EP0457320B1 - Verfahren zur teilweisen elektrolytischen Enthalogenierung von Di-und Trichloressigsäure sowie Elektrolyselösung - Google Patents
Verfahren zur teilweisen elektrolytischen Enthalogenierung von Di-und Trichloressigsäure sowie Elektrolyselösung Download PDFInfo
- Publication number
- EP0457320B1 EP0457320B1 EP91107944A EP91107944A EP0457320B1 EP 0457320 B1 EP0457320 B1 EP 0457320B1 EP 91107944 A EP91107944 A EP 91107944A EP 91107944 A EP91107944 A EP 91107944A EP 0457320 B1 EP0457320 B1 EP 0457320B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- formula
- alkyl
- another
- independently
- ppm
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 0 INC1=*CC*1 Chemical compound INC1=*CC*1 0.000 description 3
Classifications
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25B—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES FOR THE PRODUCTION OF COMPOUNDS OR NON-METALS; APPARATUS THEREFOR
- C25B3/00—Electrolytic production of organic compounds
- C25B3/20—Processes
- C25B3/25—Reduction
Definitions
- Monochloroacetic acid and its derivatives are important intermediates in industrial organic synthesis. They are used for the production of adhesives, pesticides or pharmaceutical products.
- the production of monochloroacetic acid by chlorinating acetic acid is always associated with the formation of di- and trichloroacetic acid.
- electrochemical dehalogenation is also available for removing di- and trichloroacetic acid from the product mixture (EP-B 0 241 685).
- the latter dehalogenation is carried out using graphite cathodes in the presence of small amounts of metal salts with a hydrogen overvoltage of at least 0.4 V (at a current density of 4000 A / m 2 ), preferably in water-containing, acidic electrolytes.
- This process has a high selectivity because the thermodynamically favored reduction of protons to hydrogen takes place at the cathode at low concentrations of the partially dehalogenated di- and trichloroacetic acid. In this way, undesired dehalogenation of the monochloroacetic acid is avoided, but the di- and trichloroacetic acid are also dehalogenated only with poor current efficiency.
- This method is not suitable for dehalogenation down to a very low concentration level of di- and trichloroacetic acid, since an increasing proportion of the electrical charge is used for the reduction of protons to hydrogen.
- An economical implementation of dehalogenation to monochloroacetic acid at a low concentration of di- and trichloroacetic acid has therefore only been insufficient to date (comparative example).
- the task was therefore to selectively dehalogenate di- and trichloroacetic acid with very extensive conversion to monochloroacetic acid, that is to say not completely.
- Nekrasov et al. investigated the dehalogenation of trichloroacetic acid and monochloroacetic acid in the presence of tetramethylammonium or tetraethylammonium salt in a non-protic electrolyte (Nekrasov et al., Elektrokhimiya 1988, 24, 560-563). The effects they observed in no way suggest that ammonium salts in an aqueous electrolyte could inhibit the above-mentioned undesired reduction of protons to hydrogen.
- di- and trichloroacetic acid can be dehalogenated continuously or discontinuously to form monochloroacetic acid with very extensive conversion in divided electrolysis cells if electrolysis is carried out in aqueous solutions in which, in addition to metal salts, a hydrogen overvoltage of at least 0.4 V (at a Current density of 4000 A / m 2 ) quaternary ammonium and / or phosphonium salts are still dissolved.
- Another object is an electrolysis solution for the partial dehalogenation of tri- and / or dichloroacetic acid, which contains at least one of these two acids, one or more metal salts with a hydrogen overvoltage of at least 0.4 V (at a current density of at least 4000 A / m 2 ) contains at least one compound selected from the group of compounds of the formula I to V.
- the compounds of the formulas I to V are used in concentrations of 1 to 5000 ppm, preferably 10 to 1000 ppm, but in particular 50 to 500 ppm.
- the metal salts with a hydrogen overvoltage of at least 0.4 V are generally the soluble salts of Cu, Zn, Cd, Hg, Sn, Pb, Ti, Bi, V, Ta and / or Ni, preferably the soluble salts of Cu, Zn, Cd, Sn, Hg and Pb.
- CI-, Br-, SO 4 2- , NO 3 - or CH 3 COO- are preferably used as anions, the anion being chosen so that a soluble metal salt is formed (for example PbN0 3 ).
- the salts can be added directly to the electrolysis solution or, e.g. by adding oxides or carbonates - or by adding the metals themselves, such as Zn, Cd, Sn, Pb, Ni - in the solution.
- the salt concentration in the catholyte is expediently set to about 0.1 to 5000 ppm, preferably to about 10 to 1000 ppm.
- the starting material for the process is di- and / or trichloroacetic acid or mixtures thereof with monochloroacetic acid which are inevitably formed in the acetic acid chlorination.
- the proportion by weight of di- and trichloroacetic acid in the total amount of chlorinated acetic acids is less than 10% by weight. This proportion by weight can easily be less than 5% by weight, or even less than 2% by weight, which was particularly surprising.
- the catholyte can also contain mineral acids (eg HCl, H 2 S0 4 etc.).
- the anolyte is preferably an aqueous mineral acid, in particular an aqueous hydrochloric acid or sulfuric acid.
- the same material as that for the cathode can generally be used as the anode material.
- other customary electrode materials which, however, must be inert under the electrolysis conditions, for example titanium, coated with titanium dioxide and doped with a noble metal oxide, such as e.g. Ruthenium dioxide.
- Cation exchange membranes made of perfluorinated polymers with carboxyl and / or sulfonic acid groups are generally used to divide the cells into the anode and cathode compartments. It is also generally possible to use anion exchange membranes which are stable in the electrolyte, diaphragms made of polymers or inorganic materials.
- the electrolysis temperature should generally be below 100 ° C, preferably between 10 and 90 ° C.
- the electrolysis can be carried out either continuously or batchwise.
- a continuous process is preferred, especially at a low concentration of di- and trichloroacetic acid.
- chloride is constantly consumed due to the anodic chlorine evolution. In general, the chloride consumption is then compensated for by continuously introducing gaseous HCl or aqueous hydrochloric acid.
- the electrolysis product is worked up in a known manner, e.g. by distillation.
- the metal salts and the quaternary ammonium and phosphonium compounds remain in the residue and can be returned to the process.
- the invention is illustrated by the following examples. Examples 1-9 are followed by a comparative example.
- the comparative example shows that under the electrolysis conditions of EP-B 0 241 685, when a dichloroacetic acid concentration of 31% (based on the total amount of dissolved acetic acids) is reached, the major part of the electrical charge is used for the reduction of protons to hydrogen .
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- Materials Engineering (AREA)
- Metallurgy (AREA)
- Electrolytic Production Of Non-Metals, Compounds, Apparatuses Therefor (AREA)
Description
- Monochloressigsäure und ihre Derivate sind wichtige Zwischenprodukte in der industriellen organischen Synthese. Sie werden zur Herstellung von Klebstoffen, Pflanzenschutzmitteln oder pharmazeutischen Produkten verwendet.
Die Herstellung von Monochloressigsäure durch Chlorieren von Essigsäure ist immer mit der Bildung von Di- und Trichloressigsäure verbunden. Zur Entfernung von Di- und Trichloressigsäure aus dem Produktgemisch steht neben der katalytischen Hydrierung der Di- und Trichloressigsäure zu Monochloressigsäure auch die elektrochemische Enthalogenierung zur Verfügung (EP-B 0 241 685). - Die letztgenannte Enthalogenierung wird unter Verwendung von Graphitkathoden in Gegenwart kleiner Mengen an Metallsalzen mit einer Wasserstoffüberspannung von mindestens 0,4 V (bei einer Stromdichte von 4000 A/m2) durchgeführt, und zwar bevorzugt in wasserhaltigen, sauren Elektrolyten.
- Dieses Verfahren hat eine hohe Selektivität, da an der Kathode bei niedrigen Konzentrationen der teilweise zu enthalogenierenden Di- und Trichloressigsäure die thermodynamisch begünstigte Reduktion von Protonen zu Wasserstoff stattfindet. Auf diese Weise wird zwar eine unerwünschte Enthalogenierung der Monochloressigsäure vermieden, jedoch werden auch die Di- und die Trichloressigsäure nur noch mit schlechter Stromausbeute enthalogeniert. Für eine Enthalogenierung bis zu einem sehr niedrigen Konzentrationsniveau der Di- und Trichloressigsäure ist dieses Verfahren nicht geeignet, da ein immer größerer Anteil der elektrischen Ladung für die Reduktion von Protonen zu Wasserstoff verbraucht wird. Eine wirtschaftliche Durchführung der Enthalogenierung zu Monochloressigsäure bei einer niedrigen Konzentration der Di- und Trichloressigsäure ist daher bisher nur unzureichend möglich (Vergleichsbeispiel).
- Es bestand somit die Aufgabe, Di- und Trichloressigsäure bei sehr weitgehendem Umsatz selektiv zu Monochloressigsäure - also nicht vollständig - zu enthalogenieren.
- Aus EP-A-0 280 120 ist nun bekannt, daß eine vollständige Entchlorierung von 3,3-Dichlor-2-fluoracryl- säure in Anwesenheit von protoniertem Dimethylanilin eintritt, insbesondere wenn die Entchlorierung diskontinuierlich durchgeführt wird.
- Nekrasov et al. untersuchten die Enthalogenierung von Trichloressigsäure und Monochloressigsäure in Anwesenheit von Tetramethylammonium- oder Tetraethylammoniumsalz in einem nicht-protischen Elektrolyten (Nekrasov et al., Elektrokhimiya 1988, 24, 560-563). Die von ihnen beobachteten Effekte legen jedoch in keiner Weise nahe, daß in einem wäßrigen Elektrolyten Ammoniumsalze die oben erwähnte unerwünschte Reduktion von Protonen zu Wasserstoff hemmen könnten.
- Es wurde nun überraschenderweise gefunden, daß man Di- und Trichloressigsäure mit sehr weitgehendem Umsatz in geteilten Elektrolysezellen kontinuierlich oder diskontinuierlich zu Monochloressigsäure enthalogenieren kann, wenn man in wäßrigen Lösungen elektrolysiert, in denen neben Metallsalzen mit einer Wasserstoffüberspannung von mindestens 0,4 V (bei einer Stromdichte von 4000 A/m2) noch quartäre Ammonium- und/oder Phosphoniumsalze gelöst sind.
- Ein Gegenstand der Erfindung ist daher ein Verfahren zur teilweisen Enthalogenierung von Tri- und Dichloressigsäure zu Monochloressigsäure durch Elektrolyse wäßriger Lösungen dieser Säuren in geteilten Zellen in Anwesenheit eines oder mehrerer Metallsalze mit einer Wasserstoffüberspannung von mindestens 0,4 V (bei einer Stromdichte von 4000 A/m2) unter Verwendung von Kohlenstoffkathoden, gekennzeichnet durch Zusatz von mindestens einer Verbindung ausgewählt aus der Gruppe der Verbindungen der Formel I bis V,





- X Stickstoff oder Phosphor,
- R1 bis R21, gleich oder verschieden unabhängig voneinander, Wasserstoff, geradkettiges oder verzweigtes C1-C18-Alkyl, C3-C18-Cycloalkyl oder C1-C18-Alkyl-Aryl, wobei der Arylrest 6 bis 12 Kohlenstoffatome hat, und die Reste R2 bis R16 außerdem noch unabhängig voneinander folgende Bedeutung haben können:
- R2 eine Gruppe der Formel -((CH2)n-O)m-R, wobei für R dieselben Reste infrage kommen wie für R1, aber R1 und R unabhängig voneinander sind, wobei n eine ganze Zahl von 1 bis 12 und ebenso m eine ganze Zahl von 1 bis 12 ist,
- R3 und R4 zusammen, R5 und R6 zusammen und/oder R7 und R8 zusammen unabhängig voneinander eine Kette von 2 bis 8 CH2-Gruppen oder eine Gruppe der Formel -CH2(Z)CH2-mit Z = Stickstoff, Sauerstoff, Schwefel,
- R12 und R13 zusammen, R13 und R14 zusammen, R14 und R15 zusammen und/oder R15 und R16 zusammen unabhängig voneinander eine Gruppe der Formel

- Y eine Gruppe der Formel -(CH2)p- oder -CH2-[O-(CH2)p]q-O-(CH2)2-, wobei p eine ganze Zahl von 1 bis 12 und q eine ganze Zahl von 0 bis 6 ist und
- A- eines der Anionen OH-, F-, CI-, Br-, J-, S04 2-, HS04-, NO3-, CH3C00-, BF4- oder CH3OSO3-.
- Ein weiterer Gegenstand ist eine Elektrolyselösung zur teilweisen Enthalogenierung von Tri- und/oder Dichloressigsäure, welche mindestens eine dieser beiden Säuren, ein oder mehrere Metallsalze mit einer Wasserstoffüberspannung von mindestens 0,4 V (bei einer Stromdichte von mindestens 4000 A/m2) und noch mindestens eine Verbindung ausgewählt aus der Gruppe der Verbindungen der Formel I bis V enthält.
- Bevorzugt sind, Verbindungen der Formel I, bei denen unabhängig voneinander
- R1 bis R4 = Wasserstoff oder C1-C16-Alkyl ist,
sowie Verbindungen der Formel III, bei denen - R11 = C4-C16-Alkyl und
- R12 bis R16 unabhängig voneinander = H oder C4-C16-Alkyl ist.
- Ferner sind Verbindungen der Formel II bevorzugt, bei denen unabhängig voneinander
- R5 bis R10 = C4-C6-Alkyl, Cyclohexyl oder geradkettiges und geradzahliges C8-C16-Alkyl ist.
- Besonders bevorzugt sind
- A) Verbindungen der Formel I, bei denen X = Stickstoff oder Phosphor, R1 = C1-C3-Alkyl und unabhängig voneinander R2 bis R4 = C1-C4-Alkyl ist,
- B) Verbindungen der Formel III, bei denen R11 = C8-C16-Alkyl und R12 bis R16 = H ist
- Die Verbindungen der Formeln I bis V werden in Konzentrationen von 1 bis 5000 ppm, vorzugsweise 10 bis 1000 ppm insbesondere aber 50 bis 500 ppm verwendet.
- Als Metallsalze mit einer Wasserstoffüberspannung von mindestens 0,4 V (bei einer Stromdichte von 4000 A/m2) werden im allgemeinen die löslichen Salze von Cu, Zn, Cd, Hg, Sn, Pb, Ti, Bi, V, Ta und/oder Ni, vorzugsweise die löslichen Salze von Cu, Zn, Cd, Sn, Hg und Pb eingesetzt. Als Anionen werden vorzugsweise CI-, Br-, SO4 2-, NO3- oder CH3COO- verwendet, wobei das Anion so gewählt wird, daß ein lösliches Metallsalz entsteht (z.B. PbN03).
- Die Salze können der Elektrolyselösung direkt zugesetzt werden oder auch z.B. durch Zugabe von Oxiden oder Carbonaten - oder durch Zugabe der Metalle selbst, wie bei Zn, Cd, Sn, Pb, Ni - in der Lösung erzeugt werden.
- Die Salzkonzentration im Katholyten wird zweckmäßig auf etwa 0,1 bis 5000 ppm, vorzugsweise auf etwa 10 bis 1000 ppm, eingestellt.
- Beim erfindungsgemäßen Verfahren tritt im allgemeinen eine außerordentlich geringe Wasserstoffentwicklung an der Kathode auch bei sehr niedrigen Konzentrationen der mehrfach chlorierten Essigsäuren ein, ohne daß sich bei Dauerbetrieb die hohe Selektivität der Elektrolyse verschlechtert. Das erfindungsgemäße Verfahren ist daher außerordentlich wirtschaftlich, was nach dem Stand der Technik in keiner Weise zu erwarten war. Auch eine kontinuierliche Verfahrensweise bei niedrigen Konzentrationen der Ausgangsverbindungen führt nur in sehr geringem Umfang zu Essigsäure.
- Als Ausgangsmaterial für das Verfahren werden Di- und/oder Trichloressigsäure oder deren bei der Essigsäurechlorierung zwangsläufig entstehenden Mischungen mit Monochloressigsäure verwendet.
- Im allgemeinen können, insbesondere als Katholyt, wäßrige Lösungen der chlorierten Essigsäuren in allen möglichen Konzentrationen (ca. 1 bis ca. 95 Gew.-%) verwendet werden.
- Besonders vorteilhaft ist es, wenn der Gewichtsanteil der Di- und Trichloressigsäure an der Gesamtmenge der chlorierten Essigsäuren kleiner als 10 Gew.-% ist. Dabei kann dieser Gewichtsanteil ohne weiteres kleiner als 5 Gew.-%, oder sogar kleiner als 2 Gew.-% sein, was besonders überraschend war.
Der Katholyt kann zusätzlich noch Mineralsäuren (z.B. HCI,H2S04 etc.) enthalten. - Der Anolyt ist vorzugsweise eine wäßrige Mineralsäure, insbesondere eine wäßrige Salzsäure oder Schwefelsäure.
- Als Kohlenstoffkathoden kommen im Prinzip alle üblichen Kohle-Elektrodenmaterialien in Frage wie z.B. Elektrodengraphite, imprägnierte Graphitwerkstoffe oder auch glasartiger Kohlenstoff.
- Als Anodenmaterial kann im allgemeinen das gleiche Material wie für die Kathode verwendet werden. Darüberhinaus ist auch der Einsatz anderer üblicher Elektrodenmaterialien möglich, die jedoch unter den Elektrolysebedingungen inert sein müssen, beispielsweise Titan, beschichtet mit Titandioxid und dotiert mit einem Edelmetalloxid, wie z.B. Rutheniumdioxid.
- Zur Teilung der Zellen in Anoden- und Kathodenraum werden im allgemeinen Kationenaustauschermembranen aus perfluorierten Polymeren mit Carboxyl- und /oder Sulfonsäuregruppen benutzt. Auch die Verwendung von im Elektrolyten stabilen Anionenaustauschermembranen, Diaphragmen aus Polymeren oder anorganischen Werkstoffen ist im allgemeinen möglich.
Die Elektrolysetemperatur soll im allgemeinen unter 100 °C liegen, vorzugsweise zwischen 10 und 90 °C. - Die Elektrolyse kann sowohl kontinuierlich als auch diskontinuierlich durchgeführt werden. Bevorzugt ist ein kontinuierliches Verfahren vor allem bei niedriger Konzentration der Di- und Trichloressigsäure.
- Wird als Anolyt wäßrige Salzsäure verwendet, dann wird durch die anodische Chlorentwicklung ständig Chlorid verbraucht. Im allgemeinen wird dann der Chloridverbrauch durch kontinuierliches Einleiten von gasförmigem HCI oder von wäßriger Salzsäure ausgeglichen.
- Die Aufarbeitung des Elektrolyseproduktes erfolgt auf bekannte Weise, z.B. durch Destillation. Die Metallsalze und die quartären Ammonium- und Phosphoniumverbindungen bleiben dabei im Rückstand und können wieder in den Prozeß zurückgeführt werden.
- Die Erfindung wird nun durch die folgenden Beispiele näher erläutert. Nach den Beispielen 1-9 folgt noch ein Vergleichsbeispiel. Aus dem Vergleichsbeispiel geht hervor, daß unter den Elektrolysebedingungen des EP-B 0 241 685 bereits beim Erreichen einer Dichloressigsäure-Konzentrationen von 31 % (bezogen auf die Gesamtmenge der gelösten Essigsäuren) der Hauptanteil der elektrischen Ladung für die Reduktion von Protonen zu Wasserstoff verbraucht wird.
- Umlaufzelle mit 0,0015 m2 Elektrodenfläche;
- Elektrodenabstand 5 mm
- Elektroden: imprägnierter Graphit ODiabon (der Fa. Sigri, Meitingen, Deutschland)
- Kationenaustauschermembran: ONafion 324 (der Fa. DuPont, Wilmington, Del., USA, 2-Schichtenmembran aus Copolymerisaten aus Perfluorsulfonylethoxyvinylether und Tetrafluorethylen. Auf der Kathodenseite befindet sich eine Schicht mit dem Äquivalentgewicht 1300, auf der Anodenseite eine solche mit dem Äquivalentgewicht 1100)
- Abstandshalter: Polyethylennetze
- Durchfluß: 100 I/h
- Temp.: 30 - 42 °C
- Anolyt: konzentrierte Salzsäure, kontinuierlich ergänzt durch gasförmiges HCI
- Katholyt: 800 g Wasser, 350 g Monochloressigsäure, 7 g Dichloressigsäure (im Beispiel 2 Trichloressigsäure). Die Di- bzw. Trichloressigsäure wird dem Katholyten in gleichbleibenden Mengen, bis zum Erreichen der in der Tabelle angegebenen Menge, im Abstand von ca. 10 Minuten zugeführt. Die Konzentrationen des Metallsalzes und der jeweils eingesetzten Verbindung der Formel 1 bzw. III sind aus der Tabelle ersichtlich.
- Wie Elektrolysezelle 1, aber mit folgenden Änderungen: Elektrodenfläche: 0,02 m2
- Kationenaustauschermembran: ONafion 423 (Fa. DuPont, 1-Schichtenmembran aus Copolymerisaten aus Perfluorsulfonylethoxyvinylether und Tetrafluorethylen mit einem Äquivalentgewicht von 1200)
- Durchfluß: 400 I/h
- Katholyt: 2400 g Wasser, 1050 g Monochloressigsäure, 60 g Dichloressigsäure. Die Konzentrationen des Metallsalzes und der Verbindung der Formel 1 sind aus der Tabelle ersichtlich.
- Elektrolyse nach EP-B 0 241 685
- Elektrolysebedingungen wie bei den Beispielen 1 bis 8 mit Ausnahme von:
- Katholyt: 2 kg Wasser, 0,4 kg Dichloressigsäure, 532 ppm CdC12
- Stromdichte: 4000 A/m2
- Zellspannung: 4,5 V
- Ladungsverbrauch. 145 Ah
- Elektrolyseergebnis:
- Dichloressigsäure:0,1 kg (= 31,1 Gew.-%)
- Monochloressigsäure: 0,221 kg (= 68,9 Gew.-%)
- Während der Elektrolyse wurden 36 % der Ladungsmenge für die Reduktion von Protonen zu Wasserstoff verbraucht. Die Wirtschaftlichkeit des erfindungsgemäßen Verfahrens wird bei Gegenüberstellung des Vergleichsbeispiels und Beispiel 6 besonders deutlich. Im Beispiel 6 beträgt der Anteil der elektrischen Ladung, die für die Reduktion von Protonen zu Wasserstoff verbraucht wird, bei einem Dichloressigsäureanteil von 1 Gew.-% nur 2,1 %.


Beim erfindungsgemäßen Verfahren wird mindestens eine Verbindung der Formel I oder II oder III oder IV oder V oder es werden beliebige Gemische von Verbindungen der Formeln I, II, III, IV und V in der Elektrolyse eingesetzt.
Claims (22)

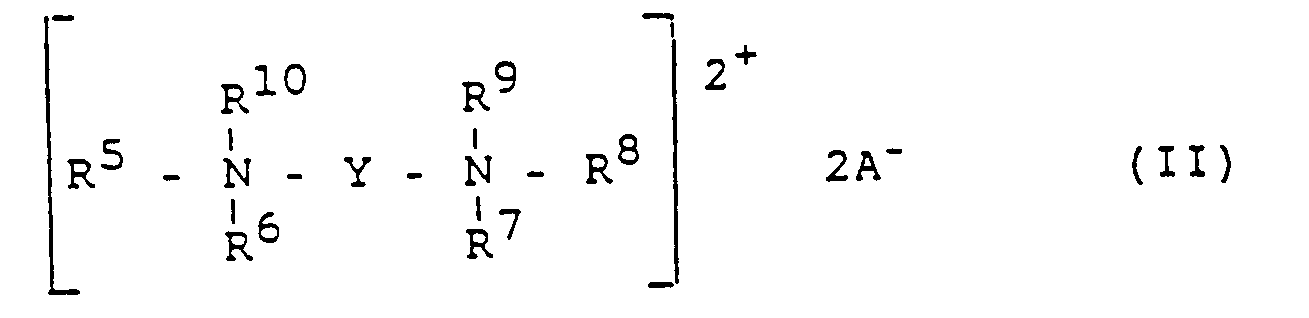










Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE4016063 | 1990-05-18 | ||
| DE4016063A DE4016063A1 (de) | 1990-05-18 | 1990-05-18 | Verfahren zur teilweisen elektrolytischen enthalogenierung von di- und trichloressigsaeure sowie elektrolyseloesung |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0457320A1 EP0457320A1 (de) | 1991-11-21 |
| EP0457320B1 true EP0457320B1 (de) | 1994-12-07 |
Family
ID=6406747
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP91107944A Expired - Lifetime EP0457320B1 (de) | 1990-05-18 | 1991-05-16 | Verfahren zur teilweisen elektrolytischen Enthalogenierung von Di-und Trichloressigsäure sowie Elektrolyselösung |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US5362367A (de) |
| EP (1) | EP0457320B1 (de) |
| JP (1) | JPH0593290A (de) |
| BR (1) | BR9102050A (de) |
| CA (1) | CA2042862A1 (de) |
| DE (2) | DE4016063A1 (de) |
| FI (1) | FI912381A7 (de) |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7083707B2 (en) * | 2001-07-27 | 2006-08-01 | Canon Kabushiki Kaisha | Decomposition apparatus and decomposition method |
| US7169287B2 (en) * | 2001-07-27 | 2007-01-30 | Canon Kabushiki Kaisha | Decomposition apparatus and decomposition method |
| US7910769B2 (en) | 2004-09-02 | 2011-03-22 | Eastman Chemical Company | Optimized liquid-phase oxidation |
| US7568361B2 (en) | 2004-09-02 | 2009-08-04 | Eastman Chemical Company | Optimized liquid-phase oxidation |
| US7692036B2 (en) | 2004-11-29 | 2010-04-06 | Eastman Chemical Company | Optimized liquid-phase oxidation |
| US7572936B2 (en) | 2004-09-02 | 2009-08-11 | Eastman Chemical Company | Optimized liquid-phase oxidation |
| US7692037B2 (en) | 2004-09-02 | 2010-04-06 | Eastman Chemical Company | Optimized liquid-phase oxidation |
| US7381836B2 (en) | 2004-09-02 | 2008-06-03 | Eastman Chemical Company | Optimized liquid-phase oxidation |
| US7371894B2 (en) | 2004-09-02 | 2008-05-13 | Eastman Chemical Company | Optimized liquid-phase oxidation |
| US7741515B2 (en) | 2004-09-02 | 2010-06-22 | Eastman Chemical Company | Optimized liquid-phase oxidation |
| US7504535B2 (en) | 2004-09-02 | 2009-03-17 | Eastman Chemical Company | Optimized liquid-phase oxidation |
| US7589231B2 (en) | 2004-09-02 | 2009-09-15 | Eastman Chemical Company | Optimized liquid-phase oxidation |
| US7572932B2 (en) | 2004-09-02 | 2009-08-11 | Eastman Chemical Company | Optimized liquid-phase oxidation |
| US8956990B2 (en) | 2010-03-26 | 2015-02-17 | Dioxide Materials, Inc. | Catalyst mixtures |
| US9012345B2 (en) | 2010-03-26 | 2015-04-21 | Dioxide Materials, Inc. | Electrocatalysts for carbon dioxide conversion |
| US10173169B2 (en) | 2010-03-26 | 2019-01-08 | Dioxide Materials, Inc | Devices for electrocatalytic conversion of carbon dioxide |
| US9957624B2 (en) | 2010-03-26 | 2018-05-01 | Dioxide Materials, Inc. | Electrochemical devices comprising novel catalyst mixtures |
| US20110237830A1 (en) | 2010-03-26 | 2011-09-29 | Dioxide Materials Inc | Novel catalyst mixtures |
| US9790161B2 (en) | 2010-03-26 | 2017-10-17 | Dioxide Materials, Inc | Process for the sustainable production of acrylic acid |
| US9566574B2 (en) * | 2010-07-04 | 2017-02-14 | Dioxide Materials, Inc. | Catalyst mixtures |
| US9815021B2 (en) | 2010-03-26 | 2017-11-14 | Dioxide Materials, Inc. | Electrocatalytic process for carbon dioxide conversion |
| US10647652B2 (en) | 2013-02-24 | 2020-05-12 | Dioxide Materials, Inc. | Process for the sustainable production of acrylic acid |
| US10774431B2 (en) | 2014-10-21 | 2020-09-15 | Dioxide Materials, Inc. | Ion-conducting membranes |
| US10975480B2 (en) | 2015-02-03 | 2021-04-13 | Dioxide Materials, Inc. | Electrocatalytic process for carbon dioxide conversion |
| CN109763138A (zh) * | 2017-11-09 | 2019-05-17 | 山东润博生物科技有限公司 | 一种3,6-二氯水杨酸的制备方法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5476521A (en) * | 1977-11-30 | 1979-06-19 | Chlorine Eng Corp Ltd | Preparation of monochloroacetic acid |
| JPS5724333A (en) * | 1980-07-18 | 1982-02-08 | Koei Chem Co Ltd | Production of quaternary ammonium acidic sulfate salt |
| US4707230A (en) * | 1985-09-23 | 1987-11-17 | Tracer Technologies, Inc. | Electrochemical dehalogenation of organic compounds |
| DE3607446A1 (de) * | 1986-03-07 | 1987-09-10 | Hoechst Ag | Verfahren zur enthalogenierung von chlor- und von bromessigsaeuren |
| US4892944A (en) * | 1987-05-13 | 1990-01-09 | Mitsubishi Petrochemical Co., Ltd. | Process for producing quaternary salts |
| DE58904307D1 (de) * | 1988-03-19 | 1993-06-17 | Hoechst Ag | Verfahren zur herstellung von ungesaettigten halogenierten kohlenwasserstoffen. |
-
1990
- 1990-05-18 DE DE4016063A patent/DE4016063A1/de not_active Withdrawn
-
1991
- 1991-05-16 EP EP91107944A patent/EP0457320B1/de not_active Expired - Lifetime
- 1991-05-16 DE DE59103750T patent/DE59103750D1/de not_active Expired - Fee Related
- 1991-05-16 JP JP3111938A patent/JPH0593290A/ja not_active Withdrawn
- 1991-05-16 FI FI912381A patent/FI912381A7/fi unknown
- 1991-05-17 BR BR919102050A patent/BR9102050A/pt not_active Application Discontinuation
- 1991-05-17 CA CA002042862A patent/CA2042862A1/en not_active Abandoned
-
1993
- 1993-10-19 US US08/139,337 patent/US5362367A/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| DE4016063A1 (de) | 1991-11-21 |
| DE59103750D1 (de) | 1995-01-19 |
| JPH0593290A (ja) | 1993-04-16 |
| FI912381A7 (fi) | 1991-11-19 |
| CA2042862A1 (en) | 1991-11-19 |
| EP0457320A1 (de) | 1991-11-21 |
| BR9102050A (pt) | 1991-12-24 |
| FI912381A0 (fi) | 1991-05-16 |
| US5362367A (en) | 1994-11-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0457320B1 (de) | Verfahren zur teilweisen elektrolytischen Enthalogenierung von Di-und Trichloressigsäure sowie Elektrolyselösung | |
| EP0012215B1 (de) | 2-Hydroxybutansulfonsaures Cholin und dessen Verwendung als Leitsalz | |
| EP0627020B1 (de) | Elektrochemisches verfahren zur herstellung von glyoxylsäure | |
| DE3704915A1 (de) | Elektrochemisches verfahren zum austausch von halogenatomen in einer organischen verbindung | |
| DE19846636A1 (de) | Elektrochemische Synthese von Perfluoralkylfluorophosphoranen | |
| DE2603144A1 (de) | Betriebsverfahren fuer eine elektrolytische zelle mit drei abteilungen zur herstellung von alkalimetallhydroxiden | |
| EP0334796B1 (de) | Verfahren zur Herstellung von ungesättigten halogenierten Kohlenwasserstoffen | |
| EP0241685B1 (de) | Verfahren zur Enthalogenierung von Chlor- und von Bromessigsäuren | |
| EP0308838B1 (de) | Verfahren zur Herstellung von fluorierten Acrylsäuren und ihren Derivaten | |
| EP0355726B1 (de) | Verfahren zur elektrolytischen Decarboxylierung einer Perfluorcarbonsäure oder deren löslichen Salzen und anschliessende Dimerisierung der dabei entstehenden Radikale | |
| DE3522304C2 (de) | Verfahren zur Herstellung von Carbonsäuren durch elektrochemische Reduktion | |
| DE4217338C2 (de) | Elektrochemisches Verfahren zur Reduktion von Oxalsäure zu Glyoxylsäure | |
| DE10031563B4 (de) | Verfahren zur Herstellung von perfluororganischen Verbindungen durch elektrochemische Fluorierung | |
| DE10031565B4 (de) | Verfahren zur Herstellung von perfluorierten organischen Verbindungen durch elektrochemische Fluorierung | |
| EP0326855B1 (de) | Verfahren zur Herstellung von Fluormalonsäure und ihren Derivaten | |
| AT394214B (de) | Funktionalisierung von jodpolyfluoralkanen durch elektrochemische reduktion | |
| DE4029068A1 (de) | Verfahren zur herstellung von halogenierten acrylsaeuren | |
| DE2701453A1 (de) | Verfahren zur elektrolytischen oxydation von dialkyldithiocarbamaten zu tetraalkylthiuramdisulfiden | |
| DE69706668T2 (de) | Verfahren zur herstellung von tetraalkyl 1,2,3,4-butantetracarboxylaten | |
| EP0293856B1 (de) | Verfahren zur Herstellung fluorierter Vinylether | |
| DE102010029272A1 (de) | Verfahren zur elektrochemischen Herstellung von Isophoron | |
| DE2434921B2 (de) | Elektrolysezelle und Verfahren zur Elektrolyse ionisierbarer chemischer Verbindungen | |
| DE102009001168A1 (de) | Verfahren zur Herstellung von Hypophosphiten und Phosphiten | |
| DE4205423C1 (de) | Elektrochemisches Verfahren zur Herstellung von Glyoxylsäure | |
| DE2802260A1 (de) | Verfahren zur herstellung eines tetraalkylthiuramdisulfids |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): BE DE ES FR GB IT NL |
|
| 17P | Request for examination filed |
Effective date: 19920514 |
|
| 17Q | First examination report despatched |
Effective date: 19930430 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): BE DE ES FR GB IT NL |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRE;WARNING: LAPSES OF ITALIAN PATENTS WITH EFFECTIVE DATE BEFORE 2007 MAY HAVE OCCURRED AT ANY TIME BEFORE 2007. THE CORRECT EFFECTIVE DATE MAY BE DIFFERENT FROM THE ONE RECORDED.SCRIBED TIME-LIMIT Effective date: 19941207 Ref country code: ES Free format text: THE PATENT HAS BEEN ANNULLED BY A DECISION OF A NATIONAL AUTHORITY Effective date: 19941207 Ref country code: NL Effective date: 19941207 Ref country code: BE Effective date: 19941207 Ref country code: FR Effective date: 19941207 Ref country code: GB Effective date: 19941207 |
|
| REF | Corresponds to: |
Ref document number: 59103750 Country of ref document: DE Date of ref document: 19950119 |
|
| EN | Fr: translation not filed | ||
| NLV1 | Nl: lapsed or annulled due to failure to fulfill the requirements of art. 29p and 29m of the patents act | ||
| GBV | Gb: ep patent (uk) treated as always having been void in accordance with gb section 77(7)/1977 [no translation filed] |
Effective date: 19941207 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed | ||
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19960201 |