(54) Título: COMPOSTO QUÍMICO DERIVADO OU ANÁLOGO DE CAROTENÓIDE, COMPOSIÇÃO FARMACÊUTICA, E, USO DO COMPOSTO (51) IntCI.: C07C 403/24; C07D 207/16; C07D 307/58; C07D 265/30; C07F 9/117; (...).
(30) Prioridade Unionista: 03/07/2003 US 60/485,304; 05/05/2003 US 60/467,973; 22/05/2003 US 60/472,831; 28/05/2003 US 60/473,741; 29/07/2002 US 60/399,194.
(73) Titular(es): CARDAX PHARMACEUTICALS, INC..
(72) Inventor(es): SAMUEL FOURNIER LOCKWOOD; SEAN CMALLEY; DAVID G. WATUMULL; LAURA Μ. HIX; HENRY JACKSON; GEOFF NADOLSKI.
(86) Pedido PCT: PCT US2003023706 de 29/07/2003 (87) Publicação PCT: WO 2004/011423 de 05/02/2004 (85) Data do Início da Fase Nacional: 31/01/2005 (57) Resumo: COMPOSTO QUÍMICO, COMPOSIÇÃO FARMACÊUTICA, MÉTODOS PARA SINTETIZAR UM COMPOSTO QUÍMICO, PARA TRATAR UM DANO DE ISQUEMIA-REPERFUSÃO, UMA DOENÇA DO FÍGADO, ARRITMIA E CÉLULA(S) CANCEROSA(S) E PRÉ-CANCEROSA(S), PARA AUMENTAR A EXPRESSÃO DE CONEXINA 43 E UMA DOENÇA COM UMA COMPOSIÇÃO QUÍMICA COMPREENDENDO UM DERIVADO DE CAROTENÓIDE E PARA REDUZIR UMA PROTEÍNA CREATIVA. Método para inibir e/ou melhorar a ocorrência de doenças associadas com espécies de oxigênio reativas espécies de nitrogênio reativas, radicais e/ou não radicais em um indivíduo, assim um indivíduo é administrado com um análogo estrutural de carotenóide, seja sozinho seja em combinação com outro análogo de carotenóide, ou formulação de co-antioxidante. O análogo ou combinação de análogos é administrado de modo que o risco do indivíduo de experimentar doenças associadas com espécies de oxigênio reativas, espécies de nitrogênio reativas, radicais e/ou não radicais pode ser assim reduzido. O análogo ou combinação de análogos pode ser administrado a um indivíduo para a inibição e/ou melhora de dano de isquemia-reperfusão. O análogo ou combinação de análogos pode ser administrado a um indivíduo para a inibição e/ou melhora da doença do fígado. O análogo ou combinação de análogos pode ser (...).
“COMPOSTO QUÍMICO DERIVADO OU ANÁLOGO DE CAROTENÓIDE, COMPOSIÇÃO FARMACÊUTICA, E, USO DO COMPOSTO”
FUNDAMENTOS DA INVENÇÃO
1. Campo da Invenção
A invenção no geral diz respeito aos campos da química medicinal ou sintética. Mais especificamente, a invenção diz respeito à síntese e ao uso de análogos de carotenóide.
2. Descrição da Técnica relevante
A doença cardiovascular (CVD) e especificamente a doença da artéria coronária (CAD), permanecem a causa principal de morte nos Estados Unidos e no mundo. A CVD é uma causa principal de mortalidade e morbidez no mundo. Reduções de pequenas a moderadas no risco cardiovascular, que leva às visitas ao departamento de emergência e hospitalizações quanto às síndromes coronárias agudas diminuídas, podem produzir benefícios clínicos e de saúde pública substanciais.
A pesquisa extensiva com antioxidantes tem mostrado que eles são agentes terapêuticos eficazes na prevenção primária e secundária de doença cardiovascular. A CVD permanece a causa principal de morte para todas as raças nos U.S.; agora, aproximadamente 60 milhões de americanos têm alguma forma de CVD. A probabilidade de vida nos U.S. aumentaria em quase 7 anos se a CVD pudesse ser eliminada. O número absoluto de mortes devido à CVD tem caído desde 1996; entretanto, ela permanece a única causa maior de morte nos Estados Unidos, com uma carga anual total para serviços “segue-se a página 2”
Petição 870180067735, de 03/08/2018, pág. 10/15
médicos de mais do que 300 bilhões de dólares (incluindo ataque cardíaco e acidente vascular cerebral).
A isquemia é a falta de um fornecimento de sangue oxigenado adequado a um tecido particular. A isquemia forma a base de muitos estados de doença agudos ou crônicos incluindo, mas não limitado a:
• Infarto do miocárdio ou MI • Angina instável • Angina do peito estável • Reoclusão abrupta a seguir da angioplastia coronária transluminal percutânea (PTCA) • Acidente vascular cerebral trombótico (85% do número total de acidentes vasculares cerebrais) • Oclusão vascular embólica • Insuficiência vascular periférica • Transplante de órgão • Trombose de veia profunda ou DVT • Oclusão por sonda de demora
A isquemia também pode vir a ser um problema em procedimentos eletivos tais como: transplante de órgão programado; cirurgia de enxerto de desvio da artéria coronária programada (CABG); e angioplastia coronária transluminal percutânea programada (PTCA). Comum a cada um destes cenários é o fenômeno de dano de reperfusão: a produção de espécies de oxigênio reativo (ROS) na reintrodução de fluxo de sangue oxigenado a uma área anteriormente isquêmica, com o dano ao tecido adicional paradóxico subsequente. Em particular, o(s) uso(s) de terapia trombolítica no infarto do miocárdio agudo (AMI) e acidente vascular cerebral trombótico agudo - assim como a revascularização cirúrgica com PTCA - são tipicamente associados com a reperfusão do miocárdio e/ou cérebro isquêmicos. O resultado clínico é melhorado com a obtenção de desobstrução precoce depois da trombose aguda, entretanto, não sem custo (isto é, “dano de reperfusão”).
A terapia corrente permite a reperfusão com agentes farmacológicos, incluindo ativador de plasminogênio do tipo de tecido recombinante (r-TPA), Anistreplase (APSAC), estreptocinase e urocinase. Estudos recentes mostram o melhor resultado clínico depois que a AMI ocorre com reperfusão cirúrgica precoce. Entretanto, a reperfusão cirúrgica é disponível em apenas 15 a 20 por cento dos centros de saúde nos Estados r
Unidos e muito menos a nível mundial. E provável, portanto, que a reperfusão farmacológica permanecerá clinicamente relevante e importante num futuro próximo. A terapia trombolítica é mal sucedida na reperfusão de cerca de 20 % das artérias infartadas. Das artérias que são reperfundidas com sucesso, aproximadamente 15 % obstrui novamente de modo abrupto (dentro de 24 horas). As medidas de inflamação sistêmica (por exemplo, níveis de soro de proteína reativa C ou CRP) correlaciona-se fortemente com a reobstrução clínica nestes pacientes. A recuperação miocárdica parece ser máxima em uma “janela terapêutica” de 2 a 6 horas subsequente à ruptura de placa aguda e à trombose. No acidente vascular cerebral trombótico ou tromboembólico agudo, esta janela terapêutica é ainda mais estreita, no geral menor do que 3 horas após a trombose. O ativador de plasminogênio do tipo de tecido recombinante administrado dentro de 3 horas do acidente vascular cerebral isquêmico melhora significantemente o resultado clínico, mas aumenta o risco de hemorragia.
Durante um período de isquemia, muitas células passam por mudanças bioquímicas e patológicas associadas com a anoxia mas permanecem potencialmente viáveis. Estas células potencialmente viáveis são portanto o “campo de batalha” no período de reperfusão. A isquemia cria mudanças no tecido afetado, com o resultado final potencial de faixa de contração e/ou necrose de coagulação de miocárdio em risco. As mudanças patológicas no miocárdio isquêmico incluem, mas não são limitados a:
Produção de radical livre e ROS
Perda de ATP e ressíntese de ATP defeituosa
Perda do fosfato da creatina
Perda do potássio extracelular
Perda da capacidade de gerar tensão ativa do miocárdio
Intumescimento celular
Acidose
Perda de homeostase iônica • Desorganização estrutural • Instabilidade elétrica e arritmogênese • Peroxidação de membrana lipídica • Glutationa e outro esgotamento de antioxidante endógeno/exógeno (incluindo vitaminas C e E e carotenóides)
O salvamento do miocárdio isquêmico que não atingiu irreversivelmente o patamar da necrose é o foco de intervenção no dano de reperfusão.
As junções de intervalo são um tipo único de junção intercelular encontrado na maioria dos tipos de célula animal. Elas formam canais aquosos que interconectam os citoplasmas de células adjacentes e permite a troca intercelular direta de componentes citoplásmicos pequenos (menores do que aproximadamente 1 quiloDalton). As junções de intervalo são criadas através do espaço celular interveniente pelo encurtamento de dois hemicanais (“conéxons”) contribuído por cada célula adjacente. Cada hemicanal é um oligômero de seis moléculas de conexina.
A conexina 43 foi o segundo gene da conexina descoberto e ela codifica uma das conexinas mais amplamente expressadas em linhagens de célula estabelecidas e tecidos. As junções de intervalo formadas pela conexina 43 foram implicadas no desenvolvimento, função cardíaca e controle do crescimento.
Uma manifestação comum de CVD é a arritmia cardíaca. A arritmia cardíaca é no geral, considerada um distúrbio da atividade elétrica do coração que se manifesta como uma anormalidade na freqüência cardíaca ou no ritmo cardíaco. Pacientes com uma arritmia cardíaca pode experimentar uma ampla variedade de sintomas que variam de palpitações até desmaio (“síncope”).
A conexina principal no sistema cardiovascular é a conexina 43. A coordenação da junção de intervalo de respostas celulares entre as células da parede vascular, em particular das células endoteliais, é considerado ser crítico para a modulação local do tônus vasomotor e para a manutenção da homeostase circulatória. Controlar a super regulagem da conexina 43 também pode ajudar na manutenção da estabilidade elétrica no tecido cardíaco. Manter a estabilidade elétrica no tecido cardíaco pode beneficiar a saúde de centenas de milhares de pessoas por ano com algum tipo de doença cardiovascular [por exemplo, doença cardíaca isquêmica (IHD) e arritmia] e pode prevenir a ocorrência de morte cardíaca súbita em pacientes com alto risco para arritmia.
O câncer é no geral considerado ser caracterizado pelo crescimento descontrolado, anormal de células. A conexina 43, como anteriormente mencionada, também está associada com o controle do crescimento celular. O controle do crescimento pela conexina 43 é provável devido à associação da conexina 43 com a comunicação de junção de intervalo. A manutenção, restauração ou aumentos na comunicação funcional da junção de intervalo inibe a proliferação de células transformadas. Portanto, a super regulagem e/ou controle da disponibilidade da conexina 43 pode inibir potencialmente e/ou melhorar a disseminação de células cancerosas.
O dano hepático crônico, independente da etiologia, pode levar a um espectro progressivo de patologia da inflamação aguda e crônica, para estágios precoces de fibrose e finalmente para cirrose e doença hepática de
estágio terminal (ESRD). Uma cascata de eventos inflamatórios secundários ao dano inicial, incluindo a liberação de citocinas e a formação de espécies de oxigênio reativo (ROS), ativa as células estreladas hepáticas (HSC). As HSC produzem componentes da matriz extracelular (ECM), incluindo colágeno e são críticos no processo que gera a fibrose hepática.
A doença hepática de estágio terminal [manifestada como cirrose ou carcinoma hepatocelular (HCC)] é a oitava causa principal de morte relacionada com doença nos Estados Unidos. A inflamação crônica no fígado que resulta da infecção viral, abuso do álcool, toxicidade induzida por medicamento, sobrecarga de ferro e cobre e muitos outros fatores podem iniciar a fibrose hepática. Subprodutos do dano hepatocelular ativam as células de Kupffer, que depois liberam várias citocinas, ROS (incluindo em particular o ânion de superóxido) e outros fatores parácrinos e autócrinos que por sua vez atuam nas células estreladas hepáticas (HSC). Acredita-se agora que a célula central na cascata fíbrogenética seja a HSC, o tipo de célula responsável pela produção de ECM. Evidência in vitro demonstra que a ROS pode induzir células HSC. Níveis elevados de marcadores indiretos de estresse oxidativo (por exemplo, espécies reativas em ácido tiobarbitúrico ou TBARS) são observadas em todos os pacientes com doença hepática crônica. Além disso, os níveis de glutationa, glutationa peroxidase, superóxido dismutase, carotenóides e α-tocoferol (vitamina E) são significantemente mais baixos em pacientes com doença hepática crônica. Fornecer estes antioxidantes endógenos e/ou exógenos reverte muitos dos sinais de doença hepática crônica, incluindo ambos os marcadores substitutos para o processo de doença, assim como medições diretas da fibrose hepática. Portanto, eles são agentes potentes prováveis para a intervenção terapêutica na doença hepática.
SUMÁRIO
Em algumas formas de realização, a administração de análogos

estruturais de carotenóides pode inibir e/ou melhorar a ocorrência de doenças em pacientes. As doenças que podem ser tratadas com análogos estruturais de carotenóides podem incluir qualquer doença que envolve a produção de espécies de oxigênio reativo e/ou outras espécies radicais (por exemplo 5 oxigênio singleto, uma espécie de oxigênio reativo mas não um radical). Em algumas formas de realização, análogos solúveis em água de carotenóides podem ser usados para tratar uma doença que envolva a produção de espécies de oxigênio reativo. A oxidação de DNA, proteínas e lipídeos pela espécie de oxigênio reativo e outro radical e espécies que não radicais foi implicada em 10 um hospedeiro de doenças humanas. Os radicais podem ser a causa primária para as seguintes condições, podem tomar o corpo mais suscetível a outros fatores que iniciam doença, podem inibir as defesas endógenas e processos de reparo e/ou podem realçar a progressão de doença(s) incipiente(s). A administração de análogos estruturais de carotenóides por uma pessoa 15 habilitada na técnica - incluindo a consideração das farmacocinéticas e farmacodinâmicas da liberação de medicamentos terapêuticos - é esperado inibir e/ou melhorar as ditas condições de doença. Na primeira categoria estão aquelas condições de doença em que um único órgão é primeiro afetado e para o qual evidência existe de que os radicais e/ou não radicais estão 20 envolvidos na patologia da doença. Estes exemplos não devem ser considerados como limitantes e condições de doença adicionais serão óbvias àqueles habilitados na técnica.
• Cabeça, Olhos, Ouvidos, Nariz e Garganta: degeneração macular relacionada com a idade (ARMD), descolamento retinal, doença 25 retinal hipertensiva, uveíte, coroidite, vitreíte, hemorragia ocular, dano retinal degenerativo, cataratogênese e cataratas, retinopatia de prematuridade, doença de Meuniere, ototoxicidade induzida por medicamento (incluindo aminoglicosídeo e toxicidade da furosemida), otite infecciosa e idiopática, otite média, sinusite infecciosa e alérgica, câncer da cabeça e pescoço;
• Sistema Nervoso Central (cérebro e medula espinal): demência senil (incluindo a demência de Alzheimer), doença de NeumanPick, reações de neurotoxina, efeitos de oxigênio hiperbárico, doença de Parkinson, trauma da medula cerebral e espinal, dano cerebrovascular hipertensivo, acidente vascular cerebral (tromboembólico, trombótico e hemorrágico), encefalite e meningite infecciosa, encefalomielite alérgica e outras doenças desmielinantes, esclerose lateral amiotrófica (ALS), esclerose múltipla, lipofuscinose ceróide neuronal, síndrome de ataxia-telangiectasia, sobrecarga de alumínio, ferro e outro(s) metal(is) pesado(s), 0 carcinoma/malignidade cerebral primária e metástases cerebrais;
• Cardiovascular: arteriosclerose, aterosclerose, doença vascular periférica, infartação miocárdica, angina estável crônica, angina instável, dano cirúrgico idiopático (durante CABC3, PTCA), doença cardíaca inflamatória [como medida e influenciada pela proteína reativa C (CRP) e mieloperoxidase (MPO)], oxidação de lipoproteína de alta densidade (oxLDL), cardiomiopatias, arritmia cardíaca (isquêmico e induzida após a infartação miocárdica), insuficiência cardíaca congestiva (CHF), toxicidade medicamentosa (incluindo adriamicina e doxorrubicina), doença de Keshan (deficiência de selênio), tripanossomíase, cardiomiopatia, estase e dano 20 venosos (incluindo trombose de veia profunda ou DVT), tromboflebite;
• Pulmonar: asma, doença das vias aéreas reativa, doença pulmonar obstrutiva crônica (COPD ou enfisema), hiperoxia, efeitos de oxigênio hiperbárico, efeitos da inalação da fumaça de cigarro, efeitos de poluentes oxidantes ambientais, síndrome da angústia respiratória aguda (ARDS), displasia broncopulmonar, pneumoconiose de poeira mineral, toxicidade pela adriamicina, toxicidade pela bleomicina, toxicidades pelo paraquat e outros pesticidas, pneumonite química, fibrose intersticial pulmonar idiopática, pneumonia infecciosa (incluindo fungica), sarcoidose, asbestose, câncer pulmonar (célula pequena e grande), infecção pelo antrax,
exposição à toxina antrax;
• Renal: doença renal hipertensiva, doença renal de estágio final, doença renal diabética, glomerulonefrite infecciosa, síndrome neffótica, glomerulonefiite alérgica, reações de hipersensibilidade do tipo I a IV, rejeição a aloenxerto renal, doença da membrana de base antiglomerular neffítica, neírotoxicidade por metal pesado, nefrotoxicidade induzida por medicamento (incluindo aminoglicosídeo, furosemida e anti-inflamatório não esteroidal), rabdomiólise, carcinoma renal;
• Hepática: dano hepático pelo tetracloreto de carbono, dano 10 hepático por endotoxina e lipopolissacarídeo, infecção viral crônica (incluindo a infecção da hepatite), hepatite infecciosa (etiologia não viral), hemacromatose, doença de Wilson, dose excessiva de acetaminofeno, insuficiência cardíaca congestiva com congestão hepática, cirrose (incluindo as de etiologia alcoólica, viral e idiopática), carcinoma hepatocelular, 15 metastases hepática;
• Gastrointestinal: doença do intestino inflamatório (incluindo doença de Crohn, colite ulcerative e síndrome do intestino irritável), carcinoma do cólon, polipose, diverticulite infecciosa, megacólon tóxico, gastrite (incluindo a infecção pelo Helicobacter pylori), carcinoma gástrico, esofagite (incluindo o esôfago de Barrett), doença do refluxo gastroesofágico (GERD), doença de Whipple, doença de cálculo biliar, pancreatite, abetalipoproteinemia, gastroenterite infecciosa, desinteria, toxicidade de antiinflamatório não esteroidal induzida por medicamento;
• Hematopoiética/Reumatológica: envenenamento por Pb 25 (chumbo), supressão da medula óssea induzida por medicamento, fotooxidação pela protoporfirina, linfoma, leucemia, porfiria(s), infecção parasítica (incluindo a malária), anemia de célula falciforme, talassemia, favismo, anemia perniciosa, anemia de Fanconi, anemia pós-infecciosa, púrpura trombocitopênica idiopática, síndrome da deficiência autoimune (AIDS);
• Genitourinária: prostatite infecciosa, carcinoma prostático, hipertrofia prostática benigna (BPH), uretrite, orquite, torsão testicular, cervicite, carcinoma cervical, carcinoma ovariano, carcinoma uterino, vaginite, vaginismo;
• Musculoesqueletal: osteoartrite, artrite reumatóide,
- tendinite, distrofia muscular, doença de disco degenerativo, doença da junta degenerativa, dano ao músculo esqueletal induzido por exercício, síndrome do * túnel carpal, síndrome de Guillan-Barre, doença de Paget do osso, espondilite — 10 aquilosante, formação óssea heterotópica; e • Tegumentar: dano pela radiação solar (incluindo queimadura do sol), dano térmico, dermatite química e de contato (incluindo dermatite de Rhus), psoríase, síndrome de Bloom, leucoplasia (particularmente oral), dermatite infecciosa, sarcoma de Kaposi.
Na segunda categoria estão as condições de órgão múltiplo cuja patologia tem sido ligada de maneira convincente de algum modo ao dano de radical e que não de radical: envelhecimento, incluindo a deficiência imune relacionada com a idade e distúrbios de envelhecimento prematuro, câncer, doença cardiovascular, doença cerebrovascular, dano por radiação, dano mediado pelo álcool (incluindo a síndrome de Wemicke-Korsakoff), dano de isquemia-reperfusão, doença inflamatória e auto-imune, toxicidade medicamentosa, doença amilóide, síndromes de sobrecarga (ferro, cobre, etc.), insuficiência de órgão de sistema múltiplo e endotoxemia/sépse.
As doenças, que podem ser tratadas com análogos de carotenóide estrutural, podem incluir, mas não são limitados à inflamação cardiovascular, infecção de hepatite C, câncer (carcinoma hepatocelular e prostático), degeneração macular, artrite reumatóide, acidente vascular cerebral, doença de Alzheimer e/ou osteoartrite. Em uma forma de realização, a administração de análogos solúveis em água de carotenóides a um paciente

pode inibir e/ou melhorar a ocorrência de dano de reperfusão em pacientes. Em algumas formas de realização, os análogos solúveis em água e outros análogos de carotenóide estrutural podem ser administrados a um paciente sozinhos ou em combinação com outros análogos de carotenóide estrutural. A ocorrência de dano de reperfusão em um paciente humano que está sofrendo ou sofreu ou está predisposto a sofrer infartação miocárdica, acidente vascular cerebral, doença vascular periférica, oclusão venosa ou arterial, transplante de órgão, cirurgia de enxerto de desvio da artéria coronária, angioplastia coronária transluminal percutânea e parada cardiovascular e/ou morte podem ser inibidos ou melhorados pela administração de quantidades terapêuticas de análogos solúveis em água e/ou outros análogos de carotenóide estrutural ao paciente.
Os análogos de carotenóide estrutural “solúveis em água” são aqueles análogos que podem ser formulados em solução aquosa, sozinhos ou com excipientes. Os análogos de carotenóide solúveis em água podem incluir aqueles compostos e derivados sintéticos que formam auto-montagens moleculares e podem ser mais apropriadamente chamados de análogos de carotenóide “dispersáveis em água”. Os análogos de carotenóide solúveis em água e/ou “dispersáveis em água” podem ser a(s) forma(s) de realização preferida(s) em alguns aspectos da corrente invenção.
Em uma forma de realização, a administração de análogos solúveis em água de carotenóides a um paciente pode inibir e/ou melhorar alguns tipos de doença cardiovascular associada com a arritmia cardíaca. Em algumas formas de realização, análogos solúveis em água de carotenóides podem ser administrados a um paciente sozinho ou em combinação com outros análogos de carotenóide. Os análogos de carotenóide podem ajudar na manutenção da estabilidade elétrica no tecido cardíaco. A ajuda na manutenção da estabilidade elétrica no tecido cardíaco pode inibir e/ou melhorar alguns tipos de doença cardiovascular, incluindo em particular
morte cardíaca súbita atribuível à arritmia cardíaca letal.
Em uma forma de realização, a administração de análogos solúveis em água de carotenóides a um paciente pode inibir e/ou melhorar a ocorrência da doença hepática no paciente. Em algumas formas de realização, análogos solúveis em água de carotenóides podem ser administrados a um paciente sozinho ou em combinação com outros análogos de carotenóide. A doença hepática pode ser uma doença hepática crônica tais como, por exemplo, infecção de hepatite C.
Em uma forma de realização, a administração de análogos solúveis em água de carotenóides a um paciente pode inibir e/ou melhorar a proliferação e propagação de célula(s) iniciada(s), transformada(s) e/ou cancerosa(s). Em algumas formas de realização, análogos solúveis em água de carotenóides podem ser administrados a um paciente sozinho ou em combinação com outros análogos de carotenóide. Os análogos de carotenóide podem inibir a taxa de proliferação de células iniciadas com carcinógeno. Os análogos de carotenóide podem aumentar a expressão de conexina 43. O aumento da expressão da conexina 43 pode aumentar, manter ou restaurar a comunicação intercelular de junção de intervalo e assim inibir o crescimento de células iniciadas por carcinógeno.
As formas de realização podem ser ainda direcionadas às composições farmacêuticas que compreendem combinações de análogos de carotenóide estrutural aos ditos pacientes. A composição de um análogo de carotenóide estrutural injetável de astaxantina pode ser particularmente útil nos métodos terapêuticos aqui descritos. Já em uma outra forma de realização, um análogo estrutural de astaxantina injetável é administrado com outros análogos estruturais de astaxantina e/ou outros carotenóides análogos estruturais ou na formulação com outros antioxidantes e/ou excipientes que promovam o propósito pretendido. Em algumas formas de realização, um ou mais dos análogos estruturais de astaxantina são solúveis em água.
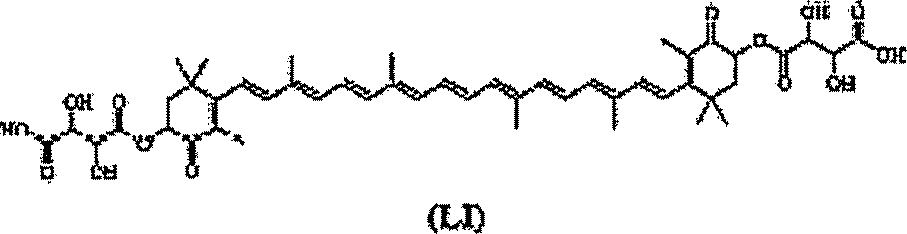
Em uma forma de realização, um composto químico incluindo um carotenóide pode ter a estrutura geral (I):
R3 R3 R3 R3 R3 R3 R3 R3 R3
R3 R3 R3 R3 R3 R® R3 R3 R3 (1).
Cada R3 pode ser independentemente hidrogênio ou metila. R1 e R2 podem ser independentemente H, um alceno cíclico com um ou mais
- 5 substituintes ou um anel cíclico incluindo um ou mais substituintes. Em algumas formas de realização, os substituintes podem ser pelo menos parcialmente hidrofílicos. Estes derivados de carotenóide podem ser usados em uma composição farmacêutica. Em uma forma de realização, uma composição farmacêutica que inclua análogos estruturais de carotenóide tendo 10 a estrutura geral (I) podem ser usados para tratar o dano de reperfusão.
Como aqui usado, os termos “derivado do sal dissódico de astaxantina dissuccinato”, “dAST”, “Cardax”, “Cardax®”, “rac” e “derivado de dissuccinato de astaxantina (ADD)” representa nomenclatura variável para o uso do derivado dissódico de astaxantina dissuccinato em vários 15 estereoisômeros e formulações aquosas e representam presentemente formas de realização preferidas mas não obstante ilustrativas para o uso pretendido deste análogo de carotenóide estrutural. O derivado de astaxantina dissuccinato diácido (astaCOOH) é a forma protonada do derivado utilizado para os estudos de fotólise cintilante para a comparação direta com 20 astaxantina não esterificada, “racêmica” (isto é, a mistura de estereoisômeros). “Cardax-C” é o derivado do sal dissódico de dissuccinato de di-vitamina C (derivado XXIII) utilizado nos experimentos de remoção de ânion superóxido ensaiado pela produção de imagem de ressonância paramagnética de elétron (EPR).
DESCRIÇÃO RESUMIDA DOS DESENHOS
A descrição resumida acima assim como outros objetivos, características e vantagens dos métodos e aparelhos da presente invenção será
mais completamente avaliada por referência à seguinte descrição detalhada das formas de realização presentemente preferidas mas não obstante ilustrativas de acordo com a presente invenção quando consideradas em conjunção com os desenhos anexos.
A FIG. 1 é uma representação gráfica de diversas estruturas de carotenóide “precursoras” como encontradas na natureza;
A FIG. 2 representa um efeito do derivado do sal dissódico de astaxantina dissuccinato sobre o ânion de superóxido de espécie de oxigênio reativo quando monitorado usando a produção de imagem de ressonância paramagnética de elétron (EPR);
A FIG. 3 representa um efeito de um derivado do sal dissódico de astaxantina dissuccinato/solução de vitamina C livre no ânion de superóxido na espécie de oxigênio reativo como monitorado usando a produção de imagem de ressonância paramagnética de elétron (EPR);
A FIG. 4 descreve uma representação gráfica de uma redução relativa do tamanho do infarto em ratos Sprague-Dawley machos com pré tratamento usando uma formulação intravenosa de derivado do sal dissódico de astaxantina dissuccinato (Cardax);
A FIG. 5 representa a estrutura química do derivado éster do sal dissódico de dissuccinato todo-trans (todo-E) de meso-astaxantina (3R,3’S- ou 3S,3’R-diidróxi-p,3-caroteno-454’-diona; dAST) sintetizado para o estudo corrente (mostrado como o bolanfífilo dianiônico todo-E);
A FIG. 6 representa o espectro de absorção ultravioleta-visível de dAST em etanol a 25° C (comprimento de célula 1 cm, c = 1,05 x 10-5 M). Os coeficientes de absorção molar são mostrados em parênteses. A segunda curva derivada do espectro de absorção indica a posição exata de picos na região próxima do UV e a estrutura fina vibracional oculta da faixa principal;
A FIG. 7 representa o espectro de absorção de dAST em tampão de Ringer (pH 7,4, comprimento de célula 1 cm, c = 1,85 x 10-5 M, t
= 37° C). Os coeficientes de absorção molar são indicados;
A FIG. 8 representa o espectro CD e UV/Vis induzido obtido pela titulação de albumina sérica bovina (HSA) com dAST em solução tampão de Ringer (pH 7,4) em relações IR baixas. A concentração de HSA foi 5 1,6 x 10-4 M e o ligando foi adicionado como alíquotas de solução de estoque de DMSO (comprimento de célula 1 cm, t = 37° C). As curvas medidas em valores L/P diferentes são mostrados. Inserções: coeficientes de absorção dicróica circular molar (Δε em M-^cm-1) e os coeficientes de absorção molar (ε em M-^cm-1) de CD induzido CD e faixas de absorção calculadas com base LH 0 na concentração meso-carotenóide total na solução;
A FIG. 9 representa o espectro CD e UV/Vis induzido obtido pela titulação de HSA com dAST em solução tampão de Ringer (pH 7,4) acima da relação L/P de 1. A concentração de HSA foi de 2,3 x 10-4 M e o ligando foi adicionado como alíquotas de solução de estoque de DMSO 15 (comprimento de célula 1 cm, t = 37° C). As curvas medidas em valores L/P de 1,2, 2,0, 2,9, 4,1, 5,7 e 7,4 são mostrados. As intensidades de CD aumentam em paralelo com a concentração de ligando;
A FIG. 10 representa o espectro CD e UV/Vis induzido obtido pela titulação de HSA com dAST em solução de tampão de fosfato 0,1 M pH 20 7,4 acima da relação L/P de 1. A concentração de HSA foi de 2,2 x 10-4 M e o ligando foi adicionado como alíquotas da solução de estoque de DMSO (comprimento de célula 1 cm, t = 37° C). As curvas medidas em valores L/P de 1,2, 2,0, 2,9, 4,1, 5,7, 9,0, 10,6 e 13,1 são mostrados. As intensidades de CD aumentam em paralelo com a concentração de ligando;
A FIG. 11 representa a ilustração de arranjos quirais da esquerda para a direita de duas moléculas meso-carotenóides para as quais as interações excitônicas produzem efeitos de Cotton de positivo de comprimento de onda longo e negativo de comprimento de onda curto no espectro CD. As moléculas de cor cinza se situam atrás do plano do papel;
A FIG. 12 representa (figura superior): extinção da fluorescência de HSA pela medida dAST em solução de tampão de fosfato 0,1 M pH 7,4 a 37° C. As concentrações inicial e final de HSA e o ligando foram variadas entre 4,2 x 10-6 M - 4,0 x 10-6 M e 1,3 x 10-6 M - 1,4 x 10-5 M, respectivamente. As relações L/P são mencionadas nas curvas. (Figura inferior): efeito de DMSO sozinho sobre a fluorescência intrínseca de HSA. Condições experimentais são como no texto;
A FIG. 13 representa a estrutura cristalográfica de raio X de HSA isento de ácido graxo. Subdomínios e os dois sítios de ligação de medicamento primários de HSA são indicados. As barras pontilhadas representam dimensões espaciais da fenda de interdomínio e o asterisco indica a posição de Trp214. A distância inter-atômica entre os átomos de carbono quiral 3 e 3’ da molécula dAST é de 28 Â;
A FIG. 14 representa que a mistura estatística de estereoisômeros do derivado do sal dissódico de astaxantina dissuccinato (“rac” nas Legendas da Figura) induz a comunicação de junção de intervalo funcional em células de fibroblasto embriônico de murino (10T1/2). As culturas confluentes foram tratadas durante 4 dias como descrito no texto, depois ensaiado quanto a capacidade para transferir o corante fluorescente Amarelo de Lúcifer. As setas indicam a célula injetada com Amarelo de Lúcifer;
A FIG. 15 A representa a expressão da proteína da conexina 43 em células tratadas com a mistura de estereoisômeros dos derivados do sal dissódico de astaxantina dissuccinato como avaliado pela análise de Western blot quantitativa. Acredita-se que as faixas superiores sejam para representar as formas fosforiladas da proteína montada nas junções de intervalo; faixas inferiores proteína não montada (Saez, 1998). Linha 1: 1:2 etanol (EtOH)/ H2O (solvente apenas controle negativo); Linha 2: TTNPB, um retinóide sintético, em acetona a 10-8 M (controle positivo); Linha 3: Acetato de retinila

em acetona a 10-5 M (controle positivo); Linha 4: Mistura estatística (“rac”) de estereoisômeros do derivado do sal dissódico de astaxantina dissuccinato a IO-5 M liberado em uma formulação 1:2 de EtOH/ H2O; Linha 5: derivado 3R,3’R do sal dissódico de astaxantina dissuccinato a 10-5 M liberado em uma formulação 1:2 de EtOH/ H2O; Linha 6: derivado 3S,3’S do sal dissódico de astaxantina dissuccinato a 10-5 M liberado em uma formulação 1:2 de EtOH/ H2O; e Linha 7: Meso derivado do sal dissódico de astaxantina dissuccinato a 10-5 M liberado em uma formulação 1:2 de EtOH/ H2O;
A FIG. 15B representa uma imunomancha tingida com Azul
L10 de Coomassie para demonstrar a carga de proteína igual de todas as faixas. Isto confirma que as diferenças na imunorrotulação não são um artefato devido à variabilidade na proteína total carregada e/ou transferida para a membrana;
A FIG. 15C representa a análise digital de níveis de indução relativos de expressão da proteína da conexina 43 pelo(s) derivado(s) do sal dissódico de astaxantina dissuccinato versus os controles positivo e tratados apenas com solvente. As linhas como na FIG. 15A. A indução de dobra é normalizada para controlar os níveis de expressão Cx43 nos controles negativos tratados com EtOH/H2O 1:2 ajustados a uma unidade arbitrária =
1,0;
A FIG. 15D representa a curva de resposta de dose da expressão da proteína Cx43 em células de fibroblasto embriônico de murino (10T 1/2) tratadas com a mistura estatística de estereoisômeros dos derivados do sal dissódico de astaxantina dissuccinato como avaliado pela análise de 25 Western blot quantitativa. Acredita-se que as faixas superiores representem as formas fosforiladas da proteína montada nas junções de intervalo; as faixas inferiores a proteína não montada. Linha 1: EtOH/H2O 1:2 (controle negativo apenas de solvente). Linha 2: TTNPB em acetona a 10-8 M (controle positivo). Linha 3: derivado do sal dissódico de astaxantina dissuccinato (“rac”) a 10-5 M liberado em uma formulação 1:2 de EtOH/ H2O. Linha 4: derivado do sal dissódico de astaxantina dissuccinato (“rac”) a 5 x 10-6 M liberado em uma formulação 1:2 de EtOH/ H2O. Linha 5: derivado do sal dissódico de astaxantina dissuccinato (“rac”) a 10-6 M liberado em uma formulação 1:2 de EtOH/ H2O;
A FIG. 15E representa a análise digital de níveis de indução relativos da expressão da proteína da conexina 43 pela mistura estatística de estereoisômeros do derivado do sal dissódico de astaxantina dissuccinato versus controles positivo e tratados apenas com solvente. Linhas como na FIG. 15D. A indução de dobra é normalizada para controlar os níveis de expressão de Cx43 nos controles tratados com EtOH/H2O 1:2 ajustados a uma unidade arbitrária = 1,0;
A FIG. 16 representa que a mistura estatística de estereoisômeros do derivado do sal dissódico de astaxantina dissuccinato aumenta a montagem de placas de junção imunorreativas Cx43. As culturas confluentes de células 10T1/2 foram tratadas durante 4 dias como descrito acima com a mistura estatística de estereoisômeros do derivado do sal dissódico de astaxantina dissuccinato: (1) a 10-5 M em EtOH/ H2O 1:2; (2) com EtOH/ H2O 1:2 como controle negativo apenas de solvente; ou (3) TTNPB a 10-8 M em solvente de tetraidrofurano (THF) como controle positivo. As células foram imunotingidas com um anticorpo Cx43 como descrito no texto. Painel A: a mistura estatística de estereoisômeros do derivado do sal dissódico de astaxantina dissuccinato a 10-5 M em EtOH/ H2O 1:2; Painel C: 1:2 EtOH/ H2O como controle de solvente; Painel E: TTNPB a 10-5 M em solvente de tetraidrofurano (THF) como controle positivo. Os painéis B, D e F: análise digital dos painéis A, C e E, respectivamente, demonstrando pixéis acima de um patamar ajustado fixo positivo para a intensidade fluorescente. Setas amarelas: placas de junção imunorreativas; setas vermelhas: posição de núcleos celulares. Observar o número maior e a intensidade de placas imunorreativas juncionais nas culturas tratadas com a mistura estatística de estereoisômeros do derivado do sal dissódico de astaxantina dissuccinato em comparação com os controles tratados apenas com solvente. As placas juncionais mostradas nos painéis C e D representam placas infreqüentes observadas nos controles; a maioria das células nestas culturas foram negativas quanto ao tingimento com Cx43;
A FIG. 17 representa os 4 estereoisômeros do diéster dissuccinato dissódico de astaxantina sintetizado para os estudos correntes (mostrado como os isômeros geométricos todo-E); a mistura de estereoisômeros ou estereoisômeros individuais, foram usados em aplicações separadas (ver as legendas da Figura);
A FIG. 18 representa a inibição percentual média do sinal do ânion de superóxido como detectado pelo aprisionamento de spin DEPMPO pelos derivados de dissuccinato dissódico de astaxantina em formulação aquosa pura. Mistura = mistura estatística de estereoisômeros [3S,3’S, meso (3R,3’S e 3’R,3S), 3R,3’R em uma relação 1:2:1]. Cada derivado em formulação aquosa foi padronizado para controlar o sinal de EPR detectado sem a adição de composto (ajustado a 0 % de inibição por convenção). Observar a ausência de inibição de superóxido pela formulação de 3S,3’S em água. Em cada caso, a formulação aquosa é menos potente do que a formulação correspondente em EtOH (FIG. 19);
A FIG. 19 representa a inibição percentual média do sinal do ânion de superóxido como detectado pelo aprisionamento de spin DEPMPO pelos derivados de dissuccinato dissódico de astaxantina em formulação etanólica. Mistura = mistura estatística de estereoisômeros [3S,3’S, meso (3R,3’S e 3’R,3S), 3R,3’R em uma relação 1:2:1], As soluções de estoque de mistura, meso e 3R,3’R foram 1:2 etanol/água (33 1/3 % de EtOH); a solução de estoque de 3S,3’S foi 1:1 etanol/água (50 % EtOH). A concentração final de EtOH no ensaio de teste de neutrófilo isolado foi de 0,3 % e 0,5 %, respectivamente. Cada derivado na formulação etanólica foi padronizado para controlar o sinal de EPR detectado sem a adição do composto (ajustado a 0 % de inibição por convenção);
A FIG. 20 representa a inibição percentual média de sinal do ânion de superóxido como detectado pelo aprisionamento de spin DEPMPO pela mistura de estereoisômeros do derivado de dissuccinato dissódico de astaxantina (testado em formulação 1:2 EtOH/Água; concentração de EtOH final no ensaio de neutrófilo isolado 0,3 %). Conforme a concentração do derivado aumenta, a inibição aumenta em uma maneira não linear, dependente da dose. A 3 mM, a inibição quase completa de sinal do ânion de superóxido é observada (95,0 % de inibição);
A FIG. 21 representa a inibição percentual média de sinal do ânion de superóxido como detectado pelo aprisionamento de spin DEPMPO pelo derivado do sal de dicloreto de astaxantina dilisina. Este derivado foi altamente solúvel em água (> 50 mg/ml) e não requereu um co-solvente para a excelente capacidade de extinção de radical neste ensaio. Comparar a inibição do ânion de superóxido deste derivado com aquela representada na Figura 20, para um derivado que forma as montagens supramoleculares em formulação aquosa pura;
A FIG. 22 representa uma plotagem padrão da concentração de astaxantina livre, não esterificada versus o tempo para plasma depois da gavagem oral de dose única em camundongos pretos. Apenas a astaxantina livre, não esterificada é detectada no plasma, corroborando a desesterificação completa do análogo carotenóide no intestino de mamífero, como foi anteriormente descrito;
A FIG. 23 representa uma plotagem padrão da concentração de astaxantina livre, não esterificada versus o tempo para o fígado depois da gavagem oral de dose única em camundongos pretos. Apenas a astaxantina livre, não esterificada é detectada no fígado, também corroborando (ver a
Figura 22 para plasma) a desesterificação completa do análogo de carotenóide no intestino de mamífero, como foi descrito anteriormente. Em cada ponto no tempo, os níveis hepáticos de astaxantina livre, não esterificada são maiores do que aqueles observados no plasma, uma nova descoberta sugere a liberação sólido-órgão vastamente melhorada de carotenóide livre no novo veículo de emulsão usado neste estudo;
A FIG. 24 representa o efeito do derivado de astaxantina dissuccinato dissódico a 500 mg/kg pela gavagem oral em dano hepático induzido por lipopolissacarídeo (LPS) em camundongos (como medido pela elevação na alanina aminotransferase sérica ou ALT). Três (3) animais foram testados em cada grupo. Os animais de controle receberam apenas solução salina (controles tratados com imitação; porção esquerda da figura) ou emulsão sem derivado de astaxantina dissuccinato dissódico (controles de veículo). Os animais tratados com imitação que recebem o novo derivado não demonstraram nenhuma efeito nos níveis de fundo de ALT; os camundongos que recebem a emulsão oral com o novo derivado a 500 mg/kg apresentaram níveis induzidos reduzidos de ALT, indicando proteção contra a necrose hepática depois do insulto de LPS;
A FIG. 25 descreve uma representação gráfica de uma redução relativa do tamanho do infarto em ratos Sprague-Dawley machos com pré tratamento usando uma formulação intravenosa de derivado do sal dissódico de astaxantina dissuccinato (Cardax®). Uma relação linear entre a dose e a redução do tamanho do infarto foi observada. Os níveis de tamanho da redução do infarto aproximam-se daqueles observados com o pré condicionamento isquêmico;
A FIG. 26 descreve uma representação gráfica de uma redução relativa de tamanho do infarto em ratos Sprague-Dawley machos com pré tratamento usando um derivado do sal dissódico de astaxantina dissuccinato formulação intravenosa (Cardax®);
-φ
A FIG. 27 representa a absorção transitória versus a demora para o derivado de astaxantina discuccinato diácido (astaCOOH) usando a fotólise cintilante. O experimento foi realizado em acetonitrila (MeCN) usando nitronaftalina (NN) como fotossensibilizador. Os espectros obtidos demonstram que o derivado de astaxantina dissuccinato diácido comporta-se identicamente à astaxantina racêmica livre, não esterifícada como um extintor de radical (formação do cátion do radical de carotenóide), identificando o derivado como um “medicamento brando” ativo que gera astaxantina livre, não esterifícada in vivo depois da liberação tanto oral quanto intravenosa;
_10 A FIG. 28 representa a absorção transitória versus a demora para o composto de referência de astaxantina racêmica livre, não esterifícada (asta)] usando fotólise cintilante. O experimento foi realizado em acetonitrila s (MeCN) usando a nitronaftalina (NN) como fotossensibilizador. Os espectros obtidos são quase super aproveitáveis em relação àqueles obtidos para o derivado de astaxantina dissuccinato diácido (astaCOOH), sugerindo as propriedades que formam radical-cátion idênticos para ambos os compostos;
A FIG. 29 descreve uma representação pictórica de um Western blot de um gel de poliacrilamida com anticorpo anti-conexina 43;
A FIG. 30 descreve uma representação pictórica de imagens ^20 densitométricas quantitativas de Western blots com anticorpos anti-conexina seguido pela quimioluminescência HRP em um, formador de imagem Biorad;
A FIG. 31 representa um gráfico de indução de dobra relativa da expressão de conexina 43 pelo controle positivo (TTNPB, retinóide sintético potente) e compostos de teste (derivado do sal dissódico de astaxantina dissuccinato em quatro formulações de água e/ou etanol (EtOH)/água: H2O-10-5, H2O-10-6, H2O-10-7 e EtOH/H2O-10-5) versus controle de água estéril (H2O) em 96 horas após a dosagem;
A FIG. 32 representa um gráfico de níveis médios de
astaxantina livre, não esterificada em plasma e fígado depois de onze (11) dias de gavagem oral de 500 mg/kg de derivado de astaxantina dissuccinato dissódico (ADD) em veículo de emulsão aos camundongos pretos. Tanto os níveis de pico quanto os de depressão no plasma e fígado obtidos foram > 200 nM, considerados ser protetivos contra a tensão oxidativa e dano hepático in vivo. Os níveis de pico obtidos no fígado em 6 horas após a 11a dose foram aproximadamente 9 vezes os níveis protetivos necessários (1760 nM);
A FIG. 33 representa a inibição percentual média de sinal do ânion de superóxido como detectado pelo aprisionamento de spin DEPMPO pelo derivado do sal dissódico de dissuccinato di-vitamina C [derivado (XXIII)]. Conforme a concentração do derivado aumenta, a inibição aumenta em uma maneira dependente da dose. Em 60 μΜ, a inibição quase completa de sinal do ânion de superóxido é observada. Este derivado também foi altamente solúvel em água e foi introduzido no ensaio de teste sem um cosolvente (ver a Figura 21). O novo derivado foi comparável na eficácia de extinção de radical para a formulação do derivado do sal dissódico de astaxantina dissuccinato em uma formulação 1:2 com vitamina C (ver a Figura 3), sugerindo propriedades de “medicamento brando”, ativo para este derivado. Esta estratégia de derivado co-antioxidante aumentou a potência de remoção de radical relativa (quando comparado com o derivado do sal dissódico de astaxantina dissuccinato) em 50 vezes;
A FIG. 34 representa efeitos de astaxantina livre, não esterificada (como a mistura todo-trans de estereoisômeros) na transformação neoplástica induzida por MCA em células de fibroblasto embriônico de camundongo (10T1/2). A astaxantina livre, não esterificada é produzida rapidamente in vivo depois da administração oral e intravenosa de novos derivados de carotenóide e é detectado em alta concentração tanto no plasma quanto nos órgãos sólidos (ver as Figuras 22 e 23). A astaxantina livre, não esterificada demonstrou níveis de redução de transformação neoplástica (100
%) acima de qualquer outro carotenóide testado neste ensaio em concentrações similares, demonstrando a utilidade aumentada deste composto para aplicações de quimioprevenção do câncer;
A FIG. 35 representa uma comparação de uma placa tratada com astaxantina para controlar placas (ver a descrição para a Figura 34);
A FIG. 36 representa uma comparação de astaxantina (como a mistura de estereoisômeros) com os carotenóides anteriormente testado neste laboratório usando este ensaio (ver a descrição para a Figura 34);
A FIG. 37 descreve uma representação gráfica de uma redução relativa de tamanho do infarto em coelhos New Zealand machos com pré tratamento usando uma formulação intravenosa de derivado do sal dissódico de astaxantina dissuccinato (Cardax®). Quando comparado com o tamanho da redução do infarto observado na mesma dose e programa de pré tratamento idêntico em roedores, um aumento de 38 % no tamanho da redução do infarto foi observado no modelo de coelho; e
A FIG. 38 descreve uma representação gráfica de uma redução relativa de níveis em circulação de proteína Creactive plasmática (CRP) em coelhos New Zealand machos com pré tratamento usando um derivado de formulação intravenosa de astaxantina dissuccinato dissódico (Cardax®). Os coelhos de controle (apenas injeção de solução salina) estimulados para a resposta de fase aguda com 1 % de óleo de croton pela injeção subcutânea apresentaram um aumento médio de 23,5 % nos níveis de CRP circulantes a partir da base de referência (amostra venosa tirada no momento da reperfusão). Ao contrário, os animais tratados com Cardax® (50 mg/kg) demonstraram uma redução média nos níveis de CRP circulantes a partir da base de referência (-15,7 %), demonstrando os efeitos anti-inflamatórios potentes do Cardax®.
DESCRIÇÃO DETALHADA
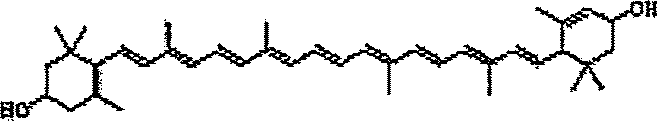
Carotenóides “precursores” no geral podem se referir àqueles compostos naturais utilizados como esqueleto de partida para a síntese de análogo de carotenóide estrutural. Derivados de carotenóide podem ser derivados de um carotenóide que ocorre naturalmente. O carotenóide que ocorre naturalmente pode incluir licopeno, licofila, licozantina, astaxantina, beta-caroteno, luteína, zeaxantina e/ou cantaxantina para citar alguns.
Os carotenóides são um grupo de pigmentos naturais produzidos principalmente pelas plantas, levedura e microalgas. A família de compostos relacionados agora inclui mais do que 600 membros descritos, exclusivo os isômeros Z e E. Cinquenta (50) foram descobertos nos soros ou tecidos humanos. Os seres humanos e outros animais não podem sintetizar carotenóides de novo e devem obtê-los da sua dieta. Todos os carotenóides compartilham características químicas comuns, tais como uma estrutura poliisoprenóide, uma cadeia de polieno longa que forma o cromóforo e simetria semelhante em tomo da ligação dupla central. A ligação de ponta a ponta de duas moléculas de geranilgeranil difosfato C2o produz o esqueleto de carbono C40 precursor. Carotenóides sem grupos funcionais oxigenados são chamados de “carotenos”, que refletem a sua natureza de hidrocarboneto; carotenos oxigenados são conhecidos como “xantofilas.” A ciclização em uma ou ambas as extremidades da molécula produz 7 grupos finais identificados (estruturas representativas mostradas na FIG. 1).
As funções do carotenóide documentadas na natureza incluem coleta de luz, fotoproteção e coloração protetiva e relacionada com o sexo em organismos microscópicos, mamíferos e pássaros, respectivamente. Uma observação relativamente recente foi o papel protetivo dos carotenóides contra as doenças relacionadas com a idade em seres humanos como parte de uma rede de antioxidante complexo dentro das células. Este papel é ditado pelas relações íntimas entre as propriedades fisicoquímicas de carotenóides individuais e as suas funções in vivo em organismos. O sistema longo de alternar ligações duplas e únicas na parte central da molécula (deslocando os elétrons orbitais n no comprimento inteiro da camada de polieno) confere a forma molecular distinta, reatividade química e propriedades absorvedoras de luz dos carotenóides. Adicionalmente, o isomerismo em tomo das ligações químicas C=C produz estruturas moleculares distintamente diferentes que podem ser isoladas como compostos separados (conhecidos como isômeros Z (“cis”) e E (“trans”) ou geométricos). Dos mais do que 600 carotenóides descritos, um número ainda maior dos isômeros mono-Z e poli-Z teoricamente possíveis são algumas vezes encontrados na natureza. A presença de uma ligação dupla Z cria impedimento estérico maior entre átomos de hidrogênio vizinhos e/ou grupos metila, de modo que os isômeros Z são no geral menos termodinamicamente estáveis e mais quimicamente reativos, do que a forma todo-E correspondente. A configuração todo-E é uma molécula estendida, linear e rígida. Os isômeros Z são, ao contrário, moléculas não simples, lineares (os chamados isômeros de “cadeia curvada”). A presença de qualquer Z na camada de polieno cria uma molécula de cadeia curvada. A tendência dos isômeros Z para cristalizar ou agregar é muito menor do que os todo-E e os isômeros Z são mais facilmente solubilizados, absorvidos e transportados in vivo do que as suas contrapartes de todo-E. Isto tem implicações importantes para a dosagem enteral (por exemplo, oral) e parenteral (por exemplo, intravenosa, infra-arterial, intramuscular e subcutânea) em mamíferos.
Os carotenóides com centros quirais podem existir como a configuração R (rectus) ou S (sinister). Como um exemplo, a astaxantina (com 2 centros quirais nos carbonos 3 e 3’) podem existir como 4 estereoisômeros possíveis: 3S, 3’S; 3R, 3’S e 3S, 3’R (formas meso); ou 3R, 3’R. As proporções relativas de cada um dos estereoisômeros pode variar pela fonte natural. Por exemplo, a farinha da microalgal Haematococcus pluvialis é 99 % 3S, 3’S astaxantina e é provavelmente a fonte evolucionária humana predominante de astaxantina. Fontes de krill (3R,3’R) e levedura produzem
composições estereoisoméricas diferentes do que a fonte nucroalgal. A astaxantina sintética, produzida por grandes fabricantes tais como a Hoffinann-LaRoche AG, Buckton Scott (USA) ou BASF AG, é fornecida como misturas de isômeros geométricos definidos de uma mistura 5 estereoisomérica 1:2:1 [3S, 3’S; 3R, 3’S, 3’R,3S (meso); 3R, 3’R] de astaxantina livre, não esterificada. A astaxantina de fonte natural do peixe salmonídeo é predominantemente um único estereoisômero (3S,3’S), mas contém uma mistura de isômeros geométricos. A astaxantina da fonte natural Haematococcus pluvialis pode conter aproximadamente 50 % de isômeros Z. _ 10 Como estabelecido acima, a mudança conformacional Z pode levar a uma interferência estérica maior entre as duas partes da molécula de carotenóide, tomando-a menos estável, mais reativa e mais suscetível à reatividade em ' tensões de oxigênio baixas. Em uma tal situação, em relação à forma todo-E, as formas Z: (1) podem ser degradadas primeiro; (2) podem reprimir melhor o 15 ataque de células pela espécie de oxigênio reativo tal como o ânion de superóxido; e (3) pode preferencialmente diminuir a formação de radicais. Especialmente, as formas Z podem ser no início termodinamicamente _ favorecidas para proteger as porções lipofílicas da célula e da membrana celular de destruição. É importante observar, entretanto, que a forma todo-E 20 de astaxantina, diferente do β-caroteno, retém significante biodisponibilidade oral assim como capacidade antioxidante na forma de suas substituições de diidróxi e diceto nos anéis β-ionona e foi demonstrado ter eficácia aumentada em relação ao β-caroteno na maioria dos estudos. A forma todo-E de astaxantina também foi postulado ter o efeito estabilizante de membrana 25 máximo em células in vivo. Portanto, é provável que a forma todo-E de astaxantina em misturas naturais e sintéticas de estereoisômeros também seja extremamente importante nos mecanismos antioxidantes e pode ser a forma mais adequada para preparações farmacêuticas particulares.
O(s) mecanismo(s) antioxidante(s) de carotenóides e em particular de astaxantina, inclui(em) a extinção de oxigênio singleto, remoção de radical direta e ruptura de cadeia de peroxidação de lipídeo. A camada de polieno do carotenóide absorve a energia excitada de oxigênio singleto, estabilizando eficazmente a transferência de energia pelo deslocamento ao longo da cadeia e dissipa a energia para o ambiente local como calor. A transferência de energia da clorofila no estado de tripleto (em plantas) ou outras porfirinas e proto-porfirinas (em mamíferos) para os carotenóides ocorre muito mais facilmente do que a transferência de energia alternativa ao oxigênio para formar o oxigênio singleto (!O2) altamente reativo e destrutivo. Os carotenóides também podem aceitar a energia de excitação do oxigênio singleto se algum deva ser formado in situ e mais uma vez dissipa a energia como valor para o ambiente local. Esta capacidade de extinção de oxigênio singleto tem implicações significantes na isquemia cardíaca, degeneração macular, porfiria e outros estados de doença em que a produção de oxigênio singleto tem efeitos nocivos. No mecanismo de extinção física a molécula de carotenóide pode ser regenerada (mais ffeqüentemente) ou ser perdida. Os carotenóides também são excelentes oxidantes rompedores de cadeia, um mecanismo importante na inibição da peroxidação de lipídeos. A astaxantina pode doar um hidrogênio (H’) ao radical de ácido graxo poliinsaturado (PUFA) instável, interrompendo a reação em cadeia. Os radicais peroxila também podem ser, além da camada de polieno dos carotenóides, a causa imediata para a terminação da cadeia de peróxido do lipídeo. A dose apropriada de astaxantina mostrou suprimir completamente a reação em cadeia do radical peroxila em sistemas de lipossoma. A astaxantina compartilha com a vitamina E o seu sistema de defesa antioxidante duplo de extinção do oxigênio singleto e de remoção de radical direta e na maioria dos casos (e particularmente na tensão de oxigênio baixa in vivo) é superior à vitamina E como descontaminante de radical e extintor físico de oxigênio singleto.
Os carotenóides e em particular a astaxantina, são descontaminantes de radical direto e extintores de oxigênio singleto potentes e possuem todas as qualidades desejáveis de tais agentes terapêuticos para a inibição ou melhora do dano de perfosão. A síntese de novos derivados de 5 carotenóide com propriedades de “medicamento brando” (isto é, atividade na forma derivatizada), com ligações cliváveis, fisiologicamente relevantes, às pró-porções, pode gerar níveis significantes de carotenóides livres tanto no plasma quanto nos órgãos sólidos. No caso da astaxantina livre, não esterificada, isto é uma forma de realização particularmente útil _ 10 (características específicas para a astaxantina livre, não esterificada abaixo):
• Lipídeo solúvel na forma natural; pode ser modificado para se tomar mais solúvel em água • Peso molecular de 597 Daltons [tamanho < 600 daltons (Da) facilmente cruza a barreira hematoencefálica ou BBB] · Característica de camada longa de polieno dos carotenóides eficazes na extinção de oxigênio singleto e ruptura da cadeia de peroxidação de lipídeo í · Nenhuma atividade de pró-vitamina A em mamíferos (eliminando os problemas de hipervitaminose A e toxicidade de retinóide em 20 seres humanos).
A administração de antioxidantes que são extintores de oxigênio singleto potentes e descontaminantes de radical diretos, particularmente do ânion de superóxido, devem limitar a íibrose hepática e a progressão da cirrose por afetar a ativação de células estreladas hepáticas 25 precoces no caminho fibrogenético. A redução no nível de ROS pela administração de um antioxidante potente pode ser portanto crucial na prevenção da ativação tanto das células HSC quanto das de Kupffer. Este efeito antioxidante protetivo parece estar espalhado através da faixa de antioxidantes terapêuticos potenciais, incluindo agentes solúveis em água (por
exemplo, vitamina C, glutationa, resveratrol) e lipofílico (por exemplo, vitamina E, β-caroteno, astaxantina). Portanto, uma estratégia de derivado de co-antioxidante em que agentes solúveis em água e lipofílicos são sinteticamente combinados é uma forma de realização particularmente útil.
A vitamina E é no geral considerada o antioxidante de referência. Quando comparados com a vitamina E, os carotenóides são mais eficiente na extinção do oxigênio singleto em solventes orgânicos homogêneos orgânicos e nos sistemas de lipossoma. Eles são oxidantes rompedores de cadeia melhores também em sistemas lipossômicos. Eles têm demonstrado eficácia e potência aumentada in vivo. Eles são particularmente eficazes na tensão de oxigênio baixa e em concentração baixa, tomando-os agentes extremamente eficazes nas condições de doença em que a isquemia é uma parte importante do dano e patologia de tecido. Estes carotenóides também têm um tropismo natural para o fígado depois da administração oral. Portanto, a administração terapêutica de carotenóides deve fornecer um benefício maior na limitação da fibrose do que a vitamina E.
Problemas relacionados com o uso de alguns carotenóides e análogos de carotenóide estrutural incluem: (1) as misturas isoméricas complexas, incluindo contaminantes que não carotenóide, fornecidas em fontes naturais e sintéticas que levam a aumentos caros nos testes de segurança e eficácia requeridos por agências tais como o FDA; (2) biodisponibilidade limitada na administração a um paciente; e (3) a indução diferencial de enzimas de citocromo P450 (esta família de enzimas exibe diferenças específicas de espécie que devem ser levadas em consideração quando da extrapolação de trabalhos com animal para estudos humanos).
Em uma forma de realização, o carotenóide precursor pode ter uma estrutura de qualquer carotenóide que ocorre naturalmente. Alguns exemplos de carotenóide que ocorrem naturalmente que podem ser usados como compostos precursores são mostrados na FIG. 1.
Em algumas formas de realização, os derivados de carotenóide podem incluir compostos tendo a estrutura (I):
R3 R? Rp R3 R3 R3 R3 R3 R3
R3 R3 R3 Ra R3 R® R3 R3 R3 (D_
R3 pode ser independentemente hidrogênio, metila, alquila, alquenila ou substituintes aromáticos. RI e R2 podem ser independentemente '5 H, um alceno cíclico com pelo menos um substituinte ou um anel cíclico com pelo menos um substituinte tendo a estrutura geral (II):
onde n pode ser entre 4 a 10 átomos de carbono. W é o substituinte. O substituinte pode ser pelo menos parcialmente hidrofílico. Um substituinte hidrofílico pode ajudar a aumentar a solubilidade em água de um 10 derivado de carotenóide. Em algumas formas de realização, um derivado de carotenóide pode ser pelo menos parcialmente solúvel em água. O anel cíclico pode incluir pelo menos um centro quiral. O alceno cíclico pode incluir pelo menos um centro quiral. O anel cíclico pode incluir pelo menos um grau de insaturação. Em algumas formas de realização do anel cíclico, o anel cíclico 15 pode ser aromático. O anel cíclico pode incluir um substituinte. O substituinte pode ser hidrofílico. Em algumas formas de realização, o anel cíclico pode incluir, por exemplo (a), (b) ou (c):
y y y /X (a). XX (Jb). ou - XX (c).
Em algumas formas de realização, o substituinte pode incluir, por exemplo, um ácido carboxílico, um aminoácido, um éster, um alcanol, 20 uma amina, um fosfato, um succinato, um glicinato, um éter, um glicosídeo, um açúcar ou um sal de carboxilato.
Em algumas formas de realização, cada substituinte W pode independentemente incluir -XR. Cada X pode independentemente incluir O, N ou S. Em algumas formas de realização, cada substituinte W pode independentemente compreender aminoácidos, ésteres, carbamatos, amidas, carbonatos, álcool, fosfatos ou sulfonatos. Em algumas formas de realização do substituinte, o substituinte pode incluir, por exemplo de (d) até (pp):
(2).
OH Ο l χΗ 'ζ
Η (»)>
O^OTI° . OH ζΧχθΗ
OH OH
onde cada R é, por exemplo, independentemente -alquila34
NR!3+, -aromático-NRV, -alquila-CO2-, -aromático-CO2-, -aminoácido-NH3 +, -aminoácido fosforilado-NH3 +, polietileno glicol, dextrano, H, alquila ou arila. Em algumas formas de realização, os substituintes podem incluir qualquer combinação de (d) até (pp). Em algumas formas de realização, os 5 substituintes negativamente carregados podem incluir metais alcalinos, um metal ou uma combinação de metais alcalinos diferentes em uma forma de realização com mais do que um substituinte negativamente carregado, como contra íons. Os metais alcalinos podem incluir, mas não são limitados a, sódio, potássio e/ou lítio.
Embora a estrutura acima e as estruturas subsequentes, representem alquenos na configuração E isto não deve ser considerado como limitante. Os compostos aqui debatidos podem incluir formas de realização onde os alcenos estão na configuração Z ou incluem alcenos em uma combinação das configurações Z e E dentro da mesma molécula. Os compostos aqui representados podem converter naturalmente entre a configuração Z e E e/ou existir em equilíbrio entre as duas configurações.
Em uma forma de realização, um composto químico pode incluir um derivado de carotenóide tendo a estrutura (III)
Cada Y pode ser independentemente O ou H2. Cada R pode ser ‘20 independentemente OR1 ou R1.
Cada R1 pode ser independentemente -alquila-NR23+, aromático-NR23+, -alquila-CO2-, -aromático-CO2-, -aminoácido-NH3+, aminoácido fosforilado-NH3+, polietileno glicol, dextrano, H, alquila, peptídeos, poli-lisina ou arila. Além disso, cada R2 pode ser 25 independentemente H, alquila ou arila. O derivado de carotenóide pode incluir pelo menos um centro quiral.
Em uma forma de realização específica onde Y é H2, derivado de carotenóide tem a estrutura (IV)
específica
Em uma forma de realização derivado de carotenóide tem a estrutura (V) onde Y é Ο,
químico pode
Em uma forma de realização, um composto incluir um derivado de carotenóide tendo a estrutura (VI)
Cada Y pode ser independentemente O ou H2. Cada R pode ser independentemente H, alquila ou arila. O derivado de carotenóide pode incluir pelo menos um centro quiral. Em uma forma de realização específica Y pode ser H2, o derivado de carotenóide tendo a estrutura (VII)
Em uma forma de realização específica derivado de carotenóide tem a estrutura (VIII) onde Y é O, o
Em uma forma de realização, um composto incluir um derivado de carotenóide tendo a estrutura (IX) .(VIII). químico pode
(IX).
Cada Y pode ser independentemente O ou H2. Cada R’ pode ser CH2. n pode ser de 1 a 9. Cada X pode ser independentemente
OH
O O 9 9 HCXA.OH , >^O'R, s <0H > ou
Cada R podem ser independentemente -alquila-NRV, aromático-NRV, -alquila-CO2-, -aromático-CO2-, -aminoácido-NH3 +, aminoácido fosforilado-NH3 +, polietileno glicol, dextrano, H, alquila ou arila. Cada R1 podem ser independentemente H, alquila ou arila. O derivado de carotenóide pode incluir pelo menos um centro quiral.
Em uma forma de realização derivado de carotenóide tem a estrutura (X) específica onde Y é H2, o
(X).
específica onde Y é O, o
Em uma forma de realização
Em uma forma de realização, um composto químico pode incluir um derivado de carotenóide tendo a estrutura (XII)
Cada Y pode ser independentemente O ou H2. O derivado de carotenóide pode incluir pelo menos um centro quiral. Em uma forma de
realização específica Y pode ser H2, o derivado de carotenóide tendo a estrutura (XIII)
Em uma forma de realização específica onde Y é O, o derivado de carotenóide tem a estrutura (XIV)
incluir um derivado de carotenóide de éster do ácido succínico tendo a estrutura (XV)
Em algumas formas de realização, um composto químico pode incluir um derivado de carotenóide de éster do ácido succínico sal dissódico
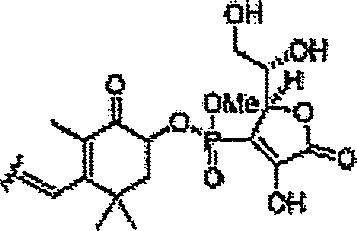
Em algumas formas de realização, um composto químico pode incluir um derivado de carotenóide com um co-antioxidante, em particular um ou mais análogos da vitamina C (isto é, o ácido L ascórbico) ligado a um carotenóide. Algumas formas de realização podem incluir derivados do ácido carboxílico e/ou carboxilato de vitamina C ligados a um carotenóide (por exemplo, a estrutura (XVII))
CXVIIJ.
Carbohydr. Res. 1978, 60, 251 a 258, divulga a oxidação em
C-6 de ácido ascórbico como representado na EQN. 5.
(5)
Condições de C- Esterificaçâo
Algumas formas de realização podem incluir a vitamina C e/ou análogos da vitamina C ligados a um carotenóide. A vitamina C pode ser 5 ligada ao carotenóide por intermédio de uma ligação de éter (por exemplo, estrutura (XVIII))
Algumas formas de realização podem incluir análogos de dissuccinato de vitamina C ligados a um carotenóide (por exemplo, a estrutura
Alguns formas de realização podem incluir soluções ou preparações farmacêuticas de carotenóides e/ou derivados de carotenóide combinadas com co-antioxidantes, em particular vitamina C e/ou análogos da vitamina C. As preparações farmacêuticas podem incluir uma relação de cerca de 2:1 de vitamina C para carotenóide respectivamente.
Em algumas formas de realização, um carotenóide (por exemplo, astaxantina) pode ser ligado à vitamina C formando uma ligação de éter. A ligação de éter pode ser formada usando a reação de Mitsunobu como naEQN. 1.
Em algumas formas de realização, a vitamina C pode ser seletivamente esterificada. A vitamina C pode ser seletivamente esterificada na posição C-3 (por exemplo, EQN. 2). J. Org. Chem. 2000, 65, 911 a 913, divulga a esterificação seletiva em C-3 de ácido ascórbico não protegido com álcoois primários.
R = Me, 77% Propila, 63 % Octila, 72 % alila, 72 % (2) benzila, 64 %
Em algumas formas de realização, um carotenóide pode ser ligado à vitamina C. A vitamina C pode ser ligada ao carotenóide na posição de diol C-6, C-5 como representado nas EQNS. 3 e 4 formando um acetal.
Em algumas formas de realização, um carotenóide pode ser ligado a uma porção solúvel em água (por exemplo, vitamina C) com um ligador de glioxilato como representado na EQN. 6. Tetrahedron 1989, 22, 15 6987 a 6998, divulga formações de acetal similares.
Em algumas formas de realização, um carotenóide pode ser ligado a uma porção solúvel em água (por exemplo, vitamina C) com um ligador de glioxilato como representado na EQN. 7. J. Med. Chem. 1988, 31, 1363 a 1368, divulga o cloreto do ácido glioxílico.
Em algumas formas de realização, um carotenóide pode ser ligado a uma porção solúvel em água (por exemplo, vitamina C) com um ligador de fosfato como representado na EQN. 8. Carbohydr. Res. 1988, 176, 73 a 78, divulga o L-ascorbato 6-fosfato.
Em algumas formas de realização, um carotenóide pode ser ‘10 ligado a uma porção solúvel em água (por exemplo, vitamina C) com um ligador de fosfato como representado na EQN. 9. Carbohydr. Res. 1979, 68, 313 a 319, divulga o derivado 6-bromo da vitamina C. Carbohydr. Res. 1988, 176, 73 a 78, divulga a reação do derivado 6-bromo da vitamina C com fosfatos.
Em algumas formas de realização, um carotenóide pode ser ligado a uma porção solúvel em água (por exemplo, vitamina C) com um ligador de fosfato como representado na EQN. 10. J. Med Chem. 2001, 44, 1749 a 1757 e J. Med Chem. 2001, 44, 3710 a 3720, divulgam o derivado de cloreto de alila e a sua reação com nucleófilos, incluindo fosfatos, sob 5 condições básicas brandas.
k.
Em algumas formas de realização, um carotenóide pode ser ligado a uma porção solúvel em água (por exemplo, vitamina C) com um ligador de fosfato como representado na EQN. 11. A vitamina C pode ser ligada ao carotenóide usando esterificação seletiva em C-3 do ácido ascórbico não protegido com álcoois primários.
p-OKa +
Em algumas formas de realização, um composto químico pode incluir um derivado de carotenóide incluindo um ou mais aminoácidos (por exemplo, lisina) e/ou aminoácidos análogos (por exemplo, sal do ácido clorídrico de lisina) ligados a um carotenóide [por exemplo, estrutura (XX)].
Em algumas formas de realização, um derivado de carotenóide pode incluir:













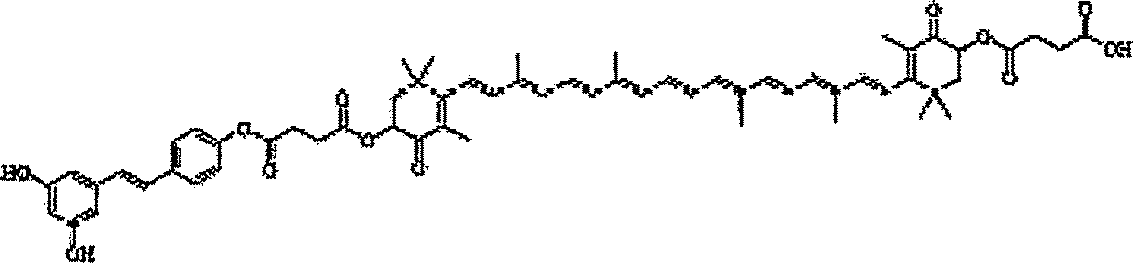

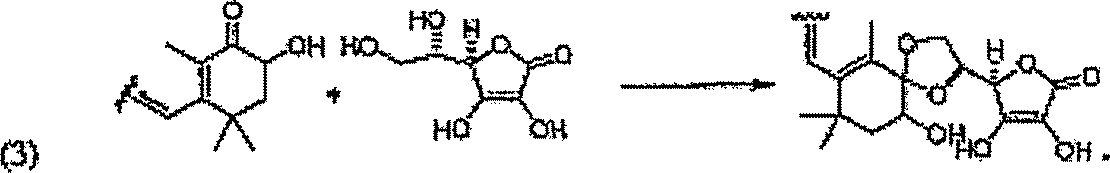
Em uma forma de realização, os derivados de carotenóide podem ser sintetizados a partir dos carotenóides que ocorrem naturalmente. Os carotenóides podem incluir as estruturas de 2A a 2E representadas na FIG. 1. Em algumas formas de realização, os derivados de carotenóide podem ser 5 sintetizados a partir de um carotenóide que ocorre naturalmente incluindo um ou mais substituintes de álcool. Em outras formas de realização, os derivados de carotenóide podem ser sintetizados a partir de um derivado de um carotenóide que ocorre naturalmente incluindo um ou mais substituintes de álcool. A síntese pode resultar em um estereoisômero único. A síntese pode resultar em um isômero geométrico único do derivado de carotenóide. A síntese/seqüência sintética podem incluir quaisquer etapas de purificação ou isolação anteriores realizadas no carotenóide precursor. Um exemplo pode incluir, mas não é limitado a, um derivado de carotenóide 3S,3’S todo-E, onde o carotenóide precursor é a astaxantina. A seqüência sintética pode incluir a proteção e subsequentemente a desproteção de várias funcionalidades do precursor de carotenóide e/ou substituinte. Os álcoois podem ser desprotonados com uma base. O álcool desprotonado pode ser reagido com um substituinte precursor com um bom grupo de partida. A base pode incluir qualquer base não nucleofílica conhecida por uma pessoa habilitada na técnica tal como, por exemplo, dimetilaminopiridina. O álcool desprotonado pode atuar como um nucleófilo que reage com o precursor substituinte, deslocando o grupo de partida. Os grupos de partida podem incluir, mas não são limitados a, Cl, Br, tosila, brosila, mesila ou trifila. Estes são apenas uns poucos exemplos de grupos de partida que podem ser usados, muitos mais são conhecidos e podem estar evidentes a uma pessoa habilitada na técnica. Em algumas formas de realização, nem sempre pode ser necessário desprotonar o álcool, dependendo do grupo de partida utilizado. Em outros exemplos o grupo de partida pode ser interno e pode ser subsequentemente incluído na estrutura final do derivado de carotenóide, um exemplo não limitante pode incluir anidridos ou éteres cíclicos deslocados. Por exemplo, o álcool desprotonado pode ser reagido com anidrido succínico. Em uma forma de realização, o éster do ácido dissuccínico de astaxantina pode ser ainda convertido ao sal dissódico. Os exemplos de sequências sintéticas para a preparação de algumas das formas de realização específicas representadas são

descritos na seção de Exemplos. O exemplo representado abaixo é um exemplo não limitante genérico de uma sequência sintética para a preparação de derivados de carotenóide.
Dano de Isquemia-Reperfusão (I/R): Características
Fisiopatológicas
A reperfusão de miocárdio isquêmico resulta em alterações celulares e locais significantes no tecido em risco que exacerba o dano criado pelo insulto isquêmico. Especificamente, o dano vascular e microvascular, a disjunção endotelial, a necrose celular acelerada e a ativação de granulócito 10 ocorrem subsequentes à reperfusão. O dano vascular e microvascular resulta da ativação de complemento, da interação de proteína reativa C circulante e localizada com Clq e fosfocolina nas células expostas formando o complexo de ataque à membrana (MAC) com a morte celular e permeabilidade endotelial aumentada que seguem, a geração de ânion de superóxido (O2-) 15 pelo endotélio afetado e leucócitos ativados, microêmbolos, liberação de citocina (em particular ILA-6) e ativação de plaquetas com a ativação de receptor IlblIIa e liberação subsequente de ADP e serotonina. A disfunção endotelial segue, com a geração subsequente de ânion de superóxido pelo endotélio disfuncional, danificando ainda o endotélio afetado em um ciclo de regeneração positivo. Foi mostrado que a isquemia-reperfusão resultam em dano precoce e grave à vasculatura, que ainda compromete a sobrevivência de miócito. A ativação de granulócito também ocorre durante a reperfusão. A ativação e desgranulação desta linhagem celular resulta na liberação de mieloperoxidase (MPO), elastases, proteases e espécies de radical e que não radical derivadas de oxigênio (os mais importantes ânion de superóxido, hipoclorito, oxigênio singleto e peróxido de hidrogênio depois do “explosão respiratória”). As espécies de radical e que não radical derivadas de oxigênio (por exemplo oxigênio singleto) são implicadas em muitos dos danos associados com a isquemia e reperfusão e a peroxidação de lipídeo tem sido claramente mostrado ser uma sequela da reperfusão como medida pela formação de substâncias reativas ao ácido tiobarbitúrico (TBARS), malondialdeído (MDA) ou dieno conjugados.
O insulto isquêmico tanto ao endotélio dos vasos coronários quanto ao próprio miocárdio cria condições que favorecem a produção de espécies de radical e outras que não radical derivadas de oxigênio capazes de danificar tecido aqui coletivamente aludidas como espécie de oxigênio reativo (“ROS”). O sistema xantina desidrogenase-xantina oxidase com base no endotélio em seres humanos é uma fonte do ânion de superóxido (O2-). O miocárdio humano carece deste sistema enzimático. No tecido saudável, 90 % da enzima existe como a forma de desidrogenase (D); ela é convertida na forma de oxidase (O) no tecido isquêmico. A forma (O), usando oxigênio molecular como o aceitador de elétron, produz o ânion de superóxido O2- no endotélio coronário. O ânion de superóxido está depois disponível para criar dano ao tecido adicional no ambiente local. O ânion de superóxido não é a espécie de radical mais reativa ou destrutiva nos sistemas biológicos por si só. Entretanto, ele é a fonte de alguns radicais de vida mais curta ou mais longa,

mais prejudiciais e/ou ROS tais como o radical hidroxila, peróxido de hidrogênio, oxigênio singleto e radicais peroxila. Como tal, ele pode ser considerado o radical “central” no dano I/R. As reações biológicas do radical de superóxido para formar estes oxidantes importantes são mostradas abaixo:
(1) o ânion de superóxido pode aceitar um único elétron (“redução monovalente”), produzindo peróxido (O2-2). Ligado com 2 prótons, o peróxido forma então peróxido de hidrogênio (H2O2). O H2O2 se difunde facilmente através das membranas celulares e não pode ser facilmente excluído do citoplasma, onde ele pode reagir com componentes celulares ou ativar as cascatas centrais inflamatórias tais como o fator nuclear capa-B (NFcapa-B), que também são implicadas no dano inflamatório adicional na dano I/R.
(2) o ânion de superóxido tipicamente reage com ele mesmo para produzir peróxido de hidrogênio e oxigênio (“dismutação”). A dismutação de superóxido pode ser espontânea ou catalisada pela enzima superóxido dismutase (SOD), uma reação que resulta na formação de SOD oxidado:
O2- + 2 H2O2 + 3O2 (3) o ânion de superóxido pode servir como um agente redutor e doa um único elétron (“redução monovalente”) a um cátion metálico. Por exemplo, nas duas etapas de processo abaixo, o ferro férrico (Fe3+) é reduzido e subsequentemente atua como um catalisador para converter peróxido de hidrogênio (H2O2) no radical hidroxila (HO-).
O2- + Fe3+ —> 3O2 + Fe2 + (etapa 1)
O ferro ferroso (Fe2+), o cátion metálico reduzido, subsequentemente catalisa a ruptura da ligação oxigênio-oxigênio do peróxido de hidrogênio. Isto produz um radical hidroxila (HO’) e um íon hidróxido (HO-). A reação é conhecida como a reação Fenton, particularmente importante no dano de reperfosão onde a compartimentalização do ferro e/ou cobre foi perdida (tipicamente através da hemólise de células sanguíneas vermelhas, RBCs): Fe2+ + H2O2 -» Fe3+ + HO’ + HO- (etapa 2)
Os radicais de hidroxila facilmente cruzam as membranas celulares. O dano pelo radical hidroxila é “limitado pela taxa de difusão”, isto é, a distância 3-dimensional em que o dano pode ser inflingido está relacionada com a taxa do radical de difusão. O radical hidroxila é um ROS particularmente tóxico. Os radicais de hidroxila podem adicionar aos substratos orgânicos (representados por R na reação abaixo) e formar um aduto hidroxilado que é ele próprio um radical. No caso de dano por isquemia-reperfusão, os ácido graxos poliinsaturado (PUFAs) nas membranas endoteliais e miocíticas são particularmente suscetíveis ao dano pelo radical hidroxila:
HO’ + R —> HOR’ (aduto hidroxilado)
O aduto formado acima pode ainda oxidar na presença de cátions metálicos ou oxigênio molecular. Isto resulta em produto(s) oxidado(s), produto(s) estável(is). No primeiro caso, o elétron extra é transferido para o íon metálico e no segundo caso, para oxigênio (formando superóxido). Dois radicais adutos também podem reagir entre si formando produtos oxidados, estáveis e reticulados mais água. Este é um processo importante na oxidação de proteínas de membrana:
HOR’ + HOR’ -> R-R + 2 H2O
Além disso, os radicais de hidroxila podem oxidar substratos orgânicos abstraindo-se elétrons de tais moléculas:
HO‘ + R->R’ + OH25 O substrato oxidado (R‘) é um radical. Tais radicais podem reagir com outras moléculas em uma reação em cadeia. Os carotenóides são rompedores de cadeia pela peroxidação de lipídeo particularmente eficientes. Em um exemplo, a reação com oxigênio no estado mais estável produz radicais peroxila (ROO’):
R' + 3O2-»ROO*.
Os radicais de peroxila são muito reativos. Eles podem reagir com outros substratos orgânicos em uma reação em cadeia:
ROO' + RH -> ROOH + R*
As reações em cadeia são comuns no dano oxidativo de PUF As e outros lipídeos de membrana suscetíveis. A medição da taxa de consumo de oxigênio é uma indicação do início e progresso da reação em cadeia. É importante mencionar que, em sistemas de modelo lipossômico, a astaxantina livre, não esterificada na dose apropriada é capaz da supressão completa da reação em cadeia e acompanhar o consumo de oxigênio.
(4) o ânion de superóxido pode reagir com o radical hidroxila (HO‘) para formar oxigênio singleto (*0^). O oxigênio singleto não é um radical, mas é altamente reativo e prejudicial em sistemas biológicos cardíacos. O oxigênio singleto foi implicado na destruição das proteínas ligadas a membrana tais como a 5’-nucleotidase, importante na manutenção ou restauração de concentrações locais de compostos vasodilatadores tais como adenosina (mostrado ser eficaz em seres humanos para a redução do tamanho do infarto):
O2 + HO'-^1O2* + HO- (5) o ânion de superóxido também pode reagir com o radical de óxido nítrico (N0‘), produzindo peroxinitrito (OONO-). Peroxinitrito é uma molécula altamente reativa e prejudicial em sistemas biológicos.
O2- + NO'-»OONOOs leucócitos polimorfonucleares (PMNs), em particular neutrófilos e macrófagos ativados são uma fonte rica de espécies radicais e que não radicais derivadas de oxigênio. O sistema de NADPH-oxidase localizado nas membranas celulares fagocíticas é uma fonte importante de radicais a seguir do estímulo. Os PMNs e macrófagos ativados rapidamente consomem oxigênio na “explosão respiratória” e o convertem no ânion de superóxido e subsequentemente peróxido de hidrogênio (H2O2), assim como quantidades signifícantes de oxigênio singleto. Os PMNs são adicionalmente uma fonte de hipoclorito, uma outra espécie de oxigênio reativo prejudicial. Embora importante na atividade celular fagocítica na infecção, no ambiente 5 local durante a isquemia e reperfusão, outra dano celular ocorre conforme este ROS ataca células hospedeiras normais e danificadas na área local.
Os neutrófilos são uma fonte primária de radicais de oxigênio durante a reperfusão depois da isquemia miocárdica prolongada, particularmente em modelos animal de infartação experimental. Muitos <_10 estudos anteriores documentaram a formação de radical oxigênio durante a isquemia-reperfusão, mas poucos trataram da(s) fonte(s) de tais radicais in vivo ou examinaram a geração de radical no contexto da isquemia miocárdica * prolongada. Os neutrófilos são recrutados em grandes quantidades dentro do tecido anteriormente isquêmico e são considerados induzir a dano pela 15 liberação local de vários mediadores, sobretudo radicais de oxigênio. Anteriormente, a contribuição de neutrófilos ativados para o dano de reperfusão e salvamento miocárdico potencial permanecia incerta. Uma metodologia foi desenvolvida para detectar radicais, em particular ânion de superóxido, sem interferir com os mecanismos transportados pelo sangue de 20 geração de radical.
Cães de peito aberto e fechado passaram pela cateterização da aorta e sino coronário (Duilio et al. 2001). Nenhum produto químico foi infundido. Ao invés, o sangue foi drenado em seringas pré enchidas com um aprisionamento de spin e analisado pela espectroscopia de ressonância 25 paramagnética de elétron (EPR). Depois de 90 minutos de oclusão da artéria coronária, a concentração transcardíaca de radicais de oxigênio se elevou várias vezes 10 minutos depois do refluxo e permaneceu significantemente elevada por pelo menos 1 hora. Os radicais foram principalmente derivados de neutrófilos, em particular ânion de superóxido. Estes radicais exibiram redução acentuada depois da administração de (1) inibidores da neutrófilo NADPH-oxidase e (2) um anticorpo monoclonal (RI5,7) contra a molécula de adesão CD18 de neutrófilo. A primeira intervenção foi planejada para reduzir a explosão respiratória de neutrófilo e a segunda para reduzir o recrutamento de neutrófilos ao(s) sítio(s) de dano de reperfusão. A redução da geração de radical pelo anticorpo monoclonal RI 5,7 também foi associada com um tamanho do infarto significantemente menor e com uma diminuição concomitante nas áreas que não de refluxo. Foi demonstrado pela primeira vez que os neutrófilos ativados foram uma fonte principal de oxidantes em corações reperfundidos in vivo depois de isquemia prolongada, que este fenômeno foi duradouro e que as intervenções anti-neutrófilo puderam eficazmente prevenir o aumento na concentração transcardíaca de radicais de oxigênio durante a reperfusão. Nestes modelos de animal de infartação experimental, a falta de patologia pré existente antes da oclusão da artéria coronária pode enfatizar excessivamente a contribuição do recrutamento neutrofílico e ativação para a dano I/R; de fato, na situação aterosclerótica humana, os macrófagos ativados e linfócitos T ativados que já se situavam na “área em risco” também pode contribuir substancialmente para a dano I/R.
A isquemia causa o esgotamento de ATP em células na área afetada. Ao nível da cadeia de transporte de elétron mitocondrial, que normalmente “vaza” aproximadamente 5 % dos elétrons processados no tecido saudável, outro vazamento de espécies de oxigênio parcialmente reduzidas (em particular O2-) é favorecido quando a cadeia respiratória tomase largamente reduzida. Isto ocorre primariamente durante a isquemia. O efeito líquido no ambiente celular local é uma ponta no equilíbrio da situação redóx de anti-oxidante para pró oxidante, que é ao mesmo tempo menos capaz de absorver insulto(s) de radical adicionais sem dano celular adicional.
Prevenção de Dano por Isquemia-Reperfusão: Agentes Farmacológicos Usados em Testes de Animal e/ou Humanos Anteriores
Os seguintes compostos foram avaliados, em modelos de animal ou em testes humanos limitados, como agentes terapêuticos para a redução da dano por isquemia-reperfusão e/ou salvamento miocárdico durante o infarto do miocárdio agudo (AMI). A maioria é de antioxidantes biológicos.
• Superóxido dismutase (e derivados ou miméticos) • Catalase • Glutationa e glutationa peroxidase • Inibidores da xantina oxidase • Vitaminas B, C, E (e derivados) • Antagonistas de cálcio • Inibidores de ACE • Compostos de sulfidrila tiol (em particular N-acetilcisteína) • Queladores de ferro (desferioxamina) • Anti-inflamatórios (por exemplo, ibuprofen) • Fosfocreatina • Ν-2-mercaptopropionil glicina (MPG) • Probucol (e derivados) • Melatonina • Coenzima Q-10
O trabalho seminal por Singh e colaboradores na índia anteriormente demonstrou que pacientes humanos que se apresentavam com infarto do miocárdio agudo foram exauridos em antioxidantes endógenos e que a suplementação com coquetéis antioxidantes e/ou monoterapia com coenzima Q10 (um antioxidante lipofílico potente) foi útil para se obter tanto o salvamento miocárdico quanto a melhora nos pontos finais clínicos tradicionalmente duros (tais como as mortes cardíacas totais e a reinfartação não fatal) em 30 dias após a AMI. O teste com AMISTAD demonstrou a utilidade da adenosina como um agente de salvamento miocárdico em 3 grupos separados de pacientes. RheothRx® (um agente reológico) foi também eficaz como um agente de salvamento em testes humanos, mas foi abandonado secundário à toxicidade renal. Mais recentemente, a Medicure, Inc. demonstrou a utilidade de um derivado da vitamina B para o salvamento miocárdico em um estudo piloto de Fase II pequeno em colaboração com o 5 Duke Clinicai Research Institute. Por este motivo, o problema “translacional” (da eficácia em modelos de animal de infartação experimental para a eficácia clínica humana) identificada nas revisões anteriores por dano I/R é agora melhor entendida. Entretanto, a janela de oportunidade comercial ainda existe, visto que nenhum agente foi especificamente aprovado para o uso humano 10 como um agente de salvamento.
Regulagem no Tempo de Tratamento Para a Dano Miocárdica por Isquemia-Reperfusão
Como debatido acima, a reperfusão inicial das infartações miocárdicas agudas (primariamente com reperfusão farmacológica ou 15 cirúrgica) faz parar a morte celular devido à isquemia, mas paradoxicamente causa outra dano - mais provável pelos mecanismos oxidantes. Horwitz et al. (1999) identificou a janela de oportunidade durante a qual antioxidantes devem estar presentes nas concentrações terapêuticas para prevenir o dano de reperfusão durante 90 minutos de isquemia e 48 horas de reperfusão 20 subsequente, em 57 cães. A análise estatística no teste focalizou na identificação de componentes do número de membros do grupo responsável pelas diferenças no tamanho do infarto e revelou que a duração de tratamento foi um determinante principal. Se começado em qualquer tempo dentro da primeira hora de reperfusão, infusões de mais do que ou igual a 3 horas 25 acentuadamente diminuíram o tamanho do infarto. Duilio et al. (2001) ainda elucidou esse problema pela demonstração de que o consumo de oxigênio reflexivo da reação em cadeia do radical peroxila começa 10 minutes depois da reperfusão e que a atividade do radical permanece elevada durante pelo menos a primeira hora de reperfusão em um modelo canino. Singh et al.
(1996) anteriormente demonstrou em pacientes humanos que o salvamento miocárdico e a melhora de pontos finais clínicos duros (reinfartação não fatal, morte) foi possível começando a terapia antioxidante em média 13 horas após a MI e continuando por 28 dias. Portanto, antioxidantes plasmáticos com meias vidas longas podem ser particularmente apropriados para este cenário, visto que eles podem ser administrados como uma dose de carga e deixados consumir no plasma por todo o período pós AMI inicial crítico (0 a 24 horas). As meia vidas plasmáticas de carotenóides administrados oralmente varia de aproximadamente 21 horas para as xantofilas (carotenóides “oxigenados” incluindo astaxantina, capsantina, luteína e zeaxantina) a 222 horas para os carotenos (carotenóides de “hidrocarboneto” tais como licopeno). A diferença significante na meia vida de antioxidante plasmático (7 minutos) no teste por Horwitz et al. (1999), para a superóxido dismutase e seus miméticos nos estudos humanos, versus uma meia vida de aproximadamente 21 horas para as xantofilas e aproximadamente 9 dias para os carotenos, salientando as vantagens farmacocinéticas e cardioproteção potencial contra dano I/R pelos carotenóides em AMI em seres humanos.
Avaliação Crítica dos Antioxidantes no Dano de Reperfusão: Estudos Humanos
Os níveis médios de vitaminas A, C, E e β-caroteno foram significantemente reduzidos em pacientes apresentando com AMI, comparados com pacientes de controle em um estudo conduzido por Singh et al. (1994). Os peróxidos lipídicos foram significantemente elevados nos pacientes AMI. As relações inversas entre AMI e os níveis de plasma baixos de vitaminas permaneceram significantes depois do ajuste para fumantes e diabéticos nestes pacientes. Similarmente, 38 pacientes com AMI foram estudados por Levy et al. (1998) e exibiram níveis significantemente diminuídos de vitaminas A, E e β-caroteno comparado com pacientes de controle saudáveis igualados em idade. Depois da trombólise, os produtos de peroxidação de lipídeo aumentaram significantemente no soro de pacientes tratados. A terapia trombolítica também causou uma diminuição signifícante nos níveis de vitamina E do plasma. Estes estudos descritivos indicam que na apresentação com AMI, é provável que os níveis de soro de vitaminas antioxidantes serão diminuídos nos pacientes passam por um evento coronário agudo. A intervenção farmacológica com compostos antioxidantes no cenário agudo provável pode remediar as deficiências nas vitaminas antioxidantes e na situação antioxidante corporal total.
Os testes com a intervenção humana em perspectiva com antioxidantes no cenário da prevenção primária e/ou secundária de CVD são similarmente limitados, mas foram largamente bem sucedidos. Quatro dos cinco estudos humanos recente fortemente sustentam a eficácia da vitamina E na redução do risco de doença cardíaca e padrão de complicação. O estudo Prevenção Secundária com Antioxidantes de Doença Cardiovascular em doença Renal de Estágio Final, em pacientes com doença renal signifícante, revelou uma redução de 70 % na MI não fatal em pacientes administrados com 800 M por dia de fonte natural de vitamina E. Similarmente, como aqui mencionado, vários agentes foram agora aplicados com sucesso para aplicações em salvamento miocárdico em seres humanos.
A liberação de um composto molecular de peso baixo intravenosamente na inibição ou melhora em cenário agudo do dano I/R requerirá uma avaliação da sua imunogenicidade. A incidência de reações do tipo transfusão e outras reações adversas para a infusão rápida do composto devem ser minimizadas. Os compostos com um peso molecular < 1000 Da, por exemplo aspirina, progesterona e astaxantina, são provavelmente não imunogênicos a menos que complexados com um carregador. Conforme o peso molecular aumenta até entre 1000 e 6000 Da, por exemplo insulina e ACTH, o composto pode ou não ser imunogênico. Conforme o peso molecular aumenta a > 6000 Da, o composto é provável ser imunogênico.
Além disso, os lipídeos são raramente imunogênicos, mais uma vez a menos que complexado a um carregador. A astaxantina, como um carotenóide de xantofila, é altamente solúvel em lipídeo na forma natural. Ela também é pequena no tamanho (597 Da). Portanto, um análogo estrutural de astaxantina 5 injetável tem uma probabilidade baixa de imunogenicidade na formulação adequado e é um composto particularmente desejável para a indicação terapêutica corrente.
Prevenção de Arritmia: Agentes Farmacológicos Usados em Testes de Animal Anteriores
Estudos conduzidos por Gutstein et al. (2001) avaliaram camundongos geneticamente modificados incapazes de expressar a conexina no miocárdio [camundongos (CKO) abatidos em condicional Cx43]. Gutstein et al. descoberto que a despeito da estrutura cardíaca e desempenho contrátil normais, os camundongos CKO Cx43 desenvolveram a morte 15 cardíaca súbita, evidentemente da(s) taquicardia(s) letal(is) ventricular(es) espontânea(s). Estes dados sustentam o papel crítico do canal de junção de intervalo e conexina 43, em particular, na manutenção da estabilidade elétrica cardíaca. A Conexina 43, que é capaz de ser induzida pelos carotenóides, é a conexina mais amplamente expressada em tecidos humanos. Os carotenóides 20 e análogos estruturais de carotenóide, portanto, podem ser usados para o tratamento da arritmia.
Prevenção de Câncer: Agentes Farmacológicos Usados em
Testes com Animais Anteriores
Os carotenóides foram avaliados, principalmente em modelos 25 de animal, quanto o seu valor terapêutico possível na prevenção e tratamento do câncer. Anteriormente as propriedades antioxidantes dos carotenóides foram o foco dos estudos direcionados na direção dos carotenóides e o seu uso na prevenção do câncer. Estudos conduzidos por Bertram et al. (1991) apontaram para o fato de que embora os carotenóides fossem antioxidantes, esta propriedade particular não pareceu ser o fator principal responsável pela sua atividade como agente quimiopreventivo contra o câncer. Foi, entretanto, descoberto que a atividade dos carotenóides estava fortemente correlacionada com a sua capacidade para super regular a comunicação de junção de 5 intervalo. Foi postulado que as junções de intervalo servem como conduítes para os sinais antiproliferativos gerados pelas células normal inibidas no crescimento. A conexina 43, que é capaz de ser induzida pelos carotenóides, é a conexina mais amplamente expressada em tecidos humanos. A super regulação da conexina 43, portanto, pode ser o mecanismo pelo qual os 10 carotenóides são úteis na quimioprevenção de câncer em seres humanos e outros animais. E recentemente, um estudo humano por Nishino et al. (2003) demonstrou que um coquetel de carotenóides (10 mg de licopeno, 5 mg de cada um de a- e β-caroteno) administrados pela administração oral crônica foi eficaz na quimioprevenção de carcinoma hepatocelular em pacientes 15 cirróticos de alto risco no Japão. É provável, então, que carotenóides quimiopreventivos do câncer mais potentes (tais como astaxantina), que acumulam mais dramaticamente no fígado, serão formas de realização particularmente úteis.
Uso de Carotenóides para o Tratamento de Dano por 20 Isquemia-Reperfiisão, Doença Hepática, Arritmia e Câncer
Como aqui usado os termos “inibir” e “melhorar” são no geral definidos como a prevenção e/ou redução das conseqüências negativas de um estado de doença. Assim, os métodos e composições aqui descritos podem ter valor tanto como uma modalidade aguda quanto uma crônica (profilática).
Como aqui usado o termo “dano por isquemia-reperfusão” é no geral definido como a patologia atribuída à reoxigenação de tecido anteriormente isquêmico (crônica ou agudamente isquêmico), que inclui a doença vascular aterosclerótica e tromboembólico e as suas enfermidades relacionadas. Em particular, as doenças ou processos principais incluindo a infartação miocárdica, acidente vascular cerebral, doença vascular periférica, oclusão venosa ou arterial, transplante de órgão, cirurgia de enxerto de desvio da artéria coronária, angioplastia coronária transluminal percutânea e parada cardiovascular e/ou morte são incluídas, mas não são observados como limitantes quanto a outros processos patológicos que envolve a reperfusão de tecido isquêmico nas suas patologias individuais.
Como aqui usado o termo “arritmia” é no geral definida como qualquer variação do ritmo normal do batimento cardíaco, incluindo a arritmia sinusal, batimento prematuro, bloqueio cardíaco, fibrilação atrial, íluter atrial, taquicardia ventricular, fibrilação ventricular, pulso altemante e taquicardia paroxística. Como aqui usado o termo “arritmia cardíaca” é no geral definida como uma perturbação da atividade elétrica do coração que se manifesta como uma anormalidade na frequência cardíaca ou no ritmo cardíaco. A arritmia é mais comumente relacionada com a doença cardiovascular e em particular, doença cardíaca isquêmica.
Como aqui usado o termo “câncer” é no geral considerado ser caracterizado pelo crescimento não controlado, anormal das células. Em particular, o câncer pode se referir ao tecido em um estado de doença incluindo células iniciadas com carcinogênio e transformadas com carcinogênio.
Como aqui usado os termos “análogos de carotenóide estruturais” podem ser no geral definidos como carotenóides e os seus análogos estruturais biologicamente ativos. Os análogos típicos incluem moléculas que demonstram função equivalente ou melhorada biologicamente útil e relevante, mas que diferem estruturalmente dos compostos precursores. Os precursores de carotenóide são selecionados dos mais de 600 carotenóides que ocorrem naturalmente descritos na literatura e seus estéreo-isômeros e isômeros geométricos. Tais análogos podem incluir, mas não são limitados a, ésteres, éteres, carbonatos, amidas, carbamatos, ésteres e éteres de fosfato, sulfatos, éteres de glicosídeo, com ou sem espaçadores (ligadores).
Como aqui usado os termos “a combinação sinergística de mais do que um análogo estrutural de carotenóides” pode ser no geral definido como qualquer composição incluindo um análogo de carotenóide 5 estrutural combinado com um ou mais outros análogos de carotenóide estruturais ou co-antioxidantes, como derivados ou em soluções e/ou formulações.
Como aqui usado os termos “paciente” podem ser no geral definidos como todos os mamíferos, em particular seres humanos.
Como aqui usado os termos “administração” podem ser no geral definidos como a administração das composições farmacêuticas ou vendido diretamente (OTC) ou nutracêuticas por quaisquer meios que alcance » o seu propósito pretendido. Por exemplo, a administração pode incluir as vias parenteral, subcutânea, intravenosa, intracoronária, retal, intramuscular, infra15 peritoneal, transdérmica ou bucal. Alternativa ou concorrentemente, a administração pode ser pela via oral. A dosagem administrada será dependente da idade, saúde, peso e estado de doença do receptor, tipo de tratamento concorrente, se algum, a frequência de tratamento e a natureza do efeito desejado. Quaisquer técnicas aqui descritas direcionadas na direção da inibição do dano por isquemia-reperfusão também podem ser aplicados à inibição ou melhora de uma doença hepática, um exemplo não limitante sendo a infecção da Hepatite C. As técnicas aqui descritas direcionadas na direção da inibição e/ou melhora do dano por isquemia-reperfusão também podem ser aplicadas à inibição e/ou melhora da arritmia. As técnicas aqui descritas direcionadas na direção da inibição e/ou melhora do dano por isquemiareperfusão também podem ser aplicadas à inibição e/ou melhora do câncer.
Uma forma de realização pode incluir a administração de análogos de carotenóide estrutural sozinhos ou em combinação a um paciente tal que a ocorrência de dano por isquemia-reperfusão seja desse modo inibida
e/ou melhorada. Os análogos de carotenóide estrutural podem ser derivados solúveis em água e/ou dispersáveis em água. Os derivados de carotenóide podem incluir quaisquer substituintes que substancialmente aumente a solubilidade em água dos carotenóides que ocorrem naturalmente. Os derivados de carotenóide podem reter e/ou melhorar as propriedades antioxidantes do carotenóide precursor. Os derivados de carotenóide podem reter as propriedades não tóxicas do carotenóide precursor. Os derivados de carotenóide podem ter biodisponibilidade aumentada, em relação ao carotenóide precursor, na administração a um paciente. O carotenóide precursor pode ser de ocorrência natural.
Uma outra forma de realização pode incluir a administração de uma composição compreendida da combinação sinergística de mais do que um análogo estrutural de carotenóides a um paciente tal que a ocorrência de dano de isquemia-reperfusão é desse modo reduzido. A composição pode ser uma mistura “racêmica” (isto é mistura das formas estereoisoméricas potenciais) de derivados de carotenóide. Incluídas também estão as composições farmacêuticas compreendidas de análogos estruturais de carotenóides em combinação com um carregador farmaceuticamente aceitável (por exemplo, albumina sérica bovina). Em uma forma de realização, análogos estruturais de carotenóides podem ser complexados com albumina sérica bovina (isto é, HSA) em um solvente. A HSA pode atuar como um carregador farmaceuticamente aceitável.
Em algumas formas de realização, as composições podem incluir todas as composições de 1,0 grama ou menos de um análogo de carotenóide estrutural particular, em combinação com 1,0 grama ou menos de um ou mais outros análogos de carotenóide estrutural e/ou co-antioxidantes, em uma quantidade que seja eficaz para se obter o seu propósito pretendido. Embora as necessidades do paciente individual variem, a determinação de faixas ótimas de quantidades eficazes de cada componente está com a habilidade da técnica. Tipicamente, um análogo de carotenóide estrutural pode ser administrado a mamíferos, em particular seres humanos, oralmente em uma dose de 5 a 100 mg por dia com referência ao peso corporal do mamífero ou ser humano tratado quanto ao dano de isquemia-reperfusão.
Tipicamente, um análogo de carotenóide estrutural pode ser administrado aos mamíferos, em particular seres humanos, parenteralmente em uma dose dentre 5 a 500 mg por dia com referência ao peso corporal do mamífero ou ser humano tratados quanto ao dano de reperfusão. Em outras formas de realização, cerca de 100 mg de um análogo de carotenóide estrutural é — 10 administrado oral ou parenteralmente para tratar ou prevenir o dano por isquemia-reperfusão.
A dose oral unitária pode compreender de cerca de 0,25 mg a cerca de 1,0 grama ou cerca de 5 a 25 mg, de um análogo de carotenóide estrutural. A dose parenteral unitária pode incluir de cerca de 25 mg a 1,0 15 grama ou entre 25 mg e 500 mg, de um análogo de carotenóide estrutural. A dose intracoronária unitária pode incluir de cerca de 25 mg a 1,0 grama ou entre 25 mg e 100 mg, de um análogo de carotenóide estrutural. As dose unitárias podem ser administradas uma ou mais vezes diariamente, em dias alternados, em dose de carga ou na forma de bolo ou titulada em uma solução 20 parenteral para marcador(es) comumente aceito(s) ou novo(s) marcador(es) substituto(s) bioquímico(s) ou pontos finais clínicos como é com a habilidade da técnica.
Além de administrar um análogo de carotenóide estrutural como um produto químico puro, os compostos podem ser administrados como 25 parte de uma preparação farmacêutica contendo carregador(es) farmaceuticamente aceitável(is), preservantes, excipientes e auxiliares que facilitem o processamento do análogo de carotenóide estrutural que pode ser farmaceuticamente usado. As preparações, particularmente aquelas preparações que podem ser oralmente administradas e que podem ser usadas para o tipo preferido de administração, tais como tabletes, géis moles, pastilhas, drágeas e cápsulas e também preparações que podem ser administradas retalmente, tais como supositórios, assim como soluções adequadas para a administração pela injeção ou oralmente, podem ser preparadas em faixas de dose que fornecem biodisponibilidade similar como descrito acima, junto com o excipiente.
As preparações farmacêuticas podem ser fabricadas em uma maneira que é por si só conhecida por uma pessoa habilitada na técnica, por exemplo, por meio de processos de mistura convencional, granulação, fabricação de drágea, encapsulação em gel mole, dissolução, extração ou liofilização. Assim, as preparações farmacêuticas para o uso oral podem ser obtidas combinando-se os compostos ativos com excipientes sólidos e semisólidos e preservantes e/ou co-antioxidantes adequados. Opcionalmente, a mistura resultante pode ser triturada e processada. A mistura resultante de grânulos pode ser usada, depois da adição de auxiliares adequados, se desejado ou necessário, para se obter tabletes, géis moles, pastilhas, cápsulas ou núcleos de drágea.
Os excipientes adequados podem ser enchedores tais como sacarídeos (por exemplo, lactose, sacarose ou manose), álcoois de açúcar (por exemplo, manitol ou sorbitol), preparações de celulose e/ou fosfatos de cálcio (por exemplo, fosfato tricálcico ou hidrogeno fosfato de cálcio). Além disso, aglutinantes podem ser usados tais como pasta de amido (por exemplo, amido de milho, amido de trigo, amido de arroz, amido de batata, gelatina, tragacanto, metil celulose, hidroxipropilmetil celulose, carboximetil celulose sódica e/ou polivinil pirrolidona). Os agentes de desintegração podem ser adicionados (por exemplo, os amidos mencionados acima) assim como carboximetil-amido, polivinil pirrolidona reticulada, ágar ou ácido algínico ou um sal deste (por exemplo, alginato de sódio). Auxiliares são, todos acima, agentes reguladores de fluxo e lubrificantes (por exemplo, sílica, talco, ácido esteárico ou sais destes, tais como estearato de magnésio ou estearato de cálcio e/ou polietileno glicol ou PEG). Os núcleos de drágea são fornecidos com revestimentos adequados que, se desejado, são resistentes aos sucos gástricos. As cápsulas de gelatina mole (“géis moles”) são fornecidas com 5 revestimentos adequados, que, tipicamente, contém gelatina e/ou corante(s) comestíveis adequados. Cápsulas de gelatina isentas de componente animal e kosher podem ser particularmente adequadas para as formas de realização aqui descritas para a ampla disponibilidade de uso e consumo. Para este propósito, as soluções de sacarídeo concentradas podem ser usadas, que 10 podem opcionalmente conter goma arábica, talco, polivinil pirrolidona, polietileno glicol (PEG) e/ou dióxido de titânio, soluções de laca e solventes orgânicos adequados ou misturas de solvente, incluindo sulfóxido de dimetila (DMSO), tetraidroíurano (THF), acetona, etanol ou outros solventes e cosolventes adequados. De modo a produzir revestimentos resistentes aos sucos 15 gástricos, soluções de preparações de celulose adequadas tais como acetilcelulose ftalato ou hidroxipropilmetil-celulose ftalato, podem ser usadas. Material corante ou pigmentos podem ser adicionados aos revestimentos de tabletes ou drágeas ou cápsulas de gelatina mole, por exemplo, para a identificação ou de modo a caracterizar as combinações de doses de composto 20 ativo ou para distinguir os conteúdos da cápsula para o uso em estudos clínicos ou outros.
Outras preparações farmacêuticas que podem ser usadas oralmente incluem cápsulas de encaixe feitas de gelatina, assim como cápsulas moles, termicamente seladas feitas de gelatina e um plasticizante tal 25 como glicerol ou sorbitol. As cápsulas de encaixe podem conter os compostos ativos na forma de grânulos que podem ser misturados com enchedores tais como, por exemplo, lactose, aglutinantes tais como amidos, e/ou lubrificantes tais como talco ou estearato de magnésio e, opcionalmente, estabilizadores e/ou preservantes. Nas cápsulas moles, os compostos ativos podem ser dissolvidos ou colocados em suspensão em líquidos adequados, tais como óleos graxos tais como óleo de farelo de arroz ou óleo de amendoim ou óleo de palma ou parafina líquida. Em outras formas de realização, estabilizadores e preservantes podem ser adicionados.
As preparações farmacêuticas possíveis que podem ser usadas retalmente incluem, por exemplo, supositórios, que consistem de uma combinação dos compostos ativos com uma base de supositório. As bases de supositório adequadas são, por exemplo, triglicerídeos naturais ou sintéticos ou hidrocarbonetos de parafina. Além disso, também é possível usar cápsulas 10 de gelatina retais que consistem de uma combinação dos compostos ativos com uma base. Os materiais de base possíveis incluem, por exemplo, triglicerídeos líquidos, polietileno glicóis ou hidrocarbonetos de parafina.
As formulações adequadas para a administração parenteral incluem, mas não são limitados às soluções aquosas dos compostos ativos na 15 forma solúvel em água e/ou dispersável em água, por exemplo, sais, ésteres, carbonatos, ésteres ou éteres de fosfato, sulfatos, éteres de glicosídeo, solúveis em água, juntos com espaçadores e/ou ligadores. Além disso, as suspensões dos compostos ativos como suspensões de injeção oleosas apropriadas podem ser administradas, particularmente adequadas para a injeção intramuscular. Os 20 solventes lipofílicos adequados, co-solventes (tais como DMSO ou etanol) ou veículos incluindo óleos graxos, por exemplo, óleo de farelo de arroz ou óleo de amendoim e/ou óleo de palma ou ésteres de ácido graxo sintéticos, por exemplo, oleato de etila ou triglicerídeos, podem ser usados. As suspensões de injeção aquosas podem conter substâncias que aumentam a viscosidade da 25 suspensão incluindo, por exemplo, carboximetil celulose sódica, sorbitol, dextrano e/ou ciclodextrinas. As ciclodextrinas (por exemplo, β-ciclodextrina) podem ser usadas especificamente para aumentar a solubilidade em água para a injeção parenteral do análogo de carotenóide estrutural. As formulações lipossômicas, em que misturas do análogo de carotenóide estrutural com, por exemplo, fosfatidilcolina da gema do ovo (E-PC), podem ser feitas para injeção. Opcionalmente, a suspensão também pode conter estabilizadores, por exemplo, antioxidantes tais como BHT ou preservantes, tais como álcool benzílico.
EXEMPLOS
Tendo agora descrito a invenção, a mesma será mais facilmente entendida por referência aos seguintes exemplos, que são fornecidos por via de ilustração e não são intencionados a serem limitantes da presente invenção.
Com respeito à síntese e caracterização de compostos aqui descritos, os reagentes foram adquiridos de fontes comerciais e usados como recebidos a menos que de outro modo indicado. Os solventes para as reações e isolações foram de grau reagente e usados sem purificação a menos que de outro modo indicado. Todas as seguintes reações foram realizadas sob atmosfera de nitrogênio (N2) e foram protegidos da luz direta. A astaxantina “racêmica” (como a mistura de estereoisômeros 3S,3’S, meso e 3R,3’R em uma relação de 1:2:1) foi adquirida de Divi’s Laboratories, Ltd (Buckton r
Scott, índia). A luteína e a zeaxantina “racêmicas” foram adquiridas da Indofine Chemical Co., Inc. A cromatografia de camada fina (TLC) foi realizada em placas Uniplate Silica gel GF 250 mícrons. A análise de HPLC para o controle em processo (IPC) foi realizada em um cromatógrafo líquido Varian Prostar Série 210 com um Alltech Rocket, Platinum-C18, 100 Â, 3 pm, 7 x 53 mm, PN 50523; Temperatura: 25° C; Fase móvel: (A = água; B = 10 % de diclorometano/metanol), 40 % de A/60 % de B (partida); gradiente linear a 100 % de B em 8 min; mantendo 100 % de B durante 4 min, gradiente linear para 40 % de A/60 % de B durante 1 min; Taxa de fluxo: 2,5 ml/min; Pressão de partida: 2050 PSI; Comprimento de onda do Detector PDA: 474 nm. A RMN foi registrada em um Bruker Advance 300 e a espectroscopia de massa foi tirada em um espectrômetro ThermoFinnigan AQA. A LC/MS foi registrada em um sistema Agilent 1100 LC/MSD VL ESI; coluna: Zorbax Eclipse XDB-C18 de Rapid Resolution (4,6 x 75 mm, 3,5 pm, USUT002736); temperatura: 25° C; pressão de partida: 107 bar; taxa de fluxo: 1,0 ml/min.; fase móvel (% de A = 0,025 % de TFA em H2O, % de B = 0,025 % de TFA em acetonitrila) Método 1 (compostos 8 a 21, 23 a 27, 30, 31): 70 % de A/ 30 % de B (partida), gradiente escalonado a 50 % de B em 5 min., gradiente escalonado para 98 % de B durante 8,30 min., mantido a 98 % de B durante 15,20 min., gradiente escalonado para 30 % de B durante 15,40 min.; Método 2 (compostos 28, 29): 70 % de A/ 30 % de B (partida), gradiente escalonado para 50 % de B durante 4 min., gradiente escalonado para 90 % de B durante 7,30 min., gradiente escalonado para 98 % de B durante 10,30 min., mantido a 98 % de B durante 15,20 min., gradiente escalonado para 30 % de B durante 15,40 min.; Método 3 (composto 22): 70 % de A/ 30 % de B (partida), gradiente escalonado para 50 % de B durante 5 min., gradiente escalonado para 98 % de B durante 8,30 min., mantido a 98 % de B durante 25,20 min., gradiente escalonado para 30 % de B durante 25,40 min.; Detector PDA: 470 nm; LRMS: modo +, ESI.

Astaxantina (2E). Tempo de retenção na HPLC: 11,629 min., 91,02 % (AUC); LRMS (ESI) m/z (intensidade relativa): 598 (M+ + 2H) (60), 597 (M+ + H) (100); Tempo de retenção na HPLC: 12,601 min., 3,67 % (AUC); LRMS (ESI) m/z (intensidade relativa): 597 (M+ + H) (100); Tempo de retenção na HPLC: 12,822 min., 5,31 % (AUC); LRMS (ESI) m/z (intensidade relativa): 597 (M+ + H) (100).
(XXX)
Luteína (XXX). Tempo de retenção na HPLC: 12,606 min.,
100 % (AUC); LRMS (ESI) m/z (intensidade relativa): 568 (M+) (100).
Zeaxantina (XXXI). Tempo de retenção na HPLC: 12,741 min., 100 % (AUC); LRMS (ESI) m/z (intensidade relativa): 568 (M) (100).
Exemplo 1: Síntese de XV (o éster do Ácido Dissuccínico de Astaxantina (Éster mono-(4-{18-[4-(3-carbóxi-propionilóxi)-2,6,6-trimetil-3oxo-cicloex-1 -enil]-3,7,12,16-tetrametil-octadeca-1,3,5,7,9,11,13,15,17nonaenil}-3,5,5-trimetil-2-oxo-cicloex-3-enílico) do ácido succínico))

A uma solução de astaxantina 2E (6,0 g, 10,05 mmoles) em DCM (“diclorometano”) (50 ml) na temperatura ambiente foi adicionado DIPEA (“N,N-diisopropiletilamina”) (35,012 ml, 201 mmoles), anidrido succínico (10,057 g, 100,5 mmoles) e DMAP (“4-(dimetilamino)piridina”) (0,6145 g, 5,03 mmoles). A mistura de reação foi agitada na temperatura ambiente durante 48 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC1 1 M (60 ml/ 10 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram secadas em Na2SÜ4 e concentradas para produzir o dissuccinato de astaxantina (XV) (100 %) Tempo de retenção na HPLC: 10,031 min., 82,57 % (AUC); LRMS (ESI) m/z (intensidade relativa): 798 (M+ + 2H) (52), 797 (M+ + H) (100); Tempo de retenção na HPLC: 10,595 min, 4,14 % (AUC); LRMS (ESI) m/z (intensidade relativa): 797 (M+ + H) (40), 697 (100); Tempo de retenção na HPLC: 10,966 min, 5,68 % (AUC); LRMS (ESI) m/z (intensidade relativa): 797 (M+ + H) (100), 679 (31); Tempo de retenção na HPLC: 11,163 min, 7,61 % (AUC); LRMS (ESI) m/z (intensidade relativa): 797 (M+ + H) (38), 679 (100) e nenhuma astaxantina 2E detectável.
Exemplo 2: Síntese de XVI (o Sal dissódico do éster do Ácido Dissuccínico de Astaxantina (Éster mono-(4-{18-[4-(3-carbóxi-propionilóxi)2,6,6-trimetil-3-oxo-cicloex-1 -enil]-3,7,12,16-tetrametiloctadeca1,3,5,7,9,11,13,15,17-nonaenil} -3,5,5-trimetil-2-oxo-cicloex-3-enílico) do

O éster do ácido dissuccínico de astaxantina (2 g, 2,509 mmoles) e 200 ml de etanol foi agitada na temperatura ambiente sob nitrogênio em um frasco de fundo redondo de 500 ml. Etóxido de sódio (340 mg, 5,019 mmoles, Acros #A012556101) foi adicionado como um sólido em i 10 uma única porção e a solução foi deixada agitar durante a noite. No dia seguinte, o precipitado foi separado por filtração e lavado com etanol seguido por cloreto de metileno para produzir um sólido púrpura, o sal dissódico do éster do ácido dissuccínico de astaxantina, XVI [1,41 g, 67 %] e foi colocado em uma linha de alto vácuo para secar. !H-RMN (Metanol-d4) δ 6,77 - 6,28 15 (14 H, m), 5,53 (2 H, dd, J = 12,6, 6,8), 2,68 - 2,47 (8 H, m), 2,08 - 1,88 (22
H, m), 1,37 (6 H, s), 1,24 (6H, s); 13C RMN (CDC13) δ 196,66, 180,80, 175,01, 163,69, 144,12, 141,38, 138,27, 136,85, 136,12, 135,43, 132,35, 129,45, 126,22, 124,71, 72,68, 44,09, 38,63, 34,02, 32,34, 31,19, 26,86, 14,06, 13,19, 12,91; Espectroscopia de massa +ESI, 819,43 sal monossódico, * 20 797,62 éster do ácido dissuccínico de astaxantina; HPLC 7,41 min (99,84 %).
Exemplo 3: Síntese do éster BocLys(Boc)OH de Astaxantina (XXI).
HO'
2) HC14M em 1,4-dioxano
1) SsçUslBóelOH. CSJAP i> sjnb.
HPLC: Coluna: Waters Symmetry Cl8 3,5 mícrons 4,6 mm x 150 mm; Temperatura: 25° C; Fase móvel: (A = 0,025 % de TFA em H2O; B = 0,025 % de TFA em MeCN), 95 % de A/5 % de B (partida); gradiente linear para 100 % de B durante 12 min, mantido durante 4 min; gradiente 5 linear para 95 % de B/5 % de A durante 2 min; gradiente linear para 95 % de A/5 % de B durante 4 min; Taxa de fluxo: 2,5 ml/min; Comprimento de onda do detetor: 474 nm.
A uma mistura de astaxantina 2E (11,5 g, 19,3 mmoles) e BocLys(Boc)OH (20,0 g, 57,7 mmoles) em cloreto de metileno (500 ml) 10 foram adicionados 4-dimetilaminopiridina (DMAP) (10,6 g, 86,6 mmoles) e 1,3-diisopropilcarbodiimida (“DIC”) (13,4 g, 86,7 mmoles). O frasco de fundo redondo foi coberto com folha de alumínio e a mistura foi agitada na temperatura ambiente sob nitrogênio durante a noite. Depois de 16 horas, a reação foi incompleta pela HPLC e TLC. Um adicional de 1,5 equivalente de 15 DMAP e DIC foram adicionados à reação e depois de 2 horas, a reação foi completa pela HPLC. A mistura foi depois concentrada a 100 ml e um sólido branco de (1,3-diisopropiluréia) foi separado por filtração. O filtrado foi submetido à cromatografia cintilante através de gel de sílica (10%a50% Heptano/EtOAc) para dar o produto desejado como um sólido vermelho 20 escuro (XXI) (28,2 g, > 100 % de rendimento). !H RMN (DMSO-d6) δ 7,24 (2 H, t, J = 6,3 Hz), 6,78 (2 H, d, 5,0 Hz), 6,57 - 6,27 (14 H, m), 5,50 - 5,41 (2
H, m), 3,99 - 3,97 (2 H, d, 6,0 Hz), 2,90 (4 H, m), 2,03 (4 H, m), 2,00 (6 H, s),
I, 97 (6 H, s), 1,82 (6 H, s), 1,70 - 1,55 (4 H, m), 1,39 - 1,33 (36 H, m), 1,24 1,13 (8 H, m), 1,01 - 0,99 (6 H, m), 0,86 - 0,83 (6 H, m). HPLC: 21,3 min (24,6 % de AUC)); 22,0 min (48,1 % (AUC)); 22,8 min (20,6 % (AUC)).
TLC (1:1 Heptano/EtOAc: Rf 0,41; Rf 0,5; Rf 0,56). A análise de LC/MS foi realizada em um sistema Agilent 1100 LC/MSD VL ESI pela injeção de fluxo no modo positivo; Fase móvel: A = 0,025 % de TFA em H2O; B = 0,025 % de TFA m MeCN, 10 % A/90 % B (partida); Pressão de partida: 10 bar; Detector
PDA 470 nm. +ESI, m/z = 1276,1 (M + Na).
Exemplo 4: Síntese do Sal de Tetracloridreto do Éster de Dilisinato de Astaxantina (XX).
Uma mistura de éster DiBocLys(Boc) de astaxantina (XXI) (20,0 g, 16,0 mmoles) e HC1 em 1,4-dioxano (4,00 M, 400 ml, 1,60 mol, 100 eq) foi agitada na temperatura ambiente sob uma atmosfera de nitrogênio. O frasco de fundo redondo foi coberto com folha de alumínio e a reação foi agitada durante 1 hora, tempo no qual a reação foi completa pela HPLC. O composto do título precipitou e foi coletado pela filtração, lavado com éter (3 x 100 ml) e secado (14,7 g, 92 %, 91,6 % de pureza pela HPLC). Uma porção (13,5 g) do sólido bruto foi dissolvido em 500 ml de uma mistura 1:2 de metanol/cloreto de metileno e agitada sob nitrogênio. Éter dietílico (168 ml) foi depois adicionado às gotas e o sólido precipitado foi coletado pela filtração para produzir o produto desejado como um sólido vermelho escuro (8,60 g, 63,7 % de rendimento). ’H RMN (DMSO-d6) δ 8,65 (6 H, s), 8,02 (6
H, s), 6,78 - 6,30 (14 H, m), 5,59 - 5,51 (2 H, m), 4,08 (2 H, m), 2,77 (4 H, m), 2,09 - 2,07 (4 H, m), 2,01 (6 H, s), 1,97 (6 H, s), 1,90 - 1,86 (4 H, m),
I, 84 (6 H, s), 1,61 - 1,58 (8 H, m), 1,37 (6 H, s), 1,22 (6 H, s). HPLC: 7,8 min (97,0 % (AUC)). A análise de LC/MS foi realizada em um sistema Agilent 1100 LC/MSD VL ESI com Zorbax Eclipse XDB-C18 Rapid Resolution 4,6 x 75 mm, 3,5 mícrons, USUT002736; Temperatura: 25° C; Fase móvel: (% de A = 0,025 % de TFA em H2O; % B = 0,025 % de TFA em MeCN), 70 % de A/30 % de B (partida); gradiente linear para 50 % de B durante 5 min, gradiente linear para 100 % de B durante 7 min; Taxa de fluxo: 1,0 ml/min; Pressão de partida: 108 bar; Detector PDA 470 nm. Espectrometria de massa +ESI, m/z = 853,9 (M + Hf), m/z = 875,8 (M + Na+); LC 4,5 min.
Exemplo 5: Síntese do 6-Éster Bis-(Ácido Ascórbico 2-OTBS) de Dissuccinato de astaxantina (XXII).
HPLC: Coluna: Waters Symmetry C18 3,5 mícrons 4,6 mm x 150 mm; Temperatura: 25° C; Fase móvel: (A = 0,025 % de TFA em água; B = 0,025 % de TFA em acetonitrila), 95 % de A/5 % de B (partida); gradiente linear para 100 % de B durante 5 min, mantido durante 10 min; gradiente linear para 95 % de B durante 2 min; gradiente linear para 95 % de A/5 % de B durante 3 min; Taxa de fluxo: 1,0 ml/min; Comprimento de onda do detetor: 474 nm.
A uma solução agitada de dissuccinato de astaxantina (XV) (20,00 g, 25,1 mmoles) em 600 ml de diclorometano foi adicionado 4dimetilaminopiridina (DMAP) (6,13 g, 50,2 mmoles), ácido ascórbico 2-Oterc-butil-dimetilsilila (OTBS) (XXVI) (21,86 g, 75,3 mmoles) e cloridreto de l-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI-HC1) (12,02 g, 62,75 mmoles). Depois de 14 horas, a mistura de reação foi submetida à cromatografia cintilante através de gel de sílica (1,0 kg de gel de sílica, eluente 0,5 % de HOAc/5 % de MeOH/EtOAc). A fração 10 foi concentrada para produzir um sólido vermelho escuro (6,47 g, 19,2 % de rendimento, 58 % de pureza AUC pela HPLC). O produto bruto foi submetido à cromatografia cintilante através de gel de sílica (600 g de gel de sílica, eluente 0,25 % de HOAc/5 % de MeOH/EtOAc). As frações 6 a 10 foram concentradas sob vácuo para produzir um sólido vermelho escuro (1,50 g, 4,4 % de rendimento, 94,8 % de pureza AUC pela HPLC ’Η-ΙΤΜΝ (CDC13) δ 11,13 (2H, s), 6,78 - 6,28 (14 H, m), 5,43 (2 H, dd, J = 12,2, 7,1 Hz), 5,34 (2H, s), 4,78 (2H, d, J = 5,4 Hz), 4,11 - 4,07 (6H, m), 2,69 - 2,65 (8 H, m),
2,05 - 1,97 (22 H, m), 1,81 (6 Η, s), 1,33 (6 Η, s), 0,92 (18 Η, s), 0,15 (6 Η, s), 0,14 (6Η, s); HPLC 13,4 min [94,8 % (AUC)]; Espectroscopia de massa -ESI, m/z = 1340,6 (M+).
Exemplo 6: Síntese do 6-Éster do Ácido Bis-Ascórbico de Dissuccinato de astaxantina (XX).
A uma solução agitada do 6-éster de bis-(ácido ascórbico 2OTBS) de dissuccinato de astaxantina (XXII) (100 mg, 0,075 mmol) em THF (5 ml) a 0o C foi adicionado BF-Et3N (121 μΐ, 0,745 mmol). A reação foi agitada durante 1 hora a 0o C depois aquecido até a temperatura ambiente. A reação foi agitada 2,5 horas antes de ser resfriada bruscamente vertendo-se em um funil de separação contendo 5 ml de IPAC e 5 ml de água. A camada aquosa foi removida e a camada orgânica foi lavada com água (2x5 ml). Os solventes orgânicos foram removidos pela evaporação rotativa para dar um sólido vermelho escuro, que foi usado sem purificação. ÍH-RMN (CDC13) δ 11,12 (2 H, s), 8,40 (2 H, s), 6,87 - 6,28 (14 H, m), 5,43 - 5,32 (4 H, m), 4,69 (s, 2H), 4,09 (s, 4H), 3,99 (s, 2H), 2,68 - 2,50 (m, 8H), 2,00 - 1,76 (22 H, m), 1,36 - 1,19 (12 H, m); HPLC 8,9 min [80,7 % (AUC)]; Espectroscopia de massa +ESI, m/z = 1113,2 (M + H1-).
Exemplo 7: Síntese do Sal Sódico do 6-Éster do Ácido BisAscórbico de Dissuccinato de astaxantina (XXIII).
A uma solução agitada do 6-éster do ácido bis-ascórbico bruto de dissuccinato de astaxantina (XIX) (0,075 mmol) em acetona (5 ml) na temperatura ambiente foi adicionado trietilortoformiato (62 μΐ, 0,373 mmol). A solução foi agitada 15 min, depois uma solução de 2-etilexanoato de sódio em acetona (93 μΐ, 0,019 mmol, 0,20 M) foi adicionado às gotas. O precipitado resultante foi removido pela filtração. O filtrado foi esfriado a 0o C e tratado com 2-etilexanoato de sódio adicional em acetona (373 μΐ, 0,075 mmol, 0,20 Μ). A reação foi agitada durante 5 min depois o material sólido foi coletado pela filtração, lavado com acetona (5 ml) e secado sob alto vácuo para dar um sólido vermelho escuro (27,8 mg, 32,2 % de rendimento): HPLC 8,9 min [88,2 % (AUC)J, Espectroscopia de massa +APCI, m/z = 1113,3 (M + 3H - 2Na*).
Exemplo 8: Síntese do Éster Dicicloexilmetilílico de Dissuccinato de astaxantina (XXIV).
HPLC: Coluna: Alltech Rocket, Platinum-C 18, 100 A, 3 pm, 7 x 53 mm; Temperatura: 25° C; Fase móvel: (A = 0,025 % de TFA em água; B = 0,025 % de TFA em acetonitrila), 70 % de A/30 % de B (partida); mantido durante 40 segundos; gradiente linear para 50 % de B durante 4 min 20 segundos; gradiente linear para 100 % de B durante 1 min 30 segundos, mantido durante 4 min 40 segundos; gradiente linear para 70 % de A/30 % de B em 20 segundos; Taxa de fluxo: 2,5 ml/min; Comprimento de onda do detetor: 474 nm.
A uma solução agitada do dissuccinato de astaxantina (XV) (100 mg, 0,125 mmol) e N,N-dimetilaformamida (6,0 ml) em um frasco de fundo redondo de 25 ml foi adicionado carbonato de césio (90,0 mg, 0,275 mmol) na temperatura ambiente sob N2 e coberto com folha de alumínio. A reação foi agitada durante 15 minutos depois bromometil cicloexano (52,0 μ, 0,375 mmol) foi adicionado. Depois de 2 dias, a reação foi resfriada bruscamente pela adição de 4 ml de uma solução saturada de bicarbonato de sódio e diluída com 50 ml de diclorometano. A solução diluída foi lavada duas vezes com 25 ml de água antes da secagem em sulfato de sódio anidro. A solução orgânica foi filtrada e o solvente foi removido pela evaporação rotativa. O resíduo bruto foi purificado pela cromatografia cintilante (10 a 50 % EtOAc/heptano) para produzir um sólido vermelho escuro (40,2 mg, 32,5 % de rendimento): ^-RMN (CDC13) δ 7,03 - 6,17 (14 H, m), 5,54 (2 H, dd, J = 12,9, 6,7 Elz), 3,92 (4 H, d, J = 6,4 Hz), 2,82 - 2,63 (8 H, m), 2,08 - 1,92 (14 H, m), 1,90 (6 H, s), 1,75 - 1,62 (14 H, m), 1,34 - 1,20 (22 H, m); HPLC 8,9 min [83,9 % (AUC)]; TLC (3:7 EtOAc/heptano: Rf 0,38); Espectroscopia de massa +ESI, m/z = 989,6 (Μ + H4).
Exemplo 9: Síntese de Ácido Ascórbico 2-OTBS-5,6Isopropiledina (XXV).
HPLC: Alltech Rocket, Platinum-C18, 100A, 3 pm, 7 x 53 mm, PN 50523; Temperatura: 25° C; Fase móvel: (A = 0,025 % de TFA em água; B = 0,025 % de TFA em acetonitrila), 90 % de A/10 % de B (partida); gradiente linear para 30 % de B durante 3 min; gradiente linear para 90 % de B durante 3 min, mantido durante 2 min; gradiente linear para 90 % de A/10 % de B durante 1 min, depois mantido durante 1 min; Taxa de fluxo: 2,5 ml/min; Comprimento de onda do detetor: 256 nm.
A uma solução agitada de ácido ascórbico 5,6-isopropiledina (100,0 g, 463 mmoles) em 1,00 litro de THF foi adicionado o cloreto de tercbutildimetilsilila (TBSCI) (76,7 g, 509 mmoles) na temperatura ambiente seguido pela adição de Ν,Ν-diisopropiletilamina (DIPEA) (161 ml, 925 mmoles) durante 30 min. A reação foi agitada 14 horas na temperatura ambiente, depois concentrada sob vácuo. A mistura foi dissolvida em éster metil terc-butila (MTBE) (1,00 litro) e extraído com carbonato de potássio 1 M (1,00 litro) em um funil de separação. A camada aquosa foi extraída mais uma vez com MTBE (1,00 litro) e o pH da camada aquosa foi ajustado até o pH 6 usando HC1 2 N. A camada aquosa foi extraída duas vezes com acetato de isopropila (IPAC) (1,00 litro) e concentrada para produzir um sólido branco amarelado (150,4 g, 98 % de rendimento): ^-RMN (DMSO d6) δ 11,3 (1 H, s), 4,78 (1 H, d, J = 2,0 Hz), 4,41 - 4,36 (1 H, m), 4,11 (1 H, dd, J = 8,4, 7,4 Hz), 3,92 (1 H, dd, J = 8,4, 6,0), 1,24 (3 H, s), 1,23 (3 H, s), 0,92 (9 H, s), 0,14 (6H, s); HPLC 5,9 min [91,6 % (AUC)J; Espectroscopia de massa -ESI, m/z = 329,2 (M-H-).
Exemplo 10: Síntese de Ácido Ascórbico 2-OTBS (XXVI).
A uma solução agitada de ácido ascórbico 2-OTBS-5,6isopropiledina (XXV) (150,4 g, 455 mmoles) em 1,50 litro de diclorometano na temperatura ambiente foi adicionado propanoditiol (54,0 ml, 546 mmoles) sob nitrogênio. A solução foi esfriada a -45° C e depois BF3-OEt2 (58,0 ml, 455 mmoles) foi adicionado às gotas em uma taxa que manteve a temperatura abaixo de -40° C. Depois de 1 hora, a reação foi completa pela HPLC. A reação foi resfriada bruscamente vertendo-se a mistura de reação fria em um funil de separação contendo 1,00 litro de IP AC e 500 ml de uma solução saturada de cloreto de amônio e 500 ml de água. A camada orgânica foi concentrada a um sólido branco. De modo a purgar o propano ditiol, o sólido foi novamente transformado em pasta fluida em diclorometano (250 ml) durante 2 horas e heptano (1,00 litro) foi adicionado e agitado durante 1 hora. A mistura foi concentrada sob vácuo até um volume de 500 ml. A mistura foi filtrada e secada sob vácuo para produzir um sólido branco (112,0 g, 85 % de rendimento): lH-RMN (DMSO d6) δ 11,0 (1 H, s), 4,89 (2 H, s), 4,78 (1 H, d, J = 1,2 Hz), 3,82 - 3,80 (1 H, m), 3,45 - 3,42 (2 H, m), 0,923 (9 H, s), 0,14 (6H, s); HPLC 4,9 min [92,0 % (AUC)]; Espectroscopia de massa -ESI, m/z = 289,0 (Μ - H-).
Exemplo 11: Síntese do Éster de bis-dimetilfosfato de
Astaxantina (XXVII).
HPLC: Waters Symmetry C18, 3 pm, 4,6 x 150 mm, WAT200632, Temperatura: 25° C; Fase móvel: (A = água; B =10 % DCM/ MeOH), 10 % de A/90 % de B (partida); gradiente linear para 100 % de B durante 9 min; mantido 100 % de B durante 11 min, gradiente linear para 10 5 % de A/90 % de B durante 1 min; Taxa de fluxo: 1,0 ml/min; Comprimento de onda do detetor: 474 nm.
A uma mistura de astaxantina 2E (500 mg, 0,84 mmol) e metil imidazol (0,50 ml, 6,27 mmoles) em cloreto de metileno a 37° C foi adicionado bromofosfato de dimetila (2 M, 5,04 ml) (Ding, 2000). Depois de 10 24 horas, a reação não foi completa pela HPLC e bromofosfato de dimetila (2
M, 5,04 ml) foi adicionado. Depois de 48 horas, a reação não foi completa pela HPLC e bromofosfato de dimetila (2 M, 5,04 ml) foi adicionado. Depois de 72 horas, a reação foi completa pela HPLC. A reação foi diluída com cloreto de metileno (20 ml) e resfriada bruscamente com água (20 ml). As 15 camadas foram separadas e a camada aquosa foi extraída mais uma vez com 20 ml de cloreto de metileno. As camadas orgânicas foram combinadas e concentradas sob vácuo para produzir 2,69 g (> 100 % de rendimento). JH RMN (CDC13) δ 6,58 - 6,14 (14 H, m), 5,05 - 4,95 (2 H, m), 3,91 - 3,60 (12 H, m), 2,11 - 2,04 (4 H, m), 2,04 - 1,92 (12 H, m), 1,85 (6 H, s), 1,26 (6 H, s), 20 1,15 (6 H, s). HPLC: 4,29 min (86,7 % de AUC)). Fase móvel: A = 0,025 % de TFA em H2O; B = 0,025 % de TFA em acetonitrila, 10 % de A/90 % de B (partida); Detector PDA 474 nm. +ESI, m/z = 813,62 (M + 1).
Exemplo 12: Síntese do Éster BocProOH de Astaxantina (XXVIII).
Análise de LC/MS: a análise de LC/MS foi realizada em um sistema Agilent 1100 LC/MSD VL ESI com Zorbax Eclipse XDB-C18 Rapid Resolution 4,6 x 75 mm, 3,5 pm, USUT002736; Temperatura: 25° C; Fase móvel: (% de A = 0,025 % de TF A em H2O; % de B = 0,025 % de TFA em MeCN), 70 % de A/30 % de B (partida); gradiente linear para 50 % de B durante 5 min, gradiente linear para 98 % de B durante 3 min, mantido a 98 % de B durante 17 min; Taxa de fluxo: 1,0 ml/min; Pressão de partida: 108 bar; Detector PDA 470 nm, 373 nm, 214 nm. LRMS: modo +, ESI.
A uma mistura de astaxantina 2E (5,00 g, 8,38 mmoles) e BocProOH (10,8 g, 50,3 mmoles) em cloreto de metileno (500 ml) foram adicionados 4-dimetilaminopiridina (DMAP) (6,14 g, 50,3 mmoles) e 1,3diisopropilcarbodiimida (DIC) (7,79 ml, 50,3 mmoles). A mistura foi agitada na temperatura ambiente sob nitrogênio durante a noite. Depois de 16 horas, a reação foi completa pela TLC. A mistura foi depois concentradas até a secura e o resíduo bruto foi transformado em pasta fluida com 100 ml de éter dietílico e filtrada através de uma almofada de Celite. O filtrado foi submetido à cromatografia cintilante através de gel de sílica (Et2O) para dar o produto desejado como um sólido vermelho escuro (8,56 g, > 100 % de rendimento). LC: 17,5 min [23,1 % de AUC)]; 18,2 min [45,1 % (AUC)]; 19,4 min [22,0 % (AUC)]. TLC (3:2 EtOAc/Hexano: Rf 0,51; Rf 0,55; Rf 0,59). MS +ESI, m/z = 1013,8 (M + Na+).
Exemplo 13: Síntese do Sal de Dicloridreto do Éster de
Diprolinato de Astaxantina (XXIX).
Uma mistura de éter dietílico (130 ml) e EtOH (48,9 ml, 838 mmoles) foi esfriado a -78° C sob uma atmosfera de nitrogênio. Cloreto de acetila (82,0 ml, 838 mmoles) foi adicionado às gotas à mistura esfriada
durante 30 minutos. A reação foi removida do banho de esfriamento e deixada aquecer lentamente até a temperatura ambiente. Os conteúdos do frasco foram vertidos em um frasco de fundo redondo separado contendo o éster DiBocPro de astaxantina (XXVIII) (8,31 g, 8,38 mmoles) e uma barra agitadora. O frasco foi coberto com folha de alumínio e a reação foi agitada na temperatura ambiente sob nitrogênio durante a noite. Depois de 16 horas a reação foi completa pela LC. O composto do título precipitou e foi coletado pela filtração, lavado com éter (3 x 100 ml) e secado (6,37 g, 88,0 % de rendimento bruto, 75,2 % de pureza pela LC). LC: 8,00 min [75,2 % (AUC)]. MS +ESI, m/z = 791,7 (Μ + H*).
Exemplo 14: Síntese de Dissuccinato de Luteína (XXXII).
A uma solução de luteína (XXX) (0,010 g, 0,018 mmol) em DCM (2 ml) na temperatura ambiente foi adicionado DIPEA (0,063 ml, 0,360 mmol), anidrido succínico (0,036 g, 0,360 mmol) e DMAP (0,021 g, 0,176 mmol). A mistura de reação foi agitada na temperatura ambiente durante 48 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC1 1 M (6 ml/ 1 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram secadas em Na2SO4 e concentradas para produzir o dissuccinato de luteína (XXXII) (93,09 %) Tempo de retenção na HPLC: 11,765 min., 93,09 % (AUC); LRMS (ESI) m/z (intensidade relativa): 769 (M+) (24), 651 (100) e nenhuma luteína detectável.
r ,
Exemplo 15: Síntese de Esteres do Acido Succínico de Zeaxantina (XXXIII, XXXIV).
(XXXIII e XXXIV)
A uma solução de zeaxantina (XXXI) (0,010 g, 0,018 mmol) em DCM (2 ml) na temperatura ambiente foi adicionado DIPEA (0,063 ml, 0,360 mmol), anidrido succínico (0,036 g, 0,360 mmol) e DMAP (0,021 g, 0,176 mmol). A mistura de reação foi agitada na temperatura ambiente durante 48 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC1 1 M (6 ml/ 1 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram secadas em Na2SO4 e concentradas para produzir monossuccinato de zeaxantina (XXXIII) (2,86 %) Tempo de retenção na HPLC: 12,207 min, 2,86 % (AUC); LRMS (HSI) m/z (intensidade relativa): 669 (M+ + H) (53), 668 (M+) (100), dissuccinato de zeaxantina (XXXIV) (97,14 %) Tempo de retenção na HPLC: 11,788 min, 67,42 % (AUC); LRMS (HSI) m/z (intensidade relativa): 792 (M++ Na) (42), 769 (M+) (73), 651 (100); Tempo de retenção na HPLC: 13,587 min, 11,19% (AUC); LRMS (HSI) m/z (intensidade relativa): 792 (M+ + Na) (36), 769 (M+) (38), 663 (100); Tempo de retenção na HPLC: 13,894 min, 18,53 % (AUC); LRMS (HSI) m/z (intensidade relativa): 769 (M+) (62), 663 (77), 651 (100) e nenhuma zeaxantina detectável
Exemplo 16: Síntese de Ésteres do Ácido Aconítico de astaxantina (XXXV, XXXVI).
(XXXV e XXXVI)
A uma solução de astaxantina 2E (0,100 g, 0,168 mmol) em DCM/DMF (‘TSl,N-dimetilformamida”) (4 ml /2 ml) na temperatura ambiente foi adicionado DIPEA (0,878 ml, 5,04 mmoles), anidrido cis-aconítico (0,2622 g, 1,68 mmol) e DMAP (0,4105 g, 3,36 mmoles). A mistura de reação foi agitada na temperatura ambiente durante 36 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC1 1 M

(20 ml/ 3 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram concentradas para produzir o monoéster aconítico (XXXV) (13,25 %) Tempo de retenção na HPLC: 10,485 min., 4,95 % (AUC); LRMS (HSI) m/z (intensidade relativa): 777 (M+ + Na + 2H) (57), 623 (100); Tempo de retenção na HPLC: 10,722 min., 8,30 % (AUC); LRMS (HSI) m/z (intensidade relativa): 777 (M++ Na + 2H) (6), 709 (100), o diéster aconítico (XXXVI) (27,67 %) tempo de retenção na HPLC: 9,478 min., 15,44 % (AUC); LRMS (HSI) m/z (intensidade relativa): 933 (M+ + Na + 2H) (10), 831 (100); Tempo de retenção na HPLC: 9,730 min., 12,23 % (AUC); LRMS (HSI) m/z (intensidade relativa): 913 (Μ* + 4H) (4), 843 (100) e astaxantina (44,40 %).
Exemplo 17: Síntese de Ésteres do ácido cítrico de astaxantina (XXXVII, XXXVIII).

A uma suspensão de ácido cítrico (0,5149 g, 2,86 mmoles) em DCM (8 ml) na temperatura ambiente foi adicionado DIPEA (1,167 ml, 0,6,70 mmol), DIC (0,525 ml, 3,35 mmoles), DMAP (0,4094 g, 3,35 mmoles) e astaxantina (0,200 g, 0,335 mmol). A mistura de reação foi agitada na temperatura ambiente durante 36 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC1 1 M (20 ml/3 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram concentradas para produzir o monoéster do ácido cítrico (XXXVII) (26,56 %) Tempo de retenção na HPLC: 9,786 min., 17,35 % (AUC); LRMS (ESI) m/z (intensidade relativa): 773 (M+ + 3H) (14), 771 (M+ + H) (100); Tempo de retenção na HPLC: 9,989 min., 9,21 % (AUC); LRMS (ESI) m/z (intensidade relativa): 773 (Nf + 3H) (50), 771 (Μ* + H) (100), diéster do ácido cítrico (XXXVIII) (7,81 %) Tempo de retenção na HPLC: 8,492 min., 3,11 % (AUC); LRMS (ESI) m/z (intensidade relativa): 968 (M* + Na) (75), 967 (100), 946 (M++ H) (37); Tempo de retenção na HPLC: 8,708 min., 2,43 % 5 (AUC); LRMS (ESI) m/z (intensidade relativa): 968 (M+ + Na) (95), 946 (M+ + H) (100); Tempo de retenção na HPLC: 8,952 min., 2,27 % (AUC); LRMS (ESI) m/z (intensidade relativa): 946 (M++ H) (19), 500 (100) e astaxantina (21,26%).
Exemplo 18: Síntese de monoéster do ácido 10 dimetilaminobutírico de astaxantina (XXXIX).

A uma suspensão de cloridreto do ácido 4-(dimetilamino)butírico (0,2816 g, 1,68 mmol) em DCM/ DMF (3 ml /3 ml) na temperatura ambiente foi adicionado DIPEA (0,878 ml, 5,04 mmoles), HOBT (“1hidroxibenzotriazol”)-H2O (0,3094 g, 2,02 mmoles), DMAP (0,4105 g, 3,36 15 mmoles) e astaxantina (0,100 g, 0,168 mmol). A mistura de reação foi agitada na temperatura ambiente durante 36 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC1 1 M (20 ml/ 3 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram concentradas para produzir o monoéster do ácido 4-(dimetilamino)butírico 20 (XXXIX) (24,50 %) Tempo de retenção na HPLC: 9,476 min., 20,32 % (AUC); LRMS (ESI) m/z (intensidade relativa): 732 (M+ + Na) (13), 729 (100); Tempo de retenção na HPLC: 9,725 min., 4,18 % (AUC); LRMS (ESI) m/z (intensidade relativa): 732 (M+ + Na) (50), 729 (100) e astaxantina (61,21 %).
Exemplo 19: Síntese de Monoéster Glutationa de astaxantina (L).

A uma suspensão de glutationa reduzida (0,5163 g, 1,68 mmol) em DCM/ DMF (3 ml /3 ml) na temperatura ambiente foi adicionado DIPEA (0,878 ml, 5,04 mmol), HOBT-H2O (0,3094 g, 2,02 mmoles), DMAP (0,4105 g, 3,36 mmoles), DIC (0,316 ml, 2,02 mmoles) e astaxantina 2E (0,100 g, 0,168 mmol). A mistura de reação foi agitada na temperatura ambiente durante 36 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC11 M (20 ml/ 3 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram concentradas para produzir o monoéster de glutationa (L) (23,61 %) Tempo de retenção na HPLC: 9,488 min., 16,64 % (AUC); LRMS (ESI) m/z (intensidade relativa): 886 (M) (13), 810 (54), 766 (100); Tempo de retenção na HPLC: 9,740 min., 3,57 % (AUC); LRMS (ESI) m/z (intensidade relativa): 886 (M+) (24), 590 (78), 546 (100); Tempo de retenção na HPLC: 9,997 min., 3,40 % (AUC); LRMS (ESI) m/z (intensidade relativa): 886 (M+) (25), 869 (85), 507 (100) e astaxantina (68,17 %).
Exemplo 20: Síntese de diéster do ácido tartárico de astaxantina (LI).
A uma suspensão de ácido (L)-tartárico (0,4022 g, 2,68 mmoles) em DCM/ DMF (5 ml /5 ml) na temperatura ambiente foi adicionado DIPEA (1,167 ml, 0,6,70 mmol), HOST-H2O (0,5131 g, 3,35 mmoles), DMAP (0,4094 g, 3,35 mmoles) e astaxantina 2E (0,200 g, 0,335 mmol). A mistura de reação foi agitada na temperatura ambiente durante 36 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com
salmoura/ HC1 1 M (20 ml/3 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram concentradas para produzir o diéster do ácido tartárico (LI) (18,44 %) Tempo de retenção na HPLC: 9,484 min., 14,33 % (AUC); LRMS (ESI) m/z (intensidade relativa): 884 (M+ + Na + H) (100), 5 815 (72), 614 (72); Tempo de retenção na HPLC: 9,732 min., 4,11 % (AUC);
LRMS (ESI) m/z (intensidade relativa): 883 (M+ + Na) (100), 539 (72) e ' astaxantina (67,11 %).
Exemplo 21: Síntese de monoéster de sorbitol de dissuccinato de astaxantina (LII).
A uma solução de dissuccinato de astaxantina (XV) (0,200 g,
0,251 mmol) em DMF (10 ml) na temperatura ambiente foi adicionado DIPEA (1,312 ml, 7,53 mmoles), HOBT-H2O (0,4610 g, 3,01 mmoles), DMAP (0,6133 g, 5,02 mmoles) e (D)-sorbitol (0,4572 g, 2,51 mmoles). A mistura de reação foi agitada na temperatura ambiente durante 36 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC1 1 M (20 ml/ 3 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram concentradas para produzir o monoéster de sorbitol (LII) (3,52 %) Tempo de retenção na HPLC: 9,172 min., 3,52 % (AUC); LRMS (ESI) m/z (intensidade relativa): 984 (M+ + Na) (28), 503 (100) e dissuccinato de astaxantina (91,15%).
Exemplo 22: Síntese de Diéster de sorbitol de dissuccinato de astaxantina (LIII).
A uma solução de dissuccinato de astaxantina (XV) (0,100 g,
0,125 mmol) em DCM/ DMF (3 ml /3 ml) na temperatura ambiente foi adicionado DIPEA (0,656 ml, 3,76 mmoles), HOBT-H2O (0,2313 g, 1,51 mmol), DMAP (0,3067 g, 2,51 mmoles), DIC (0,236 ml, 1,51 mmol) e (D)sorbitol (0,2286 g, 1,25 mmol). A mistura de reação foi agitada na temperatura ambiente durante 36 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC1 1 M (20 ml/ 3 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram concentradas para produzir diéster de sorbitol (LIII) (44,59 %) Tempo de retenção na HPLC: 8,178 min., 11,58 % (AUC); LRMS (ESI) m/z (intensidade relativa): 1148 (M+ + Na) (40), 545 (100); Tempo de retenção na HPLC: 8,298 min., 33,01 % (AUC); LRMS (ESI) m/z (intensidade relativa): 1148 (M+ + Na) (20), 545 (100) e nenhum dissuccinato de astaxantina detectável.
Exemplo 23: Síntese de Carbamatos de morfolina de astaxantina (LIV, LV).
(LIV e LV)
A uma solução de astaxantina 2E (0,100 g, 0,168 mmol) em DCM/ DMF (3 ml/3 ml) na temperatura ambiente foi adicionado DIPEA (0,878 ml, 5,04 mmoles), DMAP (0,4105 g, 3,36 mmoles) e cloreto de 4morfolino carbonila (0,196 ml, 1,68 mmol). A mistura de reação foi agitada na temperatura ambiente durante 36 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC1 1 M (20 ml/ 3 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram concentradas para produzir monocarbamato de 4-morfolina (LIV) (33,17 %) Tempo de retenção na HPLC: 11,853 min., 29,01 % (AUC); LRMS (ESI) m/z (intensidade relativa): 710 (M+) (100); Tempo de retenção na HPLC: 13,142 min., 1,37 % (AUC); LRMS (ESI) m/z (intensidade relativa): 710 (M+) (100); Tempo de retenção na HPLC: 13,383 min., 2,79 % (AUC); LRMS (ESI) m/z (intensidade relativa): 710 (M) (100), dicarbamato de 4-morfolina (LV) (33,42 %) Tempo de retenção na HPLC: 12,049 min., 29,71 % (AUC); LRMS (ESI) m/z (intensidade relativa): 824 (M++ H) (54), 823 (M+) (100); Tempo de retenção na HPLC: 13,761 min., 1,29 % (AUC); LRMS (ESI) m/z (intensidade relativa): 823 (M+) (100), 692 (75); Tempo de retenção na HPLC: 14,045 min., 2,42 % (AUC); LRMS (ESI) m/z (intensidade relativa): 823 (M+) (100), 692 (8) e astaxantina (22,10 %).
Exemplo 24: Síntese de Monocarbonato de manitol de astaxantina (LVII).
(LVII)
A uma solução de astaxantina 2E (0,100 g, 0,168 mmol) em
DCM (4 ml) a 0o C foi adicionado DIPEA (0,585 ml, 3,36 mmoles) e cloroformiato de 1,2,2,2-tetracloroetila (0,103 ml, 0,672 mmol). A mistura de reação foi agitada a 0o C durante 2 horas, depois na temperatura ambiente durante 1,5 hora, tempo no qual (D)-manitol (0,3060 g, 1,68 mmol), DMF (3 ml) e DMAP (0,2052 g, 1,68 mmol) foram adicionados à reação. A mistura de reação foi agitada na temperatura ambiente durante 24 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura (20 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram concentradas para produzir monocarbonato de manitol (LVII) (10,19 %) Tempo de retenção na HPLC: 9,474 min., 10,19 % (AUC); LRMS (ESI) m/z (intensidade relativa): 827 (M++ Na) (50), 804 (M+) (25), 725 (58), 613 (100) e astaxantina (53,73 %).
Exemplo 25: Síntese de Diéster do ácido (Dimetilamino)butírico de astaxantina (LVIII).
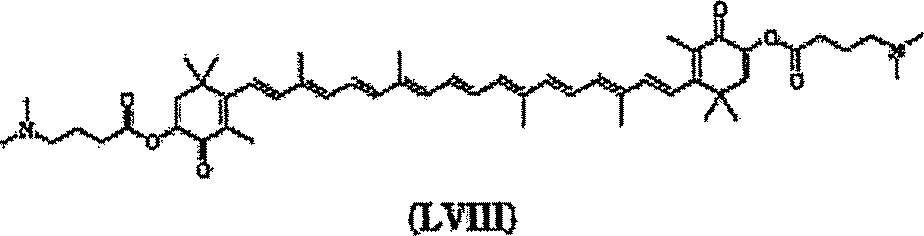
A uma suspensão de cloridreto do ácido 4-(dimetilamino)butírico (0,2816 g, 1,68 mmol) em DCM/ DMF (3 ml /3 ml) na temperatura ambiente foi adicionado DIPEA (0,878 ml, 5,04 mmoles), DMAP (0,4105 g, 3,36 mmoles), HOBT-H2O (0,3094 g, 2,02 mmoles), DIC (0,316 ml, 2,02 mmoles) e astaxantina 2E (0,100 g, 0,168 mmol). A mistura de reação foi agitada na temperatura ambiente durante 36 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC1 1 M (20 ml/ 3 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram concentradas para produzir o diéster do ácido (dimetilamino)butírico (LVIII) (77,70 %) Tempo de retenção na HPLC: 7,850 min., 56,86 % (AUC); LRMS (ESI) m/z (intensidade relativa): 824. (M+ + H) (64), 823 (M+) (100); Tempo de retenção na HPLC: 8,443 min., 3,87 % (AUC); LRMS (ESI) m/z (intensidade relativa): 823 (M+) (5), 641 (20), 520 (100); tempo de retenção na HPLC: 9,021 min., 16,97 % (AUC); LRMS (ESI) m/z (intensidade relativa): 824 (M+ + H) (58), 823 (M+) (100) e nenhuma astaxantina detectável.
Exemplo 26: Síntese de monoéter benzílico de astaxantina (LIX).
A uma solução de astaxantina 2E (0,100 g, 0,168 mmol) e brometo de benzila (0,400 ml, 3,36 mmoles) em DCM/ DMF (3 ml/ 3 ml) a 0o C foi adicionado KHMDS (“bis(trimetilsilil)amida de potássio”) (6,72 ml; 0,5 M em tolueno, 3,36 mmoles). A mistura de reação foi agitada a 0o C durante 1 hora e depois deixada aquecer até a temperatura ambiente. A mistura foi agitada na temperatura ambiente durante 24 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC1 1 M (20 ml/ 3 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram concentradas para produzir o monoéter benzílico (LIX) (15,06 %) Tempo de retenção na HPLC: 12,705 min, 15,06 % (AUC); LRMS (ESI) m/z (intensidade relativa): 686 (M+) (93), 597 (100) e astaxantina (67,96 %).
Exemplo 27: Síntese de monoéter manitol de astaxantina (LX).
(LX)
A uma solução de astaxantina 2E (0,200 g, 0,335 mmol) em DCM (15 ml) na temperatura ambiente foi adicionado HBr a 48 % (10 ml) e H2O (30 ml). A camada aquosa foi extraída com DCM e as camadas orgânicas combinadas foram secadas em Na2SÜ4 e concentradas para produzir o derivado de brometo de astaxantina como um óleo vermelho escuro. A uma solução do brometo bruto em DCM/ DMF (6 ml /6 ml) na temperatura ambiente foi adicionado DIPEA (1,58 ml, 9,09 mmoles), DMAP (0,3702 g, 3,03 mmoles) e (D)-manitol (0,5520 g, 3,03 mmoles). A mistura de reação foi agitada na temperatura ambiente durante 24 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC1 1 M (20 ml/ 3 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram concentradas para produzir o monoéter de manitol (LX) (4,40 %) Tempo de retenção na HPLC: 9,479 min, 4,40 % (AUC); LRMS (ESI) m/z (intensidade relativa): 783 (M+ + Na) (64), 710 (66), 653 (100) e astaxantina (79,80 %).
Exemplo 28: Síntese de Monoamida de
Tris(hidroximetil)aminometano de dissuccinato de astaxantina (LXI).
A uma solução de dissuccinato de astaxantina (XV) (0,100 g, 0,125 mmol) em DCM/ DMF (3 ml /3 ml) na temperatura ambiente foi adicionado DIPEA (0,653 ml, 3,75 mmoles), DMAP (0,3054 g, 2,50 mmoles), HOBT-H2O (0,2297 g, 1,50 mmol) e tris(hidroximetil)aminometano (0,1514 g, 1,25 mmol). A mistura de reação foi agitada na temperatura ambiente durante 36 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC1 1 M (20 ml/ 3 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram concentradas para produzir a monoamida de tris(hidroximetil)aminometano (LXI) (4,40 %) Tempo de retenção na HPLC: 9,521 min., 3,50 % (AUC); LRMS (ESI) m/z (intensidade relativa): 923 (M+ + Na) (36), 900 (M) (80), 560 (100); Tempo de retenção na HPLC: 9,693 min., 0,90 % (AUC); LRMS (ESI) m/z (intensidade relativa): 923 (M+ + Na) (11), 813 (33), 500 (100) e dissuccinato de astaxantina (84,34 %).
Exemplo 29: Síntese de Tris(hidroximetil)aminometano diamida de dissuccinato de astaxantina (LXII).

A uma solução de dissuccinato de astaxantina (XV) (0,100 g, 0,125 mmol) em DCM/ DMF (3 ml /3 ml) na temperatura ambiente foi adicionado DIPEA (0,653 ml, 3,75 mmoles), DMAP (0,3054 g, 2,50 mmoles), HOBT-H2O (0,2297 g, 1,50 mmol), DIC (0,235 ml, 1,50 mmol) e tris(hidroximetil)aminometano (0,1514 g, 1,25 mmol). A mistura de reação foi agitada na temperatura ambiente durante 36 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC1 1 M (20 ml/ 3 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram concentradas para produzir a tris(hidroxinietil)aininometano diamida (LXII) (66,51 %) Tempo de retenção na HPLC: 8,086 min., 19,34 % (AUC); LRMS (ESI) m/z (intensidade relativa): 1026 (M+ + Na) (22), 1004 (M+ + H) (84), 1003 (M+) (100), 502 (83); Tempo de retenção na HPLC: 8,715 min., 47,17 % (AUC); LRMS (ESI) m/z (intensidade relativa): 1004 (M+ + H) (71), 1003 (M+) (100), 986 (62) e dissuccinato de astaxantina (18,61 %).
Exemplo 30: Síntese de Monoéster de adenosina de dissuccinato de astaxantina (LXIII).

A uma solução de dissuccinato de astaxantina (XV) (0,100 g, 0,125 mmol) em DCM/ DMF (3 ml /3 ml) na temperatura ambiente foi adicionado DIPEA (0,653 ml, 3,75 mmoles), DMAP (0,3054 g, 2,50 mmoles), HOBT-H2O (0,1914 g, 1,25 mmol) e (-)-adenosina (0,3341 g, 1,25 mmol). A mistura de reação foi agitada na temperatura ambiente durante 48 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC1 1 M (20 ml/ 3 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram concentradas para produzir o monoéster de adenosina (LXIV) (21,13 %) Tempo de retenção na HPLC: 9,005 min., 2,43 % (AUC); LRMS (ESI) m/z (intensidade relativa): 1047 (M+ + H) (36), 1046 (M+) (57), 524 (100); Tempo de retenção na HPLC: 9,178 min., 10,92 % (AUC); LRMS (ESI) m/z (intensidade relativa): 1047 (M++ H) (80), 1046 (M+) (100), 829 (56), 524 (94); Tempo de retenção na HPLC: 9,930 min., 7,78 % (AUC); LRMS (ESI) m/z (intensidade relativa): 1046 (M+) (100), 524 (34) e dissuccinato de astaxantina (58,54 %).
Exemplo 31: Síntese de diéster de maltose de dissuccinato de astaxantina (LXIV).
A uma solução de dissuccinato de astaxantina (XV) (0,100 g, 0,125 mmol) em DCM/ DMF (3 ml /3 ml) na temperatura ambiente foi adicionado DIPEA (0,653 ml, 3,75 mmoles), DMAP (0,3054 g, 2,50 mmoles), HOBT-H2O (0,2297 g, 1,50 mmol), DIC (0,235 ml, 1,50 mmol) e (D)-maltose-H2O (0,4504 g, 1,25 mmol). A mistura de reação foi agitada na temperatura ambiente durante 36 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC1 1 M (20 ml/ 3 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram concentradas para produzir diéster de maltose (LXTV) (25,22 %) Tempo de retenção na HPLC: 7,411 min., 12,53 % (AUC); LRMS (ESI) m/z (intensidade relativa): 1468 (M+ + Na) (18), 1067 (16), 827 (100); Tempo de retenção na HPLC: 7,506 min., 12,69 % (AUC); LRMS (ESI) m/z (intensidade relativa): 1468 (M+ + Na) (52), 827 (76), 745 (100) e dissuccinato de astaxantina (22,58 %).
Exemplo 32: Síntese de Esteres de Resveratrol de dissucinato de astaxantina (LXV, LXVI).
(LXVeLXVI)
A uma solução de dissuccinato de astaxantina (XV) (0,100 g,
0,125 mmol) em DCM/ DMF (3 ml /3 ml) na temperatura ambiente foi adicionado DIPEA (0,653 ml, 3,75 mmoles), DMAP (0,3054 g, 2,50 mmoles), HOBT-H2O (0,2297 g, 1,50 mmol), DIC (0,235 ml, 1,50 mmol) e resveratrol (0,2853 g, 1,25 mmol). A mistura de reação foi agitada na temperatura ambiente durante 24 horas, tempo no qual a reação foi diluída com DCM, resfriada bruscamente com salmoura/ HC1 1 M (20 ml/ 3 ml) e depois extraída com DCM. As camadas orgânicas combinadas foram concentradas para produzir monoéster de resveratrol (LXV) (1,12 %) Tempo de retenção na HPLC: 10,039 min., 1,12 % (AUC); LRMS (ESI) m/z (intensidade relativa): 1009 (Μ* + 2H) (18), 1007 (M+) (21), 637 (100), diéster de resveratrol (LXVI) (60,72 %) tempo de retenção na HPLC: 10,324 min., 15,68 % (AUC); LRMS (ESI) m/z (intensidade relativa): 1217 (M+) (28), 1007 (100), 609 (69), 504 (85); Tempo de retenção na HPLC: 10,487 min, 29,26 % (AUC); LRMS (ESI) m/z (intensidade relativa): 1218 (M++ H) (80), 1217 (M+) (100), 609 (60); Tempo de retenção na HPLC: 10,666 min, 15,78 % (AUC); LRMS (ESI) m/z (intensidade relativa): 1218 (M++ H) (84), 1217 (M1-) (100), 609 (71) e nenhum dissuccinato de astaxantina detectável.
Determinação rigorosa de solubilidade em água do derivado de astaxantina dissuccinato dissódico (XVI):
Um total de 30 mg de amostra (derivado de astaxantina dissuccinato dissódico, como a mistura todo-trans de estereoisômeros 3S,3’S, meso e 3R,3’R em uma relação 1:2:1) foram adicionados a 2 ml de água filtrada estéril (0,2 μΜ Millipore®) desionizada (DI) em um tubo de centrífuga de vidro de 15 ml. O tubo foi enrolado em folha de alumínio e a mistura foi agitada durante 2 horas, depois centrifugada a 3500 rpm durante 10 minutos. A solução aquosa foi filtrada através de um dispositivo de filtro descartável de PVDF de 0,45 mícron. Uma volume de 1 ml de filtrado foi depois apropriadamente diluído com água DI e a concentração da solução foi medida a 480 nM usando uma curva de calibração de quatro pontos preparada a partir da amostra fresca. Depois de levar as diluições em conta, a concentração da solução saturada do derivado de astaxantina dissuccinato dissódico foi de 8,64 mg/ml.
Dados Experimentais para a Inibição e/ou Melhora de Doença:
Comparação da Capacidade de Formar Radical-Cátion:
Astaxantina Livre, Não Esterificada e Astaxantina Disuccinato Diácido
Usando Fotólise Cintilante:
A FIG. 27 e a FIG. 28 representa os resultados da análise espectral depois da fotólise cintilante da formação de tripleto e estados de radical livre de cátion de carotenóide para a astaxantina livre, não esterificada e o derivado diácido de dissuccinato de astaxantina foram obtidos. A formação do radical de cátion de carotenóide é uma medida do comportamento biofísico potencial do novo derivado como um antioxidante. Se um derivado retém o comportamento antioxidante da astaxantina livre, não esterificada, então todas as aplicações terapêuticas anteriormente documentadas (isto é precedentes da literatura) para a astaxantina podem ser razoavelmente assumida para o novo derivado, incluindo pelo menos a extinção de oxigênio singleto, ruptura de cadeia de peroxidação de lipídeo, e/ou remoção de radical direto.
Carotenóides irradiantes (car) diretamente não resultam na formação de tripletos de carotenóide (3car); um fotossensibilizador é necessário. Neste experimento, a nitronaftalma (NN) foi usada como o fotossensibilizador. Depois da irradiação, o sensibilizador excitado (NN*) forma um tripleto sensibilizador (3NN). Quando 3NN encontra um carotenóide, a energia e as reações de transferência de elétron com 3NN ocorrem. Os radicais de 3car e cátion carotenóide relativamente estáveis resultantes (car+) são detectados pelas faixas de absorção características. Solventes não polares (por exemplo hexano) favorecem a formação de 3car e solventes mais polares (álcoois, água) favorecem a formação do car+. O radical aniônico do sensibilizador (NN’-) não é tipicamente observado por causa de um coeficiente de absorção baixo.
NN’- + car' +
ΝΝ ΝΝ*~* 3ΝΝ + Car ΝΝ + 3car
A. Espectros com ácido dissuccínico de astaxantina (astaCOOH).
Espectros de absorção transitória de astaCOOH em acetonitrila (MeCN), sensibilizador NN.
Os picos negativos no espectro demonstram o esgotamento do estado mais estável de NN e astaCOOH. O pico positivo a 550 nm mostra a formação do tripleto de astaCOOH; o pico positivo a 850 nm mostra a formação do radical catiônico de astaCOOH. O car declina muito rapidamente. Depois de 15 ps, metade do 3car desapareceu e depois de 50 ps, nenhum 3car é deixado. O car'+ é estável dentro deste período de tempo.
B. Espectros com o composto de referência [astaxantina livre, não esterificada (asta)].
Espectros de absorção transitória de asta em acetonitrila (MeCN), sensibilizador NN.
O espectro de asta é aproximadamente idêntico àquele de astaCOOH. Depois de 50 ps, o 3car desapareceu. Durante este período de tempo, o car’+ é estável. Os picos negativo e positivo nos espectros de absorção para astaCOOH e asta são passíveis de sobreposição.
Breve Discussão dos Resultados de Fotólise Cintilante:
Parece haver pouca diferença entre o derivado de dissuccinato diácido de astaxantina (astaCOOH) e astaxantina livre, não esterificada (asta) durante os experimentos de fotólise cintilante. O astaCOOH comporta-se como a asta nos experimentos de fotólise cintilante. Portanto, a esterificação de astaxantina livre com Ácido Succínico não altera as propriedades fotofisicas e o tempo de vida do radical catiônico. Ambos os compostos foram fotoestáveis durante os experimentos de fotólise cintilante. O derivado de dissuccinato de astaxantina retém o potencial antioxidante potente de
Μastaxantina e é ativo no estado esterificado. Ele pode ser portanto considerado um medicamento “brando” (ativo como a entidade modificada) e não um pró medicamento, para aplicações terapêuticas, conferindo a(s) propriedade(s) valiosa(s) de atividade descontaminante de radical de fase dupla para este derivado (isto é remoção de radical de fase aquosa e lipídica).
Indução da Expressão da Proteína Conexina 43:
Os métodos para a cultura celular, Western blotting, análise . densitométrica quantitativa e avaliação de proteína total são descritos em detalhes em Rogers et al. (1990), com as modificações sugeridas em Bertram (1999). Em resumo, as células CH3/10T1/2 de fibroblasto embriônico de camundongo foram tratadas com as seguintes formulações em um sistema de cultura celular de 4 ml com meios contendo 2 % de soro de bezerro:
1. TTNPB [ácido p-(E)-2-(5,6,7,8-tetraidro-5,5,8,8-tetrametil- o
2-naftil) propenil benzóico] 10- M em acetona [controle positivo para a regulagem excessiva da conexina 43 (4 μΐ em 4 ml]
2. Derivado do sal dissódico de astaxantina dissuccinato/H2O a
10-5 M (40 μΐ em 4 ml)
3. Derivado do sal dissódico de astaxantina dissuccinato/ H2O a 10-6 M (4 μΐ em 4 ml)
4. Derivado do sal dissódico de astaxantina dissuccinato/ H2O a 10-7 M (diluição 1:10 e 4 μΐ em 4 ml)
5. Derivado do sal dissódico de astaxantina dissuccinato
H2O/etanol [EtOH] formulação a 10-5 M (40 μΐ em 4 ml)
6. Derivado do sal dissódico de astaxantina dissuccinato H2O /EtOH formulação a 10-6 M (4 μΐ em 4 ml)
7. Controle de H2O estéril (40 μΐ em 4 ml)
8. controle de H2O estéril/EtOH (20 μΐ de EtOH, 20 μΐ de H2O em 4 ml)
9. Controle do meio (4 ml)
As células foram colhidas depois de 96 horas de incubação com os compostos de teste e soluções de controle. Todas as soluções de meio foram idênticas na cor, entretanto depois do tratamento com o derivado do sal c dissódico de astaxantina dissuccinato em ambas as diluições 10-, a cor mudou subjetivamente mudou para uma cor laranja-vermelho. As células tratadas com TTNPB apareceram esfriadas com microscópio ótico, evidência de diferenciação para miócitos, um resultado esperado neste sistema de cultura celular. Depois da colheita e pelotização das células, os tubos contendo ambas as soluções a 10-5 de derivados do sal dissódico de astaxantina dissuccinato foram vermelho brilhante; ambos os tubos da diluição 10-6 foram de uma cor rosa. Como anteriormente documentado para outros carotenóides coloridos, esta foi uma evidência subjetiva para a captação celular dos compostos de teste.
As células foram depois lisadas e 50 pg de cada proteína foi submetida à eletroforese em um gel de poliacrilamida a 10 %. O gel foi depois transferido a um filtro de nitrocelulose. A proteína total foi ensaiada com fingimento de Azul de Coomassie (FIG. 29; linhas 6, 7 e 9 foram esfregadas secundário à transferência do gel e não foram incluídas na comparação quantitativa [FIG. 31)]. A western blotting foi realizada com anticorpos anti-conexina 43 seguido pela quimioluminescência HRP em um formador de imagem Biorad (FIG. 30). O gel original foi riscado uma vez e a Western blot repetida duas vezes antes da visualização. Os resultados foram normalizados para o controle da Linha 8 (EtOH/E^O), que demonstra a expressão constitutiva de fondo da proteína conexina 43 em uma condição de controle (nenhum composto de teste). Os resultados da indução da conexina 43 relativa pelos compostos positivos e de teste de controle são mostrados na FIG. 31.
Debate Resumido de Resultados de Cx43.
Todas as formulações de derivados do sal dissódico de

astaxantina dissuccinato testadas induziram a expressão da proteína da conexina 43 em relação aos níveis expressados constitutivamente em controles de água e etanol/água (FIG. 31). A probabilidade de detectar uma indução da expressão da proteína conexina 43 em 5 condições de teste 5 separadas na ausência de um efeito de tratamento verdadeiro (controle de hipótese nula μι = média de tratamento μ2) é 1 em 25 ou p = 0,03. Os - derivados do sal dissódico de astaxantina dissuccinato formulados em água induziram a expressão da proteína da conexina 43 em cada condição de teste (de IO-5 a ΙΟ-7 Μ). A diminuição na combinação do derivado do sal dissódico 10 de astaxantina dissuccinato/água mais baixa testada sugere a dependência da dose na resposta induzida. A indução relativa foi aumentada na única condição de teste avaliada com uma concentração etanólica final em média de 0,5 %. Esta descoberta é altamente sugestiva da biodisponibilidade aumentada desta formulação, visto que o etanol é conhecido por reduzir a agregação de 15 derivados do sal dissódico de astaxantina dissuccinato em soluções aquosas.
As soluções de derivados do sal dissódico de astaxantina dissuccinato em água em concentrações maiores do que 10-7 e em combinações de etanol/água a 10-5 parecem ter níveis de indução mais altos que o controle TTNPB positivo. TTNPB é um retinóide altamente potente, isto é eficaz na indução da ''ior'' Q expressão da conexina 43 no ponto de tempo de 96 horas a 10- M.
Indução da comunicação de junção de intervalo (GJC) intercelular em fibroblastos de murino pelos derivados do sal dissódico de astaxantina dissuccinato:
Uma série de experimentos foram realizados para avaliar a 25 capacidade dos derivados do sal dissódico de astaxantina dissuccinato para induzir a comunicação de junção de intervalo (GJC) em uma linha imortalizada de fibroblastos de murino. Os estudos foram conduzidos:
(1) ao nível funcional para medir a comunicação célula/célula pela transferência de corante aumentada entre as células confluentes na J65 cultura de monocamada;
(2) ao nível molecular como medido pela capacidade destes compostos para induzir a expressão da proteína conexina 43 (Cx43). Cx43 é a unidade estrutural dos canais intercelulares nestes fibroblastos o que permite a
GJC;
(3) ao nível celular como mostrado pela capacidade dos derivados do sal dissódico de astaxantina dissuccinato para aumentar o número e o tamanho das placas imunorreativas em Cx43 nas regiões da membrana plasmática em contato direto com as células adjacentes.
(1) Ensaios de Comunicação. Os experimentos foram realizados para avaliar a capacidade do derivado do sal dissódico de astaxantina dissuccinato [como uma mistura estatística dos estereoisômeros todo-trans (todo-E), S,S’, meso e RR’ em relação de 1:2:1] para realçar a comunicação intercelular juncional de intervalo (GJC) entre células de
C3H/10T1/2 de fibroblasto embriônico de camundongo. Anteriormente esta capacidade foi altamente correlacionada com a capacidade dos carotenóides para inibir a transformação neoplástica induzida pelo carcinogênio (Zhang, 1992). Além disso, a comunicação de junção mediada pela Cx43 entre miócitos cardíacos é responsável pela transferência de sinais que mantém contrações sincronizadas e impedem as arritmias cardíacas (Peters, 1995).
A permeabilidade de junção foi ensaiada pela microinjeção do corante fluorescente Amarelo de Lúcifer CH (Sigma, St. Louis, MO) nas células confluentes individuais essencialmente como descrito anteriormente (Zhang, 1994). Em resumo, as culturas confluentes de células C3H/10T1/2 25 foram tratadas durante 4 dias com: (1) o derivado do sal dissódico de astaxantina dissuccinato (1 x 10-5 M) dissolvido em uma formulação 1:2 de etanol/água (EtOH/ H2O); (2) um retinóide sintético, TTNPB (1 x 10-8 M) dissolvido em tetraidrofurano como um controle positivo; ou (3) células tratadas com EtOH/ H2O 1:2 como um controle negativo. As células únicas

em cada placa foram identificadas sob as óticas de contraste de fase e pressão injetada usando uma agulha de microinjeção (Eppendorf, Hamburg, Alemanha) carregada com o corante fluorescente Amarelo de Lúcifer como uma solução a 10 %. A agulha foi controlada por um micromanipulador remoto e as células e o microscópio foram posicionados em uma mesa antivibração pneumática. A injeção bem sucedida de Amarelo de Lúcifer foi confirmada pela iluminação curta com luz UV, que causa a fluorescência amarela do Amarelo de Lúcifer. Este corante é suficientemente pequeno para passar através das junções de intervalo e é eletricamente carregado e pode assim apenas entrar nas células adjacentes à célula injetada se elas estiverem em comunicação juncional. Depois de 2 minutos para permitir a transferência juncional, as imagens digitais foram tomadas sob iluminação UV. O número de células fluorescentes adjacentes à célula injetada foi mais tarde determinada pela análise de imagem digital usando um método de patamar de densidade imparcial e o programa de software SigmaScan (Jandel Scientific). Este número de células em comunicação foi usado como um índice da comunicação juncional, como descrito anteriormente (Hossain, 1993).
Os resultados desta análise demonstraram que o derivado do sal dissódico de astaxantina dissuccinato (1 x 10-5 M) dissolvido em uma formulação de EtOH/ H2O 1:2 efícazmente aumentou o grau de comunicação juncional em relação àquele observado em controles tratados com EtOH/ H2O 1:2. Das 22 células tratadas microinjetadas 15 (56 %) foram funcionalmente ligadas pelas junções de intervalo, ao contrário a apenas 3 das 11 (27 %) células de controle. Estas diferenças foram estatisticamente diferentes (p < 0,04; Teste t de Student emparelhado). As fotomicrografias representativas são mostradas na FIG. 14:
Painel A: tratamento com a mistura estatística de estereoisômeros do sal dissódico de astaxantina dissuccinato a 1 x 10-5 M em
EtOH/H2O 1:2;
100
Painel C: controle negativo de solvente EtOH/ H2O 1:2;
Painel E: TTNPB a 1 x 10-8 M em tetraidrofiirano como solvente, controle positivo; e
Os painéis B, D, F: análise digital de micrográficos A, C, E, respectivamente, demonstrando pixéis acima de um patamar ajustado positive para a fluorescência do Amarelo de Lúcifer. Porque os núcleos celulares têm a maior parte do volume, eles acumulam a maior parte do Amarelo de Lúcifer e exibem a fluorescência máxima.
(2) Estudos Moleculares. Tanto a mistura de estereoisômeros do derivado do sal dissódico de astaxantina dissuccinato quanto as formas enantioméricas purificadas do derivado do sal dissódico de astaxantina dissuccinato (as formas S,S’, meso e R,R’ a > 90 % de pureza pela HPLC) aumentam a expressão da proteína Cx43 em fibroblastos de murino como avaliado pela imuno(Westem) blotting essencialmente como descrito (Zhang, 1992 e 1994). Em resumo, as células C3H/10T1/2 fibroblasto embriônico de camundongo foram cultivadas em meio basal de Eagle com sais de Earle (Atlanta Biologicals, Atlanta, GA), suplementado com 5 % de soro fetal bovino (Atlanta Biologicals, Atlanta, GA) e 25 pg/ml de sulfato de gentamicina (Sigma, St. Louis, MO) e incubadas a 37° C em 5 % de CO2. No 7o dia depois de semear em placas de 100 milímetros (mm), as células confluentes foram tratadas durante quatro dias com os derivados do sal dissódico de astaxantina dissuccinato e depois colhidas e analisadas quanto a indução da proteína Cx43 como descrito. O teor de proteína foi medido usando o kit de Reagente de Ensaio de Proteína (Pierce Chemical Co., Rockford, IL) de acordo com as instruções do fabricante. Os lisados de célula contendo 100 pg de proteína foram analisados pela Western blotting usando o kit e aparelho de western blotting NuPage (Invitrogen, Carlsbad, CA) e a proteína Cx43 detectado usando um anticorpo policlonal de coelho (Zymed, San Francisco, CA) incitado contra um polipeptídeo sintético que corresponde
101 ao domínio de terminal C da Cx43 de camundongo, ser humano e rato. As faixas imunorreativas em Cx43 foram visualizadas pela quimioluminescência usando um anticorpo secundário conjugado a HRP anti-coelho (Pierce Chemical Co., Rockford, IL). As imagens digitais foram obtidas com uma câmara CCD esfriada e a densitometria quantitativa foi depois realizada (BioRad, Richmond, CA). A carga de proteína igual das linhas foi confirmada pelo tingimento com a mancha de proteína de azul de Coomassie e a análise de imagem digital.
Neste experimento os derivados do sal dissódico de astaxantina dissuccinato foram adicionados às culturas celulares em uma formulação de etanol/ H2O 1:2 a 1 χ ΙΟ-5 Μ. A mistura estatística de ' estereoisômeros e formas enantioméricas purificadas demonstrou a expressão aumentada de Cx43 em comparação com as culturas celulares tratadas com etanol/ H2O 1:2 sozinho (FIG. 15A e FIG. 15B). O tratamento com a mistura estatística de estereoisômeros do derivado do sal dissódico de astaxantina dissuccinato evocou o nível de indução mais alto de Cx43 de todos os derivados testados. Estes níveis de indução foram diversas vezes menores do que os níveis de indução observado com os retinóides de ácido tetraidrotetrametilnaftil propenilbenzóico (TTNPB) (Hoffman-LaRoche, Nutley, NJ) e acetato de retinila (Sigma, St. Louis, MO) incluídos como controles positivos; Esta diferença de potência relativa é compatível com estudos anteriores.
(3) Estudos Celulares. A mistura estatística de estereoisômeros do derivado do sal dissódico de astaxantina dissuccinato aumenta a montagem de Cx43 em células 10T1/2 de murino tratadas nas regiões de contato de célula/célula compatíveis com a formação das junções de intervalo funcionais.
Neste experimento a expressão e montagem de Cx43 em placas foram avaliadas pelo tingimento imunofluorescente. Os procedimentos foram essencialmente como descrito em Zhang (1992). Em resumo, as
102 culturas confluentes de células C3H/10T1/2 foram cultivadas em lâminas de 4 câmaras plásticas Permanox (Nalge Nunc International, Naperville, IL) e tratadas durante 4 dias com: (1) o derivado do sal dissódico de astaxantina dissuccinato (mistura estatística de estereoisômeros) dissolvido em uma formulação 1:2 EtOH/ H2O; (2) o retinóide TTNPB a 1 x 10-8 M em tetraidrofurano como um controle positivo; ou (3) EtOH/ H2O 1:2 como um controle de solvente. As células foram fixadas com metanol a -20° C durante a noite, lavado em tampão, bloqueadas em albumina sérica bovina a 1 % (Sigma, St, Louis, MO) em PBS e incubadas com o anticorpo policlonal de coelho anti-Cx43 (Zymed, São Francisco, CA) como em (2) acima e detectado com anti-coelho secundário conjugado com Alexa568 (Molecular Probes, Eugene, OR). As lâminas foram iluminadas com luz a 568 nm e as imagens foram adquiridas em um comprimento de onda de 600 nm usando o microscópio ótico Zeiss Axioscope e uma câmara CCD esfriada Roper Scientific. As lâminas tratadas com o controle de retinóide TTNPB e a mistura estatística de estereoisômeros do derivado do sal dissódico de astaxantina dissuccinato a 1 x 10-5 M exibiram a montagem de Cx43 immunorreativo em placas nas regiões da membrana celular no contato direto com as células adjacentes. Tal montagem é compatível com a localização e formação de placas das junções de intervalo, conhecidas por serem formadas pela agregação de junções de intervalo individuais múltiplas nas populações celulares que são conectadas por intermédio de junção (Perkins, 1997). Nas culturas tratadas com solvente como controle, tais placas imunorreativas foram infrequentes e foram menores do que aquelas detectadas em células tratadas com a mistura estatística de estereoisômeros do derivado do sal dissódico de astaxantina dissuccinato ou com TTNPB como controle positivo. A freqüência destas placas e o seu tamanho são compatíveis com as diferenças funcionais na permeabilidade da junção de intervalo como detectadas pelos experimentos de transferência de corante Amarelo de Lúcifer
103 descritos na seção 1 e FIG. 14 (TTNPB > mistura estatística de estereoisômeros do sal dissódico de astaxantina dissuccinato > controle de solvente) e com o grau de indução de Cx43 como detectado nos experimentos de imunomancha descritos na seção 2 e FIG. 15. As fotomicrografias representativas são mostradas na FIG. 16.
Inibição da transformação neoplástica induzida por carcinógeno pela astaxantina livre, não esterificada em fibroblastos de murino:
A astaxantina livre, não esterificada é gerada no intestino de mamífero depois da administração oral de astaxantina esterificada. Apenas a astaxantina livre é encontrada no plasma e órgãos sólidos de mamífero. Isto foi mais uma vez demonstrado em estudos farmacocinéticos de dose oral única e múltipla; os resultados são aqui descritos. A atividade de esterase inerente da albumina sérica e a ação de esterases promíscuas no soro e órgãos sólidos rapidamente gera astaxantina livre, não esterificada depois da administração parenteral do derivado de astaxantina dissuccinato dissódico (XVI). Os experimentos de fotólise cintilante também demonstraram que o derivado de astaxantina dissuccinato dissódico e a astaxantina não esterificada, livre têm comportamento antioxidante idêntico em termos de formação do radical de cátion carotenóide. Um experimento foi realizado para avaliar a capacidade da astaxantina livre, não esterificada (o produto de divagem final in vivo do derivado do sal dissódico de astaxantina dissuccinato (XVI), testado como a mistura todo-trans de estereoisômeros 3S,3S’, meso e 3R,3’R em uma relação 1:2:1) para inibir a transformação neoplástica no modelo de cultura de célula C3H1OT172 desenvolvido no laboratório do antigo Charles Heidelberger (Reznikoff, 1973). Este sistema de cultura celular foi mostrado imitar eficazmente o início e os eventos de transformação da formação de tumor em animais como um todo (Bertram, 1985). Nestas células, o tratamento com o hidrocarboneto policíclico
104 carcinogênico 3-metilcolantreno (MCA) produz um evento de iniciação em uma pequena proporção de células tratadas que leva 5 semanas mais tarde à transformação morfológica nestas células, exibido pela presença de focos transformados. A injeção destas células transformadas em camundongos 5 singênicos resulta na formação de sarcomas no sítio de injeção que demonstra a natureza carcinogênica da transformação (Reznikoff, 1973). Este ensaio foi • adaptado à detecção de agentes preventivos de câncer (Bertram, 1989) e retinóides e carotenóides preventivos de câncer demonstraram inibir a transformação neste sistema (Bertram, 1991; Pung, 1988; e Merriman, 1979).
Este experimento foi conduzido de acordo com os protocolos estabelecidos anteriormente (Bertram, 1991 e Pung, 1988). Em resumo, as células 10T , derivados de fibroblasto embriônicos de camundongos, foram semeados em uma densidade de 103 células/ placas de 60 mm em Meio Basal de Eagle (BME) (Atlanta Biologicals, Atlanta, GA), suplementado com 4 % 15 de soro fetal bovino (Atlanta Biologicals, Atlanta, GA) e 25 Lig/ml sulfato de gentamicina (Sigma, St. Louis, MO). As células foram tratadas 24 horas mais tarde com 5,0 μg/ml de MCA (Sigma, St. Louis, MO) em acetona ou com 0,5 % de Acetona (a concentração final) como um controle. Os meios foram trocados 24 Horas depois do tratamento MCA. As células foram tratadas com 20 astaxantina em TI-IF ou com acetato de retinol em acetona 7 dias mais tarde e re-tratadas a cada 7 dias durante 4 semanas. Outras placas foram tratadas com os controles de solvente apropriados. Depois de 5 semanas da partida do experimento, as células foram fixadas com metanol e tingida com coloração de Giemsa a 10 % (Sigma, St. Louis, MO) e classificado quanto aos focos 25 tipo II e tipo III como per (Reznikoff, 1973).
Os resultados desta análise demonstraram que o tratamento de semanas com astaxantina causaram uma diminuição dependente da concentração nos números de foco transformado induzido por MCA em comparação com as células tratadas com MCA e com THF como um controle
105
ÀU de solvente (representado na FIG. 34). A FIG. 34 representa efeitos de astaxantina livre, não esterificada (como a mistura todo-características de estereoisômeros) na transformação neoplástica induzida por MCA. O gráfico representa um total de 68 culturas tratadas com astaxantina a 3 χ ΙΟ-6 Μ, 1 x
10-6 M e 3 χ 10-7 M, liberado em um veículo de THF de 0,3 %, 0,1 % e 0,03 %, respectivamente. Os controles foram como segue: um total de 16 placas » não receberam carcinogênio e foram tratadas com 0,05 % de solvente de etanol; os controles não exibiram quaisquer eventos de transformação. Um total de 20 placas foram tratadas com MCA e 1 % de solvente de THF, V10 produzindo uma taxa de transformação de 0,92 foco/placa. A redução percentual (% de redução) de transformação nas placas tratadas com * astaxantina foi calculada por uma comparação de focos/placa médio de cada tratamento com os controles tratados com MCA. As estatísticas dedutíveis foram realizadas usando o teste t de Student emparelhado; os valores P calculados de 0,00004, 0,00001 e 0,00006, respectivamente, foram obtidos. P< 0,05 foi considerado significante. O tratamento com 3 χ 10-6 M de astaxantina resultou na supressão completa do fenótipo transformado (FIG.
35). A FIG. 35 representa uma comparação de placa tratada com astaxantina para controlar placas. As placas representativas tratadas com: A, nenhum
MCA com controle de solvente; B, MCA 5,0 pg/ml com 1 % de THF como controle de solvente; C, MCA com 3 χ 10-6 M de astaxantina (como a mistura todo-trans de estereoisômeros) em THF. É notável que este nível de inibição excedeu em muito aquele relatado anteriormente para todos os outros carotenóides testados usando protocolos idênticos (Bertram, 1991). Uma comparação dos dados correntes com os dados anteriormente relatados para a redução percentual na transformação neoplástica nas concentrações testadas revelou que a astaxantina é um inibidor muito mais potente de transformação do que o β-caroteno ou a cantaxantina (FIG. 36). A FIG. 36 representa uma comparação de astaxantina (como a mistura de estereoisômeros) para os
106

carotenóides anteriormente testados. Os dados foram compilados comparando-se a redução percentual de focos/placa neoplasticamente transformados induzidos por MCA nas culturas tratadas com astaxantina com a redução percentual de focos/placa de dados anteriormente relatados pelo laboratório Bertram depois do tratamento com β-caroteno e cantaxantina (Bertram, 1991) usando protocolos idênticos. A redução percentual na concentração mais alta testada anteriormente (1 x 10-5 M) é reportada aqui para β-caroteno e cantaxantina; esta concentração mais alta de astaxantina não foi utilizada por causa da atividade medida maior da astaxantina em concentrações mais baixas. Estes estudos demonstram o potencial para a porção de astaxantina clivada do derivado sintetizado ser um agente quimiopreventivo contra o câncer altamente eficaz, depois da administração tanto oral quanto parenteral. Ligado com os dados farmacocinéticos de acúmulo hepático também aqui relatado (depois das estratégias de dose tanto única quanto múltipla), o uso deste composto forma uma forma de realização particularmente útil.
Inibição de Espécies de Oxigênio Reativo:
Em um experimento, os neutrófilos foram isolados em um gradiente de Percoll a partir de sangue integral de um voluntário humano. Os neutrófilos isolados foram depois recolocados em suspensão em solução salina tamponada com fosfato e estimulados ao máximo com éster de forbol para induzir a explosão respiratória e a produção de ânion de superóxido. À solução de neutrófilos humanos ativados, o derivado do sal dissódico de astaxantina dissuccinato foi adicionado em várias concentrações e o sinal de superóxido [como medido com a produção de imagem de ressonância paramagnética de elétron (EPR)] foi subsequentemente medido. O derivado do sal dissódico de astaxantina dissuccinato (como a mistura de estereoisômeros) reduziu a medida de sinal do ânion de superóxido em uma maneira dependente da dose (FIG. 2); a supressão quase completa do sinal do
107 ânion de superóxido foi obtida na concentração de 3 mM. A FIG. 2 demonstra o sinal de superóxido forte depois da ativação nos controles, depois os resultados de titulação com o derivado do sal dissódico de astaxantina dissuccinato de 100 μΜ a 3 mM. O derivado do sal dissódico de astaxantina dissuccinato testado a 100 μΜ descontaminou 28 % do sinal total. A 3 mM, quase nenhum sinal de superóxido permaneceu. Estes resultados demonstram que a cardioproteção no dano por isquemia-reperfosão, como foi demonstrado com as outras intervenções anti-neutrófilo descritas acima, também pode ser obtida com o novo derivado de carotenóide aqui descrito, além de reduzir o sinal do ânion de superóxido importante no dano por isquemia-reperfusão, também é provável que o salvamento miocárdico possa ser obtido com o novo derivado de carotenóide descrito, visto que o ânion de superóxido desempenha um papel principal no dano e morte ao tecido durante a isquemia miocárdica prolongada.
A FIG. 3 representa um efeito de uma solução de derivado do sal dissódico de astaxantina dissuccinato/Vitamina C na espécie de oxigênio reativo (ânion de superóxido) como monitorado usando a produção de imagem de EPR. A solução incluiu uma mistura de cerca de 2 a cerca de 1 de vitamina C para o derivado de sal dissódico de astaxantina dissuccinato respectivamente. A solução do derivado do sal dissódico de astaxantina dissuccinato/Vitamina C reduziu a medida de sinal do ânion de superóxido em uma maneira dependente da dose (FIG. 3); a supressão completa do sinal do ânion de superóxido foi obtida na concentração de 0,02 μΜ. A FIG. 3 demonstra o sinal de superóxido forte depois da ativação nos controles, depois os resultados de titulação com a solução do derivado do sal dissódico de astaxantina dissuccinato/Vitamina C de 0,01 μΜ a 0,02 μΜ.
Em um terceiro experimento, os neutrófilos foram mais uma vez isolados em um gradiente Percoll de sangue integral de um segundo voluntário humano. Os neutrófilos isolados foram depois recolocados em
108 suspensão em solução salina tamponada com fosfato e estimulados ao máximo com éster de forbol para induzir a explosão respiratória e a produção de ânion de superóxido. À solução de neutrófilos humanos ativados, o derivado do sal de cloridreto de astaxantina dilisinato (XX) foi adicionado em quatro (4) concentrações e o sinal de superóxido (como medido com a produção de imagem de EPR) foi subsequentemente medido. O derivado do sal de cloridreto de astaxantina dilisinato também reduziu o sinal do ânion de superóxido medido em uma maneira dependente da dose (FIG. 21), de aproximadamente 5 % de redução a 1 μΜ a 98 % de redução a 3 mM. Mais uma vez, a supressão quase completa do sinal do ânion de superóxido foi obtida na concentração de 3 mM. Este derivado de carotenóide novo apresentou eficácia de remoção em concentração baixa (1 pm), assim como a capacidade para concentrações aumentadas do derivado neste ensaio in vitro até eliminar quase completamente o sinal do ânion de superóxido. A atividade deste novo derivado in vitro como um descontaminante aquoso mais uma vez sugere que os novos derivados (astaxantina dissuccinato dissódico, sal de cloridreto de astaxantina dilisina) atuarão como medicamentos brandos (isto é ativos como os novos derivados inteiros, não clivados) e não como pró medicamentos (inativos até a divagem em astaxantina livre) in vivo. A solubilidade aquosa deste derivado (XX) foi maior do que 50 mg/ml, demonstrando a utilidade dos métodos da presente invenção para aumentar a solubilidade em água dos precursores de carotenóide (neste caso a astaxantina), de solubilidade em água inerente aproximadamente zero até a faixa de mg/ml alta.
Remoção de Ânion de Superóxido Direta por um Derivado de
Astaxantina Dissuccinato Dissódico: Eficácia Relativa de Estereoisômeros
Individuais versus a Mistura Estatística de Estereoisômeros pela Produção de
Imagem de Ressonância Paramagnética de Elétron
Materiais
109
A mistura estatística [1:2:1 de estereoisômeros 3S,3’S, meso (3S,3’R e 3’S,3R) e 3R,3’R] não esterificada, todo-E de astaxantina foi adquirida da Buckton Scott (índia) e usada como fornecida (> 95 % de pureza pela HPLC). A astaxantina foi dissolvida em sulfóxido de dimetila grau HPLC (DMSO; Sigma-Aldrich, St. Louis, MO). Os derivados de astaxantina dissuccinato dissódico foram testados separadamente em nove formulações: mistura estatística de estereoisômeros (como para a astaxantina, acima, uma mistura 1:2:1 de todo-E; rotulado como “mistura” em todas as tabelas e figuras); 3S,3’S e 3R,3’R (isômeros óticos ou enantiômeros); e meso (mistura de 3S,3’R e 3’S,3R; diastereômeros do par enantiomérico). Todos os derivados de dissuccinato foram sintetizados a > 90 % de pureza pela HPLC. Os derivados de dissuccinato foram primeiro testados nas concentrações finais apropriadas em solução aquosa pura (água desionizada) das soluções de estoque de 10 mM. Cada um dos quatro derivados de dissuccinato foram depois testados a partir de soluções de estoque preparadas em uma mistura 1:2 de etanol (a concentração final de EtOH na solução de estoque 33 1/3 %; a concentração final no ensaio de neutrófilo isolado 0,3 %; etanol grau HPLC, Sigma-Aldrich, St. Louis, MO) a 10 mM. O derivado 3S,3’S também foi testado a partir de uma solução de estoque de concentração a 50 % de EtOH (a concentração final no ensaio de neutrófilo isolado 0,5 %). A formulação etanólica dos derivados de dissuccinato foi mostrada desagregar completamente as montagens supramoleculares que formam na solução aquosa pura, fornecendo soluções monoméricas dos derivados imediatamente antes da introdução no ensaio de teste. Os controles negativos de etanol sozinho (as concentrações de 0,3 % e 0,5 % de EtOH final no ensaio de neutrófilo isolado) e controle positivo mimético de superóxido dismutase (concentração final de 10 pM; Metaphore® Pharmaceuticals, Inc., St. Louis, MO) também foram realizados.
Um derivado de carotenóide [Éster mono-(4-{18-[4-(3110

carbóxi-propionilóxi)-2,6,6-trimetil-3 -oxo-cicloex-1 -enil]-3,7,12,16tetrametil-octadeca-1,3,5,7,9,11,13,15,17-nonaenil} -3,5,5-trimetil-2-oxocicloex-3-enílico) do Ácido Succínico; FIG. 17] e as suas formas estereoisoméricas foram sintetizadas, derivados de dissuccinato dissódico de astaxantina, na forma todo-trans (todo-E). Os derivados são moléculas quirais simétricas com 2 centros quirais nas posições de carbono 3 e 3’, que compreendem 4 estereoisômeros: 3R,3’R e 3S,3’S (isômeros óticos ou enantiômeros), assim como as formas meso diastereoméricas (3R,3’S e 3’R,3S). A mistura estatística de estereoisômeros sintetizados a partir da fonte comercial de astaxantina contém as formas estereoisoméricas 3R,3’R, meso (3R,3’S e 3’R,3S) e 3S,3’S em uma relação de 1:2:1. Todos os estereoisômeros individuais e a mistura estatística foram sintetizadas a > 90 % de pureza pela HPLC, permitindo a comparação direta da eficácia individual destas formas como descontaminante direto de radicais. As formas todo-E dos estereoisômeros usados neste estudo foram moléculas lineares, rígidas (bolaanfífilos) devido à falta da(s) configuração(s) cis (ou Z) na camada de polieno do material espaçador.
Os diésteres de dissuccinato sódico de astaxantina demonstram “dispersibilidade” em água aumentada em relação ao composto precursor de astaxantina. As dispersibilidades em água dos estereoisômeros individuais e da mistura estatística foram todas maiores do que 8 mg/ml (aproximadamente 10 mM), permitindo que eles fossem introduzidos no sistema de teste aquoso tamponado sem um co-solvente. A tendência para os precursores de carotenóide tais como astaxantina (Salares, 1977), assim como os novos derivados de carotenóide (por exemplo derivados de capsantina) (Zsila, 2001 e Bikadi, 2002) para formar montagens supramoleculares em solução aquosa também foi observado para os derivados testados no estudo corrente. A automontagem supramolecular resulta em agregados de tamanho significante em solução aquosa e impede a interação direta máxima de moléculas agregadas
111 com espécies radicais. Portanto, uma comparação do comportamento de remoção direta dos novos derivados de astaxantina foi conduzida tanto na formulação aquosa pura assim como com o co-solvente etanol. Nas soluções de estoque, uma concentração 1:2 de EtOH/água mostrou desagregar 5 completamente a mistura estatística, derivados meso e 3R,3’R; uma solução de estoque etanólica a 50 % foi requerida para desagregar completamente o isômero 3S,3’S. A capacidade descontaminante dos compostos também foi testada em relação aos controles negativo (isto é veículo de etanol) e positivo [miméticos da superóxido dismutase (SOD), astaxantina racêmica livre em 10 DMSO].
Purificação e Preparação de Leucócitos
Os leucócitos polimorfonucleares humanos (PMNs) foram isolados de sangue venoso recém amostrados de um único voluntário (S.F.L.) pela centrifugação de gradiente de densidade de Percoll, que produziu PMNs 15 com uma pureza de > 95 %. Cada 10 ml de sangue integral foram misturados com 0,8 ml de EDTA 0,1 M e 25 ml de solução salina. O sangue diluído foi disposto em camadas em 9 ml de Percoll a uma densidade específica de 1,080 g/ml. Depois da centrifugação a 400 x g durante 20 nm a 20° C, a célula plasmática, mononuclear e as camadas de Percoll foram removida. Os 20 eritrócitos foram lisados pela adição de 18 ml de água gelada durante 30 s, seguido por 2 ml de tampão 10X PIPES (25 mM de PIPES, 110 mM de NaCl e 5 mM de KC1, titulado ao pH 7,4 com NaOH). As células foram pelotizadas a 4° C, o sobrenadante foi decantado e o procedimento foi repetido. Depois da segunda lise hipotônica, as células foram lavadas duas vezes com tampão 25 PAG (tampão PIPES contendo 0,003 % de albumina sérica bovina e 0,1 % de glicose). Posteriormente, PMNs foram contados pela microscopia ótica em um hemocitômetro. A pelota final foi depois colocada em suspensão em tampão PAG-CM (tampão PAG com 1 mM de CaCl2 e 1 mm de MgCl2).
Medições de EPR
112
Todas as medições EPR foram realizadas usando um espectrômetro de Bruker ER 300 EPR operando na faixa X com uma cavidade TMiio. A freqüência de microonda foi medida com um contador de microonda Modelo 575 (EIP Microwave, Inc, San Jose, CA). Para medir a geração O2 a partir das PMNs estimuladas em éster de forbol (PMA), estudos de aprisionamento de spin EPR foram realizados usando DEPMPO (Oxis, Portland, OR) a 10 mM. 1 X 106 PMN’s foram estimuladas com PMA (1 ng/ml) e carregadas em tubos capilares para as medições de EPR. Para determinar a capacidade de remoção de radical da astaxantina “racêmica”, não esterificada, livre em DMSO e dos derivados de sal dissódico de dissuccinato em cada uma das nove formulações, as PMN’s foram pré incubadas durante 5 minutos com o composto seguido pelo estímulo com PMA como anteriormente descrito. Os ajustes de instrumento usados nos experimentos de aprisionamento de spin foram como segue: amplitude de modulação, 0,32 G; constante de tempo, 0,16 s; tempo de varredura, 60 s; freqüência de modulação, 100 kHz; potência da microonda, 20 miliwatts; e freqüência de microonda, 9,76 GHz. As amostras foram colocadas em uma célula plana de EPR de quartzo e os espectros foram registrados. Os sinais componentes no espectro foram identificados e quantificados como relatado (Lee, 2000).
Análise Estatística
As análises estatísticas foram realizadas com o pacote de software estatístico NCSS (NCSS 2001 e PASS 2002, Kaysville, UT). Todos os testes de estatística foram realizados a um α = 0,05.
Debate Resumido dos Resultados de EPR:
Os miméticos de SOD potentes produzidos pela Metaphore,
Inc. serviram como um controle positivo no início do estudo. Como foi observado repetidamente no laboratório Zweier, a dose de 10 μΜ em veículo de apenas água quase eliminou completamente o sinal do ânion de superóxido
113 como detectado com DEPMPO (97 % de inibição; Tabela 1). Um controle negativo de etanol sozinho (a concentração final de 0,3 %) também foi avaliada, visto que o etanol mostra atividade de remoção menor nestes sistemas; inibição de 5,7 % foi observada nesta concentração. Esta quantidade 5 de inibição não foi subtraída das formulações contendo etanol nos dados descritivos na Tabela 1, visto que a utilidade do próprio veículo de dosagem (derivado de dissuccinato dissódico em EtOH) na remoção direta estava sendo avaliada neste estudo. A astaxantina livre, não esterificada em DMSO (100 pm) foi avaliada como um padrão de referência para a comparação direta com 10 os novos derivados sintetizados para este estudo; a inibição média da astaxantina/veículo de DMSO foi de 28 % (Tabela 1).
A FIG. 18 mostra a capacidade de remoção relativa de cada um dos 4 estereoisômeros (mistura e 3 estereoisômeros individuais) em água, a uma concentração final de 100 μΜ. Exceto para o enantiômero 3R,3’R 15 (28,7 % de inibição), todas as outras formulações de derivado novas apresentaram capacidade de remoção diminuída em relação à formulação de astaxantina/DMSO (faixa -2,0 % a 19,3 % de inibição; Tabela 1). Como pode ser observado, a formulação 3S,3’S não exibiu qualquer atividade de remoção média. Quando introduzida no sistema de teste de neutrófilo isolado na 20 formulação etanólica, entretanto, em cada caso a capacidade de remoção aumentou em relação àquela do mesmo derivado formulado em água (FIG. 19; faixa de 38,0 % a 42,5 %). É importante mencionar que o derivado 3S,3’S foi formulado em 50 % de EtOH para esta comparação. Uma tendência voltada para a capacidade de remoção aumentada com relação à astaxantina 25 em DMSO foi observada para os novos derivados em formulação etanólica, mas depois da subtração da capacidade de remoção média do veículo de etanol (a concentração final no ensaio de teste de 0,3 %), a tendência não foi significante (NS). Além disso, nenhuma diferença significante na capacidade de remoção média foi observada entre as 4 formulações de novos derivados
114 %| testados em etanol (FIG. 19).
A FIG. 20 mostra os resultados da titulação de inibição do sinal de superóxido aumentando-se as concentrações da mistura de estereoisômeros de astaxantina dissuccinato dissódico na formulação 5 etanólica. Conforme a concentração foi aumentada de 100 μΜ para 3 mM, a inibição quase completa de sinal de superóxido foi observada (95,0 % de inibição na dose de 3 mM; Tabela 1 e FIG. 18). A curva de dose-resposta não foi linear. Ajustando a inibição percentual e a dose testada, o derivado de dissuccinato dissódico foi entre uma e duas ordens de magnitude menos — 10 potente do que o mimético de SOD usado como um controle positivo no estudo corrente (Tabela 1). A Tabela 1 representa estatísticas descritivas para as várias formulações de derivados de dissuccinato dissódico de astaxantina testadas no estudo corrente. Tamanhos de amostra de 3 ou maiores foram avaliados para cada formulação, com a exceção de 3S, 3’S em solução de 15 estoque de EtOH a 50 % (N = 2) e mimético de SOD (controle positivo, N = 1) avaliada no início do estudo.
TABELA 1
|
Amostra |
Solven te |
Concentração |
N |
Média (% de inibição |
Desvi 0 padrã 0 |
SEM |
Min |
Max |
Faixa |
|
Astaxanti |
DMSO |
0,1 mM |
4 |
28,0 |
7,6 |
3,8 |
20 |
35 |
15 |
|
na Mistura |
Água |
0,1 mM |
3 |
19,3 |
0,6 |
0,3 |
19 |
20 |
1 |
|
Mistura |
EtOH |
0,1 mM |
3 |
38,0 |
8,7 |
5,0 |
32 |
48 |
16 |
|
Mistura |
EtOH |
0,5 mM |
3 |
60,1 |
7,2 |
4,2 |
56 |
69 |
13 |
|
Mistura |
EtOH |
1,0 mM |
3 |
78,0 |
8,2 |
4,7 |
71 |
87 |
16 |
|
Mistura |
EtOH |
3,0 mM |
3 |
95,0 |
4,9 |
2,8 |
89 |
98 |
9 |
|
Meso |
Água |
0,1 mM |
3 |
15,7 |
5,9 |
3,4 |
9 |
20 |
11 |
|
Meso |
EtOH |
0,1 mM |
4 |
42,5 |
3,4 |
1,7 |
38 |
46 |
8 |
|
3R,3’R |
Água |
0,1 mM |
3 |
28,7 |
15,0 |
8,7 |
13 |
43 |
30 |
|
3R,3’R |
EtOH |
0,1 mM |
5 |
40,8 |
7,5 |
3,3 |
30 |
50 |
20 |
|
3S,3S’ |
Água |
0,1 mM |
3 |
-2,0 |
4,4 |
2,5 |
-7 |
1 |
8 |
|
3S,3S’ |
EtOH |
0,1 mM |
6 |
21,3 |
4,9 |
2,0 |
15 |
29 |
14 |
|
3S,3S’ |
EtOH |
0,1 mM |
2 |
38 |
1,4 |
1,0 |
37 |
39 |
2 |
|
Controle |
(50 %)
Água |
0,0 mM |
10 |
0,0 |
ND |
ND |
ND |
ND |
ND |
|
Controle |
EtOH |
0,3 % |
3 |
5,7 |
2,5 |
1,5 |
3 |
8 |
5 |
115 final
Mimético Água 10 μΜ 1 97,0 ND ND ND ND ND de SOD
Debate Resumido dos Resultados de EPR.
A astaxantina é um antioxidante lipofílico potente que normalmente exerce as suas propriedades antioxidantes em membranas celulares ricas em lipídeo, lipoproteínas e outro tecidos (Britton, 1995).
5- Derivados de astaxantina - com utilidade aumentada como agentes dispersáveis em água têm a capacidade descontaminar diretamente o ânion de superóxido de fase aquosa produzido isolando-se neutrófilos humanos depois do estímulo da explosão respiratória.
As formulações aquosas puras dos novos derivados foram •10 menos potentes do que as formulações etanólicas em termos de capacidade de remoção direta. A montagem supramolecular dos derivados de carotenóide A solúveis em água em alguns solventes (por exemplo, água) pode explicar a sua falta de potência nestes solventes. A agregação é do tipo “conjunto de cartões” helicoidal, com agregados maiores do que 240 nm no tamanho 15 formando na solução aquosa pura. Aumentar a concentração iônica de soluções tampão pode aumentar tanto o tamanho quanto a estabilidade destes agregados. A capacidade de remoção de radical destes agregados será diminuída em relação às soluções monoméricas dos mesmos compostos; de fato, nenhuma capacidade de remoção foi observada para o estereoisômero •20 3S,3’S dissolvido em água (Tabela 1, FIG. 18). Cuidado deve ser tomado na preparação de formulações para teste in vitro e in vivo, visto que a montagem supramolecular limita o número de moléculas disponíveis para a interação com as espécies radicais. O tamanho dos agregados também deve ser levado em conta, visto que os agregados contendo tantas moléculas quanto 106 e 25 atingindo 300 nm ou mais no tamanho foram descritos (Bakadi, 2002).
A titulação da dose do derivado de astaxantina dissuccinato dissódico para 3 mM (como a mistura de estereoisômeros em EtOH/Água 1:2) demonstrou a supressão quase completa do sinal do ânion de superóxido
116 (95 % de inibição), como medido com o aprisionamento de spin DEPMPO (FIG. 20). A curva de resposta de dose foi não linear, requerendo doses crescentes para a supressão quase completa de sinal de radical (FIG. 20). Na concentração mais baixa testada (100 μΜ), aproximadamente 40 % do sinal foi inibido. A potência do derivado de astaxantina dissuccinato dissódico nesta dose pode ser comparada diretamente com os miméticos de superóxido dismutase (SOD) usados como um controle positivo no estudo corrente (97 % de inibição a 10 μΜ). Os resultados mostram que como um descontaminante de radical de fase aquosa, o derivado de astaxantina dissuccinato dissódico é uma a duas ordens de magnitude menos potente do que o mimético de SOD. Entretanto, in vivo, estes derivados decompõe-se até a astaxantina livre, que se toma ativa nas membranas ricas em lipídeo de células [incluindo as membranas mitocondriais e nucleares (Goto, 2001)], fornecendo portanto proteção dupla (remoção de fase aquosa e lipídica), não obtenível com miméticos de proteínas e enzima solúveis em água. A astaxantina livre, não esterificada (quando fornecida como uma dieta suplementar a 0,02 % da alimentação p/p) é cardioprotetiva contra o insulto de exercício estrênuo mediado por ROS tanto para o músculo esqueletal quanto para o cardíaco (Aoi et al. 2003). Portanto, esta característica (isto é a remoção de radical de fase dupla) deve fornecer utilidade adicional para esta classe de compostos como agentes terapêuticos clínicos nestas indicações para as quais a prevenção de radical e espécie de oxigênio reativos é importante (Cross, 1987).
O estudo demonstra pela primeira vez a remoção direta de ânion de superóxido detectado pela espectroscopia de EPR por um novo grupo de derivados de carotenóide. Os compostos foram descobertos formar montagens supramoleculares na solução aquosa pura. A formação de montagens supramoleculares pode limitar a sua potência de remoção em relação às soluções monoméricas dos mesmos compostos. Nenhuma diferença
117 significante na capacidade de remoção foi observada entre os 4 estereoisômeros potenciais dos novos compostos. Os estudos de variação de dose revelaram que a concentração de derivado pode ser aumentada até a supressão quase completa do sinal do ânion de superóxido induzido. Como agentes terapêuticos potenciais in vivo, esta classe de compostos pode ser usada como um descontaminante tanto de fase aquosa quanto de fase lipídica, que deve encontrar ampla aplicação naquelas condições de doença agudas e crônicas para as quais os descontaminantes de radical potentes têm demonstrado eficácia.
Remoção de ânion de superóxido direta pelo derivado de astaxantina dissuccinato dissódico di-vitamina C:
Em um experimento de produção de imagem de ressonância paramagnética de elétron (EPR), os neutrófilos foram isolados em um gradiente Percoll a partir do sangue integral de um voluntário humano. Os neutrófilos isolados foram depois recolocados em suspensão em solução salina tamponada com fosfato e estimulados ao máximo com éster de forbol para induzir a explosão respiratória e a produção de ânion de superóxido. A solução de neutrófilos humanos ativados, o derivado de astaxantina dissuccinato dissódico di-vitamina C (XXIa) (nome semi-sistemático 4-[18(4-{3-[2-(3,4-diidróxi-5-oxo-2,5-diidrofuran-2-il)-2-hidróxi-etoxicarbonil]propionilóxi} -2,6,6-trimetil-2-oxo-cicloex-1 -enil)3,7,12,16-tetrametiloctadeca-1,3,5,7,9,11,13,15,17-nonaenil]-3,5,5-trimetila-2-oxo-ciclohex-3enílico éster 2-(3,4-diidróxi-5-oxo-2,5-diidrofuran-2-il)-2-hidróxi-etílico do Ácido Succínico) foi adicionado em várias concentrações e o sinal de superóxido (como medido com a produção de imagem de EPR) foi subsequentemente medido. O derivado de astaxantina dissuccinato dissódico di-vitamina C (XXIII) reduziu a medida de sinal do ânion de superóxido em uma maneira dependente da dose (FIG. 33); a supressão completa do sinal do ânion de superóxido foi obtida na concentração de 60 μΜ. Isto representa um
118 aumento de 50 vezes na potência em relação ao derivado de astaxantina dissuccinato dissódico (XVI) também sintetizado para a série corrente de experimentos. A pureza do derivado como testado foi de 88 % (pela área de HPLC sob a curva ou AUC). O novo derivado de carotenóide - planejado para 5 ser um “medicamento brando” pela esterificação na posição 6-OH de cada vitamina C - preservou a função antioxidante das moléculas de vitamina C individuais. A potência do derivado (XXIII) aproximou-se daquela da formulação de astaxantina dissuccinato dissódico (XVI) com vitamina C livre em uma relação molar de 1:2 (que suprimiu completamente o sinal do ânion 10 de superóxido em uma formulação de 20 μΜ/40 μΜ derivado de astaxantina dissuccinato dissódico (XVI)/ vitamina C livre). O derivado (XXIII), que gera 2 moles de vitamina C livre e 1 mol de astaxantina livre, não esterificada para cada mol de derivado in vivo pode ser particularmente preferido para certas indicações clínicas. O derivado (XXIII) também provavelmente apresentará 15 eficácia aumentada naquelas situações clínicas em que a remoção de fase aquosa (pelo derivado precursor intacto, assim como vitamina C livre) assim como a remoção de fase lipídica (pela astaxantina livre, não esterificada) são importantes para a redução na patologia atribuível a ROS e outras espécies de dano pelos radicais.
Tamanho da Redução do Infarto em Ratos Sprague-Dawley
Machos:
A FIG. 4, FIG. 25 e FIG. 26 descrevem representações gráficas da redução de tamanho do infarto em ratos Sprague-Dawley machos. Os ratos Sprague-Dawley machos foram pré tratados com o derivado do sal 25 dissódico de astaxantina dissuccinato (como a mistura de estereoisômeros) em solução aquosa antes de realizar uma oclusão e indução de um infarto do miocárdio. Os ratos Sprague-Dawley machos (175 a 200 gramas) foram anestesiados com 100 mg/kg de Inactina, instrumentados e o coração exposto. A artéria coronária esquerda teve uma sutura colocada em tomo dela e foi
119 /0/= submetida a 30 minutos de oclusão total da artéria coronária seguido por 2 horas de reperfusão, tempo no qual o tamanho do infarto foi medido nos corações excisados dos animais. Os corações foram lavados em tampão e incubados em solução de fingimento de cloreto de trifeniltetrazólio (TTC) mantida a 37° C em tampão de fosfato no pH de 7,40. O tamanho do infarto (IS) foi expresso como uma % da área em risco (IS/AAR, %). A pressão de sangue sistêmico, a freqüência cardíaca, os gases do sangue e a temperatura do corpo foram monitoradas por todo o experimento e a temperatura e gases do sangue foram estritamente controlados aos níveis fisiológicos normais. 25,
50 ou 75 mg/kg do derivado do sal dissódico de astaxantina dissuccinato ou veículo de solução salina estéril foi administrado I.V. pela injeção na veia da cauda a cada dia durante 4 dias antes do experimento de infarto e determinação do tamanho do infarto.
Descrição Resumida dos resultados de Salvamento.
O tamanho da redução do infarto e o salvamento miocárdico correspondente, aumentaram linear e significantemente, com a dose (P = 0,001**). Na dose máxima testada, 75 mg/kg, o salvamento miocárdico médio foi de 56 %, que se aproxima daquele obtenível com as estratégias pré condicionantes isquêmicas. As limitações de volume para a injeção I.V. de dose única neste rato impede o teste de doses mais altas; entretanto, a correlação linear significante (P < 0,001**; r2 = 0,67) entre os níveis isentos de plasma não esterificados de astaxantina e IS/AAR, % sugeriu que as doses de aproximadamente 120 a 125 mg/kg, 100 % de salvamento podería ser obtida. Esta é a primeira demonstração de cardioproteção por um novo derivado de carotenóide.
Farmacocinéticas, biodisponibilidade aumentada e distribuição de tecido alvo aumentadas do derivado de astaxantina dissuccinato dissódico oralmente administrado:
Farmacocinéticas do Plasma
120
Os parâmetros farmacocinéticos da dose oral única (incluindo Qmax> Tmax, AUC(0-72) Vd e depuração) do derivado de astaxantina dissuccinato dissódico foram determinados em camundongos C57BI/6 machos. Os animais foram administrados com o derivado oralmente em uma dose máxima única (500 mg/kg) mostrada em estudos anteriores ser eficaz na prevenção do dano secundário à administração de CC14 em ratos Sprague-Dawley (100 mg/kg de peso corporal nestes estudos). As amostras para a análise de HPLC de níveis de astaxantina livre no plasma e fígado foram obtidas nos seguintes pontos de tempo, de pelo menos 3 animais por ponto de tempo:
M10 Tempo 0 [imediatamente antes da dosagem do composto de teste], 2, 4, 6, 8,12,16, 24,48 e 72 horas depois da ingestão.
Amostras adicionais, com N < 3, foram tomadas em outros intervalos (10, 14 e 36 horas; Tabelas 2 e 3). Os níveis de astaxantina livre não esterificadas foram determinadas neste estudo visto que os ésteres de 15 carotenóide são completamente clivados no intestino de mamífero para carotenóide livre, que se movem passivamente através de enterócito.
Descrição Resumida dos Métodos Experimentais: Farmacocinéticas do Plasma
Camundongos C57BL/6 machos, aproximadamente 25 g, foram alojados em gaiola (três camundongos/gaiola) e alimentados com ração de camundongo padrão (Purina Mouse Chow, Ralston Purina, St. Louis) e água ad libitum por pelo menos cinco dias antes do início do experimento. O derivado de astaxantina dissuccinato dissódico foi misturado com os seguintes componentes para fazer uma emulsão adequada para gavagem oral:
· Água filtrada estéril (0,2 mícron Millipore);
• Óleo de oliva (Bertolli USA, Inc., Secaucus, NJ);
• Lecitina de soja, Tipo IV-S (Sigma-Aldrich Co., St. Louis, MO; número do catálogo P3644).
O derivado de astaxantina dissuccinato dissódico demonstra
121 solubilidade em água de aproximadamente 8,64 mg/ml em formulação aquosa pura. Na emulsão descrita acima, a solubilidade foi aumentada para aproximadamente 50 mg/ml, permitindo a dosagem de até 500 mg/kg pela gavagem nestes animais. Este aumento signifícante de 6 vezes na solubilidade 5 na dosagem de veículo facilitou enormemente os estudos de gavagem nestes camundongos pequenos.
Os métodos para preparar a emulsão foram como segue:
(1) Adicionar 80 mg de lecitina de soja (Sigma catálogo P3644) a 5,0 ml de água. Turbilhonar intermitentemente por aproximadamente 30 minutos em um tubo de centrífuga de 15 ml até que a suspensão fique uniforme;
(2) Adicionar 2,5 ml de óleo de oliva na temperatura ambiente e turbilhonar. Isto produz uma suspensão amarela uniforme, espessa, turva. Este material de emulsão pode ser armazenado na temperatura ambiente ou no refrigerador a 4o C. Se armazenado, turbilhonar imediatamente antes de adicionar o derivado de dissuccinato dissódico na Etapa 3 (abaixo);
(3) Adicionar o derivado de astaxantina dissuccinato dissódico a 50 mg/ml diretamente à emulsão. O composto facilmente entra em uma suspensão uniforme nesta concentração. Turbilhonar imediatamente antes da gavagem para garantir a suspensão uniforme; e (4) O material tem o potencial para coagular a agulha de gavagem de camundongo. Enxaguar a agulha de gavagem depois de cada 2 gavagens.
A emulsão foi administrada pela gavagem oral a 500 mg/kg de peso corporal em uma dose única. O alimento foi retirado de todas as gaiolas na noite antes do experimento. Uma hora depois da administração da emulsão, alimento e água foram recolocadas a todos os animais.
Os métodos para a amostragem de sangue integral e tecido, extração de amostra e análise de HPLC foi descrita em detalhes (Osterlie,
122
2000). Em resumo, o sangue integral foi coletado nos tubos de Vacutainer® contendo EDTA e plasma subsequentemente preparado pela centrifugação a
4o C, 1500 x g durante 20 minutos. As amostras de plasma foram depois aliquotadas e subtamente congeladas em nitrogênio antes do transporte e análise de HPLC.
Acúmulo em Tecido
A concentração de astaxantina livre também foi determinada nos mesmos pontos de tempo como para as amostras de plasma, no fígado. Os fígados foram removidos de cada animal no estudo de farmacocinética depois do sacrifício e subtamente congelados em nitrogênio líquido. O tecido hepático foi preparado para a análise de HPLC como descrito (Jewell, 1999). Portanto, o exame simultâneo do acúmulo hepático de astaxantina livre foi realizado nos mesmos pontos de tempo como as análises de plasma.
Descrição Resumida dos Métodos Experimentais: Acúmulo no Fígado de Astaxantina Livre
Até 300 mg de fígado de cada animal foi subtamente congelado em nitrogênio líquido. A homogeneização e extração de tecido foram realizadas com uma mistura de clorofórmio/metanol/água, de acordo com os métodos de Jewell (1999). O acúmulo de astaxantina livre, não esterificada no fígado foi depois avaliado pela HPLC como descrito acima para as amostras de plasma.
Debate Resumido dos Resultados Farmacocinéticos.
As tabelas de resumo dos níveis plasmáticos e hepáticos de astaxantina no(s) intervalo(s) de amostragem apropriado(s) são mostradas como as Tabelas 2 e 3. As áreas de astaxantina livre não esterificada plasmática e hepática sob a curva vs. tempo (AUC’s) são também incluídas nas Tabelas 2 e 3. Os resultados demonstram que para cada intervalo de amostragem, os níveis de astaxantina livre no fígado são iguais ou maiores do que no plasma. Esta liberação específica de tecido melhorada ao fígado é sem
123 precedentes na literatura; de fato, os níveis hepáticos da astaxantina livre são tipicamente mais baixos do que os níveis correspondentes no plasma nos pontos de tempo equivalente pós dose (Kurihara, 2002). Assim, o derivado de astaxantina dissuccinato dissódico na emulsão descrita acima é um veículo 5 superior para a liberação de concentrações terapêuticas de carotenóide livre aos tecidos de interesse depois da dosagem oral.
TABELA 2: Níveis Plasmáticos de Astaxantina Livre, Não Esterificada
|
Tempo |
Amostra |
AstanM |
Asta mg/kg |
Média mg/kg |
Desvio padrão |
|
0 |
PK01 |
0,00 |
0,00 |
|
|
| |
PK03 |
0,00 |
0,00 |
|
|
| |
PK06 |
0,00 |
0,00 |
|
|
| |
PK15 |
0,00 |
0,00 |
|
|
| |
PK16
PK20 |
0,00 |
0,00 |
0,0 |
|
|
2 |
PK10 |
38,04 |
0,02 |
|
|
| |
PK12 |
0,00 |
0,00 |
|
|
| |
PK21 |
0 |
0 |
|
|
| |
PK22 |
0 |
0 |
|
|
| |
PK27 |
0 |
0 |
|
|
| |
PK34 |
0 |
0 |
|
|
| |
PK42 |
311,73 |
0,19 |
|
|
| |
PK43 |
74,08 |
0,04 |
|
|
| |
PK48 |
48,41 |
0,03 |
|
|
| |
PK59 |
318,83 |
0,19 |
0,0 |
0,07 |
|
4 |
PK07 |
46,18 |
0,03 |
|
|
| |
PK11 |
115,63 |
0,07 |
|
|
| |
PK14 |
20,97 |
0,01 |
|
|
| |
PK17 |
40,57 |
0,02 |
|
|
| |
PK23 |
214,95 |
0,13 |
|
|
| |
PK24 |
179,33 |
0,11 |
|
|
| |
PK28
PK44 |
80,48 |
0,05 |
|
|
| |
PK45 |
67,16 |
0,04 |
|
|
| |
PK57 |
119,02 |
0,07 |
|
|
| |
PK58 |
147,85 |
0,09 |
0,06 |
0,039 |
|
6 |
PK13 |
40,57 |
0,02 |
|
|
| |
PK18 |
605,01 |
0,36 |
|
|
| |
PK25 |
262,73 |
0,16 |
|
|
| |
PK26
PK32 |
377,14 |
0,22 |
|
|
| |
PK46 |
739,91 |
0,44 |
|
|
| |
PK60 |
167,39 |
0,1 |
|
|
| |
PK61 |
131,74 |
0,08 |
0,19 |
0,15 |
|
8 |
PK36
PK47 |
435,17 |
0,26 |
|
|
124
| |
PK49
PK62
PK68
PK69
PK70 |
371,11
148.98
405
306,86
29.98 |
0,22
0,09
0,24
0,18
0,02 |
0,16 |
0,09 |
|
10 |
PK31 |
|
|
|
|
|
12 |
PK37 |
|
|
|
|
| |
PK63 |
37,19 |
0,02 |
|
|
| |
PK64 |
10,93 |
0,01 |
|
|
| |
PK67 |
8,12 |
0 |
|
|
| |
PK71 |
53,19 |
0,03 |
|
|
| |
PK72 |
7,66 |
0 |
|
|
| |
PK73 |
8,46 |
0,01 |
0,01 |
0,01 |
|
14 |
PK51 |
0 |
0 |
|
|
| |
PK52 |
3,14 |
0 |
|
|
|
16 |
PK65 |
8,44 |
0,01 |
|
|
| |
PK66 |
10,47 |
0,01 |
|
|
| |
PK75 |
28,24 |
0,02 |
|
|
| |
PK76 |
4,51 |
0 |
0,01 |
0,00 |
|
24 |
PK29 |
0 |
0 |
|
|
| |
PK35 |
18,03 |
0,01 |
|
|
| |
PK39 |
13,93 |
0,01 |
|
|
| |
PK50 |
1,51 |
0 |
|
|
| |
PK53 |
0 |
0 |
0,00 |
0,005 |
|
36 |
PK38 |
21,37 |
0,01 |
0,01 |
|
|
48 |
PK30 |
0 |
0 |
|
|
| |
PK33 |
0 |
0 |
|
|
| |
PK54 |
22,71 |
0,01 |
|
|
| |
PK55 |
0 |
0 |
0,00 |
0,005 |
|
72 |
PK40
PK41 |
1,7 |
0 |
|
|
| |
PK56 |
0 |
0 |
|
|
| |
PK74 |
1,92 |
0 |
|
|
TABELA 3: Níveis Hepáticos de Astaxantina Livre, NãoEsterifícada
|
Tempo |
Amostra |
AstanM |
Asta mg/kg |
Média mg/kg |
Desvio padrão |
|
0 |
PK01 |
0,00 |
0,00 |
|
|
| |
PK03 |
0,00 |
0,00 |
|
|
| |
PK06 |
0,00 |
0,00 |
|
|
| |
PK15 |
0,00 |
0,00 |
|
|
| |
PK16 |
7,67 |
0,00 |
|
|
| |
PK20 |
8,18 |
0,00 |
0,0 |
|
|
2 |
PK10 |
139,37 |
0,08 |
|
|
| |
PK12 |
30,66 |
0,02 |
|
|
| |
PK21 |
414,34 |
0,25 |
|
|
| |
PK22 |
725,87 |
0,43 |
|
|
| |
PK27 |
294,07 |
0,18 |
|
|
| |
PK34 |
165,32 |
0,1 |
|
|
| |
PK42 |
689,36 |
0,41 |
|
|
| |
PK43 |
129,66 |
0,08 |
|
|
125
| |
ΡΚ48
ΡΚ59 |
244,5
564,28 |
0,15
0,34 |
0,2 |
0,14 |
|
4 |
ΡΚ07 |
103,07 |
0,06 |
|
|
| |
ΡΚ11 |
243,4 |
0,15 |
|
|
| |
PK14 |
89,18 |
0,05 |
|
|
| |
PK17 |
1565,15 |
0,93 |
|
|
| |
PK19 |
1373,34 |
0,82 |
|
|
| |
PK23 |
2558,63 |
1,52 |
|
|
| |
PK24 |
4701,95 |
2,8 |
|
|
| |
PK28 |
1023,78 |
0,61 |
|
|
| |
PK44 |
359,73 |
0,21 |
|
|
| |
PK45 |
211,35 |
0,13 |
|
|
| |
PK57 |
322,06 |
0,19 |
|
|
| |
PK58 |
500,82 |
0,3 |
0,64 |
0,812 |
|
6 |
PK13 |
374,28 |
0,22 |
|
|
| |
PK18 |
2970,44 |
1,77 |
|
|
| |
PK25 |
3515,52 |
2,1 |
|
|
| |
PK26 |
2087,8 |
1,24 |
|
|
| |
PK32 |
687,99 |
0,41 |
|
|
| |
PK46 |
1070,13 |
0,64 |
|
|
| |
PK60 |
974,69 |
0,58 |
|
|
| |
PK61 |
841,37 |
0,5 |
0,93 |
0,69 |
|
8 |
PK36 |
1290,15 |
0,77 |
|
|
| |
PK47 |
230,88 |
0,14 |
|
|
| |
PK49 |
1115,86 |
0,67 |
|
|
| |
PK62 |
1247 |
0,74 |
|
|
| |
PK68 |
1263,31 |
0,75 |
|
|
| |
PK69 |
1036,29 |
0,62 |
|
|
| |
PK70 |
1518,27 |
0,9 |
0,63 |
0,24 |
|
10 |
PK31 |
1303,06 |
0,78 |
0,78 |
|
|
12 |
PK37 |
3225,35 |
1,92 |
|
|
| |
PK63 |
921,74 |
0,55 |
|
|
| |
PK64 |
713,97 |
0,43 |
|
|
| |
PK67 |
410,93 |
0,24 |
|
|
| |
PK71 |
1382,45 |
0,82 |
|
|
| |
PK72 |
567,95 |
0,34 |
|
|
| |
PK73 |
716,89 |
0,43 |
0,46 |
0,57 |
|
14 |
PK51 |
141,9 |
0,08 |
|
|
| |
PK52 |
179,51 |
0,09 |
0,08 |
0,00 |
|
16 |
PK65 |
240,6 |
0,14 |
|
|
| |
PK66 |
340,38 |
0,2 |
|
|
| |
PK75 |
788,66 |
0,47 |
|
|
| |
PK76 |
499,84 |
0,3 |
0,27 |
0,14 |
|
24 |
PK29 |
440,72 |
0,26 |
|
|
| |
PK35 |
321,14 |
0,19 |
|
|
| |
PK39 |
155,42 |
0,09 |
|
|
| |
PK50 |
156,61 |
0,09 |
|
|
| |
PK53 |
89,18 |
0,05 |
0,13 |
0,08 |
|
36 |
PK38 |
658,41 |
0,39 |
0,3 |
|
|
48 |
PK30 |
106,07 |
0,06 |
|
|
| |
PK33 |
116,79 |
0,07 |
|
|
126
| |
PK54
PK55 |
17,81
28,79 |
0,01
0,02 |
0,0 |
0,029 |
|
72 |
PK40 |
33,52 |
0,02 |
|
|
| |
PK41 |
11,66 |
0,01 |
|
|
| |
PK56 |
9,21 |
0,01 |
|
|
| |
PK74 |
19,31 |
0,01 |
0,013 |
0,005 |
O pré tratamento (15 dias a 6 semanas) é frequentemente requerido quando carotenóides tais como a astaxantina são fornecidos no veículo oral ou na alimentação para se alcançar níveis eficazes nos estudos de dano ao fígado (Kang, 2001; Kim, 1997; Aoi et al. 1993). Neste caso, os níveis terapêuticos (200 nm ou acima) foram obtidos com uma dose única.
A Cmax (Tabela 4) de 0,9 mg/litro também é sem precedentes em roedores, animais que absorvem apenas uma pequena porcentagem da dose oral de carotenóides. É significante que estes níveis plasmáticos e hepáticos de carotenóide livre foram obtidos depois de apenas uma única dose de composto no veículo de emulsão. Nos seres humanos, Osterlie et al. (2000) descreveu níveis plasmáticos Cmax de 1,3 mg/litro depois de uma única dose de 100 mg (aproximadamente 1,1 mg/kg dose oral) de astaxantina livre, não esterificada em veículo de óleo de oliva. Os seres humanos tipicamente absorvem de 40 a 50 % da dose oral de carotenóide quando fornecido em veículo graxo, como oposto a uns poucos pontos percentuais para os roedores. Portanto, o estudo corrente demonstra a obtenção de aproximadamente 70 % do Cmax em seres humanos com o veículo de emulsão desenvolvido para roedores, aumentando enormemente a utilidade deste derivado para os estudos de hepato-proteção.
127
TABELA 4: Parâmetros de pK
|
Parâmetro |
Fígado |
|
Plasma |
|
*Cmax (mg/1) **Tmax (h) Eliminação da meia-vida (h) Taxa de eliminação (1/h) ***AUC(0-72) (mg h/1) ***AUCoo (mg h/1) Depuração oral (L/h) Volume de distribuição (L/kg) |
0,9
6
11,655
0,059
15.8
15.9
15,856
263.9 |
|
0,2
6
3,938
0,176
1,2
1,2
216,822
1232,1 |
* Concentração máxima ** Tempo na concentração máxima *** Área sob a curva
Redução do tamanho do infarto e níveis em circulação experimentais de proteína reativa C em coelhos depois da administração parenteral de Cardax® (derivado de astaxantina dissuccinato dissódico):
A influência da administração parenteral do derivado de astaxantina dissuccinato dissódico (XVI) sobre o tamanho do infarto induzido 10 e níveis induzidos de proteína reativa C (CRP) circulante em coelhos foi investigada usando os métodos de Barrett et al. (2002) com modificações leves. O propósito do estudo corrente foi investigar a capacidade do derivado de astaxantina dissuccinato dissódico (XVI) para reduzir a inflamação como medida pela CRP no cenário da isquemia miocárdica/dano de reperfusão _ 15 experimental no coração do coelho. Foi sugerido que a CRP, habitualmente usada como um marcador para a resposta inflamatória aguda (“fase aguda”), pode de fato ter um efeito pró inflamatório mediado através da ativação da cascata de complemento. A isquemia miocárdica/dano de reperfusão, que é acompanhado por um aumento na formação de radicais de oxigênio (ROS), 20 também foi mostrado ativar o sistema de complemento. Foi demonstrado que (1) o aumento endógeno no CRP plasmático secundário a uma lesão inflamatória remota foi associada com um aumento no dano de tecido miocárdico secundário à isquemia e reperfusão regional; (2) este aumento no dano (manifestado como tamanho do infarto aumentado) foi mediado pela 25 atividade de complemento; e (3) a CRP foi um “influenciador” e não
128 meramente uma medida indireta de inflamação sistêmica, neste sistema. Portanto, a redução dos níveis de CRP circulantes, junto com a(s) redução(ões) no tamanho do infarto anteriormente mencionado com Cardax® em roedores, pode formar uma modalidade terapêutica anti-inflamatória 5 poderosa no cenário da síndrome coronariana aguda.
Em resumo, coelhos brancos New Zealand machos (2,25 a 2,5 kg) foram usados para o estudo. A resposta inflamatória de fase aguda foi induzida pela injeção subcutânea de quatro alíquotas (0,5 ml cada) de 1 % de óleo de croton em óleo de milho começando no segundo dia de pré tratamento _ 10 com Cardax®. Cardax® (a 50 mg/kg IV pela injeção na veia da orelha) em água ou volumes iguais de solução salina estéril foram dados uma vez ao dia durante 4 dias antes da infartação experimental no dia 5. O curso de tempo de aumento nos níveis de CRP circulantes foi obtido como descrito anteriormente (Barret et al. 2002), usando um método com base em ELISA com anticorpos de CRP anti-coelho. No dia final do experimento (dia 5: aproximadamente 24 horas depois da última infusão de medicamento), os coelhos foram anestesiados com uma mistura de xilazina (3 mg/kg) e cetamina (35 mg/kg) seguido pelo pentobarbital (90 mg/kg) intramuscularmente. O pentobarbital adicional foi administrado como 20 necessário para manter a anestesia. Depois da traqueotomia, os coelhos foram ventilados com ar ambiente e o coração foi exposto por intermédio de uma toracotomia esquerda. O coração foi depois sustentado em um tala pericardial e uma ligadura de seda 3-0 foi colocada em tomo da artéria coronária descendente anterior esquerda. A artéria foi ocluída durante 30 minutos exercendo-se tração na ligadura e subsequentemente reperfusada durante 180 minutos. Um pouco antes de completar o protocolo, uma amostra de sangue venoso foi obtida para a determinação de CRP plasmática.
Na conclusão da fase de reperfusão do protocolo, os corações foram removidos e canulados pela aorta no aparelho de perfusão de
129
Langendorff. Os corações foram depois perfundidos com um tampão de Krebs-Henseleit modificado durante 10 a 15 minutos (20 a 25 ml/minuto). Na conclusão deste período, os corações foram perfundidos com 80 ml de 0,4 % de cloreto de 2,3,5-trifeniltetrazólio (TTC) a 37° C para a determinação da área em risco (AAR). A artéria coronária circunflexa esquerda foi depois ligada na mesma área como o foi durante a preparação cirúrgica/infartação experimental. Neste tempo, a bomba de perfosão foi interrompida e 3,0 ml de corante azul de Evan foi injetada lentamente nos corações através de um orifício de sidearm conectado à cânula aórtica. A solução foi deixada distribuir através do coração por aproximadamente 30 segundos. Os corações foram depois cortados em seis seções transversais nos ângulos retos em relação ao eixo vertical. O ventrículo direito, o ápice e o tecido atrial foram descartados. O tecido demarcado por uma cor púrpura /azul representou a região perfundida pela distribuição da artéria coronária não relacionada com o infarto. Ambas as superfícies de cada seção transversal foram traçadas sobre folhas de acetato claras que foram escaneadas e subsequentemente digitalizadas para calcular a área de infarto. A área total em risco foi expressada como uma porcentagem do ventrículo esquerdo. O tamanho do infarto foi depois expressado como uma porcentagem da área em risco.
O tamanho médio do infarto nos animais de controle e nos animais tratados com Cardax® é mostrado na FIG. 37. Os níveis de CRP circulante nos animais de controle e animais tratados com Cardax® (mostrados como a diferença média entre os níveis de base de referência e níveis induzidos no momento da reperfusão) são mostrados na FIG. 38. As reduções no tamanho do infarto de aproximadamente 55,4 % foram observados nos coelhos tratados com Cardax®; a área isquêmica em risco foi similar em ambos os grupos. Similarmente, o aumento médio nos níveis de CRP circulantes nos controles (+23,5 %) em relação à base de referência foi completamente abolidos nos animais tratados com Cardax®, até os níveis
130 médios abaixo daqueles observados na base de referência (-15,7 %). Visto que a CRP é tanto um influenciador na síndrome coronária aguda - que resulta em um tamanho do infarto aumentado na presença de níveis elevados deste reagente de fase aguda - quanto um prognosticador independente forte de risco cardiovascular na prevenção primária e secundária de pacientes cardíacos - as reduções nos níveis desta proteína em circulação forma uma forte modalidade terapêutica.
A administração oral de astaxantina dissuccinato dissódico reduz as elevações da alanina aminotransferase (ALT) produzidas pelos lipopolissacarídeo (LPS) em camundongos:
O seguinte estudo avalia a utilidade da administração oral do derivado de astaxantina dissuccinato dissódico para efeitos hepatoprotetivos em um modelo de dano hepático induzido pelo LPS em camundongos.
Breve Descrição dos Métodos Experimentais:
Camundongos ICR machos de três meses de idade foram tratados com LPS e galactosamina de modo a induzir o dano hepático (Leist, 1995). Os camundongos foram primeiro submetidos à gavagem oral com uma emulsão de óleo de oliva /água/ lecitina (10 ml/kg ou 0,3 ml para um camundongo de 30 gramas) ou a mesma emulsão contendo o derivado de astaxantina dissuccinato dissódico (50 mg/ml) para uma dose final de astaxantina dissuccinato dissódico de 500 mg/kg. Duas horas mais tarde os camundongos foram injetados intraperitonealmente (IP) com solução salina (10 ml/kg) ou uma solução de LPS de E. coli (3 mg/kg, Sigma número do catálogo L3755) e D-galactosamina (700 mg/kg). Os animais foram sacrificados pela asfixia com dióxido de carbono (CO2) 5 horas depois da injeção IP e o plasma foi depois coletado para a determinação de ALT.
Descrição Resumida de Resultados dos Danos Induzidos por
LPS.
Estes resultados iniciais demonstraram que o derivado de
131 astaxantina dissuccinato dissódico não teve nenhum efeito sobre o ALT plasmático nos animais injetados com solução salina (controle tratado com imitação de dano hepático). Nos animais de controle submetidos à gavagem apenas com emulsão (sem o derivado), houve um aumento em mais do que 3 vezes no ALT. Nos animais que receberam a emulsão com derivado de astaxantina dissuccinato dissódico a 500 mg/kg incluídos, a elevação de ALT foi substancialmente reduzida (N = 3 animais por grupo), demonstrando a eficácia do composto na redução de ALT, um marcador sérico de necrose de hepatócito nestes animais. Visto que o dano hepático induzido por LPS é mediado por ROS (incluindo o radical óxido nítrico NO*) e inflamação sistêmica substancial ocorre depois do insulto de LPS, para o qual a astaxantina livre, não esterificada é protetiva (Ohgami et al. 2003), a utilidade do novo derivado para as indicações clínicas nas quais tal inflamação é promovida representa uma forma de realização particularmente útil.
Acúmulo de astaxantina livre no plasma e fígado depois da administração oral de dose múltipla nos camundongos pretos:
Neste estudo farmacocinético, com métodos como aqui descritos, onze (11) doses orais diárias individuais do derivado de astaxantina dissuccinato dissódico (500 mg/kg) foram administrados pela gavagem oral no veículo de emulsão aos camundongos pretos e o acúmulo de astaxantina livre no plasma e fígado foi medido em três (3) animais na Cmax e Tmax prováveis (6 horas). As Cmax e Tmax prováveis (6 horas) foram deduzidas de amostras plasmáticas e hepáticas no estudo farmacocinético oral de dose única anterior. O acúmulo de astaxantina livre, não esterificada no plasma e fígado depois das doses de emulsão única foi avaliado. A concentração de plasma médio para todos os animais testados foi de 381 nM. A concentração hepática média para todos os animais testados foi 1735 nM. No estudo de dose única, em média, um nível protetivo (ajustado na ED50 de antioxidante para a astaxantina livre, não esterificada de 200 nM) foi obtido tanto no
132 plasma quanto no fígado; a concentração hepática média obtida foi quase 9 vezes o nível protetivo.
No estudo de dose múltipla, tanto os níveis de pico quanto de depressão foram tomados (os níveis de pico tomados 6 horas depois da dosagem no Cmax provável; níveis de depressão obtidos 6 horas depois da Cmax ou 12 horas após a dose). Os níveis de pico médio no plasma no pico e depressão, respectivamente, foram 485 nM e 231 nM; os níveis de pico médio hepáticos no pico e depressão, respectivamente, foram 1760 nM e 519 nM. Mais uma vez, em cada caso os níveis protetivos foram obtidos e mantidos até 11 dias após a dosagem múltipla; no caso do fígado, níveis de quase 9 vezes o nível protetivo foram obtidos. Mais uma vez, em cada ponto de tempo depois da dosagem múltipla, o acúmulo no fígado foi maior do que aquele observado no plasma, demonstrando a utilidade aumentada deste veículo de dosagem para alvejar este órgão sólido (FIG. 32). Também está evidente a partir deste conjunto de dados que a administração crônica do derivado de astaxantina dissuccinato dissódico será eficaz na hepatoproteção.
Acúmulo de astaxantina livre no miocárdio (coração) e cérebro depois da administração oral de dose única em camundongos pretos:
Uma dose máxima única do derivado de astaxantina dissuccinato dissódico (500 mg/kg) foi dada pela gavagem oral no veículo de emulsão a camundongos pretos e o acúmulo de astaxantina livre, não esterificada foi medido em quatro (4) animais na Cmax e Tmax provável (6 horas), como deduzido a partir das amostras plasmáticas e hepáticas no estudo anterior. O acúmulo de astaxantina livre, não esterificada no coração depois de uma dose única foi excelente (média +/- SEM de 4 animais = 693,25 +/272 nM) e se igualou àquela observada com o acúmulo de astaxantina livre, não esterificada no fígado. Mais uma vez, em cada ponto de tempo, o acúmulo no coração foi maior do que aquele observado no plasma, demonstrando a utilidade aumentada deste veículo de dosagem para alvejar
133 órgãos sólidos. O acúmulo de astaxantina livre, não esterifícada no CNS (cérebro) foi menos surpreendente (média +/- SEM de 4 animais = 3,6 +/-1,7 nM), sugerindo que a penetração da barreira hematoencefálica (BBB) foi possível, mas que a administração de dose múltipla, crônica pode ser necessária para se alcançar níveis protetivos para estas aplicações no CNS (doença de Alzheimer, acidente vascular cerebral, etc.).
Interação do Sal do Derivado de Dissuccinato Dissódico de meso-Astaxantina com Albumina Sérica Bovina (HSA):
A solubilidade aquosa deficiente da maioria dos carotenóides de caroteno e a vasta maioria de xantofilas limita o seu uso como extintores de oxigênio singleto e descontaminantes de radical de fase aquosa. As modificações químicas que aumentam a solubilidade e/ou dispersibilidade evidente dos carotenóides têm encontrado aplicação na ciência básica assim como na pesquisa clínica. Entretanto, a tendência para os precursores de carotenóide e novos derivados para formar montagens supramoleculares em solução aquosa garante a avaliação compreensiva de tal comportamento antes de movimentar em ensaios in vitro e in vivo da eficácia de tais compostos.
A FIG. 5 representa um derivado de carotenóide, o derivado de sal de dissuccinato dissódico (dAST) de meso-astaxantina sintética (3R,3’Sdiidróxi-p,p-caroteno-4,4’-diona), na forma todo-trans (todo-E). A xantofila C4o simétrica usada para gerar o novo derivado tem dois centros quirais nas posições 3 e 3’. Em solução aquosa a xantofila C40 não exibe nenhuma atividade ótica, visto que estes estereocentros têm configurações absolutas opostas e intemamente compensam entre si. As moléculas de carotenóide naturais que possuem funcionalidade carboxílica se ligam preferencialmente à albumina sérica bovina (HSA), a proteína mais abundante no sangue. Visto que a albumina que se liga fortemente influencia as atividades bioquímicas in vivo potenciais de um dado composto, dicroísmo circular (CD), ultravioletavisível (UV/Vis) e espectroscopia de fluorescência foram usados para
134 caracterizar a interação deste novo derivado de carotenóide com HSA isento de ácido graxo. A ligação de proteína e as propriedades de agregação foram investigadas deste carotenóide simétrico ligado através da esterificação direta a uma porção com grupos finais de carboxilato, formando um bolanfífilo 5 dianiônico rígido, de cadeia longa, altamente insaturado. Foi verificado que na solução tampão na ausência da proteína, o moo-carotenóide formou agregados do tipo II intimamente empacotado (card-pack) não exibindo nenhum efeito de Cotton CD (CE). Nas relações molares de ligando/proteína (L/P), entretanto, o meso-carotenóide imediata e preferencialmente associou—10 se com HSA na maneira monomérica, sugerindo que as interações químicas secundárias (forças de van der Waals, ligação de hidrogênio) que permitem a montagem supramolecular em solução aquosa foram superadas em um ambiente biologicamente relevante. Acima da relação molar de 1: 1 de ligando/ proteína as moléculas de meso-carotenóide mais uma vez deram 15 origem ao agregado; a agregação observada nestas relações foi quiral, resultando em uma estrutura supramolecular que mostra atividade CD do tipo exciton intensa.
Descrição Resumida de Métodos Experimentais
Os novo derivado dAST foi sintetizado a partir da astaxantina 20 cristalina [3R,3’R, 3R,3’S, 3S,3’S (25:50:25)], uma mistura estatística de estereioisômeros obtidos comercialmente (Buckton Scott, índia). Os estereoisômeros de astaxantina foram separados pela cromatografia líquida de alta pressão (HPLC), permitindo a síntese do derivado do sal dissuccinato meso-dissódico purificado para testar no estudo corrente. A forma todo-trans 25 (todo-E) do meso estereoisômero usado foi uma molécula linear, rígida devido à falta da(s) configuração(ões) cis (ou Z) na camada de polieno do material espaçador (FIG. 5). O derivado do sal de dissuccinato dissódico de meóO-astaxantina sintética foi sintetizado com sucesso a > 99 % de pureza pela HPLC.
135
Materiais
Albumina sérica bovina essencialmente isenta de ácido graxo (catálogo Nfi A-1887, lote N2 14H9319) foi obtida da Sigma e usada como fornecida. Água duplamente destilada e sulfóxido de dimetila grau espectroscópico (DMSO, Scharlau Chemie S.A., Barcelona, Espanha) e etanol (Quimiolab, Budapest, Hungria) foram usados. Todos os outros produtos químicos foram de grau analítico.
Preparação de solução de estoque de dAST
Depois da dissolução do meso-carotenóide em DMSO, 100 μΐ de solução de DMSO foram adicionados a 2 ml de etanol em uma cubeta retangular com 1 cm de extensão de trajetória. O espectro de absorção foi registrado entre 260 e 650 nm. A concentração foi calculada a partir do valor de absorção de luz no Ámax (ε478 ητη = 116,570 M-^cm-1).
Preparação de soluções HSA
Para a preparação da amostra espectroscópica, HSA foi dissolvido em soluções tampão de Ringer pH 7,4 ou fosfato 0,1 M pH 7,4. A concentração de albumina foi calculada com o valor de £1%ΐαη = 5,31, usando os dados de absorbância experimentalmente obtidos a 279 nm. O peso molecular de HSA foi definido como 66500 Da.
Dicroísmo Circular e Espectroscopia de Absorção UV/Vis
Os espectros de CD e UV foram registrados em um espectropolarímetro Jasco J-715 a 25 ± 0,2 e 37 ± 0,2° C em uma cubeta retangular com extensão de trajetória de 1 cm. O controle de temperatura foi fornecido por um termostato Peltier equipado com agitação magnética. Todos os espectros foram acumulados três vezes com uma largura de faixa de 1,0 nm e uma resolução de 0,5 nm em uma velocidade de varredura de 100 nm/min. A CD induzida foi definida como a CD da mistura dAST-HSA menos a CD de HSA sozinha nos mesmos comprimentos de onda e é expressada como a elipticidade em miligraus (mdeg).
136
Titulação de CD/UV/Vis de HSA com dAST em soluções tampões de Ringer pH 7,4 e fosfato 0,1 M a 37° C
Tampão Ringer. Valores L/P de 0,007 a 0,10: 2 ml de 1,6 x 104 M de solução HSA foram colocados na cubeta com 1 cm extensão de trajetória e quantidades pequenas da solução de estoque de ligando (c = 2,2 x 10-4) foram adicionados com uma pipeta automática em alíquotas de 10 pL. Tampão de Ringer, valores de L/P de 0,82 a 13,13: 2 ml de 2,3 x 10-6 M de solução de HSA foram colocados na cubeta com 1 cm extensão de trajetória e volumes em μΐ da solução de estoque de ligando (c = 3,9 x 10-4) foram adicionados com uma pipeta automática. Tampão de fosfato, valores L/P de 0,82 a 13,10: 2 ml de 2,2 x 10-6 M de solução de HSA foram colocados na cubeta com extensão de trajetória de 1 cm e volumes em μΐ da solução de estoque de ligando (c = 3,6 x 10-1) foram adicionados com uma pipeta automática.
Medição da fluorescência intrínseca de HSA nã presença de dAST ml de 4,2 x 10-6 M de solução de HSA foram preparados em uma célula retangular de 1 cm em 0,1 M pH 7,4 de tampão de fosfato. 1,3 x 10-4 e 3,3 x 10-4 M de soluções em DMSO de meso-carotenóide foram consecutivamente adicionados em volumes de μΐ à cubeta na câmara de amostra do espectropolarímero Jasco J-715. A solução de amostra resultante foi excitada entre 240 e 360 nm em incrementos de comprimento de onda de 0,5 nm. A intensidade de fluorescência total foi coletada a cada comprimento de onda com um detetor de fotomultiplicador Hamamatsu tipo H5784 montado em um ângulo reto em relação à fonte luminosa. Na solução de amostra, as concentrações inicial e final de HSA e dAST foram 4,2 x 10-6 M a 4,0 x 10-6 M e 1,3 x 10-7 M a 1,4 x 10-5 M, respectivamente. A relação molar de mesO-carotenóide/HSA foi variada entre 0,03 e 3,53. Durante as medições da fluorescência, a concentração de DMSO final não excedeu 5 % v/v. Um
137 experimento de controle também foi realizado, em que a fluorescência de
HSA durante a adição de 20, 50 e 100 μΐ de DMSO à solução foi medida.
Debate Resumido dos Resultados da Espectroscopia de
UV/Vis e CD
Propriedades espectrais de UV/Vis e CD de dAST em etanol e solução tampão aquosa
Por causa do seu sistema 7E estendido, dAST exibiu absorção de luz intensa no espectro visível (FIG. 6). A faixa de absorção na forma de sino principal centralizada a 481,5 nm foi devido ao dipolo eletrônico de v^lO energia mais baixa deixado, uma transição π -> π* polarizada ao longo do eixo longo da camada de polieno. Na temperatura ambiente, a falta de estrutura fina é típica para os carotenóides contendo um ou mais grupos carbonila conjugados. Entretanto, as sub-faixas vibracionais estavam de fato presentes abaixo desta curva, como revelado pelo segundo derivado do 15 espectro (FIG. 6). Adicionalmente, na região próxima ao UV, outras transições estavam presentes. De acordo com os cálculos teóricos realizados em modelos de polieno, o momento de transição eletrônica (μ) da faixa moderadamente intensa em tomo de 300 nm é polarizada paralelo ao eixo longo da molécula de dAST. Ao mesmo tempo, a faixa a 371 nm μ é 20 orientado ao longo do eixo de simetria Cz duplicado do sistema conjugado.
As transições η -> π* fracas dos grupos carbonila foram obscurecidas pelas outras faixas. Como esperado, o composto meso-carotenóide não mostrou quaisquer faixas CD em etanol visto que os efeitos dos dois centros quirais opostos (3R,3’S) cancelaram um ao outro (dados não mostrados).
Em solução tampão de Ringer, a faixa de absorção principal de dAST mudou, exibindo uma mudança para azul grande (2541,6 cm-1) assim como estreitamento de largura de faixa (FIG. 7). Estas mudanças espectrais indicaram a formação dos chamados agregados “card-paclF, em que as moléculas foram mantidas juntas em proximidade imediata (dentro de uns
138 poucos ângstrons) tanto pela exclusão do ambiente aquoso quanto pelas interações da ligação de H. Como um resultado, as funções de onda no estado excitado das camadas de polieno foram deslocalizadas inter-molecularmente, permitindo que a interação da ressonância de exciton ocorra entre as 5 moléculas vizinhas. Esta interação resultou em um pico de exciton de alta energia no espectro UV/Vis. Devido às interações estéricas desfavoráveis que surgem entre os grupos finais volumosos, o alinhamento em paralelo das camadas de polieno não é deixada; os eixos longos das moléculas separadas ao invés fecham um ângulo de camada intermolecular definido. Em tais casos, „10 agregados de carotenóide formados por monômeros quirais também exibem efeitos de Cotton induzidos (CE) devido ao arranjo intermolecular quiral determinado pelos centros assimétricos. Ao contrário, o composto de mesocarotenóide não demonstrou nenhuma atividade ótica no estado agregado em solução (dados não mostrados) devido à falta de quiralidade líquida das 15 moléculas.
Propriedades óticas de dAST na presença de albumina sérica bovina nas relações molares de ligando/proteína baixas
Na adição de dAST à solução de HSA preparada em solução de tampão de Ringer no pH 7,4, duas faixas de CD induzidas, opostamente sinalizadas, definidas, apareceram entre 300 e 450 nm com um ponto de interseção zero a 367 nm (FIG. 8). Os insertos da figura mostram as intensidades dos efeitos de Cotton induzidos e a faixa de absorção principal em relações L/P diferentes (os valores Δε e ε são calculados com respeito à concentração de masO-carotenóide total). As magnitudes do CEs aumentaram com a concentração crescente do ligando, entretanto, a sua forma e posições de comprimento de onda permanecera, inalteradas. Como mencionado acima, existem duas transições abaixo de 450 nm que poderíam ser responsáveis pela atividade ótica observada. A faixa de absorção em tomo de 300 nm tem simetria de transição B e os momentos de transição elétrica e magnética
139 correspondentes são perpendiculares ao eixo de simetria duplo ao longo da camada de polieno. Os momentos de transição elétrica e magnética da faixa a 372,5 nm são polarizados paralelos ao eixo C2, a sua simetria de transição é A. É razoável assumir que na ligação da proteína, estas mudanças de faixa 5 para comprimentos de onda mais longos devido à mudança do microambientes que circunda a camada de polieno. Foi bem estabelecido que os espectros CD de carotenóides nos quais as porções cromofóricas pertencem ao grupo de ponto C2 conformam-se à regra C2: se o sistema conjugado gobal adquire quiralidade da esquerda para a direita (isto é ângulos diédricos em 10 tomo das ligações 6-7 e 6’-7’ são negativos), então as transições de simetria A levam a CE negativa e as transições de simetria B levam a CE positiva (FIG.
8). Portanto, o meso-carotenóide se liga a HSA em uma maneira tal que o ambiente da proteína fixa os anéis terminais em uma conformação quiral bem definida que resulta nas faixas de CD induzidas negativa e positiva 15 observadas. As configurações absolutas dos centros 3 e 3’ quirais não determinam a propriedade quiróptica da molécula; ao invés, o ambiente da proteína assimétrica da molécula de albumina (por intermédio das interações químicas não covalentes) determina a atividade observada. Ao contrário do comportamento do agregado nas soluções aquosas descritas acima, as 20 moléculas BAST na agregam na solução de HSA nestas relações L/P, como demonstrado pela retenção da faixa de absorção visível na forma de sino e ligeiramente mudada para vermelho (FIG. 8). Assim, tanto a absorção de UV/Vis quanto os espectros CD indicam que a ligação das moléculas de meso-carotenóide à HSA ocorre na forma monomérica.
Propriedades óticas de dAST na presença de HSA acima das relações de L/P de 1:1
Uma quantidade crescente de dAST foi adicionada às soluções de HSA preparadas com tampão de Ringer pH 7,4 ou tampão de fosfato 0,1 M pH 7,4 para se obter relações L/P maiores do que 1. Tanto o espectro de
140 absorção de CD quanto de UV/Vis exibiram mudanças profundas durante a adição do ligando (FIG. 9 e FIG. 10). Além da faixa de absorção visível mudada para azul um novo par de faixas CD positiva-negativa apareceu em tomo de 480 e 420 nm, respectivamente. Estas CE’s não exibiram nenhuma 5 estrutura vibracional fina e suas amplitudes cresceram com a concentração crescente do ligando. Entretanto, houveram algumas diferenças notáveis entre o espectro obtido nas soluções de tampão de Ringer e de tampão de fosfato:
*· a) A faixa de absorção principal mudou para um comprimento de onda mais baixo (434,5 nm) no tampão de Ringer. O valor correspondente 10 foi 451,5 nm no tampão de fosfato.
b) O desvio do ponto de interseção zero de CEs do máximo da faixa de absorção foi três vezes maior na solução de tampão de Ringer (441,6 cm-1) do que na solução de tampão de fosfato (148,4 cm-1).
c) Acima de um valor de L/P de 8, as intensidades das faixas
CD não mais aumentam na solução de Ringer. Ao contrário, a(s) amplitude(s) das faixas CD continuaram a aumentar com a relação L/P crescente no tampão de fosfato, mesmo em um valor L/P de 13.
d) Na mesma relação L/P, faixas CD mais intensas foram medidas no tampão de fosfato (FIG. 9 e FIG. 10). O fato de que estas faixas
CD opostamente sinalizadas aparecerem apenas acima da relação L/P de 1: 1 sugere fortemente que elas se originaram de interações intermoleculares quirais entre meso-moléculas de carotenóide adjacentes. Quando dois momentos de dipolo de transição elétrica são similares em energia, encontram-se próximos um ao outro no espaço e formam uma série quiral, a 25 sua interação é manifestada como ligação de exciton quiral: o espectro CD mostra uma parelha bissinalizada emparelhada com a posição espectral da faixa de absorção correspondente, cujo sinal é determinado pelo sentido absoluto de entrelaçamento entre os dois dipolos. De acordo com a regra de quiralidade de exciton, um entrelaçamento positivo corresponde a um
141 comprimento de onda CE longo positivo e um CE negativo no comprimento de onda mais curto e vice versa. No nosso caso, a direção do momento de dipolo de transição é conhecido; ele é polarizado ao longo do eixo longo da camada de polieno. Assim, as moléculas de meso-carotenóide vizinhas são 5 dispostas em uma maneira tal que os seus eixos longos formem um ângulo de camada intermolecular positiva (horário). Os arranjos quirais de duas cadeias conjugadas mostradas na FIG. 11 satisfazem a última condição; neste caso, uma faixa positiva de comprimento de onda longo e uma negativa de comprimento de onda curto podem aparecer no espectro CD. Entretanto, o v, 10 comportamento espectroscópico da faixa de absorção ajuda a diferenciar entre estes arranjos espaciais. Devido às interações Coulômbicas desfavoráveis entre os momentos de dipolo de transição de moléculas de meso-carotenóide vizinhas no caso de a e b (FIG. 11), a máxima absorção muda para energias mais altas; se a forma c existe, então a faixa de absorção amplia e o seu 15 máximo muda para energias mais baixas. Consequentemente, as moléculas dAST formam um arranjo quiral da esquerda para a direita caso este em que os eixos longos de monômeros de meso-carotenóide formam um ângulo agudo, positivo (FIG. 11, a e b).
O seguinte cenário é proposto para a origem da disposição 20 quiral das moléculas de ligando. A albumina parece necessária para a atividade ótica induzida e, em primeiro lugar, é tentador assumir que existe um sítio de ligação grande no HSA capaz de acomodar duas moléculas de meso-carotenóide. Nos valores de L/P baixos a albumina pode ligar-se apenas em um único ligando; nas concentrações L/P mais altas, um segundo 25 monômero meso-carotenóide pode ser complexado. Como estabelecido acima, entretanto, as magnitudes de CEs continuam a aumentar para valores de L/T bastante altos (FIG. 10), caso este em que um único sítio de ligação já deve estar saturado. Uma resolução para este problema assume que HSA tenha um padrão assimétrico no qual a auto-montagem quiral é começada. As
142
primeiras poucas moléculas de meso-carotenóide se ligam a HSA no arranjo da esquerda para a direita e monômeros de meso-carotenóide subsequentes se formam nesta arquitetura quiral. Neste cenário, HSA fornece a primeira etapa essencial, a iniciação quiral (“semeadura quiral”); depois disto a automontagem continua automaticamente. É muito importante observar, entretanto, que sem seus grupos finais quirais apenas umas poucas moléculas dAST podem ser mantidas no arranjo da esquerda para a direita no sítio de ligação de HSA. Os centros quirais 3 e 3’ desempenham um papel decisivo em permitir que os agregados formem a auto-montagem quiral nas moléculas de HSA. Na ausência da proteína, as moléculas de meso-carotenóide forma montagens à esquerda e à direita em um grau equivalente, devido à falta de discriminação quiral.
Como listado acima, as diferenças espectrais entre as curvas CD medidas nas soluções de tampão de fosfato e Ringer sugeriram a influência da concentração do sal na estabilidade dos agregados (FIG. 9 e FIG. 10). A osmolaridade e a concentração iônica do tampão de Ringer foi mais alta do que a do tampão de fosfato. As porções succínicas foram ionizadas no pH 7,4 em ambas as soluções tampão e a repulsão eletrostática surgiu tanto dentro quanto entre os agregados. Os íons de sal positivamente carregados são capazes de diminuir a sua repulsão e portanto contribuem para uma estabilidade e tamanho crescentes dos agregados na presença destes cátions. Durante a titulação de HSA com dAST acima da relação L/P de 1: 1, tanto os agregados quirais quanto os aquirais foram simultaneamente formados; entretanto, apenas os agregados quirais foram associados com HSA, enquanto os agregados aquirais não o foram. Os espectros de CD obtidos na solução tampão de Ringer (FIG. 9) sugeriram que os agregados aquirais foram melhor estabilizados neste tampão de osmolaridade mais alta devido ao efeito de varredura dos íons salinos. As moléculas de ligando adicionadas preferencialmente associadas com agregados existentes, que
143 ífc?
resultaram nas amplitudes das faixas de CD atingiram um platô e tomou-se constante em contraste com o tampão de fosfato.
Extinção da fluorescência de HSA na adição de BAST
O único resíduo de triptofano (Trp214) localizado na altura do subdomínio IIA é amplamente responsável pela fluorescência intrínseca de HSA. O espectro de emissão de fluorescência de HSA se sobrepõe com o espectro de absorção do meso-carotenóide. Portanto, as medições espectroscópicas de fluorescência foram obtidas depois da adição incrementai de dAST em DMSO a uma solução de HSA. Os resultados claramente 10 demonstraram que as moléculas de meso-carotenóide foram capazes de extinguir eficazmente a fluorescência intrínseca de HSA (FIG. 12). O DMSO usado para preparar a solução de estoque de dAST exibiu um efeito na fluorescência HSA intrínseca (FIG. 12). Em uma relação L/P de 0,7, a intensidade de fluorescência da base de referência diminuiu em 50 %. O 15 fenômeno observado sugeriu que uma molécula de zweso-carotenóide foi ligada na vicinidade de Trp214, que forma parte da parede em uma das duas cavidades de ligação principal de HSA (sítio I, subdomínio IIA; FIG. 13). Entretanto, nem o sítio I nem o sítio II (subdomínio ΠΙΑ) - ambos túneis de ligação de ácido graxo hidrofóbicos - são capazes de acomodar a molécula de 20 dAST longa, rígida (FIG. 13). Com base na similaridade estrutural, uma segunda possibilidade é que a dAST se ligue a outros sítios de ligação de ácido graxo (C18, C20) de cadeia longa de HSA, que foram bem caracterizados pela cristalografia de raio x de alta resolução. No caso de carotenóides de cadeia aberta mais curta, não tendo nenhum grupo final 25 volumoso, esta possibilidade pode ser provável. Entretanto, a camada de polieno do derivado de meso-carotenóide por si só mede 28 Â (entre os átomos de carbono quirais 3 e 3’). A despeito da sua mobilidade conformacional, as porções de succinato requerem espaço adicional, que aumentam o comprimento eficaz da molécula para 48 Â. A inspeção
144
cuidadosa da estrutura cristalina de HSA sugere que a fenda longa, estreita entre os domínios I e III podem ser adequados para a ligação de uma molécula de meso-carotenóide (FIG. 13). A fenda de interdomínio é ampla e a sua extremidade estreita é fechada ao resíduo de triptofano (Trp214; * na FIG. 13) que pode fornecer uma explicação estrutural para a extinção da fluorescência observada na ligação da molécula de meso-carotenóide à fenda de interdomínio de HSA. Além disso, pode ser assumido que a associação das moléculas dAST adicionais ao único na fenda de interdomínio induz mudanças conformacionais significantes de HSA resultando na ampliação da fenda central. Esta seria a razão do porque a extinção da fluorescência não para em uma relação de L/P = 1 mas mantém-se forte conforme os CEs aumentam (FIG. 13).
Debate dos Resultados da Espectroscopia UV/Vis e CD
Como uma consequência da exclusão do ambiente aquoso e das ligações de hidrogênio intermoleculares, o derivado do sal de dissuccinato dissódico da meso-astaxantina aquiral, sintético formou agregados do tipo card-pack, oticamente inativos nas soluções de tampão aquoso, como indicado pela mudança para azul grande das faixas de absorção visíveis principais versus a faixa observada na solução etanólica. Na presença de um excesso de ácido graxo isento de HSA, o maso-carotenóide parece estar preferencialmente associado com HSA na maneira monomérica. Estes resultados sugerem que as forças de van der Waal’s e ligações de hidrogênio fracas que permitem a montagem supramolecular em solução aquosa serão rapidamente superadas em um ambiente biologicamente relevante. A concentração de albumina no sangue humano in vivo é de aproximadamente 0,6 mM, sugerindo que nas doses de até 500 mg, o meso-carotenóide (peso molecular 841 Da) associar-se-á com a albumina na maneira monomérica (excluindo a ligação não específica potencial adicional às células e lipoproteínas sanguíneas circulantes, que podem aumentar a dose não
145 agregante potencial). A ligação das moléculas de meso-carotenóide exibiu faixas de CD induzidas que foram adequadamente explicadas por uma conformação helicoidal da esquerda para a direita do sistema conjugado. A extinção gradual da fluorescência de HSA na presença de concentrações 5 crescentes de dAST reforçou a noção de que a formação de complexos de carotenóide-albumina foi responsável por esta extinção e sugeriu a proximidade espacial entre o ligando ligado e o resíduo de triptofano 214 de HSA. Com base nos dados espectroscópicos, o comprimento molecular da molécula de dAST e a estrutura cristalina nem caracterizada de HSA, o sítio 10 de ligação foi como tentativa atribuído à fenda de interdomínio localizada entre os domínios I e III.
Parece haver um par de faixas positivo-negativo no espectro de CD acima da relação L/P de 1: 1 de meso-carotenóide para HSA. Esta descoberta foi atribuída à ligação do exciton quiral intermolacular entre as 15 camadas de polieno do masO-carotenóide disposta na montagem da esquerda para a direita. Os dados experimentais sugeriram que HSA atua como um padrão quiral no qual a auto-montagem começa e subsequentemente continua controlada pela quiralidade dos grupos finais das moléculas de mesocarotenóide. As diferenças entre os espectros de CD bissinalizados obtidos 20 nas soluções de tampão de fosfato e de Ringer no pH 7,4 indicam que a automontagem é influenciada pela osmolaridade e concentração iônica da solução. Com osmolaridade crescente, a estabilidade dos agregados é realçada presumivelmente devido à triagem eletrostática das funções de carboxilato succínico negativamente carregadas pelos cátions salinos. Outras 25 modificações e formas de realização alternativas de vários aspectos da invenção estarão evidentes àqueles habilitados na técnica em vista desta descrição. Consequentemente, esta descrição deve ser interpretada apenas como ilustrativa e é para o propósito de divulgar àqueles habilitados na técnica a maneira geral de realizar a invenção. Deve ser entendido que as
146 formas da invenção mostradas e aqui descritas devem consideradas como as formas de realização presentemente preferidas. Os elementos e materiais podem ser substituídos por aqueles ilustrados e aqui descritos, partes e processos podem ser invertidos e certas características da invenção podem ser 5 independentemente utilizadas, todos como estaria evidente a uma pessoa habilitada na técnica depois de ter o benefício desta descrição da invenção. Mudanças podem ser feitas nos elementos aqui descritos sem divergir do espírito e escopo da invenção como descrito nas seguintes reivindicações.