本发明的碱性蓄电池用非烧结式正极活性物质由氢氧化镍粒子,较好的是还由覆盖在前述氢氧化镍粒子表面的钴的氧化物组成,这种钴的氧化物属于六方晶、斜方晶、单斜晶中任何一种结晶体系,具有层状结构,(003)面的晶面距离为5.5-7.0埃,而且包含钴的平均价数大于3.0的γ-羟基氧化钴。
γ-羟基氧化钴是使2价的氢氧化钴在KOH或NaOH、水以及空气(氧)共存的气氛下进行激烈的氧化反应后得到的高次氧化物。由于γ-羟基氧化钴与历来电池内部的电化学氧化反应得到的钴的氧化物(主要是β-羟基氧化钴和2价钴的氧化物的混合物)和公知的由碱存在下的空气氧化反应得到的2.9价钴的氧化物(在结晶构造方面几乎与β-羟基氧化钴相同)相比,其导电性能优良,所以能够提高氢氧化镍的放电利用率。
又,由于γ-羟基氧化钴具有较高导电性,所以即使以过度放电或短路状态保存,也能够保证大多数氢氧化镍和基板框架之间的电流导通,因此,能够延迟前述的在基板框架近旁位置进行的集中的还原反应。而且,γ-羟基氧化钴在其自身被氧化成极高次状态的同时,和β-羟基氧化钴等相比,它在碱性电解液中的还原电位较低,所以不会很容易地被还原到2.0价的状态(电解液中钴处于能够溶解的状态)。
从以上的理由可看出,能够更进一步抑制以过度放电、短路状态保存时正极容量的劣化。
本发明的理想状态是在γ-羟基氧化钴的结晶中至少含有K+以及Na+中的一种。
包含在前述γ-羟基氧化钴中的K+及/或Na+对应于钴的原子比,较好的是在0.20-0.33的范围内。
γ-羟基氧化钴有包含K+型和包含Na+型2种(任何一种骨架形成的结晶构造都相同),从理想的结构式推测出M/Co(M为K或Na中的任何一种)的mol比为0.33。但是,因为很难得到与理想结构相当的氧化物,所以M/Co实际在上述范围内。
含有γ-羟基氧化钴的高次钴氧化物对应于氢氧化镍粒子的重量比,较好的是在0.02-0.10的范围内。高次氧化物的量少于0.02,则不能很好地保持氢氧化镍粒子和基板框架之间的电流导通,而高次氧化物的量过大,氢氧化镍的量则会相对减少,不能制造高能量密度的正极。
具有钴氧化物被覆层的氢氧化镍粒子的平均粒径为5-20μm,较好的是被覆层的厚度在0.2μm以下,粒子能够基本全部被覆盖。如果氢氧化镍粒子的平均粒径在上述范围之外,由于松密度降低等原因,使正极的能量密度也降低。如果被覆层的厚度超过0.2μm,则全部活性物质中钴氧化物所占的量过多,氢氧化镍的量就会相对减少,仍然是正极的能量密度降低。如果平均粒径以及被覆层的厚度在前述的范围内,而且氢氧化镍的集电能力达到最大,则被覆层正好完全覆盖粒子,此时作为活性物质粒子是最好的。
本发明的另一观点是具有钴氧化物被覆层的氢氧化镍粒子利用漫反射法测定的可视区域(波长400-700nm)的光谱反射率在3.5%以上,在波长450nm附近(蓝色)具有4.0%以上的最大光谱反射率,而且被覆层中钴的平均价数大于3.0。活性物质粒子的颜色和形成被覆层的钴氧化物的电子状态密切相关,呈现上述颜色的钴氧化物的电子传导性相当高。这是因为上述的颜色反映了γ-羟基氧化钴所特有的价数高于3.0的高次电子状态(存在4价钴)的缘故。
本发明的制造碱性蓄电池用正极活性物质的方法由三个步骤组成,即第1步,在装有微波加热装置和混合搅拌装置的容器内,一边混合具有氢氧化钴被覆层的氢氧化镍粒子(被覆了Co(OH)2氢氧化镍粒子)一边在其中滴加碱性水溶液,得到表面被碱性水溶液润湿的湿润粒子;第2步,密封前述的容器,以微波照射为主,对前述的湿润粒子一边进行加热,一边继续混合,使前述的湿润粒子升温到规定温度;第3步,达到规定温度后,解除密封,使容器内空气流通,一边控制前述微波的输出功率使粒子温度保持一定,一边继续混合,直到前述湿润粒子完全干燥。
被覆了Co(OH)2的氢氧化镍粒子在碱性水溶液共存的气氛下发生氧化反应时,一般认为形成被覆层的氢氧化钴的反应机理为以下2个步骤:
第1步,氢氧化钴根据式(1)的反应在存在于粒子被覆层表面的碱性水溶液中溶解,形成钴的络离子(HCoO2 -),钴的络离子如反应式(2)所示的,和氧接触被氧化,以高次钴氧化物的形式在粒子表面析出。
第2步,氢氧化钴在碱和氧共存的气氛中一边生成水,一边如反应式(3)所示的,进行固相反应生成高次钴氧化物,即不溶解就被氧化,形成高次钴氧化物。反应式(4)表示在上述气氛中氧的消耗。
上述的式子中为了简略,用CoOOH(形式上为3.0价)表示生成物,由于反应能够很好地被控制,所以能够氧化成比3.0价更高次的γ-羟基氧化钴。这种情况下,反应大致上不会有变化,但反应式会比上述略微复杂一些。
仔细观察以上的2个步骤会发现,第1个步骤的进行依赖于氢氧化钴在碱性水溶液中的溶解性(式1)。但是,在浓度为30重量%的KOH水溶液中,60℃时的溶解度也没有超过数百ppm,而且溶解速度也不那么大。所以,为使反应速度加快,应该将反应气氛设置在高温下。但是,在高温下进行反应时,因为周围湿度太低等缘故,如果溶液蒸发后干燥枯竭,就不能生成式(1)的络离子,反应会中止。另一方面,式(2)的氧化反应中使生成的钴络离子和氧(空气)充分接触是很重要的,如果处于周围缺乏氧的高温环境下,就会发生以下式(5)所示的副反应,生成导电性较差的Co3O4(Co价数为2.67)。
第2步中如果在碱共存下,加热氢氧化钴,由反应式(3)生成高次钴氧化物。此时在氧的共存下同时发生式(4)所示的反应,式(3)的反应继续进行。能够使此反应顺利进行的关键在于将反应体系置于高温、提高OH-的浓度(式3)、提高O2的浓度(式4),以及适当除去反应体系中生成的水。如果过量地除去水,即过分干燥,因为碱类不能生成OH-离子,式(3)的反应就会中止。相反地,没有充分除去水分,则氢氧化钴附近的O2的浓度会相对降低,使式(4)的反应不能顺利进行,其结果是发生式(6)所示的副反应来取代式(3)的反应。
从以上的说明可以明显看出,不论哪一个步骤,处理时水和氧(空气)的状态起到了相当重要的作用。从这个观点看,要使被覆了Co(OH)2的氢氧化镍粒子在碱共存的气氛下的氧化反应能够最有效地进行,就需要对于表面存在适量碱性水溶液,成绳索状(按照化学工业上分类,在粒子表面存在足够的溶液,而且具有通气性的湿润状态)的粒子,在高温下一边很好地控制水和氧的量一边对其进行处理。
所以,本发明的第1步是一边混合被覆了Co(OH)2的氢氧化镍粒子,一边在其中滴加碱性水溶液,形成均一的绳索状湿润粒子。在后续的第2步和第3步中用微波进行加热是利用微波照射湿润粒子,以分子标准使电介质(此时为碱性水溶液)振荡,产生分子冲突和摩擦热来达到加热目的的方法。第2步中通过在密封气氛下一边混合一边用微波加热,几乎不引起加热不均就能够将湿润粒子迅速升温到规定温度。接着在第3步中解除密封,送入空气的目的是在有效地将氢氧化钴氧化反应中生成的过剩的水排出反应体系的同时,如有必要还能提供给反应体系足够的氧。这时,通过蒸发过剩的水,除去蒸发潜热,一边监控粒子温度一边利用微波加热补充热量,使反应体系内的温度维持在规定范围内。
作为氧化进行时的加热方法,如果使用微波照射,由于经碱性水溶液润湿的钴被覆层部分被微波强烈且有选择地加热,所以和其他加热手段相比,被覆层部分的氧化效率提高了,被覆层的钴被完全氧化达到超过3.0价的状态。而且,使湿润粒子达到完全干燥状态并不能容易地识别同一氧化反应的终点,这是因为在粒子表面存在的水之间可能会继续进行氧化反应的缘故。如果使粒子达到完全干燥状态就可以说处理全部完成了。
在前述容器的内壁部分中装有加热装置,从第1步到第3步之间,较好的是利用前述的加热装置辅助性地对容器内壁面进行加热。这种加热对防止蒸发的水分或碱性气雾在容器内壁面上结露,防止容器壁上附着粒子,以及防止粒子凝结或固化都很有效。
前述的被覆了Co(OH)2的氢氧化镍粒子的平均粒径为5-20μm,被覆层几乎完全覆盖了粒子,其厚度在0.2μm以下,而且,较好的是前述粒子的BET比表面积为5-12m2。如果平均粒径不在上述范围内,由于松密度降低等原因,正极的能量密度不够高。如果被覆层的厚度过大,则决定正极容量的氢氧化镍的量就会相对减少,也就是说不能提供高能量密度的正极。此外,由于氢氧化镍母粒子的集电较容易,所以被覆层最好的是处于粒子被完全覆盖的状态。又,BET比表面积过大或过小时,因为粒子的润湿性有较大的变化,所以在处理时很难利用规定的碱性水溶液使粒子变为绳索状结构。从这个观点看,被覆了Co(OH)2的氢氧化镍粒子的BET比表面积最好的是在上述的范围内。
本发明的制造活性物质的方法中,前述第2以及第3步的加热温度较好的是在90-130℃的范围内。氧化反应的速度受温度的影响很大,设定的温度不足90℃时,氧化反应进行得较慢,每批需要数小时。同时,粒子容易附着在容器内壁,这不是我们希望的。另一方面,温度超过130℃,反应就过于激烈,会损伤被覆层内部的氢氧化镍。从以上观点看,加热温度较好的是设定在90-130℃的范围内。
前述的碱性水溶液为KOH或NaOH的水溶液,其浓度大于40重量%,而且其滴加量较好的是对应于被覆了Co(OH)2的氢氧化镍粒子的干燥重量,使碱性溶质的重量比在0.04-0.06的范围内。
本发明制造方法中的氢氧化钴的氧化反应是在碱性水溶液沸点附近进行的,所以碱性水溶液中水分的蒸发速度较大。氧化反应的第1步是将氢氧化钴溶解于碱性水溶液中,生成钴的络离子,然后此络离子和氧反应生成高次钴氧化物。所以,处理时必须于高温下在粒子表面存在一定量的碱性水溶液。换言之,即使是在高温(90-130℃)条件下,如果碱性水溶液蒸发太快,反应也不能充分进行。从这个观点看,碱性水溶液的浓度越大其沸点就越高,则蒸发速度就较慢,这样就适应了处理时的要求。此外,从第2步氧化反应的式(3)也能够明显看出,OH-浓度较高的一边氧化反应进行得较充分,也就是说,碱性水溶液浓度大的情况较好。综上所述,所用的碱性水溶液的浓度应该大于40重量%。
碱性水溶液的滴加量应在上述范围内是因为上述的量最适合将湿润粒子引向适当的绳索状结构,而且能够将粒子的凝集控制在最小限度的缘故。前述的粒子的凝集是在处理过程中,碱性溶质和空气中的二氧化碳反应,生成碳酸盐,碳酸盐在粒子干燥过程中作为粘合剂作用而引起的。碱性水溶液的滴加量太小,则氧化反应不完全,未反应的组分残留下来,另一方面,如果量太大会使粒子大量凝集,使反应不能顺利进行。
以下,在实验结果的基础上,对本发明的具体例子进行详细说明。
实施例1
1.高次钴氧化物的制造
在氢氧化钠水溶液中慢慢地加入1mol/L的硫酸钴水溶液,于35℃,一边将水溶液的pH维持在12,一边搅拌,使氢氧化钴析出。将其水洗后真空干燥,作为氢氧化钴标准试样。利用X射线衍射测得该氢氧化钴标准试样具有β型的结晶构造(JCPDS(Joint Committee on Diffractuin Standards)无机物文件编号:30-443),用电子扫描显微镜(SEM)观察同一试样,发现试样为约0.2μm大小的六角板状粒子,又用BET法确认同一试样的比表面积为25m2/g。
然后,在干燥气氛下(干燥箱中)粉碎粒状的高纯度氢氧化钾试剂,以足够的量和上述氢氧化钴标准试样混合。接着,将混合后的试样从干燥箱中取出,放到110℃的加热容器中,往里面送空气。在这个过程中因为混合试样受到了氢氧化钾潮解的影响而变成含有少量空气中水分的绳索状,并在这种状态下进行氧化反应。将试样在加热容器中放置12小时,使氧化反应进行完全。然后取出试样,充分水洗后于80℃,干燥3小时,得到本发明的高次钴氧化物x。此外,除了将氢氧化钾试剂换成粒状高纯度的氢氧化钠试剂之外,其他的操作都与上述相同,制得本发明的高次钴氧化物y。
2.正极的制作
作为正极母粒子的氢氧化镍粒子可以利用以下周知的方法合成。即以硫酸镍为主成分,调制出含有对应于镍的硫酸钴以及硫酸锌中的钴以及锌的mol比分别只有0.02以及0.05的量的水溶液。利用氨水一边调整溶液的pH值,一边在该水溶液中慢慢滴加氢氧化钠水溶液,使球状的氢氧化镍固溶体粒子析出。水洗得到的固溶了Co以及Zn的固溶体氢氧化镍粒子,然后干燥,就得到了正极母粒子。该固溶体氢氧化镍粒子的松密度约为2.0g/cc,平均粒径为10μm。
然后,在100重量份所得的固溶体氢氧化镍粒子中添加7.0重量份前述的高次钴氧化物x和适量的纯水,使其混合分散。得到活性物质的悬浮液。将该活性物质悬浮液填充到多孔度为95%、厚度为1.3mm的发泡镍多孔体基板中,在80℃的干燥器中使其干燥。然后,利用滚筒挤压,再将其厚度压延为0.7mm。最后按照规定的大小切断,就得到了对应于高次钴氧化物x的镍正极。
对高次钴氧化物y进行同样的处理,制成相应的镍正极。此外,在100重量份固溶体氢氧化镍粒子中添加7.0重量份前述的氢氧化钴标准试样和纯水,使其混合分散,得到活性物质的悬浮液,以下的操作都与前述相同,制成比较例的镍正极。
3.电池的制造和对正极的评估
将上述方法制造的3种镍正极分别和氢包藏合金组成的负极、由经亲水化处理的聚丙烯纤维非织造材料制成的隔板、以及在氢氧化钾的31重量%水溶液中使LiOH·H2O按照40g/L的比例溶解的电解液组合起来,制成尺寸为4/5A、公称容量为1600mAh的镍-氢蓄电池。按照以下①、②所述的方法对各种电池进行测试,评估各种镍正极的利用率以及短路保存后的容量恢复率。
①利用率
以0.1CmA的充电速度分别对上述的3种电池充电,时间为15小时,再以0.2CmA的放电速度放电,使电池的电压降低到1.0V,循环进行5次,测定第5次循环后的电池容量。将测出的电池容量除以正极的理论容量(正极中填充的氢氧化镍粒子的重量乘以伴随接受或释放1个电子进行的氢氧化镍的充放电反应时假定的电容量289mAh/g得出的值)就可算出各种镍正极的利用率。
②短路保存后的容量恢复率
以1CmA的充电速度对进行了①的评估后的电池充电,时间为1.2小时,再以1CmA的放电速度放电,使电池的电压降低到1.0V,循环进行5次,测定第5次循环后的电池容量。然后用1Ω的电阻使电池短路,在此状态下于45℃气氛中保存2周。再次以1CmA的充电速度对经过保存的电池充电,时间为1.2小时,再以1CmA的放电速度放电,使电池的电压降低到1.0V,循环进行5次,测定第5次的电池容量。将短路保存后的电池容量值除以短路保存前的电池容量值,就得到各种镍正极在短路保存后的容量恢复率。
按照正极添加剂的种类区分各种电池的评估结果,并将其记录在表1中。
表1
|
正极添加剂的种类 |
利用率(%) |
短路保存后的容量恢复率(%) |
|
钴氧化物x |
99.2 |
98.8 |
|
钴氧化物y |
99.4 |
98.6 |
|
氢氧化钴 |
81.8 |
75.5 |
从表1可明显看出,本发明的使用了高次钴氧化物x、y的正极和使用了氢氧化钴标准试样的比较例相比,显示出特别高的利用率以及容量恢复率。所以,为了弄清楚其中的原因,下面对上述的钴氧化物x、y进行详细地解析。
4.对高次钴氧化物的解析
4.1.高次钴氧化物x
4.1.1.对结晶构造、价数、组成的解析
图1表示的是本发明的高次钴氧化物x的X射线衍射图。使用的X射线源是Cu的Kα射线。根据图中所标的记号(黑色实心圆圈)可以确认在衍射角2θ为10°-70°的范围内有12个峰。根据JCPDS无机物质文件的检索,对应于峰所在位置的化合物进行定性分析。其结果是在作为钴氧化物登记的化合物中不存在峰所在位置的化合物,但和作为镍的高次氧化物而众所周知的γ-羟基氧化镍(文件编号:6-75)的衍射图形十分一致。γ-羟基氧化镍在著名的文献,例如P.Oliva等的J.power Sources,8(1982)229-255中记载的,模拟性地(几何结构)属于六方晶系,具有层状结构,与β型氢氧化物相比,晶格向c轴方向伸长,是镍的价数高于3.0的高次氧化物。而且,还可以知道此γ-羟基氧化镍的结晶内包含碱性阳离子。
然后,按照以下的注意事项对钴氧化物x的钴平均价数进行评估。首先,将要进行测定的钴氧化物x和硫酸亚铁铵FeSO4(NH4)2SO4混合,并将其溶解于浓盐酸中。在这一过程中,比2价高的Co离子将Fe2+氧化为Fe3+,自身被还原成Co2+。然后,利用ICP发光分析对所得的水溶液中的Co2+进行定量(定量值为1)。另一方面,通过用过锰酸钾水溶液进行的氧化还原滴定,求出先前生成的Fe3+的量(定量值2)。由这两个值,根据式2.0+(定量值2)/(定量值1),求出钴氧化物x中钴的平均价数为3.42。
另外,和以上操作不同,将钴氧化物x溶解于浓盐酸中,对所得的水溶液进行ICP发光分析,发现在同一氧化物中存在K+,其含量用对应于钴的mol比表示为约相当于0.28。
以上的解析结果暗示如果高次钴氧化物x和镍氧化物的命名相同时,可以说它是属于[γ型氧化物]的。钴和镍的原子编号只相差1,离子半径也相似,而且2价氢氧化物具有相同的结晶构造,这些都表示两者在结晶学上极为相似。所以,本发明者们认为在钴氧化物中也存在和镍氧化物相同的γ型高次氧化物的结晶构造,高次钴氧化物被定义为[γ型羟基氧化钴]。假定这种γ型羟基氧化钴为六方晶系的层状结构(从c轴方向看出的钴层的排列为ABC重复结构),在作为X射线衍射图的图1所示的12个峰中,对于强度较大,且能够大致准确读出衍射角的6个面(hkl),汇总其晶面距离和附加指数。结果如表2所示。
表2
|
hkl |
dobs(埃) |
I/IOobs |
|
003 |
6.85 |
100 |
|
006 |
3.42 |
35 |
|
101 |
2.43 |
20 |
|
102 |
2.39 |
25 |
|
105 |
2.10 |
22 |
|
110 |
1.42 |
13 |
4.1.2.过去文献的调查和对其的观察
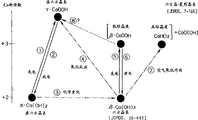
表2所示的结构和作为P.Benson等人关于钴电极的研究发表在Electrochim.Acta,9(1964)275-280.上的“anodic CoOOH(I)”得到的生成物极为相似。他们在同一文献中提出将含有“anodic CoOOH(I)”、在晶面距离为6.8埃附近提供较强X射线衍射峰的钴氧化物定义为“β-CoOOH”,以区别通过空气氧化氢氧化钴等得到的导电性较差的氧化物“CoHO2”。“CoHO2”氧化物具有六方晶-菱面晶结构,获得六方晶的单位晶格时的c轴的长度为13.13埃,c轴方向的晶面距离为其1/3,即4.38埃。但是,如果仿照现在的氢氧化镍及其氧化物被广泛接受的H.Bode等的分类(Electrochim.Acta,11(1996)1079-1087.),以具有匹配性的形式对钴进行分类,则被认为沿c轴方向晶格延伸的前述“anodic CoOOH(I)”和表2记载的高次钴氧化物x应该命名为“γ-CoOOH”(γ-羟基氧化钴)。
作为对碱性蓄电池用非烧结式正极的钴添加剂的研究,押谷等人在《汤浅时报》65(1988)28-40中的报道和氢氧化镍的H.Bode等的分类相同,表明2价氢氧化钴中存在c轴方向晶面距离延伸的[α型]和不延伸的[β型]。而且,这些添加在正极中的2价钴氧化物在最初充电时就被氧化,成为c轴方向的晶面距离为4.4埃的氧化物,由于这些氧化物具有导电性,所以具有有效的功能。和前述的P.Benson人等不同,押谷等人把上述具有导电性的氧化物称为“β-CoOOH”,这种命名和镍的H.Bode等的分类相匹配,也被广泛接受。即历来的普遍理解是“β-CoOOH”是指和β-Co(OH)2的c轴长(4.65埃)相比,没有c轴方向晶面距离的延伸,而且具有导电性的羟基氧化钴。这种“β-CoOOH”可能和被P.Benson等称为“CoHO2”的氧化物(JCPDS文件中记为CoO(OH),编号:7-169,c轴方向的晶面距离为4.384埃)具有同样的结晶结构,而且其结晶性能较低。
按照P.Benson等人的报告记为CoO(OH)的氧化物,由于微晶自身的电子传导性较差,所以其结晶性能较好(称为“CoHO2”),导电性非常差。但是,调整了制造条件,使结晶性能降低,即变为被记作“β-CoOOH”型的结晶时,微晶自身变小和微晶-微晶间的界面(此界面具有电子传导面的功能)增加相结合,显现出其导电性。近年有好几例关于这一点的报道,例如第37届电池研讨会演讲提要集,P371(1996)报道的结晶性能较低的CoO(OH)(是在碱性电解液中,将CoO电化学氧化后得到的,其中含有混入物Co3O4)的电化学行为主要是具有导电性的“β-CoOOH”的充放电现象。
4.1.3关于钴氧化物的反应的分类
将上述关于钴氧化物,特别是氢氧化物的分类模仿H.Bode等的简图并加以总结就得到了图2。历来作为碱性蓄电池用非烧结式正极使用的2价钴氧化物有α-Co(OH)2、β-Co(OH)2、CoO三种。α-Co(OH)2在碱性电解液中通过反应式③能够很容易地转变为β-Co(OH)2,CoO虽然在图2中没有表示,但也能够在碱性电解液中发生化学变化,转变为β-Co(OH)2。所以,前述的3种2价钴氧化物在反应结束时,初期充电前是以β-Co(OH)2的形式存在的。它们在初期充电时通过反应式⑤被氧化,转变为具有导电性的β-CoOOH。但是,因为一般上述β-Co(OH)2的结晶性较高,其中大部分在进行固相反应⑤的充电反应时氧化(电子离开氢氧化钴及吸引质子的反应)的反应点较少,在结晶内部残存了没有导电性的未反应的氢氧化钴。其残存量依赖于初期充电时的充电电流的大小、周围的温度、还有电解液的组成等条件。
这样形成的含有β-CoOOH的钴氧化物能够通过反应式⑥还原(放电),但在一般电池的充放电电位区域并不发生这种还原反应。所以,含有β-CoOOH的钴氧化物具有使氢氧化镍粒子的充放电顺利进行的导电网络的作用。
另一方面,本发明的高次钴氧化物x,即γ-CoOOH是通过β-Co(OH)2的剧烈氧化得到的,所以可以认为是通过图2中的式④的反应历程生成的。此外,还知道了由于β-NiOOH的过度放电,氢氧化镍容易生成γ-NiOOH,但可推测对于钴来讲,类似的反应过程(图中的反应式⑧)不易发生。这是因为作为β-CoOOH基本结构的CoO(OH)结晶和β-NiOOH相比,其氧和氧之间含有较强的氢键,为从结晶内部引出质子,使钴达到更高次的氧化状态,就需要非常大能量的缘故。这一方面钴和镍是不同的。
此外,还有2点在图2中未显示出来的钴氧化物和镍氧化物不同的地方。第1是2价钴氧化物对浓碱水溶液中的溶解性,另一点是存在尖晶石结构Co3O4(四氧化三钴)。就前者而言,2价钴氧化物虽然溶解度不大,但可以钴络离子HCoO2的形式溶解在浓碱性水溶液中。但从图2的③、④、⑤等的反应并不能知道这一行为进行到了何种程度。而后者,即钴氧化物中特有的Co3O4具有很好的热力学稳定性。所以,图2所示的氢氧化物在高温下铸烧都会转变为Co3O4,而且,在④、⑤、⑦的氧化反应中,根据不同的条件容易生成价数为2.67的Co3O4,由此妨碍了转变为更高次状态的氧化反应的进行。这样钴氧化物和镍氧化物在图2所示的能够进行极为相似的分类的反面,还有几点微妙的不同之处。
然后,以图2为例,对上述P.Benson等的文献中记载的实验结果进行说明。首先,从文献中记载的“蓝色”和其X射线衍射结果(c轴方向的晶面距离:8.4埃)可以判断出作为实验始发物质的合成氢氧化钴是α-Co(OH)2。在KOH水溶液中将其电化学氧化生成“anodic CoOOH(I)”的反应相当于①的反应。所示的“anodicCoOOH(II)”相当于在γ-CoOOH中混合一部分的β-CoOOH以及Co3O4。根据这样的α→γ的机理,也能够生成γ-CoOOH。但是,在碱性蓄电池用非烧结式正极中添加α-Co(OH)2时,从向电池中注入溶液到进行初期充电为止需要一定的时间,所以,在这期间进行③的反应,不易进行①的反应。另一方面,文献中记载的将氢氧化钴放置在KOH水溶液中得到的“CoHO2”就是始发物质α-Co(OH)2通过③的反应生成β-Co(OH)2,然后经溶液中的溶解氧进行⑦的氧化反应而得到的。其中记载的氧化物颜色的变化情况是:蓝色→白色→桃红色→茶色,α-Co(OH)2为蓝色,β-Co(OH)2为白色→桃红色,CoHO2为茶色,图2和文献记载的结果基本相同。
这样更说明了图2所示的线图和历来报道的钴氧化物(特别是氢氧化物)的行为完全没有矛盾的地方。而且,本发明的高次钴氧化物x是比3价更高的高次γ-CoOOH,这是一种在历来碱性蓄电池用正极中不存在的物质。
4.1.4.关于结晶结构的考察
图3表示的是假想的γ-羟基氧化钴的结晶结构模拟图。参考有关文献后发现这种结晶结构基本和γ-羟基氧化镍的结晶结构相同。如图3所示,γ-羟基氧化钴模拟地属于准六方晶系,是具有层状结构的钴氧化物,晶格沿c轴方向延伸。沿c轴方向可见的钴层周期排列为ABC的重复结构,如果以钴-氧-氧-钴的形式显示,则变成ACCBAACBB的重复结构。所以,仅仅着眼于此,也能够获得几何结构的菱面晶体的单位晶格。如果利用表2所示的晶面距离实测值和一般的有效离子半径:rco=0.68埃(3价-6的配位:0.685埃和4价-6配位:0.67埃的代表值)ro=1.25埃(O2-的2价-6配位:1.26埃和OH-的2价-6配位:1.23埃的代表值),根据几何学计算方法计算图3中的三角棱柱状中配置的6个氧离子或氢氧化物离子之间产生的空隙,估计可以容纳半径为1.64埃的圆球。如图中虚线圆球所示那样,这个空隙的一部分被钾离子占据。从这个现象可以理解为钾离子的有效半径rk=1.52埃(注:由于三角棱柱状6配位的代表值几乎不知道,所以一般用八面体6配位的值代替)和通过计算得到的空隙大小十分相近。而且,水分子也和空隙大小接近,所以也可以想象空隙的一部分被水分子填充。
结晶内部在图3中以虚线圆球所示的以外的位置上也存在几何学上等价的空隙,因为不能精密判定钾离子等实际占据的位置,所以在模拟图中容易看见的范围内表示空隙位置。而且,图3表示的是没有倾斜的正确的六方晶结构的模拟图,在以上钾离子等占据结晶内部的同时,还有以下现象发生,例如,a轴长度从图4左面所示的状态略微改变成图4右面的状态,成为所谓斜方晶;c轴的角度从图5左面的状态变为图5右面的状态,成为略偏离90℃的单斜晶。但是,对这一点进行严密的解释和说明是相当困难的。
4.1.5.对电化学现象的解析
为了掌握γ-羟基氧化钴的电化学现象,将高次钴氧化物x填充到焊接了导线的1cm×1cm的发泡镍多孔体基板中,然后挤压制成试样用电极,在以下的条件下,通过循环伏安测量法进行测定。
电解液:在浓度为31重量%的KOH水溶液中,按照40g/L的比例溶解LiOH·H2O后得到的溶液(20℃时的比重为1.31)
相对电极:氢包藏合金负极(预先和别的氢氧化镍电极组合,反复充放电,充分活化后得到的电极)
参照电极:Hg/HgO
扫描条件:从浸渍电位到+500mV扫描后,以1mV/秒的扫描速度从+700mV(氧化侧)开始到-700mV(还原侧)进行扫描。
周围温度:20℃
结果如图6所示,以a、b标示的是较大的充放电峰,表示相同氧化物具有较高的导电性。比600mV附近更高的电位区域中的阳极电流在试验用电极上伴有氧产生。从前述图2的线图考虑,图6中的阴极电流峰a对应于由γ-羟基氧化钴还原为α-氢氧化钴的反应(放电),阳极电流峰b则对应于其相反的氧化反应(充电)。这种γ/α系统充放电的电位和历来的β/β系统相比,降低了50-100mV。这与已知的γ-羟基氧化镍现象相同。
4.2.高次钴氧化物y
4.2.1.结晶结构、价数、组成、电化学现象的解析
本发明的高次钴氧化物y的X射线衍射图如图7所示。如记号(黑色圆点)所示,在衍射角2θ从10°-70°的范围内可以确认至少有9个峰。所以,根据JCPDS无机物质文献对对应于这些峰的位置的化合物进行定性分析后发现,它与Na0.6CoO2(文献编号为30-1181)的衍射图极为一致。Na0.6CoO2是C.Fouassier等在J.Solid State Chem.,6(1973)532-537中发表的化合物,因为它属于具有层状结构的模拟的六方晶(严密地说是斜方晶体),他们从钴-氧层的排列考虑,所示的Na0.6CoO2的结晶结构:钴-氧-氧-钴的排列ACCBAACBB和图3所示的相同。所以,高次钴氧化物y的基本结构具有图3所示的结构。
然后,利用与4.1.1.所示的相同的方法,测定高次钴氧化物y的平均价数、所含的碱性金属离子(此时为Na+)的量。其结果是,相同氧化物的钴平均价数为3.39,钠对应于钴的摩尔比约为0.31。从这些值可以判断出钴氧化物y不是前述的Na0.6CoO2。因为前述的Na0.6CoO2是由四氧化三钴(Co3O4)和过氧化钠(Na2O2)的混合物烧制而成的,所以可以理解为高次钴氧化物y是以β-氢氧化钴为反应出发材料,完全利用别的方法制得的。钴氧化物y的平均价数以及碱性金属离子的含量和4.1.1.所示的高次钴氧化物x的值极为相近。
为了掌握高次钴氧化物y的电化学现象,以与4.1.5.所示的相同的条件,利用循环伏安测量法进行解析。其结果如图8所示,它与图6所示的高次钴氧化物x几乎具有相同的充放电行为。这个试验的结果表明钴氧化物y和钴氧化物x具有同样的高导电性。与此同时,还表明在高次钴氧化物y和碱性电解液之间有可逆的质子转移,这表示在高次钴氧化物y中有质子存在。从这个观点看,钴氧化物y是“羟基氧化钴”的一种。
以上解析结果有力证明钴氧化物y与钴氧化物x相比,如果除去c轴方向的层间距离小这一点不同的地方,则两者具有基本相同的结晶结构、物理性质、电化学行为。即高次钴氧化物y也可以称为“γ-羟基氧化钴”。因此,本发明者们认为高次钴氧化物y也是γ-羟基氧化钴的一种,可以定义为Na型γ-羟基氧化钴。如果假定这种Na型γ-羟基氧化钴具有与图3相同的层状结构,在作为X射线衍射图的图7所示的9个峰中,对于比较强度大、能够基本正确地读出衍射角的6个面,与表2同样地汇总晶面距离和附加指数(作为六方晶时),结果如表3所示。
表3
|
hkl |
dobs(埃) |
I/IOobs |
|
003 |
5.57 |
100 |
|
006 |
2.78 |
10 |
|
101 |
2.42 |
19 |
|
102 |
2.35 |
32 |
|
105 |
1.97 |
20 |
|
110 |
1.41 |
13 |
4.2.2.关于结晶结构的考察
然后,对Na型γ-羟基氧化钴的结晶结构进行考察。已经在4.1.4.中由图3所示,γ-羟基氧化钴模拟地属于准六方晶系,是具有层状结构的钴氧化物,晶格沿c轴方向延伸,沿c轴方向可见的钴-氧-氧-钴的形式为ACCBAACBB的重复结构。所以,对于Na型γ-羟基氧化钴,以表3所示的晶面距离和4.1.4.中所用的有效离子半径为基础,根据几何学计算估计的三角棱柱状配置的6个氧离子或氢氧化物离子之间产生的空隙的半径约为1.15埃。这个值与钠的有效离子半径rNa=1.16埃(6配位)基本一致,空隙被钠离子占据。
图3表示的是没有倾斜的正确的六方晶结构的模拟图,完全如4.1.4.所述的,这个结构经a轴长和c轴角度的微妙变化,可以变为斜方晶单斜晶体的结构。对此详情虽然不明了,但可能是因为图1和图7的X射线衍射峰的强度比不同,即从表2和表3的I/IOobs的差异可以推测反映出以上情况。而且,从4.1.4.所示的计算结果和上述计算结果可以明显看出γ-羟基氧化钴的(003)面的晶面距离基本上受占据空隙的碱性阳离子的大小的强力支配。
5.γ-羟基氧化钴的物理性质和正极特性间的关系
如上所述,本发明的高次钴氧化物x以及高次钴氧化物y都属于γ-羟基氧化钴。以目前为止的考察结果为基础,总结其与β-羟基氧化钴的明显区别得到以下结论:模拟地属于准六方晶系,更严密地说是属于六方晶、斜方晶、单斜晶中的任何一种结晶系;是具有图3所示的层状结构的钴氧化物;与β-Co(OH)2相比其晶格沿c轴方向延伸(具体地说为5.5-7.0埃);而且钴的价数达到高于3.0的高次状态。由于γ-羟基氧化物是钴的价数高于3.0的主要原因,且微晶的钴层显现出电子传导性,所以认为其具有比β-羟基氧化钴更高的导电性。所以,以γ-羟基氧化钴为导电剂的碱性蓄电池用非烧结式正极中的氢氧化镍粒子的利用率得到了提高。
此外,γ-羟基氧化钴的电化学行为如图6或图8所示的,γ/α系统充放电的电位与历来的β/β充放电系统相比,降低了约50-100mγ。这种γ-羟基氧化钴的电化学行为与3.中所述的短路保存后的容量恢复率有密切的关系。即由于γ-羟基氧化钴的导电性较高,所以即使在短路保存时也能保持大多数氢氧化镍和基板框架之间的电流导通。γ-羟基氧化钴在其自身被氧化成超过3.0价的状态的同时,如前述的那样由于碱性电解液中的还原电位比β-羟基氧化钴低,所以不能容易地被还原到2.0价状态(电解液中溶解的状态)。所以,能够抑制历来的正极中短路保存时因为在基板附近集中进行还原反应而引起的对导电网络的破坏。这样以γ-羟基氧化钴为导电剂的碱性蓄电池用非烧结式正极和历来的正极相比,其短路保存后的容量恢复率更高。
如上所述,3.中所述的碱性蓄电池用非烧结式正极的特性和4.中阐明的γ-羟基氧化钴的各种特性有很明显的关联。
根据X射线衍射,可以基本观察出上述的高次钴氧化物x、y基本为单相。通过别的试验,在KOH存在下使用被氧化的γ-羟基氧化钴(这些是氧化处理时形成的碱性索状微小的水分含量不同的原因)作为导电剂,基本上可以得到相同的正极特性。γ-羟基氧化钴的X射线衍射结果如图9所示,具有作为一部分混合相的β-羟基氧化钴。而且,虽然在X射线衍射图中没有显示,但在KOH存在下被氧化的γ-羟基氧化钴中存在作为混合相的极少的Co3O4;在NaOH存在下被氧化的γ-羟基氧化钴(Na型γ-羟基氧化钴)中存在一部分作为混合相的β-羟基氧化钴;以及在Na型γ-羟基氧化钴中存在作为混合相的极少的Co3O4,都可以得到同样的试验结果。即如果以γ-羟基氧化钴为主要成分,存在一部分这样的不纯物质对正极特性的影响不大。
在关于γ-羟基氧化镍的文献中,有作为理想结构式的K(NiO2)3和与之略有差异的结构式如Ni0.75K0.25OOH等。γ-羟基氧化钴也具有同样的理想结构式:M(CoO2)3或Co0.75M0.25OOH(M为K或Na中的任何一种),M/Co的摩尔比的上限预计为0.33,这与4.1.1.和4.2.1.中所得的实测值相差不大。但是,很少能得到完全理想结构的氧化物。上述含有一部分不纯物的γ-羟基氧化钴的M/Co值,不论含有哪一种不纯物都约为0.20(实测值)。所以,相同比率在0.20-0.33的范围内,则可以认为正极特性没有发生变化。
此外,制备1.的试样时的氧化中使用了将氢氧化钴标准试样和碱性粒状试剂的粉末混合,一边送空气一边加热的方法,这种方法适合单独氧化微粒子氢氧化钴(粒径为0.2μm、BET比表面积为25m2/g,对溶液的润湿性很大)时使用。因为这种方法最易于制作适合于氧化的碱性绳索状物。所以,如以下的实施例2所示的,被覆了Co(OH)2的氢氧化镍粒子(粒径、比表面积不同,对溶液的润湿性也完全不同)的氧化处理方法以及细微的处理条件和上述不同。
实施例2
6.制造被覆了Co(OH)2的氢氧化镍粒子
按照以下的步骤制造作为原料的被覆了Co(OH)2的氢氧化镍粒子。首先,作为母离子的氢氧化镍粒子是使用与实施例1相同的方法得到的将钴以及锌固溶了的氢氧化镍粒子。
然后,将前述的固溶体氢氧化镍粒子投入硫酸钴水溶液中,再在其中慢慢地添加氢氧化钠水溶液,于35℃一边将其pH维持在12,一边继续搅拌,在固溶体粒子的表面析出氢氧化钴,得到被覆了Co(OH)2的氢氧化镍粒子。将氢氧化钴的被覆量调整到与被覆了Co(OH)2的氢氧化镍粒子的总重量的比例为5.0重量%。将所得的被覆了Co(OH)2的氢氧化镍粒子水洗后真空干燥。
用激光衍射型粒子分布计测定确认被覆了Co(OH)2的氢氧化镍粒子的平均粒径为10μm。而且,用电子扫描显微镜(SEM)以及电子透射显微镜进行观察,确认被覆层的氢氧化钴的厚度为0.1μm,完全覆盖了粒子。同一粒子的BET比表面积约为7m2/g。
7.对被覆了Co(OH)2的氢氧化镍粒子进行氧化处理
7.1.反应装置的说明
然后,对上述的被覆了Co(OH)2的氢氧化镍粒子进行氧化处理。首先,对处理时使用的装置进行详细地说明。
图10表示本实施例使用的氧化处理装置的大致构成的截面图。1表示用不锈钢制成的容器。容器1上方的开口部用不锈钢制成的盖子2密封。在容器1以及盖子2的壁面上都装有夹套,当热水或水蒸气等通过夹套内部时,可以对内壁面加热保温。容器1的底部装有扇动叶轮3,容器1的侧面在叶轮3的端部上方装有切碎叶轮4。利用这些叶轮混合搅拌装入容器中的粒子。在这些叶轮的轴部,如虚线所示设有空气入口5、6,可以向容器1内送入压缩空气。盖子2上设有排风口7,利用排风阀9的开启可将通过袋滤器8的空气排出容器外。在图中虽然没有表示出来,但袋滤器8还具备防止结露的加热功能以及防止堵塞的粒子散落的功能(脉冲气体方式)。在排风口7的附近装有温湿度计10,主要是用来测定被排出的空气的湿度以监控容器内粒子的湿润情况。盖子上有向容器内的滴加碱性水溶液的滴加管11贯通,装入液体容器12中的规定量的碱性水溶液可以通过滴加阀13的开启滴加到容器中。微波发生装置14通过将微波导入容器内的微波导管15与容器1连通,微波如图中的虚线标记所示照射,对容器内的粒子进行加热。而且,微波发生装置14为了将容器壁上设置的接触式温度计16(测定粒子的温度)的指示值保持在设定温度,可通过自动控制装置调整其输出功率。还有,在图中虽然没有表示,但为了防止以排风口为首的各部位装置内微波的泄漏,用金属网眼屏蔽,加以保护。17为取出处理后粒子用的排出槽,取出粒子时,打开隔板就与容器内部连通。
图11表示比较例的氧化处理装置(间歇式流动造粒装置)的大致构成。20表示圆筒状的装置筐体,在其下部圆锥状部分21的壁上设有夹套,在夹套中通入热水或水蒸气能够对内部进行加热。在筐体21的底部设有搅拌粒子的搅拌叶轮22,在叶轮22的下部设有开启了大量小孔的整流板23。送风机24通过配管26将经加热器25加热的空气供入筐体内。所供的加热空气经整流板23整流后使经叶轮22搅拌的粒子流动。筐体20的上方设有喷入碱性水溶液的喷雾喷嘴27。这个喷雾喷嘴27通过在喷入碱性水溶液时输送压缩空气的配管28以及输送碱性水溶液的配管29和储存碱性水溶液的装置30连通。筐体20的顶部通过配管31连接排风机32。为了防止粒子流动时微粒子排出筐体外部的袋式过滤器33还具备在图中没有表示出来的防止孔堵塞的粒子散落的功能(脉冲气体方式)。在筐体20的圆锥部分21上设有接触式温度计34,以监控流动着的粒子的温度。
7.2.加热氧化处理的顺序
实施例的活性物质
处理装置使用的是深江工业株式会社制造的“微波干燥机FMD”(商品名)。关于它的操作在图10中已经进行了详细的说明。
首先,在容器1以及盖子2的壁面上设置的夹套中通入110℃的水蒸气,保持容器内的温度。在其中投入6.0kg被覆了Co(OH)2的氢氧化镍粒子。然后盖紧盖子2,打开排风阀9,使扇动叶轮以及切碎叶轮4转动,混合搅拌粒子,通过滴加管11在容器内添加碱性水溶液。所用的碱性水溶液是浓度为45重量%的氢氧化钾水溶液(室温),滴加量为700g。接着关闭滴加阀13,继续混合3分钟,就完成了步骤1。
在步骤1中粒子基本转变为均一的绳索状的湿润粒子。然后,停止空气的供给,转动排风阀9使容器内基本保持密封的状态,通过微波导管15一边照射微波(频率为2450MHz)一边继续混合粒子,并对湿润粒子加热。湿润粒子在步骤1结束时已由来自容器壁的热量加热到70℃,再继续微波照射3分钟,直至达到设定温度(此时为110℃)。以上的粒子温度变化由容器内壁上设置的接触式温度计16监控。至此完成了步骤2。
接着,在开启排风阀9的同时,通过扇动叶轮轴部以及切碎叶轮轴部的空气入口5以及6向容器内部送入空气(温度为50℃,湿度为0.01kgH2O/kgdry-air),其间还继续混合湿润粒子,而且微波在照射的同时还控制温度并保持在接触式温度计16设定的110℃的指示值。在这种状态下处理20分钟后粒子将完全干燥,反应也随之结束。通过设置在排风口的温湿度计10监控对粒子的干燥状态进行判定。至此完成了步骤3。
所得的粒子不是凝集的粒子,而是均一的被覆层,被氧化成带有蓝色金属光泽的活性物质。从制品排出槽17取出这种粒子,为除去过剩的碱性组分,对其进行水洗以及干燥,最终得到正极活性物质。水洗、干燥时几乎观察不到粒子颜色的变化。将其作为活性物质粒子A。
此外,除了用浓度为45重量%的氢氧化钠水溶液代替氢氧化钾水溶液之外,其他一切操作都与上述相同,制得活性物质粒子B。这种粒子也呈现蓝色的金属光泽。
比较例的活性物质
另一方面,用比较例的装置对前述的被覆了Co(OH)2的氢氧化镍粒子进行氧化处理。所用的装置为Fuji Paudal株式会社制造的“New Marumerizer,ModelNQ”(商品名)。由图11对使用此本装置的氧化处理操作进行详细的说明。
首先,在筐体圆锥部分21的壁面上的夹套中通入80℃的热水,以保持筐体内的温度,在其中投入和实施例1所用相同的被覆了Co(OH)2的氢氧化镍粒子950g。然后,转动搅拌叶轮22,使加热器25、送风机24、以及排风机32进入工作状态,使筐体内产生上升的热气流。此时,通过整流板23的热风一边加热投入被覆了Co(OH)2的氢氧化镍粒子一边使其流动。在这种粒子处于流动状态下,利用喷雾喷嘴27喷入碱性水溶液。所用的碱性水溶液为25重量%的氢氧化钾水溶液,喷雾量为200g/10分钟。到喷雾结束这段时间内碱性水溶液浸透到粒子的表面,通过和热风作用使被覆层的氢氧化钴转变为高次氧化物。喷雾结束后再用约15分钟使粒子在热风中流动,使处理圆满结束。在这一连串的操作中,为使处理温度保持在设定值(80),利用接触式温度计34对粒子温度进行监控,调整热风条件等。然后,暂停装置,从筐体中取出粒子,为了除去过剩的碱,对粒子进行水洗以及干燥,就得到了正极活性物质。将此作为正极活性物质C。这种粒子呈黑色。
此外,除了用浓度为45重量%的氢氧化钠水溶液代替氢氧化钾水溶液之外,其他的操作都与上述相同,制得活性物质粒子D。这种活性物质呈暗茶色。
8.电池的制作以及对其进行的评估
为了确认通过上述方法制得的A-D的正极活性物质的各种特性,按照以下所述的顺序制作电池,并对其进行评估。
首先,分别在试样A-D中添加适量的纯水,配制成活性物质糊剂,将这些活性物质糊剂按规定量填充到多孔度为95%、厚度为1.3mm的发泡镍多孔体基板内。然后,将其放置在80℃的干燥器中烘干后,用滚筒挤压,并将厚度压延为0.7mm,按规定的大小将其切断,制成对应于4种活性物质A-D的镍正极。
将这4种镍正极与和实施例1相同的氢包藏合金负极、离析器、以及碱性电解液组合,制成尺寸为4/5A、公称容量为1600mAh的镍-氢蓄电池。按照实施例1中①所示的方法测定各电池的各种正极的利用率。然后用以下③中所示的方法对各正极在短路保存后的容量恢复率进行评估。
③短路保存后的容量恢复率
以1CmA的充电速度对进行了①的评估后的电池充电,时间为1.2小时,再以1CmA的放电速度放电,使电池的电压降低到1.0V,循环进行5次,测定第5次循环后的电池容量。然后用1Ω的电阻使电池短路,以此状态于45℃气氛中保存。保存时间以3天(短期)、2周(中期)、1个月(长期)作为3个不同的标准,分别使用不同的电池进行测定。再次以1CmA的充电速度对保存后的电池充电,时间为1.2小时,再以1CmA的放电速度放电,使电池的电压降低到1.0V,循环进行5次,测定第5次循环后的电池容量。根据以上测定结果,将短路保存后的电池容量值除以短路保存前的电池容量值,就得到各种正极活性物质在3个不同的保存期间短路保存后的容量恢复率。
对各种正极活性物质的评估结果如表4所示。
表4
|
正极活性 |
利用率 |
短路保存后的容量恢复率(%) |
|
物质的种类 |
(%) |
3天(短期) |
2周(中期) |
1个月(长 |
|
A |
105.8 |
99.7 |
99.2 |
98.4 |
|
B |
105.9 |
99.6 |
99.3 |
98.9 |
|
C |
98.2 |
99.2 |
93.3 |
90.6 |
|
D |
99.1 |
98.2 |
91.1 |
88.5 |
利用本发明的以微波加热为主的氧化处理方法得到的正极活性物质A、B和比较例的活性物质C、D相比,其利用率更高,其值大于100%。即氢氧化镍的充放电伴有多于1个电子的电子转移反应。又,对于短路保存后的容量恢复率,也是A、B优于C、D,特别是长期保存的情况,其差异更明显。这是因为使用了活性物质粒子A、B的正极的导电网络的集电性能要比C、D更胜一筹的缘故,同时也启示了在A-D的活性物质粒子间形成被覆层的钴氧化物的物理性质有很大的差异。
为了说明原因,对上述4种活性物质粒子以及与它们有关的钴氧化物进行以下所述的粉末解析。
9.粉末解析
9.1.活性物质粒子的解析(其1)
首先,为了明确试验时用肉眼能够观察到的活性物质粒子A-D颜色的差异,用分光测色计(Minolta公司制造的CM-3500d),根据漫反射法进行颜色的测定。测定时,在粉体试样测定用玻璃器皿(Minolta分散制造的CM-A128)中投入足够量的各种活性物质粒子,不用挤压固定,以粉末状态测定。即使挤压固定后光谱也几乎没有变化。各种条件设定如下:
照明·受光光学系统:d/8(扩散照明·8°方向受光),SCE(除去正反射光)
光源:D65
视野:10°
测定径:直径约为30mm(使用Minolta公司制造的玻璃器皿用TARGET MASKCM-A127)
利用校正玻璃(Minolta公司制造的CM-A129)除去玻璃器皿的反射光。
反射光谱的测定结果如图12所示。用肉眼就能够识别出带有蓝色光泽的本发明的活性物质粒子A以及B在全部区域的反射率较高,即在全部区域都较明亮,在波长450nm附近显示出极大反射率。所以显示为蓝色。用肉眼能够识别的黑色的比较例活性物质C在全部区域的反射率较低,用肉眼能够识别的暗茶色的比较例活性物质D在波长较大的红色区域的反射较强。用这种方法对肉眼能够感觉到的微弱颜色差别的粒子可以明确地进行定量。
呈现出以上颜色差别的原因可能有以下2个。
(1)反映了活性物质粒子的粒径、比表面积,钴氧化物层的被覆状态等、粒子形状的差异。
(2)反映了形成被覆层的钴氧化物的电子状态的差异。
为了证明(1)的可能性,对4种活性物质粒子A-D进行粒径以及粒度分布的测定(使用激光衍射式粒度分布计)、比表面积的测定(BET法)、利用电子扫描显微镜(SEM)观察粒子表面、利用电子透射显微镜(TEM)观察粒子的截面、以及利用电子探测微量分析器(EPMA)观察粒子表面·截面的Co分布状态。其结果是几乎观察不到A-D活性物质粒子在粒径、比表面积、钴氧化物层被覆状态等方面的差异,各种活性物质粒子中不论哪一种的平均粒径都是10μm,具有同一粒度分布,其表层完全被厚度约为0.1μm的钴氧化物覆盖。所以,活性物质粒子的颜色差异并不是因为(1)的原因。
对(2)的原因进行判断
活性物质粒子颜色的差异是因为(2)的原因造成的,即如果反映了形成粒子被覆层的钴氧化物的电子状态的差异,那么活性物质粒子的电导率的值也应该有差异。
为了证明以上的观点,将A-D的各种活性物质粒子制成片状,通过直流4端子法测定电导率。其结果是A和B的电导率几乎相等,顺序为A、B>C>D,呈蓝色的活性物质粒子A、B中任何一种的值都约为呈黑色的C的5倍,为呈暗茶色的D的10倍以上。所以,前述的粒子颜色的差异反映了形成被覆层的钴氧化物的电子状态的差异,如图12所示的呈蓝色的活性物质粒子(更具体讲,是可视区域的光谱反射率在3.5%以上,而且在波长450nm附近具有光谱反射率在4.0%以上的极大值的活性物质粒子)的钴氧化物的电子传导性极高。所以,正极的利用率大大提高。此外,使用活性物质A、B制成的电池经数次循环充放电后,电池分解,从正极分离出来的活性物质粒子(水洗、干燥后的物质)也呈蓝色,即确认了前述的测色结果。也就是说,电池内进行氢氧化镍的充放电反应时,形成被覆层的钴氧化物的电子状态几乎没有变化,保持了较高的电子传导性。
然后,为了明确前述形成活性物质粒子的被覆层的钴氧化物的构造,进行X射线衍射的测定。但是,前述的被覆层仅以约0.1μm这样极薄的厚度覆盖在粒子表面,在衍射图上实际只反映了作为母粒子的氢氧化镍,而不能充分观测出与形成被覆层的钴氧化物有关的衍射峰。
所以,将前面实施例1准备的氢氧化钴标准试样放置在氢氧化钾或氢氧化钠、水、空气(氧)共存的气氛中,利用各种条件使其氧化,对其进行X射线衍射的测定和利用分光测色计进行颜色的测定。
9.2.各种钴氧化物的解析和在此基础上的考察
①在KOH存在下氧化得到的γ-羟基氧化钴:制造方法和实施例1所述的相同,X射线衍射图为图1。呈现出蓝色光泽,利用分光测色计测得的反射光谱为图13。与实施例1一样,以下将其记为钴氧化物x。
②在NaOH存在下氧化得到的γ-羟基氧化钴:制造方法和实施例1所述的相同,X射线衍射图为图7。呈现出蓝色光泽,利用分光测色计测得的反射光谱为图14。与实施例1一样,以下将其记为钴氧化物y。
③CoO(OH)
(3-1)结晶性较高的物质,即CoHO2:是将氢氧化钴浸在30重量%的氢氧化钠水溶液中,然后于80℃进行15小时加热处理得到的氧化物。以下将其记为钴氧化物z。处理后经水洗·干燥测得的X射线衍射图为图15的上半部分。用肉眼观察出氧化物z的颜色为茶色,分光测色计测得的反射光谱如图16所示。利用实施例1所述的滴定法测得的钴价数为2.98。
(3-2)结晶性较低的物质,即β-CoOOH:是将氢氧化钴含浸在25重量%的氢氧化钠水溶液中,然后于80℃进行6小时加热处理得到的氧化物。以下将其记为钴氧化物u。处理后再经水洗·干燥测得的X射线衍射图为图15的下半部分。可以观察到本来应该只在20°(4.4埃)附近的(003)面的衍射峰经过一些位移出现在19°(4.6埃)附近,其衍射图形基本与氧化物z的相同。位移的原因现在还不十分清楚。用肉眼观察到的氧化物u的颜色为黑色,利用分光测色计测得的反射光谱由图16表示。利用实施例1所述的滴定法测得的钴价数为2.91。
④Co3O4:是将氢氧化钴含浸在30重量%的氢氧化钠水溶液中,然后于100℃进行6小时加热处理得到的氧化物。以下将其记为钴氧化物v。处理后经水洗·干燥测得的试样X射线衍射图为图17。Co3O4和一般的市售试剂相比,其结晶性很低。用肉眼观察到的氧化物v的颜色为黑色,利用分光测色计测得的反射光谱由图18表示。利用实施例1所述的滴定法测得的钴价数为2.62。此外,用氢氧化钾代替氢氧化钠也能够得到基本相同的结果。
将上述5种钴氧化物分别制成片状,用直流4端子法测定电导率,x和y的值基本相同,顺序为x、y>v>z。即γ-羟基氧化钴(氧化物x以及y)和其他物质相比具有较高的电子传导性。这是钴的价数超过3.0达到高次的主要原因,因为钴氧化物具有电子传导性。
然后,利用分光测色计观察γ-羟基氧化钴的反射光谱:考察图13和图14(蓝色的金属光泽)。这些光谱基本上是在粉末漫反射基础上得到的,其原理是图19所示的模式,经结晶表面反射的光a和透过结晶内部的光b合在一起测定得到。在可视区域具有吸光度较大的吸收带的过渡金属化合物中,利用漫反射法发现其吸收带的光非常强,被选择反射(即反射光a在化合物的吸收光谱附近),这种光已经被观察到了。典型的例子是经电荷转移过渡在绿色区域(530nm)具有强吸收带的过锰酸钾:KMnO4,它为水溶液时,可以看到透射光,显红紫色(绿色的补色),为细小结晶时,作为吸收带的绿色区域的光被选择反射,可看到带有光泽的绿色。在绿色区域产生漫反射光谱的反射率极大值。
在J.Electrochem.Soc.,136(1989)613-619中D.A.Corrigan等揭示了γ-羟基氧化镍在波长450nm(蓝色)的区域具有较强的吸收带,它是归属于金属-氧之间的电荷转移过渡的物质,而且和γ-羟基氧化镍中存在的4价镍有关。所以,假设利用漫反射法测定γ-羟基氧化镍的光谱,根据上述原理,在波长450nm的区域出现反射率的极大值,这就证明了4价镍的存在。
根据以上的考察结果,现在虽然不能将细小的部分也解释清楚,但本发明者们对γ-羟基氧化钴作出了与γ-羟基氧化镍同样的解释,即在450nm附近以金属-氧之间的电荷转移过渡为基础存在吸收带。这个吸收带因为吸光度较大,所以利用漫反射法得到的对应于450nm附近的光非常强,被选择反射,使同一氧化物带有蓝色的金属光泽。它作为图13、图14中标识的反射率的极大值被定量地表示出来。模仿前述的D.A.Corrigan等人的解释,这个结果也可以说反映了在γ-羟基氧化钴中存在4价钴。
将活性物质粒子A、B的反射光谱(图12中的A、B)和γ-羟基氧化钴的反射光谱(图13以及图14)中反射率的极大位置的类似性、任何一种都具有极高的电子传导性等实测结果组合在一起,可以认为形成本发明的活性物质粒子A、B的被覆层的钴氧化物是以γ-羟基氧化钴为主成分的。经过同样考察后还可以推测出形成前述比较例的活性物质粒子C、D的被覆层的钴氧化物为上述标识为z、u、v的价数在3.0以下的氧化物。
如上所述,形成被覆层的钴氧化物的氧化状态有差异是因为氧化处理方法的不同造成的。如前述的,被覆了Co(OH)2的氢氧化镍粒子的被覆层的氧化反应有2个步骤,不论是哪一个步骤,体系内的水和氧(空气)的状态都会使氧化反应的进行程度受很大影响。要使氧化反应进行完全,很重要的一点就是在表面具有碱性水溶液的条件下,利用一定的时间,使绳索状粒子在较高温度、多湿环境中完全干燥。
在比较例的制造方法(使用了间歇式流动造粒装置的方法)中,为使粒子流动化,应该随时送入大量的热风,使粒子流动化的原因是被碱性水溶液润湿的粒子和空气接触的面积较大,粒子表面的碱性水溶液干燥非常快的缘故。即在充分进行前述的步骤1以及步骤2反应之前,粒子表面的碱性水溶液枯竭的倾向很大。所以,虽然能够容易地抑制处理时伴有的粒子成块现象、装置内壁上粒子的附着,但是,要使氢氧化钴被覆层完全氧化成高次状态却很困难。
从以上的观点看,本发明的制造方法能够创造出以微波作为加热手段,使碱性水溶液迅速达到高温,必然适合于步骤1以及2反应的进行、在高温多湿下也能确保充分氧量的环境。将环境控制在上述条件下,就能够使氢氧化钴被覆层被完全氧化,得到价数高于3.0的高次γ-羟基氧化钴。
如上所述,利用本发明的制造方法,能够将被覆了Co(OH)2的氢氧化镍粒子的被覆层有效地转变为γ-羟基氧化钴。
9.3.活性物质粒子的解析(其2)
然后,使本发明的活性物质粒子A以及B溶解于酸,利用ICP发光分析测定活性物质中含有的碱性金属(钾或钠)的量。利用电子探测微量分析器(EPMA)观察活性物质粒子的截面,活性物质粒子A、B中,碱性金属只存在于被覆层部分,而不存在于内部的母粒子。前述的ICP发光分析的测定值反映了形成被覆层的钴氧化物中含有的碱性金属的量。以这点为基础,可以估计出形成活性物质粒子A、B的被覆层的钴氧化物中所含有的碱性金属的量,任何一种对应于钴的摩尔比都约为0.27。这些解析结果与前述的形成活性物质粒子A、B的被覆层的钴氧化物的主成分为γ-羟基氧化钴是不矛盾的。
接着,为了掌握本发明的活性物质粒子的电化学现象,将活性物质粒子A、分别填充到焊接了导线的1cm×1cm的发泡内多孔体基板中,挤压后制成试验用电极,在以下条件下,利用循环伏安测量法进行测定。
电解液:在浓度为31重量%的KOH水溶液中,按照40g/L的比例溶解LiOH·H2O后得到的溶液(20℃时的比重为1.31)
相对电极:氢包藏合金负极(事先和别的氢氧化镍电极组合并充放电,充分活化后得到的电极)
参照电极:Hg/HgO
扫描条件:从浸渍电位到+600mV扫描后,以3mV/秒的扫描速度从+600mV(氧化侧)开始到-800mV(还有侧)进行扫描。
周围温度:20℃
将2种活性物质粒子的测定结果合并在一起,用图20表示。处于比+400mV更高电位的阳极电流峰对应于作为母粒子的氢氧化镍的充电(与氧的产生同时进行),在+300mV附近的阴极电流峰对应于氢氧化镍的放电。所以,用a、b标识的峰对应于被覆层的钴氧化物的氧化还原峰。从图可以明显看出,本发明的活性物质粒子A中的钴氧化物和比较例的活性物质粒子C中的钴氧化物相比,其充放电电位降低了50-100mV。对活性物质粒子B进行同样的试验,显示出与活性物质粒子A相同的结果。这些现象与实施例1所示的图6以及图8的现象完全相同,从这一点也可以充分证明形成活性物质粒子A、B的被覆层的钴氧化物是以γ-羟基氧化钴为主成分的。
10.本发明的活性物质粒子的物理性质与正极特性间的关系
如上所述,形成本发明的活性物质粒子A、B的被覆层的钴氧化物是以γ-羟基氧化钴为主成分的。由于γ-羟基氧化钴是使钴的价数达到3.0以上等现象的主要原因,且显现出电子传导性,所以,具有比β-羟基氧化钴等氧化物更高的导电性。因此,使用了本发明的活性物质粒子A、B的碱性蓄电池用非烧结式正极比使用了比较例的活性物质粒子C、D的正极的利用率高。此外,使用了活性物质粒子A、B的正极的利用率比使用了实施例1的氧化物x、y的正极的利用率高是因为作为活性物质粒子A、B的原料为被覆了Co(OH)2的氢氧化镍粒子,显现出以下(1)、(2)两种效果的缘故。
(1)由于作为导电剂的钴氧化物的分散性较高,所以,离开正极基板框架位置的大多数氢氧化镍粒子的集电现象良好。
(2)氧化处理时使作为母粒子的氢氧化镍粒子和被覆层间的界面连接,使分散粒子的集电容易进行。
形成活性物质粒子A、B的被覆层的γ-羟基氧化钴的导电性较高,所以即使在短路保存时,大多数的氢氧化镍也能够较好地进行集电。而且,γ-羟基氧化钴在其自身被氧化成超过3.0的状态的同时,其充放电的电位比历来的钴氧化物降低了50-100mV,所以不容易被还原为2.0的状态(溶解于电解液的状态)。所以,历来的正极能够抑制短路保存时因为在基板附近集中进行还原反应而引起对导电网络的破坏。使用了活性物质粒子A、B的碱性蓄电池用非烧结式正极和历来的正极相比,其短路保存后的容量恢复率更高。
如上所述,8.中所述的碱性蓄电池用非烧结式正极的特性和9.中阐明的活性物质粒子A、B的各种特性有很明显的关联。
此外,在日本专利公开公报平8-148145号中记载了关于活性物质粒子的细孔分布的几篇报道。为了说明细孔分布与本发明的活性物质粒子的关系,利用上述实施例使用的活性物质粒子A-D的BET法进行细孔分布的测试,其结果如表5所示。
表5
|
正极活性物质的种类 |
制作条件等 |
直径在60埃以上的细孔空间的比例 |
|
A |
实施例中KOH水溶液共存下的氧化 |
18.5% |
|
B |
实施例中NaOH水溶液共存下的氧化 |
17.9% |
|
C |
比较例中KOH水溶液共存下的氧化 |
22.5% |
|
D |
比较例中KOH水溶液共存下的氧化 |
19.7% |
以前的公报中记载了存在于活性物质粒子中的全部细孔空间体积总和的20%以上被直径在60埃以上的细孔占据,这一点对利用率的提高相当重要。从表5可明显看出,本发明的活性物质粒子A、B基本上不符合以上记载的的情况。但是,即使勉强地制备细孔分布符合上述记载的试样(改变被覆了Co(OH)2的氢氧化镍粒子的合成条件、或其后的氧化处理条件中的任一个,能够制作成功),其利用率也不会明显上升。所以,本发明者们认为以前公报中记载的活性物质粒子的细孔分布与利用率并没有特别的关联。
此外,本实施例在制备被覆了Co(OH)2的氢氧化镍粒子时,使用了液相法形成被覆层,但此时的被覆条件等并不限定在以上所述的条件中,也可以使用将氢氧化镍粒子和氢氧化钴粉末混合,在机械混合时利用剪切力和冲击力,在粒子的表面覆盖上氢氧化钴的方法(机械混合法)来制备本发明的活性物质粒子。
如上所述,本发明提供了一种碱性蓄电池,这种碱性蓄电池由于γ-羟基氧化钴的价数高,且具有较高的导电性,所以在获得较高正极利用率的同时,在过度放电、短路状态中放置、长期保存、高温保存后,还能够维持高容量。