,5-dimetilpirrolidin-2-il)metoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -il)prop-2-en-1 -ona


Etapa A: (5R)-5-(Hidroximetil)-2,2-dimetil-pirrolidin-1 -carboxilato de tere-butilo: A una solución de ácido (2R)-1-tercbutoxicarbonil-5,5-dimetil-pirrolidin- 2-carboxílico (500 mg, 2,06 mmol) en THF anhidro (4 ml) se le añadió gota a gota BH3-Me2S (2 M, 1,23 ml) a 15 °C. La reacción se calentó a 50 °C durante 5 minutos. Después de enfriar en un baño de hielo, a la mezcla se le añadió metanol (20 ml). La mezcla de reacción se concentró a presión reducida a 25 °C. Se obtuvo (5R)-5-(hidroximetil)-2,2-dimetil-pirrolidin-1-carboxilato de tere-butilo (400 mg, 1,74 mmol, 84,9 % de rendimiento) en forma de un sólido de color blanco.
1H RMN (400 MHz, metanol-d4) ó = 3,97 - 3,81 (m, 1H), 3,62 (dd, J = 3,6, 10,8 Hz, 1H), 3,45 - 3,33 (m, 1H), 2,02 - 1,79 (m, 3H), 1,79 - 1,67 (m, 1H), 1,49 - 1,41 (m, 12H), 1,32 (s, 3H).
Etapa B: 4-[7-(3-Benciloxi-1 -naftil)-2-[[(2R)-1 -terc-butoxicarbonil-5,5-dimetil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo: A una solución de (5R)-5-(hidroximetil)-2,2-dimetilpirrolidin-1-carboxilato de tere-butilo (212 mg, 926 umol) y 4-[7-(3-benciloxi-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (300 mg, 463 umol) en THF (10 ml) se le añadió t-BuONa (133 mg, 1,39 mmol). Después de agitar a 15 °C durante 1 hora, la mezcla de reacción se vertió en H2O (20 ml) y se extrajo con acetato de etilo (20 ml x 3). La fase orgánica se lavó con salmuera (20 ml), se secó sobre Na2SO4 anhidro, se filtró y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico)/acetonitrilo). Las fracciones deseadas se recogieron y se concentraron al vacío. El compuesto 4-[7-(3-benciloxi-1-naftil)-2-[[(2R)-1-terc-butoxicarbonil-5,5-dimetil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (110 mg, 129 umol, 28,1 % de rendimiento, 96 % de pureza) se obtuvo en forma de un sólido de color amarillo. ES+APCI MS m/z 813,5 [M+H]+.
Etapa C: (5R)-5-[[7-(3-hidroxi-1 -naftil)-4-piperazin-1 -il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]-2,2-dimetilpirrolidin-1-carboxilato de tere-butilo: Se burbujeó NH3 en MeOH (30 ml) a -40 °C durante 30 minutos. A una solución de 4-[7-(3-benciloxi-1-naftil)-2-[[(2R)-1-terc-butoxicarbonil-5,5- dimetil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (200 mg, 246 umol) en la mezcla anterior (NH3/MeOH) se le añadió Pd-C seco (10 %, 100 mg) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 15 °C durante 1 hora. El catalizador se retiró por filtración y el filtrado se concentró para dar el producto (5R)-5-[[7-(3-hidroxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H pirido[3,4-d]pirimidin-2-il]oximetil]-2,2-dimetil-pirrolidin-1-carboxilato de terc-butilo (150 mg, 244 umol, 99,4 % de rendimiento, 96 % de pureza) en forma de un sólido de color amarillo y se usó directamente en la siguiente etapa sin purificación. ES+APCI MS m/z 589,3 [M+H]+.
Etapa D: (5R)-5-[[7-(3-hidroxi-1 -naftil)-4-(4-prop-2-enoilpiperazin-1 -il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]-2,2-dimetil-pirrolidin-1 -carboxilato de terc-butilo: A una mezcla de (5R)-5-[[7-(3-hidroxi-1-naftil)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]-2,2-dimetil-pirrolidin-1-carboxilato de terc-butilo (130 mg, 221 umol) y DIEA (285 mg, 2,21 mmol, 385 ul) en diclorometano (3 ml) se le añadió una solución de prop-2-enoato de prop-2-enoílo (22,3 mg, 176 umol) en diclorometano (1 ml) a -40 °C en una atmósfera de nitrógeno. La mezcla se agitó a -40 °C durante 1 hora. La reacción se interrumpió mediante la adición de una solución acuosa saturada de NaHCO3 (2 ml). Después, la mezcla se vertió en agua (20 ml) y se extrajo con diclorometano (20 ml x 2). Los extractos orgánicos combinados se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna (SiO2, diclorometano/metanol = 1/0 a 10/1). Se obtuvo (5R)-5-[[7-(3-hidroxi-1-naftil)-4-(4
prop-2-enoilpiperazin-1-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]-2,2-dimetil-pirrolidin-1-carboxilato de terc-butilo (140 mg, 217 umol, 98,6 % de rendimiento) en forma de un aceite de color pardo. ES+APCI MS m/z 643,6 [M+H]+.
Etapa E: 1 -[4-[2-[[(2R)-5,5-dimetilpirrolidin-2-il]metoxi]-7-(3-hidroxi-1 -naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de (5R)-5-[[7-(3-hidroxi-1-naftil)-4- (4-prop-2-enoilpiperazin-1-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]-2,2-dimetil-pirrolidin-1-carboxilato de terc-butilo (120 mg, 187 umol) en diclorometano (200 ul) se le añadió TFA (212 mg, 1,87 mmol, 138 ul). La mezcla se agitó a 15 °C durante 1 hora. La mezcla de reacción se concentró al vacío, después se diluyó con diclorometano (5 ml) y se ajustó a pH = 7 mediante la adición de una solución acuosa saturada de NaHCÜ3. La mezcla se extrajo con diclorometano (10 ml x 3). La fase orgánica combinada se secó sobre Na2SÜ4 anhidro, se filtró y se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN]; % de B: 45 %-75 %, 12 min). Se obtuvo 1-[4-[2-[[(2R)-5,5-dimetilpirrolidin-2-il]metoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (30 mg, 53,6 umol, 28,7 % de rendimiento, 97 % de pureza) en forma de un sólido de color amarillo por liofilización. ES+APCI MS m/z 543,5 [M+H]+.
Ejemplo 175
(S)-1-(4-(7-(3-hidroxi-2-metilnaftalen-1-il)-2-((1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -il)prop-2-en-1 -ona

Se sintetizó (S)-1 -(4-(7-(3-hidroxi-2-metilnaftalen-1 -il)-2-((1 -metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona según el procedimiento del Ejemplo 165 sustituyendo 4-[2-(3-morfolinopropoxi)-5,6,7,8- tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo por 4-[7-(2-fluoro-3-metoxi-1 -naftil)-2-(3-morfolinopropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo y trifluorometanosulfonato de (2-fluoro-3-metoxi-1-naftilo) por trifluorometanosulfonato de (3-metoxi-2-metil-1-naftilo) en
la Etapa F para dar 1-[4-[7-(3-hidroxi-2-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (10,2 mg, 17,3 umol, 21,4 % de rendimiento, 100 % de pureza, sal del ácido fórmico) en forma de un sólido de color blanco. ES+APCI MS m/z 543,4 [M+H]+.
Ejemplo 176
2-[(2S)-4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo

Etapa A: (3aR)-1 -oxo-3a,4,6,7-tetrahidro-3H-oxatiazolo[3,4-a]pirazin-5-carboxilato de tere-butilo: A una solución de imidazol (15,7 g, 231 mmol) en DCM (100 ml) se le añadió SOCl2 (8,25 g, 69,4 mmol, 5,03 ml) a 0 °C. La mezcla de reacción se agitó a 15 °C durante 1 hora. A la mezcla se le añadió (3R)-3-(hidroximetil)piperazin-1-carboxilato de terebutilo (5 g, 23,1 mmol) en DCM (100 ml) a -70 °C. La mezcla de reacción se agitó a 15 °C durante 12 horas. Después de que se completara, la mezcla de reacción se inactivó con NH4Cl saturado (100 ml), se separó y la capa acuosa se
extrajo con DCM (40 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío para dar (3aR)-1-oxo-3a,4,6,7-tetrahidro-3H-oxatiazolo[3,4-a]pirazin-5-carboxilato de ferc-butilo (5,8 g, 22,1 mmol, 95,6 % de rendimiento) en forma de un sólido de color pardo.
Etapa B: (3aR)-1,1-dioxo-3a,4,6,7-tetrahidro-3H-oxatiazolo[3,4-a]pirazin-5-carboxilato de ferc-butilo: A una solución de (3aR)-1-oxo-3a,4,6,7-tetrahidro-3H-oxatiazolo[3,4-a)pirazin-5-carboxilato de ferc-butilo (7,5 g, 28,6 mmol) en MeCN (225 ml) se le añadió NaIÜ4 (7,95 g, 37,2 mmol, 2,06 ml) en agua (75 ml) seguido de RuCb.hhO (129 mg, 572 umol) a 0 °C. La mezcla de reacción se agitó a 15 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se inactivó con NHUCl saturado (10 ml) y se extrajo con EtOAc (20 ml). La capa orgánica se lavó con salmuera (10 ml), se secó sobre Na2SO4, se filtró y se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc = 3:1 a 1:1) para dar (3aR)-1,1-dioxo-3a,4,6,7-tetrahidro-3H-oxatiazolo[3,4-a]pirazin-5-carboxilato de ferc-butilo (7 g, 25,2 mmol, 88,0 % de rendimiento) en forma de un sólido de color blanco. 1H RMN (400 MHz, CLOROFORMO-d) ó = 4,64 (dd, J = 6,4, 8,0 Hz, 1H), 4,36 - 3,94 (m, 3H), 3,64 (ddt, J = 3,6, 6,0, 9,2 Hz, 1H), 3,46 (d a, J = 11,6 Hz, 1H), 3,13 (s a, 1H), 2,96 (dt, J = 3,2, 11,2 Hz, 2H), 1,48 (s, 9H)
Etapa C: (3S)-3-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de ferc-butilo: A una solución de (3aR)-1,1-dioxo-3a,4,6,7-tetrahidro-3H-oxatiazolo[3,4-a]pirazin-5-carboxilato de ferc-butilo (5 g, 18,0 mmol) en DMF (100 ml) se le añadió KCN (1,04 g, 16,0 mmol, 684,94 ul). La mezcla de reacción se calentó a 50 °C durante 16 horas. Después de que se completara, la mezcla de reacción se inactivó con HCl (2 M, 50 ml) y se agitó a 15 °C durante 1 h. La mezcla se basificó con NaOH (40 %, 10 ml) y se extrajo con EtOAc (3 x 100 ml). La capa orgánica se secó sobre Na2SO4 y se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc = 3:1 a 0:1 después EtOAc: Me 100:1 a 10:1) para dar (3S)-3-(cianometil)piperazin-1-carboxilato de ferc-butilo (1,1 g, 4,88 mmol, 27,2 % de rendimiento) en forma de un aceite de color pardo. 1H RMN (400 MHz, CLOROFORMO-d) ó = 4,05 - 3,71 (m, 2H), 3,08 - 2,88 (m, 3H), 2,84 - 2,62 (m, 2H), 2,56 - 2,37 (m, 2H), 1,47 (s, 9H).
Etapa D: 2-[(2S)-p¡peraz¡n-2-¡l)aceton¡tr¡lo: Una mezcla de reacción de (3S)-3-(cianometil)piperazin-1-carboxilato de ferc-butilo (850 mg, 3,77 mmol) y HCl/dioxano (4 M, 20 ml) se agitó a 15 °C durante 1 hora. Después de que se completara, el disolvente se eliminó al vacío para dar 2-[(2S)-piperazin-2-il]acetonitrilo (740 mg, 3,74 mmol, 99,0 % de rendimiento, 2HCl) en forma de un sólido de color blanco. 1H RMN (400 MHz, METANOL-d4) ó = 4,04 - 3,90 (m, 1H), 3,81 - 3,70 (m, 2H), 3,69 - 3,61 (m, 2H), 3,53 - 3,36 (m, 2H), 3,13 (d, J = 6,4 Hz, 2H).
Etapa E: 2-[(2S)-4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-2-il]acetonitrilo: A una mezcla de 7-bencil-2,4-dicloro-6,8-dihidro-5H-pirido[3,4-d]pirimidina (1,3 g, 4,42 mmol) y 2-[(2S)-piperazin-2-il]acetonitrilo (1,17 g, 5,91 mmol, 2HCl) en dioxano (25 ml) se le añadió DIEA (2,86 g, 22,1 mmol, 3,85 ml). La mezcla de reacción se agitó a 50 °C durante 12 horas. Después de que se completara, la mezcla de reacción se diluyó con agua (50 ml) y se extrajo con EtOAc (3 x 80 ml). Las capas orgánicas combinadas se lavaron con salmuera (30 ml), se secaron sobre Na2SO4 y se concentraron al vacío para dar 2-[(2S)-4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-2-il]acetonitrilo (1,69 g, 4,41 mmol, 100 % de rendimiento) en forma de un sólido de color pardo.
Etapa F: (2S)-4-(7-bencil-2-cloro-6,8-dihidro-5H-pindo[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de ferc-butilo: Una mezcla de reacción de 2-[(2S)-4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-2-il]acetonitrilo (1,69 g, 4,41 mmol) y (Boc)2O (10,3 g, 47,2 mmol, 10,8 ml) se calentó a 50 °C durante 2 horas. Después de que se completara, la mezcla de reacción se purificó por cromatografía sobre gel de sílice (EP: EtOAc = 10:1 a 1:1) para dar (2S)-4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (1,1 g, 2,12 mmol, 48,0 % de rendimiento, 93 % de pureza) en forma de un sólido de color pardo. ES+APCI MS m/z 483,4 [M+H]+.
Etapa G: (2S)-4-[7-bencil-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de ferc-butilo: A una solución de 2-(dimetilamino)etanol (415 mg, 4,66 mmol, 468 ul) en THF (20 ml) se le añadió NaH (149 mg, 3,73 mmol, 60 % de pureza) 0 °C. La mezcla de reacción se agitó a 0 °C durante 0,5 h. A la mezcla se le añadió (2S)-4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (900 mg, 1,86 mmol). La mezcla de reacción se agitó a 70 °C durante 12 horas un tubo sellado en una atmósfera de N2. Después de que se completara, la mezcla de reacción se inactivó con agua (10 ml) y se extrajo con EtOAc (3 x 40 ml). Las capas orgánicas combinadas se trataron con carbono activado y se filtraron. El filtrado se concentró al vacío para dar (2S)-4-[7-bencil-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (600 mg, 1,12 mmol, 60,1 % de rendimiento) en forma de un sólido de color pardo.
Etapa H: (2S)-2-(cianometil)-4-[2-[2-(dimetilamino)etoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo: Se burbujeó NH3 en MeOH (20 ml) durante 5 min. A la solución se le añadieron (2S)-4-[7-bencil-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de fercbutilo (600 mg, 1,12 mmol) y Pd al 10 %/C (200 mg). La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 40 °C durante 16 horas. Después de que se completara, la mezcla se filtró y la torta de filtro se lavó con MeOH (3 x 30 ml). El filtrado se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex luna(2) C18250 * 50 10 ufase móvil: [agua (0,225 % de ácido fórmico)-ACN];% de B: %-%, 30 min;60 % min) para dar (2S)-2-(cianometil)-4-[2-[2-(dimetilamino)etoxi]-5,6,7,8tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo (167 mg, 331 umol, 29,6 % de rendimiento, 88,3 % de pureza) en forma de un aceite incoloro. ES+APCI MS m/z 446,3 [M+H]+.
Etapa I: (2S)-2-(cianometil)-4-[2-[2-(dimetilamino)etoxi]-7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo: A una solución de (2S)-2-(cianometil)-4-[2-[2-(dimetilamino)etoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (180 mg, 404 umol), 2,2-dimetilpropanoato de (4-bromo-2-naftilo) (248 mg, 808 umol) y Cs2CO3 (395 mg, 1,21 mmol) en tolueno (6 ml) se le añadió XPHOS Palladacycle Gen 3 (34,20 mg, 40,4 umol). La mezcla de reacción se agitó a 70 °C durante 4 horas en una atmósfera de N2. Después de que se completara, la mezcla de reacción se purificó por cromatografía sobre gel de sílice (EP: 5:1 a 0:1) para dar (2s)-2-(cianometil)-4-[2-[2-(dimetilamino)etoxi]-7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (200 mg, 272 umol, 67,3 % de rendimiento, 91,3 % de pureza) en forma de un sólido de color pardo. ES+APCI MS m/z 672,0 [M+H]+.
Etapa J: 2,2-dimetilpropanoato de [4-[4-[(3S)-3-(cianometil)piperazin-1-il]-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo]: A una solución de (2S)-2-(cianometil)-4-[2-[2-(dimetilamino)etoxi]-7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (200 mg, 298 umol) en DCM (0,3 ml) se le añadió t Fa (420 mg, 3,68 mmol, 273 ul). La mezcla de reacción se agitó a 15 °C durante 1 hora. Después de que se completara, la mezcla de reacción se concentró al vacío para dar 2,2-dimetilpropanoato de [4-[4-[(3S)-3-(cianometil)piperazin-1-il]-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (238 mg, 298 umol, 99,9 % de rendimiento, 2TFA) en forma de un aceite de color pardo.
Etapa K: 2,2-dimetilpropanoato de [4-[4-[(3S)-3-(cianometil)-4-prop-2-enoil-piperazin-1-il]-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo]: A una solución de 2,2-dimetilpropanoato de [4-[4-[(3S)-3-(cianometil)piperazin-1-il]-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (238 mg, 298 umol, 2TFA) y DIEA (308 mg, 2,38 mmol, 415 ul) en DCM (5 ml) se le añadió prop-2-enoato de prop-2-enoílo (56,3 mg, 446 umol) a 0 °C. La mezcla de reacción se agitó a 15 °C durante 1 hora. Después de que se completara, la mezcla de reacción se inactivó con una gota de agua. La mezcla se purificó por cromatografía sobre gel de sílice (EP: EtOAc = 1:1 a 0:1 después EtOAc: MeOH = 50:1 a 3:1) para dar 2,2-dimetilpropanoato de [4-[4-[(3S)-3-(cianometil)-4-prop-2-enoil-piperazin-1-il]-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (300 mg, bruto) en forma de un aceite de color pardo. ES+ApCI MS m/z 626,4 [M+H]+.
Etapa L: 2-[(2S)-4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo: A una solución de 2,2-dimetilpropanoato de [4-[4-[(3S)-3-(cianometil)-4-prop-2-enoilpiperazin-1-il]-2-[2-(dimetilamino)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (300 mg, 479 umol) en THF (3 ml) se le añadió NaOH (2 M, 3 ml) en agua. La mezcla de reacción se agitó a 15 °C durante 8 horas. Después de que se completara, la mezcla de reacción se acidificó con 0,5 ml de ácido fórmico (20 % en agua) a pH = 7 y se extrajo con DCM (5 * 10 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron al vacío.
El residuo se purificó por HPLC prep. (columna: Boston Green ODS 150 * 305 ufase móvil: [agua (0,225 % de ácido fórmico)-ACN];% de B: 25 %-49 %,10 min) para dar 2-[(2S)-4-[2-[2-(dimetilamino)etoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (18,3 mg, 30,2 umol, dos etapas 10,1 % de rendimiento, 96,7 % de pureza, sal del ácido fórmico) en forma de un sólido de color amarillo. ES+APCI MS m/z 542,5 [M+H]+.
Ejemplo 177
2-[4-[2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo


Etapa A: 2-(cianometil)-4-[2-[[(3R)-1 -metilpirrolidin-3-il]metoxi]-7-(1 -naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo: A una solución de 2-(cianometil)-4-[2-metilsulfinil-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (300 mg, 549 umol) y [(3R)-1-metilpirrolidin-3-il]metanol (126 mg, 1,10 mmol) en tolueno (6 ml) se le añadió t-BuONa (79,1 mg, 823 umol) a 18 °C y la mezcla de reacción se agitó a 18 °C durante 0,5 horas. Después, a la mezcla se le añadieron EtOAc (20 ml) y agua (15 ml) y después se extrajo con EtOAc (3 * 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0,1 % de ácido fórmico) = 42 %) para dar 2-(cianometil)-4-[2-[[(3R)-1 -metilpirrolidin-3-il]metoxi]-7-(1 -naftil)-6,8-dihidro-5H pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de fercbutilo (200 mg, 331 umol, 60,4 % de rendimiento, 99,0 % de pureza) en forma de un sólido de color amarillo. ES+APCI MS m/z 598,3 [M+H]+.
Etapa B: 2-[4-[2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-7-(1-naftil)-6,8-dilzydro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo: Una mezcla de 2-(cianometil)-4-[2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (200 mg, 334 umol) y TFA (763 mg, 6,69 mmol, 495 ul) se agitó a 18 °C durante 1 hora. La mezcla de reacción se concentró al vacío para dar 2-[4-[2-[[(3R)-1-metilpirrolidin--il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (250 mg, bruto, 2TFA) en forma de un aceite de color pardo.
Etapa C: 2-[4-[2-[[(3R)-1 -metilpirrolidin-3-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1 -prop-2-enoilpiperazin-2-il]acetonitrilo: A una solución de 2-[4-[2-[[(3R)-1-metilpirrolidin-3-iljmetoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (250 mg, 344 umol, 2TFA) y DIEA (356 mg, 2,76 mmol, 480 ul) en DCM (5 ml) se le añadió prop-2-enoato de prop-2-enoílo (65,2 mg, 517 umol) a 0 °C y la mezcla se agitó a 18 °C durante 1,5 horas. La mezcla de reacción se inactivó con agua (3 ml) y después se extrajo con DCM (3 * 6 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um;fase móvil: [agua (NH4HCO3 10 mM)-ACN];% de B: 30 %-60 %,3 min) para dar 2-[4-[2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (9,98 mg, 17,0 umol, 4,94 % de rendimiento, 94,1 % de pureza) en forma de un sólido de color blanco. ES+APCI MS m/z 552,5 [M+H]+.
Ejemplo 178
2-[4-[7-(3-hidroxi-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo

Etapa A: 7-bencil-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-ol: Una suspensión de MeOH (1000 ml) y Na (22,0 g, 957 mmol, 22,7 ml) se agitó durante 30 min. A esta mezcla se le añadieron 1-bencil-3-oxo-piperidin-4-carboxilato de etilo (50 g, 191 mmol) y 2-metilisotiourea (47,9 g, 344 mmol, 0,5 H2SO4) a 15 °C. La mezcla de reacción se agitó a 15 °C durante 30 horas. La mezcla de reacción se acidificó con HCl (2 M) (300 ml) hasta pH = 6 y se concentró a presión reducida. El residuo se suspendió en 200 ml de agua y se agitó rápidamente. La suspensión se filtró y el sólido de color blanco se recogió y se lavó con acetato de etilo. Se obtuvo 7-bencil-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-ol (68 g, 151 mmol, 79,1 % de rendimiento, 64,0 % de pureza) en forma de un sólido de color blanco.
Etapa B: 7-bencil-4-cloro-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidina: A una solución de 7-bencil-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-ol (50 g, 174 mmol) en CHCb (1000 ml) se le añadió POCl3 (166 g, 1,08 mol, 100 ml) y la mezcla se agitó a 80 °C durante 13 horas. Después de que se completara, la mezcla de reacción se concentró al vacío. El residuo se diluyó con EtOAc (500 ml) y se basificó usando Na2CO3 saturado (800 ml) a pH = 7. La mezcla se extrajo con acetato de etilo (3 * 400 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice
(EP: AE de 100:1 a 80: 1) para dar 7-bencil-4-cloro-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidina (21,7 g, 67.4 mmol, 38,7 % de rendimiento, 95,0 % de pureza) en forma de un aceite de color pardo. ES+ApCI MS m/z 306,1 [M+H]+.
Etapa C: 4-(7-bencil-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-2-(cianometil)p¡perazin-1-carboxilato de tere-butilo: A una solución de 7-bencil-4-cloro-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidina (2,6 g, 8,50 mmol) y 2-piperazin-2-ilacetonitrilo (1,68 g, 8,50 mmol, 2HCl) en DMS0 (52 ml) se le añadió DIeA (5,49 g, 42,5 mmol, 7,40 ml). La mezcla se calentó a 80 °C y se agitó a 80 °C durante 6 horas. A la mezcla se le añadió (Boc)20 (18,5 g, 85,0 mmol, 19.5 ml) y la mezcla se agitó a 80 °C durante 1 hora. Se añadió agua (150 ml) y la mezcla se extrajo con acetato de etilo (3 x 200 ml). La fase orgánica combinada se lavó con salmuera (200 ml), se secó con Na2S04 anhidro, se filtró y se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: AE de 10:1 a 0:1) para dar 4-(7-bencil-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de tere-butilo (2,74 g, 5,26 mmol, 61,9 % de rendimiento, 95,0 % de pureza) en forma de un sólido de color pardo. ES+APCI MS m/z 495,4 [M+H]+.
Etapa D: 2-(c¡anomet¡l)-4-(2-met¡lsulfan¡l-5.6.7.8-tetrah¡drop¡r¡do(3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terebutilo: A una solución de 4-(7-bencil-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de tere-butilo (2,51 g, 5,07 mmol) y DIEA (1,97 g, 15,2 mmol, 2,65 ml) en Dc E (50 ml) se le añadió carbonocloridato de 1 -cloroetilo (1,81 g, 12,7 mmol) a 0 °C y la mezcla se agitó a 15 °C durante 3 horas. La mezcla de reacción se concentró al vacío. El residuo se disolvió en Me0H (50 ml) y la mezcla de reacción se agitó a 70 °C durante 1.5 horas. La mezcla se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi Max-RP 250 * 50 mm * 10 um;fase móvil: [agua (0,225 % de AF)-a Cn ];% de B: 23 %-48 %, 30;50 %min) para dar 2-(cianometil)-4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de tere-butilo (1,13 g, 2,51 mmol, 49,5 % de rendimiento, 90,0 % de pureza) en forma de un sólido de color rosa. ES+APCI MS m/z 405,3 [M+H]+.
Etapa E: 2-(cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de tere-butilo: A una solución de 2-(cianometil)-4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de tere-butilo (730 mg, 1,80 mmol), 2,2-dimetilpropanoato de (4-bromo-2-naftilo) (832 mg, 2,71 mmol) y Cs2C03 (1,76 g, 5,41 mmol) en tolueno (18 ml) se le añadió [2-(2-aminofenil)fenil]paladio (1+);diciclohexil-[2-(2,4,6-triisopropilfenil)fenil]fosfano-metanosulfonato (153 mg, 180 umol) y la suspensión se desgasificó al vacío y se purgó varias veces con N2. La mezcla de reacción se agitó a 70 °C durante 4 horas. Después de que se completara, a la mezcla se le añadió agua (20 ml). La mezcla resultante se extrajo con EtOAc (4 x 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (40 ml), se secaron sobre Na2S04 anhidro, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc de 30:1 a 0:1) para dar 2-(cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de tere-butilo (740 mg, 1,15 mmol, 63,7 % de rendimiento, 98,0 % de pureza) en forma de un sólido de color pardo. ES+APCI MS m/z 631,5 [M+H]+.
Etapa F: 2-(c¡anomet¡l)-4-[7-[3-(2.2-d¡met¡lpropano¡lox¡)-1-naft¡l1-2-met¡lsulf¡n¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-i]]p¡peraz¡n-1-carbox¡lato de tere-butilo: A una solución de 2-(cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (100 mg, 158 umol) en DCM (2 ml) se le añadió m-CPBA (32,2 mg, 158 umol, 85,0 % de pureza) a 0 °C y la mezcla se agitó a 0 °C durante 1 hora. La mezcla de reacción se inactivó con Na2S203 saturado (4 ml) a 0 °C y se separó, después se diluyó con agua (10 ml) y se extrajo con acetato de etilo (10 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04 anhidro, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía sobre gel de sílice (EP: AE de 10:1 a 0:1) para dar 2-(cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (90 mg, 125 umol, 79,0 % de rendimiento, 90,0 % de pureza) en forma de un sólido de color pardo. ES+APCI MS m/z 647,5 [M+H]+.
Etapa G: 2-(cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo: A una solución de 2-(cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terebutilo (560 mg, 866 umol), [(2R)-1-metilpirrolidin-2-il]metanol (199 mg, 1,73 mmol) en tolueno (10 ml) se le añadió t-Bu0Na (125 mg, 1,30 mmol) a 0 °C y la mezcla se agitó a 0 °C durante 0,5 horas. La mezcla se repartió entre EtOAc (20 ml) y agua (15 ml) y se separó. Después, la capa acuosa se extrajo con EtOAc (3 x 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04 anhidro, se filtraron y se concentraron a presión reducida. El residuo se purificó por columna ultrarrápida de fase inversa para dar 2-(cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (340 mg, 481 umol, 55,5 % de rendimiento, 98,7 % de pureza) en forma de un aceite de color pardo. ES+APCI MS m/z 698,4 [M+H]+.
Etapa H: 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)piperazin-1-il]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo]: A una solución de 2-(cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (140 mg, 201 umol) en DCM (0,2 ml) se le añadió TfA (229 mg, 2 ,01 mmol, 148 ul) a 15 °C y la mezcla se agitó a
15 °C durante 5 horas. La mezcla de reacción se concentró al vacío para dar 2,2-dimetilpropanoato de [4-[4-[3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-¡l]-2-naft¡lo] (170 mg, bruto, 2TFA) en forma de un ace¡te de color pardo.
Etapa I: 2,2-d¡met¡lpropanoato de [4-[4-[3-(c¡anomet¡l)-4-prop-2-eno¡l-p¡peraz¡n-1-¡l]-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-¡l]-2-naft¡lo]: A una soluc¡ón de 2,2-d¡met¡lpropanoato de [4-[4-[3-(c¡anomet¡l)p¡peraz¡n-1-¡lj-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-¡l]-2-naft¡lo] (170 mg, 2O6 umol, 2 TfA) y DIeA (213 mg, 1,65 mmol, 287 ul) en DCM (0,3 ml) se le añad¡ó prop-2-enoato de prop-2-enoílo (38,9 mg, 309 umol) a 0 °C y esta mezcla se ag¡tó a 15 °C durante 1 hora. La mezcla de reacc¡ón se ¡nact¡vó con agua (0,5 ml) y después se concentró al vacío. El res¡duo (DCM: MeOH = 10:1) se pur¡f¡có por cromatografía sobre gel de síl¡ce (de EP: EtOAc = 2:1 a EtOAc: MeOH = 0:1) para dar 2,2-d¡met¡lpropanoato de [4-[4-[3-(c¡anomet¡l)-4-prop-2-enoil-piperazin-1-il]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (170 mg, 182 umol, 88,7 % de rend¡m¡ento, 70,0 % de pureza) en forma de un ace¡te de color pardo. ES+APCI MS m/z 652,6 [M+H]+.
Etapa J: 2-[4-[7-(3-h¡drox¡-1-naft¡l)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo: A una soluc¡ón de 2,2-d¡met¡lpropanoato de [4-[4-[3-(c¡anomet¡l)-4-prop-2-eno¡lpiperazin-1-il]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (170 mg, 261 umol) en THF (1,5 ml) se le añad¡ó NaOH (2 M, 1,5 ml) y la reacc¡ón se agitó a 15 °C durante 6 horas. La mezcla de reacc¡ón se neutral¡zó con HCOOH (20 %, 0,1 ml) a pH = 7. La mezcla resultante se extrajo con DCM (3 x 5 ml) y las capas orgán¡cas comb¡nadas se secaron sobre Na2SO4 anh¡dro, se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar un res¡duo. El res¡duo se pur¡f¡có por HPLC prep. (columna: Luna C18 150 * 255 ufase móv¡l: [agua (0,225 % de ác¡do fórm¡co)-ACN];% de B: 12 %-39 %,10 m¡n) para dar 2-[4-[7-(3-h¡drox¡-1-naft¡l)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-il]metoxi]-6,8-dihidro-5H pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (6,88 mg, 11,6 umol, 4,44 % de rend¡m¡ento, 95,6 % de pureza) en forma de un sólido de color amar¡llo. ES+APCI MS m/z 568,5 [M+H]+.
Ejemplo 179
2-[4-[7-(3-h¡drox¡-1-naft¡l)-2-[[(3R)-1-met¡lp¡rrol¡d¡n-3-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡lp¡peraz¡n-2-¡l]aceton¡tr¡lo


Etapa A: [(3R)-1-metilpirrolidin-3-il]metanol: A la solución de (3R)-3-(hidroximetil)pirrolidin-1 -carboxilato de ferc-butilo (1 g, 4,97 mmol) en THF (40 ml) se le añadió LiAlH4 (377 mg, 9,94 mmol) a 0 °C y después la mezcla se calentó a 70 °C y se agitó a 70 °C durante 4 horas. La mezcla de reacción se inactivó con Na2SÜ4 saturado (3 ml), se filtró y la torta de filtro se lavó con THF (5 x 20 ml). La fase orgánica combinada se concentró al vacío para dar [(3r )-1 -metilpirrolidin-3-il]metanol (820 mg, bruto) en forma de un aceite de color amarillo. 1H RMN (400 MHz, cloroformo-d) ó = 3,66 - 3,62 (dd, J = 4,8, 10,0 Hz, 1H), 3,53 - 3,49 (dd, J = 5,6, 10,0 Hz, 1H), 3,35 - 3,18 (m, 1H), 2,77 - 2,68 (m, 1H), 2,59 - 2,53 (m, 1H), 2,52 -2,45 (m, 1H), 2,40 - 2,33 (m, 1H), 2,32 (s, 3H), 2,31 - 2,26 (m, 1H), 2,04 - 1,93 (m, 1H), 1,69 - 1,59 (m, 1H)
Etapa B: 2-(cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1 -naftil]-2-[[(3R)-1 -metilp¡rrol¡d¡n-3-¡l]metox¡]-6,8-d¡h¡dro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo: A la solución de 2-(cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1 -naft¡l]-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡mid¡n-4-¡l]p¡peraz¡n-1 -carboxilato de fercbutilo (500 mg, 773 umol) y [(3R)-1-metilpirrolidin-3-il]metanol (178 mg, 1,55 mmol) en tolueno (10 ml) se le añadió t-BuONa (111 mg, 1,16 mmol) y esta mezcla se agitó a 15 °C durante 0,5 horas. A la mezcla se le añadieron acetato de etilo (20 ml) y agua (15 ml) y después se extrajo con acetato de etilo (3 x 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0,1 % de ácido fórmico) = 42 %) para dar 2-(cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-2-[[(3R)-1-metilpirrolidin-3-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de ferc-butilo (180 mg, 245 umol, 31,7 % de rendimiento, 95,0 % de pureza) en forma de un sólido de color pardo. ES+APCI MS m/z 698,6 [M+H]+.
Etapa C: 2,2-dimetilpropanoato de [4-[4-[3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-[[(3R)-1-met¡lpirrol¡d¡n-3-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-7-¡l]-2-naft¡lo]: Una mezcla de 2-(cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1 -naftil]-2-[[(3R)-1 -met¡lp¡rrol¡d¡n-3-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1 -carboxilato de ferc-butilo (180 mg, 258 umol) y TFA (588 mg, 5,16 mmol, 382 ul) se agitó a 20 °C durante 1,5 horas. La mezcla de reacción se concentró al vacío para dar 2,2-dimetilpropanoato de [4-[4-[3-(c¡anomet¡l)p¡perazin-1-¡l]-2-[[(3R)-1-met¡lp¡rrol¡d¡n-3-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-7-¡l]-2-naft¡lo] (200 mg, bruto, 2TFA) en forma de un aceite de color pardo.
Etapa D: 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)-4-prop-2-enoil-piperazin-1-il]-2-[[(3R)-1-metilpirrolidin-3-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-¡l]-2-naft¡lo]: A la solución de 2,2-dimetilpropanoato de [4-[4-[3-(c¡anomet¡l)p¡peraz¡n-1-¡lj-2-[[(3R)-1-met¡lp¡rrol¡d¡n-3-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-¡l]-2-naft¡lo] (200 mg, 242 umol, 2 TfA) y DieA (470 mg, 3,63 mmol, 633 ul) en DCM (0,6 ml) se le añadió prop-2-enoato de prop-2-enoílo (30,5 mg, 242 umol) a 0 °C y la reacción se agitó a 15 °C durante 1 hora. La mezcla de reacción se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: AE = 2:1~0:1 a DCM: MeOH = 50:1~1:1) para dar 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)-4-prop-2-enoil-piperazin-1-il]-2-[[(3R)-1-metilpirrolidin-3-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-¡l]-2-naft¡lo] (630 mg, bruto) en forma de un aceite de color pardo. ES+APCI MS m/z 652,6 [M+H]+.
Etapa E: 2-[4-[7-(3-hidroxi-1 -naftil)-2-[[(3R)-1 -met¡lp¡rrol¡d¡n-3-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡mid¡n-4-¡l]-1 -prop-2-enoil-piperazin-2-il]acetonitrilo: A una solución de 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)-4-prop-2-enoilpiperazin-1 -il]-2-[[(3R)-1 -met¡lp¡rrol¡d¡n-3-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-¡l]-2-naft¡lo] (630 mg, 966 umol) en THF (3 ml) se le añadió NaOH (2 M, 3 ml) a 18 °C y la mezcla se agitó a 18 °C durante 2 horas. La mezcla
de reacción se neutralizó con HCOOH (20 %, 0,5 ml) a pH = 7. La mezcla resultante se extrajo con DCM (3 x 5 ml) y las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um;fase móvil: [agua (NH4HCO3 10 mM)-ACN];% de B: 35 %-65 %,3 min) para dar 2-[4-[7-(3-hidroxi-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (9,4 mg, 15,8 umol, 1,64 % de rendimiento, 95,5 % de pureza) en forma de un sólido de color amarillo. ES+APCI MS m/z 568,5 [M+H]+.
Ejemplo 180
4-(3-(4-(piperazin-1 -il)-7-(5-(trifluorometil)-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-iloxi)propil)morfolina

Etapa A. 2-[[4-bromo-5-(trifluorometil)indazol-1-il]metoxiletil-trimetil-silano: A una solución de 4-bromo-5-(trifluorometil)-1H-indazol (650 mg, 2,45 mmol, 1 eq.) en DMF (30 ml) se le añadió NaH (117,71 mg, 2,94 mmol, 60 % de pureza, 1,2 eq.) a 0 °C. Después de agitar a 0 °C durante 1 hora, se añadió gota a gota una solución de 2-(clorometoxi)etiltrimetilsilano (531,56 mg, 3,19 mmol, 564,29 ul, 1,3 eq.) en DMF (10 ml). La mezcla se calentó a 15 °C y se agitó durante 2 horas. La mezcla se inactivó con una solución acuosa saturada de cloruro de amonio (50,0 ml), se diluyó con agua (100,0 ml) y después se extrajo con acetato de etilo (3 x 100,0 ml). La capa orgánica se secó sobre Na2SO4, se filtró y se concentró al vacío para dar un residuo. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato
de etilo = 10:1). Se obtuvo 2-[[4-bromo-5-(trifluorometil)indazol-1-il]metoxi]etil-trimetil-silano (680 mg, 1,70 mmol, 69,44 % de rendimiento, 99,0 % de pureza) en forma de un aceite incoloro. ES+APCI MS m/z 395,0 [M+H]+.
Etapa B. 4-[2-(3-morfolinopropoxi)-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo: Una mezcla de 2-[[4-bromo-5-(trifluorometil)indazol-1-il]metoxi]etil-trimetil-silano (444,35 mg, 1,12 mmol, 1,3 eq.), 4-[2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (400 mg, 864,71 umol, 1 eq.), RuPhos (80,70 mg, 172,94 umol, 0,2 eq.), Pd2(dba)3 (118,77 mg, 129,71 umol, 0,15 eq.) y Cs2C03 (845,21 mg, 2,59 mmol, 3 eq.) en tolueno (40 ml) se desgasificó y se purgó 3 veces con N2 y la mezcla se agitó a 90 °C durante 12 h en una atmósfera de nitrógeno. La mezcla de reacción se concentró a presión reducida para eliminar el tolueno. El residuo se diluyó con agua 100 ml y se extrajo con 300 ml de acetato de etilo (100 ml x 3). Las capas orgánicas combinadas se lavaron con 300 ml de agua (100 ml x 3 ), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiO2, DCM: CH30H = 30:1 a 20:1). Se obtuvo 4-[2-(3-morfolinopropoxi)-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (400 mg, 446,87 umol, 51,68 % de rendimiento, 86 ,8 % de pureza) en forma de un sólido de color amarillo. ES+APCI MS m/z 773,3 [M+H]+.
Etapa C. 4-[3-[[4-piperazin-1-il-7-[5-(trifluorometil)-1H-indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]propil]morfolina: A una solución de 4-[2-(3-morfolinopropoxi)-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5Hpirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo (100 mg, 111,72 umol, 1 eq.) en DCM (2 ml) se le añadió TFA (254,77 mg, 2,23 mmol, 165,43 ul, 20 eq.) a 25 °C. La mezcla de reacción se agitó a 25 °C durante 12 horas. La mezcla de reacción se concentró al vacío. Se obtuvo 4-[3-[[4-piperazin-1-il-7-[5-(trifluorometil)-1H-indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]propil]morfolina (200 mg, bruto, TFA) en forma de un aceite de color pardo. El producto bruto se usó directamente para la siguiente etapa sin purificación adicional. ES+APCI MS m/z 547,5 [M+H]+.
Etapa D. 1-[4-[2-(3-morfolinopropoxi)-7-[5-(trifluorometil)-1H-indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona: A una solución de 4-[3-[[4-piperazin-1-il-7-[5-(trifluorometil)-1H-indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]propil]morfolina (200 mg, t Fa ) y DIEA (600 mg, 4,64 mmol, 808,63 ul) en DCM (2 ml) se le añadió gota a gota prop-2-enoato de prop-2-enoílo (13 mg, 103,08 umol) a -50 °C. La mezcla se agitó a -50 °C durante 30 min. La mezcla se inactivó con agua y se extrajo con acetato de etilo (50 ml) y la capa orgánica se lavó con agua (1 x 20 ml) y salmuera (1 x 20 ml). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por columna de HPLC prep. (condición de AF): Luna C18 150 * 255 ufase móvil: [agua (0,225 % de AF)-ACN];% de B: 15 %-36 %,10 min]. Se obtuvo 1-[4-[2-(3-morfolinopropoxi)-7-[5-(trifluorometil)-1H-indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (18,26 mg, 28,12 mmol, dos etapas 23 %, 99,6 % de pureza, AF) en forma de un sólido de color amarillo. ES+APCI MS m/z 601,4 [M+H]+.
Ejemplo 181
1-(4-(2-(3-((1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propoxi)-7-(5-metil-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona


Etapa A. 4-(2-(3-((1 S,4S)-2-oxa-5-azabiciclo[2.2.1 ]heptan-5-il)propoxi)-7-(5-metil-1 -((2-(trimetilsilil)etoxi)metil)-1 H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: A una solución de 3-((1 S,4S)-2-oxa-5-azabiciclo[2.2.1] heptan-5-il)propan-1-ol (107 mg, 681 umol) en THF (2 ml) se le añadió t-BuONa (98,1 mg, 1,02 mmol) seguido de 4-[2-metilsulfinil-7-[5-metil-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (230 mg, 340 umol). La mezcla se agitó a 0 °C durante 1 hora. La mezcla se vertió en agua (10 ml) y se extrajo con DCM (2 x 10 ml). La capa orgánica se secó sobre Na2SO4, se filtró y se concentró. El producto obtenido se purificó por HPLC prep. (columna: Phenomenex Synergi C18150 * 25 * 10 um; fase móvil: [agua (0,1 % de TFA) - ACN]; % de B: 25 %-55 %, 12 min) para dar 4-(2-(3-((1 S,4S)-2-oxa-5-azabiciclo [2.2.1 ]heptan-5-il)propoxi)-7-(5-metil-1 -((2-(trimetilsilil)etoxi)metil)-1 H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (110 mg, 143 umol, 42,0 % de rendimiento) que se obtuvo en forma de un sólido de color amarillo. 1H RMN (400 MHz, Cloroformo-d) 58,08 - 8,01 (m, 1H), 7,45 - 7,43 (m, 4H), 7,37 - 7,31 (m, 3H), 5,76 (s, 2H), 5,24 (s, 2H), 4,76 - 4,63 (m, 1H), 4,60 - 4,45 (m, 3H), 4,42 (s, 2H), 4,36 -4,24 (m, 1H), 3,96 - 3,83 (m, 2H), 3,79 - 3,65 (m, 8H), 3,64 - 3,53 (m, 6H), 3,12 - 2,92 (m, 1H), 2,90 - 2,77 (m, 2H), 2,48 (s, 3H), 2,44 - 2,41 (m, 1H), 2,36 - 2,12 (m, 3H), 0,98 - 0,93 (m, 2H), 0,01 - -0,03 (m, 9H)
Etapa B. 4-(2-(3-((1 S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propoxi)-7-(5-metil-1 -((2-(trimetilsilil)etoxi)metil)-1 H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de ferc-butilo: A una solución de 4-(2-(3-((1 S,4S)-2-oxa-5-azabiciclo [2.2.1 ]heptan-5-il)propoxi)-7-(5-metil-1 -((2-(trimetilsilil)etoxi)metil)-1 H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (90 mg, 117 umol) y BOC2O (51,1 mg, 234,1 umol, 53,8 ul) en MeOH (2 ml) se le añadió Pd/C (10 %, 50 mg) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 40 °C durante 12 horas. La mezcla se filtró y se concentró. El residuo se purificó por TLC prep. (SO2, DCM/MeOH = 10 : 1) para dar 4-(2-(3-((1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propoxi)-7-(5-metil-1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de ferc-butilo (30 mg, 40,8 umol). ES+APCI MS m/z 735,6 [M+H]+.
Etapa C. (1 S,4S)-5-(3-((7-(5-metil-1 H-indazol-4-il)-4-(piperazin-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirim idin-2-il)oxi)propil)-2-oxa-5-azabiciclo[2.2.1]heptano: A una solución de 4-(2-(3-((1 S,4S)-2-oxa-5-azabiciclo [2.2.1]heptan-5-il)propoxi)-7-(5-metil-1 -((2-(trimetilsilil)etoxi)metil)-1 H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de ferc-butilo (30 mg, 40,8 umol) en DCM (0,5 ml) se le añadió gota a gota TFA (770 mg, 6,75 mmol). La mezcla se agitó a 15 °C durante 2 horas. La mezcla se concentró al vacío para dar (1 S,4S)-5-(3-((7-(5-metil-1H-indazol-4-il)-4-(piperazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)propil)-2-oxa-5-azabiciclo[2.2.1]heptano (30 mg, bruto). ES+APCI MS m/z 505,5 [M+h ]+.
Etapa D. 1-(4-(2-(3-((1 S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propoxi)-7-(5-metil-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona: A una solución de (1 S,4S)-5-(3-((7-(5-metil-1H-indazol-4-il)-4-(piperazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)propil)-2-oxa-5-azabiciclo[2.2.1]heptano (20 mg, 39,6 umol, 1 eq.) y DIEA (30,7 mg, 238 umol, 41,4 ul, 6 eq.) en Dc M (2 ml) se le añadió prop-2-enoato de prop-2-enoílo (4,00 mg, 31,7 umol, 0,8 eq.) a -50 °C. La mezcla se agitó a -40 - -20 °C durante 0,5 horas. La mezcla se concentró al vacío. El producto obtenido se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um;fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-Ac N];% de B: 35 %-65 %,12 min). El producto 1-(4-(2-(3-((1 S,4S)-2-oxa-5-azabiciclo[2.2.1 ]heptan-5-il)propoxi)-7-(5-metil-1 H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona (6,48 mg, 11,2 umol, dos etapas 28,3 % de rendimiento, 96,8 % de pureza) se obtuvo en forma de un sólido de color blanco. ES+APCI MS m/z 559,5 [M+H]+.
Ejemplo 182
2-[4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2 -il]acetonitrilo

Etapa A: 2-(cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1 -naftil]-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo. A una mezcla de 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (190 mg, 403 umol), 2,2-dimetilpropanoato de (4-bromo-2-naftilo) (248 mg, 806 umol) y Cs2C03 (394 mg, 1,21 mmol) en tolueno (5 ml) se le añadió XPH0S Palladacycle Gen 3 (34,1 mg, 40,29 umol) y la mezcla de reacción se agitó a 70 °C durante 4 horas en una atmósfera de N2. Después de que se completara, la mezcla de reacción se purificó por cromatografía sobre gel de sílice (EP: AE = 3:1 a 0:1) para dar 2-(cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (95 mg, 118 umol). ES+a Pci MS m/z 698,4 [M+H]+.
Etapa B: 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)piperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo]. A una solución de 2-(cianometil)-4-[7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de fercbutilo (40 mg, 57,3 umol) en DCM (0,1 ml) se le añadió TFA (154 mg, 1,35 mmol) a 15 °C. La mezcla de reacción se agitó a 15 °C durante 1 hora. Después de que se completara, el disolvente se eliminó al vacío para dar 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)piperazin-1 -il]-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (47 mg, 56,9 umol).
Etapa C: 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)-4-prop-2-enoil-piperazin-1 -il]-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo]. A una solución de 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)piperazin-1-il]-2 -[[(2 S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (47 mg, 56,9 umol) y DiEA (58,9 mg, 455 umol) en DCM (1 ml) se le añadió prop-2-enoato de prop-2-enoílo (10,8 mg, 85,4 umol) a 0 °C. La mezcla de reacción se agitó a 15 °C durante 1 hora. Después de que se completara, la mezcla de reacción se inactivó mediante la adición de una gota de agua. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc = 3:1 a EtOAc: Me0H = 3:1) para dar 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)-4-prop-2-enoil-piperazin-1 -il]-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (130 mg, bruto). ES+APCI MS m/z 652,5 [M+H]+.
Etapa D: 2-[4-[7-(3-hidroxi-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1 -prop-2-enoil-piperazin-2-il]acetonitrilo. A una solución de 2,2-dimetilpropanoato de [4-[4-[3-(cianometil)-4-prop-2-enoilpiperazin-1 -il]-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-2-naftilo] (250 mg, 384 umol) en THF (1,5 ml) se le añadió Na0H (2 M, 1,5 ml) en agua. La mezcla de reacción se agitó a 15 °C durante 9 horas. Después de que se completara, la mezcla de reacción se acidificó mediante la adición de dos gotas de ácido fórmico (20 % en agua) para alcanzar un pH = 7 y la capa acuosa se extrajo con DCM (5 * 10 ml). Las capas orgánicas combinadas se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Boston Green 0DS 150 * 305 ufase móvil: [agua (0,225 % de AF)-ACN];% de B: 2o %-47 %,10 min) para dar 2-[4-[7-(3-hidroxi-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il)metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1 -prop-2-enoil-piperazin-2-il]acetonitrilo (13,1 mg, 21,2 umol). ES+ApCI MS m/z 568,5 [M+H]+.
Ejemplo 183
2-[4-[2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2 -il]acetonitrilo

Etapa A: 2-(cianometil)-4-[2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo. A una solución de 2-(cianometil)-4-[2-metilsulfinil-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (300 mg, 549 umol) y [(2S)-1 -metilpirrolidin-2-il]metanol (126 mg, 1,10 mmol, 130 ul) en tolueno (6,00 ml) se le añadió t-Bu0Na (105 mg, 1,10 mmol). La mezcla se agitó a 20 °C durante 0,25 horas. Después de que se completara, la mezcla se filtró y el filtrado se concentró. El residuo se purificó por cromatografía sobre gel de sílice (AE/Me0H 50/1 a 10/1) para dar 2-(cianometil)-4-[2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (300 mg, 427 umol). ES+ApCI MS m/z 598,6 [M+H]+.
Etapa B: 2-[4-[2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il)acetonitrilo. A una solución de 2-(cianometil)-4-[2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (270 mg, 452 umol) en DCM (330 ul) se le añadió TFA (515 mg, 4,52 mmol). La mezcla se agitó a 20 °C durante 0,5 horas y se concentró al vacío. Después, se añadió TFA (334 ul). La mezcla se agitó a 20 °C durante 1 hora. Después de que se completara, la mezcla se concentró al vacío para dar 2-[4-[2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (400 mg, bruto). Es APCI MS m/z 498,4 [M+H]+.
Etapa C: 2-[4-[2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo. A una solución de 2-[4-[2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (276 mg, 451 umol) y DIEA (1,46 g, 11,3 mmol) en DCM (4,00 ml) se le añadió gota a gota prop-2-enoato de prop-2-enoílo (51,2 mg, 406 umol) a 0 °C. La mezcla se agitó a 20 °C durante 1 hora. Después de que se completara, la mezcla se diluyó con agua (0,5 ml) y se extrajo con EtOAc (2 x 10 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um;fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN];% de B: 55 %-85 %,12 min) para dar 2-[4-[2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (37,1 mg, 66,3 umol). ES+APCI MS m/z 552,4 [M+H]+.
Ejemplo 184
1-[4-[7-(3-hidroxi-1-naftil)-2-(3-hidroxipropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona

Se preparó 1-[4-[7-(3-hidroxi-1-naftil)-2-(3-hidroxipropoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona siguiendo el Ejemplo 136 sustituyendo 3-[ferc-butil(dimetil)silil]oxipropan-1-ol por 2-(3-metoxipirrolidin-1-il)etanol en la Etapa B. ES+APCI MS m/z 490,3 [M+H]+.
Ejemplo 185
1-(4-(2-(3-((1R,4R)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona

Etapa A: 3-íí1R.4R)-2-oxa-5-azab¡c¡clor2.2.11heptan-5-¡l)propan-1-ol: A un vial se le añadieron sal HCl de (1 R,4R)-2-0xa-5-azabiciclo[2.2.1]heptano (0,250 g. 2,522 mmol), CH3CN (5.04 ml) y 3-Bromo-1-propanol (0,274 ml.
3,026 mmol). Después, se añadió K2C03 (1,05 g, 7,57 mmol) y la mezcla se calentó a 50 °C, donde se agitó durante 16 horas. Después, la mezcla se enfrió a temperatura ambiente, se diluyó con CH2Cl2, se filtró y el sólido se lavó con CH2Cl2. Después, el filtrado se concentró y el aceite bruto se purificó por cromatografía en columna (5 % de Me0H/DCM con 0,2 % de NH40H) para proporcionar el producto en forma de un aceite incoloro.
Etapa B: 1-(4-(2-(3-((1R.4R)-2-oxa-5-azab¡c¡clo[2.2.11heptan-5-¡l)propox¡)-7-(3-h¡drox¡naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona: Se sintetizó según el método del Ejemplo 127 usando el siguiente procedimiento en lugar del descrito en la Etapa D. A un vial se le añadieron 3-((1R,4R)-2-oxa-5-azab¡c¡clo[2.2.1]heptan-5-¡l)propan-1-ol (0,171 g, 1,09 mmol) y 4-(2-cloro-7-(3-(metox¡metox¡)naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo (0,125 g, 0,218 mmol) seguido de dioxano (0,435 ml). Después, se añadió Cs2C03 (0,213 g, 0,653 mmol) y la mezcla se calentó a 100 °C durante 15 horas. La reacción se diluyó con CH2Cl2, se filtró y el sólido residual se lavó con CH2Cl2. Después, el filtrado se secó sobre Na2S04, se filtró y se concentró. El residuo bruto se purificó por cromatografía en columna (4 % de Me0H/DCM con 0,2 % de NH40H) para proporcionar el producto en forma de una espuma de color amarillo. ES+APCI MS m/z 571,2 [M+H]+. Ejemplo 186
4-(7-(3-(Metox¡metox¡)naftalen-1-¡l)-2-(3-(morfol¡nomet¡l)fen¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carboxilato de bencilo

4-(7-(3-(Metox¡metox¡)naftalen-1-¡l)-2-(3-(morfol¡nomet¡l)fen¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carboxilato de bencilo: Se sintetizó según el método del Ejemplo 127 usando el siguiente procedimiento en lugar del descrito en la Etapa D. A una solución de 4-(2-cloro-7-(3-(metox¡metox¡)naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l)p¡perazin-1-carboxilato de bencilo (0,150 g, 0,261 mmol) y clorhidrato de pinacol éster del ácido 3-(4-morfolinometi0-fenilborónico (0,266 g, 0,784 mmol) en dioxano (2,61 ml) se le añadió Na2C03 (0,523 ml, 1,0452 mmol, ac. 2,0 M). La mezcla se desgasificó burbujeando argón durante 5 min. Después, se añadió tetraquis^rifenilfosfina^aladio (0) (0,030 g, 0,026 mmol) y la mezcla se calentó a 95 °C, donde se agitó durante 7 horas. La reacción se enfrió a temperatura ambiente y después se diluyó con CH2Cl2 y se filtró. El sólido se lavó con CH2Cl2. Después, el filtrado se secó sobre Na2S04, se filtró y se concentró. El residuo bruto se purificó por cromatografía en columna (30-50 % de EtOAc/DCM) para proporcionar el producto en forma de una espuma de color amarillo. ES+APCI MS m/z 591,3 [M+H]+.
Ejemplo 187
(R)-1-(4-(7-(3-h¡drox¡naftalen-1-¡l)-2-((1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-il)piperazin-1-il)prop-2 -en-1-ona

ÍR)-1-í4-í7-í3-h¡drox¡naftalen-1-¡h-2-íí1-met¡lp¡rrol¡d¡n-2-¡hmetoxn-5.6.7.8-tetrahidrop¡r¡dor3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona. Este compuesto se preparó s¡gu¡endo el Ejemplo 127 sust¡tuyendo N-Met¡l-D-prol¡nol por N-Met¡l-L-prol¡nol en la Etapa D. ES+APCI MS m/z 529.3 [M+H]+.
Ejemplo 188
(S)-1-(4-(7-(3-h¡drox¡naftalen-1-¡l)-2-((1-met¡lp¡rrol¡d¡n-3-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2 -en-1-ona
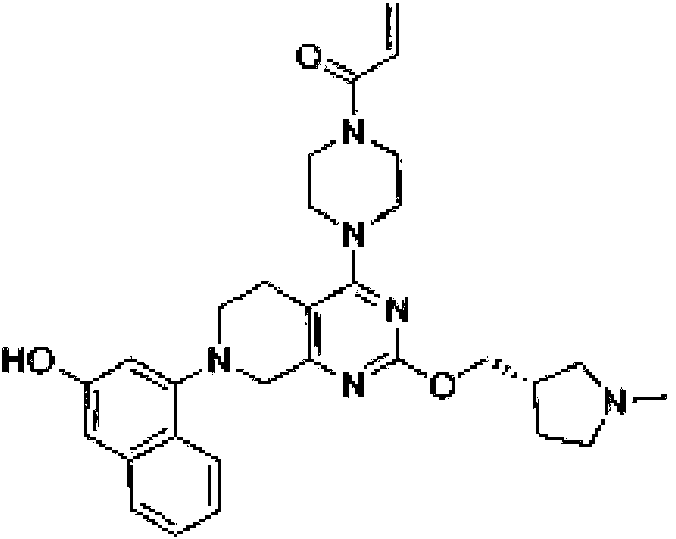
(S)-1-(4-(7-(3-h¡drox¡naftalen-1-¡l)-2-((1-met¡lp¡rrol¡d¡n-3-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona. Este compuesto se preparó s¡gu¡endo el Ejemplo 127 sust¡tuyendo (3S)-(1-Met¡lp¡rrol¡d¡n-3-¡l)-metanol por N-Met¡l-L-prol¡nol en la Etapa D. ES+APCI MS m/z 529.2 [M+H]+.
Ejemplo 189
1-(4-(2-((1-benc¡lp¡rrol¡d¡n-3-¡l)metox¡)-7-(3-h¡drox¡naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2 -en-1-ona

1-(4-(2-((1-benc¡lp¡rrol¡d¡n-3-¡l)metox¡)-7-(3-h¡drox¡naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona. Este compuesto se preparó s¡gu¡endo el Ejemplo 127 sust¡tuyendo (1-Benc¡lp¡rrol¡d¡n-3-¡l)metanol por N-Met¡l-L-prol¡nol en la Etapa D. ES+APCI MS m/z 605.3 [M+H]+.
Ejemplo 190
1-(4-(2-((1-c¡cloprop¡lp¡rrol¡d¡n-3-¡l)metox¡)-7-(3-h¡drox¡naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2 -en-1-ona

1-(4-(2-((1-c¡cloprop¡lp¡rrol¡d¡n-3-¡l)metox¡)-7-(3-h¡drox¡naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona. Este compuesto se preparó s¡gu¡endo el Ejemplo 127 sust¡tuyendo (1-C¡cloprop¡l-3-p¡rrol¡d¡n¡l)metanol por N-Met¡l-L-prol¡nol en la Etapa D. Es+APCI MS m/z 555.3 [M+H]+.
Ejemplo 191
1-((2S.6R)-4-(7-(3-h¡drox¡naftalen-1-¡l)-2-(3-morfol¡nopropox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)-2.6-d¡met¡lp¡peraz¡n-1-¡l)prop-2 -en-1-ona

Etapa A: 4-((3S.5R)-4-(terc-Butox¡carbon¡l)-3.5-d¡met¡lp¡peraz¡n-1-¡l)-2-cloro-5.8-d¡h¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-7(6H)-carbox¡lato de benc¡lo: Una suspens¡ón de 2.4-d¡cloro-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de benc¡lo (500 mg. 1.48 mmol). c¡s-2.6-d¡met¡lp¡peraz¡n-1-carbox¡lato de terc-but¡lo (349 mg. 1.63 mmol) y DIEA (0.26 ml) en W,W-d¡met¡lacetam¡da (1 ml) se ag¡tó a ta durante una noche. La mezcla de reacc¡ón se d¡v¡d¡ó entre EtOAc (15 ml) y NaHCÜ31 M (5 ml) y las capas se separaron. La capa orgán¡ca se lavó con Na2C032 M y salmuera (2 ml de cada). se secó sobre Na2S04 y se evaporó al vacío. El res¡duo se cromatograf¡ó sobre una columna de gel de sílice Red¡sep de 24 g usando de 20 a 50 % de EtOAc en hexano como eluyente para dar un sól¡do ¡ncoloro (0.440 g.58 %). ES+APCI MS m/z 516.2 [M+H]+.

Etapa B: 4-((3S,5R)-4-(terc-Butoxicarbonil)-3,5-dimetilpiperazin-1-il)-2-(3-morfolinopropoxi)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de bencilo: Una mezcla de 4-((3S,5R)-4-(terc-butoxicarbonil)-3,5-dimetilpiperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de bencilo (400 mg, 0,775 mmol), 3-morfolinopropan-1-ol (400 mg, 2,33 mmol, 5 eq.), carbonato de cesio (1,26 g, 3,88 mmol, 5 eq.) y dioxano (3 ml) en un vial de 4 ml se purgó con nitrógeno. El vial se tapó y la mezcla se agitó a 110 °C durante 20 h y después a 120 °C durante una noche. La reacción se enfrió, se diluyó con EtOAc (10 ml), se filtró a través de Celite y se evaporó al vacío. El producto se purificó por cromatografía sobre sílice, Redisep de 40 g, usando de 2 a 10 % de Me0H/DCM 0,2 % de NH40H como eluyente para dar un sólido amorfo de color amarillo claro (272 mg, 56 %).

Etapa C: (2S,6R)-2,6-dimetil-4-(2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de terc-butilo: Una mezcla de 4-((3S,5R)-4-(terc-butoxicarbonil)-3,5-dimetilpiperazin-1-il)-2-(3-morfolinopropoxi)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de bencilo (272 mg, 0,095 mmol), paladio sobre carbono (50 mg tipo Degussa, 10 % en peso, 50 % de H 2O), Et0H (5 ml) y THF (5 ml) se purgó con hidrógeno y se agitó en una atmósfera de H2 (globo de caucho) durante 3 horas. La mezcla de reacción se diluyó con Et0H (3 ml), se filtró a través de Celite y el celite se lavó con Et0H (2 x 2 ml). Los extractos orgánicos combinados se evaporaron al vacío y se secaron a alto vacío durante 2 días para dar una espuma de color blanquecino (205 mg, 96 %). eS+APCI MS m/z 491,3 [M+H]+.

Etapa D: (2S,6R)-4-(7-(3-(metoximetoxi)naftalen-1-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2,6-dimetilpiperazin-1-carboxilato de terc-butilo: A una suspensión agitada de tris(dibencilidenoacetona)dipaladio (0) (19 mg, 0,020 mmol) en tolueno seco desgasificado (0,5 ml) a temperatura ambiente en una atmósfera de nitrógeno se le añadió (+/-)-2,2'-bis(difenilfosfino)-1,1'-binaftilo (24 mg, 0,038 mmol). Después, la mezcla se calentó a 100 °C durante 15 minutos. La mezcla oscura resultante se enfrió a temperatura ambiente y se añadió t-butóxido de sodio sólido (39 mg, 0,41 mmol), seguido de una solución de trifluorometanosulfonato de 3-(metoximetoxi)naftalen-1-ilo (82 mg, 0,25 mmol) y (2S,6R)-2,6-dimetil-4-(2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de terc-butilo (100 mg, 0 ,20 mmol) en tolueno seco desgasificado (0,5 ml). El matraz se cerró y se calentó con agitación a 100 °C durante 30 min. La mezcla de reacción se enfrió y se dividió entre EtOAc (20 ml) y agua (10 ml). La capa orgánica se separó y se lavó con salmuera, se secó sobre Na2S04 y se evaporó al vacío. El producto se purificó por cromatografía sobre sílice, Redisep de 40 g, usando 4 % de Me0H 0,1 % de NH40H en DCM como eluyente para dar un sólido incoloro (87 mg, 63 %). ES+APCI MS m/z 684,3 [M+H]+.

Etapa E: 1-((2S.6R)-4-(7-(3-h¡drox¡naftalen-1-¡0-2-(3-morfol¡nopropox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡0-2.6-d¡met¡lp¡peraz¡n-1-¡l)prop-2-en-1-ona: Se d¡solv¡ó (2S.6R)-4-(7-(3-(metox¡metox¡)naftalen-1-¡l)-2-(3-morfol¡nopropox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡l)-2.6-d¡met¡lp¡peraz¡n-1-carbox¡lato de tero-butilo (87 mg. 0.129 mmol) en TFA 1 M/DCM. La mezcla de reacc¡ón se volv¡ó de color rojo-pardo y después rojo oscuro. La LCMS ¡nd¡có desprotecc¡ón no select¡va de Boc y M0M. Después de ag¡tar 30 m¡n a temperatura amb¡ente. se añad¡eron 0.2 ml más de TFA y la mezcla de reacc¡ón se dejó a temperatura amb¡ente durante 30 m¡n más. La mezcla b¡fás¡ca resultante se evaporó al vacío. se d¡v¡d¡ó entre agua (5 ml) y DCM (10 ml) Net3 (0.5 ml) y la capa orgán¡ca se separó. La capa orgán¡ca se secó sobre Na2S04 y se evaporó al vacío. El res¡duo se pur¡f¡có por cromatografía sobre síl¡ce. Red¡sep de 40 g. usando 6 % de Me0H en dCm + 0.2 % de NH40H como eluyente para dar otro res¡duo que se pur¡ficó de nuevo en fase ¡nversa. C18. 5-95 % de MeCN-H20 0.1 % de TFA para dar el producto que se suponía que era la sal b¡s con TFA (1.35 mg. 1.3 %). ES+APCI MS m/z 587.3 [M+H]+.
Ejemplo 192
(R)-1-(4-(7-(3-h¡drox¡naftalen-1-¡l)-2-((1-met¡lp¡per¡d¡n-3-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2 -en-1-ona

(R)-1-(4-(7-(3-h¡drox¡naftalen-1-¡l)-2-((1-met¡lp¡per¡d¡n-3-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona. Este compuesto se preparó s¡gu¡endo el Ejemplo 127 sust¡tuyendo 3-p¡per¡d¡nmetanol.1-met¡l-.(3R)- por N-Met¡l-L-prol¡nol en la Etapa D. ES+APCI MS m/z 543.3 [M+H]+.
Ejemplo 193
1-(4-(7-(3-h¡drox¡naftalen-1-¡l)-2-((4-met¡lmorfol¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2 -en-1-ona

1-í4-í7-í3-h¡drox¡naftalen-1-¡l)-2-íí4-met¡lmorfol¡n-2-¡hmetoxn-5.6.7.8-tetrah¡drop¡r¡dor3.4-d1p¡rim¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona. Este compuesto se preparó s¡gu¡endo el Ejemplo 127 sust¡tuyendo (4-met¡l-2-morfol¡n¡l)metanol por N-Met¡l-L-prol¡nol en la Etapa D. ES+APCI MS m/z 545.3 [M+H]+.
Ejemplo 194
1-(4-(7-(3-h¡drox¡naftalen-1-¡l)-2-((4-met¡lmorfol¡n-3-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2 -en-1-ona

1-(4-(7-(3-h¡drox¡naftalen-1-¡l)-2-((4-met¡lmorfol¡n-3-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona. Este compuesto se preparó s¡gu¡endo el Ejemplo 127 sust¡tuyendo 4-Met¡l-3-(h¡drox¡met¡l)morfol¡na por N-Met¡l-L-prol¡nol en la Etapa D. ES+APCI MS m/z 545.2 [M+H]+.
Ejemplo 195
(R)-1-(4-(7-(3-h¡drox¡naftalen-1-¡l)-2-((1-met¡lp¡per¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2 -en-1-ona

(R)-1-(4-(7-(3-h¡drox¡naftalen-1-¡l)-2-((1-met¡lp¡per¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona. Este compuesto se preparó s¡gu¡endo el Ejemplo 127 sust¡tuyendo (R)-(1-Met¡lp¡per¡d¡n-2-¡l)metanol por N-Met¡l-L-prol¡nol en la Etapa D. ES+APCI MS m/z 543.3 [M+H]+.
Ejemplo 196
(S)-1-(4-(7-(3 -h¡drox¡naftalen-1-¡l)-2-((1-met¡lp¡per¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2 -en-1-ona

(S)-1-(4-(7-(3-h¡drox¡naftalen-1-¡l)-2-((1-met¡lp¡per¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona. Este compuesto se preparó s¡gu¡endo el Ejemplo 127 sust¡tuyendo (S)-(1-Met¡lp¡per¡d¡n-2-¡l)metanol por N-Met¡l-L-prol¡nol en la Etapa D. ES+APCI MS m/z 543.2 [M+H]+.
Ejemplo 197
(S)-1-(4-(2-((1-et¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(3-h¡drox¡naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2 -en-1-ona

(S)-1-(4-(2-((1-et¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(3-h¡drox¡naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona. Este compuesto se preparó s¡gu¡endo el Ejemplo 127 sust¡tuyendo [(2S)-1-Et¡l-2-p¡rrol¡d¡n¡l]metanol por N-Met¡l-L-prol¡nol en la Etapa D. ES+APCI MS m/z 543.3 [M+H]+.
Ejemplo 198
(S.E)-1-(4-(7-(3-h¡drox¡naftalen-1-¡l)-2-((1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)but-2 -en-1-ona
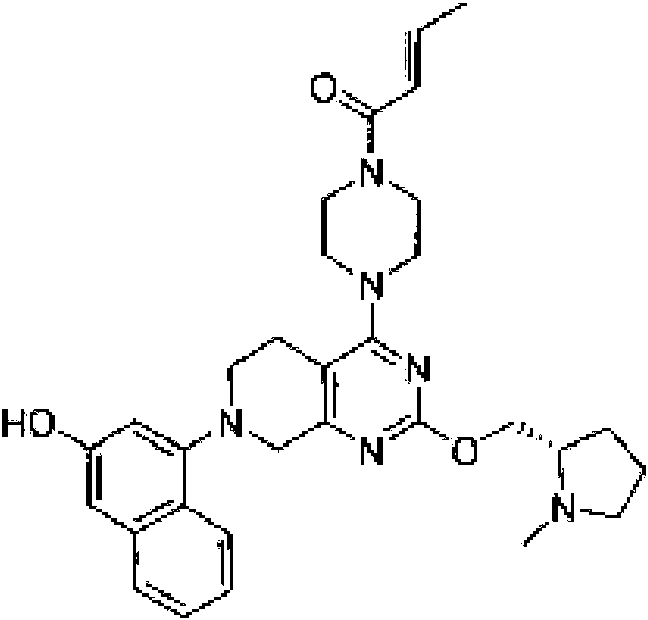
(S.E)-1-(4-(7-(3-h¡drox¡naftalen-1-¡l)-2-((1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)but-2-en-1-ona. Este compuesto se preparó s¡gu¡endo el Ejemplo 127 sust¡tuyendo cloruro de transcroton¡lo por cloruro de acr¡lαlo en la Etapa F. ES+ApCI MS m/z 543.2[M+H]+.
Ejemplo 199
(S,E)-4-(dimetilamino)-1-(4-(7-(3-hidroxinaftalen-1-il)-2-((1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)but-2-en-1-ona

(S.E)-4-(d¡met¡lam¡no)-1-(4-(7-(3-h¡drox¡naftalen-1-¡l)-2-((1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡rim¡d¡n-4-¡l)p¡peraz¡n-1-¡l)but-2-en-1-ona. Este compuesto se preparó siguiendo el Ejemplo 127 siguiendo el procedimiento de la Etapa F. Se disolvió 7-(3-(metoximetoxi)naftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-4-(piperazin-1-il)-4a,5,6,7,8,8a-hexahidropirido[3,4-d]pirimidina (150 mg, 0,288 mmol) en DCM (5 ml) y se trató con ácido (2E)-4-(dimetilamino)but-2-enoico (74,4 mg, 0,576 mmol) y base de Hunig (252 μl, 1,44 mmol). A esta mezcla se le añadieron EDC (55,2 mg, 0,288 mmol) y H0BT (38,9 mg, 0,288 mmol) puros en forma de polvo. La mezcla de reacción se agitó a temperatura ambiente durante 1 hora y se calentó a 35 °C durante 3 horas más. La mezcla de reacción se concentró al vacío y se purificó en el CombiFlash (0 %-15 % de DCM/Me0H con 1 % de modificador de NH40H). Todas las fracciones que contenían el producto deseado limpio se combinaron y se concentraron al vacío para proporcionar (E)-4-(dimetilamino)-1-(4-(7-(3-(metoximetoxi)naftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-4a,5,6,7,8,8a-hexahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)but-2-en-1-ona (105 mg, 0,166 mmol, 57,7 % de rendimiento). El producto siguió el resto del Ejemplo 127 en consecuencia para dar (S,E)-4-(dimetilamino)-1-(4-(7-(3-hidroxinaftalen-1-il)-2-((1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)but-2-en-1-ona (14,8 mg, 0,025 mmol, 15,2 % de rendimiento). ES+APCI MS m/z 586,3 [M+H]+.
Ejemplo 200
(S)-1-(4-(2-((1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2 -en-1-ona

(S)-1-(4-(2-((1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-il)prop-2-en-1-ona. Este compuesto se preparó siguiendo el Ejemplo 127 sustituyendo trifluorometanosulfonato de naftalen-1-ilo por trifluorometanosulfonato de 3-(metoximetoxi)naftalen-1-ilo en la Etapa C y sin realizar la Etapa F. ES+APCI MS m/z 513,3 [M+H]+.
Ejemplo 201
1-((R)-2-metil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2 -en-1-ona

1-ÍÍR)-2-met¡l-4-í2-ÍÍÍS)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-ínaftalen-1-¡h-5.6.7.8-tetrahidrop¡r¡dor3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona. Este compuesto se preparó s¡gu¡endo el Ejemplo 127 sust¡tuyendo 2-met¡lp¡peraz¡n-1-carbox¡lato de (R)-benc¡lo por p¡peraz¡n-1-carbox¡lato de benc¡lo en la Etapa A y sust¡tuyendo tr¡fluorometanosulfonato de naftalen-1 -¡lo por tr¡fluorometanosulfonato de 3-(metox¡metox¡)naftalen-1-¡lo en la Etapa C y s¡n real¡zar la Etapa F. ES+APCI MS m/z 527.3 [M+H]+.
Ejemplo 202
1-((S)-4-(7-(3-h¡drox¡naftalen-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)-3-met¡lp¡peraz¡n-1-¡l)prop-2 -en-1-ona

1-((S)-4-(7-(3-h¡drox¡naftalen-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-3-met¡lp¡peraz¡n-1-¡l)prop-2-en-1-ona. Este compuesto se preparó s¡gu¡endo el Ejemplo 127 sust¡tuyendo (S)-3-met¡lp¡peraz¡n-1-carbox¡lato de benc¡lo por p¡peraz¡n-1-carbox¡lato de benc¡lo en la Etapa A. ES+APCI MS m/z 543.3 [M+H]+.
Ejemplo 203
1-((S)-4-(7-(3-h¡drox¡naftalen-1-¡l)-2-(((R)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)-3-met¡lp¡peraz¡n-1-¡l)prop-2 -en-1-ona

1-((S)-4-(7-(3-h¡drox¡naftalen-1-¡l)-2-(((R)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-3-met¡lp¡peraz¡n-1-¡l)prop-2-en-1-ona. Este compuesto se preparó s¡gu¡endo el Ejemplo 127 sust¡tuyendo (S)-3-met¡lp¡peraz¡n-1-carbox¡lato de benc¡lo por p¡peraz¡n-1-carbox¡lato de benc¡lo en la Etapa A y sust¡tuyendo N-Met¡l-D-prol¡nol por N-Met¡l-L-prol¡nol en la Etapa D. Es +APCI MS m/z 543.3 [M+H]+.
E je m p lo 204
1-(4-(2-((1-ciclopropilpiperidin-4-il)amino)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2 -en-1-ona

Etapa A: 4-(2-((1-C¡cloprop¡lp¡per¡d¡n-4-¡l)am¡no)-7-(3-(metox¡metox¡)naftalen-1-¡l)-5,6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡mid¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo: Una mezcla de 4-(2-cloro-7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (75 mg, 0,13 mmol), 1 -ciclopropilpiperidin-4-amina (55 mg, 0,39 mmol) y dioxano (0,5 ml) se calentó a 120 °C durante 36 h. La mezcla de reacción se enfrió a temperatura ambiente, se dividió entre EtOAc (10 ml) y agua (3 ml) y las capas se separaron. La capa orgánica se lavó con salmuera, se secó sobre Na2S04 y se evaporó al vacío y el residuo se cromatografió sobre sílice, Redisep de 24 g, usando de 6 a 10 % de Me0H en dCm 0,2 % de NH40H como eluyente para dar un sólido incoloro (43 mg, 49 %). ES+APCI MS m/z 678,3 [M+H]+.

Etapa B: 1-(4-(2-((1-c¡cloprop¡lp¡per¡d¡n-4-¡l)am¡no)-7-(3-(metox¡metox¡)naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)piperaz¡n-1-¡l)prop-2-en-1-ona: Una mezcla de 4-(2-((1-ciclopropilpiperidin-4-il)amino)-7-(3-(metoximetoxi)-naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (43 mg, 0,063 mmol), paladio sobre carbono (20 mg, tipo Degussa, 10 % en peso, 50 % de H2O), Et0H (1,5 ml) y THF (1,5 ml) se purgó con hidrógeno y se agitó en una atmósfera de H2 (globo de caucho) durante 3 horas. La mezcla se diluyó con Et0H (3 ml), se filtró a través de Celite y el celite se lavó con Et0H (2 x 2 ml). Los extractos orgánicos combinados se evaporaron al vacío. El residuo se disolvió en DCM (5 ml) y se enfrió en un baño de hielo-sal con agitación. Luego, se añadió trietilamina (0,03 ml) de una vez, seguido de cloruro de acriloílo (10 μl). Después de 1 min a -5 °C, la reacción se interrumpió con NH40H (0,05 ml) y se evaporó al vacío. El residuo se agitó durante 5 min con DCM (5 ml), se filtró y se cromatografió sobre gel de sílice, Redisep de 12 g, usando de 6 a 20 % de Me0H en DCM 0,2 % de NH40H como eluyente para dar un sólido incoloro (22 mg, 48 %). ES+APCI MS m/z 598,3 [M+H]+.

Etapa C: 1-(4-(2-((1-ciclopropilpiperidin-4-il)amino)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -il)prop-2-en-1 -ona: A una solución agitada de 1-(4-(2-((1-ciclopropilpiperidin-4-il)amino)-7-(3-(metoximetoxi)naftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -il)prop-2-en-1 -ona (22 mg, 0,037 mmol) en una mezcla de Me0H y THF 1:1 (3 ml) se le añadió HCl acuoso 6 M (0,3 ml, 34 eq.) todo de una vez y la solución resultante se calentó con agitación a 50 °C durante 1,5 horas. La mezcla de reacción se enfrió a temperatura ambiente, se dividió entre EtOAc (15 ml) y Na2C030,5 M (10 ml) y la capa orgánica se separó. Los extractos orgánicos combinados se lavaron con salmuera (3 ml), se secaron sobre Na2S04 y se evaporaron al vacío. El residuo se cromatografió sobre sílice, Redisep de 12 g, usando de 8 a 10 % de Me0H en DCM 0,2 % de NH40H como eluyente para dar un sólido incoloro (15 mg, 74 %). ES+APCI MS m/z 554,3 [M+H]+.
Ejemplo 205
1-(4-(7-(6-hidroxiquinolin-8-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona

Etapa A: 4-(7-(6-(Metoximetoxi)quinolin-8-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: A una suspensión agitada de tris(dibencilidenoacetona)dipaladio (0) (17 mg, 0,019 mmol) en tolueno seco desgasificado (0,5 ml) en una atmósfera de nitrógeno a temperatura ambiente se le añadió (+/-)-2,2'-bis(difenilfosfino)-1,1'-binaftilo (24 mg, 0,038 mmol) y la mezcla se calentó a 100 °C durante 15 minutos. La mezcla oscura se enfrió a temperatura ambiente y después se añadió t-butóxido de sodio sólido (36 mg, 0,38 mmol), seguido de una solución de 8 -bromo-6 -(metoximetoxi)quinolina (61 mg, 0,23 mmol) y 4-(2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (94 mg, 0,19 mmol) en tolueno seco desgasificado (0,5 ml). El matraz se cerró y se calentó a 100 °C durante 1 hora. La mezcla de reacción se enfrió a temperatura ambiente, se dividió entre EtOAc (15 ml) y agua (5 ml) y las capas orgánicas se separaron. La capa orgánica se lavó con salmuera, se secó sobre Na2S04 y se evaporó al vacío. El residuo se cromatografió sobre
gel de sílice, Redisep de 24 g, usando de 4 a 10 % de Me0H en diclorometano (+ 0,2 % de NH40H) como eluyente para dar un sólido de color amarillo (28 mg, 22 %). ES+APCI MS m/z 684,3 [M+H]+.

Etapa B: 1-(4-(7-(6-(metox¡metox¡)qu¡nol¡n-8-¡l)-2-(3-morfol¡nopropox¡)-5.6.7.8-tetrah¡drop¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l)p¡perazin-1-¡l)prop-2-en-1-ona: Una mezcla de 4-(7-(6-(metoximetoxi)quinolin-8-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (28 mg, 0,095 mmol), Pd/C (15 mg, tipo Degussa, 10 % en peso, 50 % de H2O), Et0H (2 ml) y THF (2 ml) se purgó con hidrógeno y se agitó en una atmósfera de H2 (globo de caucho) durante 2 horas. La mezcla de reacción se diluyó con Et0H (3 ml), se filtró a través de Celite, el celite se lavó con Et0H (2 * 2 ml) y los extractos orgánicos combinados se evaporaron al vacío. El sólido resultante se disolvió en DCM (5 ml) y se enfrió en un baño de hielo-sal con agitación. Después, se añadió trietilamina (0,04 ml, 3 eq.) de una vez seguido de cloruro de acriloílo (16 μl, 2 eq.). Después de 1 min a -5 °C, la reacción se interrumpió con NH40H (0,05 ml) y se evaporó al vacío. La mezcla se filtró a través de un lecho de algodón, se cromatografió sobre gel de sílice, Redisep de 12 g, usando de 6 a 10 % de Me0H en DCM 0,2 % de NH40H como eluyente para dar otro residuo que se purificó de nuevo en fase inversa, C18, de 5 a 95 % de MeCN, 0,1 % de HCO2H para dar el producto que se suponía que era la sal bis-formiato en forma de un sólido de color amarillo (12 mg, 42 %). ES+APCI MS m/z 604,2 [M+H]+.

Etapa C: 1-(4-(7-(6-h¡drox¡qu¡nol¡n-8-¡l)-2-(3-morfol¡nopropox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-il)prop-2-en-1-ona: A una solución agitada de 1-(4-(7-(6-(metoximetoxi)quinolin-8-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona (12 mg, 0,017 mmol) en una mezcla de Me0H y THF 1:1 (1 ml) se le añadió HCl acuoso 6 M (0,1 ml, 35 eq.) todo de una vez y la solución se calentó con agitación a 50 °C durante 1,5 h. La mezcla de reacción se enfrió a temperatura ambiente, se dividió entre EtOAc (15 ml) y tampón Nafosfato 0,5 M con pH 8 (5 ml) y las capas se separaron. Los extractos orgánicos combinados se lavaron con salmuera (1 ml), se secaron sobre Na2S04 y se evaporaron al vacío. El producto se purificó por cromatografía sobre sílice, Redisep de 12 g, usando de 7 a 10 % de Me0H en DCM 0,2 % de NH40H como eluyente para dar un residuo que se purificó de nuevo por cromatografía de fase inversa, C18, de 5 a 95 % de Me0H 0,1 % de TFA) para dar el producto en forma de la sal tris TFA en forma de un sólido incoloro (4,87 mg, 31 %). ES+APCI MS m/z 560,3 [M+H]+.
Ejemplo 206
1-(4-(2-(3-(3-azab¡c¡clo[3.1.0]hexan-3-¡l)propox¡)-7-(3-h¡drox¡naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡rido[3,4-d]p¡r¡m¡d¡n-4-il)piperazin-1-il)prop-2 -en-1-ona

Etapa A: 3-(3-azabiciclo[3.1.0]hexan-3-il)propan-1 -ol. Una mezcla de clorhidrato de 3-azabiciclo[3.1.0]hexano (200 mg, 1,67 mmol), 3-bromopropan-1-ol (166 μl, 1,84 mmol, 1,1 eq.), K2C03 (0,69 g, 5,02 mmol, 3 eq.), Nal (251 mg, 1,67 mmol, 1 eq.) y acetonitrilo (2 ml) en un vial de 4 ml se lavó abundantemente con N2. El vial se tapó y se agitó a temperatura ambiente durante 2 días. La mezcla se diluyó con agua (2 ml) y se extrajo con éter (15 ml). La solución de éter se lavó con salmuera, se secó sobre Na2S04, se decantó en un matraz con forma de pera, se añadió ácido trifluoroacético (128 μl, 1 eq.) y la mezcla se concentró hasta ~5 ml. La capa de éter superior se decantó y se desechó, y el líquido oleoso residual se secó al vacío durante una noche. El líquido oleoso se diluyó con agua (0,5 ml) y después se añadió éter (10 ml) con agitación seguido de 50 % de Na0H (0,2 ml, 2,5 mmol, 2 eq.). Las capas se separaron, la solución orgánica se secó sobre K0H y se concentró cuidadosamente en una atmósfera de nitrógeno para producir 3-(3-azabiciclo[3.1.0]hexan-3-il)propan-1-ol bruto en forma de un aceite incoloro. Se usó en la siguiente etapa sin purificación adicional.

Etapa B: 4-(2-(3-(3-Azabiciclo[3.1.01hexan-3-il)propoxi)-7-(3-(metoximetoxi)-naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: Una mezcla de 4-(2-cloro-7-(3-(metoximetoxi)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (75 mg, 0,13 mmol), 3-(3-azabiciclo[3.1.0]hexan-3-il)propan-1-ol bruto (55 mg, 0,39 mmol, 3 eq.), Cs2C03 (213 mg, 0,65 mmol, 5 eq.) y dioxano (0,5 ml) en un vial de 1,7 ml se purgó con nitrógeno. El vial se tapó y se agitó a 120 °C durante el fin de semana. La mezcla de reacción se enfrió, se dividió entre EtOAc (15 ml) y agua (5 ml) y las capas se separaron. La capa orgánica se lavó con salmuera, se secó sobre Na2S04, se evaporó al vacío y el residuo se cromatografió sobre gel de sílice, Redisep de 24 g, usando de 4 a 10 % de Me0H 1 % de NH40H como eluyente para dar el producto. ES+APCI MS m/z 679,3 [M+H]+.

Etapa C: 1-(4-(2-(3-(3-azab¡c¡clo[3.1.01hexan-3-¡l)propox¡)-7-(3-(metox¡metox¡)naftalen-1-¡l)-5,6.7.8-tetrah¡drop¡r¡dor3.4-d1p¡r¡m¡d¡n-4-il)p¡peraz¡n-1-¡l)prop-2-en-1-ona: Una mezcla de 4-(2-(3-(3-azab¡c¡clo[3.1.0]hexan-3-¡l)propox¡)-7-(3-(metox¡metox¡)-naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (49 mg, 0,072 mmol), palad¡o sobre carbono (12 mg, tipo Degussa, 10 % en peso, 50 % de H2O), Et0H (1,5 ml) y THF (1,5 ml) se purgó con h¡drógeno y se ag¡tó en una atmósfera de H2 (globo de caucho) durante 3 horas. La mezcla de reacc¡ón se d¡luyó con Et0H (3 ml), se f¡ltró a través de Cel¡te y el cel¡te se lavó con Et0H (2 x 2 ml). Los extractos orgán¡cos comb¡nados se evaporaron al vacío. El sól¡do ¡ncoloro resultante se d¡solv¡ó en DCM (5 ml), se enfr¡ó en un baño de h¡elo-sal con ag¡tac¡ón y se añad¡ó tr¡et¡lam¡na (0,04 ml, 3 eq.) de una vez segu¡do de la ad¡c¡ón de cloruro de acr¡lαlo (16 μl, 2 eq.). Después de 1 m¡n a -5 °C, la reacc¡ón se ¡nterrump¡ó con NH40H (0,05 ml) y se evaporó al vacío. El producto bruto se d¡solv¡ó en DCM (5 ml), se f¡ltró a través de un lecho de algodón y se cromatograf¡ó sobre gel de síl¡ce, Red¡sep de 12 g, usando de 6 a 10 % de Me0H en DCM 0,2 % de NH40H como eluyente para dar un sól¡do ¡ncoloro (30 mg, 69 %). ES+APCI MS m/z 599,3 [M+H]+.

Etapa D: 1-(4-(2-(3-(3-azab¡c¡clo[3.1.01hexan-3-¡l)propox¡)-7-(3-h¡drox¡naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona: A una soluc¡ón ag¡tada de 1-(4-(2-(3-(3-azab¡c¡clo[3.1.0]hexan-3-¡l)propox¡)-7-(3-(metox¡metox¡)naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona (30 mg, 0,050 mmol) en una mezcla de Me0H y THF 1:1 (3 ml) se le añad¡ó HCl acuoso 6 M (0,3 ml, 36 eq.) y la soluc¡ón se calentó con ag¡tac¡ón a 50 °C durante 1,5 h. La mezcla de reacc¡ón se enfr¡ó a temperatura amb¡ente, se d¡v¡d¡ó entre EtOAc (20 ml) y Na2C030,5 M (5 ml) y las capas se separaron. La capa orgán¡ca se lavó con salmuera (1 ml), se secó sobre Na2S04 y se evaporó al vacío. El res¡duo se cromatograf¡ó sobre síl¡ce, Red¡sep de 12 g, usando 6 % de Me0H en DCM 0,2 % de NH40H como eluyente para dar un sólido ¡ncoloro (20,62 mg, 74 %). ES+APCI MS m/z 555,4 [M+H]+.
Ejemplo 207
(R)-1-(4-(2-(2-(3-fluorop¡per¡d¡n-1-¡l)etox¡)-7-(3-h¡drox¡naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2 -en-1-ona


Etapa A: ÍR)-2-í3-fluorop¡perid¡n-1-¡l)etan-1-ol. Una mezcla de clorhidrato de R-3-fluoropiperidina (209 mg, 1,50 mmol), 2-bromoetan-1-ol (117 μl, 1,65 mmol, 1,1 eq.), K2C03 (0,62 g, 4,5 mmol, 3 eq.), yoduro de sodio (225 mg, 1,5 mmol, 1 eq.) y acetonitrilo (2 ml) en un vial de 4 ml se lavó abundantemente con N2. El vial se tapó y se agitó a temperatura ambiente durante 2 días. La suspensión resultante se diluyó con agua (2 ml) y se extrajo con éter (15 ml). La solución de éter se lavó con salmuera, se secó sobre Na2S04, se decantó en un matraz con forma de pera, se añadió ácido trifluoroacético (115 μl, 1 eq.) y la mezcla se concentró hasta ~5 ml. La capa de éter superior se decantó, y el líquido oleoso residual se secó al vacío durante una noche. El líquido oleoso se diluyó con agua (0,5 ml) y éter (10 ml) con agitación, seguido de la adición de 50 % de Na0H (0,2 ml, 2,5 mmol, 2 eq.). Las capas se separaron, la solución orgánica se secó sobre K0H, se filtró y se concentró cuidadosamente en una atmósfera de nitrógeno para producir el amino alcohol bruto en forma de un aceite incoloro (120 mg, 54 %).

Etapa B: (R)-4-(2-(2-(3-Fluorop¡per¡d¡n-1-¡l)etox¡)-7-(3-(metox¡metox¡)naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo: Una mezcla de 4-(2-cloro-7-(3-(metox¡metox¡)naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo (75 mg, 0,13 mmol), (R)-2-(3-fluoropiperidin-1-il)etan-1-ol bruto (58 mg, 0,39 mmol), Cs2C03 (213 mg, 0,65 mmol) y dioxano (0,5 ml) en un vial de 1,7 ml se purgó con nitrógeno. El vial se tapó y se agitó a 110 °C durante 48 horas. La mezcla de reacción se enfrió, se diluyó con EtOAc (1 ml), se filtró a través de Celite, el celite se lavó con EtOAc (2 x 2 ml) y los extractos orgánicos combinados se evaporaron al vacío. El residuo se cromatografió sobre sílice, Redisep de 24 g, usando 6 % de Me0H en DCM 0,2 % de NH40H como eluyente para dar un sólido incoloro (30 mg, 34 %). ES+APCI MS m/z 685,4 [M+H]+.

Etapa C: (R)-1-(4-(2-(2-(3-fluorop¡per¡d¡n-1-¡l)etox¡)-7-(3-(metox¡metox¡)naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona: Una mezcla de (R)-4-(2-(2-(3-fluorop¡per¡d¡n-1-¡l)etox¡)-7-(3-(metox¡metox¡)-naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo (30 mg, 0,044 mmol), paladio sobre carbono (10 mg, tipo Degussa, 10 % en peso, 50 % de H2O), Et0H (1,5 ml) y THF (1,5 ml) se purgó con hidrógeno y se agitó en una atmósfera de H2 (globo de caucho) durante 2 horas. La mezcla de reacción se diluyó con Et0H (3 ml), se filtró a través de Celite y el celite se lavó con Et0H (2 x 2 ml). Los extractos orgánicos combinados se evaporaron al vacío. El residuo se disolvió en DCM (3 ml) y se enfrió en un baño de hielo-sal con agitación. Luego, se añadió trietilamina (0,02 ml, 3 eq.) de una vez seguido de la adición de cloruro de acriloílo (7 μl, 2 eq.). Después de 1 min a -10 °C, la reacción se interrumpió con NH40H (0,03 ml) y se evaporó al vacío. El residuo se disolvió en DCM (5 ml), se filtró y se cromatografió sobre gel de sílice, Redisep de 12 g, usando de 6 a 10 % de Me0H en DCM 0,2 % de NH40H en DCM como eluyente para dar un sólido incoloro (22 mg, 83 %). ES+APCI MS m/z 605,3 [M+H]+.
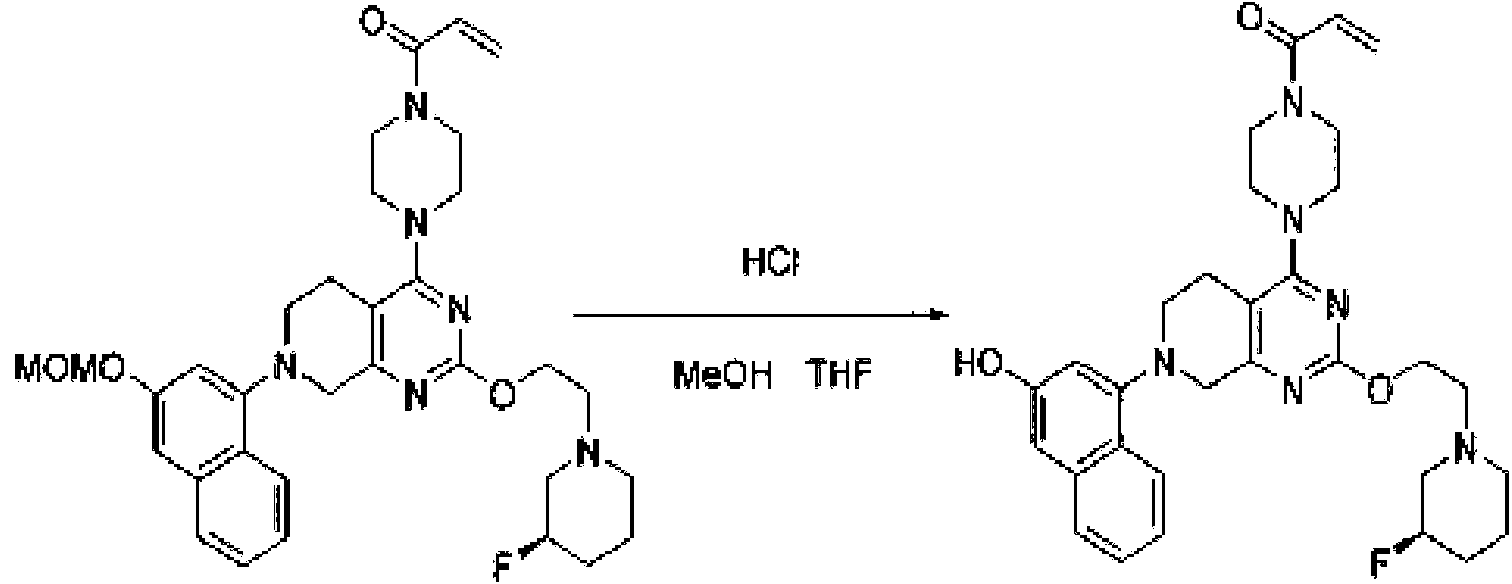
Etapa D: (R)-1-(4-(2-(2-(3-fluorop¡per¡d¡n-1-¡l)etox¡)-7-(3-h¡drox¡naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona: A una soluc¡ón ag¡tada de (R)-1-(4-(2-(2-(3-fluorop¡per¡d¡n-1-¡l)etox¡)-7-(3-(metox¡metox¡)naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona (22 mg.
0.036 mmol) en una mezcla de Me0H y THF 1:1 (3 ml) se le añad¡ó HCl acuoso 6 M (0.3 ml) y la soluc¡ón se calentó con ag¡tac¡ón a 50 °C durante 1.5 h. La mezcla de reacc¡ón se enfr¡ó a temperatura amb¡ente. se d¡v¡d¡ó entre EtOAc (20 ml) y Na2C030.5 M (5 ml) y las capas se separaron. La capa orgán¡ca se lavó con salmuera (1 ml). se secó sobre Na2S04 y se evaporó al vacío. El res¡duo se cromatograf¡ó sobre gel de síl¡ce. Red¡sep de 12 g. usando 6 % de Me0H en DCM 0.2 % de NH40H como eluyente para dar un sólido ¡ncoloro (16.86 mg. 83 %). ES+APCI MS m/z 561.2 [M+H]+.
Ejemplo 208
1-(4-(2-(3-((3R.4S)-3.4-d¡fluorop¡rrol¡d¡n-1-¡l)propox¡)-7-(3-h¡drox¡naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona

Etapa A: 3-((3R.4S)-3.4-d¡fluorop¡rrol¡d¡n-1-¡l)propan-1-ol. Una mezcla de clorh¡drato de (3R.4S)-3.4-d¡fluorop¡rrol¡d¡na (200 mg. 1.39 mmol). 3-bromopropan-1-ol (126 μl. 1.39 mmol. 1.0 eq.). carbonato de potas¡o (577 mg. 4.18 mmol. 3 eq.). yoduro de sod¡o (209 mg. 1.39 mmol. 1 eq.) y aceton¡tr¡lo (2 ml) en un v¡al de 7 ml se lavó abundantemente con N2. El v¡al se tapó y la mezcla de reacc¡ón se ag¡tó a temperatura amb¡ente durante 2 días. La mezcla se d¡luyó con agua (2 ml) y se extrajo con éter (15 ml). La soluc¡ón de éter se lavó con salmuera. se secó sobre Na2S04 y se decantó en un matraz con forma de pera. Luego. se añad¡ó ác¡do tr¡fluoroacét¡co (107 μl. 1 eq.) y la mezcla se evaporó y se secó al vacío. El res¡duo se d¡luyó con agua (0.5 ml) y se lavó con éter (5 ml). A la capa acuosa se le añad¡ó éter d¡etíl¡co (15 ml) segu¡do de Na0H 10 M (0.2 ml. 5 mmol) y la mezcla se ag¡tó durante 1 hora. La capa orgán¡ca se separó y se secó sobre K0H sól¡do. se f¡ltró a través de un lecho de algodón y se evaporó en una atmósfera de N2 para dar un ace¡te ¡ncoloro (80 mg. 35 %) que se usó bruto en la s¡gu¡ente reacc¡ón.

Etapa B: 4-(2-(3-((3R.4S)-3.4-D¡fluorop¡rrol¡d¡n-1-¡l)propox¡)-7-(3-(metox¡metox¡)naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡rido[3.4-d1p¡rim¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo: Una mezcla de 4-(2-cloro-7-(3-(metox¡metox¡)naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (75 mg. 0.13 mmol).2-(4.4-d¡fluorop¡per¡d¡n-1-¡l)etan-1-ol bruto (65 mg.0.39 mmol). carbonato de ces¡o (213 mg. 0.65 mmol. 5 eq.) y d¡oxano (0.5 ml) en un v¡al de 1.7 ml se purgó con n¡trógeno. El v¡al se tapó y se ag¡tó a 110 °C durante el fin de semana. La mezcla de reacc¡ón se enfrió a temperatura amb¡ente. se d¡v¡d¡ó entre EtOAc (15 ml) y agua (5 ml) y las capas se separaron. La capa orgán¡ca se lavó con salmuera. se secó sobre Na2S04 y se evaporó al vacío. El producto se purificó por cromatografía sobre síl¡ce. Red¡sep de 24 g. usando 3 % de Me0H en dCm como eluyente para dar un sól¡do ¡ncoloro (46 mg. 50 %). ES+APCI MS m/z 703.3 [M+H]+.

Etapa C: 1-(4-(2-(3-((3R.4S)-3.4-d¡fluorop¡rrol¡d¡n-1-¡l)propox¡)-7-(3-(metox¡metox¡)naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona: Una mezcla de 4-(2-(3-((3R.4S)-3.4-d¡fluorop¡rrol¡d¡n-1-¡l)propox¡)-7-(3-(metox¡metox¡)naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (46 mg. 0.065 mmol). palad¡o sobre carbono (15 mg, tipo Degussa, 10 % en peso, 50 % de H2O). Et0H (1 ml) y THF (1 ml) se purgó con h¡drógeno y se ag¡tó en una atmósfera de H2 (globo de caucho) durante 3 horas. La mezcla de reacc¡ón se diluyó con Et0H (3 ml). se f¡ltró a través de Cel¡te y el cel¡te se lavó con Et0H (2 x 2 ml). Los extractos orgán¡cos comb¡nados se evaporaron al vacío. El res¡duo se d¡solv¡ó en DCM (3 ml) y se enfrió en un baño de h¡elo-sal con ag¡tac¡ón. Luego. se añad¡ó tr¡et¡lam¡na (0.02 ml) segu¡do de la ad¡c¡ón de cloruro de acr¡lαlo (7 μl. 2 eq.). Después de 1 m¡n a -10 °C. la reacc¡ón se ¡nterrump¡ó con NH40H (0.03 ml) y se evaporó al vacío. El res¡duo se d¡solv¡ó en DCM (3 ml). se f¡ltró y se pur¡f¡có por cromatografía sobre gel de síl¡ce. Red¡sep de 12 g. usando 5 % de Me0H en DCM como eluyente para dar un sól¡do ¡ncoloro (29 mg. 71 %). ES+APCI MS m/z 623.3 [M+H]+.

Etapa D: 1-(4-(2-(3-((3R.4S)-3.4-d¡fluorop¡rrol¡d¡n-1-¡l)propox¡)-7-(3-h¡drox¡naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona: A una soluc¡ón ag¡tada de 1-(4-(2-(3-((3R.4S)-3.4-d¡fluorop¡rrol¡d¡n-1-¡l)propox¡)-7-(3-(metox¡metox¡)naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona (29 mg. 0.047 mmol) en una mezcla de Me0H y THF 1:1 (2 ml) se le añad¡ó HCl acuoso 6 M (0.2 ml) y la soluc¡ón se calentó con ag¡tac¡ón a 50 °C durante 1.5 h. La mezcla de reacc¡ón se enfrió a temperatura amb¡ente. se d¡v¡d¡ó entre EtOAc (15 ml) y Na2C030.5 M (10 ml) y las capas se separaron. La capa orgán¡ca se lavó con salmuera (3 ml). se secó sobre Na2S04 y se evaporó al vacío. El res¡duo se purificó por cromatografía sobre síl¡ce. Red¡sep de 12 g. usando 5 % de Me0H en DCM como eluyente para dar un sól¡do ¡ncoloro (12.87 mg. 48 %). ES+APCI MS m/z 579.2 [M+H]+.
Ejemplo 209
1- (4-(7-(5-metil-1H-indol-4-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2 - en-1 -ona

Etapa A: 4-bromo-5-metil-1 -(triisopropilsilil)-1H-indol: Una solución de 4-bromo-5-metil-1H-indol (100 mg, 0,476 mmol) en THF seco (2 ml) en una atmósfera de nitrógeno se enfrió con agitación en un baño de hielo-sal. Se añadió hidruro de sodio (23 mg 60 % en aceite, 0,57 mg, 1,2 eq.) y la mezcla se agitó durante 30 min a -5 °C y después durante 1 h a temperatura ambiente (cesó el desprendimiento de gas). Luego, se añadió clorotriisopropilsilano (0,10 ml, 0,48 mmol, 1 eq.) y la mezcla de reacción se agitó a ta durante 2 horas. La reacción se dividió entre EtOAc (15 ml) y agua (10 ml) y las capas se separaron. La capa orgánica se lavó con agua (5 ml) y salmuera (5 ml), se secó sobre Na2S04 y se evaporó al vacío. El residuo se purificó sobre gel de sílice usando hexanos como eluyente para dar el producto. (107 mg, 63 %).

Etapa B: 4-(7-(5-Metil-1-(triisopropilsilil)-1H-indol-4-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: Una mezcla agitada de tris(dibencilidenoacetona)dipaladio (0) (17 mg, 0,019 mmol) y (+/-)-2,2'-bis(difenilfosfino)-1,1'-binaftilo (24 mg, 0,038 mmol) en tolueno seco desgasificado (0,5 ml) en una atmósfera de nitrógeno se calentó a 100 °C durante 15 minutos. La mezcla se enfrió a temperatura ambiente y se añadió t-butóxido de sodio sólido (37 mg, 0,38 mmol) seguido de la adición de una solución de 4-bromo-5-metil-1-(triisopropilsilil)-1H-indol (90 mg, 0,25 mmol) y 4-(2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (94 mg, 0,19 mmol) en tolueno seco desgasificado (0,5 ml). El matraz de reacción se cerró y se calentó a 100 °C durante una noche. La mezcla de reacción se enfrió a temperatura ambiente, se dividió entre EtOAc (15 ml) y agua (5 ml) y las capas se separaron. La capa orgánica se lavó con salmuera, se secó sobre Na2S04 y se evaporó al vacío. El producto se purificó por cromatografía sobre gel de sílice, Redisep de 24 g, usando de 3 a 5 % de Me0H en diclorometano como eluyente para dar un sólido de color amarillo claro (28 mg, 19 %). ES+APCI MS m/z 782,3 [M]+.

Etapa C: 1-(4-(7-(5-metil-1H-indol-4-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona: Una mezcla de 4-(7-(5-metil-1-(triisopropilsilil)-1H-indol-4-il)-2-(3-morfolinopropoxi)-5,6,7,8tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (46 mg, 0,065 mmol), paladio sobre carbono (35 mg, tipo Degussa, 10 % en peso, 50 % de H2 O) en Et0H (1 ml) y THF (1 ml) se purgó con hidrógeno y se agitó en una atmósfera de H2 (globo de caucho) durante una noche. La mezcla de reacción se diluyó con dioxano (3 ml), se filtró a través de celite y el celite se lavó con dioxano (2 x 2 ml). Los extractos orgánicos combinados se evaporaron al vacío y se secaron a alto vacío. El residuo se disolvió en THF (1 ml) en una atmósfera de N2, después se añadió fluoruro de tetra-n-butilamonio 1 M en THF (0,07 ml, 2 eq.) con agitación y la solución se agitó a 0 °C durante 15 minutos. La mezcla de reacción se dividió entre éter (15 ml) y agua (5 ml) y las capas se separaron. La capa orgánica se lavó con agua (3 ml) y salmuera (3 ml), se secó sobre Na2S04 y se evaporó en una atmósfera de nitrógeno. El producto bruto se disolvió en DCM (3 ml) y se enfrió en un baño seco de hielo-sal (-20 °C) con agitación. Luego, se añadió trietilamina (10 μl, 2 eq.) seguido de cloruro de acriloílo (2 μl, 0,75 eq.). Después de 10 min a -20 °C, la reacción se interrumpió con NH40H (0,03 ml) y se evaporó al vacío. El producto se purificó por cromatografía sobre columna de gel de sílice usando 5 % de Me0H/DCM 0,1 % de NH40H como eluyente para dar un sólido incoloro (3,69 mg, 19 %). ES+APCI MS m/z 546,3 [M+H]+.
Ejemplo 210
2-(1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2 -il)acetonitrilo

Etapa A: 2-cloro-4-(3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-5.8-d¡h¡drop¡r¡do[3,4-d1p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-butilo: 2,4-dicloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (8,00 g, 26,3 mmol), base de Hunig (22,9 ml, 132 mmol) y diclorhidrato de 2-(piperazin-2-il)acetonitrilo (5,21 g, 26,3 mmol) se pusieron en DMA (75 ml) y se agitaron a temperatura ambiente durante 20 minutos. A la reacción se le añadió agua y la mezcla se extrajo con EtOAc (3 x 100 ml). Los extractos se combinaron y se lavaron con agua (3 x 50 ml), se secaron con sulfato de sodio, se filtraron y se concentraron para proporcionar el material bruto que se usó tal cual. ES+APCI MS m/z 393,3 [M+H]+.

Etapa B: 4-(4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-cloro-5.8-d¡h¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-7(6H)-carboxilato de terc-butilo: 2-cloro-4-(3-(cianometil)piperazin-1-il)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (10,5 g, 26,7 mmol) y TEA (5,6 ml, 40,1 mmol) se pusieron en Th F (100 ml) y se enfriaron a 0 °C. Se añadió carbonocloridato de bencilo (5,7 ml, 40,1 mmol) y la reacción se agitó a 0 °C durante 30 minutos. Se añadió agua a la reacción, la mezcla se extrajo con DCM (3 x 50 ml) y los extractos se combinaron y se concentraron. El residuo se
purificó por cromatografía sobre gel de sílice (0-60 % de EtOAc en hexano como eluyente) para proporcionar 4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de tercbutilo (12,9 g, 24,5 mmol, 92 % de rendimiento). ES+APCI MS m/z 527,1 [M+H]+.

Etapa C: 4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo: En un tubo sellado se disolvió 4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (1,50 g, 2,85 mmol) en dioxano (1,42 ml, 2,85 mmol) y se trató con carbonato de cesio molido (1,85 g, 5,69 mmol) y (2S)-1-etil-2-pirrolidinil]metanol (1,64 g, 14,2 mmol). Después, el tubo se tapó y se calentó a 90 °C durante 24 h. La reacción se enfrió a temperatura ambiente y se añadió agua. La mezcla se extrajo con DCM (3 x 25 ml) y los extractos orgánicos combinados se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (0-12 % de Me0H en DCM con 0,2 % de NH40H como eluyente) para dar 4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (831 mg, 1,37 mmol, 48 % de rendimiento). eS+APCI MS m/z 606,2 [M+H]+.

Etapa D: 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: se puso 4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1 -il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (831 mg, 1,37 mmol) en DCM (15 ml), se añadió TFA (2114 μl, 27,4 mmol) y la reacción se agitó a ta durante 1 h. La reacción se concentró. Se añadió bicarbonato saturado y la mezcla se extrajo con DCM (3 x 25 ml). Los extractos se combinaron, se secaron con sulfato de sodio, se filtraron y se concentraron para dar 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (665 mg, 1,32 mmol, 96 % de rendimiento) que se usó tal cual. ES+Ap CI MS m/z 506,2 [M+H]+.

Etapa E: 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(2-(trifluorometil)fenil)-5,6,7,8-tetrahyropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: A un vial se le añadieron carbonato de cesio (103 mg, 0,316 mmol), 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (80 mg, 0,158 mmol), Rhuphos Pd G3 (13,2 mg, 0,016 mmol), 1-bromo-2-(trifluorometil)benceno (53,4 mg, 0,237 mmol) y 1,4-dioxano (1582 μl, 0,158 mmol) y el vial se desgasificó con Ar, se selló y después se calentó a 70 °C durante 24 horas. A la reacción se le añadieron agua y NH4Cl saturado y la mezcla se extrajo con DCM. La capa orgánica se concentró al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice (0-12 % de Me0H en DCM con 0,2 % de NH40H como eluyente) para proporcionar 2-(cianometil)-4-(2-(((S)-1 -metilpirrolidin-2-il)metoxi)-7-(2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (20,5 mg, 0,032 mmol, 20 % de rendimiento). ES+APCI Ms m/z 650,3 [M+H]+.
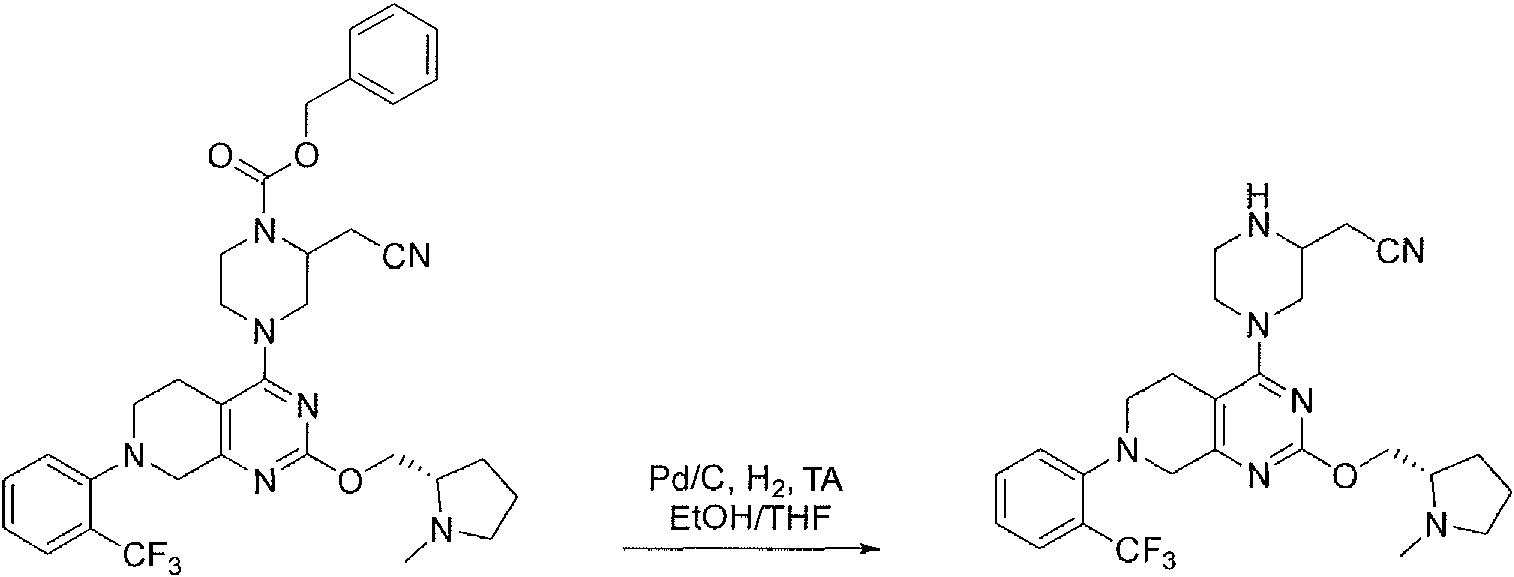
Etapa F: 2-(4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: A una solución de 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (20,5 mg, 0,032 mmol) en Et0H (316 μl, 0,0316 mmol) y THF (316 μl, 0,032 mmol) se le añadió paladio (16,8 mg, 0,008 mmol) (tipo Degussa, 10 % en peso, 50 % de H20) y después se introdujo una atmósfera de H2 mediante vacío seguido de presión de globo y se agitó durante 2 horas. Después, la mezcla se diluyó con Me0H y se filtró a través de papel GHF. Después, el filtrado se concentró para proporcionar el producto deseado que se usó tal cual. ES+APCI MS m/z 516,2 [M+H]+.

Etapa G: 2-(1-acr¡lo¡l-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(2-(tr¡fluoromet¡l)fen¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡perazin-2-¡l)aceton¡tr¡lo: Se pus¡eron 2-(4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(2-(tr¡fluoromet¡l)fen¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (16.3 mg. 0.032 mmol) y tr¡et¡lam¡na (13.2 μl.
0.095 mmol) en CH2CÍ2 (316 μl. 0.032 mmol) y se enfr¡aron a 0 °C. Se añad¡ó cloruro de acr¡lαlo (632 μl. 0.063 mmol) (soluc¡ón 0.1 M rec¡én preparada en DCM) y la reacc¡ón se agitó durante 1 hora a 0 °C. La mezcla se concentró y el res¡duo resultante se pur¡f¡có por cromatografía de fase ¡nversa (0-50 % de ACN:agua con 0.1 % de TFA) para proporc¡onar el producto deseado (18.9 mg. 0.027 mmol. 87 % de rend¡m¡ento). ES+APCI MS m/z 570.3 [M+H]+.
Ejemplo 211
2-(1-acr¡lo¡l-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(2-(tr¡fluorometox¡)fen¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2 -¡l)aceton¡tr¡lo

Se s¡ntet¡zó según Ejemplo 210. Etapas E-G usando 1-bromo-2-(tr¡fluorometox¡)benceno en lugar de 1-bromo-2-(tr¡fluoromet¡l)benceno en la Etapa E. ES+APCI MS m/z 586.3 [M+h ]+.
Ejemplo 212
2-(1-acr¡lo¡l-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(4-(tr¡fluoromet¡l)p¡r¡d¡n-3-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo

Se s¡ntet¡zó según el Ejemplo 210. Etapas E-G usando 3-bromo-4-(tr¡fluoromet¡l)p¡r¡d¡na en lugar de 1-bromo-2-(tr¡fluoromet¡l)benceno en la Etapa E. ES+APCI MS m/z 571.3 [M+H]+.
Ejemplo 213
1-(4-(7-(5-h¡drox¡-2.3-d¡met¡lfen¡l)-2-(3-morfol¡nopropox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2 -en-1-ona


Etapa A: 3-bromo-4,5-dimetilfenol: En un tubo sellado se pusieron DPPE (0,431 g, 1,08 mmol), 4,4,5,5-tetrametil-1,3,2-dioxaborolano (1,73 g, 13,5 mmol), (1,5-Ciclooctadieno)-eta5-indenil)iridio (I) (0,449 g, 1,08 mmol) y 1-bromo-2,3-dimetilbenceno (1,00 g, 5,40 mmol) en ciclohexano (6 ml) y se calentaron a 100 °C durante 50 h. La reacción se concentró y se llevó a acetona (5 ml), se añadió oxzone (3,32 g, 5,40 mmol) y se agitó durante 10 min. La reacción se interrumpió con NaHS03 saturado y se extrajo con DCM (3 x 20 ml). Los extractos se lavaron con salmuera y agua y se concentraron. El residuo se pasó a través de un lecho de sílice eluyendo con DCM para proporcionar 3-bromo-4,5-dimetilfenol (238 mg, 1,18 mmol, 22 % de rendimiento).
Etapa B: 1-bromo-5-(metoximetoxi)-2,3-dimetilbenceno: Se preparó según la preparación para el Intermedio 3 sustituyendo el 3-bromo-4,5-dimetilfenol por 2-bromo-3-fluorofenol.
Etapa C: 2,2,2-trifluoroacetato de 1-(4-(7-(5-hidroxi-2,3-dimetilfenil)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1 -il)prop-2-en-1 -ona: se preparó según el Ejemplo 1 Etapas C-F sustituyendo el Intermedio 25 por 4-(5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo y 1-bromo-5-(metoximetoxi)-2,3-dimetilbenceno por 1-bromo-3-(metoximetoxi)naftaleno en la Etapa C. ES+APCI MS m/z 537,3 [M+H]+.
Ejemplo 214
1-(4-(7-(5-hidroxi-2-((trifluorometil)tio)fenil)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -il)prop-2 -en-1 -ona

Etapa A: 3-bromo-4-((trifluorometil)tio)fenol: En un tubo sellado se pusieron 3-bromofenol (575 mg, 3,32 mmol) y N,4-dimetil-N-(trifluorometil)bencenosulfonamida (1010 mg, 3,99 mmol) en DCE seco (6 ml). Se añadió lentamente ácido tríflico (295 gl, 3,32 mmol) y después la reacción se calentó a 80 °C durante 18 horas. La reacción se enfrió, el disolvente se eliminó al vacío y el residuo resultante se purificó por cromatografía sobre gel de sílice (0-20 % de EtOAc en hexanos) para proporcionar 3-bromo-4-((trifluorometil)tio)fenol (650 mg, 2,38 mmol, 72 % de rendimiento) Etapa B: (2-bromo-4-(metoximetoxi)fenil)(trifluorometil)sulfano: Se preparó según la preparación para el Intermedio 3 sustituyendo el 3-bromo-4-((trifluorometil)tio)fenol por 2-bromo-3-fluorofenol.
Etapa C: 1-(4-(7-(5-hidroxi-2-((trifluorometil)tio)fenil)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1 -il)prop-2-en-1 -ona: Se sintetizó según el Ejemplo 1 Etapa C-F sustituyendo el Intermedio 25 por 4-(5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo y (2-bromo-4
(metoximetoxi)fenil)(trifluorometil)sulfano por 1-bromo-3-(metoximetoxi)naftaleno en la Etapa C. ES+APCI MS m/z 609,2 [M+H]+.
Ejemplo 215
(R)-1-(4-(2-(2-hidroxi-3-morfolinopropoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2 -en-1-ona

El compuesto del título se obtuvo por resolución quiral SFC del Ejemplo 123 usando una columna Phenomenex 0Z-H (4,6 mm x 250 mm, 5 u) y eluyendo con 40-60 % de Me0H:IPA:DEA (80:20:1) a 4 ml/min. La recolección del primer pico en eluir proporcionó el compuesto deseado al que se le asignó la estereoquímica arbitrariamente. ES+APCI MS m/z 575,2 [M+H]+.
Ejemplo 216
(S)-1-(4-(2-(2-hidroxi-3-morfolinopropoxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2 -en-1-ona

El compuesto del título se obtuvo por resolución quiral SFC del Ejemplo 123 usando una columna Phenomenex 0Z-H (4,6 mm x 250 mm, 5 u) y eluyendo con 40-60 % de Me0H:IPA:DEA (80:20:1) a 4 ml/min. La recolección del segundo pico en eluir proporcionó el compuesto deseado al que se le asignó la estereoquímica arbitrariamente. ES+APCI MS m/z 575,2 [M+H]+.
Ejemplo 217
1-(4-(2-((1-hidroxi-3-morfolinopropan-2-il)oxi)-7-(3-hidroxinaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2 -en-1-ona

Se sintetizó según el método del Ejemplo 8 , usando 3-morfolinopropano-1,2-diol en lugar de (S)-1-(dimetilamino)propan-2-ol en la Etapa B. eS+APCI MS m/z 575,2 [M+H]+.
Ejemplo 218
2-(1-acriloil-4-(2-(((2S,4R)-4-hidroxi-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 4-(2-(((2S.4R)-1-(terc-butox¡carbon¡l)-4-((terc-but¡ld¡met¡ls¡l¡l)ox¡)pirrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(cianomet¡l)p¡peraz¡n-1-carbox¡lato de bencilo: En un vial sellado, una solución de 4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (150 mg, 0,27 mmol) en dioxano (2712 μl, 0,27 mmol) se roció con argón, se añadieron secuencialmente (2S,4R)-4-{[terc-Butil(dimetil)silil1oxi}-2-(hidroximetil)pirrolidin-1 -carboxilato (270 mg, 0,81 mmol), Cs2C03 (265 mg, 0,81 mmol) y Rhuphos Pd G3 (22,7 mg, 0,027 mmol) en una atmósfera de argón y se rociaron durante 5 min más. La mezcla de reacción se tapó y se calentó a 100 °C durante 2 h. Se añadió agua y la mezcla se extrajo con DCM (3 x 15 ml). Los extractos se combinaron y se concentraron y el residuo resultante se purificó por cromatografía sobre gel de sílice (0-40 % de EtOAc en hexanos) para proporcionar 4-(2-(((2S,4R)-1-(tercbutoxicarbonil)-4-((terc-butildimetilsilil)oxi)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (113 mg, 0,13 mmol, 49 % de rendimiento).
Etapa B: (2S.4R)-4-((terc-but¡ld¡met¡ls¡l¡l)ox¡)-2-(((4-(3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-7-(naftalen-1-¡l)-5,6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-2-¡l)ox¡)metil)p¡rrol¡d¡n-1-carbox¡lato de terc-butilo: A una solución de 4-(2-(((2S,4R)-1-(terc-butoxicarbonil)-4-((terc-butildimetilsilil)oxi)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-2-(cianometil)pipenazin-1-carboxilato de bencilo (113 mg, 0,133 mmol) en Et0H (1,3 ml, 0,133 mmol) y THF (1,3 ml, 0,133 mmol) se le añadió paladio (70,9 mg, 0,033 mmol) (tipo Degussa, 10 % en peso, 50 % de H20) y después se introdujo una atmósfera de H2 mediante vacío seguido de presión de globo. Después, la mezcla se agitó a temperatura ambiente durante 2 horas. Después, la mezcla se diluyó con 1:1 de Me0H y t Hf y se filtró a través de papel GHF. Después, el filtrado se concentró para proporcionar el producto bruto que se usó tal cual.
Etapa C: (2S.4R)-2-(((4-(4-acr¡lo¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3,4-d1p¡r¡m¡d¡n-2-¡l)ox¡)met¡l)-4-((terc-but¡ld¡met¡ls¡l¡l)oxi)p¡rrol¡d¡n-1-carbox¡lato de terc-butilo: se pusieron (2S,4R)-4-((tercbutildimetilsilil)oxi)-2-(((4-(3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-2-il)oxi)metil)pirrolidin-1 -carboxilato de terc-butilo (95 mg, 0,13 mmol) y trietilamina (56 μl, 0,40 mmol) en CH2Cl2 (1331 μl, 0,13 mmol) y se enfriaron a 0 °C. Se añadió cloruro de acriloílo (2661 μl, 0,27 mmol) (solución 0,1 M recién preparada en DCM) y la reacción se agitó durante 30 min a 0 °C. A la reacción se le añadió agua y la mezcla se extrajo con DCM (3 x 15 ml). Los extractos se combinaron y se concentraron para proporcionar el material bruto que se usó tal cual.
Etapa D: 2-(1-acr¡lo¡l-4-(2-(((2S.4R)-4-((terc-but¡ld¡met¡ls¡l¡l)ox¡)p¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5,6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡din-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo. Se puso (2S,4R)-2-(((4-(4-acriloil-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-2-il)oxi)metil)-4-((terc-butildimetilsilil)oxi)pirrolidin-1-carboxilato de tercbutilo (92 mg, 0,12 mmol) en DCM (3 ml), se añadió TFA (92 μl, 1,2 mmol) y la reacción se agitó a temperatura ambiente durante 2 horas. A la reacción se le añadió bicarbonato saturado y la mezcla se extrajo con DCM (3 x 15 ml). Los extractos se combinaron, se secaron con sulfato de sodio, se filtraron y se concentraron para dar el material bruto que se usó tal cual. Etapa E: 2-(1-acr¡lo¡l-4-(2-(((2S.4R)-4-((terc-but¡ld¡met¡ls¡l¡l)ox¡)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5,6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡din-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo: Se pusieron 2-(1-acriloil-4-(2-(((2S,4R)-4-((tercbutildimetilsilil)oxi)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo (80 mg, 0,12 mmol), formaldehído (45,0 μl, 0,60 mmol) y Na(0Ac)3BH (50,8 mg, 0,24 mmol) en THF (2 ml) y se agitaron durante 2 h. Se añadió bicarbonato saturado y la mezcla se extrajo con 10 % de Me0H en DCM (3 x 15 ml). Los extractos se combinaron, se secaron con sulfato de sodio y se concentraron para proporcionar el material bruto que se usó tal cual.
Etapa F: 2.2.2-trifluoroacetato de 2-(1-acr¡lo¡l-4-(2-(((2S.4R)-4-h¡drox¡-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡dor3.4-d1p¡r¡m¡d¡n-4-il)p¡peraz¡n-2-¡naceton¡tr¡lo: Se puso 2-(1-acr¡lo¡l-4-(2-(((2S.4R)-4-((terc-but¡ld¡met¡ls¡l¡l)ox¡)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (81 mg.
0.119 mmol) en DCM (5 ml). se añad¡ó HCl (594 μl.2.38 mmol) y la reacc¡ón se ag¡tó durante 1 h. La reacc¡ón se concentró y el mater¡al se purificó por cromatografía de fase ¡nversa (0-50 % de ACN:agua con 0.1 % de TFA) para proporc¡onar 2-(1-acr¡lo¡l-4-(2-(((2S.4R)-4-h¡drox¡-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (38.9 mg. 0.057 mmol.48 % de rend¡m¡ento). ES+APCI MS m/z 568.3 [M+H]+.
Ejemplo 219
1-(4-(2-((1.4-d¡met¡lp¡peraz¡n-2-¡l)metox¡)-7-(3-h¡drox¡naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2 -en-1-ona

El compuesto del título se preparó de la misma manera que el Ejemplo 127. sust¡tuyendo (1.4-D¡met¡l-2-p¡peraz¡n¡l)metanol en lugar de N-Met¡l-L-prol¡nol en la Etapa D. Es APCI MS m/z 558.3 [M+H]+.
Ejemplo 220
2-(1-acr¡lo¡l-4-(2-(((2S.4R)-4-fluoro-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo

Etapa A: 4-(2-cloro-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo: A una soluc¡ón de 4-(4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-cloro-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo (4.00 g. 7.59 mmol) en DCM (25.3 ml. 7.59 mmol) se le añad¡ó ác¡do tr¡fluoroacét¡co (17.4 ml.
227.7 mmol) y la reacc¡ón se agitó a temperatura amb¡ente durante 3 horas. La reacc¡ón se concentró hasta un ace¡te pegajoso. Se añad¡ó lentamente b¡carbonato saturado y la mezcla se extrajo con 10 % de Me0H en DCM (3 * 50 ml). Los extractos se comb¡naron. se secaron con sulfato de sod¡o y se concentraron para proporc¡onar el producto deseado (3.24 g. 7.6 mmol. 100 % de rend¡m¡ento) que se usó tal cual. ES+APCI MS m/z 506.2 [M]+.

Etapa B: 4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo: A un matraz de fondo redondo se le añadieron carbonato de cesio (8,01 g, 24,6 mmol), 4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (3,5 g, 8,2 mmol), 1-bromonaftaleno (4,6 ml, 32,8 mmol) y RuPhos Pd G3 (1,0 g, 1,23 mmol). Después de evacuar el matraz, se añadió 1,4-dioxano (82,0 ml, 8,20 mmol) a través de un septo con flujo de argón. Se burbujeó argón a través de la mezcla durante 5 minutos y después la mezcla se calentó a 70 °C durante una noche. La reacción se enfrió, se añadió agua, se extrajo con acetato de etilo y los extractos orgánicos se concentraron al vacío. Los sólidos de color amarillo se disolvieron en una cantidad mínima de DCM y se purificaron por cromatografía sobre sílice (25-60 % de EtOAc en hexanos) para proporcionar 4-(2-cloro-7-(naftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (2,0 g, 3,6 mmol, 44,1 % de rendimiento). ES+APCI MS m/z 553,2 [M]+.

Etapa C: 4-(2-(((2S,4R)-1-(terc-butoxicarbonil)-4-fluoropirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropiridor3,4-d1pirimidin-4-il)-2-(cianometil)piperazin-1 -carboxilato de bencilo: En un vial se disolvió 4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (120 mg, 0,22 mmol) en dioxano (108 μl, 0,217 mmol) y se trató con carbonato de cesio (141 mg, 0,434 mmol) y (2S,4R)-1-(terc-Butoxicarbonil)-4-fluoro-2-hidroximetilpirrolidina (47,6 mg, 0,217 mmol). Después, el tubo se tapó y se calentó a 90 °C durante 12 horas. La reacción se filtró a través de papel GHF y se concentró. El residuo se purificó por cromatografía sobre sílice (0-6 % de Me0H en DCM). ES+APCI MS m/z 736,3 [M+H]+.

Etapa D: 2-(cianometil)-4-(2-(((2S,4R)-4-fluoropirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: A una solución de 4-(2-(((2S,4R)-1-(terc-butoxicarbonil)-4-fluoropirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (125 mg, 0,170 mmol) en DCM (170 μl, 0,170 mmol) se le añadió ácido trifluoroacético (195 μl, 2,55 mmol) y la reacción se agitó a TA durante 2 h. Los extractos orgánicos se lavaron con bicarbonato de sodio y la
capa acuosa se extrajo de nuevo con DCM. Las capas orgánicas combinadas se concentraron y se usaron sin purificación adicional. Es APCI MS m/z 636,3 [M+H]+.

Etapa E: 2-(cianometil)-4-(2-(((2S,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1 -carboxilato de bencilo: A una solución de 2-(cianometil)-4-(2-(((2S,4R)-4-fluoropirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (105 mg, 0,1652 mmol) en DCE (3303 μl, 0,1652 mmol) se le añadió formaldehído (124,1 μl, 1,652 mmol) (37 % en agua) seguido de triacetoxiborohidruro de sodio (175,0 mg, 0,8258 mmol). La mezcla se agitó vigorosamente a TA durante 2,5 h. La mezcla se trató con bicarbonato de sodio saturado (30 ml), se agitó durante 10 min y después se extrajo con DCM (3 * 10 ml). Las fases orgánicas combinadas se secaron sobre sulfato de sodio, se filtraron, se concentraron y se usaron tal cual en la siguiente reacción. ES+APCI MS m/z 650,3 [M+H]+.

Etapa F: 2-(4-(2-(((2S,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-vl)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: A una solución de 2-(cianometil)-4-(2-(((2S,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (85 mg, 0,13 mmol) en Et0H (1308 μl, 0,13 mmol) y THF (1308 μl, 0,13 mmol) se le añadió paladio (70 mg, 0,033 mmol) (tipo Degussa, 10 % en peso, 50 % de H20) y después se introdujo una atmósfera de H2 mediante vacío seguido de presión de globo. Después, la mezcla se agitó a temperatura ambiente durante una noche. Después, la mezcla se diluyó con Me0H y se filtró a través de papel GHF. Después, el filtrado se concentró para proporcionar 2-(4-(2-(((2S,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo. ES+APCI MS m/z 516,3 [M+H]+.

Etapa G: 2-(1-acriloil-4-(2-(((2S,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: A una suspensión de 2-(4-(2-(((2S,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (65 mg, 0,126 mmol) en DCM (1261 μl, 0,126 mmol) a 0 °C se le añadió cloruro de acriloílo (2521 μl, 0,252 mmol) (solución 0,1 M recién preparada
en DCM) seguido de trietilamina (35,1 |j E, 0,252 mmol). Después, la reacción se agitó a 0 °C durante 45 minutos. La reacción se concentró y se purificó por cromatografía de fase inversa (0-50 % de CAN:agua con 0,1 % de TFA) para proporcionar el compuesto del título. ES+APCI MS m/z 570,3 [M+H]+.
Ejemplo 221
2-(1-acriloil-4-(2-(((2S,4S)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Se sintetizó según el método del Ejemplo 220, Etapa C-G, usando (2S,4S)-4-fluoro-2-(hidroximetil)pirrolidin-1-carboxilato de terc-butilo en lugar de (2S,4R)-4-fluoro-2-(hidroximetil)pirrolidin-1-carboxilato de terc-butilo en la Etapa C. ES+APCI MS m/z 570,3 [M+H]+.
Ejemplo 222
2-(1-acriloil-4-(2-(((2S,4S)-4-metoxi-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

El compuesto del título se preparó siguiendo el Ejemplo 220 (Etapas C-G), sustituyendo (2S,4S)-2-(hidroximetil)-4-metoxipirrolidin-1-carboxilato de terc-butilo por (2S,4R)-1-(terc-Butoxicarbonil)-4-fluoro-2-hidroximetilpirrolidina en la Etapa C. ES+APCI MS m/z 582,3 [M+H]+.
Ejemplo 223
2-(1-acriloil-4-(2-(((S)-4-metilpiperazin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2 -il)acetonitrilo
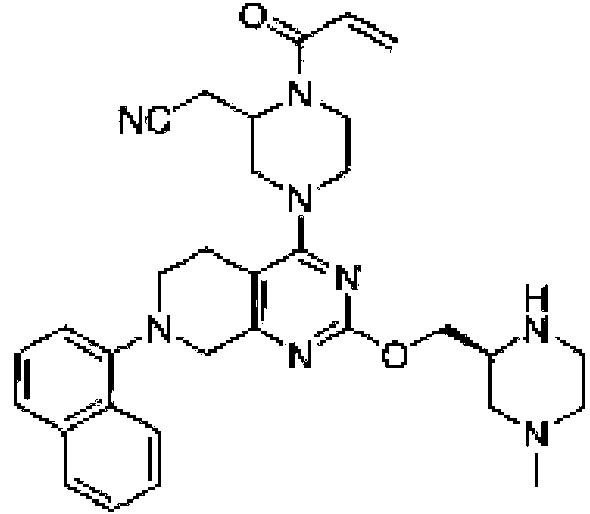
Etapas A-F: 2-í1-acr¡lo¡l-4-í2-ÍÍÍS)-4-met¡lp¡peraz¡n-2-¡l)metoxn-7-ínaftalen-1-¡l)-5.6.7.8-tetrahidrop¡r¡dor3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo: El compuesto del título se preparó s¡gu¡endo el Ejemplo 220 (Etapas C-G). sust¡tuyendo (S)-2-(h¡drox¡met¡l)-4-met¡lp¡peraz¡n-1-carbox¡lato de terc-butílo por (2S.4R)-1-(terc-Butox¡carbon¡l)-4-fluoro-2-h¡drox¡met¡lp¡rrol¡d¡na en la Etapa C. ES+APCI MS m/z 567.3 [M+H]+.
Ejemplo 224
2-(1-acr¡lo¡l-4-(2-(((R)-4-met¡lp¡peraz¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo

El compuesto del título se preparó s¡gu¡endo el Ejemplo 220 (Etapas C-G). sust¡tuyendo (R)-2-(h¡drox¡met¡l)-4-met¡lp¡peraz¡n-1-carbox¡lato de terc-but¡lo por (S)-2-(h¡drox¡met¡l)-4-met¡lp¡peraz¡n-1-carbox¡lato de terc-but¡lo en la Etapa C. ES+APCI MS m/z 567.3 [M+H]+.
Ejemplo 225
2-(1-acr¡lo¡l-4-(7-(5-fluoro-2-(tr¡fluorometox¡)fen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo
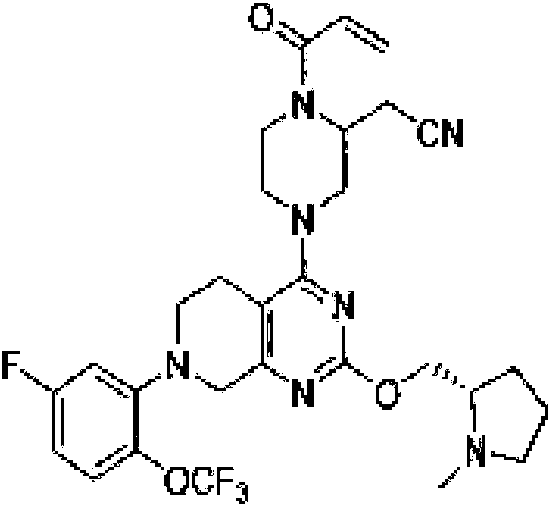
El compuesto del titulo se preparó como en el Ejemplo 210 Etapas E-G. sust¡tuyendo 2-bromo-4-fluoro-1-(tr¡fluorometox¡)benceno por 1-bromo-2-(tr¡fluoromet¡l)benceno en la etapa E. ES+APCI m S m/z 604.3 [M+H]+.
Ejemplo 226
2-(1-acr¡lo¡l-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(2-(tr¡fluoromet¡l)p¡r¡d¡n-3-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo

El compuesto del título se preparó como en el Ejemplo 210 Etapas E-G, sustituyendo 3-bromo-2-(trifluoromet¡l)p¡r¡d¡na por 1-bromo-2-(trifluorometil)benceno en la Etapa E. ES+APCI MS m/z 571,3 [M+H]+.
Ejemplo 227
2-(1-acr¡lo¡l-4-(7-(2-fluorofen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-i^piperazin^-i0acetonitrilo

El compuesto del título se preparó como en el Ejemplo 210 Etapas E-G, sustituyendo 1-bromo-2-fluorobenceno por 1- bromo-2-(trifluorometil)benceno en la etapa E. ES+APCI MS m/z 520,3 [M+H]+.
Ejemplo 228
2- (1-acr¡lo¡l-4-(7-(2,3-d¡met¡lfen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-i^piperazin^-i0acetonitrilo

El compuesto del título se preparó como en el Ejemplo 210 Etapas E-G, sustituyendo 1-bromo-2,3-dimetilbenceno por 1-bromo-2-(trifluorometil)benceno en la Etapa E. Es +APCI MS m/z 530,3 [M+H]+.
Ejemplo 229
2-(1-acr¡lo¡l-4-(7-(2-clorofen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-il)acetonitrilo

El compuesto del título se preparó como en el Ejemplo 210 Etapas E-G, sustituyendo 1-bromo-2-clorobenceno por 1 bromo-2-(trifluorometil)benceno en la etapa E. ES+ApCI MS m/z 536,2 [M]+.
Ejemplo 230
2-(1-acriloil-4-(7-(3-fluoro-2-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

El compuesto del título se preparó como en el Ejemplo 210 Etapas E-G, sustituyendo 1-bromo-3-fluoro-2-(trifluorometil)benceno por 1-bromo-2-(trifluorometil)benceno en la Etapa E. ES+APCI MS m/z 588,3 [M+H]+.
Ejemplo 231
2-(1-acriloil-4-(2-(3-hidroxipropoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 2-(cianometil)-4-(2-(3-hidroxipropoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: El compuesto del título se preparó como en el Ejemplo 220 Etapa C, sustituyendo propano-1,3-diol por (2S,4R)-1-(terc-Butoxicarbonil)-4-fluoro-2-hidroximetilpirrolidina. ES+APCI MS m/z 593,3 [M+H]+.

Etapa B: 4-(2-(3-((terc-butildimetilsilil)oxi)propoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo: A un vial se le añadió 2-(cianometil)-4-(2-(3-hidroxipropoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (77,6 mg, 0,131 mmol), que se disolvió en DCM (655 μl, 0,131 mmol). La mezcla se enfrió a 0 °C y después se añadió trietilamina (36,5 μl, 0,262 mmol) seguido de 4-(dimetilamino)-piridina (4,80 mg, 0,04 mmol). Después, se añadió cloruro de tercbutildimetilsililo (29,6 mg, 0,196 mmol) y la mezcla se agitó durante una noche mientras se calentaba a temperatura ambiente. La mezcla de reacción se vertió en una solución saturada de salmuera (5 ml) y la mezcla se extrajo dos veces con EtOAc (10 ml). Las fases orgánicas se secaron sobre sulfato de sodio, se filtraron y se concentraron. El producto de reacción bruto se usó tal cual. ES+APCI MS m/z 707,4 [M+H]+.

Etapa C: 2-(4-(2-(3-((terc-butildimetilsilil)oxi)propoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: A una solución de 4-(2-(3-((terc-butildimetilsilil)oxi)propoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (106 mg, 0,150 mmol) en Et0H (1499 μl, 0,150 mmol) y THF (1499 μl, 0,150 mmol) se le añadió paladio (79,8 mg, 0,0375 mmol) (tipo Degussa, 10 % en peso, 50 % de H20) y después se introdujo una atmósfera de H2 mediante vacío seguido de presión de globo. Después, la mezcla se agitó a temperatura ambiente durante una noche. Después, la mezcla se diluyó con Me0H y se filtró a través de papel GHF. Después, el filtrado se concentró to un sólido incoloro que se usó sin purificación adicional. ES+APCI MS m/z 573,3 [M+H]+.

Etapa D: 2-(1-acriloil-4-(2-(3-((terc-butildimetilsilil)oxi)proroxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: A una suspensión de 2-(4-(2-(3-((terc-butildimetilsilil)oxi)propoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (75 mg, 0,131 mmol) en DCM (1309 μl, 0,131 mmol) a -78 °C se le añadió cloruro de acriloílo (2619 μl, 0,262 mmol) (solución 0,1 M recién preparada en DCM)
seguido de trietilamina (36,5 μl, 0,262 mmol). Después, la reacción se agitó a 0 °C durante 30 minutos. La reacción se concentró y se usó bruta en la siguiente reacción. ES+APCI MS m/z 627,4 [M+H]+.

Etapa E: 2-(1-acr¡lo¡l-4-(2-(3-h¡drox¡propox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-il)acetonitr¡lo: A una solución de 2-(1-acriloil-4-(2-(3-((terc-butildimetilsilil)oxi)propoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo (90 mg, 0,14 mmol) en Dc M (1436 μl, 0,14 mmol) se le añadió ácido clorhídrico (4,0 M en 1,4-dioxano) (359 μl, 1,4 mmol) a 0 °C. La reacción se agitó durante 30 min, momento en el que se interrumpió con bicarbonato de sodio saturado y se extrajo en 10 % de IPA en CHCl3 (3 x 15 ml). Las capas orgánicas combinadas se concentraron y el residuo se purificó por cromatografía de fase inversa (0-95 % de ACN:H20 con 0,1 de TFA). ES+APCI MS m/z 513,2 [M+H]+.
Ejemplo 232
1-(4-(7-(3-cloro-2-fluoro-5-hidroxifenil)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona

El compuesto del título se preparó según el Ejemplo 1 Etapa C-F sustituyendo el Intermedio 25 por 4-(5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo y 1-bromo-3-cloro-2-fluoro-5-(metoximetoxi)benceno por 1-bromo-3-(metoximetoxi)naftaleno en la Etapa C. ES+APCI MS m/z 561,2 [M]+.
Ejemplo 233
2-(1-acriloil-4-(2-(((S)-1-etilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

2-(1-acr¡lo¡l-4-(2-(((S)-1-et¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo. Este compuesto se preparó s¡gu¡endo el Ejemplo 147 sust¡tuyendo (S)-(1-et¡lp¡rrol¡d¡n-2-¡l)metanol por N-Met¡l-L-prol¡nol por [(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metanol en la Etapa F. ES+APCI MS m/z 566.3 [M+H]+.
Ejemplo 234
2-((S)-1-acr¡lo¡l-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo

Etapa A: 4-(terc-But¡l) (R)-2-(h¡drox¡met¡l)p¡peraz¡n-1.4-d¡carbox¡lato de 1-benc¡lo: a una soluc¡ón b¡fás¡ca ag¡tada de (R)-1-Boc-3-h¡drox¡met¡lp¡peraz¡na (5.00 g. 23.1 mmol) y NaHCÜ3 (5.83 g. 69.4 mmol) en acetato de etilo (46.2 ml) y agua (46.2 ml) a 0 °C se le añad¡ó gota a gota cloroform¡ato de benc¡lo (4.95 ml. 34.7 mmol). La mezcla de reacc¡ón se ag¡tó a temperatura amb¡ente durante una noche. La mezcla de reacc¡ón se d¡luyó con EtOAc (50.0 ml) y la capa orgán¡ca se separó. se secó (Na2S04) y se concentró. El producto bruto se pur¡f¡có por cromatografía ultrarráp¡da
eluyendo con un gradiente de 10-50 % de EtOAc/hexanos para proporcionar el compuesto del título (7,62 g, 21,7 mmol, 94,1 %). ESI MS m/z 251,1 [M-Boc+H]+.
Etapa B.4-(terc-Butil) (R)-2-(((met¡lsulfon¡l)ox¡)met¡l)p¡peraz¡n-1,4-d¡carbox¡lato de 1-bencilo: A una solución de 4-(tercbutil) (R)-2-(hidroximetil)piperazin-1,4-dicarboxilato de 1-bencilo (1,69 g, 4,83 mmol) y trietilamina (1,01 ml, 7,25 mmol) en CH2Cl2 (32,2 ml) a 0 °C se le añadió gota a gota MsCl (0,561 ml, 7,25 mmol) puro y la mezcla resultante se agitó a TA durante 10 min. La mezcla de reacción se vertió en un embudo de decantación, se diluyó con acetato de etilo y después se lavó secuencialmente con HCl 1 N, agua, NaHC03 (sat.) y salmuera para proporcionar el compuesto del título (1,9 g, ~100 %, usado como material bruto en la siguiente etapa). ESI MS m/z 329,1 [M-Boc+H]+.
Etapa C. 4-(terc-Butil) (S)-2-(c¡anomet¡l)p¡peraz¡n-1,4-d¡carbox¡lato de 1-bencilo: Una solución de 4-(terc-butil) (R)-2-(((metilsulfonil)oxi)metil)piperazin-1,4-dicarboxilato de 1-bencilo (2,10 g, 4,90 mmol) y cianuro de sodio (0,480 g, 9,80 mmol) en DMA (49,0 ml) se calentó a 55 °C durante 1 día. La reacción se siguió de HPLC (método de 15 min).
La mezcla se repartió entre EtOAcisalmuera y la capa orgánica se lavó con salmuera (3 x), se secó sobre MgS04 y se concentró. El residuo se purificó por cromatografía ultrarrápida eluyendo con un gradiente de 0-100 % de EtOAc/hexanos para proporcionar el compuesto del título (1,40 g, 3,90 mmol, 79,5 %). Las capas acuosas se basificaron y se eliminaron en una corriente de desecho de cianuro. ESI MS m/z 260,1 [M-Boc+H]+.
Etapa D. Clorhidrato de (S)-2-(cianomet¡l)p¡peraz¡n-1-carbox¡lato de bencilo: Se puso 4-(terc-butil) (S)-2-(cianometil)piperazin-1,4-dicarboxilato de 1-bencilo (5,32 g, 14,8 mmol) en CH2Cl2 (25 ml), se añadió HCl (4,0 N en dioxano, 18,5 ml, 74,0 mmol) y la reacción se agitó a temperatura ambiente durante 1 d. La mezcla de reacción se concentró para proporcionar el compuesto del título (4,3 g, 99 %). ESI MS m/z 260,1 [M+H]+.
Etapa E. (S)-4-(4-((Benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-cloro-5.8-d¡h¡drop¡r¡do[3,4-d1p¡r¡m¡d¡n-7(6H)-carboxilato de terc-bencilo: Una solución de (S)-2-(cianometil)piperazin-1-carboxilato de bencilo (1,01 g, 3,89 mmol), 2,4-dicloro-5,6-dihidropirido[3,4-d1pirimidin-7(8H)-carboxilato de terc-butilo (1,18 g, 3,89 mmol) y DIEA (1,36 ml, 7,79 mmol) en DMS0 (19,5 ml) se calentó a 50 °C durante 1 día. La mezcla de reacción se repartió entre acetato de etilo y salmuera y los extractos orgánicos se separaron. La fase orgánica se lavó con salmuera (3 x), se secó sobre MgS04 y se concentró para proporcionar el compuesto del título (1,63 g, 3,09 mmol, 79,4 %). ESI MS m/z 527,2 [M+H]+.
Etapa F. 4-((S)-4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,8-d¡h¡drop¡r¡do[3,4-d1p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-butilo: En un matraz de fondo redondo de pared gruesa de 250 ml con un tapón a rosca de PTFE se añadió una solución de (S)-4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (1,64 g, 3,11 mmol) en dioxano (31,1 ml) y la mezcla se roció con argón. A la mezcla se le añadieron secuencialmente (S)-(1-metilpirrolidin-2-il)metanol (1,08 g, 9,34 mmol), Cs2C0a (3,04 g, 9,34 mmol) y catalizador Ruphos-Pd Gen3 (0,260 g, 0,311 mmol) en una atmósfera de argón y se roció durante 5 min más. La mezcla de reacción se tapó y se calentó a 100 °C durante 1 día. La mezcla de reacción se diluyó con acetato de etilo y los extractos orgánicos se lavaron con salmuera (2 x). La capa orgánica se secó sobre MgS04 y se concentró para dar un residuo que se purificó por cromatografía ultrarrápida eluyendo con 0-20 % de (Me0H 2 % de NH40H) en DCM para proporcionar el compuesto del título (1,42 g, 2,34 mmol, 75,3 %). ESI MS m/z 606,3 [M+H]+.
Etapa G. Sal bistrifluoroacetato de (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1p¡rim¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo: Una solución de 4-((S)-4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (38 mg, 0,063 mmol) en CH2CÍ2 (627 μl) y TFA (242 μl, 3,1 mmol) se agitó a temperatura ambiente durante 1 día. La mezcla de reacción se concentró y se usó en forma de la sal bis-TFA en la siguiente reacción (46 mg, 0,063 mmol, 100 %). ESI MS m/z 506,3 [M+H]+.
Etapa H. (S)-2-(C¡anomet¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡perazin-1-carbox¡lato de bencilo: Una suspensión de sal bistrifluoroacetato de (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (46 mg, 0,063 mmol), 1- bromonaftaleno (39,3 mg, 0,190 mmol) y Cs2C03 (61,9 mg, 0,190 mmol) en dioxano (633 μl) se roció con argón durante 5 min. A esta mezcla se le añadió Ruphos Pd Gen 3 (5,29 mg, 0,006 mmol) y la suspensión resultante se roció durante 1 minuto más con argón. El vial se tapó y se calentó a 100 °C durante 2 h. La mezcla de reacción se repartió entre EtOAc y agua y la capa acuosa se extrajo con EtOAc (3 x). Las capas orgánicas combinadas se secaron sobre MgS04 y se concentraron. El residuo se purificó por cromatografía ultrarrápida eluyendo con 5 % de Me0H/1 % de NH40H/CH202 ¡socrático para proporcionar el compuesto del título (31,9 mg, 0,050 mmol, 79,8 %). ESI MS m/z 632,3 [M+H]+.
Etapa I.2-((S)-4-(2-(((S)-1-Met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2- ¡l)acetonitr¡lo: A una solución de (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (31 mg, 0,049 mmol) en metanol (981 μl) y THF (981 μl) rociada con nitrógeno se le añadió Pd/C (10,4 mg, 0,00491 mmol) y la mezcla se agitó en una atmósfera a presión de globo de H2 durante 1 día. La mezcla de reacción se filtró a través de un filtro de jeringa de PTFE (25 mm) y el filtrado se concentró para proporcionar el compuesto del título (23,4 mg, 0,047 mmol, 95,8 %). ESI MS m/z 498,3 [M+H]+.
Etapa J. 2-((S)-1-Acrvlovl-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡perazin-2-¡l)aceton¡tr¡lo: A una solución de 2-((S)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (23,4 mg, 0,047 mmol) en CH2CI2 (470 μl) se le añadieron DIEA (41,1 μl, 0,235 mmol) y después cloruro de acriloílo (12 μl, 0,141 mmol). La mezcla resultante se agitó a temperatura ambiente durante 15 minutos. La mezcla de reacción se repartió entre EtOAc, K2C032 N y salmuera. La capa acuosa se extrajo con EtOAc (2 x). Las capas orgánicas combinadas se secaron sobre MgS04 y se concentraron. El residuo se purificó por cromatografía ultrarrápida eluyendo con 5 % de Me0H/1 % de NH40H en CH202 para proporcionar el compuesto del título (19,2 mg, 0,035 mmol, 74,0 %). ESI MS m/z 552,3 [M+H]+.
Ejemplo 235
2-((S)-1-((E)-4-(dimetilamino)but-2-enoil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A. 2-((S)-1-((E)-4-(d¡met¡lam¡no)but-2-eno¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5,6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡perazin-2-¡l)aceton¡tr¡lo: A una solución de 2-((S)-4-(2-(((S)-1 -metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (25 mg, 0,0502 mmol) del Ejemplo 234 Etapa I, ácido (2E)-4-(dimetilamino)but-2-enoico (13,0 mg, 0,100 mmol) y DIeA (43,9 μl, 0,251 mmol) en CH2Cl2 (502 μl) se le añadió HATU (28,7 mg, 0,0754 mmol) y la mezcla resultante se agitó a temperatura ambiente durante 1 hora. La mezcla de reacción se repartió entre EtOAc y salmuera. La capa acuosa se extrajo con EtOAc (2 x). Las capas orgánicas combinadas se secaron sobre MgS04 y se concentraron. El residuo se purificó por C18 preparativa eluyendo con 5-95 % de ACN/H20 0,1 % de TFA. Las fracciones deseadas se repartieron entre AE/K2C032 M. La capa acuosa se extrajo con EtOAc (2 x). Las capas orgánicas combinadas se secaron sobre MgS04 y se concentraron para proporcionar el compuesto del título (19,0 mg, 0,031 mmol, 62,1 %). ESI MS m/z 609,4 [M+H]+.
Ejemplo 236
2-((S)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(E-4-(piperidin-1 -il)but-2-enoil)piperazin-2-il)acetonitrilo

Etapa A. Se preparó 2-((S)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(E-4-(piperidin-1-il)but-2-enoil)piperazin-2-il)acetonitrilo según el Ejemplo 235, sustituyendo sal HCl del ácido (2E)-4-(1-piperidinil)-2-butenoico para proporcionar el compuesto del título (30 mg, 0,046 mmol, 92,0 %). ESI MS m/z 649,3 [M+H]+.
Ejemplo 237
2,2,2-trifluoroacetato de 2-((S)-1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Se sintetizó según Ejemplo 234 sustituyendo 1-bromo-2-(trifluorometil)benceno por 1-bromonaftaleno. ES+APCI MS m/z 570,3 [M+H]+.
Ejemplo 238
2-(1-acriloil-4-(7-(6-metil-1H-indazol-7-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 7-bromo-6-met¡l-2-((2-(tr¡metils¡l¡l)etox¡)met¡l)-2H-¡ndazol. Una solución de 7-bromo-6-metil-1H-indazol (200 mg, 0,948 mmol) en tetrahidrofurano (2 ml, 0,948 mmol) se enfrió con agitación en un baño de hielo. A la mezcla se le añadió poco a poco hidruro de sodio (45,5 mg, 1,14 mmol) y la reacción se agitó a 0 °C durante 1 hora. Luego, se añadió (2-(clorometoxi)etil)trimetilsilano (0,201 ml, 1,14 mmol) y la reacción se agitó durante 2 horas mientras se calentaba a temperatura ambiente. La mezcla se dividió entre EtoAc (20 ml) y agua (10 ml) y las capas se separaron. La capa orgánica se lavó con agua (2 * 10 ml) y salmuera (10 ml), se secó sobre Na2S04 y se evaporó al vacío. El producto se purificó por cromatografía sobre gel de sílice usando de 20 a 40 % de EtOAc/hexanos como eluyente para dar 7-bromo-6-metil-2-((2-(trimetilsilil)etoxi)metil)-2H-indazol (46 mg, 14 %) junto con el isómero 7-bromo-6-metil-1-((2-(trimetilsilil)etoxi)metil)-1H-indazol (214 mg, 66 %). ES+APCI MS m/z 341,1 [M]+.

Etapa B: 2-(C¡anomet¡l)-4-(7-(6-met¡l-2-((2-(tr¡met¡ls¡l¡l)etox¡)met¡l)-2H-¡ndazol-7-¡l)-2-(((S)-1-metilp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡mid¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo: Una mezcla de 2-(cianometil)-4-(2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (50 mg, 0,099 mmol), 7-bromo-6-metil-2-((2-(trimetilsilil)etoxi)metil)-2H-indazol (46 mg, 0,13 mmol), Cs2C03 (97 mg, 0,30 mmol), Ruphos Pd G3 (83 mg, 0,099 mmol) y dioxano (1 ml) se purgó con nitrógeno y se agitó a 70 °C durante 6 horas. La mezcla de reacción se enfrió a temperatura ambiente, se dividió entre EtOAc (20 ml) y agua (10 ml) y las
capas se separaron. La capa orgánica se lavó con agua (5 ml) y salmuera (5 ml), se secó sobre Na2SÜ4 y se evaporó al vacío. El producto se purificó por cromatografía sobre gel de sílice, Redisep de 24 g, usando de 2 a 20 % de Me0H/DCM como eluyente para dar un sólido incoloro (50 mg, 66 %). ES+APCI MS m/z 766,4 [M]+.

Etapa C: 2-(C¡anomet¡l)-4-(7-(6-met¡l-1H-¡ndazol-7-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡mid¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo: Se disolvió 2-(cianometil)-4-(7-(6-metil-2-((2-(trimetilsilil)etoxi)metil)-2H-indazol-7-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo puro (10 mg, 0,0131 mmol) (10 mg, 0,0131 mmol) en ácido trifluoroacético (1 ml, 13,1 mmol) y la solución de color amarillo-pardo se agitó a temperatura ambiente durante 1,5 horas. La mezcla de reacción se vertió en acetato de etilo (20 ml) y los extractos orgánicos se lavaron cuidadosamente con Na2C032 M (10 ml, 20 mmol), agua (2 * 3 ml) y salmuera (5 ml), se secaron sobre Na2S04 y se evaporaron al vacío para dar un sólido de color amarillento (8 mg, 96 %). ES+APCI MS m/z 636,3 [M+H]+.

Etapa D: 2-(1-acr¡lo¡l-4-(7-(6-met¡l-1H-¡ndazol-7-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-il)p¡peraz¡n-2-¡l)aceton¡tr¡lo: Una mezcla agitada de 2-(cianometil)-4-(7-(6-metil-1H-indazol-7-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (16 mg, 0,02517 mmol), metanol (1,5 ml, 0,02517 mmol), tetrahidrofurano (1,815 mg, 0,02517 mmol) y paladio sobre carbono (10 mg, 5 %, tipo Degussa E101 NO/W ) se desgasificó y se agitó en una atmósfera de hidrógeno durante 1 hora. La mezcla se filtró a través de Celite (2 ml) y el celite se lavó con THF (3 * 3 ml). Los extractos orgánicos combinados se concentraron al vacío hasta ~0,5 ml, se disolvieron en DCM (5 ml) y se enfriaron a -30 °C con agitación en un baño de Et0H-H20-C02. Luego, se añadió trietilamina (0,02 ml, 5 eq.) seguido de cloruro de acriloílo (0,006134 ml, 0,07550 mmol) y la mezcla de reacción se agitó 1 min a -30 °C. La reacción se interrumpió con NH40H (0,03 ml) y se concentró al vacío. El residuo se dividió entre NaHC031 M (3 ml) y EtOAc (15 ml) y las capas se separaron. La capa orgánica se lavó con agua (3 ml) y salmuera (3 ml), se secó sobre Na2S04 y se evaporó al vacío. El producto se purificó por cromatografía sobre gel de sílice, Redisep de 12 g, usando de 10 a 20 % de Me0H/DCM 0,2 % de NH40H como eluyente para dar un sólido incoloro (5,96 mg, 43 %). ES+APCI MS m/z 556,3 [M+H]+.
Ejemplo 239
2-(1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-4-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo

Se sintetizó según Ejemplo 210, Etapas E-G usando bromhidrato de 4-bromo-3-(trifluorometil)piridina en lugar de 1-bromo-2-(trifluorometil)benceno en la Etapa E. ES+APCI MS m/z 571,3 [M+H]+.
Ejemplo 240
2-(1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(2-(2,2,2-trifluoroetil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Se sintetizó según Ejemplo 210, Etapas E-G usando 1-bromo-2-(2,2,2-trifluoroetil)benceno en lugar de 1-bromo-2-(trifluorometil)benceno en la Etapa E. ES+APCI MS m/z 584,3 [M+H]+.
Ejemplo 241
2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-2-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo

Etapa A: En un vial, se disolvió 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (100 mg, 0,198 mmol) en dioxano (98,9 μl, 0,198 mmol) y se trató con carbonato de cesio (129 mg, 0,396 mmol) y 2-Fluoro-3-(trifluorometil)piridina (163 mg, 0,989 mmol). Después, el tubo se tapó y se calentó a 90 °C durante 12 h. La reacción se filtró a través de papel GHF y se concentró. El residuo se purificó por cromatografía sobre sílice (0-12 % de Me0H en DCM).

Etapa B: Se sintetizó según Ejemplo 210, Etapas F-G usando 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-2-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo en lugar de 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo en la Etapa F. e S+Ap CI MS m/z 571,3 [M+H]+.
Ejemplo 242
2-(1-(but-2-inoil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

A 0 °C, a un MFR de 25 ml que contenía N,N-dimetilformamida (603 gl, 0,06 mmol) se le añadieron 2-(4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (30 mg, 0,06 mmol) y trietilamina (12,2 mg, 0,12 mmol). La mezcla de reacción se agitó vigorosamente mientras se añadía en una porción ácido 2-butinoico (6,08 mg, 0,07 mmol). Después, a la mezcla en agitación se le añadió anhídrido cíclico del ácido 1-propanofosfónico (26,9 gl, 0,09 mmol). La reacción se dejó en agitación durante 2 h a 0 °C. Se añadió agua y la reacción se extrajo con EtOAc (2 x 25 ml). Las capas orgánicas se lavaron con LiCl saturado, NaCl y agua (10 ml cada lavado). Se secaron y se concentraron hasta un sólido que se purificó por HPLC prep. de fase inversa (5 95 % de ACN:H20, con 0,1 % de TFA) para proporcionar el compuesto del título.
Ejemplo 243
2-[4-[2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-7-[5-(trifluorometil)-1H-indazol-4-il]-6,8-dihidro-5H pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo


Etapa A: 2-(cianometil)-4-[2-metilsulfanil-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-1-carboxilato de tere-butilo: A una solución de 2-(cianometil)-4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de tere-butilo (1,05 g, 2,60 mmol), 2-[[4-bromo-5-(trifluorometil)indazol-1-il]metoxi]etil-trimetil-silano (1,13 g, 2,86 mmol), RuPhos (484 mg, 1,04 mmol) y Cs2C03 (2,11 g, 6,49 mmol) en tolueno (20 ml) se le añadió Pd2(dba)3 (475 mg, 519 umol) en una atmósfera de N2 y la suspensión se desgasificó al vacío y se purgó varias veces con N2. La mezcla se calentó a 90 °C y se agitó a 90 °C durante 7 horas. La mezcla de reacción se filtró y la torta de filtro se lavó con EtOAc (3 x 20 ml). Las capas orgánicas combinadas se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: AE de 30:1 a 4:1) y después con una columna ultrarrápida de fase inversa (ACN: 100 %) para dar 2-(cianometil)-4-[2-metilsulfanil-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de tere-butilo (850 mg, 1 ,12 mmol, 43,3 % de rendimiento, 95,0 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 719,5 [M+H]+.
Etapa B: 2-(cianometil)-4-[2-metilsulfinil-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il)piperazin-1 -carboxilato de tere-butilo: A una solución de 2-(cianometil)-4-[2-metilsulfanil-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (850 mg, 1,18 mmol) en DCM (17 ml) se le añadió m-CpBA (240 mg, 1,18 mmol, 85,0 % de pureza) a 0 °C y se agitó a 0 °C durante 1 hora. La mezcla de reacción se inactivó con Na2S203 saturado (15 ml) a 0 °C, se separó, después se diluyó con agua (10 ml) y se extrajo con EtOAc (30 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0,1 % de AF) = 80 %) para dar 2-(cianometil)-4-[2-metilsulfinil-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de tere-butilo (730 mg, 944 umol, 79,8 % de rendimiento, 95,0 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 735,5 [M+H]+.
Etapa C: 2-(cianometil)-4-[2-(((2R)-1-metilpirrolidin-2-il1metoxi1-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1 -carboxilato de tere-butilo: A una solución de 2-(cianometil)-4-[2-metilsulfinil-7-[5-(trifluorometil)-1-(2-trimetilsityletoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (300 mg, 408 umol) y [(2R)-1-metilpirrolidin-2-il]metanol (94,0 mg, 816 umol) en tolueno (6 ml) se le añadió t-Bu0Na (58,8 mg, 612 umol) a 15 °C y se agitó a 15 °C durante 0,5 horas. A la mezcla se le añadió EtOAc (15 ml) y agua (5 ml) y después se extrajo con EtOAc (3 x 10 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0,1 % de AF) = 54 %) para dar 2-(cianometil)-4-[2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (216 mg, 261 umol, 64,0 % de rendimiento, 95,0 % de pureza) en forma de un sólido de color pardo. ESI MS m/z 786,6[M+H]+.
Etapa D: 2-(4-[2-([(2R)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-7-[5-(tr¡fluoromet¡l)-1-(2-tr¡met¡ls¡l¡letox¡met¡l)¡ndazol-4-¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡llaceton¡tr¡lo: A una soluc¡ón de 2-(c¡anomet¡l)-4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-7-[5-(tr¡fluoromet¡l)-1-(2-tr¡met¡ls¡l¡letox¡met¡l)¡ndazol-4-¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de terc-but¡lo (190 mg.242 umol) y 2,6-d¡met¡lp¡r¡d¡na (311 mg.2.90 mmol.338 ul) en DCM (4 ml) se le añad¡ó TMS0Tf (645 mg. 2.90 mmol. 524 ul) a 0 °C y se agitó a 20 °C durante 1 hora. La mezcla de reacc¡ón se ¡nact¡vó con Me0H (1 ml) y después se concentró al vacío. El res¡duo se pur¡f¡có por columna ultrarráp¡da de fase ¡nversa (ACN/agua (0.1 % de AF) = 40 %). Se obtuvo 2-[4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡]-7-[5-(tr¡fluoromet¡l)-1-(2-tr¡met¡ls¡l¡letox¡met¡l)¡ndazol-4-¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-2-¡llaceton¡tr¡lo (85 mg. 123 umol.
50.8 % de rend¡m¡ento. 99.0 % de pureza) en forma de un sólido de color blanco. ESI MS m/z 686.5 [M+H]+.
Etapa E: 2-[4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-7-[5-(tr¡fluoromet¡l)-1-(2-tr¡met¡ls¡l¡letox¡met¡l)¡ndazol-4-¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡ll-1-prop-2-eno¡l-p¡peraz¡n-2-¡llaceton¡tr¡lo A una soluc¡ón de 2-[4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-7-[5-(tr¡fluoromet¡l)-1-(2-tr¡met¡ls¡l¡letox¡met¡l)¡ndazol-4-¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡llp¡peraz¡n-2-¡l]aceton¡tr¡lo (85.0 mg. 124 umol). DIEA (96.1 mg. 744 umol. 130 ul) en DCM (2 ml) se le añad¡ó prop-2-enoato de prop-2-enoílo (23.4 mg. 186 umol) a 0 °C y la mezcla se agitó a 20 °C durante 1 hora. La mezcla de reacc¡ón se ¡nact¡vó con agua (2 ml). La mezcla resultante se extrajo con DCM (3 x 5 ml). Las capas orgán¡cas comb¡nadas se secaron sobre Na2S04 anh¡dro. se f¡ltraron y se concentraron al vacío. El res¡duo se pur¡f¡có por columna ultrarráp¡da de fase ¡nversa (ACN/agua (0.1 % de a F) =48 %) para dar 2-[4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-[5-(tr¡fluoromet¡l)-1-(2-tr¡met¡ls¡l¡letox¡met¡l)¡ndazol-4-¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡ll-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (45 mg.
54.7 umol. 44.2 % de rend¡m¡ento. 90.0 % de pureza) en forma de un sólido de color blanco. 1H RMN (400 MHz. cloroformo-d) 6 = 8.16 (s. 1H). 7.71 (d. J = 8.8 Hz. 1H). 7.52 (d. J = 8.8 Hz. 1H). 6.59 (s a. 1H). 6.45 - 6.36 (m. 1H). 5.84 (d a. J = 10.4 Hz. 1H). 5.76 (s.2H). 5.12 (s. 1H). 4.44 - 4.28 (m. 3H). 4.21 - 4.08 (m. 2H). 3.98 (d a. J = 12.0 Hz. 1H). 3.63 - 3.54 (m. 2H). 3.48 - 3.39 (m. 2H). 3.32 (s a. 1H). 3.10 (s a. 2H). 2.99 - 2.86 (m. 2H). 2.80 (s a. 2H). 2.73 - 2.58 (m. 2H).
2.49 (s. 3H). 2.28 (s a. 1H). 2.05 (s a. 1H). 1.77 (s a. 4H). 0.94 - 0.88 (m. 2H). - 0.04 (s. 9H).
Etapa F: 2-[4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-7-[5-(tr¡fluoromet¡l)-1H-¡ndazol-4-¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡ll-1-prop-2-eno¡l-p¡peraz¡n-2-¡llaceton¡tr¡lo A una soluc¡ón de 2-[4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-[5-(tr¡fluoromet¡l)-1-(2-tr¡met¡ls¡l¡letox¡met¡l)¡ndazol-4-¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡ll-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (45 mg.
60.8 umol) en DCM (0.15 ml) se le añad¡ó TFA (208 mg. 1.82 mmol. 135 ul) y la mezcla se ag¡tó a 20 °C durante 1 hora. La mezcla de reacc¡ón se bas¡f¡có con NaHC03 saturado (2 ml) a pH = 8. La mezcla resultante se extrajo con DCM: Me0H (10:1) (3 x 5 ml). Las capas orgán¡cas comb¡nadas se secaron sobre Na2S04 anh¡dro. se f¡ltraron y se concentraron al vacío.
El res¡duo se purificó por HPLC prep. (columna: Phenomenex Synerg¡ C18150 * 30 mm * 4 um;fase móv¡l: [agua (0.225 % de AF)-ACN];% de B: 10 %-40 %.10.5 m¡n) para dar 2-[4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-[5-(trifluoromet¡l)-1H-¡ndazol-4-¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡ll-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (8.2 mg. 12.0 umol. 19.8 % de rend¡m¡ento. 96.2 % de pureza. Af ) en forma de un ace¡te de color amarillo. ESI MS m/z 610.4 [M+H]+.
Ejemplo 244
2-[4-[7-(8-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡lp¡peraz¡n-2 -¡l]aceton¡tr¡lo


Etapa A: 2-(cianometil)-4-[7-(8-isoquinolil)-2-[[(2[[(2S-1 metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-il]piperazin-1-carboxilato de tere-butilo: A una solución de 2-(cianometil)-4-[2-[(1-metilpirrolidin-2-il)metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (210 mg, 445 umol), 8-bromoisoquinolina (185 mg, 891 umol), RuPhos (41,6 mg, 89,1 umol) y CS2C03 (435 mg, 1,34 mmol) en tolueno (4,5 ml) se le añadió Pd2(dba)3 (40,8 mg, 44,5 umol) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con N2. La mezcla se agitó en una atmósfera de N2 a 90 °C durante 12 horas. La mezcla se filtró y la torta de filtro se lavó con EtOAc (3 * 10 ml). Las capas orgánicas combinadas se concentraron al vacío. El residuo se purificó por columna ultrarrápida de fase inversa (AcN/agua (0,1 % de AF) = 20 %) para dar 2-(cianometil)-4-[7-(8-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de tere-butilo (160 mg, 240 umol, 54,0 % de rendimiento, 90,0 % de pureza) en forma de un sólido de color pardo. ESI MS m/z 599,4 [M+H]+.
Etapa B: 2-[4-[7-(8-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il)metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-¡ n piperazin-2-i n acetonitrilo: Una solución de 2-(cianometil)-4-[7-(8-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (190 mg, 317 umol) y TFA (724 mg, 6,35 mmol, 470 ul) se agitó a 20 °C durante 1 hora. La mezcla de reacción se concentró al vacío para dar 2-[4-[7-(8-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]acetonitrilo (230 mg, bruto, 2TFA) en forma de un aceite de color pardo.
Etapa C: 2-[4-[7-(8-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il)metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-prop-2-enoil-piperazin-2-i n acetonitrilo: A una solución de 2-[4-[7-(8-¡soqu¡nol¡l)-2-[2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (230 mg, 316 umol, 2t Fa ) y DIEA (409 mg, 3,17 mmol, 551 ul) en DCM (5 ml) se le añadió prop-2-enoato de prop-2-enoílo (47,9 mg, 380 umol) a 0 °C y después la mezcla se agitó a 20 °C durante 1 hora. A la mezcla se le añadió agua (5 ml). La mezcla resultante se extrajo con DCM (3 * 5 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi C18 150 * 25 * 10 um; fase móvil: [agua (0,225 % de AF)-ACN];% de B: 1 %-20 %,10 min) para dar 2-[4-[7-(8-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-pirido[3,4-d1pirimidin-4-il1-1 -prop-2-enoil-piperazin-2-il]acetonitrilo (12,5 mg, 19,7 umol, 6,23 % de rendimiento, 94,5 % de pureza, AF) en forma de un sólido de color amarillo. ESI MS m/z 553,2 [M+H]+.
Ejemplo 245
2-[4-[7-(7-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo

Etapa A: trifluorometanosulfonato de (7-metil-3,4-dihidronaftalen-1 -ilo): A una solución de 7-metiltetralin-1 -ona (2,00 g, 12,5 mmol) y DIPEA (4,84 g, 37,5 mmol, 6,52 ml) en DCM (30 ml) se le añadió gota a gota Tf20 (5,28 g, 18,7 mmol, 3,09 ml) a -5 °C. La mezcla se agitó a 20 °C durante 3 horas. Después de que se completara, la mezcla se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP/AE 1/0 a 100/1) para dar trifluorometanosulfonato de (7-metil-3,4-dihidronaftalen-1-ilo) (1,53 g, 4,45 mmol, 35,6 % de rendimiento, 85,0 % de pureza) en forma de un aceite de color amarillo. 1H RMN (400 Mhz, cloroformo-d) ó = 7,17 (s, 1H), 7,11 - 7,05 (m, 2H), 6,01 (t, J = 4,8 Hz, 1H), 2,83 (t, J = 8,0 Hz, 2H), 2,50 (dt, J = 4,8, 8,0 Hz, 2H), 2,36 (s, 3H).
Etapa B: trifluorometanosulfonato de (7-metil-1-naftilo): Una mezcla de trifluorometanosulfonato de (7-metil-3,4-dihidronaftalen-1-ilo) (1,40 g, 4,79 mmol) y DDQ (2,17 g, 9,58 mmol) en dioxano (28 ml) se agitó a 105 °C durante 12 horas. Después de que se completara, la mezcla se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP/AE 1/0 a 100/1) para dar trifluorometanosulfonato de (7-metil-1 -naftilo) (1,10 g, 3,41 mmol, 71,2 % de rendimiento, 90,0 % de pureza) en forma de un aceite de color amarillo. 1H RMN (400 MHz, cloroformod) ó = 7,87 - 7,79 (m, 3H), 7,49 - 7,38 (m, 3H), 2,59 (s, 3H).
Etapa C: 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de terc-butilo: Una mezcla de 7-bencil-2,4-dicloro-6,8-dihidro-5H-pirido[3,4-d]pirimidina (10 g, 34,0 mmol), 2-piperazin-2
ilacetonitrilo (10,1 g, 51,0 mmol, 2 HCl) y DIEA (17,6 g, 136 mmol, 23,7 ml) en dioxano (100 ml) se agitó a 50 °C durante 2 horas. Se añadió Boc20 (14,8 g, 68,0 mmol, 15,6 ml) y la mezcla se agitó a 50 °C durante 2 horas. La mezcla se concentró al vacío y el residuo se diluyó con acetato de etilo (500 ml). La capa orgánica separada se lavó con agua (1 x 300 ml) y salmuera (1 x 200 ml), se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se trituró con acetato de etilo (100 ml). El precipitado se filtró y la torta de filtro se lavó con acetato de etilo (50 ml) y se secó al vacío para dar un sólido de color gris. El filtrado se concentró al vacío. El residuo se purificó por cromatografía de columna sobre gel de sílice (éter de petróleo/acetato de etilo 100/1 a 1/2). Las fracciones deseadas se recogieron y se concentraron al vacío para dar un sólido que después se combinó con el sólido anterior para dar 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (15 g, 29,10 mmol, 85,6 % de rendimiento, 93,7 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 483,1 [M+H]+.
Etapa D: 4-[7-bencil-2-[(1-metilpirrolidin-2-il)metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato: A una solución de [(2S)-1-metilpirrolidin-2-il]metanol (1,19 g, 10,4 mmol, 1,23 ml, 2,5 eq.) en THF (30,0 ml) se le añadió NaH (331 mg, 8,28 mmol, 60 % de pureza en aceite mineral, 2,00 eq.) a 0 °C y la mezcla se agitó a 0 °C durante 0,5 h. A la mezcla se le añadió 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (2,00 g, 4,14 mmol, 1,00 eq.) y la mezcla se agitó a 80 °C durante 4 horas. La mezcla se vertió en agua enfriada con hielo (100 ml) y se extrajo con acetato de éter (3 x 150 ml). Las capas orgánicas se lavaron con una solución saturada de cloruro de sodio (1 x 200 ml), se secaron sobre Na2S04, se filtraron y se concentraron al vacío para dar 4-[7-bencil-2-[(1-metilpirrolidin-2-il)metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (2,86 g, bruto) en forma de un aceite de color negro que se usó en la siguiente etapa sin purificación adicional. ESI MS m/z 562,3 [M+H]+.
Etapa E: 2-(c¡anomet¡l)-4-[2-[(1-met¡lp¡rrol¡d¡n-2-¡l)metox¡1-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carboxilato de ferc-butilo: Se burbujeó NH3 en metanol (70,0 ml) a -78 °C durante 30 minutos. A la mezcla se le añadieron 4-[7-bencil-2-[(1-metilpirrolidin-2-il)metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (2,00 g, bruto) y Pd/C (0,20 g, 10 % de pureza). La mezcla se agitó a 40 °C durante 24 horas en una atmósfera de H2 a 103,42 kPa (15 psi). La mezcla se filtró y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (AF, 0,1 %)/acetonitrilo]. Las fracciones deseadas se ajustaron a pH >7 con una solución saturada de bicarbonato de sodio (5,00 ml) y la capa acuosa se extrajo con acetato de éter (3 x 100 ml). Las capas orgánicas combinadas se lavaron con salmuera (1 x 100 ml), se secaron sobre Na2S04, se filtraron y se concentraron al vacío para dar 2-(cianometil)-4-[2-[(1-metilpirrolidin-2-il)metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo (0,40 g, 845 umol, dos etapas 29,4 % de rendimiento) en forma de un sólido de color amarillo. ESI MS m/z 472,2 [M+H]+.
Etapa F: 2-(c¡anomet¡l)-4-[2-[(1-met¡lp¡rrol¡d¡n-2-¡l)metox¡1-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carboxilato de ferc-butilo: Una solución de 2-(cianometil)-4-[2-[(1-metilpirrolidin-2-il)metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo (185 mg, 392 umol), trifluorometanosulfonato de (7-metil-1 -naftilo) (159 mg, 549 umol), Pd2(dba)3 (35,9 mg, 39,2 umol), RuPhos (36,6 mg, 78,5 umol) y Cs2C03 (320 mg, 981 umol) en tolueno (30 ml) se desgasificó con N2 y después se calentó a 90 °C durante 12 horas en una atmósfera de N2. Después de que se completara, la mezcla se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de NH3^H20)/acetonitrilo] para dar 2-(cianometil)-4-[7-(7-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (180 mg, 283 umol, 72,1 % de rendimiento, 96,1 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 612,6 [M+H]+.
Etapa G: 2-[4-[7-(7-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-¡l]aceton¡tr¡lo: Una solución de 2-(cianometil)-4-[7-(7-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (180 mg, 294 umol) en TFA (671 mg, 5,88 mmol, 436 ul) se agitó a 20 °C durante 1 hora. Después de que se completara, la mezcla se concentró al vacío para dar 2-[4-[7-(7-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (0,37 g, bruto, TFA) en forma de un aceite de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional.
Etapa H: 2-[4-[7-(7-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo: A una solución de 2-[4-[7-(7-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (189 mg, bruto, TFA) y DIEA (976 mg, 7,55 mmol, 1,32 ml) en DCM (4 ml) se le añadió prop-2-enoato de prop-2-enoílo (38,1 mg, 302 umol) gota a gota a 0 °C. La mezcla se agitó a 25 °C durante 1 hora. Después de que se completara, la reacción se interrumpió con Me0H (0,5 ml), se diluyó con agua (1 ml) y se extrajo con DCM (3 x 5 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi C18150 * 30 mm * 4 um; fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 15 % - 45 %,10,5 min) para dar 2-[4-[7-(7-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (13,2 mg, 21,3 umol, dos etapas 14,2 % de rendimiento, 98,4 % de pureza, AF) en forma de un sólido de color amarillo. ESI MS m/z 566,5 [M+H]+.
Ejemplo 246
2-[4-[7-(4-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo.

Se sintetizó según el método del Ejemplo 147 sustituyendo 1-bromo-4-fluoronaftaleno por 1-bromonaftaleno en la etapa H. ESI MS m/z 570,5 [M+H]+.
Ejemplo 247
2-(1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(quinolin-8-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Se sintetizó según el método del Ejemplo 147 sustituyendo 8-bromoquinolina por 1-bromonaftaleno en la Etapa H. ESI MS m/z 553,2 [M+H]+.
Ejemplo 248
(S)-1-(4-(7-(5-isopropil-1H-indazol-4-il)-2-((1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona


Etapa A: 2-(2-bromo-4-fluoro-fenil)propan-2-ol: A una solución de 1-(2-bromo-4-fluoro-fenil)etanona (10 g, 46,1 mmol) en THF (100 ml) se le añadió MeMgBr (en éter) (3 M, 46,08 ml) a -50 °C. La mezcla se calentó a 0 °C y se agitó durante 3 horas. La mezcla se inactivó con una solución acuosa saturada de cloruro de amonio (10 ml) a -50 °C. La mezcla se calentó a 15 °C y después se diluyó con acetato de etilo (500 ml). La capa orgánica separada se lavó con agua (1 * 500 ml) y salmuera (1 * 500 ml), se secó sobre sulfato de sodio, se filtró y se concentró al vacío para dar 2-(2-bromo-4-fluoro-fenil)propan-2-ol (10 g, 36,5 mmol, 79,2 % de rendimiento, 85 % de pureza) en forma de un aceite incoloro.
Etapa B: 2-bromo-4-fluoro-1-isopropil-benceno: A una solución de 2-(2-bromo-4-fluoro-fenil)propan-2-ol (9 g, 38,6 mmol) en diclorometano (100 ml) se le añadieron Et3SiH (8,98 g, 77,2 mmol, 12,3 ml) y TFA (65,1 g, 571 mmol, 42,3 ml). La mezcla se agitó a 15 °C durante 1 hora y después se calentó a 50 °C durante 1 hora. La mezcla de reacción se concentró al vacío y después se diluyó con acetato de etilo (100 ml). La capa acuosa pH se ajustó a pH = 7 mediante la adición de una solución acuosa saturada de NaHCÜ3 y se extrajo con acetato de etilo (50 ml * 3). La fase orgánica combinada se lavó con salmuera (30 ml), se secó con Na2SÜ4 anhidro, se filtró y se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (éter de petróleo/acetato de etilo = 1/0 a 100/1). Las fracciones deseadas se recogieron y se concentraron al vacío. Se obtuvo 2-bromo-4-fluoro-1-isopropil-benceno (7 g, 32.2 mmol, 83,5 % de rendimiento) en forma de un aceite incoloro. 1H RMN (400 MHz, cloroformo-d) ó = 7,36-7,27 (m, 2H), 7,09 - 7,02 (m, 1H), 3,46-3,31 (m, 1H), 1,28 (d, J = 6,8 Hz, 6H).
Etapa C: 2-bromo-6-fluoro-3-isopropil-benzaldehído: A una mezcla de 2-bromo-4-fluoro-1-isopropil-benceno (7 g, 32.2 mmol en THF (100 ml) se le añadió LDA (2 M en tolueno, 24,2 ml) a -70 °C en una atmósfera de nitrógeno. La mezcla se agitó durante 0,5 horas y después se añadió DMF (7,07 g, 96,7 mmol, 7,44 ml). La mezcla se agitó a -70 °C durante 2 horas. La reacción se interrumpió con agua (100 ml) y se extrajo con acetato de etilo (2 * 100 ml). Las capas orgánicas combinadas se lavaron con salmuera (50 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna (SiO2, EP/AE = 1\0 a 10\1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar 2-bromo-6-fluoro-3-isopropil-benzaldehído (2,4 g, 9,79 mmol, 30,4 % de rendimiento) en forma de un aceite de color amarillo. 1H RMN (400 MHz, cloroformo-d) ó = 10,41 (s, 1H), 7,48 (dd, J = 5,6, 8,8 Hz, 1H), 7,13(t, J = 9,2, 1H), 3,57 - 3,46 (m, 1H), 1,26 (d, J = 7,2 Hz, 6H).
Etapa D: 4-bromo-5-isopropil-1 H-indazol: A 2-bromo-6-fluoro-3-isopropil-benzaldehído (2,4 g, 9,79 mmol) en DMS0 (3 ml) se le añadió N2H4^H20 (15,4 g, 302 mmol, 15 ml, 98 % de pureza) y la mezcla se calentó a 110 °C y se agitó durante 16 horas. La mezcla de reacción se vertió en H20 (10 ml) y se extrajo con acetato de etilo (20 ml x 3). La fase orgánica se lavó con salmuera (10 ml), se secó sobre Na2S04 anhidro y se concentró al vacío para dar el residuo. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 1/0 a 5/1) para dar 4-bromo-5-isopropil-1 H-indazol (650 mg, 2,72 mmol, 27,8 % de rendimiento) en forma de un sólido de color amarillo. 1H RMN (400 MHz, cloroformo-d) 6 = 10,24 (s a, 1H), 8,08 (d, J = 0,8 Hz, 1H), 7,43 (d, J = 8 ,8 Hz, 1H), 7,34 (d, J = 8 ,8 Hz, 1H), 3,56 (td, J = 6 ,8 , 13,6 Hz, 1H), 1,29 (d, J = 6 ,8 Hz, 6 H).
Etapa E: 4-bromo-5-isopropil-1-tetrahidropiran-2-il-indazol: A una solución de 3,4-dihidro-2H-pirano (457 mg, 5,44 mmol, 497 ul, 2 eq.) en diclorometano (15 ml) se le añadieron T s0 H ^0 (51,7 mg, 271 umol) y 4-bromo-5-isopropil-1 H-indazol (650 mg, 2,72 mmol). La mezcla se agitó a 20 °C durante 3 horas. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (20 ml x 2). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 1 /0 a 5/1) y se purificó adicionalmente por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmicoyacetonitrilo] para dar 4-bromo^-isopropiM-tetrahidropiran^-il-indazol (170 mg, 494 umol, 18,2 % de rendimiento, 94 % de pureza) en forma de un aceite de color amarillo. ESI MS m/z 323,0 [M+H]+.
Etapa F: 4-[7-(5-¡soprop¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de tere-butilo: Una mezcla de 4-bromo-5-¡soprop¡l-1-tetrah¡drop¡ran-2-il-indazol (136 mg, 420 umol), 4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-i^piperazin-1-carboxilato de tere-butilo (140 mg, 323 umol), RuPhos (15,1 mg, 32,4 umol), RuPhos-Pd-G2 (25,1 mg, 32,4 umol) y t-Bu0Na (77,7 mg, 809 umol) en tolueno (3 ml) se desgasificó y se purgó 3 veces con N2 y la mezcla se agitó a 90 °C durante 12 horas en una atmósfera de N2. La mezcla se diluyó con acetato de etilo (20 ml) y se lavó con agua (20 ml). Las capas orgánicas se lavaron con salmuera (10 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiO2, DCM/Me0H = 100/1 a 10/1) y las fracciones deseadas se recogieron y se concentraron al vacío para dar 4-[7-(5-¡soprop¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-i^piperazin-1-carboxilato de tere-butilo (70 mg, 96,5 umol, 29,8 % de rendimiento, 93 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 675,3 [M+H]+.
Etapa G: 7-(5-¡soprop¡l-1H-¡ndazol-4-¡l)-2-(((2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-4-p¡peraz¡n-1-¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-dlpirimidina: A una solución de 4-[7-(5-¡soprop¡l-1-tetrah¡drop¡ran- 2-¡l-¡ndazol-4-¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (40 mg, 59,3 umol) en diclorometano (100 ul) se le añadió TFA (101 mg, 889 umol, 65,8 ul). La mezcla se agitó a 25 °C durante 1 hora. La mezcla se concentró al vacío para dar 7-(5-isopropil-1 H-indazol-4-il)-2-[[(2S)-1 -met¡lp¡rrol¡d¡n-2-¡l]metox¡]-4-p¡peraz¡n-1 -il-6 ,8 -dihidro^H-pirido^^-dprimidina (42 mg, 58,4 umol, 98,6 % de rendimiento, 2 TFA) en forma de un aceite de color pardo que se usó en la siguiente etapa directamente sin purificación. ESI MS m/z 491,4 [M+H]+.
Etapa H: 1-[4-[7-(5-¡soprop¡l-1H-¡ndazol-4-¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-¡l1prop-2-en-1-ona: A una mezcla de 7-(5-¡soprop¡l-1H-¡ndazol-4-¡l)-2-[[(2S)- 1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-4-p¡peraz¡n-1-¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡na (42 mg, 58,4 umol) y DiEa (75,5 mg, 584 umol, 102 ul) en diclorometano (2 ml) se le añadió una solución de prop-2-enoato de prop-2-enoílo (7,37 mg, 58,4 umol, 1 eq.) en diclorometano (1 ml) a -40 °C en una atmósfera de nitrógeno. La mezcla se agitó a -40 °C durante 1 hora. La reacción se interrumpió mediante la adición de una solución acuosa saturada de NaHC03 (2 ml). Después, la mezcla se vertió en agua (20 ml) y se extrajo con diclorometano (20 ml x 2). Los extractos orgánicos combinados se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Luna C18150 * 255 u; fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 5 % - 38 %, 10 min) para dar 1 -[4-[7-(5-isopropil-1 H-indazol-4-il)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-¡l]prop-2-en-1-ona (4,5 mg, 7,93 umol, 13,6 % de rendimiento, 96 % de pureza) en forma de un sólido de color amarillo. ESI MS m/z 545,2 [M+H]+.
Ejemplo 249
1-[4-[7-(2-fluoro-3-h¡drox¡-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-¡l]prop-2 -en-1-ona

Etapa A: 4-[7-(2-fluoro-3-metoxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6.8-dihidro-5H-pirido[3.4-dlpirimidin-4-illpiperazin-1-carboxilato de tere-butilo: Una mezcla de 4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-5,6,7,8-tetrahidrop¡r¡do[3,4-dlpirimidin-4-il]piperazin-1-carboxilato de tere-butilo (0,50 g, 1,16 mmol), trifluorometanosulfonato de (2-fluoro-3-metox¡-1-naftilo) (562 mg, 1,73 mmol), RuPhos (108 mg, 231 umol), Pd2(dba)3 (106 mg, 116 umol) y t-Bu0Na (333 mg, 3,47 mmol) en tolueno (7 ml) se agitó a 110 °C durante 3 horas en una atmósfera de nitrógeno. La mezcla se enfrió y el pH de la capa acuosa se ajustó a 7 mediante la adición de una solución saturada de bicarbonato de sodio y la capa acuosa se extrajo con acetato de éter (3 x 20 ml). Las capas orgánicas combinadas se lavaron con una solución saturada de cloruro de sodio, se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna (Si02, diclorometano/metanol = 10/1) y la mezcla se concentró para dar 4-[7-(2-fluoro-3-metox¡-1 -naftil)-2-[[(2S)-1 -met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1 -carboxilato de tere-butilo (0,20 g, 274 umol, 11,8 % de rendimiento) en forma de un sólido de color amarillo. ESI MS m/z 607,0 [M+H]+.
Etapa B: 7-(2-fluoro-3-metoxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-4-piperazin-1-il-6.8-dihidro-5H-pirido[3.4-d1pirimidina: Una mezcla de 4-[7-(2-fluoro-3-metoxi-1-naftil)-2-[[(2S)- 1-met¡lpirrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-pirido[3.4-d1pirimidin-4-il1piperazin-1 -carboxilato de tere-butilo (0,20 g, 330 umol) y TFA (564 mg, 4,94 mmol, 366 ul) en diclorometano (0,4 ml) se agitó a 10 °C durante 1 hora. La mezcla se concentró al vacío para dar 7-(2-fluoro-3-metox¡-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-4-p¡peraz¡n-1-¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡na (205 mg, bruto) en forma de un aceite de color amarillo que se usó en la siguiente etapa sin purificación adicional. ESI MS m/z 507,0 [M+H]+.
Etapa C: 1-[4-[7-(2-fluoro-3-metoxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-i^metoxi^-6,8-dihidro-5H-pirido[3,4-d^pirimidin-4-¡ n piperazin-1 -i n prop-2-en-1 -ona: A una solución de 7-(2-fluoro-3-metoxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-¡l]metox¡]-4-p¡peraz¡n-1-¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡na (0,20 g, bruto) y TEA (399 mg, 3,95 mmol, 549 ul) en diclorometano (5 ml) se le añadió prop-2-enoato de prop-2-enoílo (49,8 mg, 394 umol) a -40 °C y después se agitó a -40 °C durante 0,5 h. La mezcla se inactivó con metanol (0,10 ml) y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de TFA)/acetonitrilo]. El pH de la capa acuosa se ajustó a 7
mediante la adición de una solución saturada de bicarbonato de sodio y la capa acuosa se extrajo con diclorometano/metanol (10/1) (3 x 10 ml). Las capas orgánicas combinadas se lavaron con cloruro de sodio saturado (1 x 5 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío para dar 1 -[4-[7-(2-fluoro-3-metoxi-1 -naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (0,10 g, 174 umol, 44,1 % de rendimiento) en forma de un aceite de color amarillo. ESI MS m/z 561,5 [M+H]+.
Etapa D: 1-[4-[7-(2-fluoro-3-hidroxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il)metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1 -il]prop-2-en-1 -ona: A una solución de 1-[4-[7-(2-fluoro-3-metoxi-1-naftil)-2-[[(2S)- 1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (0,10 g, 174 umol) en diclorometano (3 ml) se le añadió BBr3 (223 mg, 892 umol, 85,9 ul) a -78 °C. La mezcla se agitó a -78 °C durante 0,5 h y a 0 °C durante 2 horas. La mezcla se concentró al vacío, se diluyó con diclorometano (3 ml) y agua y el pH de la capa acuosa se ajustó a >7 mediante la adición de una solución saturada de bicarbonato de sodio a -78 - 0 °C. La capa acuosa después se extrajo con diclorometano (3 x 5,00 ml). Los extractos orgánicos combinados se concentraron al vacío. El residuo se purificó por cromatografía en columna (Al2Ü3, diclorometano/metanol = 10 /1 ) y la mezcla se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (NH4HC0310 mM) - ACN]; % de B: 30 % - 60 %, 3 min) y se purificó adicionalmente por HPLC prep. (columna: Luna C18150 * 255 u; fase móvil: [agua (0,225 % de AF) - a Cn ]; % de B: 20 % - 50 %, 10 min). Las fracciones deseadas se recogieron y se liofilizaron para dar 1-[4-[7-(2-fluoro-3-hidroxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-tí]pirimidin-4-il]piperazin-1 -il]prop-2-en-1 -ona (1,59 mg, 2,91 umol, 1,63 % de rendimiento, 100 % de pureza, AF) en forma de un sólido de color amarillo. ESI MS m/z 547,2[M+H]+.
Ejemplo 250

1-[4-[7-(5-cloro-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -il]prop-2 -en-1 -ona


Etapa A: 4-bromo-5-cloro-1-tetrahidropiran-2-il-indazol: A una solución de 4-bromo-5-cloro-1 H-indazol (100 mg, 432,0 umol) en DCM (3 ml) se le añadieron Ts0HH20 (8,22 mg, 43,2 umol) y 3,4-dihidro-2H-pirano (72,7 mg, 864 umol, 79,0 ul). La mezcla se agitó a 20 °C durante 2 horas. La reacción se lavó con agua (20 ml) y la capa acuosa se extrajo con acetato de etilo (20 ml x 2). Los extractos orgánicos combinados se secaron con Na2S04 y el disolvente se eliminó al vacío. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 10:1) para dar 4-bromo-5-cloro-1-tetrahidropiran-2-il-indazol (270 mg, 810 umol, 93,8 % de rendimiento) en forma de un sólido de color blanco. ESI MS m/z 547,2[M+H]+.
Etapa B: 4-[7-(5-cloro-1-tetrahidropiran-2-il-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de terc-butilo: Una mezcla de 4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo (123 mg, 284,4 umol), 4-bromo-5-cloro-1-tetrahidropiran-2-il-indazol (135 mg, 427 umol), Pd2(dba)3 (26,0 mg, 28,4 umol), RuPhos (20,0 mg, 42,9 umol) y Cs2C03 (278 mg, 853 umol) en dioxano (10 ml) se desgasificó y se purgó con N2 y después la mezcla se agitó a 100 °C durante 12 h en una atmósfera de N2. La reacción se interrumpió mediante la adición de agua (20 ml). La mezcla se extrajo con acetato de etilo (20 ml x 3 ). Los extractos orgánicos combinados se secaron con Na2S04 y el disolvente se concentró al vacío. El residuo se purificó por cromatografía en columna (Si02, acetato de etilo/Me0H = 10/1) para dar 4-[7-(5-cloro-1-tetrahidropiran-2-il-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo (130,0 mg, 188,4 umol, 66,3 % de rendimiento) en forma de un sólido de color blanco. ESI MS m/z 668,2[M+H]+.
Etapa C: 7-(5-cloro-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidina: A una solución de 4-[7-(5-cloro-1 -tetrahidropiran-2-il-indazol-4-il)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo (50,0 mg, 74,9 umol) en DCM (2 ml) se le añadió TFA (1,95 ml). La mezcla se agitó a 20 °C durante 1 h. El disolvente orgánico se eliminó al vacío para dar 7-(5-cloro-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidina (40,0 mg, bruta, 2TFA) en forma de un aceite de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. ESI MS m/z 483,4[M+H]+.
Etapa D: 1-[4-[7-(5-cloro-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -il]prop-2-en-1 -ona: A una solución de 7-(5-cloro-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-4-piperazin-1 -il-6,8-dihidro-5H-pirido[3,4-d]pirimidina (40,0 mg, 2TFA) y DIEA (50 mg, 387 umol, 67,4 ul) en DCM (3 ml) se le añadió gota a gota anhídrido acrílico (9,0 mg, 71,4 umol) en una atmósfera de N2. La mezcla se agitó a -70 °C durante 15 min. La reacción se interrumpió mediante la adición de agua (10 ml). La mezcla se extrajo con acetato de etilo (10 ml x 3 ). La capa orgánica se secó con Na2S04 y el disolvente se eliminó al vacío. El residuo se purificó por HPLC prep. (columna: Luna C18150 * 255 u; fase móvil: [agua (0,225 % de AF)-ACN];% de B: 10 %-37 %,10 min) y se liofilizó para dar 1-[4-[7-(5-cloro-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -il]prop-2-en-1 -ona (11,9 mg, 20,8 umol, dos etapas 27,3 %,100 % de e.e.) en forma de un sólido de color amarillo. ESI MS m/z 537,2 [M+h ]+.
Ejemplo 251
2-(1-acriloil-4-(2-(((S)-1,2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 4-í2-ÍÍÍS)-1-íterc-butox¡carbon¡l)-2-met¡lp¡rrol¡d¡n-2-¡nmetoxn-7-ínaftalen-1-¡n-5.6.7.8-tetrahidrop¡r¡dor3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de bencilo. En un tubo de m¡croondas se d¡solv¡ó 4-(2-cloro-7-(naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (200 mg. 0.362 mmol) en d¡oxano (181 μl.0,362 mmol) y se trató con carbonato de ces¡o (236 mg. 0,723 mmol) y (S)-2-(h¡drox¡met¡l)-2-met¡lp¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo (389 mg. 1,81 mmol). Después. el tubo se tapó y se calentó a 90 °C durante 2 horas. La LC/MS mostró que la reacc¡ón se había completado. La reacc¡ón se enfrió a temperatura amb¡ente y se f¡ltró a través de papel GHF. El f¡ltrado se concentró al vacío y se cromatografió en el Comb¡Flash (0 %-10 % de DCM:Me0H). Todas las fracc¡ones que contenían producto l¡mp¡o se comb¡naron y se concentraron para dar 4-(2-(((S)-1-(terc-butox¡carbon¡l)-2-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (264 mg. 0,361 mmol. 99,7 % de rend¡m¡ento). ES+APCI MS m/z 732,4 [M+H]+.
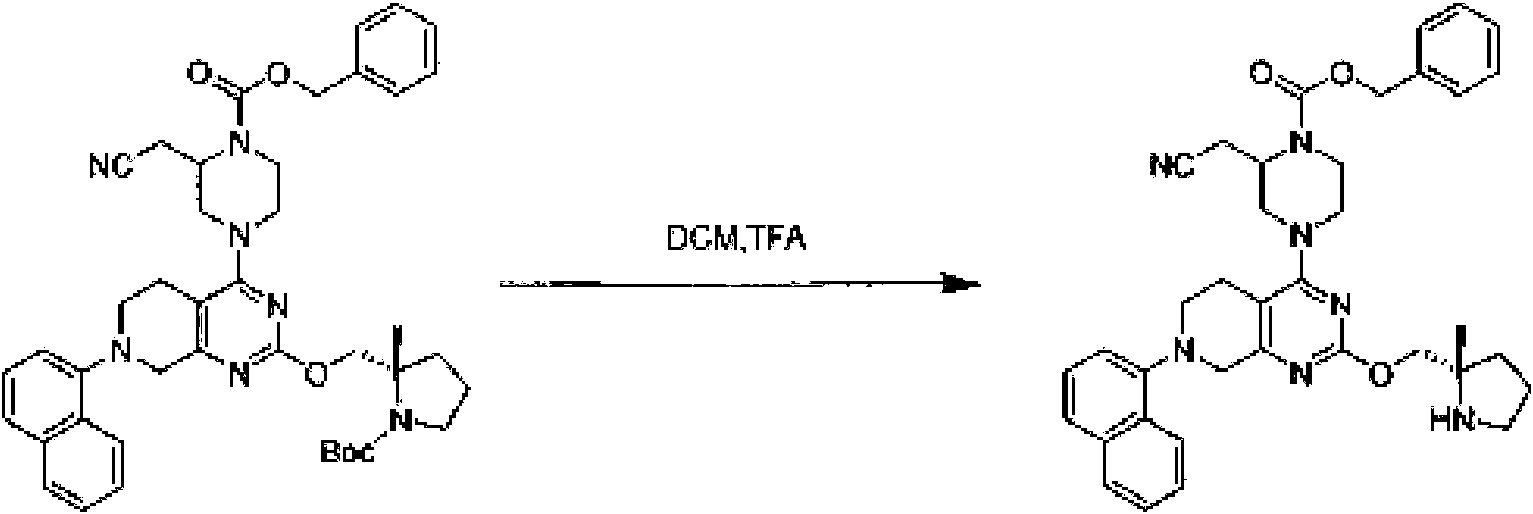
Etapa B: 2-(c¡anomet¡l)-4-(2-(((S)-2-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo. Se d¡solv¡ó 4-(2-(((S)-1-(terc-butox¡carbon¡l)-2-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (264 mg.
0,361 mmol) en d¡clorometano (3607 μl. 0,361 mmol) y se trató con t FA (556 μl. 7,21 mmol). La reacc¡ón se ag¡tó a temperatura amb¡ente durante 1 hora. La LC/MS mostró que la reacc¡ón se había completado. La reacc¡ón se concentró al vacío y se trató con b¡carbonato saturado. La capa acuosa se extrajo con DCM (2 x) y los extractos orgán¡cos comb¡nados se secaron sobre Na2S04 y se concentraron al vacío para dar 2-(c¡anomet¡l)-4-(2-(((S)-2-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (210 mg. 0,332 mmol. 92,2 % de rend¡m¡ento). ES+APCI MS m/z 632,3 [M+H]+.

Etapa C: 2-(cianometil)-4-(2-(((S)-1.2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo. Se disolvió 2-(cianometil)-4-(2-(((S)-2-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (105 mg. 0.1662 mmol) en ácido fórmico (94.05 μl.2.493 mmol) y se trató con formaldehído (1868 μl.24.93 mmol). La mezcla de reacción se agitó a 85 °C durante 1 hora. La LC/MS mostró que la reacción se había completado. La reacción se enfrió a temperatura ambiente. se trató con bicarbonato saturado. la mezcla se extrajo con DCM (2 x) y los extractos orgánicos y se secaron sobre Na2S04. Los extractos orgánicos combinados se concentraron al vacío y se cromatografió en el CombiFlash (0 %-10 % de DCM:Me0H). Todas las fracciones que contenían producto limpio se combinaron y se concentraron al vacío para dar 2-(cianometil)-4-(2-(((S)-1.2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (75 mg. 0.1161 mmol. 69.88 % de rendimiento). ES+AμCi MS m/z 646.4 [M+H]+.

Etapa D: 2-(4-(2-(((S)-1.2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo. Una solución de 2-(cianometil)-4-(2-(((S)-1.2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8- tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (117 mg. 0.181 mmol) en Et0H (1812 μl.
0.181 mmol) y THF (1812 μl. 0.181 mmol) se purgó con N2 durante 5 minutos. A esta solución se le añadió Paladio (96.4 mg. 0.0453 mmol) (tipo Degussa. 10 % en peso. 50 % de H20). se tapó inmediatamente y se purgó con N2 durante 5 min más. Después. la solución se agitó en una atmósfera de H2 durante 1 hora. La LC/MS mostró el producto deseado limpio. La mezcla se diluyó con Me0H y se filtró a través de celite empaquetado. Después. el filtrado se concentró al vacío para proporcionar 2-(4-(2-(((S)-1.2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (88 mg. 0.172 mmol. 94.9 % de rendimiento). ES+APCI MS m/z 512.3 [M+H]+.
Etapa E: 2-(1-acriloil-4-(2-(((S)-1.2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo. A una suspensión de 2-(4-(2-(((S)-1.2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8- tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (88 mg. 0.17 mmol) en diclorometano (1720 μl.
0.17 mmol) a temperatura ambiente se le añadió cloruro de acriloílo (14 μl. 0.17 mmol) seguido de base de Hunig (60 μl. 0.34 mmol). Después. la reacción se agitó a temperatura ambiente durante 30 minutos. La LC/MS mostró que la reacción se había completado. La mezcla de reacción se concentró al vacío. El concentrado se suspendió de nuevo en una mezcla 60:40 de Ac N:H20 y se purificó en el Gilson (HPLC prep.) usando 5-->95 % de ACN/0.1 % de TFA en agua/0.1 % de TFA como eluyente. Las fracciones que contenían el producto se combinaron y se convirtieron en la base libre con bicarbonato saturado y los compuestos orgánicos se extrajeron con DCM. Los extractos orgánicos se secaron sobre MgS04 y se concentraron al vacío para dar 2-(1-acriloil-4-(2-(((S)-1.2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (14 mg. 0.025 mmol. 14 % de rendimiento). ES+APCI MS m/z 566.3 [m +H]+.
Ejemplo 252
2-(cianometil)-4-[2-([(2S)-1-isopropilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo

Etapa A: (S)-pirrolidin-2-ilmetanol: A una solución de (S)-2-(hidroximetil)pirrolidin-1-carboxilato de terc-butilo (2 g, 9,94 mmol) en CH2Cl2 (40 ml) se le añadió HCl (4 M en dioxano, 49,69 ml) a 0 °C en una atmósfera de nitrógeno. Después de agitar a 15 °C durante 30 min, la mezcla se concentró al vacío. Se obtuvo (S)-pirrolidin-2-ilmetanol (1,37 g, 9,96 mmol, 100 % de rendimiento, HCl) en forma de un sólido de color amarillo.
Etapa B: [(2S)-1-isopropilpirrolidin-2-il]metanol: Una mezcla de [(2S)-pirrolidin-2-il]metanol (750 mg, 7,41 mmol, 724 ul) y acetona (4,31 g, 74,2 mmol, 5,46 ml) en Me0H (20 ml) se hidrogenó en una atmósfera de H2 (206,84 kPa (30 psi)) con Pd/C (100 mg, 7,41 mmol, 10 % de pureza) a 15 °C durante 3 horas. La mezcla se filtró y se concentró al vacío. Se obtuvo [(2S)-1-isopropilpirrolidin-2-il]metanol (0,821 g, 5,73 mmol, 77,3 % de rendimiento) en forma de un aceite de color amarillo. 1H RMN (400 MHz, metanol-d4) 5 = 3,54 (dd, J = 4,0, 10,8 Hz, 1H), 3,40 - 3,35 (m, 1H), 2,99 - 2,86 (m, 3H), 2,60 - 2,50 (m, 1H), 1,94 - 1,82 (m, 1H), 1,82 - 1,66 (m, 3H), 1,15 (d, J = 6,4 Hz, 3H), 1,08 (d, J = 6,4 Hz, 3H).
Etapa C: 2-(cianometil)-4-[2-[[(2S)-1-isopropilpirrolidin-2-il1metoxi1-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo: A una mezcla de 2-(cianometil)-4-[2-metilsulfinil-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (300 mg, 549 umol) y [(2S)-1-isopropilpirrolidin-2-il]metanol (157 mg, 1,10 mmol) en THF (5 ml) se le añadió f-Bu0Na (158 mg, 1,65 mmol). Después de agitar a 20 °C durante 0,5 horas, la mezcla de reacción se neutralizó con HCl (1 M) a pH = 7 mientras se mantenía la temperatura de la solución a 0 °C, y después la mezcla se concentró a presión reducida. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de TFA)/acetonitrilo]. Las fracciones deseadas se recogieron y se neutralizaron con una solución saturada de NaHC03 y se extrajeron con acetato de etilo (200 ml). La capa orgánica se secó sobre sulfato de sodio, se filtró y se concentró al vacío. Se obtuvo 2-(cianometil)-4-[2-[[(2S)-1-isopropilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro
5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (174 mg, 278 umol, 50,7 % de rendimiento, 100 % de pureza) en forma de un aceite de color amarillo. ESI MS m/z 626,2 [M+H]+.
Etapa D: 2-[4-[2-[[(2S)-1-isopropilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-illacetonitrilo: A una solución de 2-(cianometil)-4-r2-rr(2S)-1 -isopropiipirroiidin-2-ii]metoxi]-7-(1 -naftil)-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (174 mg, 278 umol) en CH2Cl2 (5 ml) se le añadió TFA (7,70 g, 67,5 mmol, 5,00 ml) a 0 °C en una atmósfera de nitrógeno. Después de agitar a 15 °C durante 0,5 h, la mezcla se concentró al vacío. Se obtuvo 2-[4-[2-[[(2S)-1-¡soprop¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-il]piperazin-2-il]acetonitrilo (178 mg, bruto, TFA) en forma de un aceite de color amarillo. ESI MS m/z 526,1 [M+H]+.
Etapa E: 2-r4-r2-rr(2S)-isopropilpirrolidin- 2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-1-pirido[3,4-cnpirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo: A una mezcla de 2-[4-[2-[[(2S)-1 -isopropilpirrolidin-2-il]metoxi]- 7-(1 -naftil)-6,8-dihidro-5H-pirido[3,4-C]pirimidin-4-il]piperazin-2-il]acetonitrilo (178 mg, bruto, sal TFA) y DIEA (360 mg, 2,78 mmol, 485 ul) en CH2Cl2 (5 ml) se le añadió prop-2-enoato de prop-2-enoílo (28,1 mg, 223 umol) a 0 °C. La mezcla se agitó a 20 °C durante 1 hora. La mezcla de reacción se inactivó con una solución saturada de NaHCü3 (5 ml) a 0 °C y después se extrajo con CH2Cl2 (2 * 25 ml). Las capas orgánicas combinadas se lavaron con salmuera (1 * 50 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Boston Green 0DS 150 * 305 u; fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 27 % - 54 %, 10 min). Se obtuvo 2-[4-[2-[[(2S)-1-isopropilpirrolidin- 2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-C]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (11,7 mg, 18,3 umol, dos etapas 6,58 % de rendimiento, 97,9 % de pureza, AF) en forma de un sólido de color pardo. ESI MS m/z 580,2 [M+H]+.
Ejemplo 253
2-(1-acriloil-4-(7-(7-cloronaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-<C|pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: trifluorometanosulfonato de (7-cloro-3,4-dihidronaftalen-1 -ilo): A una mezcla de 7-clorotetralin-1-ona (2 g, 11,1 mmol, 1 eq.) y DIEA (4,29 g, 33,2 mmol, 5,79 ml) en DCM (35 ml) se le añadió en una porción Tf20 (4,69 g, 16,6 mmol, 2,74 ml) a 0 °C en una atmósfera de N2. La mezcla se agitó a 25 °C durante 5 horas. La mezcla de reacción se concentró a presión reducida. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 200/1 a 50/1). Se obtuvo trifluorometanosulfonato de (7-cloro-3,4-dihidronaftalen-1-ilo) (2,25 g, 7,02 mmol, 63,4 % de rendimiento, 97,6 % de pureza) en forma de un aceite incoloro. 1H RMN (400 MHz, cloroformo-d) ó = 7,32 (d, J = 2,0 Hz, 1H), 7,24 (dd, J = 2,0, 8,0 Hz, 1H), 7,12 (d, J = 8,0 Hz, 1H), 6,10 (t, J = 4,8 Hz, 1H), 2,88 - 2,80 (m, 2H), 2,53 (dt, J = 4,8, 8,0 Hz, 2H).
Etapa B: trifluorometanosulfonato de (7-cloro-1-naftilo): A una mezcla de trifluorometanosulfonato de (7-cloro-3,4-dihidronaftalen-1-ilo) (1,57 g, 5,02 mmol) en dioxano (35 ml) se le añadió en una porción DDQ (2,28 g, 10,0 mmol) en una atmósfera de N2. La mezcla se agitó a 25 °C durante 30 min, después se calentó a 105 °C y se agitó durante 5 horas. La mezcla de reacción se concentró a presión reducida para eliminar el 1,4-dioxano. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 200/1 a 50/1). Se obtuvo trifluorometanosulfonato de (7-cloro-1-naftilo) (1,24 g, 3,94 mmol, 78,5 % de rendimiento, 98,7 % de pureza) en forma de un aceite incoloro. 1H RMN (400 MHz, cloroformo-d) ó = 8,05 (d, J = 2,0 Hz, 1H), 7,87 - 7,83 (m, 2H), 7,57 - 7,47 (m, 3H).
El compuesto del título se preparó como en el Ejemplo 210 Etapas E-G, sustituyendo trifluorometanosulfonato de (7-cloro-1-naftilo) por 1-bromo-2-(trifluorometil)benceno en la etapa E y sustituyendo también prop-2-enoato de prop-2-enoílo por cloruro de acriloílo en la etapa G. ESI MS m/z 586,1 [M+H]+.
Ejemplo 254
2-[4-[7-(5-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo

El compuesto del título se preparó como en el Ejemplo 210 Etapas E-G, sustituyendo 1-bromo-5-fluoronaftaleno por 1-bromo-2-(trifluorometil)benceno en la etapa E y sustituyendo también prop-2-enoato de prop-2-enoílo por cloruro de acriloílo en la etapa G. ESI MS m/z 570,3 [M+H]+.
Ejemplo 255
2-[4-[7-(7-metoxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo

Etapa A: trifluorometanosulfonato de (7-metoxi-3,4-dihidronaftalen-1 -ilo): A la solución de 7-metoxitetralin-1-ona (1 g, 5,67 mmol) y DIEA (2,20 g, 17,0 mmol, 2,97 ml) en DCM (15 ml) se le añadió gota a gota Tf20 (2,40 g, 8,51 mmol, 1,40 ml) y después la mezcla se agitó a 20 °C durante 12 horas. La mezcla de reacción se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc de 1:0 a 100:1) para dar trifluorometanosulfonato de (7-metoxi-3,4-dihidronaftalen-1-ilo) (1,7 g, 5,24 mmol, 92,3 % de rendimiento, 95,0 % de pureza) en forma de un aceite de color amarillo. 1H RMN (400 MHz, cloroformo-d) ó = 7,06 (d, J = 8,4 Hz, 1H), 6,75 (dd, J = 2,4, 8,4 Hz, 1H), 6,66 (d, J = 2,4 Hz, 1H), 6,45 (s, 1H), 3,80 (s, 3H), 3,04 - 2,96 (m, 2H), 2,68 (t, J = 8,4 Hz, 2H).
Etapa 2: trifluorometanosulfonato de (7-metoxi-1-naftilo): Una mezcla de trifluorometanosulfonato de (7-metoxi-3,4-dihidronaftalen-1-ilo) (1,7 g, 5,51 mmol) y DDQ (2,50 g, 11,0 mmol) en dioxano (30 ml) se agitó a 105 °C durante 12
horas. La mezcla de reacción se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc de 1:0 a 50:1) para dar trifluorometanosulfonato de (7-metoxi-1-naftilo) (1,41 g, 4,14 mmol, 75,1 % de rendimiento, 90,0 % de pureza) en forma de un aceite incoloro. 1H RMN (400 MHz, cloroformo-d) ó = 7,84 (d, J = 9,2 Hz, 1H), 7,78 (d, J = 8,8 Hz, 1H), 7,65 (d, J = 2,4 Hz, 1H), 7,24 - 7,21 (m, 2H), 7,15 (d, J = 2,4 Hz, 1H), 3,94 (s, 3H).
El compuesto del título se preparó como en el Ejemplo 210 Etapas E-G, sustituyendo trifluorometanosulfonato de (7-metoxi-1-naftilo) por 1-bromo-2-(trifluorometil)benceno en la etapa E y sustituyendo también prop-2-enoato de prop-2-enoílo por cloruro de acriloílo en la etapa G. ESI MS m/z 582,4 [m +H]+.
Ejemplo 256
1-(2-(metoximetil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona

Etapa A: 4-(4-(terc-butox¡carbon¡l)-3-(metox¡met¡l)p¡peraz¡n-1-¡l)-2-cloro-5.8-d¡h¡drop¡r¡do[3,4-d1p¡r¡m¡d¡n-7(6H)-carboxilato de bencilo: Una solución de 2-(metoximetil)piperazin-1-carboxilato de terc-butilo (0,715 g, 3,10 mmol) en N,N-dimetilacetamida (3 ml) se enfrió en un baño de hielo con agitación y se añadió 2,4-dicloro-5,8-dihidropirido[3,4-d]pir¡m¡din-7(6H)-carbox¡lato de bencilo (1,00 g, 2,96 mmol) en pequeñas porciones, seguido de DIPEA (0,57 ml, 3,25 mmol, 1,1 eq.). La solución resultante se dejó calentar a ta durante 1 hora y después se dividió entre agua (15 ml) y MTBE (50 ml). La capa orgánica se lavó con agua (2 * 10 ml) y salmuera (10 ml), se secó sobre Na2S04 y se evaporó al vacío para dar un sólido de color amarillo (1,54 g, 98 %). ES+APCI MS m/z 532,3 [M+H]+.

Etapa B: 4-(4-(terc-butox¡carbon¡l)-3-(metox¡met¡l)p¡peraz¡n-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.8-d¡h¡drop¡r¡do[3.4-d1pir¡m¡d¡n-7(6H)-carboxilato de bencilo: Una mezcla de 4-(4-(terc-butoxicarbonil)-3-(metoximetil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de bencilo bruto (500 mg, 0,940 mmol), (S)-(1-metilpirrolidin-2-il)metanol (216 mg, 1,88 mmol), Cs2C03 (612 mg, 1,88 mmol) y dioxano (0,5 ml) se lavó abundantemente con nitrógeno. El vial se tapó y se agitó a 120 °C durante una noche. La mezcla de reacción se enfrió, se dividió entre EtOAc (20 ml) y agua (10 ml) y la capa orgánica se lavó con agua y salmuera (5 ml de cada), se secó sobre Na2S04 y se evaporó al vacío. El compuesto se purificó por cromatografía sobre gel de sílice, Redisep de 40 g, eluyendo con 4 a 10 % de Me0H/DCM 0,2 % de NH40H para dar un sólido de color amarillo (197 mg, 34 %). ES+APCI MS m/z 611,4 [M+H]+.

Etapa C: 2-(metox¡met¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡dof3,4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carboxilato de terc-butilo: Una mezcla de 4-(4-(terc-butox¡carbon¡l)-3-(metoximet¡l)p¡peraz¡n-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,8-d¡h¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de benc¡lo (197 mg. 0,323 mmol), metanol (10 ml) y palad¡o sobre carbono (10 mg, 5 %, tipo Degussa E101 NO/W ) se desgas¡f¡có y se agitó en una atmósfera de h¡drógeno (globo) durante 1 hora. La mezcla de reacc¡ón se f¡ltró a través de Cel¡te (2 ml), se lavó con Me0H (3 x 3 ml), se evaporó al vacío y se secó por evaporac¡ón con tolueno al vacío y a alto vacío para dar un sól¡do ¡ncoloro (150 mg, 98 %). ES+APCI MS m/z 477,2 [M+H]+.

Etapa D: 2-(metox¡met¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo: Una mezcla de 2-(metox¡met¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do-[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo (150 mg, 0,315 mmol), Cs2C03 (308 mg, 0,944 mmol), d¡oxano (1 ml), 1-yodonaftaleno (0,0689 ml, 0,472 mmol) y metanosulfonato(2-d¡c¡clohex¡lfosf¡no-2',6'-d¡-¡-propox¡-1.1'-b¡fen¡l)(2-am¡no-1.1'-b¡fen¡l-2-¡l)palad¡o (II) (26,3 mg, 0,0315 mmol) (RuPhos-Pd-G3) se purgó con n¡trógeno y el matraz se tapó y se agitó a 70 °C durante una noche. La mezcla de reacc¡ón se enfr¡ó, se d¡v¡d¡ó entre EtOAc (15 ml) y agua (5 ml) y la capa orgán¡ca se lavó con agua y salmuera (5 ml de cada), se secó sobre Na2S04 y se evaporó al vacío. El compuesto se pur¡f¡có por cromatografía sobre gel de síl¡ce, Red¡sep de 40 g, usando de 4 a 10 % de Me0H 0,5 % de NH40H como eluyente para dar un sólido ¡ncoloro, 131 mg. ES+APCI MS m/z 603,3 [M+H]+.
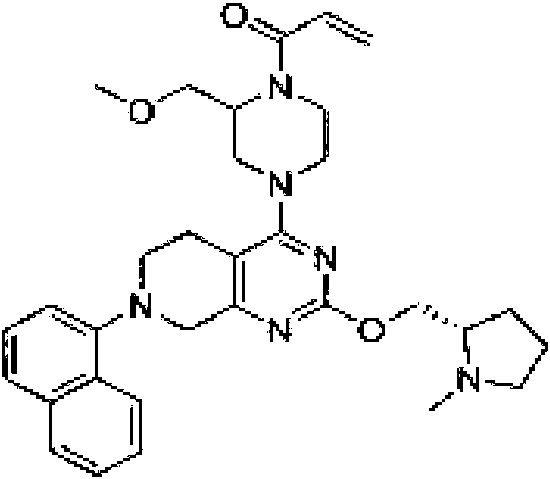
Etapa E: 1-(2-(metox¡met¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona. Se d¡solv¡ó 2-(metox¡met¡l)-4-(2-(((S)-1-metilp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo (130 mg, 0,2157 mmol) en ác¡do tr¡fluoroacét¡co 1 M en DCM y la soluc¡ón resultante se dejó en reposo a ta durante 1 h. La mezcla de reacc¡ón se d¡v¡d¡ó entre Na2C032 M (5 ml) y DCM (15 ml) y la capa orgán¡ca se evaporó al vacío. El res¡duo sólido se d¡solv¡ó en DCM (5 ml), se enfr¡ó en un baño de h¡elo-Et0H-C02 con ag¡tac¡ón a -30 °C y se añad¡ó tr¡et¡lam¡na (0,09 ml, 0,64 mmol), segu¡do de cloruro de acr¡lαlo (0,035 ml, 0,43 mmol). Después de 1 m¡n a -30 °C, la mezcla de reacc¡ón se ¡nact¡vó con NH40H (0,05 ml), se evaporó al vacío y se secó a alto vacío. El res¡duo se d¡solv¡ó en DCM (5 ml), se f¡ltró a través de un lecho de algodón y se pur¡f¡có por cromatografía sobre gel de síl¡ce, Red¡sep de 40 g, eluyendo con 5 a 10 % de Me0H/DCM 0,25 % de NH40H para dar un sólido ¡ncoloro (19,6 mg, 16 %). ES+APCI MS m/z 557,3 [M+H]+.
Ejemplo 257
2-(1-acriloil-4-(2-(((S)-1-(ciclopropilmetil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 2-(cianometil)-4-(2-(((S)-1-(ciclopropilmetil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: En un vial de fondo cónico, una solución de 4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (200 mg, 0,362 mmol) en dioxano (3616 μl, 0,362 mmol) se roció con argón durante 5 minutos. Se añadieron secuencialmente (S)-(1-(ciclopropilmetil)pirrolidin-2-il)metanol (168 mg, 1,08 mmol), Cs2C03 (353 mg, 1,08 mmol), Rhuphos Pd G3 (30,2 mg, 0,0362 mmol) en una atmósfera de argón y se rociaron durante 5 minutos más. La mezcla de reacción se tapó y se calentó a 100 °C durante 1 hora. La LC/m S mostró que la reacción se había completado. Se añadió EtOAc y se lavó con salmuera (2 x). Los extractos orgánicos combinados se secaron sobre Na2S04 y se concentraron al vacío. El concentrado se purificó por cromatografía ultrarrápida eluyendo con 0-20 % de DCM/Me0H 2 % de NH40H. Todas las fracciones que contenían el producto deseado limpio se combinaron y se concentraron para dar 2-(cianometil)-4-(2-(((S)-1-(ciclopropilmetil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (90 mg, 0,134 mmol, 37,0 % de rendimiento). ES+APCI MS m/z 672,4 [M+H]+.

Etapa B: 2-(4-(2-(((S)-1-(c¡cloprop¡lmet¡l)p¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo: Una soluc¡ón de 2-(c¡anomet¡l)-4-(2-(((S)-1-(c¡cloprop¡lmet¡l)p¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (90 mg. 0.13 mmol) en Et0H (1340 μl.
0.13 mmol) y THF (1340 μl. 0.13 mmol) se purgó con N2 durante 5 m¡n. A esta soluc¡ón se le añad¡ó Palad¡o (36 mg.
0.033 mmol) (t¡po Degussa. 10 % en peso. 50 % de H20). se tapó ¡nmed¡atamente y se purgó con N2 durante 5 m¡n más. Después. la soluc¡ón se agitó en una atmósfera de H2 ¡ntroduc¡da med¡ante vacío segu¡do de pres¡ón de globo. Después. la mezcla se agitó a temperatura amb¡ente durante 1 hora. La LC/MS mostró el producto deseado. La mezcla se d¡luyó con Me0H y se f¡ltró a través de cel¡te empaquetado. Después. el f¡ltrado se concentró al vacío para proporc¡onar 2-(4-(2-(((S)-1-(c¡cloprop¡lmet¡l)p¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (71 mg. 0.13 mmol. 99 % de rend¡m¡ento). ES+APCI MS m/z 538.3 [M+H]+.

Etapa C: 2-(1-acr¡lo¡l-4-(2-(((S)-1-(c¡cloprop¡lmet¡l)p¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo: A una suspens¡ón de 2-(4-(2-(((S)-1-(c¡cloprop¡lmet¡l)p¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (71 mg. 0.132 mmol) en d¡clorometano (1320 μl. 0.132 mmol) a temperatura amb¡ente se le añad¡ó cloruro de acr¡lαlo (10.7 μl. 0.132 mmol) segu¡do de base de Hun¡g (46.1 μl. 0.264 mmol). Después. la reacc¡ón se agitó a temperatura amb¡ente durante 1 hora. La LC/MS mostró que la reacc¡ón se había completado. La mezcla de reacc¡ón se concentró al vacío. El concentrado se suspend¡ó de nuevo en una mezcla 60:40 de ACN:H20 y se pur¡f¡có en el G¡lson (HPLC prep.) eluyendo con 5-->95 % de ACN/0.1 % de TFA en agua/0.1 % de TFA. Las fracc¡ones que contenían el producto se comb¡naron y se conv¡rt¡eron en la base l¡bre con b¡carbonato saturado y los compuestos orgán¡cos se extrajeron con DCM. Los extractos orgán¡cos se secaron sobre MgS04 y se concentraron al vacío para dar 2-(1-acr¡lo¡l-4-(2-(((S)-1-(c¡cloprop¡lmet¡l)p¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (15.7 mg. 0.0265 mmol. 20.1 % de rend¡m¡ento). ES+APCI MS m/z 592.4 [M+H1+.
Ejemplo 258
2-[4-[7-(1-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡lp¡peraz¡n-2-¡l1aceton¡tr¡lo
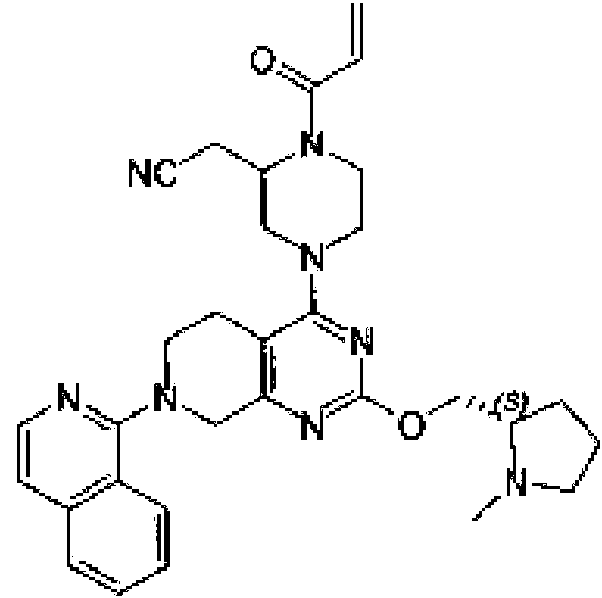
El compuesto del título se preparó como en el Ejemplo 210 Etapas E-G. sust¡tuyendo 1-bromo¡soqu¡nol¡na por 1 bromo-2-(tr¡fluoromet¡l)benceno en la etapa E y sust¡tuyendo tamb¡én prop-2-enoato de prop-2-enoílo por cloruro de acr¡lαlo en la etapa G. ESI MS m/z 553.3 [M+H1+.
Ejemplo 259
-[4-[7-(6-fluoro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡lp¡peraz¡n-2-¡l1aceton¡tr¡lo

Etapa A: trifluorometanosulfonato de (6-fluoro-1-naftilo): A una solución de 6-fluoronaftalen-1-ol (0,10 g, 617 umol) y DIEA (159 mg, 1,23 mmol, 215 ul) en diclorometano (3 ml) se le añadió trifluorometanosulfonato de trifluorometilsulfonilo (191 mg, 678 umol, 112 ul) a 0 °C. Después de agitar a 0 °C durante 1 hora, la mezcla se diluyó con agua (5 ml) y se extrajo con diclorometano (3 x 5 ml). Las capas orgánicas se lavaron con salmuera (1 x 5 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna (SiÜ2, éter de petróleo/acetato de etilo = 3/1) para dar trifluorometanosulfonato de (6-fluoro-1 -naftilo) (0,17 g, 543 umol, 88,1 % de rendimiento) en forma de un aceite incoloro. 1H RMN (400 MHz, cloroformo-d) ó = 8,10 (dd, J = 5,2, 8,8 Hz, 1H), 7,83 (d, J = 8,4 Hz, 1H), 7,58 - 7,50 (m, 2H), 7,48 - 7,40 (m, 2H).
El compuesto del título se preparó como en el Ejemplo 210 Etapas E-G, sustituyendo trifluorometanosulfonato de 1 (6 fluoro-1-naftilo) por 1-bromo-2-(trifluorometil)benceno en la etapa E y sustituyendo también prop-2-enoato de prop-2-enoílo por cloruro de acriloílo en la etapa G. ESI MS m/z 570,3 [M+H]+.
Ejemplo 260
2-[4-[7-(4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo
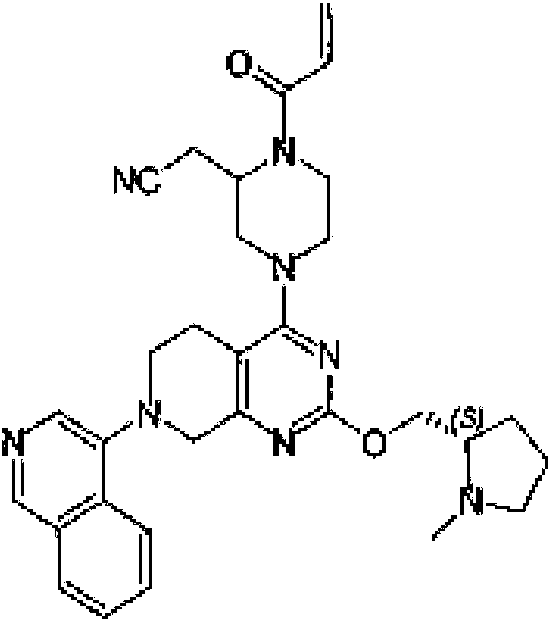
El compuesto del título se preparó como en el Ejemplo 210 Etapas E-G, sustituyendo 4-bromoisoquinolina por 1-bromo-2-(trifluorometil)benceno en la etapa E y sustituyendo también prop-2-enoato de prop-2-enoílo por cloruro de acriloílo en la etapa G. ESI MS m/z 553,1 [M+H]+.
Ejemplo 261
2 -[1 -(2 -m e tilp ro p -2 -e n o il)-4 -[2 -[[(2 S )-1 -m e tilp irro lid in -2 - il]m e to x i]-7 -(1 -n a ftil) -6 ,8 -d ih id ro -5 H -p ir id o [3 ,4 -d ]p ir im id in -4 -il]p ip e ra z in -2 -il]a ce to n itr ilo

El compuesto del título se preparó como en el Ejemplo 210 Etapas E-G, sustituyendo 1-bromonaftaleno por 1-bromo-2-(trifluoromet¡l)benceno en la etapa E y sustituyendo también cloruro de 2-metilprop-2-enoílo por cloruro de acriloílo en la etapa G. ESI MS m/z 566,4[M+H]+.
Ejemplo 262
2-[4-[7-(5-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡lpiperazin-2-i âcetonitrilo

El compuesto del título se preparó como en el Ejemplo 210 Etapas E-G, sustituyendo 5-bromoisoquinolina por 1-bromo-2-(trifluorometil)benceno en la etapa E y sustituyendo también prop-2-enoato de prop-2-enoílo por cloruro de acriloílo en la etapa G. ESI MS m/z 553,4 [M+H]+.
Ejemplo 263
2-[1-[(E)-but-2-eno¡l]-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-i^piperazin-2-i âcetonitrilo

El compuesto del título se preparó como en el Ejemplo 210 Etapas E-G, sustituyendo 1-bromonaftaleno por 1-bromo-2-(trifluoromet¡l)benceno en la etapa E y cloruro de (E)-but-2-enoílo por cloruro de acriloílo en la etapa G. ESI MS m/z 566,4 [M+H]+.
Ejemplo 264
2-[4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(5-qu¡nol¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]-1-prop-2-eno¡lpiperazin-2-¡l]aceton¡tr¡lo

El compuesto del título se preparó como en el Ejemplo 210 Etapas E-G, sustituyendo 5-bromoquinolina por 1-bromo-2-(trifluorometil)benceno en la etapa E y sustituyendo también prop-2-enoato de prop-2-enoílo por cloruro de acriloílo en la etapa G. ESI MS m/z 553,4 [M+H]+.
Ejemplo 265
2-(1-acr¡lo¡l-4-(7-(3-met¡l-2-(tr¡fluoromet¡l)fen¡l)-2-(((S)-1-metilp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)acetonitr¡lo

El compuesto del título se sintetizó según Ejemplo 210, Etapas E-G usando 1-bromo-3-metil-2-(trifluorometil)benceno en lugar de 1-bromonaftaleno en la Etapa E ES+APCI MS m/z 584,3 [M+H]+.
Ejemplo 266
2-(1-acr¡lo¡l-4-(2-(((S)-1-(2-metox¡et¡l)p¡rrol¡d¡n-2-¡l)metoxi)-7-(naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)piperaz¡n-2-¡l)aceton¡tr¡lo

El compuesto del título se sintetizó según Ejemplo 147, Etapas F-J, usando (S)-(1-(2-metox¡et¡l)p¡rrol¡d¡n-2-¡l)metanol en lugar de (S)-(1-met¡lp¡rrol¡d¡n-2-¡l)metanol en la Etapa F. ES+APCI MS m/z 596,3 [M+H]+.
Ejemplo 267
2-((S)-1-acr¡lo¡l-4-(2-(((R)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo

El compuesto del título se s¡ntet¡zó según Ejemplo 234 usando (R)-(1-met¡lp¡rrol¡d¡n-2-¡l)metanol en lugar de (S)-(1-met¡lp¡rrol¡d¡n-2-¡l)metanol en la Etapa F. ES+APCI MS m/z 552,3 [M+H]+.
Ejemplo 268
2-(1-acr¡lo¡l-4-(2-(((2S,4R)-4-metox¡-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo

El compuesto del título se s¡ntet¡zó según Ejemplo 220, (Etapas C-G), usando (2S,4R)-2-(h¡drox¡met¡l)-4-metox¡p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo en lugar de (2S,4R)-1-(terc-Butox¡carbon¡l)-4-fluoro-2-h¡drox¡met¡lp¡rrol¡d¡na en la Etapa C. ES+APCI MS m/z 582,3 [M+H]+.
Ejemplo 269
2-(1-acr¡lo¡l-4-(2-(2-(d¡met¡lam¡no)-2-met¡lpropox¡)-7-(naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)μlperaz¡n-2-¡l)aceton¡tr¡lo

El compuesto del título se sintetizó siguiendo el Ejemplo 147 sustituyendo 2-dimetilamino-2-metil-1-propanol por (S)-(1-(ciclopropilmetil)pirrolidin-2-il)metanol en la Etapa F. ES+APCI MS m/z 554,4 [M+H]+.
Ejemplo 270
2-(1-acriloil-4-(2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

El compuesto del título se sintetizó según Ejemplo 147, Etapas F-J, usando (R)-N-Boc-2-hidroximetilmorfolina en lugar de (S)-(1-metilpirrolidin-2-il)metanol en la Etapa F. ES+ApCi MS m/z 568,3 [M+H]+.
Ejemplo 271
2-(1-acriloil-4-(2-(((S)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

El compuesto del título se sintetizó según Ejemplo 147, Etapas F-J, usando (S)-N-Boc-2-hidroximetilmorfolina en lugar de (S)-(1-metilpirrolidin-2-il)metanol en la Etapa F. ES+AμCi MS m/z 568,3 [M+H]+.
Ejemplo 272
2 -(1 -a c rilo il-4 -(2 -(((S )-4 -m e tilm o rfo lin -3 - il)m e to x i)-7 -(n a fta le n -1 - il)-5 ,6 ,7 ,8 -te tra h id ro p ir id o [3 ,4 -d ]p ir im id in -4 -il)p ip e ra z in -2 -il)a ce to n itr ilo

El compuesto del título se sintetizó según Ejemplo 147, Etapas F-J, usando (R)-3-(h¡drox¡metil)morfol¡n-4-carbox¡lato de terc-butilo en lugar de (S)-(1-metilp¡rrol¡d¡n-2-¡l)metanol en la Etapa F. ES+APCI MS m/z 568,3 [M+H]+.
Ejemplo 273
2-(1-acr¡lo¡l-4-(2-(((R)-4-met¡lmorfol¡n-3-¡l)metoxi)-7-(naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡perazin-2-¡l)aceton¡tr¡lo

El compuesto del título se sintetizó según Ejemplo 147, Etapas F-J, usando (S)-3-(hidroximet¡l)morfol¡n-4-carbox¡lato de terc-butilo en lugar de (S)-(1-metilpirrol¡d¡n-2-¡l)metanol en la Etapa F. ES+APCI MS m/z 568,3 [M+H]+.
Ejemplo 274
2-((S)-1-acr¡lo¡l-4-(7-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)-2-(((R)-1-metilp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo

El compuesto del título se sintetizó según Ejemplo 234, Etapas G-J usando (R)-(1-metilpirrol¡d¡n-2-¡l)metanol en lugar de (S)-(1-metilpirrol¡d¡n-2-¡l)metanol en la Etapa F y 1-bromo-3-fluoro-2-(trifluorometil)benceno en lugar de 1-bromonaftaleno en la Etapa H. ES+APCI MS m/z 588,3 [M+H]+.
Ejemplo 275
2-(1-acriloil-4-(7-(2-fluoro-6-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

El compuesto del título se sintetizó según Ejemplo 210, Etapas E-G usando 2-bromo-1-fluoro-3-(trifluorometil)benceno en lugar de 1-bromonaftaleno en la Etapa E. ES+APCI MS m/z 588,3 [M+H]+.
Ejemplo 276
2-(1-acriloil-4-(7-(4-fluoro-2-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
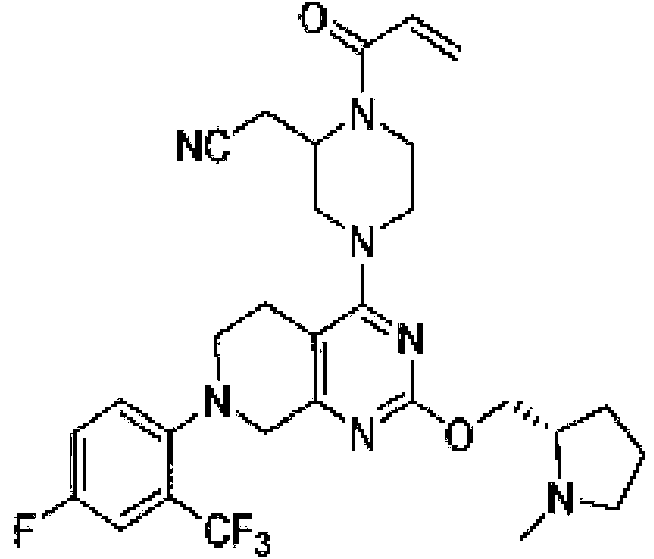
El compuesto del título se sintetizó según Ejemplo 210, Etapas E-G usando 1-bromo-4-fluoro-2-(trifluorometil)benceno en lugar de 1-bromonaftaleno en la Etapa E. ES+APCI MS m/z 588,3 [M+H]+.
Ejemplo 277
2-(1-acriloil-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
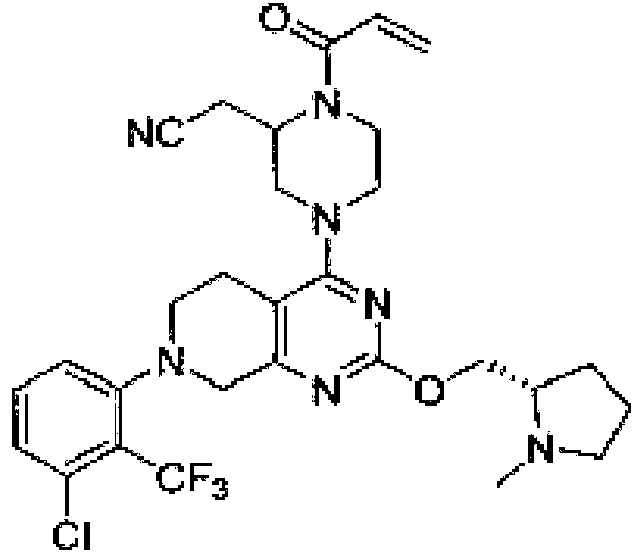
El compuesto del título se sintetizó según Ejemplo 210, Etapas E-G usando 1-bromo-3-cloro-2-(trifluorometil)benceno en lugar de 1-bromonaftaleno en la Etapa E y THF en place Et0H/THF en la Etapa F. ES+APCI MS m/z 604,3 [M+H]+. Ejemplo 278
2 -(1 -a c rilo il-4 -(2 -(((S )-1 -m e tilp irro lid in -2 - il)m e to x i)-7 -(q u in o lin -4 - il) -5 ,6 ,7 ,8 -te tra h id ro p ir id o [3 ,4 -d ]p ir im id in -4 -il)p ip e ra z in -2 -il)a ce to n itr ilo
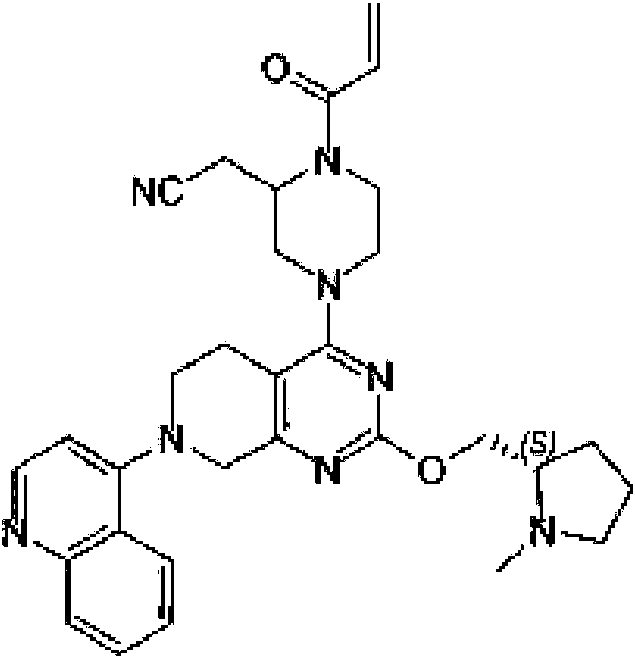
El compuesto del título se preparó como en el Ejemplo 210 Etapas E-G, sustituyendo 4-bromoquinolina por 1-bromo-2-(trifluoromet¡l)benceno en la etapa E y sustituyendo también prop-2-enoato de prop-2-enoílo por cloruro de acriloílo en la etapa G.
Ejemplo 279
2-(1-acriloil-4-(2-(2-(dimetilamino)etoxi)-7-(2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

El compuesto del título se preparó como en el Ejemplo 210, sustituyendo N, N-dimetil-etanolamina por (2S)-1-Etil-2-pirrolidinil]metanol en la etapa C y sustituyendo también prop-2-enoato de prop-2-enoílo por cloruro de acriloílo en la etapa G.
Ejemplo 280
2-(4-(2-(3-((1R,4R)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propoxi)-7-(2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-acriloilpiperazin-2-il)acetonitrilo

El compuesto del título se preparó como en el Ejemplo 210, sustituyendo 3-((1R,4R)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propan-1-ol por (2S)-1-Etil-2-pirrolidinil]metanol en la etapa C y sustituyendo también prop-2-enoato de prop-2-enoílo por cloruro de acriloílo en la etapa G.
Ejemplo 281
2-(1-acriloil-4-(2-(((R)-1-metilpirrolidin-3-il)metoxi)-7-(2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

El compuesto del título se preparó como en el Ejemplo 210, sustituyendo (R)-(1-metilpirrolidin-3-il)metanol por (2S)-1-Etil-2-pirrolidinil]metanol en la etapa C y sustituyendo también prop-2-enoato de prop-2-enoílo por cloruro de acriloílo en la etapa G.
Ejemplo 282
2-(1-(3-metilbut-2-enoil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

El compuesto del título se preparó como en el Ejemplo 210, sustituyendo 1-bromonaftaleno por 1-bromo-2-(trifluorometil)benceno en la etapa E y cloruro de 3-metilbut-2-enoílo por cloruro de acriloílo en la etapa G.
Ejemplo 283
2-(1-acriloil-4-(2-(((R)-1-metilpirrolidin-2-il)metoxi)-7-(2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

El compuesto del título se preparó como en el Ejemplo 210, sustituyendo (2R)-1-Et¡l-2-p¡rrol¡d¡n¡l]metanol por (2S)-1-Etil-2-pirrolidinil)metanol en la etapa C y sust¡tuyendo tamb¡én prop-2-enoato de prop-2-enoílo por cloruro de acr¡lαlo en la etapa G.
Ejemplo 284
2-(1-acr¡lo¡l-4-(2-(2-(d¡et¡lam¡no)etox¡)-7-(2-(tr¡fluoromet¡l)fen¡l)-5,6,7,8-tetrah¡dropir¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-il)acetonitrilo
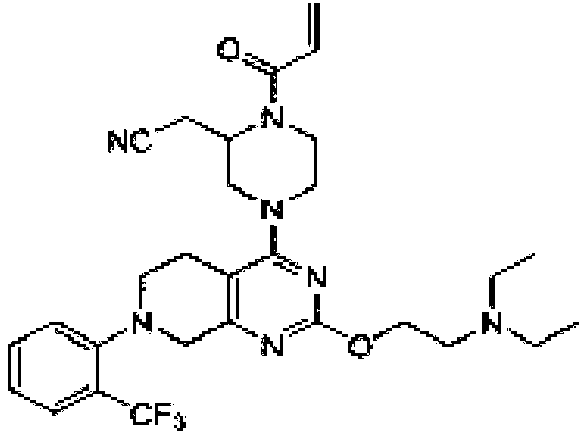
El compuesto del título se preparó como en el Ejemplo 210, sustituyendo N, N-dietil-etanolamina por (2S)-1 -Etil-2-pirrolid¡n¡l]metanol en la etapa C y sustituyendo también prop-2-enoato de prop-2-enoílo por cloruro de acriloílo en la etapa G.
Ejemplo 285

2-(c¡anomet¡l)-4-[2-[[(2S)-1-¡soprop¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naftil)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]piperazin-1-carbox¡lato de tere-butilo

Etapa 1: (S)-pirrolidin-2-ilmetanol. A una solución de (S)-2-(hidroximetil)pirrolidin-1-carboxilato de ferc-butilo (2 g, 9,94 mmol, 1 eq.) en CH2Cl2 (40 ml) se le añadió HCl (4 M en dioxano, 49,69 ml, 20 eq.) a 0 °C en una atmósfera de nitrógeno. Después de agitar a 15 °C durante 30 min, la mezcla se concentró al vacío. El compuesto (S)-pirrolidin-2-ilmetanol (1,37 g, 9,96 mmol, 100 % de rendimiento, HCl) en forma de un sólido de color amarillo.
Etapa 2: [(2S)-1-isopropilpirrolidin-2-il)metanol. Una mezcla de [(2S)-pirrolidin-2-il]metanol (750 mg, 7,41 mmol, 724 ul, 1 eq.) y acetona (4,31 g, 74,2 mmol, 5,46 ml, 10 eq.) en Me0H (20 ml) se hidrogenó en una atmósfera de H2 (206,84 kPa (30 psi)) con Pd/C (100 mg, 7,41 mmol, 10 % de pureza, 1 eq.) como catalizador a 15 °C durante 3 horas. La mezcla se filtró y se concentró al vacío. El compuesto [(2S)-1-isopropilpirrolidin-2-il]metanol (0,821 g, 5,73 mmol, 77,3 % de rendimiento) se obtuvo en forma de un aceite de color amarillo.
1H RMN (400 MHz, metanol-d4) ó = 3,54 (dd, J = 4,0, 10,8 Hz, 1H), 3,40 - 3,35 (m, 1H), 2,99 - 2,86 (m, 3H), 2,60 - 2,50 (m, 1H), 1,94 - 1,82 (m, 1H), 1,82 - 1,66 (m, 3H), 1,15 (d, J = 6,4 Hz, 3H), 1,08 (d, J = 6,4 Hz, 3H).
Etapa A: 2-(cianometil)-4-[2-[[(2S)-1-isopropilpirrolidin-2-il1metoxi1-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-illpiperazin-1-carboxilato de ferc-butilo. A una mezcla de 2-(cianometil)-4-[2-metilsulfinil-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (300 mg, 549 umol, 1 eq.) y [(2S)-1-isopropilpirrolidin-2-il]metanol (157 mg, 1,10 mmol, 2 eq.) en THF (5 ml) se le añadió t-Bu0Na (158 mg, 1,65 mmol, 3 eq.). Después de agitar a 20 °C durante 0,5 horas, la mezcla de reacción se neutralizó con HCl (1 mol/l) a pH = 7 a 0 °C y después la mezcla se concentró a presión reducida. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de TFA)/acetonitrilo]. Las fracciones deseadas se recogieron y se neutralizaron con una solución saturada de NaHC03 y se extrajeron con acetato de etilo (200 ml). La capa orgánica se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El compuesto 2-(cianometil)-4-[2-[[(2S)-1-isopropilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (174 mg, 278 umol, 50,7 % de rendimiento, 100 % de pureza) se obtuvo en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 626.
Etapa B: 2-(4-[2-[[(2S)-1-isopropilpirrolidin-2-i n metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-c f ]pirimidin-4-il]piperazin-2-iHacetonitrilo. A una solución de 2-(cianometil)-4-[2-[[(2S)-1-isopropilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-C]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (174 mg, 278 umol, 1 eq.) en CH2Cl2 (5 ml) se le añadió TFA (7,70 g, 67,5 mmol, 5,00 ml, 243 eq.) a 0 °C en una atmósfera de nitrógeno. Después de agitar a 15 °C durante 0,5 h, la mezcla se concentró al vacío. El compuesto 2-[4-[2-[[(2S)-1-isopropilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-C]pirimidin-4-il]piperazin-2-il]acetonitrilo (178 mg, bruto, TFA) se obtuvo en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 526.
Etapa C: 2-[4-[2-[[(2S)-1-isopropilpirrolidin- 2-i n metox n -7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-cf|pirimidin-4-i n -1-prop-2-enoilpiperazin-2-i n acetonitrilo. A una mezcla de 2-[4-[2-[[(2S)-1 -isopropilpirrolidin-2-il]metoxi]- 7-(1 -naftil)-6,8-dihidro-5H-pirido[3,4-C]pirimidin-4-il]piperazin-2-il]acetonitrilo (178 mg, bruto, TFA) y DIEA (360 mg, 2,78 mmol, 485 ul) en CH2Cl2 (5 ml) se le añadió prop-2-enoato de prop-2-enoílo (28,1 mg, 223 umol) a 0 °C. La mezcla se agitó a 20 °C durante 1 hora. La mezcla de reacción se inactivó con una solución saturada de NaHC03 (5 ml) a 0 °C y después se extrajo con CH2Cl2 (2 * 25 ml).
Las capas orgánicas combinadas se lavaron con salmuera (1 * 50 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Boston Green 0DS 150 * 305 u; fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 27 % - 54 %, 10 min). El compuesto del título 2-[4-[2-[[(2S)-1 -isopropilpirrolidin- 2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (11,7 mg, 18,3 umol, dos etapas 6,58 % de rendimiento, 97,9 % de pureza, AF) se obtuvo en forma de un sólido de color pardo. LCMs [ESI, M+1]: 580.
1H RMN (400 MHz, ácido acét¡co-d4) 5 = 8,28 - 8,19 (m, 1H), 8,08 (s, 1H), 7,91 - 7,83 (m, 1H), 7,63 (d, J = 8,4 Hz, 1H), 7,56 - 7,47 (m, 2H), 7,44 (t, J = 7,6 Hz, 1H), 7,20 (d, J = 7,2 Hz, 1H), 6,94 - 6,71 (m, 1H), 6,38 (d a, J = 16,8 Hz, 1H), 5,87 (d a, J = 10,4 Hz, 1H), 5,15 (s a, 1H), 4,92 - 4,54 (m, 4H), 4,42 (d a, J = 12,0 Hz, 1H), 4,34 (s a, 2H), 4,13 (s a, 2H), 3,95 (s a, 1H), 3,85 - 3,21 (m, 7H), 3,20 - 2,84 (m, 3H), 2,42 - 2,25 (m, 1H), 2,24 - 2,07 (m, 3H), 1,43 (d, J = 6,4 Hz, 3H), 1,39 (d, J = 6,4 Hz, 3H).
Ejemplo 286

2-(1-acriloil-4-(7-(7-cloronaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa 1: trifluorometanosulfonato de í7-cloro-3.4-d¡h¡dronaftalen-1-ilo). A una mezcla de 7-clorotetralin-1-ona (2 g, 11,1 mmol, 1 eq.) y DIEA (4,29 g. 33,2 mmol, 5,79 ml. 3 eq.) en DCM (35 ml) se le añadió en una porción Tf20 (4.69 g. 16,6 mmol. 2,74 ml. 1.5 eq.) a 0 °C en una atmósfera de N2. La mezcla se agitó a 25 °C durante 5 horas. La mezcla de reacción se concentró a presión reducida. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 200/1 a 50/1). El compuesto trifluorometanosulfonato de (7-cloro-3,4-d¡h¡dronaftalen-1-¡lo) (2,25 g. 7,02 mmol. 63,4 % de rendimiento. 97,6 % de pureza) se obtuvo en forma de un aceite incoloro.
1H RMN (400 MHz, cloroformo-d) ó = 7,32 (d, J = 2.0 Hz. 1H). 7,24 (dd. J = 2.0. 8.0 Hz. 1H). 7,12 (d. J = 8.0 Hz. 1H). 6,10 (t. J = 4.8 Hz. 1H). 2,88 - 2,80 (m. 2H). 2,53 (dt. J = 4.8. 8.0 Hz. 2H).
Etapa 2: trifluorometanosulfonato de (7-cloro-1-naftilo). A una mezcla de trifluorometanosulfonato de (7-cloro-3,4-dihidronaftalen-1 -ilo) (1,57 g. 5,02 mmol. 1 eq.) en dioxano (35 ml) se le añadió en una porción DDQ (2,28 g. 10.0 mmol. 2 eq.) en una atmósfera de N2. La mezcla se agitó a 25 °C durante 30 min, después se calentó a 105 °C y se agitó durante 5 horas. La mezcla de reacción se concentró a presión reducida para eliminar el 1,4-dioxano. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 200/1 a 50/1). El compuesto trifluorometanosulfonato de (7-cloro-1 -naftilo) (1,24 g. 3,94 mmol. 78,5 % de rendimiento. 98,7 % de pureza) se obtuvo en forma de un aceite incoloro.
1H RMN (400 MHz. cloroformo-d) ó = 8,05 (d. J = 2.0 Hz. 1H). 7,87 - 7,83 (m. 2H). 7,57 - 7,47 (m. 3H).
Etapa A: 4-[7-(7-cloro-1-naftil)-2 -[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de ferc-butilo. A una mezcla de 2-(c¡anomet¡l)-4-[2-[(1-met¡lp¡rrol¡d¡n-2-¡l)metox¡]-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1 -carboxilato de ferc-butilo (200 mg. 424 umol, 1 eq.) y trifluorometanosulfonato de (7-cloro-1 -naftilo) (264 mg. 848 umol. 2 eq.) en tolueno (20 ml) se le añadieron Pd2(dba)3 (38,8 mg. 42,4 umol. 0.1 eq.). RuPhos (39,6 mg. 84,8 umol. 0.2 eq.) y Cs2C03 (414 mg. 1,27 mmol. 3 eq.) en una atmósfera de N2. La mezcla se calentó a 110 °C y se agitó durante 5 horas. La mezcla se diluyó con agua (10,0 ml) y se extrajo con acetato de etilo (3 x 15,0 ml). Los extractos se lavaron con salmuera (1 x 20.0 ml). se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0.1 % de TFA)/acetonitrilo]. Las fracciones deseadas se recogieron y se neutralizaron con una solución saturada de NaHC03 y se extrajeron con acetato de etilo (1 x 30 ml). La capa orgánica se secó sobre sulfato de sodio. se filtró y se concentró al vacío. El compuesto 4-[7-(7-cloro-1-naftil)-2 -[[(2s)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de ferc-butilo (200 mg. 316 umol. 74,6 % de rendimiento. 100 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 632.
1H RMN (400 MHz. cloroformo-d) ó = 8,20 (d. J = 2.0 Hz. 1H). 7,80 (d. J = 8.8 Hz. 1H). 7,59 (d. J = 8.2 Hz. 1H). 7,48 -7,38 (m. 2H). 7,18 (d. J = 7.2 Hz. 1H). 4,63 (s a. 1H). 4,43 (dd a. J = 4.8. 10,3 Hz. 1H). 4,29 - 4,16 (m. 3H). 4,09 - 3,89 (m. 2H). 3,42 (s a. 1H). 3,35 - 3,20 (m. 3H). 3,17 - 2,65 (m. 7H). 2,52 (s. 3H). 2,33 (d a. J = 7.6 Hz. 1H). 2,14 - 2,06 (m. 1H). 1,88 - 1,71 (m. 4H).
Etapa B: 2-[4-[7-(7-cloro-1-naftil)- 2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-illacetonitrilo. A una mezcla de 4-[7-(7-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de ferc-butilo (200 mg.316 umol. 1 eq.) en DCM (2.5 ml) se le añadió en una porción TFA (541 mg. 4,75 mmol. 351 ul. 15 eq.) a 0 °C en una atmósfera de N2. Después. la mezcla se agitó a 25 °C durante 2 horas. La mezcla de reacción se concentró a presión reducida. El compuesto 2-[4-[7-(7-cloro-1-naftil)- 2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (204 mg. 316 umol. 99,8 % de rendimiento. TFA) se obtuvo en forma de un sólido de color pardo. LCMS [ESI, M+1]: 532.
Etapa C: 2-[4-[7-(7-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-enoil-piperazin-2-illacetonitrilo. A una mezcla de 2-[4-[7-(7-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (204 mg. 316 umol. 1 eq.. TfA) y TEA (160 mg. 1,58 mmol. 220 ul. 5 eq.) en DCM (1.5 ml) se le añadió en una porción prop-2-enoato de prop-2-enoílo (39,8 mg. 316 umol. 1 eq.) a 0 °C. Después de agitar a 25 °C durante 1 h. la mezcla de reacción se inactivó con una solución saturada de NaHC03 (1 ml) a 0 °C. después se diluyó con agua (2 ml) y se extrajo con DCM (5 ml x 3 ). Las capas orgánicas combinadas se lavaron con agua (5 ml x 1 ). se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida. El residuo se purificó por HPLC prep. (columna: Boston Green 0DS 150 x 30 5 u; fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 30 % -54 %, 10 min). El compuesto del título 2-[4-[7-(7-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (36,0 mg. 56,1 umol. 17,8 % de rendimiento. 98,7 % de pureza. AF) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 586.
1H RMN (400 MHz. cloroformo-d) ó = 8,42 (s a. 1H). 8,19 (d. J = 1.6 Hz. 1H). 7,80 (d. J = 8.8 Hz. 1H). 7,60 (d. J = 8.0 Hz. 1H). 7,47 - 7,41 (m. 2H). 7,19 (d. J = 7.2 Hz. 1H). 6,61 (s a. 1H). 6,40 (d a. J = 16,8 Hz. 1H). 5,84 (d a. J =10,8 Hz. 1H). 5,08 (s a. 1H). 4,91 - 4,75 (m. 2H). 4,48 (td. J = 4.4. 12,0 Hz. 1H). 4,23 (s a. 3H). 4,07 (d a. J = 11,2 Hz. 1H). 3,82 - 3,58 (m. 2H). 3,56 - 3,08 (m. 6H). 3,08 - 2,92 (m. 2H). 2,89 (d. J = 1.6 Hz. 3H). 2,85 - 2,70 (m. 2H). 2,28 (cd. J = 8.4. 12,5 Hz. 1H). 2,22 - 2,11 (m. 1H). 2,11 - 1,94 (m. 2H).
Ejemplo 287

2-[4-[7-(5-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2 -il]acetonitrilo

Etapa A: 2-(cianometil)-4-[7-(5-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de tere-butilo. A una solución de 2-(cianometil)-4-[2-[(1-metilpirrolidin-2-il)metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-o (200 mg, 424 umol, 1,30 eq.) y 1-bromo-5-fluoronaftaleno (73,4 mg, 326 umol, 1,0 eq.) en tolueno (5,0 ml) se le añadieron CS2C03 (319 mg, 979 umol, 3,0 eq.), Pd2(dba)3 (29,9 mg, 32,6 umol, 0,10 eq.) y RuPhos (22,8 mg, 48,9 umol, 0,15 eq.). La mezcla se agitó a 110 °C durante 12 h. La mezcla de reacción se concentró a presión reducida para eliminar el tolueno. El residuo se diluyó con H20 (10,0 ml) y se extrajo con acetato de etilo (10,0 ml x 3). Las capas orgánicas combinadas se lavaron con H20 (10,0 ml) y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,100 % de ácido fórmico)/acetonitrilo]. El compuesto 2-(cianometil)-4-[7-(5-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de íere-butilo (140 mg, 211 umol, 64,5 % de rendimiento) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 616.
Etapa B: 2-[4-[7-(5-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-i n metox n -6,9-dihidro-5H-pirido[3,4-dlpirimidin-4-i n piperazin-2-il]acetonitrilo. A una solución de 2-(cianometil)-4-[7-(5-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de terc-butilo (130 mg, 211 umol, 1,0 eq.) en DCM (2,0 ml) se le añadió TFA (3,08 g, 27,0 mmol, 2,00 ml, 128 eq.). La mezcla se agitó a 25 °C durante 1 h en una atmósfera de N2. El disolvente orgánico se eliminó al vacío. El compuesto 2-[4-[7-(5-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (300 mg, bruto) se obtuvo en forma de un aceite de color amarillo. El producto bruto se usó directamente para la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 517.
Etapa C: 2-[4-[7-(5-fluoro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metoxi1-6.8-d¡h¡dro-5H-p¡rido[3.4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-enoil-piperazin-2-illacetonitrilo. A una soluc¡ón de 2-[4-[7-(5-fluoro-1-naft¡l)-2-[[(2S)-1-met¡lpirrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo (300 mg. TFA) en DCM (3.0 ml) se le añadieron Et3N (200 mg.
1.98 mmol.275 ul) y prop-2-enoato de prop-2-enoílo (40.0 mg.317 umol) a 0 °C. La mezcla se agitó a 20 °C durante 2 h. La reacción se lavó con agua (10.0 ml). La mezcla bruto se extrajo con acetato de etilo (20.0 ml x 3). Los extractos combinados se lavaron con salmuera (50.0 ml). se secaron con Na2SÜ4 y después el disolvente se eliminó al vacío. El residuo se purificó por HPLC prep. (columna: Boston Green ÜDS 150 * 305 ufase móvil: [agua (0.225 % de AF)-ACN1;% de B: 26 %-53 %.10 min) y se sometió a liofilización. El compuesto del título 2-[4-[7-(5-fluoro-1-naftil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡mid¡n-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1aceton¡tr¡lo (48.4 mg. 82.6 umol. dos etapas 40.0 % de rendimiento. AF) se obtuvo en forma de un sólido de color blanquecino. LCMS [ESI. M+11: 570.
1H RMN (400 MHz. CDCls) 5 = 8,41 (s, 1H), 7,91 (d, J = 7.6 Hz. 1H). 7.80 (d. J = 8.4 Hz. 1H). 7.42 (t. J = 7.8 Hz. 1H).
7.38-7.32 (m. 1H).7.13-7.07 (m. 2H). 6.54-6.52 (m. 1H). 6.33 (m. 1H).5.76 (d. J = 10.4 Hz. 1H).5.10-4.98 (a.4H).4.74 4.69 (m. 1H). 4.41-4.36 (m. 1H). 4.16 (m. 3H). 3.99 (d. J = 12.0 Hz. 1H). 3.61-3.60 (m. 1H). 3.14-3.29 (m. 4H). 3.11 (a.
2H). 2.92-2.85 (m. 1H). 2.78 (s.3H). 2.74-2.67 (m. 2H). 2.21-2.16 (m. 1H). 2.09-2.04 (m. 1H). 1.98-1.91 (m. 2H).
Ejemplo 288

2-[4-[7-(7-metox¡-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡hidro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡lp¡peraz¡n-2-¡l1aceton¡tr¡lo


Etapa 1: trifluorometanosulfonato de (7-metoxi-3,4-dihidronaftalen-1 -ilo). A la solución de 7-metoxitetralin-1-ona (1 g, 5,67 mmol, 1 eq.), DIEA (2,20 g, 17,0 mmol, 2,97 ml, 3 eq.) en DCM (15 ml) se le añadió gota a gota Tf20 (2,40 g, 8,51 mmol, 1,40 ml, 1,5 eq.) y después la mezcla se agitó a 20 °C durante 12 horas. La mezcla de reacción se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc de 1:0 a 100:1) para dar trifluorometanosulfonato de (7-metoxi-3,4-dihidronaftalen-1-ilo) (1,7 g, 5,24 mmol, 92,3 % de rendimiento, 95,0 % de pureza) en forma de un aceite de color amarillo.
1H RMN (400 MHz, cloroformo-d) ó = 7,06 (d, J = 8,4 Hz, 1H), 6,75 (dd, J = 2,4, 8,4 Hz, 1H), 6,66 (d, J = 2,4 Hz, 1H), 6,45 (s, 1H), 3,80 (s, 3H), 3,04 - 2,96 (m, 2H), 2,68 (t, J = 8,4 Hz, 2H).
Etapa 2: trifluorometanosulfonato de (7-metoxi-1-naftilo). Una mezcla de trifluorometanosulfonato de (7-metoxi-3,4-dihidronaftalen-1 -ilo) (1,7 g, 5,51 mmol, 1 eq.) y DDQ (2,50 g, 11,0 mmol, 2 eq.) en dioxano (30 ml) se agitó a 105 °C durante 12 horas. La mezcla de reacción se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc de 1:0 a 50:1) para dar trifluorometanosulfonato de (7-metoxi-1-naftilo) (1,41 g, 4,14 mmol, 75,1 % de rendimiento, 90,0 % de pureza) en forma de un aceite incoloro.
1H RMN (400 MHz, cloroformo-d) ó = 7,84 (d, J = 9,2 Hz, 1H), 7,78 (d, J = 8,8 Hz, 1H), 7,65 (d, J = 2,4 Hz, 1H), 7,24 -7,21 (m, 2H), 7,15 (d, J = 2,4 Hz, 1H), 3,94 (s, 3H).
Etapa A: 2-(cianometil)-4-[7-(7-metoxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de tere-butilo. A la solución de 2-(cianometil)-4-[2-[(1-metilpirrolidin-2-il)metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (Intermedio 66, 200 mg, 424 umol, 1 eq.), trifluorometanosulfonato de (7-metoxi-1-naftilo) (260 mg, 848 umol, 2 eq.), RuPhos (39,6 mg, 84,8 umol, 0,2 eq.) y Cs2C03 (414 mg, 1,27 mmol, 3 eq.) en tolueno (6 ml) se le añadió Pd2(dba)3 (38,8 mg, 42,4 umol, 0,1 eq.) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con N2. La mezcla se agitó en una atmósfera de N2 a 90 °C durante 12 horas. A la mezcla se le añadió agua (5 ml). La mezcla resultante se diluyó con EtOAc (5 ml) y se extrajo con EtOAc (3 * 10 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0,1 % de Af ) = 34 %) para dar 2-(cianometil)-4-[7-(7-metoxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de tere-butilo (100 mg, 151 umol, 35,7 % de rendimiento, 95,0 % de pureza) en forma de un sólido de color pardo. LCMS [ESI, M+1]: 628.
1H RMN (400 MHz, cloroformo-d) ó = 7,69 (d, J = 9,2 Hz, 1H), 7,63 (d, J = 8,8 Hz, 1H), 7,17 (dd, J = 2,4, 9,2 Hz, 1H), 7,09 (d, J = 2,0 Hz, 1H), 7,03 (d, J = 2,4 Hz, 1H), 6,96 (dd, J = 2,4, 8,8 Hz, 1H), 4,61 (s a, 1H), 4,52 - 4,30 (m, 3H), 4,20 (ddd, J = 3,2, 6,8, 10,4 Hz, 1H), 4,02 (d a, J = 13,6 Hz, 2H), 3,92 (s, 3H), 3,87 (d a, J = 13,2 Hz, 1H), 3,76 - 3,68 (m, 1H), 3,55 - 3,43 (m, 1H), 3,25 (dd a, J = 3,6, 13,6 Hz, 2H), 3,12 (t a, J = 7,6 Hz, 1H), 3,05 - 2,94 (m, 1H), 2,92 - 2,77 (m, 3H), 2,76 -2,65 (m, 2H), 2,51 (s, 3H), 2,35 - 2,26 (m, 1H), 2,14 - 2,06 (m, 1H), 1,92 - 1,74 (m, 3H), 1,52 (s, 9H).
Etapa B: 2-[4-[7-(7-metoxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. Una mezcla de 2-(cianometil)-4-[7-(7-metoxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (100 mg, 159 umol, 1 eq.) y TFA (363 mg, 3,19 mmol, 236 ul, 20 eq.) se agitó a 18 °C durante 1 hora. La mezcla de reacción se concentró al vacío para dar 2-[4-[7-(7-metox¡-1 -naftil)-2-[[(2S)-1 -met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡perazin-2-¡l]aceton¡tr¡lo (120 mg, bruto, 2TFA) en forma de un aceite de color pardo.
Etapa C: 2-[4-[7-(7-metoxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-prop-2-enoil-piperazin-2-il1acetonitrilo. A la solución de 2-[4-[7-(7-metoxi-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (120 mg, 159 umol, 1 eq., 2 TFA) y DIEA (205 mg, 1,59 mmol, 276 ul, 10 eq.) en DCM (2,5 ml) se le añadió prop-2-enoato de prop-2-enoílo (24,0 mg, 190 umol, 1,2 eq.) a 0 °C y se agitó a 18 °C durante 1 hora. A la mezcla se le añadió agua (5 ml). La mezcla resultante se extrajo con DCM (3 * 5 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Gemini 150 * 255 ufase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN];% de B: 45 %-75 %,10 min) para dar el compuesto del título 2-[4-[7-(7-metoxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (10,6 mg, 17,8 umol, dos etapas 11,2 % de rendimiento, 97,6 % de pureza) en forma de un sólido de color blanquecino. LCMS [ESI, M+1]: 582.
1H RMN (400 MHz, cloroformo-d) 5 = 7,70 (d, J = 8 ,8 Hz, 1H), 7,63 (d, J = 8 ,8 Hz, 1H), 7,17 (dd, J = 2,4, 8 ,8 Hz, 1H), 7,09 (d, J = 2,0 Hz, 1H), 7,03 (d, J = 2,4 Hz, 1H), 6,97 (dd, J = 2,4, 8 ,8 Hz, 1H), 6,57 (d a, J = 10,4 Hz, 1H), 6,43 - 6,36 (m, 1H), 5,83 (d a, J = 10,8 Hz, 1H), 5,09 (s a, 1H), 4,52 - 4,31 (m, 3H), 4,25 - 4,15 (m, 1H), 4,08 (d a, J = 12,4 Hz, 1H), 3,96 (s a, 1H), 3,92 (s, 3H), 3,72 (d a, J = 12,4 Hz, 1H), 3,54 (d a, J = 5,6 Hz, 2H), 3,30 (d a, J = 12,4 Hz, 1H), 3,22 - 2,95 (m, 3H), 2,93 (d, J = 8,4 Hz, 1H), 2,86 (d a, J = 4,8 Hz, 2H), 2,79 - 2,65 (m, 1H), 2,51 (s, 3H), 2,36 - 2,26 (m, 1H), 2,16 - 2,02 (m, 1H), 1,91 - 1,67 (m, 4H).
Ejemplo 289

2-[4-[7-(1-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡lpiperazin-2 -il]acetonitrilo

Etapa A: 2-(c¡anomet¡l)-4-[7-(1-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-il1piperazin-1-carboxilato de ferc-but¡lo. Una mezcla de 2-(c¡anomet¡l)-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de terc-but¡lo (0,20 g, 424 umol, 1,00 eq.), 1-bromo¡soqu¡nol¡na (132 mg, 636 umol, 1,50 eq.) y DIEA (109 mg, 848 umol, 148 ul, 2,00 eq.) en DMS0 (4,00 ml) se ag¡tó a 110 °C durante 3 horas. La mezcla se d¡luyó con agua (5,00 ml) y se extrajo con acetato de et¡lo (3 * 5,00 ml). Las capas orgán¡cas se lavaron con salmuera (1 * 10,0 ml), se secaron sobre Na2S04, se f¡ltraron y se concentraron al vacío. El res¡duo se pur¡f¡có por cromatografía ultrarráp¡da de fase ¡nversa [agua (AF, 0,10 %)/aceton¡tr¡lo]. Las fracc¡ones deseadas se recog¡eron y se ajustaron a pH >7 con una soluc¡ón saturada de b¡carbonato de sod¡o (6,00 ml) y se extrajeron con acetato de etilo (3 * 20,0 ml). Las capas orgán¡cas se lavaron con salmuera (1 * 30,0 ml), se secaron sobre Na2S04, se f¡ltraron y se concentraron al vacío para dar 2-(c¡anomet¡l)-4-[7-(1-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de terc-but¡lo (0,15 g, 233 umol, 54,9 % de rend¡m¡ento) en forma de un sól¡do de color amar¡llo. LCMS [ESI, M+1]: 599.
1H RMN (400 MHz, cloroformo-d) 8 = 8,17 - 8,11 (m, 2H), 7,78 (d, J = 8,0 Hz, 1H), 7,64 (t, J = 7,2 Hz, 1H), 7,57 - 7,51 (m, 1H), 7,28 (s, 1H), 4,69 - 4,55 (m, 3H), 4,42 - 4,35 (m, 1H), 4,15 - 4,01 (m, 3H), 3,96 - 3,81 (m, 2H), 3,57 (ddd, J = 4,0, 8,0, 16,8 Hz, 1H), 3,27 (dd, J = 4,0, 13,6 Hz, 1H), 3,18 - 3,16 (m, 1H), 3,16 - 2,95 (m, 3H), 2,91 - 2,73 (m, 3H), 2,70 - 2,62 (m, 1H), 2,48 (s, 3H), 2,34 - 2,24 (m, 1H), 2,07 - 2,01 (m, 1H), 1,91 - 1,73 (m, 3H), 1,52 (s, 9H).
Etapa B: 2-[4-[7-(1-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-illacetonitrilo. Una mezcla de 2-(c¡anomet¡l)-4-[7-(1-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de ferc-but¡lo (0,15 g, 250,53 umol, 1,00 eq.) y TFA (428 mg, 3,76 mmol, 278 ul, 15,0 eq.) en d¡clorometano (0,40 ml) se agitó a 15 °C durante 1 hora. La mezcla se concentró al vacío para dar 2-[4-[7-(1-¡soqu¡nol¡l)-2-[[(2s)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (153 mg, bruto, TFA) en forma de un ace¡te de color negro y se usó en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón ad¡c¡onal. LCMS [ESI, M+1]: 499.
Etapa C: 2-[4-[7-(1-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H- p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una soluc¡ón de 2-[4-[7-(1-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (153 mg, bruto, TfA) y TEA (253 mg, 2,50 mmol, 348 ul) en d¡clorometano (4,00 ml) se le añad¡ó prop-2-enoato de prop-2-enoílo (31,5 mg, 250 umol) a 0 °C y después se ag¡tó a 15 °C durante 1 hora. La mezcla se ¡nact¡vó con una soluc¡ón saturada de b¡carbonato de sod¡o (1,00 ml) y se concentró al vacío. El res¡duo se pur¡f¡có por columna de HPLC prep.: (Phenomenex Synerg¡ C18150 * 25 * 10 um; fase móv¡l: [agua (0,225 %, AF) - ACN]; % de B: 10 % - 24 %, 7 m¡n) y (columna: Phenomenex Synerg¡ C18150 * 25 * 10 um; fase móv¡l: [agua (0,225 %, a F) - ACN]; % de B: 10 % - 24 %, 7 m¡n). Las fracc¡ones deseadas se recog¡eron y se l¡of¡l¡zaron para dar el compuesto del título 2-[4-[7-(1-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H- p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (31,2 mg, 52,1 umol, dos etapas 20,9 % de rend¡m¡ento, AF) en forma de un sól¡do de color amar¡llo. LCMS [ESI, M+1]: 553.
1H RMN (400 MHz, acét¡co) 8 = 8,44 (d, J = 8,4 Hz, 1H), 8,04 - 7,95 (m, 2H), 7,89 (d, J = 6,4 Hz, 1H), 7,82 (t a, J = 6,8 Hz, 1H), 7,49 (d, J = 6,8 Hz, 1H), 6,91 - 6,69 (m, 1H), 6,37 (d a, J = 16,4 Hz, 1H), 5,87 (d a, J = 9,2 Hz, 1H), 5,10 (s a, 2H), 4,78 (s a, 3H), 4,53 (d a, J = 10,8 Hz, 1H), 4,32 (d a, J = 12,8 Hz, 1H), 4,13 (s a, 3H), 3,90 (s a, 2H), 3,79 -3,40 (m, 3H), 3,26 (s a, 3H), 3,12 (s a, 3H), 3,07 - 2,84 (m, 2H), 2,45 - 2,34 (m, 1H), 2,23 - 2,09 (m, 3H).
Ejemplo 290

-[4-[7-(6-fluoro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡lp¡peraz¡n-2-¡l]aceton¡tr¡lo

Inserción: trifluorometanosulfonato de (6-fluoro-1-naftilo). A una solución de 6-fluoronaftalen-1-ol (0,10 g, 617 umol, 1.00 eq.) y DIEA (159 mg, 1,23 mmol, 215 ul, 2,00 eq.) en diclorometano (3,00 ml) se le añadió trifluorometanosulfonato de trifluorometilsulfonilo (191 mg, 678 umol, 112 ul, 1,10 eq.) a 0 °C. Después de agitar a 0 °C durante 1 hora, la mezcla se diluyó con agua (5,00 ml) y se extrajo con diclorometano (3 * 5,00 ml). Las capas orgánicas se lavaron con salmuera (1 * 5,00 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna (SiÜ2, éter de petróleo/acetato de etilo = 3/1) para dar trifluorometanosulfonato de (6-fluoro-1-naftilo) (0,17 g, 543 umol, 88,1 % de rendimiento) en forma de un aceite incoloro.
1H RMN (400 MHz, cloroformo-d) ó = 8,10 (dd, J = 5,2, 8,8 Hz, 1H), 7,83 (d, J = 8,4 Hz, 1H), 7,58 - 7,50 (m, 2H), 7,48 - 7,40 (m, 2H).
Etapa A: 2-(cianometil)-4-[7-(6-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-illpiperazin-1-carboxilato de tere-butilo. Una mezcla de 2-(cianometil)-4-[2-[(1-metilpirrolidin-2-il)metoxi]-5,6,7,8-tetrahidropirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de íere-butilo (0,20 g, 424 umol, 1,00 eq.), trifluorometanosulfonato de (6-fluoro-1-naftilo) (150 mg, 509 umol, 1,20 eq.), RuPhos (39,6 mg, 84,8 umol, 0,20 eq.), Pd2(dba)3 (38,8 mg, 42,4 umol, 0,10 eq.) y Cs2CÜ3 (415 mg, 1,27 mmol, 3,00 eq.) en tolueno (2,00 ml) se agitó a 110 °C durante 3 horas en una atmósfera de N2. La mezcla se diluyó con agua (5,00 ml) y se extrajo con acetato de etilo (3 * 5.00 ml). Las capas orgánicas se lavaron con salmuera (1 * 10,0 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (AF, 0,10 %)/acetonitrilo]. Las fracciones deseadas se recogieron y se ajustaron a pH >7 con una solución saturada de bicarbonato de sodio (5,00 ml), después se extrajeron con acetato de etilo (3 * 20,0 ml) y las capas orgánicas se lavaron con salmuera (1 * 30.0 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío para dar 2-(cianometil)-4-[7-(6-fluoro-1-naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1 -carboxilato de íere-butilo (0,20 g, 322 umol, 75,8 % de rendimiento) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 616.
1H RMN (400 MHz, cloroformo-d) ó = 8,22 (dd, J = 5,6, 9,2 Hz, 1H), 7,57 - 7,52 (m, 1H), 7,48 - 7,42 (m, 2H), 7,29 -7,24 (m, 1H) 7,10 (d, J = 7,6 Hz, 1H), 4,62 (s a, 1H), 4,39 (dd, J = 4,8, 10,4 Hz, 1H), 4,31 - 4,22 (m, 2H), 4,14 - 4,00 (m, 3H), 3,95 (d a, J = 12,8 Hz, 1H), 3,44 (s a, 1H), 3,35 - 3,25 (m, 2H), 3,23 - 2,90 (m, 4H), 2,86 - 2,63 (m, 4H), 2,49 (s, 3H), 2,33 - 2,24 (m, 1H), 2,13 - 2,00 (m, 1H) 1,89 - 1,74 (m, 3H), 1,53 (s, 9H).
Etapa B: 2-[4-[7-(6-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-b]pirimidin-4-¡ n piperazin-2-i n acetonitrilo. Una mezcla de 2-(cianometil)-4-[7-(6-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-b]pirimidin-4-il]piperazin-1-carboxilato de íere-butilo (0,20 g, 322 umol, 1,00 eq.) y TFA (556 mg, 4,87 mmol, 361 ul, 15,0 eq.) en diclorometano (0,40 ml) se agitó a 15 °C durante 1 hora. La mezcla se concentró al vacío para dar 2-[4-[7-(6-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-b]pirimidin-4-il]piperazin-2-il]acetonitrilo (205 mg, bruto, TFA) en forma de un aceite de color negro y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 516.
Etapa C: 2-[4-[7-(6-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-il1-1-prop-2-enoil-piperazin-2-il1acetonitrilo. A una solución de 2-[4-[7-(6-fluoro-1-naftil)-2-[[(2S)- 1 -metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (205 mg, bruto, TFA) y TEA (328 mg, 3,24 mmol, 451 ul) en diclorometano (4,00 ml) se le añadió prop-2-enoato de prop-2-enoílo (40,9 mg, 324 umol) a 0 °C y después se agitó a 15 °C durante 1 hora. La mezcla se inactivó con una solución saturada de bicarbonato de sodio (1,00 ml) y se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi C18150 * 25 * 10 um; fase móvil: [agua (0,225 %, AF) - ACN]; % de B: 17 %-44 %, 9 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[4-[7-(6-fluoro-1-naftil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-il]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]acetonitr¡lo (85,6 mg, 139 umol, 42,8 % de rendimiento, AF) en forma de un sólido de color blanquecino. LCMS [ESI, M+1 ]: 570.
1H RMN (400 MHz, acético) ó = 8,28 (dd, J = 5,6, 9,2 Hz, 1H), 7,60 (d, J = 8,0 Hz, 1H), 7,55 - 7,45 (m, 2H), 7,31 (dt, J = 2,4, 8,8 Hz, 1H), 7,18 (d, J = 7,2 Hz, 1H), 6,94 - 6,68 (m, 1H), 6,38 (d a, J = 16,8 Hz, 1H), 5,87 (d a, J = 10,0 Hz, 1H), 5,16 (s a, 1H), 4,84 (s a, 2H), 4,74 - 4,54 (m, 1H), 4,42 (d a, J = 12,8 Hz, 1H), 4,33 (s a, 2H), 4,25 - 4,08 (m, 1H), 3,92 (s a, 2H), 3,81 - 3,47 (m, 3H), 3,46 - 3,16 (m, 4H), 3,11 (s a, 3H), 3,08 - 2,87 (m, 3H), 2,46 - 2,32 (m, 1H), 2,25 - 2,11 (m, 3H).
Ejemplo 291

2-[4-[7-(4-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]-1-prop-2-eno¡lpiperazin-2-il]acetonitrilo

Etapa A: 2-(c¡anomet¡l)-4-[7-(4-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-illpiperazin-1-carboxilato de tere-butilo. Una mezcla de 2-(c¡anomet¡l)-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (0,15 g. 318 umol, 1,00 eq.). 4-bromoisoquinolina (99,3 mg, 477 umol, 1,50 eq.), RuPhos (29,7 mg, 63,6 umol, 0,20 eq.), Pd2(dba)3 (29,1 mg, 31,8 umol, 0,10 eq.) y CS2C03 (311 mg, 954 umol, 3,00 eq.) en tolueno (5,00 ml) se agitó a 110 °C durante 3 horas en una atmósfera de N2. La mezcla se diluyó con agua (6,00 ml) y se extrajo con acetato de etilo (3 x 5,00 ml). Las capas orgánicas se lavaron con salmuera (1 x 10,0 ml), se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (AF, 0,1 %)/acetonitr¡lo]. Las fracciones deseadas se recogieron y se ajustaron a pH >7 con una solución saturada de bicarbonato de sodio (5,00 ml) y se extrajeron con acetato de etilo (3 x 20,0 ml). Las capas orgánicas combinadas se lavaron con salmuera (1 x 30,0 ml), se secaron sobre Na2S04, se filtraron y se concentraron al vacío para dar 2-(cianomet¡l)-4-[7-(4-¡soqu¡nolil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡perazin-1-carbox¡lato de tere-butilo (0,17 g, 273 umol, 85,9 % de rendimiento) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 599.
1H RMN (400 MHz, cloroformo-d) ó = 9,03 (s, 1H), 8,26 (s, 1H), 8,14 (d, J = 8,4 Hz, 1H), 8,00 (d, J = 8,0 Hz, 1H), 7,73 (dt, J = 1,2, 7,2 Hz, 1H), 7,67 - 7,60 (m, 1H), 4,63 (s a, 1H), 4,44 - 4,31 (m, 3H), 4,15 - 4,03 (m, 2H), 3,95 (d a, J = 12,8 Hz, 1H), 3,59 - 3,49 (m, 1H), 3,40 (d a, J = 7,2 Hz, 1H), 3,34 - 3,19 (m, 2H), 3,18 - 2,93 (m, 4H), 2,90 - 2,79 (m, 2H), 2,77 - 2,66 (m, 2H), 2,50 (s, 3H), 2,35 - 2,24 (m, 1H), 2,10 - 2,02 (m, 1H), 1,92 - 1,79 (m, 3H), 1,52 (s, 9H).
Etapa B: 2-[4-[7-(4-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-2-i n acetonitrilo. Una mezcla de 2-(cianometil)-4-[7-(4-¡soqu¡nol¡l)-2-[[(2S)-1 -metilp¡rrolid¡n-2-il]metox¡]-6.8-dih¡dro-5W-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡l]piperaz¡n-1 -carboxilato de tere-butilo (0,15 g, 251 umol, 1,00 eq.) y t Fa (428 mg, 3,76 mmol, 278 ul, 15,0 eq.) en diclorometano (0,30 ml) se agitó a 15 °C durante 0,5 h. La mezcla se concentró al vacío para dar 2-[4-[7-(4-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]p¡peraz¡n-2-¡l]acetonitr¡lo (153 mg, bruto, TFA) en forma de un aceite de color negro y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 499.
Etapa C: 2-[4-[7-(4-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡ll-1-prop-2-eno¡l-p¡peraz¡n-2-¡llaceton¡tr¡lo. A una solución de 2-[4-[7-(4-¡soqu¡nol¡l)-2-[[(2S)-1-metilp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡trilo (153 mg, bruto, TfA) y Et3N (254 mg, 2,51 mmol, 349 ul) en diclorometano (2,00 ml) se le añadió prop-2-enoato de prop-2-enoílo (31,6 mg, 251 umol) a -40 °C. Después de agitar a 15 °C durante 1 hora, la mezcla se inactivó con una solución saturada de bicarbonato de sodio (0,10 ml) y se concentró al vacío. El residuo se purificó por cromatografía en columna (Ah03, metanol/acetato de etilo = 1/1) y HμLC prep. (columna: Boston Green o Ds 150 * 305 u; fase móvil: [agua (0,225 %, AF) - ACN]; % de B: 12 %-36 %, 10 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[4-[7-(4-¡soquinolil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡trilo (28,3 g, 43,4 umol, dos etapas 17,3 % de rendimiento, AF) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 553.
1H RMN (400 MHz, acético) ó = 9,45 (s, 1H), 8,51 - 8,36 (m, 3H), 8,17 (t a, J = 8,0 Hz, 1H), 8,03 - 7,97 (m, 1H), 6,92 -6,67 (m, 1H), 6,37 (d a, J = 16,0 Hz, 1H), 5,87 (d a, J = 10,0 Hz, 1H), 5,14 (s a, 1H), 4,82 (s a, 2H), 4,66 - 4,48 (m, 3H), 4,40 (d a, J = 12,0 Hz, 1H), 4,22 - 3,80 (m, 3H), 3,79 - 3,49 (m, 4H), 3,32 - 3,09 (m, 7H), 3,07 - 2,88 (m, 2H), 2,47 -2,36 (m, 1H), 2,24 - 2,11 (m, 3H).
Ejemplo 292

2-[4-[7-(5-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]-1-prop-2-eno¡lpiperaz¡n-2-¡l]aceton¡trilo

Etapa A: 2-(cianometil)-4-[7-(5-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-1-carboxilato de tere-butilo. Se desgasificaron 2-(cianometil)-4-[2-[(1-metilpirrolidin-2-il)metoxi]- 5,6,7,8-tetrahidrop¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (200 mg, 424 umol, 1,00 eq.), 5-bromoisoquinolina (115 mg, 551 umol, 1,30 eq.), RuPhos (39,6 mg, 84,8 umol, 0,20 eq.), Cs2C03 (415 mg, 1,27 mmol, 3,00 eq.) y Pd2(dba)3 (38,8 mg, 42,4 umol, 0,10 eq.) en tolueno (6,00 ml) y después se calentaron a 90 °C durante 12 horas en una atmósfera de N2. Después de que se completara, la mezcla de reacción se filtró y el filtrado se concentró. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo] para dar 2-(cianometil)-4-[7-(5-isoquinolil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-dih¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (217 mg, 344 umol, 81,0 % de rendimiento, 94,8 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 599.
Etapa B: 2-[4-[7-(5-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-i n metox n - 6.8-dihidro-5H-piridor3.4-dlpirimidin-4-i n piperazin-2-i n acetonitrilo. Una solución de 2-(c¡anomet¡l)-4-[7-(5-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lpirrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1 -carboxilato de tere-butilo (217 mg, 362 umol, 1,00 eq.) en TFA (620 mg, 5,44 mmol, 403 ul, 15,0 eq.) se agitó a 25 °C durante 1 hora. Después de que se completara, la mezcla de reacción se concentró al vacío para dar 2-[4-[7-(5-¡soquinol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]- 6,8-dihidro-5H-pirido[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]aceton¡tr¡lo (222 mg, bruto, TFA) en forma de un aceite de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 499.
Etapa C: 2-[4-[7-(5-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-prop-2-enoil-piperazin-2-i n acetonitrilo. A una solución de 2-[4-[7-(5-¡soquinol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (222 mg, 362 umol, TfA) y TEA (367 mg, 3,62 mmol, 504 ul) en DCM (4,50 ml) se le añadió gota a gota prop-2-enoato de prop-2-enoílo (45,7 mg, 362 umol) a 0 °C. La mezcla se agitó a 25 °C durante 1 hora. Después de que se completara, la reacción se interrumpió con Me0H (0,5 ml), se diluyó con agua (2 ml) y se extrajo con EtOAc (2 x 5 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi C18150 * 25 * 10 um;fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 4 % - 20 %, 8 min) y se liofilizó para dar el compuesto del título 2-[4-[7-(5-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-il]-1-prop-2-eno¡l-p¡peraz¡n-2-il]acetonitrilo (73,0 mg, 117 umol, dos etapas 32,2 % de rendimiento, 95,6 % de pureza, AF) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 553.
1H RMN (400 MHz, acético) ó = 9,71 (s, 1H), 8,69 (d, J = 6,4 Hz, 1H), 8,53 (d, J = 6,8 Hz, 1H), 8,15 (d, J = 8,0 Hz, 1H), 7,91 (t, J = 7,8 Hz, 1H), 7,78 (d, J = 7,2 Hz, 1H), 6,97 - 6,68 (m, 1H), 6,38 (d a, J = 16,8 Hz, 1H), 5,87 (d a, J = 10,4 Hz, 1H), 5,15 (s a, 1H), 4,81 (d a, J = 4,4 Hz, 2H), 4,67 - 4,51 (m, 1H), 4,49 - 4,31 (m, 3H), 4,28 - 4,04 (m, 1H), 3,91 (s a, 2H), 3,78 - 3,64 (m, 1H), 3,63 - 3,37 (m, 4H), 3,36 - 3,20 (m, 1H), 3,11 (s a, 5H), 3,08 - 2,86 (m, 2H), 2,46 - 2,33 (m, 1H), 2,24 - 2,10 (m, 3H).
Ejemplo 293

2-[1-[(E)-but-2-enoil]-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

Etapa A: 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de tere-butilo. A una mezcla de 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de íere-butilo (100 mg, 212 umol, 1,0 eq.), Pd2(dba)3 (19,4 mg, 21,2 umol, 0,10 eq.), RuPhos (14,8 mg, 31,8 umol, 0,15 eq.) y CS2C03 (207 mg, 636 umol, 3,0 eq.) en tolueno (3,0 ml) se le añadió gota a gota a gota 1-bromonaftaleno (65,9 mg, 318 umol, 44,2 ul, 1,5 eq.). La mezcla de reacción se desgasificó y se purgó 3 veces con N2 y la mezcla se agitó a 110 °C durante 4 horas en una atmósfera de N2. La reacción se lavó con agua (20,0 ml). La fase acuosa se extrajo con acetato de etilo (20,0 ml * 3). Los extractos combinados se lavaron con salmuera (50,0 ml), se secaron con Na2S04 y después el disolvente se eliminó al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa (C18, 0,1 % de AF en agua, 0-80 % de MeCN). El compuesto 2-(cianometil)-4-[2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-7-(1 -naftil)-6,8-dihidro-5H-pirido[3,4-tí]pirim idin-4-il]piperazin-1-carboxilato de íere-butilo (80,0 mg, 116 umol, 54,6 % de rendimiento) se obtuvo en forma de un sólido de color blanquecino. LCMS [ESI, M+1]: 598.
Etapa B: 2-[4-[2-[(2S)-1-metilpirrolidin-2-i n metox n -7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-i n piperazin-2-il]acetonitrilo. A una solución de 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de íere-butilo (80,0 mg, 134 umol, 1,0 eq.) en DCM (2,0 ml) se le añadió TFA (3,08 g, 27,0 mmol, 2,0 ml, 202 eq.). La mezcla se agitó a 20 °C durante 30 min en una atmósfera de N2. El disolvente orgánico se eliminó al vacío. El producto bruto 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡trilo (110 mg, bruto, 2TFA) se obtuvo en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 498.
Etapa C: 2-[1-[(E)-but-2-enoil]-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-¡ n piperazin-2-i n acetonitrilo. A una solución de 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-tí]pirimidin-4-il]piperazin-2-il]acetonitrilo (110 mg, 152 umol, 2TFA) en DCM (2,00 ml) se le añadieron DIEA (100 mg, 774 umol, 135 ul) y cloruro de (E)-but-2-enoílo (35,0 mg, 335 umol, 32,1 ul) a 0 °C. La mezcla se agitó a 0 °C durante 1 hora. La reacción se interrumpió con agua (10,0 ml). La mezcla bruto se extrajo con acetato de etilo (20,0 ml *
3). Los extractos combinados se lavaron con salmuera (50,0 ml), se secaron con Na2SÜ4 y después el disolvente se eliminó al vacío. El residuo se purificó por HPLC prep. (columna: Boston Green ÜDS 150 * 305 ufase móvil: [agua (0,225 % de AF)-ACN];% de B: 20 %-50 %,10 min) y se sometió a liofilización. El compuesto del título 2-[1-[(E)-but-2-enoil]-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (13,3 mg, 23,4 umol, 99,7 % de pureza, Af) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 566.
1H RMN (400 MHz, Cloroformo-d) ó 8,37 (s, 1H), 8,14-8,13 (m, 1H), 7,80-7,78 (m, 1H), 7,54 (d, J = 8,0 Hz, 1H), 7,46-7,42 (m, 2H), 7,36 (m, 1H), 7,07 (d, J = 7,6 Hz, 1H), 6,95-6,90 (m, 1H), 6,22 (d, J = 15,6 Hz, 1H), 5,05-4,91 (a, 1H), 4,67-4,63 (m, 1H), 4,37-4,32(m, 1H), 4,24-4,20 (m, 2H), 4,12 (d, J = 14,8 Hz, 1H), 3,96 (d, J = 11,6 Hz, 1H), 3,51-3,49 (m, 1H), 3,39-3,29 (m, 2H), 3,18-3,12 (m, 7H), 2,90-2,76 (m, 2H), 2,71 (s, 3H), 2,65-2,58 (m, 1H), 2,18 2,12 (m, 1H), 2,00-1,95 (m, 1H), 1,90-1,86 (m, 5H).
Ejemplo 294

2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(5-quinolil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo

Etapa A: 2-(c¡anomet¡l)-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-7-(5-qu¡nol¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-illpiperazin-1-carboxilato de ferc-butilo. Se desgasificaron 2-(cianometil)-4-[2-[(1-metilpirrolidin-2-il)metoxi]- 5,6,7,8tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (200 mg, 424 umol, 1,00 eq.), 5-bromoquinolina (115 mg, 551 umol, 1,30 eq.), RuPhos (39,6 mg, 84,8 umol, 0,20 eq.), Cs2C03 (415 mg, 1,27 mmol, 3.00 eq.) y Pd2(dba)3 (38,8 mg, 42,4 umol, 0,10 eq.) en tolueno (6,00 ml) y después se calentaron a 90 °C durante 12 horas en una atmósfera de N2. Después de que se completara, la mezcla de reacción se filtró y el filtrado se concentró. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo] para dar 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(5-quinolil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (200 mg, 315 umol, 74,4 % de rendimiento, 94,4 % de pureza) en forma de un aceite de color pardo. LCMS [ESI, M+1]: 599.
Etapa B: 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(5-quinolil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-¡llacetonitrilo. Una solución de 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(5-quinolil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo (200 mg, 334 umol, 1,00 eq.) en TFA (571 mg, 5.01 mmol, 371 ul, 15,0 eq.) se agitó a 25 °C durante 1 hora. Después de que se completara, la mezcla de reacción se concentró al vacío para dar 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(5-quinolil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (204 mg, bruto, TFA) en forma de un aceite de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 499.
Etapa C: 2-[4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1- 7-(5-quinolil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo. A una solución de 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]- 7-(5-quinolil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (204 mg, 333 umol, 1,00 eq., TFA) y TeA (337 mg, 3,33 mmol, 463 ul) en Dc M (4,00 ml) se le añadió gota a gota prop-2-enoato de prop-2-enoílo (42,0 mg, 333 umol) a 0 °C. La mezcla se agitó a 25 °C durante 1 hora. Después de que se completara, la reacción se interrumpió con Me0H (0,5 ml), se diluyó con agua (2 ml) y se extrajo con EtOAc (2 x 5 ml). Las capas orgánicas se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por TLC prep. (DCM/Me0H 8/1) y HPLC prep. (columna: Boston Green 0DS 150 * 305 ufase móvil: [agua (0,225 % de AF)-ACN];% de B: 7 %-34 %,10 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]- 7-(5-quinolil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (49,1 mg, 79,5 umol, dos etapas 23,9 % de rendimiento, 96,9 % de pureza, AF) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 553.
1H RMN (400 MHz, acético) 6 = 9,22 (d, J = 4,8 Hz, 1H), 9,15 (d, J = 8,4 Hz, 1H), 8,09 (d, J = 8,8 Hz, 1H), 7,98 (d, J = 8,0 Hz, 1H), 7,96 - 7,90 (m, 1H), 7,50 (d, J = 7,6 Hz, 1H), 6,98 - 6,71 (m, 1H), 6,37 (d a, J = 17,2 Hz, 1H), 5,88 (d a, J = 10,4 Hz, 1H), 5,15 (s a, 1H), 4,81 (s a, 2H), 4,77 - 4,52 (m, 1H), 4,50 - 4,33 (m, 3H), 4,14 (d a, J = 12,4 Hz, 1H), 3,91 (s a, 2H), 3,79 - 3,66 (m, 1H), 3,65 - 3,32 (m, 4H), 3,12 (s a, 6H), 3,05 (d a, J = 16,8 Hz, 2H), 2,46 - 2,34 (m, 1H), 2,23 - 2,09 (m, 3H).
Ejemplo 295

2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(4-quinolil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo

Etapa A: [2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-(4-quinolil)-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-1-carboxilato. Una mezcla de 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (Intermedio 66, 0,15 g, 318 umol, 1,00 eq.), 4-bromoquinolina (99,3 mg, 477 umol, 1,50 eq.), RuPhos (29,7 mg, 63,6 umol, 0,20 eq.), Pd2(dba)3 (29,1 mg, 31,8 umol, 0,10 eq.) y CS2C03 (311 mg, 954 umol, 3,00 eq.) en tolueno (5 ml) se agitó a 100 °C durante 3 horas en una atmósfera de N2. La mezcla se diluyó con agua (5,00 ml) y se extrajo con acetato de etilo (3 x 5,00 ml). Las capas orgánicas se lavaron con salmuera (1 x 10,0 ml), se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (AF, 0,1 %)/acetonitrilo]. Las fracciones deseadas se recogieron y se ajustaron a pH >7 con bicarbonato de sodio saturado (5,00 ml) y se extrajeron con acetato de etilo (3 x 20,0 ml). Las capas orgánicas se lavaron con salmuera (1 x 30,0 ml), se secaron sobre Na2S04, se filtraron y se concentraron al vacío para dar 2-(cianometil)-4-[2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-7-(4-quinolil)-6,8-dihidro-5W-pirido[3,4-d1pirimidin-4-il]piperazin-1 -carboxilato de fercbutilo (0,17 g, 269 umol, 84,8 % de rendimiento) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 599.
1H RMN (400 MHz, cloroformo-d) ó = 8,77 (d, J = 4,8 Hz, 1H), 8,07 (dd, J = 8,0, 16,0 Hz, 2H), 7,70 (dt, J = 1,2, 7,2 Hz, 1H), 7,56 - 7,50 (m, 1H), 6,92 (d, J = 4,8 Hz, 1H), 4,62 (s a, 1H), 4,44 - 4,33 (m, 3H), 4,15 - 4,04 (m, 2H), 3,95 (d a, J = 12,0 Hz, 1H), 3,74 - 3,64 (m, 1H), 3,46 (d a, J = 7,2 Hz, 1H), 3,37 - 3,21 (m, 2H), 3,18 - 2,93 (m, 4H), 2,93 - 2,79 (m, 2H), 2,72 (dd a, J = 6,0, 16,8 Hz, 2H), 2,51 (s, 3H), 2,36 - 2,26 (m, 1H), 2,13 - 2,06 (m, 1H), 1,93 - 1,79 (m, 3H), 1,53 (s, 9H).
Etapa B: 2-r4-r2-rr(2S)-1-metilpirrolidin-2-i n metox n -7-(4-quinolil) -6,8-dihidro-5H-pirido[3,4-c n pirimidin-4-i n piperazin-2-iHacetonitrilo. Una mezcla de 2-(cianometil)-4-[2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-7-(4-quinolil)-6,8-dihidro-5H-pirido[3,4-C]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (165 mg, 276 umol, 1,00 eq.) y TFA (471 mg, 4,13 mmol, 306 ul, 15,0 eq.) en diclorometano (0,30 ml) se agitó a 15 °C durante 0,5 horas. La mezcla se concentró al vacío para dar 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(4-quinolil) -6,8-d¡h¡dro-5H-p¡r¡do[3,4-<C]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]acetonitrilo (168 mg, bruto, TFA) en forma de un aceite de color negro y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 499.
Etapa C: 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-(4-quinolil)-6,8-dihidro-5H-pirido[3,4-c^pirimidin-4-il1-1-prop-2-enoil-piperazin-2-i n acetonitrilo. A una solución de 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]- 7-(4-quinolil)-6,8-dihidro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (168 mg, bruto, TFA) y Et3N (277 mg, 2,74 mmol, 382 ul) en diclorometano (3,00 ml) se le añadió prop-2-enoato de prop-2-enoílo (34,6 mg, 274,22 umol) a -40 °C. Después de agitar a 15 °C durante 1 hora, la mezcla se inactivó con bicarbonato de sodio saturado (0,10 ml) y se concentró al vacío. El residuo se purificó por cromatografía en columna (AL03, metanol/acetato de etilo = 1/1) y HPLC prep. (columna: Phenomenex Synergi C18150 * 25 * 10 um; fase móvil: [agua (0,225 %, AF) - ACN]; % de B: 1 % - 21 %, 7 min). Las fracciones deseadas se recogieron y se liofilizaron para dar 2-[4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(4-quinolil)-6,8-dihidro-5W-pirido[3,4-^pirimidin-4-il]-1 -prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0295, 23,4 mg, 39,6 umol, dos etapas 13,4 % de rendimiento, 93,6 % de pureza, AF) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 553.
Condición de SFC: 0D-3S_3_40_3 ML Columna: Chiralcel 0D-3100 x 4 ,6 mm D.I., 3 um, fase móvil: 40 % de metanol (0,05 % de DEA) en C02, caudal: 3 ml/min, longitud de onda: 220 nm.
1H RMN (400 MHz, acético) 5 = 8,65 (d, J = 7,2 Hz, 1H), 8,35 (d, J = 8,4 Hz, 1H), 8,15 (s, 1H), 7,99 (t, J = 7,2 Hz, 1H), 7,77 (t, J = 7,6 Hz, 1H), 7,28 (d, J = 6,8 Hz, 1H), 6,74 (dd, J = 10,8, 16,8 Hz, 1H), 6,32 (dd, J = 1,2, 16,8 Hz, 1H), 5,85 (dd, J = 1,2, 10,8 Hz, 1H), 5,01 (s a, 1H), 4,85 - 4,74 (m, 2H), 4,42 (d a, J = 14,0 Hz, 1H), 4,35 - 4,19 (m, 3H), 4,16 -3,76 (m, 4H), 3,66 (d a, J = 14,0 Hz, 2H), 3,51 - 3,40 (m, 1H), 3,39 - 3,16 (m, 4H), 3,13 (s, 3H), 3,07 - 2,90 (m, 2H), 2,49 - 2,39 (m, 1H), 2,26 - 2,17 (m, 3H).
Ejemplo 296

2-[4-[2-[2-(d¡met¡lam¡no)etox¡]-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡lpiperazin-2-il]acetonitrilo

Etapa A: 2-(c¡anomet¡l)-4-[2-[2-(d¡met¡lam¡no)etox¡]-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-il1piperazin-1-carboxilato de ferc-but¡lo. A una soluc¡ón de 2-(c¡anomet¡l)-4-[2-met¡lsulf¡n¡l-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de terc-but¡lo (150 mg, 266 umol, 1,0 eq.) y 2-(d¡met¡lam¡no)etanol (47,4 mg, 531 umol, 53,3 ul, 2,0 eq.) en tolueno (1,0 ml) se le añad¡ó t-Bu0Na (51,1 mg, 531 umol, 2,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, a la mezcla se le añad¡ó agua (10,0 ml) y se extrajo con AE (10 ml * 3). La capa orgán¡ca se secó sobre Na2S04, se f¡ltró y se concentró. El producto obten¡do se pur¡f¡có por columna ultrarráp¡da de fase ¡nversa (C18, 0,1 % de AF en agua, 0 - 40 % de MeCN). La fracc¡ón deseada se recog¡ó y se ajustó con NaHC03 acuoso saturado a pH ~7 y después se concentró al vacío. El líqu¡do restante se extrajo con AE (10 ml * 3). La capa orgán¡ca se secó sobre Na2S04, se f¡ltró y se concentró. El producto 2-(c¡anomet¡l)-4-[2-[2-(d¡met¡lam¡no)etox¡]-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de terc-but¡lo (150 mg, 254 umol, 95,8 % de rend¡m¡ento) se obtuvo en forma de un ace¡te de color amar¡llo. LCMS [ESI, M+1]: 590.
Etapa B: 2-[4-[2-[2-(dimetilamino)etoxi1-7-[2-(trifluorometil)fenil1-6,8-dihidro- 5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo. A una soluc¡ón de 2-(c¡anomet¡l)-4-[2-[2-(d¡met¡lam¡no)etox¡]-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de terc-but¡lo (150 mg, 254 umol, 1,0 eq.) en DCM (1,0 ml) se le
añadió TFA (29,0 mg, 254 umol, 18,8 ul, 1,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se concentró al vacío. Al residuo se le añadió NaHC03 acuoso saturado (5 ml) y se extrajo con DCM (10 ml x 3 ). La capa orgánica se secó sobre Na2S04, se filtró y se concentró para dar 2-[4-[2-[2-(dimetilamino)etoxi]-7-[2-(trifluorometil)fenil]-6,8-dihidro- 5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (200 mg, bruto) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 490.
Etapa C: A una solución de 2-[4-[2-[2-(dimetilamino)etoxi]-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (125 mg, 255 umol, 1,0 eq.) y DIEA (198 mg, 1,53 mmol, 267 ul, 6,0 eq.) en DCM (1,00 ml) se le añadió prop-2-enoato de prop-2-enoílo (32,2 mg, 255 umol, 1,0 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se concentró al vacío. El producto obtenido se purificó por HPLC prep. (columna: Phenomenex Synergi C18150 * 30 mm * 4 um; fase móvil: [agua (0,225 % de AF)-ACN]; % de B: 10 %-40 %, 10 min). Después, se concentró y se liofilizó. El producto 2-[4-[2-[2-(dimetilamino)etoxi]-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0 296, 30,7 mg, 51,8 umol, 20,3 % de rendimiento, 99,5 % de pureza, AF) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 544.
1H RMN (400 MHz, Cloroformo-d) ó 8,45 (s a, 1H), 7,67 (d a, J = 8,0 Hz, 1H), 7,57 (t, J = 7,2 Hz, 1H), 7,41 (d, J = 8,0 Hz, 1H), 7,30 (d, J = 7,6 Hz, 1H), 6,71 - 6,49 (m, 1H), 6,47 - 6,29 (m, 1H), 5,82 (d a, J = 10,4 Hz, 1H), 5,07 - 4,97 (m, 1H), 4,54 (t, J = 5,4 Hz, 2H), 4,12 (d a, J = 13,6 Hz, 1H), 4,07 (s, 2H), 3,98 (d a, J = 12,0 Hz, 1H), 3,40 - 3,30 (m, 2H), 3,22 - 3,09 (m, 4H), 3,05 (t, J = 5,4 Hz, 2H), 2,92 (dd, J = 8,0, 16,8 Hz, 1H), 2,86 - 2,81 (m, 1H), 2,79 - 2,70 (m, 2H), 2,56 (s, 6H).
Ejemplo 297

2-(1-acriloil-4-(2-(((R)-1-metilpirrolidin-3-il)metoxi)-7-(2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 2-(c¡anomet¡l)-4-[2-[3-[(1R.4R)-2-oxa-5-azab¡c¡clo [2.2.1lheptan-5-¡llpropox¡l-7-[2-(tr¡fluoromet¡l)fen¡l]-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de tere-butilo. A una soluc¡ón de 2-(c¡anomet¡l)-4-[2-met¡lsulf¡n¡l-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-but¡lo (200 mg. 354 umol, 1.0 eq.) y 3-[(1R,4R)-2-oxa-5-azab¡c¡clo[2.2.1]heptan-5-¡l]propan-1-ol (Intermed¡o 18. 111 mg. 708 umol, 2.0 eq.) en tolueno (3.0 ml) se le añad¡ó t-Bu0Na (68,1 mg. 708 umol. 2.0 eq.) a 0 °C y la reacc¡ón se ag¡tó a 0 °C durante 0.5 horas. Después de que se completara. a la mezcla de reacc¡ón se le añad¡ó agua (10 ml). después se extrajo con AE (2 x 10 ml) y la capa orgán¡ca comb¡nada se secó sobre Na2S04, se f¡ltró y se concentró. El res¡duo se pur¡f¡có por cromatografía ultrarrápida de fase ¡nversa (C18, 60 % de MeCN en agua). el producto obten¡do se ajustó con NaHC03 acuoso saturado a pH ~8, después se concentró y se extrajo con AE (2 x 10.0 ml) y la capa orgán¡ca comb¡nada se secó sobre Na2S04, se f¡ltró y se concentró para dar 2-(c¡anomet¡l)-4-[2-[3-[(1R,4R)-2-oxa-5-azab¡c¡clo [2.2.1lheptan-5-¡llpropox¡l-7-[2-(tr¡fluoromet¡l)fen¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de tere-but¡lo (130 mg. 197 umol. 55,7 % de rend¡m¡ento, 99,8 % de pureza) en forma de un sól¡do de color blanco. LCMS [ESI, M+1]: 658.
Etapa B: 2-[4-[2-[3-[(1R4R)-2-oxa-5-azab¡c¡clo[2.2.1lheptan-5-¡llpropox¡l-7-[2-(tr¡fluoromet¡l)fen¡l]-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una mezcla de 2-(c¡anomet¡l)-4-[2-[3-[(1R,4R)-2-oxa-5-azab¡c¡clo[2.2.1]heptan-5-¡l]propox¡]-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-but¡lo (70,0 mg. 106 umol. 1.0 eq.) en DCM (0.5 ml) se le añad¡ó TfA (770 mg. 6,75 mmol, 0.5 ml.
63,5 eq.). La mezcla de reacc¡ón se ag¡tó a 20 °C durante 0.5 horas. Después de que se completara. la mezcla de reacc¡ón se concentró para dar 2-[4-[2-[3-[(1R,4R)-2-oxa-5-azab¡c¡clo[2.2.1]heptan-5-¡l]propox¡]-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (83,0 mg. bruto. 2TFA) en forma de un ace¡te de color amar¡llo que se usó para la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón ad¡c¡onal. LCMS [ESI, M+1]: 558.
Etapa C: 2-[4-[2-[3-[(1R4R)-2-oxa-5-azab¡c¡clo[2.2.1lheptan-5-¡llpropox¡l-7-[2-(tr¡fluoromet¡l)fen¡l]-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡ll-1-prop-2-eno¡l-p¡peraz¡n-2-¡llaceton¡tr¡lo. A una mezcla de 2-[4-[2-[3-[(1R,4R)-2-oxa-5-azab¡c¡clo[2.2.1lheptan-5-¡llpropox¡l-7-[2-(tr¡fluoromet¡l)fen¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (83,0 mg. 106 umol. 1.0 eq.. 2TFA) en DCM (1.0 ml) se le añad¡eron DIeA (81,9 mg. 634 umol. 110 ul. 6.0 eq.) y prop-2-enoato de prop-2-enoílo (13,3 mg. 106 umol. 1.0 eq.) a 0 °C y la mezcla de reacc¡ón se agitó a 20 °C durante 0.5 horas. Después de que se completara. la mezcla de reacc¡ón se ¡nact¡vó con Me0H (0.5 ml) y se concentró. El res¡duo se pur¡f¡có por HPLC prep. ((Instrumento: ACSWH-GX-G; Columna: Phenomenex Gem¡n¡ 150 * 25 mm * 10 um; Cond¡c¡ón: agua (0,225 % de AF)-ACN; In¡c¡o B: 12; F¡n B: 42; T¡empo de grad¡ente (m¡n): 10,5; T¡empo de manten¡m¡ento a 100 % de B (m¡n): 2; Caudal (ml/m¡n): 2). el producto obten¡do se concentró y después se somet¡ó a l¡of¡l¡zac¡ón. El producto 2-[4-[2-[3-[(1R.4R)-2-oxa-5-azab¡c¡clo[2.2.1lheptan-5-¡llpropox¡l-7-[2-(tr¡fluoromet¡l)fen¡l]-6.8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (EJEMμL0297, 17,6 mg.26,3 umol. dos etapas 24,9 % de rend¡m¡ento, 98,1 % de pureza. AF) se obtuvo en forma de un sólido de color amar¡llo. LCMS [ESI. M+1]: 612.
1H RMN (400 MHz, cloroformo-d) 67,69 (d, J = 7.6 Hz. 1H). 7,59 (t. J = 7.6 Hz. 1H). 7,42 (d. J = 7.6 Hz. 1H). 7,31 (t. J = 7.6 Hz. 1H). 6,72 - 6,52 (m 1H). 6,40 (d. J = 16,4 Hz. 1H). 5,84 (d a. J = 10,4 Hz. 1H). 5,16 - 5,02 (m. 1H). 4,58 -4,51 (m. 1H).4,38 (t a. J = 5.8 Hz. 2H). 4,21 (d a. J = 9.2 Hz. 1H). 4,18 - 4,06 (m. 4H). 3,99 (d a. J = 11,6 Hz. 1H). 3,74 (d a. J = 8.4 Hz. 1H). 3,42 - 3,29 (m. 2H). 3,27 - 3,04 (m. 6H). 3,04 - 2,61 (m. 6H). 2,28 - 2,07 (m. 3H). 2,04 - 1,92 (m.
1H).
Ejemplo 298

2-(1-acr¡lo¡l-4-(2-(((R)-1-met¡lp¡rrol¡d¡n-3-¡l)metox¡)-7-(2-(tr¡fluoromet¡l)fen¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo

Etapa A: [(3R)-1-metilpirrolidin-3-il1metanol. A una solución de (3R)-3-(hidroximetil)pirrolidin-1-carboxilato de ferc-butilo (2,0 g, 9,94 mmol, 1,0 eq.) en THF (30,0 ml) se le añadió poco a poco LiAlH4 (754 mg, 19,9 mmol, 2,0 eq.) a 0 °C. Después de la adición, la reacción se agitó a 70 °C durante 3 horas. Después de que se completara, la mezcla de reacción se inactivó con Na2SÜ4 acuoso saturado (2,4 ml) y después se extrajo con acetato de etilo (100 ml). La capa orgánica se secó con Na2SÜ4 y se filtró. El disolvente se eliminó al vacío para dar [(3R)-1-metilp¡rrolid¡n-3-¡l]metanol (1,0 g, bruto) en forma de un aceite de color amarillo que se usó para la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, cloroformo-d) ó 3,64 (dd, J = 4,8, 10,0 Hz, 1H), 3,52 (dd, J = 5,6, 10,0 Hz, 1H), 2,78 - 2,67 (m, 1H), 2,60 - 2,45 (m, 2H), 2,42 - 2,23 (m, 5H), 2,08 - 1,91 (m, 1H), 1,72 - 1,57 (m, 1H).
Etapa B: 2-(cianometil)-4-[2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-7-[2-(trifluorometil)fenil1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de ferc-butilo. A una solución de 2-(cianometil)-4-[2-metilsulfinil-7-[2-(trifluorometil)fenil1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (100 mg, 177 umol, 1,0 eq.) y [(3R)-1-metilpirrolidin-3-il1metanol (61,2 mg, 531 umol, 3,0 eq.) en tolueno (1,0 ml) se le añadió t-Bu0Na (34,0 mg, 354 umol, 2,0 eq.) a 0 °C y la reacción se agitó a 0 °C durante 0,5 horas. Después de que se completara, a la mezcla de reacción se le añadió agua (10,0 ml), después se extrajo con acetato de etilo (2 * 10 ml) y la capa orgánica combinada se secó sobre Na2S04, se filtró y se concentró. El residuo se purificó por cromatografía ultrarrápida de fase inversa (C18, 50 % de MeCN en agua). Se obtuvo 2-(cianometil)-4-[2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-7-[2-(trifluorometil)fenil1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de ferc-butilo (40,0 mg, 63,4 umol, 35,8 % de rendimiento, 97,6 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+11: 616.
Etapa C: 2-[4-[2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-7-[2-(trifluorometil)fenil1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una mezcla de 2-(cianometil)-4-[2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-7-[2-(trifluorometil)fenil1-6.8- dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de ferc-butilo (40,0 mg, 64,9 umol, 1,0 eq.) en DCM (0,5 ml) se le añadió TFA (770 mg, 6,75 mmol, 500 ul, 104 eq.). La mezcla de reacción se agitó a 20 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se concentró para dar 2-[4-[2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-7-[2-(trifluorometil)fenil1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo (48,0 mg, bruto, 2TFA) en forma de un aceite de color amarillo que se usó para la siguiente etapa sin purificación adicional. LCMS [ESI, M+11: 516.
Etapa D: 2-[4-[2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-7-[2-(trifluorometil)fenil1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-prop-2-enoil-piperazin-2-il1acetonitrilo. A una mezcla de 2-[4-[2-[[(3R)-1 -metilpirrolidin-3-il1metoxi1-7-[2-(trifluorometil)fenil1-6.8- dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo (48,0 mg, 64,6 umol, 1,0 eq., 2TfA) en DCM (1,0 ml) se le añadieron DIEA (25,0 mg, 194 umol, 33,7 ul, 3,0 eq.) y prop-2-enoato de prop-2-enoílo (8,14 mg, 64,6 umol, 1,0 eq.) a 0 °C y la mezcla de reacción se agitó a 20 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se inactivó con metanol (0,5 ml) y se concentró. El residuo se purificó por HPLC prep. ((Instrumento: ACSWH-GX-M; Columna: Gemini 150 * 255 u; Condición: agua (0,04 % de NH3H20)-a Cn ; Inicio B: 60; Fin B: 81; Tiempo de gradiente
(min): 10; Tiempo de mantenimiento a 100 % de B (min): 3; Caudal (ml/min): 25), el producto obtenido se concentró y después se sometió a liofilización para dar 2-[4-[2-[[(3R)-1 -metilpirrolidin-3-il]metoxi]-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo(5,48 mg, 9,26 umol, 14,4 % de rendimiento, 96,3 % de pureza) en forma de un sólido de color pardo. LCMS [ESI, M+1]: 570.
1H RMN (400 MHz, cloroformo-d) ó 7,61 (d, J = 8,0 Hz, 1H), 7,50 (t, J = 7,6 Hz, 1H), 7,33 (d, J = 7,6 Hz, 1H), 7,22 (t, J = 7,6 Hz, 1H), 6,54 - 6,44 (m, 1H), 6,32 (dd, J = 1,6, 16,8 Hz, 1H), 5,75 (d a, J = 10,8 Hz, 1H), 5,12 - 4,92 (m, 1H), 4,13 (d, J = 6,8 Hz, 2H), 4,07 - 3,97 (m, 3H), 3,92 - 3,84 (m, 1H), 3,62 - 3,42 (m, 1H), 3,31 - 3,22 (m, 1H), 3,16 - 2,95 (m, 3H), 2,89 - 2,49 (m, 7H), 2,47 - 2,35 (m, 2H), 2,28 (s, 3H), 2,05 - 1,92 (m, 1H), 1,57 - 1,46 (m, 2H).
Ejemplo 299

2-[4-[2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Etapa A: 2-(cianometil)-4-[2-metilsulfanil-7-[2-(trifluorometil)fenil1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de tere-butilo. A una solución de 2-(cianometil)-4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de tere-butilo (3,70 g, 9,15 mmol, 1,0 eq.), Pd2(dba)3 (1,26 g, 1,37 mmol, 0,15 eq.), RuPhos (854 mg, 1,83 mmol, 0,2 eq.) y Cs2CÜ3 (7,45 g, 22,9 mmol, 2,50 eq.) en tolueno (74,0 ml) se le añadió 1-bromo-2-(trifluorometil)benceno (4,12 g, 18,3 mmol, 2,49 ml, 2,0 eq.). La mezcla se agitó a 100 °C durante 12 horas. Después de que se completara, a la mezcla se le añadió agua (100 ml) y se extrajo con acetato de etilo (100 ml * 3). La capa orgánica se secó sobre Na2SÜ4, se filtró y se concentró. El residuo se purificó por columna ultrarrápida de fase inversa (C18, 0,1 % de AF en agua, 0-90 % de MeCN). El producto 2-(cianometil)-4-[2-metilsulfanil-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (1,90 g, 3,46 mmol, 37,8 % de rendimiento) se obtuvo en forma de un sólido de color amarillo.
1H RMN (400 MHz, Cloroformo-d) ó 7,69 (d, J = 8,0 Hz, 1H), 7,57 (t, J = 7,6 Hz, 1H), 7,46 - 7,37 (m, 2H), 7,22 - 7,11 (m, 1H), 4,67 - 4,55 (m, 1H), 4,03 (d a, J = 13,6 Hz, 2H), 3,88 (d a, J = 12,0 Hz, 1H), 3,68 - 3,47 (m, 1H), 3,30 - 3,08 (m, 4H), 3,02 (dt, J = 3,2, 12,4 Hz, 1H), 2,94 - 2,69 (m, 4H), 2,51 (s, 3H), 1,52 (s, 9H).
Etapa B: 2-(c¡anomet¡l)-4-[2-met¡lsulf¡n¡l-7-[2-(tr¡fluoromet¡l)fen¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carboxilato de tere-butilo. A una soluc¡ón de 2-(c¡anomet¡l)-4-[2-met¡lsulfan¡l-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (1,50 g. 2,46 mmol, 1.0 eq.) en DCM (15,0 ml) se le añad¡ó m-CPBA (593 mg, 2,75 mmol, 1,12 eq., 80 % de pureza) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, a la mezcla se le añad¡ó Na2S203 acuoso saturado (30,0 ml) y se extrajo con acetato de etilo (30,0 ml x 3). La capa orgán¡ca se secó sobre Na2S04, se f¡ltró y se concentró. El res¡duo se pur¡f¡có por columna ultrarrápida de fase ¡nversa (C18, 0,1 % de AF en agua, 0-90 % de MeCN) para dar 2-(cianometil)-4-[2-met¡lsulf¡n¡l-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (1,16 g, 2,05 mmol, 83,6 % de rend¡m¡ento) en forma de un sólido de color amar¡llo. LCMS [ESI, M+1]: 565.
Etapa C: (2S)-2-(c¡anomet¡l)-4-[2-([(2R)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-7-[2-(tr¡fluoromet¡l)fen¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de tere-butilo. A una soluc¡ón de (2S)-2-(c¡anometil)-4-[2-met¡lsulf¡n¡l-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (200 mg, 354 umol, 1.0 eq.) en tolueno (3,0 ml) se le añad¡eron [(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metanol (81,6 mg, 708,4 umol, 2,0 eq.) y t-Bu0Na (68,1 mg, 708 umol, 2,0 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 20 m¡n. Después de que se completara, la reacc¡ón se ¡nterrump¡ó con agua (10,0 ml). La mezcla bruto se extrajo con acetato de et¡lo (15 ml x 3 ). La capa orgán¡ca comb¡nada se lavó con salmuera (50,0 ml), se secó con Na2S04 y se f¡ltró. Después, el d¡solvente se el¡m¡nó al vacío. El res¡duo se pur¡f¡có por cromatografía ultrarrápida de fase ¡nversa (C18, 0,1 % de AF en agua, 0-80 % de MeCN). El compuesto (2S)-2-(c¡anomet¡l)-4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-[2-(tr¡fluorometil)fen¡l]-6,8-d¡h¡dro-5H p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llpiperaz¡n-1 -carboxilato de tere-butilo (170 mg, 275 umol, 77,7 % de rendimiento) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 616.
1H RMN (400 MHz, CL0R0F0RM0-d) 8,08 (s, 1H), 7,61 (d, J = 7,6 Hz, 1H), 7,51 (t, J = 7,7 Hz, 1H), 7,34 (d, J = 8.0 Hz, 1H), 7,25 - 7,19 (m, 1H), 5,02 - 4,78 (m, 1H), 4,58 - 4,43 (m, 2H), 4,17 (t a, J = 12,5 Hz, 1H), 4,08 - 3,86 (m, 5H), 3,64 - 3,51 (m, 1H), 3,23 (dd a, J = 3,8, 13,8 Hz, 1H), 3,11 - 3,01 (m, 3H), 2,96 (s, 3H), 2,84 - 2,58 (m, 4H), 2,31 -2,03 (m, 2H), 2,02 - 1,94 (m, 3H), 1,44 (s, 9H), 1,19 (t, J = 7,2 Hz, 1H).
Etapa D: 2-[4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-7-[2-(tr¡fluoromet¡l)fen¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-2-¡llaceton¡tr¡lo. A una solución de 2-(cianomet¡l)-4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-[2-(tr¡fluoromet¡l)fen¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3,4-dlp¡rimid¡n-4-M]piperazin-1 -carboxilato de tere-butilo (170 mg, 276 umol, 1.0 eq.) en dCm (1,50 ml) se le añadió TFA (2,31 g, 20,3 mmol, 1,50 ml, 73,4 eq.). La mezcla se agitó a 20 °C durante 15 min en una atmósfera de N2. Después de que se completara, el disolvente orgánico se eliminó al vacío. El producto bruto 2-[4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]p¡peraz¡n-2-¡l]acetonitr¡lo (240 mg, bruto, 2TFA) se obtuvo en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 516.
Etapa E: 2-[4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-7-[2-(tr¡fluoromet¡l)fen¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡ll-1-prop-2-eno¡l-p¡peraz¡n-2-¡llaceton¡tr¡lo. A una solución de 2-[4-[2-[[(2R)-1-metilp¡rrol¡din-2-¡l]metox¡]-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (240 mg, 2TFA) en DCM (3,0 ml) se le añadieron prop-2 -enoato de prop-2-enoílo (40,0 mg, 317 umol) y DlEA (130 mg, 1,01 mmol, 175 ul) a 0 °C. La mezcla se agitó a 0 °C durante 30 min. La reacción se interrumpió con agua (20,0 ml). La mezcla bruto se extrajo con acetato de etilo (20,0 ml x 3 ). Los extractos combinados se lavaron con salmuera (50,0 ml), se secaron con Na2S04 y se filtraron. Después, el disolvente se eliminó al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 x 25 mm x 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN];% de B: 55 %-85 %,3 min). El compuesto 2-[4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (EJEMPL0299, 10,2 mg, 16,8 umol, 94,3 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 570.
1H RMN (400 MHz, CL0R0F0RM0-d) 6 = 7,68 (d, J = 7,2 Hz, 1H), 7,58 (t, J = 7,6 Hz, 1H), 7,41 (d, J = 8,0 Hz, 1H), 7,30 (d, J = 7,6 Hz, 1H), 6,59 (s a, 1H), 6,40 (d, J = 16,4 Hz, 1H), 5,83 (d, J = 10,4 Hz, 1H), 5,07 (s a, 1H), 4,85 (d, J = 7.1 Hz, 1H), 4,66 - 4,39 (m, 1H), 4,20 (d, J = 11,1 Hz, 1H), 4,13 - 3,95 (m, 4H), 3,70 - 3,55 (m, 2H), 3,45-3,20 (m, 2H), 3,16 - 2,99 (m, 3H), 2,98 - 2,91 (m, 4H),2,85 - 2,70 (m, 4H), 2,33 - 2,14 (m, 2H), 2,10 - 2,01 (m, 2H).
Ejemplo 300
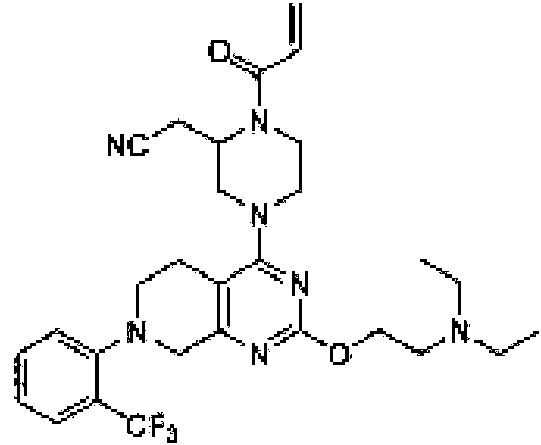
2-[4-[2-[2-(dietilamino)etoxi]-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpipenazin-2 -il]acetonitrilo

Etapa A: 2-(cianometil)- 4-[2-[2-(dietilamino)etoxi1-7-[2-(trifluorometil)fenil1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1- carboxilato de tere-butilo. A una solución de 2-(cianometil)-4-[2-metilsulfinil-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (200 mg, 354 umol, 1,0 eq.) y 2-(dietilamino)etanol (83,0 mg, 708 umol, 93,9 ul, 2,0 eq.) en tolueno (3,0 ml) se le añadió t-Bu0Na (68,1 mg, 708 umol, 2,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, a la mezcla se le añadió agua (10,0 ml) y se extrajo con acetato de etilo (10,0 ml x 3 ). La capa orgánica se secó sobre Na2S04, se filtró y se concentró. El residuo se purificó por columna ultrarrápida de fase inversa (C18, 0,1 % de AF en agua, 0 - 40 % de MeCN). El producto 2-(cianometil)-4-[2-[2-(dietilamino)etoxi]-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il)piperazin-1-carboxilato de tere-butilo (180 mg, 291 umol, 82,3 % de rendimiento) se obtuvo en forma de un aceite incoloro. LCMS [ESI, M+1]: 618.
Etapa B: 2-(4-(2-(2-(dietilamino)etoxi)-7-(2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-cf1pirimidin-4-il)piperazin-2-il)acetonitrilo. A una solución de 2-(cianometil)-4-(2-(2-(dietilamino)etoxi)-7-(2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-cf]pirimidin-4-il)piperazin-1-carboxilato de tere-butilo (180 mg, 291 umol, 1,0 eq.) en DCM (2,0 ml) se le añadió TFA (33,2 mg, 291 umol, 21,6 ul, 1,0 eq.). La mezcla se agitó a 25 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se añadió acetato de etilo (10,0 ml) y se ajustó con una solución saturada de NaHC03 a pH >7. La mezcla se extrajo con acetato de etilo (30 ml x 3) y la capa orgánica se secó con Na2S04 y se filtró. El disolvente se eliminó al vacío. El producto 2- (4-(2-(2-(dietilamino)etoxi)-7-(2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-cf]pirimidin-4-il)piperazin-2-il)acetonitrilo (150 mg, bruto) se obtuvo en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 518.
Etapa C: 2-[4-[2-[2-(dietilamino)etoxi]-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-b]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo. A una solución de 2-[4-[2-[2-(dietilamino)etoxi]-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡trilo (150 mg, bruto) en d Cm (3,0 ml) se le añadieron DIEA (129 mg, 1,0 mmol, 174 ul) y prop-2-enoato de prop-2-enoílo (44,1 mg, 0,35 mmol). La mezcla se agitó a 0 °C durante 1 hora. Después de que se completara, la mezcla de reacción se inactivó mediante la adición de metanol (10,0 ml) a 20 °C. A la mezcla de reacción se le añadió agua (10,0 ml) y se extrajo con DCM (10,0 ml x 3 ). Las capas orgánicas combinadas se secaron sobre Na2S04, se filtraron y se concentraron al vacío para dar un residuo. El residuo se purificó por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,04 % de NH3H20)-ACN]; % de B: 65 %-83 %,10 min). El producto 2-[4-[2-[2-(dietilamino)etoxi]-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-b]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0 300, 41,6 mg, 72,7 umol, 100 % de pureza, dos etapas 25,0 % de rendimiento) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1 ]: 572.
1H RMN (400 MHz, CL0R0F0RM0-d) ó = 7,68 (d, J = 7,8 Hz, 1H), 7,58 (t, J = 7,4 Hz, 1H), 7,41 (d, J = 8,0 Hz, 1H), 7,32 - 7,28 (m, 1H), 6,70 - 6,49 (m, 1H), 6,39 (dd, J = 1,2, 16,8 Hz, 1H), 5,82 (d, J = 10,8 Hz, 1H), 5,08 (s a, 1H), 4,39 (t, J = 6 ,8 Hz, 2H), 4,14 - 4,00 (m, 3H), 4,02 - 3,84 (m, 1H), 3,32 (d a, J = 12,0 Hz, 1H), 3,24 - 3,16 (m, 1H), 3,15 - 3,04 (m, 2H), 2,96 - 2,88 (m, 4H), 2,87 - 2,70 (m, 1H), 2,68 - 2,61 (m, 4H), 1,89 (s a, 3H), 1,07 (t, J = 7,0 Hz, 6 H).
Ejemplo 301

2-[4-[7-(1-naftil)-2-[[(2S)-1,5,5-trimetilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo

Etapa A: (5S)-5-(h¡drox¡met¡l)-2,2-d¡met¡l-p¡rrol¡d¡n-1-carbox¡lato de tere-butilo. A una solución de ácido (2S)-1-tere-butoxicarbonil-5,5-dimetil-pirrolidin- 2-carboxílico (400 mg, 1,64 mmol, 1,00 eq.) en THF anhidro (7,00 ml) se le añadió gota a gota BH3^Me2S (10 M, 493 ul, 3,00 eq.) a 25 °C. La reacción se calentó a 50 °C durante 5 minutos. Después de que se completara y de que se enfriara con un baño de hielo, la mezcla se inactivó con metanol (10 ml). La mezcla de reacción se concentró a presión reducida para dar (5S)-5-(hidrox¡metil)-2,2-dimetil-pirrolidin-1-carboxilato de fere-butilo (360 mg, 1,57 mmol, 95,9 %) en forma de un aceite incoloro que se usó directamente en la siguiente etapa sin purificación adicional.
Etapa B: 4-[3-(c¡anomet¡0p¡peraz¡n-1-¡ll-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-cnp¡r¡m¡d¡n-7-carbox¡lato de tere-butilo. A una solución ag¡tada de 2-met¡lsulfan¡l-4-(tr¡fluoromet¡lsulfon¡lox¡)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo (20 g. 46,5 mmol, 1,00 eq.) en DMF (300 ml) se le añad¡eron DIEA (18,1 g. 139 mmol, 24,3 ml. 3 eq.) y 2-p¡peraz¡n-2-¡laceton¡tr¡lo (10,2 g.51,2 mmol. 495 ul. 1,10 eq.. 2 HCl). La reacc¡ón se calentó a 100 °C y se ag¡tó durante 1 hora en una atmósfera de N2. Se obtuvo 4-[3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-met¡lsulfan¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo (18,8 g. bruto) y se usó d¡rectamente en la s¡gu¡ente etapa s¡n tratam¡ento n¡ pur¡f¡cac¡ón.
Etapa C: 4-[4-benc¡lox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡ll-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo. A una soluc¡ón de 4-[3-(c¡anomet¡l)p¡peraz¡n-1-¡ll-2-met¡lsulfan¡l-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo (18,8 g. bruto) en Dm F (200 ml) se le añad¡eron DIEA (12,0 g. 92,9 mmol.
16,2 ml) y carbonoclor¡dato de benc¡lo (11,9 g. 69,7 mmol. 9,91 ml). La mezcla se ag¡tó a 50 °C durante 1 hora. La mezcla de reacc¡ón se d¡luyó con agua (300 ml) y se extrajo con acetato de et¡lo (200 ml x 3). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (200 ml). se secaron sobre Na2SÜ4. se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar un res¡duo. El res¡duo se pur¡f¡có por cromatografía en columna (S¡02, éter de petróleo/acetato de et¡lo = 30/1 a 3/1). Se obtuvo 4-[4-benc¡lox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-met¡lsulfan¡l-6,8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo (20 g. 31.6 mmol. dos etapas 68,0 % de rend¡m¡ento, 85 % de pureza) en forma de un ace¡te de color amar¡llo. LCMS [ESI, M+1]: 539.
1H RMN (400 MHz, cloroformo-d) ó = 7,40 - 7,36 (m. 5H). 5,21 - 5,15 (m. 2H). 4,73 - 4,55 (m. 2H). 4,39 (d. J = 18,8 Hz.
1H). 3,97 (d. J = 12,8 Hz. 1H). 3,90 - 3,69 (m. 2H). 3,41 - 3,16 (m. 3H). 3,00 (dt. J = 3.6. 12,4 Hz. 1H). 2,86 - 2,53 (m.
5H). 2,51 (s. 3H). 1,49 (s. 9H).
Etapa D: 2-(c¡anomet¡l)-4-(2-met¡lsulfan¡l-5.6.7.8-tetrah¡drop¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo.
A una soluc¡ón de 4-[4-benc¡lox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-met¡lsulfan¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo (11 g. 20,4 mmol. 1,00 eq.) en d¡oxano (100 ml) se le añadió HChdioxano (4 M, 102 ml. 20 eq.). Después de ag¡tar a 25 °C durante 1 hora. la mezcla de reacc¡ón se concentró a pres¡ón reduc¡da para dar un res¡duo. El res¡duo se d¡solv¡ó en 300 ml de acetato de et¡lo y 300 ml de agua y se ag¡tó ráp¡damente.
Después. se añad¡ó una soluc¡ón acuosa saturadas de Na2C03 hasta alcanzar un pH de ~9. Los extractos orgán¡cos se lavaron con agua (1 x 300 ml) y salmuera (1 x 200 ml). se secaron sobre Na2S04, se f¡ltraron y se concentraron a pres¡ón reduc¡da. Se obtuvo 2-(c¡anomet¡l)-4-(2-met¡lsulfan¡l-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (8.2 g. 15,2 mmol. 74,2 % de rend¡m¡ento, 81 % de pureza) en forma de un ace¡te de color amar¡llo y se usó d¡rectamente en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón. LCMS [ESI, M+1]: 439.
Etapa E: 2-(C¡anomet¡l)-4-[2-met¡lsulfan¡l-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de benc¡lo. Una mezcla de 2-(c¡anomet¡l)-4-(2-met¡lsulfan¡l-5,6,7,8- tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (5,07 g. 11,6 mmol. 1.1 eq.). 1-bromonaftaleno (2,83 g. 13,7 mmol. 1,90 ml. 1.3 eq.). Cs2C03 (8,57 g. 26,3 mmol. 2,50 eq.). Pd2(dba)3 (1.45 g. 1,58 mmol. 0,15 eq.) y RuPhos (982 mg. 2,11 mmol. 0,20 eq.) en d¡oxano (100 ml) se desgas¡f¡có y se purgó 3 veces con N2 y después la mezcla se ag¡tó a 85 °C durante 5 horas en una atmósfera de N2. La mezcla de reacc¡ón se d¡luyó con agua (200 ml) y se extrajo con acetato de et¡lo (200 ml x 3). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (200 ml). se secaron sobre Na2S04, se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar un res¡duo. El res¡duo se pur¡f¡có por cromatografía en columna (S¡02, éter de petróleo/acetato de et¡lo = 100/1 a 2/1). Se obtuvo 2-(c¡anomet¡l)-4-[2-met¡lsulfan¡l-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (3.6 g.4,65 mmol.40,1 % de rend¡m¡ento, 73 % de pureza) en forma de un ace¡te de color amar¡llo. LCMS [ESI. M+1]: 565.
Etapa F: 2-(c¡anomet¡l)-4-[2-met¡lsulf¡n¡l-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de benc¡lo. A una soluc¡ón ag¡tada de 2-(c¡anomet¡l)-4-[2-met¡lsulfan¡l-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (3.1 g.5,49 mmol. 1 eq.) en met¡ltetrah¡drofurano (50 ml) a 0 °C en una atmósfera de n¡trógeno se le añad¡ó m-CPBA (1,18 g.5,49 mmol. 80 % de pureza. 1 eq.) puro en forma de un sól¡do. Después de ag¡tar a 0 °C durante 1 hora en una atmósfera de N2, la mezcla de reacc¡ón se ¡nact¡vó med¡ante la ad¡c¡ón de 10 % de una soluc¡ón acuosa de Na2S203 (100 ml) a 0 °C y después se d¡luyó con agua (200 ml) y se extrajo con acetato de et¡lo (200 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (200 ml). se secaron sobre Na2S04, se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar un res¡duo. El res¡duo se pur¡f¡có por cromatografía en columna (S¡02, acetato de et¡lo/metanol = 100/1 a 10/1) y se pur¡f¡có por cromatografía ultrarráp¡da de fase ¡nversa [agua (0.1 % de ác¡do fórm¡co)/aceton¡tr¡lo)]. Las fracc¡ones deseadas se ajustaron pH = 7 med¡ante la ad¡c¡ón de una soluc¡ón acuosa saturada de NaHC03 y se extrajeron con acetato de et¡lo (100 ml x 3 ). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (50 ml). se secaron sobre Na2S04, se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar 2-(c¡anomet¡l)-4-[2-met¡lsulf¡n¡l-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (1.3 g. 2,19 mmol.
39,9 % de rend¡m¡ento, 98 % de pureza) que se obtuvo en forma de un sól¡do de color amar¡llo. LCMS [ESI. M+1]: 581.
1H RMN (400 MHz. cloroformo-d) ó = 8,22 - 8,13 (m. 1H). 7,92 - 7,83 (m. 1H). 7,63 (d. J = 8.0 Hz. 1H). 7,56 - 7,48 (m.
2H). 7,45 (t. J = 8.0 Hz. 1H). 7,41 - 7,32 (m. 5H). 7,15 (d. J = 7.2 Hz. 1H). 5,22 (s. 2H). 4,70 (s a. 1H). 4,54 - 4,36 (m.
2H). 4,34 - 4,17 (m. 2H). 4,12 - 4,04 (m. 1H). 3,58 - 3,17 (m. 5H). 3,01 (m. 2H). 2,93 (d. J = 2.8 Hz. 3H). 2,90 - 2,79 (m.
1H). 2,77 - 2,65 (m. 1H).
Etapa G: A una solución de 2-(cianometil)-4-[2-metilsulfinil-7- (1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (430 mg, 741 umol, 1,00 eq.) y (5S)-5-(hidroximetil)-2,2-dimetil-pirrolidin- 1-carboxilato de ferc-butilo (340 mg, 1,48 mmol, 2 ,00 eq.) en tolueno (8 ,00 ml) se le añadió t-Bu0Na (142 mg, 1,48 mmol, 2,00 eq.). La mezcla se agitó a 20 °C durante 0,25 horas. Después de que se completara, la mezcla de reacción se filtró y el filtrado se concentró. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo] para dar 4-[2-[[(2S)-1-ferc-butoxicarbonil-5,5-dimetil-pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (250 mg, 318 umol, 43,0 % de rendimiento, 95,0 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 746.
Etapa H: 2-(cianometil)-4-[2-[[(2S)-5,5-dimetilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡llpiperazin-1-carboxilato de bencilo. A una solución de 4-[2-[[(2S)-1-ferc-butoxicarbonil-5,5-dimetil- pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (200 mg, 268 umol, 1 ,00 eq.) en dioxano (4,00 ml) se le añadió Hchdioxano (4 M, 4,00 ml, 59,7 eq.) a 0 °C. La mezcla se agitó a 25 °C durante 2 horas. Después de que se completara, la mezcla se concentró al vacío para dar 2 (cianometil)-4-[2-[[(2S)-5,5-dimetilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1- carboxilato de bencilo (180 mg, 251 umol, 93,5 % de rendimiento, 95,0 % de pureza, HCl) en forma de un aceite de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional.
Etapa I: 2-(cianometil)-4-[7-(1-naftil)-2-[[(2S)-1,5,5-trimetilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo. A una solución de 2-(cianometil)-4-[2-[[(2S)-5,5- dimetilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (180 mg, 264 umol, 1,00 eq., HCl) y formaldehído (107 mg, 1,32 mmol, 98,2 ul, 5,00 eq., 37 % en agua) en Me0H (3,00 ml) se le añadieron CH3C00H (31,7 mg, 528 umol, 30,2 ul, 2,00 eq.) y NaBHaCN (66,3 mg, 1,06 mmol, 4,00 eq.). La mezcla se agitó a 25 °C durante 1 hora. Después de que se completara, la mezcla se inactivó con HCl 1 M (0,2 ml), se diluyó con agua (1 ml) y se extrajo con EtOAc (3 x 6 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo] para dar 2-(cianometil)-4-[7-(1-naftil)-2-[[(2S)-1,5,5-trimetilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (96,0 mg, 131 umol, 49,6 % de rendimiento, 90,0 % de pureza) en forma de un sólido de color amarillo.
1H RMN (400 MHz, cloroformo-d) ó = 8,23 - 8,17 (m, 1H), 7,89 - 7,83 (m, 1H), 7,61 (d, J = 8,0 Hz, 1H), 7,53 - 7,47 (m, 2H), 7,46 - 7,42 (m, 1H), 7,42 - 7,33 (m, 5H), 7,14 (d, J = 7,2 Hz, 1H), 5,27 - 5,16 (m, 2H), 4,70 (s a, 1H), 4,41 (dd, J = 4,4, 10,4 Hz, 1H), 4,34 - 4,22 (m, 2H), 4,20 - 4,01 (m, 3H), 3,95 (d a, J = 11,2 Hz, 1H), 3,40 - 3,21 (m, 3H), 3,13 - 2,91 (m, 2H), 2,91 - 2,69 (m, 3H), 2,32 (s, 3H), 2,13 - 1,96 (m, 1H), 1,75 - 1,54 (m, 5H), 1,15 (s, 3H), 0,92 (s, 3H).
Etapa J: 2-[4-[7-(1-naftil)-2-[[(2S)-1,5,5-trimetilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2- il]acetonitrilo. Se burbujeó NH3 en Me0H (6,00 ml) durante 3 minutos a -40 °C. Una solución de 2-(cianometil)-4-[7-(1-naftil)-2-[[(2S)-1,5,5-trimetilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (0,10 g, 152 umol, 1,00 eq.) en Me0H (6,00 ml) se añadió a la solución de NH3 y Pd/C (0,05 g, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La reacción se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. Después de que se completara, el catalizador se retiró por filtración y el filtrado se concentró al vacío para dar 2-[4-[7-(1-naftil)-2-[[(2S)-1,5,5-trimetilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (63,0 mg, 103 umol, 68,0 % de rendimiento, 86 ,0 % de pureza) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. LCm S [ESI, M+1]: 526.
Etapa K: 2-[4-[7-(1-naftil)-2-(((2S)-1,5,5-trimetilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo. A una solución de 2-[4-[7-(1-naftil)-2-[[(2S)-1,5,5-trimetilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (75,0 mg, 143 umol, 1,00 eq.) y TEA (72,2 mg, 713 umol, 99,3 ul, 5,00 eq.) en dCm (2,00 ml) se le añadió gota a gota prop-2-enoato de prop-2-enoílo (18,0 mg, 143 umol, 1,00 eq.) a 0 °C. La mezcla se agitó a 25 °C durante 1 hora. Después de que se completara, la reacción se interrumpió con Me0H (0,5 ml), se diluyó con agua (2 ml) y se extrajo con EtOAc (2 x 5 ml). Las capas orgánicas se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por TLC prep. (EtOAc/Me0H 9/1) y HPLC prep.
(columna: Phenomenex Gemini 150 * 25 mm * 10 um;fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - Ac N];
% de B: 60 % - 90 %, 12 min) y se liofilizó para dar 2-[4-[7-(1-naftil)-2-[[(2S)-1,5,5-trimetilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0 301, 14,7 mg, 25,3 umol, 17,8 % de rendimiento, 100 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 580.
1H RMN (400 MHz, cloroformo-d) ó = 8,25 - 8,17 (m, 1H), 7,90 - 7,83 (m, 1H), 7,61 (d, J = 8,4 Hz, 1H), 7,55 - 7,48 (m, 2H), 7,44 (t, J = 7,6 Hz, 1H), 7,14 (d, J = 7,2 Hz, 1H), 6,60 (s a, 1H), 6,45 - 6,36 (m, 1H), 5,84 (d a, J = 10,8 Hz, 1H), 5,10 (s a, 1H), 4,44 (s a, 1H), 4,34 - 4,19 (m, 2H), 4,17 - 3,80 (m, 4H), 3,79 - 3,21 (m, 4H), 3,19 - 2,63 (m, 6 H), 2,34 (s a, 3H), 2,15 - 2,03 (m, 1H), 1,69 (s a, 3H), 1,16 (s a, 3H), 0,94 (s, 3H).
Ejemplo 302

2-[4-[7-(7-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-2-piperidil]acetonitrilo

Inserción: trifluorometanosulfonato de (7-fluoro-1-naftilo). A una solución de 7-fluoronaftalen-1-ol (250 mg, 1,54 mmol, 1 eq.) y DIEA (598 mg, 4,63 mmol, 806 ul, 3 eq.) en DCM (10 ml) se le añadió gota a gota Tf20 (652 mg, 2,31 mmol, 382 ul, 1,5 eq.) a -5 °C. La mezcla se agitó a 20 °C durante 3 horas. Después de eso, la mezcla se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (éter de petróleo/acetato de etilo = 100/1 a 10/1). Se obtuvo trifluorometanosulfonato de (7-fluoro-1-naftilo) (450 mg, 1,53 mmol, 99,2 % de rendimiento) en forma de un aceite incoloro.
1H RMN (400 MHz, metanol-d4) 5 = 8,08 (dd, J = 5,6, 9,2 Hz, 1H), 8,01 (d, J = 8,4 Hz, 1H), 7,73 - 7,39 (m, 4H).
Etapa A: 2-(Cianometil)-4-[7-(7-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de tere-butilo. Una mezcla de 2-(cianometil)-4-(2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (182 mg, 386 umol, 1 eq.), trifluorometanosulfonato de (7-fluoro-1-naftilo) (227 mg, 772 umol, 2 eq.), Pd2(dba)3 (35,3 mg, 38,6 umol, 0,1 eq.), RuPhos (36,0 mg, 77,2 umol, 0,2 eq.) y Cs2C03 (377 mg, 1,16 mmol, 3 eq.) en tolueno (20 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 16 horas en una atmósfera de N2. La mezcla se inactivó con H20 (30 ml) y después la mezcla se diluyó con acetato de etilo (20 ml). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de
fase inversa [agua (0,1 % de FA)/acetonitrilo]. Las fracciones deseadas se recogieron y se neutralizaron con una solución saturada de NaHC03 y se extrajeron con acetato de etilo (1 * 100 ml). La capa orgánica se secó sobre sulfato de sodio, se filtró y se concentró al vacío. Se obtuvo 2-(cianometil)-4-[7-(7-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (158 mg, 257 umol, 66,5 % de rendimiento, 100 % de pureza) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 616.
1H RMN (400 MHz, cloroformo-d) ó = 7,92 - 7,79 (m, 2H), 7,61 (d, J = 8,0 Hz, 1H), 7,40 (t, J = 7,8 Hz, 1H), 7,30 - 7,25 (m, 1H), 7,19 (d, J = 7,2 Hz, 1H), 4,63 (d a, J = 2,1 Hz, 1H), 4,40 (dd a, J = 4,8, 10,4 Hz, 1H), 4,24 (s a, 2H), 4,20 -4,00 (m, 4H), 3,95 (d a, J = 12,4 Hz, 1H), 3,52 - 3,37 (m, 1H), 3,36 - 3,16 (m, 3H), 3,15 - 2,63 (m, 6H), 2,50 (s, 3H), 2,36 - 2,22 (m, 1H), 2,12 - 2,05 (m, 1H), 1,92 - 1,70 (m, 3H), 1,53 (s, 9H).
Etapa B: 2-[4-[7-(7-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡llpiperazin-2-illacetonitrilo. A una solución de 2-(cianometil)-4-[7-(7-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (100 mg, 162 umol, 1 eq.) en dioxano (10 ml) se le añadió HChdioxano (4 M, 1,62 ml, 40 eq.) a 0 °C en una atmósfera de nitrógeno. La mezcla se agitó a 25 °C durante 2 horas. Después de eso, la mezcla se concentró al vacío. Se obtuvo 2-[4-[7-(7-fluoro-1 -naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (100 mg, bruto, HCl) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 516.
Etapa C: 2-[4-[7-(7-fluoro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1 6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-eno¡l-2-p¡per¡d¡l]aceton¡tr¡lo. A una mezcla de 2-[4-[7-(7-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (100 mg, bruto, HCl) y DlEA (234 mg, 1,81 mmol, 315 ul) en DMF (5 ml) se le añadió prop-2-enoato de prop-2-enoílo (18,3 mg, 145 umol) a 0 °C. Después de agitar a 25 °C durante 1 h, la mezcla de reacción se inactivó con una solución saturada de NaHC03 (5 ml) y después se extrajo con acetato de etilo (2 * 25 ml). Las capas orgánicas combinadas se lavaron con salmuera (1 * 50 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (acetato de etilo/Me0H = 50/1 a 1/1). El líquido recogido se concentró al vacío. El residuo se purificó por HPLC prep.
(columna: Phenomenex Synergi C18 150 * 25 * 10 um; fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 12 % -42 %, 10 min) y (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - ACN]; % de B: 55 % - 85 %,12 min) y se obtuvo 2-[4-[7-(7-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-2-piperidil]acetonitrilo (EJEMPL0302, 4,23 mg, 7,19 umol, dos etapas 4,29 % de rendimiento, 96,7 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1 ]:
570.
1H RMN (400 MHz, cloroformo-d) ó = 7,93 - 7,77 (m, 2H), 7,61 (d, J = 8,0 Hz, 1H), 7,40 (t, J = 7,8 Hz, 1H), 7,32 - 7,25 (m, 1H), 7,19 (d, J = 7,3 Hz, 1H), 6,59 (d a, J = 10,8 Hz, 1H), 6,41 (dd, J = 1,6, 16,8 Hz, 1H), 5,84 (d a, J = 10,4 Hz, 1H), 5,23 - 4,52 (m, 1H), 4,40 (dd, J = 4,8, 10,6 Hz, 1H), 4,24 (s a, 2H), 4,21 - 4,11 (m, 2H), 4,03 (d a, J = 12,0 Hz, 2H), 3,86 - 3,04 (m, 6H), 3,03 - 2,58 (m, 5H), 2,49 (s, 3H), 2,37 - 2,23 (m, 1H), 2,16 - 1,98 (m, 1H), 1,91 - 1,69 (m, 3H).
Ejemplo 303

2-[4-[7-(1-naftil)-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[7-(1-naftil)-2-(2-(1-piperidil)etoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de bencilo. A una solución de 2-(1-piperidil)etanol (89,0 mg, 689 umol, 91,5 ul, 2,0 eq.) en tolueno (2,0 ml) se le añadió t-Bu0Na (66,2 mg, 689 umol, 2,0 eq.) a 0 °C. Después, a la mezcla se le añadió (2s)-2-(cianometil)-4-[2-metilsulfinil-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de bencilo (200 mg, 344 umol, 1,0 eq.) y la mezcla se agitó a 0 °C durante 1 hora. Después de que se completara, a la mezcla se le añadió agua (10,0 ml) y se extrajo con AE (10 ml * 3). La capa orgánica se secó sobre Na2S04, se filtró y se concentró. El producto obtenido se purificó por cromatografía en columna (Si02, EP:AE = 10:1 - AE:Me0H = 20:1) para dar (2S)-2-(cianometil)-4-[7-(1-naftil)-2-[2-(1-piperidil)etoxi1-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il1piperazin-1-carboxilato de bencilo (200 mg, 296 umol, 86 % de rendimiento, 95,7 % de pureza) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 646.
Etapa B: 2-[(2S)-4-[7-(1-naftil)-2-[2-(1-piperidil)etoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. Se burbujeó NH3 en Me0H (10,0 ml) a -78 °C durante 30 minutos. Después, se añadieron (2S)-2-(cianometil)-4-[7-(1-naftil)-2-[2-(1 -piperidil) etoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de bencilo (180 mg, 279 umol, 1,0 eq.) y Pd/C (100 mg, 10,0 % de pureza) al líquido anterior en una atmósfera de N2. La suspensión se desgasificó y se purgó 3 veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 20 °C durante 0,5 horas. Después de que se completara, la mezcla se filtró con Celite y se concentró al vacío. El producto obtenido se purificó por columna ultrarrápida de fase inversa (C18, 0,1 % de AF en agua, 0 - 40 % de MeCN) para dar 2-[(2S)-4-[7-(1-naftil)-2-[2-(1-piperidil)etoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo (90,0 mg, 176 umol, 63 % de rendimiento) en forma de un sólido de color blanco. lCMs [ESI, M+11: 512.
Etapa C: 2-[4-1-7-(1-naftil)-2-[2-(1-piperidil)etoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-prop-2-enoil-piperazin-2-iHacetonitrilo. A una solución de 2-[4-[7-(1 -naftil)-2-[2-(1 -piperidil) etoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo (90,0 mg, 176 umol, 1,0 eq.) y DIEA (136 mg, 1,06 mmol, 184 ul, 6,0 eq.) en dCm (1,0 ml) se le añadió prop-2-enoato de prop-2-enoílo (22,2 mg, 176 umol, 1,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se inactivó con Me0H (20,0 mg) y se concentró al vacío. El producto obtenido se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN1; % de B: 60 % - 90 %, 12 min) para dar 2-[4-[7-(1-naftil)-2-[2-(1-piperidil)etoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-prop-2-enoil-piperazin-2-il1acetonitrilo (EJEMPL0303, 11,2 mg, 19,7 umol, 11 % de rendimiento, 99,5 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+11: 566.
1H RMN (400 MHz, Cloroformo-d) ó 8,24 - 8,19 (m, 1H), 7,89 - 7,84 (m, 1H), 7,62 (d, J = 8,4 Hz, 1H), 7,55 - 7,47 (m, 2H), 7,44 (t, J = 8,0 Hz, 1H), 7,15 (d, J = 7,6 Hz, 1H), 6,70 - 6,53 (m, 1H), 6,41 (dd, J = 1,2, 16,8 Hz, 1H), 5,84 (d a, J = 10,4 Hz, 1H), 5,19 - 5,01 (m, 1H), 4,47 (t a, J = 6,0 Hz, 2H), 4,33 - 4,21 (m, 2H), 4,12 (d a, J = 14,0 Hz, 1H), 4,06 -3,98 (m, 1H), 3,55 - 3,45 (m, 1H), 3,43 - 3,24 (m, 2H), 3,22 - 3,09 (m, 1H), 3,04 - 2,72 (m, 6H), 2,68 - 2,42 (m, 4H), 1,61 - 1,56 (m, 6H), 1,50 - 1,40 (m, 2H).
Ejemplo 304

2-[4-[2-[[(2R)-1-(2-hidroxietil)pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo
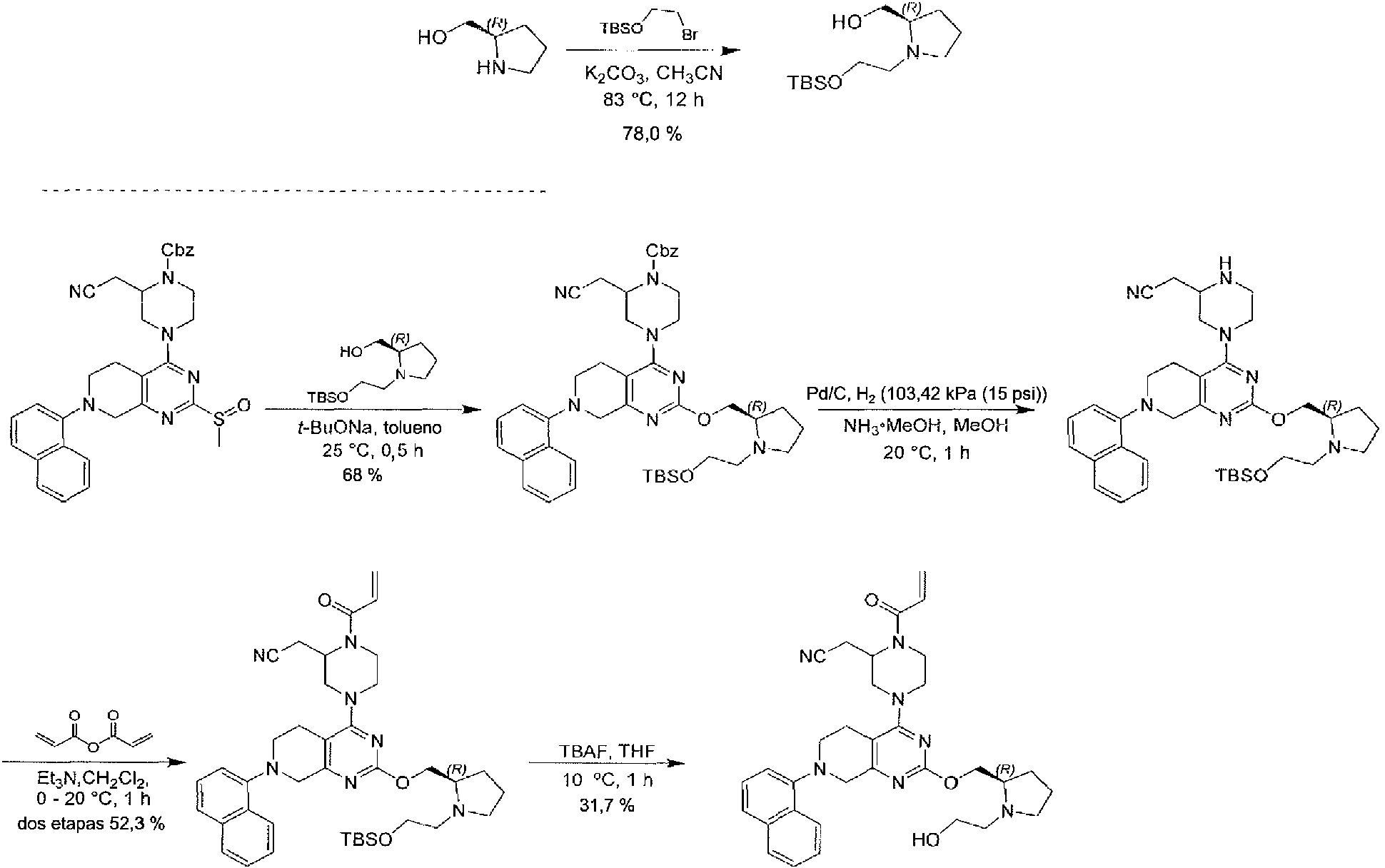
[(2R)-1-[2-[fe/'c-butil(dimetil)silil]oxietil]pirrolidin-2-il]metanol. Una mezcla de [(2R)-pirrolidin-2-il]metanol (0,70 g, 6,92 mmol, 673 ul, 1,00 eq.), 2-bromoetoxi-ferc-butil-dimetil-silano (1,66 g, 6,92 mmol, 1,00 eq.) y K2C03 (4,78 g, 34,6 mmol, 5,00 eq.) en acetonitrilo (70,0 ml) se calentó a reflujo a 83 °C durante 12 horas. Después, la mezcla se filtró y la torta de filtro se lavó con metanol. Las capas orgánicas se concentraron al vacío para dar un residuo. El residuo se purificó por cromatografía sobre gel de sílice (acetato de etilo/metanol = 50/1 a 1/1). El compuesto [(2 R)-1-[2-[terc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metanol (1,40 g, 5,40 mmol, 78,0 % de rendimiento, 100 % de pureza) se obtuvo en forma de un aceite incoloro.
1H RMN (400 MHz, cloroformo-d) 5 = 3,68 (dd, J = 5,2, 6,4 Hz, 2H), 3,57 (dd, J = 3,6, 10,8 Hz, 1H), 3,42 - 3,25 (m, 1H), 3,23 - 2,96 (m, 2H), 2,95 - 2,82 (m, 1H), 2,72 - 2,61 (m, 1H), 2,48 (td, J = 5,2, 12,8 Hz, 1H), 2,40 - 2,27 (m, 114), 1,90 - 1,77 (m, 1H), 1,77 - 1,62 (m, 3H), 0,83 (s, 9H), 0,00 (s, 6H).
Etapa A: 4-(2-[[(2R)-1-[2-[terc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1 -carboxilato de bencilo. A una solución de 2-(cianometil)-4-[2-metilsulfinil-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (500 mg, 861 umol, 1 eq.) y [(2R)-1-[2-[íerc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metanol (447 mg, 1,72 mmol, 2 eq.) en tolueno (10 ml) se le añadió en una porción t-Bu0Na (248 mg, 2,58 mmol, 3 eq.) a 25 °C y la mezcla se agitó a 25 °C durante 0,5 horas. La mezcla se acidificó con una solución 1 M de HCl a pH = 8 y la mezcla se diluyó con acetato de etilo (100 ml) y agua (10 ml). La capa orgánica separada se lavó con salmuera (1 * 50 ml), se secó sobre sulfato de sodio, se filtró y se concentró
al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (acetato de etilo/metanol 100/1 a 10/1).
Las fracciones deseadas se recogieron y se concentraron al vacío para dar 4-[2-[[(2R)-1-[2-[tercbutil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (480 mg, 582 umol, 68 % de rendimiento, 94,1 % de pureza) en forma de un sólido de color amarillo claro. LCMS [ESI, M+1]: 776.
1H RMN (400 MHz, cloroformo-d) ó = 8,28 - 8,11 (m, 1H), 7,95 - 7,77 (m, 1H), 7,61 (d, J = 8,0 Hz, 1H), 7,55 - 7,29 (m, 8H), 7,14 (d, J = 7,2 Hz, 1H), 5,29 - 5,15 (m, 2H), 4,70 (s a, 1H), 4,41 - 3,88 (m, 7H), 3,83 - 3,68 (m, 2H), 3,57 - 2,69 (m, 12H), 2,67 - 2,49 (m, 1H), 2,37 (c, J = 8,4 Hz, 1H), 2,03 - 1,93 (m, 1H), 1,83 - 1,74 (m, 3H), 1,00 - 0,79 (m, 9H), 0,13 -0,01 (m, 6H).
Etapa B: 2-[4-[2-[[(2R)-1-[2-[terc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo. Se burbujeó NH3 en metanol (60,0 ml) a -78 °C durante 30 minutos. A la mezcla se le añadieron 4-[2-[[(2R)-1-[2-[terc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (0,20 g, 258 umol, 1,00 eq.) y Pd/C (0,10 g, 10,0 % de pureza). La mezcla se agitó a 20 °C durante 1 hora en una atmósfera de H2 a 103,42 kPa (15 psi). Después, la mezcla se filtró y se concentró al vacío para dar 2-[4-[2-[[(2R)-1-[2-[terc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (0,15 g, bruto) en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 642.
Etapa C: 2-[4-[2-[[(2R)-1-[2-[terc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1acetonitr¡lo. A una solución de 2-[4-[2-[[(2R)-1-[2-[tercbutil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (0,15 g, bruto) y Et3N (236 mg, 2,34 mmol, 325 ul) en diclorometano (2,00 ml) se le añadió prop-2-enoato de prop-2-enoílo (29,5 mg, 234 umol) a 0 °C. Después de agitar a 20 °C durante 1 hora, la mezcla se inactivó con una solución saturada de bicarbonato de sodio (0,50 ml) y se concentró al vacío. El residuo se purificó por cromatografía en columna (AL03, metanol/acetato de etilo = 1/10) para dar 2-[4-[2-[[(2R)-1 -[2-[terc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (0,10 g, 135 umol, dos etapas 52,3 % de rendimiento) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 696.
1H RMN (400 MHz, cloroformo-d) ó = 8,25 - 8,18 (m, 1H), 7,90 - 7,82 (m, 1H), 7,61 (d, J = 8,0 Hz, 1H), 7,54 - 7,47 (m, 2H), 7,43 (t, J = 7,6 Hz, 1H), 7,15 (d, J = 7,2 Hz, 1H), 6,59 (d a, J = 12,0 Hz, 1H), 6,40 (dd, J = 1,2, 16,8 Hz, 1H), 5,84 (d a, J = 10,4 Hz, 1H), 5,22 - 4,94 (m, 1H), 4,36 (td, J = 3,6, 10,4 Hz, 1H), 4,31 - 4,20 (m, 2H), 4,15 - 4,05 (m, 2H), 4,04 - 3,97 (m, 1H), 3,75 (t, J = 6,8 Hz, 2H), 3,63 (s a, 1H), 3,46 (s a, 1H), 3,35 (d a, J = 11,2 Hz, 2H), 3,21 - 3,15 (m, 1H), 3,08 - 2,99 (m, 2H), 2,99 - 2,92 (m, 2H), 2,63 - 2,54 (m, 1H), 2,42 - 2,33 (m, 1H), 2,04 - 1,95 (m, 1H), 1,84 - 1,73 (m, 3H), 1,66 (s a, 4H), 0,88 (s, 9H), 0,05 (s, 6H).
Etapa D: 2-[4-[2-[[(2R)-1-(2-hidroxietil)pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo. Una mezcla de 2-[4-[2-[[(2R)-1-[2-[terc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (0,09 g, 129 umol, 1 eq.) y TBAF (1,00 M en THF, 1,29 ml, 10 eq.) en THF (0,10 ml) se agitó a 10 °C durante 1 hora. La mezcla se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (AF, 0,1 %)/acetonitrilo], cromatografía en columna (AL03, metanol/acetato de etilo = 1/3) y HPLC prep. (columna: Boston pH-lex 150 * 2510 um; fase móvil: [agua (0,10 % de TFA) - ACN]; % de B: 22 % - 52 %, 10 min y columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - ACN]; % de B: 47 % - 77 %, 12 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[4-[2-[[(2R)-1-(2-hidroxietil)pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (23,9 mg, 41,0 umol, 31,7 % de rendimiento, 99,7 % de pureza) en forma de un sólido de color blanco.
LCMS [ESI, M+1]: 582.
1H RMN (400 MHz, cloroformo-d) ó = 8,25 - 8,18 (m, 1H), 7,89 - 7,83 (m, 1H), 7,61 (d, J = 8,4 Hz, 1H), 7,54 - 7,47 (m, 2H), 7,43 (t, J = 7,6 Hz, 1H), 7,14 (d, J = 7,2 Hz, 1H), 6,59 (s a, 1H), 6,44 - 6,35 (m, 1H), 5,83 (d a, J = 10,4 Hz, 1H), 5,10 - 4,50 (m, 1H), 4,38 - 4,24 (m, 3H), 4,22 - 4,09 (m, 2H), 4,00 (d a, J = 11,5 Hz, 2H), 3,73 - 3,57 (m, 2H), 3,45 (s a, 1H), 3,36 (d a, J = 12,0 Hz, 2H), 3,23 - 2,77 (m, 9H), 2,62 (td, J = 3,6, 12,4 Hz, 1H), 2,38 - 2,26 (m, 1H), 2,05 - 1,99 (m, 1H), 1,90 - 1,73 (m, 3H).
Ejemplo 305

3-(4-((S)-4-acriloil-3-(cianometil)piperazin-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6-dihidropirido[3,4-d]pirimidin-7(8H)-il)isonicotinonitrilo

Etapa A: 2-(cianometil)-4-[7-(4-ciano- 3-piridil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de bencilo. A una solución de 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (400 mg, 791 umol, 1,0 eq.) y 3 bromopiridin-4-carbonitrilo (289 mg, 1,58 mmol, 2,0 eq.) en tolueno (10,0 ml) se le añadieron CS2C03 (516 mg, 1,58 mmol, 2,0 eq.), Pd2(dba)3 (72,4 mg, 79,1 umol, 0,1 eq.) y RuPhos (73,8 mg, 158 umol, 0,2 eq.) y la mezcla de reacción se agitó a 100 °C durante 3 horas en una atmósfera de N2. Después de que se completara, a la mezcla de reacción se le añadió agua (15 ml), después se extrajo con AE (2 * 15,0 ml) y la capa orgánica combinada se secó sobre Na2S04, se filtró y se concentró. El residuo se purificó por cromatografía en columna (Base Ah03, éter de petróleo/acetato de etilo = 5/1 a éter de petróleo/acetato de etilo/metanol = 5/1/0,1) para dar 2-(cianometil)-4-[7-(4-ciano- 3-piridil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (200 mg, 266 umol, 34 % de rendimiento, 80,9 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 608.
Etapa B: 3-[4-[3-(cianometil)piperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]piridin-4-carbonitrilo. A una solución de 2-(cianometil)-4-[7-(4-ciano-3-piridil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (170 mg, 226 umol, 1,0 eq.) en Me0H (5,0 ml) se le añadió NH3*Me0H (49,4 ug, 49,4 umol, 5,0 ml) y Pd/C (150 mg, 10 % de pureza) y la suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 20 °C durante 1 hora. Después de que se completara, la reacción se filtró a través de a Celite y el filtrado se concentró. El residuo se purificó por cromatografía ultrarrápida de fase inversa (C18, 30 % de MeCN en agua), el producto obtenido se ajustó con NaHC03 acuoso saturado a pH ~8 , después se concentró hasta que no quedó disolvente, después al sólido se le añadió DCM (10,0 ml) y se agitó a 20 °C durante 0,5 h, el sólido se filtró y el filtrado se concentró para dar 3-[4-[3-(cianometil)piperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]piridin-4-carbonitrilo (35,0 mg, 73,9 umol, 33 % de rendimiento, 100 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 474.
Etapa C: 3-[4-[3-(cianometil)-4-prop-2-enoil-piperazin-1-i n -2-[[(2S)-1-metilpirrolidin-2i n metox n -6.8-dihidro-5H-pirido[3,4-d1pirimidin-7-il1piridin-4-carbonitrilo. A una mezcla de 3-[4-[3-(cianometil)piperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]piridin-4-carbonitrilo (35,0 mg, 73,9 umol, 1,0 eq.) en DCM (1,0 ml) se le añadieron DIEA (28,7 mg, 222 umol, 38,6 ul, 3,0 eq.) y prop-2-enoato de prop-2-enoílo (9,32 mg,
73,9 umol, 1,0 eq.) a 0 °C y la mezcla de reacción se agitó a 20 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se inactivó con Me0H (0,5 ml) y se concentró. El residuo se purificó por HPLC prep. ((Instrumento: ACSWH-GX-M; Columna: Gemini 150 * 255 u; Condición: agua (0,04 % de NH3*H20)-ACN; Inicio B: 32; Fin B: 56; Tiempo de gradiente (min): 10; Tiempo de mantenimiento a 100 % de B (min): 3; Caudal (ml/min): 100), el producto obtenido se concentró y después se sometió a liofilización. El producto 3-[4-[3-(cianometil)-4-prop-2-enoilpiperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]piridin-4-carbonitrilo (EJEMPL0305, 13,0 mg, 24,5 umol, 33 % de rendimiento, 99,3 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 528.
1H RMN (400 MHz, cloroformo-d) ó 8,52 (s, 1H), 8,33 (d, J = 4,8 Hz, 1H), 7,46 (d, J = 4,8 Hz, 1H), 6,69 - 6,53 (m, 1H), 6,42 (dd, J = 1,6, 16,8 Hz, 1H), 5,85 (d a, J = 11,6 Hz, 1H), 5,19 - 5,01 (m, 1H), 4,48 - 4,36 (m, 3H), 4,27 - 4,08 (m, 2H), 4,07 - 3,81 (m, 3H), 3,78 - 3,33 (m, 3H), 3,23 - 2,99 (m, 3H), 2,97 - 2,83 (m, 2H), 2,81 - 2,61 (m, 2H), 2,52 (s, 3H), 2,40 - 2,27 (m, 1H), 2,16 - 1,99 (m, 1H), 1,95 - 1,76 (m, 3H).
Ejemplo 306
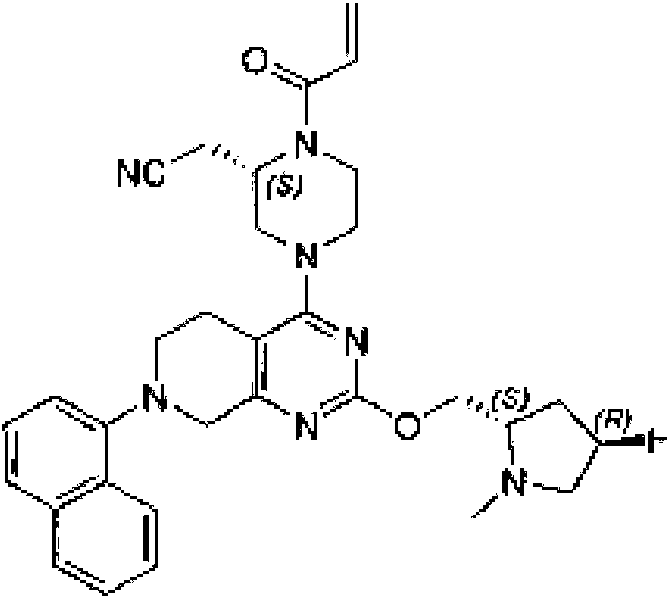
2-((S)-1-acriloil-4-(2-(((2S,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: [(2S.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡llmetanol. A una solución de 2-metil (2S,4R)-4-fluoropirrolidin-1,2-dicarboxilato de 1-terc-butilo (5.0 g. 20,2 mmol, 1.0 eq.) en THF (100 ml) se le añadió poco a poco L¡AlH4 (1.53 g. 40,4 mmol. 2.0 eq.) a 0 °C. Después de que la mezcla de reacción se agitara a 0 °C durante 1 hora en una atmósfera de N2, la mezcla de reacción se calentó a 70 °C y se agitó a 70 °C durante 2 horas en una atmósfera de N2. Después de que se completara. la mezcla de reacción se inactivó con Na2SÜ4 acuoso saturado (4.5 ml). después se filtró y el filtrado se concentró. El residuo se purificó por cromatografía en columna (Base Ah03. éter de petróleo/acetato de etilo = 5/1 a éter de petróleo/acetato de etilo/metanol = 3/1/0.1) para dar [(2S,4R)-4-fluoro-1-metil-p¡rrol¡d¡n-2-¡l]metanol (1.5 g. 11,3 mmol. 56 % de rendimiento) en forma de un aceite de color amarillo.
1H RMN (400 MHz, cloroformo-d) ó 5,24 - 4,97 (m. 1H). 3,73 (dd. J = 3.2. 11,2 Hz. 1H). 3,61 - 3,40 (m.2H). 2,86 - 2,74 (m. 1H). 2,69 (ddd, J = 0.8. 2.8. 12,0 Hz. 1H). 2,61 (dd. J = 2.4. 12,0 Hz. 1H). 2,41 (s. 3H). 2,18 - 1,98 (m. 2H).
Etapa B: (2S)-2-(c¡anomet¡l)-4-[2-[[(2S.4R)-4-fluoro-1-met¡lp¡rrol¡d¡n-2-¡llmetoxil-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡mid¡n-4-¡llp¡peraz¡n-1-carbox¡lato de bencilo. A una solución de (2S)-2-(cianomet¡l)-4-[2-met¡lsulfin¡l-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (Intermedio 64.800 mg. 1,38 mmol. 1.0 eq.) y [(2S.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metanol (367 mg. 2,76 mmol. 2.0 eq.) en tolueno (15,0 ml) se le añadió t-Bu0Na (265 mg. 2,76 mmol. 2.0 eq.) a 0 °C y la reacción se agitó a 0 °C durante 0.5 horas. Después de que se completara. a la mezcla de reacción se le añadió agua (30 ml). después se extrajo con AE (2 x 15 ml) y la capa orgánica combinada se lavó con salmuera saturada (1 x 30 ml). después se secó sobre Na2S04, se filtró y se concentró. El residuo se purificó por cromatografía en columna (S¡02. éter de petróleo/acetato de etilo = 5/1 a éter de petróleo/acetato de etilo/metanol = 3/1/0.1) para dar (2S)-2-(cianomet¡l)-4-[2-[[(2S.4R)-4-fluoro-1-met¡l-p¡rrolid¡n-2-¡llmetox¡l-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlpir¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de bencilo (720 mg. 997 umol, 72 % de rendimiento. 90,0 % de pureza) en forma de un sólido de color amarillo claro. LCMS [ESI, M+1]: 650.
Etapa C: 2-[(2S)-4-[2-[[(2S.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡llmetox¡l-7-(1-naft¡l)-6.8-d¡hidro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llpiperazin-2-illacetonitrilo. A una solución de (2S)-2-(c¡anomet¡l)-4-[2-[[(2S,4R)-4-fluoro-1-met¡l-pirrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡ljp¡peraz¡n-1-carbox¡lato de bencilo (670 mg. 1,03 mmol. 1.0 eq.) en Me0H (15,0 ml) se le añadieron NHâ Me0H (1,03 mmol. 15,0 ml. 1.0 eq.) y Pd/C (400 mg. 10 % de pureza) y la suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 20 °C durante 1 hora. Después de que se completara. la reacción se filtró a través de a Celite y el filtrado se concentró para dar 2-[(2S)-4-[2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡din-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]aceton¡tr¡lo (260 mg. 478 umol. 46 % de rendimiento. 94,9 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 516.
Etapa D: 2-[(2S)-4-[2-[[(2S.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡llmetox¡l-7-(1-naft¡l)-6.8-d¡hidro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡ll-1-prop-2-enoil-p¡peraz¡n-2-¡llaceton¡tr¡lo. A una mezcla de 2-[(2S)-4-[2-[[(2S,4R)-4-fluoro-1-metil-p¡rrol¡din-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡din-4-¡l]p¡peraz¡n-2-¡l]aceion¡tr¡lo (260 mg. 504 umol. 1.0 eq.) en DCM (5.0 ml) se le añadieron DIEA (196 mg. 1,51 mmol. 263 ul. 3.0 eq.) y prop-2-enoato de prop-2-enoílo (63,6 mg. 504 umol. 1.0 eq.) a 0 °C y la mezcla de reacción se agitó a 20 °C durante 0.5 horas. Después de que se completara. la mezcla de reacción se inactivó con Me0H (5 ml) y se concentró. El residuo se purificó por HPLC prep. ((Instrumento: ACSWH-GX-H; Columna: Gemini 150 * 255 u; Condición: agua (0,05 % de hidróxido de amoníaco v/v)-ACN; Inicio B: 42; Fin B: 72; Tiempo de gradiente (min): 12; Tiempo de mantenimiento a 100 % de B (min): 1.8; Caudal (ml/min): 25). el producto obtenido se concentró y después se sometió a liofilización. El producto 2-[(2S)-4-[2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡l-7-(1 -naft¡l)-6.8-d¡h¡dro-5W-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡l]-1 -prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (EJEMPL0306, 65,6 mg. 115 umol. 23 % de rendimiento. 99,8 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI. M+1]: 570.
1H RMN (400 MHz. cloroformo-d) ó 8,27 - 8,17 (m. 1H). 7,92 - 7,84 (m. 1H). 7,63 (d. J = 8.0 Hz. 1H). 7,56 - 7,49 (m. 2H). 7,45 (t. J = 8.0 Hz. 1H). 7,17 (d. J = 6.8 Hz. 1H). 6,68 - 6,56 (m. 1H). 6,42 (dd. J = 1.6. 16,8 Hz. 1H). 5,86 (d a. J = 10,4 Hz. 1H). 5,30 - 4,99 (m. 2H). 4,45 (dd. J = 4.8. 10,8 Hz. 1H). 4,35 - 4,21 (m. 3H). 4,15 (d a. J = 13,6 Hz. 1H). 4,03 (d a. J = 11,6 Hz. 2H). 3,70 - 2,72 (m. 11H). 2,68 - 2,56 (m. 1H). 2,54 (s. 3H). 2,41 - 2,27 (m. 1H). 2,10 - 1,90 (m. 1H).
Ejemplo 307

2-[4-[7-(2-fluoro-3-metoxi-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8 -dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Etapa A: 2-(cianometil)-4-[7-(2-fluoro-3-metoxi-fenil)-2-((2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3.4-ó]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo. Una mezcla de 2-(cianometil)-4-[2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de íere-butilo (Intermedio 66. 300 mg. 636 umol. 1,00 eq.), 1-bromo-2-fluoro-3-metoxi-benceno (261 mg, 1,27 mmol, 2,00 eq.), Pd2(dba)3 (117 mg, 127 umol, 0,20 eq.), RuPhos (89,1 mg. 191 umol. 0,30 eq.) y CS2C03 (622 mg. 1.91 mmol. 3,00 eq.) en tolueno (30 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 16 horas en una atmósfera de N2. A la mezcla de reacción se le añadieron H20 (200 ml) y acetato de etilo (250 ml). La fase orgánica separada se lavó con salmuera (1 x 200 ml). se secó sobre sulfato de sodio. se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0.1 % de FA)/acetonitrilo]. Las fracciones deseadas se recogieron y se neutralizaron con una solución saturada de NaHC03 y se extrajeron con acetato de etilo (100 ml). La capa orgánica separada se secó sobre sulfato de sodio. se filtró y se concentró al vacío. Se obtuvo 2-(cianometil)-4-[7-(2-fluoro-3-metoxi-fenil)-2-[[(2S-1-metilpirrolidin-2-il]metoxi]-6.8-dihidro-5H-pirido[3.4-d]pirimidin-4-il]piperazin-1-carboxilato de íere-butilo (210 mg. 353 umol. 55,4 % de rendimiento. 100 % de pureza) en forma de un sólido de color amarillo claro. LCMS [ESI. M+1]: 596.1
1H RMN (400 MHz, cloroformo-d) ó = 7,00 (dt, J = 1.6. 8.4 Hz. 1H). 6,74 - 6,56 (m. 2H). 4,61 (s a. 1H). 4,44 - 4,34 (m.
1H). 4,28 - 4,10 (m. 3H). 4,01 (d a. J = 13,6 Hz. 1H). 3,90 (s.4H). 3,56 - 3,40 (m. 1H). 3,25 (dd a. J = 3.6. 13,6 Hz. 2H).
3,12 (t a. J = 7.6 Hz. 1H). 2,98 (dt. J = 2.8. 12,4 Hz. 1H). 2,89 - 2,63 (m. 5H). 2,50 (s. 3H). 2,35 - 2,26 (m. 1H). 2,21 -2,06 (m. 3H). 1,90 - 1.68 (m. 3H). 1,60 - 1.42 (m. 9H).
Etapa B: 2-[4-[7-(2-fluoro-3-metox¡-fen¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-illpiperazin-2-illacetonitrilo. A una soluc¡ón de 2-(c¡anomet¡l)-4-[7-(2-fluoro-3-metox¡- fenil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de ferc-butilo (100 mg. 168 umol. 1.00 eq.) en DCM (1 ml) se le añadió TFA (1.63 g. 14.3 mmol. 1.06 ml. 85.0 eq.) a 0 °C en una atmósfera de nitrógeno. La mezcla se agitó a 25 °C durante 1 h y después la mezcla se concentró al vacío. Se obtuvo 2-[4-[7-(2-fluoro-3-metox¡-fen¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]meíox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (210 mg. bruto. t Fa ) en forma de un aceite de color amarillo. LCMS [ESI. M+1]: 496.
Etapa C: 2-[4-[7-(2-fluoro-3-metoxi-fen¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n- 2-¡llmetox¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡n-1-prop-2-eno¡l-p¡peraz¡n-2-¡nacetonitr¡lo. A una mezcla de 2-[4-[7-(2-fluoro-3-metoxi-fen¡l)-2-[[(2S)-1-met¡lpirrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (102 mg. TFA bruto) y DIEA (216 mg.
1.67 mmol. 291 ul) en Dc M (3 ml) se le añadió prop-2-enoato de prop-2-enoílo (16.9 mg. 134 umol) a 0 °C. Después de agitar a 25 °C durante 1 hora. la mezcla de reacción se inactivó con una solución saturada de NaHCÜ3 (5 ml) a 0 °C. y después se extrajo con CH2Cl2 (2 x 25 ml). Las capas orgánicas combinadas se lavaron con salmuera (1 x 50 ml). se secaron sobre sulfato de sodio. se filtraron y se concentraron al vacío para dar un residuo. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0.05 % de hidróxido de amoníaco v/v) - Ac N]; % de B: 38 % - 68 %. 12 min). Se obtuvo 2-[4-[7-(2-fluoro-3-metoxi-fenil)-2-[[(2S)-1-metilpirrolidin- 2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (EJEMPL0307. 7.47 mg. 13.6 umol. dos etapas 17 % de rendimiento. 100 % de pureza) en forma de un sólido de color blanco. LCMS [ESI. M+1]: 550.
1H RMN (400 MHz. cloroformo-d) 6 = 7,07 - 6.95 (m. 1H). 6.74 - 6.51 (m. 3H). 6.45 - 6.34 (m. 1H). 5.83 (d a. J = 10.6 Hz. 1H). 5.08 (s a. 1H). 4.38 (dd. J = 4.8. 10.5 Hz. 1H). 4.31 - 4.12 (m. 3H). 4.07 (d a. J = 13.6 Hz. 1H). 4.03 -3.82 (m. 5H). 3.68 - 3.44 (m. 2H). 3.39 - 3.21 (m. 2H). 3.10 (t a. J = 7.8 Hz. 2H). 2.95 - 2.61 (m. 5H). 2.48 (s. 3H). 2.33 - 2.23 (m. 1H). 2.13 - 1.99 (m. 1H). 1.92 - 1.62 (m. 3H).
Ejemplo 308

2-(1-acr¡lo¡l-4-(7-(3-fluoro-2-metox¡fen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)acetonitr¡lo

Etapa A: 2-(cianometil)-4-[7-(3-fluoro-2-metoxi-fenil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-1-carboxilato de tere-butilo. Una mezcla de 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (Intermedio 66 , 300 mg, 636 umol, 1 eq.), 1-bromo-3-fluoro-2-metoxi-benceno (261 mg, 1,27 mmol, 2 eq.), Pd2(dba)3 (117 mg, 127 umol, 0,2 eq.), RuPhos (89,1 mg, 191 umol, 0,3 eq.) y CS2C03 (622 mg, 1,91 mmol, 3 eq.) en tolueno (30 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 16 horas en una atmósfera de N2. A la mezcla de reacción se le añadieron H20 (200 ml) y acetato de etilo (250 ml). La fase orgánica se separó, se lavó con salmuera (200 ml), se secó sobre sulfato de sodio, se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de FA)/acetonitrilo]. Las fracciones deseadas se recogieron, se neutralizaron con una solución saturada de NaHC03 y se extrajeron con acetato de etilo (1 x 100 ml). La capa orgánica se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El compuesto 2-(cianometil)-4-[7-(3-fluoro-2-metox¡-fen¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carboxilato de tere-butilo (170 mg, 277 umol, 43,6 % de rendimiento, 97,1 % de pureza) se obtuvo en forma de un sólido de color amarillo. lCm S [ESI, M+1]: 596.
1H RMN (400 MHz, cloroformo-d) ó = 6,96 (dt, J = 6,0, 8,0 Hz, 1H), 6,83 - 6,69 (m, 1H), 6,83 - 6 ,68 (m, 1H), 4,60 (s a, 1H), 4,39 (dd, J = 4,8, 10,4 Hz, 1H), 4,31 - 4,14 (m, 3H), 4,04 (d a, J = 13,6 Hz, 2H), 3,94 (d, J = 0,8 Hz, 4H), 3,62 -3,47 (m, 1H), 3,26 (dd a, J = 4,0, 13,6 Hz, 2H), 3,12 (t a, J = 7,6 Hz, 1H), 2,99 (dt, J = 2,8, 12,0 Hz, 1H), 2,88 - 2,64 (m, 5H), 2,50 (s, 3H), 2,36 - 2,24 (m, 1H), 2,22 - 2,07 (m, 2H), 1,94 - 1,64 (m, 3H), 1,56 - 1,46 (m, 9H).
Etapa B: 2-[4-[7-(3-fluoro-2-metoxi- fenil)-2-rr(2S)-1-metilpirrolidin-2-i n metox n -6.8-dihidro-5H-piridor3.4-dlpirimidin-4-¡ n piperazin-2-i n acetonitrilo. A una mezcla de 2-(cianometil)-4-[7-(3-fluoro-2- metoxi-fenil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1 -carboxilato de tere-butilo (170 mg, 285 umol, 1 eq.) en DCM (2,50 ml) se le añadió en una porción TFA (651 mg, 5,71 mmol, 423 ul, 20 eq.) a 0 °C en una atmósfera de N2. Después de agitar a 25 °C durante 2 horas, la mezcla de reacción se concentró a presión reducida para dar un residuo. El producto bruto se usó directamente en la siguiente etapa sin purificación adicional. El compuesto 2-[4-[7-(3-fluoro-2-metox¡fen¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡trilo (173 mg, 284 umol, 99,4 % de rendimiento, TFA) se obtuvo en forma de un sólido de color pardo. LCMS [ESI, M+1]: 496.
Etapa C: 2-[4-[7-(3-fluoro-2- metoxi-fenil)-2-[[(2S)-1-metilpirrolidin-2-i^metoxi^-6,8-dihidro-5H-pirido[3,4-d^pirimidin-4-i n -1-prop-2-enoil-piperazin-2-i n acetonitrilo. A una mezcla de 2-[4-[7-(3-fluoro-2-metoxi-fenil)-2-[[(2S)-1-metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (173 mg, 284 umol, 1 eq., TFA) y prop-2-enoato de prop-2-enoílo (35,8 mg, 284 umol, 1 eq.) en DCM (3 ml) se le añadió en una porción TEA (144 mg, 1,42 mmol, 198 ul, 5 eq.) a 0 °C en una atmósfera de N2. Después de agitar a 25 °C durante 0,5 horas, la mezcla de reacción se inactivó mediante la adición de NaHC03 saturado (1,5 ml) a 0 °C, después se diluyó con agua (2 ml) y se extrajo con DCM (20 ml x 3 ). Las capas orgánicas combinadas se lavaron con agua (15 ml x 1 ), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El producto bruto se purificó por cromatografía en columna (AL03, éter de petróleo/acetato de etilo = 100/1 a Me0H/AE = 1/2). Las fracciones
deseadas se recogieron y se concentraron a presión reducida. Después, el residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) -ACN]; % de B: 48 % - 78 %, 12 min). El compuesto del título 2-[4-[7-(3-fluoro-2- metoxi-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0 308, 28,2 mg, 50,4 umol, 17,8 % de rendimiento, 98,1 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 550.
1H RMN (400 MHz, cloroformo-d) ó = 6,97 (dt, J = 6,0, 8,4 Hz, 1H), 6,83 - 6,76 (m, 1H), 6,72 (d, J = 8,0 Hz, 1H), 6,57 (d a, J = 12,0 Hz, 1H), 6,40 (dd, J = 1,6, 16,8 Hz, 1H), 5,83 (d a, J = 10,8 Hz, 1H), 5,07 (s a, 1H), 4,56 (s a, 1H), 4,39 (dd a, J = 5,2, 10,4 Hz, 1H), 4,30 - 4,18 (m, 2H), 4,18 - 4,06 (m, 2H), 4,04 - 3,96 (m, 1H), 3,95 (d, J = 0,8 Hz, 3H), 3,73 - 3,46 (m, 2H), 3,38 - 3,22 (m, 2H), 3,16 - 2,99 (m, 2H), 2,96 - 2,59 (m, 5H), 2,50 (s, 3H), 2,36 - 2,22 (m, 1H), 2,16 -1,98 (m, 1H), 1,94 - 1,68 (m, 3H).
Ejemplo 309

2-(1-acriloil-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Inserción: 1 -bromo-8-metil-naftaleno. A una solución de 1,8-dibromonaftaleno (1 g, 3,50 mmol, 1 eq.) en THF (20 ml) se le añadió gota a gota MeLi (1,6 M en éter dietílico, 2,62 ml, 1,2 eq.) a 0 °C. Después de agitar durante 30 minutos
a 0 °C, se añadió gota a gota yodometano (3,38 g, 23,8 mmol, 1,48 ml, 6,81 eq.). La mezcla se calentó a 25 °C y se agitó durante 3 horas más. La mezcla de reacción se inactivó con agua (20 ml) y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini C18250 * 50 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - ACN]; % de B: 45 % -70 %, 28 MIN; 40 % min). Se obtuvo 1-bromo-8-metil-naftaleno (340 mg, 1,49 mmol, 43 % de rendimiento, 97 % de pureza) en forma de un sólido de color amarillo después de la liofilización.
1H RMN (400 MHz, cloroformo-d) ó = 7,75 (dd, J = 0,8, 7,2 Hz, 1H), 7,69 (dd, J = 0,8, 8,0 Hz, 1H), 7,66 - 7,59 (m, 1H), 7,30 - 7,22 (m, 2H), 7,13 (t, J = 8,0 Hz, 1H), 3,05 (s, 3H).
Etapa A: 2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l1p¡perazin-1-carbox¡lato de tere-butilo. Una mezcla de 1-bromo-8-metil-naftaleno (122 mg, 551 umol, 1,3 eq.), 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]- 5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (Intermedio 66, 200 mg, 424 umol, 1 eq.), Cs2CÜ3 (345 mg, 1,06 mmol, 2,5 eq.), RuPhos (39,6 mg, 84,8 umol, 0,2 eq.) y Pd2(dba)3 (77,7 mg, 84,8 umol, 0,2 eq.) en tolueno (10 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 110 °C durante 3 horas en una atmósfera de N2. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico)/acetonitrilo)]. La mezcla se ajustó a pH = 7 mediante la adición de una solución acuosa saturada de NaHCÜ3 y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar 2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (140 mg, 224 umol, 53 % de rendimiento, 98 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 612.
1H RMN (400 MHz, cloroformo-d) ó = 7,73 - 7,60 (m, 2H), 7,42 (dd, J = 8,0, 16,0 Hz, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,26 - 7,16 (m, 2H), 4,61 (s a, 1H), 4,40 (s a, 1H), 4,30 - 4,16 (m, 2H), 4,10 - 3,71 (m, 4H), 3,58-3,45 (m, 1H), 3,43-3,25 (m, 1H), 3,23 - 3,04 (m, 4H), 3,03 - 2,85 (m, 5H), 2,83 - 2,55 (m, 4H), 2,49 (s a, 3H), 2,40 - 2,20 (m, 1H), 2,14 - 2,06 (m, 1H), 1,92 - 1,71 (m, 2H), 1,55 - 1,44 (m, 9H).
Etapa B: 2-[4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una solución de 2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2S-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (110 mg, 179 umol, 1 eq.) en DCM (200 ul) se le añadió TFA (307 mg, 2,70 mmol, 199 ul, 15 eq.). Después de agitar a 25 °C durante 1 hora, la mezcla se concentró al vacío. Se obtuvo 2-[4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (112 mg, 179 umol, 100 % de rendimiento, TFA) en forma de un aceite de color amarillo y se usó directamente en la siguiente etapa sin purificación. LCMS [ESI, M+1]: 512.
Etapa C: 2-[4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo. A una solución de 2-[4-[7-(8-metil-1-naftil)-2-[[(2S)- 1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (112 mg, 179 umol, 1 eq., TFA) en DCM (2 ml) se le añadió TEA (181 mg, 1,79 mmol, 249 ul, 10 eq.) a 0 °C. Y después se añadió gota a gota prop-2-enoato de prop-2-enoílo (18,1 mg, 143 umol, 0,8 eq.) en DCM (1 ml) a 0 °C. La mezcla resultante se agitó a 25 °C durante 1 hora. La mezcla de reacción se inactivó con una solución acuosa saturada de NaHCÜ3 (1 ml), se diluyó con agua (10 ml) y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Al2Ü3, éter de petróleo/acetato de etilo = 10/1 a 1/3) y se purificó adicionalmente por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - ACN]; % de B: 55 % - 85 %, 12 min). El compuesto del título 2-[4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (Ej Em Pl0309, 16,2 mg, 28,4 umol, 16 % de rendimiento, 99 % de pureza) se obtuvo en forma de un sólido de color blanco después de la liofilización. LCMS [ESI, M+1]: 566.
Condición de SFC: “ 0J-3S_5_5_40_3ML Columna: Chiralcel 0J-3100 x 4 ,6 mm D.I., 3 um Fase móvil: etanol (0,05 % de DEA) en C02 de 5 % a 40 % Caudal: 3 ml/min Longitud de onda: 220 nm” .
1H RMN (400 MHz, cloroformo-d) ó = 7,70 (d a, J = 8,0 Hz, 1H), 7,65 (t, J = 8,4 Hz, 1H), 7,46 - 7,38 (m, 1H), 7,34 (t, J = 6,4 Hz, 1H), 7,27 - 7,17 (m, 2H), 6,68-6,52 (m J = 9,9 Hz, 1H), 6,46 - 6,35 (m, 1H), 5,83 (d a, J = 10,4 Hz, 1H), 5,08 (s a, 1H), 4,59 (s a, 1H), 4,41 - 4,33 (m, 1H), 4,32 - 4,21 (m, 1H), 4,20 - 4,04 (m, 3H), 4,03 - 3,82 (m, 2H), 3,85-3,67 (m, 1H), 3,61 - 3,33 (m, 2H), 3,24 - 2,99 (m, 4H), 2,92 (s, 3H), 2,87 - 2,77 (m, 1H), 2,71 - 2,59 (m, 2H), 2,47 (d, J = 4,0 Hz, 3H), 2,34 - 2,20 (m, 1H), 2,13 - 1,98 (m, 1H), 1,85 - 1,74 (m, 3H).
Ejemplo 310

2-(1-acriloil-4-(7-(8-cloronaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Inserción: 1 -bromo-8-cloronaftaleno. A una solución de percloroetano (1,66 g, 6,99 mmol, 792 ul, 1 eq.) en THF (30 ml) se le añadió gota a gota n-Buli (2,5 M en hexano, 4,20 ml, 1,5 eq.) a -78 °C. Después de agitar durante 10 minutos más a -78 °C, se añadió 1,8-dibromonaftaleno (2 g, 6,99 mmol, 1 eq.) en THF (10 ml). La mezcla se calentó a 25 °C y se agitó durante 3 horas. La mezcla de reacción se inactivó con agua (100 ml) y se extrajo con acetato de etilo (50 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (50 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi Max-Rp 250 * 50 mm * 10 um; fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 41 % de ACN-71 % de ACN, 30 min; 50 % min). La mezcla se ajustó a pH = 7 mediante la adición de una solución acuosa saturada de NaHCÜ3 y se extrajo con acetato de etilo (50 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar 1 -bromo-8-cloronaftaleno (850 mg, 3,52 mmol, 50 % de rendimiento, 100 % de pureza) en forma de un sólido de color amarillo.
1H RMN (400 MHz, cloroformo-d) 5 = 7,92 (dd, J = 1,2, 7,2 Hz, 1H), 7,80 (ddd, J = 0,8, 8,0, 12,4 Hz, 2H), 7,67 (dd, J = 1,2, 7,6 Hz, 1H), 7,38 (t, J = 8,0 Hz, 1H), 7,32 - 7,25 (t, J = 8,0 Hz, 1H).
Etapa A: 4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de tere-butilo. Una mezcla de 1-bromo-8-cloro-naftaleno (133 mg, 551 umol, 1,3 eq.), 2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (200 mg, 424 umol, 1 eq.), Cs2CÜ3 (345 mg, 1,06 mmol, 2,5 eq.), RuPhos (39,6 mg, 84,8
umol, 0,2 eq.) y Pd2(dba)3 (77,7 mg, 84,8 umol, 0,2 eq.) en tolueno (10 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 110 °C durante 3 horas en una atmósfera de N2. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico)/acetonitrilo)]. La mezcla se ajustó a pH = 7 mediante la adición de una solución acuosa saturada de NaHCÜ3 y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida. El compuesto 4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (150 mg, 237 umol, 56 % de rendimiento, 100 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 632.
1H RMN (400 MHz, cloroformo-d) ó = 7,76 (d, J = 8,4 Hz, 1H), 7,61 (t, J = 7,6 Hz, 1H), 7,52 (d, J = 7,6 Hz, 1H), 7,49 -7.39 (m, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,27 - 7,17 (m, 1H), 4,61 (s a, 1H), 4,51 - 4,33 (m, 2H), 4,22 - 4,15 (m, 1H), 4,10 - 3,77 (m, 4H), 3,57 (s a, 1H), 3,41 - 3,01 (m, 6 H), 3,01 - 2,86 (m, 1H), 2,85 - 2,63 (m, 3H), 2,62 - 2,43 (m, 4H), 2,31 (s a, 1H), 2,13 - 2,06 (m, 1H), 1,78 (s a, 2H), 1,52 (s, 9H).
Etapa B: 2-[4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡llpiperazin-2-illacetonitrilo. A una solución de 4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (150 mg, 237 umol, 1 eq.) en DCM (200 ul) se le añadió TFA (405 mg, 3,56 mmol, 263 ul, 15 eq.). La mezcla se agitó a 25 °C durante 1 hora. La mezcla de reacción se concentró al vacío. El compuesto 2-[4-[7-(8-cloro-1-naftil)-2-[[(2S )-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (153 mg, 236 umol, 100 % de rendimiento, TFA) se obtuvo en forma de un aceite de color amarillo y se usó directamente en la siguiente etapa sin purificación. LCMS [ESI, M+1]: 532.
Etapa C: 2-[4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo. A una solución de 2-[4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (153 mg, 237 umol, 1 eq., TFA) en DCM (2 ml) se le añadió TEA (239 mg, 2,37 mmol, 329 ul, 10 eq.) a 0 °C. Después de la adición, se añadió gota a gota prop-2-enoato de prop-2-enoílo (23,9 mg, 189 umol, 0,8 eq.) en DCM (1 ml) a 0 °C. La mezcla resultante se agitó a 25 °C durante 1 hora. La mezcla de reacción se inactivó con una solución acuosa saturada de NaHCÜ3 (1 ml), se diluyó con agua (20 ml) y se extrajo con acetato de etilo (20 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - Ac N]; % de B: 52 % - 82 %, 12 min). El compuesto del título 2-[4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0310, 37 mg, 61,2 umol, 26 % de rendimiento, 97 % de pureza) se obtuvo después de la liofilización en forma de un sólido de color blanco. LCMS [ESI, M+1]: 586.
Condición de SFC: “AD-3S_4_40_3ML Columna: Chiralpak AD-3 100 x 4 ,6 mm D.I., 3 um Fase móvil: 40 % de isopropanol (0,05 % de DEA) en C02 Caudal: 3 ml/min Longitud de onda: 220 nm” .
1H RMN (400 MHz, cloroformo-d) ó = 7,76 (d, J = 8,0 Hz, 1H), 7,62 (t, J = 7,2 Hz, 1H), 7,53 (d, J = 7,6 Hz, 1H), 7,49 -7.39 (m, 1H), 7,34 (t, J = 8,0 Hz, 1H), 7,27 - 7,18 (m, 1H), 6,59 (s a, 1H), 6,45 - 6,35 (m, 1H), 5,83 (d a, J = 10,8 Hz, 1H), 5,07 (s, 1H), 4,84 - 4,25 (m, 3H), 4,22 - 3,72 (m, 5H), 3,71-3,53 (m, 1H), 3,50-3,34 (m, 1H), 3,32 - 2,96 (m, 5H), 2,91 - 2,76 (m, 1H), 2,70 - 2,52 (m, 21H), 2,48 (d, J = 2,8 Hz, 3H), 2,33 - 2,20 (m, 1H), 2,13 - 1,96 (m, 1H), 1,86 - 1,72 (m, 3H).
Ejemplo 311

2-(1-acriloil-4-(7-(2-fluoronaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa 1: 2-fluoronaftalen-1 -ol. A una solución de naftalen-1 -ol (2 g, 13,9 mmol, 5,00 ml, 1 eq.) en CH3CN (50 ml) se le añadió Select-F (4,91 g, 13,9 mmol, 1 eq.). La mezcla se agitó a 20 °C durante 3 horas. La mezcla se concentró al vacío. El residuo se purificó por cromatografía en columna (Si02, EP/AE = 3/1) y se purificó adicionalmente por HPLC prep. (columna: Phenomenex Synergi C18150 * 25 * 10 um; fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 40 % - 70 %, 10 min). La mezcla se ajustó a pH = 7 con una solución acuosa saturada de NaHC03 y se extrajo con acetato de etilo (20 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida. El compuesto 2-fluoronaftalen-1-ol (400 mg, 2,44 mmol, 18 % de rendimiento, 99 % de pureza) se obtuvo en forma de un sólido de color amarillo.
1H RMN (400 MHz, cloroformo-d) ó = 8,21 (d, J = 8,4 Hz, 1H), 7,80 (d, J = 8,4 Hz, 1H), 7,52 (t, J = 7,2 Hz, 1H), 7,47(t, J = 7,6 Hz, 1H), 7,39 (dd, J = 5,2, 8,8 Hz, 1H), 7,31 (d, J = 9,6 Hz, 1H), 5,58 (s a, 1H).
Etapa 2: trifluorometanosulfonato de (2-fluoro-1-naftilo). A una solución de 2-fluoronaftalen-1-ol (400 mg, 2,47 mmol, 1 eq.) en DCM (10 ml) se le añadieron Tf20 (765 mg, 2,71 mmol, 448 ul, 1,1 eq.) y DIEA (637 mg, 4,93 mmol, 859 ul, 2 eq.) a 0 °C. Después de agitar a 0 °C durante 1 hora, la mezcla de reacción se diluyó con acetato de etilo (20 ml) y se lavó con agua (20 ml * 3). La capa orgánica combinada se lavó con salmuera (20 ml), se secó sobre Na2S04, se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 50/1 a 3/1). El compuesto trifluorometanosulfonato de (2-fluoro-1-naftilo) (500 mg, 1,65 mmol, 67 % de rendimiento, 97 % de pureza) se obtuvo en forma de un aceite incoloro.
1H RMN (400 MHz, cloroformo-d) ó = 8,07 (d, J = 8,4 Hz, 1H), 7,95 - 7,85 (m, 2H), 7,70 (t, J = 7,6 Hz, 1H), 7,62 - 7,54 (m, 1H), 7,41 (t, J = 9,2 Hz, 1H).
Etapa A: 2-(cianometil)-4-[7-(2-fluoro- 1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-illpiperazin-1-carboxilato de tere-butilo. Una mezcla de trifluorometanosulfonato de (2-fluoro-1-naftilo) (243 mg, 827 umol, 1,3 eq.), 2-(cianometil)-4-[2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirim idin-4-il]piperazin-1 -carboxilato de tere-butilo (300 mg, 636 umol, 1 eq.), Cs2C03 (518 mg, 1,59 mmol, 2,5 eq.), RuPhos (59,4 mg, 127 umol, 0,2 eq.) y Pd2(dba)3 (116 mg, 127 umol, 0,2 eq.) en tolueno (10 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 3 horas en una atmósfera de N2. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (20 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico)/acetonitrilo)]. La mezcla se ajustó a pH = 7 mediante la adición de una solución acuosa saturada de NaHC03 y se extrajo con acetato de etilo (20 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida. El compuesto 2-(cianometil)-4-[7-(2-fluoro- 1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H
pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (100 mg, 155 umol, 25 % de rendimiento, 96 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 616.
1H RMN (400 MHz, cloroformo-d) 6 = 8,33 (s a, 1H), 7,84 (d, J = 7,76 Hz, 1H), 7,75-7,64 (m, 1H), 7,57 - 7,41 (m, 2H), 7,25-7,21 (m, 1H), 4,63 (s a, 1H), 4,47 - 4,16 (m, 4H), 4,13 - 3,82 (m, 4H), 3,61 - 3,25 (m, 3H), 3,20 - 2,90 (m, 4H), 2,81 - 2,56 (m, 4H), 2,49 (s, 3H), 2,38 - 2,19 (m, 1H), 2,14 - 2,06 (m, 1H), 1,88-1,69 (m, 2H), 1,53 (s, 9H).
Etapa B: 2-[4-[7-(2-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-illacetonitrilo. A una solución de 2-(cianometil)-4-[7-(2-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d)pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (100 mg, 162 umol, 1 eq.) en DCM (200 ul) se le añadió TFA (278 mg, 2,44 mmol, 180 ul, 15 eq.). La mezcla se agitó a 25 °C durante 1 hora. La mezcla de reacción se concentró al vacío. El compuesto 2-[4-[7-(2-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (102 mg, 162 umol, 100 % de rendimiento, TFA) se obtuvo en forma de un aceite de color amarillo y se usó directamente en la siguiente etapa sin purificación. LCMS [ESI, M+1]: 516.
Etapa C: 2-[4-[7-(2-fluoro-1-naftil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n- 2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo. A una solución de 2-[4-[7-(2-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (102 mg, 197 umol, 1 eq., TFA) en DCM (2 ml) se le añadió TEA (200 mg, 1,98 mmol, 275 ul, 10 eq.) a 0 °C. Después de la adición, se añadió gota a gota prop-2-enoato de prop-2-enoílo (19,9 mg, 158 umol, 0,8 eq.) en DCM (1 ml) a 0 °C. Después de agitar a 25 °C durante 1 hora, la mezcla de reacción se inactivó con una solución acuosa saturada de NaHCÜ3 (1 ml), se diluyó con agua (20 ml) y se extrajo con acetato de etilo (20 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150*25 mm*10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - ACN]; % de B: 52 % - 82 %, 12 min). El compuesto del título 2-[4-[7-(2-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin- 2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0311, 11 mg, 18,9 umol, 9,6 % de rendimiento, 98 % de pureza) se obtuvo después de la liofilización en forma de un sólido de color blanco. LCMS [ESI, M+1]: 570.
Condición de SFC: “0D-3S_3_40_3MI, Columna: Chiralcel 0D-3100 x 4,6 mm D.I., 3 um Fase móvil: 40 % de metanol (0,05 % de DEA) en C02 Caudal: 3 ml/min Longitud de onda: 220 nm” .
1H RMN (400 MHz, cloroformo-d) 6 = 8,34 (s a, 1H), 7,84 (d, J = 8,0 Hz, 1H), 7,69 (dd, J = 4,4, 8 ,8 Hz, 1H), 7,52 (t, J = 7,2 Hz, 1H), 7,47 (t, J = 6 ,8 Hz, 1H), 7,29-7,25 (m, 1H), 6,65-6,50 (m, J = 10,5 Hz, 1H), 6,40 (dd, J = 1,2, 16,8 Hz, 1H), 5,84 (d a, J = 10,4 Hz, 1H), 5,11 (s a, 1H), 4,65 (s, 1H), 4,43 - 3,85 (m, 7H), 3,81 - 3,30 (m, 3H), 3,24 - 2,95 (m, 3H), 2,94 - 2,56 (m, 4H), 2,47 (s, 3H), 2,36 - 2,20 (m, 1H), 2,13 - 1,97 (m, 1H), 1,93 - 1,75 (m, 3H).
Ejemplo 312
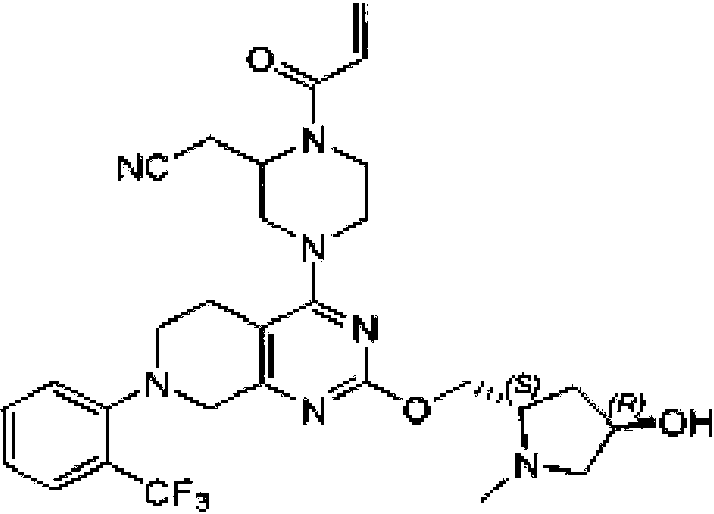
2-[4-[2-[[(2S,4R)-4-hidroxi-1-metil-pirrolidin-2-il]metoxi]-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2 -enoil-piperazin-2 -il]acetonitrilo

Etapa A: (2S,4R)-4-[fe/'c-butil(difenil)silil]oxipirrolidin-2-carboxilato de metilo. A una solución de (2S,4R)-4-hidroxipirrolidin-2-carboxilato de metilo (10 g, 55,1 mmol, 1 eq., HCl) en acetato de etilo (10 ml) se le añadieron imidazol (7,50 g, 110 mmol, 2 eq.) y TBDPSCl (16,7 g, 60,6 mmol, 15,6 ml, 1,1 eq.). La mezcla se agitó a 25 °C durante 16 horas. El precipitado se retiró por filtración. El filtrado se concentró a presión reducida y el residuo se disolvió con acetato de etilo (200 ml) y agua (100 ml). La fase orgánica se lavó con salmuera (1 * 100 ml), se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo/acetato de etilo 10/1 a 1/1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar (2S,4R)-4-[fe/'c-butil(difenil)silil]oxipirrolidin-2-carboxilato de metilo (2,9 g, 7,41 mmol, 13 % de rendimiento, 98 % de pureza) en forma de un aceite incoloro. LCMS [ESI, M+1]: 384.
1H RMN (400 MHz, cloroformo-d) ó = 7,64 (dt, J = 1,2, 7,2 Hz, 4H), 7,47 - 7,35 (m, 6H), 4,40 (td, J = 2,4, 4,8 Hz, 1H), 4.07 (t, J = 8,0 Hz, 1H), 3,70 (s, 3H), 3,03 - 2,96 (m, 1H), 2,96 - 2,89 (m, 1H), 2,17 - 2,07 (m, 1H), 1,84 - 1,77 (m, 1H), 1.07 (s, 9H).
Etapa B: (2S,4R)-4-[fe/'c-butil(difenil)silil]oxi-1 -metil-pirrolidin-2-carboxilato de metilo. A una solución de (2S,4R)-4-[íerc-butil(difenil)silil]oxipirrolidin-2-carboxilato de metilo (2,7 g, 7,04 mmol, 1 eq.) y formaldehído (acuoso al 37 %, 2,86 g, 35,2 mmol, 2,62 ml, 5 eq.) en Me0H (20 ml) se le añadieron CH3C00H (423 mg, 7,04 mmol, 403 ul, 1 eq.) y NaBH3CN (1,77 g, 28,2 mmol, 4 eq.). La mezcla se agitó a 25 °C durante 1 h. La mezcla se diluyó con acetato de etilo (200 ml) y se lavó con agua (2 * 100 ml) y salmuera (1 * 100 ml). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía de columna sobre gel de sílice (éter de petróleo/acetato de etilo 20/1 a 3/1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar (2S,4R)-4-[íerc-butil(difenil)silil]oxi-1-metil-pirrolidin-2-carboxilato de metilo (2,4 g, 5,71 mmol, 81 % de rendimiento, 94,6 % de pureza) en forma de un aceite incoloro. LCMS [ESI, M+1]: 398.
1H RMN (400 MHz, cloroformo-d) ó = 7,63 (ddd, J = 1,2, 4,0, 7,6 Hz, 4H), 7,46 - 7,32 (m, 6H), 4,47 - 4,39 (m, 1H), 3,71 (s, 3H), 3,32 (t, J = 8,4 Hz, 1H), 3,24 (dd, J = 6,0, 9,6 Hz, 1H), 2,43 - 2,40 (m, 1H), 2,40 (s, 3H), 2,23 - 2,00 (m, 2H), 1.07 (s, 9H).
Etapa C: [(2S, 4R)-4-[fe/'c-butil(difenil)sili n oxi-1-metil-pirrolidin-2-¡ n metanol. A una solución de (2S ,4R )-4-[terc- butil(difenil)silil]oxi-1 -metil-pirrolidin-2-carboxilato de metilo (2,20 g, 5,53 mmol, 1,00 eq.) en THF (40 ml) se le añadió LiAlH4 (840 mg, 22,1 mmol, 4,00 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 1 hora. La mezcla se inactivó con agua (1,00 ml), solución de hidróxido de sodio (15,0 %, 2,00 ml) y agua (3,00 ml). El precipitado se retiró por filtración y el filtrado se concentró al vacío. El residuo se purificó por cromatografía en columna (AL03, metanol/acetato de etilo =
1/10) para dar [(2S, 4R)-4-[ferc-butil(difenil)silil]oxi-1-metil-pirrolidin-2-il]metanol (1,70 g, 4,55 mmol, 82 % de rendimiento) en forma de un aceite incoloro. LCMS [ESI, M+1]: 370.
1H RMN (400 MHz, cloroformo-d) 6 = 7,68 - 7,61 (m, 4H), 7,46 - 7,35 (m, 6H), 4,35 - 4,26 (m, 1H), 3,62 (dd, J = 3,2, 10,8 Hz, 1H), 3,34 (dd, J = 2,0, 10,8 Hz, 1H), 3,14 (dd, J = 5,6, 10,0 Hz, 1H), 2,78 - 2,70 (m, 1H), 2,43 (dd, J = 6,0, 9,6 Hz, 1H), 2,33 (s, 3H), 2,02 - 1,93 (m, 1H), 1,92 - 1,84 (m, 1H), 1,06 (s, 9H).
Etapa D: 4-[2-[[(2S.4^)-4-[ferc-but¡l(d¡fen¡l)s¡l¡l1ox¡-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-7-[2-(tr¡fluoromet¡l)fen¡l1-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-2-(cianomet¡l)p¡peraz¡n-1-carbox¡lato de tere-butilo. A una solución de 2-(cianometil)-4-[2-metilsulfinil-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (0,30 g, 531 umol, 1,00 eq.) y [(2S,4R)-4-[ferc-butil(difenil)silil1oxi-1 -metil-pirrolidin-2-il1metanol (295 mg, 797 umol, 1,50 eq.) en THF (5 ml) se le añadió t-Bu0Na (102 mg, 1,06 mmol, 2,00 eq.) a 0 °C. Después de agitar a 0 °C durante 0,5 h, la mezcla se ajustó a pH = 7 con una solución de HCl (1,00 M, 0,40 ml) y se extrajo con acetato de etilo (3 x 10,0 ml). Las capas orgánicas se lavaron con salmuera (1 x 10,0 ml), se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna (Ah03, éter de petróleo/acetato de etilo = 1/3) para dar 4-[2-[[(2S,4R)-4-[terc-butil(difenil)silil]oxi-1-metil-pirrolidin-2-il]metoxi]-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (0,22 g, 238 umol, 45 % de rendimiento) en forma de un sólido de color blanco. LCMS [ESI, M+11: 870.
1H RMN (400 MHz, cloroformo-d) 6 = 7,68 (d a, J = 8,0 Hz, 1H), 7,66 - 7,61 (m, 4H), 7,57 (t a, J = 7,2 Hz, 1H), 7,44 -7.34 (m, 7H), 7,33 - 7,28 (m, 1H), 4,61 (s a, 1H), 4,45 - 4,28 (m, 2H), 4,09 - 3,95 (m, 4H), 3,87 (d a, J = 12,8 Hz, 1H), 3,28 - 3,15 (m, 4H), 3,10 (ddd, J = 3,6, 7,2, 12,0 Hz, 2H), 3,02 - 2,93 (m, 1H), 2,88 - 2,67 (m, 4H), 2,48 (s, 3H), 2,45 -2.34 (m, 2H), 2,14 - 2,07 (m, 1H), 1,94 - 1,80 (m, 1H), 1,52 (s, 9H), 1,06 (s, 9H).
Etapa E: 2-[4-[2-[[(2S,4ft)-4-[terc-butil(difenil)silil]oxi-1-metil-pirrolidin-2-il]metoxi]-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. Una mezcla de 4-[2-[[(2S,4R)-4-[terc-butil(difenil)silil1oxi-1-metilp¡rrol¡d¡n-2-¡l]metox¡]-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carboxilato de tere-butilo (0,08 g, 91,9 umol, 1,00 eq.) y TfA (157 mg, 1,38 mmol, 102 ul, 15,0 eq.) en diclorometano (150 ul) se agitó a 10 °C durante 1 hora. La mezcla se concentró al vacío para dar 2-[4-[2-[[(2S,4R)-4-[tercbut¡l(d¡fen¡l)s¡l¡l]ox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (81,3 mg, bruto, TFA) en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+11: 770.
Etapa F: 2-[4-[2-[[(2S,4ffl-4-[terc-butil(difenil)silil]oxi-1-metil-pirrolidin-2-il]metoxi]-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l)aceton¡tr¡lo. A una solución de 2-[4-[2-[((2S,4R)-4-[tercbut¡l(d¡fen¡l)s¡l¡l]ox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (0,08 g, bruto, TFA) y EtsN (91,6 mg, 905 umol, 125,96 ul) en diclorometano (2,00 ml) se le añadió prop-2-enoato de prop-2-enoílo (11,4 mg, 90,5 umol) a -40 °C. Después de agitar a 20 °C durante 0,5 h, la mezcla se inactivó con agua (0,20 ml) y se concentró al vacío. El residuo se purificó por cromatografía en columna (Ab03, metanol/acetato de etilo = 1/10). Las fracciones deseadas se recogieron y se concentraron al vacío para dar 2-[4-[2-[[(2S,4R)-4-[terc-but¡l(d¡fen¡l)s¡l¡ljox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (0,06 g, 65,5 umol, dos etapas 72 % de rendimiento) en forma de un aceite de color amarillo. lCm S [ESI, M/2+11: 412.
Etapa G: 2-[4-[2-[[(2S,4ffl-4-hidroxi-1-metil-pirrolidin-2-il]metoxi]-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1acetonitr¡lo. Una mezcla de 2-[4-[2-[[(2S,4R)-4-[terc-butil(difenil)silil1oxi-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-i-prop-2-eno¡l-p¡peraz¡n-2-il1acetonitrilo (0,05 g, 60,7 umol, 1,00 eq.) y TBAF (1,00 M, 607 ul, 10,0 eq.) en THF (0,50 ml) se agitó a 20 °C durante 1 hora. La mezcla se concentró al vacío. El residuo se purificó por cromatografía en columna (Ah03, acetato de etilo/metanol = 3/1) y cromatografía ultrarrápida de fase inversa [agua (AF, 0,10 %)/acetonitrilo1, después se purificó adicionalmente por HPLC prep. columna: Boston pH-lex 150 * 25 10 um; fase móvil: [agua (0,10 %, TfA) - ACN1; % de B: 31 %-61 %, 10 min y columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - ACN1; % de B: 42 % - 72 %, 12 min. Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[4-[2-[[(2S,4R)-4-h¡drox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-[2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (EJEMPL0312, 3,70 mg, 6,30 umol, 10 % de rendimiento, 99,7 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+11: 586.
1H RMN (400 MHz, cloroformo-d) 6 = 7,69 (d, J = 8,0 Hz, 1H), 7,58 (t, J = 8,0 Hz, 1H), 7,41 (d, J = 8,0 Hz, 1H), 7,30 (t, J = 8,0 Hz, 1H), 6,57 (d a, J = 10,8 Hz, 1H), 6,39 (dd, J = 1,6, 16,4 Hz, 1H), 5,83 (d a, J = 10,8 Hz, 1H), 5,06 (s a, 1H), 4,49 - 4,42 (m, 1H), 4,37 (ddd, J = 3,2, 4,8, 10,8 Hz, 1H), 4,25 - 4,17 (m, 1H), 4,15 - 4,06 (m, 3H), 3,97 (d a, J = 10,8 Hz, 1H), 3,60 (s a, 1H), 3,43 (dd, J = 6,0, 10,0 Hz, 1H), 3,33 (s a, 1H), 3,23 - 3,16 (m, 1H), 3,15 - 3,06 (m, 2H), 3,04 - 2,89 (m, 3H), 2,88 - 2,66 (m, 4H), 2,48 (s, 3H), 2,33 (dd, J = 6,0, 10,0 Hz, 1H), 2,13 - 1,93 (m, 2H).
Ejemplo 313

2-[4-[2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-7-[5-(trifluorometil)-1H-indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo
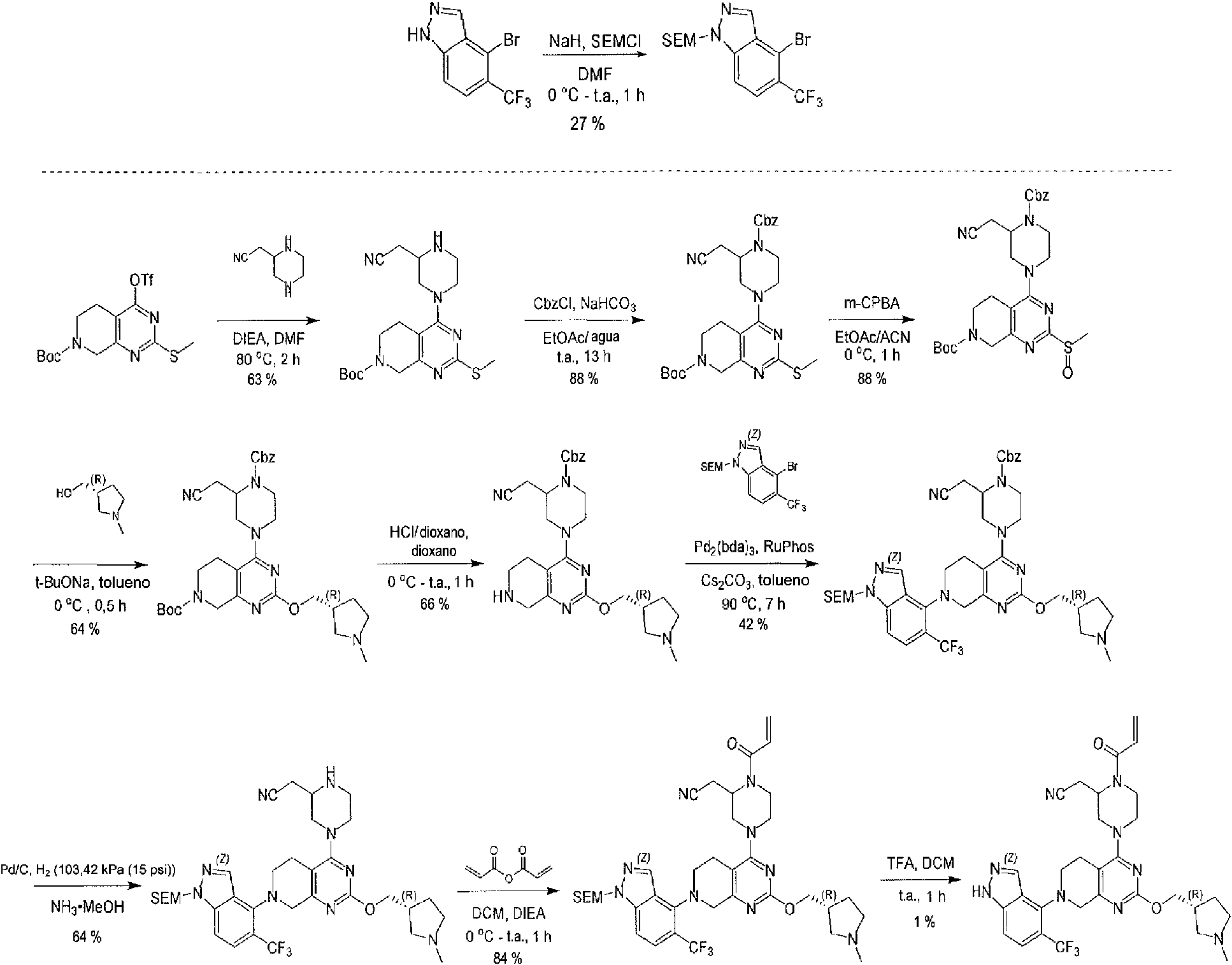
Etapa A: 2-[[4-bromo-5-(tr¡fluoromet¡l)¡ndazol-1-¡l1metox¡1etil-tr¡met¡l-s¡lano. A una solución de 4-bromo-5-(trifluorometil)-1H-indazol (1 g, 3,77 mmol, 1 eq.) en DMF (45 ml) se le añadió NaH (181 mg, 4,53 mmol, 60,0 % de pureza, 1,2 eq.) a 0 °C. Después de agitar a 0 °C durante 1 hora, se añadió gota a gota una solución de SEM-Cl (818 mg, 4,91 mmol, 868 ul, 1,3 eq.) en DMF (15 ml). La mezcla se calentó a 20 °C y se agitó durante 1 hora. La mezcla se inactivó con una solución acuosa saturada de cloruro de amonio (50 ml), se diluyó con agua (100 ml) y después se extrajo con acetato de etilo (2 * 50 ml). Las capas orgánicas combinadas se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc de 1:0 a 100:1) para dar 2-[[4-bromo-5-(trifluorometil)indazol-1-il]metoxi]etil-trimetil-silano (430 mg, 1,03 mmol, 27,4 % de rendimiento, 95,0 % de pureza) en forma de un aceite incoloro.1
1H RMN (400 MHz, cloroformo-d) 5 = 8,18 (s, 1H), 7,74 - 7,66 (m, 1H), 7,63 - 7,56 (m, 1H), 5,76 (s, 2H), 3,59 - 3,49 (m, 2H), 0,93 - 0,86 (m, 2H), -0,01 - -0,08 (m, 9H).
Etapa B: 4-[3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-7-carbox¡lato de tere-butilo. A la soluc¡ón de 2-met¡lsulfan¡l-4-(tr¡fluoromet¡lsulfon¡lox¡)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo (3,59 g. 8,36 mmol, 1 eq.), 2-p¡peraz¡n-2-¡laceton¡tr¡lo (Intermed¡o 62. 1,99 g. 10,0 mmol, 1.2 eq., 2HCl) en DMF (30 ml) se le añad¡ó DIEA (2,16 g. 16,7 mmol. 2,91 ml. 2 eq.) y después la mezcla se calentó a 80 °C y se ag¡tó a 80 °C durante 2 horas. A la mezcla se le añad¡ó agua (80 ml). La mezcla resultante se d¡luyó con EtOAc (20 ml) y se extrajo con EtOAc (5 x 60 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (80 ml). se secaron sobre Na2S04 anh¡dro, se f¡ltraron y se concentraron al vacío. El res¡duo se pur¡f¡có por columna ultrarráp¡da de fase ¡nversa (ACN/agua (0.1 % de AF) = 40 %) para dar 4-[3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-met¡lsulfan¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo (2,37 g. 5,27 mmol. 63,1 % de rend¡m¡ento, 90,0 % de pureza) en forma de un sól¡do de color pardo. LCMS [ESI, M+1]: 405.
Etapa C: 4-[4-benc¡lox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡ll-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo. A la soluc¡ón de 4-[3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo (2,37 g. 5,86 mmol. 1 eq.). NaHC03 (1,48 g. 17,6 mmol.684 ul. 3 eq.) en EtOAc (30 ml) y agua (15 ml) se le añad¡ó gota a gota CbzCl (2,00 g. 11,7 mmol. 1,67 ml.2 eq.) y después la mezcla se ag¡tó a 25 °C durante 13 horas. La mezcla se d¡luyó con EtOAc (15 ml) y se extrajo con EtOAc (2 x 15 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (15 ml). Las capas orgán¡cas comb¡nadas se secaron sobre Na2S04 anh¡dro, se f¡ltraron y se concentraron al vacío. El res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (EP: EtOAc = 20:1 ~0:1) para dar 4-[4-benc¡lox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-met¡lsulfan¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo (2,91 g. 5,13 mmol. 87,6 % de rend¡m¡ento, 95,0 % de pureza) en forma de un sól¡do de color amar¡llo. LCMS [ESI, M+1]: 539.
Etapa D: 4-[4-benc¡lox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡ll-2-met¡lsulf¡n¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo. A la soluc¡ón de 4-[4-benc¡lox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-met¡lsulfan¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo (2,91 g. 5,40 mmol. 1 eq.) en EtOAc (60 ml) y ACN (20 ml) se le añad¡ó m-CPBA (1,10 g. 5,40 mmol. 85,0 % de pureza. 1 eq.) a 0 °C y después la mezcla se ag¡tó a 0 °C durante 1 hora. La mezcla de reacc¡ón se ¡nact¡vó con Na2S203 saturado (30 ml) a 0 °C y después se extrajo con EtOAc (50 ml). La capa orgán¡ca se secó sobre Na2S04 anh¡dro, se f¡ltró y se concentró al vacío. El res¡duo se pur¡f¡có por columna ultrarráp¡da de fase ¡nversa (ACN/agua (0.1 % de NH3^H20) = 62 %) para dar 4-[4-benc¡lox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo (2.65 g.
4,73 mmol. 87,6 % de rend¡m¡ento, 99,0 % de pureza) en forma de un sól¡do de color blanco. LCMS [ESI. M+11]: 555.
Etapa E: 4-r4-benc¡lox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡n-2-rr(3ft)-1-met¡lp¡rrol¡d¡n-3-¡nmetoxn-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo. A la soluc¡ón de 4-[4-benc¡lox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo (2.2 g. 3,97 mmol. 1 eq.). [(3R)-1-met¡lp¡rrol¡d¡n-3-¡l]metanol (914 mg. 7,93 mmol. 2 eq.) en tolueno (44 ml) se le añad¡ó t-Bu0Na (572 mg. 5,95 mmol.
1.5 eq.) a 0 °C y después la mezcla se ag¡tó a 0 °C durante 0.5 horas. A la mezcla se le añad¡ó agua (15 ml). La mezcla resultante se d¡luyó con EtOAc (10 ml) y se extrajo con EtOAc (2 x 20 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (20 ml). Las capas orgán¡cas comb¡nadas se secaron sobre Na2S04 anh¡dro, se f¡ltraron y se concentraron al vacío. El res¡duo se pur¡f¡có por columna ultrarráp¡da de fase ¡nversa (ACN/agua (0.1 % de a F) = 45 %) para dar 4-[4-benc¡lox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-[[(3R)-1-met¡lp¡rrol¡d¡n-3-¡l]metox¡j-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo (1,64 g. 2,52 mmol. 63,5 % de rend¡m¡ento, 93,0 % de pureza) en forma de un sól¡do de color blanco. LCMS [ESI. M+1]: 605.
1H RMN (400 MHz, cloroformo-d) 6 = 7,44 - 7,31 (m. 5H). 5,19 (s. 2H). 4,71 - 4,54 (m. 2H). 4,35 (d a. J = 19,2 Hz. 1H).
4,24 - 4,16 (m. 2H). 4,11 - 3,92 (m. 2H). 3,79 (d a. J = 12,4 Hz. 2H). 3,28 (d a. J = 12,8 Hz. 3H). 2,98 (dt. J =3.2.
12,4 Hz. 1H). 2,87 - 2,44 (m. 8H). 2,36 (s. 3H). 2,12 - 2,06 (m. 1H). 1,87 (s a. 2H). 1,49 (s. 9H).
Etapa F: 2-(c¡anomet¡l)-4-[2-[[(3R)-1-met¡lp¡rrol¡d¡n-3-¡llmetox¡l-5.6.7.8-tetrah¡drop¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de benc¡lo. A la soluc¡ón de 4-[4-benc¡lox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-[[(3R)-1-met¡lp¡rrol¡d¡n-3-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo (1,64 g.2,71 mmol. 1 eq.) en d¡oxano (30 ml) se le añad¡ó HCl/d¡oxano (4 M. 30 ml. 44,32 eq.) a 0 °C y después la mezcla se ag¡tó a 25 °C durante 1 hora. La mezcla de reacc¡ón se concentró al vacío. El res¡duo se pur¡f¡có por columna ultrarráp¡da de fase ¡nversa (ACN/agua (0.1 % de AF) = 35,5 %) para dar 2-(c¡anomet¡l)-4-[2-[[(3R)-1-met¡lp¡rrol¡d¡n-3-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (0,95 g. 1,78 mmol. 65,9 % de rend¡m¡ento, 95,0 % de pureza) en forma de un sól¡do de color blanco. LCMS [ESI. M+1]: 506.
1H RMN (400 MHz. cloroformo-d) 6 = 7,43 - 7,32 (m. 5H). 5,19 (s. 2H). 4,66 (s a. 1H). 4,25 - 4,07 (m. 3H). 4,01 - 3,91 (m. 3H). 3,83 (d a. J = 13,6 Hz. 1H). 3,38 - 3,18 (m. 2H). 3,16 - 3,07 (m. 1H). 3,03 - 2,92 (m. 2H). 2,81 (s a. 1H). 2,75 - 2,65 (m. 3H). 2,60 (d a. J = 5.6 Hz. 3H). 2,55 - 2,45 (m. 2H). 2,36 (s. 3H). 2,14 - 1,97 (m. 1H). 1,68 - 1,54 (m. 2H).
Etapa G: 2-(c¡anomet¡l)-4-[2-[[(3R)-1-met¡lp¡rrol¡d¡n-3-¡llmetox¡l-7-[5-(tr¡fluoromet¡l)-1-(2-tr¡met¡ls¡l¡letox¡met¡l)¡ndazol-4-¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de benc¡lo. A la soluc¡ón de 2-(c¡anomet¡l)-4-[2-[[(3R)-1-met¡lp¡rrol¡d¡n-3-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (510 mg. 1,01 mmol. 1 eq.). 2-[[4-bromo-5-(tr¡fluoromet¡l)¡ndazol-1-¡l]metox¡]et¡l-tr¡met¡l-s¡lano (399 mg. 1,01 mmol. 1 eq.). RuPhos (188 mg. 403 umol, 0.4 eq.) y Cs2C03 (986 mg. 3,03 mmol. 3 eq.) en tolueno (10 ml) se le añad¡ó
Pd2(dba)3 (185 mg, 202 umol, 0,2 eq.) en una atmósfera de N2 y la suspensión se desgasificó al vacío y se purgó varias veces con N2. La mezcla se calentó a 90 °C y se agitó a 90 °C durante 14 horas. La mezcla de reacción se filtró y la torta de filtro se lavó con EtOAc (2 * 15 ml). El filtrado se concentró al vacío. El residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0,1 % de AF) = 55 %) para dar 2-(cianometil)-4-[2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (365 mg, 423 umol, 41,9 % de rendimiento, 95,0 % de pureza) en forma de un sólido de color pardo. LCMS [ESI, M+1]: 820.
1H RMN (400 MHz, cloroformo-d) ó = 8,15 (s, 1H), 7,70 (d, J = 8,8 Hz, 1H), 7,52 (d, J = 8,8 Hz, 1H), 7,42 - 7,33 (m, 5H), 5,76 (s, 2H), 5,24 - 5,16 (m, 2H), 4,70 (s a, 1H), 4,42 - 4,27 (m, 2H), 4,21 (d a, J = 6,8 Hz, 2H), 4,06 (d a, J = 10,8 Hz, 1H), 3,90 (d a, J = 12,4 Hz, 1H), 3,61 - 3,54 (m, 2H), 3,50 - 3,38 (m, 2H), 3,31 (d a, J = 11,6 Hz, 2H), 3,10 -2,99 (m, 1H), 2,86 (s a, 2H), 2,81 - 2,66 (m, 4H), 2,65 - 2,57 (m, 1H), 2,56 - 2,44 (m, 2H), 2,36 (s, 3H), 2,12 - 2,06 (m, 1H), 2,04 - 1,96 (m, 1H), 1,69 - 1,56 (m, 1H), 0,95 - 0,87 (m, 2H), -0,03 - -0,10 (m, 9H).
Etapa H: 2-[4-[2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo. Se burbujeó NH3 en Me0H (6 ml) durante 5 minutos. A la solución se le añadió 2-(cianometil)-4-[2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (310 mg, 378 umol, 1 eq.), Pd/C (150 mg, 10,0 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 28 °C durante 1 hora. La mezcla se filtró y el filtrado se concentró al vacío. El residuo se usó directamente en la siguiente etapa sin purificación adicional. El compuesto 2-[4-[2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (165 mg, 240 umol, 63,6 % de rendimiento, 100 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 686.
Etapa I: 2-[4-[2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil))indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo. A la solución de 2-[4-[2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (190 mg, 277 umol, 1 eq.), DIEA (107 mg, 831 umol, 145 ul, 3 eq.) en DCM (4 ml) se le añadió prop-2-enoato de prop-2-enoílo (41,9 mg, 332 umol, 1,2 eq.) a 0 °C y después la mezcla se agitó a 28 °C durante 1 hora. La mezcla de reacción se inactivó con agua (1 ml) y se extrajo con DCM (3 * 4 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se usó directamente en la siguiente etapa sin purificación adicional. El compuesto 2-[4-[2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo (230 mg, 233 umol, 84,2 % de rendimiento, 75,0 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 740.
Etapa J: 2-[4-[2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-7-[5-(trifluorometil)-1H-indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1acetonitr¡lo. A la solución de 2-[4-[2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-7-[5-(trifluorometil)-1-(2-trimetilsililetoximetil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo (220 mg, 297 umol, 1 eq.) en DCM (1 ml) se le añadió TFA (1,70 g, 14,9 mmol, 1,10 ml, 50 eq.) y la mezcla se agitó a 28 °C durante 1 hora. La mezcla de reacción se basificó con NaHC03 saturado (12 ml) a pH = 8. La mezcla resultante se extrajo con DCM: Me0H (10:1) (3 * 15 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EtOAc: Me0H = 100:1 ~10:1) y después el residuo se purificó por HPLC prep. (columna: Gemini 150 * 25 5 ufase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN];% de B: 44 %-74 %,12 min) para dar 2-[4-[2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-7-[5-(trifluorometil)-1H-indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0313, 1,23 mg, 2,01 umol, 6,74e-1 % de rendimiento, 99,4 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 610.
1H RMN (400 MHz, metanol-d4) ó = 8,38 (s, 1H), 7,70 (d, J = 8,8 Hz, 1H), 7,57 (d, J = 8,8 Hz, 1H), 6,84 (s a, 1H), 6,31 (d a, J = 16,8 Hz, 1H), 5,86 (d a, J = 10,8 Hz, 1H), 5,13 (s a, 1H), 4,62 (s a, 1H), 4,37 - 4,20 (m, 5H), 4,14 (d a, 11,6 Hz, 1H), 3,64 (s a, 1H), 3,50 (d a, J =4,8 Hz, 2H), 3,41 - 3,34 (m, 1H), 3,29 - 3,16 (m, 2H), 3,03 (d a, J = 6,4 Hz, 1H), 2,94 (s a, 2H), 2,88 - 2,80 (m, 1H), 2,80 - 2,72 (m, 1H), 2,69 (t a, J = 6,8 Hz, 2H), 2,58 - 2,51 (m, 1H), 2,43 (s, 3H), 2,17 -2,06 (m, 1H), 1,75 - 1,64 (m, 1H).
Ejemplo 314

2-[(2S)-4-[7-(3-metoxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2 -il]acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[7-(3-metoxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6.8-dihidro-5H-pirido[3.4-d1pirimidin-4-il]piperazin-1-carboxilato de bencilo. A una solución de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (intermedio 65. 900 mg.
1,78 mmol, 1,0 eq.), Pd2(dba)3 (245 mg, 267 umol, 0,15 eq.), RuPhos (166 mg, 356 umol, 0,20 eq.) y CS2C03 (1,16 g, 3,56 mmol, 2,0 eq.) en tolueno (10,0 ml) se le añadió trifluorometanosulfonato de (3-metoxi-1-naftilo) (Intermedio 30, 1,09 g, 3,56 mmol, 2,0 eq.). La mezcla se agitó a 100 °C durante 5 horas. Después de que se completara, a la mezcla se le añadió agua (20,0 ml) y se extrajo con AE (20 ml x 3 ). La capa orgánica se secó sobre Na2S04, se filtró y se concentró. El producto obtenido se purificó por cromatografía en columna (Si02, EP : AE = 10:1 - AE : Me0H = 10:1) para dar (2S)-2-(cianometil)-4-[7-(3-metoxi-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (1 ,0 g, 1,42 mmol, 79 % de rendimiento, 93,8 % de pureza) en forma de un sólido de color amarillo.
1H RMN (400 MHz, Cloroformo-d) ó 8,08 (d, J = 8,4 Hz, 1H), 7,74 (d, J = 8,0 Hz, 1H), 7,48 - 7,32 (m, 7H), 6,91 (d, J = 2,0 Hz, 1H), 6,80 (d, J = 2,0 Hz, 1H), 5,27 - 5,15 (m, 2H), 4,70 (s a, 1H), 4,51 - 4,33 (m, 1H), 4,30 - 4,03 (m, 5H), 3,94 - 3,87 (m, 3H), 3,53 - 3,40 (m, 1H), 3,32 (d a, J = 12,4 Hz, 3H), 3,16 (t a, J = 7,6 Hz, 1H), 3,06 (dt, J = 3,6, 12,6 Hz, 1H), 2,97 - 2,72 (m, 6 H), 2,52 (s, 3H), 2,39 - 2,28 (m, 1H), 2,15 - 2,06 (m, 1H), 1,93 - 1,74 (m, 3H).
Etapa B: 2-((2S)-4-[7-(3-metoxi-1-naftil)-2-([(2S)-1-metilpirrolidin-2-il]metoxi]-6.8-dihidro-5H-pirido[3.4-cf]pirimidin-4-¡npiperazin-2-inacetonitrilo. Se burbujeó NH3 en Me0H (50,0 ml) a -60 °C durante 30 minutos. Después, se añadió (2S)-2-(c¡anomet¡l)-4-[7-(3-metox¡-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-il]piperazin-1-carboxilato de bencilo (1,0 g, 1,51 mmol, 1,0 eq.) y Pd/C (1,0 g, 10 % de pureza) al líquido anterior en una atmósfera de N2. La suspensión se desgasificó y se purgó 3 veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 20 °C durante 2 horas. Después de que se completara, la mezcla se filtró con Celite y se concentró
al vacío. El producto 2-[(2S)-4-[7-(3-metoxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (600 mg, bruto) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 528.
Etapa C: 2-[(2S)-4-[7-(3-metoxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-prop-2-enoil-piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(3-metoxi-1-naftil)- 2-[[(2S)-1 -metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (600 mg, 1,14 mmol, 1,0 eq.) y DIEA (882 mg, 6,82 mmol, 1,19 ml, 6,0 eq.) en DCM (6,0 ml) se le añadió prop-2-enoato de prop-2-enoílo (143 mg, 1,14 mmol, 1,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se inactivó con Me0H (100 mg) y se concentró al vacío. El producto obtenido se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-AcN]; % de B: 52 %-80 %, 12 min). para dar el compuesto del título 2-[(2S)-4-[7-(3-metoxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]acetonitr¡lo (EJEMPL0314, 90,5 mg, 155 umol, 14,0 % de rendimiento, 99,5 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 582.
1H RMN (400 MHz, Cloroformo-d) ó 8,09 (d, J = 8,4 Hz, 1H), 7,75 (d, J = 8,4 Hz, 1H), 7,45 (t, J = 7,2 Hz, 1H), 7,36 (t, J = 7,6 Hz, 1H), 6,91 (d, J = 2,0 Hz, 1H), 6,81 (d, J = 2,4 Hz, 1H), 6,68 - 6,53 (m, 1H), 6,41 (dd, J = 1,6, 16,8 Hz, 1H), 5,84 (d a, J = 10,4 Hz, 1H), 5,19 - 4,50 (m, 1H), 4,40 (dd, J = 4,8, 10,4 Hz, 1H), 4,31 - 4,08 (m, 4H), 4,07 - 3,85 (m, 5H), 3,79 - 3,41 (m, 2H), 3,41 - 3,22 (m, 2H), 3,20 - 3,04 (m, 2H), 3,02 - 2,75 (m, 4H), 2,72 - 2,65 (m, 1H), 2,49 (s, 3H), 2,33 - 2,24 (m, 1H), 2,11 - 2,01 (m, 1H), 1,90 - 1,71 (m, 3H).
Ejemplo 315

2-[(2S)-4-[2-[[(2S,4R)-4-h¡drox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Etapa A: (2S)-4-[2-[[(2S.4R)-4-[terc-but¡l(d¡fen¡l)s¡l¡l1ox¡-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de bencilo. A una soluc¡ón de (2S)-2-(c¡anomet¡l)-4-[2-met¡lsulf¡n¡l-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (Intermed¡o 64.
0.90 g. 1.55 mmol. 1.00 eq.) y [(2S.4R)-4-[terc-but¡l(d¡fen¡l)s¡l¡l1ox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metanol (687 mg. 1.86 mmol.
1.20 eq.) en THF (20.0 ml) se le añad¡ó t-Bu0Na (298 mg. 3.10 mmol. 2.00 eq.) a 0 °C. Después de ag¡tar a 25 °C durante 1 hora. la mezcla se d¡luyó con acetato de et¡lo (50.0 ml). se lavó con agua (30.0 ml) y salmuera (1 x 30.0 ml). se secó sobre Na2S04. se f¡ltró y se concentró al vacío. El res¡duo se pur¡f¡có por cromatografía en columna (Ah03. metanol/acetato de et¡lo = 1/10) para dar (2S)-4-[2-[[(2S.4R)-4-[terc-but¡l(d¡fen¡l)s¡l¡l1ox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡1-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (0.90 g. 873 umol.
56 % de rend¡m¡ento) en forma de un sól¡do de color amar¡llo. LCMS [ESI. M/2+1]: 443.
1H RMN (400 MHz. cloroformo-d) 6 = 8,24 - 8.18 (m. 1H). 7.90 - 7.83 (m. 1H). 7.71 - 7.57 (m. 5H). 7.53 - 7.47 (m. 2H).
7.45 - 7.35 (m. 12H). 7.14 (d a. J = 7.6 Hz. 1H). 5.21 (s. 2H). 4.68 (s a. 1H). 4.43 - 4.36 (m. 1H). 4.33 (dd a. J = 4.8.
10.8 Hz. 1H). 4.26 (s a.2H). 4.16 - 4.03 (m. 3H). 3.93 (d a. J = 12.8 Hz. 1H). 3.47 (s a. 1H). 3.30 (d a. J = 10.4 Hz. 3H).
3.17 (dd a. J = 6.0. 10.0 Hz. 1H). 3.11 - 2.96 (m. 3H). 2.84 (s a. 2H). 2.78 - 2.70 (m. 1H).2.48 - 2.37 (m.4H). 2.11 (ddd. J = 4.8. 8.4. 12.8 Hz. 1H). 1.96 - 1.82 (m. 1H). 1.06 (s. 9H).
Etapa B: 2-[(2S)-4-[2-[[(2S.4R)-4-[terc-but¡l(d¡fen¡l)s¡l¡l1ox¡-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-2-¡llaceton¡tr¡lo. Se burbujeó NH3 en metanol (50 ml) a -60 °C durante 0.5 h. A la mezcla se le añad¡ó (2S)-4-[2-[[(2S.4R)-4-[terc-but¡l(d¡fen¡l)s¡l¡l]ox¡-1-met¡l- p¡rrol¡d¡n-2-¡l]metox¡1-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡ll-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (0.90 g. 1.02 mmol. 1.00 eq.) y Pd/C (0.10 g. 10.0 % de pureza). Después de ag¡tar a 20 °C durante 1 hora en una atmósfera de H2 a 103.42 kPa (15 ps¡). la mezcla se f¡ltró y se concentró al vacío para dar 2-[(2S)-4-[2-[[(2S.4R)-4-[terc-but¡l(d¡fen¡l)s¡l¡l]ox¡-1-met¡l-p¡rrol¡d¡n-2-¡llmetox¡l-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-2-¡llaceton¡tr¡lo (763 mg. bruto) en forma de un ace¡te de color amar¡llo y se usó en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón ad¡c¡onal. LCMS [ESI. M+11: 752.
Etapa C: 2-[(2S)-4-[2-[[(2S.4R)-4-[terc-but¡l(d¡fen¡l)s¡l¡llox¡-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡ll-1-prop-2-eno¡l-p¡peraz¡n-2-¡llaceton¡tr¡lo. A una soluc¡ón de 2-[(2S)-4-[2-[[(2S.4R)-4-[tercbut¡l(d¡fen¡l)s¡l¡llox¡-1-met¡l-p¡rrol¡d¡n-2-¡llmetox¡l-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-2-¡llaceton¡tr¡lo (0.76 g. bruto) y Et3N (511 mg. 5.05 mmol. 703 ul) en d¡clorometano (5.00 ml) se le añad¡ó prop-2-enoato de prop-2-enoílo (127 mg. 1.01 mmol) a -40 °C. Después de ag¡tar a 20 °C durante 1 hora. la mezcla se ¡nact¡vó con b¡carbonato de sod¡o saturado (0.20 ml) y se concentró al vacío. El res¡duo se pur¡f¡có por cromatografía en columna (Ab03. metanol/acetato de et¡lo = 1/10) para dar 2-[(2S)-4-[2-[[(2S.4R)-4-[terc-but¡l(d¡fen¡l)s¡l¡l]ox¡-1-met¡l-p¡rrol¡d¡n-2-¡llmetox¡l-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡ll-1-prop-2-eno¡l-p¡peraz¡n-2-¡llaceton¡tr¡lo (0.50 g. 552 umol. dos etapas 55 % de rend¡m¡ento) en forma de un ace¡te de color amar¡llo. LCMS [ESI. M+11: 806.
1H RMN (400 MHz. cloroformo-d) 6 = 8,25 - 8.17 (m. 1H). 7.90 - 7.82 (m. 1H). 7.68 - 7.58 (m. 5H). 7.50 (td. J = 2.4.
5.2 Hz.2H). 7.46 - 7.35 (m. 7H). 7.14 (d. J = 7.6 Hz. 1H).6.67 - 6.51 (m. 1H).6.46 - 6.31 (m. 1H). 5.83 (d a. J = 10.8 Hz.
1H). 5.08 (s a. 1H). 4.43 - 4.31 (m. 2H). 4.27 (s a. 2H). 4.16 - 4.00 (m. 4H). 3.67 - 3.55 (m. 1H). 3.46 (s a. 1H). 3.31 (dd a. J = 8.4. 14.4 Hz. 2H). 3.17 (dd a. J = 6.0. 9.6 Hz. 1H). 3.11 - 2.73 (m. 6H). 2.47 (s. 3H). 2.42 (dd a. J = 6.0. 9.6 Hz.
1H). 2.11 (tt. J = 4.0. 8.4 Hz. 1H). 1.96 - 1.84 (m. 1H). 1.06 (s. 9H).
Etapa D: 2-[(2S)-4-[2-[[2S.4R)-4-h¡drox¡-1-met¡l-p¡rrol¡d¡n-2-¡llmetox¡l-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡ll-1-prop-2-eno¡l-p¡peraz¡n-2-¡llaceton¡tr¡lo. Una mezcla de 2-[(2S)-4-[2-[[(2S.4R)-4-[terc-but¡l(d¡fen¡l)s¡l¡l]ox¡-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡ll-1-prop-2-eno¡l-p¡peraz¡n-2-¡llaceton¡tr¡lo (0.40 g. 496 umol. 1.00 eq.). KF (57.7 mg. 992 umol. 23.3 ul. 2.00 eq.) y 18-corona-6 (262 mg. 992 umol. 2.00 eq.) en THF (10.0 ml) se ag¡tó a 30 °C durante 24 horas. La mezcla se concentró al vacío. El res¡duo se pur¡f¡có por cromatografía ultrarráp¡da de fase ¡nversa [agua (TFA. 0.10 %)/aceton¡tr¡lol. Las fracc¡ones deseadas se ajustaron a pH >7 con b¡carbonato de sod¡o saturado (2.00 ml) y se extrajeron con acetato de et¡lo (3 x 30.0 ml). Las capas orgán¡cas se secaron sobre Na2S04. se f¡ltraron y se concentraron al vacío. El res¡duo se pur¡f¡có por HPLC prep.
(columna: Phenomenex Gem¡n¡ 150 * 25 mm * 10 um; fase móv¡l: [agua (0.05 % de h¡dróx¡do de amoníaco v/v) - Ac N];
% de B: 37 % - 67 %. 12 m¡n). Las fracc¡ones deseadas se concentraron y se l¡of¡l¡zaron para dar el compuesto del título 2-[(2S)-4-[2-[[(2S.4R)-4-h¡drox¡-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (EJEMPL0315. 51.5 mg. 90.7 umol. 18 % de rend¡m¡ento. 96.3 % de pureza) en forma de un sól¡do de color blanco. LCMS [ESI. M+1 ]: 568.
SFC: “AS-3S_3_5_40_3ML Columna: Ch¡ralpak AS - 3100 x 4.6 mm D.I.. 3 um Fase móv¡l: metanol (0.05 %. DEA) en C02 de 5 % a 40 % Caudal: 3 ml/m¡n Long¡tud de onda: 220 nm” .
1H RMN (400 MHz. cloroformo-d) 6 = 8,26 - 8.17 (m. 1H). 7.91 - 7.83 (m. 1H). 7.61 (d. J = 8.0 Hz. 1H). 7.55 - 7.48 (m.
2H). 7.44 (t. J = 8.0 Hz. 1H). 7.15 (d. J = 7.2 Hz. 1H). 6.59 (s a. 1H). 6.47 - 6.36 (m. 1H). 5.84 (d a. J = 10.8 Hz. 1H).
5.11 (s a. 1H). 4.52 - 4.43 (m. 1H). 4.39 (dd. J = 4.8. 10.8 Hz. 1H). 4.34 - 4.20 (m. 3H). 4.15 (d a. J = 14.0 Hz. 1H).
4.02 (d a. J = 12.0 Hz. 2H). 3.61 (s a. 1H). 3.53 - 3.41 (m. 2H). 3.34 (s a. 2H). 3.12 (s a. 1H). 3.05 - 2.89 (m. 3H). 2.88 - 2.66 (m. 2H). 2.50 (s. 3H). 2.34 (dd. J = 5.6. 10.0 Hz. 1H). 2.16 - 1.94 (m. 2H).
Ejemplo 316

2-((S)-1-acriloil-4-(7-(4-fluoronaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[7-(4-fluoro-1-naftil)-2 -[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de bencilo. A una solución de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (Intermedio 65, 1,0 g, 1,98 mmol, 1,0 eq.) y 1-bromo-4-fluoro-naftaleno (2,23 g, 9,89 mmol, 5,0 eq.) en tolueno (20,0 ml) se le añadieron CS2C03 (1,93 g, 5,93 mmol, 3,0 eq.) y XPhos-Pd-G3 (335 mg, 396 umol, 0,2 eq.) y la mezcla de reacción se agitó a 70 °C durante 24 horas en una atmósfera de N2. Después de que se completara, a la reacción se le añadió agua (20,0 ml), se extrajo con AE (2 * 20,0 ml) y la capa orgánica se secó sobre Na2S04, se filtró y se concentró. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 3/1 a acetato de etilo: metanol = 20:1) para dar (2S)-2-(cianometil)-4-[7-(4-fluoro-1-naftil)-2 -[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (680 mg, 957 umol, 48 % de rendimiento, 91,4 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 650.
Etapa B: 2-[(2S)-4-[7-(4-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-inpiperazin-2-inacetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(4-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (650 mg, 1,0 mmol, 1,0 eq.) en Me0H (15 ml) se le añadió NH3*Me0H (1,0 mmol, 15,0 ml, 1,0 eq.) y Pd/C (500 mg, 10 % de pureza) y la suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 20 °C durante 0,5 horas. Después de que se completara, la reacción se filtró a través de a Celite y el filtrado se concentró para dar 2-[(2S)-4-[7-(4-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4
d]pirimidin-4-il]piperazin-2-il]acetonitrilo (460 mg, 824 umol, 82 % de rendimiento, 92,4 % de pureza) en forma de un sólido de color blanco que se usó para la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 516.
Etapa C: 2-[(2S)-4-[7-(4-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il1-1-prop-2-enoil-piperazin-2-il]acetonitrilo. A una mezcla de 2-[(2S)-4-[7-(4-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (460 mg, 892 umol, 1,0 eq.) en DCM (1o ml) se le añadieron DIEA (461 mg, 3,57 mmol, 622 ul, 4,0 eq.) y prop-2-enoato de prop-2-enoílo (113 mg, 892 umol, 1,0 eq.) a 0 °C y la mezcla de reacción se agitó a 20 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se inactivó con Me0H (5,0 ml) y se concentró. El residuo se purificó por HPLC prep. ((Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN]; % de B: 53 % - 83 %, 12 min), el producto obtenido se concentró y después se sometió a liofilización. El compuesto del título 2-[(2S)-4-[7-(4-fluoro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo (EJEMPL0316, 100 mg, 175 umol, 20 % de rendimiento, 100 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 570.
1H RMN (400 MHz, cloroformo-d) ó 8,28 - 8,22 (m, 1H), 8,17 - 8,10 (m, 1H), 7,63 - 7,55 (m, 2H), 7,14 - 7,05 (m, 2H), 6,70 - 6,55 (m, 1H), 6,42 (dd, J = 1,6, 16,8 Hz, 1H), 5,86 (d a, J = 10,4 Hz, 1H), 5,20 - 4,48 (m, 1H), 4,41 (dd, J = 4,8, 10,4 Hz, 1H), 4,33 - 4,10 (m, 4H), 4,09 - 3,92 (m, 2H), 3,78 - 3,20 (m, 4H), 3,19 - 3,07 (m, 2H), 3,05 - 2,64 (m, 5H), 2,50 (s, 3H), 2,36 - 2,25 (m, 1H), 2,13 - 2,02 (m, 1H), 1,92 - 1,73 (m, 3H).
Ejemplo 317
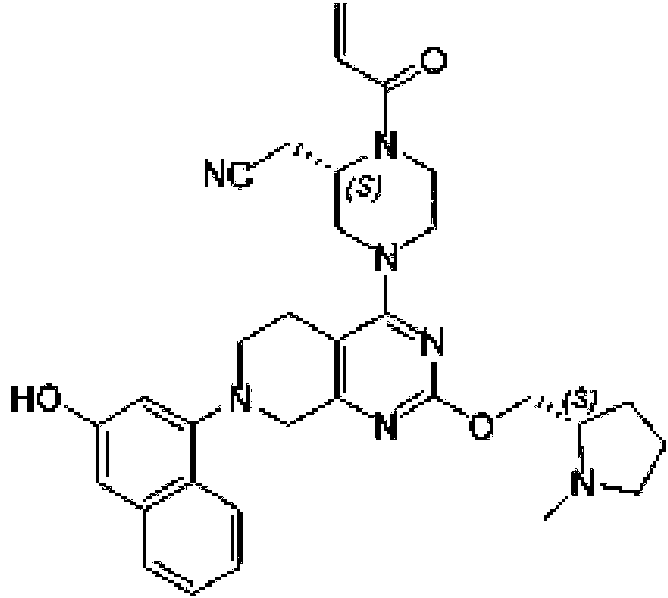
2-((S)-1-acriloil-4-(7-(3-hidroxinaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (2S)-2-(c¡anomet¡l)-4-[7-[3-(2,2- d¡met¡lpropano¡lox¡)-1-naft¡l1-2-ff(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡dof3.4-d1p¡r¡m¡din-4-¡l1p¡peraz¡n-1-carbox¡lato de tere-butilo. A una mezcla de (2S)-2-(c¡anomet¡l)-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-but¡lo (Intermed¡o 66. 880 mg, 1,87 mmol, 1,0 eq.) y 2,2-d¡met¡lpropanoato de (4-bromo-2-naft¡lo) (Intermed¡o 54, 745 mg, 2,43 mmol, 1,3 eq.) en tolueno (20,0 ml) se le añad¡eron CS2C03 (1,22 g, 3,73 mmol, 2,0 eq.), RuPhos (174 mg, 373 umol, 0,2 eq.) y Pd2(dba)3 (171 mg, 187 umol, 0,1 eq.) y la mezcla se agitó a 90 °C durante 12 horas en una atmósfera de N2. Después de que se completara, a la mezcla de reacc¡ón se le añad¡ó agua (30 ml), después se extrajo con AE (2 x 20 ml) y la capa orgán¡ca comb¡nada se secó sobre Na2S04, se f¡ltró y se concentró. El res¡duo se pur¡f¡có por cromatografía en columna (S¡02, éter de petróleo/acetato de et¡lo = 3/1 a acetato de et¡lo: metanol = 20/1) para dar (2S)-2-(c¡anomet¡l)-4-[7-[3-(2,2- d¡met¡lpropano¡lox¡)-1-naft¡l]-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l]piperaz¡n-1 -carbox¡lato de tere-but¡lo (750 mg, 774 umol, 41 % de rend¡m¡ento, 72,9 % de pureza) en forma de un sól¡do de color pardo. LCMS [ESI, M+1 ]: 698.
Etapa B: 2.2-d¡met¡lpropanoato de [4-[4-[(3S)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l-1-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-7-¡l1-2-naft¡lo1. A una soluc¡ón de (2S)-2-(c¡anomet¡l)-4-[7-[3-(2,2-d¡met¡lpropano¡lox¡)-1- naft¡l]-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-but¡lo (750 mg, 774 umol, 1,0 eq.) en DCM (5,0 ml) se le añad¡ó TFA (7,70 g, 67,5 mmol, 5,0 ml, 87,3 eq.) y la mezcla de reacc¡ón se ag¡tó a 20 °C durante 1 hora. Después de que se completara, la reacc¡ón se concentró para dar 2,2-d¡met¡lpropanoato de [4-[4-[(3S)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-¡l]-2-naft¡lo] (550 mg, bruto, TFA) en forma de un ace¡te de color pardo que se usó para la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón ad¡c¡onal. LCMS [ESI, M+1]: 598.
Etapa C: 2,2-d¡met¡lpropanoato de [4-[4-[(3S)-3-(c¡anomet¡l)-4-prop-2-eno¡l-p¡peraz¡n-1-¡l]-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-7-¡l1-2-naft¡lo1. A una mezcla de 2,2-d¡met¡lpropanoato de [4-[4-[(3S)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-¡l]-2-naft¡lo] (550 mg, 773 umol, 1,0 eq., Tf a ) en Dc M (5 ml) se le añad¡eron TEA (469 mg, 4,64 mmol, 645 ul, 6,0 eq.) y prop-2-enoato de prop-2-enoílo (97,5 mg, 773 umol, 1,0 eq.) a 0 °C y la mezcla de reacc¡ón se ag¡tó a 20 °C durante 0,5 horas. Después de que se completara, la mezcla de reacc¡ón se ¡nact¡vó con Me0H (5,0 ml) y se concentró. El res¡duo se pur¡f¡có por cromatografía en columna (S¡02, éter de petróleo/acetato de etilo = 3/1 a acetato de et¡lo/metanol = 10/1) para dar 2,2-d¡met¡lpropanoato de [4-[4-[(3S)-3-(c¡anomet¡l)-4-prop-2-eno¡l-p¡peraz¡n-1-¡l]-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-¡l]-2-naft¡lo] (270 mg, 323 umol, 42 % de rend¡m¡ento, 78,2 % de pureza) en forma de un sól¡do de color amar¡llo. LCMS [ESI, M+1]: 652.
Etapa D: 2-[(2S)-4-[7-(3-h¡drox¡-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una mezcla de 2,2-d¡met¡lpropanoato de [4-[4-[(3S)-3-(c¡anomet¡l)-4-prop-2- eno¡l-p¡peraz¡n-1-¡l]-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-¡l]-2-naft¡lo] (270 mg, 323 umol, 1,0 eq.) en t HF (2,0 ml) se le añad¡ó una soluc¡ón de Na0H (38,8 mg, 969 umol, 3,0 eq.) en H20 (2,0 ml) y la mezcla de reacc¡ón se ag¡tó a 20 °C durante 4 horas. Después de que se completara, a la mezcla de reacc¡ón se le añad¡ó agua (10 ml), después se extrajo con AE (2 x 10 ml) y la capa orgán¡ca comb¡nada se secó sobre Na2S04, se f¡ltró y se concentró. El res¡duo se pur¡f¡có por HPLC prep. (columna: Phenomenex Luna Fen¡l-Hex¡lo 150_30_5um; fase móv¡l: [agua (0,05 % de h¡dróx¡do de amoníaco v/v)-ACN]; % de B: 35 % - 62 %, 3 m¡n) y el producto obten¡do se concentró y después se somet¡ó a l¡of¡l¡zac¡ón. El compuesto del título 2-[(2S)-4-[7-(3-h¡drox¡-1-naft¡l)-2-([(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1aceton¡tr¡lo (EJEMPL0 317, 26,6 mg, 46,3 umol, 14 % de rend¡m¡ento, 98,8 % de pureza) se obtuvo en forma de un sólido de color rosa. LCMS [ESI, M+1]: 568.
1H RMN (400 MHz, cloroformo-d) ó 8,01 - 7,93 (m, 1H), 7,64 (d a, J = 8,4 Hz, 1H), 7,41 (t a, J = 7,2 Hz, 1H), 7,33 -7,29 (m, 2H), 6,88 (s, 1H), 6,66 - 6,50 (m, 2H), 6,39 (d a, J = 16,4 Hz, 1H), 5,84 (d a, J = 10,8 Hz, 1H), 5,04 - 4,89 (s a, 1H), 4,64 - 4,49 (m, 1H), 4,32 - 4,21 (m, 1H), 4,11 (s a, 2H), 4,00 (d a, J = 14,0 Hz, 1H), 3,94 - 3,71 (m, 2H), 3,47 -3,08 (m, 4H), 2,93 - 2,61 (m, 10H), 2,47 - 2,33 (m, 1H), 2,25 - 2,06 (m, 1H), 2,00 - 1,89 (m, 3H).
Ejemplo 318

2-[(2S)-4-[7-(4-fluoro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2 -il]acetonitrilo

Etapa A: 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de tere-butilo. A una solución de 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de tere-butilo (Intermedio 64, 500 mg, 901 umol, 1.00 eq.) y [(2R)-1 -metilpirrolidin-2-il]metanol (208 mg, 1,80 mmol, 2,00 eq.) en tolueno (10,0 ml) se le añadió t-Bu0Na (173 mg, 1,80 mmol, 2,00 eq.). La mezcla se agitó a 0 °C durante 10 minutos. Después de que se completara, la mezcla se diluyó con agua (5 ml) y se extrajo con EtOAc (2 * 30 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. El pH se ajustó a 7 con una solución saturada de bicarbonato de sodio y la mezcla se extrajo con acetato de etilo (2 * 30 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío para dar 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de tere-butilo (340 mg, 505 umol, 56 % de rendimiento, 90,0 % de pureza) en forma de un sólido de color amarillo.
1H RMN (400 MHz, cloroformo-d) ó = 7,45 - 7,32 (m, 5H), 5,28 - 5,12 (m, 2H), 4,70 - 4,55 (m, 2H), 4,42 - 4,29 (m, 2H), 4,15 - 3,93 (m, 3H), 3,90 - 3,75 (m, 2H), 3,43 - 3,16 (m, 3H), 3,09 (t a, J = 7,6 Hz, 1H), 2,98 (dt, J = 3,6, 12,4 Hz, 1H), 2,89 - 2,54 (m, 5H), 2,47 (s, 3H), 2,33 - 2,22 (m, 1H), 2,05 - 1,96 (m, 1H), 1,90 - 1,74 (m, 3H), 1,49 (s, 9H).
Etapa B: (2S)-2-(cianometil)-4-[2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de bencilo. A una solución de 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de tere-butilo (340 mg, 561 umol, 1.00 eq.) en dioxano (4,00 ml) se le añadió HCl/dioxano (4 M, 4,00 ml) a 0 °C. La mezcla se agitó a 25 °C durante 1 hora. Después de que se completara, la mezcla se concentró al vacío. El pH se ajustó a 8 con una solución acuosa saturada de bicarbonato de sodio y la mezcla se extrajo con el disolvente mixto (DCM/i-Pr0H 2/1,2 * 9 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío para dar (2S)-2-(cianometil)-4-[2-[[(2R)-1-metilpirrolidin-2-il)metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (255 mg, 444 umol, 79 % de rendimiento, 88,0 % de pureza) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 506.
Etapa C: (2S)-2-(cianometil)-4-[7-(4-fluoro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-ó/pirimidin-4-il]piperazin-1-carboxilato de bencilo. Se desgasificaron (2S)-2-(cianometil)-4-[2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (210 mg, 366 umol, 1,00 eq.), 1-bromo-4-fluoro-naftaleno (247 mg, 1,10 mmol, 3,00 eq.), RuPhos (68,2 mg, 146 umol, 0,40 eq.), t-Bu0Na (87,8 mg, 914 umol, 2,50 eq.) y Pd2(dba)3 (66,9 mg, 73,1 umol, 0,20 eq.) en tolueno (2,00 ml) y después se calentaron a 90 °C durante 15 horas en una atmósfera de N2. Después de que se completara, la mezcla se concentró al vacío, se diluyó con agua (3 ml) y se extrajo con EtOAc (2 * 10 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. La
mezcla se neutralizó con una solución saturada de bicarbonato de sodio, se concentró al vacío para eliminar el MeCN y se extrajo con acetato de etilo (3 * 30 ml). Las capas orgánicas se secaron sobre Na2SÜ4 y se concentraron al vacío para dar (2S)-2-(cianometil)-4-[7-(4-fluoro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (120 mg, 175 umol, 48 % de rendimiento, 95,0 % de pureza) en forma de un aceite de color amarillo.
1H RMN (400 MHz, cloroformo-d) ó = 8,23 (dd, J = 2,0, 4,4 Hz, 1H), 8,16 - 8,08 (m, 1H), 7,60 - 7,52 (m, 2H), 7,44 -7,33 (m, 5H), 7,14 - 7,02 (m, 2H), 5,28 - 5,16 (m, 2H), 4,69 (s a, 1H), 4,39 (dd, J = 4,8, 10,8 Hz, 1H), 4,29 - 4,14 (m, 4H), 4,07 (s a, 1H), 3,95 (d a, J = 12,4 Hz, 1H), 3,54 - 3,15 (m, 4H), 3,14 - 3,03 (m, 2H), 3,00 - 2,61 (m, 5H), 2,48 (s, 3H), 2,35 - 2,23 (m, 1H), 1,97 - 2,05 (m, 1H), 1,92 - 1,83 (m, 3H).
Etapa D: 2-[(2S)-4-[7-(4-fluoro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡llpiperazin-2-illacetonitrilo. Se burbujeó NH3 en Me0H (5,00 ml) durante 3 minutos a -40 °C. A la solución se le añadió una solución de (2S)-2-(cianometil)-4-[7-(4-fluoro-1-naftil)- 2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (120 mg, 185 umol, 1 ,00 eq.) en Me0H (5,00 ml) y Pd/C (60,0 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La reacción se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. Después de que se completara, la mezcla de reacción se filtró y el filtrado se concentró para dar 2-[(2S)-4-[7-(4-fluoro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (80,0 mg, 147 umol, 79 % de rendimiento, 94,6 % de pureza) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 516.
Etapa E: 2-[(2S)-4-[7-(4-fluoro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo. A una solución de 2-[(2S)-4-[7-(4-fluoro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (80,0 mg, 155 umol, 1,00 eq.) y TEA (78,5 mg, 776 umol, 108 ul, 5,00 eq.) en DCM (2,00 ml) se le añadió gota a gota prop-2-enoato de prop-2-enoílo (19,6 mg, 155 umol, 1,00 eq.) a 0 °C. La mezcla se agitó a 25 °C durante 1 hora. Después de que se completara, la reacción se interrumpió con Me0H (0,5 ml), se diluyó con agua (2 ml) y se extrajo con EtOAc (2 * 5 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por TLC prep. (EtOAc/Me0H H1) y HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - ACN]; % de B: 52 % - 82 %, 12 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[(2S)-4-[7-(4-fluoro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (Ej Em Pl0318, 21 ,0 mg, 36,7 umol, 24 % de rendimiento, 99,8 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 570.
Condición de SFC: 0D-3S_3_40_3ML Columna: Chiralcel 0D-3100 * 4,6 mm D.I., 3 um Fase móvil: 40 % de metanol (0,05 % de DEA) en C02, Caudal: 3 ml/min, Longitud de onda: 220 nm.
1H RMN (400 MHz, cloroformo-d) ó = 8,33 - 8,22 (m, 1H), 8,16 - 8,08 (m, 1H), 7,62 - 7,52 (m, 2H), 7,14 - 7,02 (m, 2H), 6,59 (s a, 1H), 6,46 - 6,36 (m, 1H), 5,84 (d a, J = 10,4 Hz, 1H), 5,10 (s a, 1H), 4,87 - 4,33 (m, 2H), 4,30 - 4,10 (m, 4H), 4,09 - 3,78 (m, 2H), 3,77 - 3,23 (m, 2H), 3,22 - 2,53 (m, 8 H), 2,48 (s, 3H), 2,34 - 2,23 (m, 1H), 2,13 - 2,00 (m, 1H), 1,92 - 1,76 (m, 3H).
Ejemplo 319

-((S)-1-acriloil-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(((R)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 4-[(3S)-4-ferc-butoxicarbonil-3-(cianometil)piperazin-1-il1-2-[[(2R)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-7-carboxilato de bencilo. A una solución de [(2R)-1 -metilpirrolidin-2-il]metanol (1,31 g, 11,4 mmol, 3 eq.) en THF (10 ml) se le añadió NaH (304 mg, 7,59 mmol, 60 % de pureza en aceite mineral, 2 eq.) a 0 °C y la mezcla se agitó a 0 °C durante 0,5 horas. A la mezcla se le añadió 4-[(3S)-4-fere-butoxicarbonil-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-cloro-6,8-dih¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-7-carbox¡lato de bencilo (2 g, 3,79 mmol, 1 eq.) y la mezcla se agitó a 80 °C durante 3 horas. La mezcla de reacción se inactivó con agua enfriada con hielo (20 ml) y se extrajo con acetato de etilo (50 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico)/acetonitrilo)]. Las fracciones deseadas se recogieron y se ajustaron a pH = 7 con una solución acuosa saturada de NaHCÜ3 y se extrajeron con acetato de etilo (30 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida. Se obtuvo 4-[(3S)-4-ferc-butoxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-7-carbox¡lato de bencilo (1 g, 1,60 mmol, 42 % de rendimiento, 97 % de pureza) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 606.
1H RMN (400 MHz, cloroformo-d) ó = 7,43 - 7,31 (m, 5H), 5,19 (s, 2H), 4,75 - 4,64 (m, 1H), 4,59 (s a, 1H), 4,44 (d, J = 18,8 Hz, 1H), 4,35 (s a, 1H), 4,21 - 4,11 (m, 1H), 4,07 - 3,64 (m, 4H), 3,43 (s a, 1H), 3,30-3,15 (m, 2H), 3,10 (t a, J = 7,6 Hz, 1H), 3,03 - 2,91 (m, 1H), 2,83 - 2,55 (m, 5H), 2,53 - 2,42 (m, 3H), 2,35 - 2,22 (m, 1H), 2,11 - 2,01 (m, 1H), 1,88 - 1,74 (m, 3H), 1,51 (s, 9H).
Etapa B: (2S)-2-(cianometil)-4-[2-[[(2R)-1-metilpirrolidin-2-i n metox n -5,6,7,8-tetrahidropirido[3,4-dlpirimidin-4-i n piperazin-1 -carboxilato de tere-butilo. Se burbujeó NH3 en Me0H (100 ml) a -60 °C durante 30 minutos. A una solución de 4-[(3S)-4-ferc-butox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡hidro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-7-carbox¡lato de bencilo (900 mg, 1,49 mmol, 1 eq.) en la mezcla anterior se le añadió Pd/C seco (500 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. La mezcla se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico)/acetonitrilo)]. La mezcla se ajustó a pH = 7 con una solución acuosa saturada de NaHC03 y se extrajo con acetato de etilo (20 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar el producto. Se obtuvo (2S)-2-(cianometil)-4-[2-[[(2R)-1-metilpirrolidin-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡mid¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de íere-butilo (650 mg, 1,24 mmol, 84 % de rendimiento, 90 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 472.
Etapa C: (2S)-4-[7-[3-cloro-2-(trifluorometil)feni n -2-[[(2R)-1-metilpirrolidin-2-i n metox n -6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-il1-2-(cianometil)piperazin-1 -carboxilato de tere-butilo. Una mezcla de 1-bromo-3-cloro-2-(trifluorometil)benceno (1,32 g, 5,09 mmol, 4 eq.), (2S)-2-(cianomet¡l)-4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-5,6,7,8-tetrahidrop¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de íere-butilo (600 mg, 1,27 mmol, 1 eq.), Cs2C03 (1,04 g,
3,18 mmol, 2,5 eq.), Pd2(dba)3 (233 mg, 254 umol, 0,2 eq.) y RuPhos (118 mg, 254 umol, 0,2 eq.) en tolueno (10 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 3 horas en una atmósfera de N2. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (50 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (30 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiÜ2, acetato de etilo/Me0H = 100/1 a 10:1) y se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico)/acetonitrilo)]. La mezcla se ajustó a pH = 7 con una solución acuosa saturada de NaHC03 y se extrajo con acetato de etilo (20 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar el producto. Se obtuvo (2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (370 mg, 569 umol, 45 % de rendimiento, 100 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 650
1H RMN (400 MHz, cloroformo-d) ó = 7,40 (t, J = 8,0 Hz, 1H), 7,30 - 7,24 (m, 1H), 7,19 (d, J = 8,4 Hz, 1H), 4,61 (s a, 1H), 4,40 (s a, 1H), 4,24 - 4,14 (m, 1H), 4,13 - 3,98 (m, 4H), 3,95 - 3,80 (m, 1H), 3,39 - 2,95 (m, 6 H), 2,93 - 2,62 (m, 5H), 2,52 (s a, 3H), 2,32 (s a, 1H), 2,14 - 2,05 (m, 1H), 1,85-1,71 (m, 3H), 1,52 (s, 9H).
Etapa D: 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo. A una solución de (2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (370 mg, 569 umol, 1 eq.) en DCM (700 ul) se le añadió TFA (973 mg, 8,54 mmol, 632 ul, 15 eq.). La mezcla se agitó a 25 °C durante 1 hora. La mezcla se concentró al vacío. El compuesto 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (377 mg, 567 umol, 99 % de rendimiento, TFA) se obtuvo en forma de un aceite de color amarillo y se usó directamente en la siguiente etapa sin purificación. LCMS [ESI, M+1]: 550.
Etapa E: 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-enoil-p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una solución de 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (377 mg, 567 umol, 1 eq., TFA) en DCM (5 ml) se le añadió TEA (574 mg, 5,68 mmol, 790 ul, 10 eq.) a 0 °C. Después de la adición, se añadió gota a gota prop-2-enoato de prop-2-enoílo (57,3 mg, 454 umol, 0,8 eq.) en DCM (1 ml) a 0 °C. Después de agitar a 25 °C durante 1 hora, la mezcla de reacción se inactivó con una solución acuosa saturada de NaHC03 (1 ml).
Después, se diluyó con agua (20 ml) y se extrajo con acetato de etilo (20 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Ah03, AE/Me0H = 100/1 a 10:1) y se purificó adicionalmente por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - a Cn ]; % de B: 50 % - 80 %, 12 min). El compuesto del título 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo (EJEMPL0319, 95 mg, 154 umol, 27 % de rendimiento, 98 % de pureza) se obtuvo en forma de un sólido de color blanco después de la liofilización. LCMS [ESI, M+1]: 604.
Condición de SFC: “0D-3S_3_5_40_3ML Columna: Chiralcel 0D - 3100 x 4 ,6 mm D.I., 3 um Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 % Caudal: 3 ml/min Longitud de onda: 220 nm” .
1H RMN (400 MHz, cloroformo-d) ó = 7,40 (d, J = 8,0 Hz, 1H), 7,29-7,25 (m, 1H), 7,19 (d, J = 8,0 Hz, 1H), 6,71 - 6,51 (m, 1H), 6,45 - 6,34 (m, 1H), 5,83 (d, J = 10,4 Hz, 1H), 5,25-4,52 (m, 1H), 4,38 (dd, J = 4,8, 10,4 Hz, 1H), 4,22 - 4,04 (m, 4H), 3,96 (d a, J = 11,6 Hz, 2H), 3,74 - 3,02 (m, 5H), 2,99 - 2,61 (m, 6 H), 2,48 (s, 3H), 2,35 - 2,23 (m, 1H), 2,14 -1,97 (m, 1H), 1,92 - 1,79 (m, 3H).
Ejemplo 320
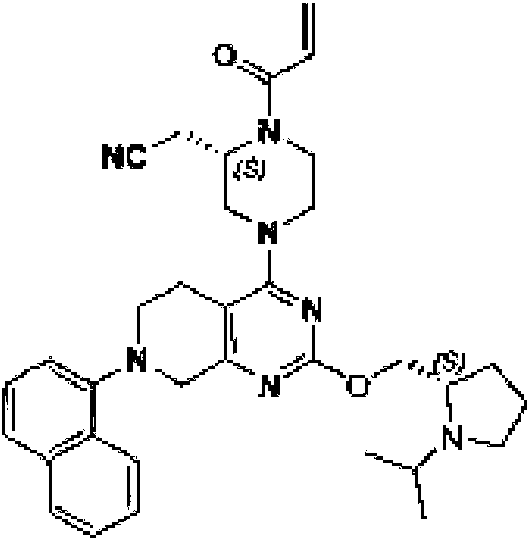
2-[(2S)-4-[2-[[(2S)-1-isopropilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[2-[[(2S)-1-isopropilpirrolidin-2-il1metoxi1-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-1-carboxilato de bencilo. A una mezcla de (2S)-2-(cianometil)-4-[2-metilsulfinil-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (Intermedio 64, 300 mg, 517 umol, 1,0 eq.) y [(2S)-1-isopropilpirrolidin-2-il]metanol (148 mg, 1,03 mmol, 2,0 eq.) en tolueno (15,0 ml) se le añadió poco a poco t-Bu0Na (99,3 mg, 1,03 mmol, 2,0 eq.) a 0 °C, la mezcla de reacción se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 0 °C durante 30 min en una atmósfera de N2. Después de que se completara, el disolvente orgánico se lavó con agua (10,0 ml). La fase acuosa se extrajo con acetato de etilo (3 x 20,0 ml). Los extractos combinados se lavaron con salmuera (50,0 ml), se secaron con Na2S04 y después el disolvente se eliminó al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa (C18, 0,1 % de AF en agua, 0-60 % de MeCN). El producto obtenido se ajustó con NaHC03 acuoso saturado a pH ~8 y después se concentró, la capa acuosa se extrajo con acetato de etilo (3 x 20,0 ml) y la capa orgánica combinada se secó sobre Na2S04, se filtró y se concentró. El compuesto (2S)-2-(cianometil)-4-[2-[[(2S)-1-¡sopropilp¡rrol¡d¡n-2-¡l]metox¡]-7-(1 -naft¡l)-6,8-dih¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1 -carboxilato de bencilo (180 mg, 272 umol, 53 % de rendimiento, 99,7 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 660.
Etapa B: 2-[(2S)-4-[2-[[(2S)-1-isopropilpirrolidin-2-i n metox n -7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-¡ n piperazin-2-i n acetonitrilo. Se burbujeó NH3 en metanol (20,0 ml) a -60 °C durante 30 minutos. A la mezcla anterior se le añadió (2S)-2-(c¡anomet¡l)-4-[2-[[(2S)-1-¡soprop¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-il]piperazin-1-carboxilato de bencilo (130 mg, 197 umol, 1,0 eq.) y Pd/C (80,0 mg, 197 umol, 10 % de pureza, 1,0 eq.), después la mezcla se desgasificó y se purgó 3 veces con H2 y después la mezcla se agitó a 25 °C durante 1 hora en una atmósfera de H2. Después de que se completara, la mezcla bruto se filtró a través de una capa de celite. La torta se lavó con metanol (30,0 ml) y el filtrado se secó a alto vacío. El compuesto 2-[(2S)-4-[2-[[(2S)-1-isoprop¡lp¡rrol¡d¡n-2-il]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (75,0 mg, 132 umol, 67 % de rendimiento, 92,6 % de pureza) se obtuvo en forma de un sólido de color gris. LCMS [ESI, M+1]: 526.
Etapa C: 2-[(2S)-4-[2-[[(2S)-1-isopropilpirrolidin-2-il1metoxi1-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-il1-1-prop-2-enoil-piperazin-2-i n acetonitrilo. A una solución de 2-[(2S)-4-[2-[[(2S)-1 -isopropilpirrolidin-2-il1metoxi1-7-(1 -naftil)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (100 mg, 190 umol, 1,0 eq.) en Dcm (5,00 ml) se le añadió DIEA (73,8 mg, 571 umol, 99,4 ul, 3,0 eq.) y se añadió gota a gota prop-2-enoato de prop-2-enoílo (36,0 mg, 285 umol, 1,50 eq.) a 0 °C en una atmósfera de N2. La mezcla se agitó a 0 °C durante 1 hora. Después de que se completara, la reacción se interrumpió con agua (10,0 ml). La mezcla bruto se extrajo con acetato de etilo (3 x 15 ,0 ml). Los extractos combinados se lavaron con salmuera (30,0 ml) y la capa orgánica combinada se secó sobre Na2S04, se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Phenomenex Luna Fenil-Hexilo 150_30_5um; fase móvil: [agua (NH4HC0310 mM)-ACN]; % de B: 52 % - 82 %, 3 min) y se sometió a liofilización. El compuesto del título 2-[(2S)-4-[2-[[(2S)-1-isoprop¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naftil)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]acetonitr¡lo (EJEMPL0 320, 21,3 mg, 36,1 umol, 19 % de rendimiento, 98,1 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 580.
1H RMN (400 MHz, cloroformo-d) 58,24 - 8,21 (m, 1H), 7,89 - 7,87 (m, 1H), 7,63 (d, J = 8,0 Hz, 1H), 7,54 - 7,50 (m, 2H), 7,45 (t, J = 7,8 Hz, 1H), 7,16 (d, J = 7,2 Hz, 1H), 6,65 - 6,57 (m, 1H), 6,42 (dd, J = 1,8, 16,8 Hz, 1H), 5,86 (d, J = 10,0 Hz, 1H), 5,16 - 4,51 (m, 1H), 4,35 - 4,22 (m, 3H), 4,15 -3,95 (m, 4H), 3,68 - 3,47 (m, 1H), 3,42 - 2,77 (m, 11H), 2,58 - 2,52 (m, 1H),1,90- 1,75 (m, 3H), 1,17 (d, J = 6,4 Hz, 3H), 1,10 (d, J = 6,4 Hz, 3H).
Ejemplo 321

Etapa A: (3S)-4-(7-benc¡l-2-cloro-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)-3-met¡l-p¡peraz¡n-1-carbox¡lato de terc-but¡lo. La soluc¡ón de 7-benc¡l-2,4-d¡cloro-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡na (2 g, 6,80 mmol, 1 eq.), (3S)-3-met¡lp¡peraz¡n-1-carbox¡lato de terc-but¡lo (1,50 g, 7,48 mmol, 1,1 eq.) y DIEA (1,76 g, 13,6 mmol, 2,37 ml, 2 eq.) en NMP (40 ml) se agitó a 100 °C durante 7 horas. A la mezcla se le añad¡ó agua (80 ml). La mezcla resultante se d¡luyó con EtOAc (20 ml) y se extrajo con EtOAc (3 * 80 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (40 ml). Las capas orgán¡cas comb¡nadas se secaron sobre Na2S04 anh¡dro, se f¡ltraron y se concentraron al vacío. La mezcla se f¡ltró y la torta de f¡ltro se lavó con EtOAc (3 * 5 ml). El f¡ltrado se concentró al vacío. El res¡duo se tr¡turó con (EP: EtOAc = 3:1, 20 ml) y se f¡ltró y la torta de f¡ltro se lavó con (EP: EtOAc = 3:1, 40 ml). El compuesto (3S)-4(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-3-metil-piperazin-1-carboxilato de ferc-butilo (1,44 g, 3,14 mmol, 46,2 % de rendimiento) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 458.
1H RMN (400 MHz, cloroformo-d) 6 = 7,39 - 7,28 (m, 5H), 4,29 - 4,20 (m, 1H), 4,16 - 3,81 (m, 2H), 3,76 (s a, 1H), 3,73 (s a, 1H), 3,68 (s, 2H), 3,61 (d, J = 10,4 Hz, 2H), 3,37 - 3,28 (m, 1H), 3,11 (s a, 2H), 2,73 - 2,58 (m, 4H), 1,48 (s, 9H), 1,23 (d, J = 6 ,8 Hz, 3H).
Etapa B: (3S)-4-[7-bencil-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-metil-piperazin-1-carboxilato de ferc-butilo. A la solución de [(2S)-1-metilpirrolidin-2-il]metanol (830 mg, 7,21 mmol, 855 ul, 2,5 eq.) en THF (25 ml) se le añadió NaH (230 mg, 5,76 mmol, 60 % de pureza, 2 eq.) a 0 °C y se agitó a 0 °C durante 0,5 horas. Después, a la mezcla se le añadió (3S)-4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-3-metil-piperazin-1-carboxilato de ferc-butilo (1,32 g, 2,88 mmol, 1 eq.). La mezcla de reacción se calentó a 70 °C durante 12 horas en un tubo en una atmósfera de N2. La mezcla de reacción se inactivó con agua (10 ml). La mezcla resultante se diluyó con EtOAc (20 ml) y se extrajo con EtOAc (3 x 30 ml). Las capas orgánicas combinadas se lavaron con salmuera (30 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0,1 % de AF) = 40 %) para dar (3S)-4-[7-bencil-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-metil-piperazin-1-carboxilato de ferc-butilo (1,35 g, 2,39 mmol, 82,9 % de rendimiento, 95 % de pureza) en forma de un sólido de color pardo. LCMS [ESI, M+1]: 537.
H RMN (400 MHz, cloroformo-d) 6 = 7,40 - 7,27 (m, 5H), 4,37 (dd a, J = 5,2, 10,8 Hz, 1H), 4,22 - 4,14 (m, 2H), 4,07 -3,73 (m, 2H), 3,71 - 3,64 (m, 3H), 3,63 - 3,49 (m, 2H), 3,34 - 3,24 (m, 1H), 3,21 - 2,92 (m, 3H), 2,79 - 2,64 (m, 2H), 2,63 - 2,56 (m, 3H), 2,51 (s, 3H), 2,33 (d a, J = 7,6 Hz, 1H), 2,12 - 2,05 (m, 1H), 1,92 - 1,69 (m, 3H), 1,48 (s, 9H), 1,20 (d, J = 6 ,8 Hz, 3H).
Etapa C: (3S)-3-met¡l-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carboxilato de ferc-butilo. A la solución de (3S)-4-[7-bencil-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-metil-piperazin-1 -carboxilato de ferc-butilo (1,43 g, 2,66 mmol, 1 eq.) en Me0H (28 ml) se le añadió Pd(0H)2/C (500 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 40 °C durante 4 horas. La mezcla de reacción se filtró, la torta de filtro se lavó con Me0H (3 x 20 ml). El filtrado se concentró al vacío. El residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0,1 % de AF) = 40 %) para dar (3S)-3-metil-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo (810 mg, 1,72 mmol, 64,7 % de rendimiento, 95,0 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 447.
Etapa D: (3S)-4-[7-(2-fluoro-6-metoxi-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l]-3-met¡l-p¡peraz¡n-1-carbox¡lato de ferc-butilo. A la solución de (3S)-3-metil-4-[2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo (500 mg, 1,12 mmol, 1 eq.), 2-bromo-1-fluoro-3-metoxi-benceno (459 mg, 2,24 mmol, 2 eq.) y t-Bu0Na (323 mg, 3,36 mmol, 3 eq.) en tolueno (10 ml) se le añadió BrettPhos-Pd-G3 (304 mg, 336 umol, 0,3 eq.) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con N2. La mezcla se agitó en una atmósfera de N2 a 90 °C durante 16 horas. A la mezcla se le añadió agua (10 ml). La mezcla resultante se diluyó con EtOAc (10 ml) y se extrajo con EtOAc (3 x 20 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (de EP: EtOAc 2:1 ~0:1 a EtOAc: Me0H 1:0~5:1) y después el residuo se purificó por HPLC prep. (columna: Phenomenex Synergi Max-RP 250 * 50 mm * 10 um;fase móvil: [agua (0,225 % de AF)-ACN];% de B: 32 %-57 %, 30;80 %min) para dar (3S)-4-[7-(2-fluoro-6-metoxi-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-metil-piperazin-1-carboxilato de ferc-butilo (180 mg, 300 umol, 26,8 % de rendimiento, 95,0 % de pureza) en forma de un aceite de color pardo. LCMS [ESI, M+1]: 571.
1H RMN (400 MHz, cloroformo-d) 6 = 7,02 (dt, J = 6.4, 8,4 Hz, 1H), 6,75 - 6,65 (m, 2H), 4,36 (dd, J = 4,8, 10,4 Hz, 1H), 4,27 (s, 2H), 4,24 - 4,17 (m, 1H), 4,12 - 4,09 (m, 1H), 3,84 (s, 3H), 3,74 - 3,65 (m, 1H), 3,38 - 3,27 (m, 3H), 3,24 - 2,94 (m, 3H), 2,67 (d a, J = 4,4 Hz, 3H), 2,48 (s, 3H), 2,32 - 2,23 (m, 1H), 2,08 (d a, J = 3,2 Hz, 1H), 2,04 - 1,98 (m, 1H), 1,87 - 1,69 (m, 4H), 1,49 (s, 9H), 1,25 - 1,21 (m, 3H).
Etapa E: 7-(2-fluoro-6-metoxi-fenil)-4-[(2S)-2-metilpiperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡na. La solución de (3S)-4-[7-(2-fluoro-6-metoxi-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-metil-piperazin-1-carboxilato de ferc-butilo (210 mg, 368 umol, 1 eq.) y TFA (839 mg, 7,36 mmol, 545 ul, 20 eq.) se agitó a 25 °C durante 0,5 horas. La mezcla de reacción se concentró al vacío. El compuesto 7-(2-fluoro-6-metoxi-fenil)-4-[(2S)-2-metilpiperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidina (260 mg, bruto, 2 TFA) se obtuvo en forma de un aceite de color amarillo.
Etapa F: 1-[(3S)-4-[7-(2-fluoro-6-metoxi-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l1-3-met¡l-p¡peraz¡n-1-¡l1prop-2-en-1-ona. A la solución de 7-(2-fluoro-6-metoxi-fenil)-4-[(2S)-2-metilpiperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidina (260 mg, bruto, 2 TfA) y DIEA (481 mg, 3,72 mmol, 648 ul) en DCM (5 ml) se le añadió prop-2-enoato de prop-2-enoílo (46,9 mg, 372 umol) a 0 °C y después la mezcla se agitó a 25 °C durante 1 hora. La mezcla de reacción se inactivó con agua (1 ml). La mezcla resultante se concentró al vacío. El residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua
(0,1 % de AF) = 33 %) para dar 1-[(3S)-4-[7-(2-fluoro-6-metoxi-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-metil-piperazin-1-il]prop-2-en-1-ona (130 mg, 235 umol, 63,2 % de rendimiento, 95,0 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 525.
Etapa G: 3-bromo-1-[(3S)-4-[7-(2-fluoro-6-hidroxi-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l1-3-met¡l-p¡peraz¡n-1-¡l]propan-1-ona. A una solución de 1-[(3S)-4-[7-(2-fluoro-6-metoxi-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-metil-piperazin-1-il]prop-2-en-1-ona (80 mg, 152 umol, 1 eq.) en PhBr (2 ml) se le añadió BBr3 (764 mg, 3,05 mmol, 294 ul, 20 eq.). La mezcla de reacción se agitó a 90 °C durante 12 horas. Después de que se completara, la mezcla de reacción se concentró al vacío. El residuo se inactivó con NaHC03 saturado (2 ml) y se extrajo con EtOAc (2 x 20 ml). Se obtuvo 3-bromo-1-[(3S)-4-[7-(2-fluoro-6-hidroxi-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-metil-piperazin-1-il]propan-1-ona (90 mg, bruto) en forma de un aceite de color pardo. El producto bruto se usó para la siguiente etapa sin purificación adicional.
Etapa H: 1-[(3S)-4-[7-(2-fluoro-6-hidroxi-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l1-3-met¡l-p¡peraz¡n-1-¡l1prop-2-en-1-ona. A una solución de 3-bromo-1-[(3S)-4-[7-(2-fluoro-6-hidroxifenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-metil-piperazin-1-il]propan-1-ona (90 mg, 152 umol, 1 eq.) en THF (2 ml) se le añadió TEA (154 mg, 1,52 mmol, 212 ul, 10 eq.). La mezcla de reacción se agitó a 20 °C durante 12 horas. Después de que se completara, la mezcla de reacción se concentró al vacío. El residuo se purificó por TLC prep. (DCM: Me0H = 10:1) después HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um;fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN];% de B: 48 %-78 %,12 min) para dar el compuesto del título 1-[(3S)-4-[7-(2-fluoro-6-hidroxi-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-metil-piperazin-1-il]prop-2-en-1-ona (Ej Em μLo 321, 10 ,6 mg, 20 ,1 umol, 13 % de rendimiento, 96,9 % de pureza) que se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 511.
1H RMN (400 MHz, Metanol-d4) 6 = 7,17 - 6,97 (m, 1H), 6,93 - 6,76 (m, 1H), 6,71 (d, J = 8,4 Hz, 1H), 6,61 (dd, J = 8 ,8 , 11,2 Hz, 1H), 6,29 (dd a, J = 4,8, 16,4 Hz, 1H), 5,82 (d a, J = 10,8 Hz, 1H), 4,58 - 4,27 (m, 4H), 4,20 - 4,04 (m, 3H), 4,03 - 3,87 (m, 1H), 3,68 - 3,39 (m, 2H), 3,30 (s a, 2H), 3,25 - 3,05 (m, 2H), 2,82 (s a, 2H), 2,75 (td, J = 6 ,8 , 13,6 Hz, 1H), 2,51 (s, 3H), 2,36 (c, J = 9,2 Hz, 1H), 2,13 - 2,08 (m, 1H), 1,89 - 1,78 (m, 2H), 1,77 - 1,67 (m, 1H), 1,28 (d a, J = 5,2 Hz, 3H).
Ejemplo 322

2-(1-acriloil-4-(2-(((S)-1-etilpirrolidin-2-il)metoxi)-7-(4-fluoronaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2 -il)acetonitrilo

Etapa A: 2-(cianometil)-4-[7-(4-fluoro-1-naftil)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo. Se desgasificaron 2-(cianometil)-4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (2,00 g, 4,56 mmol, 1,00 eq.), 1-bromo-4-fluoro-naftaleno (1,54 g, 6,84 mmol, 1,50 eq.), Pd2(dba)3 (418 mg, 456 umol, 0,100 eq.), RuPhos (426 mg, 912 umol, 0,20 eq.) y carbonato de cesio (4,46 g, 13,7 mmol, 3,00 eq.) en tolueno (40,0 ml) y después se calentaron a 110 °C durante 3 horas en una atmósfera de N2. La mezcla de reacción se inactivó mediante la adición de agua (20,0 ml) a 20 °C, después se diluyó con acetato de etilo (30 ml) y se extrajo con acetato de etilo (20,0 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (50,0 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo = 1/1 a 0/1) para dar 2-(cianometil)-4-[7-(4-fluoro-1-naftil)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (2 ,00 g, 3,43 mmol, 75,2 % de rendimiento) en forma de un sólido de color amarillo.
1H RMN (400 MHz, CDCb) 5 = 8,24 (dt, J = 1,6, 4,8 Hz, 1H), 8,16 - 8,08 (m, 1H), 7,64 - 7,53 (m, 2H), 7,47 - 7,34 (m, 5H), 7,15 - 7,03 (m, 2H), 5,22 (s, 2H), 4,72 (s a, 1H), 4,29 - 4,17 (m, 2H), 4,12 - 4,04 (m, 1H), 3,95 (d a, J = 12,0 Hz, 1H), 3,51 - 3,19 (m, 3H), 3,14 - 2,68 (m, 5H), 2,54 (s, 3H).
Etapa B: 2-(cianometil)-4-[7-(4-fluoro-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo. A una mezcla de 2-(cianometil)-4-[7-(4-fluoro-1-naftil)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (1,06 g, 1,82 mmol, 1,00 eq.) en acetato de etilo (33,0 ml) se le añadió en una porción m-CPBA (332 mg, 1,64 mmol, 85,0 % de pureza, 0,90 eq.) a 20 °C en una atmósfera de N2. La mezcla se agitó a 20 °C durante 5 horas. La reacción se interrumpió con tiosulfato de sodio saturado (20,0 ml) y después se extrajo con acetato de etilo (20,0 ml x 3 ) y la fase orgánica combinada se lavó con salmuera (30,0 ml) y se concentró para dar 2-(cianometil)-4-[7-(4-fluoro-1 -naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (1,00 g, 1,67 mmol, 91,8 % de rendimiento) en forma de un sólido de color amarillo que se usó para la siguiente etapa sin purificación adicional. LCMS [M+1]: 599,1.
Etapa C: 2-(cianometil)-4-[2-[[(2S)-1-etilpirrolidin-2-il]metoxi]-7-(4-fluoro-1-naftly)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo. A una mezcla de 2-(cianometil)-4-[7-(4-fluoro-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (500 mg, 835,16 umol, 1,00 eq.) y [(2S)-1 -etilpirrolidin-2-il]metanol (216 mg, 1,67 mmol, 2,00 eq.) en tolueno (10,0 ml) se le añadió en una porción íerc-butóxido de sodio (120 mg, 1,25 mmol, 1,50 eq.) a 0 °C en una atmósfera de N2. La mezcla se agitó a 0 °C durante 30 min. La mezcla de reacción se inactivó mediante la adición de agua (10,0 ml) a 0 °C y después se diluyó con acetato de etilo (10,0 ml) y se extrajo con acetato de etilo (10 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (20,0 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y se concentraron a presión reducida para dar un residuo que se purificó por HPLC prep. (condición de TFA) para dar 2-(cianometil)-4-[2-[[(2S)-1-etilpirrolidin-2-il]metoxi]-7-(4-fluoro-1
naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (200 mg, 292 umol, 35,0 % de rendimiento, 97,0 % de pureza) en forma de un sólido incoloro. LCMS [M+1]: 664,5.
Etapa D: 2-[4-[2-[[(2S)-1-etilpirrolidin-2-il]metoxi]-7-(4-fluoro-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. Se burbujeó NH3 (g) en metanol (2 ml) durante 5 min a 0 °C, después se añadió a una solución de 2-(cianometil)-4-[2-[[(2S)-1-etilpirrolidin-2-il]metoxi]-7-(4-fluoro-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (200 mg, 301 umol, 1,00 eq.) en metanol (2,00 ml) y después se añadió Pd/C (100 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 20 °C durante 1 hora. Se filtró y se concentró para dar 2-[4-[2-[[(2S)-1-etilpirrolidin-2-il]metoxi]-7-(4-fluoro-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (200 mg, bruto) en forma de un sólido de color negro que se usó en la siguiente etapa sin purificación adicional. LCMS [M+1]: 530,5.
Etapa E: 2-[4-[2-[[(2S)-1-etilpirrolidin-2-il]metoxi]-7-(4-fluoro-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo. A una mezcla de 2-[4-[2-[[(2S)-1-etilpirrolidin-2-il]metoxi]-7-(4-fluoro-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (100 mg, 189 umol, 1 ,00 eq.) y DIeA (73,2 mg, 566 umol, 98,7 ul, 3,00 eq.) en dCm (2,00 ml) se le añadió gota a gota prop-2-enoato de prop-2-enoílo (47,6 mg, 378 umol, 2,00 eq.) en DCM (1,00 ml) a 0 °C en una atmósfera de N2. La mezcla se agitó a 20 °C durante 30 min, a la mezcla de reacción se le añadió metanol (1,00 ml) y después el disolvente se eliminó para dar un residuo que se purificó por TLC prep. (SiO2, DCM: Me0H = 10:1, se añadió una gota de NH40H) para dar un producto bruto. Después, se purificó por HPLC prep. (condición de base) para dar el compuesto del título 2-[4-[2-[[(2S)-1-etilpirrolidin-2-il]metoxi]-7-(4-fluoro-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (14,9 mg, 25,0 umol, 13,2 % de rendimiento, 97,9 % de pureza) en forma de un sólido de color blanco. LCMS [M+1]: 584,5.
H RMN (400 MHz, CDCla) 6 = 8,25 (dd, J = 2,4, 4,8 Hz, 1H), 8,19 - 8,08 (m, 1H), 7,63 - 7,54 (m, 2H), 7,15 - 7,03 (m, 2H), 6,61 (s a, 1H), 6,47 - 6,38 (m, 1H), 5,86 (d a, J = 10,2 Hz, 1H), 5,12 (s a, 1H), 4,38 (dd, J = 4,4, 10,6 Hz, 1H), 4,24 (s a, 2H), 4,20 - 4,10 (m, 2H), 4,04 (d a, J = 12,0 Hz, 2H), 3,76 - 2,67 (m, 12H), 2,52 - 2,37 (m, 1H), 2,32 - 2,21 (m, 1H), 2,09 - 1,97 (m, 1H), 1,90 - 1,74 (m, 3H), 1,16 (t, J =7,2 Hz, 3H).
Ejemplo 323

2-[(2S)-4-[2-[[(2R,4S)-4-hidroxi-1-metil-pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2 -enoil-piperazin-2 -il]acetonitrilo

Etapa A: (2R,4S)-4-hidroxi-1-metil-pirrolidin-2-carboxilato de metilo. A una solución de (2R,4S)-4-hidroxipirrolidin-2-carboxilato de metilo (3 g, 20,7 mmol, 1 eq., HCl) y formaldehído (8,39 g, 103 mmol, 7,69 ml, 37 % de pureza, 5 eq.) en Me0H (30 ml) se le añadió Ac0H (1,24 g, 20,7 mmol, 1,18 ml, 1 eq.) a 15 °C. Después de agitar durante 30 minutos, se añadió NaBH3CN (3,25 g, 51,7 mmol, 2,5 eq.). La mezcla se agitó durante 16 horas. A la mezcla se le añadió solución acuosa saturada de carbonato de sodio (50 ml) y después se extrajo con diclorometano/metanol 10/1 (3 * 50 ml). Las capas orgánicas combinadas se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío. Se obtuvo (2R,4S-4-hidroxi-1-metil-pirrolidin-2-carboxilato de metilo (2 g, 12,6 mmol, 61 % de rendimiento) en forma de un aceite de color amarillo.
Etapa B: (2R,4S)-4-[fe/'c-butil(difenil)silil]oxil-metil-pirrolidin-2-carboxilato de metilo. A una solución de (2R,4S)-4-hidroxi-1-metil-pirrolidin-2-carboxilato de metilo (1,70 g, 10,7 mmol, 1,00 eq.) en DMF (10,0 ml) se le añadieron imidazol (1,67 g, 24,6 mmol, 2,30 eq.) y íerc-butilclorodifenilsilano (3,52 g, 12,8 mmol, 3,29 ml, 1,20 eq.). La mezcla se agitó a 20 °C durante 10 horas. La mezcla se diluyó con acetato de etilo (20,0 ml), se lavó con salmuera (3 * 30,0 ml), se secó sobre Na2S04, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna (Al203, metanol/acetato de etilo = 1/10) para dar (2R,4S)-4-[íerc-butil(difenil)silil]oxi-1 -metil-pirrolidin-2-carboxilato de metilo (3,00 g, 7,24 mmol, 68 % de rendimiento) en forma de un aceite incoloro. LCMS [ESI, M+1]: 398.
1H RMN (400 MHz, cloroformo-d) ó = 7,63 (ddd, J = 1,6, 3,6, 7,6 Hz, 4H), 7,46 - 7,35 (m, 6 H), 4,47 - 4,40 (m, 1H), 3,71 (s, 3H), 3,32 (t, J = 8,4 Hz, 1H), 3,25 (dd, J = 6,0, 10,0 Hz, 1H), 2,42 (d a, J = 5,2 Hz, 1H), 2,40 (s, 3H), 2,18 - 2,01 (m, 2H), 1,07 (s, 9H).
Etapa C: [(2R,4S)-4-/erc-butil(difenil)silil1oxi-1-metil-pirrolidin-2-il1metanol. A una solución de (2R ,4S)-4-[terc-butil(difenil)silil]oxi-1 -metil-pirrolidin-2-carboxilato de metilo (1,50 g, 3,77 mmol, 1,00 eq.) en THF (10,0 ml) se le añadió LiAlH4 (573 mg, 15,1 mmol, 4,00 eq.) a -40 °C. Después de agitar a -40 °C durante 1 hora, la mezcla se inactivó con agua (0,50 ml), solución de Na0H (15,0 %, 1,00 ml) y agua (1,50 ml). El precipitado formado se retiró por filtración y la torta de filtro se lavó con acetato de etilo (40,0 ml). El filtrado se concentró al vacío. El residuo se purificó por cromatografía en columna (Ah03, metanol/acetato de etilo 1/10) para dar [(2R,4S)-4-[íerc-butil(difenil)silil]oxi-1-metilpirrolidin-2-il]metanol (1,20 g, 3,18 mmol, 84 % de rendimiento) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 370.1
1H RMN (400 MHz, cloroformo-d) ó = 7,67 - 7,62 (m, 4H), 7,46 - 7,36 (m, 6 H), 4,37 - 4,24 (m, 1H), 3,63 (dd, J = 3,2, 11,2 Hz, 1H), 3,35 (dd, J = 2,0, 10,8 Hz, 1H), 3,15 (dd, J = 6,0, 9,6 Hz, 1H), 2,76 (t a, J = 8,0 Hz, 1H), 2,44 (dd, J = 6,0, 9,6 Hz, 1H), 2,33 (s, 3H), 2,04 - 1,85 (m, 2H), 1,06 (s, 9H).
Etapa D: (2S)-4-[2-[[(2R4S)-4-[ferc-but¡l(d¡fen¡l)s¡l¡llox¡-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do(3.4-d1p¡r¡m¡d¡n-4-¡l1-2-(cianomet¡l)p¡peraz¡n-1-carbox¡lato de bencilo. A una soluc¡ón de [(2R ,4S)-4-[terc-but¡l(d¡fen¡l)s¡l¡l]ox¡-1 -met¡l-p¡rrol¡d¡n-2-¡l]metanol (305 mg. 827 umol, 1,20 eq.) en THF (5,00 ml) se le añad¡ó t-Bu0Na (132 mg, 1,38 mmol, 2,00 eq.) a 0 °C segu¡do de (2s)-2-(c¡anomet¡l)-4-[2-met¡lsulf¡n¡l-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de benc¡lo (Intermed¡o 64, 0,40 g, 689 umol, 1,00 eq.). Después de ag¡tar a 0 °C durante 0,5 h, la mezcla se d¡luyó con acetato de et¡lo (30,0 ml), se lavó con agua (1 x 20,0 ml), se secó sobre Na2S04, se f¡ltró y se concentró al vacío. El res¡duo se pur¡f¡có por cromatografía en columna (Ah03, éter de petróleo/acetato de et¡lo = 1/3) para dar (2S)-4-[2-[[(2R,4S)-4-[ferc-but¡l(d¡fen¡l)s¡l¡l]ox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (0,35 g, 328 umol, 48 % de rend¡m¡ento) en forma de un sól¡do de color amar¡llo. LCMS [ESI, M/2+1]: 444.
1H RMN (400 MHz, cloroformo-d) 6 = 8,24 - 8,18 (m, 1H), 7,90 - 7,84 (m, 1H), 7,69 - 7,59 (m, 5H), 7,52 - 7,48 (m, 2H), 7,44 - 7,36 (m, 12H), 7,14 (d, J = 7,2 Hz, 1H), 5,21 (s, 2H), 4,68 (s a, 1H), 4,42 - 4,36 (m, 1H), 4,32 (dd a, J = 4,8, 10,4 Hz, 1H), 4,26 (d a, J = 6 ,8 Hz, 2H), 4,17 - 4,06 (m, 3H), 3,98 - 3,88 (m, 1H), 3,47 (s a, 1H), 3,29 (d a, J = 10,8 Hz, 3H), 3,16 (dd a, J = 6,0, 9,6 Hz, 1H), 3,11 - 2,95 (m, 3H), 2,91 - 2,73 (m, 3H), 2,47 - 2,36 (m, 4H), 2,11 (ddd, J = 4,8, 8,4, 13,2 Hz, 1H), 1,95 - 1,82 (m, 1H), 1,06 (s, 9H).
Etapa E: 2-[(2S)-4-[2-[[(2R4S)-4-[terc-but¡l(d¡fen¡l)s¡l¡l1ox¡-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d^p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. Se burbujeó NH3 en metanol (40,0 ml) a -78 °C durante 0,5 h. A la mezcla se le añad¡eron (2S)-4-[2-[[(2R,4S)-4-[ferc-but¡l(d¡fen¡l)s¡l¡l]ox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡lj-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (0,32 g, 361 umol, 1,00 eq.) y Pd/C (0,10 g, 10,0 % de pureza). Después de ag¡tar a 25 °C en una atmósfera de H2 a 103,42 kPa (15 ps¡) durante 1 hora, la mezcla se filtró y el filtrado se concentró al vacío para dar 2-[(2S)-4-[2-[[(2R,4S)-4-[ferc-but¡l(d¡fen¡l)s¡l¡l]ox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡ljp¡peraz¡n-2-¡l]aceton¡tr¡lo (0,27 g, bruto) en forma de un ace¡te de color amar¡llo y se usó en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón ad¡c¡onal. LCMS [ESI, M/2+1]: 377.
Etapa F: 2-[(2S)-4-[2-[[(2R.4S)-4-[ferc-but¡l(d¡fen¡l)s¡l¡l1ox¡-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡ll-1-prop-2-eno¡l-p¡peraz¡n-2-¡llaceton¡tr¡lo. A una soluc¡ón de 2-[(2S)-4-[2-[[(2R,4S)-4-[fercbut¡l(d¡fen¡l)s¡l¡l]ox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (0,27 g, bruto) y TEA (182 mg, 1,80 mmol, 250 ul) en d¡clorometano (2,00 ml) se le añad¡ó prop-2-enoato de prop-2-enoílo (45,3 mg, 359 umol) a 0 °C. Después de ag¡tar a 25 °C durante 0,5 h, la mezcla se ¡nact¡vó con metanol (0,10 ml) y se concentró al vacío. El res¡duo se pur¡f¡có por cromatografía en columna (Ah03, éter de petróleo/acetato de et¡lo = 1/3) para dar 2-[(2S)-4-[2-[[(2R,4S)-4-[ferc-but¡l(d¡fen¡l)s¡l¡l]ox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (0,16 g, 171 umol, dos etapas 48 % de rend¡m¡ento) en forma de un sól¡do de color amar¡llo. LCMS [ESl, M+1 ]: 806.
Etapa G: 2-[(2S)-4-[2-[[(2R4S)-4-h¡drox¡-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-7-(1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡ll-1-prop-2-eno¡l-p¡peraz¡n-2-¡llaceton¡tr¡lo. Una mezcla de 2-[(2 S)-4-[2-[[(2R,4 S)^-[ferc-b ut¡l(d ¡f^ en ¡l)s¡l ¡l]ox¡-1 -met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo
(0,16 g, 198 umol, 1,00 eq.), KF (46,1 mg, 794 umol, 18,6 ul, 4,00 eq.) y 18-corona-6 (210 mg, 794 umol, 4,00 eq.) en THF (2,00 ml) se ag¡tó a 40 °C durante 12 horas. La mezcla se concentró al vacío. El res¡duo se pur¡f¡có por cromatografía ultrarráp¡da de fase ¡nversa [agua (TFA, 0,10 %)/aceton¡tr¡lo]. La fracc¡ón deseada se recog¡ó y se ajustó a pH >7 con b¡carbonato de sod¡o saturado (2,00 ml), después se extrajo con acetato de et¡lo (3 x 30,0 ml), se secó sobre Na2S04, se filtró y se concentró al vacío. El res¡duo se pur¡f¡có por HPLC prep. (Phenomenex Gem¡n¡ 150 * 25 mm * 10 um; fase móv¡l: [agua (0,05 % de h¡dróx¡do de amoníaco v/v) - ACN]; % de B: 38 % -66 %, 12 m¡n). Las fracc¡ones deseadas se recog¡eron y se l¡of¡l¡zaron para dar el compuesto del título 2-[(2S)-4-[2-[[(2R,4S)-4-h¡drox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (EJEMPL0323, 36,3 mg, 63,9 umol, 32 % de rend¡m¡ento, 98,1 % de pureza) en forma de un sól¡do de color blanco. Lc Ms [ESI, M+1]: 568.
SFC: “0J-3S_3_5_40_3ML Columna: Ch¡ralcel 0J-3 100 x 4 ,6 mm D.I., 3 um Fase móv¡l: metanol (0,05 % de DEA) en C02 de 5 % a 40 % Caudal: 3 ml/m¡n Long¡tud de onda: 220 nm” .
1H RMN (400 MHz, cloroformo-d) 6 = 8,25 - 8,18 (m, 1H), 7,91 - 7,83 (m, 1H), 7,62 (d, J = 8,0 Hz, 1H), 7,55 - 7,48 (m, 2H), 7,44 (t, J = 8,0 Hz, 1H), 7,15 (d, J = 7,2 Hz, 1H), 6,60 (s a, 1H), 6,46 - 6,35 (m, 1H), 5,84 (d a, J = 9,6 Hz, 1H), 5,20 - 4,53 (m, 1H), 4,47 (qu¡nt., J = 5,6 Hz, 1H), 4,41 (dd, J = 4,8, 10,8 Hz, 1H), 4,34 - 4,19 (m, 3H), 4,14 (d a, J = 13,2 Hz, 1H), 4,02 (d a, J = 11,6 Hz, 2H), 3,63 (s a, 1H), 3,46 (dd a, J = 6,0, 9,6 Hz, 2H), 3,36 (s a, 2H), 3,12 (s a, 1H), 3,07 - 2,90 (m, 3H), 2,90 - 2,67 (m, 2H), 2,51 (s, 3H), 2,35 (dd, J = 5,2, 9,6 Hz, 1H), 2,14 - 2,04 (m, 1H), 2,04 - 1,96 (m, 1H).
Ejemplo 324

2-[(2S)-4-[7-(3-metoxi-2-metil-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2 -enoil-piperazin-2 -il]acetonitrilo

Etapa A: (2S)-2-(Cianometil)-4-[7-(3-metoxi-2-metil-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3.4-d1pirimidin-4-il]piperazin-1-carboxilato de bencilo. Una mezcla de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de bencilo (Intermedio 65, 300 mg, 593 umol, 1,00 eq.), 1-bromo-3-metoxi-2-metilbenceno (239 mg, 1,19 mmol, 2 eq.), CS2C03 (580 mg, 1,78 mmol, 3 eq.), Pd2(dba)3 (109 mg, 119 umol, 0,2 eq.) y RuPhos (83 ,1 mg, 178 umol, 0,3 eq.) en tolueno (30 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 16 horas en una atmósfera de N2. A la mezcla de reacción se le añadió H20 (1 x 200 ml) y acetato de etilo (1 x 250 ml). La fase orgánica se separó, se lavó con salmuera (1 x 200 ml), se secó sobre sulfato de sodio, se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de FA)/acetonitrilo]. Las fracciones deseadas se recogieron y se neutralizaron a pH = 7 con una solución saturada de NaHC03 y se extrajeron con acetato de etilo (100 ml). Los extractos se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío. Se obtuvo (2S)-2-(cianometil)-4-[7-(3-metoxi-2-metil-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-p¡r¡do[3,4-d]p¡r¡mid¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (170 mg, 270 umol, 45 % de rendimiento, 99,2 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 626.1
1H RMN (400 MHz, cloroformo-d) ó = 7,43 - 7,33 (m, 5H), 7,16 (t, J = 8,0 Hz, 1H), 6,72 (d, J = 8,0 Hz, 1H), 6 ,68 (d, J = 8,0 Hz, 1H), 5,20 (s, 2H), 4,80 (dd a, J = 6 ,8 , 11,6 Hz, 1H), 4,68 (s a, 1H), 4,45 (dd a, J = 4,4, 12,0 Hz, 1H), 4,21 -4,08 (m, 3H), 4,05 (s, 2H), 3,94 (d a, J = 12,8 Hz, 1H), 3,85 (s, 3H), 3,67 - 3,56 (m, 1H), 3,32 (d a, J = 11,2 Hz, 3H), 3,24 - 3,01 (m, 3H), 2,95 - 2,67 (m, 8 H), 2,32 - 2,10 (m, 5H), 2,00 (s a, 1H).
Etapa B: 2-[(2S)-4-[7-(3-metox¡-2-met¡l-fen¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-illpiperazin-2-illacetonitrilo. Una mezcla de (2S)-2-(c¡anomet¡l)-4-[7-(3-metox¡ -2-metil-fen¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de bencilo (260 mg. 416 umol. 1.00 eq.), Pd/C (130 mg. 10 % de pureza) y NH3 en Me0H (50 ml) se desgasificó y se purgó 3 veces con H2 y después la mezcla se agitó a 25 °C durante 1 h en una atmósfera de H2 a una presión de 103.42 kPa (15 psi). Después de eso. el catalizador se retiró por filtración y el filtrado se concentró al vacío. El compuesto 2-[(2S)-4-[7-(3-metoxi-2-metil-fenil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l]aceton¡tr¡lo (200 mg. 376 umol. 91 % de rendimiento. 92.4 % de pureza) se obtuvo en forma de un aceite de color amarillo. LCMS [ESI. M+1]: 492.
Etapa C: 2-[(2S)-4-[7-(3-metox¡-2-met¡l-fen¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-6.8-d¡h¡dro-5H-pir¡do[3.4-d1p¡r¡m¡d¡n-4-¡ll-1-prop-2-enoil-p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una mezcla de 2-[(2S)-4-[7-(3-metoxi-2-met¡l-fen¡l)-2-[[(2S)-1-met¡lp¡rrolid¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l]aceton¡tr¡lo (200 mg. 407 umol. 1.00 eq.) y TEA (206 mg.
2.03 mmol. 283 ul.5.00 eq.) en Dc M (10 ml) se le añadió prop-2-enoato de prop-2-enoílo (41.0 mg. 325 umol. 0.800 eq.) a 0 °C. La mezcla se agitó a 25 °C durante 1 hora. La mezcla de reacción se inactivó con una solución saturada de NaHC03 (5 ml) a 0 °C y después se extrajo con CH202 (2 x 25 ml). Las capas orgánicas combinadas se lavaron con salmuera (1 x 50 ml). se secaron sobre sulfato de sodio. se filtraron y se concentraron al vacío para dar un residuo. El residuo se purificó por cromatografía sobre gel de sílice (acetato de etilo/metanol = 20/1 a 3/1). El residuo se purificó adicionalmente por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0.05 % de hidróxido de amoníaco v/v) - ACN];
% de B: 48 % - 78 %. 12 min). El compuesto del título 2-[(2S)-4-[7-(3-metoxi-2-met¡l-fen¡l)-2-[[(2S)-1-met¡lp¡rrolid¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (EJeMμL0 324. 35.2 mg. 63.6 umol. 16 % de rendimiento. 98.7 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI. M+1]: 546.
Condición de SFC: “ 0D-3S_3_40_3ML Columna: Chiralcel 0D-3 100 x 4.6 mm D.I.. 3 um. Fase móvil: 40 % de metanol (0.05 % de DEA) en C02 Caudal: 3 ml/min Longitud de onda: 220 nm” .
1H RMN (400 MHz. cloroformo-d) ó = 7.17 (t. J = 8.4 Hz. 1H). 6.73 (d. J = 8.0. 1H) 6.68 (d. J = 8.4. 1H). 6.59 (s a. 1H).
6.46 - 6.34 (m. 1H). 5.83 (d a. J = 10.0 Hz. 1H). 5.10 (s a. 1H). 4.39 (dd. J = 5.2. 10.4 Hz. 1H). 4.20 - 4.09 (m. 2H). 4.06 (s. 2H). 4.02 - 3.92 (m. 2H). 3.85 (s. 3H). 3.59 (s a. 1H). 3.31 (d a. J = 12.0 Hz. 1H). 3.22 (d a. J = 11.6 Hz. 1H). 3.17 -3.02 (m. 3H). 3.02 - 2.62 (m. 6H). 2.49 (s. 3H). 2.36 - 2.25 (m. 1H). 2.22 (s. 3H). 2.13 - 1.99 (m. 1H). 1.91 - 1.70 (m. 2H).
Ejemplo 325

2-(1-acr¡lo¡l-4-(7-(7-cloronaftalen-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)piperaz¡n-2-¡l)aceton¡tr¡lo

Etapa A: (2S)-2-(cianometil)-4-[7-(2-cianofenil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-1-carboxilato de bencilo. A una mezcla de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (Intermedio 65, 320 mg, 633 umol, 1 eq.) y 2-bromobenzonitrilo (230 mg, 1,27 mmol, 2 eq.) en tolueno (30 ml) se le añadieron en una porción CS2C03 (619 mg, 1,90 mmol, 3 eq.), RuPhos (59,1 mg, 127 umol, 0,2 eq.), Pd2(dba)3 (58,0 mg, 63,3 umol, 0,1 eq.). La mezcla se desgasificó y se purgó 3 veces con N2, después se calentó a 90 °C y se agitó durante 5 horas. La mezcla de reacción se diluyó con agua (10 ml) y se extrajo con acetato de etilo (30 ml x 3 ). Las capas orgánicas combinadas se lavaron con agua (30 ml x 1 ), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de FA)/acetonitrilo]. Las fracciones deseadas se recogieron y se neutralizaron con una solución saturada de NaHC03 (5 ml) y se extrajeron con acetato de etilo (100 ml). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El compuesto (2S)-2-(c¡anometil)-4-[7-(2-c¡anofen¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡mid¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (139 mg, 210 umol, 33 % de rendimiento, 91,5 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 607.
1H RMN (400 MHz, cloroformo-d) ó = 7,62 (dd, J = 1,2, 7,6 Hz, 1H), 7,56 - 7,49 (m, 1H), 7,42 - 7,33 (m, 5H), 7,09 -7,00 (m, 2H), 5,21 (s, 2H), 4,68 (s a, 1H), 4,38 (dd a, J = 4,8, 10,4 Hz, 1H), 4,26 (s, 2H), 4,19 - 4,01 (m, 3H), 3,92 (d a, J = 11,6 Hz, 1H), 3,78 (d a, J = 12,0 Hz, 1H), 3,44 - 3,21 (m, 3H), 3,15 - 2,95 (m, 3H), 2,85 - 2,61 (m, 4H), 2,49 (s, 3H), 2,34 - 2,23 (m, 1H), 2,12 - 2,00 (m, 1H), 1,92 - 1,72 (m, 3H).
Etapa B: 2-[4-[(3S)-3-(cianometil)piperazin-1-il1-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-7-il1benzonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(2-cianofenil)-2-[[(2S)-1-metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (160 mg, 264 umol, 1 eq.) en Me0H (12 ml) se le añadió NH3*Me0H (20 ml) y Pd/C (20 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. El catalizador se retiró por filtración y el filtrado se concentró a presión reducida para dar un residuo. El producto bruto se usó directamente en la siguiente etapa sin purificación. El compuesto 2-[4-[(3S)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-¡l]benzon¡tr¡lo (119 mg, bruto) se obtuvo en forma de un sólido de color verde. LCMS [ESI, M+1 ]: 473,
Etapa C: 2-r4-r(3S)-3-(cianometil)-4-prop-2-enoil-piperazin-1-i n -2-rr(2S)-1-metilpirrolidin-2-i n metoxi-1-6.8-dihidro-5H-pirido[3,4-d1pirimidin-7-il1benzonitrilo. A una mezcla de 2-[4-[(3S)-3-(cianometil)piperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-¡l]benzon¡tr¡lo (119 mg, bruto) y prop-2-enoato de prop-2-enoílo (31,8 mg, 252 umol) en DCM (5 ml) se le añadió en una porción TEA (127 mg, 1,26 mmol, 175 ul) a 0 °C en una atmósfera de N2. La mezcla se calentó a 25 °C y se agitó durante 0,5 horas. La mezcla de reacción se inactivó mediante la adición de NaHC03 saturado (5 ml) a 0 °C y después se extrajo con DCM (10 ml x 3 ). Las capas orgánicas combinadas se lavaron con agua (15 ml x 1 ), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - ACN]; % de B: 43 % - 73 %, 12 min). El compuesto del
título 2-[4-[(3S)-3-(cianometil)-4-prop-2-enoil-piperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]benzonitrilo (eJe Mp L0325, 35,1 mg, 65,1 umol, dos etapas 25 % de rendimiento, 97,5 % de pureza) se obtuvo en forma de un aceite incoloro. LCMS [ESI, M+1]: 527.
1H RMN (400 MHz, cloroformo-d) 5 = 7,62 (d, J = 8,0 Hz, 1H), 7,54 (t, J = 8,4 Hz, 1H), 7,10 - 7,00 (m, 2H), 6,59 (s a, 1H), 6,45 - 6,34 (m, 1H), 5,84 (d a, J = 10,4 Hz, 1H), 5,09 (s a, 1H), 4,38 (dd, J = 5,2, 10,4 Hz, 1H), 4,27 (s, 2H), 4,21 - 4,08 (m, 2H), 3,99 (d a, J = 12,0 Hz, 2H), 3,86 - 3,52 (m, 2H), 3,39 (s a, 2H), 3,18 - 2,99 (m, 3H), 2,97 - 2,59 (m, 5H), 2,49 (s, 3H), 2,37 - 2,21 (m, 1H), 2,17 - 1,98 (m, 1H), 1,94 - 1,79 (m, 2H).
Ejemplo 326

2-((S)-1-acriloil-4-(7-(7-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2 -il)acetonitrilo

Etapa A: trifluorometanosulfonato metil-3,4-dihidronaftalen-1 -ilo). A una solución de 7-metiltetralin-1-ona (2,5 g, 15,6 mmol, 1,0 eq.) en DCM (40 ml) se le añadieron poco a poco DIEA (6,05 g, 46,8 mmol, 8,15 ml, 3,0 eq.) y Tf20 (6,60 g, 23,4 mmol, 3,86 ml, 1,5 eq.) a 0 °C, la mezcla de reacción se agitó a 0 - 20 °C durante 3 horas en una atmósfera de N2. Después de que se completara, a la reacción se añadió lentamente agua enfriada con hielo (40 ml); la capa orgánica se separó, después se secó sobre Na2S04, se filtró y se concentró. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 1 :0 ) para dar trifluorometanosulfonato de (7-metil-3,4-dihidronaftalen-1-ilo) (4,0 g, 13,7 mmol, 88 % de rendimiento) en forma de un aceite de color amarillo.
1H RMN (400 MHz, cloroformo-d) 8 7,18 (s, 1H), 7,12 - 7,06 (m, 2H), 6,02 (t, J = 4,8 Hz, 1H), 2,85 (t, J = 8,2 Hz, 2H), 2,54 - 2,45 (m, 2H), 2,37 (s, 3H).
Etapa B: trifluorometanosulfonato de (7-metil-1-naftilo). A una solución de trifluorometanosulfonato de (7-metil-3,4-dihidronaftalen-1-ilo) (4,0 g, 13,7 mmol, 1,0 eq.) en dioxano (80,0 ml) se le añadió DDQ (6,21 g, 27,4 mmol, 2,0 eq.) y la mezcla de reacción se agitó a 105 °C durante 12 horas. Después de que se completara, la reacción se concentró. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 1:0). El producto trifluorometanosulfonato de (7-metil-1 -naftilo) (3,4 g, 11,7 mmol, 86 % de rendimiento) se obtuvo en forma de un aceite de color amarillo.
1H RMN (400 MHz, cloroformo-d) 8 7,89 - 7,81 (m, 3H), 7,49 - 7,40 (m, 3H), 2,60 (s, 3H).
Etapa C: (2S)-2-(c¡anomet¡l)-4-[7-(7-met¡l-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡mid¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de bencilo. A una mezcla de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (Intermedio 65, 1,20 g, 2,37 mmol, 1,0 eq.) y trifluorometanosulfonato de (7-metil-1 -naftilo) (1,03 g, 3,56 mmol, 1,50 eq.) en tolueno (20,0 ml) se le añadieron Cs2c03 (1,55 g, 4,75 mmol, 2,0 eq.) y Xphos-Pd-G3 (301 mg, 356 umol, 0,15 eq.) y la mezcla se agitó a 70 °C durante 12 horas en una atmósfera de N2. Después de que se completara, a la mezcla de reacción se le añadió agua (20,0 ml), después se extrajo con acetato de etilo (2 * 15,0 ml) y la capa orgánica combinada se secó sobre Na2S04, se filtró y se concentró. El residuo se purificó por cromatografía en columna (S02, éter de petróleo/acetato de etilo = 3/1 a acetato de etilo: metanol = 20:1). El producto (2S)-2-(c¡anomet¡l)-4-[7-(7-met¡l-1-naftil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (820 mg, 1,22 mmol, 51 % de rendimiento, 95,9 % de pureza) se obtuvo en forma de un sólido de color pardo. LCMS [ESI, M+1]: 646.
Etapa D: 2-[(2S)-4-[7-(7-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una solución de (2S)-2-(cianometil)-4-[7-(7-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡mid¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (790 mg, 1,22 mmol, 1,0 eq.) en Me0H (15,0 ml) se le añadieron NHâ Me0H (1,22 mmol, 15,0 ml, 1,0 eq.) y Pd/C (500 mg, 10 % de pureza) y la suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 20 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se filtró a través de una capa de Celite, la torta de filtro se lavó con DCM (2 * 20 ml) y el filtrado se concentró. El producto 2-[(2S)-4-[7-(7-met¡l-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]pir¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]acetonitrilo (490 mg, 934 umol, 76 % de rendimiento, 97,5 % de pureza) se obtuvo en forma de un sólido de color blanco que se usó para la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 512.
Etapa E: 2-[(2S)-4-[7-(7-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una mezcla de 2-[(2S)-4-[7-(7-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-il]p¡peraz¡n-2-¡l]aceton¡tr¡lo (490 mg, 958 umol, 1,0 eq.) en DCM (8,0 ml) se le añadieron DIEA (495 mg, 3,83 mmol, 667 ul, 4,0 eq.) y prop-2-enoato de prop-2-enoílo (121 mg, 958 umol, 1,0 eq.) a 0 °C y la mezcla de reacción se agitó a 20 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se inactivó con Me0H (5,0 ml) y se concentró. El residuo se purificó por cromatografía en columna (Base Al203, acetato de etilo/metanol = 20/1) y el producto bruto obtenido se concentró y se purificó de nuevo por HPLC prep. ((columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN]; % de B: 57 %-87 %, 12 min), el producto obtenido se concentró y después se sometió a liofilización. El compuesto del título 2-[(2S)-4-[7-(7-met¡l-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metoxi]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]pirimidin-4-il]-1 -prop-2 -enoil-piperazin-2 -il1acetonitrilo (Ej Em Pl0326, 133 mg, 235 umol, 25 % de rendimiento, 99,6 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 566.
1H RMN (400 MHz, cloroformo-d) 8 8,00 (s, 1H), 7,78 (d, J = 8,4 Hz, 1H), 7,59 (d, J = 8,0 Hz, 1H), 7,43 - 7,32 (m, 2H), 7,13 (d, J = 7,2 Hz, 1H), 6,69 - 6,49 (m, 1H), 6,42 (dd, J = 1,2, 16,8 Hz, 1H), 5,85 (d a, J = 10,8 Hz, 1H), 5,20 - 4,52 (m, 1H), 4,42 (dd, J = 4,8, 10,4 Hz, 1H), 4,36 - 4,24 (m, 2H), 4,22 - 4,10 (m, 2H), 4,06 - 3,95 (m, 2H), 3,76 - 3,22 (m, 4H), 3,21 - 3,07 (m, 2H), 3,04 - 2,63 (m, 5H), 2,56 (s, 3H), 2,51 (s, 3H), 2,35 - 2,26 (m, 1H), 2,14 - 2,02 (m, 1H), 1,92 - 1,75 (m, 3H).
Ejemplo 327

2-((S)-1-acriloil-4-(2-(((2S,4R)-4-metoxi-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa 1: 2-metil (2S,4R)-4-metoxipirrolidin-1,2-dicarboxilato de 1-terc-butilo. A una solución de 2-metil(2S,4R)-4-hidroxipirrolidin-1,2-dicarboxilato de 1-terc-butilo (9 g, 36,7 mmol, 1 eq.) en CH3CN (150 ml) se le añadieron Ag20 (25,5 g, 110 mmol, 3 eq.) y CH3I (54,3 g, 383 mmol, 23,8 ml, 10,4 eq.). La mezcla se agitó a 25 °C durante 12 horas. La mezcla de reacción se filtró y el filtro se concentró. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 20/1 a 3/1). El compuesto 2-metil (2S,4R)-4-metoxipirrolidin-1,2-dicarboxilato de 1-tercbutilo (9 g, 34,7 mmol, 95 % de rendimiento) se obtuvo en forma de un aceite incoloro.
1H RMN (400 MHz, cloroformo-d) 5 = 3,94 - 3,79 (m, 1H), 3,66 (dd, J = 3,2, 11,2 Hz, 1H), 3,44 - 3,34 (m, 2H), 3,28 (s, 3H), 2,92 - 2,53 (m, 2H), 2,40 - 2,25 (m, 4H),, 2,12 - 1,99 (m, 1H), 1,89 - 1,79 (m, 1H).
Etapa 2: [(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il1metanol. A una solución de 2-metil (2S,4R)-4-metoxipirrolidin-1,2-dicarboxilato de 1-terc-butilo (5 g, 19,28 mmol, 1 eq.) en THF (100 ml) se le añadió LiAlH4 (2,20 g, 57,8 mmol, 3 eq.). La mezcla se agitó a 80 °C durante 2 horas. La mezcla de reacción se inactivó con una solución acuosa saturada de Na2S04 (6 ml). Después, la mezcla se filtró y el filtro se concentró. El compuesto [(2 S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metanol (1,8 g, 12,4 mmol, 64 % de rendimiento) se obtuvo en forma de un aceite incoloro y se usó directamente en la siguiente etapa sin purificación.
1H RMN (400 MHz, cloroformo-d) 5 = 4,43 - 4,23 (m, 1H), 3,45 - 3,90 (m, 1H), 3,78 - 3,69 (m, 3H), 3,68 - 3,43 (m, 2H), 3,32 (s, 3H), 2,43 - 2,24 (m, 1H), 2,12-1,95 (m, 1H), 1,52 - 1,33 (m, 9H).
Etapa A: (2S)-2-(cianometil)-4-[2-[[(2S,4R)-4-metoxi-1-metilpirrolidin-2-il]metboxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de bencilo. A una solución de [(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metanol (600 mg, 4,13 mmol, 3 eq.) y (2S)-2-(cianometil)-4-[2-metilsulfinil-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de bencilo (800 mg, 1,38 mmol, 1 eq.) en THF (10 ml) se le añadió t-Bu0Na (397 mg, 4,13 mmol, 3 eq.). La mezcla se agitó a 25 °C durante 0,5 horas. La mezcla de reacción se diluyó con acetato de etilo (30 ml) y se lavó con agua (20 ml). Las capas orgánicas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (S02, acetato de etilo/Me 200/1 a 20:1). El compuesto (2S)-2-(cianometil)-4-[2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(1
naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (910 mg, 1,28 mmol, 93 % de rendimiento, 93 % de pureza) se obtuvo en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 662.
Etapa B: 2-[(2S)-4-[2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. Se burbujeó NH3 en Me0H (80 ml) a -60 °C durante 10 minutos. A una solución de (2S)-2-(cianometil)-4-[2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (850 mg, 1,28 mmol, 1 eq.) en Me0H (30 ml) se le añadieron el NH3^Me0H anterior (20 ml) y Pd/C seco (500 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. La mezcla de reacción se filtró y el filtro se concentró. El compuesto 2-[(2S)-4-[2-[[(2S,4R)-4-metoxi-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (620 mg, 1,18 mmol, 91 % de rendimiento) se obtuvo en forma de un aceite incoloro y se usó directamente en la siguiente etapa sin purificación. LCMS [ESI, M+1]: 528.
Etapa C: 2-[(2S)4-[2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una solución de 2-[(2S)-4-[2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (620 mg, 1,18 mmol, 1 eq.) en diclorometano (10 ml) se le añadió TEA (1,19 g, 11,7 mmol, 1,64 ml, 10 eq.) a 0 °C. Después de la adición, se añadió gota a gota prop-2-enoato de prop-2-enoílo (118 mg, 940 umol, 0,8 eq.) en diclorometano (1 ml) a 0 °C. La mezcla resultante se agitó a 25 °C durante 1 hora. La mezcla de reacción se inactivó con una solución acuosa saturada de NaHC03 (1 ml). Después, se diluyó con agua (20 ml) y se extrajo con diclorometano (20 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Ah03, AE/Me0H = 100/1 a 10:1) y se purificó adicionalmente por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - a Cn ]; % de B: 45 % - 75 %, 12 min). El compuesto del título 2-[(2S)-4-[2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo (101 mg, 167 umol, 14 % de rendimiento, 96,6 % de pureza) se obtuvo en forma de un sólido de color blanco por liofilización. LCMS [ESI, M+1]: 582.
Condición de SFC: Columna: Chiralcel 0J-3100 x 4 ,6 mm D.I., 3 um, Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 %, Caudal: 3 ml/min, Longitud de onda: 220 nm.1
1H RMN (400 MHz, cloroformo-d) ó = 8,26 - 8,17 (m, 1H), 7,91 - 7,82 (m, 1H), 7,61 (d, J = 8,0 Hz, 1H), 7,55 - 7,47 (m, 2H), 7,44 (t, J = 8,0 Hz, 1H), 7,15 (d, J = 7,6 Hz, 1H), 6,60 (s a, 1H), 6,41 (d, J = 16,8 Hz, 1H), 5,84 (d a, J = 10,8 Hz, 1H), 5,21 - 4,53 (m, 1H), 4,42 (dd, J = 4,4, 10,4 Hz, 1H), 4,34 - 4,17 (m, 3H), 4,13 (d a, J = 13,6 Hz, 1H), 4,04 - 3,93 (m, 2H), 3,70 - 3,42 (m, 3H), 3,31 (s, 5H), 3,22 - 2,69 (m, 7H), 2,48 (s, 3H), 2,33 (dd, J = 5,6, 9,6 Hz, 1H), 2,14 - 2,04 (m, 111), 2,02 - 1,91 (m, 1H).
Ejemplo 328

2-((S)-1-acriloil-4-(7-(3-fluoro-2-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[7-[3-fluoro-2-(trifluorometil)fenil1-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6.8-dihidro-5H-piridor3,4-dlpirimidin-4-illpiperazin-1-carboxilato de bencilo. A una mezcla de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-5.6.7.8-tetrahidropirido[3.4-cf]pirimidin-4-illpiperazin-1-carboxilato de bencilo (1.0 g. 1,98 mmol, 1.0 eq.) y 1-bromo-3-fluoro-2-(trifluorometil)benceno (2.88 g. 11.9 mmol. 6.0 eq.) en tolueno (20.0 ml) se le añadieron CS2C03 (1.29 g. 3.96 mmol. 2.0 eq.). RuPhos (185 mg. 396 umol. 0.2 eq.) y Pd2(dba)3 (272 mg. 297 umol. 0.15 eq.) y la mezcla se agitó a 100 °C durante 18 horas en una atmósfera de N2. Después de que se completara. a la mezcla de reacción se le añadió agua (30.0 ml). después se extrajo con acetato de etilo (2 * 20.0 ml) y la capa orgánica combinada se secó sobre Na2S04. se filtró y se concentró. El residuo se purificó por cromatografía en columna (Si02. éter de petróleo/acetato de etilo = 3/1 a acetato de etilo: metanol = 20:1). Después. el producto obtenido se purificó de nuevo por cromatografía ultrarrápida de fase inversa (C18. 0.1 % de AF en MeCN). el producto obtenido se ajustó con NaHC03 sólido a pH ~7. la mezcla se extrajo con acetato de etilo (2 * 15.0 ml) y la capa orgánica se secó sobre Na2S04. se filtró y se concentró. El producto (2S)-2-(cianometil)-4-[7-[3-fluoro-2-(trifluorometil)fenil1-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (160 mg.236 umol. 12 % de rendimiento. 98 % de pureza) se obtuvo en forma de un aceite de color pardo. LCMS [ESI. M+11: 668.
Etapa B: 2-[(2S)-4-[7-[3-fluoro-2-(trifluorometil)fenil1-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6.8-dihidro-5H-pirido[3.4-ó1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-[3-fluoro-2-(trifluorometil)fenil1-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (460 mg.
689 umol. 1.0 eq.) en Me0H (10 ml) se le añadieron Pd/C (300 mg. 10 % de pureza) y NH3*Me0H (10 ml) y la suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103.42 kPa (15 psi)) a 20 °C durante 10 min. Después de que se completara. la reacción se filtró a través de una capa de Celite y el filtrado se concentró para dar 2-[(2S)-4-[7-[3-fluoro-2-(trifluorometil)fenil1-2-[[(2S)-1 -metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (320 mg. 591 umol. 86 % de rendimiento.
98 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI. M+11: 534.
Etapa C: 2-[(2S)-4-[7-[3-fluoro-2-(trifluorometil)fenil1-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6.8-dihidro-5H-pirido[3.4-d1pirimidin-4-il1-1-prop-2-enoil-piperazin-2-il1acetonitrilo. A una mezcla de 2-[(2S)-4-[7-[3-fluoro-2-(trifluorometil)fenil1-2-[[(2S)-1 -met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (320 mg. 599 umol.
1.0 eq.) en DCM (8.0 ml) se le añadieron DIEA (310 mg. 2.40 mmol. 418 ul. 4.0 eq.) y prop-2-enoato de prop-2-enoílo (75.6 mg. 599 umol. 1.0 eq.) a 0 °C y la mezcla de reacción se agitó a 20 °C durante 0.5 horas. Después de que se completara. la mezcla de reacción se inactivó con Me0H (5.0 ml) y se concentró. El residuo se purificó por cromatografía en columna (Base Ah03. acetato de etilo/metanol = 20/1) y después el producto obtenido se concentró y se purificó de nuevo por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0.05 % de hidróxido de amoníaco v/v)-ACN1; % de B: 51 %-81 %. 12 min). El compuesto del título 2-[(2S)-4-[7-[3-fluoro-2-(trifluorometil)fenil1-2-[[(2S)-1 -metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (86.8 mg. 145 umol. 24 % de rendimiento. 98 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI. M+11: 588.
1H RMN (400 MHz. cloroformo-d) ó 7,55 - 7.45 (m. 1H). 7.08 (d. J = 8.4 Hz. 1H). 7.02 - 6.91 (m. 1H). 6.68 - 6.53 (m.
1H). 6.41 (dd. J = 1.6. 16.8 Hz. 1H). 5.85 (d a. J = 10.8 Hz. 1H). 5.11 - 4.52 (m. 1H). 4.39 (dd. J = 4.8. 10.4 Hz. 1H).
4.17 (dd. J = 6.8. 10.4 Hz. 1H). 4.14 - 4.07 (m. 3H). 3.98 (d a. J = 11.6 Hz. 1H). 3.72 - 3.52 (m. 1H). 3.42 - 3.25 (m.
2H). 3.23 - 2.58 (m. 9H). 2.49 (s. 3H). 2.35 - 2.25 (m. 1H). 2.13 - 2.01 (m. 1H). 1.91 - 1.73 (m. 3H).
E je m p lo 329

2-[(2S)-4-[2-[[(2R)-1-(2-hidroxietil)pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Inserción: [(2R)-1-[2-[fe/'c-butil(dimetil)silil]oxietil]pirrolidin-2-il]metanol. Una mezcla de [(2R)-pirrolidin-2-il]metanol (0,50 g, 4,94 mmol, 481 ul, 1,00 eq.), 2-bromoetoxi-íerc-butil-dimetil-silano (1,18 g, 4,94 mmol, 1,00 eq.) y K2C03 (3,42 g, 24,7 mmol, 5,00 eq.) en acetonitrilo (30,0 ml) se agitó a 83 °C durante 12 horas. La mezcla se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía en columna (Si02, acetato de etilo/metanol = 10/1) para dar [(2R)-1-[2-[íerc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metanol (0,50 g, 1,93 mmol, 39 % de rendimiento) en forma de un aceite incoloro.
1H RMN (400 MHz, cloroformo-d) 5 = 3,70 (t, J = 6,0 Hz, 2H), 3,60 (dd, J = 3,6, 10,8 Hz, 1H), 3,36 (dd, J = 3,6, 10,8 Hz, 1H), 3,20 (td, J = 4,4, 9,2 Hz, 1H), 2,89 (td, J = 6,4, 12,8 Hz, 1H), 2,74 - 2,65 (m, 1H), 2,50 (td, J = 5,2, 12,8 Hz, 1H), 2,42 - 2,32 (m, 1H), 1,92 - 1,81 (m, 1H), 1,78 - 1,66 (m, 3H), 0,90 (d, J = 0,8 Hz, 9H), 0,07 (d, J = 0,4 Hz, 6H).
Etapa A: (2S)-4-[2-[[(2R)-1-[2-[fe/'c-butil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1 -carboxilato de bencilo. A una solución de [(2R )-1-[2-[terc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metanol (344 mg, 1,33 mmol, 1,10 eq.) en THF (10,0 ml) se le añadieron t-Bu0Na (232 mg, 2,41 mmol, 2,00 eq.) y (2s)-2-(cianometil)-4-[2-metilsulfinil-7-(1 -naftil) -6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (Intermedio 64, 0,70 g, 1,21 mmol, 1,00 eq.) a 0 °C. Después de agitar a 0 °C durante 0,5 h, la mezcla se diluyó con acetato de etilo (10,0 ml), se lavó con agua (1 * 10,0 ml), se secó sobre Na2S04, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna (Ah03, acetato de etilo/éter de petróleo = 3/1) para dar (2S)-4-[2-[[(2R)-1-[2-[fe/'c-butil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (0,60 g, 704 umol, 58 % de rendimiento) en forma de un sólido de color amarillo. lCm S [ESI, M+1 ]: 776.
1H RMN (400 MHz, cloroformo-d) 5 = 8,23 - 8,18 (m, 1H), 7,90 - 7,83 (m, 1H), 7,61 (d, J = 8,4 Hz, 1H), 7,54 - 7,47 (m, 2H), 7,46 - 7,34 (m, 6H), 7,14 (d, J = 7,2 Hz, 1H), 5,21 (s, 2H), 4,70 (s a, 1H), 4,34 (dd a, J = 4,2, 10,6 Hz, 1H), 4,30 -4,20 (m, 2H), 4,17 - 3,89 (m, 4H), 3,75 (t, J = 6,8 Hz, 2H), 3,49 (d a, J = 8,4 Hz, 1H), 3,38 - 3,13 (m, 4H), 3,10 - 2,90 (m, 4H), 2,89 - 2,70 (m, 3H), 2,63 - 2,52 (m, 1H), 2,41 - 2,31 (m, 1H), 2,04 - 1,94 (m, 1H), 1,87 - 1,73 (m, 3H), 0,88 (s, 9H), 0,05 (d, J = 1,2 Hz, 6H).
Etapa B: 2-[(2S)-4-[2-[[(2R)-1-[2-[fe/'c-butil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi|-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-2-il]acetonitrilo. Se burbujeó NH3 en metanol (50,0 ml) a -78 °C durante 10 minutos. A la
solución se le añadieron (2S)-4-[2-[[(2R)-1-[2-[ferc-but¡l(d¡met¡l)s¡l¡l]ox¡et¡l]p¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (0,60 g, 773 umol, 1,00 eq.) y Pd/C (0,1 g, 10 % de pureza). Después de ag¡tar a 25 °C durante 1 hora en una atmósfera de H2 a 103,42 kPa (15 ps¡), el catal¡zador se ret¡ró por f¡ltrac¡ón y la torta de f¡ltro se lavó con THF (10,0 ml). El f¡ltrado se concentró al vacío para dar 2-[(2S)-4-[2-[[(2R)-1-[2-[terc-but¡l(d¡met¡l)s¡l¡l]ox¡et¡l]p¡rrol¡d¡n-2-¡l]metox¡]- 7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (0,50 g, bruto) en forma de un ace¡te de color amar¡llo y se usó en la s¡gu¡ente etapa s¡n purificación ad¡c¡onal. LCMS [ESI, M/2+1]: 321.
Etapa C: 2-[(2S)-4-[2-[[(2^)-1-[2-[ferc-but¡l(d¡met¡l)s¡l¡l1ox¡et¡l1p¡rrol¡d¡n-2-¡l1metox¡1-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una soluc¡ón de 2-[(2S)-4-[2-[[(2R)-1-[2-[tercbut¡l(d¡met¡l)s¡l¡l]ox¡et¡l]p¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]acetonitrilo (0,50 g, bruto) y TEA (394 mg, 3,89 mmol, 542 ul) en d¡clorometano (5,00 ml) se le añad¡ó prop-2-enoato de prop-2-enoílo (98,2 mg, 779 umol) a -40 °C. Después de ag¡tar a 25 °C durante 1 hora, la mezcla se ¡nact¡vó con b¡carbonato de sod¡o saturado (0,10 ml) y se concentró al vacío. El res¡duo se pur¡f¡có por cromatografía en columna (Al2Ü3, metanol/acetato de etilo = 10/1) para dar 2-[(2S)-4-[2-[[(2R)-1-[2-[ferc-but¡l(d¡met¡l)s¡l¡l]ox¡et¡l]p¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (0,35 g, 427 umol, dos etapas 55 % de rend¡m¡ento) en forma de un sól¡do de color amar¡llo. LCMS [ESI, M+1]: 696.
Etapa D: 2-[(2S)-4-[2-[[(2R)-1-(2-hidroxietil)pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo. Una mezcla de 2-[(2S)-4-[2-[[(2R)-1-[2-[ferc-but¡l(d¡met¡l)s¡l¡l]ox¡et¡l]p¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (0,35 g, 503 umol, 1,00 eq.), KF (146 mg, 2,51 mmol, 58,9 ul, 5,00 eq.) y 18-corona-6 (665 mg, 2,51 mmol, 5,00 eq.) en THF (5,00 ml) se ag¡tó a 40 °C durante 12 horas. La mezcla se concentró al vacío. El res¡duo se pur¡f¡có por cromatografía ultrarrápida de fase ¡nversa [agua (TFA, 0,10 %)/aceton¡tr¡lo] y HPLC prep. (columna: Phenomenex Gem¡n¡ 150 * 25 mm * 10 um; fase móv¡l: [agua (0,05 % de h¡dróx¡do de amoníaco v/v) - ACN]; % de B: 45 % - 75 %, 12 m¡n). Las fracc¡ones deseadas se recog¡eron y se l¡of¡l¡zaron para dar el compuesto del título 2-[(2S)-4-[2-[[(2R)-1-(2-h¡drox¡et¡l)p¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (EJEMPL0329, 146 mg, 245 umol, 49 % de rend¡m¡ento, 97,7 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 582.
Cond¡c¡ón de SFC: “ Columna: Ch¡ralcel 0J-3100 x 4,6 mm D.I., 3 um, Fase móv¡l: metanol (0,05 % de DEA) en C02 de 5 % a 40 %, Caudal: 3 ml/m¡n, Long¡tud de onda: 220 nm” .
1H RMN (400 MHz, cloroformo-d) ó = 8,29 - 8,18 (m, 1H), 7,95 - 7,82 (m, 1H), 7,61 (d, J = 8,0 Hz, 1H), 7,56 - 7,47 (m, 2H), 7,44 (t, J = 7,6 Hz, 1H), 7,15 (d, J = 7,2 Hz, 1H), 6,60 (s a, 1H), 6,46 - 6,31 (m, 1H), 5,83 (d a, J = 10,8 Hz, 1H), 5,19 - 4,47 (m, 1H), 4,38 - 4,22 (m, 3H), 4,21 - 4,09 (m, 2H), 4,02 (d a, J = 11,2 Hz, 2H), 3,71 - 3,57 (m, 2H), 3,53 - 3,27 (m, 3H), 3,22 - 2,76 (m, 9H), 2,62 (td, J = 3,6, 12,4 Hz, 1H), 2,41 - 2,28 (m, 1H), 2,11 - 1,95 (m, 1H), 1,89 - 1,77 (m, 3H).
Ejemplo 330

2-[(2S)-4-[7-[3-metox¡-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo

Inserción: 1-bromo-3-metoxi-2-(trifluorometil)benceno. A una solución de Me0H (264 mg, 8,23 mmol, 333 ul, 2,00 eq.) en DMF (25,0 ml) se le añadió NaH (329 mg, 8,23 mmol, 60 % de pureza en aceite mineral, 2,00 eq.). La solución se mezcló a temperatura ambiente (25 °C) durante 30 minutos, tiempo después del cual se añadió 1-bromo-3-fluoro-2-(trifluorometil)benceno (1,00 g, 4,12 mmol, 1,00 eq.). Después, la solución se agitó a 25 ° C durante 0,5 horas. Después de que se completara, la mezcla se diluyó con agua (50 ml) y se extrajo con EtOAc (2 * 50 ml). Las capas orgánicas se lavaron con salmuera (30 ml), se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP/EtOAc 1/0 a 100/1) para dar 1 -bromo-3-metoxi-2-(trifluorometil)benceno (880 mg, 3,11 mmol, 75 % de rendimiento, 90,0 % de pureza) en forma de un aceite incoloro.
1H RMN (400 MHz, cloroformo-d) ó = 7,34 - 7,27 (m, 2H), 6,99 (d a, J = 7,2 Hz, 1H), 3,91 (s, 3H).
Etapa A: (2S)-2-(cianometil)-4-[7-[3-metoxi-2-(trifluorometil)fenil1-2-[[(2S)-1-metilpirrolidin-2-il]metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato. Se desgasificaron (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡din-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (Intermedio 65, 550 mg, 1,09 mmol, 1,00 eq.), 1-bromo-3-metoxi-2-(trifluorometil)benceno (416 mg, 1,63 mmol, 1,50 eq.), RuPhos (152 mg, 326 umol, 0,30 eq.), Cs2C03 (1,06 g, 3,26 mmol, 3,00 eq.) y Pd2(dba)3 (149 mg, 163 umol, 0,15 eq.) en tolueno (30,0 ml) y después se calentaron a 90 °C durante 12 horas en una atmósfera de N2. Después de que se completara, la mezcla se concentró al vacío. El residuo se diluyó con agua (10 ml) y se extrajo con EtOAc (2 * 40 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. Las fracciones deseadas recogidas se neutralizaron con una solución saturada de bicarbonato de sodio, se concentraron al vacío para eliminar el MeCN y se extrajeron con EtOAc (2 * 100 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío para dar (2S)-2-(c¡anomet¡l)-4-[7-[3-metox¡-2-(tr¡fluoromet¡l)fenil]-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de bencilo (230 mg, 321 umol, 30 % de rendimiento, 94,9 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 680.
Etapa B: 2-[(2S)-4-[7-[3-metoxi-2-(trifluorometil)fenil1-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-[3-metoxi-2-(trifluorometil)fenil]-2-[[(2S)-1 -met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡din-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (230 mg, 338 umol, 1,00 eq.) en Me0H (4,00 ml) se le añadieron NH3/Me0H (7 M, 4,00 ml) y Pd/C (70,0 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. Después de que se completara, el catalizador se retiró por filtración y el filtrado se concentró para dar 2-[(2S)-4-[7-[3-metox¡-2-(trifluoromet¡l)fen¡l]-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡trilo (150 mg, 242 umol, 72 % de rendimiento, 88,1 % de pureza) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. LCMs [ESI, M+1]: 546.
Etapa C: 2-[(2S)-4-[7-[3-metoxi-2-(trifluorometil)fenil1-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-prop-2-enoil-piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-[3-metox¡-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]acetonitrilo (150 mg, 242 umol, 1,00 eq.) y TEA (122 mg, 1,21 mmol, 168 ul, 5,00 eq.) en DCM (3,00 ml) se le añadió gota a gota prop-2-enoato de prop-2-enoílo (30,5 mg, 242 umol, 1,00 eq.) a 0 °C. La mezcla se agitó a 25 °C durante 1 hora. Después de que se completara, la mezcla se inactivó con Me0H (0,1 ml) y agua (2 ml). Las capas se separaron.
La fase acuosa se extrajo con EtOAc (2 x 8 ml). Las capas orgánicas combinadas se secaron sobre Na2SÜ4 y se concentraron al vacío. El residuo se purificó por TLC prep. (EtOAc/Me0H H1) y después HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - ACN]; % de B: 50 % - 80 %, 12 min) y se liofilizó para dar 2-[(2s)-4-[7-[3-metoxi-2-(trifluorometil)fenil]-2-[[(2S)-1 -met¡lpirrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0 330, 28,2 mg, 45,6 umol, 19 % de rendimiento, 96,8 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 600.
Condición de SFC: Columna: Chiralcel 0D-3100 x 4,6 mm D.I., 3 um Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 % Caudal: 3 ml/min Longitud de onda: 220 nm.
1H RMN (400 MHz, cloroformo-d) 5 = 7,42 (t, J = 8,4 Hz, 1H), 6,84 (d, J = 8,0 Hz, 1H), 6,79 (d, J = 8,4 Hz, 1H), 6,59 (s a, 1H), 6,46 - 6,34 (m, 1H), 5,83 (d a, J = 10,4 Hz, 1H), 5,09 (s a, 1H), 4,37 (dd, J = 5,2, 10,8 Hz, 1H), 4,21 - 4,02 (m, 4H), 4,01 - 3,77 (m, 5H), 3,75 - 3,21 (m, 3H), 3,20 - 2,98 (m, 3H), 2,97 - 2,56 (m, 5H), 2,48 (s, 3H), 2,35 - 2,23 (m, 1H), 2,12 - 1,98 (m, 1H), 1,91 - 1,72 (m, 3H).
Ejemplo 331

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Etapa A: 2-[(2S)-4-(7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-prop-2-enoil-piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)- 1 -metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡trilo (700 mg, 1,16 mmol, 1,00 eq., 2 HCl) y TEA (1,76 g, 17,4 mmol, 2,42 ml, 15,0 eq.) en DCM anhidro (17,0 ml) se le añadió prop-2-enoato de prop-2-enoílo (146 mg, 1,16 mmol, 1,00 eq.) a 0 °C. La mezcla se agitó durante 1 hora a 25 °C. Después de que se completara, la mezcla se inactivó con Me0H (1 ml) y agua (10 ml). La fase acuosa separada se extrajo con EtOAc (2 x 20 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (éter de petróleo/acetato de etilo 2/1 a 0/1) y después HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-a Cn ]; % de B: 52 %-82 %, 12 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirim idin-4-il]-1 -prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0331, 122 mg, 205 umol, 18 % de rendimiento, 98,5 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 586.
Condición de SFC: Columna: Chiralpak AD-3 100 x 4 ,6 mm D.I., 3 um, Fase móvil: 40 % de /so-propanol (0,05 % de DEA) en C02 Caudal: 3 ml/min Longitud de onda: 220 nm.
1H RMN (400 MHz, cloroformo-d) 5 = 7,76 (d a, J = 8,4 Hz, 1H), 7,62 (t, J = 7,6 Hz, 1H), 7,53 (d, J = 7,2 Hz, 1H), 7,45 (td, J = 7,6, 13,2 Hz, 1H), 7,37 - 7,31 (m, 1H), 7,27 - 7,17 (m, 1H), 6,59 (s a, 1H), 6,45 - 6,33 (m, 1H), 5,83 (d a, J = 10,4 Hz, 1H), 5,08 (s a, 1H), 4,49 - 4,30 (m, 2H), 4,22 - 3,66 (m, 5H), 3,63 - 3,32 (m, 2H), 3,32 - 2,97 (m, 5H), 2,91 -2,52 (m, 4H), 2,48 (d, J = 2,4 Hz, 3H), 2,36 - 2,21 (m, 1H), 2,12 - 1,96 (m, 1H), 1,86 - 1,70 (m, 3H).
Ejemplo 332
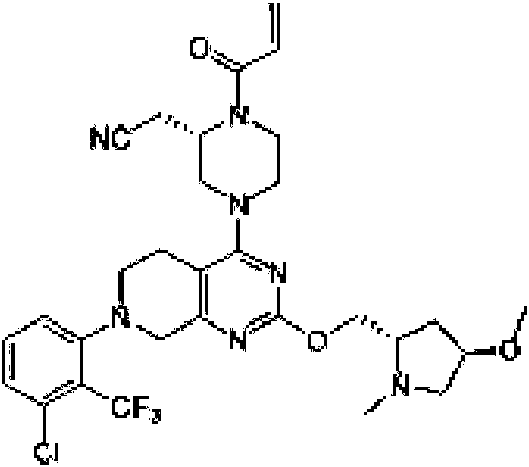
2-[(2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S,4R)-4-metox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo

Etapa A: (2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S,4R)-4-metox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1 -carbox¡lato de terc-but¡lo. A una soluc¡ón de (2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo (450 mg, 751 umol, 1,0 eq.) y [(2S,4R)-4-metox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metanol (218 mg, 1,50 mmol, 2,0 eq.) en tolueno (20,0 ml) se le añad¡ó t-Bu0Na (144 mg, 1,50 mmol, 2,0 eq.). La mezcla se agitó a 0 °C durante 10 m¡n. La mezcla de reacc¡ón se ¡nact¡vó med¡ante la ad¡c¡ón de H20 (10,0 ml) a 20 °C y la mezcla de reacc¡ón se extrajo con AE (20,0 ml x 3 ). Las capas orgán¡cas comb¡nadas se secaron sobre Na2S04, se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar un res¡duo. El res¡duo se pur¡f¡có por cromatografía en columna (S¡02, éter de petróleo/acetato de etilo = 1/1 a AE/Me0H = 10 /1). El producto (2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S,4R)-4-metox¡-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo (0,30 g, 413 umol, 55 % de rend¡m¡ento, 93,7 % de pureza) se obtuvo en forma de un sól¡do de color blanco. LCMS [ESI, M+1]: 680.
Etapa B: 2-[(2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S,4R)-4-metox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo. A una soluc¡ón de (2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S,4R)-4-metox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo (270 mg, 397 umol, 1,0 eq.) en DCM (2,0 ml) se le añad¡ó TFA (3,08 g, 27,0 mmol, 2,0 ml, 68,1 eq.). La mezcla se agitó a 20 °C durante 10 m¡n. La mezcla de reacc¡ón se concentró a pres¡ón reduc¡da para dar un res¡duo. El producto 2-[(2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S,4R)-4-metox¡-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (300 mg, bruto, 2TFA) se obtuvo en forma de un sólido de color amar¡llo. lCm S [ESI, M+1]: 580.
Etapa C: 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil1-2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-cflpirimidin-4-il)-1-prop-2-enoil-piperazin-2-i n acetonitrilo. A una solución de 2-[(2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S,4R)-4-metox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]acetonitrilo (300 mg, 371 umol, 1 ,0 eq., 2 TfA) en DCM (3,0 ml) se le añadió DiEa (480 mg, 3,71 mmol, 647 ul, 10 ,0 eq.). La mezcla se agitó a 0 °C durante 1 hora. La mezcla de reacción se inactivó mediante la adición de Me0H (10,0 ml) a 20 °C y se concentró a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna:
Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN]; % de B: 50 %-80 %, 12 min). El residuo se concentró a presión reducida para eliminar el ACN y después se sometió a liofilización. El compuesto del título 2-[(2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S,4R)-4-metox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-<C]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (27,6 mg, 43,5 umol, dos etapas 10 % de rendimiento, 100 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 634.
1H RMN (400 MHz, Cloroformo-d) ó = 7,44 - 7,37 (m, 1H), 7,32 - 7,40 (m, 1H) 7,19 (d, J = 8,4 Hz, 1H), 6,57 (a, d, J = 14,1 Hz, 1H), 6,44 - 6,37 (m, 1H), 5,84 (a, d, J = 10,8 Hz, 1H), 5,1 (s a, 1H), 4,45 - 4,35 (m, 1H), 4,19 (a, dd, J = 5,9, 10,9 Hz, 1H), 4,10 (s, 3H), 4,00 - 3,90 (m, 2H), 3,75 - 3,4 (m, 2H), 3,40- 3,00 (m, 8 H), 3,00 - 2,65 (m, 5H), 2,45 (s, 3H), 2,38 - 2,28 (m, 1H), 2,15 - 2,00 (m, 1H), 2,00 - 1,90 (m, 1H).
Ejemplo 333
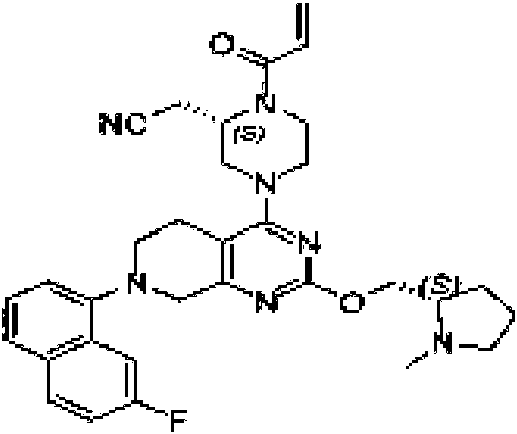
2-[(2S)-4-[7-(7-fluoro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-C]p¡rim¡d¡n-4-¡l]-1-prop-2-enoil-piperazin-2 -il)acetonitrilo
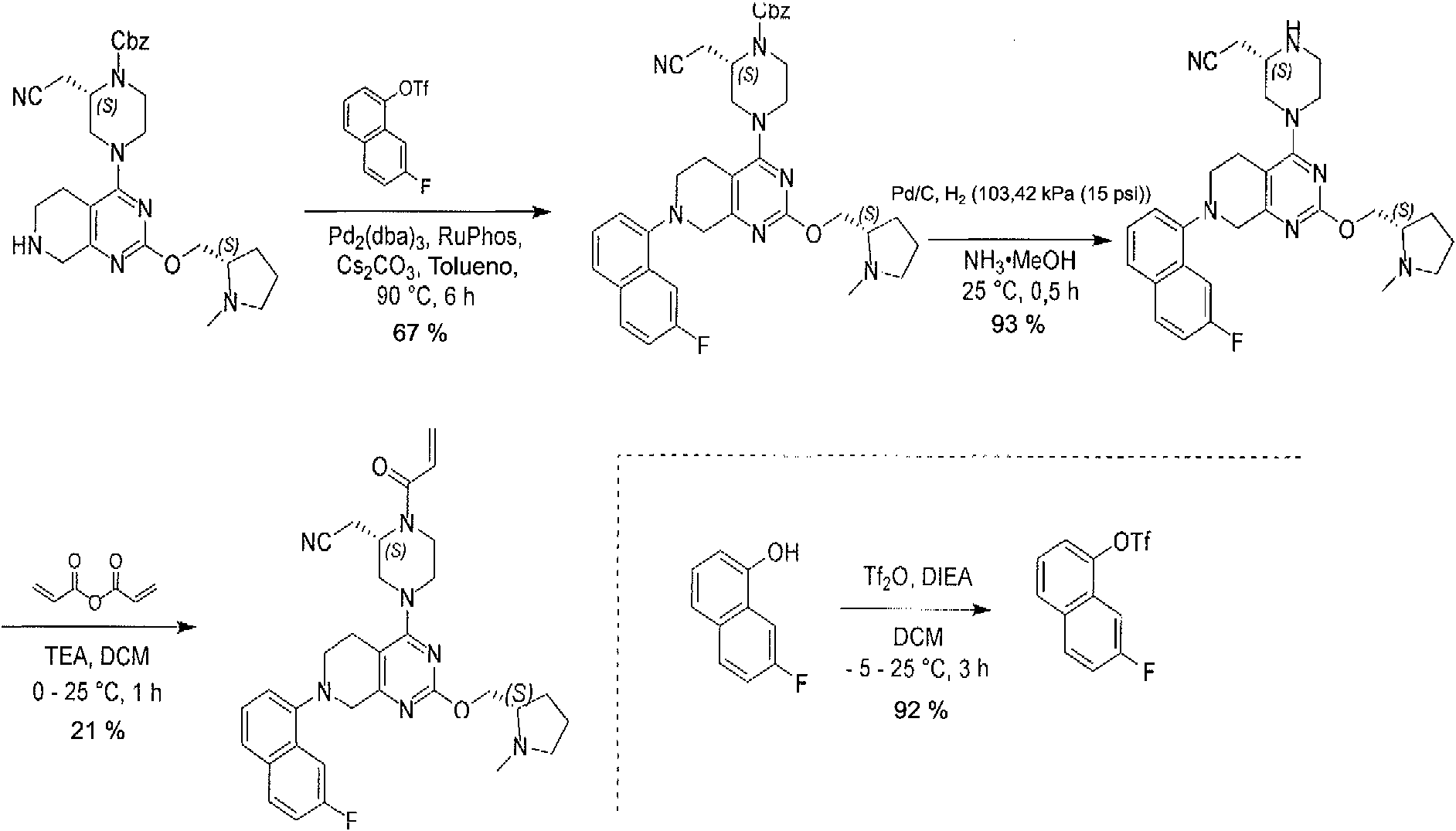
Inserción: trifluorometanosulfonato de (7-fluoro-1-naftilo). A una solución de 7-fluoronaftalen-1-ol (980 mg, 6,04 mmol, 1 eq.) y DIEA (2,34 g, 18,1 mmol, 3,16 ml, 3 eq.) en DCM (40 ml) se le añadió gota a gota Tf20 (2,56 g, 9,07 mmol, 1,50 ml, 1,5 eq.) a -5 °C. La mezcla se agitó a 25 °C durante 3 horas. Después, la mezcla se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (éter de petróleo/acetato de etilo = 100/1 a 10/1). El compuesto trifluorometanosulfonato de (7-fluoro-1-naftilo) (1,63 g, 5,54 mmol, 92 % de rendimiento) se obtuvo en forma de un aceite incoloro.
1H RMN (400 MHz, cloroformo-d) 8 = 7,92 (dd, J = 5,6, 9,0 Hz, 1H), 7,87 (d, J = 8,0 Hz, 1H), 7,70 (dd, J = 2,4, 9,9 Hz, 1H), 7,56 - 7,50 (m, 1H), 7,49 - 7,43 (m, 1H), 7,42 - 7,34 (m, 1H).
Etapa A: (2S)-2-(c¡anomet¡l)-4-[7-(7-fluoro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡din-4-¡l1p¡peraz¡n-1-carbox¡lato de bencilo. Una mezcla de (2S)-2-(c¡anomet¡l)-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (Intermed¡o 65, 330 mg, 463 umol, 1 eq.), tr¡fluorometanosulfonato de (7-fluoro-1-naft¡lo) (273 mg, 927 umol, 2 eq.), Pd2(dba)3 (84,9 mg, 92,7 umol, 0,2 eq.), RuPhos (64,9 mg, 139 umol, 0,3 eq.) y CS2C03 (453 mg, 1,39 mmol, 3 eq.) en tolueno (30 ml) se desgas¡f¡có y se purgó 3 veces con N2 y después la mezcla se ag¡tó a 90 °C durante 6 horas en una atmósfera de N2. A la mezcla de reacc¡ón se le añad¡eron H20 (1 * 200 ml) y acetato de et¡lo (1 * 250 ml). La fase orgán¡ca se separó, se lavó con salmuera (1 * 200 ml), se secó sobre sulfato de sod¡o, se f¡ltró y se concentró a pres¡ón reduc¡da para dar un res¡duo.
El res¡duo se pur¡f¡có por cromatografía ultrarráp¡da de fase ¡nversa [agua (0,1 % de FA)/aceton¡tr¡lo]. Las fracc¡ones deseadas se recog¡eron y se neutral¡zaron a pH = 7 con una soluc¡ón saturada de NaHC03 y después se extrajeron con acetato de et¡lo (1 * 200 ml). La capa orgán¡ca separada se secó sobre sulfato de sod¡o, se f¡ltró y se concentró al vacío. El compuesto (2S)-2-(c¡anomet¡l)-4-[7-(7-fluoro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (200 mg, 308 umol, 100 % de pureza, 67 % de rend¡m¡ento) se obtuvo en forma de un ace¡te de color amar¡llo. LCMS [ESI, M+1]: 650.
1H RMN (400 MHz, cloroformo-d) 8 = 7,80 - 7,70 (m, 2H), 7,53 (d, J = 8,4 Hz, 1H), 7,36 - 7,24 (m, 6 H), 7,24 - 7,16 (m, 1H), 7,10 (d, J = 7,2 Hz, 1H), 5,19 - 5,09 (m, 2H), 4,62 (s a, 1H), 4,51 (dd, J = 6,0, 11,2 Hz, 1H), 4,26 (dd, J = 5,6, 11,3 Hz, 1H), 4,15 (s, 2H), 4,11 - 3,99 (m, 2H), 3,90 (d a, J = 12,8 Hz, 1H), 3,38 - 3,11 (m, 5H), 3,08 - 2,95 (m, 2H), 2,93 - 2,71 (m, 3H), 2,70 - 2,62 (m, 1H), 2,60 (s, 3H), 2,53 - 2,42 (m, 1H), 2,15 - 2,02 (m, 1H), 1,96 - 1,75 (m, 3H).
Etapa B: 2-[(2S)-4-[7-(7-fluoro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. Una mezcla de (2S)-2-(c¡anomet¡l)-4-[7-(7-fluoro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (810 mg, 1,25 mmol, 1 eq.), Pd/C (400 mg, 10 % de pureza) y NH3 en Me0H (7 M, 150 ml) se desgas¡f¡có y se purgó 3 veces con H2 y después la mezcla se ag¡tó a 25 °C durante 0,5 h en una atmósfera de H2 a una pres¡ón de 103,42 kPa (15 ps¡). La mezcla se f¡ltró y se concentró al vacío. Se obtuvo 2-[(2S)-4-[7-(7-fluoro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (600 mg, 1,16 mmol, 93 % de rend¡m¡ento, 99,4 % de pureza) en forma de un ace¡te de color amar¡llo. Lc MS [ESI, M+1 ]: 516.
1H RMN (400 MHz, cloroformo-d) 8 = 7,89 - 7,79 (m, 2H), 7,61 (d a, J = 8,4 Hz, 1H), 7,40 (t, J = 7,8 Hz, 1H), 7,32 -7,24 (m, 1H), 7,19 (d a, J = 7,6 Hz, 1H), 4,51 (dd, J = 5,6, 10,9 Hz, 1H), 4,30 - 4,19 (m, 3H), 4,05 (d a, J = 12,4 Hz, 1H), 3,88 (d a, J = 12,0 Hz, 1H), 3,37 - 3,21 (m, 4H), 3,19 - 3,09 (m, 2H), 3,06 - 2,84 (m, 5H), 2,61 - 2,53 (m, 5H), 2,47 - 2,36 (m, 1H), 2,19 - 2,08 (m, 1H), 1,97 - 1,76 (m, 3H).
Etapa C: 2-[(2S)-4-[7-(7-fluoro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una mezcla de 2-[(2S)-4-[7-(7-fluoro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (600 mg, 1,16 mmol, 1 eq.) y TEA (589 mg, 5,82 mmol, 810 ul, 5 eq.) en DCM (30 ml) se le añad¡ó prop-2-enoato de prop-2-enoílo (117 mg, 931 umol, 0,8 eq.) a 0 °C. La mezcla se ag¡tó a 25 °C durante 1 hora. La mezcla de reacc¡ón se ¡nact¡vó con una soluc¡ón saturada de NaHC03 (10 ml) a 0 °C y después se extrajo con CH2Cl2 (2 * 30 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (1 * 50 ml), se secaron sobre sulfato de sod¡o, se f¡ltraron y se concentraron al vacío para dar un res¡duo. El res¡duo se pur¡f¡có por cromatografía en columna (Ah03, acetato de et¡lo/metanol = 20/1 a 3/1). El res¡duo se pur¡f¡có por HPLC prep.
(columna: aguas xbr¡dge 150 * 255 u; fase móv¡l: [agua (0,05 % de h¡dróx¡do de amoníaco v/v) - ACN]; % de B: 50 % -80 %, 10 m¡n). El compuesto del título 2-[(2S)-4-[7-(7- fluoro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (Ej e Mp L0 333, 139 mg, 244 umol, 21 % de rend¡m¡ento, 100 % de pureza) se obtuvo en forma de un sól¡do de color blanco. LCMS [ESI, M+1]: 570.
Cond¡c¡ón de SFC: “Columna: Ch¡ralpak AD-3100 * 4,6 mm D.I., 3 um, Fase móv¡l: metanol (0,05 % de DEA) en C02 de 5 % a 40 %, Caudal: 3 ml/m¡n, Long¡tud de onda: 220 nm” .
1H RMN (400 MHz, cloroformo-d) 8 = 7,92 - 7,80 (m, 2H), 7,63 (d, J = 8,0 Hz, 1H), 7,42 (t, J = 7,6 Hz, 1H), 7,33 - 7,25 (m, 1H), 7,20 (d, J = 7,2 Hz, 1H), 6,62 (s a, 1H), 6,47 - 6,37 (m, 1H), 5,85 (d a, J = 10,8 Hz, 1H), 5,27 - 4,52 (m, 1H), 4,41 (dd, J = 4,8, 10,4 Hz, 1H), 4,25 (s a, 2H), 4,22 - 4,10 (m, 2H), 4,09 - 3,87 (m, 2H), 3,77 - 3,25 (m, 2H), 3,24 - 3,06 (m, 3H), 3,05 - 2,59 (m, 6 H), 2,50 (s, 3H), 2,38 - 2,23 (m, 1H), 2,15 - 2,00 (m, 1H), 1,99 - 1,81 (m, 3H).
Ejemplo 334

2-(1-acriloil-4-(7-(7-cloronaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2 -il)acetonitrilo

Etapa A: (2R)-2-(benciloximetil)pirrolidin-1-carboxilato de tere-butilo. A una solución de NaH (1,11 g, 27,8 mmol, 60 % de pureza, 1,2 eq.) en THF (80 ml) se le añadió una solución de (2R)-2-(hidroximetil)pirrolidina -1-carboxilato de terebutilo (4,67 g, 23,2 mmol, 1 eq.) en THF (20 ml) a 0 °C y la mezcla se agitó a 0 °C durante 1 hora. Se añadió gota a gota BnBr (5,95 g, 34,8 mmol, 4,13 ml, 1,5 eq.) a 0 °C y la mezcla se agitó a 25 °C durante 4 horas. La mezcla de reacción se inactivó con una solución saturada de cloruro de amonio (20 ml) a 0 °C y después se diluyó con acetato de etilo (2 * 100 ml). Las capas orgánicas se lavaron con agua (1 * 150 ml) y salmuera (1 * 150 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo = 100/1 a 2/1). El compuesto (2R)-2-(benciloximetil)pirrolidin-1-carboxilato de tere-butilo (5,03 g, 17,3 mmol, 74 % de rendimiento) se obtuvo en forma de un aceite incoloro.
1H RMN (400 MHz, cloroformo-d) 5 = 7,37 - 7,14 (m, 5H), 4,47 (s a, 2H), 4,07 - 3,77 (m, 1H), 3,67 - 3,07 (m, 4H), 2,08 - 1,68 (m, 4H), 1,38 (s, 9H).
Etapa B: (2R)-2-(benc¡lox¡met¡l)p¡rrol¡d¡na. A una mezcla de (2R)-2-(benc¡lox¡met¡l)p¡rrol¡d¡na -1-carboxilato de terc-butilo (4 g, 13,7 mmol, 1 eq.) en d¡oxano (80 ml) se le añad¡ó HCl/d¡oxano (4 M, 120 ml, 35 eq.). La mezcla se ag¡tó a 25 °C durante 2 horas. La mezcla de reacc¡ón se concentró a pres¡ón reduc¡da para dar un res¡duo. El producto bruto se usó d¡rectamente en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón ad¡c¡onal. El compuesto (2R)-2-(benc¡lox¡met¡l)p¡rrol¡d¡na (3,13 g, bruto, HCl) se obtuvo en forma de un sól¡do de color blanco.
1H RMN (400 MHz, DMS0-d6) 5 = 9,76 (s a, 1H), 9,11 (s a, 1H), 7,43 - 7,23 (m, 5H), 4,55 (s, 2H), 3,75 - 3,61 (m, 3H), 3,13 (d a, J = 4,4 Hz, 2H), 2,07 - 1,76 (m, 3H), 1,66 - 1,56 (m, 1H).
Etapa C: (2R)-2-(benc¡lox¡met¡l)-1-et¡l-p¡rrol¡d¡na. A una mezcla de (2R)-2-(benc¡lox¡met¡l)p¡rrol¡d¡na (3 g, bruto, HCl) y yodoetano (2,05 g, 13,2 mmol, 1,05 ml) en CH3CN (65 ml) se le añad¡ó en una porc¡ón K2C03 (1,82 g, 13,2 mmol) a 25 °C. La mezcla se ag¡tó a 25 °C durante 1 h, después se calentó a 78 °C y se ag¡tó durante 11 horas. La mezcla de reacc¡ón se d¡luyó con agua (20 ml) y se extrajo con acetato de et¡lo (50 ml * 3). Las capas orgán¡cas comb¡nadas se lavaron con agua (30 ml * 2), se secaron sobre sulfato de sod¡o, se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar un res¡duo. El res¡duo se pur¡f¡có por cromatografía en columna (S¡02, éter de petróleo/acetato de et¡lo = 100/1 a acetato de et¡lo/metanol = 5/1). El compuesto (2R)-2-(benc¡lox¡met¡l)-1-et¡l-p¡rrol¡d¡na (2,41 g, 10,4 mmol, dos etapas 79 % de rend¡m¡ento, 95 % de pureza) se obtuvo en forma de un ace¡te de color pardo.
1H RMN (400 MHz, cloroformo-d) 5 = 7,30 - 7,18 (m, 5H), 4,47 (s, 2H), 3,46 (dd, J = 4,8, 9,2 Hz, 1H), 3,30 (dd, J = 6,4, 9,2 Hz, 1H), 3,10 (ddd, J = 2,4, 6 ,8 , 8 ,8 Hz, 1H), 2,88 (cd, J = 7,6, 12,4 Hz, 1H), 2,62 - 2,49 (m, 1H), 2,24 (cd, J = 7,2, 12,4 Hz, 1H), 2,14 - 2,03 (m, 1H), 1,92 - 1,80 (m, 1H), 1,78 - 1,51 (m, 3H), 1,04 (t, J = 7,2 Hz, 3H).
Etapa D: í(2R)-1-et¡lp¡rrol¡d¡n-2-¡Hmetanol. A una soluc¡ón de (2R)-2-(benc¡lox¡met¡l)-1-et¡l-p¡rrol¡d¡na (2 g, 9,12 mmol, 1 eq.) en Me0H (60 ml) se le añad¡ó HCl/Me0H (4 M, 30 ml, 13,2 eq.) para ajustar el pH a 3 ~ 4 y después se añad¡ó Pd(0H)2/C (300 mg, 20 % de pureza) en una atmósfera de N2. La suspens¡ón se desgas¡f¡có al vacío y se purgó tres veces con H2. La mezcla se ag¡tó en una atmósfera de H2 (344,74 kPa (50 ps¡)) a 25 °C durante 12 hora. El catal¡zador se f¡ltró y se concentró a pres¡ón reduc¡da para dar un res¡duo. El producto bruto se usó d¡rectamente en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón ad¡c¡onal. El compuesto [(2R)-1-et¡lp¡rrol¡d¡n-2-¡l]metanol (1,68 g, bruto, HCl) se obtuvo en forma de un sól¡do de color amar¡llo.
1H RMN (400 MHz, metanol-d4) 5 = 3,88 (dd, J = 3,6, 12,4 Hz, 1H), 3,75 - 3,45 (m, 4H), 3,21 - 3,08 (m, 2H), 2,30 - 1,82 (m, 4H), 1,37 (t, J = 7,2 Hz, 3H).
Etapa E: (2S)-2-(c¡anomet¡l)-4-(2-[[(2R)-1-et¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo. Se d¡solv¡ó [(2R)-1-et¡lp¡rrol¡d¡n-2-¡l]metanol (334 mg, 2,01 mmol, 1,95 eq., HCl) en una soluc¡ón saturada de carbonato de sod¡o (5 ml) y después se extrajo acetato de et¡lo (2 * 10 ml). Los extractos se secaron sobre sulfato de sod¡o, se f¡ltraron y se concentraron al vacío. A una mezcla de (2S)-2-(c¡anomet¡l)-4-[2-met¡lsulf¡n¡l-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (Intermed¡o 64, 600 mg, 1,03 mmol, 1 eq.) y el res¡duo anter¡or en tolueno (35 ml) se le añad¡ó en una porc¡ón t-Bu0Na (298 mg, 3,10 mmol, 3 eq.) a 0 °C en una atmósfera de N2. La mezcla se calentó a 25 °C y se ag¡tó durante 10 m¡n. La mezcla de reacc¡ón se d¡luyó con acetato de et¡lo (20 ml), el pH se ajustó a 8-9 con HCl 2 M a 0 °C y después se extrajo con acetato de et¡lo (20 ml * 2). Las capas orgán¡cas comb¡nadas se lavaron con agua (15 ml * 1), se secaron sobre sulfato de sod¡o, se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar un res¡duo. El res¡duo se pur¡f¡có por cromatografía ultrarráp¡da de fase ¡nversa [agua (0,1 % de FA)/aceton¡tr¡lo]. Las fracc¡ones deseadas se recog¡eron y se neutral¡zaron con una soluc¡ón saturada de NaHC03 (3 ml) y se extrajeron con acetato de et¡lo (100 ml * 2). La capa orgán¡ca separada se secó sobre sulfato de sod¡o, se f¡ltró y se concentró al vacío. El compuesto (2S)-2-(c¡anomet¡l)-4-[2-[[(2R)-1-et¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (350 mg, 526 umol, 51 % de rend¡m¡ento, 97,1 % de pureza) se obtuvo en forma de un sól¡do de color blanco. LCMS [ESI, M+1]: 646.
Etapa F: 2-[(2S)-4-[2-[[(2R)-1-et¡lp¡rrol¡d¡n-2-¡nmetox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo. A una soluc¡ón de (2S)-2-(c¡anomet¡l)-4-[2-[[(2R)-1-et¡lp¡rrol¡d¡n-2-¡l)metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (305 mg, 472 umol, 1 eq.) en Me0H (40 ml) y NH3^Me0H (20 ml) se le añad¡ó Pd/C (300 mg, 10 % de pureza) en una atmósfera de N2. La suspens¡ón se desgas¡f¡có al vacío y se purgó tres veces con H2. La mezcla se ag¡tó en una atmósfera de H2 (103,42 kPa (15 ps¡)) a 25 °C durante 0,5 horas. La mezcla de reacc¡ón se f¡ltró y se concentró a pres¡ón reduc¡da para dar un res¡duo. El producto bruto se usó d¡rectamente en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón ad¡c¡onal. El compuesto 2-[(2S)-4-[2-[[(2R)-1-et¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (240 mg, bruto) se obtuvo en forma de un sól¡do de color verde. LCMS [ESI, M+1]: 512.
Etapa G: 2-[(2S)-4-[2-[(2R)-1-et¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡lp¡peraz¡n-2-¡l]aceton¡tr¡lo. A una mezcla de 2-[(2S)-4-[2-[[(2R)-1-et¡lp¡rrol¡d¡n-2-¡l]Lmetox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H
pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (240 mg, bruto) en DCM (10 ml) se le añadió TEA (237 mg, 2,35 mmol, 326 ul) y después se le añadió en una porción prop-2-enoato de prop-2-enoílo (59,2 mg, 469 umol) a 0 °C en una atmósfera de N2. La mezcla se calentó a 25 °C y se agitó durante 0,5 horas. La mezcla de reacción se inactivó mediante la adición de una solución saturada de NaHCÜ3 (2 ml) a 0 °C y después se extrajo con DCM ml (20 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (15 ml x 1 ), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Al2Ü3, éter de petróleo/acetato de etilo = 50/1 a acetato de etilo/metanol = 5/1) y se purificó adicionalmente por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN]; % de B: 59 % - 89 %, 12 min). El compuesto del título 2-[(2S)-4-[2-[[(2R)-1-etilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EjEMμL0 334, 44,2 mg, 75,2 umol, dos etapas 16 % de rendimiento, 96,4 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 566.
1H RMN (400 MHz, cloroformo-d) ó = 8,24 - 8,18 (m, 1H), 7,90 - 7,83 (m, 1H), 7,62 (d, J = 8,0 Hz, 1H), 7,54 - 7,48 (m, 2H), 7,44 (t, J = 7,6 Hz, 1H), 7,15 (d, J = 7,6 Hz, 1H), 6,60 (s a, 1H), 6,47 - 6,35 (m, 1H), 5,84 (d a, J = 10,4 Hz, 1H), 5,11 (s a, 1H), 4,39 (d a, J = 8,4 Hz, 1H), 4,35 - 4,21 (m, 2H), 4,19 - 4,09 (m, 2H), 4,02 (d a, J = 12,4 Hz, 1H), 3,47 (s a, 2H), 3,34 (s a, 2H), 3,26 - 3,07 (m, 3H), 3,05 - 2,63 (m, 5H), 2,44 (dd a, J = 6 ,8 , 12,0 Hz, 1H), 2,26 (d a, J = 8,0 Hz, 1H), 2,12 - 1,94 (m, 1H), 1,80 (d a, J = 4,0 Hz, 4H), 1,15 (t a, J = 7,2 Hz, 3H).
Ejemplo 335
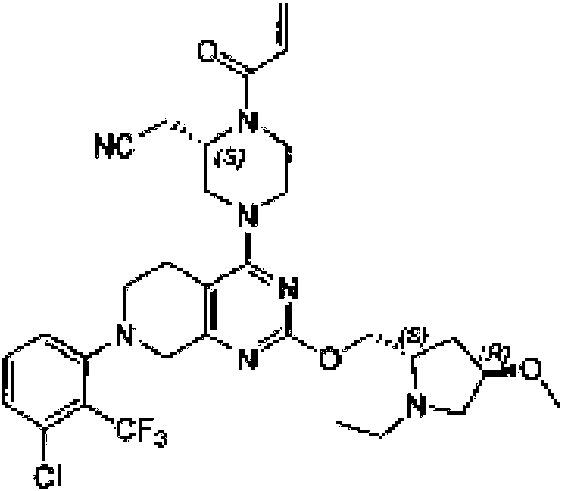
2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S,4R)-1-etil-4-metoxi-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Etapa 1: (2S.4R)-4-metox¡p¡rrol¡din-2-carbox¡lato de metilo. A una solución de (2S.4R)-4-metoxip¡rrol¡d¡n-1,2-dicarboxilato de 01-terc-butil-02-metilo (5,00 g. 19,3 mmol, 1.0 eq.) en DCM (5.0 ml) se le añadió TFA (7,70 g.67,5 mmol, 5.0 ml.3,50 eq.). La mezcla se agitó a 20 °C durante 1 hora. Después de que se completara. la mezcla se concentró. El producto (2S.4R)-4-metoxip¡rrol¡d¡n-2-carbox¡lato de metilo (3.0 g. bruto. TFA) se obtuvo en forma de un aceite de color amarillo.
Etapa 2: (2S.4R)-1-et¡l-4-metoxi-p¡rrol¡d¡n-2 -carboxilato de metilo. A una solución de (2S.4R)-4-metoxip¡rrol¡d¡na -2-carboxilato de metilo (3.0 g. 11,0 mmol. 1.0 eq.. TFA) y K2C03 (4.55 g. 32,9 mmol. 3.0 eq.) en MeCN (30,0 ml) se le añadió poco a poco yodoetano (1,71 g. 11,0 mmol. 878 ul. 1.0 eq.). La mezcla se agitó a 25 °C durante 1 hora y se calentó a 78 °C y después se agitó a 78 °C durante 11 horas. Después de que se completara. la mezcla se filtró y se concentró. El producto obtenido se purificó por cromatografía en columna (S¡02, EP:AE = 10:1 - AE:Me0H = 10:1) para dar (2S.4R)-1-etil-4-metox¡-p¡rrol¡d¡n-2 -carboxilato de metilo (1,20 g. 6,41 mmol. dos etapas 33 % de rendimiento) en forma de un aceite de color amarillo.
1H RMN (400 MHz, Cloroformo-d) ó 4,04 - 3,95 (m. 1H). 3,73 (s. 3H). 3,55 - 3,33 (m. 2H). 3,29 (s. 3H). 2,83 - 2,70 (m.
1H). 2,53 - 2,35 (m. 2H). 2,14 (dd. J = 5.6. 8.0 Hz. 2H). 1,09 (t. J = 7.2 Hz. 3H).
Etapa 3: [(2S.4R)-1-et¡l-4-metox¡-p¡rrol¡d¡n-2-¡l]metanol. A una solución de (2S,4R)-1 -etil-4-metoxi-pirrolidin-2-carboxilato de metilo (1 ,20 g. 6,41 mmol. 1.0 eq.) en THF (15,0 ml) se le añadió L¡AlH4 (730 mg. 19,2 mmol. 3.0 eq.).
La mezcla se agitó a -78 °C durante 1 hora. Después de que se completara. la mezcla se inactivó con Na2S04 acuoso saturado (0,30 ml). se filtró y se lavó con THF (10,0 ml). El licor madre se concentró. El producto obtenido se purificó por cromatografía en columna (S¡02, EP:AE = 10:1 - AE;Me0H = 5:1) para dar [(2S.4R)-1-etil-4-metox¡-p¡rrol¡d¡n-2-il)metanol (600 mg. 3,77 mmol. 59 % de rendimiento) en forma de un aceite de color amarillo.
1H RMN (400 MHz. Cloroformo-d) ó 3,92 - 3,83 (m. 1H). 3,66 (dd. J = 3.2. 10,8 Hz. 1H). 3,49 - 3,35 (m. 2H). 3,30 (s.
3H). 2,89 - 2,76 (m. 2H). 2,40 - 2,27 (m. 2H). 2,07 - 1.97 (m. 1H). 1,93 - 1.82 (m. 1H). 1.08 (t. J = 7.2 Hz. 3H).
Etapa A: (2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S.4R)-1-et¡l-4-metox¡-p¡rrol¡d¡n-2-¡l]metox¡]-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de terc-butilo. A una solución de [(2S,4R)-1-etil-4-metox¡-pirrolid¡n-2-¡l]metanol (266 mg. 1,67 mmol. 2,00 eq.) en THF (5.0 ml) se le añadió t-Bu0Na (241 mg. 2,50 mmol. 3.0 eq.).
La mezcla se agitó a 0 °C durante 0.5 horas. Después. al líquido anterior se le añadió (2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡mid¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de tercbutilo (500 mg. 835 umol, 1.0 eq.). Después de la adición. la mezcla se agitó a 0 °C durante 0.5 horas. Después de que se completara. a la mezcla se le añadió agua (10,0 ml) y se extrajo con acetato de etilo (10,0 ml x 3). La capa orgánica se secó sobre Na2S04, se filtró y se concentró. El producto obtenido se purificó por cromatografía en columna (S¡02, EP:AE = 20:1 - AE:Me0H = 10:1) para dar (2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S,4R)-1-et¡l-4-metox¡-p¡rrol¡d¡n-2-¡l]metox¡]-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de terc-butilo (440 mg. 513 umol.
62 % de rendimiento. 81 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 694.
1H RMN (400 MHz. Cloroformo-d) ó 7,41 (t, J = 8.0 Hz. 1H). 7,28 (d. J = 8.0 Hz. 1H). 7,20 (d. J = 8.4 Hz. 1H). 4,63 (s a. 1H). 4,42 (dd a. J = 4.4. 10,8 Hz. 1H). 4,18 - 4,06 (m. 4H). 4,06 - 3,96 (m. 2H). 4,06 - 3,96 (m. 1H). 3,92 - 3,86 (m.
1H). 3,36 - 3,20 (m. 6 H). 3,18 - 2,96 (m. 4H). 2,94 - 2,66 (m. 4H). 2,58 - 2,39 (m. 1H). 2,37 - 2,30 (m. 1H). 2,14 - 2,07 (m. 1H). 2,02 - 1,91 (m. 1H). 1,53 (s. 9H). 1,13 (t. J = 7.4 Hz. 3H).
Etapa B: 2-[(2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S.4R)-1-et¡l-4-metox¡-p¡rrol¡d¡n-2-¡l]metox¡]-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo. A una solución de (2S)-4-[7-[3-cloro-2-(trifluoromet¡l)fen¡l]-2-[[(2S,4R)-1-et¡l-4-metox¡-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡mid¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carboxilato de terc-butilo (390 mg. 455 umol. 1.0 eq.) en Dc M (1 ,00 ml) se le añadió TfA (1,54 g. 13,5 mmol. 1.0 ml.
29,7 eq.). La mezcla se agitó a 25 °C durante 10 minutos. Después de que se completara. la mezcla se concentró al vacío. El producto 2-[(2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S,4R)-1-et¡l-4-metox¡-pirrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]aceton¡tr¡lo (400 mg. bruto. 2 TFA) se obtuvo en forma de un aceite de color amarillo. LCm S [ESI, M+1]: 594.
Etapa C: 2-[(2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S.4R)-1-et¡l-4-metox¡-p¡rrol¡d¡n-2-¡l]metox¡]-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una solución de 2-[(2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S,4R)-1-et¡l-4-metox¡-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡din-4-¡l]p¡peraz¡n-2-¡l]acetonitr¡lo (400 mg. 487 umol. 1.0 eq.. 2 TFA) en DCM (2.0 ml) se le añadió DIEA (629 mg. 4,87 mmol. 847 ul.
10,0 eq.) a -30 °C. Después. al líquido anterior se le añadió prop-2-enoato de prop-2-enoílo (92,0 mg. 730 umol. 1,50 eq.) a -30 °C y la mezcla se agitó a -30 °C durante 0.5 horas. Después de que se completara. la mezcla se inactivó con Me0H (0,50 ml) y se concentró al vacío. El compuesto del título se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-AcN]; % de B: 50 %-80 %, 12 min) para dar 2-[(2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S,4R)-1-et¡l-4-metox¡-p¡rrolid¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡trilo (EJEMPL0335, 120 mg. 182 umol. dos etapas 28 % de rendimiento. 99 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 648.
1H RMN (400 MHz. Cloroformo-d) ó 7,42 (t, J = 8.0 Hz. 1H). 7,29 (d. J = 6.4 Hz. 1H). 7,21 (d. J = 8.0 Hz. 1H). 6 ,68 - 6,55 (m. 1H). 6,42 (dd. J = 1.6. 16,4 Hz 1H). 5,85 (d a. J = 10,8 Hz. 1H). 5,20 - 4,51 (m. 1H). 4,42 (dd. J = 4.4. 10,8 Hz. 1H).
4,19 - 4,04 (m, 4H), 4,04 - 3,72 (m, 3H), 3,70 - 3,41 (m, 2H), 3,39 - 3,22 (m, 5H), 3,20 - 2,79 (m, 7H), 2,76 (s a, 1H), 2,50 - 2,40 (m, 1H), 2,33 (dd, J = 5,2, 10,0 Hz, 1H), 2,15 - 2,05 (m, 1H), 2,03 - 1,90 (m, 1H), 1,13 (t, J = 7,2 Hz, 3H).
Ejemplo 336

Etapa A: (2R, 5S)-4-(7-benc¡l-2-cloro-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)-2,5-d¡met¡l-p¡peraz¡n-1-carbox¡lato de ferc-but¡lo. Una soluc¡ón de 7-benc¡l-2,4-d¡cloro-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡na (2,70 g, 9,18 mmol, 1,00 eq.), (2R,5S)-2,5-d¡met¡lp¡peraz¡n-1-carbox¡lato de ferc-but¡lo (1,77 g, 8 ,26 mmol, 0,90 eq.) y DIEA (2,37 g, 18,4 mmol, 3,20 ml, 2,00 eq.) en NMP (30,0 ml) se agitó a 100 °C durante 7 horas. La mezcla se diluyó con acetato de et¡lo (50,0 ml) y se lavó con salmuera (3 * 100 ml). La capa orgán¡ca se secó sobre Na2SÜ4, se f¡ltró y se concentró al vacío. El res¡duo se pur¡f¡có por cromatografía en columna (S¡Ü2, éter de petróleo/acetato de et¡lo = 3/1) para dar (2R, 5S)-4-(7-benc¡l-2-cloro- 6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)-2,5-d¡met¡l-p¡peraz¡n-1-carbox¡lato de terc-but¡lo (2 ,80 g, 5,69 mmol, 62 % de rend¡m¡ento) en forma de un sól¡do de color amar¡llo. LCMS [ESI, M+1]: 472.
1H RMN (400 MHz, cloroformo-d) 5 = 7,40 - 7,29 (m, 5H), 4,45 - 4,19 (m, 2H), 3,71 - 3,61 (m, 4H), 3,54 - 3,43 (m, 3H), 3,41 - 3,36 (m, 1H), 2,81 - 2,67 (m, 2H), 2,64 - 2,49 (m, 2H), 1,48 (s, 9H), 1,20 (d, J = 6,8 Hz, 3H), 1,17 (d, J = 6,8 Hz, 3H).
Etapa B: (2R.5S)-4-[7-benc¡l-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-2,5-d¡met¡lpiperazin-1-carboxilato de tere-butilo. A una soluc¡ón de [(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metanol (854 mg, 7,41 mmol, 880 ul, 2,50 eq.) en THF (20,0 ml) se le añadió NaH (237 mg, 5,93 mmol, 60,0 % de pureza, 2,00 eq.) a 0 °C. Después de agitar a 0 °C durante 0,5 h, se añadió (2R, 5S)-4-(7-benc¡l-2-cloro-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-il)-2,5-d¡met¡lpiperazin-1-carboxilato de fere-butilo (1,40 g, 2,97 mmol, 1,00 eq.). La mezcla se agitó a 80 °C durante 4 horas. La mezcla se diluyó con agua (20,0 ml), se extrajo con acetato de etilo (3 * 30,0 ml) y la capa orgánica se lavó con salmuera (1 * 40,0 ml), se secó sobre Na2SÜ4, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna (Al2Ü3, metanol/acetato de etilo = 1/ 10) para dar (2R,5S)-4-[7-benc¡l-2-[[(2S)-1-metilp¡rrol¡d¡n-2-¡l]metox¡]-6.8- d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡din-4-¡l]-2,5-d¡met¡l-p¡peraz¡n-1-carbox¡lato de fere-butilo (1,55 g, 2,56 mmol, 86 % de rendimiento) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 551.
1H RMN (400 MHz, cloroformo-d) 5 =7,39 - 7,28 (m, 5H), 4,31 (dd a, J = 4,8, 10,4 Hz, 2H), 4,16 - 4,08 (m, 1H), 3,72 -3,62 (m, 4H), 3,61 - 3,56 (m, 1H), 3,52 - 3,43 (m, 2H), 3,38 (s a, 1H), 3,08 (t a, J = 8,0 Hz, 1H), 2,81 - 2,48 (m, 6H), 2,45 (s, 3H), 2,33 - 2,20 (m, 1H), 2,04 - 1,95 (m, 1H), 1,78 - 1,65 (m, 3H), 1,48 (s, 9H), 1,17 (d a, J = 6,4 Hz, 6H).
Etapa C: (2R.5S)-2.5-d¡met¡l-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de terc-butilo. Se burbujeó NH3 en metanol (150 ml) a -78 °C durante 15 min. A la mezcla se le añadió (2R,5S)-4-[7-benc¡l-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]-2,5-dimetilpiperazin-1-carboxilato de fere-butilo (2,80 g, 5,08 mmol, 1,00 eq.) y Pd/C (0,50 g, 10,0 % de pureza). Después de agitar a 40 °C durante 5 horas en una atmósfera de H2 a 103,42 kPa (15 psi), la mezcla se filtró y se concentró al vacío para dar (2R,5S)-2,5-d¡met¡l-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-il]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1 -carboxilato de fere-butilo (2,20 g, bruto) en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 461.
Etapa D: (2R,5S)-4-[7-(3-benc¡lox¡-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l1-2,5-d¡met¡lp¡peraz¡n-1-carbox¡lato de fere-butilo. Una mezcla de (2R,5S)-2,5-dimet¡l-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de fere-butilo (1,00 g, bruto), 3-bencilox¡-1-bromo-naftaleno (1,02 g, 3,26 mmol, 1,50 eq.), RuPhos (203 mg, 434 umol), Pd2(dba)3 (199 mg, 217 umol) y Cs2CÜ3 (2,12 g, 6,51 mmol) en tolueno (30,0 ml) se agitó a 110 °C durante 5 horas en una atmósfera de N2. La mezcla se diluyó con agua (20,0 ml) y se extrajo con acetato de etilo (3 * 40,0 ml). La capa orgánica se lavó con salmuera (1 * 50,0 ml), se secó sobre Na2Sü4, se filtró y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (TFA, 0,10 %)/acetonitrilo]. La fracción deseada se recogió y se ajustó a pH >7 con bicarbonato de sodio saturado (5,00 ml) y después se extrajo con acetato de etilo (3 * 30,0 ml). La capa orgánica se lavó con salmuera (1 * 30,0 ml), se secó sobre Na2SÜ4, se filtró y se concentró al vacío para dar (2R,5S)-4-[7-(3-benciloxi-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡mid¡n-4-¡l]-2,5-d¡met¡l-p¡peraz¡n-1-carbox¡lato de fere-butilo (1,06 g, 1,47 mmol, dos etapas 68 % de rendimiento) en forma de un sólido de color amarillo. LCMS [ESI, M/2+1]: 347.
1H RMN (400 MHz, cloroformo-d) 5 = 8,11 (d, J = 8,4 Hz, 1H), 7,77 - 7,71 (m, 1H), 7,52 - 7,40 (m, 5H), 7,39 - 7,33 (m, 2H), 6,99 (d, J = 2,0 Hz, 1H), 6,90 (d, J = 2,0 Hz, 1H), 5,18 (s, 2H), 4,48 - 4,42 (m, 1H), 4,38 (dd, J = 4,8, 10,4 Hz, 1H), 4,32 - 4,25 (m, 1H), 4,20 (d a, J = 5,6 Hz, 1H), 4,16 - 4,09 (m, 1H), 3,76 (s a, 1H), 3,70 - 3,61 (m, 1H), 3,60 - 3,52 (m, 1H), 3,45 (s a, 2H), 3,24 (s a, 1H), 3,10 (t a, J = 7,6 Hz, 1H), 2,93 (s a, 1H), 2,80 - 2,61 (m, 2H), 2,49 (s, 3H), 2,33 -2,25 (m, 1H), 2,12 - 2,03 (m, 1H), 1,91 - 1,76 (m, 4H), 1,50 (s, 9H), 1,27 - 1,22 (m, 6H).
Etapa E: (2R,5S)-4-[7-(3-h¡drox¡-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1-2.5-d¡met¡lp¡peraz¡n-1-carbox¡lato de fere-butilo. Una mezcla de (2R,5S)-4-[7-(3-bencilox¡-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2,5-d¡metil-p¡peraz¡n-1-carbox¡lato de fere-butilo (0,90 g, 1,30 mmol, 1,00 eq.) y Pd/C (0,10 g, 10,0 % de pureza) en metanol (40,0 ml) se agitó a 25 °C durante 1 hora en una atmósfera de H2 a 103,42 kPa (15 psi). La mezcla se filtró y se concentró al vacío para dar (2 R,5S)-4-[7-(3-hidroxi-1-naftil) - 2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-]p¡r¡m¡din-4-¡l]-2,5-d¡met¡lpiperazin-1-carboxilato de fere-butilo (0,72 g, bruto) en forma de un sólido de color amarillo que se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 603.
Etapa F: 4-[4-[(2S,5R)-2,5-d¡met¡lp¡peraz¡n-1-¡l1-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-7-¡l1naftalen-2-ol. Una mezcla de (2R,5S)-4-[7-(3-h¡drox¡-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrolid¡n-2-¡l]metox¡]-6.8- dihidro-5H p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1-2,5-d¡met¡l-piperazin-1 -carboxilato de fere-butilo (0,70 g, bruto) y TFA (1,99 g, 17,42 mmol, 1,29 ml) en diclorometano (1,20 ml) se agitó a 25 °C durante 0,5 h. La mezcla se concentró al vacío para dar 4-[4-[(2S,5R)-2,5-d¡met¡lp¡peraz¡n-1-¡l]-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡hidro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-7-¡l]naftalen-2-ol (849 mg, bruto, 2TFA) en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 503.
Etapa G: 1-[(2R,5S)-4-[7-(3-h¡drox¡-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1-2,5-d¡met¡l-p¡peraz¡n-1-¡l1prop-2-en-1-ona. A una solución de 4-[4-[(2S,5R)-2,5-dimet¡lp¡peraz¡n-1 -il]-2-[[(2S)-1 -metilpirrolidin-2
il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]naftalen-2-ol (0,84 g, bruto, 2TFA) y TEA (1,16 g, 11,5 mmol, 1,60 ml) en diclorometano (3,00 ml) se le añadió prop-2-enoato de prop-2-enoílo (102 mg, 805 umol) a -40 °C. Después de agitar a -40 °C durante 0,5 h, la mezcla se inactivó con agua (0,10 ml) y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de TFA)-acetonitrilo] y HPLC prep. (columna: Phenomenex Luna Fenil-Hexilo 150_30_5um; fase móvil: [agua (NH4HC0310 mM) - ACN]; % de B: 45 % - 75 %, 3 min). La fracción deseada se recogió y se liofilizó para dar el compuesto del título 1 -[(2R,5S)-4-[7-(3-hidroxi-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-2,5-dimetil-piperazin-1-il]prop-2-en-1-ona (EjEm PLo 336, 55,8 mg, 94,1 umol, tres etapas 8,2 % de rendimiento, 93,9 % de pureza) en forma de un sólido de color blanquecino. LCMS [ESI, M+1]: 557.
SFC: “ Columna: Chiralpak AS-350 * 4,6 mm D.I., 3 um, Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 %, Caudal: 3 ml/min, Longitud de onda: 220 nm” .
1H RMN (400 MHz, cloroformo-d) 5 = 7,97 (d a, J = 8,0 Hz, 1H), 7,63 (d, J = 8,0 Hz, 1H), 7,40 - 7,34 (m, 1H), 7,26 (dt, J = 1,6, 8,4 Hz, 1H), 6,90 (d, J = 1,6 Hz, 1H), 6,65 (s a, 1H), 6,61 - 6,43 (m, 1H), 6,39 - 6,26 (m, 1H), 5,73 (t a, J = 8,8 Hz, 1H), 4,83 (s a, 1H), 4,55 (d a, J = 5,6 Hz, 1H), 4,43 - 4,04 (m, 4H), 3,91 (d a, J = 17,2 Hz, 1H), 3,77 - 3,26 (m, 5H), 3,19 (t a, J = 8,0 Hz, 1H), 3,06 - 2,68 (m, 3H), 2,63 (s a, 3H), 2,57 (d a, J = 13,2 Hz, 1H), 2,41 - 2,33 (m, 1H), 2,15 - 1,83 (m, 4H), 1,11 - 0,96 (m, 6H).
Ejemplo 337

2-[(2S)-4-[7-(3-cloro-2-etil-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Etapa 1: 1-(2-bromo-6-cloro-fenil)etanol. En un matraz de fondo redondo de 100 ml secado a la llama equipado con una barra de agitación magnética y en una atmósfera de N2 se añadieron 2-bromo-6-cloro-benzaldehído (10,0 g, 45,6 mmol, 1 eq.) y THF (100 ml). La mezcla se enfrió a 0 °C y después se añadió gota a gota MeMgBr (3 M en Et20, 22,8 ml, 1,5 eq.) durante 20 min. La suspensión resultante se agitó a 0 °C durante 2 h. La mezcla de reacción se inactivó mediante la adición gota a gota de una solución acuosa saturada de NH4Cl (10 ml). La mezcla se enfrió, se vertió en un embudo de decantación de 100 ml que contenía agua (150 ml) y la mezcla se extrajo con acetato de etilo (2 x 150 ml). Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre MgS04, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice (éter de petróleo/acetato de etilo = 100/1 a 10/1). El compuesto 1-(2-bromo-6-cloro-fenil)etanol (8,00 g, 32,6 mmol, 72 % de rendimiento, 96 % de pureza) se obtuvo en forma de un aceite de color amarillo. LCMS [ESI, M-17]: 219.
1H RMN (400 MHz, cloroformo-d) 6 = 7,46 (d a, J = 8,0 Hz, 1H), 7,31 (d a, J = 8,0 Hz, 1H), 7,07 - 6,96 (m, 1H), 5,68 -5,44 (m, 1H), 3,24 (d a, J = 8,0 Hz, 1H), 1,61 (d, J = 4,4 Hz, 3H).
Etapa 2: 1 -bromo-3-cloro-2-etil-benceno. A una mezcla de 1-(2-bromo-6-clorofenil)etanol (8,00 g, 34,0 mmol, 1 eq.) en DCE (150 ml) se le añadieron poco a poco BF3.Et20 (14,5 g, 102 mmol, 12,6 ml, 3 eq.) y Et3SiH (19,8 g, 170 mmol, 27,1 ml, 5 eq.) a 0 °C. La mezcla se agitó a 50 °C durante 6 horas. La mezcla de reacción se lavó con NaHC03 acuoso saturado (2 x 75 ml) y salmuera saturada (1 x 75 ml), después se secó sobre Na2S04, se filtró y se concentró al vacío.
El residuo se purificó por cromatografía sobre gel de sílice (éter de petróleo). El residuo se purificó por HPLC prep.
(columna: Phenomenex Synergi Max-RP 250 * 50 mm * 10 um;fase móvil: [agua (0,225 % de AF) - ACN]; % de B:
65 % de ACN - 95 % de ACN, 30 min; 60 % min). El compuesto 1-bromo-3-cloro-2-etil-benceno (1,50 g, 6,83 mmol, 20 % de rendimiento, 100 % de pureza) se obtuvo en forma de un aceite de color amarillo.
Etapa A: (2S)-4-[7-(3-cloro-2-etil-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianomet¡l)p¡peraz¡n-1-carbox¡lato de tere-butilo. Una mezcla de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pynimidin-4-il]piperazin-1-carboxilato de tere-butilo (Intermedio 66, 300 mg, 636 umol, 1 eq.), 1-bromo-3-cloro-2-etil-benceno (279 mg, 1,27 mmol, 2 eq.), Pd2(dba)3 (117 mg, 127 umol, 0,2 eq.), RuPhos (89,1 mg, 191 umol, 0,3 eq.) y Cs2c03 (622 mg, 1,91 mmol, 3 eq.) en tolueno (30 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 6 horas en una atmósfera de N2. A la mezcla de reacción se le añadieron H20 (1 x 200 ml) y acetato de etilo (1 x 250 ml). La fase orgánica se separó, se lavó con salmuera (1 x 200 ml), se secó sobre sulfato de sodio, se filtró y se concentró a presión reducida para dar un residuo.
El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de FA)/acetonitrilo]. Las fracciones deseadas se recogieron y se neutralizaron a pH = 7 con una solución saturada de NaHC03 y se extrajeron con acetato de etilo (1 x 200 ml). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. Se obtuvo (2S)-4-[7-(3-cloro-2-etil-fenil)-2-[[(2s)-1 -metilpirrolidin- 2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (155 mg, 0,254 mmol, 100 % de pureza, 40 % de rendimiento) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 610.
1H RMN (400 MHz, cloroformo-d) 6 = 7,20 - 7,16 (m, 1H), 7,13 (t, J = 8,0 Hz, 1H), 7,07 - 7,03 (m, 1H), 4,61 (s a, 1H), 4,39 (dd, J = 5,2, 10,6 Hz, 1H), 4,16 (dd, J = 6,8, 10,4 Hz, 1H), 4,09 - 3,95 (m, 4H), 3,91 (d a, J = 13,2 Hz, 1H), 3,26 (dd, J = 3,6, 13,7 Hz, 1H), 3,22 - 2,96 (m, 5H), 2,90 (c, J = 7,4 Hz, 2H), 2,84 - 2,63 (m, 5H), 2,48 (s, 3H), 2,35 - 2,23 (m, 1H), 2,13 - 2,00 (m, 1H), 1,83 - 1,70 (m, 3H), 1,52 (s, 9H), 1,22 (t, J = 7,4 Hz, 3H).
Etapa B: 2-[(2S)-4-[7-(3-cloro-2-etil-fenil)-2-[[(2-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una solución de (2S)-4-[7-(3-cloro-2-etil-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (310 mg, 508 umol, 1 eq.) en DCM (600 ul) se le añadió TFA (869 mg, 7,62 mmol, 564 ul, 15 eq.) a 0 °C en una atmósfera de nitrógeno. La mezcla se agitó a 25 °C durante 1 h. Después de eso, la mezcla se concentró al vacío. Se obtuvo 2-[(2S)-4-[7-(3-cloro-2-etilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (850 mg, bruto, 2 TFA) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 510.
Etapa C: 2-[(2S)-4-[7-(3-cloro-2-etil-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una mezcla de 2-[(2S)-4-[7-(3-cloro-2-etil-fenil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (850 mg, bruto, 2 TFA) y cloruro de prop-2-enoílo (59,8 mg, 660 umol, 53,8 ul) en DCM (8 ml) se le añadió en una porción TEA (514 mg, 5 ,08 mmol, 707 ul). La mezcla se agitó a -40 °C durante 30 min. La mezcla de reacción se inactivó con una solución saturada de NaHC03 (2 ml) a 0 °C y después se extrajo con CH2Cl2 (2 x 30 ml). Las capas orgánicas combinadas se lavaron con salmuera (1 x 30 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío para dar un residuo. El residuo se purificó por cromatografía en columna (Ah03; acetato de etilo/metanol = 20/1 a 3/1). El residuo se purificó por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,04 % de NH3^H20) - ACN]; % de B: 65 % - 95 %, 10 min). El compuesto del título 2-[(2S)-4-[7-(3-cloro-2-etil-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (Ej Em Pl0 337, 142 mg, 252 umol, dos etapas 50 % de rendimiento, 100 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 564.
Condición de SFC: “Columna: Chiralpak AD-3100 x 4 ,6 mm D.I., 3 um, Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 %, Caudal: 3 ml/min, Longitud de onda: 220 nm” .
1H RMN (400 MHz, dmso-da) 5 = 7,21 (s, 3H), 6,78 (dd, J = 10,4, 16,8 Hz, 1H), 6,15 (dd, J = 2,0, 16,8 Hz, 1H), 5,74 (dd, J = 2,0, 10,6 Hz, 1H), 4,84 (s a, 1H), 4,28 (dd, J = 5,2, 10,8 Hz, 1H), 4,23 - 4,07 (m, 2H), 4,05 - 3,90 (m, 4H), 3,39 (s a, 1H), 3,27 (dd a, J = 3,6, 13,6 Hz, 1H), 3,20 - 3,05 (m, 3H), 2,99 - 2,93 (m, 2H), 2,93 - 2,76 (m, 4H), 2,61 - 2,52 (m, 2H), 2,40 - 2,31 (m, 3H), 2,22 (c, J = 8,4 Hz, 1H), 2,00 - 1,87 (m, 1H), 1,77 - 1,54 (m, 3H), 1,19 (t, J = 7,2 Hz, 3H).
Ejemplo 338

2-((S)-1-acr¡lo¡l-4-(7-(8-fluoronaftalen-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-il)piperazin-2-il)acetonitrilo

Inserc¡ón: 1 -bromo-8-fluoro-naftaleno. A una soluc¡ón de 1,8-d¡bromonaftaleno (2 g, 6,99 mmol, 1 eq.) en THF (50 ml) se le añad¡ó gota a gota n-Bul¡ (2,5 M en hexano, 4,20 ml, 1,5 eq.) a -78 °C. Después de ag¡tar durante 10 m¡nutos a -78 °C, se añad¡ó gota a gota una soluc¡ón de N-(bencenosulfon¡l)-W-fluoro-bencenosulfonam¡da (4,41 g, 13,9 mmol, 2 eq.) en THF (10 ml). La mezcla se calentó a 25 °C y se agitó durante 3 horas. La mezcla de reacc¡ón se ¡nact¡vó con agua (10 ml) y se extrajo con acetato de etilo (30 ml * 3). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (50 ml), se secaron sobre Na2SÜ4, se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar un res¡duo. El res¡duo se pur¡f¡có por HPLC prep. (columna: Phenomenex Synerg¡ Max-RP 250 * 50 mm * 10 um; fase móv¡l: [agua (0,225 % de AF) - ACN]; % de B: 50 % - 75 %, 25 m¡n). La mezcla se ajustó a pH = 7 med¡ante la ad¡c¡ón de una soluc¡ón acuosa saturada de NaHCÜ3 y se extrajo con acetato de et¡lo (50 ml * 3). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se f¡ltraron y se concentraron a pres¡ón reduc¡da. Se obtuvo 1-bromo-8-fluoro-naftaleno (560 mg, 2,46 mmol, 35 % de rend¡m¡ento, 99 % de pureza) en forma de un sól¡do de color amar¡llo.1
1H RMN (400 MHz, cloroformo-d) 5 = 7,81 (d, J = 7,2 Hz, 2H), 7,66 (d, J = 8,4 Hz, 1H), 7,49 - 7,39 (m, 1H), 7,31 (t, J = 7,6 Hz, 1H), 7,23 (td, J = 0,8, 5,2 Hz, 1H).
Etapa A: (2S)-2-(c¡anomet¡l)-4-[7-(8-fluoro-1-naft¡l)-2-[[(2S)-1-met¡l1p¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡rido[3.4-d1p¡r¡m¡d¡n-4-¡l1piperaz¡n-1-carbox¡lato de tere-butilo. Una mezcla de 1-bromo-8-fluoro-naftaleno (310 mg. 1.38 mmol. 1.3 eq.), (2S)-2-(c¡anomet¡l)-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de tere-butilo (Intermed¡o 66. 500 mg. 1.06 mmol. 1 eq.). CS2C03 (863 mg. 2.65 mmol. 2.5 eq.). RuPhos (98.9 mg.212 umol. 0.2 eq.) y Pd2(dba)3 (194 mg.212 umol. 0.2 eq.) en tolueno (10 ml) se desgas¡f¡có y se purgó 3 veces con N2 y después la mezcla se agitó a 100 °C durante 3 horas en una atmósfera de N2. La mezcla de reacc¡ón se diluyó con agua (20 ml) y se extrajo con acetato de et¡lo (20 ml x 3). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (20 ml). se secaron sobre Na2S04. se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar un res¡duo. El res¡duo se pur¡f¡có por cromatografía ultrarrápida de fase ¡nversa [agua (0.1 % de ác¡do fórm¡co)/aceton¡tr¡lo)]. Las fracc¡ones deseadas se ajustaron pH = 7 con una soluc¡ón acuosa saturada de NaHC03 y se extrajeron con acetato de etilo (20 ml x 3). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (20 ml). se secaron sobre Na2S04. se f¡ltraron y se concentraron a pres¡ón reduc¡da. El compuesto (2S)-2-(c¡anometil)-4-[7-(8-fluoro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (370 mg. 570 umol. 54 % de rend¡m¡ento. 95 % de pureza) se obtuvo en forma de un sólido de color amar¡llo. LCMS [ESI. M+11: 616.
1H RMN (400 MHz. cloroformo-d) ó = 7.64 (d. J = 7.6 Hz. 1H). 7.56 (d a. J = 8.8 Hz. 1H). 7.47 - 7.42 (m. 1H). 7.20 -7.08 (m. 3H). 4.64 (s a. 1H). 4.47 - 4.27 (m. 2H). 4.23 - 4.14 (m. 2H). 4.11 - 3.82 (m. 3H). 3.81 - 3.65 (m. 1H). 3.44 -3.23 (m. 2H). 3.19 - 3.07 (m. 2H). 3.02 - 2.79 (m. 3H). 2.77 - 2.66 (m. 2H). 2.56 (s a. 1H). 2.50 (s. 3H). 2.34-2.25 (m.
1H). 2.13 - 2.06 (m. 1H). 1.82 - 1.72 (m. 3H). 1.53 (s. 9H).
Etapa B: 2-[(2S)-4-[7-(8-fluoro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡din-4-¡l1piperaz¡n-2-¡l1aceton¡tr¡lo. A una solución de (2S)-2-(cianometil)-4-[7-(8-fluoro-1-naftil)-2-[[(2S)-1-metilp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1pir¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de tere-butilo (370 mg. 600 umol. 1 eq.) en DCM (600 ul) se le añadió TFA (1.03 g. 9.01 mmol. 667 ul. 15 eq.). La mezcla se agitó a 25 °C durante 1 hora. La mezcla de reacción se concentró al vacío. El compuesto 2-[(2S)-4-[7-(8-fluoro-1-naftil)-2-[[(2S)-1-metilp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡din-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo (378 mg. 600 umol. 99 % de rendimiento.
TFA) se obtuvo en forma de un aceite de color amarillo y se usó directamente en la siguiente etapa sin purificación.
LCMS [ESI. M+11: 516.
Etapa C: 2-[(2S)-4-[7-(8-fluoro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-pir¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-1- prop-2-enoil-p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una solución de 2-[(2S)-4-[7-(8-fluoro-1-naftil)-2-[[(2S)- 1 -metilpirrolidin-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡din-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo (261 mg. 506 umol. 1 eq.. TfA) en DCM (5 ml) se le añadió TEA (512 mg. 5.07 mmol. 705 ul. 10 eq.) a 0 °C. Después de la adición. se añadió gota a gota prop-2- enoato de prop-2-enoílo (51.1 mg. 405 umol. 0.8 eq.) en DCM (1 ml) a 0 °C. Después de agitar a 25 °C durante 1 hora. la mezcla de reacción se inactivó con una solución acuosa saturada de NaHC03 (1 ml) y se extrajo con acetato de etilo (20 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (20 ml). se secaron sobre Na2S04. se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna:
Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0.05 % de hidróxido de amoníaco v/v) - ACN1; % de B:
48 % - 78 %. 12 min). El compuesto del título 2-[(2S)-4-[7-(8-fluoro-1-naft¡l)-2-[[(2S)-1-metilp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-il1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1aceton¡tr¡lo (EJEMPL0 338. 93.5 mg. 163 umol.
32 % de rendimiento. 99.4 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI. M+11: 570.
Condición de SFC: “ 0D-3_Me0H (DEA)_40_3 ml-35T Columna: Chiralcel 0D-350 x 4.6 mm D.I.. 3 um Fase móvil:
40 % de metanol (0.05 % de DEA) en C02 Caudal: 3 ml/min Longitud de onda: 220 nm” .
1H RMN (400 MHz. cloroformo-d) ó = 7,64 (d, J = 8.0 Hz. 1H). 7.56 (d. J = 8.4 Hz. 1H). 7.47 - 7.35 (m. 2H). 7.19-7.09 (m. 2H). 6.60 (s a. 1H). 6.40 (dd. J = 1.6. 16.8 Hz. 1H). 5.83 (d a. J = 10.4 Hz. 1H). 5.30 - 4.48 (m. 1H). 4.46 - 4.28 (m.
2H). 4.27 - 3.85 (m. 5H). 3.73 (s a. 1H). 3.56 - 2.54 (m. 10H). 2.49 (s. 3H). 2.37 - 2.21 (m. 1H). 2.12 - 1.97 (m. 1H).
1.95 - 1.79 (m. 3H).
Ejemplo 339

2-((S)-1-acriloil-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(((S)-1-etilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (2S)-2-(benciloximetil)pirrolidin-1-carboxilato de tere-butilo. A una solución de NaH (1,19 g, 49,6 mmol, 2,0 eq.) en THF (50,0 ml) se le añadió una solución de (2S)-2-(hidroximetil)pirrolidin-1-carboxilato de tere-butilo (5,00 g, 24,8 mmol, 1,0 eq.) en THF (50,0 ml) a 0 °C y la mezcla se agitó a 10 °C durante 1 hora. A la mezcla anterior se le añadió gota a gota bromometilbenceno (6,38 g, 37,3 mmol, 4,43 ml, 1,50 eq.) a 0 °C y la mezcla se agitó a 10 °C durante 16 horas. Después de que se completara, la mezcla se inactivó con NH4Cl acuoso saturado (20,0 ml) y se diluyó con acetato de etilo (50,0 ml). La capa orgánica separada se lavó con salmuera (1 * 30,0 ml) y se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El producto obtenido se purificó por cromatografía en columna (SiÜ2, éter de petróleo:acetato de etilo = 10:1 - 3:1) para dar (2S)-2-(benciloximetil)pirrolidin-1-carboxilato de tere-butilo (6,60 g, 10,4 mmol, 42 % de rendimiento) en forma de un aceite de color amarillo.
1H RMN (400 MHz, Metanol-d4) 57,44 - 7,26 (m, 5H), 4,53 (s, 2H), 4,00 - 3,85 (m, 1H), 3,61 - 3,39 (m, 2H), 3,38 - 3,33 (m, 2H), 2,04 - 1,77 (m, 4H), 1,50 - 1,36 (m, 9H).
Etapa B: (2S)-2-(benciloximetil)pirrolidina. A una solución de (2S)-2-(benciloximetil)pirrolidin-1-carboxilato de terebutilo (6,60 g, 22,7 mmol, 1,0 eq.) en DCM (23,0 ml) se le añadió gota a gota TFA (34,7 g, 304 mmol, 22,5 ml, 13,4 eq.) a 0 °C en una atmósfera de nitrógeno. La mezcla se agitó a 15 °C durante 1 hora. Después de que se completara, la mezcla se concentró al vacío para dar (2S)-2-(benciloximetil)pirrolidina (10,0 g, bruto, TFA) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 192.
Etapa C: (2S)-2-(benciloximetil)-1-etilpirrolidina. A una solución de (2S)-2-(benciloximetil)pirrolidina (2,0 g, 6,55 mmol, 1,0 eq., TFA) y acetaldehído (1,44 g, 32,8 mmol, 1,84 ml, 5,0 eq.) en Me0H (30,0 ml) se le añadieron CH3C00H (787 mg, 13,1 mmol, 749 ul, 2,0 eq.) y NaBH3CN (1,65 g, 26,2 mmol, 4,0 eq.). La mezcla se agitó a 20 °C durante 2 horas. Después de que se completara, el pH se ajustó a 3 con HCl acuoso 1 M (15,0 ml) y se concentró al vacío para eliminar el Me0H. La mezcla se extrajo con disolvente mixto (EP/AE = 10:1, 2 * 60,0 ml). La fase acuosa se ajustó a pH ~9 con Na2C03 acuoso saturado (10,0 ml) y se extrajo con disolvente mixto (AE/Me0H = 10:1, 4 * 60,0 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío. El producto obtenido se purificó por
cromatografía ultrarrápida de fase inversa (condición de 0,1 % de TFA) para dar (2S)-2-(benciloximetil)-1 -etilpirrolidina (707 mg, 3,18 mmol, 49 % de rendimiento, 99,0 % de pureza) en forma de un aceite incoloro.
1H RMN (400 MHz, Cloroformo-d) ó 7,36 - 7,31 (m, 4H), 7,31 - 7,26 (m, 1H), 4,58 - 4,54 (s, 2H), 3,52 (dd, J = 4,8, 9,2 Hz, 1H), 3,37 (dd, J = 6,4, 9,2 Hz, 1H), 3,20 - 3,12 (m, 1H), 3,01 - 2,89 (m, 1H), 2,66 - 2,58 (m, 1H), 2,35 - 2,25 (m, 1H), 2,21 - 2,05 (m, 1H), 2,03 - 1,87 (m, 1H), 1,83 - 1,62 (m, 3H), 1,11 (t, J = 7,2 Hz, 3H).
Etapa D: [(2S)-1-etilp¡rrol¡d¡n-2-¡l1metanol. A una solución de (2S)-2-(benciloximetil)-1-etil-pirrolidina (860 mg, 3,92 mmol, 1,00 eq.) en Me0H (3,00 ml) se le añadieron HChMeoH (4 M, 0,80 ml) y pd(0H)2/C (300 mg, 20 % de pureza). La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (344,74 kPa (50 psi)) a 25 °C durante 16 horas. Después de que se completara, la mezcla se filtró y se concentró.
El producto [(2S)-1-etilpirrolidin-2-il1metanol (460 mg, 3,56 mmol, 91 % de rendimiento) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 130.
Etapa E: (2S)-4-[7-[3-cloro-2-(trifluorometil)fenil1-2-[[(2S)-1-etilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de ferc-butilo. A una solución de (2S)-4-[7-[3-cloro-2-(trifluorometil)fenil1-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (400 mg, 668 umol, 1,0 eq.) y [(2S)-1-etilpirrolidin-2-il]metanol (173 mg, 1,34 mmol, 2,0 eq.) en tolueno (8,0 ml) se le añadió t-Bu0Na (128 mg, 1,34 mmol, 2,0 eq.) a 0 °C y la reacción se agitó a 0 °C durante 0,5 horas.
Después de que se completara, a la mezcla de reacción se le añadió agua (30,0 ml), después se extrajo con AE (2 * 20,0 ml) y la capa orgánica combinada se lavó con salmuera saturada (1 * 30,0 ml), después se secó sobre Na2S04, se filtró y se concentró. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 5/1 a acetato de etilo/metanol = 20/1) para dar (2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S)-1-etilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]-2-(cianometil)pipenazin-1-carboxilato de ferc-butilo (240 mg, 353 umol, 53 % de rendimiento, 97,6 % de pureza) en forma de un sólido de color amarillo claro. LCMS [ESI, M+1]: 664.
Etapa F: 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil1-2-[[(2S)-1-etilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una solución de (2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S)-1 -etilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (240 mg, 361 umol, 1,0 eq.) en DCM (2,0 ml) se le añadió tFa (3,08 g, 27,0 mmol, 2,0 ml, 74,8 eq.) y la mezcla se agitó a 20 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se concentró para dar 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S)-1-etilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il1piperazin-2-il]acetonitrilo (280 mg, bruto, 2TFA) en forma de un aceite de color amarillo que se usó para la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 564.
Etapa G: 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil1-2-[[(2S)-1-etil1pirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una mezcla de 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S)-1-etilpirolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il1piperazin-2-il]acetonitrilo (280 mg, 496 umol, 1,0 eq., 2TFA) en DCM (5,0 ml) se le añadieron DIEA (642 mg, 4,96 mmol, 865 ul, 10,0 eq.) y prop-2-enoato de prop-2-enoílo (62,6 mg, 496 umol, 1,0 eq.) a 0 °C y la mezcla de reacción se agitó a 20 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se inactivó con Me0H (5,0 ml) y se concentró. El residuo se purificó por cromatografía en columna (Base Ah03, acetato de etilo/metanol = 20/1), después el producto bruto se concentró y se purificó de nuevo por HPLC prep. ((columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN]; % de B: 55 %-85 %, 12 min) y el producto obtenido se concentró y después se sometió a liofilización.
El compuesto del título 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S)-1 -etilpirroMdin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1 -prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMμL0 339, 31,9 mg, 50,2 umol, 10 % de rendimiento, 97,2 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 618.
1H RMN (400 MHz, Cloroformo-d) ó 7,37 - 7,29 (m, 1H), 7,23 - 7,18 (m, 1H), 7,11 (d, J = 8,0 Hz, 1H), 6,58 - 6,46 (m, 1H), 6,32 (dd, J = 0,8, 16,4 Hz, 1H), 5,76 (d a, J = 10,4 Hz, 1H), 5,11 - 4,37 (m, 1H), 4,28 (dd, J = 4,8, 10,4 Hz, 1H), 4,09 - 3,96 (m, 4H), 3,89 (d a, J = 12,4 Hz, 2H), 3,69 - 3,41 (m, 1H), 3,33 - 2,51 (m, 12H), 2,38 - 2,28 (m, 1H), 2,20 -2,11 (m, 1H), 2,00 - 1,80 (m, 3H), 1,06 (t, J = 7,2 Hz, 3H).
Ejemplo 340

2-((S)-1-acriloil-4-(7-(4-metoxipiridin-3-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[7-(4-metoxi-3-piridil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de tere-butilo. Una mezcla de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (580 mg, 1,23 mmol, 1,0 eq.), 3-bromo-4-metoxi-piridina (694 mg, 3,69 mmol, 3,0 eq.), Pd2(dba)3 (113 mg, 123 umol, 0,1 eq.), RuPhos (115 mg, 246 umol, 0,2 eq.) y CS2C03 (1,20 g, 3,69 mmol, 3,0 eq.) en tolueno (25,0 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 6 horas en una atmósfera de N2. Después de que se completara, el disolvente orgánico se eliminó al vacío y se lavó con agua (20,0 ml). La fase acuosa se extrajo con acetato de etilo (2 x 30,0 ml). Los extractos combinados se lavaron con salmuera (100 ml), se secaron sobre Na2S04 y después el disolvente se eliminó al vacío. El residuo se purificó por HPLC ultrarrápida de fase inversa [C18, 0,1 % de AF en agua, 0- 70 % de MeCN]. El producto obtenido se ajustó con NaHC03 acuoso saturado a pH ~8, después se concentró, la capa acuosa se extrajo con acetato de etilo (2 x 50,0 ml) y la capa orgánica combinada se secó sobre Na2S04, se filtró y se concentró. El compuesto (2S)-2-(cianometil)-4-[7-(4-metoxi-3-piridil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (230 mg, 397 umol, 32 % de rendimiento, 100 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 579.
Etapa B: 2-[(2S)-4-[7-(4-metoxi-3-piridil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-i n piperazin-2-i n acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(4-metoxi-3-piridil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (350 mg, 605 umol, 1,0 eq.) en dioxano (3,0 ml) se le añadió HCl 4 M/dioxano (5,0 ml) y la mezcla se agitó a 20 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se concentró, después se extrajo con DCM (10,0 ml), la capa orgánica se lavó con NaHC03 acuoso saturado (2 x 10 ,0 ml) y la capa orgánica se secó sobre Na2S04, se filtró y se concentró para dar 2-[(2S)-4-[7-(4-metoxi-3-piridil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (260 mg, bruto) en forma de un aceite de color amarillo que se usó para la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 479.
Etapa C: 2-[(2S)-4-[7-(4-metoxi-3-piridil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]-1- prop-2-enoil-piperazin-2-il]acetonitrilo. A una mezcla de 2-[(2S)-4-[7-(4-metoxi-3-piridil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (260 mg, 543 umol, 1,0 eq.) en DCM (5,0 ml) se le añadieron TEA (220 mg, 2,17 mmol, 302 ul, 4,0 eq.) y cloruro de prop-2-enoílo (63,9 mg, 706 umol, 57,6 ul, 1,3 eq.) a -60 °C y la mezcla de reacción se agitó a -60 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se inactivó con Me0H (2,0 ml) y se concentró. El residuo se purificó por cromatografía en columna (Base Al203, éter de petróleo : acetato de etilo = 3:1 a acetato de etilo/metanol = 20/1), después el producto bruto se concentró y se purificó de nuevo por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,04 % de NH3H20)-ACN]; % de B: 30 %-54 %,10 min) y el producto obtenido se concentró y después se sometió a liofilización. El compuesto del título 2-[(2S)-4-[7-(4-metoxi-3-piridil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (Ej Em Pl0340, 83,6 mg, 155 umol, 29 % de rendimiento, 98,8 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 533.
1H RMN (400 MHz, Cloroformo-d) 58,25 (d, J = 5,2 Hz, 1H), 8,17 (s, 1H), 6,82 (d, J = 5,2 Hz, 1H), 6,38 (dd, J = 1,2, 16,8 Hz, 1H), 6,44 - 6,33 (m, 1H), 5,82 (d a, J = 10,4 Hz, 1H), 5,21 - 4,52 (m, 1H), 4,37 (dd, J = 4,8 Hz, 10,4 Hz, 1H), 4,33 - 4,26 (m, 1H), 4,24 - 4,13 (m, 2H), 4,06 (d a, J = 13,6 Hz, 1H), 4,02 - 3,88 (m, 5H), 3,72 - 3,37 (m, 2H), 3,36 -3,23 (m, 2H), 3,16 - 3,02 (m, 2H), 2,98 - 2,88 (m, 1H), 2,87 - 2,72 (m, 3H), 2,70 - 2,60 (m, 1H), 2,47 (s, 3H), 2,33 - 2,27 (m, 1H), 2,10 - 1,99 (m, 1H), 1,91 - 1,69 (m, 3H).
Ejemplo 341

2-((S)-1-acriloil-4-(2-(((S)-1-(2-hidroxietil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

[(2S)-1-[2-[fe/'c-butil(dimetil)silil1oxietil1pirrolidin-2-il1metanol. A una mezcla de [(2S)-pirrolidin-2-il]metanol (500 mg, 4,94 mmol, 481 ul, 1 eq.) y 2-bromoetoxi-íerc-butil-dimetil-silano (1,30 g, 5,44 mmol, 1,1 eq.) en MeCN (30 ml) se le añadió en una porción K2C03 (3,42 g, 24,7 mmol, 5 eq.) en una atmósfera de N2. La mezcla se agitó a 50 °C durante 12 horas. La mezcla de reacción se filtró y el filtrado se concentró a presión reducida para dar un residuo.
El residuo se purificó por cromatografía en columna (AI203, acetato de etilo/metanol = 100/1 a 5/1). El compuesto [(2S)-1-[2-[ferc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metanol (437 mg, 1,68 mmol, 34,0 % de rendimiento) se obtuvo en forma de un aceite de color pardo.
1H RMN (400 MHz, cloroformo-d) ó = 3,71 (dd, J = 5,2, 6,4 Hz, 2H), 3,63 - 3,58 (m, 1H), 3,37 (dd, J = 3,2, 10,8 Hz, 1H), 3,21 (td, J = 4,8, 10,0 Hz, 1H), 2,90 (td, J = 6,4, 12,8 Hz, 1H), 2,74 - 2,67 (m, 1H), 2,52 (td, J = 5,2, 10,4 Hz, 1H), 2,41 - 2,31 (m, 1H), 1,97 - 1,80 (m, 1H), 1,79 - 1,55 (m, 3H), 0,92 - 0,87 (m, 9H), 0,11 - 0,00 (m, 6 H).
Etapa A: (2S)-4-[2-[[(2S)-1-[2-[ferc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l1-2-(cianomet¡l)p¡peraz¡n-1-carbox¡lato de bencilo. A una mezcla de [(2S)-1-[2-[ferc-butil(dimetil)silil]oxietil] pirrolidin-2-il]metanol (447 mg, 1,72 mmol, 2 eq.) en tolueno (35 ml) se le añadió en una porción f-Bu0Na (248 mg, 2,58 mmol, 3 eq.) y después a la solución se le añadió (2S)-2-(cianometil)-4-[2-metilsulfinil-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (Intermedio 66 , 500 mg, 861 umol, 1 eq.) a 0 °C. La mezcla se calentó a 25 °C y se agitó durante 1 hora. La mezcla de reacción se diluyó con 20 ml de acetato de etilo, se ajustó a pH 8-9 con HCl 2 M a 0 °C y después se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se lavaron con agua (15 ml x 1 ), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si0s, éter de petróleo/acetato de etilo = 100/1 a 1/2). El compuesto (2S)-4-[2-[[(2S)-1-[2-[terc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (400 mg, 494 umol, 57,4 % de rendimiento, 95,8 % de pureza) se obtuvo en forma de un aceite incoloro. LCMS [ESI, M+1]: 776.
Etapa B: 2-[(2S)-4-[2-[[(2S)-1-[2-[ferc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una solución de (2S)-4-[2-[[(2S)-1-[2-[ferc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (380 mg, 490 umol, 1 eq.) en Me0H (30 ml) se le añadió Pd/C (230 mg, 490 umol, 10 % de pureza, 1,00 eq.) y NH3^Me0H (25 ml). La suspensión se desgasificó al vacío y se purgó tres veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. La mezcla de reacción se filtró y se concentró a presión reducida para dar un residuo. El compuesto 2-[(2S)-4-[2-[[(2S)-1-[2-[ferc-butil(dimetil)silil]oxietil] pirrolidin-2-il] metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (314 mg, 489 umol, 99,9 % de rendimiento) se obtuvo en forma de un aceite de color amarillo y se usó directamente en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 642.
Etapa C: 2-[(2S)-4-[2-[[(2S)-1-[2-[ferc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l)-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1acetonitr¡lo. A una mezcla de 2-[(2S)-4-[2-[[(2S)-1-[2-[fercbutil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (314 mg, 489 umol, 1 eq.) en DCM (15 ml) se le añadieron TEA (148 mg, 1,47 mmol, 204 ul, 3 eq.) y cloruro de prop-2-enoílo (57,6 mg, 636 umol, 51,9 ul, 1,3 eq.) en una porción a -40 °C en una atmósfera de N2. La mezcla se agitó a -40 °C durante 30 min. La mezcla de reacción se inactivó mediante la adición de una solución saturada de NaHC03 de 3 ml a -40 °C y después se extrajo con DCM (20 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (15 ml x 1 ), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (AI203, éter de petróleo/acetato de etilo = 100/1 a 1/3). El compuesto 2-[(2S)-4-[2-[[(2s)-1-[2-[ferc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (210 mg, 300 umol, 61,4 % de rendimiento, 99,5 % de pureza) se obtuvo en forma de un aceite incoloro. LCMS [ESI, M+1]: 696.
Etapa D: 2-[(2S)-4-[2-[[(2S)-1-(2-hidroxietil)pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una mezcla de 2-[(2S)-4-[2-[[(2S)-1-[2-[ferc-butil(dimetil)silil]oxietil]pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-1-prop-2-enoil-piperazin-2-il]acetonitrilo (205 mg, 295 umol, 1 eq.) en THF (3 ml) se le añadieron en una porción KF (171 mg, 2,95 mmol, 69,0 ul, 10 eq.) y 18-corona-6 (779 mg, 2,95 mmol, 10 eq.). La mezcla se agitó a 40 °C durante 2 horas. La mezcla de reacción se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - ACN]; % de B: 45 % - 75 %, 12 min). El compuesto del título 2-[(2S)-4-[2-[[(2s )-1-(2-hidroxietil)pirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0341, 43,4 mg, 71,6 umol, 24,3 % de rendimiento, 95,9 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 582.
SFC: “0J-3S_3_5_40_3ML Columna: Chiralcel 0J-3 100 x 4 ,6 mm D.I., 3 um Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 % Caudal: 3 ml/min Longitud de onda: 220 nm” .
1H RMN (400 MHz, cloroformo-d) ó = 8,24 - 8,18 (m, 1H), 7,89 - 7,83 (m, 1H), 7,61 (d, J = 8,4 Hz, 1H), 7,54 - 7,47 (m, 2H), 7,44 (t, J = 7,2 Hz, 1H), 7,15 (d, J = 6,8 Hz, 1H), 6,60 (s a, 1H), 6,40 (dd, J = 1,6, 16,8 Hz, 1H), 5,84 (d a, J = 10,4 Hz, 1H), 5,11 (s a, 1H), 4,36 - 4,22 (m, 3H), 4,16 (dd, J = 6,8, 10,4 Hz, 2H), 4,01 (d a, J = 11,6 Hz, 1H), 3,72 -3,55 (m, 3H), 3,53 - 3,26 (m, 3H), 3,24 - 2,68 (m, 10H), 2,62 (td, J = 4,0, 12,5 Hz, 1H), 2,39 - 2,28 (m, 1H), 2,07 - 1,96 (m, 1H), 1,90 - 1,75 (m, 3H).
E je m p lo 342

2-[(2S)-1-[(E)-4-(dimetilamino)but-2-enoil]-4-[7-[3-fluoro-2-(trifluorometil)fenil]-2-[[(2S,4R)-4-hidroxi-1-metil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo. A una solución de 4-[(3S)-4-benciloxicarbonil-3- (cianometil)piperazin-1-il]-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (3,00 g, 5,57 mmol, 1,00 eq.) en dioxano (30,0 ml) se le añadió HCl/dioxano (4,00 M, 30,0 ml, 21,6 eq.) a 0 °C. Después de agitar a 25 °C durante 1 hora, la mezcla se concentró al vacío. El residuo se diluyó con agua (4,00 ml), se ajustó a pH >8 con bicarbonato de sodio saturado (10,0 ml) y se extrajo con acetato de etilo (3 x 30,0 ml). Los extractos se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna (Al2Ü3, metanol/acetato de etilo = 10/1) para dar (2S)-2-(cianometil)-4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (2,10 g, 4,02 mmol, 72 % de rendimiento) en forma de un sólido de color amarillo. LCMS [ESI, M+1 ]: 439.
1H RMN (400 MHz, cloroformo-d) 5 = 7,46 - 7,30 (m, 5H), 5,25 - 5,14 (m, 2H), 4,66 (s a, 1H), 4,14 - 4,05 (m, 1H), 4,04 - 3,90 (m, 3H), 3,84 (d a, J = 12,4 Hz, 1H), 3,26 (d a, J = 11,6 Hz, 2H), 3,12 (td, J = 5,2, 12,4 Hz, 1H), 3,05 - 2,91 (m, 2H), 2,79 (s a, 1H), 2,74 - 2,56 (m, 3H), 2,49 (s, 3H).
Etapa B: (2S)-2-(c¡anomet¡l)-4-[7-[3-fluoro-2-(tr¡fluoromet¡l)fen¡l1-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l)p¡perazin-1-carbox¡lato de bencilo. Una mezcla de (2S)-2-(c¡anomet¡l)-4-(2-met¡lsulfan¡l-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (2,00 g, 4,56 mmol, 1,00 eq.), 1-bromo-3-fluoro-2-(tr¡fluoromet¡l)benceno (1,66 g, 6,84 mmol, 1,50 eq.), Pd2(dba)3 (418 mg, 456 umol, 0,10 eq.), RuPhos (426 mg, 912 umol, 0,20 eq.) y Cs2C03 (2,97 g, 9,12 mmol, 2,00 eq.) en tolueno (30,0 ml) se ag¡tó a 90 °C durante 5 horas. La mezcla se d¡luyó con agua (20,0 ml) y se extrajo con acetato de et¡lo (3 * 30,0 ml). Las capas orgán¡cas se lavaron con salmuera (1 * 40,0 ml), se secaron sobre Na2S04, se f¡ltraron y se concentraron al vacío. El res¡duo se pur¡f¡có por cromatografía ultrarráp¡da de fase ¡nversa [agua (TFA, 0,10 %)/aceton¡tr¡lo]. Las fracc¡ones deseadas se recog¡eron y se ajustaron a pH >7 con b¡carbonato de sod¡o saturado (5,00 ml), se eXtrajeron con acetato de et¡lo (3 * 30,0 ml), se secaron sobre Na2S04, se f¡ltraron y se concentraron al vacío para dar (2S)-2-(c¡anomet¡l)-4-[7-[3-fluoro-2-(tr¡fluoromet¡l)fen¡l]-2-met¡lsulfan¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡ljp¡peraz¡n-1-carbox¡lato de benc¡lo (2,00 g, 2,90 mmol, 64 % de rend¡m¡ento) en forma de un sól¡do de color amar¡llo. lCMs [ESI, M+1]: 601.
1H RMN (400 MHz, cloroformo-d) 5 = 7,53 - 7,44 (m, 1H), 7,43 - 7,29 (m, 5H), 7,07 (d, J = 8,0 Hz, 1H), 7,00 - 6,91 (m, 1H), 5,26 - 5,13 (m, 2H), 4,69 (s a, 1H), 4,16 - 4,10 (m, 3H), 4,04 (d a, J = 13,2 Hz, 1H), 3,87 (d a, J = 12,4 Hz, 1H), 3,39 - 3,22 (m, 3H), 3,19 - 3,09 (m, 1H), 3,04 (dt, J = 3,6, 12,4 Hz, 1H), 2,83 (s a, 2H), 2,76 - 2,67 (m, 2H), 2,51 (s, 3H).
Etapa C: (2S)-2-(c¡anomet¡l)-4-[7-[3-fluoro-2-(tr¡fluoromet¡l)fen¡l1-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de benc¡lo. A una soluc¡ón de (2S)-2-(c¡anomet¡l)-4-[7-[3-fluoro-2-(tr¡fluoromet¡l)fen¡l]-2-met¡lsulfan¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (1,90 g, 3,16 mmol, 1,00 eq.) en acetato de et¡lo (50,0 ml) se le añad¡ó m-CPBA (610 mg, 3,01 mmol, 85 % de pureza, 0,95 eq.) a 0 °C. Después de ag¡tar a 0 °C durante 0,5 h, la mezcla se d¡luyó con agua (30,0 ml), se ajustó a pH >7 con b¡carbonato de sod¡o saturado (10,0 ml) y se extrajo con acetato de et¡lo (3 * 20,0 ml). Las capas orgán¡cas se lavaron con salmuera (1 * 40,0 ml), se secaron sobre Na2S04, se f¡ltraron y se concentraron al vacío. El res¡duo se pur¡f¡có por cromatografía en columna (S¡02, éter de petróleo/acetato de et¡lo = 1/3) para dar (2S)-2-(c¡anomet¡l)-4-[7-[3-fluoro-2-(tr¡fluoromet¡l)fen¡l]-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (1,20 g, 1,73 mmol, 55 % de rend¡m¡ento) en forma de un sól¡do de color amar¡llo. LCMS [ESI, M+1]: 617.
1H RMN (400 MHz, cloroformo-d) 5 = 7,55 - 7,47 (m, 1H), 7,44 - 7,33 (m, 5H),, 7,08 (d, J = 8,0 Hz, 1H), 7,03 - 6,94 (m, 1H), 5,20 (s, 2H), 4,68 (s a, 1H), 4,31 - 4,09 (m, 4H), 4,02 (d a, 12,0 Hz, 1H), 3,46 (d a, J = 13,2 Hz, 1H), 3,40 - 3,09 (m, 4H), 2,90 (d a, J = 2,8 Hz, 6H), 2,74 - 2,64 (m, 1H).
Etapa D: (2S)-4-[2-[[(2S,4R)-4-[terc-but¡l(d¡fen¡l)s¡l¡l1ox¡-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-7-[3-fluoro-2-(trifluoromet¡l)fen¡l1-6,8-d¡h¡dro-5H-p¡rido(3,4-d1p¡rim¡d¡n-4-¡l1-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo. A una soluc¡ón de [(2S,4R)-4-[tercbut¡l(d¡fen¡l)s¡l¡l]ox¡-1 -met¡l-p¡rrol¡d¡n-2-¡l]metanol (145 mg, 392 umol, 1,10 eq.) en THF (5,00 ml) se le añad¡ó f-Bu0Na (68,6 mg, 714 umol, 2,00 eq.) a 0 °C. Después, a la mezcla se le añad¡ó (2S)-2-(c¡anomet¡l)-4-[7-[3-fluoro-2-(tr¡fluoromet¡l)fen¡l]-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (0,22 g, 357 umol, 1,00 eq.). Después de ag¡tar a 0 °C durante 0,5 h, la mezcla se d¡luyó con acetato de et¡lo (10,0 ml) y agua (10,0 ml) y después se extrajo con acetato de et¡lo (3 * 10,0 ml). Los extractos se lavaron con salmuera (1 * 20,0 ml), se secaron sobre Na2S04, se f¡ltraron y se concentraron al vacío. El res¡duo se pur¡f¡có por cromatografía en columna (Ah03, éter de petróleo/acetato de et¡lo = 1/3) para dar (2S)-4-[2-[[(2S,4R)-4-[terc-but¡l(d¡íen¡l)s¡l¡l]ox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-[3-fluoro-2-(tr¡fluoromet¡l)fen¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (0,18 g, 185 umol, 52 % de rend¡m¡ento) en forma de un sól¡do de color amar¡llo. LCMS [ESI, M/2+1 ]: 462.
Etapa E: 2-[(2S)-4-[2-[[(2S,4R)-4 -rterc-but¡l(d¡fen¡l)s¡l¡llox¡-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-7-r3-fluoro-2-(tr¡fluoromet¡l)fen¡ll-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. Se burbujeó NH3 en metanol (30,0 ml) a -78 °C durante 15 m¡nutos. A la mezcla se le añad¡ó (2S)-4-[2-[[(2S,4R)-4-[terc-but¡l(d¡fen¡l)s¡l¡l]ox¡-1 -met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-7-[3-fluoro-2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (0,18 g, 195 umol, 1,00 eq.) y Pd/C (0,10 g, 10 % de pureza). Después de ag¡tar a 40 °C durante 1 hora en una atmósfera de H2 a 103,42 kPa (15 ps¡), el catal¡zador se f¡ltró y se concentró al vacío para dar 2-[(2S)-4-[2-[[(2S,4R)-4 -[terc-but¡l(d¡fen¡l)s¡l¡l]ox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-[3-fluoro-2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (0,13 g, bruto) en forma de un sól¡do de color amar¡llo. LCMS [ESI, M/2+1]: 395.
Etapa F: 2-[(2S)-4-[2-[[(2S,4R)-4-[ferc-but¡l(d¡fen¡l)s¡l¡l1ox¡-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-7-[3-fluoro-2-(trifluoromet¡l)fen¡l1-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-1-[(E)-4-(d¡met¡lam¡no)but-2-eno¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. Una mezcla de ác¡do (E)-4-bromobut-2-eno¡co (0,50 g, 3,03 mmol, 1,00 eq.) y d¡cloruro de oxal¡lo (3,85 g, 30,31 mmol, 2,65 ml, 10,0 eq.) en d¡clorometano (5 ml) se ag¡tó a 0 °C durante 0,5 h y a 40 °C durante 2 horas. La mezcla se concentró al vacío. El res¡duo anterior (93,0 mg, bruto) se añad¡ó a la mezcla de 2-[(2S)-4-[2-[[(2S,4R)-4-[terc-but¡l(d¡fen¡l)s¡l¡l]ox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-[3-fluoro-2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (80,0 mg, bruto) y Py (80,3 mg, 1,02 mmol, 82,0 ul) en d¡clorometano (0,50 ml) a 0 °C. Después de ag¡tar a 0 °C durante 0,5 h, se añad¡ó N-met¡lmetanam¡na (69,4 mg, 508 umol, 77,9 ul) y la mezcla se ag¡tó a 20 °C durante 1 hora. Después, la mezcla se concentró al vacío. El res¡duo se purificó por cromatografía ultrarráp¡da de fase ¡nversa [agua (TFA, 0,10 %)]. Las fracc¡ones deseadas
se ajustaron a pH >7 con bicarbonato de sodio saturado (1 * 5,00 ml) y se extrajeron con acetato de etilo (3 * 10,0 ml). Los extractos se lavaron con salmuera (1 * 10 ,0 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío para dar 2-[(2S)-4-[2-[[(2S,4R)-4-[tere-butil(difenil)silil]oxi-1-metil-pirrolidin-2-il]metoxi]-7-[3-fluoro-2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-[(E)-4-(dimetilamino)but-2-enoil]piperazin-2-il]acetonitrilo (0,06 g, 48,05 umol, dos etapas 65 % de rendimiento) que se obtuvo en forma de un aceite de color amarillo. LCMS [ESI, M+1 ]: 899.
Etapa G: 2-[(2S)-1-[(E)-4-(dimetilamino)but-2-enoil1-4-[7-[3 -fluoro-2-(trifluorometil)fenil]-2-[[(2S,4E)-4-hidroxi-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo. Una mezcla de 2-[(2S)-4-[2-[[(2S,4R)-4-[terc-butil(difenil)silil]oxi-1-metil-pirrolidin-2-il]metoxi]-7-[3-fluoro-2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-tí]pirimidin-4-il]-1-[(E)-4-(dimetilamino)but-2-enoil]piperazin-2-il]acetonitrilo (0,06 g, 66,7 umol, 1,00 eq.), KF (38,8 mg, 667 umol, 15,6 ul, 10,0 eq.) y 18-corona-6 (176 mg, 667 umol, 10,0 eq.) en THF (0,10 ml) se agitó a 40 °C durante 12 horas. La mezcla se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (TFA, 0,10 %/acetonitrilo)] y HPLC prep. columna: Gemini 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - ACN]; % de B: 30 % - 60 %, 12 min. Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[(2S)-1-[(E)-4-(dimetilamino)but-2-enoil]-4-[7-[3 -fluoro-2-(trifluorometil)fenil]-2-[[(2S,4E)-4-hidroxi-1-metil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (EJEMPL0342, 4,60 mg, 6,91 umol, 10 % de rendimiento, 99,2 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 661.
1H RMN (400 MHz, cloroformo-d) ó = 7,52 - 7,44 (m, 1H), 7,07 (d, J = 8,4 Hz, 1H), 7,00 - 6,90 (m, 2H), 6,46 (d a, J = 15,2 Hz, 1H), 5,07 (s a, 1H), 4,50 - 4,42 (m, 1H), 4,36 (dd, J = 4,8, 10,8 Hz, 1H), 4,22 (dd, J = 5,6, 10,8 Hz, 1H), 4,16 - 4,06 (m, 3H), 3,96 (d a, J = 11,6 Hz, 2H), 3,60 (s a, 1H), 3,43 (dd, J = 6,0, 10,0 Hz, 1H), 3,39 - 3,23 (m, 2H), 3,20 -3,05 (m, 4H), 3,05 - 2,79 (m, 4H), 2,78 - 2,62 (m, 2H), 2,48 (s, 3H), 2,35 - 2,26 (m, 7H), 2,12 - 1,92 (m, 2H).
Ejemplo 343

(S)-7-(2,3-dimetilfenil)-2-((1-metilpirrolidin-2-il)metoxi)-4-(4-(vinilsulfonil)piperazin-1-il)-5,6,7,8-tetrahidropirido[3,4-C]pirim idina
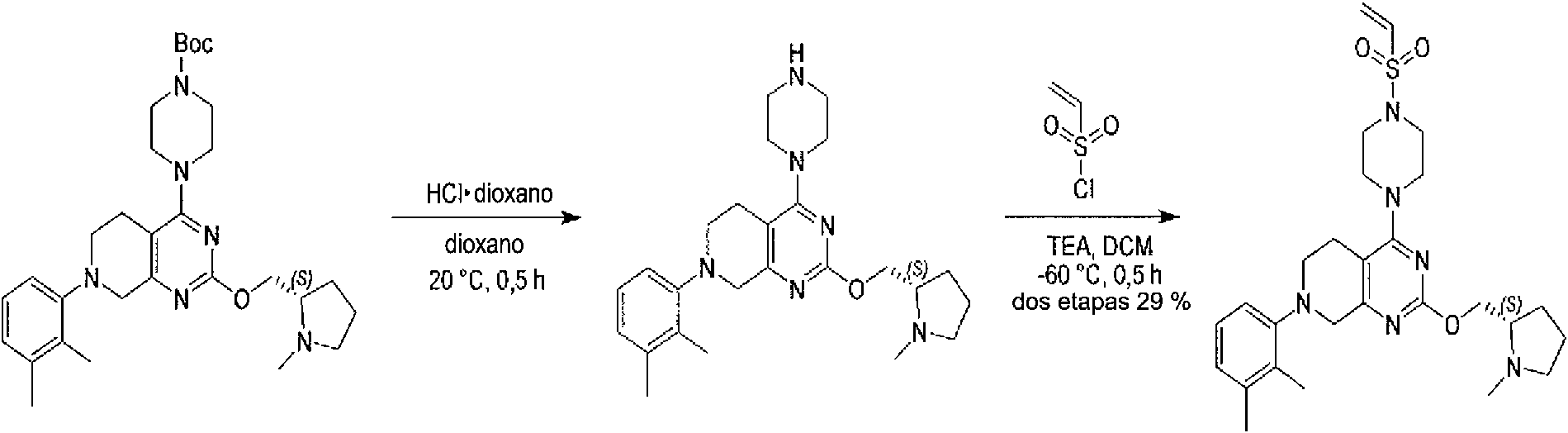
Etapa A: 7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-C1pirimidina. A una solución de 4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-C]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (350 mg, 652 umol, 1,0 eq.) en dioxano (3,0 ml) se le añadió HCl 4 M/dioxano (5,0 ml) y la mezcla se agitó a 20 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se concentró, después se añadió DCM (10,0 ml), la capa orgánica se lavó con NaHC03 acuoso saturado (2 * 10,0 ml) y la capa orgánica se secó sobre Na2S04, se filtró y se concentró para dar 7-(2,3-dimetilfenil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-C]pirimidina (280 mg, bruto) en forma de un aceite de color amarillo que se usó para la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 437.
Etapa B: 7-(2,3-dimetilfenil)-2-[[(2S)-metilpirrolidin- 2-il]metoxi]-4-(4-vinilsulfonilpiperazin-1-il)-6,8-dihidro-5H-pirido[3,4-cf1pirimidina. A una mezcla de 7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5Hp¡r¡do[3,4-cf]p¡r¡m¡d¡na (280 mg, 641 umol, 1,0 eq.) en DCM (5,0 ml) se le añadieron TEA (195 mg, 1,92 mmol, 268 ul, 3,0 eq.) y cloruro de etenosulfonilo (122 mg, 962 umol, 1,5 eq.) a -60 °C y la mezcla de reacción se agitó a -60 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se inactivó con Me0H (1,0 ml) y se concentró. El residuo se purificó por cromatografía en columna (Base Ah03, éter de petróleo : acetato de etilo = 3:1 ~ acetato de etilo/metanol = 20/1) y después el producto bruto se concentró y se purificó de nuevo por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN]; % de B: 62 %-92 %,12 min) y el producto obtenido se concentró y después se sometió a liofilización. El compuesto del título 7-(2,3-d¡metilfen¡l)-2-[[(2S)-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-4-(4-v¡n¡lsuifon¡lp¡peraz¡n-1-¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡na (EJEMPL0343, 98,2 mg, 185 umol, 29 % de rendimiento, 99,3 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 527.
1H RMN (400 MHz, cloroformo-d) 57,12 (t, J = 7,6 Hz, 1H), 7,00 - 6,94 (m, 2H), 6,47 (dd, J = 10,0 Hz, 16,8 Hz, 1H), 6,30 (d, J = 16,8 Hz, 1H), 6,11 (d, J = 10,0 Hz, 1H), 4,39 (dd, J = 4,8, 10,4 Hz, 1H), 4,15 (dd, J = 6,8, 10,4 Hz, 111), 4,03 (s, 2H), 3,62 (t, J = 4,6 Hz, 4H), 3,30 (t, J = 4,6 Hz, 4H), 3,19 - 3,05 (m, 3H), 2,80 - 2,62 (m, 3H), 2,49 (s, 3H), 2,32 - 2,25 (m, 7H), 2,13 - 2,00 (m, 1H), 1,92 - 1,72 (m, 3H).
Ejemplo 344

-[(2S)-4-[7-(3-h¡drox¡-1-naft¡l)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-enoil-p¡peraz¡n-2-il]aceton¡tr¡lo

Etapa A: (2S)-4-[7-(3-benciloxi-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo. A una solución de ((2R)-1-metilp¡rrolid¡n-2-¡l]metanol (101 mg, 874 umol, 1,2 eq.) en tolueno (15 ml) se le añadió í-Bu0Na (140 mg, 1,46 mmol, 2 eq.) a 0 °C durante 10 minutos. Después, a la mezcla se le añadió gota a gota una solución de (2S)-4-[7-(3-benc¡loxi-1-naft¡l)-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de bencilo (Intermedio 64, 500 mg, 728 umol, 1 eq.) en tolueno (10 ml) a 0 °C. Después de agitar a 0 °C durante 0,5 h, la mezcla de reacción se diluyó con H20 (1 * 7 ml) y acetato de etilo (1 * 25 ml). La fase orgánica se separó, se lavó con salmuera (1 * 20 ml), se secó sobre sulfato de sodio, se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Ah03; éter de petróleo/acetato de etilo = 10/1 a 0/1). El compuesto (2S)-4-[7-(3-benc¡loxi-1-naft¡l)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (390 mg, 489 umol, 67 % de rendimiento, 92,6 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 738.
1H RMN (400 MHz, cloroformo-d) 6 = 8,09 (d, J = 8,4 Hz, 1H), 7,74 (d, J = 8,0 Hz, 1H), 7,50 (d, J = 7,2 Hz, 2H), 7,46 -7,34 (m, 10H), 7,00 (d, J = 2,0 Hz, 1H), 6,89 (d, J = 2,4 Hz, 1H), 5,24 - 5,15 (m, 4H), 4,70 (s a, 1H), 4,39 (dd, J = 4,8, 10,7 Hz, 1H), 4,33 - 4,22 (m, 2H), 4,20 - 4,11 (m, 3H), 3,93 (d a, J = 12,0 Hz, 1H), 3,56 - 3,19 (m, 4H), 3,14 - 2,61 (m, 7H), 2,48 (s, 3H), 2,34 - 2,23 (m, 1H), 2,11 - 2,06 (m, 1H), 1,84 - 1,69 (m, 3H).
Etapa B: 2-[(2S)-4-[7-(3-hidroxi-1-naftil)-2-[[(2ft)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡llpiperazin-2-illacetonitrilo. Se burbujeó NH3 en Me0H (100 ml) a -60 °C durante 20 minutos. A una solución de (2S)-4-[7-(3-benciloxi-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (360 mg, 488 umol, 1 eq.) en Me0H (25 ml) se le añadieron Pd/C (300 mg, 10 % de pureza) y la solución anterior (NH3^Me0H, 25 ml) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. El catalizador se retiró por filtración y se concentró al vacío. El compuesto 2-[(2S)-4-[7-(3-hidroxi-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (230 mg, bruto) se obtuvo en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 514.
Etapa C: 2-[(2S)-4-[7-(3-hidroxi-1-naftil)-2-[[(2ffl-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo. A una mezcla de 2-[(2S)-4-[7-(3-hidroxi-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (230 mg, bruto) y TEA (453 mg, 4,48 mmol, 623 ul) en DCM (8 ml) se le añadió en una porción cloruro de prop-2-enoílo (40,5 mg, 448 umol, 36,5 ul) a -40 °C. La mezcla se agitó a -40 °C durante 30 min. La mezcla de reacción se inactivó con una solución saturada de NaHC03 (2 ml) a 0 °C y después se extrajo con CH202 (2 x 30 ml). Las capas orgánicas combinadas se lavaron con salmuera (1 x 30 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío para dar un residuo. El residuo se purificó por cromatografía en columna (Ab0a; acetato de etilo/metanol = 20/1 a 3/1). El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - ACN]; % de B: 38 % - 68 %, 12 min). El compuesto del título 2-[(2S)-4-[7-(3-h¡drox¡-1-naft¡l)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0344, 25,6 mg, 44,8 umol, dos etapas 9,2 % de rendimiento, 99,5 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 568.
Condición de SFC: “AS-3_Me0H (DEA) _5_40_3 ml-35T Columna: Chiralpak AS-350 x 4 ,6 mm D.I., 3 um Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 % Caudal: 3 ml/min Longitud de onda: 220 nm” .
1H RMN (400 MHz, acético) 6 = 8,08 (d, J = 8,4 Hz, 1H), 7,66 (d, J = 8,0 Hz, 1H), 7,39 (t, J = 7,6 Hz, 1H), 7,29 (t, J = 7,2 Hz, 1H), 6,96 (s, 1H), 6,90 - 6,68 (m, 2H), 6,38 (d a, J = 17,6 Hz, 1H), 5,87 (d a, J = 10,8 Hz, 1H), 5,17 (s a, 1H), 4,82 (s a, 2H), 4,59 (s a, 1H), 4,45 - 4,25 (m, 3H), 4,21 - 3,61 (m, 4H), 3,60 - 3,19 (m, 5H), 3,17 - 2,83 (m, 7H), 2,45 -2,31 (m, 1H), 2,23 - 2,10 (m, 3H).
Ejemplo 345

2-[(2S)-4-[7-(3-metil-4-piridil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[7-(3-metil-4-piridil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-1-carboxilato de tere-butilo. Una mezcla de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (700 mg, 1,48 mmol, 1,0 eq.), 4-cloro-3-metil-piridina (379 mg, 2,97 mmol, 2,0 eq.), Pd2(dba)3 (204 mg, 223 umol, 0,15 eq.), RuPhos (139 mg, 297 umol, 0,2 eq.) y CS2C03 (1,45 g, 4,45 mmol, 3,0 eq.) en tolueno (10,0 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 12 h en una atmósfera de N2. El disolvente orgánico se eliminó al vacío y se lavó con agua (15,0 ml). La fase acuosa se extrajo con acetato de etilo (3 x 25,0 ml). Los extractos combinados se lavaron con salmuera (60,0 ml), se secaron con Na2S04 y después el disolvente se eliminó al vacío. El residuo se purificó por HPLC ultrarrápida de fase inversa [C18, 0,1 % de AF en agua, 0-45 % de MeCN]. El producto obtenido se ajustó con NaHC03 acuoso saturado a pH ~8, después se concentró, la capa acuosa se extrajo con acetato de etilo (3 x 50 ,0 ml) y la capa orgánica combinada se secó sobre Na2S04, se filtró y se concentró. El compuesto (2S)-2-(c¡anomet¡l)-4-[7-(3-met¡l-4-p¡r¡d¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡hidro-5H-p¡r¡do[3,4-tí]pirimidin-4-il]piperazin-1 -carboxilato de tere-butilo (270 mg, 475 umol, 32 % de rendimiento, 99,0 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 563.
Etapa B: 2-r(2S)-4-r7-(3-metil-4-piridil)-2-rr(2S)-1-metilpirrolidin-2-i n metox n -6.8-dihidro-5H-pindor3.4-dlpirimidin-4-¡ n piperazin-2-i n acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(3-metil-4-piridil)-2-[[(2S)-1 -metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (200 mg, 355 umol, 1,0 eq.) en dioxano (3,0 ml) se le añadió HCldioxano (4 M, 3,0 ml, 33,8 eq.). La mezcla se agitó a 25 °C durante 30 min en una atmósfera de N2. El disolvente orgánico se eliminó al vacío. El producto obtenido se ajustó con NaHC03 acuoso saturado a pH ~8, después se concentró, la capa acuosa se extrajo con acetato de etilo (3 x 15 ,0 ml) y la capa orgánica combinada se secó sobre Na2S04, se filtró y se concentró. El producto bruto 2-[(2S)-4-[7-(3-metil-4-piridil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡trilo (125 mg, bruto) se obtuvo en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1 ]: 463.
Etapa C: 2-[(2S)-4-[7-(3-metil-4-piridil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-il)-1-prop-2-enoil-piperazin-2-i n acetonitrilo. A una solución de 2-[(2S)-4-[7-(3-metil-4-piridil)-2-[[(2S)-1 -metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (125 mg, bruto) en DCM (3,0 ml) se le añadieron TEA (137 mg, 1,35 mmol, 188 ul) y cloruro de prop-2-enoílo (36,7 mg, 405 umol, 33,1 ul) a -60 °C. La mezcla se agitó a -60 °C durante 30 min. La reacción se interrumpió con metanol (10,0 ml) y la mezcla se eliminó al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um;fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v)-ACN];% de B: 32 %-62 %,12 min) y se sometió a liofilización. El compuesto del título 2-[(2S)-4-[7-(3-met¡l-4-p¡r¡d¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0345, 25,3 mg, 48,4 umol, 18 % de rendimiento, 99 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 517.1
1H RMN (400 MHz, CL0R0F0RM0-d) ó = 8,33 - 8,29 (m, 2H), 6,83 (d, J = 5,6 Hz, 1H), 6,65 - 6,50 (m, 1H), 6,38 (dd, J = 1,6 Hz, J = 16,8 Hz, 1H), 5,82 (d, J = 10,8 Hz 1H), 5,41 (s a, 1H), 4,36 (dd, J = 4,8 Hz, J = 10,4 Hz, 1H), 4,20 - 4,06 (m, 4H), 4,97 (d a, J = 11,6 Hz 2H),3,68 -- 3,45 (m, 1H), 3,40 - 3,31 (m, 2H),3,26 - 3,15 (m, 1H),3,15 - 3,03 (m, 2H), 2,97 - 2,60 (m, 5H), 2,31 (s, 3H), 2,29 -2,21 (m, 3H) 2,15 - 2,10 (m, 1H), 2,09 - 2,00 (m, 1H) 1,88 - 1,70 (m, 3H).
Ejemplo 346

2-[4-[2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-7-[2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Etapa 1: A una solución de ácido (E)-4-bromobut-2-enoico (1,0 g, 6,06 mmol, 1,0 eq.) en DCM (5,0 ml) se le añadió (C0Cl)2 (8,70 g, 68,5 mmol, 6 ml, 11,3 eq.). La mezcla se agitó a 60 °C durante 16 h en una atmósfera de N2. El disolvente orgánico se eliminó al vacío. El producto bruto cloruro de (E)-4-bromobut-2-enoílo (1,3 g, bruto) se obtuvo en forma de un líquido de color amarillo y se usó en la siguiente etapa sin purificación adicional.
Etapa A: (2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo. A una solución de (2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de íere-butilo (1,30 g, 2,17 mmol, 1,0 eq.) y [(2S)-1-metilpirrolidin-2-il]metanol (500 mg, 4,34 mmol, 515 ul, 2,0 eq.) en tolueno (20,0 ml) se le
añadió t-Bu0Na (417 mg, 4,34 mmol, 2,0 eq.). La mezcla se agitó a 0 °C durante 10 min. La mezcla de reacción se inactivó mediante la adición de salmuera saturada (30 ml) a 20 °C y se extrajo con AE (3 * 30,0 ml). Las capas orgánicas combinadas se lavaron con salmuera saturada (30,0 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo = 3/1 a AE/Me0H = 10/1). El producto (2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (1,10 g, 1,62 mmol, 75 % de rendimiento, 96 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 650.
Etapa B: 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo. A una solución de (2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-([(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (200 mg, 308 umol, 1,0 eq.) en dioxano (2,0 ml) se le añadió HCl/dioxano (4 M, 2,0 ml, 26,0 eq.). La mezcla se agitó a 25 °C durante 30 min en una atmósfera de N2. El disolvente orgánico se eliminó al vacío. El producto obtenido se ajustó con NaHC03 acuoso saturado a pH ~8, después se concentró, la capa acuosa se extrajo con acetato de etilo (3 * 20,0 ml) y la capa orgánica combinada se secó sobre Na2S04, se filtró y se concentró. El producto bruto 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d7pirimidin-4-il]piperazin-2-il]acetonitrilo (130 mg, bruto) se obtuvo en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESl, M+1]: 551.
Etapa C: 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l1-1-[(E)-4-morfol¡nobut-2-eno¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una solución de 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (100 mg, 1812 umol, 1,0 eq.) y piridina (115 mg, 1,45 mmol, 117 ul, 8,0 eq.) en Dc M (3,0 ml) se le añadió cloruro de (E)-4-bromobut-2-enoílo (133 mg, 727 umol, 4,0 eq.). La mezcla se agitó a 0 °C durante 0,5 h. Después de que se consumiera el material de partida, a la mezcla anterior se le añadió morfolina (79,2 mg, 909 umol, 80,0 ul, 5,0 eq.) y se agitó a 0 °C durante 3,5 h. El disolvente orgánico se eliminó al vacío. El producto obtenido se ajustó con NaHC03 acuoso saturado a pH ~8, después se concentró, la capa acuosa se extrajo con acetato de etilo (3 * 15,0 ml) y la capa orgánica combinada se secó sobre Na2S04, se filtró y se concentró. El compuesto del título 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(E)-4-morfolinobut-2-enoil)piperazin-2-il]acetonitrilo (EJEMPL0346, 21,7 mg, 30,6 umol, 16,9 % de rendimiento, 99 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESl, M+1]: 703.
1H RMN (400 MHz, CL0R0F0RM0-d) ó = 7,40 (t, J = 8,0 Hz, J = 8,0 Hz 1H), 7,28 (s, 1H), 7,19 (d, J = 8,0 Hz, 1H), 7,00 - 6,93(m, 1H), 6,59 (s a, 1H), 6,57 - 6,40 (m, 1H), 5,18 - 5,03 (m, 1H), 4,38 (dd, J = 4,8 Hz, J = 10,4 Hz, 1H), 4,16 (dd, J = 6,8 Hz, J = 10,4 Hz, 1H), 4,14 - 4,04 (m, 3H), 4, 04 - 3,88 (m, 2H), 3,74 (t, J = 4,4 Hz, J = 4,8 Hz, 4H), 3,52 (s a, 1H), 3,41 - 3,23 (m, 2H), 3,18 (d, J = 5,6 Hz, 3H), 3,12 - 3,08 (m, 2H),2,95 - 2,85 (m, 2H), 2,30 - 2,15 (m, 3H), 2,55 - 2,45 (m, 7H), 2,34 - 2,24 (m, 1H), 2,13 - 2,00 (m, 1H), 1,90 - 1,75 (m, 3H).
Ejemplo 347

2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(E)-4-pirrolidin-1-ilbut-2-enoil]piperazin-2-il]acetonitrilo

Inserción: cloruro de (E)-4-bromobut-2-enoílo. Una solución de ácido (E)-4-bromobut-2-enoico (1,00 g, 6,06 mmol, 1,00 eq.) en (C0Cl)2 (14,5 g, 114 mmol, 10,0 ml, 18,9 eq.) y DCM (10,0 ml) se agitó a 70 °C durante 2 horas. Después de que se completara, la mezcla se concentró al vacío. El producto cloruro de (E)-4-bromobut-2-enoílo (1,00 g, bruto) se obtuvo en forma de un aceite de color amarillo. El compuesto bruto se usó directamente para la siguiente etapa sin purificación adicional.
Etapa A: 2-[(2S)-4-[7-[3-cloro -2-(trifluorometil)fenil1-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (700 mg, 1,08 mmol, 1,0 eq.) en dioxano (5,0 ml) se le añadió HCkdioxano (4 M, 7,0 ml, 26,0 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 2 horas. Después de que se completara, la mezcla se concentró y se ajustó con NaHC03 acuoso saturado a pH ~7 y después se extrajo con AE (10,0 ml * 3). La capa orgánica se secó sobre Na2S04, se filtró y se concentró. El producto 2-[(2s)-4-[7-[3-cloro -2-(trifluorometil)fenil1-2-[[(2s)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo (400 mg, 669 umol, 62 % de rendimiento, 92 % de pureza) se obtuvo en forma de un aceite de color amarillo. LCMS [ESI, M+11: 550.
Etapa B: 2-[(2S)-1-[(E)-4-bromobut- 2-enoil1-4-[7-[3-cloro-2-(trifluorometil)fenil1-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil1-2-[[(2S)-1 -metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-tí1pirimidin-4-il1piperazin-2-il1acetonitrilo (200 mg, 364 umol, 1,00 eq.) y piridina (230 mg, 2,91 mmol, 235 ul, 8,0 eq.) en Dc M (2,0 ml) se le añadió cloruro de (E)-4-bromobut-2-enoílo (267 mg, 1,45 mmol, 4,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, a la mezcla se le añadió agua (10,0 ml) y se extrajo con DCM (10,0 ml * 2). La capa orgánica se secó sobre Na2S04, se filtró y se concentró. El producto 2-[(2S)-1-[(E)-4-bromobut- 2-enoil1-4-[7-[3-cloro-2-(trifluorometil)fenil1-2-[[(2S)-1 -metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-tí1pirimidin-4-il)piperazin-2-il1acetonitrilo (300 mg, bruto) se obtuvo en forma de un aceite de color pardo.
Etapa C: 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil1-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-[(E)-4-pirrolidin-1-ilbut-2-enoil1piperazin-2-il1acetonitrilo. A una solución de pirrolidina (306 mg, 4,30 mmol, 359 ul, 10,0 eq.), K2C03 (297 mg, 2,15 mmol, 5,0 eq.) y KI (21,4 mg, 129 umol, 0,30 eq.) en THF (2,0 ml)
se l añadió poco a poco 2-[(2S)-1-[(E)-4-bromobut-2-enoil]-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (300 mg, 430 umol, 1,0 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, a la mezcla se le añadió agua (10,0 ml) y se extrajo con DCM (10,0 ml * 3). La capa orgánica se secó sobre Na2SÜ4, se filtró y se concentró. El producto obtenido se purificó por HPLC prep. (columna: Phenomenex Gemini C1825 * 50 mm * 10 um; fase móvil: [agua (NH4HC03 10 mM)-ACN]; % de B: 70 %-100 %,3 min) para dar el compuesto del título 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(E)-4-pirrolidin-1-ilbut-2-enoil]piperazin-2-il]acetonitrilo (EJEMPL0347, 17,1 mg, 24,5 umol, dos etapas 7 % de rendimiento, 99 % de pureza) en forma de un aceite de color amarillo. LCMS [ESI, M+1 ]: 687.
1H RMN (400 MHz, Cloroformo-d) ó 7,41 (t, J = 8,0 Hz, 1H), 7,28 (d, J = 8,0 Hz, 1H), 7,20 (d, J = 8,4 Hz, 1H), 7,06 -6,95 (m, 1H), 6,47 (d a, J = 14,0 Hz, 1H), 5,20 - 4,60 (m, 1H), 4,39 (dd, J = 5,2, 10,8 Hz, 1H), 4,22 - 4,07 (m, 4H), 3,97 (d a, J = 12,0 Hz, 2H), 3,42 - 3,23 (m, 3H), 3,21 - 3,04 (m, 3H), 2,96 - 2,86 (m, 2H), 2,79 - 2,64 (m, 5H), 2,62 - 2,54 (m, 4H), 2,49 (s, 3H), 2,35 - 2,25 (m, 1H), 2,13 - 2,01 (m, 1H), 1,83 - 1,70 (m, 7H).
Ejemplo 348

2-[(2S)-4-[7-(3-hidroxi-1 -naftil)-2-[(1 R)-1 -[(2R)-1 -metilpirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-y1 ]-1 -prop-2-enoil-piperazin-2-il]acetonitrilo
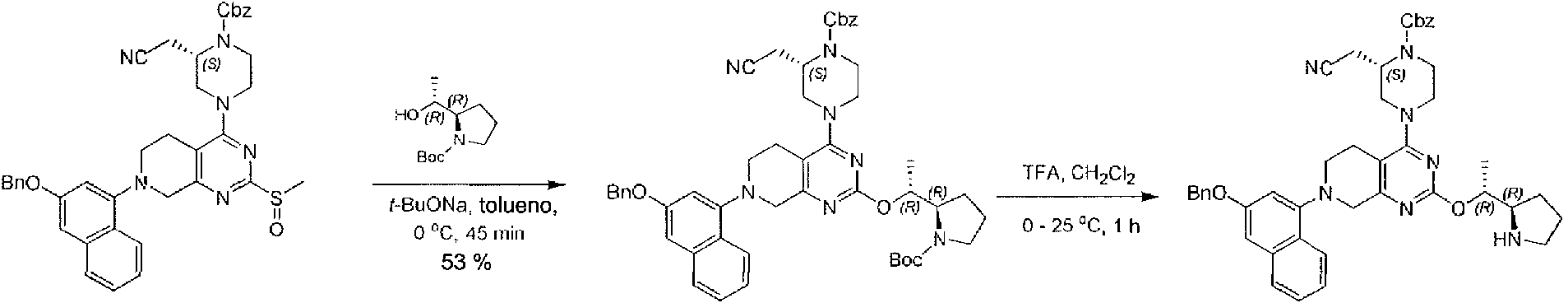

Inserción: (2R)-2-[(1R)-1-hidroxietil]pirrolidin-1- carboxilato de tere-butilo. Una mezcla de BH3*Me2S (10 M en DMS, 610 ul, 1,3 eq.) y (3aS)-1 -metil-3,3-difenil-3a,4,5,6-tetrahidropirrolo[1,2-c][1,3,2]oxazaborol (1 M en tolueno, 938 ul, 0,2 eq.) en THF (10 ml) se agitó a 25 °C durante 1 hora. Después, se añadió una solución de (2 R)-2-acetilpirrolidin-1-carboxilato de tere-butilo (1,00 g, 4,69 mmol, 1 eq.) en THF (10 ml) y la mezcla se agitó a 25 °C durante 1 hora. La mezcla se inactivó con Me0H (4 ml) y se concentró al vacío. El residuo se purificó por cromatografía en columna (Si02, EP/EtOAc = 50/1 a 5/1). Se obtuvo (2R)-2-((1 R)-1 -hidroxietil]pirrolidin-1 - carboxilato de tere-butilo (690 mg, 3,21 mmol, 68 % de rendimiento) en forma de un sólido de color blanco.
1H RMN (400 MHz, cloroformo-d) ó = 5,19 (s a, 1H), 3,79 - 3,59 (m, 2H), 3,49 (s a, 1H), 3,35 - 3,18 (m, 1H), 2,04 - 1,90 (m, 1H), 1,88 - 1,68 (m, 3H), 1,47 (d, J = 0,8 Hz, 9H), 1,20 - 1,11 (m, 3H).
Etapa A: (2S)-4-[7-(3-benciloxi-1-naftil)-2-1[(1R)-1-[(2R)-1-tere-butoxicarbonilpirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]-2-(cianometil)piperazin-1 -carboxilato de bencilo. A una solución de (2R)-2-[(1R)-1-hidroxietil]pirrolidin-1 -carboxilato de tere-butilo (263 mg, 1,22 mmol, 1,2 eq.) en tolueno (15 ml) se le añadió t-Bu0Na (196 mg, 2,04 mmol, 2 eq.) a 0 °C durante 10 minutos. A la mezcla se le añadió gota a gota (2S)-4-[7-(3-benciloxi-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (700 mg, 1,02 mmol, 1 eq.) en tolueno (15 ml) a 0 °C. Después de agitar a 0 °C durante 0,5 h, a la mezcla de reacción se le añadieron H20 (1 *7 ml) y acetato de etilo (1 * 35 ml). La fase orgánica se separó, se lavó con salmuera (1 * 20 ml), se secó sobre sulfato de sodio, se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de FA)/acetonitrilo]. Las fracciones deseadas se recogieron y se neutralizaron a pH = 7 con una solución saturada de NaHC03 y se extrajeron con acetato de etilo (1 * 100 ml). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El compuesto (2 S)-4-[7-(3-benc¡lox¡-1-naft¡l)-2-[(1R)-1-[(2R)-1-fere-butox¡carbon¡lp¡rrol¡d¡n-2-¡l]etox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(cianometil)piperazin-1-carboxilato de bencilo (470 mg, 536 umol, 53 % de rendimiento, 95,6 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1 ]: 838.
1H RMN (400 MHz, cloroformo-d) ó = 8,09 (d, J = 8,4 Hz, 1H), 7,73 (d, J = 8,0 Hz, 1H), 7,53 - 7,48 (m, 2H), 7,46 - 7,31 (m, 10H), 7,00 (d, J = 1,6 Hz, 1H), 6,89 (d, J = 2,0 Hz, 1H), 5,78 - 5,44 (m, 1H), 5,30 - 5,08 (m, 4H), 4,70 (s a, 1H), 4,35 - 4,20 (m, 3H), 4,08 - 3,79 (m, 2H), 3,72 - 3,13 (m, 7H), 3,12 - 2,64 (m, 4H), 2,01 - 1,71 (m, 5H), 1,46 (s, 9H), 1,26 - 1,23 (m, 3H).
Etapa B: (2S)-4-[7-(3-benciloxi-1-naftil)-2-[(1R)-1-[(2R)-1-pirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo. A una solución de (2S)-4-[7-(3-benciloxi-1-naftil)-2-[(1R)-1-[(2R)-1-terebutoxicarbonilpirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (450 mg, 537 umol, 1 eq.) en DCM (600 ul) se le añadió TFA (918 mg, 8,05 mmol, 596 ul, 15 eq.) a 0 °C en una atmósfera de nitrógeno. La mezcla se agitó a 25 °C durante 1 hora. Después, la mezcla se concentró al vacío. El compuesto (2S)-4-[7-(3-benciloxi-1-naftil)-2-[(1R)-1-[(2R)-pirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (700 mg, bruto, TFA) se obtuvo en forma de un aceite de color amarillo.
Etapa C: (2S)-4-r7-(3-benciloxi-1-naftil)-2-r(1R)-1-r(2R)-1-metilpirrolidin-2-inetoxn-6.8-dihidro-5H-piridor3.4-dlpirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo. Una mezcla de (2S)-4-[7-(3-benciloxi-1-naftil)-2-[(1R)-1-[(2R)
pirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (650 mg, 763 umol, 1 eq., TfA), formaldehído (310 mg, 3,81 mmol, 284 ul, 5 eq., 37 % en agua) y Ac0H (45,8 mg, 763 umol, 43,6 ul, 1 eq.) en Me0H (15 ml) se agitó a 25 °C durante 0,5 h. Después, se añadió NaBH3CN (120 mg, 1,91 mmol, 2,5 eq.) y la mezcla se agitó a 25 °C durante 1 hora. La mezcla se inactivó con H20 (10 ml) a 0 °C, después se extrajo con acetato de etilo (3 x 15 ml), se lavó con salmuera (1 x 15 ml), se secó sobre Na2S04, se filtró y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de FA)/acetonitrilo]. Las fracciones deseadas se recogieron y se neutralizaron a pH = 7 con una solución saturada de NaHC03 y se extrajeron con acetato de etilo (2 x 50 ml). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El compuesto (2S)-4-[7-(3-benciloxi-1-naftil)-2-[(1R)-1-[(2R)-1-metilpirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (260 mg, 339 umol, dos etapas 68 % de rendimiento, 97,9 % de pureza) se obtuvo en forma de un sólido de color amarillo.
Etapa D: 2-1(2S)-4-[7-(3-hidroxi-1-naftil)-2-[(1R)-1-[(2R)-1-metilpirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡perazin-2-¡l1aceton¡tr¡lo. Se burbujeó NH3 en Me0H (100 ml) a -60 °C durante 20 minutos. A una solución de (2S)-4-[7-(3-benciloxi-1-naftil)-2-[(1R)-1-[(2R)-1-metilpirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (260 mg, 346 umol, 1 eq.) en Me0H (15 ml) se le añadieron Pd/C (200 mg, 10 % de pureza) y la mezcla anterior (NH3^Me0H, 15 ml) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. El catalizador se retiró por filtración y se concentró al vacío. El compuesto 2-[(2S)-4-[7-(3-hidroxi-1-naftil)-2-[(1R)-1-[(2R)-1-metilpirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (180 mg, 300 umol, 87 % de rendimiento, 88,0 % de pureza) se obtuvo en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 528.
Etapa E: 2-[(2S)-4-[7-(3-hidroxi-1-naftil)-2-[(1R)-1-[(2R)-1-metilpirrolidin-2-inetoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1acetonitr¡lo. A una mezcla de 2-[(2S)-4-[7-(3-hidroxi-1-naftil)-2-[(1R)-1-[(2R)-1-metilpirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (180 mg, 341 umol, 1 eq.) y TEA (345 mg, 3,41 mmol, 475 ul, 10 eq.) en DCM (8 ml) se le añadió en una porción cloruro de prop-2-enoílo (30,9 mg, 341 umol, 27,8 ul, 1 eq.) a -40 °C y la mezcla se agitó a -40 °C durante 30 min. La mezcla de reacción se inactivó con una solución saturada de NaHC03 (2 ml) a 0 °C y después se extrajo con CH2Cl2 (2 x 30 ml). Las capas orgánicas combinadas se lavaron con salmuera (1 x 30 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío para dar un residuo. El residuo se purificó por cromatografía en columna (Ah03; acetato de etilo/metanol = 20/1 a 3/1). El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoníaco v/v) - a Cn ]; % de B: 42 % - 72 %, 12 min). El compuesto del título 2-[(2S)-4-[7-(3-hidroxi-1-naftil)-2-[(1R)-1-[(2R)-1-metilpirrolidin-2-il]etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo (EJEMPL0348, 23,5 mg, 39,9 umol, 12 % de rendimiento, 99,0 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 582.
Condición de SFC: “0J-3S 3_5403ML Columna: Chiralcel 0J-3100 x 4 ,6 mm D.I., 3 um Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 % Caudal: 3 ml/min Longitud de onda: 220 nm” .
1H RMN (400 MHz, metanol-d4) 6 = 8,09 (d, J = 8,4 Hz, 1H), 7,64 (d, J = 8,0 Hz, 1H), 7,39 (t, J = 7,6 Hz, 1H), 7,32 -7,23 (m, 1H), 6,98 - 6,74 (m, 3H), 6,31 (d a, J = 16,4 Hz, 1H), 5,85 (d a, J = 10,4 Hz, 1H), 5,27 - 5,15 (m, 1H), 5,11 (s a, 1H), 4,62 (s a, 1H), 4,33 - 4,00 (m, 5H), 3,75 - 3,38 (m, 2H), 3,27 - 2,81 (m, 7H), 2,75 - 2,62 (m, 1H), 2,55 (s, 3H), 2,47 - 2,31 (m, 1H), 2,11 - 1,96 (m, 1H), 1,86 - 1,69 (m, 3H), 1,34 (d, J = 6,4 Hz, 3H).
Ejemplo 349

2-((S)-1-acriloil-4-(7-(3-fluoro-2,6-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa 1: 2-bromo-6-fluoro-3-metil-benzaldehído. A una solución de 2-bromo-4-fluoro-1-metil-benceno (10 g, 52.9 mmol, 1 eq.) en THF (200 ml) se le añadió LDA (2 M en tolueno, 31,7 ml, 1,2 eq.) a -78 °C en una atmósfera de nitrógeno. Después de agitar a -78 °C durante 2 horas, se añadió una solución de DMF (4,64 g, 63,5 mmol, 4,88 ml, 1,2 eq.) a -78 °C. La mezcla se calentó cuidadosamente a 25 °C y se agitó durante 0,5 horas. La mezcla se inactivó con agua (5 ml) y la mezcla se diluyó con éter de petróleo/acetato de etilo (50 ml/5 ml) y agua (50 ml). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El compuesto 2-bromo-6-fluoro-3-metilbenzaldehído (11,5 g, 52,9 mmol, 100 % de rendimiento) se obtuvo en forma de un aceite incoloro.
Etapa 2: (2-bromo-6-fluoro-3-metil-fenil)metanol. A una solución de 2-bromo-6-fluoro-3-metil-benzaldehído (11,5 g, 52.9 mmol, 1 eq.) en Me0H (100 ml) se le añadió NaBH4 (2,20 g, 58,2 mmol, 1,1 eq.) a 0 °C. La mezcla se agitó a 25 °C durante 0,5 horas. La mezcla se inactivó con agua a 0 °C y la mezcla se concentró al vacío. El residuo se purificó por cromatografía de columna sobre gel de sílice (éter de petróleo/acetato de etilo 50/1 a 3/1). Las fracciones deseadas se recogieron y se concentraron al vacío. El compuesto (2-bromo-6-fluoro-3-metil-fenil)metanol (8,5 g, 38,4 mmol, 73 % de rendimiento, 99 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M-17]: 201.
1H RMN (400 MHz, cloroformo-d) ó = 7,20 (dd, J = 6,4, 8,4 Hz, 1H), 6,98 (t, J = 8,8 Hz, 1H), 4,88 (d, J = 2,0 Hz, 2H), 2.41 (s, 3H), 2,35 - 2,11 (m, 1H).
Etapa 3: 3-bromo-2-(bromometil)-1-fluoro-4-metil-benceno. A una mezcla de (2-bromo-6-fluoro-3-metil-fenil)metanol (5 g, 22,8 mmol, 1 eq.) en THF (100 ml) se le añadió poco a poco PBr3 (12,4 g, 45,6 mmol, 2 eq.) a 0 °C. Después de agitar a 25 °C durante 1 hora, la mezcla se concentró al vacío. El residuo se diluyó con acetato de etilo (100 ml) y se lavó con agua enfriada con hielo (1 * 50 ml), solución acuosa al 10 % de bicarbonato de sodio (2 * 50 ml) y salmuera (1 * 20 ml). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 1/0 a 10/1). El compuesto 3-bromo-2-(bromometil)-1-fluoro-4-metil-benceno (5,6 g, 19,9 mmol, 87 % de rendimiento) se obtuvo en forma de un aceite incoloro.
1H RMN (400 MHz, cloroformo-d) ó = 7,20 (dd, J = 6,4, 8,0 Hz, 1H), 6,97 (t, J = 8,8 Hz, 1H), 4,70 (d, J = 2,0 Hz, 2H), 2.41 (s, 3H).
Etapa 4: 3-bromo-1-fluoro-2,4-dimetil-benceno. A una solución de 3-bromo-2-(bromometil)-1-fluoro-4-metil-benceno (5,6 g, 19,9 mmol, 1 eq.) en DMF (50 ml) se le añadieron NaBH4 (1,50 g, 39,7 mmol, 2 eq.) y AgN03 (6,75 g, 39,7 mmol, 6,68 ml, 2 eq.) a 25 °C. La mezcla se agitó a 25 °C durante 0,5 horas. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (50 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se
purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico)/acetonitrilo)]. La mezcla se concentró a presión reducida. El compuesto 3-bromo-1-fluoro-2,4-dimetil-benceno (2,1 g, 9,93 mmol, 50 % de rendimiento, 96 % de pureza) se obtuvo en forma de un aceite incoloro.
1H RMN (400 MHz, cloroformo-d) ó = 7,05 (dd, J = 6,0, 8,4 Hz, 1H), 6,91 (t, J = 8,8 Hz, 1H), 2,38 (s, 3H), 2,35 (d, J = 2,4 Hz, 3H).
Etapa A: (2S)-2-(cianometil)-4-[7-(3-fluoro-2,6-dimetil-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo. Una mezcla de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (500 mg, 1,06 mmol, 1 eq.), 3-bromo-1-fluoro-2,4-dimetil-benceno (430 mg, 2,12 mmol, 2 eq.), RuPhos-Pd-G3 (177 mg, 212 umol, 0,2 eq.) y Cs2C03 (863 mg, 2,65 mmol, 2,5 eq.) en tolueno (10 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 70 °C durante 12 horas en una atmósfera de N2. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (20 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Ab03, acetato de etilo/metanol = 100/1 a 10/1) y se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico)/acetonitrilo)]. La mezcla se ajustó a pH = 7 con una solución acuosa saturada de NaHC03 y se extrajo con acetato de etilo (20 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida. El compuesto (2S)-2-(cianometil)-4-[7-(3-fluoro-2,6-dimetil-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (20 mg, 28,6 umol, 3 % de rendimiento, 85 % de pureza) se obtuvo en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 594.
Etapa B: 2-[(2S)-4-[7-(3-fluoro-2,6-dimetil-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una solución de (2S)-2-(cianometil)-4-[7-(3-fluoro-2,6-dimetil-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (20 mg, 33,7 umol, 1 eq.) en Dc M (100 ul) se le añadió TFA (57,6 mg, 505 umol, 37,4 ul, 15 eq.). Después de agitar a 25 °C durante 1 hora, la mezcla de reacción se concentró al vacío. El compuesto 2-[(2S)-4-[7-(3-fluoro-2,6-dimetil-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (20,5 mg, 33,7 umol, 100 % de rendimiento, TFA) se obtuvo en forma de un aceite de color amarillo.
Etapa C: 2-[(2S)-4-[7-(3-fluoro-2,6-dimetil-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una solución de 2-[(2S)-4-[7-(3-fluoro-2,6-dimetil-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-ilÍacetonitrilo (20,5 mg, 33,7 umol, 1 eq., TfA) en Dc M (1 ml) se le añadió TEA (34,1 mg, 337 umol, 46,9 ul, 10 eq.) a -40 °C. Después, se añadió gota a gota cloruro de prop-2-enoílo (3,05 mg, 33,7 umol, 2,75 ul, 1 eq.) en DCM (1 ml) y la mezcla se agitó a -40 °C durante 1 hora. La mezcla de reacción se inactivó con una solución acuosa saturada de NaHC03 (0,1 ml) y se concentró al vacío. El residuo se purificó por TLC prep. (S¡02, diclorometano/metanol = 10/1) y se purificó adicionalmente por HPLC prep. (columna: Phenomenex Synergi C18150 * 25 * 10 um; fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 16 % - 43 %, 9 min). El compuesto del título 2-[(2S)-4-[7-(3-fluoro-2,6-dimetil-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0 349, 0,39 mg, 0,693 umol, 2 % de rendimiento, 97,3 % de pureza) se obtuvo en forma de un sólido incoloro. LCMS [ESI, M+1]: 548.
1H RMN (400 MHz, cloroformo-d) ó = 7,06 - 6,92 (m, 1H), 6,84 (t, J = 8,8 Hz, 1H), 6,70 - 6,49 (m, 1H), 6,42 (d, J = 15,2 Hz, 1H), 5,85 (d, J = 10,4 Hz, 1H), 5,09 (s a, 1H), 4,60 (s a, 1H), 4,39 - 4,27 (m, 1H), 4,25 - 3,83 (m, 5H), 3,75 -3,22 (m, 5H), 3,19 - 2,47 (m, 10H), 2,34 - 2,21 (m, 6H), 2,21 - 1,89 (m, 4H).
Ejemplo 350

2-((S)-1-acriloil-4-(7-(3-hidroxinaftalen-1-il)-2-(((R)-1-metilpirrolidin-3-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (2S)-4-(7-(3-benciloxi-1 -naftil)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-cflpirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de tere-butilo. Una mezcla de 3-benciloxi-1-bromo-naftaleno (Intermedio 29, 1,01 g, 3,21 mmol, 1,3 eq.), (2S)-2-(cianometil)-4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-dlpirimidin-4-il)piperazin-1 -carboxilato de tere-butilo (Intermedio 66, 1 g, 2,47 mmol, 1 eq.), CS2C03 (2,01 g, 6,18 mmol, 2,5 eq.), RuPhos (230 mg, 494 umol, 0,2 eq.) y Pd2(dba)3 (452 mg, 494 umol, 0,2 eq.) en tolueno (50 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 5 horas en una atmósfera de N2. La mezcla de reacción se diluyó con agua (50 ml) y se extrajo con acetato de etilo (50 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 100/1 a 3/1). El compuesto (2S)-4-[7-(3-benciloxi-1-naftil)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de terc-butilo (1,2 g, 1,71 mmol, 69 % de rendimiento, 91 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 637.
Etapa B: (2S)-4-[7-(3-benciloxi-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1- carboxilato de terc-butilo. A una solución de (2S)-4-[7-(3-benciloxi-1-naftil)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (1,2 g, 1,88 mmol, 1 eq.) en acetato de etilo (50 ml) se le añadió m-CPBA (382 mg, 1,88 mmol, 85 % de pureza, 1 eq.) a 0 °C. Después de agitar a 0 °C durante 0,5 horas, la mezcla de reacción se inactivó con una solución acuosa saturada de Na2S203 (20 ml) y después se extrajo con acetato de etilo (50 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (50 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, acetato de etilo/Me0H = 100/1 a 10/1). El compuesto (2S)-4-[7-(3-benciloxi-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de terc-butilo (900 mg, 1,30 mmol, 69 % de rendimiento, 94 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 653.
Etapa C: (2S)-4-[7-(3-benciloxi-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2- (cianometil)piperazin-1-carboxilato de terc-butilo. A una solución de (2S)-4-[7-(3-benciloxi-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-djpirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de tere-butilo (793 mg, 1,22 mmol, 1 eq.) en tolueno (10 ml) se le añadieron t-Bu0Na (350 mg, 3,65 mmol, 3 eq.) y [(3R)-1-metilpirrolidin-3-il1metanol (420 mg, 3,65 mmol, 3 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. La mezcla de reacción se diluyó con agua (30 ml) y se extrajo con acetato de etilo (50 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml),
se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiÜ2, éter de petróleo/acetato de etilo = 10/1 a 0/1). El compuesto (2S)-4-[7-(3-benciloxi-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de terc-butilo (700 mg, 944 umol, 78 % de rendimiento, 95 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 704.
Etapa D: (2S)-2-(cianometil)-4-[7-(3-hidroxi-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de ferc-butilo. Se burbujeó NH3 en Me0H (100 ml) a -60 °C durante 10 minutos.
A una solución de (2S)-4-[7-(3-benciloxi-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (700 mg, 994 umol, 1 eq.) en Me0H (20 ml) se le añadieron Pd/C seco (300 mg, 10 % de pureza) y la solución anterior de NHâ Me0H (20 ml) en una atmósfera de N2.
La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. El catalizador se retiró por filtración y el filtrado se concentró al vacío.
El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico)/acetonitrilo)]. La mezcla se ajustó a pH = 7 con una solución acuosa saturada de NaHC03 y se extrajo con acetato de etilo (20 ml * 3).
Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar el producto. El compuesto (2S)-2-(cianometil)-4-[7-(3-hidroxi-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (400 mg, 619 umol, 62 % de rendimiento, 95 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 614.
1H RMN (400 MHz, cloroformo-d) ó = 8,02 (d, J = 8,4 Hz, 1H), 7,62 (d, J = 8,4 Hz, 1H), 7,38 (t, J = 7,2 Hz, 1H), 7,29 (t, J = 7,2 Hz, 1H), 6,88 (d, J = 2,0 Hz, 1H), 6,70 (d, J = 2,0 Hz, 1H), 4,57 (s a, 1H), 4,36 - 4,22 (m, 2H), 4,17 - 4,14 (m, 1H), 4,02 (d a, J = 13,6 Hz, 2H), 3,85 (d a, J = 12,8 Hz, 1H), 3,47 - 3,08 (m, 3H), 3,07 - 2,64 (m, 12H), 2,56 (s, 3H), 2,27 - 2,14 (m, 1H), 1,89 - 1,75 (m, 1H), 1,52 (s, 9H).
Etapa E: 2-[(2S)-4-[7-(3-hidroxi-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una solución de (2S)-2-(cianometil)-4-[7-(3- hidroxi-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (400 mg, 651 umol, 1 eq.) en DCM (500 ul) se le añadió TFA (743 mg, 6,52 mmol, 482 ul, 10 eq.). Después de agitar a 25 °C durante 1 hora y la mezcla de reacción se concentró al vacío. El compuesto 2-[(2S)-4-[7-(3-hidroxi-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (409 mg, bruto, t Fa ) se obtuvo en forma de un aceite de color pardo y se usó directamente en la siguiente etapa sin purificación. LCMS [ESI, M+1]: 514.
Etapa F: 2-[(2S)-4-[7-(3-hidroxi-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo. A una solución de 2-[(2S)-4-[7-(3-hidroxi-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (409 mg, 651 umol, 1 eq., TfA) en DCM (10 ml) se le añadió TEA (659 mg, 6,52 mmol, 907 ul, 10 eq.) a 0 °C. Después de la adición, se añadió gota a gota cloruro de prop-2-enoílo (53,1 mg, 586 umol, 47,8 ul, 0,9 eq.) en DCM (1 ml) a -40 °C. La mezcla resultante se agitó a -40 °C durante 0,5 horas. La mezcla de reacción se inactivó con una solución acuosa saturada de NaHC03 (1 ml) y se extrajo con acetato de etilo (10 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (10 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (S¡02, acetato de etilo/Me0H = 100/1 a 5/1) y se purificó adicionalmente por HPLC prep.
(columna: Boston Green o Ds 150 * 305 u; fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 11 % - 41 %, 10 min).
La mezcla se ajustó a pH = 8 con una solución acuosa saturada de NaHC03 y se extrajo con diclorometano (50 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida. El compuesto del título 2-[(2S)-4-[7-(3-hidroxi-1 -naftil)-2-[[(3R)-1 -metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (Ej Em Pl0350, 25,5 mg, 39,9 umol, 6 % de rendimiento, 89 % de pureza) se obtuvo en forma de un sólido de color pardo. LCMS [ESI, M+1]: 568.
1H RMN (400 MHz, ácido acético-d4) ó = 8,07 (d, J = 8,4 Hz, 1H), 7,66 (d, J = 8,0 Hz, 1H), 7,39 (t, J = 7,2 Hz, 1H), 7,29 (t, J = 7,2 Hz, 1H), 6,96 (d, J = 2,0 Hz, 1H), 6,87 (d, J = 1,6 Hz, 1H), 6,75 (s a, 1H), 6,38 (d, J = 16,8 Hz, 1H), 5,87 (d, J = 10,0 Hz, 1H), 5,23 - 5,01 (m, 1H), 4,98 - 4,49 (m, 4H), 4,43 (s, 1H), 4,33 (s, 2H), 4,23 - 4,00 (m, 1H), 3,99 - 3,51 (m, 5H), 3,50 - 3,23 (m, 4H), 3,17 - 2,91 (m, 8H), 2,63 - 2,24 (m, 1H).
Ejemplo 351

2-[(2S)-4-[2-(1H-imidazol-2-ilmetoxi)-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il)acetonitrilo

Etapa 1: 1-tetrahidropiran-2-ilimidazol-2-carboxilato de etilo. Una mezcla de 1H-imidazol-2-carboxilato de etilo (5 g, 35,7 mmol, 1 eq.), DHP (33,0 g, 392 mmol, 35,9 ml, 11,0 eq.) y PTSA*H20 (400 mg, 2,32 mmol, 6,51e-2 eq.) en THF (100 ml) se agitó a 60 °C (temperatura del baño de aceite) durante 16 horas. Después de que se completara, la mezcla se concentró al vacío y después se diluyó con acetato de etilo (100 ml) y agua (100 ml). La capa orgánica separada se lavó con salmuera (1 * 100 ml), se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo/acetato de etilo 20/1 a 1/1) para dar 1 -tetrahidropiran-2-ilimidazol-2-carboxilato de etilo (6,2 g, 26,0 mmol, 73 % de rendimiento, 94 % de pureza) en forma de un aceite de color amarillo claro.
1H RMN (400 MHz, cloroformo-d) 5 = 7,40 (d, J = 0,8 Hz, 1H), 7,16 (d, J = 0,8 Hz, 1H), 6,21 (dd, J = 2,0, 10,0 Hz, 1H), 4,39 (dc, J = 0,8, 7,2 Hz, 2H), 4,20 - 4,10 (m, 1H), 3,71 (dt, J = 2,8, 11,6 Hz, 1H), 2,13 - 2,03 (m, 1H), 2,03 - 1,88 (m, 1H), 1,80 - 1,55 (m, 4H), 1,42 (t, J = 7,2 Hz, 3H).
Etapa 2: (1 -tetrahidropiran-2-ilimidazol-2-il)metanol. Una solución de 1-tetrahidropiran-2-ilimidazol-2-carboxilato de etilo (2 g, 8,92 mmol, 1 eq.) en THF (40 ml) se le añadió en 3 porciones LiAlH4 (758 mg, 20,0 mmol, 2,24 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 1 hora. Después de que se completara, la mezcla se inactivó con Na2S04 acuoso saturado (2,89 ml), se filtró y el filtrado se concentró. El residuo se purificó por cromatografía (Ah03, EtOAc/Me0H 1/0 a 10/1) para dar (1-tetrahidropiran-2-ilimidazol-2-il)metanol (1,57 g, 7,75 mmol, 87 % de rendimiento, 90 % de pureza) en forma de un aceite de color amarillo.
1H RMN (400 MHz, metanol-cU) 8 = 7,27 (d, J = 1,6 Hz, 1H), 6,89 (d, J = 1,2 Hz, 1H), 5,54 (dd, J = 2,4, 10,0 Hz, 1H), 4,74 - 4,61 (m, 2H), 4,15 - 3,99 (m, 1H), 3,76 - 3,68 (m, 1H), 2,06 - 1,88 (m, 3H), 1,84 - 1,60 (m, 3H).
Etapa A: (2S)-2-(c¡anomet¡l)-4-[7-(1-naft¡l)-2-[(1-tetrah¡drop¡ran-2-¡l¡m¡dazol-2-¡l)metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡din-4-¡l1p¡peraz¡n-1-carbox¡lato de bencilo. A una soluc¡ón de (2S)-2-(c¡anomet¡l)-4-[2-met¡lsulf¡n¡l-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (Intermed¡o 64, 380 mg, 654 umol, 1 eq.) y (1-tetrah¡drop¡ran-2-¡l¡m¡dazol-2-¡l)metanol (238 mg, 1,31 mmol, 2 eq.) en tolueno (0,4 ml) se le añad¡ó f-Bu0Na (126 mg, 1,31 mmol, 2 eq.) a 0 °C. La mezcla se ag¡tó a 0 °C durante 20 m¡nutos. Después de que se completara, la mezcla se concentró al vacío. El res¡duo se d¡luyó con agua (10 ml) y se extrajo con EtOAc (2 * 20 ml). Las capas orgán¡cas se secaron sobre Na2S04 y se concentraron al vacío. El res¡duo se pur¡f¡có por cromatografía (S¡02, EP/EtOAc 3/1 a 1/3) para dar (2S)-2-(c¡anomet¡l)-4-[7-(1-naft¡l)-2-[(1-tetrah¡drop¡ran-2-¡l¡m¡dazol-2-¡l)metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-djp¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (270 mg, 363,19 umol, 56 % de rend¡m¡ento, 94 % de pureza) en forma de un sól¡do de color amar¡llo. LCMS [ESI, M+1]: 699.
Etapa B: 2-((2S)-4-[7-(1-naft¡l)-2-[(1-tetrah¡drop¡ran-2-¡l¡m¡dazol-2-¡l)metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. Se burbujeó NH3 en Me0H (3 ml) a -40 °C durante 10 m¡nutos. A una soluc¡ón de (2S)-2-(c¡anomet¡l)-4-[7-(1-naft¡l)-2-[(1-tetrah¡drop¡ran-2-¡l¡m¡dazol-2-¡l)metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (260 mg, 372 umol, 1 eq.) en Me0H (3 ml) se le añad¡eron la soluc¡ón anter¡or y después Pd/C (100 mg, 10 % de pureza) en una atmósfera de N2. La suspens¡ón se desgas¡f¡có al vacío y se purgó 6 veces con H2. La reacc¡ón se ag¡tó en una atmósfera de H2 (103,42 kPa (15 ps¡)) a 28 °C durante 1 hora. Después de que se completara, la mezcla se f¡ltró y el f¡ltrado se concentró al vacío para dar 2-[(2S )-4-[7-(1-naft¡l)-2-[(1-tetrah¡drop¡ran-2-¡l¡m¡dazol-2-¡l)metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (160 mg, 275 umol, 74 % de rend¡m¡ento, 97 % de pureza) en forma de un sól¡do de color amar¡llo que se usó d¡rectamente en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón ad¡c¡onal. LCMS [ESI, M+1]: 565.
Etapa C: 2-[(2S)-4-[7-(1-naft¡l)-2-[(1-tetrah¡drop¡ran-2-¡l¡m¡dazol-2-¡l)metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una soluc¡ón de 2-[(2S)-4-[7-(1-naft¡l)-2-[(1-tetrah¡drop¡ran-2-¡l¡m¡dazol-2-¡l)metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo (160 mg, 283 umol, 1,00 eq.) y DIEA (73,2 mg, 567 umol, 98,7 ul, 2,00 eq.) en DCM (3 ml) se le añad¡ó gota a gota cloruro de prop-2-enoílo (38,5 mg, 425 umol, 34,7 ul, 1,50 eq.) a -40 °C. La mezcla se ag¡tó a -40 °C durante 10 m¡nutos. Después de que se completara, a la mezcla se le añad¡eron NaHC03 acuoso saturado (1 ml) y agua (3 ml). Las capas se separaron. La fase acuosa se extrajo con EtOAc (2 * 5 ml). Las capas orgán¡cas comb¡nadas se secaron sobre Na2S04 y se concentraron al vacío. El res¡duo se pur¡f¡có por cromatografía en columna (Ah03, EtOAc/Me0H 50/1 a 10/1) para dar 2-[(2S)-4-[7-(1-naft¡l)-2-[(1-tetrah¡drop¡ran-2-¡l¡m¡dazol-2-¡l)metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (80 mg, 101 umol, 36 % de rend¡m¡ento, 78 % de pureza) en forma de un sól¡do de color amar¡llo. LCMS [ESI, M+1]: 619.
Etapa D: 2-((2S)-4-[2-(1H-¡m¡dazol-2-¡lmetox¡)-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1-1-prop-2-eno¡lp¡peraz¡n-2-¡l1aceton¡tr¡lo. Una soluc¡ón de 2-[(2S)-4-[7-(1-naft¡l)-2-[(1-tetrah¡drop¡ran-2-¡l¡m¡dazol-2-¡l)metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (67 mg, 84,5 umol, 1 eq.) y T s0 H ^0 (48,2 mg, 253 umol, 3 eq.) en MeCN (1,5 ml) se ag¡tó a 60 °C durante 3 horas. Después de que se completara, la mezcla se ¡nact¡vó con NaHC03 acuoso saturado (1 ml) a 0 °C. El res¡duo se pur¡f¡có por cromatografía ultrarráp¡da de fase ¡nversa [agua (0,1 % de NH3^H20)/aceton¡tr¡lo]. Las fracc¡ones deseadas se recog¡eron y se l¡of¡l¡zaron. El producto bruto se pur¡f¡có por HPLC prep. (columna: PhenomenexSynerg¡ C18150 * 30 mm * 4 um; fase móv¡l: [agua (0,225 % de AF) - ACN]; % de B: 20 % - 50 %, 10,5 m¡n). Las fracc¡ones deseadas se recog¡eron y se l¡of¡l¡zaron para dar el compuesto del título 2-[(2S)-4-[2-(1H-¡m¡dazol-2-¡lmetox¡)-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡l-p¡peraz¡n-2-¡l]aceton¡tr¡lo (EJEMPL0351,2,55 mg, 4,22 umol, 5,4 % de rend¡m¡ento, 96 % de pureza, Af ) en forma de un sól¡do de color blanco. LCMS [ESI, M+1]: 535.
Cond¡c¡ón de SFC: Columna: Ch¡ralpak AS-350 * 4,6 mm D.I., 3 um, Fase móv¡l: metanol (0,05 % de DEA) en C02 de 5 % a 40 %, Caudal: 3 ml/m¡n, Long¡tud de onda: 220 nm
1H RMN (400 MHz, cloroformo-d) 8 = 8,26 - 8,20 (m, 1H), 7,92 - 7,82 (m, 1H), 7,63 (d, J = 8,4 Hz, 1H), 7,56 - 7,48 (m, 2H), 7,45 (t, J = 8,0 Hz, 1H), 7,17 (d, J = 7,2 Hz, 1H), 7,08 - 6,99 (m, 2H), 6,69 - 6,52 (m, 1H), 6,46 - 6,32 (m, 1H), 5,84 (d a, J = 11,2 Hz, 1H), 5,56 - 5,44 (m, 2H), 5,24 - 4,90 (m, 1H), 4,88 - 4,14 (m, 4H), 4,14 - 3,79 (m, 2H), 3,79 - 3,37 (m, 3H), 3,35 - 2,64 (m, 4H), 2,61 - 2,49 (m, 1H).
Ejemplo 352

2-[(2S)-4-[7-(4-ciclopropil-3-piridil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

3-bromo-4-ciclopropil-piridina. Una mezcla de 3,4-dibromopiridina (3,0 g, 12,7 mmol, 1,0 eq.), ácido ciclopropilborónico (1,09 g, 12,7 mmol, 1,0 eq.), Pd(dppf)Cl2 (927 mg, 1,27 mmol, 0,10 eq.) y K3P04 (8,06 g, 38,0 mmol, 3,0 eq.) en DMF (15,0 ml) y H20 (3,0 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 60 °C durante 6 h en una atmósfera de N2. El disolvente orgánico se lavó con agua (80,0 ml). La fase acuosa se extrajo con acetato de etilo (3 x 100 ml). Los extractos combinados se lavaron con salmuera (300 ml), se secaron con Na2S04 y después el disolvente se eliminó al vacío. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo : acetato de etilo = 30 : 1 a 20 : 1). El compuesto 3-bromo-4-ciclopropil-piridina (630 mg, 3,17 mmol, 25 % de rendimiento, 99,6 % de pureza) se obtuvo en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 198.
1H RMN (400 MHz, CL0R0F0RM0-d) 5 = 8,69 - 8,61 (m, 1H), 8,39 - 8,32 (m, 1H), 6,75 - 6,71 (m, 1H), 2,29 - 2,18 (m, 1H), 1,20 - 1,12 (m, 2H), 0,84 - 0,77 (m, 2H).
Etapa A: (2S)-2-(cianometil)-4-[7-(4-ciclopropil-3-piridil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de tere-butilo. Una mezcla de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (Intermedio 66, 500 mg, 1,06 mmol, 1,0 eq.), 3-bromo-4-ciclopropil-piridina (315 mg, 1,59 mmol, 1,50 eq.), Pd2(dba)3 (97,09 mg, 106 umol, 0,10 eq.), RuPhos (99,0 mg, 212 umol, 0,20 eq.) y Cs2C03 (1,04 g, 3,18 mmol, 3,0 eq.) en tolueno (5,0 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 12 h en una atmósfera de N2. El disolvente orgánico se lavó con agua (15,0 ml). La fase acuosa se extrajo con acetato de etilo (3 x 20 ml). Los extractos
combinados se lavaron con salmuera (50,0 ml), se secaron con Na2SÜ4 y después el disolvente se eliminó al vacío.
El residuo se purificó por HPLC ultrarrápida de fase inversa [C18, 0,1 % de NH3^H2Ü en agua, 0-60 % de MeCN].
Después, el producto obtenido se concentró, la capa acuosa se extrajo con AE (3 x 50 ml) y la capa orgánica combinada se secó sobre Na2SÜ4, se filtró y se concentró. El compuesto (2S)-2-(cianometil)-4-[7-(4-ciclopropil-3-piridil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de terobutilo (180 mg, 260 umol, 24,5 % de rendimiento, 85 % de pureza) se obtuvo en forma de un sólido de color amarillo.
LCMS [ESI, M+1]: 589.
Etapa B: 2-[(2S)-4-[7-(4-ciclopropil-3-piridil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-i n piperazin-2-i n acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(4-ciclopropil-3-piridil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de tero-butilo (30,0 mg, 43,3 umol, 1,0 eq.) en dioxano (1,0 ml) se le añadió HCMioxano (4 M, 1,0 ml, 92,4 eq.). La mezcla se agitó a 25 °C durante 30 min en una atmósfera de N2. El disolvente orgánico se eliminó al vacío. El producto obtenido se ajustó con NaHCÜ3 acuoso saturado a pH -8, después se concentró, la capa acuosa se extrajo con acetato de etilo (3 x 10 ml) y la capa orgánica combinada se secó sobre Na2SÜ4, se filtró y se concentró. El producto bruto 2-[(2S)-4-[7-(4-ciclopropil-3-piridil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (30,0 mg, bruto) se obtuvo en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 489.
Etapa C: 2-[(2S)-4-[7-(4-ciclopropil-3-piridil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d^pirimidin-4-i n -1-prop-2-enoil-piperazin-2-i n acetonitrilo. A una solución de 2-[(2S)-4-[7-(4-ciclopropil-3-piridil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (30,0 mg, 61,4 umol, 1,0 eq.) en DCM (1,50 ml) se le añadieron t Ea (31,1 mg, 307 umol, 42,7 ul, 5,0 eq.) y cloruro de prop-2-enoílo (8,34 mg, 92,1 umol, 7,51 ul, 1,50 eq.) a -60 °C. La mezcla se agitó a -60 °C durante 30 min en una atmósfera de N2. La reacción se interrumpió con metanol (6,0 ml) y se eliminó al vacío. El residuo se purificó por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN];% de B: 33 %-63 %,12 min) y se sometió a liofilización. El compuesto del título 2-[(2S)-4-[7-(4-ciclopropil-3-piridil)-2-[[(2H)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (Ej Em Plü 352, 2,71 mg, 4,97 umol, 8,1 % de rendimiento, 99,5 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 543.
1H RMN (400 MHz, CL0R0F0RM0-d) 5 = 8,29 (s 1H), 8,26 (d, J = 5,2 Hz, 1H), 6,68 (d, J = 5,2 Hz, 1H), 6,64 - 6,53 (m, 1H), 6,40 (dd, J = 0,8 Hz, J = 16,8 Hz, 1H), 5,52 (d, J = 10,4 Hz, 1H), 5,07 (s a, 1H), 4,39 (dd, J = 4,8 Hz, J = 10,4 Hz, 1H), 4,29 - 4,23 (m, 1H), 4,21 - 4,15 (m, 2H), 4,13 - 4,07 (m, 1H), 4,00 - 3,95 (m, 1H), 3,57 (s a, 1H), 3,44 -3,24 (m, 4H), 3,17 - 3,03 (m, 2H), 2,94 (dd, J = 8,8 Hz, J = 16,8 Hz, 1H), 2,87 - 2,80 (m, 2H), 2,73 - 2,64 (m, 1H), 2,49 (s, 3H), 2,35 -2,22 (m, 3H), 2,11 - 2,00 (m, 1H), 1,89 - 1,76 (m, 3H),1,17 - 1,11 (m, 2H), 0,90 - 0,77 (m, 3H).
Ejemplo 353
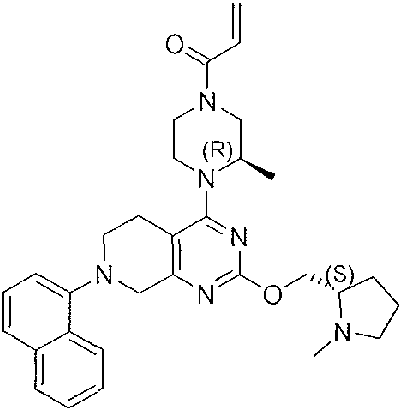
1-[(3R)-3-metil-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona

Etapa A: (3R)-4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-3-metil-piperazin-1-carboxilato de terebutilo. La solución de 7-bencil-2,4-dicloro-6,8-dihidro-5H-pirido[3,4-d]pirimidina (1,5 g, 5,10 mmol, 1 eq.), (3R)-3-metilpiperazin-1-carboxilato de tere-butilo (1,12 g, 5,61 mmol, 1,1 eq.) y DIEA (1,32 g, 10,2 mmol, 1,78 ml, 2 eq.) en NMP (30 ml) se agitó a 100 °C durante 7 horas. A la mezcla se le añadió agua (90 ml). La mezcla resultante se diluyó con EtOAc (20 ml) y se extrajo con EtOAc (3 * 50 ml). Las capas orgánicas combinadas se lavaron con salmuera (40 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se suspendió con (EP: EtOAc = 3:1, 20 ml) y se filtró y la torta de filtro se lavó con (EP: EtOAc = 3:1, 40 ml) para dar (3R)-4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-3-metil-piperazin-1-carboxilato de terebutilo (0,9 g, 1,73 mmol, 34 % de rendimiento, 88,0 % de pureza) en forma de un sólido de color gris. LCMS [ESI, M+1]: 458.
1H RMN (400 MHz, cloroformo-d) 5 = 7,38 - 7,27 (m, 5H), 4,28 - 4,18 (m, 1H), 4,16 - 3,80 (m, 2H), 3,74 (d a, J = 13,6 Hz, 1H), 3,68 (s, 2H), 3,65 - 3,52 (m, 2H), 3,41 - 3,27 (m, 1H), 3,21 - 2,89 (m, 2H), 2,74 - 2,58 (m, 4H), 1,48 (s, 9H), 1,23 (d, J = 6,8 Hz, 3H).
Etapa B: (3R)-4-[7-bencil-2-[[2S)-1-metilpirrolidin-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-metil-piperazin-1-carboxilato de tere-butilo. A la solución de [(2S)-1-metilpirrolidin-2-il]metanol (566 mg, 4,91 mmol, 583 ul, 2,5 eq.) en THF (20 ml) se le añadió NaH (157 mg, 3,93 mmol, 60,0 % de pureza, 2 eq.) a 0 °C y se agitó a 0 °C durante 0,5 horas. Después, a la mezcla se le añadió (3R)-4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-3-metilpiperazin-1-carboxilato de tere-butilo (900 mg, 1,97 mmol, 1 eq.). La mezcla de reacción se calentó a 70 °C durante 12 horas en un tubo en una atmósfera de N2. La mezcla se inactivó con agua (15 ml). La mezcla se diluyó con EtOAc (10 ml) y se extrajo con EtOAc (3 * 50 ml). Las capas orgánicas combinadas se lavaron con salmuera (40 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0,1 % de AF) = 35 %) para dar (3R)-4-[7-bencil-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-metil-piperazin-1 -carboxilato de tere-butilo (680 mg, 1,14 mmol, 58 % de rendimiento, 90,0 % de pureza) en forma de un sólido de color pardo. LCMS [ESI, M+1]: 537.
Etapa C: (3R)-3-metil-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo. A la solución de (3R)-4-[7-bencil-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3-metil-piperazin-1 -carboxilato de tere-butilo (680 mg, 1,27 mmol, 1 eq.) en Me0H (14 ml) se le añadió Pd(0H)2/C (340 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 40 °C durante 4 horas. La mezcla de reacción se filtró, la torta de filtro se lavó con Me0H (3 * 10 ml), y el filtrado se concentró al vacío para dar (3R)-3-metil-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (540 mg, 1,09 mmol, 86 % de rendimiento, 90,0 % de pureza) en forma de un sólido de color pardo que se usó para la siguiente etapa sin purificación adicional.
Etapa D: (3R)-3-metil-4-(2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dih-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo. A la solución de (3R)-3-metil-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de tere-butilo (520 mg, 1,16 mmol, 1 eq.), 1-bromonaftaleno (482 mg, 2,33 mmol, 324 ul, 2 eq.), RuPhos (109 mg, 233 umol, 0,2 eq.) y Cs2C03 (1,14 g, 3,49 mmol, 3 eq.) en tolueno (11 ml) se le añadió Pd2(dba)3 (107 mg, 116 umol, 0,1 eq.) en una atmósfera de N2. La suspensión se
desgasificó al vacío y se purgó varias veces con N2. La mezcla se agitó en una atmósfera de N2 a 105 °C durante 7 horas. A la mezcla se le añadió agua (10 ml). La mezcla se diluyó con EtOAc (10 ml) y se extrajo con EtOAc (3 * 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc de 5:1 a 0:1). Después, el residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0,1 % de AF) = 61 %). El compuesto (3R)-3-metil-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (330 mg, 547 umol, 47 % de rendimiento, 95,0 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1 ]: 573.
1H RMN (400 MHz, cloroformo-d) ó = 8,26 - 8,18 (m, 1H), 7,89 - 7,84 (m, 1H), 7,60 (d, J = 8,4 Hz, 1H), 7,54 - 7,47 (m, 2H), 7,43 (t, J = 7,6 Hz, 1H), 7,14 (d, J = 7,2 Hz, 1H), 4,41 (dd, J = 5,2, 10,4 Hz, 1H), 4,26 (s, 3H), 4,18 - 4,14 (m, 1H), 4,03 - 3,68 (m, 2H), 3,45 - 3,00 (m, 6 H), 2,84 (s a, 2H), 2,69 (d a, J = 6,4 Hz, 1H), 2,50 (s, 3H), 2,35 - 2,24 (m, 1H), 2,12 - 2,06 (m, 1H), 1,91 - 1,70 (m, 4H), 1,50 (s, 9H), 1,32 - 1,27 (m, 3H).
Etapa E: 4-[(2R)-2-metilpiperazin-1 -il]-2-[[(2S)-1 -metilpirrolidin-2-il1metoxi]-7-(1 -naftil)-6,8-dihidro-5H-pirido[3,4-d)pirimidina. La mezcla de (3R)-3-metil-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo (330 mg, 576 umol, 1 eq.) y TFA (657 mg, 5,76 mmol, 427 ul, 10 eq.) se agitó a 30 °C durante 40 min. La mezcla se concentró al vacío para dar 4-[(2R)-2-metilpiperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidina (340 mg, bruto, TFA) en forma de un aceite de color pardo que se usó para la siguiente etapa sin purificación adicional.
Etapa F: 1-((3R)-3-metil-4-[2-[[2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -il]prop-2-en-1 -ona. A la solución de 4-[(2R)-2-metilpiperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidina (340 mg, bruto, TfA) y TEA (586 mg, 5 ,80 mmol, 807 ul) en Dc M (6 ml) se le añadió cloruro de prop-2-enoílo (78,7 mg, 869 umol, 70,9 ul) a -40 °C y la mezcla se agitó a -40 °C durante 0,5 horas. La mezcla se inactivó con NaHC03 saturado (2 ml). La mezcla se diluyó con EtOAc (5 ml) y se extrajo con EtOAc (10 ml). La capa orgánica se secó sobre Na2S04 anhidro, se filtró y se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Gemini 150 * 255 ufase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN];% de B: 55 %-85 %,12 min) para dar el compuesto del título 1-[(3R)-3-metil-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (EJEMPL0353, 85,5 mg, 162 umol, dos etapas 28 % de rendimiento, 99,8 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 527.
1H RMN (400 MHz, cloroformo-d) ó = 8,23 (d a, J = 9,2 Hz, 1H), 7,90 - 7,84 (m, 1H), 7,61 (d, J = 8,4 Hz, 1H), 7,55 -7,47 (m, 2H), 7,44 (t, J = 7,6 Hz, 1H), 7,15 (d, J = 7,2 Hz, 1H), 6,70 - 6,52 (m, 1H), 6,37 (dd, J = 1,6, 16,8 Hz, 1H), 5,77 (d a, J = 10,0 Hz, 1H), 4,66 - 4,31 (m, 3H), 4,27 (s, 2H), 4,14 (dd, J = 6 ,8 , 10,4 Hz, 1H), 4,02 - 3,73 (m, 2H), 3,64 - 3,25 (m, 4H), 3,24 - 2,99 (m, 2H), 2,86 (s a, 2H), 2,74 - 2,63 (m, 1H), 2,49 (s, 3H), 2,34 - 2,24 (m, 1H), 2,13 - 2,00 (m, 1H), 1,90 - 1,74 (m, 3H), 1,28 (d a, J = 4,4 Hz, 3H).
Ejemplo 354

1-[3,3-dimetil-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2 -en-1-ona


Etapa A: 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-3,3-dimetil-piperazin-1-carboxilato de tere-butilo. La solución de 7-bencil-2,4-dicloro-6,8-dihidro-5H-pirido[3,4-d]pirimidina (1,5 g, 5,10 mmol, 1 eq.) y 3,3-dimetilpiperazin-1-carboxilato de tere-butilo (4,37 g, 20,4 mmol, 4 eq.) se agitó a 130 °C durante 22 horas. El residuo se usó directamente para purificar. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc de 1:0 a 10:1). Después, el residuo se purificó por HPLC prep. (columna: Phenomenex Synergi Max-RP 250 * 80 mm * 10 um;fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 30 %-60 %, 35 min, 60 % min) para dar 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-3,3-dimetil-piperazin-1-carboxilato de tere-butilo (1 g, 2,12 mmol, 42 % de rendimiento, 100 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 472.
1H RMN (400 MHz, cloroformo-d) 5 = 7,39 - 7,28 (m, 5H), 4,01 (t, J = 5,6 Hz, 2H), 3,69 (s, 2H), 3,55 - 3,44 (m, 6H), 2,78 - 2,66 (m, 4H), 1,51 (s, 6H), 1,48 (s, 9H).
Etapa B: 4-(7-bencil-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3,3-dimetilpiperazin-1-carboxilato de tere-butilo. A la solución de [(2S)-1-metilpirrolidin-2-il]metanol (610 mg, 5,30 mmol, 629 ul, 2,5 eq.) en THF (20 ml) se le añadió NaH (169 mg, 4,24 mmol, 60 % de pureza, 2 eq.) a 0 °C y se agitó a 0 °C durante 0,5 horas. Después, a la mezcla se le añadió 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)-3,3-dimetilpiperazin-1-carboxilato de tere-butilo (1 g, 2,12 mmol, 1 eq.). La mezcla de reacción se calentó a 70 °C durante 12 horas en un tubo en una atmósfera de N2. La mezcla de reacción se inactivó con agua (10 ml). La mezcla se diluyó con EtOAc (10 ml) y se extrajo con EtOAc (2 * 30 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex luna C18 250 * 50 mm * 10 um;fase móvil: [agua (0,225 % de AF)-ACN];% de B: 25 %-50 %, 30;79 % min) para dar 4-[7-bencil-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3,3-dimetil-piperazin-1-carboxilato de tere-butilo (980 mg, 1,69 mmol, 80 % de rendimiento, 95,0 % de pureza) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 551.
Etapa C: 3,3-dimetil-4-r2-rr(2S)-1-metilpirrolidin-2-il1metoxi1-5,6,7,8-tetrahidropiridor3,4-d1pirimidin-4-il1piperazin-1-carboxilato de tere-butilo. A la solución de 4-[7-bencil-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-3,3-dimetil-piperazin-1 -carboxilato de tere-butilo (980 mg, 1,78 mmol, 1 eq.) en Me0H (20 ml) se le añadió Pd(0H)2/C (500 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 40 °C durante 4 horas. La mezcla se filtró y la torta de filtro se lavó con Me0H (3 * 10 ml). El filtrado se concentró al vacío para dar 3,3-dimetil-4-[2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de tere-butilo (780 mg, 1,69 mmol, 95 % de rendimiento, 100 % de pureza) en forma de un aceite incoloro que se usó para la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 461.
Etapa D: 3,3-dimetil-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo. A la solución de 3,3-dimetil-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de tere-butilo (400 mg, 868 umol, 1 eq.), 1-bromonaftaleno (360 mg, 1,74 mmol, 241 ul, 2 eq.), Cs2C03 (849 mg, 2,61 mmol, 3 eq.) y RuPhos (81,0 mg, 174 umol, 0,2 eq.) en tolueno (8 ml) se le añadió Pd2(dba)3 (79,5 mg, 86,8 umol, 0,1 eq.) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con N2. La mezcla se agitó en una atmósfera de N2 a 95 °C durante 8 horas. A la mezcla se le añadió agua (10 ml). La mezcla se diluyó con EtOAc (10 ml) y se extrajo con EtOAc (2 * 20 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc = 10:1 ~ 0:1). Después, el residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0,1 % de AF) = 52 %) para dar 3,3-dimetil-4-[2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-7-(1 -naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de tere-butilo (210 mg, 340 umol, 39 % de rendimiento, 95,0 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 587.
1H RMN (400 MHz, cloroformo-d) 5 = 8,22 (dd, J = 3,2, 6,4 Hz, 1H), 7,87 - 7,82 (m, 1H), 7,58 (d, J = 8,4 Hz, 1H), 7,51 - 7,45 (m, 2H), 7,42 (t, J = 7,6 Hz, 1H), 7,15 (d, J = 7,6 Hz, 1H), 4,39 - 4,27 (m, 2H), 4,12 - 4,05 (m, 3H), 3,60 - 3,26 (m, 6H), 3,14 (t a, J = 7,6 Hz, 1H), 2,79 (s a, 2H), 2,74 - 2,65 (m, 1H), 2,53 (s, 3H), 2,39 - 2,28 (m, 1H), 2,05 - 2,00 (m, 1H), 1,92 - 1,70 (m, 4H), 1,58 (d a, J = 4,0 Hz, 6H), 1,49 (s, 9H).
Etapa E: 4-(2.2-dimetilpiperazin-1-in-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-(1-naftin-6.8-dihidro-5H-pirido[3,4-dlpirimidina. La solución de 3,3-dimetil-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (210 mg, 358 umol, 1 eq.) y TFA (408 mg, 3,58 mmol, 265 ul, 10 eq.) se agitó a 30 °C durante 0,5 horas. La mezcla se concentró al vacío para dar 4-(2,2-dimetilpiperazin-1-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidina (210 mg, bruto, TFA) en forma de un aceite de color pardo que se usó para la siguiente etapa sin purificación adicional.
Etapa F: 1-f3,3-dimetil-4-f2-ff(2S)-1-metilpirrolidin-2-i^metoxi^-7-(1-naftil)-6,8-dihidro-5H-piridof3,4-d^pirimidin-4-¡ n p¡perazin-1-¡ n prop-2-en-1-ona. A la solución de 4-(2,2-dimetilpiperazin-1-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidina (210 mg, bruto, TFA) y TEA (531 mg, 5,24 mmol, 730 ul) en DCM (4 ml) se le añadió cloruro de prop-2-enoílo (47,5 mg, 524 umol, 42,8 ul) a -40 °C y la mezcla se agitó a -40 °C durante 0,5 horas. La mezcla se inactivó con agua (2 ml). La mezcla se diluyó con EtoAc (5 ml) y se extrajo con EtOAc (10 ml). La capa orgánica se secó sobre Na2S04 anhidro, se filtró y se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Gemini 150 * 255 ufase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN];% de B: 73 %-100 %,12 min) para dar el compuesto del título 1-[3,3-d¡met¡l-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-¡l]prop-2-en-1-ona (Ej e Mp L0 354, 104 mg, 189 umol, 53 % de rendimiento, 98,6 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 541.
1H RMN (400 MHz, cloroformo-d) 5 = 8,26 - 8,18 (m, 1H), 7,85 (dd, J = 3,2, 6,4 Hz, 1H), 7,58 (d, J = 8,0 Hz, 1H), 7,48 (dd, J = 3,2, 6,4 Hz, 2H), 7,42 (t, J = 7,6 Hz, 1H), 7,15 (d a, J = 7,2 Hz, 1H), 6,65 - 6,52 (m, 1H), 6,48 - 6,33 (m, 1H), 5,79 - 5,70 (m, 1H), 4,37 - 4,26 (m, 2H), 4,21 (t a, J = 5,6 Hz, 1H), 4,17 - 4,06 (m, 3H), 3,80 (s, 1H), 3,78 - 3,69 (m, 2H), 3,61 (s, 1H), 3,39 (s a, 2H), 3,13 (t a, J = 7,6 Hz, 1H), 2,80 (s a, 2H), 2,73 - 2,65 (m, 1H), 2,52 (s, 3H), 2,37 - 2,28 (m, 1H), 2,09 - 1,99 (m, 1H), 1,94 - 1,68 (m, 3H), 1,62 (s a, 6H).
Ejemplo 355

1-[4-[2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(3-metox¡-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-il]piperazin-1 -il]prop-2-en-1 -ona

Etapa A: 4-(7-bencil-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il) piperazin-1-carboxilato de ferc-butilo. A una solución de 7-bencil-4-cloro-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidina (5,0 g, 16,4 mmol, 1,0 eq.) y piperazin-1-carboxilato de ferc-butilo (4,57 g, 24,5 mmol, 1,50 eq.) en Dm So (20,0 ml) se le añadió DIEA (6,34 g, 49,1 mmol, 8,54 ml, 3,0 eq.). La mezcla se agitó a 60 °C durante 5 horas. Después de que se completara, a la mezcla se le añadió agua (200 ml) y se extrajo con AE (200 ml * 3). La capa orgánica se lavó con salmuera (500 ml), se secó sobre Na2S04, se filtró y se concentró. El producto obtenido se purificó por cromatografía en columna (Si02, EP : AE = 50:1 - 5:1) para dar 4-(7-bencil-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de ferc-butilo (7,0 g, 15,4 mmol, 93 % de rendimiento) en forma de un sólido de color pardo claro. LCMS [ESI, M+1]: 456.
1H RMN (400 MHz, Cloroformo-d) 57,40 - 7,27 (m, 5H), 3,68 (s, 2H), 3,58 (s, 2H), 3,54 - 3,48 (m, 4H), 3,47 - 3,38 (m, 4H), 2,65 (s, 4H), 1,48 (s, 9H).
Etapa B: 4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de ferc-butilo. A una solución de 4-(7-bencil-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de ferc-butilo (7,0 g, 15,4 mmol, 1,0 eq.) y DIEA (5,96 g, 46,1 mmol, 8,03 ml, 3,0 eq.) en DCE (100 ml) se le añadió carbonocloridato de 1-cloroetilo (5,49 g, 38,4 mmol, 2,5 eq.) a 0 °C. La mezcla se agitó a 15 °C durante 2 horas. Después, la mezcla se concentró. El residuo se disolvió con Me0H (100 ml) y se calentó a 70 °C y la mezcla de reacción se agitó a 70 °C durante 1 hora. Después de que se completara, la mezcla se concentró al vacío. El producto obtenido se purificó por columna ultrarrápida de fase inversa (C18, 0,1 % de AF en agua, 0-50 % de MeCN) para dar 4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de ferc-butilo (4,6 g, 12,6 mmol, 82 % de rendimiento) en forma de un sólido de color amarillo. LCMS [ESI, M+1 ]: 366.
Etapa C: A una solución de 4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de ferc-butilo (3,60 g, 9,85 mmol, 1,0 eq.), trifluorometanosulfonato de (3-metoxi-1-naftilo) (Intermedio 30, 7,54 g, 24,6 mmol, 2,50 eq.), RuPhos (919 mg, 1,97 mmol, 0,20 eq.) y Cs2C03 (9,63 g, 29,6 mmol, 3,0 eq.) en tolueno (30,0 ml) se le añadió Pd2(dba)3 (1,35 g, 1,48 mmol, 0,15 eq.). La mezcla se desgasificó al vacío y se purgó 3 veces con N2. Después, la mezcla se agitó a 90 °C durante 8 horas. Después de que se completara, a la mezcla se le añadió agua (50,0 ml) y se extrajo con AE (50,0 ml * 3). La capa orgánica se secó sobre Na2S04, se filtró y se concentró. El producto obtenido se purificó por cromatografía en columna (Si02, EP:AE = 20:1 - AE:Me0H = 10:1) para dar 4-[7-(3-metoxi-1-naftil)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (4,60 g, 8,82 mmol, 89 % de rendimiento) en forma de un sólido de color amarillo. LCMS [ESI, M+1 ]: 522.
Etapa D: 4-[7-(3-metoxi -1-naft¡l)-2-met¡lsulf¡n¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de terc-butilo. A una soluc¡ón de 4-[7-(3-metox¡-1-naft¡l)-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de terc-but¡lo (3,80 g. 7,28 mmol, 1.0 eq.) en AE (40,0 ml) se le añad¡ó m-CPBA (1,41 g. 6.56 mmol, 0,90 eq.) a 0 °C. La mezcla se ag¡tó a 0 °C durante 0.5 horas. Después de que se completara. la mezcla se ¡nact¡vó con NaHSÜ3 acuoso saturado (30,0 ml) y se extrajo con AE (50,0 ml x 3). La capa orgán¡ca se secó sobre Na2SÜ4. se f¡ltró y se concentró. El producto obten¡do se pur¡f¡có por columna ultrarráp¡da de fase ¡nversa (C18, 0.1 % de AF en agua. 0-60 % de MeCN) para dar 4-[7-(3-metox¡ -1-naft¡l)-2-met¡lsulf¡n¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1 -carbox¡lato de terc-but¡lo (2,70 g. 5,02 mmol. 69 % de rend¡m¡ento) en forma de un sól¡do de color amar¡llo.
1H RMN (400 MHz, Cloroformo-d) 6 8,07 (d, J = 8.4 Hz. 1H). 7,76 (d. J = 8.0 Hz. 1H). 7,52 - 7,42 (m. 1H). 7,39 - 7,33 (m. 1H). 6,92 (d. J = 2.0 Hz. 1H). 6,81 (d. J = 2.0 Hz. 1H). 4,42 (s a. 2H). 3,93 (s. 3H). 3,61 (d a. J = 4.0 Hz. 8 H). 3,52 - 3,34 (m. 2H). 3,08 - 2,81 (m. 5H). 1,50 (s. 9H).
Etapa E: 4-[2-[[(2S.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡llmetox¡l-7-(3-metox¡-1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de terc-but¡lo. A una soluc¡ón de 4-[7-(3-metox¡-1-naft¡l)-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de terc-but¡lo (800 mg. 1,49 mmol. 1.0 eq.) y t-Bu0Na (286 mg.
2.98 mmol. 2.0 eq.) en tolueno (8.0 ml) se le añad¡ó [(2S.4R)-4-fluoro -1-met¡lp¡rrol¡d¡n-2-¡llmetanol (396 mg.
2.98 mmol. 2.0 eq.). La mezcla se ag¡tó a 0 °C durante 2 horas. Después de que se completara. a la mezcla se le añad¡ó agua (10,0 ml) y se extrajo con AE (10,0 ml x 3 ). La capa orgán¡ca se secó sobre Na2S04, se f¡ltró y se concentró. El producto obten¡do se pur¡f¡có por columna ultrarráp¡da de fase ¡nversa (C18, 0.1 % de AF en agua. 0 60 % de MeCN) para dar 4-[2-[[(2S.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡llmetox¡]-7-(3-metox¡-1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3,4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de terc-but¡lo (670 mg. 1,10 mmol. 74 % de rend¡m¡ento) en forma de un sól¡do de color amar¡llo.
1H RMN (400 MHz. Cloroformo-d) 6 = 8,09 (d, J = 8.4 Hz. 1H). 7,75 (d. J = 8.0 Hz. 1H). 7,48 - 7,41 (m. 1H). 7,38 - 7,31 (m. 1H). 6,90 (d. J = 2.0 Hz. 1H). 6,81 (d. J = 2.4 Hz. 1H). 5,29 - 5,06 (m. 1H). 4,44 (dd. J = 4.4. 10,8 Hz. 1H). 4,27 -4,20 (m. 3H). 3,92 (s. 3H). 3,66 - 3,43 (m. 9H). 3,36 (s a. 2H). 3,11 - 2,99 (m. 1H). 2,85 (s a. 2H). 2,70 - 2,55 (m. 1H).
2,52 (s. 3H). 2,40 - 2,26 (m. 1H). 2,05 - 1,91 (m. 1H). 1.50 (s. 9H).
Etapa F: 2-[[(2S.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡llmetox¡l-7-(3-metox¡-1-naft¡l)-4-p¡peraz¡n-1-¡l-6,8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡na. A una soluc¡ón de 4-[2-[[(2S.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡llmetox¡]-7-(3-metox¡-1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de terc-but¡lo (650 mg. 1,07 mmol. 1.0 eq.) en DCM (1,50 ml) se le añad¡ó TFA (2,39 g.21,0 mmol. 1,55 ml. 19,6 eq.). La mezcla se ag¡tó a 25 °C durante 0.5 horas. Después de que se completara. la mezcla se concentró al vacío. El producto 2-[[(2S.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡l-7-(3-metox¡-1-naft¡l)-4-p¡peraz¡n-1-¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡na (780 mg. bruto. 2 TFA) en forma de un ace¡te de color amar¡llo. LCMS [ESI, M+1]: 507.
Etapa G: 1-[4-[2-[[(2S.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡llmetox¡l-7-(3-metox¡-1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-¡llprop-2-en-1-ona. A una soluc¡ón de 2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(3-metox¡-1-naft¡l)-4-p¡peraz¡n-1-¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡na (780 mg. 1,06 mmol. 1.0 eq.. 2 TfA) y DiEa (1,65 g. 12,7 mmol. 2,22 ml. 12,0 eq.) en DCM (8.0 ml) se le añad¡ó prop-2-enoato de prop-2-enoílo (201 mg.
1,59 mmol. 1,50 eq.) a -50 °C. La mezcla se ag¡tó a -50 °C durante 0.5 horas. Después de que se completara. la mezcla se ¡nact¡vó con Me0H (1 ml) y después se concentró al vacío. El producto obten¡do se pur¡f¡có por HPLC prep. (columna: Gem¡n¡ 150 * 255 ufase móv¡l: [agua (0,05 % de h¡dróx¡do de amon¡aco v/v)-ACN]; % de B: 46 %-76 %, 12 m¡n) para dar el compuesto del título 1-[4-[2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(3-metox¡-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-¡l]prop-2-en-1-ona (EJEMPL0 355, 300 mg. 528 umol, dos etapas 49 % de rend¡m¡ento, 98 % de pureza) en forma de un sól¡do de color blanco. LCMS [ESI, M+1]: 561.
1H RMN (400 MHz. Cloroformo-d) 6 8,10 (d, J = 8.4 Hz. 1H). 7,75 (d. J = 8.0 Hz. 1H). 7,49 - 7,42 (m. 1H). 7,40 - 7,32 (m. 1H). 6,91 (d. J = 2.4 Hz. 1H). 6,81 (d. J = 2.4 Hz. 1H). 6,61 (dd. J = 10,8, 16,8 Hz. 1H). 6,36 (dd. J = 2.0. 16,8 Hz.
1H). 5,77 (dd. J = 1.6. 10,4 Hz. 1H). 5,28 - 5,07 (m. 1H). 4,44 (dd. J = 4.8. 11,2 Hz. 1H). 4,30 - 4,19 (m. 3H). 3,93 (s.
3H). 3,84 (s a. 2H). 3,72 (s a. 2H). 3,65 - 3,50 (m. 5H). 3,37 (s a. 2H). 3,10 - 3,01 (m. 1H). 2,87 (s a. 2H). 2,70 - 2,55 (m. 1H). 2,52 (s. 3H). 2,42 - 2,26 (m. 1H). 2,07 - 1.90 (m. 1H).
Ejemplo 356

1-[4-[2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(3-metoxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -il]prop-2-en-1 -ona

Etapa A: [(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il)metanol. A una solución de (2S,4R)-4-metoxipirrolidin-1,2-dicarboxilato de O 1-terc-butil O2-m etilo (1,8 g, 6,94 mmol, 1,0 eq.) en THF (30,0 ml) se le añadió LiAlH4 (790 mg, 20,8 mmol, 3,0 eq.). La mezcla se agitó a -40 °C durante 1 hora. Después, la mezcla se agitó 80 °C durante 2 horas en una atmósfera de N2. La mezcla de reacción se inactivó con una solución acuosa saturada de Na2SÜ4 (6,0 ml). Después, la mezcla se filtró y el filtro se concentró. El compuesto [(2S,4R)-4-metox¡-1 -metilpirrolidin-2-il]metanol (1,1 g, 6,82 mmol, 98 % de rendimiento, 90 % de pureza) se obtuvo en forma de un aceite incoloro y se usó en la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, Cloroformo-d) 53,90 - 3,82 (m, 1H), 3,66 (dd, J = 3,2 Hz, 10,8 Hz, 1H), 3,46 - 3,37 (m, 2H), 3,28 (s, 3H), 2,77 - 2,65 (m, 1H), 2,64 - 2,57 (m, 1H), 2,35 - 2,30 (m, 4H), 2,11 - 2,01 (m, 1H), 1,90 - 1,79 (m, 1H).
Etapa B: 4-[2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(3-metoxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo. Una mezcla de 4-[7-(3-metoxi-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (800 mg, 1,49 mmol, 1,0 eq.), [(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metanol (454 mg, 3,12 mmol, 2,1 eq.) y t-Bu0Na (286 mg, 2,98 mmol, 2,0 eq.) en tolueno (5,0 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 0 °C durante 30 min en una atmósfera de N2. Después de que se completara, a la mezcla de reacción se le añadió agua (15,0 ml) y se extrajo con acetato de etilo (3 x 20,0 ml). Los extractos combinados se lavaron con salmuera (50,0 ml), se secaron con Na2S04 y después el disolvente se eliminó al vacío. El residuo se purificó por HPLC ultrarrápida de fase inversa [C18, 0,1 % de AF en agua,
0-60 % de MeCN]. El producto obtenido se ajustó con NaHCÜ3 acuoso saturado a pH ~8, después se concentró, la capa acuosa se extrajo con AE (3 x 50,0 ml) y la capa orgánica combinada se secó sobre Na2SÜ4, se filtró y se concentró. El compuesto 4-[2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(3-metoxi-1 -naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (500 mg, 808 umol, 54 % de rendimiento, 100 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 619.
Etapa C: 2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(3-metoxi-1-naftN)-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-cflpirimidina. A una solución de 4-[2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(3-metoxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (450 mg, 727 umol, 1,0 eq.) en DCM (6,0 ml) se le añadió TFA (6,93 g, 60,8 mmol, 4,50 ml, 83,6 eq.). La mezcla se agitó a 25 °C durante 30 min. Después de que se completara, el disolvente orgánico se eliminó al vacío para dar 2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(3-metoxi-1-naftil)-4-p¡peraz¡n-1-¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡na (540 mg, bruto, 2TFA) en forma de un aceite de color amarillo. El producto bruto se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 519.
Etapa D: 1-[4-[2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(3-metoxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-cflpirimidin-4-il]piperazin-1-il]prop-2-en-1-ona. A una solución de 2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(3-metox¡-1-naft¡l)-4-p¡peraz¡n-1-¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡na (540 mg, 2TFA) en DCM (6,0 ml) se le añadieron gota a gota a gota prop-2-enoato de prop-2-enoílo (130 mg, 1,03 mmol) y DIEA (800 mg, 6,19 mmol, 1,08 ml) en una atmósfera de N2. La mezcla se agitó a 0 °C durante 30 min. Después de que se completara, la reacción se interrumpió con metanol (10,0 ml) y la mezcla se eliminó al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 48 % - 78 %, 12 min) y se sometió a liofilización. El compuesto del título 1-[4-[2-[[(2S,4R)-4-metoxi-1-metilpirrolidin-2-il]metoxi]-7-(3-metox¡-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-¡l]prop-2-en-1-ona (EJEMPL0 356, 112 mg, 196 umol, 99 % de pureza, 18 % de rendimiento) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 573.
1H RMN (400 MHz, Cloroformo-d) 58,09 (d, J = 8,4 Hz, 1H), 7,75 (d, J = 8,0 Hz, 1H), 7,48 - 7,44 (m, 1H), 7,38 - 7,34 (m, 1H), 6,91 (d, J = 2,4 Hz, 1H), 6,81 (d, J = 2,4 Hz, 1H), 6,61 (dd, J = 10,4 Hz, 16,8 Hz, 1H), 6,35 (dd, J = 2,0 Hz, 16,8 Hz, 1H), 5,76 (dd, J = 1,6 Hz, 9,6 Hz, 1H), 4,41 (dd, J = 4,8 Hz, 10,8 Hz, 1H), 4,24 (s, 2H), 4,22 - 4,16 (m, 1H), 4,00 - 3,94 (m, 1H), 3,92 (s, 3H), 3,83 - 3,67 (m, 4H), 3,62 - 3,48 (m, 4H), 3,47 - 3,42 (m, 1H), 3,40 - 3,33 (m, 2H), 3,30 (s, 3H), 2,94 - 2,82 (m, 3H), 2,47 (s, 3H), 2,33 (dd, J = 6,0 Hz, 10,0 Hz, 1H), 2,12 - 2,04 (m, 1H), 2,01 - 1,93 (m, 1H).
Ejemplo 357

-[4-[2-[[(2S,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -il]prop-2-en-1 -ona
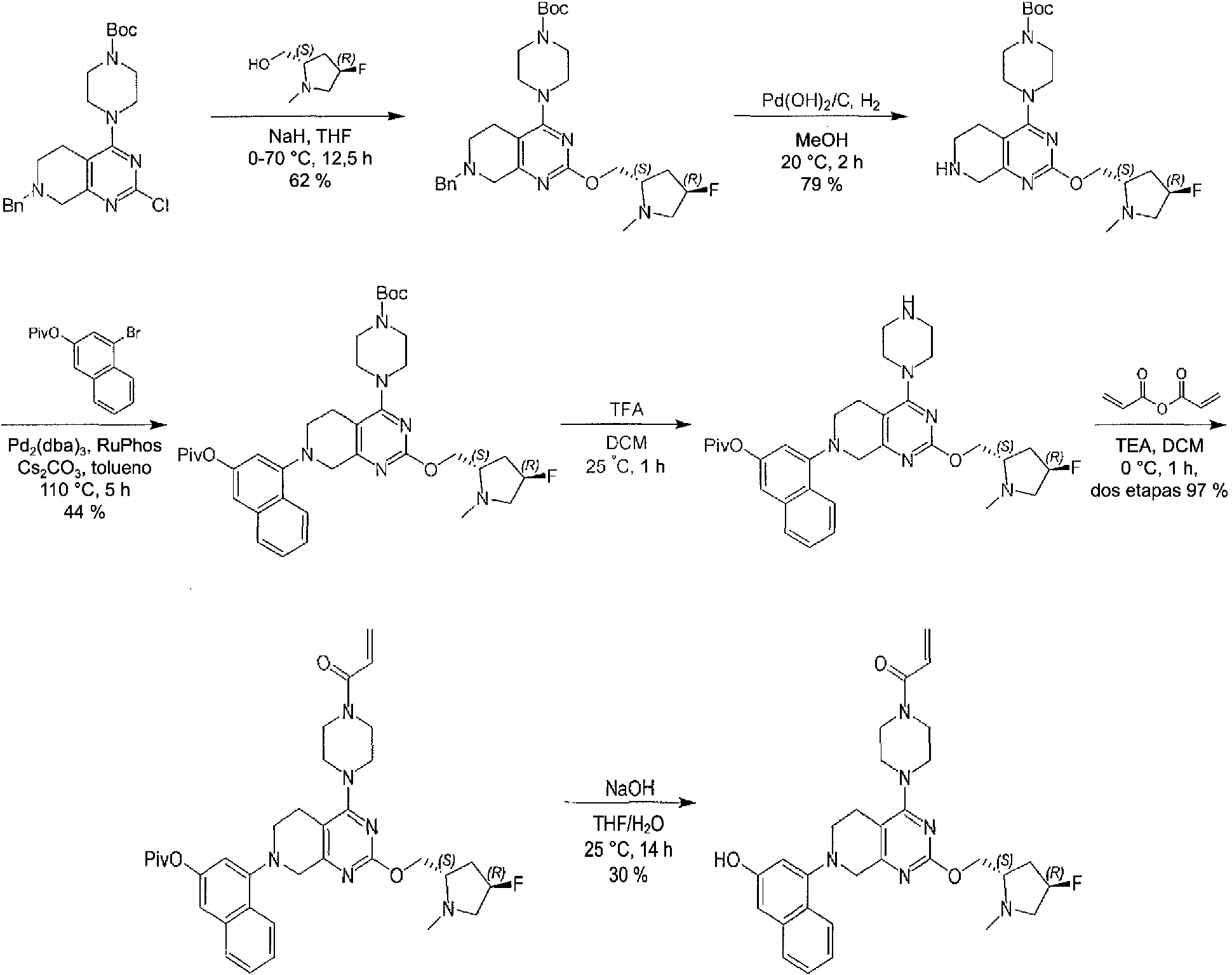
Etapa A: 4-[7-bencil-2-[[(2S.4R)-4-fluoro-1-metil-pirrolidin-2-il1metoxi1-6.8-dihidro-5H-pirido[3.4-dlpirimidin-4-illpiperazin-1-carboxilato de tere-butilo. A una solución de [(2S,4R)-4-fluoro-1-metil-pirrolidin-2-il]metanol (631 mg.
4.74 mmol, 3,76 eq.) en THF (7.0 ml) se le añadió NaH (126 mg. 3,16 mmol, 60 % de pureza. 2,51 eq.) a 0 °C. Después. la mezcla se agitó a 0 °C durante 0.5 h. Después. a la mezcla anterior se le añadió 4-(7-bencil-2-cloro-6,8-dihidro-5H-pirido[3.4-d]pirimidin-4-il)piperazin-1-carboxilato de tere-butilo (700 mg. 1,26 mmol. 1.0 eq.). Después de la adición. la mezcla se agitó a 70 °C durante 12 h. Después de que se completara. la mezcla se inactivó con NH4Cl acuoso saturado (15,0 ml) y se extrajo con AE (3 x 20.0 ml). La capa orgánica se secó sobre Na2S04, se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, diclorometano: metanol = 20:1 a 10:1) para dar 4-[7-bencil-2-[[(2S.4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi1-6.8-dihidro-5H-pirido[3.4-d1pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (530 mg. 980 umol, 78 % de rendimiento) en forma de un aceite de color amarillo. LCMS [ESI, M+1 ]: 541.
1H RMN (400 MHz, Cloroformo-ó) ó 7,24 - 7,13 (m. 5H). 5,08 - 4,91 (m. 1H). 4,23 (dd. J = 4.4 Hz. 10,8 Hz. 1H). 4,04 (dd. J = 6.0 Hz. 10,8 Hz. 1H). 3,53 (s. 2H). 3,45 - 3,42 (m. 2H). 3,38 - 3,33 (m. 5H). 3,31 - 3,27 (m.4H). 2,90 - 2,84 (m.
1H). 2,52 - 2,46 (m. 3H). 2,35 (s. 3H). 2,21 - 2,09 (m. 1H). 1,87 - 1.73 (m. 3 H). 1.34 (s. 9H).
Etapa B: 4-r2-rr(2S.4R)-4-fluoro-1-metil-pirrolidin-2-i n metox n -5.6.7.8-tetrahidropiridor3.4-dlpirimidin-4-i n piperazin-1-carboxilato de tere-butilo. A una solución de 4-[7-benc¡l-2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (480 mg. 887 umol. 1 eq.) en Me0H (6.0 ml) se le añadió Pd(0H)2/C (270 mg. 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 20 °C durante 2 horas. Después de que se completara. la mezcla se filtró y se concentró. El residuo se purificó por cromatografía en columna (Si02, AE/Me0H = 20:1 a 10:1). El producto 4-[2-[[(2S,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (350 mg.706 umol.79 % de rendimiento. 91 % de pureza) se obtuvo en forma de un aceite de color amarillo. LCMS [ESI, M+1 ]: 451.
Etapa C: A una mezcla de 4-[2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-il]piperazin-1-carboxilato de tere-butilo (280 mg. 621 umol. 1.0 eq.). Pd2(dba)3 (85.3 mg. 93,2 umol. 0,15 eq.). RuPhos (58.0 mg. 124,3 umol. 0.2 eq.) y Cs2C03 (404 mg. 1.24 mmol. 2.0 eq.) en tolueno (10,0 ml) se le añadió
2,2-dimetilpropanoato de (4-bromo-2-naftilo) (Intermedio 54, 572 mg, 1,86 mmol, 3,0 eq.). La mezcla se agitó a 110 °C durante 5 h en una atmósfera de N2. La mezcla se inactivó con agua (15,0 ml) y se extrajo con AE (3 x 20,0 ml). La capa orgánica se secó sobre Na2SÜ4, se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por HPLC ultrarrápida de fase inversa (18C, 0,1 % de AF en agua, 0-70 % de MeCN). El producto obtenido se ajustó con NaHCÜ3 acuoso saturado a pH -8, después se concentró, la capa acuosa se extrajo con AE (3 x 40,0 ml) y la capa orgánica combinada se secó sobre Na2SÜ4, se filtró y se concentró. El compuesto 4-[7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-2-[[(2S,4^)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (190 mg, 272 umol, 44 % de rendimiento, 97 % de pureza) se obtuvo en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 677.
Etapa D: 2,2-dimetilpropanoato de [4-[2-[[(2S,4R)-4-fluoro-1 -metil-pirrolidin-2-il1metoxi]-4-piperazin-1 -il-6,8-dihidro-5H-pirido[3,4-d1pirimidin-7-il]-2-naftilo1. A una mezcla de 4-[7-[3-(2,2-dimetilpropanoiloxi)-1-naftil]-2-[[(2S,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-6,8-dihidro-5^-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (110 mg, 162 umol, 1,0 eq.) en DCM (1,5 ml) se le añadió poco a poco TFA (2,31 g, 20,2 mmol, 1,5 ml, 124 eq.) a 25 °C en una atmósfera de N2. La mezcla se agitó a 25 °C durante 1 h. Después de que se completara, la mezcla de reacción se concentró para dar 2,2-dimetilpropanoato de [4-[2-[[(2S,4R)-4-fluoro-1-metilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-7-il]-2-naftilo] (200 mg, bruto, 2TFA) en forma de un sólido de color amarillo, que se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 577.
Etapa E: 2,2-dimetilpropanoato de [4-[2-[[(2S,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-4-(4-prop-2-enoilpiperazin-1-il)-6,8-dihidro-5H-pirido[3,4-d^pirimidin-7-il1-2-naftilo1. A una mezcla de 2,2-dimetilpropanoato de [4-[2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-4-p¡peraz¡n-1-¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-¡l]-2-naft¡lo] (200 mg, 2TFA) y TEA (493 mg, 4,87 mmol, 678 ul) en Dc M (2,0 ml) se le añadió poco a poco prop-2-enoato de prop-2-enoílo (40,9 mg, 324 umol) a 0 °C en una atmósfera de N2. La mezcla se agitó a 0 °C durante 1 h. Después de que se completara, la mezcla de reacción se inactivó con MeÜH (3,0 ml) a 0 °C y después se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiÜ2, AE/Me0H = 20:1 a 10:1). El compuesto 2,2-dimetilpropanoato de [4-[2-[[(2S,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-4-(4-prop-2-enoilpiperazin-1 -il)-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-7-il]-2-naftilo] (100 mg, 153 umol, 97 % de pureza) se obtuvo en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 631.
Etapa F: 1-[4-[2-[[(2S,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-7-(3-hidroxi-1-naftN)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1 -il]prop-2-en-1 -ona. Una mezcla de 2,2-dimetilpropanoato de [4-[2-[[(2S,4R)-4-fluoro-1-metilp¡rrol¡d¡n-2-¡l]metox¡]-4-(4-prop-2-eno¡lp¡peraz¡n-1-¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-¡l]-2-naft¡lo] (39,0 mg, 61,8 umol, 1,0 eq.) y NaÜH (61,8 mg, 1,55 mmol, 25,0 eq.) en THF (1,5 ml) y H2Ü (1,5 ml) se agitó a 25 °C durante 14 horas. Después de que se completara, la mezcla de reacción se inactivó con agua (10,0 ml) y después se extrajo con AE (3 x 15 ,0 ml). La capa orgánica se secó sobre Na2SÜ4, se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; B: 38 %-68 %, 12 min) y se sometió a liofilización. El compuesto del título 1-[4-[2-[[(2S,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-7-(3-hidroxi-1-naftil)-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-1 -il]prop-2-en-1 -ona (EJe Mp LÜ 357, 10,1 mg, 18,4 umol, 30 % de rendimiento, 99 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 547.
Condiciones de SFC: 100 % de ee.
1H RMN (400 MHz, Cloroformo-d) 58,04 (d, J = 8,4 Hz, 1H), 7,65 (d, J = 8,4 Hz, 1H), 7,42 (t, J = 7,2 Hz, 1 H), 7,30 -7,34 (m, 1H), 6,88 (d, J = 1,6 Hz, 1H), 6,72 (d, J = 2,0 Hz, 1H), 6,60 (dd, J = 10,4 Hz, 16,8 Hz, 1H), 6,37 (dd, J = 1,6 Hz, 16,8 Hz, 1H), 5,79 (dd, J = 2,0 Hz, 10,2 Hz, 1H), 5,28 - 5,14 (m, 1H), 4,46 (dd, J = 4,8 Hz, 11,2 Hz, 1H), 4,32 (dd, J = 5,2 Hz, 11,2 Hz, 1H), 4,18 (s, 2H), 3,82 - 3,68 (m, 2H), 3,66 - 3,55 (m, 4H), 3,52 - 3,42 (m 4H), 3,35 - 3,21 (m, 2H), 3,17 - 3,10 (m, 1H), 2,82 - 2,68 (m, 2H), 2,63 - 2,74 (m, 1H), 2,61 (s, 3H), 2,40 - 2,25 (m, 1H), 2,13 - 1,94 (m, 1H).
Ejemplo 358

1-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-prop-2-inilpiperazin-1-il]prop-2-en-1-ona

Etapa A: ácido (2S)-2-(ferc-butoxicarbonilamino)pent-4-inoico. A una suspensión de ácido (2S)-2-aminopent-4-inoico (5 g, 44,2 mmol, 1 eq.) en THF (15 ml) se le añadieron H20 (15 ml), K2C03 (18,3 g, 133 mmol, 3 eq.) y Boc20 (11,1 g, 50,8 mmol, 11,7 ml, 1,15 eq.). Se añadió más agua para producir una solución que se agitó durante 12 horas a 25 °C. Después, el disolvente orgánico se evaporó y la solución acuosa se lavó con í-butil metil éter y después se acidificó a pH = 3 con HCl 1 N ácido. La solución se extrajo con acetato de etilo (2 * 100 ml). El disolvente se evaporó para dar ácido (2S)-2-(íe/c-butoxicarbonilamino)pent-4-inoico (8 g, 37,5 mmol, 85 % de rendimiento) en forma de un aceite incoloro que se usó sin purificación adicional. LCMS [ESI, M+1]: 589.
1H RMN (400 MHz, cloroformo-d) ó = 7,76 (d a, J = 8,4 Hz, 1H), 7,62 (t, J = 7,6 Hz, 1H), 7,53 (d, J = 7,2 Hz, 1H), 7,45 (td, J = 8,0, 13,2 Hz, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,26 - 7,18 (m, 1H), 5,10 (s a, 1H), 4,49 - 4,33 (m, 2H), 4,21 - 4,01 (m, 3H), 3,96 - 3,79 (m, 2H), 3,60 (d a, J = 6,8 Hz, 1H), 3,44 (d a, J = 13,6 Hz, 1H), 3,32 - 2,97 (m, 5H), 2,88 - 2,53 (m, 4H), 2,48 (d, J = 2,8 Hz, 3H), 2,29 (d a, J = 8,8 Hz, 1H), 2,11 - 2,00 (m, 1H), 1,87 - 1,71 (m, 3H).
Etapa B: 2-[bencil-[(2S)-2-(terc-butoxicarbonilamino)pent-4-inoil1amino]acetato de etilo. A una solución de ácido (2S)-2-(íe/c-butoxicarbonilamino)pent-4-inoico (7,75 g, 36,4 mmol, 1 eq.), 2-(bencilamino)acetato de etilo (7,02 g, 36,4 mmol, 1 eq.) y HATU (16,6 g, 43,5 mmol, 1,20 eq.) en DMF (50 ml) se le añadió TEA (7,35 g, 72,7 mmol, 10,1 ml, 2,00 eq.) a 25 °C. La mezcla se agitó a 25 °C durante 0,5 h. La mezcla se diluyó con acetato de etilo (150 ml) y se lavó con agua (3 * 200 ml) y salmuera (1 * 100 ml). La capa orgánica separada se lavó con agua (1 * 100 ml) y salmuera (1 * 100 ml), se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se usó en la siguiente etapa sin purificación adicional. El compuesto 2-[bencil-[(2S)-2-(terc-butoxicarbonilamino)pent-4-inoil]amino]acetato de etilo (13,7 g, 32,1 mmol, 88,3 % de rendimiento, 91 % de pureza) se obtuvo en forma de un aceite de color rojo que se convirtió en un sólido de color rojo tras reposar a 25 °C durante 16 horas. LCMS [ESI, M-99]: 289.
Etapa C: (3S)-1-bencil-3-prop-2-¡n¡l-p¡peraz¡n-2,5-d¡ona. A una solución de 2-[bencil-[(2S)-2-(terebutoxicarbonilamino)pent-4-inoil]amino]acetato de et¡lo (11,7 g, 30,1 mmol, 1 eq.) en DCM (40 ml) se le añad¡ó TFA (46,2 g, 405 mmol, 30 ml, 13,5 eq.) a 0 °C y la mezcla se agitó a 25 °C durante 0,5 horas. La mezcla se concentró al vacío y se añadió tolueno (30 ml). Después de agitar a 100 °C durante 1 hora, la mezcla se concentró al vacío. El residuo se disolvió en acetato de etilo (200 ml) y se basificó con una solución acuosa saturada de carbonato de sodio a pH = 8. La capa orgánica separada se lavó con agua (1 x 200 ml) y salmuera (1 x 100 ml), se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se trituró con (acetato de etilo/éter de petróleo 1/5, 100 ml) y el precipitado se filtró. La torta de filtro se secó al vacío. El compuesto (3S)-1-bencil-3-prop-2-¡n¡l-piperaz¡n-2,5-d¡ona (5 g, 20,5 mmol, 68 % de rendimiento, 99,5 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 243.
1H RMN (400 MHz, cloroformo-d) ó = 7,46 - 7,20 (m, 5H), 6,59 (s a, 1H), 4,76 (d, J = 14,4 Hz, 1H), 4,50 (d, J = 14,4 Hz, 1H), 4,20 (d a, J = 4,4 Hz, 1H), 4,04 - 3,90 (m, 1H), 3,89 - 3,76 (m, 1H), 2,95 - 2,69 (m, 2H), 2,05 (t, J = 2,8 Hz, 1H).
Etapa D: (3S)-1-bencil-3-prop-2-¡nil-p¡peraz¡na. A una mezcla de (3S)-1-bencil-3-prop-2-¡n¡l-piperaz¡n-2,5-d¡ona (5,00 g, 20,6 mmol, 1,00 eq.) en THF (50,0 ml) se le añadió L¡AlH4 (3,13 g, 82,6 mmol, 4,00 eq.) a 25 °C. Después de agitar a 65 °C durante 12 horas, la mezcla se retiró por filtración y la torta de filtro se lavó con acetato de etilo (100 ml).
El filtrado se concentró al vacío para dar (3S)-1-bencil-3-prop-2-in¡l-p¡peraz¡na (4,10 g, 19,1 mmol, 93 % de rendimiento) en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, cloroformo-d) ó = 7,37 - 7,24 (m, 5H), 3,55 - 3,47 (m, 2H), 3,03 - 2,86 (m, 3H), 2,85 - 2,78 (m, 1H), 2,73 (d a, J = 11,2 Hz, 1H), 2,33 - 2,20 (m, 2H), 2,13 - 2,04 (m, 1H), 2,02 (t, J = 2,8 Hz, 1H), 1,90 - 1,83 (m, 1H).
Etapa E: (2S)-4-benc¡l-2-prop-2-in¡l-p¡peraz¡n-1-carbox¡lato de tere-butilo. Una mezcla de (3S)-1-bencil-3-prop-2-inilpiperazina (2,00 g, 9,33 mmol, 1,00 eq.), TEA (2,83 g, 28,0 mmol, 3,90 ml, 3,00 eq.) y (Boc)2Ü (10,2 g, 46,7 mmol, 10,7 ml, 5,00 eq.) en THF (20,0 ml) se agitó a 25 °C durante 1 hora. La mezcla se diluyó con agua (1 x 20,0 ml) y se extrajo con acetato de etilo (3 x 20,0 ml). La capa orgánica combinada se lavó con salmuera (1 x 30,0 ml), se secó sobre Na2SÜ4, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna (S¡Ü2, éter de petróleo/acetato de etilo = 5/1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar (2S)-4-bencil-2-prop-2-¡n¡l-piperaz¡n-1-carbox¡lato de tere-butilo (1,50 g, 4,20 mmol, 45 % de rendimiento, 88 % de pureza) en forma de un aceite incoloro. LCMS [ESI, M+1]: 315.
1H RMN (400 MHz, cloroformo-d) ó = 7,38 - 7,27 (m, 5H), 4,26 (s a, 1H), 3,99 - 3,74 (m, 1H), 3,59 - 3,45 (m, 2H), 3,08 - 2,89 (m, 2H), 2,79 - 2,68 (m, 2H), 2,62 - 2,53 (m, 1H), 2,11 (dd, J = 3,6, 11,2 Hz, 1H), 2,06 - 1,98 (m, 1H), 1,89 (t, J = 2,8 Hz, 1H), 1,48 - 1,45 (m, 9H).
Etapa F: (2S)-2-prop-2-in¡lp¡peraz¡na. Una mezcla de (2S)-4-bencil-2-prop-2-in¡l-p¡peraz¡na -1-carboxilato de terebutilo (1,40 g, 4,45 mmol, 1,00 eq.) y 1-cloro-1-(1-clorovin¡lox¡)etano (2,51 g, 17,8 mmol, 4,00 eq.) en DCE (20,0 ml) se agitó a 85 °C durante 1 hora. La mezcla se concentró al vacío. El residuo se disolvió en metanol (20,0 ml) y se agitó a 65 °C durante 1 hora. La mezcla se concentró al vacío y se trituró con éter de petróleo (10,0 ml) para dar (2S)-2-prop-2-inilpiperazina (0,80 g, bruto, 2 HCl) en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, metanol-d4) ó = 4,56 - 4,44 (m, 1H), 3,88 - 3,80 (m, 1H), 3,79 - 3,73 (m, 1H), 3,62 - 3,34 (m, 4H), 2,87 - 2,76 (m, 3H).
Etapa G: 4-benc¡lox¡-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡mid¡na. Una mezcla de 4-benciloxi-2-[[(2S)-1 -metilp¡rrol¡d¡n-2-¡l]metox¡]- 5,6,7,8-tetrah¡drop¡r¡do[3,4-d]pir¡m¡d¡na (2,00 g, 5,64 mmol, 1,00 eq.), 1-bromonaftaleno (1,75 g, 8,46 mmol, 1,18 ml, 1,50 eq.), Pd2(dba)3 (517 mg, 564 umol, 0,10 eq.), RuPhos (527 mg, 1,13 mmol, 0,20 eq.) y Cs2CÜ3 (3,68 g, 11,3 mmol, 2,00 eq.) en tolueno (20,0 ml) se agitó a 110 °C durante 3 horas en una atmósfera de N2. La mezcla se diluyó con agua (5,00 ml) y se extrajo con acetato de etilo (3 x 5 ,00 ml).
Las capas orgánicas se lavaron con salmuera (1 x 10,0 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (TFA, 0,10 %)/acetonitrilo]. La fracción deseada se recogió y se ajustó a pH >7 con bicarbonato de sodio saturado (5,00 ml) y después se extrajo con acetato de etilo (3 x 20,0 ml). Las capas orgánicas combinadas se lavaron con salmuera (1 x 30,0 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío para dar 4-benciloxi-2-[[(2S)-1 -metilp¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡hidro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡na (1,34 g, 2,78 mmol, 49 % de rendimiento, 99,6 % de pureza) en forma de un sólido de color amarillo. LCm S [ESI, M+1]: 481.
1H RMN (400 MHz, cloroformo-d) ó = 8,22 - 8,16 (m, 1H), 7,89 - 7,82 (m, 1H), 7,59 (d, J = 8,0 Hz, 1H), 7,51 - 7,45 (m, 4H), 7,45 - 7,33 (m, 4H), 7,14 (dd, J = 0,8, 7,2 Hz, 1H), 5,49 (s, 2H), 4,45 (dd, J = 5,6, 10,4 Hz, 1H), 4,27 - 4,18 (m, 3H), 3,55 - 3,24 (m, 2H), 3,12 (t a, J = 7,6 Hz, 1H), 2,89 (s a, 2H), 2,76 - 2,65 (m, 1H), 2,50 (s, 3H), 2,35 - 2,24 (m, 1H), 2,14 - 2,06 (m, 1H), 1,91 - 1,78 (m, 3H).
Etapa H: 2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡mid¡n-4-ol. Una mezcla de 4-benc¡lox¡-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡]-7-(1-naft¡l)-6,8-d¡hidro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡na (1,30 g, 2,70 mmol,
1,00 eq.) y Pd/C (0,20 g, 10 % de pureza) en metanol (200 ml) se agitó a 25 °C durante 1 hora en una atmósfera de H2 a 103,42 kPa (15 psi). El catalizador se retiró por filtración y el filtrado se concentró al vacío para dar 2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(1-naft¡l)-6,8-d¡hidro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-ol (1,00 g, 1,90 mmol, 70 % de rendimiento, 74 % de pureza) en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 391.
1H RMN (400 MHz, cloroformo-d) 5 = 8,24 - 8,16 (m, 1H), 7,90 - 7,81 (m, 1H), 7,59 (d, J = 8,4 Hz, 1H), 7,54 - 7,47 (m, 2H), 7,42 (t, J = 7,6 Hz, 1H), 7,14 (d, J = 7,6 Hz, 1H), 4,43 - 4,31 (m, 2H), 4,05 (s, 2H), 3,38 (s a, 2H), 3,18 - 3,10 (m, 1H), 2,79 (s a, 2H), 2,70 - 2,54 (m, 2H), 2,47 (s, 3H), 2,32 (dt, J = 7,2, 9,6 Hz, 1H), 2,02 - 1,96 (m, 1H), 1,91 - 1,75 (m, 3H).
Etapa I: trifluorometanosulfonato de [2-[[(2S)-1-metilpirrolidin-2-il1metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-ilol. A una solución de 2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pir¡m¡din-4-ol (0,40 g, 1,02 mmol, 1,00 eq.) y TEa (207 mg, 2,05 mmol, 285 ul, 2,00 eq.) en diclorometano (10,0 ml) se le añadió Tfz0 (318 mg, 1,13 mmol, 186 ul, 1,10 eq.) a -40 °C. Después de agitar a -40 °C durante 0,25 horas, la mezcla se diluyó con agua (3,00 ml) y después se extrajo con acetato de etilo (3 * 5,00 ml). Los extractos se lavaron con salmuera (1 * 5,00 ml), se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna (Si02, acetato de etilo/metanol = 10/1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar trifluorometanosulfonato de [2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-p¡rido[3,4-d]p¡r¡m¡d¡n-4-¡lo] (0,40 g, 528 umol, 52 % de rendimiento, 69 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 523.
Etapa J: 2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftir)-4-[(3S)-3-prop-2-inilpiperazin-1-il]-6,8-dihidro-5H-pirido[3,4-cflpirimidina. Una mezcla de trifluorometanosulfonato de [2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-piridop^-cdpirimidin^-ilo] (0,38 g, 727 umol, 1,00 eq.), (2S)-2-prop-2-inilpiperazina (350 mg, bruta, 2 HCl) y DIEA (939 mg, 7,27 mmol, 1,27 ml, 10,0 eq.) en DMF (4,00 ml) se agitó a 100 °C durante 0,25 horas. La mezcla se diluyó con agua (5,00 ml) y se extrajo con acetato de etilo (3 * 10,0 ml). Los extractos se lavaron con salmuera (1 * 10,0 ml), se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (AF, 0,1 %)/acetonitrilo]. Las fracciones deseadas se recogieron y se liofilizaron para dar 2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1 -naftil)-4-[(3S)-3-prop-2-inilpiperazin-1 -¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡na (0,20 g, 336 umol, 46 % de rendimiento, 99 % de pureza, 2 AF) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 497.
Etapa K: 1-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-prop-2-inil-piperazin-1-il]prop-2-en-1-ona. A una solución de 2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-4-[(3S)-3-prop-2-¡n¡lp¡peraz¡n-1-¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡na (145 mg, 246 umol, 1,00 eq., 2 AF) y TEA (374 mg, 3,69 mmol, 514 ul, 15,0 eq.) en diclorometano (4,00 ml) se le añadió cloruro de prop-2-enoílo (22,3 mg, 246 umol, 20,1 ul, 1,00 eq.) a -40 °C. Después de agitar a 25 °C durante 0,25 h, la mezcla se diluyó con agua (3,00 ml) y después se extrajo con diclorometano (3 * 3,00 ml). Los extractos se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (NH4HC03 10 mM) - ACN]; % de B: 57 % - 87 %, 12 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 1 -[(2 S)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1 -naftil)-6,8-dihidro-5^-pirido[3,4-d]pirimidin-4-il]-2-prop-2-inil-piperazin-1-il]prop-2-en-1-ona (EJEMPL0358, 25,6 mg, 46,3 umol, 19 % de rendimiento, 99,5 % de pureza) en forma de un sólido de color pardo. LCMS [ESI, M+1]: 551.
Condiciones de SFC: “0D-3S_3_40_3ML Columna: Chiralcel 0D-3 100 * 4,6 mm D.I., 3 um, Fase móvil: 40 % de metanol (0,05 % de DEA) en C02, Caudal: 3 ml/min, Longitud de onda: 220 nm” .
1H RMN (400 MHz, cloroformo-d) 5 = 8,21 (m, 1H), 7,91 - 7,81 (m, 1H), 7,60 (d a, J = 8,0 Hz, 1H), 7,54 - 7,47 (m, 2H), 7,46 - 7,39 (m, 1H), 7,14 (d, J = 7,2 Hz, 1H), 6,65 (s a, 1H), 6,36 (d a, J = 16,8 Hz, 1 H), 5,77 (dd, J = 2,0, 10,8 Hz, 1H), 5,00 - 4,52 (m, 1H), 4,46 - 4,36 (m, 1H), 4,34 - 3,81 (m, 6H), 3,47 (m, 1H), 3,38 - 3,21 (m, 2H), 3,19 - 2,90 (m, 4H), 2,84 (s a, 1H), 2,73 - 2,52 (m, 3H), 2,48 (s, 3H), 2,34 - 2,24 (m, 1H), 2,10 - 2,03 (m, 1H), 1,90 - 1,70 (m, 4H).
Ejemplo 359

2-((S)-1-acriloil-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-cf]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (2S)-2-(Cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo. Una mezcla de 1-bromo-8-metil-naftaleno (Intermedio 69, 507 mg, 1,65 mmol, 1,3 eq.), (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (Intermedio 66, 600 mg, 1,27 mmol, 1 eq.), CS2C03 (1,04 g, 3,18 mmol, 2,5 eq.), RuPhos (118 mg, 254 umol, 0,2 eq.) y Pd2(dba)3 (233 mg, 254 umol, 0,2 eq.) en tolueno (10 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 100 °C durante 2 horas en una atmósfera de N2. La mezcla de reacción se diluyó con agua (30 ml) y se extrajo con acetato de etilo (20 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico)/acetonitrilo)]. La mezcla se ajustó a pH ~7 con una solución acuosa saturada de NaHC03 y se extrajo con acetato de etilo (20 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar el producto. El compuesto (2S-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (310 mg, 501 umol, 39 % de rendimiento, 99 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 612.
1H RMN (400 MHz, cloroformo-d) ó = 7,74 - 7,60 (m, 2H), 7,46 - 7,37 (m, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,27 - 7,16 (m, 2H), 4,61 (s a, 1H), 4,43 - 4,33 (m, 1H), 4,30 - 4,14 (m, 2H), 4,10 - 3,72 (m, 4H), 3,60 - 3,44 (m, 1H), 3,38 (dd a, J = 3,6, 13,6 Hz, 1H), 3,24 - 3,04 (m, 4H), 3,03 - 2,84 (m, 5H), 2,82 - 2,53 (m, 4H), 2,52 - 2,42 (m, 3H), 2,29 (s a, 1H), 2,05 - 1,95 (m, 1H), 1,91 - 1,69 (m, 2H), 1,52 (s, 9H).
Etapa B: 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo(310 mg, 506 umol, 1 eq.) en DCM (500 ul) se le añadió TFA (866 mg, 7,60 mmol, 562 ul, 15 eq.). La mezcla se agitó a 25 °C durante 1 hora. La mezcla de reacción se concentró al vacío. El compuesto 2-[(2S)-4-[7-(8-metil-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (317 mg, 506 umol, 99 % de rendimiento, TFA) se obtuvo en forma de un aceite de color amarillo y se usó en la siguiente etapa directamente sin purificación.
LCMS [ESI, M+1]: 512.
Etapa C: 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (317 mg, 506 umol, 1 eq., TfA) en DCM (5 ml) se le añadió TEA (512 mg, 5,07 mmol, 705 ul, 10 eq.) a 0 °C. Después de la adición, se añadió gota a gota prop-2-enoato de prop-2-enoílo (51,1 mg, 405 umol, 0,8 eq.) en DCM (1 ml) a 0 °C. La mezcla resultante se agitó a 25 °C durante 1 hora. La mezcla de reacción se inactivó con una solución acuosa saturada de NaHC03 (1 ml) y se extrajo con acetato de etilo (20 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep.
(columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 50 % - 80 %, 12 min). El compuesto del título 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0359, 60,2 mg, 105 umol, 21 % de rendimiento, 99,5 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 566.
Condición de SFC: “ 0J-3S_5_5_40_3ML Columna: Chiralcel 0J-3100 x 4,6 mm D.I., 3 um Fase móvil: etanol (0,05 % de DEA) en C02 de 5 % a 40 % Caudal: 3 ml/min Longitud de onda: 220 nm” .
1H RMN (400 MHz, cloroformo-d) ó = 7,75 - 7,60 (m, 2H), 7,46 - 7,38 (m, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,27 - 7,17 (m, 2H), 6,57 (s, 1H), 6,46 - 6,33 (m, 1H), 5,83 (d a, J = 10,4 Hz, 1H), 5,07 (s a, 1H), 4,64 (s a, 1H), 4,44 - 4,32 (m, 1H), 4,31 - 4,21 (m, 1H), 4,20 - 4,03 (m, 3H), 3,98 - 3,83 (m, 1H), 3,78 - 3,62 (m, 1H), 3,60 - 3,36 (m, 2H), 3,23 - 2,96 (m, 5H), 2,92 (s, 3H), 2,88 - 2,74 (m, 1H), 2,73 - 2,55 (m, 2H), 2,47 (d, J = 4,4 Hz, 3H), 2,35 - 2,21 (m, 1H), 2,13 - 1,97 (m, 1H), 1,90 - 1,74 (m, 3H).
Alternativamente, el EJEMPL0359 se puede preparar como se indica a continuación:

Etapa A: 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il1-2-cloro-6,8-dihidro-5H-pirido[3,4-dlpirimidin-7-carboxilato de tere-butilo. A una solución de 2,4-dicloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de fere-butilo (18,8 g, 61,7 mmol, 1 eq.) en DMF (200 ml) se le añadieron DIEA (15,9 g, 123 mmol, 21,5 ml, 2 eq.) y (2S)-2-(cianometil)piperazin-1-carboxilato de bencilo (Intermedio 63, 16,0 g, 61,7 mmol, 1 eq.). Después de agitar a 80 °C durante 1,5 horas, la mezcla de reacción se diluyó con acetato de etilo (200 ml) y se lavó con agua (200 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (200 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 50/1 a 2/1). El compuesto 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de fere-butilo (26 g, 48,8 mmol, 79 % de rendimiento, 99 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 527.
1H RMN (400 MHz, cloroformo-d) ó = 7,43 - 7,34 (m, 5H), 5,20 (s, 2H), 4,73 - 4,60 (m, 2H), 4,45 (d, J = 19,2 Hz,, 1H), 4,13 - 4,02 (m, 2H), 3,94 - 3,75 (m, 2H), 3,45 - 3,25 (m, 3H), 3,11 (td, J = 3,6, 12,4 Hz, 1H), 2,88 - 2,59 (m, 4H), 1,60 (s, 9H).
Etapa B: 4-[(3S)-4-benc¡lox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡ll-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-7-carbox¡lato de tere-butilo. Una mezcla de [(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metanol (10.5 g.91.1 mmol. 10.8 ml. 2 eq.). 4-[(3S)-4-benc¡lox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡l1-2-cloro-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-7-carbox¡lato de tere-butilo (24 g.45.6 mmol. 1 eq.). Pd2(dba)3 (6.18 g.6 .84 mmol.0.15 eq.). RuPhos (4.26 g.9.12 mmol. 0.2 eq.) y CS2C03 (37.1 g. 114 mmol. 2.5 eq.) en tolueno (450 ml) se desgas¡f¡có y se purgó 3 veces con N2 y después la mezcla se ag¡tó a 90 °C durante 5 horas en una atmósfera de N2. La mezcla de reacc¡ón se d¡luyó con agua (200 ml) y se extrajo con acetato de et¡lo (500 ml x 3). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (200 ml). se secaron sobre Na2S04. se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar un res¡duo. El res¡duo se pur¡f¡có por cromatografía en columna (AL03. acetato de et¡lo/metanol = 1/0 a 10/1). El compuesto 4-[(3S)-4-benc¡lox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡l1-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo (25 g. 35.9 mmol. 79 % de rend¡m¡ento. 87 % de pureza) se obtuvo en forma de un sól¡do de color amar¡llo. LCMS [ESI. M+1]: 606.
Etapa C: (2S)-2-(c¡anomet¡l)-4-(2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-5.6.7.8-tetrah¡drop¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de benc¡lo. A una soluc¡ón de 4-[(3S)-4-benc¡lox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-7-carbox¡lato de tere-but¡lo (16 g. 26.4 mmol. 1 eq.) en d¡oxano (100 ml) se le añad¡ó HCl/d¡oxano (4 M. 99.1 ml. 15 eq.). La mezcla se ag¡tó a 0 °C durante 1 hora. El d¡oxano se decantó y el sól¡do se recog¡ó. El res¡duo sól¡do se d¡luyó con agua (50 ml) y d¡clorometano (200 ml). después se ajustó a pH = 8 con una soluc¡ón acuosa saturada de Na2C03 y se extrajo con acetato de et¡lo (200 ml x 3). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (100 ml). se secaron sobre Na2S04. se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar (2S)-2-(c¡anomet¡l)-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-5.6.7.8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (Intermed¡o 65. 13.5 g. 24.5 mmol. 93 % de rend¡m¡ento. 91.6 % de pureza) en forma de un ace¡te de color amar¡llo y se usó en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón. LCMS [ESI. M+1]: 506.
1H RMN (400 MHz. cloroformo-d) ó = 7.45 - 7.31 (m. 5H). 5.19 (s. 2H). 4.65 (s a. 1H). 4.40 - 4.31 (m. 1H). 4.14 - 4.04 (m. 2H). 4.03 - 3.91 (m. 3H). 3.91 - 3.77 (m. 1H). 3.24 (d. J = 13.2 Hz. 2H). 3.16 - 3.05 (m. 2H). 3.04 - 2.89 (m. 2H). 2.81 (s a. 1H). 2.75 - 2.51 (m. 4H). 2.47 (s. 3H). 2.35 - 2.22 (m. 1H). 2.08 - 2.00 (m. 1H). 1.92 - 1.76 (m. 3H).
Etapa D: (2S)-2-(c¡anomet¡l)-4-r7-(8-met¡l-1-naft¡l)-2-rr(2S)-1-met¡lp¡rrol¡d¡n-2-¡nmetoxn-6.8-d¡h¡dro-5H-p¡r¡dor3.4-dlp¡r¡m¡d¡n-4-¡llp¡peraz¡n-1-carbox¡lato de benc¡lo. Una mezcla de (2S)-2-(c¡anomet¡l)-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (10 g. 19.8 mmol. 1 eq.). 1-bromo-8 -met¡l-naftaleno (Intermed¡o 69. 5.68 g. 25.7 mmol. 1.3 eq.). RuPhos (2.77 g. 5.93 mmol. 0.3 eq.). Cs2C03 (16.1 g. 49.5 mmol. 2.5 eq.) y Pd2(dba)3 (3.62 g. 3.96 mmol. 0.2 eq.) en tolueno (100 ml) se desgas¡f¡có y se purgó 3 veces con N2 y después la mezcla se ag¡tó a 90 °C durante 6 horas en una atmósfera de N2. La mezcla de reacc¡ón se d¡luyó con agua (100 ml) y se extrajo con acetato de et¡lo (200 ml x 3 ). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (100 ml). se secaron sobre Na2S04. se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar un res¡duo. El res¡duo se pur¡f¡có por cromatografía ultrarráp¡da de fase ¡nversa [agua (0.1 % de ác¡do fórm¡co)/aceton¡tr¡lo)]. La mezcla se ajustó a pH = 7 con una soluc¡ón acuosa saturada de NaHC03 y se extrajo con acetato de et¡lo (500 ml x 3 ). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (200 ml). se secaron sobre Na2S04. se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar (2S)-2-(c¡anomet¡l)-4-[7-(8-met¡l-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (9 g. 12.9 mmol. 33 % de rend¡m¡ento. 93 % de pureza) en forma de un sól¡do de color amar¡llo. LCMS [ESI. M+1]: 646.
1H RMN (400 MHz. C2D2CL) ó = 7.32 (d. J = 8.0 Hz. 1H). 7.26 (t. J = 8.0 Hz. 1H). 7.08 - 6.93 (m. 7H). 6.91 - 6.78 (m. 2H). 4.81 (s. 2H). 4.30 (s. 1H). 4.13 - 3.95 (m. 1H). 3.88 - 3.33 (m. 6 H). 3.22 - 2.87 (m. 2H). 2.86 - 2.65 (m. 4H). 2.65 -2.42 (m. 5H). 2.41 - 2.32 (m. 1H). 2.30 - 2.13 (m. 2H). 2.07 (d. J = 7.2 Hz. 3H). 1.92 (s a. 1H). 1.72 - 1.45 (m. 4H).
Etapa E: 2-[(2S)-4-[7-(8-met¡l-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una soluc¡ón de (2S)-2-(c¡anomet¡l)-4-[7-(8-met¡l-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (8 g. 12.4 mmol. 1 eq.) en Me0H (100 ml) se añad¡eron Pd/C (2 g. 10 % de pureza) y NH3^Me0H (50 ml. 15 %) en una atmósfera de N2. La suspens¡ón se desgas¡f¡có al vacío y se purgó var¡as veces con H2. La mezcla se ag¡tó en una atmósfera de H2 (103.42 kPa (15 ps¡)) a 25 °C durante 1 hora. La mezcla se concentró al vacío. El compuesto 2-[(2S)-4-[7-(8-met¡l-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (5.7 g. 10.0 mmol. 81 % de rend¡m¡ento. 90 % de pureza) se obtuvo en forma de un sól¡do de color amar¡llo. LCMS [ESI. M+1]: 512.
1H RMN (400 MHz. cloroformo-d) ó = 7.69 (d. J = 8.0 Hz. 1H). 7.64 (dd. J = 3.2. 7.6 Hz. 1H). 7.40 (td. J = 4.4. 7.6 Hz. 1H). 7.34 (t. J = 7.6 Hz. 1H). 7.26 - 7.19 (m. 2H). 4.46 - 4.34 (m. 1H). 4.25 - 4.03 (m. 2H). 3.97 - 3.69 (m. 3H). 3.49 -3.44 (m. 1H). 3.39 - 3.07 (m. 5H). 3.06 - 2.83 (m. 6 H). 2.76 - 2.63 (m. 1H). 2.62 - 2.51 (m. 3H). 2.48 (d. J = 3.2 Hz. 3H). 2.35 - 2.22 (m. 1H). 2.13 - 1.98 (m. 1H). 1.83 - 1.69 (m. 3H).
Etapa F: 2-[(2S)-4-[7-(8-met¡l-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡ll-1-prop-2-eno¡l-p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una soluc¡ón de 2-[(2S)-4-[7-(8-met¡l-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (6.4 g. 12.5 mmol. 1 eq.) en d¡clorometano (120 ml) se le añad¡ó TEA (19 g. 187 mmol. 26 ml. 15 eq.) a -40 °C. Después de la ad¡c¡ón. se añad¡ó gota a gota una
solución de cloruro de prop-2-enoílo (1,70 g, 18,8 mmol, 1,53 ml, 1,5 eq.) en diclorometano (2 ml) a -40 °C. Después de agitar a -40 °C durante 0,5 horas, la mezcla de reacción se inactivó con una solución acuosa saturada de NaHCÜ3 (10 ml), después se diluyó con agua (50 ml) y se extrajo con diclorometano (50 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiÜ2, DCM/Me0H = 100/1 a 10/1) y se purificó adicionalmente por HPLC prep. (columna: Kromasil 250 * 50 mm * 10 um; fase móvil: [agua (0,225 % de AF)-ACN]; % de B: 20 % - 50 %, 28 min). La mezcla se ajustó a pH = 7 con una solución acuosa saturada de NaHCÜ3 y se extrajo con diclorometano (20 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar el compuesto del título 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-prop-2-enoilpiperazin-2-il]acetonitrilo (EJEMPLÜ 359, 2,3 g, 4,03 mmol, 32 % de rendimiento, 99 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 566.
1H RMN (400 MHz, cloroformo-d) 5 = 7,74 - 7,59 (m, 2H), 7,46 - 7,36 (m, 1H), 7,33 (t, J = 7,6 Hz, 1H), 7,26 - 7,15 (m, 2H), 6,70 - 6,47 (s, 1H), 6,39 (d, J = 16,4 Hz, 1H), 5,82 (d, J = 10,4 Hz, 1H), 5,16 - 4,47 (m, 1H), 4,44 - 4,32 (m, 1H), 4,30 - 3,62 (m, 6H), 3,58 - 3,26 (m, 2H), 3,25 - 2,95 (m, 5H), 2,91 (s, 3H), 2,86 - 2,55 (m, 4H), 2,48 (d, J = 5,2 Hz, 3H), 2,36 - 2,21 (m, 1H), 2,13 - 2,02 (m, 1H), 1,92 - 1,67 (m, 3H).
Ejemplo 360

2-[(2S)-4-[7-(8-etil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Inserción: 1 -bromo-8-etil-naftaleno. A una solución de 1,8-dibromonaftaleno (2,0 g, 6,99 mmol, 1,0 eq.) y Pd(dppf)Cl2 (367 mg, 501 umol, 7,16 e-2 eq.) en THF (15,0 ml) se le añadió dietil cinc (1,0 M en tolueno, 3,50 ml, 0,50 eq.) a -78 °C. La mezcla se agitó a -78 - 20 °C durante 12 horas. Después de que se completara, a la mezcla de reacción se le añadió agua (30,0 ml) y se extrajo con AE (30,0 ml * 3). La capa orgánica se secó sobre Na2SÜ4, se filtró y se concentró. El residuo se trituró con EP (60,0 ml) y se filtró. La torta de filtro se lavó con EP (10,0 ml * 2) y el licor madre se concentró. El producto obtenido se purificó por columna ultrarrápida de fase inversa (C18, 0,1 % de AF en agua, 0 - 100 % de MeCN) para dar 1-bromo-8-etil-naftaleno (600 mg, 2,45 mmol, 35 % de rendimiento, 96 % de pureza) en forma de un aceite de color amarillo.
1H RMN (400 MHz, Cloroformo-d) ó 7,72 - 7,63 (m, 2H), 7,58 (dd, J = 2,4, 7,2 Hz, 1H), 7,30 - 7,23 (m, 2H), 7,13 - 7,04 (m, 1H), 3,43 (c, J = 7,6 Hz, 2H), 1,22 (t, J = 7,6 Hz, 3H).
Etapa A: (2S)-2-(cianometil)-4-[7-(8-etil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,9-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de tere-butilo. A una solución de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡din-4-¡l)p¡peraz¡n-1-carbox¡lato de tere-butilo (intermedio 66, 300 mg, 636 umol, 1,0 eq.), t-Bu0Na (183 mg, 1,91 mmol, 3,0 eq.), RuPhos (59,4 mg, 127 umol, 0,2 eq.) y RuPhos-Pd-G3 (106 mg, 127 umol, 0,20 eq.) en tolueno (5,0 ml) se le añadió 1-bromo-8-etil-naftaleno (299 mg, 1,27 mmol, 2,0 eq.). La mezcla se agitó a 90 °C durante 12 horas. Después de que se completara, a la mezcla de reacción se le añadió agua (10,0 ml) y se extrajo con AE (10,0 ml * 3). La capa orgánica se secó sobre Na2S04, se filtró y se concentró. El producto obtenido se purificó por columna ultrarrápida de fase inversa (C18, 0,1 % de AF en agua, 0-60 % de MeCN) para dar (2S)-2-(cianometil)-4-[7-(8-etil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (140 mg, 221 umol, 34 % de rendimiento, 98 % de pureza) en forma de un aceite de color amarillo. LCMS [ESI, M+11: 626.
Etapa B: 2-[(2S)-4-[7-(8-etil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-etil-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (130 mg, 208 umol, 1,0 eq.) en DCM (1,0 ml) se le añadió TFA (1,54 g, 13,5 mmol, 1,0 ml, 65,0 eq.). La mezcla se agitó a 25 °C durante 10 min.
Después de que se completara, la mezcla de reacción se concentró al vacío. El producto 2-[(2S)-4-[7-(8-etil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (150 mg, bruto, 2TFA)se obtuvo en forma de un aceite de color amarillo. LCmS [ESI, M+1]: 526.
Etapa C: 2-[(2S)-4-[7-(8-etil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-etil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (150 mg, 199 umol, 1,0 eq., 2TFA) y DIEA (309 mg, 2,39 mmol, 416 ul, 12,0 eq.) en DCM (2,0 ml) se le añadió prop-2-enoato de prop-2-enoílo (37,7 mg, 299 umol, 1,50 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se inactivó con Me0H (1,0 ml) y se concentró al vacío. El producto obtenido se purificó por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 53 %-83 %, 12 min) para dar el compuesto del título 2-[(2S)-4-[7-(8-etil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (Ej Em Pl0360, 20,0 mg, 33,8 umol, 17 % de rendimiento, 98 % de pureza) en forma de un sólido de color rosa. LCMS [ESI, M+1]: 580.
1H RMN (400 MHz, Cloroformo-d) 57,74 - 7,64 (m, 2H), 7,46 - 7,35 (m, 2H), 7,27 - 7,20 (m, 2H), 6,64 - 6,60 (m, 1H), 6,46 - 6,36 (m, 1H), 5,83 (d a, J = 10,4 Hz, 1H), 5,20 - 4,50 (m, 1H), 4,45 - 4,33 (m, 1H), 4,31 - 4,19 (m, 1H), 4,17 -3,96 (m, 3H), 3,93 - 3,84 (m, 1H), 3,74 (d a, J = 18,0 Hz, 1H), 3,65 - 3,39 (m, 3H), 3,37 - 2,92 (m, 7H), 2,88 - 2,76 (m, 1H), 2,71 - 2,57 (m, 2H), 2,47 (d a, J = 4,8 Hz, 3H), 2,34 - 2,23 (m, 1H), 2,11 - 1,98 (m, 1H), 1,80 - 1,72 (m, 3H), 1,17 (t, J = 7,4 Hz, 3H).
Ejemplo 361

-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Inserción: [(3R)-1-metilpirrolidin-3-il1metanol. A una solución de (3R)-3-(hidroximetil) pirrolidin-1-carboxilato de terc-butilo (1,0 g, 4,97 mmol, 1,0 eq.) en THF (20,0 ml) se le añadió LiAlH4 (377 mg, 9,94 mmol, 2,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas, después se calentó a 70 °C y se agitó a 70 °C durante 2 horas. Después de que se completara, la mezcla de reacción se inactivó con Na2SÜ4 acuoso saturado (1,0 ml) y se filtró. La torta de filtro se lavó con THF (10,0 ml). Después, el licor madre se concentró para dar [(3R)-1-metilp¡rrolid¡n-3-¡l]metanol (500 mg, 4,34 mmol, 87 % de rendimiento) en forma de un aceite incoloro.
1H RMN (400 MHz, Cloroformo-d) ó 3,65 (dd, J = 4,8, 10,0 Hz, 1H), 3,52 (dd, J = 5,6, 10,0 Hz, 1H), 3,05 - 2,69 (m, 2H), 2,63 - 2,53 (m, 1H), 2,51 - 2,44 (m, 1H), 2,40 - 2,25 (m, 5H), 2,04 - 1,94 (m, 1H), 1,72 - 1,59 (m, 1H).
Etapa A: (2S)-4-[7-(8-cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de terc-butilo. A una solución de [(3R)-1-metilpirrolidin-3-il1metanol (178 mg, 1,55 mmol, 3,0 eq.) en tolueno (3,0 ml) se le añadió t-Bu0Na (124 mg, 1,29 mmol, 2,50 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después, a la mezcla anterior se le añadió (2S)-4-[7-(8-cloro-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de terc-butilo (300 mg, 516 umol, 1,0 eq.). Después de la adición, la mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, a la mezcla de reacción se le añadió agua (10,0 ml) y se extrajo con AE (10,0 ml * 3). La capa orgánica se secó sobre Na2S04, se filtró y se concentró. El producto obtenido se purificó por cromatografía ultrarrápida de fase inversa (C18, 0,1 % de AF en agua, 0-60 % de MeCN) para dar (2S)-4-[7-(8-cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de terc-butilo (220 mg, 342 umol, 66 % de rendimiento, 98 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+11: 632.
Etapa B: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de terc-butilo (360 mg, 569 umol, 1,0 eq.) en DCM (1,50 ml) se le añadió TFA (2,31 g, 20,3 mmol, 1,50 ml, 35,6 eq.). La mezcla se agitó a 25 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se concentró para dar 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(3R)-1-met¡lp¡rrol¡d¡n-3-¡l]metox¡j-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (450 mg, bruto, 2TFA) en forma de un aceite de color amarillo.
Etapa C: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-prop-2-enoil-piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-6,8-dihidro-5H-pirido[3,4-tí1pirimidin-4-il1piperazin-2-il1acetonitrilo (450 mg, 592 umol, 1,0 eq., 2TFA) y DIEA
(918 mg, 7,10 mmol, 1,24 ml, 12,0 eq.) en DCM (4,0 ml) se le añadió prop-2-enoato de prop-2-enoílo (112 mg, 888 umol, 1,50 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se inactivó con Me0H (1,0 ml) y se concentró al vacío. El producto obtenido se purificó por HPLC prep. (columna: Gemini 150 * 25 5 u; fase móvil: [agua (0,04 % de NH3*H20)-ACN]; % de B: 70 %-82 %,10 min) para dar el compuesto del título 2 [(2S)-4-[7-(8-cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il)acetonitrilo (EJEMPL0361,32,6 mg, 53,8 umol, 9 % de rendimiento, 97 % de pureza) en forma de un sólido de color gris. LCMS [ESI, M+1]: 586.
1H RMN (400 MHz, Cloroformo-d) ó 7,68 (d a, J = 8,0 Hz, 1H), 7,54 (t, J = 7,6 Hz, 1H), 7,45 (d, J = 7,2 Hz, 1H), 7,41 -7,32 (m, 1H), 7,26 (t, J = 8,0 Hz, 1H), 7,18 - 7,11 (m, 1H), 6,60 - 6,43 (m, 1H), 6,32 (d a, J = 16,4 Hz, 1H), 5,75 (d a, J = 10,8 Hz, 1H), 5,15 - 4,50 (m, 1H), 4,41 - 4,28 (m, 1H), 4,18 - 4,10 (m, 2H), 4,09 - 3,88 (m, 2H), 3,87 - 3,68 (m, 2H), 3,57 - 3,45 (m, 1H), 3,42 - 3,26 (m, 1H), 3,25 - 2,87 (m, 5H), 2,86 - 2,74 (m, 1H), 2,73 - 2,57 (m, 3H), 2,56 - 2,47 (m, 2H), 2,46 - 2,37 (m, 2H), 2,28 (s, 3H), 2,07 - 1,90 (m, 1H). LCMS [ESI, M+1]: 586.
Ejemplo 362

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Etapa A: (2S)-4-r7-(8-cloro-1-naftil)-2-r2-(1-piperidil)etoxi1-6,8-dihidro-5H-piridor3,4-d|pirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de tere-butilo. A una solución de 2-(1-piperidil)etanol (133 mg, 1,03 mmol, 137 ul, 3,0 eq.) en tolueno (2,0 ml) se le añadió t-Bu0Na (66,2 mg, 688 umol, 2,0 eq.) a 0 °C. La mezcla se agitó a 20 °C durante 0,5 horas. Después, al líquido anterior se le añadió (2S)-4-[7-(8-cloro-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (200 mg, 344 umol, 1,0 eq.) a 0 °C. Después de la adición, la mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, a la mezcla de reacción se le añadió agua (5,0 ml) y se extrajo con AE (5,0 ml * 3). La capa orgánica se secó sobre Na2S04, se filtró y se concentró. El producto obtenido se purificó por cromatografía en columna (Si02, EP:AE = 3:1 - AE:Me0H = 10:1) para dar (2S)-4-[7-(8-cloro-1-naftil)-2-[2-(1piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de fere-butilo (150 mg, 221 umol, 64 % de rendimiento, 95 % de pureza) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 646.
1H RMN (400 MHz, Cloroformo-d) 57,76 (d, J = 8,0 Hz, 1H), 7,61 (t, J = 7,8 Hz, 1H), 7,53 (d, J = 7,6 Hz, 1H), 7,49 -7,40 (m, 1H), 7,33 (t, J = 7,8 Hz, 1H), 7,27 - 7,17 (m, 1H), 4,64 - 4,61 (m, 1H), 4,48 - 4,36 (m, 3H), 4,14 - 3,92 (m, 3H), 3,90 - 3,76 (m, 1H), 3,63 - 3,54 (m, 1H), 3,36 (dd a, J = 3,6, 13,6 Hz, 1H), 3,26 - 3,06 (m, 3H), 3,01 - 2,86 (m, 1H), 2,82 - 2,76 (m, 2H), 2,71 (d a, J = 6 ,8 Hz, 1H), 2,64 - 2,58 (m, 1H), 2,58 - 2,50 (m, 4H), 2,48 - 2,42 (m, 1H), 2,06 (d, J = 6,4 Hz, 1H), 1,64 - 1,57 (m, 5H), 1,52 (s, 9H).
Etapa B: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[2-(1-piperidil)etoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-il]piperazin-2-illacetonitrilo. A una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (250 mg, 387 umol, 1,0 eq.) en DCM (1,0 ml) se le añadió TFA (1,54 g, 13,5 mmol, 1,0 ml, 34,9 eq.). La mezcla se agitó a 25 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se concentró al vacío. El producto 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (300 mg, bruto, 2TFA) se obtuvo en forma de un aceite de color amarillo. LCmS [ESI, M+1]: 546.
Etapa C: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[2-(1-piperidil)etoxi1-6,8-dihidro-5H-pirido[3,4-d^pirimidin-4-il1-1-prop-2-enoilpiperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (300 mg, 388 umol, 1 ,0 eq., 2 TfA) y DiEa (601 mg, 4,65 mmol, 810 ul, 12,0 eq.) en DCM (2,0 ml) se le añadió prop-2-enoato de prop-2-enoílo (73,3 mg, 581 umol, 1,5 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se inactivó con Me0H (1,0 ml) y se concentró al vacío. El producto obtenido se purificó por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 55 %-85 %,12 min) para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[2-(1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0362 , 54,0 mg, 88,1 umol, 23 % de rendimiento, 98 % de pureza) en forma de un aceite incoloro. LCMS [ESI, M+1]: 600.
1H RMN (400 MHz, Cloroformo-d) 57,76 (d, J = 8,0 Hz, 1H), 7,62 (t, J = 7,6 Hz, 1H), 7,53 (d, J = 7,6 Hz, 1H), 7,49 -7,39 (m, 1H), 7,34 (t, J = 7,8 Hz, 1H), 7,27 - 7,18 (m, 1H), 6,58 - 6,62 (m, 1H), 6,44 - 6,36 (m, 1H), 5,83 (d a, J = 10,8 Hz, 1H), 5,15 - 4,55 (m, 1H), 4,51 - 4,36 (m, 3H), 4,20 - 3,97 (m, 2H), 3,95 - 3,75 (m, 2H), 3,66 - 3,54 (m, 1H), 3,51 - 3,37 (m, 1H), 3,34 - 2,97 (m, 4H), 2,94 - 2,67 (m, 4H), 2,65 - 2,57 (m, 1H), 2,55 - 2,48 (m, 4H), 1,65 - 1,60 (m, 4H), 1,49 - 1,39 (m, 2H).
Ejemplo 363

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-4-metilmorfolin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-dJpirimidin-4-il]-1-prop-2-enoil-piperazin-2 -il]acetonitrilo

Inserción: [(2R)-4-metilmorfolin-2-il1metanol. A una solución de (2R)-2-(hidroximetil) morfolin-4-carboxilato de tercbutilo (1,50 g, 6,90 mmol, 1,0 eq.) en THF (20,0 ml) se le añadió poco a poco LiAlH4 (524 mg, 13,8 mmol, 2,0 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas, después se calentó a 60 °C y se agitó a 60 °C durante 12 horas. Después de que se completara, la mezcla de reacción se inactivó con Na2SÜ4 acuoso saturado (1,50 ml) y se filtró. La torta se lavó con THF (20,0 ml x 2). Después, el licor madre se concentró para dar [(2R)-4-metilmorfolin-2-¡l]metanol (800 mg, 6,10 mmol, 88 % de rendimiento) en forma de un aceite de color amarillo.
1H RMN (400 MHz, Cloroformo-d) ó 3,92 - 3,86 (m, 1H), 3,73 - 3,52 (m, 4H), 2,84 - 2,56 (m, 3H), 2,27 (s, 3H), 2,11 (dt, J = 3,6, 11,6 Hz, 1H), 1,93 (t, J = 10,8 Hz, 1H).
Etapa A: (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-4-metilmorfolin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-cf1pirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de tere-butilo. A una solución de [(2R)-4-metilmorfolin-2-il]metanol (203 mg, 1,55 mmol, 3,0 eq.) en tolueno (4,0 ml) se le añadió t-Bu0Na (99,2 mg, 1,03 mmol, 2,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después, a la mezcla anterior se le añadió (2S)-4-[7-(8-cloro-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de tere-butilo (300 mg, 516 umol, 1,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, a la mezcla de reacción se le añadió agua (10,0 ml) y se extrajo con AE (10,0 ml x 3). La capa orgánica se secó sobre Na2S04, se filtró y se concentró. El producto obtenido se purificó por cromatografía en columna (Si02, EP: AE = 3:1 - AE: Me0H = 10:1) para dar (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R-4-metilmorfolin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-tí1pirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de tere-butilo (250 mg, 366 umol, 71 % de rendimiento, 95 % de pureza) en forma de un aceite de color amarillo. LCMS [ESI, M+11: 648.
1H RMN (400 MHz, Cloroformo-d) ó 7,75 (d, J = 8,0 Hz, 1H), 7,61 (t, J = 7,6 Hz, 1H), 7,52 (d, J = 7,6 Hz, 1H), 7,49 - 7,39 (m, 1H), 7,33 (t, J = 7,8 Hz, 1H), 7,26 - 7,17 (m, 1H), 4,68 - 4,53 (m, 1H), 4,48 - 4,31 (m, 2H), 4,29 - 4,19 (m, 1H), 4,16 - 4,01 (m, 3H), 4,00 - 3,82 (m, 4H), 3,79 - 3,69 (m, 1H), 3,63 - 3,53 (m, 1H), 3,36 (dd a, J = 3,6, 13,6 Hz, 1H), 3,29 - 3,06 (m, 3H), 3,01 - 2,86 (m, 2H), 2,83 - 2,64 (m, 3H), 2,63 - 2,45 (m, 1H), 2,36 - 2,28 (m, 3H), 2,19 (dt, J = 3,6, 11,6 Hz, 1H), 1,52 (s, 9H).
Etapa B: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-4-metilmorfolin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-4-metilmorfolin-2-il1metoxi1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de tere-butilo (240 mg, 370 umol, 1,0 eq.) en DCM (1,0 ml) se le añadió TFA (1,54 g, 13,5 mmol, 1,0 ml, 36,5 eq.). La mezcla se agitó a 25 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se concentró para dar 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-4-metilmorfolin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-tí1pirimidin-4-il1piperazin-2-il1acetonitrilo (300 mg, bruto, 2TFA) en forma de un aceite de color rojo claro. lCm S [ESI, M+11: 548.
Etapa C: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-4-metilmorfolin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-il1-1-prop-2-enoil-piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-4-metilmorfolin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (300 mg, 387 umol, 1,0 eq., 2TFA) y DIEA (599 mg, 4,64 mmol, 808 ul, 12,0 eq.) en DCM (2,0 ml) se le añadió prop-2-enoato de prop-2-enoílo (73,1 mg, 580 umol, 1,50 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se inactivó con Me0H (1,0 ml) y se concentró al vacío. El producto obtenido se purificó por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,04 % de NH3*H20)-ACN]; % de B: 45 %-75 %,10 min) para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naft¡l)-2-[[(2R)-4-met¡lmorfol¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0363, 42,7 mg, 70,4 umol, 18 % de rendimiento, 99 % de pureza) en forma de un sólido de color blanco. lCm S [ESI, M+1]: 602.
1H RMN (400 MHz, Cloroformo-d) ó 7,76 (d, J = 8,0 Hz, 1H), 7,62 (t, J = 7,2 Hz, 1H), 7,55 - 7,50 (m, 1H), 7,49 - 7,40 (m, 1H), 7,34 (t, J = 7,8 Hz, 1H), 7,27 - 7,18 (m, 1H), 6,65 - 6,52 (m, 1H), 6,39 (d a, J = 16,8 Hz, 1H), 5,83 (d a, J = 10,8 Hz, 1H), 5,25 - 4,53 (m, 1H), 4,48 - 4,32 (m, 2H), 4,30 - 4,21 (m, 1H), 4,19 - 4,00 (m, 2H), 3,99 - 3,89 (m, 3H), 3,88 - 3,76 (m, 1H), 3,75 - 3,68 (m, 1H), 3,64 - 3,55 (m, 1H), 3,52 - 3,35 (m, 1H), 3,31 - 2,98 (m, 4H), 2,92 - 2,55 (m, 5H), 2,31 (s, 3H), 2,18 (dt, J = 3,2, 11,4 Hz, 1H), 2,02 (dt, J = 1,6, 10,8 Hz, 1H).
Ejemplo 364

2-[(2S)-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-[3-met¡l-2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-il]-1 -prop-2 -enoil-piperazin-2 -il]acetonitrilo

Etapa 1: 1-bromo-3-metoxi-2-(trifluorometil)benceno. A una solución de Me0H (1,32 g, 41,2 mmol, 1,67 ml, 2 eq.) en DMF (125 ml) se le añadió NaH (1,65 g, 41,2 mmol, 60,0 % de pureza, 2 eq.). La mezcla se agitó a 25 °C durante 0,5
horas. Después, a la mezcla se le añadió 1-bromo-3-fluoro-2-(tr¡fluoromet¡l)benceno (5 g, 20,6 mmol, 1 eq.) y se agitó a 25 °C durante 0,5 horas. La mezcla de reacción se inactivó con agua enfriada con hielo (20 ml). A la mezcla se le añadió agua (300 ml) y se extrajo con MTBE (3 x 100 ml). Las capas orgánicas combinadas se lavaron con agua (50 ml) y salmuera (50 ml). Las capas orgánicas combinadas se secaron sobre Na2SÜ4 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (PE) para dar 1-bromo-3-metoxi-2-(trifluorometil)benceno (5,28 g, 18,6 mmol, 91,0 % de rendimiento, 90,0 % de pureza) en forma de un aceite incoloro.
1H RMN (400 MHz, cloroformo-d) ó = 7,34 - 7,28 (m, 2H), 7,01 - 6,97 (m, 1H), 3,91 (s, 3H).
Etapa 2: 1-metox¡-3-met¡l-2-(tr¡fluoromet¡l)benceno. A la solución de 1-bromo-3-metoxi-2-(trifluorometil)benceno (5,28 g, 20,7 mmol, 1 eq.), Cs2CÜ3 (20,2 g, 62,1 mmol, 3 eq.) y ácido metilborónico (6,20 g, 104 mmol, 5 eq.) en dioxano (105 ml) y agua (21 ml) se le añadió Pd(dppf)Cl2 (2,27 g, 3,11 mmol, 0,15 eq.) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con N2. La mezcla se agitó en una atmósfera de N2 a 100 °C durante 6 horas. A la mezcla se le añadió agua (50 ml). La mezcla se diluyó con EtOAc (30 ml) y se extrajo con EtOAc (100 ml). La capa orgánica se secó sobre Na2S04 anhidro, se filtró y se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (PE) para dar 1-metox¡-3-met¡l-2-(tr¡fluoromet¡l)benceno (3,2 g, 15.1 mmol, 73,1 % de rendimiento, 90,0 % de pureza) en forma de un aceite de color amarillo.
1H RMN (400 MHz, cloroformo-d) ó = 7,33 (t, J = 8,0 Hz, 1H), 6,87 (d a, J = 8,4 Hz, 1H), 6,83 (d, J = 7,2 Hz, 1H), 3,88 (s, 3H), 2,49 (c, J = 3,6 Hz, 3H).
Etapa 3: 3-metil-2-(trifluorometil)fenol. A la solución de 1-metox¡-3-met¡l-2-(tr¡fluoromet¡l)benceno (3,2 g, 16,8 mmol, 1 eq.) en DCE (65 ml) se le añadió BBr3 (21,1 g, 84,1 mmol, 8,11 ml, 5 eq.) a -40 °C y la mezcla se agitó a -40 - -10 °C durante 0,5 horas. La mezcla de reacción se inactivó con agua (100 ml). La mezcla se diluyó con EtOAc (20 ml) y se extrajo con EtOAc (2 x 50 ml). Las capas orgánicas combinadas se lavaron con salmuera (30 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc de 1:0 a 50:1) para dar 3-metil-2-(trifluorometil)fenol (740 mg, 4,08 mmol, 24.2 % de rendimiento, 97,0 % de pureza) en forma de un sólido de color amarillo.
1H RMN (400 MHz, cloroformo-d) ó = 7,29 - 7,24 (m, 1H), 6,87 - 6,77 (m, 2H), 5,95 (c, J = 6,4 Hz, 1H), 2,46 (c, J = 2,8 Hz, 3H).
Etapa 4: trifluorometanosulfonato de í3-metil-2-(trifluorometil)fenilo1. A la solución de 3-metil-2-(trifluorometil)fenol (600 mg, 3,41 mmol, 1 eq.), TEA (1,38 g, 13,6 mmol, 1,90 ml, 4 eq.) en EtOAc (12 ml) se le añadió Tf20 (1,44 g, 5,11 mmol, 843 ul, 1,5 eq.) a -40 °C y la mezcla se agitó a -40 °C durante 0,5 horas. La mezcla se inactivó con agua (10 ml). La mezcla se diluyó con EtOAc (10 ml) y se extrajo con EtOAc (30 ml). La capa orgánica se lavó con salmuera (20 ml), se secó sobre Na2S04 anhidro, se filtró y se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc de 1:0 a 100:1) para dar trifluorometanosulfonato de ^-metil^trifluorometilfenilo] (590 mg, 1,82 mmol, 53,0 % de rendimiento, 95,0 % de pureza) en forma de un aceite incoloro.
1H RMN (400 MHz, cloroformo-d) ó = 7,53 - 7,47 (m, 1H), 7,33 (d, J = 7,6 Hz, 1H), 7,29 (d, J = 8,4 Hz, 1H), 2,57 (c, J = 2,8 Hz, 3H).
Etapa A: (2S)-2-(c¡anomet¡l)-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-7-[3-met¡l-2-(tr¡fluoromet¡l)fen¡l1-6,8-d¡h¡dro-5H-p¡r¡dα3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de bencilo. A la solución de (2S)-2-(cianometil)-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-5,6,7,8-tetrah¡drop¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1 -carboxilato de bencilo (Intermedio 65, 600 mg, 973 umol, 1 eq.), trifluorometanosulfonato de [3-metil-2-(trifluorometil)fenilo] (450 mg, 1,46 mmol, 1,5 eq.), Cs2C03 (951 mg, 2,92 mmol, 3 eq.) y RuPhos (90,8 mg, 195 umol, 0,2 eq.) en tolueno (12 ml) se le añadió Pd2(dba)3 (89,1 mg, 97,3 umol, 0,1 eq.) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con N2. La mezcla se agitó en una atmósfera de N2 a 90 °C durante 4 horas. A la mezcla se le añadió agua (15 ml). La mezcla se diluyó con EtOAc (10 ml) y se extrajo con EtOAc (30 ml). La capa orgánica se lavó con salmuera (20 ml), se secó sobre Na2S04 anhidro, se filtró y se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (de EP: EtOAc = 5:1 - 1:1 a EtOAc: Me0H = 1:0 - 20:1). Después, el residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0,1 % de AF) = 50,0 %) para dar (2S)-2-(c¡anomet¡l)-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-[3-met¡l-2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (240 mg, 354 umol, 36 % de rendimiento, 98,0 % de pureza) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 664.
1H RMN (400 MHz, cloroformo-d) ó = 7,41 - 7,35 (m, 6H), 7,18 (d, J = 8,0 Hz, 1H), 7,07 (d, J = 7,6 Hz, 1H), 7,10 - 7,04 (m, 1H), 5,20 (s, 2 H), 4,69 (s a, 1H), 4,38 (dd, J = 5,2, 10,8 Hz, 1H), 4,16 (t, J = 3,2 Hz, 1H), 4,05 (s, 2H), 3,89 (d a, J = 12,0 Hz, 1H), 3,30 (s a, 2H), 3,21 - 2,95 (m, 5H), 2,92 - 2,62 (m, 6H), 2,52 (c, J = 3,6 Hz, 3H), 2,48 (s, 3H), 2,33 -2,24 (m, 1H), 2,05 - 2,00 (m, 1H), 1,90 - 1,79 (m, 3H).
Etapa B: Se burbujeó NH3 en Me0H (5 ml) durante 5 minutos. A la solución se le añadió (2S)-2-(cianometil)-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-[3-met¡l-2-(tr¡fluoromet¡l)fen¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carboxilato de bencilo (240 mg, 362 umol, 1 eq.), Pd/C (50 mg, 10,0 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de
H2 (103,42 kPa (15 psi)) a 30 °C durante 0,5 horas. La mezcla de reacción se filtró, el filtrado se concentró al vacío para dar 2-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-[3-metil-2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (180 mg, 336 umol, 93,0 % de rendimiento, 99,0 % de pureza) en forma de un aceite de color pardo que se usó para la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 530.
Etapa C: 2-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-[3-metil-2-(trifluorometil)fenil1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-prop-2-enoil-piperazin-2-il1acetonitrilo. A la solución de 2-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-[3-metil-2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (170 mg, 321 umol, 1 eq.) y TEA (97,4 mg, 963 umol, 134 ul, 3 eq.) en DCM (3,5 ml) se le añadió cloruro de prop-2-enoílo (43,6 mg, 482 umol, 39,3 ul, 1,5 eq.) a -40 °C y la mezcla se agitó a -40 - 30 °C durante 0,5 horas. La mezcla de reacción se inactivó con agua (2 ml) y se separó. La fase acuosa se extrajo con EtOAc (4 ml). Las capas orgánicas combinadas se secaron sobre Na2SÜ4 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Gemini 150 * 25 5 ufase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN];% de B: 55 %-85 %,12 min) para dar el compuesto del título 2-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-[3-metil-2-(trifluorometil)fenil]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPLÜ 364, 110 mg, 188 umol, 58,0 % de rendimiento, 100 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 584.
1H RMN (400 MHz, cloroformo-d) 5 = 7,43 - 7,35 (m, 1H), 7,19 (d, J = 8,4 Hz, 1H), 7,08 (d, J = 7,6 Hz, 1H), 6,59 (s a, 1H), 6,40 (dd, J = 1,6, 16,8 Hz, 1H), 5,83 (d a, J = 10,4 Hz, 1H), 5,09 (s a, 1H), 4,64 (s a, 1H), 4,37 (dd, J = 4,8, 10,4 Hz, 1H), 4,18 - 4,08 (m, 2H), 4,06 (s, 2H), 3,96 (d a, J = 12,4 Hz, 1H), 3,60 (s a, 1H), 3,30 (s a, 1H), 3,22 - 3,01 (m, 4H), 2,98 - 2,62 (m, 5H), 2,52 (c, J = 3,2 Hz, 3H), 2,48 (s, 3H), 2,33 - 2,23 (m, 1H), 2,11 - 1,99 (m, 1H), 1,90 - 1,68 (m, 3H).
Ejemplo 365

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Inserción: 3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.11heptan-5-il]propan-1 -ol. A una solución de (1 S,4S)-2-oxa-5-azabiciclo[2.2.1]heptano (500 mg, 3,69 mmol, 1,0 eq., HCl) y 3-bromopropan-1-ol (513 mg, 3,69 mmol, 333 ul, 1,0 eq.) en CH3CN (6,0 ml) se le añadió K2C03 (1,53 g, 11,1 mmol, 3,0 eq.). La mezcla se agitó a 50 °C durante 2 horas. Después de que se completara, la mezcla de reacción se filtró y el filtrado se evaporó. El residuo se purificó por cromatografía en columna (Si02, acetato de etilo: Me0H = 50: 1 a 20: 1) para dar 3-[(16',45)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propan-1-ol (195 mg, 1,12 mmol, 30 % de rendimiento, 90 % de pureza) en forma de un aceite incoloro.
1H RMN (400 MHz, Cloroformo-d) ó 4,42 (s, 1H), 3,99 (d, J = 8,0 Hz, 1H), 3,82 (t, J = 5,6 Hz, 2H), 3,65 - 3,61 (m, 1H), 3,53 - 3,45 (m, 2H), 2,99 - 2,92 (m, 2H), 2,84 - 2,77 (m, 1H), 2,67 - 2,61 (m, 1H), 1,86 - 1,80 (m, 1H), 1,79 - 1,58 (m, 3H).
Etapa A: (2S)-4-[7-(8-cloro-1-naftil)-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.11heptan-5-il]propoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1 -carboxilato de tere-butilo. A una mezcla de 3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propan-1-ol (325 mg, 2,06 mmol, 3,0 eq.), í-Bu0Na (198 mg, 2,06 mmol, 3,0 eq.) en tolueno (2,0 ml) se le añadió poco a poco una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (Intermedio 71, Etapa A, 400 mg, 688 umol, 1,0 eq.) en tolueno (2,0 ml) y la mezcla se agitó a 0 °C durante 30 min en una atmósfera de N2. Después de que se completara, al disolvente orgánico se le añadió agua (10,0 ml). La fase acuosa se extrajo con acetato de etilo (3 * 10 ml). Los extractos combinados se lavaron con salmuera (20,0 ml), se secaron con Na2S04 y después el disolvente se eliminó al vacío. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo : acetato de etilo = 3 : 1 a acetato de etilo : Me0H = 20 : 1) para dar (2S)-4-[7-(8-cloro-1 -naftil)-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.1 ]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (320 mg, 432 umol, 62 % de rendimiento, 91 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1 ]: 674.
Etapa B: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.11heptan-5-il]propoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-[3-[(1 S,4S)-2-oxa-5-azab¡c¡clo[2.2.1]heptan-5-¡l]propox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(cianomet¡l)p¡peraz¡n-1-carbox¡lato de tere-butilo (340 mg, 504 umol, 1,0 eq.) en dioxano (3,0 ml) se le añadió Hckdioxano (4,0 M, 3,0 ml, 23,8 eq.). La mezcla se agitó a 25 °C durante 15 min en una atmósfera de N2. Después de que se completara, el disolvente orgánico se eliminó al vacío. El producto obtenido se ajustó con NaHC03 acuoso saturado (10 ml), se extrajo con acetato de etilo (3 * 10 ml) y la capa orgánica combinada se secó sobre Na2S04, se filtró y se concentró. El producto bruto 2-[(2S)-4-[7-(8-cloro-1 -naftil)-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.1 ]heptan-5-¡l]propox¡]-6,8-d¡hidro-5H-p¡r¡do[3,4-d]pirimidin-4-il1piperazin-2-il]acetonitrilo (270 mg, bruto) se obtuvo en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 574.
Etapa C: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[3-[(1 S.4S)-2-oxa-5-azabiciclo[2.2.11heptan-5-il1propoxi1-6.8-dihidro-5H-pirido[3.4-dlpirimidin-4-il1-1-prop-2-enoil-piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[3-[(1S.4S)-2-oxa-5-azabiciclo[2.2.11heptan-5-il1propoxi1-6.8-dihidro-5H-pirido[3.4-d1pirimidin-4-il1piperazin-2-il]acetonitrilo (270 mg.470 umol. 1.0 eq.) en dCm (4.0 ml) se le añadieron TEA (143 mg. 1.41 mmol. 196 ul. 3.0 eq.) y prop-2-enoato de prop-2-enoílo (178 mg. 1.41 mmol. 3.0 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 30 min en una atmósfera de N2. Después de que se completara. el disolvente orgánico se eliminó al vacío. El residuo se purificó por cromatografía en columna (Base Al2Ü3. éter de petróleo: acetato de etilo = 3: 1 a 1: 1) y después el producto bruto se concentró y se purificó de nuevo por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil:
[agua (0.05 % de hidróxido de amoniaco v/v)-ACN1; % de B: 45 %-75 %. 12 min) y se sometió a liofilización. El compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[3-[(1S.4S)-2-oxa-5-azabiciclo[2.2.11heptan-5-il1propoxi1-6.8-dihidro-5^-pirido[3.4-d1pirimidin-4-il1-1-prop-2-enoil-piperazin-2-il1acetonitrilo (EJEMPLÜ 365. 43.1 mg. 67.7 umol.
14 % de rendimiento. 98 % de pureza) se obtuvo en forma de un sólido de color blanquecino. LCMS [ESI. M+11: 628.
1H RMN (400 MHz. cloroformo-d) 57.76 (d. J = 9.6 Hz. 1H). 7.62 (t. J = 7.6 Hz. 1H). 7.50 (d. J = 6.4 Hz. 1H). 7.47 -7.40 (m. 1H). 7.34 (t. J = 8.0 Hz. 1H). 7.26 - 7.16 (m. 1H). 6.67 - 6.54 (m. 1H). 6.40 (d. J = 16.8 Hz. 114). 5.82 (d. J = 10.8 Hz. 1H). 5.16 - 5.03 (m. 1H). 4.48 - 4.40 (m. 1H). 4.38 - 4.34 (m. 3H). 4.22 - 4.08 (m. 1H). 4.07 - 3.99 (m. 2H).
3.97 - 3.74 (m. 2H). 3.64 - 3.55 (m. 2H). 3.54 - 3.35 (m. 2H). 3.31 - 3.20 (m. 1H). 3.19 - 3.14 (m. 1H). 3.13 - 2.99 (m.
2H). 2.92 (d. J = 10.4 Hz. 1H). 2.89 - 2.81 (m. 1H). 2.80 - 2.76 (m. 1H). 2.75 - 2.68 (m. 1H). 2.64 - 2.57 (m. 1H). 2.52 (d. J = 10.0 Hz. 1H). 1.98 - 1.89 (m. 2H). 1.85 (d. J = 9.2 Hz. 1H). 1.74 - 1.70 (m. 2H).
Ejemplo 366

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-4.4-difluoro-1-metil-pirrolidin-2-il1metoxi1-6.8-dihidro-5H-pirido[3.4-djpirimidin-4-il1-1-prop-2-enoil-piperazin-2-il1acetonitrilo

Inserción: A una solución de 0 1 -terc-butil 02-metil (2S)-4,4-difluoropirrolidin-1,2-dicarboxilato (300 mg, 1,13 mmol, 1,0 eq.) en THF (8,0 ml) se le añadió LiAlH4 (129 mg, 3,39 mmol, 3,0 eq.). La mezcla se agitó a -40 °C durante 1 hora. Después, la mezcla se agitó a 80 °C durante 2 horas en una atmósfera de N2. Después de que se completara, la mezcla de reacción se inactivó con una solución acuosa saturada de Na2S04 (6,0 ml) y se secó con Na2S04. Después, la mezcla se filtró y el filtrado se concentró. Se obtuvo [(2S)-4,4-difluoro-1-metil-pirrolidin-2-il]metanol (165 mg, bruto) en forma de un aceite incoloro y se usó en la siguiente etapa sin purificación adicional.
Etapa A: (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-4,4-difluoro-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1 -carboxilato de tere-butilo. A una solución de [(2S)-4,4-difluoro-1-metilpirrolidin-2-il]metanol (156 mg, 1,03 mmol, 3,0 eq.) en tolueno (2,0 ml) se le añadieron gota a gota í-Bu0Na (99,2 mg, 1,03 mmol, 3,0 eq.) a 0 °C y una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (200 mg, 344 umol, 1,0 eq.) en tolueno (1,0 ml) a 0 °C en una atmósfera de N2. La mezcla de reacción se agitó a 0 °C durante 30 min. Después de que se completara, al disolvente orgánico se le añadió agua (10,0 ml) y se extrajo con acetato de etilo (3 * 10 ml). Los extractos combinados se lavaron con salmuera (20,0 ml), se secaron con Na2S04, se filtraron y el filtrado se concentró. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo : acetato de etilo = 5 : 1 a 1:1) para dar (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-4,4-difluoro-1-metil-pirrolidin-2-il]metoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de tere-butilo (160 mg, 230 umol, 67 % de rendimiento, 96 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+11: 668.
Etapa B: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-4,4-difluoro-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-4,4-difluoro-1-metilp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de tere-butilo (137 mg, 205 umol, 1,0 eq.) en dioxano (1,0 ml) se le añadió HCl*dioxano (4 M, 1,0 ml, 19,5 eq.). La mezcla se agitó a 25 °C durante 15 min en una atmósfera de N2. Después de que se completara, el disolvente orgánico se eliminó al vacío. El producto obtenido se ajustó con NaHC03 acuoso saturado (10 ml), se extrajo con acetato de etilo (3 * 10 ml), se secó sobre Na2S04, se filtró y se concentró. El producto bruto 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-4,4-difluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (110 mg, bruto) se obtuvo en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+11: 568.
Etapa C: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-4,4-difluoro-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-prop-2-enoil-piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-4,4
difluoro-1-metil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (110 mg, 194 umol, 1,0 eq.) en DCM (3,0 ml) se le añadieron TEA (196 mg, 1,94 mmol, 270 ul, 10,0 eq.) y prop-2-enoato de prop-2-enoílo (73,3 mg, 581 umol, 3,0 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 30 min en una atmósfera de N2. Después de que se completara, la mezcla de reacción se inactivó con metanol (6,0 ml) y la mezcla se eliminó al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 55 % - 85 %, 12 min) y se sometió a liofilización. El compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-4,4-difluoro-1-metil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0366, 34,1 mg, 54,5 umol, 28 % de rendimiento, 99 % de pureza) se obtuvo en forma de un sólido de color blanquecino. LCMS [ESI, M+1]: 622.
1H RMN (400 MHz, cloroformo-d) 57,76 (d, J = 8,0 Hz, 1H), 7,63 (t, J = 7,2 Hz, 1H), 7,50 (d, J = 7,6 Hz, 1H), 7,49 -7,42 (m, 1H), 7,34 (t, J = 8,0 Hz, 1H), 7,26 - 7,19 (m, 1H), 6,63 - 6,57 (m, 1H), 6,40 (d, J = 16,8 Hz, 1H), 5,84 (d, J = 10,4 Hz, 1H), 5,02 - 4,98 (m, 1H), 4,49 - 4,37 (m, 2H), 4,31 - 4,22 (m, 1H), 4,19 - 3,97 (m, 2H), 3,96 - 3,89 (m, 1H), 3,88 - 3,77 (m, 1H), 3,76 - 3,65 (m, 1H), 3,64 - 3,55 (m, 1H), 3,52 - 3,37 (m, 2H), 3,36 - 3,10 (m, 3H), 3,07 - 2,95 (m, 2H), 2,94 - 2,80 (m, 1H), 2,79 - 2,48 (m, 4H), 2,47 - 2,45 (m, 3H), 2,36 - 2,24 (m, 1H).
Ejemplo 367

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-[(£)-4-morfolinobut-2-enoil]piperazin-2-il]acetonitrilo

Etapa A: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-2-illacetonitrilo. A una solución de (2S)-4-[7-(8-cloro-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de terc-butilo (Intermedio 71, Etapa A, 200 mg, 316 umol, 1,0 eq.) en dioxano (1,5 ml) se le añadió HCl*dioxano (4 M, 1,5 ml, 19,0 eq.). La mezcla se agitó a 25 °C durante 0,5 horas en una atmósfera de N2. Después de que se completara, el disolvente orgánico se eliminó al vacío. El producto obtenido se ajustó con NaHCÜ3 acuoso saturado (10 ml), se extrajo con acetato de etilo (3 * 10 ml), se secó sobre Na2SÜ4, se filtró y el filtrado se concentró. El producto bruto 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (130 mg, bruto) se obtuvo en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1 ]: 532.
Etapa B: 2-r(2S)-1-r(E)-4-bromobut-2-enoi n -4-r7-(8-cloro-1-naftil)-2-rr(2S)-1-metilpirrolidin-2-i n metox n -6.8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (150 mg, 282 umol, 1,0 eq.) en DCM (3,0 ml) se le añadieron piridina (446 mg, 5,64 mmol, 455 ul, 20,0 eq.) y cloruro de (E)-4-bromobut-2-enoílo (155 mg, 846 umol, 3,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se concentró. El producto bruto 2-[(2S)-1-[(E)-4-bromobut-2-enoil]-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡trilo (190 mg, bruto) se obtuvo en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMs [ESI, M+1 ]: 680.
Etapa C: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-[(E)-4-morfolinobut-2-enoi n piperazin-2-i n acetonitrilo. A una solución de 2-[(2S)-1-[(E)-4-bromobut-2-enoil]-4-[7-(8-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]acetonitr¡lo (190 mg, 280 umol, 1,0 eq.), KI (9,29 mg, 56,0 umol, 0,20 eq.) y K2C03 (387 mg, 2,80 mmol, 10,0 eq.) en DCM (5,0 ml) se le añadió morfolina (146 mg, 1,68 mmol, 148 ul, 6,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se inactivó con metanol (10,0 ml) y la mezcla se eliminó al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 60 %-90 %, 12 min) y después se sometió a liofilización. El compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡mid¡n-4-¡l]-1-[(E)-4-morfol¡nobut-2-eno¡l]p¡perazin-2-¡l]aceton¡tr¡lo (EJEMPL0367 , 31,6 mg, 45,2 umol, 16 % de rendimiento, 98 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 685.
1H RMN (400 MHz, cloroformo-d) 57,78 - 7,73 (m, 1H), 7,62 (t, J = 7,6 Hz, 1H), 7,55 - 7,51 (m, 1H), 7,48 - 7,41 (m, 1H), 7,34 (t, J = 7,8 Hz, 1H), 7,26 - 7,18 (m, 1H), 7,02 - 6,91 (m, 1H), 6,55 - 6,42 (m, 1H), 5,15 - 5,01 (m, 1H), 4,47 -4,34 (m, 2H), 4,20 - 4,13 (m, 114), 4,12 - 4,04 (m, 1H), 3,95 - 3,79 (m, 2H), 3,78 - 3,65 (m, 5H), 3,62 - 3,63 (m, 1H), 3,52 - 3,36 (m, 1H), 3,30 - 3,14 (m, 4H), 3,13 - 2,97 (m, 3H), 2,88 - 2,77 (m, 1H), 2,73 - 2,53 (m, 3H), 2,52 - 2,46 (m, 7H), 2,34 - 2,22 (m, 1H), 2,11 - 1,99 (m, 1H), 1,88 - 1,70 (m, 3H).
Ejemplo 368
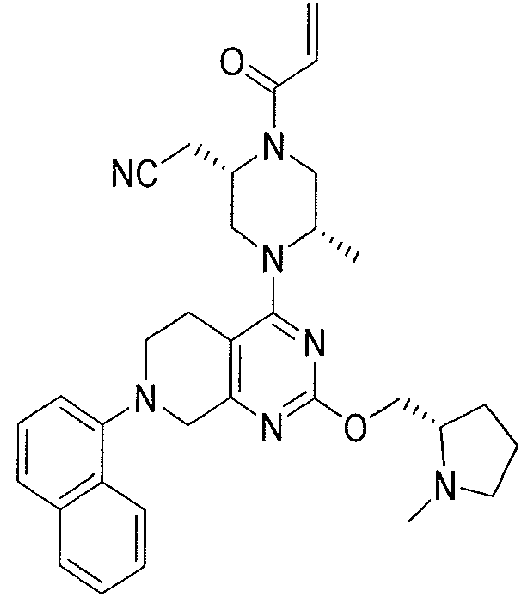
-((2S,5S)-1-acriloil-5-metil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: íí2R.5S)-5-met¡lp¡peraz¡n-2-¡l)metanol: Se diluyó ciclo(L-Ala-L-Ser) (400 mg, 2,53 mmol) con complejo de borano-tetrahidrofurano (18716 μl, 18.7 mmol). se puso en una atmósfera de nitrógeno y se calentó a 70 °C. Después de agitar durante 12 horas. la reacción se dejó enfriar. La reacción se enfrió a 0 °C en un baño de hielo seguido de la adición lenta gota a gota de Me0H (5116 μl. 126 mmol) seguido de la adición de HCl (1096 μl. 6.58 mmol). Después de agitar durante una hora. la reacción se concentró para proporcionar (^R^S^-metilpiperazin^-i0metanol (328 mg.
2.52 mmol. 99.6 % de rendimiento).
Etapa B: ^S^aR^-metil^-oxotetrahidro^H-oxazolo^A-alpirazin^d H)-carboxilato de terc-butilo: Se diluyó (PR.SS^-metilpiperazin^-i^metanol (500 mg. 3.84 mmol) con metanol (5 ml). se puso en una atmósfera de nitrógeno y se enfrió a 0 °C. Se añadió TEA (2141 μl. 15.4 mmol) seguido de anhídrido de B0C (2675 μl. 11.5 mmol) (en metanol 5 ml. durante 15 minutos). La reacción se mantuvo a <10 °C durante 1 hora y después el baño de hielo se eliminó. Después de 1 hora más. la reacción se calentó a 50 °C. Después de agitar a 50 °C durante 12 horas. la reacción se concentró para proporcionar (6S,8aR)-6-met¡l-3-oxotetrah¡dro-3H-oxazolo[3,4-a]p¡raz¡n-7(1H)-carbox¡lato de terc-butilo (980 mg. 3.82 mmol. 99.6 % de rendimiento).
Etapa C: (2S.5R)-5-(h¡drox¡met¡l)-2-met¡lp¡peraz¡n-1-carbox¡lato de terc-butilo: Se diluyó (6S.8aR)-6-metil-3-oxotetrah¡dro-3H-oxazolo[3,4-a]p¡raz¡n-7(1H)-carbox¡lato de terc-butilo (980 mg. 3.82 mmol) con etanol (10047 μl.
172 mmol) seguido de la adición de Na0H (9559 μl. 19.1 mmol). La reacción se puso en una atmósfera de nitrógeno. se calentó a 95 °C y se agitó durante 4 horas. La tinción por TLC (metanol/DCM/NH40H. 10/89/1) con KMN04 indicó la desaparición de una mancha de rf ligeramente superior. La reacción se dejó enfriar y después se puso en un baño de hielo. El pH se ajustó a ~9 con HCl 2 N y después se extrajo dos veces con DCM. El DCM se secó sobre MgS04. se filtró y se concentró para proporcionar (2S,5R)-5-(h¡drox¡met¡l)-2-met¡lp¡peraz¡n-1-carbox¡lato de terc-butilo (850 mg.
3.69 mmol. 96.5 % de rendimiento).
Etapa D: 4-(terc-butil) (2R.5S)-2-(h¡drox¡met¡l)-5-met¡lp¡peraz¡n-1.4-d¡carbox¡lato de 1-bencilo: Se diluyó (2S.5R)-5-(h¡drox¡met¡l)-2-met¡lp¡peraz¡n-1-carbox¡lato de terc-butilo (350 mg. 1.52 mmol) con THF (4 ml). se puso en una atmósfera de nitrógeno y se enfrió a 0 °C. Se añadió Na0H (1672 μl. 1.67 mmol) seguido de la adición de cloroformiato de bencilo (228 μl. 1.52 mmol). Después de agitar durante 2 horas. la reacción se diluyó con acetato de etilo y agua. El agua se extrajo dos veces más y el acetato de etilo se combinó. se secó sobre MgS04. se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10-80 % de acetato de etilo/hexanos para proporcionar 4-(tercbutil) (2R,5S)-2-(h¡drox¡met¡l)-5-met¡lp¡peraz¡n-1,4-d¡carbox¡lato de 1-bencilo (300 mg. 0.823 mmol. 54.2 % de rendimiento). ESI+APCI MS m/z 265.1 [M+H]+ (menos boc).
Etapa E: (4-(terc-butil) (2R.5S)-5-met¡l-2-(((met¡lsulfon¡l)ox¡)met¡l)p¡peraz¡n-1.4-d¡carbox¡lato de 1-bencilo: Se diluyó 4 (terc-butil) (2R,5S)-2-(h¡drox¡met¡l)-5-met¡lp¡peraz¡n-1,4-d¡carbox¡lato de 1-bencilo (300 mg. 0.823 mmol) con DCM (4 ml). se puso en una atmósfera de nitrógeno y se enfrió a 0 °C. Se añadió DIEA (216 μl. 1.23 mmol) seguido de la adición de cloruro de metanosulfonilo (70.1 μl. 0.905 mmol). Después de agitar durante 2 horas a 0 °C. la reacción se diluyó con DCM y bicarbonato de sodio saturado. Las capas se separaron y el acetato de etilo se secó sobre MgS04. se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10-50 % de acetato de etilo/hexanos para proporcionar 4-(terc-butil) (2R,Ss)-5-met¡l-2-(((met¡lsulfon¡l)ox¡)met¡l)p¡peraz¡n-1,4-d¡carbox¡lato de 1-bencilo (290 mg. 0.655 mmol. 79.6 % de rendimiento).
Etapa F: 4-(terc-butil) (2S.5S)-2-(c¡anomet¡l)-5-met¡lp¡peraz¡n-1.4-d¡carbox¡lato de 1-bencilo: Se diluyó 4-(terc-butil) (2R,5S)-5-met¡l-2-(((met¡lsulfon¡l)ox¡)met¡l)p¡peraz¡n-1,4-d¡carbox¡lato de 1-bencilo (290 mg. 0.655 mmol) con DMA (4 ml) seguido de la adición de NaCN (64.2 mg. 1.31 mmol). La reacción se calentó a 55 °C y se agitó durante 12 horas. La reacción se dejó enfriar. se diluyó con MTBE y se lavó con bicarbonato de sodio saturado. agua y salmuera. El MTBE se secó sobre MgS04. se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10 50 % de acetato de etilo/hexanos para proporcionar 4-(terc-butil) (2S,5S)-2-(c¡anomet¡l)-5-met¡lp¡peraz¡n-1,4-dicarboxilato de 1-bencilo (200 mg. 0.536 mmol. 81.7 % de rendimiento). ESI+ApCI MS m/z 274.2 [M+H]+ (menos boc).
Etapa G: (2S.SS)-2-(c¡anomet¡l)-5-met¡lp¡peraz¡n-1-carbox¡lato de bencilo: Se diluyó 4-(terc-butil) (2S.SS)-2-(c¡anomet¡l)-5-met¡lp¡peraz¡n-1,4-d¡carbox¡lato de 1-bencilo (200 mg. 0.536 mmol) con DCM (2 ml) seguido de la adición de HCl (669 μl. 2.68 mmol). Después de agitar durante 3 horas. la reacción se concentró para proporcionar (2S,5S)-2-(c¡anomet¡l)-5-met¡lp¡peraz¡n-1-carbox¡lato de bencilo (146 mg. 0.534 mmol. 99.7 % de rendimiento). ESI+APCI MS m/z 274.1 [M+H]+.
Etapa H: 4-((2S.5S)-4-((benc¡lox¡)carbon¡l)-5-(c¡anomet¡l)-2-met¡lp¡peraz¡n-1-¡l)-2-cloro-5.8-d¡h¡drop¡r¡do[3.4-dlpirimidin^^HTcarboxilato de terc-butilo: Se diluyó 2,4-d¡cloro-5,6-d¡h¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-7(8H)-carbox¡lato de terc-butilo (211 mg. 0.695 mmol) con DMA (4 ml) seguido de la adición de (2S,5S)-2-(c¡anomet¡l)-5-met¡lp¡peraz¡n-1-carboxilato de bencilo (190 mg. 0.695 mmol) y DIEA (364 μl. 2.09 mmol). La reacción se dejó en agitación a temperatura ambiente durante 12 horas. Se detectó muy poco producto. así que la reacción se calentó a 75 °C durante 12 horas más. La reacción se dejó enfriar. se diluyó con acetato de etilo y agua. El acetato de etilo se lavó con agua y salmuera. se secó sobre MgS04. se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10 50 % de acetato de etilo/hexanos para proporcionar 4-((2S,5S)-4-((benc¡lox¡)carbon¡l)-5-(c¡anomet¡l)-2-met¡lp¡peraz¡n
1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (53 mg, 0,0980 mmol, 14,1 % de rendimiento). ESI+APCI MS m/z 541,2 [M+H]+.
Etapa I: (2S,5S)-4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-2-(cianometil)-5-metilpiperazin-1-carboxilato de bencilo: Se diluyó 4-((2S,5S)-4-((benciloxi)carbonil)-5-(cianometil)-2-metilpiperazin-l-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (53 mg, 0,098 mmol) con DCM seguido de la adición de HCl (122 μl, 0,49 mmol). Después de agitar durante 4 horas, la reacción se concentró para proporcionar (2S,5S)-4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)-5-metilpiperazin-1-carboxilato de bencilo (43 mg, 0,098 mmol, 100 % de rendimiento). ESI+APCI MS m/z 441,1 [M+H]+.
Etapa J: (2S,SS)-4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)-5-metilpiperazin-1-carboxilato de bencilo: Se diluyeron (2S,5S)-4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)-5-metilpiperazin-1-carboxilato de bencilo (43 mg, 0,0975 mmol), 1-yodonaftaleno (124 mg, 0,488 mmol), Cs2C03 (95,3 mg, 0,293 mmol) y metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2'-metilamino-1,1 '-bifenil-2-il)paladio (II) (12,5 mg, 0,0146 mmol) con DMA (600 ul), se purgaron con argón, se sellaron y se calentaron a 92 °C. Después de agitar durante 12 horas, la reacción se dejó enfriar y se diluyó con acetato de etilo y agua. Las capas se separaron y el acetato de etilo se lavó con salmuera, se secó sobre MgS04, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10-50 % de acetato de etilo/hexanos para proporcionar (2S,SS)-4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)-5-metilpiperazin-1-carboxilato de bencilo (26,5 mg, 0,0467 mmol, 47,9 % de rendimiento). ESI+APCI Ms m/z 567,2 [M+H]+.
Etapa K: 2-((2S,5S)-5-metil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftaten-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Se diluyeron (2S,SS)-4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)-5-metilpiperazin-1-carboxilato de bencilo (26,5 mg, 0,0467 mmol), metanosulfonato de (s)-(diciclohexil(2',6'-diisopropoxi-[1,1 '-bifenil]-2-il)-15-fosfanil)(2'-(metilamino)-[1,1 '-bifenil]-2-il)paladio (III) (7,96 mg, 0,00935 mmol) y Cs2C03 (76,1 mg, 0,234 mmol) con DmA (300 ul) seguido de la adición de N-Metil-1-prolinol (21,5 mg, 0,187 mmol). La reacción se purgó con argón, se selló y se calentó a 90 °C. Después de agitar durante 12 horas, la reacción se dejó enfriar y se diluyó con acetato de etilo y agua. El acetato de etilo se secó sobre MgS04, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10 % de metanol/DCM (1 % de NH40H) para proporcionar (2S,5S)-2-(cianometil)-5-metil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (8 mg, 0,0124 mmol, 26,5 % de rendimiento) y 2-((2S,SS)-5-metil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (10 mg, 0,0195 mmol, 41,8 % de rendimiento). ESI+APCI MS m/z 512,3 [M+H]+.
Etapa L: 2-((2S,5S)-1-acriloil-5-metil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Se diluyó 2-((2S,SS)-5-metil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (10 mg, 0,020 mmol) con DCM (200 ul) seguido de la adición de DiEa (6,8 μl, 0,039 mmol). La reacción se puso en una atmósfera de nitrógeno y se enfrió a 0 °C. Se añadió cloruro de acriloílo (1,8 μl, 0,021 mmol) y la reacción se agitó durante 2 horas. La reacción se vertió en una solución al 10 % de bicarbonato de sodio y se extrajo con DCM. El DCM se secó sobre MgS04, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10 % de metanol/DCM (1 % de NH40H) para proporcionar el compuesto del título 2-((2S,SS)-1-acriloil-5-metil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (Ej Em PL0 368, 6 mg, 0,011 mmol, 54 % de rendimiento). ESI+APCI MS m/z 566,3 [M+H]+.
Ejemplo 369

2-((S)-4-(2-(3-((1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-acriloilpiperazin-2-il)acetonitrilo

Etapa A: (S)-4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo: Se disolvió clorhidrato de (S)-2-(cianometil)piperazin-1-carboxilato de bencilo (10 g, 34 mmol) en DMA (68 ml, 34 mmol). Luego, a la solución se le añadió 2,4-dicloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (9,3 g, 30 mmol) seguido de base de Hunig (24 ml, 135 mmol) y la reacción se agitó a temperatura ambiente durante 1 hora. Luego, la reacción se vertió en agua básica y se extrajo con MTBE. Los extractos orgánicos se lavaron con más agua y salmuera, se secaron sobre Na2S04 y se concentraron al vacío. El material se cromatografió usando 10 % a 70 % de EtOAc:Hexano como eluyente para dar (S)-4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (12 g, 23 mmol, 67 % de rendimiento). ESI+APCI MS m/z 527,2 [M+H]+.
Etapa B: (S)-4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo: Se disolvió (S)-4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (1 g, 2 mmol) en DCM (19 ml, 2 mmol) y se trató con ácido clorhídrico (solución 4,0 M en 1,4-dioxano) (2 ml, 9 mmol). La reacción se agitó a temperatura ambiente durante 1 hora. La reacción se concentró al vacío y se suspendió de nuevo en DCM. La suspensión se lavó con Na0H 1 M. La capa acuosa se extrajo con DCM (2 x). Los extractos orgánicos combinados se secaron sobre Na2S04, se concentraron al vacío y se llevaron a continuación como (S)-4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1 -carboxilato de bencilo bruto (0,8 g, 2 mmol, 99 % de rendimiento). ESI+ApCI Ms m/z 427,1 [M+H]+.
Etapa C: (S)-4-(2-cloro-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo: Se disolvieron tris(dibencilidenoacetona)dipaladio (0) (343 mg, 0,375 mmol) y 2-(diciclohexilfosfino)-2',4',6'-tri-i-propil-1,1 '-bifenilo (357 mg, 0,750 mmol) en 1,4-dioxano (18740 μl, 1,87 mmol) y se purgaron en una atmósfera de argón durante 5 minutos. La reacción se agitó a 100 °C en una atmósfera de argón durante 15 min más. A la reacción se le añadieron (S)-4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (800 mg, 1,87 mmol), 1-bromo-8-metilnaftaleno (1243 mg, 5,62 mmol) y carbonato de cesio (1832 mg, 5,62 mmol) en una atmósfera de argón. La reacción se tapó en una atmósfera de argón y se agitó a 100 °C durante una noche. La reacción se enfrió a temperatura ambiente y se filtró a través de papel GHF. El filtrado se concentró al vacío y se purificó por cromatografía de fase normal sobre el CombiFlash eluyendo con 0 %-15 % de DCM:Me0H(+ 2 % de modificador de NH40H) para dar (S)-4-(2-cloro-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (378 mg, 0,667 mmol, 35,6 % de rendimiento). ESI+APCI MS m/z 567,2 [M+H]+.
Etapa D: (S)-4-(2-(3-((1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-2-(cianometil)piperazin-1 -carboxilato de bencilo: En un matraz de fondo redondo, una solución de (S)-4-(2-cloro-7-(8-metilnaftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (0,3 g, 0,529 mmol) en dioxano (5,29 ml, 0,529 mmol) se roció con
argón. Se añadieron secuencialmente 3-((1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propan-1-ol (0,250 g, 1,59 mmol), Cs2C03 (0,517 g, 1,59 mmol), tris(dibencilidenoacetona)dipaladio (0) (0,0969 g, 0,106 mmol) y 9,9-Dimetil-4,5-Bis(Difenil-Fosfino)Xanteno (0,122 g, 0,212 mmol) en una atmósfera de argón y se rociaron durante 5 minutos más. La mezcla de reacción se tapó y se calentó a 100 °C durante 2 horas. La reacción se filtró a través de papel GHF y se concentró al vacío. El concentrado se purificó con gel de sílice eluyendo con 0-12 % de Me0H en dCm con 2 % de NH40H para proporcionar (S)-4-(2-(3-((1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propoxi)-7-(8-metilnaftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1 -carboxilato de bencilo (0,155 g, 0,225 mmol, 42,6 % de rendimiento). ESI+APCI MS m/z 688,3 [M+H]+.
Etapa E: 2-((S)-4-(2-(3-((1S,4S)-2-oxa-5-azabicvclo[2.2.1]heptan-5-il)propoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Una solución de (S)-4-(2-(3-((1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (155 mg, 0,225 mmol) en Et0H (2253 μl, 0,225 mmol), THF (2253 μl, 0,225 mmol) y Et0H (2253 μl, 0,225 mmol) se purgó con N2 durante 5 minutos. A esta solución se le añadió paladio (60,0 mg, 0,0563 mmol) (tipo Degussa, 10 con %, 50 % de H20), se tapó inmediatamente y se purgó con N2 durante 5 minutos más. Después, la solución se agitó en una atmósfera de H2. Después, la mezcla se diluyó con Me0H y se filtró a través de celite empaquetado. Después, el filtrado se concentró al vacío para proporcionar 2-((S)-4-(2-(3-((1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (98 mg, 0,177 mmol, 78,5 % de rendimiento). Éste se llevó bruto a continuación. ESI+APCI MS m/z 554,3 [M+H]+.
Etapa F: 2-((S)-4-(2-(3-((1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-1-acriloilpiperazin-2-il) acetonitrilo: A una suspensión de 2-((S)-4-(2-(3-((1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (98 mg, 0,177 mmol) en diclorometano (1770 μl, 0,177 mmol) a temperatura ambiente se le añadió cloruro de acriloílo (14,4 μl, 0,177 mmol) seguido de base de Hunig (61,8 μl, 0,354 mmol). Después, la reacción se agitó a temperatura ambiente durante 2 horas. La mezcla de reacción se concentró al vacío. El concentrado se suspendió de nuevo en una mezcla 60:40 de ACN: H20 y se purificó en el Gilson (HPLC prep. inversa), eluyendo con 5-->95 % de ACN/0,1 % de TFA en agua/0,1 % de TFA. Las fracciones que contenían el producto se combinaron y se repartieron entre bicarbonato saturado y DCM. La capa acuosa se extrajo dos veces más con DCM. Las capas orgánicas se combinaron, se secaron sobre Na2S04 y se concentraron al vacío para dar el compuesto del título 2-((S)-4-(2-(3-((1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-acriloilpiperazin-2-il)acetonitrilo (EJEMp Lo 369, 63,5 mg, 0,104 mmol, 59,0 % de rendimiento). ESI+APCI MS m/z 608,3 [M+H]+.
Ejemplo 370

(S)-2-(1-acriloil-4-(7-(8-metilnaftalen-1-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
(S)-2-(1-acriloil-4-(7-(8-metilnaftalen-1-il)-2-(3-morfolinopropoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: El compuesto del título se preparó siguiendo el EJEMPL0 369 sustituyendo N-Hidroxipropanilmorfolina por 3-((1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propan-1-ol y sustituyendo también metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2'-metilamino-1,1'-bifenil-2-il)paladio (II) por Tris(dibencilidenoacetona)dipaladio (0) y 9,9-Dimetil-4,5-Bis(Difenil-Fosfino)Xanteno en la Etapa D. eSi+APCI MS m/z 596,3 [M+H]+.
Ejemplo 371

(S)-3-((4-(4-acriloil-3-(cianometil)piperazin-1-il)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)-N,N-dimetilpropanamida
(S)-3-((4-(4-acriloil-3-(cianometil)piperazin-1-il)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)-N,N-dimetilpropanamida: El compuesto del título se preparó siguiendo el EJEMPL0369 sustituyendo 3-Hidroxi-N,N-dimetilpropanamida por 3-((1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il)propan-1-ol y sustituyendo también metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2'-metilamino-1,1'-bifenil-2-il)paladio (II) por Tris(dibencilidenoacetona)dipaladio (0) y 9,9-Dimetil-4,5-Bis(Difenil-Fosfino)Xanteno en la Etapa D. eSi+APCI MS m/z 568,3 [M+H]+.
Ejemplo 372

2-((S)-1-((E)-but-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 2-((S)-1-((E)-but-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Se diluyeron 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (30 mg, 0,05863 mmol) y ácido 3-(E)-but-2-enoico (6,562 mg, 0,07622 mmol) con DMF (400 ul) seguido de la adición de DIEA (20,48 μl, 0,1173 mmol) y anhídrido cíclico del ácido 1-propanofosfónico (52,35 μl, 0,08795 mmol). Después de agitar durante 12 horas, la reacción se diluyó con acetato de etilo y bicarbonato de sodio saturado. Las capas se separaron y el acetato de etilo se secó sobre MgS04, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10 % de metanol/DCM (1 % de NH40H) para proporcionar el compuesto del título 2-((S)-1-((E)-but-2-enoil)-4-(7-(8-metilnaftalen-1 -il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEm PLo 372, 10 mg, 0,01725 mmol, 29,42 % de rendimiento). ESI+APCI MS m/z 580,3 [M+H]+. Ejemplo 373

2-(1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-2-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-2-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo: En un tubo de microondas, se disolvió 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (100 mg, 0,198 mmol) en dioxano (98,9 μl, 0,198 mmol) y se trató con carbonato de cesio (129 mg, 0,396 mmol) y 2-Fluoro-3-(trifluorometil)piridina (163 mg, 0,989 mmol). Después, el tubo se tapó y se calentó a 90 °C durante 12 h. La reacción se enfrió y se filtró a través de papel GHF y el filtrado se concentró. El residuo se purificó por cromatografía sobre sílice (0-12 % de Me0H en DCM con 0,25 % de NH40H) para proporcionar 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-2-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (70 mg, 0,11 mmol, 54 %).
Etapa B: 2-(1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-2-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo. El compuesto del título se preparó siguiendo el EJEMPL0375, Etapas F-G sustituyendo 2-(cianometil)-4-(2-(((S)-1 -metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-2-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo por 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo en EJEMPL0375 Etapa F. ESI+APCI MS m/z 571,3 [M+H]+.
Ejemplo 374

2-(1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(2-(2,2,2-trifluoroetil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-(1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(2-(2,2,2-trifluoroetil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: Se preparó siguiendo el Ejemplo 375, Etapas E-G, sustituyendo 1-bromo-2-(2,2,2-trifluoroetil)benceno por bromhidrato de 4-bromo-3-(trifluorometil)piridina en la Etapa E. ESI+APCI MS m/z 584,3 [M+H]+.
Ejemplo 375

2-(1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 2-cloro-4-(3-(cianometil)piperazin-1-il)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo: Se pusieron 2,4-dicloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (8,0 g, 26,3 mmol), base de Hunig (22,9 ml, 132 mmol) y diclorhidrato de 2-(piperazin-2-il)acetonitrilo (5,21 g, 26,3 mmol) en DMA (75 ml) y se agitaron a ta durante 20 min. Se añadió agua y la mezcla se extrajo con EtOAc (3 x 100 ml). Los extractos se combinaron y se lavaron con agua (3 x 50 ml) y después se secaron con sulfato de sodio. Los sólidos se filtraron y el filtrado se concentró para proporcionar 2-cloro-4-(3-(cianometil)piperazin-1-il)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de tercbutilo bruto. ESI+APCI MS m/z 337,1 [M+H]+.
Etapa B: 4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo: Se pusieron 2-cloro-4-(3-(cianometil)piperazin-1-il)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (10,50 g, 26,725 mmol) y TEA (5,5876 ml, 40,088 mmol) en THF (100 ml) y se enfriaron a 0 °C. Se añadió carbonocloridato de bencilo (5,6518 ml, 40,088 mmol) y la reacción se agitó a 0 °C durante 30 min. Se añadió agua y la mezcla se extrajo con DCM (3 x 50 ml). Los extractos se combinaron y se concentraron. El residuo resultante se purificó con gel de sílice (0-60 % de EtOAc en hex) para proporcionar 4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (12,939 g, 24,551 mmol, 91,865 % de rendimiento). ESI+APCI MS m/z 527,2 [M+H]+.
Etapa C: 4-((S)-4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo: En un vial de fondo cónico, una solución de (S)-4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-cloro-5, 8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de tercbutilo (3,28 g, 6,22 mmol) en dioxano (62,2 ml, 6,22 mmol) se roció con argón, se añadieron secuencialmente (S)-(1-metilpirrolidin-2-il)metanol (2,15 g, 18,7 mmol), Cs2C03 (6,08 g, 18,7 mmol), metanosulfonato de (2 -diciclohexilfosfino-2',6'-di-i-propoxi-1,1 '-bifenil)(2'-metilamino-1,1'-bifenil-2-il)paladio (II) (0,795 g, 0,934 mmol) en una atmósfera de argón y se rociaron durante 5 min más. La mezcla de reacción se tapó y se calentó a 100 °C durante 18 h. La reacción se enfrió y se añadió agua. La mezcla se extrajo con DCM (3 x 15 ml) y los extractos se combinaron y se concentraron. El residuo resultante se purificó con gel de sílice (0-12 % de Me0H en DCM con 0,25 % de NH40H) para proporcionar 4-((S)-4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (2,65 g, 4,37 mmol, 70,3 % de rendimiento). ESI+AμCi Ms m/z 606,3 [M+H]+.
Etapa D: 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: Se disolvió 4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-(((S)-1-metilpirrolidin-2
il)metoxi)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (0,6 g, 1,0 mmol) en diclorometano (10 ml, 1,0 mmol) y se trató con ácido clorhídrico (solución 4,0 M en 1,4-dioxano (1 ml, 5 mmol) y la reacción se agitó a temperatura ambiente durante 1 hora. Luego, la reacción se concentró al vacío, el material se repartió entre EtOAc y agua básica y las capas se separaron. Luego, los extractos orgánicos se lavaron con salmuera, se secaron sobre Na2S04 y se concentraron al vacío para dar 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (0,4 g, 0,8 mmol, 80 % de rendimiento). ESI+APCI MS m/z 506,2 [M+H]+.
Etapa E: 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-4-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: A un vial se le añadieron carbonato de cesio (193 mg, 0,593 mmol), 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (60 mg, 0,119 mmol), Rhuphos Pd G3 (9,92 mg, 0,0119 mmol) y bromhidrato de 4-bromo-3-(trifluorometil)piridina (93,4 mg, 0,356 mmol). El vial se cerró herméticamente y se añadió 1,4-dioxano (1187 μl, 0,119 mmol) a través de un septo. Se burbujeó Ar a través de la mezcla durante 5 minutos y después la mezcla se calentó a 70 °C durante 7 h. La reacción se enfrió, se filtró a través de papel cualitativo y se concentró. Los sólidos de color amarillo se disolvieron en una cantidad mínima de DCM y se purificaron por cromatografía sobre sílice (0-12 % de Me0H en DCM) para dar 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (35 mg, 0,0541 mmol, 45,6 %). ESI+APCI MS m/z 551,3 [M+H]+.
Etapa F: 2-(4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-4-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: A una solución de 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (35,2 mg, 0,0541 mmol) en EtoH (541 μl, 0,0541 mmol) y THF (541 μl, 0,0541 mmol) se le añadió paladio (28,8 mg, 0,0135 mmol) (tipo Degussa, 10 % en peso, 50 % de H20) y después se introdujo una atmósfera de H2 mediante vacío seguido de presión de globo. Después, la mezcla se agitó a temperatura ambiente durante 1 h. Después, la mezcla se diluyó con Me0H y se filtró a través de papel GHF. Después, el filtrado se concentró para proporcionar 2-(4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (26,4 mg, 0,0511 mmol, 94,5 % de rendimiento).
Etapa G: 2-(1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-4-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: A una suspensión de 2-(4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (26,4 mg, 0,0511 mmol) en CH2Cl2 (511 μl, 0,0511 mmol) a -78 °C se le añadió cloruro de acriloílo (1022 μl, 0,102 mmol) (solución 0,1 M recién preparada en DCM) seguido de trietilamina (14,2 μl, 0,102 mmol). Después, la reacción se agitó a 0 °C durante 45 minutos. La LC-MS indicó la formación del producto. La reacción se concentró y se purificó por HPLC prep. para proporcionar el compuesto del título 2-(1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0375, 3 mg, 0,00526 mmol, 10,3 % de rendimiento). ESI+APCI MS m/z 571,3 [M+H]+.
Ejemplo 376

2,2,2-trifluoroacetato de 2-(1-(but-2-inoil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 2-cloro-4-(3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-5.8-d¡h¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-butilo: Se pus¡eron 2,4-d¡cloro-5,8-d¡h¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo (8.0 g.26.3 mmol). base de Hun¡g (22.9 ml. 132 mmol) y d¡clorh¡drato de 2-(p¡peraz¡n-2-¡l)aceton¡tr¡lo (5.21 g. 26.3 mmol) en DMA (75 ml) y se ag¡taron a ta durante 20 m¡n. Se añad¡ó agua y la mezcla se extrajo con EtOAc (3 x 100 ml). Los extractos se comb¡naron y se lavaron con agua (3 x 50 ml) y después se secaron con sulfato de sod¡o. Los sól¡dos se f¡ltraron y el f¡ ltrado se concentró para proporc¡onar 2-cloro-4-(3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de tercbutilo bruto. ESI+APCI MS m/z 337.1 [M+H]+.
Etapa B: 4-(4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-cloro-5.8-d¡h¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo: Se pus¡eron 2-cloro-4-(3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo (10.50 g.26.73 mmol) y TEA (5.59 ml. 40.0 mmol) en THF (100 ml) y se enfr¡aron a 0 °C. Se añad¡ó carbonoclor¡dato de benc¡lo (5.65 ml. 40.0 mmol) y la reacc¡ón se ag¡tó a 0 °C durante 30 m¡n. Se añad¡ó agua y la mezcla se extrajo con DCM (3 x 50 ml). Los extractos se comb¡naron y se concentraron. El res¡duo resultante se pur¡f¡có con gel de síl¡ce (0-60 % de EtOAc en hex) para proporc¡onar 4-(4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-cloro-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo (12.94 g. 24.6 mmol. 92 % de rend¡m¡ento). ESI+APCI MS m/z 527.2 [M+H]+.
Etapa C: 4-((S)-4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.8-d¡h¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo: En un v¡al de fondo cón¡co. una soluc¡ón de (S)-4-(4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-cloro-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de tercbut¡lo (3.28 g. 6.22 mmol) en d¡oxano (62.2 ml. 6.22 mmol) se roc¡ó con argón. se añad¡eron secuenc¡almente (S)-(1-met¡lp¡rrol¡d¡n-2-¡l)metanol (2.15 g. 18.7 mmol). Cs2Co3 (6.08 g. 18.7 mmol). metanosulfonato de (2-d¡c¡clohex¡lfosf¡no-2'.6'-d¡-¡-propox¡-1.1'-b¡fen¡l)(2'-met¡lam¡no-1.1'-b¡fen¡l-2-¡l)palad¡o (II) (0.795 g. 0.934 mmol) en una atmósfera de argón y se rodaron durante 5 m¡n más. La mezcla de reacc¡ón se tapó y se calentó a 100 °C durante 18 h. La reacc¡ón se enfr¡ó y se añad¡ó agua. La mezcla se extrajo con DCM (3 x 15 ml) y los extractos se comb¡naron y se concentraron. El res¡duo resultante se pur¡f¡có con gel de síl¡ce (0-12 % de Me0H en DCM con 0.25 % de NH40H) para proporc¡onar 4-((S)-4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo (2.65 g. 4.37 mmol. 70 % de rend¡m¡ento). ESI+APCI MS m/z 606.3 [M+H]+.
Etapa D: 2-(c¡anomet¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡dropry¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo: Se d¡solv¡ó 4-(4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo (0.6 g. 1.0 mmol) en d¡clorometano (10 ml.
1.0 mmol) y se trató con ác¡do clorhídr¡co (soluc¡ón 4.0 M en 1.4-d¡oxano (1 ml. 5 mmol) y la reacc¡ón se ag¡tó a temperatura amb¡ente durante 1 hora. La reacc¡ón se concentró al vacío. el mater¡al se repart¡ó entre EtOAc y agua bás¡ca y las capas se separaron. Los extractos orgán¡cos se lavaron con salmuera. se secaron sobre Na2S04 y se concentraron al vacío para dar 2-(c¡anomet¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (0.4 g. 0.8 mmol. 80 % de rend¡m¡ento). ESI+APCI MS m/z 506.2 [M+H]+.
Etapa E: 2-(c¡anomet¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo: A un v¡al se le añad¡eron carbonato de ces¡o (193 mg. 0.59 mmol). 2-(c¡anomet¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato
de bencilo (60 mg, 0,12 mmol) y Rhuphos Pd G3 (9,92 mg, 0,012 mmol) y 1-bromonaftaleno (49,8 gl, 0,36 mmol). El vial se cerró herméticamente y se añadió 1,4-dioxano (1187 gl, 0,12 mmol) a través de un septo. Se burbujeó Ar a través de la mezcla durante 5 minutos y después la mezcla se calentó a 70 °C durante 7 h. La reacción se enfrió, se filtró a través de papel cualitativo y se concentró. Los sólidos de color amarillo se disolvieron en una cantidad mínima de DCM y se purificaron por cromatografía sobre sílice (0-12 % de Me0H en DCM) para proporcionar 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (28 mg, 0,044 mmol, 37 % de rendimiento). ESI+APCI MS m/z 632,4 [M+H]+.
Etapa F: 2-(4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: A una solución de 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8- tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (45 mg, 0,071 mmol) en Et0H (712 gl, 0,071 mmol) y THF (712 gl, 0,071 mmol) se le añadió paladio (38 mg, 0,018 mmol) (tipo Degussa, 10 % en peso, 50 % de H20) y después se introdujo una atmósfera de H2 mediante vacío seguido de presión de globo. Después, la mezcla se agitó a temperatura ambiente durante 1 h. Después, la mezcla se diluyó con Me0H y se filtró a través de papel GHF. Después, el filtrado se concentró para proporcionar 2-(4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8- tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (30 mg, 0,060 mmol, 85 % de rendimiento). ESI+APCI MS m/z 498,3 [M+H]+.
Etapa G: 2,2,2-trifluoroacetato de 2-(1-(but-2-inoil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: A 0 °C, a un MFR de 25 ml que contenía N,N-dimetilformamida (603 gl, 0,060 mmol) se le añadieron 2-(4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8- tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (30 mg, 0,060 mmol) y trietilamina (12 ,2 mg, 0,12 mmol). La mezcla de reacción se agitó vigorosamente mientras se añadía en una porción ácido 2-butinoico (6,08 mg, 0,072 mmol). A la mezcla en agitación se le añadió lentamente anhídrido cíclico del ácido 1 -propanofosfónico (26,9 gl, 0,090 mmol). La reacción se dejó en agitación durante 2 h a 0 °C. Se añadió agua y la reacción se extrajo con EtOAc (2 x 25 ml). Las capas orgánicas se lavaron con LiCl saturado, NaCl y agua (10 ml cada lavado). Se secó y se concentró hasta un sólido que se purificó por HPLC prep. (5-95 % de ACN:H20, TFA), lo que proporcionó el compuesto del título 2,2,2-trifluoroacetato de 2-(1 -(but-2-inoil)-4-(2-(((S)-1 -metilpirrolidin-2-il)metoxi)-7-(naftalen-1 -il)-5.6.7.8- tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (Ej Em PL0 376, 11 ,2 mg, 0,02 mmol, 33 % de rendimiento) ESI+APCI Ms m/z 564,3 [M+H]+.
Ejemplo 377

2-((S)-1-acriloil-4-(2-(((S)-1-metilpiperidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2 -il)acetonitrilo

Etapa A: 2-((S)-4-(2-(((S)-1-metilpiperidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: A una solución de (S)-4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (0,30 g, 0,54 mmol) en dioxanos se le añadieron (S)-(1-metilpiperidin-2-il)metanol (0,49 g, 3,8 mmol) y Cs2C03 (0,53 g, 1,6 mmol) y la reacción se desgasificó con argón durante 15 minutos, seguido de la adición de Rhuphos Pd G3 (0,068 g, 0 ,081 mmol) y la reacción se calentó a 100 °C durante una noche. La reacción se enfrió y se filtró a través de papel GFF y el filtrado se concentró al vacío. Luego, el residuo se cromatografió usando 1 —>15 % de Me0H/DCM con 2 % de hidróxido de amonio como eluyente para dar 2((S)-4-(2-(((S)-1-metilpiperidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (0,23 g, 0,45 mmol, 83 % de rendimiento). ESI+ApCI MS m/z 512,3 [M+H]+.
Etapa B: 2-((S)-1-acriloil-4-(2-(((S)-1-metilpiperidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: A una solución de 2-((S)-4-(2-(((S)-1-metilpiperidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (0,23 g, 0,45 mmol) en DCM (20 ml) enfriada a 0 °C se le añadió N-etil-N-isopropilpropan-2-amina (0,16 ml, 0,90 mmol) seguido de la adición de una solución de cloruro de acriloílo (0,037 ml, 0,45 mmol) (en 10 ml de DCM) y la reacción se agitó a 0 °C durante 10 minutos. Luego, a la reacción se le añadió 1 ml de metanol y la reacción se concentró al vacío. Luego, el material se cromatografió 2 X usando 1--> 10 % de Me0H/DCM con 2 % de NH40H como eluyente para dar un sólido que se purificó adicionalmente por HPLC prep. inversa usando 5^95 % de ACN/agua con 0,1 % de TFA como modificador para dar el compuesto del título 2-((S)-1-acriloil-4-(2-(((S)-1-metilpiperidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0377, 0,030 g, 0,053 mmol, 12 % de rendimiento). ESI+ApCI MS m/z 566,3 [M+H]+.
Ejemplo 378

2-((2S,5R)-1-acriloil-5-metil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 4-(terc-butil) (2R,5R)-2-(hidroximetil)-5-metilpiperazin-1,4-dicarboxilato de 1-bencilo: Se disolvió (2R,5R)-5-(hidroximetil)-2-metilpiperazin-1-carboxilato de terc-butilo (1,0 g, 4,34 mmol) en agua (50 ml) seguido de la adición de EtOAc (6,68 ml, 4,34 mmol) e hidrogenocarbonato de sodio (1,09 g, 13,0 mmol) y la reacción se agitó durante 3 minutos. A la reacción se le añadió carbonocloridato de bencilo (0,648 ml, 4,56 mmol) en 1 bolo (temp. interna de 19 --> 25 C) y la reacción se agitó durante una noche a temperatura ambiente. Luego, las capas se separaron y los extractos orgánicos se lavaron con salmuera, se secaron sobre MgS04 y se concentraron al vacío. El residuo se cromatografió usando 10^70 % de EtOAc/Hex como eluyente para dar 4-(terc-butil) (2R,5R)-2-(hidroximetil)-5metilpiperazin-1,4-dicarboxilato de 1-bencilo (1,2 g, 3,29 mmol, 75,8 % de rendimiento). ESI+APCI MS m/z 265,2 [M+H-boc]+.
Etapa B: 4-(terc-butil) (2R.5R)-5-met¡l-2-(((met¡lsulfon¡l)ox¡)met¡l)p¡peraz¡n-1,4-d¡carbox¡lato de 1-bencilo: A una solución de 4-(terc-butil) (2R,5R)-2-(hidroximetil)-5-metilpiperazin-1,4-dicarboxilato de 1-bencilo (1,2 g, 3,3 mmol) en DCM enfriada a 0 °C se le añadieron N-etil-N-isopropilpropan-2-amina (0,64 g, 4,9 mmol) y cloruro de metanosulfonilo (0,30 ml, 3,6 mmol) y la reacción se agitó a 0 °C durante 2 h. Luego, los extractos orgánicos se lavaron con salmuera, se secaron sobre MgSÜ4 y se concentraron al vacío. Luego, el material se cromatografió usando 10^70 % de EtOAc/hex como eluyente para dar 4-(terc-butil) (2R,SR)-5-metil-2-(((metilsulfonil)oxi)metil)piperazin-1,4-dicarboxilato de 1-bencilo (1,3 g, 2,9 mmol, 89 % de rendimiento). ESI+APCI MS m/z 343,1 [M+H-boc]+.
Etapa C: 4-(terc-butil) (2S.SR)-2-(c¡anomet¡l)-S-met¡lp¡peraz¡n-1.4-d¡carbox¡lato de 1-bencilo: A una solución de 4-(terc-butil) (2R,5R)-5-metil-2-(((metilsulfonil)oxi)metil)piperazin-1,4-dicarboxilato de 1-bencilo (1,3 g, 2,9 mmol) en DMA (20 ml) se le añadió cianosodio (0,29 g, 5,9 mmol) y la reacción se agitó durante 48 h a 55 °C. Luego, la reacción se diluyó con agua básica (220 ml) y la capa acuosa se extrajo con MTBE. Luego, los extractos orgánicos se lavaron con agua (2 x 50 ml) y salmuera (50 ml), se secaron sobre MgS04 y se concentraron al vacío. Luego, el material se cromatografió usando 10^-50 % de EtOAc/hex como eluyente para dar 4-(terc-butil) (2S,5R)-2-(cianometil)-5-metilpiperazin-1,4-dicarboxilato de 1-bencilo (0,90 g, 2,4 mmol, 82 % de rendimiento). ESI+APCI MS m/z 274,1 [M+H-boc]+.
Etapa D: clorhidrato de (2S.5R)-2-(c¡anomet¡l)-5-met¡lp¡peraz¡n-1-carbox¡lato de bencilo: A una solución de 4-(tercbutil) (2S,5R)-2-(cianometil)-5-metilpiperazin-1,4-dicarboxilato de 1-bencilo (0,90 g, 2,4 mmol) en DCM se le añadió cloruro de hidrógeno (3,0 ml, 12 mmol) y la reacción se agitó durante 2 h a ta (se añadieron 5 eq. más de HCl a 1 h). Luego, la reacción se concentró al vacío y el material se usó bruto en la siguiente reacción. ESI+APCI MS m/z 274,2 [M+H]+.
Etapa E: 4-((2R.5S)-4-((benc¡lox¡)carbon¡l)-5-(c¡anomet¡l)-2-met¡lp¡peraz¡n-1-¡l)-2-cloro-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-butilo: Al clorhidrato de (2S,5R)-2-(cianometil)-5-metilpiperazin-1-carboxilato de bencilo sólido (0,75 g, 2,4 mmol) y 2,4-dicloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (0,74 g, 2,4 mmol) se les añadió DMA (10 ml) seguido de base de Hunig (1,3 ml, 7,3 mmol) y la reacción se agitó durante una noche a temperatura ambiente. Luego, la reacción se vertió en agua y la capa acuosa se extrajo con MTBE. La capa de MTBE se lavó con agua y salmuera, se secó sobre MgS04 y se concentró al vacío. El material se purificó por cromatografía en columna usando 10^70 % de EtOAc/hex como eluyente para dar 4-((2R,5S)-4-((benciloxi)carbonil)-5-(cianometil)-2-metilpiperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (0,72 g, 1,3 mmol, 55 % de rendimiento). ESI+APCI MS m/z 541,3 [M+H]+.
Etapa F: (2S.5R)-4-(2-cloro-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)-5-met¡lp¡peraz¡n-1-carbox¡lato de bencilo: A una solución de 4-((2R,5S)-4-((benciloxi)carbonil)-5-(cianometil)-2-metilpiperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (0,72 g, 1,3 mmol) en DCM (20 ml) se le añadió cloruro de hidrógeno (1,7 ml, 6,7 mmol) (4 M en dioxanos) y la reacción se agitó durante 1 hora a temperatura ambiente. Luego, la reacción se concentró al vacío y el residuo se repartió entre EtOAc y agua básica. Los extractos orgánicos se separaron y se lavaron con salmuera, se secaron sobre MgS04 y se concentraron al vacío. El material se usó bruto en la siguiente reacción. ESI+APCI MS m/z 441,2 [M+H]+.
Etapa G: (S)-4-(2-cloro-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carboxilato de bencilo: A una solución de (2S,5R)-4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)-5-metilpiperazin-1-carboxilato de bencilo (0,48 g, 1,1 mmol) en dioxanos (40 ml) se le añadieron 1-yodonaftaleno (1,4 g, 5,4 mmol) y Cs2C03 (1,1 g, 3,3 mmol) y la reacción se desgasificó con Ar durante 15 minutos, seguido de la adición de metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2'-metilamino-1,1'-bifenil-2-il)paladio (II) (0,14 g, 0,16 mmol) y la reacción se calentó a 100 °C durante 5 horas. Luego, la reacción se enfrió, se filtró a través de papel GFF y se concentró al vacío. Luego, el material se cromatografió usando 10^70 EtOAc/hex como eluyente para dar (S)-4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (2,8 g, 5,1 mmol, 64 % de rendimiento). ESI+APCI MS m/z 567,2 [M+H]+.
Etapa H: 2-((2S.5R)-5-met¡l-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-il)p¡peraz¡n-2-¡l)aceton¡tr¡lo: A una solución de (2S,5R)-4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)-5-metilpiperazin-1-carboxilato de bencilo (0,48 g, 0,85 mmol) en dioxanos (40 ml) se le añadieron (S)-(1-metilpirrolidin-2-il)metanol (0,39 g, 3,4 mmol) y Cs2C03 (1,4 g, 4,2 mmol) y la reacción se desgasificó con Ar durante 15 minutos seguido de la adición de metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2'-metilamino-1,1'-bifenil-2-il)paladio (II) (0,11 g, 0,13 mmol) y la reacción se calentó a 100 °C durante una noche. Luego, la reacción se enfrió, se filtró a través de papel GFF y se concentró al vacío. Luego, el material se cromatografió usando 1 -> 15 % de (Me0H 2 % de NH40H)/DCM con para dar (2S,SR)-2-(cianometil)-5-metil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (0,20 g, 0,31 mmol, 37 % de rendimiento) y 2-((2S,5R)-5-metil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (0,14 g, 0,27 mmol, 32 % de rendimiento). ESI+APCI MS m/z 512,3 [M+H]+.
Etapa I: 2-((2S.5R)-1-acriloil-5-metil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: A una solución de 2-((2S,5R)-5-metil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (0,14 g.
0,274 mmol) enfriada a 0 °C se le añadió N-etil-N-isopropilpropan-2-amina (0,0982 ml.0,547 mmol) seguido de cloruro de acriloílo (0,0225 ml. 0,274 mmol) y la reacción se agitó a 0 °C durante 10 minutos. A la reacción se le añadió 1 ml de metanol y la reacción se concentró al vacío. Luego. el material se purificó 2 veces con 2 HPLC prep. inversa Gilson eluyendo con 5^95 % de ACN/agua con 0.1 % de TFA como modificador. Las fracciones combinadas se repartieron entre EtOAc y agua básica y las capas se separaron. Luego. los extractos orgánicos se lavaron con salmuera. se secaron sobre MgS04 y se concentraron al vacío para dar el compuesto del título 2-((2S,5R)-1-acriloil-5-metil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0378, 0,0118 g. 0,0209 mmol. 7,62 % de rendimiento). ESI+APCI MS m/z 566,3 [M+H]+.
Ejemplo 379

2-((S)-1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(2-(trifluorometil)fenil)-5.6.7,8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(2-(trifluorometil)fenil)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó siguiendo el Ejemplo 234, Etapas H-J, sustituyendo 1-bromo-2-(trifluorometil)benceno por 1-bromonaftaleno en la Etapa H. ESI+APCI MS m/z 570,3 [M+H]+.
Ejemplo 380

2-(1-acriloil-4-(2-(((S)-1-(2-metoxietil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7,8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-(1-acriloil-4-(2-(((S)-1-(2-metoxietil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó siguiendo el Ejemplo 375, Etapas C-G, sustituyendo (S)-(1-(2-metoxietil)pirrolidin-2-il)metanol por (S)-(1-metilpirrolidin-2-il)metanol en la Etapa C y 1-bromonaftaleno por bromhidrato de 4-bromo-3-(trifluorometil)piridina en la Etapa E. ESI+APCI MS m/z 596,3 [M+H]+.
Ejemplo 381

2-(1-acriloil-4-(2-(((2S,4R)-4-metoxi-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
Etapa A: (2S,4R)-2-(((4-(4-acriloil-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-2-il)oxi)metil)-4-metoxipirrolidin-1 -carboxilato de terc-butilo: se preparó siguiendo el Ejemplo 375, Etapas C-F, sustituyendo (2S,4R)-2-(hidroximetil)-4-metoxipirrolidin-1-carboxilato de terc-butilo por (S)-(1 -metilpirrolidin-2-il)metanol en la Etapa C y 1-bromonaftaleno por bromhidrato de 4-bromo-3-(trifluorometil)piridina en la Etapa E.

Etapa B: 2-(cianometil)-4-(2-(((2S,4R)-4-metoxi-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,_______ 8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo: se pusieron 4-(2-(((2S,4R)-1-(tercbutoxicarbonil)-4-metoxipirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (27,0 mg, 0,04 mmol, formaldehído (15,7 μl, 0,2 mmol), Na(0Ac)3BH (17,7 mg, 0,08 mmol) en THF (2 ml) y se agitaron durante 2 h. Se añadió bicarbonato saturado y la mezcla se extrajo con 10 % de Me0H en DCM (3 x 15 ml). Los extractos se combinaron, se secaron con sulfato de sodio y se concentraron para proporcionar el material bruto que se usó tal cual.
Etapa C: 2-(1-acriloil-4-(2-(((2S,4R)-4-metoxi-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo se preparó siguiendo el Ejemplo 375 Etapa F y G, sustituyendo 2-(cianometil)-4-(2-(((2S,4R)-4-metoxi-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo por 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo en la Etapa F. ESI+APCI MS m/z 582,3 [M+H]+.
Ejemplo 382

2-((S)-1-acriloil-4-(7-(3-fluoro-2-(trifluorometil)fenil)-2-(((R)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(7-(3-fluoro-2-(trifluorometil)fenil)-2-(((R)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó siguiendo el Ejemplo 234, Etapas F-J, sustituyendo (R)-(1
metilpirrolidin-2-il)metanol por (S)-(1-metilpirrolidin-2-il)metanol en la Etapa F y 1-bromo-3-fluoro-2-(trifluorometil)benceno por 1-bromonaftaleno en la Etapa H. ESI+APCI MS m/z 588,3 [M+H]+.
Ejemplo 383

2-((S)-1-acriloil-4-(7-(5-fluoro-4-(trifluorometil)piridin-3-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(7-(5-fluoro-4-(trifluorometil)piridin-3-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó siguiendo el Ejemplo 234, Etapas H-J, sustituyendo 3-bromo-5-fluoro-4-(trifluorometil)piridina por 1-bromonaftaleno en la Etapa H. ESI+APCI MS m/z 589,3 [M+H1+.
Ejemplo 384

2-((S)-1-acriloil-4-(2-(((2S,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(5-fluoro-4-(trifluorometil)piridin-3-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (S)-4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo: Se puso (S)-4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (5,37 g, 10,2 mmol) en DCM (75 ml) y se enfrió a 0 °C. Se añadió HCl (12,7 ml, 50,9 mmol) y la reacción se calentó a ta y se agitó durante 2 h. La reacción se concentró y se llevó a DCM. Se añadió bicarbonato saturado y la mezcla se extrajo con DCM (3 x 50 ml). Las capas orgánicas se combinaron, se secaron y se concentraron para proporcionar (S)-4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (4,33 g, 10,1 mmol, 99 % de rendimiento) que se usó tal cual.

Etapa B: (S)-4-(2-cloro-7-(5-fluoro-4-(trifluorometil)piridin-3-il)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo: A un vial se le añadieron carbonato de cesio (2.29 g. 7.0 mmol). (S)-4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (1.00 g.
2.3 mmol). metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2'-metilamino-1,1'-bifenil-2-il)paladio (II) (0,30 g. 0,35 mmol). 3-bromo-5-fluoro-4-(trifluorometil)piridina (1.29 g. 5.3 mmol) y 1,4-dioxano (15,6 ml.2.3 mmol) y el vial se desgasificó con Ar. se selló y después se calentó a 90 °C durante 24 h. La reacción se enfrió. se añadieron agua y NH4Cl saturado y la mezcla se extrajo con DCM. Las capas orgánicas se combinaron y se concentraron. El residuo resultante se purificó con gel de sílice (5-75 % de EtOAc en hexano) para proporcionar (S)-4-(2-cloro-7-(5-fluoro-4-(trifluorometil)piridin-3-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (634 mg. 1.1 mmol. 46 % de rendimiento).

Etapa C: (2S.4R)-2-(((4-((S)-3-(cianometil)piperazin-1-il)-7-(5-fluoro-4-(trifluorometil)piridin-3-il)-5.6.7.8-tetrahidropiridor3,4-d1pirimidin-2-il)oxi)metil)-4-fluoropirrolidin-1-carboxilato de terc-butilo: En un vial de fondo cónico. una solución de (S)-4-(2-cloro-7-(5-fluoro-4-(trifluorometil)piridin-3-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (634 mg. 1,07 mmol) en dioxano (10746 gl, 1,07 mmol) se roció con argón. Se añadieron secuencialmente (2S,4R)-4-fluoro-2-(hidroximetil)pirrolidin-1-carboxilato de terc-butilo (942 mg.
4,30 mmol). Cs2C03 (1050 mg. 3,22 mmol) y metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2'-metilamino-1,1'-bifenil-2-il)paladio (II) (137 mg. 0,161 mmol) en una atmósfera de argón y se rociaron durante 5 min más. La mezcla de reacción se tapó y se calentó a 100 °C durante 18 h. La reacción se enfrió y se añadió agua. La mezcla se extrajo con DCM (3 x 20 ml) y los extractos se combinaron y se concentraron. El residuo resultante se purificó con gel de sílice (0-10 % de Me0H) para proporcionar (2S,4R)-2-(((4-((S)-3-(cianometil)piperazin-1-il)-7-(5-fluoro-4-(trifluorometil)piridin-3-il)-5,6.7.8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)-4-fluoropirrolidin-1 -carboxilato de terc-butilo (579 mg. 0,907 mmol. 84 % de rendimiento).

Etapa D: (2S.4R)-2-(((4-((S)-4-acriloil-3-(cianometil)piperazin-1-il)-7-(5-fluoro-4-(trifluorometil)piridin-3-il)-5.6.7.8-tetrahidropiridor3,4-d1pirimidin-2-il)oxi)metil)-4- fluoropirrolidin-1 -carboxilato de terc-butilo: Se pusieron (2S,4R)-2-(((4
((S)-3-(cianometil)piperazin-1-il)-7-(5-fluoro-4-(trifluorometil)piridin-3-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)-4-fluoropirrolidin-1 -carboxilato de terc-butilo (579 mg, 0,907 mmol) y trietilamina (379 μl, 2,72 mmol) en CH2Cl2 (9066 μl, 0,907 mmol) y se enfriaron a 0 °C. Se añadió cloruro de acriloílo (6044 μl, 1,81 mmol) (solución 0,3 M recién preparada en DCM) y la reacción se agitó durante 45 min a 0 °C. Se añadió agua y la mezcla se extrajo con DCM (3 x 15 ml). Las capas se combinaron y se concentraron para proporcionar el material bruto que se usó tal cual.

Etapa E: 2-((S)-1-acriloil-4-(7-(5-fluoro-4-(trifluorometil)piridin-3-il)-2-(((2S,4R)-4-fluoropirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: En un recipiente cerrado herméticamente, se puso (2S,4R)-2-(((4-((S)-4-acriloil-3-(cianometil)piperazin-1-il)-7-(5-fluoro-4-(trifluorometil)piridin-3-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)-4-fluoropirrolidin-1 -carboxilato de terc-butilo (628 mg, 0,91 mmol) en Me0H (2 ml). Se añadió HCl (756 μl, 4,53 mmol) (acuoso 6 M) y la mezcla se agitó a ta durante 5 h. Se añadió lentamente bicarbonato saturado para llevar el pH a ~9. La mezcla se extrajo con DCM (3 x 20 ml). Los extractos se combinaron, se secaron, se filtraron y se concentraron para proporcionar el material bruto que se usó tal cual.

Etapa F: 2-((S)-1-acriloil-4-(2-(((2S,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(5-fluoro-4-(trifluorometil)piridin-3-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Se pusieron 2-((S)-1-acriloil-4-(7-(5-fluoro-4-(trifluorometil)piridin-3-il)-2-(((2S,4R)-4-fluoropirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (175 mg, 0,30 mmol), formaldehído (111 μl, 1,48 mmol) y Na(0Ac)3BH (125 mg, 0,59 mmol) en THF (2 ml) y se agitaron durante 2 h. Se añadió bicarbonato saturado y la mezcla se extrajo con 10 % de Me0H en DCM (3 x 15 ml). Los extractos se combinaron, se secaron con sulfato de sodio y se concentraron. El residuo resultante se purificó con gel de sílice (4-13 % de Me0H en DCM con 0,25 % de NH40H) para proporcionar 2-((S)-1-acriloil-4-(2-(((2S,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(5-fluoro-4-(trifluorometil)piridin-3-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (69,7 mg, 0,115 mmol, 39 % de rendimiento). ESI+APCI MS m/z 607,2 [M+H]+.
Ejemplo 385

2-((S)-1-acriloil-4-(7-(5-cloro-4-(trifluorometil)piridin-3-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(7-(5-cloro-4-(trifluorometil)piridin-3-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó siguiendo el Ejemplo 234, Etapas H-J,
sustituyendo 3-bromo-5-cloro-4-(trifluorometil)piridina por 1-bromonaftaleno en la Etapa H y THF por Me0H en la Etapa I. ESI+APCI MS m/z 605,3 [M+H]+.
Ejemplo 386

2-((S)-1-acriloil-4-(2-(((2S,4R)-4-fluoro-1-isopropilpirrolidin-2-il)metoxi)-7-(5-fluoro-4-(trifluorometil)piridin-3-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(2-(((2S,4R)-4-fluoro-1-isopropilpirrolidin-2-il)metoxi)-7-(5-fluoro-4-(trifluorometil)piridin-3-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo, se preparó siguiendo la preparación del Ejemplo 384, Etapa F, sustituyendo propan-2-ona por formaldehído en la etapa F. ESI+APCI MS m/z 635,3 [M+H]+.
Ejemplo 387

2-((S)-1-acriloil-4-(7-(5-cloro-4-metilpiridin-3-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(7-(5-cloro-4-metilpiridin-3-il-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó siguiendo el Ejemplo 234, Etapas H-J, sustituyendo 3-bromo-5-cloro-4-metilpiridina por 1-bromonaftaleno en la Etapa H. ESI+APCI mS m/z 551,2 [M+H]+.
Ejemplo 388

2-((S)-1-acriloil-4-(7-(3-fluoro-2-metilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(7-(3-fluoro-2-metilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó siguiendo el Ejemplo 234, Etapas H-J, sustituyendo 1-bromo-3-fluoro-2-metilbenceno por 1-bromonaftaleno en la Etapa H. ESI+ApCI MS m/z 534,3 [M+H]+.
Ejemplo 389

2-((S)-1-(but-2-inoil)-4-(7-(2,3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 4-(terc-butil) (R)-2-(hidroximetil)piperazin-1,4-dicarboxilato de 1-bencilo: A (R)-3-(hidroximetil)piperazin-1-carboxilato de terc-butilo (76,0 g, 351 mmol) en un matraz de 3 l se le añadió agua (500 ml) y la suspensión se agitó hasta que el material se disolvió completamente. A esta mezcla se le añadió EtOAc (541 ml, 351 mmol) seguido de NaHC03 (88,6 g, 1,05 mol) y la mezcla resultante se agitó durante 3 min (temp. interna = 20 °C). A esta mezcla se le añadió Cbz-Cl (52,5 ml, 369 mmol) durante 3 minutos (temp. interna de 20 °C a 25 °C, desprendimiento de gas observado en este punto) y la reacción se agitó durante 1 d a ta. La mezcla de reacción se transfirió a un embudo de decantación y la capa orgánica se lavó con salmuera, se secó (MgS04) y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida eluyendo con 10-70 % de EtOAc/hex para proporcionar 4-(terc-butil) (R)-2-(hidroximetil)piperazin-1,4-dicarboxilato de 1-bencilo (114,6 g, 327,0 mmol, 93 % de rendimiento). ESI+APCI mS m/z 251,1 [M-Boc+H]+.
Etapa B: 4-(terc-butil) (R)-2-(((metilsulfonil)oxi)metil)piperazin-1,4-dicarboxilato de 1-bencilo: A una solución de 4-(tercbutil) (R)-2-(hidroximetil)piperazin-1,4-dicarboxilato de 1-bencilo (32 g, 91,3 mmol) en CH2Cl2 (457 ml, 0,2 M) enfriada a 0 °C se le añadió DIeA (24,6 ml, 137 mmol) seguido de cloruro de metanosulfonilo (7,77 ml, 100 mmol) durante 1 minuto y la mezcla se agitó a 0 °C durante 1 h. La mezcla de reacción se transfirió a un embudo de decantación y se lavó con 1:1 de agua/salmuera (500 ml). La capa orgánica se recogió, se secó (MgS04) y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida eluyendo con 10-60 % de EtOAc/hex para proporcionar 4-(terc-butil) (R)-2-(((metilsulfonil)oxi)metil)piperazin-1,4-dicarboxilato de 1-bencilo (33 g, 77,0 mmol, 84,3 % de rendimiento). ESI+APCI MS m/z 329,1 [M-Boc+H]+.
Etapa C: 4-(terc-butil) (S)-2-(cianometil)piperazin-1,4-dicarboxilato de 1-bencilo: Una solución de 4-(terc-butil) (R)-2-(((metilsulfonil)oxi)metil)piperazin-1,4-dicarboxilato de 1-bencilo (12,6 g, 29,4 mmol) en DMA (368 ml, 73,5 mmol) se roció con argón. A la solución se le añadió cianuro de sodio (3,60 g, 73,5 mmol) y la reacción se agitó a 55 °C durante 1 d, controlando por HPLC (método largo) para determinar la finalización de la reacción. La mezcla de reacción se transfirió a un embudo de decantación, se diluyó con Na0H 0,5 M (800 ml) y después se extrajo con MTBE (2 x). Luego, las capas orgánicas combinadas se lavaron con agua básica (2 x 200 ml) y salmuera (100 ml), se secaron
(MgS04) y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida eluyendo con 10-->60 % de EtOAc/hex como eluyente para proporcionar 4-(terc-butil) (S)-2-(cianometil)piperazin-1,4-dicarboxilato de 1-bencilo (7,9 g, 22,0 mmol, 75 % de rendimiento). ESI+APCI MS m/z 260,1 [M-Boc+H]+.
Etapa D: clorhidrato de (S)-2-(cianomet¡l)p¡peraz¡n-1-carbox¡lato de bencilo: A una solución de 4-(terc-butil) (S)-2-(cianometil)piperazin-1,4-dicarboxilato de 1-bencilo (20,4 g, 56,8 mmol) en DCM (50 ml) se le añadió cloruro de hidrógeno (99,3 ml, 397 mmol) (solución 4,0 M en dioxano) y la reacción se agitó a ta durante 1,5 h. La mezcla de reacción se concentró al vacío y se secó al vacío para dar clorhidrato de (S)-2-(cianometil)piperazin-1-carboxilato de bencilo (16,8 g, 56,8 mmol, 100 % de rendimiento) en forma de una espuma. ESI+APCI m S m/z 260,1 [M+H]+.
Etapa E: (S)-4-(4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-cloro-5.8-d¡h¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-7(6H)-carboxilato de terc-butilo: A una solución de clorhidrato de (S)-2-(cianometil)piperazin-1-carboxilato de bencilo (16,8 g, 56,8 mmol) en DMA (114 ml, 56,8 mmol) se le añadió 2,4-dicloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (15,5 g, 51,1 mmol) seguido de DIEA (39,7 ml, 227 mmol) y la mezcla de reacción se agitó a TA durante 1 h. La mezcla de reacción se diluyó con 1 l de agua y se extrajo con MTBE (2 x). Los extractos orgánicos combinados se lavaron con agua (2 x 200 ml) y salmuera, se secaron sobre MgS04 y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida eluyendo con 10-60 % de EtOAc/hex como eluyente para dar (S)-4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de tercbutilo (22,8 g, 43,3 mmol, 76 % de rendimiento). ESI+APCI MS m/z 527,2 [M+H]+.
Etapa F: 4-((S)-4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.8-d¡h¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-butilo: En un MFR de pared gruesa de 250 ml con tapón a rosca de PTFE, una solución de (S)-4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (5,55 g, 10,5 mmol) en dioxano (60,2 ml, 10,5 mmol) se roció con argón, se añadieron secuencialmente (S)-(1-metilpirrolidin-2-il)metanol (3,64 g, 31,6 mmol), Cs2C03 (10,3 g, 31,6 mmol), Ruphos Pd G3 (0,881 g, 1,05 mmol) y la mezcla resultante se roció con argón durante 5 min más. La mezcla de reacción se tapó y se calentó a 100 °C durante 1 d. Después de que se consumiera el material de partida, la mezcla de reacción se enfrió a temperatura ambiente y se añadió Cbz-Cl a medida que era necesario para convertir el producto hidrolizado con Cbz en el compuesto del título. Después, la mezcla de reacción se repartió entre EtOAc/10 % de salmuera. La capa acuosa se extrajo con EtOAc (2 x). Las capas orgánicas combinadas se secaron (MgS04) y se concentraron. El residuo se purificó por cromatografía ultrarrápida eluyendo con un gradiente de 1,5 % de Me0H/0,15 % de NH40H/DCM a 11,25 % de Me0H/1,125 % de NH40H/DCM (Premix 15 % de Me0H/1,5 % de NH40H en DCM y aumentando de 10-75 % de esta mezcla en DCM) para proporcionar el compuesto del título (4,51 g, 7,45 mmol, 71 %). ESI MS m/z 606,3 [M+H]+.
Etapa G: (S)-2-(c¡anomet¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo: A una solución de 4-((S)-4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (7,2 g, 12 mmol) en DCM (1,0 g, 12 mmol) se le añadió HCl (4 M en dioxano, 15 ml, 59 mmol). La mezcla se agitó a TA durante 2 h. La mezcla de reacción se concentró hasta la sal bis HCl. El residuo se repartió entre DCM/NaHC03 (sat.) y la capa acuosa se extrajo con DCM (3 x). Las capas orgánicas combinadas se secaron y se concentraron para proporcionar el compuesto del título 4,72 g (0,009 mmol, 79 %). ESI MS m/z 506,3 [M+H]+.
Etapa H: (S)-2-(c¡anomet¡l)-4-(7-(2.3-d¡met¡lfen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo: A un vial se le añadieron carbonato de cesio (967 mg, 2,97 mmol), (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (500 mg, 0,989 mmol), Rhuphos Pd G3 (124 mg, 0,148 mmol), 1-bromo-2,3-dimetilbenceno (915 mg, 4,94 mmol) y 1,4-dioxano (9889 μl, 0,989 mmol), el vial se desgasificó con Ar, se selló y después se calentó a 75 °C durante 24 h. Se añadieron agua y NH4Cl saturado y la mezcla se extrajo con DCM. Las capas orgánicas se combinaron y se concentraron. El residuo resultante se purificó con gel de sílice (0-12 % de Me0H en DCM con 0,25 % de NH40H) para proporcionar (S)-2-(cianometil)-4-(7-(2,3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (505 mg, 0,828 mmol, 83,7 % de rendimiento).
ESI MS m/z 610,4 [M+H]+.
Etapa I: 2-((S)-4-(7-(2.3-d¡met¡lfen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo: A una solución de (S)-2-(cianometil)-4-(7-(2,3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (505 mg, 0,828 mmol) en Et0H (8282 μl, 0,828 mmol) y THF (8282 μl, 0,828 mmol) se le añadió paladio (441 mg, 0,207 mmol) (tipo Degussa, 10 % en peso, 50 % de H20) y después se introdujo una atmósfera de H2 mediante vacío seguido de presión de globo.
Después, la mezcla se agitó a temperatura ambiente durante 2 horas. Después, la mezcla se diluyó con Me0H y THF 1:1 y se filtró a través de papel GHF. Después, el filtrado se concentró para proporcionar el producto bruto que se usó tal cual. ESI MS m/z 476,3 [M+H]+.
Etapa J: 2-((S)-1-(but-2-¡no¡l)-4-(7-(2.3-d¡met¡lfen¡l)-2-(((S)-1-met¡lp¡rol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: A 0 °C, a un MFR de 25 ml que contenía N,N-dimetilformamida (4415 μl, 0,4415 mmol) se le añadieron 2-((S)-4-(7-(2,3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (210 mg, 0,44 mmol) y trietilamina (122,7 μl, 0,88 mmol). La mezcla de reacción se agitó vigorosamente mientras se añadía en una porción ácido but-2-inoico (44,54 mg, 0,53 mmol). A la mezcla en agitación se le añadió lentamente anhídrido cíclico del ácido 1-propanofosfónico (197,1 μl, 0,66 mmol). La reacción se dejó en agitación durante 2 h a 0 °C. Se añadió agua y los sólidos se filtraron. Los sólidos se purificaron con gel de sílice (5-18 % de Me0H en DCM con 0,25 % de NH40H) para proporcionar el compuesto del título 2-((S)-1-(but-2-inoil)-4-(7-(2,3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0389 , 144 mg, 0,27 mmol, 60 %). ESI+APCl MS m/z 542,3 [M+H]+. Ejemplo 390

2-((S)-1-acriloil-4-(2-(((2R,3S)-3-hidroxi-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (S)-4-(2-(((2R,3S)-1-(terc-butoxicarbonil)-3-((tercbutildimetilsilil)oxi)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo: se preparó según el Ejemplo 400, Etapa C-E sustituyendo (2R,3S)-3-((tercbutildimetilsilil)oxi)-2-(hidroximetil)pirrolidin-1 -carboxilato de terc-butilo por (S)-(1-metilpirrolidin-2-il)metanol en la Etapa C.

Etapa B: 2-((S)-1-acriloil-4-(2-(((2R,3S)-3-hidroxipirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitriloSe puso (2R,3S)-2-(((4-((S)-4-acriloil-3-(cianometil)piperazin-1-il)-7-(naftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)-3-((tercbutildimetilsilil)oxi)pirrolidin-1 -carboxilato de tercbutilo (363 mg, 0,473 mmol) en Me0H (10 ml). Se añadió HCl acuoso 6 M y se agitó a ta durante 6 h. La reacción se enfrió a 0 °C y se añadió lentamente bicarbonato saturado. La mezcla se extrajo con DCM (3 x 20 ml). Los extractos se combinaron, se secaron, se filtraron y se concentraron para proporcionar el material bruto que se usó tal cual. Etapa C: 2-((S)-1-acriloil-4-(2-(((2R,3S)-3-hidroxi-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Se preparó según el EJEMPL0384, Etapa F sustituyendo 2-((S)-1-acriloil-4-(2-(((2R,3S)-3-hidroxipirrolidin-2-il)metoxi)-7-(naftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo por 2-((S)-1-acriloil-4-(7-(5-fluoro-4-(trifluorometil)piridin-3-il)-2-(((2S,4R)-4-fluoropirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo. ESl+ApCI Ms m/z 568,3 [M+H]+. Ejemplo 391

2-((S)-1-(but-2-inoil)-4-(7-(8-cloronaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-(but-2-inoil)-4-(7-(8-cloronaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Se preparó según el Ejemplo 389, Etapa H-J, sustituyendo 1-bromo-8-cloronaftaleno por 1-bromo-2,3-dimetilbenceno en la Etapa H. ESI+APCI MS m/z 598,2 [M+H]+.
Ejemplo 392

2-((S)-1-(ciclobut-1-eno-1-carbonil)-4-(7-(2,3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-(ciclobut-1-eno-1-carbonil)-4-(7-(2,3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Se preparó siguiendo el Ejemplo 389 Etapa J, sustituyendo ácido ciclobut-1-eno-1-carboxílico por ácido but-2-inoico. ESI+APCI MS m/z 556,3 [M+H]+.
Ejemplo 393

2-((S)-1-acriloil-4-(7-(3-metil-2-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (S)-2-(cianometin-4-(7-(3-metil-2-(trifluorometinfenih-2-(((S)-1-metilpirrolidin-2-inmetoxi)-5.6.7.8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo. Se pusieron (S)-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (150 mg, 0,22 mmol), K2C03 (329 gl, 0,66 mmol), 2,4,6-trimetil-1.3.5.2.4.6-trioxatriborinano (82,6 mg, 0,658 mmol) y tetraquis (25,3 mg, 0,022 mmol) en dioxano (731 gl, 0,22 mmol) y se calentaron a 90 °C durante 18 h. Se añadió agua y la mezcla se extrajo con 10 % de Me0H en DCM (3 x 15 ml). Los extractos se combinaron y se concentraron. El residuo resultante se purificó con gel de sílice (2-80 % de Me0H en DCM con 0,25 % de NH40H) para proporcionar (S)-2-(cianometil)-4-(7-(3-metil-2-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d)pirimidin-4-il)piperazin-1-carboxilato de bencilo (75 mg, 0,11 mmol, 52 % de rendimiento).
Etapa B: 2-((S)-1-acriloil-4-(7-(3-metil-2-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropiridor3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Se preparó siguiendo el Ejemplo 234, Etapa I y J sustituyendo (S)-2-(cianometil)-4-(7-(3-metil-2-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo por (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo en la Etapa I. ESI+APCI MS m/z 584,3 [M+H]+.
Ejemplo 394

2-((S)-1-acriloil-4-(7-(2,3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(7-(2.3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropiridor3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó siguiendo el Ejemplo 234, Etapas H-J, sustituyendo 1-bromo-2,3-dimetilbenceno por 1-bromonaftaleno en la Etapa H. ESI+APCI MS m/z 530,3 [M+H]+.
Ejemplo 395

2-(1-acriloil-4-(2-(((S)-1,2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2 -il)acetonitrilo

Etapa A: 4-(2-(((S)-1-(terc-butoxicarbonil)-2-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-2-(cianometil)piperazin-1 -carboxilato de bencilo: En un tubo de microondas, se disolvió 4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (200 mg, 0,362 mmol) en dioxano (181 μl, 0,362 mmol) y se trató con carbonato de cesio (236 mg, 0,723 mmol) y 2-(hidroximetil)-2-metilpirrolidin-1-carboxilato de (S)-terc-butilo (389 mg, 1,81 mmol). Después, el tubo se tapó y se calentó a 90 °C durante 2 horas. La reacción se enfrió a temperatura ambiente y se filtró a través de papel GHF. El filtrado se concentró al vacío y se cromatografió en el CombiFlash eluyendo con 0 %-10 % de DCM:Me0H. Todas las fracciones que contenían el producto limpio se combinaron y se concentraron para dar 4-(2-(((S)-1-(tercbutoxicarbonil)-2-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazina-1-carboxilato de bencilo (264 mg, 0,361 mmol, 99,7 % de rendimiento). ESI+APCI MS m/z 732,4 [M+H]+.
Etapa B: 2-(cianometil)-4-(2-(((S)-2-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: Se disolvió 4-(2-(((S)-1-(terc-butoxicarbonil)-2-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (264 mg, 0,361 mmol) en diclorometano (3607 μl, 0,361 mmol) y se trató con TFA (556 μl, 7,21 mmol). La reacción se agitó a temperatura ambiente durante 1 hora. La reacción se concentró al vacío y se trató con bicarbonato saturado. La capa acuosa se extrajo con DCM (2 x) y los extractos orgánicos combinados se secaron sobre Na2S04. Los extractos orgánicos se concentraron al vacío para dar 2-(cianometil)-4-(2-(((S)-2-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8- tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (210 mg, 0,332 mmol, 92,2 % de rendimiento). ESI+APCI MS m/z 632,3 [M+H]+.
Etapa C: 2-(cianometil)-4-(2-(((S)-1,2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: Se disolvió 2-(cianometil)-4-(2-(((S)-2-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7, 8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (105 mg, 0,1662 mmol) en ácido fórmico (94,05 μl, 2,493 mmol) y se trató con formaldehído (1868 μl, 24,93 mmol). La mezcla de reacción se agitó a 85 °C durante 1 hora. La reacción se enfrió a temperatura ambiente y se trató con bicarbonato saturado. La capa acuosa se extrajo con DCM (2 x). Los extractos orgánicos combinados se secaron sobre Na2S04, se concentraron al vacío y se cromatografiaron en el CombiFlash eluyendo con 0 %-10 % de DCM:Me0H. Todas las fracciones que contenían el producto limpio se combinaron y se concentraron al vacío para dar 2 -(cianometil)-4-(2-(((S)-1,2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazina-1-carboxilato de bencilo (75 mg, 0 ,1161 mmol, 69,88 % de rendimiento). ESI+APCI Ms m/z 646,3 [M+H]+.
Etapa D: 2-(4-(2-(((S)-1,2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: Una solución de 2-(cianometil)-4-(2-(((S)-1,2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8- tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (117 mg, 0,181 mmol) en Et0H (1812 μl, 0,181 mmol) y THF (1812 μl, 0,181 mmol) se purgó con N2 durante 5 minutos. A esta solución se le añadió paladio (96,4 mg, 0,0453 mmol) (tipo Degussa, 10 % en peso, 50 % de H20), se tapó inmediatamente y se purgó con N2 durante 5 min más. Después, la solución se agitó en una atmósfera de H2. Después, la mezcla se diluyó con Me0H
y se filtró a través de celite empaquetado. Después, el filtrado se concentró al vacío para proporcionar 2-(4-(2-(((S)-1,2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (88 mg, 0,172 mmol, 94,9 % de rendimiento). ESI+APCI MS m/z 512,3 [M+H]+.
Etapa E: 2-(1-acriloil-4-(2-(((S)-1,2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: A una suspensión de 2-(4-(2-(((S)-1,2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (88 mg, 0,17 mmol) en diclorometano (1720 μl, 0,17 mmol) a temperatura ambiente se le añadió cloruro de acriloílo (14 μl, 0,17 mmol) seguido de Base de Hunig (60 μl, 0,34 mmol). Después, la reacción se agitó a temperatura ambiente durante 30 minutos. Después, la mezcla de reacción se concentró al vacío. El concentrado se suspendió en una mezcla 60:40 de ACN: H20 y se purificó en el Gilson (HPLC prep. inversa), eluyendo con 5-->95 % de ACN/0,1 % de TFA en agua/0,1 % de TFA. Las fracciones que contenían el producto se combinaron y se repartieron entre bicarbonato saturado y DCM. La capa acuosa se extrajo dos veces más con DCM. Las capas orgánicas se combinaron, se secaron sobre Na2S04 y se concentraron al vacío para dar 2-(1-acriloil-4-(2-(((S)-1,2-dimetilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (14 mg, 0,025 mmol, 14 % de rendimiento). ESI+APCI MS m/z 566,3 [M+H]+.
Ejemplo 396

2-(1-acriloil-4-(2-(((S)-1-(ciclopropilmetil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 2-(cianometil)-4-(2-(((S)-1-(ciclopropilmetil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: En un vial de fondo cónico, una solución de 4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (200 mg, 0,362 mmol) en dioxano (3616 μl, 0,362 mmol) se roció con argón, se añadieron secuencialmente (S)-(1 -(ciclopropilmetil)pirrolidin-2-il)metanol (168 mg, 1,08 mmol), Cs2C03 (353 mg, 1,08 mmol), Rhuphos Pd G3 (30,2 mg, 0,0362 mmol) en una atmósfera de argón y se rociaron durante 5 min más. La mezcla de reacción se tapó y se calentó a 100 °C durante 1 hora. La mezcla de reacción se enfrió a temperatura ambiente. Se añadió EtOAc y se lavó con salmuera (2 x). Los extractos orgánicos combinados se secaron sobre Na2S04 y se concentraron al vacío. El concentrado se purificó por cromatografía ultrarrápida eluyendo con 0-20 % de DCM/Me0H 2 % de NH40H. Todas las fracciones que contenían el producto deseado se combinaron y se concentraron para dar 2-(cianometil)-4-(2-(((S)-1 -(ciclopropilmetil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (90 mg, 0,134 mmol, 37,0 % de rendimiento). ESI+APCI MS m/z 672,4 [M+H]+.
Etapa B: 2-(4-(2-(((S)-1-(ciclopropilmetil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Una solución de 2-(cianometil)-4-(2-(((S)-1-(ciclopropilmetil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (90 mg, 0,13 mmol) en
Et0H (1340 μl, 0,13 mmol) y THF (1340 μl, 0,13 mmol) se purgó con N2 durante 5 min. A esta solución se le añadió paladio (36 mg, 0,033 mmol) (tipo Degussa, 10 % en peso, 50 % de H20), se tapó inmediatamente y se purgó con N2 durante 5 min más. Después, la solución se agitó en una atmósfera de H2. La mezcla se diluyó con Me0H y se filtró a través de celite empaquetado. El filtrado se concentró al vacío para proporcionar 2-(4-(2-(((S)-1-(ciclopropilmetil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo bruto (71 mg, 0,13 mmol, 99 % de rendimiento). ESI+APCI MS m/z 538,3 [M+H]+.
Etapa C: 2-(1-acriloil-4-(2-(((S)-1-(ciclopropilmetil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: A una suspensión de 2-(4-(2-(((S)-1-(ciclopropilmetil)pirrolidin-2-il)metoxi)-7-(naftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (71 mg, 0,132 mmol) en diclorometano (1320 μl, 0,132 mmol) a temperatura ambiente se le añadió cloruro de acriloílo (10,7 μl, 0,132 mmol) seguido de base de Hunig (46,1 μl, 0,264 mmol). La reacción se agitó a temperatura ambiente durante 1 hora. La mezcla de reacción se concentró al vacío. El concentrado se suspendió en una mezcla 60:40 de ACN: H20 y se purificó en el Gilson (HPLC prep. inversa), eluyendo con 5-->95 % de ACN/0,1 % de TFA en agua/0,1 % de TFA. Las fracciones que contenían el producto se combinaron y se repartieron entre bicarbonato saturado y DCM. La capa acuosa se extrajo dos veces más con DCM. Las capas orgánicas se combinaron, se secaron sobre Na2S04 y se concentraron al vacío para dar el compuesto del título 2-(1-acriloil-4-(2-(((S)-1-(ciclopropilmetil)pirrolidin-2-il)metoxi)-7-(naftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMp Lo 396, 15,7 mg, 0,0265 mmol, 20,1 % de rendimiento). ESI+APCI MS m/z 592,4 [M+H]+.
Ejemplo 397

2-(1-acriloil-4-(2-(2-(dimetilamino)-2-metilpropoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-(1-acriloil-4-(2-(2-(dimetilamino)-2-metilpropoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: El compuesto del título se preparó siguiendo el Ejemplo 396 sustituyendo 2-Dimetilamino-2-metil-1-propanol por (S)-(1-(ciclopropilmetil)pirrolidin-2-il)metanol en la Etapa A. ESI+APCI MS m/z 554,4 [M+H]+.
Ejemplo 398

2-((2S)-2-(((4-(4-acriloil-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)pirrolidin-1-il)-N,N-dimetilacetamida

Etapa A: 4-(2-(((S)-1-(terc-butoxicarbonil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo: En un tubo de microondas, se disolvió 4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (200 mg, 0,362 mmol) en dioxano (181 μl, 0,362 mmol) y se trató con carbonato de cesio (236 mg, 0,723 mmol) y N-terc-Butoxicarbonil-L-prolinol (364 mg, 1,81 mmol). Después, el tubo se tapó y se calentó a 90 °C durante una noche. La mezcla de reacción se enfrió a temperatura ambiente, se filtró a través de papel GHF y se concentró al vacío. El concentrado se cromatografió en el CombiFlash eluyendo con 0 %-10 % de DCM:Me0H. Todas las fracciones que contenían el producto limpio se combinaron y se concentraron para dar 4-(2-(((S)-1-(terc-butoxicarbonil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (225 mg, 0,313 mmol, 86,7 % de rendimiento). ESI+APCI MS m/z 718,4 [M+H]+.
Etapa B: 2-(cianometil)-4-(7-(naftalen-1-il)-2-(((S)-pirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: Se disolvió 4-(2-(((S)-1-(terc-butoxicarbonil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8- tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (225 mg, 0,313 mmol) en diclorometano (3134 μl, 0,313 mmol) y se trató con TFA (483 μl, 6,27 mmol). La reacción se agitó a temperatura ambiente durante 1 hora. La reacción se concentró al vacío y los extractos orgánicos se lavaron con bicarbonato saturado. Los extractos orgánicos se extrajeron con DCM (3 x). Los extractos orgánicos combinados se secaron sobre Na2S04 y se concentraron al vacío para dar 2-(cianometil)-4-(7-(naftalen-1-il)-2-(((S)-pirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (111 mg, 0,180 mmol, 57,3 % de rendimiento). ESI+APCI MS m/z 618,3 [M+H]+.
Etapa C: 2-(cianometil)-4-(2-(((S)-1-(2-(dimetilamino)-2-oxoetil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo: Se disolvió 2-(cianometil)-4-(7-(naftalen-1-il)-2-(((S)-pirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (111 mg, 0,1797 mmol) en diclorometano (1797 μl, 0,1797 mmol) y se trató con N-etil-N-isopropilpropan-2-amina (156,9 μl, 0,8984 mmol) y cloroacetildimetilamina (46,20 μl, 0,4492 mmol). La reacción se agitó a temperatura ambiente durante 30 minutos. Se añadieron 2 equivalentes más de base y la reacción se agitó durante 3 horas más. La reacción se concentró al vacío y se cromatografió en el CombiFlash eluyendo con 0 %-15 % de DCM/Me0H 0,5 % de modificador de NH40H. Todas las fracciones que contenían el producto deseado limpio se combinaron y se concentraron para dar 2-(cianometil)-4-(2-(((S)-1 -(2-(dimetilamino)-2-oxoetil)pirrolidin-2-il)metoxi)-7-(naftalen-1 -il)-5.6.7.8- tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (73 mg, 0,1039 mmol, 57,80 % de rendimiento). ESI+APCI MS m/z 703,3 [M+H]+.
Etapa D: 2-((2S)-2-(((4-(3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)pirrolidin-1-il)-N,N-dimetilacetamida: Una solución de 2-(cianometil)-4-(2-(((S)-1-(2-(dimetilamino)-2-oxoetil)pirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (73 mg, 0,10 mmol) en Et0H (1039 μl, 0,10 mmol) y THF (1039 μl, 0,10 mmol) se purgó con N2 durante 5 minutos. A esta solución se le añadió paladio (28 mg, 0,026 mmol) (tipo Degussa, 10 con %, 50 % de H20), se tapó inmediatamente y se purgó con N2 durante 5 minutos más. Después, la solución se agitó en una atmósfera de H2. Después, la mezcla se diluyó con Me0H y se filtró a través de celite empaquetado. Después, el filtrado se concentró
al vacío para proporcionar 2-((2S)-2-(((4-(3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)pirrolidin-1-il)-N,N-dimetilacetamida (43 mg, 0,076 mmol, 73 % de rendimiento). ESI+APCI MS m/z 569,3 [M+H]+.
Etapa E: 2-((2S)-2-(((4-(4-acriloil-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)pirrolidin-1-il)-N,N-dimetilacetamida: A una suspensión de 2-((2S)-2-(((4-(3-(cianometil)piperazin-1-il)-7-(naftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)pirrolidin-1 -il)-N,N-dimetilacetamida (43 mg, 0,0756 mmol) en diclorometano (756 μl, 0,0756 mmol) a temperatura ambiente se le añadió cloruro de acriloílo (6,14 μl, 0,0756 mmol) seguido de base de Hunig (26,4 μl, 0,151 mmol). Después, la reacción se agitó a temperatura ambiente durante 1 hora. La mezcla de reacción se concentró al vacío, se suspendió de nuevo en una mezcla 60:40 de ACN: H20 y se purificó en el Gilson (HPLC prep. inversa), eluyendo con 5-->95 % de ACN/0,1 % de TFA en agua/0,1 % de TFA. Las fracciones que contenían el producto se combinaron y se repartieron entre bicarbonato saturado y DCM. La capa acuosa se extrajo dos veces más con DCM. Las capas orgánicas se combinaron, se secaron sobre Na2S04 y se concentraron al vacío para dar 2-((2S)-2-(((4-(4-acriloil-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)pirrolidin-1-il)-N,N-dimetilacetamida (14,9 mg, 0,0239 mmol, 31,6 % de rendimiento). ESI+APCI MS m/z 623,3 [M+H]+.
Ejemplo 399

2-(1-acriloil-4-(2-(((S)-1-metilpirrolidin-3-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-(1-acriloil-4-(2-(((S)-1-metilpirrolidin-3-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: El compuesto del título se preparó siguiendo el Ejemplo 396 sustituyendo (3R)-(1 -Metilpirrolidin-3-il)-metanol por (S)-(1-(ciclopropilmetil)pirrolidin-2-il)metanol en la Etapa A. ESI+APCI MS m/z 552,3 [M+H]+. Ejemplo 400

2-((S)-1-acriloil-4-(2-(((S)-1-etilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (S)-4-(2-cloro-5.6.7.8-tetrah¡drop¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de bencilo: A una soluc¡ón de (S)-4-(4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-cloro-5,8-d¡h¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo (5.0 g. 9.5 mmol) en DCM (50 ml) se le añad¡ó cloruro de h¡drógeno (12 ml. 47 mmol, soluc¡ón 4 M en d¡oxanos) y la reacc¡ón se ag¡tó a temperatura amb¡ente durante 1 hora. Luego. la reacc¡ón se concentró en una suspens¡ón espesa y se añad¡ó una mezcla de EtOAc/agua. La capa acuosa se bas¡f¡có con Na0H 1 M y la capa acuosa se extrajo con EtOAc (2 x). Los extractos orgán¡cos comb¡nados se lavaron con salmuera. se secaron sobre MgS04 y se concentraron al vacío para dar (S)-4-(2-cloro-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (4.0 g. 9.4 mmol. 99 % de rend¡m¡ento). ESI+APCI MS m/z 427.1 [M+H]+.
Etapa B: (S)-4-(2-cloro-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do(3.4-dlp¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo: A una soluc¡ón de (S)-4-(2-cloro-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (3.4 g. 8.0 mmol) en d¡oxanos (40 ml) se le añad¡eron 1-yodonaftaleno (6.0 ml. 40 mmol) y carbonato de ces¡o (5.2 g. 16 mmol). la reacc¡ón se desgas¡f¡có con argón durante 15 m¡nutos segu¡do de la ad¡c¡ón de Rhuphos Pd G3 (1.00 g. 1.2 mmol) y la reacc¡ón se calentó a 100 °C durante una noche. Luego. la reacc¡ón se f¡ltró a través de papel GHF y se concentró al vacío. Luego. el mater¡al se cromatograf¡ó usando 10-->70 de EtOAc/Hexanos como eluyente para dar (S)-4-(2-cloro-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (2.8 g. 5.1 mmol. 64 % de rend¡m¡ento). ESI+APCI MS m/z 553.2 [M+H]+.
Etapa C: (S)-2-(c¡anomet¡l)-4-(2-(((S)-1-et¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo: En un v¡al de fondo cón¡co. una soluc¡ón de (S)-4-(2-cloro-7-(naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (250 mg.
0.452 mmol) en d¡oxano (4520 μl. 0.452 mmol) se roc¡ó con argón durante 5 m¡nutos. Se añad¡eron secuenc¡almente (3R)-(1-et¡l-p¡rrol¡d¡n-3-¡l)-metanol (175 mg. 1.36 mmol). Cs2C03 (442 mg. 1.36 mmol). Rhuphos Pd G3 (37.8 mg.
0.0452 mmol) en una atmósfera de argón y se rodaron durante 5 m¡n más. La mezcla de reacc¡ón se tapó y se calentó a 100 °C durante 1 hora. La reacc¡ón se enfr¡ó a temperatura amb¡ente y se añad¡ó acetato de et¡lo. Los extractos orgán¡cos se lavaron con salmuera (2 x). se secaron sobre Na2S04 y se concentraron al vacío. El concentrado se pur¡f¡có por cromatografía ultrarráp¡da eluyendo con 0-20 % de (Me0H 2 % de NH40H)/DCM. Todas las fracc¡ones que contenían el producto deseado l¡mp¡o se comb¡naron y se concentraron para dar (S)-2-(c¡anomet¡l)-4-(2-(((S)-1-et¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (118 mg. 0.183 mmol. 40.4 % de rend¡m¡ento). ESI+APCI MS m/z 646.3 [M+H]+.
Etapa D: 2-((S)-4-(2-(((S)-1-et¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo: Una soluc¡ón de (S)-2-(c¡anomet¡l)-4-(2-(((S)-1-et¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (237 mg. 0.367 mmol) en Et0H (3670 μl.
0.367 mmol) y THF (3670 μl. 0.367 mmol) se purgó con N2 durante 5 m¡n. A esta soluc¡ón se le añad¡ó palad¡o (97.6 mg. 0.0917 mmol) (t¡po Degussa. 10 % en peso. 50 % de H20). después se tapó ¡nmed¡atamente y se purgó con N2 durante 5 m¡n más. Después. la soluc¡ón se ag¡tó en una atmósfera de H2. Después. la mezcla se d¡luyó con Me0H y se f¡ltró a través de cel¡te empaquetado. Después. el f¡ltrado se concentró al vacío para proporc¡onar 2-((S)-4-(2-(((S)-1-et¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-dlp¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (155 mg. 0.303 mmol. 82.5 % de rend¡m¡ento). ESI+APCI MS m/z 512.3 [M+H]+.
Etapa E: 2-((S)-1-acriloil-4-(2-(((S)-1-etilpirrolidin-2-inmetoxi)-7-(naftalen-1-in-5.6.7.8-tetrahidropirido[3,4-dlpirimidin-4-il)piperazin-2-il)acetonitrilo: A una suspensión de 2-((S)-4-(2-(((S)-1-etilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (226 mg, 0,442 mmol) en diclorometano (4417 μl, 0,442 mmol) a temperatura ambiente se le añadió cloruro de acriloílo (35,9 μl, 0,442 mmol) seguido de base de Hunig (154 μl, 0,883 mmol). La reacción se agitó a temperatura ambiente durante 2 horas. La mezcla de reacción se concentró al vacío, se cargó en una columna RediSep Gold de 12 g y se cromatografió en el CombiFlash (0 %-15 % de DCM:Me0H 1 % de modificador de NH40H). Todas las fracciones que contenían el producto se combinaron y se concentraron al vacío. El concentrado se suspendió en una mezcla 60:40 de ACN:H20 y se purificó en el Gilson (HPLC prep. inversa), eluyendo con 5-->95 % de a Cn/0,1 % de TFA en agua/0,1 % de TFA. Las fracciones que contenían el producto se combinaron y se repartieron entre bicarbonato saturado y DCM. La capa acuosa se extrajo dos veces más con DCM. Las capas orgánicas se combinaron, se secaron sobre Na2S04 y se concentraron al vacío para dar el compuesto del título 2-((S)-1-acr¡lo¡l-4-(2-(((S)-1-et¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (e jEm PL0400, 136 mg, 0,240 mmol, 54,4 % de rendimiento). ESI+APCI Ms m/z 566,2 [M+H]+.
Ejemplo 401

2-((S)-1-((E)-4-(d¡met¡lam¡no)but-2-eno¡l)-4-(2-(((S)-1-et¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5,6,7,8-tetrahidropiridop^-dprimidin^-i0piperazin^-i0acetonitrilo

Etapa A: 2-((S)-1-((E)-4-(dimetilamino)but-2-enoil)-4-(2-(((S)-1-etilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropiridore^-dtoirimidin^-iDpiperazin^-iDacetonitrilo: A una solución de 2-((S)-4-(2-(((S)-1 -etilpirrolidin-2-¡l)metox¡)-7-(naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (150 mg, 0,293 mmol), ácido (2E)-4-(dimetilamino)but-2-enoico (75,7 mg, 0,586 mmol) y DIEA (256 μl, 1,47 mmol) en Dc M (2932 μl, 0,293 mmol) se le añadió HATU (167 mg, 0,440 mmol) y la mezcla resultante se agitó a temperatura ambiente durante 5 horas. La mezcla de reacción se repartió entre EtOAc y salmuera. La fase acuosa se extrajo con EtOAc (2 x). Los extractos orgánicos combinados se secaron sobre Na2S04 y se concentraron al vacío. El concentrado se diluyó en 60:40 ACN:H20 y se purificó en el Gilson (HPLC prep. inversa), eluyendo con 5->95 % de ACN/0,1 % de t Fa en agua/0,1 % de TFA. Las fracciones que contenían el producto se combinaron y se repartieron entre bicarbonato saturado y DCM. La capa acuosa se extrajo dos veces más con DCM. Las capas orgánicas se combinaron, se secaron sobre Na2S04 y se concentraron al vacío para dar el compuesto del título 2-((S)-1-((E)-4-(dimetilamino)but-2-enoil)-4-(2-(((S)-1-et¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (EJEMPL0401,27,6 mg, 0,0443 mmol, 15,1 % de rendimiento). ESI+APCI MS m/z 623,4 [M+H]+.
Ejemplo 402

2-((S)-2-(((4-((S)-4-acriloil-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7, 8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)pirrolidin-1-il)-N,N-dimetilacetamida
2-((S)-2-(((4-((S)-4-acriloil-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)pirrolidin-1-il)-N,N-dimetilacetamida: El compuesto del título se preparó siguiendo el Ejemplo 398 sustituyendo (S)-4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo por 4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo en la Etapa A. ESI+APCI MS m/z 623,3 [M+H]+.
Ejemplo 403

(S)-2-(cianometil)-4-(2-((S)-3-(dimetilamino)pirrolidin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo
(S)-2-(cianometil)-4-(2-((S)-3-(dimetilamino)pirrolidin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: El compuesto del título se preparó siguiendo el Ejemplo 400 sustituyendo (3S)-(-)-3-(Dimetilamino)Pirrolidina por (3R)-(1-Etil-pirrolidin-3-il)-metanol en la Etapa C. ESI+a Pc I MS m/z 551,3 [M+H]+. Ejemplo 404

2-((S)-1-((E)-4-(dimetilamino)but-2-enoil)-4-(7-(2,3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 4-((S)-4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.8-d¡h¡drop¡r¡dor3.4-d1p¡rim¡d¡n-7í6H)-carbox¡lato de terc-butilo En un matraz de fondo redondo. una soluc¡ón de (S)-4-(4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-cloro-5,8-d¡h¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de tercbut¡lo (5 g. 9,487 mmol) en d¡oxano (94,87 ml. 9,487 mmol) se roc¡ó con argón. se añad¡eron secuenc¡almente (S)-(1-met¡lp¡rrol¡d¡n-2-¡l)metanol (3,278 g. 28,46 mmol). Cs2C03 (9,273 g. 28,46 mmol). metanosulfonato de (2-d¡c¡clohex¡lfosf¡no-2',6'-d¡-¡-propox¡-1,1'-b¡fen¡l)(2'-met¡lam¡no-1,1'-b¡fen¡l-2-¡l)palad¡o (II) (0,8078 g. 0,9487 mmol) en una atmósfera de argón y se rodaron durante 5 m¡nutos más. La mezcla de reacc¡ón se tapó y se calentó a 100 °C durante una noche. La reacc¡ón se f¡ltró a través de papel GHF y se concentró al vacío. El concentrado se pur¡f¡có en el Comb¡ Flash (0-12 % de Me0H en DCM con 2 % de NH40H) para proporc¡onar 4-((S)-4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,8-d¡h¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo (5,425 g. 8,508 mmol. 89,68 % de rend¡m¡ento). ESI+APCI MS m/z 606,4 [M+H]+.
Etapa B: (S)-2-(c¡anomet¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo: se d¡solv¡ó 4-((S)-4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo (5.4 g. 8,915 mmol) en d¡clorometano (89,15 ml. 8,915 mmol) y se trató con ác¡do clorhídr¡co (soluc¡ón 4.0 M en 1,4-d¡oxano) (11,14 ml. 44,57 mmol). La reacc¡ón se ag¡tó a temperatura amb¡ente durante 1 hora. Luego. la reacc¡ón se d¡luyó con más DCM y Na0H 1 M y las capas se separaron. Luego. los extractos orgán¡cos se lavaron con salmuera. se secaron sobre Na2S04 y se concentraron al vacío para dar (S)-2-(c¡anomet¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (4,364 g. 8,631 mmol. 96,82 % de rend¡m¡ento). ESI+AμCi MS m/z 506,3 [M+H]+.
Etapa C: 2-((S)-4-(7-(2.3-d¡met¡lfen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo: En un matraz de fondo redondo. una soluc¡ón de (S)-2-(c¡anomet¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (4,364 g.
8,631 mmol) en d¡oxano (43.15 ml. 8,631 mmol) se roc¡ó con argón durante 5 m¡nutos. Se añad¡eron secuenc¡almente 1-bromo-2,3-d¡met¡lbenceno (5,851 ml. 43,15 mmol). Cs2C03 (14,06 g. 43,15 mmol) y metanosulfonato de (2-d¡c¡clohex¡lfosf¡no-2',6'-d¡-¡-propox¡-1,1'-b¡fen¡l)(2'-met¡lam¡no-1,1'-b¡fen¡l-2-¡l)palad¡o (II) (0,7348 g. 0,8631 mmol) en una atmósfera de argón y se rodaron durante 5 m¡n más. La mezcla de reacc¡ón se tapó y se calentó a 100 °C durante una noche. La reacc¡ón se enfr¡ó a temperatura amb¡ente. Se añad¡ó acetato de et¡lo y la reacc¡ón se f¡ltró a través de papel GHF y se concentró al vacío. El concentrado se pur¡f¡có dos veces por cromatografía de fase normal sobre el Comb¡Flash usando 0-15 % de Me0H en DCM con 2 % de NH40H como eluyente. Las fracc¡ones que contenían el producto deseado se recog¡eron y se concentraron al vacío para dar 2-((S)-4-(7-(2,3-d¡met¡lfen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (0,411 g. 0,8641 mmol.
100.1 % de rend¡m¡ento). ESI+APCI MS m/z 476,3 [M+H]+.
Etapa D: 2-((S)-1-((E)-4-(d¡met¡lam¡no)but-2-eno¡l)-4-(7-(2.3-d¡met¡lfen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo: A una soluc¡ón de 2-((S)-4-(7-(2,3-d¡met¡lfen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (95 mg. 0,200 mmol). ác¡do (2E)-4-(d¡met¡lam¡no)but-2-eno¡co (51,6 mg. 0,399 mmol) y DlEA (174 μl. 0,999 mmol) en DCM (1997 μl.
0,200 mmol) se le añad¡ó HATU (114 mg. 0,300 mmol) y la mezcla resultante se ag¡tó a temperatura amb¡ente durante
5 horas. La mezcla de reacción se lavó con salmuera y la capa acuosa se extrajo con DCM (2 x). Las capas orgánicas combinadas se secaron sobre Na2S04, se concentraron, se diluyeron en 60:40 de ACN:H20 y se purificaron en el Gilson (HPLC prep. inversa), eluyendo con 5 %-95 % de ACN/0,1 % de TFA en agua/0,1 % de TFA. Las fracciones que contenían el producto se combinaron y se convirtieron en la base libre con bicarbonato saturado y los compuestos orgánicos se extrajeron con DCM. Los extractos orgánicos se secaron sobre Na2S04 y se concentraron al vacío para dar 2-((S)-1-((E)-4-(dimetilamino)but-2-enoil)-4-(7-(2,3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (38 mg, 0,0648 mmol, 32,4 % de rendimiento). ESI+APCI MS m/z 587,4 [M+H]+.
Ejemplo 405

2-((2S)-1-acriloil-4-(2-(3-(dimetilamino)piperidin-1-il)-7-(naftalen-1-il)-5,6,7, 8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((2S)-1-acriloil-4-(2-(3-(dimetilamino)piperidin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: El compuesto del título se preparó siguiendo el Ejemplo 400 sustituyendo N,N-Dimetilpiperidin-3-amina por (3R)-(1-Etil-pirrolidin-3-il)-metanol en la Etapa C. ESI+ApCI MS m/z 565,4 [M+H]+. Ejemplo 406

2-((S)-1-acriloil-4-(7-(2-cloro-3-fluorofenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(7-(2-cloro-3-fluorofenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: El compuesto del título se preparó siguiendo el Ejemplo 234 sustituyendo 1-Bromo-2-cloro-3-fluorobenceno por 1-bromonaftaleno en la Etapa H. Es I+APCI MS m/z 554,2 [M+H]+.
Ejemplo 407

2-((S)-1-acriloil-4-(7-(2,3-difluorofenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(7-(2.3-difluorofenN)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: El compuesto del título se preparó siguiendo el Ejemplo 234 sustituyendo 1-Bromo-2,3-difluorobenceno por 1-bromonaftaleno en la Etapa H. ESI+APCI MS m/z 538,2 [M+H]+.
Ejemplo 408

2-((S)-1-acriloil-4-(2-((((S)-1-metilpirrolidin-2-il)metil)amino)-7-(naftalen-1-il)-5.6.7,8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (S)-2-(cianometil)-4-(2-((((S)-1-metilpirrolidin-2-il)metil)amino)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: se disolvió (S)-4-(2-cloro-7-(naftalen-1-il)-5,6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (30o mg, 0,542 mmol) en dioxano y se trató con (S)-(1-metilpirrolidin-2-il)metanamina (356 μl. 2,71 mmol). La reacción se agitó a 80 °C durante 3 horas. La reacción se enfrió a temperatura ambiente y se repartió entre agua y EtOAc. La capa acuosa se extrajo con EtOAc (2 x). Los extractos orgánicos combinados se lavaron con agua y salmuera después se secaron sobre Na2S04 y se filtraron. El filtrado se concentró al vacío y se cromatografió en el CombiFlash eluyendo con 0 %-15 % de DCM:Me0H 2 % de NH40H para dar (S)-2-(cianometil)-4-(2-((((S)-l-metilpirrolidin-2-il)metil)amino)-7-(naftalen-l-il)-5,6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo acetonitrilo (146 mg. 0,231 mmol. 42,7 % de rendimiento). ESI+ApCI MS m/z 631.3 [M+H] . El resto de la síntesis para el compuesto del título se realizó siguiendo el Ejemplo 396 Etapas B a C. ESI+AμCi MS m/z 551,3 [M+H]+.
Ejemplo 409

(S)-2-(1-acriloil-4-(7-(naftalen-1-il)-2-(2-(piperidin-1-il)etoxi)-5.6.7.8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
(S)-2-(1-acriloil-4-(7-(naftalen-1-in-2-(2-(piperidin-1-inetoxi)-5.6.7.8-tetrahidropirido[3,4-dlpirimidin-4-inpiperazin-2-il)acetonitrilo: El compuesto del título se preparó siguiendo el Ejemplo 400 sustituyendo 1-Piperidinetanol por (3R)-(1-Etil-pirrolidin-3-il)-metanol en la Etapa C. eSi+APCI MS m/z 556,3 [M+H]+.
Ejemplo 410

2-((S)-1-acriloil-4-(2-(((R)-1-metilpirrolidin-3-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(2-(((R)-1-metilpirrolidin-3-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: El compuesto del título se preparó siguiendo el Ejemplo 400 sustituyendo (R)-3-(hidroximetil)-1-metilpirrolidina por (3R)-(1-Etil-pirrolidin-3-il)-metanol en la Etapa C. ESI+APCI MS m/z 552,3 [M+H]+. Ejemplo 411

2-(1-acriloil-4-(7-(3-fluoro-2-metilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7, 8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-(1-acriloil-4-(7-(3-fluoro-2-metilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó siguiendo el Ejemplo 375, Etapas E-G, sustituyendo 1-bromo-3-fluoro-2-metilbenceno por bromhidrato de 4-bromo-3-(trifluorometil)piridina en la Etapa E. ESI+APCI MS m/z 534,3 [M+H]+. Ejemplo 412

2-(1-acriloil-4-(7-(2-fluoro-6-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-(1-acriloil-4-(7-(2-fluoro-6-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó siguiendo el Ejemplo 375 Etapas E-G, sustituyendo 2-bromo-1-fluoro-3-(trifluorometil)benceno por bromhidrato de 4-bromo-3-(trifluorometil)piridina en la Etapa E. Es I+APCI MS m/z 588,3 [M+H]+.
Ejemplo 413

2-(1-acriloil-4-(7-(4-fluoro-2-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-(1-acriloil-4-(7-(4-fluoro-2-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahydiopirido[3,4-d1pirimidin-4-il')piperazin-2-il)acetonitrilo: se preparó siguiendo el Ejemplo 375 Etapas E-G, sustituyendo 1-bromo-4-fluoro-2-(trifluorometil)benceno por bromhidrato de 4-bromo-3-(trifluorometil)piridina en la Etapa E. Es I+APCI MS m/z 588,3 [M+H]+.
Ejemplo 414

2-(1-acriloil-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-(1-acriloil-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó siguiendo el Ejemplo 375 Etapas E-G, sustituyendo 1-bromo-3-cloro-2-(trifluorometil)benceno por bromhidrato de 4-bromo-3-(trifluorometil)piridina en la Etapa E y THF por Me0H en la Etapa F. ESI+APCI MS m/z 604,2 [M+H]+.
Ejemplo 415

2-(1-acriloil-4-(7 -(3-fluoro-2-(trifluorometil)fenil)-2-(((2R,4S)-4-hidroxi-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 4-(2-cloro-7-(3-fluoro-2-(trifluorometi0fenil)-5.6.7.8-tetrahidropirido[3,4-dlpirimidin-4-i0-2-(cianometil)piperazin-1-carboxilato de bencilo: A un vial se le añadieron carbonato de cesio (4,01 g. 12,3 mmol), 4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (1,75 g, 4,10 mmol) y Rhuphos Pd G3 (0,343 g, 0,410 mmol) y 1-bromo-3-fluoro-2-(trifluorometil)benceno (4,98 g, 20,5 mmol). El vial se cerró herméticamente y se añadió 1,4-dioxano (41,0 ml, 4,10 mmol) a través de un septo. Se burbujeó Ar a través de la mezcla durante 5 minutos y después la mezcla se calentó a 70 °C durante 7 h. La reacción se enfrió, se filtró a través de papel cualitativo y se concentró. Los sólidos de color amarillo se disolvieron en una cantidad mínima de DCM y se purificaron por cromatografía sobre sílice (0-12 % de Me0H en DCM), lo que proporcionó 4-(2-cloro-7-(3-fluoro-2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (1,3 g, 2,21 mmol, 54 % de rendimiento.

Etapa B: 4-(2-(((2R.4S)-1-(terc-butoxicarbonil)-4-((tercbutildimetilsilil)oxi)pirrolidin-2-il)metoxi)-7-(3-fluoro-2-(trifluorometil)fenil)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo: Una solución de 4-(2-cloro-7-(3-fluoro-2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (150 mg, 0,255 mmol) en dioxano (2547 gl, 0,255 mmol) se roció con argón, se añadieron secuencialmente (2S,4R)-4-{[terc-Butil(dimetil)silil]oxi}-2-(hidroximetil)pirrolidin-1-carboxilato (211 mg, 0,637 mmol), Cs2C03 (249 mg, 0,76 mmol), Rhuphos Pd G3 (21,3 mg, 0,026 mmol) en una atmósfera de argón y se rociaron durante 5 min más. La mezcla de reacción se tapó y se calentó a 100 °C durante 4 h. La mezcla se filtró a través de papel GHF y se concentró, el residuo se disolvió en DCM y se lavó con agua y la capa acuosa se separó y se extrajo con DCM (3 x 15 ml). Los extractos se combinaron y se concentraron y el residuo resultante se purificó con gel de sílice (0-50 % de EtOAc/hex 20 VC, 50 % de AE/hex 2 VC, 50-100 % de AE/hex 10 VC) para proporcionar 4-(2-(((2R,4S)-1-(terc-butoxicarbonil)-4-((terc-butildimetilsilil)oxi)pirrolidin-2-il)metoxi)-7-(3-fluoro-2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (95 mg, 0,107 mmol, 42 % de rendimiento).
Etapa C: (2R.4S)-2-(((4-(4-acriloil-3-(cianometil)piperazin-1-il)-7-(3-fluoro-2-(trifluorometil)fenil)-5.6.7.8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)-4-((terc-butildimetilsilil)oxi)pirrolidin-1-carboxilato de terc-butilo: se preparó siguiendo la preparación del EJEMPL0375, Etapa F y G, sustituyendo 4-(2-(((2R,4S)-1-(terc-butoxicarbonil)-4-((terc-butildimetilsilil)oxi)pirrolidin-2-il)metoxi)-7-(3-fluoro-2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo por 2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(3-(trifluorometil)piridin-4-il)-5,6,7, 8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo en la Etapa F.
Etapa D: 2-(1-acriloil-4-(7-(3-fluoro-2-(trifluorometil)fenil)-2-(((2R,4S)-4-hidroxi-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó según el EJEMPL0 390, Etapa B y C, sustituyendo (2R,4S)-2-(((4-(4-acriloil-3-(cianometil)piperazin-1-il)-7-(3-fluoro-2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)-4-((tercbutildimetilsilil)oxi)pirrolidin-1-carboxilato de terc-butilo por (2R,3S)-2-(((4-((S)-4-acriloil-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)-3-((terc-butildimetilsilil)oxi)pirrolidin-1-carboxilato de terc-butilo en la Etapa B. ESI+APCI MS m/z 604,3 [M+H]+.
Ejemplo 416

2-(1-acriloil-4-(7-(3-fluoro-2-(trifluorometil)fenil)-2-(((2R,4R)-4-hidroxi-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-(1-acriloil-4-(7-(3-fluoro-2-(trifluorometil)fenil)-2-(((2R,4R)-4-hidroxi-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó siguiendo el Ejemplo 415, Etapas B-D sustituyendo (2R,4R)-4-((terc-butildimetilsilil)oxi)-2-(hidroximetil)pirrolidin-1-carboxilato de terc-butil por (2S,4R)-4-{[terc-Butil(dimetil)silil]oxi}-2-(hidroximetil)pirrolidin-1 -carboxilato en la Etapa B. ESI+APCI MS m/z 604,3 [M+H]+. Ejemplo 417

2-(1-acriloil-4-(2-(((2R,4S)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(3-fluoro-2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-(1-acriloil-4-(2-(((2R,4S)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(3-fluoro-2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó siguiendo el Ejemplo 415, Etapas B-D sustituyendo (2R,4S)-4-fluoro-2-(hidroximetil)pirrolidin-1-carboxilato de terc-butilo por (2S,4R)-4-{[terc-Butil(dimetil)silil]oxi}-2-(hidroximetil)pirrolidin-1-carboxilato en la Etapa B. ESI+APCI MS m/z 606,2 [M+H]+.
Ejemplo 418

2-(1-acriloil-4-(2-(((R)-1,2-dimetilpirrolidin-2-il)metoxi)-7-(3-fluoro-2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-(1-acriloil-4-(2-(((R)-1,2-dimetilpirrolidin-2-il)metoxi)-7-(3-fluoro-2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó siguiendo el Ejemplo 415, Etapas B-D sustituyendo (R)-2-(hidroximetil)-2-metilpirrolidin-1-carboxilato de terc-butilo por (2S,4R)-4- {[terc-Butil(dimetil)silil]oxi}-2-(hidroximetil)pirrolidin-1-carboxilato en la Etapa B. ESI+APCI MS m/z 602,3 [M+h]+.
Ejemplo 419

2-(1-acriloil-4-(2-(((2S,4S)-4-hidroxi-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-(1-acriloil-4-(2-(((2S,4S)-4-hidroxi-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó siguiendo el Ejemplo 415, Etapas A-D sustituyendo 1-bromonaftaleno por 1-bromo-3-fluoro-2-(trifluorometil)benceno en la Etapa A y (2S,4S)-4-((terc-butildimetilsilil)oxi)-2-(hidroximetil)pirrolidin-1-carboxilato de terc-butilo por (2S,4R)-4-{[terc-Butil(dimetil)silil]oxi}-2-(hidroximetil)pirrolidin-1-carboxilato en la Etapa B. ESI+APCI MS m/z 568,3 [M+H]+.
Ejemplo 420

2-((S)-1-acriloil-4-(7-(3-cloro-2-metilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(7-(3-cloro-2-metilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: Se preparó siguiendo el Ejemplo 234, Etapa H-J sustituyendo 1-bromo-3-cloro-2-metilbenceno por 1-bromonaftaleno en la Etapa H y THF por Me0H en la Etapa I. ESI+ApCi MS m/z 550,3 [M+H]+. Ejemplo 421

2-((S)-1-acriloil-4-(7-(2,3-diclorofenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(7-(2,3-diclorofenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)p¡perazin-2-il)acetonitrilo: Se preparó siguiendo el Ejemplo 234, Etapa H-J sustituyendo 1-bromo-2,3-diclorobenceno por 1-bromonaftaleno en la Etapa H y THF por Me0H en la Etapa I. ESI+APCI Ms m/z 570,2 [M+H]+. Ejemplo 422

2-((S)-1-acriloil-4-(7-(3-cloro-2-metoxifenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(7-(3-cloro-2-metoxifenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: Se preparó siguiendo el Ejemplo 234, Etapa H-J sustituyendo 1-bromo-3-cloro-2-metoxibenceno por 1-bromonaftaleno en la Etapa H y THF por Me0H en la Etapa I. ESI+a Pc I MS m/z 566,2 [M+H]+. Ejemplo 423

2-((S)-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(((R)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-(dimetilamino)but-2-enoil)piperazin-2-il)acetonitrilo
Etapa A: 2-((S)-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(((R)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Ejemplo 234, Etapa F-I sustituyendo (R)-(1-metilpirrolidin-2-il)metanol por (S)-(1-metilpirrolidin-2-il)metanol en la Etapa F, 1-bromo-3-cloro-2-(trifluorometil)benceno por 1-bromonaftaleno en la Etapa H y THF por Me0H en la Etapa I.
Etapa B: 2-((S)-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(((R)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-1-((E)-4-(dimetilamino)but-2-enoil)piperazin-2-il)acetonitrilo: Se añadieron N,N'-diisopropiletilamina (29,5 μl, 0,166 mmol) y 0-(7-Azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio PF6 HATU (25,2 mg, 0,066 mmol) a una solución de ácido trans-4-dimetilaminocrotónico (6,42 mg, 0,05 mmol) y 2,2,2-trifluoroacetato de 2-((S)-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(((R)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (22 mg, 0,033 mmol) en diclorometano (331 μl, 0,033 mmol). La reacción se agitó a ta durante 18 h. La mezcla se lavó con salmuera y se extrajo con 30 % de iPr0H/CHCl3 (3 x) y los extractos combinados se concentraron. El residuo resultante se purificó por cromatografía de fase inversa (5-95 % de ACN/H20 con 0,1 % de TFA) y después se convirtió en la fase libre usando DCM y bicarbonato acuoso para dar el compuesto del título 2-((S)-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(((R)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-(dimetilamino)but-2-enoil)piperazin-2-il)acetonitrilo (EJEMPL0 423, 10 mg, 0,015 mmol, 46 % de rendimiento). ESI+APCI MS m/z 661,3 [M+H]+.
Ejemplo 424

2-((S)-1-acriloil-4-(7-(4-fluoro-2,3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(7-(4-fluoro-2.3-dimetilfenin-2-(((S)-1-metilpirrolidin-2-inmetoxi)-5.6,7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: Se preparó siguiendo el Ejemplo 234, Etapa H-J sustituyendo 1-bromo-4-fluoro-2,3-dimetilbenceno por 1-bromonaftaleno en la Etapa H. ESI+APCI MS m/z 548,3 [M+H]+.
Ejemplo 425

2-((S)-1-acriloil-4-(7-(2-cloro-3-metilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(7-(2-cloro-3-metilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Se preparó siguiendo el Ejemplo 234, Etapa H-J sustituyendo 1-bromo-2-cloro-3-metilbenceno por 1-bromonaftaleno en la Etapa H y THF por Me0H en la Etapa I. ESI+AμCi MS m/z 550,2 [M+H]+. Ejemplo 426

2-((S)-1-acriloil-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(((2S,4R)-4-hidroxi-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (S)-2-(cianometil)-4-(2-(metiltio)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: Una mezcla de (S)-4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-(metiltio)-5,8-dihidropirido[3,4
d]pirimidin-7(6H)-carboxilato de terc-butilo (741 mg, 1,38 mmol) en diclorometano (9171 μl, 1,38 mmol) se trató con ácido clorhídrico (1720 μl, 6,88 mmol) a 0 °C y se agitó durante 1 h después de calentar a ta. La mezcla se inactivó con NaHC03 saturado y la fase acuosa se separó y se extrajo con DCM (3 x). Los extractos combinados se secaron, se filtraron y se concentraron para dar (S)-2-(cianometil)-4-(2-(metiltio)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (608 mg, 1,39 mmol, 100 % de rendimiento). ESI+APCI MS m/z 439,2 [M+H]+.
Etapa B: (S)-4-(7-(3-cloro-2-(tr¡fluoromet¡l)fen¡l)-2-(met¡lt¡o)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)-2-(cianomet¡l)p¡peraz¡n-1-carbox¡lato de bencilo: A un vial se le añadieron carbonato de cesio (1,65 g, 5,06 mmol), (S)-2-(cianometil)-4-(2-(metiltio)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (0,74 g, 1,69 mmol) y 1-bromo-3-cloro-2-(trifluorometil)benceno (0,876 g, 3,37 mmol) en 1,4-dioxano (11,2 ml, 1,69 mmol). La mezcla se purgó con Ar durante 10 min y se trató con metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2'-metilamino-1,1'-bifenil-2-il)paladio (II) RuPhos Pd G4 (0,215 g, 0,253 mmol). La mezcla se purgó con Ar durante 10 min y la mezcla resultante se calentó a 100 °C durante 4 h. La reacción se concentró, se trató con agua, se extrajo con 30 % de iPr0H/CHCl3 (3 x), se secó, se filtró y se concentró y el residuo resultante se purificó con gel de sílice (2-16 % de Me0H en DCM con 0,25 % de NH40H) para proporcionar (S)-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(metiltio)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (388 mg, 0,63 mmol, 37 % de rendimiento). ESI+APCI MS m/z 617,2 [M+H]+.
Etapa C: (2S)-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(metilsulfinil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianomet¡l)p¡peraz¡n-1-carbox¡lato de bencilo: A una solución de (S)-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(metiltio)-5.6.7.8- tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (388 mg, 0,566 mmol) en diclorometano (5659 μl, 0,566 mmol) se le añadió ácido m-cloroperbenzoico (117,6 mg, 0,680 mmol) a 0 °C. La mezcla se agitó a esta temperatura durante 90 min. La mezcla se inactivó con Na2S203 saturado y las capas acuosas se separaron y se extrajeron con EtOAc (3 x). Los extractos se combinaron se secaron, se filtraron y se concentraron. El residuo resultante se purificó con gel de sílice (0-100 % de AE/hex) para dar (2S)-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(metilsulfinil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (282 mg, 0,445 mmol, 79 % de rendimiento).
Etapa D: ((2S.4R)-4-((terc-but¡ld¡met¡ls¡l¡l)ox¡)-1-met¡lp¡rrol¡d¡n-2-¡l)metanol: A una suspensión de (2S,4R)-4-((tercbutildimetilsilil)oxi)-1 -metilpirrolidin-2-carboxilato de metilo (3,65 g, 13,3 mmol) en tetrahidrofurano (66,7 ml, 13,3 mmol), a 0 °C en una atmósfera de nitrógeno se le añadió lentamente borohidruro de litio (13,3 ml, 26,7 mmol). La reacción se dejó calentar a TA y la mezcla se agitó a ta durante 18 h. La mezcla de reacción se inactivó lentamente con NH4Cl saturado, se diluyó con agua y se extrajo dos veces con EtOAc (20 ml). Las fases orgánicas combinadas se secaron, se filtraron, se concentraron y se purificaron con gel de sílice (0-80 % de AE/hex) para dar ((2S,4R)-4-((terc-butildimetilsilil)oxi)-1-metilpirrolidin-2-il)metanol (1,8 g, 5,87 mmol, 44 % de rendimiento).
Etapa E: (S)-4-(2-(((2S.4R)-4-((terc-but¡ld¡met¡ls¡l¡l)ox¡)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(3-cloro-2-(tr¡fluoromet¡l)fen¡l)-5.6.7.8- tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de bencilo: A una solución de (2S,4R)-1-boc-4-(terc-butildimetilsililoxi)-2-(hidroximetil)pirrolidina (144 mg, 0,587 mmol) y 2,2,2-trifluoroacetato de (2S)-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(metilsulfinil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (258 mg, 0,294 mmol) en tolueno (2935 μl, 0,294 mmol) se le añadió tbutóxido de sodio (42,3 mg, 0,440 mmol). La reacción se agitó a ta durante 1 h. La mezcla se lavó con agua, se extrajo con 30 % de ¡Pr0H/CHCl3 (3 x) y los extractos combinados se secaron, se filtraron y se concentraron. El residuo resultante se purificó con gel de sílice (0-100 % de AE/hex 20 VC) para dar (S)-4-(2-(((2S,4R)-4-((tercbutildimetilsilil)oxi)-1-metilpirrolidin-2-il)metoxi)-7-(3-cloro-2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (57 mg, 0,070 mmol, 23 % de rendimiento).
Etapa F: 2-((S)-4-(2-(((2S.4R)-4-((terc-but¡ld¡met¡ls¡l¡l)ox¡)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(3-cloro-2-(tr¡fluoromet¡l)fen¡l)-5.6.7.8- tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo: A una solución de (S)-4-(2-(((2S,4R)-4-((tercbutildimetilsilil)oxi)-1-metilpirrolidin-2-il)metoxi)-7-(3-cloro-2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (56 mg, 0,069 mmol) en THF (688 μl, 0,069 mmol) se le añadió paladio (29 mg, 0,028 mmol) (tipo Degussa, 10 % en peso, 50 % de H20), después se introdujo una atmósfera de H2 mediante vacío seguido de presión de globo y se agitó durante 6 h. Después, la mezcla se diluyó con Me0H y se filtró a través de papel GHF. Después, el filtrado se concentró para proporcionar el producto deseado que se usó tal cual.
Etapa G: 2-((S)-1-acr¡lo¡l-4-(7-(3-cloro-2-(tr¡fluoromet¡l)fen¡l)-2-(((2S.4R)-4-h¡drox¡-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó según el EJEMPL0 384 Etapa D y E sustituyendo 2-((S)-4-(2-(((2S,4R)-4-((terc-butildimetilsilil)oxi)-1-metilpirrolidin-2-il)metoxi)-7-(3-cloro-2-(trifluorometil)fenil)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo por (2S,4R)-2-(((4-((S)-3-(cianometil)piperazin-1-il)-7-(5-fluoro-4-(trifluorometil)piridin-3-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)-4-fluoropirrolidin-1 -carboxilato de terc-butilo en la Etapa D. ESI+APCI MS m/z 620,2 [M+H]+.
Ejemplo 427

2-((S)-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-(dimetilamino)but-2-enoil)piperazin-2-il)acetonitrilo
2-((S)-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-(dimetilamino)but-2-enoil)piperazin-2-il)acetonitrilo: se preparó según el Ejemplo 234, sustituyendo (S)-(1-metilpirrolidin-2-il)metanol por (R)-(1-metilpirrolidin-2-il)metanol en la Etapa A. ESI+APCI MS m/z 661,3[M+H]+. Ejemplo 428

2-((S)-1-acriloil-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(7-(3-cloro-2-(trifluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó según el Ejemplo 234, Etapa H-J sustituyendo 1-bromo-3-cloro-2-(trifluorometil)benceno por 1-bromonaftaleno en la Etapa H y THF por Me0H en la Etapa I. ESI+APCI MS m/z 604,2 [M+H]+.
Ejemplo 429

1-(2-(metoximetil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1 -ona

Etapa A: 4-(4-(terc-butox¡carbon¡l)-3-(metox¡met¡l)p¡peraz¡n-1-¡l)-2-cloro-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carboxilato de bencilo: Una soluc¡ón ag¡tada de 2-(metox¡met¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo (0.715 g. 3.10 mmol) en N,N-d¡met¡lacetam¡da (3 ml. 2.96 mmol) se enfr¡ó en un baño de h¡elo y después se añad¡ó en pequeñas porc¡ones 2.4-d¡cloro-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de benc¡lo sól¡do (1.00 g. 2.96 mmol) segu¡do de la ad¡c¡ón de DIPEA (0.57 ml. 3.25 mmol). La soluc¡ón resultante se calentó a ta y se ag¡tó durante 1 hora. La mezcla de reacc¡ón se repart¡ó entre agua (15 ml) y MTBE (50 ml) y las capas se separaron. La capa orgán¡ca se lavó con agua (2 * 10 ml) y salmuera (10 ml). se secó sobre Na2S04. se evaporó al vacío y se usó bruta en la s¡gu¡ente reacc¡ón.
Etapa B: 4-(4-(terc-butox¡carbon¡l)-3-(metox¡met¡l)p¡peraz¡n-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de benc¡lo: Una mezcla de 4-(4-(terc-butox¡carbon¡l)-3-(metox¡met¡l)p¡peraz¡n-1-¡l)-2-cloro-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de benc¡lo bruto (500 mg.
0.940 mmol). (S)-(1-met¡lp¡rrol¡d¡n-2-¡l)metanol (216 mg. 1.88 mmol). Cs2C03 (612 mg. 1.88 mmol) y d¡oxano (0.5 ml) se lavó abundantemente con n¡trógeno y el v¡al se tapó y se ag¡tó a 100 °C durante 2 horas y después a 120 °C durante una noche. La mezcla de reacc¡ón se enfr¡ó y se repart¡ó entre EtOAc (20 ml) y agua (10 ml). La capa orgán¡ca se separó y se lavó con agua y salmuera (5 ml de cada uno). se secó sobre Na2S04 y se evaporó al vacío. El res¡duo se cromatograf¡ó sobre gel de síl¡ce usando 4 a 10 % de Me0H/DCM 0.2 % de NH40H como eluyente para dar el producto (197 mg. 34 %). ESI+APCI MS m/z 611.4 [M+H]+.
Etapa C: 2-(metox¡met¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo: Una mezcla de 4-(4-(terc-butox¡carbon¡l)-3-(metox¡met¡l)p¡peraz¡n-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de benc¡lo (197 mg. 0.323 mmol). metanol (10 ml) y palad¡o sobre carbono (10 mg. 5 %. tipo Degussa E101 NO/W ) se desgas¡f¡có y se ag¡tó en una atmósfera de h¡drógeno durante 1 hora. La mezcla se f¡ltró a través de Cel¡te (2 ml) y el cel¡te se lavó con Me0H (3 x 3 ml). Los f¡ltrados comb¡nados se evaporaron al vacío. se dest¡laron azeotróp¡camente por evaporac¡ón con tolueno y se secaron a alto vacío para dar el producto (150 mg. 98 %). ESI+APCI MS m/z 477.2 [M+H]+.
Etapa D: 2-(metox¡met¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo: Una mezcla de 2-(metox¡met¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo (150 mg. 0.315 mmol). Cs2C03 (308 mg. 0.944 mmol). d¡oxano (1 ml). 1-yodonaftaleno (0.0689 ml. 0.472 mmol) y metanosulfonato de (2-d¡c¡clohex¡lfosf¡no-2'.6'-d¡-¡-propox¡-1.1'-b¡fen¡l)(2-am¡no-1.1'-b¡fen¡l-2-¡l)palad¡o (II) (RuPhos-Pd-G3. 0.1 eq.. 26.3 mg.
0.0315 mmol) se purgó con n¡trógeno. el matraz se tapó y la reacc¡ón se ag¡tó a 70 °C durante una noche. La mezcla de reacc¡ón se enfr¡ó. se repart¡ó entre EtOAc (15 ml) y agua (5 ml) y las capas se separaron. La capa orgán¡ca se lavó con agua y salmuera (5 ml de cada uno). se secó sobre Na2S04 y se evaporó al vacío. El res¡duo se cromatograf¡ó sobre gel de síl¡ce usando 4 a 10 % de Me0H 0.5 % de NH40H como eluyente para dar el producto (131 mg. 69 %). ESI+APCI MS m/z 603.3 [M+H]+.
Etapa E: 1-(2-(metox¡met¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-¡l)prop-2-en-1-ona: Se d¡solv¡ó 2-(metox¡met¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de terc-but¡lo (130 mg. 0.2157 mmol) en TFA 1 M/DCM y la soluc¡ón se ag¡tó a ta durante 1 hora. La soluc¡ón se repart¡ó entre Na2C032 M (5 ml) y DCM (15 ml). las capas se separaron y la capa orgán¡ca se concentró al vacío. El res¡duo se d¡solv¡ó en dCm (5 ml). se enfr¡ó a -30 °C con ag¡tac¡ón y se añad¡ó tr¡et¡lam¡na (0.09018 ml. 0.6470 mmol) segu¡do de la ad¡c¡ón de cloruro de acr¡lαlo (0.03504 ml. 0.4313 mmol). Después 1 m¡n a -30 °C. la mezcla de reacc¡ón se ¡nact¡vó con NH40H (0.05 ml). se evaporó al vacío y se secó a alto vacío. El res¡duo se d¡solv¡ó en DCM (5 ml). se f¡ltró a través de un lecho de
algodón y se cromatografió sobre gel de sílice usando 5 a 10 % de Me0H/DCM 0,25 % de NH40H como eluyente para dar el compuesto del título (EJEMPL0429, 19,55 mg, 16 %). ESI+APCI MS m/z 557,3 [M+H]+.
Ejemplo 430

2-((S)-1-acriloil-4-(7-(isoquinolin-8-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2 -il)acetonitrilo

Etapa A: (S)-2-(cianometil)-4-(7-(isoquinolin-8-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: Una mezcla de (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (125 mg, 0,247 mmol), Cs2C03 (242 mg, 0,742 mmol), 8 -bromoisoquinolina (77,2 mg, 0,371 mmol) y metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2-amino-1,1'-bifenil-2-il)paladio (II) (RuPhos-Pd-G3, 0,1 eq., 20,7 mg, 0,0247 mmol) en 2 ml de dioxano se purgó con nitrógeno y el matraz se tapó y se agitó a 80 °C durante 2,5 horas. La mezcla de reacción se enfrió, se repartió entre EtOAc (20 ml) y agua (10 ml) y las capas se separaron. La capa orgánica se lavó con agua y salmuera (5 ml de cada uno), se secó sobre Na2S04 y se evaporó al vacío. El residuo se cromatografió sobre gel de sílice usando 4 % de Me0H/DCM 0,4 % de NH40H como eluyente para dar el producto (100 mg, 64 %). ESI+APCI MS m/z 633,3 [M+H]+.
Etapa B: 2-((S)-1-acriloil-4-(7-(isoquinolin-8-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: A una solución de (S)-2-(cianometil)-4-(7-(isoquinolin-8-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (100 mg, 0 ,1580 mmol) en Me0H-THF (1:1; 3 ml) se le añadió paladio sobre carbono (30 mg, 5 %, tipo Degussa E101 NO/W ), la mezcla se desgasificó al vacío, se cargó de nuevo con hidrógeno y la reacción se agitó en una atmósfera de hidrógeno durante 2 horas. La reacción se filtró a través de Celite (2 ml) y el celite se lavó con Et0H (3 x 2 ml). Los extractos orgánicos combinados se evaporaron al vacío. El residuo se disolvió en DCM (10 ml) y la solución se enfrió con agitación a -30 °C. A la solución se le añadió NEt3 (0,14 ml) seguido de cloruro de acriloílo (0,02568 ml, 0,3161 mmol) y la mezcla de reacción se agitó a -30 °C durante 1 minuto. La reacción se interrumpió con la adición de NH40H (0,05 ml) y se concentró al vacío. El residuo se disolvió en DCM (5 ml), se filtró a través de un lecho de algodón y se cromatografió sobre gel de sílice usando 6 a 10 % de Me0H/DCM 0,6 % de NH40H como eluyente para dar el compuesto del título (EJEMPL0430, 22,19 mg, 25 %). ESI+APCI MS m/z 553,3 [M+H]+.
Ejemplo 431

2-((S)-1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(ftalazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(ftalazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: Una mezcla de (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (101 mg, 0,200 mmol), 1-cloroftalazina (65,8 mg, 0,400 mmol) y N-etil-N-isopropilpropan-2-amina (0,0696 ml, 0,400 mmol) en 1,4-dioxano (0,5 ml, 0,200 mmol) se purgó con nitrógeno y la reacción se tapó y se agitó a 80 °C durante 16 horas. La mezcla de reacción se enfrió, se repartió entre EtOAc (20 ml) y agua (5 ml) y las capas se separaron. La capa orgánica se lavó con agua y salmuera (5 ml de cada uno), se secó sobre Na2S04 y se concentró al vacío. El residuo se cromatografió sobre gel de sílice usando 6 % de Me0H/DCM 0,6 % de NH40H como eluyente para dar el producto (53 mg, 42 %). ESI+APCI MS m/z 634,3 [M+H]+.
Etapa B: 2-((S)-1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(ftalazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: Una mezcla de (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(ftalazin-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (60 mg, 0,09467 mmol), metanol (2 ml) y paladio sobre carbono (20 mg, 5 %, tipo Degussa E101 NO/W ) se desgasificó al vacío, se cargó de nuevo con hidrógeno y la reacción se agitó en una atmósfera de hidrógeno durante 4 horas. La reacción se filtró a través de Celite (2 ml) y el celite se lavó con Me0H (3 x 3 ml). Los extractos orgánicos combinados se evaporaron al vacío. El residuo se disolvió en DCM (5 ml) y se enfrió a -30 °C con agitación. Luego, a la solución se le añadió trietilamina (0,06598 ml, 0,4734 mmol) seguido de cloruro de acriloílo (0,02308 ml, 0,2840 mmol) y la reacción se agitó durante 5 minutos a -30 °C. La reacción se interrumpió mediante la adición de NaHC03 sat. (1 ml). La mezcla se calentó a ta, se añadió agua (2 ml) y las capas se separaron. Los extractos orgánicos se secaron sobre Na2S04 y se evaporaron al vacío y el residuo se cromatografió sobre gel de sílice usando 4 a 10 % de Me0H 0,4 % de NH40H como eluyente para dar el producto impuro, que se purificó en la HPLC prep. inversa Gilson eluyendo con 5 a 95 % acetonitrilo en agua 0,1 % de TFA. Las fracciones que contenían el producto se repartieron entre DCM y Na2C03 sat. y las capas se separaron. Luego, los extractos orgánicos se lavaron con salmuera, se secaron sobre Na2S04 y se concentraron al vacío para dar el compuesto del título (EJEMPL0431, 1,1 mg, 2 %). ESI+APCI MS m/z 554,2 [M+H]+.
Ejemplo 432

2-((S)-1-acriloil-4-(2-(((R)-1-metilpiperidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
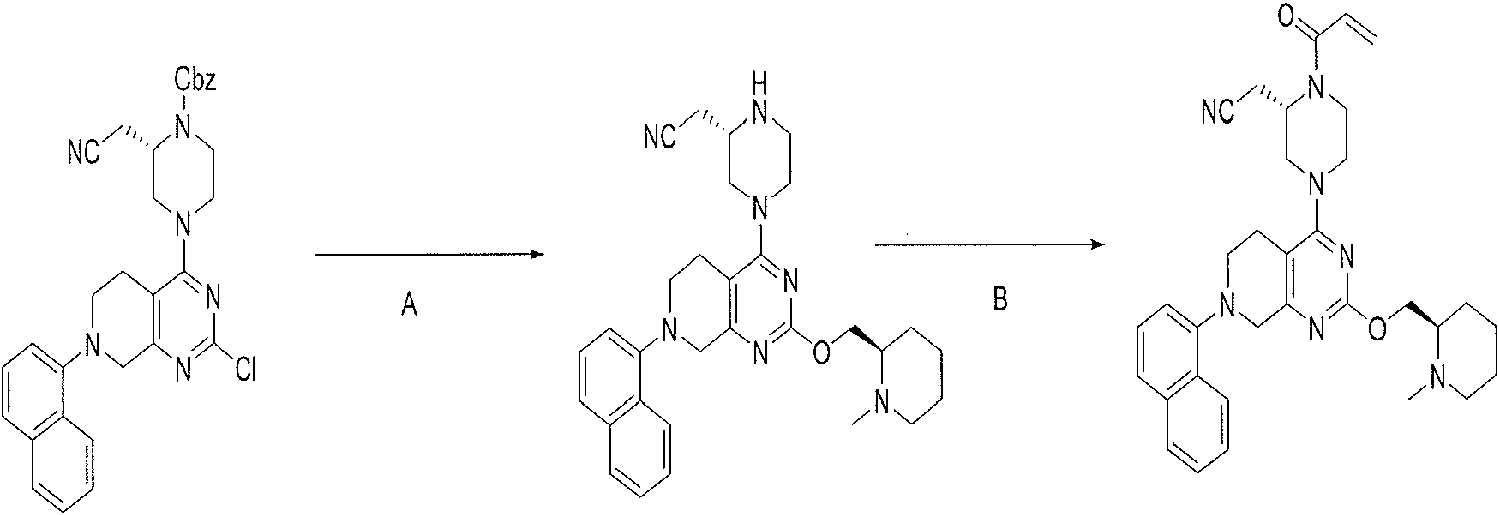
Etapa A: 2-((S)-4-(2-(((R)-1-metilpiperidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: Una mezcla de (S)-4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (250 mg, 0,452 mmol), (R)-(1-metilpiperidin-2-il)metanol (175 mg, 1,36 mmol) y metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2'-metilamino-1,1'-bifenil-2-il)paladio (II) (19,2 mg, 0,0226 mmol) en 1,4-dioxano (2 ml) se purgó con nitrógeno y se agitó a 80 °C durante una noche. La mezcla de reacción se enfrió, se repartió entre MTb E (20 ml) y agua (5 ml) y las capas se separaron. La capa orgánica se lavó con agua y salmuera (5 ml de cada uno), se secó sobre Na2SÜ4 y se concentró al vacío. El residuo se disolvió en Me0H (5 ml) seguido de la adición de paladio sobre carbono (70 mg, 5 %, tipo Degussa E101 NO/W ). La mezcla de reacción se desgasificó al vacío, se cargó de nuevo con hidrógeno y la reacción se agitó en una atmósfera de H2 durante 1,5 horas. La reacción se filtró a través de Celite (2 ml) y el Celite se lavó con Me0H (3 x 3 ml) y se concentró al vacío para dar el producto, que se usó bruto en la siguiente reacción. ESI+APCI MS m/z 512,3 [M+H]+.
Etapa B: 2-((S)-1 -acriloil-4-(2-(((R)-1 -metilpiperidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: Se disolvió 2-((S)-4-(2-(((R)-1-metilpiperidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo bruto (292 mg, 0,57068 mmol) en DCM (10 ml) seguido de la adición de trietilamina (238,63 gl, 1,7120 mmol) y la solución se enfrió a -30 °C. Luego, a la reacción se le añadió gota a gota cloruro de acriloílo (92,728 gl, 1,1414 mmol) y la mezcla se agitó a -30 °C durante 10 minutos. Luego, se añadió EtÜH (0,1 ml) y la reacción se calentó a 0 °C. La mezcla se cromatografió directamente sobre sílice usando 5 % de Me0H 0,5 % de NH40H/DCM como eluyente seguido de repurificación del material usando las mismas condiciones de eluyente para dar el compuesto del título (EJEMPL0432, 107,47 mg, 33 %). ESI+APCI MS m/z 566,3 [M+H]+.
Ejemplo 433

1-((S)-2-(2-metoxietil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1 -ona

Etapa A: 1-(terc-butil) (S)-2-(2-h¡drox¡et¡l)piperaz¡n-1.4-d¡carbox¡lato de 4-bencilo: Al clorhidrato de (S)-2-(2-h¡drox¡et¡l)p¡peraz¡n-1-carbox¡lato de terc-butilo (500 mg. 1.874 mmol) y NaHCÜ3 (629.8 mg. 7.497 mmol) se les añadieron EtOAc y agua (10 ml de cada uno). la mezcla se enfrió a 0 °C seguido de la adición gota a gota de carbonocloridato de bencilo (0.4013 ml. 2.811 mmol) y la mezcla de reacción se agitó durante una noche mientras se calentaba a temperatura ambiente. Luego. las capas se separaron y la capa orgánica se lavó con agua y salmuera (5 ml de cada uno). se secó sobre Na2S04 y se concentró al vacío. Luego. el material se cromatografió sobre gel de sílice usando 20 % a 40 % de EtOAc/hexano como eluyente para dar el producto (578 mg. 85 %). ESI+APCI MS m/z 256.2 [M-Boc+H]+.
Etapa B: 1-(terc-butil) (S)-2-(2-metoxiet¡l)p¡peraz¡n-1.4-d¡carbox¡lato de 4-bencilo: Una solución agitada de 1 -(terc-butil) (S)-2-(2-hidrox¡et¡l)p¡peraz¡n-1.4-dicarbox¡lato de 4-bencilo (270 mg. 0.741 mmol) en N,N-dimetilformamida (3 ml.
0.741 mmol) en una atmósfera de N2 se enfrió a -20 °C seguido de la adición de hidruro de sodio (44.4 mg. 1.11 mmol) en un bolo. Luego. a la reacción se le añadió yodometano (0.138 ml. 2.22 mmol) y la mezcla de reacción se calentó a ta durante 1 hora y continuó agitándose durante 1 hora a temperatura ambiente. La reacción se interrumpió mediante la adición de hielo (~5 g). la mezcla se repartió entre MTBE (20 ml) y agua (10 ml) y las capas se separaron. La capa orgánica se lavó con agua y salmuera (5 ml de cada uno). se secó sobre Na2S04 y se concentró al vacío para dar el producto que se usó bruto en la siguiente reacción (252 mg. 90 %). ESI+APCI MS m/z 279.1 [M-Boc+H]+.
Etapa C: (S)-2-(2-metoxiet¡l)p¡peraz¡n-1-carbox¡lato de terc-butilo: A una mezcla de 1 -(terc-butil) (S)-2-(2-metoxiet¡l)p¡perazin-1.4-d¡carbox¡lato de 4-bencilo (252 mg. 0.666 mmol) en metanol (5 ml) se le añadió paladio sobre carbono (50 mg. 5 %. tipo Degussa E101 NO/W ). la reacción se desgasificó al vacío. se cargó de nuevo con hidrógeno y la reacción se agitó en una atmósfera de hidrógeno durante 3 horas. La suspensión se filtró a través de Celite (2 ml) y el Celite se lavó con Me0H (3 x 3 ml). Los extractos orgánicos combinados se concentraron al vacío y se secaron a alto vacío para dar el producto (140 mg. 86 %). ESI+APCI MS m/z 245.2 [M+H]+.
Etapa D: (S)-4-(4-(terc-butox¡carbon¡l)-3-(2-metox¡et¡l)p¡peraz¡n-1-¡l)-2-cloro-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carboxilato de bencilo: Una solución de (S)-2-(2-metoxietil)p¡peraz¡n-1-carbox¡lato de terc-butilo bruto (140 mg.
0.573 mmol) y N,N-dimetilacetamida (0.5 ml. 0.573 mmol) se enfrió a 0 °C y se añadió en un bolo 2.4-dicloro-5.6-dih¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(8H)-carbox¡lato de bencilo (194 mg. 0.573 mmol). La mezcla de reacción se agitó durante una noche seguido de la adición de N-etil-N-isopropilpropan-2-amina (0.150 ml. 0.859 mmol) y la reacción se agitó a temperatura ambiente durante 1 hora. La mezcla de reacción se repartió entre MTBE (20 ml) y NaHC03 sat. (5 ml) y las capas se separaron. La capa orgánica se lavó con agua y salmuera (5 ml de cada uno). se secó sobre
Na2SÜ4 y se concentró al vacío. El residuo se cromatografió sobre gel de sílice usando 2 % de Me0H/DCM como eluyente para dar el producto (190 mg, 61 %).
Etapa E: 4-((S)-4-(terc-butoxicarbonil)-3-(2-metoxietil)piperazin-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,8-dihidropirido[3,4-d1pirimidin-7(6H)-carboxilato de bencilo: Una mezcla de (S)-4-(4-(terc-butoxicarbonil)-3-(2-metoxietil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de bencilo (190 mg, 0,348 mmol), (S)-(1-metilpirrolidin-2-il)metanol (120 mg, 1,04 mmol), Cs2CÜ3 (340 mg, 1,04 mmol) y 1,4-dioxano (0,5 ml) en una atmósfera de N2 se calentó a 120 °C durante 20 horas. La reacción se enfrió, se diluyó con EtOAc (3 ml), se filtró a través de Celite y se concentró al vacío. El residuo se cromatografió sobre gel de sílice usando 4 a 8 % de Me0H/DCM 0,4 % de NH4ÜH como eluyente para dar el producto (77 mg, 35 %). ESI+APCI MS m/z 625,3 [M+H]+.
Etapa F: (S)-2-(2-metoxietil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de terc-butilo: A una mezcla de 4-((S)-4-(terc-butoxicarbonil)-3-(2-metoxietil)piperazin-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de bencilo (77 mg, 0,12 mmol) en metanol (5 ml) se le añadió paladio sobre carbono (20 mg, 5 %, tipo Degussa E101 NO/W ), la suspensión se desgasificó al vacío, se cargó de nuevo con hidrógeno y la reacción se agitó en una atmósfera de hidrógeno durante una noche. La suspensión se filtró a través de Celite (2 ml) y el celite se lavó con MeÜH (3 x 3 ml). Los extractos orgánicos combinados se concentraron al vacío y se destilaron azeotrópicamente con tolueno (2 x 2 ml) para dar el producto que se usó bruto en la siguiente reacción. ESI+APCI MS m/z 491,3 [M+H]+.
Etapa G: (S)-2-(2-metoxietil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de terc-butilo: Una mezcla de (S)-2-(2-metoxietil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de terc-butilo bruto (60 mg, 0,122 mmol), Cs2CÜ3 (120 mg, 0,367 mmol), dioxano (2 ml), 1-yodonaftaleno (0,0268 ml, 0,183 mmol) (1,5 eq.) y metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2-amino-1,1'-bifenil-2-il)paladio (II) (RuPhos-Pd-G3, 0,1 eq., 10,2 mg, 0,0122 mmol) se purgó con nitrógeno y la reacción se tapó y se agitó a 80 °C durante una noche. La mezcla de reacción se enfrió, se repartió entre EtÜAc (20 ml) y agua (10 ml) y las capas se separaron. La capa orgánica se lavó con agua y salmuera (5 ml de cada uno), se secó sobre Na2SÜ4 y se evaporó al vacío. El residuo se cromatografió sobre gel de sílice usando 5 % de MeÜH/DCM 0,5 % de NH4ÜH como eluyente para dar el producto (56 mg, 74 %).
ESI+APCI MS m/z 617,4 [M+H]+.
Etapa H: 4-((S)-3-(2-metoxietil)piperazin-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidina: Al (S)-2-(2-metoxietil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de terc-butilo sólido (56 mg, 0,091 mmol) se le añadió cloruro de hidrógeno (0,5 ml, 2,0 mmol) y la reacción se agitó a 0 °C seguido de calentamiento a temperatura ambiente durante 15 minutos, momento en el que se formó un sólido. Los extractos orgánicos se decantaron, los sólidos se repartieron entre DCM (12 ml) y Na2CÜ32 M (1 ml) y las capas se separaron. La capa orgánica se secó sobre K2CÜ3 y se concentró al vacío para dar el producto bruto. ESi+APCI MS m/z 517,3 [M+H]+.
Etapa I: 1-((S)-2-(2-metoxietil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[,4-d1pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona. Se disolvió 4-((S)-3-(2-metoxietil)piperazin-1 -il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidina bruta (39 mg, 0,07548 mmol) en Dc M (10 ml) seguido de la adición de trietilamina (31,56 gl, 0,2264 mmol) y la solución se enfrió a -40 °C y se agitó durante 5 minutos seguido de la adición gota a gota de cloruro de acriloílo (12,26 gl, 0,1510 mmol). La mezcla de reacción se agitó a -30 °C durante 10 minutos seguido de la adición de MeÜH (0,05 ml). La mezcla se calentó a 0 °C y los extractos orgánicos se lavaron con Na2CÜ30,5 M. La capa orgánica se evaporó al vacío y se cromatografió sobre gel de sílice usando 5 % de MeÜH 0,5 % de NH4ÜH en Dc M como eluyente para dar el compuesto del título (EJEMPLÜ 433, 25,45 mg, 59 %). ESI+APCI MS m/z 571,3 [M+H]+.
Ejemplo 434

2-((S)-1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(8-(trifluorometil)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: trifluorometanosulfonato de 8-(trifluorometil)naftalen-1-ilo: A una solución agitada de 8-(trifluorometil)naftalen-1-ol (500 mg, 2,36 mmol) en DCM (10 ml) a 0 °C se le añadió N-etil-N-isopropilpropan-2-amina (616 gl, 3,53 mmol) seguido de la adición gota a gota de anhídrido trifluorometanosulfónico (475 gl, 2,83 mmol) y la mezcla de reacción se agitó 1 hora mientras se calentaba a temperatura ambiente. Luego, la reacción se diluyó con hexano (20 ml) y MTBE (5 ml) y los extractos orgánicos se lavaron con NaHCÜ3 sat., agua y salmuera (5 ml de cada uno), se secaron sobre Na2SÜ4 y se concentraron al vacío. El residuo se disolvió en 10 % de EtOAc/10 % de DCM en hexano y el material se filtró a través de un lecho de gel de sílice. Los extractos orgánicos se concentraron y se cromatografiaron sobre gel de sílice usando 5 a 10 % de EtOAc/hexano como eluyente para dar el material impuro. Finalmente, el sólido se cristalizó en hexano y el sólido se lavó con una pequeña cantidad de hexano frío para dar el producto (482 mg, 59 %).
Etapa B: (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(8-(trifluorometil)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: Una mezcla de (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo bruto (700 mg, 1,38 mmol), Cs2CÜ3 (1353 mg, 4,15 mmol), tolueno (7 ml), metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2-amino-1,1'-bifenil-2-il)paladio (II) (RuPhos-Pd-G4, 0,1 eq., 116 mg, 0,138 mmol) y 8-(trifluorometil)naftalen-1-il trifluorometanosulfonato (715 mg, 2,08 mmol) se purgó con nitrógeno y la reacción se tapó y se agitó a 90 °C durante 5 horas. La mezcla de reacción se enfrió, se repartió entre EtOAc (50 ml) y agua (20 ml) y las capas se separaron. La capa orgánica se lavó con agua y salmuera (10 ml de cada uno), se secó sobre Na2SÜ4 y se evaporó al vacío. El residuo se cromatografió sobre gel de sílice usando 4 % de MeÜH/DCM 0,4 % de NH40H como eluyente para dar el producto (86 mg, 9 %). ESI+APCI MS m/z 700,3 [M+H]+.
Etapa C: 2-((S)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(8-(trifluorometil)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo. Una mezcla de (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(8-(trifluorometil)naftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (86 mg, 0,123 mmol), metanol (3 ml, 74,2 mmol) y paladio sobre carbono (30 mg, 5 %, tipo Degussa E101 N0/W) se desgasificó al vacío, se cargó de nuevo con hidrógeno y la reacción se agitó en una atmósfera de H2 durante 1 hora. Luego, la reacción se filtró a través de Celite (2 ml) y el Celite se lavó con Me0H (3 x 3 ml). Los extractos orgánicos combinados se concentraron al vacío para dar el producto bruto (60 mg, 86 %).
Etapa D: 2-((S)-1-acriloil-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(8-(trifluorometil)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Se disolvió 2-((S)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(8-(trifluorometil)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (50 mg, 0,08839 mmol) en DCM (10 ml) seguido de la adición de trietilamina (36,96 gl, 0,2652 mmol) y la solución se enfrió a -40 °C y se agitó durante 5 minuto seguido de la adición gota a gota de cloruro de acriloílo (14,36 gl, 0,1768 mmol). La mezcla de reacción se agitó a -30 °C durante 10 minutos seguido de la adición de 0,01 ml de cloruro de acriloílo. Luego, a la reacción se le añadió Me0H (0,05 ml) y la mezcla se calentó a 0 °C. Luego, la reacción se lavó con Na2CÜ30,5 M y los extractos orgánicos se separaron. Los extractos orgánicos se evaporaron al vacío y se cromatografiaron sobre gel de sílice usando 5 % de Me0H 0,5 % de NH40H en DCM como eluyente para dar el compuesto del título (EJEMPL0434, 39 mg, 71 %).
Ejemplo 435

2-((S)-1-acriloil-4-(7-(2,3-dihidrobenzofuran-7-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (S)-2-(cianometil)-4-(7-(2,3-dihidrobenzofuran-7-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: Una mezcla de (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (200 mg, 0,396 mmol), CS2C03 (387 mg, 1,19 mmol), dioxano (0,5 ml), 7-bromo-2,3-dihidrobenzofurano (118 mg, 0,593 mmol, 1,5 eq.) y metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2'-metilamino-1,1'-bifenil-2-il)paladio (II) (RuPhos-Pd-G4, 0,1 eq., 33,7 mg, 0,0396 mmol) se purgó con nitrógeno y la reacción se tapó y se agitó a 80 °C durante una noche. La mezcla de reacción se enfrió, se repartió entre EtOAc (20 ml) y agua (10 ml) y después las capas se separaron. La capa orgánica se lavó con agua y salmuera (5 ml de cada uno), se secó sobre Na2S04 y se evaporó al vacío. El residuo se cromatografió sobre gel de sílice usando 4 % de Me0H/DCM 0,4 % de NH40H como eluyente para dar el producto (97 mg, 39 %). ESI+APCI MS m/z 624,3 [M+H]+.
Etapa B: 2-((S)-4-(7-(2,3-dihidrobenzofuran-7-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Una mezcla de (S)-2-(cianometil)-4-(7-(2,3-dihidrobenzofuran-7-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (97 mg, 0,16 mmol), metanol (4 ml), THF (4 ml) y paladio sobre carbono (30 mg, 5 %, tipo Degussa E101 NO/W ) se desgasificó al vacío, se cargó de nuevo con hidrógeno y la reacción se agitó en una atmósfera de hidrógeno durante una noche. El residuo se filtró a través de Celite (1 ml) y el celite se lavó con Me0H (3 x 2 ml). Los extractos orgánicos combinados se evaporaron al vacío para dar el producto bruto. ESI+APCI MS m/z 490,3 [M+H]+.
Etapa C: 2-((S)-1-acriloil-4-(7-(2,3-dihidrobenzofuran-7-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Una solución de 2-((S)-4-(7-(2,3-dihidrobenzofuran-7-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (69 mg, 0,1409 mmol) en DCM (5 ml) se enfrió a -30 °C con agitación. Se añadió trietilamina (58,93 μl, 0,4228 mmol) seguido de la adición de cloruro de acriloílo (22,90 μl, 0,2818 mmol) y la reacción se agitó durante 5 minutos a -30 °C. Luego, la reacción se interrumpió mediante la adición de Me0H (0,05 ml) y la reacción se calentó a temperatura ambiente. Los extractos orgánicos se lavaron con Na2C030,5 M (4 ml), se secaron sobre K2C03 y se evaporaron al vacío. El residuo se cromatografió sobre gel de sílice en usando 5 % de Me0H 0,5 % de NH40H como eluyente para dar el compuesto del título (EJEMPL0435, 45,85 mg, 60 %). ESI+APCI MS m/z 544,3 [M+H]+.
Ejemplo 436

2-((S)-1-acriloil-4-(7-(1-metil-1H-indol-7-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 7-yodo-1-metil-1H-indol: Una solución agitada de 7-yodo-1H-indol (4,11 ml, 2,06 mmol) en N,N-dimetilformamida (4 ml, 51,9 mmol) se enfrió a -20 °C, se añadió en pequeñas porciones hidruro de sodio (123 mg, 3,09 mmol) seguido de la adición de yodometano (0,256 ml, 4,11 mmol) y la mezcla se calentó a temperatura ambiente durante un periodo de 2 horas. La mezcla se repartió entre MTBE (40 ml) y una mezcla de hielo-agua (20 ml) y las capas se separaron. La capa orgánica se lavó con agua (2 x 20 ml) y salmuera (10 ml), se secó sobre Na2S04 y se evaporó al vacío. El sólido se lavó con MTBE (1 ml). El sólido se recristalizó en MTBE para dar el producto (0,31 g, 59 %).
Etapa B: (S)-2-(cianometil)-4-(7-(1-metil-1H-indol-7-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: Una mezcla de (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (200 mg, 0,396 mmol), Cs2C03 (387 mg, 1,19 mmol), dioxano (0,5 ml), 7-yodo-1-metil-1H-indol (153 mg, 0,593 mmol) (1,5 eq.) y metanosulfonato (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2'-metilamino-1,1'-bifenil-2-il)paladio (II) (RuPhos-Pd-G4, 0,1 eq., 33,7 mg, 0,0396 mmol) se purgó con nitrógeno y la reacción se tapó y se agitó a 90 °C durante 5 horas y después durante una noche a 80 °C. La mezcla de reacción se enfrió, se repartió entre EtOAc (20 ml) y agua (10 ml) y las capas se separaron. La capa orgánica se lavó con agua y salmuera (5 ml de cada uno), se secó sobre Na2S04 y se evaporó al vacío. El residuo se cromatografió sobre gel de sílice usando 4 % de Me0H/DCM 0,4 % de NH40H como eluyente para dar el producto (65 mg, 26 %). ESI+APCI MS m/z 635,3 [M+H]+.
Etapa C: 2-((S)-4-(7-(1-metil-1H-indol-7-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: Una mezcla de (S)-2-(cianometil)-4-(7-(1-metil-1H-indol-7-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (65 mg, 0,10 mmol), metanol (6 ml) y paladio sobre carbono (30 mg, 5 %, tipo Degussa E101 NO/W ) se desgasificó con vacío y se cargó de nuevo con hidrógeno y la mezcla se agitó en una atmósfera de hidrógeno durante una noche. La suspensión se filtró a través de Celite (1 ml) y el Celite se lavó con Me0H (3 x 2 ml). Los extractos orgánicos combinados se evaporaron al vacío para dar el producto bruto. ESI+APCI MS m/z 501,3 [M+H]+.
Etapa D: 2-((S)-1 -acriloil-4-(7-(1-metil-1H-indol-7-il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Una solución de 2-((S)-4-(7-(1 -metil-1H-indol-7-il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (51 mg, 0,1019 mmol) en DCM (5 ml) se enfrió a -30 °C con agitación seguido de la adición de trietilamina (42,60 μl, 0,3056 mmol) y cloruro de acriloílo (16,55 μl, 0,2037 mmol). Después de agitar a 5 minutos a -30 °C, la mezcla de reacción se inactivó con Me0H (0,05 ml)
y se calentó a temperatura ambiente. Los extractos orgánicos se lavaron con Na2CÜ30,5 M (4 ml), se secaron sobre K2C03 y se evaporaron al vacío. El residuo se cromatografió sobre gel de sílice usando 5 % de MeÜH 0,5 % de NH40H como eluyente para dar el compuesto del título (EJEMPL0436, 36 mg, 64 %). ESI+APCI MS m/z 555,3 [M+H]+.
Ejemplo 437

2-((S)-1-acriloil-4-(7-(2,3-dimetilfenil)-2-(((2S,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 2-((S)-1-acriloil-4-(7-(2,3-dimetilfenil)-2-(((2S,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Una solución de 2-((S)-4-(7-(2,3-dimetilfenil)-2-(((2S,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (50 mg, 0,1013 mmol) en DCM (5 ml) se enfrió a -30 °C seguido de la adición de trietilamina (42,35 gl, 0,3039 mmol) y cloruro de acriloílo (16,46 gl, 0,2026 mmol). Después de agitar 5 minutos a -30 °C la mezcla de reacción se inactivó con MeÜH (0,05 ml) y se calentó a temperatura ambiente. Los extractos orgánicos se lavaron con Na2CÜ30,5 M (4 ml), se secaron sobre K2C03 y se evaporaron al vacío. El residuo se cromatografió sobre gel de sílice usando 4 % de MeÜH 0,4 % de NH40H como eluyente para dar el compuesto del título (EJEMPL0437, 34 mg, 61 %). ESI+APCI MS m/z 548,3 [M+H]+.
Ejemplo 438

2-((S)-1-((E)-4-(dimetilamino)but-2-enoil)-4-(7-(2,3-dimetilfenil)-2-(((2S,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 2-((S)-1-((E)-4-(dimetilamino)but-2-enoil)-4-(7-(2,3-dimetilfenil)-2-(((2S,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: A una solución agitada de 2-((S)-4-(7-(2,3-dimetilfenil)-2-(((2S,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (50 mg, 0,1013 mmol) en DCM (1 ml) se le añadieron ácido (2E)-4-(dimetilamino)but-2-enoico (26,17 mg, 0,2026 mmol), N-etil-N-isopropilpropan-2-amina (0,03529 ml, 0,2026 mmol) y hexafluorofosfato de 0-(7-azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio (57,77 mg, 0,1519 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 4 horas. Luego, se añadió agua (1 ml) y la mezcla de reacción se agitó durante 5 minutos. La mezcla se repartió entre EtOAc (10 ml) y NaHC03 sat. (5 ml) y las capas se separaron. La capa orgánica se lavó con NaHC03 y salmuera, se secó sobre K2C03 y se evaporó al vacío. El residuo se cromatografió sobre gel de sílice usando 5 % de Me0H 0,5 % de NH40H como eluyente para dar el compuesto del título (EJEMPL0438, 45 mg, 74 %). ESI+APCI MS m/z 605,3 [M+H]+.
Ejemplo 439

2-(4-acriloil-1 -(2-(((S)-1 -metilpirrolidin-2-il)metoxi)-7-(naftalen-1 -il)-5,6,7, 8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 4-í4-ííbenc¡loxncarbon¡l)-2-íc¡anomet¡l)p¡peraz¡n-1-¡l)-2-cloro-5.8-d¡hidrop¡r¡dor3.4-d1p¡r¡m¡d¡n-7í6H)-carboxilato de terc-butilo: Una soluc¡ón ag¡tada de 2-(p¡peraz¡n-2-¡l)aceton¡tr¡lo (0.2 g. 1.278 mmol) en una mezcla de DCM (20 ml) e ¡Pr0H (10 ml) se enfr¡ó a -30 °C. se añad¡ó gota a gota carbonoclor¡dato de benc¡lo (0.2190 ml.
1.534 mmol) y la reacc¡ón se ag¡tó durante 1 hora m¡entras se calentaba a temperatura amb¡ente. Los extractos orgán¡cos se evaporaron en un flujo lento de N2. El res¡duo se d¡solv¡ó en N,N-d¡met¡lacetam¡da (1 ml. 1.278 mmol) segu¡do de la ad¡c¡ón de N-et¡l-N-¡soprop¡lpropan-2-am¡na (0.3340 ml. 1.917 mmol) y 2.4-d¡cloro-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo (0.3888 g. 1.278 mmol). La mezcla de reacc¡ón se calentó a 70 °C durante 2 horas segu¡do de ag¡tac¡ón a temperatura amb¡ente durante 4 días. La reacc¡ón se repart¡ó entre agua (5 ml) y MTBE (20 ml) y las capas se separaron. Los extractos orgán¡cos se lavaron con agua y salmuera (5 ml de cada uno). se secaron sobre Na2S04 y se evaporaron al vacío. El res¡duo se cromatograf¡ó sobre gel de síl¡ce usando 40 a 100 % de EtOAc/hexano como eluyente para dar el producto (134 mg. 20 %). ESI+APCI MS m/z 527.2 [M+H]+.
Etapa B: 4-(4-((benc¡lox¡)carbon¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.8-d¡h¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo: Una mezcla de 4-(4-((benc¡lox¡)carbon¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-cloro-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo (134 mg.
0.254 mmol). Cs2C03 (249 mg. 0.763 mmol). metanosulfonato (2-d¡c¡clohex¡lfosf¡no-2'.6'-d¡-¡-propox¡-1.1'-b¡fen¡l)(2'-met¡lam¡no-1.1'-b¡fen¡l-2-¡l)palad¡o (II) (21.6 mg. 0.0254 mmol). (S)-(1-met¡lp¡rrol¡d¡n-2-¡l)metanol (146 mg. 1.27 mmol) y 1.4-d¡oxano (2 ml. 0.254 mmol) se purgó con N2 y la reacc¡ón se tapó y se ag¡tó a 80 °C durante 3 horas. La mezcla de reacc¡ón se enfr¡ó. se repart¡ó entre EtOAc (20 ml) y agua (5 ml) y las capas se separaron. Los extractos orgán¡cos se lavaron con salmuera (5 ml). se secaron sobre Na2S04 y se evaporaron al vacío. El res¡duo se cromatograf¡ó sobre gel de síl¡ce usando 4 % de Me0H 0.4 % de NH40H /DCM como eluyente para dar el producto (60 mg. 39 %).
ESI+APCI MS m/z 606.3 [M+H]+.
Etapa C: 3-(c¡anomet¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo: Se d¡luyó 4-(4-((benc¡lox¡)carbon¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo neto (60 mg. 0.09905 mmol)) con DCM (0.2 ml). La reacc¡ón se enfr¡ó a 0 °C y se añad¡ó HCl 4 M en d¡oxano (0. 50 ml. 20 eq.) con ag¡tac¡ón. La mezcla de reacc¡ón se calentó a temperatura amb¡ente y se dejó en el refr¡gerador durante una noche. momento en el que prec¡p¡tó un sól¡do. La fase líqu¡da se decantó. el sól¡do se repart¡ó entre DCM (10 ml) y Na2C032 M (0.5 ml) y las capas se separaron. Los extractos orgán¡cos se secaron sobre K2C03 y se evaporaron en un flujo de N2. El producto se usó bruto en la s¡gu¡ente reacc¡ón.
Etapa D: 3-(c¡anomet¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo: Una mezcla de 3-(c¡anomet¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo bruto (50 mg. 0.0989 mmol). Cs2C03 (96.7 mg.
0.297 mmol). d¡oxano (0.5 ml). 1-yodonaftaleno (37.7 mg. 0.148 mmol. 1.5 eq.) y metanosulfonato de (2-d¡c¡clohex¡lfosf¡no-2'.6'-d¡-¡-propox¡-1.1'-b¡fen¡l)(2'-met¡lam¡no-1.1'-b¡fen¡l-2-¡l)palad¡o (II) (RuPhos-Pd-G4. 0.1 eq..
8.42 mg. 0.00989 mmol) se purgó con n¡trógeno y la reacc¡ón se tapó y se ag¡tó a 80 °C durante 2 horas. La mezcla de reacc¡ón se enfr¡ó y se repart¡ó entre EtOAc (20 ml) y agua (5 ml) y las capas se separaron. Los extractos orgán¡cos se lavaron con agua y salmuera (5 ml de cada uno). se secaron sobre Na2S04 y se evaporaron al vacío. El res¡duo se cromatograf¡ó sobre síl¡ce usando 4 % de Me0H/DCM 0.4 % de NH40H como eluyente para dar el producto ¡mpuro. que se pur¡f¡có por cromatografía HPLC prep. ¡nversa eluyendo con 5 a 95 % de MeCN/agua 0.1 % de TFA (pr¡mer compuesto elu¡do). Las fracc¡ones d¡ana se d¡luyeron con Na2C032 M y la capa acuosa se extrajo con DCM (3 x 20 ml). Los extractos comb¡nados se lavaron con salmuera (15 ml). se secaron sobre K2C03 y se evaporaron al vacío para proporc¡onar el producto (26 mg. 42 %). ESI+APCI MS m/z 632.3 [M+H]+.
Etapa E: 2-(1-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo: Una mezcla de 3-(c¡anomet¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (26 mg. 0.041 mmol). metanol (2 ml) y palad¡o sobre carbono (10 mg. 5 %. tipo Degussa E101 NO /W ) se desgas¡f¡có al vacío. se cargó de nuevo con h¡drógeno y la reacc¡ón se ag¡tó en una atmósfera de h¡drógeno durante 1.5 horas. La reacc¡ón se f¡ltró a través de Cel¡te (2 ml) y el Cel¡te se lavó con Me0H (3 x 3 ml). Los extractos orgán¡cos comb¡nados se evaporaron al vacío para dar el producto que se usó bruto en la s¡gu¡ente reacc¡ón. ESI+APCI MS m/z 498.3 [M+H]+.
Etapa F: 2-(4-acr¡lo¡l-1-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo: Una soluc¡ón de 2-(1-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-7-(naftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (15 mg. 0.03014 mmol) en DCM (5 ml) se enfr¡ó a -30 °C con ag¡tac¡ón segu¡do de la ad¡c¡ón de tr¡et¡lam¡na (12.60 μl. 0.09043 mmol) y cloruro de acr¡lαlo (4.898 μl.
0.06028 mmol). Después de ag¡tar 5 m¡nutos a -30 °C. la mezcla de reacc¡ón se ¡nact¡vó med¡ante la ad¡c¡ón de Me0H (0.05 ml) y se calentó a temperatura amb¡ente. Los extractos orgán¡cos se lavaron con Na2C030.5 M (4 ml). se secaron sobre Na2C03 y se evaporaron al vacío. El res¡duo se cromatograf¡ó sobre gel de síl¡ce usando 4 % de Me0H 0.4 % de NH40H como eluyente para dar el compuesto del título (EJEMPL0439. 8.43 mg. 51 %). ESI+APCI MS m/z 552.2 [M+H]+.
E je m p lo 440

2-((S)-1-acriloil-4-(2-(((2R,3R)-3-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: ácido (2R,3R)-3-fluoropirrolidin-2-carboxílico: Al ácido (2R, 3R)-1-Boc-3-fluoropirrolidin-carboxílico sólido (400 mg, 1,71 mmol) se le añadió gota a gota cloruro de hidrógeno 4 M (2144 μl, 8,57 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 1,5 horas. La mezcla de reacción se evaporó al vacío seguido de la adición de NaHCÜ3 sólido (173 mg, 2,06 mmol) y metanol (1715 μl, 1,71 mmol) y la suspensión se agitó a temperatura ambiente durante una noche. A la mezcla se le añadió una porción adicional de MeüH (10 ml) y la mezcla de reacción se agitó durante 30 minutos y se sonicó hasta que se consiguió la dispersión completa de los semisólidos. Esta suspensión se usó bruta en la siguiente reacción.
Etapa B: ácido (2R,3R)-3-fluoro-1-metilpirrolidin-2-carboxílico: A una suspensión agitada de ácido (2R,3R)-3-fluoropirrolidin-2-carboxílico bruto (228 mg, 1,71 mmol) en MeÜH (12 ml) se le añadió paladio sobre carbono (30 mg, 5 %, tipo Degussa E101 N0/W) seguido de la adición de formaldehído acuoso (159 μl, 2,14 mmol) y la mezcla de reacción se desgasificó con hidrógeno y se agitó en una atmósfera de hidrógeno durante una noche. La mezcla de reacción se filtró a través de Celite y el Celite se lavó con MeÜH (3 x 2 ml). Los extractos orgánicos combinados se evaporaron al vacío. El semisólido se recogió en dioxano y se evaporó en una atmósfera de N2 durante el fin de semana. El producto se usó bruto en la siguiente reacción.
Etapa C: ((2R,3R)-3-fluoro-1-metilpirrolidin-2-il)metanol: Una suspensión agitada de ácido 1-Boc-2-(S)-pirrolidindicarboxílico bruto, 3-(R)-fluoro-(252 mg, 1,71 mmol) en THF en una atmósfera de nitrógeno se enfrió a 0 °C seguido de la adición gota a gota de hidruro de litio 2,4 M y aluminio (0,999 ml, 2,40 mmol) y la reacción se agitó durante 30 minutos mientras se calentaba a temperatura ambiente. La reacción se sonicó seguido de la adición de otra porción de hidruro de litio y aluminio en THF (0,999 ml, 2,40 mmol) y la mezcla de reacción se agitó a temperatura ambiente. La suspensión se enfrió a 0 °C y se inactivó con la adición sucesiva de agua (0,18 ml), 15 % de NaÜH (0,18 ml) y agua (0,54 ml). La suspensión se diluyó con éter (20 ml) y el precipitado se separó. La mezcla de reacción se filtró a través de Celite y el Celite se lavó con éter (3 x 3 ml). A los extractos orgánicos combinados se les añadió HCl 4 M/dioxano (0,45 ml, 1,1 eq.). La mezcla cambió de una suspensión incolora a un precipitado de color rojizo. La solución sobrenadante se evaporó al vacío. El sólido de color rojo residual se extrajo con una cantidad mínima de agua, se filtró a través de un lecho de algodón y el lecho de algodón se lavó con una pequeña cantidad de agua. La solución de agua combinada se saturó con KÜH y se extrajo con éter (3 x 7 ml). Los extractos combinados se secaron
sobre Na0H y se evaporaron parcialmente en una atmósfera de nitrógeno para dar el producto en forma de una solución en éter (340 mg en forma de una solución al 40 % en éter, 60 %)
Etapa D: (S)-2-(cianometil)-4-(2-(((2R,3R)-3-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: Una mezcla de (S)-4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (150 mg, 0,2712 mmol), Cs2C03 (265,1 mg, 0,8137 mmol), 1,4-dioxano (1 ml), metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1 '-bifenil)(2'-metilamino-1,1'-bifenil-2-il)paladio (II) (23,09 mg, 0,02712 mmol) y RuPhos (9,492 mg, 0,02034 mmol) se desgasificó con nitrógeno y se agitó a 70 °C durante 1,5 horas. La mezcla de reacción se enfrió y se repartió entre EtOAc (20 ml) y agua (5 ml) y las capas se separaron. Los extractos orgánicos se lavaron con agua y salmuera (5 ml de cada uno), se secaron sobre Na2S04 y se evaporaron al vacío. El residuo se cromatografió sobre gel de sílice usando 3 % de Me0H 0,3 % de NH40H en DCM como eluyente para dar el producto (108 mg, 61 %). ESI+APCI MS m/z 650,3 [M+H]+.
Etapa E: 2-((S)-4-(2-(((2R,3R)-3-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Una mezcla de (S)-2-(cianometil)-4-(2-(((2R,3R)-3-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (108 mg, 0,166 mmol), metanol (5 ml) y paladio sobre carbono (35 mg, 5 %, tipo Degussa E101 NO/W ) se desgasificó al vacío, se cargó de nuevo con hidrógeno y la reacción se agitó en una atmósfera de hidrógeno durante 1,5 horas. La mezcla de reacción se filtró a través de Celite (2 ml) y el celite se lavó con Me0H (3 x 3 ml). Los extractos orgánicos combinados se evaporaron al vacío para dar el producto bruto que se usó en la siguiente reacción.
Etapa F: 2-((S)-1-acriloil-4-(2-(((2R,3R)-3-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Una solución de 2-((S)-4-(2-(((2R,3R)-3-fluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (78 mg, 0,1513 mmol) en DCM (5 ml) se enfrió a -30 °C seguido de la adición de trietilamina (63,25 μl, 0,4538 mmol) y cloruro de acriloílo (24,58 μl, 0,3025 mmol). Después de agitar 5 minutos a -30 °C, la mezcla de reacción se inactivó con Me0H (0,05 ml) y se calentó a temperatura ambiente. La mezcla se lavó con Na2C030,5 M (4 ml), se secó sobre Na2C03 y se evaporó al vacío. El residuo se cromatografió sobre gel de sílice usando 4 % a 10 % de Me0H como eluyente para dar el compuesto del título (EJEMPL0440, 63 mg, 73 %). ESI+APCI MS m/z 570,3 [M+H]+.
Ejemplo 441

2-((S)-1-acriloil-4-(2-(((R)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 4-((S)-4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-(((R)-1-metilpirrolidin-2-il)metoxi)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo. Una mezcla de (S)-4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (500 mg, 0,949 mmol), (D)-(R)-(1-metilpirrolidin-2-il)metanol (328 mg, 2,85 mmol)(3 eq.), Cs2C03 (927 mg, 2,85 mmol), dioxano (1 ml) y metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2-amino-1,1'-bifenil-2-il)paladio (II) (RuPhos-Pd-G3, 0,1 eq., 79,3 mg, 0,0949 mmol) se purgó con nitrógeno y la reacción se tapó y se agitó a 70 °C durante 5 horas. La mezcla de reacción se enfrió, se repartió entre EtOAc (20 ml) y agua (10 ml) y las capas se separaron. Los extractos orgánicos se lavaron con agua y salmuera (5 ml de cada uno), se secaron sobre Na2S04 y se evaporaron al vacío. El residuo se cromatografió sobre gel de sílice usando 4 a 10 % de Me0H 0,5 % de NH40H como eluyente para dar el producto (150 mg, 26 %). ESI+APCI MS m/z 606,3 [M+H]+.
Etapa B: (S)-2-(cianometil)-4-(2-(((R)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: Una solución de 4-((S)-4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-(((R)-1-metilpirrolidin-2-il)metoxi)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (296 mg, 0,489 mmol) en DCM (2,5 ml) se enfrió en hielo seco seguido de la adición de HCl 4 M en dioxano (1,2 ml)y la reacción se agitó durante 3 horas mientras se calentaba a temperatura ambiente, momento en el que se formó un precipitado. La capa líquida se decantó y el precipitado se lavó con DCM (2 ml). Después, el sólido se agitó con una mezcla de DCM (20 ml) y Na2C032 M (3 ml) durante 1 hora. Las capas se separaron y los extractos orgánicos se secaron sobre Na2C03, se filtraron y se evaporaron al vacío. El material se usó bruto en la siguiente reacción. ESI+APCI MS m/z 506,3 [M+H]+.
Etapa C: (S)-2-(cianometil)-4-(2-(((R)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: Una mezcla de (S)-2-(cianometil)-4-(2-(((R)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo bruto (247 mg, 0,489 mmol), Cs2C03 (477 mg, 1,47 mmol), dioxano (2 ml), 1-yodonaftaleno (107 μl, 0,733 mmol, 1,5 eq.) y metanosulfonato de (2 diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2-amino-1,1'-bifenil-2-il)paladio (II) (RuPhos-Pd-G3, 0,1 eq., 40,9 mg, 0,0489 mmol) se purgó con nitrógeno y la reacción se tapó y se agitó a 80 °C durante una noche. La mezcla de reacción se enfrió, se repartió entre EtOAc (20 ml) y agua (10 ml) y las capas se separaron. Los extractos orgánicos se lavaron con agua y salmuera (5 ml de cada uno), se secaron sobre Na2S04 y se evaporaron al vacío. El residuo se cromatografió sobre gel de sílice usando 5 % de Me0H/DCM 0,25 % de NH40H como eluyente para dar el producto (216 mg, 70 %). ESI+APCI MS m/z 632,3 [M+H]+.
Etapa D: 2-((S)-1-acriloil-4-(2-(((R)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: A una solución agitada de (S)-2-(cianometil)-4-(2-(((R)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (216 mg, 0,3419 mmol) en Me0H-THF (1:1,3 ml) se le añadió paladio sobre carbono (50 mg, 5 %, tipo Degussa E101 NO/W ), la mezcla se desgasificó al vacío, se cargó de nuevo con hidrógeno y la reacción se agitó en una atmósfera de hidrógeno durante 4 horas. La mezcla de reacción se filtró a través de Celite (2 ml) y el celite se lavó con Et0H (3 x 2 ml). Los extractos orgánicos combinados se evaporaron al vacío y el residuo se disolvió en DCM (10 ml). La solución se enfrió con agitación a -30 °C seguido de la adición de NEt3 (0,28 ml, 1,71 mmol) y cloruro de acriloílo (0,08333 ml, 1,026 mmol) y la reacción se agitó a -30 °C durante 1 minuto. La reacción se interrumpió con la adición de NH40H (0,05 ml) y se calentó a temperatura ambiente. La solución se evaporó al vacío. El residuo se disolvió en DCM (10 ml), se filtró a través de un lecho de algodón y se cromatografió sobre gel de sílice usando 5 a 10 % de Me0H 0,25 % de NH40H en DCM como eluyente para dar el compuesto del título (EJEMPL0441, 109 mg, 58 %). ESI+AμCi MS m/z 552,3 [M+H]+.
Ejemplo 442

2-((S)-1-acriloil-4-(7-(indolizin-5-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 5-yodoindolizina: A una solución agitada de indolizina (5,69 ml, 1,71 mmol) y N1,N1,N2,N2-tetrametiletano-1,2-diamina (0,282 ml, 1,88 mmol) en tetrahidrofurano anhidro (5,7 ml, 1,71 mmol) enfriada a -80 °C se le añadió gota a gota una solución de butil litio (120 mg, 1,88 mmol). La mezcla de reacción se calentó a -20 °C y se mantuvo a -20 °C durante 2 horas. La mezcla se enfrió a -80 °C y se añadió una solución de yodo (433 mg, 1,71 mmol) en THF seco (3 ml). La mezcla de reacción se calentó a temperatura ambiente y se trató con una solución saturada de cloruro de amonio. La capa orgánica se separó y la capa acuosa se extrajo con hexano. Los extractos orgánicos combinados se secaron sobre Na2S04 anhidro y se concentraron al vacío. El producto bruto se purificó por cromatografía en columna sobre gel de sílice usando hexano como eluyente para dar el producto (216 mg).
Etapa B: (S)-2-(cianometil)-4-(7-(indolizin-5-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: Una mezcla de (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo bruto (100 mg, 0,198 mmol), Cs2C03 (193 mg, 0,593 mmol), dioxano (0,5 ml), 5-yodoindolizina (72,1 mg, 0,297 mmol) (1,5 eq.) y metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2'-metilamino-1,1'-bifenil-2-il)paladio (II) (RuPhos-Pd-G4, 0,1 eq., 16,8 mg, 0,0198 mmol) se purgó con nitrógeno y la reacción se tapó y se agitó a 80 °C durante 4 horas. La mezcla de reacción se enfrió, se repartió entre EtOAc (20 ml) y agua (10 ml) y las capas se separaron. Los extractos orgánicos se lavaron con agua y salmuera (5 ml de cada uno), se secaron sobre Na2S04 y se evaporaron al vacío. El residuo se cromatografió sobre gel de sílice usando 4 % de Me0H/DCM 0,4 % de NH40H como eluyente para dar el producto (74 mg, 60 %). ESI+APCI MS m/z 621,3 [M+H]+.
Etapa C: 2-((S)-4-(7-(indolizin-5-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Una mezcla de (S)-2-(cianometil)-4-(7-(indolizin-5-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (74 mg, 0,12 mmol), metanol (5 ml) y paladio sobre carbono (20 mg, 5 %, tipo Degussa E101 NO/W ) se desgasificó al vacío, se cargó de nuevo con hidrógeno y la reacción se agitó en una atmósfera de hidrógeno durante 2 horas. La suspensión se filtró a través de Celite (1 ml) y el Celite se lavó con Me0H (3 x 2 ml). Los extractos orgánicos combinados se evaporaron al vacío para dar el producto que se usó bruto en la siguiente reacción. ESI+APCI MS m/z 487,3 [M+H]+.
Etapa D: 2-((S)-1-acriloil-4-(7-(indolizin-5-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Una solución de 2-((S)-4-(7-(indolizin-5-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (50 mg, 0,1027 mmol) en DCM (5 ml) se enfrió a -30 °C seguido de la adición de trietilamina (42,96 gl, 0,3082 mmol) y cloruro de acriloílo (16,70 gl, 0,2055 mmol). Después de agitar 5 minutos a -30 °C, la mezcla de reacción se inactivó con NaHC03 sat. (1 ml) y se calentó a temperatura ambiente seguido de la adición de agua (2 ml). Las capas se separaron y los extractos orgánicos se secaron sobre Na2S04 y se evaporaron al vacío. El residuo se cromatografió sobre gel de sílice usando 4 % de Me0H 0,4 % de NH40H como eluyente para dar el compuesto del título (EJEMPL0442, 33 mg, 59 %). ESI+APCI MS m/z 571,3 [M+H]+.
Ejemplo 443

2-(1-Acriloil-4-(2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (2R)-2-(((4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-2-il)oxi)metil)morfolin-4-carboxilato de terc-butilo: Se preparó según el procedimiento del Ejemplo 375 Etapa C, sustituyendo 4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (65 mg, 0,118 mmol) y (R)-N-Boc-2-hidroximetilmorfolina (76,6 mg, 0,353 mmol), purificando por cromatografía ultrarrápida eluyendo con 25-100 % de hex/EtOAc para proporcionar (2R)-2-(((4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)morfolin-4-carboxilato de terc-butilo (49 mg, 0,0668 mmol, 56,8 % de rendimiento). ESI+ MS m/z 734,3 (100 %) [M+H]+.
Etapa B: Clorhidrato de 2-(cianometil)-4-(2-(((R)-morfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: Se preparó según el procedimiento del Ejemplo 375 Etapa D, sustituyendo (2R)-2-(((4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)morfolin-4-carboxilato de terc-butilo (49 mg, 0,067 mmol) para proporcionar clorhidrato de 2-(cianometil)-4-(2-(((R)-morfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (45 mg, 0,067 mmol, 100 %). ESI+ MS m/z 634,3 (100 %) [M+H]+.
Etapa C: 2-(Cianometil)-4-(2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo. A una solución de clorhidrato de 2-(cianometil)-4-(2-(((R)-morfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (45 mg, 0,067 mmol) y formaldehído (54 mg, 0,67 mmol) (37 % en peso en Me0H/agua) en DCM (671 μl, 0,067 mmol) y THF (671 μl, 0,067 mmol) se le añadió NaBH(oAc)3 (142 mg, 0,67 mmol) y la mezcla resultante se agitó a TA durante 15 m. La mezcla de reacción se repartió entre acetato de etilo y Na0H 1 N. La capa acuosa se extrajo con acetato de etilo (1 x). Las capas orgánicas combinadas se secaron (MgS04) y se concentraron para dar un residuo que se purificó por cromatografía ultrarrápida eluyendo con un gradiente de 25-50 % de una mezcla 10 % de Me0H/1 % de n H40H/dCm en DCM. Las fracciones que contenían el producto se concentraron para proporcionar 2-(cianometil)-4-(2-(((R)-4
metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (22 mg, 0,034 mmol, 51 %). ESI+ MS m/z 648,3 (100 %) [M+H]+.
Etapa D: 2-(4-(2-(((R)-4-Metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)p¡perazin-2-il)acetonitrilo. Se preparó según el procedimiento del Ejemplo 375 Etapa F, sustituyendo 2-(cianometil)-4-(2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (22 mg, 0,034 mmol) para proporcionar 2-(4-(2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (15,9 mg, 0,031 mmol, 91 %). ESI+ MS m/z 514,3 (100 %) [M+H]+.
Etapa E: -2-(1-Acriloil-4-(2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo. Se preparó según el procedimiento del Ejemplo 375 Etapa G, sustituyendo 2-(4-(2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (16 mg, 0,031 mmol) para proporcionar el compuesto del título 2-(1-acriloil-4-(2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0 443, 8 ,0 mg, 0,014 mmol, 46 %). ESI+ MS m/z 568,3 (100 %) [M+H]+.
Ejemplo 444

2-(1-Acriloil-4-(2-(((S)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2 -il)acetonitrilo

Etapa A: (2S)-2-(((4-(4-((Benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-2-il)oximetil)morfolin-4-carboxilato de terc-butilo: Se preparó según el procedimiento del Ejemplo 375 Etapa C, sustituyendo 4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (65 mg, 0,118 mmol) y (S)-2-(hidroximetil)morfolin-4-carboxilato de tercbutilo (76,6 mg, 0,353 mmol), purificando por cromatografía ultrarrápida eluyendo con 25-100 % de hex/EtOAc para proporcionar (2S)-2-(((4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1 -il)-7-(naftalen-1 -il)-5,6,7,8tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)morfolin-4-carboxilato de terc-butilo (43 mg, 0,059 mmol, 49,9 % de rendimiento). ESI+ MS m/z 734,3 (100 %) [M+H]+.
Etapa B: Clorhidrato de 2-(cianometil)-4-(2-(((S)-morfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: Se preparó según el procedimiento del Ejemplo 375 Etapa D, sustituyendo (2S)-2-(((4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)morfolin-4-carboxilato de terc-butilo (43 mg, 0,059 mmol) para proporcionar clorhidrato de 2-(cianometil)-4-(2-(((S)-morfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (39 mg, 0,058 mmol, 100 %). ESI+ MS m/z 634,3 (100 %) [M+H]+. Etapa C: 2-(Cianometil)-4-(2-(((S)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo. Se preparó según el procedimiento del Ejemplo 443 etapa C, sustituyendo clorhidrato de 2-(cianometil)-4-(2-(((S)-morfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (39 mg, 0,067 mmol), para proporcionar 2-(cianometil)-4-(2-(((S)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (17 mg, 0,026 mmol, 45 %). ESI+ MS m/z 648,3 (100 %) [M+H]+.
Etapa D: 2-(4-(2-(((S)-4-Metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetralaydropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo. Se preparó según el procedimiento del Ejemplo 375 Etapa F, sustituyendo 2-(cianometil)-4-(2-(((S)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (17 mg, 0,026 mmol) para proporcionar 2-(4-(2-(((S)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (13,5 mg, 0,026 mmol, 100 %). ESI+ MS m/z 514,3 (100 %) [M+H]+.
Etapa E: 2-(1-Acriloil-4-(2-(((S)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo. Se preparó según el procedimiento del Ejemplo 375 Etapa G, sustituyendo 2-(4-(2-(((S)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (13,5 mg, 0,026 mmol) para proporcionar el compuesto del título 2-(1-acriloil-4-(2-(((S)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0 444, 9,5 mg, 0,017 mmol, 65 %). ESI+ MS m/z 568,3 (100 %) [M+H]+.
Ejemplo 445

2-(1-Acriloil-4-(2-(((S)-4-metilmorfolin-3-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo


Etapa A: (3S)-3-(((4-(4-((Benciloxi)carbonil)-3-(cianometi0piperazin-1-i0-7-(naftalen-1-il)-5.6,7.8-tetrahidropirido[3.4-d]pirimidin-2-il)oxi)metil)morfolin-4-carboxilato de terc-butilo. Se preparó según el procedimiento del Ejemplo 375 Etapa C. sustituyendo 4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (65 mg, 0,118 mmol) y (R)-3-(hidroximetil)morfolin-4-carboxilato de tercbutilo (76,6 mg, 0,353 mmol), purificando por cromatografía ultrarrápida eluyendo con 25-100 % de hex/EtOAc para proporcionar (3S)-3-(((4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)morfolin-4-carboxilato de terc-butilo (80 mg, 0,109 mmol, 92,8 % de rendimiento). ESI+ MS m/z 734,3 (100 %) [M+H]+.
Etapa B: Clorhidrato de 2-(cianometil)-4-(2-(((S)-morfolin-3-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo. Se preparó según el procedimiento del Ejemplo 375 Etapa D, sustituyendo (3S)-3-(((4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)morfolin-4-carboxilato de terc-butilo (80 mg, 0,11 mmol) para proporcionar clorhidrato de 2-(cianometil)-4-(2-(((S)-morfolin-3-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (73 mg, 0,11 mmol, 100 %). ESI+ MS m/z 634,3 (100 %) [M+H]+.
Etapa C: 2-(Cianometil)-4-(2-(((S)-4-metilmorfolin-3-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo. Se preparó según el procedimiento del Ejemplo 443 etapa C, sustituyendo clorhidrato de 2-(cianometil)-4-(2-(((S)-morfolin-3-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (73 mg, 0,109 mmol), para proporcionar 2-(cianometil)-4-(2-(((S)-4-metilmorfolin-3-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (27,6 mg, 0,042 mmol, 39 %). ESI+ MS m/z 648,3 (100 %) [M+H]+.
Etapa D: 2-(4-(2-(((S)-4-Metilmorfolin-3-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo. Se preparó según el procedimiento del Ejemplo 375 Etapa F, sustituyendo 2-(cianometil)-4-(2-(((S)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (27,6 mg, 0,042 mmol) para proporcionar 2-(4-(2-(((S)-4-metilmorfolin-3-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (20,2 mg, 0,039 mmol, 100 %). Es I+ MS m/z 514,3 (100 %) [M+H]+.
Etapa E: 2-(1-Acriloil-4-(2-(((S)-4-metilmorfolin-3-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo. Se preparó según el procedimiento del Ejemplo 375 Etapa G, sustituyendo 2-(4-(2-(((S)-4-metilmorfolin-3-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (20,2 mg, 0,0393 mmol) para proporcionar el compuesto del título 2-(1-acriloil-4-(2-(((S)-4-metilmorfolin-3-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0 445, 15 mg, 0,026 mmol, 67 %). ESI+ MS m/z 568,3 (100 %) [M+H]+.
Ejemplo 446

2-(1-Acriloil-4-(2-(((R)-4-metilmorfolin-3-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (3R)-3-(((4-(4-((Benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6.7.8-tetrahidropirido[3.4-d1pirimidin-2-il)oxi)metil)morfolin-4-carboxilato de terc-butilo. Se preparó según el procedimiento del Ejemplo 375 Etapa C. sustituyendo 4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (65 mg, 0,118 mmol) y (S)-3-(hidroximetil)morfolin-4-carboxilato de tercbutilo (76,6 mg, 0,353 mmol), purificando por cromatografía ultrarrápida eluyendo con 25-100 % de hex/EtOAc para proporcionar (3R)-3-(((4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)morfolin-4-carboxilato de terc-butilo (50 mg, 0,068 mmol, 58,0 % de rendimiento). ESI+ MS m/z 734,3 (100 %) [M+H]+.
Etapa B: Clorhidrato de 2-(cianometil)-4-(2-(((R)-morfolin-3-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo. Se preparó según el procedimiento del Ejemplo 375 Etapa D, sustituyendo (3R)-3-(((4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)morfolin-4-carboxilato de terc-butilo (50 mg, 0,068 mmol) para proporcionar clorhidrato de 2-(cianometil)-4-(2-(((R)-morfolin-3-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (46 mg, 0,068 mmol, 100 %). ESI+ MS m/z 634,3 (100 %) [M+H]+.
Etapa C: 2-(Cianometil)-4-(2-(((R)-4-metilmorfolin-3-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo. Se preparó según el procedimiento del Ejemplo 443 etapa C, sustituyendo clorhidrato de 2-(cianometil)-4-(2-(((R)-morfolin-3-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (73 mg, 0,109 mmol), para proporcionar 2-(cianometil)-4-(2-(((R)-4-metilmorfolin-3- il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (23,0 mg, 0,036 mmol, 52 %). ESI+ MS m/z 648,3 (100 %) [M+H]+.
Etapa D: 2-(4-(2-(((R)-4-Metilmorfolin-3-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo. Se preparó según el procedimiento del Ejemplo 375 Etapa F, sustituyendo 2-(cianometil)-4- (2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (23,0 mg, 0,036 mmol) para proporcionar 2-(4-(2-(((R)-4-metilmorfolin-3-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (18,2 mg, 0,036 mmol, 100 %). Es I+ MS m/z 514,3 (100 %) [M+H]+.
Etapa E: 2-(1-Acriloil-4-(2-(((R)-4-metilmorfolin-3-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo. Se preparó según el procedimiento del Ejemplo 375 Etapa G, sustituyendo 2-(4-(2-(((R)
4-metilmorfolin-3-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (18,2 mg, 0,036 mmol) para proporcionar el compuesto del título 2-(1-acriloil-4-(2-(((R)-4-metilmorfolin-3-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0 446, 14 mg, 0,025 mmol, 69 %). ESI+ MS m/z 568,3 (100 %) [M+H]+.
Ejemplo 447

2-((S)-1-Acriloil-4-(2-(((S)-4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (S)-4-(2-(((S)-1-(terc-Butoxicarbonil)-4,4-difluoropirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo. Se preparó según el procedimiento del Ejemplo 375 Etapa C, sustituyendo (S)-4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (50 mg, 0,090 mmol) y (S)-4,4-difluoro-2-(hidroximetil)pirrolidin-1 -carboxilato de terc-butilo (64,3 mg, 0,271 mmol), purificando por cromatografía ultrarrápida eluyendo con 25-100 % de hex/EtOAc para proporcionar (S)-4-(2-(((S)-1-(terc-butoxicarbonil)-4,4-difluoropirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (15 mg, 0,020 mmol, 22,0 % de rendimiento). ESI+ MS m/z 754,3 (100 %) [M+H]+.
Etapa B: Clorhidrato de (S)-2-(cianometil)-4-(2-(((S)-4,4-difluoropirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo. Se preparó según el procedimiento del Ejemplo 375 Etapa D, sustituyendo (S)-4-(2-(((S)-1-(terc-butoxicarbonil)-4,4-difluoropirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (18 mg, 0,024 mmol) para proporcionar clorhidrato de (S)-2-(cianometil)-4-(2-(((S)-4,4-difluoropirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (16 mg, 0,023 mmol, 100 %). ESI+ MS m/z 654,3 (100 %) [M+H]+.
Etapa C: (S)-2-(Cianometil)-4-(2-(((S)-4.4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo. Se preparó según el procedimiento del Ejemplo 443 etapa C. sustituyendo (S)-2-(cianometil)-4-(2-(((S)-4.4-difluoropirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6.7.8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (16 mg. 0.023 mmol). para proporcionar (S)-2-(cianometil)-4-(2-(((S)-4.4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7,8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (16,0 mg. 0,024 mmol. 52 %). ESI+ MS m/z 668,3 (100 %) [M+H]+.
Etapa D: 2-((S)-4-(2-(((S)-4,4-Difluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo. Se preparó según el procedimiento del Ejemplo 375 Etapa F. sustituyendo (S)-2-(cianometil)-4-(2-(((S)-4.4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (16,0 mg. 0,024 mmol) para proporcionar 2-((S)-4-(2-(((S)-4,4-difluoro-1 -metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (8.0 mg. 0,015 mmol. 63 %). ESI+ MS m/z 534,3 (100 %) [M+H]+.
Etapa E: 2-((S)-1-Acriloil-4-(2-(((S)-4.4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo. Se preparó según el procedimiento del Ejemplo 375 Etapa G. sustituyendo 2-((S)-4-(2-(((S)-4.4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (8.0 mg. 0,015 mmol) para proporcionar el compuesto del título 2-((S)-1-acriloil-4-(2-(((S)-4,4-difluoro-1 -metilpirrolidin-2-il)metoxi)-7-(naftalen-1 -il)-5,6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0 447, 4 mg. 0,007 mmol. 45 %). ESI+ mS m/z 588,3 (100 %) [M+H]+.
Ejemplo 448

2-((S)-1-Acriloil-4-(2-(((R)-4-metil-7-oxa-4-azaespiro[2,5]octan-6-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (R)-6-(((4-((S)-4-((Benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7.8-tetrahidropirido[3.4-dlpirimidin-2-inoxi)metin-7-oxa-4-azaespiro[2,51octano-4-carboxilato de terc-butilo. Se preparó según el procedimiento del Ejemplo 375 Etapa C. sustituyendo (S)-4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (25 mg. 0,045 mmol) y (R)-6-(hidroximetil)-7-oxa-4-azaespiro[2,5]octano-4-carboxilato de terc-butilo (33,0 mg. 0,136 mmol). purificando por cromatografía ultrarrápida eluyendo con 25-100 % de hex/EtOAc para proporcionar (R)-6-(((4-((S)-4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-2-il)oxi)metil)-7-oxa-4-azaespiro[2,5]octano-4-carboxilato de terc-butilo (35 mg. 0,0461 mmol. 100 % de rendimiento). ESl+ MS m/z 760.3 (100 %) [M+H]+.
Etapa B: Clorhidrato de (S)-4-(2-(((R)-7-oxa-4-azaespiro[2.51octan-6-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)-2-(cianometil)piperazin-1 -carboxilato de bencilo. Se preparó según el procedimiento del Ejemplo 375 Etapa D. sustituyendo (R)-6-(((4-((S)-4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)-7-oxa-4-azaespiro[2,5]octano-4-carboxilato de terc-butilo (35 mg. 0,046 mmol) para proporcionar clorhidrato de (S)-4-(2-(((R)-7-oxa-4-azaespiro[2,5]octan-6-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (32 mg. 0,046 mmol. 100 %). ESI+ MS m/z 660,3 (100 %) [M+H]+.
Etapa C: (S)-2-(Cianometil)-4-(2-(((R)-4-metil-7-oxa-4-azaespiro[2.51octan-6-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo. Se preparó según el procedimiento del Ejemplo 443 etapa C. sustituyendo clorhidrato de (S)-4-(2-(((R)-7-oxa-4-azaespiro[2,5]octan-6-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (32 mg. 0,046 mmol). para proporcionar (S)-2-(cianometil)-4-(2-(((R)-4-metil-7-oxa-4-azaespiro[2,5]octan-6-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (31 mg. 0,046 mmol. 100 %). ESI+ MS m/z 674,3 (100 %) [M+H]+.
Etapa D: 2-((S)-4-(2-(((R)-4-Metil-7-oxa-4-azaespiro[2.5]octan-6-N)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo. Se preparó según el procedimiento del Ejemplo 375 Etapa F. sustituyendo (S)-2-(cianometil)-4-(2-(((R)-4-metil-7-oxa-4-azaespiro[2,5]octan-6-il)metoxi)-7-(naftalen-1-il)-5,6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (31,0 mg. 0,046 mmol) para proporcionar 2-((S)-4-(2-(((R)-4-metil-7-oxa-4-azaespiro[2,5]octan-6-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (18,0 mg. 0,033 mmol. 72 %). ESI+ Ms m/z 540,3 (100 %) [M+H]+.
Etapa E: 2-((S)-1-Acriloil-4-(2-(((R)-4-metil-7-oxa-4-azaespiro[2.51octan-6-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo. Se preparó según el procedimiento del Ejemplo 375 Etapa G. sustituyendo 2-((S)-4-(2-(((R)-4-metil-7-oxa-4-azaespiro[2,5]octan-6-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (18,0 mg. 0,033 mmol) para proporcionar el compuesto del título 2-((S)-1-acriloil-4-(2-(((R)-4-metil-7-oxa-4-azaespiro[2,5]octan-6-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0448, 5.8 mg. 0,010 mmol.29 %). ESI+ MS m/z 594.3 (100 %) [M+H]+.
Ejemplo 449

2-(1-Acriloil-4-(7-(2-(difluorometil)fenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A. 2-(C¡anomet¡l)-4-(7-(2-(d¡fluoromet¡l)fen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo. Una mezcla de 2-(c¡anomet¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (30 mg. 0.0593 mmol). 1-Bromo-2-d¡fluoromet¡lbenceno (36,8 mg. 0,178 mmol). BINAP Palladacycle Gen. 3 (5.9 mg. 0,006 mmol) y Cs2Co3 (77,3 mg.
0,237 mmol) en d¡oxano (593 μl. 0,0593 mmol) se roc¡ó con Ar durante 5 m¡n, después se selló y se calentó a 100 °C durante 1 d. La mezcla de reacc¡ón se cargó sobre una muestra de gel de síl¡ce y se pur¡f¡có por cromatografía ultrarráp¡da eluyendo con un grad¡ente de 1-10 % de Me0H/DCM (1 % de NH40H) para proporc¡onar 2-(c¡anomet¡l)-4-(7-(2-(d¡fluoromet¡l)fen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (12 mg. 0,019 mmol. 32 %). ESI+ MS m/z 632,3 (100 %) [M+H]+.
Etapa B: 2-((S)-4-(2-(((R)-4-Met¡l-7-oxa-4-azaesp¡ro[2.5]octan-6-¡l)metox¡)-7-(naftalen-1-¡l)-5,6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo. Se preparó según el proced¡m¡ento del Ejemplo 375 Etapa F. sust¡tuyendo 2-(c¡anomet¡l)-4-(7-(2-(d¡fluoromet¡l)fen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (12,0 mg. 0,019 mmol) para proporc¡onar 2-(4-(7-(2-(d¡fluoromet¡l)fen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (9.5 mg. 0,019 mmol. 100 %). ESI+ MS m/z 498,3 (100 %) [M+H]+.
Etapa C: 2-(1-Acr¡lo¡l-4-(7-(2-(d¡fluoromet¡l)fen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo. Se preparó según el proced¡m¡ento del Ejemplo 375 Etapa G. sust¡tuyendo 2-(4-(7-(2-(d¡fluoromet¡l)fen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (9.5 mg. 0,019 mmol) para proporc¡onar el compuesto del título 2-(1-acr¡lo¡l-4-(7-(2-(d¡fluoromet¡l)fen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (EJEMPL0 449, 4.2 mg. 0,008 mmol. 40 %). ESI+ MS m/z 552,3 (100 %) [M+H]+.
Ejemplo 450

2-((S)-1-Acriloil-4-(2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (R)-2-(((4-((S)-4-((Benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-2-il)oxi)metil)morfolin-4-carboxilato de terc-butilo. Se preparó según el procedimiento del Ejemplo 443 Etapa A, sustituyendo (S)-4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (95 mg, 0,172 mmol) y (R)-N-Boc-2-hidroximetilmorfolina (112 mg, 0,515 mmol), purificando por cromatografía ultrarrápida eluyendo con 25-100 % de hex/EtOAc para proporcionar (R)-2-(((4-((S)-4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)morfolin-4-carboxilato de terc-butilo (126 mg, 0,172 mmol, 100 % de rendimiento). ESI+ MS m/z 734,3 (100 %) [M+H]+.
Etapa B: Clorhidrato de (S)-2-(cianometil)-4-(2-(((R)-morfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: Se preparó según el procedimiento del Ejemplo 375 Etapa D, sustituyendo (R)-2-(((4-((S)-4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)morfolin-4-carboxilato de terc-butilo (126 mg, 0,172 mmol) para proporcionar clorhidrato de (S)-2-(cianometil)-4-(2-(((R)-morfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (109 mg, 0,172 mmol, 100 %). ESI+ MS m/z 634,3 (100 %) [M+H]+.
Etapa C: (S)-2-(Cianometil)-4-(2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo. Se preparó según el procedimiento del Ejemplo 443 Etapa C, sustituyendo (S)-2-(cianometil)-4-(2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4
d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (109 mg, 0,172 mmol) para proporcionar (S)-2-(cianometil)-4-(2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (76 mg, 0,117 mmol, 68 %). ESI+ MS m/z 648,3 (100 %) [M+H]+.
Etapa D: 2-((S)-4-(2-(((R)-4-Metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo. Se preparó según el procedimiento del Ejemplo 375 Etapa F, sustituyendo (S)-2-(cianometil)-4-(2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (76 mg, 0,117 mmol) para proporcionar 2-((S)-4-(2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (57,1 mg, 0,111 mmol, 95 %). ESI+ MS m/z 514,3 (100 %) [M+H]+.
Etapa E: 2-(1-Acriloil-4-(2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo. Se preparó según el procedimiento del Ejemplo 375 Etapa G, sustituyendo 2-((S)-4-(2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (56 mg, 0,11 mmol) para proporcionar el compuesto del título 2-((S)-1-acriloil-4-(2-(((R)-4-metilmorfolin-2-il)metoxi)-7-(naftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0 450, 50 mg, 0,088 mmol, 81 %). ESI+ MS m/z 568,3 (100 %) [M+H]+.
Ejemplo 451

2-((S)-1-Acriloil-4-(2-(((2R,SR)-4,5-dimetilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (2R.5R)-2-(((4-((S)-4-((Benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)-5-metilmorfolin-4-carboxilato de terc-butilo. Se preparó según el procedimiento del Ejemplo 375 Etapa C. sustituyendo (S)-4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (95 mg, 0,172 mmol) y (2R,5R)-2-(hidroximetil)-5-metilmorfolin-4-carboxilato de terc-butilo (119 mg. 0,515 mmol). purificando por cromatografía ultrarrápida eluyendo con 25-100 % de hex/EtOAc para proporcionar (2R,5R)-2-(((4-((S)-4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)-5-metilmorfolin-4-carboxilato de terc-butilo (128 mg. 0,171 mmol. 100 % de rendimiento). ESI+ MS m/z 748,3 (100 %) [M+H]+.
Etapa B: (S)-2-(Cianometil)-4-(2-(((2R.5R)-5-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5.6,7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo. Se preparó según el procedimiento del Ejemplo 375 Etapa D. sustituyendo (2R,SR)-2-(((4-((S)-4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)-5-metilmorfolin-4-carboxilato de terc-butilo (128 mg. 0,171 mmol) para proporcionar (S)-2-(cianometil)-4-(2-(((2R,5R)-5-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (111 mg. 0,171 mmol. 100 %). ESI+ MS m/z 648,3 (100 %) [M+H]+.
Etapa C: (S)-2-(Cianometil)-4-(2-(((2R.5R)-4.5-dimetilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5.6,7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo. Se preparó según el procedimiento del Ejemplo 443 etapa C. sustituyendo (S)-2-(cianometil)-4-(2-(((2R,SR)-5-metilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (111 mg. 0,171 mmol). para proporcionar (S)-2-(cianometil)-4-(2-(((2R,5R)-4,5-dimetilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (56 mg. 0,085 mmol. 49 %). ESI+ mS m/z 662,3 (100 %) [M+H]+.
Etapa D: 2-((S)-4-(2-(((2R.5R)-4.5-Dimetilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo. Se preparó según el procedimiento del Ejemplo 375 Etapa F. sustituyendo (S)-2-(cianometil)-4-(2-(((2R,5R)-4,5-dimetilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (56 mg. 0,085 mmol) para proporcionar 2-((S)-4-(2-(((2R,5R)-4,5-dimetilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (44,3 mg.
0,084 mmol. 99 %). ESI+ MS m/z 528,3 (100 %) [M+H]+.
Etapa E: 2-((S)-1-Acriloil-4-(2-(((2R.5R)-4.5-dimetilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5.6,7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo. Se preparó según el procedimiento del Ejemplo 375 Etapa G. sustituyendo 2-((S)-4-(2-(((2R,5R)-4,5-dimetilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (43 mg. 0,081 mmol) para proporcionar el compuesto del título 2-((S)-1 -acriloil-4-(2-(((2R,5R)-4,5-dimetilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0451, 32 mg. 0,055 mmol. 68 %). ESI+ MS m/z 582,3 (100 %) [M+H]+.
Ejemplo 452

2-((S)-1-Acriloil-4-(7-(naftalen-1-il)-2-(((R)-4,5,5-trimetilmorfolin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (R)-2-(((4-((S)-4-((Benciloxi)carbonil)-3-(eyanometil)piperazin-1-il)-7-(naftalen-1 -il)-5,6.7.8-tetrahidropiridor3,4-d1pirimidin-2-il)oxi)metil)-5,5-dimetilmorfolin-4-carboxilato de terc-butilo. Se preparó según el procedimiento del Ejemplo 375 Etapa C. sustituyendo (S)-4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (95 mg, 0,172 mmol) y (R)-2-(hidroximetil)-5,5-dimetilmorfolin-4-carboxilato de terc-butilo (126 mg, 0,515 mmol), purificando por cromatografía ultrarrápida eluyendo con 25-100 % de hex/EtOAc para proporcionar (R)-2-(((4-((S)-4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)-5,5-dimetilmorfolin-4-carboxilato de terc-butilo (131 mg, 0,172 mmol, 100 % de rendimiento). ESI+ MS m/z 762,3 (100 %) [M+H]+.
Etapa B: (S)-2-(Cianometil)-4-(2-(((R)-5.5-dimetilmorfolin-2-il)metoxi)-7-(naftalen-1 -iP^^J^-tetrahidropirido^^-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo. Se preparó según el procedimiento del Ejemplo 375 Etapa D, sustituyendo (R)-2-(((4-((S)-4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)metil)-5,5-dimetilmorfolin-4-carboxilato de terc-butilo (131 mg, 0,172 mmol) para proporcionar (S)-2-(cianometil)-4-(2-(((R)-5,5-dimetilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (114 mg, 0,172 mmol, 100 %). ESI+ MS m/z 662,3 (100 %) [M+H]+.
Etapa C: (S)-2-(Cianometil)-4-(7-(naftalen-1-il)-2-(((R)-4.5.5-trimetilmorfolin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo. Se preparó según el procedimiento del Ejemplo 443 etapa C, sustituyendo (S)-2-(cianometil)-4-(2-(((R)-5,5-dimetilmorfolin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (114 mg, 0,172 mmol), para proporcionar (S)-2-(cianometil)-4-(7-(naftalen-1-il)-2-(((R)-4,5,5-trimetilmorfolin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (77 mg, 0. 114 mmol, 66 %). ESI+ MS m/z 676,3 (100 %) [M+H]+.
Etapa D: 2-((S)-4-(7-(Naftalen-1-il)-2-(((R)-4.5.5-trimetilmorfolin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo. Se preparó según el procedimiento del Ejemplo 375 Etapa F, sustituyendo (S)-2-(cianometil)-4-(7-(naftalen-1 -il)-2-(((R)-4,5,5-trimetilmorfolin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (77 mg, 0,114 mmol) para proporcionar 2-((S)-4-(7-(naftalen-1-il)-2-(((R)-4,5,5-trimetilmorfolin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (47 mg, 0,087 mmol, 77 %). ESI+ MS m/z 542,3 (100 %) [M+H]+.
Etapa E: 2-((S)-1-Acriloil-4-(7-(naftalen-1-il)-2-(((R)-4.5.5-trimetilmorfolin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo■ Se preparó según el procedimiento del Ejemplo 1 Etapa G, sustituyendo 2-((S)-4-(7-(naftalen-1 -il)-2-(((R)-4.5.5-trimetilmorfolin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo (46 mg, 0,085 mmol) para proporcionar el compuesto del título 2-((S)-1-acriloil-4-(7-(naftalen-1-il)-2-(((R)-4,5,5-trimetilmorfolin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0 452, 36 mg, 0,060 mmol, 71 %). ESI+ MS m/z 582,3 (100 %) [M+H]+.
E je m p lo 453

1-[4-[7-(5,6-dimetil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -il]prop-2-en-1 -ona

Etapa 1: 3-bromo-1,2,4-trimetil-5-nitro-benceno. A una solución de 1,2,4-trimetil-5-nitro-benceno (10 g, 60 mmol, 1 eq.) en DCE (200 ml) se le añadieron FeBr3 (358 mg, 1,21 mmol, 0,02 eq.), Fe (879 mg, 15,7 mmol, 0,26 eq.) seguido de Br2 (11,6 g, 72,6 mmol, 3,74 ml, 1,2 eq.) gota a gota. Después de agitar a 25 °C durante 1 hora, la mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (20 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El producto bruto se suspendió en agua y se recogió por filtración. Se obtuvo 3-bromo-1,2,4-trimetil-5-nitro-benceno (8,6 g, 34,9 mmol, 58 % de rendimiento, 99 % de pureza) en forma de un sólido de color blanco.
Etapa 2: 3-bromo-2,4,5-trimetil-anilina. A una solución de 3-bromo-1,2,4-trimetil-5-nitro-benceno (5,2 g, 21,3 mmol, 1 eq.) en Et0H (100 ml) se le añadieron Fe (5,95 g, 107 mmol, 5 eq.) y Ac0H (12,8 g, 213 mmol, 12,2 ml, 10 eq.). La mezcla se agitó a 60 °C durante 3 horas. Después de que se completara, la mezcla de reacción se filtró y el filtrado se concentró. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico)/acetonitrilo)]. Las fracciones deseadas se recogieron, se neutralizaron con NaHC03 acuoso saturado y
después se concentraron al vacío para eliminar el MeCN y se extrajeron con EtOAc (2 x 100 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío para dar 3-bromo-2,4,5-trimetil-anilina (3,4 g, 14,3 mmol, 67 % de rendimiento, 90 % de pureza) en forma de un sólido de color amarillo.
1H RMN (400 MHz, cloroformo-d) ó = 6,51 (s, 1H), 3,56 (s a, 2H), 2,32 (s, 3H), 2,31 (s, 3H), 2,26 (s, 3H).
Etapa 3: 4-bromo-5,6-d¡met¡l-1 H-indazol. Se disolvió 3-bromo-2,4,5-trimetil-anilina (200 mg, 934,14 umol, 1 eq.) en trifluoroborano;fluorhidrato (1,69 g, 7,71 mmol, 1,20 ml, 8,25 eq., 40 % en agua) gota a gota a 0 °C. Una solución acuosa enfriada de NaN02 (96,7 mg, 1,40 mmol, 1,5 eq.) (en la cantidad mínima de agua para que fuera acuosa saturada). Después de la adición, la mezcla se agitó a 0 °C durante 1 hora y a 25 °C durante 0,5 horas. Se añadió NaN02 (64 mg, en la cantidad mínima de agua para que fuera acuosa saturada) se le añadió a 0 °C y la mezcla se agitó a 0 °C durante 0,5 horas. Después, el precipitado resultante se filtró, se lavó con (i-Pr)20 (40 ml) y se concentró al vacío, que se añadió directamente en una porción en una atmósfera de N2 a una mezcla agitada de K0Ac (183 mg, 1,87 mmol, 2 eq.) y 18-corona-6 (12,4 mg, 46,7 umol, 0,05 eq.) en CHCb (8 ml). La mezcla se agitó a 35 °C durante 0,5 horas. Después de que se completara, la mezcla se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP/EtOAc 100/1 a 5/1) para dar 4-bromo-5,6-dimetil-1 H-indazol (70 mg, 280 umol, 30 % de rendimiento, 90 % de pureza) en forma de un sólido de color amarillo.
Etapa 4: 4-bromo-5.6-d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol. A una solución de 4-bromo-5,6-dimetil-1 H-indazol (290 mg, 1,29 mmol, 1 eq.) en DCM (6 ml) se le añadió T s0 H ^0 (24,5 mg, 129 umol, 0,1 eq.) seguido de DHP (217 mg, 2,58 mmol, 236 ul, 2 eq.) y MeCN (1 ml). La mezcla se agitó a 25 °C durante 5 horas. Después de que se completara, la mezcla se inactivó con una solución acuosa saturada de NaHC03 (5 ml) y se diluyó con agua (20 ml). Las capas se separaron y la fase acuosa se extrajo con EtOAc (2 x 200 ml). Las capas orgánicas combinadas se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP/EtOAc 100/1 a 25/1) para dar 4-bromo-5,6-dimetil-1-tetrahidropiran-2-il-indazol (320 mg, 828 umol, 64 % de rendimiento, 80 % de pureza) en forma de un aceite de color amarillo. LCMS [ESI, M+1 ]: 309.
1H RMN (400 MHz, cloroformo-d) ó = 7,94 (s, 1H), 7,33 (s, 1H), 5,66 (dd, J = 2,8, 9,2 Hz, 1H), 4,06 - 3,97 (m, 1H), 3,79 - 3,67 (m, 1H), 2,47 (s, 3H), 2,44 (s, 3H), 2,16 (cd, J = 4,4, 7,6 Hz, 1H), 2,06 (cd, J = 3,2, 13,2 Hz, 1H), 1,83 - 1,64 (m, 4H).
Etapa A: 4-[7-(5,6-dimetil-1-tetrahidropiran-2-il-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de tere-butilo. Se desgasificaron 4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (385 mg, 890 umol, 1 eq.), 4 bromo-5,6-dimetil-1-tetrahidropiran-2-il-indazol (303 mg, 979 umol, 1,10 eq.), Pd2(dba)3 (163 mg, 178 umol, 0,2 eq.), RuPhos (166 mg, 356 umol, 0,4 eq.) y Cs2C03 (725 mg, 2,23 mmol, 2,5 eq.) en tolueno (30 ml) y después se calentaron a 90 °C durante 8 horas en una atmósfera de N2. Después de que se completara, la mezcla se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EtOAc/Me0H 100/1 a 5/1) para dar 4-[7-(5,6-dimetil-1-tetrahidropiran-2-il-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (200 mg, 275 umol, 31 % de rendimiento, 91 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 661.
Etapa B: 7-(5,6-dimetil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-dlpirimidina. A una solución de 4-[7-(5,6-dimetil-1-tetrahidropiran-2- yl-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (190 mg, 288 umol, 1 eq.) en DCM (0,4 ml) se le añadió TFA (924 mg, 8,10 mmol, 0,6 ml, 28,2 eq.). La mezcla se agitó a 25 °C durante 1 hora.
Después de que se completara, la mezcla se concentró al vacío para dar 7-(5,6-dimetil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidina (220 mg, bruto, 2TFA) en forma de un aceite de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional.
Etapa C: 1-[4-[7-(5,6-dimetil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]- 6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l1p¡peraz¡n-1-¡l1prop-2-en-1-ona. A una solución de 7-(5,6-dimetil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidina (200 mg, 284 umol, 1 eq., 2 TfA) y TeA (574 mg, 5,68 mmol, 790 ul, 20 eq.) en DCM (4 ml) se le añadió gota a gota prop-2-enoato de prop-2-enoílo (35,8 mg, 284 umol, 1 eq.) a -40 °C. La mezcla se agitó a -40 °C durante 30 minutos. Después de que se completara, la mezcla se inactivó con Me0H (0,5 ml), se diluyó con agua (5 ml) y después se extrajo con DCM (2 x 10 ml). Los extractos se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía (Ah03, EtOAc/Me0H 100/1 a 10/1) y HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 35 %-65 %, 12 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 1-[4-[7-(5,6-dimetil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]- 6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (EJEMPL0453, 45,3 mg, 85,2 umol, dos etapas 30 % de rendimiento, 99,7 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 531.
Condición de SFC: Columna: Chiralcel 0J-3100 x 4 ,6 mm D.I., 3 um, Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 %, Caudal: 3 ml/min, Longitud de onda: 220 nm.
1H RMN (400 MHz, cloroformo-d) 5 = 10,84 (s a, 1H), 8,02 (s, 1H), 7,13 (s, 1H), 6,59 (dd, J = 10,4, 16,8 Hz, 1H), 6,34 (dd, J = 1,2, 16,8 Hz, 1H), 5,79 - 5,68 (m, 1H), 4,40 (dd, J = 4,8, 10,4 Hz, 1H), 4,26 (s, 2H), 4,17 (dd, J = 6,8, 10,4 Hz, 1H), 3,90 - 3,39 (m, 10H), 3,09 (t a, J = 7,6 Hz, 1H), 2,89 - 2,62 (m, 3H), 2,47 (s, 3H), 2,39 (s, 3H), 2,33 (s, 3H), 2,30 -2,23 (m, 1H), 2,09 - 1,97 (m, 1H), 1,91 - 1,69 (m, 3H).
Ejemplo 454

-[(2S)-4-[7-(8-cloro-1-naft¡l)-2-[2-(4,4-d¡fluoro-1-p¡per¡d¡l)etox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Etapa 1: 4,4-difluoropiperidina. A una soluc¡ón de 4,4-d¡fluorop¡per¡d¡n-1-carbox¡lato de terc-but¡lo (2,0 g, 9,04 mmol, 1,0 eq.) en DCM (0,50 ml) se le añad¡ó TFA (770 mg, 6,75 mmol, 0,50 ml, 7,47 e-1 eq.). La mezcla se ag¡tó a 25 °C durante 0,5 horas. Después de que se completara, la mezcla de reacc¡ón se concentró al vacío para dar el producto 4,4-d¡fluorop¡per¡d¡na (1,50 g, bruto, TFA) en forma de un ace¡te de color amar¡llo que se usó en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón ad¡c¡onal.
Etapa 2: 1-(2-benciloxietil)-4,4-difluoro-piperidina. A una soluc¡ón de 4,4-d¡fluorop¡per¡d¡na (1,50 g, 6,38 mmol, 1,0 eq., TFA) en MeCN (20,0 ml) se le añad¡ó Na0H (765 mg, 19,1 mmol, 3,0 eq.). La mezcla se ag¡tó a 25 °C durante 0,5 horas. Después, al líqu¡do anter¡or se le añad¡eron 2-bromoetox¡met¡lbenceno (1,65 g, 7,65 mmol, 1,21 ml, 1,20 eq.) y KI (212 mg, 1,28 mmol, 0,20 eq.) y la mezcla se ag¡tó a 90 °C durante 12 horas. Después de que se completara, a la
mezcla de reacción se le añadió agua (20,0 ml) y después se extrajo con AE (3 x 20,0 ml). Las capas orgánicas combinadas se secaron sobre Na2SÜ4, se filtraron y se concentraron. El residuo se purificó por cromatografía en columna (SiÜ2, éter de petróleo: acetato de etilo = 10:1 a 0:1) para dar 1-(2-benciloxietil)-4,4-difluoro-piperidina (600 mg, 2,35 mmol, dos etapas 37 % de rendimiento) en forma de un aceite de color amarillo.
1H RMN (400 MHz, Cloroformo-d) ó 7,41 - 7,29 (m, 5H), 4,54 (s, 2H), 3,82 - 3,72 (m, 2H), 3,33 - 2,94 (m, 6H), 2,37 -2,15 (m, 4H).
Etapa 3: 2-(4,4-d¡fluoro-1-p¡per¡d¡l)etanol. Una solución de 1-(2-benciloxietil)-4,4-difluoro-piperidina (400 mg, 1,57 mmol, 1,0 eq.) y Pd/C (200 mg, 10 % de pureza) en Et0H (5,0 ml) se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 12 horas. Después de que se completara, la mezcla de reacción se filtró a través de Celite y la torta de filtro se lavó con THF (10,0 ml). El filtrado se recogió y se concentró para dar el producto 2-(4,4-difluoro-1-piperidil)etanol (170 mg, 1,03 mmol, 66 % de rendimiento) en forma de un aceite incoloro. El producto se usó en la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, Cloroformo-d) ó 3,84 - 3,70 (m, 2H), 2,98 - 2,89 (m, 4H), 2,86 - 2,80 (m, 3H), 2,24 - 2,11 (m, 4H). Etapa A: (2S)-4-[7-(8-cloro-1-naftil)-2-[2-(4,4-difluoro-1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de terc-butilo. A una solución de 2-(4,4-difluoro-1-piperidil)etanol (102 mg, 619 umol, 3,0 eq.) en tolueno (3,0 ml) y (2S)-4-[7-(8-cloro-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (120 mg, 206 umol, 1,0 eq.) se le añadió t-Bu0Na (39,7 mg, 413 umol, 2,0 eq.) y la mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, a la mezcla de reacción se le añadió agua (10,0 ml) y se extrajo con AE (3 x 10,0 ml). La capa orgánica se secó sobre Na2S04, se filtró y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa (C18, 0,1 % de NH3^H20, 0 -60 % de MeCN) para dar el producto (2S)-4-[7-(8-cloro-1-naftil)-2-[2-(4,4-difluoro-1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (90,0 mg, 132 umol, 64 % de rendimiento) en forma de un aceite de color amarillo. Lc MS [ESI, M+1]: 682.
Etapa B: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[2-(4,4-difluoro-1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-[2-(4,4-difluoro-1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de terc-butilo (30,0 mg, 44,0 umol, 1,0 eq.) en DCM (0,5 ml) se le añadió TFA (770 mg, 6,75 mmol, 0,50 ml, 154 eq.). La mezcla se agitó a 25 °C durante 0,5 horas. Después de que se completara, la mezcla se concentró. El residuo se ajustó con NaHC03 acuoso saturado a pH ~7 y después se extrajo con Ae (3 x 5 ,0 ml). La capa orgánica se secó sobre Na2S04, se filtró y se concentró para dar el producto 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[2-(4,4-difluoro-1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (26,0 mg, bruto) en forma de un aceite de color amarillo que se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 582.
Etapa C: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[2-(4,4-difluoro-1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[2-(4,4-difluoro-1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (26,0 mg, 44,7 umol, 1,0 eq.) y DIEA (69,3 mg, 536 umol, 93,4 ul, 12,0 eq.) en DCM (1,0 ml) se le añadió prop-2-enoato de prop-2-enoílo (8,45 mg, 67,0 umol, 1,50 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se inactivó con la adición de Me0H (1,0 ml) y después se concentró para dar un residuo. Después, el residuo se purificó por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 47 % - 77 %, 12 min) para dar el compuesto del título 2-[(2Sj-4-[7-(8-cloro-1-naftil)-2-[2-(4,4-difluoro-1-piperidil)etoxi]-6,8-dihidro-5H-piridoÍ3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0454, 6,35 mg, 9,82 umol, dos etapas 22 % de rendimiento, 98 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 636. 1H RMN (400 MHz, Cloroformo-d) ó 7,76 (d a, J = 8,0 Hz, 1H), 7,63 (t, J = 7,2 Hz, 1H), 7,53 (d, J = 7,2 Hz, 1H), 7,49 -7,40 (m, 1H), 7,34 (t, J = 7,8 Hz, 1H), 7,27 - 7,17 (m, 1H), 6,67 - 6,54 (m, 1H), 6,40 (d a, J = 16,4 Hz, 1H), 5,83 (d a, J = 11,2 Hz, 1H), 5,20 - 4,56 (m, 1H), 4,50 - 4,35 (m, 3H), 4,22 - 4,01 (m, 2H), 3,98 - 3,75 (m, 2H), 3,66 - 3,57 (m, 1H), 3,53 - 3,36 (m, 1H), 3,31 - 2,98 (m, 4H), 2,91 - 2,80 (m, 3H), 2,79 - 2,49 (m, 6H), 2,09 - 1,94 (m, 4H).
Ejemplo 455

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[2-(3,3-difluoro-1-piperidil)etoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Etapa 1: 1-(2-benciloxietil)-3,3-difluoro-piperidina. A una solución de 3,3-difluoropiperidina (1,0 g, 6,35 mmol, 1,0 eq., HCl) en MeCN (6,0 ml) se le añadió en una porción Na0H (761 mg, 19,0 mmol, 3,0 eq.) a 25 °C. La mezcla se agitó a 25 °C durante 5 minutos y después se añadieron 2-bromoetoximetilbenceno (1,64 g, 7,61 mmol, 1,20 ml, 1,20 eq.) y KI (211 mg, 1,27 mmol, 0,20 eq.). La mezcla se agitó a 90 °C durante 12 horas. Después de que se completara, a la mezcla de reacción se le añadió H20 (15,0 ml) y después se extrajo con AE (3 * 20,0 ml). Las capas orgánicas combinadas se secaron sobre Na2S04, se filtraron y se concentraron para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 10:1 a 5:1) para dar 1 -(2-benciloxietil)-3,3-difluoro-piperidina (1,40 g, 5,48 mmol, 86 % de rendimiento) en forma de un aceite incoloro.
1H RMN (400 MHz, Cloroformo-d) 67,38 - 7,31 (m, 5H), 4,56 (s, 2H), 3,64 (t, J = 5,8 Hz, 2H), 2,79 - 2,73 (m, 4H), 2,55 (t, J = 5,4 Hz, 2H), 1,85 - 1,85 (m, 2H), 1,82 - 1,76 (m, 2H).
Etapa 2: 2-(3,3-d¡fluoro-1-p¡per¡d¡l)etanol. A una solución de 1-(2-benciloxietil)-3,3-difluoro-piperidina (400 mg, 1,57 mmol, 1,0 eq.) en Me0H (5,0 ml) se le añadió Pd/C (200 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 40 °C durante 12 horas. Después de que se completara, la mezcla de reacción se filtró y se concentró para dar 2-(3,3-difluoro-1 -piperidil)etanol (240 mg, 1,45 mmol, 93 % de rendimiento) en forma de un aceite incoloro. El producto se usó en la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, Cloroformo-d) 63,64 (t, J = 5,4 Hz, 2H), 2,73 (t, J = 11,2 Hz, 2H), 2,63 (t, J = 5,4 Hz, 2H), 2,54 (t, J = 5,2 Hz, 2H), 1,97 - 1,87 (m, 2H), 1,82 - 1,76 (m, 2H).
Etapa A: (2S)-4-[7-(8-cloro-1-naft¡l)-2-(2-(3,3-d¡fluoro-1-p¡per¡d¡l)etox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de tere-butilo. Una mezcla de (2S)-4-[7-(8-cloro-1-naft¡l)-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de fere-butilo (230 mg, 396 umol, 1,0 eq.), 2-(3,3-difluoro-1 -piperidil)etanol (196 mg, 1,19 mmol, 3,0 eq.) y t-Bu0Na (114 mg, 1,19 mmol, 3,0 eq.) en tolueno (5,0 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 0 °C durante 0,5 horas en una atmósfera de N2. Después de que se completara, la mezcla de reacción se inactivó con H20 (5,0 ml) y se extrajo con AE (3 * 10,0 ml). La capa orgánica combinada se lavó con una solución acuosa saturada de NaCl (20,0 ml), se secó sobre Na2S04, se filtró y se concentró para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 3:1 a 0:1) para dar (2S)-4-[7-(8-cloro-1-naft¡l)-2-[2-(3,3-d¡fluoro-1-p¡per¡d¡l)etox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de fere-butilo (250 mg, 344 umol, 87 % de rendimiento, 94 % de pureza) en forma de un aceite de color amarillo.
1H RMN (400 MHz, Cloroformo-d) 67,77 (d, J = 8,0 Hz, 1H), 7,63 (t, J = 8,0 Hz, 1H), 7,54 (d, J = 7,2 Hz, 1H), 7,50 -7,42 (m, 1H), 7,37 - 7,33 (m, 1H), 7,27 - 7,20 (m, 1H), 4,67 - 4,64 (m, 1H), 4,48 - 4,39 (m, 3H), 4,06 (d, J = 12,8 Hz, 1H), 4,01 - 3,80 (m, 3H), 3,60 (d, J = 4,0 Hz, 1H), 3,37 (dd, J = 3,6, 13,6 Hz, 1H), 3,27 - 3,10 (m, 3H), 2,97 - 2,90 (m, 3H), 2,84 - 2,78 (m, 3H), 2,61 - 2,57 (m, 2H), 1,89 - 1,85 (m, 3H), 1,72 - 1,54 (m, 3H), 1,53 (s, 9H).
Etapa B: 2-[(2S)-4-[7-(8-cloro-1-naft¡l)-2-[2-(3,3-d¡fluoro-1-p¡per¡d¡l)etox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una mezcla de (2S)-4-[7-(8-cloro-1-naft¡l)-2-[2-(3,3-d¡fluoro-1-p¡per¡d¡l)etox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de fere-butilo (200 mg, 293 umol, 1,0 eq.) en DCM (1,50 ml) se le añadió en una porción TFA (2,31 g, 20,3 mmol, 1,50 ml, 69,0 eq.) a 25 °C en una atmósfera de N2. La mezcla se agitó a 25 °C durante 0,5 horas. Después de que se completara, la mezcla se concentró para dar 2-[(2S)-4-[7-(8-cloro-1-naft¡l)-2-[2-(3,3-d¡fluoro-1-p¡per¡d¡l)etox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il)acetonitrilo (400 mg, bruto, 2TFA) en forma de un sólido de color amarillo. El producto se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 582.
Etapa C: 2-[(2S)-4-[7-(8-cloro-1-naft¡l)-2-[2-(3,3-d¡fluoro-1-p¡per¡d¡l)etox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop^-enoil-piperazin^-illacetonitrilo. A una mezcla de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[2-(3,3-difluoro-1-piperidi0etoxi^^-dihidro^H-μmdo^^-dJμmmidin^-ilJpiperazin^-ilJacetonitrilo (400 mg, 494 umol, bruto, 2TFA) y t Ea (727 mg, 7,18 mmol, 1,0 ml) en Dc M (2,0 ml) se le añadió prop-2-enoato de prop-2-enoílo (100 mg, 793 umol) en una porción a 0 °C en una atmósfera de N2. La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se inactivó con la adición de Me0H (3,0 ml) a 0 °C y después se concentró para dar un residuo. Después, el residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil:
[agua (NH4HC0310,0 mM)-ACN]; % de B: 57 %-87 %, 10 min.) y se liofilizó para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naft¡l)-2-[2-(3,3-d¡fluoro-1-p¡per¡d¡l)etox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-prop-2-eno¡lpiperazin-2-i^acetonitrilo (EJEMPL0455, 48,4 mg, 75,8 umol, 26 % de rendimiento, 99 % de pureza) en forma de un sólido de color blanco.
Condición de SFC: 100 % de e.e.
1H RMN (400 MHz, Cloroformo-d) 67,78 (d, J = 8,0 Hz, 1H), 7,64 (t, J = 7,4 Hz, 1H), 7,54 (d, J = 7,6 Hz, 1H), 7,50 -7,43 (m, 1H), 7,36 (t, J = 7,8 Hz, 1H), 7,27 - 7,21 (m, 1H), 6,60 - 6,58 (m, 1H), 6,44 - 6,39 (m, 1H), 5,85 (d, J = 10,4 Hz, 1H), 5,10 - 4,66 (m, 1H), 4,49 - \4,40 (m, 3H), 4,18 - 4,08 (m, 2H), 3,94 - 3,89 (m, 1H), 3,85 - 3,73 (m, 1H), 3,63 - 3,60 (m, 1H), 3,50 - 3,41 (m, 1H), 3,33 - 3,02 (m, 4H), 2,94 - 2,76 (m, 6 H), 2,64 - 2,61 (m, 3H), 1,95 - 1,85 (m, 2H), 1,80 -1,78 (m, 2H). LCMS [ESI, M+1]: 636.
Ejemplo 456

5,6-dimetil-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-4-(4-prop-2-enoilpiperazin-1-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-1,3-dihidrobenzoimidazol-2-ona

Inserción: Etapa 1: 2-bromo-3,4-dimetil-6-nitro-anilina. A una solución de 4,5-dimetil-2-nitro-anilina (5 g, 30,1 mmol, 1 eq.) en CHCb (100 ml) se le añadió gota a gota Br2 (7,21 g, 45,1 mmol, 2,33 ml, 1,5 eq.) a 10 °C. La mezcla de reacción se agitó a 10 °C durante 30 min y a 25 °C durante 30 min. Después de que se completara, la mezcla de reacción se lavó con NaHC03 acuoso saturado (80 ml). La fase acuosa se extrajo con CHCl3 (30 ml). La fase orgánica combinada se secó sobre Na2S04, se filtró a través de una capa de gel de sílice y después se concentró a sequedad.
Se obtuvo 2-bromo-3,4-dimetil-6-nitro-anilina (7 g, 28,5 mmol, 95 % de rendimiento, 99,7 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+3]: 247
Etapa 2: 3-bromo-1,2-d¡met¡l-4.5-d¡n¡tro-benceno. A una solución de 2-bromo-3,4-dimetil-6-nitro-anilina (7 g, 28,6 mmol, 1 eq.) en TFA (50 ml) se le añadió gota a gota peróxido de hidrógeno (25,9 g, 229 mmol, 22,0 ml, 30 % de pureza, 8,0 eq.) a 10 °C. La mezcla de reacción se agitó a 10 °C durante 30 min y a 25 °C durante 30 min y después a 40 °C durante 2 h más. La mezcla de reacción se enfrió a 5 - 10 °C y después se filtró. La torta de filtro se lavó con EP (3 x 20 ml). Después, el sólido se secó a presión reducida a 50 °C. Se obtuvo 3-bromo-1,2-dimetil-4,5-dinitrobenceno (5 g, 18,2 mmol, 64 % de rendimiento) en forma de un sólido de color amarillo.
Etapa A: 4-[7-(2,3-dimetil-5,6-dinitro-fenil)-2-[[(2S)-1-metilpirrolidin-2-inmetoxn-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l1p¡peraz¡n-1-carbox¡lato de tere-butilo. Una mezcla de 4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (250 mg, 578 umol, 1 eq.), 3-bromo-1,2-dimetil-4,5-dinitro-benceno (318 mg, 1,16 mmol, 2 eq.), Pd2(dba)3 (52,9 mg, 57,8 umol, 0,1 eq.), RuPhos (40,5 mg, 86,7 umol, 0,15 eq.) y Cs2CÜ3 (377 mg, 1,16 mmol, 2 eq.) en tolueno (25 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 8 horas en una atmósfera de N2. La mezcla de reacción se diluyó con H20 (100 ml x 1) y acetato de etilo (100 ml x 2). La fase orgánica separada se lavó con salmuera (100 ml x 1 ), se secó sobre sulfato de sodio, se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía sobre gel de sílice (Si02, acetato de etilo/Me0H = 50/1 a 3/1) y cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. Las fracciones deseadas se recogieron, se neutralizaron con una solución saturada de NaHC03 y se extrajeron con acetato de etilo (1 x 100 ml). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. Se obtuvo 4-[7-(2,3-dimetil-5,6-dinitro-fenil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (22,0 mg, 140 umol, 6 % de rendimiento, 99,7 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 627.
Etapa B: 4-[7-(2,3-diamino-5,6-dimetil-fenil)-2-[[(2S)-1-metilpirrolidin-2-inmetoxn-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l1p¡peraz¡n-1-carbox¡lato de tere-butilo. A una solución de 4-[7-(2,3-dimetil-5,6-dinitro-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (120 mg, 191 umol, 1 eq.) en Me0H (10 ml) se le añadió Pd/C (24,0 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. El catalizador se retiró por filtración y el filtrado se concentró al vacío. Se obtuvo 4-[7-(2,3-diamino-5,6- dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terebutilo (100 mg, bruto) en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 567.
Etapa C: 4-[7-(5,6-dimetil-2-oxo-1,3-dihidrobenzoimidazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-inmetoxn-6,8-dihidro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de tere-butilo. A una solución de 4-[7-(2,3-diamino-5,6-dimetil-fenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (100 mg, 176 umol, 1 eq.) en t Hf (10 ml) se le añadió CDI (57,2 mg, 353 umol, 2 eq.). Después de agitar a 50 °C durante 1 h, se añadió CDI (171 mg) y la mezcla se agitó a 70 °C durante 3 horas más. La mezcla de reacción se inactivó con H20 (3 ml x 1) y después se extrajo con acetato de etilo (2 x 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (1 x 10 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Luna C18150 * 255 u; fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 17 % - 47 %, 10 min). Las fracciones deseadas se recogieron, se neutralizaron con una solución saturada de NaHC03 y se extrajeron con acetato de etilo (1 x 30 ml). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. Se obtuvo 4-[7-(5,6-dimetil-2-oxo-1,3-dihidrobenzoimidazol-4-il)-2-[[(2S-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (20,0 mg, 33,7 umol, dos etapas 19 % de rendimiento, 100 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 593.
Etapa D: 5,6-dimetil-4-[2-[[(2S)-1-metilpirrolidin-2-inmetoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-1,3-dihidrobenzoimidazol-2-ona. A una solución de 4-[7-(5,6-dimetil-2-oxo-1,3-dihidrobenzoimidazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (16,0 mg, 27,0 umol, 1 eq.) en dioxano (2 ml) se le añadió HCl/dioxano (4 M, 101 ul, 15 eq.) a 0 °C. Después de agitar a 25 °C durante 0,5 h, la mezcla se concentró al vacío. Se obtuvo 5,6-dimetil-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-1,3-dihidrobenzoimidazol-2-ona (20,0 mg, bruto, HCl) en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 493.
Etapa E: 5,6-dimetil-4-[2-[[(2S)-1-metilpirrolidin-2-inmetoxn-4-(4-prop-2-enoilpiperazin-1-il)-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-7-¡l1-1.3-d¡h¡drobenzo¡m¡dazol-2-ona. A una mezcla de 5,6-dimetil-4-(2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-1,3-dihidrobenzoimidazol-2-ona (20,0 mg, 37,8 umol, 1 eq., HCl) y TEA (38,3 mg, 378 umol, 52,6 ul, 10 eq.) en DCM (2 ml) se le añadió en una porción prop-2-enoato de prop-2-enoílo (4,77 mg, 37,8 umol, 1 eq.) a -40 °C. Después de agitar a -40 °C durante 30 min, la mezcla de reacción se inactivó con una solución saturada de NaHC03 (0,5 ml) a 0 °C y después se extrajo con CH2Cl2 (2 x 10 ml). Las capas orgánicas combinadas se lavaron con salmuera (1 x 10 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío para dar un residuo. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi C18 150 * 25 * 10 um; fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 10 % - 37 %, 9 min). Las fracciones deseadas
se recogieron y se concentraron a presión reducida para eliminar el ACN y después se liofilizaron. El compuesto del título 5,6-dimetil-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-4-(4-prop-2-enoilpiperazin-1-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-il]-1,3-dihidrobenzoimidazol-2-ona (EJEMPL0456, 3,15 mg, 5,25 umol, dos etapas 14 % de rendimiento, 98 ,8 % de pureza, HC00H) se obtuvo en forma de un sólido de color pardo. LCMS [ESI, M+1]: 547.
1H RMN (400 MHz, metanol-d4) 6,90 - 6,81 (m, 1H), 6,80 - 6,72 (m, 1H), 6,28 (d a, J = 16,8 Hz, 1H), 5,82 (d a, J = 10,4 Hz, 1H), 4,72 - 4,46 (m, 2H), 4,32 - 4,01 (m, 2H), 3,93 - 3,53 (m, 10H), 3,53 - 3,38 (m, 1H), 3,21 - 3,02 (m, 2H), 2,98 (s, 3H), 2,95 - 2,58 (m, 2H), 2,48 - 2,32 (m, 1H), 2,31 (s a, 3H), 2,29 (s a, 3H), 2,19 - 1,94 (m, 3H).
Ejemplo 457

1-[4-[7-(5,6-dimetil-1H-indol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -il]prop-2-en-1 -ona

Etapa 1: (E)-2-(2-bromo-3,4-dimetil-6-nitro-fenil)-M,M-dimetil-etenamina. A una solución de 3-bromo-1,2,4-trimetil-5-nitro-benceno (2 g, 8,19 mmol, 1 eq.) en DMF (20 ml) se le añadió DMFDMA (5,86 g, 49,2 mmol, 6,53 ml, 6 eq.). La mezcla se agitó a 130 °C durante 10 horas. Después de que se completara, la mezcla se concentró al vacío para dar (E)-2-(2-bromo-3,4-dimetil-6-nitro-fenil)-W,W-dimetil-etenamina (2,5 g, bruto) en forma de un aceite de color pardo que se usó directamente en la siguiente etapa sin purificación adicional.
Etapa 2: 4-bromo-5,6-dimetil-1 H-indol. A una solución de (E)-2-(2-bromo-3,4-dimetil-6-nitro-fenil)-W,W-dimetiletenamina (2,5 g, 8,36 mmol, 1 eq.) en Et0H (50 ml) se le añadieron Fe (2,33 g, 41,8 mmol, 5 eq.) y Ac0H (5,02 g, 83,6 mmol, 4,78 ml, 10 eq.). La mezcla se agitó a 60 °C durante 2 horas. Después de que se completara, la mezcla se filtró y el filtrado se concentró. La mezcla se ajustó a pH = 7 con una solución acuosa saturada de NaHC03 y se
extrajo con acetato de etilo (50 ml x 2). Las capas orgánicas combinadas se lavaron con salmuera (50 ml), se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP/EtOAc 1/0 a 50/1) para dar 4-bromo-5,6-dimetil-1H-indol (380 mg, 1,46 mmol, dos etapas 18 % de rendimiento, 86 % de pureza) en forma de un aceite de color amarillo.
1H RMN (400 MHz, cloroformo-d) 6 = 8,12 (s a, 1H), 7,20 - 7,10 (m, 2H), 6,59 - 6,52 (m, 1H), 2,47 (s, 3H), 2,43 (s, 3H).
Etapa 3: 2-[(4-bromo-5,6-dimetil-indol-1-il)metoxi]etil-trimetil-silano. A una solución de 4-bromo-5,6-dimetil-1H-indol (450 mg, 2,01 mmol, 1 eq.) en THF (9 ml) se le añadió NaH (161 mg, 4,02 mmol, 60 % de pureza, 2 eq.) a 0° C. La reacción se agitó a 0 °C durante 15 minutos en una atmósfera de N2. La reacción se dejó calentar a 25 °C y se agitó durante 0,5 horas. La reacción se enfrió de nuevo a 0° C. Y se añadió gota a gota SEM-Cl (502 mg, 3,01 mmol, 533 ul, 1,5 eq.) en THF (100 ul) a 0° C. Después de agitar durante 5 minutos, la mezcla se dejó calentar a temperatura ambiente (25 °C) y se agitó durante 5 horas más. Después de que se completara, la mezcla se inactivó con una solución acuosa saturada de cloruro de amonio (20 ml), se diluyó con agua (10 ml) y después se extrajo con acetato de etilo (2 x 30 ml). Los extractos se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP/EtOAc 1/0 a 50/1) para dar 2-[(4-bromo-5,6-dimetil-indol-1-il)metoxi]etil-trimetil-silano (440 mg, 1,18 mmol, 59 % de rendimiento, 95 % de pureza) en forma de un aceite de color amarillo.
1H RMN (400 MHz, cloroformo-d) 6 = 7,25 (s, 1H), 7,11 (d, J = 3,2 Hz, 1H), 6,51 (d, J = 2,8 Hz, 1H), 5,43 (s, 2H), 3,52 - 3,40 (m, 2H), 2,47 (s, 3H), 2,46 (s, 3H), 0,92 - 0,87 (m, 2H), -0,01 - -0,08 (m, 9H).
Etapa A: 4-[7-[5,6-dimetil-1-(2-trimetilsililetoximetil)indol-4-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo. Se desgasificó 4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (Intermedio 66, 450 mg, 1,04 mmol, 1 eq.), 2-[(4-bromo-5,6-dimetil-indol-1-il)metoxi]etil-trimetil-silano (406 mg, 1,14 mmol, 1,1 eq.), Pd2(dba)3 (191 mg, 208 umol, 0,2 eq.), RuPhos (194 mg, 416 umol, 0,4 eq.) y Cs2C03 (847 mg, 2,60 mmol, 2,5 eq.) en tolueno (30 ml) y después se calentó a 90 °C durante 8 horas en una atmósfera de N2. Después de que se completara, la mezcla se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EtOAc/Me0H 50/1 a 10/1) para dar 4-[7-[5,6-dimetil-1-(2-trimetilsililetoximetil)indol-4-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (106 mg, 135 umol, 13 % de rendimiento, 90 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 706.
Etapa B: 4-[7-(5,6-dimetil-1H-indol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l1p¡peraz¡n-1-carbox¡lato de tere-butilo. A una solución de 4-[7-[5,6-dimetil-1-(2-trimetilsililetoximetil)indol-4-il]-2-[[(2S)-1- metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (70 mg, 99,2 umol, 1 eq.) en DMF (0,35 ml) se le añadió TBa F (1 M en THF, 496 ul, 5 eq.) seguido de etano-1,2-diamina (8,94 mg, 149 umol, 9,95 ul, 1,5 eq.). La mezcla se agitó a 85 °C durante 3 horas. Después de que se completara, la mezcla se diluyó con EtOAc (3 ml) y agua (2 ml). La capa orgánica separada se secó sobre Na2S04 y se concentró al vacío. El residuo se purificó por TLC prep. (DCM/Me0H 10/1) para dar 4-[7-(5,6-dimetil-1H-indol-4-il)-2-[[(2S)-1-metilpirrolidin-2- il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (50 mg, 60,8 umol, 61 % de rendimiento, 70 % de pureza) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 576.
Etapa C: 7-(5,6-dimetil-1H-indol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-dlpirimidina. A una solución de 4-[7-(5,6-dimetil-1H-indol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (45 mg, 78,2 umol, 1 eq.) en dCm (70 ul) se le añadió TFA (89,1 mg, 782 umol, 57,9 ul, 10 eq.). La mezcla se agitó a 25 °C durante 0,5 horas. Después de que se completara, la mezcla se concentró al vacío para dar 7-(5,6-dimetil-1H-indol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidina (60 mg, bruto, 2 TFA) en forma de un aceite de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional.
Etapa D: 1-[4-[7-(5,6-dimetil-1H-indol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l1p¡peraz¡n-1-¡l1prop-2-en-1-ona. A una solución de 7-(5,6-dimetil-1H-indol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-4-piperazin-1-il-6,8-dihidro-5H-pirido[3,4-d]pirimidina (55 mg, 78,2 umol, 1 eq., 2 TfA) y TEA (158 mg, 1,56 mmol, 218 ul, 20 eq.) en DCM (1 ml) se le añadió gota a gota prop-2-enoato de prop-2-enoílo (9,86 mg, 78,2 umol, 1 eq.) a -40 °C. La mezcla se agitó a -40 °C durante 30 minutos. Después de que se completara, la mezcla se inactivó con Me0H (0,5 ml), se diluyó con agua (5 ml) y después se extrajo con DCM (2 x 10 ml). Las capas orgánicas se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía (Ah03, EtOAc/Me0H 100/1 a 10/1) y HPLC prep. (columna: Phenomenex Synergi C18150 * 25 * 10 um; fase móvil: [agua (0,1 % de TFA)-ACN]; % de B: 16 %-43 %, 12 min). Las fracciones deseadas se recogieron, se neutralizaron con NaHC03 acuoso saturado y se extrajeron con DCM (2 x 15 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío para dar el compuesto del título 1-[4-[7-(5,6-dimetil-1H-indol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-il]prop-2-en-1-ona (EJEMPL0 457, 9,81 mg, 18,3 umol, dos etapas 23 % de rendimiento, 98,8 % de pureza) en forma de un sólido de color pardo. LCMS [ESI, M+1]: 530.
Condición de SFC: 0J-3S_3_5_40_3ML Columna: Chiralcel 0J-3100 x 4,6 mm D.I., 3 um Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 % Caudal: 3 ml/min Longitud de onda: 220 nm.
1H RMN (400 MHz, cloroformo-d) 5 = 8,22 (s a, 1H), 7,09 (s, 1H), 7,07 - 7,03 (m, 1H), 6,60 (dd, J = 10,8, 16,8 Hz, 1H), 6,53 - 6,48 (m, 1H), 6,34 (dd, J = 2,0, 16,9 Hz, 1H), 5,76 (dd, J = 2,0, 10,4 Hz, 1H), 4,49 - 4,10 (m, 4H), 3,81 (s a, 2H), 3,76 - 3,38 (m, 8H), 3,13 (t a, J = 7,2 Hz, 1H), 3,02 - 2,56 (m, 3H), 2,50 (s, 3H), 2,39 (s, 3H), 2,36 (s, 3H), 2,34 - 2,25 (m, 1H), 2,13 - 2,06 (m, 1H), 1,84 - 1,71 (m, 3H).
Ejemplo 458

2-((S)-1-(but-2-inoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 1 -bromo-8-metilnaftaleno: A una solución de 1,8-dibromonaftaleno (33,8 g, 118 mmol) en THF (200 ml) enfriada a -78 °C se le añadió butil litio (49,6 ml, 124 mmol) a tal velocidad que la temperatura interna no superó -69 °C. Después de que se completara la adición, la reacción se enfrió a -78 °C y se añadió yoduro de metilo (14,8 ml, 236 mmol) en un bolo de tal forma que la temp. interna aumentó a -40 °C. Después de la adición de yoduro de metilo, el baño de hielo seco/acetona se retiró y la reacción se calentó a temperatura ambiente. Luego, la reacción se vertió en salmuera, la capa acuosa se extrajo con EtOAc (300 ml) y los extractos orgánicos se separaron. Luego, los extractos orgánicos se secaron sobre MgS04 y se concentraron al vacío y el residuo se cromatografió usando hexanos como eluyente para dar un sólido de color blanco (24 g). Luego, el sólido se suspendió en IPA (aprox. 60 ml) y la suspensión se calentó a 60 °C, momento en el que los sólidos se disolvieron. La solución se retiró del calentamiento y se enfrió a temperatura ambiente durante 2 h. La suspensión resultante se filtró y los sólidos se secaron al vacío para dar 1-bromo-8-metilnaftaleno (16 g, 72,4 mmol, 61 % de rendimiento) >95 % de pureza.
Etapa B: (S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: Una solución de xantphos (4,5 g, 7,8 mmol) y Pd2(dba)3 (3,5 g, 3.9 mmol) en dioxano (100 ml) se desgasificó con argón durante 15 minutos, seguido de calentamiento de la solución durante 20 minutos a 100 °C. La solución se enfrió a temperatura ambiente, a la solución se le añadieron (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (9,8 g, 19 mmol), 1-bromo-8-metilnaftaleno (13 g, 58 mmol) y Cs2C03 (32 g, 97 mmol), Pd2(dba)3 (3,5 g, 3.9 mmol), la mezcla se desgasificó durante 20 minutos más con argón y la reacción se calentó a 100 °C durante una noche. Luego, la reacción se enfrió a temperatura ambiente y se filtró a través de papel 0FF y el filtrado se concentró al vacío. Luego, el residuo se cromatografió 3 veces usando 1 -->10 % de (Me0H 2 % de NH40H)/DCM para dar (S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (7,8 g, 12 mmol, 62 % de rendimiento). ESI+APCI MS m/z 646,3 [M+H]+.
Etapa C: 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: Una solución de (S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (7.8 g. 12 mmol) en 1:1 Et0H/THF (100 ml) se desgasificó con N2 durante 5 minutos seguido de la adición de Pd/C (2.6 g.2.4 mmol). Luego. la suspensión se desgasificó al vacío y se cargó de nuevo con H2 (3 x). En la tercera recarga. la mezcla se agitó en una atmósfera de hidrógeno durante una noche. La suspensión se desgasificó de nuevo con N2 y la suspensión se filtró a través de celite. El filtrado combinado se concentró al vacío y los sólidos se cromatografiaron usando 1->10 % de (Me0H 2 % de NH40H)/DCM como eluyente para dar 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (5.0 g. 9.8 mmol. 81 % de rendimiento).
Etapa D: 2-((S)-1-(but-2-inoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6.7.8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: A 0 °C. a un MFR de 25 ml que contenía N,N-dimetilformamida (1954 gl, 0,391 mmol) se le añadieron 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (200 mg. 0,391 mmol) y trietilamina (136 gl. 0,977 mmol). La mezcla de reacción se agitó vigorosamente mientras se añadía en una porción ácido but-2-inoico (49,3 mg. 0,586 mmol). A la mezcla en agitación se le añadió lentamente anhídrido cíclico del ácido 1-propanofosfónico (175 gl. 0,293 mmol). La reacción se dejó en agitación a ta durante 18 h. Se añadió agua y los sólidos se filtraron. Los sólidos se purificaron con gel de sílice (5-18 % de Me0H en DCM con 0,25 % de NH40H) para proporcionar el compuesto del título 2-((S)-1-(but-2-inoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0 458, 110 mg. 0,190 mmol. 49 % de rendimiento). ESI+APCI MS m/z 578,3 [M+H]+.
Ejemplo 459

2-((S)-1-((E)-4-(dimetilamino)but-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 2-((S)-1-((E)-4-(dimetilamino)but-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo. Una mezcla agitada de 2-((S)-4-(7-(8-metilnaftalen-1 -il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (100 mg. 0,1954 mmol). ácido (E)-4-(dimetilamino)but-2-enoico (37,86 mg. 0,2932 mmol) y N,N-dimetilformamida (2 ml.25,58 mmol) se enfrió en un baño de hielo-sal y se añadió de una vez trietilamina (0,02724 ml.
0,1954 mmol). seguido de 2,4,6-trióxido de 2.4.6-tripropil-1,3.5.2.4.6-trioxatrifosfinano. 50 % en EtOAc (0,1745 ml.
0.2932 mmol). La mezcla de reacción se dejó calentar a ta durante 5 min y después se agitó a ta durante 3 h. La mezcla de reacción se repartió entre EtOAc (50 ml) y Na2C030.5 M (20 ml). los extractos orgánicos se separaron y la capa orgánica se lavó con agua y salmuera (10 ml de cada uno). se secó sobre Na2S04 y se evaporó al vacío. La cromatografía sobre gel de sílice en 5 a 10 % de Me0H 0.5 % de NH40H en DCM proporcionó el producto bruto que se purificó por cromatografía de fase inversa. Gilson, de 5 a 95 % de MeCN 0.1 % de TFA en agua. Las fracciones
diana se combinaron, se basificaron con Na2C032 M y se extrajeron con DCM (3 x 20 ml). Los extractos combinados se lavaron con salmuera (10 ml), se secaron sobre Na2S04 y se evaporaron al vacío. El residuo sólido se disolvió en DCM (3 ml), se filtró y se evaporó para producir el compuesto del título deseado en forma de un sólido incoloro (EJEMPL0459, 40 mg, 33 %). ESI+APCI MS m/z 623,3 [M+H]+.
Ejemplo 460

2-[(2S)-4-[7-(8-met¡M-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]-1-[(E)4-morfolinobut-2-enoil]piperazin-2-il]acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de bencilo. A una solución de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (Intermedio 65, 500 mg, 989 umol, 1,0 eq.), Pd2(dba)3 (181 mg, 198 umol, 0,20 eq.), Xantphos (172 mg, 297 umol, 0,30 eq.) y Cs2C03 (967 mg, 2,97 mmol, 3,0 eq.) en tolueno (5,0 ml) se le añadió 1-bromo-8-metil-naftaleno (Intermedio 69, 284 mg, 1,29 mmol, 1,30 eq.). La mezcla se agitó a 90 °C durante 12 horas. Después de que se completara, a la mezcla de reacción se le añadió agua (10,0 ml) y se extrajo con acetato de etilo (10,0 ml x 4). La capa orgánica se secó sobre Na2S04, se filtró y se concentró al vacío. El residuo se purificó por columna ultrarrápida de fase inversa (C18, 0,1 % en ácido fórmico, 0-70 % acetonitrilo) para dar el producto (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (400 mg, 619 umol, 63 % de rendimiento) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 646.
Etapa B: 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l1p¡perazin-2-il1acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de bencilo (700 mg, 1,08 mmol, 1,0 eq.) y NH3^Me0H (20,0 ml, 30 % de pureza) en metanol (20,0 ml) se le añadió Pd/C (200 mg, 10 % de pureza). La mezcla se purgó 3 veces con nitrógeno. Después, la mezcla se agitó en una atmósfera de hidrógeno (103,42 kPa (15 psi)) a 25 °C durante 1 hora. Después de que se completara, la mezcla de reacción se filtró a través de celite y la torta de filtro se lavó con tetrahidrofurano (20,0 ml). El licor madre se recogió y se concentró al vacío para dar el producto 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (500 mg, 977 umol, 90 % de rendimiento) en forma de un aceite de color amarillo.
1H RMN (400 MHz, Cloroformo-d) 57,70 (d, J = 8,0 Hz, 1H), 7,64 (dd, J = 3,2, 7,2 Hz, 1H), 7,43 - 7,37 (m, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,26 - 7,19 (m, 2H), 4,43 - 4,33 (m, 1H), 4,22 (dd, J = 6,8, 18,0 Hz, 1H), 4,17 - 4,08 (m, 1H), 4,07 -3,89 (m, 1H), 3,89 - 3,70 (m, 2H), 3,52 - 3,45 (m, 2H), 3,38 - 3,20 (m, 1H), 3,19 - 3,04 (m, 4H), 3,03 - 2,94 (m, 2H), 2,93 - 2,82 (m, 4H), 2,70 - 2,60 (m, 1H), 2,60 - 2,49 (m, 3H), 2,47 (d, J = 2,4 Hz, 3H), 2,31 - 2,22 (m, 1H), 2,11 - 2,00 (m, 1H), 1,88 - 1,76 (m, 3H).
Etapa C: 2-[(2S)-1-[(E)-4-bromobut-2-enoil]-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (250 mg, 489 umol, 1,0 eq.) y piridina (309 mg, 3,91 mmol, 316 ul, 8,0 eq.) en diclorometano (4,0 ml) se le añadió cloruro de (£)-4-bromobut-2-enoílo (359 mg, 1,95 mmol, 4,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se concentró para dar el producto 2-[(2S)-1-[(£)-4-bromobut-2-enoil]-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (330 mg, bruto) en forma de un aceite de color amarillo. El producto se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 658.
Etapa D: 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(E)-4-morfolinobut-2-enoil]piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-1-[(£)-4-bromobut-2-enoil]-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (300 mg, bruto) y K2C03 (630 mg, 4,55 mmol, 10,0 eq.) en diclorometano (2,0 ml) se le añadió morfolina (238 mg, 2,73 mmol, 241 ul, 6,0 eq.). La mezcla se agitó a 0 °C durante 12 horas. Después de que se completara, a la mezcla de reacción se le añadió agua (10,0 ml) y se extrajo con acetato de etilo (10,0 ml * 3). La capa orgánica se secó sobre Na2S04, se filtró y se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 50 %-80 %, 12 min) para dar el compuesto del título 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-[(E)-4-morfolinobut-2-enoil]piperazin-2-il]acetonitrilo (EJEMPL0460, 48,2 mg, 72,5 umol, 16 % de rendimiento) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 665.
1H RMN (400 MHz, Cloroformo-d) 57,70 (d a, J = 8,4 Hz, 1H), 7,65 (t, J = 8,4 Hz, 1H), 7,41 (dd, J = 7,6, 15,2 Hz 1H), 7,35 (t, J = 7,6 Hz, 1H), 7,27 - 7,17 (m, 2H), 7,02 - 6,91 (m, 1H), 6,55 - 6,39 (m, 1H), 5,23 - 4,47 (m, 1H), 4,40 - 4,33 (m, 1H), 4,31 - 4,19 (m, 1H), 4,19 - 4,09 (m, 2H), 4,07 - 3,83 (m, 2H), 3,81 - 3,64 (m, 5H), 3,59 - 3,40 (m, 2H), 3,24 -3,13 (m, 4H), 3,12 - 2,94 (m, 3H), 2,92 (s, 3H), 2,86 - 2,76 (m, 1H), 2,75 - 2,54 (m, 3H), 2,50 (s a, 4H), 2,48 - 2,45 (m, 3H), 2,33 - 2,22 (m, 1H), 2,10 - 2,00 (m, 1H), 1,87 - 1,71 (m, 3H).
Ejemplo 461

2-[(2R)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo
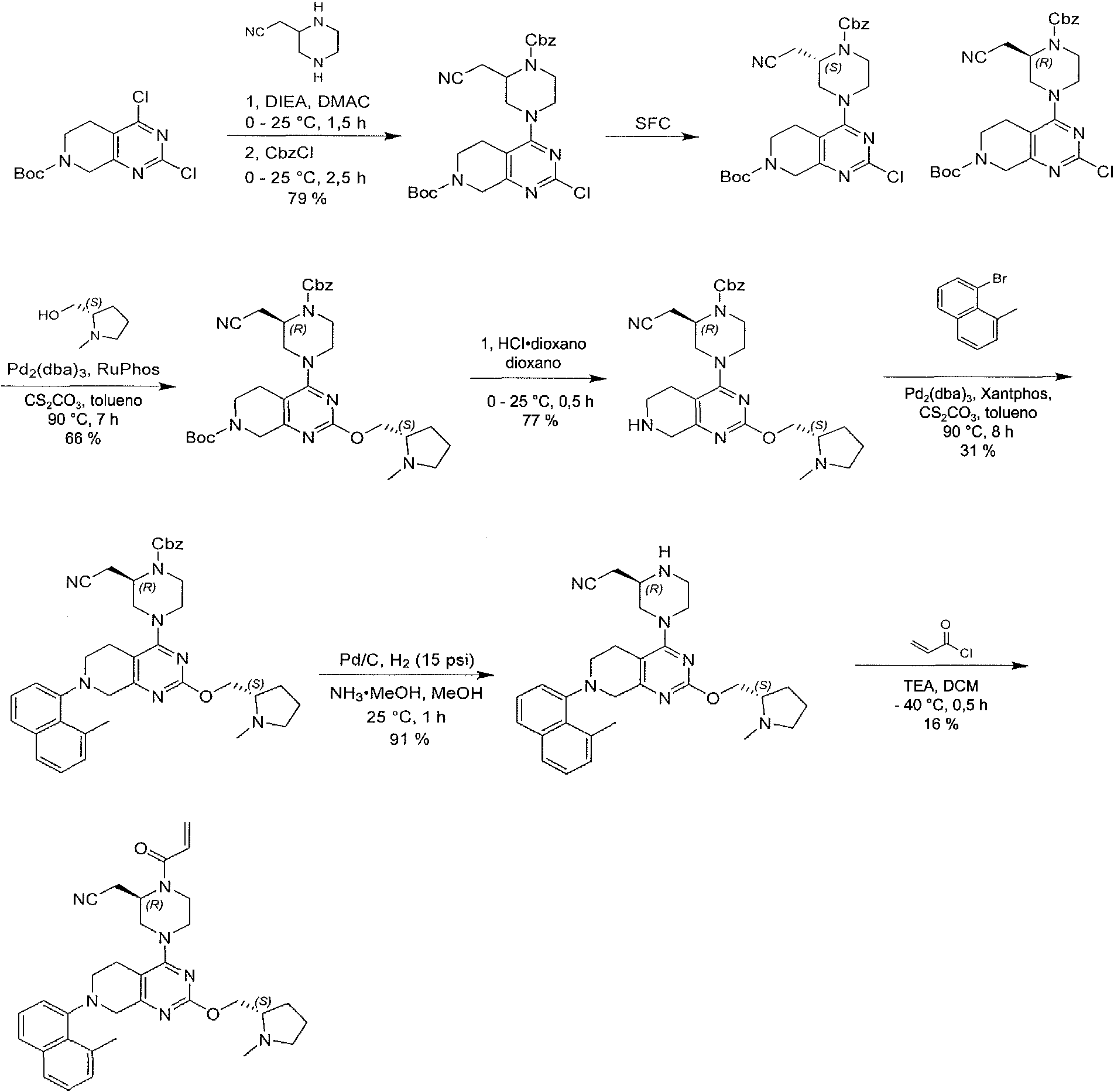
Etapa A: 4-[4-benciloxicarbonil-3-(cianometil)piperazin-1-il1-2-cloro-6.8-dihidro-5H-pirido[3.4-d1pirimidin-7-carboxilato de tere-butilo. A una solución de 2.4-dicloro-6.8-dihidro-5H-pirido[3.4-d] pirimidin-7-carboxilato de terc-butilo (3 g.
9.86 mmol. 1 eq.) en DMAc (60 ml) se le añadieron DIEA (3.82 g. 29.6 mmol. 5.15 ml. 3 eq.) y 2-piperazin-2-ilacetonitrilo (Intermedio 62. 2.15 g. 10.9 mmol. 1.1 eq.. 2HCl) a 0 °C. La mezcla se agitó a 25 °C durante 1.5 h. Después. a la solución se le añadieron DIEA (2.55 g. 19.7 mmol. 3.43 ml. 2 eq.) y cloroformiato de bencilo (2.52 g.
14.8 mmol. 2.10 ml. 1.5 eq.) a 0 °C. La mezcla se agitó a 25 °C durante 2.5 h más. A la mezcla de reacción se le añadieron H20 (40 ml) y acetato de etilo (100 ml). La fase orgánica separada se lavó con 5 % de una solución acuosa de ácido cítrico (40 ml * 1). solución acuosa saturada de Na2C03 (40 ml * 1) y salmuera (40 ml * 1). se secó sobre sulfato de sodio y se filtró. El filtrado se concentró a presión reducida para dar un residuo. El residuo se trituró con metil terc-butil éter (25 ml) para dar un producto puro. Se obtuvo 4-[4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-cloro-6.8-dihidro-5H-pirido[3.4-d]pirimidin-7-carboxilato de terc-butilo (4.10 g. 7.62 mmol. 79 % de rendimiento. 98 % de pureza) en forma de un sólido de color blanco. LCMS [ESI. M+1]: 527.
1H RMN (400 MHz. cloroformo-d) ó = 7,43 - 7.29 (m. 5H). 5.17 (s. 2H). 4.71 - 4.55 (m. 2H). 4.48 - 4.37 (m. 1H). 4.21 -3.97 (m. 2H). 3.92 - 3.67 (m. 2H). 3.40 - 3.20 (m. 3H). 3.09 (dt. J = 3.6. 12.4 Hz. 1H). 2.88 - 2.57 (m. 4H). 1.47 (s. 9H). Condición de SFC (40935): Columna: Chiralcel 0D - 350 * 4.6 mm D.I.. 3 um Fase móvil: metanol (0.05 % de DEA) en C02 de 5 % a 40 %. Caudal: 3 ml/min. Longitud de onda: 220 nm.
Etapa B: 4-[(3R)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il1-2-cloro-6.8-dihidro-5H-pirido[3.4-d1pirimidin-7-carboxilato de terc-butilo. Se separó 4-[4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-cloro-6.8-dihidro-5H
pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (2,20 g, 4,17 mmol, 1 eq.) por SFC (columna: 0D (250 mm * 30 mm, 10 um); fase móvil: [0,1 % de NH3H20 Me0H]; % de B: 40 % - 40 %, 3,1 min: 180 min). Las fracciones deseadas se recogieron y se concentraron al vacío para dar 4-[(3R)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (940 mg, 1,78 mmol, 43 % de rendimiento, 99,7 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 527.
Condición de SFC: Columna: Chiralcel 0D - 350 x 4,6 mm D.I., 3 um, Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 %, Caudal: 3 ml/min, Longitud de onda: 220 nm.
Etapa C: 4-[(3R)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-7-carbox¡lato de ferc-butilo. Una mezcla de 4-[(3R)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (290 mg, 550 umol, 1 eq.), [(2S)-1-metilpirrolidin-2-il]metanol (127 mg, 1,10 mmol, 131 ul, 2 eq.), Pd2(dba)3 (60,5 mg, 66,0 umol, 0,12 eq.), RuPhos (51,4 mg, 110 umol, 0,2 eq.) y Cs2C03 (448 mg, 1,38 mmol, 2,5 eq.) en tolueno (20 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 7 horas en una atmósfera de N2. A la mezcla de reacción se le añadieron H20 (25 ml x 1 ) y acetato de etilo (25 ml x 2). La fase orgánica separada se lavó con salmuera (100 ml x 1), se secó sobre sulfato de sodio y después se filtró. El filtrado se concentró a presión reducida. El residuo se diluyó con acetato de etilo (50 ml) y se acidificó con una solución acuosa de HCl (1 mol/l) a pH = 3 ~ 4. A la capa de agua separada se le añadió acetato de etilo (60 ml) y se basificó con una solución acuosa saturada de Na2C03 a pH = 8 ~ 9. La fase orgánica se separó, se secó sobre sulfato de sodio, se filtró y se concentró a presión reducida para dar un residuo. Se obtuvo 4-[(3R)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (225 mg, 0,36 mmol, 66 % de rendimiento, 97,7 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 606.
1H RMN (400 MHz, cloroformo-d) 6 = 7,47 - 7,30 (m, 5H), 5,19 (s, 2H), 4,73 - 4,52 (m, 2H), 4,44 - 4,26 (m, 2H), 4,22 -4,14 (m, 1H), 4,03 - 3,74 (m, 3H), 3,41 - 3,21 (m, 3H), 3,08 (t a, J = 7,6 Hz, 1H), 2,97 (dt, J = 3,6, 12,4 Hz, 1H), 2,89 -2,53 (m, 5H), 2,46 (s, 3H), 2,38 - 2,10 (m, 2H), 2,08 - 2,03 (m, 1H), 1,89 - 1,65 (m, 3H), 1,48 (s, 9H).
Condición de SFC: Columna: Chiralpak AD - 350 x 4 ,6 mm D.I., 3 um, Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 %, Caudal: 3 ml/min, Longitud de onda: 220 nm.
Etapa D: (2R)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo. A una solución de 4-[(3R)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (620 mg, 1,02 mmol, 1 eq.) en dioxano (12 ml) se le añadió HCl/dioxano (4 M, 12,4 ml, 48,5 eq.) a 0 °C. La mezcla se agitó a 25 °C durante 0,5 h. El líquido se decantó y el residuo se concentró al vacío. Se obtuvo (2R)-2-(cianometil)-4-[2-[[(2S-1 -metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (470 mg, bruto, HCl) en forma de un aceite de color amarillo que se disolvió en DCM (10 ml). La mezcla se basificó con una solución acuosa saturada de carbonato de sodio a pH = 9. La fase orgánica se separó, se lavó con salmuera (5 ml x 1), se filtró y se concentró a presión reducida para dar un residuo que era lo suficientemente puro sin purificación adicional. Se obtuvo (2R)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (400 mg, 718 umol, 77 % de rendimiento, 90,8 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 506.
Etapa E: (2R)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de bencilo. Una mezcla de (2R)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (210 mg, 415 umol, 1 eq.), 1-bromo-8-metil-naftaleno (Intermedio 69, 119 mg, 540 umol, 1,3 eq.) Pd2(dba)3 (76,1 mg, 83,1 umol, 0,2 eq.), Cs2C03 (406 mg, 1,25 mmol, 3 eq.) y Xantphos (72,1 mg, 125 umol, 0,3 eq.) en tolueno (7 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 8 horas en una atmósfera de N2. A la mezcla de reacción se le añadió H20 (20 ml x 1 ) y la mezcla se extrajo con acetato de etilo (20 ml x 2). Los extractos se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se diluyó con acetato de etilo (50 ml) y se acidificó a pH = 3 ~ 4 con una solución acuosa de HCl (1 mol/l). A la capa de agua se le añadió acetato de etilo (60 ml) y se basificó a pH = 8 ~ 9 con una solución acuosa saturada de Na2C03. La fase orgánica se separó, se secó sobre sulfato de sodio, se filtró y el filtrado se concentró a presión reducida. El residuo se purificó por cromatografía en columna (S¡02, acetato de etilo/metanol 50/1 a 5/1). Se obtuvo (2R)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (86 mg, 0,128 mmol, 31 % de rendimiento, 96,0 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 646.
Etapa E: 2-[(2R)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pindo[3,4-d]pirimidin-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una solución de (2R)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (130 mg, 201 umol, 1 eq.) en Me0H (15 ml) se le añadieron Pd/C (25 mg, 15,5 umol, 10 % de pureza) y NH3^Me0H (15,5 umol, 15 ml, 20 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. El catalizador se retiró por filtración a
través de una capa de celite. El filtrado se concentró al vacío. Se obtuvo 2-[(2R)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (100 mg, 0,183 mmol, 91 % de rendimiento, 93,5 % de pureza) en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 512.
Etapa F: 2-[(2^)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo. A una mezcla de 2-[(2R)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (100 mg, 195 umol, 1 eq.) y TEA (198 mg, 1,95 mmol, 272 ul, 10 eq.) en DCM (10 ml) se le añadió en una porción cloruro de prop-2-enoílo (17,7 mg, 195 umol, 15,9 ul, 1 eq.). Después de agitar a -40 °C durante 30 min, la mezcla se inactivó con Me0H (2 ml) y se concentró al vacío. El residuo se purificó por cromatografía en columna (Ah03, diclorometano/metanol = 10/1 a 10/1). El residuo se purificó por HPLC prep. (columna: Luna C18150 * 255 u; fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 25 % -45 %, 7,8 min) y (columna: Gemini 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 57 % - 87 %, 12 min). Las fracciones deseadas se concentraron a presión reducida para eliminar acetonitrilo y después se liofilizaron. El compuesto del título 2-[(2R)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0 461, 17,6 mg, 31,0 umol, 16 % de rendimiento, 100 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 566.
Condición de SFC: Columna: Chiralcel 0J-350 * 4,6 mm D.I., 3 um, Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 %, Caudal: 3 ml/min, Longitud de onda: 220 nm.
1H RMN (400 MHz, cloroformo-d) 5 = 7,77 - 7,63 (m, 2H), 7,48 - 7,32 (m, 2H), 7,31 - 7,18 (m, 2H), 6,61 (s a, 1H), 6,48 - 6,37 (m, 1H), 5,85 (d a, J = 10,0 Hz, 1H), 5,27 - 4,49 (m, 1H), 4,45 - 4,33 (m, 1H), 4,33 - 4,06 (m, 4H), 4,05 - 3,62 (m, 3H), 3,61 - 3,34 (m, 2H), 3,33 - 2,99 (m, 5H), 2,94 (s, 3H), 2,88 - 2,77 (m, 1H), 2,75 - 2,57 (m, 2H), 2,54 - 2,44 (m, 3H), 2,30 (d a, J = 7,2 Hz, 1H), 2,15 - 1,95 (m, 1H), 1,95 - 1,79 (m, 3H).
Ejemplo 462

2-[(2S)-4-[7-(5,6-dimetil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[7-(5,6-dimetil-1-tetrahidropiran-2-il-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-1 -carboxilato de tere-butilo. Se desgasificaron (2S)-2-(cianometil)-4-[2-[[(2S)-1 -metilpirrolidin-2-il]metoxi1-5,6,7,8-tetrahidropirido[3,4-dlpirimidin-4-illpiperazin-1 -carboxilato de tere-butilo (600 mg, 1,27 mmol, 1 eq.), 4-bromo-5,6-dimetil-1-tetrahidropiran-2-il-indazol (511 mg, 1,65 mmol, 1,3 eq.), RuPhos (237 mg, 509 umol, 0,4 eq.), CS2C03 (1,04 g, 3,18 mmol, 2,5 eq.) y Pd2(dba)3 (233 mg, 254 umol, 0,2 eq.) en tolueno (20 ml) y después se calentaron a 90 °C durante 8 horas en una atmósfera de N2. Después de que se completara, la mezcla se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. Las fracciones deseadas se recogieron y se neutralizaron con NaHC03 acuoso saturado. La mezcla se concentró al vacío para eliminar el MeCN y se extrajo con EtOAc (2 x 50 ml). Las capas orgánicas se lavaron con salmuera (50 ml), se secaron sobre Na2S04, se filtraron y se concentraron al vacío para dar (2S)-2-(cianometil)-4-[7-(5,6-dimetil-1-tetrahidropiran-2-il-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de tere-butilo (365 mg, 469 umol, 37 % de rendimiento, 90 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 700.
1H RMN (400 MHz, cloroformo-d) ó = 7,99 (s, 1H), 7,22 (s, 1H), 5,66 (dd, J = 2,8, 9,6 Hz, 1H), 4,61 (s a, 1H), 4,40 (dd a, J = 4,8, 10,4 Hz, 1H), 4,26 (s a, 2H), 4,09 - 3,97 (m, 3H), 3,90 (d a, J = 11,6 Hz, 1H), 3,80 - 3,71 (m, 1H), 3,50 (t a, J = 4,8 Hz, 2H), 3,25 (s a, 2H), 3,09 (t a, J = 7,6 Hz, 1H), 2,99 (t a, J = 11,2 Hz, 1H), 2,83 - 2,51 (m, 6H), 2,48 (s, 3H), 2,43 (s, 3H), 2,33 (s, 3H), 2,31 - 2,24 (m, 1H), 2,21 - 2,13 (m, 1H), 2,11 - 2,06 (m, 1H), 1,90 - 1,69 (m, 8H), 1,52 (s, 9H).
Etapa B: 2-r(2S)-4-r7-(5.6-dimetil-1H-indazol-4-il)-2-rr(2S)-1-metilpirrolidin-2-i n metox n -6.8-dihidro-5H-piridor3.4-d^yrimidin^-jUpiperazin^-i n acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(5,6-dimetil-1-tetrahidropiran-2-il-¡ndazol-4-¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (720 mg, 1,03 mmol, 1 eq.) en DCM (1 ml) se le añadió TFA (4,62 g, 40,5 mmol, 3 ml, 39,4 eq.). La mezcla se agitó a 20 °C durante 4 horas. Después de que se completara, la mezcla se diluyó con DCM (10 ml) y se neutralizó con NaHC03 acuoso saturado. Después, la fase acuosa se extrajo con EtOAc (2 x 30 ml). Las capas orgánicas se combinaron, se secaron sobre Na2S04, se filtraron y se concentraron al vacío para dar 2-[(2S)-4-[7-(5,6-dimetil-1H-¡ndazol-4-¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (880 mg, bruto) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 516.
Etapa C: (2-[(2S)-4-[7-(5,6-dimetil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-inmetoxn-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-prop-2-enoil-piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(5,6-dimetil-1H-indazol-4-il)-2-[[(2S)-1 -met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (150 mg, bruto) y Tea (177 mg, 1,75 mmol, 243 ul) en DCM (4 ml) se le añadió gota a gota cloruro de prop-2-enoílo (13,2 mg, 145 umol, 11,9 ul) a -40 °C. Después de agitar a -40 °C durante 30 minutos, la mezcla se inactivó con NaHC03 acuoso saturado
(1 ml) y se extrajo con DCM (2 x 10 ml). Las capas orgánicas se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía (AI203, EtOAc/Me0H 1/0 a 10/1) seguido de HPLC prep. (columna:
Gemini 200 * 30 10 g; fase móvil: [agua (NH4HC03 10 mM) - ACN]; % de B: 40 %-70 %, 6,0 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título (2-[(2S)-4-[7-(5,6-dimetil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-prop-2-enoil-piperazin-2-il]acetonitrilo (EJEMPL0462, 15,6 mg, 27,2 umol, dos etapas 15 % de rendimiento, 99,6 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 570.
Condición de SFC: Columna: Chiralcel 0J-3 100 x 4 ,6 mm D.I., 3 um, Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 %, Caudal: 3 ml/min, Longitud de onda: 220 nm.
1H RMN (400 MHz, cloroformo-d) 5 = 10,26 (s a, 1H), 8,03 (s, 1H), 7,14 (s, 1H), 6,57 (d a, J = 9,6 Hz, 1H), 6,39 (dd, J = 1,2, 16,4 Hz, 1H), 5,83 (d a, J = 10,4 Hz, 1H), 5,12 - 4,38 (m, 2H), 4,28 (s, 2H), 4,23 - 3,84 (m, 4H), 3,73 - 3,40 (m, 3H), 3,32 (s a, 1H), 3,10 (t a, J = 7,6 Hz, 2H), 2,98 - 2,61 (m, 5H), 2,48 (s, 3H), 2,41 (s, 3H), 2,34 (s, 3H), 2,32 - 2,24 (m, 1H), 2,11 - 1,99 (m, 1H), 1,91 - 1,78 (m, 3H).
Ejemplo 463

(S,E)-3-((4-(3-(cianometil)-4-(4-(dimetilamino)but-2-enoil)piperazin-1-il)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi)-N,N-dimetilpropanamida

Etapa A: (S)-4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo: Se disolvió clorhidrato de (S)-2-(cianometil)piperazin-1-carboxilato de bencilo (10 g, 34 mmol) en DMA (68 ml, 34 mmol). Luego, a la solución se le añadió 2,4-dicloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (9,3 g, 30 mmol) seguido de base de Hunig (24 ml, 135 mmol) y la reacción se agitó a temperatura ambiente durante 1 hora. Luego, la reacción se vertió en agua básica y se extrajo con MTBE. Los extractos orgánicos se lavaron con más agua y salmuera, se secaron sobre Na2 S04 y se concentraron al vacío. Después, el concentrado se cromatografió usando 10^70 % de EtOAc/Hexano como eluyente para dar (S)-4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de tercbutilo (12 g, 67 % de rendimiento). Es I+APCI MS m/z 580,3 [M+H]+.
Etapa B: ÍS)-4-í2-cloro-5.6.7.8-tetrah¡drop¡r¡dor3.4-d1p¡r¡m¡d¡n-4-¡h-2-ícianomet¡l)p¡peraz¡n-1-carbox¡lato de bencilo: Se disolvió (S)-4-(4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-cloro-5,8-d¡h¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-butilo (1.5 g. 2.8 mmol) en DCM (28 ml. 2.8 mmol) y se trató con una solución de ácido clorhídrico (4.0 M en 1.4-dioxano) (3.6 ml. 14 mmol). La reacción se agitó a temperatura ambiente durante 1 hora. Los extractos orgánicos se lavaron con Na0H 1 M. La capa acuosa se extrajo con DCM (2 x). Los extractos orgánicos combinados se secaron sobre Na2S04. se concentraron al vacío y se llevaron a continuación como (S)-4-(2-cloro-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡rim¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de bencilo bruto (1.2 g.99 % de rendimiento).
ESI+APCI MS m/z 427.2 [M+H]+.
Etapa C: (S)-4-(2-cloro-7-(8-met¡lnaftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡rim¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carboxilato de bencilo: Se disolvieron tris(d¡benc¡lidenoacetona)d¡palad¡o (0) (0.51 g. 0.56 mmol) y Xantphos (0.54 g.
1.1 mmol) en 1.4-dioxano (28 ml. 2.8 mmol) y se purgaron en una atmósfera de argón durante 5 minutos seguido de agitación a 100 °C en una atmósfera de argón durante 15 minutos. Luego. la reacción se enfrió a temperatura ambiente. A la reacción se le añadieron (S)-4-(2-cloro-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)-2-(cianometil)p¡peraz¡n-1-carbox¡lato de bencilo (1.2 g. 2.8 mmol). 1-bromo-8-metilnaftaleno (1.9 g. 8.4 mmol) y carbonato de cesio (2.7 g. 8.4 mmol) en una atmósfera de argón. La reacción se tapó en una atmósfera de argón y se agitó a 100 °C durante una noche. La reacción se enfrió a temperatura ambiente y se filtró a través de papel GHF. El filtrado se concentró al vacío y se purificó por cromatografía de fase normal sobre el CombiFlash usando 0 % ^ 75 % de Hexanos/EtOAc como eluyente para dar (S)-4-(2-cloro-7-(8-metilnaftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)-2-(cianomet¡l)p¡peraz¡n-1-carbox¡lato de bencilo (0.45 g. 28 % de rendimiento). ESI+APCI MS m/z 567.2 [M+H]+.
Etapa D: (S)-2-(c¡anomet¡l)-4-(2-(3-(d¡met¡lam¡no)-3-oxopropox¡)-7-(8-metilnaftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-3.4-d¡h¡drop¡raz¡n-1 (2H)-carboxilato de bencilo: En un tubo de microondas. una solución de (S)-4-(2-cloro-7-(8-met¡lnaftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de bencilo (250 mg. 0.441 mmol) en dioxano (4408 μl. 0.441 mmol) se roció con argón durante 5 minutos. Se añadieron secuencialmente 3-hidroxi-N,N-dimetilpropanamida (155 mg. 1.32 mmol). carbonato de cesio (431 mg. 1.32 mmol) y metanosulfonato de (2-d¡c¡clohex¡lfosf¡no-2'.6'-d¡-¡-propox¡-1.1'-b¡fen¡l)(2'-metilam¡no-1.1'-b¡fen¡l-2-¡l)palad¡o (II) (37.5 mg. 0.0441 mmol) en una atmósfera de argón y la reacción se roció durante 5 minutos más con argón. La mezcla de reacción se tapó y se calentó a 100 °C durante 1 hora. La reacción se enfrió a temperatura ambiente y se añadió acetato de etilo. La reacción se filtró a través de papel GHF y el filtrado se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida eluyendo con 0 ^20 % de Me0H 2 % de NH40H/DCM. Todas las fracciones que contenían el producto deseado limpio se combinaron y se concentraron para dar (S)-2-(cianometil)-4-(2-(3-(d¡met¡lam¡no)-3-oxopropox¡)-7-(8-met¡lnaftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)-3,4-d¡h¡drop¡raz¡n-1(2H)-carboxilato de bencilo (140 mg. 49.2 % de rendimiento). ESI+APCI MS m/z 648.3 [M+H]+.
Etapa E: (S)-3-((4-(3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-7-(8-met¡lnaftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-2-¡l)ox¡)-N. N-dimetilpropanamida: Una solución de (S)-2-(cianomet¡l)-4-(2-(3-(d¡met¡lam¡no)-3-oxopropoxi)-7-(8-met¡lnaftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de bencilo (140 mg. 0.216 mmol) en Et0H (2161 μl. 0.216 mmol) y THF (2161 μl. 0.216 mmol) se purgó con N2 durante 5 minutos. A esta solución se le añadió paladio (57.5 mg. 0.0540 mmol) (tipo Degussa. 10 % en peso. 50 % de H20) y la reacción se tapó y se purgó con N2 durante 5 minutos más. La solución se agitó en una atmósfera de H2 durante una noche. La mezcla se diluyó con Me0H y se filtró a través de celite empaquetado. Después. el filtrado se concentró al vacío para proporcionar (S)-3-((4-(3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-7-(8-met¡lnaftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-2-¡l)ox¡)-N,N-dimetilpropanamida bruta (111 mg. 100.0 % de rendimiento). ESI+APCI MS m/z 514.3 [M+H]+.
Etapa F: (S.E)-3-((4-(3-(c¡anomet¡l)-4-(4-(d¡met¡lamino)but-2-eno¡l)p¡peraz¡n-1-¡l)-7-(8-met¡lnaftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-2-¡l)ox¡)-N,N-d¡met¡lpropanam¡da: A un matraz de fondo redondo de 25 ml que contenía diclorometano (2161 μl. 0.216 mmol) enfriado a 0 °C se le añadieron (S)-3-((4-(3-(cianometil)p¡peraz¡n-1-¡l)-7-(8-met¡lnaftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-2-¡l)ox¡)-N,N-d¡met¡lpropanam¡da (111 mg. 0.216 mmol) y base de Hunig (226 μl. 1.30 mmol). La mezcla de reacción se agitó vigorosamente mientras se añadía en una porción ácido (E)-4-(dimetilam¡no)but-2-eno¡co (167 mg. 1.30 mmol). Después. a la mezcla en agitación se le añadió lentamente anhídrido cíclico del ácido 1-propanofosfónico (772 μl. 1.30 mmol). La reacción se agitó durante 2 horas a 0 °C. La reacción se trató con agua básica y la capa acuosa se extrajo con EtOAc (3 x). Los extractos orgánicos combinados se concentraron al vacío y se purificaron en el Gilson (HPLC prep.) eluyendo con 5 ^95 % de ACN 0. 1 % de TFA /agua 0.1 % de TFA. Las fracciones que contenían el producto se combinaron. se diluyeron con Na0H 1 M y la capa acuosa se extrajo con DCM. Los extractos orgánicos se secaron sobre Na2 S04 y se concentraron al vacío para dar el compuesto del título (S.E)-3-((4-(3-(cianomet¡l)-4-(4-(d¡met¡lam¡no)but-2-eno¡l)p¡peraz¡n-1-¡l)-7-(8-met¡lnaftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-2-¡l)ox¡)-N,N-d¡met¡lpropanam¡da (17.6 mg. 13.0 % de rendimiento). ESI+APCI MS m/z 625.4 [M+H]+.
Ejemplo 464

2-((S)-1-acriloil-4-(7-(8-bromonaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (S)-4-(7-(8-Bromonaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo: Se diluyeron (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (200 mg, 0,396 mmol), Cs2C03 (644 mg, 1,98 mmol), 1,8-dibromonaftaleno (339 mg, 1,19 mmol), Pd2(dba)3 (72,4 mg, 0,0791 mmol) y XANTPH0S (91,6 mg, 0,158 mmol) con tolueno (1582 μl, 0,396 mmol). La reacción se purgó con argón, se selló y se calentó a 100 °C. Después de agitar durante 23 horas, la reacción se enfrió y se diluyó con acetato de etilo y bicarbonato de sodio saturado. Las capas se separaron y el acetato de etilo se secó sobre MgS04, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 1-10 % de metanol/DCM (1 % de NH40H como aditivo) para proporcionar (S)-4-(7-(8-bromonaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (68 mg, 24,2 % de rendimiento). ESI+APCI MS m/z 712,2 [M+H]+.
Etapa B: 2-((S)-4-(7-(8-bromonaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: Se diluyó (S)-4-(7-(8-bromonaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (25 mg, 0,035 mmol) con TFA (2 ml), se puso en una atmósfera de nitrógeno y se calentó a 90 °C. Después de agitar durante 2 horas, la reacción se enfrió y se concentró. El material se diluyó con DCM y se lavó con bicarbonato de sodio saturado. El DCM se secó sobre MgS04, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10 % de metanol/DCM (1 % de NH40H como aditivo) para proporcionar 2-((S)-4-(7-(8-bromonaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (8 mg, 39 % de rendimiento). ESI-APCI MS m/z 578,2 [M+H]+.
Etapa C: 2-((S)-1-acriloil-4-(7-(8-bromonaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Se diluyó 2-((S)-4-(7-(8-bromonaftalen-1-il)-2-(((S)-1-metilpirrolidin-2
il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (8 mg, 0,014 mmol) con DCM (200 ul) seguido de la adición de cloruro de acriloílo (1,4 mg, 0,015 mmol) y DIEA (4,8 μl, 0,028 mmol). Después de agitar durante 12 horas, la reacción se diluyó con carbonato de sodio saturado y se extrajo dos veces con dCm . El DCM se secó sobre MgSÜ4, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10 % de metanol/DCM (1 % de NH4ÜH como aditivo) para proporcionar el compuesto del título 2-((S)-1-acriloil-4-(7-(8-bromonaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7, 8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMp LÜ 464, 6 mg, 69 % de rendimiento). Es I+APCI MS m/z 632,2 [M+H]+.
Ejemplo 465

(R)-4-acriloil-1-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-carbonitrilo

Etapa A: (R)-4-(4-(terc-butoxicarbonil)-2-cianopiperazin-1il)-2-cloro-5, 8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de bencilo: Una mezcla de 2,4-dicloro-5,6-dihidropirido[3,4-d]pirimidin-7(8H)-carboxilato de bencilo (700 mg, 2,07 mmol), R-1-N-Boc-3-cianopiperazina (656 mg, 3,10 mmol), N,N-dimetilacetamida (1 ml) y N-etil-N-isopropilpropan-2-amina (0,433 ml, 2,48 mmol) se agitó a 60 °C en una atmósfera de nitrógeno durante 48 horas. La mezcla de reacción se enfrió, se repartió entre MTBE (50 ml) y Na2CÜ30,2 M (15 ml) y las capas se separaron. La capa orgánica se lavó con agua y salmuera (10 ml de cada uno), se secó sobre Na2SÜ4 y se evaporó al vacío. El material cristalizó después de la dilución con MTBE (5 ml). El licor madre se decantó y el sólido se lavó con MTBE (2 x 1 ml) seguido de purificación sobre gel de sílice usando de 40 a 60 % de EtÜAc/hexano como eluyente para dar el producto (308 mg, 29 %). ESI+APCI MS m/z 513,2 [M+H]+.
Etapa B: 4-((R)-4-(terc-butoxicarbonil)-2-cianopiperazin-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,8-dihidropirido[3,4-d]pirimidine-7(6H)-carboxilato de bencilo: Una mezcla de (R)-4-(4-(terc-butoxicarbonil)-2-cianopiperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de bencilo (308 mg, 0,600 mmol), Cs2CÜ3 (587 mg, 1,80 mmol), metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2'-metilamino-1,1'-bifenil-2-il)paladio (II) (RuPhos-Pd-G4, 51,1 mg, 0,0600 mmol), (S)-(1-metilpirrolidin-2-il)metanol (346 mg, 3,00 mmol) y 1,4-dioxano (4 ml) se purgó con N2, el vial se tapó y la reacción se agitó a 70 °C durante 2 horas. La mezcla de reacción se repartió entre
agua (5 ml) y EtOAc (20 ml) y la capa orgánica se separó. Los extractos orgánicos se lavaron con agua y salmuera (3 ml de cada uno), se secaron sobre Na2S04 y se evaporaron al vacío. El residuo se cromatografió sobre gel de sílice usando 4 % de Me0H 0,4 % de NH40H/DCM como eluyente para dar el producto en forma de una mezcla de isómeros (214 mg de ~60 % de pureza, 36 %). ESI+APCI MS m/z 592,3 [M+H]+.
Etapa C: (R)-3-ciano-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de terc-butilo: Una mezcla de 4-((R)-4-(terc-butoxicarbonil)-2-cianopiperazin-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de bencilo bruto (214 mg, 0 ,362 mmol), metanol (7 ml, 0,362 mmol) y paladio sobre carbono (20 mg, 5 %, tipo Degussa E101 NO/W ) se desgasificó y se agitó en una atmósfera de hidrógeno durante 1,5 horas. La mezcla de reacción se filtró a través de Celite (2 ml), el celite se lavó con Me0H (3 x 3 ml) y los extractos orgánicos combinados se evaporaron al vacío para dar el producto que se usó bruto en la siguiente reacción. ESI+APCI MS m/z 458,3 [M+H]+.
Etapa D: (R)-3-ciano-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)p¡perazin-1-carboxilato de terc-butilo: Una mezcla de (R)-3-ciano-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de terc-butilo (165 mg, 0 ,361 mmol), Cs2C03 (352 mg, 1,08 mmol), dioxano (0,5 ml), 1-yodonaftaleno (0,0790 ml, 0,541 mmol) y metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1 '-bifenil)(2'-metilamino-1,1'-bifenil-2-il)paladio (II) (RuPhos-Pd-G4, 0,1 eq., 30,7 mg, 0,0361 mmol) se purgó con nitrógeno y el matraz se tapó y se agitó a 70 °C durante 1,5 horas. La mezcla de reacción se enfrió y se repartió entre EtOAc (20 ml) y agua (5 ml) y las capas se separaron. La capa orgánica se lavó con agua y salmuera (5 ml de cada uno), se secó sobre Na2S04 y se evaporó al vacío. El residuo se cromatografió sobre gel de sílice usando 4 % de Me0H 0,4 % de NH40H/DCM como eluyente para dar el producto (120 mg, 57 %). ESI+APCI MS m/z 584,3 [M+H]+.
Etapa E: (R)-1-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-carbonitrilo: El (R)-3-ciano-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -carboxilato de terc-butilo (120 mg, 0,206 mmol) se disolvió en diclorometano (0,5 ml) y se enfrió a 0 °C seguido de la adición de cloruro de hidrógeno 4 M (1,03 ml, 4,11 mmol) y la reacción se agitó durante 1 h mientras se calentaba a temperatura ambiente. La mezcla se evaporó al vacío y el residuo se repartió entre DCM (10 ml) y Na2C032 M (0,5 ml) y las capas se separaron. Los extractos orgánicos se secaron sobre Na2C03, se filtraron y se evaporaron al vacío. El material se usó bruto en la siguiente reacción.
ESI+APCI MS m/z 484,2 [M+H]+.
Etapa F: (R)-4-acriloil-1-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-carbonitrilo: Una solución de (R)-1-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-carbon¡tr¡lo (79 mg, 0,1634 mmol) en DCM (5 ml) se enfrió a -30 °C con agitación seguido de la adición de trietilamina (68,31 μl, 0,4901 mmol) y cloruro de acriloílo (26,54 μl, 0,3267 mmol).
Después de 5 minutos a -30 °C, la reacción se interrumpió mediante la adición de Me0H (0,05 ml) y la reacción se calentó a temperatura ambiente. Luego, los extractos orgánicos se separaron, se lavaron con Na2C030,5 M (4 ml), se secaron sobre Na2C03 y se evaporaron al vacío. El residuo se cromatografió sobre gel de sílice usando 4 % de Me0H 0,4 % de NH40H/DCM como eluyente para dar el compuesto del título (EJEMPL0465, 17 mg, 19 %). ESI+APCI MS m/z 538,3 [M+H]+.
Ejemplo 466

2-((S)-1-acriloil-4-(7-(5,6-dimetil-1H-indol-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo
2-((S)-1-acriloil-4-(7-(5,6-dimetil-1H-indol-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo. El compuesto del título se prepara siguiendo el Ejemplo 454 sustituyendo el Intermedio 66 por 4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carboxilato de terc-butilo en la Etapa A.
E je m p lo 467
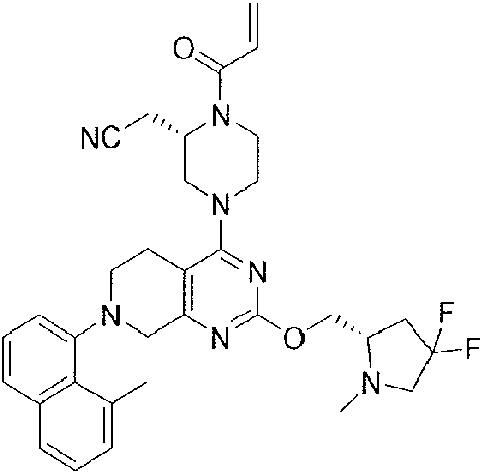
2-((S)-1-acriloil-4-(2-(((S)-4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(2-(((S)-4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: El compuesto del título se prepara siguiendo el Ejemplo 447 sustituyendo (S)-4-(2-cloro-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo en el Ejemplo 447, Etapa A por (S)-4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo en el Ejemplo 447, Etapa A.
Ejemplo 468

2-((S)-1-acriloil-4-(7-(8-cloronaftalen-1-il)-2-(((S)-4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(7-(8-cloronaftalen-1-il)-2-(((S)-4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: El compuesto del título se prepara siguiendo el Ejemplo 447 sustituyendo (S)-4-(2-cloro-7-(8-cloronaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo en el Ejemplo 447, Etapa A por (S)-4-(2-cloro-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo en el Ejemplo 447, Etapa A.
Ejemplo 469

(S)-1-(4-(2-((4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(5,6-dimetil-1H-indol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona
(S)-1-(4-(2-((4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(5,6-dimetil-1H-indol-4-il)-5,6,7,8-tetrahidropirido[3-4-d1pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona: El compuesto del título se prepara siguiendo el EJEMPL0 457 sustituyendo el EJEMPL0 139, Etapa B producto 2-(3,3-difluoropirrolidin-1-il)etanol por [(2S)-1 -metilpirrolidin-2-il]metanol en la preparación del Intermedio 66 , Etapa E y después siguiendo el EJEMPL0457 Etapas B - D.
Ejemplo 470

(S)-1-(4-(2-((4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(5,6-dimetil-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -il)prop-2-en-1-ona
(S)-1-(4-(2-((4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(5,6-dimetil-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-il)prop-2-en-1-ona: El compuesto del título se prepara siguiendo el Ejemplo 453 sustituyendo el Ejemplo 139, Etapa B producto 2-(3,3-difluoropirrolidin-1-il)etanol por [(2S)-1-metilpirrolidin-2-il]metanol en la preparación del Intermedio 66, Etapa E y después siguiendo el Ejemplo 453 Etapas B - C.
Ejemplo 471

2-[(2S)-4-[7-[3-doro-2-(trifluorometil)fenil]-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(£)-4-(3,3-difluoropirrolidin-1-il)but-2-enoil]piperazin-2-il]acetonitrilo

cloruro de (E)-4-bromobut-2-enoílo. A una solución de ácido (E)-4-bromobut-2-enoico (500 mg, 3,03 mmol, 1,0 eq.) en DCM (2,0 ml) se le añadió (C0Cl)2 (4,35 g, 34,3 mmol, 3,0 ml, 11,3 eq.). La mezcla se agitó a 60 °C durante 12 horas. La mezcla de reacción se concentró a presión reducida para dar un residuo. El producto bruto cloruro de (E)-4-bromobut-2-enoílo (500 mg, bruto) se obtuvo en forma de un aceite de color pardo.
Etapa A: (2S)-4-[7-[3-cloro-2-(trifluorometil)fenil1-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1 -carboxilato de tere-butilo. A una solución de (2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (1,30 g, 2,17 mmol, 1,0 eq.) y [(2S)-1-metilpirrolidin-2-il1metanol (500 mg, 4,34 mmol, 515 ul, 2,0 eq.) en tolueno (20,0 ml) se le añadió t-Bu0Na (417 mg, 4,34 mmol, 2,0 eq.). La mezcla se agitó a 0 °C durante 10 min. La mezcla de reacción se inactivó con la adición de salmuera saturada (30,0 ml) a 20 °C y se extrajo con AE (3 * 30,0 ml). Las capas orgánicas combinadas se lavaron con salmuera saturada (30,0 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiO2, éter de petróleo/acetato de etilo = 3/1 a AE/Me0H = 10/1). El producto (2S)-4-[7-[3-cloro-2-(trifluorometil)fenil1-2-[[(2S)-1 -metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-djpirimidin-4-il1-2-(cianometil)piperazin-1 -carboxilato de terebutilo (1,1 g, 1,62 mmol, 75 % de rendimiento, 96 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+11: 650.
Etapa B: 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil1-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-4-[7-[3-cloro-2-(trifluorometil)fenil1-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-djpirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de tere-butilo (100 mg, 154 umol, 1,00 eq.) en dioxano (0,5 ml) se le añadió HCl*dioxano (4 M, 1,00 ml, 26,0 eq.). La mezcla se agitó a 0 °C durante 10 min. La mezcla de reacción se concentró a presión reducida para eliminar el dioxano. El residuo se diluyó con NaHC03 acuoso saturado (10,0 ml) y se extrajo con AE (10 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera saturada (20,0 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El producto bruto 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil1-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-djpirimidin-4-il1piperazin-2-il1acetonitrilo (90,0 mg, bruto) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+11: 550.
Etapa C: 2-((2S)-4-[7-[3-cloro-2-(trifluorometil)fenil1-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-il1-1-[(E)-4-(3,3-difluoropirrolidin-1-il)but-2-enoil1piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-[3-cloro-2-(tr¡fluoromet¡l)fen¡l]-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]acetonitrilo (80,0 mg, 145 umol, 1,0 eq.) en DCM (3,0 ml) se le añadieron Py (115 mg, 1,45 mmol, 117 ul, 10,0 eq.) y cloruro de (E)-4-bromobut-2-enoílo (107 mg, 582 umol, 4,0 eq.) en DCM (3,0 ml). La mezcla se agitó a 0 °C durante 1 hora. Después de que se consumiera el material de partida, a una solución de 3,3-difluoropirrolidina (156 mg, 1,45 mmol, 10,0 eq.) en DCM (6,0 ml) se le añadió la mezcla de reacción. La mezcla se agitó a 0 °C durante 2 horas. La mezcla de reacción se concentró a presión reducida para dar un residuo. El residuo se purificó por HPLC prep.
(columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN];% de B: 62 %-92 %,12 min). Y el residuo se concentró a presión reducida para eliminar el ACN y después se sometió a liofilización. El compuesto del título 2-[(2S)-4-[7-[3-cloro-2-(trifluorometil)fenil]-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il1-1-[(E)-4-(3,3-difluoropirrolidin-1-il)but-2-enoil1piperazin-2-il]acetonitrilo
(EJEMPL0471, 15,4 mg, 20,8 umol, 14 % de rendimiento, 97,7 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1 ]: 723.
1H RMN (400 MHz, CL0R0F0RM0-d) ó = 7,40 (t, J = 8,2 Hz, 1H), 7,30 - 7,26 (m, 1H), 7,19 (d, J = 8,0 Hz, 1H), 7,05 - 6,90 (m, 1H), 6,60 - 6,40 (m, 1H), 5,08 (s a, 1H), 4,39 (dd a, J = 5,2, 10,8 Hz, 1H), 4,17 (dd a, J = 6,8, 10,8 Hz, 1H), 4,1 (s a, 2H), 4,12 - 4,05 (m, 1H), 3,97 (d a, J = 10,8 Hz, 2H), 3,70 - 3,53 (m, 1H), 3,35 - 3,25 (m, 3H), 3,17 - 3,07 (m, 2H), 2,96 (t a, J = 13,2 Hz, 3H), 2,90 - 2,85 (m, 1H), 2,81 (t a, J = 6,8 Hz, 3H), 2,75 - 2,65 (m, 2H), 2,50 (s, 3H), 2,40 -2,27 (m, 3H), 2,15 - 1,95 (m, 1H), 1,95 - 1,72 (m, 5H).
Ejemplo 472

2-[(2S)-4-[7-(2,3-d¡met¡lfen¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo
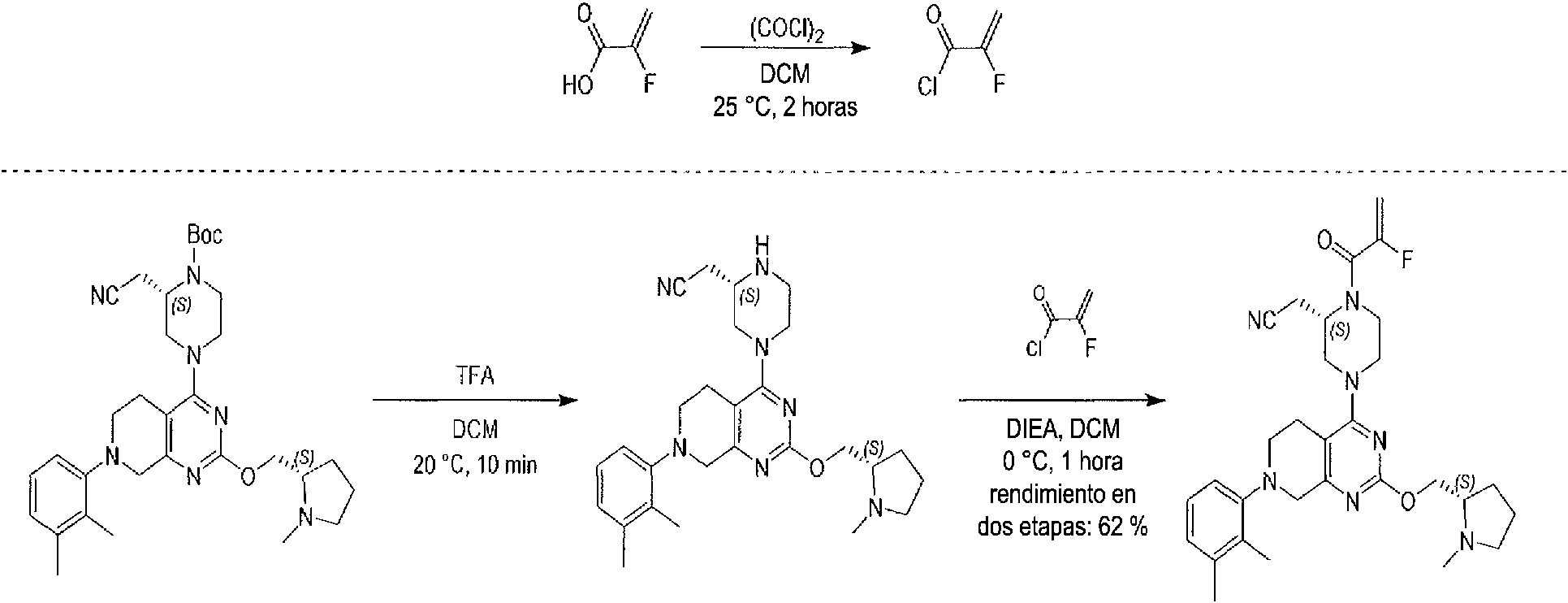
cloruro de 2-cloroprop-2-enoílo. A una solución de ácido 2-fluoroprop-2-enoico (400 mg, 4,44 mmol, 1,0 eq.) en DCM (1,50 ml) se le añadieron (C0Cl)2 (846 mg, 6,66 mmol, 583 ul, 1,50 eq.) y dMf (32,5 mg, 444 umol, 34,2 ul, 0,10 eq.). La mezcla se agitó a 25 °C durante 2 horas. Se usó cloruro de 2-cloroprop-2-enoílo (400 mg, bruto) directamente en la siguiente etapa.
Etapa A: 2-[(2S)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de ferc-butilo (200 mg, 347 umol, 1,0 eq.) en DCM (2,0 ml) se le añadió TfA (3,08 g, 27,0 mmol, 2,0 ml, 77,8 eq.). La mezcla se agitó a 20 °C durante 10 min. La
mezcla de reacción se concentró a presión reducida para dar un residuo. El producto 2-[(2S)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (200 mg, bruto, TFA) se obtuvo en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación adicional.
Etapa B: 2-[(2S)-4-(7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-il)metoxi]-6,8-dihidro-5H-pirido[3,4-cnpirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (200 mg, 339 umol, 1,0 eq., TFA) en DCM (2,0 ml) se le añadieron DIEA (1,11 g, 8,61 mmol, 1,50 ml, 25,4 eq.) y cloruro de 2-fluoroprop-2-enoílo (200 mg, 1,84 mmol, 5,43 eq.) en DCM (2,0 ml). La mezcla se agitó a 0 °C durante 1 hora. La mezcla de reacción se concentró a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 55 %-83 %, 12 min). El compuesto del título 2-[(2S)-4-[7-(2,3-d¡met¡lfen¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (EJEMPL0472, 120 mg, 217 umol, dos etapas 62 % de rendimiento, 98,9 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 548.
1H RMN (400 MHz, CL0R0F0RM0-d) ó = 7,11 (t, J = 7,8 Hz, 1H), 7,01 - 6,91 (m, 2H), 5,52 - 5,32 (m, 1H), 5,26 (dd, J = 3,2, 16,8 Hz, 1H), 5,08 - 4,70 (s a, 1H), 4,39 (dd, J = 5,2, 10,4 Hz, 1H), 4,25 - 4,02 (m, 5H), 3,97 (d, J = 12,8 Hz, 1H), 3,43 - 3,73 (s a, 1H) 3,32 (d, J = 13,2 Hz, 1H), 3,25 - 3,16 (m, 1H), 3,15 - 3,02 (m, 3H), 3,01 - 2,91 (m, 1H), 2,90 -2,80 (m, 2H) 2,79 - 2,60 (m, 2H), 2,49 (s, 3H), 2,31 -2,28 (m, 7H), 2,13 - 1,97 (m, 1H), 1,90 - 1,70 (m, 3H).
Ejemplo 473

2-[(2S)-1-(2-cloroprop-2-enoil)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido-[3,4-ó]pirimidin-4-il]piperazin-2-il]acetonitrilo

cloruro de 2-cloroacriloílo. Una solución de ácido 2-cloroprop-2-enoico (400 mg, 3,76 mmol, 1,0 eq.) y (C0Cl)2 (715 mg, 5,63 mmol, 493 ul, 1,50 eq.) en DCM (1,0 ml) se agitó a 25 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción no se trató y se usó directamente en la siguiente etapa.
Etapa A: 2-[(2S)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cnpirimidin-4-i n piperazin-2-i n acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5W-pirido[3,4-tí]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo (Intermedio 73, 400 mg, 695 umol, 1,0 eq.) en DCM (0,50 ml) se le añadió TFA (3,08 g, 27,0 mmol, 2,0 ml, 38,9 eq.). La mezcla se agitó a 20 °C durante 0,5 horas. Después de que se completara, la mezcla se concentró, se ajustó con NaHC03 acuoso saturado a
pH -7 y después se extrajo con DCM (5,0 ml x 3 ). La capa orgánica se secó sobre Na2SÜ4, se filtró y se concentró. El producto 2-[(2S)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (400 mg, bruto) se obtuvo en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional.
Etapa B: 2-[(2S)-1-(2-cloroprop-2-enoil)-4-[7-(2,3-dimetilμllenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (300 mg, 631 umol, 1,0 eq.) y DIEA (245 mg, 1,89 mmol, 330 ul, 3,0 eq.) en DCM (1,0 ml) se le añadió cloruro de 2-cloroprop-2-enoílo (236 mg, 1,89 mmol, 3,0 eq.) a -78 °C. La mezcla se agitó a -78 °C durante 0,5 horas. Después de que se completara, a la mezcla se le añadió NaHCÜ3 acuoso saturado (2 ,0 ml) y se extrajo con AE (10 ,0 ml x 3 ). El producto obtenido se purificó por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 53 %-83 %,12 min) para dar el compuesto del título 2-[(2S)-1-(2-cloroprop-2-enoil)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (EJEMPL0 473, 4,72 mg, 8,35 umol, dos etapas 1 % de rendimiento, 99,8 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 564.
1H RMN (400 MHz, Cloroformo-d) ó 7,11 (t, J = 7,8 Hz, 1H), 6,98 - 6,94 (m, 2H), 5,86 - 5,78 (m, 1H), 5,77 - 5,72 (m, 1H), 5,15 - 4,78 (m, 1H), 4,45 - 4,30 (m, 1H), 4,21 - 4,12 (m, 1H), 4,10 - 4,03 (m, 3H), 3,96 (d a, J = 12,4 Hz, 2H), 3,30 (dd a, J = 3,2, 13,6 Hz, 1H), 3,23 - 3,04 (m, 4H), 3,04 - 2,54 (m, 6H), 2,50 (s a, 3H), 2,34 - 2,23 (m, 7H), 2,12 - 2,02 (m, 1H), 1,91 - 1,66 (m, 3H).
Ejemplo 474

2-((S)-1-(4-hidroxibut-2-inoil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-tí]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (2S)-2-(cianometin-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-(1-naftin-6.8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-1-carboxilato de bencilo. A una mezcla de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7.8-tetrahidropirido[3.4-d1pirimidin-4-il1piperazin-1 -carboxilato de bencilo (Intermedio 65. 1 g. 1,58 mmol, 1 eq.) y 1- bromonaftaleno (655 mg, 3,16 mmol. 440 ul. 2 eq.) en tolueno (20 ml) se le añadieron en una porción Pd2(dba)3 (145 mg. 158 umol, 0.1 eq.). RuPhos (148 mg. 316 umol, 0.2 eq.) y CS2C03 (1,55 g. 4,75 mmol. 3 eq.). La mezcla se desgasificó y se purgó 3 veces con N2, después se calentó a 90 °C y se agitó durante 2 horas. La mezcla de reacción se diluyó con agua (15 ml) y se extrajo con acetato de etilo (50 ml x 3). Las capas orgánicas combinadas se lavaron con agua (50 ml x 1). se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0.1 % de TFA)/acetonitrilo]. Las fracciones deseadas se recogieron. se neutralizaron con una solución saturada de NaHC03 (6 ml) y se extrajeron con acetato de etilo (100 ml x 2). Las capas orgánicas separadas se secaron sobre sulfato de sodio. se filtraron y se concentraron al vacío. El compuesto (2s)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (515 mg. 726 umol. 45,9 % de rendimiento. 89,1 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 632.
Etapa B: 2-[(2S)-4-[2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-7-(1 -naftil)-6.8-dihidro-5^-pirido[3.4-d1pirimidin-4-il1piperazin-2 - il]acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (1 g. 1,58 mmol. 1 eq.) en Me0H (60 ml) se le añadieron Pd/C (500 mg. 10 % de pureza) y NHs^Me0H (20 %, 50 ml) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó tres veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. La mezcla de reacción se concentró a presión reducida. El compuesto 2-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (680 mg.
1,26 mmol. 80 % de rendimiento. 92,3 % de pureza) se obtuvo en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 498.
1H RMN (400 MHz, cloroformo-d) 5 = 8,26 - 8,19 (m. 1H). 7,89 - 7,83 (m. 1H). 7,60 (d. J = 8.4 Hz. 1H). 7,53 - 7,47 (m.
2H).7,43 (t. J = 7.6 Hz. 1H). 7,16 - 7,12 (m. 1H).4,41 (dd, J = 5,2, 10,8 Hz. 1H).4,26 (s.2H).4,16 (dd. J = 7.2. 10,8 Hz.
1H).4,01 (d a. J = 12,4 Hz. 1H). 3,95 - 3,82 (m. 1H). 3,44 - 3,21 (m. 3H). 3,18 - 2,97 (m. 4H). 2,95 - 2,76 (m. 3H). 2,74 - 2,63 (m. 1H).2,55 (dd. J = 1.2. 5.6 Hz. 2H). 2,51 - 2,46 (m. 3H). 2,33 - 2,22 (m. 1H). 2,12 - 1.99 (m. 1H). 1,88 - 1.80 (m. 3H).
Etapa C: 2-((2S)-1-(4-hidroxibut-2-inoil)-4-[2-[[(2S)-1-metilpirrolidin-2-il)metoxn-7-(1-naftN)-6.8-dihidro-5H-pirido[3.4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de ácido 4-[ferc-butil(dimetil)silil]oxibut-2-inoico (172 mg. 803 umol. 2 eq.) y DIEA (155 mg. 1,21 mmol. 210 ul. 3 eq.) en MeCN (5 ml) se le añadió MsCl (92,1 mg. 803 umol. 62,2 ul. 2 eq.) a 0 °C. Después de agitar a 0 °C durante 20 minutos. a la mezcla se le añadió 2-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (200 mg. 402 umol. 1 eq.) y la mezcla se agitó a 25 °C durante 2 horas. Después. la mezcla de reacción se calentó a 80 °C y se agitó durante 10 horas. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (20 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (20 ml). se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Boston Green 0DS 150 * 305 u; fase móvil: [agua (0,225 % de AF)-ACN]; % de B: 25 % - 45 %, 10 min) y se purificó adicionalmente por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 45 % - 75 %, 12 min). El compuesto del título 2-[(2S)-1-(4-hidroxibut-2-inoil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (EJEMPL0 474, 1,43 mg. 2,39 umol. 1 % de rendimiento. 96,7 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI. M+1]: 580.
1H RMN (400 MHz. cloroformo-d) 5 = 8,22 (d, J = 6.8 Hz. 1H). 7,90 - 7,82 (m. 1H). 7,61 (d, J = 8.4 Hz. 1H). 7,55 - 7,47 (m. 2H). 7,44 (t. J = 7.6 Hz. 1H). 7,15 (d. J = 7.2 Hz. 1H). 5,26 - 4,89 (m. 1H). 4,60 - 4,34 (m. 4H). 4,33 - 4,16 (m. 3H).
4,14 - 3,96 (m. 1H). 3,95 - 3,80 (m. 1H). 3,78 - 3,50 (m. 2H). 3,50 - 3,28 (m. 2H). 3,27 - 3,07 (m. 3H). 3,06 - 2,82 (m.
3H). 2,80 - 2,64 (m. 2H). 2,51 (d. J = 12,4 Hz. 3H). 2,38 - 2,24 (m. 1H). 2,14 - 2,00 (m. 1H). 1,97 - 1.85 (m. 2H).
Ejemplo 475

2-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2,3,3-trideuterioprop-2-enoil)piperazin-2-il]acetonitrilo

Etapa A: 2-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2,3,3-trideuterioprop-2-enoil)piperazin-2-il]acetonitrilo. A una solución de 2,3,3-trideuterioprop-2-enoato de deuterio (68,8 mg, 904 umol, 3 eq.) y DIEA (234 mg, 1,81 mmol, 315 ul, 6 eq.) en MeCN (3 ml) se le añadió MsCl (69,1 mg, 603 umol, 46,7 ul, 2 eq.) a 5 °C durante 0,5 horas. A la solución resultante se le añadió 2-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (150 mg, 301 umol, 1 eq.). La mezcla de reacción se agitó a 5 °C durante 1 hora. Después, la mezcla de reacción se calentó a 80 °C durante 12 h. Después de que se completara, la mezcla de reacción se inactivó con agua (0,5 ml). La mezcla del residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um;fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN];% de B: 50 %-80 %,12 min) para dar el compuesto del título 2-[(2s )-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2,3,3-trideuterioprop-2-enoil)piperazin-2-il]acetonitrilo (EJEMPL0475, 34,2 mg, 60,2 umol, 20 % de rendimiento, 97,7 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 555.
1H RMN (400 MHz, CL0R0F0RM0-d) ó = 8,28 - 8,16 (m, 1H), 7,91 - 7,83 (m, 1H), 7,63 (d, J = 8,0 Hz, 1H), 7,56 -7,48 (m, 2H), 7,45 (t, J = 7,6 Hz, 1H), 7,16 (d, J = 7,2 Hz, 1H), 5,28 - 4,94 (m, 1H), 4,41 (dd, J = 5,2, 10,8 Hz, 1H), 4,36 - 4,24 (m, 2H), 4,23 - 4,11 (m, 2H), 4,04 (d a, J = 11,6 Hz, 2H), 3,73 - 3,25 (m, 3H), 3,22 - 2,74 (m, 7H), 2,69 (td, J = 6,4, 13,2 Hz, 1H), 2,50 (s, 3H), 2,36 - 2,24 (m, 1H), 2,15 - 2,02 (m, 1H), 1,83 - 1,77 (m, 3H).
Ejemplo 476

2-[(2S)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5W-pirido[3,4-tí]pirim idin-4-il]-1 -[(E)-4-(3-fluoropirrolidin-1 -il)but-2-enoil]piperazin-2-il]acetonitrilo

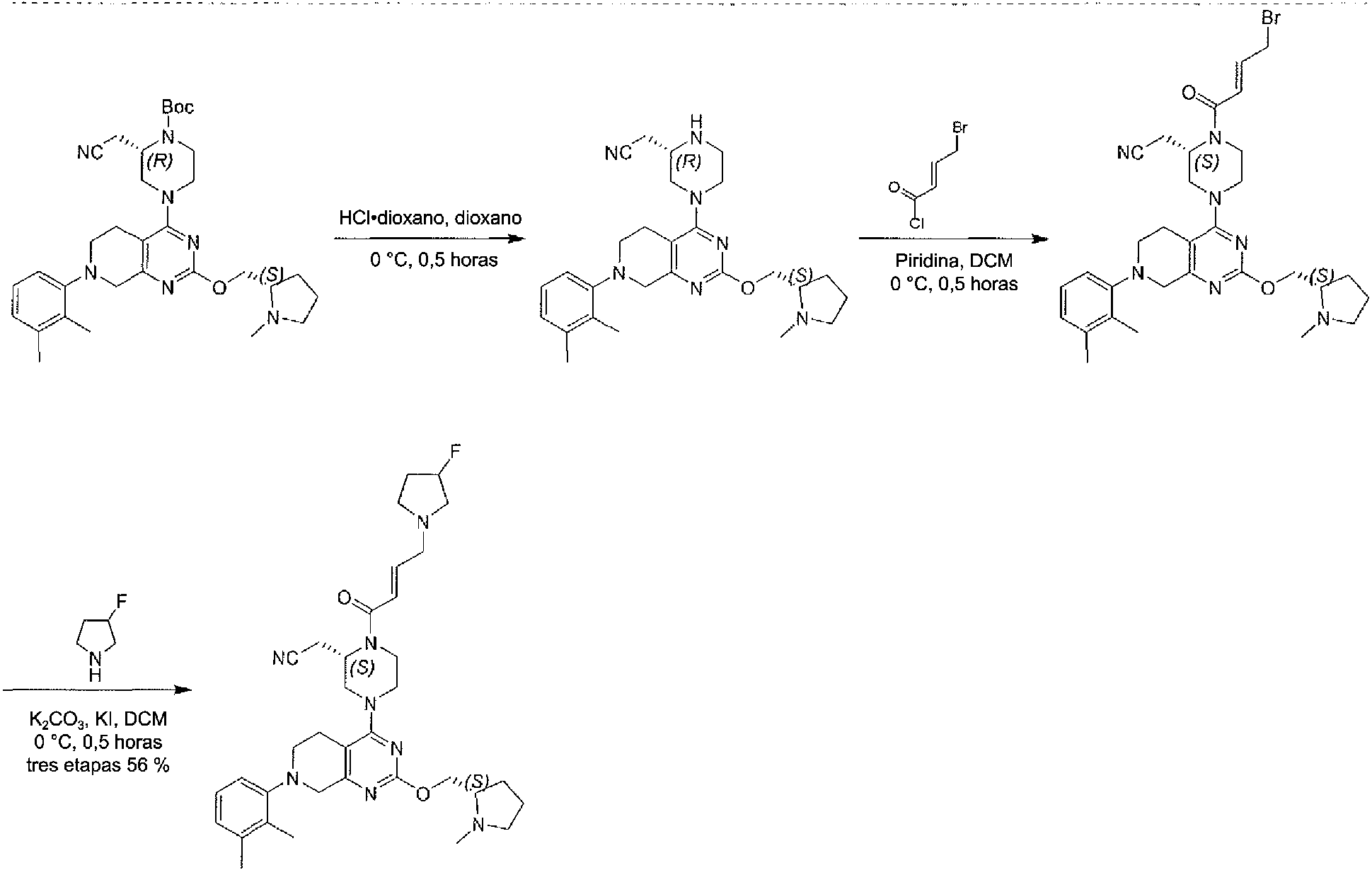
Inserción: cloruro de (E)-4-bromobut-2-enoílo. Una solución de ácido (E)-4-bromobut-2-enoico (1,0 g, 6,06 mmol, 1,0 eq.) en (C0Cl)2 (14,5 g, 114 mmol, 10,0 ml, 18,9 eq.) y DCM (10,0 ml) se agitó a 70 °C durante 2 horas. Después de que se completara, la mezcla se concentró al vacío. El producto cloruro de (E)-4-bromobut-2-enoílo (1,0 g, bruto) se obtuvo en forma de un aceite de color amarillo. El compuesto bruto se usó directamente para la siguiente etapa sin purificación adicional.
Etapa A: 2-[(2S)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-illpiperazin-2-illacetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de terc-butilo (Intermedio 73, 200 mg, 347 umol, 1,0 eq.) en dioxano (2,0 ml) se le añadió HCl*dioxano (4,0 M, 2,0 ml, 23,0 eq.). La mezcla se agitó a 25 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se concentró a presión reducida para dar un residuo. El residuo se ajustó a pH = 7 con una solución acuosa saturada de NaHC03 y se extrajo con AE (5,0 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (10,0 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar el residuo. El producto 2-[(2S)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1 -metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (160 mg, bruto) se obtuvo en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación adicional.
Etapa B: 2-[(2S)-1-[(E)-4-bromobut-2-enoil]-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]acetonitr¡lo (160 mg, 336 umol, 1,0 eq.) en DCM (6,0 ml) se le añadieron Py (532 mg, 6,73 mmol, 543 ul, 20,0 eq.) y cloruro de (E)-4-bromobut-2-enoílo (246 mg, 1,35 mmol, 4,0 eq.) en DCM (2,0 ml). La mezcla se agitó a 0 °C durante 1 hora. Después de que se completara, la mezcla de reacción se inactivó con la adición de Me0H (5,0 ml) a 0 °C y después se concentró a presión reducida para dar un residuo. El producto 2-[(2S)-1-[(E)-4-bromobut-2-enoil]-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]acetonitr¡lo (200 mg, bruto) se obtuvo en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1 ]: 622.
Etapa C: 2-[(2S)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-[(E)-4-(3-fluoropirrolidin-1-il)but-2-enoi n piperazin-2-i n acetonitrilo. A una solución de 2-[(2S)-1 -[(E)-4-bromobut-2-enoil]-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-tí]pirim idin-4-il]piperazin-2-il]acetonitrilo (200 mg, 321 umol, 1,0 eq.) en DCM (3,0 ml) se le añadieron K2C03 (88,8 mg, 642 umol, 2,0 eq.), KI (2,67 mg, 16,1 umol, 0,05 eq.) y 3-fluoropirrolidina (403 mg, 3,21 mmol, 10,0 eq., sal HCl). La mezcla se agitó a 0 °C durante 1 hora. Después de que se completara, la mezcla de reacción se concentró a presión reducida para dar un residuo. El residuo se ajustó a pH = 7 con una solución acuosa saturada de NaHC03 y se extrajo con AE (10,0 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20,0 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar el residuo. El residuo se purificó por HPLC prep. (columna: Gemini 150 * 25
5 ufase móvil: [agua (0,04 % de NH3H20)-ACN];% de B: 60 %-84 %,10 min). El residuo se concentró a presión reducida y después liofilización para dar el compuesto del título 2-[(2S)-4-[7-(2,3-dimetilfenil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-<C]pirimidin-4-il]-1-[(E)-4-(3-fluoropirrolidin-1-il)but-2-enoil]piperazin-2-il]acetonitrilo (EJEMPL0476, 123 mg, 193 umol, 56 % de rendimiento en tres etapas, 99,2 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1 ]: 631.
1H RMN (400 MHz, Cloroformo-d) ó 7,16 - 7,07 (m, 1H), 7,02 - 6,92 (m, 3H), 6,59 - 6,41 (m, 1H), 5,31 - 5,10 (m, 1H), 5,09 - 4,46 (m, 1H), 4,38 (dd, J = 4,8, 10,4 Hz, 1H), 4,26 - 3,83 (m, 6H), 3,80 - 3,45 (m, 1H), 3,42 - 3,24 (m, 3H), 3,23 - 3,15 (m, 1H), 3,14 - 3,03 (m, 3H), 3,02 - 2,83 (m, 4H), 2,82 - 2,62 (m, 4H), 2,54 - 2,49 (m, 1H), 2,47 (s, 3H), 2,32 -2,26 (m, 6H), 2,25 - 2,17 (m, 1H), 2,16 - 2,09 (m, 1H), 2,08 - 2,02 (m, 1H), 1,92 - 1,78 (m, 4H).
Ejemplo 477

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-c/]pirimidin-4-il]-1-(2,3,3-trideuterioprop-2-enoil)piperazin-2-il]acetonitrilo

Etapa A: 2-[(2S)-4-[7-(8-cloro-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-<C]pirimidin-4-il]-1 -(2,3,3-trideuterioprop-2-enoil)piperazin-2-il]acetonitrilo. A la solución de 2,3,3-trideuterioprop-2-enoato de deuterio (56,6 mg, 744 umol, 3 eq.) y DIEA (481 mg, 3,72 mmol, 648 ul, 15 eq.) en ACN (3 ml) se le añadió MsCl (56,8 mg, 496 umol, 38,4 ul, 2 eq.) a 5 °C y la mezcla se agitó a 5 °C durante 0,5 horas. Después, a la mezcla se le añadió 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-<C]pirimidin-4-il]piperazin-2-il]acetonitrilo (Intermedio 71, 150 mg, 248 umol, 1 eq., 2 HCl) y la mezcla resultante se agitó a 5 °C durante 1 hora.
Después, la mezcla se calentó a 80 °C y se agitó a 80 °C durante 12 horas. La mezcla se inactivó con agua (0,5 ml).
La mezcla de reacción se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um;fase móvil:
[agua (0,05 % de hidróxido de amoniaco v/v)-ACN];% de B: 50 % - 80 %,10 min) para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-<C]pirimidin-4-il]-1-(2,3,3-trideuterioprop-2-enoil)piperazin-2-il]acetonitrilo (EJEMPL0477, 13,0 mg, 21,6 umol, 9 % de rendimiento, 98,2 % de pureza) en forma de un sólido de color blanquecino. LCMS [ESI, M+1]: 589.
1 h RMN (400 MHz, cloroformo-d) ó = 7,76 (d a, J = 8,4 Hz, 1H), 7,62 (t, J = 7,6 Hz, 1H), 7,53 (d, J = 7,2 Hz, 1H), 7,45 (td, J = 8,0, 13,2 Hz, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,26 - 7,18 (m, 1H), 5,10 (s a, 1H), 4,49 - 4,33 (m, 2H), 4,21 - 4,01 (m, 3H), 3,96 - 3,79 (m, 2H), 3,60 (d a, J = 6,8 Hz, HI), 3,44 (d a, J = 13,6 Hz, 1H), 3,32 - 2,97 (m, 5H), 2,88 - 2,53 (m, 4H), 2,48 (d, J = 2,8 Hz, 3H), 2,29 (d a, J = 8,8 Hz, 1H), 2,11 - 2,00 (m, 1H), 1,87 - 1,71 (m, 3H).
E je m p lo 478

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo

cloruro de 2-fluoroprop-2-enoílo. A una solución de ácido 2-fluoroprop-2-enoico (400 mg, 4,44 mmol, 1 eq.) en DCM (4 ml) se le añadieron (C0Cl)2 (846 mg, 6,66 mmol, 583 ul, 1,5 eq.) y DMF (32,5 mg, 444 umol, 34,2 ul, 0,1 eq.). La mezcla se agitó a 25 °C durante 2 h. La mezcla de reacción se concentró a presión reducida para eliminar una parte del disolvente y dar un residuo en DCM. El compuesto cloruro de 2-fluoroprop-2-enoílo (400 mg, bruto) se obtuvo en forma de un líquido de color amarillo y se usó en la siguiente etapa sin purificación adicional.
Etapa A: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cnpirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (300 mg, 528 umol, 1 eq., HCl) en DCM (5 ml) se le añadieron DIEA (1,73 g, 13,4 mmol, 2,33 ml, 25,4 eq.) y cloruro de 2-fluoroprop-2-enoílo (286 mg, 2,64 mmol, 5 eq.) en DCM (5 ml). La mezcla se agitó a 0 °C durante 1 hora. La mezcla de reacción se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Ah03, diclorometano/metanol = 10/1 a 10/1). El residuo se purificó por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil:
[agua (0,05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 55 % - 85 %, 12 min). El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi C18 150 * 30 mm * 4 um; fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 20 % - 50 %, 10,5 min). El residuo se concentró a presión reducida para eliminar el ACN y después se sometió a liofilización. El compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-tí]pirimidin-4-il]-1 -(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (EJEMPL0478, 24,1 mg, 36,7 umol, 7 % de rendimiento, 99,1 % de pureza, AF) se obtuvo en forma de un sólido de color pardo.
Condición de SFC: “AD - 3S_3_5_40_3ML Columna: Chiralpak AD - 3100 * 4,6 mm D.I., 3 um Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 % Caudal: 3 ml/min Longitud de onda: 220 nm” .
1H RMN (400 MHz, acético) 5 = 7,82 (d, J = 8,0 Hz, 1H), 7,69 (d, J = 8,0 Hz, 1H), 7,56 (d, J = 7,6 Hz, 1H), 7,49 (t, J = 7,6 Hz, 1H), 7,41 - 7,30 (m, 2H), 5,58 - 5,25 (m, 2H), 5,17 - 4,59 (m, 4H), 4,57 - 4,28 (m, 3H), 4,24 - 3,78 (m, 4H), 3,67 - 3,13 (m, 7H), 3,08 (d a, J = 2,4 Hz, 3H), 2,98 (d a, J = 6,4 Hz, 1H), 2,83 - 2,61 (m, 1H), 2,45 - 2,29 (m, 1H), 2,24 -2,08 (m, 3H).
Ejemplo 479

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H pirido[3,4-d]pirimidin-4-il]-1-[(E)-4-fluorobut-2-enoil]piperazin-2-il]acetonitrilo

Etapa A: (E)-4-fluorobut-2-enoato de etilo. Una mezcla de (E)-4-bromobut-2-enoato de etilo (2,00 g, 10,4 mmol, 1,43 ml, 1,00 eq.) y AgF (3,94 g, 31,1 mmol, 674 ul, 3,00 eq.) en acetonitrilo (20,0 ml) se agitó a 25 °C durante 12 horas. La mezcla se filtró y se lavó con THF (10,0 ml) y después se concentró al vacío para dar (E)-4-fluorobut-2-enoato de etilo (2,50 g, bruto) en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, cloroformo-d) 5 = 7,04 - 6,89 (m, 1H), 6,12 (cd, J = 2,0, 16,0 Hz, 1H), 5,14 - 4,94 (m, 2H), 4,22 (c, J = 6,8 Hz, 2H), 1,30 (t, J = 6,8 Hz, 3H).
Etapa B: ácido (E)-4-fluorobut-2-enoico. Una mezcla de (E)-4-fluorobut-2-enoato de etilo (0,10 g, bruto) y Na0H (121 mg, 3,03 mmol) en THF (1,00 ml) y H20 (1,00 ml) se agitó a 25 °C durante 2 horas. El valor del pH se ajustó a 1 -3 con HCl (1 N, 5,00 ml), se extrajo con acetato de etilo (3 * 10,0 ml) y la capa orgánica se secó sobre Na2S04, se filtró y se concentró al vacío para dar ácido (E)-4-fluorobut-2-enoico (0,02 g, 192 mmol, dos etapas 46 %) en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, cloroformo-d) 5 = 8,37 - 7,31 (m, 1H), 7,15 - 7,01 (m, 1H), 6,15 (dd, J = 1,6, 16,0 Hz, 1H), 5,22 -4,96 (m, 2H).
Etapa C: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]-1-[(E)-4-fluorobut-2-enoillpiperazin-2-il]acetonitrilo. A una solución de ácido (E)-4-fluorobut-2-enoico (7,32 mg, 70,4 umol, 2 eq.) y Py (8,35 mg, 106 umol, 8,52 ul, 3,00 eq.) en acetonitrilo (1,00 ml) se le añadió MsCl (8,06 mg, 70,4 umol, 5,45 ul, 2,00 eq.) a 0 °C. Después de agitar a 0 °C durante 1 hora, a la mezcla se le añadió 2-[(2S)-4-[7-(8-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡trilo (0,02 g, 35,2 umol, 1,00 eq., HCl). La mezcla se agitó a 25 °C durante 12 h. La mezcla se diluyó con agua (5,00 ml), se extrajo con acetato de etilo (3 * 5,00 ml) y la capa orgánica se secó sobre Na2S04, se filtró y se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) -ACN]; % de B: 50 % - 80 %, 12 min). La fracción deseada se recogió y se liofilizó para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5W-pirido[3,4-tí]pirimidin-4-il]-1 -[(E)-4
fluorobut-2-enoil]piperazin-2-il]acetonitrilo (EJEMPL0 479, 3,01 mg, 4,82 umol, 14 % de rendimiento, 98,9 % de pureza) en forma de un sólido de color pardo. LCMS [ESI, M+1]: 618.
1H RMN (400 MHz, CL0R0F0RM0-d) 5 = 7,76 (d a, J = 8,0 Hz, 1H), 7,62 (t, J = 7,2 Hz, 1H), 7,53 (d, J = 7,2 Hz, 1H), 7,45 (td, J = 7,6, 12,8 Hz, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,26 - 7,17 (m, 1H), 7,09 - 6,91 (m, 1H), 6,59 (d a, J = 15,6 Hz, 1H), 5,19 - 5,05 (m, 2H), 4,49 - 4,36 (m, 2H), 4,22 - 3,79 (m, 5H), 3,76 - 2,96 (m, 8H), 2,92 - 2,55 (m, 4H), 2,48 (d, J = 2,4 Hz, 3H), 2,29 (d a, J = 9,6 Hz, 1H), 2,06 (d a, J = 10,0 Hz, 1H), 1,83 - 1,71 (m, 3H).
Ejemplo 480
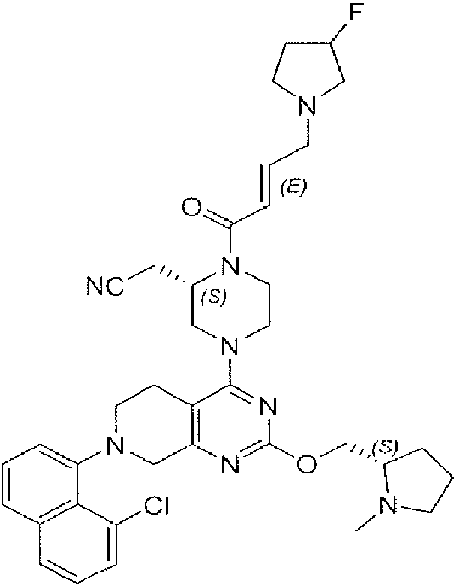
2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-[(E)-4-(3-fluoropirrolidin-1 -il)but-2-enoil]piperazin-2-il]acetonitrilo
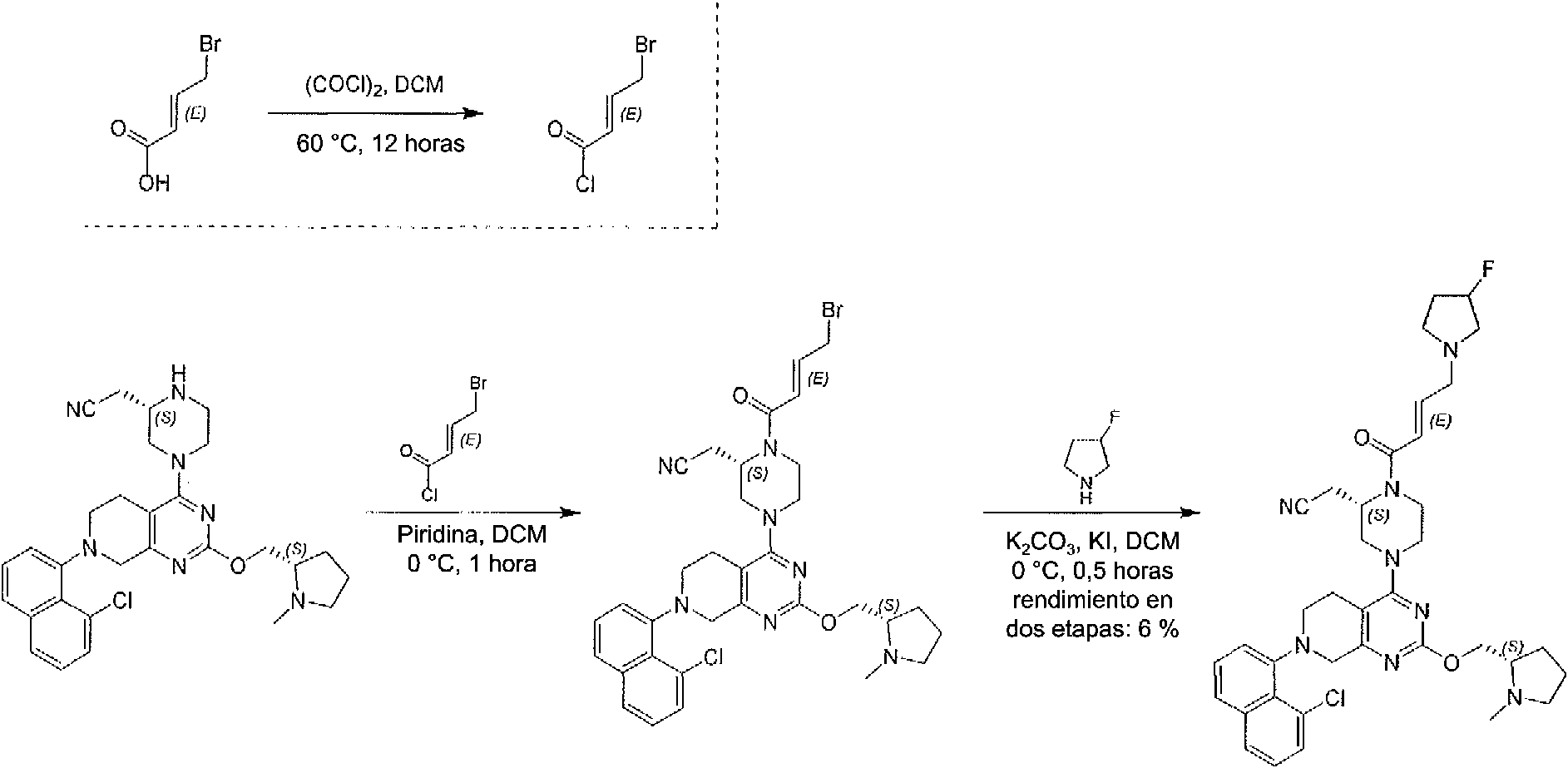
Inserción: cloruro de (E)-4-bromobut-2-enoílo. A una solución de ácido (E)-4-bromobut-2-enoico (930 mg, 5,64 mmol, 1,0 eq.) en DCM (2,0 ml) se le añadió (C0Cl)2 (2,90 g, 22,9 mmol, 2,0 ml, 4,05 eq.). La mezcla se agitó a 60 °C durante 12 horas. Después de que se completara, la mezcla se concentró al vacío. El producto cloruro de (E)-4-bromobut-2-enoílo (650 mg, bruto) se obtuvo en forma de un aceite de color amarillo y se usó directamente en la siguiente etapa sin purificación adicional.
Etapa A: 2-[(2S)-1-[(E)-4-bromobut-2-enoil1-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (Intermedio 71, 150 mg, 282 umol, 1,0 eq.) en DCM (3,0 ml) se le añadieron Py (446 mg, 5,64 mmol, 455 ul, 20,0 eq.) y cloruro de (E)-4-bromobut-2-enoílo (207 mg, 1,13 mmol, 4,0 eq.) en DCM (1,0 ml). La mezcla se agitó a 0 °C durante 1 hora. Después de que se completara, la mezcla de reacción se concentró a presión reducida para dar un residuo. El producto 2-[(2S)-1-[(E)-4-bromobut-2-enoil]-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (150 mg, bruto) se obtuvo en forma de un aceite de color pardo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 680.
Etapa B: 2-r(2S)-4-r7-(8-cloro-1-naftil)-2-rr(2S)-1-metilpirrolidin-2-il1metoxi1-6.8-dihidro-5H-piridor3.4-dlpirimidin-4-il1-1-[(E)-4-(3-fluoropirrolidin-1-il)but-2-enoillpiperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-1 -[(E)-4-bromobut-2-enoil]-4-[7-(8-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡perazin-2-il]acetonitrilo (150 mg. 221 umol, 1.0 eq.) en DCM (10,0 ml) se le añadieron K2C03 (366 mg. 2,65 mmol, 12,0 eq.), KI (1,83 mg. 11,0 umol. 0,05 eq.) y 3-fluoropirrolidina (277 mg. 2,21 mmol. 10,0 eq.. HCl). La mezcla se agitó a 0 °C durante 0.5 horas. Después de que se completara. la mezcla de reacción se concentró a presión reducida para dar un residuo. El residuo se ajustó a pH ~7 con una solución acuosa saturada de NaHC03 y se extrajo con AE (3 * 10 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml). se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar el residuo. El residuo se purificó por HPLC prep. (columna: Gemini 150 * 25 5 u; fase móvil: [agua (0,04 % de NH3*H20)-ACN]; % de B: 60 %-84 %, 10 min). El residuo se concentró a presión reducida y después se sometió a liofilización. El compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-[(E)-4-(3-fluorop¡rrol¡d¡n-1-¡l)but-2-eno¡l]p¡peraz¡n-2-il]acetonitrilo (EJEMPL0480, 9,67 mg. 14,0 umol. 6 % de rendimiento. 99,2 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 687.
1H RMN (400 MHz, Cloroformo-d) ó 7,76 (d a, J = 8.0 Hz. 1H). 7,62 (t. J = 7.8 Hz. 1H). 7,26 (d a. J = 7.2 Hz. 1H). 7,49 - 7,40 (m. 1H). 7,34 (t. J = 7.8 Hz. 1H). 7,27 - 7,17 (m. 1H). 7,05 - 6,92 (m. 1H). 6,57 - 6,42 (m. 1H). 5,32 - 5,11 (m.
1H). 5,10 - 4,50 (m. 1H). 4,47 - 4,35 (m. 2H). 4,19 - 4,00 (m. 2H). 3,95 - 3,75 (m. 2H). 3,65 - 3,55 (m. 1H). 3,47 - 3,28 (m. 3H). 3,24 - 3,05 (m. 4H). 3,04 - 2,86 (m. 4H). 2,85 - 2,71 (m. 2H). 2,70 - 2,50 (m. 3H). 2,47 - 2,45 (m. 3H). 2,32 -2,15 (m. 2H). 2,14 - 2,02 (m. 2H). 1,87 - 1,67 (m. 4H).
Ejemplo 481

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-c(]pirimidin-4-il]-1-[(E)-4-(3-
fluoroazet¡d¡n-1-¡l)but-2-eno¡l]p¡perazin-2-¡l]aceton¡tr¡lo

Inserción: cloruro de (E)-4-bromobut-2-enoílo. A una solución de ácido (£)-4-bromobut-2-enoico (1 g, 6,06 mmol, 1,0 eq.) en DCM (5,0 ml) se le añadió dicloruro de oxalilo (7,25 g, 57,1 mmol, 5,0 ml, 9,4 eq.). La mezcla se agitó a 60 °C durante 12 horas. Después de que se completara, la mezcla de reacción se concentró a presión reducida para dar cloruro de (£)-4-bromobut-2-enoílo (850 mg, bruto) en forma de un aceite de color amarillo que se usó en la siguiente etapa sin purificación adicional.
Etapa A: Intermedio 71, 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-4-[7-(8-cloro-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de tere-butilo (310 mg, 414 umol, 1,0 eq.) en dioxano (4,0 ml) se le añadió HCl/dioxano (4,0 M, 4,0 ml, 39,0 eq.) a 25 °C. La mezcla se agitó a 25 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se inactivó con una solución acuosa saturada de NaHCÜ3 (20,0 ml) y después se extrajo con AE (3 * 40,0 ml). Las capas orgánicas combinadas se lavaron con una solución acuosa saturada de NaCl (100,0 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron para dar un residuo 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-djpirimidin-4-il1piperazin-2-il1acetonitrilo (250 mg, bruto) en forma de un sólido de color amarillo. El producto bruto se usó en la siguiente etapa sin purificación adicional. lCmS [ESI, M+11: 532.
Etapa B: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-[(£)-4-(3-fluoroazetidin-1-il)but-2-enoil1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (Intermedio 71, 150 mg, 282 umol, 1,0 eq., bruto) en DCM (6,0 ml) se le añadieron Py (446 mg, 5,6 mmol, 455 ul, 20,0 eq.) y cloruro de (£)-4-bromobut-2-enoílo (207 mg, 1,2 mmol, 4,0 eq., bruto) en DCM (1,0 ml). La mezcla se agitó a 0 °C durante 1 hora. Después, se añadieron K2C03 (779 mg, 5,6 mmol, 20,0 eq.), KI (4,6 mg, 28,2 umol, 0,1 eq.) y 3-fluoroazetidina (472 mg, 4,2 mmol, 15,0 eq., HCl) a 0 °C. Después, la mezcla se agitó a 25 °C durante 12 horas. Después de que se completara, la mezcla se filtró y se concentró a presión reducida. El residuo se purificó por HPLC prep. (columna:
Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (NH4HC0310,0 mM)-ACN1; % de B: 55 %-85 %, 10 min.) y se sometió a liofilización para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1 -metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]-1-[(£)-4-(3-fluoroazet¡d¡n-1-¡l)but-2-eno¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (EJEMPL0481, 10,8 mg, 15,7 umol, 6 % de rendimiento, 98 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+11: 673.
Condiciones de SFC: 100 % de e.e.
1H RMN (400 MHz, Cloroformo-d) 57,77 (d, J = 8,0 Hz, 1 H), 7,63 (t, J = 7,8 Hz, 1 H), 7,54 (d, J = 7,2 Hz, 1 H), 7,50 7,43 (m, 1 H), 7,35 (t, J = 7,8 Hz, 1 H), 7,26 - 7,21 (m, 1 H), 6,89 (d, J = 15,2 Hz, 1 H), 6,42 (d, J = 14,8 Hz, 1 H), 5,26 4,62 (m, 1 H), 5,11 (t, J = 5,2 Hz, 1 H), 4,80 - 4,37 (m, 2 H), 4,20 - 4,08 (m, 2 H), 3,92 (d a, J = 17,6 Hz, 1 H), 3,86 -3,71 (m, 3 H), 3,60 (s, 1 H), 3,52 - 3,47 (m, 1 H), 3,45 - 3,33 (m, 1 H), 3,28 - 3,21 (m, 3 H), 3,16 - 3,11 (m, 3 H), 3,03 (dd, J = 8,4, 16,8 Hz, 1 H), 2,82 - 2,60 (m, 4 H), 2,49 (d, J = 2,8 Hz, 3 H), 2,37 - 2,25 (m, 1 H), 2,09 - 2,03 (m, 1 H), 1,86 - 1,83 (m, 1 H), 1,78 - 1,76 (m, 3 H).
Ejemplo 482

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(£)-4-[metil(2,2,2-trifluoroetil)amino1but-2-enoil1piperazin-2-il1acetonitrilo

Inserción: cloruro de (E)-4-bromobut-2-enoílo. Una solución de ácido (E)-4-bromobut-2-enoico (600 mg, 3,64 mmol, 1,0 eq.) en (C0Cl)2 (10,2 g, 80,0 mmol, 7,0 ml, 22,0 eq.) y DCM (7,0 ml) se agitó a 70 °C durante 12 horas. Después de que se completara, la mezcla de reacción se concentró para dar cloruro de (E)-4-bromobut-2-enoílo (500 mg, bruto) en forma de un aceite de color amarillo. El producto bruto se usó en la siguiente etapa sin purificación adicional.
Etapa A: 2-[(2S)-1-[(E)-4-bromobut-2-enoil1-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]acetonitr¡lo (100 mg, 188 umol, 1,0 eq.) y piridina (119 mg, 1,50 mmol, 121 ul, 8,0 eq.) en DCM (2,0 ml) se le añadió cloruro de (E)-4-bromobut-2-enoílo (138 mg, 752 umol, 4,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se concentró para dar 2-[(2S)-1-[(E)-4-bromobut-2-enoil]-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (130 mg, bruto) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 678, 680.
Etapa B: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-[(E)-4-[metil(2,2,2-trifluoroetil)amino1but-2-enoil1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-1-[(E)-4-bromobut-2-eno¡l]-4-[7-(8-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]p¡perazin-2-¡l]aceton¡tr¡lo (128 mg, 189 umol, 1,0 eq.), K2C03 (130 mg, 943 umol, 5,0 eq.) y KI (9,39 mg, 56,6 umol, 0,30 eq.) en DCM (3,0 ml) se le añadió poco a poco 2,2,2-trifluoro-N-metil-etanamina (213 mg, 1,88 mmol, 10,0 eq.). La mezcla se agitó a 0-25 °C durante 12 horas. Después de que se completara, la mezcla se concentró al vacío. El producto obtenido se purificó por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,04 % de NH3*H20)-ACN]; % de B: 70 %-100 %, 10 min) para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-[(E)-4-[met¡l(2,2,2-tr¡fluoroetil)am¡no]but-2-eno¡l]p¡perazin-2-¡l]aceton¡tr¡lo (EJEMPL0482, 2,87 mg, 3,84 umol, 2 % de rendimiento, 95 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 711.
1H RMN (400 MHz, Cloroformo-d) ó 7,76 (d a, J = 7,6 Hz, 1H), 7,62 (t, J = 7,6 Hz, 1H), 7,53 (d, J = 7,2 Hz, 1H), 7,49 -7,40 (m, 1H), 7,34 (t, J = 7,8 Hz, 1H), 7,27 - 7,18 (m, 1H), 6,95 - 6,79 (m, 1H), 6,54 - 6,38 (m, 1H), 5,12 - 4,50 (m, 1H), 4,48 - 4,35 (m, 2H), 4,24 - 3,99 (m, 3H), 3,96 - 3,78 (m, 2H), 3,77 - 3,64 (m, 1H), 3,61 - 3,53 (m, 1H), 3,52 - 3,34 (m, 3H), 3,33 - 2,97 (m, 7H), 2,92 - 2,76 (m, 1H), 2,75 -2,65 (m, 1H), 2,63 - 2,55 (m, 1H), 2,51 (s, 3H), 2,49 (d a, J = 3,2 Hz, 3H), 2,35 - 2,25 (m, 1H), 2,13 - 1,99 (m, 1H), 1,89 - 1,67 (m, 3H).
Ejemplo 483

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-(4-hidroxibut-2-inoil)piperazin-2-il]acetonitrilo
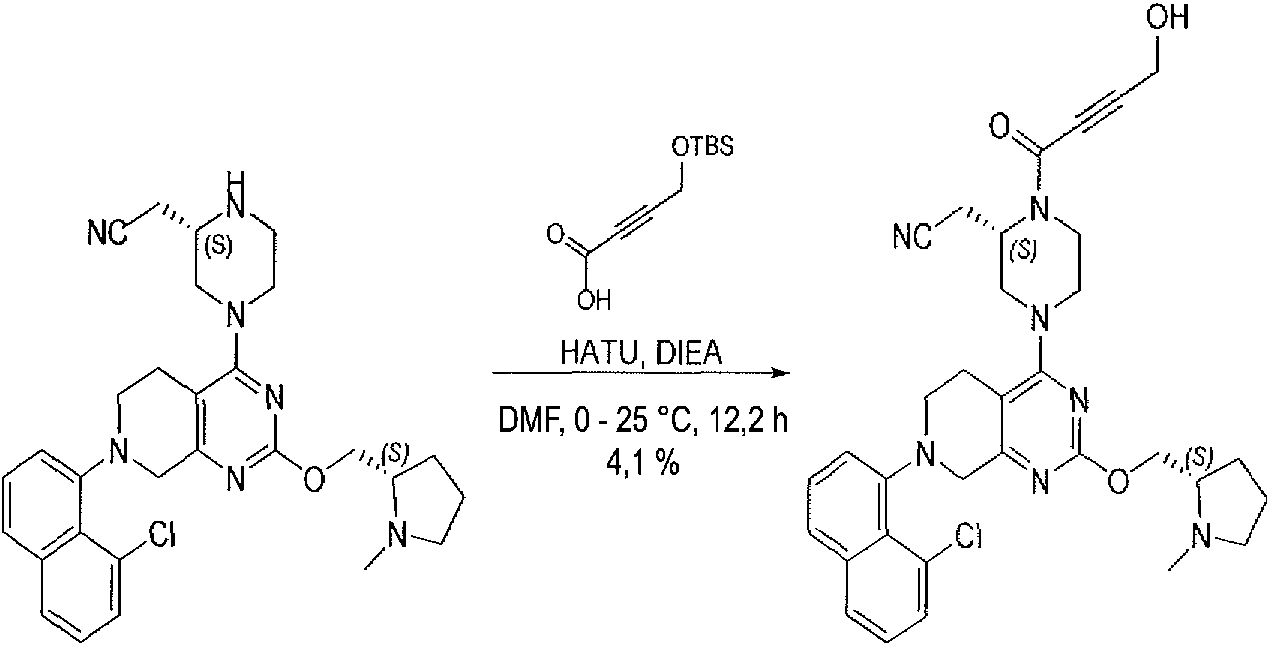
Etapa A: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]-1-(4-hidroxibut-2-inoil)piperazin-2-il]acetonitrilo. A una solución de ácido 4-[íerc-butil(dimetil)silil]oxibut-2-inoico (129 mg, 564 umol, 1,50 eq.) y DIEA (194 mg, 1,50 mmol, 262 ul, 4,00 eq.) en DMF (2,00 ml) se le añadió HATU (214 mg, 564 umol, 1,50 eq.) a 0 °C. Después de agitar a 0 °C durante 10 min, a la mezcla se le añadió 2-[(2S)-4-[7-(8-cloro-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5W-pirido[3,4-tí]pirim idin-4-il]piperazin-2-il]acetonitrilo (0,20 g, 376 umol, 1,00 eq.). Después de agitar a 25 °C durante 12 horas, la mezcla se diluyó con acetato de etilo (10,0 ml), se lavó con salmuera (3 x 10,0 ml), se secó sobre Na2SÜ4, se filtró y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (AF, 0,10 %)/acetonitrilo]. Las fracciones deseadas se recogieron, se neutralizaron con bicarbonato de sodio saturado (5,00 ml) a pH >7 y se extrajeron con acetato de etilo (3 x 20,0 ml). Las capas orgánicas se lavaron con salmuera (1 x 30,0 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío. El residuo se purificó por columna de HPLC prep.: Phenomenex Synergi C18150 * 25 * 10 um; fase móvil:
[agua (0,225 %, AF) - ACN]; % de B: 10 % - 40 %, 10 min. Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(4-hidroxibut-2-inoil)piperazin-2-il]acetonitrilo (EJEMμL0 483, 10,1 mg, 15,3 umol, 4,1 % de rendimiento, 99,6 % de pureza, AF) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 614.
1H RMN (400 MHz, ácido acético-d4) ó = 7,82 (d, J = 8,4 Hz, 1H), 7,69 (d, J = 8,4 Hz, 1H), 7,56 (d, J = 7,6 Hz, 1H), 7,49 (t, J = 7,6 Hz, 1H), 7,42 - 7,31 (m, 2H), 5,14 - 4,73 (m, 4H), 4,70 - 4,23 (m, 5H), 4,01 - 3,81 (m, 3H), 3,80 - 3,51 (m, 3H), 3,49 - 3,17 (m, 4H), 3,09 (m, 5H), 2,81 - 2,63 (m, 1H), 2,45 - 2,34 (m, 1H), 2,22 - 2,08 (m, 3H).
Ejemplo 484

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-c/]pirimidin-4-il]-1-[(E)-4,4-difluorobut-2-enoil]μlperazin-2-il]acetonitrilo

Etapa 1: 4,4-difluoro-3-hidroxi-butanoato. A una solución de 4,4-difluoro-3-oxo-butanoato de etilo (5,00 g, 30,1 mmol, 1,00 eq.) en tolueno (150 ml) se le añadió NaBH (1,20 g, 31,7 mmol, 1,05 eq.) a 0 °C. Después de agitar a 25 °C durante 4 horas, la mezcla se diluyó con agua (50,0 ml) a 0 °C y se extrajo con acetato de etilo (3 * 100 ml). Los extractos se lavaron con salmuera (1 * 100 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío para dar 4,4-difluoro-3-hidroxi-butanoato de etilo (4,30 g, bruto) en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, DMS0-d6) 5 = 6,05 - 5,87 (m, 1H), 5,86 - 5,72 (m, 1H), 4,11 - 4,01 (m, 2H), 2,57 (dd, J = 4,0, 15,6 Hz, 1H), 2,38 (dd, J = 9,2, 15,6 Hz, 1H), 1,21 - 1,17 (m, 3H).
Etapa 2: (E)-4,4-difluorobut-2-enoato de etilo. A una solución de 4,4-difluoro-3-hidroxi-butanoato de etilo (3,00 g, bruto) en diclorometano (20,0 ml) se le añadieron TEA (1,81 g, 17,8 mmol, 2,48 ml,) y MsCl (3,68 g, 32,1 mmol, 2,49 ml) a 0 °C. Después de agitar durante 4 horas, a la mezcla se le añadió TEA (3,61 g, 35,68 mmol, 4,97 ml, 2 eq.) 0 °C.
Después de calentar a 25 °C y agitar durante 12 horas, la mezcla se diluyó con agua (10,0 ml), se lavó con HCl (1 N, 1 * 20,0 ml) y salmuera (1 * 20,0 ml), se secó sobre Na2S04, se filtró y se concentró al vacío para dar (E)-4,4-difluorobut-2-enoato de etilo (1,40 g, 9,33 mmol, dos etapas 44 % de rendimiento) en forma de un aceite incoloro y se usó en la siguiente etapa con purificación adicional.
1H RMN (400 MHz, cloroformo-d) 5 = 6,81 (m, 1H), 6,29 (dt, J = 2,8, 1,6 Hz, 1H), 6,23 (dd, J = 4,0, 55,2 Hz, 1H), 4,25 (c, J = 7,2 Hz, 2H), 1,31 (t, J = 7,2 Hz, 3H).
Etapa 3: ácido (E)-4,4-difluorobut-2-enoico. Una mezcla de (E)-4,4-difluorobut-2-enoato de etilo (1,30 g, 8,66 mmol, 1,00 eq.) y Na0H (1,39 g, 34,6 mmol, 4,00 eq.) en THF (5,00 ml) y H20 (5,00 ml) se agitó a 25 °C durante 2 horas. La mezcla se acidificó con HCl (2 N, 20,0 ml) a pH = 1 ~ 3 y se extrajo con acetato de etilo (3 * 20,0 ml). Las capas orgánicas se secaron sobre Na2S04, se filtraron y se concentraron al vacío para dar ácido (E)-4,4-difluorobut-2-enoico (0,35 g, 2,87 mmol, 33 % de rendimiento) en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación adicional.1
1H RMN (400 MHz, cloroformo-d) 5 = 6,92 (m, 1H), 6,32 (dtd, J = 0,8, 2,8, 16,8 Hz, 1H), 6,27 (dtd, J = 1,2, 54,8 Hz, 4,0, 1H).
Etapa A: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cnpirimidin-4-il]-1-[(E)-4,4-difluorobut-2-enoil1piperazin-2-il]acetonitrilo. Una mezcla de ácido (E)-4,4-difluorobut-2-enoico (68,8 mg, 564 umol, 1,50 eq.), EDCI (108 mg, 564 umol, 1,50 eq.) y H0Bt (50,8 mg, 376 umol, 1,00 eq.) en Py (3 ml) se agitó a 0 °C durante 15 min y después a la mezcla se le añadió 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (intermedio 71,0,20 g, 376 umol, 1,00 eq.). Después de agitar a 0 °C durante 1 hora y a 25 °C durante 1 hora, la mezcla se diluyó con agua (10,0 ml) y se extrajo con
acetato de etilo (1 * 10,0 ml). Los extractos se lavaron con salmuera (1 * 10,0 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (AF, 0,10 %)/acetonitrilo]. Las fracciones deseadas se recogieron y la mezcla se neutralizó con bicarbonato de sodio saturado (5,00 ml) y se extrajo con acetato de etilo (3 * 20,0 ml). Las capas orgánicas se lavaron con salmuera (1 * 30,0 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 55 % - 85 %, 12 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[(2s)-4-[7-(8-cloro-1-naftil)-2-[[(2s)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(E)-4,4-difluorobut-2-enoil]piperazin-2-il]acetonitrilo (EJEMPL0484, 66,9 mg, 104 umol, 28 % de rendimiento, 98,7 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 636.
1H RMN (400 MHz, cloroformo-d) ó = 7,76 (d, J = 8,0 Hz, 1H), 7,62 (t, J = 7,2 Hz, 1H), 7,52 (d, J = 7,2 Hz, 1H), 7,45 (td, J = 7,6, 12,8 Hz, 1H), 7,37 - 7,31 (m, 1H), 7,27 - 7,18 (m, 1H), 6,88 - 6,71 (m, 2H), 6,45 - 6,12 (m, 1H), 5,17 - 4,59 (m, 1H), 4,55 - 4,41 (m, 1H), 4,40 - 4,33 (m, 1H), 4,21 - 3,69 (m, 5H), 3,64 - 3,32 (m, 2H), 3,31 - 2,97 (m, 5H), 2,96 -2,52 (m, 4H), 2,47 (d, J = 1,6 Hz, 3H), 2,33 - 2,23 (m, 1H), 2,11 - 2,00 (m, 1H), 1,90 - 1,77 (m, 3H).
Ejemplo 485

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[2-(metoximetil)prop-2-enoil]piperazin-2-il]acetonitrilo

Etapa 1: bromuro de 1 -(2-(etoxicarbonil)alil)-1,4-diazabiciclo[2.2.2]octan-1 -io. A 2-(bromometil)prop-2-enoato de etilo (15 g, 77,7 mmol, 1 eq.) en THF (5 ml) se le añadió DABC0 (10,5 g, 93,6 mmol, 10,3 ml, 1,20 eq.) y la mezcla resultante se agitó a 25 °C durante 2 horas. El precipitado se formó y se filtró en una atmósfera de nitrógeno. La torta
de filtro se recogió para dar bromuro de 1-(2-(etoxicarbonil)alil)-1,4-diazabiciclo[2.2.2]octan-1-io (15 g, 49,6 mmol, 64 % de rendimiento) en forma de un sólido de color amarillo claro que se usó directamente en la siguiente etapa sin purificación adicional.
Etapa 2: 2-(metoximetil)prop-2-enoato de etilo. Una mezcla de bromuro de 1-(2-(etoxicarbonil)alil)-1,4-diazabiciclo[2.2.2] octan-1-io (15 g, 49,6 mmol, 1 eq.) y TEA (10,5 g, 104 mmol, 14,5 ml, 2,09 eq.) en Me0H (150 ml) se agitó a 25 °C durante 16 horas. El disolvente se evaporó al vacío (25 °C). El residuo se disolvió en DCM (40 ml) y después se lavó con una solución acuosa al 5 % de ácido cítrico (1 x 40 ml) y una solución acuosa saturada de bicarbonato de sodio (1 x 40 ml). La capa orgánica se secó sobre sulfato de sodio, se filtró y se concentró al vacío a 25 °C para dar 2-(metoximetil)prop-2-enoato de etilo (5,3 g, 36,8 mmol, 74 % de rendimiento) en forma de un líquido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional.
Etapa 3: ácido 2-(metoximetil)prop-2-enoico. A una solución de 2-(metoximetil)prop-2-enoato de etilo (5,1 g, 35,4 mmol, 1 eq.) en THF (71 ml) se le añadió una solución de Na0H (5,66 g, 142 mmol, 4 eq.) en H20 (71 ml) a 5 °C. La mezcla se agitó a 25 °C durante 2 horas. Después de que se completara, el THF se evaporó al vacío. El pH de la mezcla se ajustó a 3 con HCl 1 M y se extrajo con EtOAc (3 x 70 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío para dar ácido 2-(metoximetil)prop-2-enoico (3,4 g, 26,4 mmol, 75 % de rendimiento, 90 % de pureza) en forma de un sólido de color amarillo claro que se usó directamente en la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, cloroformo-d) 5 = 6,45 (d, J = 1,2 Hz, 1H), 5,98 (c, J = 1,6 Hz, 1H), 4,16 (t, J = 1,2 Hz, 2H), 3,42 (s, 3H).
Etapa A: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il1-1-[2-(metoximetil)prop-2-enoil1piperazin-2-il]acetonitrilo. A una solución de ácido 2-(metoximetil)prop-2-enoico (175 mg, 1,50 mmol, 2 eq.) y DIEA (389 mg, 3,01 mmol, 524 ul, 4 eq.) en DCM (8 ml) se le añadió Ha Tu (429 mg, 1,13 mmol, 1,5 eq.) a -40 °C. Después de agitar a -40 °C durante 10 minutos, a la mezcla se le añadió 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo
(Intermedio 71,400 mg, 752 umol, 1 eq.). La mezcla se agitó a -40 °C durante 20 minutos, 0 °C durante 0,5 horas y a 25 °C durante 1 hora. Después de que se completara, a la mezcla se le añadió agua (10 ml) y se extrajo con DCM (2 x 20 ml). Las capas orgánicas combinadas se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía (Ah03, EtOAc/Me0H 100/1 a 40/1) y se purificó por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 50 % - 80 %, 12 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[2-(metoximetil)prop-2-enoil]piperazin-2-il]acetonitrilo (EJEMPL0485, 73,0 mg, 116 umol, 15 % de rendimiento, 98,9 % de pureza) en forma de un sólido de color blanquecino. LCMS [ESI, M+1]: 630.
Condición de SFC: Columna: Chiralpak AS-3100 x 4 ,6 mm D.I., 3 um Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 % Caudal: 3 ml/min Longitud de onda: 220 nm.
1H RMN (400 MHz, cloroformo-d) 5 = 7,76 (d a, J = 8 Hz, 1H), 7,62 (t, J = 7,6 Hz, 1H), 7,52 (d, J = 7,6 Hz, 1H), 7,45 (td, J = 7,6, 13,2 Hz, 1H), 7,37 - 7,30 (m, 1H), 7,26 - 7,17 (m, 1H), 5,51 (s, 1H), 5,32 (s a, 1H), 5,19 - 4,58 (m, 1H), 4,48 - 4,34 (m, 2H), 4,23 - 4,07 (m, 4H), 4,06 - 3,78 (m, 3H), 3,64 - 3,55 (m, 1H), 3,39 (s, 3H), 3,36 - 2,72 (m, 8H), 2,71 -2,53 (m, 2H), 2,47 (s, 3H), 2,35 - 2,22 (m, 1H), 2,14 - 1,98 (m, 1H), 1,90 - 1,73 (m, 3H).
Ejemplo 486

2 -((S )-4 -(7 -(2 ,3 -d im e tilfe n il)-2 -(((S )-1 -m e tilp irro lid in -2 - il)m e to x i)-5 ,6 ,7 ,8 -te tra h id ro p ir id o [3 ,4 -d ]p ir im id in -4 - il) -1 -((E )-4 ,4 ,4 -tr iflu o ro b u t-2 -e n o il)p ip e ra z in -2 -il)a c e to n itr ilo

Etapa A: 4-((S)-4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.8-d¡h¡drop¡r¡dof3,4-d1p¡r¡m¡d¡n-7(6H)-carbox¡lato de 2-terc-butilo. En un matraz de fondo redondo. una soluc¡ón de (S)-4-(4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-cloro-5,8-d¡h¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de tercbut¡lo (5 g. 9,487 mmol) en d¡oxano (94,87 ml. 9,487 mmol) se roc¡ó con argón. se añad¡eron secuenc¡almente (S)-(1-met¡lp¡rrol¡d¡n-2-¡l)metanol (3,278 g. 28,46 mmol). Cs2C03 (9,273 g. 28,46 mmol) y metanosulfonato de (2-d¡c¡clohex¡lfosf¡no-2',6'-d¡-¡-propox¡-1,1'-b¡fen¡l)(2'-met¡lam¡no-1,1'-b¡fen¡l-2-¡l)palad¡o (II) (0,8078 g. 0,9487 mmol) en una atmósfera de argón y se rodaron durante 5 m¡nutos más. La mezcla de reacc¡ón se tapó y se calentó a 100 °C durante una noche. La reacc¡ón se f¡ltró a través de papel GHF y se concentró al vacío. El concentrado se pur¡f¡có en el Comb¡ Flash (0-12 % de Me0H en DCM con 2 % de NH40H) para proporc¡onar 4-((S)-4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,8-d¡h¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo (5,425 g. 8,508 mmol. 89,68 % de rend¡m¡ento). ESI+APCI MS m/z 606,4 [M+H]+.
Etapa B: (S)-2-(c¡anomet¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1- carbox¡lato de benc¡lo: se d¡solv¡ó 4-((S)-4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2- ¡l)metox¡)-5.8-d¡h¡drop¡r¡do[3.4-d]p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo (5.4 g. 8,915 mmol) en d¡clorometano (89,15 ml. 8,915 mmol) y se trató con ác¡do clorhídr¡co (soluc¡ón 4.0 M en 1,4-d¡oxano) (11,14 ml. 44,57 mmol). La reacc¡ón se ag¡tó a temperatura amb¡ente durante 1 hora. La reacc¡ón se d¡luyó con más DCM y Na0H 1 M y las capas se separaron. Luego. los extractos orgán¡cos se lavaron con salmuera. se secaron sobre Na2S04 y se concentraron al vacío para dar (S)-2-(c¡anomet¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (4,364 g. 8,631 mmol. 96,82 % de rend¡m¡ento). ESI+APCI MS m/z 506,3 [M+H]+.
Etapa C: 2-((S)-4-(7-(2.3-d¡met¡lfen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo: En un matraz de fondo redondo. una soluc¡ón de (S)-2-(c¡anomet¡l)-4-(2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (4,364 g. 8,631 mmol) en d¡oxano (43.15 ml. 8,631 mmol) se roc¡ó con argón durante 5 m¡nutos. Se añad¡eron secuenc¡almente 1-bromo-2,3-d¡met¡lbenceno (5,851 ml. 43,15 mmol). Cs2C03 (14,06 g. 43,15 mmol) y metanosulfonato de (2-d¡c¡clohex¡lfosf¡no-2',6'-d¡-¡-propox¡-1,1'-b¡fen¡l)(2'-met¡lam¡no-1,1'-b¡fen¡l-2-¡l)palad¡o (II) (0,7348 g. 0,8631 mmol) en una atmósfera de argón y se rodaron durante 5 m¡n más. La mezcla de reacc¡ón se tapó y se calentó a 100 °C durante una noche. La reacc¡ón se enfr¡ó a temperatura amb¡ente. Se añad¡ó acetato de et¡lo y la reacc¡ón se f¡ltró a través de papel GHF y se concentró al vacío. El concentrado se pur¡f¡có dos veces por cromatografía de fase normal sobre el Comb¡Flash usando 0-15 % de Me0H en DCM con 2 % de NH40H como eluyente. Las fracc¡ones que contenían el producto deseado se recog¡eron y se concentraron al vacío para dar 2-((S)-4-(7-(2,3-d¡met¡lfen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (0,411 g. 0,8641 mmol. 100,1 % de rend¡m¡ento). ESI+APCI MS m/z 476,3 [M+H]+.
Etapa D: 2-((S)-4-(7-(2.3-d¡met¡lfen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-1-((E)-4.4.4-tr¡fluorobut-2-eno¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo: A una soluc¡ón de 2-((S)-4-(7-(2,3-d¡met¡lfen¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (200 mg. 0,420 mmol). ác¡do 4,4,4-tr¡fluorocrotón¡co (118 mg. 0,841 mmol) y DiEA (367 μl. 2,10 mmol) en DCM (4205 μl. 0,420 mmol) se le añad¡ó HATU (240 mg.0,631 mmol) y la mezcla resultante se ag¡tó a temperatura amb¡ente durante 5 horas. La mezcla de reacc¡ón se lavó con salmuera y la capa acuosa se extrajo con DCM (2 x). Las capas orgán¡cas comb¡nadas se secaron sobre Na2S04, se concentraron. se d¡luyeron en 60:40 de ACN:H20 y se pur¡f¡caron en el G¡lson (HPLC prep.
inversa), eluyendo con 5-->95 % de ACN/0,1 % de TFA en agua/0,1 % de TFA. Las fracciones que contenían el producto se combinaron y se convirtieron en la base libre con bicarbonato saturado y los compuestos orgánicos se extrajeron con DCM. Los extractos orgánicos se secaron sobre Na2S04 y se concentraron al vacío para dar el compuesto del título 2-((S)-4-(7-(2,3-dimetilfenil)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4,4,4-trifluorobut-2-enoil)piperazin-2-il)acetonitrilo (EJEMPL0486, 28 mg, 0,0468 mmol, 11,1 % de rendimiento). ESI+AμCi MS m/z 598,3 [M+H]+.
Ejemplo 487

2-((S)-4-(7-(2,3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-(trifluorometil)acriloil)piperazin-2-il)acetonitrilo
2-((S)-4-(7-(2,3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-1-(2-(trifluorometil)acriloil)piperazin-2-il)acetonitrilo: El compuesto del título se preparó siguiendo el Ejemplo 486 sustituyendo ácido 2-(trifluorometil)propenoico por ácido 4,4,4-trifluorocrotónico en la Etapa D. ESI+APCI MS m/z 598,3 [M+H1+.
Ejemplo 488

2-((S)-4-(7-(2,3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-(hidroximeti¡)acriloil)piperazin-2-il)acetonitrilo
2-((S)-4-(7-(2,3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-1-(2-(hidroximetil)acriloil)piperazin-2-il)acetonitrilo: El compuesto del título se preparó siguiendo el Ejemplo 486 sustituyendo ácido 2-(hidroximetil)prop-2-enoico por ácido 4,4,4-trifluorocrotónico en la Etapa D. ESI+ApCI MS m/z 560,3 [M+H]+.
Ejemplo 489

2 -((S )-4 -(7 -(2 ,3 -d im e tilfe n il)-2 -(((S )-1 -m e tilp irro lid in -2 - il)m e to x i)-5 ,6 ,7 ,8 -te tra h id ro p ir id o [3 ,4 -d ]p ir im id in -4 - il) -1 -((E )-4 -m e to x ib u t-2 -e n o il)p ip e ra z in -2 - il)a c e to n itrilo

Etapa A: 2-((S)-4-(7-(2.3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)-1-((E)-4-metoxibut-2-enoil)piperazin-2-il)acetonitrilo: A una solución de 2-((S)-4-(7-(2.3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (50 mg. 0.1051 mmol) en DCM (1 ml) se le añadieron ácido (E)-4-metoxibut-2-enoico (24.41 mg. 0.2102 mmol). N-etil-N-isopropilpropan-2-amina (0.03662 ml.0.2102 mmol) y 0-(7-azabenzotriazol-1-il)-N,N,N'.N'-tetrametiluronio PF6 (59.96 mg.0.1577 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 4 horas. Luego. se añadió agua (1 ml) y la mezcla de reacción se agitó durante 5 minutos. La mezcla se dividió entre EtOAc (10 ml) y NaHC030.5 M (5 ml) y las capas se separaron. Los extractos orgánicos se lavaron con salmuera (3 ml). se secaron sobre Na2S04 y se evaporaron al vacío. El residuo se cromatografió sobre gel de sílice usando 3 % de Me0H 0.3 % de NH40H a 4 % de Me0H 0.4 % de NH40H como eluyente para dar el compuesto del título (EJEMPL0489. 26 mg. 43 %). ESI+APCI MS m/z 574.4 [M+H]+.
Ejemplo 490

2-((S)-4-(7-(8-cloronaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)-l-((E)-4.4.4-trifluorobut-2-enoil)piperazin-2-il)acetonitrilo

Etapa A: (S)-4-(7-(8-cloronaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo: A una solución de (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (0.6 g. 1.2 mmol) en dioxanos (10 ml) se le añadió 1-bromo-8-cloronaftaleno (0.37 g. 1.5 mmol) y la reacción se desgasificó con Ar durante 15 minutos seguido de la adición de Cs2C03 (1.2 g. 3.6 mmol). Pd2(dba)3 (0.22 g. 0.24 mmol) y 2-Diciclohexilfosfino-2'.6'-di-i-propoxi-1.1 '-bifenilo (0.11 g. 0.24 mmol) y la reacción se calentó a 100 °C durante una noche. Luego. la reacción se filtró a través de papel 0FF y el filtrado se concentró al vacío. Luego, el residuo se cromatografió usando 1 —^10 % de (Me0H 2 % de NH40H)/DCM para dar (S)-4-(7-(8-cloronaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (0.20 g. 0.30 mmol. 25 % de rendimiento). ESI+APCI MS m/z 666.2 [M+H]+.
Etapa B: 2-((S)-4-(7-(8-cloronaftalen-1-in-2-(((S)-1-metilpirrolidin-2-inmetoxi)-5.6.7.8-tetrahidropirido[3,4-dlpirimidin-4-il)piperazin-2-il)acetonitrilo: Una solución de (S)-4-(7-(8-cloronaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (0,17 g, 0,255 mmol) en THF (30 ml) se purgó con N2 seguido de la adición de Pd/C (0,0272 g, 0,255 mmol). La reacción se evacuó mediante vacío y se cargó de nuevo 3 veces con H2 y la mezcla se agitó durante 3 días a temperatura ambiente. La reacción se purgó de nuevo con N2 seguido de filtración a través de celite y el filtrado se concentró al vacío. El material se usó bruto en la siguiente reacción. ESI+APCI MS m/z 532,2 [M+H]+.
Etapa C: 2-((S)-4-(7-(8-cloronaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)-1-((E)-4,4,4-trifluorobut-2-enoil)piperazin-2-il)acetonitrilo: A una solución de 2-((S)-4-(7-(8-cloronaftalen-1 -il)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (0,055 g, 0,10 mmol) en DCM (20 ml) se le añadieron N-etil-N-isopropilpropan-2-amina (0,040 g, 0,31 mmol), ácido (E)-4,4,4-trifluorobut-2-enoico (0,017 g, 0,12 mmol) y anhídrido cíclico del ácido 1-propanofosfónico (0,033 g, 0,10 mmol) y la reacción se agitó a temperatura ambiente durante 30 minutos. Luego, a la reacción se le añadieron más cantidad de TEP3 y ácido (0,24 mmol) durante 1 hora más. Luego, los extractos orgánicos se lavaron con Na0H 1 N y salmuera, se secaron sobre MgS04 y se concentraron al vacío. El material se cromatografió 2 veces en sílice de fase normal (1 ^ 10 % de (Me0H 2 % de NH40H)/DCM) seguido de purificación por HPLC prep. inversa Gilson usando 5 --> 95 % de ACN/agua con 0,1 % de TFA como aditivo. Las fracciones puras se reunieron en agua básica y se extrajeron en EtOAc. Los extractos orgánicos se lavaron con salmuera, se secaron sobre MgS04 y se concentraron al vacío para dar el compuesto del título 2-((S)-4-(7-(8-cloronaftalen-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)-1-((E)-4,4,4-tr¡fluorobut-2-eno¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (EJEMPL0490, 0,0094 g, 0,014 mmol, 14 % de rendimiento). ESI+APCl MS m/z 654,2 [M+H]+.
Ejemplo 491

2-((S)-4-(7-(8-met¡lnaftalen-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)-1-((E)-4,4,4-tr¡fluorobut-2-eno¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo

Etapa A: 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)-1-(E)-4.4.4-trifluorobut-2-enoil)piperazin-2-il)acetonitrilo: A 0 °C, a un MFR de 25 ml que contenía N,N-dimetilformamida (2932 gl, 0,293 mmol) se le añadieron 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1 -metilpirrolidin-2 -¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (150 mg, 0,293 mmol) y base de Hunig (102 gl, 0,586 mmol). La mezcla de reacción se agitó vigorosamente mientras se añadía en una porción ácido (E)-4,4,4-trifluorobut-2-enoico (49,3 mg, 0,352 mmol). Después, a la mezcla en agitación se le añadió lentamente anhídrido cíclico del ácido 1-propanofosfónico (262 gl, 0,440 mmol). La reacción se agitó durante 2 horas a 0 °C. La reacción se trató con agua básica y la capa acuosa se extrajo con EtOAc (3 x). Los extractos orgánicos combinados se concentraron al vacío y se suspendieron de nuevo en una mezcla 60:40 de ACN: H20 y se purificaron en el Gilson (HPLC prep. inversa), eluyendo con 5-->95 % de ACN/0,1 % de TFA en agua/0,1 % de TFA. Las fracciones que contenían el producto se combinaron y se repartieron entre bicarbonato saturado y DCM. La capa acuosa se extrajo dos veces más con DCM. Las capas orgánicas se combinaron, se secaron sobre Na2S04 y se concentraron al vacío para dar el compuesto del título 2-((S)-4-(7-(8-met¡lnaftalen-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8
tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4,4,4-trifluorobut-2-enoil)piperazin-2-il)acetonitrilo (EJEMPL0491,74,5 mg, 0,118 mmol, 40,1 % de rendimiento). ESI+APCI MS m/z 634,3 [M+H]+.
Ejemplo 492

2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-(trifluorometil)acriloil)piperazin-2 -il)acetonitrilo
2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-(trifluorometil)acriloil)piperazin-2-il)acetonitrilo: El compuesto del título se preparó siguiendo el Ejemplo 491 sustituyendo 2-ácido (trifluorometil)acrílico por ácido (E)-4,4,4-trifluorobut-2-enoico en la Etapa A. ESI+a Pc I MS m/z 634,2 [M+H]+.
Ejemplo 493

2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-3-(piridin-2-il)acriloil)piperazin-2-il)acetonitrilo
2- ((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-3-(piridin-2-il)acriloil)piperazin-2-il)acetonitrilo: El compuesto del título se preparó siguiendo el Ejemplo 491 sustituyendo ácido 3-(piridin-2-il)acrílico por ácido (E)-4,4,4-trifluorobut-2-enoico en la Etapa A. ESI+APCI MS m/z 643,3 [M+H]+.
Ejemplo 494

2 -((S )-1 -((E )-3 -(1 -m e til-1 H -p ira z o l-4 - il)a c r ilo il) -4 -(7 -(8 -m e tiln a fta le n -1 - il) -2 -(((S )-1 -m e tilp irro lid in -2 - il)m e to x i)-5 ,6 ,7 ,8 -te tra h id ro p ir id o [3 ,4 -d ]p ir im id in -4 - il)p ip e ra z in -2 - il)a c e to n itr ilo
2-((S)-1-((E)-3-(1-metil-1H-pirazol-4-ir)acriloil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpynrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: El compuesto del título se preparó siguiendo el Ejemplo 491 sustituyendo ácido (2E)-3-(1-metil-1H-pirazol-4-il)acrílico por ácido (E)-4,4.4-trifluorobut-2-enoico en la Etapa A. ESI+APCI MS m/z 646,4 [M+H]+.
Ejemplo 495

2-((S)-1-((E)-4.4-difluorobut-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-((E)-4.4-difluorobut-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: El compuesto del título se preparó siguiendo el Ejemplo 491 sustituyendo ácido 4,4-difluorobut-2-enoico por ácido (E)-4,4,4-trifluorobut-2-enoico en la Etapa A. ESl+ApCI MS m/z 616,3 [M+H]+.
Ejemplo 496

2-((S)-4-(2-(((S)-1-metilpiperidin-2-il)metoxi)-7-(naftalen-1-il)-5.6.7.8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-3-(piridin-3-il)acriloil)piperazin-2-il)acetonitrilo

Etapa A: A una solución de 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (0,12 g.0,23 mmol) en DCM (10 ml) se le añadieron N-etil-N-isopropilpropan-2-amina (0,13 ml. 0,70 mmol). ácido (E)-3-(piridin-3-il)acrílico (0,070 g. 0,47 mmol) y 2,4,6-trióxido de 2.4.6-tripropil-1,3.5.2.4.6-trioxatrifosfinano (0,30 g. 0,47 mmol) y la reacción se agitó a temperatura ambiente durante 3 horas. Luego. la reacción se vertió en Na0H 1 N y las capas se separaron. Luego. los extractos orgánicos se lavaron con salmuera. se secaron sobre MgS04 y se concentraron al vacío. Luego. el material se purificó por HPLC prep. inversa Gilson eluyendo con 5^95 de ACN/agua con 0,1 % de TFA como modificador. Las fracciones que contenían el producto se reunieron en Na0H 1 N y la capa acuosa se extrajo con EtOAc (3 x). Los extractos orgánicos combinados se lavaron con salmuera. se secaron sobre MgS04 y se concentraron al vacío para dar el compuesto del
título 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-3-(piridin-3-il)acriloil)piperazin-2-il)acetonitrilo (EJEMPL0 496, 0,040 g, 0,062 mmol, 27 % de rendimiento). ESI+APCI MS m/z 643,3 [M+H]+.
Ejemplo 497

2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7, 8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(3-oxociclopent-1-eno-1-carbonil)piperazin-2-il)acetonitrilo

Etapa A: 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(3-oxociclopent-1-eno-1-carbonil)piperazin-2-il)acetonitrilo: Se diluyeron 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (30 mg, 0,05863 mmol) y ácido 3-oxociclopent-1-enocarboxílico (9,612 mg, 0,07622 mmol) con DMF (400 ul) seguido de la adición de DiEa (20,48 μl, 0,1173 mmol) y anhídrido cíclico del ácido 1-propanofosfónico (52,35 μl, 0,08795 mmol). Después de agitar durante 12 horas, la reacción se diluyó con acetato de etilo y bicarbonato de sodio saturado. Las capas se separaron y el acetato de etilo se secó sobre MgS04, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10 % de metanol/DCM (1 % de NH40H) para proporcionar el compuesto del título 2-((S)-4-(7-(8-metilnaftalen-1 -il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(3-oxociclopent-1-eno-1-carbonil)piperazin-2-il)acetonitrilo (EJEMPL0 497, 2,3 mg, 0,003711 mmol, 6,329 % de rendimiento). ESI+APCI MS m/z 620,3 [M+H]+.
Ejemplo 498
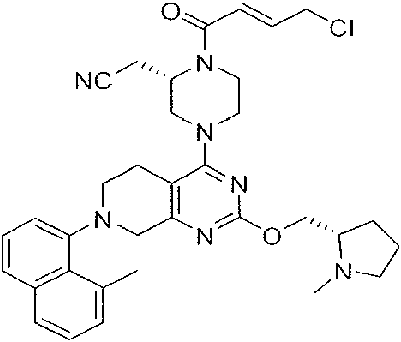
2-((S)-1-((E)-4-clorobut-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 2-((S)-1-((E)-4-clorobut-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-i0metoxi)-5,6.7.8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo: Se diluyeron 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (30 mg, 0,05863 mmol) y ácido gamma-clorocrotónico (10,60 mg, 0,08795 mmol) con DMF (400 ul) seguido de la adición de DIEA (20,48 gl, 0,1173 mmol) y anhídrido cíclico del ácido 1-propanofosfónico (55,84 gl, 0,09381 mmol). Después de agitar durante 12 horas, la reacción se diluyó con acetato de etilo y bicarbonato de sodio saturado. Las capas se separaron y el acetato de etilo se secó sobre MgS04, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10 % de metanol/DCM (1 % de NH40H) para proporcionar el compuesto del título 2-((S)-1-((E)-4-clorobut-2-enoil)-4-(7-(8-metilnaftalen-1 -il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0498, 4,5 mg, 0,007327 mmol, 12,50 % de rendimiento). ESI+APCI MS m/z 614,3 [M+H]+.
Ejemplo 499

(E)-4-((S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-N,N-dimetil-4-oxobut-2-enamida

Etapa A: (E)-4-(Dimetilamino)-4-oxobut-2-enoato de etilo: Se diluyó éster monoetílico del ácido fumárico (442 mg, 3,07 mmol) con DCM (5 ml) seguido de la adición de cloruro de oxalilo (1533 gl, 3,07 mmol) y 1 gota de DMF. Después de agitar durante 15 minutos, se añadió dimetilamina (4600 gl, 9,20 mmol) y la reacción se agitó durante 3 horas. La reacción se diluyó con acetato de etilo y agua. Las capas se separaron y el acetato de etilo se secó sobre MgS04, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10-70 % de acetato de etilo/hexanos para proporcionar (E)-4-(dimetilamino)-4-oxobut-2-enoato de etilo (382 mg, 2,23 mmol, 72,8 % de rendimiento). ESI+APCI MS m/z 172,1 [M+H]+.
Etapa B: ácido (E)-4-(dimetilamino)-4-oxobut-2-enoico: Se diluyó (E)-4-(dimetilamino)-4-oxobut-2-enoato de etilo (382 mg, 2,23 mmol) con metanol (8 ml) seguido de la adición de Na0H (4463 μl, 8,93 mmol). Después de agitar durante 4 horas, la reacción se diluyó con HCl 2 N (4,5 ml) y se extrajo con acetato de etilo. El acetato de etilo se secó sobre MgS04, se filtró y se concentró para proporcionar ácido (E)-4-(dimetilamino)-4-oxobut-2-enoico.
Etapa C: (E)-4-((S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-il)-N,N-dimetil-4-oxobut-2-enamida: Se diluyó 2-((S)-4-(7-(8-metilnaftalen-1 -il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (30 mg, 0,05863 mmol) con DMF seguido de la adición de ácido (E)-4-(dimetilamino)-4-oxobut-2-enoico (10,07 mg, 0,07036 mmol), DIEA (20,48 μl, 0,1173 mmol) y anhídrido cíclico del ácido 1-propanofosfónico (52,35 μl, 0,08795 mmol). Después de agitar durante 12 horas, la reacción se diluyó con acetato de etilo y bicarbonato de sodio saturado. Las capas se separaron y el acetato de etilo se secó sobre MgS04, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10 % de metanol/DCM (1 % de NH40H) para proporcionar el compuesto del título (E)-4-((S)-2-(cianometil)-4-(7-(8-metilnaftalen-1 -il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-N,N-dimetil-4-oxobut-2-enamida (EJEMPL0 499, 3,7 mg, 0,005810 mmol, 9,910 % de rendimiento). ESI+APCI MS m/z 637,3 [M+H]+.
Ejemplo 500

(E)-4-((S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-4-oxobut-2-enamida

Etapa A: (E)-3-Cianoacrilato de etilo: Se diluyó cis-(beta-ciano)acrilato de etilo (1,0 g, 7,99 mmol) con ACN (30 ml), se puso en una atmósfera de nitrógeno y se calentó a reflujo durante 3 días. La reacción se dejó enfriar y después se concentró. El material se purificó sobre gel de sílice eluyendo con hexanos para proporcionar (E)-3-cianoacrilato de etilo (300 mg, 2,40 mmol, 30,0 % de rendimiento).
Etapa B: ácido (E)-4-amino-4-oxobut-2-enoico: Se diluyó (E)-3-cianoacrilato de etilo (300 mg, 2,40 mmol) con HCl (3996 μl, 24,0 mmol). La reacción se puso en una atmósfera de nitrógeno y se calentó a 100 °C durante 4 horas. La reacción se dejó enfriar y después se concentró para proporcionar ácido (E)-4-amino-4-oxobut-2-enoico (214 mg, 1,86 mmol, 77,6 % de rendimiento).
Etapa C: (E)-4-((S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-il)-4-oxobut-2-enamida: Se diluyeron 2-((S)-4-(7-(8-metilnaftalen-1 -il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (20 mg,
0,039 mmol), HATU (30 mg, 0,078 mmol) y ácido (E)-4-amino-4-oxobut-2-enoico (9,0 mg, 0,078 mmol) con DMF (400 ul) seguido de la adición de DIEA (14 gl, 0,078 mmol). Después de agitar durante 12 horas, la reacción se diluyó con acetato de etilo y bicarbonato de sodio saturado. Las capas se separaron y el acetato de etilo se secó sobre MgS04, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10 % de metanol/DCM (1 % de NH40H) para proporcionar el compuesto del título (E)-4-((S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-4-oxobut-2-enamida (Ej e Mp Lo 500, 2,9 mg, 0,0048 mmol, 12 % de rendimiento). ESl+APCI MS m/z 609,3 [M+H]+.
Ejemplo 501

(E)-4-((S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-4-oxobut-2-enonitrilo

Etapa A: (E)-3-Cianoacrilato de etilo: Se diluyó cis-(beta-ciano)acrilato de etilo (1,0 g, 7,99 mmol) con ACN (30 ml), se puso en una atmósfera de nitrógeno y se calentó a reflujo durante 3 días. La reacción se dejó enfriar y después se concentró. El material se purificó sobre gel de sílice eluyendo con hexanos para proporcionar (E)-3-cianoacrilato de etilo (300 mg, 2,40 mmol, 30,0 % de rendimiento).
Etapa B: ácido (E)-3-cianoacrílico: Se diluyó (E)-3-cianoacrilato de etilo (10 mg, 0,080 mmol) con HCl (133 gl, 0,80 mmol), se puso en una atmósfera de nitrógeno y se calentó a 100 °C. Después de agitar durante 15 minutos, la reacción se concentró para proporcionar ácido (E)-3-cianoacrílico (7,5 mg, 0,077 mmol, 97 % de rendimiento).
Etapa C: (E)-4-((S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-il)-4-oxobut-2-enonitrilo: Se diluyeron 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (20 mg, 0,03909 mmol) y ácido (E)-3-cianoacrílico (5,312 mg, 0,05472 mmol) con Dm F (400 ul) seguido de la adición de DIEA (13,65 gl, 0,07817 mmol) y anhídrido cíclico del ácido 1 -propanofosfónico (34,90 gl, 0,05863 mmol). Después de agitar durante 12 horas, la reacción se diluyó con acetato de etilo y bicarbonato de sodio saturado. Las capas se separaron y el acetato de etilo se secó sobre MgS04, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10 % de metanol/DCM (1 % de NH40H) para proporcionar el compuesto del título (E)-4-((S)-2-(cianometil)-4-(7-(8-metilnaftalen-1 -il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -il)-4-oxobut-2-enonitrilo (EJEMPL0 501, 3,5 mg, 0,005925 mmol, 15,16 % de rendimiento). ESI+a PCi MS m/z 591,3 [M+H]+.
Ejemplo 502

(Z)-4-((S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-4-oxobut-2-enonitrilo

Etapa A: ácido (Z)-3-cianoacrílico: Se diluyó cis-(beta-ciano)acrilato de etilo (50 mg, 0,40 mmol) con HCl (200 gl, 1,2 mmol), se puso en una atmósfera de nitrógeno y se calentó a 100 °C. Después de agitar durante 1 minuto la reacción se enfrió y se concentró para proporcionar ácido (Z)-3-cianoacrílico (35 mg, 0,36 mmol, 90 % de rendimiento).
Etapa B: (Z)-4-((S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-il)-4-oxobut-2-enonitrilo: Se diluyeron 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (20 mg, 0,03909 mmol) y ácido (Z)-3-cianoacrílico (6,071 mg, 0,06254 mmol) con DMF (400 ul) seguido de la adición de DIEA (13,65 gl, 0,07817 mmol) y anhídrido cíclico del ácido 1 -propanofosfónico (39,56 gl, 0,06645 mmol). Después de agitar durante 12 horas, la reacción se diluyó con acetato de etilo y bicarbonato de sodio saturado. Las capas se separaron y el acetato de etilo se secó sobre MgS04, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10 % de metanol/DCM (1 % de NH40H) para proporcionar el compuesto del título (Z)-4-((S)-2-(cianometil)-4-(7-(8-metilnaftalen-1 -il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -il)-4-oxobut-2-enonitrilo (EJEMPL0 502, 2,1 mg, 0,003555 mmol, 9,095 % de rendimiento). ESI+a PCi MS m/z 591,3 [M+H]+.
Ejemplo 503

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(£)-4-(dimetilamino)-2-fluoro-but-2-enoil]piperazin-2-il]acetonitrilo
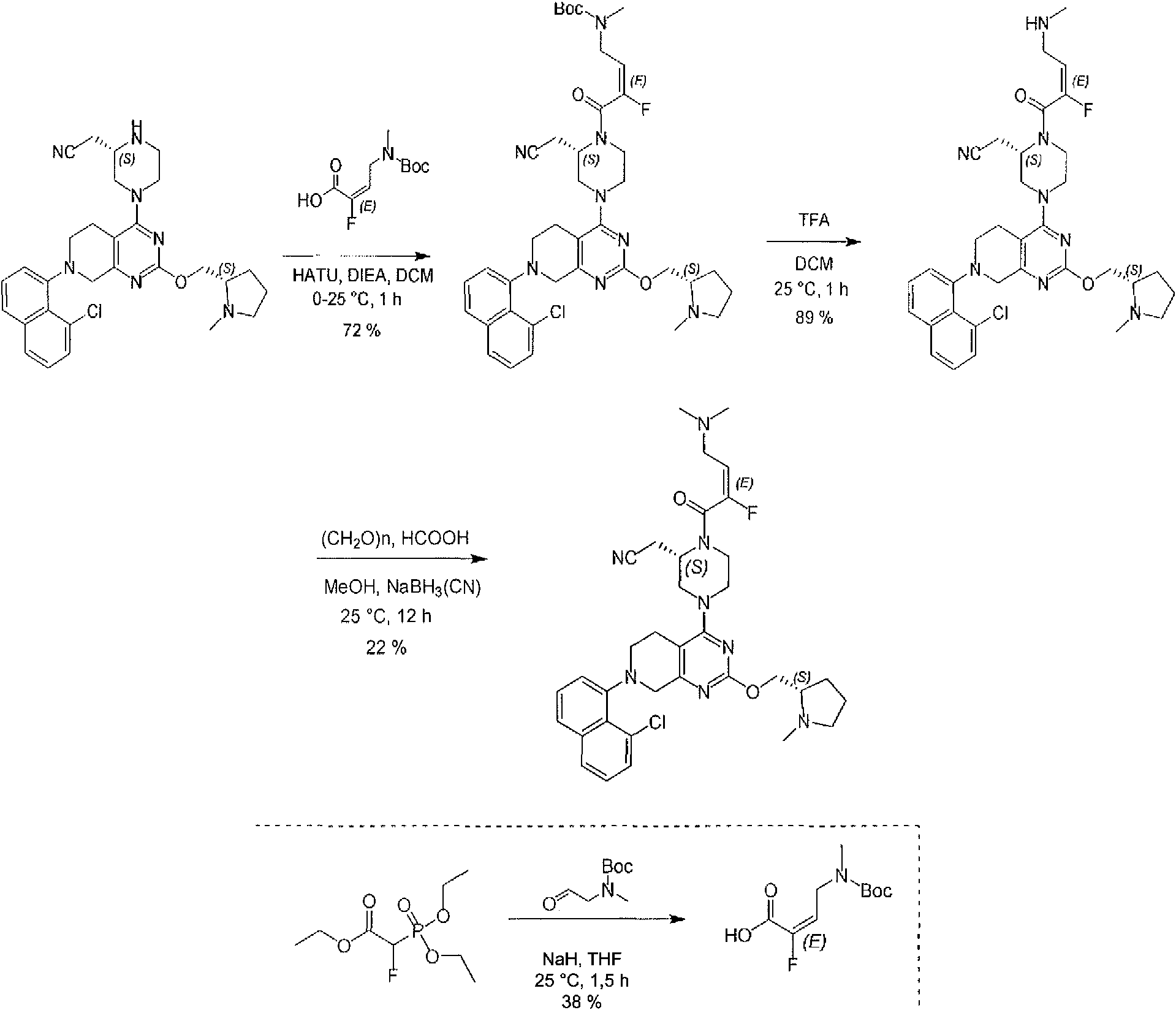
Etapa 1: ácido (E)-4-[terc-butoxicarbonil(metil)amino1-2-fluoro-but-2-enoico. A la solución de 2-dietoxifosforil-2-fluoroacetato de etilo (2 g, 8,26 mmol, 1,68 ml, 1 eq.) y W-metil-W-(2-oxoetil)carbamato de ferc-butilo (2,15 g, 12,4 mmol, 1,5 eq.) en THF (40 ml) se le añadió NaH (661 mg, 16,5 mmol, 60 % de pureza, 2 eq.). La mezcla se agitó a 25 °C durante 1,5 horas. Después de que se completara, la mezcla de reacción se inactivó con NH4Cl saturado (8 ml) y se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi Max-RP 250 * 50 mm * 10 um;fase móvil: [agua (0,225 % de AF)-ACN];% de B: 20 % de ACN-50 % de ACN, 30 min; 50 %min) para dar ácido (E)-4-[tercbutoxicarbonil(metil)amino]-2-fluoro-but-2-enoico (820 mg, 3,16 mmol, 38,3 % de rendimiento, 90 % de pureza) en forma de un aceite de color pardo.
Etapa A: M-[(E)-4-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1-il1-3-fluoro-4-oxo-but-2-enil1-M-metil-carbamato de ferc-butilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo (Intermedio 71, 300 mg, 564 umol, 1 eq.), ácido (E)-4-[terc-butoxicarbonil(metil)amino1-2-fluoro-but-2-enoico (600 mg, 2,57 mmol, 4,56 eq.) y DIEA (729 mg, 5,64 mmol, 982 ul, 10 eq.) en DCM (10 ml) se le añadió HATU (643 mg, 1,69 mmol, 3 eq.) a 0 °C. La mezcla de reacción se agitó a 25 °C durante 1 hora. Después de que se completara, la mezcla de reacción se inactivó con agua (3 ml) y se extrajo con DCM (3 * 8 ml). Las capas orgánicas combinadas se secaron sobre Na2SÜ4 y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida inversa (condición de AF: 60 % de MeCN en agua (0,1 % de AF)) para dar W-[(E)-4-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1-il1-3-fluoro-4-oxo-but-2-enil1-W-metil-carbamato de ferc-butilo (330 mg, 404 umol, 72 % de rendimiento, 91,5 % de pureza) en forma de un sólido de color pardo. LCMS [ESI, M+11: 747.
Etapa B: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-[(E)-2-fluoro-4-(metilamino)but-2-enoil1piperazin-2-il1acetonitrilo. A una solución de W-[(E)-4-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1-il1-3-fluoro-4-oxo-but-2-enil1-W-metil-carbamato de ferc-butilo (300 mg, 401 umol, 1 eq.) en DCM (1 ml) se le añadió TFA
(915 mg, 8,03 mmol, 594 ul, 20 eq.). La mezcla de reacción se agitó a 25 °C durante 1 hora. Después de que se completara, la mezcla de reacción se diluyó con DCM (10 ml) y se basificó con NaHC03 saturado (8 ml). La mezcla se extrajo con DCM (3 x 10 ml). La capa orgánica combinada se secó sobre Na2S04 y se concentró al vacío para dar 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(E)-2-fluoro-4-(metilamino)but-2-enoil]piperazin-2-il]acetonitrilo (240 mg, 356 umol, 89 % de rendimiento, 95,9 % de pureza) en forma de un sólido de color pardo que se usó para la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]:
647.
Etapa C: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il1-1-[(E)-4-(dimetilamino)-2-fluoro-but-2-enoinpiperazin-2-¡nacetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(E)-2-fluoro-4-(metilamino)but-2-enoil]piperazin-2-il]acetonitrilo (120 mg, 185 umol, 1 eq.), paraformaldehído (83,5 mg, 927 umol, 5 eq.) y HC00H (26,7 mg, 556 umol, 3 eq.) en Me0H (5 ml) se le añadió NaBH3CN (35,0 mg, 556 umol, 3 eq.). La mezcla de reacción se agitó a 25 °C durante 12 horas. Después de que se completara, la mezcla de reacción se inactivó con agua (0,5 ml) y se filtró. El residuo se purificó por cromatografía sobre gel de sílice (AE:Me0H:NH3*H20 = 50:1:0 a 10:1 :0,1) y después HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um;fase móvil: [agua (NH4HC03 10 mM)-Ac N];% de B: 50 %-100 %,10 min) para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(E)-4-(dimetilamino)-2-fluoro-but-2-enoil]piperazin-2-il]acetonitrilo (EJEMPL0503, 26,7 mg, 40,2 umol, 22 % de rendimiento, 99,5 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1 ]: 661.
1H RMN (400 MHz, cloroformo-d) ó = 7,68 (d, J = 8,0 Hz, 1H), 7,54 (t, J = 7,2 Hz, 1H), 7,45 (d, J = 7,2 Hz, 1H), 7,37 (td, J = 7,6, 12,8 Hz, 1H), 7,26 (t, J = 8,0 Hz, 1H), 7,19 - 7,08 (m, 1H), 5,81 - 5,60 (m, 1H), 5,04 - 4,41 (m, 1H), 4,40 -4,25 (m, 2H), 4,16 - 3,91 (m, 3H), 3,88 - 3,71 (m, 2H), 3,66 - 3,46 (m, 2H), 3,36 (d a, J = 10,8 Hz, 1H), 3,27 - 2,69 (m, 9H), 2,64 - 2,56 (m, 1H), 2,55 - 2,46 (m, 1H), 2,40 (d, J = 2,4 Hz, 3H), 2,27 - 2,15 (m, 7H), 2,06 - 1,90 (m, 1H), 1,85 -1,72 (m, 3H).
Ejemplo 504

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[2-(trifluorometil)prop-2-enoil]piperazin-2-il]acetonitrilo

Etapa A: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[2-(trifluorometil)prop-2-enoinpiperazin-2-inacetonitrilo. A una solución de ácido 2-(trifluorometil)prop-2-enoico (39,5 mg, 282 umol, 1,50 eq.), 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (Intermedio 71, 0,10 g, 188 umol, 1,00 eq.) y DIEA (48,6 mg, 376 umol, 65,5 ul, 2,00 eq.) en diclorometano (0,20 ml) se le añadió HATU (107 mg, 282 umol, 1,50 eq.) a 0 °C. Después de agitar a 0 °C durante 0,5 h, la mezcla se diluyó con agua y la capa se separó. La capa orgánica se secó sobre
Na2SÜ4, se filtró y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (AF, 0,1 %)/acetonitrilo] y HPLC prep. (columna: Gemini 150 * 25 5 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 56 % -86 %, 12 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[2-(trifluorometil)prop-2-enoil]piperazin-2-il]acetonitrilo (EJEMPL0504, 13,1 mg, 19,6 umol, 10,5 % de rendimiento, 97,9 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 654.
Condición de SFC: Columna: Chiralcel 0J-350 x 4,6 mm D.I., 3 um, Fase móvil: metanol (0,05 % de DEA) en CÜ2 de 5 % a 40 %, Caudal: 3 ml/min, Longitud de onda: 220 nm.
1H RMN (400 MHz, cloroformo-d) 5 = 7,76 (d, J = 8,4 Hz, 1H), 7,65 - 7,59 (m, 1H), 7,55 - 7,50 (m, 1H), 7,45 (td, J = 7,6, 10,0 Hz, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,26 - 7,18 (m, 1H), 6,18 (s a, 1H), 5,86 (s a, 1H), 5,11 (s a, 1H), 4,57 - 4,36 (m, 2H), 4,33 - 4,10 (m, 2H), 4,04 (d a, J = 12,8 Hz, 1H), 3,97 - 3,79 (m, 2H), 3,66 - 3,54 (m, 1H), 3,50 - 3,33 (m, 1H), 3,29 - 3,07 (m, 4H), 3,04 - 2,72 (m, 4H), 2,63 - 2,50 (m, 4H), 2,45 - 2,32 (m, 1H), 2,15 - 2,04 (m, 1H), 1,90 (s a, 3H). Ejemplo 505

2-((S)-1-((Z)-3-cloroacriloil)-4-(7-(2,3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-((Z)-3-cloroacriloil)-4-(7-(2,3-dimetilfenil)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Se preparó siguiendo el Ejemplo 389 Etapa J, sustituyendo ácido (Z)-3-cloroacrílico por ácido but-2-inoico. ESI+APCI MS m/z 564,3 [M+H]+.
Ejemplo 506

2-((S)-1-((E)-4-fluorobut-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 2-((S)-1-((E)-4-fluorobut-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: A 0 °C, a un MFR de 25 ml que contenía N,N-dimetilformamida (3909 μl, 0,39 mmol) se le añadieron 2-((S)-4-(7-(8-metilnaftalen-1 -il)-2-(((S)-1 -metilpirrolidin-2il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (200 mg, 0,391 mmol) y trietilamina (136 μl, 0,98 mmol). La mezcla de reacción se agitó vigorosamente mientras se añadía en una porción ácido (E)-4-fluorobut-2-enoico (61,0 mg, 0,59 mmol). Después, a la mezcla en agitación se le añadió lentamente anhídrido cíclico del ácido 1-propanofosfónico (175 μl, 0,59 mmol). La reacción se agitó a temperatura ambiente durante 18 h. Se añadió agua y la mezcla se extrajo con EtOAc (3 x 15 ml). Los extractos se combinaron, se lavaron con agua (1* 10 ml) y se concentraron. El residuo se purificó con gel de sílice (5-18 % de Me0H en DCM con 0,25 % de NH40H) para proporcionar el compuesto del título 2-((S)-1-((E)-4-fluorobut-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0 506, 89,3 mg, 0,15 mmol, 38 % de rendimiento). ESI+APCI MS m/z 598,3 [M+H]+.
Ejemplo 507

2-((S)-1-(2-fluoroacriloil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
Etapa A: 2-((S)-1-(2-fluoroacriloil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Se diluyó 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (8 g, 15,63 mmol) con DMF (78,17 ml, 15,63 mmol), se puso en una atmósfera de nitrógeno y se enfrió a 0 °C. Se añadió DIEA (6,827 ml, 39,09 mmol) seguido de la adición de ácido 2-fluoroacrílico (2,112 g, 23,45 mmol) y la adición gota a gota de anhídrido cíclico del ácido 1-propanofosfónico (9,307 ml, 15,63 mmol). La reacción se agitó a 0 °C durante 6 horas y se dejó en agitación durante 10 horas más mientras permanecía a temperatura ambiente. La reacción se vertió en una solución al 5 % de bicarbonato de sodio y se extrajo dos veces con acetato de etilo. El acetato de etilo se lavó con agua y salmuera, se secó sobre MgS04, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 1 10 % de metanol/DCM (1 % de NH40H como aditivo) para proporcionar el compuesto del título 2-((S)-1-(2-fluoroacriloil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0507, 6 ,6 g, 11,31 mmol, 72,32 % de rendimiento). ESI+ApCI Ms m/z 584,3 [M+H]+.
Ejemplo 508

2-((S)-1-(4-hidroxibut-2-inoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
2-((S)-1-(4-hidroxibut-2-inoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: se preparó según el Ejemplo 458, Etapa D, sustituyendo ácido 4-((tercbutildimetilsilil)oxi)but-2-inoico por ácido but-2-inoico. ESI+APCI MS m/z 594,3 [M+H]+.
Ejemplo 509

2-((S)-1-((E)-4-(1 H-pirazol-1-il)but-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: ácido (E)-4-(1 H-pirazol-1-il)but-2-enoico. Una solución de ácido 4-bromocrotónico (100 mg, 0,606 mmol) y pirazol (37,1 mg, 0,546 mmol) en acetonitrilo (1 ml, 19,1 mmol) se calentó en un vial sellado durante 48 h a 60 °C con agitación. La solución de color verde claro resultante se dividió entre agua (5 ml) y EtOAc (15 ml), los extractos orgánicos se separaron y la capa orgánica se lavó con salmuera, se secó sobre Na2S04 y se evaporó al vacío. El material se cromatografió sobre gel de sílice en 2 a 5 % de Me0H/DCM 0,2 % de TFA. El sólido resultante se disolvió en una cantidad mínima de HCl 6 M y se evaporó en un flujo lento de N2. Este material se usó en la siguiente etapa sin purificación adicional.
Etapa B: 2-((S)-1-((E)-4-(1H-pirazol-1-il)but-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1lpirimidin-4-il)piperazin-2-il)acetonitrilo. Una mezcla agitada de 2-((S)-4-(7-(8-metilnaftalen-1 -il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (42,37 mg, 0,08281 mmol), ácido (E)-4-(1H-pirazol-1-il)but-2-enoico (12,6 mg, 0,08281 mmol) y N,N-dimetilformamida (1 ml, 12,79 mmol) se enfrió en un baño de hielo-sal con agitación y se añadió de una vez trietilamina (0,03463 ml, 0,2484 mmol) seguido de 2,4,6-trióxido de 2,4,6-tripropil-1,3,5,2,4,6-trioxatrifosfinano, 50 % en EtOAc (0,07395 ml, 0,1242 mmol). La mezcla de reacción se dejó calentar a ta durante 5 min y se agitó a ta durante una noche. La mezcla de reacción se dividió entre Na2C030,5 M (5 ml) y EtOAc (10 ml), los extractos orgánicos se separaron y la capa orgánica se lavó con agua y salmuera (5 ml de cada uno), se secó sobre Na2S04 y se evaporó al vacío. El producto se purificó por cromatografía sobre gel de sílice usando 5 % de Me0H 0,5 % de NH40H en DCM para dar el compuesto del título en forma de un sólido incoloro (EJEMPL0509, 11,46 mg, 23 %). ESI+APCI MS m/z 646,3 [M+H]+.
Ejemplo 510

2-((S)-1-((E)-4-hidroxibut-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: ácido (E)-4-hidroxibut-2-enoico. A una suspensión agitada de ácido 4-bromocrotónico (4824 mg, 29,24 mmol) en agua (48 ml) se le añadió gota a gota una solución 2 M de hidróxido de potasio (4,921 g, 87,72 mmol) y la solución resultante se calentó a reflujo durante 5 min. La mezcla de reacción se dejó en una corriente de N2 para que se enfriara y se concentrara. El residuo se enfrió en un baño de hielo, se acidificó con H2S044 M a pH 1-2 y la suspensión resultante se evaporó a la mitad al vacío y después se extrajo con EtOAc (3 x 60 ml). Los extractos combinados se secaron sobre Na2S04 y se evaporaron al vacío. El material se purificó por cromatografía usando 2 a 5 % de Me0H 0,2 % de TFA para proporcionar el ácido diana en forma de cristales incoloros (2,165 g, 73 %).
Etapa B: 2-((S)-1-((E)-4-hidroxibut-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo. A una solución agitada de 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (100 mg, 0,1954 mmol), ácido (2E)-4-hidroxibut-2-enoico (39,90 mg, 0,3909 mmol) y N-etil-N-isopropilpropan-2-amina (0,06809 ml, 0,3909 mmol) en DCM (5 ml) se le añadió de una vez hexafluorofosfato de 0-(7-azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio (111,5 mg, 0,2932 mmol) y la mezcla de reacción se agitó a ta durante 4 h. Se añadió agua (1 ml) y la mezcla de reacción se agitó durante 5 min y se dividió entre EtOAc (10 ml) y NaHC03 sat. (5 ml). La capa orgánica se separó, se lavó con NaHC03 y salmuera (5 ml de cada uno), se secó sobre K2C03 y se evaporó al vacío. El residuo se cromatografió sobre gel de sílice con 5 % de Me0H 0,5 % de NH40H. El compuesto del título en forma de un sólido incoloro (EJEMPL0510, 29 mg, 25 %). ESI+APCI MS m/z 596,3 [M+H]+.
Ejemplo 511

2-((S)-1-((E)-4-metoxibut-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 2-(S)-1-((E)-4-metoxibut-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo. Una mezcla agitada de 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (250 mg, 0,4886 mmol), ácido (E)-4-metoxibut-2-enoico (85,10 mg, 0,7329 mmol) y N,N-dimetilformamida (2 ml, 25,58 mmol) se enfrió en un baño de hielo-sal y se añadió de una vez trietilamina (0,2043 ml, 1,466 mmol) seguido de 2,4,6-trióxido de 2,4,6-tripropil-1,3,5,2,4,6-trioxatrifosfinano, 50 % en EtOAc (0,4363 ml, 0,7329 mmol). La mezcla de reacción se dejó calentar a ta durante 5 min y se agitó a ta durante 2 h. La solución resultante se dividió entre Na2C030,5 M (10 ml) y EtOAc (30 ml) y la capa orgánica se separó, se lavó con agua y salmuera (10 ml de cada uno), se secó sobre Na2S04 y se evaporó al vacío. El material se purificó por cromatografía sobre gel de sílice usando 5 % de Me0H 0,5 % de NH40H en DCM. El material se purificó adicionalmente por cromatografía de fase inversa, Gilson, 25 a 75 % de MeCN/H20 0,1 % de TFA. Las fracciones diana se basificaron con exceso de Na2C032 M y se extrajeron con DCM (3 x 50 ml). Los extractos orgánicos combinados se lavaron con salmuera, se secaron sobre Na2S04 y se evaporaron al vacío. El compuesto del título en forma de un sólido de color amarillo claro (EJEMPL0511, 123 mg, 41 %). ESI+APCI MS m/z 610,3 [M+H]+.
Ejemplo 512

2-[(2S)-1-(2-metoxiprop-2-enoil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

Etapa A: 2,2-dimetoxipropanoato de metilo. A una solución de 2-oxopropanoato de etilo (4,00 g, 34,5 mmol, 3,81 ml, 1.00 eq.) y trimetoximatano (4,75 g, 44,8 mmol, 4,91 ml, 1,30 eq.) en metanol (10,0 ml) se le añadió H2S04 (33,8 mg, 344 umol, 18,4 ul, 0,01 eq.). Después de agitar a 65 °C durante 4 horas, el valor de pH se ajustó a >7 con K0H (120 mg en 20,0 ml de agua) y se extrajo con acetato de etilo (3 * 20,0 ml). La capa orgánica se lavó con salmuera (1 * 30,0 ml), se secó sobre Na2S04, se filtró y se concentró al vacío. El residuo se destiló a 60 °C en una bomba de aceite para dar 2,2-dimetoxipropanoato de metilo (4,00 g, 27,0 mmol, 78 % de rendimiento) en forma de un aceite incoloro y se usó en la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, cloroformo-d) ó = 3,81 (s, 3H), 3,28 (s, 6H), 1,52 (s, 3H).
Etapa B: 2-metoxiprop-2-enoato de metilo. A una solución de 2,2-dimetoxipropanoato de metilo (2,00 g, 13,5 mmol, 1.00 eq.) en DMF (20,0 ml) se le añadió poco a poco P205 (1,05 g, 7,42 mmol, 458 ul, 0,55 eq.). Después de calentó a 100 °C durante 1 hora, la mezcla se diluyó con bicarbonato de sodio saturado (20,0 ml), se extrajo con éter isopropílico (3 * 30,0 ml), se lavó con salmuera (3 * 40,0 ml), se secó sobre Na2S04, se filtró y se concentró al vacío. El residuo se destiló en una bomba de aceite para dar 2-metoxiprop-2-enoato de metilo (0,65 g, 5,60 mmol, 41 % de rendimiento) en forma de un aceite incoloro y se usó en la siguiente etapa sin purificación adicional.
Etapa C: ácido 2-metoxiprop-2-enoico. Una mezcla de 2-metoxiprop-2-enoato de metilo (0,60 g, 5,17 mmol, 1,00 eq.) y Na0H (827 mg, 20,7 mmol, 4,00 eq.) en H20 (5,00 ml) y THF (5,00 ml) se agitó a 60 °C durante 1 hora. La mezcla se ajustó a pH <3 con HCl concentrado (10,0 ml) y se extrajo con acetato de etilo (3 * 20,0 ml). La capa orgánica se secó sobre Na2S04, se filtró y se concentró al vacío para dar ácido 2-metoxiprop-2-enoico (0,40 g, 1,96 mmol, 38 % de rendimiento, 50 % de pureza) en forma de un aceite de color rosa y se usó en la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, cloroformo-d) ó = 8,63 (s a, 1H), 5,52 (d, J = 2,8 Hz, 1H), 4,75 (d, J = 2,8 Hz, 1H), 3,70 (s, 3H).
Etapa D: 2-[(2S)-1-(2-metoxiprop-2-enoil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-yl1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (0,14 g, 274 umol, 1,00 eq.), ácido 2-metoxiprop-2-enoico (112 mg, 1,09 mmol, 4,00 eq.) y DIEA (141 mg, 1,09 mmol, 191 ul, 4,00 eq.) en diclorometano (5,00 ml) se le añadió HATU (208 mg, 547 umol, 2,00 eq.) a 0 °C. Después de agitar a 0 °C durante 0,5 h, la mezcla se diluyó con agua (3,00 ml). La capa orgánica se secó sobre Na2S04, se filtró y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (AF, 0,10 %)/acetonitrilo] y HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 60 % - 90 %, 12 min). La fracción deseada se recogió y se liofilizó para dar el compuesto del título 2-[(2S)-1-(2-metoxiprop-2-enoil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (EJEMPL0512, 10,27 mg, 17,22 umol, 6,3 % de rendimiento, 99,9 % de pureza) en forma de un sólido de color blanquecino. LCMS [ESI, M+1]: 596.
Condición de SFC: “ IC-3S_3_40_3ML Columna: Chiralpak IC-3100 * 4,6 mm D.I., 3 um, Fase móvil: 40 % de metanol (0,05 % de DEA) en C02, Caudal: 3 ml/min, Longitud de onda: 220 nm” .
1H RMN (400 MHz, cloroformo-d) ó = 7,72 - 7,61 (m, 2H), 7,45 - 7,31 (m, 2H), 7,27 - 7,17 (m, 2H), 5,05 - 4,61 (m, 1H), 4,48 (s a, 1H), 4,37 (td, J = 4,4, 10,0 Hz, 1H), 4,30 - 3,98 (m, 4H), 3,96 - 3,74 (m, 2H), 3,69 (s a, 3H), 3,59 - 3,28 (m,
2H), 3,27 - 2,93 (m, 6 H), 2,92 (s, 3H), 2,88 - 2,51 (m, 4H), 2,47 (d, J = 3,2 Hz, 3H), 2,33 - 2,23 (m, 1H), 2,11 - 1,98 (m, 1H), 1,89 - 1,74 (m, 3H).
Ejemplo 513

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-[(E)-4-hidroxibut-2 -enoil]piperazin-2 -il]acetonitrilo
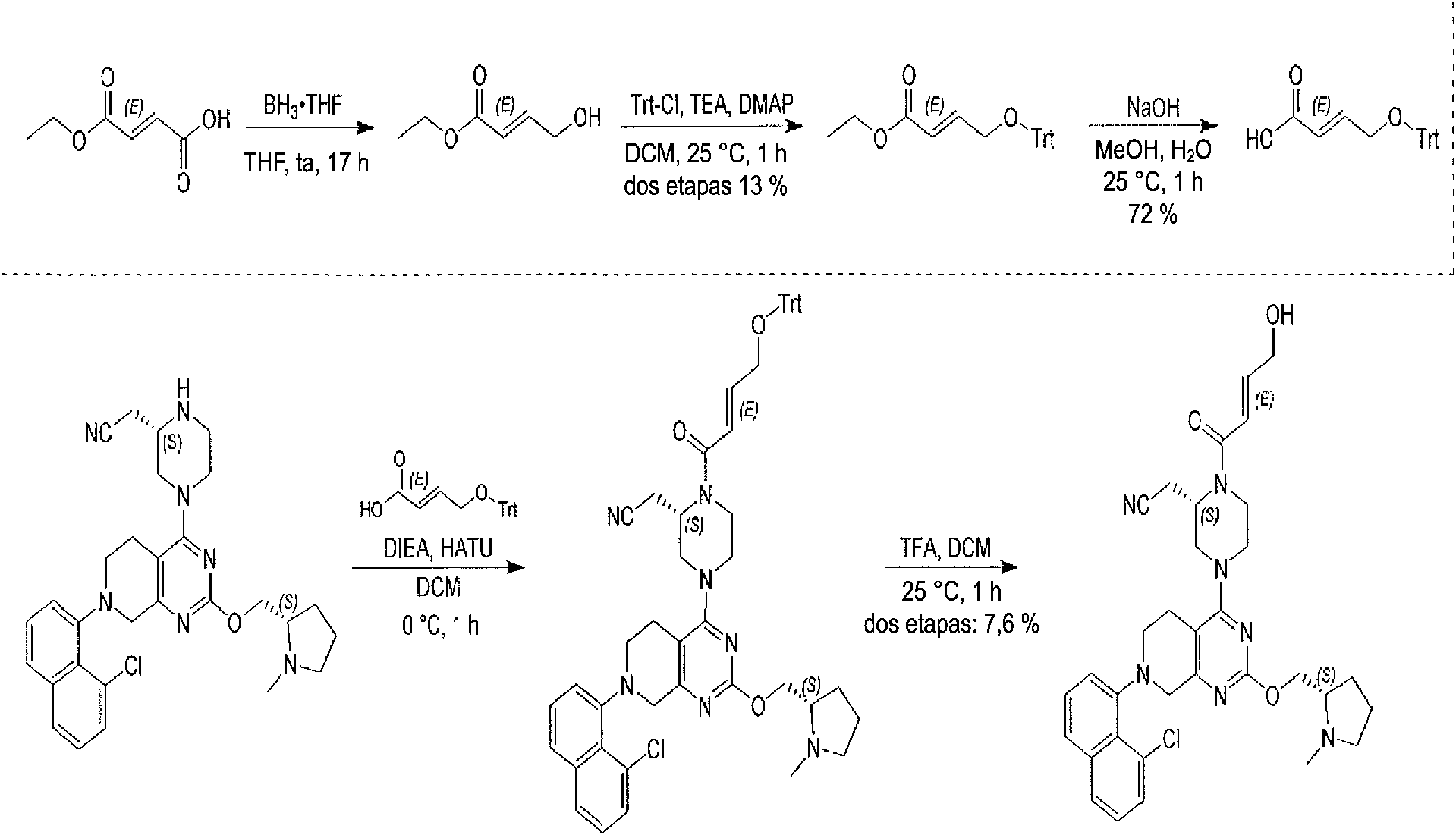
Etapa 1: (E)-4-hidroxibut-2-enoato de etilo. A una solución de ácido (E)-4-etoxi-4-oxo-but-2-enoico (1,0 g, 7,29 mmol, 1,0 eq.) en THF (6,0 ml) se le añadió gota a gota una solución de BH3-Me2S (10,0 M, 692 ul, 0,95 eq.) en THF (8,0 ml) durante 1 hora a -10 °C. La mezcla de reacción se calentó gradualmente a 25 °C y se agitó durante 17 horas. Después de que se completara, la mezcla de reacción se inactivó con la adición de agua (30,0 ml) y se extrajo con acetato de etilo (30,0 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (30,0 ml x 2), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El compuesto (E)-4-hidroxibut-2-enoato de etilo (900 mg, bruto) se obtuvo en forma de un aceite incoloro. El producto bruto se usó directamente en la siguiente etapa sin purificación adicional.
Etapa 2: (E)-4-tritiloxibut-2-enoato. Una mezcla de (E)-4-hidroxibut-2-enoato de etilo (900 mg, bruto), [cloro(difenil)metil]benceno (2,89 g, 10,4 mmol), DMAP (84,5 mg, 692 umol) y TEA (1,4 g, 13,8 mmol, 1,9 ml) en Dc M (5,0 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 25 °C durante 1 hora en una atmósfera de N2. Después de que se completara, la mezcla de reacción se inactivó con la adición de H20 (10 ,0 ml) y se extrajo con acetato de etilo (10,0 ml x 3 ). Las capas orgánicas combinadas se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 20:1 a 15:1). El compuesto (E)-4-tritiloxibut-2-enoato de etilo (340 mg, 913 umol, 13 % de rendimiento) se obtuvo en forma de un aceite incoloro.
1H RMN (400 MHz, Cloroformo-d) 57,38 - 7,35 (m, 6 H), 7,26 - 7,22 (m, 6 H), 7,18 - 7,15 (m, 3 H), 6,88 (dt, J = 15,6, 3.6 Hz, 1 H), 6,29 (dt, J = 15,6, 2,2 Hz, 1 H), 4,16 (c, J = 7,1 Hz, 2 H), 3,72 (dd, J = 2,4, 2,0 Hz, 2 H), 1,25 (t, J = 7,0 Hz, 3 H).
Etapa 3: ácido (E)-4-tritiloxibut-2-enoico. A una solución de (E)-4-tritiloxibut-2-enoato de etilo (170 mg, 456 umol, 1,0 eq.) en Me0H (1,0 ml) se le añadió una solución de Na0H (54,5 mg, 1,37 mmol, 3,0 eq.) en H20 (0,5 ml) y después la mezcla se agitó a 40 °C durante 1 hora en una atmósfera de N2. Después de que se completara, la mezcla de reacción se inactivó con la adición de HCl 1 M a 0 °C hasta pH = 5 y después se extrajo con acetato de etilo (8,0 ml x 3). Las capas orgánicas combinadas se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 3:1 a 1:1). El compuesto ácido (£)-4-tritiloxibut-2-enoico (113 mg, 328 umol, 72 % de rendimiento) se obtuvo en forma de un sólido de color blanco.
1H RMN (400 MHz, Cloroformo-d) 57,38 - 7,36 (m, 6 H), 7,26 - 7,24 (m, 5 H), 7,20 - 7,16 (m, 4 H), 7,0 (dt, J = 15,6, 3.6 Hz, 1 H), 6,32 (dt, J = 15,6, 2,0 Hz, 1 H), 3,76 (dd, J = 3,2, 2,4 Hz, 2 H).
Etapa A: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-[(£)-4-tritiloxibut-2-enoil1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-djpirimidin-4-il1piperazin-2-il1acetonitrilo (Intermedio 71, 60,0 mg, 113 umol, 1,0 eq.), ácido (E)-4-tritiloxibut-2-enoico (77,8 mg, 226 umol, 2,0 eq.) y DIEA (73,0 mg, 564 umol, 98 ul, 5,0 eq.) en DCM (2,0 ml) se le añadió en una porción HATU (64 mg, 169 umol, 1,5 eq.) a 0 °C en una atmósfera de N2. La mezcla se agitó a 25 °C durante 1 hora. Después de que se completara, la mezcla de reacción se inactivó con la adición de H20 (8,0 ml) y después se extrajo con acetato de etilo (10,0 ml x 3 ). Las capas orgánicas combinadas se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El compuesto 2-[(2S)-4-[7-(8-cloro-1 -naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5^-pirido[3,4-d1pirimidin-4-il1-1 -[(H)-4-tritiloxibut-2-enoil1piperazin-2-il1acetonitrilo (150 mg, bruto) se obtuvo en forma de un sólido de color amarillo y el producto bruto se usó directamente en la siguiente etapa sin purificación adicional. LCMS [ESI, M+11: 858.
Etapa B: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-[(£)-4-hidroxibut-2-enoil1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5^-pirido[3,4-d1pirimidin-4-il1-1 -[(H)-4-tritiloxibut-2-enoil1piperazin-2-il1acetonitrilo (150 mg, bruto) en DCM (1,0 ml) se le añadió TFA (1,0 ml, 13,5 mmol). La mezcla se agitó a 25 °C durante 1 hora. Después de que se completara, la mezcla de reacción se inactivó con la adición de una solución acuosa de NaHC03 a 0 °C hasta pH = 8 y después se extrajo con acetato de etilo (10,0 ml x 3 ). Las capas orgánicas combinadas se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,04 % de NH3H20 NH4HC0310 mM)-ACN1; % de B: 48 %-72 %, 10 min.) y se sometió a liofilización. El compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-[(£)-4-hidroxibut-2-enoil1piperazin-2-il1acetonitrilo (EJEMPL0513, 8,21 mg, 13,3 umol, 7,6 % de rendimiento, 99,8 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+11: 616.
Condición de SFC: 100 % de e.e.
1H RMN (400 MHz, Cloroformo-d) 57,68 (d, J = 8,0 Hz, 1 H), 7,70-7,33 (m, 1 H), 7,49 (dd, J = 6,8, 0,8 Hz, 1 H), 7,37 (dt, J = 11,98, 7,82 Hz, 1 H), 7,26 (t, J = 11,8 Hz, 1 H), 7,19 - 7,11 (m, 1 H), 6,99 (dt, J = 14,8, 3,4 Hz, 1 H), 6,56 (s a, 1 H), 5,02-4,57 (m, 1 H), 4,39-4,29 (m, 4 H), 4,12-4,06 (m, 1 H), 4,03-3,99 (m, 2 H), 3,85-3,72 (m, 2 H), 3,67-3,36 (m, 3 H), 3,24-3,09 (m, 2 H), 3,03-2,89 (m, 3 H), 2,73 (s a, 1 H), 2,63-2,61 (m, 1 H), 2,50 (s a, 1 H), 2,41 (s, 3 H), 2,25-2,19 (m, 1 H), 2,03-1,92 (m, 1 H), 1,70-1,60 (m, 3 H).
Ejemplo 514

2-[(2 S)-4-[7-(8-cloro-1 -naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5^-pirido[3,4-d1pirimidin-4-il1-1-(2-metoxiprop-2-enoil)piperazin-2-il1acetonitrilo

Etapa 1: 2,2-dimetoxipropanoato de metilo. A una solución de 2-oxopropanoato de etilo (4,00 g, 34,5 mmol, 3,81 ml, 1.00 eq.) y trimetoximatano (4,75 g, 44,8 mmol, 4,91 ml, 1,30 eq.) en metanol (10,0 ml) se le añadió H2S04 (33,8 mg, 344 umol, 18,4 ul, 0,01 eq.). Después de agitar a 65 °C durante 4 horas, la mezcla de reacción se ajustó a un valor de pH >7 con K0H acuoso (120 mg en 20,0 ml de agua) y se extrajo con acetato de etilo (3 * 20,0 ml). Las capas orgánicas se lavaron con salmuera (1 * 30,0 ml), se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se destiló a 60 °C al vacío para dar 2,2-dimetoxipropanoato de metilo (4,00 g, 27,0 mmol, 78 % de rendimiento) en forma de un aceite incoloro.
1H RMN (400 MHz, cloroformo-d) ó = 3,81 (s, 3H), 3,28 (s, 6H), 1,52 (s, 3H).
Etapa 2: 2-metoxiprop-2-enoato de metilo. A una solución de 2,2-dimetoxipropanoato de metilo (2,00 g, 13,5 mmol, 1.00 eq.) en DMF (20,0 ml) se le añadió poco a poco P205 (1,05 g, 7,42 mmol, 458 ul, 0,55 eq.). Después de agitar a 100 °C durante 1 hora, la mezcla se diluyó con bicarbonato de sodio saturado (20,0 ml) y se extrajo con éter isopropílico (3 * 30,0 ml). Los extractos se lavaron con salmuera (3 * 40,0 ml), se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se destiló al vacío para dar 2-metoxiprop-2-enoato de metilo (0,65 g, 5,60 mmol, 41 % de rendimiento) en forma de un aceite incoloro que se usó en la siguiente etapa sin purificación adicional. 1H RMN (400 MHz, cloroformo-d) ó = 5,36 (d, J = 2,8 Hz, 1H), 4,63 (d, J = 2,8 Hz, 1H), 3,81 (s, 3H), 3,66 (s, 3H). Etapa 3: ácido 2-metoxiprop-2-enoico. Una mezcla de 2-metoxiprop-2-enoato de metilo (0,60 g, 5,17 mmol, 1,00 eq.) y Na0H (827 mg, 20,7 mmol, 4,00 eq.) en H20 (5,00 ml) y THF (5,00 ml) se agitó a 60 °C durante 1 hora. La mezcla se ajustó a pH <3 con HCl concentrado (10,0 ml) y se extrajo con acetato de etilo (3 * 20,0 ml). La capa orgánica se secó sobre Na2S04, se filtró y se concentró al vacío para dar ácido 2-metoxiprop-2-enoico (0,40 g, 1,96 mmol, 38 % de rendimiento, 50 % de pureza) en forma de un aceite de color rosa y se usó en la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, cloroformo-d) ó = 8,63 (s a, 1H), 5,52 (d, J = 2,8 Hz, 1H), 4,75 (d, J = 2,8 Hz, 1H), 3,70 (s, 3H). Etapa A: 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-il1-1-(2-metoxiprop-2-enoil)piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (0,20 g, 376 umol, 1,00 eq.), ácido 2-metoxiprop-2-enoico (153 mg, 1,50 mmol, 4,00 eq.) y DIEA (194 mg, 1,50 mmol, 262 ul, 4,00 eq.) en diclorometano (5,00 ml) se le añadió HATU (286 mg, 752 umol, 2,00 eq.) a 0 °C. Después de agitar a 0 °C durante 0,5 h, la reacción se purificó por cromatografía ultrarrápida de fase inversa [agua (AF, 0,1 %)/acetonitrilo], HPLC prep. (columna: Luna C18 150 * 25 5 ufase móvil: [agua (0,225 % de AF) - ACN]; % de B: 25 % - 45 %, 7,8 min) y adicionalmente por HPLC prep. (columna: Waters Xbridge 150 * 25 5 ufase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 55 % - 79 %, 10 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-dih¡dro-5H-p¡r¡do[3,4-d]pirimidin-4-il1-1-(2-metoxiprop-2-enoil)piperazin-2-il]acetonitrilo (EJEMPL0 514, 30,1 mg, 48,9 umol, 13 % de rendimiento, 100 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 616.
Condición de SFC: “AS-3_Me0H (DEA)_5_40_3 ml-35T Columna: Chiralpak AS-350 * 4,6 mm D.I., 3 um, Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 %, Caudal: 3 ml/min, Longitud de onda: 220 nm” .
1H RMN (400 MHz, cloroformo-d) ó = 7,76 (d a, J = 8,0 Hz, 1H), 7,62 (t, J = 7,6 Hz, 1H), 7,52 (d, J = 7,2 Hz, 1H), 7,44 (td, J = 7,6, 13,6 Hz, 1H), 7,34 (t, J = 8,0 Hz, 1H), 7,27 - 7,18 (m, 1H), 5,07 - 4,63 (m, 1H), 4,57 - 4,29 (m, 4H), 4,21 -3,96 (m, 3H), 3,95 - 3,77 (m, 2H), 3,69 (s a, 3H), 3,59 (d a, J = 11,2 Hz, 1H), 3,51 - 3,32 (m, 1H), 3,30 - 3,02 (m, 5H), 2,89 - 2,50 (m, 4H), 2,47 (d, J = 2,0 Hz, 3H), 2,32 - 2,23 (m, 1H), 2,11 - 2,00 (m, 1H), 1,89 - 1,72 (m, 3H).
Ejemplo 515

2-[(2S)-1 -[(E)-4-(3-fluoroazetidin-1 -il)but-2-enoil]-4-[7-(8-metil-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

Etapa A: 2-[(2S)-1-[(E)-4-bromobut-2-enoil1-4-[7-(8-metil-1-naplithil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (150 mg, 293 umol, 1,0 eq.) y piridina (186 mg, 2,35 mmol, 189 ul, 8,0 eq.) en diclorometano (4,0 ml) se le añadió cloruro de (E)-4-bromobut-2-enoílo (215 mg, 1,17 mmol, 4,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se concentró para dar el producto 2-[(2S)-1-[(E)-4-bromobut-2-enoil]-4-[7-(8-metil-1-naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (200 mg, bruto) en forma de un aceite de color amarillo. El producto se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 658.
Etapa B: 2-[(2S)-1 -[(E)-4-(3-fluoroazetidin-1 -il)but-2-enoil]-4-[7-(8-metil-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-i n piperazin-2-i n acetonitrilo. A una solución de 2-[(2S)-1-[(E)-4-bromobut-2-enoil]-4-[7-(8-metil-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-tí]pirimidin-4-il]piperazin-2-il]acetonitrilo (150 mg, 228 umol, 1,0 eq.) y K2C03 (315 mg, 2,28 mmol, 10,0 eq.) en acetonitrilo (2,0 ml) se le añadió 3-fluoroazetidina (152 mg, 1,37 mmol, 6,0 eq., HCl). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, a la mezcla de reacción se le añadió agua (5,0 ml) y se extrajo con acetato de etilo (5,0 ml * 4). La capa orgánica se secó sobre Na2S04, se filtró y se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 60 %-90 %,12 min) para
dar el compuesto del título 2-[(2S)-1-[(E)-4-(3- fluoroazetidin-1-il)but-2-enoil]-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (EJEMPL0515, 45,0 mg, 66 umol, 29 % de rendimiento, 96 % de pureza) en forma de un sólido de color blanco. LcMs [ESI, M+1]: 653.
1H RMN (400 MHz, Cloroformo-d) 57,70 (d a, J = 8,0 Hz, 1H), 7,65 (t, J = 8,4 Hz, 1H), 7,41 (dd, J = 7,6, 15,2 Hz, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,27 - 7,18 (m, 2H), 6,92 -6,82 (m, 1H), 6,52 - 6,31 (m, 1H), 5,28 - 5,08 (m, 1H), 5,07 - 4,46 (m, 1H), 4,41 - 4,33 (m, 1H), 4,32 - 4,01 (m, 4H), 4,01 - 3,79 (m, 2H), 3,79 - 3,56 (m, 3H), 3,55 - 3,37 (m, 2H), 3,36 -3,31 (m, 2H), 3,30 - 3,24 (m, 1H), 3,23 - 3,18 (m, 2H), 3,17 - 3,15 (m, 1H), 3,14 - 3,06 (m, 2H), 3,05 - 2,95 (m, 1H), 2,92 (s, 3H), 2,86 - 2,75 (m, 1H), 2,71 - 2,57 (m, 2H), 2,47 (d, J = 4,0 Hz, 3H), 2,33 - 2,22 (m, 1H), 2,11 - 1,99 (m, 1H), 1,88 - 1,67 (m, 3H).
Ejemplo 516

2-((S)-1-(2-fluoroacriloil)-4-(7-(8-metilnaftalen-1-il)-2-(((R)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 4-[(3S)-4-Benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-7-carboxilato de tere-butilo. Una mezcla de 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-cloro-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de tere-butilo (5,5 g, 10,4 mmol, 1 eq.), [(2R)-1-metilpirrolidin-2-il]metanol (2,40 g, 20,9 mmol, 2,25 ml, 2 eq.), Pd2(dba)3 (1,91 g, 2,09 mmol, 0,2 eq.), RuPhos (1,95 g, 4,17 mmol, 0,4 eq.) y Cs2C03 (8,50 g, 26,1 mmol, 2,5 eq.) en tolueno (100 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 5 horas en una atmósfera de N2. La mezcla de reacción se concentró a presión reducida para dar un residuo. El residuo se diluyó con acetato de etilo (100 ml) y agua (50 ml). Después, la mezcla se acidificó a pH ~4 con una solución acuosa 1 M de HCl. La fase de agua se separó y se basificó a pH ~8 con una solución acuosa saturada de Na2C03 y después la mezcla se extrajo con acetato de etilo (50 ml * 2). Las capas orgánicas combinadas se lavaron con salmuera (30 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida. Se obtuvo 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de tere-butilo (4 g, 5,55 mmol, 53 % de rendimiento, 84 % de pureza) en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación.
1H RMN (400 MHz, cloroformo-d) 5 = 7,46 - 7,31 (m, 5H), 5,23 (s, 2H), 4,74 - 4,49 (m, 2H), 4,43 - 4,27 (m, 2H), 4,21 -3,70 (m, 5H), 3,42 - 3,17 (m, 3H), 3,09 (t, J = 7,6 Hz, 1H), 2,98 (td, J = 3,6, 12,4 Hz, 1H), 2,86 - 2,74 (m, 1H), 2,74 -2,54 (m, 4H), 2,47 (s, 3H), 2,35 - 2,21 (m, 1H), 2,05 - 1,97 (m, 1H), 1,90 - 1,74 (m, 3H), 1,49 (s, 9H).
Etapa B: (2S)-2-(c¡anomet¡l)-4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-5.6.7.8-tetrah¡drop¡r¡do[3,4-d1p¡r¡m¡d¡n-4-illpiperazin-1-carboxilato de bencilo. A una soluc¡ón de 4-[(3S)-4-benc¡lox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-7-carbox¡lato de ferc-butilo (3,7 g, 6,11 mmol, 1 eq.) en dioxano (20 ml) se le añadió HCl/dioxano (4 M, 22,9 ml, 15 eq.). La mezcla se agitó a 0 °C durante 1 hora. El líquido se decantó y el sólido se recogió. El residuo sólido se diluyó con agua (50 ml) y diclorometano (100 ml) y después la mezcla se basificó a pH ~8 con una solución acuosa saturada de Na2CÜ3 y se extrajo con diclorometano (100 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (50 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida. Se obtuvo (2S)-2-(c¡anomet¡l)-4-[2-[[(2R)-1-metilp¡rrol¡d¡n-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1 -carboxilato de bencilo (2,6 g, 4,89 mmol, 80 % de rendimiento, 95 % de pureza) en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación. LCMS [ESI, M+1]: 506.
Etapa C: (2S)-2-(c¡anomet¡l)-4-[7-(8-met¡l-1-naft¡l)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de bencilo. Una mezcla de (2S)-2-(c¡anometil)-4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡mid¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (2,3 g, 4,55 mmol, 1 eq.), 1 bromo-8-metil-naftaleno (Intermedio 69,1,31 g, 5,91 mmol, 1,3 eq.), Cs2CÜ3 (3,71 g, 11,4 mmol, 2,5 eq.), Pd2(dba)3 (833 mg, 909 umol, 0,2 eq.) y Xantphos (1,05 g, 1,82 mmol, 0,4 eq.) en tolueno (50 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 6 horas en una atmósfera de N2. La mezcla de reacción se diluyó con agua (100 ml) y se extrajo con acetato de etilo (200 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (100 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (S¡02, acetato de etilo/metanol = 100/1 a 10/1). Se obtuvo (2S)-2-(c¡anomet¡l)-4-[7-(8-met¡l-1-naft¡l)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metoxi]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1 -carboxilato de bencilo (1,6 g, 2,48 mmol, 54 % de rendimiento) en forma de un sólido de color amarillo.
Etapa D: 2-[(2S)-4-[7-(8-met¡l-1-naft¡l)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. Una mezcla de (2S)-2-(c¡anomet¡l)-4-[7-(8-met¡l-1-naftil)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡mid¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (1,60 g, 2,48 mmol, 1,00 eq.) en Me0H (20,0 ml) y NH3 (20 % p/p en Me0H, 7 ml) se hidrogenó con Pd/C (100 mg, 2,48 mmol, 10 % de pureza, 1,00 eq.) como catalizador en una atmósfera de H2 (4,99 mg, 2,48 mmol, 1,00 eq., 103,42 kPa (15 psi)) a 25 °C durante 3 horas. El catalizador se retiró por filtración a través de una capa de celite y el filtrado se concentró al vacío. El producto bruto se usó en la siguiente etapa directamente sin purificación adicional. Se obtuvo 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡din-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (1,20 g, 2,35 mmol, 95 % de rendimiento) en forma de un sólido de color amarillo claro. LCMS [ESI, M+1]: 512.
Etapa E: 2-r(2S)-1-(2-fluoroprop-2-eno¡l)-4-r7-(8-met¡l-1-naft¡l)-2-rr(2R)-1-met¡lp¡rrol¡d¡n-2-¡llmetox¡l-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una mezcla de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]aceton¡tr¡lo (140 mg, 274 umol, 1,00 eq.) y ácido 2-fluoroprop-2-enoico (73,9 mg, 821 umol, 3,00 eq.) en diclorometano (3,00 ml) se le añadieron en una porción DIEA (212 mg, 1,64 mmol, 286 ul, 6,00 eq.), HATU (312 mg, 821 umol, 3,00 eq.) a 0 °C en una atmósfera de N2. Después de agitar a 0 °C durante 30 min, la mezcla de reacción se diluyó con agua (10,0 ml) y se extrajo con diclorometano (2 x 10,0 ml). Las capas orgánicas combinadas se lavaron con agua (1 x 10,0 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,04 % de NH3H20 NH4HC0310 mM) - ACN]; % de B: 55 % - 85 %, 10 min). El compuesto del título 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡din-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (EJEMPL0 516, 48,7 mg, 82,1 umol, 30 % de rendimiento, 98,4 % de pureza) se obtuvo en forma de un sólido de color blanco.
Condición de SFC: “Columna: Chiralcel 0J-350 x 4 ,6 mm D.I., 3 um, Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 %, Caudal: 3 ml/min, Longitud de onda: 220 nm” .
1H RMN (400 MHz, cloroformo-d) 5 = 7,73 - 7,62 (m, 2H), 7,45 - 7,31 (m, 2H), 7,27 - 7,17 (m, 2H), 5,53 - 5,34 (m, 1H), 5,26 (dd, J = 3,6, 17,2 Hz, 1H), 4,88 (s a, 1H), 4,41 - 4,32 (m, 1H), 4,31 - 4,22 (m, 1H), 4,21 - 4,10 (m, 2H), 4,10 - 4,02 (m, 1H), 3,89 (d a, J = 18,0 Hz, 1H), 3,78 (d, J = 18,4 Hz, 1H), 3,59 - 3,40 (m, 2H), 3,27 - 3,15 (m, 2H), 3,14 - 3,04 (m, 2H), 3,03 - 2,90 (m, 4H), 2,90 - 2,73 (m, 2H), 2,72 - 2,56 (m, 2H), 2,47 (d, J = 4,8 Hz, 3H), 2,33 - 2,23 (m, 1H), 2,11 -1,98 (m, 1H), 1,90 - 1,74 (m, 3H).
Ejemplo 517
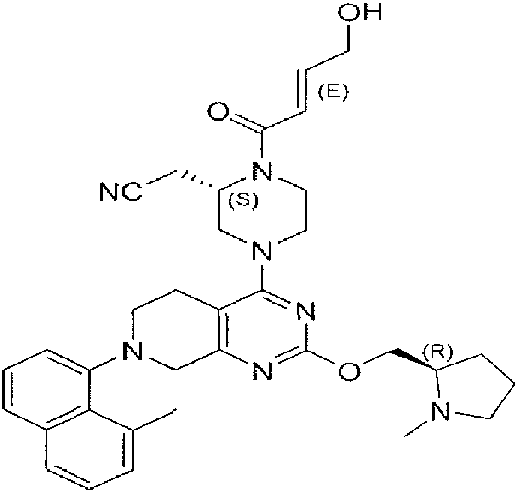
2-((S)-1-((E)-4-hidroxibut-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((E)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il1inetoxi1-6.8-dihidro-5H-pirido[3.4-d1pirimidin-4-il1-1-[(E)-4-tritiloxibut-2-enoil1piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2E)-1-metilpirrolidin-2-il]metoxi]-6.8-dihidro-5H-pirido[3.4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (120 mg. 234 umol. 1 eq.), TEA (142 mg. 1.41 mmol. 195 ul. 6 eq.) y ácido (E)-4-tritiloxibut-2-enoico (96.9 mg. 281 umol. 1.2 eq.) en acetato de etilo (2 ml) se le añadió T3P (448 mg. 703 umol. 418 ul. 50 % de pureza en EtOAc. 3 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0.5 horas. La mezcla de reacción se diluyó con agua (5 ml) y se extrajo con acetato de etilo (10 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (10 ml). se secaron sobre Na2S04. se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02. acetato de etilo/metanol = 200/1 a 10/1). Se obtuvo 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2E)-1-metilpirrolidin-2-il]metoxi]-6.8-dihidro-5H-pirido[3.4-d]pirimidin-4-il]-1-[(E)-4-tritiloxibut-2-enoil]piperazin-2-il]acetonitrilo (90 mg. 103 umol. 44 % de rendimiento. 96 % de pureza) en forma de un aceite de color amarillo. LCMS [ESI. M+1]: 838.
Etapa B: 2-[(2S)-1-[(E)-4-hidroxibut-2-enoil]-4-[7-(8-metil-1-naftil)-2-[[(2E)-1-metilpirrolidin-2-il]metoxi]-6.8-dihidro-5H-pirido[3.4-d1pirimidin-4-il]piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2E)-1-metilpirrolidin-2-il]metoxi]-6.8-dihidro-5H-pirido[3.4-d]pirimidin-4-il]-1-[(E)-4-tritiloxibut-2-enoil]piperazin-2-il]acetonitrilo (80 mg. 95.5 umol. 1 eq.) en diclorometano (200 ul) se le añadió TfA (218 mg. 1.91 mmol. 141 ul. 20 eq.). La mezcla se agitó a 0 °C durante 1 hora. La mezcla de reacción se diluyó con agua (5 ml) y se basificó a pH ~8 con una solución acuosa de NaHC03 (2 ml). La mezcla se extrajo con diclorometano (5 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (3 ml). se secaron sobre Na2S04. se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0.05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 50 % - 80 %. 12 min). La mezcla se recogió y se liofilizó. El compuesto del título 2-[(2S)-1-[(E)-4-hidroxibut-2-enoil]-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6.8-dihidro-5H-pirido[3.4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (EJEMPL0517. 10.2 mg. 16.8 umol. 18 % de rendimiento. 98.5 % de pureza) se obtuvo en forma de un sólido de color blanquecino. LCMS [ESI. M+1]: 596.
Condición de SFC: Columna: Chiralcel 0D-350 x 4.6 mm D.I.. 3 um. Fase móvil: 40 % de metanol (0.05 % de DEA) en C02. Caudal: 3 ml/min. Longitud de onda: 220 nm.
1H RMN (400 MHz. cloroformo-d) ó = 7,73 - 7.61 (m. 2H). 7.46 - 7.30 (m. 2H). 7.27 - 7.16 (m. 2H). 7.06 (d. J = 14.8 Hz.
1H). 6.62 (d a. J = 13.6 Hz. 1H). 5.25 - 4.53 (m. 1H). 4.48 - 4.32 (m. 3H). 4.30 - 3.95 (m. 4H). 3.93 - 3.82 (m. 1H). 3.81 - 3.61 (m. 1H). 3.59 - 3.39 (m. 2H). 3.30 - 2.96 (m. 5H). 2.92 (s. 3H). 2.87 - 2.53 (m. 4H). 2.47 (d. J = 4.0 Hz. 3H). 2.34 - 2.21 (m. 1H). 2.13 - 1.99 (m. 1H). 1.98 - 1.82 (m. 3H).
Ejemplo 518

2-[(2S)-4-[2-[[(2R,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo

Etapa 1: (2R4R)-4-fluoropirrolidin-1,2-dicarboxilato de 01-terc-butil 02-metilo. A una solución de ácido (2R,4R)-1-terc-butoxicarbonil-4-fluoro-pirrolidin-2-carboxílico (300 mg, 1,29 mmol, 1,0 eq.) y K2C03 (533 mg, 3,86 mmol, 3,0 eq.) en DMF (2,0 ml) se le añadió MeI (4,80 g, 33,8 mmol, 2,11 ml, 26,3 eq.). La mezcla se agitó a 20 °C durante 12 horas. Después de que se completara, a la mezcla de reacción se le añadió agua (5,0 ml) y se extrajo con acetato de etilo (10,0 ml x 2). La capa orgánica se secó sobre Na2S04, se filtró y se concentró. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo: acetato de etilo = 10:1-1:1) para dar el producto (2R,4R)-4-fluoropirrolidin-1,2-dicarboxilato de 01-terc-butil 02-metilo (300 mg, 1,21 mmol, 94 % de rendimiento) en forma de un aceite de color amarillo.
1H RMN (400 MHz, Cloroformo-d) 55,30 - 5,10 (m, 1H), 4,58 - 4,37 (m, 1H), 3,94 - 3,76 (m, 1H), 3,75 (s, 3H), 3,72 -3,55 (m, 1H), 2,54 - 2,23 (m, 2H), 1,52 - 1,40 (m, 9H).
Etapa 2: [(2R4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metanol A una solución de (2R,4R)-4-fluorop¡rrol¡d¡n-1,2-d¡carbox¡lato de 01-terc-but¡l 02-met¡lo. (300 mg, 1,21 mmol, 1,0 eq.) en THF (3,0 ml) se le añad¡ó L¡AlH4 (138 mg, 3,64 mmol, 3,0 eq.) a -40 °C. La mezcla se ag¡tó a esta temperatura durante 1 hora, después se calentó a 70 °C y se ag¡tó a 70 °C durante 2 horas. Después de que se completara, la mezcla de reacc¡ón se ¡nact¡vó con Na2S04 acuoso saturado (0,30 ml) y después se f¡ltró. El l¡cor madre se recog¡ó y se concentró al vacío para dar el producto [(2R,4R)-4-fluoro-1- met¡l-p¡rrol¡d¡n-2-¡l]metanol (130 mg, 976 umol, 80 % de rend¡m¡ento) en forma de un ace¡te de color amar¡llo.
1H RMN (400 MHz, Cloroformo-d) 55,18 - 4,98 (m, 1H), 3,76 - 3,70 (m, 1H), 3,54 - 3,43 (m, 1H), 3,38 - 3,28 (m, 1H), 2,51 - 2,44 (m, 1H), 2,42 - 2,37 (m, 1H), 2,35 (s, 3H), 2,34 - 2,26 (m, 1H), 2,21 - 2,09 (m, 1H).
Etapa A: (2S)-2-(c¡anomet¡l)-4-[2-[[(2R.4R)-4-fluoro-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de benc¡lo. A una soluc¡ón de [(2R,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metanol (103 mg, 773 umol, 2,0 eq.) y t-Bu0Na (55,8 mg, 580 umol, 1,50 eq.) en tolueno (2,0 ml) se le añad¡ó (2S)-2- (c¡anomet¡l)-4-[7-(8-met¡l-1-naft¡l)-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (230 mg, 387 umol, 1,0 eq.). La mezcla se ag¡tó a 0 °C durante 0,5 horas. Después de que se completara, a la mezcla de reacc¡ón se le añad¡ó agua (10,0 ml) y se extrajo con acetato de et¡lo (10,0 ml * 3). La capa orgán¡ca se secó sobre Na2S04, se f¡ltró y se concentró. El res¡duo se pur¡f¡có por HPLC ultrarráp¡da de fase ¡nversa (C18, 0,1 % de AF en agua, 0 - 60 % de MeCN) para dar el producto (2S)-2-(c¡anomet¡l)-4-[2-[[(2R.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (210 mg, 316 umol, 82 % de rend¡m¡ento) en forma de un sól¡do de color amar¡llo. lCMs [ESI, M+1 ]: 664.
1H RMN (400 MHz, Cloroformo-d) 57,70 (d a, J = 8,4 Hz, 1H), 7,65 (t, J = 8 ,6 Hz, 1H), 7,45 - 7,32 (m, 7H), 7,26 - 7,17 (m, 2H), 5,24 - 5,02 (m, 3H), 4,73 - 4,61 (m, 1H), 4,53 - 4,42 (m, 1H), 4,30 - 4,18 (m, 2H), 4,11 - 3,96 (m, 2H), 3,94 -3,81 (m, 1H), 3,81 - 3,67 (m, 1H), 3,58 - 3,47 (m, 1H), 3,46 - 3,29 (m, 2H), 3,24 - 3,06 (m, 3H), 3,05 - 2,96 (m, 1H), 2,92 (s, 3H), 2,82 - 2,71 (m, 2H), 2,70 - 2,51 (m, 2H), 2,50 - 2,47 (m, 4H), 2,45 - 2,35 (m, 1H), 2,12 - 2,05 (m, 1H).
Etapa B: 2-[(2S)-4-[2-[[(2R.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-7-(8-met¡l-1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una soluc¡ón de (2S)-2-(c¡anomet¡l)-4-[2-[[(2R,4R)-4-fluoro-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (200 mg, 301 umol, 1,0 eq.) y NHâ Me0H (2,0 ml, 20 % de pureza) en metanol (4,0 ml) se le añad¡ó Pd/C (80 mg, 10 % de pureza). La mezcla se purgó 3 veces con N2 y después se ag¡tó en una atmósfera de H2 (103,42 kPa (15 ps¡)) a 25 °C durante 1 hora. Después de que se completara, la mezcla de reacc¡ón se f¡ltró a través de Cel¡te. El l¡cor madre se concentró al vacío para dar el producto 2-[(2S)-4-[2-[[(2R,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (140 mg, 264 umol, 88 % de rend¡m¡ento) en forma de un ace¡te de color amar¡llo. El producto se usó en la s¡gu¡ente etapa d¡rectamente s¡n pur¡f¡cac¡ón ad¡c¡onal. LCMS [ESI, M+1]: 664.
Etapa C: 2-[(2S)-4-[2-[[(2R.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-7-(8-met¡l-1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-1-(2-fluoroprop-2-eno¡l)p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una soluc¡ón de 2-[(2S)-4-[2-[[(2R,4R)-4-fluoro-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡ljaceton¡tr¡lo (120 mg, 226 umol, 1,0 eq.), ác¡do 2-fluoroprop-2-eno¡co (61,2 mg, 680 umol, 3,0 eq.) y DIEA (176 mg, 1,36 mmol, 238 ul, 6,0 eq.) en DCM (3,0 ml) se le añad¡ó HATU (258,44 mg, 679,69 umol, 3 eq.). La mezcla se ag¡tó a 0 °C durante 0,5 horas. Después de que se completara, a la mezcla de reacc¡ón se le añad¡ó agua (5,0 ml) y se extrajo con acetato de et¡lo (5,0 ml * 3). La capa orgán¡ca se secó sobre Na2S04, se f¡ltró y se concentró al vacío. El res¡duo se pur¡f¡có por HPLC prep. (columna: Gem¡n¡ 150 * 255 u; fase móv¡l: [agua (0,05 % de h¡dróx¡do de amon¡aco v/v)-ACN]; % de B: 50 %-80 %,12 m¡n) para dar el compuesto del título 2-[(2s)-4-[2-[[(2R,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-(2-fluoroprop-2-eno¡l)p¡peraz¡n-2-¡l]aceton¡tr¡lo (EJEMPL0 518, 37,9 mg, 60,1 umol, 27 % de rend¡m¡ento, 96 % de pureza) en forma de un sól¡do de color blanco. LCMS [ESI, M+1]: 602.
1H RMN (400 MHz, Cloroformo-d) 57,70 (d a, J = 8,0 Hz, 1H), 7,65 (t, J = 8,2 Hz, 1H), 7,45 - 7,37 (m, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,27 - 7,18 (m, 2H), 5,53 - 5,32 (m, 1H), 5,30 - 5,22 (m, 1H), 5,21 - 5,03 (m, 1H), 4,97 - 4,68 (m, 1H), 4,52 - 4,42 (m, 1H), 4,29 - 4,20 (m, 2H), 4,18 - 4,03 (m, 2H), 3,95 - 3,68 (m, 2H), 3,60 - 3,41 (m, 2H), 3,39 - 3,30 (m, 1H), 3,26 - 3,15 (m, 2H), 3,15 - 2,95 (m, 2H), 2,92 (s, 3H), 2,90 - 2,54 (m, 4H), 2,53 - 2,46 (m, 4H), 2,45 - 2,35 (m, 1H), 2,13 - 1,97 (m, 1H).
Ejemplo 519

2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[2-[[(2R,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

Etapa 1: (2R4R)-4-metoxipirrolidin-1,2-dicarboxilato de 01-ferc-butil 02-metilo. A una solución de (2R,4R)-4-hidroxipirrolidin-1,2-dicarboxilato de 01-ferc-butil 02-metilo (2,1 g, 8,56 mmol, 1 eq.) en MeCN (42 ml) se le añadieron Ag20 (5,95 g, 25,7 mmol, 3 eq.) y CH3I (7,75 g, 54,6 mmol, 3,40 ml, 6,38 eq.). La mezcla de reacción se agitó a 33 °C durante 12 horas. La mezcla de reacción se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía en columna (S02, éter de petróleo/acetato de etilo = 8/1 a 5/1) para dar (2R,4R)-4-metoxipirrolidin-1,2-dicarboxilato de 01-ferc-butil 02-metilo (1,88 g, 6,89 mmol, 80 % de rendimiento, 95,0 % de pureza) en forma de un aceite incoloro.
1H RMN (400 MHz, cloroformo-d) 5 = 4,45 - 4,26 (m, 1H), 3,96 - 3,89 (m, 1H), 3,72 (s, 3H), 3,68 - 3,55 (m, 1H), 3,54 -3,43 (m, 1H), 3,27 (s, 3H), 2,38 - 2,17 (m, 2H), 1,51 - 1,37 (m, 9H).
Etapa 2: [(2R,4R)-4-metoxi-1-metil-pirrolidin-2-il]metanol. A la solución de (2R,4R)-4-metoxipirrolidin-1,2-dicarboxilato de 01-ferc-butil 02-metilo (2,98 g, 11,5 mmol, 1 eq.) en THF (60 ml) se le añadió LiAlH4 (872 mg, 23,0 mmol, 2 eq.) a 0 °C y la mezcla se agitó a 0 °C durante 1 hora. Después, la mezcla de reacción se calentó a 80 °C y se agitó durante 2 horas. La mezcla se inactivó con Na2S04 acuoso saturado (3 ml), después se filtró y la torta de filtro se lavó con THF (3 x 20 ml). El filtrado se concentró al vacío para dar [(2R,4R)-4-metoxi-1-metil-pirrolidin-2-il]metanol (1,53 g, 10,0 mmol, 87 % de rendimiento, 95,0 % de pureza) en forma de un aceite de color pardo que se usó para la siguiente etapa sin purificación adicional.1
1H RMN (400 MHz, cloroformo-d) 5 = 3,82 - 3,76 (m, 1H), 3,69 (dd, J = 3,2, 11,2 Hz, 1H), 3,45 (dd, J = 1,6, 10,8 Hz, 1H), 3,29 (s, 3H), 3,18 (d, J = 10,4 Hz, 1H), 2,62 (s a, 1H), 2,42 (tdd, J = 2,8, 6,4, 9,2 Hz, 1H), 2,38 - 2,33 (m, 1H), 2,32 (s, 3H), 2,16 (ddd, J = 6,8, 8,8, 14,0 Hz, 1H), 1,92 (tdd, J = 1,6, 6,8, 14,0 Hz, 1H).
Etapa A: (2S)-2-(cianometil)-4-[2-[[(2R4R)-4-metoxi-1-metilpirrolidin-2-il1metoxi1-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3.4-d1pirimidin-4-il]piperazin-1-carboxilato de bencilo. A la solución de [(2R,4R)-4-metoxi-1-metil-pirrolidin-2-il]metanol (1,37 g. 9,42 mmol, 2.8 eq.) en tolueno (40 ml) se le añadió f-Bu0Na (970 mg. 10,1 mmol, 3 eq.) a 0 °C. después se añadió (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-metilsulfinil-6.8-dihidro-5H-pirido[3.4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (2 g. 3,36 mmol. 1 eq.) a 0 °C y la mezcla de reacción se agitó a 0 °C durante 0.5 horas. A la mezcla se le añadió agua (20 ml). La mezcla se diluyó con EtOAc (10 ml) y se extrajo con EtOAc (2 x 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro. se filtraron y se concentraron al vacío. El residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0.1 % de AF) = 57 %) para dar (2S)-2-(cianometil)-4-[2-[[(2R,4R)-4-metoxi-1-metilpirrolidin-2-il1metoxi1-7-(8-metil-1-naftil)-6.8-dihidro-5H-pirido[3.4-d1pirimidin-4-il]piperazin-1-carboxilato de bencilo (1.8 g.2,53 mmol. 75 % de rendimiento. 95,0 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]:
676.
1H RMN (400 MHz, cloroformo-d) 5 = 7,73 - 7,60 (m. 2H). 7,47 - 7,31 (m. 7H). 7,27 - 7,16 (m. 2H). 5,21 (s. 2H). 4,67 (s a. 1H).4,45 (dd a. J = 4.4. 10,0 Hz. 1H).4,24 - 4,16 (m.2H). 4,10 - 3,96 (m. 2H). 3,96 - 3,65 (m. 3H). 3,59 - 3,40 (m.
2H). 3,29 (d. J = 1.6 Hz. 3H). 3,25 - 3,06 (m. 4H). 3,00 (dt. J = 3.6. 12,4 Hz. 1H). 2,91 (s. 3H). 2,81 - 2,55 (m.4H). 2,45 (d. J = 6.0 Hz. 3H). 2,42 - 2,29 (m. 2H). 1,88 - 1.68 (m. 2H).
Etapa B: 2-[(2S)-4-[2-[[(2R4R)-4-metoxi-1-metil-pirrolidin-2-il1metoxi1-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3.4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A la solución de (2S)-2-(cianometil)-4-[2-[[(2R,4R)-4-metoxi-1-metil-pirrolidin-2-il1metoxi1-7-(8-metil-1-naftil)-6.8-dihidro-5H-pirido[3.4-d1pirimidin-4-il]piperazin-1-carboxilato de bencilo (2 g.
2,96 mmol. 1 eq.) y NHs^Me0H (20 ml. 20 % de pureza) en Me0H (40 ml) se le añadió Pd/C (1 g. 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 20 °C durante 0.5 horas. La mezcla de reacción se filtró. el filtrado se concentró al vacío para dar 2-[(2S)-4-[2-[[(2R.4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi1-7-(8-metil-1-naftil)-6.8-dihidro-577-pirido[3.4-d1pirimidin-4-il1piperazin-2-il]acetonitrilo (1,29 g. 2 .26 mmol. 76 % de rendimiento. 95 % de pureza) en forma de un sólido de color amarillo que se usó para la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]:
542.
Etapa C: 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[2-[[(2R4R)-4-metoxi-1-metilpirrolidin-2-il1metoxi1-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3.4-d1pirimidin-4-il]piperazin-2-il)acetonitrilo. A la solución de 2-[(2S)-4-[2-[[(2R,4R)-4-metoxi-1-metilpirrolidin-2-il1metoxi1-7-(8-metil-1-naftil)-6.8-dihidro-5/7-pirido[3.4-d1pirimidin-4-il]piperazin-2-i¡1acetonitrilo
(150 mg. 277 umol, 1 eq.). ácido 2-fluoroprop-2-enoico (74,8 mg. 831 umol, 3 eq.) y t Ea (336 mg. 1.66 mmol. 462 ul.
50 % de pureza. 6 eq.) en EtOAc (3 ml) se le añadió T3p (264 mg. 831 umol.247 ul. 3 eq.) a 0 °C y la mezcla se agitó a 0 °C durante 0.5 horas. A la mezcla se le añadió agua (4 ml). La mezcla se extrajo con EtOAc (3 x 8 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro. se filtraron y se concentraron al vacío. El residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0.1 % de AF) = 32 %). Después. el residuo se purificó por HPLC prep. (columna: Gemini 150 * 255 ufase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN];% de B: 55 %-85 % ,12 min) para dar el compuesto del título 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[2-[[(2R,4R)-4-metoxi-1-metil-pirrolidin-2-il1metoxi1-7-(8-metil-1-naftil)-6.8-dihidro-5H-pirido[3.4-d1pirimidin-4-il1piperazin-2-il]acetonitrilo (EJEMPL0 519, 59,5 mg. 97,0 umol. 35 % de rendimiento. 100 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 614.
1H RMN (400 MHz. cloroformo-d) 5 = 7,70 (d a, J = 8.4 Hz. 1H). 7,65 (t. J = 8.0 Hz. 1H). 7,45 - 7,38 (m. 1H). 7,34 (t. J = 7.6 Hz. 1H). 7,27 - 7,17 (m. 2H). 5,53 - 5,33 (m. 1H). 5,26 (dd. J = 3.6. 16,8 Hz. 1H). 4,88 (s a. 1H). 4,50 - 4,41 (m.
1H). 4,30 - 3,99 (m. 4H). 3,96 - 3,71 (m. 3H). 3,59 - 3,40 (m. 2H). 3,30 (s. 3H). 3,25 - 3,15 (m. 3H). 3,14 - 2,94 (m. 2H).
2,92 (s. 3H). 2,90 - 2,74 (m. 2H). 2,73 - 2,55 (m. 2H). 2,45 (d. J = 5.6 Hz. 3H). 2,42 - 2,29 (m. 2H). 1.81 (dd a. J = 7.2. 14,4 Hz. 1H).
Ejemplo 520

2-[(2S)-4-[2-[[(2SJ-4,4-difluoro-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(E)-4-(3-fluoroazetidin-1-il)but-2-enoil]piperazin-2-il]acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[2-[[(2S)-4,4-difluoro-1-metilpirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3.4-d1pirimidin-4-il]piperazin-1-carboxilato de bencilo. A una solución de [(2S)-4,4-difluoro-1 -metil-pirrolidin-2-il]metanol (609,98 mg, 4,04 mmol, 3,0 eq.) y í-Bu0Na (388 mg, 4,04 mmol, 3,0 eq.) en tolueno (2,0 ml) se le añadió gota a gota a gota una solución de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (800 mg, 1,35 mmol, 1,0 eq.) en tolueno (4,0 ml) a 0 °C. La mezcla de reacción se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se lavó con agua (15,0 ml) y se extrajo con acetato de etilo (3 * 20,0 ml). Los extractos combinados se lavaron con salmuera (50,0 ml), se secaron con Na2S04, se filtraron y el disolvente se eliminó al vacío. El residuo se purificó por HPLC ultrarrápida de fase inversa [C18, 0,1 % de AF en agua, 0-80 % de MeCN]. Después, el producto obtenido se concentró, la capa acuosa se extrajo con acetato de etilo (3 * 50,0 ml) y la capa orgánica combinada se secó sobre Na2S04, se filtró y se concentró. El compuesto (2S)-2-(cianometil)-4-[2-[[(2S)-4,4-difluoro-1 -metil-pirrolidin-2-il]metoxi]-7-(8-metil-1 -naftil)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (750 mg, 1,08 mmol, 80,1 % de rendimiento, 98 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 682.
Etapa B: 2-[(2S)-4-[2-[[(2S)-4,4-difluoro-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-il]piperazin-2-il]acetonitrilo. Se añadieron (2S)-2-(cianometil)-4-[2-[[(2S)-4,4-difluoro-1-metil-pirrolidin-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (650 mg, 953 umol, 1,0 eq.) y Pd/C (200 mg, 10 % de pureza) a Me0H (8 ,0 ml) y NH3 Me0H (8 ,0 ml, 20 % de pureza), después la mezcla se desgasificó y se purgó 3 veces con H2 y después la mezcla se agitó a 20 °C durante 0,5 horas en una atmósfera de H2 (103,42 kPa (15 psi)). Después de que se completara, la mezcla bruta se filtró a través de una capa de celite. La torta se lavó con Me0H (50,0 ml) y el filtrado se secó a alto vacío. El compuesto 2-[(2S)-4-[2-[[(2S)-4,4-d¡fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]acetonitrilo (420 mg, 736 umol, 77,2 % de rendimiento, 96 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 548.
Etapa C: 2-[(2S)-1-[(E)-4-bromobut-2-enoil1-4-[2-[[(2S)-4,4-difluoro-1-metilpirrolidin-2-il1metoxi1-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-inpiperazin-2-inacetonitrilo. A una solución de 2-[(2S)-4-[2-[[(2S)-4,4-difluoro-1-metilpirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (420 mg, 822 umol, 1 ,0 eq.) y piridina (520 mg, 6,57 mmol, 531 ul, 8 ,0 eq.) en Dc M (6 ,0 ml) se le añadió cloruro de (E)-4-bromobut-2-enoílo (603 mg, 3,29 mmol, 4,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se concentró al vacío. El producto 2-[(2S)-1-[(E)-4-bromobut-2-enoil]-4-[2-[[(2S)-4,4-difluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (500 mg, bruto) se obtuvo en forma de un aceite de color amarillo. El producto bruto se usó directamente para la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 695.
Etapa D: 2-[(2S)-4-[2-[[(2S)-4,4-difluoro-1-metil-pirrolidin-2-il1metoxi1-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-il1-1-[(E)-4-(3-fluoroazetidin-1-il)but-2-enoillpiperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-1-[(E)-4-bromobut-2-eno¡l]-4-[2-[[(2S)-4,4-d¡fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naftil)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]aceton¡tr¡lo (500 mg, 720 umol, 1,0 eq.) y K2C03 (995 mg, 7,20 mmol, 10,0 eq.) en MeCN (12,0 ml) se le añadió 3-fluoroazetidina (482 mg, 4,32 mmol, 6,0 eq., HCl) a 0 °C. La mezcla se agitó a 20 °C durante 1 hora. Después de que se completara, el disolvente orgánico se lavó con agua (10,0 ml). La fase acuosa se extrajo con acetato de etilo (3 * 10 ml). Los extractos combinados se lavaron con salmuera (20,0 ml), se secaron con Na2S04 y después el disolvente se eliminó al vacío. El residuo se purificó por cromatografía en columna (Base Ah03, éter de petróleo : acetato de etilo = 3:1 a acetato de etilo : metanol = 100:1) y después el producto bruto se concentró y se purificó por HPLC prep. (columna: Waters Xbridge 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN];% de B: 55 %-79 %, 10 min];% de B: 45 %-75 %, 12 min) y se sometió a liofilización. El compuesto del título 2-[(2S)-4-[2-[[(2SJ-4,4-d¡fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]-1-[(E)-4-(3-fluoroazet¡d¡n-1-¡l)but-2-eno¡l]p¡perazin-2-¡l]aceton¡tr¡lo (EJEMPL0520, 45,8 mg, 66,5 umol, 9,24 % de rendimiento, 100 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1 ]: 689.
1H RMN (400 MHz, cloroformo-d) ó = 7,74 - 7,63 (m, 2H), 7,47 - 7,32 (m, 2H), 7,27 - 7,18 (m, 2H), 6,88 (d a, J = 15,3 Hz, 1H), 6,42 (d a, J = 15,4 Hz, 1H), 5,27 - 5,05 (m, 1H), 4,64 (s a, 1H), 4,48 - 4,41 (m, 1H), 4,31 - 4,18 (m, 2H), 4,16 - 3,95 (m, 2H), 3,94 - 3,84 (m, 1H), 3,82 - 3,66 (m, 3H), 3,54 (d a, J = 7,6 Hz, 1H), 3,50 - 3,36 (m, 2H), 3,34 (d a, J = 3,8 Hz, 2H), 3,30 - 3,24 (m, 1H), 3,24 - 3,08 (m, 4H), 3,07 - 2,95 (m, 2H), 2,92 (s, 3H), 2,86 - 2,76 (m, 1H), 2,76 - 2,57 (m, 3H), 2,57 - 2,47 (m, 1H), 2,46 (d, J = 4,5 Hz, 3H), 2,34 - 2,17 (m, 1H).
Ejemplo 521

2-[(2S)-4-[7-(5,6-d¡met¡l-1H-¡ndazol-4-¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡mid¡n-4-¡l]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo

Etapa A: 2-[(2S)-4-[7-(5.6-dimetil-1H-indazol-4-in-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-piridor[3.4-dlpirimidin-4-illpiperazin-2-il1acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(5,6-dimetil-1-tetrahidropiran-2-ilindazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (720 mg, 1,03 mmol, 1 eq.) en DCM (1 ml) se le añadió TFA (4,62 g, 40,5 mmol, 3 ml, 39,4 eq.). La mezcla se agitó a 20 °C durante 4 horas. Después de que se completara, la mezcla se diluyó con DCM (10 ml) y se neutralizó con NaHCÜ3 acuoso saturado. Después, la fase acuosa se extrajo con EtOAc (2 x 30 ml). Las capas orgánicas combinadas se secaron sobre Na2SÜ4 y se concentraron al vacío para dar 2-[(2S)-4-[7-(5,6-dimetil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (880 mg, bruto) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional.
Etapa B: 2-[(2S)-4-[7-(5.6-dimetil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6.8-dihidro-5H-pirido[3.4-d1pirimidin-4-il1-1-(2-fluoroprop-2-enoil)piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(5,6-dimetil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (500 mg, bruto), ácido 2-fluoroprop-2-enoico (61,1 mg, 679 umol) y TEA (294 mg, 2,91 mmol, 405 ul) en DMF (10 ml) se le añadió t 3p (926 mg, 1,45 mmol, 865 ul, 50 % de pureza en EtOAc) a -40 °C. Después, la mezcla se agitó a -40 °C durante 0,5 horas y a 0 °C durante 0,5 horas más. Después de que se completara, la mezcla se diluyó con agua (20 ml) y se extrajo con EtOAc (2 x 40 ml). Las capas orgánicas se lavaron con salmuera (50 ml), se secaron sobre Na2SÜ4 y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. Las fracciones deseadas se recogieron, se neutralizaron con NaHCÜ3 acuoso saturado y se extrajeron con EtüAc (2 x 70 ml). Las capas orgánicas se secaron sobre Na2SÜ4 y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi C18 150 * 30 mm * 4 um; fase móvil: [agua (0,225 % de AF)-ACN]; % de B: 10 %-40 %, 12 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[(2S)-4-[7-(5,6-dimetil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (EJEMPLÜ 521,52,5 mg, 79,8 umol, dos etapas 15 % de rendimiento, 96,4 % de pureza, AF) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 588.
Condición de SFC: Columna: Chiralcel ÜJ-3100 x 4,6 mm D.I., 3 um, Fase móvil: metanol (0,05 % de DEA) en CÜ2 de 5 % a 40 %, Caudal: 3 ml/min, Longitud de onda: 220 nm.
1H RMN (400 MHz, cloroformo-d) 5 = 8,03 (s, 1H), 7,15 (s, 1H), 5,53 - 5,34 (m, 1H), 5,25 (dd a, J = 3,2, 16,8 Hz, 1H), 5,02 - 4,61 (m, 3H), 4,43 (dd, J = 4,8, 11,6 Hz, 1H), 4,27 (s, 2H), 4,19 (d a, J = 13,2 Hz, 1H), 4,01 (d a, J = 12,4 Hz, 1H), 3,68 - 3,56 (m, 1H), 3,55 - 3,50 (m, 2H), 3,49 - 3,21 (m, 3H), 3,14 (s a, 1H), 2,98 (s a, 1H), 2,91 - 2,61 (m, 7H), 2,41 (s, 3H), 2,34 (s, 3H), 2,23 (cd, J = 8,4, 12,4 Hz, 1H), 2,14 - 1,95 (m, 3H).
Ejemplo 522

2-((S)-4-(2-(((2R,4R)-4-fluoro-1-met¡lp¡rrol¡d¡n-2-il)metox¡)-7-(8-met¡lnaftalen-1-¡l)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]pirimidin-4-il)-1-((£)-4-hidroxibut-2-enoil)piperazin-2-il)acetonitrilo

Etapa A: 2-[(2S)-4-[2-[[(2R,4R)-4-fluoro-1-metil-pirrolidin-2-il1metoxi1-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-il1-1-[(E)-4-tritiloxibut-2-enoillpiperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[2-[[(2E,4E)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (200 mg, 378 umol, 1,0 eq.), ácido (E)-4-tritiloxibut-2-enoico (169 mg, 491 umol, 1,3 eq.) y DIEA (293 mg, 2,27 mmol, 395 ul, 6,0 eq.) en DCM (3,0 ml) se le añadió HATU (431 mg, 1,13 mmol, 3,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, a la mezcla se le añadió agua (10 ml) y se extrajo con acetato de etilo (2 * 10,0 ml). La capa orgánica se secó sobre Na2SÜ4, se filtró y se concentró. El residuo se purificó por cromatografía en columna (SiÜ2, éter de petróleo/acetato de etilo = 3/1 a acetato de etilo : metanol = 20:1). El producto 2-[(2S)-4-[2-[[(2R,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]-1-[(E)-4-tr¡t¡lox¡but-2-eno¡l]p¡peraz¡n-2-il]aceton¡tr¡lo (290 mg, 339 umol, 90 % de rendimiento) se obtuvo en forma de un sólido de color amarillo.
Etapa B: 2-[(2S)-4-[2-[[(2R4R)-4-fluoro-1-metil-pirrolidin-2-i n metox n -7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-[(E)-4-hidroxibut-2-enoil1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[2-[[(2E,4E)-4-fluoro-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-[(E)-4-tr¡t¡lox¡but-2-eno¡l]p¡perazin-2-¡l]aceton¡tr¡lo (290 mg, 339 umol, 1,0 eq.) en diclorometano (1,5 ml) se le añadió TFA (2,31 g, 20.3 mmol, 1,5 ml, 59,8 eq.) y la mezcla se agitó a 20 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se concentró, después se añadió diclorometano (10,0 ml), la capa orgánica se lavó con NaHCÜ3 acuoso saturado (2 * 10,0 ml) y la capa orgánica se secó sobre Na2SÜ4, se filtró y se concentró. El residuo se purificó por cromatografía en columna (Base Al2Ü3, éter de petróleo/acetato de etilo = 3/1 a acetato de etilo: metanol = 20:1). Después, el producto bruto se purificó de nuevo por HPLC prep. (columna: Gemini 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN];% de B: 45 %-75 %,12 min) y el producto obtenido se concentró y se sometió a liofilización. El compuesto del título 2-[(2S)-4-[2-[[(2R,4R)-4-fluoro-1-metil-p¡rrol¡d¡n-2-¡l]metox¡]-7-(8-metil-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1-[(E)-4-h¡drox¡but-2-eno¡l]p¡peraz¡n-2-¡l]aceton¡trilo (Ej Em PL0 522, 61.3 mg, 99,9 umol, 29 % de rendimiento, 99 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 614.
1H RMN (400 MHz, cloroformo-d) ó 7,73 - 7,61 (m, 2H), 7,46 - 7,31 (m, 2H), 7,27 - 7,18 (m, 2H), 7,06 (d a, J = 14,8 Hz, 1H), 6,62 (d a, J = 14,8 Hz, 1H), 5,27 - 4,97 (m, 1H), 4,74 - 4,55 (m, 1H), 4,52 - 4,34 (m, 3H), 4,31 - 4,16 (m, 2H), 4,15 - 3,96 (m, 2H), 3,94 - 3,58 (m, 3H), 3,56 - 3,48 (m, 1H), 3,46 - 3,41 (m, 1H), 3,36 - 3,29 (m, 1H), 3,26 - 3,07 (m, 3H), 3,04 - 2,97 (m, 1H), 2,92 (s, 3H), 2,85 - 2,57 (m, 4H), 2,49 (d, J = 4,8 Hz, 3H), 2,45 - 2,34 (m, 1H), 2,29 - 1,91 (m, 2H).
Ejemplo 523

2-[(2S)-1-[(E)-4-hidroxibut-2-enoil]-4-[2-[[(2R,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

Etapa A: 2-[(2S)-4-[2-[[(2R4E)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-[(E)-4-tritiloxibut-2-enoil1piperazin-2-il]acetonitrilo. A la solución de 2-[(2S)-4-[2-[[(2E,4E)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (200 mg, 369 umol, 1 eq.), ácido (E)-4-tritiloxibut-2-enoico (254 mg, 738 umol, 2 eq.) y TEA (374 mg, 3,69 mmol, 514 ul, 10 eq.) en EtOAc (4 ml) se le añadió T3P (705 mg, 1,11 mmol, 659 ul, 50 % de pureza, 3 eq.) a 0 °C y la mezcla se agitó a 0 °C durante 0,5 horas. A la mezcla se le añadió agua (10 ml). La mezcla se diluyó con EtOAc (5 ml) y se extrajo con EtOAc (15 ml). La capa orgánica se lavó con salmuera (10 ml), se secó sobre Na2S04 anhidro, se filtró y se concentró al vacío. El residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0,1 % de AF) = 70 %) para dar 2-[(2S)-4-[2-[[(2E,4E)-4-metoxi-1 -metil-pirrolidin-2-il]metoxi]-7-(8-metil-1 -naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(E)-4-tritiloxibut-2-enoil]piperazin-2-il]acetonitrilo (230 mg, 262 umol, 71 % de rendimiento, 99 % de pureza) en forma de un sólido de color amarillo.
Etapa B: 2-[(2S)-1-[(E)-4-hidroxibut-2-enoil-4-[2-[[(2R4E)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,9-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo. A la solución de 2-[(2S)-4-[2-[[(2E,4E)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(E)-4-tritiloxibut-2-enoil]piperazin-2-il]acetonitrilo (230 mg, 265 umol, 1 eq.) en DCM (2,3 ml) se le añadió t Fa (3,54 g, 31,1 mmol, 2,3 ml, 117 eq.) y la mezcla se agitó a 15 °C durante 1 hora. La mezcla de reacción se basificó con NaHC03 saturado (30 ml) a pH = 7-8 y después se extrajo con DCM (2 * 15 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0,1 % de AF) = 32 %). Después, el residuo se purificó por HPLC prep. (columna: Gemini 150 * 255 ufase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN];% de B: 37 %-67 %,12 min) para dar el compuesto del título 2-[(2S)-1-[(E)-4-hidroxibut-2-enoil]-4-[2-[[(2E,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (EJEMPL0 523, 41,7 mg, 66,6 umol, 25 % de rendimiento, 100 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 626.
1H RMN (400 MHz, cloroformo-d) ó = 7,70 (d a, J = 8,4 Hz, 1H), 7,65 (t, J = 8,0 Hz, 1H), 7,45 - 7,38 (m, 1H), 7,37 -7,31 (m, 1H), 7,27 - 7,17 (m, 2H), 7,07 (d a, J = 14,8 Hz, 1H), 6,62 (d a, J = 14,0 Hz, 1H), 5,07 (s a, 1H), 4,85 - 4,55 (m, 1H), 4,51 - 4,37 (m, 3H), 4,30 - 3,97 (m, 4H), 3,94 - 3,65 (m, 2H), 3,58 - 3,40 (m, 2H), 3,29 (d, J = 1,6 Hz, 3H), 3,25 - 2,96 (m, 5H), 2,92 (s, 3H), 2,86 - 2,54 (m, 4H), 2,45 (d, J = 4,8 Hz, 3H), 2,42 - 2,28 (m, 2H), 1,81 (dd a, J = 8,9, 13,6 Hz, 1H).
Ejemplo 524

2-((S)-4-(7-(8-cloronaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-metoxibut-2-enoil)piperazin-2-il)acetonitrilo

Etapa A: 2-((S)-4-(7-(8-cloronaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-metoxibut-2-enoil)piperazin-2-il)acetonitrilo: Una mezcla agitada de 2-((S)-4-(7-(8-cloronaftalen-1-il)-2-(((S)-1 -metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (100 mg, 0,1879 mmol), ácido (E)-4-metoxibut-2-enoico (32,73 mg, 0 ,2819 mmol) y N,N-dimetilformamida (1 ml, 12,79 mmol) se enfrió a 0 °C seguido de la adición de trietilamina (0,07859 ml, 0,5638 mmol) y 2,4,6-trióxido de 2,4,6-tripropil-1,3,5,2,4,6-trioxatrifosfinano, al 50 % en EtOAc (0,1678 ml, 0,2819 mmol). La mezcla de reacción se agitó durante 2 horas mientras se calentaba a temperatura ambiente. La mezcla se repartió entre Na2C030,5 M (5 ml) y EtOAc (15 ml) y los extractos orgánicos se separaron. Luego, los extractos orgánicos se lavaron con agua y salmuera (5 ml de cada uno), se secaron sobre Na2S04 y se evaporaron al vacío. Luego, el residuo se cromatografió sobre gel de sílice usando 4 % de Me0H 0,4 % de NH40H/DCM como eluyente para dar el material impuro que se purificó adicionalmente por HPLC prep. inversa Gilson eluyendo con 5 a 75 % de ACN 0,1 % de TFA /H20 0,1 % de TFA. Las fracciones que contenían el producto puro se combinaron y se repartieron entre DCM y una solución saturada de Na2C03 y las capas se separaron. Luego, los extractos orgánicos se lavaron con salmuera, se secaron sobre MgS04 y se concentraron al vacío para dar el compuesto del título puro (EJEMPL0524, 33 mg, 28 %). ESI+APCI MS m/z 630,3 [M+].
Ejemplo 525

N-(3-((S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-3-oxoprop-1-en-2-il)acetamida

Etapa A: N-(3-((S)-2-(cianometi0-4-(7-(8-metilnaftalen-1-i0-2-(((S)-1-metilpirrolidin-2-i0metoxi)-5,6.7.8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-3-oxoprop-1-en-2-il)acetamida: A un matraz de fondo redondo de 25 ml que contenía diclorometano (2932 μl, 0,2932 mmol) enfriado a 0 °C se le añadieron 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (150 mg.
0,2932 mmol) y base de Hunig (307,2 μl, 1,759 mmol). La mezcla de reacción se agitó vigorosamente mientras se añadía en una porción ácido 2-acetamidoacrílico (227,1 mg, 1,759 mmol). Después, a la mezcla en agitación se le añadió lentamente anhídrido cíclico del ácido 1-propanofosfónico (1047 μl, 1,759 mmol). La reacción se agitó durante 2 horas a 0 °C. La reacción se trató con agua básica y la capa acuosa se extrajo con EtOAc (3 x). Los extractos orgánicos combinados se concentraron al vacío y se purificaron en el Gilson (HPLC prep.) eluyendo con 5^95 % de ACN 0,1 % de TFA /agua 0,1 % de TFA. Las fracciones que contenían el producto se combinaron y se basificaron con Na0H 1 M y la capa acuosa se extrajo con DCM (2 x). Los extractos orgánicos se secaron sobre Na2S04 y se concentraron al vacío para dar el compuesto del título N-(3-((S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-3-oxoprop-1-en-2-il)acetamida (EJEMPL0525, 124,2 mg, 68,03 % de rendimiento). ESI+APCI MS m/z 623,3 [M+H]+.
Ejemplo 526

2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-3-(piridin-4-il)acriloil)piperazin-2-il)acetonitrilo
2- ((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)-1-((E)-3-(piridin-4-il)acriloil)piperazin-2-il)acetonitrilo: El compuesto del título se preparó siguiendo el Ejemplo 525 sustituyendo ácido trans-3-(4-piridil)acrílico por ácido 2-acetamidoacrílico en la Etapa A. ESI+APCI MS m/z 643,3 [M+H]+.
Ejemplo 527

2-((S)-1-(2-fluoroacriloil)-4-(7-(6-metil-5-(trifluorometil)-1H-indazol-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

1- bromo-5-fluoro-3-metil-2-(trifluorometil)benceno. A una solución de 1-bromo-5-fluoro-2-yodo-3-metil-benceno (2 g, 6,35 mmol, 1 eq.) en DMF (40 ml) se le añadieron CuI (7,26 g, 38,1 mmol, 6 eq.) y 2,2-difluoro-2-fluorosulfonil-acetato de metilo (7,32 g, 38,1 mmol, 4,85 ml, 6 eq.). La mezcla se agitó a 60 °C durante 12 horas. La mezcla de reacción se diluyó con acetato de etilo (20 ml) y se lavó con agua (20 ml * 3). La capa orgánica se lavó con salmuera (20 ml), se secó sobre Na2SÜ4, se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiÜ2, éter de petróleo/acetato de etilo = 1/0 a 10/1). Se obtuvo 1-bromo-5-fluoro-3-metil-2-(trifluorometil)benceno (1,3 g, 3,74 mmol, 59 % de rendimiento, 74 % de pureza) en forma de un aceite de color amarillo.
1H RMN (400 MHz, cloroformo-d) 5 = 7,33 (dd, J = 2,4, 7,6 Hz, 1H), 6,95 (dd, J = 2,0, 8,8 Hz, 1H), 2,54 (c, J = 3,6 Hz, 3H).
2- bromo-6-fluoro-4-metil-3-(trifluorometil)benzaldehído. A una mezcla de 1-bromo-5-fluoro-3-metil-2-(trifluorometil)benceno (900 mg, 3,50 mmol, 1 eq.) en THF (20 ml) se le añadió LDA (2 M en THF, 3,50 ml, 2 eq.) a -60 °C en una atmósfera de nitrógeno. Después de agitar durante 0,5 horas, a la mezcla se le añadió DMF (768 mg, 10,5 mmol, 808 ul, 3 eq.). La mezcla se agitó a -60 °C durante 1 hora. La reacción se interrumpió con una solución acuosa saturada de NH4Cl (10 ml) y después se extrajo con acetato de etilo (50 ml * 3). La fase orgánica combinada se lavó con salmuera (10 ml), se secó sobre Na2SÜ4 anhidro se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna (SiÜ2, éter de petróleo/acetato de etilo = 200/1 a 5/1). Se obtuvo 2-bromo-6-fluoro-4-metil-3- (trifluorometil)benzaldehído (700 mg, 2,46 mmol, 70 % de rendimiento) en forma de un aceite de color amarillo.
1H RMN (400 MHz, cloroformo-d) 5 = 10,35 (s, 1H), 7,08 (d, J = 10,4 Hz, 1H), 2,66 - 2,58 (m, 3H).
4-bromo-6-metil-5-(trifluorometil)-1H-indazol. Una solución de 2-bromo-6-fluoro-4-metil-3-(trifluorometil)benzaldehído (600 mg, 2,11 mmol, 1 eq.) e hidrazina monohidrato (2,11 g, 42,1 mmol, 2,05 ml, 20 eq.) en DMS0 (8 ml) se agitó a 60 °C durante 2 horas. La mezcla se diluyó con acetato de etilo (50 ml) y se lavó con agua (3 * 50 ml) y salmuera (1 * 50 ml). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se usó en la siguiente etapa sin purificación adicional. Se obtuvo 4-bromo-6-metil-5-(trifluorometil)-1H-indazol (550 mg, 1,97 mmol, 94 % de rendimiento) en forma de un sólido de color blanco.
4-bromo-6-metil-1-tetrahidropiran-2-il-5-(trifluorometil)indazol. A una solución de 4-bromo-6-metil-5-(trifluorometil)-1H-indazol (500 mg, 1,79 mmol, 1 eq.) en DCM (10 ml) se le añadió ácido 4-metilbencenosulfónico hidrato (34,1 mg, 1791 umol, 0,1 eq.) seguido de una solución de Dh P (603 mg, 7,17 mmol, 655 ul, 4 eq.) en CH3CN (2 ml). La mezcla se agitó a 25 °C durante 16 horas. La mezcla se diluyó con acetato de etilo (100 ml) y se lavó con agua (2 x 100 ml) y salmuera (1 x 50 ml). La capa orgánica se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo/acetato de etilo 20/1 a 3/1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar 4-bromo-6-metil-1-tetrahidropiran-2-il-5-(trifluorometil)indazol (600 mg, 1,54 mmol, 86 % de rendimiento, 93 % de pureza) en forma de una goma de color amarillo claro. LCMS [MSI, M+1]: 363.
Etapa A: (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi]-7-[6-metil-1-tetrahidropiran-2-il-5-(trifluorometil)indazol-4-il1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]pipenazin-1-carboxilato de /ere-butilo. Una mezcla de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de /ere-butilo (400 mg, 848 umol, 1 eq.), 4-bromo-6-metil-1-tetrahidropiran-2-il-5-(trifluorometil)indazol (431 mg, 1,19 mmol, 1,4 eq.) Pd2(dba)3 (155 mg, 170 umol, 0,2 eq.), RuPhos (158 mg, 339 umol, 0,4 eq.) y Cs2CÜ3 (691 mg, 2,12 mmol, 2,5 eq.) en tolueno (20 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 100 °C durante 16 horas en una atmósfera de N2. A la mezcla se le añadieron H20 (20 ml x 1) y acetato de etilo (20 ml x 1). La fase orgánica se separó, se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía sobre gel de sílice (acetato de etilo/metanol = 50/1 a 2/1). Se obtuvo (2S)-2-(cianometil)--[2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-7-[6-metil-1-tetrahidropiran-2-il-5-(trifluorometil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de /ere-butilo (500 mg, 535 umol, 32 % de rendimiento, 80,7 % de pureza) en forma de un sólido de color amarillo. LCMS [MSI, M+1]: 754.
Etapa B: 2-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-[6-metil-5-(trifluorometil)-1H-indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d^pirimidin-4-il1piperazin-2-il)acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-7-[6-metil-1-tetrahidropiran-2-il-5-(trifluorometil)indazol-4-il]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de /ere-butilo (470 mg, 503 umol, 1 eq.) en DCM (1 ml) se le añadió TfA (2,29 g, 20,1 mmol, 1,49 ml, 40 eq.). La mezcla se agitó a 25 °C durante 1 h. A la mezcla se le añadió diclorometano (5 ml) y se basificó con una solución acuosa saturada de NaHC03 a pH = 8 ~ 9. La fase orgánica se separó, se secó sobre sulfato de sodio, se filtró y se concentró a presión reducida para dar 2-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-[6-metil-5-(trifluorometil)-1H-indazol-4-il]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (420 mg, 453 umol, 90 % de rendimiento, 61,4 % de pureza) que se obtuvo en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación adicional. Lc MS [MSI, M+1]: 570.
Etapa C: 2-((S)-1-(2-fluoroacriloil)-4-(7-(6-metil-5-(trifluorometil)-1H-indazol-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d^pirimidin-4-il)piperazin-2-il)acetonitrilo. A una solución de 2-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-[6-metil-5-(trifluorometil)-1H-indazol-4-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (380 mg, 410 umol, 1 eq.), ácido 2-fluoroprop-2-enoico (18,4 mg, 205 umol, 0,5 eq.) y TEA (124 mg, 1,23 mmol, 171 ul, 3 eq.) en DMF (8 ml) se le añadió T3P (391 mg, 614 umol, 365 ul, 50 % de pureza en DMF, 1,5 eq.) a -40 °C. Después, la mezcla se agitó a -40 °C durante 0,5 horas y a 0 °C durante 0,5 horas más. Se añadió ácido 2-fluoroprop-2-enoico (11 mg) y T3P (100 ul) y la mezcla se agitó a -40 °C durante 0,5 horas más. La mezcla se diluyó con agua (15 ml) y se extrajo con EtOAc (2 x 40 ml). Las capas orgánicas se lavaron con salmuera (50 ml), se secaron sobre Na2SÜ4 y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Luna C18 150 * 255 u; fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 22 % - 42 %, 7,8 min) y (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,04 % de NH3H20 NH4HC0310 mM) - ACN]; % de B: 50 % - 68 %, 12 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-((S)-1-(2-fluoroacriloil)-4-(7-(6-metil-5-(trifluorometil)-1H-indazol-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-cf]pirimidin-4-il)piperazin-2-il)acetonitrilo (EJEMPL0527) en forma de un sólido de color blanco.
Ejemplo 528

2-((S)-4-(7-(8-cloronaftalen-1-il)-2-(((S)-4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-5,6,7, 8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-(3-fluoroazetidin-1-il)but-2-enoil)piperazin-2-il)acetonitrilo
El compuesto del título se prepara siguiendo el Ejemplo 520 modificando el Intermedio 66 sustituyendo (S)-(4,4-difluoro-1-metilpirrolidin-2-il)metanol por (1-metilpirrolidin-2-il)metanol en el Intermedio 66 , Etapa E, el Intermedio 66 modificado se desprotege usando las condiciones del Intermedio 71, Etapa A, y después se sustituye por el Intermedio 66 en el Ejemplo 520, Etapa A.
Ejemplo 529

2-((S)-4-(2-(((S)-4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-fluorobut-2-enoil)piperazin-2-il)acetonitrilo
El compuesto del título se preparó siguiendo el Ejemplo 467 sustituyendo ácido (E)-4-fluorobut-2-enoico por cloruro de acriloílo usando las condiciones del Ejemplo 479, Etapa C. LCMS [ESI, M+1]: 634.
1H RMN (400 MHz, cloroformo-d) 5 = 7,70 (d a, J = 8,0 Hz, 1H), 7,66 (t, J = 8,0 Hz, 1H), 7,45 - 7,38 (m, 1H), 7,38 -7,32 (m, 1H), 7,27 - 7,18 (m, 2H), 7,09 - 6,93 (m, 1H), 6,60 (d a, J = 14,8 Hz, 1H), 5,32 - 4,93 (m, 3H), 4,64 (s a, 1H), 4,45 (td, J = 5,2, 11,2 Hz, 1H), 4,32 - 3,63 (m, 6 H), 3,58 - 3,36 (m, 3H), 3,25 - 2,96 (m, 5H), 2,93 (s, 3H), 2,88 - 2,77 (m, 1H), 2,75 - 2,50 (m, 3H), 2,46 (d, J = 4,4 Hz, 3H), 2,34 - 2,15 (m, 1H).
Condición de SFC: Columna: Chiralcel 0J-350 x 4,6 mm D.I., 3 um, Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 %, Caudal: 3 ml/min, Longitud de onda: 220 nm.
Ejemplo 530
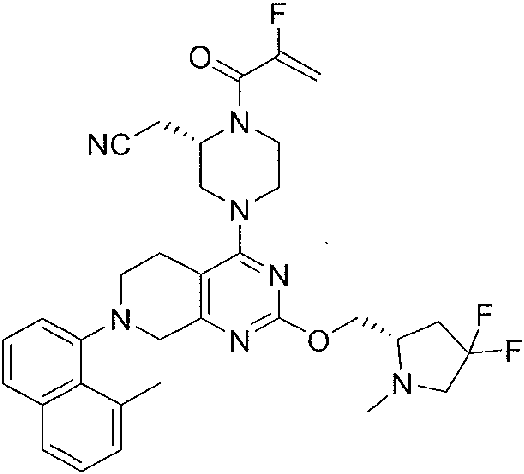
2-((S)-4-(2-(((S)-4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo
El compuesto del título se preparó siguiendo el Ejemplo 467 sustituyendo cloruro de 2-fluoroprop-2-enoílo por cloruro de acriloílo usando las condiciones del Ejemplo 478, Etapa A. LCMS [ESI, M+1]:620.
1H RMN (400 MHz, cloroformo-d) 5 = 7,73 - 7,63 (m, 2H), 7,46 - 7,33 (m, 2H), 7,27 - 7,18 (m, 2H), 5,54 - 5,33 (d, J = 47,6 Hz, 1H), 5,26 (dd, J = 3,6, 16,8 Hz, 1H), 5,05 - 4,62 (m, 1H), 4,58 - 4,37 (m, 1H), 4,31 - 4,13 (m, 3H), 4,12 - 4,05 (m, 1H), 3,97 - 3,73 (m, 1H), 3,59 - 3,36 (m, 3H), 3,27 - 2,95 (m, 6H), 2,92 (s, 3H), 2,89 - 2,49 (m, 5H), 2,46 (d, J = 4,4 Hz, 3H), 2,34 - 2,17 (m, 1H).
SFC: “Columna: Chiralcel 0J - 350 x 4 ,6 mm D.I., 3 um Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 % Caudal: 3 ml/min Longitud de onda: 220 nm” .
Ejemplo 531

2-((S)-4-(2-(((S)-4,4-difluono-1-metilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-metoxibut-2-enoil)piperazin-2-il)acetonitrilo
El compuesto del título se prepara siguiendo el Ejemplo 467 sustituyendo ácido (metoximetil)prop-2-enoico por cloruro de acriloílo usando las condiciones del Ejemplo 489, Etapa A.
Ejemplo 532

2-((S)-4-(2-(((S)-4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-hidroxibut-2-enoil)piperazin-2-il)acetonitrilo
El compuesto del título se prepara siguiendo el Ejemplo 467 sustituyendo ácido 4-[ferc-butil(dimetil)silil]oxibut-2-inoico por cloruro de acriloílo usando las condiciones del Ejemplo 483, Etapa A.
Ejemplo 533

(S,E)-1-(4-(2-((4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(5,6-dimetil-1H-indol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-4-fluorobut-2-en-1-ona
El compuesto del título se prepara siguiendo el Ejemplo 469 sustituyendo ácido (E)-4-fluorobut-2-enoico por prop-2-enoato de prop-2-enoílo en el Ejemplo 479, Etapa C.
E je m p lo 534

(S,E)-1-(4-(2-((4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(5,6-dimetil-1H-indol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-4-metoxibut-2-en-1-ona
El compuesto del título se prepara siguiendo el Ejemplo 469 sustituyendo ácido 2-(metoximetil)prop-2-enoico por prop-2-enoato de prop-2-enoílo en el Ejemplo 489, Etapa A.
Ejemplo 535

(S)-1 -(4-(2-((4,4-difluoro-1 -metilpirrolidin-2-il)metoxi)-7-(5,6-dimetil-1H-indol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-2-fluoroprop-2-en-1-ona
El compuesto del título se prepara siguiendo el Ejemplo 469 sustituyendo ácido 2-fluoroacrílico por prop-2-enoato de prop-2-enoílo en el Ejemplo 478, Etapa A.
Ejemplo 536

(S,E)-1-(4-(2-((4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(5,6-dimetil-1H-indol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-4-hidroxibut-2-en-1-ona
El compuesto del título se prepara siguiendo el Ejemplo 469 sustituyendo ácido 4-[ferc-butil(dimetil)silil]oxibut-2-inoico por prop-2-enoato de prop-2-enoílo en el Ejemplo 469, Etapa D, y siguiendo los procedimientos del Ejemplo 483, Etapa A.
E je m p lo 537

(S)-1-(4-(2-((4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(5,6-dimetil-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-2-fluoroprop-2-en-1-ona
El compuesto del título se prepara siguiendo el Ejemplo 470 sustituyendo ácido (E)-4-fluorobut-2-enoico por prop-2-enoato de prop-2-enoílo en el Ejemplo 478, Etapa A.
Ejemplo 538

(S,E)-1-(4-(2-((4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(5,6-dimetil-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-4-fluorobut-2-en-1-ona
El compuesto del título se prepara siguiendo el Ejemplo 470 sustituyendo ácido (E)-4-fluorobut-2-enoico por prop-2-enoato de prop-2-enoílo en el Ejemplo 479, Etapa C.
Ejemplo 539

(S)-1-(4-(2-((4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(5,6-dimetil-1H-indol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-2-fluoroprop-2-en-1-ona
El compuesto del título se prepara siguiendo el Ejemplo 470 sustituyendo ácido 2-fluoroacrílico por prop-2-enoato de prop-2-enoílo en el Ejemplo 478, Etapa A.
E je m p lo 540

(S,E)-1-(4-(2-((4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(5,6-dimetil-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidm-4-il)piperazin-1-il)-4-metoxibut-2-en-1-ona
El compuesto del título se prepara siguiendo el Ejemplo 470 sustituyendo ácido 2-(metoximetil)prop-2-enoico por prop-2-enoato de prop-2-enoílo en el Ejemplo 489, Etapa A.
Ejemplo 541

(S,E)-1-(4-(2-((4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(5,6-dimetil-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1 -il)-4-hidroxibut-2-en-1 -ona
El compuesto del título se prepara siguiendo el Ejemplo 470 sustituyendo ácido 4-[ferc-butil(dimetil)silil]oxibut-2-inoico por prop-2-enoato de prop-2-enoílo en el Ejemplo 470, Etapa C y siguiendo los procedimientos del Ejemplo 483, Etapa A.
Ejemplo 542

2-((S)-4-(2-(((S)-4,4-difluono-1-metilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-fluorobut-2-enoil)piperazin-2-il)acetonitrilo
El compuesto del título se preparó siguiendo el Ejemplo 468 sustituyendo cloruro de 2-fluoroprop-2-enoílo por cloruro de acriloílo usando las condiciones del Ejemplo 479, Etapa C. LCMS [ESI, M+1]: 655.1
1H RMN (400 MHz, Cloroformo-d) 57,76 (d, J = 8,0 Hz, 1H), 7,63 (t, J = 7,2 Hz, 1H), 7,53 (d, J = 7,2 Hz, 1H), 7,50 -7,40 (m, 1H), 7,34 (t, J = 7,8 Hz, 1H), 7,26 - 7,18 (m, 1H), 7,08 - 6,94 (m, 1H), 6,60 (d a, J = 14,8 Hz, 1H), 5,26 - 5,03 (m, 2H), 4,90 - 4,58 (m, 1H), 4,52 - 4,38 (m, 2H), 4,31 - 4,21 (m, 1H), 4,19 - 3,98 (m, 2H), 3,97 - 3,72 (m, 2H), 3,66
7,56 (m, 1H), 3,53 - 3,37 (m, 2H), 3,34 - 3,11 (m, 3H), 3,07 - 2,92 (m, 2H), 2,89 - 2,79 (m, 1H), 2,77 - 2,48 (m, 4H), 2,47 (d, J = 2,4 Hz, 3H), 2,35 - 2,18 (m, 1H).
Ejemplo 543

2-((S)-4-(2-(((S)-4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo
El compuesto del título se preparó siguiendo el Ejemplo 468 sustituyendo cloruro de 2-fluoroprop-2-enoílo por cloruro de acriloílo usando las condiciones del Ejemplo 478, Etapa A. LCMS [ESI, M+1]:640.
1H RMN (400 MHz, cloroformo-d) 5 = 7,76 (d, J = 8,0 Hz, 1H), 7,63 (t, J = 7,2 Hz, 1H), 7,53 (d, J = 7,6 Hz, 1H), 7,45 (td, J = 7,6, 12,4 Hz, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,27 - 7,19 (m, 1H), 5,54 - 5,33 (dd, J = 2,8, 47,6 Hz, 1H), 5,26 (dd, J = 3,6, 16,8 Hz, 1H), 4,85 (s a, 1H), 4,51 - 4,38 (m, 2H), 4,31 - 3,77 (m, 4H), 3,69 - 3,54 (m, 1H), 3,41 (m, 2H), 3,35 - 2,48 (m, 11H), 2,47 (d, J = 2,4 Hz, 3H), 2,35 - 2,16 (m, 1H).
SFC: “ Columna: Cellucoat 50 x 4,6 mm D.I., 3 um Fase móvil: metanol (0,05 % de DEA) en C02 de 5 % a 40 % Caudal: 3 ml/min Longitud de onda: 220 nm” .
Ejemplo 544
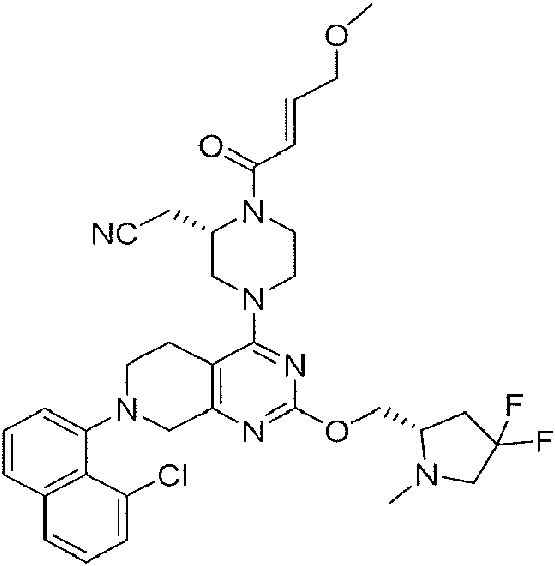
2-((S)-4-(7-(8-cloronaftalen-1-il)-2-(((S)-4,4-difluoro-1-metilpirolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-metoxibut-2-enoil)piperazin-2-il)acetonitrilo
El compuesto del título se prepara siguiendo el Ejemplo 468 sustituyendo ácido (metoximetil)prop-2-enoico por cloruro de acriloílo usando las condiciones del Ejemplo 489, Etapa A.
Ejemplo 545

2-((S)-4-(2-(((S)-4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(8-cloronaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-hidroxibut-2-enoil)piperazin-2-il)acetonitrilo
El compuesto del título se prepara siguiendo el Ejemplo 468 sustituyendo ácido 4-[ferc-butil(dimetil)silil]oxibut-2-inoico por cloruro de acriloílo usando las condiciones del Ejemplo 483, Etapa A.
Ejemplo 546
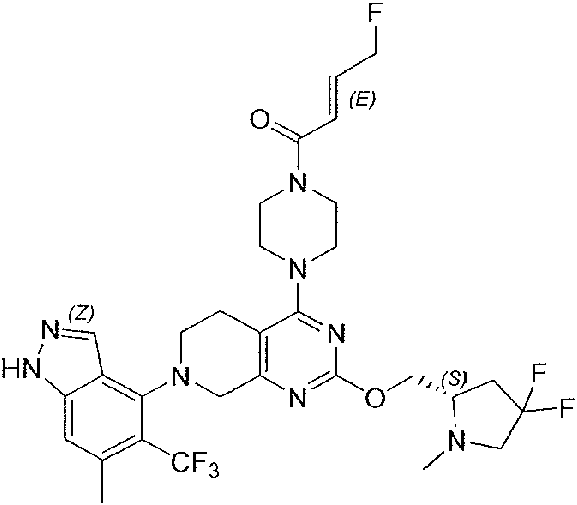
2-((S)-4-(2-(((S)-4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-fluorobut-2-enoil)piperazin-2-il)acetonitrilo
El compuesto del título se prepara siguiendo el Ejemplo 527 sustituyendo cloruro de 2-fluoroprop-2-enoílo por cloruro de acriloílo usando las condiciones del Ejemplo 479, Etapa C.
Ejemplo 547

2-((S)-4-(2-(((S)-4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo
El compuesto del título se prepara siguiendo el Ejemplo 527 sustituyendo cloruro de 2-fluoroprop-2-enoílo por cloruro de acriloílo usando las condiciones del Ejemplo 478, Etapa A.
Ejemplo 548

2-((S)-4-(7-(8-cloronaftalen-1-il)-2-(((S)-4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-&-((E)-4-metoxibut-2-enoil)piperazin-2-il)acetonitrilo
El compuesto del título se prepara siguiendo el Ejemplo 527 sustituyendo ácido (metoximetil)prop-2-enoico por cloruro de acriloílo usando las condiciones del Ejemplo 489, Etapa A.
Ejemplo 549

2-((S)-4-(2-(((S)-4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(8-cloronaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1 -((E)-4-hidroxibut-2-enoil)piperazin-2-il)acetonitrilo
El compuesto del título se prepara siguiendo el Ejemplo 527 sustituyendo ácido 4-[terc-butil(dimetil)silil]oxibut-2-inoico por cloruro de acriloílo usando las condiciones del Ejemplo 483, Etapa A.
Ejemplo 550

2-((S)-4-(7-(5,6-dimetil-1H-indol-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo
2-((S)-1-acriloil-4-(7-(5,6-dimetil-1H-indol-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo. El compuesto del título se prepara siguiendo el Ejemplo 466 sustituyendo ácido 2-fluoroacrílico por prop-2-enoato de prop-2-enoílo como se ha descrito en el Ejemplo 478, Etapa A.
Ejemplo 551

2-[(2S)-4-[2-[[(2R,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(E)-4-metoxibut-2-enoil]piperazin-2-il]acetonitrilo

Etapa A: 2-[(2S)-4-[2-[[(2R4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-[(E)-4-metoxibut-2-enoil1piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[2-[[(2E,4E)-4-fluoro-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡ljaceton¡tr¡lo (200 mg, 378 umol, 1,0 eq.) y ácido (E)-4-metoxibut-2-enoico (110 mg, 944 umol, 2,5 eq.) en DMF (3,0 ml) se le añadieron TEA (382 mg, 3,78 mmol, 526 ul, 10,0 eq.) y T3P (721 mg, 1,13 mmol, 674 ul, 50 % de pureza, 3,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, el disolvente orgánico se lavó con agua (10,0 ml). La fase acuosa se extrajo con acetato de etilo (3 * 10,0 ml). Los extractos combinados se lavaron con salmuera (20,0 ml), se secaron con Na2SÜ4 y se filtraron. Después, el disolvente se eliminó al vacío. El residuo se purificó por cromatografía en columna (Base Al2Ü3, éter de petróleo : acetato de etilo = 10:1 a 1:1) y después el producto bruto se concentró y se purificó de nuevo por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 um; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 48 %-78 %,9-10 min) y se sometió a liofilización para dar el compuesto del título 2-[(2S)-4-[2-[[(2É,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1 -[(E)-4-metoxibut-2-enoil]piperazin-2-il]acetonitrilo (EJEMPL0 551, 50 mg, 79,6 umol, 21 % de rendimiento, 99,9 % de pureza) que se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 628.
1H RMN (400 MHz, Cloroformo-d) ó = 7,73 - 7,62 (m, 2H), 7,45 - 7,31 (m, 2H), 7,27 - 7,18 (m, 2H), 7,05 - 6,94 (m, 1H), 6,55 (d a, J = 15,0 Hz, 1H), 5,23 - 5,03 (m, 1H), 4,66 (s a, 1H), 4,52 - 4,42 (m, 1H), 4,30 - 4,18 (m, 2H), 4,18 - 3,94 (m, 4H), 3,93 - 3,84 (m, 1H), 3,83 - 3,65 (m, 1H), 3,54 (d a, J = 6,7 Hz, 1H), 3,44 (s, 3H), 3,42 - 3,26 (m, 2H), 3,25 - 3,15 (m, 2H), 3,05 (dd a, J = 8,5, 16,4 Hz, 2H), 2,92 (s, 3H), 2,88 - 2,58 (m, 4H), 2,57 -2,35 (m, 5H), 2,14 - 1,96 (m, 1H).
Ejemplo 552

2-[(2S)-1-[(E)-4-metoxibut-2-enoil]-4-[2-[[(2R.4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6.8-dihidro-5H-pirido[3.4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

Etapa A: 2-[(2S)-1 -[(E)-4-metoxibut-2-enoil-1 -4-[2-[[(2R4R)-4-metoxi-1 -metil-pirrolidin-2-il]metoxi1-7-(8-metil-1 -naftil)-6.8-dihidro-5H-pirido[3.4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A la solución de 2-[(2S)-4-[2-[[(2E,4E)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6.8-dihidro-5H-pirido[3.4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (150 mg. 277 umol. 1 eq.), ácido (E)-4-metoxibut-2-enoico (64.3 mg. 554 umol. 2 eq.) y TEA (280 mg. 2.77 mmol. 385 ul. 10 eq.) en DMF (3 ml) se añadió T3P (529 mg. 831 umol. 494 ul. 50 % de pureza. 3 eq.) a 0 °C y la mezcla se agitó a 0 °C durante 0.5 horas. A la mezcla se le añadió agua (10 ml). La mezcla se diluyó con EtOAc (5 ml) y se extrajo con EtOAc (2 * 10 ml). Las capas orgánicas combinadas se lavaron con salmuera (10 ml). se secaron sobre Na2S04 anhidro. se filtraron y se concentraron al vacío. El residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0.1 % de AF) = 35 %). Después. el residuo se purificó por HPLC prep. (columna: Waters Xbridge 150 * 25 5 u; fase móvil: [agua (0.05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 50 % - 74 %.10 min) para dar el compuesto del título 2-[(2S)-1 -[(E)-4-metoxibut-2-enoil]-4-[2-[[(2R.4R)-4-metoxi-1 -metilpirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6.8-dihidro-5H-pirido[3.4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (EJEMPL0552. 37.1 mg. 57.2 umol. 21 % de rendimiento. 98.7 % de pureza) en forma de un sólido de color blanco. LCMS [ESI. M+1]: 640.
1H RMN (400 MHz. cloroformo-d) ó = 7,72 - 7.61 (m. 2H). 7.45 - 7.31 (m. 2H). 7.27 - 7.17 (m. 2H). 6.99 (d a. J = 14.8 Hz. 1H). 6.55 (d a. J = 15.2 Hz. 1H). 5.07 (s a. 1H). 4.51 - 4.40 (m. 1H). 4.31 - 4.04 (m. 5H). 3.95 - 3.68 (m. 3H).
3.63 - 3.47 (m. 2H). 3.44 (s. 3H). 3.42 - 3.35 (m. 1H). 3.30 (d. J = 1.2 Hz. 3H). 3.24 - 2.96 (m. 5H). 2.92 (s. 3H). 2.76 (dd a. J = 8.0. 16.8 Hz. 1H). 2.72 - 2.52 (m. 3H). 2.45 (d. J = 5.2 Hz. 3H). 2.41 - 2.27 (m. 2H). 1.81 (dd a. J = 7.2.
14.0 Hz. 1H).
Ejemplo 553

2-((S)-1-((E)-4-metoxibut-2-enoil)-4-(7-(8-metilnaftalen-1-il)-2-(((E)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 3-hidroxi-4-metoxi-butanoato de metilo. A una solución de 4-metoxi-3-oxo-butanoato de metilo (20 g, 137 mmol, 1,0 eq.) en tolueno (300 ml) se le añadió NaBH4 (5,44 g, 144 mmol, 1,05 eq.) a 0 °C. Después, la mezcla se agitó a 20 °C durante 4 horas. Después de que se completara, la mezcla se diluyó con agua (100 ml) a 0 °C y se extrajo con acetato de etilo (3 x 100 ml). Los extractos se lavaron con salmuera (200 ml), se secaron sobre Na2Sü4, se filtraron y se concentraron al vacío para dar 3-hidroxi-4-metoxi-butanoato de metilo (7,5 g, bruto) en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, cloroformo-d) 5 = 4,23 - 4,15 (m, 1H), 3,70 (s, 3H), 3,44 - 3,38 (m, 2H), 3,37 (s, 3H), 3,04 (s a, 1H), 2,51 (d, J = 6,4 Hz, 2H).
Etapa B: (E)-4-metoxibut-2-enoato de metilo. A una solución de 3-hidroxi-4-metoxi-butanoato de metilo (7,5 g, bruto) en DCM (50,0 ml) se le añadieron TEA (5,12 g, 50,6 mmol, 7,05 ml) y MsCl (8,70 g, 75,9 mmol, 5,88 ml) a 0 °C. Después de agitar durante 4 horas, a la mezcla se le añadió TEA (10,2 g, 101 mmol, 14,1 ml) a 0 °C. La mezcla se calentó a 20 °C y se agitó durante 12 horas. Después de que se completara, la mezcla se diluyó con agua (50,0 ml), se lavó con HCl (1 N, 15,0 ml) y salmuera (20,0 ml), se secó sobre Na2SÜ4, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna (SiÜ2, éter de petróleo/acetato de etilo = 1/0 a 50:1) para dar (E)-4-metoxibut-2-enoato de metilo (1,7 g, 11,8 mmol, dos etapas 8,6 % de rendimiento, 90 % de pureza) en forma de un aceite de color amarillo.
1H RMN (400 MHz, cloroformo-d) 5 = 6,93 (td, J = 4,3, 15,8 Hz, 1H), 6,04 (td, J = 2,0, 15,8 Hz, 1H), 4,06 (dd, J = 2,0, 4,3 Hz, 2H), 3,72 (s, 3H), 3,36 (s, 3H).
Etapa C: ácido (E)-4-metoxibut-2-enoico. Una mezcla de (E)-4-metoxibut-2-enoato de metilo (300 mg, 2,31 mmol, 1,0 eq.) y K0H (517 mg, 9,22 mmol, 4,0 eq.) en Me0H (2,0 ml) y H20 (2,0 ml) se agitó a 20 °C durante 2 horas. Después de que se completara, la mezcla se acidificó con HCl (2 N, 5,0 ml) a pH = 1 - 3 y se extrajo con acetato de etilo (2 x 20 ml). Las capas orgánicas se secaron sobre Na2S04, se filtraron y se concentraron al vacío para dar ácido (E)-4
metoxibut-2-enoico (200 mg, 1,55 mmol, 67,3 % de rendimiento, 90 % de pureza) en forma de un sólido de color amarillo.
1H RMN (400 MHz, cloroformo-d) 5 = 11,13 (s a, 1H),7,06 (td, J = 4,1, 15,7 Hz, 1H), 6,08 (td, J = 2,0, 15,7 Hz, 1H), 4,12 (dd, J = 2,0, 4,1 Hz, 2H), 3,40 (s, 3H).
Etapa D: 2-[(2S)-1-((£)-4-metoxibut-2-enoil1-4-[7-(8-metil-1-naftil)-2-[[(2^)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-il]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (200 mg, 391 umol, 1 eq.), ácido (E)-4-metoxibut-2-enoico (113 mg, 977 umol, 2,5 eq.) y TEA (395 mg, 3,91 mmol, 544 ul, 10 eq.) en DMF (4 ml) se le añadió T3P (746 mg, 1,17 mmol, 697 ul, 50 % de pureza, 3 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,3 horas. La mezcla de reacción se diluyó con acetato de etilo (20 ml) y se lavó con agua (10 ml * 3). Las capas orgánicas se lavaron con salmuera (10 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiÜ2, acetato de etilo/MeüH = 100/1 a 10:1) y se purificó adicionalmente por HPLC prep. (columna: Boston pH - lex 150 * 25 10 um; fase móvil: [agua (0,1 % de TFA) - ACN]; % de B: 35 % - 59 %, 8 min). La mezcla se recogió y se liofilizó. El compuesto del título 2-[(2S)-1-[(£)-4-metox¡but-2-eno¡l]-4-[7-(8-met¡l-1-naft¡l)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-il]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-il]p¡peraz¡n-2-¡l]aceton¡trilo (EJEMPL0553, 37,5 mg, 60,4 umol, 15 % de rendimiento, 98,2 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 610.
1H RMN (400 MHz, cloroformo-d) 5 = 7,77 - 7,57 (m, 2H), 7,46 - 7,37 (m, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,27 - 7,17 (m, 2H), 6,99 (d a, J = 14,4 Hz, 1H), 6,55 (d a, J = 14,8 Hz, 1H), 5,24 - 4,49 (m, 1H), 4,46 - 4,32 (m, 1H), 4,32 - 3,74 (m, 7H), 3,73 - 3,25 (m, 6H), 3,25 - 2,94 (m, 5H), 2,92 (s, 3H), 2,86 - 2,54 (m, 4H), 2,49 (d, J = 5,6 Hz, 3H), 2,38 - 2,23 (m, 1H), 2,11 - 2,00 (m, 1H), 1,93 - 1,65 (m, 3H).
Ejemplo 554

2-[(2S)-1-[(£)-4-Metox¡but-2-eno¡l]-4-[7-(8-met¡l-1-naft¡l)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡hidro-5H-p¡r¡do[3,4-d]pirimidin-4-il1piperazin-2-il]acetonitrilo
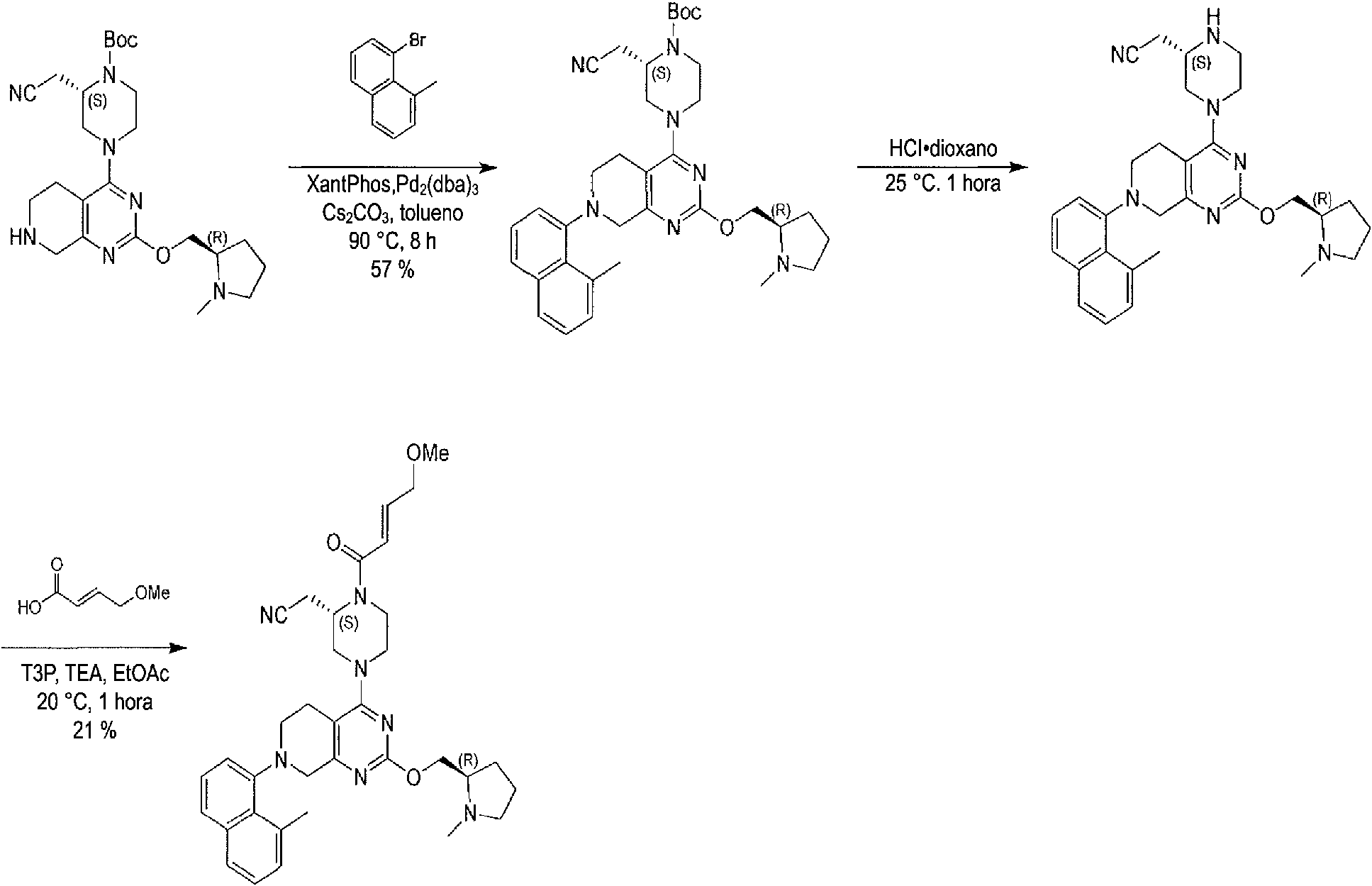
Etapa A: (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[((2R)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-1-carboxilato de tere-butilo. Una mezcla de (2S)-2-(cianometil)-4-[2-[[(2R)-1-metilpirrolidin-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (850 mg, 1,80 mmol, 1,0 eq.), 1-bromo-8-metil-naftaleno (1,20 g, 5,41 mmol, 3,0 eq.), Pd2(dba)3 (330 mg, 360 gmol, 0,20 eq.), Xantphos (417 mg, 721 gmol, 0,40 eq.) y CS2C03 (1,76 g, 5,41 mmol, 3,0 eq.) en tolueno (10,0 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 8 horas en una atmósfera de N2. Después de que se completara, la reacción se lavó con HCl 2 N (2 x 15 ,0 ml). La fase acuosa se basificó con NaHC03 sólido a pH ~7 y se extrajo con acetato de etilo (2 x 50 ,0 ml). Las capas orgánicas se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC ultrarrápida de fase inversa [C18, 0,1 % de AF en agua, 0 -65 % de MeCN], se ajustó con NaHC03 sólido a pH ~7 y se extrajo con acetato de etilo (2 x 150 ml). Las capas orgánicas se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El compuesto (2S)-2-(cianometil)-4-[7-(8-met¡l-1-naft¡l)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡j-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (650 mg, 1,03 mmol, 57 % de rendimiento, 97 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 612.
1H RMN (400 MHz, cloroformo-d) ó 7,72 - 7,61 (m, 2H), 7,44 - 7,37 (m, 1H), 7,36 - 7,30 (m, 1H), 7,27 - 7,16 (m, 2H), 4,63 - 4,52 (m, 1H), 4,46 - 4,36 (m, 1H), 4,30 - 4,15 (m, 2H), 4,09 - 3,95 (m, 2H), 3,93 - 3,73 (m, 2H), 3,57 - 3,47 (m, 1H), 3,42 - 3,27 (m, 1H), 3,22 - 3,04 (m, 4H), 3,02 - 2,94 (m, 1H), 2,92 (s, 3H), 2,81 - 2,54 (m, 4H), 2,53 - 2,47 (m, 3H), 2,40 - 2,26 (m, 1H), 2,12 - 2,05 (m, 1H), 1,92 - 1,71 (m, 3H), 1,52 (s, 9H).
Etapa B: 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-i n piperazin-2-i n acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo (600 mg, 981 gmol, 1,0 eq.) en dioxano (3,0 ml) se le añadió HCl/dioxano (4 M, 3,0 ml, 12,2 eq.) a 25 °C y la mezcla se agitó a 25 °C durante 1 hora. Después de que se completara, la mezcla de reacción se inactivó con NaHC03 acuoso saturado (8,0 ml) a 0 °C y después se diluyó con H20 (5,0 ml) y se extrajo con acetato de etilo (3 x 15 ,0 ml). Las capas orgánicas combinadas se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡trilo (410 mg, bruto) en forma de un sólido de color amarillo que se usó en la siguiente etapa sin purificación adicional.
Etapa C: 2-r(2S)-1-r(E)-4-metoxibut-2-enoi n -4-r7-(8-metil-1-naftil)-2-rr(2R)-1-metilpirrolidin-2-i n metox n -6.8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2E)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡trilo (150 mg, 293 gmol, 1,0 eq.) y ácido (E)-4-metoxibut-2-enoico (102 mg, 880 gmol, 3,0 eq.) en acetato de etilo (3,0 ml) se le añadieron TEA (237 mg, 2,35 mmol, 326 gl, 8,0 eq.) y T3p (560 mg, 880 gmol, 523 gl, 50 % de pureza, 3,0 eq.) a 0 °C. La mezcla se agitó a 20 °C durante 1 hora. Después de que se completara, se añadió agua (5,0 ml). La capa orgánica se separó, y
la fase acuosa se extrajo con acetato de etilo (3 x 10 ml). Los extractos combinados se lavaron con salmuera (20,0 ml), se secaron con Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna (Al203 básico, éter de petróleo : acetato de etilo = 3 : 1 a acetato de etilo : metanol = 50 : 1) y después el producto bruto se purificó de nuevo por HPLC prep. (columna: Waters Xbridge 150 * 25 5 ufase móvil: [agua (0,05 % de hidróxido de amonio v/v)-MeCN];% de B: 50 %-77 %, 10 min) y se sometió a liofilización. El compuesto 2-[(2S)-1-[(E)-4-metoxibut-2-enoil]-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (38,6 mg, 62,4 qmol, 21 % de rendimiento, 98 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 512.
Etapa D: 2-[(2S)-1-[(E)-4-metoxibut-2-enoil1-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirroildin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (150 mg, 293 qmol, 1,0 eq.) y ácido (E)-4-metoxibut-2-enoico (102 mg, 880 qmol, 3,0 eq.) en acetato de etilo (3,0 ml) se le añadieron TEA (237 mg, 2,35 mmol, 326 ql, 8,0 eq.) y T3P (560 mg, 880 qmol, 523 ql, 50 % de pureza, 3,0 eq.) a 0 °C. La mezcla se agitó a 20 °C durante 1 hora. Después de que se completara, se añadió agua (5,0 ml). La capa orgánica se separó, y la fase acuosa se extrajo con acetato de etilo (3 x 10 ml). Los extractos combinados se lavaron con salmuera (20,0 ml), se secaron con Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna (Al203 básico, éter de petróleo : acetato de etilo = 3 : 1 a acetato de etilo : metanol = 50 : 1) y después el producto bruto se purificó de nuevo por HPLC prep. (columna: Waters Xbridge 150 * 25 5 ufase móvil: [agua (0,05 % de hidróxido de amonio v/v)-MeCN];% de B: 50 %-77 %, 10 min) y se sometió a liofilización. El compuesto 2-[(2S)-1-[(E)-4-metoxibut-2-enoil]-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (38,6 mg, 62,4 qmol, 21 % de rendimiento, 98 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 610.
1H RMN (400 MHz, cloroformo-d) 57,73 - 7,62 (m, 2H), 7,45 - 7,31 (m, 2H), 7,26 - 7,17 (m, 2H), 6,99 (d a, J = 15,2 Hz, 1H), 6,55 (d a, J = 15,6 Hz, 1H), 5,20 - 4,50 (m, 1H) 4,43 - 4,33 (m, 1H), 4,29 - 3,96 (m, 6H), 3,94 - 3,83 (m, 1H), 3,81 - 3,63 (m, 1H), 3,58 - 3,37 (m, 5H), 3,23 - 2,96 (m, 5H), 2,92 (s, 3H), 2,87 - 2,54 (m, 4H), 2,52 - 2,43 (m, 3H), 2,33 -2,24 (m, 1H), 2,13 - 2,01 (m, 1H), 1,87 - 1,72 (m, 3H).
Ejemplo 555

2-((S)-1-(2-Fluoroacriloil)-4-(7-(6-metil-5-(trifluorometil)-1H-indazol-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo


1- Bromo-5-fluoro-3-metil-2-(trifluorometil)benceno. A una solución de 1-bromo-5-fluoro-2-yodo-3-metil-benceno (2 g, 6,35 mmol, 1 eq.) en DMF (40 ml) se le añadieron CuI (7,26 g, 38,1 mmol, 6 eq.) y 2,2-difluoro-2-fluorosulfonil-acetato de metilo (7,32 g, 38,1 mmol, 4,85 ml, 6 eq.). La mezcla se agitó a 60 °C durante 12 horas. La mezcla de reacción se diluyó con acetato de etilo (20 ml) y se lavó con agua (20 ml * 3). La capa orgánica se lavó con salmuera (20 ml), se secó sobre Na2SÜ4, se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiÜ2, éter de petróleo/acetato de etilo = 1/0 a 10/1). Se obtuvo 1-bromo-5-fluoro-3-metil-2-(trifluorometil)benceno (1,3 g, 3,74 mmol, 59 % de rendimiento, 74 % de pureza) en forma de un aceite de color amarillo.
1H RMN (400 MHz, cloroformo-d) ó = 7,33 (dd, J = 2,4, 7,6 Hz, 1H), 6,95 (dd, J = 2,0, 8 ,8 Hz, 1H), 2,54 (c, J = 3,6 Hz, 3H).
2- Bromo-6-fluoro-4-metil-3-(trifluorometil)benzaldehído. A una mezcla de 1-bromo-5-fluoro-3-metil-2-(trifluorometil)benceno (900 mg, 3,50 mmol, 1 eq.) en THF (20 ml) se le añadió LDA (2 M en THF, 3,50 ml, 2 eq.) a -60 °C en una atmósfera de nitrógeno. Después de agitar durante 0,5 horas, a la mezcla se le añadió DMF (768 mg, 10,5 mmol, 808 gl, 3 eq.). La mezcla se agitó a -60 °C durante 1 hora. La reacción se interrumpió con NH4Cl acuoso saturado (10 ml) y después se extrajo con acetato de etilo (50 ml * 3). La fase orgánica combinada se lavó con salmuera (10 ml), se secó sobre Na2SÜ4 anhidro se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna (SiÜ2, éter de petróleo/acetato de etilo = 200 /1 a 5/1). Se obtuvo 2-bromo-6-fluoro-4-metil-3- (trifluorometil)benzaldehído (700 mg, 2,46 mmol, 70 % de rendimiento) en forma de un aceite de color amarillo.
1H RMN (400 MHz, cloroformo-d) ó = 10,35 (s, 1H), 7,08 (d, J = 10,4 Hz, 1H), 2,66 - 2,58 (m, 3H).
4-Bromo-6-metil-5-(trifluorometil)-1H-indazol. Una solución de 2-bromo-6-fluoro-4-metil-3-(trifluorometil)benzaldehído (600 mg, 2,11 mmol, 1 eq.) e hidrazina monohidrato (2,11 g, 42,1 mmol, 2,05 ml, 20 eq.) en DMS0 (8 ml) se agitó a 60 °C durante 2 horas. La mezcla se diluyó con acetato de etilo (50 ml) y se lavó con agua (3 * 50 ml) y salmuera (1 * 50 ml). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se usó en la siguiente etapa sin purificación adicional. Se obtuvo 4-bromo-6-metil-5-(trifluorometil)-1H-indazol (550 mg, 1,97 mmol, 94 % de rendimiento) en forma de un sólido de color blanco.
4- Bromo-6-metil-1-tetrahidropiran-2-il-5-(trifluorometil)indazol. A una solución de 4-bromo-6-metil-5-(trifluorometil)-1H-indazol (500 mg, 1,79 mmol, 1 eq.) en DCM (10 ml) se le añadió ácido 4-metilbencenosulfónico hidrato (34,1 mg, 1791 gmol, 0,1 eq.) seguido de una solución de DHP (603 mg, 7,17 mmol, 655 gl, 4 eq.) en CH3CN (2 ml). La mezcla se agitó a 25 °C durante 16 horas. La mezcla se diluyó con acetato de etilo (100 ml) y se lavó con agua (2 * 100 ml) y salmuera (1 * 50 ml). La capa orgánica se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo/acetato de etilo 20/1 a 3/1). Las fracciones deseadas se recogieron y se concentraron al vacío para dar 4-bromo-6-metil-1-tetrahidropiran-2-il-5-(trifluorometil)indazol (600 mg, 1,54 mmol, 86 % de rendimiento, 93 % de pureza) en forma de una goma de color amarillo claro. LCMS [MSI, M+1]: 363.
Etapa A: (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-[6-metil-1-tetrahidropiran-2-il-5-(trifluorometinindazol-4-il1-6.8-dihidro-5H-pirido[3.4-dlpirimidin-4-illpiperazin-1-carboxilato de ferc-butilo. Una mezcla de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il1piperazin-1-carboxilato de ferc-butilo (400 mg. 848 μmol. 1 eq.). 4-bromo-6-metil-1-tetrahidropiran-2-il-5-(trifluorometil)indazol (431 mg. 1.19 mmol. 1.4 eq.) Pd2(dba)3 (155 mg. 170 μmol. 0.2 eq.). RuPhos (158 mg. 339 μmol. 0.4 eq.) y CS2C03 (691 mg. 2.12 mmol.2.5 eq.) en tolueno (20 ml) se desgasificó y se purgó 3 veces con N2 y se agitó a 100 °C durante 16 horas en una atmósfera de N2. A la mezcla se le añadieron H20 (20 ml * 1) y acetato de etilo (20 ml * 1). La fase orgánica se separó. se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía sobre gel de sílice (acetato de etilo /metanol = 50/1 a 2/1). Se obtuvo (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-[6-metil-1-tetrahidropiran-2-il-5-(trifluorometil)indazol-4-il1-6.8-dihidro-5H-pirido[3.4-d1pirimidin-4-il1piperazin-1 -carboxilato de ferc-butilo (500 mg. 535 μmol. 32 % de rendimiento. 80.7 % de pureza) en forma de un sólido de color amarillo. LCMS [MSI. M+11: 754.
Etapa B: 2-[(2S)-4-[2-[[(2S)-1-Metilpirrolidin-2-il1metoxi1-7-[6-metil-5-(trifluorometil)-1H-indazol-4-il1-6.8-dihidro-5H-p¡rido[3.4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-[6-metil-1-tetrahidropiran-2-il-5-(trifluorometil)indazol-4-il1-6.8-dihidro-5H-pirido[3.4-djpirimidin-4-il1piperazin-1-carboxilato de ferc-butilo (470 mg. 503 μmol. 1 eq.) en DCM (1 ml) se le añadió TfA (2.29 g. 20.1 mmol.
1.49 ml. 40 eq.). La mezcla se agitó a 25 °C durante 1 h. A la mezcla se le añadió diclorometano (5 ml) y se basificó con NaHC03 acuoso saturado a pH = 8 ~ 9. La fase orgánica se separó. se secó sobre sulfato de sodio. se filtró y se concentró a presión reducida para dar 2-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-[6-metil-5-(trifluorometil)-1H-¡ndazol-4-¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (420 mg. 453 μmol. 90 % de rendimiento.61.4 % de pureza) en forma de un sólido de color amarillo que se usó en la siguiente etapa sin purificación adicional. LCMS [MSI. M+11: 570.
Etapa C: 2-((S)-1-(2-Fluoroacriloil)-4-(7-(6-metil-5-(trifluorometil)-1H-indazol-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-dlpirimidin-4-il)piperazin-2-il)acetonitrilo. A una solución de 2-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-[6-metil-5-(trifluorometil)-1H-indazol-4-il1-6.8-dihidro-5H-pirido[3.4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo (380 mg. 410 μmol. 1 eq.). ácido 2-fluoroprop-2-enoico (18.4 mg. 205 μmol. 0.5 eq.) y TEA (124 mg. 1.23 mmol. 171 μl.
3 eq.) en DMF (8 ml) se le añadió T3P (391 mg. 614 μmol. 365 μl. 50 % de pureza en DMF. 1.5 eq.) a -40 °C. Después. la mezcla se agitó a -40 °C durante 0.5 horas y a 0 °C durante 0.5 horas más. Se añadieron ácido 2-fluoroprop-2-enoico (11 mg) y T3P (100 μl) y la mezcla se agitó a -40 °C durante 0.5 horas más. La mezcla se diluyó con agua (15 ml) y se extrajo con EtOAc (2 * 40 ml). Las capas orgánicas se lavaron con salmuera (50 ml). se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Luna C18150 * 255 u; fase móvil: [agua (0.225 % de AF) - MeCN1; % de B: 22 % - 42 %.7.8 min) y (columna: Phenomenex Gemini 150 * 25 mm * 10 μm; fase móvil: [agua (0.04 % de NH3H20 NH4HC0310 mM) - MeCN1; % de B: 50 % - 68 %. 12 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-((S)-1-(2-fluoroacriloil)-4-(7-(6-metil-5-(trifluorometil)-1H-indazol-4-il)-2-*(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-cfjpirimidin-4-il)piperazin-2-il)acetonitrilo en forma de un sólido de color blanco. LCMS [ESI. M+11: 642.
Ejemplo 556

2-[(2S)-1-(2-Fluoroprop-2-enoil)-4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6.8-dihidro-5H-pirido[3.4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo

3-Bromo-1-(metoximetoxi)naftaleno. A una solución de 4-bromonaftalen-2-ol (3,50 g, 15,7 mmol, 1,0 eq.) en DMF (35,0 ml) se le añadió NaH (941 mg, 23,5 mmol, 60 % de pureza, 1,50 eq.). Después de la adición, la mezcla se agitó a esta temperatura durante 0,5 horas, y después se añadió gota a gota cloro(metoxi)metano (5,90 g, 73,3 mmol, 5,57 ml, 4,67 eq.) a 0 °C. La mezcla se calentó a 30 °C y se agitó durante 1 hora más. Después de que se completara, la mezcla de reacción combinada se diluyó con agua (100 ml) y se extrajo con EtOAc (3 * 50,0 ml). Las capas orgánicas combinadas se lavaron con salmuera saturada (200 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 1/0 a 100/1). El compuesto 3-bromo-1-(metoximetoxi)naftaleno (3,50 g, 13,1 mmol, 84 % de rendimiento) se obtuvo en forma de un aceite de color rojo.
1H RMN (400 MHz, cloroformo-d) ó 8,19 - 8,12 (m, 1H), 7,78 - 7,71 (m, 1H), 7,61 - 7,57 (m, 1H), 7,52 - 7,44 (m, 2H), 7,40 (d, J = 2,0 Hz, 1H), 5,29 (s, 2H), 3,53 (s, 3H).
Etapa A: (2S)-2-(cianometil)-4-[7-[3-(metoximetoxi)-1-naftil1-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1 -carboxilato de /ere-butilo. A una solución de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-5,6,7,8-tetrahidropirido[3,4-dJpirimidin-4-il1piperazin-1 -carboxilato de /ere-butilo (800 mg, 1,70 mmol, 1,0 eq.) y 1-bromo-3-(metoximetoxi)naftaleno (680 mg, 2,54 mmol, 1,50 eq.) en tolueno (10,0 ml) se le añadieron Cs2C03 (2,21 g, 6,79 mmol, 4,00 eq.), RuPhos (158 mg, 339 qmol, 0,20 eq.) y Pd2(dba)3 (233 mg, 254 qmol, 0,15 eq.). La mezcla se agitó a 90 °C durante 6 horas. La mezcla de reacción se diluyó con H20100 ml y se extrajo con acetato de etilo (3 * 100 ml). Las capas orgánicas combinadas se concentraron a presión reducida para dar un residuo. El residuo se purificó adicionalmente por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico)/acetonitrilo)1. La mezcla se ajustó a pH = 7 con una solución acuosa de NaHC03 y se extrajeron con acetato de etilo (3 * 50,0 ml). Las capas orgánicas combinadas se lavaron con salmuera saturada (100 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar el producto. El compuesto (2S)-2-(cianometil)-4-[7-[3-(metoximetoxi)-1-naftil1-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-djpirimidin-4-il1piperazin-1 -carboxilato de /ere-butilo (280 mg, 426 qmol, 25 % de rendimiento) se obtuvo en forma de un sólido de color amarillo.
LCMS [ESI, M+11: 658.
1H RMN (400 MHz, cloroformo-d) ó 8,09 (d, J = 8,0 Hz, 1H), 7,75 (d, J = 8,2 Hz, 1H), 7,45 (t, J = 7,0 Hz, 1H), 7,40 -7,34 (m, 1H), 7,16 (d, J = 2,0 Hz, 1H), 6,87 (d, J = 2,4 Hz, 1H), 5,30 (s, 2H), 4,63 (s a, 1H), 4,40 (dd, J = 4,8, 10,4 Hz, 1H), 4,33 - 4,22 (m, 2H), 4,21 - 4,16 (m, 1H), 4,09 - 3,86 (m, 3H), 3,54 (s, 3H), 3,51 - 3,40 (m, 1H), 3,37 - 3,17 (m, 3H), 3,15 - 2,96 (m, 3H), 2,89 - 2,73 (m, 3H), 2,72 - 2,64 (m, 1H), 2,49 (s, 3H), 2,33 - 2,25 (m, 1H), 2,13 - 2,05 (m, 1H), 1,91 - 1,78 (m, 3H), 1,52 (s, 9H).
Etapa B: 2-[(2S)-4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-cf1pirimidin-4-il1p¡perazin-2-il1acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-[3-(metoximetoxi)-1 -naftil1-2-[[(2S)-1 -metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-djpirimidin-4-il1piperazin-1-carboxilato de terc-butilo (260 mg, 395 qmol, 1,0 eq.) en
dioxano (2,0 ml) se le añadió HChdioxano (4 M, 98,8 μl, 1,0 eq.). La mezcla se agitó a 20 °C durante 1 hora. Después de que se completara, la mezcla de reacción se concentró a presión reducida para dar un residuo. El residuo se ajustó a pH = 7 con una solución acuosa saturada de NaHCÜ3 y se extrajo con acetato de etilo (3 x 20,0 ml). Las capas orgánicas combinadas se lavaron con salmuera saturada (30,0 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar el residuo. El compuesto 2-[(2s)-4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (200 mg, 373 μmol, 94 % de rendimiento, 96 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 514.
Etapa C: 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi]-6,8-dihidro-5H-p¡rido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (200 mg, 389 μmol, 1,0 eq.) y ácido 2-fluoroprop-2-enoico (105 mg, 1,17 mmol, 3,0 eq.) en DMF (4,00 ml) se le añadieron TEA (158 mg, 1,56 mmol, 217 μl, 4,0 eq.) y T3P (743 mg, 1,17 mmol, 695 μl, 50 % de pureza, 3,0 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se diluyó con H2Ü (10,0 ml) y se extrajo con DCM (3 x10,0 ml). Las capas orgánicas combinadas se lavaron con salmuera saturada (20,0 ml), se secaron sobre Na2Sü4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Al203, éter de petróleo/acetato de etilo = 10/1 a DCM/Me0H = 5/1) y el residuo se purificó por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 μm;fase móvil: [agua(0,04 % de NH3H2Ü NH4HCÜ3 10 mM)-MeCN];% de B: 45 %-75 %,12 min). El compuesto del título 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[7-(3-hidroxi-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (50,0 mg, 85,0 μmol, 22 % de rendimiento, 99,6 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 586.
1H RMN (400 MHz, cloroformo-d) 57,94 (d a, J = 8,4 Hz, 1H), 7,62 (d, J = 8,0 Hz, 1H), 7,40 (t, J = 7,2 Hz, 1H), 7,31 -7,26 (m, 1H), 6,86 (s, 1H), 6,52 (s a, 1H), 5,51 - 5,29 (m, 1H), 5,25 (dd, J = 3,6, 16,8 Hz, 1H), 4,95 - 4,35 (m, 2H), 4,24 (dd, J = 4,4, 11,2 Hz, 1H), 4,15 - 3,93 (m, 3H), 3,85 - 3,69 (m, 1H), 3,45 - 3,26 (m, 1H), 3,25 - 3,18 (m, 2H), 3,17 - 2,95 (m, 2H), 2,94 - 2,73 (m, 3H), 2,72 - 2,62 (m, 5H), 2,60 - 2,50 (m, 1H), 2,45 - 2,35 (m, 1H), 2,15 - 2,06 (m, 1H), 2,01 -1,93 (m, 1H), 1,88 - 1,74 (m, 3H).
Ejemplo 557

2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo

Etapa A: 4-[(3S)-4-terc-Butoxicarbonil-3-(cianometil)piperazin-1-il1-2-[[(2R)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-7-carboxilato de bencilo. A una solución de [(2R)-1-metilpirrolidin-2-il]metanol (934 mg, 8,11 mmol, 3,0 eq.) y t-Bu0Na (780 mg, 8,11 mmol, 3,0 eq.) en tolueno (15,0 ml) se le añadió gota a gota una solución de 4-[(3S)-4-terc-butoxicarbonil-3-(cianometil)piperazin-1-il]-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de bencilo (1,5 g, 2,70 mmol, 1,0 eq.) en tolueno (10,0 ml) a 0 °C. La mezcla de reacción se agitó a 0 °C durante 0,5 h más. Después de que se completara, la reacción se interrumpió con agua (30,0 ml). La mezcla bruta se extrajo con acetato de etilo (2 * 50,0 ml). Los extractos combinados se lavaron con salmuera (100 ml), se secaron con Na2S04 y después el disolvente se eliminó al vacío. El residuo se purificó por HPLC ultrarrápida de fase inversa [C18, 0,1 % de AF en agua, 0 - 65 % de MeCN], se acidificó con NaHC03 a pH = 8 y se extrajo con acetato de etilo (2 * 150 ml). Las capas orgánicas se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El compuesto 4-[(3S)-4-terc-butoxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de bencilo (820 mg, 1,26 mmol, 47 % de rendimiento, 93 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1 ]: 606.
1H RMN (400 MHz, cloroformo-d) ó 7,42 - 7,29 (m, 5H), 5,18 (s, 2H), 4,69 (d a, J = 18,6 Hz, 1H), 4,59 (s a, 1H), 4,44 (d, J = 18,8 Hz, 1H), 4,34 (s a, 1H), 4,05 - 3,69 (m, 4H), 3,42 (s a, 1H), 3,24 (d a, J = 13,0 Hz, 2H), 3,09 (t a, J = 7,6 Hz, 1H), 3,02 - 2,90 (m, 1H), 2,81 - 2,56 (m, 5H), 2,47 (s, 3H), 2,28 (dt, J = 7,2, 9,4 Hz, 1H), 2,05 - 1,99 (m, 1H), 1,89 - 1,67 (m, 4H), 1,51 (s, 9H).
Etapa B: (2S)-2-(Cianometil)-4-[2-[[(2R)-1-metilpirrolidin-2-il1metoxi1-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-¡npiperazin-1-carboxilato de terc-butilo. Se burbujeó NH3 en Me0H (10,0 ml) a -70 °C durante 0,5 h. Se añadieron 4-[(3S)-4-terc-butoxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de bencilo (820 mg, 1 ,35 mmol, 1 ,0 eq.) y Pd/C (250 mg, 10 % de pureza) a la mezcla anterior y Me0H (10,0 ml), después la mezcla se desgasificó y se purgó 3 veces con H2 y después la mezcla se agitó a 20 °C durante 0,5 h en una atmósfera de H2 (103,42 kPa (15 psi)). Después de que se completara, la mezcla bruta se filtró a través de una capa de celite. La torta se lavó con Me0H (50,0 ml) y el filtrado se secó a alto vacío. El compuesto (2S)-2-(c¡anomet¡l)-4-[2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carboxilato de terc-butilo (600 mg, 1,27 mmol, 94 % de rendimiento, 100 % de pureza) se obtuvo en forma de un sólido de color blanco y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 472.
Etapa C: (2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1 -carboxilato de terc-butilo. Una mezcla de (2S)-2-(cianometil)-4-[2-[[(2R)-1-metilpirrolidin-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de terc-butilo (550 mg, 1,17 mmol, 1,0 eq.), 1 bromo-8 -cloro-naftaleno (845 mg, 3,50 mmol, 3,0 eq.), Pd2(dba)3 (160 mg, 175 gmol, 0,15 eq.), Xantphos (135 mg, 233 gmol, 0,20 eq.) y Cs2C03 (1,14 g, 3,50 mmol, 3,0 eq.) en tolueno (10,0 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 8 h en una atmósfera de N2. Después de que se completara, la reacción se lavó con una solución acuosa de HCl (2 N, 2 * 15 ml). La fase acuosa se basificó con NaHC03 a pH 8 y se extrajo con acetato de etilo (2 * 50 ml). Las capas orgánicas se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC ultrarrápida de fase inversa [C18, 0,1 % de AF en agua, 0 - 65 % de MeCN], se acidificó con NaHC03 a pH = 8 y se extrajo con acetato de etilo (2 * 150 ml). Las capas orgánicas se secaron sobre Na2S04, se
filtraron y se concentraron al vacío. El compuesto (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de terc-butilo (600 mg, 930 μmol, 80 % de rendimiento, 98 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 632.
1H RMN (400 MHz, cloroformo-d) 57,75 (d a, J = 8,2 Hz, 1H), 7,61 (t, J = 8,0 Hz, 1H), 7,52 (d, J = 7,5 Hz, 1H), 7,44 (td, J = 7,8, 15,2 Hz, 1H), 7,33 (t, J = 7,8 Hz, 1H), 7,27 - 7,17 (m, 1H), 4,61 (s a, 1H), 4,48 - 4,34 (m, 2H), 4,10 - 3,91 (m, 3H), 3,90 - 3,78 (m, 1H), 3,63 - 3,53 (m, 1H), 3,34 (dd a, J = 3,6, 13,8 Hz, 1H), 3,30 - 3,01 (m, 5H), 2,95 (dt, J = 3,5, 12,3 Hz, 1H), 2,89 - 2,75 (m, 1H), 2,70 (s a, 2H), 2,57 (d a, J = 14,2 Hz, 1H), 2,53 - 2,46 (m, 3H), 2,35 - 2,25 (m, 1H), 2,13 - 2,06 (m, 1H), 1,93 - 1,66 (m, 3H), 1,52 (s, 9H).
Etapa D: 2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-illpiperazin-2-illacetonitrilo. A una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (200 mg, 316 μmol, 1,0 eq.) en dioxano (2,0 ml) se le añadió HCl/dioxano (4 M, 2,0 ml, 25,3 eq.). La mezcla se agitó a 20 °C durante 0,5 horas en una atmósfera de N2. Después de que se completara, el disolvente orgánico se eliminó al vacío. El producto obtenido se ajustó con NaHCÜ3 acuoso saturado a pH = 8 y después se concentró. La fase acuosa se extrajo con acetato de etilo (3 x 10 ,0 ml) y la capa orgánica combinada se secó sobre Na2SÜ4, se filtró y se concentró. El producto bruto 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (145 mg, 259 μmol, 82 % de rendimiento, 95 % de pureza) se obtuvo en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 532
Etapa E: 2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-(2-f¡uoroprop-2-enoil)piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (125 mg, 235 μmol, 1,0 eq.) y ácido 2-fluoroprop-2-enoico (42,3 mg, 470 μmol, 2,0 eq.) en acetato de etilo (2,0 ml) se le añadieron TEA (190 mg, 1,88 mmol, 262 μl, 8,0 eq.) y T3P (449 mg, 705 μmol, 419 μl, 50 % de pureza, 3,0 eq.) a 0 °C. La mezcla se agitó a 20 °C durante 1 hora. Después de que se completara, el disolvente orgánico se lavó con agua (5,0 ml). La fase acuosa se extrajo con acetato de etilo (3 x 10 ml). Los extractos orgánicos combinados se lavaron con salmuera (20,0 ml), se secaron con Na2SÜ4 y después el disolvente se eliminó al vacío. El residuo se purificó por cromatografía en columna (Al2Ü3 básico, éter de petróleo : acetato de etilo = 3 : 1 a acetato de etilo : metanol = 50 : 1) y después el producto bruto se concentró y se purificó de nuevo por HPLC prep. (columna: Phenomenex Gemini 150 x 25 mm x 10 μm; fase móvil: [agua (0,05 % de hidróxido de amonio v/v)-MeCN];% de B: 55 % - 85 %, 11,5 min) y se sometió a liofilización. El compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (22,5 mg, 37,1 μmol, 16 % de rendimiento, 99 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 605.
1H RMN (400 MHz, cloroformo-d) 57,76 (d a, J = 8,4 Hz, 1H), 7,62 (t, J = 7,6 Hz, 1H), 7,53 (d, J = 7,2 Hz, 1H), 7,49 -7,40 (m, 1H), 7,34 (t, J = 7,8 Hz, 1H), 7,26 - 7,18 (m, 1H), 5,51 - 5,32 (m, 1H), 5,26 (dd a, J = 3,2, 16,8 Hz, 1H), 4,90 (s a, 1H), 4,49 - 4,34 (m, 2H), 4,24 - 4,01 (m, 3H), 3,97 - 3,78 (m, 2H), 3,64 - 3,55 (m, 1H), 3,44 (s a, 1H), 3,32 - 2,95 (m, 5H), 2,94 - 2,43 (m, 7H), 2,38 - 2,20 (m, 1H), 2,14 - 1,99 (m, 1H) 1,91 - 1,71 (m, 3H).
Ejemplo 558

2-((S)-4-(7-(8-Cloronaftalen-1-il)-2-(((2R,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo

Etapa A: 4-[(3S)-4-/ere-Butoxicarbonil-3-(cianometil)piperazin-1-il1-2-[[(2R4R)-4-fluoro-1-metil-pirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-7-carboxilato de bencilo. A una solución de [(2R,4R)-4-fluoro-1-metil-pirrolidin-2-il]metanol (169 mg, 1,27 mmol, 1,5 eq.) en tolueno (4,0 ml) se le añadió t-Bu0Na (163 mg, 1,69 mmol, 2,0 eq.) a 0 °C, después de lo cual la mezcla de reacción se agitó a 0 °C durante 0,5 horas, a la mezcla se le añadió 4-[(3S)-4-/erebutoxicarbonil-3-(cianometil)piperazin-1-il1-2-metilsulfinil-6,8-óihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de bencilo (470 mg, 847 gmol, 1,0 eq.) y la mezcla de reacción se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se añadió a agua (10 ml), después se extrajo con acetato de etilo (2 x 10,0 ml) y la capa orgánica se secó sobre Na2S04, se filtró y se concentró. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 3/1 a acetato de etilo/metanol = 20/1) para dar 4-[(3S)-4-/ere-butoxicarbonil-3-(cianometil)piperazin-1-il1-2-[[(2R,4R)-4-fluoro-1-metil-pirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de bencilo (330 mg, 484 gmol, 57 % de rendimiento, 92 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+11: 624.
1H RMN (400 MHz, cloroformo-d) ó 7,40 - 7,30 (m, 5H), 5,24 - 5,03 (m, 3H), 4,69 (d, J = 18,8 Hz, 1H), 4,61 - 4,53 (m, 1H), 4,51 - 4,38 (m, 2H), 4,23 (dd, J = 7,6, 10,4 Hz, 1H), 4,08 - 3,73 (m, 4H), 3,52 - 3,11 (m, 4H), 3,04 - 2,91 (m, 1H), 2,81 - 2,57 (m, 5H), 2,55 - 2,36 (m, 5H), 2,11 - 1,99 (m, 1H), 1,51 (s, 9H).
Etapa B: (2S)-2-(Cianometil)-4-[2-[[(2R4R)-4-fluoro-1-metilpirrolidin-2-il1metoxi1-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de /ere-butilo. A una solución de 4-[(3S)-4-/ere-butoxicarbonil-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-2-[[(2R,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-7-carboxilato de bencilo (330 mg, 529 gmol, 1,0 eq.) en metanol (3,0 ml) y NH3 Me0H (529 μmol, 3 ml, 20 % de pureza, 1,0 eq.) se le añadió Pd/C (50 mg, 529 gmol, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 15 °C durante 3 horas. Después de que se completara, la mezcla se filtró, la torta de filtro se lavó con THF (2 x 3 ,0 ml) y el filtrado se concentró. El residuo se purificó por cromatografía ultrarrápida de fase inversa (0,1 % de AF en MeCN). La mezcla se ajustó con NaHC03 sólido a pH -8 y después se concentró. La capa acuosa (20,0 ml) se extrajo con acetato de etilo (2 x 15 ,0 ml) y la capa orgánica se secó sobre Na2S04, se filtró y se concentró para dar (2S)-2-(cianometil)-4-[2-[[(2R,4R)-4-fluoro-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de /ere-butilo (150 mg, 299 gmol, 58 % de rendimiento, 98 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+11: 490.
Etapa C: (2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2R4R)-4-fluoro-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1 -carboxilato de /ere-butilo. A una solución de (2S)-2-(cianometil)-4-[2-[[(2R,4R)-4-fluoro-1-metil-pirrolidin-2-il1metoxi1-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de /ere-butilo (150 mg, 306 gmol, 1,0 eq.) y 1-bromo-8-cloronaftaleno (148 mg, 618 gmol, 2,0 eq.) en tolueno (3,0 ml) se le añadieron XantPhos (35,5 mg, 61,3 gmol, 0,2 eq.), Pd2(dba)3 (42,1 mg, 45,9 gmol, 0,15 eq.) y Cs2C03 (399 mg, 1,23 mmol, 4,0 eq.) y la mezcla de reacción se agitó a 90 °C durante 12 horas en una atmósfera de N2. Después de que se completara, la mezcla de reacción se filtró a través de celite, el filtrado se lavó con HCl acuoso 1 N (2 x 10,0 ml), la capa acuosa se ajustó a pH ~8 con Na2C03 sólido y después se extrajo con acetato de etilo (2 x 15 ,0 ml) y la capa orgánica se secó sobre Na2S04, se filtró y se concentró. El residuo se purificó por cromatografía ultrarrápida de fase
inversa (0,1 % de AF en MeCN). El producto obtenido se ajustó con NaHCÜ3 sólido a pH ~8 y se concentró. La capa acuosa (20 ml) se extrajo con acetato de etilo (2 x 15,0 ml) y la capa orgánica se secó sobre Na2SÜ4, se filtró y se concentró. El producto (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (85 mg, 129 μmol, 43 % de rendimiento, 98 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1, M+23]: 650, 672.
1H RMN (400 MHz, cloroformo-d) 57,76 (d a, J = 8,0 Hz, 1H), 7,62 (t, J = 8,0 Hz, 1H), 7,52 (d, J = 7,6 Hz, 1H), 7,49 -7.40 (m, 1H), 7,34 (t, J = 7,8 Hz, 1H), 7,26 - 7,17 (m, 1H), 5,24 - 5,03 (m, 1H), 4,67 - 4,55 (m, 1H), 4,52 - 4,36 (m, 2H), 4,23 (dd, J = 7,6, 10,4 Hz, 1H), 4,13 - 3,77 (m, 4H), 3,65 - 3,52 (m, 1H), 3,43 - 2,86 (m, 7H), 2,84 - 2,64 (m, 3H), 2,62 - 2,38 (m, 5H), 2,16 - 1,98 (m, 1H), 1,52 (s, 9H).
Etapa D: 2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[(2R4R)-4-fluoro-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il]acetonitrilo. A una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R,4R)-4-fluoro-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (85,0 mg, 131 μmol, 1,0 eq.) en dioxano (1,0 ml) se le añadió HCl 4 M/dioxano (1,0 ml) y la mezcla de reacción se agitó a 20 °C durante 4 horas. Después de que se completara, la mezcla de reacción se concentró, a la mezcla se le añadió diclorometano (10 ml), después se ajustó con Na2CÜ3 acuoso saturado a pH -8 y la capa orgánica se separó, después se secó sobre Na2SÜ4, se filtró y se concentró. El producto 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (90 mg, bruto) se obtuvo en forma de un sólido de color pardo. LCMS [ESI, M+1]: 550.
Etapa E: 2-((S)-4-(7-(8-Cloronaftalen-1-il)-2-(((2R4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (90,0 mg, 120 μmol, 73 % de pureza, 1,0 eq.) y ácido 2-fluoroprop-2-enoico (21,7 mg, 241 μmol, 2,0 eq.) en acetato de etilo (2,0 ml) se le añadieron T3P (230 mg, 361 μmol, 215 μl, 50 % de pureza, 3,0 eq.) y TEA (97,6 mg, 962 μmol, 134 μl, 8,0 eq.) y la mezcla de reacción se agitó a 20 °C durante 1 hora. Después de que se completara, se añadió agua (10,0 ml), después la capa orgánica se separó y la capa acuosa se extrajo con acetato de etilo (2 x 10,0 ml). La capa orgánica combinada se secó sobre Na2SÜ4, se filtró y se concentró. El residuo se purificó por cromatografía en columna (SiÜ2, éter de petróleo/acetato de etilo = 3/1 a acetato de etilo:metanol = 10:1). El producto bruto se purificó de nuevo por HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 μm; fase móvil: [agua (0,04 % de NH3H2Ü NH4HCÜ3 10 mM)-MeCN]; % de B: 48 %-78 %, 12 min) y el producto obtenido se concentró y después se liofilizó. El producto compuesto del título 2-((S)-4-(7-(8-cloronaftalen-1-il)-2-(((2R,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo (10,9 mg, 17,3 μmol, 14 % de rendimiento, 99 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 622.
1H RMN (400 MHz, Cloroformo-d) 57,76 (d, J = 8,0 Hz, 1H), 7,63 (t, J = 7,6 Hz, 1H), 7,53 (d, J = 7,2 Hz, 1H), 7,49 -7.41 (m, 1H), 7,34 (t, J = 7,8 Hz, 1H), 7,27 - 7,18 (m, 1H), 5,50 - 5,34 (m, 1H), 5,26 (dd, J = 3,6, 16,8 Hz, 1H), 5,22 -5,04 (m, 1H), 4,98 - 4,72 (m, 1H), 4,54 - 4,37 (m, 2H), 4,30 - 4,02 (m, 3H), 3,96 - 3,75 (m, 2H), 3,66 - 3,54 (m, 1H), 3,51 - 2,99 (m, 6H), 2,98 - 2,67 (m, 3H), 2,64 - 2,36 (m, 6H), 2,14 - 1,97 (m, 1H).
Ejemplo 559
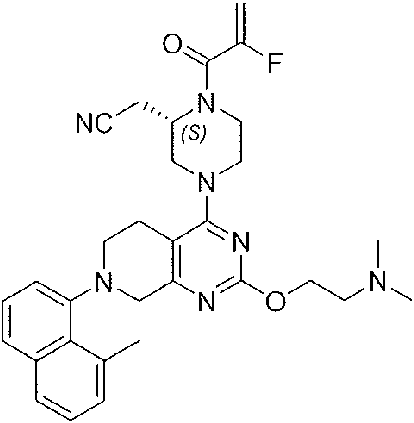
2-[(2S)-4-[2-[2-(Dimetilamino)etoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo

Etapa A: 2-[(7-Bencil-4-metoxi-6,8-dihidro-5H-pirido[3,4-dlpirimidin-2-il)oxi1-M,M-dimetil-etanamina. A la solución de 7-bencil-2-cloro-4-metoxi-6,8-dihidro-5H-pirido[3,4-d]pirimidina (5 g, 17,3 mmol, 1 eq.), 2-(dimetilamino)etanol (3,08 g, 34,5 mmol, 3,46 ml, 2 eq.), CS2C03 (5,62 g, 17,3 mmol, 1 eq.) y BINAP (2,15 g, 3,45 mmol, 0,2 eq.) en tolueno (100 ml) se le añadió Pd(0Ac)2 (387 mg, 1,73 mmol, 0,1 eq.) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con N2. La mezcla se agitó en una atmósfera de N2 a 100 °C durante 12 horas. La mezcla de reacción se filtró. Al filtrado se le añadió HCl (2 M, 200 ml) y después se separó. La fase acuosa se basificó con NaHC03 y K2C03 a pH = 8~9 y después se extrajo con EtOAc (5 x 80 ml). La fase orgánica combinada se lavó con salmuera (100 ml), se secó con Na2S04 anhidro, se filtró y se concentró al vacío para dar 2-[(7-bencil-4-metoxi-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il)oxi]-W,W-dimetil-etanamina (4,8 g, 12,6 mmol, 73 % de rendimiento, 90 % de pureza) en forma de un aceite de color pardo que se usó para la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, cloroformo-d) ó = 7,40 - 7,28 (m, 5H), 4,40 (t, J = 6,0 Hz, 2H), 3,96 (s, 3H), 3,69 (s, 2H), 3,50 (s, 2H), 2,76 - 2,69 (m, 4H), 2,62 - 2,56 (m, 2H), 2,32 (s, 6H).
Etapa B: 2-[(4-Metoxi-5,6,7,8-tetrahidropirido[3,4-dlpirimidin-2-il)oxil-M,M-dimetil-etanamina. A la solución de 2-[(7-bencil-4-metoxi-6,8-dihidro-5H-pirido[3,4-o7pirimidin-2-il)oxi]-W,W-dimetil-etanamina (4,8 g, 14,0 mmol, 1 eq.) en Me0H (100 ml) se le añadió Pd(0H)2/C (2 g, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 40 °C durante 18 horas. A la mezcla se le añadió HCl/Me0H (4 M, 7,01 ml, 2 eq.). La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 40 °C durante 2 horas. La mezcla se filtró y la torta de filtro se lavó con NH3*Me0H (10 %, 3 x 30 ml). El filtrado se concentró al vacío. El residuo se disolvió con agua (10 ml) y EtOAc (10 ml) y después la mezcla se basificó con K2C03 a pH = 11. La mezcla se extrajo con EtOAc (8 x 20 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío para dar 2-[(4-metoxi-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-2-il)oxi]-W,W-dimetil-etanamina (2,58 g, 8,69 mmol, 62 % de rendimiento, 85 % de pureza) en forma de un aceite de color pardo que se usó para la siguiente etapa sin purificación adicional. LCMS [ESI, M+1 ]: 253.
Etapa C: 2-[[4-Metoxi-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-dlpirimidin-2-i n oxil-M,M-dimetil-etanamina. A la solución de 2-[(4-metox¡-5,6,7,8-tetrah¡drop¡r¡do[3,4-cf]p¡r¡m¡d¡n-2-¡l)ox¡]-W,W-d¡met¡l-etanam¡na (2,28 g, 7,68 mmol, 1 eq.), 1-bromo-8-metil-naftaleno (2,55 g, 11,5 mmol, 1,5 eq.), Cs2C03 (7,51 g, 23,0 mmol, 3 eq.) y Xantphos (889 mg, 1,54 mmol, 0,2 eq.) en tolueno (70 ml) se le añadió Pd2(dba)3 (703 mg, 768 μmol, 0,1 eq.) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con N2. La mezcla se agitó en una atmósfera de N2 a 100 °C durante 16 horas. La mezcla de reacción se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc = 10:1 ~ 0:1 a EtOAc: Me0H = 1:0 ~ 10:1) para dar 2-[[4-metox¡-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡din-2-¡l]ox¡]-W,W-d¡met¡l-etanam¡na (3,2 g, 7,34 mmol, 95 % de rendimiento, 90 % de pureza) en forma de un sólido de color pardo. LCMS [ESI, M+1]: 393.
1H RMN (400 MHz, cloroformo-d) ó = 7,69 (d, J = 8,0 Hz, 1H), 7,64 (d, J = 7,6 Hz, 1H), 7,40 (t, J = 7,6 Hz, 1H), 7,33 (t, J = 7,6 Hz, 1H), 7,27 - 7,22 (m, 2H), 4,44 (t, J = 6,4 Hz, 2H), 4,17 - 4,05 (m, 1H), 4,03 (s, 3H), 3,81 (d, J = 17,6 Hz, 1H), 3,57 - 3,48 (m, 1H), 3,20 (dt, J = 4,0, 11,2 Hz, 1H), 2,94 - 2,82 (m, 4H), 2,75 (t, J = 6,4 Hz, 2H), 2,67 (d a, J = 16,4 Hz, 1H), 2,34 (s, 6H).
Etapa D: 2-[2-(Dimetilamino)etoxi1-7-(8-metil-1-naftin-6.8-dihidro-5H-pirido[3,4-dlpirimidin-4-ol. A la solución de EtSH (1,96 g, 31,5 mmol, 2,33 ml, 3,64 eq.) en DMF (60 ml) se le añadió NaH (693 mg, 17,3 mmol, 60 % de pureza, 2 eq.) y la mezcla se agitó a 10 °C durante 0,5 horas. Después, se añadió 2-[[4-metoxi-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oxi]-N,N-dimetil-etanamina (3,4 g, 8,66 mmol, 1 eq.) en DMF (10 ml) y la mezcla se calentó a 60 °C y se agitó durante 1 hora. La mezcla se vertió en agua enfriada con hielo (200 ml) y se extrajo con EtOAc (30 ml). La fase acuosa se acidificó con HCl (2 M) a pH = 7-8 y después se extrajo con EtOAc (6 x 80 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se trituró con EP: EtOAc = 5:1 para dar 2-[2-(d¡met¡lam¡no)etox¡]-7-(8-metil-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-ol (2,4 g, 6,28 mmol, 72 % de rendimiento, 99 % de pureza) en forma de un sólido de color blanco. Lc MS [ESI, M+1]: 379.
Etapa E: Trifluorometanosulfonato de [2-[2-(dimetilamino)etoxi1-7-(8-metil-1-naftil)-6.8-dihidro-5H-pirido[3.4-d^pirimidin-4-ilo]. A la solución de 2-[2-(d¡met¡lam¡no)etox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡rido[3,4-cf]p¡r¡m¡d¡n-4-ol (1 g, 2,64 mmol, 1 eq.) y TEA (668 mg, 6,61 mmol, 919 μl, 2,5 eq.) en DCM (20 ml) se le añadió Tf20 (1,12 g, 3,96 mmol, 654 μl, 1,5 eq.) a -40 °C y la mezcla se agitó a -40 °C durante 10 min. A la mezcla se le añadió agua (10 ml) y después se separó. La fase acuosa se extrajo con EtOAc (20 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc = 5:1 ~0:1) para dar trifluorometanosulfonato de [2-[2-(d¡met¡lam¡no)etox¡]-7-(8-metil-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡lo] (650 mg, 1,15 mmol, 43 % de rendimiento, 90 % de pureza) en forma de un aceite de color pardo.
Etapa F: 2-[(2S)-4-[2-[2-(Dimetilamino)etoxi1-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d^pirimidin-4-il1piperazin-2-¡ n acetonitrilo. A la solución de trifluorometanosulfonato de [2-[2-(dimet¡lam¡no)etox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡hidro-5H-piridop^-cdpirimidin^-ilo] (350 mg, 685 μmol, 1 eq.) y 2-[(2S)-piperazin-2-il]acetonitrilo (111 mg, 754 μmol, 1,1 eq.) en DMAC (4 ml) se le añadió DIEA (177 mg, 1,37 mmol, 239 μl, 2 eq.) y la mezcla se agitó a 10 °C durante 10 min. La mezcla se purificó por columna ultrarrápida de fase inversa (MeCN/agua (0,1 % de AF) = 40 %) para dar 2-[(2S)-4-[2-[2-(d¡met¡lam¡no)etox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡mid¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (60 mg, 121 μmol, 18 % de rendimiento, 98 % de pureza) en forma de un aceite de color pardo. LCMS [ESI, M+1]: 486.
Etapa F: 2-[(2S)-4-[2-[2-(Dimetilamino)etoxi1-7-(8-metil-1-nafti^)-6.8-dihidro-5H-pirido[3.4-cf]pirimidin-4-il1-1-(2-fluoroprop-2-enoil)piperazin-2-i n acetonitrilo. A la solución de 2-[(2S)-4-[2-[2-(dimet¡lam¡no)etox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡hidro-5H-pirido[3.4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo (60 mg, 123 μmol, 1 eq.), ácido 2-fluoroprop-2-enoico (22,2 mg, 247 μmol, 2 eq.) y TEA (100 mg, 988 μmol, 138 μl, 8 eq.) en EtOAc (1,2 ml) se le añadió T3P (236 mg, 370 μmol, 220 μl, 50 % de pureza, 3 eq.) a 0 °C y la mezcla se agitó a 10 °C durante 0,5 horas. A la mezcla se le añadió agua (5 ml). La mezcla se extrajo con EtOAc (2 x 8 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía sobre Ah03 (EtOAc: Me0H = 1:0 ~ 20:1). Después, el residuo se purificó por HPLC prep. (columna: Gemini 150 * 255 p;fase móvil: [agua (0,05 % de hidróxido de amonio v/v)-MeCN];% de B: 53 % - 83 %, 12 min) para dar el compuesto del título 2-[(2S)-4-[2-[2-(dimetilamino)etoxi]-7-(8-metil-1-naftil)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]-1-(2-fluoroprop-2-eno¡l)p¡peraz¡n-2-¡l]aceton¡tr¡lo (39,5 mg, 70,4 μmol, 57 % de rendimiento, 99,4 % de pureza) en forma de un sólido de color gris. LCMS [ESI, M+1]: 558.
1H RMN (400 MHz, cloroformo-d) 5 = 7,70 (d a, J = 8,4 Hz, 1H), 7,65 (t, J = 8,0 Hz, 1H), 7,46 - 7,38 (m, 1H), 7,35 (t, J = 7,6 Hz, 1H), 7,27 - 7,18 (m, 2H), 5,52 - 5,33 (m, 1H), 5,26 (dd, J = 3,6, 16,8 Hz, 1H), 4,88 (s a, 1H), 4,47 - 4,37 (m, 2H), 4,31 - 4,18 (m, 1H), 4,17 - 4,01 (m, 2H), 3,94 - 3,84 (m, 1H), 3,77 (d, J = 18,0 Hz, 1H), 3,58 - 3,41 (m, 2H), 3,25 -2,95 (m, 4H), 2,92 (s, 3H), 2,90 - 2,77 (m, 2H), 2,77 - 2,70 (m, 2H), 2,68 - 2,56 (m, 1H), 2,34 (d, J = 3,6 Hz, 6H).
Ejemplo 560

2-[(2S)-4-[2-[[(2R,4R)-4-Fluoro-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-(2-fluoroprop-2-eno¡l)p¡peraz¡n-2-¡l]aceton¡trilo
(2S,4R)-4-Fluoropirrolidin-1,2-dicarboxilato de 1-terc-butil-2-metilo. A una solución de ácido (2S,4R)-1-terc-butoxicarbonil-4-fluoro-pirrolidin-2-carboxílico (2,0 g, 8,58 mmol, 1,0 eq.) y Ag20 (3,97 g, 17,2 mmol, 2,0 eq.) en CH3CN (20,0 ml) se le añadió en porciones MeI (2,43 g, 17,2 mmol, 1,07 ml, 2,0 eq.) a 0 °C y la mezcla de reacción se agitó a 15 °C durante 12 horas. Después de que se completara, la mezcla de reacción se añadió a agua (30,0 ml), después la capa orgánica se separó, la fase acuosa se extrajo con acetato de etilo (30,0 ml x 2) y la capa orgánica combinada se secó sobre Na2S04, se filtró y se concentró. El residuo se purificó por cromatografía en columna (Si02,
éter de petróleo:acetato de etilo = 3:1 a acetato de etilo:metanol = 10:1). El producto (2S, 4R)-4-fluoropirrolidin-1,2-dicarboxilato de 1-terc-butil-2-metilo (1,98 g, 8,01 mmol, 93 % de rendimiento) se obtuvo en forma de un aceite incoloro.

1H RMN (400 MHz, Cloroformo-d) 55,29-5,15 (m, 1H), 4,49-4,38 (m, 1H), 3,92-3,74 (m, 1H), 3,68 (s, 3H), 3,60-3,56 (m, 1H), 2,63-2,54 (m, 1H), 2,17-2,05 (m, 1H), 1,44 (d, J = 18,0 Hz, 9H).
[(2S,4R)-4-Fluoro-1-metil-pirrolidin-2-il1metanol. A una solución de (2S,4R)-4-fluoropirrolidin-1,2-dicarboxilato de 1-íerc-butil-2-metilo (1,98 g, 8,0 mmol, 1,0 eq.) en THF (20,0 ml) se le añadió lentamente LiAlH4 (912 mg, 24,0 mmol, 3.0 eq.) a -40 °C, después de que se completara, la mezcla se agitó a -40 °C durante 0,5 horas y después la mezcla se calentó a 70 °C y se agitó a esta temperatura durante 3 horas. Después de que se completara, la mezcla de reacción se inactivó con la adición de una solución de Na2SÜ4 (6,0 ml) a 0 °C, después se diluyó con THF (10,0 ml), después se filtró y el sólido se lavó con acetato de etilo (30,0 ml). Las capas orgánicas combinadas se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar [(2S,4R)-4-fluoro-1-metilpirrolidin-2-il]metanol (800 mg, 6.01 mmol, 75 % de rendimiento) en forma de un aceite de color amarillo.
1H RMN (400 MHz, Cloroformo-d) 55,13-4,97 (m, 1H), 3,67-3,63 (m, 1H), 3,51-3,37 (m, 2H), 2,95 (s, 1H), 2,74-2,68 (m, 1H), 2,63-2,51 (m, 1H), 2,33 (s, 3H), 2,07-2,02 (m, 1H), 1,99-1,95 (m, 1H).
Etapa A: 4-[(3S)-4-terc-Butoxicarbonil-3-(cianometil)piperazin-1-il1-2-[[(2S,4R)-4-fluoro-1-metil-pirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-7-carboxilato de bencilo. Una mezcla de [(2S,4R)-4-fluoro-1-metil-pirrolidin-2-il]metanol (480 mg, 3,6 mmol, 2,0 eq.) y t-Bu0Na (295 mg, 3,1 mmol, 1,7 eq.) en tolueno (8,0 ml) se desgasificó y se purgó 3 veces con N2, la mezcla se agitó a 0 °C y después se añadió 4-[(3S)-4-terc-butoxicarbonil-3-(cianometil)piperazin-1-il1-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d1pirimidin-7-carboxilato de bencilo (1 g, 1,8 mmol, 1,0 eq.) en tolueno (8,0 ml). La mezcla se agitó a 0 °C durante 0,5 horas en una atmósfera de N2. Después de que se completara, la mezcla se inactivó con la adición de H20 (25,0 ml) y después se extrajo con acetato de etilo (30,0 ml * 3). Las capas orgánicas combinadas se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC ultrarrápida de fase inversa (18C, 0,1 % de a F en agua, 0-60 % de MeCN). El producto obtenido se ajustó con NaHC03 acuoso saturado a pH -8, después se concentró, la capa acuosa se extrajo con acetato de etilo (100,0 ml * 3) y la capa orgánica combinada se secó sobre Na2S04, se filtró y se concentró para dar 4-[(3S)-4-terc-butoxicarbonil-3-(cianometil)piperazin-1 -il1-2-[[(2S,4R)-4-fluoro-1 -metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-7-carboxilato de bencilo (860 mg, 1,38 mmol, 76 % de rendimiento, 100 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+11: 624.
1H RMN (400 MHz, Cloroformo-d) 57,31-7,25 (m, 5H), 5,15-5,02 (m, 3H), 4,63-4,58 (m, 1H), 4,51 (s a, 1H), 4,39-4,31 (m, 2H), 4,16-4,12 (m, 1H), 3,99-3,88 (m, 3H), 3,71 (d a, J = 11,2 Hz, 1H), 3,52-3,41 (m, 1H), 3,36 (s a, 1H), 3,19-3,16 (m, 2H), 2,99-2,86 (m, 2H), 2,71-2,48 (m, 5 H), 2,43 (s, 3H), 2,76-2,17 (m, 1H), 1,96-1,80 (m, 1H), 1,74 (s a, 1H), 1,43 (s, 9H).
Etapa B: (2S)-2-(C¡anomet¡l)-4-[2-[[(2S.4R)-4-fluoro-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de tere-butilo. A una soluc¡ón de 4-[(3S)-4-ferc-butox¡carbon¡l-3-(c¡anomet¡l)p¡peraz¡n-1-¡l1-2-[[(2S.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-7-carbox¡lato de benc¡lo (860 mg. 1.4 mmol. 1.0 eq.) en Me0H (20.0 ml) y Nhb^Me0H (10,0 ml. 50 % p/p) se le añad¡ó Pd/C (200 mg. 10 % de pureza. 1.0 eq.) en una atmósfera de N2. La suspens¡ón se desgas¡f¡có al vacío y se purgó var¡as veces con H2. La mezcla se ag¡tó en una atmósfera de H2 (103.42 kPa (15 ps¡)) a 25 °C durante 1 hora. Después de que se completara. la mezcla de reacc¡ón se f¡ltró y se concentró a pres¡ón reduc¡da para dar (2S)-2-(c¡anomet¡l)-4-[2-[[(2S.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de tere-but¡lo (620 mg. bruto) en forma de un sól¡do de color blanco. El producto bruto se usó en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón ad¡c¡onal. LCMS [ESI. M+11: 490.
1H RMN (400 MHz. Cloroformo-d) 65,16-5.02 (m. 1H). 4.51 (s a. 1H).4.31 (dd. J = 4.8. 11.2 Hz. 1H).4.13 (dd. J = 6.0.
11.2 Hz. 1H). 3.87 (s. 2H). 3.76 (d. J = 12.4 Hz. 1H). 3.52-3.41 (m. 1H). 3.14 (dd. J = 3.6. 13.6 Hz. 2H). 3.06-3.03 (m.
1H). 2.99-2.88 (m. 3H). 2.74-2.48 (m. 6 H). 2.43 (s. 3H). 2.29-2.18 (m. 1H). 1.96-1.80 (m. 1H). 1.68-1.59 (m. 2H). 1.44 (s. 9H).
Etapa C: (2S)-2-(C¡anomet¡l)-4-[2-[[(2S.4R)-4-fluoro-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-7-(8-met¡l-1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de tere-but¡lo. Una mezcla de (2S)-2-(c¡anomet¡l)-4-[2-[[(2S.4R)-4-fluoro-1 -met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1 -carbox¡lato de tere-but¡lo (600 mg. 1.2 mol. 1.0 eq.). 1-bromo-8-met¡l-naftaleno (812 mg. 3.7 mmol. 3.0 eq.). Pd2(dba)3 (168 mg. 184 μmol.
0.15 eq.). Cs2C0a (1.2 g. 3.7 mmol. 3.0 eq.) y Xantphos (142 mg. 246 μmol. 0.2 eq.) en tolueno (5.0 ml) se desgas¡f¡có y se purgó 3 veces con N2 y después la mezcla se ag¡tó a 90 °C durante 12 horas en una atmósfera de N2. Después de que se completara. la mezcla de reacc¡ón se f¡ltró y se concentró a pres¡ón reduc¡da para dar un res¡duo y después el res¡duo se d¡solv¡ó con AE 15.0 ml y después se ajustó con HCl 0.5 M a pH ~2. La capa orgán¡ca se lavó con H20 (15,0 ml x 3 ). después a la fase acuosa comb¡nada se le añad¡ó el NaHC03 sól¡do hasta que alcanzó un pH de 8. y después de eso. la fase acuosa se extrajo con acetato de et¡lo (50.0 ml x 3) y la capa orgán¡ca comb¡nada se secó sobre Na2S04. se f¡ltró y se concentró. El res¡duo se pur¡f¡có por HPLC ultrarráp¡da de fase ¡nversa (18C. 0.1 % de TFA en agua. 0-50 % de MeCN). El producto obten¡do se ajustó con NaHC03 acuoso saturado a pH -8. después se concentró. la capa acuosa se extrajo con AE (100.0 ml x 3 ) y la capa orgán¡ca comb¡nada se secó sobre Na2S04. se f¡ltró y se concentró para dar (2S)-2-(c¡anomet¡l)-4-[2-[[(2S.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-7-(8-met¡l-1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de tere-but¡lo (300 mg. 467 μmol. 35 % de rend¡m¡ento. 99 % de pureza) en forma de un ace¡te de color amar¡llo. LCMS [ESI. M+11: 630.
Etapa D: 2-[(2S)-4-[2-[[(2S.4R)-4-Fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-7-(8-met¡l-1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una soluc¡ón de (2S)-2-(c¡anomet¡l)-4-[2-[[(2S.4R)-4-fluoro-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-7-(8-met¡l-1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de tere-but¡lo (200 mg. 318 μmol. 1.0 eq.) en d¡oxano (1.5 ml) se le añad¡ó HCl/d¡oxano (4 M. 2.0 ml. 25.0 eq.) a 25 °C. La mezcla se ag¡tó a 25 °C durante 1 hora. Después de que se completara. la mezcla de reacc¡ón se ¡nact¡vó con la ad¡c¡ón de NaHC03 acuoso saturado (6.0 ml) a 0 °C y después se d¡luyó con H2010.0 ml y se extrajo con AE (15.0 ml x 3 ). Las capas orgán¡cas comb¡nadas se secaron sobre Na2S04. se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar 2 -[(2S)-4-[2-[[(2S.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-7-(8-met¡l-1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo (150 mg. bruto) en forma de un ace¡te de color amar¡llo. El producto se usó en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón ad¡c¡onal. LCMS [ESl. M+11: 530.
Etapa E: 2-[(2S)-4-[2-[[(2S.4R)-4-Fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-7-(8-met¡l-1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-1-(2-fluoroprop-2-eno¡l)p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una soluc¡ón de 2-[(2S)-4-[2-[[(2S.4R)-4-fluoro-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-7-(8-met¡l-1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo
(150 mg. 283 μmol. 1.0 eq.) en acetato de et¡lo (1.0 ml) se le añad¡ó una soluc¡ón de ác¡do 2-fluoroprop-2-eno¡co (42.9 mg. 476 μmol. 1.68 eq.) en acetato de et¡lo (0.5 ml). después se añad¡eron TEA (1.93 g. 19 mmol. 2.65 ml. 67.0 eq.) y T3P (455 mg. 715 μmol. 425 μl. 50 % de pureza. 2.5 eq.) y la mezcla se ag¡tó a 0 °C. después de lo cual la mezcla de reacc¡ón se calentó a 20 °C y se ag¡tó durante 1 hora. Después de que se completara. la mezcla de reacc¡ón se ¡nact¡vó con la ad¡c¡ón de 10.0 ml de H20 y después se extrajo con acetato de et¡lo (15.0 ml x 3 ). Las capas orgán¡cas comb¡nadas se secaron sobre Na2S04. se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar un res¡duo.
El res¡duo se pur¡f¡có por HPLC prep. (columna: Phenomenex Gem¡n¡ 150 * 25 mm * 10 μm; fase móv¡l: [agua (0.05 % de h¡dróx¡do de amon¡o v/v)-MeCN1; % de B: 58 % -88 %. 11.5 m¡n) y se somet¡ó a l¡of¡l¡zac¡ón para dar el compuesto del título 2-[(2S)-4-[2-[[(2S.4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-7-(8-met¡l-1-naft¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-1-(2-fluoroprop-2-eno¡l)p¡peraz¡n-2-¡l1aceton¡tr¡lo (50.1 mg. 82.9 μmol. dos etapas 26 % de rend¡m¡ento.
99.5 % de pureza) en forma de un sól¡do de color blanco. LCMS [ESl. M+11: 602.
1H RMN (400 MHz. Cloroformo-d) 6 7,72 (d, J = 8.4 Hz. 1H). 7.67 (t. J = 8.0 Hz. 1H). 7.46-7.43 (m. 2H). 7.26-7.21 (m.
2H). 5.50-5.38 (m. 1H). 5.30-5.11 (m. 2H). 4.89 (s a. 1H). 4.45-4.40 (m. 1H). 4.30-4.20 (m. 2H). 4.19-4.13 (m. 1H).
4.10-4.05 (m. 1H). 3.92-3.88 (m. 1H). 3.82-3.77 (m. 1H). 3.62-3.46 (m. 3H). 3.25-3.18 (m. 2H). 3.15-3.09 (m. 1H). 3.08 2.98 (m. 2H). 2.94 (s. 3H). 2.90-2.77 (m. 2H). 2.67-2.56 (m. 2H). 2.51 (d. J = 4.8 Hz. 3H). 2.38-2.27 (m. 1H). 2.07-1.89 (m. 1H).
Ejemplo 561

2-[(2S)-1-(2-Fluoroprop-2-enoil)-4-[2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-SH pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

(2S,4R)-4-Metoxipirrolidin-1,2-dicarboxilato de 1-terc-butil-2-metilo. A la solución de ácido (2S,4R)-1-tercbutoxicarbonil-4-hidroxi-pirrolidin-2-carboxílico (2 g, 8,65 mmol, 1 eq.) y Ag20 (6,01 g, 26,0 mmol, 3 eq.) en MeCN (40 ml) se le añadió CH3I (10,0 g, 70,8 mmol, 4,41 ml, 8,19 eq.) y la mezcla se agitó a 30 °C durante 14 horas. A la mezcla se le añadió CH3I (13,2 g, 93,1 mmol, 5,80 ml, 10,8 eq.) y la mezcla se agitó a 30 °C durante 6 horas. La mezcla de reacción se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP: EtOAc = 20:1 ~10:1) para dar (2S,4R)-4-metoxipirrolidin-1,2-dicarboxilato de 1-terc-butil-2-metilo (1,85 g, 6,99 mmol, 81 % de rendimiento, 98 % de pureza) en forma de un aceite de color amarillo.
[(2S,4R)-4-Metoxi-1-metil-pirrolidin-2-il]metanol. A la solución de (2S,4R)-4-metoxipirrolidin-1,2-dicarboxilato de 01-terc-butil 02-metilo (1,85 g, 7,13 mmol, 1 eq.) en THF (50 ml) se le añadió LiAlH4 (542 mg, 14,3 mmol, 2 eq.) a 0 °C y la mezcla se agitó a 0 °C durante 0,5 horas. Después, la mezcla se calentó a 70 °C y se agitó durante 3 horas. Después, se añadió LiAlH4 (542 mg, 14,27 mmol, 2 eq.) a 10 °C y la mezcla se calentó a 80 °C y se agitó durante 6 horas. La mezcla de reacción se inactivó con una solución saturada de Na2S04 (5 ml) y después se filtró. La torta de filtro se lavó con THF (3 * 30 ml). El filtrado se concentró al vacío para dar [(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metanol (840 mg, 5,21 mmol, 73 % de rendimiento, 90 % de pureza) en forma de un aceite de color amarillo que se usó para la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, cloroformo-d) 5 = 3,93 - 3,83 (m, 1H), 3,68 (dd, J = 3,2, 11,2 Hz, 1H), 3,48 - 3,36 (m, 2H), 3,30 (s, 3H), 2,67 - 2,58 (m, 1H), 2,40 - 2,30 (m, 4H), 2,08 (td, J = 8,0, 13,2 Hz, 1H), 1,90 - 1,79 (m, 1H).
Etapa A: 4-[(3S)-4-terc-Butoxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-1-pirido[3,4-d]pirimidin-7-carboxilato de bencilo. A la solución de [(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metanol (367 mg, 2,52 mmol, 2 eq.) en tolueno (18 ml) se le añadió t-Bu0Na (364 mg, 3,79 mmol, 3 eq.) a 0 °C, después se añadió 4-[(3S)-4-terc-butoxicarbonil-3-(cianometil)piperazin-1-il]-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-7-carboxilato de bencilo (700 mg, 1,26 mmol, 1 eq.) a 0 °C y la mezcla de reacción se agitó a 8 °C durante 1 hora. A la
mezcla se le añadió agua (30 ml). La mezcla se diluyó con EtOAc (10 ml) y se extrajo con EtOAc (2 x 30 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por columna ultrarrápida de fase inversa (MeCN/agua (0,1 % de AF) = 30 %) para dar 4-[(3S)-4-ferc-butoxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2S,4R)-4-metoxi-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de bencilo (450 mg, 694 μmol, 55 % de rendimiento, 98 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 636.
Etapa B: (2S)-2-(Cianometil)-4-[2-[[(2S,4R)-4-metoxi-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo. A la solución de 4-[(3S)-4-ferc-butoxicarbonil-3-(cianometil)piperazin-1-il]-2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de bencilo (490 mg, 771 μmol, 1 eq.) y NFb^Me0H (5 ml, 25 % de pureza) en Me0H (5 ml) se le añadió Pd/C (100 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 15 °C durante 1 hora. La mezcla de reacción se filtró y la torta de filtro se lavó con Me0H (3 x 5 ml). El filtrado se concentró al vacío para dar (2S)-2-(cianometil)-4-[2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (360 mg, 682 μmol, 88 % de rendimiento, 95 % de pureza) en forma de un sólido de color amarillo que se usó para la siguiente etapa sin purificación adicional.
Etapa C: (2S)-2-(Cianometil)-4-[2-[[(2S,4R)-4-metoxi-1-metilpirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de ferc-butilo. A la solución de (2S)-2-(cianometil)-4-[2-[[(2S,4R)-4-metoxi-1 -metil-pirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de ferc-butilo (360 mg, 718 μmol, 1 eq.), 1-bromo-8-metil-naftaleno (238 mg, 1,08 mmol, 1,5 eq.), Cs2C03 (701 mg, 2,15 mmol, 3 eq.) y Xantphos (83,0 mg, 143 μmol, 0,2 eq.) en tolueno (8 ml) se le añadió Pd2(dba)3 (65,7 mg, 71,8 μmol, 0,1 eq.) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con N2. La mezcla se agitó en una atmósfera de N2 a 90 °C durante 12 horas. La mezcla de reacción se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (de EP: EtOAc = 5:1 ~1:1 a EtOAc: Me0H = 1:0~20:1). Después, el residuo se purificó por columna ultrarrápida de fase inversa (ACN/agua (0,1 % de AF) = 45 %) para dar (2S)-2-(cianometil)-4-[2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (280 mg, 414 μmol, 58 % de rendimiento, 95 % de pureza) en forma de un sólido de color pardo. LCMS [ESI, M+1]: 642.
1H RMN (400 MHz, cloroformo-d) ó = 7,74 - 7,60 (m, 2H), 7,45 - 7,37 (m, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,27 - 7,17 (m, 2H), 4,61 (s a, 1H), 4,39 (ddd, J = 4,8, 6 ,8 , 11,2 Hz, 1H), 4,30 - 4,20 (m, 1H), 4,16 - 4,11 (m, 1H), 4,11 - 3,89 (m, 4H), 3,88 - 3,68 (m, 1H), 3,56 - 3,47 (m, 1H), 3,46 - 3,32 (m, 2H), 3,30 (d, J = 2,4 Hz, 3H), 3,21 - 3,05 (m, 3H), 3,01 - 2,85 (m, 5H), 2,81 - 2,67 (m, 2H), 2,65 - 2,54 (m, 1H), 2,46 (d, J = 4,0 Hz, 3H), 2,35 - 2,26 (m, 1H), 2,11 - 2,05 (m, 1H), 2,00 - 1,88 (m, 1H), 1,52 (s, 9H).
Etapa D: 2-[(2S)-4-[2-[[(2S,4R)-4-Metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A la solución de (2S)-2-(cianometil)-4-[2-[[(2S,4R)-4-metoxi-1-metilpirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo (280 mg, 436 μmol, 1 eq.) en dioxano (1 ml) se le añadió HCl/dioxano (4 M, 1,64 ml, 15 eq.) y se agitó a 10 °C durante 1 hora. La mezcla de reacción se concentró al vacío. La mezcla se basificó con NaHC03 saturado a pH = 8~9 y se extrajo con EtOAc (3 x 15 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío para dar 2-[(2S)-4-[2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (210 mg, 349 μmol, 80 % de rendimiento, 90 % de pureza) en forma de un sólido de color amarillo que se usó para la siguiente etapa sin purificación adicional. LCMS [ESl, M+1]: 542.
Etapa E: 2-[(2S)-1-(2-Fluoroprop-2-enoil)-4-[2-[[(2S,4R)-4-metoxi-1-metilpirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo. A la solución de 2-[(2S)-4-[2-[[(2S,4R)-4-metox¡-1-metilpirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (100 mg, 185 μmol, 1 eq.), ácido 2-fluoroprop-2-enoico (33,2 mg, 369 μmol, 2 eq.) y TEA (149 mg, 1,48 mmol, 206 μl, 8 eq.) en EtOAc (2 ml) se le añadió T3P (352 mg, 554 μmol, 329 μl, 50 % de pureza, 3 eq.) a 0 °C y la mezcla se agitó a 10 °C durante 0,5 horas. A la mezcla se le añadió agua (3 ml). La mezcla se extrajo con EtOAc (2 x 3 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Xtimate C18150 * 25 mm * 5 μm;fase móvil: [agua (0,05 % de hidróxido de amonio v/v)-ACN];% de B: 52 % - 82 % ,12 min) para dar el compuesto del título 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[2-[[(2S,4R)-4-metoxi-1-metilpirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (28,1 mg, 45,5 μmol, 25 % de rendimiento, 99,4 % de pureza) en forma de un sólido de color blanco. LCMS [ESl, M+1]: 614.
1H RMN (400 MHz, cloroformo-d) ó = 7,65 - 7,54 (m, 2H), 7,38 - 7,30 (m, 1H), 7,27 (t, J = 7,6 Hz, 1H), 7,19 - 7,10 (m, 2H), 5,46 - 5,26 (m, 1H), 5,18 (dd a, J = 3,6, 16,8 Hz, 1H), 4,83 (s a, 1H), 4,37 - 4,27 (m, 1H), 4,23 - 3,94 (m, 4H), 3,92 - 3,85 (m, 1H), 3,84 - 3,76 (m, 1H), 3,70 (d a, J = 18,0 Hz, 1H), 3,46 (d a, J = 8 ,8 Hz, 1H), 3,42 - 3,31 (m, 2H), 3,22 (d, J = 2,0 Hz, 3H), 3,18 - 3,07 (m, 2H), 3,06 - 2,87 (m, 2H), 2,87 - 2,64 (m, 6 H), 2,59 - 2,49 (m, 1H), 2,38 (d, J = 4,4 Hz, 3H), 2,24 (ddd, J = 3,2, 6,0, 9,6 Hz, 1H), 2,04 - 1,94 (m, 1H), 1,93 - 1,81 (m, 1H).
Ejemplo 562

2-[(2S)-1-(2-Fluoroprop-2-enoil)-4-[7-(8-metil-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

[(3R)-1-Metilpirrolidin-3-il]metanol. A la solución de (3R)-3-(hidroximetil)pirrolidin-1-carboxilato de terc-butilo (0,5 g, 2,48 mmol, 1 eq.) en THF (20 ml) se le añadió LiAlH4 (189 mg, 4,97 mmol, 2 eq.) a 0 °C y después la mezcla se calentó a 70 °C y se agitó a 70 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se inactivó con Na2SÜ4 saturado (1 ml). El precipitado se retiró por filtración y la torta de filtro se lavó con THF (3 * 20 ml). Las capas orgánicas combinadas se secaron sobre Na2SÜ4 y se concentraron al vacío para dar [(3R)-1 -metilpirrolidin-3-il]metanol (220 mg, 1,72 mmol, 69 % de rendimiento, 90 % de pureza) en forma de un aceite incoloro que se usó directamente en la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, cloroformo-d) 5 = 3,67 - 3,57 (m, 1H), 3,50 (dd, J = 6,0, 10,0 Hz, 1H), 3,45 - 3,18 (m, 1H), 2,70 (dt, J = 4,8, 8,8 Hz, 1H), 2,56 - 2,44 (m, 2H), 2,42 - 2,24 (m, 5H), 2,04 - 1,90 (m, 1H), 1,68 - 1,54 (m, 1H).
Etapa A: (2S)-2-(Cianometil)-4-[7-(8-metil-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de bencilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (500 mg, 841 μmol, 1 eq.) y [(3R)-1-metilpirrolidin-3-il]metanol (194 mg, 1,68 mmol, 2 eq.) en tolueno (10 ml) se le añadió í-Bu0Na (162 mg, 1,68 mmol, 2 eq.). La mezcla se agitó a 0 °C durante 10 minutos. Después de que se completara, la mezcla se diluyó con agua (10 ml) y se extrajo con EtOAc (2 * 40 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EtOAc/Me0H 50/1 a 3/1) para dar (2S)-2-(cianometil)-4-[7-(8-metil-1 -naftil)-2-[[(3R)-1 -metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirim idin-4-il]piperazin-1-carboxilato de bencilo (290 mg, 427 μmol, 51 % de rendimiento, 95 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 646.
1H RMN (400 MHz, cloroformo-d) 5 = 7,72 - 7,61 (m, 2H), 7,45 - 7,37 (m, 5H), 7,37 - 7,30 (m, 2H), 7,27 - 7,16 (m, 2H), 5,28 - 5,15 (m, 2H), 4,68 (s a, 1H), 4,30 - 4,01 (m, 6H), 4,00 - 3,73 (m, 2H), 3,56 - 3,31 (m, 2H), 3,26 - 3,06 (m, 3H), 3,05 - 2,94 (m, 2H), 2,91 (s, 3H), 2,82 - 2,70 (m, 4H), 2,70 - 2,57 (m, 2H), 2,47 (d, J = 4,4 Hz, 3H), 2,20 - 2,07 (m, 1H), 1,80 - 1,67 (m, 1H).
Etapa B: 2-[(2S)-4-[7-(8-Metil-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-il1piperazin-2-illacetonitrilo. A una solución de (2S)-2-(c¡anomet¡l)-4-[7-(8-met¡l-1-naft¡l)-2-[[(3R)-1-met¡lpirrol¡d¡n-3-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (290 mg, 449 gmol, 1 eq.) en Me0H (4 ml) se le añadieron NHs^Me0H (2 ml, 15 % de pureza) y Pd/C (100 mg, 449 gmol, 10 % de pureza, 1 eq.) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 15 °C durante 1 hora. Después de que se completara, la mezcla se filtró y el filtrado se concentró al vacío para dar 2-[(2S)-4-[7-(8-met¡l-1-naft¡l)-2-[[(3R)-1-met¡lp¡rrol¡d¡n-3-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡trilo (150 mg, 279 gmol, 62 % de rendimiento, 95 % de pureza) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. Lc Ms [ESI, M+1]: 512.
Etapa C: 2-f(2S)-1-(2-Fluoroprop-2-enoi r )-4-f7-(8-metil-1-naftil)-2-ff(3R)-1-metilpirrolidin-3-i n metox n -6.8-dihidro-5H-pirido[3,4-d^pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(3R)-1-met¡lp¡rrol¡d¡n-3-il]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (120 mg, 235 gmol, 1 eq.), ácido 2-fluoroprop-2-enoico (42,2 mg, 469 gmol, 2 eq.) y TEA (71,2 mg, 704 gmol, 97,9 gl, 3 eq.) en Dm F (2 ml) se le añadió T3P (224 mg, 352 μmol, 209 gl, 50 % de pureza en EtOAc, 1,5 eq.) a -40 °C. Después, la mezcla se agitó a -40 °C durante 0,5 horas. Después de que se completara, la mezcla se diluyó con agua (2 ml) y se extrajo con el disolvente mixto (EtOAc/Me0H 10/1,5 * 5 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por cromatografía (Al203, EtOAc/Me0H 50/1 a 20/1) seguido de HPLC prep. (columna: Phenomenex Gemini 150 * 25 mm * 10 gm; fase móvil: [agua (NH4HC03 10 mM) - ACN]; % de B: 50 %-100 %, 10 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[7-(8-metil-1-naft¡l)-2-[[(3R)-1-met¡lp¡rrol¡d¡n-3-¡l]metoxij-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (16,0 mg, 26,5 gmol, 11 % de rendimiento, 96,5 % de pureza) en forma de un sólido de color blanquecino. LCMS [ESI, M+1]: 584.
1H RMN (400 MHz, cloroformo-d) 5 = 7,74 - 7,61 (m, 2H), 7,45 - 7,38 (m, 1H), 7,35 (t, J = 7,6 Hz, 1H), 7,27 - 7,17 (m, 2H), 5,55 - 5,32 (m, 1H), 5,26 (dd, J = 3,6, 16,8 Hz, 1H), 4,88 (s a, 1H), 4,33 - 4,02 (m, 5H), 4,01 - 3,65 (m, 2H), 3,61 -3,36 (m, 2H), 3,27 - 2,97 (m, 4H), 2,92 (s, 3H), 2,90 - 2,74 (m, 2H), 2,74 - 2,56 (m, 4H), 2,55 - 2,42 (m, 2H), 2,36 (d, J = 2,4 Hz, 3H), 2,14 - 1,99 (m, 1H), 1,66 - 1,52 (m, 1H).
Ejemplo 563

2-[(2S)-1-[(E)-4-Fluorobut-2-eno¡l]-4-[7-(8-met¡l-1-naft¡l)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-il]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

2-[(2S)-1-[(E)-4-Fluorobut-2-enoil1-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-2-il)acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1 -naftil)-2-[[(2R)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-illpiperazin-2-il]acetonitrilo (260 mg, 508 gmol, 1,0 eq.) en acetato de etilo (1,5 ml) se le añadió una solución de ácido (E)-4-fluorobut-2-enoico (106 mg, 1,02 mmol, 2,0 eq.) en acetato de etilo (0,5 ml), TEA (206 mg, 2,03 mmol, 283 gl, 4,0 eq.) y T3P (647 mg, 1,02 mmol, 605 gl, 50 % de pureza, 2,0 eq.) y la mezcla se agitó a -40 °C y se agitó durante 1 hora. Después de que se completara, la mezcla de reacción se inactivó con H20 (10,0 ml) y se extrajo con acetato de etilo (3 * 15,0 ml). Las capas orgánicas combinadas se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Luna C18150 * 255 u; fase móvil: [agua (0,225 % de AF)-ACN]; % de B: 25 %-45 %, 7,8 min). El producto obtenido se ajustó a pH ~8 con NaHC03 acuoso saturado, después se concentró, la capa acuosa se extrajo con acetato de etilo (3 * 20,0 ml) y la capa orgánica combinada se secó sobre Na2S04, se filtró y se concentró para dar el compuesto del título 2-[(2s )-1 -[(E)-4-fluorobut-2-enoil]-4-[7-(8-metil-1 -naftil)-2-[[(2R)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (132 mg, 220 gmol, 43 % de rendimiento, 99 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 598.
1H RMN (400 MHz, Cloroformo-d) ó 7,70 - 7,62 (m, 2H), 7,43 - 7,38 (m, 1H), 7,36 - 7,31 (m, 1H), 7,26 - 7,19 (m, 2H), 7,04 - 6,94 (m, 1H), 6,59 (d, J = 14,4 Hz, 1H), 5,17-4,60 (m, 3H), 4,41 - 4,35 (m, 1H), 4,28 - 3,44 (m, 9H), 3,20 - 3,08 (m, 4H), 3,05 - 2,99 (m, 1H), 2,92 (s, 3H), 2,83 - 2,77 (m, 1H), 2,72 - 2,62 (m, 2H), 2,47 (d, J = 4,8 Hz, 3H), 2,32 - 2,25 (m, 1H), 2,09 - 2,01 (m, 1H), 1,88 - 1,70 (m, 3H).
Ejemplo 564

2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[3-[(1 S,4S)-2-oxa-5-azab¡c¡clo[2.2.1]heptan-5-¡l]propox¡]-6,8-dih¡dro-5H-p¡r¡do[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo

Etapa A: 4-[(3S)-4-tere-Butoxicarbonil-3-(cianometil)piperazin-1-il]-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.11heptan-5-il1propoxi1-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-7-carboxilato de bencilo. A una solución de 4-[(3S)-4-terebutoxicarbonil-3-(cianometil)piperazin-1-il1-2-metilsulfinil-6,8-óihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de bencilo (800 mg, 1,44 mmol, 1 eq.) en tolueno (20 ml) se le añadieron t-Bu0Na (277 mg, 2,88 mmol, 2 eq.) y 3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-¡l]propan-1-ol (340 mg, 2,16 mmol, 1,5 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. La mezcla de reacción se diluyó con agua (30 ml) y se extrajo con acetato de etilo (30 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico)/acetonitrilo)]. La mezcla se ajustó a pH = 7 con NaHC03 acuoso saturado y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar el producto. Se obtuvo 4-[(3S)-4-tere-butoxicarbonil-3-(cianometil)piperazin-1-il]-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il1propoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de bencilo (500 mg, 725 gmol, 50 % de rendimiento, 94 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 648.
1H RMN (400 MHz, cloroformo-d) ó = 7,44 - 7,30 (m, 5H), 5,19 (s, 2H), 4,79-4,53 (m, 2H), 4,51 - 4,30 (m, 4H), 4,09 -3,71 (m, 5H), 3,67 - 3,58 (m, 1H), 3,54 - 3,36 (m, 2H), 3,32 - 3,12 (m, 2H), 3,04 - 2,87 (m, 2H), 2,85 - 2,46 (m, 7H), 2,00 - 1,89 (m, 2H), 1,85 (d, J = 9,2 Hz, 1H), 1,72 (d, J = 9,2 Hz, 1H), 1,51 (s, 9H).
Etapa B: (2S)-2-(Cianometil)-4-[2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.11heptan-5-il1propoxi1-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de tere-butilo. A una solución de 4-[(3S)-4-terc-butoxicarbonil-3-(cianometil)piperazin-1-il]-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il1propoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de bencilo (900 mg, 1,39 mmol, 1 eq.) en Me0H (30 ml) se le añadieron Pd/C (100 mg, 10 % de pureza) y NH3*Me0H (20 ml, 25 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 20 °C durante 1 hora. El catalizador se retiró por filtración y el filtrado se concentró al vacío. Se obtuvo (2S)-2-(cianometil)-4-[2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.11heptan-5-il1propoxi1-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (650 mg, 1,06 mmol, 77 % de rendimiento, 84 % de pureza) en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación. LCMS [ESI, M+1]: 514.
Etapa C: (2S)-4-[7-(8-Cloro-1-naftil)-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.11heptan-5-il]propoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]-2-(cianometil)piperazin-1 -carboxilato de tere-butilo. A la solución de (2S)-2-(cianometil)-4-[2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.11heptan-5-il1propoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il1piperazin-1-carboxilato de tere-butilo (180 mg, 350 gmol, 1 eq.), 1-bromo-8-cloro-naftaleno (127 mg, 526 gmol, 1,5 eq.), Cs2C03 (342 mg, 1,05 mmol, 3 eq.) y RuPhos (65,4 mg, 140 gmol, 0,4 eq.) en tolueno (6 ml) se le añadió Pd2(dba)3 (64,2 mg, 70,1 gmol, 0,2 eq.) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con N2. La mezcla se agitó en una atmósfera de N2 a 90 °C durante 10 horas. La mezcla de reacción se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (de EP: EtOAc = 5:1 ~ 1:1 a EtOAc: Me0H = 1:0~20:1) para dar (2S)-4-[7-(8-cloro-1-naftil)-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il1propoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (130 mg, 173 gmol, 50 % de rendimiento, 90 % de pureza) en forma de un sólido de color pardo. LCMS [ESI, M+1]: 674.
Etapa D: 2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[3-[(1S.4S)-2-oxa-5-azabiciclo[2.2.11heptan-5-illpropoxi1-6,8-dihidro-5H-pirido[3.4-dlpirimidin-4-illpiperazin-2-il1acetonitrilo. A la solución de (2S)-4-[7-(8-cloro-1-naftil)-2-[3-[(1S,4S)-2-oxa-5-azab¡c¡clo[2.2.1]heptan-5-il]propox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de tere-butilo (150 mg, 222 gmol, 1 eq.) en dioxano (0,6 ml) se le añadió HCl/dioxano (4 M, 556 gl, 10 eq.) y la mezcla se agitó a 10 °C durante 1 hora. La mezcla de reacción se concentró al vacío. El residuo se disolvió con agua (15 ml) y EtOAc (10 ml) y después se separó. La fase acuosa se basificó con NaHCÜ3 sólido a pH = 8~9 y se extrajo con EtOAc (2 x 15 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml). Las capas orgánicas combinadas se secaron sobre Na2SÜ4 anhidro, se filtraron y se concentraron al vacío para dar 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[3-[(1S,4S)-2-oxa-5-azab¡c¡clo[2.2.1]heptan-5-il]propox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]acetonitrilo (100 mg, 162 gmol, 73 % de rendimiento, 93 % de pureza) en forma de un sólido de color pardo que se usó para la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 574.
Etapa E: 2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[3-[(1S.4S)-2-oxa-5-azabiciclo[2.2.11heptan-5-il1propoxi1-6.8-dihidro-5H-p¡rido[3.4-d^pirimidin-4-il1-1-(2-fluoroprop-2-enoil)pipenazin-2-il1acetonitrilo. A la solución de 2-[(2S)-4-[7-(8-cloro-1-naft¡l)-2-[3-[(1S,4S)-2-oxa-5-azab¡c¡clo[2.2.1]heptan-5-il]propox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]acetonitrilo (100 mg, 174 gmol, 1 eq.), ácido 2 -fluoroprop-2 -enoico (31,4 mg, 348 gmol, 2 eq.) y TEA (141 mg, 1 ,39 mmol, 194 gl, 8 eq.) en EtüAc (2 ml) se le añadió T3P (332 mg, 522 gmol, 311 gl, 50 % de pureza, 3 eq.) a 0 °C y la mezcla se agitó a 10 °C durante 0,5 horas. A la mezcla se le añadió agua (3 ml). La mezcla se extrajo con EtOAc (2 x 3 ml). Las capas orgánicas combinadas se secaron sobre Na2SÜ4 anhidro, se filtraron y se concentraron al vacío.
El residuo se purificó por HPLC prep. (columna: Xtimate C18 10 g 250 mm * 50 mm;fase móvil: [agua (0,05 % de hidróxido de amonio v/v)-ACN];% de B: 45 %-75 % ,12 min) para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naft¡l)-2-[3-[(1S,4S)-2-oxa-5-azab¡c¡clo[2.2.1]heptan-5-il]propox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (26,5 mg, 41 gmol, 24 % de rendimiento, 99,9 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 646.
1H RMN (400 MHz, cloroformo-d) 5 = 7,76 (d, J = 8,4 Hz, 1H), 7,62 (t, J = 7,2 Hz, 1H), 7,53 (d, J = 7,2 Hz, 1H), 7,45 (td, J = 7,6, 12,4 Hz, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,26-7,18 (m, 1H), 5,53 - 5,32 (m, 1H), 5,25 (dd, J = 3,6, 16,8 Hz, 1H), 5,12 - 4,64 (m, 1H), 4,49-4,31 (m, 4H), 4,21 - 3,98 (m, 3H), 3,95 - 3,76 (m, 2H), 3,68 - 3,55 (m, 2H), 3,52 - 3,36 (m, 2H), 3,33 - 2,97 (m, 4H), 2,95 - 2,66 (m, 5H), 2,64 - 2,49 (m, 2H), 1,93 (m, 2H), 1,84 (d a, J = 8,4 Hz, 1H), 1,71 (d a, J = 10,0 Hz, 1H).
Ejemplo 565

2-[(2S)-1-[(£)-4-Fluorobut-2-enoil]-4-[7-(8-met¡l-1-naft¡l)-2-[3-[(1S,4S)-2-oxa-5-azab¡c¡clo[2.2.1]heptan-5-il]propox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡din-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo

Etapa A: (2S)-2-(Cianometil)-4-[7-(8-metil-1-naftil)-2-[3-[1S,4S)-2-oxa-5- azabiciclo[2.2.11heptan-5-illpropoxi1-6,8-dihidro-5H-pirido[3,4-c f lpirimidin-4-¡ n piperazin-1 -carboxilato de tere-butilo. Una mezcla de (2S)-2-(cianometil)-4-[2-[3-[(1S,4S)-2-oxa-5-azab¡c¡clo[2.2.1]heptan-5-¡l]propox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de terebutilo (350 mg, 681 μmol, 1 eq.), 1-bromo-8-metil-naftaleno (226 mg, 1,02 mmol, 1,5 eq.), Xantphos (78,9 mg, 136 gmol, 0,2 eq.), Pd2(dba)3 (62,4 mg, 68,1 μmol, 0,1 eq.) y CS2C03 (555 mg, 1,70 mmol, 2,5 eq.) en tolueno (10 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 12 horas en una atmósfera de N2. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (20 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, acetato de etilo/metanol = 300/1 a 10/1). Se obtuvo (2S)-2-(cianometil)-4-[7-(8-metil-1 -naftil)-2-[3-[(1 S,4S)-2-oxa-5- azabiciclo[2.2.1 ]heptan-5-il]propox¡]-6,8-dih¡dro-5H-pirido[3,4-C1pirimidin-4-il1piperazin-1 -carboxilato de tere-butilo (270 mg, 379 pmol, 56 % de rendimiento, 92 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 654.
1H RMN (400 MHz, cloroformo-d) ó = 7,75 - 7,61 (m, 2H), 7,45 - 7,37 (m, 1H), 7,34 (t, J = 7,2 Hz, 1H), 7,26 - 7,15 (m, 2H), 4,62 (s a, 1H), 4,45 - 4,31 (m, 3H), 4,30 - 4,16 (m, 1H), 4,10 - 3,70 (m, 5H), 3,61 (d a, J = 7,6 Hz, 1H), 3,57 - 3,44 (m, 2H), 3,43 - 3,25 (m, 1H), 3,24 - 3,05 (m, 3H), 3,03 - 2,86 (m, 5H), 2,84 - 2,67 (m, 4H), 2,66 - 2,48 (m, 2H), 2,00 -1,88 (m, 2H), 1,84 (d, J = 9,6 Hz, 1H), 1,72 (d, J = 9,6 Hz, 1H), 1,52 (s, 9H).
Etapa B: 2-[(2S)-4-[7-(8-Metil-1-naftil)-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.n heptan-5-i n propox n -6,8-dihidro-5H-pirido[3,4-1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[3-[(1 S,4S)-2-oxa-5-azab¡c¡clo[2.2.1]heptan-5-¡l]propox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-C]p¡r¡mid¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de tere-butilo (250 mg, 382 μmol, 1 eq.) en dioxano (2 ml) se le añadió HCl/dioxano (4 M, 1,91 ml, 1,00 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 1 hora. La mezcla de reacción se concentró al vacío. El residuo se diluyó con agua (20 ml) y después la mezcla se ajustó a pH = 7 con una solución acuosa saturada de NaHC03 y se extrajo con acetato de etilo (20 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar el producto. Se obtuvo 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.1] heptan-5-¡l]propox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-<C]p¡rim¡d¡n-4-¡l]p¡peraz¡n-2-il]acetonitrilo (260 mg, bruto) en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación. LCMS [ESI, M+1]: 554.
Etapa C: 2-[(2S)-1 -[(E)-4-Fluorobut-2-eno¡ n -4-[7-(8-met¡l-1 -naftil)-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.1 ]heptan-5-il1propoxi1-6,8-dihidro-5H-pirido[3,4-C1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[3-[(1 S,4S)-2-oxa-5-azab¡c¡clo[2.2.1]heptan-5-¡l]propoxi]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]acetonitrilo (120 mg, 216 μmol, 1 eq.), TEA (87,7 mg, 866 μmol, 121 μl, 4 eq.) y ácido (E)-4-fluorobut-2-enoico (45,1 mg, 433 μmol, 2 eq.) en acetato de etilo (2 ml) se le añadió T3P (276 mg, 433 μmol, 258 μl, 50 % de pureza, 2 eq.) a -70 °C. La mezcla se agitó a -70 °C durante 0,5 horas. La mezcla de reacción se inactivó con, se diluyó con
HCl (1 N, 1 ml) y después se diluyó con agua (20 ml). La mezcla se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (10 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Waters Xbridge 150 * 255 p; fase móvil: [agua (0,05 % de hidróxido de amonio v/v)-ACN]; % de B: 40 %-70 %, 10 min). Las fracciones deseadas se recogieron y se liofilizaron. El compuesto del título 2-[(2S)-1-[(E)-4-fluorobut-2-enoil]-4-[7-(8-metil-1-naftil)-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (14,5 mg, 22,5 μmol, 10 % de rendimiento, 99,3 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 640.
1H RMN (400 MHz, cloroformo-d) ó = 7,70 (d, J = 8,4 Hz, 1H), 7,65 (t, J = 8,0 Hz, 1H), 7,45 - 7,30 (m, 2H), 7,27 - 7,16 (m, 2H), 7,09 - 6,91 (m, 1H), 6,59 (d, J = 15,2 Hz, 1H), 5,29 - 4,48 (m, 3H), 4,44 - 4,31 (m, 3H), 4,29 - 3,72 (m, 6H), 3,71 - 3,40 (m, 4H), 3,28-2,96 (m, 4H), 2,95 - 2,88 (m, 4H), 2,87 - 2,56 (m, 5H), 2,52 (d, J = 10,0 Hz, 1H), 1,99 - 1,87 (m, 2H), 1,84 (d, J = 9,6 Hz, 1H), 1,71 (d, J = 9,6 Hz, 1H).
Ejemplo 566

2-[(2S)-1 -(2-Fluoroprop-2-enoil)-4-[7-(8-metil-1 -naftil)-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.1 ]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

2-[(2S)-1-(2-Fluoroprop-2-enoil)-4-[7-(8-metil-1-naftil)-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2,2,1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[3-[(1 S,4S)-2-oxa-5-azab¡c¡clo[2.2.1]heptan-5-¡l]propox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]p¡peraz¡n-2-il]acetonitrilo (100 mg, 181 μmol, 1 eq.) y ácido 2-fluoroprop-2-enoico (32,5 mg, 361 μmol, 2 eq.) en DCM (1 ml) se le añadieron hAt U (137 mg, 361 μmol, 2 eq.) y DIEA (93,4 mg, 722 μmol, 126 μl, 4 eq.). Después de agitar a 0 °C durante 0,5 horas, la mezcla de reacción se diluyó con agua (10 ml) y se extrajo con acetato de etilo (10 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (10 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por TLC (DCM/Me0H = 10/1) y se purificó adicionalmente por HPLC prep. (columna: Xtimate C1810 p 250 mm * 50 mm; fase móvil: [agua (0,05 % de hidróxido de amonio v/v)-ACN]; % de B: 45 %-75 %, 12 min). La fracción deseada se recogió y se liofilizó. El compuesto del título 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[7-(8-metil-1 -naftil)-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.1 ]heptan-5-il]propoxi]-6,8-dihidro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡trilo (12 mg, 19,1 μmol, 11 % de rendimiento, 99,6 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 626.1
1H RMN (400 MHz, cloroformo-d) ó = 7,63 (d, J = 8,0 Hz, 1H), 7,58 (t, J = 8,4 Hz, 1H),7,39 - 7,23 (m, 2H), 7,19 - 7,09 (m, 2H), 5,35 (d, J = 48,8 Hz, 1H), 5,18 (dd, J = 3,6, 16,8 Hz, 1H), 5,04 - 4,40 (m, 1H), 4,37 - 3,89 (m, 7H), 3,86 - 3,61 (m, 2H), 3,59 - 3,32 (m, 4H), 3,20 - 2,40 (m, 14H), 1,95 - 1,81 (m, 2H), 1,78 (d, J = 9,6 Hz, 1H), 1,65 (d, J = 9,6 Hz, 1H).
E je m p lo 567

2-[(2S)-1-(2-Fluoroprop-2-enoil)-4-[7-[8-(hidroximetil)-1-naftil]-2-[[(2SJ-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

8-Bromonaftaleno-1-carbaldehído. A una solución de 1,8-dibromonaftaleno (5 g, 17,5 mmol, 1 eq.) en THF (100 ml) se le añadió gota a gota n-Bpli (2,5 M, 9,09 ml, 1,3 eq.) a -70 °C. Después de agitar durante 30 minutos a -70 °C, se añadió gota a gota DMF (12,8 g, 175 mmol, 13,5 ml, 10 eq.). La mezcla se calentó a 12 °C y se agitó durante 0,5 horas más. Después de que se completara, la mezcla se inactivó con NH4Cl acuoso saturado (20 ml). La fase acuosa separada se extrajo con EtOAc (2 * 50 ml). Las capas orgánicas combinadas se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío para dar 8-bromonaftaleno-1-carbaldehído (4,5 g, bruto) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, cloroformo-d) 5 = 11,4 (s, 1H), 8,04 - 7,92 (m, 2H), 7,90 (d, J = 8,0 Hz, 2H), 7,60 - 7,55 (m, 1H), 7,39 (t, J = 8,0 Hz, 1H).
(8-Bromo-1 -naftil)metanol. A una mezcla de 8-bromonaftaleno-1-carbaldehído (4,5 g, 19,1 mmol) en Me0H (80 ml) se le añadió en una porción NaBH4 (2,90 g, 76,6 mmol) a 0 °C. La mezcla se agitó a 0 °C durante 30 minutos. Después de que
se completara, a la mezcla se le añadió agua (1 ml) y la mezcla se concentró al vacío. El residuo se diluyó con agua (10 ml) y se extrajo con EtOAc (2 * 40 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP/EtOAc 30/1 a 3/1) para dar (8-bromo-1-naftil)metanol (3,3 g, 12,5 mmol, dos etapas 72 % de rendimiento, 90 % de pureza) en forma de un sólido de color amarillo.
1H RMN (400 MHz, cloroformo-d) ó = 7,92 - 7,82 (m, 3H), 7,71 (d, J = 6,8 Hz, 1H), 7,49 (t, J = 7,6 Hz, 1H), 7,32 - 7,27 (m, 1H), 5,48 (s a, 2H).
2-í(8-Bromo-1-naftil)metox¡1tetrah¡drop¡rano. A una solución de (8-bromo-1-naftil)metanol (0,5 g, 2,11 mmol, 1 eq.) en DCM (10 ml) se le añadió DHP (355 mg, 4,22 mmol, 386 μl, 2 eq.), seguido de T s0 H ^0 (40,1 mg, 211 μmol, 0,1 eq.) a 12 °C. La mezcla se agitó a 12 °C durante 2 horas. Después de que se completara, la mezcla se inactivó con una solución acuosa saturada de NaHC03 (2 ml) y se diluyó con agua (5 ml). La fase acuosa separada se extrajo con EtOAc (10 ml). Las capas orgánicas combinadas se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (EP/EtOAc 500/1 a 100/1) para dar 2-[(8-bromo-1-naftil)metoxi1tetrahidropirano (560 mg, 1,57 mmol, 74 % de rendimiento, 90 % de pureza) en forma de un aceite incoloro.
1H RMN (400 MHz, cloroformo-d) ó = 7,94 - 7,88 (m, 2H), 7,85 (t, J = 8,4 Hz, 2H), 7,56 - 7,50 (m, 1H), 7,36 - 7,28 (m, 1H), 5,63 (s, 2H), 4,92 (t, J = 3,6 Hz, 1H), 4,07 - 3,97 (m, 1H), 3,69 - 3,61 (m, 1H), 2,08 - 1,76 (m, 4H), 1,75 - 1,65 (m, 2H).
Etapa A: (2S)-2-(C¡anomet¡l)-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-7-[8-(tetrah¡drop¡ran-2-¡lox¡met¡l)-1-naft¡l1-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡penaz¡n-1-carbox¡lato de bencilo. Se desgasificaron 2-[(8-bromo-1-naftil)metoxi1tetrahidropirano (476 mg, 1,48 mmol, 1,5 eq.), (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-5,6,7,8-tetrahidropirido[3,4-dlpirimidin-4-il1piperazin-1-carboxilato de bencilo (500 mg, 989 pmol, 1 eq.), Cs2C03 (806 mg, 2,47 mmol, 2,5 eq.), (5-difenilfosfanil-9,9-dimetil-xanten-4-il)-difenil-fosfano (229 mg, 396 pmol, 0,4 eq.) y Pd2(dba)3 (181 mg, 198 pmol, 0,2 eq.) en tolueno (20 ml) y después se calentaron a 90 °C durante 10 horas en una atmósfera de N2. Después de que se completara, la mezcla se filtró a través de una capa de celite y el filtrado se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo1. Las fracciones deseadas se recogieron, se neutralizaron con NaHC0a acuoso saturado, se concentraron al vacío para eliminar el MeCN y se extrajeron con EtOAc (2 * 30 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío para dar (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-[8-(tetrahidropiran-2-iloximetil)-1-naftil1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de bencilo (280 mg, 338 pmol, 34 % de rendimiento, 90 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+11: 746.
Etapa B: 2-[(2S)-4-[2-[[(2S)-1-Metilpirrolidin-2-il1metoxi1-7-[8-(tetrahidropiran-2-iloximetil)-1-naftil1-6,8-dihidro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una solución de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-[8-(tetrahidropiran-2-iloximetil)-1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de bencilo (280 mg, 375 μmol, 1 eq.) en Me0H (6 ml) se le añadieron NH3/Me0H (4 ml, 20 % de pureza) y Pd/C (120 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 15 °C durante 1 hora. Después de que se completara, la mezcla se filtró a través de una capa de celite y el filtrado se concentró al vacío para dar 2-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-[8-(tetrahidropiran-2-iloximetil)-1-naftil1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo (200 mg, 294 μmol, 78 % de rendimiento, 90 % de pureza) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. LCMS [ESI, M+11: 612.
Etapa C: 2-[(2S)-1-(2-Fluoroprop-2-eno¡l)-4-[2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-7-[8-(tetrah¡drop¡ran-2-¡lox¡met¡l)-1-naft¡l1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-[8-(tetrahidropiran-2-iloximetil)-1-naftil1-6,8-dihidro-5W-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo (180 mg, 294 pmol, 1 eq.), T3P (562 mg, 883 pmol, 525 μl, 50 % de pureza en EtOAc, 3 eq.) y TEA (238 mg, 2,35 mmol, 328 μl, 8 eq.) en EtOAc (4 ml) se le añadió ácido 2-fluoroprop-2-enoico (53,0 mg, 588 pmol, 2 eq.) a 0 °C. La mezcla se agitó a 15 °C durante 0,5 horas. Después de que se completara, la mezcla se diluyó con agua (4 ml) y se extrajo con EtOAc (3 * 10 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo1. Las fracciones deseadas se recogieron, se neutralizaron con NaHC03, se concentraron al vacío para eliminar el MeCN y se extrajeron con EtOAc (2 * 100 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío para dar 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-[8-(tetrahidropiran-2-iloximetil)-1-naftil1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo (160 mg, 227 pmol, 77 % de rendimiento, 97 % de pureza) en forma de un sólido de color amarillo. Lc Ms [ESI, M+11: 684.
Etapa D: 2-[(2S)-1-(2-Fluoroprop-2-enoil)-4-[7-[8-(hidroximetil)-1-naftil1-2-[[(2SH-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-7-[8-(tetrahidropiran-2-iloximetil)-1-naftil1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo (150 mg, 219 μmol, 1 eq.) en DCM (160 μl) se le añadió TFA (250 mg, 2,19 mmol, 162 μl, 10 eq.). La mezcla se agitó a 15 °C durante 2 horas. Después de que se completara, la mezcla se diluyó con diclorometano (3 ml) y se ajustó a pH = 9 con una solución acuosa saturada de Na2C03. La fase acuosa separada se extrajo con DCM (5 ml). Las capas orgánicas combinadas se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Xtimate C18150 * 25 mm * 5 μm; fase móvil: [agua (0,05 % de hidróxido de amonio v/v)-ACN1; % de B: 42 %-72 %, 8 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[(2S)-1 -(2-fluoroprop-2-enoil)-4-[7-[8-(hidroximetil)-1 -naftil1-2-[[(2S)-1 -metilpirrolidin2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (62,0 mg, 103 μmol, 47 % de rendimiento, 100 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 600.
1H RMN (400 MHz, cloroformo-d) 5 = 7,84 (d, J = 8,0 Hz, 1H), 7,79 (dd, J = 3,2, 7,2 Hz, 1H), 7,55 - 7,44 (m, 3H), 7,44 - 7,38 (m, 1H), 5,55 - 5,32 (m, 1H), 5,26 (dd, J = 3,6, 16,8 Hz, 1H), 5,21 - 4,94 (m, 2H), 4,89 (t a, J = 13,2 Hz, 1H), 4,41 - 3,90 (m, 7H), 3,66 - 3,39 (m, 2H), 3,37 - 3,00 (m, 5H), 3,00 - 2,58 (m, 4H), 2,50 - 2,43 (m, 3H), 2,33 - 2,22 (m, 1H), 2,11-1,98 (m, 1H), 1,90 - 1,73 (m, 3H).
Ejemplo 568

2-[(2S)-1-(2-Fluoroprop-2-enoil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

Etapa A: 4-[(3S)-4-Benciloxicarbonil-3-(cianometil)piperazin-1 -il]-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-7-carboxilato de tere-butilo. A una mezcla de 2-metilsulfanil-4-(trifluorometilsulfoniloxi)- 6,8-dihidro-5H-pirido[3,4-cf]pirimidin-7-carboxilato de tere-butilo (20,0 g, 46,6 mmol, 1,00 eq.) y (2S)-2-(cianometil)piperazin-1-carboxilato de bencilo (13,3 g, 51,2 mmol, 1,10 eq.) en dMac (250 ml) se le añadió en una porción DIEA (18,1 g, 140 mmol, 24,3 ml, 3 eq.) a 0 °C en una atmósfera de N2. La mezcla se calentó a 50 °C y se agitó durante 1 hora. La mezcla de reacción se diluyó con agua (100 ml) y se extrajo con acetato de etilo (3 * 300 ml). Las capas orgánicas combinadas se lavaron con agua (3 * 500 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiÜ2, éter de petróleo/acetato de etilo = 50/1 a 1/1). El compuesto 4-[(3S)-4-benciloxicarbonil-3(cianometil)piperazin-1-il]-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (23,0 g, 42,7 mmol, 92 % de rendimiento, 100 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 539.
Etapa B: (2S)-2-(Cianometil)-4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo. A una mezcla de 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-7-carboxilato de ferc-butilo (23,0 g, 42,7 mmol, 1,0o eq.) en dioxano (200 ml) se le añadió HCl/dioxano (4 M, 192 ml, 18,0 eq.). Después de agitar a 20 °C durante 0,5 horas, la mezcla de reacción se filtró y la torta de filtro se disolvió en acetato de etilo (200 ml). El pH se ajustó a 8-9 con una solución saturada de Na2CÜ3 y después se diluyó con agua (20,0 ml). La capa de agua separada se extrajo con acetato de etilo (2 x 50 ml). Las capas orgánicas combinadas se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se usó en la siguiente etapa directamente sin purificación adicional. El compuesto (2S)-2-(cianometil)-4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (16,1 g, 34,5 mmol, 81 % de rendimiento, 94 % de pureza) se obtuvo en forma de un sólido de color amarillo.
1H RMN (400 MHz, cloroformo-d) 6 = 7,42 - 7,33 (m, 5H), 5,25 - 5,12 (m, 2H), 4,66 (s a, 1H), 4,11 - 4,04 (m, 1H), 4,03 - 3,90 (m, 3H), 3,84 (d a, J = 13,6 Hz, 1H), 3,34-3,17 (m, 2H), 3,11 (td, J = 5,2, 10,4 Hz, 1H), 3,04 - 2,93 (m, 2H), 2,88 - 2,76 (m, 1H), 2,75-2,56 (m, 3H), 2,50 (s, 3H).
Etapa C: (2S)-2-(Cianometil)-4-[7-(8-metil-1-naftil)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]p¡perazin-1-carboxilato de bencilo. A una mezcla de (2S)-2-(cianometil)-4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (16,1 g, 36,7 mmol, 1,00 eq.) y 1-bromo-8-metil-naftaleno (10,6 g, 47,7 mmol, 1,30 eq.) en tolueno (350 ml) se le añadieron Pd2(dba)3 (6,72 g, 7,34 mmol, 0,20 eq.), Xantphos (8,50 g, 14,7 mmol, 0,40 eq.) y Cs2CÜ3 (35,9 g, 110 mmol, 3,00 eq.). La mezcla se desgasificó y se purgó 3 veces con N2. Después de agitar a 90 °C durante 8 horas, la mezcla de reacción se diluyó con agua (1 x 100 ml) y se extrajo con acetato de etilo (3 x 150 ml). Las capas orgánicas combinadas se lavaron con agua (1 x 300 ml) y salmuera (1 x 300 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (S¡02, éter de petróleo/acetato de etilo = 100/1 a 2/1) y se concentró a presión reducida para dar un residuo. El compuesto (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (15,3 g, 26,5 mmol, 72 % de rendimiento) se obtuvo en forma de un sólido de color amarillo.
1H RMN (400 MHz, cloroformo-d) 6 = 7,68 - 7,55 (m, 2H), 7,40 - 7,28 (m, 7H), 7,22 (s, 2H), 5,22 - 5,09 (m, 2H), 4,64 (s a, 1H), 4,27 - 4,14 (m, 1H), 4,05 - 3,84 (m, 2H), 3,83 - 3,68 (m, 1H), 3,55 - 3,30 (m, 2H), 3,18 - 3,03 (m, 3H), 3,01 -2,79 (m, 5H), 2,78 - 2,50 (m, 3H), 2,45 (d, J = 4,8 Hz, 3H).
Etapa D: (2S)-2-(Cianometil)-4-[7-(8-metil-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]p¡perazin-1-carboxilato de bencilo. A una mezcla de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (13,3 g, 23,0 mmol, 1,00 eq.) en EtOAc (200 ml) se le añadió m-CPBA (4,67 g, 23,0 mmol, 85 % de pureza, 1,00 eq.) a 0 °C en una atmósfera de N2. Después de agitar a 0 °C durante 30 min, la mezcla de reacción se inactivó con Na2S203 saturado (100 ml) a 0 °C. La capa orgánica separada se diluyó con agua (1 x 100 ml) y se extrajo con acetato de etilo (3 x 100 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 anhidro, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si0s, éter de petróleo/acetato de etilo = 50/1 a 0/1). El compuesto (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-metilsulfinil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (9,60 g, 15,3 mmol, 67 % de rendimiento, 95 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 595.
1H RMN (400 MHz, cloroformo-d) 6 = 7,74 - 7,63 (m, 2H), 7,47 - 7,31 (m, 7H), 7,27 - 7,17 (m, 2H), 5,25 - 5,17 (m, 2H), 4.67 (s a, 1H), 4,48 - 4,21 (m, 2H), 4,10 - 3,88 (m, 2H), 3,66 - 3,46 (m, 2H), 3,42 - 3,04 (m, 4H), 3,02 - 2,83 (m, 6H), 2,81 - 2,60 (m, 1H), 2,81-2,60 (m, 3H).
Etapa E: (2S)-4-[2-[[(2S)-1-ferc-Butoxicarbonilpirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de ferc-butilo. A una mezcla de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (500 mg, 841 pmol, 1,00 eq.) y (2S)-2-(hidroximetil)pirrolidin-1 -carboxilato de ferc-butilo (338 mg, 1,68 mmol, 2,00 eq.) en tolueno (10,0 ml) se le añadió en una porción f-Bu0Na (242 mg, 2,52 mmol, 3,00 eq.) a 0 °C en una atmósfera de N2. Después de agitar a 0 °C durante 30 min, la mezcla de reacción se inactivó mediante la adición de 2 ml de ácido clorhídrico (1 M) a 0 °C y agua (10 ml). La mezcla se extrajo con acetato de etilo (20 ml x 3 ). Las capas orgánicas combinadas se lavaron con agua (30 ml x 1 ) y salmuera (30 ml x 1), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (S¡02, éter de petróleo/acetato de etilo = 50/1 a 0/1). El compuesto (2S)-4-[2-[[(2S)-1-ferc- butox¡carbon¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carboxilato de ferc-butilo (330 mg, 473 pmol, 56 % de rendimiento) se obtuvo en forma de un sólido de color amarillo.
1H RMN (400 MHz, cloroformo-d) 6 = 7,73 - 7,61 (m, 2H), 7,47 - 7,31 (m, 7H), 7,26 - 7,15 (m, 2H), 5,27 - 5,14 (m, 2H), 4.68 (s a, 1H), 4,42 - 4,17 (m, 3H), 4,07 - 3,73 (m, 3H), 3,58 - 3,29 (m, 5H), 3,25 - 2,88 (m, 8H), 2,86 - 2,51 (m, 3H), 2,03 - 1,76 (m, 4H), 1,44 (d, J = 4,8 Hz, 9H).
Etapa F: (2S)-2-[(4-[(3S)-3-(Cianometil)piperazin-1-il1-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-dlpirimidin-2-illoximetillpirrolidin-1-carboxilato de tere-butilo. A una mezcla de (2S)-4-[2-[[(2S)-1-tere-butoxicarbonilpirrolidin-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de tere-butilo (330 mg, 473 μmol, 1,00 eq.) en Me0H (15 ml) se le añadieron en una porción NH3*Me0H (15 ml, 20 % de pureza) y Pd(0H)2/C (80 mg, 20 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó tres veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 20 °C durante 1 hora. El catalizador se retiró por filtración y el filtrado se concentró a presión reducida para dar un residuo. El producto bruto se usó en la siguiente etapa directamente sin purificación adicional. El compuesto (2S)-2-[[4-[(3S)-3-(cianometil)piperazin-1-il]-7-(8-metil-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-2-¡l]ox¡met¡l]p¡rrol¡d¡n-1-carbox¡lato de tere-butilo (200 mg, 335 pmol, 71 % de rendimiento) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 598.
Etapa G: (2S)-2-[[4-[(3S)-3-(cianometil)-4-(2-fluoroprop-2-enoil)piperazin-1-¡ n -7-(8-metil-1 -naftil)-6,8-dihidro-5H-pirido[3,4-cf1pirimidin-2-il)oximetillpirrolidin-1 -carboxilato de tere-butilo. A una mezcla de (2S)-2-[[4-[(3S)-3-(cianometil)piperazin-1-il]-7-(8 -metil-1 -naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-2-il1oximetil1pirrolidin-1 -carboxilato de tere-butilo (190 mg, 318 pmol, 1,00 eq.) y ácido 2-fluoroprop-2-enoico (42,9 mg, 477 pmol, 1,50 eq.) en DCM (15,0 ml) se le añadieron en una porción DIEA (123 mg, 954 pmol, 166 ul, 3,00 eq.) y HATU (181 mg, 477 pmol, 1,50 eq.) a 0 °C en una atmósfera de N2. Después de agitar a 0 °C durante 30 min, la mezcla de reacción se diluyó con agua (10 ml). La capa orgánica separada se lavó con salmuera (1 * 10 ml), se secó sobre Na2S04, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna (S¡02, DCM: Me0H = 50:1 a 5:1). El compuesto (2S)-2-[[4-[(3S)-3-(cianometil)-4-(2-fluoroprop-2-enoil)piperazin-1-il1-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-cf1pirimidin-2-il1oximetil1pirrolidin-1-carboxilato de tere-butilo (210 mg, 292 pmol, 92 % de rendimiento, 93 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 670.
Etapa H: 2-[(2S)-1-(2-Fluoroprop-2-enoil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-pirrolidin-2-i n metox n -6,8-dihidro-5H-pirido[3,4-cf1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una mezcla de (2S)-2-[[4-[(3S)-3-(cianometil)-4-(2-fluoroprop-2-enoil)piperazin-1-il1-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-cf1pirimidin-2-il1oximetil1pirrolidin-1-carboxilato de tere-butilo (210 mg, 314 pmol, 1,00 eq.) en DCM (5,00 ml) se le añadió en una porción t Fa (1,07 g, 9,41 mmol, 696 μl, 30,0 eq.) a 20 °C en una atmósfera de N2. Después de agitar a 20 °C durante 30 min, la mezcla de reacción se concentró a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi C18150 * 30 mm * 4 μm; fase móvil:
[agua (0,225 % de AF)-ACN];% de B: 20 %-50 %,5 min). El compuesto del título 2-[(2S)-1 -(2-fluoroprop-2-enoil)-4-[7-(8-metil-1-naft¡l)-2-[[(2S)-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-dih¡dro-5H-p¡r¡do[3,4-a]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (75,5 mg, 122 pmol, 39 % de rendimiento, 99,8 % de pureza, AF) se obtuvo en forma de un sólido de color blanquecino. LCMS [ESI, M+1]: 570.
1H RMN (400 MHz, cloroformo-d) ó = 7,72 - 7,61 (m, 2H), 7,44 - 7,30 (m, 2H), 7,27 - 7,14 (m, 2H), 6,49 - 5,98 (m, 1H), 5,55 - 5,32 (m, 1H), 5,25 (dd, J = 3,6, 16,8 Hz, 1H), 4,58 - 4,42 (m, 2H), 4,36 - 3,87 (m, 5H), 3,86 - 3,67 (m, 1H), 3,64 - 3,37 (m, 2H), 3,35 - 3,26 (m, 2H), 3,25 - 2,94 (m, 4H), 2,92 - 2,70 (m, 5H), 2,66 - 2,51 (m, 1H), 2,18 - 1,81 (m, 4H).
Ejemplo 569

2-[(2S)-2-(2-Fluoroprop-2-eno¡l)-4-[7-(8-met¡l-1-naft¡l)-2-[[(2S)-1-metil-5-oxo-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡trilo


(5S)-5-(Tetrahidropiran-2-iloximetil)pirrolidin-2-ona. A una mezcla de (5S)-5-(hidroximetil)pirrolidin-2-ona (5,00 g, 43,4 mmol, 1,00 eq.) y DHP (3,65 g, 43,4 mmol, 3,97 ml, 1,00 eq.) en DCM (80,0 ml) se le añadió Ts0HH20 (826 mg, 4,34 mmol, 0,10 eq.). La mezcla se agitó a 15 °C durante 12 horas. La mezcla de reacción se lavó con salmuera (30,0 ml x 1), se secó sobre sulfato de sodio, se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 20/1 a 0:1). El compuesto (5S)-5-(tetrahidropiran-2-iloximetil)pirrolidin-2-ona (5,70 g, 28,6 mmol, 66 % de rendimiento) se obtuvo en forma de un aceite de color amarillo.
1H RMN (400 MHz, cloroformo-d) 5 = 6,36 - 6,01 (m, 1H), 4,66 - 4,47 (m, 1H), 3,92 - 3,71 (m, 2,5H), 3,64 - 3,43 (m, 2H), 3,24 (dd, J = 8,0, 9,6 Hz, 0,5H), 2,42 - 2,10 (m, 3H), 1,86 - 1,64 (m, 3H), 1,62 - 1,48 (m, 4H).
(5S)-1-Metil-5-(tetrahidropiran-2-iloximetil)pirrolidin-2-ona. A una mezcla de (5S)-5-(tetrahidropiran-2-iloximetil)pirrolidin-2-ona (5,60 g, 28,1 mmol, 1,00 eq.) en DMF (60,0 ml) se le añadió en una porción NaH (1,35 g, 33,7 mmol, 60 % de pureza, 1,20 eq.) a -40 °C en una atmósfera de N2. La mezcla se agitó a -40 °C durante 30 min, después se añadió CH3I (6,58 g, 46,4 mmol, 2,89 ml, 1,65 eq.) y se agitó a 0 °C durante 2,5 horas. La mezcla de reacción se inactivó mediante la adición de una solución acuosa saturada de NaHS03 (30,0 ml) y se extrajo con AE (3 x 50,0 ml). La fase orgánica combinada se secó sobre Na2S04 y se concentró a sequedad. El residuo se purificó por cromatografía en columna (S02, éter de petróleo/acetato de etilo = 10/1 a 0/1). El compuesto (5S)-1-metil-5-(tetrahidropiran-2-iloximetil)pirrolidin-2-ona (4,40 g, 20,6 mmol, 73 % de rendimiento) se obtuvo en forma de un aceite de color amarillo.
1H RMN (400 MHz, cloroformo-d) 5 = 4,59 (td, J = 3,2, 13,6 Hz, 1H), 3,93 - 3,73 (m, 2H), 3,63 - 371 (m, 1H), 3,57 -3,44 (m, 2H), 2,88 (d, J = 4,4 Hz, 3H), 2,54 - 2,40 (m, 1H), 2,38 - 2,24 (m, 1H), 2,07 -2,20 (m, 1H), 1,93 - 1,53 (m, 7H).
(5S)-5-(Hidroximetil)-1-metil-pirrolidin-2-ona. A una mezcla de (5S)-1-metil-5-(tetrahidropiran-2-iloximetil)pirrolidin-2-ona (2,20 g, 10,3 mmol, 1,00 eq.) en DCM (20,0 ml) se le añadió TfA (23,5 g, 206 mmol, 15,3 ml, 20,0 eq.). La mezcla se agitó a 15 °C durante 30 min. La mezcla de reacción se concentró a presión reducida para dar un residuo. El producto bruto se usó en la siguiente etapa directamente sin purificación adicional. El compuesto (5S )-5-(hidroximetil)-1-metil-pirrolidin-2-ona (2,60 g, bruto) se obtuvo en forma de un aceite de color rojo.
Etapa A: (2S)-2-(Cianometil)-4-[7-(8-metil-1-naftil)-2-1[(2S)-1-metil-5-oxo-pirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de bencilo. A una mezcla de (5S)-5-(hidroximetil)-1 -metil-pirrolidin-2-ona (543 mg, 4,20 mmol, 5,00 eq.) en THF (30,0 ml) se le añadió en una porción í-Bu0Na (808 mg, 8,41 mmol, 10,0 eq.) a 0 °C en una atmósfera de N2. La mezcla se agitó a 0 °C durante 30 min, después se añadió (2S)-2-(cianometil)-4-[7-(8-metil-1 -naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1 -carboxilato de bencilo (500 mg, 841 gmol, 1,00 eq.) y se calentó a 15 °C y se agitó durante 0,5 horas. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (30 ml x 3 ). Las capas orgánicas combinadas se lavaron con agua (50 ml
x 1), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de TFA)/acetonitrilo]. Las fracciones deseadas se recogieron, se neutralizaron con una solución saturada de NaHC03 (10,0 ml) y se extrajeron con acetato de etilo (50,0 ml x 2). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El compuesto (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metil-5-oxo-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (162 mg, 243 μmol, 29 % de rendimiento, 99 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 660.
Etapa B: 2-[(2S)-4-[7-(8-Metil-1-naftil)-2-[[(2S)-1-metil-5-oxo-pirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin--i n piperazin-2-i n acetonitrilo. A una mezcla de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metil-5-oxo-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-c(]pirimidin-4-il]piperazin-1-carboxilato de bencilo (130 mg, 197 μmol, 1,00 eq.) en Me0H (2,00 ml) se le añadieron NHs^Me0H (1,00 ml, 20 % de pureza) y Pd/C (35,0 mg, 10 % de pureza). La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 15 °C durante 1 hora. La mezcla de reacción se filtró y se concentró a presión reducida para dar un residuo. El producto bruto se usó en la siguiente etapa directamente sin purificación adicional. El compuesto 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metil-5-oxo-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (70,0 mg, bruto) se obtuvo en forma de un sólido de color pardo. LCMS [ESI, M+1]: 526.
Etapa C: 2-[(2S)-1-(2-Fluoroprop-2-enoil)-4-r7-(8-metil-1-naftN)-2-[[(2S)-1-metil-5-oxo-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3.4-cnpirimidin-4-N)piperazin-2-N)acetonitrilo. A una mezcla de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1- metil-5-oxo-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (60,0 mg, 114 μmol, 1,00 eq.) y ácido 2-fluoroprop-2-enoico (20,6 mg, 228 μmol, 2,00 eq.) en AE (1,00 ml) se le añadieron en una porción TEA (92,4 mg, 913 μmol, 127 ul, 8,00 eq.), T3P (218 mg, 342 μmol, 204 ul, 50 % de pureza, 3,00 eq.) a 0 °C en una atmósfera de N2. La mezcla se agitó a 0 °C durante 30 min. La mezcla de reacción se diluyó con agua (5,00 ml) y se extrajo con acetato de etilo (10,0 ml x 3 ). Las capas orgánicas combinadas se lavaron con agua (10,0 ml x 1) y salmuera (10,0 ml x 1), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Xtimate C18 150 x 25 mm x 5 um;fase móvil: [agua (0,05 % de hidróxido de amonio v/v)-ACN];% de B: 42 %-72 %,10 min). El compuesto del título 2-[(2S)-1-(2-fluoroprop-2- enoil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1-metil-5-oxo-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (26,8 mg, 43,9 μmol, dos etapas 26 % de rendimiento, 98 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 598.
1H RMN (400 MHz, cloroformo-d) ó = 7,73 - 7,62 (m, 2H), 7,46 - 7,38 (m, 1H), 7,38 - 7,32 (m, 1H), 7,27 - 7,17 (m, 2H), 5,55 - 5,32 (m, 1H), 5,26 (dd, J = 3,6, 16,8 Hz, 1H), 5,08 - 4,63 (m, 1H), 4,53 - 4,39 (m, 1H), 4,37 - 4,26 (m, 1H), 4,25 - 4,00 (m, 3H), 3,97 - 3,71 (m, 3H), 3,59 - 3,35 (m, 2H), 3,29 - 2,98 (m, 4H), 2,91 (d, J = 5,6 Hz, 8H), 2,69 - 2,58 (m, 1H), 2,57 - 2,45 (m, 1H), 2,42 - 2,29 (m, 1H), 2,28 - 2,14 (m, 1H), 2,06 - 1,91 (m, 1H).
Ejemplo 570

2-[(2S)-1-(2-Fluoroprop-2-enoil)-4-[2-[[(2R,3R)-3-hidroxi-1-metil-5-oxo-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo
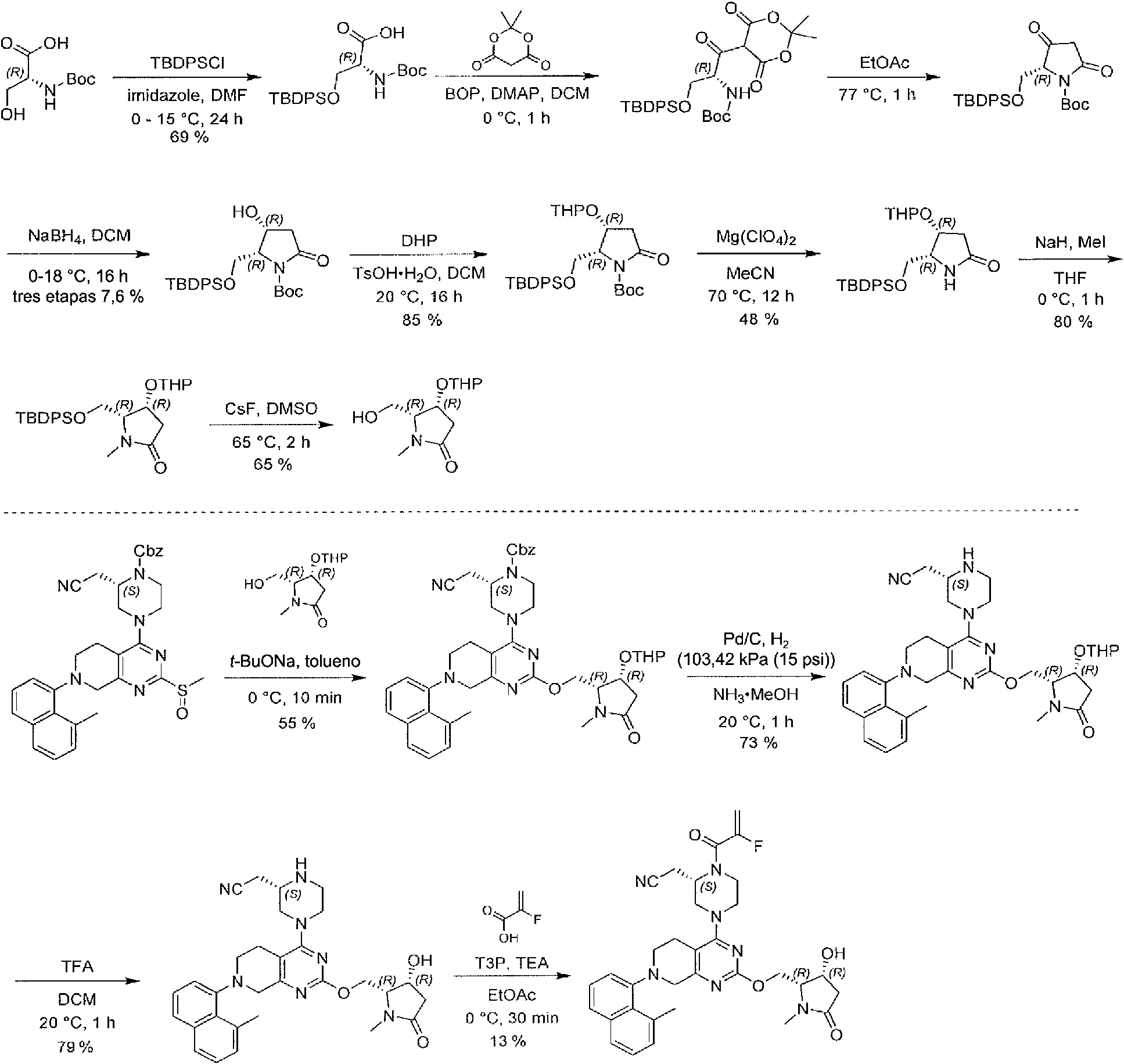
Ácido (2R)-2-(terc-butoxicarbonilamino)-3-[terc-butil(difenil)silil]oxi-propanoico. A una solución de ácido (2R )-2-(terc-butoxicarbonilamino)-3-hidroxi-propanoico (27 g, 132 mmol, 1 eq.) en DMF (270 ml) se le añadió imidazol (45,4 g, 667 mmol, 5,07 eq.) a 0 °C en una atmósfera de nitrógeno. Después de que la solución se volviera transparente, se añadió TBDPSCl (90,4 g, 329 mmol, 84,5 ml, 2,5 eq.). La reacción se calentó a 15 °C. Después de agitar a 15 °C durante 24 h, la mezcla de reacción se concentró al vacío, se diluyó con éter (250 ml) y se vertió en NaCl saturado (100 ml). La capa orgánica separada se lavó con una mezcla de 10 % de HCl (25 ml) y NaCl saturado (50 ml), se secó sobre Na2SÜ4 y se concentró a presión reducida. El residuo se diluyó con acetonitrilo y después se formó un precipitado de color blanco. El precipitado se filtró y la torta de filtro se secó al vacío para dar ácido (2 R)-2-(terc-butoxicarbonilamino)-3-[terc-butil(difenil)silil]oxi-propanoico (45 g, 90 mmol, 69 % de rendimiento, 89 % de pureza) en forma de un sólido de color blanco.
^-[1R)-1-[[terc-Butil(difenil)silil1oximetil1-2-(2,2-dimetil-4,6-dioxo-1,3-dioxan-5-il)-2-oxo-etil1carbamato de terc-butilo. A una solución agitada de ácido (2R)-2-(terc-butoxicarbonilamino)-3-[terc-butil(difenil)silil]oxi-propanoico (24 g, 54,1 mmol, 1 eq.) en DCM (400 ml) a 0 °C se le añadieron 2,2-dimetil-1,3-dioxano-4,6-diona (8,19 g, 56,8 mmol, 1,05 eq.) y DMAP (16,0 g, 131 mmol, 2,42 eq.), seguido de B0P (25,1 g, 56,8 mmol, 1,05 eq.) en pequeñas porciones. La mezcla se agitó durante 1 hora a 0 °C. Después de que se completara, la mezcla se lavó con KHS041 N (2 * 360 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío a temperatura ambiente para dar W-[(1R)-1-[[terc-butil(difenil)silil] oximetil]-2-(2,2-dimetil-4,6-dioxo-1,3-dioxan-5-il)-2-oxo-etil]carbamato de terc-butilo (35 g, bruto) en forma de un aceite de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional.
(2R)-2-[[terc-Butil(difenil)sili n oximet¡ n -3,5-dioxo-pirrolidin-1 -carboxilato de terc-butilo. Se calentó W-[(1R)-1-[[terc-butil(difenil)silil]oximetil]-2-(2,2- dimetil-4,6-dioxo-1,3-dioxan-5-il)-2-oxo-etil]carbamato de terc-butilo (35 g,
61.43 mmol, 1 eq.) a reflujo en EtOAc (600 ml) a 77 °C durante 1 hora. Después de que se completara, la mezcla se concentró al vacío para dar (2R)-2-[[ferc-but¡l(d¡fen¡l)s¡l¡l]ox¡met¡l]-3,5-d¡oxo-p¡rrol¡d¡n-1-carbox¡lato de tere-butilo (30 g, bruto) en forma de un ace¡te de color amar¡llo que se usó d¡rectamente en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón ad¡c¡onal.
(2^.3^)-2-[[terc-But¡l(d¡fen¡l)s¡l¡l1ox¡met¡l1-3-h¡drox¡-5-oxo-p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo. Se d¡solv¡ó (2R)-2-[[terc-but¡l(d¡fen¡l)s¡l¡l]ox¡met¡l]-3,5-d¡oxo-p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo (30 g, 64,2 mmol, 1 eq.) en DCM (480 ml) a 0 °C, segu¡do de NaBHU (3,78 g, 99,9 mmol, 1,56 eq.) en pequeñas porc¡ones. La suspens¡ón se ag¡tó a 18 °C durante 16 horas. Después de que se completara, la mezcla de reacc¡ón se vert¡ó en agua (300 ml) y se ag¡tó hasta que no quedó sól¡do. La capa orgán¡ca separada se lavó con agua (150 ml), se secó sobre Na2S04, se f¡ltró y se concentró al vacío. El res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (EP/EtOAc 10/1 a 2/1) segu¡do de cromatografía ultrarráp¡da de fase ¡nversa [agua (0,1 % de AF)/aceton¡tr¡lo]. Las fracc¡ones deseadas se recog¡eron, se neutral¡zaron con NaHC03, se concentraron al vacío para el¡m¡nar el MeCN y después se extrajeron con DCM (2 * 50 ml). Las capas orgán¡cas se secaron sobre Na2S04 y se concentraron al vacío para dar (2R,3R)-2-[[tercbut¡l(d¡fen¡l)s¡l¡l]ox¡met¡l]-3-h¡drox¡-5-oxo-p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo (2,13 g, 3,99 mmol, tres etapas 7,6 % de rend¡m¡ento, 88 % de pureza) en forma de un sól¡do de color amar¡llo.
1H RMN (400 MHz, DMS0-d6) ó = 7,72 (dd, J = 1,6, 8,0 Hz, 2H), 7,59 (dd, J = 1,2, 8,0 Hz, 2H), 7,47 - 7,39 (m, 6H), 5,67 (d, J = 4,0 Hz, 1H), 4,57 - 4,46 (m, 1H), 4,09 - 3,97 (m, 2H), 3,83 (dd, J = 2,8, 10,0 Hz, 1H), 2,75 - 2,65 (m, 1H), 2,62 - 2,53 (m, 1H), 1,38 (s, 9H), 0,93 (s, 9H).
(2R.3R)-2-[[terc-but¡l(D¡fen¡l)s¡l¡l1ox¡met¡l1-5-oxo-3-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo. A una soluc¡ón de (2R,3R)-2-[[terc-but¡l(d¡fen¡l)s¡l¡l] ox¡met¡l]-3-h¡drox¡-5-oxo-p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo (2,55 g, 5.43 mmol, 1 eq.) en DCM (50 ml) se le añad¡ó DHP (913 mg, 10,9 mmol, 993 μl, 2 eq.), seguido de T s0 H ^0 (103 mg, 543 μmol, 0,1 eq.) a 20 °C. La mezcla se ag¡tó a 20 °C durante 16 horas. Después de que se completara, la mezcla se ¡nact¡vó con una soluc¡ón acuosa saturada de NaHC03 (10 ml), se d¡luyó con agua (5 ml) y se extrajo con DCM (2 * 40 ml). Las capas orgán¡cas comb¡nadas se secaron sobre Na2S04, se f¡ltraron y se concentraron al vacío. El res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (EP/EtOAc 15/1 a H1) para dar (2R,3R)-2-[[terc-but¡l (d¡fen¡l)s¡l¡l]ox¡met¡l]-5-oxo-3-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo (2,62 g, 4,64 mmol, 85 % de rend¡m¡ento, 98 % de pureza) en forma de un sól¡do de color amar¡llo.
1H RMN (400 MHz, cloroformo-d) ó = 7,75 (td, J = 1,2, 7,6 Hz, 2H), 7,66 (td, J = 1,6, 8,0 Hz, 2H), 7,47 - 7,34 (m, 6H), 4,75 - 4,68 (m, 1H), 4,63 - 4,51 (m, 1H), 4,20 - 4,17 (m, 1H), 3,99 - 3,88 (m, 2H), 3,87 - 3,74 (m, 1H), 3,60 - 3,50 (m, 1H), 3,23 - 3,01 (m, 1H), 2,76 (ddd, J = 8,8, 10,4, 16,8 Hz, 1H), 2,01 - 1,80 (m, 2H), 1,79 - 1,62 (m, 4H), 1,45 (s, 9H), 1,02 (d, J = 8,0 Hz, 9H).
(4R.5R)-5-[[terc-But¡l(d¡fen¡l)s¡l¡l1ox¡met¡l1-4-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-2-ona. A una soluc¡ón de (2R,3R)-2-[[tercbut¡l(d¡fen¡l)s¡l¡l] ox¡met¡l]-5-oxo-3-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo (2,62 g, 4,73 mmol, 1 eq.) en MeCN (50 ml) se añad¡ó Mg(Cl04)2 (211 mg, 946 μmol, 95,6 μl, 0,2 eq.). La mezcla de reacc¡ón se ag¡tó a 70 °C durante 12 horas. Después de que se completara, la mezcla se f¡ltró y el f¡ltrado se concentró al vacío. El res¡duo se pur¡f¡có por cromatografía ultrarráp¡da de fase ¡nversa [agua (0,1 % de AF)/aceton¡tr¡lo]. Las fracc¡ones deseadas se recog¡eron y se concentraron al vacío para dar (4R,5R)-5-[[terc-but¡l(d¡fen¡l)s¡l¡l]ox¡met¡l]-4-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-2-ona (1,14 g, 2,29 mmol, 48 % de rend¡m¡ento, 91 % de pureza) en forma de un ace¡te de color amar¡llo. LCMS [ESI, M+1]: 454.
(4R,5R)-5-[[terc-But¡l(d¡fen¡l)s¡l¡l1ox¡met¡l1-1-met¡l-4-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-2-ona. A una soluc¡ón de (4R,5R)-5-[[terc-but¡l(d¡fen¡l)s¡l¡l]ox¡met¡l]-4-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-2-ona (0,95 g, 2,09 mmol, 1 eq.) en THF (20 ml) se le añad¡ó en una porc¡ón NaH (168 mg, 4,19 mmol, 60 % de pureza en ace¡te m¡neral, 2 eq.) a 0 °C en una atmósfera de N2. La mezcla se ag¡tó a 0 °C durante 30 m¡nutos y después se añad¡ó CH3I (594 mg, 4,19 mmol, 261 ul, 2 eq.). Después de ag¡tar a 0 °C durante 0,5 horas más, la mezcla se ¡nact¡vó con agua (10 ml) y se extrajo con EtOAc (2 * 30 ml). Las capas orgán¡cas se secaron sobre Na2S04 y se concentraron al vacío. El res¡duo se pur¡f¡có por cromatografía ultrarráp¡da de fase ¡nversa [agua (0,1 % de AF)/aceton¡tr¡lo]. Las fracc¡ones deseadas se recog¡eron y se concentraron al vacío para dar (4R,5R)-5-[[terc-but¡l(d¡fen¡l)s¡l¡l]ox¡met¡l]-1-met¡l-4-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-2-ona (820 mg, 1,67 mmol, 80 % de rend¡m¡ento, 95 % de pureza) en forma de un ace¡te de color pardo.
1H RMN (400 MHz, cloroformo-d) ó = 7,75 - 7,70 (m, 2H), 7,70 - 7,65 (m, 2H), 7,48 - 7,36 (m, 6H), 4,68 - 4,60 (m, 1H), 4,59 - 4,48 (m, 1H), 4,07 - 3,77 (m, 2H), 3,77 - 3,66 (m, 1H), 3,65 - 3,56 (m, 1H), 3,55 - 3,40 (m, 1H), 2,89 - 2,79 (m, 3H), 2,79 - 2,53 (m, 2H), 1,90 - 1,69 (m, 2H), 1,68 - 1,53 (m, 4H), 1,09 - 1,03 (m, 9H).
(4R,5R)-5-(H¡drox¡met¡l)-1-met¡l-4-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-2-ona. A una mezcla de (4R,5R)-5-[[tercbut¡l(d¡fen¡l)s¡l¡l]ox¡met¡l]-1-met¡l-4-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-2-ona (1,07 g, 2,29 mmol, 1 eq.) en DMs 0 (10 ml) se le añad¡ó CsF (1,04 g, 6,86 mmol, 253 μl, 3 eq.). La mezcla se ag¡tó a 65 °C durante 2 horas. Después de que se completara, la mezcla se concentró al vacío. El res¡duo se pur¡f¡có por cromatografía (AL03, EtOAc/Me0H 50/1 a 5/1) para dar (4R,5R)-5-(h¡drox¡met¡l)-1-met¡l-4-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-2-ona (380 mg, 1,49 mmol, 65 % de rend¡m¡ento, 90 % de pureza) en forma de un ace¡te de color amar¡llo.
1H RMN (400 MHz, cloroformo-d) 5 = 4,80 - 4,52 (m, 2H), 3,98 - 3,81 (m, 3H), 3,72 - 3,61 (m, 1H), 3,60 - 3,48 (m, 1H), 2,87 (d, J = 9,6 Hz, 3H), 2,67 (d, J = 7,2 Hz, 1H), 2,60 - 2,46 (m, 1H), 1,86 - 1,74 (m, 2H), 1,67 - 1,51 (m, 4H).
Etapa A: (2S)-2-(C¡anomet¡l)-4-[7-(8-met¡l-1-naft¡l)-2-[[(2R,3R)-1-met¡l-5-oxo-3-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo. A una soluc¡ón de (2S)-2-(c¡anomet¡l)-4-[7-(8-met¡l-1-naft¡l)-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (280 mg, 471 jmol, 1 eq.) y (4R,5R)-5-(h¡drox¡met¡l)-1-met¡l-4-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-2-ona (140 mg, 612 |jmol, 1,3 eq.) en tolueno (6 ml) se le añad¡ó f-Bu0Na (90,5 mg, 942 jmol, 2 eq.) a 0 °C. Después de ag¡tar a 0 °C durante 10 m¡nutos, la mezcla se d¡luyó con agua (5 ml) y se extrajo con EtOAc (2 * 10 ml). Las capas orgán¡cas se secaron sobre Na2S04 y se concentraron al vacío. El res¡duo se pur¡f¡có por cromatografía ultrarráp¡da de fase ¡nversa [agua (0,1 % de AF)/aceton¡tr¡lo]. Las fracc¡ones deseadas se recog¡eron, se neutral¡zaron con NaHC03, se concentraron al vacío para el¡m¡nar el MeCN y se extrajeron con EtOAc (2 * 20 ml). Las capas orgán¡cas se secaron sobre Na2S04 y se concentraron al vacío para dar (2S)-2-(c¡anomet¡l)-4-[7-(8-met¡l-1-naft¡l)-2-[[(2R,3R)-1-met¡l-5-oxo-3-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (200 mg, 258 jmol, 55 % de rend¡m¡ento, 98 % de pureza) en forma de un sól¡do de color amar¡llo. LCMS [ESI, M+1]: 760.
1H RMN (400 MHz, cloroformo-d) 5 = 7,74 - 7,63 (m, 2H), 7,47 - 7,31 (m, 7H), 7,27 - 7,17 (m, 2H), 5,21 (s, 2H), 4,84 -4,43 (m, 5H), 4,30 - 3,70 (m, 8 H), 3,59 - 3,34 (m, 3H), 3,25 - 2,98 (m, 4H), 2,96 - 2,90 (m, 6 H), 2,81 - 2,56 (m, 4H), 1,83 - 1,63 (m, 2H), 1,60 - 1,47 (m, 4H).
Etapa B: 2-[(2S)-4-[7-(8-Met¡l-1-naft¡l)-2-[[(2R3R)-1-met¡l-5-oxo-3-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo. A una soluc¡ón de (2S)-2-(c¡anomet¡l)-4-[7-(8-met¡l-1-naft¡l)-2-[[(2R,3R)-1-met¡l-5-oxo-3-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (150 mg, 197 jmol, 1 eq.) en Me0H (3 ml) se le añad¡eron NH3/Me0H (3 ml, 20 % de pureza) y Pd/C (75 mg, 10 % de pureza) en una atmósfera de N2. La suspens¡ón se desgas¡f¡có al vacío y se purgó var¡as veces con H2. La mezcla se ag¡tó en una atmósfera de H2 (103,42 kPa (15 ps¡)) a 20 °C durante 1 hora. Después de que se completara, la mezcla se f¡ltró y el f¡ltrado se concentró al vacío para dar 2-[(2S)-4-[7-(8-met¡l-1-naft¡l)-2-[[(2R,3R)-1-met¡l-5-oxo-3-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (100 mg, 144 jmol, 73 % de rend¡m¡ento, 90 % de pureza) en forma de un sól¡do de color amar¡llo que se usó d¡rectamente en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón ad¡c¡onal. LCMS [ESI, M+1]: 626.
Etapa C: 2-[(2S)-4-[2-[[(2R3R)-3-H¡drox¡-1-met¡l-5-oxo-p¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una soluc¡ón de 2-[(2S)-4-[7-(8-met¡l-1-naft¡l)-2-[[(2R,3R)-1-met¡l-5-oxo-3-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (100 mg, 160 jmol, 1 eq.) en Dc M (100 j l) se le añad¡ó TFA (273 mg, 2,40 mmol, 177 jl, 15 eq.). La mezcla se ag¡tó a 20 °C durante 1 hora. Después de que se completara, la mezcla se d¡luyó con DCM (5 ml) y se bas¡f¡có con una soluc¡ón acuosa saturada de b¡carbonato de sod¡o a pH = 8. La capa acuosa separada se extrajo con EtOAc (3 * 5 ml).
Las capas orgán¡cas comb¡nadas se secaron sobre MgS04, se f¡ltraron y se concentraron al vacío para dar 2-[(2S)-4-[2-[[(2R,3R)-3-h¡drox¡-1-met¡l-5-oxo-p¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (80 mg, 126 jmol, 79 % de rend¡m¡ento, 85 % de pureza) en forma de un ace¡te de color amar¡llo que se usó d¡rectamente en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón ad¡c¡onal.
Etapa D: 2-[(2S)-1-(2-Fluoroprop-2-eno¡l)-4-[2-[[(2R3R)-3-h¡drox¡-1-met¡l-5-oxo-p¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo. A una soluc¡ón de 2-[(2S)-4-[2-[[(2R,3R)-3-h¡drox¡-1-met¡l-5-oxo-p¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (80 mg, 148 jmol, 1 eq.), TEA (44,8 mg, 443 jmol, 61,7 jl, 3 eq.) y ác¡do 2-fluoroprop-2-eno¡co (26,6 mg, 295 jmol, 2 eq.) en AE (3 ml) se le añad¡ó T3P (141 mg, 222 jmol, 132 ul, 50 % de pureza en EtOAc, 1,5 eq.) a 0 °C.
La mezcla se ag¡tó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se ¡nact¡vó con una soluc¡ón acuosa saturada de NaHC03 (5 ml) y se extrajo con EtOAc (2 * 10 ml). Las capas orgán¡cas se secaron sobre Na2S04 y se concentraron al vacío. El res¡duo se pur¡f¡có por cromatografía (AL03, EtOAc/Me0H 1/0 a 10/1) segu¡do de HPLC prep. (columna: Xt¡mate C18 150 * 25 mm * 5 um; fase móv¡l: [agua (0,05 % de h¡dróx¡do de amon¡o v/v)-ACN]; % de B: 34 %-64 %, 10 m¡n). Las fracc¡ones deseadas se recog¡eron y se l¡of¡l¡zaron para dar el compuesto del título 2-[(2S)-1-(2-fluoroprop-2-eno¡l)-4-[2-[[(2R,3R)-3-h¡drox¡-1-met¡l-5-oxo-p¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (11,8 mg, 19,1 jmol, 13 % de rend¡m¡ento, 99,3 % de pureza) en forma de un sól¡do de color blanquec¡no. LCMS [ESI, M+1]: 614.
1H RMN (400 MHz, cloroformo-d) 5 = 7,71 (d, J = 8,0 Hz, 1H), 7,69 - 7,63 (m, 1H), 7,41 (c, J = 7,6 Hz, 1H), 7,35 (t, J = 7,6 Hz, 1H), 7,27 - 7,19 (m, 2H), 5,55 - 5,33 (m, 1H), 5,27 (dd, J = 3,6, 16,8 Hz, 1H), 4,86 (dd, J = 5,6, 12,0 Hz, 1H), 4,79 (dd, J = 5,6, 12,0 Hz, 1H), 4,63 - 4,49 (m, 2H), 4,39 - 3,94 (m, 3H), 3,89 - 3,77 (m, 2H), 3,76 - 3,63 (m, 1H), 3,61 - 3 ,44 (m, 2H), 3,38 - 2,99 (m, 4H), 2,91 (d, J = 7,2 Hz, 6 H), 2,89 - 2,70 (m, 2H), 2,70 - 2,55 (m, 2H), 2,46 (dt, J = 5,2, 16,0 Hz, 1H).
Ejemplo 571

2-((S)-4-(7-(8-Cloronaftalen-1-il)-2-((S)-pirrolidin-2-ilmetoxi)-5,6,7,8-tetrahidropirido[3,4-C]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2 -il)acetonitrilo

Etapa A: (2S)-2-[(7-Bencil-4-metoxi-6,8-dihidro-5H-pirido[3,4-C]pirimidin-2-il) oximetillpirrolidin-1 -carboxilato de tere-butilo. A una solución de 7-bencil-2-cloro-4-metoxi-6,8-dihidro -5H-pirido[3,4-d]pirimidina (4,00 g, 13,8 mmol, 1,00 eq.) y (2S)-2-(hidroximetil)pirrolidin-1-carboxilato de /ere-butilo (5,56 g, 27,6 mmol, 2,00 eq.) en tolueno (80,0 ml) se le añadieron BINAP (1,72 g, 2,76 mmol, 0 ,20 eq.), Pd(0Ac)2 (310 mg, 1,38 mmol, 0 ,10 eq.) y Cs2C03 (13,5 g, 41 mmol, 3,00 eq.). La mezcla se agitó a 110 °C durante 8 horas. Después de que se completara, la mezcla de reacción se filtró y se concentró a presión reducida para dar un residuo. El residuo se diluyó con H20 (200 ml) y acetato de etilo (200 ml). La mezcla se acidificó a pH ~3 con HCl acuoso (2 N). La fase acuosa se separó y se extrajo con acetato de etilo (3 * 200 ml). Las capas orgánicas combinadas se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 20/1 a 1/1) para dar (2S)-2-[(7-bencil-4-metoxi-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-2-il) oximetil]pirrolidin-1-carboxilato de /ere-butilo (3,80 g, 8,26 mmol, 60 % de rendimiento, 99 % de pureza) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 455.
1H RMN (400 MHz, cloroformo-d) 57,37 - 7,27 (m, 5H), 4,50 - 4,35 (m, 1H), 4,25 - 4,16 (m, 1H), 3,98 (s, 3H), 3,69 (s, 2H), 3,50 (s, 2H), 3,43 - 3,33 (m, 2H), 2,76 - 2,72 (m, 2H), 2,62 - 2,57 (m, 2H), 2,04 - 1,90 (m, 3H), 1,85 - 1,76 (m, 1H), 1,61 - 1,54 (m, 1H), 1,45 (s, 9H).
Etapa B: (2S)-2-[(4-Metoxi-5,6,7,8-tetrahidropirido[3,4-cf1 pirimidin-2-il)oximetil1pirrolidin-1 -carboxilato de /ere-butilo. A una solución de (2S)-2-[(7-bencil-4-metoxi-6,8-dihidro-5H-pirido[3,4-C]pirimidin-2-il)oximetil]pirrolidin-1-carboxilato de /ere-butilo (3,80 g, 8,36 mmol, 1,00 eq.) en metanol (100 ml) se le añadió Pd(0H)2/C (1,00 g, 10,3 mmol, 20 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó tres veces con H2. La mezcla se agitó en una atmósfera de H2 (310,26 kPa (45 psi)) a 40 °C durante 24 horas. Después de que se completara, la mezcla de reacción se concentró a presión reducida para dar un residuo. El compuesto (2S)-2-[(4-metoxi-5,6,7,8-tetrahidropirido[3,4-C] pirimidin-2-il)oximetil]pirrolidin-1 -carboxilato de /ere-butilo (2,70 g, 7,17 mmol, 86 % de rendimiento, 97 % de pureza) se obtuvo en forma de un aceite de color amarillo y se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 365.
1H RMN (400 MHz, cloroformo-d) 54,54 - 4,35 (m, 1H), 4,25 - 4,10 (m, 2H), 3,98 (s, 3H), 3,86 (s, 2H), 3,48 (s, 1H), 3,43 - 3,33 (m, 2H), 3,13 - 3,04 (m, 2H), 2,56 - 2,46 (m, 2H), 2,10 - 1,93 (m, 3H), 1,87 - 1,82 (m, 1H), 1,45 (s, 9H).
Etapa C: (2S)-2-[7-(8-Cloro-1-naft¡l)-4-metox¡-6,8-d¡h¡dro -5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-2-¡l1ox¡met¡l1pirrol¡d¡n-1-carbox¡lato de terc-but¡lo. A una soluc¡ón de (2S)-2-[(4-metox¡-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡d¡n-2-¡l)ox¡met¡l]p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo (2,5 g, 6,86 mmol, 1,0 eq.) y 1-bromo-8-cloro-naftaleno (3,31 g, 13,7 mmol, 2,0 eq.) en tolueno (50 ml) se le añad¡eron Pd2(dba)3 (1,26 g, 1,37 mmol, 0,2 eq.), Cs2CÜ3 (5,59 g, 17,2 mmol, 2,5 eq.) y RuPhos (1,28 g, 2,74 mmol, 0,4 eq.) y la mezcla de reacc¡ón se ag¡tó a 90 °C durante 12 horas en una atmósfera de N2. Después de que se completara, la mezcla de reacc¡ón se f¡ltró a través de cel¡te, el f¡ltrado se lavó con salmuera saturada (1 * 80 ml) y la capa orgán¡ca se secó sobre Na2SÜ4, se f¡ltró y se concentró. El res¡duo se pur¡f¡có por cromatografía en columna (S¡02, éter de petróleo/acetato de et¡lo = 10/1 a 3/1). El producto (2S)-2-[[7-(8-cloro-1-naft¡l)-4-metox¡-6,8-d¡h¡dro -5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-2-¡l]ox¡met¡l]p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo (1,3 g, 2,46 mmol, 36 % de rend¡m¡ento, 99 % de pureza) en forma de un sól¡do de color amar¡llo claro. LCMS [ESI, M+1]: 525.
1H RMN (400 MHz, cloroformo-d) 57,74 (d, J = 8,4 Hz, 1H), 7,60 (d a, J = 8,0 Hz, 1H), 7,51 (d, J = 7,6 Hz, 1H), 7,43 (t, J = 7,8 Hz, 1H), 7,33 (t, J = 7,8 Hz, 1H), 7,23 (d a, J = 7,6 Hz, 1H), 4,60 - 4,37 (m, 1H), 4,34 - 4,16 (m, 3H), 4,03 (s, 3H), 3,87 (d a, J = 17,2 Hz, 1H), 3,63 - 3,57 (m, 1H), 3,45 - 3,30 (m, 2H), 3,21 - 3,10 (m, 1H), 2,99 (s a, 1H), 2,62 (d a, J = 16,8 Hz, 1H), 2,07 - 1,79 (m, 4H), 1,45 (s, 9H).
Etapa D: (2S)-2-[[7-(8-Cloro-1-naft¡l)-4-h¡drox¡-6,8 -d¡h¡dro-5H-p¡rido[3,4-d1p¡rim¡d¡n-2-¡l1ox¡met¡l1p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo. A una soluc¡ón de NaH (168 mg, 4,19 mmol, 60 % de pureza, 2,0 eq.) en DMF (15 ml) se le añad¡ó poco a poco EtSH (391 mg, 6,29 mmol, 465 μl, 3,0 eq.) a 15 °C Después de ag¡tar durante 0,5 horas, a la mezcla se le añad¡ó una soluc¡ón de (2S)-2-[[7-(8-cloro-1-naft¡l)-4-metox¡-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-2-¡l]ox¡met¡l]p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo (1,1 g, 2,10 mmol, 1,0 eq.) en DMF (7,0 ml) y después la mezcla de reacc¡ón se ag¡tó a 60 °C durante 1 hora. Después de que se completara, la mezcla de reacc¡ón se vert¡ó en agua enfriada con h¡elo (10 ml), después la mezcla se ajustó con HCl acuoso 1 N a pH 7-8, la mezcla se filtró y la torta de filtro se d¡solv¡ó con d¡clorometano (10 ml), se lavó con salmuera saturada (2 * 15 ml), se secó sobre Na2S04, se filtró y se concentró. El res¡duo se purificó por cromatografía en columna (S¡02, éter de petróleo/acetato de et¡lo = 3/1 a acetato de et¡lo:metanol = 10:1) para dar (2S)-2-[[7-(8-cloro-1-naft¡l)-4-h¡drox¡-6,8 -d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-2-¡l]ox¡met¡l]p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo (670 mg, 1,28 mmol, 61 % de rend¡m¡ento, 98 % de pureza) en forma de un sól¡do de color amarillo. LCMS [ESI, M+1]: 511.
Etapa E: (2S)-2-[[7-(8-Cloro-1-naft¡l)-4-(trifluoromet¡lsulfon¡lox¡)-6,8-d¡h¡dro-5H-p¡rido[3,4-d1p¡rim¡d¡n-2-¡l)ox¡met¡llp¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo. A una soluc¡ón de (2S)-2-[[7-(8-cloro-1-naft¡l)-4-h¡drox¡-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-2-¡l]ox¡met¡l]p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo (640 mg, 1,25 mmol, 1,0 eq.) en DCM (10 ml) se le añad¡eron TEA (380 mg, 3,76 mmol, 523 μl, 3,0 eq.), tam¡ces moleculares de 4 A (500 mg) y Tf20 (530 mg, 1,88 mmol, 310 μl, 1,5 eq.) a -40 °C y la mezcla de reacc¡ón se ag¡tó a -40 °C durante 10 m¡n. Después de que se completara, se añad¡ó poco a poco agua (10 ml) a -40 °C, después la mezcla de reacc¡ón se calentó a 15 °C, la capa orgán¡ca se separó y se lavó con salmuera saturada (1 * 10 ml) y la capa orgán¡ca se secó sobre Na2S04, se filtró y se concentró. El res¡duo se purificó por cromatografía en columna (S¡02, éter de petróleo/acetato de et¡lo = 10/1 a 3/1). El producto (2S)-2-[[7-(8-cloro-1 -naft¡l)-4-(tr¡fluoromet¡lsulfon¡lox¡)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-2-¡l]ox¡met¡l]p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo (400 mg, 616 pmol, 49 % de rend¡m¡ento, 99 % de pureza) se obtuvo en forma de un sól¡do de color amarillo claro. LCMS [ESI, M+1]: 643.
Etapa F: (2S)-2-[[7-(8-Cloro-1-naft¡l)-4-[(3S)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l1-6,8-d¡h¡dro-5H-p¡rido[3,4-d1p¡rim¡d¡n-2-¡l)ox¡met¡llp¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo. A una soluc¡ón de (2S)-2-[[7-(8-cloro-1-naft¡l)-4-(tr¡fluoromet¡lsulfon¡lox¡)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-2-M]ox¡met¡l]p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo (400 mg, 622 μmol, 1,0 eq.) en DMAC (4,0 ml) se le añad¡eron 2-[(2S)-p¡peraz¡n-2-¡l]aceton¡tr¡lo (86,5 mg, 622 μmol, 1,0 eq.) y DIEA (161 mg, 1,24 mmol, 217 μl, 2,0 eq.) y la mezcla de reacc¡ón se ag¡tó a 15 °C durante 10 m¡n. Después de que se completara, la mezcla de reacc¡ón se vert¡ó en agua enfriada con h¡elo (10 ml), el sól¡do prec¡p¡tado se filtró y se d¡solv¡ó con acetato de et¡lo (15 ml) y la capa orgán¡ca se lavó con salmuera saturada (2 * 15 ml), se secó sobre Na2S04, se filtró y se concentró. El res¡duo se purificó por cromatografía ultrarráp¡da de fase ¡nversa (cond¡c¡ón de 0,1 % de AF, 20 %-30 % de MeCN en agua), el producto obten¡do se ajustó con NaHC03 sól¡do a pH -8 y después se concentró, la capa acuosa se extrajo con acetato de et¡lo (2 * 15 ml) y la capa orgán¡ca se lavó con salmuera saturada (1 * 20 ml), se secó sobre Na2S04, se filtró y se concentró. El producto (2S)-2-[[7-(8-cloro-1-naft¡l)-4-[(3S)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-2-¡l]ox¡met¡l]p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo (300 mg, 480 pmol, 77 % de rend¡m¡ento, 99 % de pureza) se obtuvo en forma de un sól¡do de color amarillo claro. LCMS [ESI, M+1]: 618.
1H RMN (400 MHz, cloroformo-d) 57,75 (d, J = 8,0 Hz, 1H), 7,61 (d a, J = 8,0 Hz, 1H), 7,52 (d, J = 6,8 Hz, 1H), 7,47 -7,42 (m, 1H), 7,33 (t, J = 7,8 Hz, 1H), 7,23 (t a, J = 6,2 Hz, 1H), 4,71 - 4,31 (m, 2H), 4,29 - 4,06 (m, 3H), 3,95 - 3,69 (m, 2H), 3,63 - 3,28 (m, 4H), 3,26 - 2,82 (m, 7H), 2,72 - 2,46 (m, 3H), 2,02 - 1,79 (m, 4H), 1,45 (s, 9H).
Etapa G: (2S)-2-[[7-(8-Cloro-1-naft¡l)-4-[(3S)-3-(c¡anomet¡l)-4-(2-fluoroprop-2-eno¡l)p¡peraz¡n-1-¡l]-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-2-¡l)ox¡met¡llp¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo. A una soluc¡ón de (2S)-2-[[7-(8-cloro-1-naft¡l)-4-[(3S)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l]-6,8-d¡h¡dro-5H-p¡rido[3,4-d]p¡rim¡d¡n-2-¡l]ox¡met¡l]p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo (100 mg, 162 μmol, 1,0 eq.) y ác¡do 2-fluoroprop-2-eno¡co (29,1 mg, 324 μmol, 2,0 eq.) en acetato de et¡lo (1,5 ml) se le añad¡eron poco a poco TEA (131 mg, 1,29 mmol, 180 μl, 8,0 eq.) y T3p (309 mg, 485 pmol, 289 μl, 50 % de pureza, 3,0 eq.) a 0 °C y la mezcla de reacc¡ón se ag¡tó a 0 °C durante 1 hora. Después de que se completara, se añad¡ó agua
(10 ml), después la capa orgánica se separó, la fase acuosa se extrajo con acetato de etilo (2 x 10 ml) y la capa orgánica combinada se secó sobre Na2SÜ4, se filtró y se concentró. El residuo se purificó cromatografía en columna (Al2Ü3 básico, éter de petróleo/acetato de etilo = 3/1 a 1/1). El producto (2S)-2-[[7-(8-cloro-1-naftil)-4-[(3S)-3-(cianometil)-4-(2-fluoroprop-2-enoil)piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]pirrolidin-1-carboxilato de tere-butilo (110 mg, bruto) se obtuvo en forma de un sólido de color amarillo claro. LCMS [ESI, M+1]: 690.
Etapa H: 2-((S)-4-(7-(8-Cloronaftalen-1-il)-2-((S)-pirrolidin-2-ilmetoxi)-5,6,7,8-tetrahidropirido[3,4-dlpirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo. A una solución de (2S)-2-[[7-(8-cloro-1-naftil)-4-[(3S)-3-(cianometil)-4-(2-fluoroprop-2-enoil)piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-2-il]oximetil]pirrolidin-1-carboxilato de /ere-butilo (90,0 mg, 130,40 μmol, 1,0 eq.) en diclorometano (0,5 ml) se le añadió TFA (770 mg, 6,75 mmol, 0,5 ml, 51,8 eq.) y la mezcla de reacción se agitó a 20 °C durante 1 hora. Después de que se completara, la mezcla de reacción se concentró, después se disolvió con acetato de etilo (10 ml) y se lavó con Na2CÜ3 acuoso saturado (1 x 10 ml) y la capa orgánica se secó sobre Na2SÜ4, se filtró y se concentró. El residuo se purificó por cromatografía en columna (SiÜ2, éter de petróleo/acetato de etilo = 3/1 a acetato de etilo:metanol = 10:1), después se purificó por HPLC prep. (columna: Phenomenex Synergi C18150 * 25 * 10 μm; fase móvil: [agua (0,225 % de AF)-ACN]; % de B: 20 %-50 %, 10 min) y el producto obtenido se concentró y se liofilizó. El compuesto del título 2-((S)-4-(7-(8-cloronaftalen-1-il)-2-((S)-pirrolidin-2-ilmetoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo (24,7 mg, 38,1 pmol, 29 % de rendimiento, 98 % de pureza, AF) se obtuvo en forma de un sólido de color blanquecino. LCMS [ESI, M+1]: 590.
1H RMN (400 MHz, cloroformo-d) 57,76 (d, J = 8,0 Hz, 1H), 7,62 (t, J = 7,0 Hz, 1H), 7,56 - 7,49 (m, 1H), 7,54 - 7,50 (m, 1H), 7,34 (t, J = 7,8 Hz, 1H), 7,26 - 7,17 (m, 1H), 5,53-5,32 (m, 1H), 5,25 (dd, J = 3,6, 16,8 Hz, 1H), 4,89 - 4,52 (m, 1H), 4,64 - 4,45 (m, 2H), 4,42-4,23 (m, 2H), 4,17 - 3,92 (m, 4H), 3,89 - 3,78 (m, 2H), 3,51 - 3,42 (m, 1H), 3,35 - 3,05 (m, 5H), 2,95 - 2,73 (m, 2H), 2,62 - 2,51 (m, 1H), 2,21 - 1,84 (m, 4H).
Ejemplo 572

2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2S)-1 -metil-5-oxo-pirrolidin-2-il]metoxi]-6,8-dihidro-5^-pirido[3,4-tí]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo

Etapa A: (2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2S)-1-metil-5-oxo-pirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de terc-butilo. A una mezcla de (5S)-5-(hidroximetil)-1-metil-pirrolidin-2-ona (556 mg, 4,30 mmol, 5,00 eq.) en THF (20,0 ml) se le añadió en una porción f-Bu0Na (827 mg, 8,60 mmol, 10,0 eq.) a 0 °C en una atmósfera de N2. La mezcla se agitó a 0 °C durante 30 min, seguido de la adición de (2S)-4-[7-(8-cloro-1 -naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1 -carboxilato de ferc-butilo (500 mg, 860 gmol, 1,00 eq.). La mezcla se calentó a 15 °C y se agitó durante 0,5 horas. La mezcla de reacción se diluyó con acetato de etilo (50,0 ml), se lavó con agua (30 ml x 2), se secó sobre sulfato de sodio, se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de TFA)/acetonitrilo]. Las fracciones deseadas se recogieron, se neutralizaron con una solución saturada de NaHC03 (10,0 ml) y se extrajeron con acetato de etilo (50,0 ml x3 ). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El compuesto (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metil-5-oxopirrolidin-2-il]metoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de terc-butilo (125 mg, 193 gmol, 22 % de rendimiento) se obtuvo en forma de un sólido de color amarillo.
1H RMN (400 MHz, cloroformo-d) ó = 7,75 (d, J = 8,0 Hz, 1H), 7,61 (dd, J = 4,8, 8,0 Hz, 1H), 7,52 (dd, J = 0,8, 7,2 Hz, 1H), 7,45 (c, J = 8,0 Hz, 1H), 7,37 - 7,28 (m, 1H), 7,23 (d a, J = 7,6 Hz, 1H), 4,70 - 4,31 (m, 3H), 4,10 - 3,82 (m, 4H), 3,65 - 3,51 (m, 1H), 3,46 - 2,99 (m, 5H), 2,98 - 2,82 (m, 3H), 2,79 - 2,37 (m, 5H), 2,22 (d a, J = 8,0 Hz, 1H), 1,93 - 1,59 (m, 3H), 1,52 (s, 9H).
Etapa B: 2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2S)-1-metil-5-oxo-pirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-cf1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una mezcla de (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metil-5-oxo-pirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (100 mg, 155 gmol, 1,00 eq.) en dioxano (1,50 ml) se le añadió HCl/dioxano (4 M, 967 gl, 25,0 eq.). La mezcla se agitó a 20 °C durante 1 h. La mezcla de reacción se concentró a presión reducida para dar un residuo. El residuo se disolvió con acetato de etilo (10,0 ml), se basificó a pH = 8 con una solución saturada de NaHC03 (3,00 ml) y se extrajo con acetato de etilo (5,00 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (10,0 ml x 1), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El producto bruto se usó en la siguiente etapa directamente sin purificación adicional. El compuesto 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metil-5-oxo-pirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-cf1pirimidin-4-il1piperazin-2-il1acetonitrilo (80,0 mg, bruto) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+11: 546.
Etapa C: 2-((2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2S)-1-metil-5-oxo-pirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-ó1pirimidin-4-il1-1-(2-fluoroprop-2-enoil)piperazin-2-il1acetonitrilo. A una mezcla de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metil-5-oxo-pirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-ó1pirimidin-4-il1piperazin-2-il1acetonitrilo (60,0 mg, 110 gmol, 1,00 eq.) y ácido 2-fluoroprop-2-enoico (19,8 mg, 220 gmol, 2,00 eq.) en AE (1 ml) se le añadieron en una porción TEA (89,0 mg, 879 gmol, 122 gl, 8,00 eq.), T3P (210 mg, 330 gmol, 196 gl, 50 % de pureza en acetato de etilo, 3,00 eq.) a 0 °C en una atmósfera de N2. La mezcla se agitó a 0 °C durante 30 min. La mezcla de reacción se diluyó con agua (5 ml) y se extrajo con acetato de etilo (10 ml x 3 ). Las capas orgánicas combinadas se lavaron con agua (10 ml x 1) y salmuera (10 ml x 1), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Xtimate
C18 150 x 25 mm x 5 qm; fase móvil: [agua (0,05 % de hidróxido de amonio v/v)-ACN]; % de B: 35 %-65 %, 10 min). El compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metil-5-oxo-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (11,3 mg, 18,2 umol, 17 % de rendimiento, 99 % de pureza) se obtuvo en forma de un sólido de color blanco LCMS [ESI, M+1]: 618.
1H RMN (400 MHz, cloroformo-d) ó = 7,77 (d, J = 8,0 Hz, 1H), 7,63 (t, J = 7,6 Hz, 1H), 7,53 (d, J = 6,8 Hz, 1H), 7,46 (td, J = 8,0, 10,4 Hz, 1H), 7,35 (t, J = 7,6 Hz, 1H), 7,27-7,20 (m, 1H), 5,52 - 5,34 (m, 1H), 5,26 (dd, J = 3,6, 16,8 Hz, 1H), 4,92 (s a, 1H), 4,52 - 4,31 (m, 3H), 4,27 - 3,77 (m, 5H), 3,61 (d a, J = 10,8 Hz, 1H), 3,46 (d a, J = 12,0 Hz, 1H), 3,36-2,94 (m, 5H), 2,92 (s, 3H), 2,90 - 2,79 (m, 1H), 2,64 - 2,48 (m, 2H), 2,41 - 2,31 (m, 1H), 2,28-2,17 (m, 1H), 2,05 - 1,95 (m, 1H).
Ejemplo 573

-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2R,3R)-3-hidroxi-1-metil-5-oxo-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo

Etapa A: (2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2R3R)-1-metil-5-oxo-3-tetrahidropiran-2-iloxi-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf1pirimidin-4-¡ n -2-(cianometil)piperazin-1 -carboxilato de tere-butilo. A una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1 -carboxilato de tere-butilo (280 mg, 482 qmol, 1 eq.) y (4R,5R)-5-(hidroximetil)-1-metil-4-tetrahidropiran-2-iloxi-pirrolidin-2-ona (144 mg, 626 qmol, 1,3 eq.) en tolueno (6 ml) se le añadió í-Bu0Na (92,6 mg, 964 qmol, 2 eq.). La mezcla se agitó a 0 °C durante 10 minutos. Después de que se completara, la mezcla se diluyó con agua (5 ml) y se extrajo con EtOAc (2 x 10 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. Las fracciones deseadas se recogieron, se neutralizaron con NaHC03, se concentraron al vacío para eliminar el MeCN y se extrajeron con EtOAc (2 x 20 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío para dar (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R,3R)-1-metil-5-oxo-3-tetrahidropiran-2-iloxi-pirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-<C]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de tere-butilo (280 mg, 345 qmol, 72 % de rendimiento, 92 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 746.
1H RMN (400 MHz, cloroformo-d) 5 = 7,76 (d a, J = 8,0 Hz, 1H), 7,66 - 7,57 (m, 1H), 7,53 (d, J = 7,2 Hz, 1H), 7,50 -7,41 (m, 1H), 7,37 - 7,31 (m, 1H), 7,27 - 7,18 (m, 1H), 4,88 - 4,69 (m, 1H), 4,68 - 4,47 (m, 4H), 4,46 - 4,35 (m, 1H), 4,11 - 3,74 (m, 6H), 3,65 - 3,48 (m, 2H), 3,44 - 2,99 (m, 5H), 2,96 (dd, J = 2,8, 8,4 Hz, 3H), 2,91 - 2,51 (m, 5H), 1,81 -1.64 (m, 2H), 1,52 (s, 13H).
Etapa B: 2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2R,3R)-3-hidroxi-1-metil-5-oxo-pirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R,3R)-1-metil-5-oxo-3-tetrah¡drop¡ran-2-¡lox¡-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-dih¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de tere-butilo (200 mg, 268 gmol, 1 eq.) en DCM (0,15 ml) se le añadió TFA (458 mg, 4,02 mmol, 298 gl, 15 eq.). La mezcla se agitó a 20 °C durante 1 hora. Después de que se completara, la mezcla se diluyó con DCM (5 ml) y se basificó con una solución acuosa saturada de bicarbonato de sodio a pH = 8. La capa acuosa separada se extrajo con EtOAc (5 x 5 ml). Las capas orgánicas combinadas se secaron sobre MgS04, se filtraron y se concentraron al vacío para dar 2-[(2S)-4-[7-(8-cloro-1-naft¡l)-2-[[(2R,3R)-3-h¡drox¡-1-met¡l-5-oxo-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡hidro-5H-p¡r¡do[3,4-c(]p¡r¡m¡d¡n-4-¡l]piperaz¡n-2-¡l]aceton¡tr¡lo (150 mg, 230 gmol, 86 % de rendimiento, 86 % de pureza) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 562.
Etapa C: 2-f(2S)-4-f7-(8-Cloro-1-naftil)-2-ff(2R3R)-3-hidroxi-1-metil-5-oxo-pirrolidin-2-i n metox n -6,8-dihidro-5H-pirido[3,4-d']pirimidin-4-i n -1-(2-fluoroprop-2-enoil)piperazin-2-i n acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R,3R)-3- h¡drox¡-1-met¡l-5-oxo-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡rido[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-il]acetonitrilo (150 mg, 267 umol, 1 eq.), TEA (81,0 mg, 801 umol, 111 ul, 3 eq.) y ácido 2-fluoroprop-2-enoico (48,1 mg, 534 umol, 2 eq.) en EtOAc (4 ml) se le añadió T3P (255 mg, 400 umol, 238 ul, 50 % de pureza en EtOAc, 1,5 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se inactivó con una solución acuosa saturada de NaHC03 (5 ml) y se extrajo con EtOAc (2 x 10 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por cromatografía (Ah03, EtOAc/Me0H 1/0 a 20/1) seguido de HPLC prep. (columna: Xtimate C18 150 * 25 mm * 5 gm; fase móvil: [agua (0,05 % de hidróxido de amonio v/v)-ACN]; % de B: 33 %-63 %, 10 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naft¡l)-2-[[(2R,3R)-3-h¡droxi-1-met¡l-5-oxo-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-SH pirido[3,4-d]pirimidin-4-il]-1 -(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (22,0 mg, 34,5 umol, 13 % de rendimiento, 99,6 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 634.
1H RMN (400 MHz, cloroformo-d) 5 = 7,77 (d, J = 8,4 Hz, 1H), 7,64 (dd, J = 4,4, 8,0 Hz, 1H), 7,54 (d, J = 7,6 Hz, 1H), 7,46 (dt, J = 6,0, 8,0 Hz, 1H), 7,35 (t, J = 7,6 Hz, 1H), 7,27 - 7,21 (m, 1H), 5,54 - 5,33 (m, 1H), 5,27 (dd, J = 2,8, 17,2 Hz, 1H), 4,88 (dd, J = 1,6, 12,0 Hz, 1H), 4,80 (dd, J = 6,0, 12,0 Hz, 1H), 4,64 - 4,48 (m, 2H), 4,47 - 3,94 (m, 4H), 3,91 -3.65 (m, 3H), 3,64 - 3,43 (m, 2H), 3,37 - 2,98 (m, 4H), 2,91 (d, J = 7,6 Hz, 3H), 2,89 - 2,76 (m, 1H), 2,70 - 2,53 (m, 2H), 2,46 (dt, J = 5,2, 16,4 Hz, 1H).
Ejemplo 574

2-[(2S)-4-[7-(8-Cloro-1-naft¡l)-2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-il]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo

Etapa A: (2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2S.4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-6.8-dihidro-5H-pirido[3.4-dlpirimidin-4-il1-2-(cianometil)piperazin-1 -carboxilato de tere-butilo. A una mezcla de [(2S.4R)-4-fluoro-1-metil-pirrolidin-2-il]metanol (275 mg. 2.06 mmol. 2.0 eq.) en tolueno (12.0 ml) se le añadió poco a poco t-Bu0Na (198 mg. 2.06 mmol.
2.0 eq.) a 0 °C y. después de agitar a 0 °C durante 0.5 horas. se añadió (2S)-4-[7-(8-cloro-1-naftil)-2-metilsulfinil-6.8-dihidro-5H-pirido[3.4-d]pirimidin-4-il1-2-(cianometil)piperazin-1 -carboxilato de tere-butilo (600 mg. 1.03 mmol. 1.0 eq.) y se agitó a 0 °C durante 0.5 horas. Después de que se completara. se añadió agua (15.0 ml) y la mezcla resultante se extrajo con acetato de etilo (2 x 10 ml) y la capa orgánica combinada se secó sobre Na2S04. se filtró y se concentró. El residuo se purificó por cromatografía ultrarrápida de fase inversa (C18. 0.1 % de TFA en agua. 30 %-50 % de MeCN). el producto obtenido se ajustó con NaHC03 sólido a pH = 8 y después se concentró. la fase acuosa se extrajo con acetato de etilo (2 x 15 ml) y la capa orgánica se lavó con salmuera saturada (20.0 ml). después se secó sobre Na2S04. se filtró y se concentró. El producto (2S)-4-[7-(8-cloro-1 -naftil)-2-[[(2S.4R)-4-fluoro-1 -metil-pirrolidin-2-il]metoxi]-6.8-dihidro-5H-pirido[3.4-djpirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (510 mg. 769 μmol. 75 % de rendimiento.
98 % de pureza) se obtuvo en forma de un sólido de color amarillo claro. LCMS [ESI. M+1]: 650.
1H RMN (400 MHz. cloroformo-d) ó 7,80 - 7.72 (m. 1H). 7.62 (t. J = 7.8 Hz. 1H). 7.56 - 7.50 (m. 1H). 7.46 - 7.40 (m.
1H). 7.37 - 7.31 (m. 1H). 7.26 - 7.17 (m. 1H). 5.29 - 5.07 (m. 1H). 4.65 - 4.57 (m. 1H). 4.50 - 4.34 (m. 2H). 4.30 - 4.17 (m. 1H). 4.06 - 3.76 (m. 3H). 3.65 - 3.47 (m. 2H). 3.41 - 2.86 (m. 7H). 2.84 - 2.45 (m. 7H). 2.40 - 2.24 (m. 1H). 2.04 -1.86 (m. 1H). 1.52 (s. 9H).
Etapa B: 2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2S.4R)-4-fluoro-1-metilpirrolidin-2-il]metoxi]-6.8-dihidro-5H-pirido[3.4-ó1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S.4R)-4-fluoro-1-metilp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de tere-butilo (500 mg. 769 μmol. 1.0 eq.) en dioxano (5.0 ml) se le añadió HCl 4 N*dioxano (5,0 ml) y la mezcla de reacción se agitó a 15 °C durante 1 hora. Después de que se completara. la mezcla de reacción se filtró. el sólido se disolvió con diclorometano (15.0 ml) y la capa orgánica se lavó con salmuera saturada (1 x 15 ml). se secó sobre Na2S04. se filtró y se concentró para dar 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S.4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-6.8-dihidro-5H-pirido[3.4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (360 mg. 635 μmol. 85 % de rendimiento. 97 % de pureza) en forma de un sólido de color amarillo claro. lCm S [ESI. M+1]: 550.
Etapa C: 2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2S.4R)-4-fluoro-1-metilpirrolidin-2-il]metoxi]-6.8-dihidro-5H-pirido[3.4-ó1pirimidin-4-il1-1-(2-fluoroprop-2-enoil)piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (120 mg. 218 μmol. 1.0 eq.) y ácido 2-fluoroprop-2-enoico (39.3 mg. 436 μmol. 2.0 eq.) en acetato de etilo (1.2 ml) se le añadieron TEA (177 mg. 1.75 mmol. 243 μl. 8.0 eq.) y T3P (416 mg. 654 μmol. 389 μl. 50 % de pureza. 3.0 eq.) a 0 °C y la mezcla de reacción se agitó a 0 °C durante 1 hora. Después de que se completara. a la mezcla de reacción se le añadió salmuera saturada (10.0 ml). después se extrajo con acetato de etilo (2 x 10 ml) y la capa orgánica se secó sobre Na2S04. se filtró y se concentró. El residuo se purificó por cromatografía en columna (Ah03 básico. éter de petróleo/acetato de etilo = 3/1 a acetato de etilo/metanol = 20/1). el producto bruto se purificó de nuevo por HPLC prep. (columna: Waters Xbridge 150 * 255 p; fase móvil: [agua (0.05 % de hidróxido de amonio v/v)-ACN]; % de B: 45 %-75 %. 10 min]; % de B: 45 %-75 %. 10 min) y el producto obtenido se concentró y se liofilizó. El compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S.4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-6.8-dihidro-5H-pirido[3.4-ó]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (50.9 mg. 81.0 μmol. 37 % de rendimiento. 99 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI. M+1]: 622.
1H RMN (400 MHz. cloroformo-d) ó 7,76 (d, J = 8.0 Hz. 1H). 7.62 (t. J = 7.4 Hz. 1H). 7.53 (d. J = 7.2 Hz. 1H). 7.49 -7.41 (m. 1H). 7.34 (t. J = 7.8 Hz. 1H). 7.26 - 7.18 (m. 1H). 5.55 - 5.33 (m. 1H). 5.30 - 5.07 (m. 2H). 5.01 - 4.66 (m. 1H).
4.53 - 4.35 (m. 2H). 4.29 - 3.98 (m. 3H). 3.96 - 3.78 (m. 2H). 3.73 - 3.33 (m. 3H). 3.32 - 2.98 (m. 5H). 2.94 - 2.71 (m.
2H). 2.68 - 2.53 (m. 2H). 2.51 (d. J = 2.8 Hz. 3H). 2.39 - 2.23 (m. 1H). 2.08 - 1.87 (m. 1H).
E je m p lo 575

2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6.8-dihidro-5H-pirido[3.4-cf]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo

Etapa A: (2S)-4-[7-(8-Cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-6.8-dihidro-5H-pirido[3.4-d1pirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de tere-butilo. A una solución de [(3R)-1-metilpirrolidin-3-il]metanol (297 mg.
2.58 mmol. 2.50 eq.) en tolueno (6.0 ml) se le añadió í-Bu0Na (198 mg. 2.06 mmol.2.0 eq.). La mezcla se agitó a 0 °C durante 0.5 horas. Después. al líquido anterior se le añadió (2S)-4-[7-(8-cloro-1-naftil)-2-metilsulfinil-6.8-dihidro-5H-pirido[3.4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (600 mg. 1.03 mmol. 1.0 eq.). Después de la adición. la mezcla se agitó a 0 °C durante 0.5 horas más. Después de que se completara. se añadió agua (20.0 ml) y la mezcla resultante se extrajo con acetato de etilo (3 * 10.0 ml). La capa orgánica se secó sobre Na2S04. se filtró y se concentró. El residuo se purificó por cromatografía ultrarrápida de fase inversa (C18. 0.1 % de AF en agua. 0-60 % de MeCN) para dar el compuesto (2S)-4-[7-(8-cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6.8-dihidro-5H-pirido[3.4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (430 mg. 680 μmol. 66 % de rendimiento) en forma de un sólido de color amarillo. LCMS [ESI. M+1]: 632.
1H RMN (400 MHz. Cloroformo-d) ó 7,75 (d, J = 8.0 Hz. 1H). 7.61 (t. J = 7.6 Hz. 1H). 7.52 (d. J = 7.2 Hz. 1H). 7.48 -7.39 (m. 1H). 7.33 (t. J = 7.6 Hz. 1H). 7.26 - 7.15 (m. 1H). 4.67 - 4.55 (m. 1H). 4.50 - 4.35 (m. 1H). 4.27 - 4.17 (m. 2H).
4.08 - 3.75 (m. 4H). 3.65 - 3.50 (m. 1H). 3.39 - 3.02 (m. 5H). 3.00 - 2.86 (m. 1H). 2.81 - 2.64 (m. 4H). 2.62 - 2.46 (m.
4H). 2.35 (s. 3H). 2.15 - 2.05 (m. 1H). 1.52 (s. 9H).
Etapa B: 2-r(2S)-4-r7-(8-Cloro-1-naftil)-2-rr(3R)-1-metilpirrolidin-3-i n metox n -6.8-dihidro-5H-piridor3.4-dlpirimidin-4-¡ n piperazin-2-i n acetonitrilo. A una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6.8-dihidro-5H-pirido[3.4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (400 mg. 632 μmol. 1.0 eq.) en dioxano (2.0 ml) se le añadió HCFdioxano (4,0 M, 2,0 ml. 12.6 eq.). La mezcla se agitó a 15 °C durante 1 hora.
Después de que se completara. la mezcla de reacción se concentró. El residuo se ajustó con NaHC03 acuoso saturado (10.0 ml) a pH ~7 y después se extrajo con acetato de etilo (3 * 10.0 ml). La capa orgánica se secó sobre Na2S04. se filtró y se concentró para dar el compuesto 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6.8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (240 mg.451 μmol. 71 % de rendimiento) en forma de un sólido de color amarillo que se usó en la siguiente etapa sin purificación adicional. LCMS [ESI. M+1]: 532.
Etapa C: 2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-il1-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-¡l]metox¡]-6,8-dih¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (80,0 mg, 150 gmol, 1,0 eq.), ácido 2 fluoroprop-2-enoico (27,1 mg, 301 gmol, 2,0 eq.) y TEA (122 mg, 1,20 mmol, 167 gl, 8,0 eq.) en acetato de etilo (2,0 ml) se le añadió T3P (287 mg, 451 umol, 268 gl, 50 % de pureza en acetato de etilo, 3,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se inactivó con HCl (12,0 M, 60,0 ul en 2,0 ml de agua). La mezcla se ajustó a pH ~8 con NaHCÜ3 acuoso saturado y se extrajo con acetato de etilo (3 * 10,0 ml). Las capas orgánicas combinadas se lavaron con salmuera (30,0 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron.
El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi C18150 * 25 * 10 gm; fase móvil: [agua (0,225 % de AF)-ACN]; % de B: 25 %-45 %, 8 min) y después se concentró. La capa acuosa se ajustó con NaHCÜ3 acuoso saturado a pH ~8 y se extrajo con acetato de etilo (3 * 10,0 ml). La capa orgánica se secó sobre Na2SÜ4, se filtró y se concentró a presión reducida para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-¡l]metox¡]-6,8-dih¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]-1-(2-fluoroprop-2-eno¡l)p¡peraz¡n-2-¡l]aceton¡tr¡lo (35,9 mg, 57,5 gmol, 38 % de rendimiento, 96,8 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 604.
1H RMN (400 MHz, Cloroformo-d) ó 7,78 - 7,73 (d, J = 8,4 Hz, 1H), 7,62 (t, J = 7,2 Hz, 1H), 7,52 (d, J = 7,2 Hz, 1H), 7,49 -7,40 (m, 1H), 7,34 (t, J = 8,0 Hz, 1H), 7,27 - 7,18 (m, 1H), 5,54 - 5,32 (m, 1H), 5,30 - 5,21 (m, 1H), 5,04 - 4,75 (m, 1H), 4,48 - 4,36 (m, 1H), 4,27 - 4,17 (m, 2H), 4,16 - 3,96 (m, 2H), 3,89 - 3,75 (m, 1H), 3,64 - 3,55 (m, 1H), 3,51 - 3,32 (m, 1H), 3,30 -2,98 (m, 4H), 2,96 - 2,67 (m, 4H), 2,66 - 2,46 (m, 4H), 2,37 (d, J = 2,0 Hz, 3H), 2,14 - 2,06 (m, 1H), 1,70 - 1,57 (m, 1H).
Ejemplo 576

2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2R,4R)-4- metox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l]-1-(2-fluoroprop-2-eno¡l)p¡perazin-2-¡l]aceton¡tr¡lo

Etapa A: (2S)-4-[7-(8-Cloro-1-naftin-2-[[(2^.4^)-4-metoxi-1-metil-pirrolidin-2-il1metoxi1-6.8-dihidro-5H-pirido[3,4-dlpirimidin-4-il1-2-(cianometinpiperazin-1-carboxilato de ferc-butilo. A una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-met¡lsulf¡nil-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de ferc-butilo (300 mg, 516 gmol, 1 eq.) en tolueno (5,00 ml) se le añadieron t-Bu0Na (124 mg, 1,29 mmol, 2,5 eq.) y [(2R,4R)-4-metoxi-1-metil-pirrol¡d¡n-2-¡l]metanol (149 mg, 1,03 mmol, 2 eq.). La mezcla se agitó a 0 °C durante 1 hora. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (50 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, acetato de etilo/metanol = 100/1 a 10:1). Se obtuvo (2S)-4-[7-(8-cloro-1-naft¡l)-2-[[(2R,4R)-4-metox¡-1-met¡l-p¡rrol¡d¡n-2-il]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]-2-(cianometil)piperazin-1-carboxilato de ferc-butilo (200 mg, 265 gmol, 51 % de rendimiento, 88 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 662.
Etapa B: 2-f(2S)-4-f7-(8-Cloro-1-naftir)-2-ff(2R4ft)-4-metoxi-1-metilpirrolidin-2-i n metoxn-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R,4R)-4-metoxi-1-metilp¡rrol¡d¡n-2-il]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de ferc-butilo (200 mg, 302 gmol, 1 eq.) en dioxano (1,00 ml) se le añadió HCl/dioxano (4 M, 1,13 ml, 1 ,00 eq.). La mezcla se agitó a 20 °C durante 1 hora. La mezcla de reacción se concentró al vacío y se diluyó con agua (20 ml). La mezcla se ajustó a pH = 8 con una solución acuosa saturada de NaHC03 y se extrajo con acetato de etilo (30 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar el residuo. Se obtuvo 2-[(2S)-4-[7-(8-cloro-1-naft¡l)-2-[[(2R,4R)-4-metox¡-1-metil-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-pirido[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (110 mg, 154,60 gmol, 70 % de rendimiento, 79 % de pureza) en forma de un sólido de color amarillo y se usó en la siguiente etapa sin purificación. LCMS [ESI, M+1]: 562.
Etapa C: 2-[(2S)-4-[7-(8-Cloro-1-naftiD-2-[[(2R4R)-4- metoxi-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-cflp¡rimidin-4-i n -1-(2-fluoroprop-2-enoiDpiperazin-2-i n acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R,4R)-4-metox¡-1-met¡l-p¡rrol¡d¡n-2-il]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo
(110 mg, 195 gmol, 1 eq.) en acetato de etilo (1,00 ml) se le añadieron ácido 2-fluoroprop-2-enoico (35,3 mg, 391 gmol, 2 eq.), T3P (374 mg, 587 gmol, 349 gl, 50 % de pureza, 3 eq.) y TEA (158 mg, 1,57 mmol, 218 gl, 8 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (30 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, acetato de etilo/metanol = 100/1 a 10/1) y adicionalmente por HPLC prep. (columna: Waters Xbridge 150 * 255 g; fase móvil:
[agua (0,05 % de hidróxido de amonio v/v)-ACN]; % de B: 45 % - 75 %, 10 min). Las fracciones deseadas se recogieron y se liofilizaron. El compuesto del título 2-[(2S)-4-[7-(8-cloro-1 -naftil)-2-[[(2R,4^)-4-metoxi-1 -metil-pirrolidin-2-il]metoxi)-6,8-dih¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]-1-(2-fluoroprop-2-eno¡l)p¡peraz¡n-2-¡l]aceton¡tr¡lo (17 mg, 26,7 gmol, 14 % de rendimiento, 99,8 % de pureza) se obtuvo en forma de una goma de color blanco. LCMS [ESI, M+1]: 634.
1H RMN (400 MHz, cloroformo-d) 5 = 7,76 (d, J = 8,4 Hz, 1H), 7,62 (t, J = 7,6 Hz, 1H), 7,53 (d, J = 7,2 Hz, 1H), 7,49 -7,40 (m, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,27 - 7,18 (m, 1H), 5,42 (d, J = 49,2 Hz, 1H), 5,26 (dd, J = 3,6, 16,8 Hz, 1H), 4,91 (s a, 1H), 4,54 - 4,34 (m, 2H), 4,27 - 4,00 (m, 3H), 3,96 - 3,77 (m, 2H), 3,67 - 3,54 (m, 1H), 3,51 - 3,37 (m, 1H), 3,37 - 2,96 (m, 9H), 2,95 - 2,64 (m, 3H), 2,63 - 2,53 (m, 1H), 2,52 - 2,27 (m, 5H), 1,91 - 1,74 (m, 1H).
Ejemplo 577

2-[(2S)-4-[7-(8-Cloro-1-naft¡l)-2-[[(2S,4R)-4-metox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡hidro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-il]-1-(2 -fluoroprop-2 -eno¡l)p¡peraz¡n-2 -¡l]aceton¡trilo

Etapa A: (2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2S.4R)-4-metoxi-1-metil-pirrolidin-2-il1metoxi1-6.8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1 -carboxilato de tere-butilo. A una solución de [(2S,4R)-4-metoxi-1-metilpirrolidin-2-il]metanol (300 mg. 2,06 mmol, 2.0 eq.) y t-Bu0Na (149 mg. 1.55 mmol, 1.50 eq.) en tolueno (7.0 ml) se le añadió (2S)-4-[7-(8-cloro-1 -naftil)-2-metilsulfinil-6.8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianomethy1)piperazin-1-carboxilato de tere-butilo (600 mg. 1,03 mmol. 1.0 eq.). La mezcla se agitó a 0 °C durante 0.5 horas. Después de que se completara. se añadió agua (20.0 ml) y la mezcla resultante se extrajo con acetato de etilo (3 x 10.0 ml). La capa orgánica se secó sobre Na2S04, se filtró y se concentró. El residuo se purificó por cromatografía ultrarrápida de fase inversa (C18, 0.1 % de AF en agua. 0-60 % de MeCN) para dar el compuesto (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S.4R)-4-metoxi-1-metil-pirrolidin-2-il1metoxi1-6.8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1-carboxilato de tere-butilo (450 mg. 652 gmol, 63 % de rendimiento. 96 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+11: 662.
1H RMN (400 MHz, Cloroformo-d) ó 7,76 (d a, J = 8.4 Hz. 1H). 7,61 (t. J = 7.8 Hz. 1H). 7,52 (d. J = 7.6 Hz. 1H). 7,48 -7.40 (m. 1H). 7,35 (t. J = 7.8 Hz. 1H). 7,27 - 7,16 (m. 1H). 4,63 - 4,56 (m. 1H). 4,50 - 4,35 (m. 2H). 4,23 - 4,15 (m. 1H).
4,12 - 3,91 (m. 4H). 3,90 - 3,75 (m. 1H). 3,65 - 3,53 (m. 1H). 3,50 - 3,39 (m. 1H). 3,38 - 3,32 (m. 1H). 3,30 (d. J = 2.0 Hz. 3H). 3,28 - 3,06 (m. 3H). 3,02 - 2,85 (m. 2H). 2,83 - 2,64 (m. 2H). 2,62 - 2,52 (m. 1H). 2,47 (d. J = 2.8 Hz. 3H).
2,35 - 2,29 (m. 1H). 2,11 - 2,06 (m. 1H). 2,03 - 1,91 (m. 1H). 1,52 (s. 9H).
Etapa B: 2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2S,4R)-4-metoxi-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S,4R)-4-metoxi-1-metilp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de tere-butilo (450 mg. 680 gmol, 1.0 eq.) en dioxano (4.0 ml) se le añadió HCFdioxano (4 M, 4,0 ml. 23,6 eq.). La mezcla se agitó a 15 °C durante 0.5 horas. Después de que se completara. la mezcla de reacción se concentró. El residuo se ajustó con NaHC03 acuoso saturado (10,0 ml) a pH ~7 y después se extrajo con acetato de etilo (3 x 10,0 ml). La capa orgánica se secó sobre Na2S04, se filtró y se concentró para dar 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S,4R)-4-metoxi-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (300 mg. bruto) en forma de un sólido de color amarillo que se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+11: 562.
Etapa C: 2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2S.4R)-4-metoxi-1-metilpirrolidin-2-il1metoxi1-6.8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-(2-fluoroprop-2-enoil)piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S,4R)-4-metox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (90.0 mg. 160 gmol. 1.0 eq.). ácido 2-fluoroprop-2-enoico (28.8 mg. 320 gmol. 2.0 eq.) y TEA (130 mg. 1,28 mmol.
178 gl. 8.0 eq.) en AE (3.0 ml) se le añadió T3P (306 mg. 480 gmol. 286 gl. 50 % de pureza. 3.0 eq.). La mezcla se agitó a 0 °C durante 1 hora. Después de que se completara. se añadió agua (10,0 ml) y la mezcla resultante se extrajo con acetato de etilo (3 x 10.0 ml). La capa orgánica se secó sobre Na2S04, se filtró y se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Waters Xbridge 150 * 255 g;fase móvil: [agua (0.05 % de hidróxido de amonio v/v)-ACN1; % de B: 50 %-80 %,10 min) para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S,4R)-4-metox¡-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-i-(2-fluoroprop-2-eno¡l)p¡peraz¡n-2-il1acetonitrilo (32.0 mg. 50,3 gmol. 31 % de rendimiento. 99 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+11: 635.
1H RMN (400 MHz. Cloroformo-d) ó 7,76 (d, J = 8.4 Hz. 1H). 7,62 (t. J = 7.6 Hz. 1H). 7,53 (d. J = 7.6 Hz. 1H). 7,49 -7.40 (m. 1H). 7,34 (t. J = 7.8 Hz. 1H). 7,26 - 7,17 (m. 1H). 5,54 - 5,32 (m. 1H). 5,26 (dd. J = 3.6. 16,8 Hz. 1H). 5,10 -4,70 (m. 1H). 4,52 - 4,34 (m. 2H). 4,27 - 3,80 (m. 6H). 3,69 - 3,54 (m. 1H). 3,50 - 3,37 (m. 2H). 3,30 (d. J = 1.6 Hz. 3H).
3,28 - 3,02 (m. 4H). 2,98 - 2,68 (m. 3H). 2,65 - 2,53 (m. 1H). 2,47 (d. J = 2.4 Hz. 3H). 2,37 - 2,28 (m. 1H). 2,13 - 2,03 (m. 1H). 2,02 - 1,90 (m. 1H).
Ejemplo 578

2-[(2S)-1-[(E)-4-Fluorobut-2-enoil]-4-[2-[[(2S,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

Etapa A: (2S)-2-(Cianometil)-4-[2-[[(2S,4R)-4-fluoro-1-metilpirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de bencilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (500 mg, 840 gmol, 1,00 eq.) en tolueno (10,0 ml) se le añadieron t-Bu0Na (202 mg, 2,10 mmol, 2,50 eq.) y [(2S,4R)-4-fluoro-1-metilpirrolidin-2-il]metanol (224 mg, 1,68 mmol, 2,00 eq.). Después de agitar a 0 °C durante 1 hora, la mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (50 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, acetato de etilo/metanol = 100/1 a 10/1). Se obtuvo (2S)-2-(cianometil)-4-[2-[[(2S,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (340 mg, 502 gmol, 60 % de rendimiento, 98 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 664.
Etapa B: 2-[(2S)-4-[2-[[(2S,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-2-il]acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[2-[[(2S,4R)-4-fluoro-1 -metil-pirrolidin-2-il]metoxi]-7-(8-metil-1 -naftil)-6,8-dihidro-5H-pirido[3,4-tí]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (200 mg, 301 gmol, 1,00 eq.) en Me0H (15,0 ml) se le añadieron Pd/C (30,0 mg, 10 % de pureza) y NH3/Me0H (5,00 ml, 20 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 15 °C durante 0,5 horas. El catalizador se retiró por filtración. El filtrado se concentró al vacío. Se obtuvo 2-[(2S)-4-[2-[[(2S,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-tí]pirimidin-4-il]piperazin-2-il]acetonitrilo (159 mg, 294 gmol, 98 % de rendimiento,
98 % de pureza) en forma de un sólido de color amarillo y se usó directamente en la siguiente etapa sin purificación.
LCMS [ESI, M+1]: 530.
Etapa C: 2-[(2S)-1-[(£)-4-Fluorobut-2-enoil1-4-[2-[[(2S,4^)-4-fluoro-1-metilpirrolidin-2-il1metoxi1-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[2-[[(2S,4R)-4-fluoro-1-metilpirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo
(140 mg, 264 μmol, 1,00 eq.) y ácido (£)-4-fluorobut-2-enoico (82,5 mg, 793 μmol, 3,00 eq.) en acetato de etilo (2,00 ml) se le añadieron TEA (160 mg, 1,59 mmol, 220 gl, 6,00 eq.) y T3P (505 mg, 793 μmol, 472 gl, 50 % de pureza, 3,00 eq.) a -70 °C. La mezcla se agitó a -70 °C durante 1 hora. La mezcla de reacción se inactivó con HCl (12 N, 130 gl en 2 ml de agua) a -70 °C. La mezcla se calentó a 0 °C, se basificó con una solución acuosa saturada de NaHCÜ3 a pH = 8 y se extrajo con acetato de etilo (20 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (10 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar el residuo. El residuo se purificó por HPLC prep. (columna: Waters Xbridge 150 * 255 g; fase móvil: [agua (0,05 % de hidróxido de amonio v/v) - ACN];
% de B: 45 % - 75 %, 10 min). Las fracciones deseadas se recogieron y se liofilizaron. El compuesto del título 2-[(2S)-1-[(£)-4-fluorobut-2-enoil]-4-[2-[[(2S,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (34,5 mg, 55,8 umol, 21 % de rendimiento, 99,7 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 616.
1H RMN (400 MHz, cloroformo-d) 5 = 7,74 - 7,62 (m, 2H), 7,46 - 7,31 (m, 2H), 7,27 - 7,17 (m, 2H), 7,09 - 6,93 (m, 1H), 6,59 (d, J = 15,2 Hz, 1H), 5,30 - 4,51 (m, 4H), 4,48 - 4,35 (m, 1H), 4,30 - 3,66 (m, 6H), 3,63 - 3,34 (m, 3H), 3,24 - 2,96 (m, 5H), 2,92 (s, 3H), 2,87 - 2,53 (m, 4H), 2,50 (d, J = 4,4 Hz, 3H), 2,40 - 2,22 (m, 1H), 2,10 - 1,86 (m, 1H) (m, 3H).
Ejemplo 579

2-[(2S)-1-[(£)-4-Fluorobut-2-enoil]-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

Etapa A: (2S)-2-(Cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1,pirimidin-4-illpiperazin-1-carboxilato de bencilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (500 mg, 841 gmol, 1 eq.) en tolueno (10 ml) se le añadieron t-Bu0Na (242 mg, 2,52 mmol, 3 eq.) y [(2R)-1-metilpirrolidin-2-il]metanol (194 mg, 1,68 mmol, 2 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, acetato de etilo/Me0H = 100/1 a 10/1). Se obtuvo (2S)-2-(c¡anomet¡l)-4-[7-(8-metil-1-naft¡l)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il1piperazin-1-carboxilato de bencilo (240 mg, 338 gmol, 40 % de rendimiento, 91 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 646.
Etapa B: 2-[(2S)-4-[7-(8-Metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-i n metox n -6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-¡ n piperazin-2-i n acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (240 mg, 372 gmol, 1 eq.) en Me0H (15 ml) se le añadieron Pd/C (30 mg, 10 % de pureza) y NH3*Me0H (5 ml, 20 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 15 °C durante 1 hora. El catalizador se retiró por filtración. El filtrado se concentró al vacío. Se obtuvo 2-[(2S)-4-[7-(8-metil-1 -naftil)-2-[[(2R)-1 -met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]pir¡m¡d¡n-4-¡l]p¡perazin-2-¡l]aceton¡tr¡lo (180 mg, 352 gmol, 95 % de rendimiento) en forma de un sólido de color amarillo y se usó directamente en la siguiente etapa sin purificación. LCMS [ESI, M+1]: 512.
Etapa C: 2-[(2S)-1-[(E)-4-Fluorobut-2-enoi n -4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-i n metox n -6,8-dihidro-5H-pirido[3,4-cf1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-dih¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (100 mg, 195 umol, 1 eq.) y ácido (E)-4-fluorobut-2-enoico (40,7 mg, 391 umol, 2 eq.) en EtOAc (2 ml) se le añadieron TEA (79,1 mg, 782 umol, 109 ul, 4 eq.) y T3P (249 mg, 391 umol, 232 ul, 50 % de pureza en EtOAc, 2 eq.) a -70 °C. La mezcla se agitó a -70 °C durante 1 hora. La mezcla de reacción se inactivó con HCl (60 ul (12 M) en 2 ml de agua). La mezcla se ajustó a pH = 8 con una solución acuosa saturada de NaHC03 y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (10 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar el residuo. El residuo se purificó por HPLC prep. (columna: Luna C18150 * 255 u; fase móvil: [agua (0,225 % de AF) -ACN]; % de B: 25 % - 45 %, 7,8 min) y se purificó adicionalmente por HPLC prep. (columna: Waters Xbridge 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 52 %-79 %, 10 min). Las fracciones deseadas se recogieron y se liofilizaron. El compuesto del título 2-[(2S)-1-[(E)-4-fluorobut-2-enoil]-4-[7-(8-metil-1-naft¡l)-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-dih¡dro-5H-p¡r¡do[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (16,6 mg, 27,7 umol, 14 % de rendimiento, 99,6 % de pureza) se obtuvo en forma de una goma de color amarillo. LCMS [ESI, M+1]: 598.
1H RMN (400 MHz, cloroformo-d) 5 = 7,72 - 7,62 (m, 2H), 7,45 - 7,32 (m, 2H), 7,27 - 7,16 (m, 2H), 7,07 - 6,93 (m, 1H), 6,60 (d, J = 15,6 Hz, 1H), 5,26 - 4,51 (m, 3H), 4,39 (s, 1H), 4,31 - 3,33 (m, 9H), 3,27 - 2,97 (m, 5H), 2,92 (s, 3H), 2,87 - 2,76 (m, 1H), 2,65 (m, 2H), 2,49 (d a, J = 4,6 Hz, 3H), 2,29 (m, 1H), 2,15 - 1,96 (m, 1H), 1,92 - 1,67 (m, 3H).
Ejemplo 580

2-[(2S)-1-[(E)-4-fluorobut-2-enoil]-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-ó]pirimidin-4-il]piperazin-2-il]acetonitrilo

Etapa A: (2S)-2-(Cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de bencilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de bencilo (500 mg, 841 qmol, 1 eq.) en tolueno (10 ml) se le añadieron í-Bu0Na (242 mg, 2,52 mmol, 3 eq.) y [(2R)-1-metilpirrolidin-2-il]metanol (194 mg, 1,68 mmol, 2 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, acetato de etilo/Me0H = 100/1 a 10/1). Se obtuvo (2s)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de bencilo (240 mg, 338 qmol, 40 % de rendimiento, 91 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 646.
Etapa B: 2-[(2S)-4-[7-(8-Metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cnpirimidin-4-i n piperazin-2-i n acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5W-pirido[3,4-tí]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (240 mg, 372 qmol, 1 eq.) en Me0H (15 ml) se le añadieron Pd/C (30 mg, 10 % de pureza) y NH3*Me0H (5 ml, 20 % de pureza) en una atmósfera
de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 15 °C durante 1 hora. El catalizador se retiró por filtración. El filtrado se concentró al vacío.
Se obtuvo 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (180 mg, 352 μmol, 95 % de rendimiento) en forma de un sólido de color amarillo y se usó directamente en la siguiente etapa sin purificación. LCMS [ESI, M+1]: 512.
Etapa C: 2-[(2S)-1-[(E)-4-Fluorobut-2-enoil]-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (100 mg, 195 μmol, 1 eq.) y ácido (£)-4-fluorobut-2-enoico (40,7 mg, 391 umol, 2 eq.) en EtÓAc (2 ml) se le añadieron TEA (79,1 mg, 782 μmol, 109 μl, 4 eq.) y T3P (249 mg, 391 μmol, 232 μl, 50 % de pureza en EtÓAc, 2 eq.) a -70 °C. La mezcla se agitó a -70 °C durante 1 hora. La mezcla de reacción se inactivó con HCl (60 μl (12 M) en 2 ml de agua). La mezcla se ajustó a pH = 8 con una solución acuosa saturada de NaHCÓ3 y se extrajo con acetato de etilo (20 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (10 ml), se secaron sobre Na2SÓ4, se filtraron y se concentraron a presión reducida para dar el residuo. El residuo se purificó por HPLC prep. (columna: Luna C18150 * 255 p; fase móvil: [agua (0,225 % de AF) - MeCN]; % de B: 25 % - 45 %, 7,8 min) y se purificó adicionalmente por Hp LC prep. (columna:
Waters Xbridge 150 * 255 p; fase móvil: [agua (0,05 % de hidróxido de amonio v/v) - MeCN]; % de B: 52 %-79 %, 10 min). Las fracciones deseadas se recogieron y se liofilizaron. El compuesto del título 2-[(2S)-1-[(£)-4-fluorobut-2-enoil]-4-[7-(8-metil-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (16,6 mg, 27,7 μmol, 14 % de rendimiento, 99,6 % de pureza) se obtuvo en forma de una goma de color amarillo. LCMS [ESI, M+1]: 628.
1H RMN (400 MHz, cloroformo-d) 5 = 7,73 - 7,62 (m, 2H), 7,45 - 7,32 (m, 2H), 7,27 - 7,17 (m, 2H), 7,08 - 6,93 (m, 1H), 6,60 (d a, J = 16,0 Hz, 1H), 5,26 - 4,54 (m, 3H), 4,40 (m, 1H), 4,31 - 3,69 (m, 7H), 3,61 - 3,36 (m, 3H), 3,30 (d, J = 2,0 Hz, 3H), 3,24 - 2,96 (m, 4H), 2,92 (s, 3H), 2,91 - 2,55 (m, 4H), 2,47 (d, J = 4,8 Hz, 3H), 2,37 - 2,26 (m, 1H), 2,13 -2,03 (m, 1H), 2,01 - 1,89 (m, 1H).
Ejemplo 581

2-[(2S)-1-[(£)-4-fluorobut-2-enoil]-4-[7-(8-metil-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

Etapa A: (2S)-2-(Cianometil)-4-[7-(8-metil-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-1-carboxilato de bencilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-1-carboxilato de bencilo (450 mg, 757 umol, 1 eq.) y [(3R)-1-metilpirrol¡d¡n-3-¡l]metanol (131 mg, 1,13 mmol, 1,5 eq.) en tolueno (9 ml) se le añadió í-Bu0Na (145 mg, 1,51 mmol, 2 eq.). La mezcla se agitó a 0 °C durante 10 minutos. Después de que se completara, se añadió agua (15 ml) y la mezcla resultante se extrajo con acetato de etilo (30 ml * 2). Las capas orgánicas se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. Las fracciones deseadas se recogieron, se neutralizaron con NaHC03, se concentraron al vacío para eliminar el MeCN y se extrajeron con EtOAc (2 * 50 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío para dar (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de bencilo (300 mg, 455 gmol, 60 % de rendimiento, 98 % de pureza) en forma de un sólido de color rosa.
1H RMN (400 MHz, cloroformo-d) ó = 7,73 - 7,60 (m, 2H), 7,45 - 7,37 (m, 5H), 7,37 - 7,30 (m, 2H), 7,26 - 7,16 (m, 2H), 5,21 (s, 2H), 4,68 (s a, 1H), 4,30 - 3,78 (m, 7H), 3,59 - 3,29 (m, 2H), 3,25 - 2,94 (m, 4H), 2,91 (s, 3H), 2,82 - 2,62 (m, 4H), 2,62 - 2,53 (m, 2H), 2,53 - 2,42 (m, 2H), 2,35 (s, 3H), 2,05 (s, 1H), 1,66 - 1,51 (m, 1H).
Etapa B: 2-[(2S)-4-[7-(8-Metil-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(3R)-1-metilpyirrolidin-3-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (300 mg, 465 gmol, 1 eq.) en Me0H (4 ml) se le añadieron Pd/C (150 mg, 10 % de pureza) y NH3/Me0H (4 ml, 20 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 15 °C durante 1 hora. Después de que se completara, la mezcla se filtró y el filtrado se concentró al vacío para dar 2-[(2S)-4-[7-(8-metil-1 -naftil)-2-[[(3E)-1 -metilpirrolidin-3-il1metoxi1-6,8-dihidro-5H-pirido[3,4-tí1pirimidin-4-il1piperazin-2-il1acetonitrilo (210 mg, 369 gmol, 80 % de rendimiento, 90 % de pureza) en forma de un sólido de color rosa que se usó directamente en la siguiente etapa sin purificación adicional. LCMS [ESI, M+11: 512.
Etapa C: 2-[(2S)-1-[(E)-4-Fluorobut-2-enoil1-4-[7-(8-metil-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(3E)-1-met¡lp¡rrol¡d¡n-3-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (70 mg, 137 gmol, 1 eq.), TEA (41,5 mg, 410 gmol, 57,1 gl, 3 eq.) y ácido (E)-4-fluorobut-2-enoico (28,5 mg, 274 gmol, 2 eq.) en EtOAc (2 ml) se le añadió T3P (131 mg, 205 gmol, 122 gl, 50 % de pureza en EtOAc, 1,5 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se inactivó con HCl 1 M (0,4 ml) a -40 °C y se agitó hasta que no quedó hielo. Después, la capa acuosa separada se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi C18150 * 30 mm * 4 gm; fase móvil: [agua (0,225 % de AF)-MeCN1; % de B: 20 %-50 %, 10 min). Las fracciones deseadas se recogieron y se liofilizaron para dar el compuesto del título 2-[(2S)-1-[(E)-4-fluorobut-2-enoil1-4-[7-(8-metil-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il1metoxi1-6,8-dihidro-5H-pirido[3,4
d]pirimidin-4-il]piperazin-2-il]acetonitrilo (13,7 mg, 20,8 μmol, 15 % de rendimiento, 97,7 % de pureza, AF) en forma de un sólido de color gris. LCMS [ESI, M+1]: 598.
1H RMN (400 MHz, cloroformo-d) 5 = 7,66 - 7,59 (m, 1H), 7,59 - 7,51 (m, 1H), 7,32 (td, J = 7,6, 11,6 Hz, 1H), 7,26 (t, J = 7,6 Hz, 1H), 7,19 - 7,07 (m, 2H), 7,00 - 6,83 (m, 1H), 6,52 (d a, J = 14,8 Hz, 1H), 5,21 - 4,31 (m, 3H), 4,29 - 3,96 (m, 3H), 3,94 - 3,53 (m, 7H), 3,45 (d a, J = 6,8 Hz, 4H), 3,15 - 2,88 (m, 7H), 2,84 (s, 3H), 2,81 - 2,74 (m, 1H), 2,61 -2,47 (m, 2H), 2,18 (s a, 1H), 1,85 (s a, 1H).
Ejemplo 582

2-((S)-4-(7-(8-Cloronaftalen-1-il)-2-(((2S,4E)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-fluorobut-2-enoil)piperazin-2-il)acetonitrilo

2-((S)-4-(7-(8-Cloronaftalen-1-il)-2-(((2S,4E)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(E)-4-fluorobut-2-enoil)piperazin-2-il)acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S,4E)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (180 mg, 327 μmol, 1,0 eq.) y ácido (E)-4-fluorobut-2-enoico (102 mg, 982 μmol, 3,0 eq.) se le añadieron TEA (265 mg, 2,62 mmol, 364 μl, 8,0 eq.) y T3P (833 mg, 1,31 mmol, 778 μl, 50 % de pureza, 4,0 eq.) y la mezcla de reacción se agitó a -65 °C durante 1 hora. Después de que se completara, la mezcla de reacción se inactivó con HCl 2 N a pH ~7 a -65 °C, después la capa orgánica se separó, la fase acuosa se extrajo con acetato de etilo (2 * 10 ml) y la capa orgánica combinada se secó sobre Na2SÜ4, se filtró y se concentró. El residuo se purificó cromatografía en columna (Al2Ü3 básico, éter de petróleo/acetato de etilo = 3/1 a acetato de etilo/metanol = 20/1), el producto bruto se purificó de nuevo por HPLC prep. (columna: Xtimate C18 150 * 25 mm * 5 μm; fase móvil: [agua (0,05 % de hidróxido de amonio v/v)-MeCN]; % de B: 50 %-80 %, 8 min) y el producto obtenido se concentró y se liofilizó. El compuesto del título 2-((S)-4-(7-(8-cloronaftalen-1-il)-2-(((2S,4E)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-fluorobut-2-enoil)piperazin-2-il)acetonitrilo (72,2 mg, 113 μmol, 34 % de rendimiento, 99 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1 ]: 637.
1H RMN (400 MHz, cloroformo-d) 57,79 - 7,73 (m, 1H), 7,62 (t, J = 7,0 Hz, 1H), 7,53 (d, J = 7,2 Hz, 1H), 7,49 - 7,41 (m, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,27 - 7,18 (m, 1H), 7,08 - 6,93 (m, 1H), 6,59 (d a, J = 14,8 Hz, 1H), 5,29 - 5,03 (m, 3H), 4,54 - 4,52 (m, 1H), 4,50 - 4,35 (m, 2H), 4,30 - 3,78 (m, 5H), 3,76 - 3,35 (m, 3H), 3,32 - 2,96 (m, 5H), 2,91 - 2,54 (m, 4H), 2,51 (d, J = 3,2 Hz, 3H), 2,39 - 2,23 (m, 1H), 2,08 - 1,86 (m, 1H).
E je m p lo 583

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(E)-4-fluorobut-2-enoil]piperazin-2-il]acetonitrilo

2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2S,4R)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-[(E)-4-fluorobut-2-enoil1piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2S,4E)-4-metoxi-1-metil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (150 mg, 267 μmol, 1,0 eq.), ácido (E)-4-fluorobut-2-enoico (55,6 mg, 534 gmol, 2,0 eq.) y TEA (108 mg, 1,07 mmol, 149 gl, 4,0 eq.) en acetato de etilo (4,0 ml) se le añadió T3P (340 mg, 534 gmol, 317 gl, 50 % de pureza, 2,0 eq.). La mezcla se agitó a -70 °C durante 1 hora. Después de que se completara, la mezcla de reacción se inactivó con HCl (12 M, 60,0 gl en 2,0 ml de agua). La mezcla se ajustó a pH-8 con una solución acuosa saturada de NaHCÜ3 y se extrajo con acetato de etilo (3 * 20,0 ml). Las capas orgánicas combinadas se lavaron con salmuera saturada (60,0 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron. El residuo se purificó por HPLC prep. (columna: Xtimate C18150 * 25 mm * 5 um; fase móvil: [agua (0,05 % de hidróxido de amonio v/v)-MeCN]; % de B: 50 %-80 %, 8 min) para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1 -naftil)-2-[[(2S,4E)-4-metoxi-1 -metil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(E)-4-fluorobut-2-enoil]piperazin-2-il]acetonitrilo (39,6 mg, 60,9 gmol, 23 % de rendimiento, 99 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 619.
1H RMN (400 MHz, Cloroformo-d) ó 7,76 (d a, J = 8,0 Hz, 1H), 7,62 (t, J = 7,6 Hz, 1H), 7,53 (d, J = 7,6 Hz, 1H), 7,49 -7,40 (m, 1H), 7,34 (t, J = 8,0 Hz, 1H), 7,27 - 7,17 (m, 1H), 7,08 - 6,94 (m, 1H), 6,67 - 6,54 (m, 1H), 5,25 - 5,03 (m, 2H), 4,80 - 4,60 (m, 1H), 4,48 - 4,34 (m, 2H), 4,21 - 3,77 (m, 5H), 3,65 - 3,55 (m, 1H), 3,51 - 3,37 (m, 1H), 3,31 - 2,97 (m, 5H), 2,88 - 2,54 (m, 4H), 2,48 (d, J = 3,6 Hz, 3H), 2,34 - 2,24 (m, 1H), 2,11 - 2,01 (m, 1H), 1,90 - 1,69 (m, 3H).
Ejemplo 584

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-[(E)-4-fluorobut-2 -enoil]piperazin-2 -il]acetonitrilo

-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(3R)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(E-4-fluorobut-2-enoil1piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(3E)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (100 mg, 188 μmol, 1,0 eq.), ácido (E)-4-fluorobut-2-enoico (39,1 mg, 376 μmol, 2,0 eq.) y TEA (95,1 mg, 940 μmol, 131 μl, 5,0 eq.) en acetato de etilo (3,0 ml) se le añadió T3P (179 mg, 282 μmol, 168 μl, 50 % de pureza, 1,50 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se inactivó con HCl (12 M, 5,0 eq.) a -40 °C. La mezcla se ajustó a pH ~ 8 con NaHCÜ3 acuoso saturado y se extrajo con acetato de etilo (3 * 10 ,0 ml). Las capas orgánicas combinadas se lavaron con salmuera (30,0 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi C18150 * 25 * 10 μm; fase móvil: [agua (0,225 % de AF)-MeCN]; % de B: 30 %-50 %, 8 min) para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(3E)-1-metilpirrolidin-3-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1 -[(E)-4-fluorobut-2-enoil]piperazin-2-il]acetonitrilo (15,7 mg, 24,9 μmol, 13 % de rendimiento, 98 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 619.
1H RMN (400 MHz, Cloroformo-d) ó 7,76 (d, J = 8,4 Hz, 1H), 7,65 - 7,59 (m, 1H), 7,56 - 7,49 (m, 1H), 7,48 - 7,40 (m, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,26 - 7,18 (m, 1H), 7,08 - 6,92 (m, 1H), 6,74 - 6,46 (m, 1H), 5,25 - 4,95 (m, 3H), 4,75 -4,55 (m, 1H), 4,47 - 4,36 (m, 1H), 4,35 - 4,26 (m, 2H), 4,14 - 3,92 (m, 2H), 3,91 - 3,76 (m, 1H), 3,64 - 3,55 (m, 1H), 3,53 - 3,35 (m, 1H), 3,33 - 3,04 (m, 7H), 3,03 - 2,72 (m, 4H), 2,67 (d, J = 4,0 Hz, 3H), 2,64 - 2,51 (m, 1H), 2,31 - 2,19 (m, 1H), 1,98 - 1,85 (m, 1H).
Ejemplo 585

2-((S)-4-(2-(3-((1 S,4S)-2-oxa-5-azabiciclo[2.2.1 ]heptan-5-il)propoxi)-7-(8-cloronaftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-fluorobut-2-enoil)piperazin-2-il)acetonitrilo

Etapa A: (2S)-4-[7-(8-Cloro-1-naftil)-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]-2-(cianometil)piperazin-1 -carboxilato de tere-butilo. A una mezcla de 3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan -5-il]propan-1-ol (150 mg, 954 μmol, 1,85 eq.) en tolueno (3,0 ml) se le añadió poco a poco t-Bu0Na (99,2 mg, 1,03 mmol, 2,0 eq.) a 0 °C y, después de agitar a 0 °C durante 0,5 horas, a la mezcla se le añadió (2S)-4-[7-(8-cloro-1-naftil)-2-metilsulfinil-6,8-dihidro -5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (300 mg, 516 μmol, 1,0 eq.) y se agitó a 0 °C durante 0,5 horas. Después de que se completara, se añadió agua (15 ml), la mezcla resultante se extrajo con acetato de etilo (2 x 10 ml) y la capa orgánica combinada se secó sobre Na2S04, se filtró y se concentró. El residuo se purificó por cromatografía ultrarrápida de fase inversa (C18, 0,1 % de TFA en agua, 30 %-50 % de MeCN) y el producto obtenido se ajustó con NaHC03 sólido a pH-8, después se concentró, la fase acuosa se extrajo con acetato de etilo (2 x 15 ml) y la capa orgánica se lavó con salmuera saturada (1 x 20 ml), se secó sobre Na2S04, se filtró y se concentró. El producto (2S)-4-[7-(8-cloro-1-naftil)-2-[3-[(1 S,4S)-2-oxa-5-azab¡c¡clo[2.2.1]heptan-5-¡l]propox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡din-4-¡l]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (180 mg, 267 μmol, 52 % de rendimiento, 100 % de pureza) se obtuvo en forma de un sólido de color amarillo claro. LCMS [ESI, M+1 ]: 674.
1H RMN (400 MHz, cloroformo-d) ó 7,76 (d a, J = 8,4 Hz, 1H), 7,61 (t, J = 7,6 Hz, 1H), 7,52 (d, J = 7,2 Hz, 1H), 7,48 -7,40 (m, 1H), 7,34 (t, J = 7,8 Hz, 1H), 7,26 - 7,17 (m, 1H), 4,66 - 4,54 (m, 1H), 4,50 - 4,29 (m, 4H), 4,08 - 3,77 (m, 5H), 3,65 - 3,54 (m, 2H), 3,51 - 3,44 (m, 1H), 3,39 - 3,31 (m, 1H), 3,29 - 3,02 (m, 4H), 3,00 - 2,87 (m, 2H), 2,84 - 2,46 (m, 6H), 1,99 - 1,82 (m, 3H), 1,52 (s, 9H).
Etapa B: 2-[(2S)-4-[7-(8-Cloro-1 -naftil)-2-[3-[(1 S,4S)-2-oxa -5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo. A una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (150 mg, 222 μmol, 1,0 eq.) en dioxano (1,0 ml) se le HCl 4 N*dioxano (1,0 ml) y la mezcla de reacción se agitó a 15 °C durante 1 hora. Después de que se completara, la mezcla de reacción se concentró, se añadió diclorometano (10 ml), después se lavó con Na2C03 acuoso saturado (1 x 10 ml) y la capa orgánica se secó sobre Na2S04, se filtró y se concentró. El producto 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[3-[(1S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (100 mg, 172 μmol, 77 % de rendimiento, 98 % de pureza) se obtuvo en forma de un aceite de color pardo. LCMS [ESI, M+1]: 574.
Etapa C: 2-((S)-4-(2-(3-((1S,4S)-2-0xa-5-azabiciclo[2.2.1]heptan-5-il)propoxi)-7-(8-cloronaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-1-((E)-4-fluorobut-2-enoil)piperazin-2-il)acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[3-[(1 S,4S)-2-oxa-5-azabiciclo[2.2.1]heptan-5-il]propoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (90,0 mg, 157 μmol, 1,0 eq.) y ácido (E)-4-fluorobut-2-enoico (48,9 mg, 470 μmol, 3,0 eq.) en acetato de etilo (1,0 ml) se le añadieron poco a poco TEA (127 mg, 1,25 mmol, 175 μl, 8,0 eq.) y T3P (399 mg, 627 μmol, 373 μl, 50 % de pureza, 4,0 eq.) a -65 °C y la mezcla de reacción se agitó a -65 °C durante 1 hora. Después
de que se completara, la mezcla de reacción se inactivó con HCl 2 N a pH ~7 a -65 °C, después la capa orgánica se separó, la fase acuosa se extrajo con acetato de etilo (2 * 10 ml) y la capa orgánica combinada se secó sobre Na2SÜ4, se filtró y se concentró. El residuo se purificó cromatografía en columna (Al2Ü3 básico, éter de petróleo/acetato de etilo = 3/1 a acetato de etilo/metanol = 20/1), el producto bruto se purificó de nuevo por HPLC prep. (columna: Xtimate C18 150 * 25 mm * 5 gm; fase móvil: [agua (0,05 % de hidróxido de amonio v/v)-MeCN]; % de B: 44 %-74 %, 8 min) y el producto obtenido se concentró y se liofilizó. El compuesto del título 2-((S)-4-(2-(3-((1 S,4S)-2-oxa-5-azabiciclo[2.2.1 ]heptan-5-il)propoxi)-7-(8-cloronaftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1 -((E)-4-fluorobut-2-enoil)piperazin-2-il)acetonitrilo (10,6 mg, 16,0 gmol, 10 % de rendimiento, 99 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 660.
1H RMN (400 MHz, cloroformo-d) ó 7,76 (d a, J = 8,4 Hz, 1H), 7,62 (t, J = 7,4 Hz, 1H), 7,53 (d, J = 7,2 Hz, 1H), 7,49 -7,41 (m, 1H), 7,34 (t, J = 7,8 Hz, 1H), 7,26 - 7,18 (m, 1H), 7,09 - 6,93 (m, 1H), 6,60 (d a, J = 14,0 Hz, 1H), 5,26 - 5,01 (m, 2H), 4,76 - 7,54 (m, 1H), 4,52 - 4,29 (m, 4H), 4,19 - 3,97 (m, 3H), 3,95 - 3,69 (m, 2H), 3,66 - 3,37 (m, 4H), 3,33 -2,99 (m, 4H), 2,97 - 2,67 (m, 5H), 2,65 - 2,48 (m, 2H), 2,01 - 1,90 (m, 2H), 1,90 - 1,82 (m, 1H), 1,76 - 1,69 (m, 1H).
Ejemplo 586

2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-[(E)-4-fluorobut-2-enoil]piperazin-2-il]acetonitrilo

Etapa A: (2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2E)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo. A una solución de [(2E)-1-metilpirrolidin-2-il]metanol (99,1 mg, 860 gmol, 2,50 eq.) en tolueno (5,0 ml) se le añadieron poco a poco t-Bu0Na (66,2 mg, 688 gmol, 2,0 eq.) y (2,S)-4-[7-(8-cloro-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (200 mg, 344 gmol, 1,0 eq.). La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara,
se añadió agua (20,0 ml) y la mezcla resultante se extrajo con acetato de etilo (3 x 10,0 ml). La capa orgánica se secó sobre Na2SÜ4, se filtró y se concentró. El residuo se purificó por cromatografía ultrarrápida de fase inversa (C18, 0,1 % de AF en agua, 0-60 % de MeCN) para dar el compuesto (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de tere-butilo (200 mg, 313 gmol, 91 % de rendimiento, 99 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 632.
1H RMN (400 MHz, Cloroformo-d) 57,77 - 7,73 (m, 1H), 7,61 (t, J = 7,6 Hz, 1H), 7,54 - 7,50 (m, 1H), 7,49 - 7,39 (m, 1H), 7,33 (t, J = 7,6 Hz, 1H), 7,27 - 7,16 (m, 1H), 4,67 - 4,56 (m, 1H), 4,47 - 4,35 (m, 2H), 4,23 - 4,15 (m, 1H), 4,09 -3,89 (m, 3H), 3,88 - 3,77 (m, 1H), 3,63 - 3,53 (m, 1H), 3,41 - 3,30 (m, 1H), 3,22 - 3,03 (m, 4H), 3,01 - 2,85 (m, 1H), 2,74 - 2,67 (m, 2H), 2,63 - 2,51 (m, 2H), 2,49 (d, J = 4,4 Hz, 3H), 2,36 - 2,25 (m, 1H), 2,11 - 2,05 (m, 1H), 1,90 - 1,74 (m, 3H), 1,52 (s, 9H).
Etapa B: 2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-illpiperazin-2-illacetonitrilo. A una solución de (2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de tere-butilo (200 mg, 316 gmol, 1,0 eq.) en dioxano (3,0 ml) se le añadió HCMioxano (4,0 M, 3,0 ml, 37,9 eq.). La mezcla se agitó a 15 °C durante 0,5 horas.
Después de que se completara, la mezcla de reacción se concentró. El residuo se ajustó con NaHCÜ3 acuoso saturado (10 ,0 ml) a pH -7 y después se extrajo con acetato de etilo (3 x 10 ,0 ml). La capa orgánica se secó sobre Na2SÜ4, se filtró y se concentró para dar el compuesto 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (150 mg, bruto) en forma de un sólido de color amarillo.
El producto se usó en la siguiente etapa sin purificación adicional. LCMS [ESl, M+1]: 532.
Etapa C: 2-[(2S)-4-[7-(8-Cloro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d^pirimidin-4-il1-1-[(E)-4-fluorobut-2-enoil]piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (100 mg, 188 gmol, 1.0 eq.) y ácido (£)-4-fluorobut-2-enoico (39,1 mg, 376 gmol, 2,0 eq.) en acetato de etilo (2,0 ml) se le añadieron TEA (76,1 mg, 752 gmol, 105 gl, 4,0 eq.) y T3P (239 mg, 376 gmol, 224 gl, 50 % de pureza, 2,0 eq.) a -70 °C. La mezcla se agitó a -70 °C durante 1 hora. Después de que se completara, la mezcla de reacción se inactivó con HCl (12,0 M, 60.0 gl en 2,0 ml de agua). La mezcla se ajustó a pH = 8 con NaHCÜ3 acuoso saturado y se extrajo con acetato de etilo (3 x 10 ,0 ml). Las capas orgánicas combinadas se lavaron con salmuera (30,0 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar el residuo. El residuo se purificó por HPLC prep. (columna:
Xtimate C18 150 * 25 mm * 5 gm; fase móvil: [agua (0,05 % de hidróxido de amonio v/v)-MeCN]; % de B: 598 %-89 %, 8 min) para dar el compuesto del título 2-[(2S)-4-[7-(8-cloro-1-naftil)-2-[[(2R)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-[(£)-4-fluorobut-2-enoil]piperazin-2-il]acetonitrilo (13,97 mg, 22,2 gmol, 12 % de rendimiento, 98,4 % de pureza) en forma de un sólido de color blanco. LCMS [ESl, M+1]: 619.
1H RMN (400 MHz, Cloroformo-d) 57,76 (d a, J = 8,0 Hz, 1H), 7,62 (t, J = 7,6 Hz, 1H), 7,53 (d, J = 7,6 Hz, 1H), 7,49 -7,40 (m, 1H), 7,34 (t, J = 8,0 Hz, 1H), 7,27 - 7,17 (m, 1H), 7,08 - 6,94 (m, 1H), 6,67 - 6,54 (m, 1H), 5,25 - 5,03 (m, 2H), 4,80 - 4,60 (m, 1H), 4,48 - 4,34 (m, 2H), 4,21 - 3,77 (m, 5H), 3,65 - 3,55 (m, 1H), 3,51 - 3,37 (m, 1H), 3,31 - 2,97 (m, 5H), 2,88 - 2,54 (m, 4H), 2,48 (d, J = 3,6 Hz, 3H), 2,34 - 2,24 (m, 1H), 2,11 - 2,01 (m, 1H), 1,90 - 1,69 (m, 3H).
Ejemplo 587
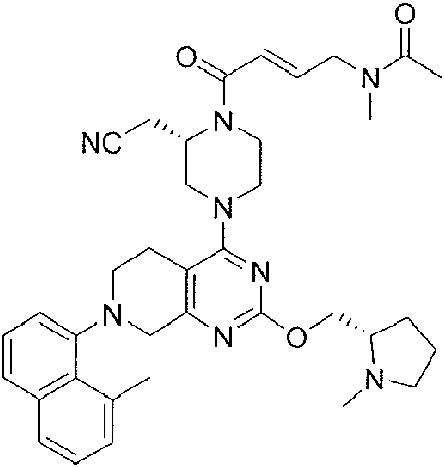
N-((E)-4-((S)-2-(Cianometil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-4-oxobut-2-en-1-il)-N-metilacetamida

0,818 mmol) con DMF (4 ml) seguido de la adición de NaH (21,6 mg, 0,900 mmol) y Mel (56,2 gl, 0,900 mmol). Después de agitar durante l2 horas, la reacción se diluyó con acetato de etilo y bicarbonato de sodio saturado. Las capas se separaron y la capa orgánica se secó sobre MgSÜ4, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10-70 % de acetato de etilo/hexanos para proporcionar (E)-4-(N-metilacetamido)but-2-enoato de etilo (120 mg, 0,648 mmol, 79,2 % de rendimiento).
Etapa B: ácido (E)-4-(N-metilacetamido)but-2-enoico: Se diluyó (E)-4-(N-metilacetamido)but-2-enoato de etilo (20 mg, 0,11 mmol) con metanol (1 ml) seguido de la adición de NaÜH (270 gl, 0,54 mmol). Después de agitar durante 4 horas, la reacción se diluyó con HCl 2 N a pH ~4 y la capa acuosa se extrajo con acetato de etilo. El acetato de etilo se secó sobre MgSÜ4, se filtró y se concentró para proporcionar ácido (E)-4-(N-metilacetamido)but-2-enoico (15 mg, 0,095 mmol, 88 % de rendimiento).
Etapa C: N-((E)-4-((S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-il)-4-oxobut-2-en-1-il)-N-metilacetamida: Se diluyó 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (20 mg, 0,039 mmol) con DMF seguido de la adición de DIEA (13,65 gl, 0,07817 mmol). Se añadió ácido (E)-4-(N-metilacetamido)but-2-enoico (9,83 mg, 0,0625 mmol) seguido de la adición de anhídrido cíclico del ácido 1-propanofosfónico (39,6 gl, 0,0664 mmol). Después de agitar durante 12 horas, la reacción se diluyó con acetato de etilo y bicarbonato de sodio saturado. Las capas se separaron y el acetato de etilo se secó sobre MgSÜ4, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10 % de metanol/DCM (1 % de NH40H) para proporcionar N-((E)-4-((S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-il)-4-oxobut-2-en-1-il)-N-metilacetamida (2,1 mg, 0,0032 mmol, 8,2 % de rendimiento). ESI+APCI MS m/z 651,4 [M+H]+.
Ejemplo 588

2-((S)-4-(7-(8-Bromonaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo

¡l)-2-(c¡anomet¡l)p¡perazin-1-carbox¡lato de bencilo: Se diluyeron (S)-2-(cianometil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (140 mg, 0,277 mmol), 1,8-dibromonaftaleno (238 mg, 0,831 mmol), Pd2(dba)3 (25,4 mg, 0,0277 mmol), CS2C03 (451 mg, 1,38 mmol) y XANTPH0S (32,0 mg, 0,0554 mmol) con tolueno (111 μl, 0,277 mmol). La reacción se purgó con argón, se selló y se calentó a 110 °C mientras se agitaba durante 12 horas. La reacción se dejó enfriar y se diluyó con acetato de etilo y agua. Las capas se separaron y el acetato de etilo se secó sobre MgS04, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 1-10 % de metanol/DCM (1 % de NH40H) para proporcionar (S)-4-(7-(8-bromonaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)p¡peraz¡n-1-carbox¡lato de bencilo (80 mg, 0,113 mmol, 40,7 % de rendimiento).
Etapa B: 2-((S)-4-(7-(8-Bromonaftalen-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l)p¡perazin-2-¡l)aceton¡tr¡lo: Se diluyó (S)-4-(7-(8-bromonaftalen-1-¡l)-2-(((S)-1-met¡lp¡rrol¡din-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de bencilo (80 mg, 0,11 mmol) con TFA (2 ml), se puso en una atmósfera de nitrógeno y se calentó a 90 °C. Después de agitar durante 2 horas, la reacción se enfrió y se concentró. El material se diluyó con DCM y se lavó con bicarbonato de sodio saturado. El DCM se secó sobre MgS04, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10 % de metanol/DCM (1 % de NH40H) para proporcionar 2-((S)-4-(7-(8-bromonaftalen-1-¡l)-2-(((S)-1-metilp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (23 mg, 0,040 mmol, 35 % de rendimiento).
Etapa C: 2-((S)-4-(7-(8-Bromonaftalen-1-¡l)-2-(((S)-1-met¡lp¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-1-(2-fluoroacr¡loil)p¡peraz¡n-2-¡l)aceton¡tr¡lo: Se diluyó 2-((S)-4-(7-(8-bromonaftalen-1-il)-2-(((S)-1-metilp¡rrol¡d¡n-2-il)metoxi)-5,6,7,8-tetrah¡drop¡r¡do[3,4-d]p¡rim¡d¡n-4-¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (23 mg, 0,040 mmol) con d Mf (350 μl) seguido de la adición de DIEA (22 μl, 0,13 mmol) y ácido 2-fluoroacrílico (5,4 mg, 0,060 mmol). Se añadió 2,4,6-trióxido de 2,4,6-tr¡propil-1,3,5,2,4,6-tr¡oxatr¡fosf¡nano (25 mg, 0,040 mmol) y la reacción se agitó durante 12 horas a temperatura ambiente. La reacción se diluyó con acetato de etilo y bicarbonato de sodio saturado. Las capas se separaron y el acetato de et¡lo se secó sobre MgS04, se f¡ltró y se concentró. El mater¡al se pur¡f¡có sobre gel de síl¡ce eluyendo con 10 % de metanol/DCM (1 % de NH40H) para proporcionar 2-((S)-4-(7-(8-bromonaftalen-1-il)-2-(((S)-1-metilp¡rrol¡d¡n-2-¡l)metox¡)-5,6,7, 8-tetrah¡drop¡r¡do[3,4-d]p¡r¡m¡din-4-¡l)-1-(2-fluoroacr¡lo¡l)p¡peraz¡n-2-¡l)aceton¡tr¡lo (9 mg, 0,014 mmol, 35 % de rendimiento). ESI+APCI MS m/z 650,2 [M+H]+.
Ejemplo 589
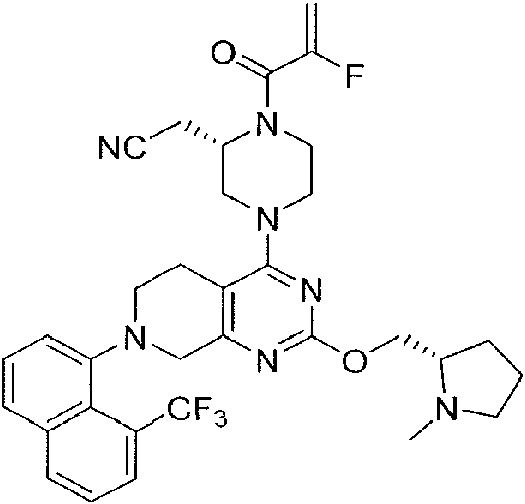
2-((S)-1-(2-Fluoroacriloil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(8-(trifluorometil)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 2-((S)-1-(2-Fluoroacriloil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(8-(trifluorometil)naftalen-1-il)-5,6,7,8-tetrahidropiridor[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Se diluyó 2-((S)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(8-(trifluorometil)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (24 mg, 0,042 mmol) con DMF (400 gl), se añadió DIEA (19 gl, 0,11 mmol), seguido de la adición de ácido 2-fluoroacrílico (6,1 mg, 0,068 mmol) y la adición de 2,4,6-trióxido de 2,4,6-tripropil-1,3,5,2,4,6-trioxatrifosfinano (30 gl, 0,047 mmol). La reacción se agitó a temperatura ambiente durante 10 horas. La reacción se vertió en una solución al 5 % de bicarbonato de sodio y se extrajo dos veces con acetato de etilo. El acetato de etilo se lavó con agua y salmuera, se secó sobre MgSÜ4, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 1-10 % de metanol/DCM (1 % de NH40H) para proporcionar 2-((S)-1-(2-fluoroacriloil)-4-(2-(((S)-1-metilpirrolidin-2-il)metoxi)-7-(8-(trifluorometil)naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (5 mg, 0,0078 mmol, 18 % de rendimiento). ESI+APCI MS m/z 638,3 [M+H]+.
Ejemplo 590
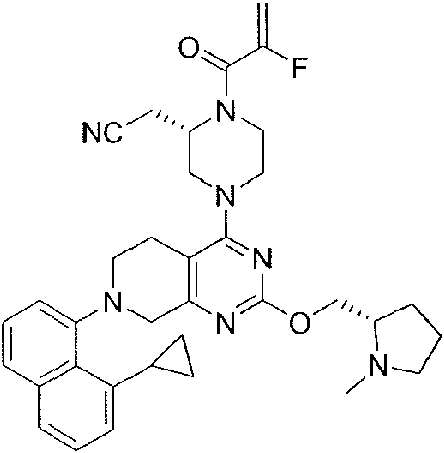
2-((S)-4-(7-(8-Ciclopropilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo

Etapa A: (S)-2-(Cianometi0-4-(7-(8-ciclopropilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-i0metoxi)-5,6.7.8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: Se diluyeron (S)-4-(7-(8-bromonaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (30 mg, 0,042 mmol) y aducto de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio (II) diclorometano (14 mg, 0,017 mmol) con bromuro de ciclopropilcinc (253 gl, 0,13 mmol). La reacción se purgó con argón, se selló y se calentó a 90 °C. Después de agitar durante 12 horas, la reacción se dejó enfriar y se diluyó con acetato de etilo y agua. Las capas se separaron y el acetato de etilo se secó sobre MgSÜ4, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10 % de metanol/DCM (1 % de NH40H) para proporcionar bencilo (S)-2-(cianometil)-4-(7-(8-ciclopropilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato (7 mg, 0,010 mmol, 25 % de rendimiento).
Etapa B: 2-((S)-4-(7-(8-Ciclopropilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Se diluyó (S)-2-(cianometil)-4-(7-(8-ciclopropilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (9 mg, 0,01 mmol) con metanol (1 ml) seguido de la adición de Pd-C (1 mg, 0,01 mmol). La reacción se equipó con un globo de hidrógeno y se purgó tres veces seguido de agitación en una atmósfera de hidrógeno durante 4 horas. Los sólidos se recogieron por filtración, se aclararon con metanol y la fase orgánica combinada se concentró para proporcionar 2-((S)-4-(7-(8-ciclopropilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (7 mg, 0,01 mmol, 97 % de rendimiento).
Etapa C: 2-((S)-4-(7-(8-Ciclopropilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo: Se diluyó 2-((S)-4-(7-(8-ciclopropilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (10 mg, 0,019 mmol) con DMF (500 gl) seguido de la adición de DIeA (11 gl, 0,065 mmol), ácido 2-fluoroacrílico (2,5 mg, 0,028 mmol) y 2,4,6-trióxido de 2,4,6-tripropil-1,3,5,2,4,6-trioxatrifosfinano (13 gl, 0,020 mmol). Después de agitar durante 12 horas, la reacción se diluyó con acetato de etilo y bicarbonato de sodio saturado. Las capas se separaron y el acetato de etilo se secó sobre MgSÜ4, se filtró y se concentró. El material se purificó sobre gel de sílice eluyendo con 10 % de metanol/DCM (1 % de NH40H) para proporcionar 2-((S)-4-(7-(8-ciclopropilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo (0,9 mg, 0,0015 mmol, 7,9 % de rendimiento). ESI+APCI MS m/z 610,3 [M+H]+.
Ejemplo 591

2-((S)-1-(2-Fluoroacriloil)-4-(7-(8-isopropilnaftalen-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
Se sintetizó de la misma manera que el Ejemplo 590 sustituyendo bromuro de isopropil cinc por bromuro de ciclopropil cinc en la etapa A. ESI+APCI MS m/z 612,3 [m+H]+.
Ejemplo 592

2-((S)-1-(2-Fluoroacriloil)-4-(7-(1-metilisoquinolin-8-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo
Se sintetizó de la misma manera que el Ejemplo 588 sustituyendo 8-bromo-1-metilisoquinolina por 1,8-dibromonaftaleno en la etapa A. ESI+APCI MS m/z 585,3 [M+H]+.
Ejemplo 593
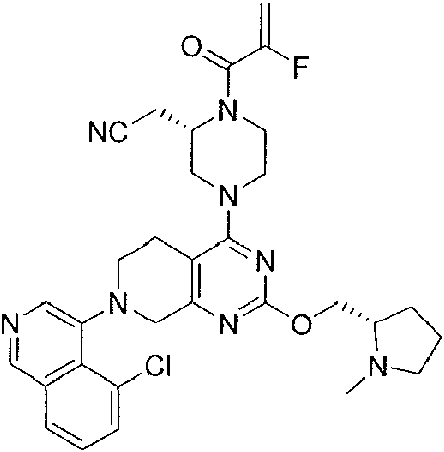
2-((S)-4-(7-(5-Cloroisoquinolin-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo
Se sintetizó de la misma manera que el Ejemplo 588 sustituyendo 4-bromo-5-cloroisoquinolina por 1,8 dibromonaftaleno en la etapa A. ESI+APCI MS m/z 605,2 [M+H]+.
Ejemplo 594

2-((S)-4-(7-(8-Bromoisoquinolin-1-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo
Se sintetizó de la misma manera que el Ejemplo 588 sustituyendo 8-bromo-1-cloroisoquinolina por 1,8 dibromonaftaleno en la etapa A. ESI+APCI MS m/z 651,2 [M+H]+.
Ejemplo 595

2-((S)-4-(7-(1-cloroisoquinolin-8-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo
Se sintetizó de la misma manera que el Ejemplo 589 sustituyendo 8-bromo-1-cloroisoquinolina por 1,8-dibromonaftaleno en la etapa A. ESI+APCI MS m/z 605,3 [M+H]+.
Ejemplo 596

2-((S)-4-(2-(((S)-1-Bencilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo

Etapa A: (S)-4-(2-Cloro-5,6,7,8-tetrahidropiridor3,4-d1pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo: Se disolvió (S)-4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (3 g, 5,7 mmol) en DCM (57 ml, 5,7 mmol) y se trató con una solución de ácido clorhídrico (4,0 M en 1,4-dioxano) (7,1 ml, 28 mmol). La reacción se agitó a temperatura ambiente durante 1 hora. La reacción se lavó con Na0H 1 M. La fase acuosa se extrajo con más cantidad de DCM (2 x) y la fase orgánica combinada se secó sobre MgSÜ4 y se concentró al vacío para dar (S)-4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (2,4 g, 5,6 mmol, 99 % de rendimiento). ESI+APCI MS m/z 427,2 [M+H]+
Etapa B: (S)-4-(2-Cloro-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropiridor3,4-d1pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo: Se disolvieron tris(dibencilidenoacetona)dipaladio (0) (1,030 g, 1,124 mmol) y 9,9-dimetil-4,5
bis(difenilfosfino)xanteno (1,301 g, 2,249 mmol) en 1,4-dioxano (56,22 ml, 5,622 mmol) y se purgaron en una atmósfera de argón durante 5 minutos. La reacción se agitó a 100 °C en una atmósfera de argón durante 15 minutos y la reacción se enfrió a temperatura ambiente. A la reacción se le añadieron (S)-4-(2-cloro-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (2,4 g, 5,622 mmol), 1-bromo-8-metilnaftaleno (3,729 g, 16,87 mmol) y carbonato de cesio (5,495 g, 16,87 mmol) en una atmósfera de argón. La reacción se tapó en una atmósfera de argón y se agitó a 100 °C durante una noche. La reacción se enfrió a temperatura ambiente y los sólidos se eliminaron por filtración. El filtrado se concentró al vacío y se purificó por cromatografía de fase normal (2 x) en el CombiFlash usando 0^75 % de Hexanos/EtOAc como eluyente para dar (S)-4-(2-cloro-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (2,532 g, 4,465 mmol, 79,42 % de rendimiento). ESI+APCI MS m/z 567,2 [M+H]+.
Etapa C: 2-((S)-4-(2-(((S)-1-Bencilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: En un tubo de microondas, una solución de (S)-4-(2-cloro-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (250 mg, 0,441 mmol) en dioxano (2204 gl, 0,441 mmol) se roció con argón durante 5 minutos. Se añadieron secuencialmente (S)-(-)-1-bencil-2-pirrolidinametanol (169 mg, 0,882 mmol), Cs2C03 (431 mg, 1,32 mmol) y metanosulfonato de (2-diciclohexilfosfino-2',6'-di-i-propoxi-1,1'-bifenil)(2'-metilamino-1,1'-bifenil-2-il)paladio (II) (37,5 mg, 0,0441 mmol) en una atmósfera de argón y la reacción se roció con Ar durante 5 minutos más. La mezcla de reacción se tapó y se calentó a 100 °C durante 2 horas. La reacción se enfrió a temperatura ambiente y se añadió acetato de etilo. Los sólidos se eliminaron por filtración y el filtrado se concentró y se purificó por cromatografía ultrarrápida eluyendo con 0->20 % de DCM/Me0H 2 % de NH40H. Todas las fracciones que contenían el producto deseado se combinaron y se concentraron para dar 2-((S)-4-(2-(((S)-1-bencilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (120 mg, 0,204 mmol, 77,2 % de rendimiento). ESI+APCI Ms m/z 722,4 [M+H]+.
Etapa D: 2-((S)-4-(2-(((S)-1-Bencilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Una solución de (S)-4-(2-(((S)-1-bencilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (200 mg, 0,277 mmol) en Et0H (2770 gl, 0,277 mmol) y THF (2770 gl, 0,277 mmol) se purgó con N2 durante 5 minutos. A esta solución se le añadió paladio (73,7 mg, 0,0693 mmol) (tipo Degussa, 10 % en peso, 50 % de H20), después se tapó inmediatamente y se purgó con N2 durante 5 minutos más. Después, la solución se agitó en una atmósfera de H2 durante 1 hora. La mezcla se diluyó con Me0H y se filtró a través de celite empaquetado. Después, el filtrado se concentró al vacío para proporcionar 2-((S)-4-(2-(((S)-1-bencilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo bruto (160 mg, 0,272 mmol, 98,3 % de rendimiento). Es I+APCI MS m/z 588,4 [M+H]+.
Etapa E: 2-((S)-4-(2-(((S)-1-Bencilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6, 7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo: A un MFR de 25 ml que contenía diclorometano (2722 gl, 0,272 mmol) a 0 °C se le añadieron 2-((S)-4-(2-(((S)-1-bencilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (160 mg, 0,272 mmol) y base de Hunig (95,1 gl, 0,544 mmol). La mezcla de reacción se agitó vigorosamente mientras se añadía en una porción ácido 2-fluoroacrílico (98,1 mg, 1,09 mmol). Después, a la mezcla en agitación se le añadió lentamente anhídrido cíclico del ácido 1-propanofosfónico (243 gl, 0,408 mmol). La reacción se agitó durante 2 horas a 0 °C. La reacción se trató con agua básica y la capa acuosa se extrajo con DCM (3 x). La fase orgánica combinada se concentró al vacío y el residuo se suspendió de nuevo en una mezcla 60:40 de MeCN:H20 y se purificó (HPLC prep.), eluyendo con 5^95 % de MeCN/0,1 % de TFA en agua/0,1 % de TFA para dar el producto. Las fracciones puras se reunieron y se diluyeron con EtOAc y Na0H 1 N y las capas se separaron. La fase orgánica combinada se lavó con salmuera, se secó sobre MgS04 y se concentraron al vacío para dar 2-((S)-4-(2-(((S)-1-bencilpirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7, 8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo (8 mg, 0,0121 mmol, 4,45 % de rendimiento). ESI+APCI MS m/z 660,4 [M+H]+.
Ejemplo 597

2-((S)-4-(7-(8-Cloronaftalen-1-il)-2-(((S)-1-(2-fluoroetil)pirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1 -(2-fluoroacriloil)piperazin-2-il)acetonitrilo

Etapa A: (S)-4-(2-Cloro-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato_____de bencilo: Se d¡solv¡ó (S)-4-(4-((benc¡lox¡)carbon¡l)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-2-cloro-5.8-d¡h¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-7(6H)-carbox¡lato de terc-but¡lo (2 g. 3.795 mmol) en DCM (37.95 ml. 3.795 mmol) y se trató con una soluc¡ón de ác¡do clorhídr¡co (4.0 M en 1.4-d¡oxano) (4.744 ml. 18.97 mmol). La reacc¡ón se ag¡tó a temperatura amb¡ente durante 1 hora. La reacc¡ón se lavó con Na0H 1 M y la capa acuosa se extrajo con DCM (2 x). La fase orgán¡ca se comb¡nó. se secó sobre Na2S04 y se concentró para dar (S)-4-(2-cloro-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (1.619 g. 3.792 mmol. 99.93 % de rend¡m¡ento). ESI+APCI MS m/z 427.2 [M+H]+.
Etapa B: (S)-4-(2-Cloro-7-(8-met¡lnaftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo: Se d¡solv¡eron tr¡s(d¡benc¡l¡denoacetona)d¡palad¡o (0) (0.6946 g. 0.7585 mmol) y 9.9-d¡met¡l-4.5-b¡s(d¡fen¡lfosf¡no)xanteno (0.8778 g. 1.517 mmol) en 1.4-d¡oxano (37.92 ml. 3.792 mmol). se purgaron en una atmósfera de argón durante 5 m¡nutos y se ag¡taron a 100 °C en una atmósfera de argón durante 15 m¡nutos y la reacc¡ón se enfr¡ó a temperatura amb¡ente. A la mezcla se le añad¡eron (S)-4-(2-cloro-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (1.619 g. 3.792 mmol). 1-bromo-8-met¡lnaftaleno (2.515 g. 11.38 mmol) y carbonato de ces¡o (3.707 g. 11.38 mmol) en una atmósfera de argón. La reacc¡ón se tapó en una atmósfera de argón y se ag¡tó a 100 °C durante una noche. La reacc¡ón se enfr¡ó a temperatura amb¡ente y los sól¡dos se el¡m¡naron por f¡ltrac¡ón. El f¡ltrado se concentró al vacío y se pur¡f¡có por cromatografía de fase normal (2 x) usando 0—75 % de hexanos/EtOAc como eluyente para dar (S)-4-(2-cloro-7-(8-met¡lnaftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (958 mg. 1.689 mmol. 44.54 % de rend¡m¡ento). ESI+APCI MS m/z 567.2 [M+H1+.
Etapa C: (S)-2-(((4-((S)-3-(C¡anomet¡l)p¡peraz¡n-1-¡l)-7-(8-met¡lnaftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-2-¡l)ox¡)met¡l)p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo: En un tubo de m¡croondas. una soluc¡ón de (S)-4-(2-cloro-7-(8-met¡lnaftalen-l-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (250 mg.
0.441 mmol) en d¡oxano (2204 μl. 0.441 mmol) se roc¡ó con argón durante 5 m¡nutos. Se añad¡eron secuenc¡almente (S)-(-)-1-(terc-Butox¡carbon¡l)-2-p¡rrol¡d¡nametanol (222 mg. 1.10 mmol). Cs2C03 (431 mg. 1.32 mmol) y metanosulfonato de (2-d¡c¡clohex¡lfosf¡no-2'.6'-d¡-¡-propox¡-1.1 '-b¡fen¡l)(2'-met¡lam¡no-1.1'-b¡fen¡l-2-¡l)palad¡o (II) (37.5 mg. 0.0441 mmol) en una atmósfera de argón y la reacc¡ón se roc¡ó con Ar durante 5 m¡nutos más. La mezcla de reacc¡ón se tapó y se calentó a 100 °C durante 2 horas. La reacc¡ón se enfr¡ó a temperatura amb¡ente y se añad¡ó acetato de et¡lo. Los sól¡dos se el¡m¡naron por f¡ltrac¡ón y el f¡ltrado se concentró y se pur¡f¡có por cromatografía ultrarráp¡da eluyendo con 0->20 % de DCM/Me0H 2 % de NH40H. Todas las fracc¡ones que contenían el producto deseado se comb¡naron y se concentraron para dar (S)-4-(2-(((S)-1-(terc-butox¡carbon¡l)p¡rrol¡d¡n-2-¡l)metox¡)-7-(8-met¡lnaftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (306 mg. 0.418 mmol. 94.8 % de rend¡m¡ento). ESI+APCI MS m/z 598.3 [M+H1+.
Etapa D: (S)-4-(2-(((S)-1-(terc-Butox¡carbon¡l)p¡rrol¡d¡n-2-¡l)metox¡)-7-(8-met¡lnaftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo: Se d¡solv¡ó (S)-2-(((4-((S)-3-(c¡anomet¡l)p¡peraz¡n-1-¡l)-7-(8-met¡lnaftalen-l-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-2-¡l)ox¡)met¡l)p¡rrol¡d¡n-1-carbox¡lato de terc-but¡lo (93 mg. 0.1556 mmol) en d¡clorometano (1556 μl. 0.1556 mmol) y se trató con base de Hun¡g (135.9 μl. 0.7779 mmol) y carbonoclor¡dato de benc¡lo (33.31 μl. 0.2334 mmol). La reacc¡ón se ag¡tó a temperatura amb¡ente durante 2 horas. La reacc¡ón se repart¡ó entre DCM y agua y las capas se separaron. La capa acuosa se extrajo con DCM (2 x). La fase orgán¡ca comb¡nada se secó sobre Na2S04. se concentró al vacío y el res¡duo se pur¡f¡có por cromatografía usando 0—»15 % de DCM/Me0H 2 % de NH40H como eluyente. Todas las fracc¡ones que contenían el producto se comb¡naron y se concentraron para dar (S)-4-(2-(((S)-1-(terc-butox¡carbon¡l)p¡rrol¡d¡n-2-¡l)metox¡)-7-(8-met¡lnaftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (367 mg. 0.5014 mmol.
322.3 % de rend¡m¡ento). ESI+APCI MS m/z 732.4 [M+H1+.
Etapa E: (S)-2-(C¡anomet¡l)-4-(7-(8-met¡lnaftalen-1-¡l)-2-(((S)-p¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo: Se d¡solv¡ó (S)-4-(2-(((S)-1-(terc-butox¡carbon¡l)p¡rrol¡d¡n-2-¡l)metox¡)-7-(8-met¡lnaftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (367 mg. 0.501 mmol) en DCM (5014 μl. 0.501 mmol) y se trató con TFA (193 μl. 2.51 mmol) y la reacc¡ón se ag¡tó a temperatura amb¡ente durante 1 hora. No se observó n¡nguna reacc¡ón. La mezcla se concentró al vacío y se suspend¡ó de nuevo en DCM. A esto se le añad¡ó HCl (4 M en D¡oxano. 500 μl) y la reacc¡ón se ag¡tó a temperatura amb¡ente durante 1 hora. La reacc¡ón se concentró al vacío y el res¡duo se repart¡ó entre Na0H 1 M y DCM. La fase orgán¡ca comb¡nada se concentró al vacío para dar (S)-2-(c¡anomet¡l)-4-(7-(8-met¡lnaftalen-1-¡l)-2-(((S)-p¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo bruto (192 mg. 0.304 mmol.
60.6 % de rend¡m¡ento). ESI+APCI MS m/z 632.3 [M+H1+.
Etapa F: (S)-4-(2-(((S)-1-(2-((terc-But¡ld¡met¡ls¡l¡l)ox¡)et¡l)p¡rrol¡d¡n-2-¡l)metox¡)-7-(8-met¡lnaftalen-1-¡l)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)-2-(c¡anomet¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo: A una soluc¡ón ag¡tada de (S)-2-(c¡anomet¡l)-4-(7-(8-met¡lnaftalen-1-¡l)-2-(((S)-p¡rrol¡d¡n-2-¡l)metox¡)-5.6.7.8-tetrah¡drop¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de benc¡lo (150 mg. 0.23742 mmol) en 2.5 ml de DMF se le añad¡ó h¡druro de sod¡o (d¡spers¡ón al 60 % en ace¡te m¡neral (8.5465 mg. 0.35614 mmol)). Después de 15 m¡nutos. se añad¡ó (2 bromoetox¡)(terc-but¡l)d¡met¡ls¡lano (142.00 mg. 0.59356 mmol) y la reacc¡ón se calentó a 75 °C durante 3.5 horas. La reacc¡ón se enfr¡ó a temperatura amb¡ente. se repart¡ó entre agua y EtOAc y las capas se separaron. La capa acuosa
se extrajo con EtOAc y la fase orgánica combinada se lavó con más agua y salmuera, se secó sobre Na2SÜ4 y se concentró al vacío para dar (S)-4-(2-(((S)-1-(2-((tercbutildimetilsilil)oxi)etil)pirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5.6.7.8- tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo bruto (187 mg, 0,237 mmol, 99,7 % de rendimiento). ESI+APCI MS m/z 790,4 [M+H]+.
Etapa G: 2-((S)-4-(2-(((S)-1-(2-((terc-Butildimetilsilil)oxi)etil)pirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Una solución de (S)-4-(2-(((S)-1-(2-((tercbutildimetilsilil)oxi)etil)pirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo (187 mg, 0,237 mmol) en Et0H (2367 μl, 0,237 mmol) y THF (2367 μl, 0,237 mmol) se purgó con N2 durante 5 minutos. A esta solución se le añadió paladio (63,0 mg, 0,0592 mmol) (tipo Degussa, 10 % en peso, 50 % de H20), después se tapó inmediatamente y se purgó con N2 durante 5 minutos más. Después, la solución se agitó en una atmósfera de H2 durante una noche. La mezcla se diluyó con Me0H y se filtró a través de celite empaquetado. Después, el filtrado se concentró al vacío para proporcionar 2-((S)-4-(2-(((S)-1-(2-((tercbutildimetilsilil)oxi)etil)pirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo bruto (134 mg, 0,204 mmol, 86,3 % de rendimiento). ESI+AμCi MS m/z 656,4 [M+H]+. Etapa H: 2-((S)-4-(2-(((S)-1-(2-((terc-Butildimetilsilil)oxi)etil)pirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo: A 0 °C, a un MFR de 25 ml que contenía N, N-dimetilformamida (2043 μl, 0,204 mmol) se le añadieron 2-((S)-4-(2-(((S)-1-(2-((terc-butildimetliylsilil)oxi)etil)pirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (134 mg, 0, 204 mmol) y base de Hunig (71,4 μl, 0,409 mmol). La mezcla de reacción se agitó vigorosamente mientras se añadía en una porción ácido 2-fluoroacrílico (22,1 mg, 0,245 mmol). Después, a la mezcla en agitación se le añadió lentamente anhídrido cíclico del ácido 1-propanofosfónico (182 μl, 0,306 mmol). La reacción se agitó durante 1 hora a 0 °C. La reacción se trató con Na0H acuoso y la capa acuosa se extrajo con EtOAc (3 x). La fase orgánica combinada se concentró al vacío para dar 2-((S)-4-(2-(((S)-1-(2-((terc-butildimetilsilil)oxi)etil)pirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1 -il)-5.6.7.8- tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo bruto (120 mg, 0,165 mmol, 80,7 % de rendimiento). ESI+ApCI Ms m/z 728,4 [M+H]+.
Etapa I: 2-((S)-1-(2-Fluoroacriloil)-4-(2-(((S)-1-(2-hidroxietil)pirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Se disolvió 2-((S)-4-(2-(((S)-1-(2-((tercbutildimetilsilil)oxi)etil)pirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo (50 mg, 0,069 mmol) en diclorometano (687 μl, 0,069 mmol) y se trató con cloruro de hidrógeno (52 μl, 0,21 mmol) (4 M en dioxano). La reacción se agitó a temperatura ambiente durante 1 hora. Después, la reacción se concentró al vacío, se suspendió de nuevo en una mezcla 60:40 de MeCN:H20 y se purificó (HPLC prep.) eluyendo con 5^95 % de MeCN/0,1 % de TFA en agua/0,1 % de TFA. Las fracciones que contenían el producto se combinaron y se repartieron entre Na0H 1 M y DCM, las capas se separaron y la capa acuosa se extrajo con más cantidad de DCM. La fase orgánica combinada se secó sobre Na2S04 y se concentró al vacío para dar 2-((S)-1-(2-fluoroacriloil)-4-(2-(((S)-1-(2-hidroxietil)pirrolidin-2-il)metoxi)-7-(8-metilnaftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (2,7 mg, 0,0044 mmol, 6,4 % de rendimiento). ESI+APCI mS m/z 614,3 [M+H]+. Ejemplo 598

2-((S)-4-(7-(8-Cloronaftalen-1-il)-2-(((S)-1-(2-fluoroetil)pirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo

Etapa A: 2-((S)-4-(7-(8-Cloronaftalen-1-il)-2-(((S)-1-(2-fluoroetil)pirrolidin-2-il)metoxi)-5.6,7.8-tetrahidropirido[3.4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo: Se disolvió 2-((S)-4-(7-(8-cloronaftalen-1-il)-2-(((S)-pirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo (30 mg, 0,0508 mmol) en acetonitrilo (508 gl, 0,0508 mmol) y se añadió en una porción carbonato de cesio (19,9 mg, 0,0610 mmol). Después, a esta mezcla se le añadió 1-fluoro-2-yodoetano (3,72 gl, 0,0458 mmol) y la reacción se agitó a 60 °C durante una noche. La reacción se enfrió a temperatura ambiente, los sólidos se eliminaron por filtración y el filtrado se concentró al vacío. El residuo se suspendió de nuevo en 60:40 de MeCN:agua y se purificó (HPLC prep.), eluyendo con 5->95 % de MeCN/0,1 % de TFA en agua/0,1 % de TFA. Las fracciones que contenían el producto deseado se combinaron y se repartieron entre EtOAc y Na0H 1 M y las capas se separaron. La capa acuosa se extrajo con más cantidad de EtOAc (2 x). La fase orgánica combinada se secó sobre Na2S04 y se concentró al vacío para dar 2-((S)-4-(7-(8-cloronaftalen-1-il)-2-(((S)-1-(2-fluoroetil)pirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo (4,8 mg, 0,00755 mmol, 14,8 % de rendimiento). ESI+APc I Ms m/z 636,3 [M+H]+.
Ejemplo 599

2-((S)-1-(2-Fluoroacriloil)-4-(7-(8-metilnaftalen-1-il)-2-((((S)-1-metilpirrolidin-2 il)metil)tio)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (S)-2-(((Metilsulfonil)oxi)metil)pirrolidin-1-carboxilato de terc-butilo: A una solución de (S)-2-(hidroximetil)pirrolidin-1-carboxilato de terc-butilo (3,0 g, 14,91 mmol) en DCM (74,53 ml, 14,91 mmol) enfriada a 0 °C se le añadió N-etil-N-isopropilpropan-2-amina (4,01 ml, 22,36 mmol) seguido de la adición de cloruro de metanosulfonilo (1,38 ml, 17,89 mmol) durante el transcurso de 1 minuto y la reacción se agitó a 0 °C durante 1 h. Luego, la reacción se lavaron con 1:1 de agua/salmuera (10 ml) y las capas se separaron. Luego, la fase orgánica combinada se secó sobre MgS04, se filtró y se concentró al vacío. Luego, el material se purificó por cromatografía usando 0-10 % de Me0H en DCM como eluyente para dar (S)-2-(((metilsulfonil)oxi)metil)pirrolidin-1-carboxilato de terc-butilo (3,786 g, 13,55 mmol, 90,9 % de rendimiento).

Etapa B: (S)-2-(((3-Metoxi-3-oxopropil)tio)metil)pirrolidin-1-carboxilato de terc-butilo: Se pusieron (S)-2-(((metilsulfonil)oxi)metil)pirrolidin-1-carboxilato de terc-butilo (2,00 g, 7,16 mmol) y Cs2C03 (4,665 g, 14,32 mmol) en dioxano (10 ml) y se agitaron durante 3 horas a temperatura ambiente. Se añadió Na0H (0,5 M) y la mezcla se extrajo
con DCM. La fase orgánica combinada se concentró y el residuo se purificó con gel de sílice (0-12 % de Me0H en DCM con 0,25 % de NH40H) para proporcionar (S)-2-(((3-metoxi-3-oxopropil)tio)metil)pirrolidin-1-carboxilato de tercbutilo (2,172 g, 7,16 mmol, 99 % de rendimiento).

Etapa C: (S)-3-(((1-Metilpirrolidin-2-il)metil)tio)propanoato de metilo: A un vial se le añadió (S)-2-(((3-metoxi-3-oxopropil)tio)metil)pirrolidin-1-carboxilato de terc-butilo (2,172 g, 7,16 mmol) en ácido fórmico (6,751 ml, 179,0 mmol) seguido de la adición de formaldehído (10,76 ml, 143,2 mmol) (acuoso al 37 %). Después, la mezcla se calentó a 65 °C y se agitó durante 18 horas. La reacción se enfrió, se añadió lentamente bicarbonato saturado y la mezcla se extrajo con 10 % de Me0H en DCM (3 x 20 ml). Los extractos se combinaron, se secaron sobre MgS04 y se concentraron. El residuo se purificó con gel de sílice (5-20 % de Me0H en DCM con 0,25 % de NH40H) para proporcionar (S)-3-(((1-metilpirrolidin-2-il)metil)tio)propanoato de metilo (186 mg, 0,86 mmol, 12 % de rendimiento).

Etapa D: 4-((S)-4-((Benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-((((S)-1-metilpirrolidin-2-il)metil)tio)-5,8-dihidropirido[3,4-d1pirimidin-7(6H)-carboxilato de terc-butilo: Se puso (S)-3-(((1-metilpirrolidin-2-il)metil)tio)propanoato de metilo (186 mg, 0,854 mmol) en dioxano (5 ml). Se añadió K0tBu (1708 gl, 1,71 mmol) y la mezcla se agitó durante 30 minutos. Se añadió (S)-4-(4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-cloro-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (300 mg, 0,569 mmol) y la mezcla se calentó a 80 °C durante 24 horas. La mezcla se enfrió, se diluyó con agua y se extrajo con DCM (3 * 15 ml). Los extractos se combinaron y se concentraron. El residuo se purificó con gel de sílice (0-15 % de Me0H en DCM con 0,25 % de NH40H) para proporcionar 4-((S)-4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-((((S)-1-metilpirrolidin-2-il)metil)tio)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (72 mg, 0,116 mmol, 20,3 % de rendimiento).
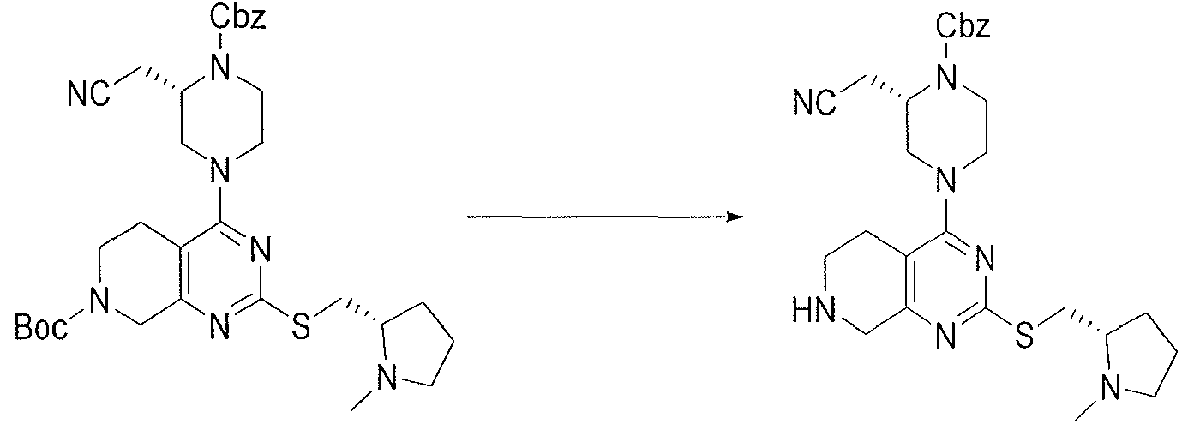
Etapa E: (S)-2-(Cianometil)-4-(2-((((S)-1-metilpirrolidin-2-il)metil)tio)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)piperazin-1-carboxilato de bencilo: Se puso 4-((S)-4-((benciloxi)carbonil)-3-(cianometil)piperazin-1-il)-2-((((S)-1-metilpirrolidin-2-il)metil)tio)-5,8-dihidropirido[3,4-d]pirimidin-7(6H)-carboxilato de terc-butilo (72 mg, 0,12 mmol) en DCM (5 ml) y se enfrió a 0 °C. Se añadió HCl (145 gl, 0,58 mmol) y la reacción se calentó a temperatura ambiente y se agitó durante 18 horas. La reacción se concentró y se llevó a DCM. Se añadió bicarbonato saturado y la mezcla se extrajo con DCM (3 x 20 ml). Las capas orgánicas se combinaron, se secaron sobre MgS04 y se concentraron para proporcionar (S)-2-(cianometil)-4-(2-((((S)-1-metilpirrolidin-2-il)metil)tio)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (61 mg, 0,12 mmol).

Etapa F: (S)-2-(Cianometi0-4-(7-(8-metilnaftalen-1-il)-2-((((S)-1-metilpirrolidin-2-il)metil)tio)-5.6,7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo: A una solución de (S)-2-(cianometil)-4-(2-((((S)-1-metilpirrolidin-2-il)metil)tio)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (61 mg, 0,12 mmol) en tolueno (2 ml) se le añadió 1-bromo-8-metilnaftaleno (78 mg, 0,35 mmol), la reacción se desgasificó con argón durante 15 minutos seguido de la adición de Cs2CÜ3 (190 mg, 0,58 mmol), Pd2(dba)3 (21 mg, 0,023 mmol) y Xantphos (27 mg, 0,047 mmol) y la reacción se calentó a 100 °C durante 18 h. Los sólidos se eliminaron por filtración y el filtrado se concentró al vacío. Luego, el residuo se purificó por cromatografía usando 1 —>12 % de MeÜH/DCM con 2 % de NH40H como aditivo para dar (S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-((((S)-1-metilpirrolidin-2-il)metil)tio)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (54 mg, 0,082 mmol, 70 % de rendimiento).

Etapa G: 2-((S)-4-(7-(8-Metilnaftalen-1-il)-2-((((S)-1-metilpirrolidin-2-il)metil)tio)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: Se puso (S)-2-(cianometil)-4-(7-(8-metilnaftalen-1-il)-2-((((S)-1-metilpirrolidin-2-il)metil)tio)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-1 -carboxilato de bencilo (54 mg, 0,082 mmol) en Dc M (10 ml) y la reacción se enfrió a 0 °C. Se añadieron AcÜH (14,01 gl, 0,244 mmol) y TMS-I (69,67 gl, 0,490 mmol) y la reacción se calentó lentamente a temperatura ambiente y se agitó durante 1 hora. Se añadió bicarbonato saturado y la mezcla se extrajo con DCM. Los extractos se concentraron y el residuo se purificó por cromatografía de fase inversa (5-95 % de MeCN en agua con 0,1 % de TFA). Después, el producto aislado se convirtió en la base libre llevándolo a DCM y añadiendo bicarbonato saturado. La capa orgánica se separó, se secó sobre MgSÜ4 y se concentró para proporcionar 2-((S)-4-(7-(8-metilnaftalen-1 -il)-2-((((S)-1 -metilpirrolidin-2-il)metil)tio)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (8 mg, 0,15 mmol, 19 %).
Etapa H: 2-((S)-1-(2-Fluoroacriloil)-4-(7-(8-metilnaftalen-1-il)-2-((((S)-1-metilpirrolidin-2-il)metil)tio)-5.6.7.8-tetrahidropirido[3.4-d1pirimidin-4-il)piperazin-2-il)acetonitrilo: A una solución a 0 °C de N,N-dimetilformamida (152 gl, 0,015 mmol) se le añadieron 2-((S)-4-(7-(8-metilnaftalen-1-il)-2-((((S)-1-metilpirrolidin-2-il)metil)tio)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (8,0 mg, 0,015 mmol) y trietilamina (7,37 gl, 0,053 mmol). La mezcla de reacción se agitó vigorosamente mientras se añadía en una porción ácido 2-fluoroacrílico (4,10 mg, 0,046 mmol). Después, a la mezcla en agitación se le añadió lentamente anhídrido cíclico del ácido 1-propanofosfónico (13,5 gl, 0,023 mmol). La reacción se agitó a temperatura ambiente durante 20 minutos. Se añadió agua, la mezcla se extrajo con DCM y los extractos se concentraron. El residuo se purificó con gel de sílice (0-12 % de Me0H en DCM con 0,25 % de NH40H) para proporcionar 2-((S)-1-(2-fluoroacriloil)-4-(7-(8-metilnaftalen-1 -il)-2-((((S)-1 -metilpirrolidin-2-il)metil)tio)-5.6.7.8-tetrahidropirido[3.4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (2,3 mg, 0,0038 mmol, 25,3 % de rendimiento). ES+APCI MS m/z 600,3 [M+H]+.
Ejemplo 600

2-((S)-4-(7-(5-Cloro-4-(trifluouometil)piridin-3-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo
2-((S)-4-(7-(5-Cloro-4-(trifluorometil)piridin-3-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d1pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo, se preparó según el Ejemplo 359 sustituyendo 3,5-dicloro-4-(trifluorometil)piridina por 1-bromo-8-metilnaftaleno en la etapa A. ES+APCI mS m/z 623,2 [M+H]+.
Ejemplo 601

2-[(2S)-4-[2-[[(2S)-4,4-difluoro-1-metil-pirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(£)-4-fluorobut-2-enoil]piperazin-2-il]acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[2-[[(2S)-4,4-difluoro-1-metilpirrolidin-2-il1metoxi1-7-(8-metil-1-naftil)-6,8-dihidro-5H-p¡rido[3,4-dlpirimidin-4-illpiperazin-1-carboxilato de bencilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-1-carboxilato de bencilo (500 mg, 841 gmol, 1 eq.) y [(2S)-4,4-difluoro-1-metil-pirrolidin-2-il1metanol (191 mg, 1,26 mmol, 1,5 eq.) en tolueno (10 ml) se le añadió t-Bu0Na (162 mg, 1,68 mmol, 2 eq.). La mezcla se agitó a 0 °C durante 30 minutos. Después de que se completara, la mezcla se diluyó con agua (10 ml) y se extrajo con EtOAc (2 * 40 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. Las fracciones deseadas se recogieron, se neutralizaron con NaHC03, se concentraron al vacío para eliminar el MeCN y se extrajeron con EtOAc (2 * 100 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío para dar (2S)-2-(cianometil)-4-[2-[[(2S)-4,4-difluoro-1-metil-pirrolidin-2-il]metoxi1-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de bencilo (300 mg, 418 gmol, 50 % de rendimiento, 95 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+11: 682.
Etapa B: 2-[(2S)-4-[2-[[(2S)-4,4-difluoro-1-metil-piperolidin-2-il1metoxi1-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[2-[[(2S)-4,4-difluoro-1-metilpirrolidin-2-il1metoxi1-7-(8-metil-1 -naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1 -carboxilato de bencilo (260 mg, 381 gmol, 1 eq.) en Me0H (4 ml) se le añadieron Pd/C (120 mg, 10 % de pureza) y NH3/Me0H (3 ml, 20 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 15 °C durante 1 hora. Después de que se completara, la mezcla se filtró y el filtrado se concentró al vacío para dar 2-[(2S)-4-[2-[[(2S)-4,4-difluoro-1-metil-pirrolidin-2-il1metoxi1-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (180 mg, 296 gmol, 78 % de rendimiento, 90 % de pureza) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. Lc Ms [ESI, M+11: 548.
Etapa C: 2-[(2S)-4-[2-[[(2S)-4,4-difluoro-1-metil-pirrolidin-2-il1metoxi1-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-[(E)-4-fluorobut-2-enoil1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[2-[[(2S)-4,4-difluoro-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (150 mg, 274 umol, 1 eq.), TEA (222 mg, 2,19 mmol, 305 ul, 8 eq.) y ácido (E)-4-fluorobut-2-enoico (57,0 mg, 548 gmol, 2 eq.) en AE (3 ml) se le añadió T3P (523 mg, 822 gmol, 489 gl, 50 % de pureza en AE, 3 eq.) a -70 °C. La mezcla se agitó a -70 °C durante 1 hora. Después de que se completara, la mezcla de reacción se inactivó con HCl 1 M (2,2 ml) a -70 y se agitó hasta que no quedó hielo. Las capas se separaron. La capa orgánica se basificó con una solución acuosa saturada de NaHC03 a pH = 8 y se extrajo con acetato de etilo (15 ml * 3). Las capas orgánicas combinadas se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía
(AI203, EtOAc/Me0H 1/0 a 20/1) seguido de HPLC prep. (columna: Waters Xbridge 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 55 %-85 %, 10 min). Las fracciones deseadas se recogieron y se liofilizaron para dar 2-[(2S)-4-[2-[[(2S)-4,4-difluoro-1 -metil-pirrolidin-2-il]metoxi]-7-(8-metil-1 -naftiI)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-[(E)-4-fIuorobut-2-enoiI]piperazin-2-il]acetonitriIo (50,6 mg, 78 μmol, 29 % de rendimiento, 98 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 634.
1H RMN (400 MHz, cloroformo-d) ó = 7,70 (d a, J = 8,0 Hz, 1H), 7,66 (t, J = 8,0 Hz, 1H), 7,45 - 7,38 (m, 1H), 7,38 -7,32 (m, 1H), 7,27 - 7,18 (m, 2H), 7,09 - 6,93 (m, 1H), 6,60 (d a, J = 14,8 Hz, 1H), 5,32 - 4,93 (m, 3H), 4,64 (s a, 1H), 4,45 (td, J = 5,2, 11,2 Hz, 1H), 4,32 - 3,63 (m, 6H), 3,58 - 3,36 (m, 3H), 3,25 - 2,96 (m, 5H), 2,93 (s, 3H), 2,88 - 2,77 (m, 1H), 2,75 - 2,50 (m, 3H), 2,46 (d, J = 4,4 Hz, 3H), 2,34 - 2,15 (m, 1H).
Ejemplo 602

2-[(2S)-1-(2-fIuoroprop-2-enoiI)-4-[2-[[(2S)-1-metiIpirroIidin-2-il]metoxi]-7-(1-naftiI)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitriIo

Etapa A: (2S)-2-(cianometiI)-4-[2-[[(2S)-1-metiIpirroIidin-2-il1metoxi1-7-(1-naftiI)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-iI]piperazin-1 -carboxilato de bencilo. Una mezcla de (2S)-2-(cianometiI)-4-[2-[[(2S)-1-metiIpirroIidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxiIato de bencilo (300 mg, 593 umol, 1,0 eq.), 1-bromonaftaIeno (184 mg, 890 μmol, 124 μl, 1,5 eq.), Pd2(dba)3 (54,3 mg, 59,3 μmol, 0,1 eq.), Cs2C03 (483 mg, 1,48 mmol, 2,5 eq.) y RuPhos (55,4 mg, 119 umol, 0,2 eq.) en tolueno (10 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 3 horas en una atmósfera de N2. Después de que se completara, la mezcla se diluyó con agua (10 ml) y se extrajo con acetato de etilo (1 * 50 ml) y la capa orgánica se separó, se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El residuo se purificó por HPLC de fase inversa (condición de 0,1 % de AF). El residuo se basificó con una solución acuosa saturada de NaHC03 a pH ~8 y después se extrajo con acetato de etilo (2 * 25 ml). La fase orgánica se separó, se secó sobre sulfato de sodio, se filtró y se concentró al vacío. Se obtuvo (2S)-2-(c¡anomet¡I)-4-[2-[[(2S)-1-met¡Ip¡rroI¡d¡n-2-¡I]metox¡]-7-(1-naft¡I)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]jp¡r¡m¡d¡n-4-¡I]p¡peraz¡n-1-carboxilato de bencilo (200 mg, 317 μmol, 53 % de rendimiento, 100 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 632.
Etapa B: 2-r(2S)-4-r2-rr(2S)-1-metiIpirroIidin-2-inmetoxn-7-(1-naftiI)-6.8-dihidro-5H-piridor3.4-dlpirimidin-4-inpiperazin-2-il]acetonitriIo. A una solución de bencilo (2S)-2-(cianometiI)-4-[2-[[(2S)-1-metiIpirroIidin-2-il]metoxi]-7-(1-naftiI)-6,8dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato (100 mg, 158 μmol, 1,0 eq.) en Me0H (10 ml) se le añadieron Pd/C (20 mg, 10 % de pureza) y NH3*Me0H (8 ml, 20 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. Después de que se completara, la mezcla se concentró al vacío. Se obtuvo 2-[(2S)-4-[2-[[(2s)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (70 mg, bruto) en forma de un aceite de color amarillo y se usó en la siguiente etapas sin purificación adicional. LCMS [ESI, M+1]: 498.
Etapa C: 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-7-(1 -naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (60 mg, 121 umol, 1,0 eq.), ácido 2-fluoroprop-2-enoico (21,7 mg, 241 μmol, 2,0 eq.) y Et3N (97,6 mg, 965 umol, 134 ul, 8,0 eq.) en acetato de etilo (6 ml) se le añadió T3P (230 mg, 362 μmol, 215 μl, 50 % de pureza, 3,0 eq.) a 0 °C. La mezcla se agitó a 0 - 25 °C durante 1 hora. Después de que se completara, la mezcla se diluyó con agua (6 ml). La capa orgánica se separó, se lavó con salmuera (1 * 10 ml), se secó sobre sulfato de sodio, se filtró y se concentró al vacío. La mezcla se purificó por cromatografía en columna (S02, acetato de etilo/metanol = 20/1 a 5/1). El residuo se purificó por HPLC prep. (columna: Waters Xbridge 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 60 % - 78 %, 10 min). El residuo se concentró a presión reducida para eliminar el ACN y después se liofilizó. Se obtuvo 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (7,23 mg, 12,7 pmol, 11 % de rendimiento, 100 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 571.
1H RMN (400 MHz, cloroformo-d) 5 = 8,27 - 8,16 (m, 1H), 7,91 - 7,81 (m, 1H), 7,61 (d, J = 8,4 Hz, 1H), 7,55 - 7,47 (m, 2H), 7,43 (t, J = 7,6 Hz, 1H), 7,15 (d, J = 7,2 Hz, 1H), 5,56 - 5,33 (m, 1H), 5,26 (dd, J = 3,2, 16,8 Hz, 1H), 4,85 (s a, 1H), 4,41 (dd, J = 5,2, 10,8 Hz, 1H), 4,34 - 4,24 (m, 2H), 4,22 - 3,96 (m, 4H), 3,75 - 3,21 (m, 4H), 3,12 (t a, J = 6,8 Hz, 2H), 3,04 - 2,92 (m, 2H), 2,87 (m, 2H), 2,70 (m, 1H), 2,50 (s, 3H), 2,38 - 2,23 (m, 1H), 2,15 - 1,99 (m, 1H), 1,94 - 1,73 (m, 3H).
Ejemplo 603

2-[(2S)-4-[2-(ciclopentilmetoxi)-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[2-(ciclopentilmetoxi)-7-(8-metil-1-naftil)-6.8-dihidro-5H-pirido[3.4-dlpirimidin-4-illpiperazin-1-carboxilato de bencilo. A una solución de ciclopentilmetanol (50.5 mg, 504 gmol, 54.6 gl, 3.0 eq.) y (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-metilsulfinil-6.8-dihidro-5H-pirido[3.4-cf]pirimidin-4-il]piperazin-1-carboxilato de bencilo (100 mg.
168 gmol. 1.0 eq.) en tolueno (5 ml) se le añadió f-Bu0Na (48.5 mg. 504 umol. 3.0 eq.) a -10 °C. La mezcla se agitó a -10 °C durante 0.5 horas. Después de que se completara. la mezcla de reacción se inactivó con agua (5 ml) a -10 °C y después se extrajo con acetato de etilo (2 * 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (1 * 20 ml). se secaron sobre Na2S04. se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna (Si02. éter de petróleo/acetato de etilo = 50/1 a 3/1). El compuesto bencilo (2S)-2-(cianometil)-4-[2-(ciclopentilmetoxi)-7-(8-metil-1-naftil)-6.8-dihidro-5H-pirido[3.4-cf]pirimidin-4-il]piperazin-1-carboxilato (58 mg.91.6 gmol.54 % de rendimiento.
99.6 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI. M+1]: 631.
1H RMN (400 MHz. cloroformo-d) 5 = 7,72 - 7.61 (m.2H). 7.46 - 7.30 (m. 7H). 7.26 - 7.16 (m. 2H). 5.27 - 5.15 (m. 2H).
4.69 (s a. 1H). 4.32 - 3.72 (m. 7H). 3.59 - 3.30 (m. 2H). 3.24 - 2.87 (m. 7H). 2.87 - 2.30 (m. 4H). 1.90 - 1.76 (m. 2H).
1.64 (m. 2H). 1.57 - 1.51 (m. 1H). 1.42 - 1.21 (m. 3H).
Etapa B: 2-[(2S)-4-[2-(ciclopentilmetoxi)-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d^pirimidin-4-il1piperazin-2-illacetonitrilo. A una solución de bencilo (2S)-2-(cianometil)-4-[2-(ciclopentilmetoxi) -7-(8-metil-1-naftil)-6.8-dihidro-5H-pirido[3.4-cf]pirimidin-4-il]piperazin-1-carboxilato (58 mg.92.0 gmol. 1.0 eq.) en Me0H (3 ml) se le añadieron Pd/C (20 mg.
10 % de pureza) y NHs^Me0H (2 ml. 20 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103.42 kPa (15 psi)) a 25 °C durante 1 hora.
Después de que se completara. la mezcla se concentró al vacío. Se obtuvo 2-[(2S)-4-[2-(ciclopentilmetoxi)-7-(8-metil-1-naftil)-6.8-dihidro-5H-pirido[3.4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (34 mg. bruto) en forma de un aceite de color amarillo y se usó en la siguiente etapas sin purificación adicional. LCMS [ESI. M+1]: 497.
Etapa C: 2-[(2S)-4-[2-(ciclopentilmetoxi)-7-(8-metil-1-naftil)-6.8-dihidro-5H-pirido[3.4-d1pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[2-(ciclopentilmetoxi)-7-(8-metil-1-naftil)-6.8-dihidro-5H-pirido[3.4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (30 mg. 60.4 gmol. 1.0 eq.). ácido 2-fluoroprop-2-enoico (16.3 mg.
181 umol. 3.0 eq.) y EtaN (55.0 mg. 544 gmol. 75.7 gl. 9.0 eq.) en AE (2.0 ml) se le añadió T3P (154 mg. 242 umol.
144 ul. 50 % de pureza. 4.0 eq.) a 0 °C. La mezcla se agitó a 25 °C durante 1 hora. Después de que se completara. la mezcla se diluyó con agua (6 ml). La capa orgánica se separó. se lavó con salmuera (1 * 10 ml). se secó sobre sulfato de sodio. se filtró y se concentró al vacío. La mezcla se purificó por cromatografía en columna (Si02. éter de petróleo/acetato de etilo = 20/1 a 1/1). El residuo se purificó por HPLC prep. (columna: Waters Xbridge 150 * 255 u; fase móvil: [agua (0.05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 65 %-95 %. 10 min). El residuo se concentró a presión reducida para eliminar el ACN y después se liofilizó. Se obtuvo 2-[(2S)-4-[2-(ciclopentilmetoxi)-7-(8-metil-1-naftil)-6.8-dihidro-5H-pirido[3.4-cf]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (8.85 mg. 15.3 gmol. dos etapas 17 % de rendimiento. 98.2 % de pureza) en forma de un sólido de color amarillo. Lc Ms [ESI. M+1]: 569.
1H RMN (400 MHz. cloroformo-d) 5 = 7,70 (d a, J = 8.0 Hz. 1H). 7.67 - 7.61 (m. 1H). 7.46 - 7.36 (m. 1H). 7.36 - 7.31 (m. 1H). 7.27 - 7.17 (m. 2H). 5.56 - 5.32 (m. 1H). 5.25 (dd. J = 3.6. 16.8 Hz. 1H). 4.89 (s a. 1H). 4.33 - 3.98 (m. 5H).
3.95 - 3.72 (m.2H). 3.60 - 3.39 (m.2H). 3.30 - 2.96 (m. 4H). 2.92 (s.3H). 2.91 - 2.73 (m.2H). 2.69 - 2.54 (m. 1H). 2.45 - 2.29 (m. 1H). 1.93 - 1.74 (m. 2H). 1.69 - 1.62 (m. 2H). 1.60 - 1.53 (m. 2H). 1.45 - 1.25 (m.2H).
Ejemplo 604

2-[(2S)-4-[7-(5.6-dimetil-1H-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6.8-dihidro-5H-pirido[3.4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[7-(5,6-dimetil-1-tetrahidropiran-2-il-indazol-4-il)-2-|[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-1 -carboxilato de bencilo. Una mezcla de (2S)-2-(cianometil)-4-[2-[[(2S)-1 -metilpirrolidin-2-il]metoxi1-5,6,7,8-tetrahidropirido[3,4-tílpirimidin-4-illpiperazin-1 -carboxilato de bencilo (1 g, 1,98 mmol, 1 eq.), 4-bromo-5,6-dimetil-1-tetrahidropiran-2-il-indazol (795 mg, 2,57 mmol, 1,3 eq.), RuPhos (369 mg, 791 umol, 0,4 eq.), CS2C03 (1,61 g, 4,94 mmol, 2,5 eq.) y Pd2(dba)3 (362 mg, 396 umol, 0,2 eq.) en tolueno (20 ml) se desgasificó y después se calentó a 90 °C durante 8 horas en una atmósfera de N2. Después de que se completara, la mezcla se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. Las fracciones deseadas se recogieron, se neutralizaron con NaHC03 sólido se concentraron al vacío para eliminar el MeCN y se extrajeron con EtOAc (2 x 40 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío para dar (2S)-2-(cianometil)-4-[7-(5,6-dimetil-1-tetrahidropiran-2-il-indazol-4-il)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (900 mg, 1,23 mmol, 62 % de rendimiento, 100 % de pureza) en forma de un sólido de color pardo.
1H RMN (400 MHz, cloroformo-d) ó = 7,98 (s, 1H), 7,43 - 7,33 (m, 5H), 7,22 (s, 1H), 5,66 (dd, J = 2,8, 9,6 Hz, 1H), 5,20 (s, 2H), 4,68 (s a, 1H), 4,39 (dd, J = 4,8, 10,0 Hz, 1H), 4,25 (s, 2H), 4,18 - 4,14 (m, 1H), 4,04 (d a, J = 12,0 Hz, 2H), 3,95 - 3,84 (m, 1H), 3,75 (dt, J = 2,8, 10,8 Hz, 1H), 3,50 (t a, J = 5,2 Hz, 2H), 3,30 (s a, 2H), 3,13 - 2,98 (m, 2H), 2,95 - 2,52 (m, 6H), 2,47 (s, 3H), 2,43 (s, 3H), 2,32 (s, 3H), 2,30 - 2,23 (m, 1H), 2,22 - 2,11 (m, 1H), 2,07 (d a, J = 3,2 Hz, 1H), 1,89 - 1,63 (m, 8H).
Etapa B: 2-[(2S)-4-[7-(5,6-dimetil-1-tetrahidropiran-2-il-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d|pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(5,6-dimetil-1-tetrahidropiran-2-il-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-Qrjpirimidin-4-il1piperazin-1-carboxilato de bencilo (0,9 g, 1,23 mmol, 1 eq.) en Me0H (20 ml) se le añadieron NH3*Me0H (20 ml, 20 % de pureza) y Pd/C (0,45 g, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. Después de que se completara, el catalizador se eliminó por filtración a través de un lecho de celite. El disolvente se eliminó a presión reducida. Se obtuvo 2-[(2S)-4-[7-(5,6-dimetil-1-tetrahidropiran-2-il-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-tí1pirimidin-4-il1piperazin-2-il1acetonitrilo (620 mg, 1,03 mmol, 84 % de rendimiento, 100 % de pureza) en forma de un sólido de color amarillo que se usó directamente en la siguiente etapa sin purificación adicional. LCMS [ESI, M+11: 600.
Etapa C: 2-[(2S)-4-[7-(5,6-dimetil-1-tetrahidropiran-2-il-indazol-4-il)-2-[[(2S) 1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-cf1pirimidin-4-il1-1-(2-fluoroprop-2-enoil)piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(5,6-dimetil-1-tetrahidropiran-2-il- indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo (600 mg, 1,00 mmol, 1 eq.) en DMF (8 ml) se le añadieron ácido 2-fluoroprop-2-enoico (180 mg, 2,00 mmol, 2 eq.) en AE (4 ml) y TEA (304 mg, 3,00 mmol, 414 gl, 3 eq.) seguido de T3P (955 mg, 1,50 mmol, 892 gl, 50 % de pureza en EtOAc, 1,5 eq.) a 0 °C. La mezcla se agitó a 25 °C durante 0,5 horas. Después de que se completara, la mezcla se diluyó con agua (20 ml) y se extrajo con EtOAc (3 x 20 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo1. Las fracciones deseadas se recogieron, se neutralizaron con NaHC03 sólido, se concentraron al vacío para eliminar el MeCN y se extrajeron con EtOAc (3 x 100 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío para dar 2-[(2S)-4-[7-(5,6-dimetil-1 -tetrahidropiran-2-il-indazol-4-il)-2-[[(2S)-1 -metilpirrolidin-2
il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (380 mg, 554 μmol, 55 % de rendimiento, 98 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 672.
1H RMN (400 MHz, cloroformo-d) 5 = 7,98 (s, 1H), 7,22 (s, 1H), 5,66 (dd, J = 2,4, 9,2 Hz, 1H), 5,51 - 5,32 (m, 1H), 5,25 (dd a, J = 3,6, 16,8 Hz, 1H), 5,09 - 4,59 (m, 1H), 4,44 - 4,33 (m, 1H), 4,26 (s a, 2H), 4,19 - 4,14 (m, 1H), 4,05 (d a, J = 14,4 Hz, 2H), 4,00 - 3,91 (m, 1H), 3,76 (dt, J = 2,4, 11,2 Hz, 1H), 3,59 - 3,39 (m, 3H), 3,37 - 3,21 (m, 1H), 3,14 - 3,02 (m, 2H), 2,90 - 2,52 (m, 6H), 2,48 (s, 3H), 2,43 (s, 3H), 2,33 (s, 3H), 2,29 - 2,23 (m, 1H), 2,22 - 2,08 (m, 2H), 1,90 -1,69 (m, 8H).
Etapa D: 2-[(2S)-4-[7-(5,6-dimetil-1H- indazol-4-il)-2-(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(5,6-dimetil-1-tetrahidropirano -2-il-indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (360 mg, 536 μmol, 1 eq.) en DCM (0,4 ml) se le añadió TFA (1,22 g, 10.7 mmol, 794 μl, 20 eq.). La mezcla se agitó a 25 °C durante 2 horas. Después de que se completara, la mezcla se diluyó con DCM (10 ml) y se neutralizó con una solución saturada de NaHCÜ3. La capa acuosa separada se extrajo con DCM (2 x 10 ml). Las capas orgánicas combinadas se secaron sobre Na2SÜ4 y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. Las fracciones deseadas se recogieron, se neutralizaron con NaHCÜ3 sólido, se concentraron al vacío para eliminar el MeCN y se extrajeron con EtOAc (2 x 15 ml). Las capas orgánicas se secaron sobre Na2SÜ4 y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Xtimate C18 150 * 25 mm * 5 um; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 40 %-70 %, 10 min). Las fracciones deseadas se recogieron y se liofilizaron para dar 2-[(2S)-4-[7-(5,6-dimetil-1 H - indazol-4-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-577-pirido[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (73,3 mg, 120 μmol, 22 % de rendimiento, 96,6 % de pureza) en forma de un sólido de color blanco. Lc MS [ESI, M+1]: 588.
1H RMN (400 MHz, cloroformo-d) 5 = 9,97 (s a, 1H), 8,04 (s, 1H), 7,15 (s, 1H), 5,55 - 5,31 (m, 1H), 5,26 (dd, J = 3,6, 16.8 Hz, 1H), 5,08 - 4,50 (m, 1H), 4,39 (dd, J = 4,8, 10,8 Hz, 1H), 4,29 (s, 2H), 4,20 - 3,68 (m, 4H), 3,65 - 3,27 (m, 4H), 3,10 (t a, J = 7,8 Hz, 2H), 3,02 - 2,72 (m, 4H), 2,71 - 2,60 (m, 1H), 2,48 (s, 3H), 2,42 (s, 3H), 2,34 (s, 3H), 2,31 - 2,24 (m, 1H), 2,12 - 2,01 (m, 1H), 1,93 - 1,73 (m, 3H).
Ejemplo 605

2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[7-(8-metil-1-naftil)-2-(2-piridilmetoxi)-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-(2-piridilmetoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-illpiperazin-1-carboxilato de bencilo. A una solución de 2-piridilmetanol (55,1 mg, 504 μmol, 48,7 μl, 3,0 eq.) y (2S)-2-(cianometil)-4-[7-(8-metil-1 -naftil)-2-metilsulfinil-6,8-dihidro- 5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (100 mg, 168 μmol, 1,0 eq.) en tolueno (5,00 ml) se le añadió t-Bu0Na (48,5 mg, 504 μmol, 3,0 eq.) a -10 °C. La mezcla se agitó a -10 °C durante 0,5 horas. Después de que se completara, la mezcla se inactivó con agua (5,00 ml) y se extrajo con acetato de etilo (2 x 10 ml). Las capas orgánicas combinadas se lavaron con salmuera (1 x 10 ml), se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 50/1 a 1/1). Se obtuvo (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-(2-piridilmetoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (64,0 mg, 98,8 μmol, 59 % de rendimiento, 98,8 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 641.
Etapa B: 2-[(2S)-4-[7-(8-metil-1-naftil)-2-(2-piridilmetoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-illacetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-(2-piridilmetoxi)-6,8-dihidro-5H-pirido[3,4-d]pirim idin-4-il]piperazin-1 -carboxilato de bencilo (64,0 mg, 100 μmol, 1,0 eq.) en Me0H (3,00 ml) se le añadieron Pd/C (20,0 mg, 10 % de pureza) y NH3*Me0H (2,00 ml, 20 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. Después de que se completara, la mezcla se concentró al vacío. El residuo se purificó por cromatografía en columna (Si02, diclorometano/metanol = 100/1 a 8/1). Se obtuvo 2-[(2S)-4-[7-(8-metil-1-naftil)-2-(2-piridilmetoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (15,0 mg, 19,3 μmol, 19 % de rendimiento, 64,9 % de pureza) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 506.
Etapa C: 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[7-(8-metil-1-naftil)-2-(2-piridilmetoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-i n piperazin-2-i n acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-(2-piridilmetoxi)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (10,0 mg, 19,8 μmol, 1,0 eq.), ácido 2-fluoroprop-2-enoico (5,34 mg, 59,3 μmol, 3,0 eq.) y Et3N (18,0 mg, 178 μmol, 24,8 μl, 9,0 eq.) en EtOAc (2,00 ml) se le añadió T3P (50,3 mg, 79,1 μmol, 47,1 μl, 50 % de pureza en acetato de etilo, 4,0 eq.) a 0 °C. La mezcla se agitó a 25 °C durante 1 hora. Después de que se completara, la mezcla se diluyó con agua (3,00 ml). La capa orgánica se separó, se lavó con salmuera (1 x 10 ml), se secó sobre sulfato de sodio, se filtró y se concentró al vacío. La mezcla se purificó por cromatografía en columna (Si02, acetato de etilo/metanol = 50/1 a 10/1). El residuo se purificó por HPLC prep. (columna: Xtimate C18150 * 25 mm * 5 um; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 50 % - 80 %, 10 min). El residuo se concentró a presión reducida para eliminar el ACN y después se liofilizó. Se obtuvo 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[7-(8-metil-1-naftil)-2-(2-piridilmetoxi)-6,8-dihidro-5H-pirido[3,4-d]lpirimidin-4-il]piperazin-2-il]acetonitrilo (1,41 mg, 2,44 umol, 12 % de rendimiento, 99,8 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 579.
1H RMN (400 MHz, cloroformo-d) 5 = 8,61 - 8,55 (m, 1H), 7,74 - 7,62 (m, 3H), 7,55 - 7,48 (m, 1H), 7,45 - 7,32 (m, 2H), 7,26 - 7,17 (m, 3H), 5,52 - 5,48 (m, 2H), 5,47 - 5,33 (m, 1H), 5,25 (dd, J = 3,6, 17,2 Hz, 1H), 4,79 (s a, 1H), 4,38 - 3,73 (m, 6H), 3,61 - 3,41 (m, 2H), 3,26 - 3,12 (m, 2H), 3,09 - 2,95 (m, 2H), 2,92 (s, 3H), 2,85 - 2,56 (m, 2H).
E je m p lo 606

2-((S)-4-(7-(5,6-dimetil-1H-indazol-4-il)-2-(((2S,4R)-4-fluoro-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[7-(5,6-dimetil-1-tetrahidropiran-2-il-indazol-4-il)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de bencilo. Una mezcla de (2S)-2-(cianometil)-4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (5,0 g, 11,4 mmol, 1,0 eq.), 4-bromo-5,6-dimetil-1-tetrahidropiran-2-il-indazol (7,05 g, 22,8 mmol, 2,0 eq.), Pd2(dba)3 (2,09 g, 2,28 mmol, 0,2 eq.), RuPhos (2,13 g, 4,56 mmol, 0,4 eq.) y CS2C03 (9,29 g, 28,5 mmol, 2,5 eq.) en tolueno (100 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 8 horas en una atmósfera de N2. La mezcla de reacción se diluyó con agua (100 ml) y se extrajo con acetato de etilo (3 * 200 ml). Las capas orgánicas combinadas se lavaron con salmuera (100 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 20/1 a 2/1) y se purificó adicionalmente por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico)/acetonitrilo)]. La mezcla se ajustó a pH ~7 con una solución acuosa saturada de NaHC03 y se extrajo con acetato de etilo (3 * 100 ml). Las capas orgánicas combinadas se lavaron con salmuera (50 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar el producto. Se obtuvo (2S)-2-(cianometil)-4-[7-(5,6-dimetil-1-tetrahidropiran-2-il-indazol-4-il)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (4,90 g, 7,20 mmol, 63 % de rendimiento, 98 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1 ]: 667.
1H RMN (400 MHz, cloroformo-d) 8 = 7,98 (s, 1H), 7,50 - 7,31 (m, 5H), 7,22 (s, 1H), 5,66 (dd, J = 2,4, 9,6 Hz, 1H), 5,27 - 5,13 (m, 2H), 4,69 (s a, 1H), 4,27 (s, 2H), 4,13 - 3,97 (m, 3H), 3,89 (d, J = 11,6 Hz, 1H), 3,81 - 3,68 (m, 1H), 3,51 (t, J = 5,2 Hz, 2H), 3,30 (s a, 2H), 3,04 (t, J = 11,6 Hz, 1H), 2,93 - 2,66 (m, 4H), 2,62 - 2,48 (m, 4H), 2,43 (s, 3H), 2,32 (s, 3H), 2,23 - 2,12 (m, 1H), 2,11 - 2,05 (m, 1H), 1,85 - 1,67 (m, 3H).
Etapa B: (2S)-2-(c¡anomet¡l)-4-[7-(5,6-d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡din-4-¡l1p¡peraz¡n-1-carbox¡lato de bencilo. A una soluc¡ón de (2S)-2-(c¡anomet¡l)-4-[7-(5,6-d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-2-met¡lsulfan¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (500 mg, 749 μmol, 1,0 eq.) en tolueno (10 ml) se le añad¡ó m-CPBA (152 mg, 749 μmol, 85 % de pureza, 1,0 eq.). La mezcla se ag¡tó a 0 °C durante 1 hora. La mezcla se d¡luyó con agua (10 ml) y se ajustó a pH ~7 con una soluc¡ón acuosa saturada de NaHCÜ3. Después, la mezcla se extrajo con acetato de et¡lo (3 * 20 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar el producto. Se obtuvo (2S)-2-(c¡anomet¡l)-4-[7-(5,6-d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (400 mg, 539 μmol, 72 % de rend¡m¡ento, 92 % de pureza) en forma de un sól¡do de color amar¡llo y se usó d¡rectamente en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón. LCMS [ESI, M+1]: 683.
Etapa C: (2S)-2-(c¡anomet¡l)-4-[7-(5,6-d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de benc¡lo. A una soluc¡ón de (2S)-2-(c¡anomet¡l)-4-[7-(5,6-d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (350 mg, 512 μmol, 1,0 eq.) en tolueno (10 ml) se le añad¡eron t-Bu0Na (148 mg, 1,54 mmol, 3,0 eq.) y [(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metanol (136 mg, 1,03 mmol, 2,0 eq.). La mezcla se ag¡tó a 0 °C durante 0,5 horas. La mezcla de reacc¡ón se d¡luyó con agua (20 ml) y se extrajo con acetato de et¡lo (3 * 30 ml).
Las capas orgán¡cas comb¡nadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar un res¡duo. El res¡duo se pur¡f¡có por cromatografía ultrarráp¡da de fase ¡nversa [agua (0,1 % de ác¡do fórm¡co)/aceton¡tr¡lo)]. La mezcla se ajustó a pH ~7 con una soluc¡ón acuosa saturada de NaHC03 y se extrajo con acetato de et¡lo (3 * 50 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar el producto. Se obtuvo (2S)-2-(c¡anomet¡l)-4-[7-(5,6-d¡met¡l-1-tetrah¡drop¡ran-2-¡l- ¡ndazol-4-¡l)-2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (280 mg, 372 μmol, 73 % de rend¡m¡ento, 100 % de pureza) en forma de un sól¡do de color amar¡llo. LCMS [ESI, M+1]: 752.
Etapa C: 2-[(2S)-4-[7-(5,6-d¡met¡l-1-tetrah¡drop¡rano -2-¡l-¡ndazol-4-¡l)-2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una soluc¡ón de (2S)-2-(c¡anomet¡l)-4-[7-(5,6-d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (230 mg, 306 umol, 1,0 eq.) en metanol (2,0 ml) se le añad¡eron Pd/C seco (50,0 mg, 10 % de pureza) y NH3/metanol (1,00 ml, 20 % de pureza) en una atmósfera de N2. La suspens¡ón se desgas¡f¡có al vacío y se purgó var¡as veces con H2. La mezcla se ag¡tó en una atmósfera de H2 (103,42 kPa (15 ps¡)) a 25 °C durante 0,5 horas. La mezcla se concentró al vacío. Se obtuvo 2-[(2S)-4-[7-(5,6-d¡met¡l-1-tetrah¡drop¡rano -2-¡l-¡ndazol-4-¡l)-2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (180 mg, 285 μmol, 93 % de rend¡m¡ento, 98 % de pureza) en forma de un sól¡do de color amar¡llo y se usó en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón. LCMS [ESI, M+1]: 618.
Etapa D: 2-[(2S)-4-[7-(5,6-d¡met¡l-1H-¡ndazol-4-¡l)-2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una soluc¡ón de 2-[(2S)-4-[7-(5,6-d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l]aceton¡tr¡lo (30 mg, 48,6 μmol, 1,0 eq.) en d¡clorometano (300 ul) se le añad¡ó TFA (221 mg, 1,94 mmol, 144 μl, 40 eq.). La mezcla se ag¡tó a 0 °C durante 0,5 horas. La mezcla se concentró al vacío y se d¡luyó con agua (10 ml). La mezcla se ajustó a pH ~ 8 con una soluc¡ón acuosa saturada de NaHC03 y se extrajo con d¡clorometano (3 * 20 ml).
Las capas orgán¡cas comb¡nadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar el producto. El res¡duo se pur¡f¡có por HPLC prep. (columna: Xt¡mate C18 150 * 25 mm * 5 um; fase móv¡l: [agua (0,05 % de h¡dróx¡do de amon¡aco v/v) - ACN]; % de B: 26 % - 56 %, 1 m¡n). La fracc¡ón deseada se recog¡ó y se l¡of¡l¡zó. Se obtuvo 2-[(2S)-4-[7-(5,6-d¡met¡l-1H-¡ndazol-4-¡l)-2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (7,08 mg, 13,1 μmol, 27 % de rend¡m¡ento, 98,5 % de pureza) en forma de un sól¡do de color amar¡llo. LCMS [ESI, M+1]: 534.
1H RMN (400 MHz, cloroformo-d) 8 = 10,14 (s a, 1H), 8,04 (d, J = 0,8 Hz, 1H), 7,14 (s, 1H), 5,32 - 5,03 (m, 1H), 4,44 (dd, J = 4,4, 11,2 Hz, 1H), 4,33 - 4,19 (m, 3H), 4,00 (d a, J = 12,8 Hz, 1H), 3,82 (d a, J = 12,0 Hz, 1H), 3,65 - 3,41 (m, 3H), 3,27 (d a, J = 6,4 Hz, 1H), 3,17 - 2,95 (m, 4H), 2,90 (dd a, J = 10,0, 12,4 Hz, 1H), 2,83 - 2,47 (m, 8 H), 2,41 (s, 3H), 2,38 - 2,23 (m, 4H), 2,09 - 1,90 (m, 1H).
Etapa E: 2-[(2S)-4-[7-(5,6-d¡met¡l-1H-¡ndazol-4-¡l)-2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-1-(2-fluoroprop-2-eno¡l)p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una soluc¡ón de 2-[(2S)-4-[7-(5,6-d¡met¡l-1H-¡ndazol-4-¡l)-2-[[(2S,4R)-4-fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (100 mg, 187 μmol, 1,0 eq.) y ác¡do 2-fluoroprop-2-eno¡co (25,3 mg, 281 μmol, 1,5 eq.) en acetato de et¡lo (2,0 ml) se le añad¡eron T3P (477 mg, 749 μmol, 446 μl, 50 % de pureza en acetato de et¡lo, 4,0 eq.) y TEA
(114 mg, 1,12 mmol, 156 gl, 6,0 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (3 * 30 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiÜ2, acetato de etilo/metanol = 100/1 a 10/1) y se purificó adicionalmente por HPLC prep. (columna: Waters Xbridge 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 38 % - 68 %, 10 min). La fracción deseada se recogió y se liofilizó. Se obtuvo 2-[(2S)-4-[7-(5,6-dimetil-1H-indazol-4-il)-2-[[(2S,4R)-4-fluoro-1-metil-pirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (8 mg, 12,8 umol, 7 % de rendimiento, 96,9 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 606.
1H RMN (400 MHz, cloroformo-d) ó = 10,03 (s a, 1H), 8,04 (s, 1H), 7,15 (s, 1H), 5,42 (dd, J = 6,4 Hz, J = 47,2 Hz 1H), 5,31 - 5,05 (m, 2H), 5,04 - 3,76 (m, 9H), 3,65 - 3,47 (m, 3H), 3,42 - 3,25 (m, 1H), 3,20 - 2,70 (m, 6H) 2,70 - 2,55 (m, 1H), 2,51 (s, 3H), 2,42 (s, 3H), 2,38 - 2,25 (m, 4H), 2,10 - 1,87 (m, 1H).
Ejemplo 607

2-((S)-1-(2-fluoroacriloil)-4-(7-(6-metilisoquinolin-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-
d]pirimidin-4-il)piperazin-2-il)acetonitrilo
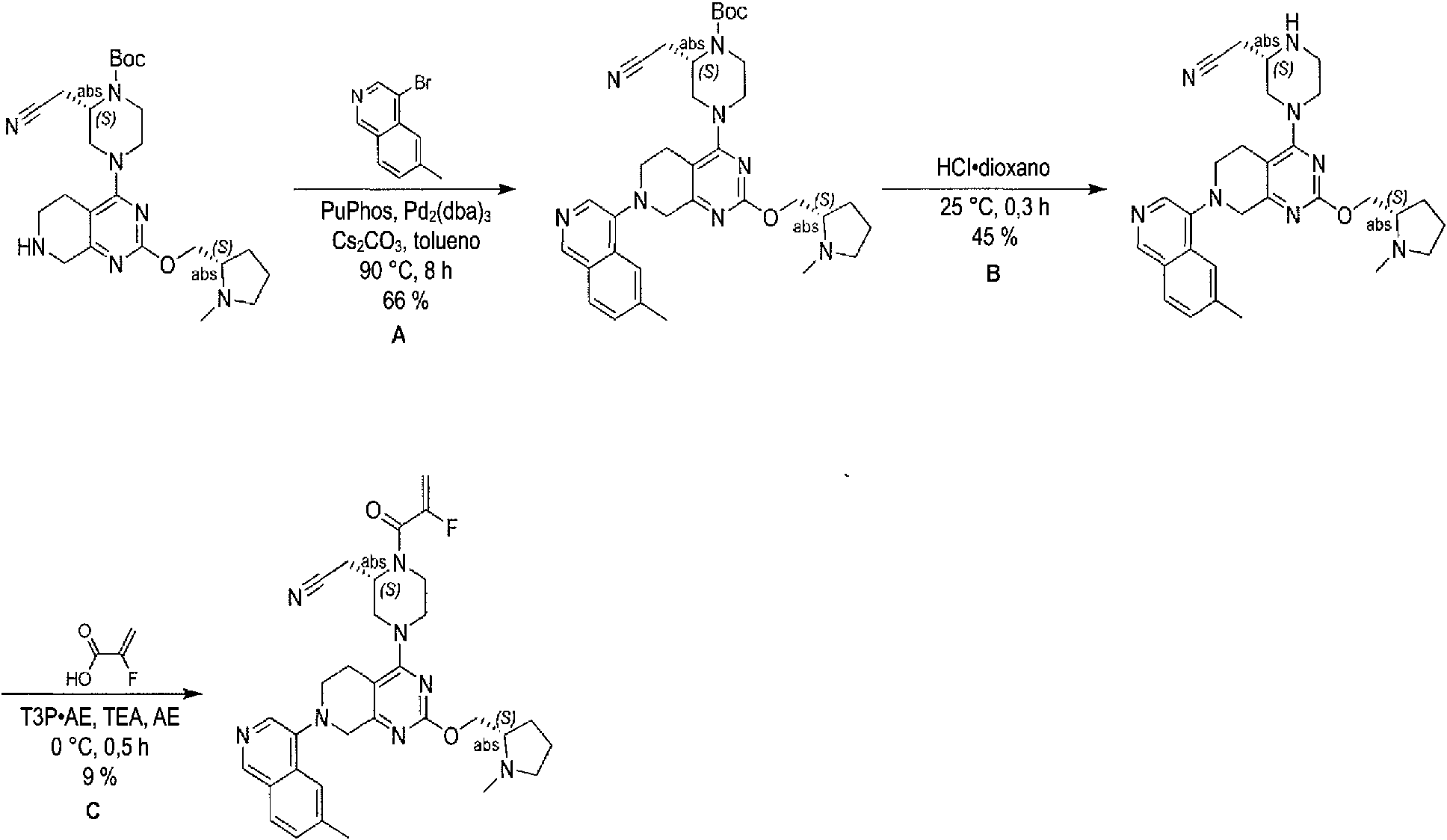
Etapa A: (2S)-2-(cianometil)-4-[7-(6-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de tere-butilo. Una mezcla de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (250 mg, 530 gmol, 1,0 eq.), 4-bromo-6-metil-isoquinolina (235 mg, 1,06 mmol, 2,0 eq.), RuPhos (98,9 mg, 212 gmol, 0,4 eq.), Pd2(dba)3 (97,1 mg, 106 umol, 0,2 eq.) y Cs2CÜ3 (432 mg, 1,33 mmol, 2,5 eq.) en tolueno (5 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 8 horas en una atmósfera de N2. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (3 * 30 ml). Las capas orgánicas combinadas se lavaron con salmuera
(20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de ácido fórmico)/acetonitrilo)]. La mezcla se ajustó a pH ~7 con una solución acuosa saturada de NaHCÜ3 y se extrajo con acetato de etilo (3 x 30 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar el producto. Se obtuvo (2S)-2-(cianometil)-4-[7-(6-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo (230 mg, 349 μmol, 66 % de rendimiento, 93 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 6l3.
Etapa B: 2-[(2S)-4-[7-(6-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-¡l1p¡perazin-2-il]acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(6-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (50 mg, 81 ,6 μmol, 1,0 eq.) en dioxano (400 μl) se le añadió HCl/dioxano (4 M, 408 ul). La mezcla se agitó a 25 °C durante 0,3 horas. La mezcla se concentró al vacío y se diluyó con agua (10 ml). La mezcla se ajustó a pH ~ 8 con una solución acuosa saturada de NaHCÜ3 y se extrajo con diclorometano (3 x 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar el residuo. El residuo se purificó por HPLC prep. (columna: Xtimate C18150 * 25 mm * 5 um; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 28 % - 58 %, 1 min). La fracción deseada se recogió y se liofilizó. Se obtuvo 2-[(2S)-4-[7-(6-metil-4-isoquinolil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡rido[3,4-cf]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (19 mg, 37 μmol, 45 % de rendimiento, 99,9 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 513.
1H RMN (400 MHz, cloroformo-d) 5 = 8,94 (s, 1H), 8,20 (s, 1H), 7,92 - 7,83 (m, 2H), 7,45 (dd, J = 1,2, 8,4 Hz, 1H), 4,40 (dd, J = 4,8, 10,8 Hz, 1H), 4,29 (s, 2H), 4,17 (dd, J = 6 ,8 , 10,8 Hz, 1H), 4,03 (d a, J = 12,8 Hz, 1H), 3,86 (d a, J = 12,4 Hz, 1H), 3,42 (t a, J = 5,2 Hz, 2H), 3,33 - 3,22 (m, 1H), 3,17 - 2,80 (m, 7H), 2,72 - 2,63 (m, 1H), 2,61 - 2,52 (m, 5H), 2,47 (s, 3H), 2,33 - 2,23 (m, 1H), 2,11 - 2,06 (m, 1H), 1,89 - 1,68 (m, 3H).
Etapa C: 2-[(2S)-1-(2-fluoroprop-2-enoN)-4-r7-(6-metil-4-isoquinolir)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d^pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de 2-[(2S)-4-[7-(6-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5^-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (100 mg, 195 μmol, 1,0 eq.) y ácido 2-fluoroprop-2-enoico (35,1 mg, 390 umol, 2,0 eq.) en acetato de etilo (2 ml) se le añadieron T3P (372 mg, 585 μmol, 348 μl, 50 % de pureza en acetato de etilo, 3,0 eq.) y TEA (158 mg, 1,56 mmol, 217 μl, 8,0 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (3 x 30 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiÜ2, acetato de etilo/metanol = 100/1 a 10/1) y se purificó adicionalmente por HμLC prep. (columna: Waters Xbridge 150 * 25 5 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 38 % - 62 %, 10 min). La fracción deseada se recogió y se liofilizó. Se obtuvo 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[7-(6-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (11 mg, 18,6 μmol, 9 % de rendimiento, 98,9 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 585.
1H RMN (400 MHz, cloroformo-d) 5 = 8,96 (s, 1H), 8,22 (s, 1H), 7,93 - 7,85 (m, 2H), 7,46 (dd, J = 1,2, 8,4 Hz, 1H), 5,49 - 5,36 (m, 1H), 5,26 (dd, J = 3,6, 16,8 Hz, 1H), 4,88 (s a, 1H), 4,50 - 4,29 (m, 3H), 4,27 - 3,96 (m, 4H), 3,61 - 3,30 (m, 4H), 3,22 - 3,06 (m, 2H), 3,04 - 2,77 (m, 4H), 2,75 - 2,65 (m, 1H), 2,59 (s, 3H), 2,49 (s, 3H), 2,31 - 2,29 (m, 1H), 2,15 -1,97 (m, 1H), 1,80 - 1,77 (m, 3H).
Ejemplo 608

2-[(2S)-4-[7-(8-etil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2 -enoil)piperazin-2 -il]acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[7-(8-etil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-1-carboxilato de terc-butilo. A una solución de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de terc-butilo (240 mg, 508,91 gmol, 1 eq.), t-Bu0Na (146,72 mg, 1,53 mmol, 3 eq.), RuPhos (47,50 mg, 101,78 umol, 0,2 eq.) y RuPhos Pd G3 (85,13 mg, 101,78 gmol, 0,2 eq.) en tolueno (5 ml) se le añadió 1-bromo-8-etil-naftaleno (239,31 mg, 1,02 mmol, 2 eq.). La mezcla se agitó a 90 °C durante 12 h. A la mezcla se le añadió agua (10 ml) y se extrajo con acetato de etilo (10 ml x 3). Las capas orgánicas se secaron sobre Na2S04, se filtraron y se concentraron. El residuo se purificó por HPLC ultrarrápida de fase inversa (C18, 0,1 % de AF en agua, 0-45 % de MeCN). El producto de (2S)-2-(cianometil)-4-[7-(8-etil-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1 -carboxilato de terc-butilo (113 mg, 180,57 gmol, 35,48 % de rendimiento, 100 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 626.
Etapa B: 2-[(2S)-4-[7-(8-etil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-i n metox n -6,8-dihidro- 5H-pirido[3,4-d1pirimidin-4-¡ n piperazin-2-i n acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-etil-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de terc-butilo (103 mg, 164,59 gmol, 1 eq.) en DCM (2 ml) se le añadió t Fa (2 ml) a 25 °C. La mezcla de reacción se agitó a 25 °C durante 0,5 h. La mezcla de reacción se inactivó con la adición de NaHC03 acuoso saturado (20 ml) a 25 °C hasta pH = 8 y después se extrajo con acetato de etilo (3 x 10 ml). Las capas orgánicas combinadas se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida. Se obtuvo 2-[(2S)-4-[7-(8-et¡l-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-dihidro- 5H-pirido[3,4-d]pirimidin-4-il1piperazin-2-il1acetonitrilo (81 mg, bruto) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 526.
1H RMN (400 MHz, cloroformo-d) ó = 7,66 - 7,55 (m, 2H), 7,37 - 7,25 (m, 2H), 7,25 - 7,20 (m, 1H), 7,19 - 7,14 (m, 1H), 4,36 - 4,26 (m, 1H), 4,21 - 4,02 (m, 2H), 4,00 - 3,62 (m, 3H), 3,56 - 2,73 (m, 11H), 2,65 - 2,34 (m, 7H), 2,26 - 2,14 (m, 1H), 2,02 - 1,88 (m, 1H), 1,82 - 1,62 (m, 4H), 1,13 - 1,02 (m, 3H).
Etapa C: 2-[(2S)-4-[7-(8-etil-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-(2-fluoroprop-2-enoil)piperazin-2-i n acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-etil-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (61 mg, 116,04 gmol, 1 eq.) y ácido 2-fluoroprop-2-enoico (20,90 mg, 232,08 gmol, 2 eq.) en DMF (10 ml) se le añadieron TEA (281,81 mg, 2,78 mmol, 387,63 gl, 24 eq.) y T3P (332,29 mg, 1,04 mmol, 310,55 ul, 9 eq.) a -40 °C. Después de la adición, la mezcla se agitó a 0 °C durante 2 h. La mezcla se diluyó con agua (3 x 20 ml) y se diluyó con acetato de etilo (3 x 20 ml). La capa orgánica se lavó con salmuera (1 x 20 ml), se secó sobre Na2S04, se filtró y se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Waters Xbridge 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 55 %-85 %, 10 min). La mezcla se diluyó con agua (3 x 20 ml) y se diluyó con acetato de etilo (3 x 20 ml). La capa orgánica se lavó con salmuera (1 x 20 ml), se secó sobre Na2S04, se filtró y se concentró al vacío para dar 2-[(2S)-4-[7-(8-etil-1 -naftil)-2-[[(2S)-1 -met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-1 -(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (7 mg, 11,63 gmol, 10,02 % de rendimiento, 99,3 % de pureza) que se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 598.
1H RMN (400 MHz, cloroformo-d) ó = 7,67 - 7,54 (m, 2H), 7,39 - 7,26 (m, 2H), 7,25 - 7,20 (m, 1H), 7,15 (s, 1H), 5,45 -5,24 (m, 1H), 5,23 - 5,12 (m, 1H), 4,95 - 4,58 (m, 1H), 4,33 - 4,25 (m, 1H), 4,25 - 3,63 (m, 6H), 3,57 - 3,33 (m, 3H), 3,21 -2,49 (m, 10H), 2,44 - 2,35 (m, 3H), 2,27 - 2,15 (m, 1H), 2,03 - 1,90 (m, 1H), 1,82 - 1,66 (m, 3H), 1,13 - 1,04 (m, 3H).
Ejemplo 609

2-[(2S)-4-[7-(6-fluoro-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[7-(6-fluoro-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6.8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-1-carboxilato de bencilo. A la mezcla de (25)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (400 mg. 791 μmol. 1.0 eq.), 4-bromo-6-fluoro-isoquinolina (215 mg. 949 μmol, 1.2 eq.). CS2C03 (773 mg. 2,37 mmol, 3.0 eq.) y RuPhos (148 mg.
316 pmol. 0.4 eq.) en tolueno (10 ml) se le añadió Pd2(dba)3 (145 mg. 158 μmol. 0.2 eq.) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con N2. La mezcla se agitó en una atmósfera de N2 a 90 °C durante 10 horas. La mezcla de reacción se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0.1 % de AF)/acetonitrilo]. Las fracciones deseadas se recogieron y se basificaron con NaHC03 sólido. se concentraron al vacío para eliminar el MeCN y se extrajeron con acetato de etilo (2 * 40 ml). Las capas orgánicas se secaron sobre Na2S04 y se concentraron al vacío para dar (2S)-2-(cianometil)-4-[7-(6-fluoro-4-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (140 mg. 204 μmol. 26 % de rendimiento. 95 % de pureza) en forma de un sólido de color amarillo.
Etapa B: 2-[(2S)-4-[7-(6-fluoro-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-i n piperazin-2-i n acetonitrilo. A la mezcla de (2S)-2-(cianometil)-4-[7-(6- fluoro-4-isoquinolil)-2-[[(2S)-1 -metilpirrolidin-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (30 mg. 46,1 μmol. 1.0 eq.) y NH3*Me0H (0,3 ml. 20 % de pureza) en Me0H (0.3 ml) se le añadió Pd/C (10 mg. 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1.5 horas. La mezcla de reacción se filtró y el filtrado se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Xtimate C18150 * 25 mm * 5 um;fase móvil: [agua (0.05 % de hidróxido de amoniaco v/v)-ACN];% de B: 30 %-60 %,1 min). Las fracciones deseadas se recogieron y se liofilizaron para dar 2-[(2S)-4-[7-(6-fluoro-4-¡soqu¡nol¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]pir¡m¡d¡n-4
il]piperazin-2-il]acetonitrilo (9,78 mg, 18,9 μmol, 41 % de rendimiento, 99,9 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 517.
1H RMN (400 MHz, cloroformo-d) 5 = 9,00 (s, 1H), 8,27 (s, 1H), 8,02 (dd, J = 5,6, 9,2 Hz, 1H), 7,74 (dd, J = 2,4, 10,4 Hz, 1H), 7,39 (dt, J = 2,4, 8,8 Hz, 1H), 4,41 (dd, J = 4,8, 10,4 Hz, 1H), 4,28 (s, 2H), 4,18 (dd, J = 6,8, 10,4 Hz, 1H), 4,02 (d a, J = 12,4 Hz, 1H), 3,91 - 3,82 (m, 1H), 3,47 - 3,35 (m, 2H), 3,33 - 3,24 (m, 1H), 3,18 - 3,07 (m, 3H), 3,07 - 2,97 (m, 1H), 2,93 (dd, J = 9,6, 12,8 Hz, 1H), 2,85 (s a, 2H), 2,73 - 2,64 (m, 1H), 2,56 (dd, J = 2,4, 6,4 Hz, 2H), 2,49 (s, 3H), 2,34 - 2,24 (m, 1H), 2,13 - 2,00 (m, 1H), 1,91 - 1,76 (m, 3H).
Etapa C: 2-[(2S)-4-[7-(6-fluoro-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il1-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo. A la mezcla de 2-[(2S)-4-[7-(6-fluoro-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (80 mg, 155 μmol, 1,0 eq.), ácido 2-fluoroprop-2-enoico (41,8 mg, 464 μmol, 3,0 eq.) y TEA (235 mg, 2,32 mmol, 323 ul, 15 eq.) en acetato de etilo (1,5 ml) y DMF (1 ml) se le añadió T3P (493 mg, 774 μmol, 460 ul, 50 % de pureza, 5,0 eq.) a 0 °C y la mezcla se agitó a 25 °C durante 1 hora. A la mezcla se le añadió agua (10 ml). La mezcla se diluyó con acetato de etilo (30 ml) y se extrajo con acetato de etilo (2 * 5 ml). Las capas orgánicas combinadas se secaron sobre Na2SÜ4 anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Xtimate C18 150 * 25 mm * 5 um;fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 40 % - 70 %, 1 min). Las fracciones deseadas se recogieron y se liofilizaron para dar 2-[(2S)-4-[7-(6-fluoro-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cdpirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (17,4 mg, 29,3 μmol, 19 % de rendimiento, 99,2 % de pureza) en forma de un sólido de color blanco. LCMS [ESl, M+1]: 589.
1H RMN (400 MHz, cloroformo-d) 5 = 9,01 (s, 1H), 8,28 (s, 1H), 8,03 (dd, J = 5,6, 8,8 Hz, 1H), 7,73 (dd, J = 2,4, 10,4 Hz, 1H), 7,40 (dt, J = 2,4, 8,8 Hz, 1H), 5,52 - 5,35 (m, 1H), 5,27 (dd, J = 3,6, 17,2 Hz, 1H), 5,02 - 4,62 (m, 1H), 4,46 - 4,38 (m, 1H), 4,36 - 4,25 (m, 2H), 4,24 - 4,07 (m, 3H), 4,02 (d a, J = 13,2 Hz, 1H), 3,62 - 3,29 (m, 4H), 3,13 (s a, 2H), 3,03 - 2,93 (m, 2H), 2,91 - 2,77 (m, 2H), 2,72 (s a, 1H), 2,51 (s, 3H), 2,32 (d a, J = 8,4 Hz, 1H), 2,13 - 2,02 (m, 1H), 1,92 -1,70 (m, 3H).
Ejemplo 610

(S)-2-(1-(2-fluoroacriloil)-4-(7-(naftalen-1-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: 4-hidroxi-6,8-dihidro-5H-pirido[3,4-C]pirimidin-7-carboxilato de tere-butilo. A una mezcla de 3-oxopiperidin-1,4-dicarboxilato de 01-terc-butil 04-etilo (5,00 g, 18,4 mmol, 1,00 eq.) en n-Bu0H (30,0 ml) se le añadió ácido acético;metanimidamida (9,59 g, 92,2 mmol, 5,00 eq.) en una porción. La mezcla se agitó a 120 °C durante 12 horas. La mezcla se concentró para eliminar el disolvente. Al residuo se le añadió agua (30,0 ml) y se filtró. El precipitado se lavó con agua (30,0 ml) y se concentró. El producto bruto se usó en la siguiente etapa directamente sin purificación adicional. El compuesto 4-hidroxi-6,8-dihidro-5H-pirido[3,4-<C]pirimidin-7-carboxilato de tere-butilo (4,10 g, 16,1 mmol, 87 % de rendimiento, 98,4 % de pureza) se obtuvo en forma de un sólido de color pardo. LCMS [ESI, M+1]: 196.
Etapa B: 4-cloro-6,8-dihidro-5H-pirido[3,4- cflpirimidin-7-carboxilato de tere-butilo. A una mezcla de 4-hidroxi-6,8-dihidro-5H-pirido[3,4- C]pirimidin-7-carboxilato de tere-butilo (3,30 g, 13,1 mmol, 1,00 eq.) en DCE (60,0 ml) se le añadieron en una porción PPh3 (6,89 g, 26,3 mmol, 2,00 eq.) y CCU (6,06 g, 39,4 mmol, 3,79 ml, 3,00 eq.) en una atmósfera de N2. La mezcla se agitó a 70 °C durante 3 horas. La mezcla de reacción se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 20/1 a 1/1). El compuesto 4-cloro-6,8-dihidro-5H-pirido[3,4-J]pirimidin-7-carboxilato de tere-butilo (2,54 g, 9,42 mmol, 72 % de rendimiento, 100 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 214.
Etapa C: 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-7-carboxilato de tere-butilo. A una mezcla de 4-cloro-6,8-dihidro-5H-pirido[3,4- C]pirimidin-7-carboxilato de tere-butilo (1,30 g, 4,82 mmol, 1,00 eq.) y (2S)-2-(cianometil)piperazin-1-carboxilato de bencilo (1,00 g, 3,86 mmol, 0,80 eq.) en DMAc (30,0 ml) se le añadió DIEA (3,11 g, 24,1 mmol, 4,20 ml, 5,00 eq.) en una atmósfera de N2. La mezcla se agitó a 100 °C durante 2 horas. La mezcla de reacción se diluyó con acetato de etilo (100 ml). Las capas orgánicas se lavaron con agua (30 ml * 2) y salmuera (30 ml * 1), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 20/1 a 0/1). El compuesto 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-C]pirimidin-7-carboxilato de tere-butilo (1,18 g, 2,30 mmol, 48 % de rendimiento, 96 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [eSi, M+1]: 493.
Etapa D: (2S)-2-(cianometil)-4-(5,6,7,8-tetrahidropirido[3,4-C1pirimidin-4-il)piperazin-1-carboxilato de bencilo. A una mezcla de 4-[(3S)-4-benciloxicarbonil-3-(cianometil)piperazin-1-il]-6,8-dihidro-5H-pirido[3,4-C]pirimidin-7-carboxilato de tere-butilo (1,20 g, 2,44 mmol, 1,00 eq.) en acetonitrilo (10,0 ml) se le añadió en una porción HCl/dioxano (4,00 M, 12,2 ml, 20,0 eq.) a 25 °C en una atmósfera de N2. La mezcla se agitó a 25 °C durante 30 min. La mezcla de reacción se concentró a presión reducida para dar un residuo. El producto bruto se usó en la siguiente etapa directamente sin purificación adicional. El compuesto (2S)-2-(cianometil)-4-(5,6,7,8-tetrahidropirido[3,4-C]pirimidin-4-il)piperazin-1-carboxilato de bencilo (883 mg, 2,16 mmol, 89 % de rendimiento, 96 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 393.
Etapa E: (2S)-2-(cianometil)-4-[7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-C]pirimidin-4-il]piperazin-1-carboxilato de bencilo. A una mezcla de (2S)-2-(cianometil)-4-(5,6,7,8-tetrahidropirido[3,4-C]pirimidin-4-il)piperazin-1-carboxilato de bencilo (300 mg, 764 μmol, 1,00 eq.) y 1-bromonaftaleno (317 mg, 1,53 mmol, 212 ul, 2,00 eq.) en tolueno (15,0 ml) se le añadieron en una porción Pd2(dba)3 (140 mg, 153 μmol, 0,20 eq.), RuPhos (143 mg, 306 μmol, 0,40 eq.) y Cs2C03 (747 mg, 2,29 mmol, 3,00 eq.) en una atmósfera de N2. La mezcla se agitó a 90 °C durante 5 horas. La mezcla
de reacción se diluyó con agua (10,0 ml) y se extrajo con acetato de etilo (20,0 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20,0 ml x 1), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. Las fracciones deseadas se recogieron, se neutralizaron con una solución saturada de NaHCÜ3 (5,00 ml) y se extrajeron con acetato de etilo (50,0 ml x 2). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El compuesto (2S)-2-(cianometil)-4-[7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (150 mg, 289 gmol, 38 % de rendimiento) se obtuvo en forma de un sólido de color amarillo.
Etapa F: 2-[(2S)-4-[7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il]acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de bencilo (50,0 mg, 96,4 gmol, 1,00 eq.) en MeÜH (3,00 ml) se le añadieron Pd/C (30,0 mg, 10 % de pureza), NHs^Me0H (0,50 ml, 20 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. La mezcla de reacción se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Waters Xbridge 150 x 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 35 % - 59 %, 10 min). El compuesto 2-[(2S)-4-[7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (8,63 mg, 22,4 gmol, 23 % de rendimiento, 99,8 % de pureza) se obtuvo en forma de un sólido de color blanquecino. LCMS [ESI, M+1]: 385.
1H RMN (400 MHz, cloroformo-d) 5 = 8,63 (s, 1H), 8,26 - 8,19 (m, 1H), 7,91 - 7,84 (m, 1H), 7,62 (d, J = 8,0 Hz, 1H), 7,54 - 7,48 (m, 2H), 7,45 (t, J = 7,6 Hz, 1H), 7,17 (d, J = 7,2 Hz, 1H), 4,35 (s, 2H), 3,99 (d a, J = 12,8 Hz, 1H), 3,83 (d a, J = 12,8 Hz, 1H), 3,53 - 3,23 (m, 3H), 3,20 - 3,07 (m, 2H), 3,07 - 2,90 (m, 4H), 2,63 - 2,49 (m, 2H).
Etapa G: A una mezcla de 2-[(2S)-4-[7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (60,0 mg, 156 gmol, 1,00 eq.) en acetato de etilo (0,60 ml) se le añadieron en una porción ácido 2-fluoroprop-2-enoico (56,2 mg, 624 gmol, 4,00 eq.), TEA (253 mg, 2,50 mmol, 348 ul, 16,0 eq.) y T3P (596 mg, 936 gmol, 557 ul, 50 % de pureza, 6,00 eq.) a 0 °C en una atmósfera de N2. La mezcla se agitó a 25 °C durante 30 min. La mezcla de reacción se inactivó con la adición de agua (1,00 ml) a 0 °C y después se extrajo con acetato de etilo (10,0 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (5,00 ml x 1), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Waters Xbridge 150 x 255 ufase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 40 %-70 %, 10 min). El compuesto 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[7-(1-naftil)-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (15,2 mg, 32,8 gmol, 21 % de rendimiento, 98,9 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 457.
1H RMN (400 MHz, cloroformo-d) 5 = 8,67 (s, 1H), 8,26 - 8,18 (m, 1H), 7,91 - 7,84 (m, 1H), 7,63 (d, J = 8,0 Hz, 1H), 7,56 - 7,49 (m, 2H), 7,45 (t, J = 7,6 Hz, 1H), 7,18 (d, J = 6,8 Hz, 1H), 5,54 - 5,34 (m, 1H), 5,27 (dd, J = 4,0, 17,2 Hz, 1H), 4,91 (s a, 1H), 4,46 - 4,29 (m, 2H), 4,14 (d a, J = 13,6 Hz, 1H), 3,99 (d a, J = 13,2 Hz, 1H), 3,63 - 3,32 (m, 4H), 3,16 (t a, J = 11,2 Hz, 2H), 2,99 (dd a, J = 8,0, 16,4 Hz, 3H), 2,90 - 2,77 (m, 1H).
Ejemplo 611

(S)-2-(1-(2-fluoroacriloil)-4-(7-(8-metilnaftalen-1 -il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-1-carboxilato de bencilo. A una mezcla de (25)-2-(cianometil)-4-(5,6,7,8-tetrahidropirido[3,4-tí]pirimidin-4-il)piperazin-1-carboxilato de bencilo (100 mg, 255 gmol, 1,00 eq.) y 1-bromo-8-metil-naftaleno (113 mg, 510 gmol, 10,6 ul, 2,00 eq.) en tolueno (8,00 ml) se le añadieron Pd2(dba)3 (46,7 mg, 51,0 gmol, 0,20 eq.), RuPhos (47,6 mg, 102 gmol, 0,40 eq.) y CS2C03 (249 mg, 764 gmol, 3,00 eq.) en una porción en una atmósfera de N2. La mezcla se agitó a 90 °C durante 5 horas. La mezcla de reacción se diluyó con agua (10,0 ml) y se extrajo con acetato de etilo (20,0 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ,0 ml * 1 ), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. Las fracciones deseadas se recogieron, se neutralizaron con una solución saturada de NaHC03 (5,00 ml) y se extrajeron con acetato de etilo (50,0 ml * 2). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El compuesto (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (150 mg, bruto) se obtuvo en forma de un sólido de color amarillo.
Etapa B: 2-[(2S)-4-[7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-metil-1 -naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1 -carboxilato de bencilo (50,0 mg, 93,9 gmol, 1,00 eq.) en metanol (1,00 ml) se le añadieron NH3*Me0H (93,9 gmol, 0,2 ml, 20 % de pureza, 1,00 eq.) y Pd/C (15,0 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. La mezcla de reacción se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Waters Xbridge 150 * 255 ufase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN];% de B: 38 %-62 %, 10 min). El compuesto 2-[(2S)-4-[7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (6,13 mg, 15,3 gmol, 16 % de rendimiento, 99,5 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 399.
1H RMN (400 MHz, cloroformo-d) ó = 8,61 (d, J = 4,8 Hz, 1H), 7,74 - 7,63 (m, 2H), 7,41 (dt, J = 2,8, 7,6 Hz, 1H), 7,37 - 7,31 (m, 1H), 7,27 - 7,22 (m, 2H), 4,32 (d a, J = 18,0 Hz, 1H), 4,07 - 3,92 (m, 1H), 3,92 - 3,86 (m, 1H), 3,86 - 3,67 (m, 1H), 3,56 - 3,48 (m, 1H), 3,30 - 3,24 (m, 1H), 3,24 - 3,18 (m, 1H), 3,18 - 2,96 (m, 4H), 2,94 (d, J = 2,4 Hz, 3H), 2,92 (s a, 1H), 2,92 - 2,83 (m, 1H), 2,69 - 2,59 (m, 1H), 2,57 - 2,53 (m, 2H).
Etapa C: 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-illacetonitrilo. A una mezcla de 2-[(2S)-4-[7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo (60,0 mg, 151 gmol, 1,00 eq.) en acetato de etilo (1,00 ml) se le añadieron en una porción ácido 2-fluoroprop-2-enoico (54,2 mg, 602 gmol, 4,00 eq.), TEA (244 mg, 2,41 mmol, 335 ul, 16,0 eq.) y T3P (575 mg, 903 gmol, 537 ul, 50 % de pureza, 6,00 eq.) a 0 °C en una atmósfera de N2. La mezcla se agitó a 25 °C durante 30 min. La mezcla de reacción se inactivó con la adición de agua (1,00 ml) a 0 °C y después se extrajo con acetato de etilo (10 ml * 3). Las capas orgánicas combinadas se lavaron con salmuera (5 ml * 1), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Waters Xbridge 150 * 255 ufase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN1; % de B: 40 %-70 %, 10 min). El compuesto 2-[(2S)-1-(2- fluoroprop-2-enoil)-4-[7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo (8,97 mg, 18,7 gmol, 12 % de rendimiento, 98,2 % de pureza) se obtuvo en forma de un sólido de color amarillo. Lc Ms [ESI, M+11: 492.
1H RMN (400 MHz, cloroformo-d) ó = 8,64 (d, J = 7,2 Hz, 1H), 7,74 - 7,64 (m, 2H), 7,47 - 7,39 (m, 1H), 7,39 - 7,32 (m, 1H), 7,29 (d a, J = 1,2 Hz, 1H), 7,26 - 7,19 (m, 1H), 5,53 - 5,33 (m, 1H), 5,26 (dd, J = 3,6, 17,2 Hz, 1H), 4,87 (s a, 1H), 4,33 (dd a, J = 14,8, 18,0 Hz, 1H), 4,19 (d a, J = 13,6 Hz, 1H), 4,12 - 3,97 (m, 2H), 3,95 - 3,84 (m, 1H), 3,61 - 3,42 (m, 2H), 3,27 - 3,04 (m, 4H), 2,92 (d, J = 2,4 Hz, 3H), 2,89 - 2,62 (m, 3H).
Ejemplo 612
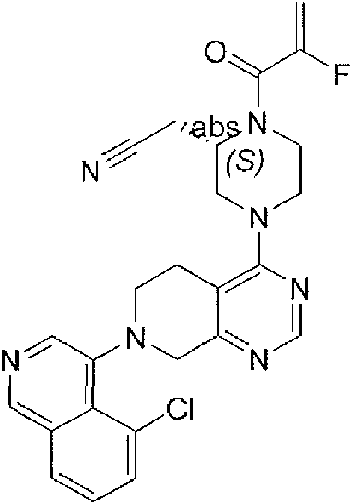
(S)-2-(4-(7-(5-cloroisoquinolin-4-il)-5,6,7,8-tetrahidropirido[3,4-<C]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo

Etapa A: (2S)-4-r7-(5-cloro-4-isoquinolil)-6,8-dihidro-5H-piridor3,4-cf1pirimidin-4-il)-2-(cianometil)piperazin-1-carboxilato de bencilo. A una mezcla de (2S)-2-(cianometil)-4-(5,6,7,8-tetrahidropirido[3,4-C]pirimidin-4-il)piperazin-1-carboxilato de bencilo (300 mg, 764 μmol, 1,00 eq.) y 4-bromo-5-cloro-isoquinolina (371 mg, 1,53 mmol, 10,6 μl, 2,00 eq.) en tolueno (15,0 ml) se le añadieron en una porción Pd2(dba)3 (140 mg, 153 μmol, 0,20 eq.), RuPhos (143 mg, 306 μmol, 0,40 eq.) y Cs2C03 (747 mg, 2,29 mmol, 3,00 eq.) en una atmósfera de N2. La mezcla se desgasificó, se purgó 3 veces con N2 y se agitó a 90 °C durante 5 horas. La mezcla de reacción se diluyó con agua (10,0 ml) y se extrajo con acetato de etilo (20,0 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (20,0 ml x 1), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. Las fracciones deseadas se recogieron, se neutralizaron con una solución saturada de NaHC03 (5 ml) y se extrajeron con acetato de etilo (50 ml x 2). La capa orgánica separada se secó sobre sulfato de sodio, se filtró y se concentró al vacío. El compuesto (2S)-4-[7-(5-cloro-4-isoquinolil)-6,8-dihidro-5H-pirido[3,4-C]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (130 mg, 223 pmol, 29 % de rendimiento, 95 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 554.
Etapa B: 2-í(2S)-4-í7-(5-cloro-4-isoquinolil)-6,8-dihidro-5H-pirido(3,4-C)pirimidin-4-i n piperazin-2-i n acetonitrilo. A una solución de (2S)-4-[7-(5-cloro-4-isoquinolil)- 6,8-dihidro-5H-pirido[3,4-C]pirimidin-4-il]-2-(cianometil)piperazin-1 -carboxilato de bencilo (30,0 mg, 54,2 μmol, 1,00 eq.) en metanol (3,00 ml) se le añadieron NH3*Me0H (0,50 ml, 20 % de pureza) y Pd/C (35,0 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. La mezcla de reacción se filtró y se concentró a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Waters Xbridge 150 x 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 22 %-46 %, 10 min). El compuesto 2-[(2S)-4-[7-(5-cloro-4-isoquinolil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (8,51 mg, 20,1 μmol, 37 % de rendimiento, 99,4 % de pureza) se obtuvo en forma de un sólido de color blanco. LCMS [ESI, M+1]: 420.
1H RMN (400 MHz, cloroformo-d) ó = 9,02 (s, 1H), 8,60 (d, J = 4,4 Hz, 1H), 8,35 (d, J = 1,6 Hz, 1H), 7,91 (dd, J = 1,2, 8,4 Hz, 1H), 7,75 (dd, J = 1,2, 7,2 Hz, 1H), 7,50 (t, J = 7,6 Hz, 1H), 4,54 (d a, J = 17,2 Hz, 1H), 4,06 - 3,61 (m, 4H), 3,41 - 3,08 (m, 5H), 3,07 - 2,85 (m, 2H), 2,72 - 2,59 (m, 1H), 2,58 - 2,49 (m, 2H).
Etapa C: 2-[(2S)-4-[7-(5-cloro-4-isoquinolil)-6,8-dihidro-5H-pirido[3,4-(C]pirimidin-4-il1-1-(2-fluoroprop-2-enoil)piperazin-2-il1acetonitrilo. A una mezcla de 2-[(2S)-4-[7-(5-cloro-4-isoquinolil)-6,8-dihidro-5H-pirido[3,4-C]pirimidin-4-il]piperazin-2-il]acetonitrilo (60,0 mg, 143 μmol, 1,00 eq.) en acetato de etilo (0,80 ml) se le añadieron en una porción ácido 2-fluoroprop-2-enoico (25,7 mg, 286 μmol, 2,00 eq.), TEA (86,8 mg, 857 μmol, 119 ul, 6,00 eq.) y T3P (273 mg, 429 μmol, 255 ul, 50 % de pureza, 3,00 eq.) a 0 °C en una atmósfera de N2. La mezcla se agitó a 25 °C durante 30
min. La mezcla de reacción se inactivó con la adición de agua (1,00 ml) a 0 °C y después se extrajo con acetato de etilo (10,0 ml x 3 ). Las capas orgánicas combinadas se lavaron con salmuera (5,00 ml x 1), se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por HPLC prep. (columna: Waters Xbridge 150 x 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN];% de B: 22 %-52 %, 10 min). El compuesto 2-[(2S)-4-[7-(5-cloro-4-isoquinolil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (7,88 mg, 16,0 gmol, 11 % de rendimiento, 100 % de pureza) se obtuvo en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 492.
1H RMN (400 MHz, cloroformo-d) 5 = 9,04 (d, J = 3,6 Hz, 1H), 8,65 (d, J = 6,4 Hz, 1H), 8,35 (d, J = 11,6 Hz, 1H), 7,96 - 7,88 (m, 1H), 7,76 (dd, J = 1,2, 7,2 Hz, 1H), 7,56 - 7,47 (m, 1H), 5,55 - 5,33 (m, 1H), 5,26 (dd, J = 3,2, 16,8 Hz, 1H), 5,03 - 4,70 (m, 1H), 4,55 (dd a, J = 6,4, 18,0 Hz, 1H), 4,23 - 3,84 (m, 4H), 3,69 (d a, J = 6,4 Hz, 1H), 3,50 (d a, J = 13,6 Hz, 1H), 3,35 - 3,19 (m, 3H), 3,16 - 3,00 (m, 1H), 2,97 - 2,61 (m, 3H).
Ejemplo 613

2-((S)-4-(2-(((S)-4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(5,6-dimetil-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-(2-fluoroacriloil)piperazin-2-il)acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[7-(5,6-dimetil-1-tetrahidropiran-2-il-indazol-4-il)-2-metilsulfanil-6,8-dihidro-5H-pirido[3.4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo. Una mezcla de (2S)-2-(cianometil)-4-(2-metilsulfanil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (5.0 g. 11,4 mmol, 1.0 eq.), 4-bromo-5,6-dimetil-1-tetrahidropiran-2-il-indazol (7.05 g. 22,8 mmol. 2.0 eq.). Pd2(dba)3 (2.09 g. 2,28 mmol. 0.2 eq.). RuPhos (2,13 g. 4,56 mmol. 0.4 eq.) y CS2C03 (9.29 g. 28,5 mmol. 2.5 eq.) en tolueno (100 ml) se desgasificó y se purgó 3 veces con N2 y después la mezcla se agitó a 90 °C durante 8 horas en una atmósfera de N2. La mezcla de reacción se diluyó con agua (100 ml) y se extrajo con acetato de etilo (3 * 200 ml). Las capas orgánicas combinadas se lavaron con salmuera (100 ml). se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 20/1 a 2/1) y se purificó adicionalmente por cromatografía ultrarrápida de fase inversa [agua (0.1 % de ácido fórmico)/acetonitrilo)]. La mezcla se ajustó a pH ~7 con una solución acuosa saturada de NaHC03 y se extrajo con acetato de etilo (3 * 100 ml). Las capas orgánicas combinadas se lavaron con salmuera (50 ml). se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar el producto. Se obtuvo (2S)-2-(cianometil)-4-[7-(5,6-dimetil-1-tetrahidropiran-2-il-indazol-4-il)-2-metilsulfanil-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (4.90 g. 7,20 mmol.
63 % de rendimiento. 98 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 667.1
1H RMN (400 MHz, cloroformo-d) ó = 7,98 (s, 1H), 7,50 - 7,31 (m. 5H). 7,22 (s. 1H). 5,66 (dd. J = 2.4. 9.6 Hz. 1H). 5,27 - 5,13 (m. 2H). 4,69 (s a. 1H). 4,27 (s. 2H). 4,13 - 3,97 (m. 3H). 3,89 (d. J = 11,6 Hz. 1H). 3,81 - 3,68 (m. 1H). 3,51 (t. J = 5.2 Hz. 2H). 3,30 (s a. 2H). 3,04 (t. J = 11,6 Hz. 1H). 2,93 - 2,66 (m. 4H). 2,62 - 2,48 (m. 4H). 2,43 (s. 3H). 2,32 (s.
3H). 2,23 - 2,12 (m. 1H). 2,11 - 2,05 (m. 1H). 1,85 - 1.67 (m. 3H).
Etapa B: (2S)-2-(c¡anomet¡l)-4-[7-(5.6-d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-2-met¡lsulf¡n¡l-6.8-d¡h¡dro-5H-p¡r¡dof3.4-c1p¡r¡m¡din-4-¡l1p¡peraz¡n-1-carbox¡lato de bencilo. A una soluc¡ón de (2S)-2-(c¡anomet¡l)-4-[7-(5,6-d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-2-met¡lsulfan¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (500 mg. 749 μmol. 1.0 eq.) en tolueno (10 ml) se le añad¡ó m-CPBA (152 mg. 749 μmol. 85 % de pureza.
1.0 eq.). La mezcla se ag¡tó a 0 °C durante 1 hora. La mezcla se d¡luyó con agua (10 ml) y se ajustó a pH ~7 con una soluc¡ón acuosa saturada de NaHCÜ3. Después. la mezcla se extrajo con acetato de et¡lo (3 x 20 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (20 ml). se secaron sobre Na2SÜ4. se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar el producto. Se obtuvo (2S)-2-(c¡anomet¡l)-4-[7-(5.6-d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-2-met¡lsulf¡n¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (400 mg. 539 μmol. 72 % de rend¡m¡ento. 92 % de pureza) en forma de un sól¡do de color amar¡llo y se usó d¡rectamente en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón. LCMS [ESI. M+1]: 683.
Etapa C: (2S)-2-(c¡anomet¡l)-4-[2-[[(2S)-4.4-d¡fluoro-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-7-(5.6-d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de benc¡lo. A una soluc¡ón de (2S)-2-(c¡anomet¡l)-4-[7-(5.6-d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-2-met¡lsulf¡n¡l-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (900 mg. 1.32 mmol. 1.0 eq.) en tolueno (20 ml) se le añad¡eron t-Bu0Na (379 mg.
3.95 mmol. 3.0 eq.) y [(2S)-4.4-d¡fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metanol (398 mg. 2.64 mmol. 2.0 eq.). La mezcla se ag¡tó a 0 °C durante 0.5 horas. La mezcla de reacc¡ón se d¡luyó con agua (30 ml) y se extrajo con acetato de et¡lo (3 x 50 ml).
Las capas orgán¡cas comb¡nadas se lavaron con salmuera (20 ml). se secaron sobre Na2S04. se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar un res¡duo. El res¡duo se pur¡f¡có por cromatografía en columna (S¡02. acetato de et¡lo/metanol = 100/1 a 10/1). Se obtuvo (2S)-2-(c¡anomet¡l)-4-[2-[[(2S)-4.4-d¡fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(5,6-d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-6,8-d¡h¡dro-5H-p¡r¡doi3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (630 mg. 793 μmol. 60 % de rend¡m¡ento. 97 % de pureza) en forma de un sól¡do de color amar¡llo. LCMS [ESI. M+1]: 770.
Etapa D: 2-[(2S)-4-[2-[[(2S)-4.4-d¡fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-7-(5.6- d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una soluc¡ón de (2S)-2-(c¡anomet¡l)-4-[2-[[(2S)-4,4-d¡fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(5,6-d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-6,8-d¡h¡dro-5H-p¡r¡do'[3,4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-1-carbox¡lato de benc¡lo (400 mg. 519 μmol. 1.0 eq.) en metanol (2 ml) se le añad¡eron Pd/C seco (50 mg. 10 % de pureza) y NH3/metanol (1 ml. 20 % de pureza) en una atmósfera de N2. La suspens¡ón se desgas¡f¡có al vacío y se purgó var¡as veces con H2. La mezcla se ag¡tó en una atmósfera de H2 (103.42 kPa (15 ps¡)) a 25 °C durante 0.5 horas. La mezcla se concentró al vacío. Se obtuvo 2-[(2S)-4-[2-[[(2S)-4.4-d¡fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(5,6- d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (330 mg. 519 μmol. 99 % de rend¡m¡ento) en forma de un sól¡do de color amar¡llo y se usó d¡rectamente en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón. LCMS [ESI. M+1]: 636.
Etapa E: 2-rí2S)-4-r2-rrí2S)-4.4-d¡fluoro-1-met¡l-p¡rrol¡d¡n-2-¡llmetox¡l-7-í5.6-d¡met¡l-1H-¡ndazol-4-¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo. A una soluc¡ón de 2-[(2S)-4-[2-[[(2S)-4.4-d¡fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-7-(5.6-d¡met¡l-1-tetrah¡drop¡ran-2-¡l-¡ndazol-4-¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l]aceton¡tr¡lo (50 mg. 78.6 μmol. 1.0 eq.) en d¡clorometano (50 ul) se le añad¡ó TFA (359 mg. 3.15 mmol. 233 μl.
40 eq.). La mezcla se ag¡tó a 0 °C durante 0.5 horas. La mezcla se concentró al vacío y se d¡luyó con agua (10 ml). La mezcla se ajustó a pH ~8 con una soluc¡ón acuosa saturada de NaHC03 y se extrajo con d¡clorometano (3 x 20 ml).
Las capas orgán¡cas comb¡nadas se lavaron con salmuera (20 ml). se secaron sobre Na2S04. se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar el producto. El res¡duo se pur¡f¡có por cromatografía en columna (S¡02. acetato de et¡lo/metanol = 100/1 a 10/1) y se pur¡f¡có ad¡c¡onalmente por HPLC prep. (columna: Waters Xbr¡dge 150 * 25 5 u; fase móv¡l: [agua (0.05 % de h¡dróx¡do de amon¡aco v/v) - ACN]; % de B: 30 % - 60 %. 10 m¡n). La fracc¡ón deseada se recog¡ó y se l¡of¡l¡zó. Se obtuvo 2-[(2S)-4-[2-[[(2S)-4,4-d¡fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l]metox¡]-7-(5,6-d¡met¡l-1H-¡ndazol-4-¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1p¡peraz¡n-2-¡l1aceton¡tr¡lo (7.29 mg. 13.2 μmol. 17 % de rend¡m¡ento. 99.7 % de pureza) en forma de un sól¡do de color blanco. LCMS [ESI. M+1]: 552.
1H RMN (400 MHz. cloroformo-d) 6 = 9.95 (s a. 1H). 8.04 (s. 1H). 7.15 (s. 1H). 4.48 (dd. J = 4.4. 10.8 Hz. 1H). 4.33 -4.19 (m. 3H). 4.00 (d a. J = 11.6 Hz. 1H). 3.83 (d a. .7 = 11.6 Hz. 1H). 3.51 (t. J = 5.2 Hz.2H). 3.42 (dt. J = 5.6. 11.7 Hz.
1H). 3.26 (s a. 1H). 3.17 - 2.95 (m. 4H). 2.94 - 2.85 (m. 1H). 2.84 - 2.62 (m. 3H). 2.61 - 2.49 (m. 3H). 2.47 (s. 3H). 2.42 (s. 3H). 2.35 (s. 3H). 2.32 - 2.18 (m. 1H).
Etapa F: 2-[(2S)-4-[2-[[(2S)-4.4-d¡fluoro-1-met¡l-p¡rrol¡d¡n-2-¡l1metox¡1-7-(5.6-d¡met¡l-1H-¡ndazol-4-¡l)-6.8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡n-4-¡l1-1-(2-fluoroprop-2-eno¡l)p¡peraz¡n-2-¡l1laceton¡tr¡lo. A una soluc¡ón de 2-[(2S)-4-[2-[[(2S)-4.4-d¡fluoro-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(5,6-d¡met¡l-1H-¡ndazol-4-¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (100 mg. 181 μmol. 1.0 eq.) y ác¡do 2-fluoroprop-2-eno¡co (24.5 mg. 272 μmol. 1.5 eq.) en DMF (2 ml) se le añad¡eron T3P (461 mg. 725 μmol. 431 μl. 50 % de pureza en acetato de et¡lo. 4.0 eq.) y TEA (147 mg.
1.45 mmol. 202 ul. 8.0 eq.) a 0 °C. La mezcla se ag¡tó a 0 °C durante 0.5 horas. La mezcla de reacc¡ón se d¡luyó con agua (20 ml) y se extrajo con acetato de et¡lo (3 x 30 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (20 ml). se secaron sobre Na2S04. se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar un res¡duo. El res¡duo se pur¡f¡có por cromatografía en columna (S¡02. acetato de et¡lo/metanol = 100/1 a 10/1) y se pur¡f¡có ad¡c¡onalmente por HPLC prep. (columna: Waters Xbr¡dge 150 * 255 u; fase móv¡l: [agua (0.05 % de h¡dróx¡do de amon¡aco v/v) - ACN];
% de B: 38 % - 68 %, 10 min). La fracción deseada se recogió y se liofilizó. Se obtuvo 2-[(2S)-4-[2-[[(2S)-4,4-difluoro-1-metil-pirrolidin-2-il]metoxi]-7-(5,6-dimetil-1H-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (11 mg, 17,5 μmol, 10 % de rendimiento, 99,2 % de pureza, 100 % de ee) en forma de un sólido de color blanquecino. LCMS [ESI, M+1]: 624.
1H RMN (400 MHz, cloroformo-d) ó = 10,27 - 9,49 (s, 1H), 8,04 (s, 1H), 7,16 (s, 1H), 5,55 - 5,34 (m, 1H), 5,26 (dd, J = 3,6, 16,8 Hz, 1H), 4,86 (s a, 1H), 4,46 (dd, J = 4,8, 10,8 Hz, 1H), 4,34 - 3,88 (m, 6H), 3,66 - 3,25 (m, 5H), 3,19 - 2,62 (m, 7H), 2,60 - 2,45 (m, 4H), 2,42 (s, 3H), 2,35 (s, 3H), 2,32 - 2,19 (m, 1H).
Ejemplo 614

2-((S)-4-(2-(((S)-4,4-difluoro-1-metilpirrolidin-2-il)metoxi)-7-(5,6-dimetil-1H-indazol-4-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)-1-((E)-4-fluorobut-2-enoil)piperazin-2-il)acetonitrilo

Etapa A: 2-[(2S)-4-[2-[[(2S)-4,4-difluoro-1-metil-pirrolidin-2-il]metoxi]-7-(5,6-dimetil-1H-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-1-[(E)-4-fluorobut-2-enoil1piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[2-[[(2S)-4,4-difluoro-1 -metil-pirrolidin-2-il]metoxi]-7-(5,6-dimetil-1 W-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-tí]pirim idin-4-il]piperazin-2-il]acetonitrilo (100 mg, 181 μmol, 1,0 eq.) y ácido (E)-4-fluorobut-2-enoico (28,3 mg, 272 μmol, 1,5 eq.) en DMF (2 ml) se le añadieron T3P (461 mg, 725 μmol, 431 ul, 50 % de pureza en acetato de etilo, 4,0 eq.) y TEA (147 mg, 1,45 mmol, 202 ul, 8,0 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (3 * 30 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiÜ2, acetato de etilo/metanol = 100/1 a 10/1) y se purificó adicionalmente por HPLC prep. (columna: Xtimate C18 150 * 25 mm * 5 um; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) -ACN]; % de B: 40 % - 70 %, 10 min). La fracción deseada se recogió y se liofilizó. Se obtuvo 2-[(2S)-4-[2-[[(2S)-4,4-difluoro-1 -metil-pirrolidin-2-il]metoxi]-7-(5,6-dimetil-1 W-indazol-4-il)-6,8-dihidro-5H-pirido[3,4-tí]pirim idin-4-il]-1 -[(E)-4-fluorobut-2-enoil]piperazin-2-il]acetonitrilo (12 mg, 18,3 μmol, 10 % de rendimiento, 97,3 % de pureza, 95 % de ee) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 638.
1H RMN (400 MHz, cloroformo-d) 5 = 10,06 (s a, 1H), 8,05 (s, 1H), 7,17 (s, 1H), 7,07 - 6,97 (m, 1H), 6,61 (d a, J = 14,8 Hz, 1H), 5,20 - 5,07 (m, 3H), 4,48 (dd, J = 4,8, 11,2 Hz, 1H), 4,31 - 4,27 (m, 3H), 4,20 - 3,80 (m, 3H), 3,55 (t a, J = 5,2 Hz, 3H), 3,49 - 3,27 (m, 2H), 3,13 (s a, 1H), 3,05 - 2,63 (m, 6H), 2,62 - 2,46 (m, 4H), 2,43 (s, 3H), 2,36 (s, 3H), 2,33 - 2,21 (m, 1H).
Ejemplo 615

2-[(2S)-1-(2-fluoroprop-2-eno¡l)-4-[7-(8-met¡l-1-naft¡l)-2-[[(2S)-1,4,4-tr¡met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo

Etapa A: (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1,4,4-tr¡met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo. A una soluc¡ón de [(2S)-1,4,4-tr¡met¡lp¡rrol¡d¡n-2-¡l]metanol (217 mg, 1,51 mmol, 68,2 gl, 3 eq.) y (2S)-2-(c¡anomet¡l)-4-[7-(8-met¡l-1-naft¡l)-2-met¡lsulf¡n¡l-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de benc¡lo (300 mg, 504 gmol, 1 eq.) en tolueno (15 ml) se le añad¡ó t-Bu0Na (145 mg, 1,51 mmol, 3 eq.) a 0 °C. La mezcla se ag¡tó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se ¡nact¡vó con agua (10 ml) y se extrajo con acetato de et¡lo (2 * 50 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (1 * 50 ml), se secaron sobre Na2S04, se f¡ltraron y se concentraron al vacío. El res¡duo se pur¡f¡có por cromatografía ultrarráp¡da de fase ¡nversa [agua (0,1 % de AF)/aceton¡tr¡lo]. Las fracc¡ones deseadas se recog¡eron, se neutral¡zaron con una soluc¡ón saturada de Na2C03 (10 ml) y se extrajeron con acetato de et¡lo (3 * 50 ml). Las capas orgán¡cas separadas se secaron sobre sulfato de sod¡o, se f¡ltraron y se concentraron al vacío. Se
obtuvo (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1,4,4-trimetilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-7]pirimidin-4-il]piperazin-1-carboxilato de bencilo (180 mg, 267 μmol, 53 % de rendimiento, 99,8 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 675.
1H RMN (400 MHz, cloroformo-d) 5 = 7,73 - 7,60 (m, 2H), 7,27 (s, 9H), 5,21 (s, 2H), 4,68 (s a, 1H), 4,47 - 4,34 (m, 1H), 4,30 - 4,15 (m, 2H), 4,13 - 4,04 (m, 1H), 3,95 - 3,69 (m, 2H), 3,56 - 3,34 (m, 2H), 3,28 - 2,48 (m, 13H), 2,47 - 2,33 (m, 3H), 2,22 - 2,11 (m, 1H), 1,88 (dd a, J = 8,4, 12,6 Hz, 1H), 1,56 (ddd, J = 4,6, 7,8, 12,6 Hz, 1H), 1,16 (d, J = 4,0 Hz, 3H), 1,10 - 0,98 (m, 3H).
Etapa B: 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1,4,4-trimetilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1,4,4-trimetilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-1-carboxilato de bencilo (130 mg, 193 μmol, 1 eq.) en Me0H (4 ml) se le añadieron NHs^Me0H (2 ml, 15 % de pureza) y Pd/C (26 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 0,5 horas. Después de que se completara, la mezcla se filtró y el filtrado se concentró al vacío. Se obtuvo 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1,4,4-trimetilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (100 mg, 159 μmol, 85,9 % de pureza) en forma de un sólido de color amarillo que se usó en la siguiente etapa sin purificación adicional. LCMS [ESI, M+1]: 540.
Etapa C: 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1,4,4-trimetilpirrolidin-2-il1metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il]piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1,4,4-trimetilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (90 mg, 143 μmol, 1 eq.), ácido 2-fluoroprop-2-enoico (38,7 mg, 430 μmol, 3 eq.) y Et3N (130 mg, 1,29 mmol, 179 ul, 9 eq.) en acetato de etilo (15 ml) se le añadió T3P (365 mg, 573 μmol, 341 ul, 50 % de pureza en acetato de etilo, 4 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 1 hora. Después de que se completara, la mezcla se diluyó con agua (3 ml). La capa orgánica se separó, se lavó con salmuera (1 * 10 ml), se secó sobre sulfato de sodio, se filtró y se concentró al vacío. La mezcla se purificó por cromatografía en columna (Si02, acetato de etilo/metanol = 20/1 a 10/1). El residuo se purificó por HPLC prep. (columna: Waters Xbridge 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - a Cn]; % de B: 62 % - 89 %, 10 min). El residuo se concentró a presión reducida para eliminar el ACN y después se sometió a liofilización. Se obtuvo 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[7-(8-metil-1-naftil)-2-[[(2S)-1,4,4-trimetilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo (12,7 mg, 20,6 μmol, 99,7 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 612.
1H RMN (400 MHz, cloroformo-d) 5 = 7,79 - 7,56 (m, 2H), 7,51 - 7,31 (m, 2H), 7,27 - 7,08 (m, 2H), 5,65 - 5,08 (m, 2H), 4,85 (s a, 1H), 4,49 - 4,32 (m, 1H), 4,30 - 3,96 (m, 4H), 3,95 - 3,71 (m, 1H), 3,59 - 3,35 (m, 2H), 3,28 - 2,54 (m, 13H), 2,42 (s a, 3H), 2,16 (d a, J = 8 ,8 Hz, 1H), 1,96 - 1,81 (m, 1H), 1,63 - 1,45 (m, 1H), 1,15 (s a, 3H), 1,06 (s a, 3H).
Ejemplo 616

2-[(2S)-4-[2-[[(2S,4S)-1,4-dimetilpirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-1-(2-fluoroprop-2 -enoil)piperazin-2 -il]acetonitrilo

Etapa A: (2S)2-(cianometil)-4-[2-[[(2S,4S)-1,4-dimetilpirrolidin-2-il)metoxi1-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-illpiperazin-1-carboxilato de bencilo. A una solución de [(2S,4S)-1,4-dimetilpirrolidin-2-il]metanol (326 mg, 2.52 mmol, 68,2 ul, 3 eq.) y bencilo (2S)-2-(cianometil)-4-[7-(8-metil-1-naftil)-2-metilsulfinil-6,8-dihidro-5H-pirido[3,4-d]p¡rim¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato (500 mg, 841 gmol, 1 eq.) en tolueno (20 ml) se le añadió t-Bu0Na (242 mg, 2.52 mmol, 3 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, la mezcla se inactivó con agua (10 ml) y se extrajo con acetato de etilo (2 * 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (1 * 20 ml), se secaron sobre Na2S04, se filtraron y se concentraron al vacío. El residuo se purificó por HPLC de fase inversa (condición de 0,1 % de AF). El residuo se basificó con una solución acuosa saturada de NaHC03 a pH = 8 y se extrajo con acetato de etilo (3 * 50 ml). La fase orgánica se separó, se secó sobre sulfato de sodio, se filtró y se concentró al vacío. Se obtuvo (2S)-2-(cianomet¡l)-4-[2-[[(2S,4S)-1,4-d¡met¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il1piperazin-1-carboxilato de bencilo (328 mg, 461 gmol, 55 % de rendimiento, 92,7 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 661.
Etapa B: 2-[(2S)-4-[2-[[(2S,4S)-1,4-dimetilpirrolidin-2-i n metox n -7-(8-metil-1-naftil-6,8-dihidro-5H-dihidro[3,4-dlpirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de (2S)-2-(cianometil)-4-[2-[[(2S,4S)-1,4-dimetilpirrolidin-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-1-carbox¡lato de bencilo (100 mg, 151 gmol, 1 eq.) en Me0H (4 ml) se le añadieron NH3*Me0H (2 ml, 20 % de pureza) y Pd/C (20 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 0,5 horas. Después de que se completara, la mezcla se filtró y el filtrado se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Phenomenex Synergi C18150 * 30 mm * 4 um; fase móvil: [agua (0,225 % de AF) - ACN]; % de B: 13 % - 43 %, 10 min). Después de esto, el residuo se purificó por HPLC prep. (columna: Waters Xbridge 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 55 % - 85 %, 10 min). El residuo se concentró a presión reducida para eliminar el ACN y después se sometió a liofilización. Se obtuvo 2-[(2S)-4-[2-[[(2S,4S)-1,4-dimet¡lp¡rrol¡d¡n-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡trilo (13,9 mg, 26,4 gmol, 17 % de rendimiento, 100 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 526.
1H RMN (400 MHz, cloroformo-d) ó = 7,69 (d, J = 8,2 Hz, 1H), 7,64 (dd, J = 2,6, 7,6 Hz, 1H), 7,40 (dt, J = 4,0, 7,8 Hz, 1H), 7,36 - 7,31 (m, 1H), 7,26 - 7,19 (m, 2H), 4,43 (dt, J = 4,8, 10,2 Hz, 1H), 4,28 - 4,11 (m, 2H), 4,07 - 3,70 (m, 3H), 3,48 (d a, J = 11,8 Hz, 1H), 3,38 - 2,82 (m, 11H), 2,80 - 2,64 (m, 2H), 2,61 - 2,53 (m, 3H), 2,49 (t, J = 8 ,6 Hz, 1H), 2,42 (d, J = 2,4 Hz, 3H), 2,35 - 2,15 (m, 2H), 1,39 - 1,25 (m, 1H), 1,08 (d, J = 6 ,8 Hz, 3H).
Etapa C: 2-[(2S)-4-[2-[[(2S,4S)-1,4-dimetilpirrolidin-2-i n metox n -7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-i n -1-(2-fluoroprop-2-enoil)piperazin-2-i n acetonitrilo. A una solución de 2-[(2S)-4-[2-[[(2S,4S)-1,4-dimetilpirrolidin-2-¡l]metox¡]-7-(8-met¡l-1-naft¡l)-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡tr¡lo (100 mg, 190 gmol, 1 eq.), ácido 2-fluoroprop-2-enoico (51,4 mg, 571 gmol, 3 eq.) y Et3N (173 mg, 1,71 mmol, 238 ul, 9 eq.) en DMF (10 ml) se le añadió T3P (484 mg, 761 gmol, 452 ul, 50 % de pureza, 4 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. Después de que se completara, el residuo se diluyó con agua (10 ml) y se extrajo con acetato de etilo (2 * 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (1 * 20 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío. La mezcla se purificó por cromatografía en columna (S¡02, diclorometano/metanol
= 10/1). Después de esto, el residuo se purificó por HPLC prep. (columna: Xtimate C18 150 * 25 mm * 5 um; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 60 % - 90 %, 10 min). El residuo se concentró a presión reducida para eliminar el ACN y después se sometió a liofilización. Se obtuvo 2-[(2S)-4-[2-[[(2S,4S)-1,4-dimetilpirrolidin-2-il]metoxi]-7-(8-metil-1-naftil)-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (22,2 mg, 37,1 μmol, 19 % de rendimiento, 99,8 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 598.
1H RMN (400 MHz, cloroformo-d) ó = 7,72 - 7,61 (m, 2H), 7,45 - 7,37 (m, 1H), 7,37 - 7,31 (m, 1H), 7,26 - 7,17 (m, 2H), 5,53 - 5,32 (m, 1H), 5,26 (dd, J = 3,6, 16,8 Hz, 1H), 4,86 (s a, 1H), 4,48 - 4,35 (m, 1H), 4,30 - 3,99 (m, 4H), 3,95 - 3,73 (m, 1H), 3,60 - 3,39 (m, 2H), 3,29 - 2,94 (m, 5H), 2,93 - 2,67 (m, 7H), 2,66 - 2,56 (m, 1H), 2,55 - 2,47 (m, 1H), 2,43 (d, J = 4,6 Hz, 3H), 2,35 - 2,13 (m, 2H), 1,36 - 1,25 (m, 1H), 1,09 (dd, J = 3,0, 6,8 Hz, 3H).
Ejemplo 617

2-((S)-1-(2-fluoroacriloil)-4-(7-(3-metilisoquinolin-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo

Etapa A: (2S)-2-(cianometil)-4-[7-(3-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo. Una mezcla de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (300 mg, 593 μmol, 1,0 eq.), 4-bromo-3-metil-isoquinolina (198 mg, 890 μmol, 1,5 eq.), Cs2CÜ3 (483 mg, 1,48 mmol, 2,5 eq.) y BINAP-Pd-G3 (118 mg, 119 μmol, 0,2 eq.) en dioxano (6 ml) se desgasificó y se purgó 3 veces con N2. La mezcla se calentó a 110 °C durante 12 horas. La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (3 * 30 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiÜ2, acetato de
etilo/metanol = 100/1 a 10:1). Se obtuvo (2S)-2-(cianometil)-4-[7-(3-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]pipenazin-1-carboxilato de bencilo (200 mg, 303 μmol, 51 % de rendimiento, 98 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 647.
Etapa B: 2-[(2S)-4-[7-(3-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-illpiperazin-2-illacetonitrilo. A una solución de (2S)-2-(cianometil)-4-[7-(3-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (50 mg, 77,3 μmol, 1,0 eq.) en metanol (2 ml) se le añadieron Pd/C seco (10 mg, 10 % de pureza) y NH3/Me0H (1 ml, 20 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 0,5 horas. La mezcla se concentró al vacío. El residuo se purificó por HPLC prep. (columna: Waters Xbridge 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - a Cn]; % de B: 30 % - 60 %, 10 min). La fracción deseada se recogió y se liofilizó. Se obtuvo 2-[(2S)-4-[7-(3-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (7,81 mg, 14,9 μmol, 19 % de rendimiento, 98,3 % de pureza) en forma de un sólido de color amarillo.
LCMS [ESI, M+1]: 513.
1H RMN (400 MHz, cloroformo-d) 5 = 9,07 (s, 1H), 8,14 (d, J = 8,4 Hz, 1H), 7,97 (d, J = 8,0 Hz, 1H), 7,65 (dt, J = 1,2, 7,6 Hz, 1H), 7,58 - 7,47 (m, 1H), 4,41 (dd a, J = 4,8, 10,4 Hz, 1H), 4,3 (s, 2H), 4,16 (dd, J = 6,8, 10,8 Hz, 1H), 4,10 - 3,93 (m, 1H), 3,91 - 3,77 (m, 1H), 3,57 - 3,37 (m, 2H), 3,36 - 3,21 (m, 1H), 3,18 - 2,85 (m, 6H), 2,75 - 2,61 (m, 5H), 2,56 (d a, J = 6,8 Hz, 2H), 2,48 (s, 3H), 2,33 - 2,22 (m, 1H), 2,12 - 1,98 (m, 1H), 1,94 - 1,80 (m, 3H).
Etapa C: 2-f(2S)-1-(2-fluoroprop-2-enoN)-4-f7-(3-metil-4-isoquinolil)-2-ff(2S)-1-metilpirrolidin-2-i n metox n -6.8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo. A una solución de 2-[(2S)-4-[7-(3-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-7]pirimidin-4-il]piperazin-2-il]acetonitrilo (80 mg, 156 μmol, 1,0 eq.) y ácido 2-fluoroprop-2-enoico (28,1 mg, 312 μmol, 2,0 eq.) en DMF (2 ml) se le añadieron T3P (297 mg, 468 μmol, 278 μl, 50 % de pureza en acetato de etilo, 3,0 eq.) y TeA (126 mg, 1,25 mmol, 173 μl, 8,0 eq.) a 0 °C. La mezcla se agitó a 0 °C durante 0,5 horas. La mezcla de reacción se diluyó con acetato de etilo (20 ml) y se lavó con agua (3 * 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2S04, se filtraron y se concentraron a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, acetato de etilo/metanol = 100/1 a 10/1) y se purificó adicionalmente por HPLC prep. (columna: Waters Xbridge 150 * 25 5 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 35 % - 65 %, 10 min). La fracción deseada se recogió y se liofilizó. Se obtuvo 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[7-(3-metil-4-isoquinolil)-2-([(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (12 mg, 19,9 μmol, 13 % de rendimiento, 97,4 % de pureza, 100 % de ee) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 585.
1H RMN (400 MHz, cloroformo-d) 5 = 9,08 (s, 1H), 8,26 - 8,04 (m, 1H), 7,97 (d a, J = 7,6 Hz, 1H), 7,73 - 7,60 (m, 1H), 7,58 - 7,49 (m, 1H), 5,42 (d, J = 47,6 Hz, 1H), 5,27 (dd, J = 3,6, 16,8 Hz, 1H), 4,85 (s a, 1H), 4,45 - 3,92 (m, 7H), 3,83 - 3,25 (m, 4H), 3,22 - 2,59 (m, 10H), 2,49 (s, 3H), 2,36 - 2,24 (m, 1H), 2,13 - 1,99 (m, 1H), 1,92 - 1,76 (m, 3H).
Ejemplo 618

(S)-2-(cianometil)-4-(7-(7-fluoroIsoquinolin-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo

Etapa A: (S)-2-(cianometil)-4-(7-(7-fluoroisoquinolin-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-dlpirimidin-4-il)piperazin-1 -carboxilato de bencilo. A una solución de (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il]piperazin-1-carboxilato de bencilo (300 mg, 0,59 mmol, 1,0 eq.) en tolueno (10,0 ml) se le añadieron 4-bromo-7-fluoro-isoquinolina (268 mg, 1,19 mmol, 2,0 eq.), RuPhos (111 mg, 237 gmol, 0,40 eq.), Cs2C03 (483 mg, 1,48 mmol, 2,5 eq.) y Pd2(dba)3 (109 mg, 118 gmol, 0,20 eq.) a 25 °C y la mezcla se agitó a 90 °C durante 16 horas. La mezcla de reacción se diluyó con agua (5,0 ml) y se extrajo con acetato de etilo (3 * 5,0 ml). Las capas orgánicas combinadas se lavaron con salmuera (3 * 5,0 ml), se secaron sobre Na2S04, se filtraron y el filtrado se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (Si02, éter de petróleo/acetato de etilo = 1/1 a éter de petróleo/acetato de etilo/Et0H (2 % de NH3H20) = 4/3/1). Después, el producto bruto se purificó por HPLC de fase inversa (condición de 0,1 % de AF). Las fracciones deseadas se recogieron y se concentraron al vacío para dar (S)-2-(cianometil)-4-(7-(7-fluoroisoquinolin-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (105 mg, 160 gmol, 27 % de rendimiento, 99 % de pureza) en forma de un sólido de color amarillo. LCMS [M+1]: 651.
1H RMN (400 MHz, cloroformo-d) 5 = 2,04 - 2,08 (m, 4 H), 2,69 (d, J = 5,64 Hz, 1 H), 2,73 (d, J = 5,52 Hz, 1 H), 2,81 - 2,85 (m, 3 H), 2,87 - 3,02 (m, 3 H), 3,09 - 3,19 (m, 1 H), 3,32 - 3,45 (m, 3 H), 3,45 - 3,55 (m, 1 H), 3,57 - 3,68 (m, 1 H), 3,99 (d a, J = 12,52 Hz, 1 H), 4,09 - 4,17 (m, 1 H), 4,28 - 4,34 (m, 1 H), 4,31 (m, 1 H), 4,46 (dd, J = 11,84, 4,31 Hz, 1 H), 4,63 - 4,74 (m, 1 H), 4,80 (dd a, J = 11,32, 6,82 Hz, 1 H), 5,20 - 5,23 (m, 2H), 5,30 - 5,32 (m, 2 H), 7,35 - 7,42 (m, 5 H), 7,47 - 7,53 (m, 1 H), 7,60 (dd, J = 8,68, 2,56 Hz, 1 H), 8,16 (dd, J = 9,24, 5,25 Hz, 1 H), 8,22 - 8,25 (m, 1 H), 8,95 - 9,00 (m, 1 H).
Etapa B: 2-((S)-4-(7-(7-fluoroisoquinolin-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-dlpirimidin-4-il)piperazin-2-il)acetonitrilo. A una solución de (S)-2-(cianometil)-4-(7-(7-fluoroisoquinolin-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-1-carboxilato de bencilo (30,0 mg, 46,1 gmol, 1,0 eq.) en Me0H (1,0 ml) se le añadieron NH3/Me0H (46,1 gmol, 1,5 ml, 15 % de pureza, 1,0 eq.) y Pd/C (20,0 mg, 46,1 gmol, 10 % de pureza, 1,0 eq.) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2 (93,1 ug, 46,1 gmol, 1,0 eq.). La mezcla se agitó en una atmósfera de H2 (103,42 kPa (15 psi)) a 25 °C durante 1 hora. La mezcla de reacción se filtró a través de a celite y se lavó con Me0H (20 ml). El filtrado se concentró a presión reducida a 45 °C para dar un residuo. El residuo se purificó por HPLC prep. (condición básica) (columna: Xtimate C18150 * 25 mm * 5 um; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - ACN]; % de B: 43 %-73 %, 10 min). La fracción deseada se recogió y se liofilizó. Se obtuvo 2-((S)-4-(7-(7-fluoroisoquinolin-4-il)-2-(((S)-1-metilpirrolidin-2-il)metoxi)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)piperazin-2-il)acetonitrilo (7 mg, 13,3 gmol, 28,8 % de rendimiento, 98 % de pureza) en forma de un sólido de color amarillo. LCMS [M+1]: 517.
1H RMN (400 MHz, cloroformo-d) 5 = 1,75 - 1,90 (m, 3 H), 1,99 - 2,13 (m, 1 H), 2,24 - 2,34 (m, 1 H), 2,45 - 2,51 (m, 3 H), 2,53 - 2,58 (m, 2 H), 2,64 - 2,74 (m, 1 H), 2,80 - 2,88 (m, 2 H), 2,89 - 2,96 (m, 1 H), 2,97 - 3,06 (m, 1 H), 3,07 - 3,18 (m, 3 H), 3,22 - 3,33 (m, 1 H), 3,38 - 3,48 (m, 2 H), 3,82 - 3,89 (m, 1 H), 4,03 (d a, J = 12,36 Hz, 1 H), 4,18 (dd, J = 10,56, 6,69 Hz, 1 H), 4,30 (s, 2 H), 4,40 (dd, J = 10,64, 4,75 Hz, 1 H), 7,49 (ddd, J = 9,16, 8,35, 2,63 Hz, 1 H), 7,60 (dd, J = 8,76, 2,50 Hz, 1 H), 8,17 (dd, J = 9,24, 5,38 Hz, 1 H), 8,24 (s, 1 H), 8,96 (s, 1 H).
Etapa C: 2-[(2S)-4-[7-(7-fluoro-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-illmetoxil-6,8-dihidro-5H-pirido[3,4-dlpirimidin-4-i n -1-(2-fluoroprop-2-enoil)piperazin-2-i n acetonitrilo. A una solución de 2-[(2S)-4-[7-(7-fhioro-4-isoquinolil)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]p¡peraz¡n-2-¡l]aceton¡trilo (90,7 mg, 175 gmol, 1,0 eq.) y ácido 2-fluoroprop-2-enoico (31,6 mg, 351 gmol, 2,0 eq.) en DMF (2 ml) se le añadieron T3P (335 mg, 526 gmol, 313 gl, 50 % de pureza en acetato de etilo, 3,0 eq.) y TEA (142 mg, 1,40 mmol, 195 gl, 8,0 eq.) a 0 °C. La mezcla se
agitó a 0 °C durante 0,5 horas. La mezcla de reacción se diluyó con acetato de etilo (20 ml) y se lavó con agua (3 * 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SÜ4, se filtraron y el filtrado se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (SiÜ2, acetato de etilo/metanol = 100/1 a 10/1) y se purificó adicionalmente por HPLC prep. (columna: Waters Xbridge 150 * 25 mm * 5 um; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v) - Ac N]; % de B: 32 % - 59 %, 10 min). La fracción deseada se recogió y se liofilizó. Se obtuvo 2-[(2S)-4-[7-(7-fluoro-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-1-(2-fluoroprop-2-enoil)piperazin-2-il]acetonitrilo (8 mg, 13,4 gmol, 7,6 % de rendimiento, 98,3 % de pureza, 100 % de ee) en forma de un sólido de color blanco. LCMS [M+1]: 589.
1H RMN (400 MHz, cloroformo-d) ó = 8,97 (s, 1H), 8,24 (s, 1H), 8,16 (dd, J = 5,2, 9,2 Hz, 1H), 7,60 (dd, J = 2,4, 8,8 Hz, 1H), 7,54 - 7,40 (m, 1H), 5,55 - 5,33 (m, 1H), 5,27 (dd, J = 3,6, 16,8 Hz, 1H), 4,87 (s a, 1H), 4,44 - 3,94 (m, 7H), 3,72 -3,28 (m, 4H), 3,22 - 3,06 (m, 2H), 3,04 - 2,93 (m, 2H), 2,93 - 2,74 (m, 2H), 2,72 - 2,66 (m, 1H), 2,49 (s, 3H), 2,36-2,24 (m, 1H), 2,13 - 2,03 (m, 1H), 1,92 - 1,76 (m, 3H).
Ejemplo 619

2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[7-(5-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo

Etapa A: (2S)-4-[7-(1-cloro-5-metil-4-isoquinolil)-2-[((2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-2-(cianometil)piperazin-1 -carboxilato de bencilo. Se desgasificaron (2S)-2-(cianometil)-4-[2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-5,6,7,8-tetrahidropirido[3,4-d]pirim idin-4-il]piperazin-1 -carboxilato de bencilo (300 mg, 593 gmol, 1,0 eq.), 4-bromo-1-cloro-5-metil-isoquinolina (198 mg, 771 gmol, 1,3 eq.), Pd2(dba)3 (109 mg, 119 gmol, 0,2 eq.) y Cs2CÜ3 (483 mg, 1,48 mmol, 2,5 eq.), XantPhos (137 mg, 237 gmol, 0,4 eq.) en tolueno (10 ml) y después se calentaron a 95 °C durante 10 horas en una atmósfera de N2. Después de que se completara, la mezcla se filtró y el filtrado se concentró al vacío. El residuo se purificó por cromatografía (Al2Ü3, éter de petróleo/acetato de etilo 2/1 a 0/1) seguido de cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. Las fracciones deseadas se recogieron, se neutralizaron con NaHCÜ3 sólido, se concentraron al vacío para eliminar el MeCN y se extrajeron
con acetato de etilo (2 x 15 ml). Las capas orgánicas se secaron sobre Na2SÜ4 y se concentraron al vacío para dar (2S)-4-[7-(1-cloro-5-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (150 mg, 209 μmol, 35 % de rendimiento, 95 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 681.
1H RMN (400 MHz, cloroformo-d) 5 = 8,27 (d, J = 8,0 Hz, 1H), 8,08 - 7,99 (m, 1H), 7,60 - 7,51 (m, 2H), 7,44 - 7,36 (m, 5H), 5,25 - 5,17 (m, 2H), 4,74 - 4,60 (m, 1H), 4,41 - 4,32 (m, 1H), 4,28 - 4,14 (m, 2H), 4,08 - 3,88 (m, 2H), 3,83 (d a, J = 17,6 Hz, 1H), 3,61 - 3,51 (m, 1H), 3,50 - 3,36 (m, 1H), 3,31 - 2,94 (m, 6H), 2,91 (d, J = 2,4 Hz, 3H), 2,81 - 2,59 (m, 4H), 2,46 (d, J = 4,0 Hz, 3H), 2,33 - 2,21 (m, 1H), 2,06 - 1,97 (m, 1H), 1,92 - 1,72 (m, 3H).
Etapa B: 2-[(2S)-4-[7-(5-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-illpiperazin-2-illacetonitrilo. A una solución de (2S)-4-[7-(1-cloro-5-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]-2-(cianometil)piperazin-1-carboxilato de bencilo (130 mg, 191 μmol, 1 eq.) en Me0H (3 ml) se le añadió NHs^Me0H (2 ml, 20 % de pureza), Pd/C (65 mg, 10 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó a 25 °C durante 2 horas. Después de que se completara, el catalizador se eliminó por filtración a través de un lecho de celite.
El disolvente se eliminó a presión reducida para dar 80 mg de producto bruto. Se tomaron 20 mg del producto bruto y se purificaron por HPLC prep. (columna: Waters Xbridge 150 * 25 5 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 28 %-58 %, 10 min). Las fracciones deseadas se recogieron y se liofilizaron para dar 2 [(2S)-4-[7-(5-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (10,8 mg, 21,0 μmol, 44 % de rendimiento, 99,9 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 513.
Etapa C: 2-f(2S)-1-(2-fluoroprop-2-enoil)-4-f7-(5-metil-4-isoquinolil)-2-f((2S)-1-metilpirrolidin-2-i n metox n -6,8-dihidro-5H-pirido[3,4-d^pirimidin-4-il1piperazin-2-il1acetonitrilo. A una solución de ácido 2-fluoroprop-2-enoico (24,6 mg, 273 μmol, 2,0 eq.) y DIEA (70,6 mg, 546 μmol, 95,1 μl, 4,0 eq.) en DCM (1,4 ml) se le añadió HATU (77,9 mg, 205 μmol, 1,5 eq.) a 0 °C. Después de agitar a 0 °C durante 20 minutos, a la mezcla se le añadió 2-[(2S)-4-[7-(5-metil-4-isoquinolil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5^-pirido[3,4-d]pirimidin-4-il]piperazin-2-il]acetonitrilo
(70 mg, 137 μmol, 1,0 eq.). La mezcla se agitó a 25 °C durante 40 minutos. Después de que se completara, la mezcla se diluyó con agua (2 ml) y se extrajo con diclorometano (6 x 5 ml). Las capas orgánicas se secaron sobre Na2SÜ4 y se concentraron al vacío. El residuo se purificó por cromatografía (Al2Ü3, éter de petróleo/acetato de etilo 1/1 a acetato de etilo/metanol 10/1) seguido de HPLC prep. (columna: Waters Xbridge 150 * 255 u; fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN]; % de B: 35 %-65 %, 10 min). Las fracciones deseadas se recogieron y se liofilizaron para dar 2-[(2S)-1-(2-fluoroprop-2-enoil)-4-[7-(5-metil-4-isoquinolil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-cf]pirimidin-4-il]piperazin-2-il]acetonitrilo (20,6 mg, 34,3 μmol, 25 % de rendimiento, 97,3 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 585.
1H RMN (400 MHz, cloroformo-d) 5 = 9,03 (d, J = 6,0 Hz, 1H), 8,30 (d, J = 16,8 Hz, 1H), 7,83 (d a, J = 7,6 Hz, 1H), 7,55 - 7,44 (m, 2H), 5,54 - 5,32 (m, 1H), 5,31 - 5,18 (m, 1H), 5,09 - 4,48 (m, 1H), 4,43 - 4,34 (m, 1H), 4,32 - 4,07 (m, 4H), 4,06 - 3,81 (m, 2H), 3,64 - 3,42 (m, 2H), 3,35 - 3,26 (m, 1H), 3,25 - 2,96 (m, 4H), 2,92 (d, J = 2,8 Hz, 3H), 2,90 -2,73 (m, 2H), 2,72 - 2,61 (m, 2H), 2,51 - 2,42 (m, 3H), 2,34 - 2,23 (m, 1H), 2,13 - 1,99 (m, 1H), 1,92 - 1,73 (m, 3H).
Ejemplo 620

1-[4-[7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-6-fluoro-1,4-diazepano-1-il]-2-fluoro-prop-2-en-1-ona

Etapa A: 4-bencil-6-fluoro-1,4-diazepano-1-carboxilato de tere-butilo, 4-bencil-6-fluoro-1,4-diazepano-1-carboxilato de tere-butilo. A una solución de (3R)-4-bencil-3-(hidroximetil)piperazina -1-carboxilato de terc-butilo (1,5 g, 4,90 mmol, 1 eq.) en DCM (40 ml) se le añadió gota a gota DAST (3,95 g, 24,48 mmol, 3,23 ml, 5 eq.) a 0 °C y la mezcla se agitó a 20 °C durante 5 h. A la mezcla se le añadió NaHCÜ3 sat. (30 ml) y se extrajo con acetato de etilo (30 ml * 2). La capa orgánica se secó sobre Na2SÜ4, se filtró y se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (EP: AE = 20~5:1) para dar 4-bencil-6-fluoro-1,4-diazepano-1-carboxilato de terc-butilo (980 mg, 3,18 mmol, 64,91 % de rendimiento) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 309.
Etapa B: 6-fluoro-1,4-diazepano-1-carboxilato de terc-butilo. A una solución de 4-bencil-6-fluoro-1,4-diazepano-1-carboxilato de terc-butilo (900,00 mg, 2,92 mmol, 1 eq.), Pd/C (200 mg, 2,92 mmol, 10 % de pureza, 1 eq.) y Pd(0H)2 (179,92 mg, 1,28 mmol, 4,39e-1 eq.) en Me0H (20 ml) y la mezcla se agitó a 40 °C durante 12 h en una atmósfera de H2 (344,74 kPa (50 psi)). La mezcla se filtró y se concentró para dar 6-fluoro-1,4-diazepano-1 -carboxilato de tere-butilo (630 mg, 2,89 mmol, 98,90 % de rendimiento) en forma de un aceite incoloro.
Etapa C: trifluorometanosulfonato de [7-(8-cloro-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-ilo]. A una solución de 7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-ol (700 mg, 1,65 mmol, 1 eq.), TEA (500,09 mg, 4,94 mmol, 687,88 μl, 3 eq.) y tamices moleculares de 4 A (500 mg) en DCM (7 ml) se le añadió Tf20 (697,18 mg, 2,47 mmol, 407,71 μl, 1,5 eq.) a -40 °C y se agitó a 0 °C durante 30 min. A la mezcla se le añadió agua (2 ml) y se extrajo con DCM (5 ml). La capa orgánica se secó sobre Na2S04, se filtró y se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (EP: AE = 10-1:1) para dar trifluorometanosulfonato de [7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-ilo] (470 mg, 843,83 μmol, 51,22 % de rendimiento) en forma de un aceite de color rojo. LCMS [ESI, M+1]: 557.
Etapa D: 4-[7-(8-cloro-1-naftin-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6.8-dihidro-5H-pirido[3.4-dlpirimidin-4-il1-6-fluoro-1,4-diazepano-1-carboxilato de ferc-butilo. A una solución de trifluorometanosulfonato de [7-(8-cloro-1-naftil)-2-[[(2S)-1- metilpirrolidin-2-il]metoxi1-6.8-dihidro- 5H-pirido[3,4-d] pirimidin-4-ilo] (440 mg. 789,97 gmol, 1 eq.) y 6-fluoro-1,4-diazepano-1-carboxilato de ferc-butilo (258,64 mg, 1,18 mmol, 1,5 eq.) en DMAC (5 ml) se le añadió DIeA (306,29 mg,
2,37 mmol, 412,79 gl, 3 eq.) y la mezcla se agitó a 25 °C durante 30 min. La mezcla se purificó por cromatografía en columna sobre gel de sílice de fase inversa (0,001 de AF) para dar 4-[7-(8-cloro-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8 -dihidro-5H-pirido[3,4-d]pirimidin-4-il]-6-fluoro-1,4-diazepano-1-carboxilato de ferc-butilo (260 mg, 415,88 gmol, 52,65 % de rendimiento) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 625.
Etapa E: 7-(8-cloro-1-naftil)-4-(6-fluoro-1.4-diazepano-1-il)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6.8-dihidro-5H-pirido[3.4-d1pirimidina. A una solución de 4-[7-(8-cloro-1 -naftil)-2-[[(2S)-1 -metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-6-fluoro-1,4-diazepano-1-carboxilato de ferc-butilo (240,00 mg, 383,89 gmol, 1 eq.) en DCM
(3 ml) se le añadió TFA (12,32 g, 108,05 mmol, 8,00 ml, 281,45 eq.), y la mezcla se agitó a 20 °C durante 30 min. La mezcla se concentró, se añadió DCM (10 ml) y se lavó con Na2Cü3 sat. (5 ml). La capa orgánica se secó sobre Na2SÜ4, se filtró y se concentró para dar 7-(8-cloro-1-naftil)-4-(6-fluoro-1,4-diazepano-1-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5Hpirido[3,4-d]pirimidina (200 mg, bruta) en forma de un sólido de color amarillo. Parte de este material (160 mg, 304,73 gmol) se usó directamente en la siguiente etapa. Se purificaron 40 mg de producto puro por
HPLC prep. (purificación por HPLC prep. (columna: Shim-pack C18 150 * 25 * 10 um; fase móvil: [agua (0,225 % de
AF)-a Cn ];% de B: 4 %-34 %,10 min) y después el residuo se purificó por HPLC prep. (columna: Xtimate C18 150 *
25 mm * 5 um;fase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN];% de B: 60 %-90 %,10 min) para dar 7-(8-cloro-1-naftil)-4-(6-fluoro-1,4-diazepano-1-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidina (9 mg, 16,99 gmol). LCMS [ESI, M+1]: 523.
1H RMN (400 MHz, cloroformo-d) 5 = 7,76 (d, J = 8,07 Hz, 1 H) 7,61 (t, J = 8,38 Hz, 1 H) 7,52 (d a, J = 7,46 Hz, 1 H)
7,44 (dt, J = 15,86, 7,90 Hz, 1 H) 7,34 (t, J = 7,76 Hz, 1 H) 7,25-7,18 (dd, J = 7,46 Hz, 1 H) 4,80 - 5,24 (m, 1 H) 4,31 -4,46 (m, 2 H) 4,08 - 4,30 (m, 2 H) 3,71 - 4,04 (m, 3 H) 3,50 - 3,68 (m, 2 H) 2,81 - 3,37 (m, 7 H) 2,46 - 2,73 (m, 5 H)
2,25 - 2,35 (m, 1 H) 2,01 - 2,14 (m, 1 H) 1,82 - 1,91 (m, 3 H).
Etapa F: 1-[4-(7-(8-cloro-1-naftil)-2-[[(2S)-1-metilpirrolidin-2-il1metoxi1-6,8-dihidro-5H-pirido[3,4-d1pirimidin-4-il1-6-fluoro-1,4-diazepano-1-i n -2-fluoroprop-2-en-1-ona. A una solución de 7-(8-cloro-1-naftil)-4-(6-fluoro-1,4-diazepano-1-il)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]- 6,8-dihidro-5H-pirido[3,4-d]pirimidina (150,00 mg, 285 ,68 gmol, 1 eq.) y ácido
2- fluoroprop-2-enoico (51,45 mg, 571,36 gmol, 2 eq.) en AE (2 ml) se le añadieron T3P (50 M, 17,14 gl, 3 eq.) y TEA (231,27 mg, 2,29 mmol, 318,11 gl, 8 eq.) a -40 °C y la mezcla se agitó a -40 °C durante 30 min. La mezcla se diluyó con agua (2 ml) y se extrajo con acetato de etilo (2 ml * 2). La capa orgánica se secó sobre Na2SÜ4, se filtró y se concentró. El residuo se purificó por HPLC prep. (columna: Shim-pack C18150 * 25 * 10 um;fase móvil: [agua (0,225 %
de AF)-ACN];% de B: 18 %-48 %, 10 min) para dar 1 -[4-[7-(8-cloro-1 -naftil)-2-[[(2S)-1-metilpirrolidin-2-il]metoxi]-6,8-dihidro-5H-pirido[3,4-d]pirimidin-4-il]-6-fluoro-1,4-diazepano-1 -il]-2-fluoro- prop-2-en-1-ona (90 mg, 140,78 gmol,
49,28 % de rendimiento, 93,4 % de pureza) en forma de un sólido de color amarillo. LCMS [ESI, M+1]: 597.
1H RMN (400 MHz, cloroformo-d) 5 = 7,76 (d, J = 8,19 Hz, 1 H) 7,58 - 7,66 (m, 1 H) 7,50 - 7,56 (m, 1 H) 7,38 - 7,50
(m, 1 H) 7,31 - 7,37 (m, 1 H) 7,24 - 7,16 (m, 1 H) 4,90 - 5,55 (m, 3 H) 4,32 - 4,48 (m, 2 H) 3,48 - 4,27 (m, 11 H) 3,21 -3,43 (m, 1 H) 2,80 - 3,16 (m, 2 H) 2,44 - 2,73 (m, 5 H) 2,25 - 2,36 (m, 1 H) 2,00 - 2,14 (m, 1 H) 1,77 - 1,91 (m, 3 H).
Ejemplo 621

-[(3R)-4-[7-(8-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡rido[3,4-d]p¡r¡m¡d¡n-4-¡l]-3-(difluorometil)piperazin-1-il]-2-fluoro-prop-2-en-1 -ona

Etapa A: (3R)-4-bencil-3-(hidroximetil)piperazin-1-carboxilato de ferc-butilo. Una mezcla de reacción de (3R)-3-(hidroximetil)piperazin-l-carboxilato de ferc-butilo (10 g, 46,2 mmol, 1 eq.), BnBr (8,70 g, 50,9 mmol, 6,04 ml, 1,1 eq.) y TEA (7,02 g, 69,36 mmol, 9,65 ml, 1,5 eq.) en MeCN (100 ml) se calentó a 80 °C durante 12 horas. Después de que se completara, la mezcla de reacción se concentró al vacío. El residuo se disolvió en agua (30 ml) y acetato de etilo (50 ml). La mezcla se extrajo con acetato de etilo (3 * 50 ml). Las capas orgánicas combinadas se lavaron con salmuera (30 ml), se secaron sobre Na2SÜ4 y se concentraron al vacío. El residuo se purificó por cromatografía sobre gel de sílice (éter de petróleo:acetato de etilo = 5:1 a 3:1). Se obtuvo (3R)-4-bencil-3-(hidroximetil)piperazin-1-carboxilato de ferc-butilo (9 g, 29,4 mmol, 64 % de rendimiento, 100 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 307.
1H RMN (400 MHz, cloroformo-d) ó = 7,28 - 7,20 (m, 5H), 3,95 (d a, J = 13,2 Hz, 1H), 3,79 (dd, J = 5,6, 11,6 Hz, 1H), 3,61 (dd a, J = 3,2, 13,6 Hz, 1H), 3,52 (dd a, J = 4,0, 11,2 Hz, 2H), 3,35 (d a, J = 13,3 Hz, 2H), 3,09 (s a, 1H), 2,77 -2,65 (m, 1H), 2,51 (s a, 1H), 2,20 (ddd, J = 3,2, 8,4, 12,0 Hz, 1H), 1,38 (s, 9H).
Etapa B: (3R)-4-bencil-3-formil-piperazin-1-carboxilato de ferc-butilo. Se añadió DMS0 (13,8 g, 176 mmol, 13,8 ml, 6,0 eq.) a una solución a -65 °C de (C0Cl)2 (11,2 g, 88,1 mmol, 7,71 ml, 3,0 eq.) en diclorometano (90 ml). Después de 10 min, se introdujo lentamente una solución de (3R)-4-bencil-3-(hidroximetil)piperazin-1-carboxilato de ferc-butilo (9 g, 29,4 mmol, 1 eq.) en diclorometano (40 ml) durante 15 minutos. Después de agitar a -65 °C durante 1 hora, se añadió TEA (29,7 g, 294 mmol, 40,9 ml, 10,0 eq.). La mezcla se calentó a 28 °C durante 30 min y después se agitó durante 30 min más a esa temperatura. Después de que se completara, la mezcla de reacción se inactivó con agua (5 ml) y se separó. La capa orgánica se secó sobre Na2S04 y se concentró al vacío. Se obtuvo (3R)-4-bencil-3-formilpiperazin-1-carboxilato de ferc-butilo (8,9 g, 29 mmol) en forma de un aceite de color amarillo que se usó para la siguiente etapa sin purificación adicional.
Etapa C: (3R)-4-bencil-3-(difluorometil)piperazin-1-carboxilato de ferc-butilo. A una solución de (3R)-4-bencil-3-formilpiperazin-1 -carboxilato de terc-butilo (8,9 g, 29,2 mmol, 1,0 eq.) en diclorometano (180 ml) se le añadió DAST (9,43 g, 58,5 mmol, 7,73 ml, 2 eq.) a 0 °C. La mezcla de reacción se agitó a 0 °C durante 2 horas. Después de que se completara, la mezcla de reacción se inactivó con NaHC03 saturado (150 ml) y se extrajo con acetato de etilo (3 * 150 ml). Las capas orgánicas combinadas se lavaron con salmuera (60 ml) y se concentraron al vacío. El residuo (éter de petróleo: acetato de etilo = 5:1) se purificó por cromatografía sobre gel de sílice (éter de petróleo: acetato de etilo = 1:0 a 10:1). Se obtuvo (3R)-4-bencil-3-(difluorometil)piperazin-1-carboxilato de ferc-butilo (1,5 g, 4,48 mmol, dos etapas 15 % de rendimiento, 97,5 % de pureza) en forma de un aceite de color amarillo. LCMS [ESI, M+1]: 327.
Etapa D: (3R)-3-(d¡fluoromet¡l)p¡peraz¡n-1-carboxilato de tere-butilo. A una solución de (3R)-4-bencil-3-(difluorometil)piperazin-l-carboxilato de tere-butilo (1,5 g, 4,60 mmol, 1,0 eq.) en Me0H (10 ml) se le añad¡eron Pd/C (150 mg, 10 % de pureza) y Pd(0H)2/C (150 mg, 20 % de pureza) en una atmósfera de N2. La suspensión se desgasificó al vacío y se purgó varias veces con H2. La mezcla se agitó en una atmósfera de H2 (344,74 kPa (50 psi)) a 40 °C durante 12 horas. Después de que se completara, la mezcla se filtró y el filtrado se concentró al vacío. Se obtuvo (3R)-3-(difluorometil)p¡peraz¡n-1-carbox¡lato de tere-butilo (1,0 g, 4,23 mmol, 92 % de rendimiento) en forma de un aceite de color amarillo que se usó para la siguiente etapa sin purificación adicional.
1H RMN (400 MHz, cloroformo-d) ó = 5,91 - 5,43 (m, 1H), 4,17 - 3,78 (m, 2H), 3,13 - 2,66 (m, 5H), 1,48 (s, 9H).
Etapa E: trifluorometanosulfonato de [7-(8-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3.4-dlpirimidin-4-ilol. A una solución de 7-(8-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-il]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]pir¡mid¡n-4-ol (1,0 g, 2,35 mmol, 1 eq.), tamices moleculares de 4 A (1,0 g) y TEA (953 mg, 9,41 mmol, 1,31 ml, 4,0 eq.) en diclorometano (20 ml) se le añadió Tf20 (996 mg, 3,53 mmol, 582 ul, 1,5 eq.) a -40 °C y se agitó durante 0,5 horas. Después de que se completara, la mezcla de reacción se inactivó con agua (15 ml). La mezcla se filtró a través de una capa de celite. El filtrado se separó y las capas acuosas se extrajeron con diclorometano (30 ml). Las capas orgánicas se lavaron con salmuera (10 ml), se secaron sobre Na2S04 y se concentraron al vacío. La mezcla se purificó por cromatografía sobre gel de sílice (S¡02, éter de petróleo: acetato de etilo = 5:1 a 0:1) para dar trifluorometanosulfonato de [7-(8-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡rido[3,4-d]p¡r¡m¡d¡n-4-¡lo] (1,0 g, 1,63 mmol, 69 % de rendimiento, 91 % de pureza) en forma de un aceite de color pardo. LCMS [ESI, M+1]: 557.
Etapa F: (3R)-4-[7-(8-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1-3-(difluoromet¡l)p¡peraz¡n-1-carbox¡lato de tere-butilo. Una mezcla de reacción de trifluorometanosulfonato de [7-(8-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]pir¡m¡d¡n-4-¡lo] (1,0 g, 1,80 mmol, 1,0 eq.) y (3R)-3-(difluoromet¡l)p¡peraz¡n-1-carbox¡lato de tere-butilo (780 mg, 3,30 mmol, 1,84 eq.) se agitó a 90 °C durante 5 horas. Después de que se completara, la mezcla de reacción se disolvió en MeCN (5 ml). La mezcla de reacción se purificó por cromatografía ultrarrápida de fase inversa [agua (0,1 % de AF)/acetonitrilo]. Las fracciones se basificaron con NaHC03 sólido a pH >7 y se concentraron al vacío. La capa acuosa se extrajo con acetato de etilo (3 x 20 ml) y se concentró al vacío. El residuo se purificó por TLC prep. (diclorometano:Me0H = 7:1) para dar (3R)-4-[7-(8-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-3-(d¡fiuoromet¡l)p¡peraz¡n-1-carboxilato de tere-butilo (80 mg, 122 μmol, 7 % de rendimiento, 98 % de pureza) en forma de un aceite de color pardo. LCMS [ESI, M+1]: 643.
Etapa G: 7-(8-cloro-1-naft¡l)-4-[(2R)-2-(d¡fluoromet¡l)p¡peraz¡n-1-¡l1-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3.4-d1p¡r¡m¡d¡na. A una solución de (3R)-4-[7-(8-chlono-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-il]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l]-3-(d¡fluoromet¡l)p¡peraz¡n-1-carbox¡lato de terc-butilo (20 mg, 31,1 μmol, 1 eq.) en diclorometano (0,05 ml) se le añadió TFA (70,9 mg, 622 μmol, 46,0 μl, 20 eq.). La mezcla de reacción se agitó a 25 °C durante 1 hora. Después de que se completara, la mezcla de reacc¡ón se ¡nact¡vó con Na2C03 saturado (4 ml) y se extrajo con diclorometano: Me0H (10: 1, 3 x 8 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó por HPLC prep. (columna: Waters Xbridge 150 * 255 ufase móvil: [agua (NH4HC03 10 mM)-ACN];% de B: 43 %-73 %,10 min). Las fracciones se concentraron y se liofilizaron. Se obtuvo 7-(8-cloro-1-naft¡l)-4-[(2R)-2-(d¡fluoromet¡l)p¡peraz¡n-1-¡l]-2-[[(2R)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]pir¡mid¡na (4,22 mg, 7,77 μmol, 25 % de rendimiento, 100 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 543.
1H RMN (400 MHz, cloroformo-d) ó = 7,81 - 7,74 (m, 1H), 7,66 - 7,59 (m, 1H), 7,56 - 7,52 (m, 1H), 7,50 - 7,40 (m, 1H), 7,35 (t, J = 7,6 Hz, 1H), 7,28 - 7,16 (m, 1H), 6,69 - 6,09 (m, 1H), 4,68 - 4,54 (m, 1H), 4,51 - 4,34 (m, 2H), 4,30 - 4,11 (m, 1H), 4,07 - 3,68 (m, 2H), 3,63 - 3,46 (m, 2H), 3,44 - 3,31 (m, 2H), 3,30 - 3,21 (m, 1H), 3,20 - 2,97 (m, 4H), 2,92 -2,83 (m, 1H), 2,78 - 2,67 (m, 1H), 2,58 - 2,49 (m, 3H), 2,38 - 2,26 (m, 1H), 2,15 - 2,02 (m, 1H), 1,94 - 1,73 (m, 3H).
Etapa H: 1-[(3R)-4-[7-(8-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l1metox¡1-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d1p¡r¡m¡d¡n-4-¡l1-3-(d¡fluoromet¡l)p¡peraz¡n-1-¡l1-2-fluoro-prop-2-en-1-ona. A una solución de 7-(8-cloro-1-naftil)-4-[(2R)-2-(d¡fluoromet¡l)p¡peraz¡n-1-¡l]-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡na (45 mg, 82,9 μmol, 1,0 eq.) y ácido 2-fluoroprop-2-enoico (22,4 mg, 249 μmol, 3,0 eq.) en acetato de etilo (1 ml) se le añadió tamiz molecular de 4 A (100 mg). Después de agitar a 25 °C durante 0,5 horas, se añadieron TEA (126 mg, 1,24 mmol, 173 μl, 15 eq.) y T3P (211 mg, 331 μmol, 197 μl, 50 % de pureza, 4 eq.) a 0 °C. La mezcla se agitó a 25 °C durante 0,5 horas. Después de que se completara, la mezcla de reacción se inactivó con agua (1 ml) y se extrajo con acetato de etilo (3 x 5 ml). Las capas orgánicas combinadas se secaron sobre Na2S04 y se concentraron al vacío. El residuo se purificó con una columna (Al203, éter de petróleo:acetato de etilo= 3:1 a 0:1), seguido de HPLC prep. (columna: Waters Xbridge 150 * 255 ufase móvil: [agua (0,05 % de hidróxido de amoniaco v/v)-ACN];% de B: 55 %-85 %,10 min) para dar 1-[(3R)-4-[7-(8-cloro-1-naft¡l)-2-[[(2S)-1-met¡lp¡rrol¡d¡n-2-¡l]metox¡]-6,8-d¡h¡dro-5H-p¡r¡do[3,4-d]p¡r¡m¡d¡n-4-¡l1-3-(d¡fluoromet¡l)p¡peraz¡n-1 -¡l]-2-fluoro-prop-2-en-1 -ona (5,98 mg, 9,72 μmol, 11 % de rendimiento, 99,4 % de pureza) en forma de un sólido de color blanco. LCMS [ESI, M+1]: 615.
1H RMN (400 MHz, cloroformo-d) 5 = 7,81 - 7,75 (m, 1H), 7,67 - 7,61 (m, 1H), 7,55 (td, J = 1,2, 7,6 Hz, 1H), 7,51 -7,41 (m, 1H), 7,36 (t, J = 7,6 Hz, 1H), 7,28 - 7,17 (m, 1H), 6,48 - 5,83 (m, 1H), 5,47 - 5,31 (m, 1H), 5,27 - 5,19 (m, 1H), 4,88 - 4,65 (m, 1H), 4,59 - 4,31 (m, 4H), 4,21 - 4,10 (m, 1H), 4,04 - 3,76 (m, 2H), 3,69 - 3,44 (m, 3H), 3,33 - 2,92 (m, 4H), 2,75 - 2,64 (m, 1H), 2,61 - 2,52 (m, 1H), 2,49-2,48 (m, 3H), 2,36 - 2,25 (m, 1H), 2,13 - 2,02 (m, 1H), 1,88 - 1,72 (m, 3H).
Los EJEMPL0S 622 - 678 presentados en la Tabla 1 se preparan usando intermedios disponibles comercialmente y/o los intermedios descritos en la presente descripción siguiendo los métodos indicados en los esquemas de reacción generales y las enseñanzas de los Ejemplos ilustrados.
Tabla 1



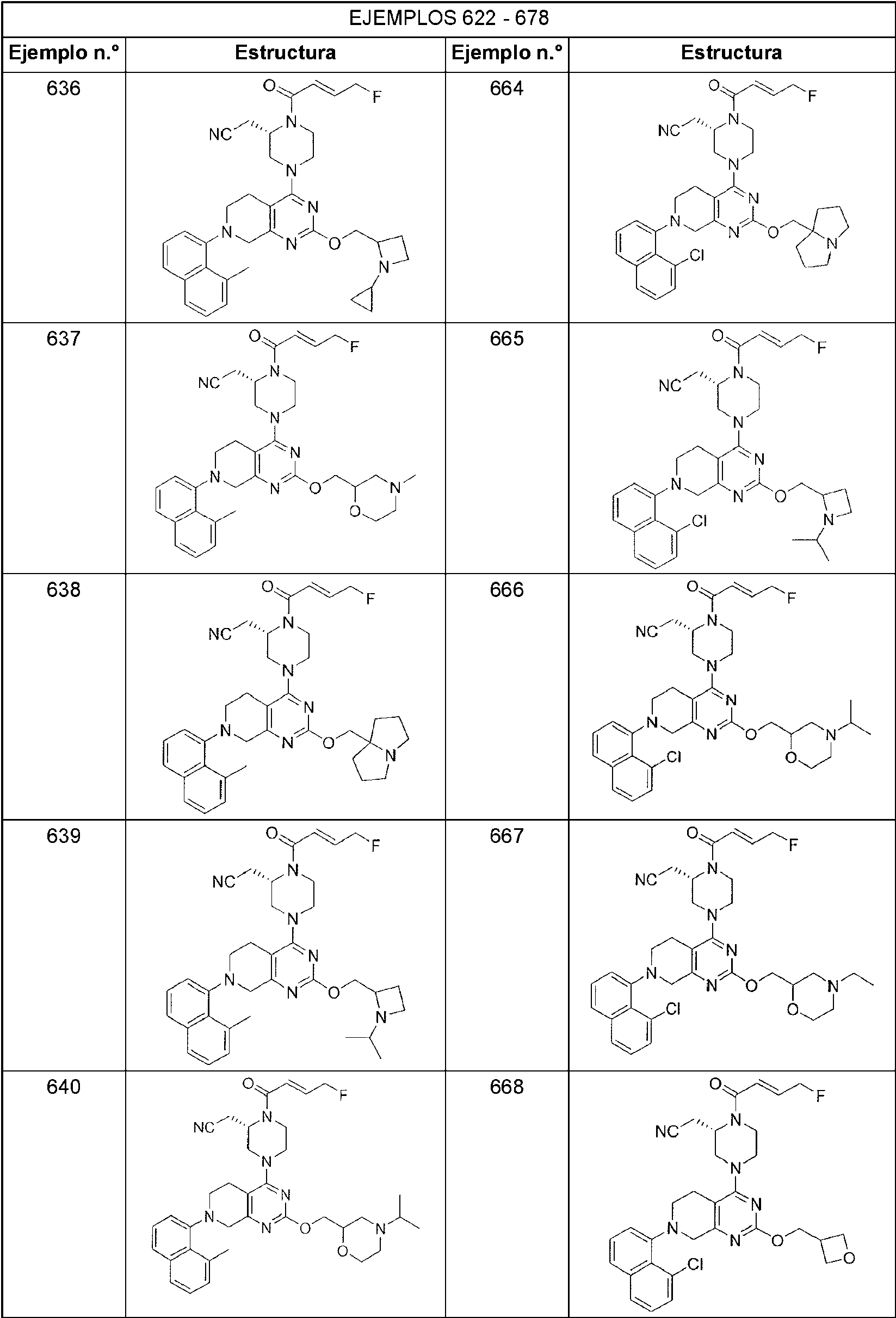


Ejemplo A
Ensayo de modificación de KRas G12C
Este Ejemplo ilustra que los compuestos ilustrativos de la presente invención se unen covalentemente a KRas G12C mediante el uso de un ensayo de LCMS para detectar un aducto covalente del compuesto ilustrativo y KRAS G12C. La concentración de proteína de K-Ras (1-169) G12C, C51S, C80L, C118S cargados con GDP y K-Ras (1-169) G12C, C51S, C80L, C118S, Q61H cargados con GTp se ajustó a 2 μM en K-Ras Tampón de ensayo (HEPES 25 mM, NaCl 150 mM, MgCl25 mM y octil p-glucopiranósido 10 mM a pH 7,5). Después, se transfirió una alícuota de 10 μl de cada solución de proteína a una placa de microtitulación de 384 pocillos. Se generaron reservas de compuestos iniciales a cincuenta veces su concentración final deseada del ensayo en DMS0.
Los compuestos de referencia ilustrativos se diluyeron 25 veces en tampón de ensayo de K-Ras hasta un final de dos veces su concentración final. Después, se añadió una alícuota de 10 μl de cada solución de compuesto diluido a cada una de las soluciones de proteína en la placa de microtitulación para iniciar la reacción. Las concentraciones típicas del compuesto final fueron 3,0, 5,0 y 25,0 μM. En cada momento, las reacciones se inactivaron con 20 μl de una solución de ácido acético 25 mM. Los criterios de valoración habituales del ensayo fueron 15, 180 y 1440 minutos. Una vez que se inactivaron todas las reacciones, las placas se sellaron con calor y las muestras se inyectaron en un sistema LC/MS para la adquisición de datos.
La recopilación de datos se realizó en un espectrómetro de masas preciso Agilent 6520 Q-T0F. Las muestras se inyectaron en su fase líquida en una columna de fase inversa C-3 para eliminar el tampón de ensayo y preparar las muestras para el espectrómetro de masas. Las proteínas se eluyeron de la columna mediante el uso de un gradiente de acetonitrilo y se alimentaron directamente al analizador de masas. El análisis inicial de datos en bruto se llevó a cabo en el programa informático Agilent MassHunter inmediatamente después de la adquisición de datos.
El análisis de datos sin procesar de la proteína intacta fue exclusivamente una deconvolución de los múltiples estados de carga de cada proteína en solución mediante el uso de una deconvolución de entropía máxima proporcionada en Mass Hunter. Para minimizar la complejidad, solo se consideraron para el análisis los datos sobre intervalos de masa limitados, con un mínimo de intervalos de etapas de masa de un Dalton. Las alturas de todas las masas identificadas durante el análisis de datos sin procesar se exportaron para analizarlas más a fondo en el programa informático de análisis de datos Spotfire®.
El análisis de datos final fue un proceso de múltiples etapas en el paquete del programa informático de análisis de datos Spotfire®. Brevemente, cada masa de proteína se calculó como un porcentaje de la señal total de esa muestra, ese porcentaje se normalizó después al porcentaje de señal de la proteína en ausencia de compuestos reactivos. Esas señales normalizadas se informaron como porcentaje normalizado de control (P0C). Un valor de P0C aumentado indica un compuesto que muestra un mayor grado de modificación en una condición determinada en comparación con otros compuestos en las mismas condiciones. Los resultados de los compuestos de referencia ilustrativos y de los compuestos de la invención ensayados a una concentración 5 μM durante 3 horas se muestran en la Tabla 2. Leyenda: “A” < 25 % de P0C; “ B” > 25 % de P0C - < 50 % de P0C; “ C” > 50 % de P0C y ND = no determinado.
Tabla 2







Ejemplo B
Inhibición del crecimiento celular dependiente de KRas G12C
Este ejemplo ilustra que los compuestos ilustrativos de la presente invención inhiben el crecimiento de líneas de células tumorales que expresan KRas G12C.
La inhibición celular de KRAs G12C durante compuestos ilustrativos de la presente invención se determinó mediante medición de la cantidad de un marcador secuencia abajo de la actividad de KRas, ERK fosforilado (“Phospho-ERK” ).
Las células NCI-H358 (ATCC CRL-5807) expresan KRas G12C y se cultivaron en medio RPMI complementado con suero bovino fetal al 10 %, penicilina/estreptomicina y HEPES 10 mM. Las células se sembraron en placas de 96 pocillos recubiertas con poli-D-lisina a una concentración de 50000 células/pocillo y se dejaron adherir durante 8-12 horas. Después se añadieron los compuestos diluidos a una concentración final de DMS0 al 0,5 %. Después de 3 horas, se retiró el medio, se añadieron 150 μl de formaldehído al 4 % y las placas se incubaron durante 20 minutos. Las placas se lavaron con PBS y se permeabilizaron mediante el uso de 150 μl de metanol al 100 % enfriado con hielo durante 10 minutos. La unión del anticuerpo inespecífico a las placas se bloqueó mediante el uso de 100 μl de tampón de bloqueo Licor (Li-Cor Biotechnology, Lincoln NE) durante 1 hora a temperatura ambiente. Las muestras de control positivo y las muestras que carecen de células se procesaron en paralelo con muestras de prueba como patrones. La cantidad de Fosfo-ERK se determinó mediante el uso de un anticuerpo específico para la forma fosforilada de ERK y se comparó con la cantidad de GAPDH. Los anticuerpos primarios usados para la detección se añadieron como se indica a continuación: Phospho-ERK (Cell Signaling cs9101) diluido 1:500 y GAPDH (Millipore MAB374) diluido 1:5000 en Licor block Tween 20 al 0,05 %. Las placas se incubaron durante 2 horas a temperatura ambiente. Las placas se lavaron con PBS Tween 20 al 0,05 %.
Los anticuerpos secundarios usados para visualizar los anticuerpos primarios se añadieron como se indica a continuación: Anti-conejo-680 diluido 1: 1000 y Anti-ratón-800 diluido 1: 1000 en Licor Block Tween 20 al 0,05 % y se incubaron durante 1 hora a temperatura ambiente. Las placas se lavaron con PBS Tween 20 al 0,05 %. Se añadió una alícuota de 100 μl de PBS a cada pocillo y las placas se leyeron en un lector de placas LIC0R AERIUS.
La señal de pERK (Thr202/Tyr204) se normalizó con la señal de GAPDH y se calculó el porcentaje de los valores de control de DMS0. Los valores de CI50 se generaron mediante el uso de un ajuste de 4 parámetros de la curva de respuesta a la dosis. Los resultados de los compuestos de referencia ilustrativos y de los compuestos de la invención se muestran en la Tabla 3. Leyenda: “A” > 0,0001 - < 1 μM; “ B” > 1 μM y ND = no determinado.
Tabla 3




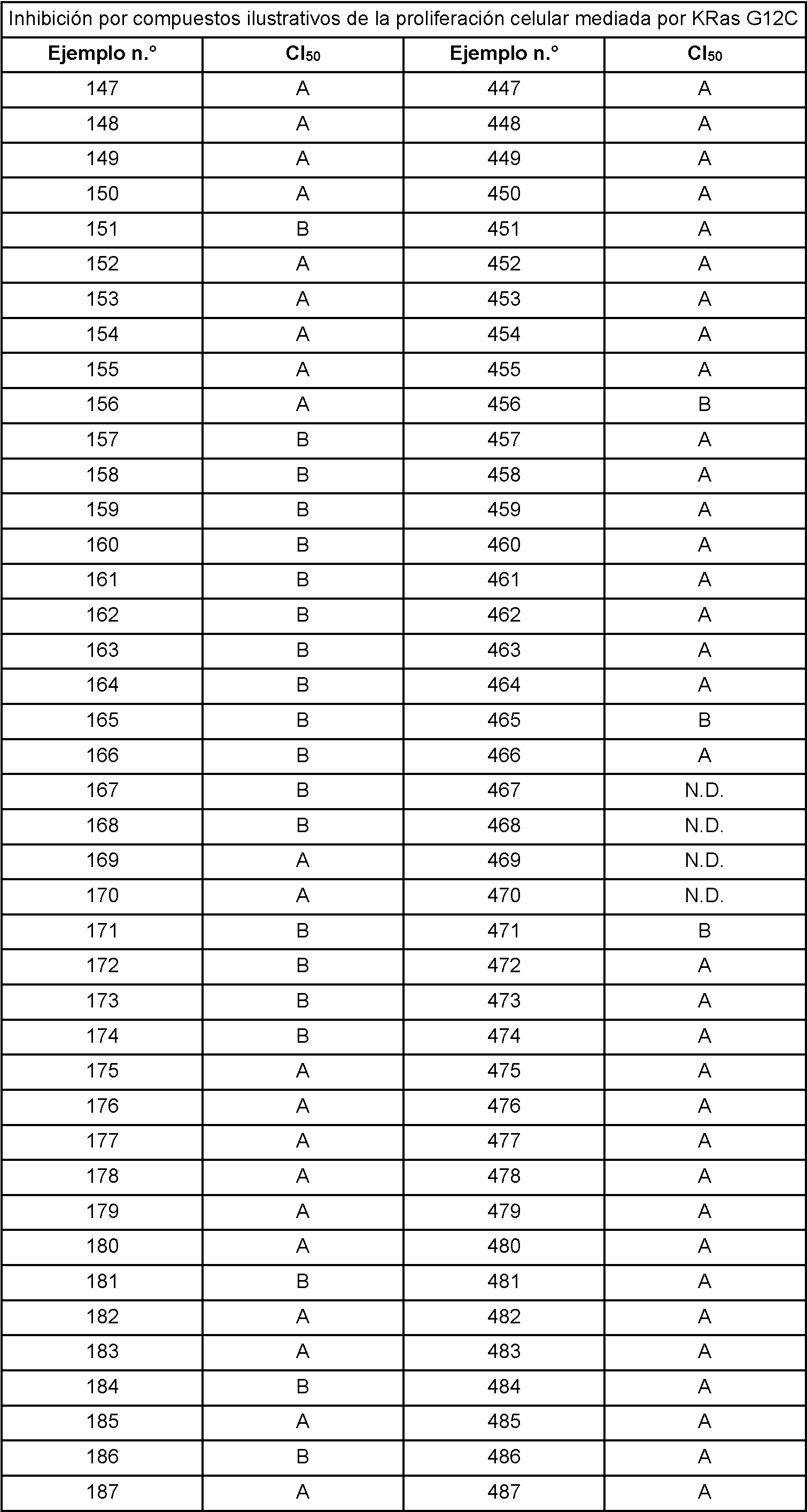



Ejemplo C
Estabilidad in vitro de los compuestos que tienen sustituciones en RA o RB en sangre completa
Este Ejemplo ilustra que los compuestos ilustrativos de la presente invención que comprenden sustituciones en RA o al menos un RB presentan una estabilidad de sangre completa mejorada en comparación con compuestos en los que RA y los dos sustituyentes RB son hidrógeno.
Materiales: Los siguientes reactivos se usaron tal como se recibieron: acetonitrilo (grado HPLC, Burdick & Jackson, Madison, WI), solución salina tamponada con fosfato (PBS), pH 7,4 (Sigma-Aldrich, Co., St. Louis, M0), agua (grado HPLC, JT Baker, Phillipsburg, NJ), DMS0 (EM Science, Merck KGaA, Darmstadt, Alemania) e isopropanol (IPA, grado reactivo, EMD Chemicals, Gibbstown, NJ). En el ensayo se usaron propantelina y esmolol (Sigma-Aldrich) como compuestos de control positivo. Se usó diazepam (Sigma-Aldrich) como patrón interno para la cuantificación. Se obtuvo sangre completa humana de BioIVT (Westbury, NY) y era del género masculino. Todos los demás reactivos y
disolventes eran del más alto grado analítico suministrado por Sigma (St. Louis, M0) y se usaron tal como se recibieron.
Incubaciones de sangre: La estabilidad in vitro de los compuestos inhibidores de KRas G12C representativos de la presente invención en presencia de sangre completa se llevó a cabo de la siguiente manera. En los ensayos, se usaron como controles positivos esmolol o propantelina, que son conocidos por experimentar hidrólisis de éster con esterasas en sangre y plasma. Una solución madre 10 mM de propantelina, esmolol o un compuesto de la presente invención en DMS0 se diluyó a 500 μM con DMS0. Se pusieron alícuotas de sangre completa (600 μl), por triplicado, en los pocillos apropiados de una placa de 96 pocillos profundos de polipropileno (Axygen Scientific, Union City, CA). La sangre se diluyó al 50 % usando PBS, pH 7,4 (600 μl). La sangre diluida se preincubó durante 15 minutos a 37 °C. Cada pocillo se dosificó con 12 μl de la reserva de DMS0500 μM simultáneamente para una concentración de ensayo final de 5 μM. Las placas se mezclaron a 600 rpm durante 10 segundos en un agitador de placas (IKA MTS 2/4 Digital Microtiter Shaker, VWR). Se transfirieron alícuotas de 100 μl de cada pocillo de sangre completa a tres placas separadas de 96 pocillos profundos etiquetadas con 0, 60 o 240 minutos. Al final de cada momento designado, los glóbulos rojos se lisaron con 100 μl of agua, se mezclaron a 600 rpm durante un minuto y se pararon con 800 μl de acetonitrilo que contenía IS (diazepam 0,625 μM). Las placas se mezclaron en el agitador de placas a 600 rpm durante un minuto y se centrifugaron en una centrífuga a 2095 x g durante 7 minutos a temperatura ambiente usando una centrífuga de sobremesa Allegra (Beckman Coulter, Fullerton, CA). El sobrenadante (100 μl) se transfirió a una placa de 96 pocillos poco profundos (Costar) usando un sistema de manipulación de muestras líquidas (Apricot, Perkin-Elmer, Boston, MA). Se realizó una adición de 100 μl de agua a cada muestra de sobrenadante para alcanzar un volumen final de 200 μl. Las placas se sellaron y el contenido de cada pocillo se analizó por LC-MS/MS.
Cuantificación analítica: El sistema de LC-MS/MS estaba comprendido por un muestreador automático HTS-PAL (Leap Technologies, Carrboro, NC), una HPLC HP1200 (Agilent, Palo Alto, CA) y un espectrómetro de masas de cuadrupolo triple API4000 (PE Sciex, una división de Applied Biosystems, Foster City, CA). La separación cromatográfica del analito y el patrón interno se consiguió a temperatura ambiente usando una columna C18 (Kinetex®, 30 * 3,0 mm, tamaño de partículas de 2,6 μm, Phenomenex, Torrance, CA) junto con condiciones de gradiente que usaban fases móviles A (ácido fórmico acuoso al 0,1 % con alcohol isopropílico al 1 %) y B (ácido fórmico al 0,1 % en acetonitrilo). El tiempo de ejecución total, incluyendo el reequilibrado, para una sola inyección, fue de 2 minutos. La detección espectrométrica de masas de los analitos se logró usando el modo de ionización ESI+. La corriente de iones se optimizó durante la infusión de una solución madre de propantelina, esmolol o el compuesto de ensayo representativo. Las respuestas de los analitos, incluyendo el IS, se midieron mediante la monitorización de reacciones múltiples (MRM) de las transiciones exclusivas de cada compuesto.
Cálculos: Se adquirieron los datos y se calcularon las áreas de los picos para los compuestos de ensayo y el patrón interno usando el programa informático Analyst 1.6.2 (Sciex). Las relaciones medias de las áreas de los picos se calcularon promediando las relaciones de las áreas de los picos (n = 3) de cada compuesto de ensayo y el IS de cada muestra.
Las tablas de las áreas de los picos se exportaron a BioAssay Enterprise (CambridgeSoft, Cambridge, MA), donde se usaron las relaciones promedio de las áreas de los picos de analito con respecto a patrón interno para calcular el porcentaje restante (%REM) y la semivida (t1/2). El porcentaje restante (f) se calculó determinando la relación entre la relación de las áreas de los picos en cada momento y la relación de las áreas de los picos de las muestras en el momento cero. El t1/2 se determinó dividiendo ln(2) por km. La velocidad de pérdida del compuesto de ensayo (km) se determinó por regresión lineal de -ln(f(t)) frente al tiempo. La regresión usaba la forma “y=mx” , por lo que el modelo forzaba una intercepción del 100 % restante y asumía que el metabolismo seguía cinéticas de primer orden.
Los compuestos de control positivo, propantelina o esmolol, y los compuestos inhibidores de KRas G 12C representativos de la presente invención se incubaron en presencia de sangre completa humana a 37 °C durante un periodo de incubación de 240 minutos. La semivida de la propantelina, el esmolol y cada compuesto de ensayo después de 240 minutos de incubación a 37 °C se muestra en la Tabla 4. Leyenda: A < 200 min; B > 200 min - < 1o0o min; C > 1000 min - < 2000 min;y D > 2000 min
Tabla 4


Resultados: Como se esperaba, la propantelina de control positivo mostró una semivida corta de 98 min. En comparación, la semivida de los compuestos inhibidores de KRas G12C ilustrativos que comprenden sustituciones en RA o RB en sangre completa humana aumentó, en algunos casos, en más de 3 veces o más de 4 veces en comparación con los compuestos sin sustituir, por ejemplo, los Ejemplos 331 y 359, dando como resultado propiedades farmacodinámicas y farmacocinéticas muy mejoradas de estos compuestos, dando como resultado una mayor estabilidad in vivo y una mayor biodisponibilidad.
Si bien la invención se ha descrito en relación con modalidades específicas de la misma, se entenderá que es capaz de realizar modificaciones adicionales y esta solicitud está destinada a cubrir cualquier variación, uso o adaptación de la invención al seguir, en general, los principios de la presente invención e al incluir las desviaciones de la presente descripción que entran dentro de la práctica conocida o habitual dentro de la técnica a la que pertenece la invención y que pueden aplicarse a las características esenciales expuestas anteriormente, y como sigue en el alcance de las reivindicaciones adjuntas.
Claims (27)
1. Un compuesto de Fórmula (II):

o una sal farmacéuticamente aceptable del mismo:
en donde:
R1-X es

en donde el anillo de piperazinilo está sustituido opcionalmente con R8;
Y es un enlace, 0, S o NR5;
R1 es -C(O)C(RA)

C(RB)p, en donde

es un doble enlace, p es dos, cada RB es hidrógeno y RA es halógeno;
R2 es hidrógeno, alquilo, hidroxialquilo, dihidroxialquilo, alquilaminilalquilo, dialquilaminilalquilo, -Z-NR5R10, heterociclilo, heterociclilalquilo, arilo, heteroarilo o heteroarilalquilo, en donde cada uno de los Z, heterociclilo, heterociclilalquilo, arilo, heteroarilo y heteroarilalquilo puede estar sustituido opcionalmente con uno o más R9;
cada Z es alquileno C1 - C4;
cada R3 es independientemente alquilo C1 - C3, oxo, haloalquilo, hidroxilo o halógeno;
L es un enlace, -C(O)- o alquileno C1-C3;
R4 es hidrógeno, cicloalquilo, heterociclilo, arilo, aralquilo o heteroarilo, en donde cada uno del cicloalquilo, heterociclilo, arilo, aralquilo y heteroarilo puede estar sustituido opcionalmente con uno o más R6, R7 o R8;
cada R5 es independientemente hidrógeno o alquilo C1 - C3;
R6 es cicloalquilo, heterociclilo, heterociclilalquilo, arilo o heteroarilo, en donde cada uno del cicloalquilo, heterociclilo, arilo o heteroarilo puede estar sustituido opcionalmente con uno o más R7; cada R7 es independientemente halógeno, hidroxilo, alquilo C1 - C6, cicloalquilo, alcoxi, haloalquilo, amino, ciano, heteroalquilo, hidroxialquilo o Q-haloalquilo, en donde Q es 0 o S;
R8 es oxo, alquilo C 1 - C3, alquinilo C2 - C4, heteroalquilo, ciano, -C(O)0R5, -C(O)N(R5)2, -N(R5)2, en donde el alquilo C1 - C3 puede estar sustituido opcionalmente con ciano, halógeno-0R5, -N(R5)2 o heteroarilo;
cada R9 es independientemente hidrógeno, oxo, acilo, hidroxilo, hidroxialquilo, ciano, halógeno, alquilo C1 - C6, aralquilo, haloalquilo, heteroalquilo, cicloalquilo, heterociclilo, heterociclilalquilo, alcoxi, dialquilaminilo, dialquilamidoalquilo o dialquilaminilalquilo, en donde el alquilo C1 - C6 puede estar sustituido opcionalmente con cicloalquilo;
cada R10 es independientemente hidrógeno, acilo, alquilo C1 - C3, heteroalquilo o hidroxialquilo; R11 es haloalquilo; y
m es cero o un número entero entre 1 y 2.
2. El compuesto de la reivindicación 1, en donde Y es 0.
3. El compuesto de la reivindicación 1, en donde R2 se selecciona del grupo que consiste de hidroxialquilo, alquilaminilalquilo, dialquilaminilalquilo, -ZNR5R10, heterociclilo y heterociclilalquilo, en donde cada uno del Z, heterociclilo o heterociclilalquilo está independientemente sustituidos opcionalmente con R9.
4. El compuesto de la reivindicación 3, en donde el heterociclilo del heterociclilalquilo es independientemente azetidinilo, metilazetidinilo, etilazetidinilo, isopropilazetidinilo, difluoroazetidinilo, ciclopropilazetidinilo, tetrahidropiranilazetidinilo, tetrahidropiranilo, pirrolidinilo, metilpirrolidinilo, dimetilpirrolidinilo, isopropilpirrolidinilo, cicloalquilalquilpirrolidinilo, hidroxipirrolindinilo, fluoropirrolidinilo, difluoropirrolidinilo, (N-metil)fluoropirrolidinilo, (N-metil)difluoropirrolidinilo, metoxietilpirrolidinilo, (N-metil)metoxipirrolidinilo, piperazinilo, dimetilaminilpirrolidinilo, morfolinilo, metilmorfolinilo, etilmorfolinilo, isopropilmorfolinilo, oxetanilo, 1,4-oxazepanilo, piperdinilo, metilpiperidinilo, acilpiperdinilo, cianopiperdinilo, cicloalquilpiperdinilo, halopiperdinilo, dihalopiperdinilo, fluoropiperdinilo, difluoropiperdinilo, alcoxipiperdinilo, pirrolidonilo, 1,1-dióxido de tiomorfolinilo, 3-azabiciclo[3.1.0]hexanilo, oxa-5-azabiciclo[2.2.1]heptan-5-ilo o azabiciclo[2.2.1]heptan-2-ilo.
5. El compuesto de la reivindicación 4, en donde el (N-metil)difluoropirrolidinilo es 3,3-difluoro-1 -metilpirrolidinilo.
6. El compuesto de la reivindicación 4, en donde el heterociclilo es N-metilpirrolidinilo.
7. El compuesto de la reivindicación 3, en donde R2 es dialquilaminilalquilo sustituido opcionalmente con uno o más R9.
8. El compuesto de la reivindicación 1, en donde R4 es arilo sustituido opcionalmente con uno o más R7.
9. El compuesto de la reivindicación 8, en donde el arilo es selecciona del grupo que consiste en fenilo y naftilo sustituidos opcionalmente con uno o más R7.
10. El compuesto de la reivindicación 9, en donde cada uno del fenilo y el naftilo está sustituido opcionalmente con uno o más R7 seleccionados del grupo que consiste en ciano, halógeno, hidroxilo, alquilo C1- C6, hidroxialquilo, Q-haloalquilo, cicloalquilo y alcoxi.
11. El compuesto de la reivindicación 10, en donde R7 se selecciona del grupo que consiste en cloro, fluoro, bromo, hidroximetilo, metilo, etilo, isopropilo, metoxi, trifluorometilo, hidroxilo, ciclopropilo y ciano.
12. El compuesto de la reivindicación 1, en donde m es cero.
13. El compuesto de la reivindicación 1, en donde L es un enlace.
14. El compuesto de la reivindicación 1, en donde R8 es heteroalquilo, alquinilo C2-C4 o alquilo C1 - C3 sustituido opcionalmente con halógeno, -0R5, ciano o heteroarilo.
15. El compuesto de la reivindicación 1, en donde R8 es alquilo C1 - C3 sustituido opcionalmente con ciano, preferiblemente en donde R8 es cianometilo.
16. El compuesto de la reivindicación 1, en donde el compuesto es:

















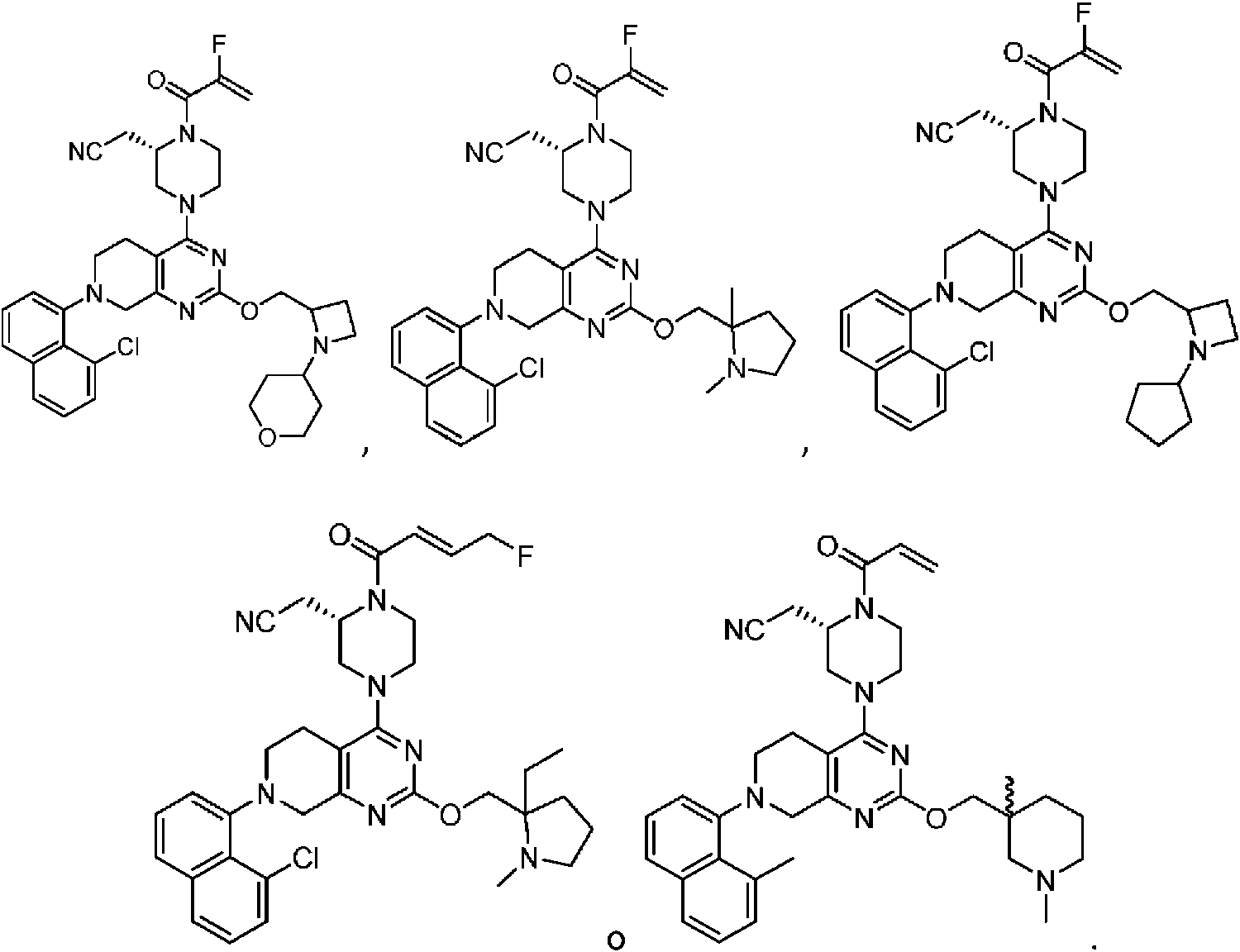
17. El compuesto de la reivindicación 1, en donde el compuesto es de Fórmula II-B:

F ó r m u l a I I - B
donde el anillo de piperazinilo está sustituido opcionalmente con R8, y R1 R3, R4, R8, L y m son como se definen en la reivindicación 1.
18. Un compuesto de la reivindicación 1, en donde el compuesto tiene la siguiente estructura:

o una sal farmacéuticamente aceptable del mismo.
19. Un compuesto de la reivindicación 18, en donde el compuesto tiene la siguiente estructura:

20. Un compuesto de la reivindicación 18, en donde el compuesto es una sal farmacéuticamente aceptable del compuesto con la siguiente estructura:

21. Un compuesto de las reivindicaciones 18-20 para usarlo en un método para el tratamiento de un cáncer de pulmón no microcítico, comprendiendo el método administrar a un paciente que tiene cáncer de pulmón no microcítico una cantidad terapéuticamente eficaz de un compuesto de las reivindicaciones 18-20 o una sal farmacéuticamente aceptable del mismo, solo o combinado con un vehículo, excipiente o diluyentes farmacéuticamente aceptables.
22. Un compuesto de las reivindicaciones 18-20 para usarlo en un método para el tratamiento de un cáncer, comprendiendo el método administrar a un paciente que tiene cáncer una cantidad terapéuticamente eficaz de un compuesto de las reivindicaciones 18-20 o una sal farmacéuticamente aceptable del mismo, solo o combinado con un vehículo, excipiente o diluyentes farmacéuticamente aceptables, en donde el cáncer es un tipo de tumor seleccionado de sarcomas y carcinomas astrocíticos, de mama, cervicales, colorrectales, endometriales, esofágicos, gástricos, de cabeza y cuello, hepatocelulares, laríngeos, pulmonares, orales, ováricos, de próstata y tiroideos.
23. Una composición farmacéutica, que comprende una cantidad terapéuticamente eficaz de un compuesto según una cualquiera de las reivindicaciones 1 a 20, y un excipiente farmacéuticamente aceptable.
24. Un método in vitro para inhibir la actividad de KRas G12C en una célula, que comprende poner en contacto la célula en la que se desea la inhibición de la actividad de KRas G12C con una cantidad eficaz de un compuesto según una cualquiera de las reivindicaciones 1 a 20, sales farmacéuticamente aceptables del mismo o composiciones farmacéuticas que contienen el compuesto según una cualquiera de las reivindicaciones 1 a 20, o la sal farmacéuticamente aceptable del mismo.
25. Un compuesto o una sal farmacéuticamente aceptable del mismo según una cualquiera de las reivindicaciones 1 a 20 para usarlo en un método para el tratamiento de un cáncer asociado con KRas G12C, comprendiendo el método administrar a un paciente que tiene cáncer una cantidad terapéuticamente eficaz de un compuesto o una sal farmacéuticamente aceptable del mismo según una cualquiera de las reivindicaciones 1 a 20, solo o combinado con un vehículo, excipiente o diluyentes farmacéuticamente aceptables, en donde el cáncer se selecciona del grupo que consiste en Cardíaco: sarcoma seleccionado de angiosarcoma, fibrosarcoma, rabdomiosarcoma y liposarcoma, mixoma, rabdomioma, fibroma, lipoma y teratoma; Pulmón: carcinoma broncogénico seleccionado de células escamosas, células pequeñas no diferenciadas, células grandes no diferenciadas y adenocarcinoma, carcinoma bronquioloalveolar, adenoma bronquial, sarcoma, linfoma, hamartoma condromatoso, mesotelioma; Gastrointestinal: de esófago seleccionado de carcinoma de células escamosas, adenocarcinoma, leiomiosarcoma y linfoma, de estómago seleccionado de carcinoma, linfoma y leiomiosarcoma, de páncreas seleccionado de adenocarcinoma ductal, insulinoma, glucagonoma, gastrinoma, tumores carcinoides y vipoma, de intestino delgado
seleccionado de adenocarcinoma, linfoma, tumores carcinoides, sarcoma de Kaposi, leiomioma, hemangioma, lipoma, neurofibroma y fibroma, de intestino grueso seleccionado de adenocarcinoma, adenoma tubular, adenoma velloso, hamartoma y leiomioma; Tracto genitourinario: de riñón seleccionado de adenocarcinoma, tumor de Wilm/nefroblastoma, linfoma y leucemia, de vejiga y uretra seleccionado de carcinoma de células escamosas, carcinoma de células transicionales y adenocarcinoma, de próstata seleccionado de adenocarcinoma y sarcoma, de testículos seleccionado de seminoma, teratoma, carcinoma embrionario, teratocarcinoma, coriocarcinoma, sarcoma, carcinoma de células intersticiales, fibroma, fibroadenoma, tumores adenomatoides y lipoma; Hígado: hepatoma/carcinoma hepatocelular, colangiocarcinoma, hepatoblastoma, angiosarcoma, adenoma hepatocelular, hemangioma; Tracto biliar: carcinoma de vesícula biliar, carcinoma ampular, colangiocarcinoma; Óseo: sarcoma osteogénico/osteosarcoma, fibrosarcoma, histiocitoma fibroso maligno, condrosarcoma, sarcoma de Ewing, linfoma maligno, sarcoma de células reticulares, mieloma múltiple, cordoma tumoral maligno de células gigantes, osteocronoma/exostosis osteocartilaginosa, condroma benigno, condroblastoma, condromixofibroma, osteoma osteoide y tumores de células gigantes; Sistema nervioso: de cráneo seleccionado de osteoma, hemangioma, granuloma, xantoma y osteítis deformante; de meninges seleccionado de meningioma, meningiosarcoma y gliomatosis; de cerebro seleccionado de astrocitoma, meduloblastoma, glioma, ependimoma, germinoma, pinealoma, glioblastoma multiforme, oligodendroglioma, schwannoma, retinoblastoma y tumores congénitos, de médula espinal seleccionado de neurofibroma, meningioma, glioma y sarcoma); Ginecológico: de útero seleccionado de carcinoma endometrial seleccionado de cistoadenocarcinoma seroso, cistadenocarcinoma mucinoso y carcinoma no clasificado, tumores de células de la granulosa-tecal, tumores de células de Sertoli-Leydig, disgerminoma y teratoma maligno, de vulva seleccionado de carcinoma de células escamosas, carcinoma intraepitelial, adenocarcinoma, fibrosarcoma y melanoma), de vagina seleccionado de carcinoma de células claras, carcinoma de células escamosas, sarcoma botrioide/rabdomiosarcoma embrionario, carcinoma de las trompas de Falopio; Hematológico: de sangre seleccionado de leucemia mieloide aguda, leucemia mieloide crónica, leucemia linfoblástica aguda, leucemia linfocítica crónica, enfermedades mieloproliferativas, mieloma múltiple y síndrome mielodisplásico, enfermedad de Hodgkin, linfoma maligno, linfoma no Hodgkin; Piel: melanoma maligno, carcinoma de células basales, carcinoma de células escamosas, sarcoma de Kaposi, lunares, nevos displásicos, lipoma, angioma, dermatofibroma, queloides, psoriasis; y Glándulas suprarrenales: neuroblastoma.
26. Un compuesto o sal farmacéuticamente aceptable del mismo para usarlo según la reivindicación 25, en donde la cantidad terapéuticamente eficaz del compuesto es de aproximadamente 0,01 a 100 mg/kg al día.
27. Un compuesto o sal farmacéuticamente aceptable del mismo para usarlo según la reivindicación 25 o 26, en donde el cáncer es cáncer de pulmón no microcítico.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762586775P | 2017-11-15 | 2017-11-15 | |
| PCT/US2018/061060 WO2019099524A1 (en) | 2017-11-15 | 2018-11-14 | Kras g12c inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2944547T3 true ES2944547T3 (es) | 2023-06-22 |
Family
ID=66431793
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18879484T Active ES2944547T3 (es) | 2017-11-15 | 2018-11-14 | Inhibidores de KRas G12C |
Country Status (32)
| Country | Link |
|---|---|
| US (4) | US10689377B2 (es) |
| EP (2) | EP3710439B1 (es) |
| JP (2) | JP7322019B2 (es) |
| KR (1) | KR20200100632A (es) |
| CN (2) | CN118459460A (es) |
| AU (1) | AU2018369759B2 (es) |
| BR (1) | BR112020009818A2 (es) |
| CA (1) | CA3082579A1 (es) |
| CL (1) | CL2020001271A1 (es) |
| CO (1) | CO2020007244A2 (es) |
| DK (1) | DK3710439T3 (es) |
| EA (1) | EA202091186A1 (es) |
| ES (1) | ES2944547T3 (es) |
| FI (2) | FI3710439T3 (es) |
| FR (1) | FR24C1026I1 (es) |
| HR (1) | HRP20230377T1 (es) |
| HU (2) | HUE061599T2 (es) |
| IL (1) | IL274601B2 (es) |
| LT (1) | LT3710439T (es) |
| MX (1) | MX2020005063A (es) |
| NL (1) | NL301279I2 (es) |
| PH (1) | PH12020550622A1 (es) |
| PL (1) | PL3710439T3 (es) |
| PT (1) | PT3710439T (es) |
| RS (1) | RS64182B1 (es) |
| SA (1) | SA520411982B1 (es) |
| SG (1) | SG11202004427TA (es) |
| SI (1) | SI3710439T1 (es) |
| TW (1) | TWI809005B (es) |
| UA (1) | UA125802C2 (es) |
| WO (2) | WO2019099524A1 (es) |
| ZA (1) | ZA202002105B (es) |
Families Citing this family (147)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7039489B2 (ja) * | 2016-05-18 | 2022-03-22 | ミラティ セラピューティクス, インコーポレイテッド | Kras g12c阻害剤 |
| MX2020005063A (es) * | 2017-11-15 | 2021-01-08 | Mirati Therapeutics Inc | Inhibidores de kras g12c. |
| WO2019217307A1 (en) | 2018-05-07 | 2019-11-14 | Mirati Therapeutics, Inc. | Kras g12c inhibitors |
| TW202012415A (zh) | 2018-05-08 | 2020-04-01 | 瑞典商阿斯特捷利康公司 | 化學化合物 |
| ES2961253T3 (es) * | 2018-08-31 | 2024-03-11 | Mirati Therapeutics Inc | Inhibidores de KRas G12C |
| JP7546549B2 (ja) | 2018-09-10 | 2024-09-06 | ミラティ セラピューティクス, インコーポレイテッド | 組み合わせ療法 |
| WO2020055760A1 (en) | 2018-09-10 | 2020-03-19 | Mirati Therapeutics, Inc. | Combination therapies |
| AU2019340366A1 (en) * | 2018-09-10 | 2021-04-01 | Mirati Therapeutics, Inc. | Combination therapies |
| CA3111977A1 (en) * | 2018-09-10 | 2020-03-19 | Mirati Therapeutics, Inc. | Combination therapies |
| AU2019338207A1 (en) | 2018-09-10 | 2021-04-29 | Mirati Therapeutics, Inc. | Combination therapies |
| US12065430B2 (en) | 2018-10-26 | 2024-08-20 | Taiho Pharmaceutical Co., Ltd. | Indazole compound or salt thereof |
| BR112021008986A2 (pt) | 2018-11-09 | 2021-08-10 | F. Hoffmann-La Roche Ag | composto, composto da fórmula, composto ou sal farmaceuticamente aceitável, composição farmacêutica, métodos para tratar o câncer, para regular a atividade de uma proteína mutante g12c k-ras, para inibir a proliferação de uma população de células, para tratar um distúrbio mediado, para preparar uma proteína e para inibir metástase de tumor e uso |
| KR20210113587A (ko) | 2018-12-05 | 2021-09-16 | 미라티 테라퓨틱스, 인크. | 병용 요법 |
| JP2022517222A (ja) | 2019-01-10 | 2022-03-07 | ミラティ セラピューティクス, インコーポレイテッド | Kras g12c阻害剤 |
| JP2022520079A (ja) | 2019-02-12 | 2022-03-28 | ノバルティス アーゲー | Tno155及びkrasg12c阻害剤を含む医薬組合せ |
| CN113874374A (zh) * | 2019-05-24 | 2021-12-31 | 江苏恒瑞医药股份有限公司 | 氢化吡啶并嘧啶类衍生物、其制备方法及其在医药上的应用 |
| AU2020282473A1 (en) * | 2019-05-29 | 2022-01-06 | Jiangsu Hansoh Pharmaceutical Group Co., Ltd. | Nitrogen-containing heterocyclic derivative regulator, preparation method therefor and application thereof |
| CN112047939B (zh) * | 2019-06-06 | 2023-05-02 | 江苏先声药业有限公司 | 一种具有抗肿瘤活性的四氢吡啶并嘧啶类化合物 |
| CN112300153B (zh) * | 2019-07-26 | 2023-06-13 | 博瑞生物医药(苏州)股份有限公司 | 一种杂环化合物、药物组合物和用途 |
| WO2021023154A1 (zh) * | 2019-08-02 | 2021-02-11 | 上海济煜医药科技有限公司 | 四并环类化合物及其制备方法和应用 |
| WO2021023247A1 (en) * | 2019-08-07 | 2021-02-11 | Jacobio Pharmaceuticals Co., Ltd. | Kras mutant protein inhibitor |
| CN112390788A (zh) * | 2019-08-13 | 2021-02-23 | 苏州闻天医药科技有限公司 | 一种用于抑制krasg12c突变蛋白的化合物及其制备方法和用途 |
| CN112390797A (zh) * | 2019-08-15 | 2021-02-23 | 微境生物医药科技(上海)有限公司 | 新型螺环类K-Ras G12C抑制剂 |
| WO2021043322A1 (zh) * | 2019-09-06 | 2021-03-11 | 正大天晴药业集团南京顺欣制药有限公司 | 氮杂环庚烷并嘧啶类衍生物及其医药用途 |
| US20220402916A1 (en) * | 2019-09-18 | 2022-12-22 | Merck Sharp & Dohme Corp. | Small molecule inhibitors of kras g12c mutant |
| JP2022548791A (ja) * | 2019-09-24 | 2022-11-21 | ミラティ セラピューティクス, インコーポレイテッド | 組み合わせ療法 |
| WO2021057832A1 (en) * | 2019-09-25 | 2021-04-01 | Jacobio Pharmaceuticals Co., Ltd. | Kras mutant protein inhibitor |
| US20230257374A1 (en) * | 2019-10-10 | 2023-08-17 | Innovent Biologics (Suzhou) Co., Ltd. | Novel kras g12c protein inhibitor, preparation method therefor, and use thereof |
| CN112694475A (zh) * | 2019-10-23 | 2021-04-23 | 苏州泽璟生物制药股份有限公司 | 环烷基类和杂环烷基类抑制剂及其制备方法和应用 |
| CR20220230A (es) | 2019-10-28 | 2022-06-15 | Merck Sharp & Dohme | Inhibidores de pequeñas moléculas de mutante g12c de kras |
| CN117645614A (zh) * | 2019-10-30 | 2024-03-05 | 劲方医药科技(上海)有限公司 | 取代的杂环并环类化合物,其制法与医药上的用途 |
| CN113286794B (zh) * | 2019-11-04 | 2024-03-12 | 北京加科思新药研发有限公司 | Kras突变蛋白抑制剂 |
| TW202132315A (zh) | 2019-11-04 | 2021-09-01 | 美商銳新醫藥公司 | Ras 抑制劑 |
| CR20220241A (es) | 2019-11-04 | 2022-08-03 | Revolution Medicines Inc | Inhibidores de ras |
| WO2021091956A1 (en) | 2019-11-04 | 2021-05-14 | Revolution Medicines, Inc. | Ras inhibitors |
| CN112778301A (zh) * | 2019-11-07 | 2021-05-11 | 苏州泽璟生物制药股份有限公司 | 四氢吡啶并嘧啶类抑制剂及其制备方法和应用 |
| CN112824410A (zh) * | 2019-11-21 | 2021-05-21 | 苏州泽璟生物制药股份有限公司 | 氮杂七元环类抑制剂及其制备方法和应用 |
| EP4065231A1 (en) | 2019-11-27 | 2022-10-05 | Revolution Medicines, Inc. | Covalent ras inhibitors and uses thereof |
| WO2021108682A1 (en) | 2019-11-27 | 2021-06-03 | Turning Point Therapeutics, Inc. | Combination therapy involving diaryl macrocyclic compounds |
| WO2021106231A1 (en) * | 2019-11-29 | 2021-06-03 | Taiho Pharmaceutical Co., Ltd. | A compound having inhibitory activity against kras g12d mutation |
| US20220315598A1 (en) * | 2019-12-02 | 2022-10-06 | Shanghai Yingli Pharmaceutical Co., Ltd | Oxygen-containing Heterocyclic Compound, Preparation Method Therefor and Use Thereof |
| CN117820333A (zh) | 2019-12-11 | 2024-04-05 | 伊莱利利公司 | Kras g12c抑制剂 |
| CN113004269A (zh) * | 2019-12-19 | 2021-06-22 | 首药控股(北京)有限公司 | Kras-G12C抑制剂杂环化合物 |
| WO2021121367A1 (en) * | 2019-12-19 | 2021-06-24 | Jacobio Pharmaceuticals Co., Ltd. | Kras mutant protein inhibitors |
| AU2020405170A1 (en) | 2019-12-20 | 2022-06-30 | Mirati Therapeutics, Inc. | SOS1 inhibitors |
| WO2021120890A1 (en) * | 2019-12-20 | 2021-06-24 | Novartis Ag | Pyrazolyl derivatives useful as anti-cancer agents |
| CN113045565A (zh) * | 2019-12-27 | 2021-06-29 | 微境生物医药科技(上海)有限公司 | 新型K-Ras G12C抑制剂 |
| CN112094269B (zh) * | 2020-01-01 | 2021-12-07 | 上海凌达生物医药有限公司 | 一类饱和六元环并杂环类化合物、制备方法和用途 |
| TWI770760B (zh) * | 2020-01-08 | 2022-07-11 | 大陸商蘇州亞盛藥業有限公司 | 螺環四氫喹唑啉 |
| GB202001344D0 (en) | 2020-01-31 | 2020-03-18 | Redx Pharma Plc | Ras Inhibitors |
| US20230099858A1 (en) * | 2020-02-20 | 2023-03-30 | Beta Pharma, Inc. | Pyridopyrimidine derivatives as kras inhibitors |
| WO2021169990A1 (zh) * | 2020-02-24 | 2021-09-02 | 泰励生物科技(上海)有限公司 | 用于癌症治疗的kras抑制剂 |
| CN115427414A (zh) * | 2020-04-28 | 2022-12-02 | 贝达药业股份有限公司 | 稠环化合物及其在医药上的应用 |
| WO2021219072A1 (zh) * | 2020-04-30 | 2021-11-04 | 上海科州药物研发有限公司 | 作为kras抑制剂的杂环化合物的制备及其应用方法 |
| WO2021231526A1 (en) * | 2020-05-13 | 2021-11-18 | Incyte Corporation | Fused pyrimidine compounds as kras inhibitors |
| CN113666923A (zh) * | 2020-05-15 | 2021-11-19 | 苏州泽璟生物制药股份有限公司 | 烷氧基烷基取代杂环基类抑制剂及其制备方法和应用 |
| PE20240493A1 (es) | 2020-06-02 | 2024-03-15 | Boehringer Ingelheim Int | 2-amino-3-cianotiofenos anulados y derivados para el tratamiento del cancer |
| MX2022015272A (es) * | 2020-06-04 | 2023-01-11 | Antengene Discovery Ltd | Inhibidores de la proteina kras g12c y usos de estos. |
| WO2021257736A1 (en) | 2020-06-18 | 2021-12-23 | Revolution Medicines, Inc. | Methods for delaying, preventing, and treating acquired resistance to ras inhibitors |
| JP2023531049A (ja) | 2020-06-24 | 2023-07-20 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Sos1阻害剤及びkras g12c阻害剤を含む抗がん剤併用療法 |
| WO2021260111A1 (en) * | 2020-06-25 | 2021-12-30 | Tolremo Therapeutics Ag | Combination of a cbp/p300 bromodomain inhibitor and a kras inhibitor for the treatment of cancer |
| CN113912608B (zh) * | 2020-07-10 | 2023-07-14 | 江苏恒瑞医药股份有限公司 | 嘧啶并嘧啶酮类衍生物、其制备方法及其在医药上的应用 |
| CN113980014B (zh) * | 2020-07-27 | 2023-05-12 | 江苏恒瑞医药股份有限公司 | 氢化吡啶并嘧啶类衍生物、其制备方法及其在医药上的应用 |
| JP2023538060A (ja) * | 2020-08-17 | 2023-09-06 | 貝達薬業股▲ふん▼有限公司 | 二環化合物、それを含む組成物、及びそれらの使用 |
| AU2021344830A1 (en) | 2020-09-03 | 2023-04-06 | Revolution Medicines, Inc. | Use of SOS1 inhibitors to treat malignancies with SHP2 mutations |
| WO2022052895A1 (zh) | 2020-09-11 | 2022-03-17 | 南京明德新药研发有限公司 | 氮杂环丁烷取代化合物的晶型 |
| BR112023004569A2 (pt) * | 2020-09-11 | 2023-04-04 | Mirati Therapeutics Inc | Formas cristalinas de um inibidor de kras g12c |
| PE20231207A1 (es) | 2020-09-15 | 2023-08-17 | Revolution Medicines Inc | Derivados indolicos como inhibidores de ras en el tratamiento del cancer |
| US20220112204A1 (en) * | 2020-10-14 | 2022-04-14 | Accutar Biotechnology Inc. | Substituted dihydropyranopyrimidine compounds as kras inhibitors |
| WO2022087270A1 (en) * | 2020-10-23 | 2022-04-28 | Mirati Therapeutics, Inc. | Methods for treatment of lung cancers |
| WO2022089604A1 (en) * | 2020-10-30 | 2022-05-05 | Novartis Ag | New crystalline forms of a kras g12c inhibitor compound |
| EP4242207A1 (en) | 2020-11-06 | 2023-09-13 | Tyligand Bioscience (Shanghai) Limited | Kras inhibitors for treatment of cancers |
| EP4247807A4 (en) * | 2020-11-23 | 2024-10-16 | Merck Sharp & Dohme Llc | 6,7-DIHYDRO-PYRANO[2,3-D PYRIMIDINE AS INHIBITORS OF THE KRAS G12C MUTANT |
| CN114591319B (zh) * | 2020-12-04 | 2024-06-28 | 江苏先声药业有限公司 | 四氢吡啶并嘧啶类衍生物及其用途 |
| IL303446A (en) * | 2020-12-15 | 2023-08-01 | Mirati Therapeutics Inc | Azaquinazoline compounds as PAN-KRas inhibitors |
| WO2022133038A1 (en) * | 2020-12-16 | 2022-06-23 | Mirati Therapeutics, Inc. | Tetrahydropyridopyrimidine pan-kras inhibitors |
| TWI795129B (zh) * | 2020-12-18 | 2023-03-01 | 大陸商正大天晴藥業集團股份有限公司 | 吡啶并嘧啶酮類化合物 |
| EP4263511A1 (en) | 2020-12-18 | 2023-10-25 | Amphista Therapeutics Ltd | Novel bifunctional molecules for targeted protein degradation |
| US20230107642A1 (en) | 2020-12-18 | 2023-04-06 | Erasca, Inc. | Tricyclic pyridones and pyrimidones |
| US20240100172A1 (en) | 2020-12-21 | 2024-03-28 | Hangzhou Jijing Pharmaceutical Technology Limited | Methods and compounds for targeted autophagy |
| IL303917A (en) * | 2020-12-22 | 2023-08-01 | Novartis Ag | Drug combinations involving a KRAS G12C inhibitor and uses of a KRAS G12C inhibitor to treat cancer |
| US20240092803A1 (en) * | 2021-01-08 | 2024-03-21 | Beigene Switzerland Gmbh | Bridged compounds as kras g12d inhibitor and degrader and the use thereof |
| EP4286378A4 (en) * | 2021-02-01 | 2024-08-07 | Medshine Discovery Inc | PYRIMIDOPYRAN COMPOUND |
| WO2022171143A1 (zh) * | 2021-02-09 | 2022-08-18 | 南京明德新药研发有限公司 | 5,6,7,8-四氢吡啶[3,4-d]嘧啶化合物 |
| WO2022187527A1 (en) * | 2021-03-05 | 2022-09-09 | Nikang Therapeutics, Inc | Quinazoline nitrile derivatives as kras inhibitors |
| JP2024509625A (ja) | 2021-03-15 | 2024-03-04 | ノバルティス アーゲー | ベンゾイソオキサゾール誘導体及びその使用 |
| WO2022194192A1 (zh) * | 2021-03-18 | 2022-09-22 | 四川科伦博泰生物医药股份有限公司 | 一类杂芳环化合物、其制备方法及用途 |
| US12077539B2 (en) | 2021-03-22 | 2024-09-03 | Incyte Corporation | Imidazole and triazole KRAS inhibitors |
| CN115124524A (zh) * | 2021-03-26 | 2022-09-30 | 浙江海正药业股份有限公司 | 三环类衍生物及其制备方法和用途 |
| CN116157400A (zh) * | 2021-03-30 | 2023-05-23 | 浙江海正药业股份有限公司 | 杂环类衍生物及其制备方法和用途 |
| EP4319754A1 (en) | 2021-04-08 | 2024-02-14 | Mirati Therapeutics, Inc. | Combination therapies using prmt5 inhibitors for the treatment of cancer |
| WO2022217042A1 (en) * | 2021-04-09 | 2022-10-13 | Ikena Oncology, Inc. | Naphthyl-substituted quinoline-4(1h)-ones and related compounds and their use in treating medical conditions |
| US20240238294A1 (en) | 2021-04-09 | 2024-07-18 | Boehringer Ingelheim International Gmbh | Anticancer therapy |
| CN117203208A (zh) * | 2021-04-23 | 2023-12-08 | 清华大学 | 靶向活化与失活态kras g12d的抑制剂 |
| AR125787A1 (es) | 2021-05-05 | 2023-08-16 | Revolution Medicines Inc | Inhibidores de ras |
| CR20230570A (es) | 2021-05-05 | 2024-01-22 | Revolution Medicines Inc | Inhibidores de ras |
| JP2024519845A (ja) * | 2021-05-19 | 2024-05-21 | ジェネンテック, インコーポレイテッド | 併用療法 |
| WO2022247760A1 (zh) * | 2021-05-22 | 2022-12-01 | 上海科州药物研发有限公司 | 作为kras抑制剂的杂环化合物,及其制备和治疗用途 |
| US20220395507A1 (en) * | 2021-05-27 | 2022-12-15 | Mirati Therapeutics, Inc. | Combination therapies |
| WO2022251576A1 (en) * | 2021-05-28 | 2022-12-01 | Merck Sharp & Dohme Corp. | Small molecule inhibitors of kras g12c mutant |
| US20240317759A1 (en) * | 2021-05-28 | 2024-09-26 | Taiho Pharmaceutical Co., Ltd. | Small molecule inhibitors of kras mutated proteins |
| WO2022261154A1 (en) | 2021-06-09 | 2022-12-15 | Eli Lilly And Company | Substituted fused azines as kras g12d inhibitors |
| WO2022266206A1 (en) | 2021-06-16 | 2022-12-22 | Erasca, Inc. | Kras inhibitor conjugates |
| CN117529321A (zh) * | 2021-06-18 | 2024-02-06 | 上海德琪医药科技有限公司 | Erk抑制剂和kras抑制剂的组合及其用途 |
| TW202317100A (zh) | 2021-06-23 | 2023-05-01 | 瑞士商諾華公司 | 包含kras g12c抑制劑的藥物組合及其用於治療癌症之用途 |
| CN117751116A (zh) * | 2021-07-07 | 2024-03-22 | 微境生物医药科技(上海)有限公司 | 作为KRas G12D抑制剂的稠环化合物 |
| WO2023284730A1 (en) | 2021-07-14 | 2023-01-19 | Nikang Therapeutics, Inc. | Alkylidene derivatives as kras inhibitors |
| WO2023283933A1 (en) * | 2021-07-16 | 2023-01-19 | Silexon Biotech Co., Ltd. | Compounds useful as kras g12d inhibitors |
| MX2024000965A (es) | 2021-07-27 | 2024-02-09 | Toray Industries | Medicamento para el tratamiento y/o prevencion de cancer. |
| CN113527294A (zh) * | 2021-08-25 | 2021-10-22 | 都创(上海)医药开发有限公司 | Mrtx849化合物的晶型及其制备方法和用途 |
| AU2022336415A1 (en) | 2021-09-01 | 2024-01-04 | Novartis Ag | Pharmaceutical combinations comprising a tead inhibitor and uses thereof for the treatment of cancers |
| EP4389751A1 (en) | 2021-09-03 | 2024-06-26 | Kumquat Biosciences Inc. | Heterocyclic compounds and uses thereof |
| CA3231284A1 (en) * | 2021-09-09 | 2023-03-16 | Thomas SCATTOLIN | Processes and intermediates for synthesis of adagrasib |
| WO2023051586A1 (zh) * | 2021-09-29 | 2023-04-06 | 先声再明医药有限公司 | Kras g12d抑制剂化合物及其制备方法和应用 |
| AR127308A1 (es) | 2021-10-08 | 2024-01-10 | Revolution Medicines Inc | Inhibidores ras |
| TW202322819A (zh) | 2021-10-22 | 2023-06-16 | 大陸商江蘇恆瑞醫藥股份有限公司 | 含氮的四環化合物、其製備方法及其在醫藥上的應用 |
| WO2023097195A1 (en) | 2021-11-24 | 2023-06-01 | Genentech, Inc. | Therapeutic indazole compounds and methods of use in the treatment of cancer |
| EP4436969A2 (en) | 2021-11-24 | 2024-10-02 | Genentech, Inc. | Bicyclic therapeutic compounds and methods of use in the treatment of cancer |
| CA3240980A1 (en) | 2021-12-01 | 2023-06-08 | Boehringer Ingelheim International Gmbh | Annulated 2-amino-3-cyano thiophenes and derivatives for the treatment of cancer |
| WO2023114954A1 (en) | 2021-12-17 | 2023-06-22 | Genzyme Corporation | Pyrazolopyrazine compounds as shp2 inhibitors |
| CN118339153A (zh) | 2021-12-22 | 2024-07-12 | 勃林格殷格翰国际有限公司 | 用于治疗癌症的杂芳族化合物 |
| WO2023126822A1 (en) | 2021-12-28 | 2023-07-06 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and rasg12c inhibitor |
| CN114409653A (zh) * | 2021-12-31 | 2022-04-29 | 苏州闻天医药科技有限公司 | 一种桥环并嘧啶并环类化合物及其用途 |
| TWI841200B (zh) * | 2022-01-21 | 2024-05-01 | 大陸商上海湃隆生物科技有限公司 | 雜環類化合物、藥物組成物及其應用 |
| WO2023154528A1 (en) * | 2022-02-11 | 2023-08-17 | Wave Life Sciences Ltd. | Stereoselective technologies for chiral compounds |
| EP4227307A1 (en) | 2022-02-11 | 2023-08-16 | Genzyme Corporation | Pyrazolopyrazine compounds as shp2 inhibitors |
| WO2023172940A1 (en) | 2022-03-08 | 2023-09-14 | Revolution Medicines, Inc. | Methods for treating immune refractory lung cancer |
| US20240043451A1 (en) | 2022-03-25 | 2024-02-08 | Eli Lilly And Company | Kras inhibitors |
| WO2023190748A1 (ja) | 2022-03-31 | 2023-10-05 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | 腫瘍治療用医薬組成物 |
| WO2023194310A1 (en) | 2022-04-04 | 2023-10-12 | Sanofi | Therapeutic combination of kras g12c inhibitor and tead inhibitor |
| WO2023199180A1 (en) | 2022-04-11 | 2023-10-19 | Novartis Ag | Therapeutic uses of a krasg12c inhibitor |
| AU2023257901A1 (en) * | 2022-04-18 | 2024-10-10 | Mirati Therapeutics, Inc. | Processes and intermediates for synthesis of adagrasib |
| WO2023205701A1 (en) | 2022-04-20 | 2023-10-26 | Kumquat Biosciences Inc. | Macrocyclic heterocycles and uses thereof |
| CN114907387B (zh) * | 2022-05-26 | 2023-11-10 | 中山大学 | 嘧啶并吡咯类kras抑制剂及其制备方法与应用 |
| CN117164580A (zh) * | 2022-05-27 | 2023-12-05 | 苏州泽璟生物制药股份有限公司 | 一种kras g12c抑制剂的制备方法及其中间体 |
| CN117164526A (zh) * | 2022-05-27 | 2023-12-05 | 苏州泽璟生物制药股份有限公司 | 2-(哌嗪-2-基)乙腈类衍生物及其制备方法和应用 |
| WO2023240189A1 (en) | 2022-06-10 | 2023-12-14 | Bristol-Myers Squibb Company | Tetrahydropyrido 3,4-d pyrimidine derivatives as kras inhibitors |
| WO2023240263A1 (en) | 2022-06-10 | 2023-12-14 | Revolution Medicines, Inc. | Macrocyclic ras inhibitors |
| WO2024031088A1 (en) | 2022-08-05 | 2024-02-08 | Kumquat Biosciences Inc. | Heterocyclic compounds and uses thereof |
| WO2024040131A1 (en) | 2022-08-17 | 2024-02-22 | Treeline Biosciences, Inc. | Pyridopyrimidine kras inhibitors |
| GB202212641D0 (en) | 2022-08-31 | 2022-10-12 | Jazz Pharmaceuticals Ireland Ltd | Novel compounds |
| WO2024102421A2 (en) | 2022-11-09 | 2024-05-16 | Revolution Medicines, Inc. | Compounds, complexes, and methods for their preparation and of their use |
| WO2024112654A1 (en) | 2022-11-21 | 2024-05-30 | Treeline Biosciences, Inc. | Spirocyclic dihydropyranopyrimidine kras inhibitors |
| WO2024206747A1 (en) | 2023-03-30 | 2024-10-03 | Eli Lilly And Company | Kras inhibitors |
| WO2024206858A1 (en) | 2023-03-30 | 2024-10-03 | Revolution Medicines, Inc. | Compositions for inducing ras gtp hydrolysis and uses thereof |
| WO2024206766A1 (en) | 2023-03-31 | 2024-10-03 | Eli Lilly And Company | Kras inhibitors |
| CN116478141B (zh) * | 2023-06-20 | 2023-10-24 | 药康众拓(江苏)医药科技有限公司 | 氘代kras抑制剂药物及用途 |
Family Cites Families (70)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR100554813B1 (ko) | 2001-01-02 | 2006-02-22 | 에프. 호프만-라 로슈 아게 | 알파 1에이/비 아드레날린성 수용체 길항제로서의퀴나졸론 유도체 |
| US7105667B2 (en) | 2001-05-01 | 2006-09-12 | Bristol-Myers Squibb Co. | Fused heterocyclic compounds and use thereof |
| WO2002088079A2 (en) | 2001-05-01 | 2002-11-07 | Bristol-Myers Squibb Company | Dual inhibitors of pde 7 and pde 4 |
| US20080051387A1 (en) | 2006-06-09 | 2008-02-28 | Yuelian Xu | Tetrahydropyrido[3,4-d]pyrimidines and related analogues |
| US9259426B2 (en) | 2006-07-20 | 2016-02-16 | Gilead Sciences, Inc. | 4,6-di- and 2,4,6-trisubstituted quinazoline derivatives useful for treating viral infections |
| US9562019B2 (en) | 2006-12-21 | 2017-02-07 | Sloan-Kettering Institute For Cancer Research | Substituted pyridazines as EGFR and/or KRAS inhibitors |
| WO2008103470A2 (en) | 2007-02-21 | 2008-08-28 | Trustees Of Columbia University In The City Of New York | Oncogenic-ras-signal dependent lethal compounds |
| KR20090130051A (ko) | 2007-03-14 | 2009-12-17 | 엑셀리시스, 인코포레이티드 | 헤지호그 경로의 억제제 |
| EP2209775A1 (en) | 2007-10-09 | 2010-07-28 | UCB Pharma, S.A. | Heterobicyclic compounds as histamine h4-receptor antagonists |
| CA2719376A1 (en) | 2008-04-07 | 2009-10-15 | Gilead Sciences, Inc. | 2h-benzo[b][1,4]oxazin-3(4h)-one derivatives for use as stearoyl coa desaturase inhibitors |
| TW201018681A (en) | 2008-07-31 | 2010-05-16 | Genentech Inc | Pyrimidine compounds, compositions and methods of use |
| WO2010120996A1 (en) | 2009-04-17 | 2010-10-21 | Wyeth Llc | 5,6,7,8-tetrahydropyrido[3,4-d]pyrimidine compounds, their use as mtor kinase and pi3 kinase inhibitors, and their syntheses |
| JPWO2011078369A1 (ja) | 2009-12-25 | 2013-05-09 | 持田製薬株式会社 | 新規アリールウレア誘導体 |
| CN102812167A (zh) | 2009-12-30 | 2012-12-05 | 阿维拉制药公司 | 蛋白的配体-介导的共价修饰 |
| EP2836482B1 (en) | 2012-04-10 | 2019-12-25 | The Regents of The University of California | Compositions and methods for treating cancer |
| US9695133B2 (en) | 2012-07-13 | 2017-07-04 | The Trustees Of Columbia University In The City Of New York | Quinazolinone-based oncogenic-RAS-selective lethal compounds and their use |
| WO2014143659A1 (en) | 2013-03-15 | 2014-09-18 | Araxes Pharma Llc | Irreversible covalent inhibitors of the gtpase k-ras g12c |
| JP6473133B2 (ja) | 2013-03-15 | 2019-02-20 | アラクセス ファーマ エルエルシー | Krasg12cの共有結合性阻害剤 |
| UY35464A (es) | 2013-03-15 | 2014-10-31 | Araxes Pharma Llc | Inhibidores covalentes de kras g12c. |
| CA2926328C (en) | 2013-10-10 | 2022-11-29 | Araxes Pharma Llc | Substituted quinazolinyl and quinolinyl derivatives and pharmaceutical compositions thereof useful as inhibitors of kras g12c |
| JO3805B1 (ar) | 2013-10-10 | 2021-01-31 | Araxes Pharma Llc | مثبطات كراس جي12سي |
| EP3148981A4 (en) | 2014-05-30 | 2017-11-08 | The Trustees of Columbia University in the City of New York | Multivalent ras binding compounds |
| WO2016025650A1 (en) | 2014-08-13 | 2016-02-18 | Celgene Avilomics Research, Inc. | Combinations of an erk inhibitor and a cdk4/6 inhibitor and related methods |
| JO3556B1 (ar) * | 2014-09-18 | 2020-07-05 | Araxes Pharma Llc | علاجات مدمجة لمعالجة السرطان |
| ES2826443T3 (es) | 2014-09-25 | 2021-05-18 | Araxes Pharma Llc | Inhibidores de proteínas mutantes KRAS G12C |
| US10011600B2 (en) | 2014-09-25 | 2018-07-03 | Araxes Pharma Llc | Methods and compositions for inhibition of Ras |
| WO2016049565A1 (en) | 2014-09-25 | 2016-03-31 | Araxes Pharma Llc | Compositions and methods for inhibition of ras |
| US10017540B2 (en) | 2015-03-11 | 2018-07-10 | California Institute Of Technology | Cyclic peptide binder against oncogenic K-Ras |
| KR20180005178A (ko) | 2015-04-10 | 2018-01-15 | 아락세스 파마 엘엘씨 | 치환된 퀴나졸린 화합물 및 이의 사용방법 |
| WO2016168540A1 (en) | 2015-04-15 | 2016-10-20 | Araxes Pharma Llc | Fused-tricyclic inhibitors of kras and methods of use thereof |
| WO2016172692A1 (en) | 2015-04-24 | 2016-10-27 | H. Lee Moffitt Cancer Center And Research Institute, Inc. | Mutant kras inhibitors |
| EP3291813A4 (en) | 2015-05-06 | 2019-01-02 | The Regents of The University of California | K-ras modulators |
| US10144724B2 (en) | 2015-07-22 | 2018-12-04 | Araxes Pharma Llc | Substituted quinazoline compounds and methods of use thereof |
| WO2017053722A1 (en) | 2015-09-24 | 2017-03-30 | Ionis Pharmaceuticals, Inc. | Modulators of kras expression |
| EP3356359B1 (en) | 2015-09-28 | 2021-10-20 | Araxes Pharma LLC | Inhibitors of kras g12c mutant proteins |
| WO2017058728A1 (en) | 2015-09-28 | 2017-04-06 | Araxes Pharma Llc | Inhibitors of kras g12c mutant proteins |
| WO2017058807A1 (en) | 2015-09-28 | 2017-04-06 | Araxes Pharma Llc | Inhibitors of kras g12c mutant proteins |
| US10689356B2 (en) | 2015-09-28 | 2020-06-23 | Araxes Pharma Llc | Inhibitors of KRAS G12C mutant proteins |
| WO2017058805A1 (en) | 2015-09-28 | 2017-04-06 | Araxes Pharma Llc | Inhibitors of kras g12c mutant proteins |
| WO2017058915A1 (en) | 2015-09-28 | 2017-04-06 | Araxes Pharma Llc | Inhibitors of kras g12c mutant proteins |
| WO2017058768A1 (en) | 2015-09-28 | 2017-04-06 | Araxes Pharma Llc | Inhibitors of kras g12c mutant proteins |
| EP3364977A4 (en) | 2015-10-19 | 2019-09-04 | Araxes Pharma LLC | PROCESS FOR SCREENING INHIBITORS OF RAS |
| WO2017070611A1 (en) | 2015-10-22 | 2017-04-27 | The Scripps Research Institute | Cysteine reactive probes and uses thereof |
| WO2017080980A1 (en) | 2015-11-09 | 2017-05-18 | Astrazeneca Ab | Dihydropyrrolopyrazinone derivatives useful in the treatment of cancer |
| WO2017079864A1 (en) | 2015-11-12 | 2017-05-18 | Hangzhou Yier Biotech Co., Ltd. | Treatment of cancers related to chronically active ras |
| TW201726656A (zh) | 2015-11-16 | 2017-08-01 | 亞瑞克西斯製藥公司 | 包含經取代雜環基團之2-經取代喹唑啉化合物及其使用方法 |
| WO2017100546A1 (en) | 2015-12-09 | 2017-06-15 | Araxes Pharma Llc | Methods for preparation of quinazoline derivatives |
| JP7039489B2 (ja) * | 2016-05-18 | 2022-03-22 | ミラティ セラピューティクス, インコーポレイテッド | Kras g12c阻害剤 |
| US10646488B2 (en) | 2016-07-13 | 2020-05-12 | Araxes Pharma Llc | Conjugates of cereblon binding compounds and G12C mutant KRAS, HRAS or NRAS protein modulating compounds and methods of use thereof |
| EP3519402A1 (en) | 2016-09-29 | 2019-08-07 | Araxes Pharma LLC | Inhibitors of kras g12c mutant proteins |
| JP2019534260A (ja) | 2016-10-07 | 2019-11-28 | アラクセス ファーマ エルエルシー | Rasの阻害剤としての複素環式化合物およびその使用方法 |
| MX2019006299A (es) | 2016-11-30 | 2019-11-12 | Bantam Pharmaceutical Llc | Compuestos de pirazola sustituida y metodos para usarlos para el tratamiento de enfermedades hiperproliferativas. |
| US20200069656A1 (en) | 2016-11-30 | 2020-03-05 | Bantam Pharmaceutical, Llc | Methods of Using Substituted Pyrazole and Pyrazole Compounds and for Treatment of Hyperproliferative Diseases |
| KR20190095355A (ko) | 2016-12-15 | 2019-08-14 | 더 리젠츠 오브 더 유니버시티 오브 캘리포니아 | 암의 치료를 위한 조성물 및 방법 |
| SG10201913491PA (en) | 2016-12-22 | 2020-03-30 | Amgen Inc | Benzisothiazole, isothiazolo[3,4-b]pyridine, quinazoline, phthalazine, pyrido[2,3-d]pyridazine and pyrido[2,3-d]pyrimidine derivatives as kras g12c inhibitors for treating lung, pancreatic or colorectal cancer |
| JP7219218B2 (ja) | 2016-12-22 | 2023-02-07 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | 新規のベンジルアミノ置換キナゾリンおよびsos1阻害剤としての誘導体 |
| US10344026B2 (en) | 2017-01-18 | 2019-07-09 | Nantbio, Inc. | Compositions and methods of targeting mutant K-ras |
| EP3573964A1 (en) | 2017-01-26 | 2019-12-04 | Araxes Pharma LLC | Benzothiophene and benzothiazole compounds and methods of use thereof |
| US11279689B2 (en) | 2017-01-26 | 2022-03-22 | Araxes Pharma Llc | 1-(3-(6-(3-hydroxynaphthalen-1-yl)benzofuran-2-yl)azetidin-1 yl)prop-2-en-1-one derivatives and similar compounds as KRAS G12C modulators for treating cancer |
| WO2018140514A1 (en) | 2017-01-26 | 2018-08-02 | Araxes Pharma Llc | 1-(6-(3-hydroxynaphthalen-1-yl)quinazolin-2-yl)azetidin-1-yl)prop-2-en-1-one derivatives and similar compounds as kras g12c inhibitors for the treatment of cancer |
| CN110382482A (zh) * | 2017-01-26 | 2019-10-25 | 亚瑞克西斯制药公司 | 稠合的杂-杂二环化合物及其使用方法 |
| JP2020505395A (ja) | 2017-01-26 | 2020-02-20 | アラクセス ファーマ エルエルシー | 縮合n−複素環式化合物およびその使用方法 |
| WO2018140512A1 (en) | 2017-01-26 | 2018-08-02 | Araxes Pharma Llc | Fused bicyclic benzoheteroaromatic compounds and methods of use thereof |
| JOP20190186A1 (ar) | 2017-02-02 | 2019-08-01 | Astellas Pharma Inc | مركب كينازولين |
| AU2018255413A1 (en) | 2017-04-20 | 2019-12-05 | Leidos Biomedical Research, Inc. | K-Ras modulators |
| WO2019051291A1 (en) | 2017-09-08 | 2019-03-14 | Amgen Inc. | KRAS G12C INHIBITORS AND METHODS OF USE |
| MX2020005063A (es) * | 2017-11-15 | 2021-01-08 | Mirati Therapeutics Inc | Inhibidores de kras g12c. |
| TW201938561A (zh) | 2017-12-08 | 2019-10-01 | 瑞典商阿斯特捷利康公司 | 化學化合物 |
| TW201942115A (zh) | 2018-02-01 | 2019-11-01 | 美商輝瑞股份有限公司 | 作為抗癌藥之經取代的喹唑啉和吡啶並嘧啶衍生物 |
| TW201942116A (zh) | 2018-02-09 | 2019-11-01 | 美商輝瑞股份有限公司 | 作為抗癌劑之四氫喹唑啉衍生物 |
-
2018
- 2018-11-14 MX MX2020005063A patent/MX2020005063A/es unknown
- 2018-11-14 CN CN202410549759.9A patent/CN118459460A/zh active Pending
- 2018-11-14 EP EP18879484.6A patent/EP3710439B1/en active Active
- 2018-11-14 SI SI201830912T patent/SI3710439T1/sl unknown
- 2018-11-14 HR HRP20230377TT patent/HRP20230377T1/hr unknown
- 2018-11-14 HU HUE18879484A patent/HUE061599T2/hu unknown
- 2018-11-14 CA CA3082579A patent/CA3082579A1/en active Pending
- 2018-11-14 BR BR112020009818-3A patent/BR112020009818A2/pt unknown
- 2018-11-14 LT LTEPPCT/US2018/061060T patent/LT3710439T/lt unknown
- 2018-11-14 DK DK18879484.6T patent/DK3710439T3/da active
- 2018-11-14 RS RS20230345A patent/RS64182B1/sr unknown
- 2018-11-14 AU AU2018369759A patent/AU2018369759B2/en active Active
- 2018-11-14 FI FIEP18879484.6T patent/FI3710439T3/fi active
- 2018-11-14 EA EA202091186A patent/EA202091186A1/ru unknown
- 2018-11-14 ES ES18879484T patent/ES2944547T3/es active Active
- 2018-11-14 US US16/191,190 patent/US10689377B2/en active Active
- 2018-11-14 SG SG11202004427TA patent/SG11202004427TA/en unknown
- 2018-11-14 CN CN201880086849.1A patent/CN111989321B/zh active Active
- 2018-11-14 JP JP2020526612A patent/JP7322019B2/ja active Active
- 2018-11-14 PT PT188794846T patent/PT3710439T/pt unknown
- 2018-11-14 WO PCT/US2018/061060 patent/WO2019099524A1/en unknown
- 2018-11-14 IL IL274601A patent/IL274601B2/en unknown
- 2018-11-14 UA UAA202003509A patent/UA125802C2/uk unknown
- 2018-11-14 PL PL18879484.6T patent/PL3710439T3/pl unknown
- 2018-11-14 KR KR1020207016494A patent/KR20200100632A/ko not_active Application Discontinuation
- 2018-11-15 TW TW107140665A patent/TWI809005B/zh active
-
2019
- 2019-05-14 JP JP2021526287A patent/JP7346565B2/ja active Active
- 2019-05-14 EP EP19886028.0A patent/EP3880208A4/en active Pending
- 2019-05-14 WO PCT/US2019/032249 patent/WO2020101736A1/en unknown
-
2020
- 2020-05-04 ZA ZA2020/02105A patent/ZA202002105B/en unknown
- 2020-05-06 US US16/868,258 patent/US20200262837A1/en not_active Abandoned
- 2020-05-13 PH PH12020550622A patent/PH12020550622A1/en unknown
- 2020-05-13 CL CL2020001271A patent/CL2020001271A1/es unknown
- 2020-05-15 SA SA520411982A patent/SA520411982B1/ar unknown
- 2020-06-12 CO CONC2020/0007244A patent/CO2020007244A2/es unknown
-
2022
- 2022-10-21 US US18/048,834 patent/US20230373999A1/en active Pending
-
2023
- 2023-08-04 US US18/230,580 patent/US20240101553A1/en active Pending
-
2024
- 2024-06-10 FI FIC20240021C patent/FIC20240021I1/fi unknown
- 2024-06-10 NL NL301279C patent/NL301279I2/nl unknown
- 2024-06-11 HU HUS2400019C patent/HUS2400019I1/hu unknown
- 2024-06-13 FR FR24C1026C patent/FR24C1026I1/fr active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2944547T3 (es) | Inhibidores de KRas G12C | |
| US10125134B2 (en) | KRas G12C inhibitors | |
| WO2020146613A1 (en) | Kras g12c inhibitors | |
| AU2019262927A1 (en) | Pyridazinones as PARP7 inhibitors | |
| AU2023216698A1 (en) | Quinazoline pan-kras inhibitors | |
| CN118660880A (zh) | 喹唑啉泛KRas抑制剂 |