ES2717436T3 - Compuestos novedosos - Google Patents
Compuestos novedosos Download PDFInfo
- Publication number
- ES2717436T3 ES2717436T3 ES14734147T ES14734147T ES2717436T3 ES 2717436 T3 ES2717436 T3 ES 2717436T3 ES 14734147 T ES14734147 T ES 14734147T ES 14734147 T ES14734147 T ES 14734147T ES 2717436 T3 ES2717436 T3 ES 2717436T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- pharmaceutically acceptable
- treatment
- formula
- agents
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- GGOYPZXHENWDBT-SQNIBIBYSA-N CC(C)[C@@H](C(N(C[C@@H](C1)O)[C@@H]1C(NCc(cc1)ccc1-c1c(C)nc[s]1)=O)=O)N Chemical compound CC(C)[C@@H](C(N(C[C@@H](C1)O)[C@@H]1C(NCc(cc1)ccc1-c1c(C)nc[s]1)=O)=O)N GGOYPZXHENWDBT-SQNIBIBYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/06—Tripeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/55—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound the modifying agent being also a pharmacologically or therapeutically active agent, i.e. the entire conjugate being a codrug, i.e. a dimer, oligomer or polymer of pharmacologically or therapeutically active compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06034—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms
- C07K5/06043—Leu-amino acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06078—Dipeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0812—Tripeptides with the first amino acid being neutral and aromatic or cycloaliphatic
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Gastroenterology & Hepatology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Description
DESCRIPCIÓN
Compuestos novedosos
Campo de la invención
La presente invención se refiere a compuestos, composiciones, combinaciones y medicamentos que contienen dichos compuestos y procesos para su preparación. La invención también se refiere a dichos compuestos, combinaciones, composiciones y medicamentos, por ejemplo, como inhibidores de la actividad del receptor de estrógeno, por ejemplo, degradando el receptor de estrógeno, para su uso en el tratamiento de enfermedades y afecciones mediadas por el receptor de estrógeno, en particular, para su uso en el tratamiento de cáncer de mama.
Antecedentes de la invención
El receptor de estrógeno (ER) es un miembro de la familia del receptor de hormonas nucleares y funciona como un factor de transcripción activado por ligando implicado con la regulación positiva y negativa de la expresión génica. La hormona natural para el receptor de estrógeno es el B17-estradiol (E2) y metabolitos estrechamente relacionados. La unión de estradiol al receptor de estrógenos provoca una dimerización del receptor y el dímero, a su vez, se une a elementos de respuesta de estrógeno (ERE) sobre el ADN. El complejo ERADN recluta otros factores de transcripción responsables de la transcripción de ADN corriente abajo desde el ERE en ARNm que finalmente se traduce en proteína. De forma alternativa, la interacción de ER con ADN puede ser indirecta a través de la intermediación de otros factores de transcripción, a saber, fos y jun. Puesto que la expresión de una gran cantidad de genes está regulada por el receptor de estrógenos y puesto que el receptor de estrógenos está expresado en muchos tipos de células, la modulación del receptor de estrógenos a través de la unión de o bien hormonas naturales o bien ligandos ER sintéticos puede tener efectos productos sobre la fisiología y patofisiología del organismo.
Varias enfermedades tienen su etiología y/o patología mediada por el ER. De forma colectiva, estas enfermedades se denominan enfermedades dependientes de estrógenos. Los estrógenos son importantes para el desarrollo sexual en hembras. Además, los estrógenos tienen un papel importante en mantener la densidad ósea, regulación de niveles de lípidos en sangre y parece que tienen efectos neuroprotectores. Por consiguiente, una producción de estrógenos reducida en mujeres post-menopáusicas está asociada con una cantidad de enfermedades tales como osteoporosis, ateroesclerosis, depresión y trastornos cognitivos. En cambio, determinadas enfermedades proliferativas tales como cáncer de mama y de útero y endometriosis están estimuladas por los estrógenos y, por lo tanto, los antiestrógenos (es decir, antagonistas de estrógenos) tiene utilidad en la prevención y tratamiento de estos tipos de trastornos. Existen dos formas distintas del receptor de estrógenos, normalmente denominado como a y p, cada una codificada por un gen separado [ESR1 y ESR2, respectivamente).
Ambos ER se expresan ampliamente en distintos tipos de tejidos, sin embargo, existen algunas diferencias notables en sus patrones de expresión. El ERa se encuentra en el endometrio, células de cáncer de mama, células de estroma de ovario y el hipotálamo. En machos, la proteína ERa se encuentra en el epitelio de los distintos conductos. La expresión de la proteína ERp se ha documentado en riñón, cerebro, hueso, corazón, pulmones, mucosa intestinal, próstata y células endoteliales. Por lo tanto, el desarrollo de ligandos selectivos puede, por lo tanto, preservar los aspectos beneficiosos del estrógeno.
El cáncer de mama es la neoplasia más común que afecta a mujeres en el mundo entero, y la incidencia de la enfermedad va en aumento. Los estrógenos, en particular, actúan como factores de crecimiento endocrino para al menos un tercio de los cánceres de mama, y privar al tumor de este estímulo es una terapia reconocida para la enfermedad avanzada. En mujeres premenopáusicas, esto se logra mediante la ablación de la función del ovario a través de medios quirúrgicos, radioterapéuticos o médicos y, en mujeres postmenopáusicas, mediante el uso de inhibidores de la aromatasa.
Un enfoque alternativo a la retiración de estrógenos es antagonizar estrógenos con antiestrógenos. Estos son fármacos que se unen y compiten por los receptores de estrógenos (ER) presentes en tejido sensible a estrógenos. Antiestrógenos no esteroideos convencionales, tales como tamoxifeno, compiten eficazmente por la unión de ER pero su eficacia se ve a menudo limitada por el agonismo parcial que muestran, que da como resultado, un bloqueo incompleto de la actividad mediada por estrógenos. Un antiestrógeno específico o "puro" con alta afinidad para Er y sin ningún efecto agonista, puede tener ventajas sobre los antiestrógenos no esteroideos convencionales en el tratamiento de enfermedades dependientes de estrógenos. El Fulvestrant es el primero de una nueva clase de antiestrógenos puros potentes y está completamente libre del agonista parcial, actividad tipo estrógenos, asociada con antiestrógenos actualmente disponibles como el tamoxifeno.
Sería deseable investigar otros enfoques para antagonizas el receptor ER.
Un enfoque sería desarrollar reguladores negativos de ER o degradadores que den como resultado la reducción de la expresión de ER o bien en el nivel de transcripción o bien en el nivel proteico.
Hay disponibles varios métodos para la manipulación de niveles proteicos, incluidas moléculas quiméricas dirigidas a la proteólisis (PROTAC) que contienen un ligando que reconoce la proteína diana unida a un ligando que se une a una ligasa de ubiquitina E3 específica. Sería deseable tener una molécula pequeña que pueda unirse simultáneamente a ER y una ligasa de ubiquitina E3 específica y que promueva la ubiquitinación de ER y conduzca a la degradación de
ER mediante el Proteosoma. Una ligasa de ubiquitina E3 específica adecuada es el supresor tumoral de von Hippel-Lindau (VHL). Cyrus et al., ChemMedChem. 2010; 5(7): 979-985 describe la optimización del emplazamiento del enlazador en el diseño de PROTAC dirigidas al receptor de estrógenos. Burke et al., J. Med. Chem., 2004, 47(5): 1193-1206 describe conjugados de doxorubicina-formaldehído dirigidos al cáncer de mama. Willson et al., J. Med. Chem., 1994, 37(11): 1550-1552 describe un estrógeno no esteroideo. El documento WO 2013/106643 describe compuestos para la degradación potenciada de proteínas dirigidas mediante una ligasa de ubiquitina E3.
Los presentes inventores han identificado compuestos que son capaces de inhibir la función receptores de estrógenos incluidos compuestos que degradan el receptor de estrógenos.
Sumario de la invención
En un aspecto se proporciona un compuesto de fórmula (I):

En la que
L es un grupo de unión, en donde el grupo de unión es un grupo alquileno de cadena lineal de 8-16 átomos de carbono en donde uno o más átomos de carbono en el grupo de unión se sustituye por un miembro del grupo seleccionado independientemente de -O-, -NH-, -N(CH3)-,

R1 es alquilo Ci -6 lineal o ramificado o cicloalquilo C3-6
R2 es 4-metiltiazol-5-il, oxazol-5-il o una sal farmacéuticamente aceptable de los mismos.
En un aspecto adicional de la presente invención, se proporciona un compuesto de fórmula (I), o una sal farmacéuticamente aceptable del mismo para su uso en la terapia, en particular en el tratamiento de enfermedades y afecciones mediadas por el receptor de estrógeno.
En un aspecto adicional de la presente invención, se proporciona una composición farmacéutica que comprende un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo y uno o más vehículos, diluyentes y excipientes farmacéuticamente aceptables.
En general, descrito en el presente documento, se proporciona un método de tratamiento de enfermedades y afecciones mediadas por el receptor de estrógeno en un sujeto que comprende la administración de una cantidad terapéuticamente eficaz de un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo.
En general, descrito en el presente documento, se proporciona el uso de un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo en la fabricación de un medicamento para su uso en el tratamiento de enfermedades y afecciones mediadas por el receptor de estrógeno.
En un aspecto adicional, se proporciona una combinación que comprende un compuesto de fórmula (I) o una sal
farmacéuticamente aceptable del mismo y al menos un agente terapéutico adicional.
En un aspecto adicional se proporciona una combinación que comprende compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo y al menos un agente terapéutico adicional para su uso en la terapia, en particular, para su uso en el tratamiento de enfermedades y afecciones mediadas por el receptor de estrógeno.
En un aspecto adicional de la invención, se proporciona una combinación que comprende compuesto de fórmula (I), o una sal farmacéuticamente aceptable del mismo y al menos un agente terapéutico adicional para su uso en el tratamiento de enfermedades y afecciones mediadas por el receptor de estrógeno.
En general, descrito en el presente documento, se proporciona un método de tratamiento de enfermedades y afecciones mediadas por el receptor de estrógeno que comprende la administración a un ser humano que lo necesita una cantidad terapéuticamente eficaz de una combinación que comprende compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo y al menos un agente terapéutico adicional.
En general, descrito en el presente documento, se proporciona el uso de una combinación que comprende compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo y al menos un agente terapéutico adicional en la fabricación de un medicamento el tratamiento de enfermedades y afecciones mediadas por el receptor de estrógeno.
En un aspecto adicional, se proporciona una combinación que comprende un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo y al menos un agente antineoplásico.
En un aspecto adicional se proporciona una combinación que comprende compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo y al menos un agente antineoplásico para su uso en la terapia, en particular, para el tratamiento de enfermedades y afecciones mediadas por el receptor de estrógeno.
En un aspecto adicional, se proporciona una combinación que comprende un compuesto de fórmula (I), o sal farmacéuticamente aceptable del mismo y al menos un agente antineoplásico para su uso en el tratamiento de enfermedades y afecciones mediadas por el receptor de estrógeno.
En general, descrito en el presente documento, se proporciona el uso de una combinación que comprende un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo y al menos un agente antineoplásico en la fabricación de un medicamento el tratamiento de enfermedades y afecciones mediadas por el receptor de estrógeno. En general, descrito en el presente documento, se proporciona un método de tratamiento de enfermedades y afecciones mediadas por el receptor de estrógeno que comprende la administración a un ser humano que lo necesita una cantidad terapéuticamente eficaz de una combinación que comprende compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo y al menos un agente antineoplásico.
En un aspecto adicional se proporciona una composición farmacéutica que comprende una combinación que comprende un compuesto de fórmula (I) o una sal farmacéuticamente aceptable de la misma y al menos un agente terapéutico adicional, en particular, al menos un agente antineoplásico y uno o más vehículos, diluyentes y excipientes farmacéuticamente aceptables.
En general, descrito en el presente documento, se proporciona un método de degradación del receptor de estrógeno que comprende la administración a un ser humano que lo necesita una cantidad terapéuticamente eficaz de un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable del mismo.
Descripción detallada de la invención
Tal como se usa en el presente documento "un compuesto de la invención" incluye todos los solvatos, complejos, polimorfos, derivados radiomarcados, estereoisómeros e isómeros ópticos de los compuestos de fórmula (I) y sales del mismo.
Tal como se usa en el presente documento "halo" significa fluoro (-F), cloro (-Cl), bromo (-Br) o yodo (-I).
Tal como se usa en el presente documento, el término "cantidad eficaz" significa que la cantidad de un fármaco o agente farmacéutico que provocará la respuesta biológica o médica de un tejido, sistema, animal o ser humano que se busca, por ejemplo, por un investigador o clínico. Además, el término "cantidad terapéuticamente eficaz" significa que cualquier cantidad que, en comparación con un sujeto correspondiente que no ha recibido tal cantidad, da como resultado un tratamiento, cura, prevención o mejora aumentada de una enfermedad, trastorno o efecto secundario o una disminución en la tasa de avance de una enfermedad o un trastorno. La expresión también incluye dentro de su alcance cantidades eficaces para potenciar la función fisiológica normal.
Tal como se usa en el presente documento, el término "farmacéuticamente aceptable" se refiere a aquellos compuestos, materiales, composiciones y formas de dosificación que se encuentran, dentro del alcance del juicio médico, adecuado para su uso en contacto con los tejidos de seres humanos y animales sin una toxicidad, irritación u otro problema o complicación excesiva, proporcional con una relación beneficio/riesgo razonable.
Los compuestos de la invención pueden existir en la forma sólida o líquida. En forma sólida, el compuesto de la invención puede existir en un continuo de estados sólidos que varían desde completamente amorfo a completamente cristalino. El término "amorfo" se refiere a un estado en el que el material carece de un intervalo de orden largo al nivel molecular y, dependiendo de la temperatura, puede presentar las propiedades físicas de un sólido o un líquido. Normalmente dichos materiales no proporcionan modelos de difracción de rayos X distintivos y, aunque presentan las propiedades de un sólido, se describen más formalmente como un líquido. Tras calentamiento, se produce un cambio de las propiedades de sólido a líquido que se caracteriza por un cambio de estado, normalmente de segundo orden (‘transición vítrea'). El término "cristalino" se refiere a una fase sólida en la que el material tiene una estructura interna ordenada de manera regular al nivel molecular y proporciona un modelo de difracción de rayos X distintivo con picos definidos. Dichos materiales, cuando se calienten de forma suficiente presentarán también las propiedades de un líquido, pero el cambio de sólido a líquido se caracteriza por un cambio de fase, normalmente de primer orden (‘punto de fusión').
El compuesto de fórmula (I) puede existir también en formas solvatadas y no solvatadas. Tal como se usa en el presente documento, el término "solvato" se refiere a un complejo de estequiometría variable formado por un soluto (en esta invención, un compuesto de fórmula (I) o una sal] y un disolvente. Dichos disolventes con los fines de la invención no pueden interferir con la actividad biológica del soluto. El experto en la materia apreciará que se pueden formar solvatos farmacéuticamente aceptables para compuestos cristalinos en los que se incorporan moléculas de disolvente en la red cristalina durante la cristalización. Las moléculas de disolvente incorporadas pueden ser moléculas de agua o no acuosas tales como etanol, isopropanol, DMSO, ácido acético, etanolamina y moléculas de acetato de etilo. Las redes cristalinas incorporadas con moléculas de agua se denominan normalmente como "hidratos". Los hidratos incluyen hidratos estequiométricos así como composiciones que contienen cantidades variables de agua. La presente invención incluye todos tales solvatos.
Los compuestos de la invención pueden tener la capacidad de cristalizarse en más de una forma, una característica, que se conoce como polimorfismo, y se entiende que tales formas polimórficas ("polimorfos") se encuentran dentro del alcance de la invención. El polimorfismo, en general, se puede producir como una respueta a cambios en la temperatura o presión o ambos y también puede resultar de variaciones en el proceso de cristalización. Los polimorfos pueden distinguirse por diversas características físicas conocidas en la técnica, tales como patrones de difracción de rayos X, solubilidad y punto de fusión.
También cabe destacar que los compuestos de fórmula (I) pueden formar tautómeros. Se entiende que todos los tautómeros y mezclas de tautómeros de los compuestos de la presente invención están incluidos dentro del alcance de los compuestos de la presente invención.
Tal como se usa en el presente documento, el término "inhibidor de recetor de estrógeno" o "inhibidor" se refiere a cualquier compuesto o tratamiento capaz de inhibir o reducir la expresión o actividad del receptor de estrógeno. El inhibidor es preferentemente selectivo.
En un aspecto R2 es 4-metiltiazol-5-il, oxazol-5-il.
En un aspecto R2 es 4-metiltiazol-5-il;
El grupo de unión es un grupo alquileno de cadena lineal de 8-16 átomos de carbono en donde uno o más átomos se sustituye por un grupo cada uno seleccionado independientemente de -O-, -NH-, -N(CH3)-,

En un aspecto, el grupo de unión es de fórmula (ii)
-(R3CH2CH2)x OCH2 (ii)
En la que cada R3 se selecciona independientemente entre -O-, -NH-, -N(CH3)- o

y x es 2-4. En un aspecto al menos un R3 es N(CH)
Mientras que los aspectos para cada variable se han enumerado, en general, anteriormente por separado para cada variable, la presente invención incluye aquellos compuestos en los que varios o cada aspecto en fórmula (I) se selecciona de cada uno de los aspectos enumerados anteriormente. Por lo tanto, la presente invención está prevista
para que incluya todas las combinaciones de aspectos para cada variable.
Ejemplos de compuestos de la presente invención incluyen los siguientes:
(2S,4R)-1-((S)-17-(4-((Z)-1,2-Difenilbut-1 -en-1-il)fenoxi)-2-isopropil-15-metil-4-oxo-6,9,12-trioxa-3,15-diazaheptadecan-1-oil)-4-hidroxi-N-(4-(4-metiltiazol-5-il)bencil)pirrolidina-2-carboxamida

(2S,4R)-1 -((S)-2-(ferc-ButM)-17-(4-((Z]-1,2-difenilbut-1 -en-1 -il)fenoxi)-15-metil-4-oxo-6,9,12-trioxa-3,15-diazaheptadecan-1-oil)-4-hidroxi-N-(4-(4-metiltiazol-5-il)bencil)pirrolidina-2-carboxamida

Los compuestos de Fórmula (I) pueden estar en la forma de una sal. Normalmente, las sales de la presente invención son sales farmacéuticamente aceptables. Las sales abarcadas dentro de la expresión "sales farmacéuticamente aceptables" se refieren a sales no tóxicas de los compuestos de esta invención. Para una revisión de las sales adecuadas véase Berge et al., J. Pharm. Sci. 1977, 66,1-19.
Sales farmacéuticamente aceptables pueden incluir sales de adición ácida. Una sal de adición de ácido farmacéuticamente aceptable se puede formar mediante la reacción de un compuesto de fórmula (I) con un ácido inorgánico u orgánico (tal como bromhídrico, clorhídrico, sulfúrico, nítrico, fosfórico, p-toluenosulfónico, bencenosulfónico, metanosulfónico, etanosulfónico, naftalenosulfónico tal como 2-naftalenosulfónico), opcionalmente en un disolvente adecuado tal como un disolvente orgánico, para proporcionar la sal que se aísla normalmente, por ejemplo, mediante cristalización y filtración. Una sal de adición de ácido farmacéuticamente aceptable de un compuesto de fórmula (I) puede comprender o ser, por ejemplo, una sal de hidrobromuro, hidrocloruro, sulfato, nitrato, fosfato, p-toluenosulfonato, bencenosulfonato, metanosulfonato, etanosulfonato, naftalenosulfonato (por ejemplo, 2-naftalenosulfonato).
Se pueden usar otras sales no farmacéuticamente aceptables, por ejemplo, trifluoroacetato, por ejemplo, en el aislamiento de compuestos de la invención y se incluyen dentro del alcance de la presente invención.
La invención incluye dentro de su alcance todas las formas estequiométricas y no estequiométricas posibles de los compuestos de fórmula (I).
Mientras que es posible que, durante su uso en la terapia, el compuesto de la invención se pueda administrar como un químico puro, es posible presentar el ingrediente activo como una composición farmacéutica. Por consiguiente, la invención proporciona adicionalmente composiciones farmacéuticas que comprenden un compuesto de la invención y uno o más vehículos, diluyentes o excipientes farmacéuticamente aceptables. El/los vehículo(s), diluyente(s) o excipiente(s) ha(n) de ser aceptable(s) en el sentido de ser compatible(s) con los demás ingredientes de la composición y no perjudicial(es) para el receptor del mismo/de los mismos. De acuerdo con otro aspecto de la invención, también se proporciona un proceso para la preparación de una composición farmacéutica que incluye el agente, o sales farmacéuticamente aceptables del mismo, con uno o más vehículos, diluyentes o excipientes farmacéuticamente
aceptables. La composición farmacéutica puede ser para su uso en el tratamiento y/o profilaxis de cualquiera de las afecciones que se describen en el presente documento.
Las composiciones farmacéuticas pueden presentarse en formas farmacéuticas unitarias que contienen una cantidad predeterminada de principio activo por dosis unitaria. Composiciones de dosificación unitaria preferentes son aquellas que contienen una dosis diaria o sub-dosis o una fracción adecuada de las mismas de un principio activo. Tales dosis unitarias pueden administrarse, por lo tanto, una o más de una vez al día. Tales composiciones farmacéuticas pueden prepararse mediante cualquier método bien conocido en la materia de la farmacia.
Las composiciones farmacéuticas se pueden adaptar para su administración mediante cualquier vía adecuada, por ejemplo, mediante la vía oral (incluida bucal o sublingual), rectal, inhalada, intranasal, tópica (incluida bucal, sublingual o transdérmica), vaginal o parenteral (incluida subcutánea, intramuscular, intravenosa o intradérmica). Tales composiciones pueden prepararse mediante cualquier método conocido en la materia de la farmacia, por ejemplo, poniendo en asociación el principio activo con el/los vehículo(s) o excipiente(s).
Las composiciones farmacéuticas adaptadas para su administración oral pueden presentarse como unidades por separado tales como cápsulas o comprimidos; polvos o gránulos; soluciones o suspensiones en líquidos acuosos o no acuosos; espumas comestibles o batidos; o emulsiones líquidas de aceite en agua o emulsiones líquidas de agua en aceite.
Por ejemplo, para una administración oral en forma de comprimido o cápsula, el componente de fármaco activo se puede combinar con un vehículo inerte no tóxico farmacéuticamente aceptable oral, tal como etanol, glicerol, agua y similares. Los polvos se preparan triturando el compuesto hasta un tamaño fino adecuado y mezclando con un vehículo triturado de manera similar, tal como un hidrato de carbono comestible como, por ejemplo, almidón o manitol. También pueden estar presentes aromatizantes, conservantes, dispersantes y un agente colorante.
Las cápsulas se fabrican mediante la preparación de una mezcla en polvo, como se ha descrito anteriormente, y el llenado de cubiertas de gelatina formadas. Pueden añadirse a la mezcla en polvo emolientes y lubricantes, tales como sílice coloidal, talco, estearato de magnesio, estearato de calcio o polietilenglicol sólido antes de la operación de relleno. Se puede añadir también un agente disgregante o solubilizante tal como agar-agar, carbonato de calcio, o carbonato de sodio para mejorar la disponibilidad del medicamento cuando se ingiere la cápsula.
Además, cuando se desee o sea necesario, también se pueden incorporar a la mezcla aglutinantes, lubricantes, agentes disgregantes y agentes colorantes. Los aglutinantes adecuados incluyen almidón, gelatina, azúcares naturales tales como glucosa o beta-lactosa, edulcorantes de maíz, gomas naturales y sintéticas, tales como acacia, tragacanto o alginato de sodio, carboximetilcelulosa, polietilenglicol, ceras y similares. Los lubricantes usados en estas formas de dosificación incluyen oleato de sodio, estearato de sodio, estearato de magnesio, benzoato de sodio, acetato de sodio, cloruro sódico y similares. Los desintegrantes incluyen, sin limitación, almidón, metilcelulosa, agar, bentonita, goma xantana y similares. Los comprimidos se formulan, por ejemplo, mediante la preparación de una mezcla en polvo, la granulación o el pegado, añadiendo un lubricante y un disgregante, y el prensado en comprimidos. Se prepara una mezcla en polvo mezclando el compuesto, convenientemente triturado, con un diluyente o una base según lo descrito anteriormente y, opcionalmente, con un aglutinante tal como carboximetilcelulosa, un alginato, gelatina, pirrolidona polivinílica, un retardante de soluciones, tal como parafina, un acelerador de la reabsorción, tal como una sal cuaternaria y/o un agente de absorción, tal como bentonita, caolín o fosfato dicálcico. La mezcla en polvo se puede granular mediante la humectación con un aglutinante tal como jarabe, pasta de almidón, mucílago de acacia o soluciones de materiales celulósicos o poliméricos, y el prensado a través de un tamiz. Como una alternativa a la granulación, la mezcla en polvo puede procesarse en la máquina para formar comprimidos y el resultado son aglomerados formados de manera imperfecta que se rompen en gránulos. Los gránulos pueden lubricarse para impedir que se adhieran a los moldes para la formación de comprimidos mediante la adición de ácido esteárico, una sal de estearato, talco o aceite mineral. A continuación, la mezcla lubricada se comprime formando comprimidos. Los compuestos de la presente invención también pueden combinarse con un vehículo inerte de flujo libre y se comprimen en forma de comprimidos directamente sin pasar por los pasos de granulación o aglomeración. Se puede proporcionar un revestimiento protector transparente u opaco que consiste de un revestimiento sellador de shellac, un revestimiento de azúcar o material polimérico, y un revestimiento de cera pulida. Se pueden añadir colorantes a estos revestimientos para distinguir diferentes dosificaciones unitarias.
Se pueden preparar fluidos orales tales como soluciones, jarabes, y elixires en formas farmacéuticas unitarias de tal manera que una cantidad dada contiene una cantidad predeterminada del compuesto. Se pueden preparar jarabes disolviendo el compuesto en una solución acuosa con sabor adecuada, mientras que los elixires se preparan mediante el uso de un vehículo alcohólico no tóxico. Las suspensiones se pueden formular dispersando el compuesto en un vehículo no tóxico. También pueden añadirse solubilizantes y emulsionantes, tales como alcoholes isoestearílicos etoxilados y éteres de polioxietileno sorbitol, conservantes, aditivos aromatizantes, tales como aceite de menta o edulcorantes naturales o sacarina u otros edulcorantes artificiales y similares.
Cuando sea apropiado, las formulaciones de dosis unitarias para una administración oral pueden estar microencapsuladas. La composición también se puede preparar para prolongar o mantener la liberación como, por
ejemplo, mediante el revestimiento o la introducción de material particulado en polímeros, cera o similares.
Los compuestos de fórmula (I) y las sales, los solvatos y sus derivados fisiológicamente funcionales también se pueden administrar en forma de sistemas de liberación de liposomas tales como pequeñas vesículas unilamelares, grandes vesículas unilamelares y vesículas multilamelares. Los liposomas se pueden formar a partir de varios fosfolípidos tales como colesterol, estearilamina o fosfatidilcolinas.
Las composiciones farmacéuticas adaptadas para administración transdérmica pueden presentarse como parches individuales previstos para permanecer en contacto íntimo con la epidermis del receptor durante un periodo prolongado de tiempo.
Las composiciones farmacéuticas adaptadas a la administración tópica pueden formularse como pomadas, cremas, suspensiones, lociones, polvos, soluciones, pastas, geles, pulverizados, aerosoles o aceites.
Para los tratamientos oculares o de otros tejidos externos, por ejemplo, la boca y piel, las formulaciones se aplican preferentemente como una pomada o crema tópica. Cuando se formulan en forma de pomada, el principio activo se puede emplear con una base bien de parafina o de pomada hidromiscible. De modo alternativo, el principio activo puede formularse en una crema con una base de crema de aceite en agua o como una base de agua en aceite.
Las composiciones farmacéuticas adaptadas para administraciones tópicas al ojo incluyen gotas oculares, en las que el principio activo se disuelve o suspende en un vehículo adecuado, especialmente un disolvente acuoso.
Las formulaciones farmacéuticas adaptadas a la administración tópica en la boca incluyen grageas, pastillas y enjuagues bucales.
Las composiciones farmacéuticas adaptadas para administración rectal pueden presentarse como supositorios o como enemas.
Las formas de dosificación para su administración nasal o mediante inhalación se pueden preparar convenientemente como aerosoles, soluciones, suspensiones, gotas o polvos secos.
Las formulaciones farmacéuticas adaptadas a la administración vaginal se pueden presentar como pesarios, tampones, cremas, geles, pastas, espumas o formulaciones en pulverizado.
Las formulaciones farmacéuticas adaptadas a la administración parenteral incluyen soluciones inyectables estériles acuosas y no acuosas que pueden contener antioxidantes, tampones, bacteriostatos y solutos que convierten la formulación en isotónica con la sangre del receptor deseado; y suspensiones estériles acuosas y no acuosas que pueden incluir agentes de suspensión y agentes espesantes. Las composiciones se pueden presentar en recipientes de una sola dosis o de múltiples dosis, por ejemplo, en ampollas selladas y viales, y se pueden almacenar en un estado criodesecado (liofilizado) que únicamente requiera la adición del vehículo líquido estéril, por ejemplo, de agua para inyecciones inmediatamente antes de su uso. Se pueden preparar soluciones y suspensiones para inyección extemporáneas a partir de polvos, gránulos y comprimidos estériles.
Se debe entender que además de los ingredientes mencionados anteriormente en particular, las formulaciones pueden incluir otros agentes convencionales en la técnica, teniendo en cuenta el tipo de formulación en cuestión, por ejemplo, aquéllas adecuadas para una administración oral pueden incluir agentes aromatizantes.
Una cantidad terapéuticamente eficaz de un compuesto de la presente invención dependerá de un número de factores incluyendo, por ejemplo, la edad y el peso del sujeto, la afección exacta que requiere tratamiento y su gravedad, la naturaleza de la formulación y la vía de administración y, finalmente, se hará según el criterio del médico o veterinario. En particular, el sujeto a tratar es un mamífero, en particular, un ser humano.
El agente se puede administrar en una dosis diaria. Esta cantidad se puede proporcionar en una única dosis al día o en cualquier cantidad (tla como, dos, tres, cuatro, cinco o seis) sub-dosis al día de modo que la dosis diaria total sea la misma.
De modo adecuado, la cantidad del compuesto de la invención administrado de acuerdo con la presente invención será una cantidad seleccionada de 0,01 mg a 1 g al día (calculado como el compuesto libre o sin sal).
Hemos encontrado que los compuestos definidos en la presente invención, o sal farmacéuticamente aceptable de los mismos, o composiciones farmacéuticas que los contienen, son capaces de degradar el receptor de estrógeno.
Por consiguiente, los compuestos de la presente invención se espera que sean agentes potencialmente útiles en el tratamiento de enfermedades o afecciones médicas mediadas solo o en parte por el receptor de estrógeno.
En general, se describen en el presente documento métodos de tratamiento o prevención de enfermedades, trastornos o afecciones mediadas por el receptor de estrógenos. El método puede comprender la administración a un sujeto, por ejemplo, un sujeto que lo necesite, una cantidad terapéuticamente eficaz de un compuesto de la invención.
De este modo, en un aspecto se proporciona un compuesto de la invención para su uso en terapia.
De este modo, en un aspecto se proporciona un compuesot de la invención para su uso en el tratamiento de enfermedades, trastornos o condiciones mediadas por el receptor de estrógeno.
En general, descrito en el presente documento, se proporciona el uso de un compuesto de la invención en la fabricación de un medicamento para el tratamiento de enfermedades, trastornos o afecciones mediadas por el receptor de estrógeno.
En general, descrito en el presente documento, se proporciona un método de tratamiento de enfermedades, trastornos o afecciones mediadas por el receptor de estrógeno en un mamífero que comprende la administración de una cantidad terapéuticamente eficaz de un compuesto de la invención.
El compuesto de la invención es útil en el tratamiento de afecciones asociadas con el receptor de estrógeno. Una "afección asociada con el receptor de estrógeno", tal como se usa en el presente documento, denota una afección o trastorno que puede tratarse modulando la función o actividad de un receptor de estrógeno en un sujeto, en el que el tratamiento comprende la prevención, alivio parcial o cura de la afección o trastorno. La modulación se puede producir localmente, por ejemplo, dentro de determinados tejidos del sujeto o más ampliamente por todo un sujeto que está siendo tratado de tal afección o trastorno.
En un aspecto la enfermedad o afección mediada por estrógeno es cáncer de mama.
Los compuestos de la presente invención pueden usarse en combinación con o incluir uno o más otros agentes terapéuticos y se pueden administrar o bien secuencialmente o bien simultáneamente a través de cualquier vía conveniente en composiciones farmacéuticas combinadas o por separado. La cantidad terapéuticamente eficaz de los agentes terapéuticos adicionales de la presente invención dependerá de una cantidad de factores entre los que se incluye, por ejemplo, la edad y peso del mamífero, la afección exacta que requiere el tratamiento, la gravedad de la afección, la naturaleza de la formulación y la vía de administración. Finalmente, la cantidad terapéuticamente eficaz será según el criterio del médico o veterinario que lo atienda. Los tiempos relativos de administración se seleccionarán para conseguir el efecto terapéutico combinado deseado.
Los compuestos de la presente invención y agente(s) terapéutico(s) adicional(es) se puede(n) emplear en combinación mediante la administración simultánea en una composición farmacéutica unitaria que incluye ambos compuestos. De modo alternativo, la combinación se puede administrar por separado en composiciones farmacéuticas por separado, incluyendo cada una uno de los compuestos de un modo secuencial en el que, por ejemplo, el compuesto de la invención se administrar en primer lugar y el otro, en segundo lugar, y viceversa. Tal administrar secuencial puede ser en un tiempo cercano (por ejemplo, simultáneamente) o en un tiempo distante. Además, no importa si los compuestos se administran en la misma forma de dosificación, por ejemplo, un compuesto se puede administrar por vía tópica y el otro compuesto se puede administrar por vía oral. De forma adecuada, ambos compuestos se administrar por vía oral.
Las combinaciones se pueden presentar como un kit de combinación. Por el término "kit de combinación" o "partes de kit" tal como se usa en el presente documento se refiere a la composición o composiciones farmacéutica(s) que se usa(n) para administrar la combinación de acuerdo con la invención. Cuando ambos compuestos se administran simultáneamente, el kit de combinación puede contener ambos compuestos en una única composición farmacéutica, tal como un comprimido, o en composiciones farmacéuticas por separado. Cuando los compuestos no se administran simultáneamente, el kit de combinación contendrá cada compuesto en composiciones farmacéuticas por separado o bien en un envase individual o en composiciones farmacéuticas por separado en envases separados.
El kit de combinación también se puede proporcionar mediante instrucción, tal como dosificación e instrucciones de administración. Tales instrucciones de dosificación y administración pueden ser del tipo que se proporcionan a un doctor, por ejemplo, mediante una etiqueta de producto de fármaco o pueden ser del tipo que se proporcionan a un doctor tales como instrucciones a un paciente.
Cuando la combinación se administra por separado de un modo secuencial en la que una se administra en primer lugar y la otra en segundo lugar, o viceversa, tal administración secuencial puede ser cercana en el tiempo o distante en el tiempo. Por ejemplo, se incluye la administración del otro agente de varios minutos a varias docenas de minutos después de la administración del primer agente, y la administración del otro agente de varias horas a varios días después de la administración del primer agente, en el que el lapso de tiempo no queda limitado, por ejemplo, un agente se puede administrar una vez al día y, el otro agente, se puede administrar 2 o 3 veces al día, o un agente se puede administrar una vez a la semana y el otro agente se puede administrar una vez al día y similares.
Resultará claro a un experto en la materia que, cuando sea necesario, el/los otro(s) agente(s) terapéutico(s) se puede(n) usar en forma de sales, por ejemplo, como sales de metal alcalino o amina como sales de adición de ácido, o profármacos, o como ésteres, por ejemplo, ésteres de alquilo inferior, o como solvatos, por ejemplo, hidratos, para
optimizar la actividad y/o estabilidad y/o características físicas, tales como solubilidad o el ingrediente terapéutico. También resultará claro que, cuando sea necesario, los ingredientes terapéuticos se pueden usar en forma ópticamente pura.
Cuando se combina en la misma composición se apreciará que los dos compuestos deben ser estables y compatibles entre sí y los otros componentes de la composición y se puedan formular para su administración. Cuando se formula por separado, se pueden proporcionar en cualquier composición adecuada, convenientemente, de tal modo como se conoce para tales compuestos en la materia.
Cuando el compuesto de fórmula (I) se usa en combinación con un segundo agente terapéutico activo frente a la misma enfermedad, afección o trastorno, la dosificación de cada compuesto puede diferir de cuando el compuesto se usa solo. Se apreciarán fácilmente dosis adecuadas por los expertos en la materia.
En una realización, el compuesto de compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo se puede emplear con otros métodos terapéuticos de tratamiento del cáncer. En particular, en terapia neoplásica, se prevé la terapia combinada con otros agente quimioterapéuticos, hormonales, agentes de anticuerpos así como tratamiento quirúrgicos y/o radiación distintos de los mencionados anteriormente.
En una realización, la terapia anticáncer adicional es la cirugía y/o la radioterapia.
En una realización, la terapia contra el cáncer adicional es al menos un agente antineoplásico adicional.
Puede utilizarse en la combinación cualquier agente antineoplásico que tenga actividad frente a un tumor susceptible de tratarse. Los agentes antineoplásicos típicos útiles en la presente invención incluyen, pero sin limitación, agentes antimicrotúbulos tales como diterpenoides y alcaloides de la vinca; complejos de coordinación de platino; agentes alquilantes tales como mostazas nitrogenadas, oxazafosforinas, alquilsulfonatos, nitrosoureas, y triazenos; agentes antibióticos tales como antraciclinas, actinomicinas y bleomicinas; inhibidores de la topoisomerasa II, tales como epipodofilotoxinas; antimetabolitos tales como análogos de purina y pirimidina y compuestos antifolato; inhibidores de la topoisomerasa I tales como camptotecinas; hormonas y análogos hormonales; inhibidores de la vía de transducción de señales; inhibidores de la angiogénesis de la tirosina quinasa no receptor; agentes inmunoterapéuticos; agentes proapoptóticos e inhibidores de la señalización del ciclo celular.
Agentes antimicrotúbulos o antimitóticos:
Los agentes antimicrotúbulos o antimitóticos son agentes específicos de fase activos frente a los microtúbulos de las células tumorales durante la fase M o de la mitosis del ciclo celular. Los ejemplos de agentes antimicrotúbulos incluyen, pero sin limitación, diterpenoides y alcaloides de la vinca.
Los diterpenoides, que se derivan de fuentes naturales, son agentes anticancerígenos específicos de fase que actúan en las fases G2/M del ciclo celular. Se considera que los diterpenoides estabilizan la subunidad p-tubulina de los microtúbulos, mediante la unión con esta proteína. Entonces, el desensamblaje de las proteínas parece estar inhibido, la mitosis detenida, siguiendo la muerte celular. Los ejemplos de diterpenoides incluyen, pero sin limitación, paclitaxel y su análogo docetaxel.
Paclitaxel, 5p,20epoxi-1,2a,4,7p,10p,13a-hexa-hidroxitax-11-en-9-ona 4,10-diacetato 2-benzoato 13-éster con (2R,3S)-N-benzoil-3-fenilisoserina; es un producto diterpeno natural aislado del tejo del Pacífico Taxus brevifolia y está disponible en el comercio como una solución inyectable TAXOL®. Es un miembro de la familia de taxanos de los terpenos. Paclitaxel se ha aprobado en los Estados Unidos para el uso clínico en el tratamiento del cáncer de ovario resistente (Markman et al., Yale Journal of Biology and Medicine, 64:583, 1991; McGuire et al., Ann. Intern, Med., 111:273,1989) y para el tratamiento de cáncer de mama (Holmes et al., J. Nat. Cancer Inst, 83:1797,1991.) Es un candidato potencial para el tratamiento de las neoplasias en la piel (Einzig et. al., Proc. Am. Soc. Clin. Oncol., 20:46) y los carcinomas de cabeza y cuello (Forastire et. al., Sem. Oncol., 20:56, 1990). El compuesto también muestra potencial para el tratamiento de la poliquistosis renal (Woo et. al., Nature, 368:750. 1994), el cáncer de pulmón y la malaria. El tratamiento de los pacientes con paclitaxel produce la supresión de la médula ósea (múltiples linajes celulares, Ignoff, RJ et. al, Cancer Chemotherapy Pocket Guide, 1998) relacionado con la duración de la dosificación por encima de una concentración umbral (50 nM) (Kearns, C.M. et. al., Seminars in Oncology, 3(6) p.16-23,1995).
El docetaxel, (2R,3S)-N-carboxi-3-fenilisoserina, N-terc-butil éster, 13-éster con 4-acetato de 2-benzoato de 5p-20-epoxi-1,2a,4,7p,10p,13a-hexahidroxitax-11-en-9-ona, trihidrato; está disponible en el comercio como una solución inyectable como TAXOTERE®. Docetaxel está indicado para el tratamiento del cáncer de mama. El docetaxel es un derivado semisintético de paclitaxel q.v, preparado usando un precursor natural, 10-desacetil-bacatina III, extraído de la aguja del árbol del tejo europeo.
Los alcaloides de la vinca son agentes antineoplásicos específicos de fase obtenidos de la vinca. Los alcaloides de la vinca actúan en la fase M (mitosis) del ciclo celular uniéndose de forma específica a la tubulina. Por consiguiente, la molécula de tubulina unida no es capaz de polimerizar a microtúbulos. Se cree que la mitosis está detenida en la
metafase con la posterior muerte celular. Los ejemplos de alcaloides de la vinca incluyen, pero sin limitación, vinblastina, vincristina, y vinorelbina.
La vinblastina, sulfato de vincaleucoblastina, está disponible en el comercio como Velban® como una solución inyectable. Si bien, tiene una posible indicación como terapia de segunda línea de diversos tumores sólidos, está indicada principalmente para el tratamiento de cáncer testicular y diversos linfomas que incluyen enfermedad de Hodgkin y linfomas linfocíticos e histiocíticos. La mielosupresión es el efecto secundario limitante de la dosis de la vinblastina.
La vincristina, vincaleucoblastina, 22-oxo-, sulfato, está disponible en el mercado como ONCOVIN® como una solución inyectable. La vincristina está indicada para el tratamiento de las leucemias agudas y también ha encontrado uso en los regímenes de tratamiento de los linfomas malignos de Hodgkin y no Hodgkin. La alopecia y los efectos neurológicos son los efectos secundarios más comunes de la vincristina y, en un menor grado, pueden producirse efectos de mielosupresión y mucositis gastrointestinal.
La vinorelbina 3',4'-dideshidro-4'-desoxi-C'-norvincaleucoblastina [ R-(R*,R*)-2,3-dihidroxibutanodioato (1:2) (sal)], disponible en el comercio como una solución inyectable de tartrato de vinorelbina (NAVELBINE®), es un alcaloide de la vinca semisintético. La vinorelbina está indicada como agente único o en combinación con otros agentes quimioterapéuticos, tales como cisplatino, en el tratamiento de diversos tumores sólidos, en particularmente cáncer pulmonar de pulmón de células no pequeñas, mamario avanzado y prostático refractario a hormonas. La mielosupresión es el efecto secundario limitante de la dosis más común de la vinorelbina.
Complejos de coordinación de platino:
Los complejos de coordinación de platino son agentes contra el cáncer no específicos de fase, que son interactivos con el ADN. Los complejos de platino entran en las células tumorales, incorporan agua y forman entrecruzamientos intra e intercatenarios con el ADN, provocando efectos biológicos adversos para el tumor. Los ejemplos de complejos de coordinación de platino incluyen, pero sin limitación, cisplatino y carboplatino.
El cisplatino, cis-diaminodicloroplatino, está disponible en el comercio como Platinol® como una solución inyectable. El cisplatino está indicado principalmente en el tratamiento del cáncer testicular y ovárico metastásico, y el cáncer de vejiga avanzado.
El carboplatino, platino, diamina [1,1-ciclobutano-dicarboxilato de dimetilo (2-)-0,0'], está disponible en el comercio como PARAPLATIN® como una solución inyectable. El carboplatino está indicado principalmente en el tratamiento de primera y segunda línea del carcinoma ovárico avanzado.
Agentes alquilantes:
Los agentes alquilantes son agentes específicos contra el cáncer no de fase y son fuertes electrófilos. Típicamente, los agentes alquilantes forman enlaces covalentes, mediante alquilación, al ADN a través de restos nucleófilos de la molécula de a Dn tales como grupos fosfato, amino, sulfhidrilo, hidroxilo, carboxilo e imidazol. Tal alquilación altera las funciones del ácido nucleico, conduciendo a la muerte celular. Los ejemplos de agentes alquilantes incluyen, pero no se limitan a, mostazas de nitrógeno tales como ciclofosfamida, melfalán y clorambucilo; sulfonatos de alquilo tales como busulfán; nitrosoureas tales como carmustina; y triazenos tales como dacarbazina.
El ciclofosfamida, 2-[bis(2-cloroetil)amino]tetrahidro-2H-1,3,2-oxazafosforina 2-óxido monohidrato, está disponible comercialmente como una solución inyectable o como comprimidos como CYTOXAN®. La ciclofosfamida está indicada como un agente individual o en combinación con otros agentes quimioterapéuticos, en el tratamiento de linfomas malignos, mieloma múltiple y leucemias.
El melfalan, 4-[bis(2-cloroetil)amino]-L-fenilalanina, está disponible comercialmente como una solución inyectable o como comprimidos como ALKERAN®. El melfalán está indicado para el tratamiento paliativo del mieloma múltiple y del carcinoma de ovario epitelial no resecable. La supresión de la médula ósea es el efecto secundario limitante de la dosis más común del melfalán.
El clorambucil, ácido 4-[bis(2-cloroetil)amino]bencenobutanoico, está disponible comercialmente como comprimidos LEUKERAN®. Clorambucil está indicado para el tratamiento paliativo de la leucemia linfática crónica, y linfomas malignos tales como el linfosarcoma, el linfoma folicular gigante y la enfermedad de Hodgkin.
El busulfán, dimetanosulfonato de 1,4-butanodiol, está disponible comercialmente como comprimidos MYLERAN®. El busulfán está indicado para el tratamiento paliativo de la leucemia mielógena crónica.
La carmustina, 1,3-[bis(2-cloroetil)-1-nitrosourea, está disponible comercialmente como viales individuales de material liofilizado como BiCNU®. La carmustina está indicada para el tratamiento paliativo como agente individual o en combinación con otros agentes para tumores cerebrales, mieloma múltiple, enfermedad de Hodgkin y linfomas no
Hodgkin.
La dacarbazina, 5-(3,3-dimetil-1-triazeno)-imidazol-4-carboxamida, está disponible comercialmente como viales individuales de material como DTIC-Dome®. La dicarbazina está indicada para el tratamiento del melanoma maligno metastásico y, en combinación con otros agentes, para el tratamiento de segunda línea de la enfermedad de Hodgkin.
Antibióticos anti-neoplásicos:
Los antibióticos anti-neoplásicos son agentes no específicos de fase, que se unen o intercalan con el ADN. Típicamente, dicha acción resulta en complejos de ADN estables o en rotura de hebra, lo que interrumpe la función normal de los ácidos nucleicos conduciendo a la muerte celular. Los ejemplos de agentes antibióticos antineoplásicos incluyen, pero no se limitan a, actinomicinas tales como dactinomicina, antrociclinas tales como daunorrubicina y doxorrubicina; y bleomicinas.
La dactinomicina, conocida también como actinomicina D, está disponible comercialmente en forma inyectable como COSMEGEN®. La dactinomicina está indicada para el tratamiento del tumor de Wilm y el rabdomiosarcoma.
La daunorubicina, clorhidrato de (8S-cis-)-8-acetil-10-[(3-amino-2,3,6-trideoxi-a-L-lixohexopiranosil)oxi]-7,8,9,10-tetrahidro-6,8,11-trihidroxi-1-metoxi-5,12 naftacediona, está disponible comercialmente como una forma inyectable liposomal como DAUNOXOME® o como un inyectable como CERUBIDINE®. La doxorrubicina está indicada para la inducción de la remisión en el tratamiento de la leucemia no linfocítica aguda y el sarcoma de Kaposi asociado con el VIH avanzado.
La doxorubicina, clorhidrato de (8S, 10S)-10-[(3-amino-2,3,6-trideoxi-a-L-lixohexopiranosil)oxi]-8-glicoloil,7,8,9,10-tetrahidro-6,8,11-trihidroxi-1-metoxi-5,12 naftacenediona, está disponible comercialmente como una forma inyectablecomo RUBEX® o ADRIAMYCIN RDF®. La doxorrubicina está indicada principalmente para el tratamiento de la leucemia linfoblástica aguda y la leucemia mieloblástica aguda, pero también es un componente útil en el tratamiento de algunos tumores sólidos y linfomas.
La bleomicina, una mezcla de antibióticos glicopeptídicos citotóxicos aislados a partir de una cepa de Streptomyces veiticillus, está disponible comercialmente como BLENOXANE®. La bleomicina está indicada como un tratamiento paliativo, como un agente individual o en combinación con otros agentes, de carcinoma de células escamosas, linfomas y carcinomas testiculares.
Inhibidores de la topoisomerasa II:
Los inhibidores de la topoisomerasa II incluyen, pero no se limitan a, epipodofilotoxinas.
Las epipodofilotoxinas son agentes antineoplásicos específicos de fase obtenidos de la mandrágora. Normalmente, las epipodofilotoxinas afectan a las células en las fases S y G2 del ciclo celular mediante la formación de un complejo ternario con la topoisomerasa II y el ADN, provocando rupturas de las cadenas de ADN. Las rupturas de las cadenas se acumulan y lo siguiente es la muerte celular. Los ejemplos de epipodofilotoxinas incluyen, pero no se limitan a, etopósido y tenipósido.
El etopósido, 4'-desmetil-epipodofilotoxina 9[4,6-0-(R)-etiliden-p-D-glucopiranósido], está disponible comercialmente como una solución inyectable o cápsulas como VePESID® y es conocido comúnmente como VP-16. El etopósido está indicado como único agente o en combinación con otros agentes quimioterapéuticos en el tratamiento de los cánceres testiculares y de pulmón no microcítico.
El tenipósido, 4-desmetil-epipodofilotoxina 9[4,6-0-(R)-tieniliden-p-D-glucopiranósido], está disponible comercialmente como una solución inyectable como VUMON® y es conocido comúnmente como VM-26. El tenipósido está indicado como único agente o en combinación con otros agentes quimioterapéuticos en el tratamiento de la leucemia aguda en niños.
Agentes neoplásicos antimetábolitos:
Los agentes neoplásicos de antimetabolito son agentes antineoplásicos específicos de fase que actúan en la fase S (síntesis de ADN) del ciclo celular, inhibiendo la síntesis de ADN o inhibiendo la síntesis de bases purínicas o pirimidínicas y, por lo tanto, limitando la síntesis de ADN. Por consiguiente, la fase S no avanza y sigue la muerte celular. Los ejemplos de agentes antineoplásicos antimetabolitos incluyen, pero no se limitan a, fluorouracilo, metotrexato, citarabina, mecaptopurina, tioguanina y gemcitabina.
El 5-fluorouracilo, 5-fluoro-2,4-(1H,3H)pirimidinodiona, está disponible comercialmente como fluorouracilo. La administración de 5-fluorouracilo conduce a la inhibición de la síntesis de timidilato y también se incorpora tanto en el ARN como el ADN. Normalmente el resultado es la muerte celular. El 5-fluorouracilo está indicado como agente único o en combinación con otros agentes quimioterapéuticos en el tratamiento de carcinomas de mama, colon, recto,
estómago y páncreas. Otros análogos de fluoropirimidina incluyen 5-fluoro desoxiuridina (floxuridina) y 5-fluorodesoxiuridina monofosfato.
La citarabina, 4-amino-1-p-D-arabinofuranosil-2 (IH)-pirimidinona, está disponible en el mercado como CYTOSAR-U® y se lo conoce comúnmente como Ara-C. Se cree que la citarabina presenta especificidad de fase celular en la fase S, inhibiendo la elongación de la cadena de ADN mediante la incorporación terminal de citarabina en la cadena de ADN que se está sintetizando. La citarabina está indicada como único agente o en combinación con otros agentes quimioterapéuticos en el tratamiento de la leucemia aguda. Otros análogos de citidina incluyen 5-azacitidina y 2',2'-difluorodesoxicitidina (gemcitabina).
La mercaptopurina, 1,7-dihidro-6H-purina-6-tiona monohidrato, está disponible comercialmente como PURINETHOL®. La mercaptopurina presenta especificidad de fase celular en la fase S inhibiendo la síntesis de ADN mediante un mecanismo aún no especificado. La mercaptopurina está indicada como único agente o en combinación con otros agentes quimioterapéuticos en el tratamiento de la leucemia aguda. Un análogo de la mercaptopurina útil es la azatioprina.
La tioguanina, 2-amino-1,7-dihidro-6H-purina-6-tiona, está disponible comercialmente como TABLOID®. La tioguanina presenta especificidad de fase celular en la fase S inhibiendo la síntesis de ADN mediante un mecanismo todavía no especificado. La tioguanina está indicada como único agente o en combinación con otros agentes quimioterapéuticos en el tratamiento de la leucemia aguda. Otros análogos de purina incluyen pentostatina, eritrohidroxinoniladenina, fosfato de fludarabina y cladribina.
La gemcitabina, monoclorhidrato de 2'-desoxi-2',2'-difluorocitidina (isómero p), está disponible comercialmente como GEMZAR®. La gemcitabina presenta especificidad de fase celular en la fase S y por bloqueo de la progresión de las células a través del límite G1/S. La gemcitabina está indicada en combinación con cisplatino en el tratamiento del cáncer de pulmón no microcítico localmente avanzado y sola en el tratamiento del cáncer de páncreas localmente avanzado.
El metotrexato, ácido N-[4[[(2,4-diamino-6-pteridinil)metil]metilamino]benzoil]-L-glutámico, está disponible comercialmente como metotrexato sódico. El metotrexato exhibe efectos de fase celular específicamente en la fase S mediante la inhibición de la síntesis, reparación y/o replicación de ADN a través de la inhibición de la reductasa de ácido dihidrofólico que es necesario para la síntesis de nucleótidos de purina y timidilato. El metotrexato está indicado como un agente individual o en combinación con otros agentes de quimioterapia en el tratamiento de coriocarcinoma, leucemia meníngea, linfoma no Hodgkin y carcinomas de mama, cabeza, cuello, ovario y vejiga.
Inhibidores de la topoisomerasa I:
Las camptotecinas, incluidos la camptotecina y los derivados de camptotecina, están disponibles o en desarrollo como inhibidores de la Topoisomerasa I. Se cree que la actividad citotóxica de las camptotecinas está relacionada con su actividad inhibidora de la Topoisomerasa I. Los ejemplos de camptotecinas incluyen, pero no se limitan a, irinotecán, topotecán y las diversas formas ópticas de 7-(4-metilpiperazino-metileno)-10,11-etilendioxi-20-camptotecina descritas a continuación.
El irinotecán HCl, clorhidrato de (4S]-4,11-dietil-4-hidroxi-9-[(4-piperidinopiperidino)carboniloxi]-1H-pirano[3',4',6,7]indolizino[1,2-b]quinolina-3,14(4H,l2H)-diona, está disponible comercialmente como la solución inyectable CAMPTOSAR®. El irinotecán es un derivado de la camptotecina que se une, junto con su metabolito activo s N-38, al complejo topoisomerasa I-ADN. Se cree que la citotoxicidad se produce como resultado de rupturas irreparables de la doble cadena provocadas por la interacción con el complejo ternario topoisomerasa I: ADN:irintecan o complejo ternario SN-38 con enzimas de replicación. El irinotecán está indicado para el tratamiento del cáncer metastásico del colon o el recto.
El topotecán HCl, (S)-10-[(dimetilamino)metil]-4-etil-4,9-dihidroxi-1H-pirano[3',4',6,7]indolizino[1,2-b]quinolina-3,14-(4H,12H)-dionemonoclorhidrato, está disponible comercialmente como la solución inyectable HYCAMTIN®. El topotecán es un derivado de la camptotecina que se une al complejo topoisomerasa I-ADN e impide el religamiento de las rupturas monocatenarias provocadas por la Topoisomerasa I en respuesta a la tensión torsional de la molécula de ADN. El topotecán está indicado para el tratamiento de segunda línea del carcinoma metastásico de ovario y el cáncer de pulmón microcítico.
Hormonas y análogos hormonales:
Las hormonas y los análogos hormonales son compuestos útiles para el tratamiento de cánceres en los que existe una relación entre la hormona (u hormonas) y el crecimiento, y/o la falta de crecimiento del cáncer. Ejemplos de hormonas y análogos hormonales útiles en el tratamiento del cáncer incluyen, aunque no de forma limitante a, adrenocorticoesteroides tales como prednisona y prednisolona que son útiles en el tratamiento de linfoma maligno y leucemia aguda en niños; aminoglutetimida y otros inhibidores de la aromatasa tales como anastrozol, letrazol, vorazol, y exemestano útiles en el tratamiento del carcinoma adrenocortical y carcinoma de mama dependiente de hormonas
que contiene receptores de estrógenos; progestinas tales como acetato de megestrol útiles en el tratamiento de cáncer de mama dependiente de hormonas y carcinoma de endometrio; estrógenos, andrógenos, y antiandrógenos tales como flutamida, nilutamida, bicalutamida, acetato de ciproterona y 5a-reductasas tales como finasterida y dutasterida, útiles en el tratamiento de carcinoma prostático y la hipertrofia benigna de próstata; anti-estrógenos tales como tamoxifeno, torremifeno, raloxifeno, droloxifeno, yodoxifeno, así como moduladores selectivos de los receptores estrogénicos (SERM) como los descritos en las Patentes de los EE.UU. n.° 5.681.835, 5.877.219, y 6.207.716, útiles en el tratamiento del carcinoma de mama dependiente de hormonas y otros cánceres susceptibles, y la hormona liberadora de gonadotropina (GnRH) y sus análogos que estimulan la liberación de la hormona luteinizante (LH) y/o la hormona folículo estimulante (FSH) para el tratamiento de carcinoma prostático, por ejemplo, agonistas y antagonistas de LHRH tales como acetato de goserelina y luprolida.
Inhibidores de la vía de transducción de señales:
Los inhibidores de la vía de transducción de señales son aquellos inhibidores que bloquean o inhiben un proceso químico que evoca un cambio intracelular. Como se usa en el presente documento, este cambio es proliferación celular o diferenciación. Los inhibidores de la vía de transducción de señales útiles en la presente invención incluyen, pero no se limitan a, inhibidores de tirosina quinasas receptoras, tirosina quinasas no receptoras, bloqueadores de dominios SH2/SH3, serina/treonina quinasas, fosfatidil inositol-3-OH quinasas, señalización de mioinositol y oncogenes Ras.
Varias proteína tirosina quinasas catalizan la fosforilación de restos tirosil específicos en diversas proteínas implicadas en la regulación del crecimiento celular. Tales proteína tirosina quinasas pueden clasificarse en líneas generales como quinasas receptoras o no receptoras.
Los receptores de tirosina quinasa son proteínas de transmembrana que tienen un dominio de unión al ligando extracelular, un dominio de transmembrana, y un dominio tirosina quinasa. Las tirosina quinasas receptoras están implicadas en la regulación del crecimiento celular y en general se denominan receptores de factores de crecimiento. Se ha demostrado que una activación inapropiada o descontrolada de muchas de estas quinasas, es decir, la actividad quinasa anómala del receptor de factores de crecimiento, por ejemplo mediante la expresión aumentada o la mutación, da como resultado el crecimiento celular descontrolado. Por consiguiente, la actividad anómala de tales quinasas se ha vinculado al crecimiento de tejidos malignos. Por consiguiente, los inhibidores de tales quinasas podrían proporcionar métodos de tratamiento para el cáncer. Los receptores del factor de crecimiento incluyen, por ejemplo, receptor de factor de crecimiento epidérmico (EGFr), receptor del factor de crecimiento derivado de las plaquetas (PDGFr), erbB2, erbB4, receptor del factor de crecimiento endotelial vascular (VEGFr), tirosina quinasa con dominios de homología de factores de crecimiento tipo inmunoglobulina y epidérmico (TIE-2), receptor del factor de crecimiento de insulina-I (IGF-I), factor estimulante de colonias de macrófagos (cfms), BTK, ckit, cmet, receptores de factor de crecimiento de fibroblastos (FGF), receptores Trk (TrkA, TrkB, y TrkC), receptores de efrina (eph) y el protooncogén RET. Varios inhibidores de receptores de crecimiento están en desarrollo e incluyen antagonistas de ligandos, anticuerpos, inhibidores de tirosina quinasa y oligonucleótidos anti-sentido. Los receptores del factor de crecimiento y agentes que inhiben la función del receptor del factor de crecimiento se describen, por ejemplo, Kath, John C., Exp. Opin. Opin. Ther. Patents (2000) 10(6):803-818; Shawver et at DDT Vol 2, No. 2 February 1997; and Lofts, F. J. et al,”Growth factor receptors as targets", New Molecular Targets for Cancer Chemotherapy, ed. Workman, Paul and Kerr, David, CRC press 1994, Londres.
Las tirosina quinasas, que no son quinasas del receptor del factor de crecimiento se denominan tirosina quinasas no receptor. Las tirosina quinasas no receptor para usar en la presente invención, que son blancos o blancos potenciales de fármacos anticáncer, incluyen cSrc, Lek, Fyn, Yes, Jak, cAbI, FAK (quinasa de adhesión focal), tirosina quinasa de Brutons y Bcr-Abl. Tales quinasas no receptoras y agentes que inhiben la función de tirosina quinasa no receptor se describen en Sinh, S. and Corey, S.J., (1999) Journal of Hematotherapy and Stem Cell Research 8 (5): 465 - 80; and Bolen, J.B., Brugge, J.S., (1997) Annual review of Immunology. 15: 371-404.
Los bloqueadores de dominio SH2/SH3 son agentes que alteran el dominio de unión de SH2 o SH3 en varias enzimas o proteínas adaptadoras que incluyen, subunidad p85 de PI3-K, quinasas de la familia Src, moléculas adaptadoras (Shc, Crk, Nck, Grb2) y Ras-GAP. Los dominios SH2/SH3 son dianas para fármacos contra el cáncer y se discuten en Smithgall, T.E. (1995), Journal of Pharmacological and Toxicological Methods. 34(3) 125-32.
Los inhibidores de la serina/treonina quinasa que incluyen bloqueadores de la cascada de MAP quinasa, que incluyen bloqueadores de Raf quinasas (rafk), quinasa regulada por mitógenos o en forma extracelular (MEK), y quinasas reguladas en forma extracelular (ERK), y bloqueadores de los miembros de la familia de proteína quinasa C que incluye bloqueadores de PKC (alfa, beta, gamma, épsilon, mu, lambda, iota, zeta). Familia de las quinasas IkB (IKKa, IKKb), quinasas de la familia PKB, miembros de la familia quinasa akt, y quinasas del receptor de TGF beta. Tales serina/treonina quinasas y sus inhibidores se describen en Yamamoto, T., Taya, S., Kaibuchi, K., (1999), Journal of Biochemistry. 126 (5) 799-803; Brodt, P, Samani, A., and Navab, R. (2000), Biochemical Pharmacology, 60. 1101 1107; Massague, J., Weis-Garcia, F. (1996) Cancer Surveys. 27:41-64; Philip, P.A., and Harris, A.L. (1995), Cancer Treatment and Research. 78: 3-27, Lackey, K. et at Bioorganic and Medicinal Chemistry Letters, (10), 2000, 223-226; U.S. Patent No. 6.268.391; and Martínez-llacaci, L., et al, Int. J. Cancer (2000), 88(1), 44-52.
Los inhibidores de los miembros de la familia de fosfatidilinositol-3 quinasa que incluyen bloqueadores de PI3-quinasa, ATM, ADN-PK, y Ku también pueden ser útiles en la presente invención. Tales quinasas se analizan en Abraham, R.T. (1996), Current Opinion in Immunology. 8 (3) 412-8; Canman, C.E., Lim, D.S. (1998), Oncogene 17 (25) 3301-3308; Jackson, S.P. (1997), International Journal of Biochemistry and Cell Biology. 29 (7):935-8; and Zhong, H. et al, Cancer Res, (2000) 60(6), 1541-1545.
También son útiles en la presente invención los inhibidores de la señalización de Mio-inositol tales como los bloqueantes de la fosfolipasa C y los análogos de Mioinositol. Tales inhibidores de señal se describen en Powis, G., and Kozikowski A., (1994) New Molecular Targets for Cancer. Chemotherapy ed., Paul Workman and David Kerr, CRC press 1994, Londres.
Otro grupo de inhibidores de las rutas de transducción de señales son los inhibidores del oncogén Ras. Tales inhibidores incluyen los inhibidores de farnesiltransferasa, geranio-geranil transferasa, y CAAX proteasas así como oligonucleótidos antisentido, ribozimas e inmunoterapia. Se ha demostrado que tales inhibidores bloquean la activación de ras en células que contienen ras mutante tipo silvestre, de este modo actúan como agentes antiproliferación. La inhibición del oncogén Ras se analiza en Scharovsky, O.G., Rozados, V.R., Gervasoni, S.I. Motor, P. (2000), Journal of Biomedical Science. 7(4) 292-8; Ashby, M.N. (1998), Current Opinion in Lipidology. 9 (2) 99 - 102; and BioChim. Biophys. Acta, (19899) 1423(3):19-30.
Tal como se ha mencionado anteriormente, los antagonistas de anticuerpo para la unión de ligando de la quinasa receptora también pueden servir como inhibidores de la transducción de señales. Este grupo de inhibidores de las rutas de transducción de señales incluyen el uso de anticuerpos humanizados frente al dominio de unión a ligando extracelular del receptor de tirosina quinasas. Por ejemplo anticuerpo específico EGFR Imclone C225 (véase, Green, M.G. et al, Monoclonal Antibody Therapy for Solid Tumors, Cancer Treat. Rev., (2000), 26(4), 269-286); anticuerpo erbB2 Herceptin® (ver Tyrosine Kinase Signalling in Breast cancer:erbB Family Receptor Tyrosine Kniases, Breast cancer Res., 2000, 2(3), 176-183); y anticuerpo específico 2CB VEGFR2 (véase, Brekken, R.A. et al, Selective Inhibition of VEGFR2 Activity by a monoclonal Anti-VEGF antibody blocks tumor growth in mice, Cancer Res. (2000) 60, 5117-5124).
Agentes antiangiogénicos:
(i) También pueden ser útiles los agentes antiangiogénicos, incluyendo los inhibidores de la angiogénesis MEK de no receptor. Los agentes antiangiogénicos como los que inhiben los efectos del factor del crecimiento endotelial vascular, (por ejemplo, el anticuerpo de factor de crecimiento celular endotelial antivascular bevacizumab [Avastin™], y compuestos que trabajan mediante otros mecanismos (por ejemplo, linomida, inhibidores de la función la integrina avp3, endostatina y angiostatina);
Agentes inmunoterapéuticos:
También pueden ser útiles en combinación con los compuestos de fórmula (I) los agentes utilizados en los regímenes inmunoterapéuticos. Los enfoques inmunoterapéuticos, incluidos, por ejemplo, los enfoques ex-vivo e in-vivo para aumentar la inmunogenicidad de células tumorales del paciente, tales como transfección con citoquinas tales como interleucina 2, interleucina 4 o factor de estimulación de granulocito-macrófago, enfoques para disminuir la anergía de linfocitos T, enfoques que usan células inmunes transfectadas tales como células dentríticas transfectadas por citoquinas, enfoques que usan líneas de células tumorales transfectadas por citoquinas y enfoques que usan anticuerpos antiidiotípicos
Agentes proapoptóticos:
Los agentes usados en regímenes proapoptóticos (por ejemplo, oligonucleótidos antisentido bcI-2) también se pueden utilizar en la combinación de la presente invención.
Inhibidores de la señalización del ciclo celular
Los inhibidores de la señalización del ciclo celular inhiben moléculas implicadas en el control del ciclo celular. Una familia de proteína quinasas llamada quinasas dependientes de ciclina (las CDK) y su interacción con una familia de proteínas denominadas ciclinas, controla la progresión a través del ciclo celular eucariota. La activación coordinada y la inactivación de distintos complejos ciclina/CDK son necesarias para la progresión normal a través del ciclo celular. Están en desarrollo varios inhibidores de la señalización del ciclo celular. Por ejemplo, ejemplos de quinasas dependientes de ciclina, que incluye CDK2, CDK4, y CDK6 e inhibidores de estos se describen, por ejemplo, Rosania et al, Exp. Opin. Ther. Patents (2000) 10(2):215- 230.
En una realización, la combinación de la presente invención comprende un compuesto de fórmula I o sal o solvato de la misma y al menos un agente antineoplásico seleccionado entre agentes antimicrotúbulos, complejos de coordinación de platino, agentes alquilantes, agentes antibióticos, inhibidores de la topoisomerasa II, antimetabolitos, inhibidores de la topoisomerasa I, hormonas y análogos hormonales, inhibidores de la vía de transducción de señales,
inhibidores de la angiogénesis MEK de tirosina no receptor, agentes inmunoterapéuticos, agentes proapoptóticos e inhibidores de la señalización del ciclo celular.
En una realización, la combinación de la presente invención comprende un compuesto de fórmula I o una sal o solvato de la misma y al menos un agente antineoplásico que es un agente antimicrotúbulos seleccionado entre diterpenoides y alcaloides de vinca.
En una realización adicional, al menos un agente antineoplásico es  un diterpenoide.
un diterpenoide.
En una realización adicional, al menos un agente antineoplásico es un alcaloide de vinca.
un alcaloide de vinca.
En una realización, la combinación de la presente invención comprende un compuesto de fórmula I o una sal o solvato de la misma y al menos un agente antineoplásico, que es un complejo de coordinación de platino.
En una realización adicional, al menos un agente antineoplásico es paclitaxel, carboplatin, o vinorelbine.
En una realización adicional, al menos un agente antineoplásico es carboplatin.
carboplatin.
En una realización adicional, al menos un agente antineoplásico es vinorelbine.
vinorelbine.
En una realización adicional, al menos un agente antineoplásico es paclitaxel.
En una realización, la combinación de la presente invención comprende un compuesto de fórmula I y sales o solvatos de la misma y al menos un agente antineoplásico, que es un inhibidor de la vía de transducción de señales.
En una realización adicional, el inhibidor de la vía de transducción de señales es un inhibidor de una quinasa de receptor de factor de crecimiento VEGFR2, TIE2, PDGFR, BTK, erbB2, EGFr, IGFR-1, TrkA, TrkB, TrkC, o c-fms. En una realización adicional el inhibidor de la vía de transducción de señales es un inhibidor de una serina/treonina quinasas de rafk, akt, o PKC-zeta.
En una realización adicional, el inhibidor de la vía de transducción de señales es un inhibidor de una treonina quinasas de no receptor seleccionada entre la familia src de quinasas.
En una realización adicional, el inhibidor de la vía de transducción de señales es un inhibidor de c-src.
En una realización adicional el inhibidor de la vía de transducción de señales es un inhibidor de oncogen Ras seleccionado entre inhibidores de la fernesil transferasa y geranilgeranil transferasa.
En una realización adicional, el inhibidor de la vía de transducción de señales es un inhibidor de una serina/treonina quinasa de no receptor seleccionada entre el grupo que consiste en PI3K.
En una realización adicional el inhibidor de la vía de transducción de señales es un inhibidor EGFr/erbB2 dual, por ejemplo, N-{3-Cloro-4-[(3-fluorobencil)oxi]fenil}-6-[5-({[2-(metanesulfonil) etil]amino}metil)-2-furil]-4-quinazolinamina (estructura a continuación):

En una realización, la combinación de la presente invención comprende un compuesto de fórmula I o una sal o solvato de la misma y al menos un agente antineoplásico, que es un inhibidor de la señalización del ciclo celular.
En una realización adicional, el inhibidor de la señalización del ciclo celular es un inhibidor de CDK2, CDK4 o CDK6. Componentes particulares de terapia de combinación incluyen combinaciones con otros antiestrógenos que incluyen tamoxifen y/o fulvestrant.
En una realización, el mamífero en los métodos y usos de la presente invención es un ser humano.
Métodos de síntesis generales
Los compuestos de fórmula general (I) se pueden preparar mediante métodos conocidos en la técnica de síntesis orgánica tal como se indica en los Ejemplos específicos descritos a continuación. En todos los métodos, se comprende bien que se pueden emplear grupos protectores para grupos sensibles o reactivos cuando sea necesario de acuerdo con los principios generales de la química. Los grupos protectores se manipulan según procedimientos estándar de síntesis orgánica (T. W. Green y P. G. M. Wuts (1999) Protective Groups in Organic Synthesis, 3a edición, John Wiley & Sons). Estos grupos se eliminan en una etapa conveniente de la síntesis del compuesto usando procedimientos que son fácilmente evidentes para las personas con conocimientos en la técnica. La selección de los procedimientos, así como las condiciones de reacción y el orden de su ejecución, deberán ser consistentes con la preparación de los compuestos de Fórmula (I).
Parte experimental
Abreviaturas:
DCM: diclorometano.
DIPEA: N, N-diisopropiletilamina.
DMF: N,N-dimetilformamida. h: hora.
HATU: hexafluorofosfato de 2-(7-aza-1H-benzotriazol-1-il)-1,1,3,3-tetrametiluronio.
HPLC: cromatografía líquida de alto rendimiento.
LCMS: cromatografía líquida-espectroscopia de masas
min: minutos.
RMN: Resonancia magnética nuclear.
TR: tiempo de retención.
tBu: terc-butóxido.
TFA: ácido trifluoroacético.
THF: tetrahidrofurano.
Método A de LCMS:
El análisis se llevó a cabo en una columna Acquity UPLC BEH C18 [50 mm x 2,1 mm diámetro interior 1,7 |jm diámetro de empaquetado) a 40 °C.
Los disolventes empleados fueron:
A = 0,1 % v/v solución de ácido fórmico en agua.
B = 0,1 % v/v solución de ácido fórmico en acetonitrilo.
El gradiente se empleó de la siguiente manera:

La detección UV fue una señal promedio de la longitud de onda de 210 nm a 350 nm y los espectros de masas se registraron en un espectrómetro de masas usando ionización por electropulverización de barrido alterno en modo positivo y negativo.
Método B de LCMS:
El análisis se llevó a cabo en una columna Acquity UPLC BEH C18 (50 mm x 2,1 mm diámetro interior 1,7 jm diámetro de empaquetado) a 40 °C.
Los disolventes empleados fueron:
A = 10 mM de bicarbonato de amonio en agua ajustado a pH 10 con solución de amoníaco.
B = acetonitrilo.
El gradiente se empleó de la siguiente manera:

La detección UV fue una señal promedio de la longitud de onda de 210 nm a 350 nm y los espectros de masas se registraron en un espectrómetro de masas usando ionización por electropulverización de barrido alterno en modo positivo y negativo.
Lo que sigue ilustra las fases móviles y gradientes usados cuando los compuestos se sometieron a purificación mediante HPLC autopreparativa dirigida por masa.
HPLC autopreparativa dirigida por masa (Modificador de ácido fórmico)
El análisis se llevó a cabo en una columna Sunfire C18 (150 mm x 30 mm diámetro interior, 5 |jm diámetro de empaquetado) a temperatura ambiente.
Los disolventes empleados fueron:
A = 0,1 % v/v solución de ácido fórmico en agua.
B = 0,1 % v/v solución de ácido fórmico en acetonitrilo.
HPLC autopreparativa dirigida por masa (Modificador de ácido trifluoroacético)
El análisis se llevó a cabo en una columna Sunfire C18 (150 mm x 30 mm diámetro interior, 5 jm diámetro de empaquetado) a temperatura ambiente.
Los disolventes empleados fueron:
A = 0,1 % v/v solución de ácido trifluoroacético.
B = 0,1 % v/v solución de ácido trifluoroacético en acetonitrilo.
HPLC autopreparativa dirigida por masa (Modificador de bicarbonato de amonio)
El análisis se llevó a cabo en una columna XBridge C18 (150 mm x 30 mm diámetro interior, 5 jm diámetro de empaquetado) a temperatura ambiente.
Los disolventes empleados fueron:
A = 10 mM de bicarbonato de amonio en agua ajustado a pH 10 con solución de amoníaco.
B = acetonitrilo.
Para cada una de las purificaciones autopreparativas dirigidas por masa, independientemente del modificador usado, el gradiente empleado dependió del tiempo de retención del compuesto particular que se estuviera sometiendo a purificación tal como se registró en la LCMS analítica y fue del siguiente modo:
Para los compuestos con un tiempo de retención de LCMS analítico inferior a 0,6 minutos se usó el siguiente gradiente:

Para compuestos con un tiempo de retención de LCMS analítico de entre 0,6 y 0,9 minutos se usó el siguiente gradiente:
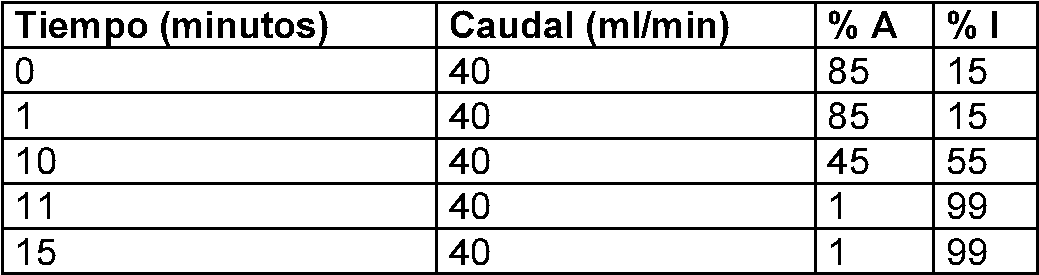
Para compuestos con un tiempo de retención de LCMS analítico de entre 0,9 y 1,2 minutos se usó el siguiente gradiente:

Para compuestos con un tiempo de retención de LCMS analítico de entre 1,2 y 1,4 minutos se usó el siguiente gradiente:

Para compuestos con un tiempo de retención de LCMS analítico superior a 1,4 minutos (LCMS método A) o superior a 3,6 minutos (LCMS método B) se usó el siguiente gradiente:

La detección UV fue una señal promedio de la longitud de onda de 210 nm a 350 nm y los espectros de masas se registraron en un espectrómetro de masas usando ionización por electropulverización de barrido alterno en modo positivo y negativo.
Los nombres químicos se generaron usando ACD Name Pro versión 6.02 de Advanced Chemistry Development, Inc.

Se puede preparar (Z)-2-(4-(1,2-Difenilbut-1-en-1-il)fenoxi)-N-metiletanamina de acuerdo con el proceso descrito por Dreaden, Erik C. et al. Bioconjugate Chem. 2009, 20, 2247-2253.
1-fenil-2,5,8,11-tetraoxatridecan-13-oato de ferc-Butilo

Se añadió terc-butóxido de potasio (disponible en el mercado, por ejemplo, en Aldrich) (7,71 g, 68,7 mmol) a una solución agitada de 2-(2-(2-(benciloxi)etoxi)etoxi)etanol (disponible en el mercado, por ejemplo, en Fluorochem) (15 g, 62,4 mmol) en terc-butanol (200 ml) y la mezcla de reacción se agitó a temperatura ambiente durante 2 h. La mezcla de reacción se enfrió a 0 °C, se añadió 2-bromoacetato de terc-butilo (disponible en el mercado, por ejemplo, por Aldrich) (16,59 ml, 112 mmol) y la mezcla se agitó a temperatura ambiente durante la noche. Se añadió d Cm (300 ml) y se lavó la fase orgánica con agua (300 ml) y, a continuación, salmuera (2 x 200 ml). El extracto orgánico se secó
usando una frita hidrófoba y se concentró a presión reducida para proporcionar el producto crudo como un aceite de color amarillo. El producto se purificó mediante cromatografía sobre sílice usando una elución de gradiente del 0 % al 100 % de metil terc-butil éter en ciclohexano para proporcionar el compuesto de título (13,3 g, 37,5 mmol, 60 % de rendimiento). LCMS (Método A) TR= 1,10 min, ES+ve m/z 372,4 [M+NH4]+.
2-(2-(2-(2-hidroxietoxi) etoxi) etoxi)acetato de ferc-Butilo

Se agitó una mezcla de 1-fenil-2,5,8,11-tetraoxatridecan-13-oato de terc-butilo (13,3 g, 37,5 mmol) y paladio sobre carbono (10 % p/p, 11,38 g, 10,69 mmol) en etanol (200 ml) a temperatura ambiente con una atmósfera de hidrógeno durante 1,5 h. El paladio sobre carbono se filtró a través de celita y el filtrado se evaporó a presión reducida para proporcionar el compuesto de título (9,74 g, 36,8 mmol, 98 % de rendimiento) como un aceite de color amarillo. RMN de 1H (400 MHz, DMSO-ds) 5 = 4,54 (s, 1h ), 3,99 (s, 2H), 3,60 - 3,40 (m, 12H), 1,43 (s, 9H).
2-(2-(2-(2-(tosiloxi)etoxi)etoxi)etoxi)acetato de terc-Butilo

Se añadió tosilcloruro (diponible en el mercado, por ejemplo, por Aldrich) (11,94 g, 62,6 mmol) a una solución enfriada (0° C) de 2-(2-(2-(2-hidroxietoxi)etioxi)etoxi)acetato de terc-butilo (9,74 g, 36,8 mmol) en piridina (150 ml). La reacción se agitó a temperatura ambiente durante 16 h. La mezcla de reacción se dividió entre acetato de etilo (300 ml) y HCI acuoso (2M, 300 ml). El extracto orgánico se lavó co HCI acuoso adicional (2M, 300 ml), K2CO3 saturado (100 ml) y salmuera (100 ml). El extracto orgánico se secó usando MgSO4 y se concentró a presión reducida para proporcionar el compuesto de título (10,3 g, 24,6 mmol, 67 % de rendimiento) como un aceite de color amarillo. Lc m S (Método A) TR= 1,14 min, ES+ve 436,2 [M+NH4]+.
(Z)-ferc-Butil 1 -(4-(1,2-difenilbut-1 -en-1 -il)fenoxi)-3-metil-6,9,12-trioxa-3-azatotradecan-14-oato

Se calentó una mezcla de (Z)-2-(4-(1,2-difenilbut-1-en-1-il)fenoxi)-N-metiletanamina (141 mg, 0,394 mmol), 2-(2-(2-(2-(tosiloxi)etoxi)etoxi)etoxi)acetato de terc-butilo (0,187 ml, 0,592 mmol) y K2CO3 (545 mg, 3,94 mmol) en DMF (5 ml) a 85 °C durante 16 h. La reacción se enfrió a temperatura ambiente y se dividió entre EtOAc (30 ml) y agua (30 ml). El extracto orgánico se secó usando una frita hidrófoba y se concentró a presión reducida. El producto se purificó mediante cromatografía sobre sílice usando una elución de gradiente del 0 % al 100 % de EtOAc en ciclohexano seguido por un 0 % a 25 % de metanol en diclorometano para proporcionar el compuesto de título (141 mg, 0,234 mmol, 59 % de rendimiento) como un vidrio incoloro. LCMS (Método B) TR= 1,63 min, ES+ve m/z 604,2 [M+H]+.
Ácido (Z)-1 -(4-(1,2-Difenilbut-1 -en-1 -il)fenoxi)-3-metil-6,9,12-trioxa-3-azatotradecan-14-oico

Se agitó una mezcla de (Z)-terc-butil 1-(4-(1,2-difenilbut-1-en-1-il)fenoxi)-3-metil-6,9,12-trioxa-3-azatotradecan-14-oato (141 mg, 0,234 mmol), y TFA (1 ml, 12.98 mmol) in DCM (1 ml) a temperatura ambiente durante 1,5 h. La mezcla de reacción se evaporó a presión reducida y el resto se sometió, a continuación, a purificación mediante HPLC preparativa automática dirigida por masa (modificador de carbonato de amonio) para proporcionar el compuesto de título (81 mg, 0,148 mmol, 63 % de rendimiento) como un vidrio incoloro. LCMS (Método b ) TR= 1,14 min, ES+ve m/z 548,2 [M+H]+.
(2S,4R}-terc- Butil 2-((4-bromobencil)carbamoil)-4-hidroxipirrolidina-1-carboxilato
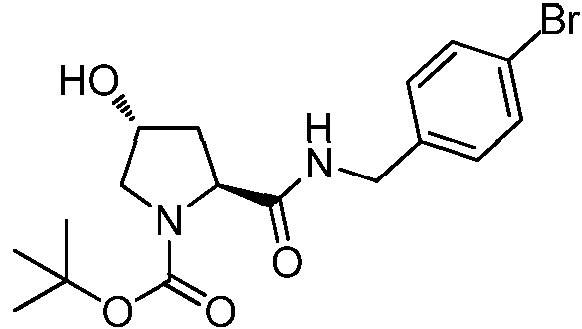
Se trató una mezcla enfriada en hielo de ácido (2S,4R)-1-(terc-butoxicarbonil)-4-hidroxipirrolidina-2-carboxilico (disponible en el mercado, por ejemplo, por Aldrich) (7,95 g, 34,4 mmol) y (4-bromofenil)metanamina (disponible en el mercado, por ejemplo, por FluroChem) (6,4 g, 34,4 mmol) en DMF (200 ml) con DIPEA (18,02 ml, 103 mmol) y, a continuación con HATU (14,39 g, 37,8 mmol) y la mezcla se agitó a temperatura ambiente durante 30 minutos. La reacción se inactivó con agua (200 ml) y se extrajo con acetato de etilo (2 x 200 ml). Las capas orgánicas combinadas se lavaron con bicarboanto de socio acuoso saturado (2 x 300 ml), agua (100 ml), salmuera (200 ml), se secó sobre sulfato de magnesio y se evaporó hasta sequedad. El producto se purificó mediante cromatografía sobre sílice usando una elución de gradiente del 0 % al 10 % de metanol en DCM para proporcionar el compuesto de título (12,9 g, 32,3 mmol, 94 % de rendimiento). LCMS (Método A) TR= 0,87 min, ES+ve m/z 399,2/401,2 [M+H]+.
(2S,4R)-terc-butil4-hidroxi-2-((4-(4-metiltiazol-5-il)bencil)carbamoil)pirrolidina-1-carboxilato

Se agitó una mezcla de (2S,4R)-terc-butil 2-((4-bromobencil)carbamoil)-4-hidroxipirrolidina-1-carboxilato (12,9 g, 32,3 mmol), 4-metiltiazol (disponible en el mercado, por ejemplo, por Aldrich) (5,88 ml, 64,6 mmol), acetato de paladio (II) (disponible en el mercado, por ejemplo, por Aldrich) (0,145 g, 0,646 mmol) y acetato de potasio (6,34 g, 64,6 mmol) en N-metil-2- pirrolidona (80 ml) a 120 °C con nitrógeno durante 18 horas. Se añadió agua (100 ml) y el producto se extrajo con acetato de etilo (4 x 300 ml). La fase orgánica combinada se lavó con salmuera (5 x 200 ml), se secó sobre sulfato de magnesio y se evaporó a sequedad. El producto se purificó mediante cromatografía sobre sílice usando una elución de gradiente del 0 % al 10 % de metanol en DCM para proporcionar el compuesto de título (8 g, 19,2 mmol, 59 % de rendimiento). LCMS (Método A) TR - 0,75 min, ES+ve m/z 418,4 [M+H]+.
(2S,4R)-4-Hidroxi-N-(4-(4-metMtMazol-5-M)bencM)pirroMdma-2-carboxamida, hidrocloruro

Se disolvió (2S,4R)-terc-Butil 4-hidroxi-2-((4-(4-metiltiazol-5-il)bencil)carbamoil)pirrolidina-1-carboxilato (8 g, 19,16 mmol) en metanol (30 ml) y DCM (20 ml) y se trató con HC1 en dioxano (4M, 8,08 ml, 32,3 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 2 horas. Se retiró el disolvente en presión reducida y el resto se trituró con DCM, filtró y secó a presión reducida para producir el compuesto de título (6,7 g, 18,9 mmol, 99 % de rendimiento). LCMS (Método A) TR= 0,49 min, ES+ve m/z 318,3 [M+H]+.
ferc-Butil((S)-1-((2S,4R)-4-hidroxi-2-((4-(4-metiltiazol-5-il)bencil)carbamoil)pirrolidin-1-il)-3-metil-1-oxobutan-2-il)carbamato

Se trató una mezcla agitada de (2S,4R)-4-hidroxi-N-(4-(4-metiltiazol-5-il)bencil)pirrolidina-2-carboxamida, hidrocloruro (125 mg, 0,35 mmol) y ácido (S)-2-((terc-butoxicarbonil)amino)-3- metilbutanoico (disponible en el mercado, por ejemplo, por Aldrich) (77 mg, 0,35 mmol) en DMF (0,9 ml) con DIPEA (0,22 ml, 1,3 mmol) y, a continuación con HATU (134 mg, 0,35 mmol) y la mezcla se agitó a temperatura ambiente durante 1 hora. La mezcla de reacción se sometió directamente a purificación por HPLC preparativa automática dirigida por masa (modificador de ácido fórmico) para proporcionar el compuesto de título (120 mg, 0,232 mmol, 72 % de rendimiento). LCMS (Método A) TR= 0,87 min, ES+ve m/z 517,3 [M+H]+.
(25,4R)-1 -((S)-2-Amino-3-metilbutanoil)-4-hidroxi-N-(4-(4-metiltiazol-5-il)bencil)pirrolidina-2-carboxamida, hidrocloruro

Se trató una solución de terc-butil((S)-1-((2S,4R)-4-hidroxi-2-((4-(4-metiltiazol-5-il)bencil)carbamoil)pirrolidin-l-il)-3-metil-l-oxobutan-2-il)carbamato (287 mg, 0,56 mmol) en THF (5 ml) con HCl en 1,4-dioxan (4M, 10 ml, 40 mmol) y se agitó a temperatura ambiente durante 2 horas. La mezcla se evaporó a sequedad para proporcionar el compuesto del título (224 mg, 0,49 mmol, cuantiativo). LCMS (Método A) TR= 0,55 min, ES+ve m/z 417,3 [M+H]+.
(2S,4R)-1 -((S)-2-Amino-3,3-dimetilbutanoil)-4-hidroxi-IV-(4-(4-metiltiazol-5-il)bencil)pirrolidina-2-carboxamida, hidrocloruro

Se trató una mezcla agitada de (2S,4R)-4-hidroxi-N-(4-(4-metiltiazol-5-il)bencil)pirrolidina-2-carboxamida, hidrocloruro (70 mg, 0,20 mmol) y ácido (S)-2-((terc-butoxicarbonil)amino)-3,3- dimetilbutanoico (disponible en el mercado, por ejemplo, por Fluka) (50 mg, 0,22 mmol) en DMF (1 ml) con DIPEA (0,14 ml, 0,79 mmol) y, a continuación con HATU (90 mg, 0,24 mmol) y se agitó a temperatura ambiente durante 30 minutos. La mezcla de reacción se sometió directamente a purificación por HPLC preparativa automática dirigida por masa (modificador de ácido fórmico) para proporcionar el producto protegido por boc intermediario. A continuación, el intermediario se disolvió en una mezcla de diclorometano (0,5 ml) y metanol (0,1 ml) y se trató con HCI en 1,4-dioxano (4M, 0,25 ml, 1,0 mmol). Después de agitar a temperatura ambiente durante 1 hora, la mezcla de reacción se evaporó a sequedad y el resto se trituró a un sólido con diclorometano y se secó al vacío para proporcionar el compuesto de título (76 mg, 0,163 mmol, 82 % de rendimiento). LCMS (Método A) TR= 0,58 min, ES+ve m/z 431,2 [M+h ]+.
Ejemplo 1
(2S,4R)-1 -((S)-17-(4-((Z)2-Difenilbut-1-en-I-il)fenoxi)-2-isopropil-15-metil-4-oxo-6,9,12-trioxa-3,15-diazaheptadecan-I-oil)-4-hidroxi-N-(4-(4-metiltiazol-5-il)bencil)pirrolidina-2-carboxamida N26699-42

Se añadió HATU (26 mg, 0,068 mmol) a una mezcla de (2S,4R)-1-((S)-2-amino-3-metilbutanoil)- 4-hidroxi-N-(4-(4-metiltiazol-5-il)bencil)pirrolidina-2-carboxamida, hidrocloruro, (25 mg, 0,055 mmol), ácido (Z)-1-(4-(l,2-difenilbut-1-en-1-il)fenoxi)-3-metil-6,9,12-trioxa-3-azatetradecan-14-oico (27 mg, 0,049 mmol) y DIPEA (0,05 ml, 0,286 mmol) en DMF (0,8 ml). La reacción se agitó a temperatura ambiente durante 10 minutos. La solución se sometió directamente a purificación por HPLC preparativa automática dirigida por masa (modificador de carbonato de amonio) para proporcionar el compuesto de título (36 mg, 0,038 mmol, 77 % de rendimiento). LCMS (Método B) TR= 1,44 min, ES+ve m/z 946,2 [M+H]+.
Ejemplo 2
(25,4R)-1 -((S)-2-(terc-Butil)-17-(4-((Z)-1,2-difenilbut-1 -en-1 -il)fenoxi)-15-metil-4-oxo-6,9,12-trioxa-3,15-diazaheptadecan-I-oil)-4-hidroxi-M-(4-(4-metiltiazol-5-il)bencil)pirrolidina-2-carboxamida N26699-42

Se añadió HATU (26 mg, 0,068 mmol) a una mezcla de (2S,4R)-1-((S)-2-amino-3,3- dimetilbutanoil)-4-hidroxi-N-(4-(4-metiltiazol-5-il)bencil)pirrolidina-2-carboxamida, hidrocloruro (25 mg, 0,054 mmol), ácido (Z)-1-(4-(1,2-difenilbut-1-en-1-il)fenoxi)-3-metil-6,9,12-trioxa-3-azatetradecan-14-oico (27 mg, 0,049 mmol) y DIPEA (0,05 ml, 0,286 mmol) en DMF (0,8 ml). La reacción se agitó a temperatura ambiente durante 10 minutos. La solución se sometió directamente a purificación por HPLC preparativa automática dirigida por masa (modificador de carbonato de amonio) para proporcionar el compuesto de título (14 mg, 0,014 mmol, 30 % de rendimiento). LCMS (Método B) TR= 1,48 min, ES+ve m/z 960,2 [M+H]+.
Degradación de receptor de estrógeno alfa (Era) y ensayo de formación de imágenes de recuento celular
Se evaluaron compuestos para su degradación de Era y los efectos de recuento celular en una línea celular de MCF-7 usando una formación de imágenes de alto contenido. Se dispensaron 50ul de MCF-7 de suspensión celular en medio a cada pocillo de pared negra, con fondo claro, placas recubiertas con PDL, que contenían una concentración definida de compuesto de ensayo disuelto en DMSO que cubría un intervalo de concentración de 0,03uM a 30uM. Se incubaron células en presencia del compuesto durante 24 horas a 37 °C, 5 % de CO2 antes de que se fijaran las células. Después de su incubación con la solución fijadora (4 % de formaldehído) las paredes se aspiraron y se añadió una solución que contenía detergente para permeabilizar las células seguido por la adición de solución de bloqueo que contenía un 1 % de BSA (albúmina de suero bovino) para bloquear los sitios de unión no específicos. Después de una incubación adicional durante 1 hora esta solución se aspiró de los pocillos y se añadió el anticuerpo específico de Era diluido en solución de bloqueo a una concentración de 1ug/ml (anti ERa, cat no sc-543, Santa Cruz). Después de la incubación con el anticuerpo durante 2 horas, las células se lavaron con una solución a base de PBS antes de la adición de un anticuerpo de cabra fosfoespecífico Alexa fluor 488 anti-conejo secundario a una concentración de 2ug/ml (cat no11008, Invitrogen) y una colocación de tinte nuclear Floechst33342 a una concentración de lug/ml (cat no FI3570, invitrogen). Después de una incubación adicional durante 1 hora, las células se volvieron a lavar con solución a base de PBS. Las placas se sometieron a formación de imágenes, a continuación y la intensidad de la teinción de Era en el núcleo y se midió el recuento celular. Se expresó la actividad de degradación de Era con respecto a DMSO, proporcionando un 0 % de degradación y una molécula de degradador interno clasificadas como que proporcionan un 100 % de actividad. Se expresó la reducción de recuento celular con respecto a DMSO, clasificada como un 0 % de reducción.
Los ejemplos mostraron evidencia de degradación de Era en este ensayo con respecto al control de DMSO a una concentración de 1uM.
Claims (15)
1. Un compuesto de fórmula (I):

en la que
L es un grupo de unión, en donde
el grupo de unión es un grupo alquileno de cadena lineal de 8-16 átomos de carbono en donde uno o más átomos de carbono en el grupo de unión estan sustituidos con un miembro del grupo seleccionado independientemente de -O-, -NH-, -N[CH3)-,

R1 es alquilo Ci -6 lineal o ramificado o cicloalquilo C3-6
R2 es 4-metiltiazol-5-il u oxazol-5-il o una sal farmacéuticamente aceptable de los mismos.
2. Un compuesto o una sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1 en donde R2 es 4-metiltiazol-5-il;
3. Un compuesto o una sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1 o la reivindicación 2- en el que el grupo de unión es de fórmula (ii)
-(R3CH2CH2)xOCH2- (ii)
en donde cada R3 se selecciona independientemente entre -O-, -NH-, -N(CH3)- o

y X es 2-4.
4. Un compuesto o una sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 3 en donde al menos un R3 es -N(CH3).
5. Un compuesto de fórmula (I) de acuerdo con la reivindicación 1 que es
(2S,4R)-1-((S)-17-(4-((Z)-1,2-Difenilbut-1-en-1-il)fenoxi)-2-isopropil-15-metil-4-oxo-6,9,12-trioxa-3,15
diazaheptadecan-1-oil)-4-hidroxi-N-(4-(4-metiltiazol-5-il)bencil)pirrolidina-2-carboxamida

(2S,4R)-1-((S)-2-(íerc-Butil)-17-(4-((Z)-1,2-difenilbut-1-en-1-il)fenoxi)-15-metil-4-oxo-6,9,12-trioxa-3,15-diazaheptadecan-1-oil)-4-hidroxi-N-(4-(4-metiltiazol-5-il)bencil)pirrolidina-2-carboxamida

y sales farmacéuticamente aceptables de los mismos.
6. Un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo de acuerdo con las reivindicaciones 1-5 para un uso en un método de tratamiento por terapia.
7. Un compuesto de acuerdo con la reivindicación 6 para su uso en el tratamiento de al menos cáncer de mama, cáncer uterino o endometriosis.
8. Un compuesto de acuerdo con la reivindicación 7 para su uso en el tratamiento de cáncer de mama.
9. Una combinación que comprende un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo de acuerdo con las reivindicaciones 1-5 y al menos un agente terapéutico adicional.
10. Una combinación de acuerdo con la reivindicación 9, en la que al menos un agente terapéutico adicional incluye al menos un agente antineoplásico.
11. Una combinación de acuerdo con la reivindicación 9 o la reivindicación 10 para su uso en un método de tratamiento por terapia.
12. Una combinación de acuerdo con la reivindicación 11 para su uso en el tratamiento de al menos cáncer de mama, cáncer uterino o endometriosis.
13. Una combinación de acuerdo con la reivindicación 12 para su uso en el tratamiento de cáncer de mama.
14. Una composición farmacéutica que comprende un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo de acuerdo con las reivindicaciones 1-5 y uno o más de vehículos, diluyentes y excipientes farmacéuticamente aceptables.
15. Una composición farmacéutica que comprende una combinación de acuerdo con la reivindicación 9 o la reivindicación 10 y uno o más de vehículos, diluyentes, excipientes farmacéuticamente aceptables.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1311891.4A GB201311891D0 (en) | 2013-07-03 | 2013-07-03 | Novel compound |
| PCT/EP2014/063901 WO2015000868A1 (en) | 2013-07-03 | 2014-07-01 | Novel compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2717436T3 true ES2717436T3 (es) | 2019-06-21 |
Family
ID=48999421
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14734147T Active ES2717436T3 (es) | 2013-07-03 | 2014-07-01 | Compuestos novedosos |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US9993514B2 (es) |
| EP (1) | EP3016967B1 (es) |
| ES (1) | ES2717436T3 (es) |
| GB (1) | GB201311891D0 (es) |
| HK (1) | HK1224312A1 (es) |
| TR (1) | TR201904340T4 (es) |
| WO (1) | WO2015000868A1 (es) |
Families Citing this family (50)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117736134A (zh) | 2012-01-12 | 2024-03-22 | 耶鲁大学 | 通过e3泛素连接酶增强靶蛋白质及其他多肽降解的化合物和方法 |
| GB201311891D0 (en) | 2013-07-03 | 2013-08-14 | Glaxosmithkline Ip Dev Ltd | Novel compound |
| GB201311888D0 (en) | 2013-07-03 | 2013-08-14 | Glaxosmithkline Ip Dev Ltd | Novel compounds |
| US10071164B2 (en) | 2014-08-11 | 2018-09-11 | Yale University | Estrogen-related receptor alpha based protac compounds and associated methods of use |
| US20170327469A1 (en) | 2015-01-20 | 2017-11-16 | Arvinas, Inc. | Compounds and methods for the targeted degradation of androgen receptor |
| CN107428734A (zh) | 2015-01-20 | 2017-12-01 | 阿尔维纳斯股份有限公司 | 用于雄激素受体的靶向降解的化合物和方法 |
| GB201504314D0 (en) * | 2015-03-13 | 2015-04-29 | Univ Dundee | Small molecules |
| CA2979070A1 (en) | 2015-03-18 | 2016-09-22 | Arvinas, Inc. | Compounds and methods for the enhanced degradation of targeted proteins |
| JP6792256B2 (ja) * | 2015-05-27 | 2020-11-25 | 学校法人関西文理総合学園 | ペプチド |
| HUE056199T2 (hu) | 2015-05-29 | 2022-01-28 | Eisai R&D Man Co Ltd | Tetraszubsztituált alkénvegyületek és alkalmazásuk |
| WO2016197114A1 (en) | 2015-06-05 | 2016-12-08 | Arvinas, Inc. | Tank-binding kinase-1 protacs and associated methods of use |
| US10772962B2 (en) | 2015-08-19 | 2020-09-15 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of bromodomain-containing proteins |
| KR20180097530A (ko) | 2015-11-02 | 2018-08-31 | 예일 유니버시티 | 단백질분해 표적화 키메라 화합물(Proteolysis Targeting Chimera compound) 및 그의 제조 및 사용 방법 |
| JP2019514878A (ja) * | 2016-04-20 | 2019-06-06 | グラクソスミスクライン、インテレクチュアル、プロパティー、ディベロップメント、リミテッドGlaxosmithkline Intellectual Property Development Limited | Ripk2阻害剤を含むコンジュゲート |
| GB201610156D0 (en) | 2016-06-10 | 2016-07-27 | Otsuka Pharma Co Ltd | Cliptac compositions |
| JP2019525955A (ja) * | 2016-07-12 | 2019-09-12 | アキュター バイオテクノロジー インコーポレイテッド | 新規化合物およびその使用 |
| GB2554071A (en) * | 2016-09-14 | 2018-03-28 | Univ Dundee | Small molecules |
| EP3512842B1 (en) | 2016-09-15 | 2024-01-17 | Arvinas, Inc. | Indole derivatives as estrogen receptor degraders |
| JP6899993B2 (ja) * | 2016-10-04 | 2021-07-07 | 国立医薬品食品衛生研究所長 | 複素環化合物 |
| KR102173463B1 (ko) | 2016-10-11 | 2020-11-04 | 아비나스 오퍼레이션스, 인코포레이티드 | 안드로겐 수용체의 표적 분해용 화합물 및 방법 |
| CA3042260C (en) | 2016-11-01 | 2023-10-03 | Arvinas, Inc. | Tau-protein targeting protacs and associated methods of use |
| EP3544956A1 (en) * | 2016-11-24 | 2019-10-02 | Eisai R&D Management Co., Ltd. | Tetrasubstituted alkene compounds and their use |
| KR102425785B1 (ko) | 2016-11-28 | 2022-07-28 | 에자이 알앤드디 매니지먼트 가부시키가이샤 | 인다졸 유도체의 염 및 이의 결정 |
| BR112019011200B1 (pt) | 2016-12-01 | 2021-12-28 | Arvinas Operations, Inc | Derivados de tetrahidronaftaleno e tetrahidroisoquinolina como degradadores do receptor de estrogênio |
| EP3559002A4 (en) | 2016-12-23 | 2021-02-17 | Arvinas Operations, Inc. | CHEMERICAL MOLECULES TARGETING EGFR PROTEOLYSIS AND RELATED METHODS OF USE |
| US11173211B2 (en) | 2016-12-23 | 2021-11-16 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of rapidly accelerated Fibrosarcoma polypeptides |
| US10806737B2 (en) | 2016-12-23 | 2020-10-20 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of fetal liver kinase polypeptides |
| JP2020504741A (ja) | 2016-12-23 | 2020-02-13 | アルビナス・オペレーションズ・インコーポレイテッドArvinas Operations, Inc. | Raf(急速進行性線維肉腫)ポリペプチドの標的分解のための化合物および方法 |
| US11191741B2 (en) | 2016-12-24 | 2021-12-07 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of enhancer of zeste homolog 2 polypeptide |
| WO2018140809A1 (en) | 2017-01-26 | 2018-08-02 | Arvinas, Inc. | Modulators of estrogen receptor proteolysis and associated methods of use |
| WO2019084026A1 (en) | 2017-10-24 | 2019-05-02 | Genentech, Inc. | (4-HYDROXYPYRROLIDIN-2-YL) -HETEROCYCLIC COMPOUNDS AND METHODS OF USE |
| US11065231B2 (en) | 2017-11-17 | 2021-07-20 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of interleukin-1 receptor- associated kinase 4 polypeptides |
| US10519152B2 (en) | 2017-12-21 | 2019-12-31 | Astrazeneca Ab | Compounds and their use in treating cancer |
| WO2019148055A1 (en) | 2018-01-26 | 2019-08-01 | Yale University | Imide-based modulators of proteolysis and methods of use |
| AU2019249231B2 (en) | 2018-04-04 | 2022-04-21 | Arvinas Operations, Inc. | Modulators of proteolysis and associated methods of use |
| EP3831811A4 (en) | 2018-07-31 | 2022-04-20 | Fimecs, Inc. | HETEROCYCLIC COMPOUND |
| CN112912376A (zh) | 2018-08-20 | 2021-06-04 | 阿尔维纳斯运营股份有限公司 | 用于治疗神经变性疾病的具有E3泛素连接酶结合活性并靶向α-突触核蛋白的蛋白水解靶向嵌合(PROTAC)化合物 |
| CN117050002A (zh) * | 2018-11-21 | 2023-11-14 | 冰洲石生物科技公司 | 具有雌激素受体α降解活性的新型化合物及其用途 |
| CN111205282B (zh) * | 2018-11-21 | 2023-12-01 | 上海科技大学 | Er蛋白调节剂及其应用 |
| KR20210136973A (ko) * | 2019-01-03 | 2021-11-17 | 더 리젠츠 오브 더 유니버시티 오브 미시간 | 에스트로겐 수용체 단백질 분해제 |
| US11547759B2 (en) | 2019-01-30 | 2023-01-10 | Montelino Therapeutics, Inc. | Bi-functional compounds and methods for targeted ubiquitination of androgen receptor |
| WO2020160295A1 (en) | 2019-01-30 | 2020-08-06 | Montelino Therapeutics, Llc | Bi-functional compounds and methods for targeted ubiquitination of androgen receptor |
| WO2021011913A1 (en) | 2019-07-17 | 2021-01-21 | Arvinas Operations, Inc. | Tau-protein targeting compounds and associated methods of use |
| CN118638043A (zh) | 2019-12-19 | 2024-09-13 | 阿尔维纳斯运营股份有限公司 | 用于雄激素受体的靶向降解的化合物和方法 |
| WO2021133886A1 (en) | 2019-12-23 | 2021-07-01 | Accutar Biotechnology | Combinations of estrogen receptor degraders and cyclin-dependent kinase inhibitors for treating cancer |
| EP4146642A1 (en) | 2020-05-09 | 2023-03-15 | Arvinas Operations, Inc. | Methods of manufacturing a bifunctional compound, ultrapure forms of the bifunctional compound, and dosage forms comprising the same |
| US11986532B2 (en) | 2021-04-16 | 2024-05-21 | Arvinas Operations, Inc. | Modulators of BCL6 proteolysis and associated methods of use |
| US11981672B2 (en) | 2021-09-13 | 2024-05-14 | Montelino Therapeutics Inc. | Bi-functional compounds and methods for targeted ubiquitination of androgen receptor |
| CN115028679B (zh) * | 2022-08-11 | 2022-11-15 | 深圳湾实验室 | 一种具有Cyclophilin A降解活性的PROTAC化合物及其制备方法与应用 |
| US11957759B1 (en) | 2022-09-07 | 2024-04-16 | Arvinas Operations, Inc. | Rapidly accelerated fibrosarcoma (RAF) degrading compounds and associated methods of use |
Family Cites Families (229)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4256108A (en) | 1977-04-07 | 1981-03-17 | Alza Corporation | Microporous-semipermeable laminated osmotic system |
| US4160452A (en) | 1977-04-07 | 1979-07-10 | Alza Corporation | Osmotic system having laminated wall comprising semipermeable lamina and microporous lamina |
| US4265874A (en) | 1980-04-25 | 1981-05-05 | Alza Corporation | Method of delivering drug with aid of effervescent activity generated in environment of use |
| US4522811A (en) | 1982-07-08 | 1985-06-11 | Syntex (U.S.A.) Inc. | Serial injection of muramyldipeptides and liposomes enhances the anti-infective activity of muramyldipeptides |
| US5007790A (en) | 1989-04-11 | 1991-04-16 | Depomed Systems, Inc. | Sustained-release oral drug dosage form |
| US5169645A (en) | 1989-10-31 | 1992-12-08 | Duquesne University Of The Holy Ghost | Directly compressible granules having improved flow properties |
| US6316003B1 (en) | 1989-12-21 | 2001-11-13 | Whitehead Institute For Biomedical Research | Tat-derived transport polypeptides |
| US5292638A (en) | 1990-12-07 | 1994-03-08 | The Regents Of The University Of California | Method of determining functional estrogen receptors for prognosis of cancer |
| US5582837A (en) | 1992-03-25 | 1996-12-10 | Depomed, Inc. | Alkyl-substituted cellulose-based sustained-release oral drug dosage forms |
| ATE173159T1 (de) | 1992-03-25 | 1998-11-15 | Depomed Systems Inc | Auf hydroxyethylzellulose basierende oralen arzneidosisformen mit verzoegerter wirkstoffabgabe |
| DE69411003T2 (de) | 1993-04-07 | 1998-10-08 | Pfizer Inc., New York, N.Y. | Cycloalkylhydroxyharnstoffe und ihre verwendung als lipoxy-genase-inhibitoren |
| US5681835A (en) | 1994-04-25 | 1997-10-28 | Glaxo Wellcome Inc. | Non-steroidal ligands for the estrogen receptor |
| US20030036654A1 (en) | 1994-08-18 | 2003-02-20 | Holt Dennis A. | Synthetic multimerizing agents |
| US5510357A (en) | 1995-02-28 | 1996-04-23 | Eli Lilly And Company | Benzothiophene compounds as anti-estrogenic agents |
| GB9603095D0 (en) | 1996-02-14 | 1996-04-10 | Zeneca Ltd | Quinazoline derivatives |
| AU728263B2 (en) | 1996-04-24 | 2001-01-04 | Medivir Uk Ltd | Auto-deconvoluting combinatorial libraries |
| CA2204082A1 (en) | 1996-05-03 | 1997-11-03 | Michael William John Urquhart | Pharmaceutical compounds |
| WO1997047285A1 (en) | 1996-06-10 | 1997-12-18 | Depomed, Inc. | Gastric-retentive oral controlled drug delivery system with enhanced retention properties |
| US5972389A (en) | 1996-09-19 | 1999-10-26 | Depomed, Inc. | Gastric-retentive, oral drug dosage forms for the controlled-release of sparingly soluble drugs and insoluble matter |
| HRP970566A2 (en) | 1996-10-30 | 1998-08-31 | Jones Deborah Defeo | Conjugates useful in the treatment of prostate canser |
| ZA982877B (en) | 1997-04-09 | 1999-10-04 | Lilly Co Eli | Treatment of central nervous system disorders with selective estrogen receptor modulators. |
| DE69841549D1 (de) | 1997-05-14 | 2010-04-22 | Sloan Kettering Inst Cancer | Verfahren und zubereitungen zur zerstörung bestimmter proteine |
| US6635280B2 (en) | 1997-06-06 | 2003-10-21 | Depomed, Inc. | Extending the duration of drug release within the stomach during the fed mode |
| CA2290624C (en) | 1997-06-06 | 2006-12-05 | John W. Shell | Gastric-retentive oral drug dosage forms for controlled release of highly soluble drugs |
| AU740597B2 (en) | 1997-07-10 | 2001-11-08 | Merck & Co., Inc. | Conjugates useful in the treatment of prostate cancer |
| DE69832715T2 (de) | 1997-07-12 | 2007-01-11 | Cancer Research Technology Ltd. | Cyclin-abhängige-kinase inhibierende purinderivate |
| GB9716557D0 (en) | 1997-08-06 | 1997-10-08 | Glaxo Group Ltd | Benzylidene-1,3-dihydro-indol-2-one derivatives having anti-cancer activity |
| US6576469B1 (en) | 1997-09-10 | 2003-06-10 | President And Fellows Of Harvard College | Inducible methods for repressing gene function |
| US6323219B1 (en) | 1998-04-02 | 2001-11-27 | Ortho-Mcneil Pharmaceutical, Inc. | Methods for treating immunomediated inflammatory disorders |
| ATE473759T1 (de) | 1998-05-22 | 2010-07-15 | Univ Leland Stanford Junior | Bifunktionelle moleküle sowie darauf basierende therapien. |
| WO2000022110A2 (en) | 1998-10-09 | 2000-04-20 | President And Fellows Of Harvard College | Targeted proteolysis by recruitment to ubiquitin protein ligases |
| US6306663B1 (en) | 1999-02-12 | 2001-10-23 | Proteinex, Inc. | Controlling protein levels in eucaryotic organisms |
| WO2000050445A1 (en) | 1999-02-26 | 2000-08-31 | Oklahoma Medical Research Foundation | Novel component of von hippel-lindau tumor suppressor complex and scf ubiquitin ligase |
| US6858709B1 (en) | 1999-02-26 | 2005-02-22 | Oklahoma Medical Research Foundation | Component of von Hippel-Lindau tumor suppressor complex and SCF ubiquitin ligase |
| JP2002543129A (ja) | 1999-05-05 | 2002-12-17 | メルク エンド カムパニー インコーポレーテッド | 抗微生物剤としての新規なプロリン類 |
| US7087386B2 (en) | 1999-06-11 | 2006-08-08 | The Burnham Institute | Nucleic acid encoding proteins involved in protein degradation, products and methods related thereto |
| US6921763B2 (en) | 1999-09-17 | 2005-07-26 | Abbott Laboratories | Pyrazolopyrimidines as therapeutic agents |
| CA2388700A1 (en) | 1999-10-19 | 2001-04-26 | Merck & Co., Inc. | Conjugates useful in the treatment of prostate cancer |
| EP1242057A2 (en) | 1999-11-02 | 2002-09-25 | DepoMed, Inc. | Pharmacological inducement of the fed mode for enhanced drug administration to the stomach |
| CA2396782A1 (en) | 2000-02-04 | 2001-08-09 | Depomed, Inc. | Shell-and-core dosage form approaching zero-order drug release |
| US6740495B1 (en) | 2000-04-03 | 2004-05-25 | Rigel Pharmaceuticals, Inc. | Ubiquitin ligase assay |
| US6488962B1 (en) | 2000-06-20 | 2002-12-03 | Depomed, Inc. | Tablet shapes to enhance gastric retention of swellable controlled-release oral dosage forms |
| PE20020354A1 (es) | 2000-09-01 | 2002-06-12 | Novartis Ag | Compuestos de hidroxamato como inhibidores de histona-desacetilasa (hda) |
| US7208157B2 (en) | 2000-09-08 | 2007-04-24 | California Institute Of Technology | Proteolysis targeting chimeric pharmaceutical |
| EP1322750A4 (en) | 2000-09-08 | 2004-09-29 | California Inst Of Techn | PROTEOLYSIC CHIMERAL PHARMACEUTICAL |
| US6451808B1 (en) | 2000-10-17 | 2002-09-17 | Depomed, Inc. | Inhibition of emetic effect of metformin with 5-HT3 receptor antagonists |
| WO2002066512A1 (en) | 2001-02-16 | 2002-08-29 | E.I. Dupont De Nemours And Company | Angiogenesis-inhibitory tripeptides, compositions and their methods of use |
| GB0106953D0 (en) | 2001-03-20 | 2001-05-09 | Univ Aberdeen | Neufofibrillary labels |
| MXPA03008560A (es) | 2001-03-22 | 2004-06-30 | Abbot Gmbh & Co Kg | Pirazolopirimidinas como agentes terapeuticos. |
| JP2004532259A (ja) | 2001-05-29 | 2004-10-21 | デポメッド ディベロップメント リミティド | 胃食道逆流症及び夜間に胃酸分泌が回復する現象の治療方法 |
| HN2002000136A (es) | 2001-06-11 | 2003-07-31 | Basf Ag | Inhibidores de la proteasa del virus hiv, compuestos que contienen a los mismos, sus usos farmaceuticos y los materiales para su sintesis |
| WO2003011854A1 (en) | 2001-08-03 | 2003-02-13 | Vertex Pharmaceuticals Incorporated | Pyrazole-derived kinase inhibitors and uses thereof |
| GB0119249D0 (en) | 2001-08-07 | 2001-10-03 | Novartis Ag | Organic compounds |
| JP4445262B2 (ja) | 2001-10-09 | 2010-04-07 | アムジェン インコーポレイテッド | 抗炎症剤としてのイミダゾール誘導体 |
| US20030133927A1 (en) | 2001-10-10 | 2003-07-17 | Defeo-Jones Deborah | Conjugates useful in the treatment of prostate cancer |
| US6723340B2 (en) | 2001-10-25 | 2004-04-20 | Depomed, Inc. | Optimal polymer mixtures for gastric retentive tablets |
| US20030091630A1 (en) | 2001-10-25 | 2003-05-15 | Jenny Louie-Helm | Formulation of an erodible, gastric retentive oral dosage form using in vitro disintegration test data |
| CA2409552A1 (en) | 2001-10-25 | 2003-04-25 | Depomed, Inc. | Gastric retentive oral dosage form with restricted drug release in the lower gastrointestinal tract |
| EP1438027A1 (en) | 2001-10-25 | 2004-07-21 | DepoMed, Inc. | Methods of treatment using a gastric retained losartan dosage |
| TWI312285B (en) | 2001-10-25 | 2009-07-21 | Depomed Inc | Methods of treatment using a gastric retained gabapentin dosage |
| US7053046B2 (en) | 2001-12-21 | 2006-05-30 | Mcgrath Kevin | Peptide activators of VEGF |
| US6682759B2 (en) | 2002-02-01 | 2004-01-27 | Depomed, Inc. | Manufacture of oral dosage forms delivering both immediate-release and sustained-release drugs |
| US20060128632A1 (en) | 2002-07-02 | 2006-06-15 | Sharma Sushil K | Peptide inhibitors of smac protein binding to inhibitor of apoptosis proteins (iap) |
| US20040053324A1 (en) | 2002-08-30 | 2004-03-18 | Brian Wong | Assays and compositions for identifying agents that modulate the activity of deubiquitinating agents |
| US20040114258A1 (en) | 2002-12-17 | 2004-06-17 | Harris Richard Alexander | Device and method for combining dynamic mathematical expressions and other multimedia objects within a document |
| US7470824B2 (en) | 2002-12-25 | 2008-12-30 | Idemitsu Kosan Co., Ltd. | Adamantane derivative and process for producing the same |
| EP2260846B1 (en) | 2003-03-27 | 2018-11-28 | Lankenau Institute for Medical Research | Novel methods for the treatment of cancer |
| US20050008640A1 (en) | 2003-04-23 | 2005-01-13 | Wendy Waegell | Method of treating transplant rejection |
| EP1651595A2 (en) | 2003-05-30 | 2006-05-03 | Rigel Pharmaceuticals, Inc. | Ubiquitin ligase inhibitors |
| US20070281907A1 (en) | 2003-12-22 | 2007-12-06 | Watkins William J | Kinase Inhibitor Phosphonate Conjugates |
| US7932382B2 (en) | 2004-01-16 | 2011-04-26 | The Regents Of The University Of Michigan | Conformationally constrained Smac mimetics and the uses thereof |
| WO2005094818A1 (en) | 2004-03-23 | 2005-10-13 | Genentech, Inc. | Azabicyclo-octane inhibitors of iap |
| SG152225A1 (en) | 2004-04-07 | 2009-05-29 | Novartis Ag | Inhibitors of iap |
| CA2569016C (en) | 2004-06-02 | 2012-11-27 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compound |
| US7244851B2 (en) | 2004-07-02 | 2007-07-17 | Genentech, Inc. | Inhibitors of IAP |
| BRPI0513020A (pt) | 2004-07-08 | 2008-04-22 | Warner Lambert Co | moduladores de andrÈgenio, seus usos, composição farmacêutica, formulação farmacêutica tópica e artigo de fabricação |
| JP5704785B2 (ja) | 2004-07-19 | 2015-04-22 | ザ・ジョンズ・ホプキンス・ユニバーシティ | 免疫抑制のためのflt3阻害剤 |
| US7294748B2 (en) | 2004-10-19 | 2007-11-13 | Wake Forest University Health Sciences | Sulfenic acid-reactive compounds and their methods of synthesis |
| WO2006062685A2 (en) | 2004-11-11 | 2006-06-15 | Affymax, Inc. | Novel peptides that bind to the erythropoietin receptor |
| GB0426661D0 (en) * | 2004-12-06 | 2005-01-05 | Isis Innovation | Pyrrolidine compounds |
| DK1836201T4 (da) | 2004-12-20 | 2013-11-11 | Genentech Inc | Pyrrolidininhibitorer af IAP. |
| CN103083644B (zh) | 2005-02-25 | 2014-05-28 | 泰特拉洛吉克药业公司 | Iap二聚体抑制剂 |
| CN100383139C (zh) | 2005-04-07 | 2008-04-23 | 天津和美生物技术有限公司 | 可抑制细胞释放肿瘤坏死因子的哌啶-2,6-二酮衍生物 |
| WO2006113942A2 (en) | 2005-04-20 | 2006-10-26 | Schering Corporation | Method of inhibiting cathepsin activity |
| WO2006122150A1 (en) | 2005-05-10 | 2006-11-16 | Incyte Corporation | Modulators of indoleamine 2,3-dioxygenase and methods of using the same |
| FR2885904B1 (fr) | 2005-05-19 | 2007-07-06 | Aventis Pharma Sa | Nouveaux derives du fluorene, compositions les contenant et utilisation |
| US20070155730A1 (en) | 2005-08-26 | 2007-07-05 | Methylgene, Inc. | Benzodiazepine And Benzopiperazine Analog Inhibitors Of Histone Deacetylase |
| JP5175191B2 (ja) | 2005-08-30 | 2013-04-03 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | グリコピラノシル置換ベンジルベンゼン誘導体、該化合物を含有する医薬品及びその使用と製造方法 |
| KR101443651B1 (ko) | 2005-10-18 | 2014-09-23 | 얀센 파마슈티카 엔.브이. | Flt-3 키나제의 억제 방법 |
| US7705022B2 (en) | 2005-10-27 | 2010-04-27 | Lankenau Institute For Medical Research | IDO inhibitors and methods of use thereof |
| KR20080095895A (ko) | 2006-03-03 | 2008-10-29 | 노파르티스 아게 | N―포르밀 히드록실아민 화합물 |
| WO2007101347A1 (en) | 2006-03-07 | 2007-09-13 | Aegera Therapeutics Inc. | Bir domain binding compounds |
| MX2008012492A (es) | 2006-03-29 | 2008-12-12 | Univ California | Compuestos de diariltiohidantoina. |
| EP2004170A1 (en) | 2006-04-11 | 2008-12-24 | Ramot at Tel-Aviv University Ltd. | Treatment of hematological malignancies with fts and a bcr-abl tyrosine kinase inhibitor |
| WO2007125405A2 (en) | 2006-05-01 | 2007-11-08 | Pfizer Products Inc. | Substituted 2-amino-fused heterocyclic compounds |
| WO2007129195A2 (en) | 2006-05-04 | 2007-11-15 | Pfizer Products Inc. | 4-pyrimidine-5-amino-pyrazole compounds |
| KR101071516B1 (ko) | 2006-05-05 | 2011-10-10 | 더 리젠츠 오브 더 유니버시티 오브 미시간 | 2가 smac 모방체 및 그의 용도 |
| JP2009544620A (ja) | 2006-07-20 | 2009-12-17 | リガンド・ファーマシューティカルズ・インコーポレイテッド | 自己免疫疾患及び炎症のためのプロリン尿素ccr1アンタゴニスト |
| US20100056495A1 (en) | 2006-07-24 | 2010-03-04 | Tetralogic Pharmaceuticals Corporation | Dimeric iap inhibitors |
| ES2403546T3 (es) | 2006-11-03 | 2013-05-20 | Pharmacyclics, Inc. | Sonda de actividad de la tirosina-cinasa de Bruton y procedimiento de utilización |
| US8518891B2 (en) | 2006-11-29 | 2013-08-27 | Longqin Hu | Chemotherapeutic conjugates and methods of use |
| KR20090118103A (ko) | 2007-03-07 | 2009-11-17 | 얀센 파마슈티카 엔.브이. | 에스트로겐 관련 수용체-알파 조절제로서 치환된 페녹시 n-알킬화 티아졸리딘디온 |
| US7846953B2 (en) | 2007-03-07 | 2010-12-07 | Janssen Pharmaceutica, Nv | Substituted phenoxy thiazolidinediones as estrogen related receptor-α modulators |
| WO2008115804A1 (en) | 2007-03-16 | 2008-09-25 | Lankenau Institute For Medical Research | Novel ido inhibitors and methods of use thereof |
| US20080233850A1 (en) | 2007-03-20 | 2008-09-25 | 3M Innovative Properties Company | Abrasive article and method of making and using the same |
| MX2009010667A (es) | 2007-04-12 | 2010-02-24 | Joyant Pharmaceuticals Inc | Dimeros y trimeros mimeticos de smac utiles como agentes anti-cancer. |
| CA2683318C (en) | 2007-04-13 | 2012-10-30 | The Regents Of The University Of Michigan | Diazo bicyclic smac mimetics and the uses thereof |
| NZ580226A (en) | 2007-04-30 | 2012-11-30 | Genentech Inc | Dimer compounds as inhibitors of iap |
| WO2008144925A1 (en) | 2007-05-30 | 2008-12-04 | Aegera Therapeutics Inc. | Iap bir domain binding compounds |
| JP5282091B2 (ja) | 2007-07-25 | 2013-09-04 | ブリストル−マイヤーズ スクイブ カンパニー | トリアジンキナーゼ阻害剤 |
| JP2010535213A (ja) | 2007-07-31 | 2010-11-18 | アンドロサイエンス コーポレーション | アンドロゲン受容体分解(ard)エンハンサーを含む組成物ならびに皮膚疾患および脱毛の予防的または治療的処理方法 |
| EP2058312A1 (en) | 2007-11-09 | 2009-05-13 | Universita' degli Studi di Milano | SMAC mimetic compounds as apoptosis inducers |
| US20100056524A1 (en) | 2008-04-02 | 2010-03-04 | Mciver Edward Giles | Compound |
| WO2010107485A1 (en) | 2009-03-17 | 2010-09-23 | The Trustees Of Columbia University In The City Of New York | E3 ligase inhibitors |
| US8691187B2 (en) | 2009-03-23 | 2014-04-08 | Eli Lilly And Company | Imaging agents for detecting neurological disorders |
| WO2010141805A1 (en) | 2009-06-05 | 2010-12-09 | Janssen Pharmaceutica Nv | Heterocyclic amides as modulators of trpa1 |
| WO2011005510A2 (en) | 2009-06-22 | 2011-01-13 | Cerapedics, Inc. | Peptide conjugates and uses thereof |
| AU2010273220B2 (en) | 2009-07-13 | 2015-10-15 | President And Fellows Of Harvard College | Bifunctional stapled polypeptides and uses thereof |
| JP2012254939A (ja) | 2009-10-07 | 2012-12-27 | Astellas Pharma Inc | オキサゾール化合物 |
| EP2784076A1 (en) | 2009-10-28 | 2014-10-01 | Joyant Pharmaceuticals, Inc. | Dimeric SMAC mimetics |
| US20110164191A1 (en) | 2010-01-04 | 2011-07-07 | Microvision, Inc. | Interactive Projection Method, Apparatus and System |
| US8198300B2 (en) | 2010-04-29 | 2012-06-12 | Universidad De Chile | Method for preventing tau protein aggregation and treating Alzheimer's disease with a quinoline derivative compound |
| EP2569434B1 (en) | 2010-05-14 | 2019-09-04 | Dana-Farber Cancer Institute, Inc. | Compositions and methods for treating leukemia and related disorders |
| BR122014024883A2 (pt) | 2010-05-14 | 2019-08-20 | Dana-Farber Cancer Institute, Inc. | Compostos no tratamento de neoplasia |
| WO2011160016A2 (en) | 2010-06-17 | 2011-12-22 | The Trustees Of Columbia University In The City Of New York | E3 binding pockets and identification and use of e3 ligase inhibitors |
| EP2588129A4 (en) | 2010-06-30 | 2014-07-09 | Univ Brandeis | ON SMALL MOLECULES TARGETED PROTEIN REMOVAL |
| EP2590656B1 (en) | 2010-07-07 | 2017-11-15 | Ardelyx, Inc. | Compounds and methods for inhibiting phosphate transport |
| EP2593457B1 (en) | 2010-07-13 | 2017-08-23 | F. Hoffmann-La Roche AG | Pyrazolo[1,5a]pyrimidine and thieno[3,2b]pyrimidine derivatives as irak4 modulators |
| US9050334B2 (en) | 2010-07-16 | 2015-06-09 | Innov88 Llc | MIF inhibitors and their uses |
| AU2011305315A1 (en) | 2010-09-24 | 2013-03-28 | The Regents Of The University Of Michigan | Deubiquitinase inhibitors and methods for use of the same |
| NZ609957A (en) | 2010-11-01 | 2015-08-28 | Celgene Avilomics Res Inc | Heterocyclic compounds and uses thereof |
| CN102477033A (zh) | 2010-11-23 | 2012-05-30 | 苏州波锐生物医药科技有限公司 | 苯并噻酚类化合物及其在制备预防和/或治疗乳腺癌骨质疏松症药物中的用途 |
| AR084070A1 (es) | 2010-12-02 | 2013-04-17 | Constellation Pharmaceuticals Inc | Inhibidores del bromodominio y usos de los mismos |
| AU2011338615B2 (en) | 2010-12-07 | 2017-07-27 | Yale University | Small-molecule hydrophobic tagging of fusion proteins and induced degradation of same |
| US20140243282A1 (en) | 2010-12-31 | 2014-08-28 | Satish Reddy Kallam | Methods and compositions for designing novel conjugate therapeutics |
| EP2688872A4 (en) | 2011-03-22 | 2014-08-27 | Merck Sharp & Dohme | AMIDOPYRAZOLHEMMER OF INTERLEUKIN RECEPTOR-MEDIATED KINASES |
| WO2012142498A2 (en) | 2011-04-13 | 2012-10-18 | Innovimmune Biotherapeutics, Inc. | Mif inhibitors and their uses |
| NO2694640T3 (es) | 2011-04-15 | 2018-03-17 | ||
| EP2709622A4 (en) | 2011-05-17 | 2015-03-04 | Plexxikon Inc | CINEMA MODULATION AND INDICATIONS THEREFOR |
| WO2012162627A1 (en) | 2011-05-26 | 2012-11-29 | The General Hospital Corporation | Treatment of angiogenic- or vascular-associated diseases |
| EP2721031B1 (en) | 2011-06-17 | 2016-01-20 | Constellation Pharmaceuticals, Inc. | Bromodomain inhibitors and uses thereof |
| WO2013042137A1 (en) | 2011-09-19 | 2013-03-28 | Aurigene Discovery Technologies Limited | Bicyclic heterocycles as irak4 inhibitors |
| AU2012316055B2 (en) | 2011-09-27 | 2016-05-12 | Amgen Inc. | Heterocyclic compounds as MDM2 inhibitors for the treatment of cancer |
| EP2773207B1 (en) | 2011-10-31 | 2018-03-07 | Merck Sharp & Dohme Corp. | Aminopyrimidinones as interleukin receptor-associated kinase inhibitors |
| WO2013071039A1 (en) | 2011-11-09 | 2013-05-16 | Ensemble Therapeutics | Macrocyclic compounds for inhibition of inhibitors of apoptosis |
| WO2013071035A1 (en) | 2011-11-09 | 2013-05-16 | Ensemble Therapeutics | Macrocyclic compounds for inhibition of inhibitors of apoptosis |
| CN103172648B (zh) | 2011-12-20 | 2016-05-25 | 上海迪诺医药科技有限公司 | 三杂环衍生物、制备方法及应用 |
| CA2863259A1 (en) | 2012-01-10 | 2013-07-18 | Nimbus Iris, Inc. | Irak inhibitors and uses thereof |
| CN117736134A (zh) | 2012-01-12 | 2024-03-22 | 耶鲁大学 | 通过e3泛素连接酶增强靶蛋白质及其他多肽降解的化合物和方法 |
| WO2013106646A2 (en) | 2012-01-12 | 2013-07-18 | Yale University | Compounds and methods for the inhibition of vcb e3 ubiquitin ligase |
| JP6096219B2 (ja) | 2012-01-13 | 2017-03-15 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | キナーゼ阻害剤として有用なトリアゾリルまたはトリアジアゾリル置換されたピリジル化合物 |
| WO2013106614A1 (en) | 2012-01-13 | 2013-07-18 | Bristol-Myers Squibb Company | Triazolyl-substituted pyridyl compounds useful as kinase inhibitors |
| ES2686500T3 (es) | 2012-01-13 | 2018-10-18 | Bristol-Myers Squibb Company | Compuestos de piridilo sustituidos con heterocíclicos, útiles como inhibidores de cinasas |
| AU2013250378B2 (en) | 2012-04-17 | 2016-01-14 | Fujifilm Corporation | Nitrogen-containing heterocyclic compound or salt thereof |
| US20150119435A1 (en) | 2012-05-11 | 2015-04-30 | Yale University | Compounds useful for promoting protein degradation and methods using same |
| WO2013175417A1 (en) | 2012-05-24 | 2013-11-28 | Novartis Ag | Pyrrolopyrrolidinone compounds |
| CN107540594A (zh) | 2012-05-30 | 2018-01-05 | 霍夫曼-拉罗奇有限公司 | 取代的吡咯烷‑2‑甲酰胺 |
| WO2014011712A1 (en) | 2012-07-10 | 2014-01-16 | Bristol-Myers Squibb Company | Iap antagonists |
| ES2742285T3 (es) | 2012-07-31 | 2020-02-13 | Novartis Ag | Marcadores asociados con la sensibilidad a inhibidores del doble minuto humano 2 (MDM2) |
| EP2882740B1 (en) | 2012-08-09 | 2017-03-01 | Bristol-Myers Squibb Company | Iap antagonists |
| TWI586668B (zh) | 2012-09-06 | 2017-06-11 | 第一三共股份有限公司 | 二螺吡咯啶衍生物之結晶 |
| WO2014047024A1 (en) | 2012-09-18 | 2014-03-27 | Bristol-Myers Squibb Company | Iap antagonists |
| KR101738063B1 (ko) | 2012-09-21 | 2017-05-19 | 아로그 파마슈티칼스, 인코퍼레이티드 | 구조적으로 활성인 인산화된 flt3 키나제의 억제 방법 |
| WO2014055461A1 (en) | 2012-10-02 | 2014-04-10 | Bristol-Myers Squibb Company | Iap antagonists |
| WO2014058691A1 (en) | 2012-10-08 | 2014-04-17 | Merck Sharp & Dohme Corp. | Inhibitors of irak4 activity |
| WO2014066834A1 (en) | 2012-10-26 | 2014-05-01 | The University Of Chicago | Synergistic combination of immunologic inhibitors for the treatment of cancer |
| CA2930030A1 (en) | 2012-11-09 | 2014-05-15 | Ensemble Therapeutics Corporation | Macrocyclic compounds for inhibition of inhibitors of apoptosis |
| AR094116A1 (es) | 2012-12-20 | 2015-07-08 | Merck Sharp & Dohme | Imidazopiridinas sustituidas como inhibidores de hdm2 |
| EP2934535B1 (en) | 2012-12-20 | 2017-07-19 | Merck Sharp & Dohme Corp. | Substituted pyrrolopyrimidines as hdm2 inhibitors |
| EP2752191A1 (en) | 2013-01-07 | 2014-07-09 | Sanofi | Compositions and methods using hdm2 antagonist and mek inhibitor |
| BR112015016282A2 (pt) | 2013-01-07 | 2017-07-11 | Arog Pharmaceuticals Inc | crenolanibe para tratamento de distúrbios proliferativos de flt3 mutado |
| GB201311910D0 (en) | 2013-07-03 | 2013-08-14 | Glaxosmithkline Ip Dev Ltd | Novel Compounds |
| NL2011274C2 (en) | 2013-08-06 | 2015-02-09 | Illumicare Ip B V 51 | Groundbreaking platform technology for specific binding to necrotic cells. |
| JP6266659B2 (ja) | 2013-02-28 | 2018-01-24 | アムジエン・インコーポレーテツド | 癌の治療のための安息香酸誘導体mdm2阻害剤 |
| AU2014236812B2 (en) | 2013-03-14 | 2018-03-01 | Amgen Inc. | Heteroaryl acid morpholinone compounds as MDM2 inhibitors for the treatment of cancer |
| EP2970334B1 (en) | 2013-03-15 | 2018-05-23 | Biogen MA Inc. | Macrocyclic compounds as irak4 inhibitors for the treatment of inflammatory diseases |
| TR201810399T4 (tr) | 2013-03-15 | 2018-08-27 | Gilead Sciences Llc | İndolamin 2,3-dioksijenaz (ıdo) inhibitörleri. |
| EP3016932B1 (en) | 2013-07-01 | 2019-02-27 | Bristol-Myers Squibb Company | Ido inhibitors |
| GB201311888D0 (en) | 2013-07-03 | 2013-08-14 | Glaxosmithkline Ip Dev Ltd | Novel compounds |
| GB201311891D0 (en) | 2013-07-03 | 2013-08-14 | Glaxosmithkline Ip Dev Ltd | Novel compound |
| ES2707961T3 (es) | 2013-07-11 | 2019-04-08 | Bristol Myers Squibb Co | Inhibidores de IDO |
| WO2015006524A1 (en) | 2013-07-12 | 2015-01-15 | Bristol-Myers Squibb Company | Iap antagonists |
| CN103570726B (zh) | 2013-07-15 | 2016-04-06 | 上海天慈生物谷生物工程有限公司 | N-烷基色胺酮衍生物及其制备方法和应用 |
| ES2648876T3 (es) | 2013-07-23 | 2018-01-08 | Bayer Pharma Aktiengesellschaft | Dihidropirido[3,4-b]pirazinonas sustituidas como inhibidores duales de proteínas BET y quinasas tipo polo |
| CN105593224B (zh) | 2013-07-31 | 2021-05-25 | 恒元生物医药科技(苏州)有限公司 | 作为溴结构域抑制剂的新型喹唑啉酮类化合物 |
| US20150051208A1 (en) | 2013-08-14 | 2015-02-19 | Boehringer Ingelheim International Gmbh | Pyridinones |
| EP3039020B1 (en) | 2013-08-27 | 2017-07-19 | Bristol-Myers Squibb Company | Ido inhibitors |
| US10463658B2 (en) | 2013-10-25 | 2019-11-05 | Videra Pharmaceuticals, Llc | Method of inhibiting FLT3 kinase |
| US9428513B2 (en) | 2013-11-07 | 2016-08-30 | Boehringer Ingelheim International Gmbh | Triazolopyrazine |
| CN105899505B (zh) | 2013-11-08 | 2018-08-28 | 武田药品工业株式会社 | 用于治疗自身免疫病症的吡唑 |
| AR098343A1 (es) | 2013-11-08 | 2016-05-26 | Incyte Holdings Corp | Proceso para la síntesis de un inhibidor de indolamina 2,3-dioxigenasa |
| CN103570727B (zh) | 2013-11-12 | 2015-08-19 | 复旦大学 | 一种n-苄基色胺酮衍生物及其制备方法和应用 |
| TWI742513B (zh) | 2013-11-18 | 2021-10-11 | 美商弗瑪治療公司 | 作為bet溴域抑制劑之四氫喹啉組成物 |
| UA115388C2 (uk) | 2013-11-21 | 2017-10-25 | Пфайзер Інк. | 2,6-заміщені пуринові похідні та їх застосування в лікуванні проліферативних захворювань |
| WO2015081203A1 (en) | 2013-11-26 | 2015-06-04 | Incyte Corporation | Bicyclic heterocycles as bet protein inhibitors |
| US9458156B2 (en) | 2014-12-23 | 2016-10-04 | Bristol-Myers Squibb Company | Tricyclic compounds as anticancer agents |
| TW201609693A (zh) | 2014-01-03 | 2016-03-16 | 必治妥美雅史谷比公司 | 雜芳基取代之菸鹼醯胺化合物 |
| JP2017505337A (ja) | 2014-01-10 | 2017-02-16 | アウリジーン ディスカバリー テクノロジーズ リミテッド | Irak4阻害剤としてのインダゾール化合物 |
| CU24389B1 (es) | 2014-01-13 | 2019-04-04 | Aurigene Discovery Tech Ltd | Compuestos de heterociclilo bicíclico como inhibidores de irak4 |
| JP6351306B2 (ja) | 2014-03-06 | 2018-07-04 | キヤノン株式会社 | 画像処理装置、画像処理装置の制御方法およびプログラム |
| US20150259288A1 (en) | 2014-03-14 | 2015-09-17 | City Of Hope | 5-bromo-indirubins |
| MA39838B1 (fr) | 2014-04-04 | 2019-05-31 | Pfizer | Composés bicycliques hétéroaryle ou aryle fusionnés et leur utilisation en tant qu'inhibiteurs irak4 |
| KR20170002446A (ko) | 2014-04-14 | 2017-01-06 | 아비나스 인코포레이티드 | 단백질분해의 이미드-기초된 조절인자 및 연관된 이용 방법 |
| US20160058872A1 (en) | 2014-04-14 | 2016-03-03 | Arvinas, Inc. | Imide-based modulators of proteolysis and associated methods of use |
| US20170166598A1 (en) | 2014-05-13 | 2017-06-15 | Ariad Pharmaceuticals, Inc. | Heteroaryl compounds for kinase inhibition |
| MX2016015005A (es) | 2014-05-15 | 2017-09-28 | Iteos Therapeutics | Derivados de pirrolidina-2,5-diona, composiciones farmaceuticas y metodos para usar como inhibidores ido1. |
| TW201613916A (en) | 2014-06-03 | 2016-04-16 | Gilead Sciences Inc | TANK-binding kinase inhibitor compounds |
| WO2016011390A1 (en) | 2014-07-18 | 2016-01-21 | Biogen Ma Inc. | Irak4 inhibiting agents |
| US10071164B2 (en) | 2014-08-11 | 2018-09-11 | Yale University | Estrogen-related receptor alpha based protac compounds and associated methods of use |
| WO2016037026A1 (en) | 2014-09-05 | 2016-03-10 | Merck Patent Gmbh | Cyclohexyl-ethyl substituted diaza- and triaza-tricyclic compounds as indole-amine-2,3-dioxygenase (ido) antagonists for the treatment of cancer |
| EP3200789B1 (en) | 2014-09-30 | 2019-11-06 | Merck Sharp & Dohme Corp. | Inhibitors of irak4 activity |
| US9943516B2 (en) | 2014-09-30 | 2018-04-17 | Merck Sharp & Dohme Corp. | Inhibitors of IRAK4 activity |
| EP3200788B1 (en) | 2014-09-30 | 2019-09-18 | Merck Sharp & Dohme Corp. | Inhibitors of irak4 activity |
| US9969749B2 (en) | 2014-09-30 | 2018-05-15 | Merck Sharp & Dohme Corp. | Inhibitors of IRAK4 activity |
| UY36390A (es) | 2014-11-05 | 2016-06-01 | Flexus Biosciences Inc | Compuestos moduladores de la enzima indolamina 2,3-dioxigenasa (ido), sus métodos de síntesis y composiciones farmacéuticas que los contienen |
| UY36391A (es) | 2014-11-05 | 2016-06-01 | Flexus Biosciences Inc | Compuestos moduladores de la enzima indolamina 2,3-dioxigenasa (ido1), sus métodos de síntesis y composiciones farmacèuticas que las contienen |
| CN107428734A (zh) | 2015-01-20 | 2017-12-01 | 阿尔维纳斯股份有限公司 | 用于雄激素受体的靶向降解的化合物和方法 |
| MA41598A (fr) | 2015-02-25 | 2018-01-02 | Constellation Pharmaceuticals Inc | Composés thérapeutiques de pyridazine et leurs utilisations |
| EP3268004B1 (en) | 2015-03-12 | 2019-12-18 | Merck Sharp & Dohme Corp. | Pyrrolopyridazine inhibitors of irak4 activity |
| WO2016144846A1 (en) | 2015-03-12 | 2016-09-15 | Merck Sharp & Dohme Corp. | Pyrazolopyrimidine inhibitors of irak4 activity |
| US10155765B2 (en) | 2015-03-12 | 2018-12-18 | Merck Sharp & Dohme Corp. | Carboxamide inhibitors of IRAK4 activity |
| US10329295B2 (en) | 2015-03-12 | 2019-06-25 | Merck Sharp & Dohme Corp. | Pyrrolotriazine inhibitors of IRAK4 activity |
| EP3268003B1 (en) | 2015-03-12 | 2020-07-29 | Merck Sharp & Dohme Corp. | Thienopyrazine inhibitors of irak4 activity |
| GB201504314D0 (en) | 2015-03-13 | 2015-04-29 | Univ Dundee | Small molecules |
| CA2979070A1 (en) | 2015-03-18 | 2016-09-22 | Arvinas, Inc. | Compounds and methods for the enhanced degradation of targeted proteins |
| CA2985642A1 (en) | 2015-05-15 | 2016-11-24 | Gilead Sciences, Inc. | Benzimidazole and imadazopyridine carboximidamide compounds having activity as inhibitors of indoleamine 2,3-dioxygenase |
| MX2018000360A (es) | 2015-07-10 | 2018-06-11 | Arvinas Inc | Moduladores basados en mdm2 de proteolisis y metodos de uso asociados. |
| WO2017011590A1 (en) | 2015-07-13 | 2017-01-19 | Arvinas, Inc. | Alanine-based modulators of proteolysis and associated methods of use |
| US10772962B2 (en) | 2015-08-19 | 2020-09-15 | Arvinas Operations, Inc. | Compounds and methods for the targeted degradation of bromodomain-containing proteins |
-
2013
- 2013-07-03 GB GBGB1311891.4A patent/GB201311891D0/en not_active Ceased
-
2014
- 2014-07-01 EP EP14734147.3A patent/EP3016967B1/en active Active
- 2014-07-01 ES ES14734147T patent/ES2717436T3/es active Active
- 2014-07-01 US US14/898,832 patent/US9993514B2/en active Active
- 2014-07-01 TR TR2019/04340T patent/TR201904340T4/tr unknown
- 2014-07-01 WO PCT/EP2014/063901 patent/WO2015000868A1/en active Application Filing
-
2016
- 2016-11-03 HK HK16112689.3A patent/HK1224312A1/zh unknown
Also Published As
| Publication number | Publication date |
|---|---|
| TR201904340T4 (tr) | 2019-05-21 |
| HK1224312A1 (zh) | 2017-08-18 |
| US20160136230A1 (en) | 2016-05-19 |
| EP3016967A1 (en) | 2016-05-11 |
| WO2015000868A1 (en) | 2015-01-08 |
| EP3016967B1 (en) | 2019-02-06 |
| US9993514B2 (en) | 2018-06-12 |
| GB201311891D0 (en) | 2013-08-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2717436T3 (es) | Compuestos novedosos | |
| ES2697686T3 (es) | Derivados de benzotiofeno como inhibidores del receptor de estrógenos | |
| US10336744B2 (en) | 1AP E3 ligase directed proteolysis targeting chimeric molecules | |
| ES2930157T3 (es) | Combinación que comprende un inhibidor de MEK y un inhibidor de B-Raf | |
| JP4458746B2 (ja) | 癌の治療方法 | |
| ES2822665T3 (es) | Terapias de combinación para el cáncer | |
| ES2432414T3 (es) | Compuestos químicos | |
| WO2014108452A1 (en) | Proteolysis targeting chimeras (protacs) directed to the modulation of the estrogen receptor | |
| ES2608053T3 (es) | Inhibidores de ácido graso sintasa | |
| JP2014515345A (ja) | 脂肪酸シンターゼ阻害剤としてのピリミジノン誘導体 | |
| JP6355724B2 (ja) | がんを治療するためのアフレセルチブと組み合わせたエンザルタミド | |
| JP6563558B2 (ja) | 組み合わせ薬物療法 | |
| JP2021155446A (ja) | 組合せ物 | |
| ES2651331T3 (es) | Inhibidores de la sintasa de ácidos grasos | |
| US20190151319A1 (en) | Combination comprising a btk inhibitor and an akt inhibitor |