EP0241685A1 - Verfahren zur Enthalogenierung von Chlor- und von Bromessigsäuren - Google Patents
Verfahren zur Enthalogenierung von Chlor- und von Bromessigsäuren Download PDFInfo
- Publication number
- EP0241685A1 EP0241685A1 EP87102846A EP87102846A EP0241685A1 EP 0241685 A1 EP0241685 A1 EP 0241685A1 EP 87102846 A EP87102846 A EP 87102846A EP 87102846 A EP87102846 A EP 87102846A EP 0241685 A1 EP0241685 A1 EP 0241685A1
- Authority
- EP
- European Patent Office
- Prior art keywords
- electrolysis
- cells
- salts
- acids
- divided
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 238000000034 method Methods 0.000 title claims abstract description 27
- KDPAWGWELVVRCH-UHFFFAOYSA-N bromoacetic acid Chemical class OC(=O)CBr KDPAWGWELVVRCH-UHFFFAOYSA-N 0.000 title claims abstract description 11
- 230000008569 process Effects 0.000 title claims description 17
- 238000005868 electrolysis reaction Methods 0.000 claims abstract description 40
- 150000003839 salts Chemical class 0.000 claims abstract description 24
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 19
- 239000001257 hydrogen Substances 0.000 claims abstract description 19
- 229910052751 metal Inorganic materials 0.000 claims abstract description 19
- 239000002184 metal Substances 0.000 claims abstract description 19
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims abstract description 18
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims abstract description 18
- 239000002253 acid Substances 0.000 claims abstract description 16
- 229910052799 carbon Inorganic materials 0.000 claims abstract description 13
- 150000007513 acids Chemical class 0.000 claims abstract description 12
- 239000000243 solution Substances 0.000 claims abstract description 11
- 150000002739 metals Chemical class 0.000 claims abstract description 10
- 239000007864 aqueous solution Substances 0.000 claims abstract description 7
- 239000007772 electrode material Substances 0.000 claims abstract description 6
- 229910052802 copper Inorganic materials 0.000 claims abstract description 5
- 229910052745 lead Inorganic materials 0.000 claims abstract description 5
- 229910052719 titanium Inorganic materials 0.000 claims abstract description 5
- 229910052709 silver Inorganic materials 0.000 claims abstract description 4
- 229910052793 cadmium Inorganic materials 0.000 claims abstract description 3
- 229910052804 chromium Inorganic materials 0.000 claims abstract description 3
- 229910052737 gold Inorganic materials 0.000 claims abstract description 3
- 229910052715 tantalum Inorganic materials 0.000 claims abstract description 3
- 229910052725 zinc Inorganic materials 0.000 claims abstract description 3
- 229910052726 zirconium Inorganic materials 0.000 claims abstract description 3
- JXTHNDFMNIQAHM-UHFFFAOYSA-N dichloroacetic acid Chemical compound OC(=O)C(Cl)Cl JXTHNDFMNIQAHM-UHFFFAOYSA-N 0.000 claims description 39
- 229960005215 dichloroacetic acid Drugs 0.000 claims description 18
- 238000005695 dehalogenation reaction Methods 0.000 claims description 13
- 239000012528 membrane Substances 0.000 claims description 13
- 239000000463 material Substances 0.000 claims description 8
- 229920000642 polymer Polymers 0.000 claims description 6
- 238000005341 cation exchange Methods 0.000 claims description 5
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 4
- SIEILFNCEFEENQ-UHFFFAOYSA-N dibromoacetic acid Chemical compound OC(=O)C(Br)Br SIEILFNCEFEENQ-UHFFFAOYSA-N 0.000 claims description 4
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical group OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 claims description 4
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 2
- 125000000542 sulfonic acid group Chemical group 0.000 claims description 2
- HLHNOIAOWQFNGW-UHFFFAOYSA-N 3-bromo-4-hydroxybenzonitrile Chemical group OC1=CC=C(C#N)C=C1Br HLHNOIAOWQFNGW-UHFFFAOYSA-N 0.000 claims 1
- 229960004319 trichloroacetic acid Drugs 0.000 claims 1
- 230000007797 corrosion Effects 0.000 abstract description 8
- 238000005260 corrosion Methods 0.000 abstract description 8
- 239000000460 chlorine Substances 0.000 abstract description 6
- 229910052801 chlorine Inorganic materials 0.000 abstract description 6
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 abstract description 5
- 230000015572 biosynthetic process Effects 0.000 abstract description 3
- 229910052759 nickel Inorganic materials 0.000 abstract 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 27
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 23
- FOCAUTSVDIKZOP-UHFFFAOYSA-N chloroacetic acid Chemical compound OC(=O)CCl FOCAUTSVDIKZOP-UHFFFAOYSA-N 0.000 description 12
- 235000011054 acetic acid Nutrition 0.000 description 9
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 8
- SZVJSHCCFOBDDC-UHFFFAOYSA-N iron(II,III) oxide Inorganic materials O=[Fe]O[Fe]O[Fe]=O SZVJSHCCFOBDDC-UHFFFAOYSA-N 0.000 description 8
- 239000000203 mixture Substances 0.000 description 8
- 239000010406 cathode material Substances 0.000 description 6
- -1 halogen ions Chemical class 0.000 description 6
- 238000006243 chemical reaction Methods 0.000 description 5
- 230000000052 comparative effect Effects 0.000 description 5
- 238000011161 development Methods 0.000 description 5
- 239000003792 electrolyte Substances 0.000 description 5
- 239000010439 graphite Substances 0.000 description 5
- 229910002804 graphite Inorganic materials 0.000 description 5
- 229910052500 inorganic mineral Inorganic materials 0.000 description 5
- 239000011707 mineral Substances 0.000 description 5
- 229920000557 Nafion® Polymers 0.000 description 4
- 229910052736 halogen Inorganic materials 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 4
- 229910000978 Pb alloy Inorganic materials 0.000 description 3
- 239000004698 Polyethylene Substances 0.000 description 3
- 125000000218 acetic acid group Chemical class C(C)(=O)* 0.000 description 3
- 239000010405 anode material Substances 0.000 description 3
- 239000008151 electrolyte solution Substances 0.000 description 3
- 150000002367 halogens Chemical class 0.000 description 3
- 229920000573 polyethylene Polymers 0.000 description 3
- SQGYOTSLMSWVJD-UHFFFAOYSA-N silver(1+) nitrate Chemical compound [Ag+].[O-]N(=O)=O SQGYOTSLMSWVJD-UHFFFAOYSA-N 0.000 description 3
- 125000006850 spacer group Chemical group 0.000 description 3
- 239000010936 titanium Substances 0.000 description 3
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- 229910006069 SO3H Chemical group 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 2
- 150000001450 anions Chemical class 0.000 description 2
- 230000031709 bromination Effects 0.000 description 2
- 238000005893 bromination reaction Methods 0.000 description 2
- 238000005660 chlorination reaction Methods 0.000 description 2
- 239000003245 coal Substances 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000005342 ion exchange Methods 0.000 description 2
- 238000007086 side reaction Methods 0.000 description 2
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 description 2
- 235000011149 sulphuric acid Nutrition 0.000 description 2
- 229910052718 tin Inorganic materials 0.000 description 2
- 230000004580 weight loss Effects 0.000 description 2
- HQLVOUOBRKMDMY-UHFFFAOYSA-N 2-ethenylperoxyethanesulfonyl fluoride Chemical compound FS(=O)(=O)CCOOC=C HQLVOUOBRKMDMY-UHFFFAOYSA-N 0.000 description 1
- 229910021607 Silver chloride Inorganic materials 0.000 description 1
- 239000003929 acidic solution Substances 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 230000005611 electricity Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 229910021397 glassy carbon Inorganic materials 0.000 description 1
- 239000007770 graphite material Substances 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 229910010272 inorganic material Inorganic materials 0.000 description 1
- 239000011147 inorganic material Substances 0.000 description 1
- 239000003014 ion exchange membrane Substances 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 150000004706 metal oxides Chemical class 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 229910000510 noble metal Inorganic materials 0.000 description 1
- MUMZUERVLWJKNR-UHFFFAOYSA-N oxoplatinum Chemical compound [Pt]=O MUMZUERVLWJKNR-UHFFFAOYSA-N 0.000 description 1
- 150000002926 oxygen Chemical class 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Substances [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 1
- 229910003446 platinum oxide Inorganic materials 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- ADZWSOLPGZMUMY-UHFFFAOYSA-M silver bromide Chemical compound [Ag]Br ADZWSOLPGZMUMY-UHFFFAOYSA-M 0.000 description 1
- HKZLPVFGJNLROG-UHFFFAOYSA-M silver monochloride Chemical compound [Cl-].[Ag+] HKZLPVFGJNLROG-UHFFFAOYSA-M 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- BFKJFAAPBSQJPD-UHFFFAOYSA-N tetrafluoroethene Chemical group FC(F)=C(F)F BFKJFAAPBSQJPD-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25B—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES FOR THE PRODUCTION OF COMPOUNDS OR NON-METALS; APPARATUS THEREFOR
- C25B3/00—Electrolytic production of organic compounds
- C25B3/20—Processes
- C25B3/25—Reduction
Definitions
- Chloro and bromoacetic acids are the mono-, di- and tri-haloacetic acids of the formulas CH2ClCOOH CH2BrCOOH CHCl2COOH CHBr2COOH CCl3COOH CBr3COOH
- the partial dehalogenation of the triple and the double halogenated acetic acids is desirable or necessary, for example, if the intention is to obtain the monohalogenated acetic acids by chlorination or bromination of acetic acid in the highest possible yields.
- a current density of about 500 to 700 A / m2 is used.
- the electrolysis temperature is below 100 ° C.
- the yields of the desired partially or completely dehalogenated products should be between 95 and 100% of theory lie.
- the following mixture is electrolyzed:
- the electrolysis of the mixture was carried out according to the information in the example mentioned in the form of a 60% aqueous solution using magnetite cathodes and carbon anodes at an average voltage of 3.25 V and a current density of 500 to 600 A / m2 at 65 ° C up to the dehalogenation of the di- and trichloroacetic acids up to the monohalogen stage.
- the yield of monochloroacetic acid is given as almost quantitative.
- Example 4 the electrolysis is continued until complete dehalogenation - that is to say halogen-free acetic acid.
- the dehalogenation essential for this process is a reduction reaction taking place at the cathode.
- the following reaction equation can be given: CHCl2COOH + 2 H+ + 2 e ⁇ CH2ClCOOH + HCl
- the reaction of the aggressive haloacetic acids at the cathode has a considerable corrosive effect on the cathode material, as was also shown by our own electrolysis experiments using magnetite and lead cathodes. Corrosion is hardly serious on carbon cathodes.
- a disadvantage of all of the cathode materials mentioned here, however, is that increasing the current density leads to increasing hydrogen evolution at the cathode, and the electrodes are covered in a long-term test for 600 hours with a coating which makes cleaning the cathode necessary, which of course is economical the procedure significantly affected.
- the halogen ions formed on the cathode are discharged at the anode; in the case of chlorine ions: 2 Cl ⁇ ⁇ Cl2 + 2 e
- the anodically formed halogen can easily come into contact with the product dehalogenated at the cathode and "react" back to the starting product; e.g. CH2ClCOOH + Cl2 ⁇ CHCl2COOH + HCl This "back reaction” can be prevented by carrying out the electrolysis in divided electrolysis cells.
- the catholyte is an aqueous solution of dichloroacetic acid + HCl and / or H2SO4 with a conductivity above 0.01 Ohm Oh1 ⁇ cm ⁇ 1.
- Graphite, lead, lead alloys and titanium with a coating of oxides of platinum metals are mentioned as anode materials;
- An aqueous mineral acid solution serves as the anolyte, with oxygen acids being preferred as mineral acids because here there is no chlorine but only oxygen evolution: H2O ⁇ 1/2 O2 + 2 H+ + 2 e
- the ion exchange capacity in grams of dry weight of the exchanger is required for the membrane material resin indicated that are necessary to neutralize 1 gram equivalent of base.
- the exchange capacity should be 500 to 1500, preferably 500 to 1000, for membrane material with SO3H groups 500 to 1800, preferably 1000 to 1500.
- this object could be achieved by using such aqueous solutions of chloro- or bromoacetic acids as starting electrolysis solutions which still contain one or more salts of metals with a hydrogen overvoltage of at least 0.4 V (at a current density of 4000 A / m2) included dissolved.
- the subject of the invention is therefore a process for the dehalogenation of chloroacids and bromoacetic acids by electrolysis of aqueous solutions of these acids using carbon cathodes and anodes also of carbon or of other conventional electrode materials in undivided or divided (electrolysis) cells, which is characterized in that that the aqueous electrolysis solutions in the undivided cells and in the cathode compartment of the divided cells still contain one or more salts of metals with a hydrogen overvoltage of at least 0.4 V (at a current density of 4000 A / m2) dissolved.
- the salts of metals with a hydrogen overvoltage of at least 0.4 V are mainly the soluble salts of Cu, Ag, Au, Zn, Cd, Hg, Sn, Pb, Ti, Zr, Bi, V, Ta, Cr and / or Ni, preferably only the soluble Cu and Pb salts, in question.
- the most common anions of these salts are mainly Cl ⁇ , Br ⁇ , SO42 ⁇ , NO3 ⁇ and CH3OCO ⁇ .
- these anions cannot be combined in the same way with all of the above-mentioned metals, because in some cases this results in salts which are difficult to dissolve (such as AgCl and AgBr; here, the soluble salt is primarily AgNO3).
- the salts can be added directly to the electrolysis solution or, e.g. by adding oxides, carbonates etc. - in some cases also the metals themselves (if soluble) - can be generated in the solution.
- the salt concentration in the electrolyte of the undivided cell and in the catholyte of the divided cell is expediently set to about 0.1 to 5000 ppm, preferably to about 10 to 1000 ppm.
- This modification of the known methods ensures an extraordinary corrosion resistance of the electrodes, combined with the possibility of working at a current density that is about 10 times higher (up to about 8000 A / m2), without deposits forming on the electrodes even during prolonged continuous operation; the process is therefore extremely economical and progressive.
- Trichloric and dichloroacetic acid and tribromoic and dibromoacetic acid are preferably used as starting compounds for the process; the electrolysis is preferably carried out only up to the monohalogen stage (monochloro or monobromoacetic acid).
- aqueous solutions can be used as the electrolyte (in the undivided cell) or catholyte (in the divided cell) the starting haloacetic acids of all possible concentrations (approx. 1 to 95%) can be used.
- the solutions can also contain mineral acids (eg HCl, H2SO4 etc.) and must contain the content of certain metal salts according to the invention.
- the anolyte (in the divided cell) is preferably an aqueous mineral acid, in particular aqueous hydrochloric acid and sulfuric acid.
- carbon cathodes e.g. Electrode graphites, impregnated graphite materials and also glassy carbon.
- the metal on which the metal salt added according to the invention is based is deposited on the cathode, which leads to a change in the properties of the cathode.
- the cathodic current density can be increased to values of up to about 8000 A / m2, preferably up to about 6000 A / m2, without excessive hydrogen evolution and a progress of the dehalogenation reaction beyond the desired stage occurring as side reactions.
- the metal deposited on the cathode is repeatedly partially dissolved by the acidic solution surrounding the cathode and then deposited again, etc. There is no disruptive deposit formation on the cathode.
- the same material as for the cathode can be used as the anode material.
- other conventional electrode materials which, however, must be inert under the electrolysis conditions.
- a preferred such other common electrode material is titanium, coated with TiO2 and doped with a noble metal oxide such as e.g. Platinum oxide.
- Preferred anolyte liquids are aqueous mineral acids such as aqueous hydrochloric acid or aqueous sulfuric acid,
- aqueous hydrochloric acid is preferable if one works in divided cells and there are other possible uses for the anodically formed chlorine; otherwise the use of aqueous sulfuric acid is cheaper.
- the implementation in the divided cells is preferred.
- the same ion exchange membranes as those described in the aforementioned JP-A-54 (1979) -76521 are suitable for dividing the cells into the anode and cathode compartments; d.s. thus those made of perfluorinated polymers with carboxyl and / or sulfonic acid groups, preferably also with the ion exchange capacities specified in JP-A.
- diaphragms made of other perfluorinated polymers or inorganic materials that are stable in the electrolyte.
- the electrolysis temperature should be below 100 ° C; it is preferably between about 5 and 95 ° C., in particular between about 40 and 80 ° C.
- a method of operation in divided electrolysis cells with discontinuous execution of the cathode reaction and continuous operation of the anode reaction is particularly expedient. If the anolyte contains HCl, Cl ⁇ is constantly consumed by the anodic chlorine development, which can be compensated for by the continuous addition of gaseous HCl or aqueous hydrochloric acid.
- the electrolysis product is worked up in a known manner, for example by distillation.
- the metal salts remain behind here and can be returned to the process.
- the electrolytic cell used in all (invention and comparative) examples was a split (plate and frame) circulation cell.
- Electrodes Electrode graphite EH (from Sigri, Meitingen)
- Cation exchange membrane (R) Nafion 315 (from DuPont); it is a 2-layer membrane made from copolymers of perfluorosulfonylethoxy vinyl ether + tetrafluoroethylene. There is a layer with the equivalent weight 1300 on the cathode side and one with the equivalent weight 1100 on the anode side.
- Spacers polyethylene nets Flow: 500 l / h Temp.: 25 - 40 ° C Current density: 4000 A / m2 Terminal voltage: 8 - 4.8 V
- Anolyte conc. HCl, continuously supplemented by gaseous HCl
- the composition of the catholyte and the electrolysis result are shown in the following table:
- Electrodes Electrode graphite EH (from Sigri, Meitingen)
- Cation exchange membrane (R) Nafion 324 (from DuPont) it is a 2-layer membrane of the same composition as Nafion 315, only with slightly thinner layers.
- Spacers polyethylene nets Flow: 1.6 m3 / h Temp .: 25-60 ° C
- Anolyte conc.
- Anode Electrode graphite EH (from Sigri, Meitingen)
- Cathode completely and densely coated stainless steel with magnetite
- Cation exchange membrane (R) Nafion 324 (from DuPont)
- Spacers polyethylene nets Flow: 500 l / h Temp .: 39 ° C
- Anolyte conc. HCl, continuously supplemented by gaseous HCl It became a catholyte with the composition 1.15 kg of monochloroacetic acid 1.28 kg dichloroacetic acid 0.24 kg acetic acid 1.43 kg water electrolyzed at a current density of 2000 A / m2.
- the terminal voltage was 3.2 V.
- the proportion of the current that was used for the development of hydrogen was 14.3%.
- the hydrogen evolution decreased briefly, but then rose again.
- 270 Ah 28% of the electricity was used for hydrogen development, after 350 Ah the value was 45% and then rose to approx. 80%.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- Materials Engineering (AREA)
- Metallurgy (AREA)
- Electrolytic Production Of Non-Metals, Compounds, Apparatuses Therefor (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Electrodes For Compound Or Non-Metal Manufacture (AREA)
- Transition And Organic Metals Composition Catalysts For Addition Polymerization (AREA)
Abstract
Description
- Chlor- und Brom-Essigsäuren sind die Mono-, Di- und Tri-halogenessigsäuren der Formeln
CH₂ClCOOH CH₂BrCOOH
CHCl₂COOH CHBr₂COOH
CCl₃COOH CBr₃COOH
Für manche Zwecke ist es erforderlich, die bei bestimmten Prozessen anfallenden Chlor- und Brom-Essigsäuren vollständig oder teilweise zu enthalogenieren. Die teilweise Enthalogenierung der 3-fach und der 2-fach halogenierten Essigsäuren ist z.B. dann wünschenswert bzw. notwendig, wenn beabsichtigt ist, die monohalogenierten Essigsäuren durch Chlorierung bzw. Bromierung von Essigsäure in möglichst hohen Ausbeuten zu erhalten. Bei der Chlorierung und Bromierung der Essigsäure entstehen nämlich - auch wenn man nicht mehr Halogen als zur Monohalogenierung notwendig verwendet - immer auch mehr oder weniger bedeutende Mengen der Di- sowie gegebenenfalls auch noch der Tri-halogenessigsäure, was natürlich die Ausbeute der gewünschten Monohalogenverbindung beeinträchtigt. - Es wurden daher bereits verschiedene Verfahren entwickelt, um die 2- und 3-fach halogenierten Essigsäuren zu enthalogenieren und die Enthalogenierung auch bei der Monohalogenstufe anzuhalten. Nach dem z.B. in der DE-B 848 807 beschriebenen Verfahren erfolgt diese Enthalogenierung auf elektrochemischem Weg durch Elektrolyse der entsprechenden Mischungen oder Lösungen in ungeteilten Elektrolysezellen. Als Kathodenmaterialien werden Kohle, Acheson-Graphit, Blei und Magnetit, als Anodenmaterialien Kohle und Magnetit namentlich genannt. Die Gegenwart indifferenter Stoffe oder anorganischer Verunreinigungen der Ausgangs-Halogenessig säuren soll sich hier nicht störend bemerkbar machen.
- Nach den Beispielen wird bei einer Stromdichte von etwa 500 bis 700 A/m² gearbeitet. Die Elektrolysetemperatur liegt unterhalb 100°C.
- Die Stoffausbeuten an den gewünschten teilweise- oder auch vollständig - enthalogenierten Produkten sollen zwischen 95 und 100 % d.Th. liegen.
- Etwa nach Beispiel 2 wird folgendes Gemisch elektrolysiert:

unter Verwendung von Magnetit-Kathoden und Kohle-Anoden bei einer Spannung von im Mittel 3,25 V und einer Stromdichte von 500 bis 600 A/m²
bei 65°C
bis zur Enthalogenierung der Di- und Tri-Chloressigsäuren bis zur Monohalogenstufe. Die Ausbeute an Monochloressigsäure wird als nahezu quantitativ angegeben. - In Beispiel 4 wird die Elektrolyse noch bis zur vollständigen Enthalogenierung - d.i. bis zur halogenfreien Essigsäure - weitergeführt.
- Die für diesen Prozeß wesentliche Enthalogenierung ist eine an der Kathode stattfindende Reduktionsreaktion. Etwa für die teilweise Enthalogenierung der Dichloressigsäure bis zur Stufe der Monochloressigsäure kann folgende Reaktionsgleichung angegeben werden:
CHCl₂COOH + 2 H⁺ + 2 e → CH₂ClCOOH + HCl
Die Reaktion der aggressiven Halogenessigsäuren an der Kathode wirkt auf das Kathodenmaterial in erheblichem Maß korrodierend, wie auch durch eigene Elektrolyseversuche unter Verwendung von Magnetit- und von Blei-Kathoden gezeigt werden konnte. An Kohle-Kathoden ist die Korrosion kaum gravierend. Nachteilig für sämtliche hier genannten Kathodenmaterialien ist jedoch, daß bei einer Erhöhung der Stromdichte in zunehmenden Maß Wasserstoffentwicklung an der Kathode erfolgt, und die Elektroden im Dauerversuch über 600 h mit einem Belag bedeckt werden, der die Reinigung der Kathode erforderlich macht, was natürlich die Wirtschaftlichkeit des Verfahrens erheblich beeinträchtigt. - An der Anode findet zumindest teilweise die Entladung der an der Kathode gebildeten Halogenionen statt; im Falle von Chlorionen also:
2 Cl⁻ → Cl₂ + 2 e
In den ungeteilten Zellen gemäß der vorerwähnten DE-B kann das anodisch gebildete Halogen mit dem an der Kathode enthalogenierten Produkt leicht in Kontakt kommen und wieder zum Ausgangsprodukt "zurückreagieren"; z.B.
CH₂ClCOOH + Cl₂ → CHCl₂COOH + HCl
Diese "Rückreaktion" kann durch die Durchführung der Elektrolyse in geteilten Elektrolysezellen verhindert werden. Die zum Zeitpunkt der Anmeldung der vorerwähnten DE-B (im Jahr 1942) bekannten Diaphragmen-Materialien (für die Teilung der Zellen in Kathoden- und Anodenraum) hielten je doch der Einwirkung der aggressiven Halogenessigsäuren und des mindestens ebenso aggressiven Halogens insbesondere in der Wärme nicht lange Stand. Deswegen werden in der genannten DE-B auch geteilte Elektrolysezellen als ungeeignet für die elektrolytische Enthalogenierung von Halogenessigsäuren beurteilt. - Mit der Entwicklung der chemisch und thermisch außerordentlich stabilen Membranmaterialien aus perfluorierten Polymeren in neuerer Zeit ist jedoch auch die Durchführung der Elektrolyse mit aggressiven Reagentien in geteilten Zellen möglich geworden.
- Ein Verfahren zur elektrochemischen Enthalogenierung von Dichloressigsäure bis zur Stufe der Monochloressigsäure in geteilten Elektrolysezellen ist in der JP-A-54 (1979)-76521 beschrieben; als Membranmaterialien werden hier speziell Kationenaustauschermembranen aus perfluorierten Polymeren mit noch COOH- oder SO₃H-Gruppen am Polymerengerüst verwendet.
- Bei diesem Verfahren dienen Blei oder Bleilegierungen als Kathoden-Werkstoffe; der Katholyt ist eine wässrige Lösung von Dichloressigsäure + HCl und/oder H₂SO₄ mit einer Leitfähigkeit über 0,01 Ohm⁻¹ · cm⁻¹.
- Als Anodenmaterialien sind Graphit, Blei, Bleilegierungen sowie Titan mit einem Überzug von Oxiden der Platinmetalle genannt; als Anolyt dient eine wässrige Mineralsäurelösung, wobei Sauerstoffsäuren als Mineralsäuren bevorzugt sind, weil hier keine Chlor-, sondern nur Sauerstoffentwicklung erfolgt:
H₂O → 1/2 O₂ + 2 H⁺ + 2 e
Für das Membranmaterial wird die erforderlich Ionenaustauschkapazität in Gramm Trockengewicht des Austauscher harzes angegeben, die nötig sind, um 1 Grammäquivalent Base zu neutralisieren. Für Membranmaterial mit Carboxylgruppen soll die Austauschkapazität zu 500 bis 1500, vorzugsweise 500 bis 1000,
für Membranmaterial mit SO₃H-Gruppen 500 bis 1800, vorzugsweise 1000 bis 1500, betragen. - Die Stromdichten bewegen sich in ähnlichen Größenordnungen wie diejenigen des Verfahrens der vorher erwähnten DE-B 848 807. Bei einer Konzentration der Dichloressigsäure unter 25 % soll die Stromdichte unter 10 A/dm² = 1000 A/m²,
bei einer Dichloressigsäurekonzentration unter 15 % unterhalb 800 A/m² und
bei einer Dichloressigsäurekonzentration unter 10 % unterhalb 400 A/m² liegen. - Selbst die hier als Kathoden bevorzugten reinen Bleikathoden unterliegen noch einer erheblichen Korrosion. Bei der Elektrolyse mit einer Kathode aus 99,99 %igem Blei und einer Elektrodenfläche von 1 dm² sowie einer Stromdichte von 4 A/dm² = 400 A/m² soll in 4 Stunden ein Gewichtsverlust der Kathode von 59,6 mg eingetreten sein.
- Für verschiedene Bleilegierungen wird unter den gleichen Bedingungen folgender Gewichtsverlust angegeben:
Pb + 4 % Sn: 62,3 mg
Pb + 6 % Sn: 64 mg
Pb + 1,8 % Ag: 112,4 mg
Nach den Beispielen liegen die Stromausbeuten durchweg um 95 % und darüber. - Obwohl die bekannten elektrochemischen Verfahren zur teilweisen oder vollständigen Enthalogenierung von Chlor- und Bromessigsäuren verschiedene Vorteile besitzen, sind sie doch insbesondere hinsichtlich der Korrosionsbeständigkeit der Kathodenmaterialien und der relativ niedrigen Stromdichten noch verbesserungsbedürftig; es bestand daher die Aufgabe, die bekannten Verfahren noch vor allem bezüglich der Kathodenmaterialien und der Stromdichten zu verbessern und die Verfahren damit noch wirtschaftlicher zu machen.
- Diese Aufgabe konnte erfindungsgemäß dadurch gelöst werden, daß man als Ausgangs-Elektrolyselösungen solche wässrigen Lösungen der Chlor- bzw. Bromessigsäuren verwendet, die noch ein oder mehrere Salze von Metallen mit einer Wasserstoffüberspannung von mindestens 0,4 V (bei einer Stromdichte von 4000 A/m²) gelöst enthalten.
- Erfindungsgegenstand ist daher ein Verfahren zur Enthalogenierung von Chlor- und von Bromessigsäuren durch Elektrolyse wässriger Lösungen dieser Säuren unter Verwendung von Kohlenstoffkathoden und von Anoden ebenfalls aus Kohlenstoff oder aus anderen üblichen Elektrodenmaterialien in ungeteilten oder in geteilten (Elektrolyse-)Zellen, das dadurch gekennzeichnet ist, daß die wässrigen Elektrolyselösungen in den ungeteilten Zellen sowie im Kathodenraum der geteilten Zellen noch ein oder mehrere Salze von Metallen mit einer Wasserstoffüberspannung von mindestens 0,4 V (bei einer Stromdichte von 4000 A/m²) gelöst enthalten.
- Als Salze von Metallen mit einer Wasserstoffüberspannung von mindestens 0,4 V (bei einer Stromdichte von 4000A/m²) kommen hauptsächlich die löslichen Salze von Cu, Ag, Au, Zn, Cd, Hg, Sn, Pb, Ti, Zr, Bi, V, Ta, Cr und/oder Ni, vorzugsweise nur die löslichen Cu- und Pb-Salze, in Frage. Die gängigsten Anionen dieser Salze sind hauptsächlich Cl⁻, Br⁻, SO₄²⁻, NO₃⁻ und CH₃OCO⁻. Diese Anionen können aber nicht in gleicher Weise mit allen vorerwähnten Metallen kombiniert werden, weil hier in einigen Fällen schwer lösliche Salze resultieren (wie z.B. AgCl und AgBr; hier kommt als lösliches Salz in erster Linie AgNO₃ in Frage).
- Die Salze können der Elektrolyselösung direkt zugesetzt oder auch z.B. durch Zugabe von Oxiden, Carbonaten etc. - in eienigen Fällen auch der Metalle selbst (sofern löslich) - in der Lösung erzeugt werden.
- Die Salzkonzentration im Elektrolyten der ungeteilten Zelle sowie im Katholyten der geteilten Zelle wird zweckmäßig auf etwa 0,1 bis 5000 ppm, vorzugsweise auf etwa 10 bis 1000 ppm, eingestellt.
- Durch diese Abänderung der bekannten Verfahren ist eine außerordentliche Korrosionsbeständigkeit der Elektroden, verbunden mit der Möglichkeit des Arbeitens bei um den Faktor etwa 10 höheren Stromdichten (bis etwa 8000 A/m²) gewährleistet, ohne daß sich auch bei längerem Dauerbetrieb Beläge auf den Elektroden bilden; das Verfahren ist daher außerordentlich wirtschaftlich und fortschrittlich.
- Es war nach dem Stand der Technik in keiner Weise zu erwarten, daß durch die Kombination von Kohlekathoden und der Gegenwart bestimmter Metallsalze in der Elektrolyt- bzw. Katholyt-Lösung eine derartige Erhöhung der Wirtschaftlichkeit des Verfahrens - insbesondere durch die Möglichkeit des Arbeitens mit höheren Stromdichten ohne die Bildung von Belägen auf den Elektroden - erzielt wird.
- Als Ausgangsverbindungen für das Verfahren werden vorzugsweise Trichlor- und Dichloressigsäure sowie Tribrom- und Dibromessigsäure, insbesondere nur Trichlor- und/oder Dichloressigsäure verwendet; die Elektrolyse wird hier vorzugsweise nur bis zur Monohalogenstufe (Monochlor- bzw. Monobromessigsäure) geführt.
- Die Fortführung der Elektrolyse bis zur (völlig enthalogenierten) Essigsäure ist natürlich möglich, aber nicht bevorzugt.
- Als Elektrolyt (in der ungeteilten Zelle) bzw. Katholyt (in der geteilten Zelle) können im Prinzip wässrige Lösungen der Ausgangs-Halogenessigsäuren aller möglichen Konzentrationen (ca. 1 bis 95 %) verwendet werden. Die Lösungen können auch noch Mineralsäuren (z.B. HCl, H₂SO₄ etc.) und müssen den erfindungsgemäßen Gehalt an bestimmten Metallsalzen enthalten.
- Der Anolyt (in der geteilten Zelle) ist bevorzugt eine wässrige Mineralsäure, insbesondere wässrige Salzsäure und Schwefelsäure.
- Als Kohlenstoffkathoden kommen im Prinzip alle möglichen Kohle-Elektrodenmaterialien in Frage wie z.B. Elektrodengraphite, imprägnierte Graphitwerkstoffe und auch glasartiger Kohlenstoff.
- Während der Elektrolyse scheidet sich auf der Kathode das dem erfindungsgemäß zugesetzten Metallsalz zugrundeliegende Metall ab, was zu einer Veränderung der Eigenschaften der Kathode führt. Dadurch kann die kathodische Stromdichte auf Werte bis zu etwa 8000 A/m², vorzugsweise bis zu etwa 6000 A/m², erhöht werden, ohne daß als Nebenreaktionen zu starke Wasserstoffentwicklung und ein Fortgang der Enthalogenierungsreaktion über die gewünschte Stufe hinaus auftreten. Das auf der Kathode abgeschiedene Metall wird von der die Kathode umgebenden sauren Lösung immer wieder teilweise aufgelöst und dann wieder abgeschieden usw. Eine störende Belagbildung auf der Kathode findet nicht statt.
- Als Anodenmaterial kann das gleiche Material wie für die Kathode verwendet werden. Darüberhinaus ist auch der Einsatz anderer üblicher Elektrodenmaterialien, die jedoch unter den Elektrolysebedinungen inert sein müssen, möglich. Ein bevorzugtes derartiges anderes übliches Elektrodenmaterial ist Titan, beschichtet mit TiO₂ und dotiert mit einem Edelmetalloxid wie z.B. Platinoxid.
- Bevorzugte Anolyt-Flüssigkeiten sind wässrige Mineralsäuren wie z.B. wässrige Salzsäure oder wässrige Schwefelsäure, Hierbei ist der Einsatz der wässrigen Salzsäure dann vorzuziehen, wenn man in geteilten Zellen arbeitet und für das anodisch gebildete Chlor anderweitige Verwendungsmöglichkeiten existieren; andernfalls ist der Einsatz der wässrigen Schwefelsäure günstiger.
- Von den beiden Möglichkeiten der Elektrolysezellen, in denen das erfindungsgemäße Verfahren ausgeführt werden kann - ungeteilte und geteilte Zellen - ist die Durchführung in den geteilten Zellen bevorzugt. Zur Teilung der Zellen in Anoden- und Kathodenraum kommen hier die gleichen Ionenaustauschermembranen in Frage wie sie auch in der vorerwähnten JP-A-54 (1979)-76521 beschrieben sind; d.s. also solche aus perfluorierten Polymeren mit Carboxyl- und/oder Sulfonsäuregruppen, vorzugsweise auch mit den in der JP-A angegebenen Ionenaustauschkapazitäten. Auch die Verwendung von im Elektrolyten stabilen Diaphragmen aus anderen perfluorierten Polymeren oder anorganischen Werkstoffen ist im Prinzip möglich.
- Die Elektrolysetemperatur soll unter 100°C liegen; vorzugsweise liegt sie zwischen etwa 5 und 95°C, insbesondere zwischen etwa 40 und 80°C.
- Es ist möglich, die Elektrolyse sowohl kontinuierlich als auch diskontinuierlich durchzuführen. Besonders zweckmäßig ist eine Arbeitsweise in geteilten Elektrolysezellen mit diskontinuierlicher Ausführung der Kathodenreaktion und kontinuierlichem Betrieb der Anodenreaktion. Wenn der Anolyt HCl enthält, wird durch die anodische Chlorentwicklung ständig Cl⁻ verbraucht, was durch laufende Ergänzung von gasförmigem HCl oder von wässriger Salzsäure auszugleichen ist.
- Die Aufarbeitung des Elektrolyseproduktes erfolgt auf bekannte Weise, z.B. durch Destillation. Die Metallsalze bleiben hier im Rückstand und können wieder in den Prozeß zurückgeführt werden.
- Die Erfindung wird nun durch die folgenden Beispiele näher erläutert. Nach den (Erfindungs-)Beispielen A folgen noch einige Vergleichsbeispiele B, aus denen hervorgeht, daß an Magnetitkathoden (anstelle von Kohlenstoffkathoden) auch in Gegenwart etwa eines Bleisalzes in der Elektrolytlösung, nicht unerhebliche Korrosion und bei höheren Stromdichten auch beträchtliche Wasserstoffentwicklung erfolgt. Ein weiteres Vergleichsbeispiel mit einer Kohlenstoffkathode, aber ohne den erfindungsgemäßen Zusatz eines Metallsalzes zur Elektrolytlösung, zeigt, daß hier bereits bei nicht zu hohen Stromdichten in erheblichem Ausmaß Wasserstoff gebildet wird; setzt man der Elektrolytlösung dagegen noch etwa ein Bleisalz zu, so unterbleibt die Wasserstoffentwicklung und die Stromdichte kann erhöht werden.
- Die in sämtlichen (Erfindungs- und Vergleichs-) Beispielen verwendte Elektrolysezelle war eine geteilte (Platten- und Rahmen-) Umlaufzellen.
- Umlaufzelle mit 0,02 m² Elektrodenfläche, Elektrodenabstand 4 mm
Elektroden: Elektrodengraphit EH (der Firma Sigri, Meitingen)
Kationenautauschermembran: (R)Nafion 315 (der Firma DuPont); es handelt sich um eine 2-Schichtenmembran aus Copolymerisaten aus Perfluorsulfonylethoxyvinylether + Tetrafluorethylen. Auf der Kathodenseite befindet sich eine Schicht mit dem Äquivalentgewicht 1300, auf der Anodenseite eine solche mit dem Äquivalentgewicht 1100.
Abstandhalter: Polyethylennetze
Durchfluß : 500 l/h
Temp. : 25 - 40°C
Stromdichte : 4000 A/m²
Klemmenspannung: 8 - 4,8 V
Anolyt : konz. HCl, kontinuierlich ergänzt durch gasförmige HCl
Die Zusammensetzung des Katholyten und das Elektrolyseergebnis sind aus der folgenden Tabelle ersichtlich: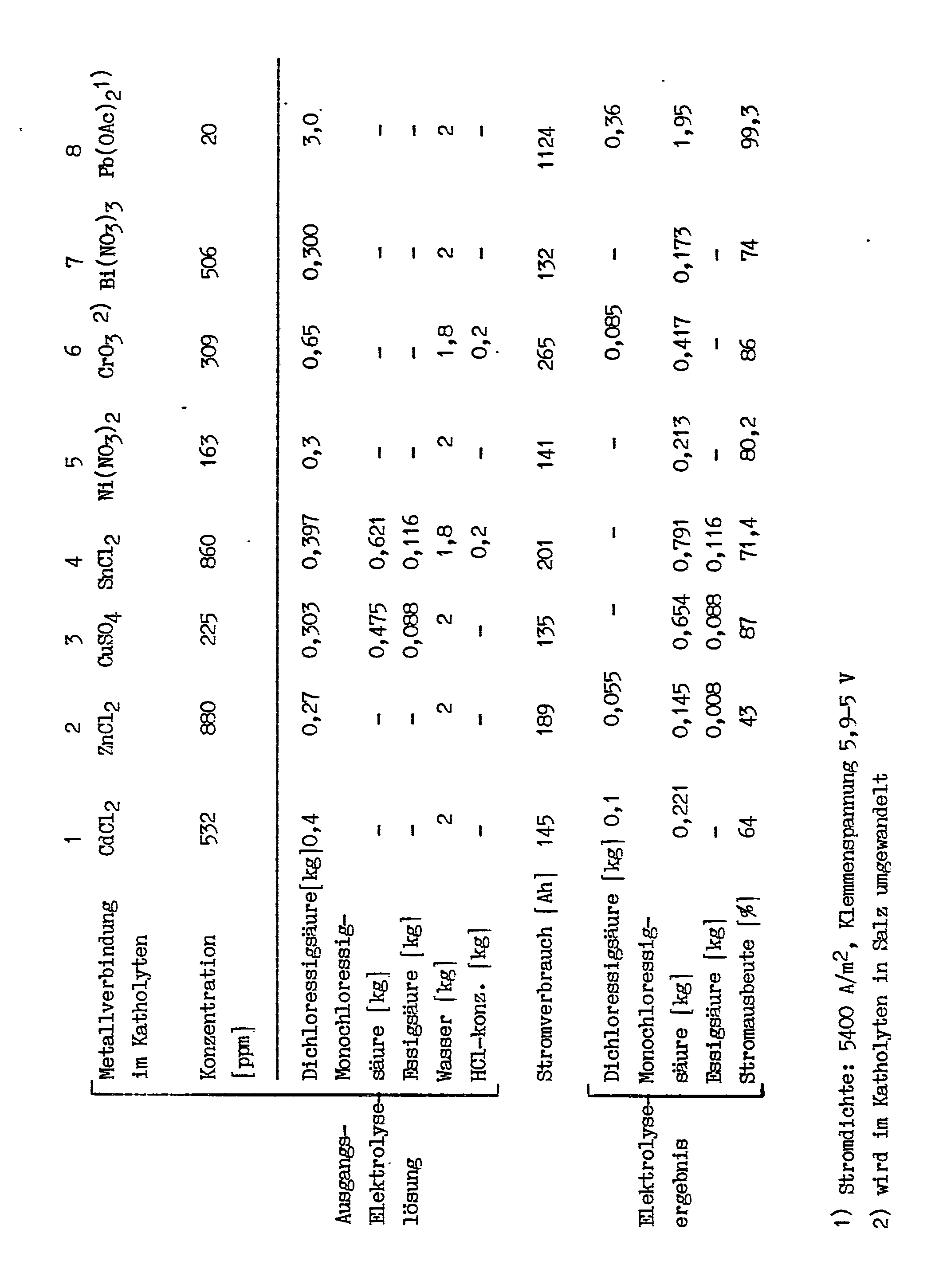
- Umlaufzelle mit 0,25 m² Elektrodenfläche, Elektrodenabstand 4 mm
Elektroden: Elektrodengraphit EH (der Firma Sigri, Meitingen)
Kationenaustauschermembran: (R)Nafion 324 (der Firma DuPont) es handelt sich um eine 2-Schichtenmembran der gleichen Zusammensetzung wie Nafion 315, lediglich mit etwas dünneren Schichten.
Abstandhalter: Polyethylennetze
Durchfluß: 1,6 m³/h
Temp.: 25-60°C
Stromdichte: 4000 A/m²
Klemmenspannung: 6-4,5 V
Anolyt: konz. HCl, kontinuierlich ergänzt durch gasförmige HCl
Ausgangskatholyt:
9,03 kg Dichloressigsäure
14,29 kg Monochloressigsäure
3,18 kg Essigsäure
13,20 kg Wasser
4 g CuSO₄·6H₂O (≙ 25 ppm Cu²⁺)
Elektrolyseergebnis:
20,79 kg Monochloressigsäure
0,15 kg Dichloressigsäure
3,18 kg Essigsäure
17,2 kg Wasser
2,52 kg HCl
Stromverbrauch: 5361 Ah
Stromausbeute: 68,2% - Umlaufzelle mit 0,02m² Elektrodenfläche, Elektrodenabstand 6 mm
Anode: Elektrodengraphit EH (der Firma Sigri, Meitingen)
Kathode: mit Magnetit vollständig und dicht beschichteter Edelstahl
Kationenaustauschermembran: (R)Nafion 324 (der Firma DuPont)
Abstandhalter: Polyethylennetze
Durchfluß: 500 l/h
Temp.: 39°C
Anolyt: konz. HCl, kontinuierlich ergänzt durch gasförmige HCl
Es wurde ein Katholyt mit der Zusammensetzung
1,15 kg Monochloressigsäure
1,28 kg Dichloressigsäure
0,24 kg Essigsäure
1,43 kg Wasser
bei einer Stromdichte von 2000 A/m² elektrolysiert. Die Klemmenspannung betrug 3,2 V. Der Anteil des Stroms, der für die Entwicklung von Wasserstoff verbraucht wurde, lag bei 14,3%.
Nach der Zugabe von 0,75 g Pb(OAc)₂·2 H₂O (100 ppm Pb²⁺) ging die Wasserstoffentwicklung kurzzeitig zurück, stieg dann aber wieder an.
Nach 270 Ah wurden 28% des Stroms für Wasserstoffentwicklung verbraucht, nach 350 Ah lag der Wert bei 45% und stieg dann weiter auf ca. 80%.
Nach einem Ladungsverbrauch von 752 Ah erhielt man einen Elektrolyten mit der Zusammensetzung:
1,77 kg Monochloressigsäure
0,42 kg Dichloressigsäure
0,27 kg Essigsäure
1,93 kg Wasser
0,24 kg HCl
0,0105 kg Eisen als Fe³⁺/Fe²⁺ (aus dem Magnetit)
0,4·10⁻³ kg Blei als Pb²⁺
Die Stromausbeute für diese geringfügige Abreicherung der Dichloressigsäure betrug nur 44%. An der Magnetitschicht der Kathode wurden schwere Korrosionsschäden festgestellt. Die Korrosionsrate betrug 14 mgFe/Ah. - Unter den in den Erfindungsbeispielen (A) 1 - 8 beschriebenen Bedingungen, aber ohne den Zusatz eines Metallsalzes, wurde ein Katholyt mit der Zusammensetzung
5,72 kg Monochloressigsäure
1,98 kg Dichloressigsäure
2 kg Essigsäure
4,4 kg H₂O·HCl
bei einer Stromdichte von 1250 A/m² elektrolysiert. Die Klemmenspannung betrug 3,9 V, Nach einem Stromverbrauch von 1104 Ah stieg der Anteil des Stroms, der für die Entwicklung von Wasserstoff verbaucht wurde auf 49 %. - Nach Zugabe von 10 g Pb(NO₃)₂ (≙ 400 ppm Pb²⁺) zum Katholyten fand keine Wasserstoffentwicklung mehr statt. Die Stromdichte konnte auf 4000 A/m² erhöht werden (Klemmenspannung 4,1 V; Temperatur 52°C). Die Nebenreaktion der Wasserstoffentwicklung setzte bei einer Dichloressigsäure-Konzentration von 3 % wieder ein. Die Stromausbeute für die Reduzierung des Dichloressigsäure-Anteils auf 0,15 kg betrug 97,2 %.
Claims (6)
dadurch gekennzeichnet, daß die wässrigen Elektrolyselösungen in den ungeteilten Zellen sowie im Kathodenraum der geteilten Zellen noch ein oder mehrere Salze von Metallen mit einer Wasserstoffüberspannung von mindestens 0,4 V (bei einer Stromdichte von 4000 A/m²) gelöst enthalten.
und daß man die Elektrlyse nur bis zur Monohalogenstufe führt.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AT87102846T ATE48657T1 (de) | 1986-03-07 | 1987-02-27 | Verfahren zur enthalogenierung von chlor- und von bromessigsaeuren. |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19863607446 DE3607446A1 (de) | 1986-03-07 | 1986-03-07 | Verfahren zur enthalogenierung von chlor- und von bromessigsaeuren |
| DE3607446 | 1986-03-07 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0241685A1 true EP0241685A1 (de) | 1987-10-21 |
| EP0241685B1 EP0241685B1 (de) | 1989-12-13 |
Family
ID=6295698
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP87102846A Expired EP0241685B1 (de) | 1986-03-07 | 1987-02-27 | Verfahren zur Enthalogenierung von Chlor- und von Bromessigsäuren |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US4707226A (de) |
| EP (1) | EP0241685B1 (de) |
| JP (1) | JPS62214189A (de) |
| AT (1) | ATE48657T1 (de) |
| AU (1) | AU583980B2 (de) |
| BR (1) | BR8701046A (de) |
| CA (1) | CA1313362C (de) |
| DD (1) | DD258424A5 (de) |
| DE (2) | DE3607446A1 (de) |
| FI (1) | FI79863C (de) |
| HU (1) | HUT43023A (de) |
| IL (1) | IL81785A (de) |
| MX (1) | MX168882B (de) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0334796A1 (de) * | 1988-03-19 | 1989-09-27 | Hoechst Aktiengesellschaft | Verfahren zur Herstellung von ungesättigten halogenierten Kohlenwasserstoffen |
| EP0457320A1 (de) * | 1990-05-18 | 1991-11-21 | Hoechst Aktiengesellschaft | Verfahren zur teilweisen elektrolytischen Enthalogenierung von Di-und Trichloressigsäure sowie Elektrolyselösung |
| WO1993017151A1 (de) * | 1992-02-22 | 1993-09-02 | Hoechst Aktiengesellschaft | Elektrochemisches verfahren zur herstellung von glyoxylsäure |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3731914A1 (de) * | 1987-09-23 | 1989-04-06 | Hoechst Ag | Verfahren zur herstellung von fluorierten acrylsaeuren und ihren derivaten |
| DE3802745A1 (de) * | 1988-01-30 | 1989-08-03 | Hoechst Ag | Verfahren zur herstellung von fluormalonsaeure und ihren derivaten |
| US5348629A (en) * | 1989-11-17 | 1994-09-20 | Khudenko Boris M | Method and apparatus for electrolytic processing of materials |
| DE4217338C2 (de) * | 1992-05-26 | 1994-09-01 | Hoechst Ag | Elektrochemisches Verfahren zur Reduktion von Oxalsäure zu Glyoxylsäure |
| JP2003205221A (ja) * | 2001-11-12 | 2003-07-22 | Canon Inc | 有機塩素化合物の処理方法及びそれに用いる装置、土壌の修復方法及びそれに用いる装置 |
| US8236565B2 (en) * | 2006-05-26 | 2012-08-07 | Dh Technologies Development Pte. Ltd. | Tagging reagents and methods for hydroxylated compounds |
| CN114409025A (zh) * | 2021-12-17 | 2022-04-29 | 浙江工业大学 | 一种维生素b12修饰电极催化电解三溴乙酸脱溴的方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE848807C (de) * | 1942-03-12 | 1952-09-08 | Lech Chemie Gersthofen | Verfahren zur elektrolytischen Reduktion von Chlor- oder Bromessigsaeuren |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR1471108A (fr) * | 1965-03-13 | 1967-02-24 | Ajinomoto Kk | Méthode électrolytique de conversion des groupes polychlorométhyle de composés organiques en groupe monochlorométhyle |
| JPS5476521A (en) * | 1977-11-30 | 1979-06-19 | Chlorine Eng Corp Ltd | Preparation of monochloroacetic acid |
| US4588484A (en) * | 1985-02-28 | 1986-05-13 | Eli Lilly And Company | Electrochemical reduction of 3-chlorobenzo[b]thiophenes |
| US4585533A (en) * | 1985-04-19 | 1986-04-29 | Exxon Research And Engineering Co. | Removal of halogen from polyhalogenated compounds by electrolysis |
-
1986
- 1986-03-07 DE DE19863607446 patent/DE3607446A1/de active Granted
-
1987
- 1987-02-27 AT AT87102846T patent/ATE48657T1/de not_active IP Right Cessation
- 1987-02-27 EP EP87102846A patent/EP0241685B1/de not_active Expired
- 1987-02-27 DE DE8787102846T patent/DE3761151D1/de not_active Expired - Fee Related
- 1987-03-03 DD DD87300407A patent/DD258424A5/de not_active IP Right Cessation
- 1987-03-05 FI FI870972A patent/FI79863C/fi not_active IP Right Cessation
- 1987-03-05 HU HU87940A patent/HUT43023A/hu unknown
- 1987-03-05 IL IL81785A patent/IL81785A/xx unknown
- 1987-03-05 US US07/021,991 patent/US4707226A/en not_active Expired - Fee Related
- 1987-03-06 BR BR8701046A patent/BR8701046A/pt unknown
- 1987-03-06 AU AU69778/87A patent/AU583980B2/en not_active Ceased
- 1987-03-06 CA CA000531325A patent/CA1313362C/en not_active Expired - Fee Related
- 1987-03-06 MX MX005489A patent/MX168882B/es unknown
- 1987-03-06 JP JP62050439A patent/JPS62214189A/ja active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE848807C (de) * | 1942-03-12 | 1952-09-08 | Lech Chemie Gersthofen | Verfahren zur elektrolytischen Reduktion von Chlor- oder Bromessigsaeuren |
Non-Patent Citations (1)
| Title |
|---|
| CHEMICAL ABSTRACTS, Band 98, 1983, Seite 492, Zusammenfassung Nr. 24482h, Columbus, Ohio, US; G. HORANYI: "Electrocatalytic reduction of some halogenated derivatives of methane and acetic acid at a platinized platinum electrode in acid medium", & J. ELECTROANAL. CHEM. INTERFACIAL ELECTROCHEM. 1982, 140(2), 329-46 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0334796A1 (de) * | 1988-03-19 | 1989-09-27 | Hoechst Aktiengesellschaft | Verfahren zur Herstellung von ungesättigten halogenierten Kohlenwasserstoffen |
| US5026460A (en) * | 1988-03-19 | 1991-06-25 | Hoechst Aktiengesellschaft | Process for the preparation of unsaturated halogenated hydrocabons |
| EP0457320A1 (de) * | 1990-05-18 | 1991-11-21 | Hoechst Aktiengesellschaft | Verfahren zur teilweisen elektrolytischen Enthalogenierung von Di-und Trichloressigsäure sowie Elektrolyselösung |
| US5362367A (en) * | 1990-05-18 | 1994-11-08 | Hoechst Aktiengesellschaft | Partial electrolytic dehalogenation of dichloroacetic and trichloroacetic acid and electrolysis solution |
| WO1993017151A1 (de) * | 1992-02-22 | 1993-09-02 | Hoechst Aktiengesellschaft | Elektrochemisches verfahren zur herstellung von glyoxylsäure |
| US5474658A (en) * | 1992-02-22 | 1995-12-12 | Hoechst Ag | Electrochemical process for preparing glyoxylic acid |
Also Published As
| Publication number | Publication date |
|---|---|
| JPS62214189A (ja) | 1987-09-19 |
| DE3607446A1 (de) | 1987-09-10 |
| CA1313362C (en) | 1993-02-02 |
| IL81785A (en) | 1990-03-19 |
| FI870972A0 (fi) | 1987-03-05 |
| ATE48657T1 (de) | 1989-12-15 |
| MX168882B (es) | 1993-06-14 |
| AU583980B2 (en) | 1989-05-11 |
| IL81785A0 (en) | 1987-10-20 |
| HUT43023A (en) | 1987-09-28 |
| DE3607446C2 (de) | 1987-12-03 |
| DE3761151D1 (de) | 1990-01-18 |
| FI79863C (fi) | 1990-03-12 |
| BR8701046A (pt) | 1988-01-05 |
| AU6977887A (en) | 1987-09-10 |
| US4707226A (en) | 1987-11-17 |
| DD258424A5 (de) | 1988-07-20 |
| FI870972L (fi) | 1987-09-08 |
| FI79863B (fi) | 1989-11-30 |
| EP0241685B1 (de) | 1989-12-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69707320T2 (de) | Verfahren zur Elektrolyse von wässrigen Lösungen von Salzsäure | |
| EP2765223B1 (de) | Elektrokatalysator, Elektrodenbeschichtung und Elektrode zur Herstellung von Chlor | |
| EP0627020B1 (de) | Elektrochemisches verfahren zur herstellung von glyoxylsäure | |
| EP0457320B1 (de) | Verfahren zur teilweisen elektrolytischen Enthalogenierung von Di-und Trichloressigsäure sowie Elektrolyselösung | |
| DE2620589C3 (de) | Aktivierte Kathode zur Verwendung bei der Elektrolyse wäßriger Lösungen | |
| DE2936033C2 (de) | ||
| DE3704915A1 (de) | Elektrochemisches verfahren zum austausch von halogenatomen in einer organischen verbindung | |
| EP0241685B1 (de) | Verfahren zur Enthalogenierung von Chlor- und von Bromessigsäuren | |
| DE2949379C2 (de) | Verfahren zur Herstellung von Glyoxalsäure durch Oxidation von Glyoxal | |
| DE2251262C2 (de) | Verfahren zur kontinuierlichen Aluminiumherstellung durch Elektrolyse von Aluminiumchlorid | |
| DE2909593C2 (de) | ||
| EP0308838B1 (de) | Verfahren zur Herstellung von fluorierten Acrylsäuren und ihren Derivaten | |
| DE2648479A1 (de) | Elektrode fuer elektrolytische prozesse | |
| DE19527642A1 (de) | Verfahren zur elektrolytischen Reduktion einer Disulfid-Verbindung | |
| DE2819964C2 (de) | Metallisches Diaphragma | |
| DE4217338C2 (de) | Elektrochemisches Verfahren zur Reduktion von Oxalsäure zu Glyoxylsäure | |
| DE2924601A1 (de) | Verfahren zum behandeln eines metallkoerpers | |
| DE2952646A1 (de) | Verfahren zur elektrolyse einer waessrigen alkalimetallchloridloesung | |
| DE3443338C2 (de) | Kathode zur Herstellung von Elektrolyt-Mangandioxid | |
| DE60121337T2 (de) | Verfahren zur verbesserung einer elektrode | |
| DE2550224A1 (de) | Anodenanordnung und bipolare elektrolysiervorrichtung mit einer derartigen anodenanordnung | |
| EP0548141A1 (de) | Verfaharen zur herstellung von halogenierten acrylsäuren | |
| DE2213528A1 (de) | Verfahren zur Beseitigung verbrauchter Überzüge von metallischen Elektroden | |
| DE4217336C2 (de) | Elektrochemisches Verfahren zur Herstellung von Glyoxylsäure | |
| AT247093B (de) | Verfahren und Bad zur Herstellung von haftfähigen Niederschlägen aus Platin mit verbesserten Überspannungseigenschaften |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AT BE CH DE FR GB IT LI NL SE |
|
| 17P | Request for examination filed |
Effective date: 19880127 |
|
| 17Q | First examination report despatched |
Effective date: 19890228 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE CH DE FR GB IT LI NL SE |
|
| REF | Corresponds to: |
Ref document number: 48657 Country of ref document: AT Date of ref document: 19891215 Kind code of ref document: T |
|
| ITF | It: translation for a ep patent filed | ||
| REF | Corresponds to: |
Ref document number: 3761151 Country of ref document: DE Date of ref document: 19900118 |
|
| ET | Fr: translation filed | ||
| GBT | Gb: translation of ep patent filed (gb section 77(6)(a)/1977) | ||
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed | ||
| ITTA | It: last paid annual fee | ||
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 19930118 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 19930120 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: SE Payment date: 19930121 Year of fee payment: 7 Ref country code: GB Payment date: 19930121 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: AT Payment date: 19930127 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 19930205 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 19930228 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 19930416 Year of fee payment: 7 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Effective date: 19940227 Ref country code: AT Effective date: 19940227 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Effective date: 19940228 Ref country code: LI Effective date: 19940228 Ref country code: CH Effective date: 19940228 Ref country code: BE Effective date: 19940228 |
|
| BERE | Be: lapsed |
Owner name: HOECHST A.G. Effective date: 19940228 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Effective date: 19940901 |
|
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee | ||
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 19940227 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Effective date: 19941031 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19941101 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| EUG | Se: european patent has lapsed |
Ref document number: 87102846.0 Effective date: 19940910 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES;WARNING: LAPSES OF ITALIAN PATENTS WITH EFFECTIVE DATE BEFORE 2007 MAY HAVE OCCURRED AT ANY TIME BEFORE 2007. THE CORRECT EFFECTIVE DATE MAY BE DIFFERENT FROM THE ONE RECORDED. Effective date: 20050227 |