WO2014136448A1 - ブロック共重合体およびその製造方法 - Google Patents
ブロック共重合体およびその製造方法 Download PDFInfo
- Publication number
- WO2014136448A1 WO2014136448A1 PCT/JP2014/001207 JP2014001207W WO2014136448A1 WO 2014136448 A1 WO2014136448 A1 WO 2014136448A1 JP 2014001207 W JP2014001207 W JP 2014001207W WO 2014136448 A1 WO2014136448 A1 WO 2014136448A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cyclic
- block copolymer
- polyphenylene sulfide
- molecular weight
- polyphenylene
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G81/00—Macromolecular compounds obtained by interreacting polymers in the absence of monomers, e.g. block polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/02—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids
- C08G69/08—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from amino-carboxylic acids
- C08G69/14—Lactams
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/42—Polyamides containing atoms other than carbon, hydrogen, oxygen, and nitrogen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/48—Polymers modified by chemical after-treatment
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G75/00—Macromolecular compounds obtained by reactions forming a linkage containing sulfur with or without nitrogen, oxygen, or carbon in the main chain of the macromolecule
- C08G75/14—Polysulfides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L71/00—Compositions of polyethers obtained by reactions forming an ether link in the main chain; Compositions of derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/10—Definition of the polymer structure
- C08G2261/12—Copolymers
- C08G2261/126—Copolymers block
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2650/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G2650/28—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule characterised by the polymer type
- C08G2650/38—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule characterised by the polymer type containing oxygen in addition to the ether group
- C08G2650/40—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule characterised by the polymer type containing oxygen in addition to the ether group containing ketone groups, e.g. polyarylethylketones, PEEK or PEK
Definitions
- the present application includes Japanese Patent Application No. 2013-040404 and Japanese Patent Application No. 2013-044005 filed on March 6, 2013, and Japanese Patent Application No. 2013-2003 filed on September 30, 2013.
- the priority based on No. 204070 is claimed, and all the contents described in these Japanese applications are incorporated.
- the present invention relates to a block copolymer. More specifically, the present invention relates to a polyarylene sulfide copolymer using cyclic polyarylene sulfide as a raw material and a method for producing the same.
- Polyarylene sulfide (hereinafter sometimes abbreviated as PAS) is a resin having properties suitable as engineering plastics such as excellent heat resistance, barrier properties, chemical resistance, electrical insulation properties, moist heat resistance, and flame resistance.
- PAS polyarylene sulfide
- PPS polyphenylene sulfide
- PPS resin can be molded into various molded parts, films, sheets, fibers, etc. by injection molding and extrusion molding, and various electric / electronic parts, mechanical parts and automobiles. Widely used in fields that require heat resistance and chemical resistance, such as parts.
- PPS resins are inferior in terms of impact resistance, toughness and molding processability compared to engineering plastics such as nylon, polycarbonate, and polybutylene terephthalate. Attempts have been made to combine these.
- As a typical compounding method there are a method of blending PAS and another different polymer to make a polymer alloy, a method of chemically bonding PAS and a different polymer, and making a block copolymer.
- block copolymerization can form a homogeneous and fine phase separation structure as compared with other composite methods, and thus various studies have been made as a PPS modification method.
- Patent Document 1 discloses a PPS copolymer containing a polysulfone component block, and this copolymer has improved bending strength and impact resistance as compared with a blend mixture.
- this copolymer is produced by a technique of first synthesizing a polysulfone having chloro groups at both ends and then performing polycondensation of PPS in the presence of polysulfone having a chloro end.
- this method has a problem that homopolymer formation / mixing and uneven block composition distribution cannot be avoided, and problems still remain to obtain a homogeneous copolymer.
- Examples of other copolymer components with PPS include polyphenylene sulfide ketone (Patent Documents 2 and 3), polyethersulfone (see, for example, Patent Document 4 and Non-Patent Document 1), polysulfide sulfone (see, for example, Patent Document 5), and polyphenylene.
- Ether ether ketone see, for example, Patent Documents 6 and 7
- polyetherimide see, for example, Patent Document 8
- liquid crystal polyester see, for example, Patent Document 9 and Non-Patent Document 2
- polyester see, for example, Patent Documents 10 and 11). It is disclosed.
- Any copolymer having these copolymer components is a method of copolymerizing different types of polymers by polycondensation in the presence of PPS having a terminal functional group or a homopolymer of a copolymer component other than PPS, Alternatively, it is produced by a method in which polymer ends are linked and copolymerized. Therefore, in principle, problems such as generation / mixing of homopolymers and uneven block composition remain.
- Patent Document 12 As a copolymer containing a PPS component block, a copolymer with a polyacrylate ester is disclosed in Patent Document 12. According to the disclosed literature, heating PPS partially having a disulfide bond and an acrylate ester in the presence of a radical initiator causes a radical chain transfer reaction to the disulfide bond. A copolymer with an acid ester is obtained. On the other hand, problems related to homopolymer mixing and block composition have not been solved, and purification operations are required to remove homopolymers, and there are many problems in industrial applications such as poor stability of disulfide bonds. Was the current situation.
- the PPS component block having a narrower molecular weight distribution than that produced by the conventional method, or a copolymer other than the PPS component block It is necessary to obtain an ingredient block.
- a method for producing PPS having a narrow molecular weight distribution a method in which a cyclic arylene sulfide oligomer is used as a raw material and heat polymerization is carried out in the presence or absence of a catalyst or an initiator is disclosed (see, for example, Patent Documents 13 to 15).
- Japanese Patent Laid-Open No. 61-225218 Japanese Patent Laid-Open No. 2-133428 JP-A-2-229857 JP 2004-168834 A JP-A-2-235929 JP-A-2-228325 JP 2004-168834 A Japanese Unexamined Patent Publication No. 64-045333 JP-A-11-222527 JP-A-4-31725 JP-A-5-295346 JP-T-4-505182 JP-A-5-163349 Japanese Patent Laid-Open No. 5-301962 International Publication No. 2007/034800
- the inventors of the present invention have arrived at the present invention as a result of intensive studies for solving the above problems. That is, the present invention has been made to solve at least a part of the problems described above, and can be realized as the following forms.
- a block copolymer comprising a polyarylene sulfide component block made from cyclic polyarylene sulfide as a block constituting the block copolymer, and the remaining blocks constituting the block copolymer are A block copolymer using a cyclic compound as a raw material.
- cyclic polyarylene sulfide represented by the following general formula (i):
- polyphenylene sulfide component block represented by the general formula (ii) and 95 to 5% by weight of at least one polymer component block selected from polyamide, polyester, polycarbonate, polysulfone and polyphenylene ether ether ketone %, The block copolymer according to (3) above.
- the maximum peak molecular weight measured by size exclusion chromatography is in the range of 2,000 or more and less than 2,000,000 and has a unimodal molecular weight distribution in the above range.
- the block copolymer according to any one of (6).
- a method for producing a block copolymer (A) ring-opening polymerization of cyclic polyphenylene sulfide in the presence of an initiator; (B) ring-opening polymerization of a cyclic compound different from cyclic polyarylene sulfide in the presence of an initiator; With A block copolymer in which the product obtained in one of the steps (a) and (b) is mixed with a cyclic compound which is a raw material in the other step, and the other step is performed. Manufacturing method.
- the cyclic compound used in the step (b) is any one kind of cyclic compound selected from cyclic amide, cyclic ester, cyclic polycarbonate, cyclic polysulfone, and cyclic polyphenylene ether ether ketone.
- a method for producing a block copolymer is any one kind of cyclic compound selected from cyclic amide, cyclic ester, cyclic polycarbonate, cyclic polysulfone, and cyclic polyphenylene ether ether ketone.
- the said one process is a manufacturing method of the block copolymer as described in said (8) or (9) which is the said (a) process.
- a block copolymer containing a homogeneous polyarylene sulfide component block having no unimodal molecular weight distribution and no homopolymer can be obtained.
- the block copolymer of the embodiment of the present invention includes at least two polymer component blocks, at least one of which is a polyarylene sulfide component block made from cyclic polyarylene sulfide.
- the “polyarylene sulfide component block” constituting the copolymer of the embodiment of the present invention is a component block constituting the block copolymer, and the repeating unit constituting this component block is substantially a cyclic polyarylene.
- the number of repeats of the polyarylene sulfide component block is not particularly defined, but can be exemplified by 1 or more, for example, preferably 2 or more, and more preferably 5 or more. The number of repetitions can be exemplified by 10,000 or less, preferably 8,000 or less, and more preferably 5,000 or less.
- the number of repetitions can be adjusted by the molar ratio of the cyclic polyarylene sulfide as a raw material to the polymerization initiator and / or the catalyst.
- This number of repeats is the number average of polystyrene-reduced polyarylene sulfide component blocks of known absolute molecular weight obtained by size exclusion chromatography (SEC) measurement using a solvent capable of dissolving polyarylene sulfide as an eluent.
- the molecular weight can be obtained by dividing the molecular weight by the repeating unit molecular weight of the polyarylene sulfide component block.
- the content of the polyarylene sulfide component block contained in the copolymer of the embodiment of the present invention is preferably 5% by weight or more, more preferably 10% by weight or more based on the whole copolymer. . Moreover, it is preferable that it is 95 weight% or less, and 90 weight% or less is further more preferable. Within this range, a uniform copolymer is easily obtained, and properties derived from the polyarylene sulfide component block such as high chemical resistance are easily developed.
- component block other than the polyarylene sulfide component block is a component block constituting the block copolymer, and the repeating unit constituting the component block is other than the cyclic polyarylene sulfide.
- the component block which is the structure which opened the cyclic compound of this is meant.
- the polyarylene sulfide component block and the other component block may be linked via a structure other than the repeating unit of each block.
- the terminal structures derived from the repeating unit of each block may be directly connected to each other.
- a plurality of blocks having the same repeating unit may be present in the same copolymer molecule.
- the cyclic polyarylene sulfide used as a raw material for the polyarylene sulfide component block constituting the copolymer in the embodiment of the present invention is mainly composed of a repeating unit of the formula, — (Ar—S) — (where Ar is an arylene group).
- the cyclic compound preferably contains 80 mol% or more of the repeating unit.
- Ar includes units represented by the following formulas (A) to (K), among which the formula (A) is particularly preferable.
- R1 and R2 are substituents selected from hydrogen, an alkyl group having 1 to 12 carbon atoms, an alkoxy group having 1 to 12 carbon atoms, an arylene group having 6 to 24 carbon atoms, and a halogen group; And R2 may be the same or different)
- this repeating unit is a main constituent unit, it can contain a small amount of branching units or crosslinking units represented by the following formulas (L) to (N).
- the copolymerization amount of these branched units or cross-linked units is preferably in the range of 0 to 1 mol% with respect to 1 mol of — (Ar—S) — units.
- cyclic polyarylene sulfides include cyclic polyphenylene sulfide (hereinafter sometimes abbreviated as cyclic PPS) containing 80 mol% or more, preferably 90 mol% or more of p-phenylene sulfide units as main structural units.
- the cyclic polyphenylene sulfide referred to here is a monomer or mixture of a cyclic compound such as the following general formula (v).
- the cyclic PPS contains at least 50% by weight of the cyclic compound of the formula (v), preferably 70% by weight or more, more preferably 80% by weight or more, and even more preferably 90% by weight or more. .
- m is preferably 4 or more, more preferably 8 or more. Since cyclic compounds having m of 7 or less tend to have low reactivity, it is advantageous to set m to 8 or more from the viewpoint that a polyphenylene sulfide component block can be obtained in a short time.
- the number of repetitions m is preferably 50 or less, more preferably 25 or less, and even more preferably 15 or less.
- the conversion of the cyclic PPS to the polyphenylene sulfide component block by heating is preferably performed at a temperature equal to or higher than the temperature at which the cyclic PPS melts.
- m is set in the above range from the viewpoint that conversion of cyclic PPS to a polyphenylene sulfide component block can be performed at a lower temperature. This is advantageous.
- the cyclic compound of the formula (v) contained in the cyclic PPS may be either a single compound having a single repeating number or a mixture of cyclic compounds having different repeating numbers.
- a mixture of cyclic compounds having different numbers of repetitions tends to have a lower melting temperature than a single compound having a single number of repetitions. Therefore, the use of a mixture of cyclic compounds having different numbers of repetitions is preferable because the temperature at the time of conversion to a polyphenylene sulfide component block can be further reduced.
- the component other than the cyclic compound of the formula (v) in the cyclic PPS is particularly preferably a polyphenylene sulfide oligomer.
- the polyphenylene sulfide oligomer is an oligomer having a repeating unit of the formula — (Ph—S) — as a main constituent unit (where Ph is a phenylene group), and preferably contains 80 mol% or more of the repeating unit. It is a linear homo-oligomer or co-oligomer.
- the polyphenylene sulfide oligomer can contain a small amount of branching or crosslinking units.
- the copolymerization amount of these branched units or cross-linked units is preferably in the range of 0 to 1 mol% with respect to 1 mol of — (Ph—S) — units.
- the polyphenylene sulfide oligomer may be any of a random copolymer, a block copolymer and a mixture thereof containing the above repeating unit. Although there is no restriction
- the upper limit of the molecular weight of the cyclic PPS used for the production of the polyphenylene sulfide component block of the embodiment of the present invention is preferably 10,000 or less, more preferably 5,000 or less, and further preferably 3,000 or less in terms of weight average molecular weight.
- the lower limit is preferably 300 or more, preferably 400 or more, and more preferably 500 or more in terms of weight average molecular weight.
- Cyclic PPS having a weight average molecular weight within the above range tends to be easily converted to a polyarylene sulfide component block from the viewpoint of melt viscosity and ring composition.
- Such a cyclic polyphenylene sulfide can be produced by, for example, a technique disclosed in International Publication No. 2008/105438.
- the cyclic compound used as a raw material for the block component other than the polyphenylene sulfide component block may be a cyclic polymer or oligomer having a repeating unit, or a low molecular cyclic compound such as lactam or lactone.
- cyclic polymer cyclic polyamide, polyester, polyacetal, polycarbonate, polyphenylene ether, polyarylate, polysulfone, polyketone, polyimide, polyamideimide, polyetherimide, polyurethane, polyurea, polyphenylene ether ether ketone and derivatives thereof are preferable. Used.
- lactam, lactone, cyclic polyamide, polyester, polycarbonate, polysulfone, and polyphenylene ether ether ketone are more preferable.
- the molecular weight of the cyclic polymer or oligomer that can be used is not particularly limited, the number average molecular weight is preferably less than 100,000, more preferably less than 50,000, more preferably from the viewpoint of the melt viscosity of the cyclic product itself or the resulting copolymer. 1,000 or less is particularly preferable.
- the cyclic amide used in the embodiment of the present invention is not particularly limited as long as it is a cyclic compound having an amide bond as a bond constituting the ring.
- lactams such as ⁇ -caprolactam, ⁇ -heptalactam, ⁇ -octalactam, ⁇ -undecalactam and ⁇ -laurolactam
- cyclic polyhexamethylene adipamide cyclic polypentamethylene adipamide
- cyclic polyhexamethylene examples include bacamide, cyclic polyhexamethylene dodecamide, cyclic polyhexamethylene terephthalamide, cyclic polyhexamethylene isophthalamide, cyclic polyxylylene adipamide, cyclic copolymers thereof, and mixtures thereof.
- These cyclic amides may have a substituent.
- ⁇ -caprolactam, ⁇ -undecalactam, and ⁇ -laurolactam are preferable.
- the cyclic ester is not limited as long as it is a cyclic compound having an ester bond as a bond constituting the ring.
- cyclic esters may have a substituent.
- cyclic polyalkylene terephthalates such as ⁇ -caprolactone, ⁇ -butyrolactone, lactide, glycolide, cyclic polyethylene terephthalate, cyclic polypropylene terephthalate, and cyclic polybutylene terephthalate are preferably used.
- Such a cyclic polyalkylene terephthalate can be produced, for example, by the method disclosed in Japanese Patent Application Laid-Open No. 2003-82081.
- a cyclic polybutylene terephthalate is commercially available as CBT (registered trademark) manufactured by Cyclics. Is possible.
- Cyclic polycarbonate is a cyclic compound having a carbonate bond as a bond constituting the ring.
- the compounds used in the embodiment of the present invention include aliphatic cyclic carbonates such as ethylene carbonate and propylene carbonate and derivatives thereof, 2,2-bis (4-hydroxyphenyl) propane (bisphenol A), bis (4- Hydroxyphenyl) methane, 1,1-bis (4-hydroxyphenyl) ethane, 1,1-bis (4-hydroxyphenyl) cyclohexane, 2,2-bis (4-hydroxy-3,5-dimethylphenyl) propane, 2,2-bis (4-hydroxy-3,5-dibromophenyl) propane, 2,2-bis (hydroxy-3-methylphenyl) propane, bis (4-hydroxyphenyl) sulfide, bis (4-hydroxyphenyl) Sulfone, hydroquinone, resorcinol, 4,6-dimethyl- , 4,6-tri (4-hydroxyphenyl) heptene, 2,4,6
- a cyclic polycarbonate derived from 2,2-bis (4-hydroxyphenyl) propane (bisphenol A) is particularly preferably used.
- a cyclic polycarbonate can be produced, for example, by the method described in Macromolecules, 24, 3035-3044 (1991).
- the cyclic polysulfone preferably used in the embodiment of the present invention is not limited as long as it is a cyclic compound having a sulfonyl bond as a bond constituting the ring, but is preferably a cyclic polysulfone having a diphenylsulfone structure as a structural unit.
- a cyclic polysulfone compound in which diphenyl sulfone structures are connected by ether bonds and a cyclic polysulfone compound in which diphenyl sulfone structures and divalent aromatic dioxy structures are alternately connected are preferably used.
- the divalent aromatic dioxy structure includes hydroquinone, catechol, resorcin, 4,4′-biphenol, 2,2-bis (4-hydroxyphenyl) propane, and 2,2-bis (4-hydroxyphenyl) methane.
- Bis (4-hydroxyphenyl) alkanes such as 2,2-bis (4-hydroxyphenyl) ethane, dihydroxyphenyl ethers such as 4,4′-dihydroxydiphenyl ether, and structures derived from these structural isomers
- structures derived from shea ether and most preferably, a structure derived from 2,2-bis (4-hydroxyphenyl propane) (bisphenol -A).
- the cyclic polyphenylene ether ether ketone preferably used in the embodiment of the present invention is a cyclic compound represented by the above general formula (vi) having paraphenylene ketone and paraphenylene ether as repeating structural units.
- the number of repetitions n in the general formula (vi) is preferably 2 or more.
- the number of repetitions n is preferably 40 or less, more preferably 20 or less, and still more preferably 15 or less.
- the conversion of the cyclic polyphenylene ether ether ketone composition to the polyphenylene ether ether ketone component block is preferably performed at a temperature equal to or higher than the temperature at which the cyclic polyphenylene ether ether ketone composition melts.
- the melting point of the cyclic polyphenylene ether ether ketone composition tends to increase as the number of repetitions n increases, the cyclic polyphenylene ether ether ketone is melted at a low temperature and converted to a polyphenylene ether ether ketone component block at a lower temperature.
- the cyclic polyphenylene ether ether ketone represented by the general formula (vi) may be either a single compound having a single repeating number or a mixture of cyclic compounds having different repeating numbers.
- a cyclic compound having a different number of repetitions tends to have a lower melting point than a single compound having a single number of repetitions, and the use of a mixture of cyclic compounds having different numbers of different repetitions leads to a polyphenylene ether ether ketone component block. This is preferable because the temperature during the conversion of can be lowered.
- Cyclic polyphenylene ether ether ketone having each repeating number n can be analyzed by component separation by high performance liquid chromatography.
- composition of the cyclic polyphenylene ether ether ketone that is, the weight fraction of the cyclic polyphenylene ether ether ketone having each repeating number n contained in the cyclic polyphenylene ether ether ketone is the peak area of each cyclic polyphenylene ether ether ketone in the high performance liquid chromatography. It is possible to calculate from the ratio.
- linear polyphenylene ether ether ketone can be mainly cited.
- the cyclic polyphenylene ether ether ketone composition is a composition containing 60% by weight or more of cyclic polyphenylene ether ether ketone, more preferably 65% by weight or more, and more preferably 70% by weight or more of cyclic polyphenylene ether ether ketone. More preferably, it is more preferably 75% by weight or more.
- a polyphenylene ether ether ketone component block can be easily obtained in a short processing time.
- the weight fraction of the cyclic polyphenylene ether ether ketone contained in the cyclic polyphenylene ether ether ketone composition can be calculated from the peak area ratio of each cyclic polyphenylene ether ether ketone in high performance liquid chromatography.
- Such a cyclic polyphenylene ether ether ketone can be produced by, for example, a technique disclosed in International Publication No. 2011/081080.
- the copolymer according to the embodiment of the present invention preferably has a maximum peak molecular weight measured by size exclusion chromatography (SEC) of 2,000 or more, more preferably 3,000 or more, It is particularly preferably 5,000 or more.
- the maximum peak molecular weight is preferably less than 2,000,000, more preferably less than 1,000,000, and particularly preferably less than 500,000.
- the maximum peak molecular weight is in the above range and has a unimodal molecular weight distribution in the above range. If the maximum peak molecular weight is within the above range, the physical properties and molding processability of the resulting copolymer will be good.
- the maximum peak molecular weight here means the molecular weight corresponding to the maximum value of the chromatogram obtained by measurement using a SEC device equipped with a differential refractive index detector, the horizontal axis represents the retention time, and the vertical axis represents the substrate concentration.
- the retention time at which the substrate concentration takes the maximum value is obtained, and this value is retained with the molecular weight obtained by measurement of a standard substance having a known absolute molecular weight (polystyrene is used in this embodiment). It can be obtained by substituting into the relational expression with time.
- the measurement temperature of SEC can be exemplified by the range of 50 to 250 ° C., and may be different for each process constituting the SEC apparatus such as a column and a detector.
- the column temperature is 210 ° C.
- the pre-temperature bath temperature is 250 ° C.
- the pump temperature chamber temperature is 50 ° C.
- the detector temperature is 210 ° C.
- the molecular weight distribution that is, the value obtained by dividing the weight average molecular weight by the number average molecular weight can be measured by SEC in the same manner as the maximum peak molecular weight.
- the molecular weight distribution value is preferably 1.1 or more.
- the molecular weight distribution value is preferably 10.0 or less, more preferably 8.0 or less, and particularly preferably 5.0 or less.
- the copolymer of the embodiment of the present invention preferably exhibits a unimodal molecular weight distribution in a molecular weight range of 2,000 or more and less than 2,000,000.
- unimodal means that the chromatogram obtained in the SEC measurement has a single positive maximum value.
- the molecular weight of the copolymer of the embodiment of the present invention is 2,000 or more, preferably 4,000 or more, more preferably 5,000 or more in terms of number average molecular weight.
- number average molecular weight there is no restriction
- limiting in particular in the upper limit of a number average molecular weight Less than 2,000,000 can be illustrated as a preferable range, More preferably, it is less than 500,000, More preferably, it is less than 200,000.
- the number average molecular weight is in this preferred range, high mechanical properties and chemical resistance can be easily obtained, and molding processability is also good, which is preferable.
- the copolymer according to the embodiment of the present invention is a method for producing a copolymer by simultaneously synthesizing at least two polymer component blocks constituting the copolymer, or a polymer obtained by synthesizing at least one polymer component block. It can be produced by any sequential production method using component blocks as raw materials or initiators. Of these, a sequential production method is preferably employed from the viewpoint of reducing the incorporation of the homopolymer into the copolymer.
- the manufacturing method of the copolymer of embodiment of this invention is described.
- the polymer component block constituting the copolymer of the embodiment of the present invention is produced by ring-opening polymerization of a cyclic compound.
- the ring-opening polymerization can be performed at least in the presence of an initiator.
- an initiator is not limited as long as it is a compound capable of initiating a ring-opening polymerization reaction.
- a photopolymerization initiator, a radical polymerization initiator, a cationic polymerization initiator, an anionic polymerization initiator, a transition metal A known initiator such as a catalyst can be used. Of these, anionic polymerization initiators are preferred.
- the initiator here includes a polymer compound having a reactive terminal capable of initiating ring-opening polymerization.
- An anionic polymerization initiator generally has a nucleophilic property, and an active species proceeds with polymerization of an anion by using a nucleophilic addition of the initiator to a monomer as an initiation reaction.
- the anionic ring-opening polymerization of a cyclic compound in an embodiment of the present invention the cyclic compound is opened by nucleophilic addition of an anionic polymerization initiator, and a polymer having an anionic terminal is generated.
- An anion ring-opening polymerization proceeds due to the ring-opening reaction, and a case where an anion is generated in a cyclic compound, that is, a monomer, and a nucleophilic addition is performed at the end of the polymer.
- the anionic polymerization initiator is not limited as long as it is a compound capable of generating an anionic species, but inorganic metal salts such as inorganic alkali metal salts and inorganic alkaline earth metal salts, or organic alkali metal salts and organic alkalis.
- Organic metal salts such as earth metal salts can be exemplified.
- examples of inorganic alkali metal salts and inorganic alkaline earth metal salts include alkali metal halides such as sodium fluoride, potassium fluoride, cesium fluoride, and lithium chloride, lithium hydride, sodium hydride, and potassium hydride.
- Alkali metal hydrides lithium hydroxide, sodium hydroxide, potassium hydroxide, cesium hydroxide, magnesium hydroxide, calcium hydroxide, and hydroxides of alkali metals or alkaline earth metals such as barium hydroxide, lithium sulfide, Alkali metal sulfides such as sodium sulfide and potassium sulfide; alkali metal hydrosulfides such as lithium hydrosulfide, sodium hydrosulfide and potassium hydrosulfide; and alkali metal carbonates such as lithium carbonate, sodium carbonate and potassium carbonate Etc.
- alkali metal halides and alkali metal sulfides are preferably used.
- an ionic compound represented by the formula (vii) is desirable.
- o is an integer from 0 to 1
- p is an integer from 1 to 4.
- R represents an alkyl group having 1 to 20 carbon atoms such as methyl, propyl, n-butyl, isobutyl, s-butyl, t-butyl, and derivatives thereof, phenyl, naphthyl, 4-phenylphenyl, 3- Aryl groups such as phenylphenyl, 2-phenylphenyl, 4-phenoxyphenyl, 3-phenoxyphenyl, 2-phenoxyphenyl, 4-benzophenyl, 3-benzophenyl, 2-benzophenyl, anthryl, fluorenyl and their derivatives, and Polymer chains such as polyphenylene sulfide, polyamide, polyester, polycarbonate and polysulfone are preferred.
- an aryl group, its derivative (s), and a polymer chain can illustrate preferably.
- X include an oxygen atom, a sulfur atom, a carboxy group, an amide group, and a carbonate group, and a functional group composed of a combination thereof may be used.
- M is preferably a monohalide of an alkali metal such as lithium, sodium, potassium and cesium, or an alkaline earth metal such as magnesium and calcium.
- Examples of the ionic compound as described above include sodium methoxide, potassium methoxide, lithium methoxide, cesium methoxide, sodium ethoxide, potassium ethoxide, lithium ethoxide, cesium ethoxide, sodium n-butoxide, potassium n -Butoxide, lithium n-butoxide, cesium n-butoxide, sodium s-butoxide, potassium s-butoxide, lithium s-butoxide, cesium s-butoxide, sodium tert-butoxide, potassium tert-butoxide, lithium tert-butoxide, cesium t -Butoxide, sodium n-propoxide, potassium n-propoxide, lithium n-propoxide, cesium n-propoxide, sodium isopropoxide, potassium isopropoxide, lithi Metal alkoxides such as alkali metal salts of aliphatic alcohols having 1 to 20 carbon atoms such as muisoprop
- the initiator used in the embodiment of the present invention may be used alone or in combination of two or more.
- the amount of initiator to be used varies depending on the molecular weight of the target polymer component block and, when a catalyst is used together with the initiator, the type of catalyst.
- the amount of the initiator is usually 0.001 mol% or more as an anion concentration capable of generating an initiator with respect to 1 mol of the main repeating unit constituting the target polymer component block. It is preferably at least mol%, more preferably at least 0.01 mol%. Moreover, what is necessary is just 20 mol% or less, 15 mol% or less is preferable and 10 mol% or less is more preferable. By adding the initiator amount within this preferable range, ring-opening polymerization tends to proceed in a short time.
- tin, stannous chloride, stannous bromide, stannous iodide, stannous sulfate, stannic oxide, stannous myristate, stannous octoate as catalysts capable of promoting the ring-opening reaction
- Tin compounds such as tin 2-ethylhexylate, tin stearate, tetraphenyltin, tin methoxide, tin ethoxide, tin propoxide, tin butoxide, and 1,3-dichloro-1,1,3,3-tetrabutyldistanoxane
- Aluminum compounds such as aluminum, aluminum oxide, aluminum acetylacetonate, aluminum isopropoxide, and aluminum-imine complex titanium tetrachloride, titanium, tetramethyl orthotitanate, tetraethyl orthotitanate, tetrapropyl orthotitanate, orthotitanium Tet
- the copolymer of the embodiment of the present invention is prepared by the polymer component block produced in the presence of a polymer component block having a reactive terminal capable of initiating anionic, cationic or other ring-opening polymerization. It is preferably produced by ring-opening polymerization of a cyclic compound having a structure different from the repeating structural unit.
- the manufacturing method of the copolymer of embodiment of this invention includes the process of following (a) and (b).
- (A) A step of ring-opening polymerization of the cyclic polyphenylene sulfide represented by the general formula (v) in the presence of an initiator.
- (B) A step of ring-opening polymerization of any one cyclic compound selected from cyclic amide, cyclic ester, cyclic polycarbonate, cyclic polysulfone and cyclic polyphenylene ether ether ketone in the presence of an initiator.
- the steps (a) and (b) may be independent of each other, and the polymer component block produced in one of the steps (a) and (b) is used as an initiator for the other step. It may be used as From the viewpoint of obtaining a uniform copolymer free from homopolymer contamination, a production method using a polymer component block produced in one of the steps (a) and (b) as an initiator in the other step is preferred. It can be illustrated.
- the heating temperature for ring-opening polymerization of the cyclic polyphenylene sulfide is not particularly limited, but is preferably a temperature at which the cyclic polyphenylene sulfide melts.
- the heating temperature is lower than the melting temperature of the cyclic polyphenylene sulfide, a long time tends to be required to obtain the polyphenylene sulfide component block.
- the temperature at which the cyclic polyphenylene sulfide melts varies depending on the composition and molecular weight of the cyclic polyphenylene sulfide and the environment during heating, and thus cannot be uniquely indicated.
- the cyclic polyphenylene sulfide can be analyzed with a differential scanning calorimeter. By analyzing, it is possible to grasp the melting solution temperature. As a minimum of heating temperature, 180 ° C or more can be illustrated, preferably 200 ° C or more, more preferably 220 ° C or more, still more preferably 240 ° C or more. In this temperature range, the cyclic polyphenylene sulfide is melted and the polyphenylene sulfide component block can be obtained in a short time.
- heating temperature 400 degrees C or less can be illustrated, Preferably it is 380 degrees C or less, More preferably, it is 360 degrees C or less.
- the reaction time (heating time) for ring-opening polymerization of the cyclic polyphenylene sulfide is various characteristics such as the content of the cyclic compound in the cyclic polyphenylene sulfide used, the number of repetitions (m), and the molecular weight. It varies depending on conditions such as the polymerization initiator to be used, the type of catalyst, and the heating temperature, but cannot be defined uniformly. However, it is preferable to set the undesirable side reaction as described above as much as possible. Examples of the heating time include 0.01 hours or longer, and 0.05 hours or longer is preferable.
- the cyclic polyphenylene sulfide tends to be sufficiently converted into a polyphenylene sulfide component block.
- 100 hours or less can be illustrated, 20 hours or less are preferable and 10 hours or less are more preferable.
- the heating time may be 2 hours or less, and examples thereof include 1 hour or less, 0.5 hours or less, 0.3 hours or less, or 0.2 hours or less. When the time is less than 100 hours, adverse effects on the properties of the resulting polyphenylene sulfide component block due to undesirable side reactions tend to be suppressed.
- the heating of the cyclic polyphenylene sulfide can be performed under a condition that does not substantially contain a solvent.
- the temperature can be raised in a short time, the reaction rate is high, and the polyphenylene sulfide component block tends to be easily obtained in a short time.
- the condition containing substantially no solvent means that the solvent in the cyclic polyphenylene sulfide is 10% by weight or less, and more preferably 3% by weight or less.
- the heating may be performed in a mold for producing a molded product as well as by a method using a normal polymerization reaction apparatus, or using an extruder or a melt kneader. You may go. Any apparatus provided with a heating mechanism can be used without particular limitation, and known methods such as a batch method and a continuous method can be employed.
- the atmosphere for heating the cyclic polyphenylene sulfide is preferably a non-oxidizing atmosphere, and is preferably performed under reduced pressure. In the case of performing under reduced pressure conditions, it is preferable that the atmosphere in the reaction system is once changed to a non-oxidizing atmosphere and then the reduced pressure conditions. This tends to suppress the occurrence of undesirable side reactions such as cross-linking reactions and decomposition reactions between cyclic polyphenylene sulfides, between polyphenylene sulfide component blocks generated by heating, and between polyphenylene sulfide component blocks and cyclic polyphenylene sulfide.
- the non-oxidizing atmosphere means that the oxygen concentration in the gas phase in contact with the cyclic polyphenylene sulfide is 5% by volume or less, preferably the oxygen concentration is 2% by volume or less, and does not substantially contain oxygen. More preferably.
- an inert gas atmosphere such as nitrogen, helium, and argon is preferable.
- a nitrogen atmosphere is particularly preferable from the viewpoints of economy and ease of handling.
- the reduced pressure condition means that the pressure in the system for performing the reaction is lower than atmospheric pressure.
- the upper limit of the pressure in the system is preferably 50 kPa or less, more preferably 20 kPa or less, and even more preferably 10 kPa or less.
- An example of the lower limit is 0.1 kPa or more, and 0.2 kPa or more is more preferable.
- the pressure under the reduced pressure condition is at least the preferred lower limit, the low molecular weight cyclic compound containing cyclic polyphenylene sulfide is less likely to be volatilized.
- the pressure is less than the preferred upper limit, undesirable side reactions such as crosslinking reactions tend not to occur. Therefore, it becomes easy to obtain a polyphenylene sulfide component block having the above-described characteristics by adopting the above-described preferable reduced pressure conditions.
- the heating temperature at the time of ring-opening polymerization of the cyclic amide compound is a temperature at which the raw material cyclic amide and the polyamide component block or copolymer produced as a result of the ring-opening polymerization are uniformly melted. If there is no restriction in particular.
- the heating temperature is equal to or higher than the homogenization temperature of the cyclic amide compound and the polyamide component block or copolymer, a polyamide component block having a high degree of polymerization tends to be easily obtained in a short time. Therefore, the heating temperature is preferably 100 ° C. or higher, and more preferably 120 ° C. or higher.
- the homogenizing temperature varies depending on the structure and molecular weight of the cyclic amide compound and the environment during heating, and cannot be uniquely indicated.
- the cyclic amide compound and the polymerization initiator are placed in a test tube under a nitrogen atmosphere. It is possible to grasp by enclosing and observing the test tube while heating. If the heating temperature is too high, undesirable side reactions such as gelation due to decomposition or crosslinking of the cyclic amide compound in the composition tend to occur, and a polyamide component block or copolymer cannot be obtained or obtained. The properties of the resulting polyamide component block may deteriorate. Therefore, the heating temperature is preferably 380 ° C. or lower, more preferably 360 ° C.
- the reaction may be performed at a constant temperature, or the temperature may be changed stepwise or continuously.
- volatilization during reaction of a cyclic amide compound is easy to be suppressed and sufficient reaction is easy to be obtained, it is preferable.
- the ring-opening polymerization temperature of the cyclic ester compound or the cyclic carbonate compound is such that the cyclic compound as a raw material and the polymer component block or copolymer generated as a result of the ring-opening polymerization are melted uniformly. If it is temperature, there will be no restriction
- the temperature at which the cyclic ester or cyclic carbonate melts varies depending on the type and molecular weight of the cyclic product and the environment at the time of heating, but it cannot be uniquely indicated. For example, the cyclic product should be analyzed with a differential scanning calorimeter. It is possible to grasp the melting solution temperature.
- heating temperature 80 degreeC or more can be illustrated, Preferably it is 100 degreeC or more, More preferably, it is 120 degreeC or more, More preferably, it is 150 degreeC or more. Above this temperature, ring-opening polymerization tends to proceed, and a high conversion and high molecular weight polymer is likely to be obtained.
- 400 degrees C or less can be illustrated, Preferably it is 380 degrees C or less, More preferably, it is 360 degrees C or less, More preferably, it is 340 degrees C or less. Below this temperature, it is easy to suppress undesirable side reactions such as depolymerization and gelation due to the backbiting reaction. Further, when the obtained polyester component block or polycarbonate component block is used in the step (a), it is preferable because a uniform copolymer is easily obtained.
- the temperature at the time of ring-opening polymerization of the cyclic sulfone compound varies depending on the kind and molecular weight of the cyclic product and the environment at the time of heating. It is possible to grasp by observing the melting or softening behavior with an optical microscope equipped with a stage.
- heating temperature 80 degreeC or more can be illustrated, Preferably it is 100 degreeC or more, More preferably, it is 120 degreeC or more, More preferably, it is 150 degreeC or more. Above this temperature, ring-opening polymerization tends to proceed, and a high conversion and high molecular weight polymer is likely to be obtained.
- heating temperature 400 degrees C or less can be illustrated, Preferably it is 380 degrees C or less, More preferably, it is 360 degrees C or less, More preferably, it is 340 degrees C or less. Further, it is preferable to use the obtained polysulfone component block in the step (a) because a uniform copolymer can be easily obtained.
- the temperature at the time of ring-opening polymerization of the cyclic polyphenylene ether ether ketone varies depending on the kind and composition of the cyclic product, the molecular weight, and the environment at the time of heating. For example, it is possible to grasp the melting point by analyzing the cyclic compound with a differential scanning calorimeter. As a minimum of heating temperature, 180 ° C or more can be illustrated, preferably 200 ° C or more, more preferably 220 ° C or more, still more preferably 240 ° C or more. In this temperature range, the cyclic compound is melted and a copolymer can be obtained in a short time.
- the temperature is too high, undesirable side reactions such as cross-linking reactions and decomposition reactions between raw materials and products tend to occur, and the properties of the resulting copolymer may deteriorate. It is desirable to avoid a temperature at which such an undesirable side reaction is remarkably generated.
- As an upper limit of heating temperature 400 degrees C or less can be illustrated, Preferably it is 360 degrees C or less. Below this temperature, adverse effects on the properties of the resulting copolymer due to undesired side reactions tend to be suppressed, and a copolymer having the properties described above can be obtained.
- the product obtained in the step (a) is used in the step (b), so that It is preferable because a simple copolymer can be easily obtained.
- the reaction time varies depending on various characteristics such as the type and molecular weight of the cyclic compound used, the type of the polymerization initiator and catalyst used, and the conditions such as the heating temperature. It is preferable to set so that undesirable side reactions do not occur as much as possible. Examples of the heating time include 0.01 hours or longer, and 0.05 hours or longer is preferable. Moreover, 100 hours or less can be illustrated, 20 hours or less are preferable and 10 hours or less are more preferable.
- the ring-opening polymerization of the cyclic compound can be performed under a condition that does not substantially contain a solvent.
- the temperature can be raised in a short time, the reaction rate is high, and the polymer component block tends to be easily obtained in a short time.
- the condition containing substantially no solvent means that the solvent in the cyclic compound is 10% by weight or less, and more preferably 3% by weight or less.
- the heating may be performed in a mold for producing a molded product as well as by a method using a normal polymerization reaction apparatus, or using an extruder or a melt kneader. You may go. Any apparatus provided with a heating mechanism can be used without particular limitation, and known methods such as a batch method and a continuous method can be employed.
- the cyclic compound is preferably heated in a non-oxidizing atmosphere, and preferably under reduced pressure.
- the atmosphere in the reaction system is once changed to a non-oxidizing atmosphere and then the reduced pressure conditions.
- the non-oxidizing atmosphere means that the oxygen concentration in the gas phase in contact with the cyclic compound is 5% by volume or less, preferably the oxygen concentration is 2% by volume or less, and does not substantially contain oxygen.
- an inert gas atmosphere such as nitrogen, helium, and argon is preferable.
- the reduced pressure condition means that the pressure in the system for performing the reaction is lower than atmospheric pressure.
- the upper limit of the pressure in the system is preferably 50 kPa or less, more preferably 20 kPa or less, and even more preferably 10 kPa or less.
- An example of the lower limit is 0.1 kPa or more, and 0.2 kPa or more is more preferable.
- the heating of the cyclic compound in the step (a) and / or the step (b) can be performed in the presence of a fibrous substance.
- the fibrous substance is a thin thread-like substance, and an arbitrary substance having a structure elongated like a natural fiber is preferable.
- reinforcing fibers composed of long fibers, which makes it possible to highly reinforce the polymer.
- wetting means that the physical state is good and maintained so that substantially no air or other gas is trapped between a fluid substance such as a molten resin and a solid substrate such as a fibrous compound. Means there is a contact made. The lower the viscosity of the fluid material, the better the wetting with the fibrous material.
- the cyclic compound of the embodiment of the present invention has a remarkably low viscosity when melted compared to a general thermoplastic resin, it tends to be easily wetted with the fibrous material.
- the cyclic compound and the copolymer are converted into a polymer by the method for producing a copolymer according to the embodiment of the present invention, so that the fibrous material and the copolymer have good wetting.
- the formed composite material structure can be easily obtained.
- the fibrous material is preferably a reinforcing fiber composed of long fibers, and the reinforcing fiber used in the embodiment of the present invention is not particularly limited.
- the reinforcing fibers that are suitably used include fibers having good heat resistance and tensile strength that are generally used as high-performance reinforcing fibers.
- the reinforcing fiber includes glass fiber, carbon fiber, graphite fiber, aramid fiber, silicon carbide fiber, alumina fiber, and boron fiber.
- carbon fiber and graphite fiber which have good specific strength and specific elastic modulus and are recognized to make a great contribution to weight reduction, can be exemplified as the best.
- any type of carbon fiber or graphite fiber can be used as the carbon fiber or graphite fiber depending on the application, but a high strength and high elongation carbon fiber having a tensile strength of 450 kgf / mm 2 and a tensile advance of 1.6% or more. Is the most suitable.
- the length is preferably 5 cm or more. In the range of this length, it becomes easy to sufficiently develop the strength of the reinforcing fiber as a composite material.
- Carbon fiber and graphite fiber may be used by mixing other reinforcing fibers. The shape and arrangement of the reinforcing fibers are not limited.
- the reinforcing fibers can be used in a single direction, a random direction, a sheet shape, a mat shape, a fabric shape, and a braided shape.
- an array in which reinforcing fibers are aligned in a single direction is most suitable, but a cloth (fabric) array that is easy to handle is also available. Suitable for embodiments of the invention.
- the conversion of the cyclic compound into a copolymer or polymer component block can be performed in the presence of a filler.
- a filler include non-fibrous glass, non-fibrous carbon, and inorganic fillers such as calcium carbonate, titanium oxide, and alumina.
- the copolymer containing the polyarylene sulfide component block according to the embodiment of the present invention is characterized by having heat resistance and solvent resistance characteristics of the polyarylene sulfide and high physical properties derived from the uniform micro-separation structure by blocking. It can be used for various uses such as parts, electrical / electronic parts, building members, various containers, daily necessities, household goods and sanitary goods, as well as fibers, films and sheets.
- the chromatogram obtained by the SEC measurement has a single peak when the molecular weight is in the range of 2,000 to 2,000,000. It was evaluated as sex.
- Cyclic polyphenylene sulfide A stainless steel autoclave equipped with a stirrer was prepared by using 140.3 g (1.20 mol) of a 48 wt% aqueous solution of sodium hydrosulfide and 96% sodium hydroxide. A 48 wt% aqueous solution 125.0 g (1.44 mol), N-methyl-2-pyrrolidone (NMP) 6,150 g, and p-dichlorobenzene (p-DCB) 180.8 g (1.23 mol) were charged. . The reaction vessel was sufficiently purged with nitrogen, and then sealed under nitrogen gas.
- the temperature was raised from room temperature to 200 ° C. over about 1 hour.
- the pressure in the reaction vessel was 0.35 MPa as a gauge pressure.
- the temperature was raised from 200 ° C. to 270 ° C. over about 30 minutes.
- the pressure in the reaction vessel at this stage was 1.05 MPa as a gauge pressure.
- the content obtained was analyzed by gas chromatography and high performance liquid chromatography. As a result, the consumption rate of monomer p-DCB was 93%, and cyclic PPS was assumed when all sulfur components in the reaction mixture were converted to cyclic PPS. The production rate was found to be 18.5%.
- 5,000 g of the obtained contents were diluted with about 15,000 g of ion-exchanged water, and then filtered through a glass filter having an average opening of 10 to 16 ⁇ m.
- the filter-on component was dispersed in about 3,000 g of ion-exchanged water, stirred at 70 ° C. for 30 minutes, and subjected to the same filtration again three times in total to obtain a white solid. This was vacuum-dried at 80 ° C. overnight to obtain a dry solid.
- the obtained solid was subjected to Soxhlet extraction with chloroform at 70 ° C. for about 5 hours to separate low molecular weight components contained in the solid content.
- this white powder has p-phenylene sulfide unit as the main constituent unit based on mass spectrum analysis of the components separated by high performance liquid chromatography (apparatus: Hitachi M-1200H) and molecular weight information by MALDI-TOF-MS.
- the cyclic polyphenylene sulfide was found to contain about 98% by weight of a cyclic compound having 4 to 13 repeating units.
- this white powder is referred to as cyclic polyphenylene sulfide (cPPS) of Reference Example 1.
- cPPS cyclic polyphenylene sulfide
- the content obtained was analyzed by gas chromatography and high performance liquid chromatography. As a result, the consumption rate of monomer p-DCB was 96%, and cyclic PPS was assumed when all sulfur components in the reaction mixture were converted to cyclic PPS. The production rate was found to be 16%.
- the obtained contents (500 g) were diluted with about 1500 g of ion exchange water, and then filtered through a glass filter having an average opening of 10 to 16 ⁇ m.
- the filter-on component was dispersed in about 300 g of ion-exchanged water, stirred at 70 ° C. for 30 minutes, and filtered again in the same manner three times to obtain a white solid. This was vacuum dried at 80 ° C. overnight to obtain a dry solid.
- the obtained solid was subjected to Soxhlet extraction with chloroform at 70 ° C. for about 5 hours to separate low molecular weight components contained in the solid content.
- this white powder has p-phenylene sulfide unit as the main constituent unit based on mass spectral analysis of the components separated by high performance liquid chromatography (apparatus; Hitachi M-1200H) and molecular weight information by MALDI-TOF-MS.
- the cyclic polyphenylene sulfide containing about 96% by weight of a cyclic compound having 4 to 13 repeating units was found.
- this white powder is referred to as cyclic polyphenylene sulfide (cPPS) of Reference Example 2.
- cPPS cyclic polyphenylene sulfide
- Reference Example 2 As a result of SEC measurement, the cyclic polyphenylene sulfide of Reference Example 2 was completely dissolved in 1-chloronaphthalene at room temperature, and the weight average molecular weight was 900.
- Reference Example 3 Cyclic Polycarbonate
- the cyclic polycarbonate of Reference Example 3 was synthesized by the method described in Macromolecules, 24, 3037 (1991). Into a 1 L three-necked eggplant flask equipped with a stirring blade and a condenser tube, 200 mL of dichloromethane, 7 mL of distilled water, 3 mL of a 9.75 M aqueous sodium hydroxide solution and 2.4 mL of triethylamine were added. While maintaining the mixed solution at 30 ° C.
- the organic layer was concentrated to dryness, and the resulting crude product was subjected to precipitation using acetone as a good solvent and water as a poor solvent, and the resulting product was vacuum-dried at 80 ° C. for 12 hours to obtain a white solid 36. 0.2 g (yield 71.2%) was obtained.
- the obtained white solid was found to be a 2- to 15-mer cyclic polycarbonate by analysis by 1 H-NMR, FT-IR and high performance liquid chromatography.
- this white solid is referred to as the cyclic polycarbonate of Reference Example 3.
- Reference Example 4 Cyclic Polysulfone
- the cyclic polysulfone of Reference Example 4 was synthesized by the method described in JP-A-3-088828. 1,500 mL of dimethyl sulfoxide and 800 mL of toluene were added to a 5 L three-necked eggplant flask equipped with a stirring blade, a condenser tube and a Dean-Stark apparatus, and the mixed solution was heated to reflux at 135 ° C. with stirring.
- the reaction solution was concentrated to 200 mL, then dropped into about 3000 mL of water, and the resulting precipitate was collected by filtration and washed several times with methanol.
- the obtained white solid was dried by heating at 80 ° C. for 6 hours. 20 g of the obtained crude product was put into 100 mL of dimethylformamide, and the resulting white precipitate was filtered off and washed with methanol. The product was dried in a vacuum dryer at 80 ° C. for 12 hours to obtain 5.2 g (yield 22.4%) of a white solid.
- the obtained white solid was found to be a 4 to 6-mer cyclic polysulfone by analysis by 1 H-NMR, FT-IR and high performance liquid chromatography.
- this white solid is referred to as cyclic polysulfone of Reference Example 4.
- the temperature was raised from room temperature to 140 ° C. and held at 140 ° C. for 1 hour. Thereafter, the temperature was raised to 180 ° C. and held at 180 ° C. for 3 hours. Thereafter, the temperature was raised to 230 ° C. and held at 230 ° C. for 5 hours, and the contents were recovered after cooling to near room temperature.
- acetic acid aqueous solution 150 g of a 1% by weight acetic acid aqueous solution was added to 50 g of the above-obtained contents. After stirring to form a slurry, the mixture was heated to 70 ° C. and stirring was continued for 30 minutes. The slurry was filtered through a glass filter (average pore size 10 to 16 ⁇ m) to obtain a filter-on component. The filter-on component was dispersed in 50 g of ion-exchanged water, stirred at 70 ° C. for 30 minutes, and subjected to the same filtration again three times in total to obtain a white solid. This was vacuum-dried at 80 ° C. overnight to obtain a dry solid.
- the obtained dry solid was charged into a cylindrical filter paper, and Soxhlet extraction was performed for about 5 hours using chloroform as a solvent to separate low molecular weight components contained in the dry solid.
- cyclic polyphenylene ether ether ketone in the cyclic polyphenylene ether ether ketone composition were linear polyphenylene ether ether ketone oligomers.
- this white powder is also referred to as cyclic polyphenylene ether ether ketone (cPEEK) of Reference Example 5.
- the test tube was taken out from the electric annular furnace and cooled to room temperature under nitrogen to obtain polyphenylene sulfide (A-1) having sodium thiophenoxide at one end.
- A-1 polyphenylene sulfide having sodium thiophenoxide at one end.
- the melting point was 280 ° C.
- the number average molecular weight was 14,800
- the weight average molecular weight was 27,800
- the maximum peak molecular weight was 16,900.
- the test tube was taken out from the electric annular furnace and cooled to room temperature under nitrogen to obtain polyphenylene sulfide (A-2) having sodium thiophenoxide at one end.
- A-22 polyphenylene sulfide having sodium thiophenoxide at one end.
- the melting point was 280 ° C.
- molecular weight measurement using SEC the number average molecular weight was 15,000, the weight average molecular weight was 28,200, and the maximum peak molecular weight was 17,100.
- Polyphenylene sulfide component block (A-4) A polyphenylene sulfide (A-4) having sodium thiophenoxide at both ends was prepared in the same manner as in Reference Example 6 except that 48.5 mg (0.19 mmol) of 4,4′-biphenyldithiol disodium salt was used as a polymerization initiator. ) As a result of measuring the melting point using a differential scanning calorimeter, the melting point was 282 ° C. As a result of SEC measurement, the number average molecular weight was 28,700, the weight average molecular weight was 68,900, and the maximum peak molecular weight was 31,100.
- Polyphenylene sulfide component block (A-5) A polyphenylene sulfide (A-5) having sodium thiophenoxide at both ends was prepared in the same manner as in Reference Example 7 except that 9.7 mg (0.04 mmol) of 4,4′-biphenyldithiol disodium salt was used as a polymerization initiator. ) As a result of melting point measurement using a differential scanning calorimeter, the melting point was 275 ° C. As a result of molecular weight measurement using SEC, the number average molecular weight was 28,700, the weight average molecular weight was 72,500, and the maximum peak molecular weight was 32,000.
- the obtained content 50 g was diluted with about 150 g of ion-exchanged water, and then filtered through a glass filter having an average opening of 10 to 16 ⁇ m to obtain a filter-on component.
- the filter-on component was dispersed in about 300 g of ion-exchanged water, stirred at 70 ° C. for 30 minutes, and filtered again in the same manner three times to obtain a white solid. This was vacuum-dried overnight at 80 ° C. to obtain a polyphenylene sulfide component block (B-2) as a dry solid.
- the melting point was 280 ° C.
- the number average molecular weight of the obtained polymer was 24,500, the weight average molecular weight was 66,200, and the maximum peak molecular weight was 37,200.
- Polyester component block (C-2) In a test tube equipped with a stirring blade, a decompression adapter and a vacuum stirrer, 1.2 g of cyclic polybutylene terephthalate (CBT (registered trademark) 100 manufactured by Cyclics) and 122 mg (0.92 mmol) of sodium thiophenoxide as a polymerization initiator were charged. The inside of the system was replaced with nitrogen, and then heated to 250 ° C. using an electric annular furnace in a nitrogen atmosphere. After 5 minutes from the start of heating, it was confirmed that the contents had melted, and then stirring was started. The reaction was performed for 3.0 hours from the start of heating.
- CBT cyclic polybutylene terephthalate
- the test tube was removed from the electric annular furnace and cooled to room temperature under nitrogen to obtain polyester (C-2) having thiophenoxide at one end.
- the melting point was 228 ° C.
- the test tube was taken out from the electric annular furnace and cooled to room temperature under nitrogen to obtain polycarbonate (C-3) having thiophenoxide at one end.
- the melting point was 251 ° C.
- the test tube was taken out of the electric annular furnace and cooled to room temperature under nitrogen to obtain polyphenylene ether ether ketone (C-5) having thiophenoxide at one end.
- C-5 polyphenylene ether ether ketone having thiophenoxide at one end.
- the melting point was 341 ° C.
- Example 1 Polyphenylene sulfide-polyamide block copolymer 1 g of freeze-pulverized fine powder of polyphenylene sulfide component block (A-1) obtained in Reference Example 6 and 1 g (5.1 mmol) of ⁇ -laurolactam were mixed with a stirring blade. In a test tube equipped with a vacuum stirrer, a nitrogen introduction tube and a reflux tube, the atmosphere was replaced with nitrogen three times. While stirring under a nitrogen stream, the temperature was raised from room temperature to 200 ° C. in 30 minutes, and then raised to 250 ° C. in 60 minutes. Next, the temperature was raised from 250 ° C. to 300 ° C. in 30 minutes and held at 300 ° C. for 2 hours.
- Example 2 Polyphenylene sulfide-polyamide block copolymer A polyphenylene sulfide-polyamide block copolymer was obtained in the same manner as in Example 1 except that (A-4) was used as the polyphenylene sulfide component block.
- (A-4) was used as the polyphenylene sulfide component block.
- the number average molecular weight was 35,200
- the maximum peak molecular weight was 38,100
- the chromatogram was unimodal.
- the coarse separation structure resulting from the homopolymer was not observed and was uniform.
- the weight fraction of soluble components in hexafluoroisopropanol was 39% by weight. The results are shown in Table 1.
- Example 3 Polyphenylene sulfide-polyamide block copolymer 1 g of freeze-pulverized powder of polyamide component block (C-1) obtained in Reference Example 13 and 1 g (9.1 mmol) of cyclic polyphenylene sulfide obtained in Reference Example 1 was placed in a test tube equipped with a stirring blade, a vacuum stirrer, a nitrogen introduction tube and a reflux tube, and purged with nitrogen three times. While stirring under a nitrogen stream, the temperature was raised from room temperature to 200 ° C. in 30 minutes, and then raised to 250 ° C. in 60 minutes. Next, the temperature was raised from 250 ° C. to 300 ° C. in 30 minutes and held at 300 ° C. for 2 hours.
- Example 4 Polyphenylene sulfide-polybutylene terephthalate block copolymer 1 g of freeze-pulverized fine powder of polyphenylene sulfide component block (A-1) obtained in Reference Example 6 and cyclic polybutylene terephthalate (CBT (registered trademark) manufactured by Cyclics) ) 100) 1.2 g (5.45 mmol) was placed in a test tube equipped with a stirring blade, a vacuum stirrer and a nitrogen inlet tube, and purged with nitrogen three times. The temperature was raised from room temperature to 200 ° C. in 30 minutes under a nitrogen stream, and then heated to 250 ° C. in 60 minutes while stirring.
- CBT cyclic polybutylene terephthalate
- Example 5 Polyphenylene sulfide-polybutylene terephthalate block copolymer A polyphenylene sulfide-polybutylene terephthalate block copolymer was obtained in the same manner as in Example 4 except that (A-4) was used as the polyphenylene sulfide component block. It was. As a result of SEC measurement of the obtained polymer, the number average molecular weight was 34,000, the maximum peak molecule was 37,600, and the chromatogram was unimodal. As a result of evaluating the uniformity at the time of melting with an optical microscope, the coarse separation structure resulting from the homopolymer was not observed and was uniform. The weight fraction of soluble components in hexafluoroisopropanol was 30% by weight. The results are shown in Table 2.
- Example 6 Polyphenylene sulfide-polybutylene terephthalate block copolymer 1 g of freeze-pulverized fine powder of polybutylene terephthalate component block (C-2) obtained in Reference Example 14 and 1 g of cyclic polyphenylene sulfide of Reference Example 1 (9. 16 mmol) was placed in a test tube equipped with a stirring blade, a vacuum stirrer, and a nitrogen introduction tube, and purged with nitrogen three times. The temperature was raised from room temperature to 200 ° C. in 30 minutes under a nitrogen stream, and then heated to 250 ° C. in 60 minutes while stirring. Next, the temperature was raised from 250 ° C. to 280 ° C. in 15 minutes to complete the reaction.
- the contents of the test tube were discharged into water, and the resulting brown solid was vacuum dried at 50 ° C. for 12 hours to obtain a polyphenylene sulfide-polybutylene terephthalate copolymer.
- the number average molecular weight was 23,100
- the maximum peak molecular weight was 26,500
- the chromatogram was unimodal.
- the coarse phase separation structure due to the homopolymer was not observed and was uniform.
- the weight fraction of soluble components in hexafluoroisopropanol was 15% by weight. The results are shown in Table 2.
- Example 7 Polyphenylene sulfide-polycarbonate block copolymer 1 g of freeze-pulverized fine powder of polyphenylene sulfide component block (A-1) obtained in Reference Example 6 and 1.2 g of cyclic polycarbonate obtained in Reference Example 3 (4 72 mmol) was placed in a test tube equipped with a stirring blade, a vacuum stirrer, and a nitrogen introduction tube, and purged with nitrogen three times. The temperature was raised from room temperature to 200 ° C. in 30 minutes under a nitrogen stream, and then heated to 250 ° C. in 60 minutes while stirring. Next, the temperature was raised from 250 ° C. to 300 ° C. in 45 minutes to complete the reaction.
- the contents of the test tube were discharged into water, and the resulting brown solid was vacuum dried at 80 ° C. for 12 hours to obtain a polyphenylene sulfide-polycarbonate copolymer.
- the number average molecular weight was 22,200
- the maximum peak molecular weight was 24,900
- the chromatogram was unimodal.
- the coarse phase separation structure due to the homopolymer was not observed and was uniform.
- the weight fraction of soluble components in dichloromethane was 12% by weight. The results are shown in Table 3.
- Example 8 Polyphenylene sulfide-polycarbonate block copolymer A polyphenylene sulfide-polycarbonate block copolymer was obtained in the same manner as in Example 7 except that (A-4) was used as the polyphenylene sulfide component block.
- the number average molecular weight was 35,700
- the maximum peak molecular weight was 39,300
- the chromatogram was unimodal.
- the coarse separation structure resulting from the homopolymer was not observed and was uniform.
- the weight fraction of soluble components in dichloromethane was 20% by weight. The results are shown in Table 3.
- Example 9 Polyphenylene sulfide-polycarbonate block copolymer 1.2 g of freeze-pulverized fine powder of the polycarbonate component block (C-3) obtained in Reference Example 15 and 1 g of cyclic polyphenylene sulfide obtained in Reference Example 1 (9 .16 mmol) was placed in a test tube equipped with a stirring blade, a vacuum stirrer, and a nitrogen introduction tube, and purged with nitrogen three times. The temperature was raised from room temperature to 200 ° C. in 30 minutes under a nitrogen stream, and then heated to 250 ° C. in 60 minutes while stirring. Next, the temperature was raised from 250 ° C. to 320 ° C. in 45 minutes to complete the reaction.
- the contents of the test tube were discharged into water, and the resulting brown solid was vacuum dried at 80 ° C. for 12 hours to obtain a polyphenylene sulfide-polycarbonate copolymer.
- the number average molecular weight was 22,200
- the maximum peak molecular weight was 24,900
- the chromatogram was unimodal.
- the coarse phase separation structure due to the homopolymer was not observed and was uniform.
- the weight fraction of soluble components in dichloromethane was 5% by weight. The results are shown in Table 3.
- Example 10 Polyphenylene sulfide-polysulfone block copolymer 1 g of freeze-pulverized fine powder of polyphenylene sulfide component block (A-1) obtained in Reference Example 6 and 1.2 g of cyclic polyethersulfone obtained in Reference Example 3 (5.17 mmol) was placed in a test tube equipped with a stirring blade, a vacuum stirrer, and a nitrogen introduction tube, and purged with nitrogen three times. The temperature was raised from room temperature to 280 ° C. in a nitrogen stream in 60 minutes, and then heated to 320 ° C. in 60 minutes while stirring. The contents of the test tube were discharged into water, and the resulting brown solid was vacuum dried at 80 ° C.
- Example 11 Polyphenylene sulfide-polysulfone block copolymer A polyphenylene sulfide-polysulfone block copolymer was obtained in the same manner as in Example 10 except that (A-4) was used as the polyphenylene sulfide component block.
- (A-4) was used as the polyphenylene sulfide component block.
- the number average molecular weight was 34,200
- the maximum peak molecular weight was 38,100
- the chromatogram was unimodal.
- the coarse separation structure resulting from the homopolymer was not observed and was uniform.
- the soluble component in N-methyl-2-pyrrolidone was 25% by weight. The results are shown in Table 4.
- Example 12 Polyphenylene sulfide-polysulfone block copolymer 1.2 g of freeze-pulverized fine powder of the polysulfone component block (C-4) obtained in Reference Example 16 and 1 g of cyclic polyphenylene sulfide obtained in Reference Example 1 (9 .16 mmol) was placed in a test tube equipped with a stirring blade, a vacuum stirrer, and a nitrogen introduction tube, and purged with nitrogen three times. The temperature was raised from room temperature to 280 ° C. in a nitrogen stream in 60 minutes, and then heated to 320 ° C. in 60 minutes while stirring. The contents of the test tube were discharged into water, and the resulting brown solid was vacuum dried at 80 ° C.
- Example 13 Polyphenylene sulfide-polyphenylene ether ether ketone block copolymer 0.92 g of freeze-pulverized fine powder of polyphenylene sulfide component block (A-2) obtained in Reference Example 7 and cyclic polyphenylene obtained in Reference Example 5 0.08 g of ether ether ketone was charged into a glass test tube (24 mm diameter) equipped with a stirring blade, a vacuum stirrer, and a nitrogen introduction tube. After the inside of the test tube was purged with nitrogen at room temperature and normal pressure, the pressure was reduced to about 0.4 kPa using a vacuum pump.
- test tube was placed in an electric annular furnace adjusted to 320 ° C., and the test tube was kept at about 0.4 kPa with a vacuum pump and stirred at 50 rpm while devolatilizing. For 90 minutes. Thereafter, the test tube was taken out from the furnace and cooled to room temperature to obtain a brown solid polymer.
- the consumption rate of cyclic polyphenylene sulfide was 99%
- the consumption rate of cyclic polyphenylene ether ether ketone was 96%.
- the melting point was 281 ° C. and 335 ° C.
- the cooling crystallization temperature was 176 ° C.
- Example 14 Polyphenylene sulfide-polyphenylene ether ether ketone block copolymer 0.7 g of freeze-pulverized fine powder of polyphenylene sulfide component block (A-2) obtained in Reference Example 7 and cyclic polyphenylene obtained in Reference Example 5 A copolymer was synthesized in the same manner as in Example 13 except that 0.3 g of the ether ether ketone composition was used.
- the consumption rate of cyclic polyphenylene sulfide was 98. %
- the consumption rate of cyclic polyphenylene ether ether ketone was 95%.
- the melting point was 278 ° C. and 328 ° C.
- the cooling crystallization temperature was 220 ° C.
- Example 16 Polyphenylene sulfide-polyphenylene ether ether ketone block copolymer
- the freeze-pulverized fine powder of the polyphenylene sulfide component block (A-5) obtained in Reference Example 10 was used.
- a copolymer was synthesized.
- Example 17 Polyphenylene sulfide-polyphenylene ether ether ketone ketone block copolymer Freely pulverized fine powder 0.30 g of polyphenylene ether ether ketone component block (C-5) obtained in Reference Example 17 and obtained in Reference Example 2 0.70 g of cyclic polyphenylene sulfide was charged into a glass test tube (24 mm diameter) equipped with a stirring blade, a vacuum stirrer, and a nitrogen introduction tube. After the inside of the test tube was purged with nitrogen at room temperature and normal pressure, the pressure was reduced to about 0.4 kPa using a vacuum pump.
- test tube was placed in an electric annular furnace adjusted to 350 ° C., and the test tube was kept at about 0.4 kPa with a vacuum pump and stirred at 50 rpm while devolatilizing. For 90 minutes. Thereafter, the test tube was taken out from the furnace and cooled to room temperature to obtain a brown solid polymer.
- the consumption rate of cyclic polyphenylene sulfide was 99. %
- the consumption rate of cyclic polyphenylene ether ether ketone was 95%.
- the melting point was 269 ° C. and 313 ° C.
- the cooling crystallization temperature was 183 ° C.
- Example 18 Polyphenylene sulfide-polyphenylene ether ether ketone block copolymer 0.5 g of the freeze-pulverized fine powder of the polyphenylene sulfide component block (A-2) obtained in Reference Example 7 and the cyclic product obtained in Reference Example 5 A copolymer was synthesized in the same manner as in Example 13 except that 0.5 g of the polyphenylene ether ether ketone composition was used.
- the consumption rate of cyclic polyphenylene sulfide was 98. %
- the consumption rate of cyclic polyphenylene ether ether ketone was 95%.
- the melting point was 272 ° C. and 333 ° C.
- the cooling crystallization temperature was 226 ° C.
- Example 19 Method for producing polyphenylene sulfide-polyphenylene ether ether ketone block copolymer 0.3 g of the cyclic polyphenylene sulfide of Reference Example 1, 0.7 g of the cyclic polyphenylene ether ale ketone composition obtained in Reference Example 5, and 1 g of a powder prepared by mixing 5 mol% of sodium benzenethiolate with a compound comprising a repeating unit of the formula-(Ph-S)-, which is the main structural unit of cyclic polyphenylene sulfide, was added to a glass test tube (24 mm diameter) equipped with a stirrer. The inside of the test tube was purged with nitrogen at room temperature and normal pressure. A test tube was placed in an electric furnace adjusted to 360 ° C. and heated for 60 minutes while stirring at 50 rpm, and then the test tube was taken out and cooled to room temperature to obtain a brown solid.
- the consumption rate of cyclic polyphenylene sulfide was 92. %
- the consumption rate of cyclic polyphenylene ether ether ketone was 100%.
- the melting point and the cooling crystallization temperature using a differential scanning calorimeter the melting point was 302 ° C. and the cooling crystallization temperature was 225 ° C.
- the number average molecular weight was 8,300, and the maximum peak molecular weight was 11,900.
- the temperature was raised from room temperature to 140 ° C. while stirring at 400 rpm, and held at 140 ° C. for 1 hour. Thereafter, the temperature was raised to 180 ° C. and held at 180 ° C. for 3 hours. Thereafter, the temperature was raised to 230 ° C. and held at 230 ° C. for 5 hours, and then cooled to room temperature to recover the contents.
- the melting point was 280 ° C., 286 ° C., 335 ° C., and the cooling crystallization temperature was 220, 234, 269 ° C. there were.
- the SEC measurement three types of polymer-derived peaks can be confirmed, and the number average molecular weight is 7,100, 10,400, 17,700, and the maximum peak molecular weight is 9,900, 13,100, in order from the lowest molecular weight. 22,400.
- the homogeneity at the time of melting with an optical microscope a coarse separation structure due to the homopolymer was observed and was not uniform.
- the weight fraction of the soluble component in p-chlorophenol was 20% by weight. The results are shown in Table 5.
- the melting point was 278 ° C., 304 ° C., and 336 ° C.
- the cooling crystallization temperature was 220, 226, and 259 ° C.
- the SEC measurement three types of polymer-derived peaks can be confirmed, and the number average molecular weights are 6,900, 12,300, 15,300, and the maximum peak molecular weights are 11,100, 15,400, 18 in descending order of molecular weight. 800.
- a coarse separation structure due to the homopolymer was observed and was not uniform.
- the weight fraction of soluble components in p-chlorophenol was 34% by weight. The results are shown in Table 5.
- a copolymer containing a polyarylene sulfide component block made of cyclic polyarylene sulfide as a raw material and a block made of a cyclic compound other than cyclic polyarylene sulfide as a raw material has a simple chromatogram obtained in SEC measurement. From the optical microscope measurement at the time of melting, a coarse phase-separated structure derived from a homopolymer is not observed, indicating that the block copolymer has a very uniform composition. It can be inferred that this is the result of quantitatively introducing reactive ends into the polyarylene sulfide component block by using a cyclic product as a raw material. Such effects were not obtained with a polyarylene sulfide component block not using a cyclic product as a raw material, and a copolymer having a non-uniform composition was obtained.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Other Resins Obtained By Reactions Not Involving Carbon-To-Carbon Unsaturated Bonds (AREA)
Abstract
Description

式中、mは4~50の整数であり、前記環状ポリフェニレンスルフィドは、同一のmを有する環状ポリフェニレンスルフィド、または、異なるmを有する複数種類の環状ポリフェニレンスルフィドの混合物であるブロック共重合体。

式中、xは1以上の整数であるブロック共重合体。
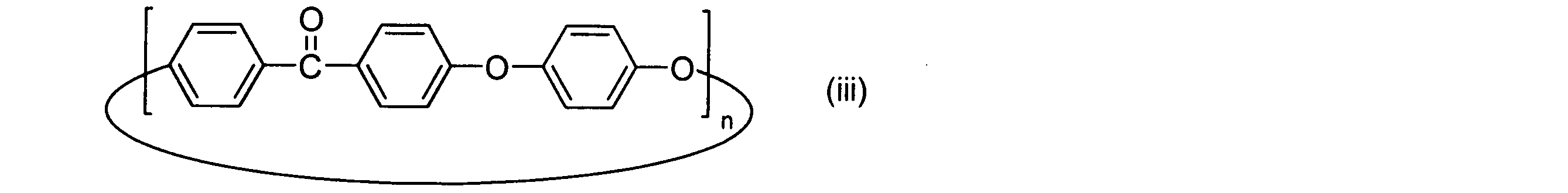
式中、nは2~40の整数であり、前記環状ポリフェニレンエーテルエーテルケトンは、同一のnを有する環状ポリフェニレンエーテルエーテルケトン、または、異なるnを有する複数種類の環状ポリフェニレンエーテルエーテルケトンの混合物であるブロック共重合体。
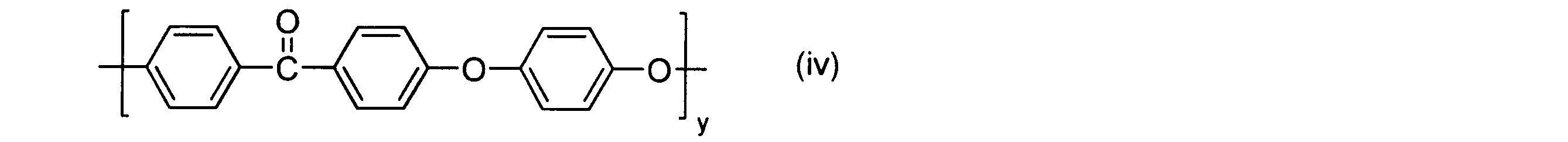
(a)環状ポリフェニレンスルフィドを、開始剤存在下で開環重合する工程と、
(b)環状ポリアリーレンスルフィドとは異なる環状化合物を、開始剤存在下で開環重合する工程と、
を備え、
前記(a)工程と前記(b)工程のうちの一方の工程で得られた生成物を、他方の工程の原料である環状化合物と混合して、前記他方の工程を行なう、ブロック共重合体の製造方法。
本発明の実施形態のブロック共重合体は、少なくとも2つのポリマー成分ブロックを含み、そのうち少なくも1つは環状ポリアリーレンスルフィドを原料とするポリアリーレンスルフィド成分ブロックである。
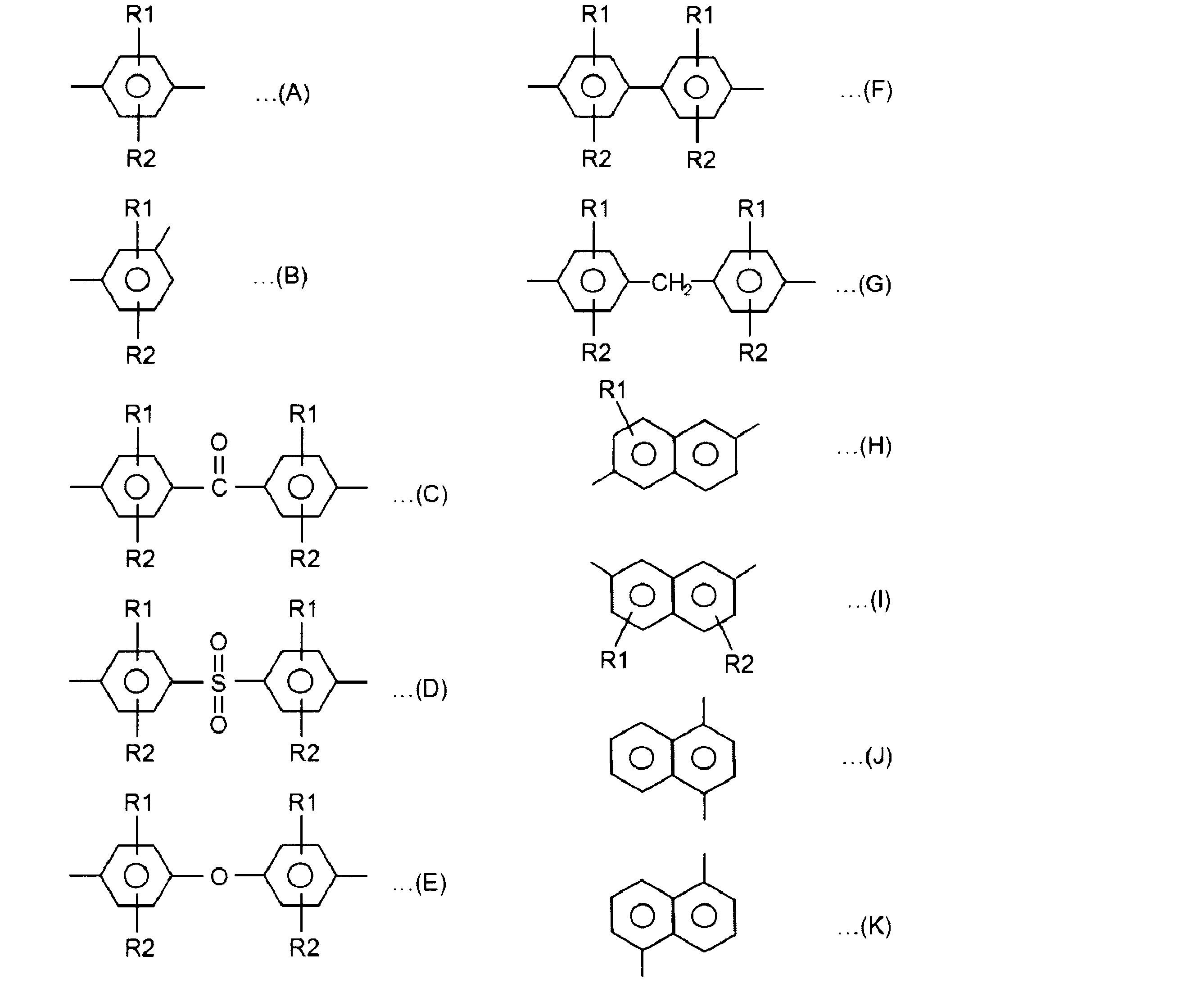

特に好ましい環状ポリアリーレンスルフィドとしては、主要構成単位としてp-フェニレンスルフィド単位を80モル%以上、好ましくは90モル%以上含有する、環状ポリフェニレンスルフィド(以下環状PPSと略する場合がある)が挙げられる。ここで言う環状ポリフェニレンスルフィドとは、下記一般式(v)のごとき環状化合物の単量体もしくは混合物である。環状PPSは、(v)式の環状化合物を少なくとも50重量%以上含むものであり、70重量%以上含むことが好ましく、80重量%以上含むことがより好ましく、90重量%以上含むものがさらに好ましい。

ポリフェニレンスルフィド成分ブロック以外のブロック成分の原料として用いる環状化合物としては、繰り返し単位を有する環状ポリマーまたはオリゴマーであっても、ラクタム、ラクトンなどの低分子環状化合物であってもよい。例えば環状のポリマーとしては、環状のポリアミド、ポリエステル、ポリアセタール、ポリカーボネート、ポリフェニレンエーテル、ポリアリレート、ポリスルホン、ポリケトン、ポリイミド、ポリアミドイミド、ポリエーテルイミド、ポリウレタン、ポリウレア、ポリフェニレンエーテルエーテルケトンおよびそれらの誘導体が好ましく用いられる。その中でも、ラクタム、ラクトン、環状のポリアミド、ポリエステル、ポリカーボネート、ポリスルホンおよびポリフェニレンエーテルエーテルケトンがさらに好適である。使用できる環状ポリマーまたはオリゴマーの分子量に特に制限はないが、環状物そのものや得られる共重合体の溶融粘度の観点から、数平均分子量100,000未満が好ましく、50,000未満がさらに好ましく、10,000以下が特に好ましい。

本発明の実施形態に記載の共重合体は、サイズ排除クロマトグラフィー(SEC)により測定される最大ピーク分子量が、2,000以上であることが好ましく、3,000以上であることがさらに好ましく、5,000以上であることが特に好ましい。また、最大ピーク分子量が2,000,000未満であることが好ましく、1,000,000未満であることがさらに好ましく500,000未満であることが特に好ましい。さらに、最大ピーク分子量は、上記の範囲にあり、かつ前記範囲で単峰性の分子量分布を有することが好ましい。最大ピーク分子量が前記範囲内であれば、得られる共重合体の物性や成形加工性が良好となる。ここでの最大ピーク分子量とは、示差屈折率検出器を備えたSEC装置を用いた測定によって得られるクロマトグラムの最大値に対応する分子量を意味し、横軸を保持時間、縦軸を基質濃度としたクロマトグラムの場合、基質濃度が最大値をとる保持時間を求め、その値を、絶対分子量が既知の標準物質(本実施形態ではポリスチレンが使用される)の測定により得られた分子量と保持時間との関係式に代入することにより求めることが可能である。本実施形態では、SECの測定条件としては、1-クロロナフタレンを溶離液として用い、共重合体を0.05重量%の濃度で溶解させている。SECの測定温度は50~250℃の範囲が例示でき、カラムや検出器などSEC装置を構成する工程毎に異なっていてもよい。本実施形態では、カラム温度210℃、プレ恒温槽温度250℃、ポンプ恒温槽温度50℃、検出器温度210℃としている。
本発明の実施形態の共重合体は、共重合体を構成する少なくとも2つのポリマー成分ブロックを同時に合成し共重合体を製造する方法、または少なくとも1つのポリマー成分ブロックを合成し、得られたポリマー成分ブロックを原料あるいは開始剤として用いる逐次的な製造方法のいずれによってでも製造することが可能である。このうち、共重合体中へのホモポリマーの混入低減という観点から、逐次的な製造方法が好ましく採用される。以下、本発明の実施形態の共重合体の製造方法を記述する。
(a)前記一般式(v)で表される環状ポリフェニレンスルフィドを、開始剤存在下で開環重合する工程。
(b)環状アミド、環状エステル、環状ポリカーボネート、環状ポリスルホンおよび環状ポリフェニレンエーテルエーテルケトンから選ばれるいずれか1種の環状化合物を、開始剤の存在下で開環重合する工程。
環状ポリフェニレンスルフィド、環状ポリフェニレンスルフィド成分ブロック、その他の成分ブロック、共重合体の各平均分子量、および最大ピーク分子量は、サイズ排除クロマトグラフィー(SEC)によりポリスチレン換算で算出した。測定条件を以下に示す。
装置:株式会社センシュー科学 SSC-7100
カラム:株式会社センシュー科学 GPC3506
溶離液:1-クロロナフタレン
検出器:示差屈折率検出器
カラム温度:210℃
プレ恒温槽温度:250℃
ポンプ恒温槽温度:50℃
検出器温度:210℃
流速:1.0mL/min
試料注入量:300μL
リンカム社製の顕微鏡用加熱ステージTH-600PM上に、ポリマーをカバーグラスに挟んで静置した。昇温速度90℃/分で25℃から300℃に昇温し、5分間滞留後の相分離状態を、株式会社ニコン インストルメンツカンパニー社製の位相差光学顕微鏡OPTIPHOT XF-Ph(倍率50倍)で観察した。溶融時均一性の評価は以下の基準で判断した。
均一:相分離サイズが1μm以下の均一な一相状態
不均一:数~数十μmオーダーで粗大相分離している状態
共重合体中の環状ポリフェニレンスルフィドまたは環状ポリフェニレンエーテルエーテルケトンの重量分率、および環状ポリフェニレンエーテルエーテルケトン組成物中の含有環状ポリフェニレンエーテルエーテルケトン組成は、以下のようにして求めた。共重合体あるいは組成物を、p-クロロフェノール/1-クロロナフタレン=6/4(v/v)混合溶媒に220℃加熱下で溶解させ、テトラヒドロフラン(THF)で希釈し、孔径0.45μmのメンブレンフィルターで濾過したものを、高速液体クロマトグラフィーにより下記条件にて測定して算出した。
装置 :株式会社島津製作所製 LC-10Avpシリーズ
カラム :Mightysil RP-18GP150-4.6
検出器 :フォトダイオードアレイ検出器(UV=270nmを使用)
カラム温度 :40℃
サンプル濃度:0.02重量%テトラヒドロフラン(THF)溶液
移動相 :THF/0.1重量%トリフルオロ酢酸水溶液
(共重合体中の環状ポリフェニレンスルフィドまたは環状ポリフェニレンエーテルエーテルケトンの消費率)(%)=100-(共重合体中の環状ポリフェニレンスルフィドまたは環状ポリフェニレンエーテルエーテルケトンの重量分率)(%)
示差走査型熱量分析装置として、セイコー電子工業株式会社製のロボットDSC RDC220を用い、窒素雰囲気下、得られたポリマーの熱的特性を測定した。下記測定条件を用い、融点としてはSecond Runの吸熱ピークの値を用い、降温結晶化温度としてはFirst Runの発熱ピークの値を用いた。
(First Run)
・50℃×1分 ホールド
・50℃から380℃へ昇温,昇温速度20℃/分
・昇温後×1分 ホールド
・50℃へ降温,降温速度20℃/分
(Second Run)
・50℃×1分 ホールド
・ 50℃から380℃へ昇温,昇温速度20℃/分
得られた共重合体の耐薬品性評価のため、共重合体500mgに、ヘキサフルオロイソプロパノール、ジクロロメタン、N-メチル-2-ピロリドンおよびp-クロロフェノールから選択される1種の溶媒を20mL加え、80℃で2時間撹拌して、不溶成分を除去した。さらに溶媒を真空除去により除いて、溶媒中に溶解していた可溶成分を得て、共重合体中の可溶成分の重量分率を算出した。得られた可溶成分の重量分率が低いほど、共重合体は耐薬品性に優れていると言える。
攪拌機を具備したステンレス製オートクレーブに、水硫化ナトリウムの48重量%水溶液を140.3g(1.20モル)、96%水酸化ナトリウムを用いて調製した水酸化ナトリウムの48重量%水溶液125.0g(1.44モル)、N-メチル-2-ピロリドン(NMP)6,150g、及びp-ジクロロベンゼン(p-DCB)180.8g(1.23モル)を仕込んだ。反応容器内を十分に窒素置換した後、窒素ガス下に密封した。
攪拌機を具備したステンレス製オートクレーブに、水硫化ナトリウムの48重量%水溶液を28.06g(0.24モル)、96%水酸化ナトリウムを用いて調製した水酸化ナトリウムの48重量%水溶液25.00g(0.29モル)、N-メチル-2-ピロリドン(NMP)1167g、及びp-ジクロロベンゼン(p-DCB)36.16g(0.25モル)を仕込んだ。反応容器内を十分に窒素置換した後、窒素ガス下に密封した。
参考例3の環状ポリカーボネートは、Macromolecules,24巻,3037頁(1991年)に記載の方法で合成した。
撹拌翼および冷却管を備えた1L三口ナスフラスコに、ジクロロメタン200mL、蒸留水7mL、9.75M水酸化ナトリウム水溶液3mLおよびトリエチルアミン2.4mLを投入した。混合溶液を30℃に保ち、400rpmで撹拌しながらビスフェノールAビス(クロロホルメート)の1.0Mジクロロメタン溶液200mLを、チューブポンプを用いて5.7mL/分の速度で系内に添加した。同時に9.75M水酸化ナトリウム水溶液59mLを滴下ロートを用いて25分かけて滴下し、トリエチルアミン2.4mLを、シリンジポンプを用いて28分かけて滴下した。ビスフェノールAビス(クロロホルメート)を添加終了後、10分間撹拌を行った後、撹拌を停止し有機層を取り出した。有機層を1.0M塩酸で洗浄後、蒸留水で3回洗浄した。有機層を濃縮乾固し、得られた粗生成物を良溶媒としてアセトン、貧溶媒として水を用いた沈殿操作を行い、得られた生成物を80℃で12時間真空乾燥し、白色固体36.2g(収率71.2%)を得た。得られた白色固体は、1H-NMR、FT-IRおよび高速液体クロマトグラフィーによる分析により、2~15量体の環状ポリカーボネートであることが分かった。以下、この白色固体を、参考例3の環状ポリカーボネートと呼ぶ。
参考例4の環状ポリスルホンは、特開平3-088828に記載の方法で合成した。
撹拌翼、冷却管およびディーン・スターク装置を備えた5L三口ナスフラスコに、1,500mLのジメチルスルホキシドおよび800mLのトルエンを加え、混合溶液を撹拌しながら135℃で加熱還流した。加熱還流しながら、ビス(4-フルオロフェニル)スルホンの0.5Mジメチルスルホキシド溶液100mLと、ビス(4-ヒドロキシフェニル)スルホンジナトリウム塩の0.5M水溶液100mLを、0.33mL/分の速度でメタロールポンプを用いて同時に添加した。留出する水/トルエン共沸物をディーン・スターク装置で留去すると同時に、濃度を一定に保つために留出したトルエンを追加しながら、15時間加熱を続けた。反応混合物を室温まで冷却した後、5mLの無水酢酸を添加した。反応溶液を200mLに濃縮した後、約3000mLの水中に滴下し、生じた沈殿物をろ過により回収し、メタノールで数回洗浄した。得られた白色固体を80℃で6時間加熱乾燥した。得られた粗生成物20gを100mLのジメチルホルムアミドに投入し、生じた白色沈殿物をろ別し、メタノールで洗浄した。生成物を80℃の真空乾燥機で12時間乾燥し、白色固体5.2g(収率22.4%)を得た。得られた白色固体は、1H-NMR、FT-IRおよび高速液体クロマトグラフィーによる分析により、4~6量体の環状ポリスルホンであることが分かった。以下、この白色固体を、参考例4の環状ポリスルホンと呼ぶ。
攪拌機を具備したステンレス製オートクレーブに、4,4'-ジフルオロベンゾフェノン10.91g(0.05モル)、ヒドロキノン5.51g(0.05モル)、無水炭酸カリウム6.91g(0.05モル)、およびN-メチル-2-ピロリドン486gを仕込んだ。反応容器内を十分に窒素置換した後、窒素ガス下に密封した。
撹拌翼、減圧アダプターおよびバキュームスターラを備えた試験管に、参考例1の環状ポリフェニレンスルフィド5g(フェニレンスルフィド繰り返し単位46.2mmol)と、重合開始剤としてのナトリウムチオフェノキシド122mg(0.92mmol)を仕込み、系内を窒素置換した後、窒素雰囲気下で電気環状炉を用いて320℃に加熱した。加熱開始5分後、内容物が溶融しているのを確認してから撹拌を開始し、加熱開始から1.5時間反応を行った。反応終了後、試験管を電気環状炉から取り出し、窒素下で室温まで冷却し、片末端にナトリウムチオフェノキシドを有するポリフェニレンスルフィド(A-1)を得た。示差走査型熱量分析装置を用いた融点測定の結果、融点は280℃であった。SEC測定の結果、数平均分子量は14,800、重量平均分子量は27,800、最大ピーク分子量は16,900であった。
撹拌翼、減圧アダプターおよびバキュームスターラを備えた試験管に、参考例2の環状ポリフェニレンスルフィド1g(フェニレンスルフィド繰り返し単位9.16mmol)と、重合開始剤としてのナトリウムチオフェノキシド24.2mg(0.18mmol)を仕込み、系内を窒素置換した後、窒素雰囲気下で電気環状炉を用いて320℃に加熱した。50rpmで撹拌しながら1時間反応を行った。反応終了後、試験管を電気環状炉から取り出し、窒素下で室温まで冷却し、片末端にナトリウムチオフェノキシドを有するポリフェニレンスルフィド(A-2)を得た。示差走査型熱量分析装置を用いた融点測定の結果、融点280℃であった。SECを用いた分子量測定の結果、数平均分子量は15,000、重量平均分子量は28,200、最大ピーク分子量は17,100であった。
重合開始剤としてナトリウムチオフェノキシド12.11mg(0.09mmol)を用いた以外は参考例7と同様の方法で、片末端にナトリウムチオフェノキシドを有するポリフェニレンスルフィド(A-3)を得た。示差走査型熱量分析装置を用いた融点測定の結果、融点282℃であった。SECを用いた分子量測定の結果、数平均分子量は20,300、重量平均分子量は50,600、最大ピーク分子量は23,100であった。
重合開始剤として4,4'-ビフェニルジチオールジナトリウム塩48.5mg(0.19mmol)を用いた以外は参考例6と同様の方法で、両末端にナトリウムチオフェノキシドを有するポリフェニレンスルフィド(A-4)を得た。示差走査型熱量分析装置を用いた融点測定の結果、融点282℃であった。SEC測定の結果、数平均分子量は28,700、重量平均分子量は68,900、最大ピーク分子量は31,100であった。
重合開始剤として4,4'-ビフェニルジチオールジナトリウム塩9.7mg(0.04mmol)を用いた以外は参考例7と同様の方法で、両末端にナトリウムチオフェノキシドを有するポリフェニレンスルフィド(A-5)を得た。 示差走査型熱量分析装置を用いた融点測定の結果、融点は275℃であった。SECを用いた分子量測定の結果、数平均分子量は28,700、重量平均分子量は72,500、最大ピーク分子量は32,000であった。
攪拌機を具備したステンレス製オートクレーブに、水硫化ナトリウムの48重量%水溶液を5.85g(0.05モル)、96%水酸化ナトリウムを用いて調製した水酸化ナトリウムの48重量%水溶液5.21g(0.06モル)、N-メチル-2-ピロリドン(NMP)1080g、および参考例1で合成したクロロホルム抽出残渣(ポリフェニレンスルフィド)108gを仕込み、窒素置換を3回繰り返した後、400rpmで撹拌を行いながら250℃まで45分で昇温した。その後、250℃で1時間反応させて、ポリフェニレンスルフィドの解重合による末端チオラート化を行った。反応液を冷却後、脱イオン水5000g中に投入し、沈殿物をろ別した。沈殿物を80℃の脱イオン水で数回洗浄し、得られた白色固体を80℃で8時間真空乾燥を行い、ポリフェニレンスルフィド(B-1)を得た。示差走査型熱量分析装置を用いた融点測定の結果、融点277℃であった。SEC測定の結果、得られたポリマーの数平均分子量は6,200、重量平均分子量は16,100、最大ピーク分子量は7,500であった。
攪拌機を具備したステンレス製オートクレーブに、水硫化ナトリウムの48重量%水溶液を28.06g(0.24モル)、96%水酸化ナトリウムを用いて調製した水酸化ナトリウムの48重量%水溶液25.00g(0.29モル)、N-メチル-2-ピロリドン(NMP)123g、及びp-ジクロロベンゼン(p-DCB)33.81g(0.23モル)を仕込んだ。反応容器を室温・常圧下にて窒素ガス下に密閉した後、400rpmで撹拌しながら、室温から200℃まで約1時間かけて昇温した。次いで200℃から250℃まで約30分かけて昇温した。250℃で2時間保持した後、室温近傍まで急冷してから内容物を回収した。
撹拌翼を備えた試験管に、ω-ラウロラクタム1g(5.1mmol)と、重合開始剤としてのカリウムエトキシド109mg(0.38mmol)とN-アセチル-ε-カプロラクタム1.0mg(6.44×10-3mmol)とを仕込み、系内を窒素置換した後、窒素雰囲気下で電気環状炉を用いて200℃に加熱した。加熱開始5分後、内容物が溶融しているのを確認してから撹拌を開始し、加熱開始から3.0時間反応を行った。反応終了後、試験管を電気環状炉から取り出し、窒素下で室温まで冷却し、ポリアミド(C-1)を得た。示差走査型熱量分析装置を用いた融点測定の結果、融点は174℃であった。
撹拌翼、減圧アダプターおよびバキュームスターラを備えた試験管に、環状ポリブチレンテレフタレート(Cyclics社製CBT(登録商標)100)1.2gと重合開始剤としてナトリウムチオフェノキシド122mg(0.92mmol)を仕込み、系内を窒素置換した後、窒素雰囲気下で電気環状炉を用いて250℃に加熱した。加熱開始5分後、内容物が溶融しているのを確認してから撹拌を開始し、加熱開始から3.0時間反応を行った。反応終了後、試験管を電気環状炉から取り出し、窒素下で室温まで冷却し、片末端にチオフェノキシドを有するポリエステル(C-2)を得た。示差走査型熱量分析装置を用いた融点測定の結果、融点は228℃であった。
撹拌翼、減圧アダプターおよびバキュームスターラを備えた試験管に、参考例3の環状ポリカーボネート5gと、重合開始剤としてのナトリウムチオフェノキシド109mg(0.44mmol)を仕込み、系内を窒素置換した後、窒素雰囲気下で電気環状炉を用いて300℃に加熱した。加熱開始5分後、内容物が溶融しているのを確認してから撹拌を開始し、加熱開始から0.5時間反応を行った。反応終了後、試験管を電気環状炉から取り出し、窒素下で室温まで冷却し、片末端にチオフェノキシドを有するポリカーボネート(C-3)を得た。示差走査型熱量分析装置を用いた融点測定の結果、融点は251℃であった。
撹拌翼、減圧アダプターおよびバキュームスターラを備えた試験管に、参考例4の環状ポリスルホン5gと、重合開始剤としてのナトリウムチオフェノキシド109mg(0.44mmol)を仕込み、系内を窒素置換した後、窒素雰囲気下で電気環状炉を用いて300℃に加熱した。加熱開始5分後、内容物が溶融しているのを確認してから撹拌を開始し、加熱開始から0.5時間反応を行った。反応終了後、試験管を電気環状炉から取り出し、窒素下で室温まで冷却し、片末端にチオフェノキシドを有するポリスルホン(C-4)を得た。
撹拌翼、減圧アダプターおよびバキュームスターラを備えた試験管に、参考例5で得られた環状ポリフェニレンエーテルエーテルケトン5gと、重合開始剤としてナトリウムチオフェノキシド46mg(0.35mmol)を仕込んだ。系内を窒素置換した後、窒素雰囲気下で電気環状炉を用いて340℃に加熱した。加熱開始5分後、内容物が溶融しているのを確認してから撹拌を開始し加熱開始から1.0時間反応を行った。反応終了後、試験管を電気環状炉から取り出し、窒素下で室温まで冷却し、片末端にチオフェノキシドを有するポリフェニレンエーテルエーテルケトン(C-5)を得た。示差走査型熱量分析装置を用いた融点測定の結果、融点は341℃であった。
攪拌機を具備したステンレス製オートクレーブに、4,4'-ジフルオロベンゾフェノン11.46g(52.5mmol)、ヒドロキノン5.51g(50mmol)、無水炭酸カリウム6.91g(50mmol)、およびN-メチル-2-ピロリドン58gを仕込んだ。反応容器を室温・常圧下にて窒素ガス下に密閉した後、400rpmで撹拌しながら、室温から140℃まで昇温し140℃で1時間保持した。その後、180℃まで昇温して180℃で3時間保持した。その後、230℃にまで昇温して230℃で5時間保持した後、室温近傍まで冷却してから内容物を回収した。
参考例6で得られたポリフェニレンスルフィド成分ブロック(A-1)の凍結粉砕微粉末1gおよびω-ラウロラクタム1g(5.1mmol)を、撹拌翼、バキュームスターラ、窒素導入管および還流管を備えた試験管に入れ、3回窒素置換した。窒素気流下で撹拌しながら、室温から200℃まで30分で昇温し、その後250℃まで60分で昇温した。次いで250℃から30分で300℃まで昇温し、300℃で2時間保持した。撹拌を止め試験管の内容物を水中に吐出し、得られた褐色固体を80℃で12時間真空乾燥し、ポリフェニレンスルフィド-ポリアミドブロック共重合体を得た。得られたポリマーのSEC測定を行った結果、数平均分子量は22,500、最大ピーク分子量は24,800であり、クロマトグラムは単峰性であった。溶融時の均一性を光学顕微鏡により評価した結果、ホモポリマーに起因する粗大な相分離構造は観察されず、均一であることが分かった。ヘキサフルオロイソプロパノールへの可溶成分の重量分率は34重量%であった。結果を表1に示す。
ポリフェニレンスルフィド成分ブロックとして(A-4)を用いた以外は実施例1と同様の方法でポリフェニレンスルフィド-ポリアミドブロック共重合体を得た。得られたポリマーのSEC測定の結果、数平均分子量は35,200、最大ピーク分子量は38,100であり、クロマトグラムは単峰性であった。溶融時の均一性を光学顕微鏡で評価した結果、ホモポリマーに起因する粗大な分離構造は観察されず均一であった。ヘキサフルオロイソプロパノールへの可溶成分の重量分率は39重量%であった。結果を表1に示す。
参考例13で得られたポリアミド成分ブロック(C-1)の凍結粉砕粉末1gおよび参考例1で得られた環状ポリフェニレンスルフィド1g(9.1mmol)を攪拌翼、バキュームスターラ、窒素導入管および還流管を備えた試験管に入れ、3回窒素置換した。窒素気流下で撹拌しながら、室温から200℃まで30分で昇温し、その後250℃まで60分で昇温した。次いで250℃から30分で300℃まで昇温し、300℃で2時間保持した。撹拌を止め試験管の内容物を水中に吐出し、得られた褐色固体を80℃で12時間真空乾燥し、ポリフェニレンスルフィド-ポリアミドブロック共重合体を得た。得られたポリマーのSEC測定を行った結果、数平均分子量は42,600、最大ピーク分子量は44,800であり、クロマトグラムは単峰性であった。溶融時の均一性を光学顕微鏡により評価した結果、ホモポリマーに起因する粗大な相分離構造は観察されず、均一であることが分かった。ヘキサフルオロイソプロパノールへの可溶成分の重量分率は22重量%であった。結果を表1に示す。
ポリフェニレンスルフィド成分ブロックとして(B-1)を用いた以外は実施例1と同様の方法で共重合体の合成を行った。得られたポリマーのSEC測定の結果、数平均分子量は9,500、最大ピーク分子量は11,500であり、クロマトグラムは、残存のホモポリフェニレンスルフィドに起因するピークが存在しており多峰性であった。溶融時の均一性を光学顕微鏡で評価した結果、ホモポリマーに起因する粗大な分離構造が観察され、不均一であった。ヘキサフルオロイソプロパノールへの可溶成分の重量分率は68重量%であった。結果を表1に示す。
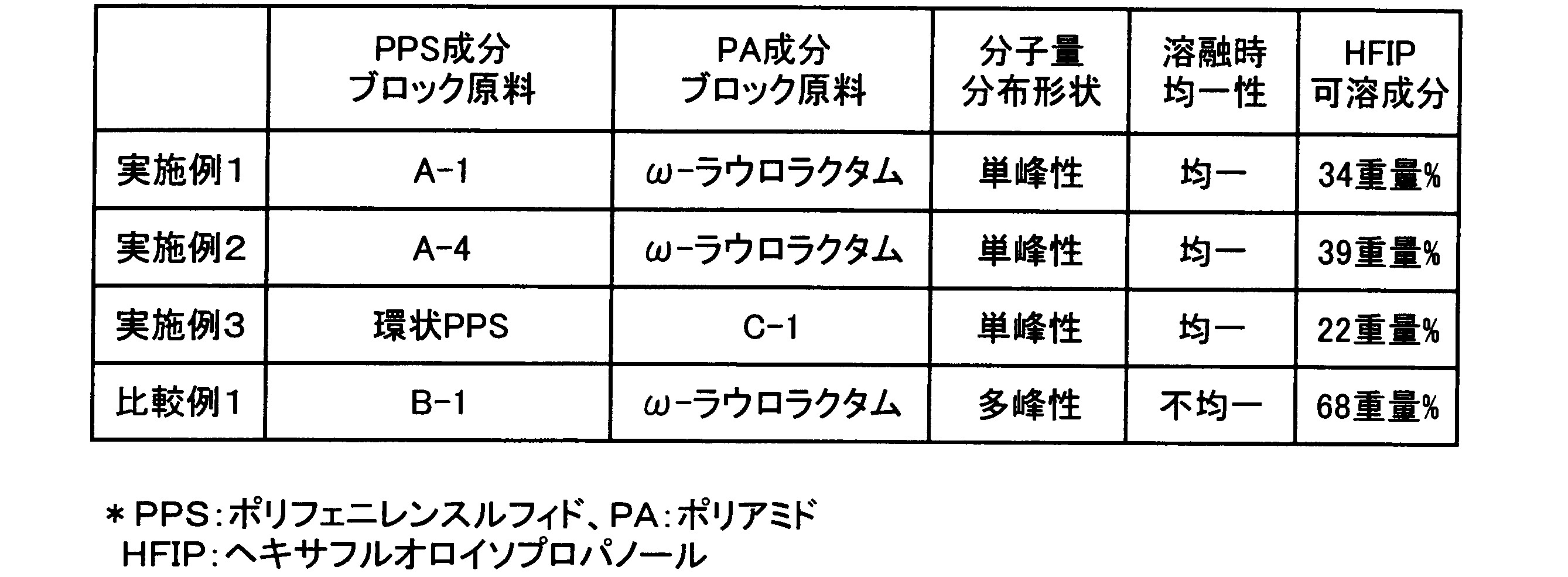
参考例6で得られたポリフェニレンスルフィド成分ブロック(A-1)の凍結粉砕微粉末1gおよび環状ポリブチレンテレフタレート(Cyclics社製CBT(登録商標)100)1.2g(5.45mmol)を、撹拌翼、バキュームスターラおよび窒素導入管を備えた試験管に入れ、3回窒素置換した。窒素気流下で室温から200℃まで30分で昇温し、その後撹拌を行いながら250℃まで60分で昇温した。次いで250℃から15分で280℃まで昇温し、反応を完結させた。試験管の内容物を水中に吐出し、得られた褐色固体を50℃で12時間真空乾燥し、ポリフェニレンスルフィド-ポリブチレンテレフタレート共重合体を得た。得られたポリマーのSEC測定を行った結果、数平均分子量は19,100、最大ピーク分子量は23,400であり、クロマトグラムは単峰性であった。溶融時の均一性を光学顕微鏡により評価した結果、ホモポリマーに起因する粗大な相分離構造は観察されず、均一であった。ヘキサフルオロイソプロパノールへの可溶成分の重量分率は22重量%であった。結果を表2に示す。
ポリフェニレンスルフィド成分ブロックとして(A-4)を用いた以外は実施例4と同様の方法でポリフェニレンスルフィド-ポリブチレンテレフタレートブロック共重合体を得た。得られたポリマーのSEC測定の結果、数平均分子量は34,000、最大ピーク分子は量37,600であり、クロマトグラムは単峰性であった。溶融時の均一性を光学顕微鏡で評価した結果、ホモポリマーに起因する粗大な分離構造は観察されず均一であった。ヘキサフルオロイソプロパノールへの可溶成分の重量分率は30重量%であった。結果を表2に示す。
参考例14で得られたポリブチレンテレフタレート成分ブロック(C-2)の凍結粉砕微粉末1gおよび参考例1の環状ポリフェニレンスルフィド1g(9.16mmol)を、撹拌翼、バキュームスターラおよび窒素導入管を備えた試験管に入れ、3回窒素置換した。窒素気流下で室温から200℃まで30分で昇温し、その後、撹拌を行いながら250℃まで60分で昇温した。次いで250℃から15分で280℃まで昇温し、反応を完結させた。試験管の内容物を水中に吐出し、得られた褐色固体を50℃で12時間真空乾燥し、ポリフェニレンスルフィド-ポリブチレンテレフタレート共重合体を得た。得られたポリマーのSEC測定を行った結果、数平均分子量は23,100、最大ピーク分子量は26,500であり、クロマトグラムは単峰性であった。溶融時の均一性を光学顕微鏡により評価した結果、ホモポリマーに起因する粗大な相分離構造は観察されず、均一であった。ヘキサフルオロイソプロパノールへの可溶成分の重量分率は15重量%であった。結果を表2に示す。
ポリフェニレンスルフィド成分ブロックとして(B-1)を用いた以外は実施例4と同様の方法で共重合体の合成を行った。得られたポリマーのSEC測定の結果、数平均分子量は8,700、最大ピーク分子量は10,300であり、クロマトグラムは、残存のホモポリフェニレンスルフィドに起因するピークが存在しており多峰性であった。また溶融時の均一性を光学顕微鏡で評価した結果、ホモポリマーに起因する粗大な分離構造が観察され、不均一であった。ヘキサフルオロイソプロパノールへの可溶成分の重量分率は61重量%であった。結果を表2に示す。

参考例6で得られたポリフェニレンスルフィド成分ブロック(A-1)の凍結粉砕微粉末1gおよび参考例3で得られた環状ポリカーボネート1.2g(4.72mmol)を、撹拌翼、バキュームスターラおよび窒素導入管を備えた試験管に入れ、3回窒素置換した。窒素気流下で室温から200℃まで30分で昇温し、その後撹拌を行いながら250℃まで60分で昇温した。次いで250℃から45分で300℃まで昇温し、反応を完結させた。試験管の内容物を水中に吐出し、得られた褐色固体を80℃で12時間真空乾燥し、ポリフェニレンスルフィド-ポリカーボネート共重合体を得た。得られたポリマーのSEC測定を行った結果、数平均分子量は22,200、最大ピーク分子量は24,900であり、クロマトグラムは単峰性であった。溶融時の均一性を光学顕微鏡により評価した結果、ホモポリマーに起因する粗大な相分離構造は観察されず、均一であった。ジクロロメタンへの可溶成分の重量分率は12重量%であった。結果を表3に示す。
ポリフェニレンスルフィド成分ブロックとして(A-4)を用いた以外は実施例7と同様の方法でポリフェニレンスルフィド-ポリカーボネートブロック共重合体を得た。得られたポリマーのSEC測定の結果、数平均分子量は35,700、最大ピーク分子量は39,300であり、クロマトグラムは単峰性であった。溶融時の均一性を光学顕微鏡で評価した結果、ホモポリマーに起因する粗大な分離構造は観察されず均一であった。ジクロロメタンへの可溶成分の重量分率は20重量%であった。結果を表3に示す。
参考例15で得られたポリカーボネート成分ブロック(C-3)の凍結粉砕微粉末1.2gおよび参考例1で得られた環状ポリフェニレンスルフィド1g(9.16mmol)を、撹拌翼、バキュームスターラおよび窒素導入管を備えた試験管に入れ、3回窒素置換した。窒素気流下で室温から200℃まで30分で昇温し、その後撹拌を行いながら250℃まで60分で昇温した。次いで250℃から45分で320℃まで昇温し、反応を完結させた。試験管の内容物を水中に吐出し、得られた褐色固体を80℃で12時間真空乾燥し、ポリフェニレンスルフィド-ポリカーボネート共重合体を得た。得られたポリマーのSEC測定を行った結果、数平均分子量は22,200、最大ピーク分子量は24,900であり、クロマトグラムは単峰性であった。溶融時の均一性を光学顕微鏡により評価した結果、ホモポリマーに起因する粗大な相分離構造は観察されず、均一であった。ジクロロメタンへの可溶成分の重量分率は5重量%であった。結果を表3に示す。
ポリフェニレンスルフィド成分ブロックとして(B-1)を用いた以外は実施例7と同様の方法で共重合体の合成を行った。得られたポリマーのSEC測定の結果、数平均分子量は10,600、最大ピーク分子量は11,900であり、クロマトグラムは残存のホモポリフェニレンスルフィドに起因するピークが存在しており多峰性であった。また溶融時の均一性を光学顕微鏡で評価した結果、ホモポリマーに起因する粗大な分離構造が観察され、不均一であった。ジクロロメタンへの可溶成分の重量分率は45重量%であった。結果を表3に示す。

参考例6で得られたポリフェニレンスルフィド成分ブロック(A-1)の凍結粉砕微粉末1gおよび参考例3で得られた環状ポリエーテルスルホン1.2g(5.17mmol)を、撹拌翼、バキュームスターラおよび窒素導入管を備えた試験管に入れ、3回窒素置換した。窒素気流下で室温から280℃まで60分で昇温し、その後撹拌を行いながら320℃まで60分で昇温した。試験管の内容物を水中に吐出し、得られた褐色固体を80℃で12時間真空乾燥し、ポリフェニレンスルフィド-ポリスルホン共重合体を得た。得られたポリマーのSEC測定を行った結果、数平均分子量は21,800、最大ピーク分子量は24,400であり、クロマトグラムは単峰性であった。溶融時の均一性を光学顕微鏡により評価した結果、ホモポリマーに起因する粗大な相分離構造は観察されず、均一であった。N-メチル-2-ピロリドンへの可溶成分は18重量%であった。結果を表4に示す。
ポリフェニレンスルフィド成分ブロックとして(A-4)を用いた以外は実施例10と同様の方法でポリフェニレンスルフィド-ポリスルホンブロック共重合体を得た。得られたポリマーのSEC測定の結果、数平均分子量は34,200、最大ピーク分子量は38,100であり、クロマトグラムは単峰性であった。溶融時の均一性を光学顕微鏡で評価した結果、ホモポリマーに起因する粗大な分離構造は観察されず均一であった。N-メチル-2-ピロリドンへの可溶成分は25重量%であった。結果を表4に示す。
参考例16で得られたポリスルホン成分ブロック(C-4)の凍結粉砕微粉末1.2gおよび参考例1で得られた環状ポリフェニレンスルフィド1g(9.16mmol)を、撹拌翼、バキュームスターラおよび窒素導入管を備えた試験管に入れ、3回窒素置換した。窒素気流下で室温から280℃まで60分で昇温し、その後撹拌を行いながら320℃まで60分で昇温した。試験管の内容物を水中に吐出し、得られた褐色固体を80℃で12時間真空乾燥し、ポリフェニレンスルフィド-ポリスルホン共重合体を得た。得られたポリマーのSEC測定を行った結果、数平均分子量は21,800、最大ピーク分子量は24,400であり、クロマトグラムは単峰性であった。溶融時の均一性を光学顕微鏡により評価した結果、ホモポリマーに起因する粗大な相分離構造は観察されず、均一であった。N-メチル-2-ピロリドンへの可溶成分は10重量%であった。結果を表4に示す。
ポリフェニレンスルフィド成分ブロックとして(B-1)を用いた以外は実施例10と同様の方法で共重合体の合成を行った。得られたポリマーのSEC測定の結果、数平均分子量は9,800、最大ピーク分子量は11,100であり、クロマトグラムは残存のホモポリフェニレンスルフィドに起因するピークが存在しており多峰性であった。溶融時の均一性を光学顕微鏡で評価した結果、ホモポリマーに起因する粗大な分離構造が観察され、不均一であった。N-メチル-2-ピロリドンへの可溶成分は47重量%であった。結果を表4に示す。

参考例7で得られたポリフェニレンスルフィド成分ブロック(A-2)の凍結粉砕微粉末0.92gおよび参考例5で得られた環状ポリフェニレンエーテルエーテルケトン0.08gを、撹拌翼、バキュームスターラおよび窒素導入管を備えたガラス製試験管(24mm径)に仕込んだ。試験管内を室温、常圧下で窒素置換した後、真空ポンプを用いて約0.4kPaに減圧した。約0.4kPaに減圧した約10秒後、320℃に温調した電気環状炉内に試験管を設置し、真空ポンプによって試験管内を約0.4kPaに保ち脱揮をしながら50rpmで撹拌し、90分間加熱した。その後、炉内から試験管を取り出し室温まで冷却し、茶褐色固体のポリマーを得た。
参考例7で得られたポリフェニレンスルフィド成分ブロック(A-2)の凍結粉砕微粉末0.7gと参考例5で得られた環状ポリフェニレンエーテルエーテルケトン組成物0.3gを用いた以外は実施例13と同様の方法で共重合体の合成を行った。
参考例8で得られたポリフェニレンスルフィド成分ブロック(A-3)の凍結粉砕微粉末を用いた以外は実施例13と同様の方法で共重合体の合成を行った。得られたポリマーを1-クロロナフタレン/p-クロロフェノール=6/4(v/v)混合溶媒に220℃で溶解させ、高速液体クロマトグラフィー測定を行った結果、環状ポリフェニレンスルフィドの消費率は97%、環状ポリフェニレンエーテルエーテルケトンの消費率は93%であった。示差走査型熱量分析装置を用いた融点および降温結晶化温度測定の結果、融点は281℃、325℃の2種であり、降温結晶化温度は221℃であった。SEC測定の結果、数平均分子量は40,500、最大ピーク分子量は46,100であり、クロマトグラムは単峰性であった。溶融時の均一性を光学顕微鏡で評価した結果、ホモポリマーに起因する粗大な分離構造は観察されず均一であった。p-クロロフェノールへの可溶成分の重量分率は0重量%であった。結果を表5に示す。
参考例10で得られたポリフェニレンスルフィド成分ブロック(A-5)の凍結粉砕微粉末を用いた以外は実施例13と同様の方法で共重合体の合成を行った。得られたポリマーを1-クロロナフタレン/p-クロロフェノール=6/4(v/v)混合溶媒に220℃で溶解させ、高速液体クロマトグラフィー測定を行った結果、環状ポリフェニレンスルフィドの消費率は99%、環状ポリフェニレンエーテルエーテルケトンの消費率は95%であった。示差走査型熱量分析装置を用いた融点および降温結晶化温度測定の結果、融点は269℃、313℃の2種であり、降温結晶化温度は183℃であった。SEC測定の結果、数平均分子量は31,300、最大ピーク分子量は35,400であり、クロマトグラムは単峰性であった。溶融時の均一性を光学顕微鏡で評価した結果、ホモポリマーに起因する粗大な分離構造は観察されず均一であった。p-クロロフェノールへの可溶成分の重量分率は0重量%であった。結果を表5に示す。
参考例17で得られたポリフェニレンエーテルエーテルケトン成分ブロック(C-5)の凍結粉砕微粉末0.30gおよび参考例2で得られた環状ポリフェニレンスルフィド0.70gを、撹拌翼、バキュームスターラおよび窒素導入管を備えたガラス製試験管(24mm径)に仕込んだ。試験管内を室温、常圧下で窒素置換した後、真空ポンプを用いて約0.4kPaに減圧した。約0.4kPaに減圧した約10秒後、350℃に温調した電気環状炉内に試験管を設置し、真空ポンプによって試験管内を約0.4kPaに保ち脱揮をしながら50rpmで撹拌し、90分間加熱した。その後、炉内から試験管を取り出し室温まで冷却し、茶褐色固体のポリマーを得た。
参考例7で得られたポリフェニレンスルフィド成分ブロック(A-2)の凍結粉砕微粉末0.5gと、参考例5で得られた環状ポリフェニレンエーテルエーテルケトン組成物0.5gとを用いた以外は、実施例13と同様の方法で共重合体の合成を行った。
参考例1の環状ポリフェニレンスルフィド0.3g、参考例5で得られた環状ポリフェニレンエーテルエールケトン組成物0.7g、および、環状ポリフェニレンスルフィドの主要構成単位である式-(Ph-S)-の繰り返し単位からなる化合物に対してナトリウムベンゼンチオラートを5モル%混合した粉末1gを、攪拌機を具備したガラス製試験管(24mm径)に仕込み、試験管内を室温、常圧下で窒素置換した。360℃に温調した電気炉内に試験管を設置し、50rpmで撹拌しながら60分間加熱した後、試験管を取り出し室温まで冷却し、茶褐色固体を得た。
攪拌機を具備したステンレス製オートクレーブに、参考例12で得られたポリフェニレンスルフィド成分ブロック(B-2)51.9g(0.18mmol)、参考例18で得られたポリフェニレンエーテルエーテルケトン成分ブロック(D-1)20.0g(0.18mol)、およびN-メチル-2-ピロリドン50mLを仕込んだ。
攪拌機を具備したステンレス製オートクレーブに、水硫化ナトリウムの48重量%水溶液を28.06g(0.24モル)、96%水酸化ナトリウムを用いて調製した水酸化ナトリウムの48重量%水溶液25.00g(0.29モル)、N-メチル-2ピロリドン120mL、及びp-ジクロロベンゼン33.81g(0.23モル)を仕込んだ。

Claims (15)
- ブロック共重合体であって、前記ブロック共重合体を構成するブロックとして、環状ポリアリーレンスルフィドを原料とするポリアリーレンスルフィド成分ブロックを含み、前記ブロック共重合体を構成する残りのブロックが、環状ポリアリーレンスルフィドとは異なる環状化合物を原料とするブロック共重合体。
- 請求項1に記載のブロック共重合体であって、
前記環状ポリアリーレンスルフィドが、下記一般式(i)

式中、mは4~50の整数であり、前記環状ポリフェニレンスルフィドは、同一のmを有する環状ポリフェニレンスルフィド、または、異なるmを有する複数種類の環状ポリフェニレンスルフィドの混合物であるブロック共重合体。 - 請求項1または2に記載のブロック共重合体であって、
前記ポリアリーレンスルフィド成分ブロックが、下記一般式(ii)

前記残りのブロックが、ポリアミド、ポリエステル、ポリカーボネート、ポリスルホンおよびポリフェニレンエーテルエーテルケトンから選ばれる少なくとも1種のポリマー成分ブロックであり、
式中、xは1以上の整数であるブロック共重合体。 - 請求項3に記載のブロック共重合体であって、
前記残りのブロックは、下記一般式(iii)
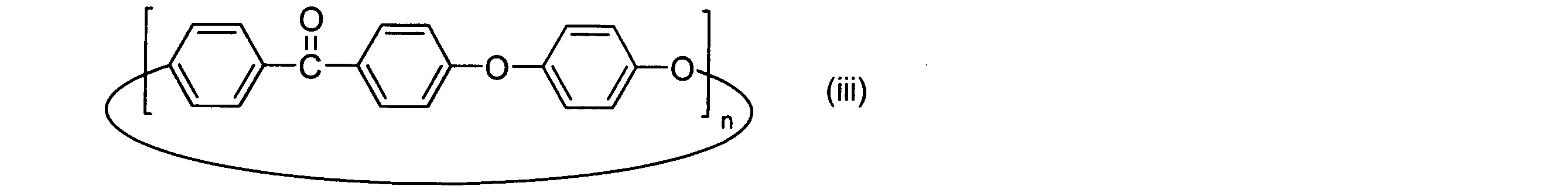
式中、nは2~40の整数であり、前記環状ポリフェニレンエーテルエーテルケトンは、同一のnを有する環状ポリフェニレンエーテルエーテルケトン、または、異なるnを有する複数種類の環状ポリフェニレンエーテルエーテルケトンの混合物であるブロック共重合体。 - 前記一般式(ii)で表されるポリフェニレンスルフィド成分ブロック5~95重量%および、ポリアミド、ポリエステル、ポリカーボネート、ポリスルホンおよびポリフェニレンエーテルエーテルケトンから選ばれる少なくとも1種のポリマー成分ブロック95~5重量%からなる、請求項3に記載のブロック共重合体。
- 請求項5に記載のブロック共重合体であって、
前記一般式(ii)で表されるポリフェニレンスルフィド成分ブロック5~95重量%および、下記一般式(iv)

式中、yは1以上の整数であるブロック共重合体。 - サイズ排除クロマトグラフィー(SEC)により測定される最大ピーク分子量が2,000以上2,000,000未満の範囲にあり、かつ前記範囲で単峰性の分子量分布を有する請求項1~6のいずれか1項に記載のブロック共重合体。
- ブロック共重合体の製造方法であって、(a)環状ポリアリーレンスルフィドを、開始剤存在下で開環重合する工程と、
(b)環状ポリアリーレンスルフィドとは異なる環状化合物を、開始剤存在下で開環重合する工程と、
を備え、
前記(a)工程と前記(b)工程のうちの一方の工程で得られた生成物を、他方の工程の原料である環状化合物と混合して、前記他方の工程を行なう、ブロック共重合体の製造方法。 - 前記(b)工程で用いられる環状化合物が、環状アミド、環状エステル、環状ポリカーボネート、環状ポリスルホンおよび環状ポリフェニレンエーテルエーテルケトンから選ばれるいずれか1種の環状化合物である請求項8に記載のブロック共重合体の製造方法。
- 前記一方の工程は前記(a)工程である、請求項8または9に記載のブロック共重合体の製造方法。
- 前記(b)工程で用いられる環状化合物が、環状ポリフェニレンエーテルエーテルケトンである、請求項10に記載のブロック共重合体の製造方法。
- 前記一方の工程は前記(b)工程である、請求項8または9に記載のブロック共重合体の製造方法。
- 前記(b)工程で用いられる環状化合物が、環状アミド、環状エステル、環状ポリカーボネートおよび環状ポリスルホンから選ばれる少なくとも1種である、請求項12に記載のブロック共重合体の製造方法。
- 前記一方の工程で用いる前記開始剤が金属塩である、請求項8~13いずれか1項に記載のブロック共重合体の製造方法。
- 前記(a)工程および前記(b)工程で進行する前記開環重合がアニオン開環重合である、請求項8~14いずれか1項に記載のブロック共重合体の製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201480012072.6A CN105008427B (zh) | 2013-03-06 | 2014-03-05 | 嵌段共聚物及其制造方法 |
| US14/770,382 US9718928B2 (en) | 2013-03-06 | 2014-03-05 | Block copolymer and production method of the same |
| JP2014513820A JP6252470B2 (ja) | 2013-03-06 | 2014-03-05 | ブロック共重合体の製造方法 |
| EP14759952.6A EP2966107A4 (en) | 2013-03-06 | 2014-03-05 | BLOCK COPOLYMER AND MANUFACTURING METHOD THEREFOR |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013-044005 | 2013-03-06 | ||
| JP2013-044004 | 2013-03-06 | ||
| JP2013044005 | 2013-03-06 | ||
| JP2013044004 | 2013-03-06 | ||
| JP2013204070 | 2013-09-30 | ||
| JP2013-204070 | 2013-09-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014136448A1 true WO2014136448A1 (ja) | 2014-09-12 |
Family
ID=51490981
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/001207 WO2014136448A1 (ja) | 2013-03-06 | 2014-03-05 | ブロック共重合体およびその製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US9718928B2 (ja) |
| EP (1) | EP2966107A4 (ja) |
| JP (1) | JP6252470B2 (ja) |
| CN (1) | CN105008427B (ja) |
| WO (1) | WO2014136448A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015098916A1 (ja) * | 2013-12-25 | 2015-07-02 | 東レ株式会社 | ポリフェニレンスルフィドブロック共重合体、その製造方法およびポリフェニレンスルフィド多孔質体の製造方法 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7126702B2 (ja) * | 2015-05-12 | 2022-08-29 | ア ラ カルト メディア,インコーポレイテッド | 電子機器を遠隔で金銭的対価と引き換えに収集するシステム及び方法 |
| KR102262524B1 (ko) * | 2017-11-16 | 2021-06-07 | 한화솔루션 주식회사 | 폴리에테르케톤케톤 제조방법 및 이에 의해 제조된 폴리에테르케톤케톤 |
| US11440997B2 (en) | 2018-01-31 | 2022-09-13 | Toray Industries, Inc. | Polyarylene sulfide copolymer and method of producing the same |
| WO2020150970A1 (zh) * | 2019-01-24 | 2020-07-30 | 大连理工大学 | 一种碱助催化烷基硫醚插入双(三)硫酯端基聚合物制备嵌段共聚物的聚合方法 |
| US20220267542A1 (en) * | 2019-07-31 | 2022-08-25 | Toray Industries, Inc. | Fiber-reinforced polyarylene sulfide copolymer composite substrate, method of manufacturing same, and molded article including same |
Citations (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS61225218A (ja) | 1985-03-30 | 1986-10-07 | Dainippon Ink & Chem Inc | ブロツク共重合体 |
| JPS6445433A (en) | 1987-06-01 | 1989-02-17 | Gen Electric | Polyarylenesulfide block copolymer, manufacture and use |
| JPH02133428A (ja) | 1988-11-14 | 1990-05-22 | Toray Ind Inc | ブロック共重合体の製造方法 |
| JPH02228325A (ja) | 1989-01-13 | 1990-09-11 | Bayer Ag | ポリエーテルエーテルケトン―ポリアリ―レンスルフイドブロツクコポリマー |
| JPH02229857A (ja) | 1988-11-15 | 1990-09-12 | Kureha Chem Ind Co Ltd | 耐熱性熱可塑性樹脂組成物 |
| JPH02235929A (ja) | 1989-03-09 | 1990-09-18 | Diafoil Co Ltd | ポリフェニレンスフィド共重合体フィルム |
| JPH038828A (ja) | 1989-05-31 | 1991-01-16 | Hagiwara Kogyo Kk | 熱可塑性樹脂多層モノフィラメントとその製造方法 |
| JPH0388828A (ja) | 1989-08-14 | 1991-04-15 | Dow Chem Co:The | 環状ポリ(アリールエーテル)オリゴマー、その製造法、および環状ポリ(アリールエーテル)オリゴマーの重合法 |
| JPH04505182A (ja) | 1990-02-08 | 1992-09-10 | イーストマン コダック カンパニー | 連鎖移動によるビニル及びアリーレンスルフィドセグメントを含むブロックコポリマー |
| JPH04311725A (ja) | 1991-04-09 | 1992-11-04 | Kureha Chem Ind Co Ltd | アリーレンチオエーテル系オリゴマーおよび共重合体の製造方法 |
| JPH05105757A (ja) * | 1991-10-15 | 1993-04-27 | Tosoh Corp | アミノ基含有ポリアリ−レンスルフイドの製造方法 |
| JPH05163349A (ja) | 1991-08-06 | 1993-06-29 | Tosoh Corp | ポリアリーレンスルフィドおよびその製造方法 |
| JPH05295346A (ja) | 1991-04-30 | 1993-11-09 | Kureha Chem Ind Co Ltd | ポリアリーレンスルフィドの接着用樹脂及び接着剤 |
| JPH05301962A (ja) | 1992-04-24 | 1993-11-16 | Tosoh Corp | 新規なポリアリーレンスルフィドの製造方法 |
| JPH11222527A (ja) | 1997-09-24 | 1999-08-17 | Ticona Gmbh | ポリアリーレンスルフィド及び芳香族ポリエステルからのコポリマー |
| JP2003082081A (ja) | 2001-09-13 | 2003-03-19 | Toray Ind Inc | ポリエステル環状オリゴマーの製造方法、およびポリエステルの製造方法 |
| JP2004168834A (ja) | 2002-11-18 | 2004-06-17 | Idemitsu Petrochem Co Ltd | ポリアリーレンスルフィド/ポリエーテルブロック共重合体及びその製造方法、並びに樹脂組成物 |
| JP2004182753A (ja) * | 2002-11-29 | 2004-07-02 | Dainippon Ink & Chem Inc | ポリアリーレンスルフィド系共重合体の製造方法 |
| WO2007034800A1 (ja) | 2005-09-22 | 2007-03-29 | Toray Industries, Inc. | ポリアリーレンスルフィドおよびその製造方法 |
| WO2008105438A1 (ja) | 2007-02-28 | 2008-09-04 | Toray Industries, Inc. | 環式ポリアリーレンスルフィドの製造方法 |
| WO2011081080A1 (ja) | 2009-12-28 | 2011-07-07 | 東レ株式会社 | 環式ポリフェニレンエーテルエーテルケトン組成物およびその製造方法 |
| WO2012057319A1 (ja) * | 2010-10-29 | 2012-05-03 | 東レ株式会社 | ポリアリーレンスルフィドの製造方法およびポリアリーレンスルフィド |
| WO2012081455A1 (ja) * | 2010-12-13 | 2012-06-21 | 東レ株式会社 | 樹脂組成物ならびにそれを用いた複合硬化物およびその製造方法 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4532502A (en) | 1980-04-11 | 1985-07-30 | Sony Corporation | Apparatus for selectively transferring data between registers |
| US4678831A (en) | 1985-03-30 | 1987-07-07 | Dainippon Ink And Chemicals, Inc. | Block copolymers and resin compositions |
| EP0527055B1 (en) | 1991-08-06 | 1997-11-05 | Tosoh Corporation | Process for the preparation of polyarylene sulfide |
| JP5597908B2 (ja) * | 2007-03-22 | 2014-10-01 | 東レ株式会社 | 成形材料 |
-
2014
- 2014-03-05 JP JP2014513820A patent/JP6252470B2/ja not_active Expired - Fee Related
- 2014-03-05 WO PCT/JP2014/001207 patent/WO2014136448A1/ja active Application Filing
- 2014-03-05 EP EP14759952.6A patent/EP2966107A4/en not_active Withdrawn
- 2014-03-05 US US14/770,382 patent/US9718928B2/en not_active Expired - Fee Related
- 2014-03-05 CN CN201480012072.6A patent/CN105008427B/zh not_active Expired - Fee Related
Patent Citations (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS61225218A (ja) | 1985-03-30 | 1986-10-07 | Dainippon Ink & Chem Inc | ブロツク共重合体 |
| JPS6445433A (en) | 1987-06-01 | 1989-02-17 | Gen Electric | Polyarylenesulfide block copolymer, manufacture and use |
| JPH02133428A (ja) | 1988-11-14 | 1990-05-22 | Toray Ind Inc | ブロック共重合体の製造方法 |
| JPH02229857A (ja) | 1988-11-15 | 1990-09-12 | Kureha Chem Ind Co Ltd | 耐熱性熱可塑性樹脂組成物 |
| JPH02228325A (ja) | 1989-01-13 | 1990-09-11 | Bayer Ag | ポリエーテルエーテルケトン―ポリアリ―レンスルフイドブロツクコポリマー |
| JPH02235929A (ja) | 1989-03-09 | 1990-09-18 | Diafoil Co Ltd | ポリフェニレンスフィド共重合体フィルム |
| JPH038828A (ja) | 1989-05-31 | 1991-01-16 | Hagiwara Kogyo Kk | 熱可塑性樹脂多層モノフィラメントとその製造方法 |
| JPH0388828A (ja) | 1989-08-14 | 1991-04-15 | Dow Chem Co:The | 環状ポリ(アリールエーテル)オリゴマー、その製造法、および環状ポリ(アリールエーテル)オリゴマーの重合法 |
| JPH04505182A (ja) | 1990-02-08 | 1992-09-10 | イーストマン コダック カンパニー | 連鎖移動によるビニル及びアリーレンスルフィドセグメントを含むブロックコポリマー |
| JPH04311725A (ja) | 1991-04-09 | 1992-11-04 | Kureha Chem Ind Co Ltd | アリーレンチオエーテル系オリゴマーおよび共重合体の製造方法 |
| JPH05295346A (ja) | 1991-04-30 | 1993-11-09 | Kureha Chem Ind Co Ltd | ポリアリーレンスルフィドの接着用樹脂及び接着剤 |
| JPH05163349A (ja) | 1991-08-06 | 1993-06-29 | Tosoh Corp | ポリアリーレンスルフィドおよびその製造方法 |
| JPH05105757A (ja) * | 1991-10-15 | 1993-04-27 | Tosoh Corp | アミノ基含有ポリアリ−レンスルフイドの製造方法 |
| JPH05301962A (ja) | 1992-04-24 | 1993-11-16 | Tosoh Corp | 新規なポリアリーレンスルフィドの製造方法 |
| JPH11222527A (ja) | 1997-09-24 | 1999-08-17 | Ticona Gmbh | ポリアリーレンスルフィド及び芳香族ポリエステルからのコポリマー |
| JP2003082081A (ja) | 2001-09-13 | 2003-03-19 | Toray Ind Inc | ポリエステル環状オリゴマーの製造方法、およびポリエステルの製造方法 |
| JP2004168834A (ja) | 2002-11-18 | 2004-06-17 | Idemitsu Petrochem Co Ltd | ポリアリーレンスルフィド/ポリエーテルブロック共重合体及びその製造方法、並びに樹脂組成物 |
| JP2004182753A (ja) * | 2002-11-29 | 2004-07-02 | Dainippon Ink & Chem Inc | ポリアリーレンスルフィド系共重合体の製造方法 |
| WO2007034800A1 (ja) | 2005-09-22 | 2007-03-29 | Toray Industries, Inc. | ポリアリーレンスルフィドおよびその製造方法 |
| WO2008105438A1 (ja) | 2007-02-28 | 2008-09-04 | Toray Industries, Inc. | 環式ポリアリーレンスルフィドの製造方法 |
| WO2011081080A1 (ja) | 2009-12-28 | 2011-07-07 | 東レ株式会社 | 環式ポリフェニレンエーテルエーテルケトン組成物およびその製造方法 |
| WO2012057319A1 (ja) * | 2010-10-29 | 2012-05-03 | 東レ株式会社 | ポリアリーレンスルフィドの製造方法およびポリアリーレンスルフィド |
| WO2012081455A1 (ja) * | 2010-12-13 | 2012-06-21 | 東レ株式会社 | 樹脂組成物ならびにそれを用いた複合硬化物およびその製造方法 |
Non-Patent Citations (5)
| Title |
|---|
| JOURNAL OF APPLIED POLYMER SCIENCE, vol. 61, 1996, pages 1607 - 1614 |
| JOURNAL OF POLYMER SCIENCE PART A: POLYMER CHEMISTRY, vol. 36, 1998, pages 2707 - 2713 |
| MACROMOLECULES, vol. 24, 1991, pages 3035 - 3044 |
| MACROMOLECULES, vol. 24, 1991, pages 3037 |
| See also references of EP2966107A4 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015098916A1 (ja) * | 2013-12-25 | 2015-07-02 | 東レ株式会社 | ポリフェニレンスルフィドブロック共重合体、その製造方法およびポリフェニレンスルフィド多孔質体の製造方法 |
| US10040910B2 (en) | 2013-12-25 | 2018-08-07 | Toray Industries, Inc. | Polyphenylene sulfide block copolymer, method for manufacturing same, and method for manufacturing polyphenylene sulfide porous body |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2966107A4 (en) | 2016-08-17 |
| US20160009868A1 (en) | 2016-01-14 |
| JPWO2014136448A1 (ja) | 2017-02-09 |
| EP2966107A1 (en) | 2016-01-13 |
| US9718928B2 (en) | 2017-08-01 |
| CN105008427A (zh) | 2015-10-28 |
| JP6252470B2 (ja) | 2017-12-27 |
| CN105008427B (zh) | 2017-03-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6252470B2 (ja) | ブロック共重合体の製造方法 | |
| CN102652133B (zh) | 环式聚苯醚醚酮组合物及其制造方法 | |
| TWI549993B (zh) | 環狀聚伸芳基硫醚之製造方法 | |
| EP2634206A1 (en) | Polyarylene sulfide production method and polyarylene sulfide | |
| JP2007530763A (ja) | ブロックコポリマーの調製の改良方法及びそれから調製されるブロックコポリマー | |
| US5869599A (en) | Free radical ring opening for polymerization of cyclic oligomers containing an aromatic sulfide linkage | |
| JP5633655B1 (ja) | 環式ポリフェニレンエーテルエーテルケトン組成物および線状ポリフェニレンエーテルエーテルケトンの製造方法、ポリフェニレンエーテルエーテルケトンの製造方法 | |
| JP2019119810A (ja) | ポリフェニレンスルフィド樹脂組成物およびその製造方法 | |
| JP5115371B2 (ja) | 環式ポリアリーレンスルフィド混合物およびその製造方法 | |
| JP5966559B2 (ja) | ポリフェニレンエーテルエーテルケトンおよび環式ポリフェニレンエーテルエーテルケトン組成物の回収方法 | |
| JP2015124225A (ja) | ポリフェニレンスルフィドブロック共重合体の製造方法 | |
| JP6024458B2 (ja) | ポリフェニレンエーテルエーテルケトンの製造方法 | |
| JP2014196459A (ja) | ブロック共重合体およびその製造方法 | |
| US10640645B2 (en) | Polyisoindolinone compositions, methods of manufacture, and compositions and articles formed therefrom | |
| US20210189064A1 (en) | Polyisoindolinones, methods of manufacture, and compositions and articles formed therefrom | |
| JP2018024851A (ja) | ポリアリーレンスルフィドおよびその製造方法 | |
| JP6221311B2 (ja) | ポリフェニレンエーテルエーテルケトンの製造方法 | |
| JP5316727B1 (ja) | 環式ポリフェニレンエーテルエーテルケトン組成物の回収方法およびそれを用いたポリフェニレンエーテルエーテルケトンの製造方法 | |
| Chen et al. | The preparation and characterization of novel cocylic (arylene disulfide) oligomers | |
| JP6354339B2 (ja) | ポリアリーレンスルフィドの製造方法 | |
| JP2017105981A (ja) | ポリアリーレンスルフィドの製造方法 | |
| JP2020037672A (ja) | 環状ポリフェニレンエーテルエーテルケトン組成物およびそれを転化してなるポリフェニレンエーテルエーテルケトンの製造方法 | |
| JP2013010345A (ja) | ポリフェニレンエーテルエーテルケトンの回転成形方法とその成形体 | |
| JP5867155B2 (ja) | 環式ポリフェニレンエーテルエーテルケトン組成物の回収方法 | |
| JP2015105303A (ja) | ポリアリーレンスルフィドの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2014513820 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14759952 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014759952 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14770382 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |