-
Hintergrund der Erfindung
-
1. Gebiet der Erfindung
-
Die
vorliegende Erfindung betrifft ein Verfahren zur Umwandlung von
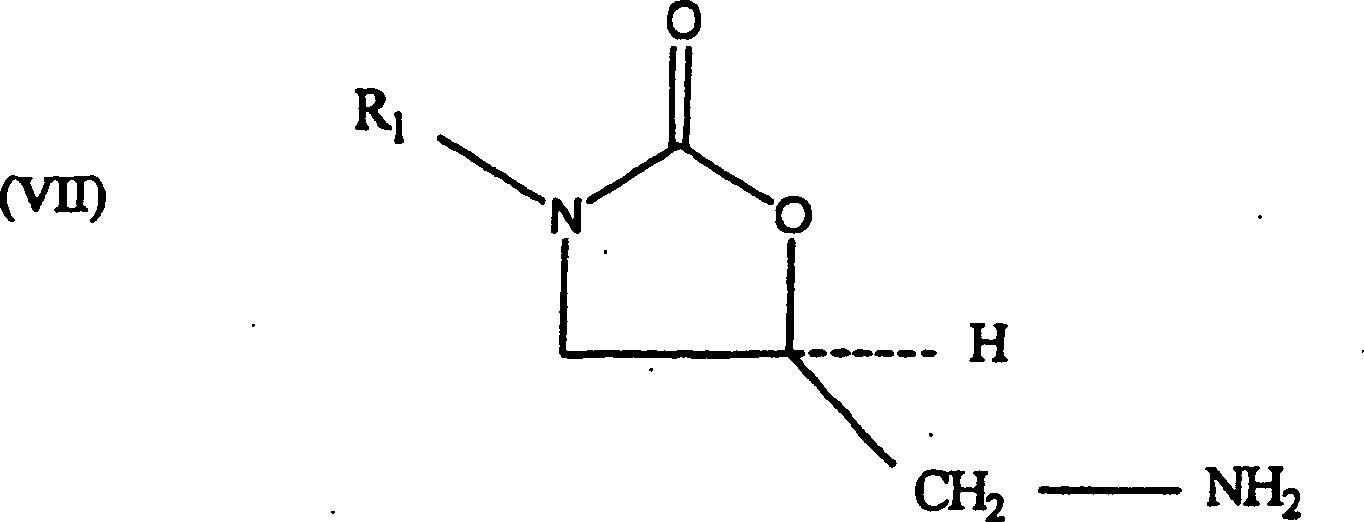
5-Hydroxymethyl-substituierten Oxazolidinonalkoholen (III) in die
entsprechenden 5-Aminomethyl-substituierten Oxazolidinonamine (VII),
die bei der Herstellung von antibakteriellen Oxazolidinon-Pharmazeutika
(VIII) verwendbar sind.
-
2. Beschreibung des Standes
der Technik
-
Die
US-Patente 5 164 510 ,
5 182 403 und
5 225 565 offenbaren 5'-Indolinyloxazolidinone,
3-(5'-Indazolyl)oxazolidinone,
3-(mit kondensiertem
Ring substituierte)-Phenyloxazolidinone, die jeweils als antibakterielle
Mittel verwendbar sind.
-
Die
US-Patente 5 231 188 und
5 247 090 offenbaren verschiedene
tricyclische Oxazolidinone mit [6.5.5]- und [6.6.5]-kondensiertem Ring,
die als antibakterielle Mittel verwendbar sind.
-
Die
WO93/09103 offenbart Mono-
und Dihalogenphenyloxazolidinon-Antibiotika, die als pharmazeutische
Mittel wegen deren antibakterieller Wirkung verwendbar sind.
-
Die
US-Patente 4 062 862 und
4 236 012 offenbaren ein
Verfahren zur Herstellung von Oxazolidinonen, das die Umsetzung
eines Epoxids mit einem primären
(ohne einen Substituenten am Stickstoffatom) Carbamat in Gegenwart
eines Katalysators umfasst. Das Verfahren "wird vorzugsweise bei einer Temperatur
von 100° bis
150° über mehrere
Stunden durchgeführt".
-
Das
kanadische Patent 681 830 offenbart
ein Verfahren zur Herstellung von Oxazolidinonen, das die Umsetzung
eines Arylethers von Glycidol mit einem primären Carbamat in Gegenwart eines
alkalischen Katalysators (vorzugsweise Lithiumamid oder Lithiumhydroxid)
umfasst. Das Verfahren wurde in dem "bevorzugten Temperaturbereich von 150° bis 165°" durchgeführt. Die
Produkte sind Arylether von 5-Hydroxymethyl-substituierten Oxazolidinonen
und die Ausbeuten sind schlecht (40–78%).
-
J.
Am. Chem. Soc., 64, 1291 (1942) und das
US-Patent 3 547 951 offenbaren ein
Verfahren zur Umwandlung von primären Alkoholen in Amine, das
eine Behandlung mit Methansulfonylchlorid unter Bildung des Mesylats
und das anschließende
Inkontaktbringen des Mesylats mit wasserfreiem Ammoniak bei Umgebungstemperatur
in einem verschlossenen Reaktionsgefäß unter hohem Druck umfasst.
-
Es
ist auch bekannt, dass die Mesylate von primären Alkoholen mit wässrigem
Ammoniak unter Bildung der entsprechenden primären Amine reagieren, doch sind
eine hohe Temperatur und ein hoher Druck (85 psig) erforderlich.
Normalerweise kann dieses Verfahren in üblichen, allgemein verwendbaren
Reaktoren nicht durchgeführt
werden und es muss in für
hohen Druck ausgelegten Spezialreaktoren durchgeführt werden.
-
Die
WO95/07271 offenbart die
Ammonolyse von Oxazolidinonmesylaten.
-
Das
US-Patent 4 476 136 offenbart
ein Verfahren zur Umwandlung von 5-Hydroxymethyl-substituierten
Oxazolidinonen (III) in die entsprechenden 5(S)-Aminomethyl-substituierten
Oxazolidinone (VII), wobei das Verfahren eine Behandlung mit Methansulfonylchlorid
und anschließend
mit Kaliumphthalimid und anschließend mit Hydrazin umfasst.
Diese Reaktionsfolge ergibt Nebenprodukte, die von dem gewünschten
Produkt schwierig abzutrennen sind.
-
J.
Med. Chem., 32, 1673 (1989) und Tetrahedron 45, 1323 (1989) offenbaren
ein Verfahren zur Umwandlung von 5-Hydroxymethyl-substituierten Oxazolidinonen
in die entsprechenden 5S-Acetamidomethyl-substituierten Oxazolidinone,
wobei das Verfahren eine Behandlung mit Methansulfonylchlorid oder
Tosylchlorid und anschließend
mit Natriumazid und anschließend
mit Trimethylphosphit oder Platindioxid/Wasserstoff und anschließend Essigsäureanhydrid
oder Acetylchlorid unter Bildung des gewünschten 5(S)-Acetamidomethyl-substituierten
Oxazolidinons umfasst. Es ist bekannt, dass Natriumazid eine Explosionsgefahr
darstellt.
-
Das
US-Patent 5 210 303 offenbart
die Umwandlung von verschiedenen substituierten Benzylchloriden
in die entsprechenden Benzylamine durch Erhitzen mit wässrigem
Ammoniak in Gegenwart aromatischer Aldehyde zur Unterdrückung einer
Dialkylierung. Die dialkylierte Verunreinigung ist generell schwierig
zu entfernen, siehe Chem. Lett., 1057 (1978).
-
Zusammenfassung der Erfindung
-
Ein
Aspekt der vorliegenden Erfindung ist ein Verfahren zur Herstellung
eines 5-Aminomethyl-substituierten Oxazolidinonamins der Formel
(VII) aus einem 5-Hydroxymethyl-substituierten Oxazolidinonalkohol der
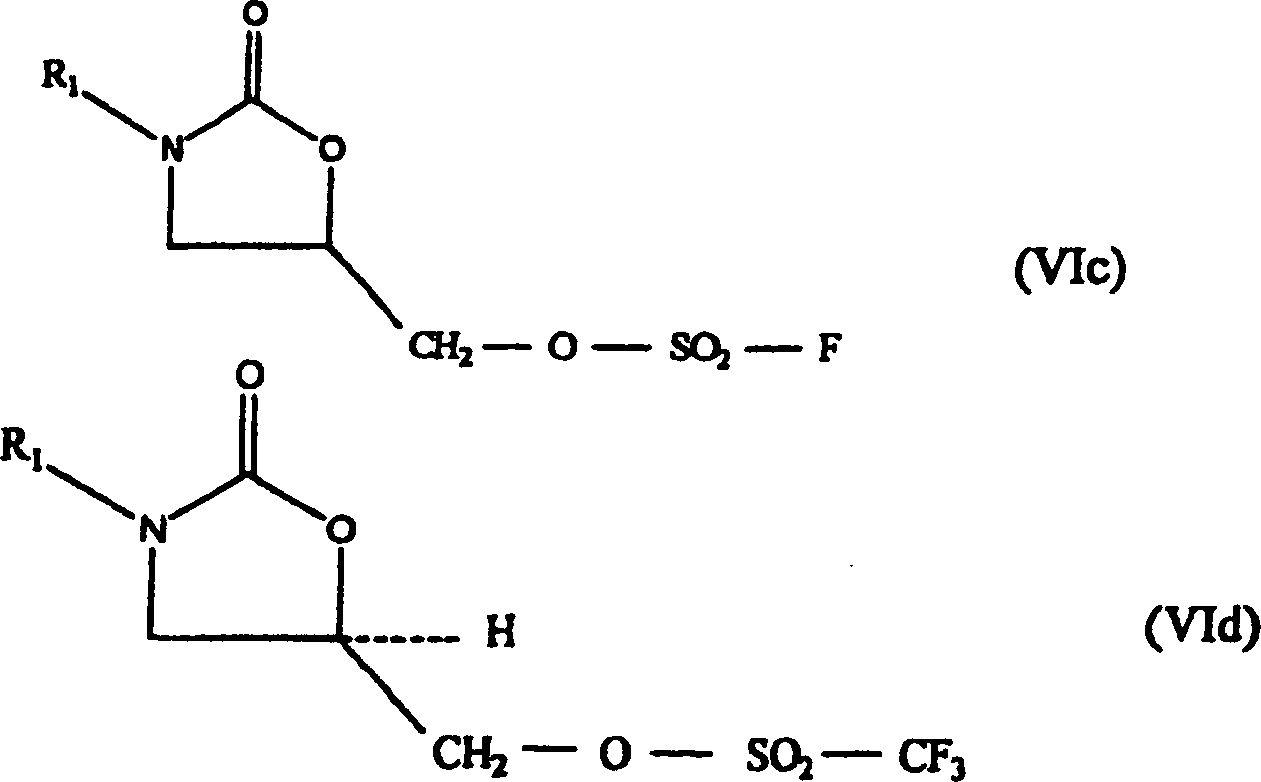
Formel (III). Dieses Verfahren erfolgt über ein Oxazolidinonsulfonat
der Formel (VIa–VId);
diese Sulfonate sind neu und stellen einen weiteren Aspekt der vorliegenden
Erfindung dar.
-
Das
Verfahren der Erfindung (alle Variablen wie in Anspruch 1 definiert)
umfasst:
- (1) das Inkontaktbringen einer Verbindung
der Formel (III) mit einem Sulfonylierungsmittel, das aus den Formeln
Va–Vd ausgewählt
ist, unter Herstellung des Oxazolidinonsulfonats und
- (2) das Inkontaktbringen des Oxazolidinonsulfonats (VIa–VId) mit Ammoniak in einem verschlossenen System
bei nicht mehr als 60°C
und bei einem Druck von weniger als 30 psig (207 kPa).
-
Detaillierte Beschreibung
der Erfindung
-
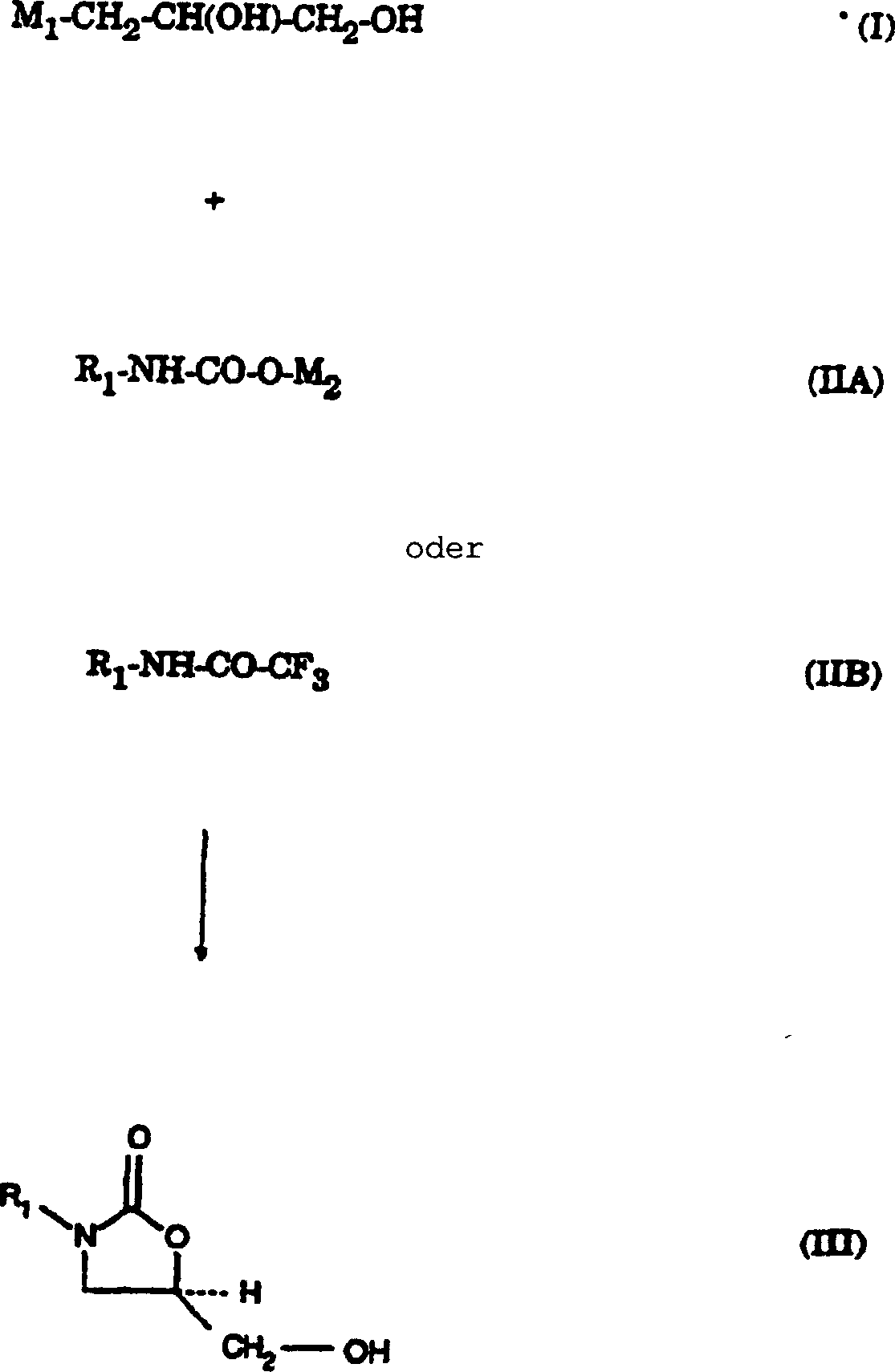
Ein
Verfahren zur Herstellung der 5-Hydroxymethyl-substituierten Oxazolidinonalkohole
(III) kann entweder die nicht-cyclischen
(S)-, (R)-Dihydroxyverbindungen der Formel (I) oder ein beliebiges
Gemisch derselben oder (S)-, (R)-Glycidol
(IV) oder ein Gemisch zur Kopplung mit dem Carbamat (IIA) oder einem
Trifluoracetamid der Formel (IIB) verwenden.
-
Die
5-Hydroxymethyl-substituierten Oxazolidinonalkohole (III) sind günstige Zwischenprodukte
zur Herstellung von 5-Aminomethyl-substituierten
Oxazolidinonaminen (VII), die zur Herstellung von pharmazeutisch
verwendbaren 5-Acylamidomethyl-substituierten Oxazolidinon(VIII)-Antibiotika
acyliert werden können. Wegen
eines Enantiomerenzentrums können
5(R)-, 5(S)-Acylamidomethyl-substituierte Oxazolidinone (VIII) und
Gemische derselben hergestellt werden. Das (S)-Enantiomer des 5-Acylamidomethyl-substituierten
Oxazolidinons (VIII) weist antibakterielle Aktivität auf, das
(R)-Enantiomer nicht.
Das 5(S)-Aminomethyl-substituierte Oxazolidinonamin(VII)-enantiomer
wird aus dem 5-(R)-Hydroxymethyl-substituierten
Oxazolidinonalkohol(III)-enantiomer, das aus der (S)-Dihydroxyverbindung
(I) oder dem (S)-Glycidol (IV) produziert wird, hergestellt. Daher
ist die gewünschte
und bevorzugte Enantiomerenfolge die Verwendung von einer enantiomerenreinen
(S)-Dihydroxyverbindung (I) oder (S)-Glycidol (IV) unter Bildung
des (R)-5-Hydroxymethyl-substituierten Oxazolidinonalkohols (III),
der zur Bildung von enantiomerenreinem (S)-5-Aminomethyl-substituiertem Oxazolidinonamin
(VII) verwendet wird, das in das enantiomerenreine (S)-5-Acylamidomethyl-substituierte Oxazolidinon
(VIII) umgewandelt wird. Jedoch ist dem Fachmann ohne weiteres klar,
dass die identischen Verfahrensstufen mit den entgegengesetzten
Enantiomerenformen ohne weiteres durchgeführt werden können und
an jedem Punkt in dem Verfahren eine nicht gewünschte Enantiomerenkonfiguration
in die gewünschte umgewandelt
werden kann. Daher wird eine Verwendung der Chemie des beanspruchten
Verfahrens mit beliebigen der Enantiomerenformen als den beanspruchten
Verfahren äquivalent
betrachtet.
-
Die
Dihydroxyverbindungen M1-CH2-CH(OH)-CH2-OH der Formel (I) und die Glycidolverbindungen C*H2-C*H-CH2-OH der
Formel (IV), wobei die durch ein * bezeichneten Kohlenstoffatome
jeweils an das gleiche Sauerstoffatom (-O-) unter Bildung eines
dreigliedrigen Rings gebunden sind, sind dem Fachmann geläufig oder
können
ohne weiteres aus bekannten Verbindungen durch dem Fachmann geläufige Verfahren
hergestellt werden. Vorzugsweise ist das Hydroxyausgangsmaterial
die Dihydroxyverbindung (I). vorzugsweise sind die Dihydroxyverbindung
(I) und das Glycidol(IV) das (S)-Enantiomer. Vorzugsweise steht
M1 für
Cl-; vorzugsweise ist die Dihydroxyverbindung (I) die von Anspruch
5, die im Handel gekauft werden kann.
-
Die
Carbamate, R1-NH-CO-O-M2 der
Formel (IIA) und das Trifluoracetamid R1-NH-CO-CF3 der Formel (IIB) sind entweder dem Fachmann
bekannt oder können
aus bekannten Verbindungen durch dem Fachmann geläufige Verfahren
ohne weiteres hergestellt werden. Die Natur der Abgangsgruppe M2 ist nicht wichtig, da sie im Laufe der
Reaktion verloren geht, was dem Fachmann geläufig ist. Verwendbare Gruppen
M2 (Abgangsgruppen) sind solche, in denen
-O-M2 eine Base ist, deren Säure einen
pka-Wert zwischen etwa 8 und etwa 24 aufweist.
Bevorzugte Gruppen M2 umfassen
C1-C20-Alkyl,
C3-C7-Cycloalkyl,
Φ-, das optional
mit einem oder zwei Resten von C1-C3-Alkyl oder F-, Cl-, Br-, I- substituiert
ist,
CH2=CH-CH2-,
CH3-CH=CH-CH2-,
(CH3)2C=CH-CH2-,
CH2=CH-,
Φ-CH=CH-CH2-,
Φ-CH2-, das optional an Φ- mit einem oder zwei Resten
von -Cl, C1-C4-Alkyl,
-NO2, -CN, -CF3 substituiert
ist,
9-Fluorenylmethyl,
(Cl)3C-CH2-,
2-Trimethylsilylethyl,
Φ-CH2-CH2-,
1-Adamantyl,
(Φ)2CH-,
CH≡C-C(CH3)2-,
2-Furanylmethyl,
Isobornyl,
wobei
stärker
bevorzugte Abgangsgruppen C1-C4-Alkyl
oder Benzyl sind. Jede andere Abgangsgruppe, die in ähnlicher
Weise funktioniert, wird als den oben angegebenen äquivalent
betrachtet. Das Carbamat (IIA) und Trifluoracetamid (IIB) tragen
die aromatische/heteroaromatische Gruppe (R1-)
des 5-Hydroxymethyl-substituierten Oxazolidinonalkohols (III). Vorzugsweise
ist R1 Phenyl, das mit einem -F und einer
substituierten Aminogruppe substituiert ist; noch besser ist R1 3-Fluor-4-[4-(benzyloxycarbonyl)-1-piperazinyl]phenyl
oder 3-Fluor-4-(4-morpholinyl)phenyl. In Abhängigkeit von den speziellen
Substituenten in R1 kann es vorkommen, dass die
Gruppen durch dem Fachmann bekannte Maßnahmen, um unerwünschte Nebenreaktionen
zu verhindern, geschützt
werden müssen,
wie dies dem Fachmann geläufig
ist. Beispielsweise ist es, wenn der R1-Substituent eine
freie primäre
oder sekundäre
Hydroxygruppe aufweist, nicht notwendig, jedoch bevorzugt, diesen
mit einer Alkoholschutzgruppe bei der Bildung der 5-Hydroxymethyl-substituierten
Oxazolidinonalkohole (III) zu schützen. Der ungeschützte Alkohol
stört generell
bei der Reaktion der Dihydroxyverbindung (I) oder des Glycidols
(IV) mit dem Carbamat (IIA) oder Trifluoracetamid (IIB) unter Bildung
des 5-Hydroxymethyl-substituierten Oxazolidi nonalkohols (III) nicht.
Jedoch stört
ein ungeschützter
Alkohol generell bei der Umwandlung des 5-Hydroxymethyl-substituierten Oxazolidinonalkohols
(III) in die entsprechenden 5-Aminomethyl-substituierten Oxazolidinonamine
(VII), da es sehr schwierig oder unmöglich ist, einen primären oder
sekundären
Alkohol an der R1-Funktionalität in Gegenwart
eines anderen primären
oder sekundären
Alkohols selektiv zu schützen. Geeignete
Alkoholschutzgruppen sind dem Fachmann geläufig, wobei C1-C5-Alkyl, Φ-CH2-, CH3-O-CH2-, CH3-, CH3-S-CH2, Φ-CH2-O-CH2-, Tetrahydropyranyl,
CH3CH(-O-C2H5)-, p-Methoxybenzyl,
p-Methoxyphenyl, p-Nitrobenzyl, (Φ)3-C-,
(CH3)3Si-, [CH3-CH(CH3)]3Si-, Φ(CH3)2Si- bevorzugt
sind. Diese Schutzgruppen werden durch dem Fachmann geläufige Mittel
entfernt. Wenn beispielsweise R1 einen Hydroxy-substituenten enthält, muss
dieser während
der Umwandlung des 5-Hydroxymethyl-substituierten Oxazolidinonalkohols
(III) in das 5-Aminomethyl-substituierte Oxazolidinonamin (VII)
oder das 5-Acylamidomethyl-substituierte Oxazolidinon (VIII) geschützt werden.
Wenn der R1-Substituent einen freien primären oder
sekundären
Aminosubstituenten enthält,
muss dieser während
der Bildung der 5-Hydroxymethyl-substituierten Oxazolidinonalkohole
(III) nicht geschützt
werden, jedoch während
der Umwandlung der 5-Hydroxymethyl-substituierten Oxazolidinonalkohole
(III) in die entsprechenden 5-Aminomethyl-substituierten
Oxazolidinonamine (VII) und die 5-Acylamidomethyl-substituierten Oxazolidinone
(VIII) geschützt
werden. Der Grund hierfür
liegt darin, dass die Aminogruppe generell eine unerwünschte Nebenreaktion
während
einer oder mehreren der Stufen, die an der Umwandlung der 5-Hydroxymethyl-substituierten
Oxazolidinonalkohole (III) in die entsprechenden 5-Acylamidomethyl-substituierten
Oxazolidinone (VIII) beteiligt sind, eingeht. Daher ist es bevorzugt,
einen freien Aminosubstituenten in der R1-Funktionalität vor der
Umsetzung der Dihydroxyverbindung (I) oder des Glycidols (IV) mit
dem Carbamat (IIA) oder Trifluoracetamid (IIB) zu schützen. Aminoschutzgruppen
sind dem Fachmann sehr geläufig. Bevorzugte
Aminoschutzgruppen umfassen:
- (I) C1-C4-Alkyl,
- (II) Φ-CH2-,
- (III) (Φ)3C-,
- (IV) Ra-CO-, wobei Ra für (A) H-,
(B) C1-C4-Alkyl,
(C) C5-C7-Cycloalkyl, (D) (C1-C5-Alkyl)-O-, (E) Cl3C-CH2-O-, (F) H2C=CH-CH2-O-, (G) Φ-CH=CH-CH2-O-, (H) Φ-CH2-O-,
(I) p-Methoxyphenyl-CH2-O-, (J) p-Nitrophenyl-CH2-O-, (K) Φ-O-, (L) CH3-CO-CH2-, (M) (CH3)3SiO- steht,
- (V) Rb -SO2-,
worin Rb für (A) (C1-Alkyl)-,
(B) Φ-,
(C) p-Methylphenyl- und (D) Φ-CH2- steht. Eine bevorzugte Aminoschutzgruppe
ist Benzyloxycarbonyl, das durch katalytische Hydrierung entfernt
werden kann, wie dem Fachmann geläufig ist. An der Verwendung
von Schutzgruppen in diesen Reaktionen oder der Natur der speziellen
Schutzgruppen ist nichts Neues. All dieses ist dem Fachmann geläufig. Die
Schutzgruppen können
nach der letzten Reaktion, in der der geschützte Substituent beeinflusst
werden kann, entfernt werden oder weitergeführt werden und nach Folgereaktionen
entfernt werden, wie dies dem Fachmann geläufig ist. Beispielsweise kann
die Schutzgruppe vorzugsweise bis zur Durchführung der Acylierungsstufe
am Ende weitergeführt
werden, bevor sie entfernt wird, wie dies dem Fachmann geläufig ist.
Optional kann der R1-Substituent nach der
Bildung der 5-Acylamidomethyl-substituierten Oxazolidinone (VIII)
in Abhängigkeit davon,
welche chemischen Reaktionen erforderlich sind, modifiziert werden,
wie dies dem Fachmann geläufig
ist.
-
Die
Reaktion von entweder Dihydroxyverbindungen (I) oder Glycidol (IV)
mit entweder den Carbamaten (IIA) oder Trifluoracetamiden (IIB)
ergibt die gleichen 5-Hydroxymethyl-substituierten Oxazolidinonalkohole (III).
Die Wahl, ob eine Dihydroxyverbindung (I) oder ein Glycidol (IV)
zur Herstellung eines speziellen 5-Hydroxymethyl-substituierten
Oxazolidinonalkohols (III) zu verwenden ist, muss auf einer Basis
von Fall zu Fall getroffen werden. Kein Ausgangsmaterial ist in
allen Fällen
bevorzugt; es besteht kein generell bevorzugter Weg allein auf der
Basis der Chemie. Die Entscheidung umfasst die kommerzielle Verfügbarkeit
des speziellen Ausgangsmaterials, dessen chemische und Enantiomerenreinheit,
dessen Kosten usw., was dem Fachmann geläufig ist.
-
Ein
Verfahren ist die Umsetzung der Dihydroxyverbindung (I) oder des
Glycidols (IV) mit dem Carbamat (IIA) oder Trifluoracetamiden (IIB)
in Gegenwart von Lithiumkationen (Li+) und
einer Base, deren konjugierte Säure
einen pKa-Wert von größer als etwa 8 aufweist.
-
Das
Verfahren erfordert etwa ein Moläquivalent
von entweder der Dihydroxyverbindung (I) oder dem Glycidol (IV)/Äquivalent
von Carbamat (IIA) oder Trifluoracetamiden (IIB). Die Reaktion erfordert
eine Base, deren Natur unkritisch ist, sofern sie zur Deprotonierung
des Carbamats (II) stark genug ist. Verwendbare Basen sind solche,
deren konjugierte Säure
einen pKa-Wert von größer als etwa 8 aufweist. Bevorzugte
Basen umfassen Verbindungen, die ausgewählt sind aus der Gruppe von:
Alkoxyverbindungen
mit einem bis sieben Kohlenstoffatomen,
Carbonat,
Methyl-,
sek-Butyl- und tert-Butylcarbanionen,
Tri(alkyl)aminen, wobei
die Alkylgruppe 1 bis 4 Kohlenstoffatome aufweist,
einer konjugierten
Base des Carbamats (II),
DBU,
DBN,
N-Methyl-piperidin,
N-Methyl-morpholin,
2,2,2-Trichlorethoxid
und
Cl3C-CH2-O–;
wobei am stärksten
bevorzugte Basen diejenigen sind, wobei die Base Alkoxy mit 4 bis
5 Kohlenstoffatomen ist. Vorzugsweise sind die Alkoholbasen mit
4 und 5 Kohlenstoffen tert-Amylat oder tert-Gutoxid. Natrium- oder
Kalium-basen in
Kombination mit einem Lithiumsalz (wie Lithiumchlo rid oder Lithiumbromid) können verwendet
werden, wobei das Lithiumkation und die Base in situ gebildet werden.
-
Die
Natur des Lösemittels
ist unkritisch. Verwendbare Lösemittel
umfassen cyclische Ether, wie THF, Amide, wie DMF und DMAC, Amine,
wie Triethylamin, Acetonitril, und Alkohole, wie tert-Amylalkohol
und tert-Butylalkohol. Die Wahl eines Lösemittels hängt von der Löslichkeit
des Carbamats (IIA) oder Trifluoracetamids (IIB) ab, was dem Fachmann
geläufig
ist.
-
Wenn
das Ausgangsmaterial die Dihydroxyverbindungen (I) sind, kann es
vorteilhaft sein, die Dihydroxyverbindung (I) mit einem Cyclisierungsmittel
vor dem Inkontaktbringen mit dem Carbamat (IIA) oder Trifluoracetamid
(IIB) umzusetzen. Der Ausdruck "Cyclisierungsmittel" bezeichnet eine
Base, die die Dihydroxyverbindung (I) mit Glycidol (IV) cyclisiert.
Verwendbare Cyclisierungsmittel umfassen Basen, deren Säure einen pKa-Wert von größer als etwa 7 aufweist; bevorzugte
Cyclisierungsmittel sind Natrium-, Kalium- oder Lithiumbutoxid,
Natrium- oder Kaliumhydroxid, Kaliumcarbonat, DBU, Lithium-, Natrium-
und Kaliumamylat; noch besser ist Kalium-tert-butoxid. Vorzugsweise wird die
Reaktion bei < 100°, noch günstiger
bei < 70°, noch besser bei < 50° und stark
bevorzugt bei < 25° durchgeführt. Die
Reaktion kann bei Raumtemperatur (etwa 20 bis etwa 25°) durchgeführt werden.
Bei etwa 20° sind
etwa 8 h zum Erreichen einer vollständigen Umsetzung (in DMAC) erforderlich.
Wenn eine schnellere Reaktion gewünscht wird, kann die Reaktion
bei höherer
Temperatur durchgeführt
werden. Wie oben angegeben, ist eine Differenzierung zwischen primären Alkoholen
und sekundären Alkoholen
schwierig. In der Cyclisierungsreaktion wird ein einfacher Alkohol
gebildet. Beispielsweise wird Benzylalkohol gebildet, wenn ein Benzylcarbonat
den Cyclisierungsbedingungen unterworfen wird. Ein Entfernen dieses
Alkohols ist für
eine erfolgreiche Umwandlung des Alkohols in ein Amin notwendig.
Dies wird durch Kristallisation unter Verwendung von Ethylacetat/Heptan (1/2)
erreicht. Der Benzylalkohol bleibt in der Lösung und der gewünschte Oxazolidinonalkohol
wird als Feststoff isoliert.
-
Das
Reaktionsschema C offenbart die Verfahren der Umwandlung der 5-Hydroxymethyl-substituierten Oxazolidinonalkohole
(III) in die entsprechenden 5-Aminomethyl-substituierten Oxazolidinonamine
(VII). Die Situation des Schützens
der Alkohol- und/oder
Aminogruppen an der R1-Funktionalität wurde
oben diskutiert. Die 5-Hydroxymethyl-substituierten Oxazolidinonalkohole
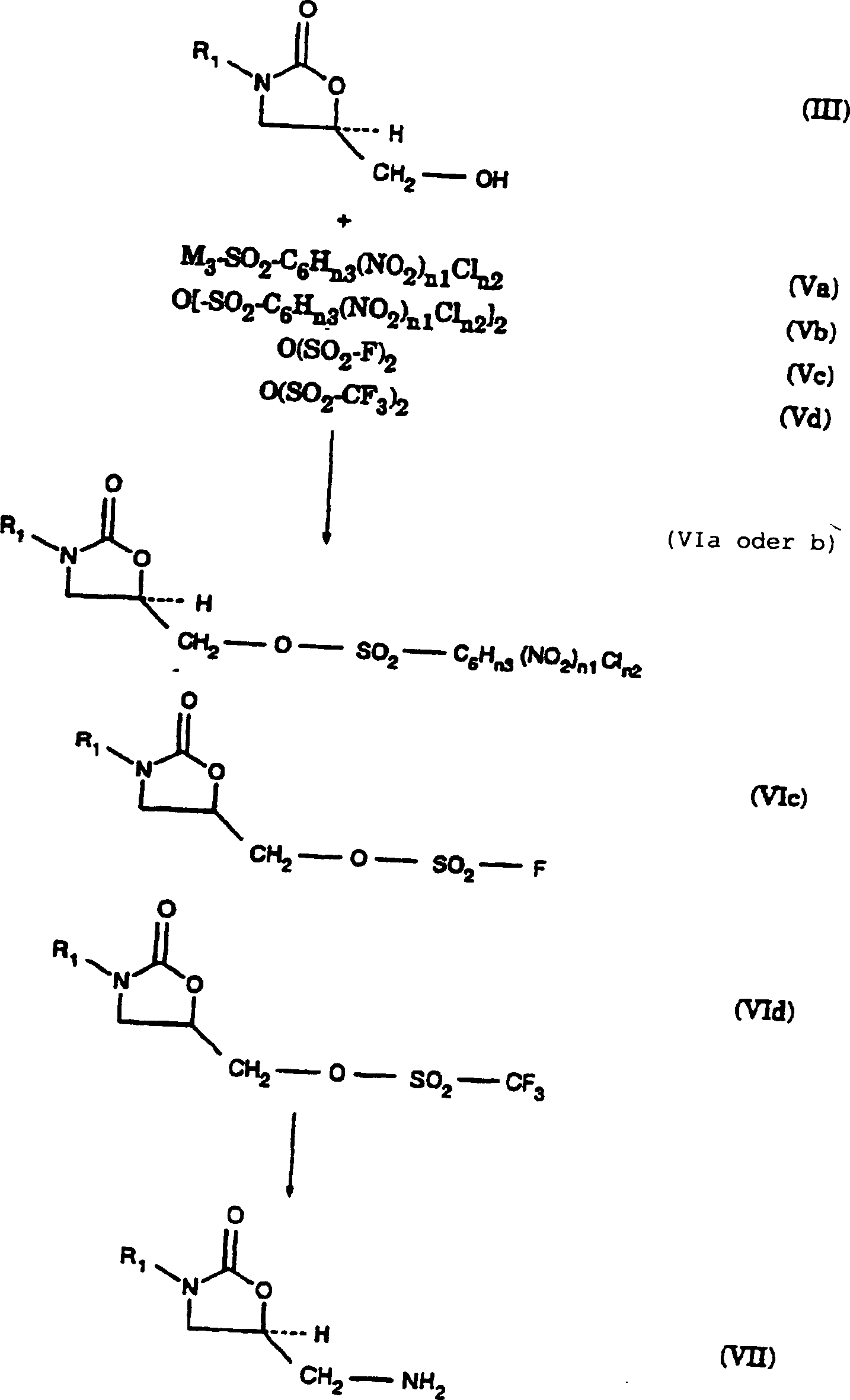
(III) werden mit einem Sulfonylierungsmittel (Va–Vd) von vier Typen in Kontakt gebracht. Diese
sind M3-SO2-C6Hn3(NO2)n1Cln2 (Va), O[-SO2-C6Hn3(NO2)n1Cln2]2 (Vb), O(SO2-F)2 (Va) und O(SO2-CF3)2 (Vd).
M3 ist eine Abgangsgruppe, die Cl- oder Br- umfasst;
vorzugsweise ist M3 Cl-. Die 5-Hydroxymethyl-substituierten
Oxazolidinone (III) werden mit einem Sulfonylierungsmittel (Va–Vd) in Kontakt gebracht, wobei ein Oxazolidinonsulfonat(VIa–VId)-Zwischenprodukt gebildet wird.
-
Die
Sulfonierungsreaktion der Umwandlung der 5-Hydroxymethyl-substituierten
Oxazolidinone (III) in die entsprechenden Oxazolidinonsulfonate
(VI) wird durch Inkontaktbringen der 5-Hydroxymethyl-substituierten
Oxazolidinone (III) mit mindestens einem Moläquivalent des Sulfonylierungsmittels
(Va–Vd) in Gegenwart einer Base in einem inerten
Lösemittel
bei etwa 0° durchgeführt. Verwendbare
Basen umfassen Triethylamin, Tributylamin, Diisopropylethylamin,
DABCO, DBU, DBN, n-Butyllithium, Ethylmagnesiumchlorid und die Äquivalente
derselben; bevorzugt ist Triethylamin. Inerte Lösemittel umfassen die meisten
organischen Lösemittel, wie
Methylenchlorid, THF, DMA, DMF, Ethylacetat und Äquivalente derselben; bevorzugt
ist Methylenchlorid.
-
Die
Ammonolysereaktion der Umwandlung der Oxazolidinonsulfonate (VI)
in die entsprechenden 5-Aminomethyl-substituierten Oxazolidinonamine
(VII) wird unter offenen oder nicht herme tisch verschlossenen Bedingungen
oder unter hermetisch verschlossenen Bedingungen durchgeführt, obwohl
sie vorzugsweise unter hermetisch verschlossenen Bedingungen durchgeführt wird.
-
In
jedem Fall wird die Ammonolysereaktion durch Inkontaktbringen der
Oxazolidinonsulfonate (VI) mit Ammoniak (vorzugsweise wässrigem)
vorzugsweise mit einem Lösemittel
oder Lösemittelgemisch
durchgeführt.
Bevorzugte Lösemittel
sind diejenigen, die sowohl die Oxazolidinonsulfonate (VI) als auch
das wässrige Ammoniak
lösen,
da durch Lösen
der beiden der Kontakt zwischen diesen sichergestellt wird. Jedoch
ist das Verfahren auch mit Lösemitteln
durchführbar,
die die Oxazolidinonsulfonate (VI) nur partiell lösen, der
Nachteil besteht darin, dass die Reaktion generell langsamer ist.
Für den
Fall der m-Nitrobenzolsulfonate ist das bevorzugte Lösemittel
ein Gemisch von Acetonitril/Isopropanol oder THF/Isopropanol. Das
System wird unter verminderten Druck gesetzt. Das System wird dann
verschlossen oder hermetisch verschlossen und das Ammoniak (vorzugsweise
wässriges
Ammoniak) wird zugesetzt und auf weniger als 50°, vorzugsweise weniger als 40°, vorzugsweise
etwa 38° (etwa
3 psig) erhitzt. Bei etwa 38–40° beträgt der Druck
etwa 0 bis etwa 10 psig, was gut unter dem Ceiling-Druckwert von allgemein
verwendbaren Reaktoren liegt. Unter diesen Bedingungen beträgt bei etwa
60° der
psig-Wert etwa 20. Vorzugsweise wird die Ammonolysereaktion bei
einem Druck von etwa 0 bis etwa 20 psig, noch günstiger etwa 0 bis etwa 5 psig
und bei etwa 60° oder
weniger durchgeführt. Alternativ
wird die Reaktion in einem offenen System unter Refluxieren durchgeführt. In
diesem Fall ist die Temperatur etwas niedriger und die Reaktion
benötigt
etwas länger
bis zu einer vollständigen
Durchführung. Das
Ammoniak kann entweder wässrig,
alkoholisch oder wasserfrei sein; jedoch ist wässriges Ammoniak bevorzugt.
-
Alternativ
kann das Inkontaktbringen mit wässrigem
Ammoniak in Gegenwart eines aromatischen Aldehyds (IX, Ar-CHO),
vorzugsweise von Salicylaldehyd, durchgeführt werden. Die 5-Aminomethyl-substituierten
Oxazolidinonamine (VII) und der Aldehyd (IX) bilden eine Schiff-Base
der Formel (Oxazolidinon-N=CH-Ar), die dann mit einer wässrigen
Säure,
wie dem Fachmann geläufig
ist, hydrolysiert wird, wobei die gewünschten 5-Aminomethyl-substituierten
Oxazolidinonamine (VII) erhalten werden. Der aromatische Aldehyd
(IX) ist zur Unterdrückung
einer Dimerbildung verwendbar.
-
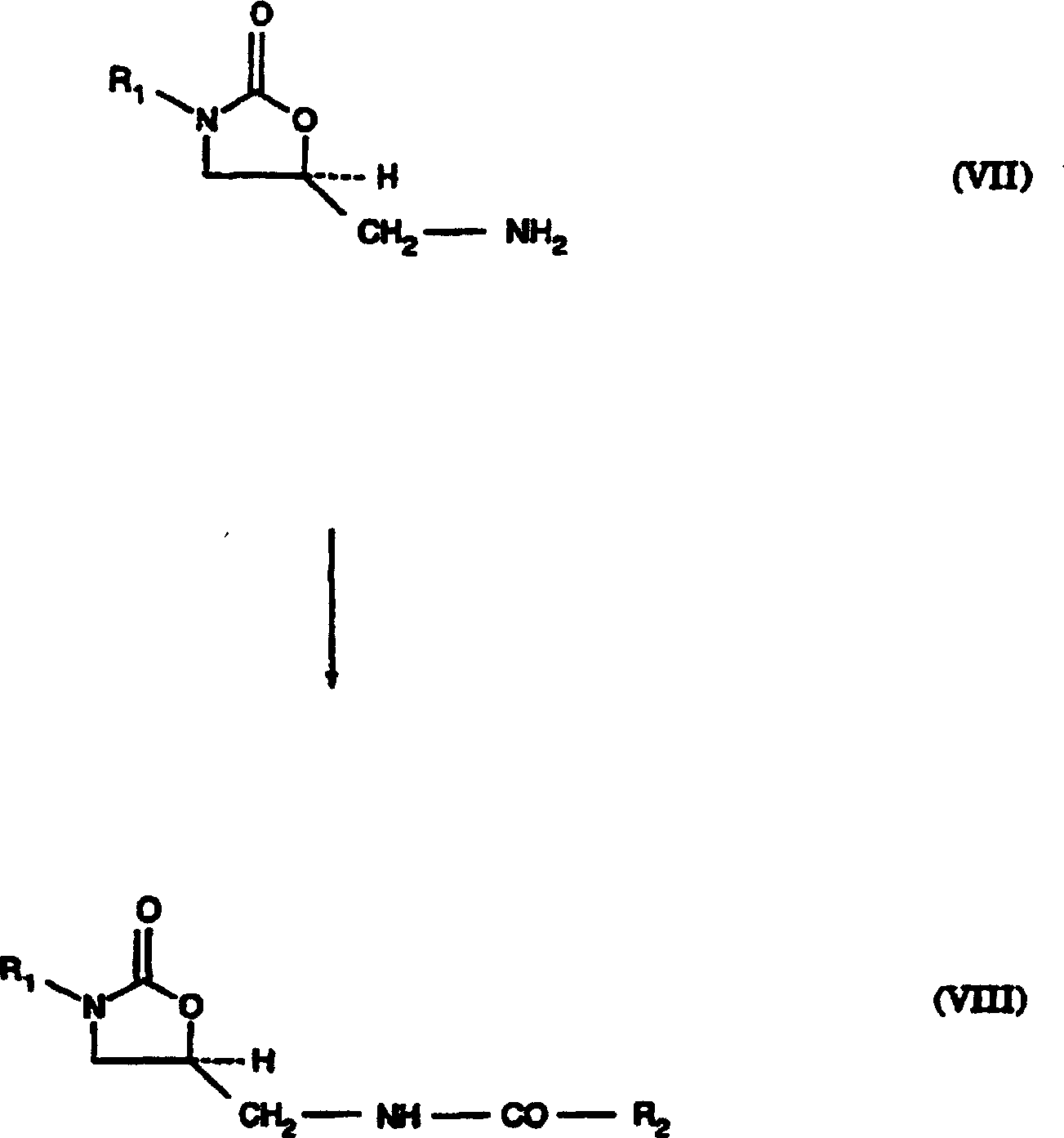
Die
5-Aminomethyl-substituierten Oxazolidinonamine (VII) werden durch
bekannte Mittel, wie Acylhalogenide oder Acylanhydride, acyliert,
wobei das entsprechende 5-Acylamidomethyl-substituierte Oxazolidinon
(VIII) gebildet wird, siehe Reaktionsschema D. Eine Alkohol- oder
Aminoschutzgruppe muss nach der Herstellung der 5-Acylamidomethyl-substituierten
Oxazolidinone (VIII) entfernt werden. Jedoch können sie in Abhängigkeit
von den in Frage kommenden speziellen Substituenten, wie dem Fachmann
geläufig
ist, früher
in der Reaktionsfolge entfernt werden.
-
Die
5-Acylamidomethyl-substituierten Oxazolidinone (VIII) sind als antibakterielle
Pharmazeutika bekannt. R2 ist aus der Gruppe
von -H, C1-C12-Alkyl,
das optional mit einem oder mehreren Halogen substituiert ist, (C3-C7)Cyclo(C5-C9)alkyl oder -O-R2a, worin R2a für C1-C6-Alkyl steht,
ausgewählt.
Vorzugsweise steht R2 für C1-Alkyl.
-
Definitionen und Übereinkünfte
-
Die
folgenden Definitionen und Erklärungen
gelten für
die durchgängig
in diesem gesamten Dokument einschließlich sowohl der Beschreibung
als auch der Ansprüche
verwendeten Ausdrücke.
-
I. Übereinkünfte für Formeln
und Definitionen von Variablen
-
Die
chemischen Formeln verschiedener Verbindungen oder Molekülfragmente
in der Beschreibung und den Ansprüchen können variable Substituenten
zusätzlich
zu ausdrücklich
definierten Strukturmerkmalen enthalten. Diese variablen Substituenten
werden durch einen Buchstaben oder einen Buchstaben mit einem anschließenden Zahlenindex,
beispielsweise "Z1" oder "Ri", wobei "i" eine ganze Zahl ist, angegeben. Diese variablen
Substituenten sind entweder einwertig oder zweiwertig, d. h. sie
stehen für
eine Gruppe, die über eine
oder zwei chemische Bindungen an der Formel hängt. Beispielsweise steht eine
Gruppe Z1 für eine zweiwertige Variable,
wenn sie an der Formel CH3-C(=Z1)H
hängt.
Die Gruppen Ri und Rj stehen
für einwertige
variable Substituenten, wenn sie an der Formel CH3-CH2-C(Ri)(Rj)-H hängen.
Wenn chemische Formeln in linearer Form, wie oben, gezeichnet sind,
sind in Klammern enthaltene variable Substituenten an das unmittelbar links
des in Klammern eingeschlossenen variablen Substituenten stehende
Atom gebunden. Wenn zwei oder mehr aufeinanderfolgende variable
Substituenten in Klammern eingeschlossen sind, ist jeder der aufeinanderfolgenden
variablen Substituenten an das links unmittelbar vorher stehende
Atom, das nicht in Klammern eingeschlossen ist, gebunden. Deshalb
sind in der obigen Formel sowohl Ri als
auch Rj an das vorhergehende Kohlenstoffatom
gebunden. Außerdem
werden bei einem beliebigen Molekül mit einem etablierten System
der Kohlenstoffnummerierung, wie Steroiden, diese Kohlenstoffatome
mit Ci, wobei "i" die
der Kohlenstoffatomnummer entsprechende ganze Zahl ist, bezeichnet.
Beispielsweise bezeichnet C6 die Position
6 oder die Kohlenstoffatomnummer in dem Steroidkern gemäß der traditionellen
Bezeichnung durch Fachleute auf dem Gebiet der Steroidchemie. In ähnlicher
Weise bedeutet der Ausdruck "R6" einen
variablen Substituenten (entweder einwertig oder zweiwertig) an
der C6-Position.
-
In
linearer Form gezeichnete chemische Formeln oder Teile derselben
bedeuten Atome in einer linearen Kette. Das Symbol "-" bedeutet im allgemeinen eine Bindung
zwischen zwei Atomen in der Kette. Daher bedeutet CH3-O-CH2-CH(Ri)-CH3 eine 2-substituierte
1-Methoxypropanverbindung. In ähnlicher
Weise bedeutet das Symbol "=" eine Doppelbindung,
z. B. CH2=C(Ri)-O-CH3 und
das Symbol "=" eine Dreifachbindung,
z. B. HC≡C-CH(Ri)-CH2-CH3. Carbonylgruppen
werden auf eine von zwei Arten dargestellt: -CO- oder -C(=O)-, wobei
die erstere der Einfachheit wegen bevorzugt ist.
-
Chemische
Formeln von cyclischen (Ring)Verbindungen oder Molekülfragmenten
können
in linearer Form dargestellt werden. Daher kann die Verbindung 4-Chlor-2-methylpyridin
in linearer Form durch N*=C(CH3)-CH=CCl-CH=C*H
mit der Übereinkunft,
dass die mit einem Stern (*) markierten Atome aneinander gebunden
sind, was zur Bildung eines Rings führt, dargestellt werden. In ähnlicher
Weise kann das cyclische Molekülfragment
4-(Ethyl)-1-piperazinyl durch -N*-(CH2)2-N(C2H5)-CH2-C*H2 dargestellt
werden.
-
Eine
starre cyclische (Ring)Struktur für beliebige Verbindungen legt
hierbei für
Substituenten, die an jedem der Kohlenstoffatome der starren cyclischen
Verbindung hängen,
eine Orientierung bezüglich
der Ringebene fest. Bei gesättigten
Verbindungen, die zwei an einem Kohlenstoffatom, das Teil eines
cyclischen Systems ist, hängende
Substituenten besitzen, -C(X1)(X2)-, können
die zwei Substituenten relativ zum Ring entweder in axialer oder äquatorialer
Position sein und zwischen axial/äquatorial wechseln. Die Position
der zwei Substituenten relativ zum Ring und zueinander bleibt jedoch
fixiert. Zwar kann jeder der zwei Substituenten zeitweilig in der
Ebene des Rings (äquatorial)
im Gegensatz zu oberhalb oder unterhalb der Ebene (axial) liegen,
doch ist ein Substituent immer oberhalb des anderen. In solche Verbindungen
beschreibenden chemischen Strukturformeln wird ein Substituent (X1), der "unterhalb" eines anderen Substituenten
(X2) liegt, als in alpha(α)-Konfiguration
vorliegend angegeben und durch eine Verbindung mit dem Kohlenstoffatom
durch eine unterbrochene, gestrichelte oder punktierte Linie, d.
h. das Symbol "---" oder "..." angegeben. Der entsprechende
Substituent (X2), der "oberhalb" des anderen (X1)
gebunden ist, wird als in beta(β)-Konfiguration
vorliegend angegeben und durch eine Verbindung mit dem Kohlenstoffatom
durch eine nicht-unterbrochene Linie angegeben.
-
Wenn
ein variabler Substituent zweiwertig ist, können die Valenzen bei der Festlegung
der Variablen zusammengenommen oder getrennt oder beides genommen
werden. Beispielsweise kann eine an einem Kohlenstoffatom hängende Variable
Ri, wie -C(=Ri)-,
zweiwertig sein und als Oxo oder Keto (wodurch eine Carbonylgruppe
(-CO-) gebildet wird) oder als zwei getrennt gebundene einwertige
variable Substituenten α-Ri-j und β-Ri-k definiert sein. Wenn eine zweiwertige
Variable Ri als aus zwei einwertigen variablen
Substituenten bestehend definiert wird, besteht die zur Festlegung
der zweiwertigen Variablen verwendete Übereinkunft aus der Form "α-Ri-j:β-Ri-k" oder
einer Variante derselben. In diesem Fall hängen sowohl α-Ri-j als auch β-Ri-k an
dem Kohlenstoffatom unter Bildung von -C(α-Ri-j)(β-Ri-k)-. Beispielsweise sind, wenn die zweiwertige
Variable R6, -C(=R6)-,
als aus zwei einwertigen variablen Substituenten bestehend definiert
wird, die zwei einwertigen variablen Substituenten α-R6-1:β-R6-2, ... α-R6-9:β-R6-10 usw., wobei -C(α-R6-1)(β-R6-2)-, ... -C(α-R6-9)(β-R6-10) usw. erhalten wird. In ähnlicher
Weise sind für
die zweiwertige Variable R11, -C(=R11)-, zwei einwertige variable Substituenten α-R11-1:β-R11-2. Für
einen Ringsubstituenten, für
den getrennte α-
und β-Orientierungen
nicht existieren (beispielsweise aufgrund des Vorliegens einer Kohlenstoff-Kohlenstoff-Doppelbindung im
Ring), und für
einen an ein Kohlenstoffatom, das nicht Teil eines Rings ist, gebundenen
Substituenten wird die obige Übereinkunft dennoch
verwendet, wobei jedoch die Bezeichnungen α und β weggelassen werden.
-
Genau
wie eine zweiwertige Variable als zwei getrennte einwertige variable
Substituenten definiert werden kann, können zwei getrennte einwertige
variable Substituenten zusammengenommen zur Bildung einer zweiwertigen
Variablen definiert werden. Beispielsweise können in der Formel -C1(Ri)H-C2-(Rj)H-
(C1 und C2 definieren
willkürlich
ein erstes bzw. zweites Kohlenstoffatom) Ri und
Rj zusammengenommen so definiert werden,
dass sie (1) eine zweite Bindung zwischen C1 und
C2 oder (2) eine zweiwertige Gruppe, wie
Oxa (-O-), bilden und die Formel dadurch ein Epoxid beschreibt.
Wenn Ri und Rj zur
Bildung einer komplexeren Einheit, beispielsweise der Gruppe -X-Y-
zusammengenommen sind, ist die Orientierung der Einheit derart,
dass C1 in der obigen Formel an X und C2 an Y gebunden ist. Daher bedeutet die Bezeichnung "... Ri und
Ri bilden zusammengenommen -CH2-CH2-O-CO-..." nach der Übereinkunft ein Lacton, in
dem das Carbonyl an C2 gebunden ist. Die
Bezeichnung "...
Rj und Ri bilden
zusammengenommen -CO-O-CH2-CH2-" bedeutet jedoch
nach der Übereinkunft
ein Lacton, in dem das Carbonyl an C1 gebunden
ist.
-
Der
Kohlenstoffatomgehalt variabler Substituenten wird auf eine von
zwei Arten angegeben. Das erste Verfahren verwendet ein Präfix zum
Gesamtnamen der Variablen, wie "C1-C4", wobei sowohl "1" als auch "4" ganze
Zahlen sind, die die minimale und maximale Anzahl von Kohlenstoffatomen
in der Variablen angeben. Das Präfix
ist von der Variablen durch einen Zwischenraum (in der deutschen Übersetzung:
-) getrennt. Beispielsweise steht "C1-C4-Alkyl" für Alkyl
mit 1–4
Kohlenstoffatomen (einschließlich
isomerer Formen derselben, wenn nicht ausdrücklich das Gegenteil angegeben
ist). Jedes Mal wenn dieses einzige Präfix angegeben ist, gibt das
Präfix
den Gesamtkohlenstoffatomgehalt der zu definierenden Variablen an.
Daher beschreibt C2-C4-Alkoxycarbonyl
eine Gruppe CH3-(CH2)n-O-CO-, wobei n 0, 1 oder 2 ist. Nach dem
zweiten Verfahren wird der Kohlenstoffatomgehalt von lediglich jedem
Teil der Definition getrennt angegeben, indem die "Ci-Cj"-Bezeichnung in in
Klammern gesetzt und unmittelbar (kein Zwi schenraum) vor den zu
definierenden Teil der Definition gesetzt wird. Nach dieser optionalen Übereinkunft
hat (C1-C3)Alkoxycarbonyl
die gleiche Bedeutung wie C2-C4-Alkoxycarbonyl, da
sich "C1-C3" lediglich
auf den Kohlenstoffatomgehalt der Alkoxygruppe bezieht. In ähnlicher
Weise definieren zwar sowohl C2-C6-Alkoxyalkyl als auch (C1-C3)-Alkoxy(C1-C3)alkyl Alkoxyalkylgruppen,
die 2 bis 6 Kohlenstoffatome enthalten, doch unterscheiden sich
die zwei Definitionen, da nach der ersten Definition entweder der
Alkoxyteil oder der Alkylteil allein 4 oder 5 Kohlenstoffatome enthalten
kann, während
die letztere Definition jede dieser zwei Gruppen auf 3 Kohlenstoffatome
beschränkt.
-
Wenn
die Ansprüche
einen ziemlich komplexen (cyclischen) Substituenten enthalten, steht
am Ende der den speziellen Substituenten bezeichnenden Phrase eine
Angabe (in Klammern), die der gleichen Bezeichnung in einem der
Reaktionsschemata entspricht, in denen auch die chemische Strukturformel
des speziellen Substituenten angegeben ist.
-
II. Definitionen
-
Alle
Temperaturen sind in Grad Celsius angegeben.
- DC bezeichnet
Dünnschichtchromatographie.
- THF bezeichnet Tetrahydrofuran.
- DMF bezeichnet Dimethylformamid.
- DBU bezeichnet 1,8-Diazabicyclo[5.4.0]undec-7-en.
- DBN bezeichnet 1,5-Diazabicyclo[4.3.0]non-5-en.
- DABCO bezeichnet 1,4-Diazabicyclo[2.2.0]octan.
- DMA bezeichnet Dimethylacetamid.
- Kochsalzlösung
bezeichnet eine wässrige
gesättigte
Natriumchloridlösung.
- Chromatographie (Säulen-
und Flashchromatographie) bezeichnet die Reinigung/Trennung von
Verbindungen, ausgedrückt
als (Träger;
Eluens). Es ist klar, dass die entsprechenden Fraktionen gepoolt
und konzentriert werden, wobei die gewünschte(n) Verbindung(en) erhalten
werden.
- IR bezeichnet Infrarotspektroskopie.
- CMR bezeichnet C-13-Kernresonanzspektroskopie, wobei die chemischen
Verschiebungen in ppm (δ)
von TMS zu niederem Feld angegeben sind.
- NMR bezeichnet (Proton)kernresonanzspektroskopie, wobei die
chemischen Verschiebungen in ppm (δ) von Tetramethylsilan zu niederem
Feld angegeben sind.
- -Φ bezeichnet
Phenyl (C6H5).
- [α]D25 bezeichnet den Rotationswinkel von linear
polarisiertem Licht (spezifische optische Drehung) bei 25° mit der
Natrium-D-Linie (589 Å).
- MS bezeichnet Massenspektrometrie, ausgedrückt als m/e, m/z oder Masse/Ladungseinheit.
- [M+H]+ bezeichnet das positive Ion einer
Mutterverbindung plus ein Wasserstoffatom. EI bezeichnet Elektronenstoß. CI bezeichnet
chemische Ionisierung. FAB bezeichnet Beschuss mit schnellen Atomen.
- HRMS bezeichnet hochaufgelöste
Massenspektrometrie.
- Pharmazeutisch akzeptabel bezeichnet die Eigenschaften und/oder
Substanzen, die für
den Patienten aus pharmakologischer/toxikologischer Sicht und für den herstellenden
pharmazeutischen Chemiker aus physikalischer/chemischer Sicht im
Hinblick auf Zusammensetzung, Formulierung, Stabilität, Patientenakzeptanz
und Bioverfügbarkeit
akzeptabel sind.
-
Wenn
Lösemittelpaare
verwendet werden, sind die Verhältnisse
verwendeter Lösemittel
als Volumen/Volumen (V/V) angegeben.
-
Wenn
die Löslichkeit
eines Feststoffs in einem Lösemittel
verwendet wird, ist das Verhältnis
von Feststoff zu Lösemittel
als Gewicht/Volumen (Gew/V) angegeben.
- NNNNNN-NN-N bezeichnet
die Registrierungsnummern von Chemical Abstracts Service (CAS, Columbus, Ohio),
wobei jedes "N" eine ganze Zahl
von 0 bis 9 ist, jedoch voranstehende Nullen im sechsstelligen Teil
der Nummer weggelassen sind. Registrierungsnummern werden einer
speziellen chemischen Verbindung durch CAS-Kriterien zugeordnet,
vorausgesetzt, die Existenz der Verbindung wurde ermittelt und sie
wurde irgendwie charakterisiert. Verbindungen, die ab etwa 1967
bis zur Gegenwart veröffentlicht
sind, sind öffentlich
registriert und die Registrierungsnummer ist der Schlüssel, um
Verweisstellen in der CAS-Datenbank für eine derartige registrierte
Verbindung zu finden. Die CAS-Datenbank steht bei mehreren Datenbankbetreibern,
wie STN International, System Development Corporation (SDC) Orbit
Search Service, Lockheed Dialog, Bibliographic Retrieval Systems,
Questrel und dergleichen öffentlich
zur Verfügung.
CAS-Registrierungsnummern sind in den "Beispielen" für
einige der Verbindungen, die registriert sind, angegeben.
- "psig" bezeichnet den "Manometerdruck", der gleich dem
Druck (in psi) minus 1 Atmosphäre
(14,7 psi) ist.
-
Beispiele
-
Es
wird angenommen, dass ein Fachmann ohne weiteren Arbeitsaufwand
unter Verwendung der vorhergehenden Beschreibung die vorliegende
Erfindung in vollem Ausmaß durchführen kann.
Die folgenden detaillierten Beispiele beschreiben, wie die verschiedenen
Verbindungen hergestellt und/oder die verschiedenen Verfahren der
Erfindung durchgeführt
werden können
und sie sollen lediglich der Erläuterung
dienen und in keinster Weise Beschränkungen der vorhergehenden
Offenbarung darstellen. Dem Fachmann sind ohne weiteres entsprechende
Variationen der Verfahren sowohl im Hinblick auf die Reaktionsteilnehmer
als auch die Reaktionsbedingungen und -techniken klar.
-
Beispiel 1
-
(R)-[N-3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methanol
(III)
-
Ein
Gemisch von N-Carbobenzoxy-3-fluor-4-(N-carbobenzoxypiperazinyl)anilin
(II, J. Med. Chem., 39(3), 673 (1996), 100 g von zu 98,4% reinem
Material, 0,2133 mol) in DMAC (300 ml) wird auf 0° gekühlt. In einem
getrennten Kolben wird ein Gemisch von tert-Amylalkohol (75 ml,
60,37 g, 0,685 mol, 3,23 eq) und Heptan (75 ml) auf –10° gekühlt und
mit n-Butyllithium
in Heptan (290 ml, 203 g einer 14,4%igen (Gew/V) Lösung, die
29,2 g oder 0,456 mol = 2/15 eq n-Butyllithium enthält) behandelt, wobei die Temperatur
unter 10° gehalten wird.
Das Lithium-tert-amylatgemisch wird dann zu dem N-Carbobenzoxy-3-fluor-4-(N-carbobenzoxypiperazinyl)anilin
(II) unter Halten der Temperatur unter 10° gegeben.
-
Pures
S-(+)-3-Chlor-1,2-propandiol (I, CAS Nr. 60827-45-4, 22 ml, 29,1
g, 0,263 mol, 1,24 eq) wird dann zugegeben, wobei mit einer kleinen
Menge Heptan gespült
wird. Das Reaktionsgemisch wird dann bei 20–25° gerührt und durch DC (Methanol/Methylenchlorid;
5/95) überwacht,
bis die Reaktion vollständig
ist. Das Reaktionsgemisch wird dann zu einem Gemisch von Essigsäure (40
ml, 42,0 g, 0,699 mol, 3,29 eq) in Methanol (700 ml) und Wasser
(700 ml) gegeben. Die gebildete Aufschlämmung wird bei 20–25° 30 min gerührt, auf 0° gekühlt, bei
0° 30 min
gerührt
und filtriert. Der Kuchen wird mit Methanol/Wasser (50/50) gewaschen
und unter vermindertem Druck getrocknet, wobei die Titelverbindung
erhalten wird. DC (Methylenchlorid/Methanol, 95/5) Rf =
0,43.
-
Beispiel 2
-
(R)-[N-3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methanol
(III)
-
tert-Amylalkohol
(0,967 g, 10,97 mmol, 2,571 eq) wird auf –10° gekühlt. Butyllithium (4,3 ml,
2,5 M in Hexanen, 10,8 mmol, 2,5 eq) wird unter Rühren zugegeben,
während
die Temperatur bei weniger als 5° gehalten
wird.
-
N-Carbobenzoxy-3-fluor-4-(N-carbobenzoxypiperazinyl)anilin
(II, 1,9780 g, 4,267 mmol, 1,000 eq) und Dimethylacetamid (6,2 ml)
werden gemischt, gerührt
und auf –25° gekühlt, wobei
eine dünne
Aufschlämmung erhalten
wird. Das Lithium-tert-amylatgemisch
wird dann zu dem N-Benzyloxycarbonyl-3-fluor-4-((4-benzyloxycarbonyl)-1-piperazinyl)anilin(II)-Gemisch gegeben,
während
weniger als –20° beibehalten
werden. Das gebildete Gemisch wird auf 0° erwärmt und mit S-(+)-3-Chlor-1,2-propandiol
(I, 0,5672 g, 5,131 mmol, 1,20 eq) versetzt. Das erhaltene Gemisch
wird auf 21° erwärmt und
7,5 h gerührt.
-
Das
Reaktionsgemisch wird zu einem Gemisch von Methanol (28 ml) und
Eisessig (0,73 ml, 12,75 mmol) bei 20–22° gegeben. Die erhaltene Aufschlämmung wird
dann auf –30° gekühlt und
das Produkt wird durch Vakuumfiltration gewonnen und mit Methanol
von –30° gewaschen.
Die Feststoffe werden in einem Stickstoffstrom getrocknet, wobei
die Titelverbindung erhalten wird.
DC (Elutionsmittel Chloroform/Methanol,
90/10), Rf = 0,67. CMR (CDCl3)
43,91, 46,39, 50,58, 62,60, 67,29, 72,89, 107,21, 107,56, 113,85,
119,36, 127,92, 128,09, 128,52, 133,51, 133,65, 136,05, 136,17,
136,57, 153,91, 154,80, 155,25 und 157,17 δ; NMR (CDCl3)
7,43, 7,31-7,37, 7,09, 6,88, 5,15, 4,67-4,90, 3,89-3,99, 3,67-3,74,
3,66, 3,25 und 2,98 δ MS
(CI, m/e) = 430 (100%, P + 1).
-
Beispiel 3
-
(R)-[N-3-(3-Fluor-4-(4-morpholinylphenyl)-2-oxo-5-oxazolidinyl]methanol
(III)
-
Tetrahydrofuran
(3,0 ml) und tert-Amylalkohol (0,66 ml, 6,03 mmol, 2,00 eq) werden
gemischt. Butyllithium (1,8 ml, 2,5 M in Hexanen, 4,55 mmol, 1,5
eq) wird unter Rühren
und unter Halten bei weniger als 2,5° zugegeben.
-
N-Carbobenzoxy-3-fluor-4-morpholinylanilin
(II, J. Med. Chem., 39(3), 673 (1996), 0,9942 g, 3,009 mmol, 1,000
eq) und Tetrahydrofuran (3,5 ml) werden als Gemisch gerührt und
gekühlt.
Das Lithium-tert-amylatgemisch wird dann zu dem Carbamat(II)-Gemisch
unter Halten einer Temperatur von weniger als 8° gegeben und mit Tetrahydrofuran
(1 ml) hineingespült.
-
Tetrahydrofuran
(3,2 ml) und S-(+)-3-Chlor-1,2-propandiol (I, 0,299 ml, 3,58 mmol,
1,19 eq) werden gemischt. Das Gemisch wird auf –16° gekühlt und mit Kalium-tert-butoxid
(3,2 ml, 1,0 M in Tetrahydrofuran, 3,2 mmol, 1,07 eq) versetzt,
während
die Temperatur bei weniger als –10° gehalten
wird. Die erhaltene Aufschlämmung
wird bei –14
bis 0° 1
h gerührt
und dann zu dem Lithiumanionengemisch gegeben, während beide Gemische bei 0° gehalten
werden, und dann mit THF (2 ml) gespült. Die erhaltene Aufschlämmung wird
bei 20–23° 2 h gerührt, dann
auf 6° gekühlt und
mit einem Gemisch von Citronensäuremonohydrat
(0,4459 g, 2,122 mmol, 0,705 eq) in Wasser (10 ml) versetzt. Die
erhaltenen flüssigen
Phasen werden getrennt und die untere wässrige Phase wird mit Ethylacetat
(12 ml) gewaschen. Die organischen Schichten werden vereinigt und
Lösemittel
wird unter vermindertem Druck entfernt, bis ein Nettogewicht von
9,73 g verbleibt. Heptan (10 ml) und Wasser (5 ml) werden zugegeben
und das Lösemittel
wird unter vermindertem Druck entfernt, bis ein Gesamtvolumen von
5 ml verbleibt. Das ausgefallene Produkt wird durch Vakuumfiltration
gewonnen und mit Wasser (7 ml) gewaschen. Die Feststoffe werden
in einem Stickstoffstrom getrocknet, wobei die Titelverbindung erhalten
wird.
DC (Chloroform/Methanol, 95/5), Rf =
0,23; CMR (CDCl3) 46,42, 51,01, 62,58, 73,07,
107,29, 107,64, 113,94, 118,80, 118,85, 128,28, 128,61, 133,15,
133,29, 136,26, 136,38, 153,82, 154,92 und 157,08 δ; NMR (CDCl3) 7,42, 7,32-7,37, 7,10, 4,67-4,75, 3,90-4,00,
3,86, 3,70-3,73, 3,44 und 3,03 δ;
MS (EI, m/e) = 296.
-
Alternativ
kann das rohe Produkt mit Methylenchlorid extrahiert werden. Das
Lösemittel
wird unter vermindertem Druck entfernt. Die Feststoffe werden in
heißem
Ethylacetat erneut gelöst,
Heptan wird zugegeben und das Gemisch wird gekühlt und die Titelverbindung
wird gewonnen.
-
Beispiel 4
-
(R)-[N-3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methanol
(III)
-
Eine
Lösung
von tert-Amylalkohol (75 ml, 60,3 g, 0,68 m) und Heptan (75 ml)
wird gerührt
und auf –10° gekühlt. Das
Gemisch wird mit n-Butyllithium in Heptan (1,6 M, 0,46 m, 290 ml) über einen
Zeitraum von 30 min unter Halten bei einer Temperatur von < 10° behandelt.
Nach 30 min wird das Gemisch von Lithium-tert-amylat zu einem Gemisch
von N-Carbobenzoxy-3-fluor-4-(N-carbobenzoxypiperazinyl)anilin
(II, 100 g, 0,22 m) und Dimethylacetamid (300 ml) bei 0° unter Halten
einer Temperatur von < 10° gegeben.
Das Gemisch wird 30 min gerührt,
dann mit S-(+)-3-Chlor-1,2-propandiol (I, 22 ml, 0,26 m) behandelt.
Die Kühlung
wird entfernt und das Gemisch wird sich auf 20–25° erwärmen gelassen. Die Reaktion
wird durch DC überwacht
und nach etwa 1 h als vollständig
erachtet. Das Reaktionsgemisch wird in ein Gemisch von Methanol
(700 ml), Wasser (700 ml) und Essigsäure (40 ml) gegossen und 30
min bei 20-25° gerührt, dann
30 min unter Kühlen auf
0° gerührt. Das
Gemisch wird filtriert, mit wässrigem
Methanol (50/50) gewaschen und unter vermindertem Druck bei 45° getrocknet,
wobei die Titelverbindung erhalten wird. DC (Silicagel; Methanol/Methylenchlorid, 5/95)
Rf = 0,5 (90,3% Ausbeute).
-
Beispiel 5
-
3-Nitrobenzolsulfonatester von (R)-[N-3-[3-fluor-4-(N-1-(4-carbobenzoxy)piperazinyl]-phenyl]-2-oxo-5-oxazolidinyl]methanol
(VI)
-
Ein
Gemisch von (R)-[N-3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methanol
(III, Beispiel 1, 43 g, 0,1 m) und Methylenchlorid (500 ml) wird
mit Triethylamin (32 ml, 0,23 m) behandelt und auf –5° gekühlt. Zu
diesem Gemisch wird ein Gemisch von 3-Nitrobenzolsulfonylchlorid
(CAS Nr. 121-51-7, 32 g, 0,14 ml) in Methylenchlorid (60 ml) unter
Halten einer Temperatur von < 10° über einen Zeitraum
von 1 h gegeben. Die Reaktion wird durch DC überwacht und nach 45 min als
vollständig
erachtet. Das Gemisch wird mit Methylenchlorid (500 ml) verdünnt und
dann mit Wasser (2 × 600
ml) gewaschen. Die organische Phase wird dann mit Salzsäure (1 N,
400 ml) gewaschen und zu einem dicken Rückstand eingeengt. Der Rückstand
wird mit Methanol (200 ml) verdünnt
und 1,5 h gerührt.
Die Feststoffe werden abfiltriert, mit Methanol gewaschen und unter
einem vermindertem Druck bei 40° über Nacht
getrocknet, wobei die Titelverbindung erhalten wird.
DC (Silicagel;
Methanol/Methylenchlorid, 5/95) Rf = 0,75.
-
Beispiel 6
-
(S)-N-[[3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]-phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamid
(VII)
-
Eine
Aufschlämmung
des 3-Nitrobenzolsulfonatesters von (R)-[N-3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]-phenyl]-2-oxo-5-oxazolidinyl]methanol
(VI, Beispiel 5, 50 g, 0,081 ml), Isopropanol (250 ml), Acetonitril
(400 ml) und wässrigem
Ammoniumhydroxid (29%iges Ammoniak, bezogen auf das Gewicht, 500 ml)
wird bei 40° 3,5
h erhitzt. Das Gemisch wird dann mit weiterem wässrigem Ammoniak (100 ml) behandelt und
20 h gerührt.
Die Reaktion wird durch DC überwacht
und an diesem Zeitpunkt als vollständig erachtet. Das Gemisch
wird unter vermindertem Druck mit Wärme eingeengt und in Methylenchlorid/Wasser
(1250 ml/750 ml) suspendiert. Die Phasen werden getrennt und die
organische Phase wird eingeengt, wobei ein Rückstand erhalten wird.
-
Der
Rückstand
wird in Methylenchlorid (2 l) gelöst und mit Triethylamin (20
ml, 0,14 m) behandelt. Das Gemisch wird dann mit Essigsäureanhydrid
(10 ml, 0,11 m) bei 20–25° über 10 min
behandelt. Die Acetylierung wird durch DC überwacht und nach 15 min als
vollständig
erachtet. Das organische Gemisch wird mit Wasser (2 × 400 ml)
gewaschen und dann zu einem Feststoff eingeengt. Die Feststoffe
werden aus Ethanol (400 ml) umkristallisiert, filtriert und unter
vermindertem Druck getrocknet, wobei die Titelverbindung erhalten
wird. DC (Silicagel; Methanol/Methylenchlorid, 5/95) Rf =
0,6.
-
Beispiel 7
-
(S)-N-[[3-[3-Fluor-4-(1-piperazinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamidhydrochlorid
(Zwischenprodukt)
-
Ein
Gemisch von (S)-N-[[3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]-phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamid
(VII, Beispiel 6, 35 kg, 74,5 mol), Palladium-auf-Kohle (5%, 10
kg, 50% wasserfeucht), Methanol (550 l) und Tetrahydrofuran (250
l) wird bei 22 bis 42° in
einer Wasserstoffatmosphäre
von 42–50
psi gerührt.
Nach 31 h zeigte eine DC-Analyse eine vollständige Umsetzung und die Wasserstoffatmosphäre wurde
durch Stickstoff ersetzt. Der Katalysator wird durch Filtration
entfernt und das Filtrat wird unter Vakuum auf 100 l eingeengt.
Zu dem erhaltenen Gemisch, das auf 2° gekühlt wurde, wird Methanol (50
l) und dann ein Gemisch von Methanol (100 l) und Acetylchlorid (6,04
kg, 77 mol) bei –2° bis 6° gegeben.
Das erhaltene Gemisch wird 90 min gerührt und dann unter Vakuum auf
60 l eingeengt, mit Aceton (100 l) verdünnt und des Weiteren auf 100
l eingeengt. Die erhaltene Aufschlämmung wird mit Aceton (200
l) verdünnt
und 15 h bei 16° gerührt. Die
Feststoffe werden auf einem Filter gewonnen, mit Aceton (50 l) gewaschen
und unter vermindertem Druck bei 20–25° getrocknet, wobei das gewünschte Produkt
erhalten wird. Es wird in Methanol (56 l) bei 53° gelöst, mit Aceton (150 l) verdünnt, 30
min bei 48° gerührt, dann
auf 15° gekühlt und
18 h gerührt.
Die Feststoffe werden auf einem Filter gewonnen, mit Aceton (50
l) gewaschen und unter vermindertem Druck bei 20–25° getrocknet, wobei die Titelverbindung
erhalten wird.
NMR (CDCl3) 7,56-7,45,
7,31, 7,12-6,86, 4,79, 4,09-4,0, 3,81, 3,62, 3,40-3,11 und 2,01 δ.
-
Beispiel 8
-
(S)-N-[[3-[3-Fluor-4-[4-hydroxyacetyl)-1-piperazinyl]-phenyl]-2-oxo-5-oxazolidinyl]methyl}-acetamid-sesquihydrat
(VIII)
-
Zu
einem gerührten
Gemisch von (S)-N-[[3-[3-Fluor-4-[1-piperazinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamidhydrochlorid
(Beispiel 7, 16,2 kg, 43,5 mol), Tetrahydrofuran (205 kg) und Triethylamin
(10,1 kg, 100 mol) wird Acetoxyacetylchlorid (6,5 kg, 47,8 mol)
in Tetrahydrofuran (11,1 kg) über
35 min gegeben, wobei die Temperatur bei 22–23° gehalten wird. Nach 40 min,
wobei an diesem Zeitpunkt eine DC- und HPLC-Analyse die vollständige Bildung
des Acetoxyacetamidzwischenprodukts zeigte, wird das Gemisch unter
vermindertem Druck auf 30 l eingeengt, mit Methanol (100 l) verdünnt und
auf 30 l eingeengt. Zu dem Rückstand
werden Methanol (25 l) und eine wässrige Kaliumcarbonatlösung (5,6
kg in 56 l) gegeben. Das erhaltene Gemisch wird 20 h bei 22–25° gerührt, wobei
an diesem Zeitpunkt eine DC- und HPLC-Analyse zeigt, dass die Reaktion vollständig ist.
Der pH-Wert wird
mit Salzsäure
(4 N, 14,3 l) auf 7–7,5
eingestellt. Das Gemisch wird 18 h bei 15–22°, dann 3 h bei 2–5° gerührt. Die
Feststoffe werden auf einem Filter gewonnen, mit Wasser (68 l) gewaschen
und bei 20–25° mit rückgeführtem Stickstoff
getrocknet, wobei das gewünschte
Produkt erhalten wird. Das rohe Produkt wird in Wasser (225 l) bei
60–70° gelöst, durch
ein 0,6-μm-Filter
geklärt,
mit einer Wasserspülung
(55 l) verdünnt
und 17 h bei 15° gerührt. Die
Feststoffe werden auf einem Filter gewonnen, mit Wasser bei 15° gewaschen
und bei 45° mit
rückgeführtem Stickstoff
auf einen Wassergehalt von 0,33% getrocknet. Diese Feststoffe werden
in einer Lösung
von Ethylacetat (143 l), Methanol (65 l) und Wasser (1,95 l) bei
60–65° gelöst. Die
Lösung
wird auf 15–25° gekühlt und
16 h zur Kristallisation gerührt.
Die Feststoffe werden auf einem Filter gewonnen, mit Ethylacetat
(75 l) gewaschen und mit Stickstoff von 45° getrocknet, wobei das gewünschte Produkt
erhalten wird. Das Produkt wird zwei weitere Male aus Wasser (147
1, dann 133 l) bei 60–70° umkristallisiert,
jedes Mal durch ein 0,6-μm-Filter
geklärt
und mit Wasser (40 l und 30 l) gespült. Die Feststoffe werden auf
dem Filter bei 30° mit
rückgeführtem Stickstoff
getrocknet, wobei nach Deagglomeration durch eine Mühle die
Titelverbindung als das Sesquihydrat (6,45 % Wasser) erhalten wird.
DC
(Silicagel; Methanol/Methylenchlorid, 5/95) Rf =
0,45; [α]D = –20° (c = 1,0,
Ethanol).
-
Beispiel 9
-
3-Nitrobenzolsulfonatester von (R)-[N-3-[3-Fluor-4-(N-1-(4-carbobenzoxy)piperazinyl]-phenyl]-2-oxo-5-oxazolidinyl]methanol
(VI)
-
Zu
einer Aufschlämmung
von (R)-[N-3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methanol
(III, Beispiel 1, 5,086 g, 11,86 mmol) in Methylenchlorid (50 ml)
und Triethylamin (2,0 ml, 14,38 mmol) bei 0° wird tropfenweise über 6 min
eine Lösung
von 3-Nitrobenzolsulfulfonylchlorid (V) in Methylenchlorid (0,356
M, 33,4 ml, 11,89 mmol) gegeben. Nach Rühren während 3,25 h werden weitere
3,4 ml (1,21 mmol) der 0,356 M Lösung
von 3-Nitrobenzolsulfonylchlorid
(V) zugegeben. Nach Rühren während 1,75
h wird Salzsäure
(1 N, 50 ml) zugegeben. Die Pha sen werden getrennt und die wässrige Phase wird
mit Methylenchlorid extrahiert. Die vereinigten organischen Phasen
werden mit Kochsalzlösung
gewaschen, über
Magnesiumsulfat getrocknet und eingeengt. Das Konzentrat wird aus
heißem
Methylenchlorid/Methanol umkristallisiert, wobei die Titelverbindung
erhalten wird.
Fp = 155–157°; NMR (CDCl3, 400 MHz) 8,72, 8,51, 8,23, 7,81, 7,35,
7,01, 6,91, 5,17, 4,85, 4,44, 4,39, 4,09, 3,85, 3,68 und 3,01 δ; CMR (CDCl3, 100 MHz) 44,26, 46,81, 50,91, 67,64, 69,54,
69,91, 107,85, 114,32, 119,85, 123,55, 128,30, 128,47, 128,91, 129,15,
131,51, 133,71, 136,99, 137,70, 148,71, 153,62, 155,57 und 155,88 δ; IR (Mineralölverreibung)
1744, 1703, 1528, 1520, 1367, 1347 und 1192 cm–1;
MS (EI, M/Z) 614, 411, 107, 91, 79, 65 und 56; [a]D = –78° (c = 0,9812,
CHCl3); DC (Ethylacetat/Hexan, 3/1) Rf = 0,43.
-
Beispiel 10
-
2-Nitrobenzolsulfonatester von (R)-[N-3-[3-Fluor-4-(N-1-(4-carbobenzoxy)piperazinyl]-phenyl]-2-oxo-5-oxazolidinyl]methanol
(VI)
-
Nach
dem allgemeinen Verfahren von Beispiel 5 (für den 3-Nitrobenzolsulfonylester, (VI)) und
unter Durchführung
unkritischer Variationen wird (R)-[N-3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methanol
(III, Beispiel 1, 1,106 g, 2,578 mmol) mit Triethylamin (0,54 ml,
3,882 mmol) und 2-Nitrobenzolsulfonylchlorid handelsüblicher
Qualität
(V, 679 mg, 3,064 mmol) behandelt, wobei die Titelverbindung erhalten
wird.
NMR (CDCl3, 400 MHz) 8,15, 7,82,
7,37, 7,06, 6,94, 5,17, 4,89, 4,59, 4,50, 4,10, 3,98, 3,69 und 3,03 δ; IR (Mineralölverreibung)
1757, 1697, 1517, 1445, 1423, 1376, 1237 und 1188 cm–1;
MS (EI, M/Z; relative Häufigkeit): 614
(18,3, M+), 91 (100), 69 (23,8) und 56 (52,9);
DC (Ethylacetat/Hexan, 3/1) Rf = 0,31.
-
Beispiel 11
-
2,4-Dinitrobenzolsulfonatester von (R)-[N-3-[3-Fluor-4-(N-1-(4-carbobenzoxy)piperazinyl]-phenyl]-2-oxo-5-oxazolidinyl]methanol
(VI)
-
Nach
dem allgemeinen Verfahren von Beispiel 5 (für den 3-Nitrobenzolsulfonylester, (VI)) und
unter Durchführung
unkritischer Variationen wird (R)-[N-3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methanol
(III, Beispiel 1, 1,094 g, 2,550 mmol) mit Triethylamin (0,55 ml,
3,950 mmol) und 2,4-Ditrobenzolsulfonylchlorid handelsüblicher
Qualität
(833 mg, 3,124 mmol) behandelt, wobei die Titelverbindung erhalten
wird.
NMR (CDCl3, 400 MHz) 8,59, 8,38,
7,35, 7,02, 5,17, 4,88, 4,74, 4,58, 4,10, 3,98, 3,71 und 3,05 δ; IR (Mineralölverreibung)
1756, 1697, 1554, 1541, 1517, 1351, 1237 und 1189 cm–1;
MS (FAB, M/Z, relative Häufigkeit) 660
(21,3, [M+H]+), 659 (24,2 M+),
102 (76,5) und 91 (100); DC (Ethylacetat/Hexan, 3/1) Rf =
0,41.
-
Beispiel 12
-
(R)-[N-3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methanol-4-chlorbenzolsulfonatester
(VI)
-
Zu
einer Aufschlämmung
von (R)-[N-3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methanol
(III, Beispiel 1, 3,450 g, 8,034 mmol) in Methylenchlorid (40 ml)
und Triethylamin (2,55 ml, 18,3 mmol) bei –12° wird 4-Chlorbenzolsulfonylchlorid
(V, Aldrich Chemcial Co. – Handelsqualität, 2,298
g, 10,88 mmol) als Feststoff auf einmal gegeben. Das Gemisch wird
in einem Bad von 0° 2,5
h gerührt,
dann mit Wasser (2 × 35
ml) und 1 N Salzsäure
(35 ml) gewaschen. Die organischen Extrakte werden auf ein Gesamtvolumen
von 20 ml eingeengt und Methanol (50 ml) wird zugegeben. Der Niederschlag
wird durch Vakuumfiltration gewonnen, mit Methanol gewaschen, getrocknet
und erneut in Methylenchlorid (55 ml) gelöst. Das Gemisch wird auf eine Aufschlämmung des
Gewichts von 32 g eingeengt und mit Methanol (11 ml) versetzt. Der
Niederschlag wird durch Vakuumfiltration gewonnen, mit Methanol
gewaschen und getrocknet. Die Feststoffe werden dann in Methylenchlorid
(58 ml) gelöst
und säulenchromatographiert
(Silicasäule,
93 g 40–63 μ); Elution
mit jeweils 450 ml der folgenden Ethylacetat/Cyclohexan-Gemische
25/75, 35/65, 45/55, 55/45; Gewinnung der letzten 50% des Eluats).
Das gewonnene Eluat wird auf 200 ml eingeengt und mit 200 ml Heptan
versetzt. Der Niederschlag wird durch Vakuumfiltration gewonnen
und getrocknet, wobei die Titelverbindung erhalten wird;
DC
(Silicagel; Methanol/Chloroform, 5/95) Rf =
0,53;
MS (FAB, M/Z) = 604,7 (100 [P+H]+);
NMR (DMSO-d6, 300 MHz) 7,93, 6,7, 7,75,
7,48-7,32, 7,12-7,03, 5,12, 4,93-4,92,
4,40, 4,09, 3,69, 3,57 und 2,96 δ;
CMR (DMSO-d6, 75 MHz) 43,51, 45,84, 50,22,
66,33, 69,75, 70,75, 106,63, 114,08, 119,83, 127,59, 127,87, 128,43,
129,62, 130,00, 133,31, 133,63, 135,52, 136,84, 139,63, 153,54,
154,40 und 154,62 δ.
-
Beispiel 13
-
(R)-[N-3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methanol-2,5-dichlorbenzolsulfonatester
(VI)
-
Zu
einer Aufschlämmung
von (R)-[N-3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methanol
(III, Beispiel 1, 3,439 g, 8,008 mmol) in Methylenchlorid (40 ml)
und Triethylamin (2,55 ml, 18,3 mmol) bei –8° wird 2,5-Dichlorbenzolsulfonylchlorid
(V, Aldrich Chemical Co. – Handelsqualität, 2,675
g, 10,90 mmol) als Feststoff auf einmal gegeben. Das Gemisch wird
in einem Bad von 0° 2,5
h gerührt,
dann mit Wasser (2 × 35
ml) und 1 N Salzsäure
(35 ml) gewaschen. Die organischen Extrakte werden dann auf 12,0
g eingeengt und dieses wird säulenchromatographiert
(Silicasäule,
108 g, 40–63 μ; Elution
mit jeweils 450 ml der folgenden Ethylacetat/Cyclohexangemische 10/90,
20/80, 30/70, 40/60 und 60/40, Gewinnung der letzten 20% des Eluats).
Das gewonnene Eluat wird eingeengt und mit 300 ml Methanol versetzt.
Der Niederschlag wird durch Vakuumfiltration gewonnen, mit Methanol
gewaschen und getrocknet, wobei die Titelverbindung erhalten wird.
DC
(Silicagel; Methanol/Chloroform, 5/95) Rf =
0,66; MS (FAB, M/Z) = 638,6 (100%, [P+H]+);
NMR (CDCl3, 300 MHz) 8,04, 7,57-7,32, 7,06,
6,91, 5,16, 4,89-4,47, 4,42, 4,08, 3,93, 3,67 und 3,01 δ; CMR (CDCl3, 75 MHz), 43,93, 45,51, 50,56, 67,26, 69,16,
69,46, 107,55, 113,98, 119,41, 127,92, 128,10, 128,54, 131,21, 131,46, 132,97,
133,44, 133,50, 134,68, 135,15, 136,45, 136,61, 153,36, 155,22 und
155,53 δ.
-
Beispiel 14
-
(R)-[N-3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methanol-4-nitrobenzolsulfonatester
(VI)
-
Zu
einer Aufschlämmung
von (R)-[N-3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methanol
(III, Beispiel 1, 3,437 g, 8,003 mmol) und 4-Nitrobenzolsulfonylchlorid (V, 75 %
reines technisches Material, Aldrich Chemical Co. – Handelsqualität, 3,077
g, 10,41 mmol) in Methylenchlorid (32 ml) bei 0° wird Triethylamin (2,23 ml,
16,0 mmol) gegeben. Das Gemisch wird in einem Bad von 0° 1 h gerührt, dann
wird Wasser (1 ml) zugegeben und das Gemisch bei 20–25° 30 min gerührt. Methylenchlorid
(75 ml) wird zugegeben und das Gemisch wird mit Salzsäure (5%,
50 ml) und dann Natriumbicarbonat (5%, 50 ml) gewaschen und auf
Magnesiumsulfat getrocknet. Die organischen Extrakte werden dann
eingeengt und das Konzentrat wird in siedendem Ethylacetat/Cyclohexan
(1/1, 10 ml) aufgenommen und säulenchromatographiert
(Silicagel, 4 cm × 6'', 40–63 μ; Elution mit etwa jeweils 400
ml der folgenden Ethylacetat/Cyclohexan-Gemische 20/80, 30/70, 40/60, 50/50,
60/40 und 70/30, Gewinnung der letzten etwa 45% des Eluats). Die
entsprechenden Fraktionen werden kombiniert und zu einem Feststoff
einge engt, der in 70 ml Methylenchlorid und 50 ml Ethylacetat gelöst wird.
Das Gemisch wird zweimal auf 50 ml eingeengt und nach dem Einengen
mit Cyclohexan (50 ml) versetzt. Der Niederschlag wird durch Vakuumfiltration
gewonnen, mit Cyclohexan gewaschen und getrocknet, wobei die Titelverbindung
erhalten wird.
DC (Silicagel, Ethylacetat/Cyclohexan, 60/40)
Rf = 0,37;
NMR (CDCl3,
300 MHz) 8,36, 8,07, 7,38-7,29, 7,03, 6,89, 5,15, 4,86-4,80, 4,39,
4,07, 3,80, 3,67 und 3,00 δ; CMR
(CDCl3, 75 MHz) 43,85, 46,34, 50,45, 67,20,
69,17, 69,57, 107,64, 113,88, 119,34, 124,63, 127,85, 128,05, 128,49,
129,26, 132,67, 136,48, 136,57, 140,75, 150,95, 153,29, 155,14 und
155,40 δ.
-
Beispiel 15
-
(S)-[N-3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methylamin
(VII)
-
Unter
Stickstoff werden bei 40° der
3-Nitrobenzolsulfonatester von (R)-[N-3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methanol
(VI, Beispiel 5, 1,0099 g, 1,643 mmol), Isopropanol (5,6 ml), Acetonitril
(9,0 ml), Benzaldehyd (0,50 ml, 4,92 mmol) und wässriges Ammoniak (29,8 Gew.-%,
9,5 ml, 148,6 mmol) gemischt. Das Gemisch wird bei 40° 21,5 h gerührt, dann
unter vermindertem Druck eingeengt. Toluol (13,3 ml und Ethanol
(6,0 ml) werden zugegeben und das Gemisch wird in einem Bad von
70° erwärmt. Citronensäuremonohydrat
(2,433 g, 11,58 mmol) wird dann über
3,5 h zugegeben und die Phasen werden bei 64° getrennt. Die organische Phase
wird mit Wasser (2,5 ml) bei 64° gewaschen.
Die vereinigten wässrigen
Schichten werden mit Toluol (10 ml) bei 64° gewaschen. Toluol (10 ml) wird
dann zu dem wässrigen Gemisch
gegeben und das Gemisch wird auf 0° gekühlt. Der Niederschlag wird
durch Vakuumfiltration gewonnen, mit Toluol von 0° (10 ml)
und Wasser von 0° (10
ml) gewaschen und zu einem Feststoff getrocknet. Ein Teil dieses
Feststoffs (0,7301 g) wird in Wasser (10 ml) und Methylenchlorid
(10 ml) aufgeschlämmt
und der pH-Wert wird mit wässrigem
Natriumhydroxid (50%, 0,3915 g, 4,90 mmol) bei –4 bis –2° von 2,78 auf 13,92 eingestellt.
Das Gemisch wird auf 20–25° erwärmt und
unter Rühren
0,5 h ultraschallbehandelt. Methylenchlorid (55 ml), gesättigtes
wässriges
Natriumchlorid (5 ml) und Wasser (35 ml) werden zugegeben und die
Phasen werden getrennt. Die wässrige
Phase wird zweimal mit Methylenchlorid (25 ml) gewaschen und die
vereinigten organischen Phasen werden auf Natriumsulfat getrocknet,
filtriert und unter vermindertem Druck eingeengt. Toluol (5 ml)
wird zugegeben und anschließend
erfolgt eine langsame Zugabe von Heptan (25 ml). Der erhaltene Niederschlag
wird durch Vakuumfiltration gewonnen, mit Heptan (20 ml) gewaschen
und getrocknet, wobei die Titelverbindung erhalten wird.
DC
(Silicagel; Methanol/Chloroform, 10/90) Rf =
0,32; MS (EI), M/Z (relative Intensität) = 428 (28%, M+),
252 (15%), 92 (32%), 91 (100%); NMR (CDCl3,
300 MHz), 7,46, 7,38-7,27,
7,12, 6,90, 5,16, 4,69-4,60, 3,98, 3,80, 3,67, 3,09, 3,00-2,92 und
1,30 δ;
CMR (CDCl3, 75 MHz) 43,94, 44,89, 47,60,
50,63, 67,23, 73,84, 107,29, 113,72, 119,37, 127,92, 128,07, 128,52,
133,79, 136,05, 136,64, 154,57, 155,19 und 155,61 δ.
-
Beispiel 16
-
(R)-[N-3-[3-Fluor-4-morpholinylphenyl]-2-oxo-5-oxazolidinyl]methanol-4-nitrobenzolsulfonatester
(VI)
-
Zu
einer Aufschlämmung
von (R)-[N-3-[3-Fluor-4-morpholinylphenyl]-2-oxo-5-oxazolidinyl]methanol (III,
Beispiel 3, 43,0 g, 145 mmol) und Triethylamin (36 g, 355 mmol)
in Methylenchlorid (450 ml) bei 0° wird
ein Gemisch von 4-Nitrobenzolsulfonylchlorid
(V, 32 g, 145 mmol) in Methylenchlorid (55 ml) gegeben. Das Gemisch
wird in einem Bad von 0° 30
min gerührt
und dann mit Salzsäure
(10%, 200 ml) gequencht. Die organische Phase wird abgetrennt und
die wässrige
Phase wird erneut mit Methylenchlorid (200 ml) extra hiert. Die vereinigten
organischen Extrakte werden dann eingeengt und säulenchromatographiert (Silicagel,
4 cm × 6'', 40–63 μ; Methanol/Methylenchlorid 1–2/98–99, etwa
8 l). Die entsprechenden Fraktionen werden vereinigt und eingeengt,
wobei die Titelverbindung erhalten wird.
Rf =
0,2; NMR (CDCl3, 300 MHz) 8,73, 8,54, 8,23,
7,82, 7,33, 7,04, 6,91, 4,86, 4,42, 4,12, 3,86 und 3,05 δ; CMR (CDCl3, 75 MHz, partiell) 46,42, 50,89, 66,87,
69,09, 69,45, 107,45, 113,95, 118,84, 123,14, 128,73, 131,08, 133,28
und 137,27 δ.
-
Beispiel 17
-
(S)-[N-3-[3-Fluor-4-morpholinylphenyl]-2-oxo-5-oxazolidinyl]methylamin-salicylaldehydimin
-
Ein
Gemisch von (R)-[N-3-(3-Fluor-4-(4-morpholinylphenyl)-2-oxo-5-oxazolidinyl]methanol-4-nitrobenzolsulfonatester
(VI, Beispiel 16, 20,608 g, 42,803 mmol), Isopropanol (149 ml),
Acetonitril (245 ml), Salicylaldehyd (13,7 ml, 129 mmol) und wässrigem
Ammoniak (30%, 257 ml, 4,02 mol) wird auf 40° erhitzt und 24 h bei 39–42° gerührt. Das
Gemisch wird dann auf –22° gekühlt und
der Niederschlag wird durch Vakuumfiltration gewonnen, mit Wasser
(10 ml) gewaschen und getrocknet, wobei die Titelverbindung erhalten
wird.
DC (Silicagel; Methanol/Chloroform, 5/95) Rf =
0,79; SIMS (m/z, relative Intensität) = 399 (M+,
51) 234 (11), 196 (11), 149 (22), 135 (100), 134 (47); NMR (300
MHz, CDCl3) 8,44, 7,41, 7,33-6,87, 4,96-4,88,
4,12, 3,94-3,84 und 3,04 δ;
CMR (CDCl3, 75 MHz) 48,21, 50,99, 61,94,
66,95, 71,30, 107,68, 114,12, 117,02, 118,43, 118,82, 119,01, 131,93,
133,04, 136,51, 154,24, 155,47, 160,78 und 168,87 δ.
-
Beispiel 18
-
(S)-N-[[3-(3-Fluor-4-morpholinylphenyl)-2-oxo-5-oxazolidinyl]methyl]acetamid
(VIII)
-
(S)-[N-3-[3-Fluor-4-morpholinylphenyl]-2-oxo-5-oxazolidinyl]methylamin-salicylaldehydimin
(Beispiel 17, 1,0068 g, 2,521 mmol) wird in Wasser (10 ml) und 37%iger
wässriger
Salzsäure
(0,417 ml, 5,04 mmol) aufgeschlämmt
und 15 h bei 20–25° gerührt. Toluol
(10 ml) wird zugegeben und die Phasen werden getrennt; dann wird
die organische Phase mit Salzsäure
(1 M, 5 ml) gewaschen und die vereinigten wässrigen Phasen werden mit Toluol
(10 ml) gewaschen. Die Toluolwaschflüssigkeit wird mit Salzsäure (1 M,
5 ml) zurückextrahiert.
Die vereinigten wässrigen
Phasen werden dann mit wässrigem
Natriumhydroxid (50%, 1,83 g, 22,9 mmol) auf einen pH-Wert von 13,0
eingestellt. Zu der erhaltenen Aufschlämmung werden dann Methylenchlorid
(10 ml) und Natriumchlorid (1 g) gegeben und die Phasen werden getrennt.
Die wässrige
Phase wird dann mit Methylenchlorid (10 ml) gewaschen. Zu den kombinierten
organischen Phasen wird dann Essigsäureanhydrid (0,472 ml, 5,00
mmol) unter Beibehalten von 24–27° gegeben.
Das Gemisch wird 40 min gerührt
und dann mit Wasser (5 ml) versetzt. Die Phasen werden getrennt
und die wässrige
Phase wird mit Methylenchlorid (5 ml) gewaschen. Die vereinigten
organischen Phasen werden eingeengt und mit Ethylacetat (25 ml)
versetzt. Das Gemisch wird auf 70° erwärmt und
dann wird das erhaltene Gemisch langsam auf –25° gekühlt. Der Niederschlag wird
durch Vakuumfiltration gewonnen, mit Ethylacetat von –25° (5 ml) gewaschen
und getrocknet, wobei die Titelverbindung erhalten wird.
HPLC
der Hauptkomponente (99,93 Flächenprozent
bei Detektion bei 254 nm) Retentionszeit = 0,97 min, Säule = Zorbax
RX-C8, 250 × 4,6
mm, mobile Phase = 650 ml Acetonitril, 1,85 ml Triethylamin, 1,30
ml Essigsäure und
ausreichend Wasser zum Auffüllen
auf 1000 ml; Durchflussrate = 3 ml/min.
-
Beispiel 19
-
(R)-[N-3-[3-Fluor-4-[N-1-(4-carbobenzoxy)piperazinyl]phenyl]-2-oxo-5-oxazolidinyl]methanol
(III)
-
Ein
Gemisch von N-Carbobenzoxy-3-fluor-4-(N-carbobenzoxypiperazinyl)anilin
(II, 2,014 g, 4,345 mmol) und THF (10 ml) wird auf –20° gekühlt. In
einem getrennten Kolben wird eine Lösung von tert-Amylalkohol (0,71
ml, 6,48 mmol) in THF (10 ml) bei –33° mit n-Butyllithium in Heptan
(13,65 Gew.-%, 2,53 g, 5,38 mmol) behandelt, während das Gemisch bei weniger
als –20° gehalten
wird. Die erhaltene Lithium-tert-amylatlösung wird dann zu dem N-Carbobenzoxy-3-fluor-4-(N-carbobenzoxypiperazinyl)anilingemisch
unter Aufrechterhalten von weniger als –20° gegeben und mit THF (4 ml)
hineingespült.
Zu dem erhaltenen Gemisch bei –20° wird dann
S-Glycidol (IV, 0,3360 g, 4,536 mmol) gegeben. Das Gemisch wird
dann 1,5 h bei –20°, dann 17
h bei –16°, 4 h bei –11°, dann 2
h bei –1° gerührt. Ein
HPLC-Assay zeigte dann, dass die Hauptkomponente eine mit der Titelverbindung
konsistente Retentionszeit aufwies (90,4 Flächenprozent bei Detektion bei
254 nm; Retentionszeit = 1,30 min; Säule = Zorbax RX-C8, 250 × 4,6 mm;
mobile Phase = 650 ml Acetonitril, 1,85 ml Triethylamin, 1,30 ml
Essigsäure
und die Zugabe von ausreichend Wasser zum Auffüllen auf 1000 ml; Durchflussrate
= 3 ml/min) wie auch DC (Silicagel; Methanol/Chloroform 10/90) Rf = 0,60.
-
Beispiel 20
-
(R)-[N-3-[3-Fluor-4-morpholinylphenyl]-2-oxo-5-oxazolidinyl]methanol-4-nitrobenzolsulfonatester
(VI)
-
Nach
dem allgemeinen Verfahren von Beispiel 16 und unter Durchführen unkritischer
Variationen, jedoch ausgehend von 4-Nitrobenzolsulfonylchlorid,
wird die Titelverbindung erhalten.
-
Beispiel 21
-
(R)-[N-3-[3-Fluor-4-morpholinylphenyl]-2-oxo-5-oxazolidinyl]methanol-2-nitrobenzolsulfonatester
(VI)
-
Nach
dem allgemeinen Verfahren von Beispiel 16 und unter Durchführen unkritischer
Variationen, jedoch ausgehend von 2-Nitrobenzolsulfonylchlorid,
wird die Titelverbindung erhalten.
-
Beispiel 22
-
(R)-[N-3-[3-Fluor-4-morpholinylphenyl]-2-oxo-5-oxazolidinyl]methanol-2,4-dinitrobenzolsulfonatester
(VI)
-
Nach
dem allgemeinen Verfahren von Beispiel 16 und unter Durchführen unkritischer
Variationen, jedoch ausgehend von 2,4-Dinitrobenzolsulfonylchlorid,
wird die Titelverbindung erhalten.
-
Beispiel 23
-
(R)-[N-3-[3-Fluor-4-morpholinylphenyl]-2-oxo-5-oxazolidinyl]methanol-4-chlorbenzolsulfonatester
(VI)
-
Nach
dem allgemeinen Verfahren von Beispiel 16 und unter Durchführen unkritischer
Variationen, jedoch ausgehend von 4-Chlorbenzolsulfonylchlorid,
wird die Titelverbindung erhalten.
-
Beispiel 24
-
(R)-[N-3-[3-Fluor-4-morpholinylphenyl]-2-oxo-5-oxazolidinyl]methanol-2,5-dichlorbenzolsulfonatester
(VI)
-
Nach
dem allgemeinen Verfahren von Beispiel 16 und unter Durchführen unkritischer
Variationen, jedoch ausgehend von 2,5-Dichlorbenzolsulfonylchlorid,
wird die Titelverbindung erhalten. Reaktionsschema
A
-
Reaktionsschema B
-
- C*H2-C*H-CH2-OH (IV)wobei
die durch ein * bezeichneten Kohlenstoffatome jeweils an das gleiche
Sauerstoffatom (-O-) unter Bildung eines dreigliedrigen Rings oder
Epoxids gebunden sind
-
-
-

 steht; worin X1 für H oder F steht; worin X2 für H oder F steht; worin Q1 für:
steht; worin X1 für H oder F steht; worin X2 für H oder F steht; worin Q1 für: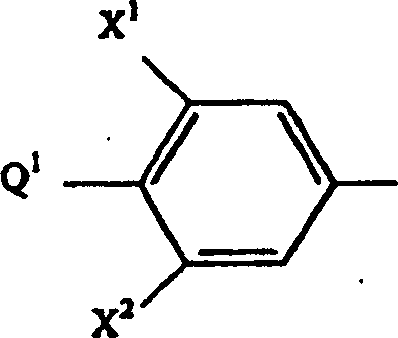
 steht; Q1 und X2 zusammengenommen für:
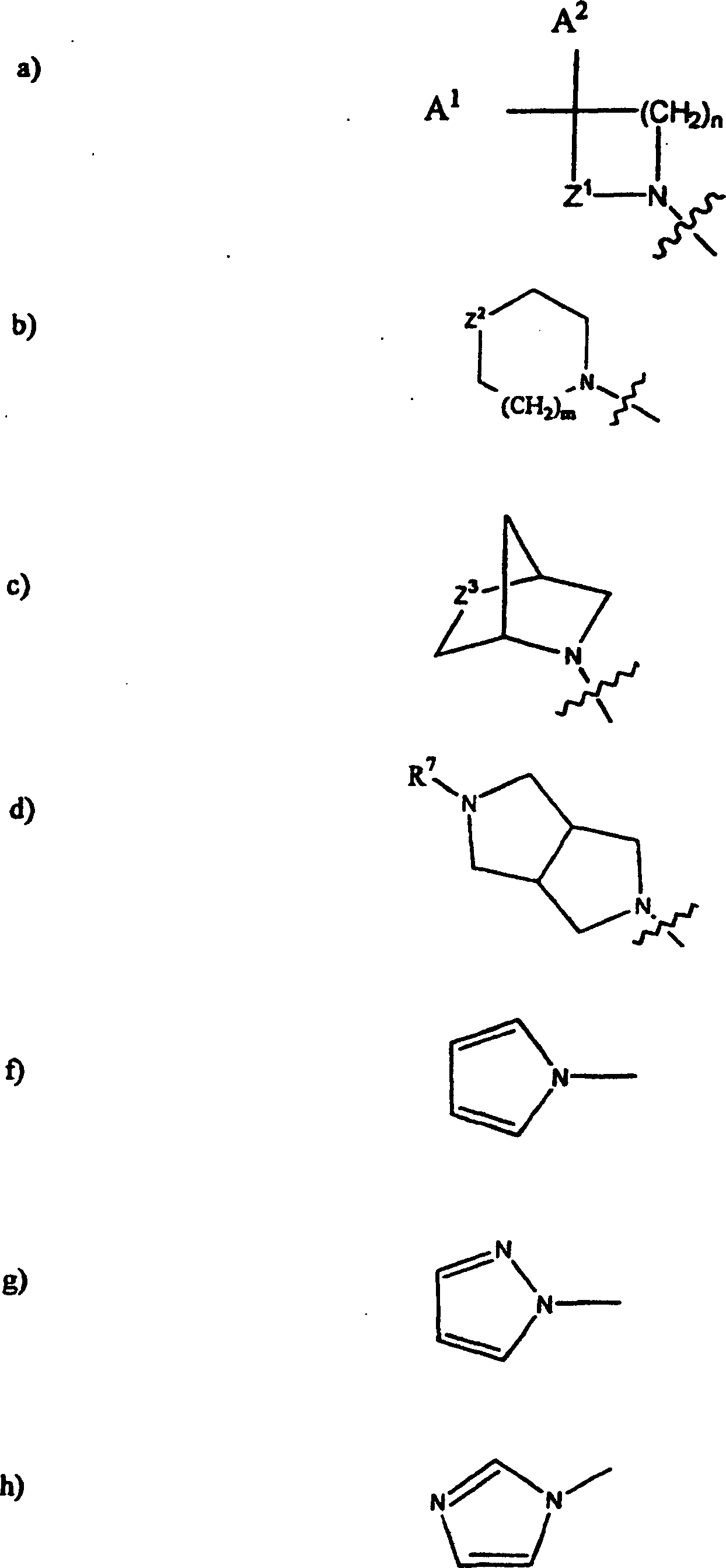
steht; Q1 und X2 zusammengenommen für: stehen; worin Z1 für: a) -CH2-, b) -CH(R4)-CH2-, c) -C(O)- oder d) -CH2CH2CH2- steht; worin Z2 für: a) -O2S-, b) -O-, c) -N(R7)-, d) -OS- oder e) -S- steht; worin Z3 für: a) -O2S-, b) -O-, c) -OS- oder d) -S- steht; worin A1 für: a) H oder b) CH3 steht; worin A2 für: a) H, b) HO-, c) CH3-, d) CH3O-, e) R2O-CH2-C(O)-NH-, f) R3O-C(O)-NH-, g) (C1-C2)Alkyl-O-C(O)-, h) HO-CH2-, i) CH3O-NH-, j) (C1-C3)Alkyl-O2C-, k) CH3-C(O)-, l) CH3-C(O)-CH2-,
stehen; worin Z1 für: a) -CH2-, b) -CH(R4)-CH2-, c) -C(O)- oder d) -CH2CH2CH2- steht; worin Z2 für: a) -O2S-, b) -O-, c) -N(R7)-, d) -OS- oder e) -S- steht; worin Z3 für: a) -O2S-, b) -O-, c) -OS- oder d) -S- steht; worin A1 für: a) H oder b) CH3 steht; worin A2 für: a) H, b) HO-, c) CH3-, d) CH3O-, e) R2O-CH2-C(O)-NH-, f) R3O-C(O)-NH-, g) (C1-C2)Alkyl-O-C(O)-, h) HO-CH2-, i) CH3O-NH-, j) (C1-C3)Alkyl-O2C-, k) CH3-C(O)-, l) CH3-C(O)-CH2-,
 steht; A1 und A2 zusammengenommen für : a) -O-CHR12-CH2-O-, b) O=, c) R14-N= stehen; worin R2 für: a) H, b) CH3-, c) Phenyl-CH2- oder d) CH3C(O)- steht; worin R3 für: a) (C1-C3)Alkyl oder b) Phenyl steht; worin R4 für: a) H oder b) HO- steht; worin R5 für: a) H, b) (C1-C3)Alkyl, c) CH2=CH-CH2- oder d) CH3-O-(CH2)2- steht; worin R6 für: a) CH3-C(O)-, b) H-C(O)-, c) Cl2CH-C(O)-, d) HOCH2-C(O)-, e) CH3SO2-,
steht; A1 und A2 zusammengenommen für : a) -O-CHR12-CH2-O-, b) O=, c) R14-N= stehen; worin R2 für: a) H, b) CH3-, c) Phenyl-CH2- oder d) CH3C(O)- steht; worin R3 für: a) (C1-C3)Alkyl oder b) Phenyl steht; worin R4 für: a) H oder b) HO- steht; worin R5 für: a) H, b) (C1-C3)Alkyl, c) CH2=CH-CH2- oder d) CH3-O-(CH2)2- steht; worin R6 für: a) CH3-C(O)-, b) H-C(O)-, c) Cl2CH-C(O)-, d) HOCH2-C(O)-, e) CH3SO2-, g) F2CHC(O)-,
g) F2CHC(O)-, i) H3C-C(O)-O-CH2-C(O)-, j) H-C(O)-O-CH2-C(O)-,
i) H3C-C(O)-O-CH2-C(O)-, j) H-C(O)-O-CH2-C(O)-, l) HC≡CH-CH2O-CH2-C(O)- oder m) Phenyl-CH2-O-CH2-C(O)- steht; worin R7 für a) R2O-C(R10)(R11)-C(O)-, b) R3O-C(O)-, c) R8-C(O)
l) HC≡CH-CH2O-CH2-C(O)- oder m) Phenyl-CH2-O-CH2-C(O)- steht; worin R7 für a) R2O-C(R10)(R11)-C(O)-, b) R3O-C(O)-, c) R8-C(O) f) H3C-C(O)-(CH2)2-C(O)-, g) R9-SO2-,
f) H3C-C(O)-(CH2)2-C(O)-, g) R9-SO2-,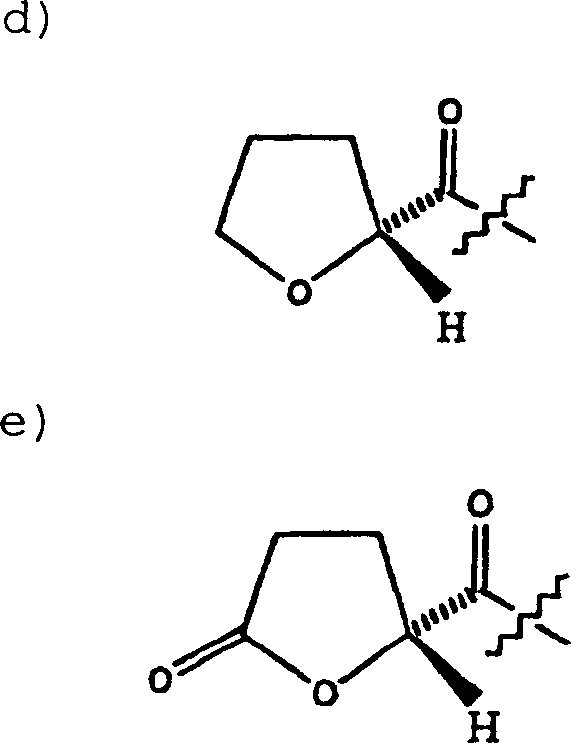 i) HO-CH2-C(O)-, j) R16-(CH2)2-, k) R13-C(O)-O-CH2-C(O)-, l) (CH3)2N-CH2-C(O)-NH-, m) NC-CH2- oder n) F2CH-CH2- steht; worin R8 für: a) H, b) (C1-C4)Alkyl, c) Aryl-(CH2)p-, d) ClH2C-, e) Cl2HC-, f) FH2C-, g) F2HC- oder h) (C3-C6)Cycloalkyl steht; worin R9 für: a) -CH3, b) -CH2Cl, c) -CH2CH=CH2, d) Aryl oder e) -CH2CN steht; worin R10 für H oder CH3 steht; worin R11 für H oder CH3 steht; worin R12 für: a) H, b) CH3O-CH2O-CH2- oder c) HOCH2- steht; worin R13 für: a) CH3-, b) HOCH2-, c) (CH3)2N-Phenyl oder d) (CH3)2N-CH2- steht; worin R14 für: a) HO-, b) CH3O-, c) H2N-, d) CH3O-C(O)-O-, e) CH3-C(O)-O-CH2-C(O)-O-, f) Phenyl-CH2-O-CH2-C(O)-O-, g) HO-(CH2)2-O-, h) CH3O-CH2-O-(CH2)2-O- oder i) CH3O-CH2-O- steht; worin R15 für: a) H oder b) Cl steht; worin R16 für: a) HO-, b) CH3O- oder c) F steht; worin m 0 oder 1 ist; worin n 1 bis 3 ist; worin p 0 oder 1 ist; worin Aryl ein Phenyl ist, das mit null (0) oder einem (1) der folgenden Reste substituiert ist: a) -F, b) -Cl, c) -OCH3, d) -OH, e) -NH2, f) -(C1-C4)Alkyl, g) -O-C(O)-OCH3 oder h) -NO2 und geschützte Formen desselben, mit der Maßgabe, dass jedes primäre oder sekundäre Amin in R1 geschützt ist, wobei das Verfahren (1) das Inkontaktbringen eines 5-Hydroxymethyl-substituierten Oxazolidinonalkohols der Formel (III)
i) HO-CH2-C(O)-, j) R16-(CH2)2-, k) R13-C(O)-O-CH2-C(O)-, l) (CH3)2N-CH2-C(O)-NH-, m) NC-CH2- oder n) F2CH-CH2- steht; worin R8 für: a) H, b) (C1-C4)Alkyl, c) Aryl-(CH2)p-, d) ClH2C-, e) Cl2HC-, f) FH2C-, g) F2HC- oder h) (C3-C6)Cycloalkyl steht; worin R9 für: a) -CH3, b) -CH2Cl, c) -CH2CH=CH2, d) Aryl oder e) -CH2CN steht; worin R10 für H oder CH3 steht; worin R11 für H oder CH3 steht; worin R12 für: a) H, b) CH3O-CH2O-CH2- oder c) HOCH2- steht; worin R13 für: a) CH3-, b) HOCH2-, c) (CH3)2N-Phenyl oder d) (CH3)2N-CH2- steht; worin R14 für: a) HO-, b) CH3O-, c) H2N-, d) CH3O-C(O)-O-, e) CH3-C(O)-O-CH2-C(O)-O-, f) Phenyl-CH2-O-CH2-C(O)-O-, g) HO-(CH2)2-O-, h) CH3O-CH2-O-(CH2)2-O- oder i) CH3O-CH2-O- steht; worin R15 für: a) H oder b) Cl steht; worin R16 für: a) HO-, b) CH3O- oder c) F steht; worin m 0 oder 1 ist; worin n 1 bis 3 ist; worin p 0 oder 1 ist; worin Aryl ein Phenyl ist, das mit null (0) oder einem (1) der folgenden Reste substituiert ist: a) -F, b) -Cl, c) -OCH3, d) -OH, e) -NH2, f) -(C1-C4)Alkyl, g) -O-C(O)-OCH3 oder h) -NO2 und geschützte Formen desselben, mit der Maßgabe, dass jedes primäre oder sekundäre Amin in R1 geschützt ist, wobei das Verfahren (1) das Inkontaktbringen eines 5-Hydroxymethyl-substituierten Oxazolidinonalkohols der Formel (III) worin R1 wie oben definiert ist, mit einem Sulfonylierungsmittel, das aus den Formeln Va–Vd ausgewählt ist,
worin R1 wie oben definiert ist, mit einem Sulfonylierungsmittel, das aus den Formeln Va–Vd ausgewählt ist,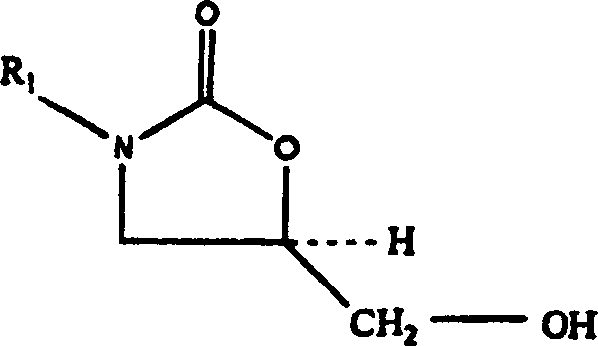
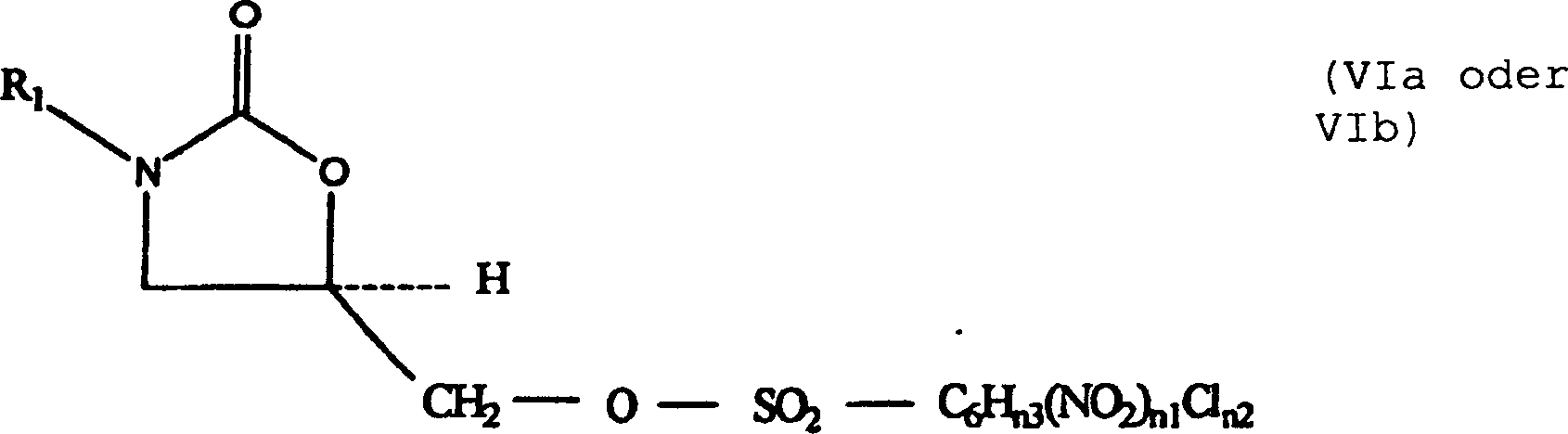 mit Ammoniak in einem verschlossenen System bei einer Temperatur von nicht mehr als 60°C und einem Druck von weniger als 207 kPa (30 psig) umfasst.
mit Ammoniak in einem verschlossenen System bei einer Temperatur von nicht mehr als 60°C und einem Druck von weniger als 207 kPa (30 psig) umfasst.