CN1205196C - 选择性衍生紫杉烷的方法 - Google Patents
选择性衍生紫杉烷的方法 Download PDFInfo
- Publication number
- CN1205196C CN1205196C CNB98801341XA CN98801341A CN1205196C CN 1205196 C CN1205196 C CN 1205196C CN B98801341X A CNB98801341X A CN B98801341XA CN 98801341 A CN98801341 A CN 98801341A CN 1205196 C CN1205196 C CN 1205196C
- Authority
- CN
- China
- Prior art keywords
- taxan
- hydroxyl
- dab
- acidylate
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 238000000034 method Methods 0.000 title claims abstract description 79
- 230000008569 process Effects 0.000 title claims abstract description 36
- 238000001212 derivatisation Methods 0.000 title abstract description 11
- 229940123237 Taxane Drugs 0.000 title abstract description 5
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 144
- 238000006243 chemical reaction Methods 0.000 claims description 72
- OVMSOCFBDVBLFW-VHLOTGQHSA-N 5beta,20-epoxy-1,7beta,13alpha-trihydroxy-9-oxotax-11-ene-2alpha,4alpha,10beta-triyl 4,10-diacetate 2-benzoate Chemical compound O([C@@H]1[C@@]2(C[C@H](O)C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)O)C(=O)C1=CC=CC=C1 OVMSOCFBDVBLFW-VHLOTGQHSA-N 0.000 claims description 47
- 239000011541 reaction mixture Substances 0.000 claims description 42
- 239000003795 chemical substances by application Substances 0.000 claims description 37
- 150000002170 ethers Chemical class 0.000 claims description 14
- 150000001875 compounds Chemical class 0.000 claims description 13
- 238000003381 deacetylation reaction Methods 0.000 claims description 12
- 150000002148 esters Chemical class 0.000 claims description 12
- 239000003513 alkali Substances 0.000 claims description 11
- 238000005917 acylation reaction Methods 0.000 claims description 10
- 229930014667 baccatin III Natural products 0.000 claims description 9
- 239000002841 Lewis acid Substances 0.000 claims description 8
- 150000007517 lewis acids Chemical class 0.000 claims description 8
- 150000008065 acid anhydrides Chemical group 0.000 claims description 7
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 claims description 6
- 238000005886 esterification reaction Methods 0.000 claims description 5
- 229910021591 Copper(I) chloride Inorganic materials 0.000 claims description 4
- XPDWGBQVDMORPB-UHFFFAOYSA-N Fluoroform Chemical compound FC(F)F XPDWGBQVDMORPB-UHFFFAOYSA-N 0.000 claims description 4
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical compound [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 claims description 4
- 239000002243 precursor Substances 0.000 claims description 4
- 229940045803 cuprous chloride Drugs 0.000 claims description 3
- 230000032050 esterification Effects 0.000 claims description 3
- 235000005074 zinc chloride Nutrition 0.000 claims description 3
- 239000011592 zinc chloride Substances 0.000 claims description 3
- LPUQGPXGMKKLMY-UHFFFAOYSA-N 1,3-oxazolidine-2-carbonyl 1,3-oxazolidine-2-carboxylate Chemical compound N1CCOC1C(=O)OC(=O)C1NCCO1 LPUQGPXGMKKLMY-UHFFFAOYSA-N 0.000 claims description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical class NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 claims description 2
- 229910052768 actinide Inorganic materials 0.000 claims description 2
- 150000001255 actinides Chemical class 0.000 claims description 2
- JXLHNMVSKXFWAO-UHFFFAOYSA-N azane;7-fluoro-2,1,3-benzoxadiazole-4-sulfonic acid Chemical compound N.OS(=O)(=O)C1=CC=C(F)C2=NON=C12 JXLHNMVSKXFWAO-UHFFFAOYSA-N 0.000 claims description 2
- BOXVSFHSLKQLNZ-UHFFFAOYSA-K dysprosium(iii) chloride Chemical compound Cl[Dy](Cl)Cl BOXVSFHSLKQLNZ-UHFFFAOYSA-K 0.000 claims description 2
- 229910052747 lanthanoid Inorganic materials 0.000 claims description 2
- 150000002602 lanthanoids Chemical class 0.000 claims description 2
- ICAKDTKJOYSXGC-UHFFFAOYSA-K lanthanum(iii) chloride Chemical compound Cl[La](Cl)Cl ICAKDTKJOYSXGC-UHFFFAOYSA-K 0.000 claims description 2
- 229910044991 metal oxide Inorganic materials 0.000 claims description 2
- 150000004706 metal oxides Chemical group 0.000 claims description 2
- HPGGPRDJHPYFRM-UHFFFAOYSA-J tin(iv) chloride Chemical compound Cl[Sn](Cl)(Cl)Cl HPGGPRDJHPYFRM-UHFFFAOYSA-J 0.000 claims description 2
- CKLHRQNQYIJFFX-UHFFFAOYSA-K ytterbium(III) chloride Chemical compound [Cl-].[Cl-].[Cl-].[Yb+3] CKLHRQNQYIJFFX-UHFFFAOYSA-K 0.000 claims description 2
- KNWCDYSKJSREAQ-UHFFFAOYSA-N 1,3-oxazolidine-2-carboxylic acid Chemical compound OC(=O)C1NCCO1 KNWCDYSKJSREAQ-UHFFFAOYSA-N 0.000 claims 1
- IMSODMZESSGVBE-UHFFFAOYSA-N 2-Oxazoline Chemical compound C1CN=CO1 IMSODMZESSGVBE-UHFFFAOYSA-N 0.000 claims 1
- 125000003460 beta-lactamyl group Chemical group 0.000 claims 1
- GSWAOPJLTADLTN-UHFFFAOYSA-N oxidanimine Chemical group [O-][NH3+] GSWAOPJLTADLTN-UHFFFAOYSA-N 0.000 claims 1
- YWLXLRUDGLRYDR-UHFFFAOYSA-N 10-deacetylbaccatin Chemical compound CC(=O)OC12COC1CC(O)C(C(C(O)C1=C(C)C(O)CC3(O)C1(C)C)=O)(C)C2C3OC(=O)C1=CC=CC=C1 YWLXLRUDGLRYDR-UHFFFAOYSA-N 0.000 abstract description 53
- 238000002360 preparation method Methods 0.000 abstract description 16
- 229960001592 paclitaxel Drugs 0.000 abstract description 12
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 abstract description 12
- 229930012538 Paclitaxel Natural products 0.000 abstract description 11
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 176
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 103
- 125000000217 alkyl group Chemical group 0.000 description 100
- 235000019439 ethyl acetate Nutrition 0.000 description 87
- -1 silyl acid amides Chemical class 0.000 description 80
- 239000000243 solution Substances 0.000 description 78
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 74
- MTZQAGJQAFMTAQ-UHFFFAOYSA-N ethyl benzoate Chemical compound CCOC(=O)C1=CC=CC=C1 MTZQAGJQAFMTAQ-UHFFFAOYSA-N 0.000 description 73
- 125000001072 heteroaryl group Chemical group 0.000 description 41
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 36
- 229910052799 carbon Inorganic materials 0.000 description 35
- 238000005160 1H NMR spectroscopy Methods 0.000 description 34
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 34
- 229910052739 hydrogen Inorganic materials 0.000 description 34
- 239000001257 hydrogen Substances 0.000 description 34
- 239000000741 silica gel Substances 0.000 description 34
- 229910002027 silica gel Inorganic materials 0.000 description 34
- 229960001866 silicon dioxide Drugs 0.000 description 34
- 125000004423 acyloxy group Chemical group 0.000 description 25
- 238000005406 washing Methods 0.000 description 25
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 24
- 150000002431 hydrogen Chemical class 0.000 description 24
- 238000003818 flash chromatography Methods 0.000 description 23
- 239000007787 solid Substances 0.000 description 22
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 20
- 238000004458 analytical method Methods 0.000 description 20
- 238000003756 stirring Methods 0.000 description 20
- 238000001291 vacuum drying Methods 0.000 description 20
- 239000012074 organic phase Substances 0.000 description 19
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 18
- 230000004224 protection Effects 0.000 description 18
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 16
- 229910052757 nitrogen Inorganic materials 0.000 description 16
- 229920006395 saturated elastomer Polymers 0.000 description 16
- 238000001035 drying Methods 0.000 description 15
- 238000006884 silylation reaction Methods 0.000 description 15
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 14
- 230000006837 decompression Effects 0.000 description 14
- 239000000203 mixture Substances 0.000 description 14
- 150000001241 acetals Chemical class 0.000 description 13
- 150000001721 carbon Chemical group 0.000 description 13
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 12
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 12
- OIFBSDVPJOWBCH-UHFFFAOYSA-N Diethyl carbonate Chemical compound CCOC(=O)OCC OIFBSDVPJOWBCH-UHFFFAOYSA-N 0.000 description 11
- 238000001914 filtration Methods 0.000 description 11
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 11
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 10
- 239000012043 crude product Substances 0.000 description 10
- 125000000468 ketone group Chemical group 0.000 description 10
- 239000011734 sodium Substances 0.000 description 10
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 9
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- 125000003118 aryl group Chemical group 0.000 description 9
- 125000004432 carbon atom Chemical group C* 0.000 description 9
- 239000000460 chlorine Substances 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- 125000002252 acyl group Chemical group 0.000 description 8
- 150000001720 carbohydrates Chemical class 0.000 description 8
- 235000014633 carbohydrates Nutrition 0.000 description 8
- 230000003197 catalytic effect Effects 0.000 description 8
- GVNVAWHJIKLAGL-UHFFFAOYSA-N 2-(cyclohexen-1-yl)cyclohexan-1-one Chemical compound O=C1CCCCC1C1=CCCCC1 GVNVAWHJIKLAGL-UHFFFAOYSA-N 0.000 description 7
- 101150065749 Churc1 gene Proteins 0.000 description 7
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 7
- 102100038239 Protein Churchill Human genes 0.000 description 7
- 125000003342 alkenyl group Chemical group 0.000 description 7
- 239000002585 base Substances 0.000 description 7
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 7
- 125000006239 protecting group Chemical group 0.000 description 7
- 230000009257 reactivity Effects 0.000 description 7
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 6
- 239000007864 aqueous solution Substances 0.000 description 6
- 238000005660 chlorination reaction Methods 0.000 description 6
- DCFKHNIGBAHNSS-UHFFFAOYSA-N chloro(triethyl)silane Chemical compound CC[Si](Cl)(CC)CC DCFKHNIGBAHNSS-UHFFFAOYSA-N 0.000 description 6
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 6
- 239000002994 raw material Substances 0.000 description 6
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 5
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical group O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 5
- 125000000304 alkynyl group Chemical group 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 239000000284 extract Substances 0.000 description 5
- 238000000605 extraction Methods 0.000 description 5
- 229910052736 halogen Inorganic materials 0.000 description 5
- 150000002367 halogens Chemical class 0.000 description 5
- 150000002460 imidazoles Chemical class 0.000 description 5
- 229910052760 oxygen Inorganic materials 0.000 description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 5
- 238000010926 purge Methods 0.000 description 5
- 125000001424 substituent group Chemical group 0.000 description 5
- DKPFODGZWDEEBT-QFIAKTPHSA-N taxane Chemical class C([C@]1(C)CCC[C@@H](C)[C@H]1C1)C[C@H]2[C@H](C)CC[C@@H]1C2(C)C DKPFODGZWDEEBT-QFIAKTPHSA-N 0.000 description 5
- OCEKXXOSQITQEJ-UHFFFAOYSA-N 2,2,2-trifluoro-n-triethylsilylacetamide Chemical class CC[Si](CC)(CC)NC(=O)C(F)(F)F OCEKXXOSQITQEJ-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 4
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 4
- 238000006555 catalytic reaction Methods 0.000 description 4
- 125000000753 cycloalkyl group Chemical group 0.000 description 4
- 238000000354 decomposition reaction Methods 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- 125000000524 functional group Chemical group 0.000 description 4
- 125000001183 hydrocarbyl group Chemical group 0.000 description 4
- 239000001301 oxygen Substances 0.000 description 4
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 150000003839 salts Chemical class 0.000 description 4
- 235000002639 sodium chloride Nutrition 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- 150000003952 β-lactams Chemical class 0.000 description 4
- PNVPNXKRAUBJGW-UHFFFAOYSA-N (2-chloroacetyl) 2-chloroacetate Chemical compound ClCC(=O)OC(=O)CCl PNVPNXKRAUBJGW-UHFFFAOYSA-N 0.000 description 3
- NRKYWOKHZRQRJR-UHFFFAOYSA-N 2,2,2-trifluoroacetamide Chemical class NC(=O)C(F)(F)F NRKYWOKHZRQRJR-UHFFFAOYSA-N 0.000 description 3
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 3
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 3
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- 125000003545 alkoxy group Chemical group 0.000 description 3
- 125000004429 atom Chemical group 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 3
- 239000006227 byproduct Substances 0.000 description 3
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- KWYZNESIGBQHJK-UHFFFAOYSA-N chloro-dimethyl-phenylsilane Chemical compound C[Si](C)(Cl)C1=CC=CC=C1 KWYZNESIGBQHJK-UHFFFAOYSA-N 0.000 description 3
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical compound C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 description 3
- 125000006182 dimethyl benzyl group Chemical group 0.000 description 3
- 239000011737 fluorine Substances 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 125000002541 furyl group Chemical group 0.000 description 3
- 238000006206 glycosylation reaction Methods 0.000 description 3
- 230000033444 hydroxylation Effects 0.000 description 3
- 238000005805 hydroxylation reaction Methods 0.000 description 3
- OVMSOCFBDVBLFW-IAGPSASASA-N molport-003-665-782 Chemical compound O([C@@H]1[C@@]2(C[C@H](O)C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3(C21)OC(C)=O)=O)OC(=O)C)O)C(=O)C1=CC=CC=C1 OVMSOCFBDVBLFW-IAGPSASASA-N 0.000 description 3
- BABPEPRNSRIYFA-UHFFFAOYSA-N silyl trifluoromethanesulfonate Chemical compound FC(F)(F)S(=O)(=O)O[SiH3] BABPEPRNSRIYFA-UHFFFAOYSA-N 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 229940063683 taxotere Drugs 0.000 description 3
- 125000001544 thienyl group Chemical group 0.000 description 3
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 2
- LJCZNYWLQZZIOS-UHFFFAOYSA-N 2,2,2-trichlorethoxycarbonyl chloride Chemical compound ClC(=O)OCC(Cl)(Cl)Cl LJCZNYWLQZZIOS-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- VPBOBLLNWVCKLT-UHFFFAOYSA-N N-silyl-N-(trifluoromethyl)acetamide Chemical compound C(C)(=O)N([SiH3])C(F)(F)F VPBOBLLNWVCKLT-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- 235000016408 Podocarpus macrophyllus Nutrition 0.000 description 2
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 2
- 239000005864 Sulphur Substances 0.000 description 2
- 244000162450 Taxus cuspidata Species 0.000 description 2
- 235000009065 Taxus cuspidata Nutrition 0.000 description 2
- QYKIQEUNHZKYBP-UHFFFAOYSA-N Vinyl ether Chemical compound C=COC=C QYKIQEUNHZKYBP-UHFFFAOYSA-N 0.000 description 2
- GARJITDFQFOYKD-UHFFFAOYSA-N [N-]=[N+]=[N-].[Li+].[Si+4].[N-]=[N+]=[N-].[N-]=[N+]=[N-].[N-]=[N+]=[N-].[N-]=[N+]=[N-] Chemical compound [N-]=[N+]=[N-].[Li+].[Si+4].[N-]=[N+]=[N-].[N-]=[N+]=[N-].[N-]=[N+]=[N-].[N-]=[N+]=[N-] GARJITDFQFOYKD-UHFFFAOYSA-N 0.000 description 2
- 239000003377 acid catalyst Substances 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 229910052783 alkali metal Inorganic materials 0.000 description 2
- 125000005910 alkyl carbonate group Chemical group 0.000 description 2
- SRBFZHDQGSBBOR-LECHCGJUSA-N alpha-D-xylose Chemical compound O[C@@H]1CO[C@H](O)[C@H](O)[C@H]1O SRBFZHDQGSBBOR-LECHCGJUSA-N 0.000 description 2
- 125000003368 amide group Chemical group 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 125000001231 benzoyloxy group Chemical group C(C1=CC=CC=C1)(=O)O* 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N biotin Natural products N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- 229910052796 boron Inorganic materials 0.000 description 2
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 description 2
- YHASWHZGWUONAO-UHFFFAOYSA-N butanoyl butanoate Chemical compound CCCC(=O)OC(=O)CCC YHASWHZGWUONAO-UHFFFAOYSA-N 0.000 description 2
- 125000004106 butoxy group Chemical group [*]OC([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 150000001244 carboxylic acid anhydrides Chemical class 0.000 description 2
- 239000000571 coke Substances 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- 238000010908 decantation Methods 0.000 description 2
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 239000004210 ether based solvent Substances 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 238000005907 ketalization reaction Methods 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 239000012452 mother liquor Substances 0.000 description 2
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 2
- PIJWXSSJROBBTA-UHFFFAOYSA-N n-[tert-butyl(dimethyl)silyl]-2,2,2-trifluoroacetamide Chemical class CC(C)(C)[Si](C)(C)NC(=O)C(F)(F)F PIJWXSSJROBBTA-UHFFFAOYSA-N 0.000 description 2
- DGTNSSLYPYDJGL-UHFFFAOYSA-N phenyl isocyanate Chemical compound O=C=NC1=CC=CC=C1 DGTNSSLYPYDJGL-UHFFFAOYSA-N 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 239000011574 phosphorus Substances 0.000 description 2
- 238000003825 pressing Methods 0.000 description 2
- 150000003141 primary amines Chemical class 0.000 description 2
- WYVAMUWZEOHJOQ-UHFFFAOYSA-N propionic anhydride Chemical compound CCC(=O)OC(=O)CC WYVAMUWZEOHJOQ-UHFFFAOYSA-N 0.000 description 2
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 239000012266 salt solution Substances 0.000 description 2
- 229910052710 silicon Inorganic materials 0.000 description 2
- 239000010703 silicon Substances 0.000 description 2
- 239000008247 solid mixture Substances 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 2
- 125000003396 thiol group Chemical group [H]S* 0.000 description 2
- 229960003487 xylose Drugs 0.000 description 2
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- VAYTZRYEBVHVLE-UHFFFAOYSA-N 1,3-dioxol-2-one Chemical compound O=C1OC=CO1 VAYTZRYEBVHVLE-UHFFFAOYSA-N 0.000 description 1
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 1
- VZAWCLCJGSBATP-UHFFFAOYSA-N 1-cycloundecyl-1,2-diazacycloundecane Chemical compound C1CCCCCCCCCC1N1NCCCCCCCCC1 VZAWCLCJGSBATP-UHFFFAOYSA-N 0.000 description 1
- RRQYJINTUHWNHW-UHFFFAOYSA-N 1-ethoxy-2-(2-ethoxyethoxy)ethane Chemical compound CCOCCOCCOCC RRQYJINTUHWNHW-UHFFFAOYSA-N 0.000 description 1
- HCKNRHBSGZMOOF-UHFFFAOYSA-N 1-methoxy-2-methylperoxyethane Chemical compound COCCOOC HCKNRHBSGZMOOF-UHFFFAOYSA-N 0.000 description 1
- HOCDENBQBXLRAW-UHFFFAOYSA-N 2,2,2-trifluoro-n-methyl-n-triethylsilylacetamide Chemical compound CC[Si](CC)(CC)N(C)C(=O)C(F)(F)F HOCDENBQBXLRAW-UHFFFAOYSA-N 0.000 description 1
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-Lutidine Substances CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 1
- LTFMAUZHNCYYHZ-UHFFFAOYSA-N 2-methylpropyl propyl carbonate Chemical compound CCCOC(=O)OCC(C)C LTFMAUZHNCYYHZ-UHFFFAOYSA-N 0.000 description 1
- ATVJXMYDOSMEPO-UHFFFAOYSA-N 3-prop-2-enoxyprop-1-ene Chemical compound C=CCOCC=C ATVJXMYDOSMEPO-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- XVMSFILGAMDHEY-UHFFFAOYSA-N 6-(4-aminophenyl)sulfonylpyridin-3-amine Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=N1 XVMSFILGAMDHEY-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical group CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 1
- 229930190007 Baccatin Natural products 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 239000004641 Diallyl-phthalate Substances 0.000 description 1
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- XISCNCUWNKVZOD-UHFFFAOYSA-N O1C(NCC1)C(=O)O.O1C=NCC1 Chemical compound O1C(NCC1)C(=O)O.O1C=NCC1 XISCNCUWNKVZOD-UHFFFAOYSA-N 0.000 description 1
- HJVBNJPABYKSFN-UHFFFAOYSA-N S(=O)(=O)(O)C=1C(OC=CC1)OC1OC=CC=C1S(=O)(=O)O Chemical compound S(=O)(=O)(O)C=1C(OC=CC1)OC1OC=CC=C1S(=O)(=O)O HJVBNJPABYKSFN-UHFFFAOYSA-N 0.000 description 1
- 229910052772 Samarium Inorganic materials 0.000 description 1
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 1
- 241001116498 Taxus baccata Species 0.000 description 1
- 229930003756 Vitamin B7 Natural products 0.000 description 1
- XXFXTBNFFMQVKJ-UHFFFAOYSA-N [diphenyl(trityloxy)methyl]benzene Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(C=1C=CC=CC=1)OC(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 XXFXTBNFFMQVKJ-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000002877 alkyl aryl group Chemical group 0.000 description 1
- 125000004647 alkyl sulfenyl group Chemical group 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 150000001491 aromatic compounds Chemical class 0.000 description 1
- 125000005104 aryl silyl group Chemical group 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- CHIHQLCVLOXUJW-UHFFFAOYSA-N benzoic anhydride Chemical compound C=1C=CC=CC=1C(=O)OC(=O)C1=CC=CC=C1 CHIHQLCVLOXUJW-UHFFFAOYSA-N 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- FFBHFFJDDLITSX-UHFFFAOYSA-N benzyl N-[2-hydroxy-4-(3-oxomorpholin-4-yl)phenyl]carbamate Chemical compound OC1=C(NC(=O)OCC2=CC=CC=C2)C=CC(=C1)N1CCOCC1=O FFBHFFJDDLITSX-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- WOIVNLSVAKYSKX-UHFFFAOYSA-N benzyl nitrate Chemical compound [O-][N+](=O)OCC1=CC=CC=C1 WOIVNLSVAKYSKX-UHFFFAOYSA-N 0.000 description 1
- FHRRJZZGSJXPRQ-UHFFFAOYSA-N benzyl phenylmethoxycarbonyl carbonate Chemical compound C=1C=CC=CC=1COC(=O)OC(=O)OCC1=CC=CC=C1 FHRRJZZGSJXPRQ-UHFFFAOYSA-N 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- QUDWYFHPNIMBFC-UHFFFAOYSA-N bis(prop-2-enyl) benzene-1,2-dicarboxylate Chemical compound C=CCOC(=O)C1=CC=CC=C1C(=O)OCC=C QUDWYFHPNIMBFC-UHFFFAOYSA-N 0.000 description 1
- JKJWYKGYGWOAHT-UHFFFAOYSA-N bis(prop-2-enyl) carbonate Chemical compound C=CCOC(=O)OCC=C JKJWYKGYGWOAHT-UHFFFAOYSA-N 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- USKRHFHDJIMKIB-UHFFFAOYSA-N butyl 2-methylpropyl carbonate Chemical compound CCCCOC(=O)OCC(C)C USKRHFHDJIMKIB-UHFFFAOYSA-N 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- ZFTFAPZRGNKQPU-UHFFFAOYSA-L carboxylato carbonate Chemical compound [O-]C(=O)OC([O-])=O ZFTFAPZRGNKQPU-UHFFFAOYSA-L 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- AOGYCOYQMAVAFD-UHFFFAOYSA-N chlorocarbonic acid Chemical compound OC(Cl)=O AOGYCOYQMAVAFD-UHFFFAOYSA-N 0.000 description 1
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- VUPKGFBOKBGHFZ-UHFFFAOYSA-N dipropyl carbonate Chemical class CCCOC(=O)OCCC VUPKGFBOKBGHFZ-UHFFFAOYSA-N 0.000 description 1
- POLCUAVZOMRGSN-UHFFFAOYSA-N dipropyl ether Chemical compound CCCOCCC POLCUAVZOMRGSN-UHFFFAOYSA-N 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- ZOOODBUHSVUZEM-UHFFFAOYSA-N ethoxymethanedithioic acid Chemical compound CCOC(S)=S ZOOODBUHSVUZEM-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- UQEAIHBTYFGYIE-UHFFFAOYSA-N hexamethyldisiloxane Chemical compound C[Si](C)(C)O[Si](C)(C)C UQEAIHBTYFGYIE-UHFFFAOYSA-N 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- NSPJNIDYTSSIIY-UHFFFAOYSA-N methoxy(methoxymethoxy)methane Chemical compound COCOCOC NSPJNIDYTSSIIY-UHFFFAOYSA-N 0.000 description 1
- CXHHBNMLPJOKQD-UHFFFAOYSA-M methyl carbonate Chemical compound COC([O-])=O CXHHBNMLPJOKQD-UHFFFAOYSA-M 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- VAOCPAMSLUNLGC-UHFFFAOYSA-N metronidazole Chemical compound CC1=NC=C([N+]([O-])=O)N1CCO VAOCPAMSLUNLGC-UHFFFAOYSA-N 0.000 description 1
- 229960000282 metronidazole Drugs 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 1
- 125000006502 nitrobenzyl group Chemical group 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000002894 organic compounds Chemical group 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- LCLOXRAKDJBSMN-UHFFFAOYSA-N pentyl hydrogen carbonate Chemical compound CCCCCOC(O)=O LCLOXRAKDJBSMN-UHFFFAOYSA-N 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- CAEWJEXPFKNBQL-UHFFFAOYSA-N prop-2-enyl carbonochloridate Chemical compound ClC(=O)OCC=C CAEWJEXPFKNBQL-UHFFFAOYSA-N 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 239000002534 radiation-sensitizing agent Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- KZUNJOHGWZRPMI-UHFFFAOYSA-N samarium atom Chemical compound [Sm] KZUNJOHGWZRPMI-UHFFFAOYSA-N 0.000 description 1
- DCKVNWZUADLDEH-UHFFFAOYSA-N sec-butyl acetate Chemical compound CCC(C)OC(C)=O DCKVNWZUADLDEH-UHFFFAOYSA-N 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 229920002994 synthetic fiber Polymers 0.000 description 1
- XKXIQBVKMABYQJ-UHFFFAOYSA-N tert-butyl hydrogen carbonate Chemical compound CC(C)(C)OC(O)=O XKXIQBVKMABYQJ-UHFFFAOYSA-N 0.000 description 1
- FGTJJHCZWOVVNH-UHFFFAOYSA-N tert-butyl-[tert-butyl(dimethyl)silyl]oxy-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)O[Si](C)(C)C(C)(C)C FGTJJHCZWOVVNH-UHFFFAOYSA-N 0.000 description 1
- DZLFLBLQUQXARW-UHFFFAOYSA-N tetrabutylammonium Chemical compound CCCC[N+](CCCC)(CCCC)CCCC DZLFLBLQUQXARW-UHFFFAOYSA-N 0.000 description 1
- 150000003527 tetrahydropyrans Chemical class 0.000 description 1
- LGSAOJLQTXCYHF-UHFFFAOYSA-N tri(propan-2-yl)-tri(propan-2-yl)silyloxysilane Chemical compound CC(C)[Si](C(C)C)(C(C)C)O[Si](C(C)C)(C(C)C)C(C)C LGSAOJLQTXCYHF-UHFFFAOYSA-N 0.000 description 1
- UTXPCJHKADAFBB-UHFFFAOYSA-N tribenzyl(chloro)silane Chemical compound C=1C=CC=CC=1C[Si](CC=1C=CC=CC=1)(Cl)CC1=CC=CC=C1 UTXPCJHKADAFBB-UHFFFAOYSA-N 0.000 description 1
- WILBTFWIBAOWLN-UHFFFAOYSA-N triethyl(triethylsilyloxy)silane Chemical compound CC[Si](CC)(CC)O[Si](CC)(CC)CC WILBTFWIBAOWLN-UHFFFAOYSA-N 0.000 description 1
- ILWRPSCZWQJDMK-UHFFFAOYSA-N triethylazanium;chloride Chemical compound Cl.CCN(CC)CC ILWRPSCZWQJDMK-UHFFFAOYSA-N 0.000 description 1
- FJKIXWOMBXYWOQ-UHFFFAOYSA-N vinyl ethyl ether Natural products CCOC=C FJKIXWOMBXYWOQ-UHFFFAOYSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000011735 vitamin B7 Substances 0.000 description 1
- 235000011912 vitamin B7 Nutrition 0.000 description 1
- 239000012991 xanthate Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D305/00—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms
- C07D305/14—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
- C07F7/1872—Preparation; Treatments not provided for in C07F7/20
- C07F7/1892—Preparation; Treatments not provided for in C07F7/20 by reactions not provided for in C07F7/1876 - C07F7/1888
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epoxy Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Soil Working Implements (AREA)
- Fats And Perfumes (AREA)
- Percussion Or Vibration Massage (AREA)
- Plural Heterocyclic Compounds (AREA)
- Saccharide Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
本发明涉及制备紫杉醇和其它紫杉烷的方法,该方法通过10-DAB的C(7)和C(10)羟基的选择性衍生反应实现,本发明尤其涉及采用新步骤的新方法,在该步骤中C(10)羟基先于C(7)羟基被保护或被衍生,以及提供C(7)和C(10)衍生的10-DAB化合物。
Description
发明背景
本发明一般性涉及制备紫杉醇和其它紫杉烷的方法,特别涉及紫杉烷的C(7)或C(10)羟基被选择性地衍生的方法。
从taxus baccata L.即英国紫杉的针叶中提取的10-DAB(1)已经成为生产紫杉醇和Taxotere的关键原料,二者都是有效的抗癌药物。将10-DAB转变成紫杉醇,Taxotere_和其它有抗肿瘤活性的紫杉烷需要C(7)和C(10)羟基进行保护或衍生作用,然后酯化C(13)羟基,使适当的侧链连到该位置上。
1 R10=R7=H
2 R10=H,R7=TES
到目前为止,制备紫杉醇和紫杉醇类似物的步骤都是基于Senilh等人的观察(C. R. Acad. Sci.Paris,II,1981,293,501),即10-DAB的4个羟基与吡啶中乙酸酐的反应性能相互之间的关系为C(7)-OH>C(10)-OH>C(13)-OH>C(1)-OH。Denis等人报道(《美国化学学会杂志》(J.Am.Chem.Soc.),1988,110,5917)了用三乙基甲硅烷基氯的吡啶选择性地甲硅烷基化10-DAB的C(7)羟基,以85%的产率得到7-三乙基甲硅烷基-10-脱乙酰基浆果赤霉素(III)(2)。基于这些报道,在这些需要区分C(7)和C(10)羟基的方法(例如,由10-DAB制备紫杉醇)中,必须在C(10)羟基被保护或衍生之前保护(或衍生)C(7)羟基。例如,可以通过下列步骤制备紫杉醇:首先,用三乙基甲硅烷基氯处理10-DAB以保护C(7)羟基,然后,乙酰化C(10)羟基,通过酯化C(13)羟基来连接侧链,最后,除去保护基。
众所周知,有多个取代基连接C(10)或C(7)氧的紫杉烷显示出抗癌活性。为了提供更有效的合成这些原料的方法,获得更有效和更高选择性的保护或衍生C(10)和C(7)羟基的方法是非常有益的。
发明概述
因此,本发明目的之一是提供通过10-DAB和其它紫杉烷的C(7)或C(10)羟基的选择性衍生作用制备紫杉醇和其它紫杉烷的高效方法,特别是在C(7)羟基被保护或衍生之前就将C(10)羟基保护或衍生的方法,以及提供C(7)或C(10)衍生的紫杉烷。
因此,简而言之,本发明涉及酰基化紫杉烷的C(10)羟基的方法。该方法包括形成含有紫杉烷和相对于1当量紫杉烷碱含量不足1当量的酰化剂的反应混合物,然后使紫杉烷与酰化剂反应,形成C(10)酰化紫杉烷。
本发明还涉及甲硅烷基化有C(10)羟基的紫杉烷的C(10)羟基的方法。该方法包括用甲硅烷酰胺或双甲硅烷酰胺处理紫杉烷,形成C(10)甲硅烷基化紫杉烷。
本发明还涉及将10-酰氧基-7-羟基紫杉烷的C(7)羟基转变成缩醛或缩酮的方法。该方法包括在酸催化剂存在下用缩酮化剂处理10-酰氧基-7-羟基紫杉烷,形成C(10)缩酮化紫杉烷。
本发明还涉及有下面结构的紫杉烷:
其中
M是金属或含有铵;
R1是氢,羟基,保护的羟基,或与R14或R2一起形成碳酸酯;
R2是酮基,-OT2,酰氧基,或与R1一起形成碳酸酯;
R4是-OT4或酰氧基;
R7是-OSiRJRKRL;
R9是氢,酮基,-OT9,或酰氧基;
R10是氢,酮基,-OT10,或酰氧基;
R13是羟基,保护的羟基,酮基,或MO-;
R14是氢,-OT14,酰氧基,或与R1一起形成碳酸酯;
RJ,RK,RL分别是烃基,取代的烃基或杂芳基,但是,条件是,如果各RJ,RK和RL是烷基,则RJ,RK和RL中至少有一个是有至少4个碳原子的碳骨架结构;及
T2,T4,T9,T10和T14分别是氢或羟基保护基。
本发明的其它目的和特征将在下文中陆续出现和指出。
优选具体实例详述
在其它目的中,本发明能够选择性地衍生紫杉烷的C(10)羟基而不用先保护C(7)羟基。换句话说,人们发现,以前报道的关于C(7)和C(10)羟基的反应性可以颠倒,即在某些条件下C(10)羟基的反应性变得比C(7)羟基的反应性更强。
虽然本发明可以被用来选择性地衍生在C(7)或C(10)位有羟基的紫杉烷,但本发明的最大优点在于选择性地衍生在C(7)和C(10)位有羟基的紫杉烷,即7,10-二羟基紫杉烷。总之,可以根据本发明进行选择性衍生的7,10-二羟基紫杉烷对应于下式结构:
其中
R1是氢,羟基,保护的羟基,或与R14或R2一起形成碳酸酯;
R2是酮基,-OT2,酰氧基,或与R1一起形成碳酸酯;
R4是-OT4或酰氧基;
R9是氢,酮基,-OT9或酰氧基;
R13是羟基,保护的羟基,酮基,或

R14是氢,-OT14,酰氧基,或与R1一起形成碳酸酯;
T2,T4,T9和T14分别是氢或羟基保护基;
X1是-OX6,-SX7或-NX8X9;
X2是氢,烃基,取代的烃基或杂芳基;
X3和X4分别是氢,烃基,取代的烃基或杂芳基;
X5是-X10,-OX10,-SX10,-NX8X10或-SO2X11;
X6是烃基,取代的烃基,杂芳基,羟基保护基或增加紫杉烷衍生物的水溶性的官能团;
X7是烃基,取代的烃基,杂芳基或巯基保护基;
X8是氢,烃基或取代的烃基;
X9是氨基保护基;
X10是烃基,取代的烃基或杂芳基;
X11是烃基,取代的烃基,杂芳基,-OX10或-NX8X14;及
X14是氢,烃基,取代的烃基或杂芳基。
C(10)的选择性衍生作用
根据本发明的方法,我们已经发现,紫杉烷的C(10)羟基可以在没有碱,优选没有胺碱存在的情况下被选择性地酰化。因此,如果已经有胺碱如吡啶,三乙胺,二甲氨基吡啶和2,6-二甲基吡啶存在,则优选它们以相对低的浓度存在于反应混合物中。换句话说,如果在反应混合物中有碱存在,则胺碱与紫杉烷的摩尔比最好小于1∶1,更优选小于10∶1,最优选小于100∶1。
可用于紫杉烷的C(10)羟基的选择性酰化反应的酰化剂包括酸酐,二碳酸酯,硫代二碳酸酯和异氰酸酯。总之,酸酐,二碳酸酯和硫代二碳酸酯对应于结构式4,异氰酸酯对应于结构式5。

其中,R1是-ORa,-SRa或Ra;R2是-OC(O)Rb,-OC(O)ORb,-OC(O)SRb,-OPORbRc或-OS(O)2Rb;R3是烃基,取代的烃基或杂芳基;Ra,Rb,Rc分别是烃基,取代的烃基或杂芳基。例如,合适的羧酸酐酰化剂包括乙酸酐,氯乙酸酐,丙酸酐,苯甲酸酐和其它含有取代的或未取代的烃基或杂芳基部分的羧酸酐;合适的二碳酸酯酰化剂包括二碳酸二苄酯,二碳酸二烯丙酯,二碳酸二丙酯和其它含有取代的或未取代的烃基或杂芳基部分的二碳酸酯;及合适的异氰酸酯酰化剂,包括异氰酸苯酯和其它含有取代的或未取代的烃基或杂芳基部分的异氰酸酯。另外,虽然用作酰化剂的酸酐,二碳酸酯和硫代二碳酸酯可以是混杂的,但一般优选为对称的;即选择分子对称的R1和R2(例如,如果R1是Ra,则R2是-OC(O)Rb,且Ra与Rb相同)。
虽然,对于许多酰化剂来说,紫杉烷的C(10)羟基的酰化反应都会以适当的速率进行,但我们发现,如果反应混合物中有Lewis酸存在,反应速率会有所增加。显然,对Lewis酸的浓度没有严格限制,但迄今所获得的实验结果表明,其用量既可以是化学计算量,也可以是催化量。总之,可用的Lewis酸包括元素周期表(美国化学学会格式)中IB,IIB,IIIB,IVB,VB,VIB,VIIB,VIII,IIIA,IVA,镧系和锕系元素的三氟甲磺酸盐和卤化物。优选的Lewis酸包括氯化锌,氯化锡,三氯化铈,氯化亚铜,三氯化镧,三氯化镝和三氯化镱。如果酰化剂是酸酐或二碳酸酯,氯化锌或三氯化铈是特别优选的。如果酰化剂是异氰酸酯,氯化亚铜是特别优选的。
用于选择性酰化反应的溶剂优选醚溶剂,如四氢呋喃,但其它溶剂如乙醚或二甲氧基乙烷也可以使用。
进行C(10)选择性酰化反应的温度没有被严格限制,但是,一般优选在室温或较高温度进行,以使反应能以足够高的速率进行。
为了说明本发明,涉及二碳酸二苄酯,二碳酸二烯丙酯,乙酸酐,氯乙酸酐和异氰酸苯酯的酰化反应将在下面反应流程1-5中加以说明。在这一系列反应流程中,在C(10)位上被选择性酰化的紫杉烷是10-脱乙酰基浆果赤霉素III。但是,应该理解,这些反应流程只是为了说明本发明,而且,一般含有C(10)羟基的其它紫杉烷,特别是其它7,10-二羟基紫杉烷都可以用本发明的这些酰化剂和其它酰化剂选择性地酰化。
流程1
流程2
流程3

流程4
流程5

本发明另一方面是紫杉烷的C(10)羟基可以被选择性地甲硅烷基化。一般来说,甲硅烷基化剂选自甲硅烷基酰胺和双甲硅烷基酰胺。优选的甲硅烷基酰胺和双甲硅烷基酰胺分别对应于结构式6和7:
其中,RD,RE,RF,RG和RH分别为烃基,取代的烃基或杂芳基。优选的甲硅烷基化剂选自三(烃基)甲硅烷基三氟甲基乙酰胺和双三(烃基)甲硅烷基三氟甲基乙酰胺,其中烃基部分是取代的或未取代的烷基或芳基。例如,优选的甲硅烷基酰胺和双甲硅烷基酰胺包括N,O-双(三甲基甲硅烷基)三氟乙酰胺,N,O-双(三乙基甲硅烷基)三氟乙酰胺,N-甲基-N-三乙基甲硅烷基三氟乙酰胺和N,O-双(叔丁基二甲基甲硅烷基)三氟乙酰胺。
甲硅烷基化剂既可以单独使用,也可以与催化量的碱如碱金属碱结合使用。一般来说,碱金属酰胺如酰化锂催化剂,特别是六甲基乙硅叠氮化锂(lithium hexamethyldisilazide)是优选的。
用于选择性甲硅烷基化反应的溶剂优选醚溶剂,如四氢呋喃,但其它溶剂如乙醚或二甲氧基乙烷也可以使用。
对进行C(10)选择性甲硅烷基化反应的温度没有严格限制,但是,一般优选在0℃或较高温度进行。
涉及N,O-双(三甲基甲硅烷基)三氟乙酰胺和N,O-双(三乙基甲硅烷基)三氟乙酰胺的C(10)选择性甲硅烷基化反应将在下面反应流程6-7中加以说明。在这些反应流程中,在C(10)位被选择性甲硅烷基化的紫杉烷是10-脱乙酰基浆果赤霉素III。但是,应该理解,这些反应流程只是为了说明本发明,而且,一般含有C(10)羟基的其它紫杉烷,特别是其它7,10-二羟基紫杉烷都可以用本发明的这些甲硅烷基化剂和其它的甲硅烷基化剂选择性地甲硅烷基化。
流程6

流程7
按照本文所述方法衍生7,10-二羟基紫杉烷的C(10)羟基之后,可以迅速保护C(7)羟基,或者在C(1)和C(13)羟基(如果C(14)存在,也包括C(14)羟基)存在下选择性地衍生C(7)羟基。
C(7)的选择性衍生作用
C(10)酰化或甲硅烷基化的紫杉烷的C(7)羟基的选择性酰化反应可以用任何一种常规酰化剂实现,所说酰化剂包括(但不限于)取代的或未取代的羧酸衍生物,例如,羧酸卤化物,酸酐,二碳酸酯,异氰酸酯和卤代甲酸酯。例如,浆果赤霉素III,10-酰基-10-脱乙酰基浆果赤霉素III或10-三烃基甲硅烷基-10-脱乙酰基浆果赤霉素III的C(7)羟基可以用二碳酸二苄酯,二碳酸二烯丙酯,氯甲酸2,2,2-三氯乙酯,氯甲酸苄酯或其它常用酰化剂选择性酰化。
C(10)酰化或甲硅烷基化的紫杉烷的C(7)羟基的酰化反应一般比7,10-二羟基紫杉烷(如10-DAB)的C(7)的酰化反应更有效和更具选择性,也就是说,一旦C(10)羟基被酰化或甲硅烷基化,剩下的C(7),C(13)和C(1)羟基(如果C(14)存在,也包括C(14)羟基)的反应性就会明显不同。这些酰化反应可以任意在有或没有碱存在下进行。
C(10)羟基已经被酰化或甲硅烷基化的紫杉烷的C(7)羟基的选择性酰化反应的实例示于反应流程8-11。在这些反应流程中,在C(7)位被选择性酰化的紫杉烷是浆果赤霉素III或10-三乙基甲硅烷基-10-脱乙酰基浆果赤霉素III。但是,应该理解,这些反应流程仅为了说明本发明,而且,在C(10)位有其它酰基和甲硅烷基部分的紫杉烷以及在其它紫杉烷环位的其它取代基都可以用本发明的这些或其它酰化剂对C(7)羟基进行选择性酰化。
流程8
流程9
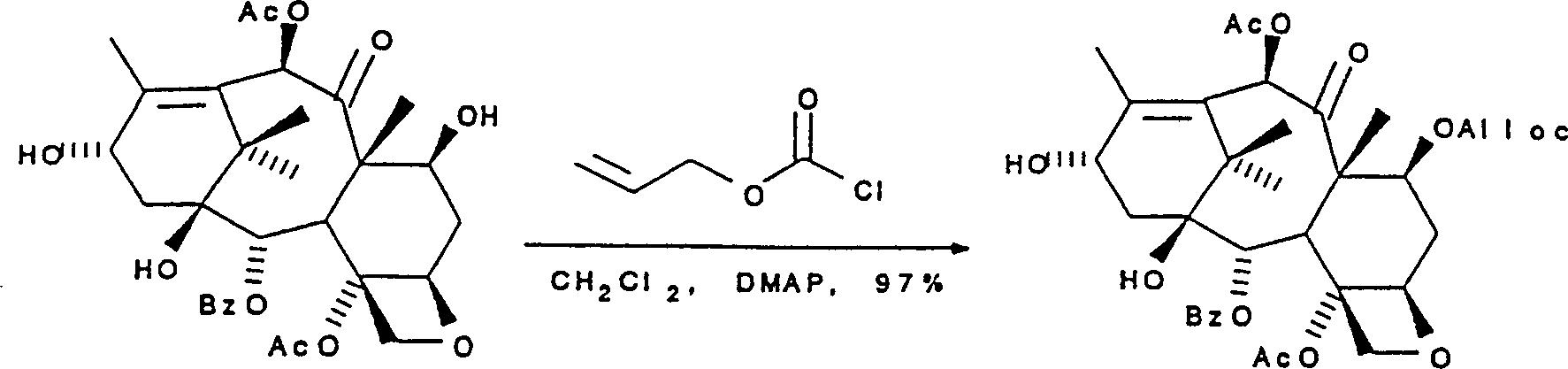
流程10

流程11
或者,C(10)酰化的紫杉烷衍生物的C(7)羟基可以用任何一种羟基保护基如缩醛,缩酮,甲硅烷基和可除去的酰基保护基有选择地保护。例如,C(7)羟基可以用任何一种常用甲硅烷基化剂甲硅烷基化,所说的甲硅烷基化剂包括(但不限于)三(烃基)甲硅烷基卤化物和三(烃基)甲硅烷基三氟甲磺酸酯。这些化合物的烃基部分可以是取代的或未取代的,优选取代的或未取代的烷基或芳基。例如,浆果赤霉素III的C(7)羟基可以用甲硅烷基化剂如三苄基甲硅烷基氯化物,三甲基甲硅烷基氯化物,三乙基甲硅烷基氯化物,二甲基异丙基甲硅烷基氯化物,二甲基苯基甲硅烷基氯化物等有选择地甲硅烷基化。
C(10)酰化的紫杉烷的C(7)羟基的甲硅烷基化反应一般比7,10-二羟基紫杉烷(如10-DAB)的甲硅烷基化反应更有效和更具选择性,也就是说,一旦C(10)羟基被酰化,剩下的C(7),C(13)和C(1)羟基(如果C(14)存在,也包括C(14)羟基)的反应性就会明显不同。C(7)的甲硅烷基化反应可以在很宽的条件范围下进行,包括在有或没有胺碱存在下进行。
C(10)已经被酰化的紫杉烷的C(7)羟基的选择性甲硅烷基化反应的实例示于反应流程12-15。在这些反应流程中,在C(7)位被选择性甲硅烷基化的紫杉烷是浆果赤霉素III或10-脱乙酰基浆果赤霉素III的另一种C(10)-酰氧基衍生物。但是,应该理解,这些反应流程仅为了说明本发明,而且,其它紫杉烷可以用本发明的这些或其它甲硅烷基化剂进行选择性甲硅烷基化。
流程12
流程13
流程14

流程15

或者,C(10)酰化的紫杉烷的C(7)羟基可以用任何一种常用试剂有选择地保护,所说试剂包括(但不限于)普通缩醛,缩酮,乙烯醚,并在酸性催化剂存在下进行。这些试剂(无论是缩醛,缩酮,乙烯醚,还是其它)在本文中都被称作“缩酮化剂”,在T.W.Greene的《有机合成中的保护基》(Protective Groups in Organic Synthesis),John Wiley andSons,1981中有所论述。所用的酸催化剂可以是有机酸或无机酸,如甲苯磺酸或樟脑磺酸,用量至少是催化量。例如,浆果赤霉素III的C(7)羟基可以用2-甲氧基丙烯有选择地缩酮化。制备缩醛和缩酮的其它适当试剂包括甲基乙烯醚,乙基乙烯醚,四氢吡喃等。
C(10)酰化的紫杉烷的C(7)取代基的选择性缩酮化反应一般比10-DAB的缩酮化反应更有效和更具选择性,也就是说,一旦C(10)羟基被酰化,剩下的C(7),C(13)和C(1)羟基(如果C(14)存在,也包括C(14)羟基)的反应性将会极为不同。
浆果赤霉素III的C(7)缩酮的选择性形成反应的实例在反应流程16中说明。但是,应该理解,该反应流程仅为了说明本发明,而且,其它紫杉烷可以用本发明的这些或其它缩酮化剂进行选择性缩酮化。
流程16
在适当的条件下,含有C(10)羟基的紫杉烷的C(7)羟基可以被选择性地甲硅烷基化。显然,这些甲硅烷基化反应不限于带有烷基(有3个或更少碳原子)的甲硅烷基。
紫杉烷的C(7)羟基一般可以用甲硅烷基化剂选择性地甲硅烷基化,所说的甲硅烷基化剂含有-SiRJRKRL部分,其中RJ,RK和RL分别是取代的或未取代的烃基或杂芳基,条件是任何取代基都不是羟基。在本发明一个具体实例中,如果各RJ,RK和RL均为烷基,则至少RJ,RK和RL之一包括一个至少有4个碳原子的碳骨架结构(即碳链或碳环)。合适的甲硅烷基化剂包括甲硅烷基卤化物和甲硅烷基三氟甲磺酸酯,例如,三(烃基)甲硅烷基卤化物和三(烃基)甲硅烷基三氟甲磺酸酯。这些甲硅烷基化剂的烃基取代基部分可以是取代的或未取代的,优选取代的或未取代的烷基或芳基。
C(7)羟基的选择性甲硅烷基化反应可以在溶剂如二甲基甲酰胺(“DMF”)或吡啶中并在胺碱如咪唑或吡啶存在下进行。反应流程17-20描述了10-DAB的C(7)羟基的甲硅烷基化反应过程,即分别用叔丁基二甲基甲硅烷基氯化物,三苄基甲硅烷基氯化物,二甲基-异丙基甲硅烷基氯化物和二甲基苯基甲硅烷基氯化物处理10-DAB,以高产率甲硅烷基化C(7)羟基。令人惊讶的是,Denis等人(《美国化学学会杂志》(J.Am.Chem.Soc.),1988,110)的报告认为在这些条件下的甲硅烷基化反应不可能选择性地形成7-TBS-10-DAB。
流程17

流程18
流程19

流程20
本发明的方法可还被用于用不同的甲硅烷基保护基保护7,10-二羟基紫杉烷的C(7)和C(10)羟基。通过选择在不同的条件下可以除去的基团可以对C(7)和C(10)羟基进行衍生反应。因此,这些反应增加了整个方法的灵活性,并使许多单个保护反应的产率比当前使用的方法的产率高。例如,从C(10)除去三乙基甲硅烷基保护基比从C(7)除去叔丁基二甲基甲硅烷基保护基更容易,而从C(7)除去二甲基苯基甲硅烷基保护基比从C(10)除去叔丁基二甲基甲硅烷基保护基更容易。7-叔丁基二甲基甲硅烷基-10-三乙基甲硅烷基-10-DAB和7-二甲基苯基甲硅烷基-10-叔丁基二甲基甲硅烷基-10-DAB的制备如反应流程21和22所示。
流程21
流程22

本文公开的方法可用于大量源自天然或合成材料的不同的紫杉烷,用来制备多种多样可进一步衍生的紫杉烷中间体。例如,本发明方法可有效地用来在引入C(13)β-酰氨基酯的C(13)侧链前体与紫杉烷的偶合反应之前,以及在紫杉烷的核心上的多个位置有可变取代基的紫杉烷的制备反应之前保护C(7)和C(10)羟基官能团。
C(13)侧链前体与紫杉烷的连接反应可以用多种已知技术进行。例如,可以将适当取代的β-内酰胺,噁唑啉,噁唑烷羧酸,噁唑烷羧酸酐或异丝氨酸衍生物与有C(13)羟基,金属氧化物或氧化铵取代基的三环-或四环紫杉烷反应,形成如《紫杉醇:科学与应用》,编者:M.Suffness,CRC出版社(Boca Rotan,FL),1995,第V章,97-121页,所述的在C(13)位有β-酰氨基酯取代基的化合物。例如,从10-DAB合成紫杉醇的反应如反应流程23所示。应该注意到,虽然β-内酰胺和10-DAB被用于该反应流程,但其它侧链前体和其它紫杉烷被取代也不背离本发明精神。
流程23
反应流程23所示方法比目前已知的任何其它方法都有效得多,这是因为10-DAB的C(10)羟基的三氯化铈催化的乙酰化反应和接下来进行的C(7)羟基的甲硅烷基化反应的高产率和高选择性。该合成反应分为4步,总产率89%。
反应流程24和25用来说明带有附带取代基的C(7)羟基和游离C(10)羟基的紫杉烷的制备方法。本发明方法提供了很大的灵活性,使得连接C(7)羟基的取代基既可以在连接C(13)侧链之前也可以在其之后就位。
反应流程24概括了紫杉烷的制备方法,已经发现所说紫杉烷是极好的化疗用放射致敏剂,而且该反应流程对在引入C(13)侧链之前将取代基连接到C(7)羟基作了说明。根据反应流程7的方法,10-DAB首先被转化成10-TES-10-DAB。然后,通过用羰基二咪唑处理将C(7)羟基转化为中间体咪唑化物。然后,不用分离,将咪唑化物中间体与甲硝唑醇反应,得到7-metro-10-TES-10-DAB。将7-metro-10-TES-10-DAB与要在C(13)位引入侧链的β-内酰胺偶合,然后通过用HF和吡啶处理除去C(10)和C(2’)位的TES基团。
流程24
反应流程25概括了用于识别蛋白质的紫杉烷的制备方法,该蛋白质与紫杉烷形成生物缀合物。它说明了在引入C(13)侧链之后取代基在C(7)羟基上连接的方案。根据反应流程7和11的方法,10-DAB首先被转化成7-对硝基苄氧羰基-10-TES-10-DAB。用TES保护的β-内酰胺连接C(13)侧链,然后,通过用氢和钯催化剂处理选择性地除去对硝基苄氧羰基保护基,生成2’,10-(双)-TES-紫杉醇。然后,将C(7)羟基与羰基二咪唑反应,并将所得咪唑化物与1,4-二氨基丁烷反应,得到伯胺。伯胺与生物素的羟基琥珀酰亚胺酯的反应完成了生物素酰胺基团在C(7)基团上的连接。最后,用HF的吡啶溶液处理除去C(10)和C(2’)上的TES保护基。
流程25
保护的紫杉烷衍生物或用于这种保护的紫杉烷衍生物制备的中间体或原料可以进一步被修饰以提供在紫杉烷的各位置上的可变取代基。
分别具有C(2)和/或C(4)取代基而非苯甲酰氧基和乙酰氧基的紫杉烷可以从浆果赤霉素III,10-DAB和在PCT专利申请WO 94/01223中更全面论述的其它紫杉烷制备。一般来说,C(2)和C(4)酰氧基取代基用氢化铝锂或其它合适的还原剂处理,在C(2)和C(4)位形成羟基,然后将其,例如,与羧酸卤化物(任选在用1,2-碳酸酯保护基与C(1)羟基一起保护C(2)羟基之后进行)反应,得到所要的C(2)和C(4)衍生物。
具有C(7)取代基而非上述羟基和酰氧基的紫杉烷可以从浆果赤霉素III,10-DAB和在PCT专利申请WO 94/17050中更全面论述的其它紫杉烷制备。例如,可以进行C(7)黄原酸酯的氢化锡还原反应,生成相应的C(7)二氢紫杉烷。或者,通过在室温下和THF溶液中用2-氯-1,1,2-三氟三乙胺处理C(13)-三乙基甲硅烷基保护的浆果赤霉素III来制备C(7)氟取代的紫杉烷。其它带有游离C(7)羟基的浆果赤霉素衍生物的行为类似。或者,通过用含有过量三乙胺盐酸盐的甲磺酰氯和三乙胺的二氯甲烷溶液处理浆果赤霉素III可以制备7-氯浆果赤霉素III。
具有C(9)取代基而非酮基的紫杉烷可以从浆果赤霉素III,10-DAB和在PCT专利申请WO 94/20088中更全面论述的其它紫杉烷制备。紫杉烷的C(9)酮基取代基一般被选择性还原,生成相应的带有硼氢化物(优选四丁基硼氢化铵(Bu4NBH4)或三乙氧基硼氢化物)的C(9)β-羟基衍生物。然后,可以用羟基保护基在C(7)位保护C(9)β-羟基衍生物,用本文所述的酰化C(7)羟基的方法的可以酰化C(9)羟基。或者,7-保护的-9β-羟基衍生物与KH的反应引起乙酸酯基团(或其它酰氧基)从C(10)向C(9)迁移和羟基基团从C(9)向C(10)迁移,从而得到10-脱乙酰基衍生物,可以根据本文其它部分所述的方法将其酰化。
具有C(10)取代基而非本文所述羟基,酰氧基或保护羟基的紫杉烷可以按照PCT专利申请WO 94/15599和其它文献中更全面论述的方法制备。例如,具有C(10)酮基取代基的紫杉烷可以通过10-脱乙酰基紫杉烷的氧化反应制备。在C(10)位被二氢取代的紫杉烷通过C(10)羟基或酰氧基取代的紫杉烷与二碘化钐反应制备。
具有C(14)取代基而非氢的紫杉烷也可以制备。用于这些化合物制备的原料可以是,例如,羟基化紫杉烷(14-羟基-10-脱乙酰基浆果赤霉素III),已经发现可以从紫杉针叶中提取它(
C&EN,36-37页,1993年4月12日)。这种有上述各种C(2),C(4),C(7),C(9),C(10),C3’和C5’官能团的羟基化紫杉烷的衍生物也可以用这种羟基化紫杉烷制备。另外,如
C&EN中所述10-DAB的C(14)羟基可以与C(1)羟基一起被转化成1,2-碳酸酯,或者,可以将其转化成本文所述与C(2),C(4),C(9)和C(10)取代基有关的各种酯或其它官能团。
因此,本发明方法能够制备有下式结构的紫杉烷:

其中
M含有铵或是金属;
R1是氢,羟基,保护的羟基,或与R14或R2一起形成碳酸酯;
R2是酮基,-OT2,酰氧基,或与R1一起形成碳酸酯;
R4是-OT4或酰氧基;
R7是氢,卤素,-OT7,或酰氧基;
R9是氢,酮基,-OT9,或酰氧基;
R10是氢,酮基,-OT10,或酰氧基;
R7,R9和R10分别有α或β立体化学构型;
R13是羟基,保护的羟基,酮基,MO-或

R14是氢,-OT14,酰氧基,或与R1一起形成碳酸酯;
T2,T4,T7,T9,T10和T14分别是氢或羟基保护基。
X1是-OX6,-SX7或-NX8X9;
X2是氢,烃基,取代的烃基或杂芳基;
X3和X4分别是氢,烃基,取代的烃基或杂芳基;
X5是-X10,-OX10,-SX10,-NX8X10或-SO2X11;
X6是氢,烃基,取代的烃基,杂芳基,羟基保护基或增加紫杉烷衍生物的水溶性的官能团;
X7是烃基,取代的烃基,杂芳基或巯基保护基;
X8是氢,烃基或取代的烃基;
X9是氨基保护基;
X10是烃基,取代的烃基或杂芳基;
X11是烃基,取代的烃基,杂芳基,-OX10或-NX8X14;及
X14是氢,烃基,取代的烃基或杂芳基。
在本发明一个具体实例中,紫杉烷的取代基(非C(7),C(10)和C(13)取代基)对应于浆果赤霉素III或10-DAB上的取代基。即R14是氢,R9是酮基,R4是酰氧基,R2是苯甲酰氧基,R1是羟基。在本发明另一具体实例中,相对于C(13)侧链紫杉烷有不同于紫杉醇或Taxotere_的结构,并且至少有一个其它取代基。例如,R14可以是羟基;R2可以是羟基,-OCOZ2或-OCOOZ22,其中Z2是氢,烃基,取代的烃基或杂芳基,而Z22是烃基,取代的烃基或杂芳基;R4可以是羟基,-OCOZ4或-OCOOZ44,其中Z4是氢,烃基,取代的烃基或杂芳基,而Z44是烃基,取代的烃基或杂芳基;R7可以是氢,羟基,-OCOZ7或-OCOOZ77,其中Z7是氢,烃基,取代的烃基或杂芳基,而Z77是氢,烃基,取代的烃基或杂芳基;R9可以是氢,羟基,-OCOZ9或-OCOOZ99,其中Z9是氢,烃基,取代的烃基或杂芳基,而Z99是氢,烃基,取代的烃基或杂芳基;及R10可以是氢,羟基,-OCOZ10或-OCOOZ1010,其中Z10是氢,烃基,取代的烃基或杂芳基,而Z1010是氢,烃基,取代的烃基或杂芳基。
在优选的具体实例中,紫杉烷有下式结构:
其中,P10是酰基,该酰基至少含有3个碳原子或2个碳原子和1个氮原子、氧原子或硫原子。换句话说,-OP10不同于乙酰氧基。优选地,P10是-(C=O)RA,-(C=O)ORB,或-(C=O)NRC,其中,RA是取代的或未取代的烃基或杂芳基,所说的未取代的烃基含有至少2个碳原子;RB和RC分别是取代的或未取代的烃基。更优选地,RA是取代的或未取代的烷基或芳基,所说的未取代的烷基含有至少2个碳原子;RB和RC分别是取代的或未取代的烷基或芳基。
在本发明另一具体实例中,紫杉烷有下式结构:
其中,P7和P10分别是取代的或未取代的酰基。在此具体实例中,优选R7与R10不同。
定义
本文所用术语“选择性”和“选择性衍生反应”是指所要产物形成的量远远超过其它任何副产物。优选地,所要产物与其它任何副产物的摩尔比至少为9∶1,更优选地,所要产物与其它任何副产物的摩尔比至少为20∶1。
此外,“Ph”指苯基;“Bz”指苯甲酰基;“Bn”指苄基;“Me”指甲基;“Et”指乙基;“iPr”指异丙基;“tBu”指叔丁基;“Ac”指乙酰基;“TES”指三乙基甲硅烷基;“TMS”指三甲基甲硅烷基;“TBS”指Me2t-BuSi-;“CDI”指羰基二咪唑;“BOM”指苄氧基甲基;“DBU”指二氮杂双环十一烷;“DMAP”指对二甲氨基吡啶;“LHMDS”或“LiHMDS”指六甲基乙硅叠氮化锂;“DMF”指二甲基甲酰胺;“10-DAB”指10-脱乙酰基浆果赤霉素III;“Cbz”指苄氧羰基;“Alloc”指烯丙氧羰基;“THF”指四氢呋喃;“BOC”指苄氧羰基;“PNB”指对硝基苄基;“Troc”指2,2,2-三氯乙氧羰基;“EtOAc”指乙酸乙酯;“THF”指四氢呋喃;“保护的羟基”指-OP,其中P是羟基保护基;及“羟基保护基”包括(但不限于)有2-10个碳原子的缩醛,有2-10个碳原子的缩酮,以及醚类,如甲醚,叔丁醚,苄醚,对甲氧基苄醚,对硝基苄醚,烯丙醚,三苯甲基醚,甲氧基甲醚,甲氧基乙氧基甲醚,乙氧基乙醚,甲氧基丙醚,四氢吡喃基醚,四氢硫代吡喃基醚;及三烷基甲硅烷基醚如三甲基甲硅烷基醚,三乙基甲硅烷基醚,二甲基芳基甲硅烷基醚,三异丙基甲硅烷基醚和叔丁基二甲基甲硅烷基醚;酯类如苯甲酰酯,乙酰酯,苯基乙酰酯,甲酰酯,单-,二-和三卤代乙酰酯如氯乙酰酯,二氯乙酰酯,三氯乙酰酯,三氟乙酰酯;及碳酸酯,包括(但不限于)有1-6个碳原子的烷基碳酸酯,如碳酸甲酯,碳酸乙酯,碳酸正丙酯,碳酸异丙酯,碳酸正丁酯,碳酸叔丁酯,碳酸异丁酯和碳酸正戊酯;有1-6个碳原子并被一个或多个卤原子取代的烷基碳酸酯,如2,2,2-三氯乙氧基甲基碳酸酯和2,2,2-三氯乙基碳酸酯;有2-6个碳原子的链烯基碳酸酯,如乙烯碳酸酯和烯丙基碳酸酯;有3-6个碳原子的环烷基碳酸酯,如环丙基碳酸酯,环丁基碳酸酯,环戊基碳酸酯和环己基碳酸酯;任意在苯环上被一个或多个C1-6烷氧基或硝基取代的苯基或苄基碳酸酯。另一些羟基保护基可在T.W.Greene的《有机合成中的保护基》,John Wiley and Sons,1981,和第2版,1991,中查到。
本文所述“烃”和“烃基”部分是元素碳和氢构成的有机化合物或基团。这些部分包括烷基,链烯基,炔基和芳基部分。这些部分还包括被其它脂肪烃或环烃取代的烷基,链烯基,炔基和芳基部分,并包括烷芳基,链烯芳基和炔芳基。优选这些部分含有1-20个碳原子。
本文所述烷基优选主链上含有1-6个碳原子且至多20个碳原子的低级烷基。它们可以是直链,支链或环形烷基,包括甲基,乙基,丙基,异丙基,丁基,己基等。它们可以被脂肪烃或环烃基团取代。
本文所述链烯基优选主链上含有2-6个碳原子且至多20个碳原子的低级链烯基。它们可以是直链或支链链烯基,包括乙烯基,丙烯基,异丙烯基,丁烯基,异丁烯基,己烯基等。它们可以被脂肪烃或环烃基团取代。
本文所述炔基优选主链上含有2-6个碳原子且至多20个碳原子的低级炔基。它们可以是直链或支链炔基,包括乙炔基,丙炔基,丁炔基,异丁炔基,己炔基等。它们可以被脂肪烃或环烃基团取代。
本文所述芳基部分含有6-20个碳原子并包括苯基。它们可以是被本文定义的各种取代基取代的烃基。苯基是较优选的芳基。
本文所述杂芳基部分是类似于芳香化合物或基团并总共含有5-20个原子,通常有5或6个环原子,并至少有是1个非碳原子的杂环化合物或基团,例如,呋喃基,噻吩基,吡啶基等。杂芳基部分可以被烃基,杂原子取代的烃基或含有选自氮,氧,硅,磷,硼,硫和卤素的杂原子的取代基取代。这些取代基包括羟基;低级烷氧基如甲氧基,乙氧基,丁氧基;卤素如氯或氟;醚类;缩醛;缩酮;酯类;杂芳基如呋喃基或噻吩基;烷酰氧基(alkanoxy);酰基;酰氧基;硝基;氨基;及酰氨基。
本文所述取代的烃基部分是至少被1个非碳和非氢原子取代的烃基部分。在所说部分中碳链原子被氮,氧,硅,磷,硼,硫和卤素等杂原子取代。这些取代基包括羟基;低级烷氧基如甲氧基,乙氧基,丁氧基;卤素如氯或氟;醚类;缩醛;缩酮;酯类;杂芳基如呋喃基或噻吩基;烷酰氧基(alkanoxy);酰基;酰氧基;硝基;氨基;及酰氨基。
本文所述酰基部分和酰氧基部分包括烃基,取代的烃基或杂芳基部分。它们一般分别有结构式-C(O)G和-C(O)G,其中G是取代的或未取代的烃基,烃基氧基,烃基氨基,烃基硫基或杂芳基。
本文所述缩酮部分具有结构式
其中X31,X32,X33和X34分别是烃基,取代的烃基或杂芳基部分。它们可以任意被本文所定义的各种取代基取代。优选的缩酮部分是取代的或未取代的烷基或链烯基,更优选取代的或未取代的低级(C1-C6)烷基。另外,这些缩酮可以包括糖类或取代的糖类,并且包括从糖类或取代的糖类如葡萄糖和木糖制成的缩酮部分。当缩酮部分被当作C(7)羟基保护基而掺入本发明的紫杉烷时,X31或X32代表紫杉烷部分。
本文所述缩醛部分具有结构式
其中X31,X32和X33分别是烃基,取代的烃基或杂芳基部分。它们可以任意被本文定义的各种非羟基取代基取代。优选的缩醛部分是取代的或未取代的烷基或链烯基,更优选取代的或未取代的低级(C1-C6)烷基。另外,这些缩醛部分可以包括糖类或取代的糖类,并且包括从糖类或取代的糖类如葡萄糖和木糖制成的缩醛部分。当缩醛部分被当作C(7)羟基保护基而掺入本发明的紫杉烷时,X31或X32代表紫杉烷部分。
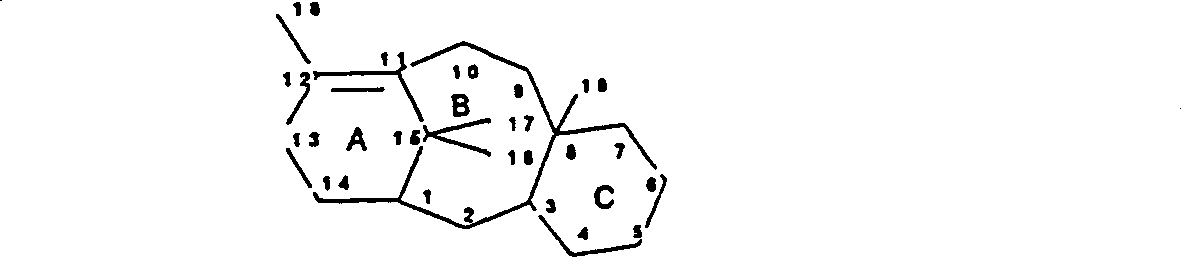
本文所用术语“紫杉烷”指含有A,B和C环(环位编号如图所示)的化合物:
下列实施例用于说明本发明。
实施例1
A.C(10)羟基的选择性酰化反应
10-Cbz-10-DAB
室温和N2下向10-DAB(30mg,0.055mmol)的THF(1mL)溶液中加入焦碳酸二苄酯(dibenzyl pyrocarbonate)(320mg,1.1mmol,20当量)。将反应混合物在室温下搅拌24h。加入EtOAc(10mL),然后用短硅胶柱快速过滤溶液。用EtOAc(100mL)洗涤硅胶柱,减压浓缩溶液。剩余物经快速柱色谱纯化,用EtOAc∶己烷(1∶1)洗脱,并真空干燥过夜,得到37mg(产率98%)10-Cbz-10-DAB为无色固体。
mp 205-206℃;[α]Hg-63°(CHCl3,c=0.41);1H NMR(400MHz,CDCl3)δ1.11(s,3H,Me17),1.13(s,3H,Me16),1.58(s,1H,1-OH),1.71(s,3H,Me19),1.89(ddd,J=14.7,10.9,2.3Hz,1H,H6b),2.00(d,J=5.1Hz,1H,13-OH),2.08(d,J=1.0,3H,Me18),2.28(s,3H,4-Ac),2.30(m,2H,H14a,H14b),2.43(d,J=4.1Hz,1H,7-OH),2.58(ddd,J=14.7,9.6,6.6Hz,1H,H6a),3.88(d,J=6.9Hz,1H,H3),4.19(d,J=8.6Hz,1H,H20b),4.31(d,J=8.6Hz,1H,H20a),4.44(ddd,J=10.9,6.6,4.1Hz,1H,H7),4.89(m,1H,H13),4.98(dd,J=9.6,2.3Hz,1H,H5),5.23(d,J=12.1,1H,CHH′OC(O)),5.26(d,J=12.1,1H,CHH′OC(O)),5.65(d,J=6.9Hz,1H,H2),6.19(s,1H,H10),7.35-7.44(m,5H,PhCH2O),7.48(dd,J=8.1,7.6Hz,2H,苯甲酸酯,m),7.60(tt,J=7.6,1.0Hz,1H,苯甲酸酯,p),8.11(d,J=8.1,1.0Hz,2H,苯甲酸酯,o)ppm.13CNMR(75MHz,CDCl3)δ9.1(Me(19)),15.3(Me(18)),20.7(4-Ac),22.3,26.7(Me16,Me17),35.5(C(6)),38.6(C(14)),42.5(C(15)),46.1(C(3)),58.7(C(8)),67.9(C(13)),70.5(OCH2Ph),72.2,75.0,76.5(C(7),C(2),C(20)),79.0,79.1(C(1),C(10)),80.9(C(4)),84.5(C(5)),128.6,128.8,129.7,130.3,131.9,133.8(OCH2Ph,苯甲酸酯),135.1(C(11)),147.5(C(12)),155.6(OC(O)O),167.4(苯甲酸酯),171.0(4-Ac),204.7(C(9))ppm.
C37H42O12·1/2H2O元素分析:
计算值:C,64.62;H,6.30;
实测值:C,64.34;H,6.31。
10-Alloc-10-DAB
室温和N2下向10-DAB(30mg,0.055mmol)的THF(1mL)溶液中加入焦碳酸二烯丙酯(366mL,2.2mmol,40当量)。将反应混合物在室温下搅拌48h。TLC分析结果表明有未反应的原料伴随所要的产物存在。加入EtOAc(20mL),然后用短硅胶柱快速过滤溶液。用EtOAc(100mL)洗涤硅胶柱,并浓缩溶液。剩余物经快速柱色谱纯化,用EtOAc∶己烷(1∶1)洗脱,并真空干燥过夜,得到23mg 10-Alloc-10-DAB为无色固体,产率为67%(由转换70%产率为95%换算而来)。回收的10-DAB 9mg(30%)。
10-alloc-10-DAB:mp 201-203℃;[α]Hg-81°(CHCl3,c=0.53);1H NMR(400MHz,CDCl3)δ1.11(s,3H,Me17),1.12(s,3H,Me16),1.60(s,1H,1-OH),1.69(s,3H,Me19),1.87(ddd,J=14.7,11.0,2.1Hz,1H,H6b),2.05(d,J=5.1Hz,1H,13-OH),2.08(d,J=1.2,3H,Me18),2.28(s,3H,4-Ac),2.29(m,2H,H14a,H14b),2.47(d,J=4.2Hz,1H,7-OH),2.57(ddd,J=14.7,9.6,6.7Hz,1H,H6a),3.86(d,J=7.0Hz,1H,H3),4.16(d,J=8.4Hz,1H,H20b),4.31(d,J=8.4Hz,1H,H20a),4.44(ddd,J=11.0,6.7,4.2Hz,1H,H7),4.70(br d,J=5.9Hz,2H,CHH′=CHCH2O),4.90(m,1H,H13),4.97(dd,J=9.6,2.1Hz,1H,H5),5.32(dd,J=10.4,1.2Hz,1H,CHH′=CHCH2O),5.42(dd,J=17.2,1.2Hz,1H,CHH′=CHCH2O),5.63(d,J=7.0Hz,1H,H2),5.98(ddt,J=17.2,10.4,5.9Hz,1H,CHH′=CHCH2O),6.16(s,1H,H10),7.48(dd,J=8.1,7.5Hz,2H,苯甲酸酯,m),7.60(tt,J=7.5,1.2Hz,1H,苯甲酸酯,p),8.11(d,J=8.1,1.2Hz,2H,苯甲酸酯,o)ppm;13C NMR(75MHz,CDCl3)δ9.1(Me(19)),15.3(Me(18)),20.7(4-Ac),22.3,26.7(Me16,Me17),35.5(C(6)),38.6(C(14)),42.5(C(15)),46.1(C(3)),58.7(C(8)),67.9(C(13)),69.3(CH2=CHCH2O),72.1,75.0,76.5(C(7),C(2),C(20)),79.0,79.1(C(1),C(10)),80.9(C(4)),84.5(C(5)),119.6(CH2=CHCH2O),128.8,129.7,130.3,133.8(苯甲酸酯),131.4,131.9(CH2=CHCH2O,C(11)),147.5(C(12)),155.4(OC(O)O),167.4(苯甲酸酯),170.9(4-Ac),204.7(C(9))ppm.
C33H40O12元素分析:
计算值:C,63.05;H,6.41;
实测值:C,62.77;H,6.48。
B.用ZnCl2进行C(10)羟基的选择性酰化反应
浆果赤霉素III
室温和N2下向10-DAB(100mg,0.184mmol)的THF(6mL)溶液中加入乙酸酐(6.5mL)和ZnCl2/THF溶液(0.5M,726mL,0.368mmol,2当量)的混合物。将反应混合物在室温下搅拌4h。反应混合物用EtOAc(100mL)稀释,并先后用饱和NaHCO3水溶液(40mL×3)和盐水洗涤。有机相用Na2SO4干燥,减压浓缩。剩余物经快速柱色谱纯化,用EtOAc∶己烷(1∶1)洗脱,并真空干燥,得到100mg(产率93%)浆果赤霉素III为无色固体。mp 237-238℃分解(参考值236-238℃分解)。[α]Hg-63°(CH3OH,c=0.45)(ref[α]D-54°,CH3OH);1H NMR(400MHz,CDCl3)δ1.11(s,6H,Me16,Me17),1.61(s,1H,1-OH),1.67(s,3H,Me19),1.87(ddd,J=14.7,10.9,2.1Hz,1H,H6b),2.05(d,J=3.8Hz,1H,13-OH),2.05(s,3H,Me18),2.24(s,3H,10-Ac),2.28(s,3H,4-Ac),2.30(m,2H,H14a,H14b),2.47(d,J=4.2Hz,1H,7-OH),2.57(ddd,J=14.7,9.4,6.7Hz,1H,H6a),3.89(d,J=7.0Hz,1H,H3),4.16(d,J=8.4Hz,1H,H20b),4.31(d,J=8.4Hz,1H,H20a),4.47(ddd,J=10.9,6.7,4.2Hz,1H,H7),4.90(m,1H,H13),4.99(dd,J=9.4,2.1Hz,1H,H5),5.63(d,J=7.0Hz,1H,H2),6.33(s,1H,H10),7.48(dd,J=7.8,7.8Hz,2H,苯甲酸酯,m),7.61(dd,J=7.8,7.4Hz,1H,苯甲酸酯,p),8.11(d,J=7.4Hz,2H,苯甲酸酯,o)ppm.13C NMR(100MHz,CDCl3)δ9.4(Me(19)),15.6(Me(18)),20.9(4-Ac,10-Ac),22.6,27.0(Me16,Me17),35.6(C(6)),38.6(C(14)),42.7(C(15)),46.1(C(3)),58.8(C(8)),68.0(C(13)),72.3,75.0,76.2,76.4(C(7),C(2),C(10),C(20)),79.1(C(1)),80.9(C(4)),84.5(C(5)),128.6,129.4,130.1,133.7(苯甲酸酯),132.0(C(11)),146.3(C(12)),167.1(苯甲酸酯),170.7,171.3(10-Ac,4-Ac),204.1(C(9))ppm.
10-氯乙酰基-10-DAB
室温和N2下用注射器向10-DAB(116mg,0.21mmol)的THF(3mL)溶液中加入氯乙酸酐(2.8g,16.3mmol,78当量)和ZnCl2/THF溶液(0.5M,0.85L,0.42mmol,2当量)的混合物。将反应混合物在室温下搅拌5h。将反应混合物倒入EtOAc(200mL)和饱和NaHCO3水溶液(100mL)的混合物。分离有机相,水相用EtOAc(100mL×3)萃取。合并有机相,用Na2SO4干燥,过滤并减压浓缩。剩余物经快速柱色谱纯化,用EtOAc∶己烷(1∶1)洗脱,并真空干燥过夜,得到123mg(产率93%)10-氯乙酰基-10-DAB为无色固体。mp 231-233℃分解。[α]Hg-66°(EtOAc,c=0.45);1H NMR(400MHz,CDCl3)δ1.11(s,3H,Me17),1.12(s,3H,Me16),1.63(s,1H,1-OH),1.69(s,3H,Me19),1.89(ddd,J=14.6,10.9,2.1Hz,1H,H6b),2.07(d,J=5.2Hz,1H,13-OH),2.09(d,J=1.2,3H,Me18),2.12(d,J=4.5Hz,1H,7-OH),2.29(s,3H,4-Ac),2.30(m,2H,H14a,H14b),2.58(ddd,J=14.6,9.7,6.7Hz,1H,H6a),3.88(d,J=7.0Hz,1H,H3),4.16(d,J=8.3Hz,1H,H20b),4.27(br s,2H,ClCH2),4.31(d,J=8.3Hz,1H,H20a),4.44(ddd,J=10.9,6.7,4.5Hz,1H,H7),4.90(m,1H,H13),4.98(dd,J=9.7,2.1Hz,1H,H5),5.64(d,J=7.0Hz,1H,H2),6.41(s,1H,H10),7.49(dd,J=7.9,7.4Hz,2H,苯甲酸酯,m),7.61(tt,J=7.4,1.3Hz,1H,苯甲酸酯,p),8.11(d,J=7.9,1.3Hz,2H,苯甲酸酯,o)ppm.13C NMR(75MHz,CDCl3)δ9.3(Me(19)),15.3(Me(18)),20.6(4-Ac),22.3,26.7(Me16,Me17),35.8(C(6)),38.6(C(14)),40.5(ClCH2),42.6(C(15)),46.2(C(3)),58.8(C(8)),68.0(C(13)),72.0,75.0,75.9(C(7),C(2),C(110),C(20)),79.0(C(1)),80.9(C(4)),84.4(C(5)),128.8,129.7,130.3,133.9(苯甲酸酯),131.8(C(11)),147.1(C(12)),167.4,167.7(ClCH2C(O)O,苯甲酸酯),171.0(4-Ac),203.7(C(9))ppm.
C31H37ClO11·H2O元素分析:
计算值:C,58.26;H,6.15;
实测值:C,58.26;H,6.07。
10-丙酰基-10-DAB
室温和N2下向10-DAB(47mg,0.086mmol)的THF(2mL)溶液中加入丙酸酐(4mL)和ZnCl2/THF溶液(0.5M,350mL,0.173mmol,2当量)的混合物。将反应混合物在室温下搅拌14h。反应混合物用EtOAc(150mL)稀释,并先后用饱和NaHCO3水溶液(50mL×3)和盐水彻底洗涤。有机相用Na2SO4干燥,减压浓缩。剩余物经快速柱色谱纯化,用EtOAc∶己烷(1∶1)洗脱,并真空干燥,得到48mg(产率93%)10-丙酰基-10-DAB为白色固体。mp 212-213℃分解。[α]Hg-96°(CHCl3,c=0.78);1H NMR(400MHz,CDCl3)δ1.11(s,6H,Me16,Me17),1.24(t,J=7.6Hz,3H,CH3CH2),1.60(s,1H,1-OH),1.67(s,3H,Me19),1.87(ddd,J=14.7,10.9,2.2Hz,1H,H6b),2.05(d,J=5.1Hz,1H,13-OH),2.06(d,J=1.3Hz,3H,Me18),2.28(s,3H,4-Ac),2.30(d,J=7.5Hz,2H,H14a,H14b),2.51(d,J=4.1Hz,1H,7-OH),2.55(q,J=7.6Hz,2H,CH3CH2),2.57(ddd,J=14.7,9.5,6.7Hz,1H,H6a),3.90(d,J=6.9Hz,1H,H3),4.16(dd,J=8.4,0.8Hz,1H,H20b),4.31(d,J=8.4Hz,1H,H20a),4.48(ddd,J=10.9,6.7,4.1Hz,1H,H7),4.90(m,1H,H13),4.99(dd,J=9.5,2.2Hz,1H,H5),5.63(d,J=6.9Hz,1H,H2),6.34(s,1H,H10),7.48(dd,J=8.1,7.4Hz,2H,苯甲酸酯,m),7.61(tt,J=7.4,1.3Hz,1H,苯甲酸酯,p),8.11(dd,J=8.3,1.3Hz,2H,苯甲酸酯,o)ppm.13C NMR(75MHz,CDCl3)δ8.8(CH3CH2),9.2(Me(19)),15.2(Me(18)),20.7(4-Ac),22.3,26.8,27.4(Me16,Me17,CH3CH2),35.5(C(6)),38.7(C(14)),42.6(C(15)),46.1(C(3)),58.7(C(8)),67.9(C(13)),72.3,75.1,76.1,76.5(C(7),C(2),C(10),C(20)),79.1(C(1)),80.9(C(4)),84.5(C(5)),128.7,129.7,130.3,133.8(苯甲酸酯),132.3(C(11)),146.5(C(12)),167.4(苯甲酸酯),170.9,174.9(4-Ac,10-C(O)O),204.6(C(9))ppm.
C32H40O11元素分析:
计算值:C,63.99;H,6.71;
实测值:C,63.81;H,6.80。
C.用CeCl3进行C(10)羟基的选择性酰化反应
总过程
N2下向10-DAB的THF(每毫摩尔10-DAB 20mL)溶液中加入CeCl3和适当的酸酐或焦碳酸酯(用量如表1所示)。将反应混合物在25℃搅拌并用TLC分析监测。当分析结果显示反应已完成(反应时间见表1)时,用EtOAc稀释反应混合物,并用饱和NaHCO3水溶液洗涤3次。合并的碳酸氢盐洗液用EtOAc萃取3次。合并有机相并用Na2SO4干燥,蒸发溶剂。粗产物经快速柱色谱纯化。如果需要,可以通过从EtOAc/己烷重结晶进一步纯化。
表1 CeCl3催化的10-DAB酰化反应

10-丁酰基-10-DAB mp 145-149℃;[α]Hg-86.6°(CHCl3,c=1);1H NMR(500MHz,CDCl3)δ8.13-8.11(2H,m),7.62(1H,m),7.51-7.48(2H,m),6.35(1H,s),5.64(1H,d,J 7.0Hz),4.99(1H,d,J 7.7Hz),4.90(1H,m),4.48(1H,m),4.31(1H,d,J 8.3Hz),4.18(1H,d,J8.3Hz),3.91(1H,d,J 7.0Hz),2.60-2.42(4H,m),2.36-2.26(2H,m),2.28(3H,s),2.06(3H,d,J 1.0Hz),1.88(1H,ddd,J 1.9,10.9,13.0Hz),1.76(2H,hex,J7.4Hz),1.68(3H,s),1.12(6H,s)和1.04(3H,t,J7.4Hz);13C NMR(100MHz,CDCl3)δ204.2,173.9,170.6,167.1,146.2,133.7,132.0,130.1,129.4,128.6,84.5,88.9,79.1,76.5,76.0,75.0,72.3,68.0,58.8,46.2,42.7,38.7,37.1,36.2,35.6,30.6,27.0,22.6,20.9,18.4,17.8,15.5和9.4;
C33H42O11元素分析:
计算值:C,64.48;H,6.89;
实测值:C,63.67;H,7.01。
10-异丁酰基-10-DAB mp 143℃;[α]Hg-62.6°(CHCl3,c=0.075);1H NMR(CDCl3,500MHz):δ8.12(2H,d,J7.3Hz),7.62(1H,m),7.51-7.48(2H,m),6.33(1H,s),5.65(1H,d,J 7.3Hz),5.00(1H,d,J 7.9Hz),4.91(1H,m),4.48(1H,ddd,J 4.3,6.7,11.0Hz),4.31(1H,d,J8.6Hz),4.18(1H,d,J 8.6Hz),3.91(1H,d,J 7.3Hz),2.74(1H,pent,J 6.7Hz),2.57(1H,m),2.51(1H,d,J4.3Hz),2.31(1H,m),2.28(3H,s),2.06(3H,s),2.01(1H,d,J 5.5Hz),1.90(1H,ddd,J 2.3,11.0,14.6Hz),1.68(3H,s),1.60(1H,s),1.51(3H,s),1.33(3H,d,J6.7Hz),1.26(3H,d,J 6.7Hz),1.13(3H,s)和1.12(3H,s);13C NMR(100MHz,CDCl3)δ204.1,177.2,170.6,167.1,146.2,133.7,132.1,130.1,129.4,128.6,95.5,84.5,80.9,79.1,76.5,75.8,74.9,72.3,68.0,58.8,46.2,42.7,38.7,35.6,34.1,27.0,22.6,20.9,19.2,18.7,15.5和9.4;
C33H42O11·0.5H2O元素分析:
计算值:C,64.48;H,6.89;
实测值:C,63.05;H,6.70。
10-苯甲酰基-10-DAB 1H NMR(CDCl3,500MHz):δ8.15-8.11(4H,m),7.64-7.6(2H,m),7.52-7.48(4H,m),6.62(1H,s),5.7(1H,d,J 7.1Hz),5.02(1H,d,J7.7Hz),4.94(1H,m),4.57(1H,ddd,J 4.4,7.1,11.0Hz),4.33(1H,d,J 8.2Hz),4.20(1H,d,J 8.3Hz),3.99(1H,d,J 6.6Hz),2.62(1H,ddd,J 6.6,9.3,14.8),2.55(1H,d,J 4.4Hz),2.35(2H,m),2.30(3H,s),2.13(3H,d,J1.1Hz),2.03(1H,d,J 4.9Hz),1.91(1H,ddd,J 2.2,11.0,13.2Hz),1.71(3H,s),1.65(1H,s),1.25(3H,s)和1.21(3H,s);13C NMR(100MHz,CDCl3)δ204.0,170.7,167.1,166.5,146.5,133.7,133.6,132.0,130.1,129.9,129.4,129.3,128.7,128.5,84.5,80.9,79.1,76.5,75.0,72.4,68.1,58.8,46.3,42.8,38.7,35.8,29.7,27.2,22.6,21.2,15.6和9.5;
C35H40O11元素分析:
计算值:C,66.66;H,6.22;
实测值:C,66.46;H,6.19。
10-反巴豆酰基-10-DAB 1H NMR(CDCl3,500MHz):δ8.13(2H,d,J 7.1Hz),7.62(1H,m),7.51-7.48(2H,m),7.11(1H,m),6.42(1H,s),6.02(1H,dq,J 1.7,15.4Hz),5.66(1H,d,J 7.1Hz),4.99(1H,dd,J 2.0,9.6Hz),4.91(1H,t,J 7.6Hz),4.50(1H,dd,J 7.1,10.8Hz),4.31(1H,d,J 8.3Hz),4.19(1H,d,J 8.3Hz),3.93(1H,d,J7.1Hz),2.61-2.55(2H,m),2.33-2.31(2H,m),2.28(3H,s),2.07(3H,d,J 1.5Hz),1.95(3H,dd,J 1.6,6.8Hz),1.89(1H,ddd,J 2.3,11.0,13.4Hz),1.68(3H,s),1.15(3H,s)和1.14(3H,s);13C NMR(75MHz,CDCl3)δ212.4,181.0,170.8,167.3,166.5,146.4,133.8,132.3,130.2,129.5,128.7,121.9,116.0,84.7,84.6,80.9,79.2,77.2,75.9,75.1,72.4,68.1,58.8,46.1,42.7,38.6,35.6,27.0,20.9,18.0,15.4和9.3;
C33H40O11元素分析:
计算值:C,64.69;H,6.58;
实测值:C,63.93;H,6.61。
10-环丙酰基-10-DAB 1H(CDCl3,500MHz):δ8.12(2H,d,J 7.3Hz),7.62(1H,t,J 7.5Hz),7.49(2H,t,J 7.7Hz),6.35(1H,s),5.65(1H,d,J 7.0Hz),4.99(1H,app-d,J 8.2Hz),4.91(1H,m),4.46(1H,ddd,J 4.1,6.8,10.8Hz),4.31(1H,d,J 8.1Hz),4.18(1H,d,J8.1Hz),3.90(1H,d,J 7.0Hz),2.56(1H,m),2.51(1H,d,J 4.1Hz),2.31(2H,m),2.07(3H,d,J 1.0Hz),2.00(1H,d,J 4.9Hz),1.87(1H,ddd,J 2.1,10.8,14.6Hz),1.79(1H,ddd,J 3.4,7.9,12.4Hz),1.68(3H,s),1.60(1H,s),1.16-1.14(2H,m),1.13(6H,s)和1.01-0.97(2H,m);13C NMR(100MHz,CDCl3)δ204.3,175.2,170.6,167.1,146.4,133.7,132.0,130.1,129.4,128.6,84.5,80.9,79.1,76.5,76.0,74.9,72.4,68.0,58.8,46.2,42.7,38.6,35.6,34.0,27.0,25.6,24.9,22.6,21.0,15.6,13.1,9.4和9.1;
C33H40O11元素分析:
计算值:C,64.69;H,6.58;
实测值:C,64.47;H,6.66。
10-乙氧羰基 -10-DAB. mp 214-215℃;[α]Hg-81°(CHCl3,c=0.35);1H NMR(500MHz,CDCl3)δ1.13(s,3H,Me17),1.14(s,3H,Me16),1.38(t,J=7.1Hz,3H,CH3CH2),1.59(s,1H,1-OH),1.70(s,3H,Me19),1.88(ddd,J=14.6,10.5,2.1Hz,1H,H6b),2.00(d,J=5.0Hz,1H,13-OH),2.10(d,J=1.4Hz,3H,Me18),2.28(s,3H,4-Ac),2.30(m,2H,H14a,H14b),2.46(d,J=4.2Hz,1H,7-OH),2.57(ddd,J=14.6,9.6,6.7Hz,1H,H6a),3.88(d,J=6.9Hz,1H,H3),4.18(d,J=8.2Hz,1H,H20b),4.31(d,J=8.2Hz,1H,H20a),4.23-4.33(m,2H,CH3CH2),4.44(ddd,J=10.5,6.7,4.2Hz,1H,H7),4.90(m,1H,H13),4.98(dd,J=9.6,2.1Hz,1H,H5),5.65(d,J=6.9Hz,1H,H2),6.17(s,1H,H10),7.48(dd,J=8.2,7.3Hz,2H,苯甲酸酯,m),7.60(tt,J=7.3,1.4Hz,1H,苯甲酸酯,p),8.11(d,J=8.2,1.4Hz,2H,苯甲酸酯,o)ppm;13C NMR(75MHz,CDCl3)δ9.2,14.0,15.5,20.8,22.4,26.7,35.4,38.5,42.4,46.0,58.6,65.0,67.7,72.2,74.9,76.4,78.7,79.0,80.6,84.4,128.7,129.4,130.1,131.5,133.7,147.5,155.4,167.1,170.8,204.7ppm.
10-甲氧羰基 -10-DAB. mp 218-219℃;[α]Hg-83°(CHCl3,c=0.58);1H NMR(500MHz,CDCl3)δ1.12(s,3H,Me17),1.13(s,3H,Me16),1.59(s,1H,1-OH),1.70(s,3H,Me19),1.88(ddd,J=14.7,10.8,1.8Hz,1H,H6b),2.00(d,J=5.0Hz,1H,13-OH),2.10(d,J=1.4Hz,3H,Me18),2.28(s,3H,4-Ac),2.30(m,2H,H14a,H14b),2.40(d,J=4.1Hz,1H,7-OH),2.57(ddd,J=14.7,9.7,6.6Hz,1H,H6a),3.87(d,J=6.9Hz,1H,H3),3.88(s,3H,MeOC(O)),4.18(d,J=8.4Hz,1H,H20b),4.31(d,J=8.4Hz,1H,H20a),4.44(ddd,J=10.8,6.6,4.1Hz,1H,H7),4.90(m,1H,H13),4.98(dd,J=9.7,1.8Hz,1H,H5),5.65(d,J=6.9Hz,1H,H2),6.17(s,1H,H10),7.48(t,J=8.2,7.3Hz,2H,苯甲酸酯,m),7.61(tt,J=7.3,1.4Hz,1H,苯甲酸酯,p),8.11(d,J=8.2,1.4Hz,2H,苯甲酸酯,o)ppm;13C NMR(75MHz,CDCl3)δ9.2,15.5,20.7,22.4,26.7,35.5,38.5,42.4,46.0,55.4,58.6,65.0,67.7,72.1,74.8,76.4,78.9,79.0,80.6,84.4,128.7,129.4,130.1,131.4,133.7,147.5,155.9,167.1,170.8,204.6ppm.
10-tBoc-10-DAB.mp 193-194℃;[α]Hg-82°(CHCl3,c=0.33);1H NMR(500MHz,CDCl3)δ1.13(s,6H,Me17,Me16),1.48(s,9H,tBuO),1.58(s,1H,1-OH),1.69(s,3H,Me19),1.88(ddd,J=14.9,11.0,2.2Hz,1H,H6b),1.99(d,J=5.0Hz,1H,13-OH),2.08(d,J=1.4Hz,3H,Me18),2.28(s,3H,4-Ac),2.30(m,2H,H14a,H14b),2.56(ddd,J=14.9,9.6,6.9Hz,1H,H6a),2.68(d,J=3.6Hz,1H,7-OH),3.88(d,J=6.9Hz,1H,H3),4.19(d,J=8.2Hz,1H,H20b),4.31(d,J=8.2Hz,1H,H20a),4.46(ddd,J=11.0,6.9,3.6Hz,1H,H7),4.90(m,1H,H13),4.99(dd,J=9.6,2.2Hz,1H,H5),5.64(d,J=6.9Hz,1H,H2),6.11(s,1H,H10),7.48(t,J=7.8Hz,2H,苯甲酸酯,m),7.60(tt,J=7.8,1.3Hz,1H,苯甲酸酯,p),8.11(dd,J=7.8,1.3Hz,2H,苯甲酸酯,o)ppm;13CNMR(75MHz,CDCl3)δ9.2,15.6,20.9,22.4,26.8,27.5,35.3,38.5,42.5,45.9,58.7,67.9,72.3,74.7,76.4,78.0,79.2,80.8,83.8,84.5,128.7,129.4,130.1,131.8,133.7,147.3,154.0,167.2,170.8,205.0ppm.
D.C(10)羟基的选择性氨基甲酰基化反应
10-DAB的C(10)羟基的选择性氨基甲酰基化反应的总过程
在0℃和N2下将0.061mmol(1.1摩尔当量)异氰酸酯的2mL THF溶液加到10-DAB(30mg,0.055mmol)和CuCl(5.5mg,0.055mmol)的混合物中。将反应混合物按表2所示时间搅拌。之后,将反应升至25℃,并按表2所示时间继续搅拌。加入饱和氯化铵水溶液淬灭反应,反应混合物用EtOAc萃取3次。合并有机相并用饱和碳酸氢钠水溶液洗涤,Na2SO4干燥,蒸发溶剂,得到白色固体。粗产物用快速柱色谱纯化,EtOAc/己烷(2∶1)洗脱剂洗脱。
表2 10-DAB的氨基甲酰基化反应
10-乙基氨基甲酰基-10-DAB mp 241-243℃;[α]Hg-92.0°(CHCl3,c=0.5);1H NMR(400MHz,CDCl3)δ8.13(2H,d,J7.1Hz),7.63(1H,m),7.52-7.48(2H,m),6.27(1H,s),5.63(1H,d,J 6.9Hz),5.01(1H,dd,J 1.9,9.6Hz),4.97(1H,m),4.91(1H,m),4.50(1H,ddd,J 3.7,6.5,10.5Hz),4.31(1H,d,J 8.3Hz),4.17(1H,d,J 8.3Hz),3.88(1H,d,J 7.0Hz),3.32-3.25(2H,m),3.10(1H,d,J3.7Hz),2.56(1H,ddd,J 6.8,9.8,14.8Hz),2.31(1H,m),2.29(3H,s),2.09(3H,s),1.88(1H,ddd,J 2.2,11.0,13.3Hz),1.67(3H,s),1.60(1H,s),1.19(3H,t,J7.2Hz)和1.10(6H,s);
C32H40NO11元素分析:
计算值:C,62.43;H,6.71;
实测值:C,61.90;H,6.77。
10-丁基氨基甲酰基-10-DAB [α]Hg-89.6°(CHCl3,c=0.25);1H NMR(500MHz,CDCl3)δ8.12(2H,d,J 7.3Hz),7.61(1H,m),7.51-7.45(2H,m),6.27(1H,s),5.64(1H,d,J 6.7Hz),5.00(1H,d,J 8.0Hz),4.91(1H,m),4.49(1H,m),4.31(1H,d,J 8.5Hz),4.19(1H,d,J 8.5Hz),3.89(1H,d,J 6.7Hz),3.25-3.23(2H,m),3.04(1H,m),2.56(1H,ddd,J 6.7,9.7,14.7Hz),2.30(1H,d,J7.9Hz),2.28(3H. s),2.09(3H,s),1.99(1H,d,J4.9Hz),1.88(1H,ddd,J 2.5,11.0,13.4Hz),1.68(3H,s),1.59(1H,s),1.55(2H,b),1.42-1.37(2H,m),1.11(6H,s)和0.95(3H,t,J 7.6Hz);
C34H44NO11元素分析:
计算值:C,63.44;H,7.05;
实测值:C,62.64;H,7.01。
10-苯基氨基甲酰基-10-DAB mp 178-180℃;[α]Hg-93.0°(CHCl3,c=0.5);1H NMR(400Hz,CDCl3)δ8.13(2H,d,J 6.9Hz),7.63(1H,t,J 7.4Hz),7.51(2H,t,J 7.6Hz),7.42(1H,d,J 7.8Hz),7.36-7.32(2H,m),7.12(1H,t,J7.4Hz),6.87(1H,b),6.38(1H,s),5.66(1H,d,J7.0Hz),5.02(1H,app d,J 7.8Hz),5.93(1H,m),4.52(1H,ddd,J 3.8,6.5,10.5Hz),4.33(1H,d,J 8.3Hz),4.18(1H,d,J 8.3Hz),3.91(1H,d,J 7.0Hz),2.83(1H,d,J 4.0Hz),2.59(1H,ddd,J 6.5,9.4,14.5Hz),2.33(1H,m),2.29(3H,s),2.12(3H,d,J 1.4Hz),2.04(1H,d,J 5.1Hz),1.89(1H,ddd,J 2.2,11.0,14.4Hz),1.69(3H,s),1.62(1H,s),1.15(3H,s)和1.13(3H,s).
10-烯丙基氨基甲酰基-10-DAB mp 165-170℃;[α]Hg-80.0°(CHCl3,c=0.25);1H NMR(500MHz,CDCl3)δ8.12(2H,d,J 7.3Hz),7.62(1H,m),7.51-7.48(2H,m),6.27(1H,s),5.89(1H,m),5.62(1H,d,J 6.7Hz),5.31(1H,s),5.19(1H,d,J 9.8Hz),5.08(1H,m),5.00(1H,d,J7.9Hz),4.90(1H,m),4.49(1H,ddd,J,3.7,6.1,10.4Hz),4.31(1H,d,J 8.5Hz),4.17(1H,d,J 8.5Hz),3.88-3.86(2H,m),3.03(1H,d,J 3.7Hz),2.55(1H,ddd,J 6.7,9.8,15.9Hz),2.30(1H,m),2.29(3H,s),2.08(3H,s),2.06(1H,app d,J 4.9Hz),1.87(1H,ddd,J1.8,11.0,14.0Hz),1.67(3H,s),1.58(1H,s)和1.09(6H,s);
C33H40NO11元素分析:
计算值:C,63.15;H,6.58;
实测值:C,61.73;H,6.45。
E.C(10)羟基的选择性甲硅烷基化反应
10-TMS-10-DAB
在0℃和N2下向10-DAB(100mg,0.18mmol)的THF(10mL)溶液中慢慢加入N,O-双(三甲基甲硅烷基)三氟乙酰胺(1.0mL,3.7mmol,20当量)。将反应混合物在0℃搅拌5h。加入EtOAc(20mL),然后用短硅胶柱过滤溶液。用EtOAc(100mL)洗涤硅胶柱,减压浓缩溶液。剩余物经快速柱色谱纯化,用EtOAc/己烷(1∶1)洗脱并真空干燥过夜,得到103mg(产率91%)10-TMS-10-DAB为白色固体。
mp 189-191℃;[α]Hg-70°(CHCl3,c=0.55);1H NMR(400MHz,CDCl3)δ0.18(s,9H,Me3Si),1.06(s,3H,Me17),1.16(s,3H,Me16),1.31(d,J=8.6Hz,1H,7-OH),1.56(s,1H,1-OH),1.68(s,3H,Me19),1.79(ddd,J=14.4,11.1,2.1Hz,1H,H6b),1.97(d,J=4.9Hz,1H,13-OH),2.03(d,J=1.3Hz,3H,Me18),2.27(m,2H,H14a,H14b),2.28(s,3H,4-Ac),2.58(ddd,J=14.4,9.6,7.5Hz,1H,H6a),4.01(d,J=7.2Hz,1H,H3),4.16(d,J=8.2Hz,1H,H20b),4.25(ddd,J=11.1,8.6,7.5Hz,1H,H7),4.30(d,J=8.2Hz,1H,H20a),4.84(m,1H,H13),4.97(dd,J=9.6,2.1Hz,1H,H5),5.27(s,1H,H10),5.64(d,J=7.2Hz,1H,H2),7.47(dd,J=8.2,7.5Hz,2H,苯甲酸酯,m),7.60(tt,J=7.5,1.2Hz,1H,苯甲酸酯,p),8.11(dd,J=8.2,1.2Hz,2H,苯甲酸酯,o)ppm.13C NMR(75MHz,CDCl3)δ0.2(Me3S),9.7(Me(19)),14.4(Me(18)),19.6(4-Ac),22.4,26.6(Me16,Me17),37.1(C(6)),38.6(C(14)),42.6(C(15)),47.2(C(3)),57.8(C(8)),68.0(C(13)),72.0,75.1,76.1,76.8(C(7),C(2),C(10),C(20)),78.9(C(1)),81.2(C(4)),84.3(C(5)),128.8,130.3,133.7(苯甲酸酯),137.0(C(11)),139.0(C(12)),167.4(苯甲酸酯),171.0(4-Ac),209.5(C(9))ppm.
C32H44O10Si·1/2H2O元素分析:
计算值:C,61.42;H,7.25;
实测值:C,61.61;H,7.12。
10-TES-10-DAB
在0℃和N2下向10-DAB(85mg,0.16mmol)的THF(3mL)溶液中分别慢慢加入N,O-双(三乙基甲硅烷基)三氟乙酰胺(484mL,1.56mmol,10当量)和催化量的LiHMDS/THF溶液(1M,5mL,0.005mmol)。将反应混合物在0℃搅拌5分钟。加入EtOAc(10mL),然后用短硅胶柱过滤溶液。用EtOAc(100mL)洗涤硅胶柱,减压浓缩溶液。剩余物经快速柱色谱纯化,用EtOAc/己烷(1∶2)洗脱并真空干燥过夜,得到98mg(产率95%)10-TES-10-DAB为白色固体。mp234-235℃ dec;[α]Hg-69°(CHCl3,c=0.95);IR 3690,2958,1714,1602cm-1;1H NMR(500MHz,CDCl3)δ0.68(m,6H,(CH3CH2)3Si),1.00(t,J=7.9,9H,(CH3CH2)3Si),1.08(s,3H,Me17),1.19(s,3H,Me16),1.29(d,J=8.4Hz,1H,7-OH),1.55(s,1H,1-OH),1.69(s,3H,Me19),1.79(ddd,J=14.4,11.0,2.0Hz,1H,H6b),1.92(d,J=5.0Hz,1H,13-OH),2.03(d,J=1.0Hz,3H,Me18),2.27(s,3H,4-Ac),2.29(m,2H,H14a,H14b),2.59(ddd,J=14.4,9.5,6.7Hz,1H,H6a),4.02(d,J=7.2Hz,1H,H3),4.18(d,J=8.5Hz,1H,H20b),4.23(ddd,J=11.0,8.4,6.7Hz,1H,H7),4.30(d,J=8.5Hz,1H,H20a),4.86(m,1H,H13),4.97(dd,J=9.5,2.0Hz,1H,H5),5.28(s,1H,H10),5.66(d,J=7.2Hz,1H,H2),7.47(dd,J=7.9,7.9Hz,2H,苯甲酸酯,m),7.59(tt,J=7.9,1.0Hz,1H,苯甲酸酯,p),8.11(dd,J=7.9,1.0Hz,2H,苯甲酸酯,o)ppm.13C NMR(75MHz,CDCl3)δ4.9,6.5(TES),9.7(Me(19)),14.3(Me(18)),19.6(4-Ac),22.4,26.6(Me16,Me17),37.1(C(6)),38.6(C(14)),42.6(C(15)),47.3(C(3)),57.9(C(8)),67.9(C(13)),71.9,75.1,76.1,76.7(C(7),C(2),C(10),C(20)),78.9(C(1)),81.2(C(4)),84.3(C(5)),128.7,129.9,130.3,133.7(苯甲酸酯),137.0(C(11)),138.8(C(12)),167.4(苯甲酸酯),171.0(4-Ac),209.5(C(9))ppm.
C35H50O10Si·H2O元素分析:
计算值:C,62.11;H,7.74;
实测值:C,62.45;H,7.74。
实施例2
制备7-甲硅烷基-10-TES-10-DAB的总过程
在0℃和N2下向7-三乙基甲硅烷基-10-DAB,7-叔丁基二甲基甲硅烷基-10-DAB或7-二甲基异丙基甲硅烷基-10-DAB的THF溶液中分别慢慢加入N,O-双(三乙基甲硅烷基)三氟乙酰胺(5当量)和催化量的LiHMDS/THF溶液(5mol%)。将反应混合物在0℃搅拌15分钟。加入EtOAc(10mL),然后用短硅胶柱过滤溶液。用EtOAc(100mL)洗涤硅胶柱,减压浓缩溶液。剩余物经快速柱色谱纯化,用EtOAc/己烷(1∶2)洗脱并真空干燥过夜,分别得到7,10-双(三乙基甲硅烷基)-10-DAB(产率95%),7-叔丁基二甲基甲硅烷基-10-三乙基甲硅烷基-10-DAB(产率98%)或7-二甲基异丙基甲硅烷基-10-三乙基甲硅烷基-10-DAB(产率94%)。
7-二甲基苯基甲硅烷基-10-TBS-10-DAB
在0℃和N2下向7-二甲基苯基甲硅烷基-10-DAB(35mg,0.052mmol)的THF(2mL)溶液中分别慢慢加入N,O-双(叔丁基二甲基甲硅烷基)三氟乙酰胺(337μL,1.09mmol,20当量)和催化量的LiHMDS/THF溶液(1M,6μL,0.006mmol)。将反应混合物在0℃搅拌4h,然后在室温再搅拌4h。加入EtOAc(10mL),然后用短硅胶柱过滤溶液。用EtOAc(100mL)洗涤硅胶柱,减压浓缩溶液。剩余物经快速柱色谱纯化,用EtOAc/己烷(1∶2)洗脱并真空干燥过夜,得到39mg(产率92%)7-二甲基苯基甲硅烷基-10-叔丁基二甲基甲硅烷基-10-DAB。
实施例3
C(7)羟基的选择性甲硅烷基化反应
7-TBS-10-DAB
在室温和N2下向10-DAB(38mg,0.070mmol),咪唑(190mg,2.79mmol,40当量)和叔丁基二甲基甲硅烷基氯(210mg,1.40mmol,20当量)的混合物中加入DMF(0.1mL)。将反应混合物在室温剧烈搅拌24h。加入EtOAc(20mL),然后用短硅胶柱过滤溶液。用EtOAc(200mL)洗涤硅胶柱,减压浓缩溶液。剩余物经快速柱色谱纯化,用10%EtOAc-CH2Cl2洗脱并真空干燥过夜,得到41mg(产率90%)7-TBS-10-DAB为白色固体。
mp 222-223℃;[α]Hg-51°(CHCl3,c=0.36);1HNMR(400MHz,CDCl3)δ0.05,0.06(2s,6H,Me2Si),0.83(s,9H,Me3C),1.09(s,6H,Me16,Me17),1.57(s,1H,1-OH),1.75(s,3H,Me19),1.87(ddd,J=14.4,10.6,2.0Hz,1H,H6b),2.01(d,J=5.0Hz,1H,13-OH),2.09(d,J=1.3,3H,Me18),2.28(m,2H,H14a,H14b),2.29(s,3H,4-Ac),2.46(ddd,J=14.4,9.6,6.7Hz,1H,H6a),3.96(d,J=6.9Hz,1H,H3),4.16(d,J=8.3Hz,1H,H20b),4.24(d,J=2.2Hz,1H,10-OH),4.31(d,J=8.3Hz,1H,H20a),4.38(dd,J=10.6,6.7Hz,1H,H7),4.88(m,1H,H13),4.96(dd,J=9.6,2.0Hz,1H,H5),5.15(d,J=2.0Hz,1H,H10),5.60(d,J=6.9Hz,1H,H2),7.47(dd,J=8.1,7.5Hz,2H,苯甲酸酯,m),7.60(tt,J=7.5,1.3Hz,1H,苯甲酸酯,p),8.10(d,J=8.1,1.3Hz,2H,苯甲酸酯,o)ppm.13C NMR(75MHz,CDCl3)δ-5.8,-3.8(Me2Si),9.7(Me(19)),14.8(Me(18)),17.6(Me3C),19.3(4-Ac),22.4,26.7(Me16,Me17),25.4(Me3C),37.4(C(6)),38.7(C(14)),42.7(C(15)),47.0(C(3)),58.0(C(8)),68.0(C(13)),73.1,74.7,75.0(C(7),C(2),C(10),C(20)),78.9(C(1)),80.9(C(4)),84.3(C(5)),128.8,129.8,130.3,133.8(苯甲酸酯),135.7(C(11)),141.9(C(12)),167.4(苯甲酸酯),171.2(4-Ac),210.8(C(9))ppm.
C35H50O10Si元素分析:
计算值:C,63.80;H,7.65;
实测值:C,63.72;H,7.70。
7-二甲基苯基甲硅烷基-10-DAB
在-20℃和N2下向10-DAB(54mg,0.099mmol)的THF(3mL)溶液中加入吡啶(0.6mL)和二甲基苯基甲硅烷基氯(250mL,1.49mmol,15当量)。将反应混合物在-20℃搅拌2h。加入EtOAc(10mL)和饱和NaHCO3水溶液(0.5mL),然后迅速用短硅胶柱过滤溶液。用EtOAc(100mL)洗涤硅胶柱,减压浓缩溶液。剩余物经快速柱色谱纯化,用EtOAc∶CH2Cl2(1∶10)洗脱并真空干燥过夜,得到62mg(产率92%)7-二甲基苯基甲硅烷基-10-DAB为白色固体。mp219-220℃;[α]Hg-28°(CHCl3,c=0.27);1H NMR(400MHz,CDCl3)δ0.35,0.37(2 s,6H,Me2Si),1.05(s,3H,Me17),1.06(s,3H,Me16),1.54(s,1H,1-OH),1.73(d,J=1.1,3H,Me18),1.76(s,3H,Me19),1.90(ddd,J=14.4,10.6,2.1Hz,1H,H6b),1.93(d,J=5.0Hz,1H,13-OH),2.23(m,2H,H14a,H14b),2.25(s,3H,4-Ac),2.43(ddd,J=14.4,9.6,6.8Hz,1H,H6a),3.86(d,J=7.0Hz,1H,H3),4.10(d,J=2.1Hz,1H,10-OH),4.16(d,J=8.3Hz,1H,H20b),4.28(d,J=8.3Hz,1H,H20a),4.31(dd,J=10.6,6.8Hz,1H,H7),4.81(m,1H,H13),4.84(d,J=2.1Hz,1H,H10),4.90(dd,J=9.6,2.1Hz,1H,H5),5.59(d,J=7.0Hz,1H,H2),7.41,7.53(2m,5H,C6H5),7.46(dd,J=8.0,7.5Hz,2H,苯甲酸酯,m),7.55(tt,J=7.5,1.2Hz,1H,苯甲酸酯,p),8.09(d,J=8.0,1.2Hz,2H,苯甲酸酯,o)ppm.13C NMR(75MHz,CDCl3)δ-1.8,-1.1(Me2Si),9.8(Me(19)),14.4(Me(18)),19.4(4-Ac),22.3,26.7(Me16,Me17),37.2(C(6)),38.6(C(14)),42.6(C(15)),46.7(C(3)),58.0(C(8)),68.0(C(13)),73.2,74.7,75.0(C(7),C(2),C(10),C(20)),78.8(C(1)),80.8(C(4)),84.3(C(5)),128.3,128.8,129.8,130.2,130.3,133.65,133.74(PhSi,苯甲酸酯),135.4(C(11)),142.1(C(12)),167.4(苯甲酸酯),171.0(4-Ac),210.9(C(9))ppm.
C37H46O10Si·1/2H2O元素分析:
计算值:C,64.61;H,6.89;
实测值:C,64.72;H,6.81。
7-二甲基异丙基甲硅烷基-10-DAB
在-10℃和N2下向10-DAB(97mg,0.18mmol)的吡啶(1mL)溶液中加入二甲基异丙基甲硅烷基氯(580mL,3.57mmol,20当量)。将反应混合物在-10℃搅拌3h。加入EtOAc(10mL),然后迅速用短硅胶柱过滤溶液。用EtOAc(150mL)洗涤硅胶柱,减压浓缩溶液。剩余物经快速柱色谱纯化,用EtOAc∶己烷(1∶2)洗脱并真空干燥过夜,得到107mg(产率93%)7-二甲基异丙基-10-DAB为白色固体。
mp 229-230℃;[α]Hg-56°(CHCl3,c=0.62);1HNMR(400MHz,CDCl3)δ0.05,0.06(2s,6H,Me2Si),0.70(m,1H,CHSi),0.90,0.92(2 dd,J=7.4,1.7,6H,Me2CH),1.09(s,6H,Me16,Me17),1.56(s,1H,1-OH),1.74(s,3H,Me19),1.89(ddd,J=14.4,10.6,2.1Hz,1H,H6b),1.99(d,J=5.0Hz,1H,13-OH),2.09(d,J=1.4,3H,Me18),2.28(d,J=7.9,2H,H14a,H14b),2.29(s,3H,4-Ac),2.44(ddd,J=14.4,9.7,6.7Hz,1H,H6a),3.96(d,J=7.3Hz,1H,H3),4.17(d,J=8.3Hz,1H,H20b),4.24(d,J=2.2Hz,1H,10-OH),4.31(d,J=8.3Hz,1H,H20a),4.38(dd,J=10.6,6.7Hz,1H,H7),4.85(m,1H,H13),4.95(dd,J=9.7,2.1Hz,1H,H5),5.15(d,J=2.2Hz,1H,H10),5.61(d,J=7.3Hz,1H,H2),7.47(dd,J=8.2,7.5Hz,2H,苯甲酸酯,m),7.60(tt,J=7.5,1.4Hz,1H,苯甲酸酯,p),8.10(d,J=8.2,1.4Hz,2H,苯甲酸酯,o)ppm.13C NMR(75MHz,CDCl3)δ-4.6,-3.3(Me2Si),9.7(Me(19)),14.8,14.9(CHSi,Me(18)),16.4,16.5(Me2CH),19.4(4-Ac),22.4,26.7(Me16,Me17),37.3(C(6)),38.7(C(14)),42.7(C(15)),47.0(C(3)),58.0(C(8)),68.0(C(13)),73.1,74.7,75.0(C(7),C(2),C(10),C(20)),78.9(C(1)),80.9(C(4)),84.3(C(5)),128.8,129.8,130.3,133.7(苯甲酸酯),135.7(C(11)),142.0(C(12)),167.4(苯甲酸酯),171.1(4-Ac),210.8(C(9))ppm.
C34H48O10Si·H2O元素分析:
计算值:C,61.61;H,7.60;
实测值:C,61.30;H,7.35。
7-三苄基甲硅烷基-10-DAB
N2下向10-DAB(62mg,0.11mmol),咪唑(280mg,4.11mmol,36当量)和三苄基甲硅烷基氯(364mg,1.14mmol,10当量)的混合物中加入DMF(0.4mL)。将反应混合物在室温搅拌3h。加入EtOAc(30mL),然后用短硅胶柱过滤溶液。用EtOAc(150mL)洗涤硅胶柱,减压浓缩溶液。剩余物经快速柱色谱纯化两次,第一次用EtOAc∶己烷(1∶2)洗脱,第二次用EtOAc∶CH2Cl2洗脱,并真空干燥过夜,得到88mg(产率91%)7-三苄基甲硅烷基-10-DAB为白色固体。
mp161-163℃;IR 3690,2928,2890,1712,1600cm-1;[α]Hg-46°(CHCl3,c=0.46);1H NMR(400MHz,CDCl3)δ1.10(s,3H,Me17,Me16),1.56(s,1H,1-OH),1.71(ddd,J=14.2,10.9,2.0Hz,1H,H6b),1.74(s,3H,Me19),2.00(d,J=5.1Hz,1H,13-OH),2.07(ddd,J=14.2,9.6,6.6Hz,1H,H6a),2.10(d,J=1.2,3H,Me18),2.12(s,6H,(PhCH2)3Si),2.27(d,J=7.5Hz,2H,H14a,H14b),2.27(s,3H,4-Ac),3.99(d,J=7.0Hz,1H,H3),4.16(d,J=8.5Hz,1H,H20b),4.18(d,J=2.2Hz,1H,10-OH),4.28(d,J=8.5Hz,1H,H20a),4.58(dd,J=10.9,6.6Hz,1H,H7),4.81(dd,J=9.6,2.0Hz,1H,H5),4.89(m,1H,H13),5.21(d,J=2.2Hz,1H,H10),5.61(d,J=7.0Hz,1H,H2),6.93,7.09,7.20(3m,15H,(PhCH2)3Si),7.48(dd,J=8.1,7.5Hz,2H,苯甲酸酯,m),7.61(tt,J=7.5,1.3Hz,1H,苯甲酸酯,p),8.10(d,J=8.1,1.3Hz,2H,苯甲酸酯,o)ppm.13C NMR(75MHz,CDCl3)δ9.9(Me(19)),15.0(Me(18)),19.5(4-Ac),22.4,26.7(Me16,Me17),23.6(Si(CH2Ph)3),36.9(C(6)),38.7(C(14)),42.7(C(15)),46.8(C(3)),58.0(C(8)),68.0(C(13)),74.4,74.9,75.0(C(7),C(2),C(10),C(20)),78.8(C(1)),80.8(C(4)),84.1(C(5)),124.9,128.7,128.8,129.1,129.8,130.3,133.8,137.8(Si(CH2Ph)3,苯甲酸酯),135.5(C(11)),142.2(C(12)),167.4(苯甲酸酯),170.9(4-Ac),210.8(C(9))ppm.
C50H56O10Si·1/2H2O元素分析:
计算值:C,70.32;H,6.73;
实测值:C,70.11;H,6.57。
实施例4
10-酰基-10-DAB的选择性酰化反应
10-Alloc-7-对硝基苄氧羰基-10-DAB
在0℃和N2下向10-Alloc-10-DAB(33mg,0.053mmol)和DMAP(19.3mg,0.16mmol,3当量)的二氯甲烷(4mL)混合物中加入氯甲酸对硝基苄酯(23mL,0.11mmol,2当量)的二氯甲烷溶液(1mL)。将反应混合物在0℃搅拌4h。加入EtOAc(10mL),然后用短硅胶柱迅速过滤溶液。用EtOAc(100mL)洗涤硅胶柱并减压浓缩溶液。剩余物经快速柱色谱纯化,用EtOAc∶己烷(1∶2)洗脱并真空干燥过夜,得到34mg(产率92%)10-Alloc-7-对硝基苄氧羰基-10-DAB为无色固体。
7-苄氧羰基浆果赤霉素III
在室温和N2下向搅拌的浆果赤霉素III(100mg,0.168mmol)的二氯甲烷溶液中先后加入4-二甲氨基吡啶(204mg,1.68mmol)和氯甲酸苄酯(240mL,1.68mmol)。将反应混合物在室温搅拌,并用TLC监视反应的进展情况。约4h后反应结束。加入EtOAc(10mL)稀释混合物,然后将溶液转移到装有50mL 50%EtOAc/己烷的分液漏斗中。用饱和NaHCO3洗涤混合物,然后分离有机相。用20mL 50%EtOAc/己烷洗涤水相,合并的有机相用盐水洗涤,MgSO4干燥,然后减压浓缩。粗产物通过短硅胶柱后得到115mg(产率95%)白色固体。m.p.245-248℃;[α]25 D-60.5°c(C=0.007,CHCl3).1H NMR(CDCl3,400MHz)δ8.10(d,J=9.6Hz,2H,o-苯甲酸酯),7.60-6.8(m,8H,苯甲酸酯,Bn),6.45(s,1H,H10),5.63(d,J=6.9Hz,1H,H2b),5.56(dd,J=10.6,7.2Hz,1H H7),5.56(dd,J=18.5,12.0Hz,2H,Bn),4.97(d,J=10.6,1H,H5),4.87(m,1H,H13),4.31(d,J=10.5,1H,H20a),4.15(d,J=10.5,1H,H20b),4.02(d,J=6.9,1H,H3),2.61(m,1H,H6a),2.30(m,2H,H14′s),2.29(s,3H,4Ac),2.18(s,3H,10Ac),2.15(br s,3H,Me18),2.08(d,J=5.2Hz,13OH),1.94(m,1H,6b),1.79(s,3H,Me19),1.58(s,1H,1OH),1.14(s,3H,Me16),1.09(s,3H,Me17).
7-烯丙氧羰基浆果赤霉素III
在室温和N2下向搅拌的浆果赤霉素III(30mg,0.051mmol)的二氯甲烷溶液(1mL)中先后加入4-二甲氨基吡啶(62.3mg,0.51mmol)和氯甲酸烯丙酯(54mL,0.51mmol)。将反应混合物在室温搅拌,并用TLC监视反应的进展情况。约1.5h后反应结束。加入EtOAc(5mL)稀释混合物,然后将溶液转移到装有50mL 50%EtOAc/己烷的分液漏斗中。用饱和NaHCO3洗涤混合物,然后分离有机相。用10mL 50%EtOAc/己烷洗涤水相,合并的有机相用盐水洗涤,MgSO4干燥,然后减压浓缩。粗产物通过短硅胶柱后得到33.1mg(产率97%)白色固体。m.p.239-244℃;[α]25 D-61.5c(0.01,CHCl3)1H NMR(CDCl3,500MHz)δ8.12(d,J=8.3Hz,2H,o--苯甲酸酯),7.66-7.45(m,3H,苯甲酸酯),6.43(s,1H,H10),5.97(m,1H,int.烯丙基),5.64(d,J=7.0Hz,1H,H2b),5.54(dd,J=10.5,7.0Hz,1H,H7),5.28(m,2H,ext.allyl),4.97(d,J=9.6Hz,1H,H5),4.87(m,1H,H13),4.67(m,2H,CH2烯丙基),4.31(d,J=8.5Hz,1H,H20a),4.17(d,J=8.5,1H,H20b),4.02(d,J=7.0,1H,H3),2.64(m,1H,H6a),2.30(d,J=8.0Hz,2H,H14′s),2.29(s,3H,4Ac),2.16(s,3H,10Ac),2.15(br s,3H,Me18),2.01(d,J=5Hz,13OH),1.96(m,1H,6b),1.81(s,3H,Me19),1.58(s,1H,1OH),1.15(s,3H,Me16),1.02(s,3H,Me17).
实施例5
10-酰基-10-DAB的选择性缩酮化反应
7-MOP浆果赤霉素III
在-20℃和N2下向浆果赤霉素III(101mg,0.172mmol)的THF(8mL)溶液中先后加入2-甲氧基丙烯(0.66mL,6.89mmol,40当量)和催化量甲苯磺酸(0.1M的THF溶液,43μL,0.004mmol,0.025当量)。将反应混合物在-20℃搅拌3h。TLC分析结果显示原料耗尽,形成唯一的主要产物是所要物质的产物。加入三乙胺(0.5mL),将溶液暖至室温,用EtOAc(100mL)稀释,饱和NaHCO3水溶液洗涤,Na2SO4干燥,并减压浓缩。剩余物经真空干燥过夜,得到112mg(产率99%)粗产物。将粗产物从EtOAc/己烷重结晶,得到105mg(产率93%)7-MOP浆果赤霉素III为白色晶体。
mp 181-183℃;1H NMR(500MHz,C6D6)δ1.01(s,3H,Me17),1.11(br s,1H,13-OH),1.28(s,3H,Me16),1.39,1.78(2 s,6H,Me2CO),1.62(s,1H,1-OH),1.78(s,3H,10-Ac),1.92(s,3H,4-Ac),2.09(s,3H,Me18),2.12(s,3H,Me19),2.14(ddd,J=15.0,10.9,2.2Hz,1H,H6b),2.18(dd,J=15.6,9.4Hz,1H,H14b),2.31(dd,J=15.6,7.0Hz,1H,H14b),2.97(s,3H,MeO),3.15(ddd,J=15.0,9.9,6.7Hz,1H,H6a),4.08(d,J=7.0Hz,1H,H3),4.24(m,1H,H13),4.33(d,J=8.3Hz,1H,H20b),4.41(d,J=8.3Hz,1H,H20a),4.78(dd,J=10.9,6.7Hz,1H,H7),4.97(dd,J=9.9,2.2Hz,1H,H5),5.95(d,J=7.0Hz,1H,H2),6.79(s,1H,H10),7.15(m,3H,苯甲酸酯,m,p),8.28(d,J=8.0Hz,2H,苯甲酸酯,o)ppm;
C35H46O12元素分析:
计算值:C,63.82;H,7.04;
实测值:C,63.72;H,7.07。
实施例6
10-甲硅烷基-10-DAB的选择性酰化反应
7-乙酰基-10-TES-10-DAB
在0℃和N2下向搅拌的10-TES-10-DAB(65mg,0.098mmol)的二氯甲烷(4mL)溶液中先后加入DMAP(36mg,0.296mmol,3当量)和乙酸酐(14mL,0.148mmol,1.5当量)。将反应混合物在0℃搅拌4.5h,这时,TLC分析结果显示原料已完全耗尽。然后将反应混合物用短硅胶柱过滤,硅胶用EtOAc(100mL)洗涤,并减压浓缩溶液。剩余物经快速柱色谱纯化,用EtOAc∶己烷(1∶2)洗脱并真空干燥过夜,得到65.7mg(产率95%)7-乙酰基-10-TES-10-DAB。
1H NMR(CDCl3,400MHz),δ0.60(m,6H,(CH3CH2)3Si),0.97(t,J=7.9Hz,9H,(CH3CH2)3Si),1.05(s,3H,Me17),1.18(s,3H,Me16),1.56(s,1H,1-OH),1.79(s,3H,Me19),1.83(ddd,J=14.5,10.3,2.0Hz,1H,H6b),1.97(m,1H,13-OH),2.00(s,3h,7-Ac),2.07(d,J=1.3Hz,3H,Me18),2.26(m,2H,H14a,H14b),2.29(s,3H,4-Ac),2.57(ddd,J=14.5,9.5,7.3Hz,1H,H6a),4.06(d,J=7.0Hz,1H,H3),4.17(d,J=8.2Hz,1H,H20b),4.31(d,J=8.2Hz,1H,H20a),4.84(m,1H,H13),4.94(dd,J=9.5,2.0Hz,1H,H5),5.29(s,1H,H10),5.46(dd,J=10.3,7.3Hz,1H,H7),5.65(d,J=7.0Hz,1H,H2),7.47(m,2H,苯甲酸酯,m),7.60(m,1H,苯甲酸酯,p),8.11(d,J=8.0Hz,2H,苯甲酸酯,o)ppm.
7-Troc-10-TES-10-DAB
N2下向10-TES-10-DAB(40mg,0.061mmol)和DMAP(72mg,0.61mmol,10当量)的二氯甲烷(2mL)混合物中加入氯甲酸三氯乙酯(24mL,0.184mmol,3当量)。将反应混合物在室温搅拌,并通过TLC分析监视反应的进展情况。0.5h后,TLC分析显示10-TES-10-DAB几乎完全耗尽,并形成作为唯一主要斑点的产物。加入甲醇(5mL),溶液迅速通过短硅胶柱进行过滤。用EtOAc(100mL)洗涤硅胶柱并减压浓缩溶液。剩余物经快速色谱纯化,用EtOAc∶CH2Cl2(1∶10)洗脱并真空干燥过夜,得到49mg(产率97%)7-Troc-10-TES-10-DAB为白色固体。
1H NMR(500MHz,CDCl3)δ0.61(m,6H,(CH3CH2)3Si),0.99(t,J=7.9,9H,(CH3CH2)3Si),1.08(s,3H,Me17),1.20(s,3H,Me16),1.56(s,1H,1-OH),1.84(s,3H,Me19),1.96(d,J=4.9Hz,1H,13-OH),2.01(ddd,J=14.4,10.5,2.0Hz,1H,H6b),2.08(d,J=1.2,3H,Me18),2.29(m,2H,H14a,H14b),2.29(s,3H,4-Ac),2.68(ddd,J=14.4,9.5,7.3Hz,1H,H6a),4.08(d,J=6.7Hz,1H,H3),4.18(d,J=8.5Hz,1H,H20b),4.32(d,J=8.5Hz,1H,H20a),4.43(d,J=11.9Hz,1H,CHH′OC(O)),4.86(m,1H,H13),4.95(dd,J=9.3,2.0Hz,1H,H5),4.98(d,J=11.9Hz,1H,CHH′OC(O)),5.33(s,1H,H10),5.37(dd,J=10.5,7.3Hz,1H,H7),5.67(d,J=6.7Hz,1H,H2),7.48(dd,J=7.9,7.3Hz,2H,苯甲酸酯,m),7.60(tt,J=7.3,1.2Hz,1H,苯甲酸酯,p),8.11(dd,J=7.9,1.2Hz,2H,苯甲酸酯,o)ppm.
7-对硝基苄氧羰基-10-TES-10-DAB
N2下向10-TES-10-DAB(40mg,0.061mmol)和DMAP(72mg,0.61mmol,10当量)的无水氯仿(2mL)混合物中加入氯甲酸对硝基苄酯(131mg,0.61mmol,10当量)。将反应混合物在室温搅拌,并通过TLC分析监视反应的进展情况。45分钟后,TLC分析显示10-TES-10-DAB几乎完全耗尽,并形成作为唯一主要斑点的产物。加入甲醇(10mL),溶液迅速通过短硅胶柱进行过滤。用EtOAc(100mL)洗涤硅胶柱并减压浓缩溶液。剩余物经快速色谱纯化,用EtOAc∶CH2Cl2(1∶10)洗脱并真空干燥过夜,得到48.3mg(产率95%)7-对硝基苄氧羰基-10-TES-10-DAB为白色固体。1H NMR(500MHz,CDCl3)δ0.60(m,6H,(CH3CH2)3Si),0.95(t,J=7.9,9H,(CH3CH2)3Si),1.08(s,3H,Me17),1.19(s,3H,Me16),1.55(s,1H,1-OH),1.83(s,3H,Me19),1.93(ddd,J=14.3,10.4,2.2Hz,1H,H6b),1.96(d,J=4.9Hz,1H,13-OH),2.09(d,J=1.2,3H,Me18),2.29(m,2H,H14a,H14b),2.29(s,3H,4-Ac),2.65(ddd,J=14.3,9.3,7.3Hz,1H,H6a),4.08(d,J=7.0Hz,1H,H3),4.18(d,J=8.6Hz,1H,H20b),4.31(d,J=8.6Hz,1H,H20a),4.86(m,1H,H13),4.95(dd,J=9.3,2.2Hz,1H,H5),5.06(d,J=13.4Hz,1H,CHH′OC(O)),5.31(d,J=13.4Hz,1H,CHH′OC(O)),5.33(s,1H,H10),5.36(dd,J=10.4,7.3Hz,1H,H7),5.66(d,J=7.0Hz,1H,H2),7.48(dd,J=7.4,7.3Hz,2H,苯甲酸酯,m),7.53(d,J=8.9Hz,2H,NO2C6H4),7.59(tt,J=7.3,1.2Hz,1H,苯甲酸酯,p),8.12(dd,J=7.4,1.2Hz,2H,苯甲酸酯,o),8.23(d,J=8.9Hz,2H,NO2C6H4)ppm.
7-Cbz-10-TES-10-DAB
N2下向10-TES-10-DAB(40mg,0.061mmol)和DMAP(440mg,3.64mmol,60当量)的无水氯仿(2mL)混合物中每隔10分钟用注射器慢慢加入四等份氯甲酸苄酯(4×130mL,3.64mmol,60当量)。将反应混合物在室温搅拌,并通过TLC分析监视反应的进展情况。2h后,TLC分析显示10-TES-10-DAB几乎完全耗尽,并形成作为唯一主要斑点的产物。加入甲醇(10mL),将溶液倒入乙酸乙酯(100mL),并依次用饱和NaHCO3水溶液,H2O和盐水洗涤。溶液用Na2SO4干燥,然后减压浓缩。剩余物经快速色谱纯化,用EtOAc∶CH2Cl2(1∶10)洗脱并真空干燥过夜,得到45mg(产率93%)7-Cbz-10-TES-10-DAB为白色固体。1H NMR(500MHz,CDCl3)δ0.62(m,6H,(CH3CH2)3Si),0.97(t,J=7.9,9H,(CH3CH2)3Si),1.07(s,3H,Me17),1.20(s,3H,Me16),1.55(s,1H,1-OH),1.81(s,3H,Me19),1.91(ddd,J=14.3,10.5,2.1Hz,1H,H6b),1.96(d,J=4.9Hz,1H,13-OH),2.10(d,J=1.2,3H,Me18),2.28(m,2H,H14a,H14b),2.28(s,3H,4-Ac),2.64(ddd,J=14.3,9.5,7.3Hz,1H,H6a),4.08(d,J=7.0Hz,1H,H3),4.17(d,J=8.5Hz,1H,H20b),4.30(d,J=8.5Hz,1H,H20a),4.86(m,1H,H13),4.95(dd,J=9.5,2.1Hz,1H,H5),5.01(d,J=12.2Hz,1H,CHH′OC(O)),5.24(d,J=12.2Hz,1H,CHH′OC(O)),5.34(s,1H,H10),5.37(dd,J=10.5,7.3Hz,1H,H7),5.65(d,J=7.0Hz,1H,H2),7.32-7.37(m,5H,PhCH2O),7.47(dd,J=8.3,7.3Hz,2H,苯甲酸酯,m),7.59(tt,J=7.3,1.3Hz,1H,苯甲酸酯,p),8.12(dd,J=8.3,1.3Hz,2H,苯甲酸酯,o)ppm.
实施例7
10-酰基-10-DAB的选择性甲硅烷基化反应
7-二甲基异丙基甲硅烷基浆果赤霉素III
在0℃和N2下向搅拌的浆果赤霉素III(30mg,0.051mmol)的吡啶(0.6mL)溶液中加入氯二甲基异丙基硅烷(160μL,1.02mmol)。将反应混合物在0℃搅拌,并用TLC监视反应的进展情况。约1.5h后反应结束。加入EtOAc(5mL),然后将溶液转移到装有50mL 50%EtOAc/己烷的分液漏斗中。用饱和NaHCO3洗涤混合物,然后分离有机相。用10mL 50%EtOAc/己烷洗涤水相,合并的有机相用饱和氯化钠洗涤,MgSO4干燥,然后减压浓缩。粗产物通过短硅胶柱后得到33.9mg(产率97%)白色固体。m.p.204-207℃;[α]25 D-58.6℃(0.009,CHCl3).1HNMR(CDCl3,500MHz),δ8.10(d,J=8.4Hz,2H,o-苯甲酸酯),7.60-7.20(m,3H,苯甲酸酯),6.4(s,1H,H10),5.64(d,J=7.1Hz,1H,H2b),4.95(d,J=4.9Hz,1H,H5),4.84(m,1H,H13),4.44(dd,J=10.4,6.8Hz,1H,H7),4.30(d,J=8.3Hz,1H,H20a),4.14(d,J=8.3Hz,1H,H20b),4.15(d,J=7.2Hz,1H,H3),2.49(m,1H,H6a),2.23(m,2H,H14′s),2.28(s,3H,4Ac),2.18(br s,3H,Me18),2.17(s,3H,10Ac),2.01(d,J=5.0Hz,13OH),1.86(m,1H,6b),1.69(s,3H,Me19),1.61(s,1H,1OH),1.20(s,3H,Me16),1.05(s,3H,Me17),0.87(d,J=7.1Hz,6H,i-pr),0.73(m,1H,i-pr),0.09(s,6H,Me2Si).
7-二甲基苯基甲硅烷基浆果赤霉素III
在-10℃和N2下向搅拌的浆果赤霉素III(20mg,0.034mmol)的THF(1.25mL)溶液中先后加入氯二甲基苯基硅烷(68μL,0.41mmol)和吡啶(250mL,3.1mmol)。将反应混合物在℃搅拌,并用TLC监视反应的进展情况。约1h后反应结束。加入EtOAc(5mL),然后将溶液转移到装有30mL50%EtOAc/己烷的分液漏斗中。用饱和NaHCO3洗涤混合物,然后分离有机相。用10mL 50%EtOAc/己烷萃取水相,合并的有机相用饱和氯化钠洗涤,MgSO4干燥,然后减压浓缩。粗产物通过短硅胶柱后得到24.1mg(产率98%)白色固体。m.p.
210-213℃;[α]25 D-58.3.5℃(0.005,CHCl3)1H NMR(CDCl3,500MHz)δ8.35(d,J=8.5Hz,2H,o-苯甲酸酯),7.627.25(m,8H,苯甲酸酯,苯基),6.42(s,1H,H10),5.64(d,J=6.9Hz,1H,H2b),4.84(m,1H,H5),4.81(m,1H,H13),4.46(dd,J=10.6,6.9Hz,1H,H7),4.21(d,J=8.5Hz,1H,H20a),4.14(d,J=8.5Hz,1H,H20b),3.85(d,J=6.9Hz,1H,H3),2.34(m,1H,H6a),2.26(d,J=8Hz,2H,H14′s),2.24(s,3H,4Ac),2.15(s,3H,10Ac),2.02(br d,J=1Hz,3H,Me 18),1.93(d,J=5Hz,1H,13OH),1.77(m,1H,6b),1.72(s,3H,Me19),1.59(s,1H,1OH),1.20(s,3H,Me16),1.05(s,3H,Me17),0.446(s,3H,MeSi),0.335(s,3H,MeSi).
7-二甲基苯基甲硅烷基-10-丙酰基-10-DAB
在-10℃向搅拌的10-丙酰基-10-DAB(0.200g,0.333mmol)的THF(12mL)溶液中加入氯二甲基苯基硅烷(0.688mL,4.00mmol),然后滴加吡啶(2.48mL,30.64mmol)。将反应混合物搅拌90分钟。加入EtOAc(20mL),然后将溶液转移到装有100mL 50% EtOAc/己烷的分液漏斗中。用饱和NaHCO3洗涤混合物,然后分离有机相。用30mL 50% EtOAc/己烷萃取水相,合并的有机相用饱和氯化钠洗涤,Na2SO4干燥,然后真空浓缩。粗固体产物经快速柱色谱纯化,用50%EtOAc/己烷洗脱,得到0.242g(产率99%)7-二甲基苯基甲硅烷基-10-丙酰基-10-DAB为固体。1H NMR(CDCl3,500MHz),δ0.34,0.45(2s,6H,Me2Si),1.05(s,3H,Me17),1.20(t,J=7.5Hz,3H,CH3CH2),1.21(s,3H,Me16),1.60(s,1H,1-OH),1.72(s,3H,Me19),1.78(ddd,J=14.5,10.0,2.0Hz,1H,H6b),2.04(m,1H,13-OH),2.05(s,3H,Me18),2.27(m,2H,H14a,H14b),2.25(s,3H,4-Ac),2.34(ddd,J=14.5,9.5,7.0Hz,1H,H6a),2.42,2.49(2dq,J=16.5,7.5Hz,6H,CH3CH2),3.87(d,J=7.5Hz,1H,H3),4.14(d,J=8.0Hz,1H,H20b),4.27(d,J=8.0Hz,1H,H20a),4.47(dd,J=10.0,7.0Hz,1H,H7),4.82(m,1H,H13),4.85(dd,J=9.5,2.0Hz,1H,H5),5.64(d,J=7.5Hz,1H,H2),6.44(s,1H,H10),7.32-7.36,7.55-7.57(2m,5H,PhSi),7.46(m,2H,苯甲酸酯,m),7.59(m,1H,苯甲酸酯,p),8.10(d,J=8.0Hz,2H,苯甲酸酯,o)ppm.
7-二甲基苯基甲硅烷基-10-环丙羰基-10-DAB
在-10℃和N2下向10-环丙羰基-10-DAB(680mg,1.1mmol)的THF(25mL)溶液中加入搅拌的吡啶(3.5mL),然后加入氯二甲基苯基硅烷(1.8mL,11mmol)。搅拌溶液直到反应完成。然后用饱和NaHCO3(20mL)淬灭。用EtOAc(2×250mL)萃取混合物。合并的有机相用盐水(2×10mL)洗涤,干燥并过滤。真空浓缩滤液,剩余物经快速色谱纯化,用EtOAc∶己烷(1∶4)洗脱,得到816mg(产率约100%)7-二甲基苯基甲硅烷基-10-环丙羰基-10-DAB。
1H NMR(CDCl3,500MHz),δ0.32,0.43(2s,6H,Me2Si),0.91,1.00,1.17(3m,5H,环丙基),1.07(s,3H,Me17),1.21(s,3H,Me16),1.73(s,3H,Me19),1.74(s,1H,1-OH),1.78(ddd,J=14.4,10.5,2.1Hz,1H,H6b),2.04(m,1H,13-OH),2.05(d,J=1.5Hz,3H,Me18),2.24(s,3H,4-Ac),2.26(m,2H,H14a,H14b),2.34(ddd,J=14.4,9.5,6.7Hz,1H,H6a),3.87(d,J=7.0Hz,1H,H3),4.15(d,J=8.2Hz,1H,H20b),4.26(d,J=8.2Hz,1H,H20a),4.46(dd,J=10.5,6.7Hz,1H,H7),4.82(m,1H,H13),4.85(dd,J=9.5,2.1Hz,1H,H5),5.65(d,J=7.0Hz,1H,H2),6.44(s,1H,H10),7.32-7.36,7.55-7.57(2m,5H,PhSi),7.46(m,2H,苯甲酸酯,m),7.59(m,1H,苯甲酸酯,p),8.10(d,J=8.0Hz,2H,苯甲酸酯,o)ppm.
实施例8
7-对硝基苄氧羰基-10-DAB
在室温和N2下向10-Alloc-7-对硝基苄氧羰基-10-DAB(34mg,0.048mmol)的THF(1mL)溶液中先后加入甲酸(19mL,0.48mmol,10当量)和丁胺(47mL,0.48mmol,10当量)的THF(1mL)溶液和Pd(PPh3)4。将反应混合物在室温搅拌0.5h。加入EtOAc(10mL),将溶液快速通过短硅胶柱过滤。硅胶用EtOAc(100mL)洗涤,并减压浓缩溶液。剩余物经快速柱色谱纯化,用EtOAc∶己烷(1∶2)洗脱并真空干燥过夜,得到28mg(产率93%)7-对硝基苄氧羰基-10-DAB为无色固体。
[α]Hg-38°(CHCl3,c=0.48);1HNMR(400MHz,CDCl3)δ1.06(s,3H,Me16),1.09(s,3H,Me17),1.55(s,1H,1-OH),1.86(s,3H,Me19),2.01(ddd,J=14.4,10.7,2.0Hz,1H,H6b),2.03(d,J=5.1Hz,1H,13-OH),2.09(d,J=1.3,3H,Me18),2.28(m,2H,H14a,H14b),2.30(s,3H,4-Ac),2.62(ddd,J=14.4,9.5,7.3Hz,1H,H6a),3.89(d,J=2.0Hz,1H,10-OH),4.08(d,J=6.9Hz,1H,H3),4.20(d,J=8.4Hz,1H,H20b),4.34(d,J=8.4Hz,1H,H20a),4.88(m,1H,H13),4.96(dd,J=9.5,2.0Hz,1H,H5),5.19(d,J=13.3,1H,CHH′OC(O)),5.26(d,J=13.3,1H,CHH′OC(O)),5.36(dd,J=10.7,7.3Hz,1H,H7),5.40(d,J=2.0Hz,1H,H10),5.64(d,J=6.9Hz,1H,H2),7.48(dd,J=8.1,7.5Hz,2H,苯甲酸酯,m),7.52(d,J=8.7,2H,NO2C6H4),7.61(tt,J=7.5,1.3Hz,1H,苯甲酸酯,p),8.10(d,J=8.1,1.3Hz,2H,苯甲酸酯,o),8.26(d,J=8.7,2H,NO2C6H4)ppm.13C NMR(75MHz,CDCl3)δ10.5(Me(19)),14.6(Me(18)),19.4(4-Ac),22.2,26.4(Me16,Me17),33.2(C(6)),38.7(C(14)),42.4(C(15)),46.5(C(3)),56.5(C(8)),67.9,68.3(C(13),OCH2Ph-NO2-p),74..7,75.2,76.8(C(7),C(2),C(10),C(20)),78.8(C(1)),80.4(C(4)),83.6(C(5)),124.1,128.4,128.9,130.3,133.9(OCH2Ph-NO2-p,苯甲酸酯),135.0(C(11)),142.4,143.0(OCH2Ph-NO2-p,C(12)),154.2(OC(O)O),167.3(苯甲酸酯),171.1(4-Ac),211.6(C(9))ppm.
实施例9
用CyCl3催化10-DAB的C(10)羟基的选择性酯化反应
N2下向10-DAB(30mg,0.055mmol)和DyCl3(1.3mg,相对于10-DAB为10mol%)的固体混合物中加入丁酸酐(0.55mmol)的THF(1.32mL)溶液。将所得悬浮液在室温搅拌直到TLC分析(2∶1 EtOAc/己烷)结果断定反应完成。加入EtOAc稀释反应混合物,并用饱和NaHCO3溶液洗涤3次。将碳酸氢盐洗涤液合并,用EtOAc萃取3次。合并的有机相用Na2SO4干燥,然后蒸发溶剂。粗产物用己烷研磨,滗析母液。从EtOAc/己烷结晶后得到10-丁酰基-10-DAB,与从CeCl3催化的反应中分离的一致。
实施例10
用YbCl3催化10-DAB的C(10)羟基的选择性酯化反应
N2下向10-DAB(30mg,0.055mmol)和YbCl3(1.3mg,相对于10-DAB为10mol%)的固体混合物中加入丁酸酐(0.55mmol)的THF(1.32mL)溶液。将所得悬浮液在室温搅拌直到TLC分析(2∶1 EtOAc/己烷)结果断定反应完成。加入EtOAc稀释反应混合物,并用饱和NaHCO3溶液洗涤3次。将碳酸氢盐洗涤液合并,用EtOAc萃取3次。合并的有机相用Na2SO4干燥,然后蒸发溶剂。粗产物用己烷研磨,滗析母液。从EtOAc/己烷结晶后得到10-丁酰基-10-DAB,与从CeCl3催化的反应中分离的一致。
从上面所述可以看出,本发明的几个目的都实现了。
不脱离本发明范围下可以在上述组合中进行各种变化,也就是说,包括在本文描述中的所有事物都应被理解为是为了说明本发明而非限制本发明。
Claims (13)
1.酰化紫杉烷的C(10)羟基的方法,该方法包括用酰化剂处理紫杉烷,形成C(10)酰化的紫杉烷,在反应混合物中对应于每当量紫杉烷含有少于一当量的碱。
2.权利要求1的方法,其中与酰化剂反应的紫杉烷有C(7)和C(10)羟基,而且酰化剂选择性地与C(10)羟基反应。
3.权利要求1的方法,其中与酰化剂反应的紫杉烷是10-脱乙酰基浆果赤霉素III。
4.权利要求1,2或3的方法,其中酰化剂选自酸酐,二碳酸酯,硫代二碳酸酯和异氰酸酯。
5.权利要求4的方法,其中反应混合物含有Lewis酸。
6.权利要求1,2或3的方法,其中反应混合物含有Lewis酸。
7.权利要求6的方法,其中Lewis酸选自IB,IIB,IIIB,IVB,VB,VIIB,VIII,IIIA,IVA,镧系和锕系元素的卤化物或三氟甲磺酸盐。
8.权利要求7的方法,其中Lewis酸选自氯化锌,氯化锡,三氯化铈,氯化亚铜,三氯化镧,三氯化镝和三氯化镱。
9.权利要求1的方法,其中C(10)酰化的紫杉烷还含有C(7)羟基,而且,该方法还包括用甲硅烷基化剂处理C(10)酰化的紫杉烷,将C(7)羟基甲硅烷基化。
10.权利要求9的方法,其中C(10)酰化的紫杉烷是浆果赤霉素III。
11.权利要求1的方法,其中C(10)酰化的紫杉烷还含有C(7)羟基,而且,该方法还包括用酰化剂处理C(10)酰化的紫杉烷,将C(7)羟基酰化。
12.权利要求11的方法,其中C(10)酰化的紫杉烷是浆果赤霉素III。
13.权利要求1的方法,其中C(10)酰化的紫杉烷还含有C(13)羟基,金属氧化物或氧化铵取代基,而且,该方法还包括通过用选自β-内酰胺,噁唑啉,噁唑烷羧酸,噁唑烷羧酸酐和异丝氨酸衍生物的侧链前体处理C(10)酰化的紫杉烷来酯化C(10)酰化的紫杉烷的步骤。
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US5600097P | 1997-08-18 | 1997-08-18 | |
| US60/056,000 | 1997-08-18 | ||
| US8126598P | 1998-04-09 | 1998-04-09 | |
| US60/081,265 | 1998-04-09 | ||
| US09/063,477 US7288665B1 (en) | 1997-08-18 | 1998-04-20 | Process for selective derivatization of taxanes |
| US09/063,477 | 1998-04-20 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB2005100641228A Division CN100351246C (zh) | 1997-08-18 | 1998-08-17 | 选择性衍生紫杉烷的方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1239476A CN1239476A (zh) | 1999-12-22 |
| CN1205196C true CN1205196C (zh) | 2005-06-08 |
Family
ID=27368950
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2006101492418A Pending CN1974563A (zh) | 1997-08-18 | 1998-08-17 | 选择性衍生紫杉烷的方法 |
| CNB2005100641228A Expired - Fee Related CN100351246C (zh) | 1997-08-18 | 1998-08-17 | 选择性衍生紫杉烷的方法 |
| CNB98801341XA Expired - Fee Related CN1205196C (zh) | 1997-08-18 | 1998-08-17 | 选择性衍生紫杉烷的方法 |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2006101492418A Pending CN1974563A (zh) | 1997-08-18 | 1998-08-17 | 选择性衍生紫杉烷的方法 |
| CNB2005100641228A Expired - Fee Related CN100351246C (zh) | 1997-08-18 | 1998-08-17 | 选择性衍生紫杉烷的方法 |
Country Status (25)
| Country | Link |
|---|---|
| US (5) | US7288665B1 (zh) |
| EP (6) | EP0956284B1 (zh) |
| JP (1) | JP2001504864A (zh) |
| KR (1) | KR100596989B1 (zh) |
| CN (3) | CN1974563A (zh) |
| AT (3) | ATE341540T1 (zh) |
| AU (1) | AU744987B2 (zh) |
| BR (1) | BR9806145A (zh) |
| CA (2) | CA2597799A1 (zh) |
| CY (1) | CY1105399T1 (zh) |
| CZ (1) | CZ296580B6 (zh) |
| DE (3) | DE69837684T2 (zh) |
| DK (3) | DK1170293T3 (zh) |
| ES (3) | ES2286073T3 (zh) |
| HU (1) | HUP0300425A3 (zh) |
| ID (1) | ID21654A (zh) |
| IL (3) | IL129386A0 (zh) |
| NO (4) | NO325139B1 (zh) |
| NZ (1) | NZ335156A (zh) |
| PL (3) | PL193674B1 (zh) |
| PT (3) | PT956284E (zh) |
| RU (1) | RU2225402C2 (zh) |
| SG (1) | SG97986A1 (zh) |
| WO (1) | WO1999009021A1 (zh) |
| ZA (1) | ZA987394B (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008116388A1 (fr) * | 2007-03-27 | 2008-10-02 | Dalian Institute Of Chemical Physics, Chinese Academy Of Sciences | Procédé de fabrication de taxol, de baccatine et de dérivés de ces substances |
| WO2008116387A1 (fr) * | 2007-03-27 | 2008-10-02 | Dalian Institute Of Chemical Physics, Chinese Academy Of Sciences | Procédé de fabrication de taxol et de dérivés de taxol |
Families Citing this family (72)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5750736A (en) * | 1996-07-11 | 1998-05-12 | Napro Biotherapeutics, Inc. | Method for acylating 10-deacetylbaccatin III selectively at the c-10 position |
| US7288665B1 (en) | 1997-08-18 | 2007-10-30 | Florida State University | Process for selective derivatization of taxanes |
| IT1308636B1 (it) * | 1999-03-02 | 2002-01-09 | Indena Spa | Procedimento per la preparazione di tassani da 10-desacetilbaccatinaiii. |
| AR030188A1 (es) | 2000-02-02 | 2003-08-13 | Univ Florida State Res Found | Compuestos de taxano sustituidos con esteres en el c7; composiciones farmaceuticas que los contienen y proceso para tratar un sujeto mamifero que sufre de una condicion que responde a los taxanos |
| BR0104352A (pt) | 2000-02-02 | 2002-01-02 | Univ Florida State Res Found | Taxanos substituìdos por c10 carbamoilóxi como agentes antitumorais |
| WO2001057028A1 (en) | 2000-02-02 | 2001-08-09 | Florida State University Research Foundation, Inc. | C7 carbamoyloxy substituted taxanes as antitumor agents |
| RO121271B1 (ro) | 2000-02-02 | 2007-02-28 | Florida State University Research Foundation, Inc. | Taxani substituiţi cu carbonat la c7, ca agenţiantitumorali |
| MXPA01009924A (es) | 2000-02-02 | 2003-08-01 | Univ Florida State Res Found | Taxanos de acetato heteosustituido en c10 como agentes antitumorales. |
| US6649632B2 (en) | 2000-02-02 | 2003-11-18 | Fsu Research Foundation, Inc. | C10 ester substituted taxanes |
| MXPA01009922A (es) | 2000-02-02 | 2003-07-14 | Univ Florida State Res Found | Taxanos de acetato heterosustituido en c7 como agentes antitumorales. |
| US6660866B2 (en) | 2000-02-02 | 2003-12-09 | Psu Research Foundation, Inc. | C10 carbonate substituted taxanes |
| US6362217B2 (en) * | 2000-03-17 | 2002-03-26 | Bristol-Myers Squibb Company | Taxane anticancer agents |
| IT1320107B1 (it) * | 2000-11-28 | 2003-11-18 | Indena Spa | Procedimento per la preparazione di derivati tassanici. |
| IT1319682B1 (it) * | 2000-12-06 | 2003-10-23 | Indena Spa | Procedimento di sintesi del paclitaxel. |
| US7351542B2 (en) | 2002-05-20 | 2008-04-01 | The Regents Of The University Of California | Methods of modulating tubulin deacetylase activity |
| DK2286795T3 (en) | 2002-06-26 | 2017-02-06 | Syncore Biotechnology Co Ltd | PROCEDURE FOR PREPARING A Cationic LIPOSOMAL PREPARATION INCLUDING A LIPOPHIL COMPOUND |
| CN101039957A (zh) | 2003-05-16 | 2007-09-19 | 因特缪恩公司 | 合成的趋化因子受体配体及其使用方法 |
| CN1303077C (zh) * | 2004-01-16 | 2007-03-07 | 桂林晖昂生化药业有限责任公司 | 合成紫杉烷的制备工艺 |
| SV2006002010A (es) | 2004-02-13 | 2006-08-23 | Univ Florida State Res Found | Taxanos sustituidos con esteres de ciclopentilo en c10 |
| PA8625301A1 (es) | 2004-03-05 | 2006-06-02 | Univ Florida State Res Found | Taxanos con sustituyente lactiloxilo en el c7 |
| KR20070013300A (ko) * | 2004-05-14 | 2007-01-30 | 이뮤노젠 아이엔씨 | 박카틴 ⅲ 화합물의 합성 방법 |
| WO2006089209A2 (en) | 2005-02-17 | 2006-08-24 | The Board Of Trustees Of The Leland Stanford Junior University | Txr1 and enhanced taxane sensitivity based on the modulation of a pathway mediated thereby |
| US7667055B2 (en) * | 2005-06-10 | 2010-02-23 | Florida State University Research Foundation, Inc. | Processes for the production of polycyclic fused ring compounds |
| CA2610908A1 (en) * | 2005-06-10 | 2006-12-21 | Florida State University Research Foundation, Inc. | Processes for the preparation of paclitaxel |
| EP3056196A1 (en) | 2006-03-22 | 2016-08-17 | SynCore Biotechnology CO., LTD | Treatment of triple receptor negative breast cancer |
| US7847111B2 (en) * | 2006-06-19 | 2010-12-07 | Canada Inc. | Semi-synthetic route for the preparation of paclitaxel, docetaxel, and 10-deacetylbaccatin III from 9-dihydro-13-acetylbaccatin III |
| PL210984B1 (pl) * | 2006-10-31 | 2012-03-30 | Inst Farmaceutyczny | Sposób wytwarzania docetakselu |
| WO2008106491A2 (en) * | 2007-02-27 | 2008-09-04 | University Of Utah Research Foundation | Peptides that interact with topoisomerase i and methods thereof |
| JP4833126B2 (ja) * | 2007-03-26 | 2011-12-07 | Ntn株式会社 | 潤滑剤劣化検出装置および検出装置付き軸受 |
| WO2009040829A1 (en) * | 2007-09-27 | 2009-04-02 | Benzochem Lifescience (P) Limited | A novel process for the preparation of docetaxel |
| US8242166B2 (en) | 2008-03-31 | 2012-08-14 | Florida State University Research Foundation, Inc. | C(10) ethyl ester and C(10) cyclopropyl ester substituted taxanes |
| CA2724550C (en) | 2008-05-22 | 2017-01-03 | Kereos, Inc. | Combination antitumor therapy |
| US20110172293A1 (en) | 2008-07-08 | 2011-07-14 | Fish Jason E | Methods and Compositions for Modulating Angiogenesis |
| CN101863861A (zh) * | 2009-04-16 | 2010-10-20 | 山东靶点药物研究有限公司 | 一种简便高效地制备紫杉醇类似物Larotaxel的方法 |
| US8791279B2 (en) * | 2010-12-13 | 2014-07-29 | Yung Shin Pharm. Ind. Co., Ltd. | Process for preparing taxoids from baccatin derivatives using lewis acid catalyst |
| WO2012131698A1 (en) | 2011-04-01 | 2012-10-04 | Shilpa Medicare Limited | Process for preparing docetaxel and its hydrate |
| US20130090484A1 (en) * | 2011-10-11 | 2013-04-11 | Scinopharm Taiwan Ltd. | Process for making an intermediate of cabazitaxel |
| CN103159704B (zh) * | 2011-12-12 | 2015-06-10 | 上海佰霖国际贸易有限公司 | 卡巴他赛中间体及卡巴他赛中间体的制备方法 |
| KR102165464B1 (ko) | 2012-07-19 | 2020-10-14 | 레드우드 바이오사이언스 인코포레이티드 | Cd22에 대해 특이적인 항체 및 이들의 사용 방법 |
| EP2916835A4 (en) | 2012-11-12 | 2016-07-27 | Redwood Bioscience Inc | COMPOUNDS AND METHOD FOR PRODUCING A CONJUGATE |
| EP2956175B1 (en) | 2013-02-15 | 2017-10-04 | The Regents of the University of California | Chimeric antigen receptor and methods of use thereof |
| JP6410790B2 (ja) | 2013-03-14 | 2018-10-24 | ザ ボード オブ トラスティーズ オブ ザ レランド スタンフォード ジュニア ユニバーシティー | ミトコンドリアアルデヒドデヒドロゲナーゼ−2調節因子およびその使用方法 |
| CN103145654A (zh) * | 2013-04-12 | 2013-06-12 | 江苏斯威森生物医药工程研究中心有限公司 | 一种多西他赛的半合成方法 |
| WO2014191989A1 (en) * | 2013-05-26 | 2014-12-04 | Idd Therapeutics Ltd. | Conjugate of a taxane and biotin and uses thereof |
| EP3074010A4 (en) | 2013-11-27 | 2017-10-25 | Redwood Bioscience, Inc. | Hydrazinyl-pyrrolo compounds and methods for producing a conjugate |
| CN106458910B (zh) | 2014-02-19 | 2020-11-06 | 亚倍生技制药公司 | 线粒体醛脱氢酶2(aldh2)结合多环酰胺和其用于治疗癌症的用途 |
| CN110845616A (zh) | 2014-03-21 | 2020-02-28 | 艾伯维公司 | 抗-egfr抗体及抗体药物偶联物 |
| EP3373977A4 (en) | 2015-11-12 | 2019-07-17 | The Board of Trustees of the Leland Stanford Junior University | CELLULAR PENETRATION-RICH GUANIDINIC OLIGOPHOSPHOTRIESTERS FOR THE DELIVERY OF MEDICATION AND PROBE |
| TWI760319B (zh) | 2015-12-30 | 2022-04-11 | 杏國新藥股份有限公司 | 乳癌治療 |
| EP3231421A1 (en) | 2016-04-11 | 2017-10-18 | Greenaltech, S.L. | Uses of a carotenoid in the treatment or prevention of stress induced conditions |
| CA3019398A1 (en) | 2016-04-26 | 2017-11-02 | R.P. Scherer Technologies, Llc | Antibody conjugates and methods of making and using the same |
| LT3463308T (lt) | 2016-06-01 | 2022-02-10 | Servier IP UK Limited | Polialkilenoksido-asparaginazės vaisto formos ir jų gamybos būdai bei panaudojimas |
| MX2018015285A (es) | 2016-06-08 | 2019-09-18 | Abbvie Inc | Anticuerpos anti-b7-h3 y conjugados de anticuerpo y farmaco. |
| JP2019526529A (ja) | 2016-06-08 | 2019-09-19 | アッヴィ・インコーポレイテッド | 抗b7−h3抗体及び抗体薬物コンジュゲート |
| AU2017279550A1 (en) | 2016-06-08 | 2019-01-03 | Abbvie Inc. | Anti-B7-H3 antibodies and antibody drug conjugates |
| EP3468599A2 (en) | 2016-06-08 | 2019-04-17 | AbbVie Inc. | Anti-cd98 antibodies and antibody drug conjugates |
| JP2019524651A (ja) | 2016-06-08 | 2019-09-05 | アッヴィ・インコーポレイテッド | 抗cd98抗体及び抗体薬物コンジュゲート |
| TWI852903B (zh) | 2017-01-05 | 2024-08-21 | 杏國新藥股份有限公司 | 胰臟癌治療 |
| US20190367621A1 (en) | 2017-01-18 | 2019-12-05 | F1 Oncology, Inc. | Chimeric antigen receptors against axl or ror2 and methods of use thereof |
| EP3388082A1 (en) | 2017-04-13 | 2018-10-17 | Galera Labs, LLC | Combination cancer immunotherapy with pentaaza macrocyclic ring complex |
| BR112020015520A2 (pt) | 2018-01-31 | 2021-02-02 | Galera Labs, Llc | terapia de combinação para câncer com complexo de anel macrocíclico pentaaza e agente anticâncer à base de platina |
| CN112368011A (zh) | 2018-04-11 | 2021-02-12 | 俄亥俄州创新基金会 | 缓释微粒用于眼用药物递送的方法和组合物 |
| JP2021530507A (ja) | 2018-07-18 | 2021-11-11 | マンザニータ ファーマシューティカルズ,インク. | 神経細胞に抗がん剤を送達するためのコンジュゲート、その使用方法及び製造方法 |
| AU2020253449A1 (en) | 2019-04-02 | 2021-11-18 | Kenjockety Biotechnology, Inc. | Efflux pump-cancer antigen multi-specific antibodies and compositions, reagents, kits and methods related thereto |
| MX2022009355A (es) | 2020-01-29 | 2022-09-02 | Kenjockety Biotechnology Inc | Anticuerpo anti-mdr1 y uso del mismo. |
| WO2021247423A1 (en) | 2020-06-04 | 2021-12-09 | Kenjockety Biotechnology, Inc. | Abcg2 efflux pump-cancer antigen multi-specific antibodies and compositions, reagents, kits and methods related thereto |
| WO2021247426A1 (en) | 2020-06-04 | 2021-12-09 | Kenjockety Biotechnology, Inc. | Anti-abcg2 antibodies and uses thereof |
| US20230272117A1 (en) | 2020-09-02 | 2023-08-31 | Kenjockety Biotechnology, Inc. | Anti abcc1 antibodies and uses thereof |
| EP4244257A1 (en) | 2020-11-13 | 2023-09-20 | Kenjockety Biotechnology, Inc. | Anti-mrp4 (encoded by abcc4 gene) antibodies and uses thereof |
| UY39610A (es) | 2021-01-20 | 2022-08-31 | Abbvie Inc | Conjugados anticuerpo-fármaco anti-egfr |
| CA3238353A1 (en) | 2021-12-13 | 2023-06-22 | William Robert ARATHOON | Anti-abcb1 antibodies |
| WO2023159220A1 (en) | 2022-02-18 | 2023-08-24 | Kenjockety Biotechnology, Inc. | Anti-cd47 antibodies |
Family Cites Families (63)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US569666A (en) * | 1896-10-20 | Brake-beam | ||
| FR2601676B1 (fr) | 1986-07-17 | 1988-09-23 | Rhone Poulenc Sante | Procede de preparation du taxol et du desacetyl-10 taxol |
| USRE34277E (en) | 1988-04-06 | 1993-06-08 | Centre National De La Recherche Scientifique | Process for preparing taxol |
| FR2629818B1 (fr) * | 1988-04-06 | 1990-11-16 | Centre Nat Rech Scient | Procede de preparation du taxol |
| JPH01309997A (ja) * | 1988-06-09 | 1989-12-14 | Kanto Kasei Kogyo Kk | 耐食性に優れた銅−ニッケル−クロム光沢電気めっき方法およびそれにより得られためっき皮膜 |
| US4960790A (en) | 1989-03-09 | 1990-10-02 | University Of Kansas | Derivatives of taxol, pharmaceutical compositions thereof and methods for the preparation thereof |
| US5175315A (en) * | 1989-05-31 | 1992-12-29 | Florida State University | Method for preparation of taxol using β-lactam |
| US5136060A (en) | 1989-11-14 | 1992-08-04 | Florida State University | Method for preparation of taxol using an oxazinone |
| FR2678930B1 (fr) * | 1991-07-10 | 1995-01-13 | Rhone Poulenc Rorer Sa | Procede de preparation de derives de la baccatine iii et de la desacetyl-10 baccatine iii. |
| US5489601A (en) | 1991-09-23 | 1996-02-06 | Florida State University | Taxanes having a pyridyl substituted side-chain and pharmaceutical compositions containing them |
| US5714513A (en) | 1991-09-23 | 1998-02-03 | Florida State University | C10 taxane derivatives and pharmaceutical compositions |
| SG46582A1 (en) | 1991-09-23 | 1998-02-20 | Univ Florida State | 10-Desacetoxytaxol derivatives |
| US5229526A (en) * | 1991-09-23 | 1993-07-20 | Florida State University | Metal alkoxides |
| US5284865A (en) | 1991-09-23 | 1994-02-08 | Holton Robert A | Cyclohexyl substituted taxanes and pharmaceutical compositions containing them |
| US5654447A (en) | 1991-09-23 | 1997-08-05 | Florida State University | Process for the preparation of 10-desacetoxybaccatin III |
| US5399726A (en) | 1993-01-29 | 1995-03-21 | Florida State University | Process for the preparation of baccatin III analogs bearing new C2 and C4 functional groups |
| US5990325A (en) | 1993-03-05 | 1999-11-23 | Florida State University | Process for the preparation of 9-desoxotaxol, 9-desoxobaccatin III and analogs thereof |
| US5430160A (en) | 1991-09-23 | 1995-07-04 | Florida State University | Preparation of substituted isoserine esters using β-lactams and metal or ammonium alkoxides |
| FR2687145B1 (fr) | 1992-02-07 | 1994-03-25 | Rhone Poulenc Rorer Sa | Nouveaux anhydrides d'acides, leur preparation et leur emplot et |
| US5200534A (en) | 1992-03-13 | 1993-04-06 | University Of Florida | Process for the preparation of taxol and 10-deacetyltaxol |
| US5416225A (en) | 1992-03-30 | 1995-05-16 | Sloan-Kettering Institute For Cancer Research | Total synthesis of taxol |
| US5254703A (en) | 1992-04-06 | 1993-10-19 | Florida State University | Semi-synthesis of taxane derivatives using metal alkoxides and oxazinones |
| US5440056A (en) | 1992-04-17 | 1995-08-08 | Abbott Laboratories | 9-deoxotaxane compounds |
| CA2130578A1 (en) | 1992-04-17 | 1993-10-28 | Geewananda P. Gunawardana | Taxol derivatives |
| US5269442A (en) * | 1992-05-22 | 1993-12-14 | The Cornelius Company | Nozzle for a beverage dispensing valve |
| US5470866A (en) | 1992-08-18 | 1995-11-28 | Virginia Polytechnic Institute And State University | Method for the conversion of cephalomannine to taxol and for the preparation of n-acyl analogs of taxol |
| WO1994005282A1 (en) | 1992-09-04 | 1994-03-17 | The Scripps Research Institute | Water soluble taxol derivatives |
| US5478854A (en) | 1992-10-01 | 1995-12-26 | Bristol-Myers Squibb Company | Deoxy taxols |
| FR2696458B1 (fr) | 1992-10-05 | 1994-11-10 | Rhone Poulenc Rorer Sa | Procédé de préparation de dérivés du taxane. |
| FR2696460B1 (fr) | 1992-10-05 | 1994-11-25 | Rhone Poulenc Rorer Sa | Procédé de préparation de dérivés du taxane. |
| US5412116A (en) | 1992-11-06 | 1995-05-02 | Hauser Chemical Research, Inc. | Oxidation of glycoside substituted taxanes to taxol or taxol precursors and new taxane compounds formed as intermediates |
| FR2698363B1 (fr) | 1992-11-23 | 1994-12-30 | Rhone Poulenc Rorer Sa | Nouveaux dérivés du taxane, leur préparation et les compositions qui les contiennent. |
| US5380751A (en) | 1992-12-04 | 1995-01-10 | Bristol-Myers Squibb Company | 6,7-modified paclitaxels |
| FR2698871B1 (fr) | 1992-12-09 | 1995-02-24 | Rhone Poulenc Rorer Sa | Nouveau taxoïdes, leur préparation et les compositions pharmaceutiques qui les contiennent. |
| US5646176A (en) | 1992-12-24 | 1997-07-08 | Bristol-Myers Squibb Company | Phosphonooxymethyl ethers of taxane derivatives |
| US5948919A (en) * | 1993-02-05 | 1999-09-07 | Napro Biotherapeutics, Inc. | Paclitaxel synthesis from precursor compounds and methods of producing the same |
| FR2702212B1 (fr) | 1993-03-02 | 1995-04-07 | Rhone Poulenc Rorer Sa | Nouveaux taxoïdes, leur préparation et les compositions pharmaceutiques qui les contiennent. |
| US5547981A (en) | 1993-03-09 | 1996-08-20 | Enzon, Inc. | Taxol-7-carbazates |
| US5703247A (en) | 1993-03-11 | 1997-12-30 | Virginia Tech Intellectual Properties, Inc. | 2-Debenzoyl-2-acyl taxol derivatives and methods for making same |
| US5475011A (en) | 1993-03-26 | 1995-12-12 | The Research Foundation Of State University Of New York | Anti-tumor compounds, pharmaceutical compositions, methods for preparation thereof and for treatment |
| US5336684A (en) | 1993-04-26 | 1994-08-09 | Hauser Chemical Research, Inc. | Oxidation products of cephalomannine |
| IL109926A (en) * | 1993-06-15 | 2000-02-29 | Bristol Myers Squibb Co | Methods for the preparation of taxanes and microorganisms and enzymes utilized therein |
| US5405972A (en) * | 1993-07-20 | 1995-04-11 | Florida State University | Synthetic process for the preparation of taxol and other tricyclic and tetracyclic taxanes |
| FR2718137B1 (fr) | 1994-04-05 | 1996-04-26 | Rhone Poulenc Rorer Sa | Procédé de préparation de trialcoylsilyl-7 baccatine III. |
| US6100411A (en) * | 1994-10-28 | 2000-08-08 | The Research Foundation Of State University Of New York | Taxoid anti-tumor agents and pharmaceutical compositions thereof |
| JPH10508022A (ja) * | 1994-10-28 | 1998-08-04 | ザ リサーチ ファウンデーション オブ ステート ユニバーシティ オブ ニューヨーク | タキソイド誘導体、それらの製造、およびそれらの抗腫瘍薬としての使用 |
| US5489589A (en) | 1994-12-07 | 1996-02-06 | Bristol-Myers Squibb Company | Amino acid derivatives of paclitaxel |
| JPH08253465A (ja) * | 1995-03-17 | 1996-10-01 | Dai Ichi Seiyaku Co Ltd | 四環性化合物 |
| MA23823A1 (fr) * | 1995-03-27 | 1996-10-01 | Aventis Pharma Sa | Nouveaux taxoides, leur preparation et les compositions qui les contiennent |
| US5847170A (en) * | 1995-03-27 | 1998-12-08 | Rhone-Poulenc Rorer, S.A. | Taxoids, their preparation and pharmaceutical compositions containing them |
| US5780653A (en) | 1995-06-07 | 1998-07-14 | Vivorx Pharmaceuticals, Inc. | Nitrophenyl, 10-deacetylated substituted taxol derivatives as dual functional cytotoxic/radiosensitizers |
| US5760251A (en) | 1995-08-11 | 1998-06-02 | Sepracor, Inc. | Taxol process and compounds |
| FR2743074B1 (fr) * | 1995-12-27 | 1998-03-27 | Seripharm | Procede de protection selective des derives de la baccatine et son utilisation dans la synthese des taxanes |
| US5688977A (en) | 1996-02-29 | 1997-11-18 | Napro Biotherapeutics, Inc. | Method for docetaxel synthesis |
| ES2151277T3 (es) * | 1996-05-22 | 2000-12-16 | Protarga Inc | Composiciones que comprenden conjugados de acido cis-docosahexaenoico y taxotere. |
| US5773461A (en) | 1996-06-06 | 1998-06-30 | Bristol-Myers Squibb Company | 7-deoxy-6-substituted paclitaxels |
| FR2750989B1 (fr) | 1996-07-09 | 1998-09-25 | Rhone Poulenc Rorer Sa | Procede de monoacylation d'hydroxy taxanes |
| US5750736A (en) | 1996-07-11 | 1998-05-12 | Napro Biotherapeutics, Inc. | Method for acylating 10-deacetylbaccatin III selectively at the c-10 position |
| US5811452A (en) | 1997-01-08 | 1998-09-22 | The Research Foundation Of State University Of New York | Taxoid reversal agents for drug-resistance in cancer chemotherapy and pharmaceutical compositions thereof |
| US6017935A (en) * | 1997-04-24 | 2000-01-25 | Bristol-Myers Squibb Company | 7-sulfur substituted paclitaxels |
| DK0875508T3 (da) | 1997-05-02 | 2004-01-26 | Pharmachemie Bv | Fremgangsmåde til fremstilling af baccatin lll og derivater ud fra 10-deacetylbaccatin lll |
| US7288665B1 (en) | 1997-08-18 | 2007-10-30 | Florida State University | Process for selective derivatization of taxanes |
| US5914411A (en) | 1998-01-21 | 1999-06-22 | Napro Biotherapeutics, Inc. | Alternate method for acylating 10-deacetylbaccatin III selectively at the C-10 position |
-
1998
- 1998-04-20 US US09/063,477 patent/US7288665B1/en not_active Expired - Fee Related
- 1998-08-17 WO PCT/US1998/017016 patent/WO1999009021A1/en active Application Filing
- 1998-08-17 NZ NZ335156A patent/NZ335156A/en not_active IP Right Cessation
- 1998-08-17 ES ES01203287T patent/ES2286073T3/es not_active Expired - Lifetime
- 1998-08-17 EP EP98942080A patent/EP0956284B1/en not_active Expired - Lifetime
- 1998-08-17 BR BR9806145-3A patent/BR9806145A/pt not_active Application Discontinuation
- 1998-08-17 DE DE69837684T patent/DE69837684T2/de not_active Expired - Lifetime
- 1998-08-17 DK DK01203288T patent/DK1170293T3/da active
- 1998-08-17 AU AU90211/98A patent/AU744987B2/en not_active Ceased
- 1998-08-17 DK DK01203287T patent/DK1170292T3/da active
- 1998-08-17 DE DE69836072T patent/DE69836072T2/de not_active Expired - Lifetime
- 1998-08-17 AT AT98942080T patent/ATE341540T1/de not_active IP Right Cessation
- 1998-08-17 CZ CZ0133299A patent/CZ296580B6/cs not_active IP Right Cessation
- 1998-08-17 SG SG200006873A patent/SG97986A1/en unknown
- 1998-08-17 ZA ZA987394A patent/ZA987394B/xx unknown
- 1998-08-17 PT PT98942080T patent/PT956284E/pt unknown
- 1998-08-17 IL IL12938698A patent/IL129386A0/xx not_active IP Right Cessation
- 1998-08-17 RU RU99110384/04A patent/RU2225402C2/ru not_active IP Right Cessation
- 1998-08-17 EP EP01203288A patent/EP1170293B1/en not_active Expired - Lifetime
- 1998-08-17 EP EP01203286A patent/EP1193262A1/en not_active Withdrawn
- 1998-08-17 EP EP05108674A patent/EP1671960A2/en not_active Withdrawn
- 1998-08-17 CN CNA2006101492418A patent/CN1974563A/zh active Pending
- 1998-08-17 AT AT01203287T patent/ATE360624T1/de not_active IP Right Cessation
- 1998-08-17 EP EP07104179A patent/EP1795528A1/en not_active Withdrawn
- 1998-08-17 JP JP51349899A patent/JP2001504864A/ja not_active Ceased
- 1998-08-17 CA CA002597799A patent/CA2597799A1/en not_active Abandoned
- 1998-08-17 PL PL98377990A patent/PL193674B1/pl not_active IP Right Cessation
- 1998-08-17 PL PL98332723A patent/PL332723A1/xx not_active IP Right Cessation
- 1998-08-17 KR KR1019997003388A patent/KR100596989B1/ko not_active IP Right Cessation
- 1998-08-17 ES ES01203288T patent/ES2259646T3/es not_active Expired - Lifetime
- 1998-08-17 ES ES98942080T patent/ES2274579T3/es not_active Expired - Lifetime
- 1998-08-17 AT AT01203288T patent/ATE326455T1/de not_active IP Right Cessation
- 1998-08-17 CN CNB2005100641228A patent/CN100351246C/zh not_active Expired - Fee Related
- 1998-08-17 PL PL377991A patent/PL201641B1/pl not_active IP Right Cessation
- 1998-08-17 PT PT01203288T patent/PT1170293E/pt unknown
- 1998-08-17 CN CNB98801341XA patent/CN1205196C/zh not_active Expired - Fee Related
- 1998-08-17 DE DE69834584T patent/DE69834584T2/de not_active Expired - Fee Related
- 1998-08-17 ID IDW990217A patent/ID21654A/id unknown
- 1998-08-17 DK DK98942080T patent/DK0956284T3/da active
- 1998-08-17 EP EP01203287A patent/EP1170292B1/en not_active Expired - Lifetime
- 1998-08-17 HU HU0300425A patent/HUP0300425A3/hu unknown
- 1998-08-17 CA CA002268662A patent/CA2268662A1/en not_active Abandoned
- 1998-08-17 PT PT01203287T patent/PT1170292E/pt unknown
-
1999
- 1999-04-16 NO NO19991838A patent/NO325139B1/no not_active IP Right Cessation
- 1999-06-17 US US09/335,408 patent/US6191287B1/en not_active Expired - Lifetime
-
2000
- 2000-06-07 US US09/588,933 patent/US6291691B1/en not_active Expired - Fee Related
- 2000-09-28 US US09/672,270 patent/US6706896B1/en not_active Expired - Fee Related
-
2005
- 2005-06-09 US US11/148,833 patent/US20050272944A1/en not_active Abandoned
- 2005-09-29 NO NO20054513A patent/NO20054513L/no unknown
- 2005-09-29 NO NO20054512A patent/NO20054512L/no unknown
- 2005-09-29 NO NO20054514A patent/NO20054514L/no unknown
-
2006
- 2006-06-26 CY CY20061100861T patent/CY1105399T1/el unknown
-
2007
- 2007-07-01 IL IL184321A patent/IL184321A0/en unknown
- 2007-07-01 IL IL184320A patent/IL184320A0/en unknown
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008116388A1 (fr) * | 2007-03-27 | 2008-10-02 | Dalian Institute Of Chemical Physics, Chinese Academy Of Sciences | Procédé de fabrication de taxol, de baccatine et de dérivés de ces substances |
| WO2008116387A1 (fr) * | 2007-03-27 | 2008-10-02 | Dalian Institute Of Chemical Physics, Chinese Academy Of Sciences | Procédé de fabrication de taxol et de dérivés de taxol |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1205196C (zh) | 选择性衍生紫杉烷的方法 | |
| CN1067682C (zh) | 抗肿瘤化合物、药物组合物、其制备方法以及其用于治疗的方法 | |
| CN1046261C (zh) | 紫杉醇及其衍生物的合成 | |
| CN1062561C (zh) | 7-卤代和7β,8β-亚甲基-紫杉醇,抗肿瘤用途以及含其药物组合物 | |
| CN1301986C (zh) | 前列腺素及其类似物的制备方法 | |
| CN1094940C (zh) | 具有抗肿瘤活性的紫杉酚衍生物 | |
| CN1200731A (zh) | 半合成塔三烷用的中间体及其制备方法 | |
| CN1075718A (zh) | 金属醇盐 | |
| CN1798759A (zh) | 制备具有端二烯基的环孢素"a"类似物的方法 | |
| CN1703409A (zh) | 新紫杉烷和用途及制备方法 | |
| CN101048394A (zh) | 太平洋紫杉醇向多烯紫杉醇的半合成转化 | |
| CN1139925A (zh) | Δ12,13-异紫杉酚类似物、其抗肿瘤的用途和含有这些化合物的药物组合物 | |
| CN1205184C (zh) | β-内酰胺 | |
| CN1097416A (zh) | 新的β-内酰胺类、制备紫杉烷的方法及带侧链紫杉烷类 | |
| CN1151740A (zh) | 7-醚-紫杉酚类似物、抗肿瘤药物用途及含有它们的药物组合物 | |
| CN100341853C (zh) | 带烷基取代的侧链的紫杉烷及含该化合物的药物组合物 | |
| CN1942460A (zh) | 紫杉烷衍生物的一锅合成及其向太平洋紫杉醇和多烯紫杉醇的转化 | |
| CN1286823C (zh) | 用于制备紫杉烷的噁唑烷中间体 | |
| CN1244569C (zh) | C-10位紫杉烷衍生物及其药物组合物 | |
| CN1221539C (zh) | 紫杉烷衍生物 | |
| CN1101346A (zh) | 碳2塔三烷衍生物和含有该衍生物的药物组合物 | |
| CN1179955C (zh) | 制备紫杉醇及其衍生物的方法 | |
| CN1068821A (zh) | 苯并二氢吡喃衍生物 | |
| CN1090625C (zh) | 选择性保护浆果赤霉素衍生物的方法及其在合成紫杉烷中的应用 | |
| CN1147491C (zh) | 新的吖啶羧酸酯化合物、其制备方法及其药物组合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20050608 Termination date: 20090917 |