Spirocyclic Dihydropyranopyrimidine KRas Inhibitors CROSS-REFERENCE TO RELATED APPLICATIONS This application claims priority to U.S. Provisional Application Serial Nos.63/426,950, filed November 21, 2022; 63/456,235, filed March 31, 2023; 63/515,290, filed July 24, 2023; 63/533,346, filed August 17, 2023; 63/535,006, filed August 28, 2023; 63/542,188, filed October 3, 2023; and 63/545,531, filed October 24, 2023, each of which is incorporated by reference in its entirely herein. DESCRIPTION OF THE TEXT FILE SUBMITTED ELECTRONICALLY This application contains a Sequence Listing which has been submitted electronically in XML format. The Sequence Listing XML is incorporated herein by reference. The XML file, created on November 3, 2023, is named TRLN-008-007WO1_ST26_SL.xml and is 2,080 bytes in size. TECHNICAL FIELD This disclosure provides compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, that inhibit a KRas GTPase (e.g., a KRas GTPase that has a dysregulation (referred to herein as a dysregulated KRas protein)). In some embodiments, the KRas protein is a dysregulated KRas protein that has a mutation (referred to herein as a mutant KRas protein). These compounds are useful, for example, for treating a disease, disorder, or condition in which increased and/or sustained (e.g., excessive) KRas activation, such as KRas activation associated with a mutant KRas protein, contributes to the pathology and/or symptoms and/or progression of the disease, disorder, or condition (e.g., cancer) in a subject (e.g., a human). This disclosure also provides compositions containing compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b),
(II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, as well as methods of using and making the same. BACKGROUND The KRAS gene is frequently dysregulated (e.g., mutated or amplified) in various human cancers. Oncogenic mutations in KRas typically occur at hotspots in the protein such as at amino acids positions 12, 13, and 61. In some cases, a mutation can lead to maintenance of KRas activation (GTP-bound state), e.g., due to a deficiency of intrinsic GTPase activity and/or insensitivity for GTPase-activating proteins (GAPs) and consequent increased KRas signaling. Specifically, some of the most common protein mutations include those at position 12 (referred to herein as G12X) such as G12A, G12C, G12D, G12R, G12S, and G12V; position 13 (referred to herein as G13X) such as G13C, G13D, and G13V; and Q61 (referred to herein as Q61X), such as Q61E, Q61H, Q61K, Q61L, Q61P, and Q61R. KRas is widely recognized as a target for the design and development of therapies that can specifically bind and inhibit KRas signaling in cancer cells but had long been considered to be undruggable. Currently, there are few approved KRas-targeted therapies. In some embodiments, the KRas protein is a dysregulated KRas protein that has a mutation (referred to herein as a mutant KRas protein). These compounds are useful, for example, for treating a disease, disorder, or condition in which increased and/or sustained (e.g., excessive) KRas activation, such as KRas activation associated with a mutant KRas protein, contributes to the pathology and/or symptoms and/or progression of the disease, disorder, or condition (e.g., cancer) in a subject (e.g., a human). This disclosure also provides compositions containing the same as well as methods of using and making the same. SUMMARY This disclosure provides compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d),
(V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, that inhibit a KRas protein (e.g., a dysregulated KRas protein, such as a mutant KRas protein). These compounds are useful, for example, for treating a disease, disorder, or condition in which increased KRas activation, such as KRas activation associated with a mutant KRas protein or KRas activation associated with KRas amplification, contributes to the pathology and/or symptoms and/or progression of the disease, disorder, or condition (e.g., cancer) in a subject (e.g., a human). This disclosure also provides compositions containing compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, as well as methods of using and making the same. Provided herein are compounds of Formula (A):
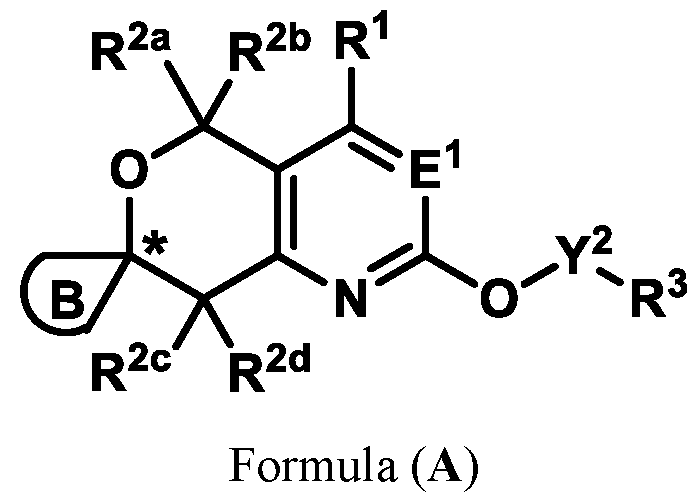
or pharmaceutically acceptable salts thereof, wherein: E
1 is selected from the group consisting of N, CH, and CR
4, wherein R
4 is selected from the group consisting of: CN, halo, C1-3 alkyl, C1-3 haloalkyl, and C3-6 cycloalkyl; R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)
2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and
(iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; R
2a and R
2b are independently selected from the group consisting of: H, C1-3 alkyl, C1-
3 haloalkyl, and C
3-6 cycloalkyl; or R
2a and R
2b taken together with the ring carbon atom to which each is attached form a C3-6 cycloalkyl ring or a 4-6 membered heterocyclyl ring; R
2c and R
2d are independently selected from the group consisting of: H, halo, CN, C
1-3 alkyl, C1-3 haloalkyl, and C3-6 cycloalkyl; or R
2c and R
2d taken together with the ring carbon atom to which each is attached form a C
3-6 cycloalkyl ring or a 4-6 membered heterocyclyl ring; Ring B is selected from the group consisting of:
, ,
, wherein: the * marks the ring carbon atom common to both Ring
X
1 is selected from the group consisting of a bond, S(O)
0-2, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2;
b1 is 0, 1, or 2; R
9 is selected from the group consisting of: H, OH, NR
dR
e, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C
1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; or a pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C
1-6 alkoxy, C
1-6 haloalkoxy, C
1-6 alkyl, and C
1-6 haloalkyl, or a pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C
1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
(m) C(=O)N(R
f)
2; (n) S(O)0-2(C1-6 alkyl); (o) S(O)0-2(C1-6 haloalkyl); (p) S(O)
1-2N(R
f)
2; and (q) C1-6 alkyl, C2-6 alkenyl, or C2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)
0-2-, C(=O), and C
1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)
2, S(O)
0-2(C
1-6 alkyl), S(O)
0-2(C
1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)N(R
f)
2, S(O)
1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, C1-3 haloalkyl, C
3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-.
Also provided herein are compounds of Formula (I):
Formula (I) or pharmaceutically acceptable salts thereof, wherein: R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)
2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; Ring B is selected from the group consisting of:
, ,
, wherein: the * marks the ring carbon atom common to both Ring
X
1 is selected from the group consisting of a bond, S(O)0-2, CH2, CHR
L, C(R
L)2, and O;
X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; b1 is 0, 1, or 2; R
9 is selected from the group consisting of: H, OH, NH2, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; or a pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-6 cycloalkyl ring; Y
2 is a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or a pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)
2, S(O)
0-2(C
1-6 alkyl), S(O)
0-2(C
1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)N(R
f)
2, S(O)
1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, and C1-
3 haloalkyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-. Also provided herein are compounds of Formula (II):
Formula (II) or pharmaceutically acceptable salts thereof, wherein: R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; X
1 is selected from the group consisting of a bond, S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; b1 is 1 or 2; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo,
C
1-6 alkoxy, C
1-6 haloalkoxy, C
1-6 alkyl, and C
1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C
1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
each R
b is independently selected from the group consisting of: -(L
b)
b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3
alkyl)-, -S(O)
0-2-, C(=O), and C
1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)N(R
f)
2, S(O)
1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, C1-3 haloalkyl, C
3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-. Also provided herein are compounds of Formula (III):
Formula (III) or pharmaceutically acceptable salts thereof, wherein: R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein 30 the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms;
and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; X
1 is selected from the group consisting of a bond, S(O)
0-2, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; R
9 is selected from the group consisting of: H, NR
dR
e, -OH, and halo; b4 is 0 or 1; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C
1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of: (a) halo;
each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with
1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, C
1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. Also provided herein are compounds of Formula (IV):
Formula (IV) or pharmaceutically acceptable salts thereof, wherein: X
1 is selected from the group consisting of a bond, S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that at least one of X
1, X
2, and X
3 is CHR
L or C(R
L)2; further provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)
0-2; b1 is 0, 1 or 2; R
9 is selected from the group consisting of: H, OH, NR
dR
e, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C
1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)
2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms;
30 and
(iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; Y
2 is a bond or a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
(o) S(O)
0-2(C
1-6 haloalkyl); (p) S(O)1-2N(R
f)2; and (q) C1-6 alkyl, C2-6 alkenyl, or C2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C
1-6 alkyl), S(O)
1-2(C
1-6 haloalkyl), S(O)
1-2N(R
f)
2, and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, C
1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. Also provided herein are compounds of Formula (V): 30
Formula (V) or pharmaceutically acceptable salts thereof, wherein: X
1 is selected from the group consisting of a bond, S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that 2-3 of X
1, X
2, and X
3 are independently CHR
L or C(R
L)2; one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; and each additional R
L is independently selected from the group consisting of: C1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; b1 is 0, 1 or 2; R
9 is selected from the group consisting of: H, OH, NR
dR
e, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; Y
2 is a bond or a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl;
R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g;
each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)
2, S(O)
0-2(C
1-6 alkyl), S(O)
0-2(C
1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)N(R
f)
2, S(O)
1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, C1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-. Also provided herein are compounds of Formula (VI):
Formula (VI) or pharmaceutically acceptable salts thereof, wherein: R
1 is a 4-10 membered heterocyclyl substituted with -CN, –(C1-3 alkylene)-CN, or – (C3-6 cycloalkylene)-CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7; wherein each R
7 is independently selected from the group consisting of R
a and R
b; X
1 is selected from the group consisting of S(O)
0-2, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; 30 b1 is 0, 1, or 2;
R
9 is selected from the group consisting of: H, NR
dR
e, -OH, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C
1-6 alkoxy, C
1-6 haloalkoxy, C
1-6 alkyl, and C
1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
(n) S(O)
0-2(C
1-6 alkyl); (o) S(O)0-2(C1-6 haloalkyl); (p) S(O)1-2N(R
f)2; and (q) C
1-6 alkyl, C
2-6 alkenyl, or C
2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)
0-2-, C(=O), and C
1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)
2, S(O)
0-2(C
1-6 alkyl), S(O)
0-2(C
1-6 haloalkyl), and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)N(R
f)
2, S(O)
1- 2(C
1-6 alkyl), S(O)
1-2(C
1-6 haloalkyl), S(O)
1-2N(R
f)
2, and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, C1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. Also provided herein are pharmaceutical compositions comprising a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g.,
Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier. Provided herein are methods for treating cancer in a subject in need thereof, the methods comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as provided herein. Also provided herein are methods for treating cancer in a subject in need thereof, the methods comprising (a) determining that the cancer has a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation)); and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as provided herein. Provided herein are methods of treating a KRas-associated disease or disorder (e.g., a mutant KRas-associated disease or disorder (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer)) in a subject, the methods comprising administering to a subject identified or diagnosed as having a KRas-associated disease or disorder a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-
b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as provided herein. This disclosure also provides methods of treating a KRas-associated disease or disorder (e.g., a mutant KRas-associated disease or disorder (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer)) in a subject, the methods comprising: determining that the disease or disorder in the subject is a KRas-associated disease or disorder (e.g., a mutant KRas-associated disease or disorder (e.g., a KRas G12D-associated disease or disorder, a KRas G12R-associated disease or disorder, or a KRas G12V-associated disease or disorder)); and administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as provided herein. Further provided herein are methods of treating a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R- associated cancer, or a KRas G12V-associated cancer)) in a subject, the methods comprising administering to a subject identified or diagnosed as having a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R- associated cancer, or a KRas G12V-associated cancer)) a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as provided herein. This disclosure also provides methods of treating a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R- associated cancer, or a KRas G12V-associated cancer)) in a subject, the methods comprising:
determining that the cancer in the subject has a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation)); and administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as provided herein. To facilitate understanding of the disclosure set forth herein, a number of terms are provided. Generally, the nomenclature used herein and the laboratory procedures in organic chemistry, medicinal chemistry, and pharmacology described herein are those well-known and commonly employed in the art. Unless defined otherwise, all technical and scientific terms used herein generally have the same meaning as commonly understood by one of ordinary skill in the art to which this disclosure belongs. Each of the patents, applications, published applications, and other publications that are mentioned throughout the specification and the attached appendices are incorporated herein by reference in their entireties. In the case of conflict between the present disclosure and any content incorporated by reference, the present disclosure controls. The details of one or more embodiments of the invention are set forth in the accompanying drawings and the description below. Other features and advantages of the invention will be apparent from the description and drawings, and from the claims. DETAILED DESCRIPTION This disclosure provides compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, that inhibit a KRas protein (e.g., a dysregulated KRas protein, such as a mutant KRas protein). These compounds are useful, e.g., for treating a disease, disorder, or condition associated with a KRas dysregulation (e.g., a KRas
mutation or amplification) in which increased and/or sustained (e.g., excessive) KRas activation contributes to the pathology and/or symptoms and/or progression of the disease, disorder, or condition (e.g., cancer) in a subject (e.g., a human). These compounds can also be useful, e.g., for treating a disease, disorder, or condition in which a mutant KRas protein (e.g., a resistance mutation) confers intrinsic resistance to one or more KRas inhibitors (e.g., a KRas inhibitor selective for a KRas G12C mutant protein), or to a non-KRas-targeted therapeutic agent. See, e.g., Misale, et al., Nature 486.7404 (2012): 532-536, doi: 10.1038/nature11156 and Awad, et al., New England Journal of Medicine 384.25 (2021): 2382-2393, doi: 10.1056/NEJMoa2105281. This disclosure also provides compositions containing the compounds provided herein as well as methods of using and making the same. Ras family genes (e.g., KRAS, NRAS, and HRAS) were the first oncogenes identified and are some of the most commonly mutated of all discovered oncogenes. See, e.g., Hunter et al. Mol Cancer Res. 2015;13(9):1325-35, doi: 10.1158/1541-7786.MCR-15-0203. The Ras family are guanine nucleotide binding proteins generally found at the inner leaflet of the cell membrane. A wild type Ras protein becomes activated when bound to GTP, but it is inactive when bound to GDP. Normally, growth factors bind to extracellular receptors to induce nucleotide exchange with the help of guanine nucleotide exchange factors (GEF) (e.g., Son of sevenless homolog 1 (SOS1)). These GEFs allow GDP to dissociate from a Ras protein and GTP to bind. Ras proteins can interact with effector proteins such as cRAF when bound to GTP. Hydrolysis of GTP to form GDP can deactivate Ras proteins, and the hydrolysis can be achieved through the intrinsic GTPase activity, which may be enhanced by binding to a GTPase activating protein (GAP). There are 3 major Ras proteins in humans: KRas, HRas, and NRas. Some oncogenic KRas missense mutations can prevent or slow GTP hydrolysis and result in the accumulation of KRas in the active state. Signaling pathways associated with KRas are persistently activated in many cancers, where they participate in cellular growth and proliferation, differentiation, protein synthesis, glucose metabolism, cell survival, and inflammation. Mutant KRas proteins often have altered Raf affinity and/or altered intrinsic GTPase activity. See, for example, Table 1 reproduced from Hunter et al. Mol Cancer Res. 2015;13(9):1325-35, doi: 10.1158/1541-7786.MCR-15-0203. These changes and other factors can contribute to increased KRas signaling in mutant KRas proteins. Table 1


KRas inhibitors are described in, for example, International Publication Nos. WO 2023/154766; WO 2023/143623; WO 2022/240971; WO 2020/236940; WO 2022/115439; WO 2023/086383; WO 2021/093758; WO 2022/135546; WO 2021/139748; WO 2022/251576; and WO 2023/025116. Additional examples of KRas inhibitors are described in, for example, International Publication Nos. WO 2022/132200; WO 2022/133038; WO 2023/150284; WO 2022/261154; WO 2023/183585; WO 2023/099592; WO 2023/099623; WO 2023/099624; WO 2023/099608; WO 2022/250170; WO 2022/173870; WO 2022/236578; WO 2022/237649; WO 2022/248885; WO 2022/256459; WO 2022/258974; WO 2022/266015; WO 2023/018809; WO 2023/018810; WO 2023/018812; WO 2023/020518; WO 2023/020519; WO 2023/020521; WO 2023/020523; WO 2023/046135; WO 2023/061294; WO 2023/097227; WO 2023/114733; WO 2023/137223; WO 2023/141300; WO 2023/138583; WO 2023/159086; WO 2023/159087; WO 2023/173016; WO 2023/173017; WO 2023/179703; WO 2023/125627; WO 2022/216762; and CN 116143806. Compound Embodiments Provided herein are compounds of Formula (A):
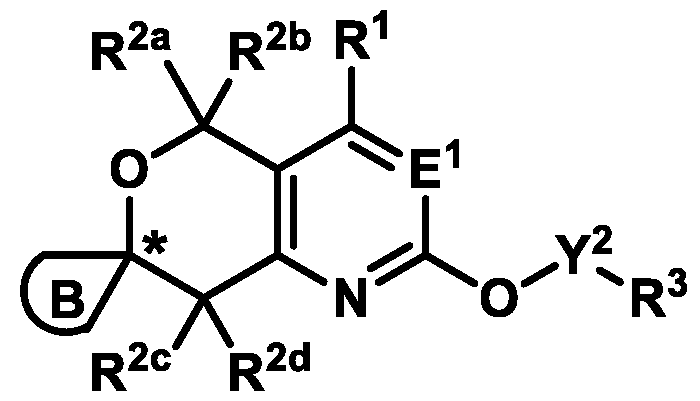
Formula (A) or pharmaceutically acceptable salts thereof, wherein: E
1 is selected from the group consisting of N, CH, and CR
4, wherein R
4 is selected from the group consisting of: CN, halo, C1-3 alkyl, C1-3 haloalkyl, and C3-6 cycloalkyl;
R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; R
2a and R
2b are independently selected from the group consisting of: H, C
1-3 alkyl, C
1- 3 haloalkyl, and C
3-6 cycloalkyl; or R
2a and R
2b taken together with the ring carbon atom to which each is attached form a C
3-6 cycloalkyl ring or a 4-6 membered heterocyclyl ring; R
2c and R
2d are independently selected from the group consisting of: H, halo, CN, C
1-3 alkyl, C1-3 haloalkyl, and C3-6 cycloalkyl; or R
2c and R
2d taken together with the ring carbon atom to which each is attached form a C
3-6 cycloalkyl ring or a 4-6 membered heterocyclyl ring; Ring B is selected from the group consisting of:
, ,
, wherein:
the * marks the ring carbon atom common to both Ring
X
1 is selected from the group consisting of a bond, S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; b1 is 0, 1, or 2; R
9 is selected from the group consisting of: H, OH, NR
dR
e, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; or a pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C
1-6 alkoxy, C
1-6 haloalkoxy, C
1-6 alkyl, and C
1-6 haloalkyl, or a pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C
1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
(e) -C
1-6 alkoxy; (f) -C1-6 haloalkoxy; (g) -NR
dR
e; (h) C(=O)C
1-6 alkyl; (i) C(=O)C1-6 haloalkyl; (j) C(=O)OH; (k) C(=O)OC
1-6 alkyl; (l) C(=O)OC1-6 haloalkyl; (m) C(=O)N(R
f)2; (n) S(O)
0-2(C
1-6 alkyl); (o) S(O)
0-2(C
1-6 haloalkyl); (p) S(O)1-2N(R
f)2; and (q) C1-6 alkyl, C2-6 alkenyl, or C2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h;
each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, C
1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. In some embodiments, the compounds of Formula (A) are compounds of Formula (I):
Formula (I) or pharmaceutically acceptable salts thereof. In some embodiments of Formula (I), R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)
2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and
selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; Ring B is selected from the group consisting of:
the * marks the ring carbon atom common to both Ring
X
1 is selected from the group consisting of a bond, S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)
0-2; b1 is 0, 1, or 2; R
9 is selected from the group consisting of: H, OH, NH2, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; or a pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-6 cycloalkyl ring; Y
2 is a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or a pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of: (a) halo; (b) cyano; (c) -OH; (d) oxo; (e) -C
1-6 alkoxy; (f) -C1-6 haloalkoxy;
(g) -NR
dR
e; (h) C(=O)C1-6 alkyl; (i) C(=O)C1-6 haloalkyl; (j) C(=O)OH; (k) C(=O)OC1-6 alkyl; (l) C(=O)OC1-6 haloalkyl; (m)C(=O)N(R
f)
2; (n) S(O)0-2(C1-6 alkyl); (o) S(O)0-2(C1-6 haloalkyl); (p) S(O)
1-2N(R
f)
2; and (q) C
1-6 alkyl, C
2-6 alkenyl, or C
2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)
0-2-, C(=O), and C
1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1-
2(C
1-6 alkyl), S(O)
1-2(C
1-6 haloalkyl), S(O)
1-2N(R
f)
2, and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, and C
1- 3 haloalkyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. In some embodiments of Formula (I), R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl substituted with -OH, -(C
1-3 alkylene)-OH, -CN, or – (C1-3 alkylene)-CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and
wherein b2 is 0, 1, or 2, and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; Ring B is selected from the group consisting of:
, wherein: the * marks the ring carbon atom common to both Ring
X
1 is selected from the group consisting of a bond, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, and O, provided that no more than one of X
1, X
2, and X
3 is O; b1 is 0, 1, or 2; R
9 is selected from the group consisting of: H, OH, NH2, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c;
Y
2 is a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or a pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
y y y y y each R
b is independently selected from the group consisting of: -(L
b)
b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3
alkyl)-, -S(O)
0-2-, C(=O), and C
1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)
2, S(O)
0-2(C
1-6 alkyl), S(O)
0-2(C
1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)N(R
f)
2, S(O)
1- 2(C
1-6 alkyl), S(O)
1-2(C
1-6 haloalkyl), S(O)
1-2N(R
f)
2, and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, and C1- 3 haloalkyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. In some embodiments of Formula (A) (e.g., Formula (I)), it is provided that when X
1 is a bond; and b1 is 0, then: (1) R
9 is OH or NR
dR
e (e.g., OH or NH
2); or (2) Y
2 is a straight- chain C1-6 alkylene optionally substituted with one R
Y. In some embodiments of Formula (A) (e.g., Formula (I)), it is provided that one or more of (1)-(4) applies: (1) R
9 is OH or NR
dR
e; (2) b1 is 1 or 2; (3) X
1 is CH
2; and/or (4) Y
2 is a straight-chain C
1-6 alkylene optionally substituted with one R
Y. In some embodiments of Formula (A) (e.g., Formula (I)), R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl substituted with -OH, -(C1-3 alkylene)-OH, -CN, or – (C1-3 alkylene)-CN on a ring carbon atom, wherein the heterocyclyl is further optionally
substituted with 1-3 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and
wherein b2 is 0, 1, or 2, and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; and Ring B is selected from the group consisting of:
, wherein: X
1 is selected from the group consisting of a bond, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)2, and O, provided that no more than one of X
1, X
2, and X
3 is O; b1 is 0, 1, or 2; R
9 is selected from the group consisting of: H, OH, NH
2, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c. For avoidance of doubt, the * in each alternative of Ring B represents the * in Formula (A) (e.g., in Formula (I)). In some embodiments, the ring carbon atom labelled with * in Formula (A) (e.g., Formula (I)) has (S)-stereochemistry. In some embodiments, the ring carbon atom labelled with * in Formula (A) (e.g., Formula (I)) has (R)-stereochemistry.
In some embodiments of Formula (A) (e.g., Formula (I)), Ring
. In some embodiments of Formula (A) (e.g., Formula (I)), Ring
. In some embodiments of Formula (A) (e.g., Formula (I)), X
1 is a bond. In some embodiments of Formula (A) (e.g., Formula (I)), X
1 is selected from the group consisting of: CH
2, CHR
L, and C(R
L)
2. For example, X
1 can be CH
2. In some embodiments of Formula (A) (e.g., Formula (I)), X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, and C(R
L)
2. For example, X
2 and X
3 can both be CH
2. In some embodiments of Formula (A) (e.g., Formula (I)), CH2; and X
3 is selected from the group consisting of: CHR
L and C(R
L)2. In some embodiments of Formula (A) (e.g., Formula (I)), X
2 is CH
2; and X
3 is CHR
L. For example, X
2 can be CH2; and X
3 can be CHMe. In some embodiments of Formula (A) (e.g., Formula (I)), one of X
2 and X
3 is -O-; and the other of X
2 and X
3 is selected from the group consisting of: CH
2, CHR
L, and C(R
L)
2. For example, X
2 can be -O-; and X
3 can be CH2 or CHMe. In some embodiments of Formula (A) (e.g., Formula (I)), X
1 is CHR
L; X
2 is CH2; and X
3 is CHR
L, wherein the pair of R
L taken together with the ring atoms connecting them form a C
3-6 cycloalkyl ring (e.g., C
4 cycloalkyl ring). In some embodiments of Formula (A) (e.g., Formula (I)), Ring
X
1 is CH
2; and X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, and C(R
L)2. In some embodiments, X
2 and X
3 are both CH2. In some embodiments, X
2 is CH2; 25 and X
3 is CHR
L (e.g., CHMe).
In some embodiments of Formula (A) (e.g., Formula (I)), Ring
X
1 is CH2; one of X
2 and X
3 is -O-; and the other of X
2 and X
3 is selected from the group consisting of: CH
2, CHR
L, and C(R
L)
2. In some embodiments, X
2 is -O-; and X
3 is CH
2 or CHMe. In some embodiments of Formula (A) (e.g., Formula (I)), each R
L is independently C
1- 3 alkyl optionally substituted with 1-3 R
c. In some embodiments of Formula (A) (e.g., Formula (I)), Ring
X
1 is CHR
L; X
2 is CH2; and X
3 is CHR
L, wherein the pair of R
L taken together with the ring atoms connecting them form a C3-6 cycloalkyl ring (e.g., C4 cycloalkyl ring). For example, Ring B can be:
. In some embodiments of Formula (A) (e.g., Formula (I)), R
9 is para to -X
3-. In some embodiments, the compounds of Formula (I) are compounds of Formula (I- a1):
Formula (I-a1) or pharmaceutically acceptable salts thereof, wherein: X
1 is a bond or CH2;
X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, and C(R
L)2; and b1 is 0, 1, or 2 (e.g., 0 or 1). In some embodiments, the compounds of Formula (I) are compounds of Formula (I- b1):
or pharmaceutically acceptable salts thereof, wherein: X
1 is a bond or CH2; X
2 and X
3 are independently selected from the group consisting of: -O-, CH
2, CHR
L, and C(R
L)
2; and b1 is 0, 1, or 2. In some embodiments, the compounds of Formula (I) are compounds of Formula (I- b1):
Formula (I-c1) or pharmaceutically acceptable salts thereof, wherein: X
1 is a bond or CH
2; X
2 and X
3 are independently selected from the group consisting of: -O-, CH2, CHR
L, and C(R
L)2; and b1 is 0, 1, or 2. In some embodiments of Formula (I-a1), (I-b1), or (I-c1), X
1 is a bond. In some embodiments of Formula (I-a1), (I-b1), or (I-c1), X
1 is CH
2.
In some embodiments of Formula (I-a1), (I-b1), or (I-c1), X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2. In some embodiments of Formula (I-a1), (I-b1), or (I-c1), X
2 and X
3 are both CH2. In some embodiments of Formula (I-a1), (I-b1), or (I-c1), X
2 is CH
2; and X
3 is selected from the group consisting of: CHR
L and C(R
L)2. For example, X
2 can be CH2; and X
3 can be CHMe. In some embodiments of Formula (I-a1), (I-b1), or (I-c1), X
1 is CH
2; and X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2. In some embodiments, X
2 and X
3 are both CH2. In some embodiments, X
2 is CH2; and X
3 is selected from the group consisting of: CHR
L and C(R
L)
2. In some embodiments of Formula (I-a1), (I-b1), or (I-c1), X
1 is CH
2; one of X
2 and X
3 is -O-; and the other of X
2 and X
3 is selected from the group consisting of: CH2, CHR
L, and C(R
L)2. In some embodiments, X
2 is -O-; and X
3 is CH2 or CHMe. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), R
9 is selected from the group consisting of -OH, -NH2, and halo. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), R
9 is selected from the group consisting of -OH and -NR
dR
e. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), R
9 is selected from the group consisting of -OH and -NH
2. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), R
9 is -OH. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), R
9 is -NH
2. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), R
9 is halo (e.g., -Br). In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), R
9 is H. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), b1 is 0 or 1. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), b1 is 1 or 2. For example, b1 can be 1. For example, b1 can be 2.
In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), b1 is 1 or 2; and each R
10 is independently selected from the group consisting of: - Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), b1 is 1; and R
10 is -CN. In some embodiments, b1 is 1; R
10 is ortho to R
9; and R
10 is -CN. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), b1 is 1 or 2; and each R
10 is independently -Cl or -F. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), b1 is 1 or 2; 1-2 occurrence(s) of R
10 is ortho to R
9; and each R
10 is independently -Cl or -F. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), b1 is 0. In some embodiments of Formula (A) (e.g., Formula (I)), Ring B is selected from the
wherein: X
2 is -O- or -CH2-; X
3 is -CH
2- or -CHR
L-, wherein R
L is C
1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments of Formula (A) (e.g., Formula (I)), Ring B is selected from the group consisting of:
, wherein: X
2 is -O- or -CH2- ; X
3 is -CH
2- or -CHR
L-, wherein R
L is C
1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3
alkyl optionally substituted with 1-3 R
c. In some embodiments of Formula (A) (e.g., Formula (I)), Ring B is selected from the
In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl substituted with -OH, -(C
1-3 alkylene)-OH, -(C
3-6 cycloalkylene)-OH, -CN, –(C
1-3 alkylene)-CN, or –(C
3-6 cycloalkylene)-CN, on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)
2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, or 2, and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), R
1 is 4-10 membered heterocyclyl optionally substituted with 1-4 R
7. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), R
1 is a 4-10 membered heterocyclyl substituted with -OH, -(C1-3 alkylene)-OH, - CN, or –(C1-3 alkylene)-CN on a ring carbon atom, wherein the heterocyclyl is further
optionally substituted with 1-3 R
7. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), R
1 is a 4-10 (e.g., 6, 7, or 8) membered heterocyclyl substituted with -OH or - CH
2CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7, and wherein the heterocyclyl contains one ring nitrogen atom and 0-2 additional ring heteroatoms each independently selected from the group consisting of: O and S(O)
0-2. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))
further optionally substituted with 1-2 R
7 at one or more ring carbon atoms. For example, R
1 can
In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), R
1 is a 7-10 (e.g., 7) membered heterocyclyl optionally substituted with 1-4 R
7. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), R
1 is a 7-10 (e.g., 7) membered heterocyclyl having one ring nitrogen atom, one ring oxygen atom, and no additional ring heteroatoms, wherein the 7-10 membered heterocyclyl is optionally substituted with 1-4 R
7. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))
optionally substituted with 1-4 R
7 at one or more ring carbon atoms. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C1-3 alkyl optionally substituted with 1-3 R
c, wherein: each R
c is independently selected from the group consisting of: -F, -OH, and -CN. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1),
or (I-c1))), R
1 is an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms. For example, R
1 can
In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))
, wherein b2 is 0, 1, or 2, and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7. In some embodiments, A
2 is CH. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))
example, R
1 can
. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))
selected from the group consisting of: C(=O)N(R
f)
2, C(O)N(C
1-3 alkyl)R
b1, -C(O)N(H)R
b1, R
b1, and C(O)R
b1. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))
R
7 is selected from the group consisting of:
(a) C(=O)N(R
f)
2, wherein each R
f is independently H or C
1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is 4-10 membered heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), Y
2 is -CH2-. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), R
3 is a 4-10 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1), or (I-c1))), R
3 is a bicyclic 7-10 membered heterocyclyl optionally substituted with 1-6 R
a. In some embodiments of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-b1),
In some embodiments, the compounds of Formula (I) are compounds of Formula (I- a2) or (I-b2):
Formula (I-a2)
or pharmaceutically acceptable salts thereof, wherein: R
1 is selected from the group consisting of:
; X
1 is a bond or CH2; X
2 and X
3 are independently selected from the group consisting of: O, CH
2, CHR
L, and C(R
L)2; and b1 is 0, 1, or 2 (e.g., 0 or 1). In some embodiments, the compounds of Formula (I) are compounds of Formula (I- a3) or (I-b3):
or pharmaceutically acceptable salts thereof, wherein: R
1 is a 7-10 membered heterocyclyl having one ring nitrogen atom, one ring oxygen
atom, and no additional ring heteroatoms, wherein the 7-10 membered heterocyclyl is optionally substituted with 1-4 R
7; X
1 is CH2; X
2 and X
3 are independently selected from the group consisting of: O, CH
2, CHR
L, and C(R
L)2; and b1 is 0, 1, or 2. In some embodiments, the compounds of Formula (I) are compounds of Formula (I- a4) or (I-b4):
Formula (I-b4) or a pharmaceutically acceptable salt thereof, wherein: b3 is 0, 1, 2, or 3; X
1 is CH
2; X
2 and X
3 are independently selected from the group consisting of: O, CH
2, CHR
L, and C(R
L)2; and b1 is 0, 1, or 2. In some embodiments of Formula (I-a4) or (I-b4), b3 is 0. In some embodiments of Formula (I-a4) or (I-b4), b3 is 1 or 2; and each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C
1-3 alkyl optionally substituted with 1-3 R
c, wherein each R
c is independently selected from the group consisting
25 of: -F, -OH, and -CN.
In some embodiments, the compounds of Formula (I) are compounds of Formula (I- a5) or (I-b5):
Formula (I-b5) or a pharmaceutically acceptable salt thereof, wherein: R
7 is selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
1 is CH
2; X
2 and X
3 are independently selected from the group consisting of: O, CH
2, CHR
L, and C(R
L)2; and b1 is 0, 1, or 2. In some embodiments of Formula (I-a5) or (I-b5), R
7 is selected from the group consisting of: (a) C(=O)N(R
f)
2, wherein each R
f is independently H or C
1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is 4-10 membered heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom.
In some embodiments of Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I-b4), (I-a5), or (I-b5), X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)
2. In some embodiments of Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I-b4), (I-a5), or (I-b5), X
2 and X
3 are both CH2. In some embodiments of Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I-b4), (I-a5), or (I-b5), X
2 is CH2; and X
3 is CHR
L. For example, X
2 can be CH2; and X
3 can be CH(Me). In some embodiments of Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I-b4), (I-a5), or (I-b5), X
2 is -O-; and X
3 is selected from the group consisting of: CH
2, CHR
L, and C(R
L)
2. For example, X
2 can be -O-; and X
3 can be CH
2 or CH(Me). In some embodiments of Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I-b4), (I-a5), or (I-b5), R
9 is -NH
2. In some embodiments of Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I-b4), (I-a5), or (I-b5), R
9 is -OH. In some embodiments of Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I-b4), (I-a5), or (I-b5), b1 is 1. In some embodiments of Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I- b4), (I-a5), or (I-b5), b1 is 2. In some embodiments of Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I-b4), (I-a5), or (I-b5), b1 is 0. In some embodiments of Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I-b4), (I-a5), or (I-b5), each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments of Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I-b4), (I-a5), or (I-b5), b1 is 1 or 2; and each R
10 is independently selected from the group consisting of: - Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments of Formula (I-a2), (I-a3), (I-a4), or (I-a5), the
30
moiety is selected from the group consisting of:
,
, wherein: X
2 is -O- or -CH2-; X
3 is -CH
2- or -CHR
L-, wherein R
L is C
1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments of Formula (I-a2), (I-a3), (I-a4), or (I-a5), the
X
3 is -CH
2- or -CHR
L-, wherein R
L is C
1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments of Formula (I-a2), (I-a3), (I-a4), or (I-a5), the
In some embodiments of Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I-b4), (I-a5), or (I-b5), Y
2 is -CH
2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. In some embodiments of Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I-b4), (I-a5),
In some embodiments of Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I-b4), (I-a5), or (I-b5), the ring carbon atom labelled with * in Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I-b4), (I-a5), or (I-b5) has (S)-stereochemistry. In some embodiments of Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I-b4), (I-a5), or (I-b5), the ring carbon atom labelled with * in Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I-b4), (I-a5), or (I-b5) has (R)-stereochemistry. In some embodiments of Formula (I-a2), (I-b2), (I-a3), (I-b3), (I-a4), (I-b4), (I-a5),
In some embodiments, the compounds of Formula (A) (e.g., Formula (I)) are selected from the group consisting of compounds depicted in Table C1, or pharmaceutically acceptable salts thereof. Table C1
142a 143 143a 143b 144

Note 1: In some compounds as filed in U.S. Provisional Application Serial No. 63/515,290 (e.g., Compounds Nos. 143a, 143b, 149b, and 149c), a stereogenic center was previously denoted with a “*,” this stereogenic center was resolved, but its absolute configuration was not assigned. Herein, the compound structures have been redrawn with enhanced stereochemical notation. See Note 2. Note 2: In certain compounds of Table C1, one or more stereogenic centers are denoted with the “V3000 enhanced stereochemical notation” (see: support.collaborativedrug.com/hc/en- us/articles/360020872171-Advanced-Stereochemistry-Registration-Atropisomers-Mixtures- Unknowns-and-Non-Tetrahedral-Chirality, accessed on December 23, 2022 and Accelrys Chemical Representation Guide, Accelrys Software Inc., 2014, each of which is incorporated by reference herein in its entirety). Using this stereochemical notation, certain stereogenic centers are denoted with “abs”, “&x”, or “orx”, wherein x is an integer (e.g., 1 or 2). For avoidance of doubt, the stereochemical notations in Table C1 have the following meaning: When a structure does not contain any wedged or hashed bonds (i.e., each stereogenic center is undefined), then each stereogenic center can independently adopt a (R) or (S) stereochemical configuration. For avoidance of doubt, such structures also encompass mixtures of stereoisomers. For example,
p , , or a mixture
. When a structure contains a stereogenic center or a plurality of stereogenic centers that is depicted with wedges and hashes (i.e., one or more stereogenic center is defined), the following notations are used:
(1) When a defined stereogenic center is denoted with “abs” or when the defined stereogenic center is not denoted with an enhanced stereochemical notation (e.g., “abs”, “&x”, or “orx”), the defined stereogenic center has the absolute configuration as depicted by the
structural formula. For example, both of the structures and refer to (S)-(1-methylpyrrolidin-2-yl)methanol. (2) When a defined stereogenic center is denoted with “orx” in a structural formula, the defined stereogenic center has been resolved but the configuration at the defined
stereogenic center has not been determined. For example, the structure refers to one stereoisomer selected from the group consisting of (S)-(1-methylpyrrolidin-2-yl)methanol and (R)-(1-methylpyrrolidin-2-yl)methanol. (3) When a stereogenic center is undefined (i.e., no wedged or hashed bonds attached to the undefined stereogenic center) in a structural formula having at least one defined stereogenic center (i.e., having a wedged and/or hashed bond attached to the at least one defined stereogenic center), a mixture of stereoisomers differing at the undefined stereogenic center is
represented. For example, the structure represents a mixture of
(4) When two or more defined stereogenic centers are denoted with “orx” in a structural formula, each of these defined stereogenic centers has been resolved but the configurations at the defined stereogenic centers have not been determined. Specifically: a. For any pair of defined stereogenic centers denoted with “orx” in a structural formula, when the numerical parts in the notation are different (e.g., two defined stereogenic centers denoted with “or1” and “or2” respectively), each defined stereogenic center should be independently interpreted according to “(2)” supra.
For example, the structure
refers to one stereoisomer selected ,
b. For any pair of defined stereogenic centers denoted with “orx” in a structural formula, when the numerical part in the notation is identical (e.g., two defined stereogenic centers are each denoted with “or1”), the structural formula refers to one stereoisomer having the relative stereochemistry at these stereogenic centers as depicted in the structural formula, but the absolute configurations of these stereogenic centers have not been determined. For example, the structure
. another example, the structure
refers to one of the “anti” stereoisomers:
. (5) When two or more defined stereogenic centers are denoted with “&x” in a structural formula, the structural formula refers to a mixture of stereoisomers that differ in the configuration at the defined stereogenic centers. Specifically: a. For any pair of defined stereogenic centers denoted with “&x” in a structural formula, when the numerical parts in the notation are different (e.g., two defined stereogenic centers denoted with “&1” and “&2” respectively), the structural
formula refers to a mixture of stereoisomers at these two defined stereogenic centers, wherein the configuration at each of the defined stereogenic centers can vary independently of one another. For example, the structure
refers to a mixture of four stereoisomers:
,
b. For any pair of defined stereogenic centers denoted with “&x” in a structural formula, when the numerical part in the notation is identical (e.g., two defined stereogenic centers are each denoted with “&1”), the structural formula refers to a mixture of stereoisomers at these two defined stereogenic centers, wherein the relative configurations are as depicted in the structural formula. For example, the structure
refers to a mixture of “syn” stereoisomers:
. another example, the
.
In some embodiments, the compounds of Formula (A) (e.g., Formula (I)) are selected from the group consisting of compounds depicted in Table C1s of U.S. Provisional Application Serial Nos. 63/426,950, filed November 21, 2022; 63/456,235, filed March 31, 2023; 63/515,290, filed July 24, 2023; 63/533,346, filed August 17, 2023; 63/535,006, filed August 28, 2023; 63/542,188, filed October 3, 2023; and 63/545,531, filed October 24, 2023, or pharmaceutically acceptable salts thereof, each of the Table C1s is incorporated by reference in its entirety herein. In some embodiments, the compounds of Formula (A) (e.g., Formula (I)) are compounds of Formula (II):
Formula (II) or pharmaceutically acceptable salts thereof, wherein: R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; X
1 is selected from the group consisting of a bond, S(O)
0-2, CH
2, CHR
L, C(R
L)
2, and O;
X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; b1 is 1 or 2; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
(k) C(=O)OC
1-6 alkyl; (l) C(=O)OC1-6 haloalkyl; (m) C(=O)N(R
f)2; (n) S(O)
0-2(C
1-6 alkyl); (o) S(O)0-2(C1-6 haloalkyl); (p) S(O)1-2N(R
f)2; and (q) C
1-6 alkyl, C
2-6 alkenyl, or C
2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)
2, S(O)
0-2(C
1-6 alkyl), S(O)
0-2(C
1-6 haloalkyl), and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1-
2(C
1-6 alkyl), S(O)
1-2(C
1-6 haloalkyl), S(O)
1-2N(R
f)
2, and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, C
1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-.
In some embodiments, the compounds of Formula (II) are compounds of Formula (II-
Formula (II-a) or pharmaceutically acceptable salts thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments, the compounds of Formula (II) are compounds of Formula (II- b):
Formula (II-b) or pharmaceutically acceptable salts thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments, the compounds of Formula (A) (e.g., Formula (I)) are compounds of Formula (III):
Formula (III) or pharmaceutically acceptable salts thereof, wherein: R
1 is selected from the group consisting of:
(i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; X
1 is selected from the group consisting of a bond, S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; R
9 is selected from the group consisting of: H, NR
dR
e, -OH, and halo; b4 is 0 or 1; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and
(b) -NR
dR
e; each R
a is independently selected from the group consisting of:
each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl),
and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1-
2(C
1-6 alkyl), S(O)
1-2(C
1-6 haloalkyl), S(O)
1-2N(R
f)
2, and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, C1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. In some embodiments of Formula (III), R
9 is selected from the group consisting of: NR
dR
e, -OH, and halo. In some embodiments of Formula (III), R
9 is NR
dR
e. In some embodiments of Formula (III), R
9 is -NH2; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)) or Formula (III), X
1 is CH2 or CHR
L. In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)) or Formula (III), X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, and C(R
L)
2. In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)) or Formula (III), X
1 is CH2; and X
2 and X
3 are both CH2. In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)) or Formula (III), at least one of X
1, X
2, and X
3 is selected from the group consisting of: CHR
L and C(R
L)2. In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)) or Formula (III), one of X
1, X
2, and X
3 (e.g., X
3) is selected from the group consisting of: CHR
L and C(R
L)
2. In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)) or Formula (III), X
1 is CH2; and X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2, provided that 1-2 (e.g., one) of X
2 and X
3 is independently CHR
L or C(R
L)2. In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)) or Formula (III), X
1 is CH2; X
2 is CH2; and X
3 is CHR
L. In some embodiments, each R
L is independently selected from the group consisting of: CH3, CF3, CHF2, and CH2F.
In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)) or Formula (III), one of X
2 and X
3 is -O-; and the other of X
2 and X
3 is selected from the group consisting of: CH2, CHR
L, and C(R
L)2. In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)) or Formula (III), X
2 is -O-; and X
3 is selected from the group consisting of: CH2, CHR
L, and C(R
L)2. In some embodiments, each R
L is independently selected from the group consisting of: CH3, CF3, CHF2, and CH
2F. In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)) or Formula (III), each R
L is independently selected from the group consisting of: CH3, CF3, CHF2, and CH2F. In some embodiments of Formula (II), the
moiety is selected from
b4 is 0 or 1; X
2 is -O- or -CH2-; X
3 is -CH
2- or -CHR
L-, wherein R
L is C
1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c.
. In some embodiments of Formula (III), the
moiety is
, wherein: b4 is 0 or 1; X
2 is -O- or -CH2-; X
3 is -CH2- or -CHR
L-, wherein R
L is C1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. ,
In some embodiments, the compounds of Formula (A) (e.g., Formula (I)) are compounds of Formula (IV):
Formula (IV) or pharmaceutically acceptable salts thereof, wherein: X
1 is selected from the group consisting of a bond, S(O)
0-2, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that at least one of X
1, X
2, and X
3 is CHR
L or C(R
L)2; further provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; b1 is 0, 1 or 2; R
9 is selected from the group consisting of: H, OH, NR
dR
e, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; Y
2 is a bond or a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or
one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and
each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)
2, S(O)
0-2(C
1-6 alkyl), S(O)
0-2(C
1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)N(R
f)
2, S(O)
1- 2(C
1-6 alkyl), S(O)
1-2(C
1-6 haloalkyl), S(O)
1-2N(R
f)
2, and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, C1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. In some embodiments of Formula (IV), R
9 is selected from the group consisting of: OH, NR
dR
e, and halo. For example, R
9 can be NR
dR
e. In some embodiments, the compounds of Formula (IV) are compounds of Formula (IV-
Formula (IV-a) or pharmaceutically acceptable salts thereof, wherein: each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c.
In some embodiments, the compounds of Formula (IV) are compounds of Formula (IV- b):
Formula (IV-b) or pharmaceutically acceptable salts thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments of Formula (IV) (e.g., Formula (IV-a) or (IV-b)), X
1 is CH
2. In some embodiments of Formula (IV) (e.g., Formula (IV-a) or (IV-b)), X
2 is CH
2; and X
3 is CHR
L. In some embodiments of Formula (IV) (e.g., Formula (IV-a) or (IV-b)), X
2 is -O-; and X
3 is selected from the group consisting of: CHR
L and C(R
L)
2. In some embodiments of Formula (IV) (e.g., Formula (IV-a) or (IV-b)), each R
L is independently selected from the group consisting of: CH3, CF3, CHF2, and CH2F. In some embodiments, the compounds of Formula (IV) are compounds of Formula (IV- c):
Formula (IV-c) or pharmaceutically acceptable salts thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments, the compounds of Formula (A) (e.g., Formula (I)) are
compounds of Formula (V):
Formula (V) or pharmaceutically acceptable salts thereof, wherein: X
1 is selected from the group consisting of a bond, S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that 2-3 of X
1, X
2, and X
3 are independently CHR
L or C(R
L)
2; one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-6 cycloalkyl ring; and each additional R
L is independently selected from the group consisting of: C
1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; b1 is 0, 1 or 2; R
9 is selected from the group consisting of: H, OH, NR
dR
e, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)
2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b;
Y
2 is a bond or a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein:
b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)
2, S(O)
0-2(C
1-6 alkyl), S(O)
0-2(C
1-6 haloalkyl), and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1-
2(C
1-6 alkyl), S(O)
1-2(C
1-6 haloalkyl), S(O)
1-2N(R
f)
2, and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, C
1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. In some embodiments, the compounds of Formula (V) are compounds of Formula (V-
Formula (V-a) or pharmaceutically acceptable salts thereof, wherein: each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c.
In some embodiments, the compounds of Formula (V) are compounds of Formula (V- b):
Formula (V-b) or pharmaceutically acceptable salts thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments of Formula (V) (e.g., Formula (V-a) or (V-b)), X
2 is -O- or - CH
2- (e.g., -CH
2-). In some embodiments of Formula (V) (e.g., Formula (V-a) or (V-b)), X
1 is CHR
L; and X
3 is CHR
L, wherein the pair of R
L on different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-4 cycloalkyl ring. In some embodiments of Formula (V) (e.g., Formula (V-a) or (V-b)), X
1 is CHR
L; and X
3 is C(R
L)2, wherein the pair of R
L on different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-4 cycloalkyl ring, and the remaining R
L is C
1-2 alkyl optionally substituted with 1-3 F. In some embodiments, the compounds of Formula (V) are compounds of Formula (V- c) or Formula (V-d):
Formula (V-c)
Formula (V-d) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; the pair of R
L1 taken together with the ring atom(s) connecting them form a C3-4 cycloalkyl ring, and R
L2 is C
1-2 alkyl optionally substituted with 1-3 F. In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)), Formula (III), Formula (IV) (e.g., Formula (IV-a), (IV-b), or (IV-c)), or Formula (V) (e.g., Formula (V-a), (V-b), (V-c), or (V-d)), Y
2 is -CH2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)), Formula (III), Formula (IV) (e.g., Formula (IV-a), (IV-b), or (IV-c)), or Formula (V) (e.g., Formula (V-a),
In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)), Formula (III), Formula (IV) (e.g., Formula (IV-a), (IV-b), or (IV-c)), or Formula (V) (e.g., Formula (V-a),
In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)), Formula (III), Formula (IV) (e.g., Formula (IV-a), (IV-b), or (IV-c)), or Formula (V) (e.g., Formula (V-a), (V-b), (V-c), or (V-d)), R
1 is
, wherein b2 is 0, 1, or 2, and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7. In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)), Formula (III), Formula (IV) (e.g., Formula (IV-a), (IV-b), or (IV-c)), or Formula (V) (e.g., Formula (V-a),
In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)), Formula (III), Formula (IV) (e.g., Formula (IV-a), (IV-b), or (IV-c)), or Formula (V) (e.g., Formula (V-a),
selected from the group consisting of: C(=O)N(R
f)
2, C(O)N(C
1-3 alkyl)R
b1, -C(O)N(H)R
b1, R
b1, and C(O)R
b1. In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)), Formula (III), Formula (IV) (e.g., Formula (IV-a), (IV-b), or (IV-c)), or Formula (V) (e.g., Formula (V-a), (V-b), (V-c), or (V-d)), R
7 is selected from the group consisting of: (a) C(=O)N(R
f)
2, wherein each R
f is independently H or C
1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is 4-10 membered heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. In some
embodiments, R
7 is C(=O)N(R
f)
2, wherein each R
f is independently H or C
1-3 alkyl optionally substituted with 1-3 R
h. In some embodiments, R
7 is R
b1, wherein the R
b1 is 5-6 membered heteroaryl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-2 R
g. In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)), Formula (III), Formula (IV) (e.g., Formula (IV-a), (IV-b), or (IV-c)), or Formula (V) (e.g., Formula (V-a),
wherein R
7a and R
7b are independently selected R
7. In some embodiments, R
7a is selected from the group consisting of: C(=O)N(R
f)
2, C(O)N(C
1-3 alkyl)R
b1, -C(O)N(H)R
b1, R
b1, and C(O)R
b1; and R
7b is -halo, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments, R
7a is selected from the group consisting of: (a) C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C
1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C
3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is 4-10 membered heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. In some embodiments, R
7a is R
b1, wherein the R
b1 is 5-6 membered heteroaryl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-2 R
g. In some embodiments, R
7b is halo (e.g., -Cl). In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)), Formula (III), Formula (IV) (e.g., Formula (IV-a), (IV-b), or (IV-c)), or Formula (V) (e.g., Formula (V-a), (V-b), (V-c), or (V-d)), R
1 is a 7-10 (e.g., 7) membered heterocyclyl having one ring nitrogen atom, one ring oxygen atom, and no additional ring heteroatoms, wherein the 7-10 membered heterocyclyl is optionally substituted with 1-4 R
7.
In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)), Formula (III), Formula (IV) (e.g., Formula (IV-a), (IV-b), or (IV-c)), or Formula (V) (e.g., Formula (V-a),
optionally substituted with 1-4 R
7 at one or more ring carbon atoms. For example, R
1 can
. In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)), Formula (III), Formula (IV) (e.g., Formula (IV-a), (IV-b), or (IV-c)), or Formula (V) (e.g., Formula (V-a), (V-b), (V-c), or (V-d)), each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C1-3 alkyl optionally substituted with 1-3 R
c, wherein: each R
c is independently selected from the group consisting of: -F, -OH, and -CN. In some embodiments of Formula (II) (e.g., Formula (II-a) or (II-b)), Formula (III), Formula (IV) (e.g., Formula (IV-a), (IV-b), or (IV-c)), or Formula (V) (e.g., Formula (V-a), (V-b), (V-c), or (V-d)),
optionally substituted with 1-4 R
7 at one or more ring carbon atoms, wherein each R
7 is independently selected from the group consisting of: -OH; - CN; -F; and C
1-3 alkyl optionally substituted with 1-3 R
c, wherein: each R
c is independently selected from the group consisting of: -F, -OH, and -CN. In some embodiments, the compounds of Formula (II) are compounds of Formula (II- a1) or (II-b1):
Formula (II-a1)
Formula (II-b1) or pharmaceutically acceptable salts thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; b3 is 0, 1, 2, or 3; X
1 is CH
2; and X
2 and X
3 are independently selected from the group consisting of: O, CH2, CHR
L, and C(R
L)2. In some embodiments, the compounds of Formula (III) are compounds of compound of Formula (III-1):
Formula (III-1) or pharmaceutically acceptable salts thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; b3 is 0, 1, 2, or 3; X
1 is CH2; and X
2 and X
3 are independently selected from the group consisting of: O, CH
2, CHR
L, and C(R
L)2;.
In some embodiments of Formula (III-1), R
9 is NR
dR
e (e.g., -NH
2). In some embodiments, the compounds of Formula (IV) are compounds of Formula (IV- a1) or (IV-b1):
Formula (IV-b1) or pharmaceutically acceptable salts thereof, wherein: b4 is 0 or 1; b1 is 0, 1, or 2; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; b3 is 0, 1, 2, or 3; X
1 is CH2; one of X
2 and X
3 is independently selected from the group consisting of: CHR
L and C(R
L)
2; and the other of X
2 and X
3 is CH
2 or O. In some embodiments of Formula (IV-a1) or (IV-b1), X
2 is CH2; and X
3 is CHR
L. In some embodiments of Formula (IV-a1) or (IV-b1), R
9 is NR
dR
e (e.g., -NH
2). In some embodiments, the compounds of Formula (V) are compounds of Formula (V-
a1) or (V-b1):
Formula (V-b1) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; b1 is 0, 1, or 2; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; b3 is 0, 1, 2, or 3; X
2 is -O- or -CH2-; X
1 is CHR
L; and X
3 is CHR
L or C(R
L)
2, wherein: one pair of R
L on different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-4 cycloalkyl ring; and the remaining R
L if present is C1-2 alkyl optionally substituted with 1-3 F. In some embodiments of Formula (II-a1), (II-b1), (III-1), (IV-a1), (IV-b1), (V-a1), or (V-b1), b3 is 0. In some embodiments of Formula (II-a1), (II-b1), (III-1), (IV-a1), (IV-b1), (V-a1), or (V-b1), b3 is 1 or 2; and each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C1-3 alkyl optionally substituted with 1-3 R
c, wherein each R
c is independently
selected from the group consisting of: -F, -OH, and -CN. In some embodiments, the compounds of Formula (II) are compounds of Formula (II- a2) or (II-b2):
Formula (II-b2) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
1 is CH
2; and X
2 and X
3 are independently selected from the group consisting of: O, CH
2, CHR
L, and C(R
L)2. In some embodiments, the compounds of Formula (III) are compounds of Formula (III-2):
Formula (III-2) or pharmaceutically acceptable salts thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)
2, C(O)N(C
1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
1 is CH
2; and X
2 and X
3 are independently selected from the group consisting of: O, CH
2, CHR
L, and C(R
L)2. In some embodiments of Formula (III-2) R
9 is NR
dR
e (e.g., -NH
2). In some embodiments, the compounds of Formula (IV) are compounds of compound of Formula (IV-a2) or (IV-b2):
Formula (IV-a2)
Formula (IV-b2) or pharmaceutically acceptable salts thereof, wherein: b4 is 0 or 1; b1 is 0, 1, or 2; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
1 is CH
2; one of X
2 and X
3 is independently selected from the group consisting of: CHR
L and C(R
L)2; and the other of X
2 and X
3 is CH
2 or O. In some embodiments of Formula (IV-a2) or (IV-b2), X
2 is CH2; and X
3 is CHR
L. In some embodiments, the compounds of Formula (V) are compounds of Formula (V- a2) or (V-b2):
Formula (V-a2)
Formula (V-b2) or pharmaceutically acceptable salts thereof, wherein: b4 is 0 or 1; b1 is 0, 1, or 2; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)
2, C(O)N(C
1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
2 is -O- or -CH2-; X
1 is CHR
L; and X
3 is CHR
L or C(R
L)
2, wherein: one pair of R
L on different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-4 cycloalkyl ring; and the remaining R
L if present is C
1-2 alkyl optionally substituted with 1-3 F. In some embodiments of Formula (II-a2), (II-b2), (III-2), (IV-a2), (IV-b2), (V-a2), or (V-b2), R
7 is selected from the group consisting of: (a) C(=O)N(R
f)
2, wherein each R
f is independently H or C
1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is 4-10 membered heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. In some embodiments of Formula (II-a2), (II-b2), (III-2), (IV-a2), (IV-b2), (V-a2), or (V-b2), R
7 is R
b1, wherein the R
b1 is 5-6 membered heteroaryl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-2 R
g. In some embodiments of Formula (II-a1), (II-b1), (III-1), (IV-a1), (IV-b1), (V-a1),
(V-b1), (II-a2), (II-b2), (III-2), (IV-a2), (IV-b2), (V-a2), or (V-b2), Y
2 is -CH
2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. In some embodiments of Formula (II-a1), (II-b1), (III-1), (IV-a1), (IV-b1), (V-a1),

. In some embodiments, the compound of Formula (II) is selected from the group consisting of Compound Nos.139, 139a, 139b, 139c, 158, 158a, 158b, 158c, 160, 160a, 161, 161a, 161b, 161c, 164, 164a, 164b, 170, 170a, 171, 171a, 171b, 171c, 176, 176a, 176b, 176c, 176d, 176e, 178, 178a, 178b, 179, 179a, 179b, 179d, 179e, 179f, 180, 180a, 180b, 180c, 181, 181a, 183, 183a, 185, 185a, 190, 190a, 190b, 190c, 191, 191a, 191b, 191c, 191d, 192, 192a, 192b, 192c, 192d, and 192e, as depicted in Table C1, or a pharmaceutically acceptable salt thereof. For example, the compound of Formula (II) can be selected from the group consisting of Compound Nos.139, 139a, 139b, 139c, 158, 158a, 158b, 158c, 160, 160a, 161, 161a, 164, 164a, 164b, 170, 170a, 171, 171a, 176, 176a, 176b, 176c, 176d, 178, 178a, 178b, 179, 179a,
180, 180a, 181, and 181a as depicted in Table C1, or a pharmaceutically acceptable salt thereof. In some embodiments, the compound of Formula (III) is selected from the group consisting of Compound Nos.158, 158a, 158b, 158c, 161, 161a, 161b, 161c, 176, 176a, 176b, 176c, 176d, 176e, 177, 177a, 178, 178a, 178b, 179, 179a, 179b, 179d, 179e, 179f, 180, 180a, 180b, 180c, 184, 184b, 184c, 192, 192a, 192b, 192c, 192d, 192e, 194, and 194a, as depicted in Table C1, or a pharmaceutically acceptable salt thereof. For example, the compound of Formula (III) can be selected from the group consisting of Compound Nos.158, 158a, 158b, 158c, 161, 161a, 176, 176a, 176b, 176c, 176d, 177, 177a, 178, 178a, 178b, 179, 179a, 180, and 180a as depicted in Table C1, or a pharmaceutically acceptable salt thereof. In some embodiments, the compound of Formula (IV) is selected from the group consisting of Compound Nos.124, 124a, 124b, 124c, 124d, 124e, 124f, 125, 125a, 130, 130a, 130b, 130c, 131, 131a, 131b, 133, 133a, 133b, 133c, 134, 134a, 138, 138a, 148, 148a, 162, 162a, 162b, 162c, 162d, 163, 163a, 163b, 175, 175a, 176, 176a, 176b, 176c, 176d, 176e, 177, 177a, 178, 178a, 178b, 179, 179a, 179b, 179d, 179e, 179f, 182, 182a, 182b, 182c, 182d, 193, and 193a, as depicted in Table C1, or a pharmaceutically acceptable salt thereof. For example, the compound of Formula (IV) can be selected from the group consisting of Compound Nos. 124, 124a, 124b, 125, 125a, 130, 130a, 131, 131a, 133, 133a, 133b, 134, 134a, 138, 138a, 148, 148a, 162, 162a, 163, 163a, 175, and 175a as depicted in Table C1, or a pharmaceutically acceptable salt thereof. In some embodiments, the compound of Formula (V) is selected from the group consisting of Compound Nos. 172, 172a, 172b, and 172c as depicted in Table C1, or a pharmaceutically acceptable salt thereof. In some embodiments, compounds of Formula (A) (e.g., Formula (I)) are compounds of Formula (VI):

or pharmaceutically acceptable salts thereof, wherein: R
1 is a 4-10 membered heterocyclyl substituted with -CN, –(C
1-3 alkylene)-CN, or –
(C
3-6 cycloalkylene)-CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7; wherein each R
7 is independently selected from the group consisting of R
a and R
b; X
1 is selected from the group consisting of S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; b1 is 0, 1, or 2; R
9 is selected from the group consisting of: H, NR
dR
e, -OH, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
(e) -C
1-6 alkoxy; (f) -C1-6 haloalkoxy; (g) -NR
dR
e; (h) C(=O)C
1-6 alkyl; (i) C(=O)C1-6 haloalkyl; (j) C(=O)OH; (k) C(=O)OC
1-6 alkyl; (l) C(=O)OC1-6 haloalkyl; (m) C(=O)N(R
f)2; (n) S(O)
0-2(C
1-6 alkyl); (o) S(O)
0-2(C
1-6 haloalkyl); (p) S(O)1-2N(R
f)2; and (q) C1-6 alkyl, C2-6 alkenyl, or C2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h;
each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, C
1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. In some embodiments of Formula (VI), R
1 is a 4-10 membered heterocyclyl substituted with -CN or –(C
1-3 alkylene)-CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7. In some embodiments of Formula (VI), R
1 is a 6-8 membered heterocyclyl substituted with -CN or –(C1-3 alkylene)-CN on a ring carbon atom, wherein: the heterocyclyl has one ring nitrogen atom and 0-1 ring oxygen atom; and the heterocyclyl is further optionally substituted with 1-3 R
7. In some embodiments of Formula (VI), each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C
1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments of Formula (VI), R
1 is selected from the group consisting of:
, wherein b3 is 0, 1, or 2. In some embodiments, each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C
1-3 alkyl optionally substituted with 1-3 R
c (e.g., C1-3 alkyl optionally substituted with 1-3 -F). In some embodiments, b3 is 0. For example, R
1 can
Formula (VI). In some embodiments of Formula (VI), R
9 is -NR
dR
e or OH (e.g., -NH
2). In some embodiments, the compounds of Formula (VI) are compounds of Formula (VI- a):
Formula (VI-a) or pharmaceutically acceptable salts thereof, wherein: each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments of Formula (VI-a), b1 is 1 or 2. In some embodiments, the compounds of Formula (VI) are compounds of Formula (VI- b):
Formula (VI-b) or pharmaceutically acceptable salts thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments of Formula (VI-b), R
9 is -NR
dR
e. In some embodiments, the compounds of Formula (VI) are compounds of Formula (VI- c):
Formula (VI-c)
or pharmaceutically acceptable salts thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments of Formula (VI) (e.g., Formula (VI-a), (VI-b), or (VI-c)), X
1 is CH
2 or CHR
L. In some embodiments of Formula (VI) (e.g., Formula (VI-a), (VI-b), or (VI-c)), X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2. In some embodiments of Formula (VI) (e.g., Formula (VI-a), (VI-b), or (VI-c)), X
1 is CH
2; and X
2 and X
3 are both CH
2. In some embodiments of Formula (VI) (e.g., Formula (VI-a), (VI-b), or (VI-c)), at least one (e.g., one) of X
1, X
2, and X
3 is selected from the group consisting of: CHR
L and C(R
L)2. In some embodiments of Formula (VI) (e.g., Formula (VI-a), (VI-b), or (VI-c)), X
1 is CH2; and X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2, provided that 1-2 of X
2 and X
3 is independently CHR
L or C(R
L)2. In some embodiments of Formula (VI) (e.g., Formula (VI-a), (VI-b), or (VI-c)), X
1 is CH
2; X
2 is CH
2; and X
3 is CHR
L. In some embodiments of Formula (VI) (e.g., Formula (VI-a), (VI-b), or (VI-c)), X
1 is CH
2; one of X
2 and X
3 is -O-; and the other of X
2 and X
3 is selected from the group consisting of: CH
2, CHR
L, and C(R
L)
2. In some embodiments of Formula (VI) (e.g., Formula (VI-a), (VI-b), or (VI-c)), X
1 is CH2; X
2 is -O-; and X
3 is selected from the group consisting of: CH2, CHR
L, and C(R
L)2. In some embodiments of Formula (VI) (e.g., Formula (VI-a), (VI-b), or (VI-c)), each R
L is independently selected from the group consisting of: CH3, CF3, CHF2, and CH2F. In some embodiments, the compounds of Formula (VI) are compounds of Formula (VI- d):
Formula (VI-d)
or pharmaceutically acceptable salts thereof, wherein: X
1 is selected from the group consisting of S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that at least one of X
1, X
2, and X
3 is CHR
L or C(R
L)
2; and further provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments, the compounds of Formula (VI) are compounds of Formula (VI-
Formula (VI-e) or pharmaceutically acceptable salts thereof, wherein: X
1 is selected from the group consisting of S(O)
0-2, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that 2-3 of X
1, X
2, and X
3 are independently CHR
L or C(R
L)2; one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-6 cycloalkyl ring; and each additional R
L is independently selected from the group consisting of: C1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments of Formula (VI-d) or (VI-e), R
9 is NH
2. In some embodiments of Formula (VI-d) or (VI-e), b1 is 1 or 2.
In some embodiments of Formula (VI-d) or (VI-e), the
selected from the group consisting of:
b4 is 0 or 1; X
2 is -O- or -CH
2-; X
3 is -CH2- or -CHR
L-, wherein R
L is C1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments of Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e)), Y
2 is -CH
2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. In some embodiments of Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d),
In some embodiments of Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d),
In some embodiments, the compound of Formula (VI) is selected from the group consisting of Compound Nos.149, 149a, 149b, 149c, 173, 173a, 174, 174a, 186, 186a, 186b, 186c, 187a, 187a, 188, 188a, 189, 189a, 191, 191a, 191b, 191c, 191d, 192, 192a, 192b, 192c,
192d, 192e, 195, and 195a, as depicted in Table C1, or a pharmaceutically acceptable salt thereof. Certain examples of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))) compounds were synthesized using methods involving resolution of stereoisomeric mixture(s) (e.g., SFC separation of stereoisomers). In Table C1, the resolved stereogenic centers in these compounds are labelled with the “or1” and/or “or2” enhanced stereochemical notations. In some instances, the stereoisomeric resolutions were performed during the last step of the synthesis, thereby providing the individual stereoisomers of the Formula (A) compounds. Alternatively, in some other instances, the resolutions were performed on an intermediate or starting material, wherein each of the constituent stereoisomers of the intermediate or starting material could be separately subjected to the subsequent steps of the synthesis to provide the respective Formula (A) compounds as separate stereoisomers. A person of ordinary skill in the art would understand that, under either approach for stereoisomeric resolution, stereoisomers having both (R)- and (S)-configurations at a resolved stereogenic center are provided. See Table C3, wherein Table C1 compounds whose stereoisomers contain the or1 and/or or2 stereochemical notations are provided in non-stereogenic form, followed by the respective stereoisomers having the (R)- and (S)-configurations. Table C3
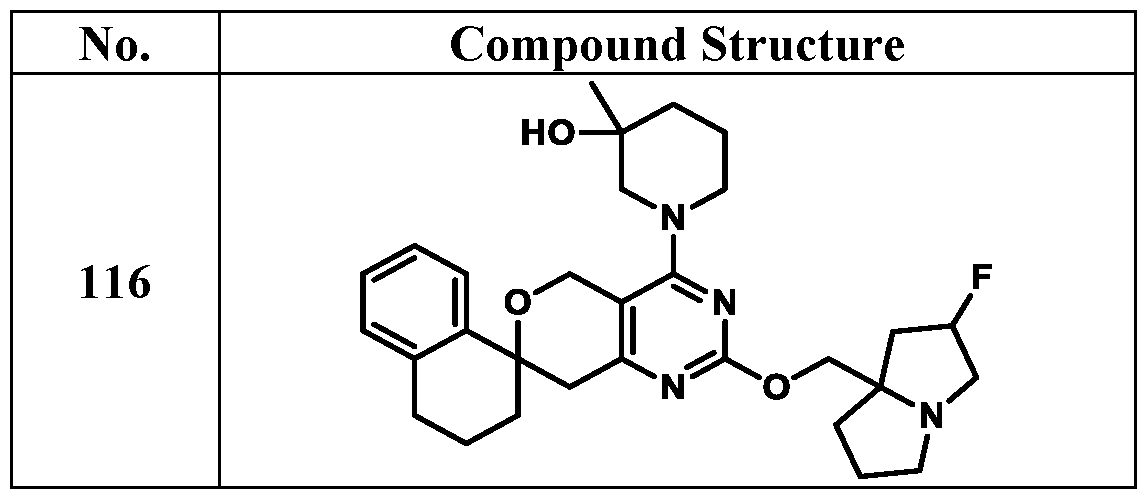
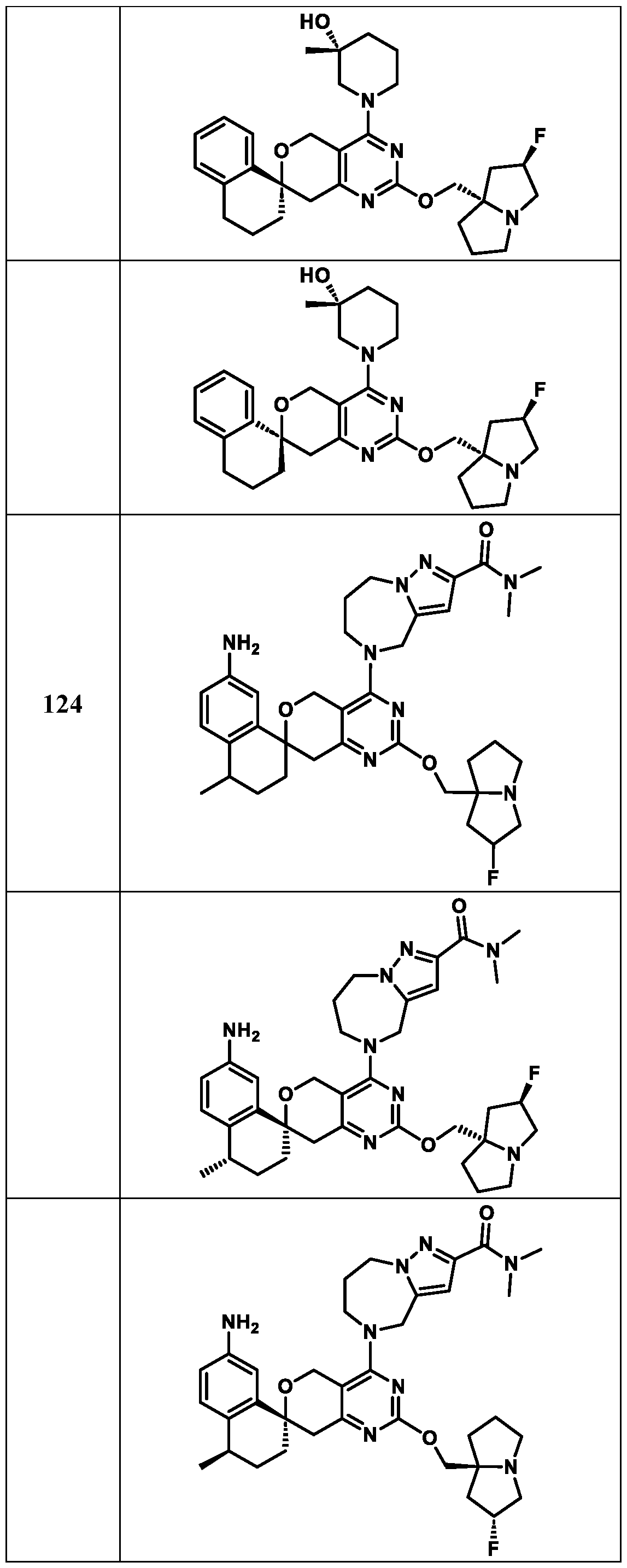

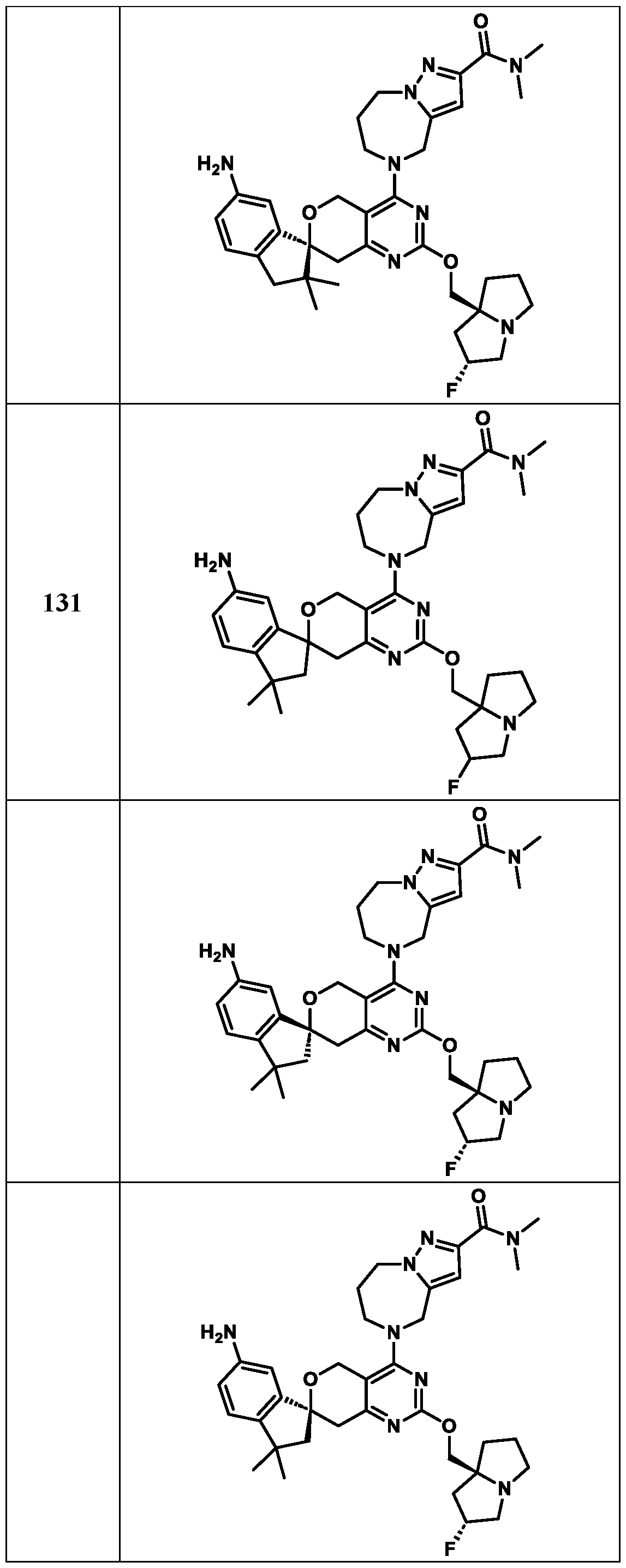


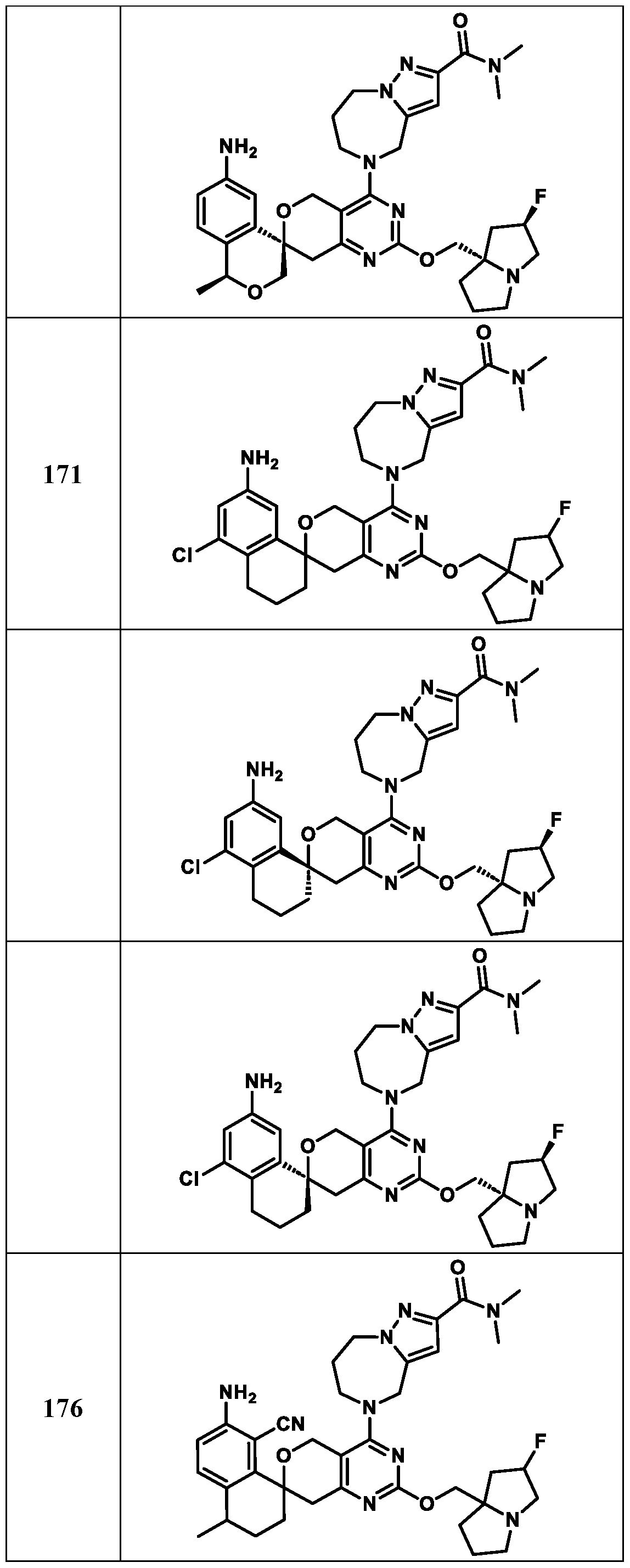
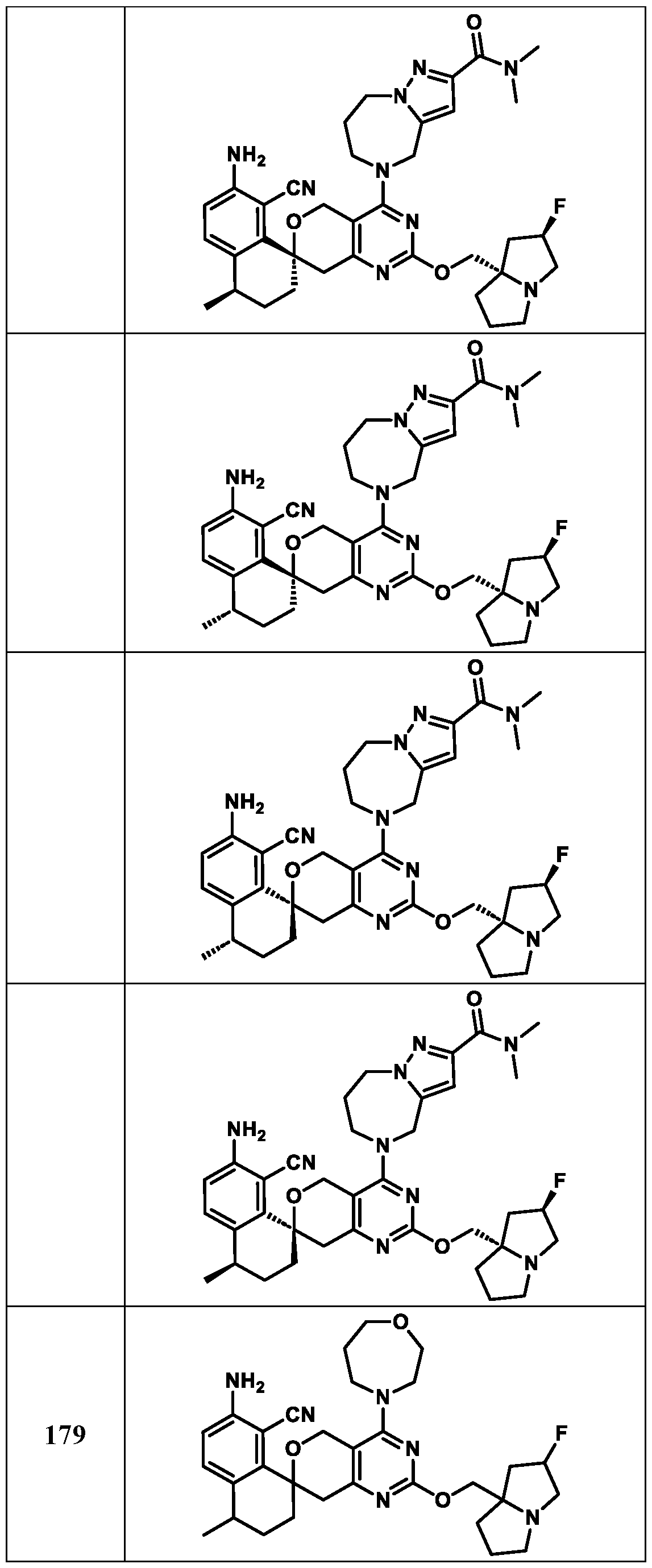
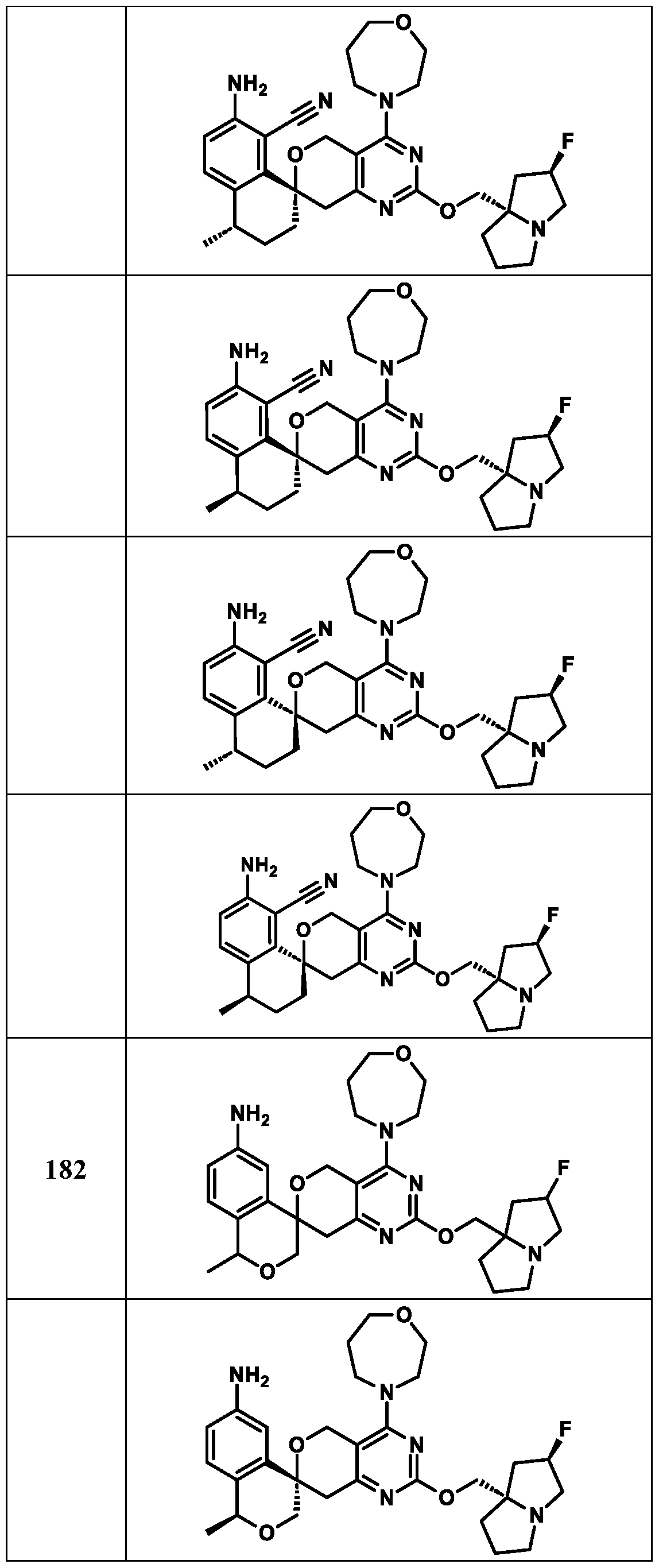
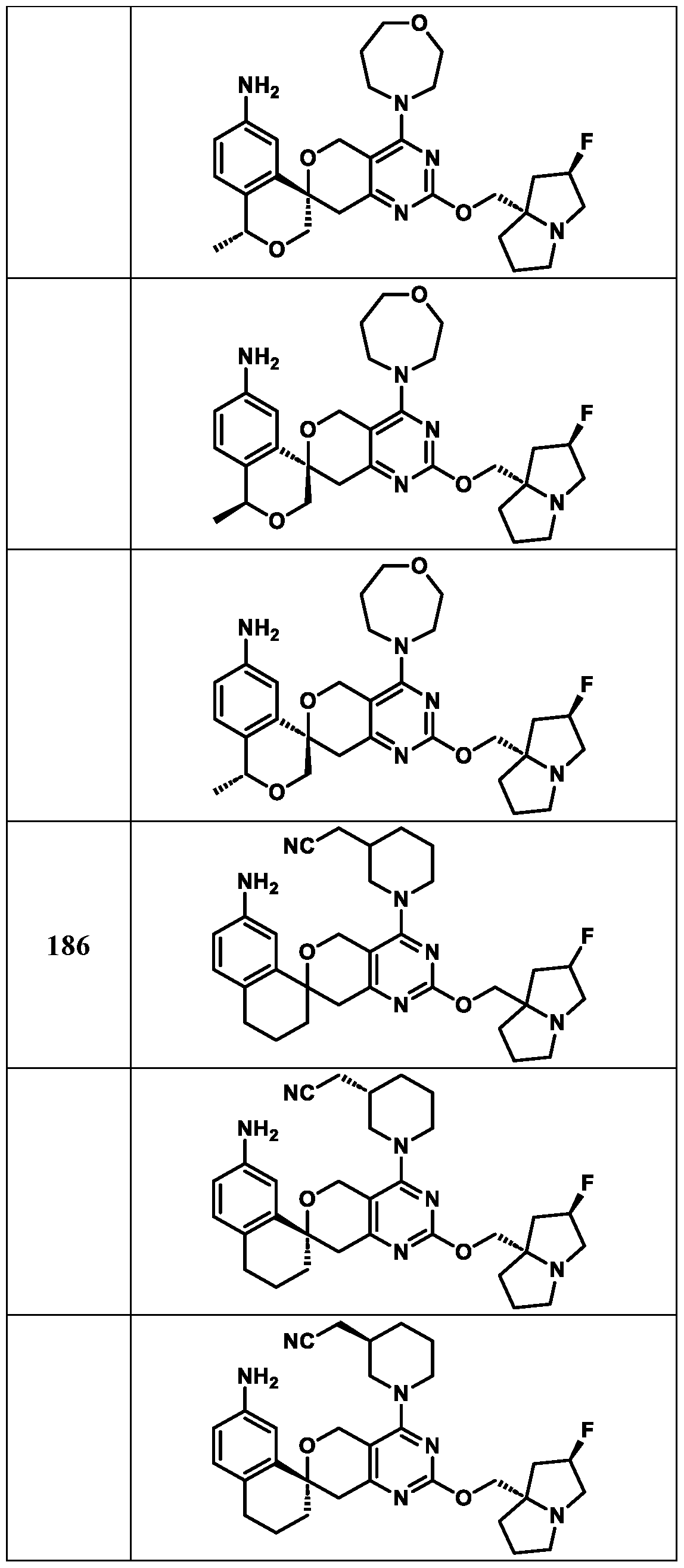
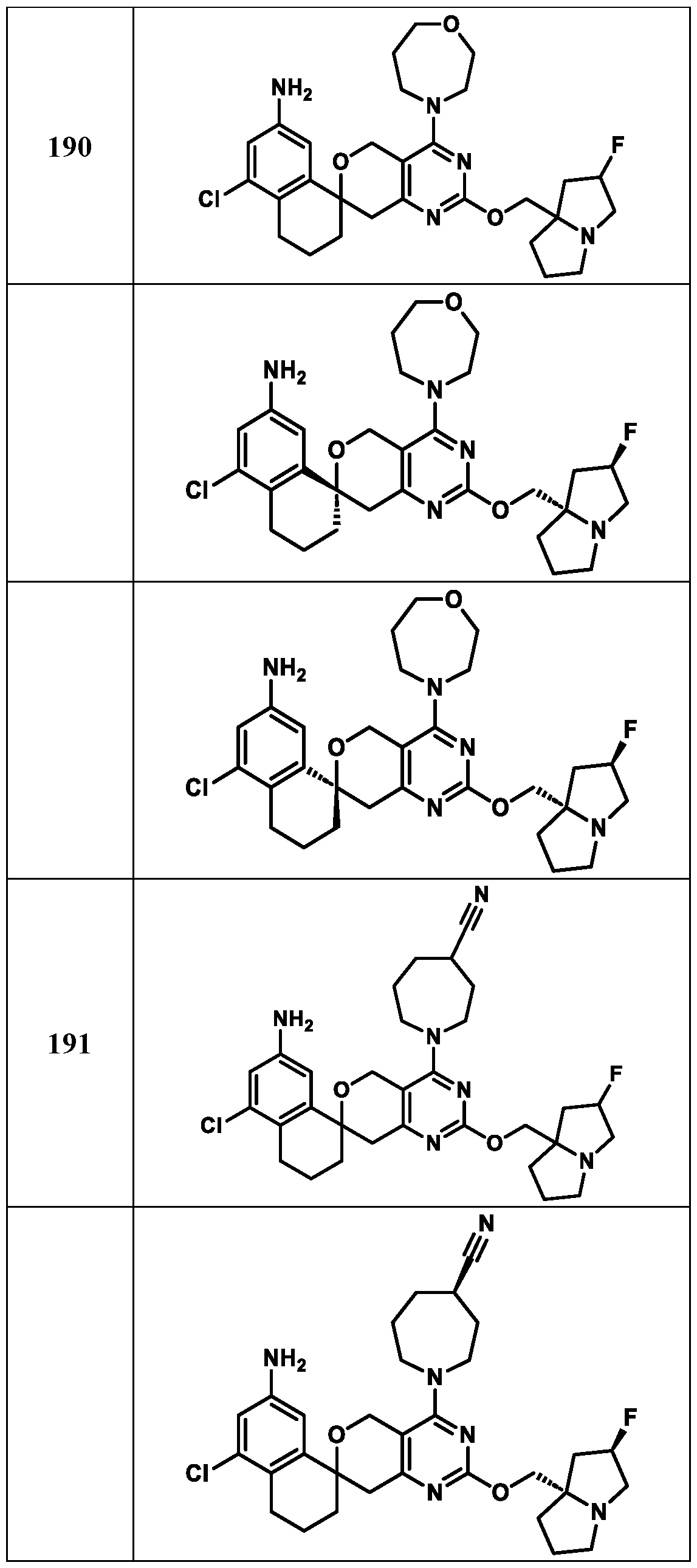

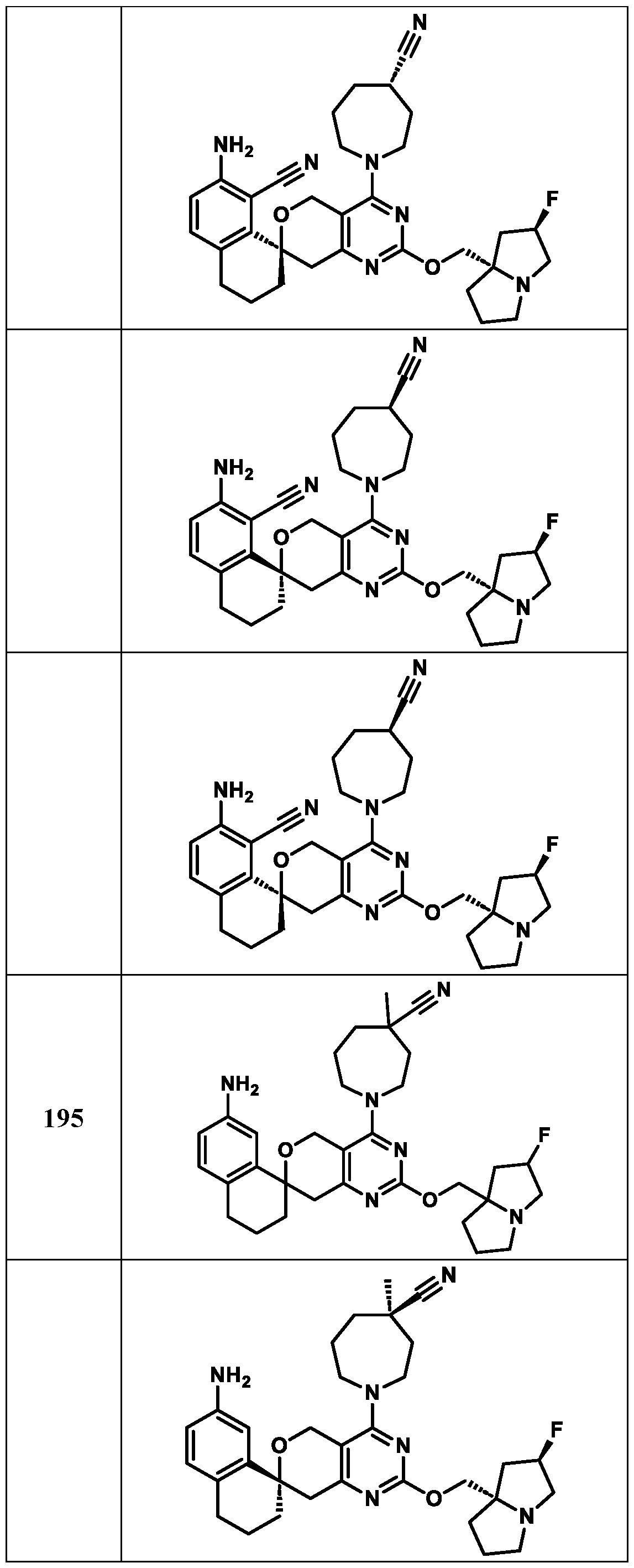
Also provided herein is a KRas protein (e.g., a dysregulated KRas protein (e.g., a mutated KRas protein)) non-covalently bound with a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof. In some embodiments, Gly10 of the KRas protein interacts non-covalently with the R
1 group of the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, (e.g., via a hydrogen bond between the carbonyl of Gly10 and an OH group of R
1 and/or via a water mediated interaction between the NH group of Gly10 and an OH group of R
1). Without wishing to be bound by theory, in some embodiments, the interaction between Gly10 of the KRas protein and the R
1 group of the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof , facilitates the inhibition of interaction between the KRas protein and Raf-RBD. In some embodiments, Arg68 of the KRas protein interacts
non-covalently with the R
1 group of the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, (e.g., via a hydrogen bond between the guanidine of Arg68 and a CN group of R
1). Without wishing to be bound by theory, in some embodiments, the interaction between Arg68 of the KRas protein and the R
1 group of the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, facilitates the inhibition of interaction between the KRas protein and Raf-RBD. Chemical definitions The term “halo” refers to fluoro (F), chloro (Cl), bromo (Br), or iodo (I). The term “oxo” refers to a divalent doubly bonded oxygen atom (i.e., “=O”). As used herein, oxo groups are attached to carbon atoms to form carbonyls. The term “alkyl” refers to a saturated acyclic hydrocarbon radical that may be a straight chain or branched chain, containing the indicated number of carbon atoms. For example, C
1-10 indicates that the group may have from 1 to 10 (inclusive) carbon atoms in it. Alkyl groups can either be unsubstituted or substituted with one or more substituents. Non-limiting examples include methyl, ethyl, iso-propyl, tert-butyl, n-hexyl. The term “saturated” as used in this context means only single bonds present between constituent carbon atoms and other available valences occupied by hydrogen and/or other substituents as defined herein. The term “haloalkyl” refers to an alkyl, in which one or more hydrogen atoms is/are replaced with an independently selected halo (e.g., -CF
3, -CHF
2, or -CH
2F). The term “alkoxy” refers to an -O-alkyl radical (e.g., -OCH3). The term “alkylene” refers to a divalent alkyl (e.g., -CH2-). Similarly, terms such as
“cycloalkylene” and “heterocyclylene” refer to divalent cycloalkyl and heterocyclyl respectively. For avoidance of doubt, in “cycloalkylene” and “heterocyclylene”, the two radicals can be on the same ring carbon atom (e.g., a geminal diradical such
) or on different ring atoms (e.g., ring carbon and/or nitrogen atoms (e.g., vicinal ring carbon and/or nitrogen atoms)
The term “alkenyl” refers to an acyclic hydrocarbon chain that may be a straight chain or branched chain having one or more carbon-carbon double bonds. The alkenyl moiety contains the indicated number of carbon atoms. For example, C
2-6 indicates that the group may have from 2 to 6 (inclusive) carbon atoms in it. Alkenyl groups can either be unsubstituted or substituted with one or more substituents. The term “alkynyl” refers to an acyclic hydrocarbon chain that may be a straight chain or branched chain having one or more carbon-carbon triple bonds. The alkynyl moiety contains the indicated number of carbon atoms. For example, C2-6 indicates that the group may have from 2 to 6 (inclusive) carbon atoms in it. Alkynyl groups can either be unsubstituted or substituted with one or more substituents. The term “aryl” refers to a 6-20 carbon mono-, bi-, tri- or polycyclic group wherein at least one ring in the system is aromatic (e.g., 6-carbon monocyclic, 10-carbon bicyclic, or 14- carbon tricyclic aromatic ring system); and wherein 0, 1, 2, 3, or 4 atoms of each ring may be substituted by a substituent. Examples of aryl groups include phenyl, naphthyl, tetrahydronaphthyl, and the like. The term “cycloalkyl” as used herein refers to mono-, bi-, tri-, or polycyclic saturated or partially unsaturated hydrocarbon groups having, e.g., 3 to 20 ring carbons, preferably 3 to 15 ring carbons, and more preferably 3 to 12 ring carbons or 3-10 ring carbons or 3-6 ring carbons, wherein the cycloalkyl group may be optionally substituted. The term “saturated” as used in this context means only single bonds present between constituent carbon atoms. Examples of saturated cycloalkyl groups include, without limitation, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl. Partially unsaturated cycloalkyl may have any degree of unsaturation provided that one or more double bonds is present in the cycloalkyl, none of the rings in the ring system are aromatic, and the partially unsaturated 30 cycloalkyl group is not fully saturated overall. Examples of partially unsaturated cycloalkyl
include, without limitation, cyclopentenyl, cyclohexenyl, cycloheptenyl, and cyclooctenyl. Cycloalkyl may include multiple fused and/or bridged rings. Non-limiting examples of fused/bridged cycloalkyl includes: bicyclo[1.1.0]butyl, bicyclo[2.1.0]pentyl, bicyclo[1.1.1]pentyl, bicyclo[3.1.0]hexyl, bicyclo[2.1.1]hexyl, bicyclo[3.2.0]heptyl, bicyclo[4.1.0]heptyl, bicyclo[2.2.1]heptyl, bicyclo[3.1.1]heptyl, bicyclo[4.2.0]octyl, bicyclo[3.2.1]octyl, bicyclo[2.2.2]octyl, and the like. Cycloalkyl also includes spirocyclic rings (e.g., spirocyclic bicycle wherein two rings are connected through just one atom). Non-limiting examples of spirocyclic cycloalkyls include spiro[2.2]pentyl, spiro[2.5]octyl, spiro[3.5]nonyl, spiro[3.5]nonyl, spiro[3.5]nonyl, spiro[4.4]nonyl, spiro[2.6]nonyl, spiro[4.5]decyl, spiro[3.6]decyl, spiro[5.5]undecyl, and the like. The term “heteroaryl”, as used herein, means a mono-, bi-, tri- or polycyclic group having 5 to 20 ring atoms, alternatively 5, 6, 9, 10, or 15 ring atoms; wherein at least one ring in the system contains one or more heteroatoms independently selected from the group consisting of N, O, S (inclusive of oxidized forms such as:
(inclusive of oxidized forms such as: ) and at least one ring in the system is aromatic (but does not have to be a ring which contains a heteroatom, e.g. tetrahydroisoquinolinyl, e.g., tetrahydroquinolinyl). Heteroaryl groups can either be unsubstituted or substituted with one or more substituents. Examples of heteroaryl include thienyl, pyridinyl, furyl, oxazolyl, oxadiazolyl, pyrrolyl, imidazolyl, triazolyl, thiodiazolyl, pyrazolyl, isoxazolyl, thiadiazolyl, pyranyl, pyrazinyl, pyrimidinyl, pyridazinyl, triazinyl, thiazolyl benzothienyl, benzoxadiazolyl, benzofuranyl, benzimidazolyl, benzotriazolyl, cinnolinyl, indazolyl, indolyl, isoquinolinyl, isothiazolyl, naphthyridinyl, purinyl, thienopyridinyl, pyrido[2,3-d]pyrimidinyl, pyrrolo[2,3-b]pyridinyl, quinazolinyl, quinolinyl, thieno[2,3-c]pyridinyl, pyrazolo[3,4- b]pyridinyl, pyrazolo[3,4-c]pyridinyl, pyrazolo[4,3-c]pyridinyl, pyrazolo[4,3-b]pyridinyl, tetrazolyl, chromanyl, 2,3-dihydrobenzo[b][1,4]dioxinyl, benzo[d][1,3]dioxolyl, 2,3- dihydrobenzofuranyl, tetrahydroquinolinyl, 2,3-dihydrobenzo[b][1,4]oxathiinyl, isoindolinyl, and others. In some embodiments, the heteroaryl is selected from thienyl, pyridinyl, furyl, pyrazolyl, imidazolyl, isoindolinyl, pyranyl, pyrazinyl, and pyrimidinyl. For purposes of clarification, heteroaryl also includes aromatic lactams, aromatic cyclic ureas, or vinylogous analogs thereof, in which each ring nitrogen adjacent to a carbonyl is tertiary (i.e., all three valences are occupied by non-hydrogen substituents), such as one or more of pyridonyl (e.g.,
imidazolonyl (e.g.,
wherein each ring nitrogen adjacent to a carbonyl is tertiary (i.e., the oxo group (i.e., “=O”) herein is a constituent part of the heteroaryl ring). The term “heterocyclyl” refers to a mono-, bi-, tri-, or polycyclic saturated or partially unsaturated ring system with 3-15 ring atoms (e.g., 5-8 membered monocyclic, 8-12 membered bicyclic, or 11-15 membered tricyclic ring system) having 1-3 heteroatoms if monocyclic, 1-6 heteroatoms if bicyclic, or 1-9 heteroatoms if tricyclic or polycyclic, the heteroatoms selected from O, N, S (inclusive of oxidized forms such as:
and P (inclusive of oxidized forms such as:

) (e.g., carbon atoms and 1-3, 1-6, or 1-9 heteroatoms of N, O, S, or P if monocyclic, bicyclic, or tricyclic, respectively), wherein 0, 1, 2 or 3 atoms of each ring may be substituted by a substituent. The term “saturated” as used in this context means only single bonds present between constituent ring atoms and other available valences occupied by hydrogen and/or other substituents as defined herein. Examples of saturated heterocyclyl groups include piperazinyl, pyrrolidinyl, dioxanyl, morpholinyl, tetrahydrofuranyl, and the like. Partially unsaturated heterocyclyl groups may have any degree of unsaturation provided that one or more double bonds is present in the heterocyclyl, none of the rings in the ring system are aromatic, and the partially unsaturated heterocyclyl group is not fully saturated overall. Examples of partially unsaturated heterocyclyl groups include, without limitation, tetrahydropyridyl, dihydropyrazinyl, dihydropyridyl, dihydropyrrolyl, dihydrofuranyl, dihydrothiophenyl. Heterocyclyl may include multiple fused and bridged rings. Non-limiting examples of fused/bridged heteorocyclyl includes: 2-azabicyclo[1.1.0]butyl, 2-azabicyclo[2.1.0]pentyl, 2- azabicyclo[1.1.1]pentyl, 3-azabicyclo[3.1.0]hexyl, 5-azabicyclo[2.1.1]hexyl, 3-25 azabicyclo[3.2.0]heptyl, octahydrocyclopenta[c]pyrrole, 3-azabicyclo[4.1.0]heptyl, 7-
azabicyclo[2.2.1]heptyl, 6-azabicyclo[3.1.1]heptyl, 7-azabicyclo[4.2.0]octyl, 2- azabicyclo[2.2.2]octyl, 3-azabicyclo[3.2.1]octyl, 2-oxabicyclo[1.1.0]butyl, 2- oxabicyclo[2.1.0]pentyl, 2-oxabicyclo[1.1.1]pentyl, 3-oxabicyclo[3.1.0]hexyl, 5- oxabicyclo[2.1.1]hexyl, 3-oxabicyclo[3.2.0]heptyl, 3-oxabicyclo[4.1.0]heptyl, 7- oxabicyclo[2.2.1]heptyl, 6-oxabicyclo[3.1.1]heptyl, 7-oxabicyclo[4.2.0]octyl, 2- oxabicyclo[2.2.2]octyl, 3-oxabicyclo[3.2.1]octyl, and the like. Heterocyclyl also includes spirocyclic rings (e.g., spirocyclic bicycle wherein two rings are connected through just one atom). Non-limiting examples of spirocyclic heterocyclyls include 2-azaspiro[2.2]pentyl, 4- azaspiro[2.5]octyl, 1-azaspiro[3.5]nonyl, 2-azaspiro[3.5]nonyl, 7-azaspiro[3.5]nonyl, 2- azaspiro[4.4]nonyl, 6-azaspiro[2.6]nonyl, 1,7-diazaspiro[4.5]decyl, 7-azaspiro[4.5]decyl 2,5- diazaspiro[3.6]decyl, 3-azaspiro[5.5]undecyl, 2-oxaspiro[2.2]pentyl, 4-oxaspiro[2.5]octyl, 1- oxaspiro[3.5]nonyl, 2-oxaspiro[3.5]nonyl, 7-oxaspiro[3.5]nonyl, 2-oxaspiro[4.4]nonyl, 6- oxaspiro[2.6]nonyl, 1,7-dioxaspiro[4.5]decyl, 2,5-dioxaspiro[3.6]decyl, 1- oxaspiro[5.5]undecyl, 3-oxaspiro[5.5]undecyl, 3-oxa-9-azaspiro[5.5]undecyl and the like. As used herein, when a ring is described as being “partially unsaturated”, it means the ring has one or more additional degrees of unsaturation (in addition to the degree of unsaturation attributed to the ring itself; e.g., one or more double or triple bonds between constituent ring atoms), provided that the ring is not aromatic. Examples of such rings include: cyclopentene, cyclohexene, cycloheptene, dihydropyridine, tetrahydropyridine, dihydropyrrole, dihydrofuran, dihydrothiophene, and the like. For the avoidance of doubt, and unless otherwise specified, for rings and cyclic groups (e.g., aryl, heteroaryl, heterocyclyl, heterocycloalkenyl, cycloalkenyl, cycloalkyl, and the like described herein) containing a sufficient number of ring atoms to form bicyclic or higher order ring systems (e.g., tricyclic, polycyclic ring systems), it is understood that such rings and cyclic groups encompass those having fused rings, including those in which the points of fusion are located (i) on adjacent ring atoms (e.g., [x.x.0] ring systems, in which 0 represents a zero atom bridge (e.g.,

(ii) a single ring atom (spiro-fused ring systems)
,
(iii) a contiguous array of ring atoms (bridged ring systems
In addition, atoms making up the compounds of the present embodiments are intended to include all isotopic forms of such atoms. Isotopes, as used herein, include those atoms having the same atomic number but different mass numbers. By way of general example and without limitation, isotopes of hydrogen include tritium and deuterium, and isotopes of carbon include
13C and
14C. In addition, the compounds generically or specifically disclosed herein are intended to include all tautomeric forms. Thus, by way of example, a compound containing the moiety:
encompasses the tautomeric form containing the moiety:

. Similarly, a pyridinyl or pyrimidinyl moiety that is described to be optionally substituted with hydroxyl encompasses pyridone or pyrimidone tautomeric forms. The compounds provided herein may encompass various stereochemical forms. The compounds also encompass diastereomers as well as optical isomers, e.g., mixtures of enantiomers including racemic mixtures, as well as individual enantiomers and diastereomers, which arise as a consequence of structural asymmetry in certain compounds. Unless otherwise indicated, when a disclosed compound is named or depicted by a structure without specifying the stereochemistry and has one or more chiral centers, it is understood to represent all possible stereoisomers of the compound. Methods of Treatment Indications Provided herein are methods for inhibiting a KRas protein. For example, provided herein are inhibitors of a KRas protein (e.g., a dysregulated KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, or a KRas G12V mutant protein))) useful for treating or preventing diseases or disorders associated with the KRas dysregulation (i.e., a KRas-associated disease or disorder), such as a cardiovascular disease, an inflammatory and/or autoimmune disease, or a cancer (e.g., a KRas-associated cancer).
The term "KRas-associated disease or disorder" as used herein refers to diseases or disorders associated with or having a dysregulation of a KRAS gene, a KRas protein, or the expression or activity or level of any (e.g., one or more) of the same (e.g., any of the types of dysregulations of a KRAS gene, a KRas protein, or the expression or activity or level of any of the same described herein). Non-limiting examples of a KRas-associated disease or disorder include, for example, cancer, a cardiovascular disease (e.g., arteriovenous malformations), endometriosis, and an inflammatory and/or autoimmune disease (e.g., a nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis). See, e.g., Adashek et al. Genome Med. 2020; 12: 16, doi: 10.1186/s13073-020-0714-y; Niemela et al. Blood. 2011; 117(10):2883-6, doi: 10.1182/blood-2010-07-295501; Nosan et al. Croat Med J.2013; 54(6): 574–578, doi: 10.3325/cmj.2013.54.574; and Messina et al. Small GTPases 11.5 (2020): 312- 319, 10.1080/21541248.2018.1502591. The term “mutant KRas-associated disease or disorder” as used herein refers to diseases or disorders associated with or having a KRas mutation (e.g., a KRAS gene having a mutation corresponding to a mutation in a KRas protein and/or a KRas protein having a mutation). Non- limiting examples of a mutant KRas-associated disease or disorder include, for example, cancer, a cardiovascular disease (e.g., arteriovenous malformations), endometriosis, and an inflammatory and/or autoimmune disease (e.g., a nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis). See, e.g., Adashek et al. Genome Med. 2020; 12: 16, doi: 10.1186/s13073-020-0714-y; Niemela et al. Blood.2011; 117(10):2883-6, doi: 10.1182/blood- 2010-07-295501; Nosan et al. Croat Med J. 2013; 54(6): 574–578, doi: 10.3325/cmj.2013.54.574; and Messina et al. Small GTPases 11.5 (2020): 312-319, 10.1080/21541248.2018.1502591. The phrase “dysregulation of a KRAS gene, a KRas protein, or the expression or activity or level of any of the same” refers to a genetic mutation (e.g., a mutation in a KRAS gene that results in the expression of a KRas protein that includes a deletion of at least one amino acid as compared to a wild type KRas protein, a mutation in a KRAS gene that results in the expression of a KRas protein with one or more point mutations as compared to a wild type KRas protein, a mutation in a KRAS gene that results in the expression of a KRas protein with at least one inserted amino acid as compared to a wild type KRas protein, a gene duplication that results in an increased level of KRas protein in a cell, or a mutation in a regulatory sequence (e.g., a promoter and/or enhancer) that results in an increased level of KRas protein in a cell); an alternative spliced version of a KRas mRNA that results in a KRas protein having a deletion
of at least one amino acid in the KRas protein as compared to the wild type KRas protein; or increased expression (e.g., increased levels) of a wild type KRas protein in a mammalian cell due to aberrant cell signaling and/or dysregulated autocrine/paracrine signaling (e.g., as compared to a control non-cancerous cell). As an example, a dysregulation of a KRAS gene, a KRas protein, or expression or activity, or level of any of the same, can be a mutation in a KRAS gene that encodes a KRas protein that has low GTPase activity and/or has increased signaling activity as compared to a protein encoded by a KRAS gene that does not include the mutation. As another example, a dysregulation of a KRAS gene, a KRas protein, or expression or activity, or level of any of the same, can be a KRas amplification. In some embodiments, a KRas amplification is an amplification of the wild type KRas. In some embodiments, a KRas amplification is an amplification of a mutant KRas. A “dysregulated KRas protein” as used herein refers to (i) a KRas protein having a mutation (e.g., a deletion of at least one amino acid as compared to a wild type KRas protein, one or more point mutations as compared to a wild type KRas protein, or an insertion of at least one amino acid as compared to a wild type KRas protein); (ii) a KRas protein resulting from a gene duplication event, e.g., of the gene encoding the KRas protein (e.g., the wild type KRas protein), thus resulting in an increased level and/or activity of the KRas protein (e.g., the wild type KRas protein) in a cell; (iii) a KRas protein resulting from a mutation in a regulatory sequence (e.g., a promoter and/or enhancer) that can also result in an increased level and/or activity of the KRas protein (e.g., the wild type KRas protein) in a cell); (iv) a KRas protein resulting from an alternative spliced version of a KRas mRNA that results in a KRas protein having a deletion of at least one amino acid in the KRas protein as compared to the wild type KRas protein); or (v) a KRas protein resulting from increased expression (e.g., increased levels) of a wild type KRas protein in a mammalian cell due to aberrant cell signaling and/or dysregulated autocrine/paracrine signaling (e.g., as compared to a control non-cancerous cell). In some embodiments, a dysregulated KRas protein is a dysregulated human KRas protein. A “mutant KRas protein” as used herein refers to a KRas protein including a substitution, an insertion, a deletion, a truncation and/or a fusion relative to the wild type human KRas sequence shown in SEQ ID NO:1. For example, a mutant human KRas protein includes a substitution at any amino acid position (relative to SEQ ID NO: 1). A “KRas G12X mutant protein” as used herein refers to a KRas protein including substitution of a glycine to any other amino acid at the twelfth amino acid position (relative to SEQ ID NO: 1).
A “KRas G12A mutant protein” as used herein refers to a KRas protein including a glycine to alanine substitution at the twelfth amino acid position (relative to SEQ ID NO: 1). A “KRas G12C mutant protein” as used herein refers to a KRas protein including a glycine to cysteine substitution at the twelfth amino acid position (relative to SEQ ID NO: 1). A “KRas G12D mutant protein” as used herein refers to a KRas protein including a glycine to aspartic acid substitution at the twelfth amino acid position (relative to SEQ ID NO: 1). A “KRas G12R mutant protein” as used herein refers to a KRas protein including a glycine to arginine substitution at the twelfth amino acid position (relative to SEQ ID NO: 1). A “KRas G12S mutant protein” as used herein refers to a KRas protein including a glycine to serine substitution at the twelfth amino acid position (relative to SEQ ID NO: 1). A “KRas G12V mutant protein” as used herein refers to a KRas protein including a glycine to valine substitution at the twelfth amino acid position (relative to SEQ ID NO: 1). A “KRas G13X mutant protein” as used herein refers to a KRas protein including substitution of a glycine to any other amino acid at the thirteenth amino acid position (relative to SEQ ID NO: 1). A “KRas G13C mutant protein” as used herein refers to a KRas protein including a glycine to cysteine substitution at the thirteenth amino acid position (relative to SEQ ID NO: 1). A “KRas G13D mutant protein” as used herein refers to a KRas protein including a glycine to aspartic acid substitution at the thirteenth amino acid position (relative to SEQ ID NO: 1). A “KRas G13V mutant protein” as used herein refers to a KRas protein including a glycine to valine substitution at the thirteenth amino acid position (relative to SEQ ID NO: 1). A “KRas Q61X mutant protein” as used herein refers to a KRas protein including substitution of a glutamine to any other amino acid at the sixty-first amino acid position (relative to SEQ ID NO: 1). A “KRas Q61E mutant protein” as used herein refers to a KRas protein including a glutamine to glutamic acid substitution at the sixty-first amino acid position (relative to SEQ ID NO: 1). A “KRas Q61H mutant protein” as used herein refers to a KRas protein including a glutamine to histidine substitution at the sixty-first amino acid position (relative to SEQ ID NO: 1).
A “KRas Q61K mutant protein” as used herein refers to a KRas protein including a glutamine to lysine substitution at the sixty-first amino acid position (relative to SEQ ID NO: 1). A “KRas Q61L mutant protein” as used herein refers to a KRas protein including a glutamine to leucine substitution at the sixty-first amino acid position (relative to SEQ ID NO: 1). A “KRas Q61P mutant protein” as used herein refers to a KRas protein including a glutamine to proline substitution at the sixty-first amino acid position (relative to SEQ ID NO: 1). A “KRas Q61R mutant protein” as used herein refers to a KRas protein including a glutamine to arginine substitution at the sixty-first amino acid position (relative to SEQ ID NO: 1). A “KRas inhibitor” as used herein includes any compound exhibiting KRas protein inactivation activity (e.g., inhibiting or decreasing KRas signaling activity). In some embodiments, a KRas inhibitor as described herein has an IC50 value of 1 µM or less in a nucleotide exchange assay as described herein, an IC50 value of 1 µM or less in a Raf kinase interaction assay as described herein, or both. In some embodiments, a KRas inhibitor inhibits the signaling activity of a wild type KRas protein. In some embodiments, a KRas inhibitor inhibits the signaling activity of a dysregulated KRas protein, for example, resulting in a decrease in activated Raf or other downstream effectors, such as ERK. In some embodiments, a KRas inhibitor inhibits the signaling activity of a mutant KRas protein. In some embodiments, a KRas inhibitor inhibits both the signaling activity of a wild-type KRas protein and the signaling activity of one or more mutant KRas proteins and can be termed a “pan KRas inhibitor”. In some embodiments, a KRas inhibitor inhibits one or more mutant KRas proteins, and such a KRas inhibitor can be termed a “mutant KRas inhibitor”, and also termed by the mutant(s) it inhibits. For example, a KRas inhibitor that inhibits KRas G12R mutant protein could be termed a “KRas G12R inhibitor”. As another example, a KRas inhibitor that inhibits both KRas G12C mutant protein and KRas G12D mutant protein could be termed a “KRas G12C inhibitor” and/or a “KRas G12D inhibitor”. In some embodiments, a “mutant KRas inhibitor” inhibits two or more mutant KRas proteins and can be termed a “pan mutant KRas inhibitor”. In some embodiments, a pan mutant KRas inhibitor inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas
G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. For example, a “KRas G12X inhibitor” can inhibit two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. As yet another example, a KRas inhibitor that inhibits a KRas G13D mutant protein could be termed a “KRas G13D inhibitor”. In some embodiments, a KRas inhibitor can inhibit a KRas protein having one or more mutations, and such a KRas inhibitor can be termed a “mutant KRas inhibitor” whether or not the mutant KRas inhibitor also inhibits wild type KRas protein. In some embodiments, a KRas inhibitor is a mutant KRas inhibitor. In some embodiments, a KRas inhibitor is an allosteric inhibitor. Compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I- a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, are KRas inhibitors. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a mutant KRas inhibitor. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas
proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12V mutant protein, or a combination thereof. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12D mutant protein, a KRas G12V mutant protein, or both. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12R mutant protein, a KRas G12V mutant protein, or both. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1),
(V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12D mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12X inhibitor. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g.,
Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits four or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a),
(VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits five or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or
(II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits four or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12A mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12D mutant protein, a KRAS G12V mutant protein, or both. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4),
(I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12A mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12C mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, does not inhibit a KRas G12C mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12D mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, does not inhibit a KRas G12D mutant protein. In
some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12S mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G13X inhibitor. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula
(VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G13C mutant protein, a KRas G13D mutant protein, and a KRas G13V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G13C mutant protein, a KRas G13D mutant protein, and a KRas G13V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G13C mutant protein, a KRas G13D mutant protein, and a KRas G13V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G13C mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G13D mutant protein.
In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G13V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas Q61X inhibitor. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas mutant protein. In some embodiments, a compound of Formula
(A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits four or more mutant KRas proteins selected from the group consisting of: a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits five or more mutant KRas proteins selected from the group consisting of: a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI)
(e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas Q61E mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas Q61H mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas Q61K mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas Q61L mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula
(II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas Q61P mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12X mutant protein, a KRas G13X mutant protein, and a KRas Q61X mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12X mutant protein, a KRas G13X mutant protein, and a KRas Q61X mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-
b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant human KRas proteins selected from the group consisting of: a KRas G12X mutant protein, a KRas G13X mutant protein, and a KRas Q61X mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12X mutant protein, a KRas G13X mutant protein, and a KRas Q61X mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas
G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits four or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-
c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, five or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12A mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II)
(e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12V mutant protein, a KRas G13D mutant protein, and a KRas Q61H mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12V mutant protein, a KRas G13D mutant protein, and a KRas Q61H mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12V mutant protein, a KRas G13D mutant protein, and a KRas Q61H mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one
or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12D mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein. In some embodiments, compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c),
(V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12D mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12D mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein. In some such embodiments, the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, are useful for treating a bladder cancer. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12V mutant protein, and a KRas G13D mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or
(V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12V mutant protein, and a KRas G13D mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12V mutant protein, and a KRas G13D mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-
b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12C mutant protein, a KRas G12D mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12D mutant protein, a KRas G12V mutant protein, or both. In some such embodiments, the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, are useful for treating a cervical cancer. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-
b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt
thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein. In some such embodiments, the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, are useful for treating a colorectal cancer. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))),
or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas Q61H mutant protein, and a KRas Q61L mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas Q61H mutant protein, and a KRas Q61L mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas Q61H mutant protein, and a KRas Q61L mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one
or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12D mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2),
or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12D mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12A mutant protein, a KRas G12D mutant protein, and a KRas G12V mutant protein. In some such embodiments, the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, are useful for treating an endometrial cancer. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, and a KRas Q61H mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-
2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, and a KRas Q61H mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, and a KRas Q61H mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D
mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12D mutant protein, a KRas G12V mutant protein, or both. In some such embodiments, the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or pharmaceutically acceptable salts thereof, are useful for treating an esophageal or stomach cancer. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant
protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas Q61E mutant protein, a KRas Q61H mutant protein, a KRas Q61K mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1),
(IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant
protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein. In some such embodiments, the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, are useful for treating a leukemia. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant
protein, a KRas G12R mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas a KRas Q61K mutant protein, a KRas Q61L mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas a KRas Q61K mutant protein, a KRas Q61L mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G13D mutant protein, a KRas G13V mutant protein, a KRas a KRas Q61K mutant protein, a KRas Q61L mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, and a KRas G12R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula
(II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12C mutant protein, a KRas G12D mutant protein, and a KRas G12R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12C mutant protein, a KRas G12D mutant protein, and a KRas G12R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12D mutant protein, and a KRas G12R mutant protein, or both. In some such embodiments, the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, are useful for treating a melanoma. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-
a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas Q61H mutant protein, and a KRas Q61L mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas Q61H mutant protein, and a KRas Q61L mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas G13D mutant protein, a KRas Q61H mutant protein, and a KRas Q61L mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-
d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12D mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g.,
Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12D mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12A mutant protein, a KRas G12D mutant protein, and a KRas G12V mutant protein. In some such embodiments, the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, are useful for treating a lung cancer (e.g., non-small cell lung cancer). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas Q61H mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or
(II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas Q61H mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas G13C mutant protein, a KRas Q61H mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula
(VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula
(II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein. In some such embodiments, the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, are useful for treating a pancreatic cancer. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant
protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, a KRas G12V mutant protein, a KRas Q61L mutant protein, a KRas Q61P mutant protein, and a KRas Q61R mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula
(VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits three or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits one or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits two or more mutant KRas proteins selected from the group consisting of: a KRas G12A mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12A mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein. In some such embodiments, the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d),
(V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, are useful for treating a testicular cancer (e.g., seminoma). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can bind to a KRas protein in the GTP-bound state. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can bind selectively to a KRas protein in the GTP-bound state. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can bind to a KRas protein in the GDP-bound state. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can bind selectively to a KRas protein in the GDP-bound state.
An exemplary sequence of mature human KRas protein is shown below (UniProtKB entry P01116) (SEQ ID NO: 1) MTEYKLVVVG AGGVGKSALT IQLIQNHFVD EYDPTIEDSY RKQVVIDGET CLLDILDTAG QEEYSAMRDQ YMRTGEGFLC VFAINNTKSF EDIHHYREQI KRVKDSEDVP MVLVGNKCDL PSRTVDTKQA QDLARSYGIP FIETSAKTRQ RVEDAFYTLV REIRQYRLKK ISKEEKTPGC VKIKKCIIM As used herein, “selective” or “selectively”, when referring to an assayed compound, indicates at least a 5-fold (e.g., at least a 10-fold, at least a 25-fold, at least a 50-fold, or at least a 100-fold) superior performance in an assay (e.g., binding affinity and/or potency) for a specified condition with reference to a comparator protein variant in the assay. For example, if a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, binds “selectively” to a KRas G12X mutant protein over the wild type KRas protein as determined by a surface plasmon resonance (SPR) assay, then the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, has at least a 5-fold (e.g., at least a 10-fold, at least a 25-fold, at least a 50-fold, or at least a 100-fold) smaller K
D value for any one or more KRas mutant proteins selected from the group consisting of the KRas G12X mutant proteins than for the wild type KRas protein when measured by the SPR assay. As a further example, if a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-
c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, “selectively” reduces the viability the KRas G12V mutant protein-expressing cells over the cells expressing KRas G12C protein as determined by a cell proliferation assay, then the compound has at least a 5-fold (e.g., at least a 10-fold, at least a 25-fold, at least a 50-fold, or at least a 100-fold) EC50 value for the KRas G12V mutant protein-expressing cells than for the KRas G12C protein-expressing cells when measured by the cell proliferation assay. In another example, if a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, “selectively” inhibits a KRas G13X mutant protein over the wild type KRas protein as determined by a Raf kinase interaction assay, then the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, has at least a 5-fold (e.g., at least a 10-fold, at least a 25-fold, at least a 50-fold, or at least a 100-fold) smaller IC50 value for the KRas G13X protein than for the wild type KRas protein when measured by the Raf kinase interaction assay. As a further example, if a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, “selectively” inhibits the KRas G12R mutant protein over the wild type KRas protein as determined by a nucleotide exchange assay, then the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g.,
Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, has at least a 5-fold (e.g., at least a 10-fold, at least a 25-fold, at least a 50-fold, or at least a 100-fold) smaller IC50 value for the KRas G12R mutant protein than for the wild type KRas protein when measured by the nucleotide exchange assay. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a pan mutant KRas inhibitor (i.e., can inhibit two or more mutant KRas proteins (e.g., two or more of a KRas G12A mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, and a KRas G12V mutant protein)). For example, such a compound can inhibit each mutant KRas protein (e.g., two or more mutant KRas proteins) with an IC
50 of less than 1 µM (e.g., less than 750 nM, less than 500 nM, or less than 200 nM). As another example, such a compound can inhibit ERK phosphorylation in cell lines each expressing a mutant KRas protein with an independent IC
50 of less than 1 µM (e.g., less than 750 nM, less than 500 nM, or less than 200 nM) in at least of the two cell lines. For example, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can inhibit ERK phosphorylation in a cell line expressing a KRas G12R mutant protein with an IC50 of less than 1 µM, and the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g.,
Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can inhibit ERK phosphorylation in a cell line expressing a KRas G12V mutant protein with an IC
50 of less than 1 µM. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a pan KRas inhibitor (i.e., the compound can inhibit wild type KRas and one or more mutant KRas proteins). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, does not inhibit certain KRas proteins (e.g., wild type KRas or one or more dysregulated KRas proteins). For example, such a compound can inhibit the interaction between a KRas protein it does not inhibit (e.g., a dysregulated KRas protein) and one or more Raf proteins with an IC
50 of 1 µM or greater than 1 µM (e.g., greater than 2 µM, greater than 5 µM, greater than 10 µM, or greater than 30 µM). As another example, such a compound can inhibit ERK phosphorylation in cell lines expressing the KRas protein it does not inhibit (e.g., a dysregulated KRas protein) with an IC
50 of 1 µM or greater than 1 µM (e.g., greater than 2 µM, greater than 5 µM, greater than 10 µM, or greater than 30 µM). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits a KRas G12D mutant protein and a KRas
G12V mutant protein. In some such embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in a cell line expressing a KRas G12D mutant protein (e.g., AGS, ASPC1, GP2D, LS180, Panc04.03, HPAFII, Panc02.03, A427, and HPAC) with an IC50 that is within about 10-fold, i.e., within about 10-fold less or within about 10-fold more (e.g., within about 9-fold less or within about 9-fold more, within about 8-fold less or within about 8-fold more, within about 7-fold less or within about 7-fold more, within about 6-fold less or within about 6-fold more, within about 5-fold less or within about 5-fold more, or within about 2-fold less or within about 2-fold more) of the IC
50 measured for inhibition of ERK phosphorylation by the compound in a cell line expressing a KRas G12V mutant protein (e.g., SW620, H727, CFPAC1, CAPAN1, RKN, H441, and SW480). For example, if the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in a cell line expressing a KRas G12D mutant protein with an IC50 of about 150 nM, then the IC
50 measured for inhibition of ERK phosphorylation by the compound in a cell line expressing a KRas G12V mutant protein would be within about 10-fold more than about 150 nM, thus ranging from about 150 nM to about 1500 nM, or within about 10-fold less than 150 nM, thus ranging from about 15 nM to about 150 nM. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically
acceptable salt thereof, can inhibit ERK phosphorylation in a GP2d cell line with an IC
50 that is within about 10-fold, i.e., within about 10-fold less or within about 10-fold more (e.g., within about 9-fold less or within about 9-fold more, within about 8-fold less or within about 8-fold more, within about 7-fold less or within about 7-fold more, within about 6-fold less or within about 6-fold more, within about 5-fold less or within about 5-fold more, or within about 2-fold less or within about 2-fold more) of the IC50 measured for inhibition of ERK phosphorylation by the compound in a SW620 cell line. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in a cell line expressing a KRas G12D mutant protein (e.g., AGS, ASPC1, GP2D, LS180, Panc04.03, HPAFII, Panc02.03, A427, and HPAC) with an IC50 that is within about 10-fold less (e.g., within about 9-fold less, within about 8-fold less, within about 7-fold less, within about 6-fold less, within about 5-fold less, or within about 2-fold less) than the IC
50 measured for inhibition of ERK phosphorylation by the compound in a cell line expressing a KRas G12V mutant protein (e.g., SW620, H727, CFPAC1, CAPAN1, RKN, H441, and SW480). For example, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can inhibit ERK phosphorylation in a GP2d cell line with an IC
50 that within about 10-fold less (e.g., within about 9-fold less, within about 8-fold less, within about 7-fold less, within about 6-fold less, within about 5-fold less, or within about 2-fold less) than the IC50 measured for inhibition of ERK phosphorylation by the compound in a SW620 cell line. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-
b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in a cell line expressing a KRas G12D mutant protein (e.g., AGS, ASPC1, GP2D, LS180, Panc04.03, HPAFII, Panc02.03, A427, and HPAC ) with an IC50 that is within about 10-fold more (e.g., within about 9-fold more, within about 8-fold more, within about 7- fold more, within about 6-fold more, within about 5-fold more, or within about 2-fold more) than the IC50 measured for inhibition of ERK phosphorylation by the compound in a cell line expressing a KRas G12V mutant protein (e.g., SW620, H727, CFPAC1, CAPAN1, RKN, H441, and SW480). For example, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can inhibit ERK phosphorylation in a GP2d cell line with an IC
50 that is within about 10-fold more (e.g., within about 9-fold more, within about 8-fold more, within about 7-fold more, within about 6-fold more, within about 5- fold more, or within about 2-fold more) than the IC50 measured for inhibition of ERK phosphorylation by the compound in a SW620 cell line. In some such embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in a cell line expressing a KRas G12V mutant protein (e.g., SW620, H727, CFPAC1, CAPAN1, RKN, H441, and SW480) with an IC50 of less than 250 nM (e.g., less than 200 nM, less than 150 nM, less than 125 nM, less than 100 nM, less than 75 nM, less than 50 nM, less than 30 nM). For example, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-
1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can inhibit ERK phosphorylation in a SW620 cell line with an IC50 of less than 250 nM (e.g., less than 200 nM, less than 150 nM, less than 125 nM, less than 100 nM, less than 75 nM, less than 50 nM, less than 30 nM). In some such further embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in a cell line expressing a KRas G12D mutant protein (e.g., AGS, ASPC1, GP2D, LS180, Panc04.03, HPAFII, Panc02.03, A427, and HPAC) with an IC50 of less than 250 nM (e.g., less than 200 nM, less than 150 nM, less than 125 nM, less than 100 nM, less than 75 nM, less than 50 nM, less than 30 nM). For example, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can inhibit ERK phosphorylation in a GP2d cell line with an IC
50 of less than 250 nM (e.g., less than 200 nM, less than 150 nM, less than 125 nM, less than 100 nM, less than 75 nM, less than 50 nM, less than 30 nM). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in a cell line expressing a KRas G12D mutant protein (e.g., AGS, ASPC1, GP2D, LS180, Panc04.03,
HPAFII, Panc02.03, A427, and HPAC) with an IC
50 that is within about 10-fold, i.e., within about 10-fold less or within about 10-fold more (e.g., within about 9-fold less or within about 9-fold more, within about 8-fold less or within about 8-fold more, within about 7-fold less or within about 7-fold more, within about 6-fold less or within about 6-fold more, within about 5- fold less or within about 5-fold more, or within about 2-fold less or within about 2-fold more) of the IC50 measured for inhibition of ERK phosphorylation by the compound in a cell line expressing a KRas G12V mutant protein (e.g., SW620, H727, CFPAC1, CAPAN1, RKN, H441, and SW480), wherein the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in the cell line expressing a KRas G12V mutant protein (e.g., SW620, H727, CFPAC1, CAPAN1, RKN, H441, and SW480) with an IC50 of less than 250 nM (e.g., less than 200 nM, less than 150 nM, less than 125 nM, less than 100 nM, less than 75 nM, less than 50 nM, less than 30 nM). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in the cell line expressing a KRas G12D mutant protein (e.g., AGS, ASPC1, GP2D, LS180, Panc04.03, HPAFII, Panc02.03, A427, and HPAC) with an IC50 of less than 250 nM (e.g., less than 200 nM, less than 150 nM, less than 125 nM, less than 100 nM, less than 75 nM, less than 50 nM, less than 30 nM). For example, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-
a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can inhibit ERK phosphorylation in a GP2d cell line with an IC50 that is within about 10-fold, i.e., within about 10-fold less or within about 10-fold more (e.g., within about 9-fold less or within about 9-fold more, within about 8-fold less or within about 8-fold more, within about 7-fold less or within about 7-fold more, within about 6- fold less or within about 6-fold more, within about 5-fold less or within about 5-fold more, or within about 2-fold less or within about 2-fold more) of the IC
50 measured for inhibition of ERK phosphorylation by the compound in a SW620 cell line, wherein the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in a SW620 cell line with an IC50 of less than 250 nM (e.g., less than 200 nM, less than 150 nM, less than 125 nM, less than 100 nM, less than 75 nM, less than 50 nM, less than 30 nM). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in a GP2d cell line with an IC
50 of less than 250 nM (e.g., less than 200 nM, less than 150 nM, less than 125 nM, less than 100 nM, less than 75 nM, less than 50 nM, less than 30 nM). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in a cell line expressing a KRas G12D mutant protein (e.g., AGS, ASPC1, GP2D, LS180,
Panc04.03, HPAFII, Panc02.03, A427, and HPAC) with an IC
50 that is within about 10-fold less (e.g., within about 9-fold less, within about 8-fold less, within about 7-fold less, within about 6-fold less, within about 5-fold less, or within about 2-fold less) than the IC50 measured for inhibition of ERK phosphorylation by the compound in a cell line expressing a KRas G12V mutant protein (e.g., SW620, H727, CFPAC1, CAPAN1, RKN, H441, and SW480), wherein the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in the cell line expressing a KRas G12V mutant protein (e.g., SW620, H727, CFPAC1, CAPAN1, RKN, H441, and SW480) with an IC
50 of less than 250 nM (e.g., less than 200 nM, less than 150 nM, less than 125 nM, less than 100 nM, less than 75 nM, less than 50 nM, less than 30 nM). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in the cell line expressing a KRas G12D mutant protein (e.g., AGS, ASPC1, GP2D, LS180, Panc04.03, HPAFII, Panc02.03, A427, and HPAC) with an IC
50 of less than 250 nM (e.g., less than 200 nM, less than 150 nM, less than 125 nM, less than 100 nM, less than 75 nM, less than 50 nM, less than 30 nM). For example, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can inhibit ERK phosphorylation in a GP2d cell line with an IC50 that is within about 10-fold less (e.g., within about 9-fold less, within about 8-fold
less, within about 7-fold less, within about 6-fold less, within about 5-fold less, or within about 2-fold less) than the IC50 measured for inhibition of ERK phosphorylation by the compound in a SW620 cell line, wherein the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in a SW620 cell line with an IC
50 of less than 250 nM (e.g., less than 200 nM, less than 150 nM, less than 125 nM, less than 100 nM, less than 75 nM, less than 50 nM, less than 30 nM). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in a GP2d cell line with an IC50 of less than 250 nM (e.g., less than 200 nM, less than 150 nM, less than 125 nM, less than 100 nM, less than 75 nM, less than 50 nM, less than 30 nM). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in a cell line expressing a KRas G12D mutant protein (e.g., AGS, ASPC1, GP2D, LS180, Panc04.03, HPAFII, Panc02.03, A427, and HPAC) with an IC50 that is within about 10-fold more (e.g., within about 9-fold more, within about 8-fold more, within about 7-fold more, within about 6- fold more, within about 5-fold more, or within about 2-fold more) than the IC
50 measured for inhibition of ERK phosphorylation by the compound in a cell line expressing a KRas G12V mutant protein (e.g., SW620, H727, CFPAC1, CAPAN1, RKN, H441, and SW480), wherein
the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in the cell line expressing a KRas G12V mutant protein (e.g., SW620, H727, CFPAC1, CAPAN1, RKN, H441, and SW480) with an IC50 of less than 250 nM (e.g., less than 200 nM, less than 150 nM, less than 125 nM, less than 100 nM, less than 75 nM, less than 50 nM, less than 30 nM). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in the cell line expressing a KRas G12D mutant protein (e.g., AGS, ASPC1, GP2D, LS180, Panc04.03, HPAFII, Panc02.03, A427, and HPAC) with an IC50 of less than 250 nM (e.g., less than 200 nM, less than 150 nM, less than 125 nM, less than 100 nM, less than 75 nM, less than 50 nM, less than 30 nM). For example, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can inhibit ERK phosphorylation in a GP2d cell line with an IC
50 that is within about 10-fold more (e.g., within about 9-fold more, within about 8- fold more, within about 7-fold more, within about 6-fold more, within about 5-fold more, or within about 2-fold more) than the IC50 measured for inhibition of ERK phosphorylation by the compound in a SW620 cell line, wherein the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula
(III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in a SW620 cell line with an IC50 of less than 250 nM (e.g., less than 200 nM, less than 150 nM, less than 125 nM, less than 100 nM, less than 75 nM, less than 50 nM, less than 30 nM). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits ERK phosphorylation in a GP2d cell line with an IC
50 of less than 250 nM (e.g., less than 200 nM, less than 150 nM, less than 125 nM, less than 100 nM, less than 75 nM, less than 50 nM, less than 30 nM). The ability of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, to bind to a KRas protein can be measured, for example, by a direct determination method (e.g., surface plasmon resonance or isothermal titration calorimetry); by radio labelling the compound prior to binding, isolating the compound/protein complex, and determining the amount of radio label bound; or by running a competition experiment where new compounds are incubated with the protein bound to known radioligands. As another example, the occupancy of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c),
(VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can be determined using a proximity-based technique, such as time-resolved Fluorescence Resonance Energy Transfer (FRET); for instance, using a labeled probe that binds mutually exclusively with the inhibitor, and using an antibody that binds to a position on the protein separate from where the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, binds (for example, an antibody that binds to an N-terminal tag). It will be understood that the antibody and probe can be tagged with any appropriate FRET pair. See, e.g., International Publication Nos. WO 2021/041671, WO 2021/120890, and U.S. Publication No. US 2021/0179633. In some cases, binding affinities (e.g., as measured by dissociation constant KD) of the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof with a KRas protein (e.g., a wild type KRas protein or a mutant KRas protein) in the GDP-bound and/or GTP-bound state can be measured using methods known in the art (e.g., using SPR (e.g., using one or more methods described herein (e.g., using the methods described in Example B1 or in Example B5 herein))). Binding affinity with the KRas protein in the GDP-bound state can be measured by loading the KRas protein with GDP (e.g., at the concentrations described in Example B1 or in Example B5). Binding affinity with the KRas protein in the GTP-bound state can be measured by loading the KRas protein with GMPPNP (e.g., at the concentrations described in Example B1). Another exemplary assay for determining the potency of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a),
(IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, includes measuring the effect of the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, on cell proliferation. Cell proliferation assays can be performed in a number of formats, including 2D and 3D. Similarly, a cell proliferation assay can be performed with any appropriate cell line, including, for example, PSN1, HUPT3, KP2, CAL62, TCCPAN2, PK8, PATC50, H358, MIAPaCa-2, CALU1, AGS, A427, ASPC1, HKA1, KMS20, A549, LS123, HTK, H727, H441, HCT116, H460, A375, NCI-H1993, PC9, MKN1, NCI-H211, NCI-H424, and/or NCI-H526. In some embodiments, the cell line can be AGS, A427, ASPC1, H727, and/or H441. As an illustrative example, a 3D cell proliferation assay can include growing cells in a 3D medium, contacting the cells with a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, measuring the cellular proliferation using an appropriate reagent (e.g., CELLTITERGLO® 3D), and then comparing the signal from the experiment with the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, to the signal from a control experiment (e.g., lacking a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-
b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)))). As another illustrative example, a 2D cell proliferation assay can include plating cells onto a growth surface, optionally letting the cells grow for a period of time, contacting the cells with a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, measuring the cellular proliferation using an appropriate reagent (e.g., CELLTITERGLO®), and then comparing the signal from the experiment with a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, to the signal from a control experiment (e.g., lacking a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof). See, e.g., Example B7 herein. In some embodiments, cellular proliferation can be assessed using a platform for live cell imaging (e.g., an INCUCYTE® SX5 Live-Cell Analysis Instrument). See also, e.g., U.S. Publication No. US 2021/0179633, US 2021/0230142, and US 2019/0284144. As another example, the potency and/or efficacy of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-
b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can be evaluated in an animal model, for example, a xenograft model (e.g., using an established cancer cell line such as H727, H441, AGS, A427 and/or ASPC1 or a patient-derived xenograft (PDX) model). See, e.g., U.S. Publication No. US 2021/0179633. Additional assays can include, for example, assays based on hydrogen exchange (HX) mass spectrometry. Such assays can be useful, for example, to evaluate whether a compound (e.g., a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I- a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof) stabilizes the GTP-bound state or GDP-bound state of a KRas protein (e.g., a dysregulated KRas protein, e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, or a KRas G12V mutant protein)). In such assays, the rate of hydrogen exchange of the backbone amide hydrogens can be measured for a KRas protein (e.g., a dysregulated KRas protein, e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, or a KRas G12V mutant protein)) bound to a non-hydrolyzable GTP mimic (GMPPNP), GDP, or a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof. See, e.g., Lim et al. Angew Chem Int Ed Engl.2014; 53(1): 199–204, doi: 10.1002/anie.201307387. In some embodiments, potency of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1),
(IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, as provided herein can be determined by EC
50 value. A compound with a lower EC
50 value, as determined under substantially similar conditions, is a more potent inhibitor relative to a compound with a higher EC50 value. In some embodiments, an EC50 value can be determined (e.g., using a KRas- dependent phosphorylation level (e.g., a phosphoERK level (sometimes called a “pERK” level)) or using a cell viability assay) in cells (e.g., in tumor cells, (e.g., cell lines such as PSN1, HUPT3, KP2, CAL62, TCCPAN2, PK8, PATC50, H358, MIAPaCa-2, CALU1 AGS, A427, ASPC1, HKA1, KMS20, A549, LS123, HTK, H727, HCT116, H460, A375, NCI-H1993, PC9, MKN1, NCI-H211, NCI-H424, and/or NCI-H526) expressing a KRas protein, such as a dysregulated KRas protein (e.g., a mutant KRas protein or an amplified KRas protein), or a fragment thereof). In some embodiments, potency of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, as provided herein can also be determined by IC
50 value. A compound with a lower IC
50 value, as determined under substantially similar conditions, is a more potent inhibitor relative to a compound with a higher IC50 value. In some embodiments, an IC50 value can be determined (e.g., using a KRas- dependent phosphorylation level (e.g., a phosphoERK level) or using a cell viability assay), in cells (e.g., in tumor cells, (e.g., cell lines such as PSN1, HUPT3, KP2, CAL62, TCCPAN2, PK8, PATC50, H358, MIAPaCa-2, CALU1 AGS, A427, ASPC1, HKA1, KMS20, A549, LS123, HTK, H727, HCT116, H460, A375, NCI-H1993, PC9, MKN1, NCI-H211, NCI-H424, and/or NCI-H526) expressing a KRas protein, such as a dysregulated KRas protein (e.g., a mutant KRas protein or an amplified KRas protein), or a fragment thereof). In some embodiments, measuring the potency of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-
b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, includes measuring the phosphorylation of a downstream kinase, such as ERK (e.g., ERK1 and/or ERK2) or MEK. Such assays can be used to measure the inhibition of KRas signaling activity, for instance, in a cell line (e.g., PSN1, HUPT3, KP2, CAL62, TCCPAN2, PK8, PATC50, H358, MIAPaCa-2, CALU1 AGS, A427, ASPC1, HKA1, KMS20, A549, LS123, HTK, H727, H441, HCT116, H460, A375, NCI-H1993, PC9, MKN1, NCI-H211, NCI-H424, and/or NCI- H526 (e.g., AGS, A427, ASPC1, H727, and/or H441)). For example, cells can be contacted with a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II- b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof for a period of time, then lysed or permeabilized, and total ERK or MEK and phosphoERK or phosphoMEK content can be determined (e.g., using antibodies, or a kit, such as Invitrogen InstantOne ERK1/ERK2 (Phospho) [pT202/pY204]/[pT185/pY187] ELISA, MesoScale Discovery p/t ERK1/2, AlphaScreen SUREFIRE® p-ERK1/2 (Thr202/Tyr204), or an HTRF® Phospho-ERK (Thr202/Tyr204) cellular kit (CisBio)). In some embodiments, multiple concentrations of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof can be used to construct a dose response curve. See, e.g., Example B6 herein. See, e.g., International Publication No. WO 2021/041671, U.S. Publication Nos. US 2021/0122764, US 2018/0334454, US 2021/0179633, US 2018/0334454, and US 2019/0144444. An exemplary ERK phosphorylation protocol follows. In some embodiments, an ERK phosphorylation assay can be carried out using the AlphaLisa SUREFIRE® Ultra Multiplex Phospho/Total ERK1/2 (Thr202/Tyr204) Assay Kit. In a plate (e.g., a white, opaque-
bottom Perkin Elmer CulturPlate-384 (product number 6007680)), cells are seeded at the desired concentration one day prior to treatment with compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, and incubated overnight in a standard 37 °C, 5% CO2 humidified incubator. The cells can be any cells of interest, such as MIAPACA2 (KRas G12C), H358 (KRas G12C), AGS (KRas G12D), ASPC1 (KRas G12D), GP2D (KRas G12D), LS180 (KRas G12D), Panc04.03 (KRas G12D), HPAFII (KRas G12D), Panc02.03 (KRas G12D), A427 (KRas G12D), HPAC (KRas G12D), TCCPAN2 (KRas G12R), PSN1 (KRas G12R), KP2 (KRas G12R), LS123 (KRas G12S), SW620 (KRas G12V), H727 (KRas G12V), CFPAC1 (KRas G12V), CAPAN1 (KRas G12V), RKN (KRas G12V), H441 (KRas G12V), SW480 (KRas G12V), PACADD159 (KRas G12V/G12S), HS766T (KRas Q61H), H460 (KRas Q61H), PANC0213 (KRas Q61R), or A3735 (KRas WT). The day after seeding, compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, are dispensed into the treatment plates (e.g., using a Tecan D300e compound printer in 9-point DRC format (1:3 dilution), 10- ^M top concentration, in triplicate). Treatment plates are then returned to a standard 37 °C, 5% CO
2 humidified incubator for the pre-determined treatment time. Following compound treatment, all media is removed from the treatment plate(s), and the cells are subsequently lysed (e.g., using 1X Lysis Buffer in accordance with manufacturer protocol). Next, the Acceptor Mix (prepared in accordance with manufacturer’s protocol) is added to each well of the assay plate and incubated on an orbital shaker at room temperature for 2 hours. Following incubation with the Acceptor Mix, the Donor Mix (prepared in accordance with manufacturer protocol) is added to each well of the assay plate, covered to protect from light, and incubated on an orbital shaker at room temperature overnight. Assay plates are read the following day (e.g., on a BMG
Labtech PHERAstar FSX microplate reader). Data are then analyzed by calculating the ratio of ERK1/2-phosphorylation relative to Total ERK1/2 for each individual well. 615 ^^ ^^ ^^ ^^ ^^ ^^ ^^ ^^ ( ^^ ^^ ^^ ^^1/2) ^^ ^^ ^^ ^^ ^^ ^^ ^^ ^^ ^^1/2 = 545 ^^ ^^ ^^ ^^ ^^ ^^ ^^ ^^ ( ^^ ^^ ^^ ^^ ^^ ^^ ^^ ^^1/2) The replicate ratios for each concentration are averaged and normalized to a DMSO control or other corresponding co-treatment before performing a variable slope (4-parameter), non-linear regression curve fit for each compound of interest. Data can be reported as IC50 values. In some embodiments, the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, inhibit ERK phosphorylation in a cell line expressing a KRas protein (e.g., a dysregulated KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, or a KRas G12V mutant protein))) with an IC50 of less than 1 µM (e.g., less than 750 nM, less than 500 nM, or less than 200 nM). In some embodiments, the compounds inhibit ERK phosphorylation in a cell line expressing the KRas protein (e.g., a dysregulated KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, or a KRas G12V mutant protein))) with an IC50 of less than 200 nM (e.g., less than 150 nM, less than 200 nM, less than 100 nM, less than 10 nM, less than 1 nM). For example, the compounds can inhibit ERK phosphorylation in a cell line expressing the KRas protein (e.g., a dysregulated KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, or a KRas G12V mutant protein))) with an IC
50 of 0.1 nM to 100 nM, 0.1 nM to 50 nM, 1 nM to 50 nM, or 1 nM to 20 nM. In some cases, a KRas A59G mutant protein (e.g., as a single mutant or as a double mutant with another mutation of interest, e.g., KRas G12X) can be used to “lock” the KRas protein in the GTP-bound state (e.g., by abrogating the GTPase activity of the protein); such an assay can be useful, for example, to determine the affinity of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a),
(IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof for the GTP-bound state and/or to determine the effect of the compound on downstream signaling (e.g., interaction with an RBD and/or the phosphorylation of a downstream kinase, such as ERK), potentially independent of the GTP cycling of the KRas protein. See, e.g., Hall, et al. Proceedings of the National Academy of Sciences 99.19 (2002): 12138-12142, doi: 10.1073/pnas.192453199; Lu, et al. Biochemistry 57.3 (2018): 324-333, doi: 10.1021/acs.biochem.7b00974; and Lim, Shuhui, et al. Chemical Science 12.48 (2021): 15975- 15987, doi: 10.1039/D1SC05187C. In some embodiments, the potency of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, as a KRas inhibitor can be evaluated by its effect on the nucleotide exchange of GDP for GTP. For example, nucleotide exchange can be measured via the increase in fluorescence of protein-bound N- methylanthraniloyl (MANT)-GDP upon the addition of an excess amount of a non- hydrolyzable GTP analog such as guanosine-5'-[(β,γ)-imido]triphosphate (GppNHp, sometimes also referred to as GMPPNP), when exchange is inhibited. See, e.g., Kanie and Jackson, Bio Protoc. 2018; 8(7): e2795, doi: 10.21769/BioProtoc.2795. As another example, nucleotide exchange can be measured via the decrease in fluorescence of an incubated mixture of KRas protein-bound fluorophore-tagged GDP (e.g., Bodipy-GDP (e.g., EDA-GTP-DY- 647P1)) and a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, followed by treatment with unlabeled GTP. In such an assay, an exchange of fluorophore-tagged GDP (e.g., Bodipy-GDP) for unlabeled GTP
results in a reduced TR-FRET signal. As another example, nucleotide exchange can be measured via the increase in fluorescence of an incubated mixture of KRas protein-bound GDP and a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II- b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, followed by treatment with labeled GTP. In such an assay, an exchange of GDP for labeled GTP results in an increased FRET signal. See, e.g., International Publication No. WO 2020/085493 and U.S. Publication Nos. US 2021/0122764, US 2021/0269434, and US 2018/0334454. In some embodiments of nucleotide exchange assays, a guanine nucleotide exchange factor (e.g., SOS1) can be added to accelerate nucleotide exchange. Inhibition of SOS1-catalyzed exchange of GDP for GTP on the KRas protein by compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof can be measured using methods known in the art (e.g., using one or more methods described herein (e.g., using methods described in Example B2 herein)). Additional examples of in vitro assays include assays that determine inhibition of the GTPase activity of KRas protein. In some embodiments, the potency of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can be evaluated by its effect on GTPase activity (or lack thereof, as a decrease in GTPase activity is generally believed to be associated with aberrant signaling). For example, GTPase activity of a KRas protein can be
measured using a phosphate assay system that continuously measures phosphate release. In some embodiments, a purine nucleoside phosphorylase-based (PNP) assay can be used to measure GTPase activity of a KRas protein. See, e.g., Hunter et al. Mol Cancer Res. 2015; 13(9):1325-35, doi: 10.1158/1541-7786.MCR-15-0203. In some embodiments, an enzyme- linked immunosorbent assay (ELISA) can be used to measure the effect of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, on the GTPase activity of a KRas protein (e.g., a dysregulated KRas protein, e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, or a KRas G12V mutant protein)), for example, by detecting a change in the amount of GST-Ras- RBD that binds to the KRas protein following pull-down and antibody detection of the complex. See, e.g., US 2021/0179633. An exemplary SOS1-catalyzed nucleotide exchange assay protocol follows. GST-KRas G12R (1-169) loaded with GDP nucleotide is mixed with Anti-GST (Cisbio) antibody in assay buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 0.005% NP40, 1% DMSO) to produce a 1.5x solution.10µL of the 1.5x KRas-Ab solution is added to wells of a black, low-volume 384-well assay plate. Compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or pharmaceutically acceptable salts thereof, are added to wells using acoustic transfer technology. A 10-point dose response of each compound is performed with a 30 µM top dose. The KRas/Ab-compound mixture is incubated 1 hour at room temperature. A 3x solution of SOS1 (564-1049) and EDA-GTP-DY-647P1 (Jena Bioscience) is prepared in assay buffer.5 µL of the SOS1-labeled GTP solution is added to the wells to initiate the nucleotide exchange reaction. The final concentration of KRas G12R and SOS1 are 10 nM and 200 nM, respectively. Time resolved fluorescence is read on a PHERAstar plate reader equipped with a
filter module with excitation = 337 nm and emission 1 = 620 nm, emission 2 = 665 nm. The HTRF signal is calculated as the ratio of fluorescence intensity [emission 665 nm]/[emission 620 nm]. IC50 values are calculated using a four-parameter, variable response sigmoidal dose response curve fit in Graphpad Prism software. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits SOS1-catalyzed exchange of GDP for GTP on the KRas protein with an IC50 of less than 1 µM (e.g., less than 750 nM, less than 500 nM, or less than 200 nM). In some embodiments, the compounds inhibit SOS1-catalyzed exchange of GDP for GTP on the KRas protein with an IC
50 of less than 200 nM (e.g., less than 150 nM, less than 200 nM, less than 100 nM, less than 10 nM, less than 1 nM, less than 0.1 nM, or less than 0.01 nM). For example, the compounds can inhibit SOS1-catalyzed exchange of GDP for GTP on the KRas protein with an IC
50 of 0.001 nM to 500 nM, 0.005 nM to 100 nM, 0.025 nM to 100 nM, 0.1 nM to 50 nM, or 0.1 nM to 10 nM. Additional assays for evaluating the potency of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can also include, for example, a RAF kinase interaction assay. Such assays can be used to measure the affinity of KRas:nucleotide complexes for the Ras Binding Domain (RBD) of a RAF protein kinase (e.g., as impacted by a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))),
or a pharmaceutically acceptable salt thereof). For example, FLAG tagged KRas protein can be preloaded with the GTP analogue GppNHp and then incubated with biotinylated Raf-RBD to form complexes. A competition assay can then be performed by adding untagged KRas protein preloaded with GppNHp, which had been preloaded with various test molecules, over a range of concentrations. The proximity-dependent signal after addition of streptavidin donor and anti-flag acceptor beads (e.g., ALPHASCREEN® beads) can be measured to determine the affinity of the KRas protein for the Raf kinase. See, e.g., Hunter et al. Mol Cancer Res. 2015; 13(9):1325-35, doi: 10.1158/1541-7786.MCR-15-0203; Lim et al. Angew Chem Int Ed Engl. 2014; 53(1): 199–204, doi: 10.1002/anie.201307387; and Durrant, et al. Molecular Cancer Therapeutics 20.9 (2021): 1743-1754, doi: 10.1158/1535-7163.MCT-21-0175. As another example, for compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, that may bind selectively to the GTP-state, His- tagged KRas protein can be preloaded with the GTP analogue GppNHp and then incubated with a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II- b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, to form complexes. A competition assay can then be performed by adding Raf-RBD. The proximity-dependent signal after addition of Alpha detection reagents, compared to the signal from the same experiment using GDP instead of GppNHP, can be used to determine an IC
50 value. See, e.g., International Publication No. WO 2021/085653. It will be understood that in many cases, tagging technologies (e.g., FLAG tag, His tag, biotinylation) may be altered in an assay by one of skill in the art. In some embodiments, a RAF kinase interaction assay can be coupled with a nucleotide exchange assay; for example, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula
(II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can be incubated with a KRas protein (e.g., a dysregulated KRas protein, e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, or a KRas G12V mutant protein)) and GDP, then GTP (and optionally, a GEF such as SOS1) can be introduced. Then, RAF (e.g., cRAF) acceptor beads (e.g., GST-tagged acceptor beads) can be incubated with the KRas mixture, followed by introduction of donor beads (e.g., glutathione donor beads) and measurement using ALPHASCREEN® technology. As an alternative to ALPHASCREEN® technology, any appropriate FRET pair can be used to perform homogenous time resolved fluorescence. See, e.g., U.S. Publication Nos. US 2018/0334454 and US 2021/0230142. Another exemplary assay to measure the affinity of KRas:nucleotide complex for a RBD is to incubate cells with a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, lyse the cells, then pull down non-RBD-bound KRas using an immobilized RBD. See, e.g., U.S. Publication No. US 2019/0233440. As another example, the effect of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I- a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, on the interaction between KRas and Raf-RBD can be evaluated using HiBiT and/or NANOBIT™ technology, wherein two parts of an enzyme are fused to or inserted into two proteins of interest (e.g., KRas and Raf-RBD); when the two proteins of interest are in proximity, the two parts of the enzyme complement each other to complete an enzyme that has signaling activity (e.g., that produces luminescence). In some
such assays, the affinity of the two parts of the enzyme can be tuned, for example, to reduce or eliminate signal based on proximity driven by the two parts of the enzyme. See, e.g., Schwinn, et al. ACS Chemical Biology 13.2 (2018): 467-474, doi: 10.1021/acschembio.7b00549. Similarly, the effect of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, on the interaction between KRas and Raf-RBD can be evaluated using NANOBRET™ technology, wherein two parts of signaling system (e.g., a protein and a ligand) are fused to or inserted into two proteins of interest (e.g., KRas and Raf- RBD); when the two proteins of interest are in proximity, the two parts of the signaling system have signaling activity (e.g., producing fluorescence). See, e.g., Durrant, et al. Molecular Cancer Therapeutics 20.9 (2021): 1743-1754, doi: 10.1158/1535-7163.MCT-21-0175. In some embodiments, a RAF kinase interaction assay can be used to determine if a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is selective for a KRas protein (e.g., a dysregulated KRas protein, e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, or a KRas G12V mutant protein)) in the GDP-bound state or the GTP-bound state. Inhibition of the interaction between the KRas protein and Raf-RBD by compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts
thereof, can be measured using methods known in the art (e.g., using one or more methods described herein (e.g., using methods described in Example B3 or Example B4 herein)). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, modulates the interaction between the KRas protein and one or more Raf proteins. In some embodiments, the compounds inhibit the interaction between the KRas protein and Raf-RBD with an IC
50 of less than 1 µM (e.g., less than 750 nM, less than 500 nM, or less than 200 nM). In some embodiments, the compounds inhibit the interaction between the KRas protein and Raf-RBD with an IC50 of less than 200 nM (e.g., e.g., less than 150 nM, less than 200 nM, less than 100 nM, less than 10 nM, less than 1 nM, less than 0.1 nM, or less than 0.01 nM). For example, the compounds inhibit the interaction between the KRas protein and Raf-RBD with an IC50 from 0.001 nM to 500 nM, from 0.005 nM to 100 nM, from 0.025 nM to 100 nM, from 0.1 nM to 50 nM, or from 0.1 nM to 10 nM. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, inhibits the interaction between the KRas protein and Raf-RBD with an IC50 of less than 1 µM in the absence of cyclophilin A (e.g., less than 750 nM, less than 500 nM, or less than 200 nM). In some embodiments, the compounds inhibit the interaction between the KRas protein and Raf-RBD with an IC
50 of less than 200 nM in the absence of cyclophilin A (e.g., e.g., less than 150 nM, less than 200 nM, less than 100 nM, less than 10 nM, less than 1 nM, less than 0.1 nM, or less than 0.01 nM). For example, the compounds inhibit the interaction between the KRas protein and Raf-RBD with an IC
50 from 0.001 nM to 500 nM, from 0.005 nM to 100 nM, from 0.025 nM to 100 nM, from 0.1 nM to 50 nM, or from 0.1 nM to 10 nM in the absence of cyclophilin A.
Another exemplary assay for evaluating the potency of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, includes measuring the phosphorylation of a downstream kinase, such as ERK (e.g., ERK1 and/or ERK2) or MEK. Such assays can be used to measure the inhibition of KRas signaling activity, for instance, in a cell line (e.g., PSN1, HUPT3, KP2, CAL62, TCCPAN2, PK8, PATC50, H358, MIAPaCa-2, CALU1 AGS, A427, ASPC1, HKA1, KMS20, A549, LS123, HTK, H727, HCT116, H460, A375, NCI-H1993, PC9, MKN1, NCI-H211, NCI-H424, and/or NCI-H526). For example, cells can be contacted with a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, for a period of time, then lysed or permeabilized, and total ERK or MEK and phosphoERK or phosphoMEK content can be determined (e.g., using antibodies, or a kit, such as Invitrogen InstantOne ERK1/ERK2 (Phospho) [pT202/pY204]/[pT185/pY187] ELISA or MesoScale Discovery p/t ERK1/2). In some embodiments, multiple concentrations of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can be used to construct a dose response curve. In some embodiments, the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-
a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, inhibit ERK phosphorylation in a cell line expressing a KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, or a KRas G12V mutant protein)) with an IC50 of less than 1 µM (e.g., less than 750 nM, less than 500 nM, or less than 200 nM). In some embodiments, the compounds inhibit ERK phosphorylation in a cell line expressing a KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, or a KRas G12V mutant protein)) with an IC50 of less than 200 nM (e.g., less than 150 nM, less than 200 nM, less than 100 nM, less than 10 nM, less than 1 nM). For example, the compounds can inhibit ERK phosphorylation in a cell line expressing a KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, or a KRas G12V mutant protein)) with an IC50 from 0.1 nM to 100 nM, from 0.1 nM to 50 nM, from 1 nM to 50 nM, or from 1 nM to 20 nM. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can selectively inhibit one or more mutant KRas proteins over wild type KRas protein. The selectivity between wild type KRas protein and a mutant KRas protein as described herein can be measured using cellular proliferation assays where cell proliferation is dependent on signaling activity. For example, HEK293 cells transfected with a suitable version of wild type KRas, or HEK293 cells transfected with KRas containing one or more mutations as described herein (e.g., a G12D mutation, a G12R mutation, or a G12V mutation) can be used. Proliferation assays are performed at a range of inhibitor concentrations (e.g., 10 µM, 3 µM, 1.1 µM, 330 nM, 110 nM, 33 nM, 11 nM, 3 nM, 1 nM) and an EC50 is calculated. See also the assays described in International Publication Nos. WO 2021/120890; WO 2021/041671; and U.S. Publication Nos. US 2021/0130369; US 2021/0179633; US 2018/0334454; and US 2021/0122764.
The pharmacokinetic parameters of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can be evaluated in an animal model, for instance, a mouse model, a rat model, a dog model, or a nonhuman primate (e.g., cynomolgus monkey) model. Pharmacokinetics parameters, including clearance (CL), volume of distribution (V
d), maximum plasma concentration (C
max), time of maximum plasma concentration (t
max), half-life (t
1/2), area under the curve (AUC), and oral bioavailability (%F) can be calculated using, e.g., a non-compartmental model. In some embodiments, a reference compound (e.g., a first KRas inhibitor (e.g., MRTX1133)) may be used as a comparator. See, e.g., Example 3 (“Pharmacokinetic experiments in mice”) of International Publication No. WO 2023/098425. Certain pharmacokinetic parameters of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can be evaluated in hepatocytes, such as in mouse, rat, dog, nonhuman primate (e.g., cynomolgus monkey), or human hepatocytes. Pharmacokinetics parameters, including clearance (CL) and half-life (t
1/2), can be calculated. In some embodiments, a reference compound (e.g., a first KRas inhibitor (e.g., MRTX1133)) may be used as a comparator. See, e.g., Example VI (“Liver microsomal metabolically stability”) of International Publication No. WO 2023/284881. In some embodiments, the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or
(VI-e))), or pharmaceutically acceptable salts thereof, can exhibit potent and selective inhibition of a dysregulated KRas protein (e.g., a wild-type KRas protein and/or a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable salt thereof, can selectively inhibit a dysregulated KRas protein (e.g., a wild-type KRas protein and/or a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) over another GTPase or non-GTPase target. In some embodiments, the compounds provided herein can exhibit nanomolar potency against a KRas protein (e.g., a wild-type KRas protein and/or a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) with minimal activity against related GTPases (e.g., wild type NRas protein, and/or wild type HRas protein). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit greater inhibition of a KRas protein (e.g., a wild-type KRas protein and/or a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) relative to inhibition of a related GTPase (e.g., wild type NRas protein, and/or wild type HRas protein). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b),
(II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit at least 2-fold, 3-fold, 5-fold, 10-fold, 25-fold, 50-fold, or 100-fold greater inhibition of a KRas protein (e.g., a wild-type KRas protein and/or a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) relative to inhibition of a related GTPase (e.g., wild type NRas protein, and/or wild type HRas protein). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit up to 10000-fold greater inhibition of a KRas protein (e.g., a wild-type KRas protein and/or a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) relative to inhibition of a related GTPase (e.g., wild type NRas protein, and/or wild type HRas protein). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit from about 2-fold to about 10-fold greater inhibition of a KRas protein (e.g., a wild-type KRas protein and/or a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) relative to inhibition of a related GTPase (e.g., wild type NRas protein, and/or wild type HRas protein). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula
(I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit from about 10-fold to about 100-fold greater inhibition of a KRas protein (e.g., a wild-type KRas protein and/or a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) relative to inhibition of a related GTPase (e.g., wild type NRas protein, and/or wild type HRas protein). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit from about 100-fold to about 1000-fold greater inhibition of a KRas protein (e.g., a wild-type KRas protein and/or a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) relative inhibition of a related GTPase (e.g., wild type NRas protein, and/or wild type HRas protein). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit from about 1000-fold to about 10000-fold greater inhibition of a KRas protein (e.g., a wild-type KRas protein and/or a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) relative to inhibition of a related GTPase (e.g., wild type NRas protein, and/or wild type HRas protein).
In some embodiments, the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, can exhibit potent and selective inhibition of a dysregulated KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable salt thereof, can selectively inhibit a dysregulated KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) over another GTPase or non-GTPase target. In some embodiments, the compounds provided herein can exhibit nanomolar potency against a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) with minimal activity against related GTPases (e.g., wild type KRas protein, wild type NRas protein, and/or wild type HRas protein). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit greater inhibition of a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V
mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein)) relative to inhibition of wild type KRas protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit at least 2-fold, 3-fold, 5-fold, 10-fold, 25-fold, 50-fold, or 100-fold greater inhibition of a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein)) relative to inhibition of wild type KRas protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit up to 10000-fold greater inhibition of a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein)) relative to inhibition of wild type KRas protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit from about 2-fold to about 10-fold greater inhibition of a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein)) relative to inhibition of wild type KRas protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-
a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit from about 10-fold to about 100-fold greater inhibition of a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein)) relative to inhibition of wild type KRas protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit from about 100-fold to about 1000-fold greater inhibition of a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein)) relative to inhibition of wild type KRas protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit from about 1000-fold to about 10000-fold greater inhibition of a mutant KRas protein (e.ga KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein)) relative to inhibition of wild type KRas protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-
a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit nanomolar potency against a dysregulated KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) with minimal activity against wild type NRas protein and/or wild type HRas protein In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit greater inhibition of a dysregulated KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) relative to inhibition of wild type NRas protein and/or wild type HRas protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit at least 2-fold, 3-fold, 5-fold, 10-fold, 25-fold, 50-fold or 100-fold greater inhibition of a dysregulated KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) relative to inhibition of wild type NRas protein and/or wild type HRas protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI)
(e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit up to 1000-fold greater inhibition of a dysregulated KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) relative to inhibition of wild type NRas protein and/or wild type HRas protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit up to 10000-fold greater inhibition of a dysregulated KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) relative to inhibition of wild type NRas protein and/or wild type HRas protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit from about 2-fold to about 10-fold greater inhibition of a dysregulated KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) relative to inhibition of wild type NRas protein and/or wild type HRas protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt
thereof, can exhibit from about 10-fold to about 100-fold greater inhibition of a dysregulated KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) relative to inhibition of wild type HRas protein and/or wild type NRas protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit from about 100-fold to about 1000-fold greater inhibition of a dysregulated KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) relative to inhibition of wild type NRas protein and/or wild type HRas protein. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can exhibit from about 1000-fold to about 10000-fold greater inhibition of a dysregulated KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, and/or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein and/or a KRas G12V mutant protein))) relative to inhibition of wild type NRas protein and/or wild type HRas protein. Compounds Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II- b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, are useful for treating diseases and disorders including cardiovascular
disease (e.g., arteriovenous malformations or Noonan syndrome), endometriosis, an inflammatory and/or autoimmune disease (e.g., a nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis), and proliferative disorders such as cancers, including hematological cancers and solid tumors (e.g., advanced solid tumors). In some embodiments, the diseases and disorders are KRas-associated diseases and disorders (e.g., mutant KRas- associated diseases or disorders (e.g., KRas G12D-, KRas G12R-, or G12V-associated diseases or disorders)). In certain embodiments, compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, are useful for preventing diseases and disorders as defined herein (for example, a cardiovascular disease, endometriosis, and an inflammatory and/or autoimmune disease, or cancer). In some embodiments of any of the methods or uses described herein, the inflammatory and/or autoimmune disease is RAS-associated autoimmune leukoproliferative disease. See, e.g., Niemela et al. Blood.2011; 117(10):2883-6, doi: 10.1182/blood-2010-07-295501. In some embodiments, the subject has been identified or diagnosed as having a cancer with a KRas dysregulation (e.g., a KRas mutation or amplification) (e.g., as determined using a regulatory agency-approved, e.g., FDA-approved, assay or kit). In some embodiments, the subject has a cancer (e.g., a tumor sample) that has a KRas dysregulation (e.g., a KRas mutation or amplification) (e.g., as determined using a regulatory agency-approved assay or kit). The subject can be a subject with a cancer (e.g., one or more tumor samples) that is positive for a KRas dysregulation (e.g., a KRas mutation or amplification) (e.g., identified as positive using a regulatory agency-approved, e.g., FDA-approved, assay or kit). The subject can be a subject whose cancer (e.g., a tumor sample) has a KRas dysregulation (e.g., a KRas mutation or amplification) (e.g., where the cancer (e.g., tumor sample) is identified as such using a regulatory agency-approved, e.g., FDA-approved, kit or assay). In some embodiments, the subject is suspected of having a mutant KRas-associated cancer. In some embodiments, the subject has a clinical record indicating that the subject has a cancer (e.g., a tumor sample) that has a KRas dysregulation (e.g., a KRas mutation or amplification) (and optionally the clinical
record indicates that the subject should be treated with any of the compounds and/or compositions provided herein). In some such embodiments, the cancer (e.g., a tumor sample) has a KRas mutation selected from the group consisting of: a KRas G12X mutation, a KRas G13X mutation, and a KRas Q61X mutation. In some embodiments, a KRas mutation is selected from the group consisting of: a KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C mutation, a KRas G13D mutation, a KRas G13V mutation, a KRas Q61E mutation, a KRas Q61H mutation, a KRas Q61K mutation, a KRas Q61L mutation, a KRas Q61P mutation, and a KRas Q61R mutation. In some embodiments, a KRas mutation is selected from the group consisting of: a KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G12S mutation, and a KRas G12V mutation. In some embodiments, the cancer (e.g., a tumor sample) has a KRas mutation selected from the group consisting of: a KRas G12A mutation, a KRas G12D mutation, a KRas G12R mutation, and a KRas G12V mutation. In some embodiments, the cancer (e.g., a tumor sample) has a KRas G12D mutation, a KRas G12R mutation, or KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation). In some embodiments, the cancer (e.g., a tumor sample) has a KRas G12D mutation or a KRas G12V mutation. In some embodiments, the cancer (e.g., a tumor sample) has a KRas G12D mutation. In some embodiments, the cancer (e.g., a tumor sample) has a KRas G12R mutation. In some embodiments, the cancer (e.g., a tumor sample) has a KRas G12V mutation. The term “KRas-associated cancer” as used herein refers to cancers associated with or having a dysregulation of a KRAS gene, a KRas protein, or the expression or activity or level of any (e.g., one or more) of the same (e.g., any of the types of dysregulations of a KRAS gene, a KRas protein, or the expression or activity or level of any of the same described herein). Non- limiting examples of a KRas-associated cancer are described herein. The term “mutant KRas-associated cancer” as used herein refers to cancers associated with or having a KRas mutation (e.g., a KRAS gene having a mutation corresponding to a mutation in a KRas protein and/or a KRas protein having a mutation). Non-limiting examples of a mutant KRas-associated cancer are described herein. The term “KRas G12X-associated cancer” as used herein refers to cancers associated with or having a KRas G12X mutation (e.g., a KRAS gene having a mutation corresponding to a G12X mutation in a KRas protein and/or a KRas protein having a G12X mutation). Non-
limiting examples of a KRas G12X-associated cancer are described herein. The term “KRas G12A-associated cancer” as used herein refers to cancers associated with or having a KRas G12A mutation (e.g., a KRAS gene having a mutation corresponding to a G12A mutation in a KRas protein and/or a KRas protein having a G12A mutation). Non- limiting examples of a KRas G12A-associated cancer are described herein. The term “KRas G12C-associated cancer” as used herein refers to cancers associated with or having a KRas G12C mutation (e.g., a KRAS gene having a mutation corresponding to a G12C mutation in a KRas protein and/or a KRas protein having a G12C mutation). Non- limiting examples of a KRas G12C-associated cancer are described herein. The term “KRas G12D-associated cancer” as used herein refers to cancers associated with or having a KRas G12D mutation (e.g., a KRAS gene having a mutation corresponding to a G12D mutation in a KRas protein and/or a KRas protein having a G12D mutation). Non- limiting examples of a KRas G12D-associated cancer are described herein. The term “KRas G12R-associated cancer” as used herein refers to cancers associated with or having a KRas G12R mutation (e.g., a KRAS gene having a mutation corresponding to a G12R mutation in a KRas protein and/or a KRas protein having a G12R mutation). Non- limiting examples of a KRas G12R-associated cancer are described herein. The term “KRas G12S-associated cancer” as used herein refers to cancers associated with or having a KRas G12S mutation (e.g., a KRAS gene having a mutation corresponding to a G12S mutation in a KRas protein and/or a KRas protein having a G12S mutation). Non- limiting examples of a KRas G12S-associated cancer are described herein. The term “KRas G12V-associated cancer” as used herein refers to cancers associated with or having a KRas G12V mutation (e.g., a KRAS gene having a mutation corresponding to a G12V mutation in a KRas protein and/or a KRas protein having a G12V mutation). Non- limiting examples of a KRas G12V-associated cancer are described herein. The term “KRas G13X-associated cancer” as used herein refers to cancers associated with or having a KRas G13X mutation (e.g., a KRAS gene having a mutation corresponding to a G13X mutation in a KRas protein and/or a KRas protein having a G13X mutation). Non- limiting examples of a KRas G13X-associated cancer are described herein. The term “KRas G13C-associated cancer” as used herein refers to cancers associated with or having a KRas G13C mutation (e.g., a KRAS gene having a mutation corresponding to a G13C mutation in a KRas protein and/or a KRas protein having a G13C mutation). Non- limiting examples of a KRas G13C-associated cancer are described herein.
The term “KRas G13D-associated cancer” as used herein refers to cancers associated with or having a KRas G13D mutation (e.g., a KRAS gene having a mutation corresponding to a G13D mutation in a KRas protein and/or a KRas protein having a G13D mutation). Non- limiting examples of a KRas G13D-associated cancer are described herein. The term “KRas G13V-associated cancer” as used herein refers to cancers associated with or having a KRas G13V mutation (e.g., a KRAS gene having a mutation corresponding to a G13V mutation in a KRas protein and/or a KRas protein having a G13V mutation). Non- limiting examples of a KRas G13V-associated cancer are described herein. The term “KRas Q61X-associated cancer” as used herein refers to cancers associated with or having a KRas Q61X mutation (e.g., a KRAS gene having a mutation corresponding to a Q61X mutation in a KRas protein and/or a KRas protein having a Q61X mutation). Non- limiting examples of a KRas Q61X-associated cancer are described herein. The term “KRas Q61E-associated cancer” as used herein refers to cancers associated with or having a KRas Q61E mutation (e.g., a KRAS gene having a mutation corresponding to a Q61E mutation in a KRas protein and/or a KRas protein having a Q61E mutation). Non- limiting examples of a KRas Q61E-associated cancer are described herein. The term “KRas Q61H-associated cancer” as used herein refers to cancers associated with or having a KRas Q61H mutation (e.g., a KRAS gene having a mutation corresponding to a Q61H mutation in a KRas protein and/or a KRas protein having a Q61H mutation). Non- limiting examples of a KRas Q61H-associated cancer are described herein. The term “KRas Q61K-associated cancer” as used herein refers to cancers associated with or having a KRas Q61K mutation (e.g., a KRAS gene having a mutation corresponding to a Q61K mutation in a KRas protein and/or a KRas protein having a Q61K mutation). Non- limiting examples of a KRas Q61K-associated cancer are described herein. The term “KRas Q61L-associated cancer” as used herein refers to cancers associated with or having a KRas Q61L mutation (e.g., a KRAS gene having a mutation corresponding to a Q61L mutation in a KRas protein and/or a KRas protein having a Q61L mutation). Non- limiting examples of a KRas Q61L-associated cancer are described herein. The term “KRas Q61P-associated cancer” as used herein refers to cancers associated with or having a KRas Q61P mutation (e.g., a KRAS gene having a mutation corresponding to a Q61P mutation in a KRas protein and/or a KRas protein having a Q61P mutation). Non- limiting examples of a KRas Q61P-associated cancer are described herein. The term “KRas Q61R-associated cancer” as used herein refers to cancers associated
with or having a KRas Q61R mutation (e.g., a KRAS gene having a mutation corresponding to a Q61R mutation in a KRas protein and/or a KRas protein having a Q61R mutation). Non- limiting examples of a KRas Q61R-associated cancer are described herein. Such mutations can be associated with the development of a variety of cancers. See, e.g., Hunter et al. Mol Cancer Res. 2015;13(9):1325-35, doi: 10.1158/1541-7786.MCR-15- 0203. Provided herein are methods of treating a cancer in a subject in need of such treatment, the methods comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof. In some embodiments, the subject is treatment naïve with respect to the cancer. In some embodiments, the subject has received one or more lines of previous therapy for the cancer. Also provided herein are methods of treating a cancer in a subject in need thereof, the methods comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof, as a monotherapy. In some embodiments, the subject is treatment naïve with respect to the cancer. In some embodiments, the subject has received one or more lines of previous therapy for the cancer. Provided herein is use of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d),
(V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof, for the treatment of cancer, for example, any of the cancers provided herein. Provided herein is use of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof, as a medicament for the treatment of cancer, for example, any of the cancers provided herein. Provided herein is use of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the treatment of cancer, for example, any of the cancers provided herein. Provided herein is a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof, for use as a medicament. Also provided herein is a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-
d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof, for use as a medicament for the treatment of cancer, for example, any of the cancers provided herein. Provided herein is a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof, for use in treating a cancer, for example, any of the cancers provided herein. As used herein, “monotherapy”, when referring to a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, means that the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is the only therapeutic agent or therapy (e.g., anticancer agent or therapy) administered to the subject during the treatment cycle (e.g., no additional targeted therapeutics, anticancer agents, chemotherapeutics, or checkpoint inhibitors are administered to the subject during the treatment cycle). As a person of ordinary skill in the art would understand, monotherapy does not exclude the co-administration of medicaments for the treatment of side effects or general symptoms associated with the cancer or treatment, such as pain, rash, edema, photosensitivity, pruritis, skin discoloration, hair brittleness, hair loss, brittle nails, cracked nails, discolored nails, swollen cuticles, fatigue, weight loss, general malaise, shortness of breath, infection, anemia, or gastrointestinal symptoms, including nausea,
diarrhea, and lack of appetite. As used herein, “the subject has previously received one or more therapeutic agents or therapies for the cancer” means that the subject has been previously administered one or more therapeutic agents or therapies (e.g., anticancer agent or therapy) for the cancer other than a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, during a prior treatment cycle. In some embodiments, the subject cannot tolerate the one or more therapeutic agents or therapies previously administered for the cancer. In some embodiments, the subject did not respond to the one or more therapeutic agents or therapies previously administered for the cancer. In some embodiments, the subject did not adequately respond to one or more therapeutic agents or therapies previously administered for the cancer. In some embodiments, the subject has stopped responding to the one or more therapeutic agents or therapies previously administered for the cancer. In some embodiments, a lack of response, an inadequate response, or a discontinued response can be determined by objective criteria (e.g., tumor volume, or by criteria such as RECIST 1.1). In some embodiments, a lack of response, an inadequate response, or a discontinued response can be determined by the subject’s physician. As used herein, “the subject is treatment naïve with respect to the cancer” means that the subject has not been previously administered one or more therapeutic agents or therapies for the cancer. For any of the solid tumors described herein, the solid tumors can be primary tumors or metastatic (or secondary) tumors. As used herein, “primary” tumors are those located at the site where the tumor began to grow (i.e., where it originated). As used herein, “metastatic” (or “secondary”) tumors are those that have spread to other parts of body from the original tumor site. In some embodiments, the metastatic or secondary tumors are the same type of cancer as the primary tumor. In some embodiments, the metastatic or secondary tumors are not genetically identical to the primary tumor. Provided herein is a method of treating a cancer in a in a subject in need of such treatment, the method comprising a) detecting a KRas dysregulation (e.g., a KRas mutation
(e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation)) or amplification) in a sample from the subject (e.g., detecting a KRAS gene having a mutation corresponding to a mutation in KRas protein and/or detecting a KRas protein having a mutation, a KRAS gene copy number increase, and/or an increase in KRas mRNA or protein expression); and b) administering a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof. Also provided herein is a method of treating a KRas- associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. For example, provided herein are methods for treating a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))) in a subject in need of such treatment, the methods comprising a) detecting a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation)) or amplification) in a sample from the subject (e.g., detecting a KRAS gene having a mutation corresponding to a mutation in KRas protein and/or detecting a KRas protein having a mutation, a KRAS gene copy number increase, and/or an increase in KRas mRNA or protein expression); and b) administering a therapeutically effective amount of a compound of Formula
(A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof. In some embodiments of any of the methods or uses described herein, the cancer (e.g., KRas-associated cancer (e.g., mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer)))) is breast cancer (e.g., breast invasive carcinoma, breast invasive ductal carcinoma), central or peripheral nervous system tissue cancer (e.g., brain cancer (e.g., astrocytoma, glioblastoma, glioma, oligoastrocytoma)), endocrine or neuroendocrine cancer (e.g., adrenal cancer (e.g., adrenocortical carcinoma, pheochromocytoma, paraganglioma), multiple neuroendocrine type I and type II tumors, parathyroid cancer, pituitary tumors, thyroid cancer (e.g., papillary thyroid cancer)), eye cancer (e.g., uveal cancer (e.g., uveal melanoma)), gastrointestinal cancer (e.g., anal cancer, bile duct cancer (e.g., cholangiocarcinoma), colorectal cancer (e.g., colon adenocarcinoma, rectal adenocarcinoma, mucinous adenocarcinoma, mucinous carcinoma), esophageal cancer (e.g., esophageal adenocarcinoma), gallbladder cancer, gastrointestinal carcinoid tumor, gastrointestinal stromal tumor, liver cancer (e.g., hepatocellular carcinoma, intrahepatic bile duct cancer), pancreatic cancer (e.g., pancreatic adenocarcinoma, pancreatic islet cell cancer), small intestine cancer, or stomach cancer (e.g., stomach adenocarcinoma, signet ring cell carcinoma of the stomach)), genitourinary cancer (e.g., bladder cancer (e.g., bladder urothelial carcinoma), kidney cancer (e.g., renal clear cell carcinoma, renal papillary cell carcinoma, kidney chromophobe), prostate cancer (e.g., prostate adenocarcinoma), testicular cancer (e.g., testicular germ cell tumors, seminoma), or ureter cancer), gynecologic cancer (e.g., cervical cancer (e.g., cervical squamous cell carcinoma, endocervical adenocarcinoma, mucinous carcinoma), ovarian cancer (e.g., serous ovarian cancer, ovarian serous cystadenocarcinoma), uterine cancer (e.g., uterine carcinosarcoma, uterine endometrioid carcinoma, uterine serous carcinoma, uterine papillary serous carcinoma, uterine corpus endometrial carcinoma), or vulvar cancer), head and neck cancer (e.g., ear cancer (e.g., middle ear cancer), head and neck squamous cell carcinoma, nasal cavity cancer, oral cancer, pharynx cancer (e.g., hypopharynx cancer, nasopharynx cancer, oropharyngeal cancer), hematological cancer (e.g., leukemia (e.g.,
chronic lymphocytic leukemia (CLL), acute lymphocytic leukemia (ALL) (e.g., Philadelphia chromosome positive ALL), acute myeloid leukemia (AML) (e.g., acute promyelocytic leukemia (APL)), chronic myeloid leukemia (CML)), lymphoma (e.g., Hodgkin lymphoma (e.g., nodular lymphocyte predominant Hodgkin lymphoma (NLPHL)), non-Hodgkin lymphoma (e.g., Burkitt lymphoma (BL), diffuse large B-cell lymphoma (DLBCL), diffuse histiocytic lymphoma (DHL), follicular lymphoma (FL), intravascular large B-cell lymphoma (IVLBCL), mantle cell lymphoma (MCL), small lymphocytic lymphoma (SLL))), or myeloma (e.g., multiple myeloma)), Li-Fraumeni tumors, mesentery cancer (e.g., omentum cancer, peritoneal cancer), pleural cancer, respiratory cancer (e.g., larynx cancer, lung cancer (e.g., lung squamous cell carcinoma, lung adenocarcinoma, mesothelioma, non-small cell lung cancer (NSCLC)), tracheal cancer), sarcoma (e.g., bone cancer (e.g., osteosarcoma, chondrosarcoma) or soft tissue sarcoma (Ewing sarcoma, leiomyosarcoma, myxofibrosarcoma, rhabdomyosarcoma)), skin cancer (e.g., melanoma), thymus cancer (e.g., thymoma), or a combination thereof. In some embodiments, the cancer (e.g., KRas-associated cancer (e.g., mutant KRas- associated cancer)) is a hematological cancer, a soft tissue cancer, bile duct cancer, bladder cancer, brain cancer, breast cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, rectal cancer, ovarian cancer, pancreatic cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer, thymus cancer, thyroid cancer, urothelial cancer, or uterine cancer. In some embodiments, the cancer (e.g., a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12X-associated cancer))) is a hematological cancer, bile duct cancer, brain cancer, breast cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, pancreatic cancer, prostate cancer, rectal cancer, testicular cancer (e.g., seminoma), skin cancer, stomach cancer, thymus cancer, thyroid cancer, urothelial cancer, or uterine cancer. In some embodiments, the cancer is a hematological cancer, brain cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, pancreatic cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, thymus cancer, urothelial cancer, or uterine cancer. In some embodiments, the cancer is a KRas G12D-associated cancer. In some embodiments, the cancer is a hematological cancer, bladder cancer, bile duct cancer, colon cancer, ovarian cancer, pancreatic cancer, prostate cancer, rectal cancer, skin
cancer, or testicular cancer. In some embodiments, the cancer is a KRas G12R-associated cancer. In some embodiments, the cancer is a hematological cancer, bladder cancer, bile duct cancer, brain cancer, breast cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, ovarian cancer, pancreatic cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer (e.g., seminoma), thymus cancer, or uterine cancer. In some embodiments, the cancer is a G12V- associated cancer. In some embodiments, the cancer (e.g., a KRas-associated cancer (e.g., a mutant KRas- associated cancer (e.g., a KRas G13X-associated cancer))) is a hematological cancer, a soft tissue cancer, cervical cancer, colon cancer, endometrial cancer, liver cancer, lung cancer, pancreatic cancer, rectal cancer, skin cancer, stomach cancer, or urothelial cancer. In some embodiments, the cancer (e.g., a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas Q61X-associated cancer)) is bladder cancer, colon cancer, lung cancer, ovarian cancer, rectal cancer, thyroid cancer, or uterine cancer. In some embodiments, the cancer (e.g., a KRas-associated cancer (e.g., a cancer associated with KRas amplification (e.g., a cancer associated with wild-type KRas amplification)) is colorectal cancer, gastric cancer, gastroesophageal cancer, head and neck squamous carcinoma, or lung cancer (e.g., NSCLC). See, e.g., the public database cBioPortal. In some embodiments, the cancer (e.g., a KRas-associated cancer (e.g., mutant KRas- associated cancer)) is pancreatic cancer or metastatic pancreatic cancer. In some embodiments, the cancer (e.g., a KRas-associated cancer (e.g., mutant KRas-associated cancer)) is pancreatic ductal adenocarcinoma (PDAC). In some such embodiments, the pancreatic cancer is a KRas G12R-associated cancer. In some embodiments, the cancer (e.g., a KRas-associated cancer (e.g., mutant KRas- associated cancer)) is advanced-stage lung adenocarcinoma. In some embodiments, the cancer is a solid tumor. In some embodiments, the solid tumor has a KRas dysregulation (e.g., a KRas mutation or amplification). For example, the solid tumor has a KRas mutation selected from the group consisting of: a KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G12S mutation, and a KRas G12V mutation. In some embodiments, the solid tumor has a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation. In some embodiments, the solid tumor has a KRas G12D mutation or a KRas G12V mutation. In some embodiments, the solid
tumor has a KRas G12D mutation. In some embodiments, the solid tumor has a KRas G12R mutation. In some embodiments, the solid tumor has a KRas G12V mutation. Also provided herein is a method of treating a solid tumor in a subject in need thereof, the method comprising: (a) detecting a KRas dysregulation (e.g., a KRas mutation or amplification) in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Also provided herein is a method of treating a solid tumor in a subject in need thereof, the method comprising: (a) detecting a KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G12S mutation, or a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a solid tumor in a subject in need thereof, the method comprising: (a) detecting a KRas G12D or a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described
herein. Additionally provided herein is a method of treating a solid tumor in a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a solid tumor in a subject in need thereof, the method comprising: (a) detecting a KRas G12R mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a solid tumor in a subject in need thereof, the method comprising: (a) detecting a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some embodiments, the cancer is a bladder cancer. In some embodiments, the bladder cancer has a KRas dysregulation (e.g., a KRas mutation or amplification). For example, the bladder cancer has a KRas mutation selected from the group consisting of: a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G12V mutation, a KRas
G13D mutation, and a KRas Q61H mutation. In some embodiments, the bladder cancer has a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation. In some embodiments, the bladder cancer has a KRas G12D mutation or a KRas G12V mutation. In some embodiments, the bladder cancer has a KRas G12D mutation. In some embodiments, the bladder cancer has a KRas G12R mutation. In some embodiments, the bladder cancer has a KRas G12V mutation. Also provided herein is a method of treating a bladder cancer in a subject in need thereof, the method comprising: (a) detecting a KRas dysregulation (e.g., a KRas mutation or amplification) in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Also provided herein is a method of treating a bladder cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G12V mutation, a KRas G13D mutation, or a KRas Q61H mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a bladder cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12D or a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-
1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a bladder cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a bladder cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12R mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a bladder cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition
as described herein. In some embodiments, the cancer is a cervical cancer. In some embodiments, the cervical cancer has a KRas dysregulation (e.g., a KRas mutation or amplification). For example, the cervical cancer has a KRas mutation selected from the group consisting of: a KRas G12C mutation, a KRas G12D mutation, a KRas G12V mutation, and a KRas G13D mutation. In some embodiments, the cervical cancer has a KRas G12D mutation or a KRas G12V mutation. In some embodiments, the cervical cancer has a KRas G12D mutation. In some embodiments, the cervical cancer has a KRas G12V mutation. Also provided herein is a method of treating a cervical cancer in a subject in need thereof, the method comprising: (a) detecting a KRas dysregulation (e.g., a KRas mutation or amplification) in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Also provided herein is a method of treating a cervical cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12C mutation, a KRas G12D mutation, a KRas G12V mutation, or a KRas G13D mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a cervical cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation or a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-
b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a cervical cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a cervical cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II- b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some embodiments, the cancer is a colorectal cancer. In some embodiments, the colorectal cancer has a KRas dysregulation (e.g., a KRas mutation or amplification). For example, the colorectal cancer has a KRas mutation selected from the group consisting of: a KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C mutation, a KRas G13D mutation, a KRas G13V mutation, a KRas Q61E mutation, a KRas Q61H mutation, a KRas Q61K mutation, a KRas Q61L mutation, a KRas Q61P mutation, and a KRas Q61R mutation. In some embodiments, the colorectal cancer has a KRas G12D mutation, a KRas
G12R mutation, or a KRas G12V mutation. In some embodiments, the colorectal cancer has a KRas G12D mutation or a KRas G12V mutation. In some embodiments, the colorectal cancer has a KRas G12D mutation. In some embodiments, the colorectal cancer has a KRas G12V mutation. Also provided herein is a method of treating a colorectal cancer in a subject in need thereof, the method comprising: (a) detecting a KRas dysregulation (e.g., a KRas mutation or amplification) in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Also provided herein is a method of treating a colorectal cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C mutation, a KRas G13D mutation, a KRas G13V mutation, a KRas Q61E mutation, a KRas Q61H mutation, a KRas Q61K mutation, a KRas Q61L mutation, a KRas Q61P mutation, or a KRas Q61R mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a colorectal cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation or a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or
(II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a colorectal cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a colorectal cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12R mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II- b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a colorectal cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt
thereof, or a pharmaceutical composition as described herein. In some embodiments, the cancer is an endometrial cancer. In some embodiments, the endometrial cancer has a KRas dysregulation (e.g., a KRas mutation or amplification). For example, the endometrial cancer has a KRas mutation selected from the group consisting of: a KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C mutation, a KRas G13D mutation, a KRas G13V mutation, a KRas Q61H mutation, and a KRas Q61L mutation. In some embodiments, the endometrial cancer has a KRas G12D mutation or a KRas G12V mutation. In some embodiments, the endometrial cancer has a KRas G12D mutation. In some embodiments, the endometrial cancer has a KRas G12V mutation. Also provided herein is a method of treating an endometrial cancer in a subject in need thereof, the method comprising: (a) detecting a KRas dysregulation (e.g., a KRas mutation or amplification) in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Also provided herein is a method of treating an endometrial cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C mutation, a KRas G13D mutation, a KRas G13V mutation, a KRas Q61H mutation, or a KRas Q61L mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating an
endometrial cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation or a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating an endometrial cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating an endometrial cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II- b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some embodiments, the cancer is an esophageal or stomach cancer. In some embodiments, the esophageal or stomach cancer has a KRas dysregulation (e.g., a KRas mutation or amplification). For example, the esophageal or stomach cancer has a KRas mutation selected from the group consisting of: a KRas G12C mutation, a KRas G12D
mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C mutation, a KRas G13D mutation, and a KRas Q61H mutation. In some embodiments, the esophageal or stomach cancer has a KRas G12D mutation or a KRas G12V mutation. In some embodiments, the esophageal or stomach cancer has a KRas G12D mutation. In some embodiments, the esophageal or stomach cancer has a KRas G12V mutation. Also provided herein is a method of treating an esophageal or stomach cancer in a subject in need thereof, the method comprising: (a) detecting a KRas dysregulation (e.g., a KRas mutation or amplification) in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Also provided herein is a method of treating an esophageal or stomach cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12C mutation, a KRas G12D mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C mutation, a KRas G13D mutation, or a KRas Q61H mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating an esophageal or stomach cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation or a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula
(IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating an esophageal or stomach cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating an esophageal or stomach cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some embodiments, the cancer is a leukemia. In some embodiments, the leukemia has a KRas dysregulation (e.g., a KRas mutation or amplification). For example, the leukemia has a KRas mutation selected from the group consisting of: a KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C mutation, a KRas G13D mutation, a KRas G13V mutation, a KRas Q61E mutation, a KRas Q61H mutation, a KRas Q61K mutation, a KRas Q61L mutation, a KRas Q61P mutation, and a KRas Q61R mutation. In some embodiments, the leukemia has a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation. In some embodiments, the leukemia has a KRas G12D mutation or a KRas G12V mutation. In
some embodiments, the leukemia has a KRas G12D mutation. In some embodiments, the leukemia has a KRas G12R mutation. In some embodiments, the leukemia has a KRas G12V mutation. Also provided herein is a method of treating a leukemia in a subject in need thereof, the method comprising: (a) detecting a KRas dysregulation (e.g., a KRas mutation or amplification) in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Also provided herein is a method of treating a leukemia in a subject in need thereof, the method comprising: (a) detecting a KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C mutation, a KRas G13D mutation, a KRas G13V mutation, a KRas Q61E mutation, a KRas Q61H mutation, a KRas Q61K mutation, a KRas Q61L mutation, a KRas Q61P mutation, or a KRas Q61R mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a leukemia in a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation or a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-
a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a leukemia in a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a leukemia in a subject in need thereof, the method comprising: (a) detecting a KRas G12R mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a leukemia in a subject in need thereof, the method comprising: (a) detecting a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein.
In some embodiments, the cancer is a melanoma. In some embodiments, the melanoma has a KRas dysregulation (e.g., a KRas mutation or amplification). For example, the melanoma has a KRas mutation selected from the group consisting of: a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G13D mutation, a KRas G13V mutation, a KRas a KRas Q61K mutation, a KRas Q61L mutation, and a KRas Q61R mutation. In some embodiments, the melanoma has a KRas G12D mutation or a KRas G12R mutation. In some embodiments, the melanoma has a KRas G12D mutation. In some embodiments, the melanoma has a KRas G12R mutation. Also provided herein is a method of treating a melanoma in a subject in need thereof, the method comprising: (a) detecting a KRas dysregulation (e.g., a KRas mutation or amplification) in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Also provided herein is a method of treating a melanoma in a subject in need thereof, the method comprising: (a) detecting a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G13D mutation, a KRas G13V mutation, a KRas a KRas Q61K mutation, a KRas Q61L mutation, or a KRas Q61R mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a melanoma in a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I)
(e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a melanoma in a subject in need thereof, the method comprising: (a) detecting a KRas G12R mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some embodiments, the cancer is a lung cancer (e.g., non-small cell lung cancer). In some embodiments, the lung cancer (e.g., non-small cell lung cancer) has a KRas dysregulation (e.g., a KRas mutation or amplification). For example, the lung cancer (e.g., non-small cell lung cancer) has a KRas mutation selected from the group consisting of: a KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C mutation, a KRas G13D mutation, a KRas Q61H mutation, and a KRas Q61L mutation. In some embodiments, the lung cancer has a KRas G12D mutation or a KRas G12V mutation. In some embodiments, the lung cancer has a KRas G12D mutation. In some embodiments, the lung cancer has a KRas G12V mutation. Also provided herein is a method of treating a lung cancer (e.g., non-small cell lung cancer) in a subject in need thereof, the method comprising: (a) detecting a KRas dysregulation (e.g., a KRas mutation or amplification) in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-
a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Also provided herein is a method of treating a lung cancer (e.g., non-small cell lung cancer) in a subject in need thereof, the method comprising: (a) detecting a KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C mutation, a KRas G13D mutation, a KRas Q61H mutation, or a KRas Q61L mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a lung cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation or a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a lung cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-
a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a lung cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some embodiments, the cancer is a pancreatic cancer. In some embodiments, the pancreatic cancer has a KRas dysregulation (e.g., a KRas mutation or amplification). For example, the pancreatic cancer has a KRas mutation selected from the group consisting of: a KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C mutation, a KRas Q61H mutation, and a KRas Q61R mutation. In some embodiments, the pancreatic cancer has a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation. In some embodiments, the pancreatic cancer has a KRas G12D mutation or a KRas G12V mutation. In some embodiments, the pancreatic cancer has a KRas G12D mutation. In some embodiments, the pancreatic cancer has a KRas G12R mutation. In some embodiments, the pancreatic cancer has a KRas G12V mutation. Also provided herein is a method of treating a pancreatic cancer in a subject in need thereof, the method comprising: (a) detecting a KRas dysregulation (e.g., a KRas mutation or amplification) in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition
as described herein. Also provided herein is a method of treating a pancreatic cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C mutation, a KRas Q61H mutation, or a KRas Q61R mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a pancreatic cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a pancreatic cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation or a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a pancreatic cancer in
a subject in need thereof, the method comprising: (a) detecting a KRas G12D mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II- b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a pancreatic cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12R mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a pancreatic cancer in a subject in need thereof, the method comprising: (a) detecting a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some embodiments, the cancer is a testicular cancer (e.g., seminoma). In some embodiments, the testicular cancer (e.g., seminoma) has a KRas dysregulation (e.g., a KRas mutation or amplification). For example, the testicular cancer (e.g., seminoma) has a KRas mutation selected from the group consisting of: a KRas G12A mutation, a KRas G12R
mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas Q61L mutation, a KRas Q61P mutation, and a KRas Q61R mutation. In some embodiments, the testicular cancer (e.g., seminoma) cancer has a KRas G12R mutation or a KRas G12V mutation. In some embodiments, the testicular cancer (e.g., seminoma) cancer has a KRas G12R mutation. In some embodiments, the testicular cancer (e.g., seminoma) cancer has a KRas G12V mutation. Also provided herein is a method of treating a testicular cancer (e.g., seminoma) in a subject in need thereof, the method comprising: (a) detecting a KRas dysregulation (e.g., a KRas mutation or amplification) in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Also provided herein is a method of treating a testicular cancer (e.g., seminoma) in a subject in need thereof, the method comprising: (a) detecting a KRas G12A mutation, a KRas G12R mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas Q61L mutation, a KRas Q61P mutation, or a KRas Q61R mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II- b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a testicular cancer (e.g., seminoma) in a subject in need thereof, the method comprising: (a) detecting a KRas G12R mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II- b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)),
Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Additionally provided herein is a method of treating a testicular cancer (e.g., seminoma) in a subject in need thereof, the method comprising: (a) detecting a KRas G12V mutation in a sample from the subject, and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II- b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. Also provided herein is a method of treating a bladder cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some embodiments, the method further comprises determining that the bladder cancer has a KRas mutation selected from the group consisting of: a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G12V mutation, a KRas G13D mutation, and a KRas Q61H mutation. In some such embodiments, the cancer is a KRas G12C-associated cancer, a KRas G12D-associated cancer, a KRas G12R-associated cancer, a KRas G12V-associated cancer, a KRas G13D-associated cancer, or a KRas Q61H-associated cancer. In some aspects of this embodiment, the cancer is a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer. In some embodiments, the cancer is a KRas G12D- associated cancer. In some embodiments, the cancer is a KRas G12R-associated cancer. In some embodiments, the cancer is a KRas G12V-associated cancer. In some embodiments, the
compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G12D inhibitor, a KRas G12R inhibitor, a KRas G12V inhibitor, a KRas G13D inhibitor, a KRas Q61H inhibitor, or two or more thereof. In some aspects of this embodiment, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12R inhibitor, and/or a KRas G12V inhibitor. In some aspects of this embodiment, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12V inhibitor, or both. Also provided herein is a method of treating a cervical cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some embodiments, the method further comprises determining that the cervical cancer has
a KRas mutation selected from the group consisting of: a KRas G12C mutation, a KRas G12D mutation, a KRas G12V mutation, and a KRas G13D mutation. In some embodiments, the cancer is a KRas G12C-associated cancer, a KRas G12D-associated cancer, a KRas G12V- associated cancer, or a KRas G13D-associated cancer. In some embodiments, the cancer is a KRas G12D-associated cancer or a KRas G12V-associated cancer. In some embodiments, the cancer is a KRas G12D-associated cancer. In some embodiments, the cancer is a KRas G12V- associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G12D inhibitor, a KRas G12V inhibitor, a KRas G13D inhibitor, or two or more thereof. In some aspects of this embodiment, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12V inhibitor, or both. Also provided herein is a method of treating a colorectal cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some embodiments, the method further comprises determining that the colorectal cancer has a KRas mutation selected from the group consisting of: a KRas G12A mutation, a KRas G12C
mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C mutation, a KRas G13D mutation, a KRas G13V mutation, a KRas Q61E mutation, a KRas Q61H mutation, a KRas Q61K mutation, a KRas Q61L mutation, a KRas Q61P mutation, and a KRas Q61R mutation. In some embodiments, the cancer is a KRas G12A-associated cancer, a KRas G12C-associated cancer, a KRas G12D- associated cancer, a KRas G12R-associated cancer, a KRas G12S-associated cancer, a KRas G12V-associated cancer, a KRas G13C-associated cancer, a KRas G13D-associated cancer, a KRas G13V-associated cancer, a KRas Q61E-associated cancer, a KRas Q61H-associated cancer, a KRas Q61K-associated cancer, a KRas Q61L-associated cancer, a KRas Q61P- associated cancer, or a KRas Q61R-associated cancer. In some aspects of this embodiment, the cancer is a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V- associated cancer. In some embodiments, the cancer is a KRas G12D-associated cancer or a KRas G12V-associated cancer. In some embodiments, the cancer is a KRas G12D-associated cancer. In some embodiments, the cancer is a KRas G12R-associated cancer. In some embodiments, the cancer is a KRas G12V-associated cancer. In some embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12C inhibitor, a KRas G12D inhibitor, a KRas G12R inhibitor, a KRas G12S inhibitor, a KRas G12V inhibitor, a KRas G13C inhibitor, a KRas G13D inhibitor, a KRas G13V inhibitor, a KRas Q61E inhibitor, a KRas Q61H inhibitor, a KRas Q61K inhibitor, a KRas Q61L inhibitor, a KRas Q61P inhibitor, a KRas Q61R inhibitor, or two or more thereof. In some aspects of this embodiment, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12R inhibitor, and/or a KRas G12V
inhibitor. In some aspects of this embodiment, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12V inhibitor, or both. Also provided herein is a method of treating an endometrial cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some embodiments, the method further comprises determining that the endometrial cancer has a KRas mutation selected from the group consisting of: a KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C mutation, a KRas G13D mutation, a KRas G13V mutation, a KRas Q61H mutation, and a KRas Q61L mutation. In some embodiments, the cancer is a KRas G12A-associated cancer, a KRas G12C-associated cancer, a KRas G12D-associated cancer, a KRas G12S-associated cancer, a KRas G12V-associated cancer, a KRas G13C-associated cancer, a KRas G13D-associated cancer, a KRas G13V-associated cancer, a KRas Q61H- associated cancer, or a KRas Q61L-associated cancer. In some embodiments, the cancer is a KRas G12D-associated cancer or a KRas G12V-associated cancer. In some embodiments, the cancer is a KRas G12D-associated cancer. In some embodiments, the cancer is a KRas G12V- associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-
c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12C inhibitor, a KRas G12D inhibitor, a KRas G12S inhibitor, a KRas G12V inhibitor, a KRas G13C inhibitor, a KRas G13D inhibitor, a KRas G13V inhibitor, a KRas Q61H inhibitor, a KRas Q61L inhibitor, or two or more thereof. In some aspects of this embodiment, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12V inhibitor, or both. Also provided herein is a method of treating an esophageal or stomach cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some embodiments, the method further comprises determining that the esophageal or stomach cancer has a KRas mutation selected from the group consisting of: a KRas G12C mutation, a KRas G12D mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C mutation, a KRas G13D mutation, and a KRas Q61H mutation. In some embodiments, the cancer is a KRas G12C-associated cancer, a KRas G12D-associated cancer, a KRas G12S-associated cancer, a KRas G12V-associated cancer, a KRas G13C- associated cancer, a KRas G13D-associated cancer, or a KRas Q61H-associated cancer. In some embodiments, the cancer is a KRas G12D-associated cancer or a KRas G12V-associated cancer. In some embodiments, the cancer is a KRas G12D-associated cancer. In some embodiments, the cancer is a KRas G12V-associated cancer. In some embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b),
(II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G12D inhibitor, a KRas G12S inhibitor, a KRas G12V inhibitor, a KRas G13C inhibitor, a KRas G13D inhibitor, a KRas Q61H inhibitor, or two or more thereof. In some aspects of this embodiment, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12V inhibitor, or both. Also provided herein is a method of treating a leukemia in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some embodiments, the method further comprises determining that the leukemia has a KRas mutation selected from the group consisting of: a KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C mutation, a KRas G13D mutation, a KRas G13V mutation, a KRas Q61E mutation, a KRas Q61H mutation, a KRas Q61K mutation, a KRas Q61L mutation, a KRas Q61P mutation, and a KRas Q61R mutation. In some embodiments, the cancer is a KRas G12A-associated cancer, a KRas G12C-associated cancer, a KRas G12D- associated cancer, a KRas G12R-associated cancer, a KRas G12S-associated cancer, a KRas G12V-associated cancer, a KRas G13C-associated cancer, a KRas G13D-associated cancer, a KRas G13V-associated cancer, a KRas Q61E-associated cancer, a KRas Q61H-associated
cancer, a KRas Q61K-associated cancer, a KRas Q61L-associated cancer, a KRas Q61P- associated cancer, or a KRas Q61R-associated cancer. In some aspects of this embodiment, the cancer is a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V- associated cancer. In some embodiments, the cancer is a KRas G12D-associated cancer or a KRas G12V-associated cancer. In some embodiments, the cancer is a KRas G12D-associated cancer. In some embodiments, the cancer is a KRas G12R-associated cancer. In some embodiments, the cancer is a KRas G12V-associated cancer. In some embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12C inhibitor, a KRas G12D inhibitor, a KRas G12R inhibitor, a KRas G12S inhibitor, a KRas G12V inhibitor, a KRas G13C inhibitor, a KRas G13D inhibitor, a KRas G13V inhibitor, a KRas Q61E inhibitor, a KRas Q61H inhibitor, a KRas Q61K inhibitor, a KRas Q61L inhibitor, a KRas Q61P inhibitor, a KRas Q61R inhibitor, or two or more thereof. In some aspects of this embodiment, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12R inhibitor, and/or a KRas G12V inhibitor. In some aspects of this embodiment, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12V inhibitor, or both.
Also provided herein is a method of treating a melanoma in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some embodiments, the method further comprises determining that the melanoma has a KRas mutation selected from the group consisting of: a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G13D mutation, a KRas G13V mutation, a KRas Q61K mutation, a KRas Q61L mutation, and a KRas Q61R mutation. In some embodiments, the cancer is a KRas G12C-associated cancer, a KRas G12D-associated cancer, a KRas G12R- associated cancer, a KRas G13D-associated cancer, a KRas G13V-associated cancer, a KRas Q61K-associated cancer, a KRas Q61L-associated cancer, or a KRas Q61R-associated cancer. In some embodiments, the cancer is a KRas G12D-associated cancer or a KRas G12R-associted cancer. In some embodiments, the cancer is a KRas G12D-associated cancer. In some embodiments, the cancer is a KRas G12R-associated cancer. In some embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G12D inhibitor, a KRas G12R inhibitor, a KRas G13D inhibitor, a KRas G13V inhibitor, a KRas Q61K inhibitor, a KRas Q61L inhibitor, a KRas Q61R inhibitor, or two or more thereof. In some aspects of this embodiment, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or
(V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12R inhibitor, or both. Also provided herein is a method of treating a lung cancer (e.g., NSCLC) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some embodiments, the method further comprises determining that the lung cancer (e.g., NSCLC) has a KRas mutation selected from the group consisting of: KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C-asociated cancer, a KRas G13D mutation, a KRas Q61H mutation, and a KRas Q61L mutation. In some embodiments, the cancer is a KRas G12A- associated cancer, a KRas G12C-associated cancer, a KRas G12D-associated cancer, a KRas G12S-associated cancer, a KRas G12V-associated cancer, a KRas G13C-asociated cancer, a KRas G13D-associated cancer, a KRas Q61H-associated cancer, or a KRas Q61L-associated cancer. In some embodiments, the cancer is a KRas G12D-associated cancer or a KRas G12V- associated cancer. In some embodiments, the cancer is a KRas G12D-associated cancer. In some embodiments, the cancer is a KRas G12V-associated cancer. In some embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12C inhibitor, a KRas G12D inhibitor, a KRas G12S inhibitor, a KRas G12V inhibitor, a KRas G13C-asociated cancer, a KRas G13D inhibitor, a KRas Q61H inhibitor, a KRas Q61L inhibitor, or two or more thereof. In some aspects of this embodiment, the compound of Formula (A) (e.g., Formula (I) (e.g.,
Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, a KRas G12D inhibitor, a KRas G12V inhibitor, or both. Also provided herein is a method of treating a pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some embodiments, the method further comprises determining that the pancreatic cancer has a KRas mutation selected from the group consisting of: a KRas G12A mutation, a KRas G12C mutation, a KRas G12D mutation, a KRas G12R mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas G13C-asociated cancer, and a KRas Q61H mutation. In some embodiments, the cancer is a KRas G12A-associated cancer, a KRas G12C-associated cancer, a KRas G12D-associated cancer, a KRas G12R-associated cancer, a KRas G12S-associated cancer, a KRas G12V-associated cancer, a KRas G13C-asociated cancer, or a KRas Q61H- associated cancer. In some aspects of this embodiment, the cancer is a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer. In some embodiments, the cancer is a KRas G12D-associated cancer or a KRas G12V-associated cancer. In some embodiments, the cancer is a KRas G12D-associated cancer. In some embodiments, the cancer is a KRas G12R-associated cancer. In some embodiments, the cancer is a KRas G12V-associated cancer. In some embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-
b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12C inhibitor, a KRas G12D inhibitor, a KRas G12R inhibitor, a KRas G12S inhibitor, a KRas G12V inhibitor, a KRas G13C inhibitor, a KRas Q61H inhibitor, or two or more thereof. In some aspects of this embodiment, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12R inhibitor, and/or a KRas G12V inhibitor. In some aspects of this embodiment, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12V inhibitor, or both. Also provided herein is a method of treating a testicular cancer (e.g., seminoma) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some embodiments, the method further comprises determining that the testicular cancer (e.g., seminoma) has a KRas mutation selected from the group consisting of: a KRas G12A mutation, a KRas G12R mutation, a KRas G12S mutation, a KRas G12V mutation, a KRas Q61L mutation, a KRas Q61P mutation, and a KRas Q61R mutation. In some
embodiments, the cancer is a KRas G12A-associated cancer, a KRas G12R-associated cancer, a KRas G12S-associated cancer, a KRas G12V-associated cancer, a KRas Q61L-associated cancer, a KRas Q61P-associated cancer, or a KRas Q61R-associated cancer. In some aspects of this embodiment, the cancer is a KRas G12R-associated cancer or a KRas G12V-associated cancer. In some embodiments, the cancer is a KRas G12R-associated cancer. In some embodiments, the cancer is a KRas G12V-associated cancer. In some embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12R inhibitor, a KRas G12S inhibitor, a KRas G12V inhibitor, a KRas Q61L inhibitor, a KRas Q61P inhibitor, a KRas Q61R inhibitor, or two or more thereof. In some aspects of this embodiment, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas G12V inhibitor, or both. In some embodiments, the KRas-associated cancer is bladder cancer, breast cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, liver cancer, skin cancer (e.g., melanoma), lung cancer (e.g., NSCLC), or pancreatic cancer. In some embodiments, the cancer has a KRas G12C mutation or a KRas G12D mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G12D inhibitor,
or both. Also provided herein is a method of treating a KRas G12C-associated cancer or a KRas G12D-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is bladder cancer, breast cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, liver cancer, skin cancer (e.g., melanoma), lung cancer (e.g., NSCLC), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G12D inhibitor, or both. Also provided herein is a method of treating bladder cancer, breast cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, liver cancer, skin cancer (e.g., melanoma), lung cancer (e.g., NSCLC), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12C-associated cancer or a KRas
G12D-associated cancer, or both. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G12D inhibitor, or both. In some embodiments, the KRas-associated cancer is bladder cancer, breast cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, leukemia, lung cancer (e.g., NSCLC), pancreatic cancer, or kidney cancer. In some embodiments, the cancer has a KRas G12D mutation or a KRas G12V mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12V inhibitor, or both. Also provided herein is a method of treating a KRas G12D-associated cancer or a KRas G12V-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is bladder cancer, breast cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, leukemia, lung cancer (e.g., NSCLC), pancreatic cancer, or kidney cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I)
(e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12V inhibitor, or both. Also provided herein is a method of treating bladder cancer, breast cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, leukemia, lung cancer (e.g., NSCLC), pancreatic cancer, or kidney cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12D-associated cancer or a KRas G12V- associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12V inhibitor, or both. In some embodiments, the KRas-associated cancer is bladder cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, liver cancer, leukemia, skin cancer (e.g., melanoma), or lung cancer (e.g., NSCLC). In some embodiments, the cancer has a KRas G12D mutation or a KRas G13D mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b),
(II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G13D inhibitor, or both. Also provided herein is a method of treating a KRas G12D-associated cancer or a KRas G13D-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is bladder cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, liver cancer, leukemia, skin cancer (e.g., melanoma), or lung cancer (e.g., NSCLC). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G13D inhibitor, or both. Also provided herein is a method of treating bladder cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, liver cancer, leukemia, skin cancer (e.g., melanoma), or lung cancer (e.g., NSCLC) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula
(V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12D-associated cancer or a KRas G13D-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G13D inhibitor, or both. In some embodiments, the KRas-associated cancer is bladder cancer, breast cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some embodiments, the cancer has a KRas G12C mutation or a KRas G12V mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G12V inhibitor, or both. Also provided herein is a method of treating a KRas G12C-associated cancer or a KRas G12V-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is
bladder cancer, breast cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G12V inhibitor, or both. Also provided herein is a method of treating bladder cancer, breast cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12C-associated cancer or a KRas G12V-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G12V inhibitor, or both. In some embodiments, the KRas-associated cancer is bladder cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, liver cancer, skin cancer (e.g., melanoma), or lung cancer (e.g., NSCLC). In some embodiments, the cancer has a KRas G12C mutation or a KRas G13D mutation. In some such embodiments, the compound of
Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G13D inhibitor, or both. Also provided herein is a method of treating a KRas G12C-associated cancer or a KRas G13D-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is bladder cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, liver cancer, skin cancer (e.g., melanoma), or lung cancer (e.g., NSCLC). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G13D inhibitor, or both. Also provided herein is a method of treating bladder cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, liver cancer, skin cancer (e.g., melanoma), or lung cancer (e.g., NSCLC) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-
b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12C-associated cancer or a KRas G13D-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G13D inhibitor, or both. In some embodiments, the KRas-associated cancer is bladder cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, leukemia, lung cancer (e.g., NSCLC), or pancreatic cancer. In some embodiments, the cancer has a KRas G12D mutation or a KRas Q61H mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas Q61H inhibitor, or both. Also provided herein is a method of treating a KRas G12D-associated cancer or a KRas Q61H-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-
a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is bladder cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, leukemia, lung cancer (e.g., NSCLC), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas Q61H inhibitor, or both. Also provided herein is a method of treating bladder cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, leukemia, lung cancer (e.g., NSCLC), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12D- associated cancer or a KRas Q61H-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas Q61H inhibitor, or both. In some embodiments, the KRas-associated cancer is bladder cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, leukemia, or lung cancer (e.g., NSCLC). In some embodiments, the cancer has a KRas G12V mutation or a KRas G13D
mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12V inhibitor, a KRas G13D inhibitor, or both. Also provided herein is a method of treating a KRas G12V-associated cancer or a KRas G13D-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is bladder cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, leukemia, or lung cancer (e.g., NSCLC). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12V inhibitor, a KRas G13D inhibitor, or both. Also provided herein is a method of treating bladder cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, leukemia, or lung cancer (e.g., NSCLC) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula
(III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12V- associated cancer or a KRas G13V-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12V inhibitor, a KRas G13D inhibitor, or both. In some embodiments, the KRas-associated cancer is bladder cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, leukemia, lung cancer (e.g., NSCLC), or pancreatic cancer. In some embodiments, the cancer has a KRas G12V mutation or a KRas Q61H mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12V inhibitor, a KRas Q61H inhibitor, or both. Also provided herein is a method of treating a KRas G12V-associated cancer or a KRas Q61H-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a
pharmaceutical composition as described herein. In some such embodiments, the cancer is bladder cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, leukemia, lung cancer (e.g., NSCLC), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12V inhibitor, a KRas Q61H inhibitor, or both. Also provided herein is a method of treating bladder cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, leukemia, lung cancer (e.g., NSCLC), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12V- associated cancer or a KRas Q61H-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12V inhibitor, a KRas Q61H inhibitor, or both. In some embodiments, the KRas-associated cancer is bladder cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some embodiments, the cancer has a KRas G12C mutation or a KRas Q61H mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g.,
Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas Q61H inhibitor, or both. Also provided herein is a method of treating a KRas G12C-associated cancer or a KRas Q61H-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is bladder cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas Q61H inhibitor, or both. Also provided herein is a method of treating bladder cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-
a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12C- associated cancer or a KRas Q61H-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas Q61H inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), pancreatic cancer, or testicular cancer (e.g., seminoma). In some embodiments, the cancer has a KRas G12S mutation or a KRas G12V mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas G12V inhibitor, or both. Also provided herein is a method of treating a KRas G12S-associated cancer or a KRas G12V-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is
colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), pancreatic cancer, or testicular cancer (e.g., seminoma). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas G12V inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), pancreatic cancer, or testicular cancer (e.g., seminoma) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12S-associated cancer or a KRas G12V-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas G12V inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), pancreatic cancer, or testicular cancer (e.g., seminoma). In some embodiments, the cancer has a KRas G12A mutation or a KRas G12S mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g.,
Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12S inhibitor, or both. Also provided herein is a method of treating a KRas G12A-associated cancer or a KRas G12S-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), pancreatic cancer, or testicular cancer (e.g., seminoma). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12S inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), pancreatic cancer, or testicular cancer (e.g., seminoma) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-
a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12A-associated cancer or a KRas G12S-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12S inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), pancreatic cancer, or testicular cancer (e.g., seminoma). In some embodiments, the cancer has a KRas G12A mutation or a KRas G12V mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12V inhibitor, or both. Also provided herein is a method of treating a KRas G12A-associated cancer or a KRas G12V-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), pancreatic cancer, or
testicular cancer (e.g., seminoma). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12V inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), pancreatic cancer, or testicular cancer (e.g., seminoma) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12A-associated cancer or a KRas G12V-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12V inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some embodiments, the cancer has a KRas G12C mutation or a KRas G12S mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-
1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G12S inhibitor, or both. Also provided herein is a method of treating a KRas G12C-associated cancer or a KRas G12S-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G12S inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or
a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12C-associated cancer or a KRas G12S-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G12S inhibitor, or both. In some embodiments, the KRas-associated cancer is bladder cancer, colorectal cancer, skin cancer (e.g., melanoma), pancreatic cancer, or prostate cancer. In some embodiments, the cancer has a KRas G12D mutation or a KRas G12R mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12R inhibitor, or both. Also provided herein is a method of treating a KRas G12D-associated cancer or a KRas G12R-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is bladder cancer, colorectal cancer, skin cancer (e.g., melanoma), pancreatic cancer, or prostate cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)),
Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12R inhibitor, or both. Also provided herein is a method of treating bladder cancer, colorectal cancer, skin cancer (e.g., melanoma), pancreatic cancer, or prostate cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12D-associated cancer or a KRas G12R- associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12R inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some embodiments, the cancer has a KRas G12D mutation or a KRas G12S mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-
a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12S inhibitor, or both. Also provided herein is a method of treating a KRas G12D-associated cancer or a KRas G12S-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12S inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12D-associated cancer or a KRas
G12S-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G12S inhibitor, or both. In some embodiments, the KRas-associated cancer is bladder cancer, colorectal cancer, ovarian cancer, pancreatic cancer, or testicular cancer (e.g., seminoma). In some embodiments, the cancer has a KRas G12R mutation or a KRas G12V mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas G12V inhibitor, or both. Also provided herein is a method of treating a KRas G12R-associated cancer or a KRas G12V-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is bladder cancer, colorectal cancer, ovarian cancer, pancreatic cancer, or testicular cancer (e.g., seminoma). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1),
(IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas G12V inhibitor, or both. Also provided herein is a method of treating bladder cancer, colorectal cancer, ovarian cancer, pancreatic cancer, or testicular cancer (e.g., seminoma) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12R-associated cancer or a KRas G12V- associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas G12V inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some embodiments, the cancer has a KRas G12S mutation or a KRas Q61H mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas Q61H inhibitor,
or both. Also provided herein is a method of treating a KRas G12S-associated cancer or a KRas Q61H-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas Q61H inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, esophageal or stomach cancer, lung cancer (e.g., NSCLC), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12S-associated cancer or a KRas Q61H-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4),
(I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas Q61H inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), ovarian cancer, or testicular cancer (e.g., seminoma). In some embodiments, the cancer has a KRas G12V mutation or a KRas Q61L mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12V inhibitor, a KRas Q61L inhibitor, or both. Also provided herein is a method of treating a KRas G12V-associated cancer or a KRas Q61L-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), ovarian cancer, or testicular cancer (e.g., seminoma). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-
b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12V inhibitor, a KRas Q61L inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), ovarian cancer, or testicular cancer (e.g., seminoma) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12V-associated cancer or a KRas Q61L-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12V inhibitor, a KRas Q61L inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some embodiments, the cancer has a KRas G12A mutation or a KRas G12C mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12C inhibitor, or both. Also provided herein is a method of treating a KRas G12A-associated cancer or a KRas
G12C-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12C inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12A-associated cancer or a KRas G12C-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2),
or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12C inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some embodiments, the cancer has a KRas G12A mutation or a KRas G12D mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12D inhibitor, or both. Also provided herein is a method of treating a KRas G12A-associated cancer or a KRas G12D-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12D inhibitor, or both.
Also provided herein is a method of treating colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12A-associated cancer or a KRas G12D-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12D inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some embodiments, the cancer has a KRas G12A mutation or a KRas G13C mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G13C inhibitor, or both. Also provided herein is a method of treating a KRas G12A-associated cancer or a KRas G13C-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-
b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G13C inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12A-associated cancer or a KRas G13C-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G13C inhibitor,
or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some embodiments, the cancer has a KRas G12A mutation or a KRas Q61H mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas Q61H inhibitor, or both. Also provided herein is a method of treating a KRas G12A-associated cancer or a KRas Q61H-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas Q61H inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of
Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12A-associated cancer or a KRas Q61H-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas Q61H inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or testicular cancer (e.g., seminoma). In some embodiments, the cancer has a KRas G12A mutation or a KRas Q61L mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas Q61L inhibitor, or both. Also provided herein is a method of treating a KRas G12A-associated cancer or a KRas Q61L-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a),
(IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or testicular cancer (e.g., seminoma). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas Q61L inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or testicular cancer (e.g., seminoma) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12A-associated cancer or a KRas Q61L- associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas Q61L inhibitor, or both. In some embodiments, the KRas-associated cancer is bladder cancer, colorectal cancer,
skin cancer (e.g., melanoma), or pancreatic cancer. In some embodiments, the cancer has a KRas G12C mutation or a KRas G12R mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G12R inhibitor, or both. Also provided herein is a method of treating a KRas G12C-associated cancer or a KRas G12R-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is bladder cancer, colorectal cancer, skin cancer (e.g., melanoma), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G12R inhibitor, or both. Also provided herein is a method of treating bladder cancer, colorectal cancer, skin cancer (e.g., melanoma), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-
b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12C-associated cancer or a KRas G12R-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G12R inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some embodiments, the cancer has a KRas G12C mutation or a KRas G13C mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G13C inhibitor, or both. Also provided herein is a method of treating a KRas G12C-associated cancer or a KRas G13C-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a
pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G13C inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12C-associated cancer or a KRas G13C-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas G13C inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, skin cancer (e.g., melanoma), or lung cancer (e.g., NSCLC). In some embodiments, the cancer has a KRas G12C mutation or a KRas Q61L mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-
a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas Q61L inhibitor, or both. Also provided herein is a method of treating a KRas G12C-associated cancer or a KRas Q61L-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, skin cancer (e.g., melanoma), or lung cancer (e.g., NSCLC). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas Q61L inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, skin cancer (e.g., melanoma), or lung cancer (e.g., NSCLC) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula
(VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12C-associated cancer or a KRas Q61L-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas Q61L inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some embodiments, the cancer has a KRas G12D mutation or a KRas G13C mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G13C inhibitor, or both. Also provided herein is a method of treating a KRas G12D-associated cancer or a KRas G13C-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-
a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G13C inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12D-associated cancer or a KRas G13C-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas G13C inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, skin cancer (e.g., melanoma), or lung cancer (e.g., NSCLC). In some embodiments, the cancer has a KRas G12D mutation or a KRas Q61L mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula
(V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas Q61L inhibitor, or both. Also provided herein is a method of treating a KRas G12D-associated cancer or a KRas Q61L-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, skin cancer (e.g., melanoma), or lung cancer (e.g., NSCLC). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas Q61L inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, skin cancer (e.g., melanoma), or lung cancer (e.g., NSCLC) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12D-associated cancer or a KRas Q61L-associated
cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas Q61L inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, skin cancer (e.g., melanoma), ovarian cancer, or testicular cancer (e.g., seminoma). In some embodiments, the cancer has a KRas G12R mutation or a KRas Q61L mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas Q61L inhibitor, or both. Also provided herein is a method of treating a KRas G12R-associated cancer or a KRas Q61L-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, skin cancer (e.g., melanoma), ovarian cancer, or testicular cancer (e.g., seminoma). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1),
(IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas Q61L inhibitor, or both. Also provided herein is a method of treating colorectal cancer, skin cancer (e.g., melanoma), ovarian cancer, or testicular cancer (e.g., seminoma) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12R-associated cancer or a KRas Q61L- associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas Q61L inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, skin cancer (e.g., melanoma), pancreatic cancer, or testicular cancer (e.g., seminoma). In some embodiments, the cancer has a KRas G12R mutation or a KRas Q61R mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas Q61R inhibitor,
or both. Also provided herein is a method of treating a KRas G12R-associated cancer or a KRas Q61R-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, skin cancer (e.g., melanoma), pancreatic cancer, or testicular cancer (e.g., seminoma). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas Q61R inhibitor, or both. Also provided herein is a method of treating colorectal cancer, skin cancer (e.g., melanoma), pancreatic cancer, or testicular cancer (e.g., seminoma) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12R-associated cancer or a KRas Q61R- associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or
(I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas Q61R inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some embodiments, the cancer has a KRas G12S mutation or a KRas G13C mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas G13C inhibitor, or both. Also provided herein is a method of treating a KRas G12S-associated cancer or a KRas G13C-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))),
or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas G13C inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12S-associated cancer or a KRas G13C-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas G13C inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, esophageal or stomach cancer, or lung cancer (e.g., NSCLC). In some embodiments, the cancer has a KRas G12S mutation or a KRas G13D mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas G13D inhibitor, or both. Also provided herein is a method of treating a KRas G12S-associated cancer or a KRas G13D-associated cancer in a subject in need of such treatment, the method comprising
administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, esophageal or stomach cancer, or lung cancer (e.g., NSCLC). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas G13D inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, esophageal or stomach cancer, or lung cancer (e.g., NSCLC) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12S-associated cancer or a KRas G13D- associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-
c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas G13D inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or testicular cancer (e.g., seminoma). In some embodiments, the cancer has a KRas G12S mutation or a KRas Q61L mutation. In some such embodiments, compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas Q61L inhibitor, or both. Also provided herein is a method of treating a KRas G12S-associated cancer or a KRas Q61L-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or testicular cancer (e.g., seminoma). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas Q61L inhibitor, or both.
Also provided herein is a method of treating colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or testicular cancer (e.g., seminoma) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12S-associated cancer or a KRas Q61L- associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas Q61L inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some embodiments, the cancer has a KRas G12V mutation or a KRas G13C mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12V inhibitor, a KRas G13C inhibitor, or both. Also provided herein is a method of treating a KRas G12V-associated cancer or a KRas G13C-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-
b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12V inhibitor, a KRas G13C inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, lung cancer (e.g., NSCLC), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12V-associated cancer or a KRas G13C-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12V inhibitor, a KRas G13C inhibitor,
or both. In some embodiments, the KRas-associated cancer is colorectal cancer, pancreatic cancer, testicular cancer (e.g., seminoma), or thyroid cancer. In some embodiments, the cancer has a KRas G12V mutation or a KRas Q61R mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12V inhibitor, a KRas Q61R inhibitor, or both. Also provided herein is a method of treating a KRas G12V-associated cancer or a KRas Q61R-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, pancreatic cancer, testicular cancer (e.g., seminoma), or thyroid cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12V inhibitor, a KRas Q61R inhibitor, or both. Also provided herein is a method of treating colorectal cancer, pancreatic cancer, testicular cancer (e.g., seminoma), or thyroid cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a
compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12V-associated cancer or a KRas Q61R-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12V inhibitor, a KRas Q61R inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, pancreatic cancer, or testicular cancer (e.g., seminoma). In some embodiments, the cancer has a KRas G12A mutation or a KRas G12R mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12R inhibitor, or both. Also provided herein is a method of treating a KRas G12A-associated cancer or a KRas G12R-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or
(V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, pancreatic cancer, or testicular cancer (e.g., seminoma). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12R inhibitor, or both. Also provided herein is a method of treating colorectal cancer, pancreatic cancer, or testicular cancer (e.g., seminoma) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12A-associated cancer or a KRas G12R-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G12R inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, endometrial cancer, or lung cancer (e.g., NSCLC). In some embodiments, the cancer has a KRas G12A mutation or a KRas G13D mutation. In some such embodiments, the compound of Formula
(A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G13D inhibitor, or both. Also provided herein is a method of treating a KRas G12A-associated cancer or a KRas G13D-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, endometrial cancer, or lung cancer (e.g., NSCLC). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G13D inhibitor, or both. Also provided herein is a method of treating colorectal cancer, endometrial cancer, or lung cancer (e.g., NSCLC) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or
(V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12A-associated cancer or a KRas G13D-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas G13D inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, pancreatic cancer, or testicular cancer (e.g., seminoma). In some embodiments, the cancer has a KRas G12A mutation or a KRas Q61R mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas Q61R inhibitor, or both. Also provided herein is a method of treating a KRas G12A-associated cancer or a KRas Q61R-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, pancreatic cancer, or testicular cancer (e.g., seminoma). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2),
(I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas Q61R inhibitor, or both. Also provided herein is a method of treating colorectal cancer, pancreatic cancer, or testicular cancer (e.g., seminoma) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12A-associated cancer or a KRas Q61R-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12A inhibitor, a KRas Q61R inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, skin cancer (e.g., melanoma), or pancreatic cancer. In some embodiments, the cancer has a KRas G12C mutation or a KRas Q61R mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-
a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas Q61R inhibitor, or both. Also provided herein is a method of treating a KRas G12C-associated cancer or a KRas Q61R-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, skin cancer (e.g., melanoma), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas Q61R inhibitor, or both. Also provided herein is a method of treating colorectal cancer, skin cancer (e.g., melanoma), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12C-associated cancer or a KRas Q61R-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-
a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12C inhibitor, a KRas Q61R inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, skin cancer (e.g., melanoma), or pancreatic cancer. In some embodiments, the cancer has a KRas G12D mutation or a KRas Q61R mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas Q61R inhibitor, or both. Also provided herein is a method of treating a KRas G12D-associated cancer or a KRas Q61R-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, skin cancer (e.g., melanoma), or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or
a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas Q61R inhibitor, or both. Also provided herein is a method of treating colorectal cancer, skin cancer (e.g., melanoma), or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12D-associated cancer or a KRas Q61R-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12D inhibitor, a KRas Q61R inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, pancreatic cancer, or testicular cancer (e.g., seminoma). In some embodiments, the cancer has a KRas G12R mutation or a KRas G12S mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas G12S inhibitor, or both. Also provided herein is a method of treating a KRas G12R-associated cancer or a KRas G12S-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A)
(e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, pancreatic cancer, or testicular cancer (e.g., seminoma). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas G12S inhibitor, or both. Also provided herein is a method of treating colorectal cancer, pancreatic cancer, or testicular cancer (e.g., seminoma) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12R-associated cancer or a KRas G12S-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically
acceptable salt thereof, is a KRas G12R inhibitor, a KRas G12S inhibitor, or both. In some embodiments, the KRas-associated cancer is bladder cancer, colorectal cancer, or skin cancer (e.g., melanoma). In some embodiments, the cancer has a KRas G12R mutation or a KRas G13D mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas G13D inhibitor, or both. Also provided herein is a method of treating a KRas G12R-associated cancer or a KRas G13D-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is bladder cancer, colorectal cancer, or skin cancer (e.g., melanoma). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas G13D inhibitor, or both. Also provided herein is a method of treating bladder cancer, colorectal cancer, or skin cancer (e.g., melanoma) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-
b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12R-associated cancer or a KRas G13D-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas G13D inhibitor, or both. In some embodiments, the KRas-associated cancer is bladder cancer, colorectal cancer, or pancreatic cancer. In some embodiments, the cancer has a KRas G12R mutation or a KRas Q61H mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas Q61H inhibitor, or both. Also provided herein is a method of treating a KRas G12R-associated cancer or a KRas Q61H-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a
pharmaceutical composition as described herein. In some such embodiments, the cancer is bladder cancer, colorectal cancer, or pancreatic cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas Q61H inhibitor, or both. Also provided herein is a method of treating bladder cancer, colorectal cancer, or pancreatic cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12R- associated cancer or a KRas Q61H-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas Q61H inhibitor, or both. In some embodiments, the KRas-associated cancer is bile duct cancer (e.g., cholangiocarcinoma), colorectal cancer, or skin cancer (e.g., melanoma). In some embodiments, the cancer has a KRas G12R mutation or a KRas Q61K mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-
1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas Q61K inhibitor, or both. Also provided herein is a method of treating a KRas G12R-associated cancer or a KRas Q61K-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is bile duct cancer (e.g., cholangiocarcinoma), colorectal cancer, or skin cancer (e.g., melanoma). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas Q61K inhibitor, or both. Also provided herein is a method of treating bile duct cancer (e.g., cholangiocarcinoma), colorectal cancer, or skin cancer (e.g., melanoma) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a
pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12R-associated cancer or a KRas Q61K- associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12R inhibitor, a KRas Q61K inhibitor, or both. In some embodiments, the KRas-associated cancer is colorectal cancer, pancreatic cancer, or testicular cancer (e.g., seminoma). In some embodiments, the cancer has a KRas G12S mutation or a KRas Q61R mutation. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas Q61R inhibitor, or both. Also provided herein is a method of treating a KRas G12S-associated cancer or a KRas Q61R-associated cancer in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is colorectal cancer, pancreatic cancer, or testicular cancer (e.g., seminoma). In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g.,
Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas Q61R inhibitor, or both. Also provided herein is a method of treating colorectal cancer, pancreatic cancer, or testicular cancer (e.g., seminoma) in a subject in need of such treatment, the method comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein. In some such embodiments, the cancer is a KRas G12S-associated cancer or a KRas Q61R-associated cancer. In some such embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is a KRas G12S inhibitor, a KRas Q61R inhibitor, or both. Also provided herein is a method for treating a subject diagnosed with or identified as having a KRas-associated cancer, e.g., any of the exemplary mutant KRas-associated cancers disclosed herein, comprising administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2))) or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof, as defined
herein. Accordingly, provided herein are methods for treating a subject diagnosed with (or identified as having) a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))) that include administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof. In some embodiments, the subject that has been identified or diagnosed as having a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))) through the use of a regulatory agency-approved, e.g., FDA-approved test or assay for identifying a dysregulation associated with KRas (e.g., a KRAS gene having a mutation corresponding to a mutation in a KRas protein and/or a KRas protein having a mutation, a KRAS gene copy number increase, and/or an increase in KRas mRNA or protein expression), in a subject or a biopsy sample from the subject or by performing any of the non-limiting examples of assays described herein. In some embodiments, the subject that has been identified or diagnosed as having a KRas-associated cancer through the use of a regulatory agency-approved, e.g., FDA-approved test or assay for identifying a KRas dysregulation (e.g., KRas mutation) in a subject or a biopsy sample from the subject or by performing any of the non-limiting examples of assays described herein. In some embodiments, the test or assay is provided as a kit. Also provided are methods for treating cancer in a subject in need thereof, the method comprising: (a) detecting a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V- associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))) in the subject; and (b) administering to the subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b),
(II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof. Some embodiments of these methods further include administering to the subject another anticancer agent (e.g., a second compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, an immunotherapy, or any of the other anticancer agents described herein). In some embodiments, the subject was previously treated with another anticancer treatment, e.g., a kinase inhibitor (e.g., an EGFR inhibitor), at least partial resection of the tumor, radiation therapy, or a combination thereof. In some embodiments, the cancer in the subject is determined to have a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation))) through the use of a regulatory agency-approved, e.g., FDA- approved test or assay for identifying a KRas mutation (e.g., a KRAS gene having a mutation corresponding to a mutation in a KRas protein and/or a KRas protein having a mutation, a KRAS gene copy number increase, and/or an increase in KRas mRNA or protein expression), in a subject or a sample (e.g., a tumor sample or blood sample) from the subject or by performing any of the non-limiting examples of assays described herein. In some embodiments, the subject that has been identified or diagnosed as having a cancer with a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation))) through the use of a regulatory agency-approved, e.g., FDA-approved test or assay for identifying a KRas dysregulation in a subject or a biopsy sample from the subject or by performing any of the non- limiting examples of assays described herein. In some embodiments, the test or assay is provided as a kit. Also provided are methods of treating a subject that include performing an assay on a sample (e.g., a tumor sample or a blood sample) obtained from the subject to determine whether
the cancer in the subject has a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation))), and administering (e.g., specifically or selectively administering) a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof, to the subject determined a cancer having a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation))). In some embodiments, provided are methods of treating a subject that include performing an assay on a sample (e.g., a tumor sample or a blood sample) obtained from the subject to determine whether the cancer in the subject has a KRas dysregulation (e.g., a KRAS gene having a mutation corresponding to a mutation in a KRas protein and/or a KRas protein having a mutation, a KRAS gene copy number increase, and/or an increase in KRas mRNA or protein expression), and administering (e.g., specifically or selectively administering) a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof, to the subject having a cancer determined to have a KRas dysregulation. Some embodiments of these methods further include administering to the subject another anticancer agent (e.g., a second compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c),
(VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or immunotherapy). In some embodiments of these methods, the subject was previously treated with another anticancer treatment, e.g., a first KRas inhibitor, a kinase inhibitor (e.g., an EGFR inhibitor), at least partial resection of a tumor, radiation therapy, or a combination thereof. In some embodiments, the subject is a subject suspected of having a KRas-associated cancer (e.g., a mutant KRas- associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V- associated cancer))), a subject presenting with one or more symptoms of a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))), or a subject having an elevated risk of developing a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D- associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))). In some embodiments, the assay utilizes next generation sequencing, pyrosequencing, or immunohistochemistry. In some embodiments, the assay is a regulatory agency-approved assay, e.g., FDA-approved kit. In some embodiments, the assay is a liquid biopsy. Additional, non-limiting assays that may be used in these methods are described herein. Additional assays are also known in the art. Also provided is a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof, for use in treating a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D- associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))) in a subject identified or diagnosed as having a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))) through a step of performing an assay (e.g., an in vitro assay) on a sample (e.g., a tumor sample or a blood sample) obtained from the subject to determine whether the cancer in the subject has a KRas
dysregulation, where the presence of a KRas dysregulation (e.g., a KRAS gene having a mutation corresponding to a mutation in a KRas protein and/or a KRas protein having a mutation, a KRAS gene copy number increase, and/or an increase in KRas mRNA or protein expression), identifies that the cancer in the subject has a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R- associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))). Also provided is the use of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, for the manufacture of a medicament for treating a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))) in a subject identified or diagnosed as having a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))) through a step of performing an assay on a sample obtained from the subject to determine whether the cancer in the subject has a KRas dysregulation (e.g., a KRAS gene having a mutation corresponding to a mutation in a KRas protein and/or a KRas protein having a mutation, a KRAS gene copy number increase, and/or an increase in KRas mRNA or protein expression) where the presence of a KRas dysregulation (e.g., a KRAS gene having a mutation corresponding to a mutation in a KRas protein and/or a KRas protein having a mutation, a KRAS gene copy number increase, and/or an increase in KRas mRNA or protein expression), identifies that the subject has a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V- associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))). Some embodiments of any of the methods or uses described herein further include recording in the subject’s clinical record (e.g., a computer readable medium) that the subject is determined to have a KRas dysregulation (e.g., a KRAS gene having a mutation corresponding
to a mutation in a KRas protein and/or a KRas protein having a mutation, a KRAS gene copy number increase, and/or an increase in KRas mRNA or protein expression), through the performance of the assay, should be administered a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof. In some embodiments, the assay utilizes next generation sequencing, pyrosequencing, or immunohistochemistry. In some embodiments, the assay is a regulatory agency-approved assay, e.g., FDA-approved kit. In some embodiments, the assay is a liquid biopsy. Also provided is a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, for use in the treatment of a cancer in a subject in need thereof, or a subject identified or diagnosed as having a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R- associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))) (e.g., any of the KRas-associated cancers described herein). Also provided is the use of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, for the manufacture of a medicament for treating a cancer in a subject identified or diagnosed as having a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated
cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))) (e.g., any of the KRas-associated cancers described herein). In some embodiments, a mutant KRas-associated cancer is a cancer that was previously identified as having no KRas mutation (e.g., KRas wild type), for example, in a cancer that was previously identified as having no KRas mutation and then, later, a KRas mutation (e.g., a resistance mutation) was identified. In some embodiments, a subject is identified or diagnosed as having a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D- associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))) through the use of a regulatory agency-approved, e.g., FDA-approved, kit for identifying a KRas dysregulation, in a subject or a biopsy sample from the subject. As provided herein, a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R- associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))) includes those described herein and known in the art. In some embodiments of any of the methods or uses described herein, the subject has been identified or diagnosed as having a cancer with a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation)) or amplification). In some embodiments of any of the methods or uses described herein, the subject has a cancer (e.g., a tumor sample) that is positive for a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation)) or amplification). In some embodiments of any of the methods or uses described herein, the subject can be a subject with a cancer (e.g., one or more tumor samples) that is positive for a KRas dysregulation (e.g., KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation))). In some embodiments of any of the methods or uses described herein, the subject is suspected of having a mutant KRas-associated cancer (e.g., a cancer that was previously identified a cancer having no KRas mutation (e.g., KRas wild type)). In some embodiments, provided herein are methods for treating a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R- associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))) in a subject in need of such treatment, the method comprising a) detecting a KRas dysregulation (e.g., a KRAS gene having a mutation
corresponding to a mutation in a KRas protein and/or a KRas protein having a mutation, a KRAS gene copy number increase, and/or an increase in KRas mRNA or protein expression) in a sample from the subject; and b) administering a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof. In some embodiments, a mutant KRas-associated cancer is characterized by a mutation that arises from treatment with a first KRas inhibitor; for example, a mutant KRas-associated cancer as described herein can include one or more KRas mutations that confer resistance to treatment with a first KRas inhibitor. For example, a subject can acquire one or more of the following KRas mutations as a resistance mutation to a KRas G12C inhibitor: G12D, G12R, G12V, G12W, G13D, Q61H, R68S, H95D, H95Q, H95R, or Y96C. See, e.g., Awad et al. N Engl J Med.2021 Jun 24;384(25):2382-2393, doi: 10.1056/NEJMoa2105281. As used herein, a “first inhibitor of KRas” or “first KRas inhibitor” is a KRas inhibitor as defined herein, but which does not include a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, as defined herein. As used herein, a “second inhibitor of KRas” or a “second KRas inhibitor” is a KRas inhibitor as defined herein, but which does not include a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof. When both a first and a second inhibitor
of KRas are present in a method provided herein, the first and second inhibitors of KRas are different. In some embodiments, the first and/or second inhibitor of KRas bind in a different location than a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I- a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III- 2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV- b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof. Exemplary first and second inhibitors of KRas are described herein. In some embodiments, a first or a second inhibitor of KRas can be a KRas G12C inhibitor. In some embodiments, a first or second inhibitor of KRas can be selected from the group consisting of sotorasib, adragrasib, ARS-853, ARS-1620, ARS-3248, ATG-012, BI 1823911, D-1553, ERAS-3490, GDC-6036, GFH925, JAB-21822, JDQ-443, LY3537982, MRTX-1257, RMC- 6291, and combinations thereof. In some embodiments, a first or second inhibitor of KRas can be selected from the group consisting of sotorasib, adragrasib, ARS-853, ARS-1620, ARS- 3248, ATG-012, BI 1823911, D-1553, ERAS-3490, GDC-6036, GFH925, JAB-21822, JDQ- 443, LY3537982, MRTX-1133, MRTX-1257, RMC-6291, RMC-6236, and combinations thereof. In some embodiments, the methods provided herein include performing an assay on a sample (e.g., a tumor sample or a blood sample) obtained from the subject to determine whether the cancer in the subject has a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation)) or amplification). In some such embodiments, the method also includes administering to a subject determined to have a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation)) or amplification) a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or
a pharmaceutically acceptable salt thereof. In some embodiments, the method includes determining that a cancer in a subject has a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation)) or amplification) via an assay performed on a sample obtained from the subject. In such embodiments, the method also includes administering to a subject a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof. Some embodiments of these methods further include administering to the subject another anticancer agent (e.g., a second compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or immunotherapy). In some embodiments of any of the methods or uses described herein, an assay is used to determine whether the cancer in the subject has a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation)) or amplification), using a sample (e.g., a tumor sample or a blood sample) from a subject can include, for example, next generation sequencing, immunohistochemistry, fluorescence microscopy, Southern blotting, Western blotting, FACS analysis, Northern blotting, and PCR-based amplification (e.g., RT-PCR and quantitative real- time RT-PCR). As is well-known in the art, the assays are typically performed, e.g., with at least one labelled nucleic acid probe or at least one labelled antibody or antigen-binding fragment thereof. Assays can utilize other detection methods known in the art for detecting a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation)) or amplification) (see, e.g., the references cited herein). In some embodiments, the sample is
tumor biopsy sample (e.g., a paraffin-embedded biopsy sample) from the subject. In some embodiments, the subject is a subject suspected of having a KRas-associated cancer (e.g., mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R- associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))), a subject having one or more symptoms of a KRas- associated cancer (e.g., mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D- associated cancer or a KRas G12V-associated cancer))), and/or a subject that has an increased risk of developing a KRas-associated cancer (e.g., mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))). In some embodiments, a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation)) or amplification) can be identified using a liquid biopsy (variously referred to as a fluid biopsy or fluid phase biopsy). See, e.g., Karachialiou et al., “Real-time liquid biopsies become a reality in cancer treatment”, Ann. Transl. Med., 3(3):36, 2016, doi: 10.3978/j.issn.2305-5839.2015.01.16. Liquid biopsy methods can be used to detect total tumor burden and/or the KRas dysregulation (e.g., the KRas mutation or amplification). Liquid biopsies can be performed on biological samples obtained relatively easily from a subject (e.g., via a simple blood draw) and are generally less invasive than traditional methods used to detect tumor burden and/or KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation)) or amplification). In some embodiments, liquid biopsies can be used to detect the presence of a KRas dysregulation (e.g., a KRas mutation or amplification) at an earlier stage than traditional methods. In some embodiments, the biological sample to be used in a liquid biopsy can include, blood, plasma, urine, cerebrospinal fluid, saliva, sputum, broncho- alveolar lavage, bile, lymphatic fluid, cyst fluid, stool, ascites, and combinations thereof. In some embodiments, a liquid biopsy can be used to detect circulating tumor cells (CTCs). In some embodiments, a liquid biopsy can be used to detect cell-free DNA. In some embodiments, cell-free DNA detected using a liquid biopsy is circulating tumor DNA (ctDNA) that is derived from tumor cells. Analysis of ctDNA (e.g., using sensitive detection techniques such as, without limitation, next-generation sequencing (NGS), traditional PCR, digital PCR, or microarray analysis) can be used to identify a KRas dysregulation (e.g., KRas mutation (e.g.,
a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation))). Also provided is a method for modulating (e.g., decreasing) KRas protein activity (e.g., dysregulated KRas protein activity (e.g., mutant KRas protein activity (e.g., KRas G12R mutant protein activity or G12V mutant protein activity))) in a cell, comprising contacting the cell with a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof. In some embodiments, the contacting is in vitro. In some embodiments, the contacting is in vivo. In some embodiments, the contacting is ex vivo. In some embodiments, the contacting is in vivo, wherein the method comprises administering an effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, to a subject having a cell having a KRas protein (e.g., a dysregulated KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein or a KRas G12V mutant protein)))). In some embodiments, the contacting is ex vivo, wherein the method comprises contacting a cell from a subject having a KRas protein (e.g., a dysregulated KRas protein (e.g., a mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein or a KRas G12V mutant protein)))) with a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a),
(VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof. In some embodiments, the cell is a cancer cell. In some embodiments, the cell is a mammalian cell. In some embodiments, the cell is a mammalian cancer cell. In some embodiments, the cancer cell is any cancer as described herein. In some embodiments, the cancer cell is a KRas-associated cancer cell (e.g., a mutant KRas-associated cancer cell (e.g., a KRas G12D-associated cancer cell, a KRas G12R-associated cancer cell, or a KRas G12V-associated cancer cell (e.g., a KRas G12D-associated cancer cell or a KRas G12V-associated cancer cell))). As used herein, the term “contacting” refers to the bringing together of indicated moieties in an in vitro system, an in vivo system, or an ex vivo system. For example, “contacting” a KRas protein with a compound provided herein includes the administration of a compound provided herein to an individual or subject, such as a human, having a KRas protein, as well as, for example, introducing a compound provided herein into a sample containing a cellular or purified preparation containing the KRas protein. Also provided herein is a method of inhibiting cell proliferation, in vitro, in vivo, or ex vivo, the method comprising contacting a cell with an effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof as defined herein. In some embodiments, the cell has a KRas dysregulation. In some embodiments, the cell has a KRas mutation. In some embodiments, the cell has a KRas G12D mutation or a KRas G12V mutation. In some embodiments, the cell has a KRas G12D mutation. In some embodiments, the cell has a KRas G12R mutation. In some embodiments, the cell has a KRas G12V mutation. In some embodiments, the cell has a KRas amplification. Further provided herein is a method of increasing cell death, in vitro, in vivo, or ex vivo, the method comprising contacting a cell with an effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g.,
Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof as defined herein. Also provided herein is a method of increasing tumor cell death in a subject. The method comprises administering to the subject a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I- a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, in an amount effective to increase tumor cell death. In some embodiments, the cell has a KRas dysregulation. In some embodiments, the cell has a KRas mutation. In some embodiments, the cell has a KRas G12D mutation or a KRas G12V mutation. In some embodiments, the cell has a KRas G12D mutation. In some embodiments, the cell has a KRas G12R mutation. In some embodiments, the cell has a KRas G12V mutation. In some embodiments, the cell has a KRas amplification. When employed as pharmaceuticals, the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, can be administered in the form of pharmaceutical compositions as described herein. Also provided herein is a method for inhibiting a KRas protein in a mammalian cell, the method comprising contacting the mammalian cell with an effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof. Also provided herein is a method for inhibiting a dysregulated KRas protein (e.g., a
mutant KRas protein (e.g., a KRas G12D mutant protein, a KRas G12R mutant protein, or a KRas G12V mutant protein (e.g., a KRas G12D mutant protein or a KRas G12V mutant protein))) in a mammalian cell, the method comprising contacting the mammalian cell with an effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof. In some embodiments, the mammalian cell is ex vivo. Also provided herein is a method of treating a subject having a cancer, wherein the method comprises: administering a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II- b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV- b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V- b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, as a monotherapy or in conjunction with a first anticancer agent to the subject who has been administered one or more doses of the first anticancer agent to the subject for a period of time. Also provided herein is a method of treating a subject having a cancer, wherein the method comprises: (a) administering one or more doses of a first anticancer agent to the subject for a period of time; (b) after (a), determining whether a cancer cell in a sample (e.g., a tumor sample or a blood sample) obtained from the subject has a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation)) or amplification); and (c) administering a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or
(II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, as a monotherapy or in conjunction with a second anticancer agent to the subject if the subject has been determined to have a cancer cell that has a KRas dysregulation (e.g., a KRas mutation or amplification); or (d) administering additional doses of the first anticancer agent of step (a) to the subject if the subject has not been determined to have a cancer cell that has a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation)) or amplification). Further provided herein is a method of treating a subject having a cancer, wherein the method comprises: (a) determining whether a cancer cell in a sample (e.g., a tumor sample or a blood sample) obtained from a subject having a cancer and previously administered one or more doses of a first anticancer agent has a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation)) or amplification); and (b) administering a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, as a monotherapy or in conjunction with a second anticancer agent to the subject if the subject has been determined to have a cancer cell that has a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation)) or amplification); or (c) administering additional doses of the first anticancer agent to the subject if the subject has not been determined to have a cancer cell that has a KRas dysregulation (e.g., KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation)) or amplification).
Also provided herein is a method of treating a subject having a cancer, wherein the method comprises: (a) determining that a cancer cell in a sample (e.g., a tumor sample or a blood sample) obtained from a subject having a cancer and previously administered one or more doses of a first anticancer agent has a KRas dysregulation (e.g., a KRas mutation (e.g., a KRas G12D mutation, a KRas G12R mutation, or a KRas G12V mutation (e.g., a KRas G12D mutation or a KRas G12V mutation)) or amplification); and (b) administering a therapeutically effective amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, as a monotherapy or in conjunction with a second anticancer agent to the subject. In some embodiments of any of the methods described herein, the first anticancer agent can be a first KRas inhibitor. When employed as pharmaceuticals, the compounds Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, can be administered in the form of pharmaceutical compositions as described herein. Combinations In any of the indications described herein, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c),
(VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can be used as a monotherapy. In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, can be used prior to administration of an additional therapeutic agent or additional therapy. For example, a subject in need thereof can be administered one or more doses of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, for a period of time and then undergo at least partial resection of the tumor. In some embodiments, the treatment with one or more doses of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, reduces the size of the tumor (e.g., the tumor burden) prior to the at least partial resection of the tumor. In some embodiments, a subject in need thereof can be administered one or more doses of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II- b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, for a period of time and under one or more rounds of
radiation therapy. In some embodiments, the treatment with one or more doses of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, reduces the size of the tumor (e.g., the tumor burden) prior to the one or more rounds of radiation therapy. In some embodiments, a subject has a cancer (e.g., a locally advanced or metastatic tumor) that is refractory or intolerant to standard therapy (e.g., administration of a chemotherapeutic agent, such as a first KRas inhibitor, a kinase inhibitor, immunotherapy, or radiation. In some embodiments, a subject has a cancer (e.g., a locally advanced or metastatic tumor) that is refractory or intolerant to prior therapy (e.g., administration of a chemotherapeutic agent, such as a first KRas inhibitor, a kinase inhibitor, immunotherapy, or radiation). In some embodiments, a subject has a cancer (e.g., a locally advanced or metastatic tumor) that has no standard therapy. In some embodiments, a subject is KRas inhibitor naïve. In some embodiments, a subject is not KRas inhibitor naïve. In some embodiments, a subject has undergone prior therapy. For example, treatment with surgery, radiation, a chemotherapeutic agent, an immunotherapy, a multi-kinase inhibitor (MKI), a KRas inhibitor, a RAF/MEK/PI3K pathway inhibitor, a MEK inhibitor, a Raf inhibitor, a YAP inhibitor, a proteasome inhibitor, a PI3K-AKT-mTOR pathway inhibitor, an ERK inhibitor, a pan-ErbB inhibitor, a MET inhibitor, a farnesyl transferase inhibitor, a FAK inhibitor, a HSP90 inhibitor, or a combination thereof. In some embodiments of any the methods described herein, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is administered in combination with a therapeutically effective amount of at least one additional therapeutic agent selected from one or more additional therapies or therapeutic (e.g.,
chemotherapeutic) agents. Non-limiting examples of additional therapeutic agents include: RAS pathway targeted therapeutic agents (e.g., Ras/RAF/MEK/PI3K pathway inhibitors or degraders, (e.g., Ras inhibitors or degraders, KRas-targeted therapeutic agents, SOS1 inhibitors or degraders, SOS1/Ras protein-protein interaction inhibitors, SHP2 inhibitors or degraders, PI3K-AKT- mTOR pathway inhibitors or degraders)), kinase-targeted therapeutics (e.g., MEK inhibitors or degraders, ERK inhibitors or degraders, Raf inhibitors or degraders (e.g., BRaf inhibitors or degraders), PI3K inhibitors or degraders, AKT inhibitors or degraders, mTOR inhibitors or degraders, CDK4/5 inhibitors or degraders, CDK4/6 inhibitors or degraders, MET inhibitors or degraders, FAK inhibitors or degraders, ErbB family inhibitors or degraders (e.g., EGFR inhibitors or degraders, Her2 inhibitors or degraders), Src inhibitors or degraders), mTORC1 inhibitors or degraders, YAP inhibitors or degraders, proteasome inhibitors or degraders, farnesyl transferase inhibitors or degraders, HSP90 inhibitors or degraders, PTEN inhibitors or degraders, signal transduction pathway inhibitors or degraders, checkpoint inhibitors, modulators of the apoptosis pathway (e.g., venetoclax, navitoclax, obataclax), chemotherapeutics, angiogenesis-targeted therapies, immune-targeted agents including immunomodulatory imide drugs (sometimes called “IMiDs” or “CELMoDs”), immunotherapy (e.g., anti-PD1, anti-PD-L1, anti-CTLA4, anti-LAG3, anti-TIM3, anti-B7-H3, anti-VISTA therapies, including antibodies (e.g., single-targeted antibodies targeting one or more of PD1, PD-L1, CTLA4, LAG3, TIM3, B7-H3, or VISTA; bispecific antibodies (including bispecific T cell engagers (BiTEs)) targeting one or more of PD1, PD-L1, CTLA4, LAG3, TIM3, B7- H3, or VISTA; and antibody-drug conjugates (ADCs) incorporating one or more of PD1, PD- L1, CTLA4, LAG3, TIM3, B7-H3, or VISTA) or antigen-binding fragments thereof, a PD-1 inhibitor, a PD-L1 inhibitor, or an ADOR2A inhibitor), cell-based therapeutics (e.g., adoptive cell therapy (e.g., CAR T therapy, cytokine-induced killer cells (CIKs), natural killer cells (e.g., CAR-modified NK cells)) or antibody-armed cell therapy), and radiotherapy. See also, e.g., the therapeutic agents listed in U.S. Publication No. US 2021/0130303. A “degrader” as used herein is a heterobifunctional molecule that induces degradation of a target protein, the degrader including a moiety that binds to the target protein and a moiety that binds to a ubiquitin E3 ligase (sometimes referred to as an E3 ligase or simply an E3), these two moieties being optionally separated by a linker. Such degraders are sometimes known as “PROTACs”. A “Ras pathway targeted therapeutic agent” as used herein includes any compound
exhibiting inactivation activity of any protein in a Ras pathway (e.g., kinase inhibition, allosteric inhibition, inhibition of dimerization, and/or induction of degradation). Non-limiting examples of a protein in a Ras pathway include any one of the proteins in the Ras-RAF-MAPK pathway or PI3K/AKT pathway such as Ras (e.g., KRas, HRas, and NRas), RAF, BRAF, MEK, ERK, PI3K, AKT, and mTOR. In some embodiments, a Ras pathway modulator can be selective for a protein in a Ras pathway, e.g., the Ras pathway modulator can be selective for Ras (also referred to as a Ras modulator). In some embodiments, a Ras modulator is a covalent inhibitor. In some embodiments, a Ras pathway targeted therapeutic agent is a “KRas pathway modulator.” A KRas pathway modulator includes any compound exhibiting inactivation activity of any protein in a KRas pathway (e.g., kinase inhibition, allosteric inhibition, inhibition of dimerization, and/or induction of degradation). Non-limiting examples of a protein in a KRas pathway include any one of the proteins in the KRas-RAF-MAPK pathway or PI3K/AKT pathway such as KRas, RAF, BRAF, MEK, ERK, PI3K, AKT, and mTOR. In some embodiments, a KRas pathway modulator is a KRas-targeted therapeutic agent. In some embodiments, the Ras pathway targeted therapeutic agent is a SOS1 inhibitor or a SHP2 inhibitor. Non-limiting examples of SOS1 inhibitors include MRTX-0902, and RMC-5845. Non-limiting examples of SHP2 inhibitors include batoprotafib (TNO-155), vociprotafib (RMC-4630), ARRY-558, BBP-398, ENT-03, ERAS-601, ET-0038, GDC-1971 (RLY-1971), GH-21, HS-10381, ICP-189, JAB-3068, JAB-3312, and SH-3809. Non-limiting examples of KRas-targeted therapeutic agents (e.g., a first KRas inhibitor or a second KRas inhibitor) include a KRas-selective inhibitor, a Ras inhibitor, and an anti- KRas antibody. In some embodiments, the KRas inhibitor is a covalent inhibitor. In some embodiments, the KRas-targeted therapeutic agent is adagrasib, divarasib (GDC-6036), sotorasib, ARS-1620, ARS-3248, ARS-853, ASP-3082, ATG-012, BI-1701963, BI-1823911, BPI-421286, D-1553, ERAS-3490, GFH-925, JAB-21822, JDQ-443, LY-3537982, MRTX- 1133, MRTX-1257, RMC-6236, RMC-6291, RSC-1255, or a combination thereof. In some embodiments, the KRas-targeted therapeutic agent is an agent that inhibits the interaction between KRas and SOS1 or SHP2. Non-limiting examples of an agent that inhibits the interaction between SOS1 and KRas include BI-3406, BI-1701963, and BAY 293. Additional KRas-targeted therapeutic agents (e.g., a first KRas inhibitor or a second KRas inhibitor) include those disclosed in International Publication Nos. WO 2021/104431; WO WO2021/119343; WO2021/113595; WO 2021/107160; WO 2016/161361; WO 2016/17262; WO 2020/035031; WO 2021/041671; WO 2016/077793; WO 2020/180768; WO
2021/092115; WO 2020/180770; U.S. Patent Nos. US 10,898,487; US 10,829,487; US 10,858,359; US 10,561,655; US 10,532,042; U.S. Publication Nos. US 2021/0101870; US 2019/0231805; US 2020/0017517; US 2020/0017511; US 2020/0147058; US 2021/0009577; and Hillig et al. PNAS.2019; 116(7): 2551-2560, doi: 10.1073/pnas.1812963116. Further non-limiting examples of Ras pathway-targeted therapeutic agents include BRAF inhibitors, MEK inhibitors, ERK inhibitors, PI3K inhibitors, AKT inhibitors, and mTOR inhibitors. In some embodiments, the BRAF inhibitor is avutometinib, dabrafenib (e.g., dabrafenib mesylate, TAFINLAR®), encorafenib (BRAFTOVI™), naporafenib, sorafenib (e.g., sorafenib tosylate), vemurafenib (ZELBORAF®), ARQ 736, AZ304, BMS-908662 (XL281), C17071479-F, CHIR-265, FORE-8394, GDC-0879, GSK2118436, HLX-208, HM95573, LGX818, LXH254, PLX-3603, PLX-4720, PLX-8394, RAF265, RO5126766, RO5185426, or a combination thereof. In some embodiments, the BRAF inhibitor is avutometinib, dabrafenib (e.g., dabrafenib mesylate), encorafenib, naporafenib, sorafenib (e.g., sorafenib tosylate), vemurafenib, C17071479-F, CHIR-265, FORE-8394, HLX-208, or a combination thereof. In some embodiments, the MEK inhibitor is avutometinib, binimetinib (MEKTOVI®, MEK162), cobimetinib (e.g., cobimetinib fumarate, COTELLIC®), mirdametinib, pimasertib, refametinib, selumetinib (e.g., selumetinib sulfate, AZD6244), trametinib (e.g., trametinib dimethyl sulfoxide, GSK-1120212 MEKINIST®), zapnometinib, hypothemycin, CI1040 (PD184352), CS3006, FCN-159, MSC1936369B, NFX-179, PD0325901, PD98059,RO5126766, SHR7390, TAK-733, WX-554, or a combination thereof. In some embodiments, the MEK inhibitor is avutometinib, binimetinib, cobimetinib (e.g., cobimetinib fumarate), mirdametinib, pimasertib, refametinib, selumetinib (e.g., selumetinib sulfate), trametinib (e.g., trametinib dimethyl sulfoxide, GSK-1120212), zapnometinib, FCN-159, NFX-179, TAK-733, or a combination thereof. In some embodiments, the ERK inhibitor is 25-OH-D3-3-BE (B3CD, bromoacetoxycalcidiol), 5-7-Oxozeaenol, 5-iodotubercidin, AEZ-131 (AEZS-131), AEZS- 136, ASN007, AZ-13767370, BL-EI-001, CC-90003, FR148083, FR-180204, FRI-20 (ON- 01060), GDC0994, GDC-0994 (RG-7482), KO-947, KO-947, LTT-462, LY-3214996, MK- 8353 (SCH900353), ONC201SCH772984, ulixertinib (BVD-523), VTX-11e, or a combination thereof. In some embodiments, the ERK inhibitor is rineterkib, ulixertinib, or a combination thereof.
In some embodiments, PI3K inhibitor is alpelisib (BYL719), apitolisib (GDC-0980), buparlisib (BKM120), copanlisib (ALIQOPA™, BAY80-6946), dactolisib (NVP-BEZ235, BEZ-235), gedatolisib (PF-05212384, PKI-587), omipalisib (GSK2126458, GSK458), pictilisib (GDC-0941), pilaralisib (XL147, SAR245408), rigosertib, serabelisib (TAK-117, MLN1117, INK 1117), sonolisib (PX-866), taselisib (GDC-0032, RG7604), voxtalisib (XL756, SAR245409), wortmannin, AMG 511, AMG319, ASN003, AZD8835, BGT-226 (NVP-BGT226), CH5132799, CUDC-907, GDC-0077, GDC-0084 (RG7666), GS-9820, GSK1059615, GSK2636771, KIN-193 (AZD-6428), LY2023414, LY294002, PF-04691502, PI-103, PKI-402, PQR309, SAR260301, SF1126, VS-5584 (SB2343), WX-037, XL-765, ZSTK474, or a combination thereof. In some embodiments, the PI3K inhibitor is alpelisib, amdizalisib, apitolisib, bimiralisib, buparlisib, copanlisib (e.g., copanlisib dihydrochloride or a hydrate of copanlisib dihydrochloride), dactolisib, dezapelisib, dordaviprone, duvelisib (e.g., a hydrate of duvelisib), eganelisib, fimepinostat, gedatolisib, idelalisib, inavolisib, leniolisib (e.g., leniolisib phosphate), linperlisib, parsaclisib, paxalisib, risovalisib, seletalisib, serabelisib, sonolisib, tenalisib, umbralisib (e.g., umbralisib tosylate), zandelisib, PF- 04691502, SHC-014748-M, TQ-B-3525, or a combination thereof. In some embodiments, the AKT inhibitor is 2-[4-(2-aminoprop-2-yl)phenyl]-3- phenylquinoxaline, 3-oxo-tirucallic acid, A-443654, A-674563, afuresertib, API-1, ARQ092, AT13148, AT7867, AZD5363, BAY 1125976, boc-Phe-vinyl ketone, CCT128930, DC120, DM-PIT-1, edelfosine, erucylphophocholine, erufosine, GSK2141795, GSK690693, H-89, ipatasertib (GDC-0068, RG7440), lactoquinomycin, miltefosine (IMPADIVO®), MK-2206, N-(4-(5-(3-acetamidophenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b] pyridin-3- yl)benzyl)-3-fluorobenzamide, NL-71-101, ONC201, OSU-A9, Perifosine (D-21266), PH- 316, PHT-427, PIT-1, SR13668, TCN, TCN-P, triciribine (Triciribine Phosphate Monohydrate), uprosertib, wortmannin, or a combination thereof. In some embodiments, the AKT inhibitor is capivasertib (AZD-5363), miransertib (e.g., miransertib mesylate), pifusertib, uprosertib, BXT-10, or a combination thereof. In some embodiments, the mTOR inhibitor is MLN0128, AZD-2014, CC-223, AZD2014, CC-115, everolimus (RAD001), temsirolimus (CCI-779), ridaforolimus (AP- 23573), sirolimus (rapamycin), or a combination thereof. In some embodiments, the mTOR inhibitor is apitolisib, bimiralisib, dactolisib, everolimus, fosciclopirox (e.g., fosciclopirox sodium), gedatolisib, onatasertib, paxalisib, sapanisertib, sirolimus, sodium 2-
hydroxylinoleate, temsirolimus, umirolimus, zandelisib, zotarolimus, BI-860585, CC-115, PF- 04691502, or a combination thereof. In some embodiments, the farnesyl transferase inhibitor is lonafarnib, tipifarnib, BMS- 214662, L778123, L744832, and FTI-277. In some embodiments, the farnesyl transferase inhibitor is lonafarnib, tipifarnib, BMS-214662, or a combination thereof. In some embodiments, a chemotherapeutic agent includes a DNA replication inhibitor (e.g., a DNA intercalator (e.g., an anthracycline)), a DNA crosslinker (e.g., cyclophosphamide, a mitomycin (e.g., mitomycin C), a platinum complex), a ribonucleotide-diphosphate reductase inhibitor (e.g., gemcitabine), or a topoisomerase inhibitor), an anti-microtubule agent (e.g., a taxane a vinca alkaloid, or eribulin), or a combination thereof. Non-limiting examples of a taxane include paclitaxel, docetaxel, abraxane, and taxotere. In some embodiments, the anthracycline is selected from daunorubicin, doxorubicin, epirubicin, idarubicin, and combinations thereof. In some embodiments, the platinum-based agent is selected from carboplatin, cisplatin, oxaliplatin, nedplatin, triplatin tetranitrate, phenanthriplatin, picoplatin, satraplatin and combinations thereof. In some embodiments, the chemotherapy is a platinum complex, a microtubule inhibitor (e.g., a microtubule destabilizer or a microtubule stabilizer), a topoisomerase inhibitor, or an antibody-drug conjugate including any thereof. In some embodiments, the platinum complex is carboplatin, cisplatin, lobaplatin, miriplatin, oxaliplatin, or a combination thereof. In some embodiments, the microtubule inhibitor is cabazitaxel, colchicine, desoxyepothilone B, docetaxel, eribulin, ixabepilone, nab-paclitaxel, paclitaxel, plinabulin, sabizabulin, tirbanibulin, vinblastine, vinflunine, vinorelbine, or a combination thereof. In some embodiments, the microtubule inhibitor is cabazitaxel, docetaxel, nab-paclitaxel, paclitaxel, or a combination thereof. In some embodiments, the topoisomerase inhibitor is aclarubicin, amsacrine, belotecan, camptothecin, daunorubicin, dexrazoxane, elliptinium, epirubicin, etoposide, gepotidacin, idarubicin, mitoxantrone, nemonoxacin, pirarubicin, pixantrone, razoxane, rubitecan, sobuzoxane, temozolomide, teniposide, topotecan, SN-38, or a combination thereof. In some embodiments, the hypomethylating agent is azacitidine, decitabine, or a combination thereof. In some embodiments, the chemotherapy is a platinum complex and a topoisomerase inhibitor (e.g., cisplatin and etoposide). In some embodiments, the antibody-drug conjugate including the microtubule inhibitor is belantamab mafodotin,
brentuximab vedotin, cofetuzumab pelidotin, disitamab vedotin, enfortumab vedotin (e.g., enfortumab vedotin-ejfv, or a biosimilar thereof), mirvetuximab soravtansine (e.g., mirvetuximab soravtansine-gynx, or a biosimilar thereof), polatuzumab vedotin, telisotuzumab vedotin, tisotumab vedotin, trastuzumab emtansine (e.g., ado-trastuzumab emtansine, or a biosimilar thereof), tusamitamab ravtansine, upifitamab rilsodotin, zilovertamab vedotin, Alpha-Her2-pAF1-AS-269, BAT-8001, TAA-013, biosimilars thereof, or a combination thereof. In some embodiments, the antibody-drug conjugate including the microtubule inhibitor is enfortumab vedotin (e.g., enfortumab vedotin-ejfv, or a biosimilar thereof). In some embodiments, the antibody-drug conjugate including the microtubule inhibitor is mirvetuximab soravtansine (e.g., mirvetuximab soravtansine-gynx, or a biosimilar thereof). In some embodiments, the antibody-drug conjugate including the microtubule inhibitor is trastuzumab emtansine (e.g., ado-trastuzumab emtansine, or a biosimilar thereof). In some embodiments, the antibody-drug conjugate including the topoisomerase inhibitor is datopotamab deruxtecan, patritumab deruxtecan, sacituzumab govitecan (e.g., sacituzumab govitecan-hziy, or a biosimilar thereof), trastuzumab deruxtecan (fam-trastuzumab deruxtecan- nxki, or a biosimilar thereof), or a combination thereof. In some embodiments, the antibody- drug conjugate including the topoisomerase inhibitor is sacituzumab govitecan (e.g., sacituzumab govitecan-hziy, or a biosimilar thereof). In some embodiments, the antibody-drug conjugate including the topoisomerase inhibitor is trastuzumab deruxtecan (e.g., fam- trastuzumab deruxtecan-nxki, or a biosimilar thereof). In some embodiments, the EGFR inhibitor is abivertinib, afatinib, alflutinib, almonertinib, amivantamab, befotertinib, bleomycetin, brigatinib, canertinib, cetuximab, dacomitinib, delphinidin, depatuxizumab, dovitinib, duligotumab, erlotinib, furmonertinib, futuximab, gefitinib, icotinib, imgatuzumab, lapatinib, lazertinib, lisocabtagene, mereletinib, mobocertinib, modotuximab, nazartinib, necitumumab, neratinib, nimotuzumab, olmutinib, osimertinib, panitumumab, pelitinib, pingyangmycin, poziotinib, pyrotinib, quercetin, sapitinib, tarloxotinib, tesevatinib, tomuzotuximab, vandetanib, varlitinib, zalutumumab, and zorifertinib. In some embodiments, the EGFR inhibitor is abivertinib, afatinib (e.g., afatinib dimaleate), alflutinib (e.g., alflutinib mesylate), almonertinib (e.g., almonertinib mesylate), befotertinib, brigatinib, canertinib, dacomitinib (e.g., dacomitinib monohydrate), dovitinib, erlotinib (e.g., erlotinib hydrochloride), gefitinib, icotinib, lapatinib (e.g., lapatinib ditosylate monohydrate), larotinib, lazertinib, limertinib, mobocertinib (e.g., mobocertinib succinate), nazartinib, neratinib (e.g., neratinib maleate), olmutinib, osimertinib (e.g., osimertinib
mesylate), pelitinib, poziotinib, pyrotinib (e.g., pyrotinib maleate), ruserontinib (SKLB-1028), sapitinib, sunvozertinib, tesevatinib, vandetanib, varlitinib, zorifertinib, BIBW-2948, BPI- 7711, HA-121-28, SH-1028, or a combination thereof In some embodiments, the PARP inhibitor is iniparib, niraparib, olaparib (LYNPARZA®), pamiparib (BGB-290), rucaparib, talazoparib, veliparib, 2X-121, ABT-767, BMN 673, BSI-201, CEP 9722, E7016, IMP4297, INO-1001, JPI-289, KU-0059436 (AZD2281), NOV1401, PF-01367338, and RBN-2397. In some embodiments, the PARP inhibitor is fuzuloparib (fluzoparib), niraparib (e.g., niraparib tosylate monohydrate), olaparib, pamiparib, rucaparib (e.g., rucaparib camsylate), saruparib (AZD5305), senaparib, stenoparib, talazoparib (e.g., talazoparib tosylate), veliparib, CEP-9722, JPI-289, NMS-03305293, or a combination thereof. In some embodiments, the PARP inhibitor is a PARP1 inhibitor. In some embodiments, the PARP1 inhibitor is saruparib (AZD5305), NMS-03305293, or a combination thereof. Non-limiting examples of immunotherapy include immune checkpoint therapies. Non- limiting examples of immune checkpoint therapies include antibodies and/or inhibitors that target CTLA-4, PD-1, PD-L1, BTLA, LAG-3, ADORA2A, TIM-3, B7-H3, VISTA, IDO, and combinations thereof. In some embodiments, the anti-CTLA4 therapy is abatacept (e.g., ORENCIA® (abatacept), or a biosimilar thereof), botensilimab, cadonilimab, erfonrilimab, gotistobart, ipilimumab (e.g., YERVOY® (ipilimumab), or a biosimilar thereof), nurulimab, quavonlimab, tremelimumab (ticilimumab) (e.g., IMIUDO® (tremelimumab), or a biosimilar thereof), volrustomig, vudalimab, zalifrelimab, BMS-986218, PSB-205, biosimilars thereof, or a combination thereof. In some embodiments, the anti-PD1 therapy is balstilimab, budigalimab, cadonilimab, camrelizumab, cemiplimab (e.g., cemiplimab-rwlc, or a biosimilar thereof), cetrelimab, dostarlimab (e.g., dostarlimab-gxly, or a biosimilar thereof), ezabenlimab, geptanolimab, ivonescimab, nivolumab (e.g., OPDIVO® (nivolumab), or a biosimilar thereof), nofazinlimab, pembrolizumab (e.g., KEYTRUDA® (pembrolizumab), or a biosimilar thereof), penpulimab, pidilizumab, pimivalimab, prolgolimab, pucotenlimab, retifanlimab (e.g., retifanlimab-dlwr, or a biosimilar thereof), rilvegostomig, rosnilimab, rulonilimab, sasanlimab, serplulimab, sintilimab (e.g., TYVYT® (sintilimab), or a biosimilar thereof), spartalizumab, tebotelimab, tislelizumab, toripalimab, volrustomig, vudalimab, zimberelimab, QL-1604, HX-009, INCB- 086550, RG-6139, BAT-1306, SG-001, biosimilars thereof, or a combination thereof.
In some embodiments, the anti-PD-L1 therapy is adebrelimab, atezolizumab (e.g., TECENTRIQ® (atezolizumab), or a biosimilar thereof), avelumab (e.g., BAVENCIO® (avelumab), or a biosimilar thereof), bintrafusp alfa, cosibelimab, danburstotug, durvalumab (e.g., IMFINZI® (durvalumab), or a biosimilar thereof), envafolimab (e.g., ENWEIDA® (envafolimab), or a biosimilar thereof), erfonrilimab, pacmilimab, socazolimab, sugemalimab (e.g., CEJEMLY® (sugemalimab), or a biosimilar thereof), A-167, APL-502, AUPM-170, BNT-311, SHR-1701, biosimilars thereof, or a combination thereof. In some embodiments, the PD-L1 inhibitor is INCB-086550. In some embodiments, the anti-LAG3 therapy is eftilagimod alfa, favezelimab, fianlimab, ieramilimab, INCAGN-02385, miptenalimab, relatlimab (e.g., relatlimab-rmbw, or a biosimilar thereof), tebotelimab, IBI-110, LBL-007, RG-6139, biosimilars thereof, or a combination thereof. In some embodiments, the ADOR2A inhibitor is etrumadenant, inupadenant, istradefylline, mefloquine (e.g., mefloquine), taminadenant, CPI-444, PBF-999, or a combination thereof. In some embodiments, the ADOR2A inhibitor is etrumadenant, inupadenant, istradefylline, mefloquine (e.g., mefloquine), taminadenant, PBF-999, or a combination thereof. In some embodiments, the anti-TIM3 therapy is cobolimab, sabatolimab (MBG-453), AZD-7789, INCAGN-02390, TQB-2618, or a combination thereof. In some embodiments, the anti-B7-H3 therapy is omburtamab, enoblituzumab, or a combination thereof. In some embodiments, the anti-VISTA therapy is onvatilimab (JNJ-61610588), HMBD-002, K01401-020, KVA-12.1, SNS-101, or a combination thereof. In some embodiments, the IDO inhibitor (e.g., IDO1 and/or IDO2 inhibitor) is 3- deazaguanine, beta-lapachone, diindolylmethane, epacadostat, indole-3-carbinol, indoximod, sertaconazole (e.g., sertaconazole nitrate), or a combination thereof. See, for example, Marin-Acevedo, et al., J Hematol Oncol. 11: 39 (2018), doi: 10.1186/s13045-018-0582-8. In some embodiments, the additional therapy or therapeutic agent is a combination of atezolizumab and nab-paclitaxel. Accordingly, also provided herein is a method of treating cancer, comprising administering to a subject in need thereof (a) a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-
c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, (b) an additional therapeutic agent, and (c) optionally at least one pharmaceutically acceptable carrier for simultaneous, separate or sequential use for the treatment of cancer, wherein the amounts of the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, and the additional therapeutic agent are together effective in treating the cancer. In some embodiments, the additional therapeutic agent(s) includes any one of the above listed therapies or therapeutic agents which are standards of care in cancers wherein the cancer has a KRas dysregulation (e.g., a KRas mutation or amplification). In some embodiments, the additional therapeutic agent(s) includes any one of the above listed therapies or therapeutic agents which are standards of care in a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))). These additional therapeutic agents may be administered with one or more doses of the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, or pharmaceutical composition thereof
, as part of the same or separate dosage forms, via the same or different routes of administration, and/or on the same or different administration schedules according to standard pharmaceutical practice known to one skilled in the art.
Also provided herein is (i) a pharmaceutical composition for treating a cancer in a subject in need thereof, which comprises (a) a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, (b) at least one additional therapeutic agent (e.g., any of the exemplary additional therapeutic agents described herein or known in the art), and (c) optionally at least one pharmaceutically acceptable carrier for simultaneous, separate or sequential use for the treatment of cancer, wherein the amounts of the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, and of the additional therapeutic agent are together effective in treating the cancer; (ii) the use of such a composition for the preparation of a medicament for the treatment of cancer; and (iii) a commercial package or product comprising such a composition for simultaneous, separate or sequential use. In some embodiments, the cancer is a KRas- associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))). Accordingly, also provided herein is a method of treating a cancer, comprising administering to a subject in need thereof (a) a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, and (b) an additional therapeutic agent, wherein the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-
a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, and the additional therapeutic agent are administered simultaneously, separately or sequentially, wherein the amounts of the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, and the additional therapeutic agent are together effective in treating the cancer. In some embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, and the additional therapeutic agent are administered simultaneously as separate dosages. In some embodiments, compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, and the additional therapeutic agent are administered as separate dosages sequentially in any order, in jointly therapeutically effective amounts, e.g., in daily or intermittently dosages. In some embodiments, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g.,
Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, and the additional therapeutic agent are administered simultaneously as a combined dosage. In some embodiments, the cancer is a KRas-associated cancer (e.g., a mutant KRas-associated cancer (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer))). The term “wild type” or “wild-type” describes a nucleic acid (e.g., a KRAS gene or a KRas mRNA) or protein (e.g., a KRas protein) sequence that is typically found in a subject that does not have a disease or disorder related to the reference nucleic acid or protein. Although a wild type nucleic acid or protein sequence is the sequence that is typically found in a subject that does not have a disease or disorder related to the reference nucleic acid or protein, it is not necessarily the case that a subject that has a disease or disorder related to the reference nucleic acid or protein lacks wild type sequence. For example, a subject with a gene duplication of the reference gene may have the wild type sequence but could still have a disease or disorder related to the reference nucleic acid or protein due to the duplication event. As another example, a subject with a disease or disorder related to the reference nucleic acid or protein may have one allele that encodes wild type protein, and another allele that encodes a mutant protein. The term “wild type KRas” or “wild-type KRas” describes a KRas nucleic acid (e.g., a KRAS gene or a KRas mRNA) or protein (e.g., a KRas protein) that is found in a subject that does not have a KRas-associated disease, e.g., a KRas-associated cancer (and optionally also does not have an increased risk of developing a KRas-associated disease and/or is not suspected of having a KRas-associated disease), or is found in a cell or tissue from a subject that does not have a KRas-associated disease, e.g., a KRas-associated cancer (and optionally also does not have an increased risk of developing a KRas-associated disease and/or is not suspected of having a KRas-associated disease). Although a wild type KRas nucleic acid or protein sequence is the sequence that is typically found in a subject that does not have a KRas-associated disease or disorder, it is not necessarily the case that a subject that has a KRas-associated disease or disorder lacks the wild type KRas sequence. For example, a subject with a KRas gene duplication may have the wild type sequence but could still have a KRas-associated disease or disorder due to the duplication event. As another example, a subject with a KRas-associated disease or disorder may have one allele that encodes wild type KRas protein, and another allele
that encodes a mutant KRas protein. As used herein, terms “treat” or “treatment” refer to therapeutic or palliative measures. Beneficial or desired clinical results include, but are not limited to, alleviation, in whole or in part, of symptoms associated with a disease or disorder or condition, diminishment of the extent of disease, stabilized (i.e., not worsening) state of disease, delay or slowing of disease progression, amelioration or palliation of the disease state (e.g., one or more symptoms of the disease), and remission (whether partial or total), whether detectable or undetectable. “Treatment” can also mean prolonging survival as compared to expected survival if not receiving treatment. As used herein, the terms “subject,” “individual,” or “patient,” are used interchangeably, refers to any animal, including mammals such as mice, rats, other rodents, rabbits, dogs, cats, swine, cattle, sheep, horses, primates, and humans. In some embodiments, the subject is a human. In some embodiments, the subject has experienced and/or exhibited at least one symptom of the disease or disorder to be treated and/or prevented. In some embodiments, the subject is a pediatric subject. The term “pediatric subject” as used herein refers to a subject under the age of 21 years at the time of diagnosis or treatment. The term “pediatric” can be further be divided into various subpopulations including: neonates (from birth through the first month of life); infants (1 month up to two years of age); children (two years of age up to 12 years of age); and adolescents (12 years of age through 21 years of age (up to, but not including, the twenty-second birthday)). Berhman RE, Kliegman R, Arvin AM, Nelson WE. Nelson Textbook of Pediatrics, 15th Ed. Philadelphia: W.B. Saunders Company, 1996; Rudolph AM, et al. Rudolph’s Pediatrics, 21st Ed. New York: McGraw-Hill, 2002; and Avery MD, First LR. Pediatric Medicine, 2nd Ed. Baltimore: Williams & Wilkins; 1994. In some embodiments, a pediatric subject is from birth through the first 28 days of life, from 29 days of age to less than two years of age, from two years of age to less than 12 years of age, or 12 years of age through 21 years of age (up to, but not including, the twenty-second birthday). In some embodiments, a pediatric subject is from birth through the first 28 days of life, from 29 days of age to less than 1 year of age, from one month of age to less than four months of age, from three months of age to less than seven months of age, from six months of age to less than 1 year of age, from 1 year of age to less than 2 years of age, from 2 years of age to less than 3 years of age, from 2 years of age to less than seven years of age, from 3 years of age to less than 5 years of age, from 5 years of age to less than 10 years of age, from 6 years of age to less than 13 years of age, from 10 years of age to
less than 15 years of age, or from 15 years of age to less than 22 years of age. The term “activating mutation” in reference to KRas describes a mutation in a KRas gene that results in the expression of a KRas protein that has decreased GTPase activity and/or increased effector activation activity, e.g., as compared to a wild type KRas protein, e.g., when assayed under identical conditions. For example, an activating mutation can be a mutation in a KRAS gene that results in the expression of a KRas protein that has one or more (e.g., two, three, four, five, six, seven, eight, nine, or ten) amino acid substitutions (e.g., any combination of any of the amino acid substitutions described herein) that has decreased GTPase activity, e.g., as compared to a wild type KRas protein, e.g., when assayed under identical conditions. In another example, an activating mutation can be a mutation in a KRAS gene that results in the expression of a KRas protein that has one or more (e.g., two, three, four, five, six, seven, eight, nine, or ten) amino acids deleted, e.g., as compared to a wild type KRas protein, e.g., when assayed under identical conditions. In another example, an activating mutation can be a mutation in a KRAS gene that results in the expression of a KRas protein that has at least one (e.g., at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, at least 10, at least 12, at least 14, at least 16, at least 18, or at least 20) amino acid inserted as compared to a wild type KRas protein, e.g., the exemplary wild type KRas protein described herein, e.g., when assayed under identical conditions. Additional examples of activating mutations are known in the art. The term “preventing” as used herein means to delay the onset, recurrence or spread, in whole or in part, of the disease or condition as described herein, or a symptom thereof. The term “regulatory agency” refers to a country's agency for the approval of the medical use of pharmaceutical agents with the country. For example, a non-limiting example of a regulatory agency is the U.S. Food and Drug Administration (FDA). The phrase “therapeutically effective amount” means an amount of compound that, when administered to a subject in need of such treatment, is sufficient to (i) treat a KRas- associated disease or disorder (e.g., a mutant KRas-associated disease or disorder (e.g., a KRas G12D-associated cancer, a KRas G12R-associated cancer, or a KRas G12V-associated cancer (e.g., a KRas G12D-associated cancer or a KRas G12V-associated cancer)), (ii) attenuate, ameliorate, or eliminate one or more symptoms of the particular disease, disorder, or condition, or (iii) delay the onset of one or more symptoms of the particular disease, disorder, or condition described herein. The amount of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula
(II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, that will correspond to such an amount will vary depending upon factors such as the particular compound, disease condition and its severity, the identity (e.g., weight) of the subject in need of treatment, but can nevertheless be routinely determined by one skilled in the art. Pharmaceutical Compositions and Administration General In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is administered as a pharmaceutical composition that includes the compound, or a pharmaceutically acceptable salt thereof, and one or more pharmaceutically acceptable excipients, and optionally one or more additional therapeutic agents as described herein. In some embodiments, the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, can be administered in combination with one or more conventional pharmaceutical excipients. Pharmaceutically acceptable excipients include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, self- emulsifying drug delivery systems (SEDDS) such as d-α-tocopherol polyethylene glycol 1000 succinate, surfactants used in pharmaceutical dosage forms such as Tweens, poloxamers or other similar polymeric delivery matrices, serum proteins, such as human serum albumin,
buffer substances such as phosphates, tris, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium- chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose- based substances, polyethylene glycol, sodium carboxymethyl cellulose, polyacrylates, waxes, polyethylene-polyoxypropylene-block polymers, and wool fat. Cyclodextrins such as α-, ^-, and γ-cyclodextrin, or chemically modified derivatives such as hydroxyalkylcyclodextrins, including 2- and 3-hydroxypropyl-β-cyclodextrins, or other solubilized derivatives can also be used to enhance delivery of compounds described herein. Dosage forms or compositions containing a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, as described herein in the range of 0.005% to 100% with the balance made up from non-toxic excipient may be prepared. The contemplated compositions may contain 0.001%-100% of a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I- c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV- a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V- c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, provided herein, in one embodiment 0.1-95%, in another embodiment 75-85%, in a further embodiment 20-80%. Actual methods of preparing such dosage forms are known, or will be apparent, to those skilled in this art; for example, see Remington: The Science and Practice of Pharmacy, 22
nd Edition (Pharmaceutical Press, London, UK.2012). Routes of Administration and Composition Components In some embodiments, the compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g.,
Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, or a pharmaceutical composition comprising a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II- a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salts thereof , can be administered to a subject in need thereof by any accepted route of administration. Acceptable routes of administration include, but are not limited to, buccal, cutaneous, endocervical, endosinusial, endotracheal, enteral, epidural, interstitial, intra-abdominal, intra-arterial, intrabronchial, intrabursal, intracerebral, intracisternal, intracoronary, intradermal, intraductal, intraduodenal, intradural, intraepidermal, intraesophageal, intragastric, intragingival, intraileal, intralymphatic, intramedullary, intrameningeal, intramuscular, intraovarian, intraperitoneal, intraprostatic, intrapulmonary, intrasinal, intraspinal, intrasynovial, intratesticular, intrathecal, intratubular, intratumoral, intrauterine, intravascular, intravenous, nasal, nasogastric, oral, parenteral, percutaneous, peridural, rectal, respiratory (inhalation), subcutaneous, sublingual, submucosal, topical, transdermal, transmucosal, transtracheal, ureteral, urethral and vaginal. In certain embodiments, a preferred route of administration is parenteral (e.g., intratumoral). In some embodiments, a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I- a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV- a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, as described herein, or a pharmaceutical composition thereof, can be administered orally to a subject in need thereof. Without being bound by any particular theory, it is believed that oral dosing (e.g., versus IV dosing) can be preferred by patients for convenience, perception of efficacy, and/or past experience.
Compositions can be formulated for parenteral administration, e.g., formulated for injection via the intravenous, intramuscular, sub-cutaneous, or even intraperitoneal routes. Typically, such compositions can be prepared as injectables, either as liquid solutions or suspensions; solid forms suitable for use to prepare solutions or suspensions upon the addition of a liquid prior to injection can also be prepared; and the preparations can also be emulsified. The preparation of such formulations will be known to those of skill in the art in light of the present disclosure. The pharmaceutical forms suitable for injectable use include sterile aqueous solutions or dispersions; formulations including sesame oil, peanut oil, or aqueous propylene glycol; and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions. In all cases the form must be sterile and must be fluid to the extent that it may be easily injected. It also should be stable under the conditions of manufacture and storage and must be preserved against the contaminating action of microorganisms, such as bacteria and fungi. The carrier also can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (for example, glycerol, propylene glycol, and liquid polyethylene glycol, and the like), suitable mixtures thereof, and vegetable oils. The proper fluidity can be maintained, for example, by the use of a coating, such as lecithin, by the maintenance of the required particle size in the case of dispersion, and by the use of surfactants. The prevention of the action of microorganisms can be brought about by various antibacterial and antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid, thimerosal, and the like. In many cases, it will be preferable to include isotonic agents, for example, sugars or sodium chloride. Prolonged absorption of the injectable compositions can be brought about by the use in the compositions of agents delaying absorption, for example, aluminum monostearate and gelatin. Sterile injectable solutions are prepared by incorporating the active compounds in the required amount in the appropriate solvent with various of the other ingredients enumerated above, as required, followed by filtered sterilization. Generally, dispersions are prepared by incorporating the various sterilized active ingredients into a sterile vehicle which contains the basic dispersion medium and the required other ingredients from those enumerated above. In the case of sterile powders for the preparation of sterile injectable solutions, the preferred methods of preparation are vacuum-drying and freeze-drying techniques, which yield a powder of the active ingredient, plus any additional desired ingredient from a previously sterile-filtered solution thereof.
Intratumoral injections are discussed, e.g., in Lammers, et al., “Effect of Intratumoral Injection on the Biodistribution and the Therapeutic Potential of HPMA Copolymer-Based Drug Delivery Systems” Neoplasia.2006, 10, 788–795, doi: 10.1593/neo.06436. Pharmacologically acceptable excipients usable in the rectal composition as a gel, cream, enema, or rectal suppository, include, without limitation, any one or more of cocoa butter glycerides, synthetic polymers such as polyvinylpyrrolidone, PEG (like PEG ointments), glycerine, glycerinated gelatin, hydrogenated vegetable oils, poloxamers, mixtures of polyethylene glycols of various molecular weights and fatty acid esters of polyethylene glycol Vaseline, anhydrous lanolin, shark liver oil, sodium saccharinate, menthol, sweet almond oil, sorbitol, sodium benzoate, anoxid SBN, vanilla essential oil, aerosol, parabens in phenoxyethanol, sodium methyl p-oxybenzoate, sodium propyl p-oxybenzoate, diethylamine, carbomers, carbopol, methyloxybenzoate, macrogol cetostearyl ether, cocoyl caprylocaprate, isopropyl alcohol, propylene glycol, liquid paraffin, xanthan gum, carboxy-metabisulfite, sodium edetate, sodium benzoate, potassium metabisulfite, grapefruit seed extract, methyl sulfonyl methane (MSM) , lactic acid, glycine, vitamins, such as vitamin A and E and potassium acetate. In certain embodiments, suppositories can be prepared by mixing a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I- b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II- b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, with suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at ambient temperature but liquid at body temperature and therefore melt in the rectum and release the active compound. In other embodiments, compositions for rectal administration are in the form of an enema. In other embodiments, the compounds described herein, or a pharmaceutical composition thereof, are suitable for local delivery to the digestive or GI tract by way of oral administration (e.g., solid or liquid dosage forms.). Solid dosage forms for oral administration include capsules, tablets, pills, powders, and granules. In such solid dosage forms, the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)),
Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI- d), or (VI-e))), or a pharmaceutically acceptable salt thereof, is mixed with one or more pharmaceutically acceptable excipients, such as sodium citrate or dicalcium phosphate and/or: a) fillers or extenders such as starches, lactose, sucrose, glucose, mannitol, and silicic acid, b) binders such as, for example, carboxymethylcellulose, alginates, gelatin, polyvinylpyrrolidinone, sucrose, and acacia, c) humectants such as glycerol, d) disintegrating agents such as agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, and sodium carbonate, e) solution retarding agents such as paraffin, f) absorption accelerators such as quaternary ammonium compounds, g) wetting agents such as, for example, cetyl alcohol and glycerol monostearate, h) absorbents such as kaolin and bentonite clay, and i) lubricants such as talc, calcium stearate, magnesium stearate, solid polyethylene glycols, sodium lauryl sulfate, and mixtures thereof. In the case of capsules, tablets and pills, the dosage form may also comprise buffering agents. Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polyethylene glycols and the like. In one embodiment, the compositions will take the form of a unit dosage form such as a pill or tablet and thus the composition may contain, along with a compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I- b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI- a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, provided herein, a diluent such as lactose, sucrose, dicalcium phosphate, or the like; a lubricant such as magnesium stearate or the like; and a binder such as starch, gum acacia, polyvinylpyrrolidine, gelatin, cellulose, cellulose derivatives, or the like. In another solid dosage form, a powder, marume, solution or suspension (e.g., in propylene carbonate, vegetable oils, PEGs, poloxamer 124 or triglycerides) is encapsulated in a capsule (gelatin or cellulose base capsule). Unit dosage forms in which one or more compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula
(II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV- b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof, provided herein or additional active agents are physically separated are also contemplated; e.g., capsules with granules (or tablets in a capsule) of each drug; two-layer tablets; two-compartment gel caps, etc. Enteric coated or delayed release oral dosage forms are also contemplated. Other physiologically acceptable compounds include wetting agents, emulsifying agents, dispersing agents or preservatives that are particularly useful for preventing the growth or action of microorganisms. Various preservatives are well known and include, for example, phenol and ascorbic acid. In certain embodiments the excipients are sterile and generally free of undesirable matter. These compositions can be sterilized by conventional, well-known sterilization techniques. For various oral dosage form excipients, such as tablets and capsules, sterility is not required. The USP/NF standard is usually sufficient. In certain embodiments, solid oral dosage forms can further include one or more components that chemically and/or structurally predispose the composition for delivery of the compound of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I-a2), (I-a3), (I-a4), (I- a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III-1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V-a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or a pharmaceutically acceptable salt thereof, to the stomach or the lower GI; e.g., the ascending colon and/or transverse colon and/or distal colon and/or small bowel. Exemplary formulation techniques are described in, e.g., Filipski, K.J., et al., Current Topics in Medicinal Chemistry, 2013, 13, 776- 802, doi: 10.2174/1568026611313070002. Examples include upper-GI targeting techniques, e.g., Accordion Pill (Intec Pharma), floating capsules, and materials capable of adhering to mucosal walls. Other examples include lower-GI targeting techniques. For targeting various regions in the intestinal tract, several enteric/pH-responsive coatings and excipients are available. These materials are typically polymers that are designed to dissolve or erode at specific pH ranges,
selected based upon the GI region of desired drug release. These materials also function to protect acid labile drugs from gastric fluid or limit exposure in cases where the active ingredient may be irritating to the upper GI (e.g., hydroxypropyl methylcellulose phthalate series, Coateric (polyvinyl acetate phthalate), cellulose acetate phthalate, hydroxypropyl methylcellulose acetate succinate, Eudragit series (methacrylic acid–methyl methacrylate copolymers), and Marcoat). Other techniques include dosage forms that respond to local flora in the GI tract, Pressure-controlled colon delivery capsule, and Pulsincap. Ocular compositions can include, without limitation, one or more of any of the following: viscogens (e.g., Carboxymethylcellulose, Glycerin, Polyvinylpyrrolidone, Polyethylene glycol); Stabilizers (e.g., Pluronic (triblock copolymers), Cyclodextrins); Preservatives (e.g., Benzalkonium chloride, ETDA, SofZia (boric acid, propylene glycol, sorbitol, and zinc chloride; Alcon Laboratories, Inc.), Purite (stabilized oxychloro complex; Allergan, Inc.)). Topical compositions can include ointments and creams. Ointments are semisolid preparations that are typically based on petrolatum or other petroleum derivatives. Creams containing the selected active agent are typically viscous liquid or semisolid emulsions, often either oil-in-water or water-in-oil. Cream bases are typically water-washable, and contain an oil phase, an emulsifier, and an aqueous phase. The oil phase, also sometimes called the “internal” phase, is generally comprised of petrolatum and a fatty alcohol such as cetyl or stearyl alcohol; the aqueous phase usually, although not necessarily, exceeds the oil phase in volume, and generally contains a humectant. The emulsifier in a cream formulation is generally a nonionic, anionic, cationic, or amphoteric surfactant. As with other carriers or vehicles, an ointment base should be inert, stable, nonirritating, and non-sensitizing. In any of the foregoing embodiments, pharmaceutical compositions described herein can include one or more one or more of the following: lipids, interbilayer crosslinked multilamellar vesicles, biodegradable poly(D,L-lactic-co-glycolic acid) [PLGA]-based or poly anhydride-based nanoparticles or microparticles, and nanoporous particle-supported lipid bilayers. Dosages The dosages may be varied depending on the requirement of the patient, the severity of the condition being treated, and the particular compound being employed. Determination of the proper dosage for a particular situation can be determined by one skilled in the medical
arts. The total daily dosage may be divided and administered in portions throughout the day or by means providing continuous delivery. In some embodiments, the compounds described herein are administered at a dosage of from about 0.001 mg/kg to about 500 mg/kg (e.g., from about 0.001 mg/kg to about 200 mg/kg; from about 0.01 mg/kg to about 200 mg/kg; from about 0.01 mg/kg to about 150 mg/kg; from about 0.01 mg/kg to about 100 mg/kg; from about 0.01 mg/kg to about 50 mg/kg; from about 0.01 mg/kg to about 10 mg/kg; from about 0.01 mg/kg to about 5 mg/kg; from about 0.01 mg/kg to about 1 mg/kg; from about 0.01 mg/kg to about 0.5 mg/kg; from about 0.01 mg/kg to about 0.1 mg/kg; from about 0.1 mg/kg to about 200 mg/kg; from about 0.1 mg/kg to about 150 mg/kg; from about 0.1 mg/kg to about 100 mg/kg; from about 0.1 mg/kg to about 50 mg/kg; from about 0.1 mg/kg to about 10 mg/kg; from about 0.1 mg/kg to about 5 mg/kg; from about 0.1 mg/kg to about 1 mg/kg; from about 0.1 mg/kg to about 0.5 mg/kg). Regimens The foregoing dosages can be administered on a daily basis (e.g., as a single dose or as two or more divided doses) or non-daily basis (e.g., every other day, every two days, every three days, once weekly, twice weeks, once every two weeks, once a month). In some embodiments, the period of administration of a compound described herein is for 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 8 days, 9 days, 10 days, 11 days, 12 days, 13 days, 14 days, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 7 weeks, 8 weeks, 9 weeks, 10 weeks, 11 weeks, 12 weeks, 4 months, 5 months, 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, 12 months, or more. In a further embodiment, a period of during which administration is stopped is for 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 8 days, 9 days, 10 days, 11 days, 12 days, 13 days, 14 days, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 7 weeks, 8 weeks, 9 weeks, 10 weeks, 11 weeks, 12 weeks, 4 months, 5 months, 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, 12 months, or more. In an embodiment, a therapeutic compound is administered to an individual for a period of time followed by a separate period of time. In another embodiment, a therapeutic compound is administered for a first period and a second period following the first period, with administration stopped during the second period, followed by a third period where administration of the therapeutic compound is started and then a fourth period following the third period where administration is stopped. In an aspect of this embodiment, the period of administration of a therapeutic compound followed by a period where administration is stopped is repeated for a determined
or undetermined period of time. In a further embodiment, a period of administration is for 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 8 days, 9 days, 10 days, 11 days, 12 days, 13 days, 14 days, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 7 weeks, 8 weeks, 9 weeks, 10 weeks, 11 weeks, 12 weeks, 4 months, 5 months, 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, 12 months, or more. In a further embodiment, a period of during which administration is stopped is for 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 8 days, 9 days, 10 days, 11 days, 12 days, 13 days, 14 days, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 7 weeks, 8 weeks, 9 weeks, 10 weeks, 11 weeks, 12 weeks, 4 months, 5 months, 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, 12 months, or more. The term “acceptable” with respect to a formulation, composition, or ingredient, as used herein, means having no persistent detrimental effect on the general health of the subject being treated. “API” refers to an active pharmaceutical ingredient. The term “excipient” or “pharmaceutically acceptable excipient” means a pharmaceutically acceptable material, composition, or vehicle, such as a liquid or solid filler, diluent, carrier, solvent, or encapsulating material. In one embodiment, each component is “pharmaceutically acceptable” in the sense of being compatible with the other ingredients of a pharmaceutical formulation, and suitable for use in contact with the tissue or organ of humans and animals without excessive toxicity, irritation, allergic response, immunogenicity, or other problems or complications, commensurate with a reasonable benefit/risk ratio. See, e.g., Remington: The Science and Practice of Pharmacy, 21st ed.; Lippincott Williams & Wilkins: Philadelphia, PA, 2005; Handbook of Pharmaceutical Excipients, 6th ed.; Rowe et al., Eds.; The Pharmaceutical Press and the American Pharmaceutical Association: 2009; Handbook of Pharmaceutical Additives, 3rd ed.; Ash and Ash Eds.; Gower Publishing Company: 2007; Pharmaceutical Preformulation and Formulation, 2nd ed.; Gibson Ed.; CRC Press LLC: Boca Raton, FL, 2009. The term “pharmaceutically acceptable salt” refers to a formulation of a compound that does not cause significant irritation to an organism to which it is administered and does not abrogate the biological activity and properties of the compound. In certain instances, pharmaceutically acceptable salts are obtained by reacting a compound described herein, with acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, salicylic acid, and the like. In some instances, pharmaceutically acceptable salts are obtained by reacting a compound
having acidic group described herein with a base to form a salt such as an ammonium salt, an alkali metal salt, such as a sodium or a potassium salt, an alkaline earth metal salt, such as a calcium or a magnesium salt, a salt of organic bases such as dicyclohexylamine, N-methyl-D- glucamine, tris(hydroxymethyl)methylamine, and salts with amino acids such as arginine, lysine, and the like, or by other methods previously determined. The term “pharmacologically acceptable salts” is not specifically limited as far as it can be used in medicaments. Examples of a salt that the compounds described herein form with a base include the following: salts thereof with inorganic bases such as sodium, potassium, magnesium, calcium, and aluminum; salts thereof with organic bases such as methylamine, ethylamine, and ethanolamine; salts thereof with basic amino acids such as lysine and ornithine; and ammonium salt. The salts may be acid addition salts, which are specifically exemplified by acid addition salts with the following: mineral acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, and phosphoric acid:organic acids such as formic acid, acetic acid, propionic acid, oxalic acid, malonic acid, succinic acid, fumaric acid, maleic acid, lactic acid, malic acid, tartaric acid, citric acid, methanesulfonic acid, and ethanesulfonic acid; acidic amino acids such as aspartic acid and glutamic acid. The term “pharmaceutical composition” refers to a mixture of a compound described herein with other chemical components (referred to collectively herein as “excipients”), such as carriers, stabilizers, diluents, dispersing agents, suspending agents, and/or thickening agents. The pharmaceutical composition facilitates administration of the compound to a subject. Multiple techniques of administering a compound exist in the art including, but not limited to: rectal, oral, intravenous, aerosol, parenteral, ophthalmic, pulmonary, and topical administration. Compound Preparation The compounds disclosed herein can be prepared in a variety of ways using commercially available starting materials, compounds known in the literature, or from readily prepared intermediates, by employing standard synthetic methods and procedures either known to those skilled in the art, or in light of the teachings herein. Standard synthetic methods and procedures for the preparation of organic molecules and functional group transformations and manipulations can be obtained from the relevant scientific literature or from standard textbooks in the field. Although not limited to any one or several sources, classic texts such as R. Larock, Comprehensive Organic Transformations, VCH Publishers (1989); L. Fieser and M. Fieser, Fieser and Fieser's Reagents for Organic
Synthesis, John Wiley and Sons (1994); Smith, M. B., March, J., March' s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5th edition, John Wiley & Sons: New York, 2001 ; and Greene, T.W., Wuts, P.G. M., Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons: New York, 1999, are useful and recognized reference textbooks of organic synthesis known to those in the art. The following descriptions of synthetic methods are designed to illustrate, but not to limit, general procedures for the preparation of compounds of the present disclosure. The synthetic processes disclosed herein can tolerate a wide variety of functional groups; therefore, various substituted starting materials can be used. The processes generally provide the desired final compound at or near the end of the overall process, although it may be desirable in certain instances to further convert the compound to a pharmaceutically acceptable salt thereof. Scheme 1 depicts an exemplary method for synthesizing certain intermediates useful in the preparation of compounds of Formula (I):
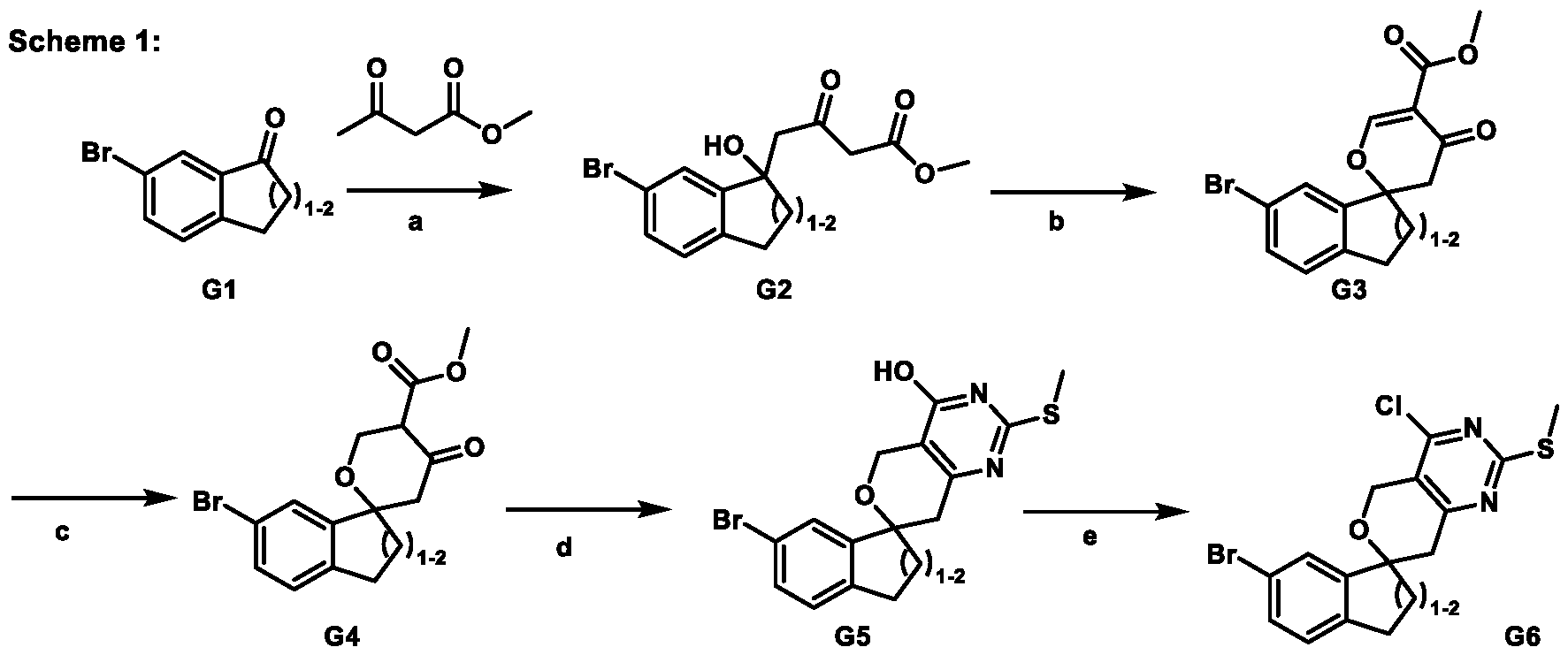
Referring to Scheme 1, methyl 3-oxobutanoate is treated with a base (e.g., NaH) followed by reaction with G1, e.g., in the presence of a second base (e.g., LDA, - 70 to 10
oC) to provide compound G2. The reaction of compound G2 with DMA-DMF in the presence of BF3 etherate (e.g., in DCM) provides compound G3 which is then reduced (e.g., with L- Selectride in THF) to provide compound G4. Reaction of G4 with thiourea (e.g., in the presence of a base such as DBU) in an appropriate solvent (e.g., acetonitrile) provides a cyclized product. Treatment of this product with methyl iodide (e.g., in the presence of aq. NaOH) then provides compound G5. Alternatively, G5 can be obtained from the reaction of
G4 with S-methylthiourea (e.g., with K
2CO
3 and DMF). The reaction of G5 with a chlorinating agent (e.g., POCl3) in an appropriate solvent (e.g., acetonitrile) under heat (e.g., reflux in acetonitrile) then provides compound G6. Scheme 2 depicts an exemplary method for synthesizing compounds G8 and G9 which are compounds of Formula (I).
Referring to Scheme 2, compound G6 is subjected to an SNAr reaction (e.g., in the presence of a base such as DIPEA using e.g., acetonitrile as a solvent, at e.g., 80
oC) to provide compound G7, wherein R
1 is as defined for Formula (I) (e.g., wherein R
1 is attached to the pyrimidine ring via a nitrogen atom). G7 is then oxidized at the thioether (e.g., with mCPBA or Oxone) and then reacted with compound of formula HO-Y
2-R
3, wherein R
3 and Y
2 are as defined for Formula (I), to provide compound G8 which is a compound of Formula (I) with R
9 = Br. Compound G8 can be transformed into compound G9 wherein R
9 is as defined for Formula (I) (e.g., R
9 is OH or NH2). For example, G8 can be treated with B2pin2 in the presence of a palladium catalyst (e.g., Pd(dppf)Cl
2 in the presence of KOAc in 1,4-dioxane) to provide a boronate compound, which can then be converted into G9 (e.g., by reacting the boronate compound with H2O2 in the presence of NaOH to provide G9 wherein R
9 = OH). Alternatively, G8 can be reacted with NH2Boc in the presence of a palladium catalyst (e.g., Pd(OAc)2 in the 20 presence of an appropriate ligand such as XantPhos and a base (e.g., Cs
2CO
3) in an appropriate
solvent (e.g., 1,4-dioxane)), wherein the resulting carbamate can be converted to G9 wherein R
9 is NH2 through removal of the Boc group. Alternatively, G8 can be converted into G9 (wherein R
9 is OH) through reaction with KOH in the presence of a palladium catalyst (e.g., Pd
2dba
3 in the presence of an appropriate ligand (e.g., tBuXPhos) in an appropriate solvent (e.g., 1,4-dioxane)). Scheme 3 depicts exemplary methods for preparing compounds of Formula (I-a1): Scheme 3
Referring to Scheme 3, (4-chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol is treated with a base (e.g., LDA (e.g., in THF at – 78
oC)), followed by a compound of Formula (SV-a1) to provide an intermediate which is converted into a compound of Formula (SIV-a1) under appropriate conditions (e.g., H
3PO
4 in toluene under reflux; or TPP and DIAD in THF; or TsCl and nBuLi in THF), wherein X
1, X
2, X
3, b1, and R
10 are as defined for Formula (I-a1); and X is halo (e.g., -Br). A compound of Formula (SIV-a1) is then reacted with R
1-H, wherein R
1 is as defined for Formula (I-a1) (e.g., R
1 is attached to the pyrimidine ring via a nitrogen atom), to provide a compound of Formula (SIII-a1) under appropriate conditions (e.g., in the presence of a base (e.g., triethylamine or N,N-diisopropylethylamine) in an appropriate solvent (e.g., EtOH, 1,4-dioxane, or DMF)). A compound of Formula (SIII-a1) is then reacted with a compound having Formula R
9-H, wherein R
9 is as defined for Formula (I-a1), to provide a
compound of Formula (SII-a1) under appropriate conditions (e.g., in the presence of a palladium catalyst and optionally a ligand and/or a base (e.g., Pd(OAc)2, XantPhos, Cs2CO3, in 1,4-dioxane at 80
oC; or BrettPhos Pd G4, Cs2CO3, in 1,4-dioxane at 80
oC)). Subsequently, the thioether group in the compound of Formula (SII-a1) is oxidized (e.g., with Oxone or mCPBA). This is followed by coupling with R
3-Y
2-OH, wherein R
3 and Y
2 are as defined for Formula (I-a1), to provide a compound of Formula (I-a1). Variations to the reaction sequence in Scheme 3 are also contemplated. For example, to provide certain compounds of Formula (I-a1) wherein R
9 is halo, step d in Scheme 3 may be bypassed (i.e., compounds of Formula (SIII-a1) are reacted with R
3-Y
2-OH under steps e and f). For example, in the preparation of certain compounds of Formula (I-a1) wherein R
9 is NH
2, the R
9 group can be protected with one or two nitrogen protecting groups (e.g., the NH
2 can be protected as NBn2 or NHBoc) during the reaction sequence e.g., in Formula (SII-a1) and/or R
9-H of step d, followed by the appropriate deprotection steps (e.g., HCl in 1,4-dioxane for NHBoc group; or Pd(OAc)
2/H
2 for NBn
2). Representative examples of nitrogen protecting groups include: acetyl (Ac), benzoyl (Bz), benzyl (Bn), benzyloxycarbonyl (Cbz), formyl, phenylsulfonyl, pivaloyl, tert-butoxycarbonyl (Boc), tert-butyl acetyl, ethyloxycarbonyl, trifluoroacetyl, triphenylmethyl (trityl), and triisopropylsilane. See: T. W. Greene and P. G. M. Wuts in “Protective groups in organic chemistry” John Wiley and Sons, 4th Edition, 2006. A person of ordinary skill in the art would understand that the final product and/or intermediates in Scheme 3 can be subjected to standard functional group transformation(s) to provide additional examples of compounds of Formula (I-a1). For example, a compound of Formula (I-a1) as provided in Scheme 3 can be converted into a second compound of Formula (I-a1) having a different R
9 group. For example, a compound of Formula (I-a1) wherein R
9 is -NHC(O)O
tBu can be converted into a second compound of Formula (I-a1) wherein R
9 is - NH2 upon treatment with an acid (e.g., HCl in 1,4-dioxane). Scheme 4 depicts exemplary methods for preparing compounds of Formula (I-b1):
Scheme 4
Referring to Scheme 4, (4-Chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol is treated with a base (e.g., LDA (e.g., in THF at – 78
oC)), followed by a compound of Formula (SV-b1) to provide an intermediate which is converted into a compound of Formula (SIV-b1) under appropriate conditions (e.g., H3PO4 in toluene under reflux; or TPP and DIAD in THF; or TsCl and nBuLi in THF), wherein X
1, X
2, X
3, b1, and R
10 are as defined for Formula (I-b1); and X is halo (e.g., -Br). A compound of Formula (SIV-b1) is then reacted with R
1-H, wherein R
1 is as defined for Formula (I-b1) (e.g., R
1 is attached to the pyrimidine ring via a nitrogen atom), to provide a compound of Formula (SIII-b1) under appropriate conditions (e.g., in the presence of a base (e.g., triethylamine or N,N-diisopropylethylamine in an appropriate solvent (e.g., EtOH, 1,4-dioxane, or DMF)). A compound of Formula (SIII-b1) is then reacted with a compound having Formula R
9-H, wherein R
9 is as defined for Formula (I-b1), to provide a compound of Formula (SII-b1) under appropriate conditions (e.g., in the presence of a palladium catalyst and optionally a ligand and/or a base (e.g., Pd(OAc)
2, XantPhos, Cs
2CO
3, in 1,4-dioxane at 80
oC; or BrettPhos Pd G4, Cs2CO3, in 1,4-dioxane at 80
oC)). Subsequently, the thioether group in the compound of Formula (SII-b1) is oxidized (e.g., with Oxone or mCPBA). This is followed by coupling with R
3-Y
2-OH, wherein R
3 and Y
2 are as defined for Formula (I-b1), to provide a compound of Formula (I-b1). Variations to the reaction sequence in Scheme 4 are also contemplated. For example,
to provide certain compounds of Formula (I-b1) wherein R
9 is halo, step d in Scheme 4 may be bypassed (i.e., compounds of Formula (SIII-b1) are reacted with R
3-Y
2-OH under steps e and f). For example, in the preparation of certain compounds of Formula (I-b1) wherein R
9 is NH
2, the R
9 group can be protected with one or two nitrogen protecting groups (e.g., the NH
2 can be protected as NBn2 or NHBoc) during the reaction sequence e.g., in Formula (SII-b1) and/or R
9-H of step d, followed by the appropriate deprotection steps (e.g., HCl in 1,4-dioxane for NHBoc group; or Pd(OAc)
2/H
2 for NBn
2). A person of ordinary skill in the art would understand that the final product and/or intermediates in Scheme 4 can be subjected to standard functional group transformation(s) to provide additional examples of compounds of Formula (I-b1). For example, a compound of Formula (I-b1) as provided in Scheme 4 can be converted into a second compound of Formula (I-b1) having a different R
9 group. For example, a compound of Formula (I-b1) wherein R
9 is -NHC(O)O
tBu can be converted into a second compound of Formula (I-b1) wherein R
9 is - NH
2 upon treatment with an acid (e.g., HCl in 1,4-dioxane). Scheme 5 depicts exemplary methods for preparing compounds of Formula (I-c1): Scheme 5
Referring to Scheme 5, (4-Chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol is
treated with a base (e.g., LDA (e.g., in THF at – 78
oC)), followed by a compound of Formula (SV-c1) to provide an intermediate which is converted into a compound of Formula (SIV-c1) under appropriate conditions (e.g., H3PO4 in toluene under reflux; or TPP and DIAD in THF; or TsCl and nBuLi in THF), wherein X
1, X
2, X
3, b1, and R
10 are as defined for Formula (I-c1); and X is halo (e.g., -Br). A compound of Formula (SIV-c1) is then reacted with R
1-H, wherein R
1 is as defined for Formula (I-c1) (e.g., R
1 is attached to the pyrimidine ring via a nitrogen atom), to provide a compound of Formula (SIII-c1) under appropriate conditions (e.g., in the presence of a base (e.g., triethylamine or N,N-diisopropylethylamine in an appropriate solvent (e.g., EtOH, 1,4-dioxane, or DMF)). A compound of Formula (SIII-c1) is then reacted with a compound having Formula R
9-H, wherein R
9 is as defined for Formula (I-c1), to provide a compound of Formula (SII-c1) under appropriate conditions (e.g., in the presence of a palladium catalyst and optionally a ligand and/or a base (e.g., Pd(OAc)2, XantPhos, Cs2CO3, in 1,4-dioxane at 80
oC; or BrettPhos Pd G4, Cs2CO3, in 1,4-dioxane at 80
oC)). Subsequently, the thioether group in the compound of Formula (SII-c1) is oxidized (e.g., with Oxone or mCPBA). This is followed by coupling with R
3-Y
2-OH, wherein R
3 and Y
2 are as defined for Formula (I-c1), to provide a compound of Formula (I-c1). Variations to the reaction sequence in Scheme 5 are also contemplated. For example, to provide certain compounds of Formula (I-c1) wherein R
9 is halo, step d in Scheme 5 may be bypassed (i.e., compounds of Formula (SIII-c1) are reacted with R
3-Y
2-OH under steps e and f). For example, in the preparation of certain compounds of Formula (I-c1) wherein R
9 is NH
2, the R
9 group can be protected with one or two nitrogen protecting groups (e.g., the NH
2 can be protected as NBn2 or NHBoc) during the reaction sequence e.g., in Formula (SII-c1) and R
9-H of step d, followed by the appropriate deprotection steps (e.g., HCl in 1,4-dioxane for NHBoc group; or Pd(OAc)
2/H
2 for NBn
2). A person of ordinary skill in the art would understand that the final product and/or intermediates in Scheme 5 can be subjected to standard functional group transformation(s) to provide additional examples of compounds of Formula (I-c1). For example, a compound of Formula (I-c1) as provided in Scheme 5 can be converted into a second compound of Formula (I-c1) having a different R
9 group. For example, a compound of Formula (I-c1) wherein R
9 is -NHC(O)O
tBu can be converted into a second compound of Formula (I-c1) wherein R
9 is - NH
2 upon treatment with an acid (e.g., HCl in 1,4-dioxane). Scheme 6 depicts an exemplary method for synthesizing certain intermediates useful
in the preparation of compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1))): Scheme 6
Referring to Scheme 6, Compound G10 is reacted with compound G11 to provide compound G12 under appropriate conditions (e.g., in the presence of a palladium catalyst (e.g., Pd(OAc)2, LiCl, LiOAc, tetrabutylammonium chloride), wherein R
9, b1, and R
10 are as defined for Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1))) (e.g., R
9 is halo such as -Br); and X
3 is CH
2, CHR
L, or C(R
L)
2 wherein R
L is as defined for Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1))) (e.g., X
3 is CH2 or CHR
L). The aldehyde group in compound G12 is subsequently oxidized (e.g., under Pinnick oxidation conditions (e.g., with NaClO
2, NaH
2PO
4)). The oxidized product is then cyclized to provide compound G13 (e.g., with AlCl
3, oxalyl chloride, DMF). Also provided herein are compounds useful, e.g., as intermediates or starting materials, in the preparation of compounds of Formula (A) (e.g., Formula (I) (e.g., Formula (I-a1), (I- a2), (I-a3), (I-a4), (I-a5), (I-b1), (I-b2), (I-b3), (I-b4), (I-b5), or (I-c1)), Formula (II) (e.g., Formula (II-a), (II-b), (II-a1), (II-b1), (II-a2), or (II-b2)), Formula (III) (e.g., Formula (III- 1) or (III-2)), Formula (IV) (e.g., Formula (IV-a), (IV-b), (IV-c), (IV-a1), (IV-b1), (IV-a2), or (IV-b2)), or Formula (V) (e.g., Formula (V-a) or (V-b), (V-a1), (V-c), (V-d), (V-b1), (V- a2), or (V-b2)), or Formula (VI) (e.g., Formula (VI-a), (VI-b), (VI-c), (VI-d), or (VI-e))), or pharmaceutically acceptable salts thereof. Provided herein are compounds of Formula (SII-a2), (SIV-a2), (SV-a2), (SVI-a2), and (SVII-a2):
Formula (SVII-a2) or salts thereof, wherein: ns is 0, 1, or 2; R
S is C
1-6 alkyl optionally substituted with 1-3 substituents independently selected from the group consisting of R
c and R
b1; R
11 is selected from the group consisting of: NH
2, NHPg, and N(Pg)
2; R
11a selected from the group consisting of: NHPg and N(Pg)2; each Pg is an independently selected nitrogen protecting group; R
12 is selected from the group consisting of: halo (e.g., -Br) and CN; b4 is 0 or 1; R
10 is selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; X
1 is selected from the group consisting of a bond, S(O)0-2, CH2, CHR
L, C(R
L)2, and O;
X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; each R
L is independently selected from the group consisting of C
1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-6 cycloalkyl ring; R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; Y
2 is a bond or a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
(a) halo; (b) cyano; (c) -OH; (d) oxo; (e) -C1-6 alkoxy; (f) -C1-6 haloalkoxy; (g) -NR
dR
e; (h) C(=O)C1-6 alkyl; (i) C(=O)C1-6 haloalkyl; (j) C(=O)OH; (k) C(=O)OC
1-6 alkyl; (l) C(=O)OC1-6 haloalkyl; (m) C(=O)N(R
f)2; (n) S(O)
0-2(C
1-6 alkyl); (o) S(O)0-2(C1-6 haloalkyl); (p) S(O)1-2N(R
f)2; and (q) C
1-6 alkyl, C
2-6 alkenyl, or C
2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1-
2(C
1-6 alkyl), S(O)
1-2(C
1-6 haloalkyl), S(O)
1-2N(R
f)
2, and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, C1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-. In some embodiments of Formula (SII-a2), (SIV-a2), (SV-a2), or (SVI-a2), R
11 is NH
2. In some embodiments of Formula (SII-a2), (SIV-a2), (SV-a2), or (SVI-a2), R
11 is NHPg or N(Pg)2, wherein each Pg is an independently selected nitrogen protecting group. Representative examples of nitrogen protecting groups include: acetyl (Ac), benzoyl (Bz), benzyl (Bn), benzyloxycarbonyl (Cbz), formyl, phenylsulfonyl, pivaloyl, tert-butoxycarbonyl (Boc), tert-butyl acetyl, ethyloxycarbonyl, trifluoroacetyl, triphenylmethyl (trityl), and triisopropylsilane. See: T. W. Greene and P. G. M. Wuts in “Protective groups in organic chemistry” John Wiley and Sons, 4th Edition, 2006. In some embodiments of Formula (SII-a2), (SIV-a2), (SV-a2), or (SVI-a2), R
11 is N(Bn)
2, wherein each Bn group is optionally substituted with 1-3 R
a. In some embodiments of Formula (SII-a2), (SIV-a2), (SV-a2), or (SVI-a2), R
11 is N(Bn)
2. In some embodiments of (SVII-a2), R
11a is NHPg (e.g., NHBoc). In some embodiments of (SVII-a2), R
11a is N(Pg)2. In some embodiments, R
11a is N(Bn)2, wherein each Bn group is optionally substituted with 1-3 R
a. For example, R
11a can be N(Bn)
2. In some embodiments of Formula (SII-a2), (SIV-a2), (SV-a2), (SVI-a2), or (SVII-a2), R
12 is -Br. In some embodiments of Formula (SII-a2), (SIV-a2), (SV-a2), (SVI-a2), or (SVII- a2), R
12 is -CN. In some embodiments of Formula (SII-a2), (SIV-a2), or (SVI-a2), the
some embodiments of Formula (SII-a2), (SIV-a2), or (SVI-a2), the
, wherein X
3 is CH
2 or CHR
L. For example, X
3 can be CH
2. For example, X
3 can be CHMe. In some embodiments of Formula (SII-a2), (SIV-a2), or (SVI-a2), the
example, X
3 can be CH
2. For example, X
3 can be CHMe. In some embodiments of Formula (SII-a2), (SIV-a2), or (SVI-a2), the
, wherein X
3 is CH
2 or CHR
L. For example, X
3 can be CH
2. For example, X
3 can be CHMe. In some embodiments of Formula (SVII-a2), the
In some embodiments of Formula (SII-a2) or Formula (SIV-a2), ns is 0. In some embodiments of Formula (SII-a2) or Formula (SIV-a2), ns is 1 or 2 (e.g., 2). In some embodiments of Formula (SII-a2) or Formula (SIV-a2), R
S is C1-6 alkyl (e.g., C1-3 alkyl (e.g., methyl)).
In some embodiments of Formula (
is selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, -C(O)N(H)R
b1, R
b1, and C(O)R
b1. For example, R
1 can
In some embodiments of Formula (SIV-a2) or (SVII-a2), R
1 is a 7-10 (e.g., 7) membered heterocyclyl having one ring nitrogen atom, one ring oxygen atom, and no additional ring heteroatoms, wherein the 7-10 membered heterocyclyl is optionally substituted with 1-4 R
7. In some embodiments of Formula (
optionally substituted with 1-4 R
7 at one or more ring carbon atoms
some embodiments of Formula (SIV-a2) or (SVII-a2), each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C
1-3 alkyl optionally substituted with 1-3 R
c, wherein each R
c is independently selected from the group consisting of: -F, -OH, and -CN. In some embodiments of Formula (SVI-a2) or (SVII-a2), Y
2 is CH2; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is
optionally substituted with 1-6 R
a. In some embodiments of Formula (SVI-a2) or (SVII-a2),
In some embodiments of Formula (SV-a2), R
L is C
1-3 alkyl optionally substituted with 1-3 R
c. In some embodiments, R
L is C1-3 alkyl (e.g., methyl). Also provided herein is a method of preparing a compound of Formula (II-a), or a pharmaceutically acceptable salt thereof, wherein the method comprises: contacting a compound of Formula (SVI-a2) with a hydroxyl activating agent followed by a compound of Formula R
1-H, to provide a compound of Formula (II-a), or a pharmaceutically acceptable salt thereof:
wherein: R
11 is NH2; R
12 is CN; b4 is 0 or 1; R
10 is selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; X
1 is selected from the group consisting of a bond, S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring;
R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; Y
2 is a bond or a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
g
(h) C(=O)C
1-6 alkyl; (i) C(=O)C1-6 haloalkyl; (j) C(=O)OH; (k) C(=O)OC
1-6 alkyl; (l) C(=O)OC1-6 haloalkyl; (m) C(=O)N(R
f)2; (n) S(O)
0-2(C
1-6 alkyl); (o) S(O)0-2(C1-6 haloalkyl); (p) S(O)1-2N(R
f)2; and (q) C
1-6 alkyl, C
2-6 alkenyl, or C
2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1-
2(C
1-6 alkyl), S(O)
1-2(C
1-6 haloalkyl), S(O)
1-2N(R
f)
2, and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, C
1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. Examples of hydroxyl activating groups include peptide coupling reagents, such as AOP, BOP, PyBOP, PyAOP, HBTU, HATU, HCTU, HBPyU, HAPyU, TFFH, TBTU, BTFFH, EDC-HCl, PyBrop, DPPA, BOP-Cl, DCC, DIC, DEPC, EEDQ, IIDQ, CIP, PfTU, PfPU, BroP and CDI. In some embodiments, the method comprises contacting a compound of Formula (SVI-a2) with PyBOP followed by a compound of Formula R
1-H. In some embodiments of the method, b4 is 0. In some embodiments of the method, X
1 is CH2; X
2 is CH2; and X
3 is CHR
L or CH2 (e.g., CHMe or CH2). In some embodiments of the method, the
moiety of Formula (
, wherein X
3 is CHR
L or CH2 (e.g., CH2). In some embodiments of the method, the
, wherein X
3 is CHR
L or CH2 (e.g., CH2). In some embodiments of the method, Y
2 is CH
2; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from
the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. For example, R
3 can
. In some embodiments of the method,
selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, -C(O)N(H)R
b1, R
b1, and C(O)R
b1.
In some embodiments of the method,
optionally substituted with 1-4 R
7 at one or more ring carbon atoms
Also provided herein is a method of preparing a compound of Formula (II-a), or a pharmaceutically acceptable salt thereof, wherein the method comprises: removing the nitrogen protecting group(s) present on R
11a of the compound of Formula (SVII-a2), to provide a compound of Formula (II-a), or a pharmaceutically acceptable salt thereof:
wherein: R
11a is NHPg or N(Pg)
2, wherein each Pg is an independently selected nitrogen protecting group; R
12 is CN; b4 is 0 or 1; R
10 is selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; X
1 is selected from the group consisting of a bond, S(O)
0-2, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)
2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7;
each R
7 is independently selected from the group consisting of R
a and R
b; Y
2 is a bond or a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, C
1-3 haloalkyl, C
3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-. In some embodiments of the method, R
11a is N(Bn)2; and removing the nitrogen protecting groups present on R
11a of the compound of Formula (SVII-a2) comprises subjecting the compound of Formula (SVII-a2) to hydrogenolysis (e.g., with Pd(OH)
2 under hydrogen atmosphere), thereby removing the Bn protecting groups. In some embodiments of the method, b4 is 0. In some embodiments of the method, X
1 is CH2; X
2 is CH2; and X
3 is CHR
L or CH2
(e.g., CHMe or CH2). In some embodiments of the method, the
moiety of Formula (
, wherein X
3 is CHR
L or CH
2 (e.g., CH2). In some embodiments of the method,
Formula
, wherein X
3 is CHR
L or CH
2 (e.g., CH
2). In some embodiments of the method, Y
2 is CH2; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. For example, R
3 can be . Also provided herein is a method of preparing a compound of Formula (II-a), or a pharmaceutically acceptable salt thereof, wherein the method comprises: (i) contacting a compound of Formula (SIV-a2) with a compound of Formula R
3-Y
2- OH; and (ii) optionally removing nitrogen protecting group(s) present on the product of step (i), to provide a compound of Formula (II-a):
wherein: ns is 1 or 2; R
S is C1-6 alkyl optionally substituted with 1-3 substituents independently selected from the group consisting of R
c and R
b1; R
11 is NH2, NHPg, or N(Pg)2, wherein each Pg is an independently selected nitrogen protecting group; R
12 is CN; b4 is 0 or 1; R
10 is selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; X
1 is selected from the group consisting of a bond, S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; each R
L is independently selected from the group consisting of C
1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-6 cycloalkyl ring; R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and
(iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; Y
2 is a bond or a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
(o) S(O)
0-2(C
1-6 haloalkyl); (p) S(O)1-2N(R
f)2; and (q) C1-6 alkyl, C2-6 alkenyl, or C2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1-
2(C
1-6 alkyl), S(O)
1-2(C
1-6 haloalkyl), S(O)
1-2N(R
f)
2, and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, C
1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. In some embodiments, the method comprises contacting a compound of Formula (SIV- a2) with a compound of Formula R
3-Y
2-OH in the presence of a base (e.g., NaH). In some embodiments, the method comprises contacting a compound of Formula (SIV-a2) with a compound of Formula R
3-Y
2-OH in the presence of a base (e.g., NaH) and in the presence of a polar aprotic solvent (e.g., THF). In some embodiments of the method, R
11 is NH2. In some embodiments of the method,
R
11 is N(Pg)
2. For example, R
11 can be N(Bn)
2. In some embodiments, R
11 is N(Bn)2; and the method comprises removing the benzyl protecting groups present on the product of step (i), e.g., via hydrogenolysis (e.g., with Pd(OH)2 under hydrogen atmosphere). In some embodiments of the method, ns is 2; and R
S is C1-3 alkyl (e.g., methyl). In some embodiments of the method, b4 is 0. In some embodiments of the method, X
1 is CH
2; X
2 is CH
2; and X
3 is CHR
L or CH
2 (e.g., CHMe or CH2). In some embodiments of the method, the
In some embodiments of the method, Y
2 is CH2; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. For example, R
3 can
.
EXAMPLES In some of the examples disclosed herein, one or more compounds in a described chemical reaction sequence (e.g., starting materials, intermediates, or products) is structurally depicted with enhanced stereochemical notation(s) at one or more defined stereogenic center(s). Examples of such notations include or1, or2, &1, &2, and the like. In some such examples, in the chemical name of the same compound, each of such defined stereogenic center(s) is assigned a tentative configuration (e.g., (R)- or (S)-) shown by the wedge/hash representation of its structural formula. However, the defined stereogenic center(s) should be understood to have configurations consistent with the enhanced stereochemical notation(s), as described herein, based e.g., on the conventions explained below. For avoidance of doubt, the chemical names of these compounds, having one or more enhanced stereochemical notation(s), adopt the following conventions: When the chemical name of a compound having only one stereogenic center contains the prefix “rel,” the stereogenic center is resolved, but its absolute configuration is either (R) or (S), thereby corresponding to an or1 enhanced stereochemical notation at the corresponding stereogenic center in its structural formula. For example, the chemical name rel-(R)-(1- methylpyrrolidin-2-yl)methanol represents one stereoisomer selected from the group consisting of:

. When one stereogenic center is labelled with an asterisk (“*”) in the chemical name of a compound having more than one stereogenic centers (e.g., when a stereogenic center is denoted as (R*)), the stereogenic center labeled with the asterisk is resolved, but its absolute configuration is either (R) or (S), thereby corresponding to an or1 enhanced stereochemical notation at the corresponding stereogenic center in its structural formula. For example, the chemical name ((2R*,4R)-1,4-dimethylpyrrolidin-2-yl)methanol represents one stereoisomer selected from the group consisting of:

. When the chemical name of a compound contains two stereogenic centers labelled with asterisks, these two stereogenic centers may have (R) or (S) configurations, either concertedly or independently, in conformity with the orx (e.g., or1 or or2) notations in its structural formula in Table C1. When two stereogenic centers are labelled or1 and or1 in a compound structure, and they are labelled with asterisks in the chemical name, then the relative stereochemistry between
the two stereogenic centers (e.g., syn or anti relationship) is as represented by the name, but the absolute configurations of the two stereogenic centers can vary concertedly (e.g., a compound designated (R*,R*) is one stereoisomer selected from (R,R) and (S,S); for avoidance of doubt, the compound designated (R*,R*) does not have (S,R) or (R,S) configurations across these stereogenic centers). For example, the chemical name of

((2R*,3S,4R*)-3-cyclopropyl-1,4-dimethylpyrrolidin-2-yl)methanol. This name, taken together with the structural formula, represents one stereoisomer selected from the group consisting
. When two stereogenic centers are labelled or1 and or2 in a compound structure, and they are labelled with asterisks in the chemical name, then the configuration of the two stereogenic centers can vary independently (e.g., a compound designated (R*,R*) is one stereoisomer selected from (R,R), (S,S), (R,S), and (S,R)). For example, the chemical name of
is ((2R*,3S,4R*)-3-cyclopropyl-1,4-dimethylpyrrolidin-2-yl)methanol. This name, taken together with the structural formula, represents one stereoisomer selected from the
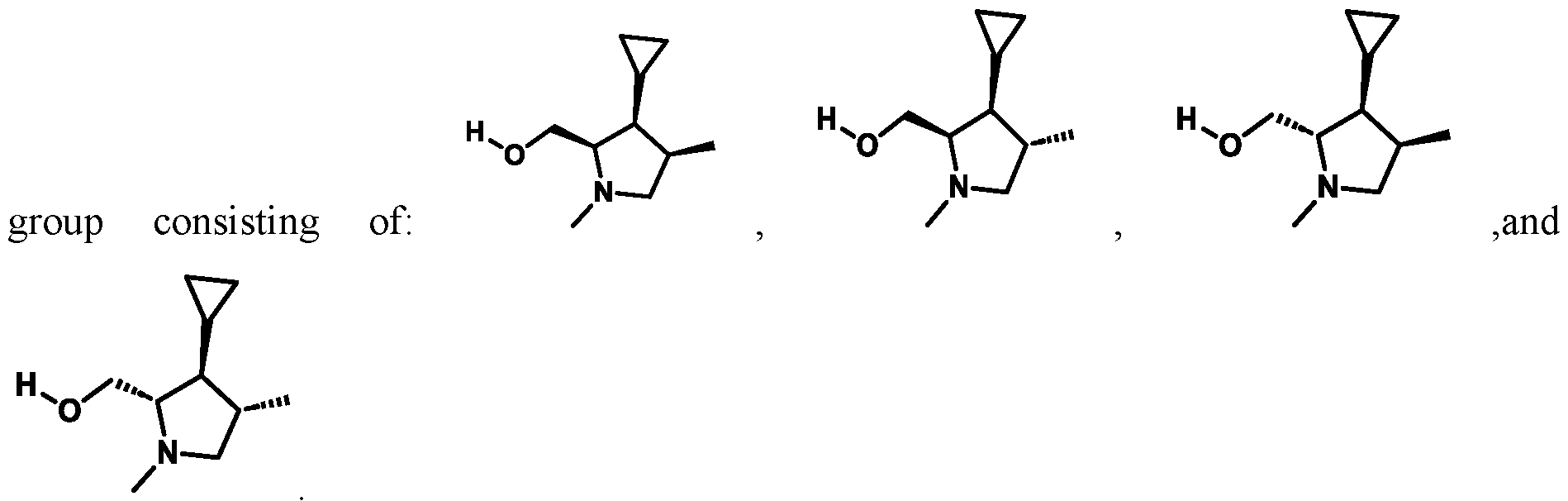
. When the chemical name of a compound contains two stereogenic centers designated “RS” and/or “SR,” a mixture of stereoisomers is provided wherein among the constituent stereoisomers these two stereogenic centers differ either concertedly or independently, in
conformity with the &x (e.g., &1 or &2) notations in Table C1 for the corresponding compound. When two defined stereogenic centers are labelled &1 and &1 in a chemical structure, and they are designated “RS” and/or “SR” in the corresponding chemical name, then a mixture of two stereoisomers is provided, wherein each constituent stereoisomer has the relative stereochemistry between these two stereogenic centers (e.g., syn or anti relationship) as depicted by the structural formula and by the name. As an example, a chemical name designated (RS,SR) represents a mixture of (R,S) and (S,R) stereoisomers, and this mixture does not include (R,R) or (S,S) stereoisomers. As a second example, a chemical name designated (SR,SR) represents a mixture of (S,S) and (R,R) stereoisomers, and this mixture does not include (R,S) or (S,R) stereoisomers. For example, the chemical name
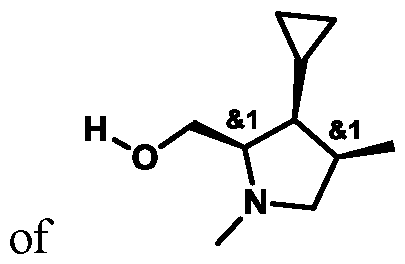
is ((2RS,3S,4RS)-3-cyclopropyl-1,4-dimethylpyrrolidin-2-yl)methanol. This name, taken together with the structural formula, represents a mixture of two stereoisomers:
. When two defined stereogenic centers are labelled &1 and &2 in a chemical structure, and they are designated “RS” and/or “SR” in the corresponding chemical name, then a mixture of four stereoisomers is provided each differing in configuration at one or both of these two stereogenic centers (e.g., a chemical name designated (RS,SR) represents a mixture of (R,S), (S,R), (R,R), and (S,S) stereoisomers). For example, the chemical name
((2RS,3S,4RS)-3-cyclopropyl-1,4-dimethylpyrrolidin-2-yl)methanol. This name, taken together with the structural formula, represents a mixture of four stereoisomers:
General Analytical Methods: Method A: VanGuard Pre-Column CSH C18, 1.7 μm, 2.1 × 5 mm, Pre-run: 1 mL/min for 0.7 minutes. Column: Acquity UPLC, CSH C18, 1.7 μm, 2.1 × 30 mm, Flow rate: 0.9 mL/min, 5 to 100% MeCN/ H
2O (+ 0.1% formic acid) for 3 minutes. Method B: VanGuard Pre-Column CSH C18, 1.7 μm, 2.1 × 5 mm, Pre-run: 1 mL/min for 0.7 minutes. Column: Acquity UPLC, CSH C18, 1.7 μm, 2.1 × 30 mm, Flow rate: 0.9 mL/min, 5 to 100% MeCN/ H2O (+ 10 mM ammonium bicarbonate) for 3 minutes. Method C: Acquity UPLC, CSH C18, 1.7 μm, 2.1 × 30 mm, Mobile phase A: 0.1% formic acid in H
2O; Mobile phase B: 0.1% formic acid in MeCN (v/v); Gradient: 95% H
2O/5% MeCN linear to 5% H2O/95% MeCN in 2.0 minutes, hold at 5% H2O/95% MeCN to 2.50 minutes. Then 5% H2O/95% MeCN linear to 95% H2O/5% MeCN in 0.5 minutes. Flow rate: 0.6 mL/min. Method D: Acquity UPLC, CSH C18, 1.7 μm, 2.1 × 30 mm, Mobile phase A: 0.1% ammonium hydroxide in H
2O; Mobile phase B: 0.1% ammonium hydroxide in MeCN (v/v); Gradient: 95% H
2O/5% MeCN linear to 5% H
2O/95% MeCN in 2.0 minutes, hold at 5% H2O/95% MeCN to 2.50 minutes. Then 5% H2O/95% MeCN linear to 95% H2O/5% MeCN in 0.5 minutes. Flow rate: 0.6 mL/min. Method E: Acquity UPLC, CSH C18, 1.7 μm, 2.1 × 30 mm, Mobile phase A: 0.1% formic acid in H2O; Mobile phase B: 0.1% formic acid in MeCN (v/v); Gradient: 95% H2O/5% MeCN linear to 5% H
2O/95% MeCN in 4.0 minutes, hold at 5% H
2O/95% MeCN to 4.50 minutes. Then 5% H2O/95% MeCN linear to 95% H2O/5% MeCN in 0.5 minutes. Flow rate: 0.6 mL/min. Method F: Poroshell SB-C18, 4.6 × 150 mm, Mobile phase A: 0.1% TFA in H
2O (v/v), Mobile phase B: 0.1% TFA in MeCN (v/v), Gradient: 30-95% Mobile phase B over 15 minutes. Method G: Chiralpak IB-U (3.0 × 100 mm, 1.6 mM, P/N 81U83)
Method H: Kinetex® EVO C182.1 × 30 mm 5 µm, Mobile phase A: 0.0375% TFA in H2O (v/v), Mobile phase B: 0.01875% TFA in MeCN (v/v), Gradient: 95% H2O/5% MeCN linear to 5% H2O/95% MeCN in 0.6 minutes, hold at 5% H2O/95% MeCN to 0.78 minutes. Flow rate: 2.0 mL/min. Then 5% H
2O/95% MeCN linear to 95% H
2O/5% MeCN in 0.02 minutes. Flow rate: 2.0 mL/min. Method I: VanGuard Pre-Column CSH C18, 1.7 μm, 3.0 × 50 mm, Pre-run: 0.65 mL/min for 2.5 minutes. Column: Acquity UPLC, CSH C18, 1.7 μm, 2.1 × 75 mm, Flow rate: 0.6 mL/min, 5 to 100% MeCN/ H2O (+ 10 mM ammonium bicarbonate) for 7.5 minutes. Method J: Column: Chiralcel OD-3R (2.1 × 100 mm, 3 μm, P/N 14893); Mobile phase A = 0.1% v/v ammonium hydroxide in H2O, 0.1% v/v ammonium hydroxide in MeCN; Gradient: 95% H2O/5% MeCN linear to 5% H2O/95% MeCN in 8.0 minutes, hold at 5% H
2O/95% MeCN to 9.00 minutes. Then 5% H
2O/95% MeCN linear to 95% H
2O/5% MeCN in 1.0 minute. Method K: Acquity UPLC, CSH C18, 1.7 μm, 2.1 x 30 mm, Mobile phase A: 0.1% TFA n H
2O; Mobile phase B: 0.1% formic acid in MeCN (v/v); Gradient: 95% H
2O/5% MeCN linear to 5% H2O/95% MeCN in 4.0 minutes, hold at 5% H2O/95% MeCN to 4.50 minutes. Then 5% H2O/95% MeCN linear to 95% H2O/5% MeCN in 0.5 minutes. Flow rate: 0.6 mL/min. Table of Abbreviations:
Example P. Synthesis of Intermediates Intermediate 1: (4-Chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol

To ethyl 4-chloro-6-methyl-2-(methylthio)pyrimidine-5-carboxylate (535 g, 2.17 mol) in toluene (1.5 L) and THF (1.5 L) at -14 °C was added DIBAL-H (4 L of 1.03 M in THF and 666 g of 25 wt% in toluene, 5.29 mol) over 66 minutes. The mixture was gradually warmed to 20 °C and held for 2 hours and then added to potassium sodium tartrate tetrahydrate (4.5 kg, 15.9 mol) in water (9 L) at 2 °C over 15 minutes. Gas evolution and exotherm continued after the transfer was complete with a maximum internal temperature of 31 °C. Ethyl acetate (3.25 L) was added, and the layers were separated. The organic layer was washed with brine (4 L). The aqueous layers were sequentially extracted with ethyl acetate (3.25 L). The combined organic layers were dried magnesium sulfate and filtered through a pad of Magnesol (380 g), washing with ethyl acetate (1.5 L). The filtrate was concentrated to a solid then triturated in ethyl acetate (800 mL) at 45-50 °C, diluted with n-heptane (2.4 L) over 20 minutes, and cooled to room temperature overnight. The slurry was filtered and washed with n-heptane, and the solids were dried under vacuum to afford (4-chloro-6-methyl-2-(methylthio)pyrimidin-5- yl)methanol (328 g, 1.60 mol) as a white solid. 1H NMR (400 MHz, CDCl
3) δ 4.80 (d, 2H), 2.64 (s, 3H), 2.59 (s, 3H), 1.90 (t, 1H). tR = 4.84 minutes (Method F). Intermediate 2: 7-Bromo-4'-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]
Step 1: 7-Bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-ol To diisopropylamine (355 mL, 2.53 mol) in THF (3.5 L) at -28 °C was added nBuLi in hexanes (1 L, 2.5 M, 2.5 mol) over 24 minutes. The mixture was held at -16 to -26 °C for 10 minutes then cooled to -71 °C. (4-Chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (255 g, 1.25 mol) in THF (1.4 L) was added over 45 minutes, rinsing with THF (100 mL) while maintaining the internal temperature below -66°C. The mixture was stirred for one hour at -70 °C, and then 7-bromo-3,4-dihydronaphthalen-1(2H)-one (280 g, 1.24 mol) in THF (1 L) was added over 37 minutes, maintaining the temperature below -70 °C. The mixture was warmed to 2 °C over two hours and then quenched with ammonium chloride (500 g) in water (1.75 L). The layers were separated, and the organic layer was washed with brine (1.5 L). The aqueous layers were sequentially extracted with ethyl acetate (1 L). The combined organic layers were dried over magnesium sulfate and then evaporated to a gummy residue. Dichloromethane (700 mL) was added, and the mixture was re-concentrated to a solid. The solid was triturated in dichloromethane (2 L) at 35°C for 20 minutes, cooled to 15 °C for 30 minutes, filtered, and washed with dichloromethane (400 mL). The solids were dried under vacuum at 35°C to afford 7-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-1,2,3,4- tetrahydronaphthalen-1-ol (360 g, 0.84 mol) as a white solid. 1H NMR (400 MHz, CDCl
3) δ7.70 (s, 1H), 7.30 (d, 1H), 7.00 (d, 1H), 4.80-4.70 (m, 2H), 4.33 (s, 1H), 3.40-3.25 (m, 3H), 2.80 (t, 2H), 2.55 (s, 3H), 2.15-1.85 (m, 3H), 1.75-1.65 (m, 1H). tR = 10.491 minutes (Method F). Step 2: 7-Bromo-4'-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] 7-Bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)- 1,2,3,4-tetrahydronaphthalen-1-ol (200 g, 465 mmol) was suspended in toluene (1 L, 5 vol). Phosphoric acid (86 wt%, 35 mL, 512 mmol) was added before heating the reaction to reflux for 27 hours. The solution was cooled to room temperature before adjusting the pH from 1 to 7 using 4 N NaOH (150 mL, 600 mmol). The reaction mixture was then diluted with ethyl acetate (500 mL, 2.5 vol) and water (500 mL, 2.5 vol). The phases were separated, and the organic phase washed further with brine (500 mL, 2.5 vol). The aqueous phases were
sequentially back-extracted with ethyl acetate (500 mL, 2.5 vol). The organic phases were combined and dried over sodium sulfate and concentrated to a neat orange oil. The crude product was dissolved in EtOAc (140 mL, 1 vol) at 50 °C. Hexane (450 mL, 3 vol) was added to the solution over 5 minutes, and the resulting crystals were filtered and washed with EtOAc/hexane (1:4, 100 mL). This process was repeated three times to afford 7-bromo-4'- chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine] (113 g, 274 mmol) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ7.55 (s, 1H), 7.35 (d, 1H), 7.05 (d, 1H), 4.80 (q, 2H), 3.15 (q, 2H), 2.90-2.78 (m, 2H), 2.60 (s, 3H), 2.07-1.92 (m, 3H), 1.85-1.70 (m, 1H). t
R = 13.149 minutes (Method F). Intermediate 2a: (S)-7-Bromo-4'-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]
The enantiomers of 7-bromo-4'-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (5.0 g) were separated by chiral SFC (Chiralpak IB-N 21mm ID × 250 mm, 5μm (serial number IBS5MJ-BV001; MeOH, 40%, backpressure = 120 psi). The first eluting peak was isolated as Intermediate 2a (1.85 g). LCMS: m/z (ESI) [M+H]
+ 413.0, tR = 4.00 minutes (Method E) Chiral HPLC: t
R = 4.78 minutes. (Method G; Gradient 5-40% MeOH over 10 minutes) 1H NMR (400 MHz, CDCl
3) δ 7.54 (d, 1H), 7.27 (dd, 1H), 6.93 (d, 1H), 4.70 (q, 2H), 3.17 – 2.85 (m, 2H), 2.82 – 2.61 (m, 2H), 2.50 (s, 3H), 1.99 – 1.79 (m, 3H), 1.78 – 1.62 (m, 1H). Intermediate 2b: (R)-7-Bromo-4'-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]
The second eluting peak was isolated as Intermediate 2b (2.05 g). LCMS: m/z (ESI) [M+H]
+ 413.0, tR = 4.00 minutes (Method E) Chiral HPLC: tR = 6.12 minutes. (Method G; Gradient 5-40% MeOH over 10 minutes) 1H NMR (400 MHz, CDCl
3) δ 7.61 (d, 1H), 7.34 (dd, 1H), 7.00 (d, 1H), 4.92 – 4.63 (m, 2H), 3.26 – 2.94 (m, 2H), 2.90 – 2.72 (m, 2H), 2.57 (s, 3H), 2.09 – 1.86 (m, 3H), 1.82 – 1.67 (m, 1H). Intermediate 3: tert-Butyl (2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-oxo-3,3',4,4',5',8'-hexahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate

Step 1: 4'-(Benzyloxy)-7-bromo-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] Phenylmethanol (367 μL, 1.5 equiv, 3.53 mmol) was added to a suspension of sodium hydride (188 mg, 60 wt%, 2 equiv, 4.71 mmol) in THF (6 mL). The mixture was stirred at room temperature for 5 minutes until gas evolution had ceased. A solution of 7-bromo-4'-chloro-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (970 mg, 1 equiv, 2.36 mmol) in THF (6 mL) was added dropwise, and the reaction mixture was stirred for 1 hour. The mixture was cooled in an ice bath and quenched by the addition of saturated aqueous ammonium chloride (20 mL) and then extracted with EtOAc (2 × 50 mL). The combined organic phases were washed with brine (50 mL), dried over sodium sulfate, and concentrated to a solid which was triturated with ethanol (60 mL) (sonication was used to break up the material). The solid was separated by filtration, washed with ethanol (10 mL), and dried to yield 4'-(benzyloxy)-7-bromo-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidine] (1.05 g, 2.17 mmol). LCMS: m/z (ESI) [M+H]
+ 485.1, tR = 2.24 minutes (Method A) Step 2: tert-Butyl (4'-(benzyloxy)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate
A suspension of 4'-(benzyloxy)-7-bromo-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (2.75 g, 1 equiv, 5.69 mmol), cesium carbonate (5.56 g, 3 equiv, 17.1 mmol), and tert-butyl carbamate (2.00 g, 3 equiv, 17.1 mmol) in 1,4-dioxane (80 mL) was degassed with nitrogen at room temperature for 5 minutes. BrettPhos Pd G4 (524 mg, 0.1 equiv, 0.57 mmol) was added, and nitrogen was bubbled through the mixture at room temperature for another 5 minutes. The reaction mixture was stirred for 5 hours, concentrated to a residue, and purified by flash chromatography (silica, 120 g silicycle cartridge, solid loading, gradient of EtOAc in DCM) to provide tert-butyl (4'- (benzyloxy)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (1.91 g). LCMS: m/z (ESI) [M+H]
+ 520.3, t
R = 2.14 minutes (Method A) Step 3: tert-Butyl (4'-(benzyloxy)-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate Tetrabutylammonium hydrogen sulfate (199 mg, 0.16 equiv, 0.59 mmol) and sodium tungstate dihydrate (121 mg, 0.1 equiv, 0.37 mmol) were added to a solution of tert-butyl (4'- (benzyloxy)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (1.90 g, 1 equiv, 3.66 mmol) in EtOAc (70 mL), and the temperature was raised to 40 °C. Hydrogen peroxide (2.61 mL, 30 wt%, 7 equiv, 25.6 mmol) was added dropwise, and the reaction mixture was stirred for 100 minutes. The reaction mixture was diluted with EtOAc (100 mL), washed with water (100 mL) and brine (100 mL), dried over sodium sulfate, and concentrated. After the material was concentrated to dryness, DCM (20 mL) was added, and the resulting material was concentrated to dryness again to yield tert-butyl (4'-(benzyloxy)-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate (2.09 g). LCMS: m/z (ESI) [M+H]
+ 552.2, tR = 1.86 minutes (Method A) Step 4: tert-Butyl (4'-(benzyloxy)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate Sodium hydride (359 mg, 60 wt%, 2.5 equiv, 8.97 mmol) was added to a solution of ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (1.43 g, 2.5 equiv, 8.97 mmol) in anhydrous THF (20 mL). This solution was stirred for 2 minutes until gas evolution
had ceased, and then a solution of tert-butyl (4'-(benzyloxy)-2'-(methylsulfonyl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (2.04 g, 1 equiv, 3.59 mmol) in anhydrous THF (20 mL) was added over 1 minute. The mixture was stirred for 2 hours at room temperature, and then water (20 mL) and brine (20 mL) were added. The mixture was extracted with DCM:MeOH 9:1 (3 ×100 mL). The combined organic phases were washed with brine (100 mL), dried over sodium sulfate, and concentrated to a residue which was purified by flash chromatography (silica, 120 g silicycle cartridge, gradient of DCM:MeOH:NH4OH 85:13.5:1.5 in DCM) to yield tert-butyl (4'-(benzyloxy)-2'-(((2R,7aS)- 2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (1.9 g). LCMS: m/z (ESI) [M+H]
+ 631.5, t
R = 1.43 minutes (Method A) Step 5: tert-Butyl (2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-oxo-3,3',4,4',5',8'-hexahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate To a solution of tert-Butyl (4'-(benzyloxy)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (1.50 g, 1 equiv, 2.14 mmol) in methanol (60 mL) was added Pd/C (114 mg, 10 wt%, 0.05 equiv, 0.11 mmol). The mixture was degassed with nitrogen, and then hydrogen was bubbled through the mixture at room temperature for 5 minutes. The reaction mixture was stirred at room temperature for 2 hours and then purged with nitrogen. The mixture was diluted with DCM (50 mL), and filtered through a syringe filter. The syringe filter was washed with DCM (3 × 10 mL). The combined liquid was concentrated to a residue which was purified by flash chromatography (silica, gradient of DCM:MeOH:NH
4OH 85:13.5:1.5 in DCM) to yield tert-butyl (2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-oxo-3,3',4,4',5',8'-hexahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (1.13 g). LCMS: m/z (ESI) [M+H]
+ 541.3, t
R = 1.04 minutes (Method A) 1H NMR (400 MHz, DMSO-d6): δ 12.29 (s, 1H), 9.17 (s, 1H), 7.51 (s, 1H), 7.23 (d, 1 H), 6.97 (d, 1H), 5.26 (d, 1H), 4.37-4.24 (m, 2H), 4.03-3.92 (m, 2H), 3.12-2.95 (m, 3H), 2.82- 2.55 (m, 5H), 2.09-1.64 (m, 10H), 1.43 (s 9H). Intermediate 4: tert-Butyl ((S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-
7a(5H)-yl)methoxy)-4'-oxo-3,3',4,4',5',8'-hexahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate

Step 1: (S)-4'-(benzyloxy)-7-bromo-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] Phenylmethanol (1.05 mL, 1.5 equiv, 10.1 mmol) was added to a suspension of sodium hydride (540 mg, 60 wt% dispersion in mineral oil, 2 equiv, 13.5 mmol) and stirred for 5 minutes at room temperature until gas evolution had ceased. A solution of (S)-7-bromo-4'- chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine] (2.78 g, 1 equiv, 6.75 mmol) in THF (12 mL) was added dropwise, resulting in the formation of a hazy solution. After 50 minutes, the mixture was cooled in an ice bath and quenched by addition of saturated aqueous ammonium chloride (40 mL). The mixture was extracted with EtOAc (2 × 100 mL). The combined organic phases were washed with brine (50 mL), dried over anhydrous sodium sulfate, and concentrated to a residue which was purified by flash column chromatography (silica, 80 g silicycle cartridge, DCM in hexanes (0-100%)) to afford (S)-4'-(benzyloxy)-7-bromo-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (3.13 g). LCMS: m/z (ESI) [M+H]
+ 485.1, tR = 2.25 minutes (Method A) 1H NMR (400 MHz, CDCl
3): δ7.62 (s, 1 H), 7.41-7.31 (m, 6 H), 6.98 (d, 1H), 5.51- 5.44 (m, 2 H), 4.78-4.65 (m, 2 H), 3.08-2.89 (m, 2 H), 2.78-2.75 (m, 2 H), 2.55 (s, 3 H), 1.97- 1.89 (m, 3 H), 1.76-1.73 (m, 1 H). Step 2: tert-Butyl (S)-(4'-(benzyloxy)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate A suspension of (S)-4'-(benzyloxy)-7-bromo-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (3.08 g, 1 equiv, 6.37 mmol), cesium carbonate (6.23 g, 3 equiv, 19.1 mmol), and tert-butyl carbamate (2.24 g, 3 equiv, 19.1 mmol) in 1,4-dioxane (90 mL) was degassed for 5 minutes. BrettPhos Pd G4 (587 mg, 0.1 equiv, 0.637
mmol) was added, and the mixture was degassed at room temperature for another 5 minutes and then stirred at 80 °C for 6 hours. The reaction mixture was concentrated to a residue which was purified by flash column chromatography (silica, 120 g silicycle cartridge, gradient of EtOAc in DCM, (0-100%) to afford tert-butyl (S)-(4'-(benzyloxy)-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (2.27 g). LCMS: m/z (ESI) [M+H]
+ 520.3, tR = 2.14 minutes (Method A) 1H NMR (400 MHz, CDCl
3): δ 7.44-7.30 (m, 7 H), 7.03 (d, 1 H), 6.37 (s, 1 H), 5.47 (s, 2 H), 4.74-4.64 (m, 2 H), 3.14-2.90 (m, 2 H), 2.79-2.76 (m, 2 H), 2.55 (s, 3 H), 1.97-1.91 (m, 3 H), 1.74-1.72 (m, 1 H), 1.49 (s, 9 H). Step 3: tert-Butyl (S)-(4'-(benzyloxy)-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate Tetrabutylammonium hydrogen sulfate (237 mg, 0.16 equiv, 0.70 mmol) and sodium tungstate dihydrate (144 mg, 0.1 equiv, 0.44 mmol) were added to a yellow solution of tert- butyl (S)-(4'-(benzyloxy)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate (2.27 g, 1 equiv, 4.37 mmol) in EtOAc (90 mL), and the temperature was raised to 40 °C. Hydrogen peroxide (3.47 g, 3.12 mL, 30 wt%, 7 equiv, 30.6 mmol) was added dropwise. The mixture was stirred at 40 °C for 80 minutes. The crude reaction mixture was diluted with EtOAc (100 mL), washed with water (100 mL), brine (100 mL), dried over anhydrous sodium sulfate, and concentrated. DCM (20 mL) was added, and the material was concentrated to dryness twice to yield tert-butyl (S)-(4'-(benzyloxy)-2'- (methylsulfonyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (2.39 g), which was used without further purification. LCMS: m/z (ESI) [M-H]- 550.3, t
R = 1.86 minutes (Method A) 1H NMR (400 MHz, CDCl3): δ 7.48-7.38 (m, 6 H), 7.19 (dd,1 H), 7.05 (d, 1H), 6.38 (s, 1H), 5.56 (s, 2H), 4.82-4.71 (m, 2H), 3.31-3.09 (m, 5H), 2.82-2.74 (m, 2H), 2.00-1.91 (m, 3H), 1.74-1.70 (m, 1H), 1.48 (s, 9H). Step 4: tert-Butyl ((S)-4'-(benzyloxy)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate Sodium hydride (433 mg, 60 wt%, dispersion in mineral oil, 2.5 equiv, 10.8 mmol) was added to a solution of ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (1.72 g,
2.5 equiv, 10.8 mmol) in anhydrous THF (24 mL) and stirred for 2 minutes at room temperature until gas evolution ceased. A solution of tert-butyl (S)-(4'-(benzyloxy)-2'-(methylsulfonyl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (2.39 g, 1 equiv, 4.33 mmol) in anhydrous THF (24 mL) was added over 1 minute and stirred for 70 minutes. A mixture of water (20 mL) and brine (20 mL) was added, and the aqueous phase was extracted with DCM:MeOH 9:1 (3 × 100 mL). The combined organic phases were washed with brine (100 mL), dried over anhydrous sodium sulfate, filtered, and concentrated to a residue, which was purified using flash column chromatography (silica, 120 g silicycle cartridge, gradient of DCM:MeOH:NH4OH 85:13.5:1.5/DCM (0-100%) to yield tert-butyl ((S)-4'- (benzyloxy)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (1.89 g). LCMS: m/z (ESI) [M+H]
+ 631.5, tR = 1.43 minutes (Method A) 1H NMR (400 MHz, CDCl3): δ 7.36-7.22 (m, 7 H), 7.04 (d, 1H), 6.37 (s, 1H), 5.44 (s, 2H), 5.26 (d, 1H), 4.72-4.63 (m, 2 H), 4.16-4.02 (m, 2 H), 3.27-2.71 (m, 8 H), 2.27-2.12 (m, 3 H), 1.98-1.82 (m, 6 H), 1.74-1.70 (m, 1 H), 1.49 (s, 9 H). 1
9F NMR (376 MHz, CDCl3): δ -173.2. Step 5: tert-Butyl ((S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-oxo-3,3',4,4',5',8'-hexahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate To a degassed solution of tert-butyl ((S)-4'-(benzyloxy)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (1.89 g, 1 equiv, 3.00 mmol) in methanol (60 mL) was added Pd/C (159 mg, 10 wt%, wet, 0.05 equiv, 0.15 mmol). The suspension was degassed for 5 minutes. Then hydrogen was bubbled through the mixture at room temperature for 5 minutes. The mixture was stirred at room temperature for 2 hours. The reaction mixture was purged with nitrogen, diluted with DCM (50 mL), and filtered. The solid was washed with DCM (3 × 10 mL), and the combined filtrates were concentrated under reduced pressure to yield tert-butyl ((S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-oxo-3,3',4,4',5',8'-hexahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (1.62 g). LCMS: m/z (ESI) [M+H]
+ 541.3, tR = 1.05 minutes (Method A)
1H NMR (400 MHz, DMSO-d
6): δ 12.29 (s, 1H), 9.17 (s, 1H), 7.51 (s, 1H), 7.23 (dd, 1H), 6.97 (d, 1H), 5.26 (d, 1H), 4.38-4.24 (m, 2H), 4.03-3.92 (m, 2H), 3.13-2.96 (m, 3H), 2.81- 2.54 (m, 5H), 2.09-1.63 (m, 10H), 1.43 (s, 9 H). 1
9F NMR (376 MHz, DMSO): δ -172.0. Intermediate 5: tert-Butyl ((S)-4'-((1H-benzo[d][1,2,3]triazol-1-yl)oxy)-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate

((1H-benzo[d][1,2,3]triazol-1-yl)oxy)tris(dimethylamino)phosphonium hexafluorophosphate(V) (972 mg, 1.1 equiv, 2.20 mmol) was slowly added to a solution of tert-butyl ((S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'- hydroxy-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (1.08 g, 1 equiv, 2.00 mmol) and DIPEA (516 mg, 2 equiv, 4.00 mmol) in DMF (20 mL). The resulting colorless solution was stirred at room temperature for 3 hours. The mixture was poured into water (20 mL) and stirred for 5 minutes. The resulting precipitate was removed by filtration, washed with water (3 × 10 mL) and dried under vacuum to yield tert- butyl ((S)-4'-((1H-benzo[d][1,2,3]triazol-1-yl)oxy)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (1.19 g). LCMS: m/z (ESI) [M+H]
+ 658.4, tR = 1.37 minutes (Method A). 1H NMR (400 MHz, DMSO-d
6): δ 9.23 (s, 1H), 8.18 (d, 1H), 7.86 (d, 1H), 7.62-7.67 (m, 2H), 7.53 (t, 1H), 7.23 (dd, 1H), 7.02 (d, 1H), 5.12 (s, 1H), 4.91 (t, 2H), 3.49-3.61 (m, 2H), 2.97-3.09 (m, 2H), 2.87 (d, 1H), 2.80-2.82 (m, 1H), 2.76-2.79 (m, 1H), 2.71-2.74 (m, 2H), 2.62-2.68 (m, 2H), 1.99 (s, 2H), 1.90 (br s, 1H), 0.85-1.68 (m, 3H), 1.64 (dd, 2H), 1.50-1.58 (m, 1H), 1.40-1.43 (m, 9H). 1
9F NMR (376 MHz, DMSO): F -172.46 to -171.99 (m).
Intermediate 6: N,N-Dibenzyl-8-bromo-4'-chloro-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine
Step 1: 7-Amino-8-bromo-3,4-dihydronaphthalen-1(2H)-one A solution of 1-bromopyrrolidine-2,5-dione (2.86 g, 1 equiv, 16.1 mmol) in DMF (20 mL) was added dropwise to a solution of 7-amino-3,4-dihydronaphthalen-1(2H)-one (2.59 g, 1 equiv, 16.1 mmol) in DMF (20 mL) at 0 °C. The resulting mixture was stirred for 4 hours at room temperature and then poured into water. The mixture was extracted with DCM three times, and the combined extracts were washed with water. The combined organic phases were dried over sodium sulfate and passed through a Celite pad. The filtrate was concentrated to a residue which was purified by column chromatography (80 g, 0-20% hexane/EtOAc) to afford 7-amino-8-bromo-3,4-dihydronaphthalen-1(2H)-one (3.31 g). LCMS: m/z (ESI) [M+H]
+ 239.9, tR = 1.02 minutes (Method A). 1H NMR (400 MHz, CDCl
3): δ 7.01 (d, 1H), 6.88 (d, 1H), 4.30 (s, 2 H), 2.86 (t, 2 H), 2.66 (t, 2H), 2.03-2.09 (m, 2 H). Step 2: 8-Bromo-7-(dibenzylamino)-3,4-dihydronaphthalen-1(2H)-one Potassium carbonate (5.36 g, 3.5 equiv, 38.8 mmol) was added to 7-amino-8-bromo- 3,4-dihydronaphthalen-1(2H)-one (2.66 g, 1 equiv, 11.1 mmol) in anhydrous DMF (55.4 mL) followed by the dropwise addition of (bromomethyl)benzene (4.71 mL, 3.5 equiv, 38.8 mmol) at room temperature. The resulting mixture was stirred at 70 °C for 16 hours. Water was added, and the aqueous layer was extracted with EtOAc three times. The combined extracts were washed with brine, dried over sodium sulfate, filtered, and evaporated to a residue, which was purified by column chromatography (80 g silica, 0-5% hexane/EtOAc) to afford 8-bromo- 7-(dibenzylamino)-3,4-dihydronaphthalen-1(2H)-one (4.25 g). LCMS: m/z (ESI) [M+H]
+ 422.2, tR = 1.99 minutes (Method A). Step 3: 8-Bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-7-(dibenzylamino)-1,2,3,4-tetrahydronaphthalen-1-ol LDA (26.9 mL, 1 M, 2.5 equiv, 26.9 mmol) was added to a solution of (4-chloro-6- methyl-2-(methylthio)pyrimidin-5-yl)methanol (2.20 g, 1 equiv, 10.8 mmol) in THF (108 mL)
at -78 °C over 15 minutes. The mixture was stirred at that temperature for 1 hour, and then a solution of 8-bromo-7-(dibenzylamino)-3,4-dihydronaphthalen-1(2H)-one (4.970 g, 1.1 equiv, 11.8 mmol) in THF (50 mL) was added over 15 minutes. The reaction mixture was stirred at that temperature for 1 hour and then quenched with aqueous ammonium chloride at -78 °C. The reaction mixture was warmed slowly to room temperature, extracted with EtOAc, and washed with brine. The combined organic layers were dried over sodium sulfate, filtered, and concentrated to a residue which was purified by flash column chromatography (120 g silica, 0- 20% hexane/EtOAc) to give 8-bromo-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-7-(dibenzylamino)-1,2,3,4-tetrahydronaphthalen-1-ol (4 g). LCMS: m/z (ESI) [M+H]
+ 626.1, t
R = 2.07 minutes (Method B). Step 4: N,N-Dibenzyl-8-bromo-4'-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine Butyllithium (5.70 mL, 1.6 M, 2.5 equiv, 9.12 mmol) was added dropwise to a solution of 8-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-7- (dibenzylamino)-1,2,3,4-tetrahydronaphthalen-1-ol (2.28 g, 1 equiv, 3.65 mmol) in THF (25.6 mL) at -65 °C. The reaction mixture was stirred for 30 minutes. A solution of 4- methylbenzenesulfonyl chloride (1.04 g, 1.5 equiv, 5.47 mmol) in THF (20 mL) was added dropwise at -65 °C, and the resulting mixture was warmed to room temperature and stirred for 2 hours. Saturated aqueous ammonium chloride solution was added, and the mixture was extracted with EtOAc three times. The organic phase was washed with brine, dried over sodium sulfate, filtered, and concentrated under reduced pressure to give a residue which was purified by flash chromatography (80 g, 0-10% hexane/EtOAc) to give N,N-dibenzyl-8-bromo-4'- chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-amine (1.7 g). LCMS: m/z (ESI) [M+H]
+ 608.0, t
R = 2.31 minutes (84%, Method B). Intermediate 7a: (R)-7-bromo-4-methyl-3,4-dihydronaphthalen-1(2H)-one
Aluminum chloride (20.8 g, 2.5 equiv., 156 mmol) was stirred rapidly as 4-methyl-3,4- dihydronaphthalen-1(2H)-one (10.0 g, 1 equiv., 62.4 mmol) was added dropwise by syringe over 20 minutes. The resulting dark-red mass was heated at 85 °C until it melted, at which point Br
2 (12.0 g, 3.86 mL, 1.2 equiv., 74.9 mmol) was added dropwise by syringe over 40 minutes. The mixture was stirred for 1 hour at 85 °C. The reaction mixture was cooled to room temperature and then added to ice-water (500 mL) and concentrated HCl (100 mL). The resulting mixture was extracted with ethyl acetate (4 × 300 mL), and the combined organic layers were filtered through Celite. The filtrate was washed with saturated aqueous sodium bicarbonate solution (250 mL), water (250 mL), and brine (250 mL), dried over sodium sulfate, filtered, and concentrated to give a residue which was purified by reverse-phase HPLC (0.1% formic acid condition) to give 7-bromo-4-methyl-3,4-dihydronaphthalen-1(2H)-one (10.5 g). LCMS: m/z (ESI) [M+H]
+ 240.9, tR = 0.462 minutes (Method H). The enantiomers of 7-bromo-4-methyl-3,4-dihydronaphthalen-1(2H)-one (5 g, 20.9 mmol) were separated by chiral SFC (Column: Chiralpak AD-350 × 4.6 mm I.D., 3 μm Mobile phase: Phase A for CO2, and Phase B for MeOH (0.05% DEA), Gradient elution: B in A from 5% to 40% Flow rate: 3 mL/min, Detector: PDA, Column Temp: 35 °C, Back Pressure: 100 bar) to afford (R)-7-bromo-4-methyl-3,4-dihydronaphthalen-1(2H)-one (Intermediate 7a) and (S)-7-bromo-4-methyl-3,4-dihydronaphthalen-1(2H)-one (Intermediate 7b). Intermediate 7a: First eluting peak, t
R = 1.027 minutes (1780 mg) 1H NMR (400 MHz, CD3OD) δ 8.03 (d, 1H), 7.69 (dd, 1H), 7.36 (d, 1H), 3.15 - 3.04 (m, 1H), 2.84 - 2.74 (m, 1H), 2.66 - 2.55 (m, 1H), 2.31 - 2.20 (m, 1H), 1.96 - 1.84 (m, 1H), 1.40 (d, 3H). Intermediate 7b: (S)-7-bromo-4-methyl-3,4-dihydronaphthalen-1(2H)-one
Intermediate 7b: Second eluting peak, tR = 1.167 minutes (1549 mg) 1H NMR (400 MHz, CD3OD) δ 8.03 (d, 1H), 7.69 (dd, 1H), 7.36 (d, 1H), 3.17 - 3.01 (m, 1H), 2.85 - 2.73 (m, 1H), 2.69 - 2.53 (m, 1H), 2.31 - 2.20 (m, 1H), 1.96 - 1.82 (m, 1H), 1.40 (d, 3H).
Intermediate 8: 4-Chloro-2-(methylthio)-5,6',7',8-tetrahydro-5'H- spiro[pyrano[4,3-d]pyrimidine-7,8'-quinoline]
Step 1: 8-((6-Chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)- 5,6,7,8-tetrahydroquinolin-8-ol LDA (2.889 g, 26.97 mL, 1 M, 2.3 equiv, 26.97 mmol) was added dropwise to (4- chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (2.400 g, 1 equiv, 11.7 mmol) in THF (40 mL) at –78 °C, while keeping the internal temperature below –70 °C. The resulting mixture was kept at –78 °C for 1.5 hours. An orange solid formed, and additional THF (5 mL) was added.6,7-Dihydroquinolin-8(5H)-one (2.071 g, 1.2 equiv, 14.07 mmol) was added dropwise as a solution in THF (20 mL), and the resulting mixture was then stirred at –78 °C for 1.5 hours. The reaction was quenched at –78 °C with saturated aqueous ammonium chloride and diluted with EtOAc. The aqueous phase was extracted with EtOAc (× 4). The combined organic layers were dried over sodium sulfate, filtered, and concentrated to a residue which was triturated with EtOAc to afford 8-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)- 5,6,7,8-tetrahydroquinolin-8-ol as a yellow solid (2.66 g). 1H NMR (400 MHz, CDCl3) δ 8.36 (dd, 1H), 7.47 (dq, 1H), 7.17 (dd, 1H), 4.91 (s, 1H), 4.85 (d, 2H), 4.36 – 4.25 (m, 1H), 3.58 (d, 1H), 3.20 (d, 1H), 2.93 – 2.85 (m, 2H), 2.46 (s, 3H), 2.24 – 2.09 (m, 2H), 2.03 – 1.93 (m, 1H), 1.91 – 1.80 (m, 1H). Step 2: 4-Chloro-2-(methylthio)-5,6',7',8-tetrahydro-5'H-spiro[pyrano[4,3- d]pyrimidine-7,8'-quinoline] Polymer-bound triphenylphosphine (2.38 g, 1.3 equiv, 3.81 mmol) was added to 8-((6- chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-5,6,7,8-tetrahydroquinolin- 8-ol (1.03 g, 1 equiv, 2.93 mmol) in THF (14 mL). The resulting mixture solidified and was cooled to –78 °C. DIAD (770 mg, 740 μL, 1.3 equiv, 3.81 mmol) was added dropwise. The cold bath was removed, and the reaction mixture was stirred for 1 hour. The reaction mixture was filtered to remove triphenylphosphine oxide and rinsed with DCM. The filtrate was
concentrated to a residue which was purified by flash chromatography (24 g silica gel cartridge with gradient elution from 0:100-EtOAc:heptane to 100:0-EtOAc:heptane) to give 4-chloro-2- (methylthio)-5,6',7',8-tetrahydro-5'H-spiro[pyrano[4,3-d]pyrimidine-7,8'-quinoline] (570 mg). 1H NMR (400 MHz, CDCl
3) δ 8.40 (dd, 1H), 7.48 (d, 1H), 7.16 (dd, 1H), 4.75 – 4.53 (m, 2H), 3.76 (dd, 1H), 3.00 – 2.75 (m, 3H), 2.58 (s, 3H), 2.26 – 2.16 (m, 1H), 2.16 – 2.06 (m, 1H), 1.93 – 1.77 (m, 2H). Intermediate 9: 6-Bromo-4'-chloro-2'-(methylthio)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]
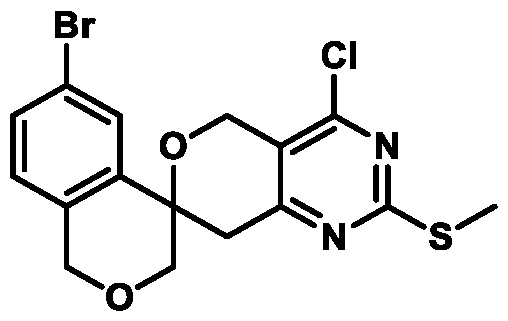
Step 1: 6-bromo-4-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)isochroman-4-ol To a dry flask was combined (4-chloro-6-methyl-2-(methylthio)pyrimidin-5- yl)methanol (2.00 g, 1 equiv, 9.77 mmol) and THF (40.0 mL). The mixture was cooled to -78 °C. LDA solution (2.62 g, 24.4 mL, 1.0 M, 2.5 equiv, 24.4 mmol) was added dropwise at -78 °C, maintaining an internal temperature below -60 °C. After 1 hour, a degassed solution of 6- bromoisochroman-4-one (2.22 g, 1 equiv, 9.77 mmol) in THF (30.0 mL) was added dropwise, and the resulting mixture was stirred at -78 °C for 10 minutes. The reaction mixture, while still cold, was then quickly quenched by being poured into a round-bottom flask containing room temperature saturated aqueous ammonium chloride (80 mL). The organic contents were then extracted with ethyl acetate (80 mL × 2) followed by dichloromethane (80 mL × 2). The combined organic layers were dried over sodium sulfate, filtered, and concentrated under reduced pressure to give a residue which was purified by column chromatography (220 g SiO2 cartridge, 0-40% ethyl acetate in heptane) to give 6-bromo-4-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)isochroman-4-ol (1.84 g). 1H NMR (400 MHz, CDCl
3) δ 7.85 (m, 1H), 7.41 (m, 1H), 6.90 (m, 1H), 5.52 (s, 1H), 4.93 – 4.68 (m, 5H), 3.64 – 3.49 (m, 3H), 3.26 (m, 1H), 3.05 (m, 1H).
Step 2: 6-Bromo-4'-chloro-2'-(methylthio)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidine] To a vial of 6-bromo-4-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)isochroman-4-ol (736 mg, 1 equiv, 1.70 mmol) was added triphenylphosphine (2.13 g, 2.0 equiv, 3.41 mmol) (polymer supported, ~1.6 mmol/g loading). The vial was degassed and filled with N2 before dry THF (20.0 mL) and dry DCM (20.0 mL) were added. To this mixture was added dropwise DIAD (345 mg, 1 equiv, 1.70 mmol) at ambient temperature. The reaction mixture was stirred for 4 hours. The reaction mixture was diluted with ethyl acetate (100 mL) and then dichloromethane (100 mL). The mixture was then filtered over Celite and sodium sulfate and concentrated to a residue which was purified by column chromatography (50 g SiO
2 cartridge, 0-40% ethyl acetate in heptane) to give 6-bromo-4'-chloro-2'- (methylthio)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine] (383 mg). 1H NMR (400 MHz, DMSO-d6) δ 7.69 (d, 1H), 7.51 (dd, 1H), 7.11 (d, 1H), 4.82 (m, 1H), 4.74 – 4.65 (m, 3H), 3.93 (m, 1H), 3.78 (m, 1H), 3.25 (m, 1H), 2.99 (m, 1H), 2.53 (s, 3H). Intermediate 10: (3R)-1-(6-Bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol

Step 1: Methyl 4-(6-bromo-1-hydroxy-2,3-dihydro-1H-inden-1-yl)-3- oxobutanoate Sodium hydride (1.00 g, 60 wt%) was suspended in THF (50 mL) and cooled to 0 ℃. Methyl 3-oxobutanoate (4.00 g) was added, maintaining an internal temperature below 10 ℃. The solution was cooled to -75 ℃, and LDA (4.00 g) was added dropwise, maintaining a temperature below -68 ℃. The reaction mixture was gradually warmed to 0 ℃, cooled to -78 ℃, followed by dropwise addition of 6-bromo-2,3-dihydro-1H-inden-1-one (5.00 g) in THF (10 mL), maintaining a temperature below -68 °C. The reaction mixture was stirred and gradually warmed to 10 °C over the course of 4 hours. The reaction was quenched with aqueous ammonium chloride and EtOAc was added to the mixture (50 mL) and further extracted with
ethyl acetate (2 × 40 mL). The organic layers were dried over sodium sulfate, filtered, and concentrated to a crude product, which was purified by column chromatography (SiO2, heptanes/EtOAc = 100/0 to 10/6) to give methyl 4-(6-bromo-1-hydroxy-2,3-dihydro-1H- inden-1-yl)-3-oxobutanoate (4.50 g). LCMS: m/z (ESI) [M-H]- 325.2, tR = 1.70 minutes (Method C). 1H NMR (400 MHz, CDCl3) δ 7.41-7.49 (m, 1H), 7.33-7.39 (m, 1H), 7.06-7.13 (m, 1H), 3.81 (s, 1H), 3.74 (s, 3H), 3.42-3.58 (m, 2H), 3.06-3.16 (m, 1H), 2.89-3.04 (m, 2H), 2.70- 2.82 (m, 1H), 2.23-2.33 (m, 2H). Step 2: Methyl 6-bromo-4'-oxo-2,3,3',4'-tetrahydrospiro[indene-1,2'-pyran]-5'- carboxylate DMA-DMF (1.8 g, 2.0 mL, 1.1 equiv, 15 mmol) was added to methyl 4-(6-bromo-1- hydroxy-2,3-dihydro-1H-inden-1-yl)-3-oxobutanoate (4.50 g, 1 equiv, 14 mmol) in DCM (100 mL) and stirred for 6 hours at room temperature. The mixture was cooled to 0 °C, followed by dropwise addition of BF3 ^OEt2 (2.1 g, 1.9 mL, 1.1 equiv, 15 mmol). After gradual warming to room temperature over 2 hours, the reaction was quenched with aqueous sodium bicarbonate and extracted with EtOAc (2 × 30 mL). Organic extracts were dried over sodium sulfate, filtered, and concentrated to a crude product, which was purified by column chromatography (SiO2, heptanes/EtOAc = 100/0 to 10/6) to give methyl 6-bromo-4'-oxo-2,3,3',4'- tetrahydrospiro[indene-1,2'-pyran]-5'-carboxylate (3.45 g). 1H NMR (400 MHz, CDCl
3) δ 8.25-8.30 (m, 1H), 7.46-7.54 (m, 2H), 7.17-7.24 (m, 1H), 3.83 (s, 3H), 3.01-3.16 (m, 2H), 2.83-2.95 (m, 1H), 2.68-2.77 (m, 1H), 2.44-2.55 (m, 1H), 2.24-2.36 (m, 1H). Step 3: Methyl 6-bromo-4'-oxo-2,3,3',4',5',6'-hexahydrospiro[indene-1,2'-pyran]- 5'-carboxylate Methyl 6-bromo-4'-oxo-2,3,3',4'-tetrahydrospiro[indene-1,2'-pyran]-5'-carboxylate (5.2 g, 1 equiv, 15 mmol) in THF (80 mL) was cooled to -78 °C followed by the addition of L- selectride (17 mL, 1 M, 1.1 equiv, 17 mmol). The mixture was stirred for 2 hours. The reaction was quenched with aqueous ammonium chloride and extracted with EtOAc (2 × 20 mL). Organic layers were dried with magnesium sulfate, filtered, and concentrated to a crude product, which was purified by column chromatography (SiO2, heptanes/EtOAc = 100/0 to 10/6) to give methyl 6-bromo-4'-oxo-2,3,3',4',5',6'-hexahydrospiro[indene-1,2'-pyran]-5'-
carboxylate (4.40 g). 1H NMR (400 MHz, CDCl3) δ 7.37-7.46 (m, 2H), 7.10-7.17 (m, 1H), 4.29-4.39 (m, 1H), 4.17-4.26 (m, 1H), 4.07-4.16 (m, 1H), 3.80 (s, 3H), 2.97-3.09 (m, 1H), 2.73-2.84 (m, 1H), 2.50-2.60 (m, 2H), 2.25-2.35 (m, 1H), 2.05-2.15 (m, 1H). Step 4: 6-Bromo-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'- pyrano[4,3-d]pyrimidin]-4'-ol Methyl 6-bromo-4'-oxo-2,3,3',4',5',6'-hexahydrospiro[indene-1,2'-pyran]-5'- carboxylate (3.00 g, 1 equiv, 9 mmol) in acetonitrile (50 mL) was treated with thiourea (0.9 g, 1.3 equiv, 0.01 mol) and DBU (2.00 g, 1.6 equiv, 0.01 mol). The mixture was heated to 75 °C for about 3 hours and then stirred at room temperature for 16 hours. Aqueous sodium hydroxide (0.6 g, 0.01 L, 1.0 M, 1.6 equiv, 0.01 mol) and iodomethane (2.00 g, 1.4 equiv, 0.01 mol) were added, and the mixture was stirred at room temperature for 2 hours. The reaction was quenched with aqueous ammonium chloride, diluted with DCM (40 mL), and extracted with DCM (2 × 30 mL). The organic layers were dried with magnesium sulfate, filtered, and concentrated to a crude product, which was purified by column chromatography (SiO2, DCM/MeOH = 100/0 to 10/2). The product was precipitated from acetonitrile to give 6-bromo-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-ol (550 mg). LCMS: m/z (ESI) [M+H]
+ 381.1, tR = 1.88 minutes 1H NMR (400 MHz, CDCl
3) δ 7.40 (dd, 1H), 7.33 (d, 1H), 7.16 (d, 1H), 4.60 (q, 2H), 3.03-3.11 (m, 1H), 2.83-2.92 (m, 3H), 2.59 (s, 3H), 2.31-2.38 (m, 1H), 2.08-2.17 (m, 1H). Step 5: 6-Bromo-4'-chloro-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] 6-Bromo-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-ol (550 mg, 1 equiv, 1.45 mmol) in acetonitrile (5 mL) was treated with POCl3 (1.78 g, 8 equiv, 11.6 mmol) and heated to 80 ℃ for 1 hour. The reaction mixture was cooled to room temperature. It was then quenched with aqueous sodium bicarbonate and extracted with EtOAc (3 × 15 mL). The organic layers were dried over sodium sulfate, filtered, and concentrated to give 6-bromo-4’-chloro-2’-(methylthio)-2,3,5’,8’-tetrahydrospiro[indene-1,7’- pyrano[4,3-d]pyrimidine] that was used without further purification. 1H NMR (400 MHz, CDCl3) δ 7.42 (dd, 1H), 7.23 (d, 1H), 7.17 (d, 1H), 4.74 (q, 2H), 3.00-3.12 (m, 3H), 2.79-2.87 (m, 1H), 2.58 (s, 3H), 2.32-2.38 (m, 1H), 2.07-2.12 (m, 1H).
Step 6: (3R)-1-(6-bromo-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene- 1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol 6-Bromo-4’-chloro-2’-(methylthio)-2,3,5’,8’-tetrahydrospiro[indene-1,7’-pyrano[4,3- d]pyrimidine] (833 mg, 1 equiv, 2.09 mmol) and (R)-3-methylpiperidin-3-ol hydrochloride (476 mg, 1.5 equiv, 3.14 mmol) in EtOH (10 mL) was treated with DIPEA (406 mg, 1.5 equiv, 3.14 mmol). The reaction mixture was heated to 70 ℃ and stirred for 2 hours. The reaction was quenched with aqueous ammonium chloride and extracted with EtOAc (2 × 20 mL). The organic layers were dried with magnesium sulfate, filtered, and concentrated to a crude product, which was purified by column chromatography (SiO
2, heptanes/EtOAc = 100/0 to 10/6) to give (3R)-1-(6-bromo-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol (572 mg). LCMS: m/z (ESI) [M+H]
+ 477.7, tR = 1.78 minutes (Method C) 1H NMR (400 MHz, CDCl
3) δ 7.38-7.44 (m, 1H), 7.18-7.31 (m, 2H), 4.35-4.66 (m, 2H), 3.72-3.85 (m, 1H), 3.52 (dd, 1H), 2.94-3.17 (m, 5H), 2.76-2.89 (m, 1H), 2.54 (s, 3H), 2.31-2.42 (m, 1H), 2.13-2.25 (m, 1H), 1.76-1.96 (m, 2H), 1.49-1.65 (m, 3H), 1.25 (s, 3H). Step 7: (3R)-1-(6-bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)-3-methylpiperidin-3-ol (3R)-1-(6-Bromo-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol (550 mg, 1 equiv, 1.15 mmol) in MeOH (6 mL) and water (3 mL) was treated with Oxone (1.42 g, 2 equiv, 2.31 mmol) and stirred for 1 hour at room temperature. The reaction was quenched with aqueous sodium bicarbonate, diluted with EtOAc, and extracted with EtOAc (2 × 15 mL). The organic layers were dried with magnesium sulfate, filtered, and concentrated to give (3R)-1-(6-bromo-2'-(methylsulfonyl)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol that was used without further purification. Sodium hydride (160 mg, 3 equiv, 4.01 mmol) was suspended in THF (3 mL) followed by the addition of (2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (639 mg, 3 equiv, 4.01 mmol). (3R)-1-(6-bromo-2'-(methylsulfonyl)-2,3,5',8'-tetrahydrospiro[indene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol (680 mg, 1 equiv, 1.34 mmol) in DMF (6 mL) was treated by dropwise addition of the prepared alkoxide mixture and the resulting
reaction mixture was stirred for 90 minutes at 50 ℃. The reaction was quenched by addition of aqueous ammonium chloride, diluted with EtOAc, and extracted with EtOAc (3 × 1 mL). The organic layers were dried with sodium sulfate, filtered, and concentrated to a crude mixture, which was purified by column chromatography (SiO
2, DCM/MeOH = 100/0 to 10/2) to give (3R)-1-(6-bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol (626 mg). LCMS: m/z (ESI) [M+H]
+ 589.5, tR = 1.43 minutes (Method C) Intermediate 11: (3R)-1-(2'-(((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol

A suspension of (3R)-1-(6-bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-3- methylpiperidin-3-ol (105 mg, 1 equiv, 179 µmol), potassium acetate (26.3 mg, 1.5 equiv, 268 µmol) and bis(pinacolato)diboron (81.7 mg, 1.8 equiv, 322 µmol) in 1,4-dioxane (2 mL) was degassed with nitrogen, followed by the addition of Pd(dppf)Cl2·CH2Cl2 (13.1 mg, 0.1 equiv, 17.9 µmol). The mixture was heated to 95 ℃ for 1 hour and purified by column chromatography (SiO2, DCM/MeOH = 100/0 to 10/1, DCM/MeOH with NH4OH = 10/2) to give (3R)-1-(2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-6-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol (83 mg). LCMS: m/z (ESI) [M+H]
+ 635.8, tR = 1.46 minutes (Method C) Intermediate 12: (3R)-1-(7-bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol

Step 1: (3R)-1-(7-bromo-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol 7-Bromo-4'-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine] (90 mg, 1 equiv, 220 µmol) and (R)-3-methylpiperidin-3-ol hydrochloride (66 mg, 2 equiv, 440 µmol) in EtOH (1.1 mL) was treated with DIPEA (57 mg, 2 equiv, 440 µmol). The reaction mixture was heated to 80 ℃ and stirred for 1 hour. The reaction was quenched with aqueous ammonium chloride and extracted with EtOAc (3 × 15 mL). The organic layers were dried with magnesium sulfate, filtered, and concentrated to give (3R)-1-(7-bromo-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol (100 mg) that was used without further purification. LCMS: m/z (ESI) [M+H]
+ 491.8, tR = 2.78 minutes (Method E) 1H NMR (400 MHz, CDCl3) δ 7.46 (dd, 1H), 7.18-7.27 (m, 1H), 6.87-6.96 (m 1H), 4.38-4.68 (m, 2H), 3.54-3.67 (m, 1H), 3.39-3.49 (m, 1H), 2.87-3.04 (m, 4H), 2.63-2.79 (m, 2H), 2.45 (s, 3H), 1.83-1.93 (m, 2H), 1.67-1.80 (m, 3H), 1.36-1.57 (m, 4H), 1.17 (s, 3H). Step 2: (3R)-1-(7-bromo-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol (3R)-1-(7-bromo-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol (100 mg, 1 equiv, 184 µmol) and Oxone (226 mg, 2 equiv, 367 µmol) in methanol (1.22 mL) was treated with water (612 µL) and stirred for 2 hours. The reaction was quenched with aqueous ammonium chloride, diluted with EtOAc (15 mL), and extracted with EtOAc (3 × 15 mL). The organic layers were dried with magnesium sulfate, filtered, and concentrated to give (3R)-1-(7-bromo-2'- (methylsulfonyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)-3-methylpiperidin-3-ol (70 mg) that was used without further purification. LCMS: m/z (ESI) [M+H]
+ 524.0, t
R = 3.08 minutes (Method E) 1H NMR (400 MHz, CDCl
3) δ 7.40 (d, 1H), 7.25 (d, 1H), 6.94 (dd, 1H), 4.47-4.75 (m, 2H), 3.80-3.59 (m, 2H), 3.23-3.16 (m, 3H), 3.15-2.91 (m, 4H), 2.83-2.62 (m, 3H), 2.06-1.79
(m, 4H), 1.77-1.64 (m, 2H), 1.64-1.48 (m, 2H), 1.17 (s, 3H). Step 3: (3R)-1-(7-bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)-3-methylpiperidin-3-ol Sodium hydride (16 mg, 3 equiv, 0.40 mmol) was suspended in THF (0.35 mL) followed by the dropwise addition of (2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methanol (64 mg, 3 equiv, 0.40 mmol) in THF (0.67 mL). (3R)-1-(7-Bromo-2'- (methylsulfonyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)-3-methylpiperidin-3-ol (70 mg, 1 equiv, 0.13 mmol) in DMF (0.67 mL) was treated by the dropwise addition of the prepared alkoxide mixture, and the resulting reaction mixture was stirred for 90 minutes at 50 ℃. The reaction was quenched by the addition of aqueous ammonium chloride, diluted with EtOAc, and extracted with EtOAc (3 × 1 mL). The organic layers were dried with sodium sulfate, filtered, and concentrated to a crude product, which was purified by column chromatography (SiO2, DCM/MeOH = 100/0 to 10/2) to give (3R)-1-(7- bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol (53 mg). LCMS: m/z (ESI) [M+H]
+ 603.5, tR = 1.43 minutes (Method C) 1H NMR (400 MHz, CDCl
3) δ 7.54 (dd, 1H), 7.40 – 7.29 (m, 1H), 7.02 (t, 1H), 5.49 – 5.13 (m, 1H), 4.70 (dd, 1H), 4.53 (dd, 1H), 4.16 (dd, 1H), 4.04 (dd, 1H), 3.77 (dd, 1H), 3.63 – 3.45 (m, 1H), 3.41 – 3.16 (m, 3H), 3.16 – 2.91 (m, 5H), 2.91 – 2.69 (m, 2H), 2.43 – 2.09 (m, 3H), 2.09 – 1.72 (m, 9H), 1.73 – 1.48 (m, 2H), 1.27 (s, 3H). Intermediate 13a: (1R,4S)-7-bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]
Step 1: (4S)-7-Bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-4-methyl-1,2,3,4-tetrahydronaphthalen-1-ol LDA solution (2.60 mL, 1 M, 2.3 equiv, 2.60 mmol) was added dropwise to a solution of (4-chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (231 mg, 1 equiv, 1.13
mmol) in THF (4 mL) at -78 °C and stirred for 1.25 hours. To this mixture was added (S)-7- bromo-4-methyl-3,4-dihydronaphthalen-1(2H)-one (2
nd eluting peak) (Intermediate 7b, supra) (270 mg, 1 equiv, 1.13 mmol) in THF (3 mL) dropwise, keeping the temperature below -70 °C. The reaction mixture was stirred at -78 °C for 1 hour, at which point the reaction was quenched by aqueous ammonium chloride (10 mL) at -78 °C with vigorous stirring. The reaction mixture was then diluted with EtOAc (15 mL), and the aqueous phase was extracted with EtOAc (3 × 25 mL). The combined organics were dried with sodium sulfate and concentrated to a residue, which was purified via column chromatography (SiO2; Gradient: 100% DCM - 100% EtOAc) to give (4S)-7-bromo-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-4-methyl-1,2,3,4-tetrahydronaphthalen-1-ol (405 mg). 1H NMR (400 MHz, CDCl
3) δ 7.67 (m, 1H), 7.36 (m, 1H), 7.11 (m, 1H), 4.81 – 4.67 (m, 2H), 3.40 – 3.16 (m, 2H), 2.93 (m, 1H), 2.55 (m, 3H), 2.18 (m, 1H), 2.06 – 1.92 (m, 1H), 1.91 – 1.62 (m, 2H), 1.32 (m, 3H). Step 2: (1R,4S)-7-bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] Phosphoric acid (658 μL, 85 wt%, 1 equiv, 9.62 mmol) was added to (4S)-7-bromo-1- ((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-4-methyl-1,2,3,4- tetrahydronaphthalen-1-ol (4.27 g, 1 equiv, 9.62 mmol) in toluene (19.2 mL). The mixture was refluxed for 2 hours. Water (50 mL) and EtOAc (50 mL) were added, and the aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organics were dried with sodium sulfate and concentrated to a residue, which was purified via chiral SFC (Column: Chiralpak IB-U (3.0 × 100 mm), 1.6 μm, P/N 81U83, flow rate: 1.2 mL/min, Mobile phase: (Methanol with 0.25% DEA)) to afford (1R,4S)-7-bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (Peak 1) (2.02 g) and (1S,4S)-7-bromo-4'- chloro-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine] (Peak 2) (1.78 g). Peak 1: tR = 1.34 minutes (Method G; 30% methanol with 0.25% DEA) LCMS: ESI [M+H]
+ = 427.0 m/z; tR = 4.14 minutes (Method E) Intermediate 13b: (1S,4S)-7-bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]
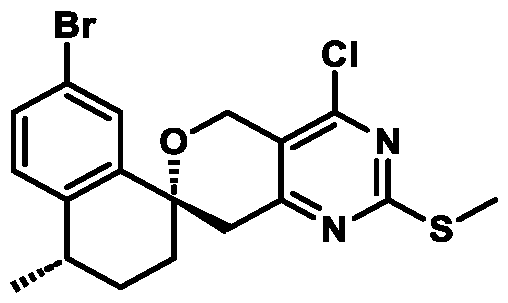
Phosphoric acid (658 μL, 85 wt%, 1 equiv, 9.62 mmol) was added to (4S)-7-bromo-1- ((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-4-methyl-1,2,3,4- tetrahydronaphthalen-1-ol (4.27 g, 1 equiv, 9.62 mmol) in toluene (19.2 mL). The mixture was refluxed for 2 hours. Water (50 mL) and EtOAc (50 mL) were added, and the aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organics were dried with sodium sulfate and concentrated to a residue, which was purified via chiral SFC (Column: Chiralpak IB-U (3.0 × 100 mm), 1.6 μm, P/N 81U83, flow rate: 1.2 mL/min, Mobile phase: (Methanol with 0.25% DEA)) to afford (1R,4S)-7-bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (Peak 1) (2.02 g) and (1S,4S)-7-bromo-4'- chloro-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine] (Peak 2) (1.78 g). Peak 2: tR = 2.06 minutes (Method G; 30% methanol with 0.25% DEA) 1H NMR (400 MHz, CDCl
3) δ 7.63 (d, 1H), 7.41 (dd, 1H), 7.19 (d, 1H), 4.78 (q, 2H), 3.12 (q, 2H), 3.00 – 2.86 (m, 1H), 2.59 (s, 3H), 2.13 – 1.87 (m, 3H), 1.48 (qd, 1H), 1.34 (d, 3H). Intermediate 14: (S)-8-bromo-7-(dibenzylamino)-4-methyl-3,4- dihydronaphthalen-1(2H)-one

Step 1: (S)-7-amino-4-methyl-3,4-dihydronaphthalen-1(2H)-one NMP (27.9 mL) and ammonium hydroxide (29.3 g, 32.4 mL, 20 equiv, 836 mmol) were delivered to a pressure vessel charged with (S)-7-bromo-4-methyl-3,4-dihydronaphthalen- 1(2H)-one (Intermediate 7b) (10.0 g, 1.00 equiv, 41.8 mmol) and copper(I) oxide (598 mg, 0.10 equiv, 4.18 mmol) under nitrogen. The pressure vessel was sealed, and the reaction mixture was stirred at 80 °C for 17 hours. The reaction mixture was cooled to ambient temperature and poured in a separatory funnel charged with water (100 mL) and EtOAc (200 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (3 x 150
mL). The combined organic extracts were dried over sodium sulfate, filtered, and concentrated to residue. The crude residue was purified by silica gel chromatography (2 to 45% heptane/EtOAc) to afford (S)-7-amino-4-methyl-3,4-dihydronaphthalen-1(2H)-one (6.79 g, 38.8 mmol). 1H NMR (400 MHz, CDCl3) δ 7.34 (d, 1H), 7.16 (d, 1H), 6.94 – 6.86 (m, 1H), 3.68 (bs, 2H), 3.07 – 2.95 (m, 1H), 2.88 – 2.72 (m, 1H), 2.64 – 2.52 (m, 1H), 2.28 – 2.16 (m, 1H), 1.93 – 1.80 (m, 1H), 1.37 (d, 3H). LCMS: m/z (ESI) [M+H]
+ 176.2, tR = 1.51 minutes (Method E) Step 2: (S)-7-amino-8-bromo-4-methyl-3,4-dihydronaphthalen-1(2H)-one A solution of N-bromosuccinimide (6.90 g, 1.0 equiv, 38.8 mmol) in DMF (25.9 mL) was delivered dropwise to a flask containing (S)-7-amino-4-methyl-3,4-dihydronaphthalen- 1(2H)-one (6.79 g, 1.00 equiv, 38.79 mmol) in DMF (51.7 mL) at 0 °C, under nitrogen. The resulting mixture was stirred at 0 °C for 5 min, then warmed to room temperature and stirred for 2 hours. The reaction mixture was quenched by the addition of water (150 mL) and EtOAc (200 mL) was added. The layers were separated and the aqueous phase was extracted with EtOAc (3 x 150 mL). The combined organic extracts were dried over sodium sulfate, filtered, and concentrated to residue. Crude material was purified by silica gel chromatography (0 to 30% heptane/EtOAc) to afford (S)-7-amino-8-bromo-4-methyl-3,4-dihydronaphthalen-1(2H)- one (7.96 g, 31.3 mmol). 1H NMR (400 MHz, CDCl
3) δ 7.07 (d, 1H), 6.92 (d, 1H), 4.15 (bs, 2H), 3.05 – 2.91 (m, 1H), 2.86 – 2.74 (m, 1H), 2.68 – 2.56 (m, 1H), 2.25 – 2.12 (m, 1H), 1.90 – 1.77 (m, 1H), 1.31 (d, 3H). LCMS: m/z (ESI) [M+H]
+ 254.1, t
R = 2.47 minutes (Method E) Step 3. (S)-8-bromo-7-(dibenzylamino)-4-methyl-3,4-dihydronaphthalen-1(2H)- one Benzyl bromide (16.1 g, 11.2 mL, 3 equiv, 94.0 mmol) and potassium carbonate (15.2 g, 3.5 equiv, 110 mmol) were sequentially delivered to a solution of (S)-7-amino-8-bromo-4- methyl-3,4-dihydronaphthalen-1(2H)-one (7.96 g, 1.0 equiv, 31.3 mmol) in MeCN (313 mL) at room temperature, were added. The resulting mixture was stirred vigorously at reflux for 18 hours at which point the reaction mixture was cooled to room temperature. The solids were filtered and rinsed with EtOAc, the filtrate was concentrated, and the remaining crude residue
was purified by silica gel chromatography (1 to 35% heptane/EtOAc) to afford (S)-8-bromo-7- (dibenzylamino)-4-methyl-3,4-dihydronaphthalen-1(2H)-one (12.5 g, 28.7 mmol). 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.19 (m, 10H), 7.09 – 6.97 (m, 2H), 4.24 – 4.10 (m, 4H), 3.03 – 2.90 (m, 1H), 2.89 – 2.76 (m, 1H), 2.73 – 2.61 (m, 1H), 2.24 – 2.12 (m, 1H), 1.90 – 1.77 (m, 1H), 1.30 (d, 3H). LCMS: m/z (ESI) [M+H]
+ 434.3, tR = 3.78 minutes (Method E) Intermediate 15: (1S,4S)-N,N-dibenzyl-8-bromo-4'-chloro-4-methyl-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- amine

Step 1: (4S)-8-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-7-(dibenzylamino)-4-methyl-1,2,3,4-tetrahydronaphthalen-1-ol LDA (1.0 M in THF/hexanes) (9.25 g, 86.3 mL, 1.0 molar, 3.01 equiv, 86.3 mmol) was delivered dropwise to a solution of (4-chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (8.83 g, 1.51 equiv, 43.1 mmol) in THF (191 mL) at -78 °C. The resulting mixture was stirred at -78 °C for 1.25 hours, followed by the dropwise addition of a solution of (S)-8-bromo-7- (dibenzylamino)-4-methyl-3,4-dihydronaphthalen-1(2H)-one (Intermediate 14) (12.5 g, 1.0 equiv, 28.7 mmol) in THF (96 mL) keeping the internal temperature below - 65 °C. The resulting solution was stirred for 1 hour at -78 °C. The reaction was quenched by the dropwise addition of HCl (2.0 M in Et2O) (3.34 g, 45.9 mL, 2.0 molar, 3.2 equiv, 91.7 mmol) at -78 °C, followed by water (200 mL). The mixture was warmed to room temperature, transferred to a separatory funnel, and the layers were separated. The aqueous phase was extracted with EtOAc (1 x 200 mL) and CH₂Cl₂ (2 x 200 mL). The combined organic extracts were dried over sodium sulfate, filtered, and concentrated to a residue which was purified by silica gel chromatography (2 → 50% CH₂CL₂/EtOAc, (product elutes around 15% EtOAc)) to afford (4S)-8-bromo-1- ((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-7-(dibenzylamino)-4- methyl-1,2,3,4-tetrahydronaphthalen-1-ol (16.05 g, 24 mmol).
LCMS: m/z (ESI) [M+H]
+ 640.2, t
R = 4.39 minutes (Method E) Step 2: (1S,4S)-N,N-dibenzyl-8-bromo-4'-chloro-4-methyl-2'-(methylthio)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine n-Butyllithium (2.5 M in hexanes) (3.35 g, 20.9 mL, 2.50 molar, 2.2 equiv, 52.3 mmol) was delivered dropwise to a solution of (4S)-8-bromo-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-7-(dibenzylamino)-4-methyl-1,2,3,4- tetrahydronaphthalen-1-ol (16.0 g, 95% wt, 1.0 equiv, 23.8 mmol) in THF (198 mL) at -78 °C. The resulting solution was stirred at -78 °C for 35 min, followed by the dropwise addition of a solution of p-toluenesulfonyl chloride (6.80 g, 1.5 equiv, 35.7 mmol) in THF (99 mL). The resulting mixture was stirred at -78 °C for 10 min and the acetone/dry ice bath was removed. The reaction mixture was stirred for another 45 min after reaching ambient temperature, then quenched by the addition of a 1:1 mixture of a saturated aqueous ammonium chloride solution and water (250 mL). The reaction mixture was transferred to a separatory funnel. EtOAc (100 mL) was added and the layers were separated. The aqueous phase was extracted with EtOAc (3 x 300 mL). The combined organic extracts were dried over sodium sulfate, filtered, and concentrated. The crude material was purified by silica gel chromatography (0 → 10% heptane/EtOAc). 12.3 g of the material isolated from column chromatography was stirred in 200 mL MeCN at 40 °C overnight, filtered, and the solid rinsed with MeCN) to afford a white solid (6.56 g). The filtrate was concentrated to a brown oil which was purified by silica gel chromatography (0 → 10% heptane/EtOAc) to give (1S,4S)-N,N-dibenzyl-8-bromo-4'-chloro- 4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-amine (1.64 g, 2.63 mmol) LCMS: m/z (ESI) [M+H]
+ 620.0, t
R = 4.63 minutes (Method E) Intermediate 16: 7-Bromo-4',5-dichloro-8-fluoro-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]

Step 1: 4-(4-Bromo-2-chloro-5-fluorophenyl)butanal A 200 mL round bottom flask was charged with 1-bromo-5-chloro-2-fluoro-4- iodobenzene (10 g, 1 equiv., 30 mmol), palladium(II) acetate (0.40 g, 0.06 equiv., 1.8
mmol), lithium acetate (4.9 g, 2.5 equiv., 75 mmol), lithium chloride (1.3 g, 1 equiv., 30 mmol), and tetrabutylammonium chloride hydrate (18 g, 2 equiv., 60 mmol). The mixture was evacuated and backfilled with nitrogen three times. DMF (75 mL) and but-3-en-1-ol (2.6 g, 3.1 mL, 1.2 equiv, 36 mmol) were then added via syringe and the mixture was stirred overnight at 70 °C. The reaction was quenched with saturated ammonium chloride solution (50 mL) and 50 mL of heptanes. The aqueous and organic layers were separated and the aqueous was extracted two more times with 50 mL of heptanes. The combined organics were washed with 50 mL of 1 M HCl and 50 mL of brine. The organic layers were dried over magnesium sulfate, and filtered. The aqueous was re-extracted with EtOAc and the organic layer was washed with 1 M HCl and brine. The organics were concentrated, re-dissolved in heptanes, dried over magnesium sulfate, filtered, and concentrated. The combined crude oil was purified using a RediSep Rf Gold 220 g silica gel cartridge (eluting with a 100% heptane to 20% EtOAc in heptane gradient) to give 4-(4-bromo-2-chloro-5-fluorophenyl)butanal (3.83 g, 13.7 mmol). 1H NMR (400 MHz, CDCl
3) δ 9.79 (m, 1H), 7.54 (m, 1H), 7.00 (m, 1H), 2.71 (m, 2H), 2.51 (m, 2H), 1.94 (m, 2H). Step 2: 4-(4-Bromo-2-chloro-5-fluorophenyl)butanoic acid Sodium chlorite (6.15 g, 5 equiv., 68.0 mmol) was added to 4-(4-bromo-2-chloro-5- fluorophenyl)butanal (3.80 g, 1 equiv., 13.6 mmol), 2-methyl-2-butene (38.1 g, 57.6 mL, 40 equiv., 544 mmol), sodium dihydrogen phosphate hydrate (18.8 g, 10 equiv., 136 mmol), water (45.3 mL) and t-BuOH (90.6 mL) at 0
°C. After 15 minutes, the reaction was brought to room temperature and stirred for 2.5 hours. The reaction mixture was then poured into a separatory funnel containing a 1:1 mixture of water and brine and CH₂Cl₂. The layers were separated. The aqueous phase was extracted with CH₂Cl₂ (3 x 50 mL). The combined organic extracts were dried over magnesium sulfate, filtered and concentrated to afford 4-(4-bromo-2-chloro-5- fluorophenyl)butanoic acid (4.40 g, 14.9 mmol; contains residual t-BuOH) which was used without further purification. 1H NMR (400 MHz, CDCl
3) δ 7.54 (m, 1H), 7.01 (m, 1H), 2.79 – 2.69 (m, 2H), 2.42 (m, 2H), 1.95 (m, 2H). Step 3: 7-Bromo-5-chloro-8-fluoro-3,4-dihydronaphthalen-1(2H)-one Oxalyl chloride (2.3 mL, 2 equiv., 27.1 mmol) was added dropwise to a solution of 4- (4-bromo-2-chloro-5-fluorophenyl)butanoic acid (4.00 g, 1 equiv., 13.5 mmol) and N,N-
dimethylformamide (105 μL, 0.1 equiv., 1.35 mmol) in DCM (54 mL) at room temperature. The resulting mixture was stirred for 2 hours then concentrated. The residue was re-dissolved in DCM (54.1 mL), followed by the addition of aluminum chloride (3.61 g, 2 equiv., 27.1 mmol) in one portion. The reaction mixture was stirred for 22 hours at ambient temperature. The reaction mixture was cooled, quenched with water, diluted with DCM, and extracted with DCM/brine (3 x 40 mL). Organics were collected, dried over magnesium sulfate, filtered and concentrated to give a residue which was taken up in DCM and purified using a RediSep Rf Gold 120 g silica gel cartridge (eluting with 0-40% EtOAc in heptanes) to afford 7-bromo-5- chloro-8-fluoro-3,4-dihydronaphthalen-1(2H)-one (2.08 g, 7.49 mmol). 1H NMR (400 MHz, CDCl
3) δ 7.74 (m, 1H), 2.96 (m, 2H), 2.66 (m, 2H), 2.15 (m, 2H). Step 4: 7-Bromo-5-chloro-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-8-fluoro-1,2,3,4-tetrahydronaphthalen-1-ol To an oven-dried 125 mL round bottom flask was added (4-chloro-6-methyl-2- (methylthio)pyrimidin-5-yl)methanol (1.11 g, 1.5 equiv., 5.41 mmol). The flask was then sealed and degassed with nitrogen. THF (27 mL) was then added, and the reaction was cooled to -78 °C. LDA (10.8 mL, 1 molar, 3 equiv., 10.8 mmol) was then added dropwise, maintaining a temperature below -73
oC by internal probe. The solution was stirred at -78 °C for 1 hour.7- bromo-5-chloro-8-fluoro-3,4-dihydronaphthalen-1(2H)-one (1.00 g, 1 equiv., 3.60 mmol) was then dissolved in THF (14 mL) and added dropwise to the reaction mixture maintaining a temperature below -73
o C. The solution was stirred for 1 hour and the product mixture was then quenched with HCl (5.77 mL, 2.0 M, 3.2 equiv., 11.5 mmol) slowly while maintaining temperature below -75
o C. The product mixture was removed from the dry ice bath warming to about -20
oC and was poured into a separatory funnel containing saturated aqueous sodium bicarbonate and EtOAc. The aqueous layer was separated and further extracted with EtOAc (2x). The organic layers were combined, dried over magnesium sulfate, filtered, and concentrated to give a yellow solid. The residue was purified on a 120 g redisep column (0- 100% EtOAc/heptanes; followed by 40% (20% MeOH, 2.5% ammonium hydroxide in DCM) in DCM) to afford 7-bromo-5-chloro-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-8-fluoro-1,2,3,4-tetrahydronaphthalen-1-ol (1.27 g, 2.63 mmol). LCMS: m/z (ESI) [M+H]
+ 481.0, tR = 2.18 minutes (Method C)
Step 5: 7-Bromo-4',5-dichloro-8-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] Phosphoric acid (170 μL, 85% wt, 1 equiv., 2.49 mmol) was added to the white suspension of 7-bromo-5-chloro-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-8-fluoro-1,2,3,4-tetrahydronaphthalen-1-ol (1.20 g, 1 equiv., 2.49 mmol) in toluene (27 mL) and the mixture was heated at 110 °C for 2 hours. The mixture was cooled to room temperature and was diluted with ethyl acetate. The product mixture was transferred to a separatory funnel and washed with saturated aqueous sodium bicarbonate and brine. The organic layer was dried over magnesium sulfate, filtered, and concentrated to give a crude residue which was purified by flash column (RediSep SiO
2 gold 80 g; eluting: ethyl acetate/heptanes, 0-60%) to give 7-bromo-4',5-dichloro-8-fluoro-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (451 mg, 0.972 mmol) LCMS: m/z (ESI) [M+H]
+ 465.0, tR = 2.29 minutes (Method C) Intermediate 17a: rel-(S)-azepane-4-carbonitrile
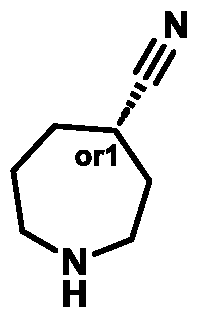
Step 1: benzyl rel-(S)-4-cyanoazepane-1-carboxylate Benzyl chloroformate (0.80 mL, 2.0 equiv, 7.0 mmol) was added to a solution of azepane-4-carbonitrile (0.35 g, 1 equiv, 2.8 mmol) and DIPEA (1.2 mL, 2.5 equiv, 7.0 mmol) in dichloromethane (1.5 mL) at room temperature. The reaction mixture was stirred for 16 hours. Afterward, the reaction mixture was concentrated to a residue, which was purified by flash chromatography (12 g Redisep Column using 25-30% hexanes in ethyl acetate) to afford benzyl 4-cyanoazepane-1-carboxylate (0.13 g). The enantiomers of benzyl 4- cyanoazepane-1-carboxylate were separated by chiral SFC (Column: i-Amylose-3; 21.2×250 mm; 5 mm; Mobile Phase A: 50% MeOH at 30 mL/min for 10 minutes) to afford benzyl rel- (S)-4-cyanoazepane-1-carboxylate (Peak 1) and benzyl rel-(R)-4-cyanoazepane-1-carboxylate (Peak 2). Peak 1: HPLC: tR = 1.74 minutes (Column: i-Amylose-3, 4.6x250 mm; 5 mm; Mobile Phase A: 50% MeOH @ 4 mL/min for 5 minutes)
Step 2: rel-(S)-azepane-4-carbonitrile Palladium hydroxide on carbon (90 mg, 20 wt%, 0.3 equiv, 0.13 mmol) was added to benzyl rel-(S)-4-cyanoazepane-1-carboxylate (Peak 1, 110 mg, 1 equiv, 0.43 mmol) in THF (3 mL) at room temperature and stirred for 3 hours under hydrogen atmosphere. The reaction mixture was purged with nitrogen, filtered through a Celite bed, concentrated, and dried to afford rel-(S)-azepane-4-carbonitrile (45 mg, 0.36 mmol). 1H NMR (400 MHz, CDCl
3): δ 3.03-2.58 (m; 5H), 2.07-1.45 (m; 6 H). Intermediate 17b: rel-(R)-azepane-4-carbonitrile
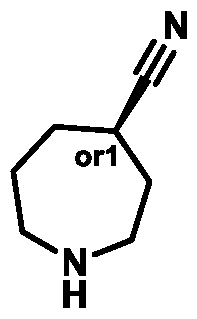
Step 1: benzyl rel-(R)-4-cyanoazepane-1-carboxylate Benzyl chloroformate (0.80 mL, 2.0 equiv, 7.0 mmol) was added to a solution of azepane-4-carbonitrile (0.35 g, 1 equiv, 2.8 mmol) and DIPEA (1.2 mL, 2.5 equiv, 7.0 mmol) in dichloromethane (1.5 mL) at room temperature. The reaction mixture was stirred for 16 hours. Afterward, the reaction mixture was concentrated to a residue, which was purified by flash chromatography (12 g Redisep Column using 25-30% hexanes in ethyl acetate) to afford benzyl 4-cyanoazepane-1-carboxylate (0.13 g). The enantiomers of benzyl 4- cyanoazepane-1-carboxylate were separated by chiral SFC (Column: i-Amylose-3; 21.2×250 mm; 5 mm; Mobile Phase A: 50% MeOH at 30 mL/min for 10 minutes) to afford benzyl rel- (S)-4-cyanoazepane-1-carboxylate (Peak 1) and benzyl rel-(R)-4-cyanoazepane-1-carboxylate (Peak 2). Peak 2: HPLC: tR = 2.16 minutes (Column: i-Amylose-3, 4.6x250 mm; 5 mm; Mobile Phase A: 50% MeOH @ 4 mL/min for 5 minutes) Step 2: rel-(R)-azepane-4-carbonitrile Palladium hydroxide on carbon (90 mg, 20 wt%, 0.3 equiv, 0.13 mmol) was added to benzyl rel-(R)-4-cyanoazepane-1-carboxylate (Peak 2, 110 mg, 1 equiv, 0.43 mmol) in THF (3 mL) at room temperature and stirred for 3 hours under hydrogen atmosphere. The reaction mixture was purged with nitrogen, filtered through a Celite bed, concentrated, and dried to afford rel-(R)-azepane-4-carbonitrile (45 mg, 0.36 mmol).
1H NMR (400 MHz, CDCl
3): δ 2.97-2.82 (m; 5H), 2.09-1.55 (m; 6 H). Intermediate 18: 7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-hydroxy-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine]-8-carbonitrile

Step 1: N,N-Dibenzyl-4'-(benzyloxy)-8-bromo-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine To a solution of phenylmethanol (511 μL, 4.94 mmol, 1.5 equiv.) in THF (32.9 mL), at room temperature, was added sodium hydride (60% dispersion in mineral oil, 264 mg, 6.59 mmol, 2.0 equiv.). The resulting mixture was stirred at room temperature for 10 minutes, followed by the portionwise addition of N,N-dibenzyl-8-bromo-4'-chloro-2'-(methylthio)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (2.00 g, 3.29 mmol). The reaction mixture was stirred at room temperature for 2 hours. The reaction was quenched by the addition of saturated aqueous ammonium chloride solution at 0 °C. The layers were separated, and the aqueous phase was extracted with EtOAc (3 x). The combined organic extracts were washed with brine, dried over sodium sulfate, filtered, and concentrated under reduced pressure to give crude material, which was purified by silica gel chromatography (80 g cartridge, 80 mL/min, hexanes/DCM: 0 to 100%) to give N,N-dibenzyl-4'-(benzyloxy)-8- bromo-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-amine (1.9 g, 2.8 mmol) as an off white solid. LCMS: m/z (ESI) [M+H]⁺ 678.4, tR = 2.46 minutes, (Method B) Step 2: 4'-(Benzyloxy)-7-(dibenzylamino)-2'-(methylthio)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile To a solution of N,N-dibenzyl-4'-(benzyloxy)-8-bromo-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (1.8 g, 2.7 mmol) in DMF (53 mL) was added CuCN (2.4 g, 27 mmol, 10 equiv.). The reaction mixture was stirred at 120 °C for 4 hours. The reaction mixture was cooled to room temperature and concentrated. The residue was partitioned between DCM and water, upon which a precipitate 30 formed. A solution of NH4OH/MeOH (1:1) was added until no solid remained, and two clear
layers were obtained. The biphasic mixture was stirred for 30 minutes, and the layers were separated. The aqueous phase was extracted with DCM (3 x). The combined organic extracts were washed with brine, dried over sodium sulfate, filtered, and concentrated under reduced pressure to give crude material, which was purified by silica gel chromatography (120 g cartridge, 110 mL/min, hexanes/EtOAc: 0 to 40%) to give 4'-(benzyloxy)-7-(dibenzylamino)- 2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8- carbonitrile (1.4 g, 2.2 mmol) as an off white solid. LCMS: m/z (ESI) [M+H]⁺ 625.4, t
R = 2.30 minutes, (Method B) 1H NMR (400 MHz, DMSO-d
6) δ 7.51 – 7.46 (m, 2H), 7.43 – 7.37 (m, 2H), 7.37 – 7.32 (m, 5H), 7.31 – 7.25 (m, 4H), 7.24 – 7.18 (m, 3H), 7.15 (d, 1H), 5.50 (d, 1H), 5.47 (d, 1H), 4.75 (d, 1H), 4.67 (d, 1H), 4.26 (d, 2H), 4.21 (d, 2H), 3.35 – 3.28 (m, 1H), 2.94 (d, 1H), 2.73 – 2.63 (m, 2H), 2.50 (s, 3H), 2.09 – 2.01 (m, 1H), 1.82 – 1.62 (m, 3H). Step 3: 4'-(Benzyloxy)-7-(dibenzylamino)-2'-(methylsulfonyl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile To a suspension of 4'-(benzyloxy)-7-(dibenzylamino)-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (1.32 g, 2.11 mmol), tetrabutylammonium hydrogen sulfate (115 mg, 338 μmol, 0.16 equiv.), and sodium tungstate dihydrate (70 mg, 0.21 mmol, 0.1 equiv.) in EtOAc (21.1 mL) was added hydrogen peroxide (1.51 mL, 30 wt%, 14.8 mmol, 7.0 equiv.). The reaction mixture was stirred at 75
oC for 1 hour. The reaction was quenched by the addition of water and a 5% sodium bisulfate aqueous solution. EtOAc was added, and the layers were separated. The aqueous phase was extracted with EtOAc (3 x). The combined organic extracts were washed with brine, dried over sodium sulfate, filtered, and concentrated to yield 4'-(benzyloxy)-7-(dibenzylamino)-2'- (methylsulfonyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8- carbonitrile (1.35 g, 2.06 mmol) which was used in the next step without further purification. LCMS: m/z (ESI) [M+H]⁺ 657.4, t
R = 2.07 minutes, (Method B) Step 4: 4'-(benzyloxy)-7-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine]-8-carbonitrile To a solution of 4'-(benzyloxy)-7-(dibenzylamino)-2'-(methylsulfonyl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (1.35 g, 2.06 mmol) in DMF (41.1 mL) at room temperature, was added ((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methanol (1.64 g, 10.3 mmol, 5 equiv.) under argon. The resulting
solution was cooled to 0 °C, followed by the dropwise addition of lithium bis(trimethylsilyl)amide (1.0 M in THF, 3.08 mL, 3.08 mmol, 1.5 equiv.). The ice/water bath was removed, and the reaction mixture was stirred for 1 hour. The reaction was quenched by the slow addition of a 1:1 mixture of saturated aqueous ammonium chloride solution and water at 0 °C. EtOAc was added, and the layers were separated. The aqueous phase was extracted with EtOAc. The combined organic extracts were dried over sodium sulfate, filtered, and concentrated under reduced pressure to give crude material, which was purified by silica gel chromatography (80 g cartridge, 60 mL/min, DCM /[DCM /MeOH/NH4OH (80:18:2)], 0 to 60%) to give 4'-(benzyloxy)-7-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine]-8-carbonitrile (1.20 g, 1.63 mmol) as an orange solid. LCMS: m/z (ESI) [M+H]⁺ 736.6, t
R = 2.24 minutes, (Method B) 1H NMR (400 MHz, DMSO-d6) δ 7.51 – 7.46 (m, 2H), 7.43 – 7.38 (m, 2H), 7.37 – 7.32 (m, 5H), 7.31 – 7.26 (m, 4H), 7.24 – 7.18 (m, 3H), 7.15 (d, 1H), 5.47 (d, 1H), 5.44 (d, 1H), 5.26 (d, 1H), 4.74 (d, 1H), 4.65 (d, 1H), 4.24 (d, 2H), 4.23 (d, 2H), 4.02 (dd, 1H), 3.93 (dd, 1H), 3.31 – 3.24 (m, 1H), 3.12 – 2.96 (m, 3H), 2.93 – 2.86 (m, 1H), 2.85 – 2.78 (m, 1H), 2.71 – 2.65 (m, 2H), 2.13 – 1.92 (m, 4H), 1.87 – 1.63 (m, 6H). Step 5: 7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-hydroxy-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine]-8-carbonitrile A flask was charged with 4'-(benzyloxy)-7-(dibenzylamino)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (1.10 g, 1.49 mmol) and MeOH (29.9 mL). The flask headspace was sparged with argon, and Pd(OH)2 on carbon (20 wt%, 2.62 g, 3.74 mmol, 2.5 equiv.) was added. The inert atmosphere was exchanged with hydrogen by degassing the reaction mixture under reduced pressure with a backflow of hydrogen from a balloon. The reaction mixture was stirred overnight under a hydrogen atmosphere at room temperature. Celite was added and the hydrogen atmosphere was exchanged with argon. The suspension was filtered over celite, and the solids were rinsed with MeOH. The filtrate was concentrated to provide crude 7-amino-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-hydroxy-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (630 mg, 1.35 mmol) as a white solid.
LCMS: m/z (ESI) [M+H]⁺ 466.4, t
R = 0.97 minutes, (Method B) 1H NMR (400 MHz, DMSO-d
6): δ 7.04 (d, 1H), 6.73 (d, 1H), 5.75 (br s, 2H), 5.41 (br d, 1H), 4.51 (d, 1H), 4.42 (d, 1H), 4.29 – 4.10 (m, 2H), 3.63 – 3.11 (m, 6H), 3.03 – 2.98 (m, 2H), 2.63 – 2.57 (m, 3H), 2.30 – 1.71 (m, 6H), 1.68 – 1.58 (m, 2H). General Procedures: General Procedure A: A mixture of tert-butyl (2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-oxo-3,3',4,4',5',8'-hexahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (1.0 eq) and PyBOP (1.5 equiv.) in N,N-dimethylformamide (0.2- 0.5 M) and DIPEA (2.5 equiv.) was stirred for 30 minutes at room temperature. The reaction was quenched with ice, and the resulting suspension was filtered. The resulting solid was dried to obtain tert-butyl (4'-((1H-benzo[d][1,2,3]triazol-1-yl)oxy)-2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate which was used without further purification. The freebase or salt form of the appropriate amine (1.5-3.0 equiv.) was added to a solution of tert-butyl (4'-((1H-benzo[d][1,2,3]triazol-1-yl)oxy)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (1.0 equiv.) and DIPEA (3.0 equiv.) in N,N-dimethylformamide or ethanol (0.2 M). The reaction mixture was stirred at 100 °C for 2 hours. The mixture was then cooled to room temperature, and quenched with ice. The resulting solid was either filtered or purified by column chromatography to afford the Boc- protected title compound. Acetonitrile (0.03 M) and p-toluenesulfonic acid (2.0 eq) was added, and the mixture stirred at 50 °C for 3 hours. Alternatively, the mixture was treated with HCl in diethyl ether at room temperature. The reaction mixture was concentrated to a residue, which was purified by flash chromatography or preparative HPLC to yield the title compound. General Procedure B: The freebase or salt form of the appropriate amine (1.5-3.0 equiv.) was added to a solution of tert-butyl ((S)-4'-((1H-benzo[d][1,2,3]triazol-1-yl)oxy)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (1.0 equiv.) in N,N-
dimethylformamide or ethanol (0.2 M) and DIPEA (3.0 equiv.). The reaction mixture was stirred at 100
oC for 2 hours then quenched with ice. The resulting solid was either filtered or purified by column chromatography to afford the Boc-protected title compound. Acetonitrile (0.03 M) and p-toluenesulfonic acid (2.0 equiv.) was added, and the mixture was stirred at 50 °C for 3 hours. Alternatively, the mixture was treated with HCl in diethyl ether at room temperature. The reaction mixture was concentrated to a residue which was purified by flash chromatography or preparative HPLC to yield the title compound. Example 1: 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 4'-((R)-3-hydroxy-3-methylpiperidin-1-yl)-2,3,5',8'-tetrahydrospiro[indene-1,7'- pyrano[4,3-d]pyrimidin]-6-ol (Compound 101a)

(3R)-1-(2'-(((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-6- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,3,5',8'-tetrahydrospiro[indene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol (80 mg, 1 equiv, 130 µmol) in THF (1 mL) and MeOH (1 mL) was treated with aqueous sodium hydroxide (5.0 mg, 0.130 mL, 1 equiv, 130 µmol) and hydrogen peroxide (24 mg, 22 µL, 35 wt%, 2 equiv, 0.25 mmol) at room temperature for 30 minutes. The reaction was quenched with aqueous sodium bicarbonate and extracted with EtOAc (3 × 10 mL). The organic extracts were dried over sodium sulfate, filtered, and dried to a crude product, which was purified by reverse-phase column chromatography (ACCQPrep, Mobile phase A = 0.1% NH4OH in water; Mobile phase B = 0.1% NH
4OH in MeCN; Gradient = 100/10 to 100/95) to give 2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-((R)-3-hydroxy-3-methylpiperidin-1-yl)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-ol (10 mg). LCMS: ESI [M+H]
+ = 525.8; t
R = 1.23 minutes. (Method C) 1H NMR (400 MHz, CDCl
3) δ 7.14 (dd, 1H), 6.77 (dt, 1H), 6.61 (dd, 1H), 5.35 – 5.11 (m, 1H), 4.57 – 4.39 (m, 2H), 4.10 (dd, 1H), 4.00 (dd, 1H), 3.79 – 3.70 (m, 1H), 3.54 (dd, 1H), 3.28 – 2.86 (m, 8H), 2.82 – 2.68 (m, 1H), 2.42 – 2.28 (m, 1H), 2.26 – 2.05 (m, 3H), 1.96 – 1.71 (m, 5H), 1.69 – 1.44 (m, 4H), 1.27 – 1.13 (m, 3H).
Example 3: (3R)-1-(6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)-3-methylpiperidin-3-ol (Compound 109a)

Step 1: tert-butyl (2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-((R)-3-hydroxy-3-methylpiperidin-1-yl)-2,3,5',8'-tetrahydrospiro[indene- 1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (3R)-1-(6-Bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-3- methylpiperidin-3-ol (300 mg, 1 equiv, 511 µmol), tert-butyl carbamate (150 mg, 2.5 equiv, 1.28 mmol), and cesium carbonate (499 mg, 3 equiv, 1.53 mmol) were taken up in 1,4-dioxane (3 mL). The vial was degassed with nitrogen and Pd(OAc)
2 (11.5 mg, 0.1 equiv, 51.1 µmol) and XantPhos (35.5 mg, 0.12 equiv, 61.3 µmol) were added. The reaction mixture was heated to 100 ℃ and purified via column chromatography (SiO2, DCM/MeOH = 100/0 to 10/2, ammonia modifier) to give tert-butyl (2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-((R)-3-hydroxy-3-methylpiperidin-1-yl)-2,3,5',8'-tetrahydrospiro[indene-1,7'- pyrano[4,3-d]pyrimidin]-6-yl)carbamate (120 mg). LCMS: ESI [M+H]
+ = 624.8; tR = 1.43min. (Method C) Step 2: (3R)-1-(6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-3- methylpiperidin-3-ol tert-butyl (2'-(((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'- ((R)-3-hydroxy-3-methylpiperidin-1-yl)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-6-yl)carbamate (120 mg, 1 equiv, 192 µmol) in acetonitrile (1 mL) was treated with HCl in 1,4-dioxane (4 N, 56.1 mg, 8 equiv, 1.54 mmol). The reaction mixture was stirred at room temperature for 1 hour, quenched with aqueous sodium bicarbonate, diluted with
EtOAc, and extracted with EtOAc (2 × 10 mL). The organic extracts were dried over sodium sulfate, filtered, and concentrated to a crude product, which was purified via reverse phase chromatography (ACCQPrep, water/MeCN, 0.1% formic acid modifier = 100/10 to 100/95) to give (3R)-1-(6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol (9 mg). LCMS: ESI [M+H]
+ = 524.5; t
R = 1.27min. (Method C) 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 7.08 (t, J = 6.9 Hz, 1H), 6.63 (d, J = 7.9 Hz, 1H), 6.51 (d, J = 10.4 Hz, 1H), 5.36 (d, J = 53.0 Hz, 1H), 4.63 – 4.45 (m, 2H), 4.45 – 4.23 (m, 2H), 3.91 – 3.50 (m, 5H), 3.30 (dd, J = 18.5, 14.5 Hz, 1H), 3.12 – 2.91 (m, 6H), 2.79 – 2.67 (m, 1H), 2.49 – 2.25 (m, 4H), 2.22 – 2.00 (m, 4H), 1.95 – 1.73 (m, 2H), 1.60 – 1.48 (m, 2H), 1.26 – 1.20 (m, 3H). Example 4: 4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-ol (Compound 106a)
Step 1: 6-bromo-4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5

][1,4]diazepin-5(6H)-yl)- 2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] 6-Bromo-4'-chloro-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidine] (0.56 g, 1 equiv, 1.4 mmol) and 5,6,7,8-tetrahydro-4H-[1,2,3]triazolo[1,5- a][1,4]diazepine, HCl (0.37 g, 1.5 equiv, 2.1 mmol) in ethanol (5 mL) were treated with DIPEA (0.54 g, 3 equiv, 4.2 mmol) followed by the addition of acetonitrile (1 mL). After 1 hour of stirring at room temperature, an additional portion of 5,6,7,8-tetrahydro-4H- [1,2,3]triazolo[1,5-a][1,4]diazepine, hydrochloride (200 mg) was added along with DIPEA (4 equiv.), and the reaction mixture was stirred for two days. The reaction was quenched with aqueous ammonium chloride, diluted with EtOAc, and extracted with EtOAc (2 × 15 mL). The
organic layers were dried over sodium sulfate, filtered, and concentrated to a crude product, which was then purified by column chromatography (SiO2, heptanes/EtOAc = 100/5 to 100/100) to give 6-bromo-4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)- 2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] (262 mg). LCMS: ESI [M+H]
+ = 501.0; tR = 1.85 minutes. (Method C) 1H NMR (400 MHz, CDCl3) δ 7.56 (s, 1H), 7.44 (d, 1H), 7.20-7.24 (m, 2H), 4.46-4.78 (m, 6H), 3.89-3.95 (m, 1H), 3.75-3.81 (m, 1H), 2.98-3.14 (m, 3H), 2.79-2.84 (m, 1H), 2.51 (s, 3H), 2.32-2.38 (m, 1H), 2.11-2.24 (m, 3H). Step 2: 6-bromo-4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5

][1,4]diazepin-5(6H)-yl)- 2'-(methylsulfonyl)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] 6-Bromo-4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'- (methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] (260 mg, 1 equiv, 521 µmol) in methanol (4 mL) and water (2 mL) was treated with Oxone (640 mg, 2 equiv, 1.04 mmol). The reaction mixture was stirred for 2 hours and then quenched with aqueous sodium bicarbonate, diluted with EtOAc, and extracted with EtOAc (2 × 15 mL). The organic layers were dried over sodium sulfate, filtered, and concentrated to give 6-bromo-4'-(7,8- dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(methylsulfonyl)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] (277 mg) which was used in the next step without further purification. LCMS: ESI [M+H]
+ = 533.4; t
R = 1.77 minutes. (Method C) Step 3: 6-bromo-4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5

][1,4]diazepin-5(6H)-yl)- 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] Sodium hydride (41.7 mg, 2 equiv, 1.04 mmol) was suspended in THF (3 mL) followed by the dropwise addition of ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (64 mg, 3 equiv, 0.40 mmol) in THF (3 mL).6-Bromo-4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(methylsulfonyl)-2,3,5',8'-tetrahydrospiro[indene-1,7'- pyrano[4,3-d]pyrimidine] (277 mg, 1 equiv, 512 µmol) in DMF (6 mL) was treated by the dropwise addition of the prepared alkoxide solution, and the mixture was stirred for 90 minutes at 50 ℃. The reaction was quenched by the addition of aqueous ammonium chloride, diluted with EtOAc, and extracted with EtOAc (3 × 1 mL). The organic layers were dried with sodium
sulfate, filtered, and concentrated to a crude product, which was purified by column chromatography ((SiO2, DCM/methanol = 100/0 to 10/2, with ammonia modifier) to give 6- bromo-4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-2,3,5',8'-tetrahydrospiro[indene-1,7'- pyrano[4,3-d]pyrimidine] (230 mg). LCMS: ESI [M+H]
+ = 612.3; tR = 1.40 minutes. (Method C) 1H NMR (400 MHz, CDCl
3) δ 7.57 (s, 1H), 7.44 (d, 1H), 7.18-7.23 (m, 2H), 5.26 (d, 1H), 4.61-4.79 (m, 4H), 4.51-4.60 (m, 1H), 4.43-4.52 (m, 1H), 4.03-4.09 (m, 1H), 3.83-3.99 (m, 2H), 3.74-3.83 (m, 1H), 3.19-3.29 (m, 2H), 3.06-3.18 (m, 3H), 2.92-3.03 (m, 2H), 2.77- 2.86 (m, 1H), 2.28-2.39 (m, 1H), 2.06-2.29 (m, 6H), 1.81-1.99 (m, 3H). Step 4: 4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-6-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] 6-Bromo-4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] (130 mg, 1 equiv, 213 µmol), potassium acetate (31.3 mg, 1.5 equiv, 319 µmol), and bis(pinacolato)diboron (97.3 mg, 1.8 equiv, 383 µmol) in 1,4-dioxane (2 mL) was degassed with nitrogen, followed by the addition of Pd(dppf)Cl
2·CH
2Cl
2 (15.6 mg, 0.1 equiv, 21.3 µmol). The reaction mixture was heated to 90 ℃ for 45 minutes and then at 100 ℃ for 2 hours. The crude product was purified via column chromatography (SiO2, DCM/methanol = 100/0 to 100/15, with ammonia modifier) to give 4'- (7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-6-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] (140 mg). LCMS: ESI [M+H]
+ = 658.8; tR = 1.50 minutes. (Method C) Step 5: 4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-ol 4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-6-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] (140 mg, 1
equiv, 213 µmol) in methanol (2 mL) and 1,4-dioxane (1 mL) was treated with aqueous NaOH (25.6 mg, 3 equiv, 639 µmol, 1 M) and hydrogen peroxide (41.4 mg, 35 wt%, 2 equiv, 426 µmol). The mixture was stirred at 0 ℃ for 30 minutes. The reaction was quenched with aqueous sodium bicarbonate, aqueous sodium thiosulfate, diluted with EtOAc, and extracted with EtOAc (3 × 10 mL). The crude product was purified via reverse-phase chromatography (ACCQPrep, water/MeCN = 100/0 to 100/60, with 0.1% formic acid modifier) to give 4'-(7,8- dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-6-ol (16 mg). LCMS: ESI [M+H]
+ = 548.5; t
R = 1.25 minutes. (Method C) 1H NMR (400 MHz, CDCl
3) δ 7.54 (d, 1H), 7.15 (dd, 1H), 6.88-6.84 (m, 1H), 6.52 (dd, 1H), 5.29 (d, 1H), 4.65-4.76 (m, 2H), 4.50-4.61 (m, 4H), 4.00-4.17 (m, 3H), 3.58-3.69 (m, 1H), 3.31-3.43 (m, 2H), 3.19-3.30 (m, 1H), 2.91- 3.18 (m, 4H), 2.71-2.79 (m, 1H), 1.90-2.37 (m, 10H). Example 6: 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 4'-((R)-3-hydroxy-3-methylpiperidin-1-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-7-ol (Compound 105a)
A solution of potassium hydroxide (15.4 mg, 85 wt%, 3 equiv, 0.233 mmol) in a 4:1 mixture of 1,4 dioxane and water (1.5 mL) was degassed with nitrogen. (3R)-1-(7-Bromo-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol (Intermediate 12, 46.8 mg, 1 equiv, 0.078 mmol) was added along with Pd
2(dba)
3 (10.7 mg, 0.15 equiv, 0.012 mmol) and t-BuXPhos (9.91 mg, 0.3 equiv, 0.023 mmol). The reaction mixture was stirred at 95 ℃ for 6 hours and quenched with aqueous sodium bicarbonate, diluted with DCM (2 mL), and extracted with DCM (3 × 10 mL). Organic extracts were dried over sodium sulfate, filtered, and concentrated to a crude product, which was purified via reverse-phase chromatography (C18, water/MeCN with 0.1% formic acid = 100/0 to 0/100) to give 2'-(((2R,7aS)-2-
fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-((R)-3-hydroxy-3-methylpiperidin-1- yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-ol (16.2 mg). LCMS: ESI [M+H]
+ = 540.0; tR = 1.25 minutes. (Method C) 1H NMR (400 MHz, CD
3OD) δ 6.98 (dd, 1H), 6.75 (dd, 1H), 6.72 – 6.64 (m, 1H), 5.71 – 5.45 (m, 1H), 4.87 – 4.58 (m, 2H), 4.50 (q, 3H), 4.04 – 3.64 (m, 5H), 3.57 – 3.38 (m, 2H), 3.38 – 3.30 (m, 2H), 3.30 – 3.17 (m, 2H), 3.04 (dd, 1H), 2.92 (d, 1H), 2.86 – 2.66 (m, 2H), 2.66 – 2.47 (m, 2H), 2.47 – 2.25 (m, 3H), 2.25 – 1.87 (m, 4H), 1.85 – 1.58 (m, 1H), 1.22 (d, 3H). Example 6a: (R)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-((R)-3-hydroxy-3-methylpiperidin-1-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-ol (Compound 105c)
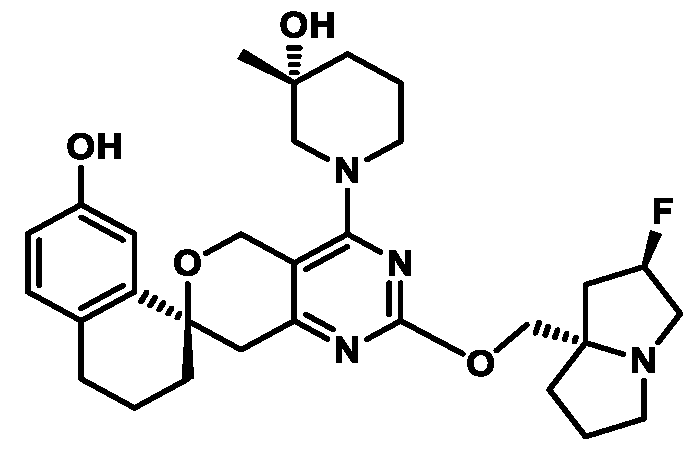
The diastereomers of 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-((R)-3-hydroxy-3-methylpiperidin-1-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-ol were separated via Chiralpak IC (20 × 250 mm, 5 µm, P/N 83345) on ACCQPrep under basic conditions (0.1% ammonium hydroxide in water, 0.1% ammonium hydroxide in MeCN), retention time 22 minutes, to provide (R)-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-((R)-3-hydroxy-3- methylpiperidin-1-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-ol (Peak 1) (4.4 mg) and (S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-((R)-3-hydroxy-3-methylpiperidin-1-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-ol (Peak 2) (5.2 mg). LCMS: ESI [M+H]
+ = 539.3 m/z; tR = 6.63 minutes. (Column: IC-U (3 × 100 mm, 1.6 μm, P/N 83U83; mobile phase A: 0.1% NH4OH in MeCN; mobile phase B: 0.1% NH4OH in water; 5% to 95% over 10 minutes) Example 6b: (S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-((R)-3-hydroxy-3-methylpiperidin-1-yl)-3,4,5',8'-tetrahydro-2H-
spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-ol (Compound 105b)
(S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-((R)-3- hydroxy-3-methylpiperidin-1-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-ol (5.2 mg) was obtained from Example 6a as Peak 2: Chiralpak IC (20 × 250 mm, 5 µm, P/N 83345) on ACCQPrep under basic conditions (0.1% ammonium hydroxide in water, 0.1% ammonium hydroxide in MeCN), retention time 26 minutes. LCMS: ESI [M+H]
+ = 539.3 m/z; tR = 7.93 minutes. (Column: IC-U (3 × 100 mm, 1.6 μm, P/N 83U83; mobile phase A: 0.1% NH
4OH in MeCN; mobile phase B: 0.1% NH
4OH in water; 5% to 95% over 10 minutes) Example 7: 4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)- yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (Compound 107a)

4'-(7,8-Dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine was prepared according to General Procedure A with 5,6,7,8-tetrahydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepine (101 mg, 1 equiv, 729 µmol) as the appropriate amine. The reaction mixture was purified via preparative HPLC (C18 column; Gradient: 5% to 60% MeCN in ammonium bicarbonate buffer) to provide 4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-
spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (47 mg). LCMS: ESI [M+H]
+ = 561.4 m/z, tR = 0.69 minutes. (Method A) 1H NMR (DMSO-d6, 400 MHz): δ 7.58 (1H, s), 6.77 (1H, d), 6.54 (1H, d), 6.45 (1H, dd), 5.26 (1H, d), 4.71-4.83 (5H, m), 4.65 (2H, m), 4.48 (1H, d), 3.91 (1H, dd), 3.83 (3H, m), 3.00-3.08 (4H, m), 2.73-2.88 (3H, m), 2.60 (1H, m), 1.65-2.14 (12H, m). 1
9F NMR (DMSO, 376 MHz): δ -172.0. Example 8: 4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)- yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-ol (Compound 108a)
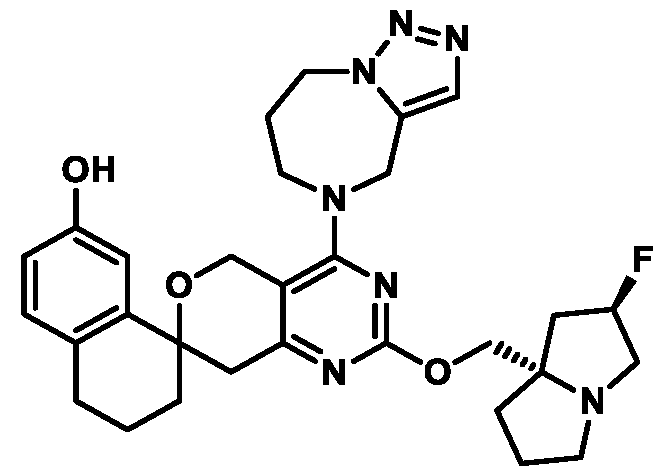
Step 1: 7-bromo-4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)- 2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine] 7-Bromo-4'-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine] (300 mg, 1 equiv, 729 µmol) and 5,6,7,8-tetrahydro-4H- [1,2,3]triazolo[1,5-a][1,4]diazepine (101 mg, 1 equiv, 729 µmol) in EtOH (3.64 mL) was treated with DIPEA (753 mg, 8 equiv, 5.83 mmol). The reaction mixture was stirred at 80 ℃ for 1 hour. The reaction was quenched with aqueous ammonium chloride and extracted with EtOAc (3 × 15 mL). The organic layers were dried with sodium sulfate, filtered, and concentrated to give 7-bromo-4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)- yl)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (256 mg) that was used without further purification. LCMS: ESI [M+H]
+ = 515.0 m/z; tR = 1.95 minutes (Method C). Step 2: 7-bromo-4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,
][1,4]diazepin-5(6H)-yl)- 2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine]
7-bromo-4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (250 mg, 1 equiv, 0.49 mmol) in methanol (4 mL) and water (2 mL) was treated with Oxone (599 mg, 2 equiv, 0.97 mmol). The reaction mixture was stirred for 4 hours. The reaction was quenched with sodium bicarbonate, diluted with EtOAc, and extracted with EtOAc (3 × 15 mL). The organic layers were dried with sodium sulfate, filtered, and concentrated to give 7- bromo-4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(methylsulfonyl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (277.6 mg) that was used without further purification. LCMS: ESI [M+H]
+ = 547.3 m/z; t
R = 1.87 minutes (Method C). Step 3: 7-bromo-4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,
][1,4]diazepin-5(6H)-yl)- 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] Sodium hydride (15 mg, 3 equiv, 0.38 mmol) was suspended in THF (0.35 mL) followed by the dropwise addition of ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methanol (60 mg, 0.38 mmol) in THF (0.63 mL). 7-Bromo-4'-(7,8-dihydro-4H- [1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (69 mg, 1 equiv, 100 µmol) in DMF (0.63 mL) was treated by the dropwise addition of the prepared alkoxide solution, and the resulting mixture was stirred for 90 minutes at 50 ℃. The reaction was quenched by the addition of aqueous ammonium chloride then diluted with EtOAc, and extracted with EtOAc (3 × 1 mL). The organic layers were dried with sodium sulfate, filtered, and concentrated to a crude product, which was purified by column chromatography (SiO
2, DCM/MeOH = 100/0 to 10/2) to give 7-bromo-4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (42 mg). LCMS: ESI [M+H]
+ = 626.0 m/z; t
R = 1.46 minutes. (Method C) Step 4: 4'-(7,8-Dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-7-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine]
7-Bromo-4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (77 mg, 1 equiv, 120 µmol), potassium acetate (18 mg, 1.5 equiv, 180 µmol), and bis(pinacolato)diboron (56 mg, 1.8 equiv, 220 µmol) in 1,4-dioxane (1.2 mL) was degassed with nitrogen, followed by the addition of Pd(dppf)Cl2·CH2Cl2 (9.0 mg, 0.1 equiv, 12 µmol). The mixture was heated to 95 ℃ overnight and filtered through a silica plug to provide crude 4'-(7,8-dihydro-4H-[1,2,3]triazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (150 mg). LCMS: ESI [M+H]
+ = 673.0 m/z; t
R = 1.49 minutes (Method C). Step 5: 4'-(7,8-Dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-ol 4'-(7,8-Dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-7-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (150 mg, 20 wt%, 1 equiv, 0.045 mmol) in methanol (425 µL) and 1,4-dioxane (213 µL) was cooled to 0 ℃ and treated with hydrogen peroxide (6.08 mg, 50 wt%, 2 equiv, 0.089 mmol). The reaction mixture was stirred for 30 minutes. The reaction was quenched with aqueous sodium bicarbonate and sodium thiosulfate. The mixture was then diluted with EtOAc, and extracted with EtOAc (3 × 10 mL). The organic layers were dried and purified via ACCQPrep HPLC (0.1% formic acid in water, 0.1% formic acid in MeCN, 4 mL/min; Gradient: 25% to 35% B over 27 minutes then 100% B to 32 minutes), to provide 4’-(7,8-dihydro-4H- [1,2,3]triazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2’-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5’,8’-tetrahydro-2H-spiro[naphthalene-1,7’-pyrano[4,3- d]pyrimidin]-7-ol (4.5 mg). LCMS: ESI [M+H]
+ = 563.3 m/z; tR = 1.27 minutes (Method C). 1H NMR (400 MHz, MeOD) δ 8.48 (s, 1H), 7.65 (s, 1H), 6.96-7.00 (m, 1H), 6.66-6.70 (m, 2H), 5.42 (d, 1H), 4.79-4.82 (m, 1H), 4.72 (m, 2H), 4.52-4.56 (m, 1H), 4.18-4.29 (m, 2H), 3.86-4.03 (m, 2H), 3.49-3.63 (m, 3H), 3.17-3.26 (m, 1H), 2.88-3.05 (m, 2H), 2.67-2.84 (m,
2H), 2.32-2.50 (m, 2H), 2.22-2.27 (m, 1H), 2.08-2.20 (m, 5H), 1.93-2.06 (m, 4H), 1.74 (m, 1H). Example 9: (3R)-1-(7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol (Compound 113a)
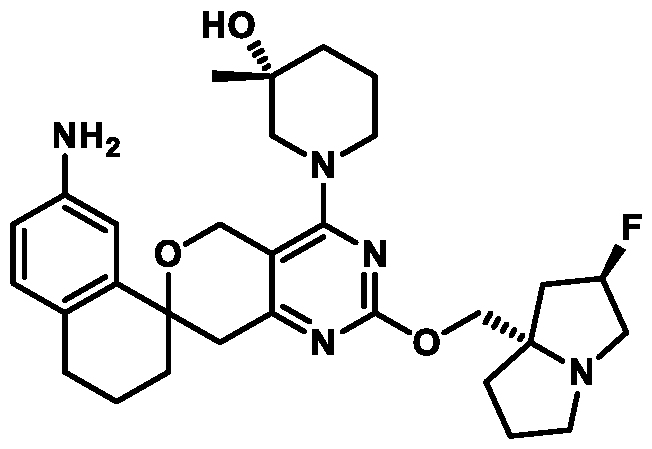
Step 1: tert-butyl (4'-((R)-3-hydroxy-3-methylpiperidin-1-yl)-2'-(methylthio)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (3R)-1-(7-bromo-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol (251 mg, 1 equiv, 0.51 mmol), tert- butyl carbamate (150 mg, 2.5 equiv, 1.3 mmol), Pd(OAc)
2 (12 mg, 0.1 equiv, 0.051 mmol), Xanthphos (44 mg, 0.15 equiv, 0.076 mmol) and cesium carbonate (500 mg, 3 equiv, 1.54 mmol) were purged with nitrogen gas in a vial followed by the addition of 1,4-dioxane (2.1 mL). The sealed flask was heated to 100 ℃ for 1 hour. The reaction was quenched with aqueous sodium bicarbonate and extracted with EtOAc (3 × 20 mL). The organic layers were dried over sodium sulfate, filtered, and concentrated to a crude product, which was purified by column chromatography (SiO2, heptanes/EtOAc = 100/0 to 0/100, 5% DCM modifier) to give tert- butyl (4'-((R)-3-hydroxy-3-methylpiperidin-1-yl)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (138 mg). LCMS: ESI [M+H]
+ = 527.8 m/z; tR = 2.65 minutes (Method E). Step 2: tert-butyl (2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-((R)-3-hydroxy-3-methylpiperidin-1-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate tert-Butyl (4'-((R)-3-hydroxy-3-methylpiperidin-1-yl)-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (138 mg, 90 wt%, 1 equiv, 0.24 mmol) in methanol (1.57 mL) and water (786 µL) was treated with Oxone (290 mg, 2 equiv, 0.47 mmol). The mixture was stirred at room temperature for 2 hours. The
reaction was quenched with aqueous ammonium chloride, diluted with EtOAc (15 mL), and extracted with EtOAc (3 × 15 mL). The organic phases were dried with sodium sulfate, filtered, and concentrated to give tert-butyl (4'-((R)-3-hydroxy-3-methylpiperidin-1-yl)-2'- (methylsulfonyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (130 mg) which was used without further purification. Sodium hydride (28 mg, 60 wt%, 3 equiv, 0.70 mmol) was suspended in THF (1.2 mL) followed by dropwise addition of ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methanol (111 mg, 3 equiv, 0.70 mmol). tert-Butyl (4'-((R)-3-hydroxy-3-methylpiperidin- 1-yl)-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (130 mg, 1 equiv, 0.23 mmol) in THF (1.2 mL) was treated by the dropwise addition of the prepared alkoxide solution, and the resulting mixture was stirred for 90 minutes at 50 ℃. The reaction was quenched by the addition of aqueous ammonium chloride then diluted with EtOAc, and extracted with EtOAc (3 × 1 mL). The organic layers were dried with sodium sulfate, filtered, and concentrated to a crude product, which was purified by reverse-phase chromatography (ACCQPrep, XSelect CSH C18 OBD Prep Column, 130 Å, 5 µm, 19 mm × 250 mm, P/N: 186005492 with acidic mobile phases (Mobile phase A = 0.1% v/v ammonium hydroxide in water, Mobile phase B = 0.1% v/v ammonium hydroxide in acetonitrile)) to give tert-butyl (2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-((R)-3-hydroxy-3-methylpiperidin-1-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (26.3 mg). LCMS: ESI [M+H]
+ = 638.7 m/z; t
R = 1.42 minutes (Method C). Step 3: (3R)-1-(7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol tert-Butyl (2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'- ((R)-3-hydroxy-3-methylpiperidin-1-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate (33.2 mg, 1 equiv, 52.1 μmol) in 1,4-dioxane (521 μL) was treated with HCl (19 mg, 4 N, 10 equiv, 521 μmol). The reaction mixture was stirred overnight. The reaction mixture was concentrated under reduced pressure to obtain (3R)-1-(7- amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol as a crude product (30 mg).
LCMS: ESI [M+H]
+ = 538.5 m/z; t
R = 1.07 minutes (Method C). 1H NMR (400 MHz, MeOD) δ 7.49 – 7.40 (d, 1H), 7.29 – 7.13 (m, 2H), 5.56 (d, 0.5H), 5.44 (d, 0.5H), 4.96 (d, 1H), 4.77 – 4.44 (m, 3H), 3.97 – 3.70 (m, 3H), 3.68 – 3.44 (m, 2H), 3.33 (dd, 2H), 3.15 (d, 1H), 3.02 (s, 2H), 2.80 (d, 2H), 2.73 – 2.31 (m, 4H), 2.29 – 2.07 (m, 3H), 2.07 – 1.82 (m, 2H), 1.82 – 1.58 (m, 4H), 1.26 – 1.10 (m, 4H). Example 9a: (R)-1-((R)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol (Compound 113b)

The diastereomers of (3R)-1-(7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]- 4'-yl)-3-methylpiperidin-3-ol were separated under basic conditions (ACCQPrep, Mobile Phase A = 0.1% v/v ammonium hydroxide in water, 0.1% v/v ammonium hydroxide in acetonitrile) on Chiralcel OD (21 × 250 mm, 5 μm, P/N 14745)) to give product (R)-1-((R)-7- amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol (Peak 1) (9.4 mg) and (R)-1-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]- 4'-yl)-3-methylpiperidin-3-ol (Peak 2) (6.9 mg). LCMS: ESI [M+H]
+= 538.3 m/z; t
R = 2.26 minutes (Method J) 1H NMR (400 MHz, CDCl3) δ 6.92 (d, 1H), 6.71 (d, 1H), 6.58 (m, 1H), 5.25 (d, 1H), 4.66 (d, 1H), 4.48 (d, 1H), 4.11 (d, 1H), 3.98 (d, 1H), 3.78 (d, 1H), 3.60 (d, 1H), 3.30 – 3.09 (m, 4H), 3.07 – 2.88 (m, 4H), 2.83 – 2.66 (m, 2H), 2.21 – 2.09 (m, 3H), 2.07 – 1.72 (m, 10H), 1.62 – 1.46 (m, 2H).1.26 (s, 3H). Example 9b: (R)-1-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylpiperidin-3-ol (Compound 113c)
(R)-1-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-3- methylpiperidin-3-ol (9.4 mg) was obtained from Example 9a, as Peak 2: (ACCQPrep, Mobile Phase A = 0.1% v/v ammonium hydroxide in water, 0.1% v/v ammonium hydroxide in acetonitrile) on Chiralcel OD (21 × 250 mm, 5 μm, P/N 14745)). LCMS: ESI [M+H]
+= 538.3 m/z; t
R = 1.82 minutes (Method J) 1H NMR (400 MHz, CDCl3) δ 6.92 (d, 1H), 6.71 (m, 1H), 6.59 (m, 1H), 5.25 (d, 1H), 4.66 (d, 1H), 4.48 (d, 1H), 4.11 (d, 1H), 3.98 (d, 1H), 3.78 (d, 1H), 3.60 (d, 1H), 3.19-3.30 (m, 2H), 3.01-3.18 (m, 2H), 2.89-3.01 (m, 4H), 2.64-2.82 (m, 2H), 2.09-2.27 (m, 3H), 1.65-2.08 (m, 10H), 1.46-1.62 (m, 2H), 1.23 (s, 3H). Example 10: 5-(7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide (Compound 114a)
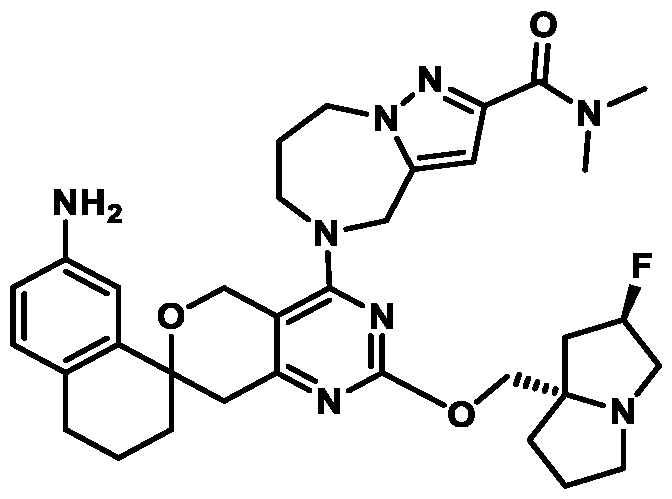
Step 1: 5-(7-bromo-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide 7-Bromo-4'-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine] (266 mg, 1 equiv, 0.65 mmol) and N,N-dimethyl-5,6,7,8-tetrahydro-
4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (404 mg, 3 equiv, 1.94 mmol) in ethanol (3.23 mL) were treated with DIPEA (418 mg, 5 equiv, 3.23 mmol). The reaction mixture was heated to 80 ℃ for 9 hours. The reaction was quenched with aqueous ammonium chloride, diluted with EtOAc (15 mL), and extracted with EtOAc (3 × 40 mL). The organic extracts were dried over sodium sulfate, filtered, and concentrated. The crude product obtained was purified by column chromatography (SiO2, DCM/MeOH = 100/0 to 100/20) to give 5-(7-bromo-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (251 mg). LCMS: ESI [M+H]
+ = 585.0 m/z; tR = 1.88 minutes. (Method C) Step 2: tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate 5-(7-Bromo-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (250.8 mg, 1 equiv, 0.43 mmol), tert-butyl carbamate (126 mg, 3 equiv, 1.07 mmol), Pd(OAc)
2 (9.65 mg, 0.1 equiv, 0.043 mmol), XantPhos (37.30 mg, 0.15 equiv, 0.065 mmol) and cesium carbonate (420 mg, 3 equiv, 1.29 mmol) in a flask were purged with nitrogen and taken up in 1,4-dioxane (4.3 mL). The reaction mixture was heated to 100 ℃ and stirred for 2.5 hours. Upon standard workup, the mixture was then purified by column chromatography (SiO
2, heptanes/EtOAc = 100/0 to 0/100, 1% MeOH modifier) to give tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)- yl)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (111 mg). 1H NMR (400 MHz, CDCl3) δ 7.28 (m, 1H), 7.07 (d, 1H), 6.60 (s, 1H), 6.44 (s, 1H), 4.71 (d, 1H), 4.48-4.61 (m, 3H), 4.41-4.48 (bs, 2H), 3.85 (s, 2H), 3.34 (s, 3H), 3.04-3.18 (m, 4H), 2.93-3.01 (m, 1H), 2.70-2.89 (m, 2H), 2.50 (s, 3H), 2.16 (s, 2H), 1.93-2.10 (m, 3H), 1.69- 1.81 (m, 1H), 1.60 (s, 1H), 1.45 (s, 9H). Step 3: tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate
tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin- 5(6H)-yl)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (100.5 mg, 1 equiv, 162 µmol) in methanol (1.44 mL) and water (721 µL) was treated with Oxone (199 mg, 2 equiv, 324 µmol). The mixture was stirred at room temperature for 2 hours. The reaction was quenched with aqueous ammonium chloride, diluted with EtOAc (15 mL), and extracted with EtOAc (3 × 15 mL). The organic phases were dried with sodium sulfate, filtered, and concentrated to give tert-butyl (4'-(2-(dimethylcarbamoyl)- 7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(methylsulfonyl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (125 mg) which was used without further purification. Sodium hydride (13.8 mg, 60 wt%, 3 equiv, 0.57 mmol) was suspended in THF (1.16 mL) followed by the dropwise addition of ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methanol (91.2 mg, 3 equiv, 573 µmol). tert-Butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro- 4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (125 mg, 1 equiv, 0.19 mmol) in THF (955 µL) was treated by the dropwise addition of the prepared alkoxide solution, and the reaction mixture was stirred for 90 minutes at 50 ℃. The reaction was quenched by addition of aqueous ammonium chloride, diluted with EtOAc, and extracted with EtOAc (3 × 1 mL). The organic layers were dried with sodium sulfate, filtered, and concentrated to give a crude product, which was purified by reverse-phase chromatography (XSelect CSH C18 OBD Prep Column, 130 Å, 5 µm, 19 mm × 250 mm, P/N: 186005492 with basic mobile phases (Mobile phase A = 0.1% v/v ammonium hydroxide in water, Mobile phase B = 0.1% v/v ammonium hydroxide in acetonitrile)) to give tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]- 7-yl)carbamate (57 mg). 1H NMR (400 MHz, CDCl
3) δ 7.32 – 7.20 (m, 1H), 7.06 (d, 1H), 6.59 (s, 1H), 6.45 (s, 1H), 5.26 (d, 1H), 4.70 (d, 1H), 4.64 – 4.50 (m, 3H), 4.47 – 4.40 (m, 2H), 4.07 (d, 1H), 3.96 (d, 1H), 3.86 – 3.80 (m, 2H), 3.33 (s, 3H), 3.27 – 3.20 (m, 2H), 3.18 – 3.05 (m, 5H), 3.01 – 2.91 (m, 2H), 2.90 – 2.78 (m, 1H), 2.78 – 2.68 (m, 1H), 2.32 – 2.12 (m, 5H), 2.08 – 1.65 (m, 8H), 1.45 (d, 9H).
Step 4: 5-(7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide tert-Butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin- 5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (57 mg, 1 equiv, 0.078 mmol) in chloroform (0.78 mL) was treated with 4 N HCl (43 mg, 15 equiv, 1.2 mmol). The mixture was stirred for 16 hours. The mixture was concentrated under reduced pressure to obtain 5-(7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (47.6 mg). LCMS: ESI [M+H]
+= 631.6 m/z; tR = 1.10 minutes (Method C) 1H NMR (400 MHz, MeOD) δ 7.42 (dd, 1H), 7.28 (d, 1H), 7.20 (dd, 1H), 6.39 (s, 1H), 5.50 (d, 1H), 5.07 – 4.83 (m, 3H), 4.73 – 4.60 (m, 3H), 4.58 – 4.44 (m, 3H), 4.34 – 4.19 (m, 1H), 3.97 – 3.70 (m, 4H), 3.39 – 3.30 (m, 1H), 3.21 (s, 3H), 3.04 (s, 2H), 2.99 (s, 3H), 2.92 – 2.73 (m, 2H), 2.72 – 2.50 (m, 2H), 2.50 – 2.35 (m, 1H), 2.31 – 1.92 (m, 9H), 1.80 – 1.68 (m, 1H). Example 10a: 5-((R)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 114b)
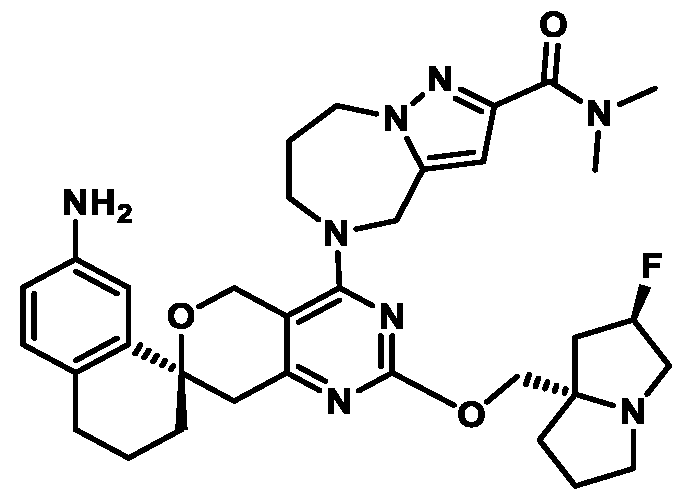
The diastereomers of 5-(7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]- 4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide were separated under basic conditions (ACCQPrep, Mobile Phase A = 0.1% v/v ammonium hydroxide in water, Mobile phase B = 0.1% v/v ammonium hydroxide in acetonitrile, Chiralcel
OD (21 × 250 mm, 5 μm, P/N 14745)) to give 5-((R)-7-amino-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Peak 1) (5.2 mg, 86% ee) and 5-((S)-7-amino- 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Peak 2) (10.6 mg, 98% ee). LCMS: ESI [M+H]
+= 631.4 m/z; tR = 6.04 minutes (Method J) 1H NMR (400 MHz, MeOD) δ 6.91 (d, 1H), 6.69 (d, 1H), 6.65 (dd, 1H), 6.54 (s, 1H), 5.30 (d, 1H), 4.82 – 4.69 (m, 3H), 4.58 – 4.45 (m, 3H), 4.15 – 4.04 (m, 2H), 4.00 – 3.87 (m, 2H), 3.30 – 3.18 (m, 4H), 3.04 (d, 5H), 2.86 (d, 1H), 2.82 – 2.62 (m, 2H), 2.35 – 1.80 (m, 13H), 1.74 (dt, 1H). Example 10b: 5-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 114c)

5-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl- 5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (10.6 mg, 98% ee) was obtained from Example 10a, as Peak 2: (ACCQPrep, Mobile phase A = 0.1% v/v ammonium hydroxide in water, Mobile phase B = 0.1% v/v ammonium hydroxide in acetonitrile, Chiralcel OD (21 × 250 mm, 5 μm, P/N 14745)). LCMS: ESI [M+H]
+= 631.4 m/z; t
R = 7.70 minutes (Method J) 1H NMR (400 MHz, MeOD) δ 6.91 (d, 1H), 6.69 (d, 1H), 6.64 (dd, 1H), 6.54 (s, 1H), 5.29 (d, 1H), 4.83 – 4.70 (m, 4H), 4.62 – 4.43 (m, 3H), 4.12 (d, 1H), 4.05 (d, 1H), 3.94 (s, 2H), 3.29 – 3.15 (m, 2H), 3.04 (d, 6H), 2.86 (d, 1H), 2.81 – 2.62 (m, 2H), 2.31 – 1.83 (m, 13H), 1.74
(dt, 1H). Example 11: (3R)-1-(2'-Amino-2-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-5,6',7',8-tetrahydro-5'H-spiro[pyrano[4,3-d]pyrimidine-7,8'- quinolin]-4-yl)-3-methylpiperidin-3-ol (Compound 110a)
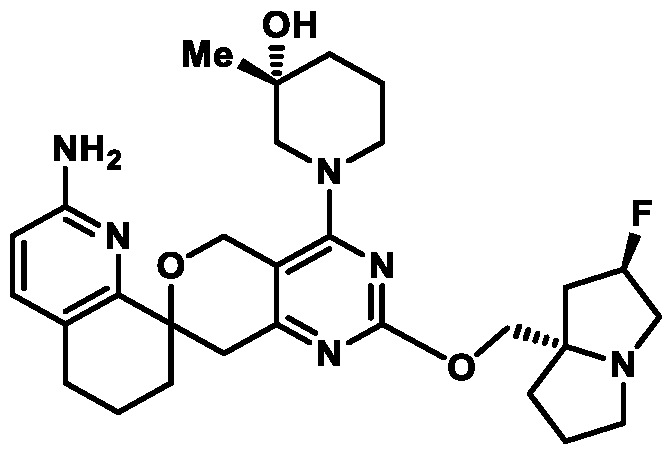
Step 1: (3R)-3-methyl-1-(2-(methylthio)-5,6',7',8-tetrahydro-5'H- spiro[pyrano[4,3-d]pyrimidine-7,8'-quinolin]-4-yl)piperidin-3-ol To a solution of 4-chloro-2-(methylthio)-5,6',7',8-tetrahydro-5'H-spiro[pyrano[4,3- d]pyrimidine-7,8'-quinoline] (260 mg, 1 equiv, 0.78 mmol) in ethanol (3.1 mL) were added (R)-3-methylpiperidin-3-ol (179 mg, 2 equiv, 1.56 mmol) and DIPEA (407 μL, 3 equiv, 2.36 mmol). The mixture was stirred at 65 °C for 17 hours. The reaction mixture was cooled to room temperature and concentrated to a residue, which was diluted with DCM and water. The aqueous layer was extracted with DCM (× 4). The combined organic layers were dried over sodium sulfate, filtered, and concentrated to give (3R)-3-methyl-1-(2-(methylthio)- 5,6',7',8-tetrahydro-5'H-spiro[pyrano[4,3-d]pyrimidine-7,8'-quinolin]-4-yl)piperidin-3-ol (280 mg) which was used without further purification. LCMS: m/z (ESI) [M+H]
+ 413.3, tR = 1.25 minutes (Method C) Step 2: 4-((R)-3-Hydroxy-3-methylpiperidin-1-yl)-2-(methylsulfonyl)-5,6',7',8- tetrahydro-5'H-spiro[pyrano[4,3-d]pyrimidine-7,8'-quinoline]-1'-oxide 3-Chlorobenzoperoxoic acid (669 mg, 70 wt%, 4 equiv, 2.72 mmol) was added to (3R)- 3-methyl-1-(2-(methylthio)-5,6',7',8-tetrahydro-5'H-spiro[pyrano[4,3-d]pyrimidine-7,8'- quinolin]-4-yl)piperidin-3-ol (280 mg, 1 equiv, 0.68 mmol) in DCM (3.4 mL) at 0 °C in one portion. The ice bath was removed, and the reaction mixture was stirred at room temperature for 1.5 hours. The reaction mixture was cooled to 0 °C, and additional 3-chlorobenzoperoxoic acid (669 mg, 70 wt%, 4 equiv, 2.72 mmol) was added in one portion. The ice bath was removed, and the reaction mixture was stirred at room temperature for 1.5 hours. The reaction
was quenched with a saturated aqueous solution of sodium thiosulfate, diluted with DCM, and then stirred for 10 minutes. The mixture was extracted with DCM. The combined organic layers were washed with saturated sodium carbonate, dried over sodium sulfate, filtered, and concentrated to a residue which was purified by flash column chromatography (RediSep Rf Gold 24 g silica gel cartridge; gradient elution from 0:100 MeOH:DCM to 15:85 MeOH:DCM) to give 4-((R)-3-hydroxy-3-methylpiperidin-1-yl)-2-(methylsulfonyl)-5,6',7',8-tetrahydro-5'H- spiro[pyrano[4,3-d]pyrimidine-7,8'-quinoline]-1'-oxide (208 mg). LCMS: m/z (ESI) [M+H]
+ 461.3, tR = 1.38 minutes (Method C) Step 3: 2-(((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4- ((R)-3-hydroxy-3-methylpiperidin-1-yl)-5,6',7',8-tetrahydro-5'H-spiro[pyrano[4,3- d]pyrimidine-7,8'-quinoline]-1'-oxide ((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (62 mg, 0.9 equiv, 0.39 mmol) was added to a stirred solution of 4-((R)-3-hydroxy-3-methylpiperidin-1-yl)-2- (methylsulfonyl)-5,6',7',8-tetrahydro-5'H-spiro[pyrano[4,3-d]pyrimidine-7,8'-quinoline]-1'- oxide (200 mg, 1 equiv, 0.43 mmol) in THF (1.7 mL). The resulting mixture was cooled to – 20 °C, and potassium tert-butoxide (868 μL, 1 M, 2 equiv, 0.87 mmol) was added dropwise. The reaction mixture was stirred at –20 °C for 1 hour and at 0 °C for 1 hour. The reaction was quenched by saturated ammonium chloride and diluted with EtOAc. The aqueous layer was extracted with EtOAc (× 2) and with DCM (× 3). The combined organic layers were dried over sodium sulfate, filtered, and concentrated to a residue, which was purified by flash column chromatography (RediSep Rf Gold 24 g silica gel cartridge; gradient elution from 0:100- premixed solution made up of 2.5% NH4OH in 20% MeOH/DCM:DCM to 60:40-premixed solution made up of 2.5% NH
4OH in 20% MeOH/DCM:DCM) to afford 2-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4-((R)-3-hydroxy-3-methylpiperidin-1- yl)-5,6',7',8-tetrahydro-5'H-spiro[pyrano[4,3-d]pyrimidine-7,8'-quinoline]-1'-oxide (160 mg). LCMS: m/z (ESI) [M+H]
+ 540.3, t
R = 1.05 minutes (Method C) Step 4: (3R)-1-(2'-Amino-2-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-5,6',7',8-tetrahydro-5'H-spiro[pyrano[4,3-d]pyrimidine-7,8'-quinolin]-4- yl)-3-methylpiperidin-3-ol Pyridine (72 μL, 10 equiv, 0.89 mmol) was added to a stirred solution of 2-(((2R,7aS)- 2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4-((R)-3-hydroxy-3-methylpiperidin-
1-yl)-5,6',7',8-tetrahydro-5'H-spiro[pyrano[4,3-d]pyrimidine-7,8'-quinoline]-1'-oxide (50 mg, 1 equiv, 0.09 mmol) in DCM (0.8 mL). Trifluoroacetic anhydride (75 μL, 6 equiv, 0.53 mmol) was added dropwise, and the resulting mixture was stirred at room temperature for 1 hour. The reaction mixture was concentrated to dryness and diluted with ethanol (0.8 mL) and ethanolamine (108 μL, 20 equiv, 1.78 mmol). This mixture was stirred at room temperature for 16 hours. The reaction mixture was concentrated to remove ethanol, and the resulting residue was diluted with DCM and water. The aqueous layer was extracted with DCM (× 4). The combined organic layers were dried over sodium sulfate, filtered, and concentrated to a residue, which was purified by flash column chromatography (RediSep Rf Gold 12 g silica gel cartridge; gradient elution from 0:100-premixed solution made up of 2.5% NH
4OH in 20% MeOH/DCM:DCM to 20:80 to 40:60 premixed solution made up of 2.5% NH
4OH in 20% MeOH/DCM:DCM; desired product elutes around 30%) to give (3R)-1-(2'-amino-2- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-5,6',7',8-tetrahydro-5'H- spiro[pyrano[4,3-d]pyrimidine-7,8'-quinolin]-4-yl)-3-methylpiperidin-3-ol (37 mg). LCMS: m/z (ESI) [M+H]
+ 539.3, tR = 1.44 minutes (Method E) 1H NMR (400 MHz, CDCl3) δ 7.20 (d, 1H), 6.40 (d, 1H), 5.36 – 5.16 (m, 1H), 4.70 – 4.53 (m, 2H), 4.26 (d, 2H), 4.13 (dd, 1H), 4.01 (t, 1H), 3.80 – 3.58 (m, 3H), 3.49 (dd, 1H), 3.35 – 3.11 (m, 3H), 3.05 – 2.87 (m, 3H), 2.78 – 2.59 (m, 3H), 2.29 – 2.06 (m, 4H), 2.06 – 1.97 (m, 1H), 1.97 – 1.69 (m, 7H), 1.60 – 1.45 (m, 2H), 1.22 (d, 3H). Example 12: 5-(6-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Compound 118a)
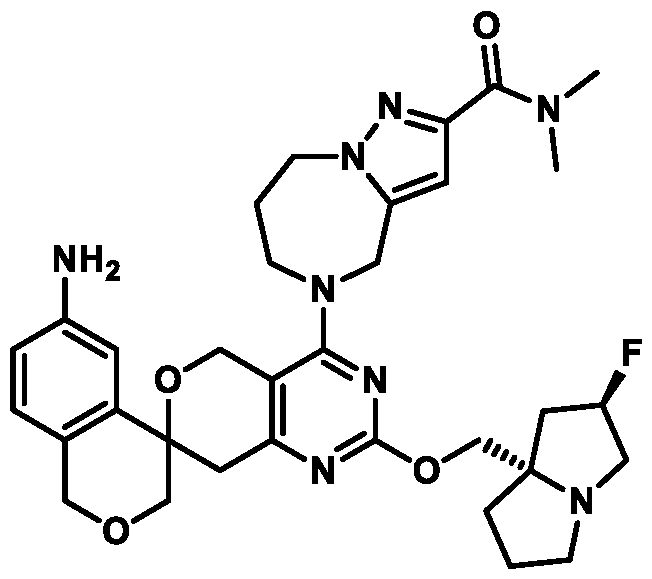
Step 1: 5-(6-bromo-2'-(methylthio)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide A mixture of 6-bromo-4'-chloro-2'-(methylthio)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidine] (200 mg, 1 equiv, 0.48 mmol), N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide, HCl (296 mg, 2.5 equiv, 1.21 mmol), MeCN (5.0 mL), and DIPEA (505 μL, 6.0 equiv, 2.90 mmol) was stirred at 65 °C for 72 hours. The reaction temperature was then increased to 80 °C for 24 hours. The reaction mixture was diluted with ethyl acetate (100 mL) and washed with water (20 mL × 2). The organic layer was then filtered over sodium sulfate and concentrated under reduced pressure to give a residue, which was purified by column chromatography (10 g SiO
2 cartridge, 0-10% methanol in dichloromethane) to afford 5-(6-bromo-2'-(methylthio)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (205 mg). 1H NMR (400 MHz, CDCl3) δ 7.50 (d, 1H), 7.42 (dd, 1H), 6.95 (d, 1H), 6.54 (s, 1H), 4.88 – 4.62 (m, 4H), 4.61 (s, 2H), 4.50 – 4.41 (m, 2H), 3.96 – 3.76 (m, 4H), 3.31 (s, 3H), 3.07 (s, 3H), 3.00 (d, 2H), 2.51 (s, 3H), 2.16 (dd, 2H), 1.59 (s, 2H). Step 2: 5-(6-Bromo-2'-(methylsulfonyl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide Oxone (980 mg, 45 wt%, 2.05 equiv., 0.718 mmol) was added to 5-(6-bromo-2'- (methylthio)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (205 mg, 1.0 equiv, 0.35 mmol) in methanol (5 mL) and water (5 mL). The mixture was stirred at room temperature for 6 hours. The reaction mixture was extracted with ethyl acetate followed by dichloromethane. The combined organic layers were washed with water, dried over sodium sulfate, and concentrated to yield 5-(6-bromo-2'-(methylsulfonyl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (197 mg) which was used without further purification. LCMS: m/z (ESI) [M+H]
+ 619.3, tR = 2.46 minutes (Method E)
Step 3: 5-(6-bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide NaH (51.0 mg, 60 wt%, 4 equiv, 1.28 mmol) was added to ((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (254 mg, 5 equiv, 1.60 mmol) in dry THF (5 mL) and stirred for 10 minutes. 5-(6-Bromo-2'-(methylsulfonyl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (197 mg, 1 equiv, 0.319 mmol) in dry THF (5 mL) was added, and the resultant mixture was stirred for 2 hours at ambient temperature. The reaction mixture was diluted with ethyl acetate (100 mL) and washed with water (20 mL × 2). The organic layer was then filtered over sodium sulfate and concentrated under reduced pressure to a residue, which was purified by column chromatography (25 g SiO2 cartridge, 0-20% methanol in dichloromethane) to give 5-(6-bromo-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (181 mg). 1H NMR (400 MHz, CDCl
3) δ 7.49 (m, 1H), 7.43 – 7.39 (m, 1H), 6.94 (m, 1H), 6.55 (s, 1H), 5.27 (m, 1H), 4.85 – 4.75 (m, 2H), 4.72 (m, 1H), 4.67 – 4.60 (m, 3H), 4.48 – 4.43 (m, 2H), 4.06 (m, 2H), 3.93 – 3.80 (m, 4H), 3.30 – 3.17 (m, 6H), 3.07 (s, 3H), 2.98 (m, 3H), 2.29 – 2.09 (m, 5H), 1.96 (m, 3H). 1
9F NMR (376 MHz, CDCl
3) δ -173.00. Step 4: tert-Butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6- yl)carbamate A degassed suspension of 5-(6-bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (30 mg, 1.0 equiv, 0.043 mmol), tert-butyl carbamate (10 mg, 2.0 equiv, 0.086 mmol), Pd(OAc)2 (1.9 mg, 0.2 equiv, 0.086 mmol), cesium carbonate (42 mg, 3.0 equiv, 0.130 mmol), and 2- (dicyclohexylphosphanyl)-2',4',6'-tris(isopropyl)biphenyl (8.2 mg, 0.4 equiv, 0.017 mmol) in dry 1,4-dioxane (1.0 mL) was stirred at 100 °C for 16 hours. The reaction mixture was diluted
with dichloromethane (100 mL), filtered over sodium sulfate
, and concentrated to a residue, which was purified by column chromatography (10 g SiO2 cartridge, 0-20% methanol in dichloromethane) to afford tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6- yl)carbamate (36 mg, 0.049 mmol). 1H NMR (400 MHz, CDCl
3) δ 7.41 – 7.33 (m, 1H), 7.29 – 7.25 (m, 1H), 7.02 – 6.91 (m, 2H), 5.39 (s, 2H), 4.87 – 4.74 (m, 2H), 4.71 – 4.55 (m, 4H), 4.48 – 4.39 (m, 2H), 4.20 (m, 2H), 3.95 (m, 1H), 3.87 – 3.73 (m, 3H), 3.52 (s, 1H), 3.32 (m, 5H), 3.06 (s, 3H), 3.01 – 2.87 (m, 2H), 2.39 – 2.20 (m, 3H), 2.14 (m, 2H), 2.00 (m, 3H), 1.43 (m, 9H). Step 5: 5-(6-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide HCl (150 μL, 4.0 M in 1,4-dioxane, 15 equiv, 0.60 mmol) was added to tert-butyl (4'- (2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (30 mg, 1 equiv, 0.041 mmol) in CDCl3 (1.0 mL). The reaction mixture was stirred at 25 °C for 5 hours and concentrated to give 5-(6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (29 mg). LCMS: m/z (ESI) [M+H]
+ 633.5, tR = 1.22 minutes (Method E) 1H NMR (400 MHz, MeOD) δ 6.85 (m, 1H), 6.70 (m, 2H), 6.61 (s, 1H), 5.42 (s, 1H), 4.80 (m, 2H), 4.76 – 4.67 (m, 4H), 4.48 (m, 2H), 4.39 – 4.29 (m, 2H), 3.90 (m, 5H), 3.68 (m, 3H), 3.07 (m, 3H), 2.94 (m, 2H), 2.66 (s, 5H), 2.50 – 2.42 (m, 2H), 2.32 (m, 1H), 2.22 (m, 2H), 2.13 (m, 3H). Example 12a: 5-((R)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'- yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Compound 118c)
The diastereomers of 5-(6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide were separated by SFC using ChiralCel OJ-H 21 × 250 mm column. Isocratic mobile phase: 35% methanol, 0.25% TEA in CO
2 at a flow rate of 70 mL/min to afford 5-((R)-6-amino-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Peak 1) and 5-((S)-6-amino-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Peak 2). LCMS: m/z (ESI) [M+H]
+ 633.3, t
R = 1.25 minutes (ChiralCel OJ-H, 4.6x100 mm, Mobile phase: 35% methanol with 0.25% DEA in CO2; 2.5 mL/min). 1H NMR (400 MHz, MeOD) δ 6.86 (m, 1H), 6.71 – 6.67 (m, 2H), 6.58 (s, 1H), 5.28 (m, 1H), 4.82 – 4.65 (m, 7H), 4.49 (m, 2H), 4.22 (m, 1H), 4.11 (m, 1H), 4.05 (m, 1H), 3.97 – 3.83 (m, 4H), 3.27 – 3.16 (m, 4H), 3.07 (s, 3H), 2.90 (m, 2H), 2.34 – 2.25 (m, 1H), 2.22 – 2.07 (m, 6H), 1.96 (m, 3H), 1.89 – 1.83 (m, 1H). Example 12b: 5-((S)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'- yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Compound 118b)
5-((S)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide was obtained from Example 12a, as Peak 2: by SFC using ChiralCel OJ-H 21 × 250 mm column. Isocratic mobile phase: 35% methanol, 0.25% TEA in CO2 at a flow rate of 70 mL/min. LCMS: m/z (ESI) [M+H]
+ 633.3, t
R = 1.99 minutes. (ChiralCel OJ-H, 4.6x100 mm, Mobile phase: 35% Methanol with 0.25% DEA in CO
2; 2.5 mL/min). 1H NMR (400 MHz, MeOD) δ 6.85 (m, 1H), 6.69 (m, 2H), 6.58 (s, 1H), 5.27 (m,1H), 4.82 – 4.65 (m, 7H), 4.49 (m, 2H), 4.13 – 4.02 (m, 2H), 3.88 (m, 5H), 3.26 – 3.14 (m, 3H), 3.07 (s, 3H), 3.03 – 2.87 (m, 4H), 2.28 – 2.06 (m, 6H), 2.01 – 1.83 (m, 4H). Example 13: 1-(7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)azepane-4-carbonitrile (Compound 149a)
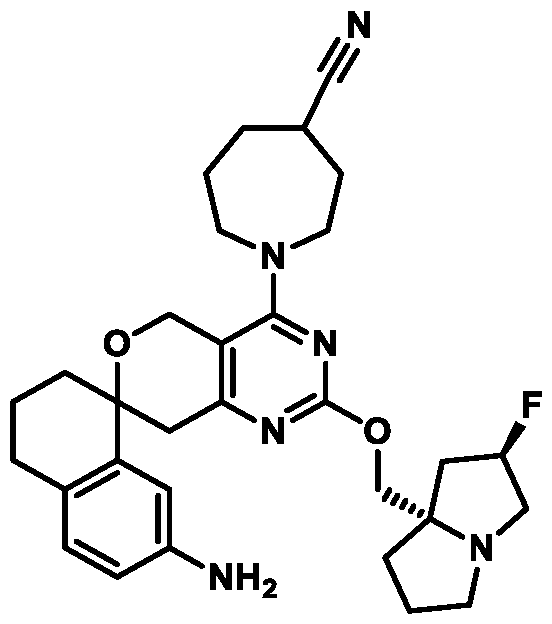
1-(7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4- carbonitrile was prepared according to General Procedure A using the p-toluenesulfonic acid salt of azepane-4-carbonitrile salt (40 mg, 3 equiv, 0.32 mmol). The material was purified by reverse-phase preparative purification with a C-18 Biotage column in basic buffer to afford 1-
(7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile (20 mg). LCMS: m/z (ESI) [M+H]
+ 547.3, t
R = 4.04 minutes (Method I) 1H NMR (400 MHz, DMSO-d6) δ 6.74 (d, 1H), 6.56 (t, 1H), 6.47 – 6.37 (m, 1H), 5.24 (d, 1H), 4.85 – 4.64 (m, 3H), 4.48 (dd, 1H), 3.99 – 3.90 (m, 1H), 3.89 – 3.78 (m, 1H), 3.68 – 3.41 (m, 4H), 3.14 – 2.93 (m, 4H), 2.90 – 2.69 (m, 3H), 2.65 – 2.51 (m, 2H), 2.14 – 2.04 (m, 2H), 2.02 – 1.67 (m, 13H), 1.62 (q, 1H). Example 13a: (S*)-1-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile (Compound 149b)

Step 1: Benzyl rel-(S)-4-cyanoazepane-1-carboxylate Benzyl chloroformate (0.80 mL, 2.0 equiv, 7.0 mmol) was added to a solution of azepane-4-carbonitrile (0.35 g, 1 equiv, 2.8 mmol) and DIPEA (1.2 mL, 2.5 equiv, 7.0 mmol) in dichloromethane (1.5 mL) at room temperature. The reaction mixture was stirred for 16 hours. Afterward, the reaction mixture was concentrated to a residue, which was purified by flash chromatography (12 g Redisep Column using 25-30% hexanes in ethyl acetate) to afford benzyl 4-cyanoazepane-1-carboxylate (0.13 g). The enantiomers of benzyl 4- cyanoazepane-1-carboxylate were separated by chiral SFC (Column: i-Amylose-3; 21.2×250 mm; 5 mm; Mobile Phase A: 50% MeOH at 30 mL/min for 10 minutes) to afford benzyl rel- (S)-4-cyanoazepane-1-carboxylate (Peak 1) and benzyl rel-(R)-4-cyanoazepane-1-carboxylate (Peak 2). Peak 1: HPLC: tR = 1.74 minutes (Column: i-Amylose-3, 4.6x250 mm; 5 mm; Mobile Phase A: 50% MeOH @ 4 mL/min for 5 minutes)
Step 2: rel-(S)-azepane-4-carbonitrile Palladium hydroxide on carbon (90 mg, 20 wt%, 0.3 equiv, 0.13 mmol) was added to benzyl rel-(S)-4-cyanoazepane-1-carboxylate (Peak 1, 110 mg, 1 equiv, 0.43 mmol) in THF (3 mL) at room temperature and stirred for 3 hours under hydrogen atmosphere. The reaction mixture was purged with nitrogen, filtered through a Celite bed, concentrated, and dried to afford rel-(S)-azepane-4-carbonitrile (45 mg, 0.36 mmol). 1H NMR (400 MHz, CDCl
3): δ 2.97-2.82 (m; 4H), 2.09-1.55 (m; 6 H). Step 3: (S*)-1-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)azepane-4-carbonitrile (S*)-1-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)azepane-4-carbonitrile was prepared according to General Procedure A using rel-(S)- azepane-4-carbonitrile (40 mg, 0.32 mmol) to afford (S*)-1-((S)-7-amino-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile (15 mg). LCMS: ESI [M+H]
+ = 547.3 m/z; t
R = 4.07 minutes. (Method I) 1H NMR (400 MHz, DMSO-d6): δ 6.74 (d, 1 H), 6.56 (d, 1 H), 6.42 (dd, 1 H), 5.24 (d, 1 H), 4.48-4.76 (m, 4 H), 3.88 (dd, 2 H), 3.40-3.66 (m, 3H), 2.70-3.04 (m, 6 H), 2.58 (s, 2 H), 1.61-2.05 (m, 16 H). Example 13b: (R*)-1-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile (Compound 149c)
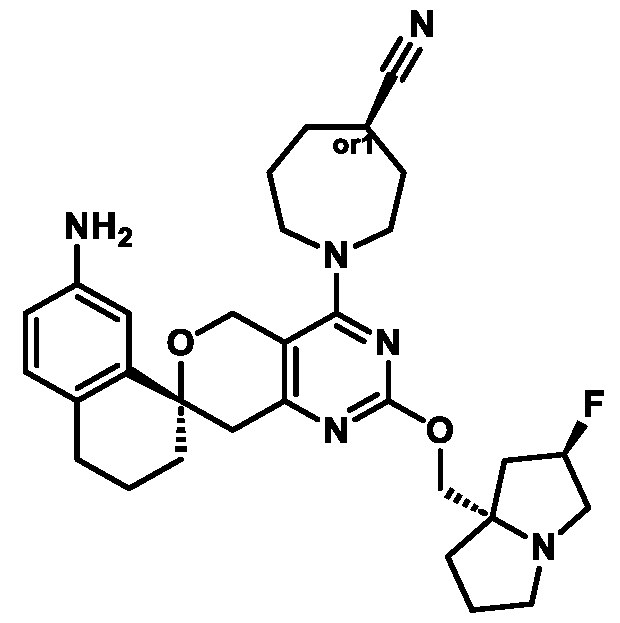
Step 1: Benzyl rel-(R)-4-cyanoazepane-1-carboxylate
Benzyl chloroformate (0.80 mL, 2.0 equiv, 7.0 mmol) was added to a solution of azepane-4-carbonitrile (0.35 g, 1 equiv, 2.8 mmol) and DIPEA (1.2 mL, 2.5 equiv, 7.0 mmol) in dichloromethane (1.5 mL) at room temperature. The reaction mixture was stirred for 16 hours. Afterward, the reaction mixture was concentrated to a residue, which was purified by flash chromatography (12 g Redisep Column using 25-30% hexanes in ethyl acetate) to afford benzyl 4-cyanoazepane-1-carboxylate (0.13 g). The enantiomers of benzyl 4- cyanoazepane-1-carboxylate were separated by chiral SFC (Column: i-Amylose-3; 21.2×250 mm; 5 mm; Mobile Phase A: 50% MeOH at 30 mL/min for 10 minutes) to afford benzyl rel- (S)-4-cyanoazepane-1-carboxylate (Peak 1) and benzyl rel-(R)-4-cyanoazepane-1-carboxylate (Peak 2). Peak 2: HPLC: t
R = 2.16 minutes (Column: i-Amylose-3, 4.6x250 mm; 5 mm; Mobile Phase A: 50% MeOH @ 4 mL/min for 5 minutes) Step 2: rel-(R)-azepane-4-carbonitrile Palladium hydroxide on carbon (90 mg, 20 wt%, 0.3 equiv, 0.13 mmol) was added to benzyl rel-(R)-4-cyanoazepane-1-carboxylate (Peak 2, 110 mg, 1 equiv, 0.43 mmol) in THF (3 mL) at room temperature and stirred for 3 hours under hydrogen atmosphere. The reaction mixture was purged with nitrogen, filtered through a Celite bed, concentrated, and dried to afford rel-(R)-azepane-4-carbonitrile (45 mg, 0.36 mmol). 1H NMR (400 MHz, CDCl
3): δ 3.03-2.58 (m; 4H), 2.07-1.45 (m; 6 H). Step 3: (R*)-1-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)azepane-4-carbonitrile (R*)-1-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)azepane-4-carbonitrile was prepared according to General Procedure A using rel-(R)- azepane-4-carbonitrile (35 mg, 0.28 mmol) to afford (R*)-1-((S)-7-amino-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile (2.5 mg). LCMS: ESI [M+H]
+ = 547.4 m/z; t
R = 4.04 minutes. (Method I)
1H NMR (400 MHz, CD
3OD): δ 6.89 (d, 1 H), 6.74 (d, 1 H), 6.63 (dd, 1 H), 5.34-5.49 (m, 1 H), 4.84-4.93 (m, 1 H), 4.61 (d, 1 H), 4.24-4.37 (m, 2 H), 3.49-3.85 (m, 6 H), 3.06-3.20 (m, 2 H), 2.84-3.05 (m, 2 H), 2.68-2.75 (m, 2 H), 1.81-2.34 (m, 17 H). Example 14: 2'-(((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-amine (Compound 136a)

2'-(((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4- oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- amine was prepared according to General Procedure A using tert-butyl (2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-oxo-3,3',4,4',5',8'-hexahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (150 mg, 0.23 mmol) and 1,4- oxazepane (35 mg, 0.34 mmol, 1.5 equiv.). The reaction mixture was purified by preparative HPLC (Gemini 5 mm NX-C18, 110 Å, 100×30 mm, 45 mL/min; 10 mM NH4CO3/MeCN; 35- 100%) to afford 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4- oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- amine (20 mg). LCMS: m/z (ESI) [M+H]
+ 524.3, tR = 1.57 minutes (Method B) 1H NMR (400 MHz, DMSO-d
6): δ 6.76 (d, 1H); 6.57 (s, 1H), 6.43 (dd, 1H), 5.19-5.34 (m, 1H), 4.80 (s, 2H), 4.58 (dd, 2H), 3.93 (dd, 1H), 3.83 (dd, 1H), 3.59-3.70 (m, 8H), 3.06 (s, 2H), 2.99 (s, 1H), 2.74-2.89 (m, 3H), 2.55-2.67 (m, 3H), 1.69-2.04 (m, 11H). Example 14a: (S)-2'-(((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-amine (Compound 136b)
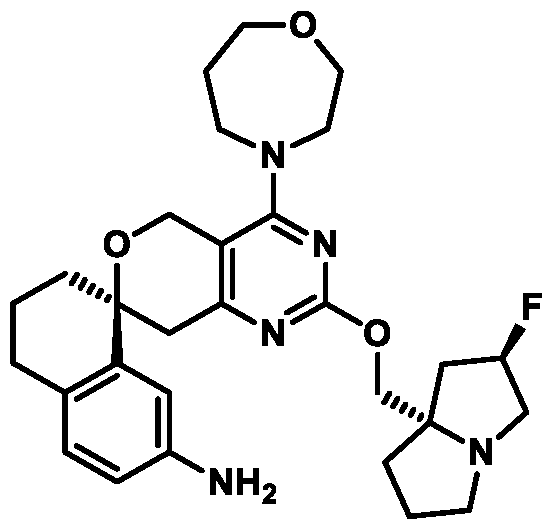
(S)-2'-(((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4- oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- amine was prepared according to General Procedure A using tert-butyl ((S)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-oxo-3,3',4,4',5',8'-hexahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (200 mg, 0.30 mmol) and 1,4- oxazepane (46 mg, 0.45 mmol, 1.5 equiv.) and purified by preparative HPLC (C18 Biotage 12 g column; 10 mM NH4CO3/MeCN) to afford (S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (31 mg). LCMS: ESI [M+H]
+ 524.4 m/z, t
R = 3.85 minutes (Method I) 1H NMR (400 MHz, DMSO-d6) δ 6.73 (d, 1H), 6.54 (d, 1H), 6.41 (dd, 1H), 5.23 (d, 1H), 4.75 (s, 2H), 4.66 (d, 1H), 4.44 (d, 1H), 3.91 (d, 1H), 3.80 (d, 1H), 3.69 (d, 2H), 3.67 – 3.46 (m, 5H), 3.15 – 2.92 (m, 3H), 2.91 – 2.68 (m, 3H), 2.56 (t, 2H), 2.17 – 1.96 (m, 2H), 1.96 – 1.52 (m, 9H). Example 15: 5-(7-Amino-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 158a)

Step 1: 5-(8-Bromo-7-(dibenzylamino)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide DIPEA (1.70 g, 2.30 mL, 8 equiv, 13.2 mmol) was added to a suspension of N,N- dibenzyl-8-bromo-4'-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-amine (1.00 g, 1 equiv, 1.65 mmol) and N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide hydrochloride (605 mg, 1.5 equiv, 2.47 mmol) in DMF (33 mL) at room temperature. The mixture was heated at 120 °C for 18 hours, cooled to room temperature, and diluted with water. The aqueous layer was extracted with EtOAc three times, and the combined organic layers were washed with brine, dried over sodium sulfate, filtered, and concentrated to a residue, which was purified by flash column chromatography (80 g, 0-50% DCM/EtOAc to 0-10% DCM/methanol) to give 5- (8-bromo-7-(dibenzylamino)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (719 mg). LCMS: m/z (ESI) [M+H]
+ 778.3, tR = 2.15 minutes (Method B). Step 2: 5-(8-Cyano-7-(dibenzylamino)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide Copper (I) cyanide (578 mg, 10 equiv, 6.46 mmol) was added to a solution of 5-(8- bromo-7-(dibenzylamino)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (503 mg, 1 equiv, 0.646 mmol) in DMF (12.9 mL) and stirred at 120 °C for 5 hours. The reaction mixture was cooled to room temperature and then concentrated to a residue, which was treated with DCM/water. A solution of concentrated NH
4OH/MeOH (1:1) was added until a biphasic solution was observed. The mixture was stirred for 30 minutes, and the aqueous layer was extracted with DCM three times. The combined organic layers were washed with brine, dried over sodium sulfate, filtered, and concentrated to yield 5-(8-cyano-7-(dibenzylamino)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (440 mg) which was used without further purification.
LCMS: m/z (ESI) [M+H]
+ 725.5, t
R = 1.94 minutes (Method B). Step 3: 5-(8-cyano-7-(dibenzylamino)-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide 3-Chlorobenzoperoxoic acid (370 mg, 85 wt%, 3 equiv, 1.82 mmol) was added to a solution of 5-(8-cyano-7-(dibenzylamino)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (440 mg, 1 equiv, 0.61 mmol) in DCM (12.1 mL) at room temperature. The resulting yellow solution was stirred at room temperature for 1 hour and then diluted with DCM. A solution of saturated sodium bicarbonate and saturated sodium thiosulfate (10:1) was added, and the mixture was stirred at room temperature for 1 hour. The organic layer was separated, and the aqueous layer was extracted with DCM. The combined organic layers were washed with saturated potassium carbonate and brine, dried over sodium sulfate, filtered, and concentrated to give 5-(8-cyano-7-(dibenzylamino)-2'- (methylsulfonyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (358 mg) as a foam, which was used without further purification. LCMS: m/z (ESI) [M+H]
+ 757.3, tR = 1.70 minutes (Method B). Step 4: 5-(8-cyano-7-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide ((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (226 mg, 3 equiv, 1.42 mmol) was added to a suspension of sodium hydride (56.8 mg, 60 wt%, 3 equiv, 1.42 mmol) in THF (9.5 mL) under argon at room temperature. The mixture was stirred at room temperature for 10 minutes. Then a solution of 5-(8-cyano-7-(dibenzylamino)-2'- (methylsulfonyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (358 mg, 1 equiv, 0.47 mmol) in THF (2 mL) was added. The resulting orange solution was stirred at room temperature for 10 minutes. The mixture was then quenched with ice water and extracted with EtOAc three times. The combined organic layers were washed with brine, dried
over sodium sulfate, filtered, and concentrated to a residue, which was purified by reverse- phase chromatography (30 g Biotage C18 silica, 0 - 60% (4 cv), 60% -100% (10 cv) MeCN/0.1% aqueous NH4HCO3) to provide 5-(8-cyano-7-(dibenzylamino)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (273 mg). LCMS: m/z (ESI) [M+H]
+ 836.5, t
R = 1.79 minutes (Method B). Step 5: 5-(7-Amino-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide Palladium (II) hydroxide (105 mg, 20 wt%, 2.5 equiv, 0.15 mmol) was added to a solution of 5-(8-cyano-7-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]- 4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (50 mg, 1 equiv, 0.06 mmol) in MeOH (1.2 mL). The mixture was then sparged with argon. The inert atmosphere was exchanged with hydrogen by degassing the reaction mixture under reduced pressure with backflow of hydrogen from a balloon. Four drops of acetic acid were added, and the black reaction mixture was stirred overnight under hydrogen atmosphere at room temperature. Celite was added, and the atmosphere was exchanged for argon. The suspension was filtered over Celite with MeOH and concentrated. The crude residue was purified by reverse-phase chromatography (6 g Biotage C18 silica, 0 - 60% (4 cv), 60% -100% (10 cv) MeCN/0.1% aqueous NH
4HCO
3) to provide 5-(7-amino-8-cyano-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (17 mg). LCMS: m/z (ESI) [M+H]
+ 656.5, t
R = 1.26 minutes (Method B). 1H NMR (400 MHz, DMSO-d6) δ 7.01 (d, 1H), 6.70 (d, 1H), 6.51 (s, 1H), 5.70 (s, 2H), 5.21 (d, 1H), 4.95 (d, 1H), 4.79 – 4.52 (m, 3H), 4.43 (t, 2H), 3.95 – 3.70 (m, 4H), 3.22 (s, 3H), 3.09 – 2.97 (m, 3H), 2.98 – 2.73 (m, 6H), 2.61 – 2.51 (m, 2H), 2.21 – 1.86 (m, 6H), 1.84 – 1.54 (m, 4H).
Example 15a: 5-((R)-7-amino-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 158b)

The diastereomers of 5-(7-amino-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide were separated by chiral preparative HPLC (Column: Gemini 5 mm NX-C18; 30 × 100 mm; Mobile phase A: 10 mM ammonium bicarbonate pH = 10.0; Mobile phase B: MeOH; Gradient: 55% to 45% B) to afford 5-((R)-7-amino-8-cyano-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Peak 1) (10 mg) and 5-((S)-7-amino-8-cyano- 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Peak 2) (8 mg). Peak 1: Chiral HPLC: t
R = 5.12 minutes. (Column: Cellulose-1, 4.6 × 250 mm; 5 mM; Mobile Phase: 40% MeOH with 0.1% NH4OH over 10 minutes.) 1H NMR (400 MHz, DMSO-d6): δ 7.04 (d, 1 H), 6.73 (d, 1 H), 6.54 (s, 1 H), 5.73 (s, 2 H), 5.34-5.10 (m, 1 H), 4.98 (d, 1 H), 4.72 (t, 2 H), 4.62 (t, 1 H), 4.45 (t, 2 H), 3.92 (d, 2 H), 3.83 (d, 2H), 3.11 (s, 1 H), 3.01-3.07 (m, 3 H), 2.99 (s, 1 H), 2.93 (s, 3 H), 2.78-2.86 (m, 2 H), 2.60 (s, 3 H), 2.14 (d, 2 H), 2.05-2.07 (m, 1 H), 1.96 (d, 3 H), 1.72-1.82 (m, 5 H). Example 15b: 5-((S)-7-amino-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-
pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 158c)
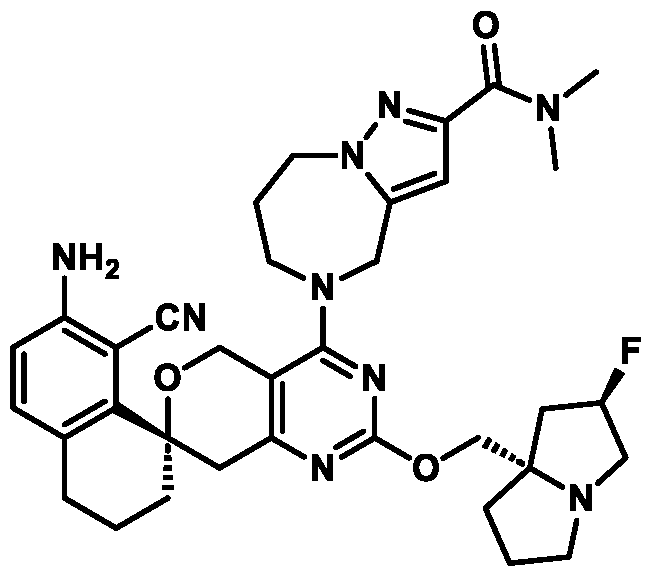
5-((S)-7-amino-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8 mg) was obtained from Example 15a, as Peak 2: Chiral HPLC: tR = 6.38 minutes. (Column: Cellulose-1, 4.6 × 250 mm; 5 mM; Mobile phase: 40% MeOH with 0.1% NH
4OH over 10 minutes.) 1H NMR (400 MHz, DMSO-d6): δ 7.04 (d, 1 H), 6.73 (d, 1 H), 6.54 (s, 1 H), 5.73 (s, 2 H), 5.34-5.07 (m, 1 H), 4.98 (d, 1 H), 4.72 (t, 2 H), 4.62 (t, 1 H), 4.45 (t, 2 H), 3.92 (d, 2 H), 3.83 (d, 2H), 3.11 (s, 1 H), 3.01-3.07 (m, 3 H), 2.99 (s, 1 H), 2.93 (s, 3 H), 2.78-2.86 (m, 2 H), 2.60 (s, 3 H), 2.14 (d, 2 H), 2.05-2.07 (m, 1 H), 1.96 (d, 3 H), 1.72-1.82 (m, 5 H). Example 16: 7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine]-8-carbonitrile (Compound 161a)
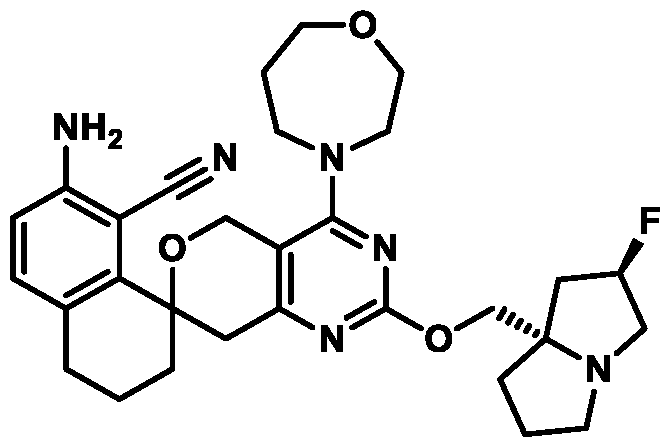
Step 1: N,N-Dibenzyl-8-bromo-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine DIPEA (1.49 mL, 8 equiv, 8.57 mmol) was added to a suspension of N,N-dibenzyl-8- bromo-4'-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-amine (650 mg, 1 equiv, 1.07 mmol) and 1,4-oxazepane (186 μL, 96 wt%, 1.5
equiv, 1.61 mmol) in DMF (21.4 mL) at room temperature. The mixture was heated at 120 °C for 2 hours, cooled to room temperature, and diluted with water. The aqueous layer was extracted with EtOAc (3 ×). The combined organic layers were washed with brine, dried over sodium sulfate, filtered, and concentrated. The resulting residue was purified by flash chromatography (24 g, 0-20% DCM/EtOAc) to give N,N-dibenzyl-8-bromo-2'-(methylthio)- 4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-amine (235 mg). LCMS: ESI [M+H]
+ = 673.2 m/z; tR = 2.12 minutes. (Method B) Step 2: 7-(Dibenzylamino)-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile Copper (I) cyanide (256 mg, 10 equiv, 2.86 mmol) was added to N,N-dibenzyl-8- bromo-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-amine (192 mg, 1 equiv, 0.29 mmol) in DMF (5.7 mL) and stirred at 120 °C for 2 hours. The reaction mixture was cooled to room temperature and concentrated to a residue which was treated with DCM/water to form a precipitate. A solution of concentrated NH
4OH/MeOH (1:1) was added until no solid was visible, and the mixture was stirred for 30 minutes. Then the aqueous layer was extracted with DCM three times. The combined organic layers were washed with brine, dried over sodium sulfate, filtered, and concentrated to afford 7-(dibenzylamino)-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (162 mg), which was used in the next step without further purification. LCMS: m/z (ESI) [M+H]
+ 618.4, tR = 1.93 minutes (Method B). Step 3: 7-(Dibenzylamino)-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile mCPBA (160 mg, 85 wt%, 3 equiv, 0.79 mmol) was added to the solution of 7- (dibenzylamino)-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (162 mg, 1 equiv, 0.26 mmol) in DCM (5.2 mL) and stirred at room temperature for 1 hour. The reaction mixture was diluted with DCM, and a solution of saturated sodium bicarbonate and saturated sodium thiosulfate (10:1) was added. The mixture was stirred at room temperature for 1 hour. The organic layer was separated, and the aqueous layer was extracted with DCM. The combined
organic layers were washed with saturated potassium carbonate followed by brine. The organic layers were dried over sodium sulfate, filtered, and concentrated to give 7-(dibenzylamino)-2'- (methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine]-8-carbonitrile (150 mg) which was used in the next step without further purification. LCMS: m/z (ESI) [M+H]
+ 650.4, tR = 1.73 minutes (Method B). Step 4: 7-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile ((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (110 mg, 3 equiv, 0.69 mmol) was added to a suspension of sodium hydride (27.7 mg, 60 wt%, 3 equiv, 0.69 mmol) in THF (4.6 mL) and stirred at room temperature for 10 minutes. A solution of 7- (dibenzylamino)-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (150 mg, 1 equiv, 0.23 mmol) in THF (0.5 mL) was added over 5 minutes. The mixture was then stirred at room temperature for 20 minutes. The reaction was quenched with ice water and extracted with EtOAc three times. The combined organic layers were washed with brine, dried over sodium sulfate, filtered, and concentrated to a residue, which was purified by reverse-phase chromatography (30 g Biotage C18 silica, 0 - 40% (4 cv), 40% -100% (8 cv) MeCN/0.1% aqueous NH
4HCO
3) to provide 7-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (40 mg). LCMS: m/z (ESI) [M+H]
+ 729.5, t
R = 1.88 minutes (Method B). Step 5: 7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine]-8-carbonitrile Palladium(II) hydroxide (94 mg, 20 wt%, 2.5 equiv, 0.13 mmol) was added to 7- (dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4- oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8- carbonitrile (39 mg, 1 equiv, 0.05 mmol) in MeOH (1.1 mL). The mixture was sparged with argon. The inert atmosphere was exchanged with hydrogen by degassing the reaction mixture
under reduced pressure and back-filled with hydrogen from a balloon. Four drops of acetic acid were added to the reaction mixture, and the black reaction mixture was stirred overnight under hydrogen atmosphere at room temperature. Celite was added, and the atmosphere was exchanged for argon. The suspension was filtered over Celite with MeOH and concentrated to a residue, which was purified by reverse-phase chromatography (6 g Biotage C18 silica, 0 - 60% (4 cv), 60% -100% (10 cv) MeCN/0.1% aqueous NH4HCO3) to provide 7-amino-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (16 mg, 29 μmol). LCMS: m/z (ESI) [M+H]
+ 549.3, t
R = 1.32 minutes (Method B). 1H NMR (400 MHz, DMSO-d
6): δ 7.04 (d, 1H), 6.73 (d, 1H), 5.74 (s, 2 H), 5.22 (d, 1 H), 4.88 (d, 1H), 4.66 (d, 1H), 3.84-3.93 (m, 2H), 3.72 (d, 6H), 3.60 (br s, 2H), 3.06-3.11 (m, 3H), 2.99 (s, 1H), 2.84 (d, 2H), 2.59 (s, 2H), 2.12 (d, 2H), 1.98 (d, 3H), 1.83 (br s, 3H), 1.71 (d, 4H). Example 16a: (S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Compound 161c)
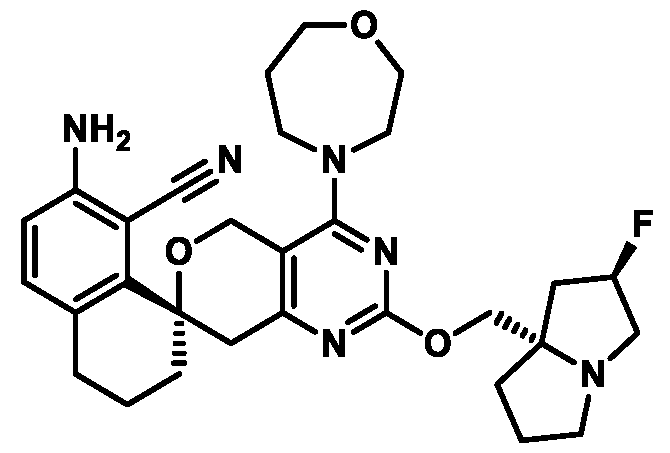
The diastereomers of 7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine]-8-carbonitrile were separated by chiral preparative HPLC (Column: Cellulose-15 ^m; 30 × 250 mm; Mobile phase A: CO2; Mobile phase B: MeOH with 0.1% DEA; Isocratic: 35% B; 100 mL/min;) to afford (S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Peak 1) (49.2 mg) and (R)-7- amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan- 4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Peak 2) (64.7 mg).
Peak 1: Chiral HPLC: tR = 7.31 minutes. (Column: Lux Cellulose-3, 4.6 × 150 mm; 5 ^M; column temperature; 30
oC; Gradient:70% water with 10mM NH4CO3/30% MeOH over 12 minutes.) 1H NMR (400 MHz, DMSO-d
6): δ 7.01 (d, 1H), 6.70 (d, 1H), 5.71 (s, 2H), 5.26 (d, 1H), 4.85 (d, 1H), 4.63 (d, 1H), 4.03 – 3.81 (m, 2H), 3.81 – 3.62 (m, 6H), 3.62 – 3.48 (m, 2H), 3.19 – 2.94 (m, 3H), 2.94 – 2.76 (m, 2H), 2.63 – 2.51 (m, 2H), 2.20 – 2.06 (m, 2H), 2.02 (s, 1H), 2.00 – 1.88 (m, 2H), 1.88 – 1.55 (m, 6H). Example 16b: (R)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Compound 161b)
(R)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'- (1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]- 8-carbonitrile (64.7 mg) was obtained from Example 16a, as Peak 2: Chiral HPLC: tR = 8.68 minutes. (Column: Lux Cellulose-3, 4.6 × 150 mm; 5 ^M; column temperature; 30
oC; Gradient:70% water with 10mM NH
4CO
3/30% MeOH over 12 minutes.) 1H NMR (400 MHz, DMSO-d6): δ 7.01 (d, 1H), 6.69 (d, 1H), 5.72 (s, 2H), 5.22 (d, 1H), 4.85 (d, 1H), 4.63 (d, 1H), 3.90 (d, 1H), 3.81 (d, 1H), 3.78 – 3.61 (m, 4H), 3.61 – 3.48 (m, 2H), 3.10 – 3.00 (m, 3H), 3.00 – 2.93 (m, 1H), 2.84 (s, 1H), 2.82 – 2.72 (m, 1H), 2.61 – 2.52 (m, 2H), 2.20 – 2.08 (m, 1H), 2.08 – 2.02 (m, 1H), 2.02 – 1.87 (m, 3H), 1.88 – 1.60 (m, 6H), 1.56 (s, 1H). Example 17: 5-(7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide (Compound 124a)

Step 1: 7-Bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-4-methyl-1,2,3,4-tetrahydronaphthalen-1-ol LDA solution (2.81 mL, 1 molar, 2.3 equiv, 2.81 mmol) was added dropwise to (4- chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (250 mg, 1 equiv, 1.22 mmol) at -78 °C, maintaining a temperature below -74 °C. The solution was stirred at -78 °C for 30 minutes.7-Bromo-4-methyl-3,4-dihydronaphthalen-1(2H)-one (321 mg, 1.1 equiv, 1.34 mmol) was dissolved in THF (0.75 mL) and added dropwise to the solution, keeping the reaction mixture below -74 °C. After 30 minutes, equal volume of saturated ammonium chloride solution was added, and the reaction mixture was stirred for 15 minutes. The reaction mixture was then extracted with EtOAc (3 × 100 mL), and then the organic layer was dried using sodium sulfate. The mixture was concentrated to a residue, which was purified by flash chromatography (ISCO silica Gold cartridge; Gradient: 100% DCM to 85:15 DCM:EtOAc) to afford 7-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-4- methyl-1,2,3,4-tetrahydronaphthalen-1-ol (320 mg). 1H NMR (400 MHz, CDCl3) δ 7.71 (m, 1H), 7.39 (m, 1H), 7.13 (m, 1H), 4.86 – 4.72 (m, 2H), 4.05 (m, 1H), 3.49 – 3.16 (m, 2H), 3.06 – 2.81 (m, 2H), 2.57 (m, 3H), 2.29 – 2.14 (m, 1H), 2.09 – 1.96 (m, 1H), 1.93 – 1.64 (m, 2H), 1.34 (m, 3H). Step 2: 7-Bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] To a 100 mL round bottom flask containing 7-bromo-1-((6-chloro-5-(hydroxymethyl)- 2-(methylthio)pyrimidin-4-yl)methyl)-4-methyl-1,2,3,4-tetrahydronaphthalen-1-ol (320 mg, 1 equiv, 0.72 mmol) in toluene (1.34 mL) was added phosphoric acid (49 μL, 85 wt%, 1 equiv, 0.72 mmol). The reaction mixture was heated to reflux for 2 hours. The solvent was removed under reduced pressure, and the resulting residue was partitioned between EtOAc (8 mL) and water (8 mL). The organic phase was separated, and the aqueous was extracted with EtOAc (3 × 4 mL). The combined organics were dried with sodium sulfate, and solvent was removed
under reduced pressure to give 7-bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (285 mg). 1H NMR (400 MHz, CDCl3) δ 7.62 (m, 1H), 7.38 (m, 1H), 7.11 (m, 1H), 5.09 – 4.58 (m, 2H), 3.22 – 2.82 (m, 3H), 2.56 (m, 3H), 2.20 – 1.85 (m, 4H), 1.31 (m, 3H). Step 3: 5-(7-Bromo-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide DIPEA (475 μL, 4 equiv, 2.72 mmol) was added to 7-bromo-4'-chloro-4-methyl-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (290 mg, 1 equiv, 0.68 mmol) and N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide, HCl (310 mg, 1.6 equiv, 1.09 mmol) in ethanol (1.36 mL). The mixture was heated to 85 °C for 16 hours. The reaction mixture was concentrated, and EtOAc (10 mL) was added. The solution was washed with water (10 mL) and brine (10 mL). The combined aqueous layers were extracted with EtOAc (20 mL), and the combined organic layers were dried over sodium sulfate and concentrated to afford 5-(7-bromo-4-methyl-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (361.1 mg). LCMS: ESI [M+H]
+ = 599.3; t
R = 1.98 minutes (Method C). Step 4: tert-Butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate A mixture of tert-butyl carbamate (142 mg, 2 equiv, 1.21 mmol), 5-(7-bromo-4-methyl- 2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (361 mg, 1 equiv, 0.60 mmol), 2-(dicyclohexylphosphanyl)-2',4',6'-tris(isopropyl)biphenyl (25.9 mg, 0.09 equiv, 0.05 mmol), and Pd(OAc)2 (4 mg, 0.03 equiv, 0.02 mmol) in 1,4-dioxane (4.8 mL) was sparged with nitrogen. The reaction mixture was heated to 100 °C for 60 minutes, cooled to room temperature, and filtered. The filtrate was concentrated to a residue, which was purified via column chromatography (SiO2, 100% DCM to 85:15 DCM:MeOH) to give tert- butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-4-
methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (274 mg). 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.09 (m, 3H), 6.73 – 6.25 (m, 2H), 4.89 – 4.37 (m, 6H), 3.87 (m, 2H), 3.34 (m, 3H), 3.21 – 2.85 (m, 6H), 2.50 (s, 3H), 2.29 – 1.82 (m, 6H), 1.45 (d, J = 1.9 Hz, 9H), 1.30 (m, 3H). Step 5: tert-Butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-4-methyl-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate Oxone (1.13 g, 45 wt%, 2.1 equiv, 0.83 mmol) was added to tert-butyl (4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-4-methyl-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (250 mg, 1 equiv, 0.39 mmol) in water (4 mL) and methanol (4 mL). The mixture was stirred for 30 minutes and then quenched by the addition of water (10 mL). EtOAc (10 mL) was added, and the layers were separated. The aqueous phase was extracted with EtOAc (3 × 5 mL), and the combined organics were dried over sodium sulfate, filtered, and concentrated to afford tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-4-methyl-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (109 mg). LCMS: (ESI) m/z = 666.5 [M+H]
+ ; t
R = 1.90 minutes. (Method C). Step 6: tert-Butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate Potassium tert-butoxide in THF (666 μL, 1 M, 2 equiv, 0.67 mmol) was added dropwise to a solution of tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-4-methyl-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (223 mg, 1 equiv, 0.33 mmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (53.0 mg, 1 equiv, 0.33 mmol) in THF (3.3 mL) at 0 °C. The reaction mixture was stirred for 40 minutes followed by the addition of water (1 mL) and EtOAc (2 mL). The aqueous layer was extracted with EtOAc (3 × 4 mL) and the combined organics were dried over sodium sulfate, filtered, and
concentrated to a residue, which was purified by flash chromatography (SiO
2; Gradient: 100% DCM to 85:15 DCM:MeOH) to give tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (77.6 mg). 1H NMR (400 MHz, CDCl3) δ 7.47 – 6.96 (m, 3H), 6.72 – 6.35 (m, 2H), 5.26 (d, J = 53.9 Hz, 1H), 4.86 – 4.34 (m, 6H), 4.13 – 3.71 (m, 4H), 3.42 – 2.86 (m, 13H), 2.29 – 1.77 (m, 12H), 1.48 – 1.38 (m, 9H), 1.30 (dd, J = 7.0, 4.8 Hz, 3H). Step 7: 5-(7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide HCl (350 μL, 4 M, 14 equiv, 1.40 mmol) was added to tert-butyl (4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)- 2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (75 mg, 1 equiv, 0.10 mmol) at 0 °C. The mixture was stirred for 30 minutes, and the reaction mixture was concentrated to afford 5-(7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4- methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide, 3HCl (68 mg). LCMS: ESI [M+H]
+ = 645.5 m/z; tR = 1.16 minutes (Method C) 1H NMR (400 MHz, MeOD) δ 7.59 – 7.42 (m, 2H), 7.39 – 7.28 (m, 1H), 6.49 (d, 1H), 5.60 (d, 1H), 5.12 – 4.88 (m, 2H), 4.81 – 4.47 (m, 5H), 4.31-4.19 (m, 1H), 4.09 – 3.83 (m, 3H), 3.49 – 3.43 (m, 2H), 3.37 – 3.32 (m, 6H), 3.17 – 3.00 (m, 6H), 2.79 – 2.52 (m, 2H), 2.53 – 1.85 (m, 9H), 1.37 (dd, 3H). 1
9F NMR (376 MHz, MeOD) δ -174.31. Example 18: 5-((1S,4S)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 124b)
Step 1: (4S)-7-Bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-4-methyl-1,2,3,4-tetrahydronaphthalen-1-ol LDA solution (2.60 mL, 1 M, 2.3 equiv, 2.60 mmol) was added dropwise to a solution of (4-chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (231 mg, 1 equiv, 1.13 mmol) in THF (4 mL) at -78 °C and stirred for 1.25 hours. To this mixture was added (S)-7- bromo-4-methyl-3,4-dihydronaphthalen-1(2H)-one (2
nd eluting peak) (Intermediate 7b supra) (270 mg, 1 equiv, 1.13 mmol) in THF (3 mL) dropwise, keeping the temperature below -70 °C. The reaction mixture was stirred at -78 °C for 1 hour, at which point the reaction was quenched by aqueous ammonium chloride (10 mL) at -78 °C with vigorous stirring. The reaction mixture was then diluted with EtOAc (15 mL), and the aqueous phase was extracted with EtOAc (3 × 25 mL). The combined organics were dried with sodium sulfate and concentrated to a residue, which was purified via column chromatography (SiO
2; Gradient: 100% DCM - 100% EtOAc) to give (4S)-7-bromo-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-4-methyl-1,2,3,4-tetrahydronaphthalen-1-ol (405 mg). 1H NMR (400 MHz, CDCl
3) δ 7.67 (m, 1H), 7.36 (m, 1H), 7.11 (m, 1H), 4.81 – 4.67 (m, 2H), 3.40 – 3.16 (m, 2H), 2.93 (m, 1H), 2.55 (m, 3H), 2.18 (m, 1H), 2.06 – 1.92 (m, 2H), 1.91 – 1.62 (m, 2H), 1.32 (m, 3H). Step 2: (4S)-7-Bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] – two resolved diastereomers Phosphoric acid (658 μL, 85 wt%, 1 equiv, 9.62 mmol) was added to (4S)-7-bromo-1- ((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-4-methyl-1,2,3,4- tetrahydronaphthalen-1-ol (4.27 g, 1 equiv, 9.62 mmol) in toluene (19.2 mL). The mixture was refluxed for 2 hours. Water (50 mL) and EtOAc (50 mL) were added, and the aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organics were dried with sodium sulfate and concentrated to a residue, which was purified via chiral SFC (Column: Chiralpak IB-U (3.0 ×
100 mm), 1.6 μm, P/N 81U83, flow rate: 1.2 mL/min, Mobile phase: (Methanol with 0.25% DEA)) to afford the two diastereomers of (4S)-7-bromo-4'-chloro-4-methyl-2'-(methylthio)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]: (1S,4S)-7-bromo-4'- chloro-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine] (Peak 1) (2.02 g) and (1R,4S)-7-bromo-4'-chloro-4-methyl-2'-(methylthio)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (Peak 2) (1.78 g). Peak 1: t
R = 1.34 minutes (Method G; 30% methanol with 0.25% DEA) LCMS: ESI [M+H]
+ = 427.0 m/z; tR = 4.14 minutes (Method E) Peak 2: tR = 2.06 minutes (Method G; 30% methanol with 0.25% DEA) 1H NMR (400 MHz, CDCl
3) δ 7.63 (d, 1H), 7.41 (dd, 1H), 7.19 (d, 1H), 4.78 (q, 2H), 3.12 (q, 2H), 3.00 – 2.86 (m, 1H), 2.59 (s, 3H), 2.13 – 1.87 (m, 3H), 1.48 (qd, 1H), 1.34 (d, 3H). Step 3: 5-((1S,4S)-7-Bromo-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide DIPEA (2.31 mL, 13.3 mmol) was added to (1S,4S)-7-bromo-4'-chloro-4-methyl-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (Peak 1) (2.02 g, 1 equiv, 3.32 mmol) and N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide, 2HCl (1.74 g, 1.5 equiv, 4.98 mmol) in ethanol (7 mL). The reaction mixture was heated to reflux for 3 hours. N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (484 mg, 0.7 equiv, 2.32 mmol) and DIPEA (810 μL, 1.4 equiv, 4.65 mmol) were then delivered sequentially. The reaction mixture was heated to reflux for an additional 45 minutes and concentrated under reduced pressure to an oil that was partitioned between water (50 mL) and EtOAc (50 mL). The aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organics were dried over sodium sulfate and concentrated to a residue, which was purified by flash chromatography (SiO
2; Gradient: 0- 100% EtOAc in DCM then switched to 5% MeOH in DCM, 40 g column) to give 5-((1S,4S)- 7-bromo-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (2.44 g). LCMS: ESI [M+H]
+ = 597.3 m/z; tR = 2.97 minutes (Method E)
Step 4: tert-Butyl ((1S,4S)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate 5-((1S,4S)-7-Bromo-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (2.44 g, 1 equiv, 2.98 mmol) was added to BrettPhos Pd G4 (411 mg, 0.15 equiv, 0.45 mmol), tert-butyl carbamate (1.75 g, 5 equiv, 14.9 mmol), and cesium carbonate (2.91 g, 3 equiv, 8.94 mmol) in 1,4-dioxane (40 mL) and sparged for 10 minutes with nitrogen. The reaction mixture was heated to 95 °C for 16 hours and then cooled to room temperature, filtered, and concentrated to a residue, which was purified by flash chromatography (SiO
2220 g: Gradient: 100% DCM to 100% EtOAc then switched to MeOH and ramped from 0-7.5%) to afford tert-butyl ((1S,4S)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro- 4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (1.61 g). LCMS: ESI [M+H]
+ = 634.3 m/; tR = 2.83 minutes (Method E) Step 5: tert-Butyl ((1S,4S)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-4-methyl-2'-(methylsulfonyl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate mCPBA (1.67 g, 70 wt%, 3 equiv, 6.77 mmol) was added to tert-butyl ((1S,4S)-4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-4-methyl-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (1.57 g, 1 equiv, 2.25 mmol) in DCM (45 mL) at 0 °C. The reaction mixture was stirred for 10 minutes at 0 °C, removed from the ice bath, and warmed to room temperature over 55 minutes. The reaction mixture was diluted with DCM (50 mL) and quenched with sodium thiosulfate (15 mL) and saturated sodium bicarbonate (125 mL). The resulting biphasic mixture was stirred for 30 minutes. The aqueous was separated and extracted with DCM (120 mL). The combined organics were washed with saturated sodium carbonate (2 × 200 mL), dried over sodium sulfate, and concentrated to afford tert-butyl ((1S,4S)-4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-4-methyl-2'- (methylsulfonyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (1.76 g). LCMS: ESI [M+H]
+ = 666.2 m/z; tR = 2.91 minutes (Method E)
Step 6: tert-Butyl ((1S,4S)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate Sodium tert-butoxide (221 mg, 1.15 mL, 2 M, 1.21 equiv, 2.30 mmol) was added dropwise to tert-butyl ((1S,4S)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-4-methyl-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (1.27 g, 1 equiv, 1.91 mmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (303 mg, 1 equiv, 1.91 mmol) in THF at 0 °C. After 10 minutes, sodium 2-methylpropan-2-olate (550 μL, 2 M, 0.58 equiv, 1.10 mmol) was added dropwise. The resulting reaction mixture was stirred for 10 minutes and then quenched with HCl (1.8 mL, 2 M in diethyl ether, 1.9 equiv, 3.6 mmol). The mixture was stirred for 5 minutes and then concentrated to afford tert-butyl ((1S,4S)-4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)- 2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (2.05 g). LCMS: ESI [M+H]
+ = 745.4 m/z; t
R = 2.09 minutes. (Method E) Step 7: 5-((1S,4S)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide Hydrogen chloride (15.9 mL, 2 M in diethyl ether, 15 equiv, 31.8 mmol) was added to tert-butyl ((1S,4S)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin- 5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4-methyl- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (2.05 g, 1 equiv, 2.12 mmol) in DCM (10.6 mL) and stirred for 2 hours. The reaction mixture was concentrated to a residue followed by the addition of DCM (30 mL). Sodium hydroxide (8.3 mL, 2 M, 16.6 mmol) was added, and the mixture was extracted with DCM (10 ×). The combined organics were dried over sodium sulfate and concentrated to a residue, which was purified by SFC (Column: Torus 2-PIC (19 × 100 mm, 5 μm, PN 186008586) Mobile phase: methanol with 0.25% DEA; Gradient: 5% to 40% at 0.6 mL/min over 10 minutes; backpressure
= 120 bar) to give 5-((1S,4S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (776 mg). LCMS: ESI [M+H]
+ = 645.3 m/z; tR = 1.44 minutes. (Method E) 1H NMR (400 MHz, CD3OD) δ 7.02 (m, 1H), 6.76 – 6.65 (m, 2H), 6.61 (s, 1H), 5.39 – 5.12 (m, 1H), 4.93 – 4.71 (m, 3H), 4.65 (m, 1H), 4.58 – 4.45 (m, 2H), 4.14 – 4.01 (m, 2H), 3.96 (m, 2H), 3.32 (s, 3H), 3.27 – 3.12 (m, 3H), 3.08 (s, 3H), 3.06 – 2.95 (m, 2H), 2.87 (s, 2H), 2.30 – 2.02 (m, 6H), 2.03 – 1.69 (m, 6H), 1.29 (m, 3H). 1
9F NMR (376 MHz, CD
3OD) δ -169.56, -176.41 (m). Example 19: 5-(6'-Amino-2-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-2',3',5,8-tetrahydro-1'H-spiro[pyrano[4,3-d]pyrimidine-7,4'- [1,3]methanonaphthalen]-4-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 172a)
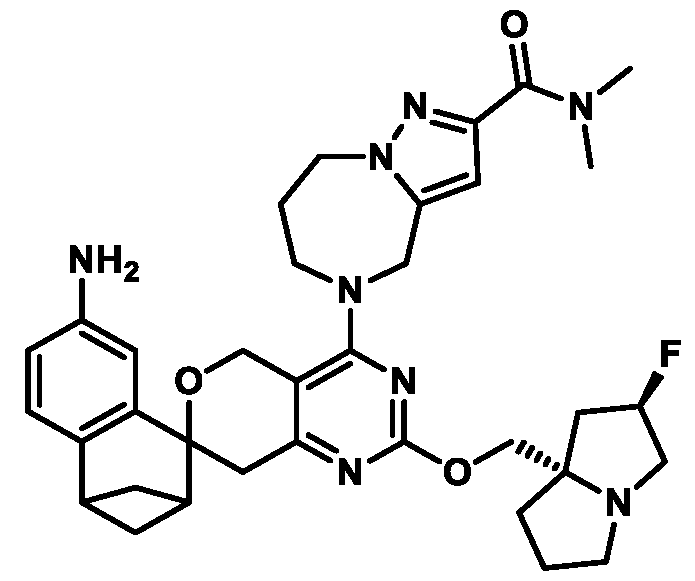
Step 1: 6-Bromo-4-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-1,2,3,4-tetrahydro-1,3-methanonaphthalen-4-ol LDA solution (11.1 mL, 1 M, 2.3 equiv, 11.1 mmol) was added to a nitrogen purged solution of (4-chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (988 mg, 1 equiv, 4.83 mmol) in THF (12.7 mL) at -78 °C. The internal temperature was maintained below -74 °C. The resulting orange solution was stirred for 15 minutes and then 6-bromo-2,3-dihydro-1,3- methanonaphthalen-4(1H)-one (1.26 g, 1.1 equiv, 5.31 mmol) was added dropwise to the solution, keeping the reaction temperature below -78 °C. The reaction mixture was stirred at - 78 °C for 45 minutes and then quenched with HCl (4 mL, 4 M in 1,4-dioxane), keeping the temperature below -50 °C. Sodium bicarbonate was then added until pH = ~7 was achieved, and the mixture was extracted with DCM (3 × 25 mL). The organic layer was then concentrated
to a residue, which was washed with MeOH giving 6-bromo-4-((6-chloro-5-(hydroxymethyl)- 2-(methylthio)pyrimidin-4-yl)methyl)-1,2,3,4-tetrahydro-1,3-methanonaphthalen-4-ol (1.33 g) which was used without further purification. 1H NMR (400 MHz, CDCl
3) δ 7.62 (d, 1H), 7.24 (dd, 1H), 6.87 (d, 1H), 4.84 – 4.67 (m, 2H), 3.51 (d, 1H), 3.31 (d, 1H), 3.03 (q, 1H), 2.54 – 2.42 (m, 5H), 2.35 – 2.25 (m, 1H), 1.88 – 1.76 (m, 1H), 1.70 – 1.57 (m, 1H). Step 2: 6'-Bromo-4-chloro-2-(methylthio)-2',3',5,8-tetrahydro-1'H- spiro[pyrano[4,3-d]pyrimidine-7,4'-[1,3]methanonaphthalene] A solution of 6-bromo-4-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-1,2,3,4-tetrahydro-1,3-methanonaphthalen-4-ol (1.33 g, 1 equiv, 3.0 mmol), phosphoric acid (346 mg, 85 wt%, 1 equiv, 3.0 mmol), and toluene (6.0 mL) was stirred at 100 °C for 1 hour. The reaction mixture was concentrated to give a cloudy oil, to which methanol (4 mL) was added. The resulting suspension was filtered to afford 6'-bromo-4-chloro- 2-(methylthio)-2',3',5,8-tetrahydro-1'H-spiro[pyrano[4,3-d]pyrimidine-7,4'- [1,3]methanonaphthalene] (940 mg). 1H NMR (400 MHz, CDCl
3) δ 7.52 (d, 1H), 7.26 (dd, 1H), 6.88 (d, 1H), 4.74 (d, 1H), 4.50 (d, 1H), 3.32 (d, 1H), 3.08 (q, 1H), 2.96 (d, 1H), 2.70 (q, 1H), 2.56 – 2.45 (m, 4H), 2.41 – 2.26 (m, 1H), 1.82 (t, 1H), 1.58 (dd, 1H). Step 3: 5-(6'-Bromo-2-(methylthio)-2',3',5,8-tetrahydro-1'H-spiro[pyrano[4,3- d]pyrimidine-7,4'-[1,3]methanonaphthalen]-4-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide DIPEA (1.93 mL, 5 equiv, 11.1 mmol) was added to 6'-bromo-4-chloro-2-(methylthio)- 2',3',5,8-tetrahydro-1'H-spiro[pyrano[4,3-d]pyrimidine-7,4'-[1,3]methanonaphthalene] (940 mg, 1 equiv, 2.2 mmol) and N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (693 mg, 1.5 equiv, 3.33 mmol) in ethanol (8.9 mL). The mixture was stirred at reflux for 16 hours. The mixture was concentrated to a viscous oil, and EtOAc (20 mL) was added. The solution was washed with saturated aqueous ammonium chloride (20 mL). The organic layer was concentrated to a residue, which was purified by flash chromatography (100% DCM to 90:10 DCM:MeOH) to afford 5-(6'-bromo-2-(methylthio)- 2',3',5,8-tetrahydro-1'H-spiro[pyrano[4,3-d]pyrimidine-7,4'-[1,3]methanonaphthalen]-4-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1.04 g).
LCMS: ESI [M+H]
+ = 595.2 m/z; t
R = 1.87 minutes (Method C) Step 4: tert-Butyl (4-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2-(methylthio)-2',3',5,8-tetrahydro-1'H-spiro[pyrano[4,3- d]pyrimidine-7,4'-[1,3]methanonaphthalen]-6'-yl)carbamate BrettPhos Pd G4 (241 mg, 0.15 equiv, 0.26 mmol) was added to a degassed suspension of 5-(6'-bromo-2-(methylthio)-2',3',5,8-tetrahydro-1'H-spiro[pyrano[4,3-d]pyrimidine-7,4'- [1,3]methanonaphthalen]-4-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (1.04 g, 1 equiv, 1.75 mmol), tert-butyl carbamate (1.03 g, 5 equiv., 8.75 mmol), and cesium carbonate (1.71 g, 3 equiv, 5.25 mmol) in 1,4-dioxane (7.0 mL). The mixture was stirred at 100 °C for 3 hours. The reaction mixture was cooled to room temperature, filtered, and concentrated to a residue which was purified by flash chromatography (SiO2, 0 to 10% MeOH in DCM), to afford tert-butyl (4-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2-(methylthio)- 2',3',5,8-tetrahydro-1'H-spiro[pyrano[4,3-d]pyrimidine-7,4'-[1,3]methanonaphthalen]-6'- yl)carbamate (834 mg). LCMS: ESI [M+H]
+ = 632.3 m/z; t
R = 1.78 minutes. (Method C) Step 5: tert-Butyl (4-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2-(methylsulfonyl)-2',3',5,8-tetrahydro-1'H-spiro[pyrano[4,3- d]pyrimidine-7,4'-[1,3]methanonaphthalen]-6'-yl)carbamate mCPBA (937 mg, 70 wt%, 3 equiv, 3.8 mmol) was added tert-butyl (4-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2-(methylthio)- 2',3',5,8-tetrahydro-1'H-spiro[pyrano[4,3-d]pyrimidine-7,4'-[1,3]methanonaphthalen]-6'- yl)carbamate (834 mg, 1 equiv, 1.27 mmol) in DCM (25 mL) at 0 °C. The reaction mixture was stirred for 10 minutes at 0 °C and then stirred at room temperature for 55 minutes. The mixture was diluted with DCM (50 mL) and quenched with aqueous sodium thiosulfate (15 mL) and saturated aqueous sodium bicarbonate (135 mL). The resulting biphasic mixture was stirred for 30 minutes, and the aqueous layer was extracted with DCM (2 × 20 mL). The combined organics were washed with aqueous NaOH (1 M, 2 × 20 mL), dried over sodium sulfate, and concentrated under reduced pressure to give tert-butyl (4-(2-(dimethylcarbamoyl)- 7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2-(methylsulfonyl)-2',3',5,8-
tetrahydro-1'H-spiro[pyrano[4,3-d]pyrimidine-7,4'-[1,3]methanonaphthalen]-6'-yl)carbamate (822 mg). LCMS: ESI [M+H]
+ = 664.7 m/z; tR = 2.77 minutes. (Method E) Step 6: 5-(6'-Amino-2-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-2',3',5,8-tetrahydro-1'H-spiro[pyrano[4,3-d]pyrimidine-7,4'- [1,3]methanonaphthalen]-4-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide NaH (50 mg, 60 wt%, 1 equiv, 1.24 mmol) was added to tert-butyl (4-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2- (methylsulfonyl)-2',3',5,8-tetrahydro-1'H-spiro[pyrano[4,3-d]pyrimidine-7,4'- [1,3]methanonaphthalen]-6'-yl)carbamate (822 mg, 1 equiv, 1.24 mmol) and ((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (197 mg, 1 equiv, 1.24 mmol) in THF at 0°C. The reaction mixture was stirred for 10 minutes, quenched with saturated aqueous ammonium chloride (2 mL), and diluted with EtOAc (5 mL). The aqueous layer was extracted with EtOAc (2 × 20 mL). The combined organics were dried over sodium sulfate and then concentrated to give a residue, which was purified by flash chromatography (SiO
2, 0% to 20% MeOH in DCM) to afford a white foam (750 mg). The foam was dissolved in DCM (3 mL) and HCl (2.5 mL, 2 M in diethyl ether, 5.0 mmol) was added. The mixture was stirred for 1 hour, and the resulting suspension was filtered to provide 5-(6'-amino-2-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-2',3',5,8-tetrahydro-1'H- spiro[pyrano[4,3-d]pyrimidine-7,4'-[1,3]methanonaphthalen]-4-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (409 mg). LCMS: ESI [M+H]
+ = 643.3 m/z; t
R = 1.13 minutes. (Method C) 1H NMR (400 MHz, MeOD) δ 7.56 (d, 1H), 7.33 – 7.20 (m, 2H), 6.76 (s, 1H), 5.61 (d, 1H), 5.25 – 5.04 (m, 2H), 5.04 – 4.94 (m, 1H), 4.90 – 4.81 (m, 3H), 4.82 – 4.71 (m, 1H), 4.72 – 4.47 (m, 2H), 4.28 – 4.08 (m, 2H), 4.08 – 3.78 (m, 3H), 3.54 – 3.42 (m, 2H), 3.42 – 3.33 (m, 5H), 3.26 (s, 1H), 3.19 – 2.99 (m, 4H), 2.84 – 2.61 (m, 2H), 2.61 – 2.46 (m, 1H), 2.43 – 2.29 (m, 3H), 2.29 – 2.14 (m, 2H), 1.86 (t, 1H), 1.69 (t, 1H). Example 20: (S)-2'-(((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-morpholino-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-amine (Compound 159a)
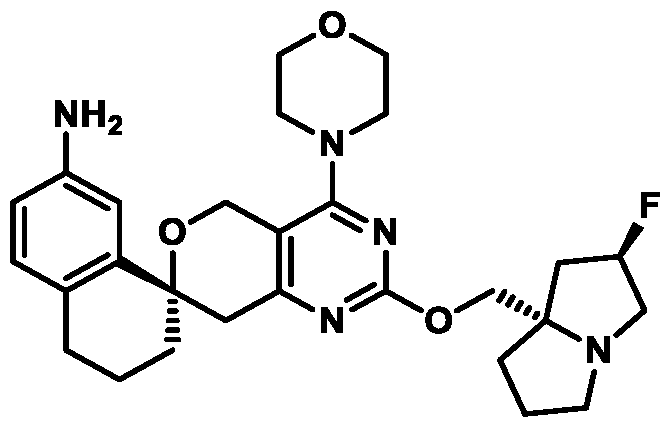
Step 1: (S)-7-Bromo-2'-(methylthio)-4'-morpholino-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] DIPEA (110 mg, 148 μL, 2 equiv, 850 μmol) was added to (S)-7-bromo-4'-chloro-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (175 mg, 1 equiv, 0.43 mmol) and morpholine (74 mg, 2 equiv, 0.85 mmol) in ethanol (850 μL). The mixture was stirred under reflux for 16 hours. The reaction mixture was then concentrated to a residue, which was purified by flash chromatography (SiO
2) to afford (S)-7-bromo-2'- (methylthio)-4'-morpholino-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine] (185 mg). LCMS: ESI [M+H]
+ = 463.8 m/z; t
R = 1.99 minutes. (Method C) Step 2: tert-Butyl (S)-(2'-(methylthio)-4'-morpholino-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate Pd(OAc)
2 (9 mg, 0.10 equiv, 0.04 mmol) was added to a solution of tert-butyl carbamate (94 mg, 2.0 equiv, 0.80 mmol), (S)-7-bromo-2'-(methylthio)-4'-morpholino- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (185 mg, 1.0 equiv, 0.400 mmol), and 2-(dicyclohexylphosphanyl)-2',4',6'-tris(isopropyl)biphenyl (57 mg, 0.30 equiv, 0.12 mmol) in 1,4-dioxane (3.2 mL). The reaction mixture was stirred at 100 °C for 12 hours, and then BrettPhos Pd G4 (10 mg) was added. The reaction mixture was stirred for an additional 6 hours. The reaction mixture was concentrated and washed with 90:10 heptanes:EtOAc to afford a solid, which was purified via flash chromatography (SiO
2) to provide tert-butyl (S)-(2'-(methylthio)-4'-morpholino-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (230 mg). LCMS: ESI [M+H]
+ = 499.8 m/z; t
R = 1.86 minutes. (Method C) Step 3: tert-Butyl (S)-(2'-(methylsulfonyl)-4'-morpholino-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate
mCPBA (223 mg, 3 equiv, 1.29 mmol) was added to tert-butyl (S)-(2'-(methylthio)-4'- morpholino-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (215 mg, 1 equiv, 0.431 mmol) in DCM (8.6 mL), and the resulting mixture was stirred at room temperature until complete conversion of the starting material was observed. The reaction mixture was diluted with saturated aqueous sodium bicarbonate and then saturated aqueous sodium thiosulfate (0.5 mL) was added. The reaction mixture was stirred for an additional 30 minutes and then extracted with DCM (3 ×). The combined organics were concentrated under reduced pressure to provide tert-butyl (S)-(2'-(methylsulfonyl)-4'- morpholino-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (133 mg). LCMS: ESI [M+H]
+ = 531.4 m/z; t
R = 1.88 minutes. (Method C) Step 4: (S)-2'-(((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 4'-morpholino-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]- 7-amine NaH (10 mg, 60 wt%, 1 equiv, 0.25 mmol) was added to a solution of ((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (40 mg, 1 equiv, 0.25 mmol) in THF (2.5 mL) at 0 °C. The mixture was stirred for 30 minutes, followed by the addition of tert-butyl (S)- (2'-(methylsulfonyl)-4'-morpholino-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate (133 mg, 1 equiv, 0.251 mmol) in THF (1.0 mL) dropwise. The reaction mixture was stirred for 10 minutes. Water (10 mL) was then added, and the solution was extracted with EtOAc (3 ×). The combined organics were concentrated to a residue, which was purified by flash chromatography (SiO2) to afford the desired protected product (60 mg). The material was then dissolved in DCM, and HCl (2 M in diethyl ether) was added. The reaction mixture was stirred for 1 hour and then concentrated under reduced pressure to provide (S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 4'-morpholino-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- amine (40 mg). LCMS: ESI [M+H]
+ = 510.3 m/z; tR = 1.06 minutes. (Method C) 1H NMR (400 MHz, MeOD) δ 7.51 (s, 1H), 7.31 (d, 1H), 7.23 (d, 1H), 5.56 (d, 1H), 4.93 (s, 2H), 4.64 (s, 2H), 3.89 (d, 10H), 3.42 (s, 1H), 3.27 (s, 6H), 3.10 (s, 1H), 2.88 (s, 2H), 2.60 (s, 1H), 2.50 – 1.95 (m, 6H), 1.85 (s, 1H).
Example 21: (1S,4S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Compound 179b)

Step 1: (1S,4S)-7-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile DIPEA (408 mg, 550 μL, 7.0 equiv., 3.16 mmol) was added to a solution of (1S,4S)- N,N-dibenzyl-8-bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (Intermediate 15, 280 mg, 1.0 equiv., 452 μmol) and 1,4-oxazepane hydrochloride (155 mg, 2.5 equiv., 1.13 mmol) in 1,4- dioxane (2.25 mL) at room temperature. The resulting mixture was stirred at 100 °C for 1 hour. The reaction was cooled to ambient temperature, quenched by the addition of water (15 mL), and diluted with EtOAc (15 mL). The reaction mixture was transferred to a separatory funnel, the layers were separated, and the aqueous layer was extracted (3 x 20 mL) with EtOAc. The combined organics were dried over sodium sulfate and filtered. The solution was concentrated in vacuo to give a crude residue (398.6 mg). This residue was taken up in DMF (8.15 mL) and added under nitrogen to a flask containing copper (I) cyanide (364 mg, 10 equiv., 4.07 mmol) at room temperature. The resulting mixture was purged with N
2 and heated to 120 °C and stirred for 1 hour. The mixture was cooled to room temperature and diluted sequentially with CH₂Cl₂ (100 mL), water (100 ml), MeOH (20 mL), and NH4OH (30 mL). The mixture was stirred until a blue biphasic mixture was observed. The reaction mixture was transferred into a separatory funnel and the two phases were separated. The aqueous layer was extracted with CH₂Cl₂ (100 mL x 3). The combined organic layers were dried over sodium sulfate, filtered, and concentrated to give 337 mg of a crude solid.
The crude solid (337 mg) was taken up in THF (4.38 mL), methanol (2.73 mL), and water (2.73 mL). To this mixture was added Oxone (908 mg, 3 equiv., 1.48 mmol). The resulting mixture was stirred for 2 hours then diluted sequentially with water (30 mL) and EtOAc (30 mL). The reaction mixture was transferred to a separatory funnel, the phases were separated, and aqueous layer was extracted with EtOAc (3 x 40 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated to give 391 mg of crude residue. To a flask containing the above crude residue (391 mg) and ((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (328 mg, 5.00 equiv., 2.06 mmol) in DMF (8.25 mL), lithium bis(trimethylsilyl)amide (1.5 M in THF) (103 mg, 410 μL, 1.5 M, 1.49 equiv., 615 μmol) was added dropwise at 0 °C under nitrogen. The ice/water bath was removed, and the reaction mixture was stirred for 3 hours. The reaction was quenched by the slow addition of a 1:1 mixture of a saturated aqueous ammonium chloride solution and water (10 mL). EtOAc (20 mL) was added and the reaction mixture transferred to a separatory funnel. The layers were separated and the aqueous phase was extracted with EtOAc (3 x 30 mL). The combined organic extracts were dried over sodium sulfate, filtered, and concentrated. Crude material was purified by silica gel chromatography (load: CH₂Cl₂, column: RediSep Rf Gold 80 g, 60 mL/min, CH₂Cl₂/[CH₂Cl₂/MeOH/NH₄OH, 75:22.5:2.5], 0 → 100% over 30 min) to afford (1S,4S)-7-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine]-8-carbonitrile (163 mg, 0.20 mmol) LCMS: m/z (ESI) [M+H]
+ 743.5, t
R = 2.61 minutes (Method E) Step 2: (1S,4S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile MeOH (3.91 mL) was delivered dropwise to a mixture of (1S,4S)-7-(dibenzylamino)- 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-4'-(1,4- oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8- carbonitrile (163 mg, 89% wt, 1 equiv., 195 µmol) and palladium(II) hydroxide (80 mg, 23 μL, 2.9 equiv., 0.57 mmol) under N2. H2 was bubbled through the reaction mixture for 10 min before the dropwise delivery of acetic acid (0.01 g, 0.01 mL, 0.9 equiv., 0.2 mmol).
Subsequently, H
2 was bubbled through the reaction mixture for 5 hours. An additional aliquot of MeOH was delivered to solution after 3 hours (3 mL), then the needle was removed and the reaction mixture was stirred overnight. The reaction mixture was filtered through celite and solvent removed under reduced pressure to give a solid residue which was purified by prep HPLC (XSelect ® CSH™ Prep, C185 µm OBD™, 19x150mm Column) (20 to 70% MeCN in H2O Ammonium Hydroxide additive 0.1%) to give (1S,4S)-7-amino-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (17 mg, 30 μmol). 1H NMR (400 MHz, CD
3OD) δ 7.12 (d, 1H), 6.77 (d, 1H), 5.36 – 5.16 (m, 1H), 5.02 (d, 1H), 4.85 – 4.81 (m, 1H), 4.13 (d, 1H), 4.03 (d, 1H), 3.95 – 3.75 (m, 6H), 3.76 – 3.66 (m, 2H), 3.27 – 3.12 (m, 4H), 3.01 – 2.96 (m, 1H), 2.93 – 2.83 (m, 2H), 2.32 – 2.15 (m, 2H), 2.14 – 1.81 (m, 9H), 1.75 – 1.66 (m, 1H), 1.24 (d, 3H). LCMS: m/z (ESI) [M+H]
+ 563.3, tR = 1.32 minutes (Method E) Example 22: 5-((S)-7-amino-5-chloro-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 164b)
Step 1: 5-(7-bromo-5-chloro-8-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-
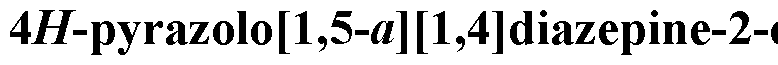
carboxamide DIPEA (1.01 mL, 6 equiv., 5.82 mmol) was added to the suspension of 7-bromo-4',5- dichloro-8-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine] (Intermediate 16, 450 mg, 1 equiv., 0.969 mmol) and N,N-dimethyl-5,6,7,8-
tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide, bis-hydrochloride (409 mg, 1.5 equiv., 1.45 mmol) in 1,4-dioxane (5 mL) and DMA (2.5 mL) at ambient temperature. The mixture was then heated at 110 °C for 18 hours. The reaction mixture was cooled to room temperature followed by removal of the solvents under reduced pressure. Water was added to the crude reaction mixture with stirring and the light brown solid was collected by filtration. The collected solids were washed with water and heptanes to give crude residue which was passed through a 24 g SiO
2 Redisep gold column (eluting with 0-30% 2.5% ammonium hydroxide 20% MeOH solution in DCM/DCM) to give 5-(7-bromo-5-chloro-8-fluoro-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (576 mg, 0.906 mmol). LCMS: m/z (ESI) [M+H]
+ 637.0, tR = 2.03 minutes (Method C) Step 2: tert-Butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-8-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate Brettphos Pd-G4 (30 mg, 0.11 equiv., 0.033 mmol), tert-butyl carbamate (38.0 mg, 1.1 equiv., 0.32 mmol) and cesium carbonate (103 mg, 1.03 equiv., 0.316 mmol) were added to an oven dried vial. The vial was evacuated and purged with nitrogen three times.5-(7-bromo-5- chloro-8-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (195 mg, 1 equiv., 0.308 mmol) in 1,4-dioxane (4.88 mL) was added. The mixture was sparged with nitrogen for 5 min at 20 °C. The sparged reaction mixture was added to the vial containing the solids and the resultant mixture was sparged for 3 minutes. Then the vial was transferred to a preheated aluminum block and heated to 80 °C for 4.5 hours. The mixture was then cooled to ambient temperature and was diluted with water. The aqueous layer was extracted with ethyl acetate (3x), and the combined organics were washed with brine, and dried over magnesium sulfate. The dried organics were filtered and concentrated to give a crude residue which was purified by flash column (gold SiO2 RediSep 24 g, (eluting with 0-30% (2.5% ammonium hydroxide 20% MeOH in DCM) in DCM) to give tert-butyl (5-chloro-4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-8-fluoro-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (202 mg, 0.300 mmol).
LCMS: m/z (ESI) [M+H]
+ 672.2, t
R = 2.03 minutes (Method C) Step 3: tert-butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-8-fluoro-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate 3-chlorobenzoperoxoic acid (200 mg, 77% wt, 3 equiv., 0.893 mmol) was added to the solution of tert-butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-8-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (200 mg, 1 equiv., 0.298 mmol) in DCM (6.0 mL) at room temperature. The resulting yellow solution was stirred at room temperature for 1 hour. The mixture was then diluted with DCM (30 mL) and saturated sodium thiosulfate (30 mL). The organic layer was separated, and the aqueous layer was extracted with DCM (3 x 30 mL). The combined organic layers were washed with saturated aqueous sodium bicarbonate solution, dried over magnesium sulfate, and concentrated to give tert-butyl (5-chloro-4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-8-fluoro-2'- (methylsulfonyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (208 mg, 0.295 mmol). LCMS: m/z (ESI) [M+H]
+ 704.2, t
R = 1.99 minutes (Method C) Step 4: tert-Butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate LiHMDS (0.55 mL, 1.0 M, 2.0 equiv., 0.55 mmol) was added dropwise to a solution of tert-butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-8-fluoro-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (194 mg, 1 equiv., 0.275 mmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (132 mg, 3 equiv., 0.826 mmol) in DMF (5.5 mL) at ambient temperature. The resulting solution was stirred at ambient temperature. After 1 hour, additional 0.30 mL LiHMDS solution (1.0 M) was added. After two hours, the reaction mixture was diluted with EtOAc (30 mL) then quenched with water (30 mL) and saturated aqueous ammonium chloride (30 mL). The aqueous phase was extracted three times with EtOAc (20 mL). The combined organic phases were washed with
brine, dried over magnesium sulfate, filtered and concentrated to give a crude residue which was then purified by flash column (SiO2 Redisep gold 40 g, eluting with 0-80% (2.5% ammonium hydroxide 20% methanol in DCM) solution in DCM) to give tert-butyl (5-chloro- 4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-8-fluoro- 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (94.0 mg, 0.12 mmol). LCMS: m/z (ESI) [M+H]
+ 783.5, t
R = 1.53 minutes (Method C) Step 5: 5-(7-Amino-5-chloro-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide Hydrogen chloride (0.40 mL, 2.0 M in ether, 6.7 equiv., 0.80 mmol) in ether was added to a solution of tert-butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (93 mg, 1 equiv., 0.120 mmol) in DCM (2.5 mL) at ambient temperature. The mixture was stirred for 3 hours. Additional hydrogen chloride solution (1.3 mL, 2.0 M, 22 equiv., 2.6 mmol) in ether was then added. The product mixture was concentrated to remove ether and was resuspended in DCM and 500 µL 2.0 M HCl in ether.500 µL of 4.0 M HCl in dioxanes was additionally added and the contents and stirred for 15 minutes. The resulting solid was collected following decantation of the solvent and was then dried to give a solid, which was taken up in 30 mL DCM and vigorously washed with saturated aqueous sodium bicarbonate solution (3 x 20 mL). The aqueous phase was extracted with further DCM (3 x 20 mL). The organic layers were combined, dried over magnesium sulfate, filtered and concentrated to afford 5-(7-amino-5-chloro-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (57.0 mg, 0.083 mmol) as a mixture of two diastereomers. LCMS: m/z (ESI) [M+H]
+ 683.3, tR = 1.35 minutes (Method C) Step 6: 5-((S)-7-amino-5-chloro-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-
pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide The diastereomers of 5-(7-amino-5-chloro-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide were separated by reverse-phase chiral prep-HPLC ACCQ- Prep Auto Purification. Chiralpak IC, 20mm ID x 250ml, 5µm particle size, part number 83345. Isocratic 35% B in A (H2O with 0.1% TFA (A) and MeCN with 0.1% TFA(B)) to afford 5-((S)- 7-amino-5-chloro-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Peak 1) (22 mg) and 5-((R)-7-amino-5-chloro-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (Peak 2) (20.5 mg). Peak 1: LCMS: m/z (ESI) [M+H]
+ > 95% dr., 683.3, tR = 2.01 min(Method K) 1H NMR (400 MHz, CDCl
3) δ 6.82 (d, J = 7.8 Hz, 1H), 6.58 (s, 1H), 5.30 (m, 1H), 4.84 – 4.51 (m, 4H), 4.45 (m, 2H), 4.12 (s, 1H), 3.81 (m, 3H), 3.50 – 3.16 (m, 7H), 3.06 (m, 4H), 2.95 – 2.84 (m, 1H), 2.83 – 2.60 (m, 2H), 2.24 (m, 6H), 1.97 (m, 6H). Example 23: 5-((R)-7-amino-5-chloro-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 164a)
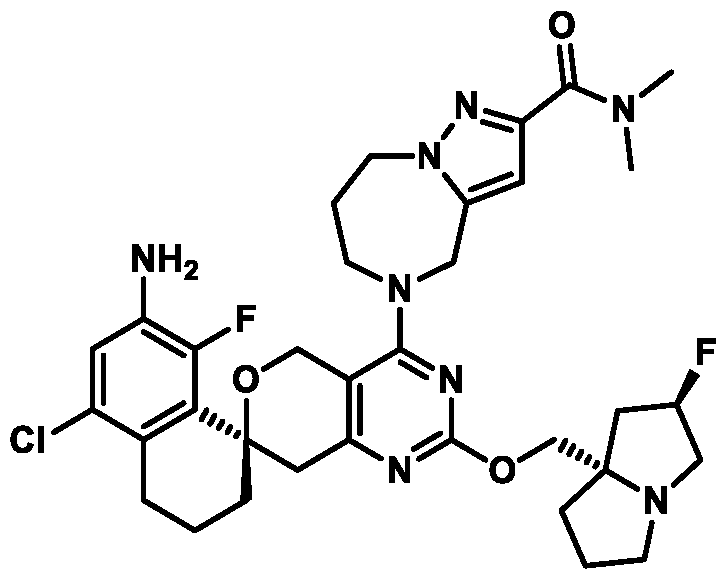
Step 1: 5-((R)-7-amino-5-chloro-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-
pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide 5-((R)-7-amino-5-chloro-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]- 4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (22 mg) was obtained from Example 22, as Peak 2: reverse-phase chiral prep-HPLC ACCQ-Prep Auto Purification. Chiralpak IC, 20mm ID x 250ml, 5µm particle size, part number 83345. Isocratic 35% B in A (H2O with 0.1% TFA (A) and MeCN with 0.1% TFA(B)). Peak 2: LCMS: m/z (ESI) [M+H]
+ > 95% dr., 683.3, tR = 1.65 minutes (Method K) 1H NMR (400 MHz, CDCl
3) δ 6.82 (d, J = 7.7 Hz, 1H), 6.56 (s, 1H), 5.29 (d, J = 53.5 Hz, 1H), 4.76 (m, 1H), 4.66 (m, 1H), 4.62 – 4.51 (m, 2H), 4.45 (m, 2H), 4.12 (s, 1H), 3.75 (m, 3H), 3.48 (m, 4H), 3.42 – 3.18 (m, 6H), 3.09 – 2.85 (m, 4H), 2.74 (m, 1H), 2.24 (m, 5H), 1.96 (m, 6H). Example 24: (R)-5-chloro-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (Compound 181a)
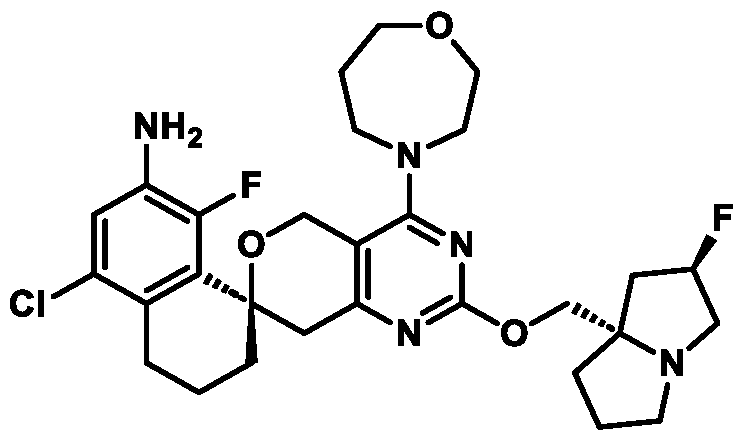
Step 1: 7-Bromo-5-chloro-8-fluoro-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] A mixture of 1,4-oxazepane (196 mg, 1.2 equiv., 1.94 mmol) 7-bromo-4',5-dichloro-8- fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine] (Intermediate 16, 750 mg, 1 equiv., 1.62 mmol), DIPEA (1.25 g, 1.69 mL, 6 equiv., 9.69 mmol) and 1,4-dioxane (20 mL) was heated at 110 °C for 4 hours. The reaction mixture was cooled to 22 °C, diluted with CHCl3 (20 mL) and water (20 mL). The organic layer was separated and the aqueous layer was extracted with CHCl
3/MeOH (4:1) (4 X 15 mL). The combined organic layer was washed with brine (3 X 10 mL), dried over sodium sulfate and filtered. The solvent was evaporated under reduced pressure and the residual mass was purified by flash column chromatography (24 g cartridge, dry injection) with EtOAc/DCM (0-30%) to
yield 7-bromo-5-chloro-8-fluoro-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (773 mg, 1.46 mmol). LCMS: m/z (ESI) [M+H]
+ 528.2, tR = 2.06 minutes (Method B) Step 2: tert-Butyl (5-chloro-8-fluoro-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate A mixture of 7-bromo-5-chloro-8-fluoro-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (305 mg, 1 equiv., 577 μmol), tert-butyl carbamate (108 mg, 1.6 equiv., 923 μmol), and cesium carbonate (564 mg, 3 equiv., 1.73 mmol) was purged with nitrogen then degassed. DMF (20 mL) was then added via syringe. The resulting mixture was degassed with nitrogen for 5 minutes then BrettPhos Pd-G4 (53.1 mg, 0.10 equiv., 57.7 μmol) was added. Then it was degassed with nitrogen for another 3 minutes. The pressure vessel was capped and heated at 80 °C for 15 hours. Upon cooling to 22 °C, the solid was removed by vacuum filtration on celite and the filtrate was collected with EtOAc (20 mL). The organic filtrate was treated with further EtOAc (60 mL) and was washed with brine (3 × 15 mL), dried over sodium sulfate and filtered. The solvent was evaporated and the crude material was purified by flash column chromatography (25 g cartridge, dry injection) with EtOAc/hexanes (10-70%) to afford tert-butyl (5-chloro-8- fluoro-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate (163 mg, 288 μmol). LCMS: m/z (ESI) [M-H]
+ 565.3, t
R = 5.67 minutes (Method B) Step 3: tert-Butyl (5-chloro-8-fluoro-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate Hydrogen peroxide (320 μL, 30% wt, 7 equiv., 3.11 mmol) was added to a mixture of tert-butyl (5-chloro-8-fluoro-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (251 mg, 1 equiv., 444 μmol), sodium tungstate dihydrate (15.0 mg, 0.10 equiv., 44.0 μmol), and hydrogen tetra(but- 1-yl)ammonium sulphate (24.0 mg, 0.16 equiv., 71.0 μmol) in EtOAc (10 mL). The resulting mixture was heated at 40 °C for 40 minutes. The reaction mixture was cooled to ambient temperature and was diluted with water (10 mL). The organics were extracted with EtOAc (4 X 15 mL). The combined organics were washed with brine (3 X 15 mL), dried over sodium sulfate and filtered. The solvent was evaporated to yield tert-butyl (5-chloro-8-fluoro-2'-
(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate (260 mg, 435 μmol). LCMS: m/z (ESI) [M-H]
+ 597.4, tR = 1.77 minutes (Method B) Step 4: tert-Butyl (5-chloro-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate Potassium tert-butoxide in THF (1.09 mL, 1.0 M, 2.5 equiv., 1.09 mmol) was added to a solution of ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (104 mg, 1.5 equiv., 653 μmol) in anhydrous THF (160 μL) at 0 °C. After 5 minutes, tert-butyl (5-chloro-8- fluoro-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (260 mg, 1 equiv., 435 μmol) was added and the contents were stirred for another 10 minutes. The reaction mixture was quenched with EtOAc (10 mL) and aqueous ammonium chloride (10 mL). The organic layer was separated, and the aqueous layer was extracted with EtOAc (4 X 15 mL). The combined organics were washed with brine (3 X 10 mL), dried over sodium sulfate and filtered. The solvent was evaporated to yield tert-butyl (5-chloro-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate which was used as crude material in the subsequent step assuming quantitative conversion to product. LCMS: m/z (ESI) [M-H]
+ 676.4, t
R = 5.39 minutes (Method I) Step 5: (R)-5-chloro-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-7-amine A mixture was prepared with tert-butyl (5-chloro-8-fluoro-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (260 mg, 1 equiv., 385 μmol), p-toluenesulfonic acid (199 mg, 3 equiv., 1.15 mmol) and acetonitrile (3 mL). The mixture was heated to 40 °C for 4 hours. The contents were then diluted with EtOAc (15 mL) and aqueous sodium bicarbonate (15 mL). The organic layer was separated, and the aqueous layer was extracted further with EtOAc (4 X 15 mL). The combined organics were washed with brine (3 X 15 mL), dried over sodium sulfate, and filtered. The solvent was
evaporated and the residue was purified by flash column chromatography (25 g cartridge, dry injection) with DCM/EtOAc (1-75%) to yield 5-chloro-8-fluoro-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (153 mg) as a mixture of diastereomers. The diastereomers were separated by reverse-phase prep-HPLC Gemini 5um NX-C18 110 Å, 100 x 30 mm (45 mL/min, 40-100% methanol in water with ammonium bicarbonate buffer, pH = 10.0) to afford (R)-5-chloro-8-fluoro-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (30 mg). Peak 1: LCMS: m/z (ESI) [M-H]
+ 576.4, t
R = 4.29 minutes (Method I) 1H NMR (DMSO-d
6, 400 MHz): δ
H 6.81 (1H, d, J = 7.7 Hz), 5.30 (1H, s), 5.15 (2H, s), 4.77 (1H, m), 4.55 (1H, m), 3.92 (1H, m), 3.83 (1H, m), 3.57-3.73 (8H, m), 2.97-3.14 (4H, m), 2.78 (2H, m), 2.60 (2H, m), 2.06 (1H, m), 1.70-1.97 (11H, m). 1
9F NMR (DMSO, 376 MHz): δ -131.2, -172.2. Example 25: 5-(7-Amino-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 176a)

Step 1: 7-Amino-4-methyl-3,4-dihydronaphthalen-1(2H)-one To a pressure vessel charged with 7-bromo-4-methyl-3,4-dihydronaphthalen-1(2H)- one (10.0 g, 1.00 equiv., 41.8 mmol) and copper (I) oxide (598 mg, 0.10 equiv., 4.18 mmol), were added NMP (27.9 mL) and ammonium hydroxide (29.3 g, 32.4 mL, 20 equiv., 836 mmol) under nitrogen. Strong gas release/bubbling was observed. The pressure vessel was rapidly sealed with a Teflon screw cap and the reaction mixture was stirred at 80 °C for 17 hours. The reaction mixture fully solubilized within the first 5 min of heating. The reaction mixture was cooled to room temperature and quenched with water (100 mL) and extracted with
EtOAc (3 x 100 mL). The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated. Crude material was purified by silica gel chromatography to afford 7-amino-4-methyl-3,4-dihydronaphthalen-1(2H)-one (6.18 g, 35.3 mmol), as a yellow solid. 1H NMR (400 MHz, CDCl
3) δ 7.32 (d, 1H), 7.14 (d, 1H), 6.87 (dd, 1H), 3.70 (s, 2H), 2.99 (m, 1H), 2.75 (m, 1H), 2.55 (m, 1H), 2.20 (m, 1H), 1.84 (m, 1H), 1.35 (d, J = 7.0 Hz, 3H). Step 2: 7-Amino-8-bromo-4-methyl-3,4-dihydronaphthalen-1(2H)-one To a solution of 7-amino-4-methyl-3,4-dihydronaphthalen-1(2H)-one (6.18 g, 1.00 equiv., 35.3 mmol) in DMF (47.0 mL) at 0 °C, was added a solution of N-bromosuccinimide (6.28 g, 1.00 equiv., 35.3 mmol) in DMF (23.5 mL) dropwise under nitrogen. The resulting mixture was stirred at 0 °C for 5 min at room temperature (ice/water bath removed) for 2.5 hours. Upon complete conversion to the desired product observed by analytical LCMS of an aliquot, the reaction was quenched by the addition of water (80 mL). EtOAc (100 mL) was added, and the layers were separated. The aqueous phase was extracted with EtOAc (3 x 100 mL). The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated. Crude material was purified by silica gel chromatography to isolate 7-amino-8- bromo-4-methyl-3,4-dihydronaphthalen-1(2H)-one (7.40 g, 29.1 mmol), as a purple solid. 1H NMR (400 MHz, CDCl
3) δ 7.07 (d, 1H), 6.91 (d, 1H), 4.30 (br s, 2H), 2.99 (m, 1H), 2.80 (m, 1H), 2.63 (m, 1H), 2.19 (m, 1H), 1.88 – 1.78 (m, 1H), 1.32 (d, J = 7.0 Hz, 3H). Step 3: 8-Bromo-7-(dibenzylamino)-4-methyl-3,4-dihydronaphthalen-1(2H)-one To a solution of 7-amino-8-bromo-4-methyl-3,4-dihydronaphthalen-1(2H)-one (4.00 g, 1.0 equiv., 15.7 mmol) in MeCN (120 mL) at room temperature, were added benzyl bromide (5.92 g, 4.12 mL, 2.2 equiv., 34.6 mmol) and potassium carbonate (6.53 g, 3.0 equiv., 47.2 mmol) sequentially. The resulting mixture was stirred vigorously at reflux for 18 hours. Analytical LCMS of an aliquot showed a 2:1 ratio of desired product and mono benzylated aniline. More potassium carbonate (3.26 g, 1.5 equiv., 23.6 mmol) and benzyl bromide (2.96 g, 2.06 mL, 1.1 equiv., 17.3 mmol) were added at room temperature and the reaction mixture was stirred at reflux for another 7.5 hours. Analytical LCMS of an aliquot showed complete conversion to the desired dibenzyl aniline product. The reaction mixture was cooled to room temperature. The solids were filtered and rinsed with EtOAc. The filtrate was concentrated. Crude material was purified by silica gel chromatography to afford 8-bromo-7- (dibenzylamino)-4-methyl-3,4-dihydronaphthalen-1(2H)-one as a white solid.
LCMS: ESI [M+H]
+= 434.1 m/z; t
R = 2.31 min (Method A) 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.31 (m, 4H), 7.30 – 7.25 (m, 4H), 7.25 – 7.19 (m, 2H), 7.06 (d, J = 8.3 Hz, 1H), 7.01 (d, J = 8.3 Hz, 1H), 4.19 (d, 2H), 4.14 (d, 2H), 3.02 – 2.92 (m, 1H), 2.83 (m, 1H), 2.67 (m, 1H), 2.23 – 2.13 (m, 1H), 1.89 – 1.79 (m, 1H), 1.31 (d, 3H). Step 4: 8-Bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-7-(dibenzylamino)-4-methyl-1,2,3,4-tetrahydronaphthalen-1-ol To a solution of (4-chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (4.81 g, 1.5 equiv., 23.5 mmol) in THF (116 mL) at -78 °C, was added LDA (1.0 M in THF/hexanes) (5.03 g, 47.0 mL, 1.0 M, 3.0 equiv., 47.0 mmol) dropwise, by addition funnel over 15 minutes. The rate of addition was set so that the internal temperature did not exceed - 72 °C. The resulting clear mixture, which became bright yellow over time, was stirred at -78 °C for 1.25 hours, followed by the dropwise addition of a solution of 8-bromo-7-(dibenzylamino)-4- methyl-3,4-dihydronaphthalen-1(2H)-one (6.80 g, 1.0 equiv., 15.7 mmol) in THF (58.0 mL) by addition funnel. The rate of dropwise addition was set so that the internal temperature did not exceed - 74 °C. The reaction mixture was stirred -78 °C for 1 hour. Upon complete consumption of the tetralone starting material observed by analytical LCMS of an aliquot, the reaction was quenched by the dropwise addition of HCl (2.0 M in Et2O) (1.83 g, 25.0 mL, 3.2 equiv., 50.1 mmol) at -78 °C, by addition funnel, followed by the addition of a 1:1 mixture of saturated aqueous ammonium chloride solution and water (100 mL). The biphasic mixture was warmed to room temperature and the layers were separated. The aqueous phase was extracted with EtOAc (3 x 100 mL). The combined organic extracts were dried over magnesium sulfate, filtered and concentrated. Crude material was purified by silica gel chromatography to afford 8-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-7- (dibenzylamino)-4-methyl-1,2,3,4-tetrahydronaphthalen-1-ol (7.87 g, 12.3 mmol), as a white solid. LCMS: ESI [M+H]
+= 640.3 m/z; t
R = 2.46 min (Method A) Step 5: N,N-Dibenzyl-8-bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine To a solution of 8-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-7-(dibenzylamino)-4-methyl-1,2,3,4-tetrahydronaphthalen-1-ol (5.85 g, 1.0 equiv.,
9.15 mmol) in THF (76.3 mL) at -78 °C, was added n-butyllithium (2.5 M in hexanes) (1.29 g, 8.06 mL, 2.2 equiv., 20.1 mmol) dropwise by syringe. The internal temperature was monitored during the addition by an internal electronic temperature probe and the addition rate was set so that the internal temperature did not exceed -72 °C. The resulting mixture, which became dark brown upon addition of n-butyllithium, was stirred at -78 °C for 35 min, followed by the dropwise addition of a solution of p-toluenesulfonyl chloride (2.62 g, 1.5 equiv., 13.7 mmol) in THF (38.1 mL) by addition funnel. The reaction mixture rapidly turned to a yellow color. The resulting mixture was stirred at -78 °C for 10 min and the acetone/dry ice bath was removed. The reaction mixture was stirred for another 1.75 hours, followed by the addition of a 1:1 mixture of a saturated aqueous ammonium chloride solution and water (100 mL). EtOAc (50 mL) was added and the layers were separated. The aqueous phase was extracted with EtOAc (3 x 75 mL). The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated. Crude material was purified by silica gel chromatography (heptane/EtOAc, 0% → 10%) to afford N,N-dibenzyl-8-bromo-4'-chloro-4-methyl-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (3.31 g, 5.33 mmol), as a white solid. LCMS: m/z ESI [M+H]⁺ = 620.14, t
R = 2.82 min (Method A) Step 6: 5-(8-Bromo-7-(dibenzylamino)-4-methyl-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl- 5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide To a suspension of N,N-dibenzyl-8-bromo-4'-chloro-4-methyl-2'-(methylthio)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (1.15 g, 1 equiv., 1.85 mmol) in MeOH (18.5 mL) at room temperature, were added DIPEA (2.39 g, 3.23 mL, 10 equiv., 18.5 mmol) and N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide, 2HCl (2.08 g, 4 equiv., 7.41 mmol) sequentially. The resulting suspension was stirred at reflux for 17 hours. Analytical LCMS showed minor conversion to the desired product, with mostly unreacted starting material. DMF (18.5 mL), DIPEA (957 mg, 1.29 mL, 4 equiv., 7.41 mmol) and N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide, 2HCl (1.04 g, 2 equiv., 3.70 mmol) were added, and the reaction mixture was heated up to 90 °C, upon which a clear brown/orange solution was obtained. The reaction mixture was stirred for another 5 hours. Analytical LCMS showed full conversion to the desired product. The volatiles were removed in vacuo and the
crude material was purified by silica gel chromatography (heptane/EtOAc, 5 → 100%) to afford 5-(8-bromo-7-(dibenzylamino)-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1.27 g, 1.60 mmol), as a white amorphous solid LCMS: m/z ESI [M+H]⁺ = 792.24, tR = 2.29 min, (Method A) Step 7: 5-(8-Bromo-7-(dibenzylamino)-4-methyl-2'-(methylsulfonyl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl- 5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide To a solution of 5-(8-bromo-7-(dibenzylamino)-4-methyl-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (635 mg, 1 equiv., 801 μmol) in DCM (8.01 mL) at 0 °C, was added 3-chloroperoxybenzoic acid (718 mg, 77% wt, 4 equiv., 3.20 mmol) in one portion. The resulting colorless solution was stirred at 0 °C for 10 min and the ice/water was removed. The reaction mixture, which became yellow over time, was stirred for another 1.5 hours. The reaction was quenched by the addition of a 3:1 mixture of a sodium bicarbonate saturated aqueous solution and a sodium thiosulfate saturated aqueous solution (8.0 mL). CH₂Cl₂ (10 mL) was added and the layers were separated. The aqueous phase was extracted with EtOAc (3 x 101 mL). The combined organic extracts were dried over magnesium sulfate, filtered and concentrated to afford 5-(8-bromo-7-(dibenzylamino)-4- methyl-2’-(methylsulfonyl)-3,4,5’,8’-tetrahydro-2H-spiro[naphthalene-1,7’-pyrano[4,3- d]pyrimidin]-4’-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide which was of sufficient purity to be used directly in the next step without further purification. LCMS: m/z ESI [M+H]⁺ = 824.34, tR = 2.19 min (Method A) Step 8: 5-(8-Bromo-7-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide To a solution of 5-(8-bromo-7-(dibenzylamino)-4-methyl-2'-(methylsulfonyl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (661 mg, 1.0 equiv., 801
μmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (166 mg, 1.3 equiv., 1.04 mmol) in THF (8.01 mL) at -20 °C, was added potassium tert-butoxide (234 mg, 2.08 mL, 1.0 M, 2.6 equiv., 2.08 mmol) dropwise. The resulting mixture was stirred at -20 °C for 30 min and at 0 °C for 30 min. The reaction was quenched by the addition of a 1:1 mixture of a saturated aqueous ammonium chloride solution and water (8.0 mL). EtOAc (10 mL) was added, and the layers were separated. The aqueous phase was extracted with EtOAc (3 x 10 mL). The combined organic extracts were dried over magnesium sulfate, filtered and concentrated. Crude material was purified by silica gel chromatography to afford 5-(8-bromo- 7-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4- methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (418 mg, 462 μmol), as a white solid. LCMS: m/z ESI, [M+2H⁺]/2 = 453.3, tR = 1.69 min (Method A) Step 9: 5-(8-Cyano-7-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide To a microwave vial charged with copper (I) cyanide (414 mg, 10 equiv., 4.62 mmol), was added a solution of 5-(8-bromo-7-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (418 mg, 1 equiv., 462 μmol) in DMF (9.25 mL) (DMF degassed prior to use by sparging nitrogen for 15 min) under nitrogen. The vial headspace was evacuated and refilled with nitrogen (3 x), sealed, and the reaction mixture was stirred at 120 °C for 2.3 hours. Analytical LCMS of a sample taken in DMSO showed full consumption of the starting material. The reaction mixture was cooled to room temperature and then concentrated under reduced pressure. The residue was partitioned between CH₂Cl₂ (75 mL) and water (40 mL), upon which a precipitate formed. A 1:1 mixture of concentrated NH4OH and MeOH (50 mL) was added, until no solid was visible (clear layers observed). The layers were separated, and then aqueous layer was extracted with CH₂Cl₂ (3 x 50 mL). The combined organic layers were washed with brine, dried over magnesium sulfate, filtered, and concentrated under reduced pressure. Crude material was purified by silica gel chromatography
to afford 5-(8-cyano-7-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (125 mg, 147 μmol) LCMS: m/z ESI [M+H]⁺ = 850.6, tR = 1.62 min (Method A) Step 10: 5-(7-Amino-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide To a vial charged with 5-(8-cyano-7-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (125 mg, 1 equiv., 147 μmol) was added palladium(II) hydroxide on carbon (20 wt%, 258 mg, 0.368 mmol, 2.5 equiv.). The vial headspace was evacuated and refilled with nitrogen, followed by the addition of MeOH (4.20 mL). The vial headspace was evacuated and refilled with hydrogen from a balloon (3 ×), followed by the addition of acetic acid (8 drops). The reaction mixture was stirred vigorously under an atmosphere of hydrogen at room temperature for 4 hours. Analytical LCMS of an aliquot showed full conversion to the desired aniline product. The vial headspace was evacuated and refilled with nitrogen. The reaction mixture was filtered through a syringe filter and the filtrate was stirred with sodium carbonate for 5 min, filtered and concentrated at room temperature to afford 5-(7-amino-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (95 mg). LCMS: m/z ESI, [M+H]⁺ = 670.4, t
R = 1.26 min (Method A) 1H NMR (400 MHz, CDCl
3) δ 7.87 – 7.55 (m, 1H), 7.32 – 7.20 (m, 1H), 7.13 (s, 1H), 5.93 – 5.64 (m, 1H), 5.63 – 5.44 (m, 1H), 5.41 – 5.24 (m, 5H), 5.09 – 4.89 (m, 2H), 4.63 – 4.46 (m, 3H), 4.45 – 4.29 (m, 1H), 3.88 – 3.74 (m, 4H), 3.74 – 3.50 (m, 6H), 3.49 – 3.32 (m, 2H), 2.98 – 2.31 (m, 11 H), 1.89 – 1.69 (m, 4H). Example 26: 5-((1S,4R)-7-amino-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-
pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 176b)
The diastereomers of 5-(7-amino-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide were separated via Chiralpak IB-N (21 x 250 mm, 5 um, P/N 88445) on ACCQPrep under isocratic cosolvent (methanol with 0.25% diethylamine) to provide 5-((1S,4S)-7-amino-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (Peak 1) (1.2 mg) and 5-((1S,4R)-7-amino-8-cyano-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Peak 2) (1.9 mg). Peak 2: Chiral HPLC: tR = 2.13 min (Chiralpak IB-U (3.0 x 100 mm, 1.6 um, P/N 81U83, isocratic 50% cosolvent) (ethanol with 0.05% diethylamine) 1H NMR (400 MHz, CD3CN) δ 7.23 (d, 1H), 6.69 (d, 1H), 6.41 (s, 1H), 5.21 (d, 1H), 5.00 – 4.88 (m, 4H), 4.80 – 4.71 (m, 2H), 4.68 – 4.51 (m, 3H), 4.45 – 4.31 (m, 3H), 3.92 – 3.79 (m, 2H), 3.55 – 3.46 (m, 3H), 3.20 – 3.13 (m, 5H), 3.10 – 2.77 (m, 9H), 1.38 – 1.13 (m, 4H), 0.88 – 0.71 (m, 4H). Example 27: 5-((1S,4S)-7-amino-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 176c)
5-((1S,4S)-7-amino-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (1.2 mg) was obtained from Example 26, as Peak 1: Chiral HPLC: t
R = 1.44 min (Chiralpak IB-U (3.0 x 100 mm, 1.6 um, P/N 81U83, isocratic 50% cosolvent) (ethanol with 0.05% diethylamine). 1H NMR (400 MHz, D
2O) δ 7.50 (d, 1H), 7.11 (d, 1H), 6.86 (s, 1H), 5.72 (d, 1H), 5.42 – 5.24 (m, 2H), 5.08 – 4.97 (m, 4H), 4.78-4.59 (s, 7H), 4.35 – 4.06 (m, 2H), 4.00 – 3.82 (m, 4H), 3.49 – 3.10 (m, 2H), 2.67 – 2.10 (m, 13H), 1.70 – 1.40 (m, 4H). Example 29: 5-(7-Amino-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 185a)
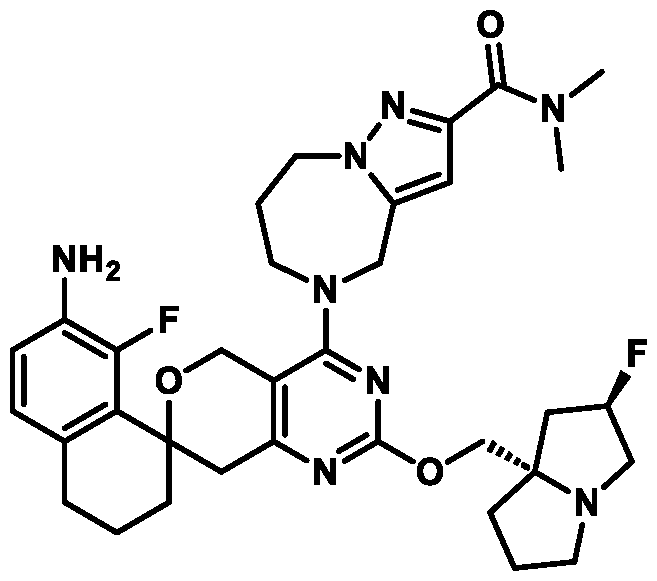
Step 1: tert-Butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate
To a 10 mL vial was added tert-butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro- 4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (95 mg, 46% wt, 1 equiv., 56 µmol) in MeOH (4 mL). The vial was evacuated and backfilled with nitrogen and then palladium on carbon (59 mg, 10% wt, 1 equiv., 56 µmol) was added. The inert atmosphere was exchanged with hydrogen by degassing the reaction mixture under reduced pressure with backflow of hydrogen from a balloon. The black reaction mixture was stirred at room temperature under hydrogen atmosphere for 72 hours. At this point the reaction mixture was filtered through celite and washed with methanol, the filtrate was concentrated and the residue was basified with aqueous potassium carbonate and extracted with DCM. The organic layer was dried over sodium sulfate, filtered and concentrated to give 73 mg of crude material which was purified by preparative HPLC (Gemini® 5 µm NX-C18110 Å, 100 x 30 mm column eluting with a gradient of 40 to 100% MeCN in 10 mM ammonium bicarbonate buffer) to yield tert-butyl (4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-8-fluoro-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (31 mg, 56 µmol) as a white solid. LCMS: m/z (ESI) [M+H]
+ 749.5 tR = 1.64 min (Method B) Step 2: 5-(7-Amino-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide To a vial was added tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (30.0 mg, 1 equiv., 40.1 µmol) in DCM (1 mL). A 2 M HCl solution in Et2O was added (200 µl, 10 equiv., 401 µmol). The resulting mixture was stirred at room temperature for 2 hours. At this point LCMS showed all the starting material had been consumed. The reaction mixture was concentrated and the residue was diluted with DCM (10 mL) and water (2 mL). The layers were basified with concentrated ammonium hydroxide until pH = 10 was reached under stirring. The layers were separated and the aqueous layer was
extracted with DCM three times (10 mL each). The combined organics were washed with brine, dried over sodium sulfate, filtered, and concentrated to give 5-(7-amino-8-fluoro-2'-(((2R,7aS)- 2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (14 mg, 22 µmol) as a white solid after lyophilization. LCMS: m/z (ESI) [M+H]
+ 649.5 t
R = 1.35 min (Method B) 1H NMR (400 MHz,CDCl3): δ 6.75 – 6.67 (m, 2H), 6.52 (s, 1H), 5.25 (d, 1H), 4.76 (d, 1H), 4.66 (d, 1H), 4.56 (s, 2H), 4.58-4.42 (m, 2H), 4.06 (d, 1H), 3.95 (d, 1H), 3.88 – 3.77 (m, 2H), 3.69 – 3.60 (m, 2H), 3.42 (d, 1H), 3.33 (s, 3H), 3.28 – 3.10 (m, 3H), 3.06 (s, 3H), 3.00 – 2.87 (m, 2H), 2.81 – 2.61 (m, 2H), 2.33 – 2.07 (m, 5H), 2.01 – 1.63 (m, 7H) Example 30: (1S,4S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-7-amine (Compound 163a)

Step 1: (4S)-7-Bromo-4-methyl-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] DIPEA (2.02 g, 2.72 mL, 16 equiv., 15.6 mmol) was delivered to a 100 mL round bottom flask containing (4S)-7-bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (416 mg, 1 equiv., 977 μmol) and 1,4- oxazepane, HCl (538 mg, 4.0 equiv., 3.91 mmol) in MeCN (6.50 mL). The mixture was heated to 100 °C for 3 hours. After 3 hours, the reaction mixture was cooled to ambient temperature and concentrated under reduced pressure. The residue was purified directly by snap silica chromatography (0-100% EtOAc in DCM) to give (4S)-7-bromo-4-methyl-2'-(methylthio)-4'- (1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (329 mg, 0.57 mmol) which was used as is in the subsequent step. LCMS: m/z (ESI) [M+H]
+ 490.4, tR = 2.84 min (Method E)
Step 2: tert-Butyl ((4S)-4-methyl-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate To a 30 mL vial conatining (4S)-7-bromo-4-methyl-2'-(methylthio)-4'-(1,4-oxazepan- 4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (329 mg, 1 equiv., 671 μmol) and tert-butyl carbamate (393 mg, 5.0 equiv., 3.35 mmol) was added cesium carbonate (656 mg, 3 equiv., 2.01 mmol), and BrettPhos Pd-G4 (92.6 mg, 0.15 equiv., 101 μmol). The reaction vial was purged with nitrogen followed by the addition of 1,4-dioxane (2.8 mL). The mixture was heated to 90 °C for 16 hours. The reaction mixture was cooled to ambient temperature, and concentrated under reduced pressure to give a residue which was dry loaded directly onto silica and purified by snap column chromatography (0 to 100% EtOAc in DCM) to give tert-butyl ((4S)-4-methyl-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (243 mg). LCMS: m/z (ESI) [M+H]
+ 527.3, t
R = 2.69 min (Method E) Step 3: tert-butyl ((4S)-4-methyl-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate Methanol (6.15 mL) and water (3.08 mL) were added to a vial containing tert-butyl ((4S)-4-methyl-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (243 mg, 1 equiv., 461 μmol) followed by Oxone (851 mg, 3 equiv., 1.38 mmol). The reaction mixture was stirred for 16 hours, and was quenched with 3 mL saturated sodium thiosulfate, and diluted with 20 mL water and 30 mL EtOAc. The organic layer was separated and the aqueous layer was further extracted with 3 x 25 mL EtOAc. The combined organics were dried over sodium sulfated and concentrated under reduced pressure to give tert-butyl ((4S)-4-methyl-2'-(methylsulfonyl)-4'- (1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]- 7-yl)carbamate (240 mg). LCMS: m/z (ESI) [M+H]
+ 559.2, t
R = 3.14 min (Method E) Step 4: tert-Butyl ((4S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate Sodium tert-butoxide (322 μL, 2.0 M in THF, 1.5 equiv., 644 μmol) was added dropwise
to a vial containg ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (61.5 mg, 0.9 equiv., 387 μmol) and tert-butyl ((4S)-4-methyl-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4- yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (240 mg, 1 equiv., 430 μmol) in THF at 0 °C. The reaction was quenched after 30 minutes with 2.0 mL saturated ammonium chloride and was then diluted with 5 mL EtOAc. The organic layer was separated and the aqueous layer was further extracted with EtOAc (2 x 20 mL) and the combined organics were dried over sodium sulfate then concentrated to a residue under reduced pressure. The residue was purified by snap column chromatography (12 g SiO2 column, 0-20% MeOH in DCM) to give tert-butyl ((4S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (140 mg). LCMS: m/z (ESI) [M+H]
+ 638.4, tR = 2.01 min (Method E) Step 5: (1S,4S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-7-amine Hydrogen chloride (3 mL, 2.0 M in ether, 30.0 equiv., 6.0 mmol) was added dropwise to a vial containing tert-butyl ((4S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate (140 mg, 1 equiv., 220 μmol) in DCM (3 mL) and was then stirred for 3 hours. Solvent was removed under vacuum to give (4S)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (140 mg, 0.22 mmol) as a mixture of diastereomers, which was subjected to chiral SFC (Chiralpak IA, 21 mm ID x 250mm, 5µm, part number 810445) eluting with 30% EtOH, 0.05% TEA, 70% CO2 isocratic co-solvent, stacked injections to afford (1S,4S)-2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (Peak 1) (31.8 mg) and (1R,4S)-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-4'-(1,4-oxazepan- 4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (Peak 2) (31.3 mg). Peak 1: LCMS: m/z (ESI) [M+H]
+ >95% dr., 538.3 tR = 3.73 minutes. (Chiralpak IA, 2.1 mm ID x 50mm, 1.6µm, part number 80U83). 30% MeOH, 0.25% DEA, 70% CO2 with
30%B isocratic co-solvent. 1H NMR (400 MHz, CDCl3) δ 7.01 (d, J = 8.2 Hz, 1H), 6.72 (m, 1H), 6.65 (m, 1H), 5.51 (d, J = 51.9 Hz, 1H), 4.77 (m, 2H), 4.57 (m, 1H), 4.43 (m, 1H), 4.13 (m, 2H), 3.86 (m, 2H), 3.78 – 3.65 (m, 6H), 3.52 (m, 1H), 3.28 – 3.18 (m, 1H), 2.92 – 2.65 (m, 5H), 2.56 – 2.36 (m, 3H), 2.23 (m, 4H), 1.98 (m, 2H), 1.89 (m, 2H), 1.75 (m, 1H), 1.26 (d, J = 7.0 Hz, 3H). Example 31: (1R,4S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-7-amine (Compound 163b)
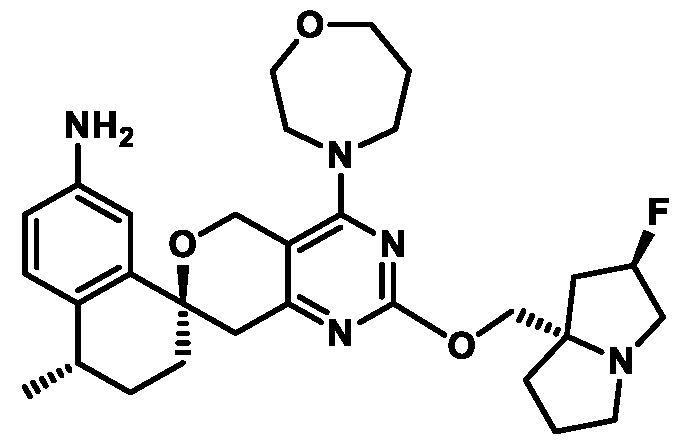
(1R,4S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4- methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-amine (31.3 mg) was obtained from Example 30, as Peak 2: Chiral SFC (Chiralpak IA, 21 mm ID x 250mm, 5µm, part number 810445) eluting with 30% EtOH, 0.05% TEA, 70% CO
2 isocratic co-solvent, stacked injections. Peak 2: LCMS: m/z (ESI) [M+H]
+ >95% dr., 538.3, t
R = 4.90 minutes. (Chiralpak IA, 2.1 mm ID x 50mm, 1.6 µm, part number 80U83).30% MeOH, 0.25% DEA, 70% CO2 with 30%B isocratic co-solvent. 1H NMR (400 MHz, CDCl
3) δ 7.06 (d, J = 8.2 Hz, 1H), 6.69 (m, 1H), 6.62 (m, 1H), 5.35 – 5.18 (m, 1H), 4.70 (m, 1H), 4.51 (m, 1H), 4.10 (m, 1H), 4.01 (m, 1H), 3.82 (m, 2H), 3.72 (m, 5H), 3.64 (m, 2H), 3.34 – 3.17 (m, 3H), 3.10 (m, 1H), 3.01 – 2.86 (m, 3H), 2.32 – 2.21 (m, 2H), 2.20 – 2.13 (m, 2H), 2.11 – 2.02 (m, 3H), 1.93 (m, 6H), 1.26 (d, J = 6.5 Hz, 3H). Example 32: 7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Compound 179a)

Step 1: 7-Amino-4-methyl-3,4-dihydronaphthalen-1(2H)-one To a pressure vessel charged with 7-bromo-4-methyl-3,4-dihydronaphthalen-1(2H)- one (10.0 g, 1.00 equiv., 41.8 mmol) and copper(I) oxide (598 mg, 0.10 equiv., 4.18 mmol), were added NMP (27.9 mL) and ammonium hydroxide (29.3 g, 32.4 mL, 20 equiv., 836 mmol) under nitrogen. The pressure vessel was sealed, and the reaction mixture was stirred at 80 °C for 17 hours. The reaction mixture was cooled down to room temperature and poured into a separatory funnel charged with water (100 mL) and EtOAc (100 mL). The layers were separated and the aqueous phase was extracted with EtOAc (3 x 100 mL). The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated. Crude material was purified by silica gel chromatography (load: CH₂Cl₂, column: RediSep Rf Gold 330 g, 200 mL/min, heptane/EtOAc, 2 → 50% over 25 min) to afford 7-amino-4-methyl-3,4- dihydronaphthalen-1(2H)-one (6.18 g), as a yellow solid. 1H NMR (400 MHz, CDCl
3) δ 7.32 (d, 1H), 7.14 (d, 1H), 6.87 (dd, 1H), 3.70 (br s, 2H), 2.99 (dqd, 1H), 2.75 (ddd, 1H), 2.55 (ddd, 1H), 2.20 (dddd, 1H), 1.84 (dddd, 1H), 1.35 (d, 3H) Step 2: 7-Amino-8-bromo-4-methyl-3,4-dihydronaphthalen-1(2H)-one To a solution of 7-amino-4-methyl-3,4-dihydronaphthalen-1(2H)-one (615 mg, 1.00 equiv., 3.51 mmol) in DMF (5.85 mL) at 0 °C, was added a solution of N-bromosuccinimide (625 mg, 1.00 equiv., 3.51 mmol) in DMF (2.92 mL) dropwise under nitrogen. The resulting mixture was stirred at 0 °C for 5 min and at room temperature (ice/water bath removed) for 2.5 hours. Upon complete conversion to the desired product observed by analytical LCMS, the reaction was quenched by the addition of water (8.0 mL). EtOAc (8.0 mL) was added, and the layers were separated. The aqueous phase was extracted with EtOAc (3 x 8.0 mL). The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated. Crude material was purified by silica gel chromatography (load: CH₂Cl₂, column: RediSep Rf Gold 40 g, 40 mL/min, heptane/EtOAc, 2 → 50% over 20 min) to afford 7-amino-8-bromo-4- methyl-3,4-dihydronaphthalen-1(2H)-one (814 mg). LCMS: m/z (ESI) [M+H]
+ 254.01, t
R = 2.37 minutes (Method E)
Step 3: 8-Bromo-7-(dibenzylamino)-4-methyl-3,4-dihydronaphthalen-1(2H)-one To a solution of 7-amino-8-bromo-4-methyl-3,4-dihydronaphthalen-1(2H)-one (4.00 g, 1.0 equiv., 15.7 mmol) in MeCN (120 mL) at room temperature, were added benzyl bromide (5.92 g, 4.12 mL, 2.2 equiv., 34.6 mmol) and potassium carbonate (6.53 g, 3.0 equiv., 47.2 mmol) sequentially. The resulting mixture was stirred vigorously at reflux for 18 hours. After which point, the reaction was cooled to room temperature and additional potassium carbonate (3.26 g, 1.5 equiv., 23.6 mmol) and benzyl bromide (2.96 g, 2.06 mL, 1.1 equiv., 17.3 mmol) were added. The reaction mixture was heated to reflux and stirred for another 7.5 hours. The reaction mixture was cooled to room temperature. The solids were filtered and rinsed with EtOAc. The filtrate was concentrated. Crude material was purified by silica gel chromatography (load: CH₂Cl₂, column: RediSep Rf Gold 330 g, 200 mL/min, heptane/EtOAc, 1 → 30% over 25 min) to afford, 8-bromo-7-(dibenzylamino)-4-methyl-3,4- dihydronaphthalen-1(2H)-one (6.81 g, 15.7 mmol). LCMS: m/z (ESI) [M+H]
+ 434.0, t
R = 2.31 minutes (Method A) Step 4: 8-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-7-(dibenzylamino)-4-methyl-1,2,3,4-tetrahydronaphthalen-1-ol To a solution of (4-chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (4.81 g, 1.5 equiv., 23.5 mmol) in THF (116 mL) at -78 °C, was added LDA (1.0 M in THF/hexanes) (5.03 g, 47.0 mL, 1.0 M, 3.0 equiv., 47.0 mmol) dropwise. The resulting clear mixture, was stirred at -78 °C for 1.25 hours, followed by the dropwise addition of a solution of 8-bromo-7- (dibenzylamino)-4-methyl-3,4-dihydronaphthalen-1(2H)-one (6.80 g, 1.0 equiv., 15.7 mmol) in THF (58.0 mL). The reaction mixture was stirred -78 °C for 1 hour. The reaction was quenched by the dropwise addition of HCl (2.0 M in Et
2O) (1.83 g, 25.0 mL, 2.0 M, 3.2 equiv., 50.1 mmol) at -78 °C, followed by the addition of a 1:1 mixture of a saturated aqueous NH₄Cl solution and water (100 mL). The biphasic mixture was warmed to room temperature and the layers were separated. The aqueous phase was extracted with EtOAc (3 x 100 mL). The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated. Crude material was purified by silica gel chromatography (load: CH₂Cl₂, column: RediSep Rf Gold 330 g, 200 mL/min, heptane/EtOAc, 2 → 50% over 27 min) to afford 8-bromo-1-((6- chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-7-(dibenzylamino)-4- methyl-1,2,3,4-tetrahydronaphthalen-1-ol (7.87 g, 12.3 mmol).
LCMS: m/z (ESI) [M+H]
+ 638.8, t
R = 2.46 minutes (Method A) Step 5: N,N-dibenzyl-8-bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine To a solution of 8-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-7-(dibenzylamino)-4-methyl-1,2,3,4-tetrahydronaphthalen-1-ol (2.00 g, 1.0 equiv., 3.13 mmol) in THF (26.1 mL) at -78 °C, was added n-butyllithium (2.5 M in hexanes) (441 mg, 2.75 mL, 2.50 M, 2.2 equiv., 6.89 mmol) dropwise. The resulting mixture was stirred at - 78 °C for 30 min, followed by the dropwise addition of a solution of p-toluenesulfonyl chloride (895 mg, 1.5 equiv., 4.69 mmol) in THF (13.0 mL). The acetone/dry ice bath was removed, and the reaction mixture was stirred for another 1.5 hours. The reaction was quenched by the addition of a 1:1 mixture of a saturated aqueous ammonium chloride solution and water (24 mL). EtOAc (20 mL) was added, and the layers were separated. The aqueous phase was extracted with EtOAc (3 x 25 mL). The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated. Crude material was purified by silica gel chromatography, column: RediSep Rf Gold 80 g, 60 mL/min, heptane/EtOAc, 0 → 10% over 25 min) to afford N,N-dibenzyl-8-bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (1.15 g, 1.85 mmol). LCMS: m/z (ESI) [M+H]
+ 620.1, tR = 4.60 minutes (Method E) Step 6: N,N-Dibenzyl-8-bromo-4-methyl-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine To a solution of N,N-dibenzyl-8-bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (1.12 g, 1.0 equiv., 1.80 mmol) and 1,4-oxazepane hydrochloride (620 mg, 2.5 equiv., 4.51 mmol) in 1,4-dioxane (9.02 mL) at room temperature, was added DIPEA (1.63 g, 2.20 mL, 7.0 equiv., 12.6 mmol). The resulting mixture was stirred at 100 °C for 4.3 hours. The volatiles were removed in vacuo and the crude material was purified by silica gel chromatography (load: CH₂Cl₂, column: RediSep Rf Gold 80 g, 60 mL/min, heptane/EtOAc, 2 → 50% over 27 min) to afford N,N- dibenzyl-8-bromo-4-methyl-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (1.17 g, 1.71 mmol). LCMS: m/z (ESI) [M+H]
+ 685.6, t
R = 1.92 minutes (Method A)
Step 7: 7-(Dibenzylamino)-4-methyl-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile To a 100 mL flask charged with copper(I) cyanide (1.53 g, 10 equiv., 17.1 mmol), was added a solution of N,N-dibenzyl-8-bromo-4-methyl-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (1.17 g, 1 equiv., 1.71 mmol) in DMF (34.1 mL) under nitrogen. The resulting mixture was stirred at 120 °C for 2.25 hours. The reaction mixture was cooled to room temperature and concentrated. Water (30 mL) and CH₂Cl₂ (30 mL) and a 1:1 mixture of concentrated NH
4OH and MeOH (30 mL) was added and the mixture was poured in a separatory funnel charged with water (50 mL), CH₂Cl₂ (50 mL) and a 1:1 mixture of concentrated NH
4OH and MeOH (270 mL). The mixture was shaken vigorously, yielding two resolved layers. The layers were separated, and the aqueous layer was extracted with CH₂Cl₂ (3 x 100 mL). The combined organic layers were washed with brine, dried over magnesium sulfate, filtered, and concentrated to give 1.39 g of crude residue. The above residue (1.39 g) was taken up in THF (28.4 mL), methanol (17.2 mL) and water (17.2 mL), subsequently, at room temperature, was added Oxone (9.02 g, 45% wt, 3 equiv., 6.60 mmol) in one portion. The resulting mixture was stirred at room temperature for 3.5 hours. Water (100 mL) and EtOAc (75 mL) were added, and the layers were separated. The aqueous phase was extracted with EtOAc (3 x 100 mL). The combined organic extracts were dried over magnesium sulfate, filtered and concentrated to give 7-(dibenzylamino)-4-methyl- 2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine]-8-carbonitrile (1.10 g) LCMS: m/z (ESI) [M+H]
+ 664.3, tR = 2.16 minutes (Method A) Step 8: 7-(Dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile To a flask charged with ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methanol (1.22 g, 5.0 equiv., 7.68 mmol) was added a solution of 7-(dibenzylamino)-4- methyl-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (1.02 g, 1 equiv., 1.54 mmol) in DMF (30.7 mL) under nitrogen, at room temperature. The resulting solution was cooled to 0 °C, followed by the dropwise addition of lithium bis(trimethylsilyl)amide (1.5 M in THF) (386 mg, 1.54 mL,
1.5 M, 1.5 equiv., 2.30 mmol). The ice/water bath was removed, and the reaction mixture was stirred for 45 minutes. The reaction was quenched by the slow addition of a 1:1 mixture of a saturated aqueous ammonium chloride solution and water (24 mL). EtOAc (50 mL) was added, and the layers were separated. The aqueous phase was extracted with EtOAc (3 x 50 mL). The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated. Crude material was purified by silica gel chromatography (load: CH₂Cl₂, column: RediSep Rf Gold 80 g, 60 mL/min, CH₂Cl₂/[CH₂Cl₂/MeOH/NH₄OH, 75:22.5:2.5], 0 → 100% over 30 min) to afford 741 mg of impure material the major component of which is consistent with the desired product. The material was further purified by reverse phase HPLC prep chromatography by splitting the purification in 4 injections (load: DMSO (4 x 2.0 mL); column: XSelect® CSH™ Prep C185 µm OBD™ 19x150 mm; mobile phase: water 0.1% FA / MeCN 0.1% FA; flow: 18.9 mL/min; gradient: 10 → 35% for 1 min then 35 → 55% over 20 min) to afford 7-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine]-8-carbonitrile (485 mg, 653 μmol). LCMS: m/z (ESI) [M+H]
+ 743.5, t
R = 1.61 minutes (Method A) Step 9: 7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile A vial was charged with 7-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (242 mg, 1 equiv., 326 μmol) and palladium(II) hydroxide on carbon (20 wt. %) (572 mg, 20% wt, 2.5 equiv., 814 μmol). The vial headspace was evacuated and refilled with nitrogen, followed by the addition of MeOH (6.51 mL). The vial headspace was evacuated and refilled with hydrogen from a balloon three times, followed by the addition of acetic acid (16 drops). The reaction mixture was stirred vigorously under an atmosphere of hydrogen (balloon) at room temperature for 2.5 hours. The vial headspace was evacuated and refilled with nitrogen. The reaction mixture was filtered through a syringe filter and the filtrate was stirred with sodium carbonate for 5 min, filtered and concentrated at room temperature to afford 7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (183 mg)
1H NMR (400 MHz, MeOD) δ 7.37 – 7.08 (m, 1H), 6.82 – 6.74 (m, 1H), 5.27 (d, 1H), 5.06 – 4.93 (m, 1H), 4.84 – 4.80 (m, 1H), 4.13 (d, J = 10.3 Hz, 1H), 4.07 – 4.00 (m, 1H), 3.91 – 3.77 (m, 5H), 3.76 – 3.67 (m, 2H), 3.29 – 3.10 (m, 4H), 3.04 – 2.86 (m, 3H), 2.33 – 1.78 (m, 12H), 1.77 – 1.67 (m, 1H), 1.31 – 1.22 (m, 3H). LCMS: m/z (ESI) [M+H]
+ 563.3, tR = 1.27 minutes (Method A) Example 33: 5-(7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-2-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide (Compound 134a)
Step 1: 7-Bromo-2-methyl-3,4-dihydronaphthalen-1(2H)-one To a cooled solution of LDA (1.0 g, 9.8 mL, 1 M, 9.8 mmol) was added 7-bromo-3,4- dihydronaphthalen-1(2H)-one (2.2 g, 9.8 mmol) in THF (10 mL) at - 78 °C. After stirring the mixture for 30 min, MeI (1.7 g, 0.73 mL, 12 mmol) was added. The mixture was gradually warmed to room temperature while stirring overnight. The mixture was treated with aqueous ammonium chloride. The mixture was extracted with EtOAc (2 x 30 mL). The pooled organic fractions were concentrated then directly subjected to purification (0-15% EtOAc/heptanes) to afford 7-bromo-2-methyl-3,4-dihydronaphthalen-1(2H)-one (466 mg). 1H NMR (400 MHz, CDCl
3) δ 8.14 (d, 1H), 7.55 (dd, 2.2 Hz, 1H), 7.12 (d, 1H), 2.98 – 2.85 (m, 2H), 2.64 – 2.51 (m, 1H), 2.20 (ddd, 1H), 1.93 – 1.82 (m, 1H), 1.26 (d, 3H). Step 2: 5-(7-Bromo-2-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide To a solution of (4-chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (360 mg, 1.76 mmol) in THF (7.80 mL) was added LDA (376 mg, 3.50 mL, 1 M, 3.51 mmol) at -70 °C.
The mixture was stirred at this temperature for 45 minutes. The mixture was added, via a cannula, to a cooled (-78
oC) solution of 7-bromo-2-methyl-3,4-dihydronaphthalen-1(2H)-one (280 mg, 1.17 mmol) in THF (3.90 mL). After stirring the mixture for 90 min, the mixture was treated with aqueous ammonium chloride and extracted with EtOAc (2 x 20 mL). The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated. The crude was subjected to purification via flash chromatography (0-20% EtOAc/heptanes) to afford the 7-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-2- methyl-1,2,3,4-tetrahydronaphthalen-1-ol (160 mg) as a clear oil which was used directly in the next step. 7-Bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-2- methyl-1,2,3,4-tetrahydronaphthalen-1-ol (150 mg, 338 μmol) in toluene (25 mL) was treated with phosphoric acid (1 drop) and the mixture heated to 100 °C for about 18 hours. The reaction was cooled to room temperature and quenched by the addition of aqueous sodium bicarbonate and EtOAc and the layers were separated. The aqueous phase was extracted with EtOAc (1 x 15 mL) and the combined organic extracts were dried over magnesium sulfate, filtered and concentrated. The crude was subjected to purification via flash chromatography (0-50% EtOAc/heptanes) to afford 7-bromo-4'-chloro-2-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (40 mg) which was combined with previous material and carried directly to the next step (100 mg total). To a slurry of N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide, 2HCl (132 mg, 2 equiv., 470 μmol) in EtOH (2 mL) were added 7-bromo-4'- chloro-2-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine] (100 mg, 235 μmol) and DIPEA (152 mg, 205 μL, 1.17 mmol). The mixture was heated to 75 °C for 16 hours. The mixture was concentrated and the crude was directly subjected to purification via flash chromatography (0-15% MeOH/DCM) to afford 5-(7- bromo-2-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (55 mg). LCMS: m/z (ESI) [M+H]
+ 597.3, tR = 1.92 minutes (Method C)
Step 3: 5-(7-Bromo-2-methyl-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide To a solution of 5-(7-bromo-2-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (160 mg, 268 μmol) in water (1.2 mL), methanol (1.2 mL) and THF (240 μL) was added Oxone (1.10 g, 45% wt,, 803 μmol) in one portion. The slurry was stirred for 2 hours. The reaction was treated with aqueous sodium thiosulfate, EtOAc (5 mL), and the layers were separated. The aqueous phase was extracted with EtOAc (1 x 15 mL) and the combined organic extracts were dried over magnesium sulfate, filtered, and concentrated. The crude was subjected to purification via flash chromatography (0-15% MeOH/DCM) to afford 5-(7-bromo-2-methyl-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (85 mg) as a white solid. LCMS: m/z (ESI) [M+H]
+ 629.2, 631.2, tR = 1.85 minutes (Method C) Step 4: 5-(7-Bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-2-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide To a solution of 5-(7-bromo-2-methyl-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (59 mg, 94 μmol) and ((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (19 mg, 0.12 mmol) in THF (2 mL) was added a solution of KOtBu (0.14 mL, 1 M, 0.14 mmol) dropwise at -40 °C. The mixture was warmed to 0 °C. After 2 hours, the mixture was treated with aqueous ammonium chloride and EtOAc (5 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (2 x 10 mL). The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated. The crude material was subjected to purification via flash chromatography (0- 20% MeOH/DCM) to afford 5-(7-Bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-2-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (55 mg).
LCMS: m/z (ESI) [M+H]
+ 708.4, 710.4, t
R = 1.45 minutes (Method C) Step 5: tert-Butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-2-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate A slurry of tert-butyl carbamate (14 mg, 0.12 mmol), 5-(7-bromo-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-2-methyl-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (55 mg, 78 μmol) and cesium carbonate (51 mg, 0.16 mmol) in 1,4-dioxane (5 mL) was degassed for 5 min followed by addition of XPhos Palladacycle G4 (14 mg, 16 μmol). The mixture was heated to 100 °C. The mixture was cooled down to room temperature and filtered through a short pad of celite. The mixture was concentrated under reduced pressure and directly subjected to purification via flash chromatography [0-40% (2.5% NH4OH and 20% MeOH in DCM)/DCM] to afford tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-2-methyl-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate. LCMS: m/z (ESI) [M+H]
+ 745.5, tR = 1.47 minutes (Method C) Step 6: 5-(7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-2-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide To a solution of tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-2-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (40 mg, 54 μmol) in 1,4-dioxane (1 mL) was added hydrogen chloride in dioxane (0.13 mL, 4 M, 0.54 mmol) at room temperature. After 1 hour, the mixture was concentrated and the crude was subjected to purification via reverse-phase chromatography (XSelect® CSH C18, 10x150 mm, Mobile phase A: 0.1% TFA in H2O (v/v), mobile phase B: 0.1% TFA in MeCN (v/v), Gradient: 0-50% mobile phase B over 25 minutes.) to afford the formic acid salt of 5-(7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-
7a(5H)-yl)methoxy)-2-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (24 mg) as a white powder. LCMS: m/z (ESI) [M+H]
+ 645.5, t
R = 1.18 minutes (Method C) 1
9F NMR (376 MHz, DMSO) δ -171.60 – -172.22 (m). Example 34: 6-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidine]-5-carbonitrile (Compound 180a)

Step 1: 6-Aminoisochroman-4-one To a pressure vessel containing 6-bromoisochroman-4-one (1.0 g, 4.4 mmol) and copper(I) oxide (63 mg, 0.44 mmol), were added NMP (1.0 mL) and ammonium hydroxide (3.4 mL, 88 mmol) under nitrogen. The pressure vessel was sealed, and the mixture was stirred at 80 °C for 17 hours. The mixture was cooled down to room temperature and poured into a separatory funnel charged with water (100 mL) and EtOAc (100 mL). The layers were separated and the aqueous phase was extracted with EtOAc (3 x 100 mL). The combined organic extracts were dried over sodium sulfate, filtered, and concentrated under reduced pressure to give a crude product which was subjected to purification via flash chromatography (0-50% EtOAc/heptanes) to afford 6-aminoisochroman-4-one (56 mg). 1H NMR (400 MHz, CDCl3) δ 7.30 (d, 1H), 7.03 (d, 1H), 6.89 (dd, 1H), 4.80 (s, 2H), 4.32 (s, 2H), 3.80 (brs, 2H). Step 2: 6-amino-5-bromoisochroman-4-one To a solution of 6-aminoisochroman-4-one (50 mg, 0.31 mmol) in DMF (1 mL) at 0 °C, was added a solution of N-bromosuccinimide (55 mg, 0.31 mmol) in DMF (0.5 mL) dropwise under nitrogen. The resulting mixture was stirred at 0 °C for 1 hour. The mixture was treated with water and extracted with EtOAc and DCM. The organic phase was dried over
sodium sulfate. The crude was subjected to purification via flash chromatography (0-55% EtOAc/heptanes) to afford 6-amino-5-bromoisochroman-4-one (75 mg) LCMS: m/z (ESI) [M+H]
+ 242.1, tR = 1.78 minutes (Method E) Step 3: 5-bromo-6-(dibenzylamino)isochroman-4-one To a solution of 6-amino-5-bromoisochroman-4-one (75.0 mg, 310 μmol) in MeCN (2 mL) at room temperature was added potassium carbonate (214 mg, 1.55 mmol) followed by dropwise addition of BnBr (180 μL, 1.55 mmol). The mixture was heated to 65 °C for 16 hours. The mixture was then treated with additional potassium carbonate (100 mg, 0.72 mmol) and BnBr (100 µL, 0.84 mmol) and was stirred at 80 °C for 2 hours. The mixture was then cooled to room temperature and treated with EtOAc and water. The phases were separated, and the organic phase was dried over sodium sulfate. The crude was subjected to purification via flash chromatography (0-40% EtOAc/heptanes) to afford 5-bromo-6-(dibenzylamino)isochroman- 4-one (105 mg) 1H NMR (400 MHz, CDCl3) δ 7.39 – 7.27 (m, 8H), 7.25 – 7.20 (m, 2H), 6.97 (d, J = 8.2 Hz, 1H), 5.30 (s, 1H), 4.77 (s, 2H), 4.35 (s, 2H), 4.24 (s, 4H). Step 4: 5-Bromo-4-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-6-(dibenzylamino)isochroman-4-ol (4-Chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (48.5 mg, 237 μmol) in THF (2.0 mL) was cooled to -78 °C. LDA solution (550 μL, 1.0 M, 545 μmol) was added dropwise, maintaining a reaction temperature at -78 °C. After stirring for 1 hour, a solution of 5-bromo-6-(dibenzylamino)isochroman-4-one (100 mg, 237 μmol) in THF (2.0 mL) was added dropwise and the reaction was stirred at -78 °C until complete consumption of the starting material was observed. Afterwards, the mixture was poured into a solution of saturated ammonium chloride (30 mL). The mixture was then extracted with EtOAc then DCM. The organic phase was dried over sodium sulfate, filtered and concentrated. The crude was subjected to purification (0-40% EtOAc/heptanes) to afford the partially purified 5-bromo-4- ((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-6- (dibenzylamino)isochroman-4-ol (81 mg), which was used without additional purification LCMS: m/z (ESI) [M+H]
+ 628.1, t
R = 3.83 minutes (Method E)
Step 5: N,N-Dibenzyl-5-bromo-4'-chloro-2'-(methylthio)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine A reaction vessel containing 5-bromo-4-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-6-(dibenzylamino)isochroman-4-ol (81.0 mg, 129 μmol and triphenylphosphine (242 mg, 388 μmol) (polymer supported, 1.6 mmol/g) was degassed with nitrogen. The mixture was then treated with dry THF (2.0 mL) and dry DCM (2.0 mL). Afterwards, DIAD (80 μL, 388 μmol) was added dropwise to the mixture. After stirring for 1 hour, the mixture was filtered over celite and rinsed with DCM. The concentrated crude was directly subjected to purification via flash chromatography (0-30% EtOAc/heptanes) to afford the partially purified product N,N-dibenzyl-5-bromo-4'-chloro-2'-(methylthio)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (39 mg). LCMS: m/z (ESI) [M+H]
+ 608.2, tR = 4.33 minutes (Method E) Step 6: N,N-Dibenzyl-5-bromo-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine To a mixture of N,N-dibenzyl-5-bromo-4'-chloro-2'-(methylthio)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (40.0 mg, 65.7 μmol) and 1,4-oxazepane hydrochloride (36.2 mg, 263 μmol) was added MeCN (1.0 mL) and DIPEA (136 mg, 183 μL, 1.05 mmol). The mixture was then stirred at 80 °C for 3 hours. Afterwards, the mixture was concentrated and directly subjected to purification via flash chromatography (0- 10% MeOH/DCM) to afford the partially purified N,N-dibenzyl-5-bromo-2'-(methylthio)-4'- (1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (39.0 mg), which was used without additional purification. LCMS: m/z (ESI) [M+H]
+ 673.5, t
R = 2.87 minutes (Method E) Step 7: N,N-dibenzyl-5-bromo-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine A mixture of N,N-dibenzyl-5-bromo-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (40.0 mg, 59.4 μmol) and Oxone (170 mg, 125 μmol) were treated with MeOH (1 mL) then water (1 mL). The mixture was then stirred at room temperature for 4 hours and 30 minutes. The mixture was treated with water (10 mL) and extracted with EtOAc (10 mL). The aqueous phase was extracted with EtOAc (3 x 20 mL). The combined organic extracts were dried over sodium sulfate, filtered,
and concentrated to afford N,N-dibenzyl-5-bromo-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)- 5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (36 mg). LCMS: m/z (ESI) [M+H]
+ 705.4, tR = 3.56 minutes (Method E) Step 8: N,N-Dibenzyl-5-bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-6-amine A reaction vessel containing N,N-dibenzyl-5-bromo-2'-(methylsulfonyl)-4'-(1,4- oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (121.0 mg, 171.5 μmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (81.9 mg, 514.4 μmol) was purged with nitrogen. DMF (2 mL) was added, followed by dropwise addition of KOtBu (340 μL, 1.0 M, 342.9 μmol). The mixture was then stirred for 25 min at room temperature. The mixture was then treated with water, followed by extraction with EtOAc. The organic layer was concentrated and directly subjected to purification via flash chromatography (0-15% MeOH/DCM) to afford N,N-dibenzyl-5-bromo-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (124 mg) LCMS: m/z (ESI) [M+H]
+ 784.6, t
R = 2.61 minutes (Method E) Step 9: 6-(Dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidine]-5-carbonitrile Copper(I) cyanide (142 mg, 10 equiv., 1.58 mmol), was added to N,N-dibenzyl-5- bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan- 4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (124 mg, 158 μmol) and dry DMF (3.0 mL) under nitrogen. The resulting mixture was stirred at 100 °C. After 2 hours, the reaction mixture was cooled to room temperature. Water (30 mL), DCM (30 mL), NH
4OH(aq) (20 mL) and methanol (20 mL) were added to the mixture. The phases were separated and the aqueous solution washed with DCM (2 x 20 mL). The organic phase was concentrated to afford 6-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidine]-5-carbonitrile (106 mg) and used immediately without additional purification. LCMS: m/z (ESI) [M+H]
+ 731.5, tR = 2.47 minutes (Method E)
Step 10: 6-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidine]-5-carbonitrile To 6-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidine]-5-carbonitrile in a reaction vial was added palladium(II) hydroxide on carbon (202 mg, 20% wt, 287 μmol). The vial headspace was evacuated and refilled with nitrogen, followed by the addition of MeOH (3 mL). The vial headspace was evacuated and refilled with hydrogen, followed by the addition of acetic acid (4 drops). The reaction mixture was stirred vigorously under an atmosphere of hydrogen at room temperature for 4 hours. The mixture was filtered over celite and washed with DCM and MeOH. The filtrate was concentrated to a residue and purified by reverse-phase chromatography (XSelect® CSH C18, 10x150 mm, Mobile phase A: 0.1% TFA in H
2O (v/v), mobile phase B: 0.1% TFA in MeCN (v/v), Gradient: 10- 35% mobile phase B over 25 minutes.) to afford 6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidine]-5-carbonitrile (5.7 mg). LCMS: m/z (ESI) [M+H]
+ 551.3, t
R = 1.38 minutes (Method E) Example 34a. (S)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidine]-5-carbonitrile (Compound 180c)

The diastereomers of 6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidine]-5-carbonitrile (4 mg, 7.26 µmol) were separated by chiral SFC (Column: Chiralpak IB-N 21 x 250 mm, 5 μm; part number 88445; Mobile phase: Phase A for CO2, and Phase B for MeOH (0.25% DEA), Isocratic elution: B in A from 30% Flow rate: 50 mL/min, Detector: PDA, Column Temp: 40 °C, Back Pressure: 120 bar) to afford (S)-6-amino-2'-
(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)- 5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]-5-carbonitrile (Peak 1) (0.64 mg) and (R)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'- (1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]-5- carbonitrile (Peak 2) (0.93 mg). Peak 1: HPLC: tR = 2.10 minutes (Method G; mobile phase A = CO2, B = Methanol; isocratic 30% B for 5 minutes. @ 40 °C.) 1H NMR (400 MHz, MeOD) δ 7.01 (d, J = 8.6 Hz, 1H), 6.79 (d, J = 8.6 Hz, 1H), 5.38 – 5.18 (m, 1H), 4.99 – 4.90 (m, 2H), 4.63 (s, 2H), 4.13 (d, J = 10.3 Hz, 1H), 4.08 – 3.99 (m, 2H), 3.92 – 3.67 (m, 7H), 3.62 (d, J = 11.1 Hz, 1H), 3.34 (s, 1H), 3.29 – 3.11 (m, 5H), 2.99 (td, J = 9.4, 5.2 Hz, 1H), 2.32 – 1.80 (m, 8H). 1
9F NMR (376 MHz, MeOD) δ -173.36 – -174.07 (m). Example 34b. (R)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidine]-5-carbonitrile (Compound 180b)
(R)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'- (1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]-5- carbonitrile (0.93 mg) was obtained from Example 34a, as Peak 2: HPLC: tR = 2.61 minutes (Method G; mobile phase A = CO
2, B = Methanol; isocratic 30% B for 5 minutes. @ 40 °C.) 1H NMR (400 MHz, MeOD) δ 7.01 (d, J = 8.6 Hz, 1H), 6.79 (d, J = 8.6 Hz, 1H), 5.28 (d, J = 53.9 Hz, 1H), 5.00 – 4.90 (m, 2H), 4.68 – 4.57 (m, 2H), 4.14 (d, J = 10.4 Hz, 1H), 4.09 – 3.98 (m, 2H), 3.92 – 3.67 (m, 7H), 3.61 (d, J = 11.2 Hz, 1H), 3.35 – 3.32 (m, 1H), 3.27 – 3.09 (m, 5H), 3.03 – 2.93 (m, 1H), 2.34 – 1.81 (m, 8H). 1
9F NMR (376 MHz, MeOD) δ -173.28 – -173.87 (m).
Example 35: 5-(7-amino-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide (Compound 139a)
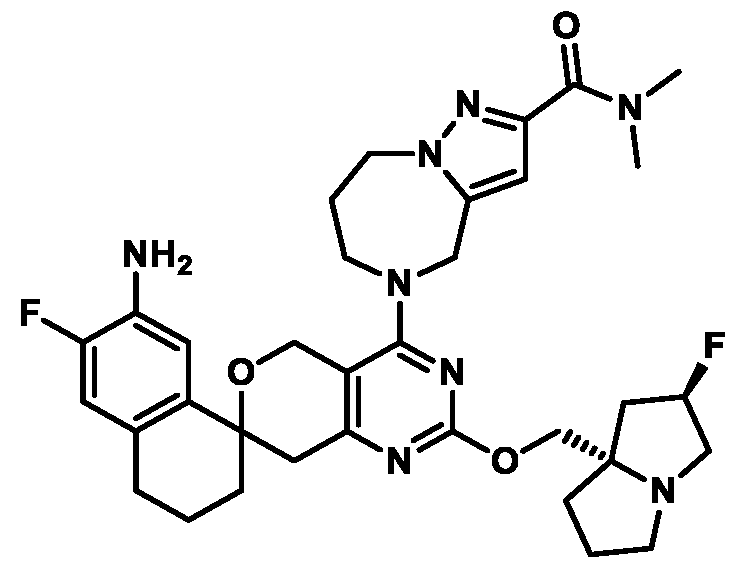
Step 1: 7-chloro-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-6-fluoro-1,2,3,4-tetrahydronaphthalen-1-ol (4-Chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (1500 mg, 7.33 mmol) was dissolved in THF (15 mL) and cooled to -78 °C. LDA solution (18.7 mL, 0.9 M, 16.9 mmol) was added dropwise while maintaining a temperature below -70 °C. The solution was stirred for 30 minutes. A solution of 7-chloro-6-fluoro-3,4-dihydronaphthalen-1(2H)-one (1.74 g, 8.8 mmol) in THF (2.3 mL) was added dropwise to the reaction mixture while maintaining a temperature below -65 °C. After 15 minutes, the reaction mixture was cannulated into a stirring solution of a saturated aqueous ammonium chloride solution. The aqueous layer was extracted with EtOAc (10 mL x 3), dried over sodium sulfate, filtered, and concentrated to a golden oil. The crude material was purified by column chromatography (SiO2, 0-20% ethyl acetate in DCM) to give 7-chloro-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-6-fluoro-1,2,3,4-tetrahydronaphthalen-1-ol (513 mg, 1.27 mmol). LCMS: m/z (ESI) [M-H
2O+H]
+ 385.3, t
R = 2.05 minutes (Method A) Step 2: 4',7-Dichloro-6-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] 7-chloro-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-6- fluoro-1,2,3,4-tetrahydronaphthalen-1-ol (513 mg, 1.27 mmol) was dissolved in toluene (25.4 mL). Phosphoric acid (147 mg, 87 μL, 1.27 mmol) was added and the reaction was heated to 90 °C for 2 hours. The mixture was cooled to 25 °C and the solvent was removed under vacuum.
EtOAc (25 mL) and water (25 mL) were added to the crude material. The aqueous layer was washed an additional three times with 25 mL of EtOAc and the organic layers were combined and dried over sodium sulfate and then concentrated to give 4',7-dichloro-6-fluoro-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (471 mg, 1.1 mmol) which was used without further purification. LCMS: m/z (ESI) [M+H]
+ 385.3, tR = 2.41 minutes (Method A) Step 3: 5-(7-Chloro-6-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide 4',7-Dichloro-6-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine] (472 mg, 1.22 mmol) and N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide, 2HCl (564 mg, 1.63 equiv., 2.00 mmol) were dissolved in ethanol (3 mL) DIPEA (853 μL, 4.89 mmol). The reaction mixture was heated to 70 °C for 12 hours. Solvent was removed under reduced pressure and the resulting brown oil was diluted with EtOAc (10 mL). The organics were washed with water (15 mL), and NaOH (aq, 1 M) (15 mL). The organics were then dried over sodium sulfate, filtered, and concentrated to a light orange solid. The crude material was purified by column chromatography (SiO2, 0- 20% MeOH in DCM) to give 5-(7-chloro-6-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (563 mg, 1.01 mmol). LCMS: m/z (ESI) [M+H]
+ 557.3, tR = 1.83 minutes (Method A) Step 4: 5-(7-chloro-6-fluoro-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide 5-(7-Chloro-6-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (563.8 mg, 1.012 mmol) and Oxone (1.53 g, 2.49 mmol) were dissolved in methanol (6.7 mL). Water (3.4 mL) was added dropwise via syringe. The reaction was stirred for 12 hours. An aqueous solution of sodium thiosulfate (15 mL) was added to the mixture, followed by EtOAc (15 mL), and the layers were separated. The aqueous phase was extracted with EtOAc (15 mL x 3). The organic extracts were combined and dried over sodium
sulfate, filtered and concentrated to yield 5-(7-chloro-6-fluoro-2'-(methylsulfonyl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (564.8 mg, 0.96 mmol). LCMS: m/z (ESI) [M+H]
+ 589.3, t
R = 1.83 minutes (Method A) Step 5: 5-(7-chloro-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide A round bottom flask containing 5-(7-chloro-6-fluoro-2'-(methylsulfonyl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (564.8 mg, 1 equiv., 959 μmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (149 mg, 0.98 equiv., 936 μmol) was evacuated and filled with N
2 three times then charged with THF (1.91 mL) and cooled to 0 °C. A 1 M solution of potassium 2-methylpropan-2-olate in THF (1.918 mL) was added dropwise to the solution. After 20 minutes EtOAc (5 mL) and water (5 mL) were added to the solution. The aqueous layer was washed with EtOAc (5 mL x 3). The organic layers were combined, dried over sodium sulfate, filtered, and concentrated to a dark orange oil. The Crude material was purified by column chromatography (SiO20-25% MeOH in DCM) to give 5-(7-chloro-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (296 mg). LCMS: m/z (ESI) [M+H]
+ 667.9, tR = 1.38 minutes (Method C) Step 6: tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate 5-(7-chloro-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (264 mg, 1 equiv., 395 μmol), potassium carbonate (169 mg, 1.22 mmol), tert-butyl carbamate (231.3 mg, 1.975 mmol), and BrettPhos Pd G4 (91 mg, 98.7 μmol) were combined under a nitrogen
atmosphere.1,4-Dioxane (2.0 mL) was added to the reaction mixture, and the mixture was stirred at 90 °C for 1 hour. The reaction was then cooled to 25 °C, filtered, and concentrated under reduced pressure. The crude material was purified by column chromatography (SiO2, 0- 50% [DCM/MeOH/NH₄OH, 75:22.5:2.5] in DCM) to give tert-butyl (4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-6-fluoro-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (158 mg, 0.21 mmol). LCMS: m/z (ESI) [M+H]
+ 749.5, tR = 1.41 minutes (Method C) Step 7: 5-(7-amino-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide tert-Butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin- 5(6H)-yl)-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (159 mg, 1 equiv., 212 μmol) was dissolved in DCM (2.1 mL) and cooled to 0 °C. HCl (4.0 M in 1,4-dioxane) (2.7 mL, 10.6 mmol) was added dropwise and the resulting mixture was stirred at 25 °C for 45 minutes. The mixture was concentrated in vacuo and the material was dried for 2 days under vacuum to yield, 5-(7-amino-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (176.4 mg, contains residual solvent), as a light yellow solid. 1H NMR (400 MHz, DMSO) δ 7.22 – 7.17 (m, 1H), 7.00 (d, J = 11.5 Hz, 1H), 6.67 (s, 1H), 5.58 (d, J = 52.5 Hz, 1H), 4.92 (d, J = 18.7 Hz, 3H), 4.71 – 4.40 (m, 5H), 3.99 (s, 2H), 3.88 – 3.62 (m, 5H), 3.48 (dd, J = 11.5, 4.5 Hz, 1H), 3.25 (s, 4H), 2.95 (s, 4H), 2.72 (s, 2H), 2.30 (d, J = 10.2 Hz, 1H), 2.19 – 1.82 (m, 9H), 1.68 (s, 1H), LCMS: m/z (ESI) [M+H]
+ 648.9, t
R = 1.24 minutes (Method C) Example 35a. 5-((R)-7-amino-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 139b)

The diastereomers of 5-(7-amino-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide were separated using preparative SFC chromatography (Column AS-5H 21 x 250 mm, Flow rate: 70 mL/min, 45% Isopropanol + 0.25% diethylamine in CO2.) to afford 5-((R)- 7-amino-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl- 5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Peak 1) (40.5 mg) and 5-((S)-7-amino-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl- 5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Peak 2) (7.8 mg). Peak 1: SFC: (Column ChiralPak AS-H 4.6 x 100 mm Column AS-5H 21 x 250 mm, Flow rate: 2.5 mL/min, 30% Isopropanol + 0.25% diethylamine in CO
2) t
R = 1.97 minutes LCMS: m/z (ESI) [M+H]
+ 649.2, tR = 1.54 minutes (Method C) Example 35b: 5-((S)-7-amino-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 139c)
5-((S)-7-amino-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (7.8 mg) was obtained from Example 35a, as Peak 2: SFC: (Column ChiralPak AS-H 4.6 x 100 mm Column AS-5H 21 x 250 mm, Flow rate: 2.5 mL/min, 30% Isopropanol + 0.25% diethylamine in CO
2) t
R = 2.76 minutes LCMS: m/z (ESI) [M+H]
+ 649.2, tR = 1.54 minutes (Method C) 1H NMR (400 MHz, DMSO-d6) δ 6.85 – 6.66 (m, 2H), 6.50 (s, 1H), 5.26 (d, 1H), 4.91 – 4.67 (m, 4H), 4.60 – 4.37 (m, 2H), 3.99 – 3.76 (m, 3H), 3.32 – 3.19 (m, 5H), 3.10 – 2.90 (m, 5H), 2.90 – 2.74 (m, 3H), 2.72 – 2.53 (m, 2H), 2.23 – 1.59 (m, 12H). Example 37: 5-(7-Amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 171a)

Step 1: 4-(4-Bromo-2-chlorophenyl)butanal But-3-en-1-ol (2.73 g, 3.25 mL, 1.20 equiv., 37.8 mmol) was added to a solution of 4- bromo-2-chloro-1-iodobenzene (10.0 g, 1.00 equiv., 31.5 mmol), palladium diacetate (424 mg, 0.06 equiv., 1.89 mmol), tetrabutylammonium chloride hydrate (18.7 g, 2.00 equiv., 63.0 mmol), lithium chloride (1.34 g, 1.00 equiv., 31.5 mmol), and lithium acetate (5.20 g, 2.50 equiv., 78.8 mmol) in DMF (75.0 mL) under N2. The reaction mixture was stirred for 24 hours at 70 °C. The product mixture was concentrated, and the residue obtained was diluted with a 1:1 mixture of saturated aqueous ammonium chloride (125 mL) and EtOAc (125 mL). The layers that formed were separated, and the aqueous phase was extracted with EtOAc (3 x 100 mL). The combined organic layers were dried over magnesium sulfate, filtered, and
concentrated. The residue obtained was purified by column chromatography (SiO
2, 0-15% EtOAc in heptanes) to afford 4-(4-bromo-2-chlorophenyl)butanal (3.09 g, 11.8 mmol) as a yellow oil. 1H NMR (400 MHz, DMSO-d
6) δ 9.68 (s, 1H), 7.70 – 7.67 (m, 1H), 7.53 – 7.48 (m, 1H), 7.32 (d, 1H), 2.68 (t, 2H), 2.50 (t, 2H), 1.87 – 1.77 (m, 2H). Step 2: 4-(4-Bromo-2-chlorophenyl)butanoic acid 2-Methyl-2-butene (33.1 g, 50.1 mL, 40 equiv., 473 mmol), sodium dihydrogen phosphate monohydrate (16.3 g, 10 equiv., 118 mmol), and sodium chlorite (5.34 g, 5.0 equiv., 59.1 mmol) were added sequentially to a solution of 4-(4-bromo-2-chlorophenyl)butanal (3.09 g, 1.0 equiv., 11.8 mmol) in a 2:1 mixture of t-BuOH (78.8 mL) and water (39.4 mL) at 0 °C. The reaction mixture was stirred vigorously for 15 min at 0 °C and for 1.5 hours at room temperature. The product mixture was directly concentrated then diluted with DCM (40 mL) and a 1:1 mixture of water and brine (40 mL). The layers that formed were separated, and the aqueous layer was extracted with DCM (3 x 40 mL). The combined organic layers were dried over magnesium sulfate, filtered and concentrated to provide 4-(4-bromo-2- chlorophenyl)butanoic acid (3.40 g, 12.0 mmol) which was used without further purification. 1H NMR (400 MHz, DMSO-d
6) δ 12.13 (s, 1H), 7.68 – 7.63 (m, 1H), 7.51 – 7.45 (m, 1H), 7.31 – 7.26 (m, 1H), 2.67 (t, 2H), 2.24 (t, J = 7.4 Hz, 2H), 1.82 – 1.72 (m, 2H). Step 3: 7-Bromo-5-chloro-3,4-dihydronaphthalen-1(2H)-one A 2.0 M solution of oxalyl chloride (2.94 g, 11.6 mL, 2 equiv., 23.1 mmol) was added slowly to a solution of 4-(4-bromo-2-chlorophenyl)butanoic acid (3.21 g, 1 equiv., 11.6 mmol) and DMF (84.5 mg, 89.6 μL, 0.1 equiv., 1.16 mmol) in DCM (60.6 mL) under N
2. The reaction mixture was stirred for 2 hours at room temperature. The reaction mixture was directly concentrated and diluted with DCM (60.6 mL). Aluminum chloride (3.08 g, 2 equiv., 23.1 mmol) was added to the mixture under N
2. The reaction mixture was stirred for 18 hours at room temperature. The product mixture was quenched with cold water (60 mL) and brine (20 mL). The layers that formed were separated, and the aqueous layer was extracted with DCM (3 x 50 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO
2, 0-60% EtOAc in heptanes) to provide 7-bromo-5-chloro-3,4-dihydronaphthalen-1(2H)-one (2.08 g, 802 mmol) as an orange solid.
1H NMR (400 MHz, DMSO-d
6) δ 8.00 (s, 1H), 7.91 (s, 1H), 2.91 (t, 2H), 2.63 (t, 2H), 2.13 – 2.03 (m, 2H). Step 4: 7-Bromo-5-chloro-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-ol A 1.0 M LDA solution (795 mg, 7.4 mL, 2.3 equiv., 7.42 mmol) was added dropwise, while maintaining a temperature below -70 °C, to a solution of (4-chloro-6-methyl-2- (methylthio)pyrimidin-5-yl)methanol (660 mg, 1 equiv., 3.22 mmol) in THF (6.7 mL) in a dry flask under N2 at -78 °C. The reaction mixture was stirred for 1.5 hours. A solution of 7-bromo- 5-chloro-3,4-dihydronaphthalen-1(2H)-one (924 mg, 1.1 equiv., 3.56 mmol) in THF (1.0 mL) was added dropwise to the reaction mixture, while maintaining a temperature of -78 °C. The reaction mixture was stirred for 30 min then poured into saturated aqueous ammonium chloride (10 mL). The aqueous layer was extracted with EtOAc (3 x 8 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO2, 0-20% EtOAc in DCM) to give 7-bromo-5-chloro- 1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-1,2,3,4- tetrahydronaphthalen-1-ol (272 mg, 586 μmol) as a white powder. 1H NMR (400 MHz, DMSO-d
6) δ 7.59 (s, 1H), 7.52 (s, 1H), 5.63 (s, 1H), 5.28 (t, 1H), 4.84 – 4.78 (m, 1H), 4.68 – 4.61 (m, 1H), 3.37 (d, 1H), 3.17 (d, 1H), 2.78 (d, 1H), 2.67 – 2.57 (m, 1H), 2.39 (s, 3H), 2.14 – 1.81 (m, 4H). Step 5: 7-Bromo-4',5-dichloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] Phosphoric acid (67.6 mg, 40 μL, 85% wt, 586 μmol) was added to a solution of 7- bromo-5-chloro-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)- 1,2,3,4-tetrahydronaphthalen-1-ol (272.1 mg, 1 equiv., 586.2 μmol) in toluene (12 mL) in a dry flask under N
2. The reaction mixture was refluxed for 2 hours. The product mixture was cooled to room temperature and was directly concentrated. The residue obtained was diluted with EtOAc (3 mL) and water (3 mL). The layers that formed were separated, and the aqueous layer was extracted with EtOAc (3 x 3 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated to give 7-bromo-4',5-dichloro-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (228 mg, 510 μmol) which was used without further purification.
1H NMR (400 MHz, DMSO-d
6) δ 7.72 – 7.65 (m, 2H), 7.60 – 7.42 (m, 1H), 4.80 (d, 1H), 4.69 (d, 1H), 4.61 (s, 1H), 4.13 (s, 1H), 3.24 (d, 1H), 3.03 (d, 1H), 2.82 – 2.72 (m, 2H), 2.68 – 2.58 (m, 1H), 2.52 (s, 3H). Step 6: 5-(7-Bromo-5-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide DIPEA (264 mg, 355 μL, 4 equiv., 2.04 mmol) was added to a solution of 7-bromo-4',5- dichloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine] (228 mg, 1 equiv., 510 μmol) and N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide, 2HCl (229 mg, 1.6 equiv., 816 μmol) in EtOH (1.3 mL). The reaction mixture was stirred for 22 hours at 70 °C. DMF (500 μL), N,N-dimethyl- 5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide, 2HCl (229 mg, 1.6 equiv., 816 μmol), and DIPEA (131.8 mg, 178 μL, 2 equiv., 1.020 mmol) were added. The reaction mixture was stirred for an additional 3.5 hours at 70 °C. The product mixture was directly concentrated, and the residue obtained was diluted with EtOAc (3 mL). The product solution was washed sequentially with water (2 mL) and a 1.0 M aqueous NaOH solution (2 mL). The washed organic layer was dried over sodium sulfate, filtered, and concentrated to give 5-(7-bromo-5-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (306 mg, 495 μmol) which was used without further purification. LCMS: m/z (ESI) [M+H]⁺ 617.1, tR = 2.06 minutes (Method C) Step 7: tert-Butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate 1,4-Dioxane (9.9 mL) that had been sparged with N
2 for 30 min was added to a mixture of 5-(7-bromo-5-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (306 mg, 1 equiv., 495 μmol), XPhos Pd G4 (128 mg, 0.3 equiv., 149 μmol), cesium carbonate (323 mg, 2 equiv., 990 μmol), and tert-butyl carbamate (69.6 mg, 1.2 equiv., 594 μmol) under N2. The reaction mixture was stirred for 18 hours at 100
°C. The product mixture was cooled to room temperature, passed through a syringe filter, and concentrated. The residue obtained was purified by column chromatography (SiO2, 0-40% DCM:MeOH:NH₄OH 75:22.5:2.5 in DCM) to give tert-butyl (5-chloro-4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (245 mg, 260 μmol) as an orange oil. LCMS: m/z (ESI) [M+H]⁺ 654.2, t
R = 1.97 minutes (Method C) Step 8: tert-Butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate mCPBA (251 mg, 77% wt, 4.28 equiv., 1.12 mmol) was added to a solution of tert-butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)- yl)-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (245 mg, 70% wt, 1 equiv., 262 μmol) in DCM (5.2 mL) at 0 °C. The reaction mixture was stirred for 10 min at 0 °C and for 1 hour at room temperature. The product mixture was diluted with DCM (2 mL) and a 3:1 mixture of saturated aqueous sodium bicarbonate and water (5 mL). The resulting biphasic mixture was stirred for 30 minutes. The organic layer was separated, and the aqueous layer was extracted with DCM (2 x 3 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated to give tert-butyl (5-chloro- 4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'- (methylsulfonyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (255 mg, 220 μmol) which was used without further purification. LCMS: m/z (ESI) [M+H]⁺ 686.3, t
R = 1.96 minutes (Method C) Step 9: tert-Butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate A 1.0 M potassium 2-methylpropan-2-olate solution (83.4 mg, 82.0 μL, 3.33 equiv., 743 μmol) was added dropwise to a solution of tert-butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8- dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (255 mg, 60% wt, 1
equiv., 223 μmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (57.7 mg, 1.63 equiv., 362 μmol) in THF (446 μL) under N2 at 0 °C. The reaction mixture was stirred for 30 minutes. EtOAc (2 mL) and water (1 mL) were added in sequence to the product solution. The aqueous layer was washed with EtOAc (3 x 2 mL). The aqueous layer was further diluted with saturated aqueous ammonium chloride (2 mL) and EtOAc (2 mL), then concentrated partially in vacuo. EtOAc (4 mL) and water (2 mL) were added. The layers that formed were separated, and the aqueous layer was extracted with EtOAc (3 x 3 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO2, 0-30% MeOH in DCM) to give tert- butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin- 5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (35.6 mg, 223 μmol) as an orange solid. LCMS: m/z (ESI) [M+H]⁺ 765.5, t
R = 1.50 minutes (Method C) Step 10: 5-(7-Amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide A 4.0 M HCl solution in 1,4-dioxane (84.8 mg, 581 μL, 4.0 M, 50 equiv., 2.33 mmol) was added dropwise to a solution of tert-butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8- dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (35.6 mg, 1 equiv., 46.5 μmol) in DCM (465 μL) at 0 °C. The reaction mixture was stirred for 30 min at room temperature. The product mixture was directly concentrated, and the residue obtained was purified by reverse phase column chromatography (C18, 0-100% MeCN in water (0.1% NH
4OH additive)) to provide 5-(7-amino-5-chloro-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (3.2 mg, 4.8 μmol) as a white solid. LCMS: m/z (ESI) [M+H]⁺ 665.4, t
R = 1.28 minutes (Method C)
1H NMR (400 MHz, CD
3CN) δ 6.66 (d, 1H), 6.61 (d, 1H), 6.41 (s, 1H), 5.39 – 5.10 (m, 1H), 4.74 (d, 1H), 4.71 – 4.56 (m, 2H), 4.52 (d, 1H), 4.46 – 4.38 (m, 2H), 4.12 (s, 2H), 3.98 (d, 1H), 3.91 (d, 1H), 3.86 – 3.80 (m, 2H), 3.23 (s, 3H), 3.13 – 3.00 (m, 3H), 2.97 (s, 3H), 2.93 – 2.81 (m, 3H), 2.76 – 2.59 (m, 2H), 2.10 – 1.98 (m, 6H), 1.91 – 1.69 (m, 6H). Example 38: 5-(7-Amino-5-chloro-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 170a)
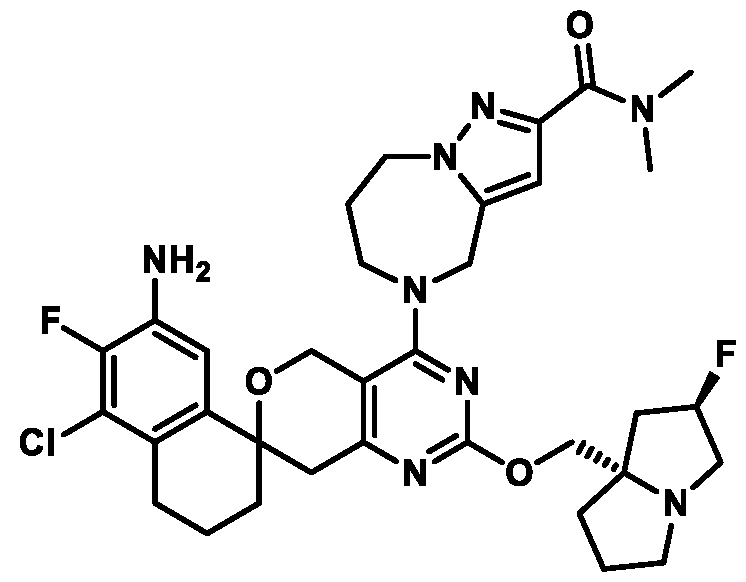
Step 1: 4-(4-Bromo-2-chloro-3-fluorophenyl)butanal A solution of but-3-en-1-ol (748 mg, 1.2 equiv., 10.4 mmol) and 1-bromo-3-chloro-2- fluoro-4-iodobenzene (2.90 g, 1 equiv., 8.65 mmol) in DMF (10 mL) was added to a solution of tetrabutylammonium chloride, hydrate (5.12 g, 2 equiv., 17.3 mmol), lithium acetate (1.43 g, 2.5 equiv., 21.6 mmol), and lithium chloride (367 mg, 1 equiv., 8.65 mmol) in DMF (50 mL) under N2. Palladium(II) acetate (116 mg, 0.06 equiv., 519 μmol) was added, and the reaction mixture was sparged with N
2 for 7 min. The mixture was then stirred for 16 hours at 70 °C. The product mixture was filtered, and the filter cake was washed with EtOAc (20 mL). The filtrate was concentrated, and the residue obtained was diluted with EtOAc (30 mL), ether (30 mL), and water (100 mL). The layers that formed were separated, and the organic layer was washed sequentially with water (4 x 30 mL), 1.0 M aqueous HCl solution (20 mL), and brine (2 x 20 mL). The washed organic layer was dried over sodium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO2, 0-50% EtOAc in Hexanes) to provide 4-(4-bromo-2-chloro-3-fluorophenyl)butanal (1.65 g, 5.90 mmol) as a yellow oil.
1H NMR (400 MHz, CDCl
3) δ 9.79 (t, 1H), 7.47 – 7.32 (m, 1H), 6.93 – 6.90 (m, 1H), 2.80 – 2.73 (m, 2H), 2.54 – 2.47 (m, 2H), 2.01 – 1.89 (m, 2H). Step 2: 4-(4-Bromo-2-chloro-3-fluorophenyl)butanoic acid Sodium chlorite (2.67 g, 5 equiv., 29.5 mmol) was added slowly to a mixture of 4-(4- bromo-2-chloro-3-fluorophenyl)butanal (1.65 g, 1 equiv., 5.90 mmol), sodium phosphate monobasic monohydrate (8.15 g, 10 equiv., 59.0 mmol), and 2-methyl-2-butene (8.28 g, 12.5 mL, 20 equiv., 118 mmol) in t-BuOH (30 mL) and water (15 mL) at 0 °C. The reaction mixture was stirred for 2 hours at room temperature. A 5.0 M aqueous HCl solution was added to reach a pH = 3. The aqueous layer was extracted with EtOAc (4 x 15 mL). The combined organic layers were washed with brine (3 x 15 mL), dried over sodium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO2, 0-5% MeOH in EtOAc) to provide 4-(4-bromo-2-chloro-3-fluorophenyl)butanoic acid (1.70 g, 5.75 mmol) as an off white solid. 1H NMR (400 MHz, CDCl3) δ 7.41 – 7.35 (m, 1H), 6.95 – 6.91 (m, 1H), 2.83 – 2.76 (m, 2H), 2.42 (t, 2H), 2.01 – 1.91 (m, 2H). Step 3: 7-Bromo-5-chloro-6-fluoro-3,4-dihydronaphthalen-1(2H)-one Oxalyl chloride (1.5 g, 1.0 mL, 2 equiv., 12 mmol) and DMF (42 mg, 45 μL, 0.10 equiv., 0.58 mmol) were added to a solution of 4-(4-bromo-2-chloro-3-fluorophenyl)butanoic acid (1.7 g, 1 equiv., 5.8 mmol) in anhydrous DCM (20 mL) at room temperature. The reaction mixture was stirred for 15 min then directly concentrated. The residue obtained was diluted with DCM (10 mL) and aluminum chloride (1.5 g, 2 equiv., 12 mmol) was added. The reaction mixture was stirred for 16 hours at room temperature. The product mixture was diluted with cold water (10 mL) and CHCl3 (10 mL). The organic layer was separated, and the aqueous layer was extracted with CHCl3 (3 x 10 mL). The combined organic layers were washed with brine (3 x 10 mL), dried over sodium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO
2, 0-50% EtOAc in Hexanes) to yield 7-bromo- 5-chloro-6-fluoro-3,4-dihydronaphthalen-1(2H)-one (1.47 g, 5.30 mmol) as an off white solid. LCMS: m/z (ESI) [M+H]
+ 279.1, t
R = 1.70 minutes (Method A).
Step 4: 7-Bromo-5-chloro-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-6-fluoro-1,2,3,4-tetrahydronaphthalen-1-ol A 1.0 M LDA solution (724 mg, 6.76 mL, 1 M, 2.5 equiv., 6.76 mmol) was added to a solution of (4-chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (553 mg, 1 equiv., 2.70 mmol) in anhydrous THF (20 mL) at -78 °C via syringe pump over 20 minutes. The reaction mixture was stirred for 1 hour. A solution of 7-bromo-5-chloro-6-fluoro-3,4- dihydronaphthalen-1(2H)-one (750 mg, 1 equiv., 2.70 mmol) in anhydrous THF (10 mL) was added via syringe pump over 15 minutes, and the reaction mixture was stirred for 1 hour at -78 °C. The product mixture was quenched with saturated aqueous ammonium chloride (20 mL) and was warmed to room temperature. The aqueous layer was extracted with EtOAc (3 x 30 mL), and the combined organic layers were washed with brine (2 x 20 mL). The washed organic layers were dried over sodium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO2, 0-40% EtOAc in DCM) to afford 7-bromo-5- chloro-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-6-fluoro- 1,2,3,4-tetrahydronaphthalen-1-ol (800 mg, 1.66 mmol) as a white solid. LCMS: m/z (ESI) [M+H]
+ 483.1, tR = 1.87 minutes (Method 1). Step 5: 7-Bromo-4',5-dichloro-6-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] Phosphoric acid (203 mg, 80% wt, 1 equiv., 1.66 mmol) was added to a suspension of 7-bromo-5-chloro-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)- 6-fluoro-1,2,3,4-tetrahydronaphthalen-1-ol (800 mg, 1 equiv., 1.66 mmol) in anhydrous toluene (30 mL). The reaction mixture was refluxed for 1 hour. The product mixture was diluted with EtOAc (40 mL) and water (40 mL). The organic layer was separated, and the aqueous layer was extracted with EtOAc (4 x 15 mL). The combined organic layers were washed with brine (3 x 15 mL), dried over sodium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO
2, 0-5% EtOAc in DCM) to afford 7-bromo-4',5-dichloro-6-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (513 mg, 1.11 mmol) as an off white solid. LCMS: m/z (ESI) [M-H]
+ 465.1, t
R = 2.26 minutes (Method A).
Step 6: 5-(7-Bromo-5-chloro-6-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide A solution of N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide hydrochloride (406 mg, 1.5 equiv., 1.66 mmol), 7-bromo-4',5-dichloro-6-fluoro- 2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (513 mg, 1 equiv., 1.11 mmol), and DIPEA (857 mg, 1.16 mL, 6 equiv., 6.63 mmol) in 1,4-dioxane (20 mL) was stirred for 48 hours at 110 °C. The product mixture was cooled to room temperature and was diluted with CHCl3 (20 mL) and water (20 mL). The organic layer was separated, and the aqueous layer was extracted with 4:1 CHCl
3:MeOH (4 x 15 mL). The combined organic layers were washed with brine (3 x 10 mL), dried over sodium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO2, 0-10% MeOH in EtOAc) to provide 5-(7-bromo-5-chloro-6-fluoro-2'-(methylthio)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl- 5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (601 mg, 945 μmol) as an off white solid. 1H NMR (400 MHz, CDCl
3) δ 7.57 (d, 1H), 6.55 (s, 1H), 4.80 (d, 1H), 4.69 – 4.63 (m, 1H), 4.61 – 4.56 (m, 1H), 4.55 – 4.41 (m, 2H), 4.03 – 3.94 (m, 1H), 3.81 – 3.68 (m, 1H), 3.49 (s, 1H), 3.33 (s, 3H), 3.20 – 2.97 (m, 5H), 2.91 – 2.64 (m, 2H), 2.50 (s, 3H), 2.34 – 2.05 (m, 4H), 1.94 – 1.75 (m, 2H). Step 7: tert-Butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-6-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate Brettphos Pd G4 (261 mg, 0.30 equiv., 283 μmol) was added to a solution of 5-(7- bromo-5-chloro-6-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (600 mg, 1 equiv., 943 μmol), cesium carbonate (922 mg, 3 equiv., 2.83 mmol), and tert-butyl carbamate (553 mg, 5 equiv., 4.72 mmol) in DMF (31.4 mL) under N2. The reaction mixture was sparged with N2 for an additional 3 minutes. The reaction mixture was stirred for 16 hours at 100 °C. The product mixture was cooled to room temperature and was filtered. The filter cake was washed with EtOAc (20 mL), and the filtrate layers were separated. The aqueous layer was extracted with EtOAc (4 x 15 mL). The combined
organic layers were washed with brine (3 x 15 mL), dried over sodium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO2, 0-10% MeOH in DCM then 100% EtOAc (1% NH4OH additive)) to provide tert-butyl (5-chloro-4'- (2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-6-fluoro-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (200 mg, 298 μmol) as an off-white solid. LCMS: m/z (ESI) [M+H]
+ 672.3, t
R = 5.20 minutes (Method I). Step 8: tert-Butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-6-fluoro-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate Hydrogen peroxide (236 mg, 213 μL, 30% wt, 7 equiv., 2.08 mmol) was added to a solution of tert-butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-6-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (200 mg, 1 equiv., 298 μmol), sodium tungstate dihydrate (9.81 mg, 0.10 equiv., 29.8 μmol), and hydrogen tetra(but-1-yl)ammonium sulphate (16.2 mg, 0.16 equiv., 47.6 μmol) in EtOAc (10 mL). The reaction mixture was stirred for 40 min at 40 °C. The product mixture was cooled to room temperature and was diluted with water (10 mL). The organic layer was separated, and the aqueous layer was extracted with EtOAc (4 x 15 mL). The combined organic layers were washed with brine (3 x 15 mL), dried over sodium sulfate, filtered, and concentrated to yield tert-butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8- dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-6-fluoro-2'-(methylsulfonyl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (186 mg, 264 μmol) which was used without further purification. LCMS: m/z (ESI) [M+H]
+ 704.3, tR = 4.55 minutes (Method I) Step 9: tert-Butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate A solution of ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (126 mg, 3 equiv., 792 μmol) in anhydrous THF (5 mL) was added slowly to a suspension of sodium
hydride (31.7 mg, 60% wt, 3 equiv., 792 μmol) in anhydrous THF (3 mL) at 0 °C. The reaction mixture was stirred for 10 minutes. A solution of tert-butyl (5-chloro-4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-6-fluoro-2'- (methylsulfonyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (186 mg, 1 equiv., 264 μmol) in anhydrous THF (2 mL) was added, and the reaction mixture was stirred for 1.25 hours at room temperature. The product solution was diluted with cold water (10 mL) and EtOAc (10 mL). The organic layer was separated, and the aqueous layer was extracted with EtOAc (3 x 10 mL). The combined organic layers were washed with brine (3 x 10 mL), dried over sodium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO
2, 2-20% 2% NH
4OH in MeOH in DCM) to provide tert-butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (172 mg, 220 μmol) as a white solid. LCMS: m/z (ESI) [M+H]
+ 783.3, tR = 1.82 minutes (Method B) Step 10: 5-(7-Amino-5-chloro-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide p-Toluenesulfonic acid monohydrate (124 mg, 3 equiv., 651 μmol) was added to a solution of tert-butyl (5-chloro-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (170 mg, 1 equiv., 217 μmol) in MeCN (5 mL). The reaction mixture was stirred for 2 hours at 60 °C. The product mixture was directly concentrated, and the residue obtained was purified by reverse phase column chromatography (C18, 0-100% MeCN in water (2% NH
4HCO
3 additive)) to give 5-(7-amino-5-chloro-6-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (111 mg, 162 μmol) as a white solid. LCMS: m/z (ESI) [M+H]
+ 683.4, tR = 4.29 minutes (Method I)
1H NMR (400 MHz, CDCl
3) δ 6.67 – 6.63 (m, 1H), 6.41 (s, 1H), 5.34 – 5.16 (m, 1H), 4.66 (d, 1H), 4.58 – 4.33 (m, 5H), 4.08 – 3.98 (m, 2H), 3.97 – 3.90 (m, 1H), 3.84 (s, 2H), 3.74 – 3.63 (m, 1H), 3.33 – 3.16 (m, 5H), 3.16 – 2.91 (m, 7H), 2.82 – 2.68 (m, 2H), 2.32 – 2.04 (m, 6H), 2.02 – 1.67 (m, 6H). Example 39: 5-((7S)-6'-amino-2-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-2',3',5,8-tetrahydro-1'H-spiro[pyrano[4,3-d]pyrimidine-7,4'- [1,3]methanonaphthalen]-4-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 172c)
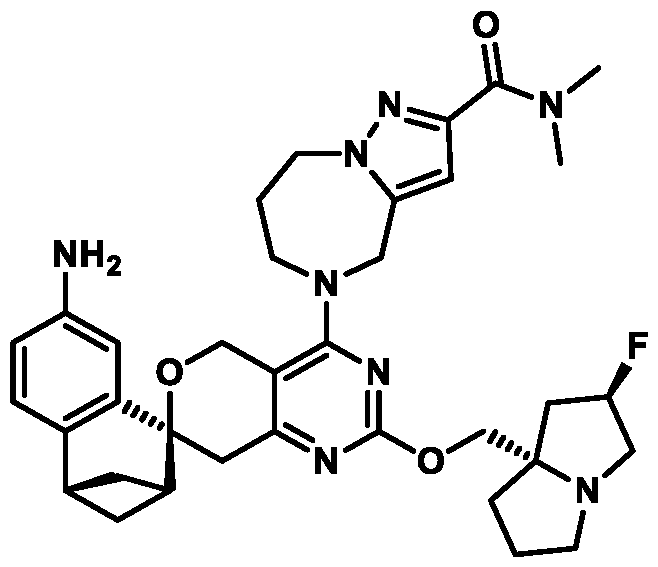
The diastereomers of 5-(6'-amino-2-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-2',3',5,8-tetrahydro-1'H-spiro[pyrano[4,3-d]pyrimidine-7,4'- [1,3]methanonaphthalen]-4-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide, 3HCl (90 mg) were purified using preparative SFC chromatography (Column AD-H 21 x 250 mm, Flow rate: 70 mL/min, 35% ethanol + 0.25% diethylamine in CO2.) to afford 5-((7S)-6'-amino-2-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-2',3',5,8-tetrahydro-1'H-spiro[pyrano[4,3-d]pyrimidine-7,4'- [1,3]methanonaphthalen]-4-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Peak 1) (37.5 mg) and 5-((7R)-6'-amino-2-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-2',3',5,8-tetrahydro-1'H- spiro[pyrano[4,3-d]pyrimidine-7,4'-[1,3]methanonaphthalen]-4-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Peak 2) (51.7 mg). Peak 1 (37.5 mg) was further purified by reverse phase chromatography (Column XSelect CSH Prep C185 µm 20 x 150 mm, Flow rate: 18.9 mL/min Mobile phase A: 0.1% TFA in water (v/v); Mobile phase B: 0.1% TFA in acetonitrile (v/v); Gradient: 90% H2O/10% MeCN linear to 70% H2O/30% MeCN) to afford 5-((7S)-6'-amino-2-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-2',3',5,8-tetrahydro-1'H-
spiro[pyrano[4,3-d]pyrimidine-7,4'-[1,3]methanonaphthalen]-4-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (7.4 mg) 1H NMR (400 MHz, MeOD) δ 7.43 (d, J = 2.1 Hz, 1H), 7.28 – 7.16 (m, 2H), 6.65 (s, 1H), 5.57 (d, J = 51.7 Hz, 1H), 4.90 (m, 3H), 4.73 (d, J = 14.4 Hz, 1H), 4.59 – 4.39 (m, 4H), 4.19 – 3.77 (m, 6H), 3.45 (m, 1H), 3.35 (s, 3H), 3.25 (m, 2H), 3.08 (s, 3H), 3.02 (m, 1H), 2.92 (d, J = 17.9 Hz, 1H), 2.75 – 2.50 (m, 4H), 2.49 – 1.97 (m, 6H), 1.86 (t, J = 8.6 Hz, 1H), 1.66 (t, J = 8.8 Hz, 1H). 1
9F NMR (376 MHz, MeOD) δ -173.75 – -174.27 (m). SFC: (Column ChiralPak AD-H 4.6 x 100 mm, Flow rate: 2.5 mL/min, 45% Ethanol + 0.25% diethylamine in CO
2) t
R = 3.09 minutes LCMS: m/z (ESI) [M+H]
+ 643.3, t
R = 1.1 minutes (Method C) Example 40: 5-((7R)-6'-amino-2-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-2',3',5,8-tetrahydro-1'H-spiro[pyrano[4,3-d]pyrimidine-7,4'- [1,3]methanonaphthalen]-4-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 172b)
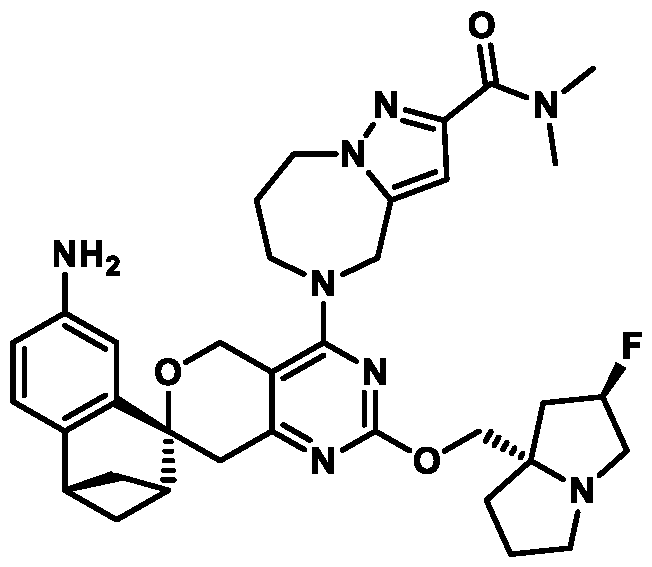
5-((7R)-6'-amino-2-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 2',3',5,8-tetrahydro-1'H-spiro[pyrano[4,3-d]pyrimidine-7,4'-[1,3]methanonaphthalen]-4-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide was obtained from Example 39, as Peak 2: preparative SFC chromatography (Column AD-H 21 x 250 mm, Flow rate: 70 mL/min, 35% ethanol + 0.25% diethylamine in CO2.) Peak 2 (51.7 mg) was further purified by reverse phase chromatography (Column XSelect CSH Prep C185 µm 20 x 150 mm, Flow rate: 18.9 mL/min Mobile phase A: 0.1% TFA in water (v/v); Mobile phase B: 0.1% TFA in acetonitrile (v/v); Gradient: 90% H2O/10% MeCN linear to 70% H2O/30% MeCN) to afford 5-((7R)-6'-amino-2-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-2',3',5,8-tetrahydro-1'H-
spiro[pyrano[4,3-d]pyrimidine-7,4'-[1,3]methanonaphthalen]-4-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8.4 mg). 1H NMR (400 MHz, CD3OD) δ 7.45 (d, J = 2.1 Hz, 1H), 7.29 – 7.18 (m, 2H), 6.66 (s, 1H), 5.57 (d, J = 51.5 Hz, 1H), 4.93 (m, 4H), 4.75 (d, J = 14.4 Hz, 1H), 4.62 – 4.42 (m, 4H), 4.24 – 3.81 (m, 4H), 3.45 (m, 1H), 3.40 – 3.17 (m, 5H), 3.08 (s, 3H), 3.03 m, 1H), 2.94 (d, J = 18.0 Hz, 1H), 2.74 – 2.50 (m, 4H), 2.49 – 1.99 (m, 6H), 1.86 (m, 1H), 1.66 (m, 1H). 1
9F NMR (376 MHz, CD
3OD) δ -173.44 – -175.02 (m). SFC: (Column ChiralPak AD-H 4.6 x 100 mm, Flow rate: 2.5 mL/min, 45% Ethanol + 0.25% diethylamine in CO2) tR = 3.52 minutes LCMS: m/z (ESI) [M+H]
+ 643.3, t
R = 1.1 minutes (Method C) Example 41: 5-(7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4,4-dimethyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide (Compound 133a)
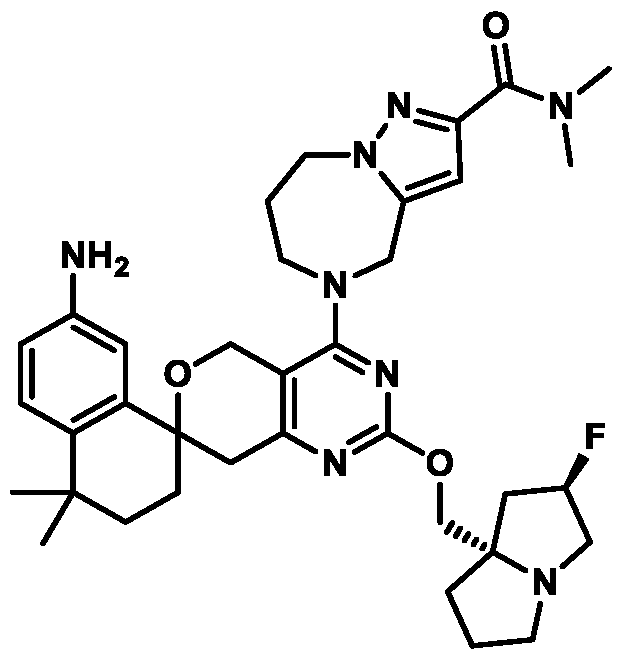
Step 1: 7-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-ol A solution of (4-chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (Intermediate 1) (1.0 g, 1 equiv., 4.9 mmol) in THF (20 mL) was cooled to -78 °C. The reaction vessel was evacuated and filled with N2 three times. LDA (1.3 g, 12 mL, 1 M, 2.5 equiv., 12 mmol) was then added over 10 minutes. The resulting mixture was stirred at that temperature for 30 minutes. A solution of 7-bromo-4,4-dimethyl-3,4-dihydronaphthalen- 1(2H)-one (1.3 g, 97% wt, 1 equiv., 4.9 mmol) in THF (5 mL) was then added using a syringe over 10 minutes. The reaction mixture was stirred at -78 °C for 30 minutes. After the indicated
time, the reaction mixture was quenched with aqueous ammonium chloride (10 mL) at -78 °C. The quenched mixture was warmed up to room temperature. The aqueous layer was extracted with ethyl acetate (3 x 50 mL). The combined organic layers were washed with brine (20 ml), dried over sodium sulfate, filtered and concentrated to give the crude product which was purified by flash column (silica gel, gradient elution from 0% EtOAc to 60% EtOAc/Hexanes) to give 7-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-4,4- dimethyl-1,2,3,4-tetrahydronaphthalen-1-ol (820 mg) as a white solid. LCMS: m/z (ESI) [M-OH]
+ 459.1, tR = 1.88 minutes (Method B) Step 2: 7-bromo-4'-chloro-4,4-dimethyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] Phosphoric acid (206 mg, 85% wt, 1 equiv., 1.79 mmol) was added to the suspension of 7-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-4,4- dimethyl-1,2,3,4-tetrahydronaphthalen-1-ol (820 mg, 1 equiv., 1.79 mmol) in toluene (16 mL). The mixture was heated at 110 °C for 3 hours. The reaction mixture was cooled to room temperature and diluted with EtOAc (50 mL). The organic layer was washed with water (20 mL), dried over sodium sulfate, filtered and concentrated to give the crude product which was purified by flash column chromatography (silica gel, gradient elution from 0% EtOAc to 50% EtOAc/Hexanes) to give 7-bromo-4'-chloro-4,4-dimethyl-2'-(methylthio)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (510 mg). LCMS: m/z (ESI) [M+H]
+ 439.1, t
R = 2.02 minutes (Method B) Step 3: 5-(7-bromo-4,4-dimethyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide DIPEA (570 mg, 99% wt, 6 equiv., 4.37 mmol) was added to the mixture of 7-bromo- 4'-chloro-4,4-dimethyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine] (320 mg, 1 equiv., 728 μmol) and N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (356 mg, 2 equiv., 1.46 mmol) in 1,4- dioxane (10 mL) and DMA (5 mL). The resulting mixture was heated at 110 °C for 18 hours. The reaction mixture was cooled to room temperature and concentrated to dryness to give the crude product. The crude product was treated with cold water (10 mL) and stirred at room temperature for 30 minutes. The precipitate was filtered and rinsed with water and ether (1 mL)
to give 5-(7-bromo-4,4-dimethyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (400 mg). LCMS: m/z (ESI) [M+H]
+ 611.3, t
R = 1.87 minutes (Method B) Step 4: tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-4,4-dimethyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate Brettphos Pd G4 (30.1 mg, 0.1 equiv., 32.7 μmol) was added to a mixture of 5-(7- bromo-4,4-dimethyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (200 mg, 1 equiv., 327 μmol) and tert-butyl carbamate (192 mg, 5 equiv., 1.64 mmol) and cesium carbonate (266 mg, 2.5 equiv., 818 μmol) in 1,4-dioxane (5 mL) under N
2. The vial was evacuated and filled with N
2 three times. The reaction mixture was heated at 110 °C for 2 hours. The reaction mixture was diluted with water (10 mL), and the aqueous layer was extracted with ethyl acetate (3 x 20 mL), washed with brine (5 mL) and dried over sodium sulfate. The combined organic extracts were filtered and concentrated to give the crude product which was purified by flash column (silica gel, gradient elution: MeOH/DCM:0-6%) to afford tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-4,4-dimethyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (201 mg) as a pale yellow solid. LCMS: m/z (ESI) [M+H]
+ 648.4, tR = 1.90 minutes (Method B) Step 5: tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-4,4-dimethyl-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate 3-chlorobenzoperoxoic acid (132 mg, 85% wt, 3 equiv., 648 μmol) was added to the solution of tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-4,4-dimethyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (140 mg, 1 equiv., 216 μmol) in DCM (5 mL). The yellow solution was stirred at room temperature for 30 minutes. The reaction mixture was diluted with DCM (10 mL), saturated aqueous sodium bicarbonate (5
mL), and aqueous sodium thiosulfate (0.5 mL). The resulting mixture was stirred at room temperature for 1 hour. The organic layer was separated, and the aqueous layer was extracted with DCM (5 mL). The combined organic layers were washed with saturated aqueous potassium carbonate (2 mL) and brine (2 mL). The organic layer was dried over sodium sulfate, filtered and concentrated to give the crude product, tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8- dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-4,4-dimethyl-2'-(methylsulfonyl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (160 mg), as a pale yellow solid, which was used in the next step without further purification. LCMS: m/z (ESI) [M+H]
+ 680.4, tR = 1.70 minutes (Method B) Step 6: tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4,4-dimethyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate ((7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (98.4 mg, 3 equiv., 618 μmol) was added to the suspension of sodium hydride (24.7 mg, 60% wt, 3 equiv., 618 μmol) in THF (6 mL) under N
2. After the mixture was stirred at room temperature for 10 min, a solution of tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-4,4-dimethyl-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (140 mg, 1 equiv., 206 μmol) in THF (3 mL) was added slowly through a syringe. The resulting brown solution was stirred at room temperature for 10 minutes. The reaction mixture was quenched with ice water (5 mL). The aqueous layer was extracted with ethyl acetate (3 x 15 mL). The combined organic layers were washed with brine (10 mL), dried over sodium sulfate, filtered and concentrated to give the crude product which was purified by flash column chromatography (silica gel, gradient elution: DCM(90%)-MeOH(9%)-NH3H2O(1%)/DCM:0-60%) to give tert-butyl (4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4,4-dimethyl-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (60 mg) as a light brown solid. LCMS: m/z (ESI) [M+H]
+ 759.5, tR = 1.80 minutes (Method B) Step 7: 5-(7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4,4-dimethyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-
d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide HCl ether solution (132 μL, 2 M, 10 equiv., 264 μmol) was added slowly to the solution of tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)- yl)-2'-(((7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4,4-dimethyl-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (20.0 mg, 1 equiv., 26.4 μmol) in DCM (0.5 mL) at room temperature. The reaction mixture was stirred at room temperature for 2 hours. The mixture was then filtered and washed with diethyl ether and dried to give 5-(7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4,4-dimethyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide, 2HCl (16 mg) as a pale yellow solid. LCMS: m/z (ESI) [M+H]
+ 659.6, tR = 1.46 minutes (Method B) 1H NMR (400 MHz, DMSO-d
6) δ 11.26 (br s, 1H), 10.23 (br s, 2H), 7.54 (d, 1H), 7.37 (d, 1H), 7.26 (m, 1H), 6.68 (s, 1H), 5.57 (d, 1H), 5.07 – 4.79 (m, 3H), 4.71 (m, 1H), 4.60 – 4.39 (m, 4H), 4.03 – 3.76 (m, 6H), 3.25 (s, 4H), 3.01 – 2.79 (m, 5H), 2.29 (d, 1H), 2.16 (s, 3H), 2.02 (d, 4H), 1.79 (d, 1H), 1.66 (m, 1H), 1.28 (d, 6H). Example 42: 5-((R)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4,4-dimethyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 133c)
The diastereomers of 5-(7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4,4-dimethyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide were separated by preparative SFC chromatography (Column Cellulose-121 x
250 mm, Flow rate: 150 mL/min, 50% Methanol + 0.1% diethylamine in CO
2.) to afford 5- ((R)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4,4- dimethyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Peak 1) (31.2 mg) and 5-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 4,4-dimethyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Peak 2) (48 mg). Peak 1: SFC: (Column Lux Cellulose-16x 150 mm, 5µm, Flow rate: 1 mL/min, 10% Ethanol in 90% Hexanes + 0.25% diethylamine) t
R = 7.38 minutes 1H NMR (400 MHz, DMSO-d
6): δ 7.00 (d, 1H), 6.60 – 6.51 (m, 2H), 6.48 (dd, 1H), 5.39 (d, 1H), 4.84 (d, 1H), 4.73 (s, 2H), 4.58 (d, 1H), 4.44 (t, 2H), 4.26 – 3.99 (m, 2H), 3.95 – 3.76 (m, 2H), 3.58 – 3.36 (m, 2H), 3.23 (s, 4H), 3.11 – 2.95 (m, 1H), 2.92 (s, 3H), 2.79 (s, 2H), 2.35 – 2.14 (m, 2H), 2.14 – 2.02 (m, 2H), 2.02 – 1.75 (m, 7H), 1.75 – 1.64 (m, 1H), 1.64 – 1.48 (m, 1H), 1.17 (d, 7H). 1
9F NMR (DMSO, 376 MHz): δ -172.7, -172.5. Example 43: 5-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4,4-dimethyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 133b)
5-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 4,4-dimethyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (48 mg) was obtained from Example 42, as Peak 2: SFC: (Column Lux Cellulose-16x 150 mm, 5µm,
Flow rate: 1 mL/min, 10% Ethanol in 90% Hexanes + 0.25% diethylamine) t
R = 44.3 minutes 1H NMR (400 MHz, DMSO-d6): δ 6.99 (d, 1H), 6.58 – 6.43 (m, 3H), 5.23 (d, 1H), 4.92 – 4.66 (m, 5H), 4.55 (d, 1H), 4.43 (t, 2H), 3.99 – 3.71 (m, 4H), 3.23 (s, 3H), 3.13 – 3.00 (m, 2H), 3.00 – 2.86 (m, 4H), 2.85 – 2.72 (m, 3H), 2.16 – 2.02 (m, 2H), 2.02 – 1.85 (m, 4H), 1.85 – 1.63 (m, 4H), 1.62 – 1.50 (m, 1H), 1.17 (d, 7H). 1
9F NMR (DMSO, 376 MHz): δF -172.1 -171.2 Example 44: 5-(6-Amino-7-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide (Compound 160a)

Step 1: 6-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-7-fluoro-2,3-dihydro-1H-inden-1-ol A 1.0 M solution of LDA in THF (722 mg, 6.74 mL, 2.3 equiv., 6.74 mmol) was added dropwise, while maintaining a temperature below -74 °C, to a solution of (4-chloro-6-methyl- 2-(methylthio)pyrimidin-5-yl)methanol (600 mg, 1 equiv., 2.93 mmol) in THF (5.90 mL) in a dry flask at -78 °C under N2. The reaction mixture was stirred for 30 min at -78 °C. A solution of 6-bromo-7-fluoro-2,3-dihydro-1H-inden-1-one (604 mg, 0.9 equiv., 2.64 mmol) in THF (5.90 mL) was added dropwise, while maintaining a temperature below -74 °C. The reaction mixture was stirred for 5 minutes. A saturated aqueous solution of ammonium chloride (6.0 mL) was added, and the product mixture was stirred for 15 minutes. The aqueous layer was extracted with EtOAc (3 x 10 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO2, 0-15% EtOAc in DCM) to provide 6-bromo-1-((6-chloro-5-
(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-7-fluoro-2,3-dihydro-1H-inden-1-ol (250 mg, 576 μmol) as a white foam. LCMS: m/z (ESI) [M+H]⁺ 433.1, tR = 3.44 minutes (Method E) Step 2: 6-Bromo-4'-chloro-7-fluoro-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] Phosphoric acid (75.8 mg, 45.0 μL, 85% wt, 1.2 equiv., 657 μmol) was added to a solution of 6-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)- 7-fluoro-2,3-dihydro-1H-inden-1-ol (250 mg, 95% wt, 1 equiv., 548 μmol) in toluene (1.10 mL). The reaction mixture was refluxed for 45 minutes. The product solution was concentrated, and the residue obtained was purified by column chromatography (SiO
2, 0-50% EtOAc in heptanes) to provide 6-bromo-4'-chloro-7-fluoro-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] (205 mg, 0.47 mmol) as a white solid. LCMS: m/z (ESI) [M+H]⁺ 415.0, t
R = 3.30 minutes (Method E) Step 3: 5-(6-Bromo-7-fluoro-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene- 1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide DIPEA (104 mg, 140 μL, 2 equiv., 801 μmol) was added to a solution of 6-bromo-4'- chloro-7-fluoro-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidine] (167 mg, 1 equiv., 401 μmol), and N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide, HCl, HCl (182 mg, 99% wt, 1.6 equiv., 641 μmol) in EtOH (1.60 mL). The reaction mixture was refluxed for 20 hours. The product mixture was diluted with EtOAc (3 mL) and sonicated for 30 minutes. The product mixture was filtered, and the filtrate was concentrated. The residue obtained was purified by column chromatography (SiO2, 0-5% MeOH in DCM) to afford 5-(6-bromo-7-fluoro-2'-(methylthio)- 2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (150 mg, 0.25 mmol) as a white foam. LCMS: m/z (ESI) [Br
81 M+H]⁺ 589.2, tR = 1.73 minutes (Method C)
Step 4: tert-Butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-7-fluoro-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'- pyrano[4,3-d]pyrimidin]-6-yl)carbamate A solution of tert-butyl carbamate (59.9 mg, 2 equiv., 512 μmol), 5-(6-bromo-7-fluoro- 2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (150 mg, 1 equiv., 256 μmol), 2-(dicyclohexylphosphanyl)-2',4',6'-tris(isopropyl)biphenyl (32.9 mg, 0.27 equiv., 69.1 μmol), PdOAc2 (5.17 mg, 0.09 equiv., 23.0 μmol) in 1,4-dioxane (1.0 mL) was sparged with N2. The sparged reaction mixture was stirred for 24 hours at 100 °C. The product mixture was directly concentrated, and the residue obtained was purified by column chromatography (SiO
2, 0-15% MeOH in DCM) to give tert-butyl (4'-(2-(dimethylcarbamoyl)- 7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-7-fluoro-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (40 mg, 64.0 μmol). LCMS: m/z (ESI) [M+H]⁺ 624.4, t
R = 2.67 minutes (Method E) Step 5: tert-Butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-7-fluoro-2'-(methylsulfonyl)-2,3,5',8'-tetrahydrospiro[indene- 1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate mCPBA (32.0 mg, 70% wt, 2 equiv., 0.13 mmol) was added to a solution of tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-7-fluoro- 2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (40.0 mg, 1 equiv., 64.0 μmol) in DCM (0.64 mL) at 0 °C. The reaction mixture was stirred for 5 min at 0 °C and for 48 hours at room temperature. The product mixture was diluted with DCM (5 mL) and an aqueous 1.0 M NaOH solution (10 mL). The layers that formed were separated, and the aqueous layer was extracted with DCM (10 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated to give tert-butyl (4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-7-fluoro-2'- (methylsulfonyl)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6- yl)carbamate (41.0 mg, 47.0 μmol) which was used without further purification. LCMS: m/z (ESI) [M+H]⁺ 656.2, tR = 1.77 minutes (Method C)
Step 6: 5-(6-Amino-7-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide NaH (2.30 mg, 1.5 equiv., 95.8 μmol) was added to a solution of tert-butyl (4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-7-fluoro-2'- (methylsulfonyl)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6- yl)carbamate (41.9 mg, 1 equiv., 63.9 μmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methanol (10.2 mg, 1 equiv., 63.9 μmol) in THF (640 μL) under N2 at 0 °C. The reaction mixture was stirred for 5 minutes. The product mixture was directly concentrated, and the residue obtained was dissolved in DCM (640 μL). A 2.0 M HCl solution in ether (11.7 mg, 160 μL, 5 equiv., 320 μmol) was added, and the reaction mixture was stirred until all the starting material was consumed. The product mixture was directly concentrated, and the residue obtained was purified by reverse phase column chromatography (C18, 5-50% MeCN in water (0.1% TFA additive)) to provide 5-(6-amino-7-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]- 4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (7.5 mg, 12.0 μmol). LCMS: m/z (ESI) [M+H]⁺ 635.6, t
R = 1.39 minutes (Method C) 1H NMR (400 MHz, MeOD) δ 7.16 – 7.01 (m, 2H), 6.60 (s, 1H), 5.69 – 5.44 (m, 1H), 4.81 – 4.69 (m, 2H), 4.62 – 4.44 (m, 5H), 4.25 – 4.10 (m, 1H), 4.06 – 3.79 (m, 4H), 3.54 – 3.39 (m, 2H), 3.36 (s, 3H), 3.20 – 3.05 (m, 6H), 2.91 – 2.80 (m, 1H), 2.75 – 2.52 (m, 2H), 2.48 – 2.03 (m, 8H). Example 45: 5-((4R)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 124d)

Step 1: (4R)-7-Bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin- 4-yl)methyl)-4-methyl-1,2,3,4-tetrahydronaphthalen-1-ol A 1.0 M solution of LDA in THF (602 mg, 5.62 mL, 2.3 equiv., 5.62 mmol) was added dropwise, while maintaining a temperature below -74 °C, to a solution of (4-chloro-6-methyl- 2-(methylthio)pyrimidin-5-yl)methanol (500 mg, 1 equiv., 2.44 mmol) in THF (6.00 mL) in a dry flask at -78 °C under N2. The reaction mixture was stirred for 30 min at -78 °C. A solution of (R)-7-bromo-4-methyl-3,4-dihydronaphthalen-1(2H)-one (Intermediate 7a, 526 mg, 0.9 equiv., 2.20 mmol) in THF (6.00 mL) was added dropwise, while maintaining a temperature below -74 °C. The reaction mixture was stirred for 5 minutes. A saturated aqueous solution of ammonium chloride (6.0 mL) was added, and the product mixture was stirred for 15 minutes. The aqueous layer was extracted with EtOAc (3 x 10 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO2, 0-15% EtOAc in DCM) to provide (4R)-7-bromo-1-((6-chloro- 5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-4-methyl-1,2,3,4- tetrahydronaphthalen-1-ol (596 mg, 1.34 mmol) as a white foam. 1H NMR (400 MHz, CDCl3) δ 7.72 – 7.65 (m, 1H), 7.40 – 7.32 (m, 1H), 7.17 – 7.01 (m, 1H), 4.84 – 4.68 (m, 2H), 4.17 – 4.04 (m, 1H), 3.54 – 3.14 (m, 2H), 3.04 – 2.79 (m, 2H), 2.54 (s, 3H), 2.27 – 2.10 (m, 1H), 2.08 – 1.92 (m, 1H), 1.89 – 1.58 (m, 2H), 1.36 – 1.26 (m, 3H). Step 2: (4R)-7-Bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] Phosphoric acid (155 mg, 91.9 μL, 85% wt, 1.2 equiv., 1.34 mmol) was added to a solution of (4R)-7-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-4-methyl-1,2,3,4-tetrahydronaphthalen-1-ol (596 mg, 1 equiv., 1.34 mmol)
in toluene (5.40 mL). The reaction mixture was stirred for 2 hours at 85 °C. The product solution was concentrated, and the residue obtained was diluted with water (20 mL) and EtOAc (20 mL). The layers that formed were separated, and the aqueous layer was extracted with EtOAc (3 x 20 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO2, 0-50% EtOAc in heptanes) to provide (4R)-7-bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (300 mg, 705 µmol) as a white foam. LCMS: m/z (ESI) [M+H]⁺ 425.1, tR = 4.13 minutes (Method E) Step 3: 5-((4R)-7-Bromo-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide DIPEA (364 mg, 491 μL, 4 equiv., 2.82 mmol) was added to a solution of (4R)-7- bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine] (300 mg, 1 equiv., 705 μmol), and N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide, HCl, HCl (320 mg, 99% wt, 1.6 equiv., 1.13 mmol) in ethanol (1.40 mL). The reaction mixture was stirred for 18 hours at 85 °C. The product mixture was directly concentrated, and the residue obtained was diluted with EtOAc (10 mL) and washed with water (2 x 20 mL) and brine (10 mL). The combined aqueous layers were extracted with EtOAc (20 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated to afford 5-((4R)-7-bromo-4-methyl-2'-(methylthio)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl- 5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (419 mg, 0.67 mmol) which was used without further purification. LCMS: m/z (ESI) [M+H]⁺ 597.2, tR = 1.91 minutes (Method C) Step 4: tert-Butyl ((4R)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate A solution of tert-butyl carbamate (165 mg, 2 equiv., 1.41 mmol), 5-((4R)-7-Bromo-4- methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-
carboxamide (420 mg, 1 equiv., 703 μmol), 2-(dicyclohexylphosphanyl)-2',4',6'- tris(isopropyl)biphenyl (30.2 mg, 0.09 equiv., 63.3 μmol), PdOAc2 (4.73 mg, 0.03 equiv., 21.1 μmol) in 1,4-dioxane (1.4 mL) was sparged with N2. The sparged reaction mixture was stirred for 12 hours at 100 °C. The product mixture was cooled to room temperature, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO2, 0-15% MeOH in DCM) to give tert-butyl ((4R)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (395 mg, 623 μmol). LCMS: m/z (ESI) [M+H]⁺ 634.3, tR = 2.80 minutes (Method E) Step 5: tert-Butyl ((4R)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-4-methyl-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate Oxone (2.02 g, 45% wt, 2.1 equiv., 1.48 mmol) was added to a solution of tert-butyl ((4R)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-4- methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (446 mg, 1 equiv., 703 μmol) in water (7.03 mL) and methanol (7.03 mL). The reaction mixture was stirred for 1 hour. The product mixture was diluted with water (10 mL) and EtOAc (10 mL). The layers that formed were separated, and the aqueous layer was extracted with EtOAc (3 x 20 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated to give tert-butyl ((4R)-4'-(2-(dimethylcarbamoyl)- 7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-4-methyl-2'-(methylsulfonyl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (468 mg, 703 μmol) which was used without further purification. LCMS: m/z (ESI) [M+H]⁺ 666.2, tR = 2.91 minutes (Method E) Step 6: tert-Butyl ((4R)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate A 2.0 M solution of potassium tert-butoxide in THF (118 mg, 527 μL, 1.5 equiv., 1.05 mmol) was added to a solution of tert-butyl ((4R)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-4-methyl-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H-
spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (468 mg, 1 equiv., 703 μmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (112 mg, 1 equiv., 703 μmol) in THF (7.03 mL) under N2. The reaction mixture was stirred for 40 min, and water (1 mL) and EtOAc (2 mL) were added. The layers that formed were separated, and the aqueous layer was extracted with EtOAc (4 x 3 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO
2, 0-15% MeOH in DCM) to provide tert-butyl ((4R)-4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)- 2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (300 mg, 403 μmol). LCMS: m/z (ESI) [M+H]⁺ 745.4, t
R = 1.44 minutes (Method C) Step 7: 5-((4R)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide A 2.0 M solution of HCl in ether (100 mg, 2.0 mL, 6 equiv., 4.00 mmol) was added to a solution of tert-butyl ((4R)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (300 mg, 1 equiv., 403 μmol) in DCM (5.00 mL). The reaction mixture was stirred for 3 hours. The product mixture was filtered and concentrated to afford 5- ((4R)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4-methyl- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl- 5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (263 mg, 408 μmol). LCMS: m/z (ESI) [M+H]⁺ 645.5, tR = 1.15 minutes (Method M) 1H NMR (400 MHz, CD
3OD) δ 7.58 – 7.45 (m, 2H), 7.34 (m, 1H), 6.52 (d, 1H), 5.70 – 5.53 (m, 2H), 5.15 – 5.00 (m, 2H), 4.85 (d, 1H), 4.75 (d, 1H), 4.57 (m, 3H), 4.13 – 3.77 (m, 4H), 3.47 – 3.38 (m, 1H), 3.32 (s, 3H), 3.17 (s, 1H), 3.10 (m, 5H), 2.82 – 1.90 (m, 13H), 1.37 (m, 3H). Example 46: 5-((1S*,4R*)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-
pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 124e)

The diastereomers of 5-(7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (260 mg, 0.7 mmol) were separated by chiral preparative SFC (Daicel Chiralpak IB, 21 mm I.D. x 250 mm, 5µm, part number 88447. Isocratic Mobile phase 45%A CO2 and 55%B MeOH with 0.25% DEA. Flow rate: 50 mL/min. Back pressure: 120 bar) to afford 5- ((1S*,4R*)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4- methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Peak 1) (tR = 5.6 min, 10.3 mg) and 5-((1R*,4R*)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (Peak 2) (9.77 min, 1.7 mg). Peak 1: LCMS: ESI [M+H]
+= 645.5 m/z; t
R = 1.23 min (Method A) 1H NMR (400 MHz, MeOD) δ 7.01 (d, 1H), 6.77 – 6.64 (m, 2H), 6.59 (s, 1H), 5.27 (d, 1H), 4.82 – 4.73 (m, 2H), 4.56 – 4.44 (m, 3H), 4.15 – 4.01 (m, 2H), 3.99 – 3.89 (m, 3H), 3.39 – 3.27 (m, 4H), 3.26 – 3.15 (m, 3H), 3.07 (s, 5H), 3.04 – 2.95 (m, 2H), 2.92 – 2.79 (m, 2H), 2.34 – 1.75 (m, 9H), 1.38 – 1.23 (m, 4H). Example 47: 5-((1R*,4R*)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 124f)
5-((1R*,4R*)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (9.77 min, 1.7 mg) was obtained from Example 46, as Peak 2: chiral preparative SFC (Daicel Chiralpak IB, 21 mm I.D. x 250 mm, 5µm, part number 88447. Isocratic Mobile phase 45%A CO
2 and 55%B MeOH with 0.25% DEA. Flow rate: 50 mL/min. Back pressure: 120 bar). Peak 2: LCMS: ESI [M+H]
+= 645.5 m/z; tR = 1.20 min (Method A) 1H NMR (400 MHz, MeOD) δ 7.06 – 6.99 (m, 1H), 6.68 – 6.62 (m, 2H), 6.48 (s, 1H), 5.23 (d, 1H), 4.78 – 4.65 (m, 2H), 4.56 – 4.38 (m, 3H), 4.12 – 3.94 (m, 2H), 3.95 – 3.84 (m, 2H), 3.29 – 3.24 (m, 4H), 3.22 – 3.09 (m, 3H), 3.07 – 3.00 (m, 4H), 3.00 – 2.80 (m, 4H), 2.78 – 2.68 (m, 2H), 2.32 – 1.75 (m, 9H), 1.40 – 1.20 (m, 4H). Example 48: 5-(6-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-2,2-dimethyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide (Compound 130a)

Step 1: 6-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-2,2-dimethyl-2,3-dihydro-1H-inden-1-ol To an oven-dried 100 mL flask was added (4-chloro-6-methyl-2-(methylthio)- pyrimidin-5-yl)methanol (250 mg, 1 equiv., 1.22 mmol). The flask was then evacuated and backfilled with nitrogen three times. THF (2.5 mL) was then added and the solution was cooled to -78 °C.1 M LDA solution (2.81 mL, 2.3 equiv., 2.81 mmol) was then added dropwise.6- Bromo-2,2-dimethyl-2,3-dihydro-1H-inden-1-one (321 mg, 1.1 equiv., 1.34 mmol) was dissolved in THF (0.75 mL) and added dropwise to the solution keeping the internal temperature below -74 °C. The reaction was stirred for 5 minutes at which point an aliquot of the mixture showed complete consumption of starting material. At this point the mixture was quenched with one volume of saturated ammonium chloride solution and the resulting mixture was stirred for 15 minutes. The mixture was then extracted three times with 100 mL of EtOAc and the combined organic layers were dried using sodium sulfate, filtered, and concentrated. The crude mixture was purified by flash column chromatography (10 g biotage silica gel cartridge, 0 to 15 % EtOAc in DCM) to yield 6-bromo-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-2,2-dimethyl-2,3-dihydro-1H-inden-1-ol (350 mg, 0.834 mmol) as a white foam. 1H NMR (400 MHz, CDCl
3): δ 7.33 – 7.28 (m, 1H), 7.08 – 7.04 (m, 1H), 6.72 – 6.68 (m, 1H), 4.68 (s, 1H), 4.58 – 4.49 (m, 1H), 4.22 – 4.15 (m, 1H), 3.18 (d, 1H), 3.08 (d, 1H), 2.78 (d, 1H), 2.68 (d, 1H), 2.48 (s, 3H), 1.24 (s, 3H), 1.01 (s, 3H) Step 2: 6-Bromo-4'-chloro-2,2-dimethyl-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] To an oven-dried 50 mL flask containing 6-bromo-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-2,2-dimethyl-2,3-dihydro-1H-inden-1-ol (350 mg, 1 equiv., 0.792 mmol) in toluene (3.2 mL), phosphoric acid (65 μL, 85 wt %, 1.2 equiv., 0.95 mmol) was added. The resulting mixture was heated to 95 °C overnight. At this point the mixture was concentrated and purified via silica gel chromatography (0 to 50 % EtOAc in heptane) to yield 6-bromo-4'-chloro-2,2-dimethyl-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] (266 mg, 0.62 mmol) LCMS: m/z (ESI) [M+H]
+ 425.1, t
R = 4.23 min (Method E)
Step 3: 5-(6-Bromo-2,2-dimethyl-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide To a flask containing 6-bromo-4'-chloro-2,2-dimethyl-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] (280 mg, 1 equiv., 658 μmol) and N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide, HCl, HCl (299 mg, 99% wt, 1.6 equiv., 1.05 mmol) in ethanol (1.32 mL), was added DIPEA (340 mg, 458 μL, 4 equiv., 2.63 mmol). The reaction mixture was heated to reflux for 1 hour, cooled to room temperature and stirred for 48 hours, and refluxed for an additional 1 hour. Solvent was removed under reduced pressure and the resulting thick oily residue was diluted with 10 mL EtOAc, transferred to a separatory funnel (using 10 mL EtOAc to rinse flask). The organic layer was washed with water (20 mL), water (10 mL), then brine (10 mL). The combined aqueous layers were extracted once with EtOAc (20 mL) and the combined organics were dried over sodium sulfate then concentrated to residue under reduced pressure to give a yellow solid foam. The solid residue was taken up in EtOAC, washed two times with 10 mL NaOH (aqueous, 1 M), dried over sodium sulfate, and concentrated under reduced pressure to give 5- (6-bromo-2,2-dimethyl-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (375.4 mg, 0.60 mmol) as a light yellow solid foam. LCMS: m/z (ESI) [M+H, Br
81]
+ 599.0, t
R = 1.97 min (Method C) Step 4: tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2,2-dimethyl-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene- 1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate To an oven dried 25 mL flask was added tert-butyl carbamate (147 mg, 2 equiv., 1.26 mmol), 5-(6-bromo-2,2-dimethyl-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (375 mg, 1 equiv., 628 μmol), 2-(dicyclohexylphosphanyl)- 2',4',6'-tris(isopropyl)biphenyl (26.9 mg, 0.09 equiv., 56.5 μmol), PdOAc2 (4.23 mg, 0.03 equiv., 18.8 μmol), and 5-(6-bromo-2,2-dimethyl-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-3-carboxamide (375 mg, 1 equiv., 628 μmol) followed by 1,4-dioxane (6.3 mL). The reaction vessel was then sparged with N2 and then 1,4-dioxane
(6.3 mL) was added. The reaction was then heated to 100 °C using an aluminum heating block monitoring the temperature using an internal temperature probe. After 60 minutes of stirring at 100 °C, the reaction was cooled to room temperature, filtered, concentrated, and purified via column chromatography (100% DCM to 85:15 DCM:MeOH) to give tert-butyl (4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2,2-dimethyl- 2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (364.9 mg, 575.7 μmol) LCMS: m/z (ESI) [M-H]- 632.4, tR = 3.10 min (Method E) Step 5: tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2,2-dimethyl-2'-(methylsulfonyl)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate To a 10 mL round bottom flask was added tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8- dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2,2-dimethyl-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (364 mg, 1 equiv., 574 μmol), oxone (741 mg, 2.1 equiv., 1.21 mmol), MeOH (5.74 mL), and water (5.74 mL). After 45 min of stirring an aliquot was taken and analyzed via HPLC showing full conversion of the starting material. The reaction was then extracted three times with DCM and the concentrated to give tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin- 5(6H)-yl)-2,2-dimethyl-2'-(methylsulfonyl)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-6-yl)carbamate (382 mg, 0.574 mmol) LCMS: m/z (ESI) [M+H]
+ 666.4, tR = 1.97 min (Method C) Step 6: tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-2,2-dimethyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-6-yl)carbamate A 10 mL round bottom flask containing tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8- dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2,2-dimethyl-2'-(methylsulfonyl)- 2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (382 mg, 1 equiv., 574 μmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (91.3 mg, 1 equiv., 574 μmol) was evacuated and filled with N2. The mixture was then charged with THF (5.7 mL) and cooled to 0 °C. To this solution, potassium tert-butoxide in THF (129 mg,
1.15 mL, 1 M, 2 equiv., 1.15 mmol) was delivered dropwise. The reaction mixture was stirred for 30 minutes after which time LCMS showed complete consumption of starting material. After 40 min, water (1 mL) and EtOAc (2 mL) was delivered to the solution, the aqueous was separated and extracted with EtOAc (4 x 3 mL) before drying the combined organics over sodium sulfate. The dried organics were filtered and concentrated to give tert-butyl (4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)- 2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-2,2-dimethyl-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (77.6 mg, 104 μmol) as an orange solid film. LCMS: m/z (ESI) [M+H]
+ 745.6, t
R = 1.49 min (Method C) Step 7: 5-(6-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-2,2-dimethyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide tert-Butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin- 5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-2,2-dimethyl- 2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (80 mg, 1 equiv., 0.11 mmol) was added to a 10 mL round bottom flask. DCM (1.1 mL) was then added followed by HCl in diethyl ether (20 mg, 0.27 mL, 2 M, 5 equiv., 0.54 mmol). The reaction was stirred for 1 hour at which point TLC indicated full consumption of the starting material. The reaction was concentrated giving 5-(6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-2,2-dimethyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide, 3HCl (70 mg, 93 μmol) as a white powder. LCMS: m/z (ESI) [M+H]
+ 645.4, tR = 1.26 min (Method C) 1H NMR (400 MHz, CDCl
3): δ 7.54 (d, 1H), 7.42 – 7.33 (m, 2H), 6.50 – 6.25 (m, 1H), 5.63 (d, 1H), 5.20 – 5.05 (m, 1H), 4.94 – 4.75 (m, 3H), 4.68 – 4.51 (m, 3H), 4.36 (d, 1H), 4.04 – 3.74 (m, 4H), 3.54 – 3.41 (m, 1H), 3.36 – 3.18 (m, 6H), 3.10 (s, 3H), 2.93 – 2.15 (m, 10H), 1.29 (s, 3H), 0.91 (s, 3H) Example 49: 5-((R*)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-2,2-dimethyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-
d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide (Compound 130b)
The diastereomers of 5-(6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-2,2-dimethyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (60.5 mg, 93.9 mmol) were separated via SFC using ChiralCel OJ-H (21 x 250 mm) under isocratic cosolvent (25% Ethanol + 0.25% diethylamine in CO
2) with a flow rate of 70 mL/min to afford 5-((R*)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-2,2-dimethyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (Peak 1) (17.5 mg) and 5-((S*)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-2,2-dimethyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (Peak 2) (18.2 mg). Peak 1: Chiral SFC: tR = 1.05 min (Chiralpak OJ-H (4.6 x 100 mm), isocratic 25% ethanol + 0.25% diethylamine in CO
2 with a flow rate of 2.5 mL/min) 1H NMR (400 MHz, CDCl3) δ 7.06 (d, 1H), 6.67 – 6.56 (m, 1H), 6.48 – 6.32 (m, 2H), 5.38 – 5.11 (m, 1H), 4.65 – 4.49 (m, 2H), 4.44 – 4.33 (m, 2H), 4.26 (d, 2H), 4.09 – 3.91 (m, 2H), 3.71 (q, 2H), 3.46 – 3.35 (m, 1H), 3.31 (s, 3H), 3.27 – 3.19 (m, 1H), 3.18 – 3.11 (m, 1H), 3.09 – 3.00 (m, 4H), 2.98 – 2.92 (m, 1H), 2.76 (d, 1H), 2.51 – 2.33 (m, 2H), 2.33 – 2.05 (m, 3H), 2.03 – 1.72 (m, 3H), 1.37 – 1.10 (m, 6H), 0.86 (s, 3H). 1
9F NMR (376 MHz, CDCl3) δ -172.9
Example 50: 5-((S*)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-2,2-dimethyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide (Compound 130c)
5-((S*)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 2,2-dimethyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (18.2 mg) was obtained from Example 49, as Peak 2: Chiral SFC: tR = 1.70 min (Chiralpak OJ-H (4.6 x 100 mm), isocratic 25% ethanol + 0.25% diethylamine in CO2 with a flow rate of 2.5 mL/min). 1H NMR (400 MHz, CDCl
3) δ 7.06 (d, 1H), 6.67 – 6.56 (m, 1H), 6.45 – 6.31 (m, 2H), 5.25 (d, 1H), 4.74 – 4.47 (m, 2H), 4.45 – 4.20 (m, 4H), 4.06 (d, 1H), 3.90 (d, 1H), 3.78 – 3.66 (m, 2H), 3.48 – 3.36 (m, 1H), 3.31 (s, 3H), 3.22 (d, 1H), 3.17 – 3.10 (m, 1H), 3.10 – 3.00 (m, 4H), 2.96 (td, 1H), 2.76 (d, 1H), 2.41 (dd, 2H), 2.30 – 2.09 (m, 3H), 2.02 – 1.71 (m, 3H), 1.27 – 1.12 (m, 6H), 0.86 (s, 3H). 1
9F NMR (376 MHz, CDCl3) δ -173.0 Example 51: 5-((1SR,3RS)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 175a)

Step 1: 4-(4-bromophenyl)-3-methylbutanal A 250 mL round bottom flask fitted with a stir bar was charged with 1-bromo-4- iodobenzene (10.0 g, 1.00 equiv., 35.3 mmol), lithium acetate (5.83 g, 2.50 equiv., 88.4 mmol), lithium chloride (1.50 g, 1.00 equiv., 35.3 mmol), tetrabutylammonium chloride hydrate (19.6 g, 2.00 equiv., 70.7 mmol) and palladium diacetate (476 mg, 0.06 equiv., 2.12 mmol). The flask headspace was evacuated and refilled with nitrogen, followed by the addition of DMF (70.7 mL) and 3-methylbut-3-en-1-ol (3.04 g, 3.57 mL, 1.00 equiv., 35.3 mmol). The resulting mixture was stirred at 70 °C for 66 hours. The volatiles were removed in vacuo and the residue was partitioned between a 1:1 mixture of a saturated aqueous ammonium chloride solution (125 mL) and EtOAc (125 mL). The layers were separated and the aqueous phase was extracted with EtOAc (3 x 100 mL). The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated. The crude material was purified by silica gel chromatography (heptane/EtOAc, 0 → 15%) to afford 4-(4-bromophenyl)-3-methylbutanal (2.51 g), as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 9.72 (s, 1H), 7.43 – 7.38 (m, 2H), 7.05 – 7.01 (m, 2H), 2.61-2.21 (m, 5H), 0.96 (d, 3H). Step 2: 4-(4-bromophenyl)-3-methylbutanoic acid To a solution of 4-(4-bromophenyl)-3-methylbutanal (2.51 g, 1.0 equiv., 10.4 mmol) in a 2:1 mixture of t-BuOH (69.4 mL) and water (34.7 mL) at 0 °C with vigorous stirring, was sequentially added 2-methyl-2-butene (29.2 g, 44.1 mL, 40 equiv., 416 mmol), sodium dihydrogen phosphate monohydrate (14.4 g, 10 equiv., 104 mmol) and sodium chlorite (4.71 g, 5.0 equiv., 52.0 mmol). The resulting mixture, which quickly became orange, was stirred vigorously at 0 °C for 15 min then at room temperature for 2 hours. Upon complete conversion observed by TLC, the reaction mixture was poured in a separatory funnel containing a 1:1 mixture of water and brine (200 mL) and CH₂Cl₂ (150 mL). The layers were separated and the
aqueous phase was extracted with CH₂Cl₂ (3 x 100 mL). The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated to afford 4-(4-bromophenyl)-3- methylbutanoic acid (2.68 g) as a colorless oil, was of sufficient purity to be used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 10.82 (br s, 1H), 7.43 – 7.38 (m, 2H), 7.06 – 7.02 (m, 2H), 2.66-2.58 (m, 1H), 2.52-2.44 (m, 1H), 2.39-2.31 (m, 1H), 2.31 – 2.22 (m, 1H), 2.22-2.15 (m, 1H), 0.97 (d, 3H). Step 3: 7-bromo-3-methyl-3,4-dihydronaphthalen-1(2H)-one To a solution of 4-(4-bromophenyl)-3-methylbutanoic acid (2.80 g, 1.0 equiv., 10.9 mmol) in dichloromethane (27.2 mL) at room temperature, were added trifluoroacetic anhydride (13.7 g, 9.08 mL, 6.0 equiv., 65.3 mmol) and triflic acid (2.45 g, 1.45 mL, 1.5 equiv., 16.3 mmol). The resulting mixture was stirred at room temperature for 1 hour. At this time the volatiles were removed in vacuo and the crude material was purified by silica gel chromatography (heptane/EtOAc, 0 → 20%) to afford 7-bromo-3-methyl-3,4- dihydronaphthalen-1(2H)-one (2.14 g), as a solid. 1H NMR (400 MHz, CDCl
3) δ 8.14 (d, 1H), 7.61-7.55 (m, 1H), 7.16-7.10 (d, 1H), 2.98- 2.90 (m, 1H), 2.79 – 2.69 (m, 1H), 2.67-2.58 (m, 1H), 2.39 – 2.27 (m, 2H), 1.15 (d, 3H). Step 4: (1SR,3RS)-7-bromo-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-3-methyl-1,2,3,4-tetrahydronaphthalen-1-ol To a solution of 7-bromo-3-methyl-3,4-dihydronaphthalen-1(2H)-one (2.75 g, 1.5 equiv., 13.4 mmol) in THF (80 mL) at -78 °C was added LDA (1.0 M in THF/hexanes) (2.88 g, 26.8 mL, 1.0 M, 3.0 equiv., 26.8 mmol) by addition funnel, dropwise. The rate of dropwise addition was set so that the internal temperature did not exceed -72 °C. The resulting clear mixture, which became dark yellow/orange over time, was stirred at -78 °C for 1 hour and was cannulated dropwise to a precooled -78 °C solution of 7-bromo-3-methyl-3,4- dihydronaphthalen-1(2H)-one (2.14 g, 1.0 equiv., 8.95 mmol) in THF (40 mL). The reaction mixture was stirred -78 °C for 1.25 hours followed by the addition of a 1:1 mixture of a saturated aqueous ammonium chloride solution and water (100 mL) at -78 °C. The mixture was warmed to room temperature and the layers were separated and the aqueous phase was extracted with EtOAc (3 x 75 mL). The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated. The crude material was purified by silica gel
chromatography (heptane/EtOAc, 1 → 40%) to afford (1SR,3RS)-7-bromo-1-((6-chloro-5- (hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-3-methyl-1,2,3,4- tetrahydronaphthalen-1-ol (2.54 g). 1H NMR (400 MHz, CDCl
3) δ 7.68 (d, 1H), 7.35-7.28 (m, 1H), 6.97 (d, 1H), 4.78 (d, 1H), 4.75 (d, 1H), 3.30 (d, 1H), 3.21 (d, 1H), 2.92-2.84 (m, 1H), 2.56 (s, 3H), 2.34 – 2.22 (m, 1H), 2.35-2.24 (m, 1H), 2.10-2.03 (m, 1H), 1.44-1.36 (m, 1H), 1.07 (d, 3H). Step 5: (1SR,3RS)-7-bromo-4'-chloro-3-methyl-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] To a 2.0-5.0 mL microwave vial charged with (1SR,3RS)-7-bromo-1-((6-chloro-5- (hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-3-methyl-1,2,3,4- tetrahydronaphthalen-1-ol (100 mg, 1.0 equiv., 225 μmol), were added toluene (2.25 mL) and phosphoric acid (26.0 mg, 15.4 μL, 85% wt, 1.0 equiv., 225 μmol) at room temperature. The vial was sealed, and the resulting suspension was stirred at reflux for 1 hour. The suspension rapidly became a clear solution upon heating. Upon complete consumption of starting material, the reaction mixture was cooled to room temperature and was quenched by the addition of water (2.0 mL), EtOAc (2.0 mL) and brine (2.0 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (3 x 4.0 mL). The combined organic extracts were dried over magnesium sulfate, filtered and concentrated to afford crude (1SR,3RS)-7- bromo-4'-chloro-3-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine] which was directly carried in the next step. Step 6: 5-((1SR,3RS)-7-bromo-3-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide To a vial charged with (1SR,3RS)-7-bromo-4'-chloro-3-methyl-2'-(methylthio)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (96.0 mg, 1 equiv., 225 μmol) and N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide, 2HCl (254 mg, 4 equiv., 902 μmol) were added MeCN (4.5 mL) and DIPEA (175 mg, 234 μL, 6 equiv., 1.35 mmol) at room temperature. The resulting suspension was stirred at 65 °C for 4 hours. LCMS analysis of a reaction aliquot at this time showed incomplete conversion. More DIPEA (87.4 mg, 117 μL, 3 equiv., 676 μmol), N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide, 2HCl (127 mg, 2 equiv., 451
μmol) and MeCN (2.0 mL) were added and the reaction mixture was stirred for another 17 hours at 65 °C. The mixture was cooled to room temperature and the volatiles were removed in vacuo. The crude material was directly purified by silica gel chromatography (heptane/EtOAc, 10 → 100%) to afford 5-((1SR,3RS)-7-bromo-3-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (159 mg) as a white solid in sufficient purity to use in the next step. LCMS: ESI [M+H]
+= 597.34 m/z; tR = 3.03 min (Method C) Step 7: 5-((1SR,3RS)-7-bromo-3-methyl-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide To a suspension of 5-((1SR,3RS)-7-bromo-3-methyl-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (159 mg, 1 equiv., 266 μmol) in THF (2.00 mL), methanol (3.20 mL) and water (3.20 mL) at room temperature, was added Oxone (1.45 g, 45% wt, 4 equiv., 1.06 mmol) in one portion. The resulting mixture was stirred at room temperature for 1 hour. TLC of an aliquot showed full conversion of the starting material. The organic volatiles were removed in vacuo. Water (15 mL) and EtOAc (15 mL) were added and the layers were separated. The aqueous phase was extracted with EtOAc (3 x 20 mL). The combined organic extracts were dried over magnesium sulfate, filtered and concentrated to give crude 5-((1SR,3RS)-7-bromo-3-methyl-2'-(methylsulfonyl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide which was directly used in the next step without further purification. Step 8: 5-((1SR,3RS)-7-bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide To a suspension of sodium hydride (60% dispersion in mineral oil) (12.7 mg, 60% wt, 2 equiv., 318 μmol) in THF (2.12 mL) under nitrogen, was added ((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (50.6 mg, 2 equiv., 318 μmol) and the
mixture was stirred at room temperature for 20 minutes. A solution of 5-((1SR,3RS)-7-bromo- 3-methyl-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (100 mg, 1 equiv., 159 μmol) in DMF (4.24 mL) was added to the above mixture. The resulting solution was stirred at 40 °C for 2 hours. The reaction mixture was cooled to room temperature. Water (2 mL) and brine (5 mL) were added. The mixture was extracted with EtOAc (3 x 7.0 mL). The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated. The crude material was purified by silica gel chromatography (CH2Cl2/[CH2Cl2/MeOH/NH4OH, 75:22.5:2.5], 0 → 30%) to afford 5-((1SR,3RS)-7-bromo- 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3-methyl-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (95 mg). LCMS: ESI [M+H]
+= 708.5 m/z; tR = 2.13 min (Method C) Step 9: tert-butyl ((1SR,3RS)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate To a vial charged with 5-((1SR,3RS)-7-bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (95.0 mg, 1 equiv., 134 μmol), tert-butyl carbamate (78.5 mg, 5 equiv., 670 μmol), BrettPhos Pd G4 (18.5 mg, 0.15 equiv., 20.1 μmol) and cesium carbonate (153 mg, 3.5 equiv., 469 μmol), was added 1,4-dioxane (4.47 mL) under nitrogen. The resulting mixture was further degassed by evacuating the vial headspace and refilling it with nitrogen three times. The mixture was stirred at 80 °C for 20 hours. The solids were filtered through a syringe filter and the filtrate was concentrated. The crude material was purified by silica gel chromatography (CH₂Cl₂/[CH₂Cl₂/MeOH/NH₄OH, 75:22.5:2.5], 0 → 50%) to afford tert-butyl ((1SR,3RS)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (69 mg). LCMS: ESI [M+2H
+] = 746.3 m/z; tR = 2.15 min (Method C)
Step 10: 5-((1SR,3RS)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide To a solution of tert-butyl ((1SR,3RS)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (69 mg, 1 equiv., 93 μmol) in CH2Cl2 (3.1 mL) at 0 °C, was added HCl (4.0 M in 1,4-dioxane) (0.17 g, 1.2 mL, 4.0 M, 50 equiv., 4.6 mmol). The resulting mixture was stirred at room temperature. Upon complete conversion of the starting material, the volatiles were removed in vacuo. The crude material was purified by reverse phase HPLC prep chromatography (XSelect® CSH™ Prep C185 µm OBD™ 19x150 mm; mobile phase: water 0.1% FA / MeCN 0.1% FA; flow: 18.9 mL/min; gradient: 5% for 1 min then 5 → 25% over 20 min) to afford 5-((1SR,3RS)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (26.0 mg). LCMS: ESI [M+H]
+= 645.44 m/z; tR = 2.15 min (Method C) 1H NMR (400 MHz, CD
3CN) δ 6.83 (d, 1H), 6.69 (d, 1H), 6.54 – 6.49 (m, 2H), 5.43 (d, 1H), 4.93 (d, 1H), 4.68 (s, 2H), 4.61 (d, 1H), 4.49 – 4.22 (m, 4H), 3.98 – 3.88 (m, 1H), 3.88 – 3.78 (m, 1H), 3.78 – 3.57 (m, 2H), 3.57 – 3.46 (m, 1H), 3.25 (s, 3H), 3.28 – 3.16 (m, 1H), 2.97 (s, 3H), 2.89-2.83 (m, 2H), 2.72-2.65 (m, 1H), 2.50 – 1.97 (m, 11H), 1.60-1.51 (m, 1H), 1.02 (d, 3H). 1
9F NMR (376 MHz, CD3CN) δ -174.12 Example 52: 8-Fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-amine (Compound 183a)
Step 1: 1-Bromo-4-(but-3-en-1-yl)-2-fluorobenzene To a 250 mL round bottom flask containing 1-bromo-4-(bromomethyl)-2- fluorobenzene (10 g, 1 equiv., 37 mmol) in THF (20 mL) was added allyl(bromo)magnesium (8.1 g, 56 mL, 1 M, 1.5 equiv., 56 mmol), at 0 °C. The reaction was warmed to room temperature then refluxed for 3 hours. The reaction was cooled to room temperature, quenched with ammonium chloride (25 mL) and diluted with EtOAc (25 mL). The organic layer was separated, and the aqueous layer was extracted with DCM (50 mL). The combined organics were dried over sodium sulfate and solvent removed under reduced pressure to give 1-Bromo- 4-(but-3-en-1-yl)-2-fluorobenzene (8.39 g, 35 mmol) as a light purple oil. 1H NMR (400 MHz, CDCl
3) δ 7.34 – 7.20 (m, 1H), 6.83 – 6.75 (m, 1H), 6.75 – 6.63 (m, 1H), 5.69 – 5.54 (m, 1H), 4.94 – 4.77 (m, 2H), 2.57 – 2.42 (m, 2H), 2.29 – 2.12 (m, 2H). Step 2: 4-(4-Bromo-3-fluorophenyl)butan-1-ol To a flask containing 1-bromo-4-(but-3-en-1-yl)-2-fluorobenzene (8.39 g, 1 equiv., 36.6 mmol) in THF (100 mL) was added borane dimethyl sulfide complex (4.17 g, 4.76 mL, 1.5 equiv., 54.9 mmol) at 0 °C. The reaction mixture was stirred for 30 min at 0 °C then warmed to room temperature and stirred overnight. After 16 hours, a solution of H
2O
2 (7.12 g, 6.41 mL, 35% wt, 2 equiv., 73.2 mmol) and NaOH (2.93 g, 73.2 mL, 1 M, 2 equiv., 73.2 mmol) was slowly delivered to the reaction mixture at 0 °C, the ice bath was removed, and the mixture was stirred for 2 hours at room temperature. The reaction was then diluted with Et
2O (50 mL) and water (50 mL). The aqueous phase was separated and extracted with Et
2O (100 mL). The combined organics were washed with brine (50 mL), dried over sodium sulfate, and solvent removed under reduced pressure to provide 4-(4-bromo-3-fluorophenyl)butan-1-ol in sufficient purity for the next step. 1H NMR (400 MHz, CDCl3) δ 7.46 – 7.37 (m, 1H), 7.01 – 6.91 (m, 1H), 6.88 – 6.82 (m, 1H), 3.84 – 3.71 (m, 1H), 3.69 – 3.52 (m, 2H), 2.72 – 2.56 (m, 2H), 1.94 – 1.47 (m, 4H). Step 3: 4-(4-Bromo-3-fluorophenyl)butanoic acid
To a flask containing 4-(4-bromo-3-fluorophenyl)butan-1-ol (5.87 g, 1 equiv., 23.8 mmol) in acetone (35 mL) was added a solution of chromium(III) oxide (3.97 g, 1.1 equiv., 26.1 mmol), sulfuric acid (7.30 g, 3.97 mL, 3.14 equiv., 74.5 mmol), and water (25 g, 25 mL, 58 equiv., 1.4 mol) dropwise. The resulting solution was stirred for 30 minutes. The solvent was then removed under vacuum and the resulting residue was diluted with water (100 mL) and EtOAc (100 mL). The aqueous phase was separated then extracted with EtOAc (200 mL). The combined organics were washed with water (4x 100 mL) then extracted with 3 M aqueous NaOH (3x 65 mL). The combined NaOH layers were acidified with 1 M HCl (800 mL) to generate a precipitate which was filtered and dried to afford 4-(4-bromo-3- fluorophenyl)butanoic acid (800 mg) as an off-white solid. 1H NMR (400 MHz, CDCl
3) δ 7.41 – 7.32 (m, 1H), 6.93 – 6.85 (m, 1H), 6.82 – 6.75 (m, 1H), 2.62 – 2.54 (m, 2H), 2.35 – 2.27 (m, 2H), 1.94 – 1.82 (m, 2H). Step 4: 7-Bromo-8-fluoro-3,4-dihydronaphthalen-1(2H)-one To a flask containing 4-(4-bromo-3-fluorophenyl)butanoic acid (850 mg, 1 equiv., 3.26 mmol) was added polyphosphoric acid (12 g, 5.8 mL, 21 equiv., 67 mmol). The reaction mixture was stirred at 110 °C for 2.5 hours, cooled to room temperature then poured into ice water (60 mL) and diluted with EtOAc (60 mL). The aqueous layer was extracted with EtOAc (3 x 50 mL). The combined organics were dried over sodium sulfate and concentrated under reduced pressure to give 7-bromo-8-fluoro-3,4-dihydronaphthalen-1(2H)-one (749.6 mg) as a brown residue that was used without further purification. 1H NMR (400 MHz, CDCl3) δ 8.22 (d, 1H), 7.00 (d, 1H), 2.95 – 2.87 (m, 2H), 2.68 – 2.59 (m, 2H), 2.19 – 2.08 (m, 2H). Step 5: 7-Bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-8-fluoro-1,2,3,4-tetrahydronaphthalen-1-ol To an oven-dried 100 mL round bottom flask was added (4-chloro-6-methyl-2- (methylthio)pyrimidin-5-yl)methanol (599 mg, 0.95 equiv., 2.93 mmol). The flask was then sealed and evacuated and backfilled with nitrogen three times. THF (8 mL) was added, and the reaction was then cooled to -78 °C. LDA solution in THF/hexanes (759 mg, 7.09 mL, 1 M, 2.3 equiv., 7.09 mmol) was then added dropwise, maintaining a temperature below -70 °C. After complete addition of the LDA, an orange heterogeneous solution formed. In order to solubilize the reaction mixture, THF (6 mL) was added, and the reaction was stirred at -78 °C for 1
hour.7-bromo-8-fluoro-3,4-dihydronaphthalen-1(2H)-one (749 mg, 1 equiv., 3.08 mmol) was then dissolved in THF (3 mL) and the solution was added dropwise to the reaction mixture maintaining a temperature below -70 °C. The solution was stirred for 1 hour then quenched with ammonium chloride (10 mL) at -78 °C. The reaction was warmed to room temperature then diluted with EtOAc (10 mL) and water (10 mL). The aqueous layer was separated and extracted with EtOAc (3 x 20 mL). The combined organics were dried over sodium sulfate, and solvent removed under reduced pressure. The remaining residue was suspended up in DCM (15 mL) and filtered. The filtrate was concentrated, then taken up again in DCM (5 mL) and filtered again. The filtrate was concentrated to provide 7-bromo-1-((6-chloro-5- (hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-8-fluoro-1,2,3,4- tetrahydronaphthalen-1-ol (975 mg) as a fine white solid. 1H NMR (400 MHz, CDCl3) δ 7.73 (d, 1H), 6.86 (d, 1H), 4.84 – 4.70 (m, 2H), 4.36 (s, 1H), 3.35 – 3.20 (m, 2H), 2.85 – 2.74 (m, 3H), 2.54 (s, 3H), 2.11 – 1.86 (m, 3H), 1.76 – 1.65 (m, 1H). Step 6: 7-Bromo-4'-chloro-8-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] To an oven-dried 100 mL flask containing 7-bromo-1-((6-chloro-5-(hydroxymethyl)- 2-(methylthio)pyrimidin-4-yl)methyl)-8-fluoro-1,2,3,4-tetrahydronaphthalen-1-ol (975 mg, 1 equiv., 2.18 mmol) in toluene (4.36 mL) was added phosphoric acid (276 mg, 164 μL, 85% wt, 1.1 equiv., 2.40 mmol). The reaction mixture was heated to reflux for 2 hours then cooled to room temperature. The mixture was concentrated under reduced pressure and added to EtOAc (50 mL) and water (50 mL). The aqueous layer was separated and extracted EtOAc (3 x 50 mL). The combined organics were dried over sodium sulfate and solvent removed under reduced pressure to provide 7-Bromo-4'-chloro-8-fluoro-2'-(methylthio)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (863 mg) in sufficient purity for the next step. 1H NMR (400 MHz, CDCl
3) δ 7.59 (d, 1H), 6.80 (d, 1H), 4.80 – 4.60 (m, 2H), 3.17 – 2.91 (m, 2H), 2.76 – 2.67 (m, 2H), 2.49 (s, 3H), 2.02 – 1.59 (m, 4H). Step 7: 7-Bromo-8-fluoro-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]
To a 30 mL vial conatining 7-bromo-4'-chloro-8-fluoro-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (362.6 mg, 1 equiv., 843.8 μmol) and 1,4-oxazepane, HCl (348.3 mg, 3 equiv., 2.531 mmol) was added DIPEA (981.5 mg, 1.32 mL, 9 equiv., 7.59 mmol) and ethanol (1.7 mL). The reaction mixture was heated to reflux for 16 hours then cooled to room temperature followed by dilution with EtOAc (20 mL) and water (20 mL). The aqueous layer was separated and further extracted with additional EtOAc (3x 20 mL). The combined organic extracts were dried over sodium sulfate then concentrated under reduced pressure to provide 7-bromo-8-fluoro-2'-(methylthio)-4'-(1,4- oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (140 mg) LCMS: ESI [M+H]
+= 496.1 m/z; t
R = 2.71 min (Method C) Step 8: tert-Butyl (8-fluoro-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate To a 30 mL vial conatining 7-bromo-8-fluoro-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (140 mg, 1 equiv., 283 μmol) and BrettPhos Pd G4 (39 mg, 0.15 equiv., 43 μmol) was added cesium carbonate (277 mg, 3 equiv., 849 μmol), and tert-butyl carbamate (166 mg, 5 equiv., 1.42 mmol). The reaction vial was purged with N2.1,4-Dioxane (2.8 mL) was added and the mixture was heated to 90 °C for 16 hours. Incomplete conversion was observed by LCMS at this time. Additional BrettPhos Pd G4 (15 mg, 0.058 equiv., 16 μmol) in 1,4-dioxane (1 mL) was added to the reaction mixture. The resulting mixture was stirred for 2 hours at 110 °C until complete conversion was observed by LCMS. Reaction was cooled to room temperature, loaded directly onto silica, and purified via SiO
2 column chromatography (0 to 100% EtOAc in DCM) to provide tert-butyl (8-fluoro-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (110 mg). LCMS: ESI [M+H]
+= 531.3 m/z; t
R = 2.59 min (Method C) Step 9: tert-Butyl (8-fluoro-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate MeOH (2.7 mL) and water (1.4 mL) were added to a vial containing tert-butyl (8- fluoro-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate (110.5 mg, 1 equiv., 208.2 μmol) and Oxone (384.0
mg, 3 equiv., 624.7 μmol). The reaction mixture was stirred for 16 hours, quenched with saturated sodium thiosulfate (3 mL), and diluted with water (20 ml) and EtOAc (30 mL). The organic layer was separated, and the aqueous layer was extracted with EtOAc (3x 25 mL). The combined organics were dried over sodium sulfate and concentrated under reduced pressure to give tert-butyl (8-fluoro-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate as a yellow residue (131.4 mg). This residue was used without further purification. LCMS: ESI [M+H]
+= 563.2 m/z; tR = 3.02 min (Method C) Step 10: tert-Butyl (8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate Sodium tert-butoxide (33.0 mg, 172 μL, 2 M, 1.5 equiv., 344 μmol) was added dropwise to a vial containing tert-butyl (8-fluoro-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (129 mg, 1 equiv., 229 μmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (36.5 mg, 1 equiv., 229 μmol) in THF at 0 °C. After 30 min, the reaction was quenched with saturated ammonium chloride (2 mL) and diluted with EtOAc (5 mL). The organic layer was separated, and the aqueous layer was extracted with additional EtOAc (40 mL). The combined organic extracts were dried over sodium sulfate then concentrated under reduced pressure to provide a solid off-white foam. The crude material was purified by silica column chromatography (eluting from 0 to 20% MeOH in DCM) to provide tert-butyl (8-fluoro-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (88 mg). LCMS: ESI [M+H]
+= 642.4 m/z; tR = 1.96 min (Method C) Step 11: 8-Fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-amine HCl in diethyl ether (17 mg, 0.23 mL, 2 M, 20 equiv., 0.47 mmol) was added dropwise to a vial containing tert-butyl (8-fluoro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-
pyrano[4,3-d]pyrimidin]-7-yl)carbamate (15 mg, 1 equiv., 23 μmol) and DCM (0.2 mL). After 3 hours, the solvent was removed under reduced pressure to provide 8-fluoro-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (8.775 mg). 1H NMR (400 MHz, MeOD) δ 7.81 – 7.59 (m, 1H), 7.21 (d, 1H), 5.60 (d, 1H), 5.13 – 4.97 (m, 1H), 4.80 – 4.58 (m, 2H), 4.15 – 3.65 (m, 10H), 3.59 – 3.40 (m, 2H), 3.12 (s, 2H), 2.99 – 2.86 (m, 2H), 2.79 – 2.57 (m, 2H), 2.52 – 2.41 (m, 1H), 2.40 – 2.27 (m, 2H), 2.27 – 2.16 (m, 2H), 2.15 – 1.95 (m, 4H), 1.93 – 1.77 (m, 2H). 1
9F NMR (376 MHz, MeOD) δ -125.2, -170.3 LCMS: ESI [M+H]
+= 542.4 m/z; t
R = 1.53 min (Method C) Example 53: 5-((1R,4S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 124c)

Step 1: (1RS,4S)-7-Bromo-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-4-methyl-1,2,3,4-tetrahydronaphthalen-1-ol To an oven-dried 100 mL round bottom flask was added (4-chloro-6-methyl-2- (methylthio)pyrimidin-5-yl)methanol (500 mg, 1 equiv., 2.44 mmol). The flask was then sealed and evacuated and backfilled with nitrogen three times. THF (5.9 mL) was then added to the mixture and the reaction was cooled to -78 °C. LDA solution in THF/hexanes (602 mg, 5.62 mL, 1 M, 2.3 equiv., 5.62 mmol) was then added dropwise, maintaining a temperature below - 74 °C. After complete addition, a dark red homogeneous solution formed, and this solution was stirred at -78 °C for 30 minutes. (S)-7-bromo-4-methyl-3,4-dihydronaphthalen-1(2H)-one (Intermediate 7b, 526 mg, 0.9 equiv., 2.20 mmol) was dissolved in THF (0.4 M) and added dropwise to the solution keeping the reaction below -74 °C. After 5 minutes, saturated
ammonium chloride solution (6 mL) was added, and the reaction was warmed to room temperature followed by stirring for an additional 15 minutes. The reaction mixture was then extracted with EtOAc (3x 100 mL) and then the organic layer was dried using sodium sulfate. The organic layers were then concentrated under reduced pressure to give the crude material. The crude material was purified by silica gel chromatography eluting with 100% DCM to 85:15 DCM:EtOAc to provide (1RS,4S)-7-bromo-1-((6-chloro-5- (hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-4-methyl-1,2,3,4- tetrahydronaphthalen-1-ol (595 mg) as a white foam. 1H NMR (400 MHz, CDCl3) δ 7.76 – 7.60 (m, 1H), 7.47 – 7.30 (m, 1H), 7.20 – 7.00 (m, 1H), 4.89 – 4.63 (m, 2H), 4.20 – 3.89 (m, 1H), 3.49 – 3.14 (m, 2H), 3.00 – 2.84 (m, 2H), 2.54 (s, 3H), 2.28 – 2.11 (m, 1H), 2.03 – 1.92 (m, 1H), 1.93 – 1.81 (m, 1H), 1.78 – 1.64 (m, 1H), 1.38 – 1.21 (m, 3H). Step 2: (1RS,4S)-7-Bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] To a 100 mL round bottom flask containing (1RS,4S)-7-bromo-1-((6-chloro-5- (hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-4-methyl-1,2,3,4- tetrahydronaphthalen-1-ol (320 mg, 1 equiv., 721 μmol) in toluene (2.88 mL), was added phosphoric acid (83.1 mg, 49.3 μL, 85% wt, 1 equiv., 721 μmol). The reaction mixture was heated to 85 °C for 2 hours. The reaction was cooled to room temperature and the solvent was removed under reduced pressure. To the resulting yellow residue was added water (20 mL) and EtOAc (20 mL). The organic phase was separated and the aqueous layer was extracted with EtOAc (3 x 10 mL). The combined organics were dried with sodium sulfate, and concentrated under reduced pressure to give crude (1RS,4S)-7-bromo-4'-chloro-4-methyl-2'-(methylthio)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] which was directly advanced to the next step as a 1:1 mixture of diastereomers. LCMS: m/z (ESI) [M+H]
+ = 427.0, t
R = 4.14 min (Method C) Step 3: 5-((1RS,4S)-7-Bromo-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide To a vial containing (1RS,4S)-7-bromo-4'-chloro-4-methyl-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (0.261 g, 1 equiv., 613
μmol) and N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide, 2HCl (428 mg, 80.6% wt, 2 equiv., 1.23 mmol) in ethanol (1.0 mL), was added DIPEA (634 mg, 854 μL, 8 equiv., 4.90 mmol). The reaction mixture was heated to reflux. The reaction was quenched after 16 hours with water (30 mL) and diluted with EtOAc (30 mL). The organic layer was separated and the aqueous layer was extracted with EtOAc (40 mL). The combined organics were dried over sodium sulfate then concentrated under reduced pressure to give an orange solid foam. The solid residue was purified by silica column chromatography (eluting with 0 to 100% EtOAc in DCM) to provide 5-((1RS,4S)-7-bromo-4- methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (117.8 mg) as a solid colorless residue. 1H NMR (400 MHz, CDCl3) δ 7.58 – 7.50 (m, 1H), 7.42 – 7.32 (m, 1H), 7.21 – 7.05 (m, 1H), 6.60 – 6.49 (m, 1H), 4.89 – 4.40 (m, 6H), 4.05 – 3.92 (m, 1H), 3.84 – 3.73 (m, 1H), 3.33 (s, 3H), 3.11 – 3.06 (m, 4H), 3.05 – 2.87 (m, 2H), 2.56 – 2.46 (m, 3H), 2.32 – 2.06 (m, 3H), 2.03 – 1.90 (m, 1H), 1.84 – 1.73 (m, 1H), 1.54 – 1.42 (m, 1H), 1.34 – 1.30 (m, 3H). Step 4: tert-Butyl ((1RS,4S)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate Tert-butyl carbamate (92 mg, 5 equiv., 0.79 mmol), 5-((1RS,4S)-7-bromo-4-methyl-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (94 mg, 1 equiv., 0.16 mmol), BrettPhos Pd G4 (22 mg, 0.15 equiv., 24 μmol), and cesium carbonate (0.15 g, 3 equiv., 0.47 mmol) were delivered to a flask and purged with N
2.1,4-Dioxane (2 mL) was delivered to a flask, sealed, and heated to 80 °C for 6 hours. The reaction was filtered, concentrated, and directly purified by silica gel chromatography to provide tert-butyl ((1RS,4S)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)- yl)-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (66 mg) which was used as is in the next reaction. LCMS: m/z (ESI) [M+H]
+ = 634.4, tR = 2.91 min (Method C)
Step 5: tert-Butyl ((1RS,4S)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-4-methyl-2'-(methylsulfonyl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate tert-Butyl ((1RS,4S)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-4-methyl-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (66 mg, 1 equiv., 0.10 mmol) and oxone (0.22 g, 3.5 equiv., 0.36 mmol) were delivered to a flask followed by methanol (2 mL) and water (1 mL). The mixture was stirred until the starting material was consumed as monitored by LCMS. The reaction was diluted with water (10 mL) and EtOAc (10 mL). The aqueous layer was separated and extracted EtOAc (3x 20 mL). The combined organics were dried over sodium sulfate and the organics were concentrated under reduced pressure to provide tert-butyl ((1RS,4S)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin- 5(6H)-yl)-4-methyl-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate (67 mg) which was advanced without further purification. LCMS: m/z (ESI) [M+H]
+ = 665.9, tR = 3.08 min (Method C) Step 6: tert-Butyl ((1RS,4S)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate Sodium tert-butoxide in THF (19 mg, 0.10 mL, 2 M, 2 equiv., 0.20 mmol) was added dropwise to a vial containing tert-butyl ((1RS,4S)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-4-methyl-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (67 mg, 1 equiv., 0.10 mmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (16 mg, 1 equiv., 0.10 mmol) in THF (1 mL) at 0 °C. The reaction was quenched after 30 min with saturated aqueous ammonium chloride (2 mL) and diluted with EtOAc (5 mL). The organic layer was separated and the aqueous layer was extracted with EtOAc (40 mL). The combined organic extracts were dried over sodium sulfate then concentrated to provide a solid residue. This crude material was purified by silica gel chromatography (eluting with 0 to 20% MeOH in DCM) to provide tert-butyl ((1RS,4S)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-
yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (36 mg) LCMS: m/z (ESI) [M+H]
+ = 744.8, tR = 2.23 min (Method C) Step 7: 5-((1R,4S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide HCl in diethyl ether (35 mg, 0.48 mL, 2 M, 20 equiv., 0.97 mmol) was added dropwise to a vial containing tert-butyl ((1RS,4S)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (36 mg, 1 equiv., 48 μmol) in DCM (1 mL). After 3 hours, the solvent was directly concentrated to provide 5-((1RS,4S)-7-amino-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide which was directly progressed for separation of diastereomers by chiral SFC. LCMS: m/z (ESI) [M+H]
+ = 645.4, tR = 1.52 min (Method C) The diastereomers of 5-((1RS,4S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide were separated by chiral preparative SFC (Daicel Chiralpak IB, 21 mm I.D. x 250 mm, 5µm, part number 88447. Isocratic Mobile phase 45%A CO2 and 55%B MeOH with 0.25% DEA. Flow rate: 50 mL/min. Back pressure: 120 bar) to afford 5- ((1R,4S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4- methyl-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Peak 2) (tR = 7.57 min, 3.7 mg) Peak 2: LCMS: ESI [M+H]
+= 645.3 m/z; t
R = 1.58 min (Method C) 1H NMR (400 MHz, MeOD) δ 7.07 (d, 1H), 6.76 – 6.63 (m, 2H), 6.52 (s, 1H), 5.28 (d, 1H), 4.83 – 4.66 (m, 3H), 4.61 – 4.45 (m, 3H), 4.18 – 4.03 (m, 2H), 4.01 – 3.86 (m, 1H), 3.37
– 3.34 (m, 1H), 3.27 – 3.13 (m, 2H), 3.11 – 3.05 (m, 3H), 3.04 – 2.96 (m, 2H), 2.95 – 2.85 (m, 2H), 2.85 – 2.73 (m, 4H), 2.38 – 1.79 (m, 10H), 1.47 – 1.23 (m, 6H). 1
9F NMR (376 MHz, MeOD) δ -168.80 – -176.94 (m). Example 54: 5-(6-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,3-dimethyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]- 4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Compound 125a)

Step 1: 6-Bromo-3,3-dimethylisochroman-4-one A vial containing 6-bromoisochroman-4-one (500 mg, 1 equiv., 2.20 mmol) was degassed followed by the addition of dry THF (10.0 mL) and MeI (641 mg, 282 μL, 2.05 equiv., 4.51 mmol). The reaction mixture was then stirred at 25 °C followed by the addition of sodium 2-methylbutan-2-olate (3.22 mL, 1.4 M in THF, 2.05 equiv., 4.51 mmol) dropwise. The reaction was then heated to 65 °C for 5 hours. The reaction mixture was diluted with ethyl acetate (100 mL) and the organic layer was washed with saturated ammonium chloride(aq) solution before drying over sodium sulfate and being concentrated under reduced pressure to give the crude residue. The residue was purified by column chromatography (25 g SiO2 cartridge, 0-40% ethyl acetate in heptane) to give 6-bromo-3,3-dimethylisochroman-4-one (274 mg, 1.07 mmol). 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 2.1 Hz, 1H), 7.65 (dd, J = 8.2, 2.1 Hz, 1H), 7.07 (d, J = 8.2 Hz, 1H), 4.87 (m, 2H), 1.46 (s, 6H). Step 2: 6-bromo-4'-chloro-3,3-dimethyl-2'-(methylthio)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine] A 40 mL vial containing (4-chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (217 mg, 1.00 equiv., 1.06 mmol) and THF (10.0 mL) was cooled to -78 °C. A LDA solution
(2.43 mL, 1.0 M in THF, 2.30 equiv., 2.43 mmol) was added dropwise while maintaining a reaction temperature at -78 °C. The reaction gradually turned yellow to dark orange over 60 minutes. A THF solution of 6-bromo-3,3-dimethylisochroman-4-one (270 mg, 1.00 equiv., 1.06 mmol) in THF (10.0 mL) was added to the reaction dropwise and the reaction was stirred at -78 °C for 15 minutes. The reaction contents (while cold) were then poured into a saturated ammonium chloride (aq) solution (30 mL). The organic layer was then extracted into ethyl acetate (2 x 50 mL) and was dried over sodium sulfate followed by concentration under reduced pressure to give 6-bromo-4-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-3,3-dimethylisochroman-4-ol (450 mg, 0.78 mmol) which was then taken into the subsequent step. Triphenylphosphine (1.84 g, 3.0 equiv., 2.94 mmol) (polymer supported, ~1.6 mmol/g loading) was added to this material and the vial was degassed and filled with N2 before dry THF (10.0 mL) and dry DCM (10.0 mL) were added. To this mixture was dropwise added DIAD (594 mg, 3.0 equiv., 2.94 mmol) at ambient temperature. The reaction was stirred for 1 hour. The reaction mixture was diluted with ethyl acetate (100 mL) and dichloromethane (100 mL) and was filtered over Celite and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (25 g SiO
2 cartridge, 0-35% ethyl acetate in heptane) to give 6-bromo-4'-chloro-3,3-dimethyl-2'-(methylthio)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine] (243 mg). 1H NMR (400 MHz, CDCl
3) δ 7.42 (m, 1H), 7.09 (m, 1H), 7.00 – 6.96 (m, 1H), 4.92 (m, 1H), 4.79 (m, 2H), 4.28 (m, 1H), 3.29 (m, 1H), 2.98 (m, 1H), 2.60 (s, 3H), 1.42 (s, 3H), 1.14 (s, 3H). Step 3: 5-(6-Bromo-3,3-dimethyl-2'-(methylthio)-4a',5',8',8a'- tetrahydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide DIPEA (300 μL, 7.62 equiv., 1.72 mmol) was added to a suspension of 6-bromo-4'- chloro-3,3-dimethyl-2'-(methylthio)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidine] (99.8 mg, 1.0 equiv., 0.226 mmol) and N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide, 2HCl (88.5 mg, 1.39 equiv., 0.315 mmol) in EtOH (2.0 mL) in a 25 mL round bottom flask equipped with a reflux condenser. The reaction was stirred at 100 °C for 10 hours. After this time, additional N,N-dimethyl-5,6,7,8-
tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide, 2HCl (22.0 mg, 0.35 equiv., 0.078 mmol) was then added, and the reaction was left to stir for additional 6 hours at 100 °C. The reaction was removed from heat and concentrated under a stream of nitrogen. Ethyl acetate (20 mL) was added, and the mixture was sonicated for 5 minutes to yield a colorless suspension. The suspension was washed with 10% citric acid (5 mL). The suspension turned to a biphasic solution. The aqueous layer was separated and extracted with ethyl acetate (10 mL). The combined organic layers were washed with 1N sodium hydroxide (5 mL) and brine (5 mL) then dried over magnesium sulfate. The organic layer was filtered, and concentrated to yield a residue which was purified by column chromatography (10 g SiO2 cartridge, 0-10% methanol in dichloromethane) to give 5-(6-bromo-3,3-dimethyl-2'-(methylthio)-4a',5',8',8a'- tetrahydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (128 mg) LCMS: m/z (ESI) [M+H]
+ 613.3, tR = 2.63 minutes. (Method E) Step 4: 5-(6-Bromo-3,3-dimethyl-2'-(methylsulfonyl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide To a vial was added 5-(6-bromo-3,3-dimethyl-2'-(methylthio)-4a',5',8',8a'- tetrahydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (128 mg, 1 equiv., 0.209 mmol), oxone (598 mg, 45% wt, 2.1 equiv., 0.438 mmol), methanol (2 mL) and water (2 mL). The vial was then capped and stirred at 25 °C for 3 hours. The reaction mixture was diluted with ethyl acetate (50 mL) and was then washed with water. The organics were then dried over sodium sulfate, filtered and concentrated under reduced pressure to give 5-(6-bromo-3,3-dimethyl-2'- (methylsulfonyl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (109 mg). LCMS: m/z (ESI) [M+H]
+ 647.2, t
R = 2.50 minutes. (Method E) Step 5: 5-(6-Bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,3-dimethyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]- 4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide A vial containing NaH (27.0 mg, 60% wt, 4 equiv., 0.675 mmol) and ((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (134 mg, 5 equiv., 0.844 mmol) was
evacuated and refilled with nitrogen three times followed by addition of dry THF (3 mL) with an exit needle. The reaction mixture was stirred for 15 minutes. A second vial was charged with 5-(6-bromo-3,3-dimethyl-2'-(methylsulfonyl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (109 mg, 1 equiv., 0.169 mmol) and was evacuated and refilled with nitrogen three times before dry THF (3 mL) was added. The alkoxide was then added dropwise at 25 °C to the previously prepared mixture and the contents were stirred at ambient temperature for 3 hours. The reaction mixture was diluted with ethyl acetate (100 mL) and washed with saturated ammonium chloride(aq) (20 mL x 2). The organic layer was then dried over sodium sulfate and was then concentrated under reduced pressure to give the crude reaction residue. The residue was purified by column chromatography (10 g SiO
2 cartridge, 0- 20% methanol in dichloromethane) to give 5-(6-bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,3-dimethyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (65 mg, 0.090 mmol). 1H NMR (400 MHz, CDCl3) δ 7.43 (m, 1H), 7.22 (s, 1H), 6.99 (m, 1H), 6.46 (s, 1H), 5.28 (m, 1H), 4.90 (m, 1H), 4.79 (m, 1H), 4.70 (m, 1H), 4.57 (m, 1H), 4.51 – 4.34 (m, 3H), 4.26 (m, 1H), 4.09 (m, 2H), 3.81 (m, 1H), 3.67 (m, 1H), 3.29 (s, 3H), 3.27 – 3.12 (m, 3H), 3.06 (s, 3H), 2.99 (m, 1H), 2.88 (m, 1H), 2.31 – 2.12 (m, 4H), 2.06 – 1.84 (m, 5H), 1.38 (d, J = 1.4 Hz, 3H), 1.16 (s, 3H). 1
9F NMR (376 MHz, CDCl
3) δ -172.96. Step 6: tert-Butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,3-dimethyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]- 6-yl)carbamate To a vial was added 5-(6-bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,3-dimethyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (50 mg, 1 equiv., 0.069 mmol), tert-butyl carbamate (16 mg, 2.0 equiv., 0.140 mmol) palladium diacetate (3.1 mg, 0.2 equiv., 0.014 mmol) cesium carbonate (67 mg, 3.0 equiv., 0.210 mmol) and 2-(dicyclohexylphosphanyl)-2',4',6'-tris(isopropyl)biphenyl (13.0 mg, 0.4 equiv., 0.028 mmol). Once the vial was degassed and refilled with nitrogen, dry 1,4-dioxane
(1.0 mL) was added via syringe. The reaction was then stirred at 100 °C for 18 hours. At this time, the reaction mixture was diluted with ethyl acetate (60 mL) and was then filtered over Celite and was concentrated under reduced pressure to give a crude residue. The residue was purified by reverse phase HPLC prep chromatography (load: DMSO (1.00 mL); 30 g column: XSelect® CSH C18 OBD™ Prep Column 130 Å, 5 µm, 10 mm x 150 mm; mobile phase: water / MeCN 0.1% FA; flow: 4 mL/min; gradient: 10 → 35% for 1 min then 35 → 75% over 15 min) to give tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,3-dimethyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6- yl)carbamate which was utilized without further purification. LCMS: m/z (ESI) [M+H]
+ 761.5, t
R = 1.92 minutes (Method E) Step 7: 5-(6-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,3-dimethyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]- 4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide To a vial was added tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,3-dimethyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidin]-6-yl)carbamate (20 mg, 1 equiv., 0.026 mmol) and CDCl3 (1.0 mL) and HCl (165 μL, 4.0 M, 25.0 equiv., 0.657 mmol). The reaction was stirred at 25 °C for 4 hours. The volatiles were then removed under reduced pressure, and the product was purified using reverse-phase prep-HPLC (AccQ Prep Purification. 5-25% MeCN/Water gradient (0.1% FA). Column info: (Waters XSelect CSH C18 QBD Prep Column, 10 mm ID x 150 mm, 5µm particle size, part number 186008238)) to give 5-(6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,3-dimethyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (1.9 mg) as a mixture of diastereomers. 1H NMR (400 MHz, MeOD) δ 6.90 (m, 1H), 6.71 (m, 1H), 6.44 (m, 2H), 5.44 (m, 1H), 4.83 – 4.57 (m, 6H), 4.51 – 4.19 (m, 5H), 4.06 – 3.96 (m, 1H), 3.77 – 3.68 (m, 1H), 3.64 – 3.50 (m, 3H), 3.30 (m, 4H), 3.24 (m, 1H), 3.08 (m, 4H), 2.88 (m, 1H), 2.46 – 2.34 (m, 2H), 2.28 – 2.13 (m, 4H), 2.00 (s, 2H), 1.35 (s, 3H), 1.15 (s, 3H). 1
9F NMR (376 MHz, MeOD) δ -173.77 – -174.07 (m).
Example 55: (1RS,4SR)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane- 4,7'-pyrano[4,3-d]pyrimidine]-5-carbonitrile (Compound 178a)

Step 1: 1-(2-Bromo-4-nitrophenyl)ethan-1-ol A solution of 1-(2-bromo-4-nitrophenyl)ethan-1-one (5.00 g, 1 equiv., 20.5 mmol) in MeOH (100 mL) was treated with sodium borohydride (1.63 g, 2.1 equiv., 43.0 mmol) portion-wise at 0 °C. The mixture was stirred for 15 minutes and was quenched at 0 °C with saturated ammonium chlorideaq (80 mL). The mixture was stirred until bubbling stopped. The mixture was then concentrated under reduced pressure. The remaining mixture was extracted with EtOAc (3 x 20 mL). The organic fraction was washed with brine (20 mL) then dried over sodium sulfate, filtered and concentrated. The crude residue was purified by column chromatography (SiO2 cartridge, 0-30% EtOAc/heptanes) to afford 1-(2-bromo-4- nitrophenyl)ethan-1-ol (4.94 g). 1H NMR (400 MHz, CDCl
3) δ 8.39 (d, J = 2.3 Hz, 1H), 8.21 (m, 1H), 7.83 (d, J = 8.6 Hz, 1H), 5.29 (m, 1H), 2.13 – 2.04 (m, 1H), 1.50 (d, J = 6.4 Hz, 3H). Step 2: 1-(1-(Allyloxy)ethyl)-2-bromo-4-nitrobenzene To an oven-dried flask was added NaH (1.37 g, 2.8 equiv., 57.0 mmol) followed by THF (14.0 mL). A separate vial was charged with 1-(2-bromo-4-nitrophenyl)ethan-1-ol (4.95 g, 1 equiv., 20.1 mmol) and THF (14.0 mL). The alcohol solution was delivered slowly to the stirring NaH mixture at 0 °C and was stirred for 30 minutes. Allyl bromide (2.5 mL, 1.4 equiv., 29.0 mmol) was added dropwise with stirring at 0 °C. The reaction was left to stir for 18 hours at room temperature. The mixture was cooled to 0 °C then poured into a mixture of saturated ammonium chloride
aq containing ice. The mixture was then extracted with diethyl ether (3 x 20 mL). The pooled organic fractions were dried over sodium sulfate, filtered and concentrated to give 1-(1-(allyloxy)ethyl)-2-bromo-4-nitrobenzene (5.30 g). 1H NMR (400 MHz, CDCl
3) δ 8.43 – 8.37 (m, 1H), 8.20 (m, 1H), 7.73 (d, J = 8.6 Hz, 1H), 5.97 – 5.83 (m, 1H), 5.28 (m, 1H), 5.20 (m, 1H), 4.89 (m, 1H), 3.95 – 3.80 (m, 2H), 1.43
(dd, J = 6.4, 0.9 Hz, 3H). Step 3: 1-Methyl-4-methylene-6-nitroisochromane To a 40 mL vial was added 1-(1-(allyloxy)ethyl)-2-bromo-4-nitrobenzene (5.00 g, 1 equiv., 17.5 mmol), Pd(OAc)2 (785 mg, 0.2 equiv., 3.49 mmol) and 1,2- bis(diphenylphosphanyl)benzene (2.26 g, 0.3 equiv., 5.07 mmol). The vial was degassed followed by the addition of dry 2-MeTHF (22.5 mL). The mixture was then stirred at 70 °C for 4 hours. The mixture was filtered through Celite using diethyl ether. The filtrate was concentrated then treated with water (20 mL) and extracted with ether (3 x 10 mL). The organic phase was washed with brine, dried over sodium sulfate, filtered, and concentrated under reduced pressure to give a residue which was purified by flash chromatography (330 g SiO
2, 0-10% EtOAc/heptanes) to afford the product 1-methyl-4-methylene-6-nitroisochromane (844 mg). 1H NMR (400 MHz, CDCl
3) δ 8.51 (m, 1H), 8.09 (m, 1H), 7.29 (m, 1H), 5.77 (m, 1H), 5.20 (m, 1H), 4.92 (m, 1H), 4.59 (m, 1H), 4.38 (m, 1H), 1.61 (d, J = 6.6 Hz, 3H). Step 4: 1-Methyl-6-nitroisochroman-4-one To a 4-dram vial containing 1-methyl-4-methylene-6-nitroisochromane (844 mg, 1 equiv., 4.11 mmol) in THF (13.7 mL), was added sodium periodate (2.11 g, 2.4 equiv., 9.87 mmol) and potassium osmate(VI)dihydrate (30.3 mg, 0.02 equiv., 0.082 mmol). Afterwards, water (6.85 mL) was added, and the mixture was stirred at 70 °C for 4 hours. The mixture was quenched with saturated sodium thiosulfate (20 mL) and was stirred for 10 minutes. The mixture was extracted with EtOAc (3 x 20 mL). The organic phase was washed with brine, dried over sodium sulfate, filtered, and concentrated to afford 1-methyl-6- nitroisochroman-4-one (0.775 g). 1H NMR (400 MHz, CDCl3) δ 8.87 (m, 1H), 8.42 (m, 1H), 7.47 (m, 1H), 5.00 (m, 1H), 4.59 (m, 1H), 4.36 (m, 1H), 1.72 (m, 3H). Step 5: 6-Amino-1-methylisochroman-4-one To a flask containing 1-methyl-6-nitroisochroman-4-one (1.55 g, 1 equiv., 7.48 mmol) in THF (3 mL), was added ammonium chloride (1.60 g, 4 equiv., 29.9 mmol) and iron (1.67 g, 4 equiv., 29.9 mmol). Afterwards, EtOH (15 mL) and H2O (5 mL) were added and the reaction mixture was stirred for 18 hours at room temperature. The mixture was filtered through
Celite with EtOAc (50 mL) and DCM (50 mL). The filtrate was concentrated then filtered over sodium sulfate. The filtrate was concentrated and subjected to purification via flash chromatography (0-40% EtOAc/heptanes) to afford 6-amino-1-methylisochroman-4-one (531 mg). 1H NMR (400 MHz, CDCl3) δ 7.33 (m, 1H), 7.06 (d, J = 8.3 Hz, 1H), 6.94 (m,1H), 4.84 (m, 1H), 4.48 (m, 1H), 4.29 – 4.21 (m, 1H), 1.60 (d, J = 6.5 Hz, 3H). Step 6: 6-Amino-5-bromo-1-methylisochroman-4-one To a solution of 6-amino-1-methylisochroman-4-one (534 mg, 1.00 equiv., 3.02 mmol) in DMF (10 mL) at 0 °C, was added a solution of N-bromosuccinimide (590 mg, 1.1 equiv., 3.32 mmol) in DMF (4 mL) dropwise under nitrogen at 0 °C. After stirring for 1.5 hours at 0 °C, additional N-bromosuccinimide (590 mg, 1.1 equiv., 3.32 mmol) in DMF (1 mL) was added at 0 °C. The contents were warmed to ambient temperature and stirred for 18 hours. The mixture was then treated with saturated sodium thiosulfate (100 mL) then stirred for 10 minutes. The organics were extracted with EtOAc (3 x 20 mL), washed with 1:1 water/brine (60 mL), dried over sodium sulfate, filtered and concentrated to give a crude product which was subjected to chromatographic purification (120 g silica column; 0-60% EtOAc/heptanes) to afford the product 6-amino-5-bromo-1-methylisochroman-4-one (832 mg). 1H NMR (400 MHz, CDCl3) δ 6.97 (m, 1H), 6.90 (m, 1H), 4.74 (m, 1H), 4.42 (m, 1H), 4.19 (m, 1H), 1.53 (d, J = 6.5 Hz, 3H). Step 7: 5-Bromo-6-(dibenzylamino)-1-methylisochroman-4-one To a solution of 6-amino-5-bromo-1-methylisochroman-4-one (772 mg, 1.00 equiv., 3.02 mmol) in MeCN (12 mL) at room temperature was added potassium carbonate (1.80 g, 4.3 equiv., 13.0 mmol) followed by the dropwise addition of benzyl bromide (2.30 g, 1.60 mL, 4.5 equiv., 13.0 mmol). The contents were stirred at 25 °C before being heated to 65 °C for 18 hours. The mixture was then cooled to room temperature and additional potassium carbonate (740 mg, 1.78 equiv., 5.35 mmol) and benzyl bromide (863 mg, 0.600 mL, 1.67 equiv., 5.04 mmol) were added. The mixture was heated for an additional 18 hours at 65 °C. The temperature was then increased to 80 °C for an additional 2 hours. The mixture was then cooled to room temperature and diluted with water (30 mL). The reaction mixture was extracted with EtOAc (3 x 20 mL). The organic fractions were dried over sodium sulfate, filtered, and concentrated and subjected to chromatographic separation (120 g SiO2 column (0-40%
EtOAc/heptanes) to afford 5-bromo-6-(dibenzylamino)-1-methylisochroman-4-one (912 mg). LCMS: m/z (ESI) [M+H]
+ 436.1, tR = 2.24 minutes (Method C) Step 8: 5-Bromo-4-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-6-(dibenzylamino)-1-methylisochroman-4-ol To an oven-dried 50 mL round bottom flask was added (4-chloro-6-methyl-2- (methylthio)pyrimidin-5-yl)methanol (556 mg, 1.30 equiv., 2.72 mmol). The flask was then sealed and degassed with nitrogen. THF (9.5 mL) was then added, and the reaction was then cooled to -78 °C. LDA (5.5 mL, 1.0 M in THF, 2.6 equiv., 5.5 mmol) was then added dropwise, maintaining a temperature below -77 °C. Additional THF (4.4 mL) was then added. The mixture was stirred at -78 °C for 1 hour. A separate solution of 5-bromo-6-(dibenzylamino)-1- methylisochroman-4-one (911.5 mg, 1 equiv., 2.09 mmol) in THF (4.4 mL) was added dropwise to the reaction mixture maintaining a temperature below -77 °C. To a separate oven- dried 50 mL round bottom flask was added additional (4-chloro-6-methyl-2- (methylthio)pyrimidin-5-yl)methanol (500 mg, 1.17 equiv., 2.44 mmol). The flask was then sealed and degassed with nitrogen. THF (9 mL) was then added, and the reaction was then cooled to -78 °C. LDA (6 mL, 1.0 M in THF, 3 equiv., 6.0 mmol) was then added dropwise, maintaining a temperature below -75 °C. Additional THF (2 mL) used to help dissolve precipitate generated on walls of reaction vessel. After 1 hour, the mixture was transferred to the main reaction vessel using a cannula. The resultant mixture was stirred for 30 minutes. The mixture was then quenched with HCl (3.13 mL, 2.0 M in ether, 3 equiv., 6.27 mmol) slowly while maintaining temperature below -75 °C. Then the mixture was poured into a solution of 20 mL saturated ammonium chloride solution. The reaction was diluted with EtOAc (20 mL) and the aqueous layer was separated and extracted with further EtOAc (3x 20 mL). The combined organics were dried over sodium sulfate, filtered, concentrated and subjected to chromatographic purification (SiO2, 0-5% EtOAc/DCM) to afford 5-bromo-4-((6-chloro-5- (hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-6-(dibenzylamino)-1- methylisochroman-4-ol (875 mg, 1.37 mmol). LCMS: m/z (ESI) [M+H]
+ 640.1, tR = 2.34 minutes (Method C) Step 9: (1SR,4SR)-N,N-dibenzyl-5-bromo-4'-chloro-1-methyl-2'-(methylthio)- 5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine and (1RS,4SR)-
N,N-dibenzyl-5-bromo-4'-chloro-1-methyl-2'-(methylthio)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine A round bottom flask containing 5-bromo-4-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-6-(dibenzylamino)-1-methylisochroman-4-ol (0.876 g, 1 equiv., 1.37 mmol), triphenylphosphine (polymer supported) (2.9 g, 3.4 equiv., 4.6 mmol) (polymer supported, ~1.6 mmol/g loading) was degassed and backfilled with nitrogen (x 3), followed by the addition of dry THF (68.3 mL) and dry DCM (68.3 mL). DIAD (760 μL, 2.86 equiv., 3.91 mmol) was added dropwise to the mixture. The mixture of N,N-dibenzyl-5- bromo-4'-chloro-1-methyl-2'-(methylthio)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidin]-6-amine was filtered over Celite with DCM and was then subjected to chromatographic purification (120 g silica cartridge, 0-5% EtOAc/heptanes) to afford separable diastereomers (1SR,4SR)-N,N-dibenzyl-5-bromo-4'-chloro-1-methyl-2'- (methylthio)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (Peak 1) (334 mg) and (1RS,4SR)-N,N-dibenzyl-5-bromo-4'-chloro-1-methyl-2'-(methylthio)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (Peak 2) (136 mg) Peak 1: LCMS: m/z (ESI) [M+H]
+ 624.1, tR = 4.43 minutes (Method E) Peak 2: LCMS: m/z (ESI) [M+H]
+ 624.1, t
R = 4.36 minutes (Method E) Step 10: (1RS,4SR)-N,N-dibenzyl-5-bromo-1-methyl-2'-(methylthio)-4'-(1,4- oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine To a vial was added (1RS,4SR)-N,N-dibenzyl-5-bromo-4'-chloro-1-methyl-2'- (methylthio)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (Step 9, Peak 2; 137.0 mg, 1.0 equiv., 0.220 mmol), 1,4-oxazepane, HCl (4.0 equiv., 0.880 μmol), MeCN (2.0 mL) and DIPEA (613 μL, 16.0 equiv., 3.52 mmol). The vial was capped and stirred at 100 °C for 3 hours. The reaction was removed from heat and concentrated in vacuo to yield a residue which was purified by column chromatography (0-10% methanol in dichloromethane) to give (1RS,4SR)-N,N-dibenzyl-5-bromo-1-methyl-2'-(methylthio)-4'-(1,4- oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (181.0 mg). LCMS: m/z (ESI) [M+H]
+ 687.2, tR = 3.47 minutes. (Method E) Step 11: (1RS,4SR)-N,N-dibenzyl-5-bromo-1-methyl-2'-(methylsulfonyl)-4'-(1,4- oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine To a vial was added (1RS,4SR)-N,N-dibenzyl-5-bromo-1-methyl-2'-(methylthio)-4'-
(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (80.0 mg, 1 equiv., 0.116 mmol), oxone (334 mg, 45% wt, 2.1 equiv., 0.244 mmol), methanol (2 mL) and water (2 mL). The vial was capped and stirred at 25 °C for 2 hours. The reaction was quenched by the addition of water (20 mL). EtOAc (20 mL) and the organic layer was separated. The aqueous phase was extracted with EtOAc (3 x 20 mL). The combined organic extracts were dried over sodium sulfate, filtered and concentrated to yield (1RS,4SR)-N,N- dibenzyl-5-bromo-1-methyl-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (54.0 mg). LCMS: m/z (ESI) [M+H]
+ 719.4, tR = 3.57 minutes (Method E) Step 12: (1RS,4SR)-N,N-dibenzyl-5-bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine To a flask charged with ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methanol (59.7 mg, 5.0 equiv., 0.375 mmol) and (1RS,4SR)-N,N-dibenzyl-5-bromo-1- methyl-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-6-amine (54.0 mg, 1 equiv., 0.0750 mmol) was added THF (1.0 mL) under nitrogen at room temperature. The resulting solution was then treated with dropwise addition of t-BuOK (150 μL, 1.0 M in THF, 2.0 equiv., 0.150 mmol). The reaction mixture was stirred for 2 hours at room temperature. At this time, the reaction mixture was concentrated under reduced pressure to give the crude residue. The residue was purified by column chromatography (25 g silica cartridge, 0-10% methanol in dichloromethane) to give (1RS,4SR)- N,N-dibenzyl-5-bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidin]-6-amine (31 mg). LCMS: m/z (ESI) [M+H]
+ 798.5, tR = 2.52 minutes (Method E) Step 13: (1RS,4SR)-6-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]-5-carbonitrile To a vial charged with copper(I) cyanide (34.0 mg, 10 equiv., 0.38 mmol), was added (1RS,4SR)-N,N-dibenzyl-5-bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-
pyrano[4,3-d]pyrimidin]-6-amine (30.0 mg, 1 equiv., 0.038 mmol) and dry DMF (1.0 mL) under nitrogen. The resulting mixture was stirred at 100 °C for 7 hours. The reaction mixture was then cooled to room temperature and was treated with dichloromethane (30 mL), water (30 mL), methanol (30 mL) and NH
4OH
aq (30 mL). The organic layer was collected and combined with further dichloromethane (3 x 20 mL) extracts. The organics were dried over sodium sulfate, filtered, concentrated and purified by column chromatography (10 g silica cartridge, (0-50% (2.5% NH
4OH, 20% MeOH in DCM) in DCM) to give (1RS,4SR)-6- (dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-1- methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]- 5-carbonitrile (207 mg). This material was subjected to the subsequent reaction without further purification. LCMS: m/z (ESI) [M+H]
+ 745.5, tR = 2.39 minutes (Method E) Step 14: (1RS,4SR)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane- 4,7'-pyrano[4,3-d]pyrimidine]-5-carbonitrile To a vial charged with (1RS,4SR)-6-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]-5-carbonitrile (30.00 mg, 1 equiv., 0.040 mmol) was added palladium(II) hydroxide on carbon (70.0 mg, 20% wt, 2.5 equiv., 0.100 mmol). To the vial was added MeOH (1 mL) followed by the addition of acetic acid (2 drops). The reaction mixture was then stirred vigorously under an atmosphere of hydrogen at room temperature for 3 hours. The reaction mixture was then filtered over Celite with dichloromethane and methanol and the volatiles were then removed in vacuo. The contents were dissolved in DMF (1.2 mL) and purified via reverse phase HPLC prep chromatography (column: XSelect® CSH C18 OBD™ Prep Column 130 Å, 5 µm, 10 mm x 150 mm; mobile phase: water 0.1% TFA / MeCN 0.1% TFA; flow: 4.7 mL/min; gradient: 5 → 45%) to yield (1RS,4SR)-6-amino-2'-(((2R,7aS)- 2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]-5-carbonitrile (1.0 mg). 1H NMR (400 MHz, CDCl
3) δ 7.08 (d, J = 8.7 Hz, 1H), 6.81 (d, J = 8.7 Hz, 1H), 5.57 (m, 1H), 4.99 (m, 2H), 4.61 – 4.46 (m, 2H), 4.04 – 3.70 (m, 13H), 3.50 – 3.40 (m, 1H), 3.23 – 3.00 (m, 3H), 2.74 – 2.65 (m, 1H), 2.63 – 2.51 (m, 2H), 2.34 (m, 3H), 2.21 – 1.93 (m, 4H),
1.48 (d, J = 6.7 Hz, 3H). LCMS: m/z (ESI) [M+H]
+ 565.3, tR = 1.46 minutes (Method E) Example 56: (1SR,4SR)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane- 4,7'-pyrano[4,3-d]pyrimidine]-5-carbonitrile (Compound 178b)

Step 1: (1SR,4SR)-N,N-dibenzyl-5-bromo-1-methyl-2'-(methylthio)-4'-(1,4- oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine To a vial was added (1SR,4SR)-N,N-dibenzyl-5-bromo-4'-chloro-1-methyl-2'- (methylthio)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (Example 55, Step 9, Peak 1; 334.5 mg, 1 equiv., 0.537 mmol) in MeCN (5.4 mL) followed by DIPEA (750 μL, 8.02 equiv., 4.31 mmol) then 1,4-oxazepane, HCl (295.5 mg, 4 equiv., 2.148 mmol) was added. The vial was capped and stirred at 100 °C. After stirring at 100 °C for 3 hours. The reaction mixture was concentrated under reduced pressure. The crude residue was dissolved in 15 mL DCM and was washed with saturated ammonium chloride (2 x 30 mL). The organic extracts were combined, dried over sodium sulfate, filtered and concentrated. The crude material was subjected to purification [25 g silica column (0-15% MeOH/DCM)] to afford (1SR,4SR)-N,N-dibenzyl-5-bromo-1-methyl-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)- 5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (341.0 mg) as an off- white solid. LCMS: tR = 2.21 min, 689.1 m/z: [M+H]
+. (Method C) Step 2: (1SR,4SR)-6-(dibenzylamino)-1-methyl-2'-(methylthio)-4'-(1,4-oxazepan- 4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]-5-carbonitrile A vial containing (1SR,4SR)-N,N-dibenzyl-5-bromo-1-methyl-2'-(methylthio)-4'-(1,4- oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (170 mg, 1 equiv., 247 μmol) was treated with copper(I) cyanide (220 mg, 10.0 equiv., 2.46
mmol) followed by a degassed solution of DMF (5 mL). The resulting mixture was degassed for 3 min followed by stirring at 120 °C for 2.5 hours. The reaction mixture was then cooled to room temperature and concentrated. Water (5 mL) and NH4OH (5 mL) followed by DCM (10 mL) were added. The mixture was sonicated to dissolve the insoluble precipitate. The fractions were separated and the aqueous solution was washed with 5:1 DCM/IPA solution (3 x 5 mL). The organic fractions were pooled together and washed with brine. Afterwards, the mixture was washed with brine (10 mL), dried over sodium sulfate, filtered over celite to afford crude material which was subjected to purification (24 g silica, 0-20% EtOAc/DCM) to afford (1SR,4SR)-6-(dibenzylamino)-1-methyl-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]-5-carbonitrile (146.0 mg) as an off- white solid. LCMS: tR = 2.06 min, 634.3 m/z: [M+H]
+. (Method C) Step 3: (1SR,4SR)-6-(dibenzylamino)-1-methyl-2'-(methylsulfonyl)-4'-(1,4- oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]-5- carbonitrile To a solution of (1SR,4SR)-6-(dibenzylamino)-1-methyl-2'-(methylthio)-4'-(1,4- oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]-5-carbonitrile (146 mg, 1 equiv., 230 μmol) in THF (3.0 mL), methanol (1.80 mL) and water (1.80 mL) at room temperature, was added Oxone (944 mg, 45% wt, 3 equiv., 691 μmol) in one portion. The resulting mixture was stirred at room temperature for 6 hours. At this time, additional Oxone (944 mg, 45% wt, 3 equiv., 691 μmol) was added and the mixture was stirred overnight at ambient temperature. Water (5 mL) was added followed by extraction of the mixture with EtOAc (4 x 5 mL). The combined organic extracts were dried over sodium sulfate, filtered and concentrated to provide (1SR,4SR)-6-(dibenzylamino)-1-methyl-2'-(methylsulfonyl)-4'-(1,4- oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]-5-carbonitrile (131.5 mg), which was used in the next step without further purification. LCMS: t
R = 2.09 min, 666.2 m/z: [M+H]
+. (Method C) Step 4: (1SR,4SR)-6-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]-5-carbonitrile To a flask charged with ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-
yl)methanol (157.2 mg, 5.0 equiv., 987.5 μmol) was added a solution of (1SR,4SR)-6- (dibenzylamino)-1-methyl-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]-5-carbonitrile (131.0 mg, 1 equiv., 197.0 μmol) in DMF (4 mL) under nitrogen, at room temperature. The resulting solution was cooled to 0 °C, followed by the dropwise addition of lithium bis(trimethylsilyl)amide (1.5 M in THF) (200 μL, 1.5 M, 1.52 equiv., 300 μmol) (temperature monitored using a electronic probe, addition rate controlled so that temperature did not raise above 1 °C). The ice/water bath was removed and the reaction mixture was stirred for 16 hours. After stirring overnight, reaction was treated with additional lithium bis(trimethylsilyl)amide (200 μL, 1.5 M, 1.52 equiv., 300 μmol) at 0 °C. After stirring for 1 hour at ambient temperature, the reaction was quenched by the slow addition of a saturated aqueous ammonium chloride solution (10 mL; temperature monitored, remained below 30 °C). The mixture was treated with EtOAc (10 mL) and the layers were separated. The aqueous phase was extracted with EtOAc (3 x 5 mL). The combined organic extracts were dried over magnesium sulfate, filtered and concentrated. Crude material was purified by flash chromatography (0-20% (2.5% NH4OH and 20%MeOH in DCM)/DCM) to afford (1SR,4SR)-6-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]-5-carbonitrile (120 mg). LCMS: tR = 1.62 min, 745.3 m/z: [M+H]
+ (Method C) Step 5: (1SR,4SR)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane- 4,7'-pyrano[4,3-d]pyrimidine]-5-carbonitrile A vial containing (1SR,4SR)-6-(dibenzylamino)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]-5-carbonitrile (120.0 mg, 1 equiv., 160.0 μmol) and palladium(II) hydroxide (281.6 mg, 20% wt, 2.5 equiv., 401.1 μmol) was purged with nitrogen gas. Afterwards, MeOH (4 mL) was added under nitrogen, then the solution was degassed further for 3 minutes. The resulting solution was bubbled with H2 for 5 min, followed by the addition of acetic acid (8 drops). The reaction mixture was stirred vigorously under an atmosphere of hydrogen at room temperature and was stirred for 2 hours. The vial headspace was evacuated and refilled with nitrogen. The reaction mixture was filtered through a syringe filter and the filtrate was stirred with sodium carbonate for 5 min, filtered
and concentrated. The crude residue was subjected to purification via flash chromatography [4 g silica gel; 0-10% (2.5% NH4OH, 20% MeOH in DCM)/DCM] to afford (1SR,4SR)-6-amino- 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-4'-(1,4- oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]-5-carbonitrile (31.0 mg) as a mixture of diastereomers. LCMS: tR = 1.24 min, 565.3 m/z: [M+H]
+ (Method C) 1H NMR (400 MHz, MeOD) δ 7.10 (d, 1H), 6.79 (d, 1H), 5.36 – 5.18 (m, 1H), 4.98 – 4.88 (m, 2H), 4.69 (m, 1H), 4.13 (m, 1H), 4.05 (m, 1H), 4.00 (m, 1H), 3.92 – 3.65 (m, 8H), 3.57 (m, 1H), 3.30 – 3.08 (m, 5H), 2.98 (m, 1H), 2.34 – 1.78 (m, 8H), 1.43 (d, 3H). Example 57: 5-(8-Cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,
][1,4]diazepine-2-carboxamide (Compound 184a)
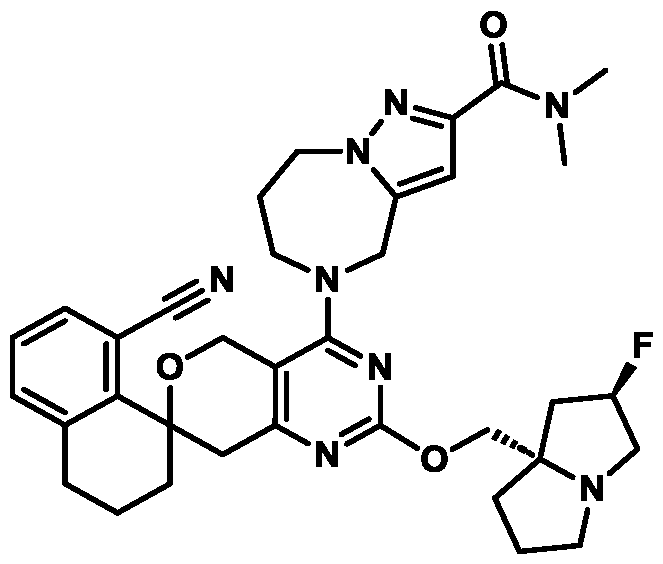
Step 1: 8-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-ol To a stirring solution of (4-chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (450 mg, 2.20 mmol) in THF (3.7 mL) at –78 °C was added LDA (4.2 mL, 1 M, 4.21 mmol) dropwise. After the resulting mixture was stirred at –78 °C for 1 hour, 8-bromo-3,4- dihydronaphthalen-1(2H)-one (412 mg, 1.83 mmol) in THF (3.7 mL) was added dropwise to the cooled mixture. The resulting mixture was then stirred at –78 °C for 1 hour. Afterwards, the mixture was treated with slow addition of HCl in ether (2.2 mL, 2 M, 4.31 mmol) at -78 °C. The reaction was warmed to room temperature then concentrated to afford a thick yellow oil. The crude was subjected to purification via flash chromatography (0-100% EtOAc/DCM) to afford the partially pure product. The partially pure product was subjected to purification via flash chromatography (0-100% EtOAc/DCM) to afford 8-bromo-1-((6-chloro-5-
(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-ol (231 mg) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.50 (d, 1H), 7.11 (d, 1H), 7.04 (dd, 1H), 4.94 (d, 1H), 4.83 (d, 1H), 4.28 (dd, 1H), 3.17 (d, 1H), 3.01 – 2.85 (m, 2H), 2.55 (s, 3H), 2.19 – 2.05 (m, 2H), 1.91 – 1.80 (m, 1H), 1.73 – 1.63 (m, 1H). Step 2: 8-Bromo-4'-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] A mixture of 8-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-ol (780 mg, 1.81 mmol) in toluene (7.3 mL) was treated with phosphoric acid (124 μL, 85% wt, 1.81 mmol) and the mixture was heated to 85 °C. After 2 hours, the solvent was removed under reduced pressure. The remaining yellow residue was treated with water and EtOAc. This process was repeated once more. The organic phase was separated and the aqueous phase was extracted with EtOAc. The combined organic fractions were dried over sodium sulfate, filtered, and concentrated. The crude was subjected to purification via flash chromatography (0-15% EtOAc/DCM) to afford 8-bromo-4'-chloro- 2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (485 mg). LCMS: m/z (ESI) [M+H]
+ 411.0, tR = 2.38 minutes (Method C) Step 3: 5-(8-Bromo-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide To a vial containing 8-Bromo-4'-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (200 mg, 486 μmol) and N,N-dimethyl- 5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide; dihydrochloride (205 mg, 729 μmol) in ethanol (1.9 mL) was added DIPEA (314 mg, 423 μL, 2.43 mmol). The reaction mixture was heated to reflux overnight. The reaction was cooled to room temperature and then concentrated to afford a crude viscous oil. The oil was dissolved in ethyl acetate (20 mL) and then washed with an aqueous solution of ammonium chloride (20 mL). The organic layer was then concentrated and subjected to purification via flash chromatography (0-10% MeOH/DCM) to afford 5-(8-bromo-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-
spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (200 mg). LCMS: m/z (ESI) [M+H]
+ 583.2, tR = 1.73 minutes (Method C) Step 4: 5-(8-Cyano-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide A vial containing 5-(8-bromo-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (170 mg, 291 μmol) was treated with copper(I) cyanide (261 mg, 2.91 mmol) followed by addition of a degassed solution of DMF (5.8 mL). The resulting mixture was degassed for 3 min followed by stirring at 120 °C. After 2 hours, the reaction mixture was cooled to room temperature and concentrated. Water (10 mL), NH4OH (10 mL) and DCM (10 mL) were added to the mixture. The fractions were separated, and the aqueous solution was washed with a 5:1 DCM/IPA solution (3 x 5 mL). The organic fractions were combined and washed with brine (10 mL). Afterwards, the mixture was dried over sodium sulfate, filtered, and concentrated to afford an oil. The crude material was subjected to purification via flash chromatography (0-100% EtOAc/DCM) to afford 5-(8-cyano-2'- (methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (130.8 mg) as a beige solid. 1H NMR (400 MHz, CDCl3) δ 7.60 (d, 1H), 7.33 (d, 1H), 7.31 – 7.24 (m, 1H), 6.56 (s, 1H), 4.97 – 4.86 (m, 2H), 4.71 – 4.56 (m, 2H), 4.55 – 4.39 (m, 2H), 4.07 – 3.98 (m, 1H), 3.79 – 3.65 (m, 1H), 3.41 – 3.27 (m, 4H), 3.13 – 3.02 (m, 3H), 2.98 (d, 1H), 2.91 – 2.80 (m, 2H), 2.49 (s, 3H), 2.36 – 2.17 (m, 2H), 2.16 – 2.04 (m, 1H), 2.02 – 1.94 (m, 1H), 1.92 – 1.72 (m, 2H). LCMS: m/z (ESI) [M+H]
+ 530.3, t
R = 1.62 minutes (Method C) Step 5: 5-(8-Cyano-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide 3-Chlorobenzoperoxoic acid (166 mg, 77% wt, 741 μmol) was added to the solution of 5-(8-cyano-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-
d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (130.8 mg, 247.0 μmol) in DCM (5 mL) at room temperature. The resulting yellow solution was stirred at room temperature for 1 hour. The mixture was treated with a saturated aqueous solution of sodium thiosulfate. After stirring for 5 min, the organic layer was separated, and the aqueous layer was extracted with DCM (3 x 5 mL). The combined organic layers were washed with saturated sodium bicarbonate solution, dried over magnesium sulfate, and concentrated to afford 5-(8-cyano-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (148.4 mg) as an off-white solid, and the material was used immediately without purification. 1H NMR (400 MHz, CDCl
3) δ 7.57 (d, 1H), 7.33 (d, 1H), 7.31 – 7.24 (m, 1H), 6.60 (s, 1H), 5.03 (d, 1H), 4.94 (d, 1H), 4.75 (d, 1H), 4.68 (d, 1H), 4.54 – 4.39 (m, 2H), 4.18 – 4.03 (m, 1H), 3.92 – 3.80 (m, 1H), 3.46 (d, 1H), 3.29 (s, 3H), 3.21 (s, 3H), 3.16 (d, 1H), 3.04 (s, 3H), 2.90 – 2.78 (m, 2H), 2.31 – 2.13 (m, 2H), 2.12 – 1.96 (m, 2H), 1.89 (t, 1H), 1.82 – 1.68 (m, 1H). LCMS: m/z (ESI) [M+H]
+ 562.3, tR = 1.63 minutes (Method C) Step 6: 5-(8-Cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide To a flask charged with ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methanol (126.2 mg, 792.7 μmol) was added a solution of 5-(8-cyano-2'-(methylsulfonyl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl- 5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (148.4 mg, 264.2 μmol) in DMF (5 mL) under nitrogen, at room temperature. The resulting solution was cooled to 0 °C, followed by the dropwise addition of lithium bis(trimethylsilyl)amide (87.8 mg, 350 μL, 1.5 M, 525 μmol). After stirring the mixture for 3 hours, the mixture was quenched by the slow addition of saturated aqueous ammonium chloride solution (10 mL). The mixture was treated with EtOAc (10 mL) and the layers were separated. The aqueous phase was extracted with EtOAc (3 x 5 mL). The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated. The crude material was subjected to purification via flash chromatography (4 g silica, 60:10:10:10, EtOAc/MeOH/EtOAc/H2O) to afford the partially pure product. The product was subjected to purification via flash chromatography (4 g silica, 0-30% (2.5%
NH
4OH and 20% MeOH in DCM)/DCM) to afford 5-(8-cyano-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H- pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (42.9 mg). 1H NMR (400 MHz, CDCl3) δ 7.58 (d, 1H), 7.31 (d, 1H), 7.29 – 7.23 (m, 1H), 6.56 (s, 1H), 5.33 – 5.14 (m, 1H), 4.91 (s, 2H), 4.66 – 4.53 (m, 2H), 4.52 – 4.36 (m, 2H), 4.06 (d, 1H), 4.01 – 3.89 (m, 2H), 3.78 – 3.64 (m, 1H), 3.37 – 3.28 (m, 4H), 3.28 – 3.15 (m, 2H), 3.15 – 3.11 (m, 1H), 3.10 – 3.03 (m, 3H), 3.01 (d, 1H), 2.99 – 2.90 (m, 1H), 2.89 – 2.78 (m, 2H), 2.32 – 2.02 (m, 6H), 2.01 – 1.68 (m, 6H). 1
9F NMR (376 MHz, CDCl
3) δ -173.06, -173.11. LCMS: m/z (ESI) [M+H]
+ 641.4, t
R = 1.33 minutes (Method C) Example 58: 5-((S)-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide (Compound 184b)
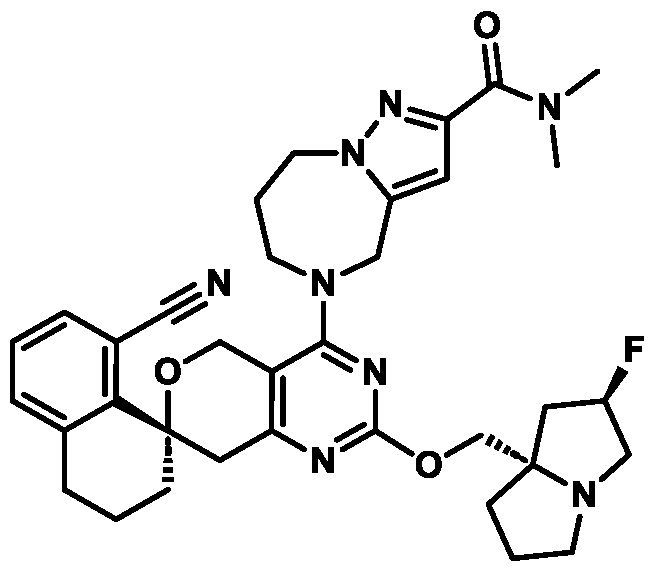
The diastereomers of 5-(8-Cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]- 4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (40 mg, 0.06 mmol) were separated by chiral SFC (Column: Chiralpak IB-N 21 x 250 mm, 5 μm; part number 88445; Mobile phase: Phase A for CO2, and Phase B for MeOH (0.25% DEA), Isocratic elution: B in A from 35% Flow rate: 50 mL/min, Detector: PDA, Column Temp: 40 °C, Back Pressure: 120 bar) to afford 5-((S)-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (Peak 1) (11.1 mg) and 5-((R)-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H-
pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (Peak 2) (9.3 mg). Peak 1: First eluting peak, t
R = 9.85 minutes 1H NMR (400 MHz, CDCl3) δ 7.59 (d, 1H), 7.32 (d, 1H), 7.29 – 7.24 (m, 1H), 6.56 (s, 1H), 5.36 – 5.15 (m, 1H), 4.92 (s, 2H), 4.68 – 4.54 (m, 2H), 4.54 – 4.37 (m, 2H), 4.07 (d, 1H), 4.01 – 3.87 (m, 2H), 3.79 – 3.65 (m, 1H), 3.39 – 3.12 (m, 7H), 3.11 – 3.04 (m, 3H), 3.04 – 2.91 (m, 2H), 2.88 – 2.79 (m, 2H), 2.33 – 2.03 (m, 6H), 2.01 – 1.69 (m, 6H). 1
9F NMR (376 MHz, CDCl3) δ -173.11. Example 59: 5-((R)-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide (Compound 184c)
5-((R)-8-cyano-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl- 5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (9.3 mg) was obtained from Example 58, as Peak 2: chiral SFC (Column: Chiralpak IB-N 21 x 250 mm, 5 μm; part number 88445; Mobile phase: Phase A for CO
2, and Phase B for MeOH (0.25% DEA), Isocratic elution: B in A from 35% Flow rate: 50 mL/min, Detector: PDA, Column Temp: 40 °C, Back Pressure: 120 bar). Peak 2: Second eluting peak, t
R = 12.78 minutes 1H NMR (400 MHz, CDCl
3) δ 7.60 (d, J = 7.3 Hz, 1H), 7.32 (d, J = 7.4 Hz, 1H), 7.29 – 7.24 (m, 2H), 6.56 (s, 1H), 5.25 (d, J = 53.7 Hz, 1H), 4.96 – 4.86 (m, 2H), 4.61 (s, 2H), 4.55 – 4.35 (m, 2H), 4.07 (d, J = 10.1 Hz, 1H), 4.00 – 3.90 (m, 2H), 3.79 – 3.67 (m, 1H), 3.40 – 3.12 (m, 7H), 3.12 – 3.03 (m, 3H), 3.03 – 2.91 (m, 2H), 2.90 – 2.76 (m, 2H), 2.31 – 2.03 (m,
6H), 2.03 – 1.68 (m, 6H). 1
9F NMR (376 MHz, CDCl3) δ -173.06. Example 60: 5-((1SR,4RS)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 162c)
Step 1: 1-(2-Bromo-4-chlorophenyl)ethan-1-ol 2-Bromo-4-chlorobenzaldehyde (5.00 g, 1 equiv, 22.8 mmol) was dissolved in THF (45.6 mL) and cooled to 0 °C. A 3 M solution of methyl magnesium bromide in diethyl ether (8.35 mL) was added dropwise. After 30 minutes, a saturated aqueous solution of ammonium chloride (50 mL) was added at 0 °C. The reaction mixture was extracted with EtOAc (3 x 20 mL). The organic phases were combine, dried with sodium sulfate, filtered and concentrated to give 1-(2-bromo-4-chlorophenyl)ethan-1-ol (5.30 g) LCMS: m/z (ESI) [M-H]- 235.0, t
R = 2.33 minutes (Method E) Step 2: 1-(1-(Allyloxy)ethyl)-2-bromo-4-chlorobenzene 1-(2-Bromo-4-chlorophenyl)ethan-1-ol (5.30 g, 1 equiv, 22.5 mmol) was dissolved in THF (100 mL) and cooled to -15 °C.60% wt sodium hydride (2.70 g) was then added to the mixture portion wise. The mixture was warmed to 0 °C for 5 minutes then stirred at room temperature for 30 minutes. After 30 minutes the reaction mixture was cooled back to 0 °C and allyl bromide (3.89 mL) was added dropwise. The mixture was then stirred at 25 °C until the starting material was completely consumed. After 18 hours, the mixture was cooled to 0 °C and then poured into a saturated aqueous solution of ammonium chloride (100 mL) containing ice (50 g). The mixture was then extracted with EtOAc (2 x 30 mL). The organic phases were combined, dried with sodium sulfate, filtered, and concentrated. The crude was purified via
flash chromatography (330 g SiO
2; 0-30% EtOAc in heptanes) to afford 1-(1-(allyloxy)ethyl)- 2-bromo-4-chlorobenzene (5.86 g) as a clear oil. 1H NMR (400 MHz, CDCl3) δ 7.53 (d, J = 2.0 Hz, 1H), 7.45 (d, J = 8.4 Hz, 1H), 7.32 (m, 1H), 5.97 – 5.83 (m, 1H), 5.26 (m, 1H), 5.18 (m, 1H), 4.81 (m, 1H), 3.96 – 3.78 (m, 2H), 1.39 (d, J = 6.4 Hz, 4H). Step 3: 6-Chloro-1-methyl-4-methyleneisochromane 1-(1-(Allyloxy)ethyl)-2-bromo-4-chlorobenzene (868 mg), triphenylphosphine (207 mg), PdOAc2 (70 mg), and cesium carbonate (2.05 g) were combined and put under a nitrogen atmosphere. Dry DMF (24 mL) was then added, and the mixture was heated to 80 °C. After 3 hours, the reaction was cooled to 25 °C and a saturated aqueous solution of NaCl (25 mL) was added. The mixture was then extracted with heptanes (3 x 10 mL). The organic phases were combined, dried with sodium sulfate, filtered, and concentrated. The crude was purified via flash chromatography (120 g SiO
2, 0-10% EtOAc in heptanes) to afford 6-chloro-1-methyl-4- methyleneisochromane (378 mg) which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 2.2 Hz, 1H), 7.15 (m, 1H), 6.98 (m, 1H), 5.51 (t, J = 1.1 Hz, 1H), 4.98 (m, 1H), 4.76 (m, 1H), 4.51 – 4.43 (m, 1H), 4.26 (m, 1H), 1.48 (d, J = 6.4 Hz, 4H). Step 4: 6-Chloro-1-methylisochroman-4-one 6-chloro-1-methyl-4-methyleneisochromane (1.02 g, 1 equiv, 5.2425 mmol) was dissolved in THF (17 mL). Sodium periodate (2.69 g, 2.4 equiv, 12.582 mmol) and potassium osmate(VI)dihydrate (38.6 mg, 0.02 equiv, 104.8 μmol) were then added followed by water (8.5 mL). The mixture was then stirred at 70 °C until all the starting material was consumed. After 3 hours, the reaction was cooled to 25 °C, and a saturated aqueous sodium thiosulfate (20 mL) solution was added. The mixture was stirred for 10 minutes. The mixture was then extracted with EtOAc (3 x 20 mL). The organic phases were combined, washed with a saturated aqueous NaCl solution, dried over sodium sulfate, filtered and concentrated. The crude was purified via flash chromatography (80 g SiO2, 0-20% EtOAc in heptanes) to afford 6-chloro- 1-methylisochroman-4-one (378 mg) which was used without further purification. 1H NMR (400 MHz, CDCl
3) δ 8.00 (d, 1H), 7.55 (dd, 1H), 7.21 (d, 1H), 4.90 (q, 1H), 4.52 (d, 1H), 4.29 (dd, 1H), 1.65 (d, 3H).
Step 5: 6-Chloro-4-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-1-methylisochroman-4-ol (4-Chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (314 mg) was dissolved in THF (7.0 mL) and cooled to -78 °C. 1 M LDA solution in THF (5.85 mL) was added dropwise keeping the mixture below -70 °C. After complete addition of the LDA solution, the reaction was stirred at -78 °C for 1 hour. 6-Chloro-1-methylisochroman-4-one (500 mg) in THF (5.0 mL) was added dropwise to the reaction mixture and the mixture was stirred at -78 °C until complete conversion of the starting material was observed. After 30 minutes the reaction mixture was poured in a saturated aqueous solution of ammonium chloride (30 mL) and the reaction mixture was extracted with EtOAc (3 x 10 mL). The organic phases were combined, dried over sodium sulfate, and then concentrated. The crude material was then purified via column chromatography (25 g SiO2, 0-20% EtOAc in heptanes) to give 6-chloro- 4-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-1-methylisochroman- 4-ol (456 mg). 1H NMR (400 MHz, CDCl3) δ 7.71 (m, 1H), 7.28 (s, 1H), 7.00 (m, 1H), 5.48 (m, 1H), 5.00 (m, 1H), 4.80 – 4.65 (m, 2H), 3.65 (m, 1H), 3.56 – 3.33 (m, 3H), 3.12 – 2.96 (m, 1H), 2.58 (m, 3H), 1.56 (m, 3H). Step 6: (1SR,4RS)-4',6-dichloro-1-methyl-2'-(methylthio)-5',8'-dihydro-6'H- spiro[isochromane-4,7'-quinazoline] and (1SR,4SR)-4',6-dichloro-1-methyl-2'- (methylthio)-5',8'-dihydro-6'H-spiro[isochromane-4,7'-quinazoline] 6-Chloro-4-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-1- methylisochroman-4-ol (456 mg) and polymer supported triphenylphosphine (2.135 g) were combined and put under a nitrogen atmosphere. THF (18 mL) and DCM (18 mL) were added followed by dropwise addition of DIAD (664.1 μL) to the reaction mixture. After 2 hours the reaction was diluted with DCM (20 mL), filtered through celite, and concentrated. The crude was then purified via column chromatography (120 g SiO
2, 0-10% EtOAc in heptanes) to afford two sets of diastereomers isolated as (1RS,4SR)-4',6-dichloro-1-methyl-2'-(methylthio)-5',8'- dihydro-6'H-spiro[isochromane-4,7'-quinazoline] (Peak 1) (192 mg) and (1SR,4RS)-4',6- dichloro-1-methyl-2'-(methylthio)-5',8'-dihydro-6'H-spiro[isochromane-4,7'-quinazoline] (Peak 2) (99.5 mg). Peak 1: LCMS: m/z (ESI) [M+H]
+ 383.1, tR = 3.80 minutes (Method E) Peak 2: LCMS: m/z (ESI) [M+H]
+ 382.6, tR = 3.61 minutes (Method E)
Step 7: 5-((1SR,4RS)-6-chloro-1-methyl-2'-(methylthio)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1SR,4RS)-4',6-dichloro-1-methyl-2'-(methylthio)-5',8'-dihydro-6'H- spiro[isochromane-4,7'-quinazoline] (99 mg, Step 6, Peak 2) and N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide, 2HCl (319.2 mg, 5.0 equiv, 1.304 mmol) were dissolved in acetonitrile (4.0 mL). DIPEA (539.5 mg, 727 μL, 16.0 equiv, 4.174 mmol) was then added and the reaction was heated to 100 °C. After heating for 22 hours the reaction was cooled to 25 °C and water (4.0 mL) was then added. The mixture was then extracted with EtOAc (2 x 10 mL) followed by DCM (10 mL). The organic phases were combined, dried over sodium sulfate, filtered, and then concentrated. The crude material was purified via column chromatography (SiO2, 0-15% MeOH in DCM) to give 5-((1SR,4RS)-6- chloro-1-methyl-2'-(methylthio)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (167 mg, 0.30 mmol, contains residual DCM). 1H NMR (400 MHz, CDCl
3) δ 7.30 (dd, 1H), 7.26 (m,1H), 7.11 (d, 1H), 6.52 (s, 1H), 4.87 (q, 1H), 4.82 – 4.55 (m, 4H), 4.52-4.38 (m, 2H), 4.08 (d, 1H), 3.93 – 3.75 (m, 2H), 3.71 (d, 1H), 3.30 (s, 3H), 3.08 (s, 3H), 3.01 (d, 1H), 2.84 (d, 1H), 2.52 (s, 3H), 228-2.03 (m, 2H), 1.61 (d, 3H). Step 8: tert-butyl ((1SR,4RS)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-1-methyl-2'-(methylthio)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate tert-Butyl carbamate (69.6 mg), 5-((1SR,4RS)-6-chloro-1-methyl-2'-(methylthio)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (165 mg), cesium carbonate (194 mg) and BrettPhos-G4 (27.8 mg, 0.1 equiv, 29.7 μmol) were dissolved in 1,4-dioxane. The reaction vessel was sealed and then the solution was sparged with nitrogen for 5 minutes. The reaction was heated to 100 °C. After 7 hours the reaction was cooled to 25 °C and then diluted with EtOAc (10 mL). The mixture was then filtered through Celite, dried over sodium sulfate, and then concentrated. The crude material was purified via column chromatography (SiO2, 0- 10% MeOH in DCM) to give tert-butyl ((1SR,4RS)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-
4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-1-methyl-2'-(methylthio)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (122 mg) which was used without further purification. 1H NMR (400 MHz, CDCl
3) δ 7.31 (d, 1H), 7.26 (s, 1), 7.10 (d, 1H), 6.75 (s, 1H), 6.49 (s, 1H), 4.87 (m, 1H), 4.74 – 4.56 (m, 4H), 4.48 – 4.37 (m, 2H), 4.07 (d, 1H), 3.92 (m, 1H), 3.85 – 3.61 (m, 2H), 3.34 (s, 3H), 3.09 (s, 3H), 3.03 (d, 1H), 2.80 (d, 1H), 2.51 (s, 3H), 2.16 (m, 4H), 1.60 (d, 3H), 1.54 (s, 9H). Step 9: tert-butyl ((1SR,4RS)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-1-methyl-2'-(methylsulfonyl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate tert-butyl ((1SR,4RS)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-1-methyl-2'-(methylthio)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-6-yl)carbamate (120 mg, 0.19 mmol) and Oxone (567 mg, 0.42 mmol) were dissolved in a 1:1 mixture of methanol (2 mL) and water (2 mL) and was stirred at 25 °C for 4 hours. After 4 hours the reaction was diluted with water (10 mL) and EtOAc (10 mL). The organic layer was separated, and the aqueous layer was washed with EtOAc (3 x 20 mL). The organic layers were combined, dried over sodium sulfate, filtered and then concentrated to give tert-butyl ((1SR,4RS)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-1-methyl-2'-(methylsulfonyl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (102 mg) which was used without further purification. LCMS: m/z (ESI) [M+H]
+ 668.5, tR = 2.55 minutes (Method E) Step 10: tert-butyl ((1SR,4RS)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-1-methyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidin]-6-yl)carbamate tert-butyl ((1SR,4RS)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-1-methyl-2'-(methylsulfonyl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-6-yl)carbamate (100.0 mg, 0.15 mmol) and ((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (25.0 mg, 0.16 mmol) were dissolved in THF (1 mL) under a nitrogen atmosphere.1 M t-BuONa solution in THF (300 μL) was then
added dropwise and the reaction was stirred for 20 minutes. After 20 minutes the reaction was concentrated in vacuo and the crude material was purified via column chromatography (24 g neutral Al2O3, 0-50% EtOAc in DCM) to give tert-butyl ((1SR,4RS)-4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)- 2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (56 mg). LCMS: m/z (ESI) [M+H]
+ 747.5, t
R = 1.87 minutes (Method E) Step 11: 5-((1SR,4RS)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-1-methyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide tert-butyl ((1SR,4RS)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-1-methyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6- yl)carbamate (56.0 mg, 0.08 mmol) was dissolved in CDCl3 (1.0 mL). A 2 M HCl solution in diethyl ether (562 uL, 1.12 mmol) was then added and the reaction mixture stirred at 25 °C for 2 hours. After 2 hours, more 2 M HCl in diethyl ether (200 uL) was added and the reaction mixture was stirred for an additional 2 hours. The reaction was then dried under a stream of nitrogen to afford 5-((1SR,4RS)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-1-methyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]- 4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (60 mg). LCMS: m/z (ESI) [M+H]
+ 647.3, t
R = 1.21 minutes (Method E) 1H NMR (400 MHz, MeOD) δ 6.97 (d, 1H), 6.72 (m, 1H), 6.65 – 6.56 (m, 2H), 5.45 – 5.24 (m, 1H), 4.87 – 4.60 (m, 5H), 4.50 (m, 2H), 4.22 – 4.09 (m, 2H), 4.08 – 3.94 (m, 2H), 3.86 (m, 1H), 3.72 (m, 1H), 3.47 – 3.33 (m, 3H), 3.31 (s, 3H), 3.07 (s, 5H), 2.94 (d, 1H), 2.75 (d, 1H), 2.42 – 1.84 (m, 7H), 1.52 (d, 3H). 1
9F NMR (376 MHz, MeOD) δ -174.29 Example 61: 5-((1R*,4S*)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-5',8'-dihydrospiro[isochromane-4,7'-
pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 162a)

The diastereomers of 5-((1SR,4RS)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (60 mg) were purified using preparative SFC chromatography (Column: Chiralpak IB N-5 (21 x 250 mm, 5 um, P/N 88445) Mobile Phase: Methanol with 0.25% TEA with isocratic 45% cosolvent at 50 mL/min over 20 min; backpressure = 120 bar) to give 5-((1R*,4S*)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-1-methyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Peak 1) (12 mg) and 5-((1S*,4R*)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-1-methyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Peak 2) (7.0 mg). Peak 1: Analytical SFC: m/z (ESI) [M+H]
+ 647.5, t
R = 1.85 minutes (Chiralpak IB-U (3.0 x 100 mm, 1.6 um, P/N 81U83)) Mobile Phase: Methanol with 0.25% DEA with isocratic 40% cosolvent at 2 mL/min over 5 min) 1H NMR (400 MHz, MeOD) δ 6.97 (d, 1H), 6.72 (dd, 1H), 6.65 – 6.56 (m, 2H), 5.45 – 5.25 (m, 1H), 4.84 – 4.63 (m, 5H), 4.50 (t, 2H), 4.22 – 4.10 (m, 2H), 4.06 – 3.96 (m, 2H), 3.86 (ddd, 1H), 3.74 (s, 1H), 3.47 – 3.33 (m, 3H), 3.31 (s, 3H), 3.19 (q, 1H), 3.10 (s, 1H), 3.07 (s, 3H), 2.94 (d, 1H), 2.75 (d, 1H), 2.43 – 1.88 (m, 7H), 1.52 (d, 3H). Example 62: 5-((1S*,4R*)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-5',8'-dihydrospiro[isochromane-4,7'-
pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 162b)
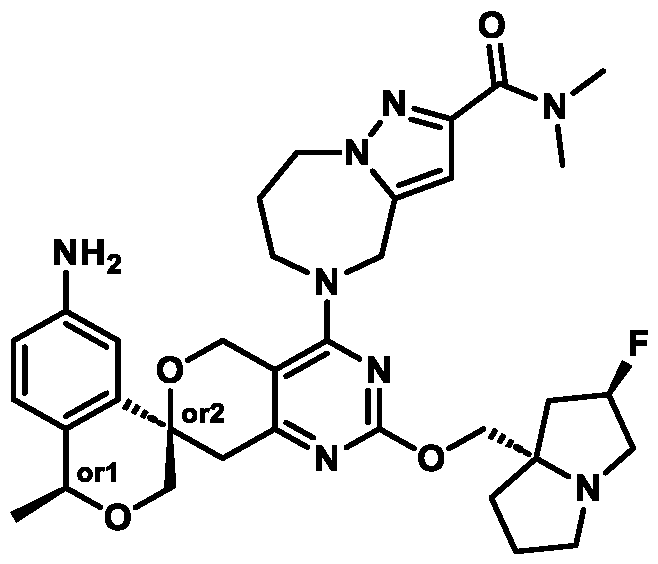
5-((1S*,4R*)-6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-1-methyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (7.0 mg) was obtained from Example 61, as Peak 2: Analytical SFC: m/z (ESI) [M+H]
+ 647.5, tR = 2.12 minutes (Chiralpak IB-U (3.0 x 100 mm, 1.6 um, P/N 81U83)) Mobile Phase: Methanol with 0.25% DEA with isocratic 40% cosolvent at 2 mL/min over 5 min) 1H NMR (400 MHz, MeOD) δ 6.85 (d, 1H), 6.61 (m, 1H), 6.55 – 6.45 (m, 2H), 5.18 (m, 1H), 4.76 – 4.52 (m, 5H), 4.39 (m, 2H), 4.08 – 3.71 (m, 5H), 3.61 (m, 1H), 3.28 – 3.05 (m, 6H), 3.00 – 2.86 (m, 5H), 2.82 (m, 1H), 2.62 (m, 1H), 2.25 – 1.68 (m, 7H), 1.41 (d, 3H). 1
9F NMR (376 MHz, MeOD) δ -172.74 – -174.61 (m). Example 63: (1SR,4RS)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-6-amine (Compound 182a)
Step 1: (1SR,4RS)-4',6-Dichloro-1-methyl-2'-(methylthio)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine]
6-Chloro-4-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-1- methylisochroman-4-ol (865 mg, 2.16 mmol) and polymer supported triphenylphosphine (4.02 g, 6.47 mmol, 1.6 mmol/g) were combined and put under a nitrogen atmosphere. THF (20 mL) and DCM (20 mL) were added followed by the dropwise addition of DIAD (664.1 μL, 6.47 mmol) to the reaction mixture. After 2 hours, the reaction was diluted with DCM (20 mL), filtered through celite, and concentrated. The crude was then purified via column chromatography (SiO
2, 0-10% EtOAc in heptanes) to give (1SR,4RS)-4',6-dichloro-1-methyl- 2'-(methylthio)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine] (163 mg) which was the second eluting peak.
1H NMR (400 MHz, CDCl
3) δ 7.33 (d, 1H), 7.29 (dd, 1H), 7.06 (d, 1H), 4.92 (q, 1H), 4.78 (s, 2H), 4.04 (d, 1H), 3.69 (d, 1H), 3.14 – 2.98 (m, 2H), 2.57 (s, 3H), 1.57 (d, 3H). Step 2: tert-Butyl ((1SR,4RS)-1-methyl-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)- 5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (1SR,4RS)-4',6-Dichloro-1-methyl-2'-(methylthio)-5',8'-dihydrospiro[isochromane- 4,7'-pyrano[4,3-d]pyrimidine] (160 mg, 0.42 mmol) was dissolved in acetonitrile (2.0 mL). DIPEA (863 mg, 1.16 mL, 16.0 equiv, 6.68 mmol) and 1,4-oxazepane, HCl (230 mg, 4 equiv, 1.67 mmol) were then added sequentially and the reaction was heated to 100 °C for 2 hours. The reaction mixture was concentrated to a residue which was purified via column chromatography (25 g SiO
2 (0-15% MeOH in DCM)) to give tert-butyl ((1SR,4RS)-1-methyl- 2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidin]-6-yl)carbamate (149 mg, 0.33 mmol). LCMS: m/z (ESI) [M+H]
+ 448.1, tR = 2.38 minutes (Method E) Step 3: tert-butyl ((1SR,4RS)-1-methyl-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)- 5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate tert-butyl ((1SR,4RS)-1-methyl-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (175 mg, 0.33 mmol) and Oxone (950 mg, 0.70 mmol) were dissolved in a 1:1 mixture of methanol (3 mL) and water (3 mL). The mixture was stirred at 25 °C for 2 hours. The reaction was diluted with water (15 mL) and EtOAc (15 mL). The organic layer was separated, and the aqueous layer was washed with EtOAc (3 x 30 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated to tert-butyl ((1SR,4RS)-1-methyl-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-
yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (191 mg, 0.34 mmol) which was used without further purification. LCMS: m/z (ESI) [M+H]
+ 561.5, tR = 2.70 minutes (Method E) Step 4: tert-butyl ((1SR,4RS)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane- 4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate tert-Butyl ((1SR,4RS)-1-methyl-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (185 mg, 0.33 mmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (55.2 mg, 0.35 mmol) were combined under nitrogen and dissolved in THF (2 mL). A 1 M t-BuONa solution in THF (660 μL, 0.66 mmol) was added dropwise. After 1 hour, the reaction mixture was concentrated in vacuo and purified via column chromatography (24 g Al2O3, 0-5% MeOH in DCM) to give the desired product contaminated with ((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methanol. This material was further purified via column chromatography (24 g Al2O3, 0-3% MeOH in DCM) to tert-butyl ((1SR,4RS)-2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (81 mg, 0.13 mmol). LCMS: m/z (ESI) [M+H]
+ 640.3, tR = 1.80 minutes (Method E) Step 5: (1SR,4RS)-2'-(((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-6-amine tert-Butyl ((1SR,4RS)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-6-yl)carbamate (80.0 mg, 1 equiv, 0.13 mmol) was dissolved in CDCl
3 (1.0 mL). A 2 M solution of HCl in diethyl ether (938 μL, 1.88 mmol) was added in one portion. The reaction was stirred at 25 °C for 40 minutes. The reaction mixture was then concentrated in vacuo to give (1SR,4RS)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-6-amine (77 mg, 0.13 mmol). LCMS: m/z (ESI) [M+H]
+ 540.3, tR = 1.08 minutes (Method E)
1H NMR (400 MHz, MeOD) δ 7.63 (m, 1H), 7.45 – 7.29 (m, 2H), 5.70 – 5.49 (m, 1H), 5.07 – 4.96 (m, 2H), 4.85 – 4.70 (m, 3H), 4.15 (m, 1H), 4.10 – 3.74 (m, 12H), 3.55 – 3.41 (m, 2H), 3.10 (s, 2H), 2.81 – 2.56 (m, 2H), 2.47 (m, 1H), 2.37 (m, 2H), 2.25 (d, J = 11.0 Hz, 1H), 2.06 (m, 2H), 1.58 (d, J = 6.6 Hz, 3H). 1
9F NMR (376 MHz, MeOD) δ -170.55 – -176.64 (m). Example 63a: (1R*,4S*)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-6-amine (Compound 182d)
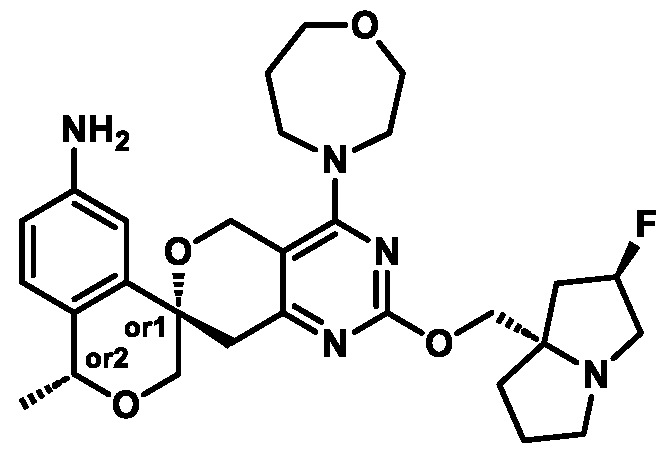
The diastereomers of (1SR,4RS)-2'-(((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-6-amine (70 mg) were purified using preparative SFC chromatography (Column: Chiralpak IB N-5 (21 x 250 mm, 5 um, P/N 88445) Mobile Phase: Methanol with 0.25% TEA with isocratic 40% cosolvent at 50 mL/min over 20 min; backpressure = 120 bar) to give (1R*,4S*)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-6-amine (Peak 1) (12 mg) and (1S*,4R*)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (Peak 2) (15 mg). Peak 1: Analytical SFC: m/z (ESI) [M+H]
+ 540.5, t
R = 1.29 minutes (Chiralpak IB-U (3.0 x 100 mm, 1.6 um, P/N 81U83)) Mobile Phase: Methanol with 0.25% DEA with isocratic 40% cosolvent at 2 mL/min over 5 min) 1
9F NMR (376 MHz, MeOD) δ -172.23 – -176.11 (m). Example 63b: (1S*,4R*)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-6-amine (Compound 182c)
(1S*,4R*)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-1- methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]- 6-amine was obtained from Example 63a as Peak 2: Analytical SFC: m/z (ESI) [M+H]
+ 540.5, t
R = 1.76 minutes (Chiralpak IB-U (3.0 x 100 mm, 1.6 um, P/N 81U83)) Mobile Phase: Methanol with 0.25% DEA with isocratic 40% cosolvent at 2 mL/min over 5 min) 1H NMR (400 MHz, MeOD) δ 6.94 (d, 1H), 6.74 – 6.65 (m, 2H), 5.42 – 5.19 (m, 1H), 4.95 – 4.48 (m, 4H), 4.16 (d, 1H), 4.08 – 3.96 (m, 2H), 3.92 – 3.63 (m, 9H), 3.29 – 3.10 (m, 3H), 3.01 (m, 1H), 2.91 (d, 1H), 2.73 (d, 1H), 2.34 – 1.78 (m, 9H), 1.50 (d, 3H).
19F NMR (376 MHz, MeOD) δ -165.00 – -183.87 (m). Example 64: (1SR,4SR)-2'-(((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-6-amine (Compound 182b)
Step 1: (1SR,4SR)-4',6-Dichloro-1-methyl-2'-(methylthio)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine] 6-Chloro-4-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-1- methylisochroman-4-ol (865 mg, 2.16 mmol) and polymer supported triphenylphosphine (4.02 g, 6.47 mmol, 1.6 mmol/g) were combined and put under a nitrogen atmosphere. THF (20 mL) and DCM (20 mL) were added followed by the dropwise addition of DIAD (664.1 μL, 6.47 mmol) to the reaction mixture. After 2 hours, the reaction was diluted with DCM (20 mL), filtered through celite, and concentrated. The crude was then purified via column
chromatography (SiO
2, 0-10% EtOAc in heptanes) to give (1SR,4SR)-4',6-dichloro-1-methyl- 2'-(methylthio)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine] (300 mg) which was the first eluting peak. 1H NMR (400 MHz, CDCl
3) δ 7.54 (d, J = 2.2 Hz, 1H), 7.28 (m, 1H), 7.04 (d, J = 8.4 Hz, 1H), 4.91 (d, J = 16.5 Hz, 1H), 4.83 (m, 1H), 4.68 (m, 1H), 4.01 (m, 1H), 3.72 (m, 1H), 3.38 (m, 1H), 3.04 – 2.84 (m, 1H), 2.56 (s, 3H), 1.52 (m, 3H), Step 2: (1SR,4SR)-6-Chloro-1-methyl-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidine] (1SR,4SR)-4',6-Dichloro-1-methyl-2'-(methylthio)-5',8'-dihydrospiro[isochromane- 4,7'-pyrano[4,3-d]pyrimidine] (150 mg, 0.39 mmol) was dissolved in acetonitrile (3.9 mL). DIPEA (408 mg, 550 μL, 8.07 equiv, 3.16 mmol) and 1,4-oxazepane, HCl (215 mg, 4 equiv, 1.57 mmol) were then added sequentially and the reaction was heated to 100 °C for 2 hours. The reaction mixture was concentrated then dissolved in DCM (10 mL). The mixture was washed with a saturated aqueous ammonium chloride solution (2 x 30 mL). The organic phase was dried over sodium sulfate, filtered and concentrated to a residue which was purified via column chromatography (25 g SiO
2 (0-15% MeOH in DCM)) to give (1SR,4SR)-6-chloro-1- methyl-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidine] (175 mg). LCMS: m/z (ESI) [M+H]
+ 449.1, t
R = 1.72 minutes (Method E) Step 3: tert-Butyl ((1SR,4SR)-1-methyl-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)- 5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate tert-Butyl carbamate (92 mg, 2.00 equiv, 782 μmol), (1SR,4SR)-6-chloro-1-methyl-2'- (methylthio)-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidine] (175.2 mg, 0.39 mmol), cesium carbonate (255 mg, 2.00 equiv, 783 μmol) and BrettPhos-G4 (37 mg, 0.10 equiv, 40 μmol) were dissolved in 1,4-dioxane (3.2 mL) under a nitrogen atmosphere. The mixture was heated to 100 °C and stirred for 14 hours. The reaction was cooled to 25 °C and diluted with EtOAc (10 mL). The reaction mixture was then filtered over celite and then concentrated in vacuo to give the crude product, which was then purified via column chromatography (4 g SiO
2, 0-20% (2.5% NH
4OH and 20% MeOH in DCM)/DCM) to give tert-butyl ((1SR,4SR)-1-methyl-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)- 5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (207 mg).
LCMS: m/z (ESI) [M+H]
+ 529.7, t
R = 1.63 minutes (Method E) Step 4: tert-Butyl ((1SR,4SR)-1-methyl-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)- 5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate tert-Butyl ((1SR,4SR)-1-methyl-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (211 mg, 0.40 mmol) and Oxone (1.203 g.0.88 mmol) were dissolved in a mixture of methanol (3.2 mL) and water (3.2 mL). After 5 hours, the reaction was diluted with EtOAc (10 mL) and the organic layer was separated. The aqueous layer was extracted with EtOAc (3 x 10 mL) and then extracted with DCM (3 x 10 mL). The organic layers were combined, dried over sodium sulfate, filtered and concentrated to give tert-butyl ((1SR,4SR)-1-methyl-2'-(methylsulfonyl)-4'-(1,4- oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (222 mg) which was used without further purification. LCMS: m/z (ESI) [M+H]
+ 561.3, t
R = 1.78 minutes (Method E) Step 5: tert-Butyl ((1SR,4SR)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane- 4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate tert-Butyl ((1SR,4SR)-1-methyl-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (222 mg, 0.40 mmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (60 mg, 0.38 mmol) were dissolved in THF (3.2 mL) under a nitrogen atmosphere. A 2 M solution of t- BuONa in THF (400 μL, 0.80 mmol) was then added to the mixture dropwise. The mixture was stirred at 25 °C for 2.5 hours. The reaction mixture was diluted with a saturated aqueous ammonium chloride solution (10 mL) and extracted with EtOAc (3 x 10 mL) and DCM (3 x 10 mL). The combined organic phases were dried over sodium sulfate, filtered and then concentrated to give the crude product contaminated with ((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methanol. The crude material was dissolved in DCM (10 mL) and the solution was washed with a saturated aqueous sodium bicarbonate solution (20 mL) followed by a saturated aqueous NaCl solution (20 mL). The organic layer was dried over sodium sulfate and then concentrated to give the crude product, which was then purified via column chromatography (SiO2, 0-20% (2.5% NH4OH and 20% MeOH in DCM)/DCM) to give tert- butyl ((1SR,4SR)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-1-
methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]- 6-yl)carbamate (140 mg). LCMS: m/z (ESI) [M+H]
+ 640.6, tR = 1.33 minutes (Method E) Step 6: (1SR,4SR)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-6-amine tert-butyl ((1SR,4SR)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-6-yl)carbamate (140 mg, 0.22 mmol) was dissolved in CDCl
3 (3 mL) and the reaction mixture was cooled to 0 °C. A 2 M solution of HCl (2.2 mL, 4.4 mmol) in diethyl ether was then added dropwise to the reaction mixture. The reaction was warmed to 25 °C and stirred for 3 hours. The mixture was concentrated to afford (1SR,4SR)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-4'-(1,4-oxazepan-4-yl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-amine (93.5 mg). 1H NMR (400 MHz, MeOD) δ 7.62 (dd, J = 9.8, 2.2 Hz, 1H), 7.49 – 7.27 (m, 2H), 5.70 – 5.51 (m, 1H), 5.05 (d, J = 14.8 Hz, 1H), 4.99 – 4.90 (m, 2H), 4.82 – 4.68 (m, 2H), 4.39 (dd, J = 11.1, 6.4 Hz, 1H), 4.06 – 3.83 (m, 10H), 3.78 (t, J = 10.6 Hz, 2H), 3.54 – 3.40 (m, 1H), 3.36 (d, J = 3.1 Hz, 1H), 3.07 (dd, J = 17.7, 3.9 Hz, 1H), 2.80 – 2.55 (m, 2H), 2.47 (q, J = 8.7 Hz, 1H), 2.35 (dq, J = 11.7, 6.9 Hz, 2H), 2.23 (s, 1H), 2.05 (qd, J = 11.6, 7.9 Hz, 2H), 1.56 (d, J = 6.4 Hz, 3H). 1
9F NMR (376 MHz, MeOD) δ -174.30 LCMS: m/z (ESI) [M+H]
+ 540.6, tR = 1.03 minutes (Method E) Example 65: 5-((1SR,4SR)-6-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 162d)

Step 1: 5-((1SR,4SR)-6-Chloro-1-methyl-2'-(methylthio)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide DIPEA (1.4 mL, 8.0 mmol) and N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (743 mg, 3.0 mmol) were added to a solution of (1SR,4SR)- 4',6-dichloro-1-methyl-2'-(methylthio)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidine] (192.5 mg, 0.50 mmol) in acetonitrile (24 mL). The reaction mixture was heated to 100 °C for 24 hours, then reaction was cooled to 25 °C and N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (300 mg) was added. The reaction mixture was heated to 100 °C for an additional 24 hours, then concentrated. The mixture was diluted with DCM (20 mL) and washed with a saturated aqueous ammonium chloride solution (2 x 30 mL). The organic layer was dried over sodium sulfate, filtered and concentrated to a residue which was purified via column chromatography (25 g SiO2, (0-15% MeOH in DCM)) to give 5-((1SR,4SR)-6-chloro-1-methyl-2'-(methylthio)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (300 mg). LCMS: m/z (ESI) [M+H]
+ 555.1, tR = 1.78 minutes (Method E) Step 2: tert-Butyl ((1SR,4SR)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-1-methyl-2'-(methylthio)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate tert-Butyl carbamate (127 mg, 1.08 mmol), 5-((1SR,4SR)-6-chloro-1-methyl-2'- (methylthio)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (300 mg, 0.54 mmol), cesium carbonate (353 mg, 1.08 mmol) and BrettPhos-G4 (51 mg, 0.05 mmol) were dissolved in 1,4-dioxane (3.6 mL) under nitrogen. The reaction mixture was then heated to 110 °C for 3 hours. After 3 hours the reaction mixture was diluted with EtOAc (10 mL), filtered
through celite and concentrated to give a residue which was purified via column chromatography (4 g SiO2, 0-5% MeOH in DCM) to give tert-butyl ((1SR,4SR)-4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-1-methyl-2'- (methylthio)-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (269 mg) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 2.2 Hz, 1H), 7.26 (m, 1H), 7.06 – 7.00 (m, 1H), 6.54 (s, 1H), 4.86 (m, 1H), 4.81 – 4.69 (m, 2H), 4.67 – 4.52 (m, 2H), 4.46 (m, 2H), 4.05 (d, J = 10.9 Hz, 1H), 3.95 – 3.77 (m, 2H), 3.72 (d, J = 10.9 Hz, 1H), 3.33 (s, 3H), 3.26 (d, J = 17.9 Hz, 1H), 3.08 (s, 3H), 2.93 (d, J = 17.9 Hz, 1H), 2.50 (s, 3H), 2.31 – 2.09 (m, 2H), 1.51 (d, J = 6.5 Hz, 3H). Step 3: tert-Butyl ((1SR,4SR)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-1-methyl-2'-(methylsulfonyl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate tert-Butyl ((1SR,4SR)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-1-methyl-2'-(methylthio)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-6-yl)carbamate (212 mg, 0.33 mmol) and Oxone (1.00 g, 0.73 mmol) were dissolved in a mixture of methanol (3.2 mL) and water (3.2 mL) and stirred for 18 hours. The reaction mixture was diluted with EtOAc (10 mL) and water (10 mL). The organic layer was separated, and the aqueous phase was extracted with EtOAc (3 x 10 mL) and DCM (3 x 10 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated to give tert-butyl ((1SR,4SR)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-1-methyl-2'-(methylsulfonyl)-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (219 mg) which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 7.46 (s, 1H), 7.23 (s, 1H), 7.04 (d, J = 8.4 Hz, 1H), 6.65 (s, 1H), 6.56 (s, 1H), 4.91 (m, 1H), 4.84 – 4.75 (m, 2H), 4.74 – 4.61 (m, 2H), 4.49 (m, 2H), 4.06 – 3.91 (m, 3H), 3.79 (d, J = 11.1 Hz, 1H), 3.33 (m, 4H), 3.26 (s, 3H), 3.12 (d, J = 19.0 Hz, 1H), 3.08 (s, 3H), 2.20 (d, J = 5.5 Hz, 2H), 1.51 (d, J = 6.5 Hz, 3H), 1.45 (s, 9H). Step 4: tert-Butyl ((1SR,4SR)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-
7a(5H)-yl)methoxy)-1-methyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidin]-6-yl)carbamate tert-Butyl ((1SR,4SR)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-1-methyl-2'-(methylsulfonyl)-5',8'-dihydrospiro[isochromane-4,7'- pyrano[4,3-d]pyrimidin]-6-yl)carbamate (216 mg, 0.32 mmol) and ((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (51.5 mg, 0.32 mmol) were dissolved in THF (3.2 mL) under a nitrogen atmosphere. A 2 M solution of t-BuONa in THF (330 μL, 0.66 mmol) was then added to the reaction mixture dropwise. After 20 minutes, the reaction mixture was diluted with a saturated aqueous ammonium chloride solution (10 mL) followed by EtOAc (10 mL). The organic layer was separated, and the aqueous layer was washed with EtOAc (3 x 10 mL) and DCM (3 x 10 mL). The combined organic layers were dried over sodium sulfate, filtered and then concentrated to a residue which was purified via column chromatography (4 g SiO2; 0-20% (2.5% NH4OH and 20% MeOH in DCM) in DCM) to give tert-butyl ((1SR,4SR)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)- yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-5',8'- dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (133 mg). 1H NMR (400 MHz, CDCl
3) δ 7.39 (s, 1H), 7.29 (d, J = 8.6 Hz, 1H), 7.01 (d, J = 8.5 Hz, 1H), 6.58 (s, 1H), 6.51 (s, 1H), 5.26 (m, 1H), 4.88 (m, 1H), 4.78 – 4.67 (m, 2H), 4.58 (m, 2H), 4.45 (m, 2H), 4.08 (d, J = 9.9 Hz, 1H), 4.03 – 3.95 (m, 2H), 3.82 (s, 2H), 3.73 (d, J = 10.9 Hz, 1H), 3.33 (s, 3H), 3.26 – 3.11 (m, 4H), 3.08 (s, 3H), 3.00 – 2.91 (m, 2H), 2.30 – 2.09 (m, 6H), 1.95 – 1.79 (m, 3H), 1.62 (d, J = 30.6 Hz, 5H), 1.48 (m, 12H). Step 5: 5-((1SR,4SR)-6-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-1-methyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide tert-Butyl ((1SR,4SR)-4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-1-methyl-5',8'-dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-6- yl)carbamate (133 mg, 0.18 mmol) was dissolved in CDCl3 (2.4 mL). A 2 M solution of HCl in diethyl ether (1.78 mL, 3.6 mmol) was added and the reaction mixture stirred at 25 °C for 3 hours. After 3 hours, the mixture was concentrated to afford 5-((1SR,4SR)-6-amino-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-1-methyl-5',8'-
dihydrospiro[isochromane-4,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (117 mg). 1H NMR (400 MHz, MeOD) δ 7.51 (dd, J = 7.1, 2.2 Hz, 1H), 7.35 – 7.23 (m, 2H), 6.62 (d, J = 3.3 Hz, 1H), 5.63 – 5.40 (m, 1H), 5.22 – 4.92 (m, 3H), 4.83 (m, 2H), 4.74 (m, 1H), 4.63 (d, J = 11.7 Hz, 1H), 4.55 – 4.37 (m, 2H), 4.27 (m, 1H), 4.04 (m, 2H), 3.83 (m, 2H), 3.73 (m, 2H), 3.44 – 3.30 (m, 1H), 3.32 – 3.24 (m, 3H), 2.99 (s, 4H), 2.69 – 2.57 (m, 1H), 2.53 (d, J = 2.9 Hz, 1H), 2.41 (m, 1H), 2.31 – 2.01 (m, 5H), 1.46 (d, J = 6.5 Hz, 3H) 1
9F NMR (376 MHz, MeOD) δ -174.41 Example 66: (1R,4S*)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Compound 179d)
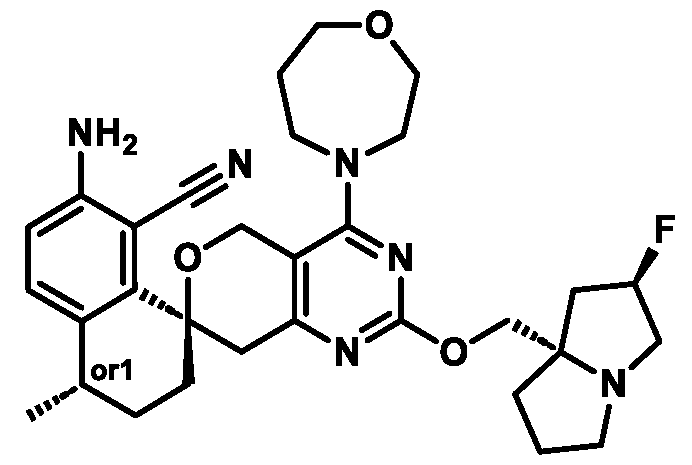
The diastereomers of 7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine]-8-carbonitrile (79 mg, 0.14 mmol) were separated via SFC (Column: Lux-Cellulose-1 (30 x 250 mm, 5 µm) Mobile Phase: Methanol with 0.1% DEA with isocratic 25% cosolvent at 150 mL/min over 30 min; backpressure = 100 bar) to give (1R,4S*)- 7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-4'- (1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]- 8-carbonitrile (Peak 1) (23.8 mg), (1S,4S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Peak 2) (22.8 mg), (1S,4R)- 7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-4'- (1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]- 8-carbonitrile (Peak 3) (7.7 mg), and (1R,4R*)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Peak 4) (6.14 mg).
Peak 1: Analytical HPLC: t
R = 6.87 minutes (Lux-Cellulose-1 (4.6 x 150mm) 50% MeCN in water (+ 10 mM ammonium bicarbonate) for 15 min at 1 mL/min over 1H NMR (400 MHz, DMSO-d6) δ 7.10 (d, J = 8.6 Hz, 1H), 6.75 (d, J = 8.6 Hz, 1H), 5.76 (s, 2H), 5.35 – 5.12 (m, 1H), 4.92 (d, J = 14.5 Hz, 1H), 4.65 (d, J = 14.4 Hz, 1H), 3.98 – 3.81 (m, 2H), 3.81 – 3.50 (m, 9H), 3.03 (dd, J = 25.6, 7.0 Hz, 4H), 2.82 (dd, J = 16.9, 8.4 Hz, 3H), 2.15 – 1.51 (m, 13H), 1.16 (d, J = 7.1 Hz, 3H). 1
9F NMR (376 MHz, DMSO) δ -168.02 – -175.60 (m). Example 67: (1S,4R)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Compound 179f)
(1S,4R)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine]-8-carbonitrile was obtained from Example 66, as Peak 3: Analytical HPLC: tR = 9.99 minutes (Lux-Cellulose-1 (4.6 x 150mm) 50% MeCN in water (+ 10 mM ammonium bicarbonate) for 15 min at 1 mL/min). 1H NMR (400 MHz, DMSO-d
6) δ 7.26 (d, J = 8.8 Hz, 1H), 6.77 (d, J = 8.7 Hz, 1H), 5.76 (s, 2H), 5.25 (d, J = 54.4 Hz, 1H), 4.86 (d, J = 14.4 Hz, 1H), 4.65 (d, J = 14.4 Hz, 1H), 3.94 (d, J = 10.3 Hz, 1H), 3.83 (d, J = 10.3 Hz, 1H), 3.79 – 3.50 (m, 7H), 3.17 – 2.94 (m, 4H), 2.91 – 2.76 (m, 2H), 2.68 (q, J = 9.3 Hz, 1H), 2.20 – 2.05 (m, 2H), 2.04 – 1.60 (m, 9H), 1.36 (q, J = 12.8 Hz, 1H), 1.20 (d, J = 6.7 Hz, 3H). 1
9F NMR (376 MHz, DMSO) δ -172.07. Example 68: (1R,4R*)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Compound 179e)
(1R,4R*)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4-methyl-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine]-8-carbonitrile (6.14 mg) was obtained from Example 66, as Peak 4: Analytical HPLC: t
R = 11.27 minutes (Lux-Cellulose-1 (4.6 x 150mm) 50% MeCN in water (+ 10 mM ammonium bicarbonate) for 15 min at 1 mL/min. 1H NMR (400 MHz, DMSO-d
6) δ 7.24 (d, J = 8.7 Hz, 1H), 6.75 (d, J = 8.8 Hz, 1H), 5.74 (s, 2H), 5.23 (d, J = 54.4 Hz, 1H), 4.85 (d, J = 14.4 Hz, 1H), 4.63 (d, J = 14.5 Hz, 1H), 3.98 – 3.46 (m, 9H), 3.16 – 2.73 (m, 6H), 2.67 (d, J = 15.2 Hz, 1H), 2.17 – 1.58 (m, 12H), 1.35 (q, J = 12.8 Hz, 1H), 1.18 (d, J = 6.7 Hz, 3H). 1
9F NMR (376 MHz, DMSO) δ -172.06 Example 69: 2-(1-((S)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)piperidin-3-yl)acetonitrile (Compound 186c)
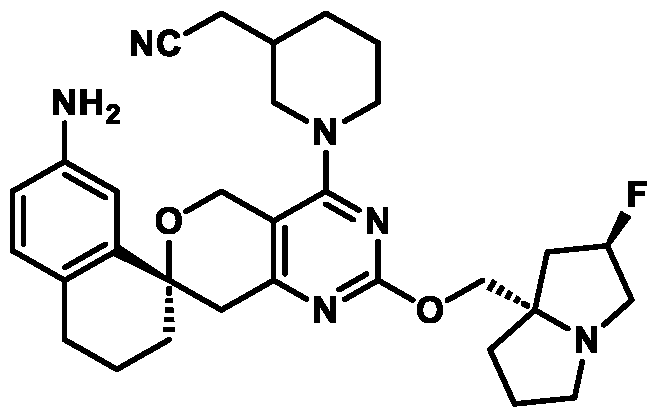
Step 1: tert-Butyl ((1S)-4'-(3-(cyanomethyl)piperidin-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate To a 2 dram vial containing a stir bar, tert-butyl ((S)-4'-((1H-benzo[d][1,2,3]triazol-1- yl)oxy)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (Intermediate 5, 80 mg, 1 equiv, 0.12 mmol) was dissolved in DMF (0.5 mL). Then, DIPEA (79 mg, 0.11 25 mL, 5 equiv, 0.61 mmol) and 3-piperidinylacetonitrile hydrochloride (39 mg, 2 equiv, 0.24
mmol) were added subsequently to the reaction mixture. The reaction mixture was heated to 90 °C for 1.5 hours. The reaction mixture was cooled to room temperature and purified directly using a C-18 Biotage column (12 g cartridge, liquid injection) reverse prep with 10 mM ammonium bicarbonate solution and acetonitrile to obtain tert-butyl ((1S)-4'-(3- (cyanomethyl)piperidin-1-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (55 mg, 85 μmol) as an off-white solid. LCMS: m/z (ESI) [M-H]
+ 636.5, tR = 1.74 min (Method B) Step 2: 2-(1-((S)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)piperidin-3-yl)acetonitrile To a 10 ml sealed vial with a septum were added tert-butyl ((1S)-4'-(3- (cyanomethyl)piperidin-1-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate (45 mg, 1 equiv, 70 μmol) and p-toluenesulfonic acid monohydrate (66 mg, 5 equiv, 0.35 mmol) in MeCN (0.5 mL) at room temperature. The reaction mixture was heated to 60 °C and stirred for 1 hour. Upon completion, the solvent was removed under reduced pressure. The crude was purified using C-18 Biotage column (12 g cartridge, liquid injection) reverse prep with 10 mM ammonium bicarbonate solution and acetonitrile to obtain 2-(1-((S)- 7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)piperidin-3- yl)acetonitrile (25 mg, 46 μmol) as a white solid. 1H NMR (400 MHz, DMSO-d
6) δ 6.76 (d1H), 6.58 – 6.52 (m, 1H), 6.47 – 6.40 (m, 1H), 5.26 (d, 1H), 4.78 (s, 2H), 4.66 (d, 1H), 4.43 – 4.33 (m, 1H), 4.02 – 3.93 (m, 1H), 3.90 – 3.77 (m, 2H), 3.68 – 3.53 (m, 1H), 3.12 – 2.72 (m, 8H), 2.67 – 2.53 (m, 4H), 2.15 – 1.44 (m, 14H), 1.37 – 1.23 (m, 1H). LCMS: m/z (ESI) [M+H]
+ 547.4 t
R = 1.54 min (Method D) Example 70: 2-((R*)-1-((S)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)piperidin-3-yl)acetonitrile (Compound 186b)
The diastereomers of 2-(1-((S)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)piperidin-3-yl)acetonitrile (17 mg) were dissolved in methanol (0.75 mL) and purified using Chiralpak IB-N (21 x 250 mm, 5 µm, P/N 88445), 40% isocratic methanol with 0.25% diethylamine to give 2-((R*)-1-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)piperidin-3-yl)acetonitrile (3.5 mg, 6.4 mmol). LCMS: m/z (ESI) [M+H]
+ 547.3, t
R = 1.56 min (Method E) 1H NMR (400 MHz, MeOD) δ 6.89 (d, 1H), 6.73 (d, 1H), 6.67 – 6.60 (m, 1H), 5.29 (d, 1H), 4.69 (d, 1H), 4.47 (d, 1H), 4.23 – 4.06 (m, 3H), 3.75 (d, 1H), 3.37 – 3.12 (m, 4H), 3.09 – 2.97 (m, 3H), 2.93 – 2.83 (m, 2H), 2.80 – 2.62 (m, 2H), 2.49 (d, 2H), 2.36 – 1.83 (m, 10H), 1.82 – 1.55 (m, 3H), 1.50 – 1.37 (m, 1H). Impurities: Grease 1H NMR (400 MHz, MeOD) δ 1.29 (m), 0.90 (m), 0.10 (s). Example 71: 2-((S*)-1-((S)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)piperidin-3-yl)acetonitrile (Compound 186a)
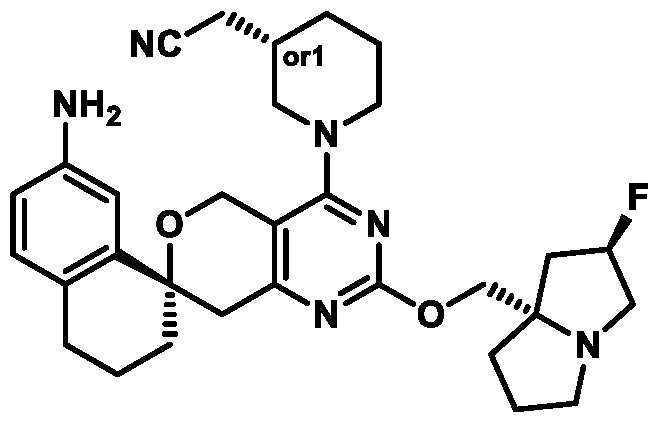
The diastereomers of 2-(1-((S)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)piperidin-3-yl)acetonitrile (17 mg) were dissolved in methanol (0.75 mL) and purified using Chiralpak IB-N (21 x 250 mm, 5 µm, P/N 88445), 40% isocratic methanol25 with 0.25% diethylamine to give 2-((S*)-1-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-
pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)piperidin-3-yl)acetonitrile (5.1 mg, 9.0 mmol). 1H NMR (400 MHz, MeOD) δ 6.88 (d, 1H), 6.75 (d, 1H), 6.66 – 6.59 (m, 1H), 5.28 (d, 1H), 4.77 (d, 1H), 4.47 (d, 1H), 4.22 – 4.07 (m, 3H), 3.71 (d, 1H), 3.23 – 2.94 (m, 6H), 2.92 – 2.81 (m, 2H), 2.76 – 2.64 (m, 2H), 2.57 – 2.41 (m, 2H), 2.35 – 1.84 (m, 11H), 1.83 – 1.72 (m, 2H), 1.65 – 1.54 (m, 1H), 1.49 – 1.38 (m, 1H). Impurities: Grease 1H NMR (400 MHz, MeOD) δ 1.36 – 1.22 (m), 0.94 – 0.82 (m), 0.10 (s). LCMS: m/z (ESI) [M+H]
+ 547.3, tR = 1.53 min (Method E) Example 72: 2-(4-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-1,4-oxazepan-2-yl)acetonitrile (Compound 174a)

Step 1: (4-Benzyl-1,4-oxazepan-2-yl)methyl methanesulfonate TEA (1.37 g, 1.89 mL, 3 equiv, 13.6 mmol) and methanesulfonyl chloride (828 mg, 560 μL, 1.6 equiv, 7.23 mmol) were added sequentially to a solution of (4-benzyl-1,4- oxazepan-2-yl)methanol (1.00 g, 1 equiv, 4.52 mmol) in THF (22.6 mL) at 0 °C. The reaction mixture was stirred for 15 min at 0 °C. The product mixture was diluted with water (20 mL) and EtOAc (40 mL), and the layers that formed were separated. The aqueous layer was extracted with EtOAc (2 x 40 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated under educed pressure to give (4-benzyl-1,4-oxazepan-2- yl)methyl methanesulfonate (1.35 g, 4.51 mmol) which was used without further purification. LCMS: m/z (ESI) [M+H]
+ 300.2, tR = 0.67 min (Method C) Step 2: 2-(4-Benzyl-1,4-oxazepan-2-yl)acetonitrile Cesium fluoride (3.42 g, 5 equiv, 22.5 mmol) and (trimethylsilylnitrile) (2.06 g, 2.7 mL, 4.6 equiv, 20.7 mmol) were added to a solution of (4-benzyl-1,4-oxazepan-2-yl)methyl
methanesulfonate (1.35 g, 1 equiv, 4.51 mmol) in DMF (22.5 mL). The reaction mixture was stirred for 4 hours at 80 °C. The product mixture was cooled to room temperature, and saturated aqueous sodium bicarbonate solution (15 mL) was added. The aqueous layer was extracted with EtOAc (3 x 40 mL). The combined organic layers were washed with brine, dried over sodium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO2, 0-20% 2.5% NH4OH/20% MeOH/77.5% DCM in DCM) to give 2-(4- benzyl-1,4-oxazepan-2-yl)acetonitrile (610 mg, 2.65 mmol). 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.14 (m, 5H), 4.07 – 3.70 (m, 3H), 3.66 – 3.54 (m, 2H), 2.88 – 2.27 (m, 6H), 1.89 – 1.74 (m, 2H). Step 3: 2-(1,4-Oxazepan-2-yl)acetonitrile Chloroformic acid 1-chloroethyl ester (484 mg, 366 μL, 1.3 equiv, 3.39 mmol) was added to a solution of 2-(4-benzyl-1,4-oxazepan-2-yl)acetonitrile (600 mg, 1 equiv, 2.61 mmol) in DCM (12 mL). The reaction mixture was stirred for 2 hours at room temperature and was concentrated. The resulting residue was diluted with methanol (12 mL) and stirred for 1.5 hours at 60 °C. The product mixture was cooled to room temperature and was concentrated under reduced pressure to give a residue which was purified by column chromatography (SiO
2, 0-60% 2.5% NH
4OH/20% MeOH/77.5% DCM in DCM) to yield 2-(1,4-oxazepan-2- yl)acetonitrile (349 mg, 2.49 mmol). 1H NMR (400 MHz, MeOD) δ 5.45 (s, 1H), 4.19 – 3.98 (m, 2H), 3.78 – 3.70 (m, 1H), 3.53 – 3.24 (m, 3H), 3.15 – 3.04 (m, 1H), 2.84 – 2.65 (m, 2H), 2.16 – 2.01 (m, 2H). Step 4: tert-Butyl ((S)-4'-((SR)-2-(Cyanomethyl)-1,4-oxazepan-4-yl)-2'-(((2R,7aS)- 2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate DIPEA (95.6 mg, 129 μL, 8 equiv, 740 μmol) was added to a solution of tert-butyl ((S)- 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-oxo-3,3',4,4',5',8'- hexahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (Intermediate 4, 50.0 mg, 1 equiv, 92.5 μmol), 2-(1,4-oxazepan-2-yl)acetonitrile (38.9 mg, 3 equiv, 277 μmol), and (benzotriazol-1-yloxy)tripyrrolidinophosphoniumhexafluorophosphate (57.8 mg, 1.2 equiv, 111 μmol) in DMF (1.85 mL). The reaction mixture was stirred for 27 hours at 50-80 °C. The product solution was cooled to room temperature and was diluted with a 1:1 mixture of water/brine (2 mL) and EtOAc (2 mL). The layers that formed were separated.
The aqueous layer was extracted with EtOAc (3 x 2 mL). The combined organic layers were washed with water (2 x 7 mL). The washed organic layers were dried over magnesium sulfate, filtered, and concentrated. The residue obtained was purified by reverse phase column chromatography (C18, 25-45% MeCN in water (0.1% FA additive)) to provide tert-butyl ((S)- 4'-((SR)-2-(cyanomethyl)-1,4-oxazepan-4-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]- 7-yl)carbamate (27.0 mg, 41.0 μmol). LCMS: m/z (ESI) [M+H]
+ 663.4, tR = 1.42 min (Method C) Step 5: 2-(4-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)-1,4-oxazepan-2-yl)acetonitrile p-Toluenesulfonic acid monohydrate (34.0 mg, 5 equiv, 184 μmol) was added to a solution of tert-butyl ((S)-4'-((SR)-2-(cyanomethyl)-1,4-oxazepan-4-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (24.0 mg, 1 equiv, 36.0 μmol) in MeCN (0.33 mL). The reaction mixture was stirred for 8 hours at 35-45 °C. The product mixture was directly concentrated under educed pressure, and diluted with EtOAc (2 mL), water (1 mL), and saturated aqueous sodium bicarbonate solution (1 mL). The layers that formed were separated, and the aqueous layer was extracted with EtOAc (3 x 2 mL). The combined organic layers were dried over magnesium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO2, 0-60% 2.5% NH4OH/20% MeOH/77.5% DCM in DCM) to afford 2-(4-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-1,4-oxazepan-2-yl)acetonitrile (16.2 mg, 28.8 μmol). LCMS: m/z (ESI) [M+H]
+ 563.3, tR = 1.08 and 1.11 min (Method C) 1H NMR (400 MHz, MeOD) δ 7.04 – 6.54 (m, 3H), 5.29 (d, 1H), 4.75 – 4.56 (m, 1H), 4.55 – 4.25 (m, 2H), 4.17 – 4.02 (m, 6H), 3.89 – 3.72 (m, 1H), 3.64 – 3.42 (m, 2H), 3.28 – 3.16 (m, 4H), 3.09 – 2.96 (m, 2H), 2.92 – 2.82 (m, 1H), 2.81 – 2.59 (m, 4H), 2.38 – 2.07 (m, 4H), 1.99 – 1.81 (m, 7H), 1.77 – 1.67 (m, 1H).
Example 73: 2-(4-((S)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-1,4-oxazepan-6-yl)acetonitrile (Compound 173a)
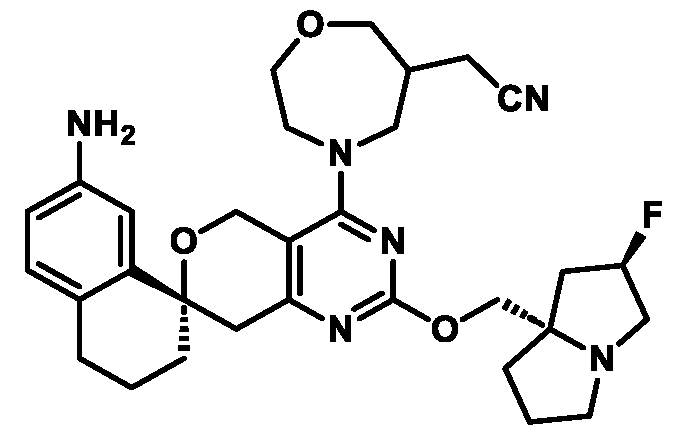
Step 1: tert-butyl 6-(cyanomethylene)-1,4-oxazepane-4-carboxylate Sodium hydride (453 mg, 60% wt, 1.22 equiv, 11.3 mmol) was added to a solution of diethyl cyanomethylphosphonate (2.06 g, 1.88 mL, 1.25 equiv, 11.6 mmol) in THF (67 mL) at 0 °C. The reaction mixture was stirred for 30 min at 0 °C. A solution of tert-butyl 6-oxo-1,4- oxazepane-4-carboxylate (2.00 g, 1 equiv, 9.29 mmol) in THF (13 mL) was added to the reaction mixture at 0 °C. The reaction mixture was stirred and warmed to room temperature over 1.5 hours. The product solution was diluted with water (100 mL). The aqueous layer was extracted with ethyl acetate (2 x 150 mL). The organic layers were combined and washed with brine (150 mL). The washed organic layers were dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The residue obtained was purified by column chromatography (SiO
2, 0-25% EtOAc in heptanes) to afford tert-butyl 6-(cyanomethylene)- 1,4-oxazepane-4-carboxylate (1.90 g, 8.00 mmol). LCMS: m/z (ESI) [M+H-Boc]
+ 139.1, tR = 1.62 and 1.64 min (Method C) Step 2: tert-Butyl 6-(cyanomethyl)-1,4-oxazepane-4-carboxylate Palladium on carbon (1.76 g, 10% wt, 0.21 equiv, 1.65 mmol) was added to a solution of tert-butyl 6-(cyanomethylene)-1,4-oxazepane-4-carboxylate (1.90 g, 1 equiv, 7.97 mmol) in methanol (93 mL) that had been sparged with nitrogen for 10 min at room temperature. The suspension was sparged again with H2 for 5 min, and the reaction mixture was stirred under a H2 atmosphere for 50 min. The product mixture was filtered and concentrated under reduced pressure. The residue obtained was purified by flash column chromatography (SiO
2, 0-80% ethyl acetate in heptanes) to afford tert-butyl 6-(cyanomethyl)-1,4-oxazepane-4-carboxylate (1.60 g, 6.70 mmol). 1H NMR (400 MHz, CDCl3) δ 4.08 – 3.23 (m, 9H), 2.48 – 2.29 (m, 2H), 1.50 – 1.43 (m, 9H).
Step 3: 2-(1,4-Oxazepan-6-yl)acetonitrile hydrochloride A solution of HCl (1.80 g, 12.0 mL, 4 M, 7.7 equiv, 48.0 mmol) in 1,4-dioxanes was added to a solution of tert-butyl 6-(cyanomethyl)-1,4-oxazepane-4-carboxylate (1.50 g, 1 equiv, 6.24 mmol) in acetone (45 mL) at room temperature. The reaction mixture was stirred for 2 hours at room temperature. The product mixture was directly concentrated to give 2-(1,4- oxazepan-6-yl)acetonitrile hydrochloride (1.10 g, 6.23 mmol) which was used without further purification. 1H NMR (400 MHz, MeOD) δ 4.18 – 3.87 (m, 3H), 3.69 – 3.58 (m, 1H), 3.53 – 3.40 (m, 2H), 3.37 – 3.21 (m, 3H), 2.76 – 2.51 (m, 2H). Step 4: tert-Butyl ((S)-4'-(6-(cyanomethyl)-1,4-oxazepan-4-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate DIPEA (95.6 mg, 129 μL, 8 equiv, 740 μmol) was added to a solution of tert-butyl ((S)- 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-oxo-3,3',4,4',5',8'- hexahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (Intermediate 4, 50.0 mg, 1 equiv, 92.5 μmol), 2-(1,4-oxazepan-6-yl)acetonitrile hydrochloride (49.0 mg, 3 equiv, 277 μmol), and (benzotriazol-1- yloxy)tripyrrolidinophosphoniumhexafluorophosphate (57.8 mg, 1.2 equiv, 111 μmol) in DMF (1.85 mL). The reaction mixture was stirred for 24 hours at 50-75 °C. The product solution was cooled to room temperature and was diluted with a 1:1 mixture of water/brine (2 mL) and EtOAc (2 mL). The layers that formed were separated. The aqueous layer was extracted with EtOAc (2 x 2 mL). The combined organic layers were dried over magnesium sulfate, filtered, and concentrated. The residue obtained was purified by column chromatography (SiO2, 0-50% 2.5% NH4OH/20% MeOH/77.5% DCM in DCM) to provide tert-butyl ((S)-4'-(6- (cyanomethyl)-1,4-oxazepan-4-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7- yl)carbamate containing residual hydroxytri(pyrrolidin-1-yl)phosphonium (80 mg, 121 μmol) which was used without further purification. LCMS: m/z (ESI) [M+H]
+ 663.3, t
R = 1.36 min (Method C)
Step 5: 2-(4-((S)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-1,4-oxazepan-6-yl)acetonitrile p-Toluenesulfonic acid monohydrate (48.0 mg, 3 equiv, 0.25 mmol) was added to a solution of tert-butyl ((S)-4'-(6-(cyanomethyl)-1,4-oxazepan-4-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (56 mg, 1 equiv, 84.0 μmol) in MeCN (0.77 mL). The reaction mixture was stirred for 3 hours at 35 °C. The product mixture was directly concentrated under reduced pressure, diluted with a 1:9 mixture of ammonium hydroxide:methanol (1.5 mL), and concentrated again. The residue obtained was purified first by column chromatography (SiO
2, 0-80% 2.5% NH
4OH/20% MeOH/77.5% DCM in DCM). The residue obtained was further purified by reverse phase column chromatography (C18, 5- 95% MeCN in water (0.1% NH4OH additive)) to provide 2-(4-((S)-7-amino-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-1,4-oxazepan-6-yl)acetonitrile (5.0 mg, 8.89 μmol). LCMS: m/z (ESI) [M+H]
+ 563.4, t
R = 1.05 and 1.09 min (Method C) 1H NMR (400 MHz, MeOD) δ 6.93 – 6.85 (m, 1H), 6.76 – 6.68 (m, 1H), 6.66 – 6.60 (m, 1H), 5.41 – 5.17 (m, 1H), 4.87 – 4.64 (m, 1H), 4.58 – 4.45 (m, 1H), 4.26 – 4.05 (m, 3H), 4.02 – 3.60 (m, 5H), 3.60 – 3.37 (m, 2H), 3.28 – 3.15 (m, 3H), 3.10 – 2.95 (m, 2H), 2.92 – 2.82 (m, 1H), 2.82 – 2.61 (m, 2H), 2.58 – 2.43 (m, 3H), 2.37 – 1.83 (m, 9H), 1.82 – 1.65 (m, 1H). Example 74: 5-(6-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,3-dimethyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide (Compound 131a)

Step 1: 6-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-3,3-dimethyl-2,3-dihydro-1H-inden-1-ol To an oven-dried 100 mL round bottom flask was added (4-chloro-6-methyl-2- (methylthio)pyrimidin-5-yl)methanol (250 mg, 1 equiv, 1.22 mmol). The flask was then sealed, evacuated, and backfilled with nitrogen three times. THF (2.5 mL) was then added and cooled to -78 °C. LDA solution (301 mg, 2.81 mL, 1 M, 2.3 equiv, 2.81 mmol) was added dropwise, maintaining a temperature below -74 °C. After complete addition of the LDA, a dark red homogeneous solution formed, and this solution was stirred at -78 °C for 30 min.6-bromo-3,3- dimethyl-2,3-dihydro-1H-inden-1-one (321 mg, 1.1 equiv, 1.34 mmol) was dissolved in THF (750 μL) and added dropwise to the solution keeping the reaction below -74 °C. Upon complete conversion of the starting material, the reaction mixture was quenched by the addition of saturated aqueous ammonium chloride solution and the mixture was stirred for 15 min. The reaction mixture was then extracted with EtOAc (3 ×100 mL) and the combined organic extracts were dried over sodium sulfate, filtered and concentrated. The crude material was loaded onto a 10 g biotage silica cartridge and purified via 100% DCM-> 85:15 DCM:EtOAc to give 6-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-3,3- dimethyl-2,3-dihydro-1H-inden-1-ol (330 mg, 0.74 mmol) as a white foam. 1H NMR (400 MHz, CDCl3) δ 7.45 (dd, 1H), 7.41 (d, 1H), 7.09 (d, 1H), 4.78 – 4.74 (m, 2H), 4.51 (s, 1H), 3.54 (d1H), 3.19 (d1H), 2.62 – 2.54 (m, 4H), 2.43 (d, 1H), 2.02 (d, 1H), 1.35 (s, 3H), 1.32 (s, 3H). Step 2: 6-bromo-4'-chloro-3,3-dimethyl-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] To an oven-dried 50 mL round-bottom flask containing 6-bromo-1-((6-chloro-5- (hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-3,3-dimethyl-2,3-dihydro-1H-inden-
1-ol (330 mg, 1 equiv, 706 μmol) in toluene (1.41 mL) was added phosphoric acid (97.7 mg, 58.0 μL, 81.2 equiv, 848 μmol). The reaction mixture was refluxed overnight. The mixture was concentrated under reduced pressure and purified via column chromatography (100% Hep -> 1:1 Heptanes:EtOAc) to afford 6-bromo-4'-chloro-3,3-dimethyl-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] (200 mg, 0.47 mmol). 1H NMR (400 MHz, CDCl3) δ 7.48 (dd, 1H), 7.21 (d, 1H), 7.12 (d, 1H), 4.75 (d, 1H), 4.69 (d, 1H), 3.21 (d, 1H), 2.99 (d, 1H), 2.58 (s, 3H), 2.31 (d, 1H), 1.91 (d, 1H), 1.39 (s, 3H), 1.31 (s, 3H). Step 3: 5-(6-bromo-3,3-dimethyl-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene- 1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide To a round bottom flask containing 6-bromo-4'-chloro-3,3-dimethyl-2'-(methylthio)- 2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] (200 mg, 1 equiv, 470 μmol) and N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (bis- HCl salt, 213 mg, 1.6 equiv, 752 μmol) in ethanol (939 μL) was added DIPEA (327 μL, 4 equiv, 1.88 mmol) and the reaction mixture was heated to reflux for about 2 hours. The reaction mixture was cooled to room temperature and the solvents were removed under reduced pressure and the resulting thick oily residue was partitioned between EtOAc (10 mL) and water (20 mL). The layers were separated, and the aqueous phase extracted with EtOAc (10 mL). The combined organic extracts were washed with brine and NaOH (1 M, 2 × 10 mL), dried over sodium sulfate, filtered and concentrated to afford 5-(6-bromo-3,3-dimethyl-2'-(methylthio)- 2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8- tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (260 mg, 0.435 mmol) as a light yellow solid foam. LCMS: m/z (ESI) [M+H]
+ 597, 599, tR = 1.95 min (Method C). Step 4: tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-3,3-dimethyl-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene- 1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate To an oven dried 25 mL round bottom flask was added tert-butyl carbamate (147 mg, 2 equiv, 1.26 mmol), 5-(6-bromo-3,3-dimethyl-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-
4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (375 mg, 1 equiv, 628 μmol), 2- (dicyclohexylphosphanyl)-2',4',6'-tris(isopropyl)biphenyl (26.9 mg, 0.09 equiv, 56.5 μmol), cesium carbonate (613 mg, 3 equiv, 1.88 mmol) and Pd(OAc)2 (4.23 mg, 0.03 equiv, 18.8 μmol). The reaction vessel was then sparged with nitrogen and then 1,4-dioxane (6.28 mL) was added. The reaction mixture was then heated to 100 °C. After 60 min and full consumption of starting material, the reaction mixture was cooled to room temperature, filtered, concentrated and purified via column chromatography (100% DCM -> 85:15 DCM:MeOH) to give tert- butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)- 3,3-dimethyl-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6- yl)carbamate (223 mg, 0.35 mmol). LCMS: m/z (ESI) [M+H]
+ 634.5, t
R = 2.95 min (Method E) Step 5: tert-Butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-3,3-dimethyl-2'-(methylsulfonyl)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate To a 10 mL round bottom flask was added tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8- dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-3,3-dimethyl-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (223 mg, 1 equiv, 352 μmol), oxone (454 mg, 2.1 equiv, 739 μmol), MeOH (3.52 mL) and water (3.52 mL). After 45 min, the reaction mixture was extracted three times with DCM and the combined organic extracts were concentrated to give tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-3,3-dimethyl-2'-(methylsulfonyl)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (234 mg, 0.35 mmol) which was used without further purification. LCMS: m/z (ESI) [M+H]
+ 666.5, tR = 1.90 min (Method C). Step 6: 5-(6-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,3-dimethyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide A 10 mL round bottom flask containing crude tert-butyl (4'-(2-(dimethylcarbamoyl)- 7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-3,3-dimethyl-2'-(methylsulfonyl)- 2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (182 mg, 1
equiv, 273 μmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (43.5 mg, 1 equiv, 273 μmol) was evacuated and filled with N2. The flask was then charged with THF (2.73 mL) and cooled to 0 °C. To this solution was delivered potassium tert-butoxide in THF (61.3 mg, 547 μL, 1 M, 2 equiv, 547 μmol) dropwise to give a dark orange then brown solution. After 40 min, 1 mL water and 2 mL EtOAc were delivered to the reaction mixture, the aqueous phase was separated, extracted with EtOAc (4 x 3 mL), and the combined organic extracts were dried over sodium sulfate and concentrated under reduced pressure. The crude material was purified via column chromatography (9:1 DCM:MeOH) to give tert-butyl (4'-(2- (dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)- 2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,3-dimethyl-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (77.6 mg, 104 μmol) which was carried forward without further purification. To a solution of tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,3-dimethyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6- yl)carbamate (70 mg, 1 equiv, 94 μmol) in DCM (0.94 mL) was added a solution of HCl (0.23 mL, 2 M, 5 equiv, 0.47 mmol) at room temperature. The reaction mixture was stirred for 1 hour and then concentrated to give 5-(6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,3-dimethyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (60 mg) as a white powder. LCMS: m/z (ESI) [M+H]
+ 645.5, tR = 1.15 min (Method C). 1H NMR (400 MHz, MeOD) δ 7.52 (d, 1H), 7.50 – 7.36 (m, 2H), 6.62 (s, 1H), 5.61 (d, 1H), 5.27 – 4.95 (m, 3H), 4.84 – 4.71 (m, 3H), 4.60 (m, 2H), 4.15 – 3.79 (m, 4H), 3.49 (m, 1H), 3.35 (s, 3H), 3.27 – 3.02 (m, 6H), 2.83 – 2.08 (m, 11H), 1.45 (s, 3H), 1.40 (s, 3H). 1
9F NMR (376 MHz, MeOD) δ -173.65 – -175.83 (m). Example 75: 5-((R*)-6-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,3-dimethyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine- 2-carboxamide (Compound 131b)
The diastereomers of 5-(6-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,3-dimethyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (48 mg) were separated by chiral SFC using ChiralCel OJ-H 21mm ID x 250 mm L (mobile phase: 30% ethanol, 0.25% diethylamine in CO
2) to give Peak 1: 5-((S*)-6-amino- 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,3-dimethyl-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8.6 mg) and Peak 2: 5-((R*)-6-amino-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,3-dimethyl-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (14.5 mg). Peak 2: HPLC tR = 2.19 min (ChiralCel OJ-H 4.6 mm ID x 100 mm L, mobile phase: 30% ethanol, 0.25% diethylamine in CO2; 2.5 mL/min) 1H NMR (400 MHz, CDCl
3) δ 7.04 (d, 1H), 6.74 – 6.67 (m, 1H), 6.50 (s, 1H), 6.41 (d, 1H), 5.36 – 5.14 (m, 1H), 4.72 – 4.58 (m, 2H), 4.58 – 4.48 (m, 2H), 4.44 – 4.34 (m, 2H), 4.30 – 4.15 (m, 1H), 4.08 – 3.98 (m, 2H), 3.90 (d, 1H), 3.54 – 3.43 (m, 1H), 3.31 (s, 3H), 3.25 – 3.18 (m, 1H), 3.15 – 3.10 (m, 1H), 3.05 (d, 4H), 3.01 – 2.91 (m, 2H), 2.47 – 2.36 (m, 1H), 2.28 – 2.08 (m, 4H), 2.05 – 1.77 (m, 5H), 1.65 (dd, 1H), 1.50 (dd, 1H), 1.31 (s, 3H), 1.26 (s, 3H). LCMS: m/z (ESI) [M+H]
+ 645.5, tR = 1.25 min (Method C). Example 76: 5-(6-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3-methyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Compound 148a)

Step 1: 6-Bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-3-methyl-2,3-dihydro-1H-inden-1-ol To an oven-dried 100 mL round bottom flask was added (4-chloro-6-methyl-2- (methylthio)pyrimidin-5-yl)methanol (500 mg, 1 equiv, 2.44 mmol). The flask was then sealed, evacuated, and backfilled with nitrogen three times. THF (4.89 mL) was then added and the mixture was cooled to -78 °C. LDA solution (602 mg, 5.62 mL, 1 M, 2.3 equiv, 5.62 mmol) was added dropwise, maintaining a temperature below -74 °C. After complete addition of the LDA a dark red homogeneous solution formed and this solution was stirred at -78 °C for 30 min.6- Bromo-3-methyl-2,3-dihydro-1H-inden-1-one (495 mg, 0.9 equiv, 2.20 mmol) was dissolved in 2 mL of THF and added dropwise to the solution keeping the reaction below -74 °C. Upon consumption of the starting material, the reaction mixture was quenched by addition of saturated aqueous ammonium chloride solution and the reaction mixture was stirred for 15 min. The reaction mixture was then extracted three times with 100 mL of EtOAc and the combined organic phases were dried over sodium sulfate, filtered and concentrated under reduced pressure to give a residue which was loaded onto a 50 g biotage silica cartridge and purified by column chromatography (100% DCM-> 85:15 DCM:EtOAc) to afford 6-bromo-1-((6-chloro- 5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-3-methyl-2,3-dihydro-1H-inden-1- ol (450 mg, 1.05 mmol). 1H NMR (400 MHz, CDCl
3) δ 7.41 (dd, 1H), 7.28 (d, 1H), 7.10 (dd, 1H), 4.73 (dd, 1H), 4.65 (dd, 1H), 4.53 (s, 1H), 3.25 (dd, 1H), 3.17 – 3.07 (m, 2H), 2.71 (dd, 1H), 2.66 – 2.60 (m, 1H), 2.54 (s, 3H), 1.69 (ddd, 1H), 1.33 (d, 3H). Step 2: 6-Bromo-4'-chloro-3-methyl-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine]
To an oven-dried 50 mL round-bottom flask containing 6-bromo-1-((6-chloro-5- (hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)-3-methyl-2,3-dihydro-1H-inden-1-ol (450 mg, 1 equiv, 995 μmol) in toluene (1.99 mL) was added phosphoric acid (138 mg, 81.7 μL, 1.2 equiv, 1.19 mmol) and the reaction was refluxed for 45 min. The solution was concentrated under reduced pressure and purified via column chromatography (100% Hep -> 1:1 Hep:EtOAc) to afford a diastereomeric mixture of 6-bromo-4'-chloro-3-methyl-2'- (methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] (265 mg, 0.64 mmol). LCMS: m/z (ESI) [M+H]
+ 410, 412, tR = 4.54 and 4.61 min (Method E). Step 3: 5-(6-Bromo-3-methyl-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene- 1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide To a flask containing 6-bromo-4'-chloro-3-methyl-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidine] (265 mg, 1 equiv, 644 μmol) and N,N- dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (bis-HCl salt, 292 mg, 1.6 equiv, 1.03 mmol) in ethanol (1.29 mL) was added DIPEA (166 mg, 224 μL, 2 equiv, 1.29 mmol). The reaction mixture was heated to reflux overnight. The reaction mixture dried up over 2 days. To this residue was added ethyl acetate (20 mL) and the mixture was sonicated for 5 min. The resulting suspension was filtered, and the filtrate was washed with water (5 mL) and brine (5 mL). The mixture was then dried over magnesium sulfate, filtered, and concentrated to yield 5-(6-bromo-3-methyl-2'-(methylthio)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro- 4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (242.8 mg, 0.42 mmol) as a pale, yellow foam. LCMS: m/z (ESI) [M+H]
+ 583.3, 585.3, tR = 3.35 and 3.48 min (Method E). Step 4: tert-Butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-3-methyl-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'- pyrano[4,3-d]pyrimidin]-6-yl)carbamate To a 10 mL oven dried round bottom flask was added tert-butyl carbamate (243.1 mg, 5 equiv, 2.075 mmol), 5-(6-bromo-3-methyl-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene- 1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-
a][1,4]diazepine-2-carboxamide (242.2 mg, 1 equiv, 415.0 μmol), cesium carbonate (405.7 mg, 3 equiv, 1.245 mmol), palladium(II) acetate (2.8 mg, 0.03 equiv, 12.45 μmol), and BrettPhos Pd G4 (57.31 mg, .15 equiv, 62.26 μmol). The reaction mixture was purged with nitrogen, sealed, and placed under nitrogen. 1,4-Dioxane (4.150 mL) was added, and the reaction mixture was then heated to 95 °C and stirred. The reaction was monitored over the course of 90 min which showed full conversion of the starting material. The reaction was then cooled to room temperature and quenched with 10 mL of aqueous sodium bicarbonate solution. The aqueous layer was extracted with DCM (3 X 10 mL) and the combined organic extracts were dried over magnesium sulfate, filtered, and concentrated under reduced pressure to give a residue which was loaded onto an Isco gold 24 g column and purified (100% DCM - 95:5 DCM:MeOH) to give tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-3-methyl-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'- pyrano[4,3-d]pyrimidin]-6-yl)carbamate (190 mg, 0.31 mmol) as a yellow foam. LCMS: m/z (ESI) [M+H]
+ 620, t
R = 2.17 and 2.22 min (Method C). Step 5: tert-Butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-3-methyl-2'-(methylsulfonyl)-2,3,5',8'-tetrahydrospiro[indene- 1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate mCPBA (303.7 mg, 70% wt, 4 equiv, 1.23 mmol) was delivered to a round bottom flask containing tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-3-methyl-2'-(methylthio)-2,3,5',8'-tetrahydrospiro[indene-1,7'- pyrano[4,3-d]pyrimidin]-6-yl)carbamate (190.9 mg, 1 equiv, 308 μmol) in DCM (3.08 mL) at 0 °C. After the addition of mCPBA, the reaction mixture was stirred for 5 min at 0 °C. The reaction mixture was removed from the ice bath and warmed to room temperature while stirring. The reaction mixture was stirred at room temperature for 2 hours. The mixture was quenched with 1 mL sodium thiosulfate and stirred for 10 min. The reaction was then diluted with 5 mL DCM and 10 mL 1 M NaOH. The aqueous phase was separated and extracted with 10 mL of DCM. The combined organic extracts were dried over sodium sulfate, filtered, and solvent removed under vacuum to give tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H- pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-3-methyl-2'-(methylsulfonyl)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (41 mg, 0.063 mmol). LCMS: m/z (ESI) [M+H]
+ 652.3, tR = 1.83 min (Method C).
Step 6: 5-(6-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3-methyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,

][1,4]diazepine-2-carboxamide A 10 mL round bottom flask containing tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8- dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)-3-methyl-2'-(methylsulfonyl)-2,3,5',8'- tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6-yl)carbamate (40 mg, 1 equiv, 61 μmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (9.8 mg, 1 equiv, 61 μmol) was evacuated and filled with N2. The flask was then charged with THF (0.61 mL) and cooled to 0 °C. To this solution was delivered potassium tert-butoxide in THF (10 mg, 46 μL, 2 M, 1.5 equiv, 92 μmol) dropwise to give a dark orange then brown solution. After 40 min, 1 mL water and 2 mL EtOAc were delivered to the reaction mixture, the aqueous layer was separated and extracted with EtOAc (4 × 3 mL) and the combined organic extracts were dried over sodium sulfate. The dried organic extracts were filtered and concentrated under reduced pressure to give tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3-methyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6- yl)carbamate. The crude tert-butyl (4'-(2-(dimethylcarbamoyl)-7,8-dihydro-4H-pyrazolo[1,5- a][1,4]diazepin-5(6H)-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3-methyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3-d]pyrimidin]-6- yl)carbamate was dissolved in 2 mL of DCM and then 2 mL of 2 M HCl in ether was added. The reaction mixture was stirred for 30 min. At which point a solid formed and was isolated by filtration giving a diastereomeric mixture of 5-(6-amino-2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-3-methyl-2,3,5',8'-tetrahydrospiro[indene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (18.1 mg, 28.7 μmol). LCMS: m/z (ESI) [M+H]
+ 631.5, tR = 1.22 min (Method C). 1H NMR (400 MHz, MeOD) δ 7.53 (m, 1H), 7.47 – 7.33 (m, 2H), 6.61 (m, 1H), 5.60 (m, 1H), 5.23 – 4.90 (m, 2H), 4.78 (m, 2H), 4.58 (m, 3H), 4.21 (m, 2H), 4.03 – 3.75 (m, 4H), 3.36 (s, 3H), 3.20 – 2.94 (m, 5H), 2.83 – 1.99 (m, 12H), 1.40 (m, 3H). 1
9F NMR (376 MHz, MeOD) δ -173.47 – -175.25 (m). Example 77: 1-((S)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-
d]pyrimidin]-4'-yl)azetidine-3-carbonitrile (Compound 188a)
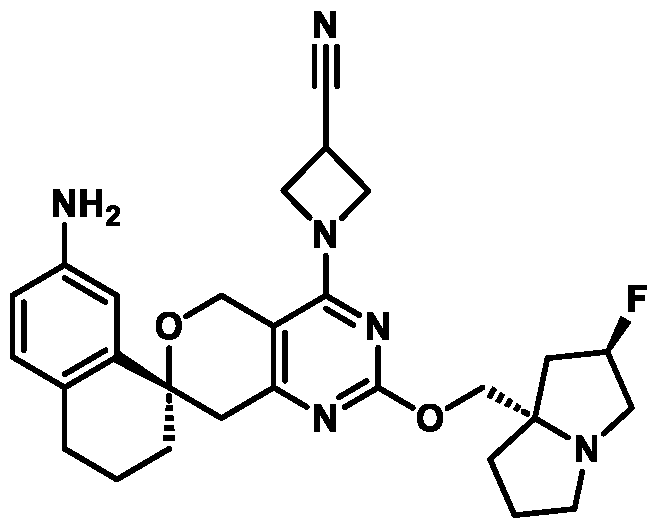
Step 1: tert-Butyl ((S)-4'-(3-carbamoylazetidin-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate tert-Butyl ((S)-4'-(3-carbamoylazetidin-1-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate was prepared following Step 1 of General Procedure B using azetidine-3-carbonitrile to afford tert-butyl ((S)-4'-(3-carbamoylazetidin-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (34 mg). 1H NMR (400 MHz, DMSO-d6) δ 9.19 (s, 1H), 7.53 (s, 1H), 7.47 (s, 1H), 7.23 (d, 1H), 7.06 (s, 1H), 6.98 (d, 1H), 5.35 – 5.11 (m, 1H), 4.67 (d, 1H), 4.58 (d, 1H), 4.32 – 4.10 (m, 4H), 3.95 (d, 1H), 3.81 (d, 1H), 3.41 – 3.34 (m, 1H), 3.13 – 3.03 (m, 2H), 2.99 (s, 1H), 2.86 – 2.75 (m, 2H), 2.73 – 2.59 (m, 3H), 2.10 – 2.05 (m, 1H), 2.03 – 1.56 (m, 9H), 1.44 (s, 9H). LCMS: m/z (ESI) [M+H]
+ 623.5, t
R = 1.48 min (Method B) Step 2: 1-((S)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)azetidine-3-carbonitrile To a stirred solution of tert-butyl ((S)-4'-(3-carbamoylazetidin-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (34 mg, 55 μmol) in DCM (1.1 mL) was added pyridine (18 μL, 0.22 mmol) and trifluoroacetic anhydride (27 μL, 0.19 mmol) at 0 °C. The reaction mixture was stirred for 1 hour at 0 °C then concentrated under reduced pressure to a residue. The residue was dissolved in MeCN (3.0 mL) and 4- methylbenzenesulfonic acid hydrate (31 mg, 0.16 mmol) was added at room temperature. The reaction mixture was warmed to 40 °C and stirred for 1 hour. The mixture was concentrated to
a residue which was purified via reverse-phase chromatography (12 g C-18; 0-85% MeCN/10 mM ammonium bicarbonate solution) to afford 1-((S)-7-amino-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)azetidine-3-carbonitrile (2.1 mg) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 6.92 (d, 1H), 6.71 (d, 1H), 6.59 (dd, 1H), 5.38 – 5.13 (m, 1H), 4.66 (d, 1H), 4.54 (d, 1H), 4.50 – 4.33 (m, 4H), 4.11 (d, 1H), 3.99 (d, 1H), 3.64 – 3.47 (m, 2H), 3.33 – 3.10 (m, 3H), 3.06 (d, 1H), 3.02 – 2.92 (m, 1H), 2.87 (d, 1H), 2.82 – 2.61 (m, 2H), 2.34 – 2.06 (m, 3H), 2.02 – 1.79 (m, 6H), 1.78 – 1.60 (m, 2H). (obscured by water) 1
9F NMR (376 MHz, CDCl
3): δ -172.14 – -174.27 (m). LCMS: m/z (ESI) [M+H]
+ 547.5, t
R = 1.50 min (Method B) Example 78: 2-((S)-1-Acetyl-4-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)piperazin-2-yl)acetonitrile (Compound 189a)

Step 1: Benzyl (S)-4-((S)-7-((tert-butoxycarbonyl)amino)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-2-(cyanomethyl)piperazine-1- carboxylate Benzyl (S)-4-((S)-7-((tert-butoxycarbonyl)amino)-2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-2-(cyanomethyl)piperazine-1-carboxylate was prepared following Step 1 of General Procedure B using benzyl (S)-2-(cyanomethyl)piperazine-1- carboxylate to afford benzyl (S)-4-((S)-7-((tert-butoxycarbonyl)amino)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-2-(cyanomethyl)piperazine-1- carboxylate (45 mg, 83%).
LCMS: m/z (ESI) [M+H]
+ 782.5, t
R = 1.91 min (Method B) Step 2: tert-Butyl ((S)-4'-((S)-3-(cyanomethyl)piperazin-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate A solution of benzyl (S)-4-((S)-7-((tert-butoxycarbonyl)amino)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-2-(cyanomethyl)piperazine-1- carboxylate (90 mg, 88% wt, 0.10 mmol) in MeOH (2.0 mL) was degassed and purged with argon three times. Pd(OH)
2/C (71 mg, 20% wt, 0.10 mmol) was added and the mixture was purged under argon for 2 min. Hydrogen was bubbled through the reaction mixture for 4 hours, then filtered through celite and the filtrate was concentrated to a residue which was purified via reverse-phase chromatography (12 g C-18; 0-85% MeCN/10 mM ammonium bicarbonate solution) to afford tert-butyl ((S)-4'-((S)-3-(cyanomethyl)piperazin-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate as an off-white solid (40 mg). LCMS: m/z (ESI) [M+H]
+ 648.5, t
R = 1.61 min (Method B) Step 3: 2-((S)-1-Acetyl-4-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)piperazin-2-yl)acetonitrile TEA (34 μL, 0.25 mmol) and acetic anhydride (5.6 μL, 59 μmol) in DCM (10 mL) were added dropwise at 0 °C to a solution of tert-butyl ((S)-4'-((S)-3-(cyanomethyl)piperazin-1-yl)- 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (40 mg, 80% wt, 49 μmol) in DCM (1.0 mL). The reaction mixture was stirred at room temperature for 3 hours. The mixture was treated with saturated aqueous sodium bicarbonate (5 mL) and extracted with DCM (3 x 10 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated to afford tert-butyl ((S)-4'-((S)-4-acetyl-3-(cyanomethyl)piperazin-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (35 mg, 31 μmol), which was used without additional purification.
The material was dissolved in MeCN (1.0 mL) and p-toluenesulfonic acid monohydrate (30 mg, 0.16 mmol) was added at room temperature. The reaction mixture was heated to 60 °C and stirred for 30 min then concentrated to a residue which was purified via reverse-phase chromatography (12 g C-18; 0-85% MeCN/10 mM ammonium bicarbonate solution) to afford 2-((S)-1-acetyl-4-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)piperazin-2-yl)acetonitrile (4.2 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 6.73 (d, 1H), 6.56 (d, 1H), 6.41 (dd, 1H), 5.23 (d, 1H), 4.71 (d, 3H), 4.60 – 4.19 (m, 2H), 3.95 (d, 1H), 3.88 – 3.41 (m, 4H), 3.23 – 3.06 (m, 1H), 3.06 – 2.93 (m, 5H), 2.93 – 2.70 (m, 5H), 2.68 – 2.52 (m, 2H), 2.10 (s, 1H), 2.07 – 1.99 (m, 2H), 1.99 – 1.87 (m, 3H), 1.87 – 1.76 (m, 4H), 1.76 – 1.52 (m, 3H). 1
9F NMR (376 MHz, DMSO) δ -171.56 – -172.63 (m). LCMS: m/z (ESI) [M+H]
+ 590.4, tR = 1.30 min (Method B) Example 79: 1-((S)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-3-methylazetidine-3-carbonitrile (Compound 187a)
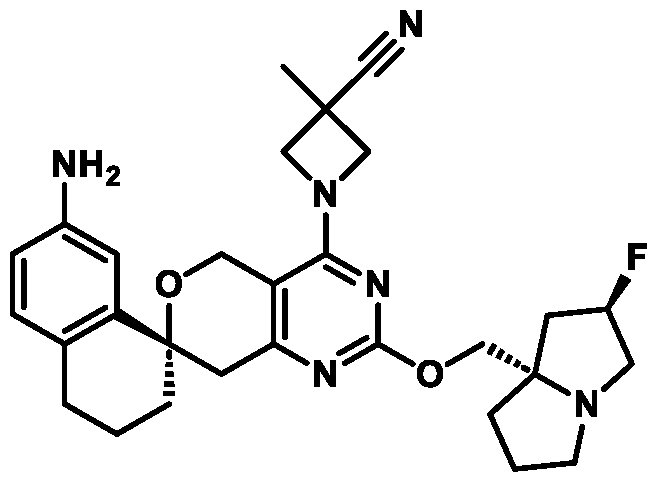
1-((S)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-3- methylazetidine-3-carbonitrile was prepared following General Procedure B using 3-cyano- 3-methylazetidine hydrochloride to afford 1-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-3-methylazetidine-3-carbonitrile (37 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 6.75 (d, 1H), 6.61 (d, 1H), 6.44 (dd, 1H), 5.36 – 5.13 (m, 1H), 4.81 (s, 2H), 4.64 (d, 1H), 4.56 (d, 1H), 4.48 (d, 2H), 4.18 – 4.10 (m, 2H), 3.95 (d, 1H), 3.81 (d, 1H), 3.14 – 3.02 (m, 2H), 3.02 – 2.94 (m, 1H), 2.91 – 2.77 (m, 2H), 2.66 (d, 1H), 2.62 – 2.54 (m, 2H), 2.11 – 1.98 (m, 2H), 1.97 – 1.92 (m, 1H), 1.92 – 1.67 (m, 7H). LCMS: m/z (ESI) [M+H]
+ 519.4, tR = 1.45 min (Method B)
Example 80: (4R*)-1-(7-amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile (Compound 191a)

Step 1: 4-(4-Bromo-2-chlorophenyl)butanal A 250 mL round bottom flask fitted with a stir bar was charged with 4-bromo-2-chloro- 1-iodobenzene (10.0 g, 31.5 mmol), palladium(II) diacetate (424 mg, 1.89 mmol, 6 mol%), tetrabutylammonium chloride hydrate (18.7 g, 63.0 mmol, 2.0 equiv.), lithium chloride (1.34 g, 31.5 mmol, 1.0 equiv.) and lithium acetate (5.20 g, 78.8 mmol, 2.5 equiv.). The flask headspace was evacuated and refilled with nitrogen, followed by the addition of DMF (75.0 mL) and but-3-en-1-ol (3.25 mL, 37.8 mmol, 1.2 equiv.). The flask headspace was evacuated and refilled with nitrogen and the resulting mixture was stirred at 70 °C for 24 hours. The mixture was cooled to room temperature and the volatiles were removed in vacuo. The residue was partitioned between water (75 mL) and EtOAc (75 mL) and the layers were separated. The aqueous phase was extracted with EtOAc (3 x 50 mL). The combined organic extracts were dried over magnesium sulfate, filtered and concentrated under reduced pressure. Crude material was purified by silica gel chromatography (load: CH₂Cl₂, column: RediSep Rf Gold 330 g, 200 mL/min, heptane/EtOAc, 0 → 20% over 25 min) to afford 4-(4-bromo-2-chlorophenyl)butanal (4.20 g, 16.1 mmol), as a colorless oil. 1H NMR (400 MHz, CDCl
3) δ 9.78 (t, 1H), 7.51 (d, 1H), 7.32 (dd, 1H), 7.08 (d, 1H), 2.75 – 2.69 (m, 2H), 2.49 (td, 2H), 1.99 – 1.90 (m, 2H). Step 2: 4-(4-Bromo-2-chlorophenyl)butanoic acid To a solution of 4-(4-bromo-2-chlorophenyl)butanal (4.20 g, 16.1 mmol) in a 2:1 mixture of t-BuOH (107 mL) and water (53.5 mL) at 0 °C under vigorous stirring, were added 2-methyl-2-butene (68.0 mL, 642 mmol, 40 equiv.), sodium dihydrogen phosphate monohydrate (22.2 g, 161 mmol, 10 equiv.) and sodium chlorite (7.26 g, 80.3 mmol, 5.0 equiv.)
sequentially. The resulting mixture was stirred vigorously at 0 °C for 15 min at room temperature for 2 hours. The reaction mixture was poured in a separatory funnel containing a 1:1 mixture of water/brine (300 mL) and CH₂Cl₂ (250 mL). The layers were separated. The aqueous phase was extracted with CH₂Cl₂ (3 x 200 mL). The combined organic extracts were dried over magnesium sulfate, filtered and concentrated to afford 4-(4-bromo-2- chlorophenyl)butanoic acid (4.46 g, 16.1 mmol) which was used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 7.51 (d, 1H), 7.32 (dd, 1H), 7.09 (d, 1H), 2.75 (dd, 2H), 2.41 (t, 2H), 1.99 – 1.90 (m, 2H). Step 3: 7-Bromo-5-chloro-3,4-dihydronaphthalen-1(2H)-one To a solution of 4-(4-bromo-2-chlorophenyl)butanoic acid (4.46 g, 16.1 mmol) and DMF (124 μL, 1.61 mmol, 0.1 equiv.) in CH2Cl2 (64.3 mL) at room temperature under nitrogen, was added oxalyl chloride (2.72 mL, 32.1 mmol, 2.0 equiv.) dropwise (strong gas release observed upon addition of oxalyl chloride). The resulting mixture was stirred for 2.0 hours. The reaction mixture was concentrated, and the residue was dissolved in CH2Cl2 (64.3 mL), followed by the addition of aluminum chloride (4.29 g, 32.1 mmol, 2.0 equiv.) in one portion. The media was stirred for 17 hours. The reaction mixture was poured slowly in a conical flask containing a 1:1 mixture of water and brine (300 mL) cooled to 0 °C. CH₂Cl₂ (200 mL) was added and the layers were separated. The aqueous phase was extracted with CH₂Cl₂ (3 x 150 mL). The combined organic extracts were washed with brine (300 mL), dried over magnesium sulfate, filtered and concentrated. Crude material was purified by silica gel chromatography (load: CH₂Cl₂, column: RediSep Rf Gold 220 g, 150 mL/min, heptane/EtOAc, 1 → 40% over 25 min) to afford 7-bromo-5-chloro-3,4-dihydronaphthalen-1(2H)-one (3.35 g, 12.9 mmol), as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.09 (d, 1H), 7.70 (d, 1H), 2.96 (t, 2H), 2.65 (dd, 2H), 2.20 – 2.12 (m, 2H). Step 4: 7-Bromo-5-chloro-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-ol To a solution of (4-chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (0.70 g, 3.4 mmol) in THF (20.0 mL) at -78 °C was added LDA (1.0 M in THF/hexanes, 7.9 mL, 7.9 mmol, 2.3 equiv.) over 15 min. The resulting mixture was stirred at -78 °C for 1 hour, followed
by the addition of a solution of 7-bromo-5-chloro-3,4-dihydronaphthalen-1(2H)-one (0.98 g, 3.8 mmol, 1.1 equiv.) in THF (10.0 mL) over 15 min. The reaction mixture was stirred at -78 °C for 1 hour. The reaction was quenched by the addition of HCl (2.0 M in Et2O, 3.0 mL) at - 78 °C and the solution was poured in a 1:1 mixture of acetonitrile and saturated aqueous ammonium chloride solution (10 mL) precooled to -78 °C. The mixture was warmed slowly to room temperature and EtOAc (100 mL) was added. The layers were separated, and the aqueous phase was extracted with EtOAc (2 x 100 mL). The combined organic extracts were washed with brine, dried over sodium sulfate, filtered and concentrated under reduced pressure to give a residue which was purified by silica gel chromatography (column: 120 g, 30 mL/min, hexane/EtOAc, 20 → 30%) to afford 7-bromo-5-chloro-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-ol (0.95 g, 2.0 mmol) as a white solid. LCMS: m/z (ESI) [M+H]⁺ = 463.1, tR = 1.84 min, (Method B). 1H NMR (400 MHz, CDCl
3) δ 7.65 (d, 1H), 7.46 (d, 1H), 4.78 (d, 1H), 4.75 (d, 1H), 3.29 (d, 1H), 3.25 (d, 1H), 2.92 – 2.82 (m, 1H), 2.73 – 2.62 (m, 1H), 2.54 (s, 3H), 2.10 – 1.93 (m, 3H), 1.74 – 1.64 (m, 1H). Step 5: 7-Bromo-4',5-dichloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] To a solution of 7-bromo-5-chloro-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-ol (0.85 g, 1.8 mmol) in toluene (15.0 mL) was added phosphoric acid (85 wt%, 0.19 mL, 2.7 mmol, 1.5 equiv.) dropwise. The resulting mixture was stirred at 110 °C for 8 hours. The reaction was quenched by the addition of water (100 mL). EtOAc (300 mL) was added, and the layers were separated. The aqueous phase was extracted with EtOAc (2 x 300 mL). The combined organic extracts were dried over sodium sulfate, filtered and concentrated under reduced pressure to give a residue which was purified by silica gel chromatography (column: 80 g, 18 mL/min, CH₂Cl₂/hexane, 25 → 45%) to afford 7-bromo-4',5-dichloro-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (0.50 g, 1.1 mmol) as a white solid. LCMS: m/z (ESI) [M+H]⁺ = 445.0, t
R = 2.25 min, (Method B)
1H NMR (400 MHz, CDCl
3) δ 7.59 (d, 1H), 7.49 (d, 1H), 4.84 (d, 1H), 4.73 (d, 1H), 3.11 (d, 1H), 3.04 (d, 1H), 2.94 – 2.85 (m, 1H), 2.72 – 2.65 (m, 1H), 2.56 (s, 3H), 2.09 – 1.92 (m, 2H), 1.92 – 1.82 (m, 1H), 1.80 – 1.68 (m, 1H). Step 6: (R*)-1-(7-Bromo-5-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile To a mixture of rel-(R)-azepane-4-carbonitrile hydrochloride (Intermediate 17b, 216 mg, 1.34 mmol, 2.0 equiv.) and 7-bromo-4',5-dichloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (300 mg, 672 μmol) in DMF (3.0 mL) at room temperature, was added DIPEA (0.35 mL, 2.0 mmol, 3.0 equiv.). The resulting mixture was stirred at 90 °C for 2 hours. The reaction mixture was purified directly by reverse phase chromatography (column: Sepaflash C-1820 g; flow: 10 mL/min; 95 to 100% MeCN/water (+ 10 mM ammonium bicarbonate)) to give (R*)-1-(7-bromo-5-chloro-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile (250 mg, 0.468 mmol) as a white solid. LCMS: m/z (ESI) [M+H]⁺ = 533.1, tR = 2.06 min, (Method B). 1H NMR (400 MHz, CDCl
3) δ 7.62 – 7.42 (m, 2H), 4.86 – 4.50 (m, 2H), 3.95 – 3.38 (m, 4H), 3.15 – 2.60 (m, 6H), 2.50 (s, 3H), 2.27 – 1.69 (m, 9H). Step 7: (R*)-1-(7-Bromo-5-chloro-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile To a suspension of (R*)-1-(7-bromo-5-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile (275 mg, 515 μmol), sodium tungstate dihydrate (17 mg, 52 μmol, 10 mol%) and tetrabutylammonium hydrogensulfate (28 mg, 82 μmol, 16 mol%) in EtOAc (10 mL) was added hydrogen peroxide (30 wt%, 368 μL, 3.61 mmol, 7.0 equiv.) at room temperature. The resulting mixture was stirred at 40 °C for 1.5 hours. The mixture was cooled to room temperature and diluted with water (20 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (4 x 50 mL). The combined organic extracts were washed with brine (3 x 15 mL), dried over sodium sulfate, filtered and concentrated to give (R*)-1-(7-bromo-5-chloro-2'-(methylsulfonyl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile (280 mg, 0.50 mmol) which was used in the next step without further purification. LCMS: m/z (ESI) [M+H]⁺ = 565.2, tR = 1.81 min, (Method B).
Step 8: (R*)-1-(7-Bromo-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile To a solution of crude (R*)-1-(7-bromo-5-chloro-2'-(methylsulfonyl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile (0.28 g, theoretical: 0.49 mmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methanol (0.39 g, 2.5 mmol, 5.0 equiv.) in DMF (9.9 mL) at 0 °C, was added LiHMDS (1.0
M in THF, 0.74 mL, 0.74 mmol, 1.5 equiv.) dropwise under nitrogen. The reaction mixture was stirred at room temperature for 5 hours. To the mixture was added saturated aqueous ammonium chloride solution and EtOAc. The layers were separated, and the aqueous phase was extracted with EtOAc (3 x 100 mL). The combined organic extracts were washed with brine (50 mL), dried over sodium sulfate, filtered and concentrated to a residue which was purified by silica gel chromatography (column: 12 g, 20 mL/min, CH₂Cl₂/MeOH, 1 → 8%) to afford (R*)-1-(7-bromo-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)azepane-4-carbonitrile (70 mg, 0.11 mmol) as a yellow solid. LCMS: m/z (ESI) [M+H]⁺ = 644.3, t
R = 1.98 min, (Method B). 1H NMR (400 MHz, CDCl3) δ 7.50 – 7.42 (m, 2H), 5.25 (br d, 1H), 4.75 (dd, 1H), 4.59 (d, 1H), 4.07 (d, 1H), 3.96 (d, 1H), 3.92 – 3.75 (m, 1H), 3.71 – 3.42 (m, 3H), 3.29 – 3.19 (m, 2H), 3.19 – 3.08 (m, 1H), 3.06 – 2.76 (m, 6H), 2.75 – 2.64 (m, 1H), 2.28 – 1.98 (m, 8H), 1.98 – 1.72 (m, 7H). Step 9: tert-Butyl (5-chloro-4'-((R*)-4-cyanoazepan-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate To a vial charged with (R*)-1-(7-bromo-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)azepane-4-carbonitrile (50 mg, 78 μmol), cesium carbonate (51 mg, 0.16 mmol, 2.0 equiv.) and tert-butyl carbamate (18 mg, 0.16 mmol, 2.0 equiv.) was added DMF (2.0 mL) at room temperature. Nitrogen was bubbled through for 5 min while stirring the reaction mixture. BrettPhos-Pd-G4 (11 mg, 12 μmol, 15 mol%) was added and nitrogen was bubbled through for 5 min while stirring the resulting mixture. The vent needle was removed,
and the reaction mixture was stirred at 120 °C for 5 hours. The volatiles were removed in vacuo. The residue was purified by reverse phase chromatography (column: Sepaflash C-1820 g; flow: 10 mL/min; 80 to 95% MeCN/water (+ 10 mM ammonium bicarbonate)) to give tert- butyl (5-chloro-4'-((R*)-4-cyanoazepan-1-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]- 7-yl)carbamate (15 mg, 22 μmol) as a white solid. LCMS: m/z (ESI) [M+H]⁺ = 681.4, t
R = 1.94 min, (Method B). Step 10: (4R*)-1-(7-amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile To a solution of tert-butyl (5-chloro-4'-((R*)-4-cyanoazepan-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (12 mg, 18 µmol) in MeCN (3.0 mL) at room temperature, was added PTSA hydrate (17 mg, 88 µmol, 5 equiv.). The resulting mixture was stirred at 70 ˚C for 3 hours. The reaction mixture was concentrated, and the residue was purified by reverse phase chromatography (column: Sepaflash C-1820 g; flow: 10 mL/min; 60 to 75% MeCN/water (+ 10 mM ammonium bicarbonate)) to give (4R*)-1-(7- amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile (6.0 mg, 10 µmol). LCMS: m/z (ESI) [M+H]⁺ = 581.3, tR = 4.56 min, (Method I). 1H NMR (400 MHz, DMSO-d6) δ 6.59 (s, 2H), 5.26 (d, 1H), 5.12 (d, 2H), 4.73 (d, 1H), 4.55 – 4.45 (m, 1H), 3.99 – 3.90 (m, 1H), 3.89 – 3.80 (m, 1H), 3.65 – 3.57 (m, 2H), 3.56 – 3.12 (m, 3H), 3.10 – 3.03 (m, 3H), 3.01 – 2.96 (m, 1H), 2.86 – 2.77 (m, 3H), 2.69 – 2.53 (m, 3H), 2.13 – 2.04 (m, 2H), 2.02 – 1.89 (m, 5H), 1.88 – 1.71 (m, 7H). 1
9F NMR (376 MHz, DMSO) δ -172.04. Example 81: 5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-amine (Compound 190a)

Step 1: 7-Bromo-5-chloro-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-ol To a solution of (4-chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (8.54 g, 41.7 mmol, 1.5 equiv.) in THF (206 mL) at -78 °C, was added LDA (1.0 M in THF/hexanes, 83.5 mL, 83.5 mmol, 3.0 equiv.) dropwise, by addition funnel (15 min). The rate of addition was set so that the internal temperature did not exceed - 72 °C. The resulting clear mixture, which became bright orange over time, was stirred at -78 °C for 1.5 hours, followed by the dropwise addition of a solution of 7-bromo-5-chloro-3,4-dihydronaphthalen-1(2H)-one (7.22 g, 27.8 mmol) in THF (103 mL) by addition funnel. The rate of dropwise addition was set so that the internal temperature did not exceed - 74 °C. The reaction mixture was stirred -78 °C for 1 hour. The reaction was quenched by the dropwise addition of HCl (2.0 M in Et2O, 44.5 mL, 89.0 mmol, 3.2 equiv.) at -78 °C, by addition funnel, followed by the addition of a 1:1 mixture of saturated aqueous ammonium chloride solution and water (150 mL). The biphasic mixture was warmed to room temperature and the layers were separated. The aqueous phase was extracted with EtOAc (3 x 150 mL). The combined organic extracts were dried over magnesium sulfate, filtered and concentrated. Crude material was purified by silica gel chromatography (load: CH₂Cl₂, column: RediSep Rf Gold 220 g, 150 mL/min, heptane/EtOAc, 1 → 40% over 27 min) to afford 7-bromo-5-chloro-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-ol (9.96 g, 21.5 mmol), as a white amorphous solid. 1H NMR (400 MHz, CDCl3) δ 7.66 (d, 1H), 7.46 (d, 1H), 4.83 – 4.72 (m, 2H), 4.43 (s, 1H), 3.30 (d, 1H), 3.25 (d, 1H), 2.88 (ddd, 1H), 2.77 – 2.63 (m, 2H), 2.54 (s, 3H), 2.11 – 1.94 (m, 3H), 1.70 (ddd, 1H). Step 2: 7-Bromo-4',5-dichloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] To a suspension of 7-bromo-5-chloro-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-ol (9.71 g, 20.9 mmol) in
toluene (105 mL) at room temperature, was added phosphoric acid (1.22 mL, 20.9 mmol, 1.0 equiv.). The resulting mixture was heated to reflux and stirred for 3 hours, upon which more phosphoric acid (608 μL, 10.5 mmol, 0.5 equiv.) was added. The mixture was stirred at reflux for an additional 2 hours. The mixture was then cooled to room temperature and the volatiles were removed in vacuo. The residue was partitioned between EtOAc (150 mL) and water (150 mL) and the biphasic mixture was stirred vigorously. A white precipitate was observed at the interface between the organic and aqueous layers. The flask containing the biphasic mixture was stored at -20 °C overnight, upon which significant precipitate formation was observed. The biphasic mixture was filtered, the solids were transferred to a vial and dried under vacuum at 55 °C for 3 hours, to yield 7-bromo-4',5-dichloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (4.68 g, 10.5 mmol). LCMS: m/z (ESI) [M+H]⁺ = 444.9, tR = 2.62 min, (Method C). 1H NMR (400 MHz, DMSO-d6) δ 7.70 (d, J = 2.1 Hz, 1H), 7.68 (d, J = 2.1 Hz, 1H), 4.80 (d, J = 16.5 Hz, 1H), 4.69 (d, J = 16.5 Hz, 1H), 3.24 (d, J = 18.0 Hz, 1H), 3.03 (d, J = 18.0 Hz, 1H), 2.77 (app dt, J = 17.5, 4.6 Hz, 1H), 2.68 – 2.57 (m, 1H), 2.52 (s, 3H), 2.00 – 1.68 (m, 4H). Step 3: 7-Bromo-5-chloro-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] To a solution of 7-bromo-4',5-dichloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (1.00 g, 2.24 mmol) and 1,4-oxazepane hydrochloride (771 mg, 5.60 mmol, 2.5 equiv.) in 1,4-dioxane (11.0 mL) at room temperature, was added DIPEA (2.73 mL, 15.7 mmol, 7.0 equiv.) slowly. The resulting mixture was stirred at 100 °C for 5 hours. DMF (4.0 mL) was added after 10 min to help obtain a clear solution. The mixture was cooled to room temperature and the volatiles were removed in vacuo. Crude material was purified by silica gel chromatography (load: CH₂Cl₂, column: RediSep Rf Gold 80 g, 60 mL/min, heptane/EtOAc, 2 → 50% over 25 min) to afford 7-bromo-5-chloro-2'- (methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine] (1.07 g, 2.09 mmol) as a solid. LCMS: m/z (ESI) [M+H]⁺ = 510.0, tR = 2.10 min, (Method C). 1H NMR (400 MHz, CDCl
3) δ 7.51 (d, 1H), 7.46 (d, 1H), 4.75 (d, 1H), 4.59 (d, 1H), 3.88 – 3.83 (m, 2H), 3.83 – 3.59 (m, 6H), 3.02 (d, 1H), 3.01 (d, 1H), 2.84 (app dt, 1H), 2.75 –
2.65 (m, 1H), 2.50 (s, 3H), 2.13 – 2.03 (m, 2H), 2.03 – 1.93 (m, 2H), 1.90 (app t, 1H), 1.86 – 1.74 (m, 1H). Step 4: tert-Butyl (5-chloro-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate A vial was charged with 7-bromo-5-chloro-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (820 mg, 1.61 mmol), tert-butyl carbamate (207 mg, 1.77 mmol, 1.1 equiv.), BrettPhos-Pd-G4 (222 mg, 241 μmol, 15 mol%) and cesium carbonate (549 mg, 1.69 mmol, 1.05 equiv.). The vial headspace was evacuated and refilled with nitrogen, followed by the addition of 1,4-dioxane (16.1 mL, degassed prior to addition by sparging with nitrogen for 30 min). The resulting mixture was further degassed by evacuating the vial headspace and refilling it with nitrogen three times. The mixture was stirred at 80 °C for 4 hours. The reaction mixture was cooled to room temperature and the solids were filtered through a syringe filter. The filtrate was concentrated under reduced pressure to give a residue which was purified by silica gel chromatography (load: CH₂Cl₂, column: RediSep Rf Gold 80 g, 60 mL/min, heptane/EtOAc, 1 → 45% over 30 min) to afford tert-butyl (5-chloro-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (793 mg, 1.45 mmol) as a solid. LCMS: m/z (ESI) [M+H]⁺ = 547.3, t
R = 2.02 min, (Method C). 1H NMR (400 MHz, CDCl
3) δ 7.56 (br s, 1H), 7.18 (d, 1H), 6.36 (br s, 1H), 4.74 (d, 1H), 4.58 (d, 1H), 3.88 – 3.81 (m, 2H), 3.80 – 3.60 (m, 6H), 3.06 (d, 1H), 2.98 (d, 1H), 2.84 (dt, 1H), 2.72 (ddd, 1H), 2.50 (s, 3H), 2.11 – 1.86 (m, 5H), 1.84 – 1.72 (m, 1H), 1.48 (s, 9H). Step 5: tert-butyl (5-chloro-2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate To a suspension of tert-butyl (5-chloro-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (923 mg, 1.69 mmol) in THF (7.0 mL), methanol (9.0 mL) and water (6.0 mL) at room temperature, was added Oxone (45 wt%, 9.22 g, 6.75 mmol, 4.0 equiv.) in one portion. The resulting mixture was stirred at room temperature for 3 hours. The mixture was poured in a separatory funnel charged with water (150 mL) and CH₂Cl₂ (150 mL). The layers were separated, and the aqueous phase was extracted with CH₂Cl₂ (3 x 100 mL). The combined organic extracts were dried over
magnesium sulfate, filtered, and concentrated to afford tert-butyl (5-chloro-2'- (methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate (932 mg, 1.61 mmol) which was used without further purification. LCMS: m/z (ESI) [M+H]⁺ = 579.3, tR = 2.17 min, (Method C). Step 6: tert-Butyl (5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate To a vial charged with crude tert-butyl (5-chloro-2'-(methylsulfonyl)-4'-(1,4-oxazepan- 4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (932 mg, theoretical: 1.61 mmol) and ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methanol (769 mg, 4.83 mmol, 3.0 equiv.) was added DMF (16.1 mL). The resulting solution was cooled to 0 °C and LiHMDS (1.0 M in THF, 3.22 mL, 3.22 mmol, 2.0 equiv.) was added dropwise. The reaction mixture was stirred at 0 °C for 1 hour and the ice/water bath was removed. The reaction mixture was stirred for another 2.5 hours. The reaction was quenched by the slow addition of a 1:1 mixture of a saturated aqueous ammonium chloride solution and water (12 mL) and the biphasic mixture was poured into a separatory funnel charged with a 1:1 mixture of a saturated aqueous ammonium chloride solution/water (50 mL) and EtOAc (50 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (3 x 50 mL). The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated. Crude material was purified by silica gel chromatography (load: CH₂Cl₂, column: RediSep Rf Gold 80 g, 60 mL/min, CH₂Cl₂/[CH₂Cl₂/MeOH/NH₄OH, 75:22.5:2.5], 0 → 40% over 30 min) to afford tert-butyl (5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate (841 mg, 1.28 mmol), as a solid. LCMS: m/z (ESI) [M+H]⁺ = 658.3, t
R = 1.57 min, (Method C). 1H NMR (400 MHz, CDCl
3) δ 7.57 (br s, 1H), 7.18 – 7.13 (m, 1H), 6.43 – 6.34 (m, 1H), 5.25 (br d, 1H), 4.74 (d, 1H), 4.58 (d, 1H), 4.07 (d, 1H), 3.94 (d, 1H), 3.83 (t, 2H), 3.76 – 3.69 (m, 4H), 3.69 – 3.62 (m, 2H), 3.27 – 3.05 (m, 3H), 3.04 – 2.97 (m, 2H), 2.97 – 2.91 (m, 1H), 2.87 – 2.78 (m, 1H), 2.77 – 2.67 (m, 1H), 2.32 – 1.85 (m, 10H), 1.85 – 1.71 (m, 2H), 1.48 (s, 9H).
Step 7: 5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-amine To a solution of tert-butyl (5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate (841 mg, 1.28 mmol) in CH2Cl2 (12.8 mL) at 0 °C, was added HCl (4.0 M in 1,4-dioxane, 6.39 mL, 25.6 mmol, 20 equiv.). The resulting mixture was stirred at room temperature for 1 hour and the volatiles were removed in vacuo. A portion of the crude (75 mg) was purified by achiral HPLC prep chromatography (load: water (3.0 mL); column: XSelect® CSH™ Prep C185 µm OBD™ 19x150 mm; mobile phase: water 0.1% FA / MeCN 0.1% FA; flow: 18.9 mL/min; gradient: 5 → 15% over 1 min then 15 → 35% over 20 min) to afford 5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-amine (45 mg, 1.28 mmol). LCMS: m/z (ESI) [M+H]⁺ = 558.3, tR = 1.81 min, 3 min method FA. 1H NMR (400 MHz, CDCl3) δ 6.69 (d, 1H), 6.68 – 6.64 (m, 1H), 5.25 (br d, 1H), 4.73 (d, 1H), 4.56 (d, 1H), 4.08 (dd, 1H), 3.96 (dd, 1H), 3.85 – 3.79 (m, 2H), 3.78 – 3.69 (m, 4H), 3.69 – 3.62 (m, 2H), 3.29 – 3.20 (m, 2H), 3.20 – 3.10 (m, 1H), 3.10 – 3.02 (m, 1H), 3.01 – 2.92 (m, 2H), 2.78 (dt, 1H), 2.68 (ddd, 1H), 2.33 – 2.10 (m, 3H), 2.09 – 1.68 (m, 11H). 1
9F NMR (376 MHz, CDCl
3) δ -173.14. Example 82: (S*)-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-7-amine (Compound 190b)
The diastereomers of 5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-amine were separated by chiral SFC prep chromatography (column: ChiralPak AS-H 20 x 250 mm; mobile phase: 40% ethanol+ 0.25% diethylamine in
CO
2, flow rate: 70 mL/min; back pressure: 100 bar; sample: 711 mg dissolved in 70 ml methanol + 500 µL diethylamine; injection: 1.5 mL; detection: 220 nm) to afford (S*)-5-chloro- 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (Peak 1, 254 mgs) and (R*)-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-amine (Peak 2; 258 mgs). Both fractions obtained were further purified by achiral HPLC prep chromatography (load: DMSO (3.0 mL); column: XSelect® CSH™ Prep C185 µm OBD™ 19x150 mm; mobile phase: water 0.1% FA / MeCN 0.1% FA; flow: 18.9 mL/min; gradient: 5 → 15% over 1 min then 15 → 35% over 20 min) to give (S*)-5-chloro-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (Peak 1, 163 mg, 292 µmol) and (R*)-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-amine (Peak 2; 170 mg, 305 µmol, 95% purity). Peak 1: LCMS: m/z (ESI) [M+H]⁺ = 558.3, tR = 1.25 min, 3 min method FA. Analytical SFC, (ESI) [M+H]⁺ = 558.3, t
R = 2.04 min, column: ChiralPak AS-H 4.6 x 100 mm; mobile phase: 20% ethanol + 0.25% diethylamine in CO
2; flow rate: 2.5 mL/min; backpressure: 140 bar; temperature: 40 °C; sample: 1.0 mg/mL; injection: 2 µL; detection: 220 nm and 254 nm. 1H NMR (400 MHz, DMSO-d
6) δ 6.60 (br s, 2H), 5.48 (d, 1H), 5.13 (br s, 1H), 4.75 (d, 1H), 4.55 (d, 1H), 4.37 – 4.13 (m, 1H), 3.77 – 3.52 (m, 12H), 3.21 – 3.08 (m, 1H), 2.91 – 2.78 (m, 2H), 2.68 – 2.52 (m, 2H), 2.43 – 2.26 (m, 2H), 2.22 – 2.13 (m, 1H), 2.12 – 1.74 (m, 9H), 1.73 – 1.60 (m, 1H). 1
9F NMR (376 MHz, DMSO) δ -172.72. Example 83: (R*)-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene- 1,7'-pyrano[4,3-d]pyrimidin]-7-amine (Compound 190c)
(R*)-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'- (1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]- 7-amine (258 mg) was obtained from Example 82, as Peak 2: achiral HPLC prep chromatography (load: DMSO (3.0 mL); column: XSelect® CSH™ Prep C185 µm OBD™ 19x150 mm; mobile phase: water 0.1% FA / MeCN 0.1% FA; flow: 18.9 mL/min; gradient: 5 → 15% over 1 min then 15 → 35% over 20 min). Peak 2: LCMS: m/z (ESI) [M+H]⁺ = 558.3, t
R = 1.24 min, 3 min method FA. Analytical SFC, (ESI) [M+H]⁺ = 558.2, tR = 2.85 min, column: ChiralPak AS-H 4.6 x 100 mm; mobile phase: 20% ethanol + 0.25% diethylamine in CO
2; flow rate: 2.5 mL/min; backpressure: 140 bar; temperature: 40 °C; sample: 1.0 mg/mL; injection: 2 µL; detection: 220 nm and 254 nm. 1H NMR (400 MHz, DMSO-d6) δ 6.60 (br s, 2H), 5.47 (d, 1H), 5.13 (br s, 1H), 4.75 (d, 1H), 4.55 (d, 1H), 4.25 (br s, 1H), 3.80 – 3.23 (m, 12H), 3.22 – 3.08 (m, 1H), 2.91 – 2.78 (m, 2H), 2.68 – 2.53 (m, 2H), 2.44 – 2.26 (m, 2H), 2.23 – 2.13 (m, 1H), 2.14 – 1.73 (m, 9H), 1.74 – 1.61 (m, 1H). 1
9F NMR (376 MHz, DMSO) δ -172.72. Example 84: (4S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-4-(trifluoromethyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (Compound 193a)

Step 1: Methyl 4-(4-bromophenyl)-5,5-difluoropent-4-enoate
To a nitrogen purged vessel containing methyl-4-(4-bromophenyl)-4-oxobutanoate (16 g, 59 mmol) and 2,2-difluoro-2-(tris(dimethylamino)phosphonio)acetate (15 g, 59 mmol) was added anhydrous toluene (120 mL) and DMA (40.0 mL). The mixture was then stirred at 100 °C for 4 hours. The mixture was cooled to room temperature then treated with EtOAc and water. The phases were separated, and the organic phase was dried over sodium sulfate, filtered and concentrated. The crude was then subjected to purification via flash chromatography (220 g silica; 0-30% EtOAc/Heptanes) to afford methyl 4-(4-bromophenyl)-5,5-difluoropent-4- enoate (10 g, 56%). 1H NMR (400 MHz, CDCl3) δ 7.51 – 7.46 (m, 2H), 7.20 – 7.13 (m, 2H), 3.63 (s, 3H), 2.77 – 2.66 (m, 2H), 2.35 (t, J = 7.8 Hz, 2H). 1
9F NMR (376 MHz, CDCl
3) δ -89.37 – -89.70 (m). Step 2: 7-Bromo-4-(trifluoromethyl)-3,4-dihydronaphthalen-1(2H)-one To a nitrogen purged vessel containing methyl 4-(4-bromophenyl)-5,5-difluoropent-4- enoate (6.0 g, 20 mmol) was added anhydrous toluene (55 mL) and DMA (18 mL). The mixture was then heated at 100 °C followed by the addition of a THF solution of tetrabutylammonium fluoride (59 mL, 1 M, 59 mmol). The mixture was cooled to room temperature then treated with water (60 mL) and EtOAc (60 mL). The phases were separated, and the aqueous phase was extracted with DCM (30 mL). The combined organic phases were dried over sodium sulfate, filtered, and concentrated. The crude was used without additional purification. A solution of methyl 4-(4-bromophenyl)-5,5,5-trifluoropentanoate (13.4 g, 41.2 mmol) in THF (51 mL) was treated with water (102 mL) then lithium hydroxide monohydrate (6.05 g, 144 mmol). The mixture was then heated at 60 °C for 1 hour. The mixture was treated with saturated aqueous sodium bicarbonate (10 mL/g) and extracted with Et
2O (2 x 10 mL/g). The aqueous solution was treated with concentrated HCl until pH = 3. The mixture was extracted with DCM (3 x 10 mL/g). The combined DCM phases were dried over sodium sulfate, filtered, and concentrated. The crude product was used without additional purification. Oxalyl chloride (9.37 mL, 107 mmol) was added slowly to a solution of crude 4-(4- bromophenyl)-5,5,5-trifluoropentanoic acid (11.1 g, 35.7 mmol)) and DMF (276 μL, 3.57 mmol) in DCM (100 mL) under nitrogen. The resulting mixture was stirred at room temperature for 2 hours. Afterwards, the mixture was concentrated, and the resulting residue was dissolved in DCM (100 mL) and treated with aluminum chloride (14.3 g, 107 mmol) at room temperature. After stirring the mixture for 3 hours, the mixture was poured slowly over
iced water (200 mL). The resulting mixture was extracted with DCM (3 x 80 mL). The organic fractions were collected, dried over sodium sulfate, filtered, and concentrated under reduced pressure to give a crude product which was purified by flash chromatography (330 g silica; 0- 30% EtOAc/heptanes) to afford 7-bromo-4-(trifluoromethyl)-3,4-dihydronaphthalen-1(2H)- one (4.39 g). 1H NMR (400 MHz, CDCl3) δ 8.25 (d, 1H), 7.70 (dd, 1H), 7.32 (d, 1H), 3.69 – 3.56 (m, 1H), 2.96 – 2.82 (m, 1H), 2.73 – 2.63 (m, 1H), 2.57 – 2.47 (m, 1H), 2.44 – 2.29 (m, 1H). The enantiomers of 7-bromo-4-(trifluoromethyl)-3,4-dihydronaphthalen-1(2H)-one (4.2 g) were purified by chiral SFC (Column: Chiralpak IG-N 21 x 250 mm, 5 μm; part number 87445; Mobile phase: Phase A for CO
2, and Phase B for MeOH (0.25% DEA), Isocratic elution: B in A from 30% Flow rate: 50 mL/min, Detector: PDA, Column Temp: 40 °C, Back Pressure: 120 bar) to afford (R)-7-bromo-4-(trifluoromethyl)-3,4-dihydronaphthalen-1(2H)- one (Peak 1, 1.404 g, 4.79 mmol) and (S)-7-bromo-4-(trifluoromethyl)-3,4- dihydronaphthalen-1(2H)-one (Peak 2, 1.80 g, 6.14 mmol). (R)-7-Bromo-4-(trifluoromethyl)-3,4-dihydronaphthalen-1(2H)-one; Peak 1: tR = 3.28 min. 1H NMR (400 MHz, CDCl
3) δ 8.24 (d, 1H), 7.69 (dd, 1H), 7.32 (d, 1H), 3.70 – 3.56 (m, 1H), 2.88 (ddd, 1H), 2.73 – 2.63 (m, 1H), 2.57 – 2.46 (m, 1H), 2.44 – 2.29 (m, 1H). 9F NMR (376 MHz, CDCl
3) δ -67.38. (S)-7-Bromo-4-(trifluoromethyl)-3,4-dihydronaphthalen-1(2H)-one; Peak 2: tR = 3.89 min. 1H NMR (400 MHz, CDCl
3) δ 8.24 (d, J = 2.2 Hz, 1H), 7.69 (dd, J = 8.3, 2.2 Hz, 1H), 7.32 (d, J = 8.2 Hz, 1H), 3.68 – 3.54 (m, 1H), 2.88 (ddd, J = 19.1, 13.7, 5.6 Hz, 1H), 2.73 – 2.63 (m, 1H), 2.58 – 2.45 (m, 1H), 2.44 – 2.29 (m, 1H). 9F NMR (376 MHz, CDCl
3) δ -67.38. Step 3: (4-Methyl-2-(methylthio)-6-(1,4-oxazepan-4-yl)pyrimidin-5-yl)methanol To a 40 mL vial was combined 1,4-oxazepane, HCl (740mg, 1.1 equiv., 5.38 mmol), (4-chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (1000 mg, 1 equiv., 4.89 mmol), MeCN (12.2 mL), and DIPEA (1.90 g, 2.55 mL, 3 equiv., 14.7 mmol) to yield a clear solution. The reaction was heated at 65 °C for 16 hours. At this point the reaction was quenched with
saturated sodium bicarbonate solution (30 mL) and extracted 5 times with EtOAc (5 x 30 mL). The organic layer was concentrated to yield (4-methyl-2-(methylthio)-6-(1,4-oxazepan-4- yl)pyrimidin-5-yl)methanol as a tan oil which was used directly in the next step (1.7 g, 6.3 mmol). 1H NMR (400 MHz, CDCl3): δ 4.58 (d, 2H), 3.92 – 3.83 (m, 6H), 3.81 – 3.75 (m, 2H), 2.48 (s, 3H), 2.45 (s, 3H), 2.10 – 2.02 (m, 2H) Step 4: 4-(5-(((tert-Butyldimethylsilyl)oxy)methyl)-6-methyl-2- (methylthio)pyrimidin-4-yl)-1,4-oxazepane To a 20 ml vial was combined (4-methyl-2-(methylthio)-6-(1,4-oxazepan-4- yl)pyrimidin-5-yl)methanol (500 mg, 1 equiv., 1.86 mmol), DCM (6.2 mL), imidazole (253 mg, 2 equiv., 3.71 mmol), DMAP (22.7 mg, 0.1 equiv., 186 μmol), and tert- butyldimethylchlorosilane (308 mg, 340 μL, 1.1 equiv., 2.04 mmol). The suspension was sealed and stirred at room temperature for 3 hours. The reaction was quenched with water (10 mL) and extracted 3 times with DCM (10 mL). The combined organics were concentrated and purified by silica gel chromatography (20% EtOAc in heptane) to yield 4-(5-(((tert- butyldimethylsilyl)oxy)methyl)-6-methyl-2-(methylthio)pyrimidin-4-yl)-1,4-oxazepane (500 mg, 1.30 mmol) as a white solid. 1H NMR (400 MHz, CDCl3): δ 4.48 (s, 2H), 3.89 – 3.74 (m, 8H), 2.47 (s, 3H), 2.40 (s, 3H), 2.07 – 1.99 (m, 2H), 0.90 (s, 9H), 0.12 (s, 6H) Step 5: 4-(5-(((tert-Butyldimethylsilyl)oxy)methyl)-6-methyl-2- (methylsulfonyl)pyrimidin-4-yl)-1,4-oxazepane To a 4 ml vial was combined 4-(5-(((tert-butyldimethylsilyl)oxy)methyl)-6-methyl-2- (methylthio)pyrimidin-4-yl)-1,4-oxazepane (500 mg, 1 equiv., 1.30 mmol), DCM (6.52 mL), and mCPBA (730 mg, 77% wt, 2.5 equiv., 3.26 mmol). The reaction was stirred at room temperature for 3 hours. The reaction was quenched with 3:1 saturated sodium bicarbonate:saturated sodium thiosulfate (5 mL) and extracted four times with DCM (5 mL). The combined organic layers were concentrated to yield 4-(5-(((tert- butyldimethylsilyl)oxy)methyl)-6-methyl-2-(methylsulfonyl)pyrimidin-4-yl)-1,4-oxazepane (500 mg, 1.20 mmol). 1H NMR (400 MHz, CDCl3): δ 4.48 (s, 2H), 3.89 – 3.81 (m, 6H), 3.79 – 3.74 (m, 2H), 2.47 (s, 3H), 2.40 (s, 3H), 2.06 – 1.99 (m, 2H), 0.90 (s, 9H), 0.12 (s, 6H)
Step 6: 4-(5-(((tert-Butyldimethylsilyl)oxy)methyl)-2-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-6-methylpyrimidin-4-yl)-1,4- oxazepane 4-(5-(((tert-Butyldimethylsilyl)oxy)methyl)-6-methyl-2-(methylsulfonyl)pyrimidin-4- yl)-1,4-oxazepane (500 mg, 1 equiv., 1.20 mmol) and ((2S,3aR)-2-fluorohexahydropentalen- 3a(1H)-yl)methanol (190 mg, 1 equiv., 1.20 mmol) were azeotroped with toluene two times to remove trace water. The resulting mixture was dissolved in THF (6.0 mL) and cooled to 0
°C. Sodium tert-pentoxide (159 mg, 1.44 mL, 1 M, 1.2 equiv., 1.44 mmol) was then added dropwise. The mixture was stirred for 1 hour at room temperature. At this point reaction mixture was quenched with HCl (4 M dioxane) (52.6 mg, 361 μL, 4 M, 1.2 equiv., 1.44 mmol), washed with sodium bicarbonate, and extracted three times with EtOAc. The combined organic layers were concentrated and purified by silica column (10% MeOH in DCM) to yield 4-(5- (((tert-butyldimethylsilyl)oxy)methyl)-2-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-6-methylpyrimidin-4-yl)-1,4-oxazepane (500 mg, 1.01 mmol) as a white solid. LCMS: m/z (ESI) [M+H]
+ 495.6, t
R = 1.40 min (Method M) Step 7: (2-(((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4- methyl-6-(1,4-oxazepan-4-yl)pyrimidin-5-yl)methanol 4-(5-(((tert-Butyldimethylsilyl)oxy)methyl)-2-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-6-methylpyrimidin-4-yl)-1,4-oxazepane (450 mg, 1 equiv., 910 μmol) was dissolved in MeOH (4.55 mL) and HCl (199 mg, 455 μL, 12 M, 6 equiv., 5.46 mmol) was added. The resulting mixture was stirred at room temperature for 16 hours. The mixture was concentrated to remove MeOH, quenched with saturated sodium bicarbonate solution, and extracted with 3:1 DCM:IPA. The combined organic layers were concentrated and suspended in EtOAc, the filtrate was then concentrated and purified on a silica gel chromatography (20 % MeOH in DCM) to yield (2-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-6-(1,4-oxazepan-4-yl)pyrimidin-5-yl)methanol (140 mg, 368 μmol) as a white solid. LCMS: m/z (ESI) [M+H]
+ 381.4, t
R = 0.68 min (Method M)
Step 8: (4S)-7-bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-4-(trifluoromethyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] To an oven-dried round bottom flask containing (2-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4-methyl-6-(1,4-oxazepan-4-yl)pyrimidin-5-yl)methanol (740 mg, 1.95 mmol) was added THF (40 mL). The mixture was cooled to -78 °C followed by the addition of LDA (4.3 mL, 1 M, 4.3 mmol) in THF/hexanes dropwise. After 15 min, additional THF (5 mL) was added. The solution was stirred at -78 °C for 1 hour. (S)-7-bromo-4- (trifluoromethyl)-3,4-dihydronaphthalen-1(2H)-one (500 mg, 1.71 mmol) in THF (5 mL) was added dropwise to the reaction mixture. The solution was stirred for 1 hour. The mixture was immediately poured into an ice solution of ammonium chloride (20 mL). The mixture was extracted with EtOAc (20 mL) and the phases were separated. The aqueous phase was extracted with EtOAc (3 x 20 mL), combined organics were dried over sodium sulfate, filtered and concentrated under reduced pressure to give crude product which was purified via flash chromatography (12 g silica; 0-20% (2.5% NH4OH and 20% MeOH in DCM)/DCM) to afford an off-white solid. Phosphoric acid (210 μL, 85% wt, 3.42 mmol) was added and the mixture was stirred at 100 °C for 3 hours. The mixture was decanted and triturated with toluene. The remaining solid was treated with saturated aqueous sodium bicarbonate solution (10 mL) and sonicated until all solids were dissolved. The mixture was extracted with EtOAc (3 x 10 mL). The combined organics were dried over sodium sulfate, filtered, and concentrated to afford (4S)-7-bromo-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4- oxazepan-4-yl)-4-(trifluoromethyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine] (571.9 mg) as a white solid. 1H NMR (400 MHz, CDCl
3) δ 7.65 (d, 1H), 7.42 (dd, 1H), 7.29 – 7.26 (m, 1H), 5.35 – 5.09 (m, 1H), 4.78 (d, 1H), 4.60 (d, 1H), 4.09 – 4.04 (m, 1H), 3.98 – 3.88 (m, 1H), 3.88 – 3.58 (m, 8H), 3.56 – 3.43 (m, 1H), 3.29 – 3.18 (m, 2H), 3.19 – 3.04 (m, 1H), 3.04 – 2.86 (m, 3H), 2.42 – 2.05 (m, 6H), 2.04 – 1.73 (m, 6H). 1
9F NMR (376 MHz, CDCl
3) δ -67.82, -67.96, -173.17. LCMS: m/z (ESI) [M+H]
+ 655.1, tR = 2.43 min (Method E) Step 9: tert-Butyl ((4S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-4-(trifluoromethyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate
A slurry of tert-butyl carbamate (378.2 mg, 3.228 mmol), (4S)-7-bromo-2'-(((2R,7aS)- 2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-4- (trifluoromethyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (571.9 mg, 872.4 μmol), cesium carbonate (1.22 g, 3.75 mmol), and BrettPhos-G4 (81.5 mg, 87.2 μmol) in 1,4-dioxane (8.4 mL) was degassed for 5 min. The mixture was heated to 85 °C for 3 hours. The mixture was concentrated then redissolved with EtOAc and filtered through celite. The filtrate was concentrated to afford a brown oil. The crude was subjected to purification via flash chromatography (0-20% (2.5% NH4OH and 20% MeOH in DCM)/DCM) to afford tert-butyl ((4S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 4'-(1,4-oxazepan-4-yl)-4-(trifluoromethyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate (422.7 mg) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.47 – 7.36 (m, 2H), 7.31 (d, 1H), 6.44 (s, 1H), 5.33 – 5.15 (m, 1H), 4.77 (d, 1H), 4.60 (d, 1H), 4.07 (d, 1H), 3.94 (d, 1H), 3.88 – 3.78 (m, 2H), 3.78 – 3.69 (m, 4H), 3.67 – 3.59 (m, 2H), 3.59 – 3.42 (m, 1H), 3.27 – 3.04 (m, 3H), 3.04 – 2.89 (m, 3H), 2.39 – 2.30 (m, 1H), 2.30 – 2.23 (m, 1H), 2.23 – 2.06 (m, 3H), 2.05 – 1.76 (m, 7H), 1.51 – 1.46 (m, 9H). 1
9F NMR (376 MHz, CDCl
3) δ -68.13, -173.15. LCMS: m/z (ESI) [M+H]
+ 692.6, t
R = 1.47 min (Method C) Step 10: (4S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-4-(trifluoromethyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine To a solution of tert-butyl ((4S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-4-(trifluoromethyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (422.7 mg, 611.0 mmol) in DCM (6.1 mL) at 0 °C, was added HCl (3.055 mL, 4 M, 12.22 mmol). The resulting mixture was stirred at room temperature for 3 hours. The mixture was concentrated and the crude was filtered with DCM then Et
2O to afford (4S)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-(1,4-oxazepan-4-yl)-4-(trifluoromethyl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-amine (146.3 mg) as a white solid HCl salt. 1H NMR (400 MHz, MeOD-d
4) δ 7.68 (d, 1H), 7.62 (d, 1H), 7.39 (dd, 1H), 5.69 – 5.51 (m, 1H), 5.07 (d, 1H), 4.79 (d, 2H), 4.72 (d, 1H), 4.11 – 3.81 (m, 11H), 3.82 – 3.72 (m, 1H),
3.51 – 3.40 (m, 1H), 3.14 (d, 1H), 3.05 (d, 1H), 2.80 – 2.55 (m, 2H), 2.50 – 2.29 (m, 4H), 2.30 – 2.14 (m, 4H), 2.09 – 1.98 (m, 2H). 1
9F NMR (376 MHz, MeOD) δ -69.16, -174.26. LCMS: m/z (ESI) [M+H]
+ 592.5, t
R = 1.21 min (Method C) Example 85: 5-((S*)-7-amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 171b)
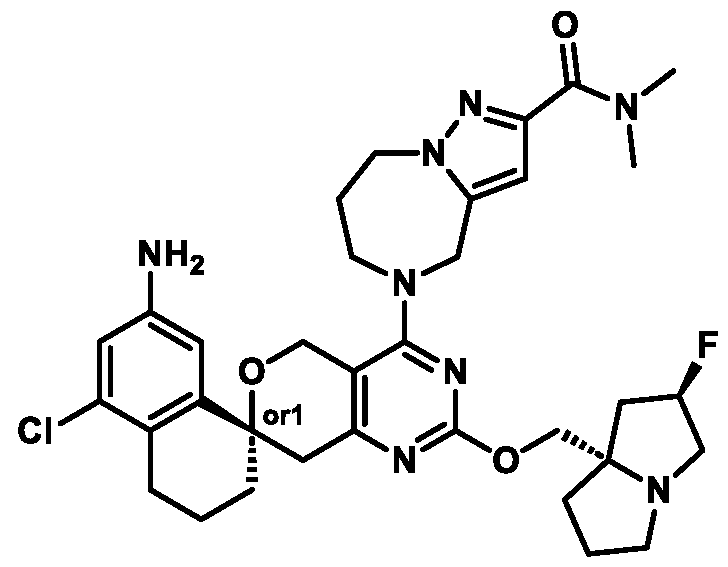
The diastereomers of 5-(7-Amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2- carboxamide (1.08 g, 1.62 mmol) were purified by chiral SFC (ChiralPak AS-H 20 x 250 mm (Mobile Phase: 45% Ethanol, 0.25% diethylamine in CO2; back pressure = 100 bar) to give 5- ((S*)-7-amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl- 5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Peak 1, 325 mg, 489 µmol) and 5-((R*)-7-amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (Peak 2, 338 mg, 508 µmol). Peak 1: LCMS: m/z (ESI) [M+H]
+ 665.3, tR = 1.82 min (ChiralPak AS-H 20 x 250 mm; Mobile Phase: 30% Ethanol, 0.25% diethylamine in CO
2; back pressure = 140 bar) 1H NMR (400 MHz, DMSO-d6) δ 6.61 – 6.57 (m, 2H), 6.50 (s, 1H), 5.37 – 5.02 (m, 3H), 4.78 (d, 1H), 4.75 – 4.65 (m, 2H), 4.54 (d, 1H), 4.49 – 4.39 (m, 2H), 3.97 – 3.75 (m, 4H), 3.24 (s, 3H), 3.13 – 3.03 (m, 2H), 3.00 (s, 1H), 2.93 (s, 3H), 2.88 – 2.76 (m, 3H), 2.68 – 2.51 (m, 2H), 2.14 – 1.61 (m, 12H).
Example 86: 5-((R*)-7-amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5- a][1,4]diazepine-2-carboxamide (Compound 171c)
5-((R*)-7-amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)- N,N-dimethyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (338 mg, 508 µmol) was obtained from Example 85, as Peak 2: LCMS: m/z (ESI) [M+H]
+ 665.3, tR = 2.32 min (ChiralPak AS-H 20 x 250 mm; Mobile Phase: 30% Ethanol, 0.25% diethylamine in CO2; back pressure = 140 bar). 1H NMR (400 MHz, DMSO-d6) δ 6.61 – 6.56 (m, 2H), 6.50 (s, 1H), 5.36 – 5.04 (m, 3H), 4.78 (d, 1H), 4.76 – 4.66 (m, 2H), 4.54 (d, 1H), 4.49 – 4.40 (m, 2H), 3.97 – 3.75 (m, 4H), 3.24 (s, 3H), 3.14 – 3.03 (m, 2H), 3.01 (s, 1H), 2.93 (s, 3H), 2.88 – 2.76 (m, 3H), 2.68 – 2.52 (m, 2H), 2.14 – 1.61 (m, 12H). Example 87: (S*)-1-(7-amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile (Compound 191b)
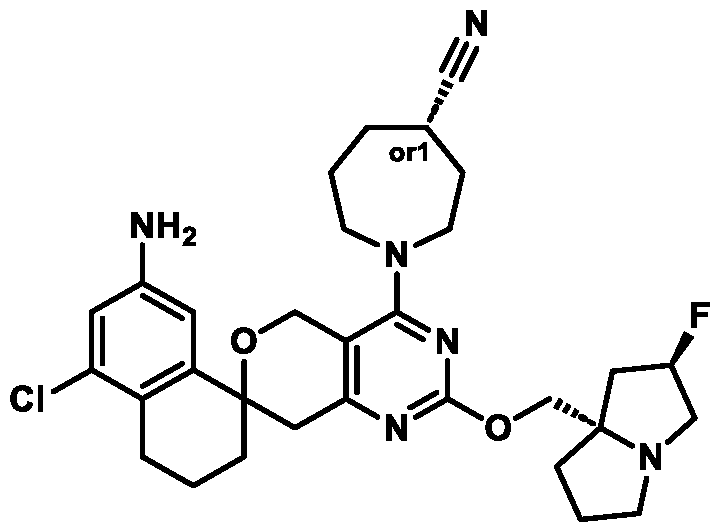
Step 1: 7-Bromo-5-chloro-1-((6-chloro-5-(hydroxymethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-ol LDA (5.42 g, 50.6 mL, 1 M in THF, 2.3 equiv, 50.6 mmol) was added over 15 min to a -78 °C solution of (4-chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (4.50 g, 1 equiv, 22.0 mmol) in THF (20 mL). The mixture was stirred at that temperature for 1 hour. A solution of 7-bromo-5-chloro-3,4-dihydronaphthalen-1(2H)-one (6.28 g, 1.1 equiv, 24.2 mmol) in THF (20 mL) was then added through a syringe over 15 min. The reaction mixture was stirred at that temperature for 1.5 hours. The reaction mixture was quenched with 2 M HCl in ether at -78 °C followed by the addition of a mixture of acetonitrile and saturated ammonium chloride. The reaction mixture was warmed to room temperature. The aqueous layer was extracted with hexanes and ethyl acetate (2:1) (2 x 300 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulphate, filtered and concentrated to give the crude product. The crude product was dissolved in ethyl acetate (10 mL), and a few drops of hexanes were added. The precipitate was filtered, washed with hexanes, and dried to afford 7- bromo-5-chloro-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)methyl)- 1,2,3,4-tetrahydronaphthalen-1-ol (7.40 g) as a light-yellow material. LCMS: m/z (ESI) [M+H]
+ 465.1, t
R = 1.87 minutes (Method B) Step 2: 7-Bromo-4',5-dichloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] 7-Bromo-5-chloro-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-ol (3.5 g, 1 equiv, 7.5 mmol) in toluene (45.0 mL) was heated for 10 min at 120 °C. Phosphoric acid (1.3 g, 0.77 mL, 85% wt, 1.5 equiv, 11 mmol) was then added dropwise to the heated solution. The reaction mixture was heated at 120 °C for 8 hours. The reaction mixture was cooled to room temperature and concentrated to dryness. Ice cold water was added, and the resulting mixture was stirred after which a precipitate formed. Further precipitation was promoted by trituration with ethyl acetate (10-15 mL). The precipitate was filtered and dried to give 7-bromo-4',5-dichloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (2.60 g) as an off-white solid. LCMS: m/z (ESI) [M+H]
+ 447.1, tR = 2.24 minutes (Method B) Step 3: 4'-(Benzyloxy)-7-bromo-5-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]
Benzyl alcohol (0.18 g, 0.17 mL, 1.5 equiv, 1.7 mmol) was added dropwise over 10 minutes to a 0 °C mixture of sodium hydride (90 mg, 60% wt, 2 equiv, 2.2 mmol) in THF (11 mL). 7-bromo-4',5-dichloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine] (0.50 g, 1 equiv, 1.1 mmol) in THF (20 mL) was added dropwise and stirred at 0 °C for 1 hour and at room temperature for 8 hours. The resulting mixture was quenched by addition of a saturated aqueous ammonium chloride (15 mL) at 0 °C. The aqueous layer was extracted with ethyl acetate (2 x 50 mL). The combined organic layers were washed with water, dried over anhydrous sodium sulfate, filtered and concentrated under vacuum to give the crude product which was purified by flash chromatography on silica gel with 90% DCM/hexanes to give 4'-(benzyloxy)-7-bromo-5-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (300 mg) as an off-white solid. LCMS: m/z (ESI) [M+H]
+ 517.1, tR = 2.38 minutes (Method B) Step 4: tert-Butyl (4'-(benzyloxy)-5-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate 4'-(Benzyloxy)-7-bromo-5-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (400 mg, 1 equiv, 772 μmol), cesium carbonate (503 mg, 2.0 equiv, 1.54 mmol), tert-butyl carbamate (181 mg, 2.0 equiv, 1.54 mmol) and N,N-dimethylformamide (10 mL) were added to a flask. Nitrogen was bubbled through the mixture at room temperature for 5 min. BrettPhos Pd G4 (71.1 mg, 0.10 equiv, 77.2 μmol) was added, and nitrogen was bubbled through the mixture at room temperature for another 5 min. The reaction vial is sealed and heated under nitrogen at 90 °C until starting material is consumed. The reaction mixture was cooled to room temperature and filtered through celite and washed with ethyl acetate and MeOH. The crude product was purified by flash chromatography on silica gel with gradient elution from 20% ethyl acetate/hexanes to 45% ethyl acetate/hexanes to afford tert-butyl (4'-(benzyloxy)-5-chloro-2'-(methylthio)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (250 mg) as a white solid. LCMS: m/z (ESI) [M+H]
+ 554.3, tR = 2.27 minutes (Method B) Step 5: tert-Butyl (4'-(benzyloxy)-5-chloro-2'-(methylsulfonyl)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate
Hydrogen peroxide (315 mg, 284 μL, 30% wt, 7 equiv, 2.78 mmol) was added to a mixture of tert-butyl (4'-(benzyloxy)-5-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (220 mg, 1 equiv, 397 μmol), sodium tungstate dihydrate (13.1 mg, 0.10 equiv, 39.7 μmol) and hydrogen tetra(but-1- yl)ammonium sulphate (21.6 mg, 0.16 equiv, 63.5 μmol) in EtOAc (10 mL). The resulting mixture was heated at 40 °C for 40 minutes. The reaction mixture was diluted with water (15 mL). The aqueous layer was extracted with ethyl acetate (2 x 50 ml). The organic layer was washed with brine (3 x 15 mL), dried over anhydrous sodium sulphate, filtered and concentrated to dryness to give tert-butyl (4'-(benzyloxy)-5-chloro-2'-(methylsulfonyl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (200 mg) which was used in the next step without further purification. LCMS: m/z (ESI) [M-H]- 584.2, tR = 2.02 minutes (Method B) Step 6: tert-Butyl (4'-(benzyloxy)-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate A solution of ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (136 mg, 2.5 equiv, 853 μmol) and NaH (40.9 mg, 60% wt, 3 equiv, 1.02 mmol) in DMF (2 mL) was stirred at room temperature for 5 minutes. Then a solution of tert-butyl (4'-(benzyloxy)-5- chloro-2'-(methylsulfonyl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (200 mg, 1 equiv, 341 μmol) in DMF (3 mL) was added dropwise. The resulting mixture was stirred at room temperature for 16 hours. The reaction mixture was cooled to 0 °C and quenched by addition of a saturated solution of ammonium chloride (5 mL). The aqueous layer was extracted with EtOAc (2 x 30 mL) then with DCM (2 x 30 mL). The combined organic layers were washed with water, dried over anhydrous sodium sulfate and concentrated under vacuum to give the crude product. The crude residue was purified by flash chromatography on silica gel with 15% to 20% Methanol/DCM/NH
4OH (9:91:1 ratio) in DCM to afford tert-butyl (4'-(benzyloxy)-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]- 7-yl)carbamate (150 mg) as yellow solid. LCMS: m/z (ESI) [M+H]
+ 665.4, t
R = 2.19 minutes (Method B)
Step 7: tert-Butyl (5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-hydroxy-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate To a solution of tert-butyl (4'-(benzyloxy)-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate (30 mg, 1 equiv, 45 μmol) in tetrahydrofuran (2 mL) was added palladium hydroxide on carbon (6.3 mg, 10% wt, 0.1 equiv, 4.5 μmol). The reaction mixture was degassed under vacuum and then stirred under hydrogen atmosphere at room temperature for 90 minutes. The reaction mixture was filtered through a pad of celite and rinsed with methanol to afford tert-butyl (5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-4'-hydroxy-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (20 mg) which was used in the next step without any further purification. LCMS: m/z (ESI) [M+H]
+ 575.3, t
R = 1.43 minutes (Method B) Step 8: tert-Butyl (5-chloro-4'-((S*)-4-cyanoazepan-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate N,N-diisopropylethylamine (13 mg, 18 μL, 2 equiv, 0.10 mmol) and benzotriazol-1- yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (28 mg, 1.2 equiv, 63 μmol) were added to a solution of tert-butyl (5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-4'-hydroxy-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate (30 mg, 1 equiv, 52 μmol) in DMF (2 mL). The reaction mixture was stirred at room temperature for 5 hours. DIPEA (14 mg, 19 μL, 2.5 equiv, 0.11 mmol) and (S)-azepane-4-carbonitrile hydrochloride (17 mg, 2.5 equiv, 0.11 mmol) in DMF (1 mL) were then added at room temperature. The resulting mixture was stirred at 90 °C for 2 hours. The reaction mixture was concentrated to give the crude product which was purified by reverse phase chromatography eluting with 85-98% acetonitrile in 10 mM ammonium bicarbonate buffer to give tert-butyl (5-chloro-4'-((S*)-4-cyanoazepan-1-yl)-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (15 mg) as an off-white solid. LCMS: m/z (ESI) [M+H]
+ 681.4, tR = 1.92 minutes (Method B)
Step 9: (S*)-1-(7-amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)azepane-4-carbonitrile 4-Methylbenzenesulfonic acid hydrate (3.9 mg, 1 equiv, 21 μmol) was added to a solution of tert-butyl (5-chloro-4'-((S*)-4-cyanoazepan-1-yl)-2'-(((2R,7aS)-2-fluorotetrahydro- 1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-7-yl)carbamate (14 mg, 1 equiv, 21 μmol) in acetonitrile (3.0 mL). The reaction mixture was heated to 70 °C and stirred for 3 hours. The reaction mixture was concentrated and purified by reverse phase chromatography with 65-75% acetonitrile (with 10 mM ammonium bicarbonate) in water to give (S*)-1-(7-amino-5-chloro-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile (3.0 mg) as an off-white solid. LCMS: m/z (ESI) [M+H]
+ 581.4, t
R = 4.55 minutes (Method B) 1H NMR (400 MHz, DMSO-d6) δ 6.59 (s, 2H), 5.26 (d, J = 54.2 Hz, 1H), 5.12 (d, J = 6.7 Hz, 2H), 4.73 (d, J = 14.6 Hz, 1H), 4.57 – 4.45 (m, 1H), 3.99 – 3.90 (m, 1H), 3.89 – 3.80 (m, 1H), 3.57 (d, J = 31.5 Hz, 4H), 3.03 (d, J = 30.4 Hz, 4H), 2.82 (s, 4H), 2.59 (d, J = 6.1 Hz, 2H), 2.08 (s, 2H), 2.00 (s, 2H), 1.94 (d, J = 8.8 Hz, 2H), 1.87 – 1.68 (m, 9H). 1
9F NMR (376 MHz, DMSO) δ -172.12. Example 88: (R*)-1-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-4-methylazepane-4-carbonitrile (Compound 195a)
Step 1: Benzyl 4-cyano-4-methylazepane-1-carboxylate A mixture of benzyl 4-cyanoazepane-1-carboxylate (2000 mg, 1 equiv., 7.74 mmol) in THF (20 mL) was added LiHMDS (1.30 g, 7.74 mL, 1 M, 1 equiv., 7.74 mmol) at -78 °C. The mixture was stirred at -78 °C for 5 mins under N
2 atmosphere. Then MeI (1.65 g, 726 μL, 1.5
equiv., 11.6 mmol) was added to the mixture under N
2. The resulting mixture was stirred at - 78 °C for 0.5 hour under N2 atmosphere. The reaction mixture was quenched by addition of ammonium chloride (30 mL) at 0 °C, and then diluted with H2O (30 mL) and extracted with ethyl acetate (30 mL × 3). The combined organic layers were washed with brine (40 mL × 2), dried over sodium sulfate, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 10/1 to 2/1) to give benzyl 4-cyano-4-methylazepane-1-carboxylate (1 g, 4 mmol) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ = 7.42 - 7.29 (m, 5H), 5.15 (d, 2H), 4.00 - 3.79 (m, 1H), 3.74 - 3.63 (m, 1H), 3.49 - 3.38 (m, 1H), 3.33 - 3.17 (m, 1H), 2.08 - 1.94 (m, 4H), 1.69 - 1.56 (m, 1H), 1.53 - 1.45 (m, 1H), 1.40 (d, 3H). The enantiomers were separated by SFC (Column: Chiralpak IG-350 × 4.6 mm I.D., 3um Mobile phase: Phase A for CO
2, and Phase B for MeOH (0.05% DEA); Gradient elution: B in A from 5% to 40% Flow rate: 3 mL/min; Detector: PDA; Column Temp: 35C; Back Pressure: 100 Bar) to yield rel-benzyl (R)-4-cyano-4-methylazepane-1-carboxylate (Peak 1 tR =1.440 min, 390 mg, 1.43 mmol) and rel-benzyl (S)-4-cyano-4-methylazepane-1-carboxylate (Peak 2 t
R =1.715 min, 400 mg, 1.47 mmol). Step 2: rel-(R)-4-methylazepane-4-carbonitrile To a solution of rel-benzyl (R)-4-cyano-4-methylazepane-1-carboxylate (340 mg, 1 equiv., 1.25 mmol) in DCM (3 mL) was added dropwise TMS-I (1.50 g, 1.02 mL, 6 equiv., 7.49 mmol) at 0 °C. The resulting mixture was stirred at 0 °C for 0.5 hour. The reaction mixture was concentrated under reduced pressure to give a residue. The residue was purified by prep- HPLC (column: Phenomenex luna C18150 × 25 mm × 10 um; mobile phase: [water (TFA)- MeCN]; B%: 0%-5%, 8 min) to give rel-(R)-4-methylazepane-4-carbonitrile, TFA (254. mg, 1.01 mmol) as a yellow solid. LCMS: m/z (ESI) [M+H]
+ 138.8, t
R = 0.099 min 1H NMR (400 MHz, CD3OD) δ = 3.49 - 3.34 (m, 3H), 3.28 - 3.19 (m, 1H), 2.29 - 2.17 (m, 2H), 2.11 - 1.97 (m, 3H), 1.83 - 1.74 (m, 1H), 1.47 (s, 3H)
Step 3: tert-Butyl ((S)-4'-((R*)-4-cyano-4-methylazepan-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate To a 2 dram vial containing a stir bar and tert-butyl ((S)-4'-((1H-benzo[d][1,2,3]triazol- 1-yl)oxy)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (80 mg, 0.12 mmol) was added dry N,N-dimethylformamide (0.7 mL). DIPEA (79 mg, 0.11 mL, 0.61 mmol) and rel-(R)-4-methylazepane-4-carbonitrile (25 mg, 0.18 mmol) were then added to the solution. After 2 hours, more rel-(R)-4-methylazepane-4-carbonitrile (10 mg, 0.015 mmol) was added to the reaction. The reaction was then heated to 90°C for 12 hours. After 12 hours the reaction mixture was cooled to room temperature. The reaction mixture was then purified via reverse phase chromatography (12 g, C-18 Biotage, 5:95 – 95:5 acetonitrile in water) to give tert-butyl ((S)-4'-((R*)-4-cyano-4-methylazepan-1-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-7-yl)carbamate (68 mg, 0.10 mmol) as an off-white solid. LCMS: m/z (ESI) [M+H]
+ = 661.1, tR = 1.86 min (Acquity UPLC, CSH C18, 1.7 μm, 2.1 x 30 mm, Mobile phase A: 10 mM ammonium carbonate in water (v/v); Mobile phase B: 10 mM ammonium carbonate in acetonitrile (v/v); Gradient: 95% H
2O/5% MeCN to 100% MeCN. Flow: 0.9 mL/min.) Step 4: (R*)-1-((S)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-4-methylazepane-4-carbonitrile tert-Butyl ((S)-4'-((R*)-4-cyano-4-methylazepan-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (69 mg, 0.10 mmol) was dissolved in MeCN (2 mL). 4-methylbenzenesulfonic acid hydrate (99 mg, 0.52 mmol) was then added to the mixture and the mixture was heated to 50 °C. After 1 hour the reaction was concentrated and a mixture of NH4OH and MeOH (1:9, 1 mL) was then added to the crude residue. The solution was then concentrated and then purified via reverse phase chromatography (13 g, C-18 RediSep 5:95 – 95:5 acetonitrile in water 10 mM ammonium carbonate buffer) to give (R*)-1-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-
7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]- 4'-yl)-4-methylazepane-4-carbonitrile (25 mg, 45 μmol) as a white solid. LCMS: m/z (ESI) [M+H]
+ = 561.1, tR = 1.57 min (Acquity UPLC, CSH C18, 1.7 μm, 2.1 x 30 mm, Mobile phase A: 10 mM ammonium carbonate in water (v/v); Mobile phase B: 10 mM ammonium carbonate in acetonitrile (v/v); Gradient: 95% H2O/5% MeCN to 100% MeCN. Flow: 0.9 mL/min.) 1H NMR (400 MHz, DMSO-d
6) δ 6.75 (m, 1H), 6.59 (m, 1H), 6.43 (m, 1H), 5.51 – 5.04 (m, 1H), 4.90 – 4.71 (m, 3H), 4.52 (m, 1H), 3.97 (m, 1H), 3.87 – 3.78 (m, 2H), 3.67 – 3.49 (m, 1H), 3.40 (m, 1H), 3.19 – 2.93 (m, 3H), 2.80 (m, 3H), 2.59 (s, 2H), 2.15 – 1.47 (m, 16H), 1.36 (s, 3H). Example 89: (S*)-1-((S)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)-4-methylazepane-4-carbonitrile (Compound 195b)

Step 1: tert-Butyl ((S)-4'-((S*)-4-cyano-4-methylazepan-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate To a 2 dram vial containing a stir bar and tert-butyl ((S)-4'-((1H-benzo[d][1,2,3]triazol- 1-yl)oxy)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'- tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (80 mg, 0.12 mmol) was added dry N,N-dimethylformamide (0.7 mL). DIPEA (79 mg, 0.11 mL, 0.61 mmol) and rel-(S)-4-methylazepane-4-carbonitrile (synthesized analogously to Step 2 of Example 88; Peak 2, 25 mg, 0.18 mmol) were then added to the solution. After 5 hours, the reaction mixture was then cooled to room temperature then purified via reverse phase chromatography (12 g, C-18 Biotage, 5:95 – 95:5 acetonitrile in water) to give tert-butyl ((S)- 4'-((S*)-4-cyano-4-methylazepan-1-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-
7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]- 7-yl)carbamate (70 mg, 0.10 mmol) as an off-white solid. LCMS: m/z (ESI) [M+H]
+ = 661.1, tR = 1.88 min (Acquity UPLC, CSH C18, 1.7 μm, 2.1 x 30 mm, Mobile phase A: 10 mM ammonium carbonate in water (v/v); Mobile phase B: 10 mM ammonium carbonate in acetonitrile (v/v); Gradient: 95% H2O/5% MeCN to 100% MeCN. Flow: 0.9 mL/min.) Step 2: (S*)-1-((S)-7-Amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)-4-methylazepane-4-carbonitrile tert-Butyl ((S)-4'-((S*)-4-cyano-4-methylazepan-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-7-yl)carbamate (70 mg, 0.11 mmol) was dissolved in MeCN (2 mL). 4-Methylbenzenesulfonic acid hydrate (62 mg, 0.33 mmol) was then added to the mixture and the mixture was heated to 50 °C. After 1 hour the reaction was concentrated and a mixture of NH4OH and MeOH (1:9, 1 mL) was then added to the crude residue. The solution was then concentrated and then purified via reverse phase chromatography (15.2 g, Santai C-18 5:95 – 95:5 acetonitrile in water 10 mM ammonium carbonate buffer) to give (S*)-1-((S)-7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]- 4'-yl)-4-methylazepane-4-carbonitrile (30.5 mg, 53 μmol) as a white solid. LCMS: m/z (ESI) [M+H]
+ = 561.1, tR = 1.58 min (Acquity UPLC, CSH C18, 1.7 μm, 2.1 x 30 mm, Mobile phase A: 10 mM ammonium carbonate in water (v/v); Mobile phase B: 10 mM ammonium carbonate in acetonitrile (v/v); Gradient: 95% H
2O/5% MeCN to 100% MeCN. Flow: 0.9 mL/min.) 1H NMR (400 MHz, DMSO-d6) δ 6.77 (m, 1H), 6.55 (m, 1H), 6.45 (m, 1H), 5.26 (m, 1H), 4.75 (s, 2H), 4.66 (m, 1H), 4.52 (m, 1H), 3.96 (m, 1H), 3.90 – 3.77 (m, 2H), 3.65 – 3.34 (m, 3H), 3.14 – 2.97 (m, 3H), 2.82 (m, 3H), 2.67 – 2.52 (m, 2H), 2.19 – 1.47 (m, 16H), 1.35 (s, 3H). Example 90: 2'-(((2R,7aS)-2-Fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine]-8-carbonitrile (Compound 194a)

Step 1: 8-Bromo-4'-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] To a stirring solution of (4-chloro-6-methyl-2-(methylthio)pyrimidin-5-yl)methanol (1.36 g, 6.66 mmol) in THF (11 mL) was added 1 M THF solution of LDA (14.2 mL, 14.2 mmol) dropwise at -78 °C. The resulting mixture was stirred at -78 °C for 1 hour. Afterwards, 8-bromo-3,4-dihydronaphthalen-1(2H)-one (1000 mg, 4.44 mmol) in THF (11 mL) was added dropwise to the reaction mixture. The resulting mixture was then stirred at –78 °C for 1 hour. The mixture was quenched at -78 °C by the slow addition of 2 M diethyl ether solution of HCl (7 mL, 14 mmol). The mixture was warmed to room temperature and the mixture was concentrated to afford a thick yellow oil. The crude was subjected to flash chromatography (80 g silica; 0-100% EtOAc/DCM) to afford 8-bromo-1-((6-chloro-5-(chloromethyl)-2- (methylthio)pyrimidin-4-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-ol, which was used without additional purification. A solution of 8-bromo-1-((6-chloro-5-(hydroxymethyl)-2-(methylthio)pyrimidin-4- yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-ol (780 mg, 1.81 mmol) in toluene (7.3 mL) was added phosphoric acid (124 μL, 85% wt, 1.81 mmol) and the mixture was heated to 85 °C. After 2 hours, the mixture was cooled to room temperature then concentrated. The crude residue was treated with water (26 mL) and EtOAc (26 mL), followed by additional rinsing with both water (26 mL) and EtOAc (26 mL). The aqueous phase was extracted with EtOAc (3 x 26 mL) and the combined organics were dried (sodium sulfate), and concentrated to a residue which was purified by flash chromatography (0-15% EtOAc/ DCM) to afford 8-bromo- 4'-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine] (485 mg, 1.18 mmol). 1H NMR (400 MHz, CDCl3) δ 7.52 (d, 1H), 7.09 (d, 1H), 7.06 – 7.00 (m, 1H), 4.91 (d, 1H), 4.73 (d, 1H), 3.94 (d, 1H), 3.05 – 2.76 (m, 3H), 2.57 (s, 3H), 2.15 – 2.01 (m, 1H), 2.00 – 1.85 (m, 2H), 1.78 – 1.65 (m, 1H). LCMS: m/z (ESI) [M+H]
+ 411.0, tR = 2.38 minutes (Method C)
Step 2: 8-Bromo-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] A mixture of 8-bromo-4'-chloro-2'-(methylthio)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (200 mg, 486 μmol) and 1,4-oxazepane hydrochloride (100 mg, 729 μmol) in ethanol (1.9 mL) was treated with DIPEA (420 μL, 2.41 mmol) and the reaction mixture was heated at reflux overnight. The reaction was cooled to room temperature and concentrated to afford a viscous oil. The oil was dissolved in EtOAc (20 mL) and then washed with aqueous ammonium chloride solution (20 mL). The organic layer was concentrated to a residue which was purified via flash chromatography (0-10% MeOH/DCM) to afford 8-bromo-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro- 2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (198 mg, 0.42 mmol). 1H NMR (400 MHz, CDCl3) δ 7.50 (d, 1H), 7.07 (d, 1H), 7.02 (d, 1H), 4.86 (d, 1H), 4.78 (d, 1H), 3.94 – 3.56 (m, 9H), 2.96 – 2.78 (m, 3H), 2.50 (s, 3H), 2.29 – 2.18 (m, 1H), 2.15 – 2.03 (m, 1H), 2.02 – 1.85 (m, 3H), 1.83 – 1.72 (m, 1H). LCMS: m/z (ESI) [M+H]
+ 476.0, tR = 1.63 minutes (Method C) Step 3: 2'-(Methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile A mixture of 8-bromo-2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine] (170 mg, 357 μmol) and copper(I) cyanide (320 mg, 3.57 mmol) in DMF (7.1 mL) was degassed for 3 min followed by stirring at 120 °C. After 2 hours, the mixture was cooled to room temperature and concentrated. Water (10 mL) and NH4OH (10 mL) were added, followed by DCM (10 mL). The mixture was sonicated to dissolve the insoluble precipitate. The phases were separated and the aqueous solution was washed with 5:1 DCM/IPA solution (3 x 5 mL). The combined organic layers were washed with brine (10 mL), dried (sodium sulfate), filtered and concentrated to an oil which was purified via flash chromatography (4 g silica, 0-20% EtOAc/DCM) to afford 2'-(methylthio)- 4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine]-8-carbonitrile (184 mg, 0.44 mmol, contains residual DCM) as an off-white solid. LCMS: m/z (ESI) [M+H]
+ 423.2, t
R = 1.54 minutes (Method C)
Step 4: 2'-(Methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile 3-Chlorobenzoperoxoic acid (286 mg, 77% wt, 1.28 mmol) was added to the solution of 2'-(methylthio)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine]-8-carbonitrile (180 mg, 426 μmol) in DCM (8.5 mL) at room temperature. The resulting mixture was stirred at room temperature for 1 hour, then treated with saturated aqueous sodium thiosulfate. After stirring for 5 min, the organic layer was separated, and the aqueous phase was extracted with DCM (3 x 5 mL). The combined organic layers were washed with saturated aqueous sodium bicarbonate solution, dried (magnesium sulfate), filtered and concentrated to afford 2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)- 3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (200 mg, 0.44 mmol, contains residual DCM). LCMS: m/z (ESI) [M+H]
+ 455.3, tR = 1.72 minutes (Method C) Step 5: 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-4'- (1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine]-8-carbonitrile A mixture of ((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methanol (207.3 mg, 1.302 mmol) and 2'-(methylsulfonyl)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (197 mg, 434 μmol) in DMF (8.7 mL) was cooled to 0 °C, followed by the dropwise addition of lithium bis(trimethylsilyl)amide (580 μL, 1.5 M, 870 μmol). The mixture was then stirred at room temperature for 2 hours. The reaction was quenched by the slow addition of a saturated aqueous NH₄Cl solution (10 mL) at 0 °C. The mixture was treated with EtOAc (10 mL) and the layers were separated. The aqueous phase was extracted with EtOAc (3 x 5 mL). The combined organic extracts were dried over magnesium sulfate, filtered and concentrated to a residue which was purified by flash chromatography (4 g silica, 60:10:10:10, EtOAc/MeOH/EtOAc/H
2O) to afford the partially pure product. The product was then subjected to purification by flash chromatography [4 g silica, 0-30% (2.5% NH
4OH and 20% MeOH in DCM)/DCM] to afford 2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-(1,4-oxazepan-4-yl)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidine]-8-carbonitrile (10.5 mg, 20 μmol) as a brown oil.
1H NMR (400 MHz, CDCl
3) δ 7.65 – 7.59 (m, 1H), 7.52 – 7.38 (m, 2H), 5.41 – 5.12 (m, 1H), 4.60 (dd, 1H), 4.28 (dd, 1H), 4.11 (dd, 1H), 4.02 – 3.92 (m, 1H), 3.92 – 3.52 (m, 9H), 3.29 – 3.16 (m, 2H), 3.18 – 3.03 (m, 2H), 3.01 – 2.90 (m, 1H), 2.88 – 2.70 (m, 1H), 2.43 – 2.09 (m, 5H), 2.07 – 1.66 (m, 8H). 1
9F NMR (376 MHz, CDCl3) δ -173.08, -173.17. Example 91: 7-amino-4'-((S*)-4-cyanoazepan-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Compound 192a)
To a solution of 7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-hydroxy-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine]-8-carbonitrile (130 mg, 279 μmol) in DMF (5.6 mL) at room temperature, were added DIPEA (97 μL, 0.56 mmol, 2.0 equiv.) and benzotriazole-1-yl-oxy-tris- (dimethylamino)-phosphonium hexafluorophosphate (148 mg, 335 μmol, 1.2 equiv.) sequentially. The reaction mixture was stirred for 2 hours, followed by the sequential addition of DIPEA (97 μL, 0.56 mmol, 2.0 equiv.) and rel-(S)-azepane-4-carbonitrile hydrochloride (112 mg, 698 μmol, 2.5 equiv.). The reaction mixture was stirred at 90
oC for 2 hours. The mixture was cooled to room temperature and concentrated under reduced pressure. The crude material was purified by reverse phase chromatography (Biotage C1812 g, 10 mL/min, 0.1% aq. NH
4HCO
3 / MeCN, 10 to 80%) to give 7-amino-4'-((S*)-4-cyanoazepan-1-yl)-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile which was submitted directly for separation of stereoisomers. LC-MS: m/z (ESI) [M+H]
+ 572.4 [M+1]
+, tR = 4.03 min (51%) and tR = 4.09 min (44%) (Method D)
Example 92: (S*)-7-amino-4'-((S*)-4-cyanoazepan-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Compound 192b)
The diastereomers of 7-amino-4'-((S*)-4-cyanoazepan-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile were separated by chiral SFC prep chromatography (column: Cellulose-1 (250 x 30 mm, 5 μm); mobile phase: 50% ethanol + 0.1% diethylamine in CO
2, flow rate: 120 mL/min; back pressure: 100 bar; heat exchanger temperature: 40 ºC; concentration: 10.2 mg/mL; injection: 2.0 mL) to yield (S*)-7-amino-4'- ((S*)-4-cyanoazepan-1-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8- carbonitrile (Peak 1) (27.8 mg, 49.6 μmol) and (R*)-7-amino-4'-((S*)-4-cyanoazepan-1-yl)-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Peak 2) (25.4 mg, 44.4 μmol). Peak 1: LC-MS: m/z (ESI) [M+H]⁺ 572.4, tR = 1.46 minutes, (Method B) 1H NMR (400 MHz, DMSO-d6) δ 7.04 (d, 1H), 6.72 (d, 1H), 5.73 (br s, 2H), 5.25 (br d, 1H), 4.90 (d, 1H), 4.70 (d, 1H), 3.94 (d, 1H), 3.85 (d, 1H), 3.78 – 3.69 (m, 1H), 3.69 – 3.60 (m, 1H), 3.59 – 3.47 (m, 2H), 3.17 – 3.04 (m, 4H), 3.03 – 2.95 (m, 1H), 2.88 – 2.77 (m, 2H), 2.63 – 2.56 (m, 2H), 2.21 – 2.11 (m, 2H), 2.11 – 2.05 (m, 1H), 2.05 – 1.58 (m, 13H). 1
9F NMR (376 MHz, DMSO-d6) δ -172.03. Example 93: (R*)-7-amino-4'-((S*)-4-cyanoazepan-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Compound 192c)
(R*)-7-amino-4'-((S*)-4-cyanoazepan-1-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine]-8-carbonitrile was obtained from Example 92, as Peak 2: SFC: column: Cellulose-1 (100 x 4.6 mm); mobile phase: 50% ethanol + 0.1% diethylamine in CO2; flow rate: 4 mL/min; backpressure: 100 bar; temperature: 40 °C; injection: 14 µL, t
R = 2.76 minutes. LC-MS: m/z (ESI) [M+H]⁺ 572.4, t
R = 1.48 minutes, (Method B) 1H NMR (400 MHz, DMSO-d
6) δ 7.04 (d, 1H), 6.72 (d, 1H), 5.73 (br s, 2H), 5.25 (br d, 1H), 4.90 (d, 1H), 4.70 (d, 1H), 3.96 (d, 1H), 3.84 (d, 1H), 3.75 – 3.64 (m, 2H), 3.62 – 3.52 (m, 1H), 3.50 – 3.40 (m, 1H), 3.13 – 2.96 (m, 5H), 2.87 – 2.77 (m, 2H), 2.64 – 2.55 (m, 2H), 2.22 – 1.61 (m, 16H). 1
9F NMR (376 MHz, DMSO) δ -172.07. Example 94: (S*)-7-Amino-4'-((R*)-4-cyanoazepan-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Compound 192d)
To a solution of 7-amino-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-4'-hydroxy-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine]-8-carbonitrile (220 mg, 473 μmol) in DMF (9.5 mL) at room temperature, were added DIPEA (0.17 mL, 0.95 mmol, 2.0 equiv.) and benzotriazole-1-yl-oxy-tris- (dimethylamino)-phosphonium hexafluorophosphate (251 mg, 567 μmol, 1.2 equiv.)
sequentially. The reaction mixture was stirred for 3 hours, followed by the sequential addition of DIPEA (0.17 mL, 0.95 mmol, 2.0 equiv.) and rel-(R)-azepane-4-carbonitrile hydrochloride (190 mg, 1.18 mmol, 2.5 equiv.). The reaction mixture was stirred at 90
oC for 10 hours. The mixture was then cooled to room temperature and concentrated under reduced pressure. The crude material was purified by reverse phase chromatography (Biotage C1830 g, 15 mL/min, 0.1% aq. NH4HCO3 / MeCN, 10 to 80%) to give 7-amino-4'-((R*)-4-cyanoazepan-1-yl)-2'- (((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile which was subjected to chiral SFC prep chromatography to yield (S*)-7-Amino-4'-((R*)-4-cyanoazepan-1-yl)-2'-(((2R,7aS)- 2-fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Peak 1) (8.8 mg, 15 μmol) and (R*)-7-amino-4'-((R*)-4-cyanoazepan-1-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine]-8-carbonitrile (Peak 2) (6.0 mg, 10 μmol) Peak 1: SFC: column: Cellulose-1 (100 x 4.6 mm); mobile phase: 50% ethanol + 0.1% diethylamine in CO2; flow rate: 4 mL/min; backpressure: 100 bar; temperature: 40 °C; injection: 14 µL, t
R = 1.71 minutes. LC-MS: m/z (ESI) [M+H]⁺ 572.3, t
R = 1.47 minutes, (Method B) 1H NMR (400 MHz, DMSO-d
6) δ 7.01 (d, 1H), 6.69 (d, 1H), 5.70 (br s, 2H), 5.22 (d, 1H), 4.87 (d, 1H), 4.66 (d, 1H), 3.91 (d, 1H), 3.84 (d, 1H), 3.71 – 3.61 (m, 2H), 3.59 – 3.48 (m, 1H), 3.47 – 3.36 (m, 1H), 3.10 – 2.91 (m, 5H), 2.85 – 2.74 (m, 2H), 2.61 – 2.53 (m, 2H), 2.19 – 2.11 (m, 1H), 2.11 – 1.57 (m, 15H). 1
9F NMR (376 MHz, DMSO-d6) δ -172.03. Example 95: (R*)-7-amino-4'-((R*)-4-cyanoazepan-1-yl)-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidine]-8-carbonitrile (Compound 192e)
(R*)-7-amino-4'-((R*)-4-cyanoazepan-1-yl)-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidine]-8-carbonitrile was obtained from Example 94, as Peak 2: SFC: tR = 2.36 minutes, Cellulose-1 (100 x 4.6 mm); mobile phase: 50% ethanol + 0.1% diethylamine in CO
2; flow rate: 4 mL/min; backpressure: 100 bar; temperature: 40 °C; injection: 14 µL. LC-MS: m/z (ESI) [M+H]⁺ 572.3, tR = 1.47 minutes (Method B) 1H NMR (400 MHz, DMSO-d6) δ 7.01 (d, 1H), 6.69 (d, 1H), 5.70 (br s, 2H), 5.22 (br d, 1H), 4.87 (d, 1H), 4.67 (d, 1H), 3.92 (d, 1H), 3.81 (d, 1H), 3.75 – 3.66 (m, 1H), 3.66 – 3.57 (m, 1H), 3.57 – 3.44 (m, 2H), 3.15 – 2.92 (m, 5H), 2.85 – 2.74 (m, 2H), 2.66 – 2.50 (m, 2H), 2.18 – 2.03 (m, 3H), 2.02 – 1.86 (m, 4H), 1.85 – 1.57 (m, 9H). 1
9F NMR (376 MHz, DMSO) δ -172.06. Example 96: (S*)-1-((R)-7-amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile (Compound 191c)
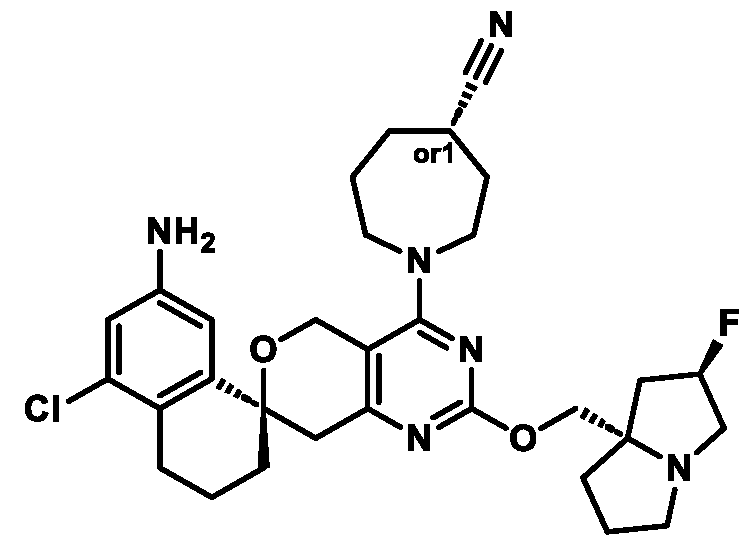
The diastereomers of (S*)-1-(7-amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3- d]pyrimidin]-4'-yl)azepane-4-carbonitrile (25 mg) were separated by chiral SFC using Lux Column 5 µm, Cellulose 1, 150 x 21 mm, 9.0 mg/mL, 13.5 mg/inj (mobile phase: 50% methanol, 0.1% diethylamine) (S*)-1-((R)-7-amino-5-chloro-2'-(((2R,7aS)-2- fluorotetrahydro-1H-pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H- spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile (Peak 1) (6.5 mg) and (S*)-1-((S)-7-amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin- 7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]- 4'-yl)azepane-4-carbonitrile (Peak 2) (8.2 mg). Peak 1: SFC: (Cellulose 1, 100 x 4.6 mm, 5 µm; mobile phase: 50% methanol, 0.1% diethylamine) t
R = 4.03 minutes
LCMS: m/z (ESI) [M+H]
+ 581.4, t
R = 4.54 minutes (Method B) 1H NMR (400 MHz, DMSO-d6) δ 6.59 (s, 2H), 5.26 (m, 1H), 5.12 (s, 2H), 4.73 (m, 1H), 4.51 (m, 1H), 3.94 (m, 1H), 3.85 (m, 1H), 3.57 (m, 4H), 3.07 (s, 3H), 2.99 (s, 1H), 2.82 (s, 3H), 2.67 – 2.55 (m, 2H), 2.08 (s, 2H), 2.02 – 1.92 (m, 5H), 1.87 – 1.70 (m, 9H). Example 97: (S*)-1-((S)-7-amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H- pyrrolizin-7a(5H)-yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'- pyrano[4,3-d]pyrimidin]-4'-yl)azepane-4-carbonitrile (Compound 191d)
(S*)-1-((S)-7-amino-5-chloro-2'-(((2R,7aS)-2-fluorotetrahydro-1H-pyrrolizin-7a(5H)- yl)methoxy)-3,4,5',8'-tetrahydro-2H-spiro[naphthalene-1,7'-pyrano[4,3-d]pyrimidin]-4'- yl)azepane-4-carbonitrile (8.2 mg) was obtained from Example 96, as Peak 2: SFC: (Cellulose 1, 100 x 4.6 mm, 5 µm; mobile phase: 50% methanol, 0.1% diethylamine) t
R = 6.43 minutes LCMS: m/z (ESI) [M+H]
+ 581.4, tR = 4.60 minutes (Method B) 1H NMR (400 MHz, DMSO-d6) δ 6.59 (s, 2H), 5.26 (m, 1H), 5.10 (s, 2H), 4.73 (m, 1H), 4.52 (m, 1H), 3.96 (m, 1H), 3.83 (m, 1H), 3.66 – 3.48 (m, 4H), 3.07 (m, 3H), 2.99 (s, 1H), 2.82 (s, 3H), 2.58 (m, 2H), 2.09 (d, 2H), 2.04 – 1.91 (m, 5H), 1.87 – 1.67 (m, 9H). Example B1. Surface Plasmon Resonance Methods herein pertain to Cytiva (formerly Biacore) instrumentation and consumables and are applicable to other SPR-based systems. A Series S Streptavidin or Neutravidin chip is docked into a Biacore 8K. The instrument is then primed into an appropriate running buffer (for example, 20 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM MgCl
2, 1 mM TCEP, 0.005% Tween-20, and 2% DMSO) supplemented with either 5 µM GDP or GMPPNP to match the nucleotide state of the KRas state being tested (GDP for KRas in the GDP-bound state; and GMPPNP for KRas in the GTP-bound state). Biotinylated-KRas loaded with the appropriate nucleotide state (e.g., GDP or GMPPNP) is then captured to between 500-2000 RU to provide adequate signal for small molecules of interest, typically in a range of 300-600 Da, as well as
to accommodate a range of potencies, when appropriate. The remaining biotin binding sites can be blocked with Biocytin. Compounds are injected over both a reference surface (Streptavidin or Neutravidin alone) as well as the active surface (KRas captured to Neutravidin or Streptavidin), and all curves are double-referenced before analysis (specifically, a signal subtraction of the reference flow cell from the active flow cell as well as a buffer subtraction). Where appropriate, a solvent correction is applied to all curves to account for differential bulk effects from DMSO on the reference and active surfaces. Data is then analyzed using the Biacore software fitting kinetics if resolvable, or fitting via a Langmuir isotherm model if no kinetics are resolvable, to report a binding affinity as a dissociation constant, KD. Example B2. SOS1-Catalyzed Nucleotide Exchange Assay KRas G12R and WT: Compounds are pre-dispensed using acoustic transfer technology into a black, low volume 384-well assay plate. A 10-point dose response of each compound is performed with a 30 µM top dose. Biotinylated KRas WT(1-169) or KRas G12R (1-169) loaded with GDP nucleotide is mixed with Streptavidin-Tb cryptate (Cisbio) in assay buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 2 mM MgCl2, and 0.005% CA680) to produce a 1.5x solution. 10 µL of the 1.5x KRas-cryptate solution is added to wells of a black, low- volume 384-well assay plate. The KRas/cryptate-compound mixture is incubated for 1 hour at room temperature. A 3x solution of SOS1 (564-1049) and EDA-GTP-DY-647P1 (Jena Bioscience) is prepared in assay buffer.5 µL of the SOS1-labeled GTP solution is added to the wells to initiate the nucleotide exchange reaction. The final concentration of KRas G12R and SOS1 are 10 nM and 200 nM, respectively. The final concentration of KRas WT and SOS1 are 20 nM and 10 nM, respectively. The plate is allowed to incubate for 1 hr, then time resolved fluorescence is read on a PHERAstar plate reader equipped with a filter module with excitation = 337 nm and emission 1 = 620 nm, emission 2 = 665 nm. The TR-FRET signal is calculated as the ratio of fluorescence intensity [emission 665 nm]/[emission 620 nm]. IC50 values are calculated using a four-parameter, variable response sigmoidal dose response curve fit in PerkinElmer Signals VitroVivo. KRas G12V [Method 1]: Biotinylated KRas G12V (aa 1-169) loaded with GDP nucleotide and Streptavidin-Europium (SA-EU, Columbia Biosciences) were preincubated in assay buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 0.01% Brij- 35, 0.3 mg/mL BSA) on ice for 30 min. Separately SOS1 (aa 546-1049) and EDA-GTP-
DY647P1 (Jena Bioscience) were preincubated in assay buffer on ice for 30 min. After pre- incubation, 5 µL buffer and 5 µL of KRas G12V-SA-Eu were added to each well of a white, low-volume, non-binding 384-well plate. Compounds were then added to respective wells with a Tecan D300e liquid dispenser. A 10-point concentration response curve for each compound was prepared with appropriate top concentration (1 µM, 10 µM, or 300 µM) and incubated for 30 min.5 µL of SOS1-EDA-GTP-D467P1 was added to start the nucleotide exchange reaction. Final concentration in the assay were 5 nM KRas G12V, 50 nM SOS1 (aa 564-1049). The reaction was allowed to proceed for 60-90 minutes after which the plate was read on a PHERAstar FSX plate reader (exc: 337 nm, em 1: 665 nm, em 2: 620 nm). After normalization of the raw data (ratio of Signal
665nm/Signal
620nm) the data was fit to a four-parameter logistic curve in GraphPad Prism (v9.4.1). The results are summarized in Table B1, below: Table B1.
Notes: (1) "++++": IC50 ≤ 100
µM < IC
50 ≤ 100 µM KRas G12V [Method 2]: Compounds were pre-dispensed via Echo liquid handling (acoustic, touch-free) to generate assay ready plates (ARP). More precisely, a 10-point concentration response curve for each compound was prepared with appropriate top concentration (1 µM, 10 µM, 100 µM). Biotinylated KRas G12V (aa 1-169) loaded with GDP nucleotide and Streptavidin-Europium (SA-EU, Columbia Biosciences) were preincubated in assay buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 5 mM MgCl
2, 1 mM DTT, 0.01% Brij-
35, 0.3 mg/mL BSA) on ice for 30 min. Separately SOS1 (aa 546-1049) and EDA-GTP- DY647P1 (Jena Bioscience) were preincubated in assay buffer on ice for 30 min. After 30 minutes of incubation for the assay components, 5 µL buffer and 5 µL of KRas G12V-SA-Eu were added to each well of the ARP. After a further 30 min, 5 µL of SOS1-EDA-GTP-D467P1 was added to start the nucleotide exchange reaction. Final concentration in the assay were 5 nM KRas G12V, 50 nM SOS1 (aa 564-1049). The reaction was allowed to proceed for 60-90 minutes after which the plate was read on a plate reader. After normalization of the raw data, the data was fit to a four-parameter logistic curve. For some compounds, data was collected at multiple sites, and the geometric mean value is presented in Table B2 below. The results are summarized in Table B2, below: Table B2.
(2) “#” indicates that an IC50 value could not be determined under the assay conditions. (3) “*”: Data revised compared to U.S. Provisional Application Serial No.63/456,235, 63/515,290, 63/533,346, and 63/535,006. (4) “**”: Data revised compared to U.S. Provisional Application Serial No.63/533,346
Example B3. KRas-cRAF Protein-Protein Interaction (PPI) Assay Compounds are pre-dispensed using acoustic transfer technology into a black, low volume 384-well assay plate. A 10-point dose response of each compound is performed with a 30µM top dose. Biotinylated KRas protein (e.g., KRas G12R(1-169)) is loaded with GppNHp (i.e., GMPPNP) nucleotide and GST-cRAF(1-149) are diluted to 90 nM and 30 nM, respectively, in assay buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 5 mM MgCl
2, and 0.005% CA680).5 µL of KRas protein is added to wells of a black, low-volume 384-well assay plate. The KRas-compound mixture is incubated for 30 minutes at room temperature.5 µL of GST- cRAF protein is added to the KRas-compound mixture and incubated for 30 minutes at room temperature. 100x stocks of Tb cryptate-labeled anti-GST antibody (Anti-GST-Tb) (Cisbio) and Streptavidin-XL655 (Cisbio) are used to make a 3x detection mixture in a total volume of 5 µL of assay buffer. The detection mixture is added to the assay wells and incubated an additional 1 hour at room temperature. Time resolved fluorescence is read on a PHERAstar plate reader equipped with a filter module with excitation = 337 nm and emission 1 = 620 nm, emission 2 = 665 nm. The TR-FRET signal is calculated as the ratio of fluorescence intensity [emission 665 nm]/[excitation 337 nm]. IC
50 values are calculated using a four-parameter, variable response sigmoidal dose response curve fit in PerkinElmer Signals VitroVivo. Example B4. KRas-cRAF Protein-Protein Interaction (PPI) Assay Compounds are pre-dispensed via Echo liquid handling (acoustic, touch-free) to generate assay ready plates (ARP). A 10-point dose response of each compound is performed with a 30µM top dose. Biotinylated KRas G12V (aa 1-169) loaded with GMPPNP nucleotide and Streptavidin-Europium (SA-EU, Columbia Biosciences) are preincubated in assay buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 0.01% Brij-35, 0.3 mg/ml BSA) on ice for 30 min. Separately GST-tagged cRAF(1-149) and anti-GST-APC antibody (Columbia Biosciences) are preincubated in assay buffer on ice for 30min. After incubation, 5 µL buffer and 5 µL of KRas G12V-SA-Eu are added to each well of the ARP. After a 30- minute equilibration phase at room temperature, 5 uL of GST-cRAF-anti-GST-APC is added to each well. The final concentrations in the assay are 10 nM KRas G12V, 10 nM cRAF. The reaction is allowed to proceed for 60-90 minutes after which the plate is read on a PHERAstar FSX plate reader (exc: 337 nm, em 1: 665 nm, em 2: 620 nm). After normalization of the raw
data (ratio of Signal
665nm/Signal
620nm) the data is fit to a four-parameter logistic curve in GraphPad Prism (v9.4.1). Example B5. Surface Plasmon Resonance (SPR) Biacore 8K buffer line was placed into 1 L of Immobilization buffer (20 mM HEPES pH 7.5 / 150 mM NaCl / 5 mM MgCl2 / 0.5 mM TCEP / 0.005% P20 / 500 nM GDP) and Change Solutions was performed. A Biacore Series S SA chip (Cytiva 29104992) was inserted and Change Solutions was performed 3 times. The chip surface was conditioned by injecting conditioning solution (40 mM NaOH / 1 mM NaCl) in four pulses of 30 seconds at 30 uL/min. These pulses were followed by pulses of running buffer injected for 30 seconds at 30 uL/min. Normalization was run using the BiaNormalize solution (Cytiva 29207950). 300 nM Biotinylated KRas G12V 1-169 was injected over all active flow cells at a flow rate of 5 uL/min until reaching a response of 1000 RUs. The buffer line was switched into Running Buffer (20 mM HEPES pH 7.5 / 150 mM NaCl / 5 mM MgCl
2 / 0.5mM TCEP / 0.005% P20 / 500 nM GDP / 2% DMSO) and Change Solutions was performed. Compounds were plated into a Greiner v-bottom 384-well Microplate (Greiner 781280) in 8-point dose-response with a log- fold dilution with a final volume of 2 uL in DMSO.98uL of non-DMSO containing buffer was backfilled into all compound wells (final volume of 100 uL with 2% DMSO). Solvent corrections of 1%, 1.5%, 2%, 2.5%, and 3% DMSO were plated into a Greiner U-bottom 96- well microplate (Greiner 650201). The SPR method was run with both flow cell and sample compartment at 20 °C with 5 startup cycles of 30 second association, and 30 second dissociation at a flow rate of 70 uL/min. An eight-point single cycle kinetics (SCK) analysis was run with 30 second association time, 1600 second dissociation time, 70 uL/min flow rate, 1600 sec stability period, and a 50% DMSO wash after injection. Solvent corrections were run at the end of the method. Results were analyzed with the Biacore Insights Evaluation Software using the Single Cycle Kinetics analysis method. Sensorgrams were fit using a 1:1 binding model. K
D values, as an average of multiple runs, if applicable, are presented in Table B3 with two significant figures. Table B3.
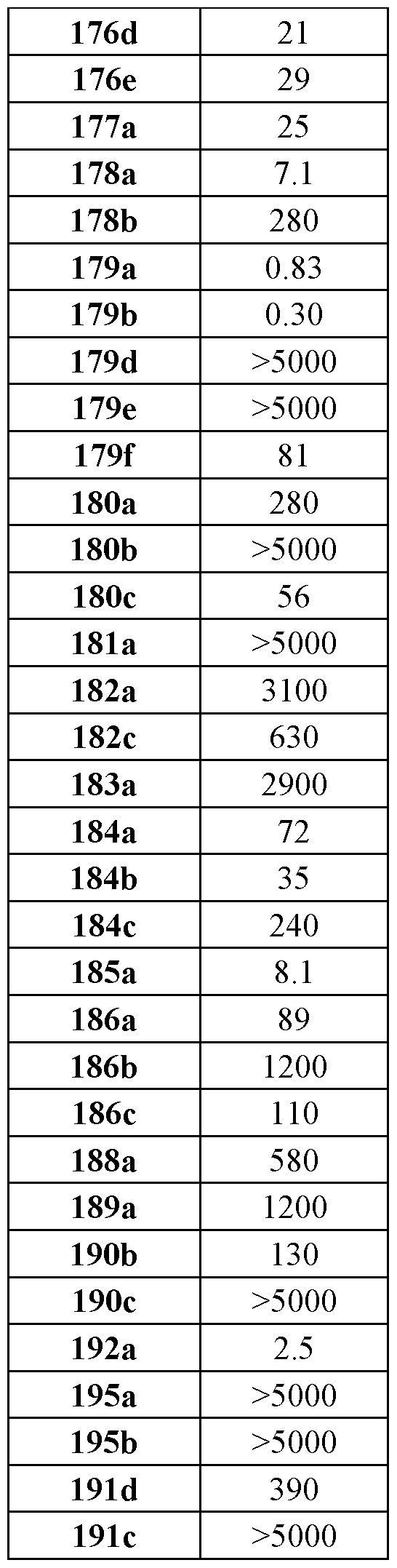
Example B6. Measurement of cellular phospho- and total-ERK1/2 In a white, opaque-bottom Revvity CulturPlate-384, SW620 [SW-620] (ATCC CCL- 227) or GP2d (Sigma, CB_95090714) cells were seeded at previously determined seeding densities such that the well would be approximately 80% confluent at the end of the assay, and incubated overnight in a standard 37 °C, 5% CO2 humidified incubator. The day after seeding, compounds were dispensed into the treatment plates using a Tecan D300e compound printer in 9-point DRC format (1:3 dilution), 10-micromolar top concentration, in triplicate. Treatment plates were then returned to a 37 °C, 5% CO2 humidified incubator for 2 hours
unless otherwise stated. Following compound treatment, phosphoERK and total ERK were measured using the Alpha SureFire Ultra Multiplex Phospho/Total ERK1/2 Assay Kit (Revvity). All media was removed from the treatment plate(s) and the cells subsequently lysed using 1X Lysis Buffer in accordance with manufacturer protocol. Next, the Acceptor Mix (prepared in accordance with manufacturers protocol) was added to each well of the assay plate and incubated on an orbital shaker at room temperature for 2 hours. Following Acceptor incubation, the Donor Mix (prepared in accordance with manufacturer protocol) was added to each well of the assay plate, covered to protect from light and incubated on an orbital shaker at room temperature overnight. Assay plates were read the following day on a BMG Labtech PHERAstar FSX microplate reader. Data were then analyzed by calculating the ratio of ERK1/2-phosphorylation relative to Total ERK1/2 for each individual well.

The replicate ratios for each concentration were averaged and normalized to DMSO control or other corresponding co-treatment before performing a variable slope (4-parameter), non-linear regression curve fit for each compound of interest. IC
50 values, as a geometric mean of multiple runs, if applicable, are presented in from this assay are presented in Table B4 with two significant figures. Table B4.

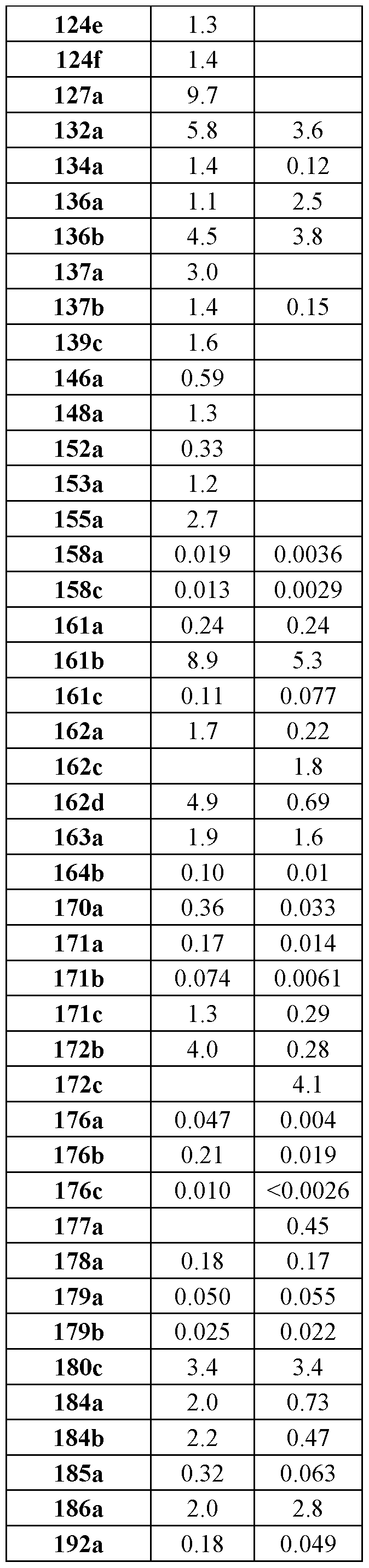
Example B7. Measurement of cellular proliferation In black, clear-bottom 384-well tissue culture treated plates, SW620 [SW-620] (ATCC CCL-227) cells were seeded at previously determined seeding densities such that the well would be approximately 80% confluent at the end of the assay. Immediately after seeding, compounds were dispensed into the treatment plates using a Tecan D300e compound printer in 9-point DRC format (1:3 dilution), 10- micromolar top concentration, in triplicate. Treatment plates were then transferred to a 37 °C, 5% CO2 humidified incubator for 72 hours unless otherwise stated. After the completion of treatment, CELLTITER-GLO® 2.0 reagent (Promega) was added directly to each well. The plates were covered to protect from light and incubated on an orbital shaker at room temperature for 30 minutes. Immediately before reading the assay plates, an opaque seal was attached to the bottom of each clear-bottom plate; assay plates were then read on a BMG Labtech PHERAstar FSX microplate reader. Data were analyzed by calculating the ratio of signal from treated wells relative to negative control-treated wells. The replicates were averaged and a variable slope (4-parameter), non-linear regression curve was fitted for each compound of interest. IC50 values, as a geometric mean of multiple runs, if applicable, are presented in from this assay are presented in Table B5 with two significant figures. Table B5.

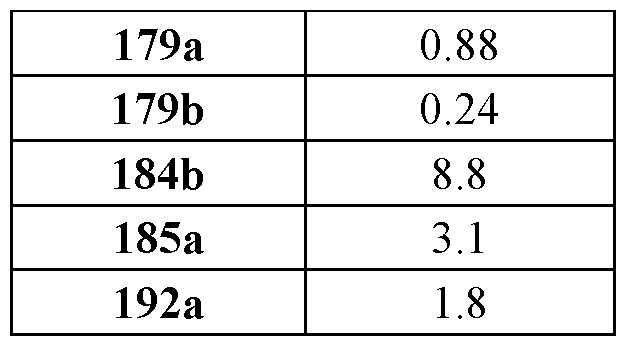
Example B8. Protein Production A KRas construct of amino acid residues 1-169, harboring the G12V mutation, a C- terminal Avi tag, and an N-terminal 6x Histidine tag followed by a tobacco etch virus (TEV) protease site was expressed in E. coli BL21(DE3). A single colony was inoculated in 100 mL LB with 100 µg/mL ampicillin and incubated at 37 °C overnight on an orbital shaker at 225 rpm. The overnight strain was inoculated into 2L TB with 100 µg/mL ampicillin at a ratio of 1:100. The culture was grown at 37 °C on an orbital shaker until the OD
600 reached 0.8. The culture was cooled to 18 °C, induced with IPTG at a final concentration of 0.2 mM and grown 16 hours at 18 °C on an orbital shaker at 180 rpm. Cells were harvested at 13,600 x g for 5 minutes at 4 °C. The KRas G12V protein was purified by affinity chromatography using NiNTA resin. The cell pellet resuspended in 20 mM Tris pH 8.0, 500 mM NaCl, 2 mM beta-mercaptoethanol supplemented with EDTA-free protease inhibitor (Roche). Cells were sonicated at a power of 350 W with 3 s on and 3 s off pulses for a total of 300 cycles. The lysate was centrifuged at 13,600 x g at 4 °C for 30 min. The supernatant was collected and centrifuged a second time with the same parameters.5 mL NiNTA resin was pre-equilibrated with 20 mM Tris pH 8.0, 500 mM NaCl, 2 mM beta-mercaptoethanol, added to the cleared lysate, and incubated for 2 h at 4 °C on a rotator. The resin was washed in succession with 20 mM Tris pH 8.0, 500 mM NaCl, 10 mM imidazole, 2 mM beta-mercaptoethanol, and then 20 mM Tris pH 8.0, 500 mM NaCl, 20 mM imidazole, 2 mM beta-mercaptoethanol. Finally, the protein was eluted from the resin using 20 mM Tris pH 8.0, 500 mM NaCl, 250 mM imidazole, 2 mM beta- mercaptoethanol. The 6x Histidine tag was removed from the KRas G12V protein by overnight incubation with TEV protease at 4 °C while dialyzing against 20 mM HEPES pH 7.5, 150 mM NaCl. The Avi tag on the C terminus was specifically labeled with biotin using recombinant BirA enzyme during the tag cleavage process. 16 mg of recombinant BirA enzyme, 1 mM biotin, 7 mM ATP, and 7.5 mM MgCl
2 was added to the 4 °C overnight reaction. KRas G12V protein was subsequently mixed with NiNTA resin pre-equilibrated with 20 mM HEPES pH 7.5, 150 mM NaCl – the cleaved and biotinylated protein was collected in the flow-through.
KRas G12V protein was subsequently loaded with nucleotide by adding 10 mM EDTA and 2 mM GDP. The mixture was incubated on a rotator overnight at 4 °C.20 mM MgCl2 was added to quench loading and stabilize the protein. KRas G12V was further purified and excess GDP nucleotide was removed by size exclusion chromatography. The protein was loaded on a Superdex 75 pg 16/600 column equilibrated with 20 mM HEPES pH 7.5, 150 mM NaCl and eluted with an isocratic flow. Biotin incorporation and GDP loading was confirmed by mass spectrometry on an Agilent 1290 Infinity II UPLC connected to an Agilent 6545XT qTOF. EXEMPLARY EMBODIMENTS P01/P02 Embodiments Embodiment 1. A compound of Formula (I):
Formula (I) or a pharmaceutically acceptable salt thereof, wherein: R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl substituted with -OH, -(C
1-3 alkylene)-OH, -CN, or – (C1-3 alkylene)-CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, or 2, and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b;
Ring B is selected from the group consisting of:
, wherein: X
1 is selected from the group consisting of a bond, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, and O, provided that no more than one of X
1, X
2, and X
3 is O; b1 is 0, 1, or 2; R
9 is selected from the group consisting of: H, OH, NH2, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; Y
2 is a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or a pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of: (a) halo; (b) cyano; (c) -OH; (d) oxo; (e) -C
1-6 alkoxy;
(f) -C
1-6 haloalkoxy; (g) -NR
dR
e; (h) C(=O)C1-6 alkyl; (i) C(=O)C
1-6 haloalkyl; (j) C(=O)OH; (k) C(=O)OC1-6 alkyl; (l) C(=O)OC
1-6 haloalkyl; (m) C(=O)N(R
f)2; (n) S(O)0-2(C1-6 alkyl); (o) S(O)
0-2(C
1-6 haloalkyl); (p) S(O)
1-2N(R
f)
2; and (q) C1-6 alkyl, C2-6 alkenyl, or C2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)
0-2-, C(=O), and C
1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)N(R
f)
2, S(O)
1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, and C1-
3 haloalkyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-. Embodiment 2. A compound of Formula (I):
Formula (I) or a pharmaceutically acceptable salt thereof, wherein: R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl substituted with -OH, -(C1-3 alkylene)-OH, -CN, or – (C1-3 alkylene)-CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, or 2, and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; Ring B is selected from the group consisting of:
, wherein:
X
1 is selected from the group consisting of a bond, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)2, and O, provided that no more than one of X
1, X
2, and X
3 is O; b1 is 0, 1, or 2; R
9 is selected from the group consisting of: OH, NH2, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; Y
2 is a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or a pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
(l) C(=O)OC
1-6 haloalkyl; (m) C(=O)N(R
f)2; (n) S(O)0-2(C1-6 alkyl); (o) S(O)
0-2(C
1-6 haloalkyl); (p) S(O)1-2N(R
f)2; and (q) C1-6 alkyl, C2-6 alkenyl, or C2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, and C
1- 3 haloalkyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-. Embodiment 3. The compound of Embodiment 1 or 2, wherein Ring B is
. Embodiment 4. The compound of any one of Embodiments 1-3, wherein X
1 is a bond. Embodiment 5. The compound of any one of Embodiments 1-3, wherein X
1 is selected from the group consisting of: CH
2, CHR
L, and C(R
L)
2. Embodiment 6. The compound of Embodiment 5, wherein X
1 is CH2. Embodiment 7. The compound of any one of Embodiments 1-6, wherein X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2. Embodiment 8. The compound of Embodiment 7, wherein X
2 and X
3 are both CH
2. Embodiment 9. The compound of any one of Embodiments 1-8, wherein R
9 is para to -X
3-. Embodiment 10. The compound of Embodiment 1 or 2, wherein the compound of Formula (I) is a compound of Formula (I-a1):
Formula (I-a1) or a pharmaceutically acceptable salt thereof, wherein: X
1 is a bond or CH
2; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, and C(R
L)2; and b1 is 0 or 1.
Embodiment 11. The compound of Embodiment 10, wherein X
2 and X
3 are both CH2. Embodiment 12. The compound of Embodiments 10 or 11, wherein X
1 is a bond. Embodiment 13. The compound of Embodiments 10 or 11, wherein X
1 is CH2. Embodiment 14. The compound of any one of Embodiments 1-13, wherein R
9 is -OH. Embodiment 15. The compound of any one of Embodiments 1-13, wherein R
9 is -NH2. Embodiment 16. The compound of any one of Embodiments 1-13, wherein R
9 is halo (e.g., -Br). Embodiment 17. The compound of any one of Embodiments 1-16, wherein b1 is 0. Embodiment 18. The compound of any one of Embodiments 1-17, wherein R
1 is a 4-10 membered heterocyclyl substituted with -OH, -(C
1-3 alkylene)-OH, -CN, or –(C
1-3 alkylene)-CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7. Embodiment 19. The compound of any one of Embodiments 1-18, wherein R
1 is a 4-10 (e.g., 6, 7, or 8) membered heterocyclyl substituted with -OH or -CH2CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7, and wherein the heterocyclyl contains one ring nitrogen atom and 0-2 additional ring heteroatoms each independently selected from the group consisting of: O and S(O)0-2. Embodiment 20. The compound of any one of Embodiments 1-19, wherein R
1 is
further optionally substituted with 1-2 R
7 at one or more ring carbon atoms.
Embodiment 21. The compound of any one of Embodiments 1-20, wherein R
1 is
Embodiment 22. The compound of any one of Embodiments 1-17, wherein R
1 is an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)
2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms. Embodiment 23. The compound of any one of Embodiments 1-17 or 22, wherein
Embodiment 24. The compound of any one of Embodiments 1-17, wherein R
1 is
, wherein b2 is 0, 1, or 2, and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7. Embodiment 25. The compound of Embodiment 24, wherein A
2 is CH. Embodiment 26. The compound of any one of Embodiments 1-17 or 24-25, wherein
. Embodiment 27. The compound of any one of Embodiments 1-26, wherein Y
2 is -CH
2-.
Embodiment 28. The compound of any one of Embodiments 1-27, wherein R
3 is a 4-10 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b. Embodiment 29. The compound of any one of Embodiments 1-28, wherein R
3 is a bicyclic 7-10 membered heterocyclyl optionally substituted with 1-6 R
a. Embodiment 30. The compound of any one of Embodiments 1-29, wherein R
3 is
optionally substituted with 1-3 R
a. Embodiment 31. The compound of any one of Embodiments 1-30, wherein R
3 is
. Embodiment 32. The compound of any one of Embodiments 1-31, wherein the ring carbon atom labelled with * in Formula (I) has (S)-stereochemistry. Embodiment 33. The compound of any one of Embodiments 1-31, wherein the ring carbon atom labelled with * in Formula (I) has (R)-stereochemistry. Embodiment 34. The compound of Embodiment 1 or 2, wherein the compound of Formula (I) is a compound of Formula (I-a2):
Formula (I-a2) or a pharmaceutically acceptable salt thereof, wherein:
R
1 is selected from the group consisting of:
; X
1 is a bond or CH
2; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, and C(R
L)2; and b1 is 0 or 1. Embodiment 35. The compound of Embodiment 34, wherein X
2 and X
3 are both CH2. Embodiment 36. The compound of Embodiments 34 or 35, wherein R
9 is -OH. Embodiment 37. The compound of Embodiment 1, wherein the compound of Formula (I) is selected from the group consisting of Compound Nos. 101, 101a, 101b, 101c, 102, 102a, 103, 103a, 103b, 103c, 104, 104a, 105, 105a, 105b, 105c, 106, 106a, 107, 107a, 108, 108a, 109, 109a, 110, 110a, 110b, 110c, 111, 111a, 111b, 111c, 113, 113a, 113b, 113c, 114, 114a, 114b, 114c, 115, 115a, 116, 116a, 116b, 117, 117a, 118, 118a, 119, 119a, 120, 120a as depicted in Table C1, or a pharmaceutically acceptable salt thereof. Embodiment 38. The compound of Embodiment 2, wherein the compound of Formula (I) is selected from the group consisting of Compound Nos. 101, 101a, 101b, 101c, 102, 102a, 103, 103a, 103b, 103c, 104, 104a, 105, 105a, 105b, 105c, 106, 106a, 107, 107a, 108, 108a, 109, 109a, 110, 110a, 110b, 110c, 111, 111a, 111b, and 111c, as depicted in Table C1, or a pharmaceutically acceptable salt thereof. Embodiment 39. A pharmaceutical composition comprising a compound of any one of Embodiments 1-38, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient. Embodiment 40. A dysregulated KRas protein non-covalently bound with a compound of any one of Embodiments 1-38, or a pharmaceutically acceptable salt thereof.
Embodiment 41. A method for treating a KRas-associated cancer in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-38, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 39. Embodiment 42. A method for treating a KRas-associated cancer in a subject in need thereof, the method comprising (a) determining that the cancer in the subject has a KRas dysregulation; and (b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-38, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 39. Embodiment 43. A method of treating a KRas-associated cancer in a subject, the method comprising administering to a subject identified or diagnosed as having a cancer having a KRas dysregulation a therapeutically effective amount of a compound of any one of Embodiments 1-38 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 39. Embodiment 44. A method of treating a KRas-associated cancer in a subject, the method comprising: (a) determining that the cancer in the subject has a KRas dysregulation; and (b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-38 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 39. Embodiment 45. The method of any one of Embodiments 41-44, wherein the KRas-associated cancer is a mutant KRas-associated cancer. Embodiment 46. The method of Embodiment 45, wherein the mutant KRas- associated cancer is a KRas G12A-associated cancer, a KRas G12C-associated cancer, a KRas G12D-associated cancer, a KRas G12R-associated cancer, a KRas G12S-associated cancer, or a KRas G12V-associated cancer. Embodiment 47. The method of Embodiment 46, wherein the mutant KRas-
associated cancer is a KRas G12R-associated cancer. Embodiment 48. The method of Embodiment 46, wherein the mutant KRas- associated cancer is a KRas G12V-associated cancer. Embodiment 49. The method of any one of Embodiments 42 or 44, wherein the step of determining that the cancer in the subject has a KRas dysregulation includes performing an assay to detect the KRas dysregulation (e.g., a KRas mutation) in a tumor sample from the subject. Embodiment 50. The method of Embodiment 49, wherein detecting the KRas dysregulation includes detecting a KRAS gene having a mutation corresponding to a substitution of glycine 12 in a KRas protein and/or a KRas protein having a substitution of glycine 12. Embodiment 51. The method of Embodiment 50, wherein the substitution of glycine 12 is a substitution to alanine, cysteine, aspartic acid, arginine, serine, or valine. Embodiment 52. The method of Embodiment 51, wherein the substitution of glycine 12 is a substitution to arginine. Embodiment 53. The method of Embodiment 51, wherein the substitution of glycine 12 is a substitution to valine. Embodiment 54. The method of any one of Embodiments 49-53, further comprising obtaining a tumor sample from the subject. Embodiment 55. The method of Embodiment 54, wherein the tumor sample is a biopsy sample. Embodiment 56. The method of any one of Embodiments 49-55, wherein the assay is selected from the group consisting of sequencing, immunohistochemistry, and enzyme-linked immunosorbent assay, and fluorescence in situ hybridization (FISH). 35 Embodiment 57. The method of Embodiment 56, wherein the sequencing is
pyrosequencing or next generation sequencing. Embodiment 58. The method of any one of Embodiments 41-57, wherein the KRas-associated cancer is selected from the group consisting of: a hematological cancer, a soft tissue cancer, bile duct cancer, bladder cancer, brain cancer, breast cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, ovarian cancer, pancreatic cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer, thymus cancer, thyroid cancer, urothelial cancer, uterine cancer, and a combination thereof. Embodiment 59. The method of Embodiment 58, wherein the KRas-associated cancer is pancreatic cancer. Embodiment 60. The method of any one of Embodiments 41-59, further comprising administering an additional therapy or therapeutic agent to the subject. Embodiment 61. The method of Embodiment 60, wherein the additional therapy or therapeutic agent is selected from the group consisting of Ras pathway targeted therapeutic agents, kinase-targeted therapeutics, mTORC1 inhibitors or degraders, YAP inhibitors or degraders, proteasome inhibitors or degraders, HSP90 inhibitors or degraders, farnesyl transferase inhibitors or degraders, PTEN inhibitors or degraders, signal transduction pathway inhibitors or degraders, checkpoint inhibitors, modulators of the apoptosis pathway, chemotherapeutics, angiogenesis-targeted therapies, immune-targeted agents, radiotherapy, and combinations thereof. Embodiment 62. A method for a method for modulating KRas protein activity in a mammalian cell, the method comprising contacting the mammalian cell with an effective amount of a compound of any one of Embodiments 1-38, or a pharmaceutically acceptable salt thereof. Embodiment 63. The method of Embodiment 62, wherein the contacting occurs in vivo. Embodiment 64. The method of Embodiment 62, wherein the contacting occurs
in vitro. Embodiment 65. The method of any one of Embodiments 62-64, wherein the mammalian cell is a mammalian cancer cell. Embodiment 66. The method of any one of Embodiments 62-65, wherein the KRas protein is a mutant KRas protein. Embodiment 67. The method of Embodiment 66, wherein the mutant KRas protein is a mutant KRas protein selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. P03 Embodiments Embodiment 1. A compound of Formula (A):
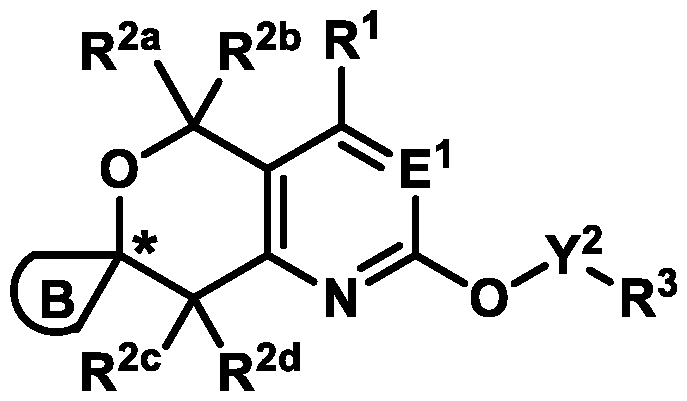
Formula (A) or a pharmaceutically acceptable salt thereof, wherein: E
1 is selected from the group consisting of N, CH, and CR
4, wherein R
4 is selected from the group consisting of: CN, halo, C
1-3 alkyl, C
1-3 haloalkyl, and C
3-6 cycloalkyl; R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)
2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently
selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; R
2a and R
2b are independently selected from the group consisting of: H, C
1-3 alkyl, C
1- 3 haloalkyl, and C3-6 cycloalkyl; or R
2a and R
2b taken together with the ring carbon atom to which each is attached form a C
3-6 cycloalkyl ring or a 4-6 membered heterocyclyl ring; R
2c and R
2d are independently selected from the group consisting of: H, halo, CN, C1-3 alkyl, C1-3 haloalkyl, and C3-6 cycloalkyl; or R
2c and R
2d taken together with the ring carbon atom to which each is attached form a C
3-6 cycloalkyl ring or a 4-6 membered heterocyclyl ring; Ring B is selected from the group consisting of:
, ,
, wherein: X
1 is selected from the group consisting of a bond, S(O)
0-2, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)
0-2; b1 is 0, 1, or 2; R
9 is selected from the group consisting of: H, OH, NR
dR
e, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; or a pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring;
Y
2 is a bond or a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or a pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein:
b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)
2, S(O)
0-2(C
1-6 alkyl), S(O)
0-2(C
1-6 haloalkyl), and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C
1-6 alkyl), S(O)
1-2(C
1-6 haloalkyl), S(O)
1-2N(R
f)
2, and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, C
1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-, provided that when X
1 is a bond; and b1 is 0, then: (1) R
9 is OH or NR
dR
e; or (2) Y
2 is a straight-chain C1-6 alkylene optionally substituted with one R
Y. Embodiment 2. The compound of Embodiment 1, wherein the compound is a compound of Formula (I):
Formula (I) or a pharmaceutically acceptable salt thereof.
30
Embodiment 3. The compound of Embodiments 1 or 2, wherein the compound is a compound of Formula (I):
Formula (I) or a pharmaceutically acceptable salt thereof, wherein: R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b;
X
1 is selected from the group consisting of a bond, S(O)0-2, CH2, CHR
L, C(R
L)2, and O;
X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; b1 is 0, 1, or 2; R
9 is selected from the group consisting of: H, OH, NH2, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; or a pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-6 cycloalkyl ring; Y
2 is a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C
1-6 alkoxy, C
1-6 haloalkoxy, C
1-6 alkyl, and C
1-6 haloalkyl, or a pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C
1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
(j) C(=O)OH; (k) C(=O)OC1-6 alkyl; (l) C(=O)OC1-6 haloalkyl; (m)C(=O)N(R
f)
2; (n) S(O)0-2(C1-6 alkyl); (o) S(O)0-2(C1-6 haloalkyl); (p) S(O)
1-2N(R
f)
2; and (q) C1-6 alkyl, C2-6 alkenyl, or C2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)
0-2-, C(=O), and C
1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)
2, S(O)
0-2(C
1-6 alkyl), S(O)
0-2(C
1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)N(R
f)
2, S(O)
1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, and C1- 3 haloalkyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-.
Embodiment 4. The compound of any one of Embodiments 1-3, wherein R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl substituted with -OH, -(C1-3 alkylene)-OH, -(C3-6 cycloalkylene)-OH, -CN, –(C
1-3 alkylene)-CN, or –(C
3-6 cycloalkylene)-CN, on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)
2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, or 2, and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7. Embodiment 5. The compound of any one of Embodiments 1-4, wherein R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl substituted with -OH, -(C
1-3 alkylene)-OH, -CN, or – (C
1-3 alkylene)-CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)
2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, or 2, and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; and
Ring B is selected from the group consisting of:
, wherein: X
1 is selected from the group consisting of a bond, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, and O, provided that no more than one of X
1, X
2, and X
3 is O; b1 is 0, 1, or 2; R
9 is selected from the group consisting of: H, OH, NH2, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 6. The compound of any one of Embodiments 1-5, wherein Ring B The compound of any one of Embodiments 1-5, wherein Ring B
. Embodiment 8. The compound of any one of Embodiments 1-7, wherein X
1 is a bond. Embodiment 9. The compound of any one of Embodiments 1-7, wherein X
1 is selected from the group consisting of: CH2, CHR
L, and C(R
L)2.
Embodiment 10. The compound of Embodiment 9, wherein X
1 is CH2. Embodiment 11. The compound of any one of Embodiments 1-10, wherein X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2. Embodiment 12. The compound of Embodiment 11, wherein X
2 and X
3 are both CH
2. Embodiment 13. The compound of Embodiment 11, wherein X
2 is CH2; and X
3 is selected from the group consisting of: CHR
L and C(R
L)
2. Embodiment 14. The compound of Embodiments 11 or 13, wherein X
2 is CH2; and X
3 is CHR
L. Embodiment 15. The compound of any one of Embodiments 11 or 13-14, wherein X
2 is CH2; and X
3 is CHMe. Embodiment 16. The compound of any one of Embodiments 1-10, wherein one of X
2 and X
3 is -O-; and the other of X
2 and X
3 is selected from the group consisting of: CH2, CHR
L, and C(R
L)2. Embodiment 17. The compound of any one of Embodiments 1-10 or 16, wherein X
2 is -O-; and X
3 is CH2 or CHMe. Embodiment 18. The compound of any one of Embodiments 1-17, wherein R
9 is para to -X
3-. Embodiment 19. The compound of Embodiments 2 or 3, wherein the compound of Formula (I) is a compound of Formula (I-a1):
Formula (I-a1) or a pharmaceutically acceptable salt thereof, wherein:
X
1 is a bond or CH
2; X
2 and X
3 are independently selected from the group consisting of: -O-, CH2, CHR
L, and C(R
L)2; and b1 is 0, 1, or 2. Embodiment 20. The compound of Embodiments 2 or 3, wherein the compound of Formula (I) is a compound of Formula (I-b1):
Formula (I-b1) or a pharmaceutically acceptable salt thereof, wherein: X
1 is a bond or CH2; X
2 and X
3 are independently selected from the group consisting of: -O-, CH
2, CHR
L, and C(R
L)2; and b1 is 0, 1, or 2. Embodiment 21. The compound of Embodiments 19 or 20, wherein X
1 is CH
2. Embodiment 22. The compound of any one of Embodiments 19-21, wherein X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, and C(R
L)
2. Embodiment 23. The compound of any one of Embodiments 19-22, wherein X
2 is CH
2; and X
3 is selected from the group consisting of: CHR
L and C(R
L)
2. Embodiment 24. The compound of any one of Embodiments 19-23, wherein X
2 is CH
2; and X
3 is CHMe. Embodiment 25. The compound of any one of Embodiments 19-22, wherein X
2 and X
3 are both CH2. Embodiment 26. The compound of any one of Embodiments 1-25, wherein R
9 is -OH or -NH2.
Embodiment 27. The compound of any one of Embodiments 1-26, wherein R
9 is -NH2. Embodiment 28. The compound of any one of Embodiments 1-27, wherein b1 is 0 or 1. Embodiment 29. The compound of any one of Embodiments 1-28, wherein b1 is 1. Embodiment 30. The compound of any one of Embodiments 1-27, wherein b1 is 2. Embodiment 31. The compound of any one of Embodiments 1-27, wherein b1 is 1 or 2; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 32. The compound of any one of Embodiments 1-27, wherein b1 is 1; and R
10 is -CN. Embodiment 33. The compound of any one of Embodiments 1-27 or 32, wherein b1 is 1; R
10 is ortho to R
9; and R
10 is -CN. Embodiment 34. The compound of any one of Embodiments 1-27, wherein b1 is 1 or 2; and each R
10 is independently -Cl or -F. Embodiment 35. The compound of any one of Embodiments 1-27 or 34, wherein b1 is 1 or 2; 1-2 occurrence(s) of R
10 is ortho to R
9; and each R
10 is independently -Cl or -F. Embodiment 36. The compound of any one of Embodiments 1-27, wherein b1 is 0. Embodiment 37. The compound of any one of Embodiments 1-6, wherein Ring B
is selected from the group consisting of:
, and
, wherein: X
2 is -O- or -CH
2-; X
3 is -CH2- or -CHR
L-, wherein R
L is C1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 38. The compound of any one of Embodiments 1-6 or 37, wherein Ring B is selected from the group consisting of:
, wherein: X
2 is -O- or -CH2-; X
3 is -CH
2- or -CHR
L-, wherein R
L is C
1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 39. The compound of any one of Embodiments 1-6 or 37, wherein
Embodiment 40. The compound of any one of Embodiments 1-39, wherein R
1 is 4-10 membered heterocyclyl optionally substituted with 1-4 R
7. Embodiment 41. The compound of any one of Embodiments 1-40, wherein R
1 is a 4-10 membered heterocyclyl substituted with -OH, -(C1-3 alkylene)-OH, -CN, or –(C1-3 alkylene)-CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7. Embodiment 42. The compound of any one of Embodiments 1-41, wherein R
1 is a 4-10 (e.g., 6, 7, or 8) membered heterocyclyl substituted with -OH or -CH
2CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7, and wherein the heterocyclyl contains one ring nitrogen atom and 0-2 additional ring heteroatoms each independently selected from the group consisting of: O and S(O)
0-2. Embodiment 43. The compound of any one of Embodiments 1-42, wherein R
1 is
further optionally substituted with 1-2 R
7 at one or more ring carbon atoms. Embodiment 44. The compound of any one of Embodiments 1-43, wherein R
1 is
Embodiment 45. The compound of any one of Embodiments 1-3 or 6-40, wherein R
1 is a 7-10 (e.g., 7) membered heterocyclyl optionally substituted with 1-4 R
7. Embodiment 46. The compound of any one of Embodiments 1-3, 6-40, or 45, wherein R
1 is a 7-10 (e.g., 7) membered heterocyclyl having one ring nitrogen atom, one ring oxygen atom, and no additional ring heteroatoms, wherein the 7-10 membered heterocyclyl is optionally substituted with 1-4 R
7. Embodiment 47. The compound of any one of Embodiments 1-3, 6-40, or 45-46,
wherein
optionally substituted with 1-4 R
7 at one or more ring carbon atoms. Embodiment 48. The compound of Embodiment 47, wherein each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C1-3 alkyl optionally substituted with 1-3 R
c, wherein: each R
c is independently selected from the group consisting of: -F, -OH, and -CN. Embodiment 49. The compound of any one of Embodiments 1-39, wherein R
1 is an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms. Embodiment 50. The compound of any one of Embodiments 1-39 or 49, wherein
Embodiment 51. The compound of any one of Embodiments 1-39, wherein R
1 is
, wherein b2 is 0, 1, or 2, and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7. Embodiment 52. The compound of Embodiment 51, wherein A
2 is CH. Embodiment 53. The compound of any one of Embodiments 1-39 or 51-52, wherein
. 25 Embodiment 54. The compound of any one of Embodiments 1-39 or 51-53,
wherein
selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, -C(O)N(H)R
b1, R
b1, and C(O)R
b1. Embodiment 55. The compound of Embodiment 54, wherein R
7 is selected from the group consisting of: (d) C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h; (e) C(O)N(C
1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C
3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (f) C(O)R
b1, wherein R
b1 is heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 56. The compound of any one of Embodiments 1-55, wherein Y
2 is -CH
2-. Embodiment 57. The compound of any one of Embodiments 1-56, wherein R
3 is a 4-10 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b. Embodiment 58. The compound of any one of Embodiments 1-57, wherein R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. Embodiment 59. The compound of any one of Embodiments 1-58, wherein R
3 is a bicyclic 7-10 membered heterocyclyl optionally substituted with 1-6 R
a. Embodiment 60. The compound of any one of Embodiments 1-59, wherein R
3 is
optionally substituted with 1-3 R
a. Embodiment 61. The compound of any one of Embodiments 1-60, wherein R
3 is
Embodiment 62. The compound of any one of Embodiments 1-61, wherein the ring carbon atom labelled with * in Formula (A) or Formula (I) has (S)-stereochemistry. Embodiment 63. The compound of any one of Embodiments 1-61, wherein the ring carbon atom labelled with * in Formula (A) or Formula (I) has (R)-stereochemistry. Embodiment 64. The compound of Embodiments 2 or 3, wherein the compound of Formula (I) is a compound of Formula (I-a2) or (I-b2):
Formula (I-b2) or a pharmaceutically acceptable salt thereof, wherein: R
1 is selected from the group consisting of:
; X
1 is a bond or CH2; X
2 and X
3 are independently selected from the group consisting of: O, CH
2, CHR
L, and
C(R
L)
2; and b1 is 0, 1, or 2 (e.g., 0 or 1). Embodiment 65. The compound of Embodiments 2 or 3, wherein the compound of Formula (I) is a compound of Formula (I-a3) or (I-b3):
or a pharmaceutically acceptable salt thereof, wherein: R
1 is a 7-10 membered heterocyclyl having one ring nitrogen atom, one ring oxygen atom, and no additional ring heteroatoms, wherein the 7-10 membered heterocyclyl is optionally substituted with 1-4 R
7; X
1 is CH2; X
2 and X
3 are independently selected from the group consisting of: O, CH2, CHR
L, and C(R
L)
2; and b1 is 0, 1, or 2 Embodiment 66. The compound of any one of Embodiments 2-3 or 64, wherein the compound of Formula (I) is a compound of Formula (I-a4) or (I-b4):
or a pharmaceutically acceptable salt thereof, wherein: b3 is 0, 1, 2, or 3; X
1 is CH
2; X
2 and X
3 are independently selected from the group consisting of: O, CH
2, CHR
L, and C(R
L)2; and b1 is 0, 1, or 2. Embodiment 67. The compound of Embodiment 66, wherein b3 is 0. Embodiment 68. The compound of Embodiment 66, wherein b3 is 1 or 2; and each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C1-3 alkyl optionally substituted with 1-3 R
c, wherein: each R
c is independently selected from the group consisting of: -F, -OH, and -CN. Embodiment 69. The compound of Embodiments 2 or 3, wherein the compound of Formula (I) is a compound of Formula (I-a5) or (I-b5):
Formula (I-b5) R
7 is selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
1 is CH2; X
2 and X
3 are independently selected from the group consisting of: O, CH2, CHR
L, and C(R
L)
2; and b1 is 0, 1, or 2. Embodiment 70. The compound of Embodiment 69, wherein R
7 is selected from the group consisting of: (a) C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C
1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C
3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 71. The compound of any one of Embodiments 64-70, wherein X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, and C(R
L)
2.
Embodiment 72. The compound of any one of Embodiments 64-71, wherein X
2 and X
3 are both CH2. Embodiment 73. The compound of any one of Embodiments 64-71, wherein X
2 is CH2; and X
3 is CHR
L. Embodiment 74. The compound of any one of Embodiments 64-71 or 73, wherein X
2 is CH2; and X
3 is CH(Me). Embodiment 75. The compound of any one of Embodiments 64-71, wherein X
2 is -O-; and X
3 is selected from the group consisting of: CH
2, CHR
L, and C(R
L)
2. Embodiment 76. The compound of any one of Embodiments 64-71 or 75, wherein X
2 is -O-; and X
3 is CH
2 or CH(Me). Embodiment 77. The compound of any one of Embodiments 64-76, wherein R
9 is -NH
2. Embodiment 78. The compound of any one of Embodiments 64-76, wherein R
9 is -OH. Embodiment 79. The compound of any one of Embodiments 64-78, wherein b1 is 1. Embodiment 80. The compound of any one of Embodiments 64-78, wherein b1 is . Embodiment 81. The compound of any one of Embodiments 64-80, wherein each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 82. The compound of any one of Embodiments 64-81, wherein 1-2 occurrence(s) of R
10 is ortho to R
9. Embodiment 83. The compound of any one of Embodiments 64-78, wherein b1 is
0. Embodiment 84. The compound of any one of 64-70, wherein the
X
2 is -O- or -CH
2-; X
3 is -CH
2- or -CHR
L-, wherein R
L is C
1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 85. The compound of any one of Embodiments 64-70 or 84, wherein the
moiety in Formula (I-a2), (I-a3), (I-a4), or (I-a5) is selected from the
, wherein: X
2 is -O- or -CH
2- ; X
3 is -CH2- or -CHR
L-, wherein R
L is C1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 86. The compound of any one of Embodiments 64-70 or 84, wherein
Embodiment 87. The compound of any one of Embodiments 64-86, wherein Y
2 is -CH
2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. Embodiment 88. The compound of Embodiment 87, wherein
,

Embodiment 89. The compound of any one of Embodiments 64-88, wherein the ring carbon atom labelled with * has (S)-stereochemistry. Embodiment 90. The compound of any one of Embodiments 1-3, wherein the compound of Formula (A) (e.g., Formula (I)) is selected from the group consisting of Compound Nos. 101, 101a, 101b, 101c, 102, 102a, 103, 103a, 103b, 103c, 104, 104a, 105,
105a, 105b, 105c, 106, 106a, 107, 107a, 108, 108a, 109, 109a, 110, 110a, 110b, 110c, 111, 111a, 111b, 111c, 113, 113a, 113b, 113c, 114, 114a, 114b, 114c, 115, 115a, 116, 116a, 116b, 117, 117a, 118, 118a, 118b, 118c, 119, 119a, 119b, 120, 120a, 120b, 121, 121a, 122, 122a, 123, 123a, 124, 124a, 124b, 125, 125a, 126, 126a, 127, 127a, 128, 128a, 128b, 129, 129a, 130, 130a, 131, 131a, 132, 132a, 133, 133a, 133b, 134, 134a, 135, 135a, 136, 136a, 136b, 137, 137a, 137b, 137c, 138, 138a, 139, 139a, 139b, 139c, 140, 140a, 141, 141a, 142, 142a, 143, 143a, 143b, 144, 144a, 145, 145a, 146, 146a, 147, 147a, 148, 148a, 149, 149a, 149b, 149c, 150, 150a, 151, 151a, 152, 152a, 153, 153a, 154, 154a, 155, 155a, 156, 156a, 157, 157a, 158, 158a, 158b, 158c, 159, 159a, 160, 160a, 161, 161a, 162, 162a, 163, 163a, 164, 164a, 164b, 165, 165a, 165b, 166, 166a, 167, 167a, 168, 168a, 169, 169a, 170, 170a, 171, 171a, 172, 172a, 173, 173a, 174, and 174a, as depicted in Table C1, or a pharmaceutically acceptable salt thereof. Embodiment 91. A pharmaceutical composition comprising a compound of any one of Embodiments 1-90, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient. Embodiment 92. A dysregulated KRas protein non-covalently bound with a compound of any one of Embodiments 1-90, or a pharmaceutically acceptable salt thereof. Embodiment 93. A method for treating a KRas-associated cancer in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-90, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 91. Embodiment 94. A method for treating a KRas-associated cancer in a subject in need thereof, the method comprising (a) determining that the cancer in the subject has a KRas dysregulation; and (b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-90, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 91. Embodiment 95. A method of treating a KRas-associated cancer in a subject, the method comprising administering to a subject identified or diagnosed as having a cancer having
a KRas dysregulation a therapeutically effective amount of a compound of any one of Embodiments 1-90 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 91. Embodiment 96. A method of treating a KRas-associated cancer in a subject, the method comprising: (a) determining that the cancer in the subject has a KRas dysregulation; and (b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-90 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 91. Embodiment 97. The method of any one of Embodiments 93-96, wherein the KRas-associated cancer is a mutant KRas-associated cancer. Embodiment 98. The method of Embodiment 97, wherein the mutant KRas- associated cancer is a KRas G12A-associated cancer, a KRas G12C-associated cancer, a KRas G12D-associated cancer, a KRas G12R-associated cancer, a KRas G12S-associated cancer, or a KRas G12V-associated cancer. Embodiment 99. The method of Embodiment 98, wherein the mutant KRas- associated cancer is a KRas G12D-associated cancer or a KRas G12V-associated cancer. Embodiment 100. The method of Embodiment 98, wherein the mutant KRas- associated cancer is a KRas G12D-associated cancer. Embodiment 101. The method of Embodiment 98, wherein the mutant KRas- associated cancer is a KRas G12R-associated cancer. Embodiment 102. The method of Embodiment 98, wherein the mutant KRas- associated cancer is a KRas G12V-associated cancer. Embodiment 103. The method of any one of Embodiments 94 or 96, wherein the step of determining that the cancer in the subject has a KRas dysregulation includes performing an assay to detect the KRas dysregulation (e.g., a KRas mutation) in a tumor sample from the
subject. Embodiment 104. The method of Embodiment 103, wherein detecting the KRas dysregulation includes detecting a KRAS gene having a mutation corresponding to a substitution of glycine 12 in a KRas protein and/or a KRas protein having a substitution of glycine 12. Embodiment 105. The method of Embodiment 104, wherein the substitution of glycine 12 is a substitution to alanine, cysteine, aspartic acid, arginine, serine, or valine. Embodiment 106. The method of Embodiment 105, wherein the substitution of glycine 12 is a substitution to aspartic acid. Embodiment 107. The method of Embodiment 105, wherein the substitution of glycine 12 is a substitution to arginine. Embodiment 108. The method of Embodiment 105, wherein the substitution of glycine 12 is a substitution to valine. Embodiment 109. The method of any one of Embodiments 103-108, comprising obtaining a tumor sample from the subject. Embodiment 110. The method of Embodiment 109, wherein the tumor sample is a biopsy sample. Embodiment 111. The method of any one of Embodiments 103-110, wherein the assay is selected from the group consisting of sequencing, immunohistochemistry, and enzyme-linked immunosorbent assay, and fluorescence in situ hybridization (FISH). Embodiment 112. The method of Embodiment 111, wherein the sequencing is pyrosequencing or next generation sequencing. Embodiment 113. The method of any one of Embodiments 93-112, wherein the KRas-associated cancer is selected from the group consisting of: a hematological cancer, a soft tissue cancer, bile duct cancer, bladder cancer, brain cancer, breast cancer, cervical cancer,
colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, ovarian cancer, pancreatic cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer, thymus cancer, thyroid cancer, urothelial cancer, uterine cancer, and a combination thereof. Embodiment 114. The method of Embodiment 113, wherein the KRas-associated cancer is pancreatic cancer. Embodiment 115. The method of any one of Embodiments 93-114, comprising administering an additional therapy or therapeutic agent to the subject. Embodiment 116. The method of Embodiment 115, wherein the additional therapy or therapeutic agent is selected from the group consisting of Ras pathway targeted therapeutic agents, kinase-targeted therapeutics, mTORC1 inhibitors or degraders, YAP inhibitors or degraders, proteasome inhibitors or degraders, HSP90 inhibitors or degraders, farnesyl transferase inhibitors or degraders, PTEN inhibitors or degraders, signal transduction pathway inhibitors or degraders, checkpoint inhibitors, modulators of the apoptosis pathway, chemotherapeutics, angiogenesis-targeted therapies, immune-targeted agents, radiotherapy, and combinations thereof. Embodiment 117. A method for a method for modulating KRas protein activity in a mammalian cell, the method comprising contacting the mammalian cell with an effective amount of a compound of any one of Embodiments 1-90, or a pharmaceutically acceptable salt thereof. Embodiment 118. The method of Embodiment 117, wherein the contacting occurs in vivo. Embodiment 119. The method of Embodiment 117, wherein the contacting occurs in vitro. Embodiment 120. The method of Embodiment 117, wherein the contacting occurs ex vivo.
Embodiment 121. The method of any one of Embodiments 117-120, wherein the mammalian cell is a mammalian cancer cell. Embodiment 122. The method of any one of Embodiments 117-121, wherein the KRas protein is a mutant KRas protein. Embodiment 123. The method of Embodiment 122, wherein the mutant KRas protein is a mutant KRas protein selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. P04 Embodiments Embodiment 1. A compound of Formula (II):
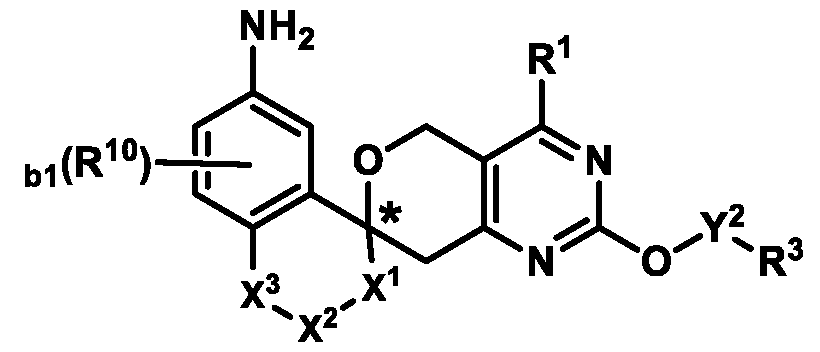
Formula (II) or a pharmaceutically acceptable salt thereof, wherein: R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)
2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; X
1 is selected from the group consisting of a bond, S(O)0-2, CH2, CHR
L, C(R
L)2, and O;
X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; b1 is 1 or 2; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
(k) C(=O)OC
1-6 alkyl; (l) C(=O)OC1-6 haloalkyl; (m) C(=O)N(R
f)2; (n) S(O)
0-2(C
1-6 alkyl); (o) S(O)0-2(C1-6 haloalkyl); (p) S(O)1-2N(R
f)2; and (q) C
1-6 alkyl, C
2-6 alkenyl, or C
2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)
2, S(O)
0-2(C
1-6 alkyl), S(O)
0-2(C
1-6 haloalkyl), and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1-
2(C
1-6 alkyl), S(O)
1-2(C
1-6 haloalkyl), S(O)
1-2N(R
f)
2, and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, C
1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. Embodiment 2. The compound of Embodiment 1, wherein the compound is a
compound of Formula (II-a):
Formula (II-a) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 3. The compound of Embodiment 1, wherein the compound is a compound of Formula (II-b):
Formula (II-b) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 4. A compound of Formula (III):
Formula (III) or a pharmaceutically acceptable salt thereof, wherein: R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7;
(ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; X
1 is selected from the group consisting of a bond, S(O)
0-2, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)
0-2; R
9 is selected from the group consisting of: H, NR
dR
e, -OH, and halo; b4 is 0 or 1; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e;
each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)1-2N(R
f)2;
each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, C
1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. Embodiment 5. The compound of Embodiment 4, wherein R
9 is -NH2; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 6. The compound of any one of Embodiments 1-5, wherein X
1 is CH2 or CHR
L. Embodiment 7. The compound of any one of Embodiments 1-6, wherein X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2. Embodiment 8. The compound of any one of Embodiments 1-7, wherein X
1 is CH2; and X
2 and X
3 are both CH2. Embodiment 9. The compound of any one of Embodiments 1-7, wherein at least one (e.g., one) of X
1, X
2, and X
3 is selected from the group consisting of: CHR
L and C(R
L)
2. Embodiment 10. The compound of any one of Embodiments 1-7 or 9, wherein X
1 is CH
2; and X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, and C(R
L)2, provided that 1-2 of X
2 and X
3 is independently CHR
L or C(R
L)2. Embodiment 11. The compound of any one of Embodiments 1-7 or 9-10, wherein X
1 is CH
2; X
2 is CH
2; and X
3 is CHR
L. Embodiment 12. The compound of any one of Embodiments 1-6, wherein one of
X
2 and X
3 is -O-; and the other of X
2 and X
3 is selected from the group consisting of: CH
2, CHR
L, and C(R
L)2. Embodiment 13. The compound of any one of Embodiments 1-6 or 12, wherein X
2 is -O-; and X
3 is selected from the group consisting of: CH2, CHR
L, and C(R
L)2. Embodiment 14. The compound of any one of Embodiments 1-13, wherein each R
L is independently selected from the group consisting of: CH
3, CF
3, CHF
2, and CH
2F. Embodiment 15. The compound of Embodiment 1, wherein the moiety is selected from the group consisting of:
, wherein: b4 is 0 or 1; X
2 is -O- or -CH2-; X
3 is -CH
2- or -CHR
L-, wherein R
L is C
1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 16. The compound of Embodiment 15, wherein is the ,
Embodiment 17. A compound of Formula (IV):
Formula (IV) or a pharmaceutically acceptable salt thereof, wherein: X
1 is selected from the group consisting of a bond, S(O)
0-2, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that at least one of X
1, X
2, and X
3 is CHR
L or C(R
L)
2; further provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; b1 is 0, 1 or 2; R
9 is selected from the group consisting of: H, OH, NR
dR
e, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; Y
2 is a bond or a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y;
each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3;
each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, C
1-3 haloalkyl, C
3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. Embodiment 18. The compound of Embodiment 17, wherein the compound is a compound of Formula (IV-a):
Formula (IV-a) or a pharmaceutically acceptable salt thereof, wherein: each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 19. The compound of Embodiments 17, wherein the compound is a
compound of Formula (IV-b):
Formula (IV-b) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 20. The compound of any one of Embodiments 17-19, wherein X
1 is CH2. Embodiment 21. The compound of any one of Embodiments 17-20, wherein X
2 is CH2; and X
3 is CHR
L. Embodiment 22. The compound of any one of Embodiments 17-20, wherein X
2 is -O-; and X
3 is selected from the group consisting of: CHR
L and C(R
L)2. Embodiment 23. The compound of any one of Embodiments 17-22, wherein each R
L is independently selected from the group consisting of: CH3, CF3, CHF2, and CH2F. Embodiment 24. The compound of any one of Embodiments 17-19, wherein the compound is a compound of Formula (IV-c):
Formula (IV-c) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c.
Embodiment 25. A compound of Formula (V):
Formula (V) or a pharmaceutically acceptable salt thereof, wherein: X
1 is selected from the group consisting of a bond, S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that 2-3 of X
1, X
2, and X
3 are independently CHR
L or C(R
L)
2; one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; and each additional R
L is independently selected from the group consisting of: C
1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; b1 is 0, 1 or 2; R
9 is selected from the group consisting of: H, OH, NR
dR
e, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)
2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b;
Y
2 is a bond or a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C
1-6 alkoxy, C
1-6 haloalkoxy, C
1-6 alkyl, and C
1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C
1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
(q) C
1-6 alkyl, C
2-6 alkenyl, or C
2-6 alkynyl, each optionally substituted with 1-6 R ; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1,
wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)
0-2-, C(=O), and C
1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)
2, S(O)
0-2(C
1-6 alkyl), S(O)
0-2(C
1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)N(R
f)
2, S(O)
1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, C1-3 haloalkyl, C
3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-. Embodiment 26. The compound of Embodiment 25, wherein the compound is a compound of Formula (V-a):
Formula (V-a) or a pharmaceutically acceptable salt thereof, wherein: each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c.
Embodiment 27. The compound of Embodiment 25, wherein the compound is a compound of Formula (IV-b):
Formula (V-b) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 28. The compound of any one of Embodiments 25-27, wherein X
2 is -O- or -CH
2- (e.g., -CH
2-). Embodiment 29. The compound of any one of Embodiments 25-28, wherein X
1 is CHR
L; and X
3 is CHR
L, wherein the pair of R
L on different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-4 cycloalkyl ring. Embodiment 30. The compound of any one of Embodiments 25-28, wherein X
1 is CHR
L; and X
3 is C(R
L)
2, wherein the pair of R
L on different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-4 cycloalkyl ring; and the remaining R
L is C1-
2 alkyl optionally substituted with 1-3 F. Embodiment 31. The compound of any one of Embodiments 25-27, wherein the compound is a compound of Formula (V-c) or Formula (V-d):
Formula (V-c)
Formula (V-d) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; the pair of R
L1 taken together with the ring atom(s) connecting them form a C
3-4 cycloalkyl ring, and R
L2 is C1-2 alkyl optionally substituted with 1-3 F. Embodiment 32. The compound of any one of Embodiments 1-31, wherein Y
2 is -CH2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. Embodiment 33. The compound of any one of Embodiments 1-32, wherein R
3 is 34. The compound of any one of Embodiments 1-33, wherein R
1 is
, erein b2 is 0, 1, or 2, and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7. Embodiment 35. The compound of any one of Embodiments 1-34, wherein R
1 is
alkyl)R
b1, -C(O)N(H)R
b1, R
b1, and C(O)R
b1. Embodiment 37. The compound of Embodiment 36, wherein R
7 is selected from the group consisting of: (a) C(=O)N(R
f)
2, wherein each R
f is independently H or C
1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 38. The compound of any one of Embodiments 35-37, wherein R
7 is C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h. Embodiment 39. The compound of any one of Embodiments 1-35, wherein R
1 is
, wherein R
7a and R
7b are independently selected R
7. Embodiment 40. The compound of Embodiment 39, wherein R
7a is selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, -C(O)N(H)R
b1, R
b1, and
C(O)R
b1; and R
7b is -halo, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 41. The compound of Embodiment 40, wherein R
7a is selected from the group consisting of: (a) C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C
1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C
3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 42. The compound of any one of Embodiments 1-33, wherein R
1 is a 7-10 (e.g., 7) membered heterocyclyl having one ring nitrogen atom, one ring oxygen atom, and no additional ring heteroatoms, wherein the 7-10 membered heterocyclyl is optionally substituted with 1-4 R
7. Embodiment 43. The compound of Embodiment 42, wherein
optionally substituted with 1-4 R
7 at one or more ring carbon atoms
Embodiment 44. The compound of Embodiment 43, wherein each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C1-3 alkyl optionally substituted with 1-3 R
c, wherein: each R
c is independently selected from the group consisting of: -F, -OH, and -CN. Embodiment 45. The compound of Embodiment 1, wherein the compound is a compound of Formula (II-a1) or (II-b1):
Formula (II-b1) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; b3 is 0, 1, 2, or 3; X
1 is CH
2; X
2 and X
3 are independently selected from the group consisting of: O, CH2, CHR
L, and C(R
L)2; and b1 is 0, 1, or 2. Embodiment 46. The compound of Embodiment 4, wherein the compound is a compound of Formula (III-1):
Formula (III-1)
or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; b3 is 0, 1, 2, or 3; X
1 is CH2; X
2 and X
3 are independently selected from the group consisting of: O, CH
2, CHR
L, and C(R
L)2; and b1 is 0, 1, or 2. Embodiment 47. The compound of Embodiment 46, wherein R
9 is NR
dR
e (e.g., - NH2). Embodiment 48. The compound of Embodiment 17, wherein the compound is a compound of Formula (IV-a1) or (IV-b1):
Formula (IV-b1) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; 25 b1 is 0, 1, or 2;
each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; b3 is 0, 1, 2, or 3; X
1 is CH
2; one of X
2 and X
3 is independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2; and the other of X
2 and X
3 is CH
2 or O. Embodiment 49. The compound of Embodiment 48, wherein X
2 is CH2; and X
3 is CHR
L. Embodiment 50. The compound of Embodiment 25, wherein the compound is a compound of Formula (V-a1) or (V-b1):
Formula (V-b1) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; b1 is 0, 1, or 2; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 25 alkyl optionally substituted with 1-3 R
c;
b3 is 0, 1, 2, or 3; X
2 is -O- or -CH2-; X
1 is CHR
L; and X
3 is CHR
L or C(R
L)2, wherein: one pair of R
L on different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-4 cycloalkyl ring; and the remaining R
L if present is C1-2 alkyl optionally substituted with 1-3 F. Embodiment 51. The compound of any one of Embodiments 45-51, wherein b3 is 0. Embodiment 52. The compound of any one of Embodiments 45-52, wherein b3 is 1 or 2; and each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C1-3 alkyl optionally substituted with 1-3 R
c, wherein each R
c is independently selected from the group consisting of: -F, -OH, and -CN. Embodiment 53. The compound of Embodiment 1, wherein the compound is a compound of Formula (II-a2) or (II-b2):
Formula (II-b2)
or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
1 is CH
2; X
2 and X
3 are independently selected from the group consisting of: O, CH2, CHR
L, and C(R
L)2; and b1 is 0, 1, or 2. Embodiment 54. The compound of Embodiment 4, wherein the compound is a compound of Formula (III-2):
Formula (III-2) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)
2, C(O)N(C
1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
1 is CH2; X
2 and X
3 are independently selected from the group consisting of: O, CH
2, CHR
L, and C(R
L)2; and b1 is 0, 1, or 2. Embodiment 55. The compound of Embodiment 54, wherein R
9 is NR
dR
e (e.g., - NH2).
Embodiment 56. The compound of Embodiment 17, wherein the compound is a compound of Formula (IV-a2) or (IV-b2):
Formula (IV-b2) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; b1 is 0, 1, or 2; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
1 is CH
2; one of X
2 and X
3 is independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2; and the other of X
2 and X
3 is CH
2 or O. Embodiment 57. The compound of Embodiment 56, wherein X
2 is CH2; and X
3 is CHR
L.
Embodiment 58. The compound of Embodiment 25, wherein the compound is a compound of Formula (V-a2) or (V-b2):
Formula (V-b2) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; b1 is 0, 1, or 2; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
2 is -O- or -CH
2-; X
1 is CHR
L; and X
3 is CHR
L or C(R
L)
2, wherein: one pair of R
L on different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-4 cycloalkyl ring; and the remaining R
L if present is C
1-2 alkyl optionally substituted with 1-3 F.
Embodiment 59. The compound of any one of Embodiments 50-58, wherein R
7 is selected from the group consisting of: (a) C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 60. The compound of any one of Embodiments 45-59, wherein Y
2 is -CH
2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. Embodiment 61. The compound of Embodiment 60, wherein

). Embodiment 62. The compound of any one of Embodiments 1-61, wherein the ring carbon atom labelled with * has (S)-stereochemistry. Embodiment 63. The compound of Embodiment 1, wherein the compound of Formula (II) is selected from the group consisting of Compound Nos.139, 139a, 139b, 139c, 158, 158a, 158b, 158c, 160, 160a, 161, 161a, 164, 164a, 164b, 170, 170a, 171, 171a, 176, 176a, 176b, 176c, 176d, 178, 178a, 178b, 179, 179a, 180, 180a, 181, and 181a of Table C1, or a pharmaceutically acceptable salt thereof. Embodiment 64. The compound of Embodiment 4, wherein the compound of Formula (III) is selected from the group consisting of Compound Nos.158, 158a, 158b, 158c, 161, 161a, 176, 176a, 176b, 176c, 176d, 177, 177a, 178, 178a, 178b, 179, 179a, 180, and 180a of Table C1, or a pharmaceutically acceptable salt thereof. 30
Embodiment 65. The compound of Embodiment 17, wherein the compound of Formula (IV) is selected from the group consisting of Compound Nos.124, 124a, 124b, 125, 125a, 130, 130a, 131, 131a, 133, 133a, 133b, 134, 134a, 138, 138a, 148, 148a, 162, 162a, 163, 163a, 175, and 175a of Table C1, or a pharmaceutically acceptable salt thereof. Embodiment 66. The compound of Embodiment 25, wherein the compound of Formula (V) is selected from the group consisting of Compound Nos.172 and 172a of Table C1, or a pharmaceutically acceptable salt thereof. Embodiment 67. A pharmaceutical composition comprising a compound of any one of Embodiments 1-66, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient. Embodiment 68. A dysregulated KRas protein non-covalently bound with a compound of any one of Embodiments 1-66, or a pharmaceutically acceptable salt thereof. Embodiment 69. A method for treating a KRas-associated cancer in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-66, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 67. Embodiment 70. A method for treating a KRas-associated cancer in a subject in need thereof, the method comprising (a) determining that the cancer in the subject has a KRas dysregulation; and (b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-66, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 67. Embodiment 71. A method of treating a KRas-associated cancer in a subject, the method comprising administering to a subject identified or diagnosed as having a cancer having a KRas dysregulation a therapeutically effective amount of a compound of any one of Embodiments 1-66 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 67. Embodiment 72. A method of treating a KRas-associated cancer in a subject, the
method comprising: (a) determining that the cancer in the subject has a KRas dysregulation; and (b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-66 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 67. Embodiment 73. The method of any one of Embodiments 69-72, wherein the KRas-associated cancer is a mutant KRas-associated cancer. Embodiment 74. The method of Embodiment 73, wherein the mutant KRas- associated cancer is a KRas G12A-associated cancer, a KRas G12C-associated cancer, a KRas G12D-associated cancer, a KRas G12R-associated cancer, a KRas G12S-associated cancer, or a KRas G12V-associated cancer. Embodiment 75. The method of Embodiment 74, wherein the mutant KRas- associated cancer is a KRas G12D-associated cancer or a KRas G12V-associated cancer. Embodiment 76. The method of Embodiment 74, wherein the mutant KRas- associated cancer is a KRas G12D-associated cancer. Embodiment 77. The method of Embodiment 74, wherein the mutant KRas- associated cancer is a KRas G12R-associated cancer. Embodiment 78. The method of Embodiment 74, wherein the mutant KRas- associated cancer is a KRas G12V-associated cancer. Embodiment 79. The method of any one of Embodiments 70 or 72, wherein the step of determining that the cancer in the subject has a KRas dysregulation includes performing an assay to detect the KRas dysregulation (e.g., a KRas mutation) in a tumor sample from the subject. Embodiment 80. The method of Embodiment 79, wherein detecting the KRas dysregulation includes detecting a KRAS gene having a mutation corresponding to a substitution of glycine 12 in a KRas protein and/or a KRas protein having a substitution of 35 glycine 12.
Embodiment 81. The method of Embodiment 80, wherein the substitution of glycine 12 is a substitution to alanine, cysteine, aspartic acid, arginine, serine, or valine. Embodiment 82. The method of Embodiment 81, wherein the substitution of glycine 12 is a substitution to aspartic acid. Embodiment 83. The method of Embodiment 81, wherein the substitution of glycine 12 is a substitution to arginine. Embodiment 84. The method of Embodiment 81, wherein the substitution of glycine 12 is a substitution to valine. Embodiment 85. The method of any one of Embodiments 79-84, comprising obtaining a tumor sample from the subject. Embodiment 86. The method of Embodiment 85, wherein the tumor sample is a biopsy sample. Embodiment 87. The method of any one of Embodiments 79-86, wherein the assay is selected from the group consisting of sequencing, immunohistochemistry, and enzyme-linked immunosorbent assay, and fluorescence in situ hybridization (FISH). Embodiment 88. The method of Embodiment 87, wherein the sequencing is pyrosequencing or next generation sequencing. Embodiment 89. The method of any one of Embodiments 79-88, wherein the KRas-associated cancer is selected from the group consisting of: a hematological cancer, a soft tissue cancer, bile duct cancer, bladder cancer, brain cancer, breast cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, ovarian cancer, pancreatic cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer, thymus cancer, thyroid cancer, urothelial cancer, uterine cancer, and a combination thereof.
Embodiment 90. The method of Embodiment 89, wherein the KRas-associated cancer is pancreatic cancer. Embodiment 91. The method of Embodiment 89, wherein the KRas-associated cancer is selected from the group consisting of: a hematological cancer, brain cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, pancreatic cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, thymus cancer, urothelial cancer, and uterine cancer. Embodiment 92. The method of Embodiment 89, wherein the KRas-associated cancer is selected from the group consisting of: the cancer is a hematological cancer, bladder cancer, bile duct cancer, brain cancer, breast cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, ovarian cancer, pancreatic cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer (e.g., seminoma), thymus cancer, and uterine cancer. Embodiment 93. The method of Embodiment 89, wherein the KRas-associated cancer is selected from the group consisting of: bladder cancer, breast cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, leukemia, lung cancer (e.g., NSCLC), pancreatic cancer, and kidney cancer. Embodiment 94. The method of any one of Embodiments 79-93, comprising administering an additional therapy or therapeutic agent to the subject. Embodiment 95. The method of Embodiment 94, wherein the additional therapy or therapeutic agent is selected from the group consisting of Ras pathway targeted therapeutic agents, kinase-targeted therapeutics, mTORC1 inhibitors or degraders, YAP inhibitors or degraders, proteasome inhibitors or degraders, HSP90 inhibitors or degraders, farnesyl transferase inhibitors or degraders, PTEN inhibitors or degraders, signal transduction pathway inhibitors or degraders, checkpoint inhibitors, modulators of the apoptosis pathway, chemotherapeutics, angiogenesis-targeted therapies, immune-targeted agents, radiotherapy, and combinations thereof. Embodiment 96. A method for a method for modulating KRas protein activity in
a mammalian cell, the method comprising contacting the mammalian cell with an effective amount of a compound of any one of Embodiments 1-90, or a pharmaceutically acceptable salt thereof. Embodiment 97. The method of Embodiment 96, wherein the contacting occurs in vivo. Embodiment 98. The method of Embodiment 96, wherein the contacting occurs in vitro. Embodiment 99. The method of Embodiment 96, wherein the contacting occurs ex vivo. Embodiment 100. The method of any one of Embodiments 96-99, wherein the mammalian cell is a mammalian cancer cell. Embodiment 101. The method of any one of Embodiments 96-99, wherein the KRas protein is a mutant KRas protein. Embodiment 102. The method of Embodiment 101, wherein the mutant KRas protein is a mutant KRas protein selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. P06 Embodiments Embodiment 1. A compound of Formula (II):

Formula (II) or a pharmaceutically acceptable salt thereof, wherein: R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7;
(ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; X
1 is selected from the group consisting of a bond, S(O)
0-2, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)
0-2; b1 is 1 or 2; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C
1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C
1-6 alkoxy, C
1-6 haloalkoxy, C
1-6 alkyl, and C
1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C
1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e;
each R
a is independently selected from the group consisting of: (a) halo; (b) cyano; (c) -OH; (d) oxo; (e) -C
1-6 alkoxy; (f) -C1-6 haloalkoxy; (g) -NR
dR
e; (h) C(=O)C
1-6 alkyl; (i) C(=O)C
1-6 haloalkyl; (j) C(=O)OH; (k) C(=O)OC1-6 alkyl; (l) C(=O)OC
1-6 haloalkyl; (m) C(=O)N(R
f)2; (n) S(O)0-2(C1-6 alkyl); (o) S(O)
0-2(C
1-6 haloalkyl); (p) S(O)
1-2N(R
f)
2; and (q) C1-6 alkyl, C2-6 alkenyl, or C2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)1-2N(R
f)2;
each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, C
1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. Embodiment 2. The compound of Embodiment 1, wherein the compound is a compound of Formula (II-a):
Formula (II-a) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 3. The compound of Embodiment 1, wherein the compound is a compound of Formula (II-b):
Formula (II-b) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and
each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 4. A compound of Formula (III):
Formula (III) or a pharmaceutically acceptable salt thereof, wherein: R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; X
1 is selected from the group consisting of a bond, S(O)
0-2, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; R
9 is selected from the group consisting of: H, NR
dR
e, -OH, and halo; b4 is 0 or 1; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; or
one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, C
1-3 haloalkyl, C
3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-. Embodiment 5. The compound of Embodiment 4, wherein R
9 is -NH2; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 6. The compound of any one of Embodiments 1-5, wherein X
1 is CH2 or CHR
L. Embodiment 7. The compound of any one of Embodiments 1-6, wherein X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2.
Embodiment 8. The compound of any one of Embodiments 1-7, wherein X
1 is CH2; and X
2 and X
3 are both CH2. Embodiment 9. The compound of any one of Embodiments 1-7, wherein at least one (e.g., one) of X
1, X
2, and X
3 is selected from the group consisting of: CHR
L and C(R
L)2. Embodiment 10. The compound of any one of Embodiments 1-7 or 9, wherein X
1 is CH2; and X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2, provided that 1-2 of X
2 and X
3 is independently CHR
L or C(R
L)2. Embodiment 11. The compound of any one of Embodiments 1-7 or 9-10, wherein X
1 is CH2; X
2 is CH2; and X
3 is CHR
L. Embodiment 12. The compound of any one of Embodiments 1-6, wherein one of X
2 and X
3 is -O-; and the other of X
2 and X
3 is selected from the group consisting of: CH
2, CHR
L, and C(R
L)2. Embodiment 13. The compound of any one of Embodiments 1-6 or 12, wherein X
2 is -O-; and X
3 is selected from the group consisting of: CH
2, CHR
L, and C(R
L)
2. Embodiment 14. The compound of any one of Embodiments 1-13, wherein each R
L is independently selected from the group consisting of: CH
3, CF
3, CHF
2, and CH
2F. Embodiment 15. The compound of Embodiment 1, wherein the moiety is selected from the group consisting of:
, wherein: b4 is 0 or 1; X
2 is -O- or -CH2-; X
3 is -CH
2- or -CHR
L-, wherein R
L is C
1-3 alkyl (e.g., methyl); and
each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 16. The compound of Embodiment 15, wherein is the
Embodiment 17. A compound of Formula (IV):
Formula (IV) or a pharmaceutically acceptable salt thereof, wherein: X
1 is selected from the group consisting of a bond, S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that at least one of X
1, X
2, and X
3 is CHR
L or C(R
L)2; further provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)
0-2; b1 is 0, 1 or 2; R
9 is selected from the group consisting of: H, OH, NR
dR
e, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C
1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c;
R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)
2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and
wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; Y
2 is a bond or a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C
1-6 alkoxy, C
1-6 haloalkoxy, C
1-6 alkyl, and C
1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C
1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of: (a) Halo; (b) cyano; (c) -OH; (d) oxo; (e) -C1-6 alkoxy; (f) -C
1-6 haloalkoxy; (g) -NR
dR
e; (h) C(=O)C1-6 alkyl;
(i) C(=O)C
1-6 haloalkyl; (j) C(=O)OH; (k) C(=O)OC1-6 alkyl; (l) C(=O)OC
1-6 haloalkyl; (m) C(=O)N(R
f)2; (n) S(O)0-2(C1-6 alkyl); (o) S(O)
0-2(C
1-6 haloalkyl); (p) S(O)1-2N(R
f)2; and (q) C1-6 alkyl, C2-6 alkenyl, or C2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, C1-3 haloalkyl, C
3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-.
Embodiment 18. The compound of Embodiment 17, wherein the compound is a compound of Formula (IV-a):
Formula (IV-a) or a pharmaceutically acceptable salt thereof, wherein: each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 19. The compound of Embodiment 17, wherein the compound is a compound of Formula (IV-b):
Formula (IV-b) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 20. The compound of any one of Embodiments 17-19, wherein X
1 is CH2. Embodiment 21. The compound of any one of Embodiments 17-20, wherein X
2 is CH2; and X
3 is CHR
L. Embodiment 22. The compound of any one of Embodiments 17-20, wherein X
2 is -O-; and X
3 is selected from the group consisting of: CHR
L and C(R
L)2.
Embodiment 23. The compound of any one of Embodiments 17-22, wherein each R
L is independently selected from the group consisting of: CH3, CF3, CHF2, and CH2F. Embodiment 24. The compound of any one of Embodiments 17-19, wherein the compound is a compound of Formula (IV-c):
Formula (IV-c) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 25. A compound of Formula (V):
Formula (V) or a pharmaceutically acceptable salt thereof, wherein: X
1 is selected from the group consisting of a bond, S(O)
0-2, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that 2-3 of X
1, X
2, and X
3 are independently CHR
L or C(R
L)
2; one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; and each additional R
L is independently selected from the group consisting of: C
1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; b1 is 0, 1 or 2;
R
9 is selected from the group consisting of: H, OH, NR
dR
e, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; R
1 is selected from the group consisting of: (iii) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)
2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; Y
2 is a bond or a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C
1-6 alkoxy, C
1-6 haloalkoxy, C
1-6 alkyl, and C
1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C
1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
(f) -C
1-6 haloalkoxy; (g) -NR
dR
e; (h) C(=O)C1-6 alkyl; (i) C(=O)C
1-6 haloalkyl; (j) C(=O)OH; (k) C(=O)OC1-6 alkyl; (l) C(=O)OC
1-6 haloalkyl; (m) C(=O)N(R
f)2; (n) S(O)0-2(C1-6 alkyl); (o) S(O)
0-2(C
1-6 haloalkyl); (p) S(O)
1-2N(R
f)
2; and (q) C1-6 alkyl, C2-6 alkenyl, or C2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)
0-2-, C(=O), and C
1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)N(R
f)
2, S(O)
1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, C1-3
haloalkyl, C
3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-. Embodiment 26. The compound of Embodiment 25, wherein the compound is a compound of Formula (V-a):
Formula (V-a) or a pharmaceutically acceptable salt thereof, wherein: each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 27. The compound of Embodiment 25, wherein the compound is a compound of Formula (IV-b):
Formula (V-b) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 28. The compound of any one of Embodiments 25-27, wherein X
2 is -O- or -CH2- (e.g., -CH2-). Embodiment 29. The compound of any one of Embodiments 25-28, wherein X
1 is CHR
L; and X
3 is CHR
L, wherein the pair of R
L on different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-4 cycloalkyl ring.
Embodiment 30. The compound of any one of Embodiments 25-28, wherein X
1 is CHR
L; and X
3 is C(R
L)2, wherein the pair of R
L on different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-4 cycloalkyl ring; and the remaining R
L is C
1- 2 alkyl optionally substituted with 1-3 F. Embodiment 31. The compound of any one of Embodiments 25-27, wherein the compound is a compound of Formula (V-c) or Formula (V-d):
or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; the pair of R
L1 taken together with the ring atom(s) connecting them form a C3-4 cycloalkyl ring, and R
L2 is C1-2 alkyl optionally substituted with 1-3 F. Embodiment 32. The compound of any one of Embodiments 1-31, wherein Y
2 is -CH
2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a.
Embodiment 33. The compound of any one of Embodiments 1-32, wherein R
3 is 34. The compound of any one of Embodiments 1-33, wherein R
1 is
, erein b2 is 0, 1, or 2, and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7. Embodiment 35. The compound of any one of Embodiments 1-34, wherein R
1 is
alkyl)R
b1, -C(O)N(H)R
b1, R
b1, and C(O)R
b1. Embodiment 37. The compound of Embodiment 36, wherein R
7 is selected from the group consisting of: (a) C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C
1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C
3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and
(c) C(O)R
b1, wherein R
b1 is heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 38. The compound of any one of Embodiments 35-37, wherein R
7 is C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h. Embodiment 39. The compound of any one of Embodiments 1-35, wherein R
1 is
, wherein R
7a and R
7b are independently selected R
7. Embodiment 40. The compound of Embodiment 39, wherein R
7a is selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, -C(O)N(H)R
b1, R
b1, and C(O)R
b1; and R
7b is -halo, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 41. The compound of Embodiment 40, wherein R
7a is selected from the group consisting of: (a) C(=O)N(R
f)
2, wherein each R
f is independently H or C
1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 42. The compound of any one of Embodiments 1-33, wherein R
1 is a 7-10 (e.g., 7) membered heterocyclyl having one ring nitrogen atom, one ring oxygen atom, and no additional ring heteroatoms, wherein the 7-10 membered heterocyclyl is optionally substituted with 1-4 R
7.
Embodiment 43. The compound of Embodiment 42, wherein
optionally substituted with 1-4 R
7 at one or more ring carbon atoms
Embodiment 44. The compound of Embodiment 43, wherein each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C1-3 alkyl optionally substituted with 1-3 R
c, wherein: each R
c is independently selected from the group consisting of: -F, -OH, and -CN. Embodiment 45. The compound of Embodiment 1, wherein the compound is a compound of Formula (II-a1) or (II-b1):
Formula (II-b1) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c;
20 b3 is 0, 1, 2, or 3;
X
1 is CH
2; and X
2 and X
3 are independently selected from the group consisting of: O, CH2, CHR
L, and C(R
L)2. Embodiment 46. The compound of Embodiment 4, wherein the compound is a compound of Formula (III-1):
Formula (III-1) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; b3 is 0, 1, 2, or 3; X
1 is CH2; and X
2 and X
3 are independently selected from the group consisting of: O, CH2, CHR
L, and C(R
L)
2. Embodiment 47. The compound of Embodiment 46, wherein R
9 is NR
dR
e (e.g., - NH2). Embodiment 48. The compound of Embodiment 17, wherein the compound is a compound of Formula (IV-a1) or (IV-b1):
25 Formula (IV-a1)
Formula (IV-b1) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; b1 is 0, 1, or 2; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; b3 is 0, 1, 2, or 3; X
1 is CH2; one of X
2 and X
3 is independently selected from the group consisting of: CHR
L and C(R
L)
2; and the other of X
2 and X
3 is CH2 or O. Embodiment 49. The compound of Embodiment 48, wherein X
2 is CH
2; and X
3 is CHR
L. Embodiment 50. The compound of Embodiment 25, wherein the compound is a compound of Formula (V-a1) or (V-b1):
Formula (V-a1)
Formula (V-b1) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; b1 is 0, 1, or 2; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; b3 is 0, 1, 2, or 3; X
2 is -O- or -CH
2-; X
1 is CHR
L; and X
3 is CHR
L or C(R
L)
2, wherein: one pair of R
L on different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-4 cycloalkyl ring; and the remaining R
L if present is C
1-2 alkyl optionally substituted with 1-3 F. Embodiment 51. The compound of any one of Embodiments 45-50, wherein b3 is 0. Embodiment 52. The compound of any one of Embodiments 45-51, wherein b3 is 1 or 2; and each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C
1-3 alkyl optionally substituted with 1-3 R
c, wherein each R
c is independently selected from the group consisting of: -F, -OH, and -CN. Embodiment 53. The compound of Embodiment 1, wherein the compound is a compound of Formula (II-a2) or (II-b2):
Formula (II-b2) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)
2, C(O)N(C
1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
1 is CH2; and X
2 and X
3 are independently selected from the group consisting of: O, CH
2, CHR
L, and C(R
L)
2. Embodiment 54. The compound of Embodiment 4, wherein the compound is a compound of Formula (III-2):
Formula (III-2) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)
2, C(O)N(C
1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
1 is CH
2; and X
2 and X
3 are independently selected from the group consisting of: O, CH
2, CHR
L, and C(R
L)2. Embodiment 55. The compound of Embodiment 54, wherein R
9 is NR
dR
e (e.g., - NH
2). Embodiment 56. The compound of Embodiment 17, wherein the compound is a compound of Formula (IV-a2) or (IV-b2):
Formula (IV-a2)
Formula (IV-b2) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; b1 is 0, 1, or 2; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)
2, C(O)N(C
1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
1 is CH
2; one of X
2 and X
3 is independently selected from the group consisting of: CHR
L and C(R
L)2; and the other of X
2 and X
3 is CH2 or O. Embodiment 57. The compound of Embodiment 56, wherein X
2 is CH
2; and X
3 is CHR
L. Embodiment 58. The compound of Embodiment 25, wherein the compound is a compound of Formula (V-a2) or (V-b2):
Formula (V-a2)
Formula (V-b2) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; b1 is 0, 1, or 2; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
2 is -O- or -CH
2-; X
1 is CHR
L; and X
3 is CHR
L or C(R
L)2, wherein: one pair of R
L on different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-4 cycloalkyl ring; and the remaining R
L if present is C1-2 alkyl optionally substituted with 1-3 F. Embodiment 59. The compound of any one of Embodiments 50-58, wherein R
7 is selected from the group consisting of: (a) C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C
1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C
3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 60. The compound of any one of Embodiments 45-59, wherein Y
2 is -CH
2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein
the heterocyclyl is optionally substituted with 1-6 R
a. Embodiment 61. The compound of Embodiment 60, wherein
Embodiment 62. The compound of any one of Embodiments 1-61, wherein the
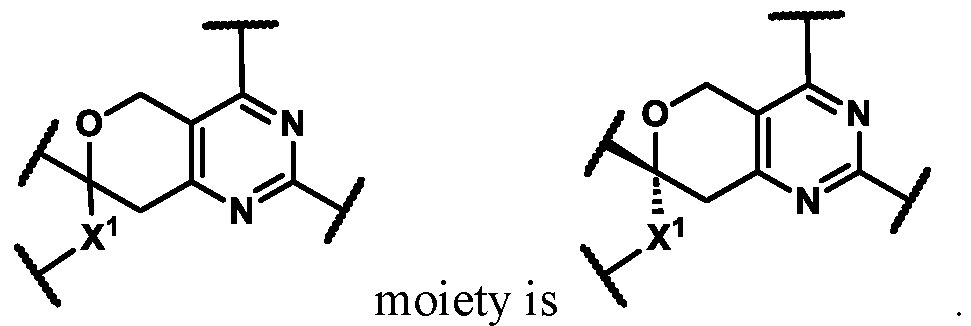
. Embodiment 63. The compound of Embodiment 1, wherein the compound of Formula (II) is selected from the group consisting of Compound Nos.139, 139a, 139b, 139c, 158, 158a, 158b, 158c, 160, 160a, 161, 161a, 161b, 161c, 164, 164a, 164b, 170, 170a, 171, 171a, 176, 176a, 176b, 176c, 176d, 176e, 178, 178a, 178b, 179, 179a, 179b, 179d, 179e, 179f, 180, 180a, 180b, 180c, 181, 181a, 183, 183a, 185, and 185a as depicted in Table C1, or a pharmaceutically acceptable salt thereof. Embodiment 64. The compound of Embodiment 4, wherein the compound of Formula (III) is selected from the group consisting of Compound Nos.158, 158a, 158b, 158c, 161, 161a, 161b, 161c, 176, 176a, 176b, 176c, 176d, 176e, 177, 177a, 178, 178a, 178b, 179, 179a, 179b, 179d, 179e, 179f, 180, 180a, 180b, 180c, 184, 184b, and 184c as depicted in Table C1, or a pharmaceutically acceptable salt thereof. Embodiment 65. The compound of Embodiment 17, wherein the compound of Formula (IV) is selected from the group consisting of Compound Nos.124, 124a, 124b, 124c, 124d, 124e, 124f, 125, 125a, 130, 130a, 130b, 130c, 131, 131a, 131b, 133, 133a, 133b, 134, 134a, 138, 138a, 148, 148a, 162, 162a, 162b, 162c, 162d, 163, 163a, 175, 175a, 176, 176a, 176b, 176c, 176d, 176e, 177, 177a, 178, 178a, 178b, 179, 179a, 179b, 179d, 179e, 179f, 182, 182a, 182b, and 182c as depicted in Table C1, or a pharmaceutically acceptable salt thereof.
Embodiment 66. The compound of Embodiment 25, wherein the compound of Formula (V) is selected from the group consisting of Compound Nos. 172, 172a, 172b, and 172c as depicted in Table C1, or a pharmaceutically acceptable salt thereof. Embodiment 67. A compound of Formula (VI):
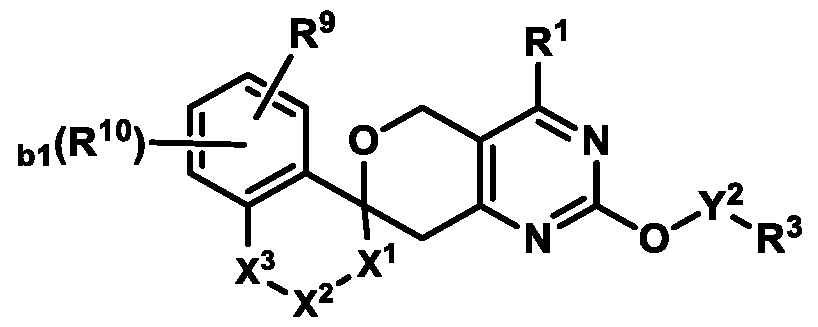
Formula (VI) or a pharmaceutically acceptable salt thereof, wherein: R
1 is a 4-10 membered heterocyclyl substituted with -CN, –(C
1-3 alkylene)-CN, or – (C3-6 cycloalkylene)-CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7; wherein each R
7 is independently selected from the group consisting of R
a and R
b; X
1 is selected from the group consisting of S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; b1 is 0, 1, or 2; R
9 is selected from the group consisting of: H, NR
dR
e, -OH, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which
is optionally substituted with 1-3 independently selected C
1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)
0-2-, C(=O), and C
1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally
substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, C1-3 haloalkyl, C
3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-. Embodiment 68. The compound of Embodiment 67, wherein R
1 is a 4-10 membered heterocyclyl substituted with -CN or –(C1-3 alkylene)-CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7. Embodiment 69. The compound of Embodiment 67 or 68, wherein R
1 is a 6-8 membered heterocyclyl substituted with -CN or –(C1-3 alkylene)-CN on a ring carbon atom, wherein: the heterocyclyl has one ring nitrogen atom and 0-1 ring oxygen atom; and the heterocyclyl is further optionally substituted with 1-3 R
7. Embodiment 70. The compound of any one of Embodiments 67-69, wherein each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 71. The compound of any one of Embodiments 67-69, wherein R
1 is
selected from the group consisting of:
, wherein b3 is 0, 1, or 2. Embodiment 72. The compound of Embodiment 71, wherein each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C
1-3 alkyl optionally substituted with 1-3 R
c (e.g., C
1-3 alkyl optionally substituted with 1-3 -F). Embodiment 73. The compound of Embodiment 71 or 72, wherein b3 is 0. Embodiment 74. The compound of any one of Embodiments 67-73, wherein R
9 is -NR
dR
e or OH (e.g., -NH2). Embodiment 75. The compound of any one of Embodiments 67-74, wherein the compound of Formula (VI) is a compound of Formula (VI-a):
Formula (VI-a) or a pharmaceutically acceptable salt thereof, wherein: each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 76. The compound of any one of Embodiments 67-75, wherein b1 is 1 or 2. Embodiment 77. The compound of any one of Embodiments 67-73, wherein the compound of Formula (VI) is a compound of Formula (VI-b):
Formula (VI-b) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 78. The compound of Embodiment 77, wherein R
9 is -NR
dR
e. Embodiment 79. The compound of any one of Embodiments 67-78, wherein the compound of Formula (VI) is a compound of Formula (VI-c):
Formula (VI-c) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 80. The compound of any one of Embodiments 67-79, wherein X
1 is CH2 or CHR
L. Embodiment 81. The compound of any one of Embodiments 67-80, wherein X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2. Embodiment 82. The compound of any one of Embodiments 67-81, wherein X
1 is CH2; and X
2 and X
3 are both CH2.
Embodiment 83. The compound of any one of Embodiments 67-81, wherein at least one (e.g., one) of X
1, X
2, and X
3 is selected from the group consisting of: CHR
L and C(R
L)2. Embodiment 84. The compound of any one of Embodiments 67-81 or 83, wherein X
1 is CH2; and X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)
2, provided that 1-2 of X
2 and X
3 is independently CHR
L or C(R
L)
2. Embodiment 85. The compound of any one of Embodiments 67-81 or 83-84, wherein X
1 is CH2; X
2 is CH2; and X
3 is CHR
L. Embodiment 86. The compound of any one of Embodiments 67-80, wherein X
1 is CH2; one of X
2 and X
3 is -O-; and the other of X
2 and X
3 is selected from the group consisting of: CH
2, CHR
L, and C(R
L)
2. Embodiment 87. The compound of any one of Embodiments 67-80 or 86, wherein X
1 is CH2; X
2 is -O-; and X
3 is selected from the group consisting of: CH2, CHR
L, and C(R
L)2. Embodiment 88. The compound of any one of Embodiments 67-87, wherein each R
L is independently selected from the group consisting of: CH3, CF3, CHF2, and CH2F. Embodiment 89. The compound of any one of Embodiments 67-73, wherein the compound is a compound of Formula (VI-d):
Formula (VI-d) or a pharmaceutically acceptable salt thereof, wherein: X
1 is selected from the group consisting of S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that at least one of X
1, X
2, and X
3 is CHR
L or C(R
L)2; and further provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)
0-2; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3
alkyl optionally substituted with 1-3 R
c. Embodiment 90. The compound of any one of Embodiments 67-73, wherein the compound is a compound of Formula (VI-e):
Formula (VI-e) or a pharmaceutically acceptable salt thereof, wherein: X
1 is selected from the group consisting of S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that 2-3 of X
1, X
2, and X
3 are independently CHR
L or C(R
L)
2; one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; and each additional R
L is independently selected from the group consisting of: C
1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 91. The compound of Embodiment 89 or 90, wherein R
9 is NH2. Embodiment 92. The compound of any one of Embodiments 89-91, wherein b1 is 1 or 2. Embodiment 93. The compound of any one of Embodiments 89-92, wherein the
moiety is selected from the group consisting of:
, wherein: b4 is 0 or 1; X
2 is -O- or -CH
2-; X
3 is -CH
2- or -CHR
L-, wherein R
L is C
1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 94. The compound of any one of Embodiments 67-93, wherein Y
2 is -CH2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. The compound of any one of Embodiments 67-94, wherein R
3 is
Embodiment 96. The compound of any one of Embodiments 67-95, wherein the
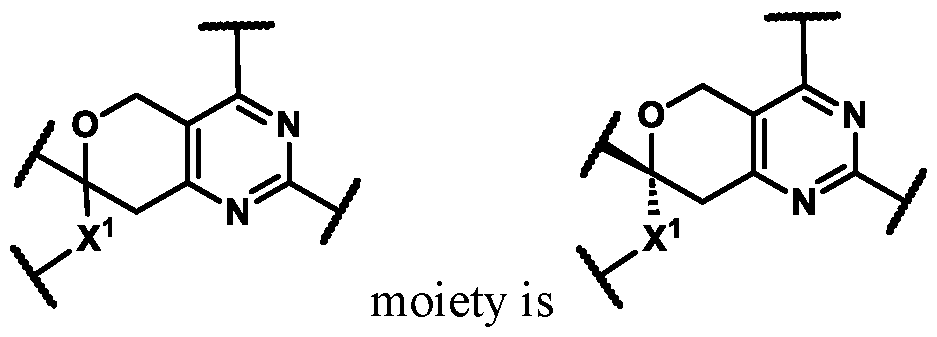
Embodiment 97. The compound of Embodiment 67, wherein the compound of Formula (VI) is selected from the group consisting of Compound Nos.149, 149a, 149b, 149c, 173, 173a, 174, 174a, 186, 186a, 186b, and 186c as depicted in Table C1, or a pharmaceutically acceptable salt thereof. Embodiment 98. A pharmaceutical composition comprising a compound of any 25 one of Embodiments 1-97, or a pharmaceutically acceptable salt thereof, and a
pharmaceutically acceptable excipient. Embodiment 99. A dysregulated KRas protein non-covalently bound with a compound of any one of Embodiments 1-97, or a pharmaceutically acceptable salt thereof. Embodiment 100. A method for treating a KRas-associated cancer in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-97, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 98. Embodiment 101. A method for treating a KRas-associated cancer in a subject in need thereof, the method comprising (a) determining that the cancer in the subject has a KRas dysregulation; and (b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-97, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 98. Embodiment 102. A method of treating a KRas-associated cancer in a subject, the method comprising administering to a subject identified or diagnosed as having a cancer having a KRas dysregulation a therapeutically effective amount of a compound of any one of Embodiments 1-97 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 98. Embodiment 103. A method of treating a KRas-associated cancer in a subject, the method comprising: (a) determining that the cancer in the subject has a KRas dysregulation; and (b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-97 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 98. Embodiment 104. The method of any one of Embodiments 100-103, wherein the KRas-associated cancer is a mutant KRas-associated cancer. Embodiment 105. The method of Embodiment 104, wherein the mutant KRas-
associated cancer is a KRas G12A-associated cancer, a KRas G12C-associated cancer, a KRas G12D-associated cancer, a KRas G12R-associated cancer, a KRas G12S-associated cancer, or a KRas G12V-associated cancer. Embodiment 106. The method of Embodiment 105, wherein the mutant KRas- associated cancer is a KRas G12D-associated cancer or a KRas G12V-associated cancer. Embodiment 107. The method of Embodiment 105, wherein the mutant KRas- associated cancer is a KRas G12D-associated cancer. Embodiment 108. The method of Embodiment 105, wherein the mutant KRas- associated cancer is a KRas G12R-associated cancer. Embodiment 109. The method of Embodiment 105, wherein the mutant KRas- associated cancer is a KRas G12V-associated cancer. Embodiment 110. The method of any one of Embodiments 101 or 103, wherein the step of determining that the cancer in the subject has a KRas dysregulation includes performing an assay to detect the KRas dysregulation (e.g., a KRas mutation) in a tumor sample from the subject. Embodiment 111. The method of Embodiment 110, wherein detecting the KRas dysregulation includes detecting a KRAS gene having a mutation corresponding to a substitution of glycine 12 in a KRas protein and/or a KRas protein having a substitution of glycine 12. Embodiment 112. The method of Embodiment 111, wherein the substitution of glycine 12 is a substitution to alanine, cysteine, aspartic acid, arginine, serine, or valine. Embodiment 113. The method of Embodiment 112, wherein the substitution of glycine 12 is a substitution to aspartic acid. Embodiment 114. The method of Embodiment 112, wherein the substitution of glycine 12 is a substitution to arginine. Embodiment 115. The method of Embodiment 112, wherein the substitution of
glycine 12 is a substitution to valine. Embodiment 116. The method of any one of Embodiments 110-115, comprising obtaining a tumor sample from the subject. Embodiment 117. The method of Embodiment 116, wherein the tumor sample is a biopsy sample. Embodiment 118. The method of any one of Embodiments 110-117, wherein the assay is selected from the group consisting of sequencing, immunohistochemistry, and enzyme-linked immunosorbent assay, and fluorescence in situ hybridization (FISH). Embodiment 119. The method of Embodiment 118, wherein the sequencing is pyrosequencing or next generation sequencing. Embodiment 120. The method of any one of Embodiments 100-119, wherein the KRas-associated cancer is selected from the group consisting of: a hematological cancer, a soft tissue cancer, bile duct cancer, bladder cancer, brain cancer, breast cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, ovarian cancer, pancreatic cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer, thymus cancer, thyroid cancer, urothelial cancer, uterine cancer, and a combination thereof. Embodiment 121. The method of Embodiment 120, wherein the KRas-associated cancer is pancreatic cancer. Embodiment 122. The method of Embodiment 120, wherein the KRas-associated cancer is selected from the group consisting of: a hematological cancer, brain cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, pancreatic cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, thymus cancer, urothelial cancer, and uterine cancer. Embodiment 123. The method of Embodiment 120, wherein the KRas-associated cancer is selected from the group consisting of: the cancer is a hematological cancer, bladder 35 cancer, bile duct cancer, brain cancer, breast cancer, cervical cancer, colon cancer, endometrial
cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, ovarian cancer, pancreatic cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer (e.g., seminoma), thymus cancer, and uterine cancer. Embodiment 124. The method of Embodiment 120, wherein the KRas-associated cancer is selected from the group consisting of: bladder cancer, breast cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, leukemia, lung cancer (e.g., NSCLC), pancreatic cancer, and kidney cancer. Embodiment 125. The method of any one of Embodiments 110-124, comprising administering an additional therapy or therapeutic agent to the subject. Embodiment 126. The method of Embodiment 125, wherein the additional therapy or therapeutic agent is selected from the group consisting of Ras pathway targeted therapeutic agents, kinase-targeted therapeutics, mTORC1 inhibitors or degraders, YAP inhibitors or degraders, proteasome inhibitors or degraders, HSP90 inhibitors or degraders, farnesyl transferase inhibitors or degraders, PTEN inhibitors or degraders, signal transduction pathway inhibitors or degraders, checkpoint inhibitors, modulators of the apoptosis pathway, chemotherapeutics, angiogenesis-targeted therapies, immune-targeted agents, radiotherapy, and combinations thereof. Embodiment 127. A method for a method for modulating KRas protein activity in a mammalian cell, the method comprising contacting the mammalian cell with an effective amount of a compound of any one of Embodiments 1-90, or a pharmaceutically acceptable salt thereof. Embodiment 128. The method of Embodiment 127, wherein the contacting occurs in vivo. Embodiment 129. The method of Embodiment 127, wherein the contacting occurs in vitro. Embodiment 130. The method of Embodiment 127, wherein the contacting occurs ex vivo.
Embodiment 131. The method of any one of Embodiments 127-130, wherein the mammalian cell is a mammalian cancer cell. Embodiment 132. The method of any one of Embodiments 127-131, wherein the KRas protein is a mutant KRas protein. Embodiment 133. The method of Embodiment 132, wherein the mutant KRas protein is a mutant KRas protein selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. P07 Embodiments Embodiment 1. A compound of Formula (II):
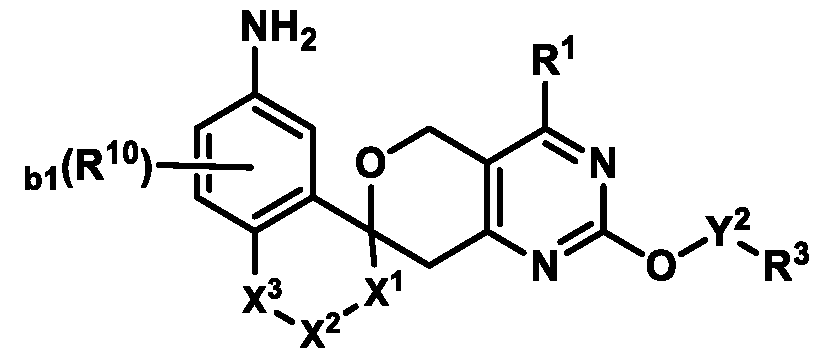
Formula (II) or a pharmaceutically acceptable salt thereof, wherein: R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b;
X
1 is selected from the group consisting of a bond, S(O)
0-2, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; b1 is 1 or 2; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C
1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
(i) C(=O)C
1-6 haloalkyl; (j) C(=O)OH; (k) C(=O)OC1-6 alkyl; (l) C(=O)OC
1-6 haloalkyl; (m) C(=O)N(R
f)2; (n) S(O)0-2(C1-6 alkyl); (o) S(O)
0-2(C
1-6 haloalkyl); (p) S(O)1-2N(R
f)2; and (q) C1-6 alkyl, C2-6 alkenyl, or C2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, C1-3 haloalkyl, C
3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-.
Embodiment 2. The compound of Embodiment 1, wherein the compound is a compound of Formula (II-a):
Formula (II-a) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 3. The compound of Embodiment 1, wherein the compound is a compound of Formula (II-b):
Formula (II-b) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 4. A compound of Formula (III):
Formula (III) or a pharmaceutically acceptable salt thereof, wherein:
R
1 is selected from the group consisting of: (iii) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)
2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; X
1 is selected from the group consisting of a bond, S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)
0-2; R
9 is selected from the group consisting of: H, NR
dR
e, -OH, and halo; b4 is 0 or 1; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C
1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C
1-6 alkoxy, C
1-6 haloalkoxy, C
1-6 alkyl, and C
1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C
1-3 alkyl; R
3 is selected from the group consisting of:
(a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of: ( ) h l
each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)
0-2-, C(=O), and C
1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, C
1-3 haloalkyl, C
3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-. Embodiment 5. The compound of Embodiment 4, wherein R
9 is -NH2; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 6. The compound of any one of Embodiments 1-5, wherein X
1 is CH2 or CHR
L. Embodiment 7. The compound of any one of Embodiments 1-6, wherein X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2. Embodiment 8. The compound of any one of Embodiments 1-7, wherein X
1 is CH2; and X
2 and X
3 are both CH2. Embodiment 9. The compound of any one of Embodiments 1-7, wherein at least one (e.g., one) of X
1, X
2, and X
3 is selected from the group consisting of: CHR
L and C(R
L)2. Embodiment 10. The compound of any one of Embodiments 1-7 or 9, wherein X
1 is CH
2; and X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, and C(R
L)2, provided that 1-2 of X
2 and X
3 is independently CHR
L or C(R
L)2.
Embodiment 11. The compound of any one of Embodiments 1-7 or 9-10, wherein X
1 is CH2; X
2 is CH2; and X
3 is CHR
L. Embodiment 12. The compound of any one of Embodiments 1-6, wherein one of X
2 and X
3 is -O-; and the other of X
2 and X
3 is selected from the group consisting of: CH2, CHR
L, and C(R
L)2. Embodiment 13. The compound of any one of Embodiments 1-6 or 12, wherein X
2 is -O-; and X
3 is selected from the group consisting of: CH2, CHR
L, and C(R
L)2. Embodiment 14. The compound of any one of Embodiments 1-13, wherein each R
L is independently selected from the group consisting of: CH
3, CF
3, CHF
2, and CH
2F. Embodiment 15. The compound of Embodiment 1, wherein the moiety is selected from the group consisting of:
, wherein: b4 is 0 or 1; X
2 is -O- or -CH2-; X
3 is -CH
2- or -CHR
L-, wherein R
L is C
1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 16. The compound of Embodiment 15, wherein is the
moiety is selected from the group consisting of:
,
Embodiment 17. A compound of Formula (IV):
Formula (IV) or a pharmaceutically acceptable salt thereof, wherein: X
1 is selected from the group consisting of a bond, S(O)
0-2, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that at least one of X
1, X
2, and X
3 is CHR
L or C(R
L)
2; further provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; b1 is 0, 1 or 2; R
9 is selected from the group consisting of: H, OH, NR
dR
e, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C
1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and
(iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; Y
2 is a bond or a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
(o) S(O)
0-2(C
1-6 haloalkyl); (p) S(O)1-2N(R
f)2; and (q) C1-6 alkyl, C2-6 alkenyl, or C2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1-
2(C
1-6 alkyl), S(O)
1-2(C
1-6 haloalkyl), S(O)
1-2N(R
f)
2, and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, C
1-3 haloalkyl, C3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. Embodiment 18. The compound of Embodiment 17, wherein the compound is a compound of Formula (IV-a):
Formula (IV-a) or a pharmaceutically acceptable salt thereof, wherein: each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 19. The compound of Embodiment 17, wherein the compound is a compound of Formula (IV-b):
Formula (IV-b) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 20. The compound of any one of Embodiments 17-19, wherein X
1 is CH2. Embodiment 21. The compound of any one of Embodiments 17-20, wherein X
2 is CH2; and X
3 is CHR
L. Embodiment 22. The compound of any one of Embodiments 17-20, wherein X
2 is -O-; and X
3 is selected from the group consisting of: CHR
L and C(R
L)2. Embodiment 23. The compound of any one of Embodiments 17-22, wherein each R
L is independently selected from the group consisting of: CH3, CF3, CHF2, and CH2F.
Embodiment 24. The compound of any one of Embodiments 17-19, wherein the compound is a compound of Formula (IV-c):
Formula (IV-c) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 25. A compound of Formula (V):
Formula (V) or a pharmaceutically acceptable salt thereof, wherein: X
1 is selected from the group consisting of a bond, S(O)
0-2, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that 2-3 of X
1, X
2, and X
3 are independently CHR
L or C(R
L)2; one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; and each additional R
L is independently selected from the group consisting of: C1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; b1 is 0, 1 or 2; R
9 is selected from the group consisting of: H, OH, NR
dR
e, and halo; each R
10 is independently selected from the group consisting of R
a and R
b;
R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)
2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and
wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; Y
2 is a bond or a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C
1-6 alkoxy, C
1-6 haloalkoxy, C
1-6 alkyl, and C
1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C
1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
(i) C(=O)C
1-6 haloalkyl; (j) C(=O)OH; (k) C(=O)OC1-6 alkyl; (l) C(=O)OC
1-6 haloalkyl; (m) C(=O)N(R
f)2; (n) S(O)0-2(C1-6 alkyl); (o) S(O)
0-2(C
1-6 haloalkyl); (p) S(O)1-2N(R
f)2; and (q) C1-6 alkyl, C2-6 alkenyl, or C2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, C1-3 haloalkyl, C
3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-.
Embodiment 26. The compound of Embodiment 25, wherein the compound is a compound of Formula (V-a):
Formula (V-a) or a pharmaceutically acceptable salt thereof, wherein: each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 27. The compound of Embodiment 25, wherein the compound is a compound of Formula (IV-b):
Formula (V-b) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 28. The compound of any one of Embodiments 25-27, wherein X
2 is -O- or -CH2- (e.g., -CH2-). Embodiment 29. The compound of any one of Embodiments 25-28, wherein X
1 is CHR
L; and X
3 is CHR
L, wherein the pair of R
L on different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-4 cycloalkyl ring. Embodiment 30. The compound of any one of Embodiments 25-28, wherein X
1 is CHR
L; and X
3 is C(R
L)2, wherein the pair of R
L on different ring carbon atom(s) taken together
with the ring atom(s) connecting them form a C
3-4 cycloalkyl ring; and the remaining R
L is C
1- 2 alkyl optionally substituted with 1-3 F. Embodiment 31. The compound of any one of Embodiments 25-27, wherein the compound is a compound of Formula (V-c) or Formula (V-d):
Formula (V-d) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; the pair of R
L1 taken together with the ring atom(s) connecting them form a C3-4 cycloalkyl ring, and R
L2 is C1-2 alkyl optionally substituted with 1-3 F. Embodiment 32. The compound of any one of Embodiments 1-31, wherein Y
2 is -CH
2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. Embodiment 33. The compound of any one of Embodiments 1-32, wherein R
3 is
4. The compound of any one of Embodiments 1-33, wherein R
1 is
, erein b2 is 0, 1, or 2, and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7. Embodiment 35. The compound of any one of Embodiments 1-34, wherein R
1 is
alkyl)R
b1, -C(O)N(H)R
b1, R
b1, and C(O)R
b1. Embodiment 37. The compound of Embodiment 36, wherein R
7 is selected from the group consisting of: (a) C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C
1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C
3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is 4-10 membered heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom.
Embodiment 38. The compound of any one of Embodiments 35-37, wherein R
7 is C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h. Embodiment 39. The compound of any one of Embodiments 1-35, wherein R
1 is
, wherein R
7a and R
7b are independently selected R
7. Embodiment 40. The compound of Embodiment 39, wherein R
7a is selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, -C(O)N(H)R
b1, R
b1, and C(O)R
b1; and R
7b is -halo, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 41. The compound of Embodiment 40, wherein R
7a is selected from the group consisting of: (a) C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C
1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C
3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is 4-10 membered heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 42. The compound of any one of Embodiments 1-33, wherein R
1 is a 7-10 (e.g., 7) membered heterocyclyl having one ring nitrogen atom, one ring oxygen atom, and no additional ring heteroatoms, wherein the 7-10 membered heterocyclyl is optionally substituted with 1-4 R
7. Embodiment 43. The compound of Embodiment 42, wherein
optionally substituted with 1-4 R
7 at one or more ring carbon atoms
Embodiment 44. The compound of Embodiment 43, wherein each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C1-3 alkyl optionally substituted with 1-3 R
c, wherein: each R
c is independently selected from the group consisting of: -F, -OH, and -CN. Embodiment 45. The compound of Embodiment 1, wherein the compound is a compound of Formula (II-a1) or (II-b1):
Formula (II-b1) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; b3 is 0, 1, 2, or 3; X
1 is CH
2; and X
2 and X
3 are independently selected from the group consisting of: O, CH2, CHR
L, and C(R
L)2.
Embodiment 46. The compound of Embodiment 4, wherein the compound is a compound of Formula (III-1):
Formula (III-1) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; b3 is 0, 1, 2, or 3; X
1 is CH
2; and X
2 and X
3 are independently selected from the group consisting of: O, CH2, CHR
L, and C(R
L)2. Embodiment 47. The compound of Embodiment 46, wherein R
9 is NR
dR
e (e.g., - NH2). Embodiment 48. The compound of Embodiment 17, wherein the compound is a compound of Formula (IV-a1) or (IV-b1):
Formula (IV-a1)
Formula (IV-b1) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; b1 is 0, 1, or 2; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; b3 is 0, 1, 2, or 3; X
1 is CH2; one of X
2 and X
3 is independently selected from the group consisting of: CHR
L and C(R
L)
2; and the other of X
2 and X
3 is CH
2 or O. Embodiment 49. The compound of Embodiment 48, wherein X
2 is CH2; and X
3 is CHR
L. Embodiment 50. The compound of Embodiment 25, wherein the compound is a compound of Formula (V-a1) or (V-b1):
Formula (V-a1)
Formula (V-b1) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; b1 is 0, 1, or 2; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; b3 is 0, 1, 2, or 3; X
2 is -O- or -CH2-; X
1 is CHR
L; and X
3 is CHR
L or C(R
L)2, wherein: one pair of R
L on different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-4 cycloalkyl ring; and the remaining R
L if present is C1-2 alkyl optionally substituted with 1-3 F. Embodiment 51. The compound of any one of Embodiments 45-50, wherein b3 is 0. Embodiment 52. The compound of any one of Embodiments 45-51, wherein b3 is 1 or 2; and each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C1-3 alkyl optionally substituted with 1-3 R
c, wherein each R
c is independently selected from the group consisting of: -F, -OH, and -CN. Embodiment 53. The compound of Embodiment 1, wherein the compound is a compound of Formula (II-a2) or (II-b2):
Formula (II-b2) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)
2, C(O)N(C
1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
1 is CH2; and X
2 and X
3 are independently selected from the group consisting of: O, CH
2, CHR
L, and C(R
L)
2. Embodiment 54. The compound of Embodiment 4, wherein the compound is a compound of Formula (III-2):
Formula (III-2) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)
2, C(O)N(C
1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
1 is CH
2; and X
2 and X
3 are independently selected from the group consisting of: O, CH
2, CHR
L, and C(R
L)2. Embodiment 55. The compound of Embodiment 54, wherein R
9 is NR
dR
e (e.g., - NH
2). Embodiment 56. The compound of Embodiment 17, wherein the compound is a compound of Formula (IV-a2) or (IV-b2):
Formula (IV-a2)
Formula (IV-b2) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; b1 is 0, 1, or 2; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)
2, C(O)N(C
1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
1 is CH
2; one of X
2 and X
3 is independently selected from the group consisting of: CHR
L and C(R
L)2; and the other of X
2 and X
3 is CH2 or O. Embodiment 57. The compound of Embodiment 56, wherein X
2 is CH
2; and X
3 is CHR
L. Embodiment 58. The compound of Embodiment 25, wherein the compound is a compound of Formula (V-a2) or (V-b2):
Formula (V-a2)
Formula (V-b2) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; b1 is 0, 1, or 2; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
2 is -O- or -CH
2-; X
1 is CHR
L; and X
3 is CHR
L or C(R
L)2, wherein: one pair of R
L on different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-4 cycloalkyl ring; and the remaining R
L if present is C1-2 alkyl optionally substituted with 1-3 F. Embodiment 59. The compound of any one of Embodiments 50-58, wherein R
7 is selected from the group consisting of: (a) C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C
1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C
3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is 4-10 membered heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 60. The compound of any one of Embodiments 45-59, wherein Y
2 is -CH
2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein
the heterocyclyl is optionally substituted with 1-6 R
a. Embodiment 61. The compound of Embodiment 60, wherein
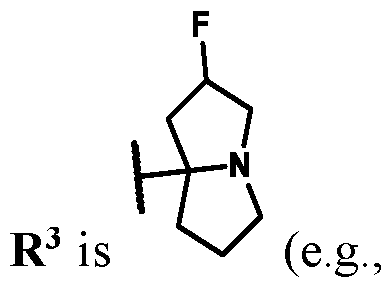

Embodiment 63. The compound of Embodiment 1, wherein the compound of Formula (II) is selected from the group consisting of Compound Nos.139, 139a, 139b, 139c, 158, 158a, 158b, 158c, 160, 160a, 161, 161a, 161b, 161c, 164, 164a, 164b, 170, 170a, 171, 171a, 176, 176a, 176b, 176c, 176d, 176e, 178, 178a, 178b, 179, 179a, 179b, 179d, 179e, 179f, 180, 180a, 180b, 180c, 181, 181a, 183, 183a, 185, and 185a as depicted in Table C1, or a pharmaceutically acceptable salt thereof. Embodiment 64. The compound of Embodiment 4, wherein the compound of Formula (III) is selected from the group consisting of Compound Nos.158, 158a, 158b, 158c, 161, 161a, 161b, 161c, 176, 176a, 176b, 176c, 176d, 176e, 177, 177a, 178, 178a, 178b, 179, 179a, 179b, 179d, 179e, 179f, 180, 180a, 180b, 180c, 184, 184b, and 184c as depicted in Table C1, or a pharmaceutically acceptable salt thereof. Embodiment 65. The compound of Embodiment 17, wherein the compound of Formula (IV) is selected from the group consisting of Compound Nos.124, 124a, 124b, 124c, 124d, 124e, 124f, 125, 125a, 130, 130a, 130b, 130c, 131, 131a, 131b, 133, 133a, 133b, 134, 134a, 138, 138a, 148, 148a, 162, 162a, 162b, 162c, 162d, 163, 163a, 175, 175a, 176, 176a, 176b, 176c, 176d, 176e, 177, 177a, 178, 178a, 178b, 179, 179a, 179b, 179d, 179e, 179f, 182, 182a, 182b, and 182c as depicted in Table C1, or a pharmaceutically acceptable salt thereof.
Embodiment 66. The compound of Embodiment 25, wherein the compound of Formula (V) is selected from the group consisting of Compound Nos. 172, 172a, 172b, and 172c as depicted in Table C1, or a pharmaceutically acceptable salt thereof. Embodiment 67. A compound of Formula (VI):

Formula (VI) or a pharmaceutically acceptable salt thereof, wherein: R
1 is a 4-10 membered heterocyclyl substituted with -CN, –(C
1-3 alkylene)-CN, or – (C3-6 cycloalkylene)-CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7; wherein each R
7 is independently selected from the group consisting of R
a and R
b; X
1 is selected from the group consisting of S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; b1 is 0, 1, or 2; R
9 is selected from the group consisting of: H, NR
dR
e, -OH, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which
is optionally substituted with 1-3 independently selected C
1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)
0-2-, C(=O), and C
1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally
substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, C1-3 haloalkyl, C
3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-. Embodiment 68. The compound of Embodiment 67, wherein R
1 is a 4-10 membered heterocyclyl substituted with -CN or –(C1-3 alkylene)-CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7. Embodiment 69. The compound of Embodiment 67 or 68, wherein R
1 is a 6-8 membered heterocyclyl substituted with -CN or –(C1-3 alkylene)-CN on a ring carbon atom, wherein: the heterocyclyl has one ring nitrogen atom and 0-1 ring oxygen atom; and the heterocyclyl is further optionally substituted with 1-3 R
7. Embodiment 70. The compound of any one of Embodiments 67-69, wherein each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 71. The compound of any one of Embodiments 67-69, wherein R
1 is
selected from the group consisting of:
, wherein b3 is 0, 1, or 2. Embodiment 72. The compound of Embodiment 71, wherein each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C
1-3 alkyl optionally substituted with 1-3 R
c (e.g., C
1-3 alkyl optionally substituted with 1-3 -F). Embodiment 73. The compound of Embodiment 71 or 72, wherein b3 is 0. Embodiment 74. The compound of any one of Embodiments 67-73, wherein R
9 is -NR
dR
e or OH (e.g., -NH2). Embodiment 75. The compound of any one of Embodiments 67-74, wherein the compound of Formula (VI) is a compound of Formula (VI-a):
Formula (VI-a) or a pharmaceutically acceptable salt thereof, wherein: each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 76. The compound of any one of Embodiments 67-75, wherein b1 is 1 or 2. Embodiment 77. The compound of any one of Embodiments 67-73, wherein the compound of Formula (VI) is a compound of Formula (VI-b):
Formula (VI-b) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 78. The compound of Embodiment 77, wherein R
9 is -NR
dR
e. Embodiment 79. The compound of any one of Embodiments 67-78, wherein the compound of Formula (VI) is a compound of Formula (VI-c):
Formula (VI-c) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 80. The compound of any one of Embodiments 67-79, wherein X
1 is CH
2 or CHR
L. Embodiment 81. The compound of any one of Embodiments 67-80, wherein X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, and C(R
L)
2. Embodiment 82. The compound of any one of Embodiments 67-81, wherein X
1 is CH
2; and X
2 and X
3 are both CH
2.
Embodiment 83. The compound of any one of Embodiments 67-81, wherein at least one (e.g., one) of X
1, X
2, and X
3 is selected from the group consisting of: CHR
L and C(R
L)2. Embodiment 84. The compound of any one of Embodiments 67-81 or 83, wherein X
1 is CH2; and X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)
2, provided that 1-2 of X
2 and X
3 is independently CHR
L or C(R
L)
2. Embodiment 85. The compound of any one of Embodiments 67-81 or 83-84, wherein X
1 is CH2; X
2 is CH2; and X
3 is CHR
L. Embodiment 86. The compound of any one of Embodiments 67-80, wherein X
1 is CH2; one of X
2 and X
3 is -O-; and the other of X
2 and X
3 is selected from the group consisting of: CH
2, CHR
L, and C(R
L)
2. Embodiment 87. The compound of any one of Embodiments 67-80 or 86, wherein X
1 is CH2; X
2 is -O-; and X
3 is selected from the group consisting of: CH2, CHR
L, and C(R
L)2. Embodiment 88. The compound of any one of Embodiments 67-87, wherein each R
L is independently selected from the group consisting of: CH3, CF3, CHF2, and CH2F. Embodiment 89. The compound of any one of Embodiments 67-73, wherein the compound is a compound of Formula (VI-d):
Formula (VI-d) or a pharmaceutically acceptable salt thereof, wherein: X
1 is selected from the group consisting of S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that at least one of X
1, X
2, and X
3 is CHR
L or C(R
L)2; and further provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)
0-2; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3
alkyl optionally substituted with 1-3 R
c. Embodiment 90. The compound of any one of Embodiments 67-73, wherein the compound is a compound of Formula (VI-e):
Formula (VI-e) or a pharmaceutically acceptable salt thereof, wherein: X
1 is selected from the group consisting of S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that 2-3 of X
1, X
2, and X
3 are independently CHR
L or C(R
L)
2; one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; and each additional R
L is independently selected from the group consisting of: C
1-3 alkoxy, -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 91. The compound of Embodiment 89 or 90, wherein R
9 is NH2. Embodiment 92. The compound of any one of Embodiments 89-91, wherein b1 is 1 or 2. Embodiment 93. The compound of any one of Embodiments 89-92, wherein the
moiety is selected from the group consisting of:
, wherein: b4 is 0 or 1; X
2 is -O- or -CH
2-; X
3 is -CH
2- or -CHR
L-, wherein R
L is C
1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 94. The compound of any one of Embodiments 67-93, wherein Y
2 is -CH2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. Embodiment 95. The compound of any one of Embodiments 67-94, wherein R
3 is
Embodiment 96. The compound of any one of Embodiments 67-95, wherein the
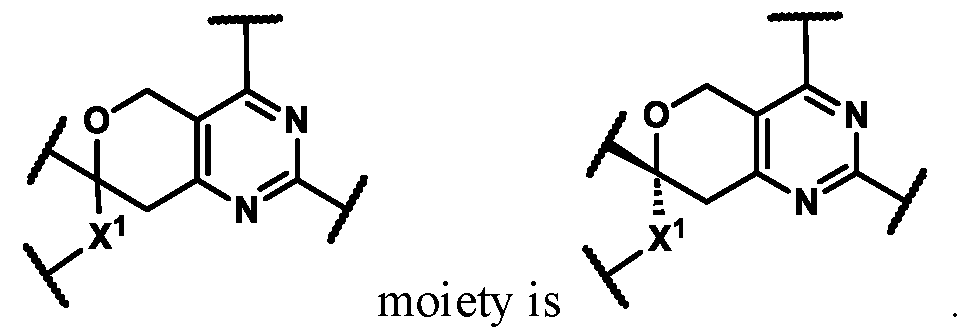
. Embodiment 97. The compound of Embodiment 67, wherein the compound of Formula (VI) is selected from the group consisting of Compound Nos.149, 149a, 149b, 149c, 173, 173a, 174, 174a, 186, 186a, 186b, and 186c as depicted in Table C1, or a pharmaceutically acceptable salt thereof. Embodiment 98. A pharmaceutical composition comprising a compound of any 25 one of Embodiments 1-97, or a pharmaceutically acceptable salt thereof, and a
pharmaceutically acceptable excipient. Embodiment 99. A dysregulated KRas protein non-covalently bound with a compound of any one of Embodiments 1-97, or a pharmaceutically acceptable salt thereof. Embodiment 100. A method for treating a KRas-associated cancer in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-97, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 98. Embodiment 101. A method for treating a KRas-associated cancer in a subject in need thereof, the method comprising (a) determining that the cancer in the subject has a KRas dysregulation; and (b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-97, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 98. Embodiment 102. A method of treating a KRas-associated cancer in a subject, the method comprising administering to a subject identified or diagnosed as having a cancer having a KRas dysregulation a therapeutically effective amount of a compound of any one of Embodiments 1-97 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 98. Embodiment 103. A method of treating a KRas-associated cancer in a subject, the method comprising: (a) determining that the cancer in the subject has a KRas dysregulation; and (b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-97 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 98. Embodiment 104. The method of any one of Embodiments 100-103, wherein the KRas-associated cancer is a mutant KRas-associated cancer. Embodiment 105. The method of Embodiment 104, wherein the mutant KRas-
associated cancer is a KRas G12A-associated cancer, a KRas G12C-associated cancer, a KRas G12D-associated cancer, a KRas G12R-associated cancer, a KRas G12S-associated cancer, or a KRas G12V-associated cancer. Embodiment 106. The method of Embodiment 105, wherein the mutant KRas- associated cancer is a KRas G12D-associated cancer or a KRas G12V-associated cancer. Embodiment 107. The method of Embodiment 105, wherein the mutant KRas- associated cancer is a KRas G12D-associated cancer. Embodiment 108. The method of Embodiment 105, wherein the mutant KRas- associated cancer is a KRas G12R-associated cancer. Embodiment 109. The method of Embodiment 105, wherein the mutant KRas- associated cancer is a KRas G12V-associated cancer. Embodiment 110. The method of any one of Embodiments 101 or 103, wherein the step of determining that the cancer in the subject has a KRas dysregulation includes performing an assay to detect the KRas dysregulation (e.g., a KRas mutation) in a tumor sample from the subject. Embodiment 111. The method of Embodiment 110, wherein detecting the KRas dysregulation includes detecting a KRAS gene having a mutation corresponding to a substitution of glycine 12 in a KRas protein and/or a KRas protein having a substitution of glycine 12. Embodiment 112. The method of Embodiment 111, wherein the substitution of glycine 12 is a substitution to alanine, cysteine, aspartic acid, arginine, serine, or valine. Embodiment 113. The method of Embodiment 112, wherein the substitution of glycine 12 is a substitution to aspartic acid. Embodiment 114. The method of Embodiment 112, wherein the substitution of glycine 12 is a substitution to arginine. Embodiment 115. The method of Embodiment 112, wherein the substitution of
glycine 12 is a substitution to valine. Embodiment 116. The method of any one of Embodiments 110-115, comprising obtaining a tumor sample from the subject. Embodiment 117. The method of Embodiment 116, wherein the tumor sample is a biopsy sample. Embodiment 118. The method of any one of Embodiments 110-117, wherein the assay is selected from the group consisting of sequencing, immunohistochemistry, and enzyme-linked immunosorbent assay, and fluorescence in situ hybridization (FISH). Embodiment 119. The method of Embodiment 118, wherein the sequencing is pyrosequencing or next generation sequencing. Embodiment 120. The method of any one of Embodiments 100-119, wherein the KRas-associated cancer is selected from the group consisting of: a hematological cancer, a soft tissue cancer, bile duct cancer, bladder cancer, brain cancer, breast cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, ovarian cancer, pancreatic cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer, thymus cancer, thyroid cancer, urothelial cancer, uterine cancer, and a combination thereof. Embodiment 121. The method of Embodiment 120, wherein the KRas-associated cancer is pancreatic cancer. Embodiment 122. The method of Embodiment 120, wherein the KRas-associated cancer is selected from the group consisting of: a hematological cancer, brain cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, pancreatic cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, thymus cancer, urothelial cancer, and uterine cancer. Embodiment 123. The method of Embodiment 120, wherein the KRas-associated cancer is selected from the group consisting of: the cancer is a hematological cancer, bladder 35 cancer, bile duct cancer, brain cancer, breast cancer, cervical cancer, colon cancer, endometrial
cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, ovarian cancer, pancreatic cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer (e.g., seminoma), thymus cancer, and uterine cancer. Embodiment 124. The method of Embodiment 120, wherein the KRas-associated cancer is selected from the group consisting of: bladder cancer, breast cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, leukemia, lung cancer (e.g., NSCLC), pancreatic cancer, and kidney cancer. Embodiment 125. The method of any one of Embodiments 110-124, comprising administering an additional therapy or therapeutic agent to the subject. Embodiment 126. The method of Embodiment 125, wherein the additional therapy or therapeutic agent is selected from the group consisting of Ras pathway targeted therapeutic agents, kinase-targeted therapeutics, mTORC1 inhibitors or degraders, YAP inhibitors or degraders, proteasome inhibitors or degraders, HSP90 inhibitors or degraders, farnesyl transferase inhibitors or degraders, PTEN inhibitors or degraders, signal transduction pathway inhibitors or degraders, checkpoint inhibitors, modulators of the apoptosis pathway, chemotherapeutics, angiogenesis-targeted therapies, immune-targeted agents, radiotherapy, and combinations thereof. Embodiment 127. A method for a method for modulating KRas protein activity in a mammalian cell, the method comprising contacting the mammalian cell with an effective amount of a compound of any one of Embodiments 1-90, or a pharmaceutically acceptable salt thereof. Embodiment 128. The method of Embodiment 127, wherein the contacting occurs in vivo. Embodiment 129. The method of Embodiment 127, wherein the contacting occurs in vitro. Embodiment 130. The method of Embodiment 127, wherein the contacting occurs ex vivo.
Embodiment 131. The method of any one of Embodiments 127-130, wherein the mammalian cell is a mammalian cancer cell. Embodiment 132. The method of any one of Embodiments 127-131, wherein the KRas protein is a mutant KRas protein. Embodiment 133. The method of Embodiment 132, wherein the mutant KRas protein is a mutant KRas protein selected from the group consisting of: a KRas G12A mutant protein, a KRas G12C mutant protein, a KRas G12D mutant protein, a KRas G12R mutant protein, a KRas G12S mutant protein, and a KRas G12V mutant protein. Exemplary Embodiments of Formula (II) Embodiment 1. A compound of Formula (II):
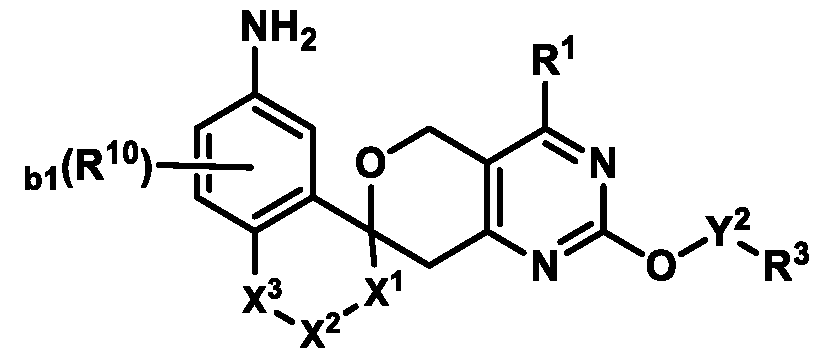
Formula (II) or a pharmaceutically acceptable salt thereof, wherein: R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and (iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b;
X
1 is selected from the group consisting of a bond, S(O)
0-2, CH
2, CHR
L, C(R
L)
2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)
2, O, and S(O)
0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)0-2; b1 is 1 or 2; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C
1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of: (a) halo; (b) cyano; (c) -OH; (d) oxo; (e) -C1-6 alkoxy; (f) -C
1-6 haloalkoxy; (g) -NR
dR
e; (h) C(=O)C1-6 alkyl;
(i) C(=O)C
1-6 haloalkyl; (j) C(=O)OH; (k) C(=O)OC1-6 alkyl; (l) C(=O)OC
1-6 haloalkyl; (m) C(=O)N(R
f)2; (n) S(O)0-2(C1-6 alkyl); (o) S(O)
0-2(C
1-6 haloalkyl); (p) S(O)1-2N(R
f)2; and (q) C1-6 alkyl, C2-6 alkenyl, or C2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C
1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, C1-3 haloalkyl, C
3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-.
Embodiment 2. The compound of Embodiment 1, wherein the compound is a compound of Formula (II-a):
Formula (II-a) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 3. The compound of Embodiment 1, wherein the compound is a compound of Formula (II-b):
Formula (II-b) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 4. The compound of any one of Embodiments 1-3, wherein X
1 is CH
2 or CHR
L. Embodiment 5. The compound of any one of Embodiments 1-4, wherein X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2. Embodiment 6. The compound of any one of Embodiments 1-5, wherein X
1 is
CH
2; and X
2 and X
3 are both CH
2. Embodiment 7. The compound of any one of Embodiments 1-5, wherein at least one (e.g., one) of X
1, X
2, and X
3 is selected from the group consisting of: CHR
L and C(R
L)
2. Embodiment 8. The compound of any one of Embodiments 1-5 or 7, wherein X
1 is CH
2; and X
2 and X
3 are independently selected from the group consisting of: CH
2, CHR
L, and C(R
L)2, provided that 1-2 of X
2 and X
3 is independently CHR
L or C(R
L)2. Embodiment 9. The compound of any one of Embodiments 1-5 or 7-8, wherein X
1 is CH
2; X
2 is CH
2; and X
3 is CHR
L. Embodiment 10. The compound of any one of Embodiments 1-4, wherein one of X
2 and X
3 is -O-; and the other of X
2 and X
3 is selected from the group consisting of: CH
2, CHR
L, and C(R
L)
2. Embodiment 11. The compound of any one of Embodiments 1-4 or 10, wherein X
2 is -O-; and X
3 is selected from the group consisting of: CH
2, CHR
L, and C(R
L)
2. Embodiment 12. The compound of any one of Embodiments 1-11, wherein each R
L is independently selected from the group consisting of: CH3, CF3, CHF2, and CH2F. Embodiment 13. The compound of Embodiment 1, wherein the
b4 is 0 or 1; X
2 is -O- or -CH2-; X
3 is -CH2- or -CHR
L-, wherein R
L is C1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3
alkyl optionally substituted with 1-3 R
c. Embodiment 14. The compound of Embodiments 1 or 13, wherein the
Embodiment 15. The compound of any one of Embodiments 1-14, wherein Y
2 is -CH
2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. Embodiment 16. The compound of any one of Embodiments 1-15, wherein R
3 is 17. The compound of any one of Embodiments 1-16, wherein R
1 is
, erein b2 is 0, 1, or 2, and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7. Embodiment 18. The compound of any one of Embodiments 1-17, wherein R
1 is
alkyl)R
b1, -C(O)N(H)R
b1, R
b1, and C(O)R
b1. Embodiment 20. The compound of Embodiment 19, wherein R
7 is selected from the group consisting of: (a) C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is 4-10 membered heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 21. The compound of any one of Embodiments 18-20, wherein R
7 is C(=O)N(R
f)
2, wherein each R
f is independently H or C
1-3 alkyl optionally substituted with 1-3 R
h. Embodiment 22. The compound of any one of Embodiments 1-18, wherein R
1 is
, wherein R
7a and R
7b are independently selected R
7. Embodiment 23. The compound of Embodiment 22, wherein R
7a is selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, -C(O)N(H)R
b1, R
b1, and C(O)R
b1; and
R
7b is -halo, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 24. The compound of Embodiment 23, wherein R
7a is selected from the group consisting of: (a) C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C
1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C
3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is 4-10 membered heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 25. The compound of any one of Embodiments 1-16, wherein R
1 is a 7-10 (e.g., 7) membered heterocyclyl having one ring nitrogen atom, one ring oxygen atom, and no additional ring heteroatoms, wherein the 7-10 membered heterocyclyl is optionally substituted with 1-4 R
7. Embodiment 26. The compound of Embodiment 25, wherein
optionally substituted with 1-4 R
7 at one or more ring carbon atoms
Embodiment 27. The compound of Embodiment 26, wherein each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C1-3 alkyl optionally substituted with 1-3 R
c, wherein: each R
c is independently selected from the group consisting of: -F, -OH, and -CN. Embodiment 28. The compound of Embodiment 1, wherein the compound is a compound of Formula (II-a1):
Formula (II-a1) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; b3 is 0, 1, 2, or 3; X
1 is CH
2; and X
2 and X
3 are independently selected from the group consisting of: O, CH2, CHR
L, and C(R
L)
2. Embodiment 29. The compound of Embodiment 28, wherein b3 is 0. Embodiment 30. The compound of any one of Embodiments 28 or 29, wherein b3 is 1 or 2; and each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C1-3 alkyl optionally substituted with 1-3 R
c, wherein each R
c is independently selected from the group consisting of: -F, -OH, and -CN. Embodiment 31. The compound of Embodiment 1, wherein the compound is a compound of Formula (II-a2):
Formula (II-a2) or a pharmaceutically acceptable salt thereof, wherein:
b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)
2, C(O)N(C
1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
1 is CH2; and X
2 and X
3 are independently selected from the group consisting of: O, CH
2, CHR
L, and C(R
L)2. Embodiment 32. The compound of Embodiment 31, wherein R
7 is selected from the group consisting of: (a) C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C
1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C
3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is 4-10 membered heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 33. The compound of any one of Embodiments 28-32, wherein Y
2 is -CH
2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. Embodiment 34. The compound of Embodiment 33, wherein
,
Embodiment 35. The compound of any one of Embodiments 1-34, wherein the

Embodiment 36. The compound of Embodiment 1, wherein the compound of Formula (II) is selected from the group consisting of Compound Nos.139, 139a, 139b, 139c, 158, 158a, 158b, 158c, 160, 160a, 161, 161a, 161b, 161c, 164, 164a, 164b, 170, 170a, 171, 171a, 176, 176a, 176b, 176c, 176d, 176e, 178, 178a, 178b, 179, 179a, 179b, 179d, 179e, 179f, 180, 180a, 180b, 180c, 181, 181a, 183, 183a, 185, and 185a as depicted in Table C1, or a pharmaceutically acceptable salt thereof. Embodiment 37. A pharmaceutical composition comprising a compound of any one of Embodiments 1-36, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient. Embodiment 38. A compound of any one of Embodiments 1-36, or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 37 for use in treatment. Embodiment 39. A compound of any one of Embodiments 1-36 or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 37 for use in the treatment of a KRAS associated cancer. Embodiment 40. A compound of any one of Embodiments 1-36 or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 37 for use in a method of treating a KRAS associated cancer, the method comprising (a) determining that the cancer in the subject has a KRas dysregulation; and (b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1- 36, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 37. Embodiment 41. A compound of any one of Embodiments 1-36 or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment
37 for use in a method of treating a KRAS associated cancer, the method comprising administering to a subject identified or diagnosed as having a cancer having a KRas dysregulation a therapeutically effective amount of a compound of any one of Embodiments 1-36 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 37. Embodiment 42. A compound of any one of Embodiments 1-36 or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 37 for use in a method of treating a KRAS associated cancer, the method comprising: (a) determining that the cancer in the subject has a KRas dysregulation; and (b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-36 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 37. Embodiment 43. The compound or pharmaceutical composition of any one of Embodiments 39-42, wherein the KRas-associated cancer is a mutant KRas-associated cancer. Embodiment 44. The compound or pharmaceutical composition of Embodiment 43, wherein the mutant KRas-associated cancer is a KRas G12A-associated cancer, a KRas G12C-associated cancer, a KRas G12D-associated cancer, a KRas G12R-associated cancer, a KRas G12S-associated cancer, or a KRas G12V-associated cancer; optionally wherein the mutant KRas-associated cancer is a KRas G12D-associated cancer or a KRas G12V-associated cancer; optionally wherein the mutant KRas-associated cancer is a KRas G12D-associated cancer; optionally wherein the mutant KRas-associated cancer is a KRas G12R-associated cancer; or optionally wherein the mutant KRas-associated cancer is a KRas G12V-associated cancer. Embodiment 45. The compound or pharmaceutical composition of any one of Embodiments 40 or 42, wherein the step of determining that the cancer in the subject has a KRas dysregulation includes performing an assay to detect the KRas dysregulation (e.g., a KRas mutation) in a tumor sample from the subject. Embodiment 46. The compound or pharmaceutical composition of Embodiment
45, wherein detecting the KRas dysregulation includes detecting a KRAS gene having a mutation corresponding to a substitution of glycine 12 in a KRas protein and/or a KRas protein having a substitution of glycine 12. Embodiment 47. The compound or pharmaceutical composition of Embodiment 46, wherein the substitution of glycine 12 is a substitution to alanine, cysteine, aspartic acid, arginine, serine, or valine; optionally wherein the substitution of glycine 12 is a substitution to aspartic acid; optionally wherein the substitution of glycine 12 is a substitution to arginine; or optionally wherein the substitution of glycine 12 is a substitution to valine. Embodiment 48. The compound or pharmaceutical composition of any one of Embodiments 39-47, wherein the KRas-associated cancer is selected from the group consisting of: a hematological cancer, a soft tissue cancer, bile duct cancer, bladder cancer, brain cancer, breast cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, ovarian cancer, pancreatic cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer, thymus cancer, thyroid cancer, urothelial cancer, uterine cancer, and a combination thereof; optionally wherein the KRas-associated cancer is pancreatic cancer; optionally wherein the KRas-associated cancer is selected from the group consisting of: a hematological cancer, brain cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, pancreatic cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, thymus cancer, urothelial cancer, and uterine cancer; optionally wherein the KRas-associated cancer is selected from the group consisting of: the cancer is a hematological cancer, bladder cancer, bile duct cancer, brain cancer, breast cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, ovarian cancer, pancreatic cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer (e.g., seminoma), thymus cancer, and uterine cancer; optionally wherein the KRas-associated cancer is selected from the group consisting of: bladder cancer, breast cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, leukemia, lung cancer (e.g., NSCLC), pancreatic cancer, and kidney cancer.
Embodiment 49. The compound or pharmaceutical composition of any one of Embodiments 39-48, the method comprises administering an additional therapy or therapeutic agent to the subject. Embodiment 50. The compound or pharmaceutical composition of Embodiment 49, wherein the additional therapy or therapeutic agent is selected from the group consisting of Ras pathway targeted therapeutic agents, kinase-targeted therapeutics, mTORC1 inhibitors or degraders, YAP inhibitors or degraders, proteasome inhibitors or degraders, HSP90 inhibitors or degraders, farnesyl transferase inhibitors or degraders, PTEN inhibitors or degraders, signal transduction pathway inhibitors or degraders, checkpoint inhibitors, modulators of the apoptosis pathway, chemotherapeutics, angiogenesis-targeted therapies, immune-targeted agents, radiotherapy, and combinations thereof. Embodiment 51. The compound or pharmaceutical composition of Embodiment 50, wherein the compound of any one of Embodiments 1-36, or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 37, and the additional therapeutic agent or therapy are administered simultaneously, separately, or sequentially. Exemplary Formula (III) Embodiments Embodiment 1. A compound of Formula (III):
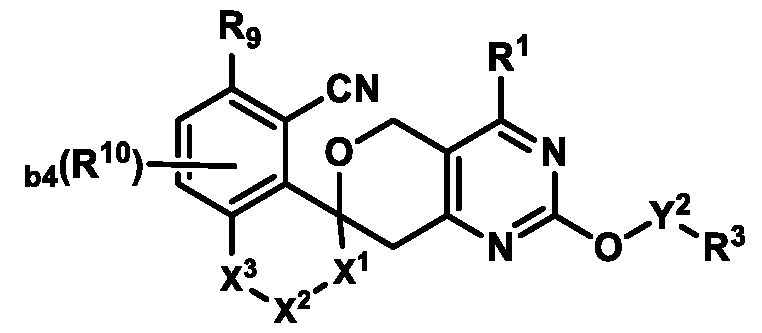
Formula (III) or a pharmaceutically acceptable salt thereof, wherein: R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and
(iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; X
1 is selected from the group consisting of a bond, S(O)0-2, CH2, CHR
L, C(R
L)2, and O; X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, C(R
L)2, O, and S(O)0-2, provided that no more than one of X
1, X
2, and X
3 is selected from the group consisting of: O and S(O)
0-2; R
9 is selected from the group consisting of: H, NR
dR
e, -OH, and halo; b4 is 0 or 1; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of C
1-3 alkoxy, -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; or one pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-6 cycloalkyl ring; Y
2 is a bond or a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or one pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C
1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of: (a) halo; (b) cyano;
(c) -OH; (d) oxo; (e) -C1-6 alkoxy; (f) -C
1-6 haloalkoxy; (g) -NR
dR
e; (h) C(=O)C1-6 alkyl; (i) C(=O)C
1-6 haloalkyl; (j) C(=O)OH; (k) C(=O)OC1-6 alkyl; (l) C(=O)OC
1-6 haloalkyl; (m) C(=O)N(R
f)
2; (n) S(O)0-2(C1-6 alkyl); (o) S(O)0-2(C1-6 haloalkyl); (p) S(O)
1-2N(R
f)
2; and (q) C1-6 alkyl, C2-6 alkenyl, or C2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)
0-2-, C(=O), and C
1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NR
dR
e, C(=O)C
1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)
2, S(O)
0-2(C
1-6 alkyl), S(O)
0-2(C
1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)N(R
f)
2, S(O)
1- 2(C1-6 alkyl), S(O)1-2(C1-6 haloalkyl), S(O)1-2N(R
f)2, and C1-6 alkyl optionally substituted with 1-3 R
h;
each R
f is independently selected from the group consisting of: H and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, C1-3 haloalkyl, C
3-5 cycloalkyl, and 4-5 membered heterocyclyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NH2, -N(H)(C1-3 alkyl), and -N(C1-3 alkyl)2-. Embodiment 2. The compound of Embodiment 1, wherein R
9 is -NH2; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 3. The compound of any one of Embodiments 1 or 2, wherein X
1 is CH2 or CHR
L. Embodiment 4. The compound of any one of Embodiments 1-3, wherein X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2. Embodiment 5. The compound of any one of Embodiments 1-4, wherein X
1 is CH2; and X
2 and X
3 are both CH2. Embodiment 6. The compound of any one of Embodiments 1-4, wherein at least one (e.g., one) of X
1, X
2, and X
3 is selected from the group consisting of: CHR
L and C(R
L)2. Embodiment 7. The compound of any one of Embodiments 1-4 or 6, wherein X
1 is CH2; and X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2, provided that 1-2 of X
2 and X
3 is independently CHR
L or C(R
L)2. Embodiment 8. The compound of any one of Embodiments 1-4 or 6-7, wherein X
1 is CH2; X
2 is CH2; and X
3 is CHR
L. Embodiment 9. The compound of any one of Embodiments 1-3, wherein one of X
2 and X
3 is -O-; and the other of X
2 and X
3 is selected from the group consisting of: CH2, CHR
L, and C(R
L)2.
Embodiment 10. The compound of any one of Embodiments 1-3 or 9, wherein X
2 is -O-; and X
3 is selected from the group consisting of: CH2, CHR
L, and C(R
L)2. Embodiment 11. The compound of any one of Embodiments 1-10, wherein each R
L is independently selected from the group consisting of: CH3, CF3, CHF2, and CH2F. Embodiment 12. The compound of any one of Embodiments 1-11, wherein Y
2 is -CH2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. Embodiment 13. The compound of any one of Embodiments 1-12, wherein R
3 is 14. The compound of any one of Embodiments 1-13, wherein R
1 is
, erein b2 is 0, 1, or 2, and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7. Embodiment 15. The compound of any one of Embodiments 1-14, wherein R
1 is
. Embodiment 16. The compound of any one of Embodiments 1-15, wherein R
1 is
selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, -C(O)N(H)R
b1, R
b1, and C(O)R
b1. Embodiment 17. The compound of Embodiment 16, wherein R
7 is selected from the group consisting of: (a) C(=O)N(R
f)
2, wherein each R
f is independently H or C
1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is 4-10 membered heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 18. The compound of any one of Embodiments 15-17, wherein R
7 is C(=O)N(R
f)
2, wherein each R
f is independently H or C
1-3 alkyl optionally substituted with 1-3 R
h. Embodiment 19. The compound of any one of Embodiments 1-15, wherein R
1 is
, wherein R
7a and R
7b are independently selected R
7. Embodiment 20. The compound of Embodiment 19, wherein R
7a is selected from the group consisting of: C(=O)N(R
f)2, C(O)N(C1-3 alkyl)R
b1, -C(O)N(H)R
b1, R
b1, and C(O)R
b1; and R
7b is -halo, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 21. The compound of Embodiment 20, wherein R
7a is selected from the group consisting of: (a) C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h;
(b) C(O)N(C
1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C
3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is 4-10 membered heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 22. The compound of any one of Embodiments 1-13, wherein R
1 is a 7-10 (e.g., 7) membered heterocyclyl having one ring nitrogen atom, one ring oxygen atom, and no additional ring heteroatoms, wherein the 7-10 membered heterocyclyl is optionally substituted with 1-4 R
7. Embodiment 23. The compound of Embodiment 22, wherein
optionally substituted with 1-4 R
7 at one or more ring carbon atoms
Embodiment 24. The compound of Embodiment 23, wherein each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C1-3 alkyl optionally substituted with 1-3 R
c, wherein: each R
c is independently selected from the group consisting of: -F, -OH, and -CN. Embodiment 25. The compound of Embodiment 1, wherein the compound is a compound of Formula (III-1):
Formula (III-1) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1;
each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; b3 is 0, 1, 2, or 3; X
1 is CH
2; and X
2 and X
3 are independently selected from the group consisting of: O, CH2, CHR
L, and C(R
L)2. Embodiment 26. The compound of Embodiment 25, wherein R
9 is NR
dR
e (e.g., - NH2). Embodiment 27. The compound of any one of Embodiments 25 or 26, wherein b3 is 0. Embodiment 28. The compound of any one of Embodiments 25-27, wherein b3 is 1 or 2; and each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C1-3 alkyl optionally substituted with 1-3 R
c, wherein each R
c is independently selected from the group consisting of: -F, -OH, and -CN. Embodiment 29. The compound of Embodiment 1, wherein the compound is a compound of Formula (III-2):
Formula (III-2) or a pharmaceutically acceptable salt thereof, wherein: b4 is 0 or 1; each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c; R
7 is selected from the group consisting of: C(=O)N(R
f)
2, C(O)N(C
1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1;
X
1 is CH
2; and X
2 and X
3 are independently selected from the group consisting of: O, CH2, CHR
L, and C(R
L)2. Embodiment 30. The compound of Embodiment 29, wherein R
9 is NR
dR
e (e.g., - NH2). Embodiment 31. The compound of any one of Embodiments 29 or 30, wherein R
7 is selected from the group consisting of: (a) C(=O)N(R
f)
2, wherein each R
f is independently H or C
1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is 4-10 membered heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 32. The compound of any one of Embodiments 29-31, wherein Y
2 is -CH
2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. Embodiment 33. The compound of Embodiment 32, wherein
Embodiment 35. The compound of Embodiment 1, wherein the compound of Formula (III) is selected from the group consisting of Compound Nos.158, 158a, 158b, 158c, 161, 161a, 161b, 161c, 176, 176a, 176b, 176c, 176d, 176e, 177, 177a, 178, 178a, 178b, 179, 179a, 179b, 179d, 179e, 179f, 180, 180a, 180b, 180c, 184, 184b, and 184c as depicted in Table C1, or a pharmaceutically acceptable salt thereof. Embodiment 36. A pharmaceutical composition comprising a compound of any one of Embodiments 1-35, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient. Embodiment 37. A compound of any one of Embodiments 1-35, or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 36 for use in treatment. Embodiment 38. A compound of any one of Embodiments 1-35 or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 36 for use in the treatment of a KRAS associated cancer. Embodiment 39. A compound of any one of Embodiments 1-35 or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 36 for use in a method of treating a KRAS associated cancer, the method comprising (a) determining that the cancer in the subject has a KRas dysregulation; and (b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1- 35, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 36. Embodiment 40. A compound of any one of Embodiments 1-35 or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 36 for use in a method of treating a KRAS associated cancer, the method comprising administering to a subject identified or diagnosed as having a cancer having a KRas dysregulation a therapeutically effective amount of a compound of any one of Embodiments
1-35 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 36. Embodiment 41. A compound of any one of Embodiments 1-35 or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 36 for use in a method of treating a KRAS associated cancer, the method comprising: (a) determining that the cancer in the subject has a KRas dysregulation; and (b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-35 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 36. Embodiment 42. The compound or pharmaceutical composition of any one of Embodiments 38-41, wherein the KRas-associated cancer is a mutant KRas-associated cancer. Embodiment 43. The compound or pharmaceutical composition of Embodiment 42, wherein the mutant KRas-associated cancer is a KRas G12A-associated cancer, a KRas G12C-associated cancer, a KRas G12D-associated cancer, a KRas G12R-associated cancer, a KRas G12S-associated cancer, or a KRas G12V-associated cancer; optionally wherein the mutant KRas-associated cancer is a KRas G12D-associated cancer or a KRas G12V-associated cancer; optionally wherein the mutant KRas-associated cancer is a KRas G12D-associated cancer; optionally wherein the mutant KRas-associated cancer is a KRas G12R-associated cancer; or optionally wherein the mutant KRas-associated cancer is a KRas G12V-associated cancer. Embodiment 44. The compound or pharmaceutical composition of any one of Embodiments 39 or 41, wherein the step of determining that the cancer in the subject has a KRas dysregulation includes performing an assay to detect the KRas dysregulation (e.g., a KRas mutation) in a tumor sample from the subject. Embodiment 45. The compound or pharmaceutical composition of Embodiment 44, wherein detecting the KRas dysregulation includes detecting a KRAS gene having a mutation corresponding to a substitution of glycine 12 in a KRas protein and/or a KRas protein
having a substitution of glycine 12. Embodiment 46. The compound or pharmaceutical composition of Embodiment 45, wherein the substitution of glycine 12 is a substitution to alanine, cysteine, aspartic acid, arginine, serine, or valine; optionally wherein the substitution of glycine 12 is a substitution to aspartic acid; optionally wherein the substitution of glycine 12 is a substitution to arginine; or optionally wherein the substitution of glycine 12 is a substitution to valine. Embodiment 47. The compound or pharmaceutical composition of any one of Embodiments 38-46, wherein the KRas-associated cancer is selected from the group consisting of: a hematological cancer, a soft tissue cancer, bile duct cancer, bladder cancer, brain cancer, breast cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, ovarian cancer, pancreatic cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer, thymus cancer, thyroid cancer, urothelial cancer, uterine cancer, and a combination thereof; optionally wherein the KRas-associated cancer is pancreatic cancer; optionally wherein the KRas-associated cancer is selected from the group consisting of: a hematological cancer, brain cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, pancreatic cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, thymus cancer, urothelial cancer, and uterine cancer; optionally wherein the KRas-associated cancer is selected from the group consisting of: the cancer is a hematological cancer, bladder cancer, bile duct cancer, brain cancer, breast cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, ovarian cancer, pancreatic cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer (e.g., seminoma), thymus cancer, and uterine cancer; optionally wherein the KRas-associated cancer is selected from the group consisting of: bladder cancer, breast cancer, cervical cancer, colorectal cancer, endometrial cancer, esophageal or stomach cancer, leukemia, lung cancer (e.g., NSCLC), pancreatic cancer, and kidney cancer.
Embodiment 48. The compound or pharmaceutical composition of any one of Embodiments 38-47, the method comprises administering an additional therapy or therapeutic agent to the subject. Embodiment 49. The compound or pharmaceutical composition of Embodiment 48, wherein the additional therapy or therapeutic agent is selected from the group consisting of Ras pathway targeted therapeutic agents, kinase-targeted therapeutics, mTORC1 inhibitors or degraders, YAP inhibitors or degraders, proteasome inhibitors or degraders, HSP90 inhibitors or degraders, farnesyl transferase inhibitors or degraders, PTEN inhibitors or degraders, signal transduction pathway inhibitors or degraders, checkpoint inhibitors, modulators of the apoptosis pathway, chemotherapeutics, angiogenesis-targeted therapies, immune-targeted agents, radiotherapy, and combinations thereof. Embodiment 50. The compound or pharmaceutical composition of Embodiment 49, wherein the compound of any one of Embodiments 1-35, or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 36, and the additional therapeutic agent or therapy are administered simultaneously, separately, or sequentially. Exemplary Formula (I) Embodiments Embodiment 1. A compound of Formula (I-a1):

Formula (I-a1) or a pharmaceutically acceptable salt thereof, wherein: R
1 is selected from the group consisting of: (i) a 4-10 membered heterocyclyl optionally substituted with 1-4 R
7; (ii) an 8-12 membered bicyclic heterocyclyl, wherein the heterocyclyl comprises an endocyclic group selected from the group consisting of C(=O)NH and S(O)2NH, and wherein the heterocyclyl is further optionally substituted with 1-3 R
7 at one or more ring carbon atoms; and
(iii)
, wherein b2 is 0, 1, 2, or 3; and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7; each R
7 is independently selected from the group consisting of R
a and R
b; Ring B is selected from the group consisting of:
,
, wherein: X
1 is a bond or CH2; X
2 and X
3 are independently selected from the group consisting of: -O-, CH2, CHR
L, and C(R
L)
2; b1 is 0, 1, or 2; R
9 is selected from the group consisting of: H, OH, NH2, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of -F, CN, and C
1-3 alkyl optionally substituted with 1-3 R
c; or a pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C
3-6 cycloalkyl ring; Y
2 is a straight-chain C1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C
1-6 alkoxy, C
1-6 haloalkoxy, C
1-6 alkyl, and C
1-6 haloalkyl, or a pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C
1-3 alkyl;
R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C
1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g;
each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1-
2(C
1-6 alkyl), S(O)
1-2(C
1-6 haloalkyl), S(O)
1-2N(R
f)
2, and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, and C
1- 3 haloalkyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. Embodiment 2. The compound of Embodiment 1, wherein X
1 is CH2. Embodiment 3. The compound of any one of Embodiments 1 or 2, wherein X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2. Embodiment 4. The compound of any one of Embodiments 1-3, wherein X
2 is CH2; and X
3 is selected from the group consisting of: CHR
L and C(R
L)2. Embodiment 5. The compound of any one of Embodiments 1-4, wherein X
2 is CH2; and X
3 is CHMe. Embodiment 6. The compound of any one of Embodiments 1-3, wherein X
2 and X
3 are both CH
2. Embodiment 7. The compound of any one of Embodiments 1-6, wherein b1 is 1. Embodiment 8. The compound of any one of Embodiments 1-6, wherein b1 is 2. Embodiment 9. The compound of any one of Embodiments 1-6, wherein b1 is 1
or 2; and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1- 3 alkyl optionally substituted with 1-3 R
c. Embodiment 10. The compound of any one of Embodiments 1-6, wherein b1 is 1; and R
10 is -CN. Embodiment 11. The compound of any one of Embodiments 1-6 or 10, wherein b1 is 1; R
10 is ortho to R
9; and R
10 is -CN. Embodiment 12. The compound of any one of Embodiments 1-6, wherein b1 is 1 or 2; and each R
10 is independently -Cl or -F. Embodiment 13. The compound of any one of Embodiments 1-6 or 12, wherein b1 is 1 or 2; 1-2 occurrence(s) of R
10 is ortho to R
9; and each R
10 is independently -Cl or -F. Embodiment 14. The compound of any one of Embodiments 1-13, wherein R
1 is 4-10 membered heterocyclyl optionally substituted with 1-4 R
7. Embodiment 15. The compound of any one of Embodiments 1-14, wherein R
1 is a 4-10 membered heterocyclyl substituted with -OH, -(C1-3 alkylene)-OH, -CN, or –(C1-3 alkylene)-CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7. Embodiment 16. The compound of any one of Embodiments 1-15, wherein R
1 is a 4-10 (e.g., 6, 7, or 8) membered heterocyclyl substituted with -OH or -CH
2CN on a ring carbon atom, wherein the heterocyclyl is further optionally substituted with 1-3 R
7, and wherein the heterocyclyl contains one ring nitrogen atom and 0-2 additional ring heteroatoms each independently selected from the group consisting of: O and S(O)
0-2. Embodiment 17. The compound of any one of Embodiments 1-14, wherein R
1 is a 7-10 (e.g., 7) membered heterocyclyl optionally substituted with 1-4 R
7. Embodiment 18. The compound of any one of Embodiments 1-14 or 17, wherein R
1 is a 7-10 (e.g., 7) membered heterocyclyl having one ring nitrogen atom, one ring oxygen 35 atom, and no additional ring heteroatoms, wherein the 7-10 membered heterocyclyl is
optionally substituted with 1-4 R
7. Embodiment 19. The compound of any one of Embodiments 1-14 or 17-18, wherein
optionally substituted with 1-4 R
7 at one or more ring carbon atoms. Embodiment 20. The compound of Embodiment 19, wherein each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C
1-3 alkyl optionally substituted with 1-3 R
c, wherein: each R
c is independently selected from the group consisting of: -F, -OH, and -CN. Embodiment 21. The compound of any one of Embodiments 1-13, wherein R
1 is
, wherein b2 is 0, 1, or 2, and A
1 and A
2 are independently selected from the group consisting of: N, CH, and CR
7. Embodiment 22. The compound of Embodiment 21, wherein A
2 is CH. Embodiment 23. The compound of any one of Embodiments 1-13 or 21-22, wherein
. Embodiment 24. The compound of any one of Embodiments 1-13 or 21-23, wherein
selected from the group consisting of: C(=O)N(R
f)
2, C(O)N(C
1-3 alkyl)R
b1, -C(O)N(H)R
b1, R
b1, and C(O)R
b1. Embodiment 25. The compound of Embodiment 24, wherein R
7 is selected from
the group consisting of: (a) C(=O)N(R
f)2, wherein each R
f is independently H or C1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C
1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C
3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 26. The compound of any one of Embodiments 1-25, wherein Y
2 is -CH
2-. Embodiment 27. The compound of any one of Embodiments 1-26, wherein R
3 is a 4-10 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b. Embodiment 28. The compound of any one of Embodiments 1-27, wherein R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a. Embodiment 29. The compound of any one of Embodiments 1-28, wherein R
3 is a bicyclic 7-10 membered heterocyclyl optionally substituted with 1-6 R
a. Embodiment 30. The compound of any one of Embodiments 1-29, wherein R
3 is
optionally substituted with 1-3 R
a. Embodiment 31. The compound of any one of Embodiments 1-30, wherein R
3 is
Embodiment 32. The compound of any one of Embodiments 1-31, wherein the ring carbon atom labelled with * in Formula (I) has (S)-stereochemistry.
Embodiment 33. The compound of any one of Embodiments 1-31, wherein the ring carbon atom labelled with * in Formula (I) has (R)-stereochemistry. Embodiment 34. A pharmaceutical composition comprising a compound of any one of Embodiments 1-33, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient. Embodiment 35. A compound of any one of Embodiments 1-33, or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 34 for use in treatment. Embodiment 36. A compound of any one of Embodiments 1-33 or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 34 for use in the treatment of a KRAS associated cancer. Embodiment 37. A compound of any one of Embodiments 1-33 or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 34 for use in a method of treating a KRAS associated cancer, the method comprising (a) determining that the cancer in the subject has a KRas dysregulation; and (b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1- 33, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 34. Embodiment 38. A compound of any one of Embodiments 1-33 or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 34 for use in a method of treating a KRAS associated cancer, the method comprising administering to a subject identified or diagnosed as having a cancer having a KRas dysregulation a therapeutically effective amount of a compound of any one of Embodiments 1-33 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 34. Embodiment 39. A compound of any one of Embodiments 1-33 or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 34 for use in a method of treating a KRAS associated cancer, the method comprising: (a) determining that the cancer in the subject has a KRas dysregulation; and
(b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-33 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 34. Embodiment 40. The compound or pharmaceutical composition of any one of Embodiments 36-39, wherein the KRas-associated cancer is a mutant KRas-associated cancer. Embodiment 41. The compound or pharmaceutical composition of Embodiment 40, wherein the mutant KRas-associated cancer is a KRas G12A-associated cancer, a KRas G12C-associated cancer, a KRas G12D-associated cancer, a KRas G12R-associated cancer, a KRas G12S-associated cancer, or a KRas G12V-associated cancer; optionally wherein the mutant KRas-associated cancer is a KRas G12D-associated cancer or a KRas G12V-associated cancer; optionally wherein the mutant KRas-associated cancer is a KRas G12D-associated cancer; optionally wherein the mutant KRas-associated cancer is a KRas G12R- associated cancer; or optionally wherein the mutant KRas-associated cancer is a KRas G12V-associated cancer. Embodiment 42. The compound or pharmaceutical composition of any one of Embodiments 37 or 39, wherein the step of determining that the cancer in the subject has a KRas dysregulation includes performing an assay to detect the KRas dysregulation (e.g., a KRas mutation) in a tumor sample from the subject. Embodiment 43. The compound or pharmaceutical composition of Embodiment 42, wherein detecting the KRas dysregulation includes detecting a KRAS gene having a mutation corresponding to a substitution of glycine 12 in a KRas protein and/or a KRas protein having a substitution of glycine 12. Embodiment 44. The compound or pharmaceutical composition of Embodiment 43, wherein the substitution of glycine 12 is a substitution to alanine, cysteine, aspartic acid, arginine, serine, or valine; optionally wherein the substitution of glycine 12 is a substitution to aspartic acid;
optionally wherein the substitution of glycine 12 is a substitution to arginine; or optionally wherein the substitution of glycine 12 is a substitution to valine. Embodiment 45. The compound or pharmaceutical composition of any one of Embodiments 36-44, wherein the KRas-associated cancer is selected from the group consisting of: a hematological cancer, a soft tissue cancer, bile duct cancer, bladder cancer, brain cancer, breast cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, ovarian cancer, pancreatic cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer, thymus cancer, thyroid cancer, urothelial cancer, uterine cancer, and a combination thereof; optionally wherein the KRas-associated cancer is pancreatic cancer. Embodiment 46. The compound or pharmaceutical composition of any one of Embodiments 36-44, the method comprises administering an additional therapy or therapeutic agent to the subject. Embodiment 47. The compound or pharmaceutical composition of Embodiment 46, wherein the additional therapy or therapeutic agent is selected from the group consisting of Ras pathway targeted therapeutic agents, kinase-targeted therapeutics, mTORC1 inhibitors or degraders, YAP inhibitors or degraders, proteasome inhibitors or degraders, HSP90 inhibitors or degraders, farnesyl transferase inhibitors or degraders, PTEN inhibitors or degraders, signal transduction pathway inhibitors or degraders, checkpoint inhibitors, modulators of the apoptosis pathway, chemotherapeutics, angiogenesis-targeted therapies, immune-targeted agents, radiotherapy, and combinations thereof. Exemplary Formula (I) Embodiments Embodiment 1. A compound of Formula (I-a4):
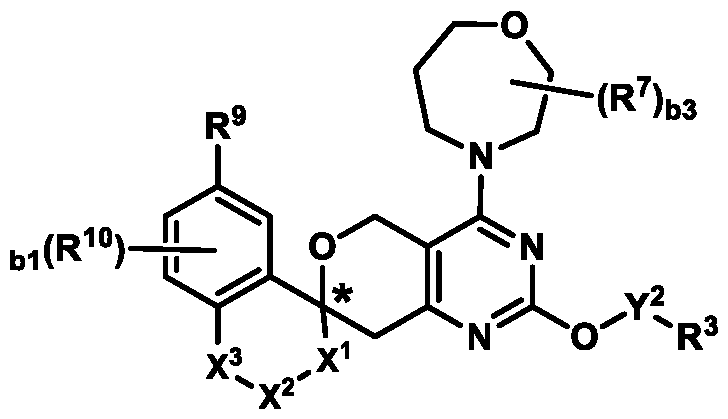
Formula (I-a4) 30 or a pharmaceutically acceptable salt thereof, wherein:
each R
7 is independently selected from the group consisting of R
a and R
b; b3 is 0, 1, 2, or 3; X
1 is CH2; X
2 and X
3 are independently selected from the group consisting of: O, CH
2, CHR
L, and C(R
L)2; b1 is 0, 1, or 2; R
9 is selected from the group consisting of: H, OH, NH
2, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; or a pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; Y
2 is a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or a pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C
3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
(h) C(=O)C
1-6 alkyl; (i) C(=O)C1-6 haloalkyl; (j) C(=O)OH; (k) C(=O)OC
1-6 alkyl; (l) C(=O)OC1-6 haloalkyl; (m)C(=O)N(R
f)2; (n) S(O)
0-2(C
1-6 alkyl); (o) S(O)0-2(C1-6 haloalkyl); (p) S(O)1-2N(R
f)2; and (q) C
1-6 alkyl, C
2-6 alkenyl, or C
2-6 alkynyl, each optionally substituted with 1-6 R
c; each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C
6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)2, S(O)0-2(C1-6 alkyl), S(O)0-2(C1-6 haloalkyl), and S(O)
1-2N(R
f)
2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC1-6 haloalkyl, C(=O)N(R
f)2, S(O)1-
2(C
1-6 alkyl), S(O)
1-2(C
1-6 haloalkyl), S(O)
1-2N(R
f)
2, and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C
1-3 alkyl, and C
1- 3 haloalkyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C1-
6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. Embodiment 2. The compound of Embodiment 1, wherein b3 is 0. Embodiment 3. The compound of Embodiment 1, wherein b3 is 1 or 2; and each R
7 is independently selected from the group consisting of: -OH; -CN; -F; and C1-3 alkyl optionally substituted with 1-3 R
c, wherein: each R
c is independently selected from the group consisting of: -F, -OH, and -CN. Embodiment 4. A compound of Formula (I-a5):
Formula (I-a5) or a pharmaceutically acceptable salt thereof, wherein: R
7 is selected from the group consisting of: C(=O)N(R
f)
2, C(O)N(C
1-3 alkyl)R
b1, - C(O)N(H)R
b1, R
b1, and C(O)R
b1; X
1 is CH2; X
2 and X
3 are independently selected from the group consisting of: O, CH2, CHR
L, and C(R
L)
2; b1 is 0, 1, or 2; R
9 is selected from the group consisting of: H, OH, NH2, and halo; each R
10 is independently selected from the group consisting of R
a and R
b; each R
L is independently selected from the group consisting of -F, CN, and C1-3 alkyl optionally substituted with 1-3 R
c; or a pair of R
L on the same or different ring carbon atom(s) taken together with the ring atom(s) connecting them form a C3-6 cycloalkyl ring; Y
2 is a straight-chain C
1-6 alkylene optionally substituted with 1-6 R
Y; each R
Y is independently selected from the group consisting of: halo, cyano, -OH, oxo, C1-6 alkoxy, C1-6 haloalkoxy, C1-6 alkyl, and C1-6 haloalkyl, or
a pair of R
Y on the same or different carbon atom(s) taken together with the atom(s) connecting them forms a C3-6 cycloalkyl ring or 4-6 membered heterocyclyl ring, each of which is optionally substituted with 1-3 independently selected C1-3 alkyl; R
3 is selected from the group consisting of: (a) 4-15 membered heterocyclyl optionally substituted with 1-6 substituents independently selected from the group consisting of: R
a and R
b; and (b) -NR
dR
e; each R
a is independently selected from the group consisting of:
each R
b is independently selected from the group consisting of: -(L
b)b-R
b1 and -R
b1, wherein: b is 1, 2, or 3; each -L
b is independently selected from the group consisting of: -O-, -N(H)-, -N(C1-3 alkyl)-, -S(O)0-2-, C(=O), and C1-3 alkylene; and
each R
b1 is independently selected from the group consisting of: C
3-10 cycloalkyl, 4-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each of which is optionally substituted with 1-3 R
g; each R
c is independently selected from the group consisting of: halo, cyano, -OH, -C1- 6 alkoxy, -C1-6 haloalkoxy, -NR
dR
e, C(=O)C1-6 alkyl, C(=O)C1-6 haloalkyl, C(=O)OC1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)OH, C(=O)N(R
f)
2, S(O)
0-2(C
1-6 alkyl), S(O)
0-2(C
1-6 haloalkyl), and S(O)1-2N(R
f)2; each R
d and R
e is independently selected from the group consisting of: H, C(=O)C1-6 alkyl, C(=O)C
1-6 haloalkyl, C(=O)OC
1-6 alkyl, C(=O)OC
1-6 haloalkyl, C(=O)N(R
f)
2, S(O)
1- 2(C
1-6 alkyl), S(O)
1-2(C
1-6 haloalkyl), S(O)
1-2N(R
f)
2, and C
1-6 alkyl optionally substituted with 1-3 R
h; each R
f is independently selected from the group consisting of: H and C1-6 alkyl optionally substituted with 1-3 R
h; each R
g is independently selected from the group consisting of: R
h, C1-3 alkyl, and C1- 3 haloalkyl; and each R
h is independently selected from the group consisting of: halo, cyano, -OH, -C
1- 6 alkoxy, -C
1-6 haloalkoxy, -NH
2, -N(H)(C
1-3 alkyl), and -N(C
1-3 alkyl)
2-. Embodiment 5. The compound of Embodiment 4, wherein R
7 is selected from the group consisting of: (a) C(=O)N(R
f)
2, wherein each R
f is independently H or C
1-3 alkyl optionally substituted with 1-3 R
h; (b) C(O)N(C1-3 alkyl)R
b1 or -C(O)N(H)R
b1, wherein: R
b1 is C3-6 cycloalkyl or 4-6 membered heterocyclyl, each of which is optionally substituted with 1-3 R
g; and (c) C(O)R
b1, wherein R
b1 is heterocyclyl optionally substituted with 1-3 R
g, wherein R
b1 is attached to the C(O) via a ring nitrogen atom. Embodiment 6. The compound of any one of Embodiments 1-5, wherein X
2 and X
3 are independently selected from the group consisting of: CH2, CHR
L, and C(R
L)2. Embodiment 7. The compound of any one of Embodiments 1-6, wherein X
2 and X
3 are both CH2. Embodiment 8. The compound of any one of Embodiments 1-6, wherein X
2 is
CH
2; and X
3 is CHR
L. Embodiment 9. The compound of any one of Embodiments 1-6 or 8, wherein X
2 is CH
2; and X
3 is CH(Me). Embodiment 10. The compound of any one of Embodiments 1-9, wherein R
9 is - NH
2. Embodiment 11. The compound of any one of Embodiments 1-10, wherein b1 is 1. Embodiment 12. The compound of any one of Embodiments 1-10, wherein b1 is . Embodiment 13. The compound of any one of Embodiments 1-12, wherein each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 14. The compound of any one of Embodiments 1-13, wherein 1-2 occurrence(s) of R
10 is ortho to R
9. Embodiment 15. The compound of any one of 1-5, wherein the
X
2 is -O- or -CH2-; X
3 is -CH2- or -CHR
L-, wherein R
L is C1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3
alkyl optionally substituted with 1-3 R
c. Embodiment 16. The compound of any one of Embodiments 1-5, wherein the
moiety in Formula (I-a4) or (I-a5) is selected from the group consisting of:
X
3 is -CH2- or -CHR
L-, wherein R
L is C1-3 alkyl (e.g., methyl); and each R
10 is independently selected from the group consisting of: -Cl, -F, -CN, and C
1-3 alkyl optionally substituted with 1-3 R
c. Embodiment 17. The compound of any one of Embodiments 1-5, wherein the
. Embodiment 18. The compound of any one of Embodiments 1-17, wherein Y
2 is -CH2-; and R
3 is a 4-10 membered heterocyclyl having one ring nitrogen atom and 0-1 additional ring heteroatom selected from the group consisting of oxygen and nitrogen, wherein the heterocyclyl is optionally substituted with 1-6 R
a.
Embodiment 19. The compound of Embodiment 18, wherein


Embodiment 20. The compound of any one of Embodiments 1-19, wherein the ring carbon atom labelled with * has (S)-stereochemistry. Embodiment 21. A pharmaceutical composition comprising a compound of any one of Embodiments 1-20, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient. Embodiment 22. A compound of any one of Embodiments 1-20, or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 21 for use in treatment. Embodiment 23. A compound of any one of Embodiments 1-20 or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 21 for use in the treatment of a KRAS associated cancer. Embodiment 24. A compound of any one of Embodiments 1-20 or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 21 for use in a method of treating a KRAS associated cancer, the method comprising (a) determining that the cancer in the subject has a KRas dysregulation; and (b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1- 20, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 21. Embodiment 25. A compound of any one of Embodiments 1-20 or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 21 for use in a method of treating a KRAS associated cancer, the method comprising
administering to a subject identified or diagnosed as having a cancer having a KRas dysregulation a therapeutically effective amount of a compound of any one of Embodiments 1-20 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 21. Embodiment 26. A compound of any one of Embodiments 1-20 or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of Embodiment 21 for use in a method of treating a KRAS associated cancer, the method comprising: (a) determining that the cancer in the subject has a KRas dysregulation; and (b) administering to the subject a therapeutically effective amount of a compound of any one of Embodiments 1-20 or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition according to Embodiment 21. Embodiment 27. The compound or pharmaceutical composition of any one of Embodiments 23-26, wherein the KRas-associated cancer is a mutant KRas-associated cancer. Embodiment 28. The compound or pharmaceutical composition of Embodiment 27, wherein the mutant KRas-associated cancer is a KRas G12A-associated cancer, a KRas G12C-associated cancer, a KRas G12D-associated cancer, a KRas G12R-associated cancer, a KRas G12S-associated cancer, or a KRas G12V-associated cancer; optionally wherein the mutant KRas-associated cancer is a KRas G12D-associated cancer or a KRas G12V-associated cancer; optionally wherein the mutant KRas-associated cancer is a KRas G12D-associated cancer; optionally wherein the mutant KRas-associated cancer is a KRas G12R- associated cancer; or optionally wherein the mutant KRas-associated cancer is a KRas G12V-associated cancer. Embodiment 29. The compound or pharmaceutical composition of any one of Embodiments 24 or 26, wherein the step of determining that the cancer in the subject has a KRas dysregulation includes performing an assay to detect the KRas dysregulation (e.g., a KRas mutation) in a tumor sample from the subject. Embodiment 30. The compound or pharmaceutical composition of Embodiment
29, wherein detecting the KRas dysregulation includes detecting a KRAS gene having a mutation corresponding to a substitution of glycine 12 in a KRas protein and/or a KRas protein having a substitution of glycine 12. Embodiment 31. The compound or pharmaceutical composition of Embodiment 30, wherein the substitution of glycine 12 is a substitution to alanine, cysteine, aspartic acid, arginine, serine, or valine; optionally wherein the substitution of glycine 12 is a substitution to aspartic acid; optionally wherein the substitution of glycine 12 is a substitution to arginine; or optionally wherein the substitution of glycine 12 is a substitution to valine. Embodiment 32. The compound or pharmaceutical composition of any one of Embodiments 23-31, wherein the KRas-associated cancer is selected from the group consisting of: a hematological cancer, a soft tissue cancer, bile duct cancer, bladder cancer, brain cancer, breast cancer, cervical cancer, colon cancer, endometrial cancer, esophageal cancer, kidney cancer, liver cancer, lung cancer, mucinous carcinoma, ovarian cancer, pancreatic cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer, thymus cancer, thyroid cancer, urothelial cancer, uterine cancer, and a combination thereof; optionally wherein the KRas-associated cancer is pancreatic cancer.. Embodiment 33. The compound or pharmaceutical composition of any one of Embodiments 23-32, the method comprises administering an additional therapy or therapeutic agent to the subject. Embodiment 34. The compound or pharmaceutical composition of Embodiment 33, wherein the additional therapy or therapeutic agent is selected from the group consisting of Ras pathway targeted therapeutic agents, kinase-targeted therapeutics, mTORC1 inhibitors or degraders, YAP inhibitors or degraders, proteasome inhibitors or degraders, HSP90 inhibitors or degraders, farnesyl transferase inhibitors or degraders, PTEN inhibitors or degraders, signal transduction pathway inhibitors or degraders, checkpoint inhibitors, modulators of the apoptosis pathway, chemotherapeutics, angiogenesis-targeted therapies, immune-targeted agents, radiotherapy, and combinations thereof.





























































































































































































































































































































































































































































































































































































































